





























































































































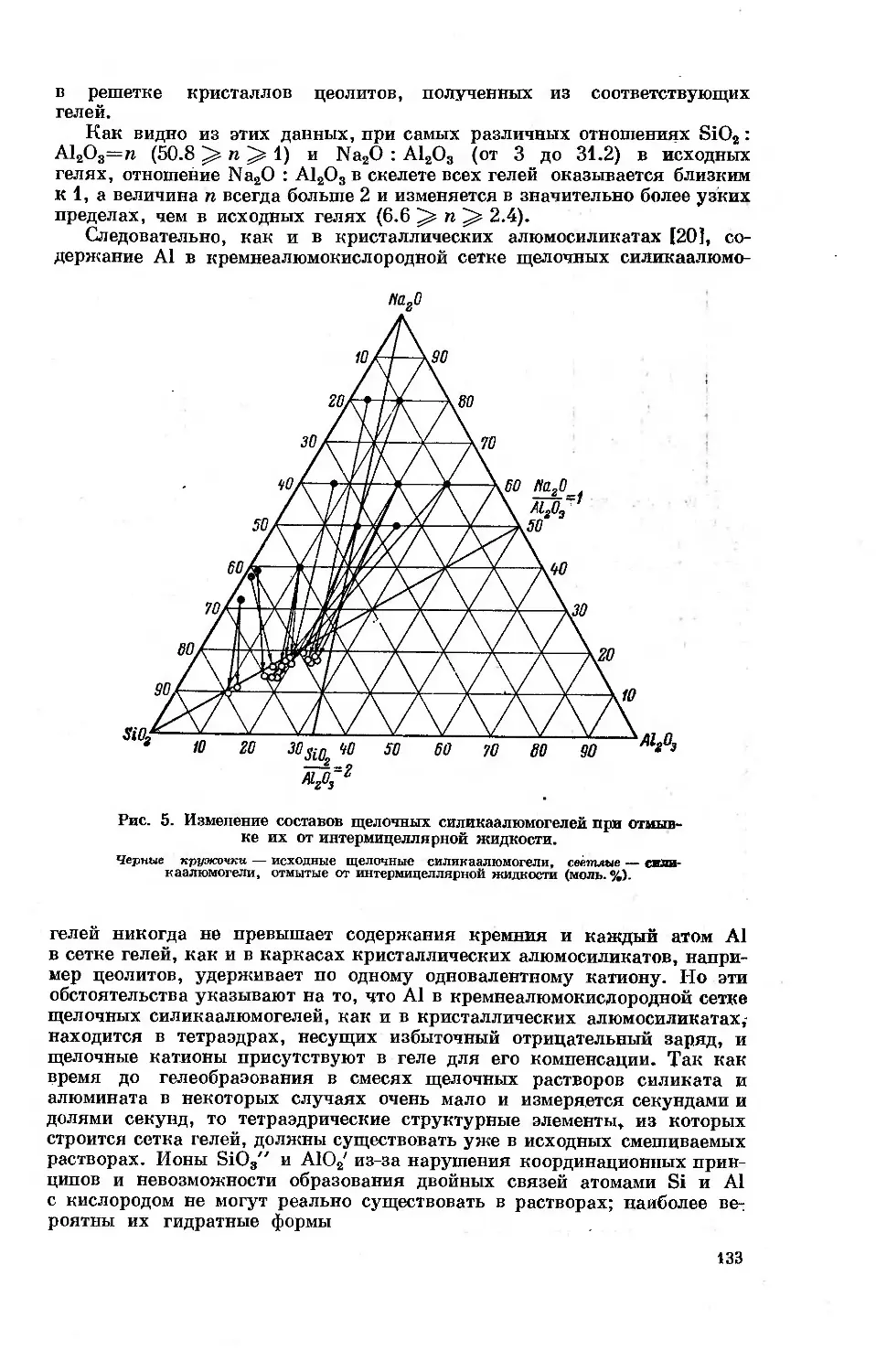





















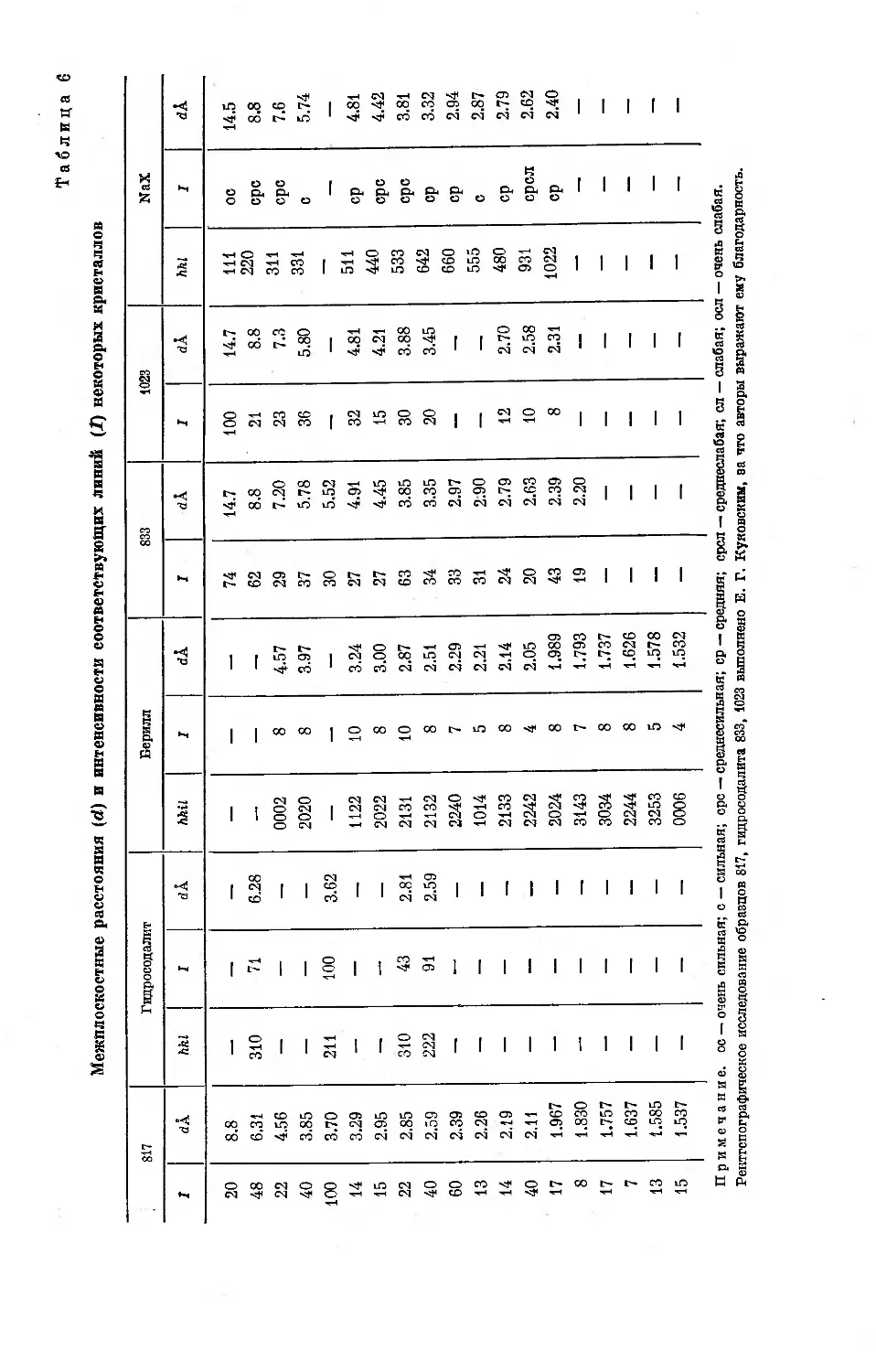







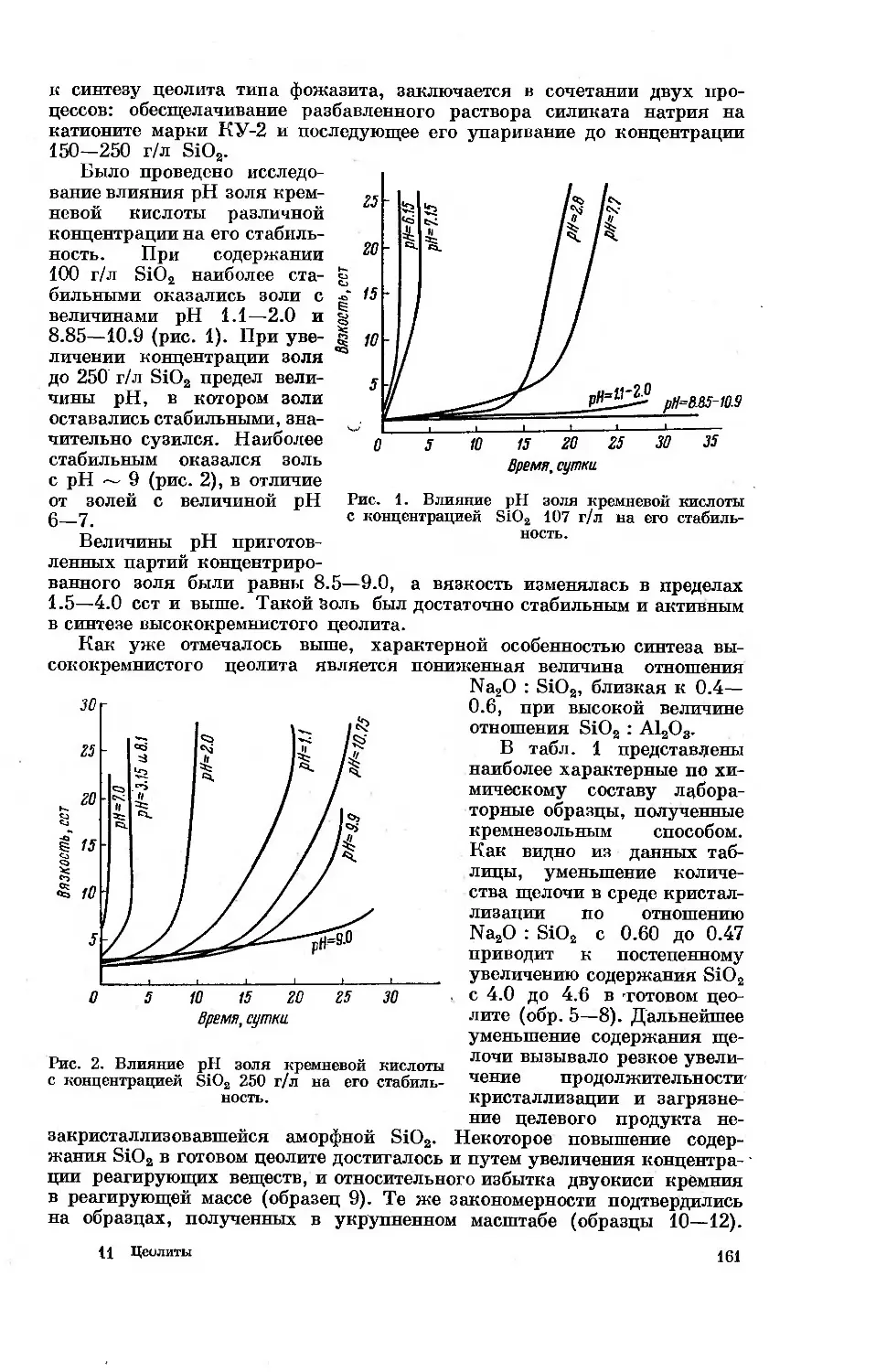



































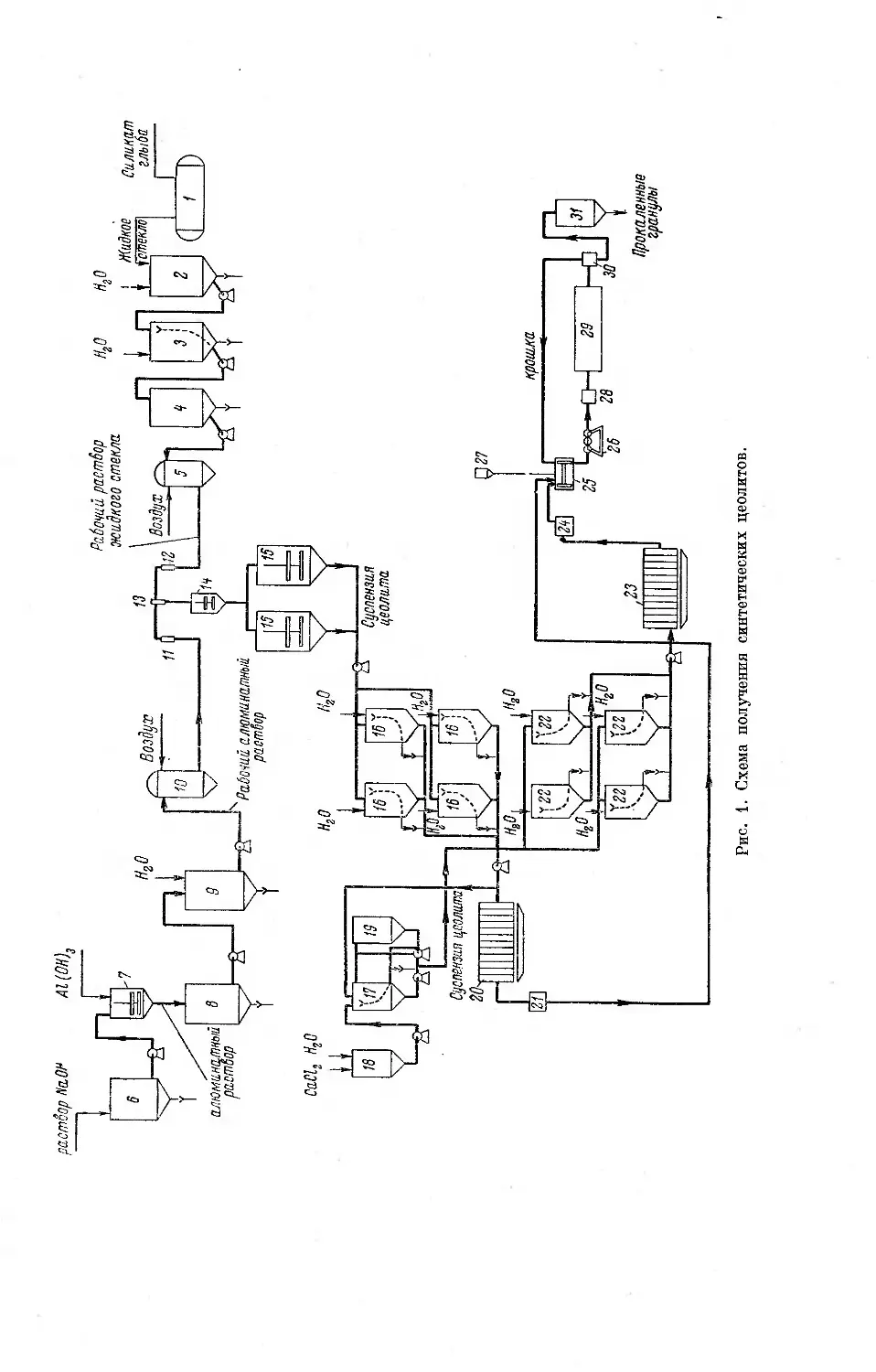



























































































































































































Автор: Дубинин М.М. Плаченов Т.Г.
Теги: химия народное хозяйство общая химия сборник статей издательство наука техническая химия цеолиты адсорбенты
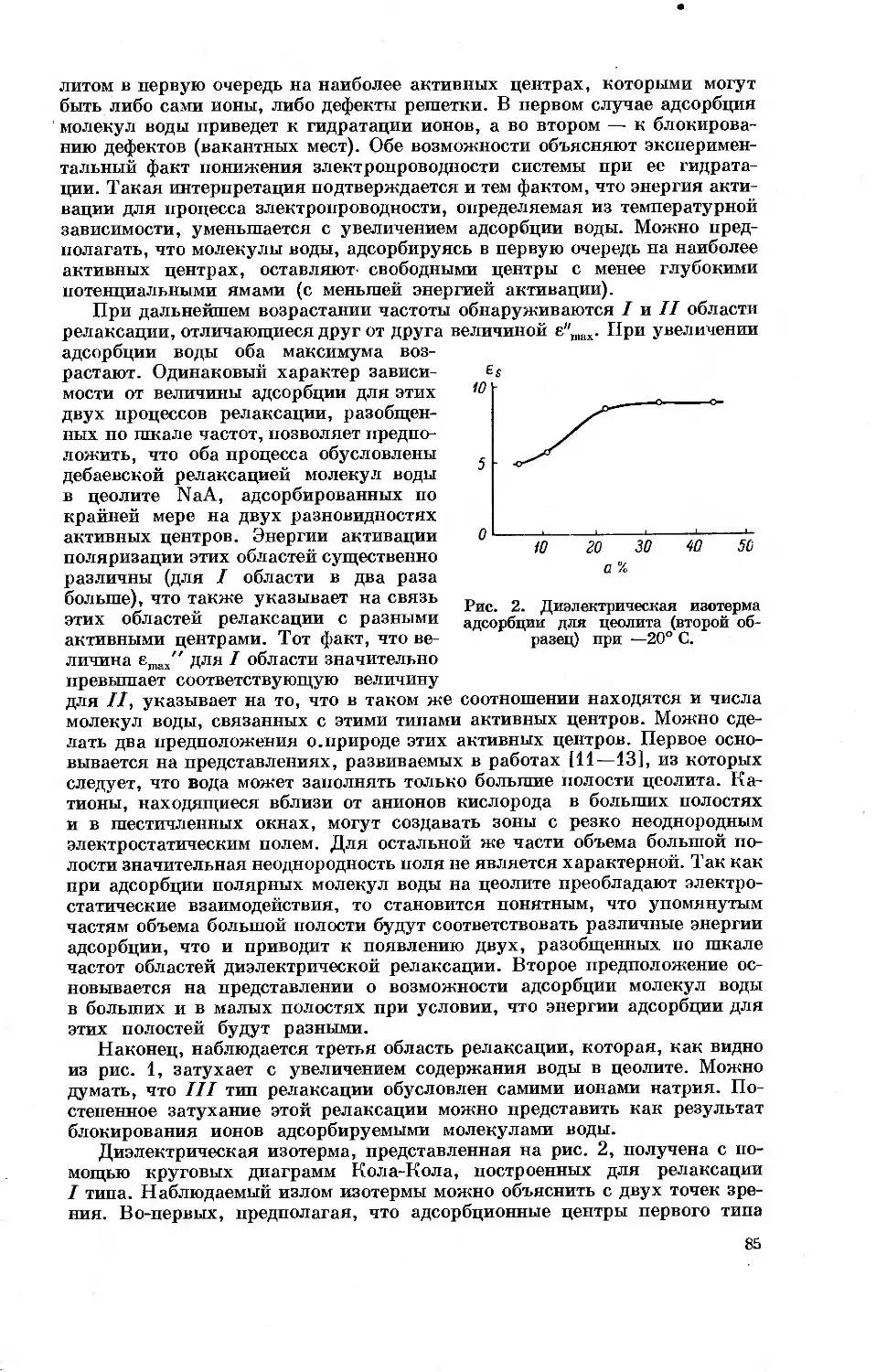
Год: 1965




ИХ СИНТЕЗ,
СВОЙСТВА
И ПРИМЕНЕНИЕ
Больше химической литературы на
vk.com/chemzone
More chemistry books you can find on
vk.com/chemzone
vk.com/chemzone
АКАДЕМИЯ НАУК СССР
ОТДЕЛЕНИЕ ОБЩЕЙ И ТЕХНИЧЕСКОЙ ХИМИИ
НАУЧНЫЙ СОВЕТ ПО СИНТЕЗУ, ИЗУЧЕНИЮ И ПРИМЕНЕНИЮ
АДСОРБЕНТОВ
ЦЕОЛИТЫ, ИХ СИНТЕЗ,
СВОЙСТВА и ПРИМЕНЕНИЕ
(МАТЕРИАЛЫ II ВСЕСОЮЗНОГО
СОВЕЩАНИЯ ПО ЦЕОЛИТАМ)
ИЗДАТЕЛЬСТВО «НАУ К А»
Москва — 1965 — Ленинград
АННОТАЦИЯ
Сборник включает материалы II Всесоюзного совещания
по цеолитам, происходившего в Ленинграде в 1964 г. В статьях
сборника освещаются вопросы теории адсорбции на цеолитах,
приводятся новые данные по синтезу цеолитов, исследованию
их свойств и применению в народном хозяйстве.
Книга рассчитана на широкий круг научных и инженерно-
технических работников, занимающихся вопросами синтеза и
использования адсорбентов и катализаторов в различных отраслях
народного хозяйства.
Отв. редакторы:
академик М. М. Д У Б И Н И Н,
доктор техн, наук, пр оф. Т. Г. ПЛАЧЕНОВ
2-5-4
201-65 (доп.)
ПРЕДИСЛОВИЕ
За последние годы в Советском Союзе проведены значительные работы
в области синтеза цеолитов, изучения их свойств и применения в народ-
ном хозяйстве. 'С целью широкого обсуждения достигнутых результатов
и координации дальнейших исследований в этой области Научный совет
по адсорбентам при Отделении общей и технической химии Академии
наук СССР признал целесообразным провести второе Всесоюзное совеща-
ние, посвященное проблеме синтетических цеолитов; это совещание состоя-
лось в Ленинграде 12—15 мая 1964 г. и подвело итоги большой работы, вы-
полненной советскими учеными, инженерами и рабочими за три года, про-
шедшие после первого совещания, проведенного в мае 1961 г.
На втором совещании были рассмотрены результаты исследований,
посвященных вопросам теории адсорбции на цеолитах, выяснению их
строения и процессов, протекающих при адсорбции на цеолитах паров и
газов. Были также рассмотрены результаты исследований по синтезу цеоли-
тов, созданию рациональной технологии их промышленного получения
и изучению их свойств. Большое внимание было уделено результатам
работ по применению синтетических цеолитов для разделения и тонкой
очистки углеводородов и других газов. Обсуждались результаты иссле-
дований по применению синтетических цеолитов в некоторых каталити-
ческих реакциях.
Широкое представительство научно-исследовательских организаций
СССР на втором Всесоюзном совещании и разнообразный круг рассматри-
ваемых совещанием вопросов свидетельствуют о большом размахе, который
получили в Советском Союзе работы по синтезу, исследованию и примене-
нию синтетических цеолитов — этих высокоэффективных сорбентов йо-
вого типа, обладающих исключительно высокой избирательной способ-
ностью и ценными каталитическими свойствами. Не вызывает сомнения,
что цеолиты найдут широкое применение при решении многочисленных
задач, поставленных нашей партией перед страной в 1963 г. декабрьским
Пленумом ЦК КПСС в связи с развитием большой и малой химии.
На совещании были представлены доклады об исследованиях, выпол-
ненных в институтах академий наук (Академии наук СССР, Украинской
ССР, Белорусской ССР, Грузинской ССР, Азербайджанской ССР), в ряде
научно-исследовательских институтов химической, нефтяной и других
отраслей промышленности и в высших учебных заведениях (Московский
государственный университет, Ленинградский технологический инсти-
тут им. Ленсовета, Московский химико-технологический институт
им. Д. И. Менделеева, Дальневосточный государственный университет, Ле-
нинградский технологический институт холодильной промышленности, Во-
ронежский сельскохозяйственный институт и др.). В совещании принимали
участие также ученые из ГДР, ЧССР, Венгерской Народной Республики.
3
Доклады в сборнике распределены по трем разделам: 1) «Теоретические
вопросы адсорбции на цеолитах и исследование их свойств»; 2) «Получение
цеолитов»; 3) «Применение цеолитов». Следует подчеркнуть, что отнесение
некоторых докладов к тому или иному разделу является несколько услов-
ным в связи с их комплексным характером. При подготовке к печати
доклады во многих случаях подвергались значительным сокращениям.
Несмотря на то, что в докладах изложены в основном результаты ори-
гинальных исследований, материал сборника дает достаточно полное
представление о проблеме в целом. Настоящий сборник может представить
интерес не только для научных работников, непосредственно работающих
в области получения и применения цеолитов, но и для широкого круга
химиков.
Сборник трудов совещания подготовлен к изданию выделенной Науч-
ным советом по адсорбентам редколлегией в составе академика М. М. Ду-
бинина (отв. редактор), профессора Т. Г. Плаченова (отв. редактор), до-
цента Г. М. Белоцерковского и мл. научного сотрудника А. Л. Аранович.
Зам. председателя Научного совета по адсорбентам
профессор Т. Г. ПЛАЧЕНОВ
ТЕОРЕТИЧЕСКИЕ ВОПРОСЫ АДСОРБЦИИ
НА ЦЕОЛИТАХ И ИССЛЕДОВАНИЕ ИХ СВОЙСТВ
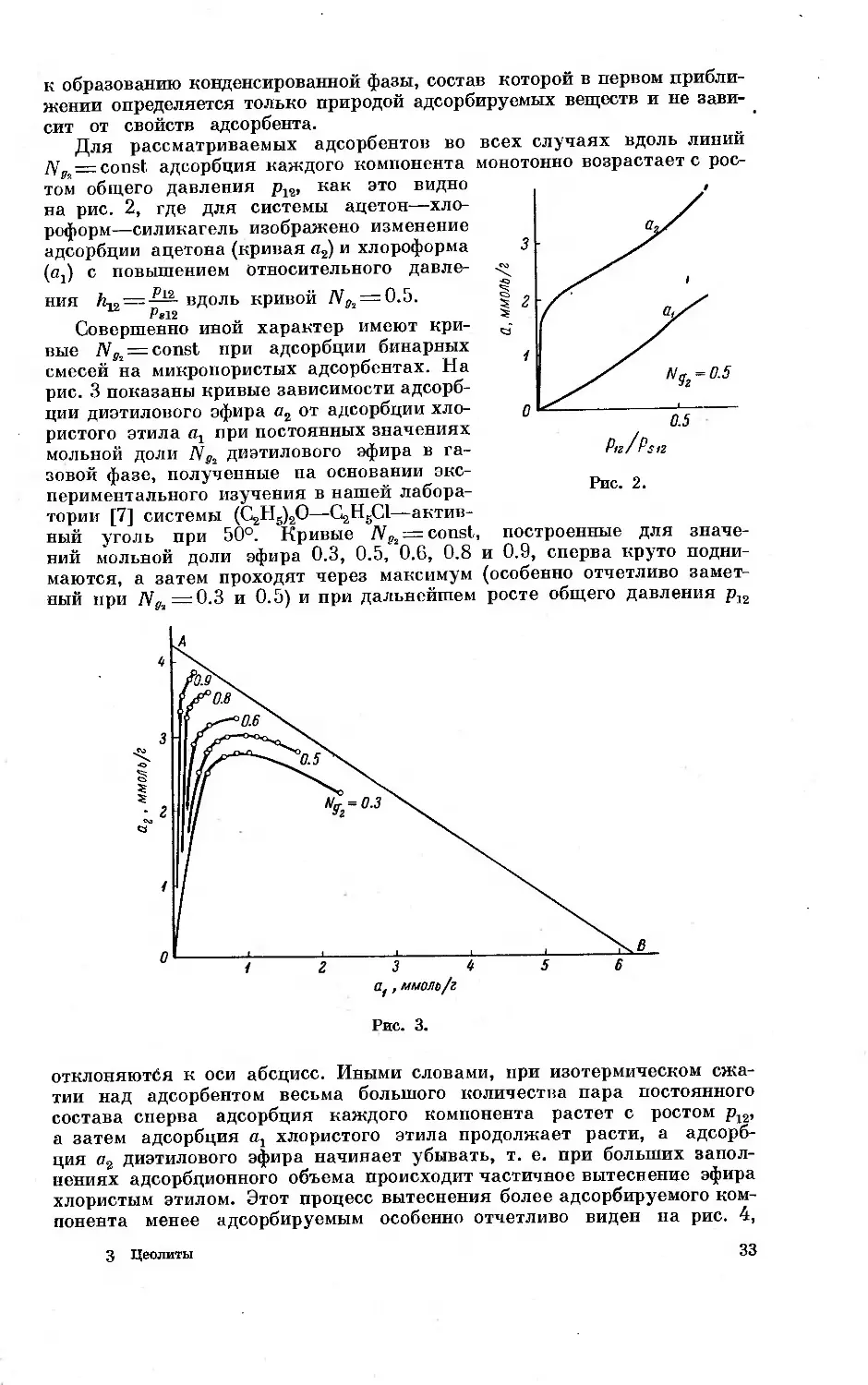
ОСОБЕННОСТИ АДСОРБЦИИ ПАРОВ РАЗЛИЧНЫХ ВЕЩЕСТВ
НА ЦЕОЛИТАХ КАК НА МИКРОПОРИСТЫХ АДСОРБЕНТАХ
М. М. Дубинин
В современной сорбционной технике для глубокой осушки, тонкой
очистки й разделения веществ в газовых и жидких фазах и рекуперации
паров летучих растворителей применяются пористые адсорбенты. По
признаку размера пор, который может быть выражен отношением эффек-
тивного радиуса пор гп к среднему радиусу адсорбируемых молекул г,
пористые адсорбенты могут быть разделены на две группы: относительно
крупнопористые адсорбенты и микропористые адсорбенты.
Для адсорбентов первой группы относительные размеры пор, l = rjr,
обычно больше 7—10. К адсорбентам этой группы принадлежат силика-
гели, крупнопористые стекла, обезвоженные гели гидратов окислов ме-
таллов и большинство природных адсорбентов. Характерной особенностью
непористых или относительно крупнопористых адсорбентов является фи-
зическая реальность понятия поверхности адсорбента. Первичный ад-
сорбционный процесс в случае паров обычно сводится к заполнению по-
верхности с образованием адсорбционных слоев. Благодаря значительному
размеру пор собственно адсорбция паров на относительно крупнопористых
адсорбентах в большинстве случаев может рассматриваться как про-
исходящая в неограниченном стенками пор адсорбционном пространстве.
Объемное заполнение пор этой разновидности адсорбентов происходит
в результате капиллярной конденсации паров, т. е. вторичного сорбцион-
ного процесса.
Для микропористых адсорбентов относительные размеры пор или по-
лостей I выражаются немногими единицами и обычно не превышают трех.
К этой группе адсорбентов принадлежат активные угли, цеолиты и мел-
копористые стекла. Для микропористых адсорбентов характерно влияние
размеров микропор на энергию адсорбционного взаимодействия, в осо-
бенности обязанного проявлению дисперсионных сил. Адсорбция паров
на этой разновидности адсорбентов происходит в ограниченном объеме
адсорбционного пространства, и его заполнение является результатом
протекания первичного адсорбционного процесса. Представление о по-
верхности микропористых адсорбентов теряет физический смысл. Допол-
нительная особенность микропористых адсорбентов заключается в про-
явлении молекулярно-ситового действия в тех случаях, когда размеры
входов в микропоры или полости адсорбентов существенно меньше раз-
меров самих микропор. Более детальное рассмотрение основных особен-
ностей адсорбции паров микропористыми адсорбентами и является пред-
метом настоящего доклада.
5
Адсорбционные свойства непористых и микропористых адсорбентов
Наиболее детально изучены различия в адсорбционных свойствах и
энергии адсорбции для микропористых и непористых углеродных адсор-
бентов. В этом случае определяющая роль в адсорбционных взаимодей-
ствиях принадлежит дисперсионным силам, для проявления которых
в микропористых системах характерно существенное увеличение энергии
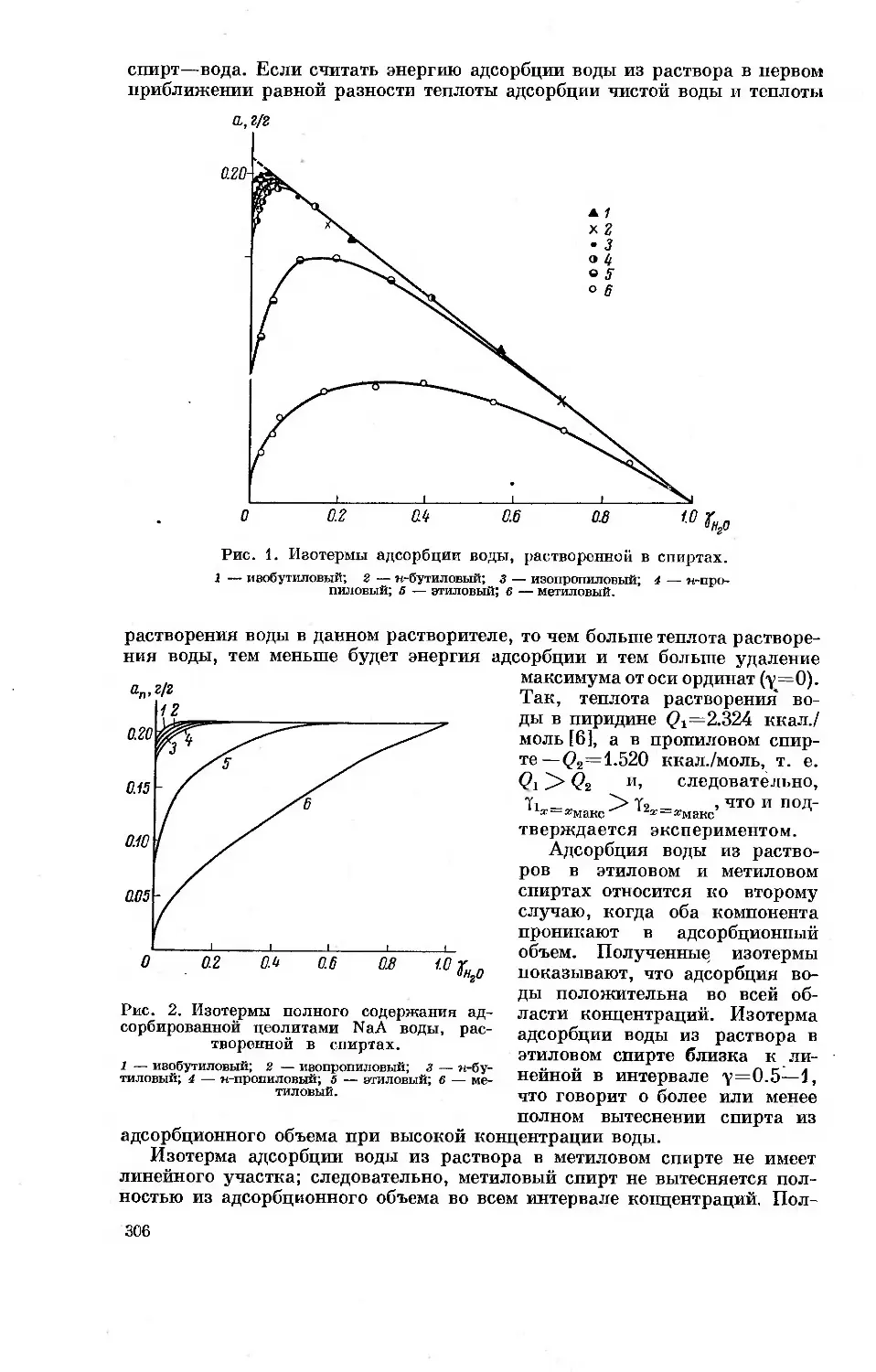
адсорбции. На рис. 1 приведены определенные калориметрически в лабо-
ратории А. В. Киселева кривые
дифференциальных теплот адсорб-
ции паров н-гексана при 20° для
микропористого углеродного ад-
сорбента (активного угля) и не-
пористой сажи, подвергнутой тер-
мической обработке при 950°.
Удельная поверхность сажи со-
ставляла 108 м2/г. В изученном
интервале величин адсорбции от
0.1 до 0.5 ммоль/г дифференциаль-
ные теплоты адсорбции для актив-
ного угля в среднем в 1.5 раза
превышают дифференциальные те-
плоты адсорбции для сажи.
Повышение энергии адсорбци-
онного взаимодействия в микро-
порах активных углей приводит
к резкому возрастанию величин
адсорбции паров в области малых
равновесных давлений. В табл. 1
приведены относительные величи-
ны адсорбции бензола при 20°
для непористого углеродного ад-
сорбента — прокаленной при 950°
сажи Сферон-6 с удельной поверх-
ностью 77 м2/г и двух образцов
активных углей АУ-1 и АУ-2, ак-
тивированных при 950° и облада-
ющих различной микропористой
структурой. Ее параметрами яв-
лялись константы уравнения ад-
сорбции теории объемного запол-
нения микропористых адсорбен-
тов: предельный объем адсорб-
ционного пространства Wo и константа В, зависящая от размеров
микропор. За единицы условно приняты величины адсорбции для равно-
весного относительного давления 0.15. Для сажи это давление примерно
отвечает образованию сплошного мономолекулярного слоя, а для актив-
ных углей — практическому завершению заполнения микропор в резуль-
тате адсорбции.
Экспериментальные данные таблицы 1 наглядно иллюстрируют, сколь
значительно возрастают величины адсорбции пара при малых равновес-
ных давлениях при переходе от непористых (или крупнопористых) угле-
родных адсорбентов к микропористым адсорбентам, в особенности с ма-
лыми значениями константы В. При давлениях порядка 10"4 или 10~5
это различие становится еще большим.
Рис. 1. Зависимость дифференциальных
теплот адсорбции н-гексана при 20° от ве-
личин адсорбции.
1 — активный уголь; 2 — непористая сажа; X —
теплота конденсации. Треугольники и кружки
обозначают параллельные опыты.
6
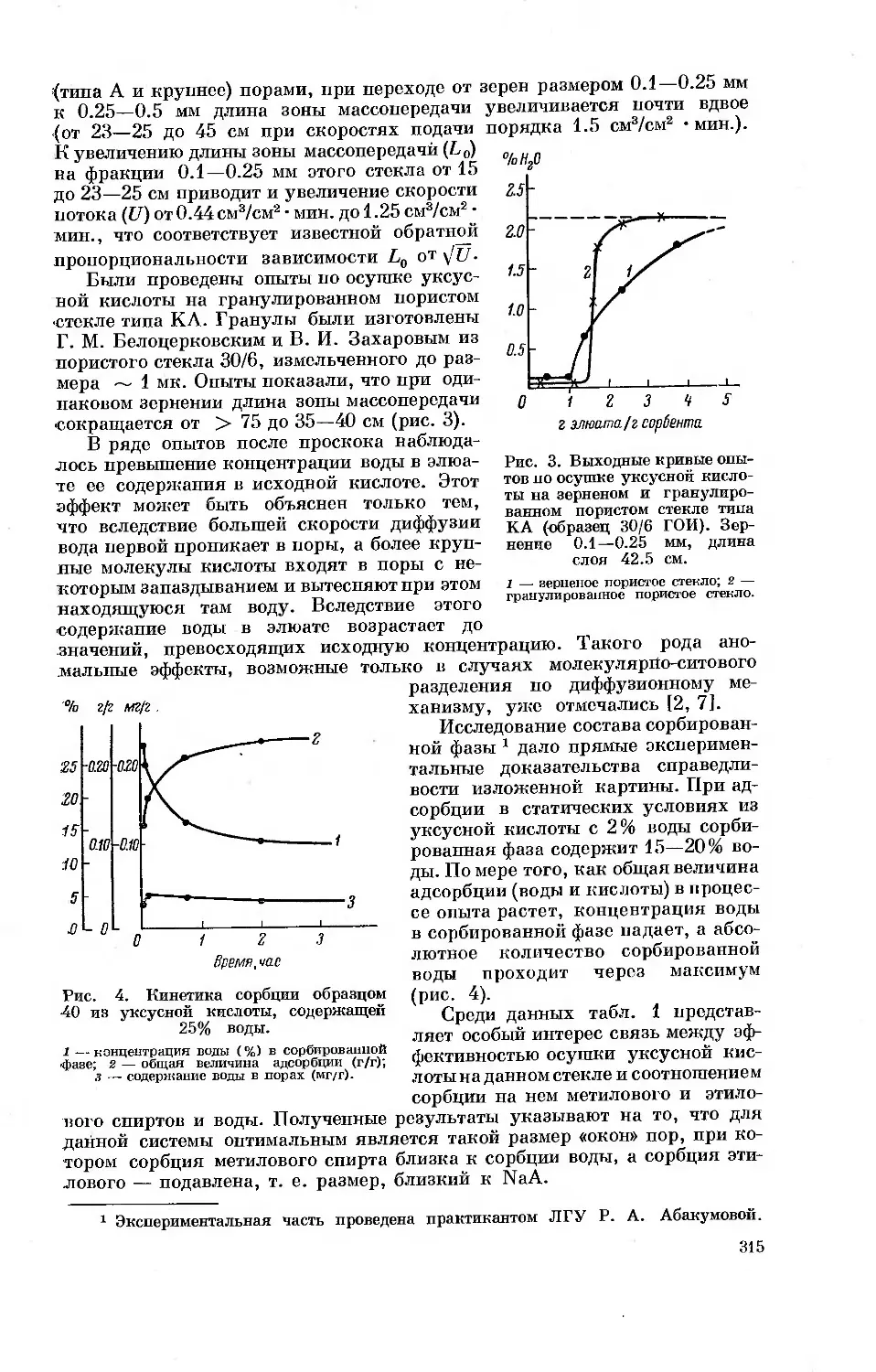
В случае цеолитов возможно сопоставление величин адсорбции пара
как на внешней поверхности, так и в полостях кристаллов цеолитов. Для
этой цели можно воспользоваться близкими по фрагментам структуры
цеолитами NaA и NaX, кристаллы которых построены из различным
образом связанных кубооктаэдричес-
ких структурных единиц. В качестве
адсорбируемого пара необходимо
выбрать пар вещества, для молекул
которого недоступны окна, ведущие
в большие полости цеолита NaA,
но вполне доступны входы в боль-
шие полости цеолита NaX. Примером
такого вещества может служить бен-
зол.
При помощи высокочувствитель-
ной весовой адсорбционной установки
А. И. Сараховым и Ю. Ф. Березкиной
были изучены изотермы адсорбции
паров бензола при 20° на трех образ-
цах дегидратированных кристаллов
NaA, отличающихся по дисперсности,
т. е. по величинам внешней удель-
ной поверхности кристаллов. В табл. 2
содержатся интерполированные по
приведенным изотермам адсорбции i
для единицы внешней поверхности к
Таблица 1
Относительные величины адсорбции
бензола при 20° для углеродных
адсорбентов
Сажа s = 77 м’/г АУ-1 В = 1.00- 10-« = 0.330 см’/г АУ-2 В = 0.407 • 10-' !У„ = 0.475 см3/г
0.001 0.10 0.48 0.75
0.010 0.29 0.73 0.89
0.025 0.44 0.83 0.94
0.050 0.66 0.89 0.97
0.100 0.83 0.95 0.99
0.150 1.00 1.00 1.00
адсорбции паров бензола
при различных равновес-
ных давлениях.
По данным табл. 2, адсорбционные свойства внешней поверхности изу-
ченных образцов кристаллов NaA практически одинаковы. При равновес-
ном относительном давлении 0.15 уже завершено образование сплошных
мономолекулярных слоев адсорбированного бензола на поверхности кри-
сталлов NaA, но еще не началась капиллярная конденсация пара в зазо-
рах между контактирующими кристалликами.
Таблица 2
Адсорбция СеН6 при 20° на внешней поверхности кристал-
лов цеолитов NaA
pIps 21, мкмоль/м2
NaA-1 s = 2.08 м2/г NaA-2 s = 3.64 м2/г NaA-3 s = 4.68 м2/г среднее значение
0.010 2.2 2.3 2.6 2.4
0.025 2.5 2.7 3.0 2.7
0.050 2.8 3.1 3.3 3.1
. 0.100 3.3 3.5 3.6 3.5
0.150 3.8 3.9 3.9 3.9
В табл. 3 сопоставляются средние значения относительных величин
адсорбции паров бензола на внешней поверхности кристаллов NaA и
в полостях кристаллов NaX, состав которых выражается отношением
SiO2/Al2O3=2.96. Изотерма адсорбции бензола для цеолита NaX была
определена Е. Ф. Полстяновым. При равновесном относительном давле-
нии 0.15 практически завершается заполнение полостей кристаллов NaX
адсорбированным бензолом, Поэтому при сопоставлении относительных
величин адсорбции бензола на внешней поверхности кристаллов NaA
7
Таблица 3
Относительные величины адсорбции бензола при 20°
цеолитами IS а А и NaX
Р1Рв NaA 21, МКМОЛЬ/М2 NaX а. мкмоль/г Относительные величины адсорбции
NaA NaX
0.010 2.4 3190 0.63 0.95
0.025 2.7 3230 0.67 0.96
0.050 3.1 3280 0.80 0.98
0.100 3.5 3320 0.90 0.99
0.150 3.9 3360 1.00 1.00
и в полостях кристаллов NaX также целесообразно принять за единицы
величины адсорбции для относительного давления 0.15, при котором
в обоих случаях практически завершается первичный адсорбционный про-
цесс.
Согласно экспериментальным данным (табл. 3), для цеолитов также
наблюдается значительное возрастание относительных величин адсорб-
Рис. 2. Схема взаимного располо-
жения кубооктаэдрических струк-
турных единиц в алюмосиликатном
скелете цеолита типа А.
специфическое взаимодействие
ции при малых равновесных давлениях
при переходе от адсорбции на внешней
поверхности кристаллов к адсорбции
в полостях цеолитов. Однако из срав-
нения относительных величин адсорб-
ции бензола на непористом углеродном
адсорбенте (табл. 1) и на внешней по-
верхности кристаллов NaA (табл. 3)
следует, что при малых давлениях от-
носительная адсорбируемость бензола
на поверхности кристаллов NaA в 1.5—
2 раза больше.
В отличие от аполярных углеродных
адсорбентов цеолиты являются гетеро-
полярными адсорбентами. Они содержат
катионы (в рассматриваемом случае
натрия), компенсирующие избыточные
отрицательные заряды алюмосиликат-
ных скелетов. Поэтому при адсорбции/
бензола на цеолитах наряду с дисперсий
онным взаимодействием проявляется!
электронных облаков л-связей молекул
бензола с ионами натрия. В результате сложения дисперсионного и спе-
цифического взаимодействия, по-видимому, и наблюдается существенное
возрастание относительной адсорбируемости бензола на цеолитах по срав-
нению с углеродными адсорбентами, для которых характерно только дис-
персионное взаимодействие.
Если поверхности кристаллов цеолитов NaA образованы плоскостями
кубических элементарных кристаллических ячеек (рис. 2), то каждая
сторона кубической ячейки с параметром аоА и с площадью аоА2 содержит
5 ионов натрия, находящихся вблизи центров окон, одно из которых ведет
в большую полость, а четыре других в малые полости. Для кристаллов
NaA среднее значение аоА=12.26 А. Из этих данных следует, что На 1 м2
внешней поверхности кристаллов содержится 3.3 • 1018 или 5.5 мкмоль/м2
ионов натрия. Таким образом, при образовании сплошного мономолеку-
8
лярного слоя бензола на внешней поверхности кристаллов NaA (2lm «
«3.5 мкмоль/м2) на одну адсорбированную молекулу бензола в среднем
приходится 1.6 ионов натрия или активных центров поверхности.
В состав элементарной ячейки цеолита NaX с отношением SiO2/Al2O3=
=2.96 входит в среднем 77.4 иона натрия. Число элементарных ячеек
в 1 г дегидратированных кристаллов цеолита составляет 4.57 • 1019.
Если рассматривать ионы натрия в полостях цеолита как активные центры
для адсорбции [1 ], то число этих центров в 1 г цеолита выразится 3-54 4021,
или 5900 мкмоль/г. Предельная величина адсорбции для цеолита
NaX составляет около 3360 мкмоль/г (табл. 3). Отсюда следует, что одной
адсорбированной молекуле бензола в среднем отвечают около 1.7 адсорб-
ционных центров, т. е. практически столько же, сколько приходится на
одну молекулу бензола в сплошном мономолекулярном слое на внешней
поверхности кристаллов NaA. Однако по данным табл. 3 относительная
адсорбируемость бензола в полостях цеолита NaX, составляющая 0.95,
значительно выше его относительной адсорбируемости 0.63 на поверхно-
сти кристаллов NaA.
Таким образом, относительная адсорбируемость паров микропористыми
адсорбентами при малых равновесных давлениях много больше их отно-
сительной адсорбируемости на непористых или крупнопористых адсор-
бентах с одинаковой или близкой химической природой. Это указывает
на повышение энергии адсорбционных взаимодействий в микропорах по
сравнению с взаимодействиями для пепористых или крупнопористых ад-
сорбентов.
Характер заполнения микропористых адсорбентов
В отличие от других микропористых адсорбентов для дегидратирован-
ных кристаллов синтетических цеолитов типов А и X мы располагаем
сведениями из независимых рентгеноструктурных данных о форме, раз-
мерах и объемах микропор [1, 2]. Несмотря на осложненный в ряде слу-
чаев характер адсорбционных взаимодействий на цеолитах, как на
гетерополярных адсорбентах, знание параметров их микропористой струк-
туры позволяет решать, на основании исследований равновесной адсорб-
ции паров различных веществ на цеолита^, ряд задач, имеющих общее
значение для развития теории адсорбции паров микропористыми адсор-
бентами.
Е. Г. Жуковской, К. О. Мурдмаа и Е. Ф. Полстяновым были изучены
в широких интервалах равновесных относительных давлений изотермы
адсорбции паров азота, кислорода, аргона, окиси углерода при —196°
и паров воды и н-пентана при 20° на цеолитах СаА и NaX (SiO2/AlaO3=
=2.96), синтезированных С. П. Ждановым в лабораторных условиях.
Вычисленные из рентгеноструктурных данных и состава цеолитов объемы
больших полостей составляли для цеолита СаА гБА = 0.278 см3/г [2] и
для цеолита NaX кБХ = 0.322 см3/г [1]. Формальные геометрические
удельные поверхности больших полостей на основании вычислений
были оценены следующими величинами: для СаА sRA = 1640 м2/г и
NaX sEx = 1400 м2/г [3]. Предельные величины адсорбции а0 надежно
определялись из экспериментальных изотерм по уравнению теории объем-
ного заполнения микропористых адсорбентов [4].
В табл. 4 приведены предельные величины адсорбции паров изучен-
ных веществ на цеолитах СаА и NaX с различными параметрами микро-
пористой структуры и отношения этих величин.
По данным табл. 4, отношения предельных величин адсорбции а0 для
цеолитов СаА и NaX в среднем составляют 0.861 +0.028, т. е. с точностью
до 3% являются постоянными. Среднее значение хорошо совпадает с от-
9
ношением вычисленных объемов больших полостей .цеолитов, составляю-
щим 0.863, и значительно отличается от отношения их формальных удель-
ных поверхностей, равного 1.17. Таким образом, для цеолитов предель-
ные величины адсорбции отвечают объемному заполнению их микропор.
Средние мольные объемы предельно адсорбированных паров v*
являются частными из деления вычисленных объемов больших полостей
цеолитов Кб на предельные величины адсорбции а0. В табл. 5 вычислен-
ные средние значения мольных объемов предельно адсорбированных ве-
ществ и* сравниваются с мольными объемами нормальных жидкостей v
для температур опытов. Для аргона при —196° мольный объем v отвечает
переохлажденной жидкости.
Таблица 4
Предельные величины адсорбции
паров на цеолитах
Вещество а0, ммоль/г СаА/NaX ctO /а0
СаА NaX
н2о . 15.50 17.96 . 0.863
n2 . . 8.27 9.55 0.866
со . . 8.61 9.71 0.886
С5Н12. 2.26 2.56 0.883
о2 . . 9.02 10.86 0.831
Аг . . 8.58 10.27 0.835
Таблица 5
Средние мольные объемы предельно
адсорбированных веществ на цеолитах
Вещество см3/моль V, см3/моль
н20 . 17.9 18.0 0.99
n2 . . 33.7 34.7 0.97
со . . 32.3 34.5 0.94
CSH12 124 115 1.08
О2 . . 30.3 26.2 .1.16
Аг . . 31.9 27.3 1.17
Отклонения мольных объемов v* для цеолитов СаА и NaX от их сред-
них значений только в отдельных случаях достигают +1.5%. По данным
табл. 5, мольные объемы адсорбированных воды, азота и окиси углерода
мало отличаются от соответствующих значений для объемных жидких
фаз. Наибольшие отклонения этих величин наблюдаются для кислорода
и аргона, что обязано особенностям упаковки молекул этих веществ в по-
лостях цеолитов, так как средние числа адсорбированных молекул из
расчета на одну полость, например для азота (отсутствие отклонений) и
аргона (большие отклонения), отличаются менее чем на 4% для цеолита
СаА. Заметим, что, согласно теории объемного заполнения микропори-
стых адсорбентов, отношения v*/v в принципе должны отличаться от еди-
ницы [5].
Среди изученных веществ наибольшее отклонение мольного объема
адсорбированного вещества (к*=31.9 см3/моль) от мольного объема нор-
мальной жидкости (к=27.3 см3/моль) характерно для аргона. Теперь
следует установить, в какой мере это аномальное значение мольного объема
аргона в адсорбированном состоянии типично для микропористых адсор-
бентов другой природы. Для этой цели Е. Г. Жуковской были изучены
изотермы адсорбции паров аргона и других веществ на уже упоминав-
шихся образцах активных углей АУ-1 и АУ-2 с практически предельными
параметрами их микропористой структуры. Для сравнения служил сили-
кагель СМ, принадлежащий к группе относительно крупнопористых ад-
сорбентов. Пористая структура этого силикагеля при относительном дав-
лении, равном единице, объемно заполнялась в результате капиллярной
конденсации паров. По предельным величинам адсорбции а0 для активных
углей были вычислены предельные адсорбционные объемы Wo, причем
для аргона принималось как экспериментальное значение мольного объема
для адсорбированного состояния v*=31.9 см3/моль, так и значение v=
10
=27.3 см3/моль для нормальной переохлажденной жидкости. Для дру-
гих паров принимались нормальные значения мольных объемов. В слу-
чае силикагеля аналогичным образом по предельным величинам сорбции
zzg вычислялись предельные сорбционные объемы vs. Результаты вычисле-
ний приведены в табл. 6.
Таблица 6
Предельные адсорбционные (сорбционные) объемы для
различных адсорбентов
Адсорбент Вещество Wo, см3/г для АУ; t>8 см’/г для СМ
для Аг в * == 31.9 см3/моль для Аг в — 27.3 см3/моль
АУ-2 0.407 • 10-8 N2 . . . Аг . . . 0.57 0.54 0.46
В = АУ-1 1.00-10-6 N2 .. . С6нс . . Аг . . . 0.33 0.33 0.34 0.29
г,п = СМ 24 А * N2 . . . Н2О . . свне . . сен12. . Аг . . . 0.61 0.61 0.61 0.61 0.69 0.59
* Эффективный радиус гт пор силикагеля с поправкой на толщину
адсорбционного слоя для максимума кривой распределения объема пор
по эффективным радиусам.
По данным табл. 6, предельные объемы адсорбционного пространства
Wo для активных углей практически одинаковы для всех изученных
паров, если для аргона при вычислении И70 принято значение мольного
объема v*, найденного из опытов с цеолитами. В случае силикагеля, поры
которого заполнены при предельной сорбции капиллярно-сконденсиро-
ванной жидкостью, жидкому аргону в сорбированном состоянии отве-
чает мольный объем и соответственно плотность нормальной переохла-
жденной жидкости. Таким образом, микропористые и относительно круп-
нопористые адсорбенты существенно различаются по свойствам предельно
адсорбированных и соответственно сорбированных веществ.
Важное значение для теории адсорбции паров микропористыми адсор-
бентами имеет исследование мольных объемов адсорбированных веществ
при различных заполнениях объема адсорбционного пространства. Эта
задача эквивалентна определению мольных объемов предельно адсорби-
рованных веществ для микропористого адсорбента, адсорбционное про-
странство которого изменяется в достаточно широких пределах. В случае
цеолитов это можно осуществить путем прогрессирующего заполнения
при обычной температуре их полостей хорошо адсорбирующимся веще-
ством с последующим определением изотерм низкотемпературной адсорб-
ции паров избранных веществ.
Е. Г. Жуковской изучены изотермы адсорбции при —196° паров азота
и аргона на цеолите NaX, содержащем различные количества предадсор-
•бированной воды, давление паров которой при температуре опытов было
неизмеримо мало. На рис. 3 приведены в качестве примера изотермы ад-
сорбции азота в линейной форме по уравнению адсорбции теории объем-
ного заполнения микропористых адсорбентов. Из графика видно, что по
точкам пересечения прямых с осью ординат могут быть вполне надежно
определены предельные величины адсорбции. На графике рис. 4 изобра-
11
жена зависимость предельных величин адсорбции а2 от количества пред-
адсорбированной воды ах на цеолите. Достаточно хорошая линейная за-
висимость в интервале заполнения предадсорбированной водой объема
0.5 L
О
W (Ц Ps/Pf
Рис. 3. Изотермы адсорбции азота при —196° в линейной форме
на цеолите NaX.
Содержание предадсорбированной воды (ммоль/г): 1 — 0; 2 — 1.43; 3 — 3.21;
4 — 4.41; 5 — 5.81 и 6 — 6.98.
полостей цеолита от 0.322 до 0.193 см3/г может быть разумно интерпрети-
рована только при предположении о постоянстве мольных объемов пред-
адсорбированной воды vt* и азота или аргона п2* :
рг„ = av* 4- а V*.
Ьл 1 1 1 2 2’
(1)
откуда следует наблюдаемая на опыте линейная зависимость а2 от а1:
Рис. 4. Зависимость предельных вели-
чин адсорбции (а2) при —196° от ко-
личества предадсорбированной воды (ог)
на цеолите NaX.
1 — аргон; 2 — авот.
БХ vi
— * — * (2)-
^2 V2
Воспользовавшись значениями в2*
для аргона и азота, приведенными
в табл. 5, мы получили из опытов с
азотом и аргоном практически одина-
ковые мольные объемы предадсорби-
рованной воды (18.6 см3/моль и
соответственно 18.8 см3/моль). Это'
служит дополнительным обосновани-
ем физической реальности уравнения
(1). В заключение следует отметить,
что изменению при адсорбции, напри-
мер азота при —196°, заполненных
объемов адсорбционного пространст-
ва от 0.322 до 0.193 см3/г отвечает
интервал равновесных относитель-
ных давлений от величины порядка
10до значений порядка 10 ~7. Показанное постоянство мольных объе-
мов адсорбированных веществ при различных заполнениях объема ад-
сорбционного пространства находится в соответствии с теорией объем-
ного заполнения при адсорбции паров микропористыми адсорбентами.
Из изложенного следует, что микропористые адсорбенты образуют
особую разновидность пористых адсорбентов со специфическими особен-
12
ностями адсорбционных взаимодействий и различным характером запол-
нения адсорбционного пространства по сравнению с непористыми или
относительно крупнопористыми адсорбентами.
ЛИТЕРАТУРА
1. М. М. Ду бинин, С. П. Жданов, Е. Г. Ж у к о в с к а я, К. О. Мурд-
м а а, Е. Ф. П о л с т я н о в, И. Е. Сакавов и Н. А. Шишаков,
Изв. АН СССР, сер. хим., 1964, 9, 1573.
2. М. М. Дубинин, С. П. Жданов, Е. Г. Ж у к о в с к а я, К. О. Мурд-
маа, Е. Ф. П о л с т я н о в, И. Е. Сакавов и Н. А. Шишаков,
Изв. АН СССР, сер. хим., 1964, 9, 1565.
3. М. М. Дубинин, Изв. АН СССР, сер. хим., 1964, 2, 209.
4. М. М. Д убинин, Е. Г. Ж у к о в с к а я, К. О. Мурдмаа л Е. Ф. По л-
с т я н о в, Изв. АН СССР, ОХН, 1962, 12, 2113.
5. Б. И. Беринг и В. В. Сер пинский, ДАН СССР, 1963, 148, 1331.
ПРИРОДА АДСОРБЦИИ ЦЕОЛИТАМИ
А. В. Киселев
Общая характеристика связи химического строения цеолитов
с их адсорбционными свойствами
Цеолиты больше известны как молекулярные сита [1]. Однако в очень
важных применениях, например в газовой хроматографии [2], при «скла-
дировании» 13] нужных молекул для их использования в различных
средах при десорбции и во многих случаях адсорбционных разделений ис-
пользуется не молекулярно-ситовое действие, но особенности адсорб-
ционного поля каналов этих пористых кристаллов, связанные с особенно-
стями их химического строения. На это обращалось внимание уже в ра-
ботах пионера в области синтеза и изучения адсорбционных свойств мно-
гих цеолитов Р. М. Бэррера и его сотрудников [1, 4]. В настоящей работе
мы рассмотрим именно химические аспекты адсорбции цеолитами. По-
скольку в этом случае речь идет о молекулах, свободно проникающих
в их каналы, наряду с геометрической структурой адсорбирующейся
молекулы важнейшую роль играет электронная структура ее звеньев —
симметрия электронной оболочки, наличие только о-связей или также и
л-связей, наличие или отсутствие свободных электронных пар, локализа-
ция дипольных моментов и т. п. Эти различия в распределении электрон-
ной плотности даже при близости геометрической структуры вызывают
глубокие различия в природе взаимодействия молекул с цеолитом, в энер-
гиях этого взаимодействия,’в состоянии как остова цеолита, так и адсор-
бируемых им молекул. Эти различия ярко проявляются при комплексном
исследовании адсорбции цеолитами адсорбционно-калориметрическими,
газо-хроматографическими и молекулярно-спектроскопическими методами.
Ради краткости изложения мы ограничимся здесь результатами, полу-
ченными для синтетических фожазитов (цеолитов типа X), хотя каче-
ственно такие же результаты получаются и для других цеолитов, каналы
которых доступны для молекул различной электронной структуры. Бла-
годаря совместной работе с С. П. Ждановым мы располагали однородными
пористыми кристаллами синтетических фожазитов различного состава.
Описание их синтезов, состава и структуры приводится в [5]. В [6] рас-
смотрена геометрическая структура фожазита, принимая за основные
единицы связанные общими кислородами тетраэдры Si04 и А104. Благо-
даря несовпадению валентности и координационного числа алюминия
13
тетраэдр А104 имеет единичный отрицательный заряд и представляет в це-
лом большой комплексный анион. Существенно, что этот отрицательный
заряд не сосредоточен на его периферии, но рассредоточен на внутренних
связях О—А1 этого комплекса. Наоборот, компенсирующий положитель-
ный заряд сосредоточен в доступных для звеньев молекул адсорбата об-
менных катионах гораздо меньшего радиуса.
Эта особенность структуры остова цеолита, в частности поверхности
больших полостей синтетических фожазитов, определяет природу ее
взаимодействия с молекулами разной электронной структуры. С этой
точки зрения цеолиты относятся ко второму типу адсорбентов предло-
женной нами классификации химии поверхности твердых тел по трем
типам [7], т. е. к адсорбентам, несущим на поверхности сосредоточенные
положительные заряды (катионы малого радиуса, протонизированные
атомы водорода гидроксильных групп кислого характера), расположенные
около рассредоточенных отрицательных зарядов (например, около боль-
ших, комплексных анионов). Во всех этих случаях молекулы, имеющие
звенья с сосредоточенной на периферии электронной плотностью, напри-
мер л-связи у азота, ненасыщенных и ароматических углеводородов
и свободные электронные пары у атомов кислорода воды, спиртов,
эфиров, кетонов или у атомов азота аммиака, аминов, пиридина и т. п.,
должны проявлять качественно сходное специфическое взаимо-
действие с центрами сосредоточения на поверхности адсорбента положи-
тельного заряда. Действительно, характер адсорбции перечисленных мо-
лекул на поверхности каналов цеолитов качественно сходен с таковым
на поверхности сульфата бария [7] (сосредоточенный положительный за-
ряд и рассредоточенный в комплексном анионе SO4 отрицательный за-
ряд) и, как мы увидим ниже, на поверхности кремнезема (или алюмосили-
катного катализатора), несущей гидроксильные группы с частично
протонизированным водородом [7, 8]. Вклады классических и квантово-
механических эффектов в энергию специфических взаимодействий коли-
чественно различаются, но качественно эти взаимодействия сходны. Водо-
родная связь является их частным случаем.
Наоборот, при адсорбции таких молекул на адсорбентах с химически
насыщенной поверхностью (на адсорбентах первого типа нашей класси-
фикации), таких как графитированные сажи [7], BN [9, 10], MoS2 [10],
политетрафторэтилен [11], окислы с поверхностью, модифицированной
прививкой алкилсилильных или алифатических групп [12], наличие
в молекулах адсорбата звеньев с сосредоточенной на периферии электрон-
ной плотностью практически не проявляется и взаимодействие остается
неспецифическим — в основном дисперсионным.
Неспецифическое взаимодействие наблюдается и в тех случаях, когда
в адсорбирующемся партнере этого взаимодействия — молекуле адсор-
бата — электронная плотность распределена наиболее симметрично (сфе-
рические электронные оболочки, о-связи). В этом случае взаимодействие
остается неспецифическим не только при адсорбции на адсорбентах пер-
вого типа (с насыщенной поверхностью), но и при адсорбции на адсорбен-
тах второго типа (с сосредоточенными на поверхности положительными
зарядами), когда к основному вкладу неспецифических дисперсионных
взаимодействий добавляется вклад классических в индукционных взаимо-
действий.
Рассмотрим теперь с этой точки зрения имеющийся эксперименталь-
ный материал по теплотам адсорбции и по инфракрасным спектрам, а
также теоретические расчеты энергии взаимодействия. Для того чтобы
выявить качественное сходство специфической адсорбции
на поверхности каналов цеолитов и на поверхности других адсорбентов
второго типа и для того чтобы выявить качественное различие
14
с неспецифической адсорбцией тех же молекул на поверхности адсорбен-
тов первого типа, мы сопоставим адсорбционные характеристики в основ-
ном для трех адсорбентов: цеолитов с «катионированной» поверхностью
и кремнеземов с гидроксилированной поверхностью, т. е. для двух спе-
цифических адсорбентов второго типа, и, наконец, для графитированных
саж (неспецифических адсорбентов первого типа).
Что касается адсорбатов, то мы выберем в основном несколько пар
молекул, сходных по занимаемой на поверхности площади или по числу
атомов углерода и по величинам общей поляризуемости, но резко разли-
чающихся по распределению электронной плотности в отдельных звеньях.
Сюда входят молекулы азота (две л-связи и вызванный ими большой квадру-
польный момент) и аргона (сферически симметричная оболочка), этилена
(л- и о-связи) и этана (только о-связи), бензола (л- и о-связи) и н-гексана
(только ст-связи), диэтилового эфира (две свободные электронные пары
у атома кислорода, большой периферический дипольный момент) и н-пен-
тана (только o'-связи). Кроме того, мы сопоставим адсорбцию молекул^
обладающих атомом кислорода с двумя свободными электронными парами:
воды и метанола.
Общий вид изотерм адсорбции цеолитами и зависимости
дифференциальной теплоты адсорбции от заполнения
Вклад в энергию адсорбции в узких каналах цеолитов даже неспеци-
фических взаимодействий так велик, что даже вблизи температур кипения
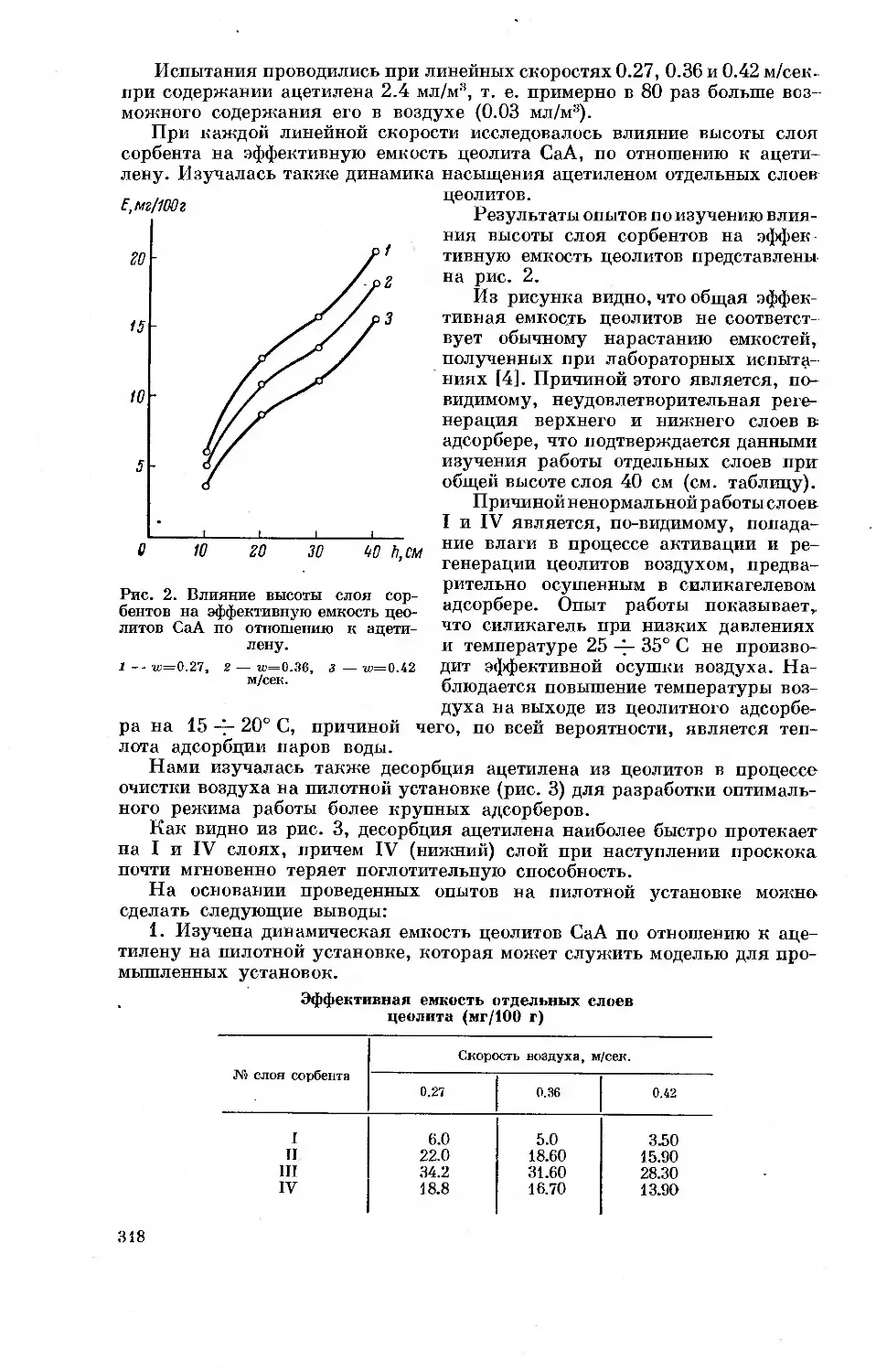
адсорбатов изотермы адсорбции поднимаются чрезвычайно круто. Из
рис. 1 видно, например, что изотерма неспецифической адсорбции насы-
щенного углеводорода — н-пентана (температура кипения 36.07° С) при
20° поднимается так круто, что, например, до равновесного давления
р=10 мм кристаллами синтетического цеолита NaX адсорбируется уже
90% от предельно адсорбируемого вблизи давления насыщенного пара ps
количества ag=2.6 ммоль/г [13]. Рис. 1 показывает, что исследовать
обратимую адсорбцию даже таких сравнительно небольших и химически
устойчивых молекул, как молекул н-пентана при заполнениях цеолита,
меньших 25%, т. е. в наиболее важной для многих практических приме-
нений области, можно лишь около 200° С [14]. В случае более тяжелых
и сложных молекул, адсорбирующихся, кроме того, специфически, для
точного измерения обратимых изотерм адсорбции цеолитами нужны
были бы еще более высокие температуры, при которых возникает воз-
можность каталитического превращения.
Поэтому наряду с изучением обратимых изотерм адсорбции мы пред-
приняли калориметрические измерения дифференциальных теплот ад-
сорбции, начиная с малых заполнений при обычной температуре. Усовер-
шенствование калориметра позволило исследовать медленно выделяю-
щиеся тепловые эффекты адсорбции цеолитами даже таких сильно и
специфически адсорбирующихся молекул, как диэтиловый эфир (теплота
адсорбции больше 20 ккал./моль). Эти исследования показали, что внут-
ренняя поверхность каналов синтезированного в лаборатории С. П. Жда-
нова Na-фожазита [13—15] довольно однородна. Дифференциальная теп-
лота адсорбции, например н-пентана, при росте заполнения до 0.6 растет
вначале медленно, а затем, благодаря усилению взаимодействий адсор-
бат—адсорбат, быстрее. В соответствии с этим изотермы адсорбции в об-
ласти заполнений до 0.75 от предельного удовлетворительно описываются
простыми уравнениями, приближенно учитывающими взаимодействие ад-
сорбат—адсорбат. Величины и теплоты адсорбции синтетическими
фожазитами обратимы, хорошо воспроизводятся для разных образ-
цов одинакового состава и представляют поэтому физико-химические кон-
15.
станты, зависящие при данной температуре и концентрации только от
свойств системы адсорбат—адсорбент. Это делает цеолиты прекрасными
•объектами для изучения природы межмолекулярных взаимодействий как
Рис. 1. Изотермы адсорбции синтетическим Na-фожазитом
пара и-пентана при 20 (1), 200 (2), 230 (3) и 250° (4).
На оси ординат адсорбция выражена в абсолютных единицах а, ммоль/г,
значения а8 и р8 для 20° отмечены стрелками. На оси абцисс — абсо-
лютное давление пара р, мм рт. ст. Черные точки — десорбция. 0 —
степень заполнения.
путем сопоставления изотерм, теплот и молекулярных спектров при ад-
сорбции цеолитами с этими эффектами для других адсорбентов, так и пу-
тем теоретического исследования энергии взаимодействия молекул ад-
сорбата с цеолитами, формы изотерм адсорбции и зависимости адсорб-
ции от температуры.
Сопоставление дифференциальных теплот адсорбции молекул,
различающихся по электронной структуре отдельных звеньев, на
катионированной поверхности каналов цеолита, на гидроксилированной
поверхности кремнезема и на графитированной саже
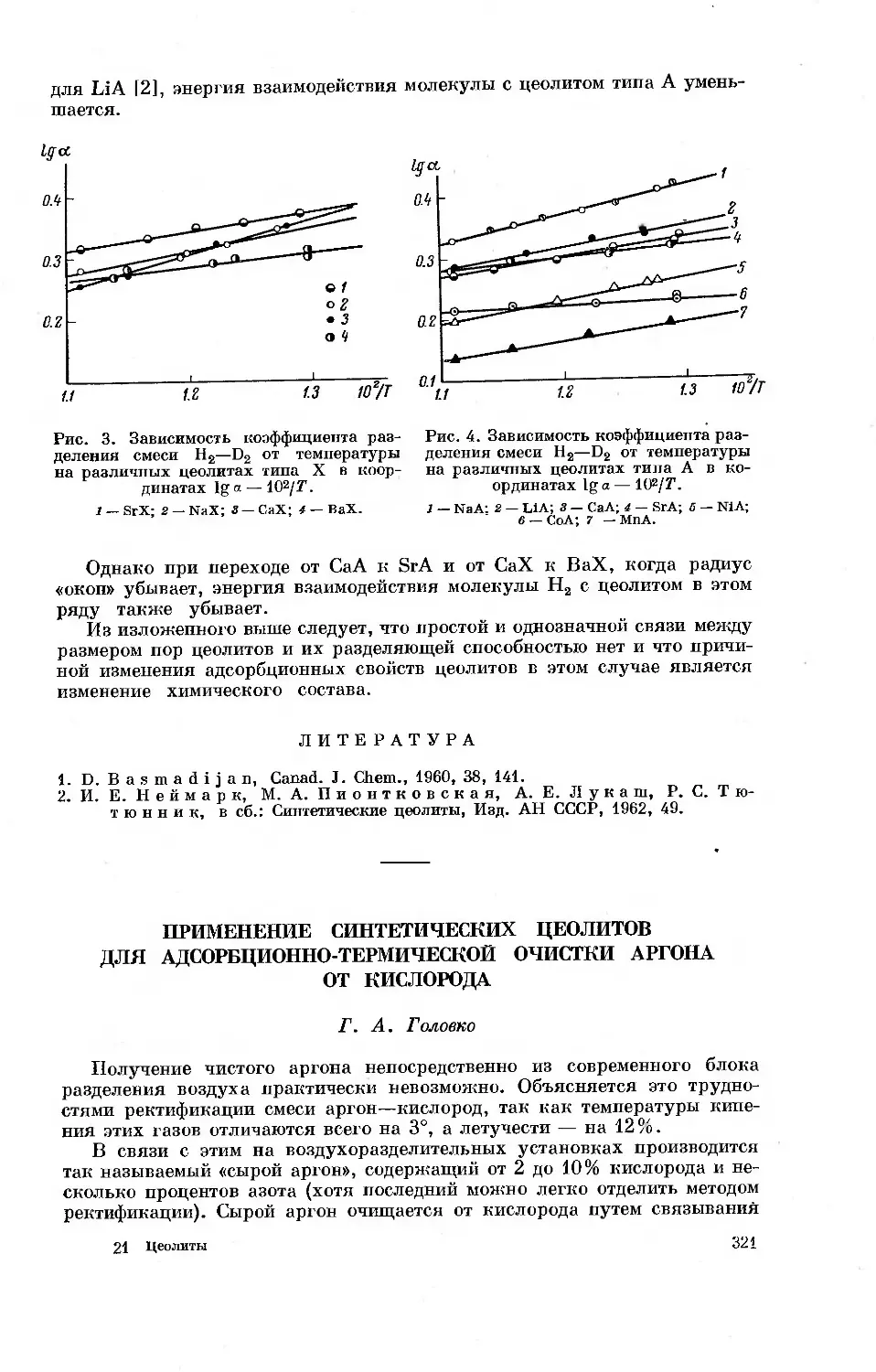
На рис. 2 приведены результаты калориметрических, изостерических
и газо-хроматографических определений дифференциальных теплот ад-
сорбции некоторых алкенов (л- и о-связи в молекуле) и алканов (только
о-связи) на катионированной поверхности синтетического Na-фожазита
[2, 16], гидроксилированной поверхности кремнезема [17] и на графити-
рованной саже [7]. В случае химически насыщенной поверхности графити-
рованной сажи теплоты адсорбции алкенов в соответствии с меньшим
числом атомов водорода в их молекулах даже ниже теплот адсорбции
соответствующих алканов. Наличие в молекулах алкенов л-связей
16
Рис. 2. Дифференциальные теплоты адсорбции и-алканов (7),
алкенов (2) и ацетилена (5) при небольших заполнениях
насыщенной поверхности графитированной сажи (а), гид-
роксилированной поверхности кремнезема (б) и катиониро-
ванной поверхности Na-фожазита (в).
п — число атомов углерода в молекуле.
О 0.5 1.0 1.5 0 025 0.50 0.75 0 0.25 0.50 0.75 1 в
Рис. 3. Зависимости дифференциальной теплоты адсорбции бензола (7) и и-гексана (2)
от величины адсорбции на графитированной саже (а), гидроксилированной поверх-
ности кремнезема (б) и катионированной поверхности Na-фожазита (в).
L — теплоты конденсации (то же на других рисунках этой статьи).
2 Цеолиты
в этом случае не проявляется, потому что другой вступающий во
взаимодействие партнер — базисная грань графита — способен только
к неспецифическому взаимодействию. В случае же адсорбции на гидро-
ксилированной поверхности кремнезема и катионированной поверхно-
сти цеолита картина резко изменяется: благодаря возможности специфи-
ческого взаимодействия л-электронных связей с протонизированным водо-
родом [8] гидроксильных групп поверхности кремнезема или с обменными
катионами поверхности цеолита теплоты адсорбции алкенов значительно
превосходят теплоты адсорбции алканов.
На рис. 3 сопоставлены зависимости дифференциальных теплот ад-
сорбции молекул бензола (ароматические л-связи) и н-гексана (только
20-
15-
10 -
25г
__I_I_I_I_ _1-U-1-1- --1--L-L
о 1 234012340 1 2 3
сс,мкмоль/ме <х.,мкмоль[м2 а,,ммоль/г
Рис. 4. Зависимости дифференциальных теплот адсорбции диэтилового эфира (1) и
н-пентана (2) от величины адсорбции на графитированной саже (а), гидроксилирован-
ной поверхности кремнезема (б) и катионированной поверхности Na-фожазита (в).
Ь — теплоты конденсации (то же на других рисунках этой статьи).
о-связи) на поверхности этих же адсорбентов. При малых заполнениях
повышенная энергия адсорбции бензола наблюдается только на гидрокси-
лированной поверхности кремнезема и катионированной поверхности
цеолита. На поверхности же графитированной сажи теплота адсорбции
бензола даже меньше теплоты адсорбции гексана [7, 15]. На рис. 4 ана-
логичное сопоставление сделано для диэтилового эфира и н-пентана [7].
Переход от адсорбции на поверхности неспецифического адсорбента —
графита, как к адсорбции на гидроксилированной поверхности кремне-
зема, так и к адсорбции на катионированной поверхности цеолита носит
качественный характер, различие же в адсорбции на гидроксилированной
и катионированной поверхностях скорее лишь количественное: в обоих
этих случаях наличие свободных электронных пар у атомов кислорода
проявляется качественно одинаково.
Наконец на рис. 5 сопоставлены теплоты адсорбции паров воды и ме-
танола, молекулы которых содержат атом кислорода с двумя свободными
электронными парами.
Теплоты адсорбции на графитированной саже отдельных молекул
воды [18] и спиртов [7, 19] удается определить лишь при очень малых
заполнениях, т. е. газо-хроматографическим методом с чувствительным
детектором, так как при больших заполнениях эти теплоты включают
энергию взаимных водородных связей. Теплоты неспецифического взаимо-
действия отдельных молекул воды и метанола с графитом значительно
меньше теплот их конденсации. Наличие в этих молекулах электроно-
18
донорных и электроно-акцепторных центров при адсорбции на графити-
рованной саже не вносит специфического вклада. Теплоты адсорбции
отдельных молекул спиртов, например, в этих случаях близки к энергии
неспецифического их взаимодействия с графитом (дисперсионного и сла-
бого индукционного). В случае же гидроксилированной поверхности
кремнезема [20, 21] и катионированной поверхности Na-фожазита (по-
дробные данные для цеолита рассматриваются в [22]) теплоты адсорбции
этих молекул включают значительный вклад специфических взаимодей-
ствий, представляющих в основном взаимодействия свободных электрон-
ных пар атомов кислорода молекул воды и спирта с протонизированными
гидроксильными группами поверхности кремнезема 1 и катионами цео-
Рис. 5. Зависимости дифференциальных теплот адсорбции паров воды (2) и
метанола (2) от величины адсорбции на графитированной саже (а), гидрокси-
лированной поверхности кремнезема (б) и катионированной поверхности Na-
фожазита (в).
Знаками с крестами отмечены газохроматографические данные.
лита. Помимо резкого качественного различия в теплотах адсорбции этих
молекул на графитированной саже и гидроксилированной поверхности
кремнезема (т. е. при переходе от неспецифической адсорбции к специфи-
ческой), мы можем отметить близость теплот адсорбции воды и метанола
на гидроксилированной поверхности кремнезема и катионированной по-
верхности цеолита. Кроме того, мы можем отметить близость теплот ад-
сорбции воды и метанола между собой. Различия характера зависимости
теплот адсорбции от заполнения связаны с различной степенью неоднород-
ности поверхностей кремнезема и цеолита и взаимодействий адсорбат—ад-
сорбат у спирта и воды [22].
В табл. 1 приведено сопоставление результатов определения теплот
адсорбции этими тремя адсорбентами для нескольких пар молекул, близ-
ких по значениям площади, занимаемой ими на поверхности в плотном
монослое, и по общей поляризуемости, но различающихся наличием или
1 При дегидроксилировании вклад специфических взаимодействий практически
исчезает [7].
2*
19
Таблица 1
Сопоставление теплот адсорбции при небольших запол-
нениях на графитированной саже, гидроксилированной
поверхности кремнезема и катионированной поверхности
цеолита (ккал./моль)
Адсорбаты Графит Гидроксили- рованный кремнезем Цеолит NaX
Азот 2.3 3.5 5.2
Аргон 2.4 2.5 3.1
Этилен 4.0 6.5 9.2
Этан 4.3 4.8 6.2
Бензол 10.1 11.0 18.0
н-Гексан 11.4 9.2 14.7
Диэтиловый эфир . . 8.9 14.9 21.0
н-11ентан 9.2 7.3 12.4
Примечание. Молекулы взятых пар веществ сильно разли-
чаются распределением электронной плотности в отдельных звеньях.
отсутствием звеньев с периферически сосредоточенной электронной плот-
ностью. Из таблицы виден резкий рост теплоты адсобции азота по срав-
нению с аргоном, этилена по сравнению с этаном, бензола по сравнению
с н-гексаном и диэтилового эфира по сравнению с н-пентаном при переходе
от адсорбции на графитированной саже (неспецифическая адсорбция)
к адсорбции на гидроксилированном кремнеземе и катионированном цео-
лите (специфическая молекулярная адсорбция).
Отметим также, что этот эффект проявляется как в случае полярных
молекул эфира, так и в случае неполярных молекул бензола, этилена и
азота. Наличие и величина дипольного момента сами по себе еще недо-
статочны для проявления специфического взаимодействия. Существенно
благоприятное распределение электронной плотности на периферии соот-
ветствующих звеньев взаимодействующих партнеров, в данном случае
молекул адсорбата и поверхности адсорбента. Это подтверждается также
тем, что теплота адсорбции нитробензола на специфических адсорбентах
второго типа меньше теплоты адсорбции анилина, несмотря на то что ди-
польный момент нитробензола в несколько раз больше, чем у анилина.
Таким образом, старые представления о полярных и неполярных молеку-
лах недостаточны для описания специфических взаимодействий [7]. Это же
относится и к адсорбентам. При сближении ионных радиусов у таких
полярных адсорбентов, как щелочно-галоидные соли, т. е. при переходе
от NaJ к NaCl, а также при уменьшении степени протонизирования гидро-
ксильных групп, т. е. при переходе от поверхностей кислых гидроокисей
(алюмосиликатных катализаторов, кремнезема) к щелочным (NaOH),
специфичность адсорбции уменьшается и даже исчезает [23, 7].
Отметим, наконец, что специфическое взаимодействие должно быть
в значительно большей степени направленным, а поэтому и более чув-
ствительным к изменению температуры, чем неспецифическое. Этот эф-
фект выявляется с помощью газовой хроматографии. Например, из ко-
лонки с цеолитом при 110° выходит вначале этан, затем пропан, а этилен
отстает благодаря сильному специфическому взаимодействию его л-элек-
тронной связи с катионами цеолита. По мере роста температуры пик эти-
лена обгоняет пик пропана [24]. Аналогичные результаты получаются
в других подобных случаях, например при адсорбции цеолитами азота
(л-связи в молекуле) и метана (только о-связи) [2].
20
Изменения инфракрасных и электронных спектров молекул в результате
специфического взаимодействия с гидроксилированной поверхностью
кремнезема и катионированной поверхностью цеолита
Специфические взаимодействия вызывают значительные изменения
в состоянии адсорбированных молекул, которые проявляются в инфра-
красном и в ультрафиолетовом спектре. Наблюдено изменение колеба-
тельного спектра ароматических молекул при их адсорбции на гидро-
ксилированной поверхности кремнезема 125]. При этом происходит
в особенности сильное изменение полосы поглощения внеплоского дефор-
мационного колебания связи СН (чувствительного к перераспределению
электронной плотности л-связей бензольного кольца 126]). Специфи-
ческая адсорбция на гидроксилированной поверхности кремнезема
исследовалась спектроскопически для двух содержащих азот произ-
водных бензола—анилина и нитробензола 127]. Адсорбция анилина вы-
зывает большое смещение максимума
полосы поглощения гидроксильных
групп поверхности, что вместе с фор-
мой контура указывает на образова-
ние сильной водородной связи типа
—S—ОН . . . NH2CeHB. Подобно это-
му положение полосы валентных ко-
лебаний свободных гидроксильных
групп поверхности кремнезема силь-
но смещается при адсорбции амми-
ака 128], а изменение спектра самого
адсорбированного аммиака меньше,
чем в том случае, когда его водород
участвует в образовании водородной
связи. К такому же выводу приводит
анализ колебательного спектра ад-
сорбированного аммиака [29].
В результате воздействия гидрок-
Рис. 6. Инфракрасные спектры Na-фо-
жазита с высоким содержанием алю-
миния и катионов в области валентных
и деформационных колебаний молекул
воды после откачки адсорбированной
воды при 25 (Z), 100 (2), 200 (.?), 300 (4) и
400° (5).
сильных групп поверхности крем-
незема на л-связи бензольного кольца происходит изменение гекса-
гональной симметрии, что в электронном спектре приводит к сдвигу
полос поглощения и к появлению запрещенной полосы электрон-
ного перехода A |,7B2k (0—0) [30]. При адсорбции отмечен сдвиг электрон-
ных полос анилина и нитробензола [30]. У анилина он происходит в ко-
ротковолновую сторону в соответствии с большой энергией образующейся
водородной связи, полоса же поглощения нитробензола смещается в длин-
новолновую сторону. Это различие соответствует меньшему потенциалу
ионизации анилина по отношению к бензолу и, наоборот, большему по-
тенциалу ионизации (т. е. более низкой электронно-донорной способно-
сти) нитробензола. Нужному для специфического взаимодействия усло-
вию отвечает наличие свободных электронных пар у атома азота в моле-
куле анилина и не отвечает распределение электронной плотности в группе
NO2 нитробензола, хотя его общий дипольный момент много больше.
Влияние адсорбционного поля цеолита на инфракрасный спектр
специфически адсорбированной цеолитом молекулы можно наблюдать
на примере адсорбции воды [311, метанола[32], бензола [33] и аммиака [28].
Полученные результаты обсуждаются подробно в [34]. Здесь мы отметим
лишь, что эти исследования привели, в частности, к заключению о раз-
личных типах связи молекул воды и метанола с цеолитом и друг с другом
21
внутри каналов цеолита. Среди этих связей наибольшее значение имеет
связь с катионами цеолита. На рис. 6 показаны некоторые из инфракрас-
ных спектров в области валентных и деформационных колебаний гидро-
ксильных групп молекул воды, полученные при постепенной десорбции
воды из кристаллов синтетического Na-фожазита с высоким содержанием
алюминия и катионов [31J. В области валентных и деформационных
колебаний групп ОН молекул воды при десорбции можно наблюдать
несколько полос, что указывает на различие состояний адсорбированных
молекул воды. Сильная связь осуществляется преимущественно через
взаимодействие свободных электронных пар атома кислорода молекулы
воды с катионами цеолита [34]. Связанные так молекулы воды по отноше-
нию к соседним (адсорбирующимся при более высоких заполнениях),
Рис. 7. Инфракрасные спект-
ры метанола CD3OD, адсорби-
рованного Na-фожазитом, при
указанных у кривых заполне-
ниях.
по-видимому, не могут ориентироваться
наиболее благоприятно для образования
сильных взаимных водородных связей. По-
этому взаимные водородные связи проявля-
ются в спектре интенсивнее при меньшем
содержании в цеолите алюминия, т. е. при
более редком расположении катионов [31].
На рис. 7 показаны инфракрасные спект-
ры CD3OD, адсорбированного Na-фожазитом
при разных заполнениях [32]. Полоса ва-
лентных колебаний О—D у адсорбированных
молекул по сравнению с молекулами в газе
(2725 см-1) смещена в длинноволновую об-
ласть и размыта. Можно заметить по край-
ней мере два различных состояния адсорби-
рованных молекул. Полосы в более корот-
коволновой области проявляются при неболь-
ших и средних заполнениях, т. е. при пре-
имущественном взаимодействии свободных
электронных пар молекул метанола с катио-
нами цеолита. Полоса в более длинноволно-
вой области проявляется при больших заполнениях, т. е. при преиму-
щественном образовании взаимных водородных связей между молекулами
метанола.
Инфракрасный спектр бензола, адсорбированного Na-фожазитом [33],
изменяется в том же направлении, что и на гидроксилированной поверх-
ности кремнезема, но более резко, в соответствии с большей энергией
специфической адсорбции бензола цеолитом.
Изменения в инфракрасном спектре аммиака при переходе из газо-
образного состояния в адсорбированное на поверхностях кремнезема [28] и
алюмосиликатного катализатора [35], несущих гидроксильные группы
с протонизированным водородом, и на катионированной поверхности цео-
лита 128], носят также сходный характер [29] и резко отличаются от
соответствующих изменений при переходе аммиака в твердое состояние.
Анализ колебательного спектра адсорбированного аммиака с помощью
вычисленных зависимостей между колебательными частотами и измене-
ниями коэффициентов матрицы силовых постоянных [29] показал, что
специфическое взаимодействие его молекулы с гидроксилированными по-
верхностями кремнезема и алюмосиликатного катализатора и с катиони-
ровапной поверхностью полостей цеолита происходит в основном за счет
взаимодействия свободной электронной пары атома азота молекулы ам-
миака с протонизированным водородом гидроксильной группы или
катионом поверхности, но не с атомами кислорода этих адсорбен-
тов.
22
Теоретические расчеты потенциальной энергии адсорбции цеолитами
Р. М. Бэррером и С. Василевским [36] был сделан расчет энергии ад-
сорбции иода цеолитами. Однако такой случай не является простейшим,
поскольку молекула иода не обладает шаровой симметрией оболочки и
имеет очень большую поляризуемость. Поэтому в работах П. Бройера,
А. А. Лопаткина и С. Шпигиль рассчитывалась энергия взаимодействия
с различными катионными формами фожазита и молекул благородных
газов и метанола. Подробнее обоснования и схема расчета излагаются
в их статье [37]. Мы остановимся здесь вкратце лишь на результатах.
Рассматривалась локализация молекулы адсорбата непосредственно
над катионом, расположенным в центре плоскости шестичленного кольца
из тетраэдров Si04 и А104 на поверхности кубооктаэдра, обращенной
в большую полость фожазита. 2 * * * Учитывалось взаимодействие со 111 бли-
жайшими атомами кислорода решетки и с центральным катионом. При-
нималось во внимание дисперсионное и поляризационное притяжение.
Полная энергия взаимодействия рассчитывалась по формуле
in in
Ф = —С0-4 2 Ч + В0-А 2 L7}2 - СК-А W-A + ВК-А d-^A + Фпол>
i i
где Co-а и Во-а—константы дисперсионного притяжения и отталкива-
ния для взаимодействия молекулы адсорбата с атомами кислорода
решетки, СК-а и Вк_А — то же для взаимодействия молекулы адсорбата
с катионом, — расстояния между центрами молекулы адсорбата и
атомов кислорода решетки, a —расстояние между центрами мо-
лекулы адсорбата и катиона. Поляризационная энергия Фпм определя-
лась из расчета напряженности электрического поля, создаваемого
в центре молекулы адсорбата зарядом катиона и небольшими отрица-
тельными зарядами, которые принимались равномерно распределенными
по всем атомам кислорода решетки.
Таблица 2
Вычисленные теоретически величины энергии адсорбции цеолитами — Фи опре-
деленные из опытов теплоты адсорбции при малых заполнениях Qo (ккал./моль)
Катионная форма цеолита Аргон Криптон Метан
—ф Qa —ф Qa —Ф Qa
LiX . . . . 3.4 3.3
NaX . . . 3.0 2.8 4.4 4.4 4.4 4.3
КХ . . . . 3.2 3.0 4.6 — — —
СаХ . . . 4.7 5.0 — — — —
SrX . . . . 3.9 3.8 — — — —
ВаХ . . . 3.2 3.3 — — — —
В табл. 2 вычисленные значения энергии взаимодействия для указан-
ной выше локализации сопоставлены с экспериментальными значениями
теплот адсорбции, экстраполированными к нулевому заполнению.
Вычисленные и экспериментальные значения для энергии неспецифи-
ческого взаимодействия близки, и, что особенно важно, вычисленные
2 Это положение катиона определяет и равновесные расстояния до атомов кисло-
рода решетки. Предварительно решением обратной задачи, т. е. исходя из экспери-
ментальных значений энергии адсорбции при малых заполнениях, было найдено, что
центр катионов небольших размеров можно принять лежащим в указанной плоскости.
23
величины правильно передают последовательность энергий неспецифиче-
ского взаимодействия для разных катионов цеолита и разных адсорбатов.
Таким образом, и в этом, более сложном по геометрии, случае неспе-
цифической адсорбции в каналах пористых кристаллов, как и в более про-
стых случаях адсорбции на графите [38—41], окиси магния [42] и неко-
торых солях [43], приближенная теория неспецифического межмолеку-
лярного взаимодействия дает удовлетворительные результаты.
В дальнейшем мы надеемся расширить сопоставление вычисленных
величин с экспериментальными, определив недостающие в табл. 2 теплоты
адсорбции цеолитами X и А. Кроме того, надо вычислить энергии взаимо-
действия для разных положений молекул относительно решетки, учесть
взаимодействия адсорбат—адсорбат и вычислить молекулярно-статическим
путем адсорбционные равновесия. Как одну из важнейших задач в этой
области мы рассматриваем создание более точной и полной теории, охва-
тывающей как неспецифические, так и специфические взаимодействия
различных молекул с цеолитами.
Выводы
Взаимодействие различных молекул с цеолитами определяется, с од-
ной стороны, особенностями химического строения каркаса цеолита —
большой концентрацией атомов кислорода, сосредоточением положитель-
ного заряда в катионах и рассредоточением отрицательного заряда внутри
алюмокислородных тетраздров, а с другой стороны — геометрической
структурой молекулы и характером распределения электронной плотно-
сти в ее звеньях. Молекулы со сферически симметричными электронными
оболочками или имеющие только сг-связи, адсорбируются цеолитами не-
специфически. Энергия адсорбции таких молекул, однако, велика глав-
ным образом за счет большой концентрации атомов кислорода в каркасе
цеолита. Энергия взаимодействия в случае неспецифической адсорбции
простейших молекул на различных катионных формах синтетического
фожазита может быть вычислена на основе приближенной теории меж-
молекулярных взаимодействий в удовлетворительном согласии с опытом.
Молекулы, обладающие звеньями с периферически сосредоточенной
электронной плотностью (л-связи, свободные электронные пары у атомов
кислорода и азота воды, аммиака, спиртов, аминов и т. п.), кроме то-
го, взаимодействуют с катионами цеолитов и специфически. Это специ-
фическое молекулярное взаимодействие резко увеличивает энергию ад-
сорбции и изменяет состояние адсорбированных молекул, что отчетливо
проявляется в их инфракрасном спектре.
ЛИТЕРАТУРА
1. R. М. В а г г е г. The Structure and Properties of Porous Materials, ed. D. Eve-
rett, F. Stone, London, 1958, 6; Brit. Chem. Engng., 1959. May, 1.
2. А. В. К и с e л e в, Ю. Л. Ч ер пень ков а, Я. И. Яшин, Нефтехимия,
1965, 5, 141.
3. В. М. В а г г е г, New Scientist, 1959, 18, 497.
4. G. L. Kington. The Structure and Properties of Porous Materials. Ed. D. Eve-
rett, F. Stone, London, 1958, 59.
5. С. П. Жданов, настоящий сборник, стр. 178.
6. А. В. Киселев, А. А. Лопаткин, Кинетика и катализ, 1963, 4, 786.
7. А. В. Киселев, ЖФХ, 1964, 38, 2753.
8. М. R. В a s i 1 a, J. Chem. Phys., 1961, 35, 1151.
9. S. Ross, W. W. Pultz, J. Coll. Sci., 1958, 13, 397.
10. Л. Д. Б e л я к о в а, А. В. К и с е л е в, Изв. АН СССР, сер. хим., 1964, 1164.
11. D. G г a h a m, J. Phys. Chem., 1962, 66,1815; А. В. К и с е л е в, М. В. С е р д о-
б о в, Колл, ж., 1963, 25, 543.
12. И. Ю. Б а б к и н, А. В. К и с е л е в, ЖФХ, 1962, 36, 2448; Б. Г. А р и с т о в,
И. Ю. Б а б к и н, А. В. К и с е л е в, Колл, ж., 1962, 24, 643.
24
13. О. М. Джигит, А. В. К и с е л е в, Г. Г. М у тти к, Колл, ж., 1963, 25, 34.
14. О. М. Д ж и г и т, С. П. Ж д а в о в, К. Н. М и к о с, настоящий сборник, стр. 46.
15. Н. Н. А в г у л ь, А. В. К и с е л е в, А. А. Л о п а т к и и, И. А. Л ы г и н а,
М. В. С е р д о б о в, Колл, ж., 1963, 25, 129.
16. R. М. Barre г, J. W. Stuart, Proc. Boy. Soc., 1959, A 24(1, 464.
17. А. Г. Б e в у с, В. П. Д p e в и и г, А. В. К и с е л е в, ЖФХ, 1964, 38, 59.
18. В. М. G а 1 е, В. А. В е е Ь е, J. Phys. Chem., 1964, 68.
19. Л. Д. Б е л я к о в а, А. В. К и с е л е в, Н. В. К о в а л е в а, ДАН СССР,
1964, 157, 646.
20. А. В. Киселев, ЖФХ, 1961, 35, 233; Вестн. Моск, унив., сер. хим., 1962,
1, 3.
21. И. Ю. Б а б к и и, А. В. К и с е л е в, ЖФХ, 1963, 37, 228; О. М. Джигит,
А. В. Киселев, Г. Г. Муттик, Колл, ж., 1961, 23, 504.
22. Н. Н. А в г у л ь, Г. Г. М у т т и к, настоящий сборник, стр. 75.
23. С. G. S с о 11. Gas Chromatography. Ed. M. van Swaay, London, 1962, 46.
24. А. В. К и с e л e в, Я. И. Яшин, ЖФХ, 1963, 37, 2614.
25. Г. А. Г а л к и н, А. В. К и с е л е в, В. И. Л ы г и н, Кинетика и катализ,
1964, 5, 1040.
26. В. D. К г о s s, V. A. F a s s е 1, М. М argosch es, J. Chem. Soc., 1956, 78,
1332.
27. В. H. А б p а м о в, А. В. К и с e л e в, В. И. Л ы г и н, ЖФХ, 1964, 38, 1044.
28. А. В. К и с е л е в, В. И. Л ы г и н, Т. И. Т и т о в а, ЖФХ, 1964, 38, 2730.
29. В. Н. А б р а М о в, А. В. К и с е л е в, В. И. Л ы г и н, ЖФХ, 1964, 38, 1867.
30. В. Н. А б р а м о в, А. В. К и с е л е в, В. И. Л ы г и н, ЖФХ, 1963, 37, 2783.
31. С. П. Ж д а н о в, В. И. Л ы г и н, Т. И. Т и т о в а, настоящий сборник, стр. 53;
С. П. Жданов, А. В. К и с е л е в, В. И. Л ы г и н, Т. И. Титова,
ЖФХ, 1964, 38, 2403.
32. А. В. К и с е л е в, Л. К у б е л к о в а, В. И. Л ы г и н, ЖФХ, 1964, 38, 2719.
33. В. Н. А б р а м о в, А. В. К и с е л е в, В. И. Л ы г и н, ЖФХ, 1963, 37, 1156.
34. В. И. Л ы г и н, настоящий сборник, стр. 58.
35. Л. М. Р о е в, В. Н. Ф и л и м о. н о в, Оптика и спектроскопия, 1958, 4, 328.
36. В. М. В а г г е г, S. W a s i 1 е w s k i, Trans. Faraday. Soc., 1961, 57, 1140.
37. П. Бройер, А. А. Лопаткин, С. Шпигиль, настоящий сборник, стр. 36.
38. Н. Н. А в г у л ь, А. А. И с и р и к я н, А. В. К и с е л е в, И. А. Л ы г и н а,
Д. П. П о ш к у с, Изв. АН СССР, ОХН, 1957, 1314; Н. Н. А в г у л ь,
А. В. К и с е л е в, И. А. Л ы г и и а, Д. П. П о ш к у с, Изв. АН СССР,
ОХН 1959 1196.
39. A. D. Crowell, В. В. S t е е 1 е, J. Chem. Phys., 1961, 34, 1347.
40. S. С г о s s, G. Р. О 1 i v i е г, Advances in Chem., 1961, 33, 309.
41. J. В. S a ms, Trans. Faraday. Soc., 1964, 60, 149.
42. А. В. К и с e л e в, Д. П. П о ш к у с, ЖФХ, 1958, 32 , 2824.
43. W. J. О г г, Trans. Faraday. Soc., 1939, 35,1247; Proc. Hoy. Soc., 1939, A 173, 349;
Усп. химии, 1941, 10, 474.
К ВОПРОСУ О ТЕОРИИ УРАВНЕНИЯ ИЗОТЕРМЫ СОРБЦИИ ПАРОВ
НА ПОРИСТЫХ СОРБЕНТАХ ТАК НАЗЫВАЕМОГО ПЕРВОГО
СТРУКТУРНОГО ТИПА
Л. В. Радушкевич
Известно, что уравнение вида
W = Wo exp [—(1)
хорошо оправдывается на опыте для сорбентов, значительно отличаю-
щихся друг от друга по своей химической природе, например для цеоли-
тов и для активных углей. Это обстоятельство, а также температурная
инвариантность приводят к выводу, что данное уравнение связано с об-
щими свойствами микропористых систем. Для создания теории сорбции
паров в таких системах возможны два взаимно не исключающих подхода,
из которых один требует представлений о модели пористой структуры,
а другой находится в связи с некоторыми положениями физической ста-
тистики. Мы здесь рассмотрим возможности второго метода.
25
1. Основой нашего взгляда является допущение об объемном запол-
нении сорбатом свободных полостей в этих системах. Чрезвычайная малость
микропор дает возможность применять к таким системам данные, извест-
ные в так йазывасмых клатратных соединениях, где устанавливаются
отношения «гостя» и «хозяина». Система вместе с сорбатом представляет
собой квазиоднородную структуру, подобную бинарной смеси, например
раствору. Если пользоваться этими представлениями, то вообще исклю-
чается понятие удельной поверхности этих систем, а следовательно, и су-
ществование внутренних поверхностей сорбата в микропорах. Ясно, что
схема последовательно заполняемых пор от наиболее активных мест к ме-
нее активным выпадает, так же как и понятие о распределении обычно
вводимого «адсорбционного потенциала». Очевидно, объем заполнения
является объемом для всех полостей взятой навески «хозяина» и потому
есть величина постоянная: IK^const. Наиболее правдоподобна следую-
щая схема процесса: при введении некоторого количества сорбента в замк-
нутый большой объем (термостат) с течением времени происходит запол-
нение объема Wo сорбатом и постепенно устанавливается равновесие,
причем, так как система квазиоднородна, то равновесное давление р
существует только снаружи и отвечает измеримому на опыте давлению
пара. В этих условиях можно говорить только о плотности сорбата внутри.
По мере дальнейшего пуска пара в систему плотность «гостя» в системе
возрастает и растет наружная упругость пара. Пределом является равно-
весное значение р=р,- Следовательно, никакого объема W по уравнению (1)
не существует вообще, а все определяется плотностью или величиной моль-
ного объема сорбата. Однако по всем данным плотность даже при неболь-
ших р/р, будет сравнительно велика и близка к плотности нормальной
жидкости.
Все эти соображения относятся к некоторой части реального сорбента
(куска), где не имеется участков со средними порами или с макропорами,
т. е. к фрагменту, состоящему из одних микропор, где уравнение (1) наи-
более оправдывается на опыте. Это может быть небольшой кусок сарано-
вого активного угля или кристалла цеолита.
Обычно уравнение (1) преобразуется в уравнение изотермы сорбции
с допущениями
а = р0И\ а0 = р0И70, (2)
где плотность р0 принимается постоянной. Теперь из изложенного
ясно, что более правильно вместо условий (2) полагать
а = рЖ0, а0=р0Ж0, (2')
и тогда уравнение (1) принимает вид:
р = рое~“е2 или v = гоеда2. (3)
Очевидно, фиктивное W выпадает из системы понятий. Раньше допу-
скалось, что при постоянных р0 и v0 величина W переменна, теперь счи-
таем, что постоянно Wo, арии переменны.
2. В поисках рациональной интерпретации уравнения (1) мы обратили
внимание на вид зависимости от е в (1) или в (3). В экспоненте здесь со-
держится квадрат величины, имеющей размерность энергии. В статистике
Гиббса в экспоненте для распределения содержится первая степень энер-
гии. Квадратичная форма, наблюдаемая на опыте с данными пористыми
системами, наводит на мысль о роли дисперсии энергии, которая, как из-
вестно, распределена по закону Гаусса.
С другой стороны, известно, что системы, аналогичные по своей струк-
туре изучаемым нами микропористым телам, проявляют ряд аномальных
свойств. Так, для рыхлых цепочечных или сетчатых структур, например
26
для активных углей, силикатов, стекол и цеолитов, известны аномалии
теплоемкостей при низких температурах, когда имеют место значительные
отступления от закона Дебая. Сейчас имеется большая литература по
этому вопросу [1]. Установлены и другие аномалии. Колебания в таких
рыхлых решетках связаны с рядом свойств этих систем, отличных от моно-
кристаллов с нормальной структурой. Ясно, что всякая жидкость, обра-
зующаяся в таких системах, как «гость у хозяина», будет участвовать
в этих колебаниях; это должно вызывать большие флюктуации различных
термодинамических величин, например полной энергии.
Наиболее полное решение задачи о флюктуации энергии в нашей си-
стеме «гость у хозяина» можно дать, рассматривая эту систему подобной
некоторому бинарному раствору. В таком случае из общей теории флюк-
туации термодинамических величин следует необходимость анализа слож-
ных квадратичных форм с учетом переменного числа частиц «растворен-
ного вещества». Переход к состоянию равновесия дается анализом знаков
у якобианов, откуда получается условие для частных производных хими-
ческих потенциалов. Однако нельзя упускать возможности в виде первого
приближения дать упрощенное решение задачи, верное для достаточно
больших величин сорбции, когда плотность сорбата в пористой системе
достаточно велика. Мы здесь рассмотрим этот упрощенный подход, не
касаясь более строгого анализа.
3. Напомним прежде всего вывод (упрощенный) важного выражения
для отклонения энергии сорбата от среднего значения. Пусть пористое
тело находится в термостате и содержит некоторое количество сорбтива.
В эту систему мы включаем только сорбат вместе с термостатом, тогда как
сорбент является лишь источником флюктуаций. Итак, имеем два тела
(\ и С2, первое из них сравнительно невелико, тогда как второе (термо-
стат) имеет практически неограниченные размеры. Оба они обмениваются
энергиями.
Для тела Сг средняя энергия равна Ev а для второго она есть £2.
Пусть небольшое количество энергии е может переходить от С2 к Сг
или обратно. Тогда
Е1 = Ё1-}-е и одновременно Е2 = Ё2 — е. (4)
Пользуясь аналогом энтропии по Гиббсу и допуская каноническое
распределение, найдем статистические веса или число комплексий
В
Р—ehT .
При изучаемом переходе е от С2 к (\ имеем изменение числа ком-
плексий, если е лежит от е до е —<7е:
dP = Р± (Ё1 + е) Р2 (Ё2 - е) de.
Разложим функции РД/^Д-е) и Р2(Ё2— е) в ряд Тэйлора. Тогда
е д In е2 d2 In Pj е д In Р2
InP^InPj+j, дЕ^ 4-2! dEl 1п р2 =1п ^2 — 7] dEi + • • (5)
Для термостата достаточно ограничиться двумя членами. Имеем
Рг = ект; Pg = ект . Поэтому
£, dlnPi 1 , _ Ё2 dlnP1 дЬЛ 1
m дЕх ~ кТ • inP^~kT' дЕг ~ дЕг ~ кТ ’
d21nPj 1 дТ
дЁ{ =~ кТИ’-дЁ^- Н»’
27
дЕг д2 In Pj д2 In Pj 1 d In P2 1
дТ —с» и дЕ2 = d£l = — kfz’c” далее: dE2 ==~kf'
Складывая уравнения (5), имеем
PiP2 = Pi--P2exp(-2^r75),
и потому
dw = dP ~ РГР2 ехр (- 2^) .
Выражение найдем, как всегда, из интеграла нормировки.
Очевидно, плотность вероятности этого Гауссова распределения есть
/ е2 \
<у = ехр^—2с„кТг)’
Здесь ё2 — 2с„кТ2 является дисперсией энергии в системе, а 8 =
= ^ё2—\/2сскТ — флюктуацией.
4. Естественно предположить, что наблюдаемые отступления плот-
ности жидкости, находящейся в пористой системе от нормального зна-
чения р0, вызываются флюктуациями энергии в этой системе, контак-
тирующей с термостатом. Это позволяет сблизить формулы (3) и (6) и,
учитывая, что плотность всегда пропорциональна числу частиц, на-
писать:
В данном случае мы имеем дело с большими флюктуациями. Вели-
чины в формуле (7) отнесем к 1 молю, и тогда мы переходим к удель-
ным значениям е и с„. Умножим числитель и знаменатель дроби в (7)
на число Авогадро, и тогда:
e7V = e1 кал ./моль • град.;
Имеем:
Ce==crZV кал./моль • град.; kN = R кал./моль • град.
Р = Ро ехР
Г Е2 1
L 2c„RT2 J
(8)
Флюктуации термодинамических величин связаны с работой флюк-
туации, поэтому приближенно допустимо принять, что Sj есть диффе-
ренциальная мольная работа при выборе состояния обычной жидкости
за стандартное состояние. Во всяком случае было бы неверным счи-
тать, что ех является некоторым «потенциалом», т. е. функцией состоя-
ния. Отсюда видим, что
Вводя зто выражение в формулу (8), находим
(9)
Особенностью и недостатком полученного соотношения является то,
что в нем учитываются свойства и поведение пара, заполняющего
сорбент, но не отражены свойства самого сорбента. Этого и следовало
ожидать, поскольку рассматривались не детали участия последнего
в обмене энергии, а только то, что сорбент предоставляет свой объем
«гостю» и является источником беспорядочных колебаний энергии
в системе. На самом деле все расчеты по вычислению дисперсии сле-
28
дует производить с бинарной системой пар—сорбент в термостате.
Впрочем, формула (8) дает возможность сравнить опытные данные
с теорией, чего нельзя упускать из вида. На основании опытных дан-
ных неоднократно были получены коэффициенты аффинности р харак-
теристических кривых. Для их получения обычно принимается условие
для двух сравниваемых веществ W1=W2. Принимая обычную формулу
для изотермы сорбции вида
Wo Г ВТ2 I р8\2-|
«=—J.
(Ю)
находим коэффициенты р для разных веществ по отношению к условно
принятому стандартному пару. Применяя уравнение (10) и графическое
построение, получаем значения р. В нашей теории раскрывается зна-
чение константы а. Из соотношения (8) видно, что
а = (2С,№)-1.
Сопоставляя (9) и (10), легко находим
где С'с — мольная теплоемкость данной жидкости, заполняющей поры
сорбента, a C't — мольная теплоемкость жидкости стандартного веще-
ства. Считая эти величины близкими к теплоемкостям нормальных
жидкостей, можно взять значения Сс из справочников, где обычно при-
водятся удельные теплоемкости се.
Тогда
ёх-
Мы сопоставили опытные значения Р для различных. веществ на ак-
тивном угле и цеолите с данными расчетов по формуле (11).
Значения коэффициента аффинности (опытные и расчетные) для
различных веществ на активном угле и цеолите
Пар м кал./г • град. Ррасч ₽оп ₽расч/Роп
Активный уголь
СзН8 44 0.58 0.896 0.715 1.25
н-С4Н10 58 0.55 1.000 0.865 1.11
72 0.527 1.097 ' 1.10 0.98
н-С-Н14 86 0.60 1.278 1.30 0.95
СС14 154 0.201 0.954 0.96 0.995
n2 28 0.474 0.65 0.32 2.03
Цеолит
^6-^12 84 0.506 1.15 1.29 0.89
C6HSN 79 0.431 1.03 2.30 0.45
Н2О 18 1.00 0.76 0.33 2.3
N2 28 0.474 0.65 0.20 3.25
Некоторые результаты в виде примеров приведены в таблице.
В графе ррасч показаны коэффициенты аффинности, рассчитанные
по формуле (11). В графе роп даны опытные значения этих коэффици-
ентов по результатам работ [2, 3]. В качестве стандартного вещества
принимался бензол, для которого М~ 78 и с„ = 0.406 кал./г • град,
(при 20° С).
29
В графе с„ приведены справочные данные для удельных теплоемкостей
жидкостей взятых веществ. Сравнивая Ррасч и (30п, можно видеть, что они
различаются между собой лишь в пределах одного порядка и по большей
части отношение их близко к единице. Все же отступления имеют место,
особенно для цеолита, что можно объяснить сложной, хотя и правильной
структурой последнего и наличием в ней ионов. Во всяком случае предло-
женная грубая схема расчета дает удовлетворительные результаты, хотя
и нуждается в уточнениях, о которых было выше сказано.
Выводы
Сделана попытка выяснить физическую сущность, лежащую в основе
уравнения изотермы сорбции для пористых сорбентов первого структурного
типа. При этом была использована теория флюктуации энергии, откуда
можно было получить значения коэффициентов аффинностей, близкие
к опытным величинам.
ЛИТЕРАТУРА
1. Н. А. Ч е р н о п л е к о в, Усп. химии, 1956, 25, 3, 288.
2. М. М. Дубинин, Е. Д. Заверина и Д. П. Тимофеев, Изв. АН СССР,
ОХН, 1957, 6, 670.
3. М. М. Д у б и н и н с сотр. Изв. АН СССР, сер. хим., 1964, 9, 1965.
ОСОБЕННОСТИ АДСОРБЦИИ СМЕСЕЙ ПАРОВ НА МИКРОПОРИСТЫХ
АДСОРБЕНТАХ
Б. П. Беринг, В. В. Берлинский, С. И. Суринова
При изучении физической адсорбции бинарных смесей газов и паров
на твердых адсорбентах удается обнаружить некоторые особенности ад-
сорбционного поведения таких многокомпонентных систем, связанные
со структурными характеристиками адсорбента. Исследование адсорбции
смесей паров позволяет в ряде случаев получить более определенные вы-
воды о механизме процесса сорбции и его связи со структурой адсорбента,
чем зто можно сделать на основании изучения изотерм адсорбции инди-
видуальных веществ. Ниже будет показано, что экспериментальные дан-
ные по адсорбции смесей паров могут служить веским дополнительным
аргументом в пользу представлений, развитых в статье М. М. ДубининаН],
о структуре адсорбентов и о принципиальном различии адсорбционных
свойств сравнительно крупнопористых или вообще непористых адсор-
бентов (силикагели, окислы, непористые кристаллы, графитированные
сажи) и микропористых адсорбентов, характеризующихся очень тонкими
порами, близкими по величине к размерам молекул (активные угли, цео-
литы).
Наиболее отчетливо различие в адсорбционном поведении смесей паров
двух веществ на крупнопористых и микропористых адсорбентах прояв-
ляется при графическом изображении зависимости величин адсорбции
а2 одного компонента смеси паров (брлее адсорбируемого в области малых
заполнений) от величины адсорбции ах другого (менее адсорбируемого)
компонента смеси при постоянном составе газовой фазы. Рассмотрение
подобных кривых привело нас к выводу, что все изученные системы можно
подразделить на два основных типа, соответствующих двум указанным
группам адсорбентов.
30
В качестве примера адсорбции бинарных смесей паров на сравни-
тельно-крупнопористых адсорбентах рассмотрим подробно изученную нами
систему ацетон—хлороформ—силикагель [2]. На рис. 1 показано семей-
ство кривых — при Ng* — const, изображающих зависимость ве-
личины адсорбции а2 ацетона от величины адсорбции ах хлороформа
(ммоль/г), при постоянной мольной доле Ng* (более адсорбируемого)
ацетона в газовой фазе. Эти кривые построены на основании экспери-
ментальных данных, полученных при температуре 35° на силикагеле
с удельной поверхностью по азоту <$’БЭТ = 580 м2/г и общим объемом
пор v„ = 0.47 см3/г. Координаты точек слегка выпуклой линии ЛВ дают
величины предельной сорб-
ции компонентов из насы-
щенного пара, а поле ОЛВ
определяет всю область
изменения величин а2 и ог
Прерывистые линии, соот-
ветствующие постоянству
относительного давления
смеси паров h12 = const,
проведенные для ряда зна-
чений Л12 от 0.05 до 0.6,
являются в хорошем при-
ближении прямыми лини-
ями. Изображенные на ри-
сунке кривые Ng* — const
проведены для значений
Ng* от 0.1 до 0.9 с интер-
валом 0.2. Каждая из
этих кривых выражает
процесс одновременной
сорбции обоих компонен-
тов при изотермическом
сжатии над адсорбентом
в равновесных условиях
весьма большого количест-
ва пара неизменного со-
става Ng* от очень малого
начального давления до
относительного давления
пара смеси веществ над
которого определенного состава Ni*. Зависимость Ng* от /V;., для би-
нарных растворов ацетона в хлороформе хорошо изучена в работе [3].
Все кривые Ng* — const на рис. 1 сперва круто поднимаются вверх,
в соответствии с резко выраженной избирательностью адсорбции аце-
тона, а затем уменьшают свой наклон и идут далее в виде прямоли-
нейных отрезков (например, CD) до пересечения с ограничивающей
линией АВ. Если вдоль линейного отрезка кривой N g* = const (напри-
мер, для Л^2 = 0.3) найти приращения адсорбции обоих компонентов Дп2
и Д<7Х между точками С и D, то отношение оказывается в пределах
7V, 1
ошибок опыта равным отношению , т. е. отношению мольных долей
п1,
обоих компонентов такого объемного раствора, который образуется
при обычной конденсации пара состава Ng* (в отсутствие адсорбента)
при давлении насыщения. Отсюда следует, что вдоль каждого линей-
ного участка CD кривой Ng* — const после точки С сорбируется рас-
й12 = 1, т. е. до давления насыщенного
равновесным объемным раствором не-
31
твор постоянного состава, причем состав этого сорбционного раствора
равен составу объемного раствора /V(1, находящегося в равно-
весии с насыщенным паром заданного состава Ng2. Угловой коэффи-
циент каждого линейного участка CD для любой кривой = const,
о « I,
равный соответствующей величине ~, однозначно определяется из
диаграммы состояния объемных растворов компонентов, и составы рас-
творов, сорбирующихся вдоль прямых CD, совершенно не зависят от
свойств адсорбента.
На основании сказанного можно заключить, что при сжатии над ад-
сорбентом пара постоянного состава в области малых давлений оба ком-
понента адсорбируются на свободной поверхности твердого тела, и здесь,
при адсорбции в первом слое, полностью проявляется специфичность дей-
ствия адсорбционного поля, определяющего коэффициент избирательно-
сти и величину адсорбции из раствора. Однако после заполнения при-
мерно одного статистического адсорбционного слоя (отмеченного на рис. 1
линией MtM2) дальнейший процесс сорбции теряет свою специфичность.
В этой области последующее сжатие пара приводит к появлению конден-
сированной фазы, состав которой резко отличен от состава заполненного
монослоя и равен составу жидкости, получающейся при обычной конден-
сации этого пара. Сорбция в области CD может протекать или по механизму
полимолекулярной адсорбции, или по механизму капиллярной конден-
сации. В рассматриваемом случае в связи с наличием области сорбцион-
ного гистерезиса на индивидуальных изотермах адсорбции в области CD,
мы, по-видимому, имеем дело с процессом капиллярной конденсации.
Отметим, что для этой системы капиллярная конденсация начинается
значительно раньше точки начала сорбционного гистерезиса,. как это
видно на рис. 1, где линия GH проведена через точки начала гистерезиса,
непосредственно определенные на индивидуальных изотермах адсорбции
компонентов и вычисленные по уравнению Кельвина для ряда значений
Ng,. Каждая из кривых 7VP,=const сохраняет линейность в некоторой
области, расположенной слева от кривой GH. Вероятно, в этой области
происходит обратимая капиллярная конденсация в зазорах между кон-
тактирующими частицами силикагеля в соответствии с механизмом, рас-
смотренным «Л. В. Радушкевичем [4].
При изучении адсорбции той же бинарной смеси веществ на другом,
более крупнопористом образце силикагеля, мы получили аналогичное
семейство кривых 7Vff.=const, отличающихся только большей протяжен-
ностью линейных участков CD в соответствии с большей величиной объема
пор и более развитой петлей гистерезиса для этого силикагеля. Наряду
с этим при обработке экспериментальных данных Бриглеба и Шольце [5]
по адсорбции смесей ацетона и хлороформа на стеклянных шариках мы
также нашли, что в области полимолекулярной адсорбции или капилляр-
ной конденсации кривые Ng,=const имеют совершенно те же наклоны,
как и для случая адсорбции на силикагеле. Качественно совершенно
такую же картину мы получили, обрабатывая данные Арнольда [6] по
адсорбции смесей паров азота и кислорода на анатазе при 78° К. Кривые
7Vffj=const, построенные по данным этой работы, обладают весьма про-
тяженной линейной областью CD, причем экспериментально определен-
ные наклоны этих прямых очень хорошо соответствуют величинам накло-
нов, вычисленным по диаграмме состояния объемных растворов в системе
кислород—азот.
На основании сказанного можно думать, что при адсорбции на непо-
ристых твердых телах и на относительно широкопористых адсорбентах,
как при полимолекулярной адсорбции, так и при капиллярной конден-
сации, сорбция во втором и последующих слоях неспецифична и приводит
32
к образованию конденсированной фазы, состав которой в первом прибли-
жении определяется только природой адсорбируемых веществ и не зави-
сит от свойств адсорбента.
Для рассматриваемых адсорбентов во всех случаях вдоль линий
Ne,— const адсорбция каждого компонента монотонно возрастаете рос-
Рис. 2.
том общего давления р12, как это видно
на рис. 2, где для системы ацетон—хло-
роформ—силикагель изображено изменение
адсорбции ацетона (кривая а2) и хлороформа
(аД с повышением относительного давле-
ния h^ — ^- вдоль кривой 7Vff2 = 0.5.
Р«12
Совершенно иной характер имеют кри-
вые N!h — const при адсорбции бинарных
смесей на микропористых адсорбентах. На
рис. 3 показаны кривые зависимости адсорб-
ции диэтилового эфира а2 от адсорбции хло-
ристого этила ctj при постоянных значениях
мольной доли Ng„ диэтилового эфира в га-
зовой фазе, полученные на основании экс-
периментального изучения в нашей лабора-
тории [7] системы (С2Н5)2О—С2Н5С1—актив-
ный уголь при 50°. Кривые 7VPl = const,
ний мольной доли эфира 0.3, 0.5, 0.6, 0.8 и
маются, а затем проходят через максимум (особенно отчетливо
ный при А'Л=0.3 и 0.5) и при дальнейшем росте общего давления р12
построенные для
0.9, сперва круто
значе-
подни-
замет-
отклоняются к оси абсцисс. Иными словами, при изотермическом сжа-
тии над адсорбентом весьма большого количества пара постоянного
состава сперва адсорбция каждого компонента растет с ростом р12,
а затем адсорбция аг хлористого этила продолжает расти, а адсорб-
ция а2 диэтилового эфира начинает убывать, т. е. при больших запол-
нениях адсорбционного объема происходит частичное вытеснение эфира
хлористым этилом. Этот процесс вытеснения более адсорбируемого ком-
понента менее адсорбируемым особенно отчетливо виден на рис. 4,
3 Цеолиты
33
где показаны изотермы адсорбции эфира (кривая о2) и хлористого этила
(кривая ctj) в сечении Ngi = 0.3. В отличие от кривых рис. 2 с ростом
общего давления р12 адсорбция хлористого этила все время растет,
тогда как адсорбция эфира проходит через максимум и затем
убывает. Для рассматриваемой системы эффект вытеснения, веро-
ятно, существует и при 7V^^>0.6, однако эта область не была нами
изучена экспериментально, так как прибор не был рассчитан для дав-
лений, больших атмосферного.
Кривые = const, проходящие через максимум, были получены
нами во всех случаях адсорбции бинарных смесей паров на активных
углях и на синтетических цеоли-
тах. В качестве иллюстрации мы
приводим на рис. 5 кривые
Nt2= const для системы хлори-
стый этил (п2)—вода (flj)—актив-
ный уголь [8] при значениях
от 0.1 до 0.4, где эффект выте-
снения водой хлористого этила
особенно отчетливо заметен. Ка-
чественно совершенно такие же
кривые Nд.. = const были получе-
ны нами для системы н-гептан —
вода—активный уголь [2], в ко-
торой при больших давлениях
также наблюдалось вытеснение
гептана водой. Наконец, на рис. 6
приведена кривая /Vft = 0.5 для
случая адсорбции смесей диэти-
лового зфира (а2) и хлороформа
(ат) на цеолите NaX при 60°
[9J, а на рис. 7 показаны изотермы адсорбции эфира (кривая
а2) и хлороформа (кривая аД в сечении 7Vff2 = 0.5. Изотерма ад-
сорбции эфира па этом рисунке также обладает отчетливым макси-
мумом и областью, соответствующей процессу вытеснения эфира хло-
роформом. На основании экспериментальных данных Бэррера и Ро-
бинса [10] по адсорбции смесей Ar-\-N2 на шабазите можно показать,
что/ и для этой системы кривые Ng„ — const имеют форму кривой (рис. 6).
34
В связи с тем, что адсорбционный объем микропористых адсорбентов
практически заполняется уже при сравнительно небольших давлениях,
а коэффициент адсорбционной избирательности обычно убывает с ростом
общего давления, формально можно показать [11], что при этих условиях
величина аг должна расти, а величина а2 должна убывать с ростом рк.
Наряду с этим возможно и другое, физически более ясное, объяснение
эффекта вытеснения. Для системы этиловый эфир—хлористый этил—ак-
тивный уголь, изученной при 50 и 71°, удалось качественно показать,
что если в области восходящих ветвей кривых ?Vff2=const дифференциаль-
ная теплота адсорбции более адсорбируемого компонента Q2 больше
теплоты адсорбции менее адсорбируемого компонента Qi, то вдоль нисхо-
дящих ветвей этих кривых разность Q2—меняет знак и в области вы-
теснения Qi > Q2. Нам представляется ве-
роятным, что обращение знака разности
Q2—после максимума на кривых Ngt=
=const происходит во всех случаях адсорб-
ции на микропористых адсорбентах, од-
нако пока мы можем рассматривать это
лишь как правдоподобную гипотезу, под-
твержденную только для одной системы.
Во всяком случае во всех без исключе-
ния примерах адсорбции бинарных смесей
паров на микропористых адсорбентах
кривые Д’ff2=const имеют форму, показан-
ную на рис. 3. Таким образом, кривые
?V^=const на рис. 1 и 3 можно счи-
тать типичными для адсорбции двойных
смесей паров на широкопористых (или не-
пористых) и микропористых адсорбентах.
Кривые рис. 3 особенно ясно указы-
вают на отсутствие процесса капилляр-
ной конденсации при заполнении практически всего адсорбционного
объема микропористых адсорбентов. Действительно, в этом случае мы не
находим линейных участков кривых Ng2—const с положительными угло-
выми коэффициентами, определяемыми из диаграммы состояния объемных
растворов компонентов. Как было показано выше, процесс капиллярной
конденсации неспецифичен, так как состав жидкости, конденсирующейся
в капиллярах, не зависит от природы адсорбента. В то же время наклоны
кривых Ng= const (рис. 3) при больших давлениях (вблизи насыщения)
остаются отрицательными и определяются одновременно свойствами
адсорбента и свойствами бинарной смеси. Иными словами, вся область
изменения переменных а2 и т. е. все адсорбционное пространство микро-
пористых адсорбентов при любых степенях заполнения обладает адсорб-
ционной специфичностью, и, следовательно, процесс поглощения вещества
микропористым адсорбентом остается адсорбционным Процессом
вплоть до предельного заполнения всего объема микропор. В этом мы
видим коренное различие адсорбционных свойств крупнопористых (или
непористых) и микропористых адсорбентов.
В заключение еще раз подчеркнем, что в свете проведенного анализа
цеолиты несомненно следует отнести к классу микропористых адсорбентов.
ЛИТЕРАТУРА
1. М. М. Дубинин, настоящий сборник, стр. 5.
2. Б. П. Беринг, Докт. дисс., М., 1957.
3. J. Z a w i d s k i, Z. Phys. Chem., 1900, 35, 129.
4. Л. В. P а д у in к e в и ч, Изв. АН СССР, ОХН, 1952, 6, 1008.
3*
35
5. G. В г i g 1 e b, H. Scholze, Monatshr. f. Chem., 1954, 85, 731.
<5. J. R. Arnold, J. Am. Chem. Soc., 1949, 71, 104.
7. Б. П. Б e p и н г, В. В. Серпинский, С. И. С у p и и о в а, ДАН СССР,
1963 153 1 129.
8. Б. П. Б е р и н г, В. В. С е р п и и с к и й, Изв. АН СССР, ОХН, 1959, 7, 1186.
9. Б. П. Б еринг, В. В. С е р п и н с к и й, С. И. С у р и н о в а, Изв. АН СССР,
сер. хим., 1964, 7, 1309.
10. R. М. В а г г е г, А. В. R о b i n s, Trans. Faraday. Soc., 1953, 49, 929.
И. Б. П. Беринг, В. В. Се рпинский, С. И. С у р и и о в а, ДАН СССР,
1964, 154, 6, 1417.
РАСЧЕТ ЭНЕРГИИ ВЗАИМОДЕЙСТВИЯ ПРОСТЫХ МОЛЕКУЛ
С РАЗЛИЧНЫМИ КАТИОННЫМИ ФОРМАМИ ЦЕОЛИТА ТИПА X
П. Бройер, А. А. Лопаткин, С. Шпигилъ
Теоретический расчет потенциальной энергии молекул в поле адсор-
бента представляет большой интерес как для теории адсорбции, так и для
теории межмолекулярных взаимодействий. Очевидно, такой расчет мо-
жет быть проведен только в том случае, если достаточно хорошо известна
геометрия поверхности.
В настоящее время опубликовано значительное число подобных рас-
четов [1 — 4]. В основном они относятся к случаям, когда адсорбент
неполярен (сажа), а молекулы адсорбата просты и также неполярны
(например, благородные газы); при этом можно ограничиться определе-
нием дисперсной энергии и энергии отталкивания. Более сложными и
требующими применения квантовой механики являются случаи взаимо-
действия полярных групп поверхности или катионов с полярными моле-
кулами или молекулами, содержащими л-электронные связи [5].
Синтетические цеолиты представляют собой кристаллические адсор-
бенты, структура которых достаточно хорошо известна. В эту структуру
входят атомы кремния и алюминия, атомы кислорода, по-видимому, не-
сущие некоторый отрицательный заряд и катионы. Имеется только одна
работа [6], где рассматривалось взаимодействие молекулы адсорбата со
значительным количеством атомов кислородного остова (влияние катионов
при этом не учитывалось). В других работах, наоборот, рассматривалось
только взаимодействие с катионами [7, 8].
В настоящей статье сделана попытка рассчитать энергию взаимодей-
ствия простых молекул с решеткой цеолитов при учете большого числа
кислородных атомов решетки и одного катиона; при этом рассчитывалась не
только дисперсионная, но и поляризационная (индукционная) составляю-
•щая, обусловленная поляризуемостью молекул адсорбата. В приведенном
ниже расчете использовалась идеализированная модель цеолита (с более
высокой симметрией), хотя возможно использование совершенно точной
модели (такая работа проводится в настоящее время).
Основные трудности и допущения
Потенциал адсорбционного взаимодействия молекулы адсорбата с кис-
лородным остовом решетки цеолита в нашем приближении выражается
формулой
(1)
где первые два члена по существу представляют собой потенциал Ден-
нарда—Джонса [9] для совокупности парных взаимодействий, а Фйвд —
36
индукционная составляющая. Суммирование проводится по парным взаимо-
действиям молекулы адсорбата с атомами кислорода решетки (без учета
взаимодействия с катионами).
Существенная трудность возникает при расчете константы дисперсион-
ного притяжения С. В работе [6] эта константа рассчитывалась по фор-
муле Кирквуда—Моллера
С19 = 6тпс2-, (2)
21 I 2» '
71У.2
где т — масса электрона, с — скорость света, и а2 и и %2 — соответ-
ственно поляризуемости и магнитные восприимчивости молекулы адсор-
бата и элемента решетки. За элемент решетки в [6] был принят атом кис-
лорода, который считался отрицательно-двухзарядным. Для такого «иона»
в решетке невозможно точно определить а и %, и поэтому приходится
пользоваться приближенными и малообоснованными значениями. Кон-
станты С для взаимодействия О2- с разными адсорбатами приведены
в табл. 1. В нашем расчете значения а и % для О2- были взяты из [6].
Таблица 1
Константы дисперсионного притяжения
Адсорбат С . ккал./моль о-адс
из второго вириального коэффициента по формуле Криквуда—Мюллера среднее значение
Ne 4.715-10-« 4.988 • 10-46 4.852 -10-46
Аг . 1.628 10-45 1.878 • 10-45 1.753 • 10-45
Кг 2.248 • 10-45 2.804 • 10-45 2.526- 10-45
Хе 3.645 • 10-45 4.553 • 10-45 4.099 • 10-45
СН4 2.545 • 10-45 2.705 • 10-45 2.625 • 10-45
Возможен другой путь приближенного определения С, а именно, по
значениям параметров е и о потенциала Леннарда—Джонса, записанного
в виде [9]
(3)
Эти параметры определяются из второго вириального коэффициента
и связаны с константой С соотношением [9]:
С = 4еа6. (4)
Константа С по уравнению (4) рассчитывается для парных взаимодей-
ствий одинаковых молекул. Для расчета С12, т. е. константы притяжения
неодинаковых молекул, мы пользовались формулой [3]:
C12 = VC11 • С22- (5)
Значения С12, рассчитанные этим способом, также приведены в табл. 1.
Первый и второй способы дают близкие значения констант. Для даль-
нейших расчетов бралось среднее арифметическое из этих двух констант,
поскольку связь кислорода в решетке цеолита не является ни гомеопо-
лярной, ни ионной. Взаимодействие молекулы адсорбата с катионом рас-
считывалось отдельно. Взаимодействие с атомами Si и А1 не учитывалось.
На рисунке представлена часть геометрической модели, использовав-
шейся при расчете. В качестве наиболее вероятного места адсорбции нами
37
Таблица 2
Аг Смещение катиона относительно плоскости шести- членного кольца ^кат А
L1X . . . 4-0.04
NaX . . . 4-0.10
КХ . . . . 4-0.09
СаХ . . . —0.18
SrX . . . 4-0.04
ВаХ . . . —0.08
принято шестичленное кольцо (мы приближенно считали, что атомы Si
и А1, образующие это кольцо, лежат в одной плоскости), выходящее
в большую полость и образованное шестью кремний- и алюмо-кисло-
родными тетраэдрами. Центр этого шестиугольника принят за начало
координат. Рассматривались различные поло-
жения катиона и молекулы адсорбата на оси
z, проходящей через центр шестиугольного
кольца, перпендикулярно его плоскости (ось
3-го порядка). Принималось также, что катион
и молекула адсорбата всегда находятся в со-
прикосновении, т. е. расстояние между ними:
*^кат—аде ^*адс “Ь ^кат» (0)
гДе гадс — ван-дер-ваальсов радиус адсорбата,
а гкат — ионный радиус катиона, и таким об-
разом они передвигаются вдоль оси z как одно
целое. Последнее условие введено в первой
части расчета потому, что точное положение
катионов неизвестно. Для определения поло-
жения катионов мы воспользовались экспери-
ментальными данными по теплотам адсорбции
катионных формах фожазита [8]. Весь расчет был
аргона на различных
проведен так, как описано ниже, только определялась не энергия адсорб-
ции (которая бралась из опыта), а величина Д;ат (см. рисунок), т. е. сме-
i
щение катиона относительно плоскости шестичленного кольца. Резуль-
таты расчета приведены в табл. 2.
Из табл. 2 видно, что смещения невелики и вполне могут быть обус-
ловлены неточностью расчета; поэтому в дальнейшем при всех расчетах
принималось, что все катионы при адсорбции всех рассмотренных.адсорба-
тов всегда лежат в плоскости шестичленного
кольца.
38
Наконец, еще одним допущением является то, что мы считали поляри-
зуемость молекул адсорбата изотропной, что не вполне верно, вследствие
наличия достаточно большого градиента электростатического поля внутри
полости цеолита. Это поле образовано как катионами, так и избыточными
отрицательными зарядами, которые возникают вследствие того, что поло-
жительная валентность А1 на единицу меньше, чем валентность Si. Эти
отрицательные заряды мы считали равномерно распределенными по всем
атомам кислорода. Средний заряд, приходящийся на атом кислорода
{мы принимаем, что он сосредоточен в центре этого атома), для цеолита
типа X с формулой 0.97 Na20 • А12О3 • 2.96 SiO2 [10] составляет
~ 0.214 е, где е — заряд электрона.
Расчет энергии адсорбции
При расчете энергии адсорбции учитывалось взаимодействие со
111 атомами кислорода, лежащими внутри шара, с радиусом —10 А.
Расстояние от центра молекулы адсорбата до центра каждого атома
кислорода рассчитывалось по формуле
£v = ^ + (2y-d,.)2, (7)
где смысл величин понятен из рис. 1. Выражение потенциала адсорб-
ционного взаимодействия принимает вид:
in in
ф = -со.адс2 +До-«дс 2 -
* 4=1 4=1
__ Г' I Z? fJ—12 | ф
v Еат-адсакат-адс « ^кат-адс^ват-адс “т" ч инд‘
Константа Во.й№ находилась из условия минимума энергии при
равновесном расстоянии, т. е.
Значения z,-0 для разных катионов и адсорбатов (при условии, что
катионы расположены в центре шестичленного кольца) рассчитывались
как сумма ионного радиуса катиона и ван-дер-ваальса радиуса
молекулы адсорбата. Величины, относящиеся к взаимодействию адсор-
бат-катион, как уже говорилось, постоянны и не зависят от Zy. Кон-
станта ^ват-адс равна (для изолированной пары катион—молекула ад-
сорбата):
С d6
о _____ кат-адскат-адс И
^кат-адс .*
где Ска1.адс рассчитывается по формуле Кирквуда—Мюллера. Были
использованы два пути расчета: точный и с применением эмпириче-
ских формул. Оба расчета дали очень близкие результаты, поэтому
в дальнейшем мы будем приводить данные только одного из них.
Точный расчет основывался на том, что, согласно формуле (7), L(j
является непрерывной функцией Zy и поэтому при дифференцировании
формулы (8) по Zy (для определения константы В0.адс) можно получить
выражение для производной в виде явной функции от Zy.
Второй путь расчета основан на применении эмпирических фор-
мул вида:
in
2 Ly = 10~А^£ == ajt (И)
»=1
2 L~^ 10-А'^+£' = bJt (12)
»=1
39
где коэффициенты А, В, А' и В' определялись графически в полуло-
гарифмических координатах, а также методом наименьших квадратов.
Вклад индукционной (или поляризационной) составляющей, Финд, рас-
считывался следующим образом. Энергия Фянд определялась формулой
Ф '— -— ТГ Ct F2
внд 2 аДс результ’
(13)
где F — абсолютная величина результирующего вектора напря-
женности электростатического поля в точке, соответствующей центру
молекулы адсорбата. Суммарный вектор получается алгебраическим
сложением проекций каждого из частных векторов на ось z. Отдель-
ные векторы определяются геометрическим положением соответствую-
щих зарядов. Выражение для абсолютной величины результирующего
вектора можно записать в виде
то
Так как
^результ z0e
кат-адс
Zy —
cos —---------т-----,
111
F — z^e j 2кате
результ z0e 3 “Г 2
♦=1 °Бвт-адс
(14)
(15)
(16)
где z0—заряд на атоме кислорода, zKaT— заряд катиона, е—заряд
электрона.
Входящую в выражение (15) сумму также можно представить с по-
мощью приближенной эмпирической формулы:
^А" +в" ъ +с"
= с.
(17)
Эта формула хорошо описывает только часть кривой (ограничен-
ный интервал изменений zj), однако можно подобрать коэффициенты
таким образом, чтобы в той области, которая требуется для расчета,
приближение было наилучшим. Окончательно для поляризационной
энергии получаем:
2 2 । ^20гкат
V/ + d2
кат-адс
z2
•‘•’кат
<г4
кат-адс
(18)
Вводя обозначения
1
— ~2 “a,Ve2zO = К1<
аатгс ' z0 zk&t
кат-адс
a e2z2
аде кат
d4
кат-адс
(19)
1
®ипД — 2 “»дсе2
40
и принимая во внимание обозначения, введенные ранее [выражения
(И) и (12)], получаем окончательно для потенциала взаимодействия
адсорбата с решеткой цеолита:
4 А
Ф (zjo) = —СО-адсау (2у0) + А' С0-^ау (Zy0) +
2К1С - (z -о) + К2 7 de - (z )\ 1
-----—r_ ____________________________— г я-a
A' In 10 \ dzy )Zj—zj0 2 кат-адс кат-адс ”
+ X1c}(z/0) + K2c/(zy()) + K8.
(20)
В этом выражении константа отталкивания Вс.адс уже заменена
с помощью выражения (9) на равновесное расстояние, поэтому вместо
Zj здесь всюду написано zyo. С помощью этой формулы были рассчи-
таны энергии взаимодействия Li-, Na-, К-, Са-, Sr- и Ва-форм
фожазита с Ne, Аг, Кг, Хе и СН4. В табл. 3 проведено сравнение
Таблица 3
Сравнение рассчитанных энергий адсорбции с экспериментальными теплотами
адсорбции
Адсорбат Катионная форма цеолита Теплота адсорбции, ккал ./моль Ссылка на литературу Рассчитанные энергии адсорбции, ккал./моль Отклонения, %
LiX . . —3.3 18] —3.44 4.2
NaX . —2.8 [8| —3.04 8.4
Лт КХ . . —3.0 [8] —3.20 6.5
АГ СаХ . -5.0 [8] —4.73 5.5
SrX . . —3.8 [8| —3.90 2.7
ВаХ . —3.3 [8] —3.22 2.5
NaX . —4.3 [11] —4.42 2.7
Кг | КХ . . —4.6 [11] —4.57 0.7
си4 NaX . —4.3 [12] —4.36 1.5
рассчитанных энергий с экспериментальными для тех случаев, для ко-
торых нам удалось найти в литературе данные по теплотам адсорбции.
При этом теплоты адсорбции экстраполировались к нулевому запол-
.нению. Из таблицы видно, что максимальное отклонение рассчитанных
энергий от экспериментальных не превышает 10%. Более того, нужно
отметить, что рассчитанные величины правильно передают наблюдаемую
экспериментально зависимость энергий от размеров и заряда катиона.
Таким образом^ несмотря на то что при расчете были сделаны доста-
точно грубые приближения, получено хорошее согласие рассчитанных
величин с экспериментальными.
В заключение следует сделать еще ряд замечаний.
1. При расчете учитывалось взаимодействие со 111 атомами кисло-
рода. Это количество достаточно для сумм и 2^О‘2’ н0 нед0"
статочно для суммы 3——. зависящей от 3-й степени и по-
Lij ГТ
этому медленнее уменьшающейся с расстоянием. Вследствие этого
поляризационная энергия рассчитана с меньшей точностью.
2. При более точном расчете поляризационной энергии нужно учиты-
вать также другие катионы, расположенные в других шестичленных коль-
цах большой полости.
41.
3. Необходимо в дальнейшем оценить энергию взаимодействия моле-
кулы адсорбата с другими участками решетки цеолита. Однако здесь воз-
никает ряд трудностей, связанных с тем, что неизвестен ван-дер-вааль-
сов радиус кислорода.
ЛИТЕРАТУРА
1. Н. Н. А в г у л ь, А. В. К и с е л е в, Изв. АН СССР, ОХН, 1957 , 230.
2. Н. Н. А в г у л ь, А. А. И с и р и к я н, А. В. К и с е л е в, И. А. Л ы г и н а,
Д. И. П о ш кус, Изв. АН СССР, ОХН, 1957, 1314.
3. A. D. С г о w е 1 1, R. В. S t е е 1 в, J. Chem. Phys., 1961, 34, 4, 1347.
4. J. R. Sams, Trans. Faraday. Soc., 1964, 60, 1.
5. Я. К о у т e ц к и, Й. Ч и ж e к, ЖФХ, 1962, 36, 1508.
•6. R. М. В а г г в г, S. Wasilewski, Trans. Faraday. Soc., 1961, 57, 1140.
7. R. M. Barret, G. С. В г a 11, J. Phys. Chem., 1959, 130, 146, 154.
8. R. M. В а г г e r, W. S. Stuart, Proc. Roy. Soc., 1959, A 249, 464.
9. Дж. Гиршфельдер, Ч. Кертисс, Р. Берд. Молекулярная теория
газов и жидкостей. ИЛ, 1961.
10. А. В. Киселев, А. А. Л о п а' т к и н, Кинетика и катализ, 1963, 4, 786.
11. В. Б о с а ч е к, настоящий сборник, стр. 103.
12. R. М. В а г т е г, J. W. Sutherland, Proc. Roy. Soc., 1956, A 237, 1211.
ИССЛЕДОВАНИЕ СПЕКТРОВ
ПРИ НИЗКИХ
ЯМР ПРОТОНОВ В ЦЕОЛИТАХ
ТЕМПЕРАТУРАХ
В. И. Квливидзе
Исследование спектров ядерного магнитного резонанса [1] адсорби-
рованного вещества позволяет получить ряд ценных сведений о подвиж-
ности молекул на поверхности, о структуре поверхностного слоя и фа-
зовых переходах в нем. Ранее [2] нами был исследован ЯМР в воде, адсор-
бированной на силикагеле. Цеолиты в отличие от таких классических
адсорбентов, как силикагели и активированные угли, представляют более
удобный модельный объект для изучения состояния адсорбированной фазы,
поскольку из рентгеновских данных нам известны структура и геометри-
ческие размеры полостей кристаллической решетки цеолита. Последнее
значительно упрощает интерпретацию наблюдаемых явлений. В рабо-
тах [3, 4, 5] было проведено изучение спектров ЯМР воды в синтетиче-
ских цеолитах. Однако в работах [3, 4] измерения проводились лишь
при комнатной температуре, а в работе [5] не исследовалась зависимость
спектров от заполнения. Исследование спектров ЯМР адсорбированного
вещества при низких температурах значительно расширяет получаемую
информацию о поведении молекул на поверхности, так как интенсивное
движение при комнатных температурах усложняет интерпретацию на-
блюдаемых спектров [6].
В настоящей работе исследовались спектры ЯМР протонов воды в цео-
лите NaX в зависимости от количества адсорбированной воды в диапазоне
температур от 90 до 230° К. Был использован синтетический цеолит NaX
состава 0.964 Na2O • А12Оа • 2.96 SiO2, полученный методом гидротер-
мального синтеза. Цеолит NaX имеет гранецентрированную кубическую
решетку [7]. Большие полости цеолита имеют объем 881 А и вмещают
при полном заполнении 30 молекул воды [8, 9]. Образцы цеолита дегидра-
тировались при температуре 350° С, а затем проводилась адсорбция воды.
Количество адсорбированной воды определялось взвешиванием ампул
с цеолитом до и после пуска паров воды. Содержание воды в образцах
менялось от 0.3 до 17.3 ммоль/г. Для улучшения теплообмена ампулы
42
с цеолитом заполнялись гелием. Спектры ЯМР снимались на спектрометре
для широких линий [2]. Криостат, в который помещался исследуемый
образец, позволял получать температуры от 90 до 273° К с точностью
до 0.5°. По полученным спектрам определялся второй момент линии погло-
щения и ширина линии (расстояние между максимумами производной).
Наблюдаемые производные сигналов поглощения ЯМР протонов воды
в цеолитах* при разных заполнениях в зависимости от температуры при-
ведены на рис. 1. Как видно из рис. 1, при 90° К спектры ЯМР для образ-
цов с содержанием воды до 7.15 ммоль/г включительно имеют вид дубле-
а б 6
Рис. 1. Производные сигналов поглощения ЯМР протонов воды в цео-
лите NaX при разных температурах.
Содержание воды (ммоль/г) : а — 2.72; б — 7.15; е — 17.3. На кривых не указан
небольшой пик в центре линии» связанный с сигналом от протонов стекла каркаса
катушки и ампулы.
тов, типичных для кристаллогидратов с жестко закрепленными молеку-
лами воды [10]. При заполнении 17.3 ммоль/г и выше характер спектра
меняется, дублетная структура исчезает и наблюдается широкая линия.
На рис. 2 приведена зависимость второго момента от количества адсорби-
рованной воды при температуре 90° К. Видно, что увеличение адсорбции
приводит к возрастанию второго момента от 22 до 32 э2.
Согласно адсорбционным измерениям [11], можно предположить, что
малые полости цеолита не заполняются водой даже при высокой упруго-
сти паров воды. Относительно заполнения больших полостей цеолита
существуют разные мнения: либо допускается, что молекулы воды рас-
пределяются по порам цеолита случайно и’на ряду с полностью заполнен-
ными порами существуют пустые, либо молекулы воды распределены по
порам равномерно. Форма спектра ЯМР при малых заполнениях и изме-
нение ее при увеличении количества воды говорят о том, что осуществляется
вторая возможность. В начальной области адсорбции молекулы воды рас-
полагаются далеко друг от друга, взаимодействуя только с каркасом цео-
лита и практически не взаимодействуя друг с другом. Об этом свидетель-
43
ствует малое значение второго момента для образца с количеством адсор-
бированной воды 2.72 ммоль/г. Пользуясь формулой Ван-Флека для
второго момента поликристаллического образца [12], мы нашли, что рас-
стояние между протонами в молекуле воды d=l.59+0.15 А. Такое же
межпротонное расстояние получено, например, для молекул воды в натро-
лите [13]. Для образцов с количеством адсорбированной воды 5.65 и
7.15 ммоль/г второй момент при 90° К равен 27 э2, что свидетельствует
о появлении межмолекулярного взаимодействия. Однако молекулы
Рис. 2. Зависимость второго момен-
та и ширины липни от содержания
адсорбированной воды при темпера-
туре 90° К (A) (Z — второй момент,
2 — ширина линии) и изотерма ад-
сорбции паров воды при комнатной
температуре на цеолите NaX (изо-
терма адсорбции получена в лабора-
тории акад. М. М. Дубинина в Ин-
ституте физической химии АН СССР
К. О. Мурдмаа) (В).
располагаются еще достаточно далеко
друг от друга, так как вклад протонов
соседних молекул во второй момент не
превышает 20 %. Дальнейшее увеличение
заполнения приводит к возрастанию
второго момента до 32 э2, при этом фор-
ма линии приближается к наблюдаемой
для льда [14]. Однако необходимо от-
метить, что имеется заметное различие
в величине вторых моментов. Для льда
при температуре 90° К второй момент
равен 36.7 э2 [14]. Уменьшение второго
момента для адсорбированной воды по
сравнению со вторым моментом для
льда наблюдалось нами и в случае
адсорбции на силикагеле [6]. Это свя-
зано, по-видимому, во первых, с боль-
шой дисперсностью образующегося льда,
а во-вторых, — с наличием дефектов
в структуре. Так как положение мо-
лекул воды в порах цеолита NaX не
определено, то точный расчет второго
момента провести нельзя. В работе [51
сделана оценка второго момента для
воды в порах цеолита NaX при пред-
положении, что молекулы воды в со-
ответствии с гипотезой Н. В. Белова
[15] занимают вершины и центр пента-
додекаэдра. Авторы [5] брали межпро-
тонное расстояние d=1.54A и получили
второй момент, равный 35 э2. Если взять d=1.59 А, которое получается
из наших данных по спектрам ЯМР при малом заполнении, то величина
второго момента будет 31 э2, что лучше согласуется с нашими данными для
образца с полным заполнением. В этом случае существенную роль играет
не только взаимодействие молекул воды с каркасом цеолита, но и взаимо-
действие молекул воды друг с другом через водородные связи.
Температурное изменение спектров ЯМР образцов с различным коли-
чеством адсорбированной воды позволяет получить дополнительные све-
дения о характере связи молекул воды в полости цеолита. Как видно иа
рис. 1, при повышении температуры изменяется форма линии сигнала
ЯМР, что связано с появлением более узкого компонента спектра, ши-
рина и интенсивность которого меняется с температурой. Оценка интеграль-
ных интенсивностей показала, что интенсивность узкой компоненты ме-
няется по экспоненциальному закону. Узкая и широкая компоненты одно-
временно существуют в широком интервале температур, причем ширина
широкой компоненты меняется незначительно вплоть до температуры
185° К для образцов с заполнением до 7.15 ммоль/г и до температуры
200° К для образца с 17.3 ммоль/г воды. Выше указанных температур
44
происходит быстрое исчезновение широкой компоненты и спектр весь су-
жается, приобретая вид, характерный для жидкостей. На рис. 3 приведена
зависимость второго момента и ширины широкой компоненты от темпера-
туры для образцов с различным содержанием воды. Увеличение количе-
ства адсорбированной воды в цеолите приводит к повышению температуры,
при которой происходит появление узкой компоненты. Ширина узкой
компоненты при температуре ее появления позволяет считать, что сначала
возникает вращательная, а затем поступательная подвижность молекул
воды, причем в этих движениях участвует лишь небольшая часть моле-
кул, что свидетельствует о неэквивалентности молекул воды с энергети-
ческой точки зрения.
Рис. 3. Зависимость второго момента (4) и зависимость ширины линии широкого
компонента (Б) от температуры для образцов с различным содержанием воды.
Количество воды (ммоль/г): 1 — 2.72; 2 — 7.15; 3 —-17.3.
Интерпретация такого изменения спектров ЯМР при повышении тем-
пературы вместе с данными для температуры 90° К приводит к следующей
предположительной картине заполнения пор. В исследуемом цеолите в боль-
шой полости находится приблизительно 8 ионов Na+, причем 4 из них распо-
ложены вблизи больших и 4 — около малых окон в полости цеолита, т. е. су-
ществуют по крайней мере два типа центров адсорбции, энергия взаимодей-
ствия которых с молекулами воды, по-видимому, различна. Для образца
с заполнением 2.72 ммоль/г, что соответствует примерно 5 молекулам
воды, в среднем на большую полость цеолита, при температуре 180° К
участвуют в движении приблизительно 20% молекул, т. е. четыре моле-
кулы закреплены неподвижно, а одна перемещается из одного положе-
ния равновесия в другое. Этот факт можно рассматривать как подтвер-
ждение неэквивалентности адсорбционных центров — ионов Na+. Энер-
гия активации для данного образца оказалась равной приблизительно
3 ккал./моль. Для образца с двенадцатью молекулами на полость (а=
= 7.15 ммоль/г) число подвижных молекул при той же температуре равно
четырем, т. е. числу молекул, которые не связаны непосредственно с ионами
Na+. Энергия активации для этого образца получилась равной
~ 4.2 ккал./моль, что указывает на появление взаимодействия между мо-
лекулами воды. Для образца с полным заполнением энергия активации
оказалась равной — 5.4 ккал./моль, что близко к энергии водородной
45
связи. Появление подвижных молекул воды (или протонов) при полном
заполнении можно объяснить наличием большого числа дефектов в струк-
туре воды, замерзшей в порах цеолита.
Выводы
1. Анализ полученных результатов показывает, что адсорбированные
молекулы воды равномерно распределяются по большим полостям цео-
лита, а в самих полостях при низких температурах они занимают опре-
деленные положения, неэквивалентные с энергетической точки зрения.
2. При малых заполнениях взаимодействие адсорбированных молекул
воды между собой мало и по величине второго момента можно определить
межпротонное расстояние в молекулах воды, которое оказалось равным
1.59+0.015 А.
3. При изменении температуры для всех образцов второй момент ме-
няется плавно и в широком интервале температур одновременно суще-
ствуют жестко закрепленные и подвижные молекулы воды. Температура
фазового перехода зависит от содержания воды в порах и лежит в диапа-
зоне 210—230° К.
В заключение автор считает своим приятным долгом выразить благо-
дарность В. Ф. Киселеву и В. В. Серпинскому за обсуждение результа-
тов работы, а также О. А. Васильевой и Ю. В. Шутовой за приготовление
образцов и помощь при обработке экспериментальных данных.
ЛИТЕРАТУРА
1. Э. Эндрю. Ядерный магнитный резонанс. ИЛ, М., 1957.
2. В. И. К в л и в и д з е, Н. М. И е в с к а я, Т. С. Е г о р о в а, В. Ф. Кисе-
лев, И. Д. С о к о л о в, Кинетика и катализ, 1962, 3, 1, 91.
3. Р. D и с г о s, X. Р а г е, Arch. Sci., 1960, 13а, 383.
4. Л. В. М а т я ш, М. А. П и о н т к о в с к а я, Л. М. Т а р а с е н к о, Р. С. Тю-
тю нн и к, Ж. структурн. химии, 1962, 3, 214.
5. С. И. Г а б у д а, Г. М.Ми х а й л о в, Ж. структурн. химии, 1963, 4, 446.
6. В. И. К в л и в и д з е, ДАН СССР, 1964, 157, 1.
7. R. М. В аггег, W. М. М е ier, Trans. Faraday. Soc., 1963, 54, 1074.
8. M. M. Д у бинин, С. П. Ж д а н о в и др., Йзв. АН СССР, сер. хим., 1964,
9, 1573.
9. М. М. Д у б и н и н, Е. Г. Ж у к о в с к а я, Изв. АН СССР, 1962, 2113.
10. G. Е. Раке, J. Chem. Phys., 1948, 16, 327.
11. М. М. Д у б и н и н и др., Изв. АН СССР, ОХН, сер. хим., 1964, 9, 1965, 2, 209.
12. J. Н. Van V 1 е с к, Phys. Rev., 1948, 74, 1168.
13. С. П. Г а б у д а, А. Г. Л у н д и н и др., Кристаллография, 1963, 8, 388.
14. К. К и га е, J. Phys. Soc. Japan, 1960, 15, 1493.
15. Н. В. Белов. Кристаллохимия силикатов с крупными катионами. Изд. АН
СССР, М., 1961.
ИЗОТЕРМЫ И ДИФФЕРЕНЦИАЛЬНЫЕ ТЕПЛОТЫ
АДСОРБЦИИ ВЕЩЕСТВ С РАЗЛИЧНОЙ ЭЛЕКТРОННОЙ
СТРУКТУРОЙ МОЛЕКУЛ РАЗНЫМИ КАТИОННЫМИ ФОРМАМИ
ЦЕОЛИТА ТИПА ФОЖАЗИТ
О. М. Джигит, С. П. Жданов, К. Н. Микос
Фиксированное расположение в пространстве силовых центров цео-
литов позволяет в принципе рассчитать потенциальную энергию адсор-
бированных молекул и вычислить термодинамические свойства системы
цеолит—адсорбат, однако для сложных молекул такой расчет очень
труден. Поэтому представляет интерес чисто экспериментально выделить
из общей энергии адсорбции вклады как универсальных взаимодействий
46
всей молекулы адсорбата со всеми силовыми центрами скелета цеолита,,
так и специфических взаимодействий активных частей молекулы адсор-
бата с катионами цеолита. Изменяя природу этих силовых центров, т. е.
изучая, например, различные катионные формы цеолитов и беря вещества,
молекулы которых имеют различное электронное строение, можно по-
дойти к выяснению природы адсорбционных взаимодействий. Исследова-
ния влияния катионов на теп-
лоты адсорбции аргона, азота,
воды, аммиака проводились
путем определения их из
изостер [1, 2], а для бензола
и н-гексана, а также н-гек-
сана и гексена — из хрома-
тографических данных [3].
В настоящей работе начато
изучение влияния природы
катиона цеолита на диффе-
ренциальные теплоты адсорб-
ции веществ, молекулы ко-
торых имеют близкую геомет-
рическую структуру, но резко
различаются по распределе-
нию электронной плотности.
В качестве такой пары ве-
ществ мы выбрали н-пентан
и диэтиловый эфир, адсорб-
Рис. 1. Изостерические теплоты адсорбции нор-
мальных углеводородов fNa-фожазитом по дан-
ным [7] (величины а пересчитаны нами и вы-
ражены в ммолях/г сухого Na-фожазита).
ция которых изучена для
ряда других адсорбентов [4—6]. Для развития термодинамики ад-
сорбции цеолитами очень важно исследование обратимых изотерм и
теплот адсорбции в области малых степеней заполнений. Однако, несмотря
на большой интерес к цеолитам, таких работ мало. Объясняется это тем,
что калориметрические измерения требуют сложной аппаратуры и тру-
доемки, а получение изотерм адсорбции затруднено из-за того, что основ-
ная часть полостей цеолитов заполняется при очень малых равновесных
давлениях, которые трудно измерить с достаточной точностью. Для повы-
шения точности измерения изотерм адсорбции и определения теплот ад-
сорбции из изостер обычно повышают температуру. На рис. 1 представ-
лены изостерические теплоты адсорбции нормальных углеводородов
Na-фожазитом [7]. Уже для бутана повышение температуры до 70°
позволяет охватить область 6 лишь от 0.8 и выше. Чтобы исследовать
область меныпих заполнений, надо или значительно повышать тем-
пературу опыта, или производить калориметрические измерения. В на-
стоящей работе мы использовали оба зти пути.
Образцы и измерения
Для выяснения воспроизводимости адсорбционных свойств поверх-
ности цеолитов мы провели в одинаковых условиях ряд независимых син-
тезов цеолита NaX и взяли для исследования три партии этого цеолита,
состав которых приведен в табл. 1.
Для исследования влияния природы катиона цеолита на теплоты ад-
сорбции н-пентана и диэтилового эфира нами был выбран цеолит типа X,
в котором окна, ведущие в большие полости, имеют достаточно большие
размеры и не препятствуют проникновению этих молекул внутрь полостей.
Различные катионные формы цеолита X были приготовлены из Na-формы
47'
путем ионного обмена в водных растворах соответствующих солей и
имели состав, указанный в табл. 2.
Все образцы цеолитов перед измерениями откачивались в течение
— 100 часов при 400°.
Измерения дифференциальных теплот адсорбции производились при
23° в изотермическом калориметре с постоянным теплообменом [8]. Более
99.5% теплового эффекта компенсировалось включением (при десорбции)
Таблица 1 Состав различных партий цеолита NaX или выключением (при адсорбции) нагревателя постоянной мощности. Критерием установления адсорбци- онного равновесия служил ровный ход калориметра. Дозирование пор- ций пара адсорбата производилось
Цеолит Na А1О, S1O2
II партия . . VII партия . . е', I партия . 0.97 1.02 0.943 1 1 1 1.48 1.5 1.38 испарением жидкости из вакуум- ной микробюретки [9]. Погрешность определения прироста адсорбции Да и кривой дифференциальных теплот адсорбции обычно не превышала 1 %. Изменение интервалов между от-
дельными измерениями от двух часов до двух суток не влияло на полу-
ченные результаты. При комнатной температуре практически невозможно
измерить в области 0.8 теплоты десорбции и десорбционную ветвь
изотермы, вследствие крайне замедленной кинетики десорбции эфира и
пентана из каналов цеолита. Поэтому для получения обратимых изотерм
адсорбции вплоть до а -> 0 пришлось сильно повысить температуру
опытов. Изотермы адсорбции н-пентана измерялись при температурах
200, 230 и 250°. Для этих измерений использовалась установка с вакуум-
ными регистрирующими весами с электромагнитной компенсацией [10].
Таблица 2
Состав различных катионных форм цеолита X
Цеолит Na м аю2 SiO2
NaX 0.943 1 1.38
LiX 0.38 0.62 1 1.38
КХ - 0.21 0.66 1 1.33
ВЬХ 0.29 0.63 1 1.31
CsX 0.37 0.55 1 1.34
М — катион, указанный в обозначении цеолита.
Примечание.
В описанную в [10] конструкцию установки были внесены следующие
изменения: на коромысле весов вместо пермаллоевого сердечника был
укреплен намагниченный стержень из сплава альнико и изменена кон-
струкция катушки электромагнита. Эти усовершенствования позволили
получить линейную зависимость тока электромагнита от изменения веса
образца, что значительно облегчило калибровку весов. Отсутствие в уста-
новке смазанных кранов и ртути обеспечивало высокую чистоту поверх-
ности откачанных адсорбентов. Давление пара адсорбата во время опыта
непосредственно не измерялось, а задавалось температурой жидкости.
Для обеспечения термостатирования жидкости с точностью не ниже 0.1°
мы применили криостат, описанный в [11], внеся некоторые изменения
в схему управления им. Этот криостат обеспечивает стабильность темпе-
ратуры с точностью +0.02°. Температура криостата измерялась медь-
48
константановой термопарой при помощи потенциометра ППТН. Работая
в интервале от —120 до +20° (соответственно от давления н-пентана
— 1 • 10 "1 2 до 400 мм), мы определяли задаваемое давление с погреш-
ностью, не превышающей Q.5%.1
Результаты и обсуждение
Изотермы адсорбции. Из рис. 2 видно, что даже при ад-
сорбции цеолитом NaX н-пентана, адсорбирующегося в основном за счет
неспецифических взаимодействий, изотерма адсорбции при 23° подни-
мается так круто, что до равновесного давления р=10 мм уже достигается
0g=0.9.2 Из рис. 2 видно, что изотермы адсорбции при 200, 230 и 250° С
обратимы вплоть до самого на-
чала. До р=10 мм при этих
температурах достигается
^0.12, 0.06 и 0.03 соответ-
ственно, и поэтому можно де-
тально исследовать начальную
часть изотерм адсорбции.
Для описания этих изотерм
мы применили приближенные
уравнения для мономолекуляр-
ной адсорбции, как не учиты-
вающие (1), так и учитывающие
(2) [12] и (3) [13] взаимодей-
ствия адсорбат—адсорбат:
6
- = к1-к1е, (1)
Рис. 2. Изотермы адсорбции н-пентапа цео-
литом NaX при 23, 200, 230 и 250°.
Черные точки здесь и далее — десорбция, в — сте-
пень заполнения.
6
р(1 — в) =к1 + к1к»е-
«’(6. Р)=Г2Гб + 1иГТГ0 — 1пр =
= 1пК14-Х26. (3)
Из рис. 3, а, на котором для примера приведена изотерма адсорбции
н-пентана цеолитом NaX при 230° в координатах уравнений (1)—(3), видно,
что все три уравнения дают прямолинейный участок в довольно широком
интервале 0. Из наклона прямых линий и отсекаемых отрезков на оси
ординат были определены константы уравнений (1)—(3), по которым был
произведен обратный расчет изотермы адсорбции (рис. 3, б, 230°).
Изотерма адсорбции, рассчитанная по уравнению Лэнгмюра, совпа-
дает с экспериментальной во всем изученном интервале давлений,3 4 а
рассчитанные по уравнениям (2) и (3) в области малых давлений идут не-
сколько ниже экспериментальной.
1 В интервале от 0 до -ф-20° криостат обычно заменялся ультратермостатом.
а
2 6g =—, где а8 — предельно адсорбируемое при 23° количество вблизи дав-
as
левия насыщенного пара рв. Предельно адсорбируемые количества при 200—250°,
вероятно, отличаются от ав для 23°, поэтому значения 6е для этих температур
не имеют точного смысла и используются нами лишь для приблизительной оценки.
3 Применимость уравнении Лэнгмюра для описания изотерм адсорбции йода
цеолитами отмечалась в [14].
4 Цеолиты
49
Используя константу уравнения Лэнгмюра для 230° и применив урав-
нения зависимости Кг от температуры
дд о q
К.- е'1 е,а = g e,iT, (4)
мы нашли значения констант для температур 200 и 250° и рассчитали
по ним соответствующие изотермы адсорбции. Из рис. 3, б видно, что они
Рис. 3. а — Изотерма адсорбции н-пентана цеолитом NaX при 230° в координатах урав-
нений: 1— (1), 2 — (2) и 3 — (3); б — изотерма адсорбции (пунктир) при 230°,
вычисленная по константам уравнений, полученным из рис. 3, а. Изотермы для
200 и 250° (пунктир) вычислены по константам уравнения Лэнгмюра, определенным
по уравнению (4) из константы для 230°.
Точки — экспериментальные данные. Внизу справа — начало изотерм в крупном масштабе.
хорошо совпадают с экспериментальными изотермами. Это свидетель-
ствует о том, что в указанной области заполнений при этих температурах
взаимодействие между молекулами адсор-
бата еще не проявляется. Очевидно, что
при понижении температуры опыта будет
возрастать взаимодействие адсорбат—ад-
сорбат и сокращаться интервал примени-
мости уравнения Лэнгмюра. Изостериче-
ские теплоты адсорбции н-пентана цеоли-
том NaX (рис. 4), рассчитанные из изотерм
адсорбции при 200—250°, в изученном
интервале а почти не меняются с запол-
нением, в соответствии с тем, что изотермы
gy адсорбции при этих температурах хорошо
а, ммоль/г описываются уравнением Лэнгмюра. При
Рис. 4. Зависимость теплоты ад-
’сорбции н-пентана цеолитом NaX,
рассчитанной из изостер в интер-
вале 200—250°, от адсорбирован-
ного количества.
малых заполнениях изостерические теп-
лоты близки к теплотам, измеренным
в калориметре при 23°. Это свидетель-
ствует о малой зависимости в этой области
теплоты адсорбции от температуры. На
рис. 4 крестиком нанесено значение на-
чальной теплоты адсорбции н-пентана, рассчитанное по наклону прямой
зависимости 1g Кг (константы уравнения Лэнгмюра) от 1/Т.
50
Воспроизводимость адсорбционных свойств разных
партий цеолита NaX. Если синтезы цеолитов NaX проводятся в оди-
наковых условиях, то состав их удовлетворительно воспроизводится.
Нами были взяты три партии NaX, состав которых приведен в табл. 1,
и для них измерены дифференциальные теплоты адсорбции н-пентана.
При сопоставлении только изотерм адсорбции (рис. 5, Г) можно сделать
вывод о невоспроизводимое™ адсорбционных свойств этих партий цео-
лита. Однако измеренные дифференциальные теплоты адсорбции показы-
вают, что это не так. Из рис. 5, В видно, что теплоты адсорбции н-пентана
в области а < 1.5 ммоль/г (6g < 0.6) для всех трех партий цеолита прак-
тически совпадают.4 В области 6, > 0.6 кривая, соответствующая образ-
Рис. 5. Зависимость дифференциальных теплот адсорб-
ции Qa н-пентана различными партиями цеолита NaX
от адсорбированного количества а (А, В) и соответ-
ствующие изотермы адсорбции (Б, Г).
L — здесь и далее теплота конденсации.
цам VII и е' (треугольники и квадраты), смещена влево. При исследовании
кристаллов VII партии NaX под микроскопом была обнаружена примесь
филипсита, в полости которого н-пентан не входит, поэтому можно было
уменьшение общей емкости цеолита этой партии отнести за счет примеси
и соответственно исправить навеску собственно NaX. То же сделано и
в случае партии е'. При этом теплоты (рис. 5, Л) и величины адсорбции
(рис. 5, Б) для образцов VII и е' совпали с соответствующими кривыми
для образца II во всей области заполнений. Это свидетельствует' о хо-
рошей воспроизводимости свойств поверхности полостей синтетических
цеолитов NaX близкого состава. Некоторые различия в общей емкости
отдельных партий NaX объясняются присутствием примесей.
Таким образом, величины теплот адсорбции являются физико-хими-
ческими константами, зависящими при данной температуре и концентра-
4 Такое же совпадение теплот адсорбции для партий II и е' получено в случае
эфира.
4*
51
ции адсорбированного вещества только от свойств системы цеолит—ад-
сорбат.
Влияние природы катионов на теплоты адсорбции.
На рис. 6 приведены дифференциальные теплоты адсорбции н-пентана и
эфира для трех катионных форм цеолита X. Из рисунка видно, что общий
характер зависимости сходен, но имеются и отличия. При малых запол-
нениях теплоты адсорбции н-пентана увеличиваются в ряду LiX, NaX,
RbX в соответствии с ростом дисперсионного потенциала по мере утяже-
ления катиона. Далее, для всех катионных форм наблюдается рост теплоты
адсорбции н-пентана с запол-
нением, обусловленный уси-
лением взаимодействия меж-
ду адсорбированными моле-
кулами. При рассмотрении
зависимости теплоты адсорб-
ции эфира от заполнения
надо иметь в виду два фак-
тора: поскольку дисперсион-
ные составляющие энергии
адсорбции эфира и н-пентана
отличаются мало (теплоты ад-
сорбции их на графитирован-
ной саже близки [6, 15]), из-
менение этой составляющей
при переходе от одной ка-
тионной формы к другой
должно быть для эфира таким
же, как для пентана. Но при
адсорбции эфира проявляется
дополнительное специфиче-
ское взаимодействие атома
кислорода в молекуле эфира
с катионом цеолита.
Катион © ® (пв)
ЧЭ^п 9.5 8.5 5
-I_____I_____I_____1______L
1 2 3
а, ммоль/г
Рис. 6. Зависимость дифференциальных теплот
адсорбции эфира и н-пентана от адсорбированного
количества для разных катионных форм цеоли-
та X.
Это специфическое взаимодействие тем больше, чем меньше радиус
катиона, т. е. чем более сосредоточен положительный заряд. Поэтому в ряду
LiX, NaX, RbX это специфическое взаимодействие уменьшается. В ре-
зультате влияния обоих этих факторов превышение теплоты адсорбции
эфира над теплотой адсорбции н-пентана в области малых .заполнений
составляет: для LiX 9.5 ккал./моль, для NaX 8.5 ккал./моль и для RbX
5.0 ккал./моль.
В области больших 6 размывание максимума теплоты адсорбции н-пен-
тана и исчезновения его для эфира в случае LiX и менее резкое падение
теплот адсорбции после максимума объясняется, по-видимому, проявле-
нием большей неоднородности поверхности Li-формы по сравнению с Na-
и Rb-формами.5 Еще большая неоднородность поверхности наблюдается
для Са-формы цеолита X [16].
Таким образом, неоднородность, связанная с наличием в цеолитах
отрицательных зарядов, рассредоточенных по многим атомам, и поло-
жительных зарядов, сосредоточенных в катионах, проявляется тем больше,
чем больше по величине и чем более сосредоточены на поверхности поло-
жительные заряды.
Обращает на себя внимание тот факт, что для всех катионных форм
цеолита X теплоты адсорбции эфира во всей области заполнений остаются
5 Большая неоднородность Li-формы по сравнению с Na-формой отмечалась
также в [1, 2].
52
больше теплот адсорбции «-пентана. Это указывает на такую локализа-
цию всех молекул эфира в больших полостях цеолита, которая обеспечи-
вает сильное специфическое взаимодействие диполя эфира с катионами
цеолита.
Большие теплоты адсорбции как эфира, так и пентана приводят к силь-
ному уменьшению энтропии при адсорбции. По сравнению с жидким со-
стоянием энтропия адсорбированного н-пентана уменьшается на 25—
28 э. е., а эфира — на 35—37 э. е. Это свидетельствует о сильном торможе-
нии адсорбированных молекул в полостях пористых кристаллов цеолита.
ЛИТЕРАТУРА
1. R. М. В а г г е г, W. S. S t u а г t, Proc. Roy. Soc., 1959, A 249, 464.
2. R. M. В a rr er, G. С. В г a 11, J. Phys. Chem. Solids, 1959, 12, 130, 146, 154.
3. R. E. E b erly, J. Phys. Chem., 1961, 65, 68; 1962, 66, 812.
4. А. А. И с и p и к я н, А. В. К и с e л e в, Б. А. Ф р о л о в, ЖФХ, 1959, 33,
389; О. М. Джигит, А. В. К и с е л е в, Г. Г. М уттик, Колл, ж., 1951,
23, 504.
5. Н. Н. А в г у л ь, А. В. К и с е л е в, И. А. Л ы г и н а, Изв. АН СССР, ОХН,
1961, 1116.
6. О. М. Джигит, А. В.. К и'с е л е в, Г. Г. М у т т и к, J. Phys. Chem., 1962,
66, 2127.
7. R. М. В а г г е г, J. W. Sutherland, Proc. Roy. Soc., 1956, A 237, 439.
8. А. В. Киселев, Г. Г. M уттик, ЖФХ, 1961, 35, 2153.
9. В. П. Д р е в и н г, А. В. К и с е л е в, Ю. А. Э л ь т е к о в, ДАН СССР, 1952,
86, 349.
10. Г. Г. М утти к, ЖФХ, 1957, 31, 263.
11. А. В. Киселев, Г. Г. М уттик, Колл, ж., 1957, 19, 562.
12. А. В. Киселев, Колл, ж., 1958, 20, 338.
13. Т. Н i 1 1, J. Chem. Phys., 1948, 14, 441; S. R о s s, W. W i n k 1 e r, J. CoU.
Sei., 1955, 10, 319, 330.
14. R. M. В arrer, S. W a s i 1 e w s k i, Trans. Faraday. Soc., 1961, 57, 1140, 1156.
15. А. В. Киселев, настоящий сборник, стр. 13.
16. О. М, Джигит, А. В. К и с е л е в, Г. Г. М у т т и к, Колл, ж., 1963, 25, 15.
ИНФРАКРАСНЫЕ СПЕКТРЫ ЦЕОЛИТОВ РАЗЛИЧНОЙ СТРУКТУРЫ
С. П. Жданов, В. И. Лыгин, Т. И. Титова
В настоящей работе метод инфракрасной спектроскопии применен для
исследования синтетических цеолитов различного состава, различных
катионных форм и различной структуры с различной степенью декатио-
нирования.
Химический состав исследованных цеолитов представлен в таблице.
Для уменьшения рассеяния инфракрасной радиации образцы цеолитов
прессовались под давлением 50 кг/мм2 в таблетки весом 5—7 мг/см2.
Термическая обработка образцов в вакууме и съемка спектров проводи-
лись в кювете, аналогичной [1] на спектрометре UR-10 фирмы Цейс.
Спектры снимались последовательно после откачки образцов при 25,
100, 200, 300, 400° С в течение 4 часов при каждой температуре.
В работе были исследованы Na-фожазиты (образцы 1—6) с переменным
отношением Si : Al от 1.41 до 2.55 (в таблице это отношение указывается
индексами у SiO2). Такое изменение в составе, согласно рентгеновским
данным, мало меняло постоянную кристаллической решетки фожазитов [2],
но почти в два раза уменьшало число обменных катионов, приходящихся
на элементарную ячейку, и приводило к закономерным изменениям в спек-
тре кристаллического каркаса фожазитов. На рис. 1 кривые 1—6 пред-
ставляют спектр колебаний Si—Al—О каркаса фожазитов по мере увели-
чения содержания Si и, соответственно, уменьшения содержания А1
53
Химический состав исследованных цеолитов
образца Тип цеолита Состав безводных кристаллов Количество катионов Na+ в элементарной ячейке
1 X Ho.o2Na0.eg(A102)1(Si02)1j1 89
2 X Но.1б^ао.84(А102)1(8102)1.28 71
3 X 11о.ы^а0.<)6(Л102)!(8!02)|.50 74
4 X H0.o6Nao.94(A102)1(Si02)1.74 69
5 Y Naj.ootAlOaMSiC^j^si 58
6 Y Но.об^ ao.84(A102)i(Si02)2.5C 51
7 X (NH4)o.74Nao.26(A102)1(Sid2)i,48 —
8 X Ho.o4Nao.i2Cao.42(A102)i(Si02)i.4g .—
9 X H0.05Wa0.02Sro.465(Al O2)i(SiO2)154 .—
10 А Ho.oSNa0.87(A162)1(Si02)1 —
И А 11о.О8^ао.92(А102)j (SiO2)i —
12 А Ho.75^so.25(Al()‘2)i(Si02)1 —
13 X Ho.2iNao.79( Al O2) i (S iO2) —
Рис. 1. Инфракрасные спектры
колебаний (Z—б) кристалличе-
ского каркаса фожазитов различ-
ного состава, снятые после обра-
ботки при 400° в течение 4 ча-
сов.
в образцах. Анализ спектра колебаний кристаллического каркаса фожа-
зитов проводился на основе сравнения со спектрами кварца [3] и алюмо-
силикатов [4]. Полосы 405, 465, 683 и сплошное поглощение выше 900 см'1
лежат в области полос поглощения кварца.
Особенно чувствительными к изменению
состава оказались полосы 568, 610 и
758 см'1. По мере увеличения содержания
Si интенсивность полосы 568 см-1 умень-
шается и она сдвигается до 582 см-1. По-
лоса 610 см'1 смещается в сторону боль-
ших частот до 640 см'1 и наиболее интен-
сивна в спектрах с максимальным содер-
жанием (Si0)</2 тетраэдров. Полоса 758 см-1
лежит в области, где у алюмосиликатов
при изоморфном замещении Si4+ на А13+
наблюдается полоса поглощения, вызван-
ная колебанием связи А1—О. Интенсив-
ность этой полосы уменьшается по мере
уменьшения содержания А! в фожазитах
(кривые 1—6), что дает основание интер-
претировать ее как полосу, принадлежа-
щую колебаниям связи А1—О внутри
алюмокислородных тетраэдров.
В соответствии с изменением химиче-
ского состава фожазитов и с изменениями
в спектре колебаний их кристаллического
остова находятся изменения в спектре
колебаний гидроксильных групп адсорби-
рованных молекул воды. На рис. 2 пред-
ставлены спектры цеолитов различного со-
става (образцы 1—6), снятые после 4 часов вакуумной обработки при ком-
натной температуре. В спектре фожазитов с максимальным содержанием
(АЮ)«/г тетраэдров, т. е. с большим числом катионов (рис. 2, 1—4), имеется
несколько полос в области валентных и деформационных колебаний гидро-
ксильных групп адсорбированных молекул воды, что указывает на суще-
ственно различные энергетические состояния адсорбированных молекул
воды в этих цеолитах. Спектр фожазитов с минимальным содержанием
54
алюмокислородных тетраэдров (рис. 2, 5, 6) беден полосами в области
валентных колебаний гидроксильных групп, что, по-видимому, связано
с уменьшением содержания катионов, влияющих на тип ассоциации мо-
лекул воды друг с другом.
Для того чтобы выявить влия-
ние компенсирующего катиона на
спектр кристаллического остова,
были исследованы различные ка-
тионные формы (Na, Sr, Са-фожа-
зиты, образцы 3, 8, 9) с постоян-
ным отношением Si : Al (рис. 3,
1, 2, 8). Спектр колебаний кри-
сталлического каркаса чувствите-
лен к заряду и радиусу обменного
катиона. Как следует из рис. 3,
различие между спектрами фожа-
зитов с двувалентными обменными
катионами (СаХ и SrX) значитель-
но меньше, чем между спектрами
каркаса фожазитов с одновалент-
ным и двувалентным компенсирую-
щим катионом. К виду обменного
катиона чувствительно положение
полосы 763 см-1, что также под-
тверждает принадлежность этой
полосы колебаниям связи А1—О.
Рис. 2. Инфракрасные спектры цеолитов
различного состава (7 —6) в области валент-
ных и деформационных колебаний гидро-
ксильных групп молекул воды, снятые
после вакуумной обработки при 25° в те-
чение 4 часов.
Исследование спектров колебаний кристаллического каркаса цеоли-
тов типа NaA (рис. 4, а) и NaX (рис. 4, б) и их декатионированных форм
Рис. 3. Инфракрасные спектры коле-
баний кристаллического каркаса фожа-
зитов с различными катионами, сня-
тые после обработки при 400°.
1 — NaX; 2 — SrX; в — СаХ.
(образцы 3, 10—13) показало, что
имеется сходство в положении целого
ряда полос поглощения (465, 570,
760 см-1) этих двух различных по
кристаллической структуре цеолитов.
Однако по общему числу полос спектр
цеолита NaX богаче, чем NaA. Кроме
того, полосы поглощения в спектре
цеолита NaA более размыты вслед-
ствие наложения широкой полосы
либрационных колебаний адсорбиро-
ванных молекул воды, которые в от-
личие от цеолита NaX не удаляются
из каналов цеолита NaA даже после
откачки при 400° в течение 4 часов.
На основании рассмотрения спек-
тров декатионированных форм цео-
литов NaA и NaX можно заклю-
чить, что декатионирование отмывкой
до 20% не вносит существенных из-
менений в спектр колебаний кристал-
лического каркаса. Это говорит о
том, что значительной перестройки
структуры при такой степени декатио-
нирования не происходит и измене-
ния, связанные с декатионированием, носят, по-видимому, локальный
характер. И только при сильном декатионировании цеолита NaA (обра-
зец 12), из которого отмывкой удалено 75% ионов Na+ (рис. 4, а, 3), на-
55
блюдается размывание полос, свидетельствующее об аморфизации струк-
туры, что согласуется с данными рентгеновского анализа этого образца.
Можно предполагать, что в процессе декатионирования при промывке
обмен катионов: образование на месте уда-
водой должен происходить
Рис. 4. Инфракрасные спектры
(7—3) цеолита NaA и его декатио-
нированных форм (а) и цеолита
NaX и его декатионированных
форм (б) в области колебаний кри-
сталлического каркаса, снятые
после обработки при 400°.
ленных катионов Na+ ионов гидроксония,
компенсирующих отрицательный заряд
алюмокислородных тетраэдров. Однако
спектральные критерии обнаружения иона
гидроксония нечетки: полосы поглощения
гидроксония лежат в области 1700 —
2800 см-1 [5], мало интенсивны и сравни-
тельно мало отличаются от полос погло-
щения адсорбированных молекул воды.
Вследствие этого или вследствие малой
концентрации иовов гидроксовия в иссле-
дованных декатионированных цеолитах не
удалось наблюдать их полос поглощения.
Гораздо большие изменения претерпе-
вает спектр колебаний кристаллического
каркаса при термическом декатионирова-
нии аммониевого фожазита. На рис. 5 пред-
ставлены спектры цеолита NH4X (образец
7) в области колебаний каркаса, в области
деформационных и валентных колебаний
адсорбированной воды и иона аммония,
снятые до откачки и с откачкой при 25,
100, 200, 300, 400°. Особенно отчетливо
полосы поглощения в области колебаний
кристаллического каркаса цеолита NH4X
проявляются после откачки при 25 и 100°, когда почти полностью удалена
вода, адсорбированная в каналах цеолита и имеющая широкую полосу
либрационных колебаний около 650 см”1 [6]. Затем после 200- и 300-гра-
дусной обработки полосы становятся более размытыми, что указывает на
Рис. 5. Инфракрасные спектры цеолита NH4X, декатионированного в результате
термической обработки.
Спектр образца до откачки (Z) и после откачки при 25° (2), 100 (.3), 200 (4), 300 (5), 400° (6).
перестройку кристаллической структуры цеолита, и уже после обработки
при 400° спектр цеолита NH4X совпадает со спектром NaX (образец 3),
прокаленного при 800°, когда происходит полная аморфизация структуры.
В соответствии с этими изменениями спектра колебаний остова нахо-
дится и изменение полосы 1452 см-1, принадлежащей деформационным
56
колебаниям иона NHt, и полосы 1645 см-1, принадлежащей деформацион-
ным колебаниям адсорбированных молекул воды. Интенсивность полосы
1452 см-1 уменьшается уже после откачки при 25°, что указывает на не-
которое разрушение иона NHt, происходящее уже при этой температуре,
и эта полоса полностью исчезает из спектра после обработки при 300°.
Интенсивность полосы 1645 см-1 заметно уменьшается после обработки
при 100° и почти полностью исчезает после обработки при 200°. В области
валентных колебаний наблюдается целый ряд широких перекрывающихся
полос адсорбированных молекул воды и иона NHt- После откачки при
25° в спектре появляется узкая полоса 3410 см"1 адсорбированного ам-
миака, образующегося при разрушении
иона NHt. При более высоких темпера-
турах вакуумной обработки адсорбции
молекул NH3 не происходит. После от-
качки при 25° в спектре появляется узкая
полоса 3660 см"1, лежащая в области ва-
лентных колебаний гидроксильных групп.
Интенсивность этой полосы увеличивается
с повышением температуры обработки и
становится максимальной при 300°, когда
происходит полное разрушение иона NHt,
и затем уменьшается после обработки при
400°. На рис. 6 представлено изменение
оптической плотности полос поглощения
деформационных колебаний молекул воды
(1645 см"1, кривая 1), иона NHJ
(1452 см-1, кривая 2), а также валентных
колебаний гидроксильных групп, обуслов-
ливающих полосу поглощения (3660 см-1,
кривая 3), в зависимости от температуры
обработки. Изменение оптической плот-
ности этих полос приблизительно отражает
изменение концентрации молекул воды,
иона аммония и гидроксильных групп, об-
условливающих полосу 3660 см-1. Как
видно из рис. 6, с повышением темпера-
туры обработки наблюдается рост коли-
Рис. 6. Изменения оптической
плотности полос деформацион-
ных колебаний молекул воды (7),
иона NHJ (8), валентных коле-
баний гидроксильных групп (3)
в зависимости от температуры
обработки.
чества гидроксильных групп (5), в то время как количество адсор-
бированных молекул воды уже при 100-градусной обработке стано-
вится незначительным (кривая 7) и практически отсутствует при 300-гра-
дусной обработке, когда количество гидроксильных групп становится
максимальным. Поэтому естественно сопоставить изменение концентра-
ции гидроксильных групп с ходом кривой 2, отражающей процесс разло-
жения иона NHt- Разрушение иона NHt сопровождается ростом числа
гидроксильных групп, которое достигает максимума при практически
полном разрушении иона NHt, происходящем при температуре 300°.
Это дало возможность предположить, что полоса 3660 см-1 принадлежит
структурным гидроксильным группам, образующимся в результате раз-
ложения иона NHf, сопровождающегося выделением протона, который
разрывает связи Si—О—А1 в фожазите с образованием структурных
гидроксильных групп. Образование структурных гидроксильных групп
можно представить происходящим по следующей схеме:
NH+
/О\ / \ НОЧ /
>А1< NSiz _> \А1 \SiZ 4-NH3.
СИ ХО /х z ХО СИ х
I I О ||
I
57
О процессе перестройки структуры, происходящем при этом, свиде-
тельствует изменение спектра колебаний кристаллического остова (рис. 4).
В то же время образование структурных гидроксильных групп в извест-
ной мере стабилизирует структуру и спектры колебаний каркаса при 200
и 300°, когда концентрация структурных гидроксильных групп наиболь-
шая, мало отличаются друг от друга. Только при 400°, когда
концентрация структурных гидроксильных групп резко падает, по-ви-
димому, вследствие смыкания их и выделения воды, происходит резкое
изменение спектра колебаний кристаллического каркаса, свидетельствую-
щее о полной аморфизации структуры.
Таким образом, приведенные данные показывают, что метод инфра-
красной спектроскопии является чувствительным методом исследования
структуры цеолитов и дает ценную информацию о влиянии состава, вида
компенсирующего катиона и декатионирования на структуру цеолита.
В дальнейшем работа будет продолжена в направлении исследования
декатионированных цеолитов типа Y.
Авторы выражают благодарность профессору А. В. Киселеву за руко-
водство работой и за обсуждение ее результатов.
ЛИТЕРАТУРА
1. Я. В. Д а в ы д о в, А. В. К и с е л е в, В. И. Л ыгин, Колл, ж., 1963, 25, 152.
2. С. П. Ждан ов, А. В. К и с е л е в, В. И. Л ыгин, Т. И. Т и т о в а, ЖФХ,
1964, 38, 2408.
3. J. R е i t z е 1, J. Chem. Phys., 1955, 23, 2407; J. Simon, H. O. Mahon,
J. Chem. Phys., 1953, 21, 23; J. M. Hunt, M. P. W i s c h e г d, L.C. В on-
ham, Analyt. Chem., 1950, 22, 1478.
4. В. А. Колесова, Оптика и спектроскопия, 1959, 6, 38.
5. С. С. F о г г i s s о, D. F. Н о г n i g, J. Chem. Phys., 1955, 23, 1464; M. Falk,
P. A. G i g u e г r e, Canad. J. Chem., 1957, 35, 1195.
6. P. A. G i g u e г г e, К. В. H a г v e у, Canad. J. Chem., 1958, 34, 798.
ИНФРАКРАСНЫЕ СПЕКТРЫ АДСОРБИРОВАННЫХ
ЦЕОЛИТАМИ МОЛЕКУЛ
В. И. Лыгин
Для изучения разделяющей способности цеолитов особый интерес
представляет исследование адсорбции ими молекул с периферическим рас-
пределением электронной плотности (л-электроны, свободные электроны),
способных к специфическому взаимодействию [1] и обладающих, кроме
того, группами, могущими образовывать водородные связи. Состояние
таких адсорбированных молекул определяется конкурирующим действием
силовых центров поверхности каналов цеолита и водородной связью со-
седних молекул друг с другом и затруднено для исследования обычными
адсорбционными методами.
В исследовании адсорбции таких молекул цеолитами большие возмож-
ности открывает использование метода инфракрасной спектроскопии,
нашедшего в настоящее время широкое применение в адсорбции [2].
Для цеолитов этот метод применен значительно меньше и в основном только
для исследования адсорбции воды [3—8J, СО2 [6], NO [9] и бензола [101.
В настоящей работе методом инфракрасной спектроскопии исследовано
состояние адсорбированных цеолитами молекул Н2О, СН3ОН, СвН5ОН
и NH3, CeH5NH2, CeH6NO2. Для простейших молекул этого ряда (Н2О,
58
NH3) произведен анализ их колебательного спектра в адсорбированном
состоянии на основе общей теории колебаний многоатомных молекул
с проведением расчетов на электронно-вычислительной машине.
Методика
В работе использовались кристаллы синтетических цеолитов без
связующего, синтезированные в лаборатории С. П. Жданова. Для умень-
шения рассеяния инфракрасного света цеолиты прессовались в таблетки
под давлением 50 кг/мм2; вес 1 см2 таблетки 5—7 мг. Термическая обра-
ботка образца в вакууме, адсорбция и съемка спектров спектрометром
UR-10 фирмы Цейса проводились в кювете, аналогичной [11].
Вода, метанол, фенол
На рис. 1, а представлено изменение спектра валентных и деформа-
ционных колебаний молекул воды, адсорбированных цеолитом NaX
при его термической дегидратации. При малых заполнениях каналов цео-
лита в спектре отчетливо наблюдаются полосы валентных колебаний гидро-
ксильных групп: узкая—3690 см-1 и две широкие — 3240 и 3385 см-1.
Как форма и ширина по-
лос, так и их положение
по отношению к соответ-
ствующим полосам погло-
щения молекул пара воды,
жидкости и кристалла
(табл. 1) указывают на
принадлежность узкой по-
лосы к свободным, мало
возмущенным, а широких
полос — к возмущенным
водородной связью гидро-
ксильным группам адсор-
бированных молекул воды.
Адсорбированные молеку-
лы испытывают также дей-
ствие катионов, влияние
которых особенно отчет-
Рис. 1. Изменение инфракрасного спектра цеолита
NaX состава Н003, Na0()7, (АЮгИ • (SiО2)1Л8 (а) и
NaA состава Н0.0з • Na097 - (A102)j • (SiO2)i (б) при
термической обработке в вакууме.
ливо проявляется на ва-
лентных колебаниях мало
возмущенных гидроксиль-
ных групп. Так, возра-
станию величины смещения
узкой полосы поглощения
валентных колебаний мо-
I — исходный; обработан в течение 4 часов; 2 — при 25°, 3 —
при 100°, 4 — при 400°.
лекул воды при переходе от цеолитов типа LiX к КХ [6, 7], т. е. увели-
чению степени возмущения гидроксильных групп, соответствует прибли-
зительно пропорциональное увеличение радиуса ионнообменного катиона
(рис. 2).
Спектр молекул воды, адсорбированных цеолитом NaA и цеолитом
NaX, различен (рис. 1, б). В спектре цеолита NaA не наблюдается отчет-
ливо узкой полосы поглощения валентных колебаний около 3700 см-1,
а широкие полосы смещены меньше, чем в цеолите X (табл. 1). Этот факт
может быть следствием участия гидроксильных групп молекул воды в об-
разовании более слабой водородной связи, чем в случае цеолита X. Та-
кое отличие спектра молекул воды, адсорбированных цеолитами X и А,
59
Таблица 1
Спектр колебаний молекул Н2О, CD3OD, C6HSOH в газообразном, жидком,
кристаллическом и адсорбированном цеолитами состояниях (см-1)
Вещество Тип колебания Пар Адсорбирован цеолитом 1 Жидкость Кристалл
NaX NaA SrX
онвяп. 3756 [12] 3690
3385 3450 3395 [13]
Н2О 3240 3290 3256 [12]
0Ндеф • 1585 [12] 1660 1665 1645 [13]
1590 1570 1595 [12]
0Г>вал • 2725 [14] 2625
2500 2550 2474 [14] 2432 [14]
'-‘^сим 2081 [14] 2082 2082 2082 [14] 2075 [14]
вал
С6н5он 0Нвал • 3200 3615 2
3400 3250 [15]
объясняется, по-видимому, различием числа и места фиксации катионов
в каналах цеолита, влияющих на расположение молекул и тем самым —
на прочность водородной связи. Спектр адсорбированных молекул воды
Дх),см’
100
50
О 1 2 rj
чувствителен также к составу цеолита [16]
и его декатионированию [17].
Положение полос поглощения колеба-
ний воды при полном заполнении и уда-
лении значительной части (рис. 1) воды
из каналов указывает на близость их со-
стояния к состоянию молекул воды в жид-
кости в первом случае и на большее, чем
в жидкости, возмущение во втором слу-
чае.
Столь же отчетливое влияние на спектр
адсорбированных молекул катионов и сте-
пени заполнения каналов цеолитов на-
блюдается при адсорбции метанола. По-
ложение полосы поглощения валентных
колебаний OD метанола при малых запол-
Рис. 2. Зависимость смещения
полосы поглощения валентного
колебании гидроксильной груп-
пы адсорбированных молекул
воды относительно полосы сво-
бодных молекул воды Дм от раз-
мера обменного катиона цеоли-
тов.
нениях каналов цеолитов в случае одно-
валентных (Na+) и двухвалентных (Sr'*'+)
обменных катионов различно (рис. 3,
табл. 1). Смещение этой полосы при уве-
личении заполнения каналов цеолита SrX
молекулами CD./JD может объясняться
большей неоднородностью поля в каналах,
чем в цеолите NaX. В обоих случаях при
больших степенях заполнения наблюдается появление «плеча» около
2400 см 1, обязанного, по-видимому, молекулам CDfiD, не испытываю-
щим влияния катионов и ориентированным вследствие этого наиболее
благоприятно для образования сильной водородной связи друг с дру-
гом [18].
1 При заполнении 6 = 0.3.
2 Раствор в СС14.
60
При адсорбции цеолитами фенола, имеющего в своем составе ядро
бензола, ^-электроны которого взаимодействуют с катионами [10], также
наиболее сильно меняются валентные ко-
лебания ОН (табл. 1).
Аммиак, анилин, нитробензол
Изменение спектра аммиака при ад-
сорбции цеолитом представлено в табл. 2.
При адсорбции анилина цеолитом полосы
валентных колебаний NH смещаются
больше, чем на 100 см4. Изменение ко-
лебаний ароматического ядра менее зна-
чительно. В противоположность этому из-
менение полос поглощения колебаний
функциональной группы NO2 в адсорби-
рованном цеолитом нитробензоле менее
значительно. Большие изменения в этом
случае испытывают валентные колебания
связи СП, что указывает на преимуще-
ственное специфическое взаимодействие
с катионами цеолита бензольного кольца.
Столь же сильное изменение спектра ва-
лентных колебаний NH аммиака, анилина
и гораздо меньшее нитробензола
наблюдается также при адсорбции их
аэросилом [19]. Такому различию в ха-
рактере специфического взаимодействия
молекул аммиака, анилина и нитробен-
Рис. 3. Спектр валентных коле-
баний гидроксильных групп ме-
танола CD^OD, адсорбированного
цеолитами NAX (а) и SrX (б).
Цифры «а кривых ^обозначают заполне-
ние каналов цеолита.'-]
зола соответствуют и изменения электрон-
ного спектра при адсорбции этих молекул
на поверхности аэросила [20]. Сходство
спектральных проявлений при адсорбции
этих молекул указывает на общность
механизма взаимодействия этих молекул
как с протонизированным водородом гидро-
ксильной группы аэросила, так и с положительным катионом цеолита.
В свою очередь различие в спектральных проявлениях адсорбции ам-
Таблица 2
Спектр аммиака в газообразном,
адсорбированном и кристаллическом
состояниях (см"1)
Тип колебания Состояние аммиака
гав [12] адсорбиро- ван NaX- цеолитом кри- сталл [13]
'«'З 3444 3370 3378
AVg . . . . — 74 66
vi 3336 3308 3223
Avj .... — 28 113
к стягиванию на атом азота несвязанной пары электронов, находящихся
в сопряжении с бензольным кольцом. В результате этого спектр колеба-
ний в замещающей группе должен приближаться к спектру колебаний
миака, анилина, с одной стороны, и
нитробензола, с другой, указывает
на особую роль свободных электрон-
ных пар атомов кислорода и азота
при адсорбции.
Перераспределение электронной
плотности в адсорбированных цео-
литом молекулах фенола и анилина,
по-видимому, аналогично случаю до-
норно-акцепторного взаимодействия
свободных электронов заместителя
ароматического ядра с донором про-
тонов [21]. Взаимодействие атома
азота с положительным катионом дол-
жно приводить в соответствии с этим
61
групп NH-соединений (например, аммиака), в которых свободные электрон-
ные пары не участвуют в сопряжении с ароматическим кольцом. Действи-
тельно, сдвиг полос поглощения связей NH адсорбированного анилина
больше, чем для адсорбированного аммиака (табл. 2), и положение его
полос поглощения близко к положению полос адсорбированного аммиака.
Колебательный спектр и силовые коэффициенты потенциальной функции
адсорбированных молекул Н2О и NH3
Некоторые сведения об изменении внутреннего силового поля адсорби-
рованных молекул можно получить путем анализа их колебательного
спектра на основе общей теории колебаний многоатомных молекул. Этот
анализ позволяет установить связь между элементами матрицы силовых
и частотами полос поглощения их спектра.
постоянных
молекулы
Рис. 4. Зависимость частот колебатель-
ного спектра воды от величин коэффициен-
тов матрицы силовых постоянных Кд и
КТ
1 — изменяется только одна постоянная; 2 — сим-
метричное изменение обеих постоянных
Такой подходограничен, поскольку
силовое поле в данном случае
характеризуется коэффициентами
матрицы силовых постоянных,
имеющих сложный физический
смысл, для валентно-силовой мо-
дели молекулы в аддитивном при-
ближении.
Вековые уравнения составля-
лись по способу М. А. Ельяше-
вича [22]. Численное решение
уравнений производилось на элек-
тронно-вычислительной машине
«Стрела». На рис. 4 представлено
рассчитанное изменение частот
нормальных колебаний молекулы
Н2О. Как следует из рисунка, при
симметричном изменении силовых
постоянных связей ОН сильно
уменьшаются частоты симметрич-
ного (v*) и асимметричного (v3)
колебаний (кривые 2). Разность
частот между ними остается рав-
ной 100—110 см-1. При асимме-
тричном изменении этих постоян-
ных частота v3 изменяется мало,
в то время как частота уменьша-
ется так же сильно, как и в случае
симметричной нагрузки на связь.
При увеличении силовой постоян-
ной К частота деформационного колебания v2 увеличивается. В соответ-
ствии с ходом кривых рис. 4 узкая полоса валентных колебаний может
быть приписана асимметричному, а каждая из широких полос (рис. 1, а,
табл. 1) — симметричному колебанию асимметрично возмущенной моле-
кулы воды. Таким образом, для этого типа состояния возможны два со-
четания частот: 3700, 3400, 1585 см-1 и 3700, 3250, 1585 см-1. Другое со-
четание экспериментальных частот (табл. 1) — 3400, 3250, 1660 см-1,
согласно ходу кривых 2 рис. 1, соответствует молекулам воды с сильным
приблизительно симметричным возмущением обеих связей. Выбор полос
поглощения для этих состояний позволяет найти тенденцию изменения
формы колебания и смещения атомов при колебаниях в адсорбированном
состоянии. Смещения атомов при колебаниях представлены на рис. 5
62
для свободных (7), асимметрично возмущенных (II) и симметрично воз-
мущенных (III) адсорбированных цеолитом молекул воды. Из рис. 5
Рис. 5. Смещение атомов свободной молекулы Н2О (/) и адсорби-
рованной цеолитом молекулы Н20 при асимметричном (11) и
симметричном (III) воздействии на связи ОН.
следует, что в случае асимметричного возмущения адсорбированных мо-
лекул воды при симметричных и асимметричных колебаниях в основном
смещается атом водорода только
одной связи.
В случае возмущения обеих связей
величины смещений водорода меньше
отличаются от смещений в газообраз-
ном состоянии. Однако колебание Vx
в большей степени, чем любое дру-
гое, перестает быть чисто валентным.
Кривые зависимости частот от
значения силовых постоянных для
аммиака (рис. 6) показывают, что
асимметричное изменение силовых
коэффициентов связи NH(Kq) и угла
между связями (К^) приводит к сня-
тию вырождения частот v3 и v4. Ве-
личина расщепления вырожденного
колебания Av3 при изменении Kq
постепенно увеличивается и затем
остается практически постоянной.
Частота колебания при асимме-
тричном изменении Kq изменяется
вначале медленнее, чем при симме-
тричном воздействии, и в дальнейшем
наклон этих кривых становится одина-
ковым.
Экспериментально наблюдается
превышение, приблизительно в два
Рис. 6. Зависимость частот колебатель-
ного спектра аммиака от величины мат-
рицы силовых постоянных Kq и Кг
1 — изменение всех постоянных, 2 — одной по-
стоянной, 3 — двух постоянных Кд.
раза, величины смещения полосы поглощения Av3 над Avt в случае ад-
сорбции аммиака цеолитом и обратное соотношение между этими величи-
нами в случае образования водородной связи в кристаллическом аммиаке
(табл. 2). Такое соотношение может быть объяснено, согласно ходу кривых 2
(рис. 6), небольшим асимметричным изменением (меньше 2%) величин Kq
63
в первом случае и большим (8—10%) во втором. В свою очередь этот факт
может быть объяснен взаимодействием катиона цеолита с атомом азота,
приводящим к небольшому изменению силовой постоянной Kq, и большим
возмущением связи NH при образовании водородной связи. Таким обра-
зом, такой анализ спектра дает подтверждение предположению об участии
свободных электронов атомов азота во взаимодействии с катионами, не
столь очевидное из вида самого спектра (табл. 2).
Выводы
Инфракрасные спектры указывают на различие состояния молекул
воды, адсорбированных цеолитом NaX и NaA. Положение полосы валент-
ных колебаний ОН молекул воды и метанола чувствительно к виду об-
менного катиона и заполнению каналов цеолита. Анализ спектров на основе
общей теории колебаний указывает на существование двух основных ти-
пов состояний адсорбированных цеолитом NaX молекул воды и на взаимо-
действие молекулы аммиака с катионом цеолита через свободную электрон-
ную пару атома азота.
ЛИТЕРАТУРА
1. А. В. Киселев, ЖФХ, 1964, 38, 12, 2753.
2. А. Н. Те р енин, в сб.: Поверхностные химические соединения и их роль
в явлениях адсорбции, М., 1957, 206; А. В. Киселев, В. И. Л ы г и н,
У си. химии, 1962, 31, 351.
3. G. J. С. F г о h n s d о г f f, G. L. К i n g t о n, Proc. Roy. Soc., 1958, 247, 469.
4. H. A. S z u m a n s k i, D. N. St a mires, G. R. Lynch, J. Opt. Soc. Am.,
1960, 50, 1323.
5. Г. В. Цицишвили, Г. Д. Багратишвили, в сб.: Синтетические
цеолиты, Изд. АН СССР,. 1962, 38.
6. L. Bertsch, Н. W. Habgood, J. Phys. Chem., 1963, 67, 1621.
7. С. П. Ж д а н о в, А. В. К и с е л е в, В. И. Л ы г и н, Т. И. Т и т о в а, ДА'Н
СССР, 1963, 150, 584.
8. Г. В. Ю х н е в и ч, Э. Э. С е н д е р о в, Геохимия, 1963, 2, 48.
9. А. Н. А л е к с е е в, В. Н. Ф и л и м о н о в, А. Н. Т е р е н и н, ДАН СССР,
1962, 147, 1392.
10. В. Н. А б р а м о в, А. В. К и с е л е в, В. И. Л ы г и н, ЖФХ, 1963, 37, 1156.
11. В. Я. Д а в ы д о в, А. В. К и с е л е в, В. И. Л ы г и н, Колл, ж., 1963, 25,
152.
12. Г. Герцберг. Колебательные и вращательные спектры многоатомных моле-
кул. ИЛ, М., 1945.
13. Р. A. Giguerre, К. В. Harvey, Can. J. Chem., 1956, 34, 789.
14. М. F а 1 k, Е. W а 11 е у, J. Chem. Phys., 1961, 34, 1554.
15. G. К ага gounis, О. Pet er, Z. Elektrochem., 1957, 61, 464.
16. С. П. Ж д а н о в, А. В. К и с е л е в, В. И. Л ы г и н, Т. И. Титов а, ЖФХ,
1964, 38, 2408.
17. С. П. Ж д а н о в, А. В. К и с е л е в, Т. И. Т и т о в а, М. Е. О в сепян,
ЖФХ, 1965 (в печати).
18. А. В. К и с е л е в, Л. К у б е л к о в а, В. И. Л ы г и н, ЖФХ, 1964, 38, 2719.
19. В. Н. А б р а м о в, А. В. К и с е л е в, В. И. Л ы г и н, ЖФХ, 1964, 38, 1044.
20. В. Н. А б р а м о в, А. В. К и с е л е в, В. И. Л ы г и н, ЖФХ, 1964, 38, 1867.
21. В. Н. Филимонов, Д. Е. Б ы с т р о в, Оптика и спектроскопия, 1962,
12, 66.
22. М. В. Волькенштейн, М. А. Ельяшевич, Б. И. Степанов.
Колебания молекул. Изд. ГТТЛ, 1949.
64
ИССЛЕДОВАНИЕ АДСОРБЦИОННОГО ВЗАИМОДЕЙСТВИЯ ВЕЩЕСТВ
С СИНТЕТИЧЕСКИМИ ЦЕОЛИТАМИ
МЕТОДОМ ИНФРАКРАСНЫХ СПЕКТРОВ ПОГЛОЩЕНИЯ
Г. Д. Багратишвили, Г. В. Цицишвили, М. Л. Кантария, Н. И. Ониашвили
За последние годы в области экспериментальных методов исследова-
ния адсорбционных процессов, помимо классических методов, широко
стали применяться новые физические и физико-химические методы, осно-
ванные на изучении оптических, электрических и магнитных свойств
адсорбата и адсорбента. В этом направлении особенно перспективным
является метод исследования инфракрасных спектров веществ в адсорби-
рованном состоянии, который впервые был применен А. И. Терениным
и сотр. [1].
В настоящей работе методом инфракрасных спектров поглощения
исследована природа поверхности различных катионзамещенных форм
цеолитов (Na, Са, Ag, NH4, Н), а также изучено взаимодействие этих
форм цеолитов с веществами различной природы (вода, тяжелая вода,
окись углерода, ацетилен и др.).
Исследование проводили на однолучевом инфракрасном спектрометре
ИКС-12 с призмами из фтористого лития и каменной соли. Термическая
обработка и съемка спектров проводились в вакуумной установке, в ко-
торой создавался вакуум 5 • 10~6 мм рт. ст.
Приготовление образцов цеолитов для съемки ИК-спектров описано
нами ранее [2, 3]. Толщина адсорбента варьировалась в пределах 10—
15 мг/см2.
Перед адсорбцией образцы цеолитов откачивались в вакууме 10 —
10“8 мм рт. ст. в течение 8—10 часов при температуре 250° С. После такой
обработки адсорбент выдерживался в .течение нескольких часов в атмо-
сфере адсорбата под давлением, соответствующим для жидкостей упруго-
сти насыщенного пара, а для газов 300—400 мм. рт. ст. Затем производи-
лась откачка адсорбата и на промежуточных стадиях откачки при задан-
ных давлениях снимались ИК-спектры.
Исследование ИК-спектров полностью обезгаженных катионзамещен-
ных форм цеолитов, в том числе и водородных, показывает, что в области
валентных колебаний гидроксильных групп не наблюдается заметного
поглощения [2, 3]. Следует отметить, что это характерно не только для
цеолитов, но и для многих других представителей алюмосиликатных
систем (монтмориллониты [4], алюмосиликатные катализаторы и др.).
Вместе с тем в ИК-спектрах окисных адсорбентов типа SiO2, А12О3, TiO2,
ZnO и др. в области около 3760 см-1 найдена острая и интенсивная полоса,
соответствующая свободным гидроксильным группам поверхности. По-
лоса взаимодействующих между собой поверхностных гидроксильных
групп приближается к полосе свободных ОН групп со стороны длинных
волн в виде сравнительно широкого крыла.
Приведенные факты свидетельствуют о том, что непроявление в ИК-
спектрах цеолитов полос ОН связей обусловлено отсутствием гидроксиль-
ных групп на их поверхности.
Особо следует разобрать вопрос о водородной форме цеолитов. В ра-
боте [5] в ИК-спектре водородных цеолитов найдена острая и интенсивная
полоса около 3550 см-1. Аналогичная полоса была обнаружена также
в спектрах аммониевой и даже натриевой форм цеолитов. В наших [2, 3]
и в других [6, 7] работах полоса при 3550 см-1 не была обнаружена.
В качестве одного из вариантов можно допустить, что в кристалли-
ческой решетке водородного цеолита атом водорода занимает место исход-
5 Цеолиты
65
на
А! А !/<
0 0 0 0 о о
Рис. 1.
.0
ного катиона и взаимодействует с анионом [АЮ4]_ в целом (рис. 1). По-
этому нет основания говорить о гидроксильной группе в обычном смысле
(SiOH или АЮН). По некоторой аналогии со спектрами минеральных
кислот, колебание водорода в группировке Н+[А1О4]“ должно проявиться
по крайней мере в области 3300—3400 см-1 в виде широкой полосы. С этой
точки зрения несколько непонятны наблюдения
Шиманского и др. [5].
Значительный интерес представляет иссле-
дование адсорбционного взаимодействия различ-
ных катионных форм цеолитов с молекулами,
которые, являясь нейтральными (без неспаренных
электронов), способны образовать с катионом
донорно-акцепторные связи. Здесь подразуме-
ваются переходные металлы и металлы первой
подгруппы периодической системы Менделеева.
Выяснение природы взаимодействия таких молекул с катионами цеолита
может пролить свет на вопрос валентного состояния последнего в алюмо-
силикатном каркасе цеолита.
По данным Брек и др. [8], адсорбция окиси углерода на цеолите типа
NaA в условиях комнатной температуры мала и составляет при давлении
700 мм рт. ст. 41 мг/г. В соответствии с этим нам не
ИК-спектры окиси углерода, адсорбированной на
цеолитах типа NaA и NaX.
В ИК-спектре цеолита AgA с адсорбированной
окисью углерода при давлении 300 мм рт. ст. и
комнатной температуре в области 2000—2200 см-1
наблюдается сильное поглощение, которое следует
отнести к молекулам окиси углерода, как адсор-
бированным на поверхности, так и находящимся
в мертвом пространстве кюветы.1 Уже минутная
откачка кюветы полностью удаляет молекулы
газообразной окиси углерода,2 и оставшееся по-
глощение полностью соответствует адсорбирован-
ным молекулам. В этом случае в ИК-спектре
наблюдается довольно узкая полоса окиси угле
рода с максимумом при 2131 см-1 (рис. 2).
Дальнейшая откачка образца и съемка спек-
тров при давлениях 101, 10 “2, 10-3 и 10 мм
рт. ст. не изменяют формы полосы при 2131 см-1,
и наблюдается лишь ее ослабление. Наконец,
в результате двухчасовой откачки полоса совсем
исчезает.
Первое, на что приходится обратить внимание
при рассмотрении ИК спектра окиси углерода
на цеолите типа AgA, это — простота спектра.
Во всем диапазоне давлений в ИК-спектрах про-
является лишь одна полоса окиси углерода с по-
лушириной около 50—60 см-1. Не относя эту
полосу к определенным видам взаимодействия в системе СО—AgA, следует
отметить, что небольшая ширина полосы при различных заполнениях по-
верхности, по-видимому, указывает на сравнительно однородную поверх-
удалось получить
Г, %
100г
75
50
25
О
2000 2200 см
Рис. 2. ИК спектры
окиси углерода, адсор-
бированной на AgA-цео-
лите.
1 — в кювете, наполненной
парами окиси углерода,
2 — сразу после откачки
окиси углерода; давление
паров окиси углерода (мм
рт. ст.), 3 — 2.5 • 10-1,
* — 1.8 • 10-*, 6 — 3.5 •
10-з, 6 _ 2.3 • 10-‘, 7 —
после 90 мин. откачки при
давлении 9.6 - 10-5 мм.
рт. ст.
1 Оптическая длина нашей кюветы около 70 мм; для получения ИК-слектра газо-
образной окиси углерода достаточно давление 100 мм рт. ст. при толщине слоя около .
100 мм.
2 Полная откачка газообразной окиси углерода из кюветы контролировалась
снятием спектра холостой кюветы.
66
ность, или, вернее говоря, на наличие однородных адсорбционных цент-
ров для окиси углерода на поверхности цеолитов, что, вероятно, обуслов-
лено их кристаллическим строением.
В отсутствии окислительных процессов для взаимодействия окиси
углерода с активной поверхностью предложены две схемы, описывающие
Мех \
/С=О I хемосор-
МеХ /
образование линейных (Me—СО) и мостиковых
бированных видов. Первый из них характеризуется полосой валент-
ного колебания СО в области 2040—2090 см-1, а второй — около
1850—1960 см-1 [9—12]. Это наблюдение основывается на исследова-
нии свободных карбонилов металлов [13].
Однако анализ литературных данных пока-
зывает, что приведенные области частот харак-
терны в основном для молекул окиси углерода,
связанных с переходными металлами. Для ме-
таллов, расположенных в периодической системе
рядом с переходными элементами, характери-
зующихся заполненными d-оболочками (Си, Ag,
Ап), частота соответствующих колебаний окиси
углерода может быть смещена и в более ко-
ротковолновую область. В самом деле, полоса
окиси углерода, хемосорбированной на меди, про-
является при 2001 см-1 [9, 14]. Литературные
данные по ИК-спектрам хемосорбированной
окиси углерода на серебре и золоте нам не уда-
лось найти.
Исходя из вышеизложенного, полосу при
20 131 см-1, наблюдаемую в ИК спектрах окиси
углерода на цеолите типа AgA, по-видимому, можно
приписать структуре типа Ag—С=О, образован-
ной за счет хемосорбции.
Химическая природа взаимодействия СО с цео-
литом типа AgA вытекает уже из самого факта
адсорбции при комнатной температуре.
Химический характер взаимодействия цеолитов
Рис. 3. ИК спектры
ацетилена, адсорбиро-
ванного на NaA-цеоли-
те. Давление С2Н2 100 мм
(2), после откачки С2Н2
при температуре 120° С
и вакууме 10~4 в течение
15 мин. (2), в течение
1 часа (5) и в течение
3 часов (4).
с молекулами ацети-
лена можно проследить также при изучении ИК-спектров ацетилена, ад-
сорбированного на цеолитах.
Инфракрасные спектры ацетилена, адсорбированного на микропори-
стом стекле [15] или на силикагеле [16] при комнатной температуре, ука-
зывают на слабую физическую адсорбцию молекул, которые могут быть
удалены с поверхности откачкой в вакууме при комнатной температуре
за 5 сек. При этом полоса валентного колебания группировки =С—Н
физически адсорбированного ацетилена смещена по отношению к парам
на 22 см-1 около 0.7% и проявляется при 3265 см"1. Это смещение факти-
чески соответствует величине, наблюдаемой для ацетилена при переходе
от паров к кристаллу (3263 см-1).
Однако при адсорбции ацетилена на окиси алюминия [16], кроме фи-
зически адсорбированных молекул, в ИК-спектраХ наблюдаются также
прочно хемосорбированные молекулы. Последние проявляются в ИК-
области спектра около 3300 см-1 (v8 колебание ацетилена) и десорбируются
с поверхности только при откачке в высоком вакууме за 10 мин. в усло-
виях температуры 300° С.
Нами получены ИК-спектры ацетилена, адсорбированного в условиях
комнатной температуры на цеолите NaA при давлении 100 мм рт. ст.,
и спектры того же образца при различной степени откачки (рис. 3). Для
5*
67
всех образцов независимо от степени заполнения поверхности наблю-
дается единственная узкая полоса валентного колебания концевой ацети-
леновой группировки =С—Н около 3218 см-1. Полное исчезновение этой
полосы наступает только при откачке образца в течение трех часов в ус-
ловиях высокого вакуума и температуры 120° С. Следует отметить, что
исчезновение полосы при 3218 см-1 не приводит к появлению каких-либо
новых полос в области СН колебаний.
Смещение полосы т3 (3218 см-1) адсорбированного на цеолите NaA
ацетилена от ее положения в парах составляет около 70 см-1 (2.1%), что
втрое больше аналогичного смещения для ацетилена на силикагеле.
Наблюдаемое спектральное поведение адсорбированного на цеолите
типа NaA ацетилена исключает диссоциативный механизм хемосорбции,
предусматривающий превращение ацетилена в результате автогидриро-
вания в этилен на чистом никеле или на системе Ni—кизельгур [17].
По-видимому, хемосорбция ацетилена на цеолите типа NaA происходит
без диссоциации с участием в ковалентных связях л-электронов кратных
связей.
ЛИТЕРАТУРА
1. А. И. Теренин, ЖФХ, 1940, 14, 1362; в сб.: Поверхностные химические
соединения и их роль в явлениях адсорбции, изд. МГУ, М., 1957, 206, 271.
2. Г. В. Цицишвили, Г. Д. Багратишвили, в сб.: Синтетические цео-
литы, Изд. АН СССР, 1962, 38.
3. Г. В. Цицишвили, Г. Д. Багратишвили, К. А. Б е ж а ш в и л и,
Д. Н. Барнабишвили, М. С. Шуакришвили, ДАН СССР,
1963, 152, 1136.
4. Г. Д. Багратишвили, Г. В. Цицишвили, Б. В. Пайлодзе,
Тез. 5-й Всесоюзн. конф, по коллоидной химии, Одесса, Изд. АН СССР, 1962, 25.
5. Н. A. S z у m a n s k i, D. N. S t a mi г es, G. R. L у n с h, J. Opt. Soc. Am.,
1960, 50. '
6. G. J. C. F г о n s d о r f f, G. L. King ton, Proc. Roy. Soc., 1958, A 247, 469.
7. С. П. Ж д а н о в, А. В. К и с e л e в, В. И. Л ы г и н, Т. И. Т и т о в а, ДАН
СССР, 1963, 150, 584.
8. D. W. В г е с к, W. G. Е v е г s о 1 е, R. М. М i 11 о n, Т. В. R е е d, Т. L.Th о-
mas, J. Am. Chem. Soc., 1956, 78, 5963.
9. P. Э й ш e н с, В. П л и с к и н, в сб.: Катализ. Исследование поверхности ка-
тализаторов, ИЛ, М., 1960, 284.
10. Р. Э й ш е н с, в сб.: Новые методы изучения гетерогенного катализа, ИЛ, М.,
1963.
11. D. Y a t е s, J. Phys. Chem., 1961, 65, 746.
12. С. Е. О’ N е i 11, D. Y a t е s, J. Phys. Chem., 1961, 65, 901.
13. Ф. Коттон, в сб.: Современная химия координационных соединений, ИЛ,
М., 1963.
14. R. Р. Е i s с h е n s, W. А. Р 1 i s k i n, S. A. F r e n c i s, J. Chem. Phys., 1954,
22, 10, 1786.
15. N. S h e p p a r d, D. Y a t e s, Trans. Faraday. Soc., 1956, 238, 69.
16. D. Yates, P. Lycchesi, J. Chem. Phys., 1961, 35, 243.
17. J. D о u g 1 a s, B. S. R a b i n о v i t c h, J. Am. Chem. Soc., 1952, 74, 2486.
ПОДВИЖНОСТЬ И ФОРМА СВЯЗИ МОЛЕКУЛ воды
В ЦЕОЛИТАХ
С. П. Габудам
Для создания микроскопической теории сорбционных явлений важ-
нейшее значение имеет исследование элементарных актов адсорбции
под которыми мы подразумеваем в основном два процесса: вступление
молекулы в связь с поверхностью адсорбента и процесс последующей диф-
фузии вдоль поверхности (или вдоль канала структуры). При этом прежде
всего представляет интерес выяснение природы и характера взаимодей-
68
ствия адсорбируемых молекул с адсорбентом, микроскопический меха-
низм диффузии.
Исследование состояния и свойств адсорбированной фазы и выяснение
влияния ее на свойства цеолитов представляет большой интерес также
и для физики. «Тончайшая пленка» адсорбированного вещества на внут-
ренней поверхности цеолита может рассматриваться как самостоятельная
фаза с особым энергетическим спектром и особыми свойствами, изучение
которых кажется многообещающим для теории и практики.
В ряде работ [1—10] нами были изучены методом ядерного магнитного
резонанса (ЯМР) свойства и состояние цеолитной воды. Метод ЯМР чув-
ствителен к положению и подвижности протонов, поэтому применение его
дало целый ряд сведений о микроскопическом характере взаимодействия
и движения молекул Н2О. Одно из ценных достоинств метода ЯМР —
чувствительность к движениям низкой частоты (104—107 гц), тогда как,
например, оптические методы чувствительны к движениям с частотой
1010—1012 гц. Поэтому применение оптических методов к исследованию
явлений, протекающих с низкой скоростью (диффузия, заторможенные
вращения), дает лишь «мгновенный снимок» системы.
Настоящая работа посвящена изложению некоторых новых результа-
тов исследования цеолитов методом ЯМР и обобщающему анализу этих
и полученных ранее в работах [1—10] данных.
Подвижность молекул воды и спектры ЯМР
Спектры гидратов обычно представляют собой дублеты (или сумму
нескольких дублетов). Расстояние (АН) между компонентами дублета
определяется статическим локальным магнитным полем, действующим
на протон. Формула Пейка для монокристалла [11] дает
3
Д// = у [ЛГ-З (3 cos2 6—1), (1)
где р. — магнитный момент протона, г — расстояние между протонами
в молекуле Н2О и 0 — угол между направлениями протон-протонного
(р—р) вектора и магнитного поля.
Если молекула реориентируется, локальное поле, действующее на
соседнее ядро и приводящее к дублетной структуре спектра, уже не яв-
ляется статическим и нужно рассматривать среднее по времени локальное
поле. В общем случае это среднее по времени поле, взятое для различных
допустимых ориентаций дипольных пар, будет меньше, чем статическое
локальное поле для жестких систем. Поэтому спектры поглощения ЯМР
в твердых телах при наличии реориентаций молекул сужаются по сравне-
нию со спектрами для жестких структур. Если молекула вращается во-
круг фиксированной в структуре кристалла оси, а угол между этой осью
и вектором р — р молекул Н2О равен спектр также имеет дублет-
ную структуру с расстоянием между компонентами дублета [12]
з
Д77 = рт-з (з cos? — 1) (3 COS2 6' — 1), (2)
где 6' — угол между осью вращения молекулы и направлением внешнего
магнитного поля.
Если ось вращения молекулы прецессирует под углом к некоторой
фиксированной в кристалле оси, т. е. молекула обладает двумя степенями
69
свободы, спектр также сохраняет формулу дублета и расстояние между
компонентами его равно [13]
3
Aff=-g-fir 3 (3 cos2 — 1) (3 cos2? — l)(3cos26"— 1), (3)
где 6" — угол между осью прецессии и магнитным полем Но.
Таким образом, с увеличением числа степеней свободы молекулы воды
дублетная форма спектра сохраняется, но расстояние между компонентами,
вообще говоря, падает. Если число степеней свободы молекулы равно
трем, спектр теряет форму дублета, но вследствие межмолекулярного
взаимодействия сохраняет конечную (1-4-2 э) ширину. Наконец, если
молекула имеет также и поступательные степени свободы (диффузия),
спектр становится таким же узким, как и у жидкостей (менее 0.1 э). Та-
ким образом, метод ЯМР позволяет детально исследовать процессы внут-
ренней подвижности молекул, определить оси вращения и углы прецессии,
а также область температур, в которой эти движения имеют место.
Здесь необходимо подчеркнуть, что метод ЯМР протонов фиксирует
положения и подвижность протонов, а не молекулы Н2О в целом. Это озна-
чает, что мы не имеем права говорить, например, о прецессии молекулы
Н2О вокруг некоторой оси, речь может идти лишь о некотором анизо-
тропном движении (р—р)-вектора в пространстве. Если вероятность
пребывания направления (р—р) — вектора в телесном угле (ф,й) обозна-
чим тг (о) еек&, можно записать:
л (?) = [1 + (cos <р) + Х4Р4 (cos <р). . .], (4)
где Р2, Р4 —поликомы Лежандра, Х2, )-4— соответствующие коэффи-
циенты. Усреднение локального магнитного поля (1) по тг(<р) дает узкий
дублет, расщепление которого может быть выражено через коэффи-
циенты в разложении (4). Случаю прецессии молекулы Н2О [формула (3)]
соответствует Х2^А0, . . . =0. Если \=у^0, зависимость дублетных
расщеплений от ориентации уже нельзя описать простой моделью пре-
цессирующих молекул Н2О.
Состояние подвижности молекул Н2О в цеолитах
Пользуясь методом, описанным выше, мы исследовали ряд цеолитов
в насыщенном состоянии, определили подвижность молекул Н2О. Резуль-
таты приведены в таблице.
Цеолит Состав Число степе- ней свободы (20° С) Число неэквива- лентных поло- жений
Натролит Na2AI,Si3O10 • 2Н,0 0 4
1 Томсонит NaCa2AlsSis02o-6H20 0 3
Сколецит CaAl2SisO10 • 3H2O 0 5
Анальцим NaAlSi2Oe • H2O 1 1
Гейландит CaAl2SieO16 • 5H2O 2 1
Дессмин CaAl2Si7O]8- 7H2O 2 1
О Гармотом BaAl2Si4O12-6H2O 2 2
Шабазит Ca2Al4Si8O24 • 13H2O 2 1
Na—морденит .... INagAlgS I40O96 ’ 24Й2О 2 р
Са—морденит .... Ca4AlgS14oOg6 * wH2O 2 р
NaA •• Nai2Al12Si19O48 - 27 H2O 4—6 1
3 NaX NaeCa2Al]0Si]4O48- 30H2O 4—6 1
NaY NagAlgSilcO4g • zzHgO 4—6 1
70
Таким образом, можно выделить в основном три большие группы цео-
литов: 1) цеолиты с отдельными молекулами воды, в которых молекулы
Н20 закреплены «жестко»; 2) цеолиты с порами диаметром 4—7 А, моле-
кулы воды в которых обладают двумя степенями свободы («прецессия»);
3) цеолиты с порами диаметром до 13 А, молекулы воды в которых обла-
дают всеми вращательными и поступательными степенями свободы. Аналь-
цим должен был бы относиться к 1-й группе цеолитов, однако кольцевая
диффузия натрия в его структуре приводит к реориентации молекул
воды в ней [4].
Наиболее интересным является факт, что в большинстве цеолитов 2-й
и 3-й групп положения всех молекул Н2О эквивалентны с точки зрения
ЯМР. В то же время известно, что молекулы воды в этих цеолитах нахо-
дятся в кристаллографически неэквивалентных положениях и, несмотря
на это, все они ведут себя одинаково — в цеолитах 2-й группы прецесси-
руют вокруг одной и той же оси с одним и тем же углом прецессии. Угол
прецессии не зависит от температуры и зависит лишь от содержания воды.
В цеолитах 3-й группы эквивалентность молекул Н20 определяется нали-
чием лишь одного времени протонной релаксации.
Эти факты заставляют считать, что взаимодействие молекул Н2О
между собой играют более важную роль, чем взаимодействия отдельных
молекул Н20 с каркасом, т. е. что взаимодействие молекул воды в цеоли-
тах 2-й и 3-й групп носит коллективный характер.
Этот вывод имеет важное значение, так как следствием его является
вывод о том, что совокупность молекул воды, поглощенных цеолитом,
взаимодействует с каркасом цеолита как единое целое. Одним из следствий
этого являются характерные кривые потери воды при нагревании цеоли-
тами 2-й и 3-й групп, на которых нет резких пиков, так как отдача воды
носит плавный характер.
Природа взаимодействия молекул воды с каркасом цеолита
Различия в состоянии молекулы воды в цеолитах тесным образом свя-
заны с геометрическими условиями вхождения молекул Н20 в структуру.
В цеолитах 1-й группы молекулы Н2О находятся или в центре тетраэдра
(вершинами которого являются два атома кислорода и два катиона),
или в плоскости треугольника, образованного двумя атомами кислорода
каркаса и одним катионом. Определение положения протонов в натролите
и томсоните показало, что молекула Н20 связана с двумя атомами кисло-
рода водородными связями О—Н ... О, тогда как с катионами связан непо-
средственно кислород молекулы воды (за счет остаточных «валентностей»).
Таким образом, молекулы Н20 в цеолитах 1-й группы связаны, в соответ-
ствии с моделью Бернала-Фаулера [14], четырьмя тетраэдрическими ва-
лентностями, силой ~ */2 единицы каждая, двумя положительными и
двумя отрицательными.
Наличие сравнительно больших групп молекул в полостях структуры
цеолитов 2-й и 3-й групп приводит к тому, что молекулы воды взаимодей-
ствуют не только с жестким каркасом и катионами, но и друг с другом.
Однако, по крайней мере в цеолитах 3-й группы, взаимодействие моле-
кулы Н20 с окружением может быть описано моделью Бернала—Фаулера.
В соответствии с предположением Н. В. Белова, в полостях структуры
цеолитов 3-й группы молекулы Н20 расположены в вершинах и центре
пентагондодекаэдра; в этом случае три из четырех «валентностей» каждой
из молекул «замкнуты» на другие молекулы Н20, и только четвертая дает
связь с каркасом. Всего у такой додекаэдрической «молекулы» Н20 X
X 20 aq имеется двадцать «валентностей», часть из которых положительна
71
(дает водородные связи с каркасом) и часть — отрицательйа (обуславли-
вающая связь с катионами).
Молекулы воды при этом должны часто (10’ сек.-1) обмениваться ме-
стами (самодиффузия), с тем, чтобы объяснить наличие узкого спектра
(4—6 степеней свободы) и единственного времени релаксации.
Размеры и форма каналов цеолитов 2-й группы не допускают образо-
вания подобных взаимно насыщенных водородными связями структур.
Можно предполагать, что особое поведение молекул Н2О в цеолитах этого
типа обусловлено наличием в кристалле сильного электрического поля,
вокруг которого «прецессируют» дипольные моменты молекул воды. Это
электрическое поле слагается из полей, создаваемых каркасом, катионами
и дипольными моментами самих молекул Н2О, т. е. должно носить само-
согласованный характер. Это подтверждается зависимостью угла пре-
цессии молекул Н2О от содержания воды. При уменьшении содержания
воды в цеолите угол прецессии растет, т. е. движение (р—р)-вектора ста-
новится более близким к изотропному, что может быть следствием умень-
шения общего силового поля.
Таким образом, можно сделать вывод, что в цеолитах второй группы
взаимодействие молекул воды между собой и с каркасом цеолита имеет,
по-видимому, электростатический характер. Параллельно ориентирован-
ные дипольные моменты молекул Н2О должны приводить к взаимному
отталкиванию между молекулами и к их стремлению как бы обособиться,
что, действительно, наблюдается в структуре гармотома.
Характерно, что все цеолиты с большими полостями в структуре при-
надлежат к кубической сингонии, т. е. в них не может иметь места резуль-
тирующее электрическое поле.
Диффузия молекул воды в цеолитах
Метод ЯМР позволяет изучить характер движения молекул Н2О
в процессе диффузии вдоль каналов структуры. Здесь также существенно,
что частота движений должна быть выше, чем 104 гц, т. е. необходимо изу-
чать эти процессы при повышенных температурах. Мы исследовали диффу-
зию молекул воды в ряде цеолитов 1-й и 2-й групп при температурах до
400° (при атмосферном давлении). В цеолитах 3-й группы процесс диффу-
зии не изучался из-за перекрывающего влияния процесса самодиффузии
молекул в пределах самих полостей.
В цеолитах первой группы удается проследить npoifecc ухода из струк-
туры отдельных неэквивалентных в структурном отношении молекул
воды [3]. Удалось показать, что при переходе из одного «равновесного»
положения в другое по крайней мере одна из четырех связей молекулы
Н2О не разрывается. Можно думать, что таким же образом движутся
молекулы воды в цеолитах 3-й группы и вдоль поверхности других твер-
дых тел. Энергия разрыва трех связей составляет — 15 ккал./моль и
двух связей — — 12.5 ккал./моль.
В цеолитах 2-й группы спектр образцов при нагревании до 150—200° С
мало меняется, только дублетное расщепление в связи с уменьшением со-
держания воды несколько падает. Поскольку в этих цеолитах потеря воды
происходит уже при температуре ~ 50° С, можно думать, что молекулы
воды переходят из одного равновесного положения в другое с частотой,
меньшей, чем 104 гц, либо что при движении вдоль канала молекулы пере-
мещаются параллельно самим себе.
Если частота движения меньше ширины линии (104 гц), спектр не
должен меняться вообще. Если же молекула движется параллельно
самой себе, меняется лишь межмолекулярная составляющая локального
поля (связанная с взаимодействием с соседними молекулами воды). При
72
этом дублетное расщепление не меняется, но уменьшается ширина от-
дельных компонент дублета (уширение компонент дублетов обусловлено
межмолекулярным взаимодействием).
В наших опытах при нагревании образцов от —30° до +100 +- 150° С
при неизменном дублетном расщеплении ширина отдельных компонент
уменьшалась примерно в полтора-два раза. Это означает, что при движе-
нии вдоль каналов структуры цеолитов 2-й группы молекулы воды пере-
мещаются параллельно самим себе (не «кувыркаются»), в соответствии
с наличием предполагаемого электрического поля.
Физические свойства цеолитов
Цеолит может рассматриваться как своеобразная матрица, насыщенная
различными веществами. Физические свойства системы цеолит+адсорбат
будут в значительной степени определяться свойствами адсорбированной
фазы, которую с известным приближением можно принимать как само-
стоятельную физическую систему (во многом аналогичную тончайшей
пленке). Ниже мы рассмотрим вопрос о возможности фазовых переходов,
связанных с наличием у молекул цеолитной воды вращательных степеней
свободы.
6
Спектры монокристалла гейландита (Н||у) при различных температурах (а) и
соответствующие им состояния и ориентации молекул Н20 (б). Ось у перпен-
дикулярна плоскости совершенной спайности, в которой находятся слои из
молекул Н2О.
При понижении температуры до —60—70° С для цеолитов 3-й группы
и до-------30—50° С для цеолитов 2-й группы спектр ЯМР начинает
расширяться, чему соответствует уменьшение подвижности («вымерзание»)
молекул Н2О. Практически все цеолиты при —100—110° С обладают
спектром ЯМР, характерным для жестких систем.
В общем случае, как показал Л. Д. Ландау [15], изменение вращатель-
ного движения молекул может приводить к фазовым переходам только
1-го рода. Это связано с тем, что в точке фазового перехода 2-го рода со-
стояние тела Должно меняться непрерывным образом, и поэтому не может
возникнуть резкое изменение характера вращательного движения. Та-
ким образом, «вращательный» фазовый переход может быть связан
лишь с изменением вероятности различных ориентаций молекул (т. е.
73
с изменением тормозящих потенциальных барьеров), причем это будет
переход 1-го рода.
Нами был исследован монокристалл гейландита. На рисунке приведены
спектры гейландита до п после перехода. При комнатной температуре
спектр соответствует прецессии всех молекул воды вокруг оси, перпенди-
кулярной плоскости слоев (ось у) с углом прецессии 24°. Результирующий
дипольный момент направлен параллельно оси у. При —183° С по крайней
мере один (р—р)-вектор направлен вдоль оси у, т. е. дипольный момент
соответствующей молекулы воды теперь направлен перпендикулярно оси у.
Таким образом, дипольный момент одной из молекул Н20 повернулся
на 90°, чему должно соответствовать резкое изменение характера (или
природы) силового поля, действующего на молекулу. Поэтому имеются
все основания считать, что в гейландите при---40° С (при этой темпера-
туре меняется характер и ширина спектра) имеет место фазовый переход
1-го рода.
Исследование других цеолитов 2-й группы приводит к аналогичным
выводам. При низких температурах, в соответствии с наличием в струк-
туре самых различных силовых полей и в соответствии с моделью Бер-
нала—Фаулера, молекулы Н20 имеют самые различные ориентации.
При высокой температуре все молекулы ведут себя одинаково. Поэтому
можно считать, что изменение характера взаимодействия молекул воды
у всех цеолитов этой группы связано с фазовым переходом. Некоторые
из этих переходов, как следует из теории И. В. Курчатова [16], могут
быть сегнетоэлектрическими. Интересно, что в одном из сегнетоэлектри-
ков — K4Fe(CN)e • ЗН2О — состояние кристаллизационной воды такое же,
как и у цеолитов второй группы. Сегнетоэлектрический фазовый переход
в этом веществе имеет место при —24° С, когда начинается процесс «вы-
мерзания» коллективного движения молекул воды.
ЛИТЕРАТУРА
1. С. П. Га бу да, ДАН СССР, 1962, 146, 870.
2. С. П. Г а б у д а, А. Г. Л у и д и н, Г. М. М и х а й л о в, К. С. А л е к с а н д-
р о в, Кристаллография, 1963, 7, 388.
3. С. П. Г а б у д а, А. Г. Л у н д и н, Г. М. М и х а й л о в, Кристаллография
(в печати).
4. С. П. Г а б у д а, Г. М. М и х а й л о в, Сб. трудов СибТИ по физике и химии,
1963, 36.
5. С. П. Г а б у д а, Г. М. Михайлов, Сб. матер, конф, по итогам н.-иссл.
работ СибТИ за 1962, 1963 гг., Красноярск, 1963, 43.
6. С. П. Г а б у д а, Г. М.Михайлов, Изв. Сиб. отд. АН СССР, 1963, 11, 3, 123.
7. С. П. Г а б у д а, А. Г. Л у н д и н, Г. М. М и х а й л о в, Геохимия, 1963,
4, 436.
8. С. П. Г а б у д а, Г. М. М и х а й л о в, К. С. А л е к с а н д р о в, ДАН СССР,
1963 152.
9. С. П. Г а бу да, А. Г. Л у н д и и, Г. М. М и х а й л о в,- К. С. Александ-
ров, Сб. матер, конф, по итогам н.-иссл. работ СибТИ за 1961, 1962 гг., Красно-
ярск, 1963, 92.
10. С. П. Г а б у д а, Г. М. М и х а й л о в, Ж. структ. химия, 1963, 4, 436.
И. G. Е. Раке, J. Chem. Phys., 1948, 16, 327.
12. Н. S. Gutowsky, G. Е. Раке, J. Chem. Phys., 1950, 18, 163.
13. S. Yano, J. Phys. Soc. Japan, 1959, 14, 942.
14. Дж. Бернал, P. Фаулер, УФН, 1934, 14, 5, 5861.
15. Л. Д. Ландау, Е. М. Лифшиц. Статистическая физика. Гостехиздат,
М., 1951.
16. И. В. Курчатов. Сегнетоэлектричество. ОНТИ, 1933.
ТЕПЛОТА АДСОРБЦИИ ПОЛЯРНЫХ МОЛЕКУЛ ЦЕОЛИТОМ NaX
Н. Н. Авгулъ, Г. Г. Муттик
Пористые кристаллы являются высокоспецифическими адсорбентами
благодаря тому, что положительные заряды их поверхности сосредото-
чены в катионах небольших размеров, а отрицательные заряды рассредо-
точены по многим атомам кислорода алюминий- и кремний-кислородных
тетраэдров. Представляет интерес на такой поверхности исследовать
адсорбцию веществ, молекулы которых содержат атомы, имеющие различ-
ную возможность специфических взаимодействий с катионами. Нами
проводились исследования изотерм и дифференциальных теплот адсорбции
аммиака, паров воды [1] и метанола синтетическим цеолитом типа Na-
фожазит, приготовленным С. П. Ждановым [2]. Полученные данные со-
поставлялись с соответствующими данными по адсорбции диэтилового
эфира [3]. Теплоты адсорбции измерялись в калориметрах с постоянным
теплообменом |4, 5], теплоты адсорбции воды измерялись в изотермиче-
ском режиме [4].
Из рис. 1, на котором представлены изотермы и теплоты адсорбции
аммиака, воды и метанола, видно, что заполнение каналов цеолита прибли-
зительно на 20% сопровождается наиболее значительным падением теп-
лот адсорбции. В дальнейшем в широком интервале заполнения происхо-
дит менее резкое снижение кривой дифференциальных теплот, а для мета-
нола имеется даже некоторый рост теплот. Наиболее сильно продолжает
снижаться кривая теплот для аммиака; в случае воды происходит ступе-
необразное снижение кривой, а в случае метанола имеется небольшой
максимум на кривой теплот адсорбции. Рис. 2, а свидетельствует о бли-
зости теплот адсорбции всех этих веществ в начальной области заполнения.
Благодаря резкой специфичности адсорбции наличие метильной группы
молекулы метанола не играет, при малых степенях заполнения, существен-
ной роли. Возможно, что в случае воды имеется некоторое повышение
теплоты адсорбции за счет проникновения молекул воды в малые полости;
это, по-видимому, не может происходить ни в случае аммиака, ни в слу-
чае метанола. С увеличением адсорбции во всех случаях уменьшается
эффективное расстояние между адсорбирующимися молекулами. В слу-
чае метанола сближение молекул ведет даже к росту теплоты адсорбции
за счет взаимодействий адсорбат—адсорбат. Молекулы аммиака, взаимо-
действуя с ионами цеолита благодаря наличию одной свободной пары
электронов, практически не имеют возможности связываться друг с дру-
гом в адсорбированном слое. Поэтому для аммиака характерно непрерыв-
ное падение теплот адсорбции по мере использования наиболее выгодных
для адсорбции участков поверхности. Адсорбция пара воды занимает
промежуточное (по сравнению с метанолом и аммиаком) положение —-
молекулы воды даже при малых степенях заполнения выбирают на поверх-
ности такие положения, которые позволяют им осуществлять весьма тес-
ное взаимодействие между собой.
Величина теплоты адсорбции диэтилового эфира в средней области за-
полнений превышает величину теплоты адсорбции метанола на 5 ккал./моль,
что близко к инкременту теплоты адсорбции, вносимому тремя группами
СН2 при адсорбции н-алканов на цеолите NaX (5.7 ккал./моль [6]). По-
следовательность расположения изотерм адсорбции воды и метанола на-
ходится в соответствии с величинами теплот адсорбции (рис. 2, б). Таким
образом, различия в теплотах адсорбции этих кислородсодержащих мо-
лекул определяются размером углеводородного радикала.
О специфичности адсорбции можно судить из сопоставления теплот
адсорбции неполярного адсорбата н-гексана [7] и метанола на цеолите
75
а., ммоль/г , клал./моль
Рис. 1. Изотермы адсорбции (внизу) и теплот адсорбции (вверху) аммиака (а), паров воды (б) и
метанола (в) цеолитом типа Na-фожазит.
Различные обозначения на кривых относятся к различным сериям опытов. То же и на рис. 2.
и неполярной поверхности графитированной сажи [8, 9] (рис. 3). Теплота
взаимодействия н-гексана в случае адсорбции на цеолите NaX благодаря
несколько более плотной упаковке атомов в решетке цеолита превышает
Рис. 2. Изотермы теплот адсорбции (а) и самой адсорбции (б)
аммиака (7), воды (2), метанола (3) и диэтилового эфира (4) Na-
фожазитом.
теплоту взаимодействия гексана с графитированной сажей приблизи-
тельно на 3.5 ккал./моль (неспецифическое взаимодействие). При малых же
величинах адсорбции метанола теплота адсорбции на NaX значительно
Рис. 3. Изотермы теплот адсорбции н-гек-
сана (а) и метанола (б) на цеолите (7) и
графитированной саже (2).
Крестом отмечены хроматографические данные [8].
превышает теплоту адсорбции на графитированной саже. Разница в вели-
чинах теплот адсорбции цеолитом по сравнению с графитированной са-
жей, достигающая 11—12 ккал./моль, указывает на наличие донорно-
акцепторного взаимодействия при адсорбции метанола и воды цеолитами.
Авторы выражают благодарность А. В. Киселеву за руководство этой
работой.
ЛИТЕРАТУРА
1. О. М. Д ж и г и т, А. В. К и с е л е в, К. Н. М и к о с и Г. Г. М у ттик, ЖФХ,
1964, 38, 1791.
2. С. П. Ж д а н о в, А. В. К и с е л е в, Л. Ф. П а в л о в а, Кинетика и катализ,
1962, 3, 421.
3. О. М. Д ж и г и т, А. В. К и с е л е в, Г. Г. М уттик, Колл, ж., 1963, 25, 34.
77
4. А. В. Кисел ев, Г. Г. М уттик, ЖФХ, 1961, 35, 2153.
5. Н. Н. А в г у л ь, Г. И. В е р е з и н, А. В. Киселев, И. А. Л ыг и на,
Г. Г. Муттик, ЖФХ, 1957, 31, 1111.
6. А. В. Киселев, настоящий сборник, стр. 13.
7. Н. Н. А в г у л ь, А. В. К и с е л е в, А. А. Л о и а т к и н, И. А. Л ы г и н а,
М. В. С е р д о б о в, Колл, ж., 1963, 125, 29.
8. Л. Д. Б е л я к о в а, А. В. Киселев, Н. В. К о в а л е в а, ДАН СССР,
1964, 157, 646.
9. Н. Н. А в г у л ь, Г. И. Б е р е з и н, А. В. К и с е л е в, И. А. Л ы г и н а, Изв.
АН СССР, ОХН, 1962, 205.
ВЛИЯНИЕ ЗАМЕСТИТЕЛЕЙ НА ТЕПЛОТУ АДСОРБЦИИ
НЕПОЛЯРНЫХ ПРОИЗВОДНЫХ НАСЫЩЕННЫХ УГЛЕВОДОРОДОВ
ГРАФИТИРОВАННОЙ САЖЕЙ И СИНТЕТИЧЕСКИМ Na-ФОЖАЗИТОМ
М. В. Сердобое
В настоящей работе производились измерения изотерм и тецлот ад-
сорбции метилциклогексана и перфторметилциклогексана на неполярной
иоверхнос-'нг графитированной сажи МТ (2700—3100°) и катионированной
поверхности синтетического натрий-фожазита, приготовленного в лабора-
тории С. П. Жданова [1]. Теплоты адсорбции этих веществ на графити-
рованной саже измерялись в динамических условиях с помощью чувстви-
Дифференциальные теплоты адсорбции перфторметилциклогек-
сана (7) и метилциклогексана (2) синтетическим натрий-фожа-
зитом (а) и графитированной сажей (б).
Квадратиками показаны результаты второй серии измерений теплот адсорб-
ций CFsCeFn на цеолите.
тельного дифференциального калориметра с непрерывным нагревом [2].
В случае натрий-фожазита теплоты определялись на калориметре с по-
стоянным теплообменом [3] в статических условиях.
Неполярные молекулы метилциклогексана и перфторметилциклогек-
сана имеют одинаковый углеродный остов, но различаются размерами
и общей поляризуемостью. Следовательно, различие их адсорбционных
свойств должно особенно резко проявляться при адсорбции на адсорбен-
тах различной полярности. Действительно (см. рисунок, б), в начальной
области заполнения поверхности теплота адсорбции метилциклогексана
графитированной сажей выше теплоты адсорбции перфторметилциклогек-
сана. Это объясняется тем, что атомы углерода адсорбированных моле-
кул перфторметилциклогексана дальше удалены от силовых центров
78
адсорбента, а влияние большей поляризуемости этих молекул при адсорб-
ции на неполярной поверхности графитированной сажи невелико. В слу-
чае адсорбции этих веществ на катионированной поверхности каналов
цеолита наблюдается обратное соотношение начальных теплот адсорбции
(см. рисунок, а) вследствие сильного поляризующего влияния обменных
катионов на фторированное соединение. Кроме эффекта поляризации,
здесь, по-видимому, могут иметь место электростатическое взаимодействие
и квантово-химическое взаимодействие (типа донорно-акцепторного) элек-
троотрицательных атомов фтора с концентрированными положительными
зарядами поверхности (обменными катионами). Качественно эти взаимо-
действия, вероятно, напоминают специфические взаимодействия непо-
лярных молекул бензола, имеющих л-электронные связи, с катионами
поверхности цеолита [4].
Такие донорно-акцепторные взаимодействия наблюдались также с по-
мощью ИКС при адсорбции хлорпроизводных метана на гидратированном
силикагеле, поверхностные гидроксильные группы которого имеют «про-
тонизированные» атомы водорода [5].
ЛИТЕРАТУРА
1. С. П. Ж д а н о в, А. В. К и с е л е в, Л. Ф. П а в л о в а, Кинетика и катализ,
1962, 3, 421.
2. Г. И. Б е р е з и н, А. В. К и с е л е в, М. В. С ер доб о в, ЖФХ, 1962, 36, 2091.
3. Н. Н. А в г у л ь, Г. И. Б е р е з и н, И. А. Л ы г и и а, Г. Г. М уттик, ЖФХ,
1957, 31, 1111.
4. Н. Н. А в г у л ь, А. В. К и с е л е в, А. А. Л о и а т к и н, И. А. Л ы г и н а,
М. В. С е р д о б о в, Колл, ж., 1963, 25, 129.
5. R. М. В a s i 1 1 a, J. Chem. Phys., 1961, 35, 1151.
ТЕРМИЧЕСКАЯ ОБРАБОТКА ГРАНУЛ
И АДСОРБЦИОННЫЕ СВОЙСТВА КРИСТАЛЛИЧЕСКИХ
И ФОРМОВАННЫХ СИНТЕТИЧЕСКИХ ЦЕОЛИТОВ NaX и СаА
Т. Н. Шишакова, М. М. Дубинин
При гранулировании синтетических цеолитов инертная в адсорб-
ционном отношении добавка связующего разбавляет массу кристалличе-
ского порошка, но адсорбционные свойства и предельные адсорбционные
объемы формованных синтетических цеолитов типов А и X обычно пони-
жены по сравнению с оцененными из рентгеноструктурных данных в боль-
шей степени, чем это соответствует такому разбавлению кристаллической
массы связующим. Этот эффект может быть вызван или свойствами самих
кристаллов, или их контактом со связующим при переметивании пасты,
формовании и термической обработке гранул.
Для выяснения основных причин неполного использования адсорб-
ционных свойств кристаллов в формованных синтетических цеолитах мы
изучали влияние промывки кристаллического порошка ИД термическую
стабильность цеолитов. Снимали изотермы н-пентана на кристаллических
образцах цеолитов NaX и СаА с различной степенью промывки порошка
после эвакуирования в течение 6 часов при температурах 350, 700,
800, 900° С (для NaX) и 350, 600° С (для СаА). Обнаружено, что увеличе-
ние степени промывки не ухудшает термическую устойчивость кристаллов
исследованных цеолитов NaX. Их предельные адсорбированные коли-
чества, предельные адсорбционные объемы и константы уравнения изо-
79
термы адсорбции B/f>2, характеризующие крутизну изотермы, остаются
постоянными для кристаллических образцов различной степени промывки,
подвергнутых предварительному прокаливанию. Кристаллический поро-
шок NaX с максимальной степенью промывки оказался наиболее терми-
чески устойчивым. Он полностью теряет свою адсорбционную активность
при 900° С, в то время как образцы с меньшей степенью промывки разру-
шаются при 850° С. Прокаливание же кристаллического порошка СаА
(600° С) нормальной заводской промывки приводит к значительному
(~ 30%) увеличению константы уравнения изотермы адсорбции 2?/р2.
Дополнительная промывка этого образца не приводит к изменению пре-
дельного адсорбированного количества и константы 2?/р2, а прокаливание
этого образца увеличивает ее на 30%.
С другой стороны, мы изучали влияние связующего и термической
обработки гранул на сорбционные свойства. Снимали изотермы н-пентана
на образцах NaX и СаА, соответствующих различным стадиям технологии
производства:
1) на кристаллическом порошке; 2) на сухих таблетках (смесь кристал-
лического порошка со связующим, высушенная при 120° С); 3) на прока-
ленных таблетках (сухие таблетки, прокаленные при 550—600° С, т. е.
готовые гранулы).
Таблетирование кристаллического порошка (добавка связующего и
прокаливание таблеток 550—600° С) цеолита NaX не приводит к изме-
нению константы уравнения изотермы адсорбции 2?/р2, а величина пре-
дельного адсорбированного количества снижается на 20%, что как раз
и соответствует разбавлению кристаллического порошка практически
инертным в адсорбционном отношении связующим. Таким образом, ясно,
что добавка связующего не влияет на термическую устойчивость кристал-
лического порошка. Прокаливание сухих таблеток (550—600° С) СаА
увеличивает константу В If? на 30%, т. е. на ту же величину, что и про-
каливание кристаллического порошка СаА, из которого изготовлены эти
таблетки. Таким образом, ясно, что термическая обработка гранул СаА
при 600е С, применяемая на производстве, может служить причиной за-
метного снижения адсорбционных свойств собственно кристаллов цеолита,
входящих в состав гранул. Причина понижения адсорбционной активно-
сти формованных синтетических цеолитов по сравнению с оцененной из
рентгеноструктурных данных в большей степени, чем это соответствует
разбавлению инертным в адсорбционном отношении связующим, вряд ли
может быть приписана влиянию связующего на адсорбционные свойства
кристаллического порошка.
Важно отметить, что эффект увеличения константы уравнения изо-
термы адсорбции В1[>2 в результате прокаливания цеолита наблюдается
только для кристаллов СаА опытно-промышленного производства. Для
кристаллов СаА, синтезированных С. П. Ждановым в лабораторных
условиях, зтот эффект отсутствует. Выяснение его основных причин
поможет улучшению качества производственных образцов цеолитов СаА.
80
О СТРУКТУРНЫХ ИЗМЕНЕНИЯХ НЕКОТОРЫХ
ПРИРОДНЫХ ЦЕОЛИТОВ ПРИ НАГРЕВАНИИ
Э. Н. Корыткова, А. Д. Федосеев
Долгое время считалось общепринятым мнение, что выделение воды
при нагревании цеолитов не вызывает никаких изменений в структурах
этих минералов. Однако более детальные исследования, проведенные
некоторыми авторами раньше [1,2] и особенно в последние годы [3, 4],
поставили под сомнение вопрос о неизменности структур цеолитов при
дегидратации.
Нами было изучено поведение ряда природных цеолитов при нагре-
вании [5, 6, 7]. В процессе этого исследования установлено, что даже
частичное обезвоживание цеолитов вызывает определенные изменения
в их кристаллических структурах. При этом не только интенсивность,
но и характер этих изменений неодинаков у различных, в том чцсле изо-
структурных, цеолитов (см. таблицу). Выявленные деформации структур
у некоторых цеолитов не являются устойчивыми. Наблюдение за обрат-
ным поглощением воды обезвоженными в разной степени цеолитами по-
казало их различную способность к регидратации. Так, полностью обез-
воженный натролит поглощает даже большее, по сравнению с исходным,
количество воды. Вероятно, его несколько расширившаяся решетка
создает благоприятные условия для такого избыточного водопоглощения.
Небольшое сжатие решетки натролита, происходящее при нагревании
до 550°, не препятствует его полной регидратации. У мезолита расшире-
ние кристаллической решетки, наблюдаемое при нагревании его до 350°,
обеспечивает полное обратное поглощение воды дегидратированным об-
разцом. При зтом решетка регидратированного мезолита сохраняет свои
увеличенные параметры. Полное обезвоживание мезолита, приводящее
к сжатию его решетки, несколько уменьшает его поглотительную спо-
собность, однако даже неполная регидратация оказывается достаточной,
для восстановления структуры минерала.
Иначе обстоит дело со сколецитом и томсонитом. Если сжатие ре-
шетки, наблюдающееся после первой стадии обезвоживания этих минера-
лов, не препятствует их полному оводнению и восстановлению структур,
то полное обезвоживание, приводящее к более значительному уменьшению
параметров решетки, а у сколецита — даже к некоторому изменению
ориентировки цепочек (в связи с увеличением угла Р), полностью лишает
дегидратированный сколецит поглотительной способности и очень резко
сокращает эту способность у томсонита. Рентгеновское исследование
дегидратированного при 480° и находившегося в течение полугода во
влажной атмосфере сколецита не обнаружило никакого изменения его
решетки по сравнению с дегидратированным образцом. У томсонита в тех
же условиях хотя и отмечено незначительное увеличение параметра а,
практически восстановления структуры также не происходит.
Обезвоженные пластинчатые цеолиты (десмин, гейландит) легко и
быстро регидратируются. Десмин, потерявший основную часть своей воды
при нагревании до 350° и претерпевший заметное сжатие кристалличе-
ской решетки, на воздухе регидратируется почти полностью. При этом
происходит восстановление его структуры. Дальнейшее нагревание при-
водит к полному обезвоживанию десмина, вызывая лишь незначительное
сжатие его решетки по оси Ь. Это сжатие, однако, не препятствует быстрому
поглощению воды дегидратированным образцом. Возможно, структуры
пластинчатых цеолитов не отличаются большой жесткостью каркаса,
что и позволяет молекулам воды легко вновь раздвигать слои тетраэдров
и располагаться между ними.
6 Цеолиты
81
Результаты рентгеновского исследования цеолитов
Параметры, А Натролит
исходный образец нагретый до 460° нагретый до 560°
дегидрати- рованный регидрати- ров энный дегидрати- рованный регидрати- рованный
а .......... 18.33+0.01 18.35 18.35 (8.29 18.30
ь 18.60+0.02 18.73 18.73 18.62 18.61
с 6.60+0.01 6.60 6.60 6.60 6.60
Мезолит
нагретый до 350° нагретый до 480°
Параметры, ИСХОДНЫЙ образец дегидрати- регидрати- дегидрати- регидрати-
А ров энный рованный рованный рованный
а .......... 56.6 +0.3 57.8 57.8 56.8 56.8
ь 6.54+0.01 6.54 6.54 6.54 6.54
с ......... 18.49+0.07 18.55 18.55 17.96 18.46
Сколецит
нагретый до 320° нагретый до 480°
X исходный
А образец дегидрати- регидрати- дегидрати- регидрати-
рованный рованный рованный рованный
а .......... 18.50+0.01 18.43 18.50 18.40 18.40
ъ 18.96+0.01 18.90 18.93 18.76 18.76
с 6.54 + 0.01 6.54 6.54 6.54 6.54
р 90°46+4 90°44 90°48 91°35 91°35
Томсонит
Параметры, А исходный образец нагретый до 230° нагретый до 370° нагретый до 440°
дегидра- тирован- ный регидра- тировэн- ный дегидра- тирован- ный регидра- тиров эн- ный дегидра- тирован- ный регидра- тирован- ный
а ....... ъ с ...... 13.00+0.01 13.02+0.01 13.18+0.01 12.96 12.99 13.18 12.98 13.00 13.18 12.93 12.95 13.18 12.96 12.98 13.18 12.88 12.94 13.18 12.91 12.95 13.18
Д е с м и в
Параметры, А исходный образец Нагретый до 330° дегидратиров эн- ный при 470°
дегидратиро- ванный регидратиро- в энный
а .... 13.68 + 0.02 13.60 13.68 13.60
ъ 18.17 + 0.03 17.95 18.05 17.88
11.31+0.02 11.25 11.30 11.25
₽ 129°23 + 20 129°39 129°27 129°24
82
Полученные экспериментальные данные, на наш взгляд, являются
достаточным доказательством изменяемости структур цеолитов в процессе
их обезвоживания. А так как выявленные структурные изменения должны
несколько менять форму и величину полостей у дегидратированных цео-
литов, такие деформации необходимо учитывать при практическом ис-
пользовании этих минералов в качестве молекулярных сит.
ЛИТЕРАТУРА
1. J. W у а г t, Compt. Rend. Acad. Sci. Paris, 1930, 191, 1343.
2. M. He y, Mineral. Mag., 1932, 23, 137.
3. J. V. Smith, Acta crystallogr., 1962, 15, 9, 835.
4. Г. В. Ю x н e в и ч, Э. Э. С e н д e p о в, Геохимия, 1963, 1, 48.
5. А. Д. Ф е д о с е е в, Э. Н. К о р ы т к о в а, Зап. Всес. мин. общ., 1963, 92, 4Г
410.
6. Э. Н. К о р ы т к о в а, А. Д. Ф е д о с е е в, Зап. Всес. мин. общ., 1964, 93, 3, 15.
7. 3. Н. К о р ы т к о в а, А. Д. Ф е д о с е е в, Изв. АН СССР, сер. хим., 1964, 11,
1925.
ИССЛЕДОВАНИЕ ДИЭЛЕКТРИЧЕСКОЙ РЕЛАКСАЦИИ ЦЕОЛИТА NaA
ПРИ АДСОРБЦИИ ВОДЫ
Б. А. Глазун, М. М. Дубинин, И. В. Жиленков, В. М. Федоров
Исследование диэлектрических свойств силикагелей [1—5] показало
возможность наблюдения диэлектрической релаксации адсорбированных
полярных молекул и попутного определения таких величин, как стати-
ческая диэлектрическая проницаемость, энергия активации поляризации
и электропроводности, некоторые термодинамические параметры, степень
неоднородности распределения активных центров. С появлением интереса
к адсорбентам, обладающим молекулярно-ситовыми свойствами, диэлектри-
ческий метод исследования был распространен и на эти объекты [6].
Целью нашей работы было первоначальное исследование диэлектриче-
ского поведения цеолита NaA опытно-промышленного производства на
основе методики, примененной в работах [1—3].
Экспериментальная часть
Мелкокристаллический цеолит NaA без связующих добавок поме-
щался в вакуумный конденсатор и дегидратировался путем длительной
откачки и нагревания до 350° С. После измерения диэлектрической про-
ницаемости е' и коэффициента диэлектрических потерь е" в диапазоне
частот от 102 до 107 гц и при одновременном ступенчатом изменении тем-
пературы от +20° до —180° цеолит дозированно сорбировал воду в коли-
честве от 1.2 до 40% от предельно адсорбированного количества, которое
для образца 1 составляло 13.2, а для образца 2 — 14.5 ммоль/г.
Наше исследование показало, что при изменении частоты, темпера-
туры и степени гидратации образца можно наблюдать три области дебаев-
ской релаксации и область диэлектрических потерь, обусловленных ге-
терогенностью и проводимостью системы. Последняя интерпретируется
в свете представлений теории Максвелла—Вагнера [7]. Ограниченность
диапазона частот, которым мы располагали в настоящем исследовании,
не позволяла наблюдать все области диэлектрической релаксации образца
при какой-либо одной температуре. Однако, пользуясь тем, что частота
дебаевской релаксации с понижением температуры уменьшается так, что
6*
83
отношение приращения логарифма частоты релаксационного максимума
к приращению обратной величины абсолютной температуры остается
постоянным, мы могли наблюдать высокочастотные релаксации в пределах
нашего диапазона частот при надлежащем понижении температуры. Ос-
новываясь на той же зависимости, можно экспериментальные данные путем
пересчета привести к стандартной температуре +20°. Следует, однако,
заметить, что возможность фазовых переходов в сорбированном веществе
Рис. 1. Расположение релаксационных макси-
мумов, связанных с наличием воды в цеолите
при температуре +20° С.
Кривая 1 соответствует дегидратированному цеолиту,
2— содержанию воды 4.6%, 3—содержанию воды
9.3% от предельного адсорбированного количества.
при значительном изменении
температуры всегда остается
причиной стремления экспе-
риментаторов расширять
верхнюю границу7 частотного
диапазона.
Величины е' и е", опреде-
ленные при постоянной тем-
пературе, но при различных
частотах, позволяют с по-
мощью круговых диаграмм
Кола-Кола [8] определять ста-
тическую диэлектрическую
проницаемость е8, непосред-
ственно не измеряемую обыч-
ными методами. Эта величина
интересна тем, что она яв-
ляется мерой поляризации
в рамках связи, определяемой
теорией. Для систем с извест-
ным расположением молекул
удовлетворительные резуль-
таты дает теория Фрёлиха [9].
Форма круговых диаграмм Кола-Кола позволяет судить и о наличии
распределения времен релаксации адсорбированных молекул и опреде-
лить параметр 1 — а, характеризующий ширину этого распределения.
Связь между логарифмом частоты релаксации и температурой, выте-
кающая из теории абсолютных скоростей реакций [10], позволяет опре-
делять энергию активации для процесса поляризации или электропро-
водности, а также свободную энергию активации и энтропию активации
адсорбированного вещества. В соответствии с высказанными соображе-
ниями, на рис. 1 в полусхематическбм виде представлена частотная зави-
симость е", приведенная к температуре +20° для разных величин адсорб-
ции. На рис. 2 изображена диэлектрическая изотерма, выражающая
зависимость е/ от величины адсорбции воды на цеолите при постоянной
температуре, а на рис. 3 — аналогичная зависимость параметра 1—а,
характеризующего ширину распределения времен релаксации адсорбиро-
ванной воды.
Обсуждение результатов опытов
На рис. 1, перемещаясь от низких частот к высоким, мы отмечаем часть
максвелл-вагнеровской релаксации (восходящие ветви), которая показы-
вает, что электропроводность (величина е" в этой области при данной
частоте пропорциональна электропроводности) порошка цеолита умень-
шается с увеличением содержания воды. Есть основание предполагать
ионный характер электропроводности дегидратированного цеолита. Из-
вестно, что в массивных кристаллах перемещение ионов возможно только
при наличии дефектов решетки, т. е. при наличии вакансий для перемеще-
ния иона. Можно предполагать, что молекулы воды адсорбируются цео-
84
литом в первую очередь на наиболее активных центрах, которыми могут
быть либо сами ионы, либо дефекты решетки. В первом случае адсорбция
молекул воды приведет к гидратации ионов, а во втором — к блокирова-
нию дефектов (вакантных мест). Обе возможности объясняют эксперимен-
тальный факт понижения электропроводности системы при ее гидрата-
ции. Такая интерпретация подтверждается и тем фактом, что энергия акти-
вации для процесса злектропроводности, определяемая из температурной
зависимости, уменьшается с увеличением адсорбции воды. Можно пред-
полагать, что молекулы воды, адсорбируясь в первую очередь на наиболее
активных центрах, оставляют- свободными центры с менее глубокими
потенциальными ямами (с меньшей энергией активации).
При дальнейшем возрастании частоты обнаруживаются I и II области
релаксации, отличающиеся друг от друга величиной е"шах. При увеличении
адсорбции воды оба максимума воз-
растают. Одинаковый характер зависи-
мости от величины адсорбции для этих
двух процессов релаксации, разобщен-
ных по шкале частот, позволяет предпо-
ложить, что оба процесса обусловлены
дебаевской релаксацией молекул воды
в цеолите NaA, адсорбированных по
крайней мере на двух разновидностях
активных центров. Энергии активации
поляризации этих областей существенно
различны (для I области в два раза
больше), что также указывает на связь
этих областей релаксации с разными
активными центрами. Тот факт, что ве-
личина к'' для I области значительно
Рис. 2. Диэлектрическая изотерма
адсорбции для цеолита (второй об-
разец) при —20° С.
превышает соответствующую величину
для II, указывает на то, что в таком же соотношении находятся и числа
молекул воды, связанных с этими типами активных центров. Можно сде-
лать два предположения о.природе этих активных центров. Первое осно-
вывается на представлениях, развиваемых в работах [11 —13], из которых
следует, что вода может заполнять только большие полости цеолита. Ка-
тионы, находящиеся вблизи от анионов кислорода в больших полостях
и в шестичленных окнах, могут создавать зоны с резко неоднородным
электростатическим полем. Для остальной же части объема большой по-
лости значительная неоднородность поля не является характерной. Так как
при адсорбции полярных молекул воды на цеолите преобладают электро-
статические взаимодействия, то становится понятным, что упомянутым
частям объема большой полости будут соответствовать различные энергии
адсорбции, что и приводит к появлению двух, разобщенных по шкале
частот областей диэлектрической релаксации. Второе предположение ос-
новывается на представлении о возможности адсорбции молекул воды
в больших и в малых полостях при условии, что энергии адсорбции для
этих полостей будут разными.
Наконец, наблюдается третья область релаксации, которая, как видно
из рис. 1, затухает с увеличением содержания воды в цеолите. Можно
думать, что III тип релаксации обусловлен самими ионами натрия. По-
степенное затухание этой релаксации можно представить как результат
блокирования ионов адсорбируемыми молекулами воды.
Диэлектрическая изотерма, представленная на рис. 2, получена с по-
мощью круговых диаграмм Кола-Кола, построенных для релаксации
I типа. Наблюдаемый излом изотермы можно объяснить с двух точек зре-
ния. Во-первых, предполагая, что адсорбционные центры первого типа
85
имеют вторичную дифференциацию. Можно, например, представить, что
ионы, локализованные вблизи шестичленных и восьмичленных кислород-
ных колец, отличаются по энергиям адсорбции. Следует заметить, что
непосредственное наблюдение релаксационного максимума I типа, до-
Рис. 3. Зависимость параметра 1—а
от содержания воды в цеолите (пер-
вый образец) при ]-10° С.
вольно сильно размытого, не позволяет
обнаружить это более тонкое раз-
личие. Во-вторых, излом на диэлек-
трической изотерме может быть след-
ствием прогрессирующего развития
связей между адсорбированными моле-
кулами воды, так как при этом будет
уменьшаться вклад полярных молекул
воды в общую поляризацию системы.
В пользу второго предположения гово-
рит уменьшение ширины распределения
времен релаксации адсорбированной
воды с увеличением адсорбции, хорошо
видное на рис. 3 (увеличение параметра
1—а означает уменьшение ширины
распределения). В согласии с этим представлением находится и обнару-
женное нами увеличение энергии активации поляризации Дп и энтро-
пии активации Де при возрастании количества адсорбированной воды, пред-
ставленное ниже (образец 2, температура —40°).
Адсорбция, % Дц, кдж/моль As, дж/град. моль
5.2 50.5 28.6
20.8 62.5 104
32.6 76.0 158
43.0 95.0 235
Возрастание энтропии и структурирование представляются как ука-
зание на то, что в более структурированной системе поворот одного ди-
поля нарушает порядок среди большего числа своих соседей.
Выводы
1. Исследовано поведение диэлектрической проницаемости и коэффи-
циента потерь цеолита NaA при разных степенях гидратации (до 40%),
при изменении температуры и частоты.
2. Установлены три области диэлектрической релаксации. Первые
две (считая в сторону возрастания частоты) соответствуют релаксации
адсорбированных молекул воды на разных типах активных центров.
Третья — соответствует, видимо, релаксации ионов цеолита. Этот тип
релаксации затухает при увеличении содержания воды в цеолите.
3. Область максвелл-вагнеровской дисперсии указывает на уменьше-
ние электропроводности системы с увеличением содержания воды, что
может быть связано с блокированием молекулами воды ионов или вакант-
ных мест решетки.
4. Диэлектрическая изотерма, изменения в энергии активации, эн-
тропии активации и ширины распределения времен релаксации дают
основание предполагать структурирование адсорбированной воды по
мере возрастания ее количества.
ЛИТЕРАТУРА
1. И. В. Ж и л е н к о в, И. Р. П е р с к и й, Л. И. Ф е д о т о в а, Колл, ж.,
1937 1 3 537.
2. И. В. Ж и л е н к о в, Изв. АН СССР, ОХН, 1957, 2, 232.
86
3. И. В. Жил ей к о в, ЖФХ, 1962, 36, 11, 2406.
4. J. В о t, S. М о n t a gn er, Compt. Rend. Acad. Sci., 1951, 233, 862.
5. M. P ol land, R. В e г n a r d, Compt. Rend. Acad. Sci., 1951, 252, 1098.
6. R. M. Barter et al., Trans. Faraday Soc., 1962, 58, 156.
7. K. Wagner, Ann. d. Phys., 1913, 5, 40, 817.
8. K. S. Cole, R. N. С о 1 e, J. Chem. Phys., 1941, 9, 341.
9. Г. Фрёлих. Теория диэлектриков. ИЛ, М., 1960.
10. С. Г л е с т о и, К. Л е й д л е р, Г. Э й р и и г. Теория абсолютных скоростей
реакций. ГИИ Л, 1948.
11. М. М. Дубинин, Иав. АН СССР, сер. хим., сообщ. 8, 1964, 2, 209.
12. М. М. Дубинин, Изв. АН СССР, сер. хим., сообщ. 9, 1964, 9, 1965.
13. М. М. Дубинин, Изв. АН СССР, сер. хим., сообщ. 10, 1964, 9, 1573.
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
НОВЫХ ИОНООБМЕННЫХ ФОРМ ЦЕОЛИТОВ
Г. В. Цицшивили
В настоящем сообщении излагается ряд вопросов, касающихся при-
роды синтетических цеолитов на основании результатов работ лаборатории
физической химии Института химии им. П. Г. Меликишвили АН Грузин-
ской ССР. В работе принимали участие сотрудники лаборатории Т. Г. Ан-
дроникашвили. К. Е. Авалиани, Г. Д. Багратишвили, Д. Н. Барнабишвили,
III. Д. Сабелашвили, Л. Я. Лаперашвили, М. Г. Адолашвили, М. В. Уру-
шадзе и ассистент кафедры физической химии Тбилисского государствен-
ного университета Е. Л. Григолия.
В последние годы широким фронтом проводятся работы по синтезу,
исследованию свойств и применению цеолитов [1]. Однако, несмотря на
это, еще многие их физико-химические свойства требуют дальнейшего
изучения. Это относится и к такому характерному для цеолитов свойству,
как селективность. Ранее нами отмечалось [2], что, по-видимому, целесооб-
разно отличать геометрическую и катионную селективность. Такие термины,
конечно, являются условными. Под геометрической селективностью сле-
дует понимать специфичность адсорбции, обусловленную основной кри-
сталлической структурой анионного каркаса цеолита. При этом, как
известно, катионы не только компенсируют избыточный отрицательный
заряд каркаса, но и сами существенно влияют на селективность этих тон-
копористых твердых тел.
Влияние ряда катионов на адсорбционную способность может быть
проиллюстрировано на материале по адсорбции паров воды. Цеолиты,
содержащие в своем составе хорошо гидратирующиеся катионы, лучше
адсорбируют пары воды, чем исходная натриевая форма. Так, например,
в то время как при р/ра=0А0 цеолит NaA (Горьковская опытная база
ВНИИЦП, серия Ц-202-85) адсорбирует 15.00 ммоль/г, шестикратно
замещепная никелевая и двенадцатикратно замещенная кобальтовая
формы соответственно адсорбируют 15.60 и 17.70 ммоль/г паров воды.
Специфические сорбционные свойства цеолитов хорошо видны на при-
мере таких форм, как серебряная, кобальтовая, никелевая.
Как известно, многие цеолиты избирательно поглощают ионы серебра
из водных растворов. Вследствие этого одним из распространенных ме-
тодов получения серебряной формы цеолитов типа А, X и Y является
ионообменное взаимодействие натриевой формы цеолитов с растворами
азотнокислого серебра. Таким путем нами были получены для исследова-
ния серебряные формы цеолитов [3]. Ранее в нашей лаборатории было
87
показано [4], что серебряная форма цеолита существенно отличается от
исходной натриевой формы по своим адсорбционно-хроматографическим
свойствам. Оказалось, что серебряные цеолиты типа X способны особенно
при повышенной температуре сравнительно прочно фиксировать водород,
окись углерода и олефиновые углеводороды. Смесь насыщенных углево-
дородов (метан, этан, пропан, бутан) хорошо делится на отдельные ком-
поненты.
Серебряная форма цеолита впервые применялась чешским исследова-
телем Янаком [5] для анализа углеводородных газов. Им, в частности,
было успешно осуществлено определение небольших количеств «-парафи-
нов в этилене.
Нами изучались хроматографические свойства серебряных цеолитов
для выяснения ряда их физико-химических свойств. Исследованы следую-
щие образцы в качестве наполнителей хроматографических колонок:
1 — серебряная форма цеолита типа X, полученная обработкой рас-
твором нитрата серебра гранулированного натриевого цеолита (Горьков-
ская опытная база ВНИИНП, серия Ц-202-208), содержание Ag,0 —
23.50%;
2 — серебряная форма цеолита X, полученная обработкой раствором
нитрата серебра кристаллического натриевого цеолита X и сгранулиро-
ванного при добавке каолина, содержание Ag2O — 22.08%;
3 — представляет собой первый образец, восстановленный в токе
водорода;
4 — представляет собой смесь из свежеприготовленной окиси серебра
и цеолита NaX. Содержание Ag2O — 19.1%.
Исследования проводились на хроматографе ХТ-2М в температурном
интервале от 24 до 240° С. В качестве газа-носителя применялись воздух
и гелий, а модельная смесь, которая делилась на хроматографической
колонке, состояла из водорода, окиси углерода, предельных и непредель-
ных углеводородных газов состава Сх—С4.
Соответствующие хроматограммы приведены на рис. 1 и 2. На основа-
нии их можно сделать следующие выводы.
Первые два образца (содержащие ионы серебра) при повышенных
температурах прочно поглощают окись углерода, водород и непредельные
углеводороды, причем это свойство проявляется при применении в ка-
честве газа-носителя как воздуха, так и гелия. Использование инертного
газа в качестве носителя исключает окисление перечисленных веществ
и свидетельствует об их фиксировании, по-видимому, за счет хемосорбции.
Третий образец — серебряная форма цеолита, восстановленная в токе
водорода. Применением в качестве газа-носителя гелия было установлено,
что молекулярный водород, окись углерода и непредельные соединения не
задерживаются хроматографической колонкой. Если в качестве газа-носи-
теля взять воздух, то наблюдается фиксирование молекулярного водорода,
окиси углерода й непредельных соединений, что, по-видимому, вызвано
окислением восстановленного серебра в ионы серебра и превращением
восстановленной серебряной формы в серебряную форму цеолита. Все
вышеприведенные факты свидетельствуют о том, что в то время как цео-
литы, содержащие атомы серебра, неспособны хорошо адсорбировать
молекулярный водород, окись углерода и непредельные соединения, эти
соединения вступают в энергичное взаимодействие с цеолитом, содержа-
щим ионы серебра, возможно, с образованием комплексов.
Исследованием хроматографических свойств четвертого образца (смесь
окиси серебра и цеолита) было найдено, что на наполнителе хроматографи-
ческой колонки происходит прочная адсорбция молекулярного водорода,
окиси углерода и непредельных углеводородов и разделение предельных
углеводородов состава Сх—С4, как на обычном цеолите. Применяя как
88
NaX +АдгО
Рис. 1. Хроматограммы разделения смеси газов на гранулах и порошке серебряных
цеолитов и смеси натриевого цеолита и окиси серебра.
Рис. 2. Хроматограмма разделения газовой смеси на восстановленном
серебряном цеолите; газ-носители гелий (о, б) и воздух (в).
Рис. 3. Изотермы адсорбции двуокиси
углерода на натриевых и серебряных
цеолитах типа А при 23 и —77° С.
наполнитель колонки только окись серебра, не удается разделить предель-
ные углеводороды состава Сх—С4. На основании полученных данных видно,
что специфические свойства серебряной формы цеолита, с одной стороны,
определяются нахождением в ее составе ионов серебра, а с другой стороны—
цеолитной кристаллической структурой адсорбента.
Определяющее влияние иона серебра на специфическую способность
цеолита энергично сорбировать непредельные соединения может быть по-
нято на основании исследований по комплексным соединениям иона се-
ребра с соединениями, включающими л-связи [6]. В результате таких
работ установлено, что соединения с л-связями (непредельные, аромати-
ческие) способны образовывать ком-
плексы с ионами серебра, причем
стойкость таких комплексов зависит
от состава и строения этих веществ.
В последнее время этим обстоятель-
ством и обусловлено применение
в качестве стационарной фазы азотно-
кислого серебра в органических рас-
творителях (гликоль, глицерин,бен-
зилцианид) в газо-жидкостной хро-
матографии.
Результаты изучения инфракрас-
ных спектров также убеждают нас
в том, что окись углерода, по-види-
мому, образует координационную хи-
мическую связь с ионом серебра.
Изучение адсорбции двуокиси
углерода на серебряной форме цео-
лита типа А показало, что замеще-
ние иона натрия на ион серебра
уменьшает ее адсорбцию. Понижение
температуры в обоих случаях спо-
собствует росту величины адсорбции
(рис. 3).
Серебряный цеолит при дегидратации (300° С) в вакууме меняет
белый цвет на кирпично-красный. Использование серебряных цеолитов
типа А для индикации паров воды было предложено чешскими исследова-
телями [7]. Они показали, что небольшое количество паров воды (40—
70 частей на миллион) может быть индицировано по переходу цвета деги-
дратированного серебряного цеолита из желто-красного в светло-желтый.
Нашими исследованиями показано, что для определения более
высоких содержаний влаги в системах могут быть применены де-
гидратированные никелевые и кобальтовые формы цеолитов типа А.
Характер изменения цвета серебряных, никелевых и кобальтовых
цеолитов зависит от степени замещения иона натрия на катионы се-
ребра, никеля и кобальта. На рис. 4 приводим данные по изменению
цвета цеолитов при нахождении цеолита в системе с фиксированной
влажностью в течение одного и 24 часов. Наибольшую чувствительность
в отношении индикации паров воды проявляет серебряная форма цеолита,
наименьшей чувствительностью характеризуется кобальтовый цеолит,
промежуточное место занимает никелевая форма цеолита. При помощи
этого набора цеолитов вода может быть индицирована в сравнительно
широком диапазоне концентраций.
Обращают на себя внимание цериевые формы цеолитов типа А и X.
Цериевые цеолиты получались нами путем замещения иона натрия на
ион церия. Натриевые цеолиты типа А и X обрабатывались 0.05 и 0.10 н.
90
растворами азотнокислого церия по ранее разработанной методике [12].
Оказалось, что даже трехкратная обработка натриевого цеолита типа А
не приводит к существенному?замещению иона натрия на ион церия.
Ч*
D
лзел
а
Ультрамариновая Сире- неоая РозоВая
СаренеВая Телесная Салатная
СВетло-оранжеВая РозоВая ЖелтоВато-белая
Темно-оранжеВая РозоВая ЖелтоВато- белая
6
УльтрамариноВая Сшэе- неоая РозоВая
СаренеВая Телесная СВетло-салатная
СВетло- оранжеВая РозоВая Желтобато-белая
Темно- оранжеВая РозоВая ЖелтоВато-белая ГрязноВато-желтая
СобА
Ад,А
г -1.5 -1 -0.5 О 0.5 1 1.5 Z
Рис. 4. Изменение цвета дегидратированных серебряных,
никелевых и кобальтовых цеолитов в зависимости от вы-
держки (а—1 час, б — 24 часа) и давления паров воды.
Возможно, особенность этого процесса обязана ситовому эффекту, так как
в растворе ионы гидратированы и в полость цеолита для замещения дол-
а,ммоль/г а. ммоль/г
Рис. 5. Изотермы адсорбции азота, воды (а) и двуокиси
углерода (б) на натриевых и цериевых цеолитах типа X.
жен попасть сильно гидратированный ион церия. Другая картина наблю-
далась для цеолитов типа X. В этом случае обмен шел гладко и* были по-
лучены цериевые цеолиты типа X различной степени замещения:
91
СехХ с содержанием Се2О3 3.90%
Се4Х » » Се2О3 14.50
СееХ » » Се2О3 17.50
Нами изучались адсорбционные свойства дегидратированных (300° С)
цериевых цеолитов по парам воды, азота и двуокиси углерода. Соответ-
ствующие изотермы адсорбции приведены на рис. 5. Как видно, замещение
иона натрия на ион церия приводит вначале к некоторому росту адсорб-
ционной способности по парам
а, ммоль/г
10\-
10 P/Ps
0
Изотермы адсорбции азота,
бензола на кадмиевых цео-
0.2 0.4 0.Б
NaX
о Cd'X
® Cd3X
Рис. 6.
воды и
литах типа X. На образце натрие-
вого цеолита изучалась адсорбция
азота и бензола.
воды, а затем по мере увеличения степени
замещения происходит ее уменьшение.
Это может быть обусловлено несколь-
кими причинами, при этом не исклю-
чено частичное разрушение решетки
цеолита под влиянием трехзарядного
катиона. Замещение иона натрия на
ион церия уменьшает адсорбционную
способность цеолита по парам азота и
двуокиси углерода, что, по-видимому,
вызвано уменьшением числа катионных
адсорбционных центров в цеолите.
Аналогичные результаты получены
при исследовании адсорбции паров азо-
та, воды и бензола на кадмиевых цео-
литах (рис. 6). Содержание CdO в CdxX,
Cd3X и Cd6X соответственно равно
7.52, 13.82 и 22.85%.
Изучение кинетики адсорбции на
цеолитах привлекает внимание иссле-
дователей. Этой области поверхностных
явлений посвящены работы Бэррера [8],
Хэбгуда [9], Д. П. Тимофеева [10],
Брэка [11], нашей лаборатории [12].
Кинетика адсорбции изучалась дина-
мическим методом из потока тщательно
очищенного и высушенного воздуха.
Кинетические исследования проводились
при разных концентрациях, но здесь
мы приводим результаты для одной
относительной концентрации p/ps=0.10.
Опыты ставились при 25° С и удельной
скорости потока куд.=0.35 л/мин. см2.
Диаметр и высота цилиндрических гра-
нул были равны 6 мм.
иона натрия на ион калия получается
При замещении в цеолите А
калиевая форма молекулярного сита. По ряду данных для этого цеолита
диаметр входа в полость близок к 3 А°, что определяет его избирательную
сорбционную способность по парам воды. В этой связи было интересно
изучить влияние иона калия на кинетические свойства цеолитов.
Исследовалась кинетика адсорбции паров воды на кристаллическом
натриевом цеолите (Горьковской опытной базы ВНИИНП, серия Ц-202-85)
и полученном из него шестикратным замещением калиевом цеолите типа А.
Результаты приведены в виде кинетических кривых a — и — — <р(\/Г),
ао
где а и а0—адсорбция для времени t и при насыщении.
Как видно из рис. 7, кинетическая кривая a=j(t) для натриевой формы
цеолита ^ежит значительно выше, чем кривая для калиевой формы. Та-
ким образом, при замещении иона натрия на ион калия происходит наряду
92
с повышением сорбционной избирательности цеолита по парам воды также
уменьшение его адсорбционной способности и существенное замедление
адсорбционного процесса. Такая особенность калиевой формы цеолита
типа А 1 сказывается и на коэффициентах диффузии. Эти величины, рас-
считанные по методу Д. П.
Тимофеева, соответственно
равны для натриевой и ка-
лиевой формы 9.3 • 10 ~7 и 5.0 •
10-7 см2 сек.-1, т. е. коэффи-
циент диффузии при замеще-
нии иона натрия на ион калия
уменьшается примерно вдвое.
Обращают на себя внима-
ние данные, полученные при
изучении кинетики адсорбции
паров воды на водородных
цеолитах типа А и X различ-
ной степени замещения. Водо-
родные цеолиты приобретают
Рис. 7. Кинетические кривые адсорбции паров
воды на натриевых и шестизамещенных калиевых
цеолитах типа А.
важное значение, так как
они обладают рядом интерес-
ных для практики свойств
(повышенная кислотостой-
кость, специфическая ката-
литическая активность и др.). Эти цеолиты могут быть получены
путем осторожного термического-разложения аммонийной формы. Такой
путь является наиболее простым, особенно для практических целей.
Однако при нагревании аммонийной формы не исключено разрушение
кристаллической решетки цеолита. В этой связи нами была изучена кине-
тика адсорбции паров воды
и бензола на водородных
цеолитах различных сте
пеней замещения, полу-
ченных из аммонийных
цеолитов.
Объектами исследова-
ния были взяты водородные
формы цеолитов типа А и
X. Процент замещения
ионов натрия ионами ам-
мония для типа А был
равен 15.27 и40% для типа
X —56.65и75%. Гранулы
аммонийных цеолитов, по-
лученные прессованием
кристаллического порош-
ка, осторожно нагревались
до 275° С. Условия проведе-
ния кинетических опытов были аналогичны предыдущим. На рис. 8 приво-
дим данные по кинетике адсорбции паров воды на натриевой и водородных
формах. Замещение иона натрия на ион аммония в цеолите А на 15% ипо-
__________।_________।_________।_________।_________।______
о во <60 гм зго т t
Рис. 8. Кинетические кривые адсорбции паров
воды на натриевом и водородных цеолитах типа А.
следующее его термическое разложение приводит к водородному цеолиту,
который близок по своим адсорбционно-кинетическим свойствам к натрие-
1 Калиевый цеолит типа А был получен в нашей лаборатории М. Понома-
ревой, а аммонийные и водородные цеолиты — К. Бежашвили.
93
вому цеолиту. Водородные цеолиты, полученные из аммонийных цеолитов
с большой степенью замещения, характеризуются меньшей адсорбционной
способностью и замедленной кинетикой сорбции паров воды. Таким обра-
зом, по-видимому, замещение иона натрия на ион аммония в цеолите типа А
свыше 15—20% способствует разрушению кристаллической решетки цео-
лита. Аналогичные данные получены для цеолита типа X, лишь с той раз-
ницей, что в этом случае замещение иона натрия на ион аммония до 50%
не оказывает существенного влияния на сорбционные свойства по парам
воды (рис. 9). Увеличение степени замещения выше 50—60% снижает
адсорбционную способность по воде, что так же, как и в случае цеолита А,
Рис. 9. Кинетические кривые адсорбции паров воды на
натриевом и водородных цеолитах типа X.
по-видимому, в определенной части обусловлено разрушением цеолито-
вой кристаллической структуры.
Изучение кинетики адсорбции паров бензола (рис. 10) и сравнение
с данными по кинетике адсорбции паров воды позволяют сделать следую-
щее заключение.
На натриевой форме цеолита типа X при одинаковых относительных
концентрациях (C7Cs=0.10) значительно быстрее адсорбируются пары
воды по сравнению с парами бензола (рис. 9—10). Другая картина на-
блюдается для водородных цеолитов. Время установления адсорбцион-
ного равновесия для паров воды и паров бензола на этих цеолитах при-
мерно одинаково. Однако пары бензола значительно меньше адсорби-
руются на водородных цеолитах, чем на натриевом цеолите типа X. Это
вероятно, обусловлено тем, что при нагревании на воздухе аммонийных
цеолитов типа X происходит частичное разрушение цеолитовой кристал-
лической решетки. Действительно, ранее нами было показано, что при
нагревании аммонийных цеолитов в вакууме получаются образцы водо-
родных цеолитов, которые адсорбируют пары бензола даже несколько
лучше, чем натриевая форма цеолита [13]. Можно допустить, что при тер-
мическом разложении аммонийных цеолитов на воздухе происходит обра-
зование в теле цеолита тонкопористых участков (областей), которые об-
условливают проявление ситового эффекта в отношении молекул бензола.
Однако здесь надо принять во внимание и данные по коэффициентам
диффузии. Расчеты показывают, что коэффициенты диффузии бензола для
94
натриевого цеолита и водородных цеолитов — НхХ, Н2Х и Н3Х соответ-
ственно равны 5.2, 7.0, 9.0 и 8.3- 10~7 см2сек.-1. Величина коэффициента
диффузии для натриевого цеолита почти в два раза больше для воды
(9.3 • 10 “7 см2сек.) по сравнению с этой величиной для бензола, что согла-
суется с более быстрым поглощением паров воды по сравнению с адсорб-
цией паров бензола. При переходе от натриевого цеолита к водородным
цеолитам коэффициенты диффузии для паров воды мало изменяются и
растут для паров бензола. Последнее обстоятельство указывает на то, что,
по-видимому, в водородных цеолитах имеются еще более открытые поры,
чем в натриевых. Таким образом,, можно допустить, что при термическом
воздействии на аммоний-
ные цеолиты типа X про-
исходит изменение пори-
стости (учитывая измене-
ние свойств адсорбцион-
ных центров).
На основании вышепри-
веденных данных можно
сделать вывод о важности
исследования кинетики
сорбции молекул разной
природы и размеров для
суждения об изменении
пористой структуры адсор-
бентов под влиянием ряда
факторов, что не может
быть осуществлено только
на основании измерений
адсорбционных изотерм.
Данные по кинетике ад-
сорбции паров свидетель-
Рис. 10. Кинетические кривые адсорбции паров
бензола на натриевом и водородных цеолитах
типа X.
ствуют о внутреннедиффузионном механизме адсорбционного процесса
на цеолитах.
Исследованием магнитных свойств синтетических цеолитов удается
получить ряд новых характеристик этих кристаллов. В этой области в по-
следние годы выполнен ряд интересных работ [14, 15].
Чистые натриевые, кальциевые и другие цеолиты, содержащие диа-
магнитные катионы, должны быть диамагнитными. В процессе обмена
диамагнитного катиона на парамагнитный катион следует ожидать пере-
хода диамагнитного кристалла в парамагнитное тело. В принципе изме-
рение магнитной восприимчивости чистых кристаллических цеолитов
может дать сведения о степени обмена, влиянии кристаллической решетки
цеолита на магнитные свойства катионов, возможном изменении валент-
ности катиона в процессе обмена и др.
Магнитное исследование цеолитов встречает ряд трудностей, одна
из которых связана с магнитохимической чистотой изучаемых веществ. Хи-
мическая или даже спектральная чистота препарата не гарантирует его «ма-
гнитохимическую чистоту». Нередко случается, что вещества, подлежащие
магнитному исследованию, содержат следы ферромагнитных примесей,
которые могут совершенно замаскировать истинное значение восприим-
чивости вещества. Наличие таких примесей обнаруживается при исследо-
вании зависимости кажущейся восприимчивости диамагнитных и парамаг-
нитных веществ от напряженности поля. Ферромагнитные примеси почти
невозможно выявить обыкновенным анализом, и если их не удается уда-
лить перекристаллизацией вещества, то для элиминирования их влияния
можно применить два метода.
95
Первый метод заключается в нагревании вещества и проведении из-
мерений при температуре, которая выше точки Кюри примеси, второй —
в проведении измерений при различных значениях напряженности поля
с последующей экстраполяцией восприимчивости к бесконечно большой
напряженности поля.
Так как к образцам цеолитов, которые изучаются магнито-химическими
методами, предъявляются высокие требования по чистоте, то нами спе-
циально был синтезирован натриевый цеолит типа А и из него методом
ионного обмена были получены цеолиты с парамагнитными катионами.
Опытные образцы, с которыми мы вели обычные, адсорбционные, хромато-
графические и другие исследования, даже после тщательной очистки не
удовлетворяют требованиям магнитохимической чистоты. Для исследова-
ния магнитной восприимчивости нами был применен метод Гуи [16].
Изучалась магнитная восприимчивость марганцевых, кобальтовых
и никелевых цеолитов при разной степени замещения иона натрия на
ион марганца, кобальта и никеля. Более подробно изучены марганцевые
цеолиты, которые, характеризуются ‘ярко выраженным парамагнетизмом.
Из чистого натриевого цеолита типа А методом ионного обмена
были приготовлены цеолиты двухвалентного марганца с различной сте-
пенью замещения. В зависимости от кратности обработки натриевого' цео-
лита раствором нитрата марганца приготовлялись однократно-, трех-
кратно- и шестикратнозамещенные цеолиты с содержанием МпО в 5.15,
11.59 и 17.20%. Изучалась магнитная восприимчивость этих цеолитов и
гидрата хлористого марганца (МпС12 • 4Н2О). Восприимчивость послед-
него соединения измерена ранее, и: ее значение сравнительно хорошо со-
впало с нашими данными. Для безводной соли удельная магнитная вос-
приимчивость по нашим измерениям равна 120.7 • 10 е, а по литератур-
ным источникам 122 • 10”® [17].
Приводим данные по магнитным моментам для иона марганца, рассчи-
танным на основании измерения магнитной восприимчивости:
Магнитный момент МгГ'+
Соединение в магнетонах Бора
Цеолит МщА...............5.90
Цеолит Мн3А..............5.97
Цеолит Мн6А..............6.66
MnCl,.....................6.00
Ион двухвалентного марганца находится в S состоянии, и его магнит-
ный момент должен соответствовать пяти неспаренным электронам, т. е.
должен быть равен 5.92 магн. Бора. Магнитный момент иона марганца
в хлористом марганце близок к этому значению и равен 6.00 магн. Бора.
Величина магнитного момента иона марганца, полученного на основании
данных по магнитной восприимчивости цеолитов, немного отличается от
теоретического значения и находится в пределах опытных значений
(5.65 4- 6.10) для этой величины [18]. Полученные данные свидетельствуют
о том, что ионы марганца в изученных образцах цеолита типа А находятся
в спинсвободном состоянии. Работа в направлении более подробного ис-
следования магнитных свойств цеолитов нами продолжается.
ЛИТЕРАТУРА
1. М. М. Дубинин в сб.: Синтетические цеолиты, Изд. АН СССР, 1962;
С. К. Hers h. Molecular Sieves. N. Y., 1961.
2. Г. В. Цицишвили, в сб.: Поверхностные явления на алюмосиликатах.
Изд. АН Груз. ССР., 1965 , 5.
3. Г. В. Ц и цишвили, Т. Г. Андроникашвили, Л. Я. Лапера-
швили, Ц. А. Геджадзе, Сообщ. АН Груз.ССР, 1962, 28, 3, 281.
96
4. Т. Г. Андроникашвили, Ш. Д. Сабелашвили, Г. В. Цици-
ш в и л и, Нефтехимия, 1962, 2, 2, 248; Г. В. Ц и ц и ш в и л и, Т. Г. А н д р о-
никашвили, Ш. Д. Сабелашвили, 3. И. Коридзе, Сообщ.
АН Груз. ССР, 1964, 35, 87.
5. J. Janak. Vapour Phase Chromatography. Ed. D. Desty, London, 1957, 247.
6. Д. В e й л a p. Химия координационных • соединений. ИЛ, М., 1960, 411.
7. М. Ра лек, П. И р у, О. Г р у б н е р, Г. Б е й е р, в сб.: Синтетические
цеолиты, Изд. АН СССР, 1962, 263.
8. R. М. В а г г е г, Proc. Roy. Soc., 1938, А 167, 392; Trans. Faraday Soc., 1949, 45,
358‘ 1953 49 1049.
9. H. W. H a b g о о d, Canad. J. Chem., 1958, 36, 1384.
10. Д. П. T и м о ф e e в, И. Г. E p а ш к о, Изв. АН СССР, OXH, 1961, 7, 1192;
Д. П. T и м о ф e e в, О. H. К а б а н о ₽ а, И. Г. E p а ш к о, А. С. Поно-
марёв, в сб.: Синтетические цеолиты, Изд. АН СССР, 1962, 24.
11. D. W. В г е с k, W. Eversole, R. М i 11 о n, Т. R е е d, Т. Thomas,
J. Am. Chem. Soc., 1956, 78, 5963.
12. Г. В. Цицишвили, Т. Г. Андроникашвили, в сб.: Синтетические
цеолиты, Изд. АН СССР, 1962, 117, 126.
13. Г. В. Цицишвили, Г. Д. Б аг р атишви л и, К. А. Б е ж а ш в и л и,
Д. Н. Б а р н а бишв и л и, М. С. Шуакришвили, ДАН СССР,
1963 152 1137.
14. С. П. Г а б у д а, ДАН СССР, 1962, 146, 840; С. П. Г а б у д а, Г. М. М и х а й-
л о в, К. С. Александров, ДАН СССР, 1963, 153, 6, 1360; Вести.
АН СССР, 1963, 3, 73.
15. J. R а Ь о et al., Actes congr. Internal. Catalyse, Paris, 1960, 61, 2055.
16. В. Клем м. Магнетохимия, M., 1939, 46.
17. Сборник физических констант под ред. Дорфмана. Л.—М., 1937, 209.
18. Современная химия координационных соединений. М., 1963, 394.
О НЕКОТОРЫХ СВОЙСТВАХ РАЗЛИЧНЫХ ИОНООБМЕННЫХ
ФОРМ ЦЕОЛИТОВ ТИПА А
М. А. Пионтковская, И. Е. Неймарк, Г. С. Шамеко,
Г. И. Денисенко, И. Н. Червецова
Нами изучались физико-химические и структурные характеристики
катионзамещенных форм цеолитов. В настоящем сообщении приводятся
результаты исследования: 1); радиационной устойчивости ионообменных
форм цеолитов; 2) термостабильности катионзамещенных цеолитов и
3) влияния катионов, компенсирующих отрицательный заряд алюмосили-
катного каркаса, на величину теплоты адсорбции низший углеводородов.
Для определения радиационной устойчивости навески цеолитов при
комнатной температуре выдерживали определенное время в поле у-излу-
чения. Источником у-лучей служил Со60 активностью 70 тысяч г • экв.
радия. Мощность дозы 1390 рад в секунду, интегральные дозы изменялись
в пределах от 0 до 108 рад. Цеолиты в запаянных ампулах в абсолютно
сухом состоянии подвергали действию у-излучения. Параллельно с этим
изучали радиационную устойчивость' цеолитов при облучении их в вод-
ной среде.
Результаты облучения цеолитов в водной среде могут представить
интерес при ионообменном разделении радиоизотопов.
Для определения физико-химических свойств облученных и необлу-
ченных цеолитов использовали весовой адсорбционный метод исследова-
ния. Изучение адсорбционной характеристики пористых кристаллов
производили по изотермам сорбции паров воды.
В табл. 1 приведены данные о заполнении первичной пористой струк-
туры катионзамещенных цеолитов после облучения их в абсолютно сухом
состоянии и в водной среде.
7 Цеолиты
97
Таблица 1
Адсорбционная емкость дегидратированных цеолитов (ммоль/г)
Образец Адсорбция паров воды при p/pg = 0.5
доза облучения цеолитов в абсолютно сухом состоянии, рад доза облучения цеолитов в водной среде, рад
0 10* 105 10? 108 0 10* 10» 107 108
MgNaA 16.4 _ 16.5 15.8 15.3 16.4 16.2 16.2 16.0 16.2
SrNaA 12.6 13.4 13.7 13.6 14.1 12.6 13.7 13.6 13.8 14.2
ZnNaA 13.5 13.4 12.9 13.1 13.4 13.5 — 12.8 13.0 13.7
CdNaA 12.4 12.6 12.6 12.8 12.5 12.4 — 12.9 12.3 12.4
При облучении катионзамещенных цеолитов, содержащих ионы ще-
лочноземельных металлов, в абсолютно сухом состоянии и в водной среде
изменений в адсорбционной структуре образцов не было обнаружено.
Как следует из табл. 1, ионообменные формы цеолитов не испытывают
структурных изменений при дозах облучения до 108 рад. Это обстоятель-
ство является весьма важным. Благодаря радиационной устойчивости
катионообменных форм цеолитов, пористые кристаллы могут найти при-
Рис. 1. Микрофтометрические кривые рентгенограмм магнийзаме-
щенного цеолита, приготовленного из натриевого образца до и
после облучения.
менение в качестве ионообменников при поглощении радиоактивных
изотопов.
Интерес представляют опыты по изучению влияния у-излучения на
ионообменные свойства молекулярных сит.
Облучение дегидратированного цеолита NaA производили в вакууми-
рованных запаянных ампулах при комнатной температуре на у-установке
с Со60, интегральные дозы изменялись в пределах от 0 до 1.53 - 107 рад.
Катионообменную емкость цеолита до и после облучения опреде-
ляли по иону магния при контактировании 2 г молекулярного сита NaA
с 100 мл 0.1 н. раствора сернокислого магния. После облучения ампулы
вскрывали в растворе сернокислого магния. В продукте обмена опреде-
ляли натрий пламенным фотометром и рассчитывали процент обмена от
обменной емкости. О структурных изменениях облученных образцов
судили по данным рентгеновских исследований.
98
Таблица 2
Влияние у-излучения Со60 на ионообменные свойства
цеолита и влагоемкость катионообменных форм
№ образца Дова облуче- ния X 10е рад Содержание Na2O в магний- замещенном цеолите Адсорбция паров воды на магний- з вмещенном цео- лите при р/р8 — 0.5, ммоль/г
12 5.10 17.3
7 0.3 5.55 17.9
10 4.6 6.0 16.8
9 7.23 5.95 17.9
11 15.3 6.0 16.2
Согласно рентгеноструктурным измерениям, молекулярное сито NaA
после облучения ионизирующим излучением не испытывает заметных
структурных изменений.
На рис. 1 приведена микрофотометрическая кривая рентгенограммы
магнийзамещенного цеолита, синтезированного из натриевого образца
после облучения дозой 1.53 • 107 рад (нижняя кривая, 77). На этом же
рисунке для сопоставления представлена микрофотометрическая кривая
магнийзамещенного цеолита, приготовленного из натриевого образца
без облучения (верхняя кривая, 12). Из рис. 1 видно, что ионообменные
формы пористых кристаллов, синтезированные из облученного и необлу-
ченного натриевого цеолита, обла-
дают идентичной структурой.
В табл. 2 приведены данные по
влиянию у-излучения Со60 на катио-
нообменные свойства молекуляр-
ных сит. Как видно из табл. 2, про-
цент Na2O в металлзамещенном цео-
лите, приготовленном из молеку-
лярного сита NaA при интеграль-
ной дозе облучения от 0 до 1.53 •
10’ рад, остается неизменным и
практически равным содержанию
Na2O в образце после обмена с
необлученным цеолитом.
Влагоемкость магнийзамещен-
ного цеолита, как это следует из
данных табл. 2 и изотерм адсорб-
ции паров воды (рис. 2), после
Рис. 2. Изотермы адсорбции паров воды
при температуре 20° С на магнийзвмещен-
ном цеолите, приготовленном из натрие-
вого образца после облучения разными
дозами ионизационного излучения.
1 — обр. 7, 0.3 • 10“ рад; 2 — обр. 9, 7.23 • 10“
рад; 3 — обр. 12, иеоблученный; 4 — обр. 10,
4.6 • 10“ рад.
облучения исходного образца дозой
до 1.53 • 107 рад, остается практически неизменной.
Таким образом, адсорбционная емкость и ионообменные свой
ства молекулярного сита NaA не изменяются при дозах облучения
1.53-107 рад. Структура пористых кристаллов катионообменных форм цео-
литов, содержащих ионы натрия и катионы щелочноземельных металлов,
не разрушаются при дозах облучения до 108 рад. Эти адсорбенты являются
устойчивыми к ионизирующему излучению. и могут быть использованы
в качестве ионообменников в производстве радиоактивных элементов и
при переработке высокоактивных растворов. Между тем как большин-
ство органических анионитов и катионитов при дозах облучения до
1.5-108 рад претерпевают глубокую деструкцию [1, 2].
В настоящем сообщении представлены результаты исследования тер-
мостабильности различных катионзамещенных форм цеолитов.
7*
99'
Зависимость сорбционных свойств цеолитов типа NaA, NaX, СаА,
СаХ от температурной обработки изучена достаточно подробно [3, 4].
Изучена термическая устойчивость кристаллических алюмосиликатов
с одновалентными ионами. Установлено [4], что катионы элементов пер-
вой группы периодической системы, компенсирующие отрицательный
заряд алюмосиликатного каркаса, оказывают влияние на термостабиль-
ность ионзамещенных форм цеолитов.
Нами была исследована термостабильность синтетических цеолитов,
содержащих в качестве компенсирующих катионов ионы тяжелых ме-
таллов и катионы элементов второй группы.
Термическая устойчивость образцов изучена в области температур
300—800° С. На дегидратированных цеолитах исследованы изотермы
адсорбции паров воды при 20° С. После нагревания в воздухе при соот-
ветствующей температуре в течение 4 часов образцы перед снятием изо-
терм сорбции прогревались в вакуумной установке при температуре 300°
в течение 4 часов. При температуре 600° С проведены дополнительные
опыты по определению термической устойчивости различных ионообмен-
ных форм цеолитов в зависимости от продолжительности прокаливания.
Таблица 3
Адсорбционная емкость ионзамещенных форм цеолитов, дегидратированных при
разных температурах (ммоль/г)
Образец Адсорбция паров воды при р/р„ = 0.5 Степень обмена в % от об- менной емкости
вакуум ’ 300° температура прокаливания, °C
500° 690° 600° в течение 12 часов 650° 700° 750° 800°
MgNaA 17.0 16.5 16.3 17.0 15.0 0 0 47.0
SrNaA 12.6 14.4 14.2 13.5 —_ 13.5 11.2 0 48.0
CuNaA 15.2 15.3 15.3 14.7 0.7 0.3 — 0 38.0
ZnNaA 13.5 13.6 13.6 13.2 — 13.3 0.17 0 64.8
CdNaA 12.4 13.1 13.3 12.9 — 12.9 0.2 0 40.6
Cob’aA 13.5 15.1 16.6 14.6 — 14.8 0.2 0 47.9
NiNaA 15.6 14.6 14.8 14.3 11.0 3.0 0.7 0 36.8
PbNaA 12.1 —. — 12.8 11.6 0 0 — 39.7
В табл. 3 приведена адсорбционная характеристика ионзамещенных
цеолитов, дегидратированных при разных температурах. Адсорбционная
емкость определена из изотерм сорбции паров воды при р/р„=0.5, т. е.
при заполнении первичной пористой структуры продуктов дегидратации.
Из данных, приведенных в табл. 3, следует, что структура металлза-
мещенных цеолитов остается неизменной при нагревании их при темпера-
туре 600° С в течение 4 часов. Увеличение продолжительности прокалива-
ния до 12 часов не вносит существенных изменений в процесс регенерации
образцов.
Как следует из данных той же таблицы, дегидратация металлзамещен-
ных цеолитов практически завершается при прокаливании их при тем-
пературе 300° С в течение 4 часов. Однако стронций- и особенно кобальт-
обменные формы регенерируются в условиях более высоких температур.
Разрушение кристаллической структуры у молекулярных сит, содержа-
щих катионы тяжелых металлов и щелочноземельных элементов, наблю-
дается при температуре прокаливания 800° С.
У магний-, стронций-, кадмий-, цинк- и кобальтзамещенных цеолитов
сохраняется структура кристаллов после прокаливания их при 700° С;
разрушение пористых кристаллов в этом температурном режиме имеет
место у медь-, никель-, свинецзамещенных цеолитов.
100
1.6 1.8 2.0 2.2 2Л 2.6 2.8 3.0 1000/Т
Рис. 3. Зависимость логарифма удельного
удерживаемого объема индивидуальных газов
от обратной температуры на магнийзамещен-
ном цеолите.
и подвергали элюированию потоком воз-
Данные о термостабильности катионообменных форм цеолитов имеют
значение при анализе результатов Исследования 'свойств молекулярных
сит в области высоких температур, в частности при изучении высокотем-
пературной адсорбции веществ на синтетических цеолитах.
В литературе имеется не много работ по изучению высокотемператур-
ной адсорбции веществ на цеолитах. А. В. Киселев с сотрудниками [5]
использовал данные высокотемпературной адсорбции веществ для опре-
деления теплот адсорбции низших углеводородов от Сх до С4 на цеолитах
5А. Т. Г. Андроникошвили и Ш. Д. Сабелашвили [6] применили газоад-
сорбционную хроматографию для разделения смеси углеводородных газов
на синтетических цеолитах
NaX, СаА при повышенных тем-
пературах. Эберли [7] опубли-
ковал работы по исследованию
высокотемпературной адсорб-
ции бензола и н-гексана на
различных ионообменных фор-
мах цеолита А.
Нами изучена высокотемпе-
ратурная адсорбция индивиду-
альных газов на различных
ионообменных формах цеолита
типа А. По данным хромато-
графических измерений опреде-
лены теплоты адсорбции и удер-
живаемый объем индивидуаль-
ных газов.
Хроматографические иссле-
дования проводили на приборе
ХТ-2М с автоматической за-
писью электронным потенцио-
метром ЭПП-09. Индивидуаль-
ный газ пропускали через ад-
сорбент, помещенный в хромато-
графическую колонку прибора,
духа. Дозировку газовой смеси производили вручную при помощи шприца.
Температуру нагрева хроматографической колонки изменяли от 20 до
360° С. Теплоты адсорбции определены из логарифмической зависимости
удельных удерживаемых объемов индивидуальных газов от обратной
температуры [8]. Величины удерживаемых объемов вычислены по выход-
ным кривым хроматографического анализа. При определении удерживае-
мого объема внесены исправления на мертвое пространство колонки и вес
адсорбента (vg, см3/г). В широкой области температур на различных ка-
тионзамещенных формах цеолитов определены удерживаемые объемы
окиси углерода и низших предельных, непредельных углеводородов.
На рис. 3 представлена зависимость логарифма удельных удерживае-
мых объемов индивидуальных газов от обратной температуры на магний-
замещенном цеолите.
Аналогичные зависимости получены на других катионзамещенных
формах молекулярных сит. Из зависимости 1g vg от МТ вычислены теп-
лоты адсорбции индивидуальных газов. В табл. 4 приведены данные по
теплотам адсорбции.
Как следует из табл. 4, теплоты адсорбции цеолитами зависят от при-
роды индивидуального газа. Для одного и того же предельного углеводо-
рода теплота адсорбции практически не зависит от ион-металла цеолита.
При увеличении в углеводороде числа углеродных атомов теплота адсорб-
101
ции возрастает на 2—3 ккал./моль на одну СН2-группу. Эти результаты
находятся в соответствии с данными [9].
Представляет интерес зависимость удельного удерживаемого объема
от числа углеродных атомов в углеводороде. На рис. 4 изображена за-
Число углеродных атомод
Рис. 4. Зависимость между
lg Vf и числом углеродных
атомов в предельных угле-
водородах при пяти темпера-
турах (°C) на магнийаамещен-
ном цеолите.
висимость между lg vg и числом углеродов
в предельных углеводородах при пяти тем-
пературах на магнийзамещенном цеолите.
При построении рис. 4 использованы как
опытные величины удельных удерживаемых
объемов, так и найденные из графиков lg vg,
1/Т путем интерполяции. В области изучен-
ных температур между lg vs и числом угле-
родных атомов в углеводороде наблюдается
линейная зависимость. С увеличением тем-
пературы опыта и уменьшением числа угле-
родов в адсорбате удельный удерживаемый
объем уменьшается. Очевидно, при погло-
щении предельных углеводородов адсорбция
в основном происходит на кислородах тетра-
эдров [5] с участием атомов углерода.
Несколько иная картина наблюдается при
изучении теплот адсорбции молекул окиси
углерода и молекул углеводородов с ненасы-
щенными связями.
Из данных, приведенных в табл. 4, сле-
дует, что при переходе от насыщенных угле-
водородов к окиси углерода и соответствую-
щим ненасыщенным углеводородам теплоты
адсорбции увеличиваются. Величина теплоты
адсорбции окиси углерода и алкенов моле-
кулярными ситами зависит от катиона, ком-
пенсирующего отрицательный заряд алюмо-
силикатного каркаса. При поглощении на
цеолитах окиси углерода и углеводородов с ненасыщенными связями
теплоты адсорбции, помимо дисперсионного взаимодействия, определяются
также ион-дипольным взаимодействием молекул адсорбата с катионами
металлов первичных пор.
Таблица 4
Теплота адсорбции индивидуальных газов у различных катионзамещенных
цеолитов
Образец Степень обмена в % от обменной емкости Q, ккал./моль
СО сн, С,Нс С2Н« CSHS С3НВ
MgNaA 47 5.2 4.0 5.9 8.6 8.8 10.8
CaNaA 78 5.9 3.6 6.1 9.0 8.4 11.4
SrNaA 48.0 3.2 3.4 6.0 11.2 3.3 —
CdNaA 93.3 5.3 2.9 6.3 9.2 6.8 —
CoNaA 62 7.6 3.4 5.0 13.1 8.1 12.5
Алюмогель — 2.1 1.9 4.45 5.0 4.9 7.2
Алюмосиликогель — 1.8 1.2 2.8 6.7 7.0 8.9
Примечания. По-видимому, при высоких температурах на кобальтовом образце процесс
выделения пропилена осложняется каталитической реакцией: на выходных кривых появляется два
пика. В силу стерических затруднений некоторые полости цеолитов SrNaA, CdNaA недоступны
для молекул пропана.
102
На поверхности алюмогеля и алюмосиликагеля теплота адсорбции
различных углеводородов (табл. 4) меньше теплот адсорбции соответствую-
щих углеводородов на поверхности цеолитов. Очевидно, катионированная
поверхность элементарных полостей благоприятствует преимущественной
адсорбции алкенов, которая, в свою очередь, определяется природойкатиона.
Таким образом, изменяя химическую природу поверхности полостей,
можно решить ряд важных практических задач разделения сложных сме-
сей газов.
Выводы
1. Структура катионзамещенных цеолитов, содержащих ионы щелоч-
ноземельных металлов, не изменяется при дозах облучения до 108 рад
2. Структура и ионообменные свойства цеолита NaA остаются практи-
чески неизменными при дозах облучения до 1.53 • 107 рад.
3. Термостабильность и минимальная температура регенерации ме-
таллзамещенных форм цеолитов зависит от катиона, компенсирующего
отрицательный заряд алюмосиликатного каркаса.
4. Теплота адсорбции молекул окиси углерода и углеводородов с не-
насыщенными связями зависит от катиона металла первичных пор.
ЛИТЕРА ТУР А
1. А. М. Семушин, И. А. К у з и в, ЖПХ, 1959, 32, 2193; И. А. Кузин,
А. М. Семуши н, ЖПХ, 1961, 34, 577; А. П. Полеводов и др , в сб.:
Химия и химическая технология. Научные доклады высшей школы, 1958, 4,
761.
2. И. А. К у з и н, А. М. Семушин и В. П. Т а у ш к а н о в, ЖПХ, 1963,
36, 914.
3. Л. И. П и г у з о в а, А. В. Агафонов и др., в сб.: Синтетические цеолиты,
Изд. АН СССР, 1962, 152.
4. И. Е. Н е й м а р к, М. А. Пионтковская, А. Е. Лукаш, Р. С. Тю-
тю н и к, в сб.: Синтетические цеолиты, Изд. АН СССР, 1962, 49.
5. А. В. К и с е л е в, Е. В. X р а п о в а, К. Д. Щ е р б а к о в а, Нефтехимия,
1962, 2, 6, 877.
6. Т. Г. А и д р о н и к о ш в и л и, 1П. Д. Сабелашвили, в сб.: Синтети-
ческие цеолиты, Изд. АН СССР, 1962.
7. Р. Е. Е b е г 1 у, J. Phys. Chem. 1961, 65, 68; 1962, 66, 812.
8. Н. М. Туркельтауб, А. А. Жуховицкий, Н. В. Поршнева,
ЖПХ, 1961, 34, 1946.
9. А. В. К и с е л е в, в сб.: Газовая хроматография, Изд. АН СССР, 1960, 45.
АДСОРБЦИЯ БЛАГОРОДНЫХ ГАЗОВ
НА ЦЕОЛИТАХ ТИПА X, МОДИФИЦИРОВАННЫХ ИОННЫМ ОБМЕНОМ
В. Босачек
При исследовании адсорбционных свойств цеолитов оказалось, что
катионы, присутствующие в кристаллической решетке цеолитов, имеют
решающее влияние на адсорбционные характеристики газов и паров [1—6].
В последней опубликованной нами работе [7] была исследована роль
катионов Na+, Li+ и К+ в цеолите типа Х..Было показано, что индивидуаль-
ная природа катиона проявляется при хроматографическом разделении
этих газов в порядке расположения элюционных кривых и эффективности
разделения компонентов смеси. Li- и Na-формы цеолита X не разделяют
достаточно эффективно Кг и N2. Калиевый цеолит полностью разделяет
на компоненты смесь кислорода, азота, криптона и ксенона, что может
103
Р. мм рт. ст
РиС. 1. Изотермы адсорбции [криптона на LiX,
NaX и КХ при температуре —78° С.
NaX и КХ
нения объема,
наклонах,но и
ной формы для
быть использовано при аналитических определениях или при очистке
благородных газов от воздуха. Хроматографическая техника позволила
также получить данные о наклонах изотерм адсорбций этих газов для
малых заполнений адсорбционного объема. Более подробное исследование
адсорбционных свойств цео
литов было сделано измере-
нием изотерм адсорбции этих
газов при нескольких темпе-
ратурах.
В качестве исходного цео-
лита был использован обра-
зец цеолита X Ц-202-96, при-
готовленный Б. А. Липкин-
дом, и цеолит МСП-30, при-
готовленный в нашей лабо-
ратории [8].
Адсорбционные измерения
были выполнены на обыкно-
венной стеклянной весовой
установке.
На рис. 1 изображены
изотермы адсорбции крипто-
на на литиевой, натриевой
й калиевой формах цеолита
X. Изотермы отложены в ко-
ординатах: давление р (мм
рт. ст.), Н — степень запол-
Изотермы адсорбции различаются не только в начальных
в постепенном изменении формы, начиная с сигмоидаль-
литиевого цеолита и кончая формой изотермы Ленгмюра
для калиевого цеолита. Аналогично ведет себя й ксенон, как показано на
рис. 2. Изменение формы изотермы
адсорбции в случае литиевого цео-
лита было обнаружено и на образце
МСП-30, так же как и на ряде образ-
цов с различным отношением лития
к калию. Сигмоидальную форму изо-
термы адсорбции объясняют обыкно-
венно большим межмолекулярным
взаимодействием, вследствие кото-
рого возможно при соответствующей
температуре протекание двумерной
конденсации [9—10], или, как в слу-
чае воды на активном угле, своеоб-
разным механизмом адсорбции [11].
Для адсорбции, при которой при-
нимают участие кроме адсорбционных
сил адсорбент—адсорбат и межмо-
лекулярные силы адсорбат—адсор-
бат, было предложено несколько мо-
делей, которые дали возможно,сть
сформулировать уравнения изотерм
адсорбции. Для описания измеренных
Гилла [12]
Рис. 2.
на LiX,
и КХ при температуре
—78° С.
Изотермы адсорбции ксенона
NaX
изотерм мы
применили уравнение
1
—----к,—
. „Vm
-V е
v
(1)
104
где Кг и К2 — постоянные, р — давление, v — адсорбированное коли-
чество, vm — предельный объем цеолита. Дальше было использовано
уравнение А. В. Киселева [13]
V
Р (vm v)
V
(2>
где Кг — постоянная, характеризующая взаимодействие адсорбент—ад-
сорбат, и Кп — постоянная, характеризующая взаимодействие адсорбат—
адсорбат. Каждое из этих уравнений имеет три постоянные, которые мы
рассчитали по методу наименьших ква-
дратов на вычислительной машине [14].
В табл. 1 приведены рассчитанные зна-
чения этих постоянных. Оба уравнения
применимы как для криптона, так и
для ксенона [кроме уравнения (2) для
ксенон—Li—X]. Значение постоянной
в уравнениях Гилла и Киселева по-
степенно возрастает от литиевой формы
через натриевую к калиевой форме.
э
Так как постоянная K1=K(pRT, то
энергия адсорбции Э возрастает в том
же ряду от литиевой к калиевой форме,
если К 0 не отличается существенно для
различных форм цеолита. Постоянную
уравнения (1) можно рассчитать из
трехмерных постоянных Ван-дер-Вааль-
са [15]. Для криптона теоретически К2
равняется 3.65 и для ксенона К2 рав-
няется 5.13. Эти значения постоянной К2
Рис. 3. Зависимость изостерических
теплот адсорбции криптона от вели-
чины адсорбции на цеолитах LiX,
NaX и КХ.
хорошо совпадают с полученными
значениями, особенно для литиевой формы.
Рост энергии адсорбции для криптона и ксенона в последовательности
LiX, NaX, КХ был подтвержден изостерическими теп лотами адсорбции,
которые были получены из изотерм адсорбции при двух температурах.
На рис. 3 показаны теплоты адсорбции криптона в зависимости от вели-
чины адсорбции. Теплоты адсорбции, соответствующие наименьшим за-
полнениям объема, когда в среднем приходится одна молекула на несколько
полостей кристалла, связаны, вероятно, с дефектами решетки и поэтому
являются нетипичными. Среднее значение теплоты адсорбции, лучше
характеризующее данный цеолит, находим в таких областях заполнений,
Таблица 1
Значения констант уравнений Гилла и Киселева для Кг и Хе на различных
цеолитах
Адсорбат Цеолит Изотерма Гилла Изотерма Киселева Опытное Теорети- ческое
к, К2 Ki Кп к,
LiX 0.00113 3.582 0.00117 4.159 5.86 4.845 3.647
Кг | NaX 0.00197 2.911 6.53 0.00244 2.549 4.78 4.48 -—
КХ 0.00308 2.267 6.02 0.00438 1.245 4.31 4.32 —
LiX 0.02393 5.05 5.56 Не применимо. 5.31 5.130
Хе | NaX 0.06035 4.45 5.20 0.058 8.102 4.07 4.91 —
КХ 0.1280 3.42 4.61 0.338 0.580 3.61 4.74 —
1.05
когда в одной полости адсорбированы 1—2 молекулы и межмолекулярные
силы еще могут оказывать существенное влияние. В этой области запол-
нений последовательность теплот адсорбции для криптона и ксенона на
LiX, NaX и КХ, полученная нами, находится в хорошем совпадении с по-
следовательностью теплот адсорбции аргона на литиевом, натриевом и
калиевом цеолитах X. На рис. 4 показаны теплоты адсорбции ксенона.
Постепенное увеличение теплоты адсорбции и относительно малая раз-
fl, ммоль/г
Рис. 4. Зависимость изостерических теп-
лот адсорбции ксенона от величины адсорб-
ции на цеолитах LiX, NaX и КХ.
ница между теплотами адсорбции
на различных цеолитах свидетель-
ствуют, вероятно, о высоком меж-
молекулярном взаимодействии де-
формирующихся молекул ксенона.
Несмотря на зто, и в случае ксе-
нона можно видеть повышение
теплот адсорбции в ряду от литие-
вой к калиевой форме цеолита
в области, где точность экспери-
мента позволяет это различить.
Из характера изотерм и из значе-
ний теплот адсорбции аргона,
криптона, ксенона и пентана [18]
вытекает, что при адсорбции не-
полярных адсорбатов литиевая
форма цеолитов типа X менее ак-
тивна, а калиевая и рубидиевая
формы более активны, чем на-
триевая; это является эксперимен-
тальным фактом. Бзррер и Стю-
арт [2] попытались рассчитать
энергию взаимодействия адсорби-
рованной молекулы с катионом и с анионным скелетом большой полости
и объяснить последовательность теплот адсорбции аргона. На основе
этих расчетов авторы считают, что в случае благородного газа
определяющей составляющей является дисперсионная энергия, которая
для лития, натрия и калия находится в прямой пропорциональной зави-
симости от теплот адсорбции на исследованных формах цеолита X. По-
ляризационная составляющая, по предположению Бэррера [2, 3], про-
является слабо и только при малых заполнениях объема.
При помощи формул, которыми пользовались Бэррер и Стюарт [2],
мы рассчитали энергию взаимодействия аргона, криптона и ксенона с ка-
тионом, окруженным шестью кислородными анионами. При этом уделя-
лось внимание определению последовательности энергий адсорбции,
Таблица 2
Значения постоянных, принятых при расчете энергии взаимодействия
Постоянные Ы+ Na+ К+ О2“ Аг Кг Хе
а • 1024, СМ3 0.029 0.180 0.840 3.89 1.63 2.48 4.00
X • 1030, см3 0.99 6.95 27.54 20.92 32.2 46.5 71.5
г, А 0.68 0.97 1.33 1.437 1.91 2.01 2.20
Примечание, а — коэффициент поляризуемости; у — диамагнитная восприимчивость;
г —радиус.
106
которые мы определили при помощи формул Леннарда—Джонеса и Кирк-
вуда—Миллера и формулы для потенциала поляризационных сил. Зна-
чении постоянных, которые мы применили, приведены в табл. 2. В табл. 3
приведены результаты расчета. В случае расчета поляризационной энер-
гии надо учитывать не только электростатическое поле катиона, но и рав-
нодействующую напряженностей всех заряженных частиц.
Таблица 3
Энергия взаимодействия аргона, криптона и ксенона с различными катионами
Гав Катион Потенциалы для индиви- дуального катиона Сумма потенциалов над модельной группой Теплота адсорбции (опытная) Q, ккал./моль
Ф -1- ф R ФР о1- ЕФ ЕФ
Li+ 0.07 6.00 1.25 10.03 2.7
Аг | Na+ 0.23 3.92 1.69 12.86 2.8
К+ 0.50 2.45 2.37 14.96 3.0
Li+ 0.09 7.84 1.89 16.70 3.8
Кг | Na+ 0.30 4.63 2.69 20.53 4.4
К+ 0.66 3.30 3.66 23.15 4.7
Li+ 0.09 9.60 1.89 30.84 5.1
Хе | Na+ 0.31 6.56 3.42 35.22 5.6
K + 0.74 4.25 4.67 37.57 5.7
Примечание. Ф^ — потенциал
Фр ~ потенциал поляризационных сил.
дисперсионных сил; Фо — потенциал отталкивания,
а
Рис. 5. Модельная группа катиона с
окружающими кислородными анионами
и адсорбированной молекулой М.
На рис. 5 изображена модель адсорбции. Размеры шестичленного окна
мы взяли из работы [16]. Поляризационная энергия рассчитывалась по
формуле
• 1 [ 6z I Т2
—фГй = v “е2 72" ’ cos ? ~ 7П ’
Z L т к J
где z — заряд кислорода, гт — ак-
туальное расстояние молекулы от
кислородов, гк — расстояние моле-
кулы от катиона при плотном кон-
такте. Для кислородного аниона яв-
ляется (по Брэггу) вероятным значе-
ние меньше 2 и больше 1 отрицатель-
ного заряда [17]. Расчет для данной
модели показал, что при величине за-
ряда кислорода больше 0.6 результи-
рующая энергия адсорбции с учетом
поляризационного эффекта совпадает
ио последовательности с эксперимен-
тальными теплотами адсорбции. Ис-
пользованная модель является, конечно, слишком простой, и нельзя по-
этому переоценивать даже качественное совпадение расчетных данных с
экспериментальными, но она все-таки служит нам в качестве иллюстрации
данной проблемы. Остается также и другое возможное объяснение по-
следовательности теплот адсорбции при помощи представления о недо-
ступности катионов для адсорбированных молекул. Кроме того, вопрос
усложняется и тем, что при температуре активации выше 400° С в цео-
лите находятся небольшие количества воды, СО и СО2, которые удаляются
при температурах выше 500° С.
107
Интересными являются также результаты изучения изменений эн-
тропий адсорбции криптона и ксенона. Энтропия была рассчитана по
методу Де Бура [15]. В качестве стандартного состояния служил газ при
температуре адсорбции и при давлении 1 атм.
На рис. 6 изображена зависимость энтропий адсорбции криптона
от величины адсорбции. Та же зависимость показана на рис. 7 для ксе-
Рис. 6. Изменение дифференциальной
энтропии адсорбции криптона в зависи-
мости от величины адсорбции на цеоли-
тах LiX, NaX и КХ.
Рис. 7. Изменение дифференциальной
энтропии адсорбции ксенона в зависи-
мости от величины адсорбции на цеоли-
тах LiX, NaX и КХ.
нона на исследованных цеолитах. На обеих диаграммах прерывистой
линией обозначено изменение энтропии, связанное с переходом моле-
кул из стандартного состояния в жидкое состояние. Как можно видеть,
адсорбированные молекулы как криптона, так и ксенона находятся в бо-
лее упорядоченном состоянии, чем в жидкости. Кроме того, видно, что
постепенное увеличение абсолютной величины AS с заполнением адсорб-
ционного пространства отвечает росту упорядоченности молекул в адсор-
бированном состоянии.
ЛИТЕРАТУРА
1. R. М. В а г г е г. Proceedings of the tenth Symposium of the Colston Research
Society. Bristol, 1958.
2. R. M. Barre r, W. I. Stuart, Proc. Roy. Soc., 1959, A249, 464.
3. R. M. В arrer, R. G. Bratt, J. Phys. Chem. Solids, 1959, 12, 130.
4. W. D. Breck, G. W. Eversole, R. M. Milton, J. Am. Chem. Soc., 1956,
78, 2338.
5. Г. В. Цицишвили, T. Г. Андроникашвили, веб.: Синтетические
цеолиты, Изд. АН СССР, 1962, 117.
6. И. Е. Ней мар к, А. И. Растрененко, В. П. Федоровская,
А. С. П л а ч и н д а, в сб.: Синтетические цеолиты, Изд. АН СССР, 1962, 46.
7. V. В о s а с е k, Coll. Czech. Chem. Commun. 1964, 29, 8, 1797.
8. P. J i r u, O. G r u b n e r, M. R a 1 e к. в сб.: Синтетические цеолиты, Изд. АН
СССР 1962 129
9. S. R о s s, Н. Clark, J. Am. Chem. Soc., 1954, 76, 4291.
10. G. J u r a, D. С r i d d 1 e, J. Phys. Chem., 1951, 55, 163.
11. M. M. Дубинин, в сб.: Поверхностные химические соединения и их роль
в явлениях адсорбции, М., 1957, 57.
12. Т. Hill, J. Chem. Phys. 1946, 14, 441.
13. А. В. Киселев, Колл, ж., 1958, 20, 338.
14. В. Босачек, Г. Грубнер, Е. Кучера, ДАН СССР, 1965, 164, 6, 637.
15. J. Н. D е Boer. The dynamical character of adsorption. Oxford, 1953.
16. L. В г о u s s a r d, D. P. Shoemaker, J. Am. Chem. Soc., 1960, 82, 1041.
17. Я. К. С ы p к и н, M. E. Д я т к и н а. Химическая связь и строение молекул.
М„ 1946.
18. О. М. Д ж и г и т, С. П. Ж д а н о в, К. Н. М и ко с, настоящий сборник,
стр. 46.
108
АДСОРБЦИЯ ПАРОВ ЦИКЛОГЕКСАНА,
БЕНЗОЛА И н-ПЕНТАНА НА ЦЕОЛИТАХ NaX и СаХ
Е. Ф. Полстяное, М. М. Дубинин
Цеолиты принадлежат к микропористым адсорбентам [1, 2]. Гетеро-
полярная природа кристаллов цеолитов и наличие в их полостях катио-
нов создают благоприятные условия для проявления дополнительных к ди-
сперсионному электростатических и специфических адсорбционных взаимо-
действий, к которым способны молекулы, обладающие неоднородным
распределением электронной плотности или кратными связями. Для мо-
лекул веществ с насыщенными валентностями и отсутствием полярных
групп, естественно, основным взаимодействием при их адсорбции на цео-
литах будет дисперсионное. В этом отношении цеолиты из-за однородно-
сти и строгой упорядоченности пористой структуры являются хорошими
объектами для исследования.
На основании непосредственных калориметрических измерений теп-
лоты адсорбции на микропористых адсорбентах для случая дисперсион-
ного взаимодействия, примерно в 1.5—2 раза превышают теплоты
адсорбции на крупнопористых или непористых адсорбентах (напри-
мер, сажах), имеющих одинаковую химическую природу. Этот эффект
может быть обоснован и теоретическим расчетом по схеме, данной в ра-
боте [3], но с учетом реальной пористой структуры адсорбента. Сле-
довательно, если считать, что теплота адсорбции на однородной плос-
кой поверхности не на много отличается от теплоты конденсации
вещества, то теплоты адсорбции веществ на цеолите, даже если будут от-
сутствовать специфические взаимодействия, должны значительно пре-
вышать теплоту конденсации адсорбируемого вещества.
Задачей данной работы являлось изучение адсорбции веществ, спо-
собных к специфическим воздействиям при адсорбции их на цеолитах
{на примере бензола), и веществ, адсорбирующихся в основном за счет
проявления дисперсионных взаимодействий (циклогексан, н-пентан).
В круг изучаемых вопросов входило рассмотрение адсорбционных рав-
новесий в широком интервале равновесных давлений и температур,
построение и анализ изостер адсорбции и, наконец, определение изостери-
ческих дифференциальных теплот адсорбции.
В практике адсорбционных исследований существуют различные ме-
тоды определения теплот адсорбции: прямые калориметрические опре-
деления, расчет по изостерам адсорбции, расчет по структурным кон-
стантам уравнений теории объемного заполнения, расчет по хромато-
граммам. Естественно, наиболее распространенным методом является
метод непосредственных калориметрических измерений. Однако этот ме-
тод требует достаточно сложной аппаратуры и ненадежен для измерения
тепловых эффектов процессов, длительных по времени. Так, например,
современные калориметры с постоянным теплообменом 14, 5] измеряют
теплоты, выделяющиеся за промежуток времени менее чем 3 часа. Если же
процесс установления адсорбционного равновесия значительно больше
указанного времени, то это, естественно, приводит к заниженным резуль-
татам измерения теплот. Более надежным в подобных случаях является
расчет дифференциальных теплот по равновесным изостерам.
1. Объектами исследования служили кристаллические цеолиты NaX
и СаХ, синтезированные в лабораторных условиях С. П. Ждановым.
Состав цеолита NaX 0.964 Na2O • А12О3- 2.96 SiO2, степень замещения иона
Na на ион Са у цеолита СаХ составляла более 60%. Адсорбируемые ве-
щества характеризовались высокой степенью чистоты.
109
Изотермы адсорбции при различных температурах в интервале от
О до 140° определялись по вакуумному методу сорбционных весов. При
калибровании установки особое внимание было уделено контролю темпе-
ратуры адсорбента и тща-
тельности установления
адсорбционного равнове
сия. Контроль адсорбцион-
ного равновесия осущест-
влялся тем, что после из-
мерения изотермы адсорб-
ции проверялось адсорб-
ционное равновесие в не-
скольких десорбционных
точках.
В качестве примера на
рис. 1 и 2 представлены
семейство изотерм адсорб-
ции циклогексана на цео-
лите NaX (рис. 1) и семей-
Рис. 1. Изотермы адсорбции паров циклогексана ство изотерм адсорбции
на цеолите NaX. бензола на цеолите NaX
(рис. 2). Изотермы построе-
ны в полулогарифмических координатах, что позволяет наиболее наглядно
представить адсорбцию во всем интервале равновесных давлений. На этих
графиках кружками обозначены адсорбционные точки, сплошными знач-
ками — десорбционные. Хорошее совпадение адсорбционной и десорб-
ционной ветвей изотермы
дает возможность сделать
вывод о том, что в дан-
ном случае имеет место фи-
зическая адсорбция и что
адсорбционное равновесие
во всех точках было об-
ратимым. Время установ-
ления адсорбционного рав-
новесия зависело от тем-
пературы (при которой
изучалась адсорбция) ад-
сорбента, адсорбируемого
вещества и изменялось от
6—7 часов, для опытов ад-
сорбции н-пентана и цик-
логексана на цеолите СаХ
в начальной области за-
а, ммоль/г
Рис. 2. Изотермы адсорбции паров бензола на цео-
лите NaX.
Iffp
полнения адсорбционного
объема (до а/а0=0.2), до
одного часа при предель-
ных заполнениях адсорб-
ционного объема. Таким образом, для начальной области заполнения
адсорбционного объема время установления адсорбционного равновесия
на цеолитах существенно превышало длительность обычных калоримет-
рических измерений.
Обращает на себя внимание своеобразная форма изотерм адсорбции
циклогексана (рис. 1). Заполнение адсорбционного объема происходит
в узком интервале равновесных давлений, а повышение температуры,
Мало изменяя форму изотермы, перемещает ее по шкале давлений в сто-
110
рону больших значений. Аналогичны по форме изотермы адсорбции
н-пентана на цеолите NaX; при адсорбции н-пентана на цеолите СаХ на-
чальной области заполнения адсорбционного объема (при одинаковых
равновесных давлениях паров ве-
ществ) соответствуют большие, чем
для цеолита NaX, величины адсорб-
ции. Следует также отметить особен-
ности адсорбции бензола по сравнению
с циклогексаном. Электронная неодно-
родность молекул бензола, обязанная
наличию л-связей в его электронном
облаке, сильно проявляется при его
адсорбции на цеолитах. Это проявле-
ние выражается и в форме изотерм ад-
сорбции. При температурах 20, 50°
для бензола в отличие от циклогекса-
на характерны пологие изотермы со
значительными величинами адсорбции
при малых равновесных давлениях.
И только при повышенных темпера-
турах (НО, 140°) изотермы становятся
более крутыми и похожими на изотер-
мы циклогексана.
2. Различие в адсорбируемости бен-
зола, с одной стороны, а циклогек-
Рис. 3. Изостеры адсорбции н-пентана
на цеолите NaX.
Величины о выражены в ммоль/г
проявляется также и в характере изостер
большинства адсорбционных систем изо-
стеры адсорбции в координатах ЦТ,
1g р обычно удовлетворительно ап-
проксимируются прямыми линиями в
довольно широком интервале темпе-
ратур; это свидетельствует о прак-
тической независимости изостериче-
ских теплот адсорбции от темпера-
туры в данном интервале температур.
В качестве такого примера могут слу-
жить изостеры адсорбции н-пентана
на цеолите NaX (рис. 3). Точки из-
остер адсорбции, вычисленные по изо-
термам, как видно из графика, хоро-
шо ложатся на прямые линии. Пря-
молинейность изостер наблюдается
также при адсорбции н-пентана на
цеолите СаХ и циклогексана на цео-
лите NaX. Если же в суммарном ад-
сорбционном взаимодействии имеет
место специфическая составляющая,
эффект которой зависит от ориента-
сана и я-пентана — с другой,
адсорбции. Известно, что для
Рис. 4. Изостеры адсорбции бензола на
цеолите NaX.
ции молекулы в поле силовых центров,
то это должно приводить к искрив-
лению изостер адсорбции. Подобным
примером является адсорбция бензола на цеолите NaX (рис. 4). Искрив-
ление таким образом изостер адсорбции наблюдается для более низких
температур и малых величин заполнения адсорбционного объема.
3. По изостерам адсорбции были вычислены дифференциальные изо-
стерические теплоты. На рис. 5 представлен график кривой измерения
ill
дифференциальных теплот адсорбции циклогексана на цеолите NaX в за-
висимости от величины адсорбции. Прерывистой линией L„ на графике
показана теплота конденсации циклогексана. Дифференциальные теплоты
адсорбции значительно превышают теплоту конденсации циклогексана.
Своеобразен также ход изменения дифференциальных теплот адсорбции.
Всю кривую теплот можно подразделить как бы на четыре участка: вна-
а, ммоль/г
чале теплоты постоянны, потом не-
сколько возрастают, затем возрастают
сильнее и, наконец, происходит спад
дифференциальных теплот. Расчеты
показали, что первому участку соот-
ветствует адсорбция, в среднем мень-
шая, чем соответствующая нахожде-
нию по одной молекуле из расчета
на одну большую полость, второму —
две, и т. д. Максимум на кривой
соответствует случаю, когда на каж-
дую полость приходится в среднем
три молекулы, т. е. тогда, когда мо-
Рис. 5. Изостерические теплоты адсорб- лекулы довольно-таки плотно распо-
ции циклогексана на цеолите NaX. ложены в полости и среднестатисти-
ческие расстояния между ними, а
также между ними и силовыми центрами полости сокращены. Этот эф-
фект повышения энергии по мере заполнения полостей адсорбированными
молекулами может быть сведен в расчетном смысле как бы к уменьшению
размеров полостей, что приводит к повышению дисперсионной составляю-
щей адсорбционного вза-
имодействия.
На рис. 6 изображены
кривые дифференциальных
теплот адсорбции н-пента-
на на цеолитах NaX и
СаХ. Кривая теплот ад-
сорбции н-пентана на цео-
лите NaX аналогична по
форме кривой циклогекса-
на на том же цеолите, но
для кривой теплот адсорб-
ции на цеолите СаХ ха-
рактерны для начального
заполнения адсорбцион- Рис- 6* Изостерические теплоты адсорбций «-пен-
ного объема более высо- тана на цеолитах NaX и СаХ-
кие теплоты адсорбции.
По-видимому, это связано с заменой иона Na на ион Са, что создает
большую энергетическую неоднородность адсорбционного пространства
цеолитов.
Полученные нами дифференциальные теплоты адсорбции н-пентана
в основном близки к тем величинам, которые получены в работе [7] по-
средством прямых калориметрических измерений. Некоторое различие
в теплотах наблюдается в начальной области заполнения адсорбционного
объема для случая адсорбции н-пентана на цеолите NaX. Понижение
теплот адсорбции в начальной области заполнения адсорбционного объема,
для которой, как нами уже упоминалось, время установления адсорб-
ционного равновесия значительно превышает временные возможности
обычных калориметрических измерений, может быть обязано недости-
жению равновесных состояний.
112
Теплоты адсорбции циклогексана и н-пентана вычислялись по тангенсу
угла наклона линейных изостер. Естественно, такой метод вычисления не
применим для искривленных изостер, как это имеет место при адсорбции
бензола на цеолите NaX (рис. 4), поэтому теплоты адсорбции бензола
вычислялись для каждого
интервала температур.
Рассчитанные изостериче-
ские теплоты адсорбции бен-
зола на цеолите NaX приве-
дены на рис. 7. Прерывисты-
ми кривыми на этом графике
показаны значения изостери-
ческих теплот, которые были
рассчитаны по эксперимен-
тальным данным и ошибка
определения которых превы-
шала 5% [6].
4. В литературе описана
адсорбция различных веществ
на цеолитах типа NaX, и для
многих из них характерен та-
Рис. 7. Изостерические теплоты адсорбции бен-
зола на цеолите NaX.
кой же ход изменения диффе-
ренциальных теплот адсорбции, как это имеет место в нашем случае при
адсорбции н-пентана и циклогексана на цеолите NaX (рис. 5 и 6). Такие же
кривые теплот адсорбции описаны в работах Бзррера и его сотрудников [7,
8], изучавших адсорбцию предельных углеводородов и их фторпроиз-
водных на цеолите типа NaX. Бэррером также отмечалось [7], что в го-
0s
------We ----------We oCFt •We
i_______I________I_______I_______I_______I________L.
0 0.1 0.2 0.3
o.-v,cm3Iz
мологическом ряду метана
возрастание максимума на
кривой теплот пропорцио-
нально возрастанию теплот
конденсации веществ. Если
же считать, что теплота кон-
денсации вещества опреде-
ляется в основном проявле-
нием сил той же природы,
что и при адсорбции, то мож-
но в первом приближении
предполагать о постоянстве от-
носительных дифференциаль-
ных теплот qlLs, которые для
микропористых адсорбентов
Рис. 8. Относительные дифференциальные теп-
лоты адсорбции и-пентана, циклогексана и фтор-
углеродов на цеолите NaX.
целесообразно представлять
в зависимости от заполненного
объема адсорбционного про-
странства. Относительные
дифференциальные теплоты адсорбции н-пентана и циклогексана изоб-
ражены на рис. 8. Точками на этом графике показаны отношения q/Ls
для трех гомологов фторуглеродов по данным Бэррера [8]. Отклонение
точек от кривых не превышает 10%. Описанная выше закономерность
также хорошо проявляется при адсорбции веществ на активных углях.
Таким образом, изучено адсорбционное равновесие паров циклоге-
ксана, н-пентана и бензола на цеолитах типа X в широком интервале
равновесных давлений и температур. Среди этих веществ адсорбция бен-
зола носит особый характер из-за способности молекул бензола к специ-
фическим взаимодействиям при их адсорбции на цеолитах.
. 8 Цеолиты
ИЗ
Обсуждение изостер адсорбции и дифференциальных теплот адсорбции
позволило отметить основные особенности адсорбции веществ при про-
явлении адсорбционных взаимодействий различной природы.
ЛИТЕРАТУРА
1. М. М. Дубинин, Изв. АН СССР, ОХН, 1962, 5, 760.
2. М. М. Д у бинин, С. П. Ж д а нов, Е. Г. Жуковой ая, К. О. Му рд-
м а а, Е. Ф. П о л с т я н о в, И. Е. С а к а в о в, Н. А. Ш и ш а к о в.
Изв. АН СССР, сер. хим., 1964, 9, 1573.
3. J. Н. D е Boer a. J. Gust ers, Z. phys. Chem., 1934, 25, 225.
4. H. H. А в г у л ь, Г. И. Б е р е з и н, А. В. К и с е л е в, И. А. Л ы г и н а,
Г. Г. М у т т и к, ЖФХ, 1957, 31, 1111.
5. Н. Н. А в г у л ь, А. В. К и с е л е в, А. А. Л о п а т к и н, И. А. Л ы г и н а,
М. В. С е р д о б о в, Колл, ж., 1963, 25, 2, 129.
6. О. М. Джигит, А. В. К и с е л е в, Г. Г. Муттик, Колл, ж., 1963, 25,
1, 34.
7. R. М. Barter, J. W. Sutherland, Proc. Roy. Soc., 1956, A 237, 439.
8. R. M. Barter, P. J. R e u с г о f t, Proc. Roy. Soc., 1960, 258.
АДСОРБЦИЯ ПАРОВ БЕНЗОЛА НА ЦЕОЛИТЕ NaX
О. Кадлец
В связи с вопросом о применимости теории объемного заполнения
микропористых адсорбентов к адсорбционным равновесиям на цеолитах
могут представить интерес результаты изучения температурной зависи-
мости изотерм адсорбции бензола на цеолите NaX.
Проведено измерение изотерм адсорбции бензола при помощи вакуум-
ной установки с кварцевыми весами типа весов Мак-Бена (чувствитель-
ность 2 мг/мм) в интервале температур 25—130° на гранулированном цео-
лите NaX (13Х — фирмы Линде), и на основании полученных результа-
тов проверена применимость теории объемного наполнения путем построе-
ния характеристических кривых. Все характеристические кривые в коор-
динатах е——RTinh (где h — относительное давление) и W=av (а — ад-
сорбция, v — мольный объем адсорбата) ложатся в пределах точности
измерений на одну кривую (рис. 1). Этот результат позволяет считать,
что в рассматриваемом случае хорошо выполняется основной постулат
теории (^г\ = 0, и при этом коэффициент объемного расширения а
бензола в адсорбированном состоянии мало отличается от той же ве-
личины для объемной фазы, а величина —RTlnh хорошо выражает диф-
ференциальную мольную работу адсорбции. В таком случае оправды-
вается применение уравнений Б. П. Беринга и В. В. Серпинского [1]
для вычисления теплот и энтропий адсорбции по одной изотерме или
по характеристической кривой. Уравнения Беринга и Серпинского для
чистой теплоты адсорбции
(д In h\
q = Q-^aRT^^T-RTiah (1)
и для дифференциальной энтропии адсорбции
/д In h\
= <2>
можно представить в форме, удобной для вычислений по характеристиче-
ской кривой:
д = е-к = -аЛг(^-)г+в, (3)
hS = aw(^\„. (4)
\(7l4 /Т
Зависимость дифференциальных теплот адсорбции Q, вычисленных
по уравнению (3), от величины заполнения адсорбционного пространства
при 20° приведена на рис. 2 (2). На этом же рисунке для сравнения при-
веден ход дифференциальных теплот адсорбции бензола на цеолите NaX
по калориметрическим измерениям при 20° (2). Из рисунка видно, что эти
данные в основных чертах согласуются между собой. Эту согласованность
также можно считать доказательством хорошего выполнения постулатов тео-
рии объемного заполнения микропор. Не исключено, что небольшие разли-
чия кривых дифференциальных теплот адсорбции, полученных калори-
метрически и вычисленных по характеристической кривой (превышающие
10%), вызваны медленностью установления адсорбционного равновесия
бензола на цеолите NaX при комнатных температурах, при которых про-
водились калориметрические измерения. В пользу этого предположения
8*
115.
свидетельствует тот факт, что адсорбционные и десорбционные точки
изотермы бензола на исследованном образце цеолита NaX, измеренные
при давлениях ниже 10~2 мм, заметно расходятся, как это видно из рис. 3,
на котором изображены в
полулогарифмических коор-
динатах адсорбционные и де-
сорбционные точки изотермы
адсорбции бензола при 20°.
Следует подчеркнуть, что в
каждом опыте наблюдение за
установлением равновесия
длилось не менее 8 часов, а
в отдельных случаях — даже
несколько дней. Этот своеоб-
разный гистерезис, очевид-
но, вызван очень малой ско-
ростью установления адсорб-
Рис. 2. Зависимость дифференциальной теплоты
адсорбции бензола при 20° от заполнения адсорб-
ционного пространства.
Кривая 1 — вычислена по характеристической кривой
бензола на цеолите NaX (Линде); кривая 2 — резуль-
тат калориметрических измерений на кристаллах цео-
лита NaX. Условные обозначения (кружки и крестики)
на кривой 2 обозначают различные серии опытов.
ционного равновесия при
низких температурах, так
как при более высоких темпе-
ратурах (80°) он полностью
исчезал. На рис. 3 сплош-
ной линией изображены ве-
личины адсорбции при 25°,
вычисленные по характери-
стической кривой. .Интересно, что десорбционные точки изотермы прак-
тически совпадают с этой кривой. Поэтому адсорбционные точки изотер-
мы, измеренные при комнатных температурах, в случае бензола на цео-
лите NaX нельзя считать равно-
Рис. 3. Адсорбционные и десорбционные точ- Рис. 4. Зависимость дифференциаль-
ки изотермы бензола при 25° в полулогариф- ной энтропии адсорбции при 20°
мических координатах. от заполнения адсорбционного про-
Сеетлые кружки — адсорбция, черные — десорбция. странства, вычисленная по харак-
теристической кривой.
Таким образом, нам кажется, что адсорбционный метод, позволяющий
проводить измерения при более высоких температурах, может давать в не-
которых случаях более правильную зависимость дифференциальных
116
теплот адсорбции от заполнения, чем калориметрический метод, в котором
измерения проводятся при комнатных температурах.
Ход дифференциальных энтропий адсорбции, вычисленных по харак-
теристической кривой при помощи уравнения (4), приведен на рис. 4.
Полученная кривая свидетельствует о растущем торможении поступа-
тельного и вращательного движений молекул бензола при постепенном
заполнении адсорбционного пространства.
ЛИТЕРАТУРА
1. Б. П. Б е р и н г, В. В. С е р и и н с к и й, ДАН СССР, 1957, 114, 1254.
2. Н. Н. А в г у л ь, А. В. К и с е л е в, А. А. Л о п а т к и н, И. А. Л ы г и и а,
М. В. С е р д о б о в, Колл, ж., 1963, 25, 129.
УДЕЛЬНАЯ ПОВЕРХНОСТЬ ВТОРИЧНЫХ ПОР
СИНТЕТИЧЕСКИХ ЦЕОЛИТОВ
А. И. Саратов, М. М. Дубинин, Ю. Ф. Березкина
Синтетические цеолиты находят практическое применение, как пра-
вило, в виде формованных гранул, таблеток или шариков. В таком виде
они, кроме собственно кристаллов цеолитов, имеющих размер несколько
микрон, содержат обычно связующее вещество, скрепляющее кристаллы
между собой. Таким образом, пористая структура цеолита образована
как структурой собственно цеолита (полости в кристаллах), называемой
первичной пористостью, так и зазорами между кристаллами, частично
заполненными связующим веществом, — структура, называемая вторич-
ной пористостью.
Первичная пористая структура цеолитов, являющаяся основной по
своему значению, определяет как адсорбционные^ так и селективные свой-
ства этих сорбентов. Вторичная пористая структура играет положитель-
ную роль как транспортная система, позволяющая адсорбируемым веще-
ствам быстро проникать в глубину гранул. В остальном ее наличие яв-
ляется вредным. Во-первых, вторичная пористая структура уменьшает
кажущийся вес формованных цеолитов и тем самым увеличивает объем
аппаратуры, использующей цеолит. Во-вторых, а это самый главный недо-
статок этой структуры, поры ее образуют неселективпую адсорбционную
поверхность, так как они доступны всем размерам молекул. Следовательно,
для того чтобы существенно не снизилось селективное действие формован-
ных цеолитов, необходимо, чтобы неселективная адсорбция во вторичных
порах была бы существенно меньше величины адсорбции в полостях цео-
литов, т. е. в первичных порах. Иными словами, удельная поверхность
вторичных пор формованных цеолитов не должна быть больше некоторой
сравнительно малой величины.
Удельная поверхность вторичных пор формованных цеолитов склады-
вается из наружной поверхности собственно кристаллов цеолитов и из
поверхности связующего вещества, которое частично закрывает собой
поверхность кристаллов. Удельная поверхность собственно кристаллов
при современной технологии производства цеолитов является величиной
сравнительно постоянной, так как определяется в основном размерами
кристаллов. Величина зтой поверхности для продукта нормального ка-
чества составляет обычно несколько квадратных метров на грамм дегидра-
тированного цеолита. При такой малой внешней поверхности кристаллов
неселективная адсорбция на их наружной поверхности не превышает
117
1% от адсорбции внутри полостей кристаллов. Следовательно, наружная
поверхность кристаллов хотя и несколько понижает селективную способ-
ность формованных цеолитов, но ее влияние достаточно мало. Иная кар-
тина получается с поверхностью связующего вещества, так как в зависи-
мости от типа связующего, его количества величина ее может доходить
до 20—30 м2/г дегидратированного формованного цеолита, что составит
в пересчете на адсорбцию 2—4% от общей адсорбционной емкости гра-
нул. В этом случае формованный цеолит существенно ухудшается как
сорбент с селективными адсорбционными свойствами.
Таким образом, удельная поверхность вторичной пористой структуры
формованных цеолитов является весьма важным параметром, определяю-
щим неселективную адсорбционную способность гранул. Однако до на-
стоящего времени этот важный параметр почти не исследовался, а его
определение не являлось обязательным при оценке формованных цеолитов
промышленного изготовления. Последнее в значительной мере объяс-
няется сложностью методов определения сравнительно малой удельной
поверхности вторичных пор.
Целью настоящей работы является попытка правильно оценить вклад
поверхности, образованной связующим, в общую удельную поверхность
формованных цеолитов и дать некоторые практические рекомендации
для упрощенных и быстрых методов определения удельной поверхности
вторичных пор или самого связующего.
Удельная поверхность вторичных пор для единицы массы полностью
дегидратированного формованного цеолита может быть определена тремя
методами: а) вдавливанием ртути, б) капиллярной конденсацией паров и
в) по изотерме адсорбции БЭТ. Первые два метода требуют точного знания
формы и размера пор, а так как это невозможно, то на практике прибегают
к упрощающим допущениям о форме пор, что, естественно, сказывается
очень сильно на получаемых результатах и может приводить к ошибкам
в 1.5—3 раза. Третий метод, адсорбционный, не требует представлений
о форме пор, но тем не менее по точности не намного лучше первых двух,
так как требует применения плохо нам известной величины — молекуляр-
ной площадки адсорбата. Однако вследствие того, что сами цеолиты и свя-
зующие представляют собой дегидратированные в стандартных условиях
алюмосиликаты, метод БЭТ будет давать удовлетворительную точность
при сравнении различных цеолитов, хотя абсолютная точность определе-
ния самой удельной поверхности может быть также невелика. Главным
недостатком всех этих трех методов является аппаратурная сложность и
длительность измерений, что делает практически невозможным их при-
менение для серийных анализов в промышленных условиях.
Экспериментальная часть
В наших исследованиях вторичной пористой структуры мы пользова-
лись адсорбционным методом БЭТ определения удельной поверхности,
используя для этого высокочувствительные вакуумные весы (чувствитель-
ность около 4 • 10 ~7 при нагрузке 1 г) и прецизионную установку объем-
ного метода.
Объектами исследования были кристаллические и гранулированные
цеолиты типа А отечественного производства и отдельно связующие ве-
щества, применявшиеся для гранулирования — глуховская и латнен-
ская глины и окись алюминия. Глины перед исследованием формовались
в гранулы диаметром 4 мм и длиной 5 мм и обжигались при 600° в течение
4 часов. Окись алюминия, полученная от Т. Г. Плаченова уже прокаленной
при 500°, подвергалась лишь прогреву до 400° в вакууме для удаления
всех поглощенных газов и паров.
118
В качестве адсорбируемого вещества для кристаллических и гранули-
рованных цеолитов служил бензол, не проникающий в полости цеолитов
типа А, а для связующих веществ — азот при его температуре кипения.
Полученные изотермы адсорбции обрабатывались по методу БЭТ для
получения величин удельных поверхностей. Результаты этих вычислений
приведены в табл. 1. Удельные поверхности были вычислены при условии,
„Таблица 1
Результаты применения уравнения БЭТ к адсорбции паров бензола
Цеолит ат, ммоль/г с Интервал применимости Р/Ра S, М!/Г
Кристаллические цеолиты
NaA I 0.0126 88 0.04—0.25 3.6
NaA II 0.0072 77 0.02—0.30 2.1
NaA ГОБ 0.0162 180 0.01—0.18 4.7
СаА ГКЗ 0.0161 21 0.03—0.38 4.7
СаА Жданова 0.0228 120 0.02—0.28 6.6
СаА ГОБ 0.0242 54 0.02—0.30 7.0
Гранулированные цеолиты
NaA I 0.0319 18 0.04—0.18 9.2
NaA II 0.1446 28 0.03—0.22 42.0
СаА ГКЗ 0.0253 23 0.02—0.28 7.3
что молекулярная площадка бензола <в=48 А2. В формованных цеолитах
NaA I связующим была глуховская глина, NaA II — окись алюминия и
СаА ГКЗ — латненская глина.
Данные по адсорбции азота на связующих при —195°, выраженные
в константах уравнения БЭТ, приведены в табл. 2. Молекулярная пло-
щадка азота принималась 16.2 А2.
Таблица 2
Результаты применения уравнений БЭТ в адсорбции азота
Свявующее ммоль/г С Интервал применимости pipe S, м’/г
Глуховская глина 0.615 74 0.04—0.25 60
Латненская глина 0.393 160 0.01—0.33 38
Окись алюминия 1.63 150 0.01—0.35 160
Обсуждение результатов опытов
Из приведенных данных по адсорбции бензола видно, что величина
удельной поверхности вторичных пор формованных цеолитов нормаль-
ного качества, к которым относятся цеолиты NaA I и СаА ГКЗ, составляет
величину, не превышающую 10 м2/г а для цеолита NaA II, в котором на-
меренно использовано связующее с большой удельной поверхностью,
а кристаллы — с малой наружной поверхностью, эта величина уже равна
42 м2/г. В то же время наружная поверхность кристаллов, из которых
сформованы эти цеолиты, имеет величину, не превышающую 5 м2/г, а для
цеолитов СаА — 7 м2/г. Следовательно, в формованных цеолитах нормаль-
ного качества величина поверхности вторичных пор, образованная свя-
зующим веществом, очень близка к наружной поверхности самих кри-
сталлов.
119
Для того чтобы выяснить, можно ли определить удельную поверхность
вторичных пор формованных цеолитов, исходя из наружной поверхности
собственно кристаллов (считая ее постоянной для всех марок цеолитов)
и из удельной поверхности чистого связующего, мы проделали следующие
расчеты: зная долю связующего в гранулах а, удельную поверхность
формованного продукта S$ и считая (в первом приближении), что по-
верхность вторичных пор состоит из свободной наружной поверхности
кристаллов Sk и поверхности связующего Sc, можно написать:
S(P = (i— а) Sk-\-а • Sc, (1)
откуда
Sc =------------ (2)
Результаты этих вычислений приведены в табл. 3, в которую для срав-
нения внесены значения удельных поверхностей связующих, измеренные
Таблица 3
Удельная поверхность связующих, полученная разными
методами
Связующее S, м2/г
вычислено по формуле (2) по адсорбпии азота
Глуховская глина 32 60
Латненская глина 18 38
Окись алюминия 286 160
по азоту. Сравнения показывают, что поверхность глуховской и латнен-
ской глин в формованных цеолитах приблизительно в два раза меньше
поверхности этих же связующих, взятых в чистом виде. Исключение
составляет окись алюминия, для которой поверхность по азоту почти
в два раза меньше, чем поверхность, вычисленная по схеме аддитивности,
что, по-видимому, объясняется различием в процессе образования твер-
дых осадков. Окись алюминия вносилась в цеолит в виде водного раствора
оксинитрата алюминия, производилось гранулирование, а затем прокали-
вание гранул. Для получения чистой окиси алюминия этот же раствор
оксинитрата алюминия выпаривался, а осадок прокаливался. Поэтому
удельная поверхность во втором случае могла получиться меньше.
Таким образом, для того чтобы оценить поверхность вторичных пор
гранулированных цеолитов с точностью до 20—50 %, что для практических
целей вполне достаточно, нужно определить лишь удельную поверхность
чистого связующего после соответствующей его обработки. Наружную
поверхность кристаллов можно не учитывать совсем, так как она в зна-
чительной мере закрыта связующим веществом. Тогда удельную поверх-
ность вторичных пор формованных цеолитов можно выразить как
Зф=8с-а.
Для исследованных нами цеолитов такие вычисления дают для NaA I —
5^=12 м2/г, NaA II — 5дз=22.4 м2/г и СаА ГКЗ S$=7.6 м2/г, т. е., если
исключить нехарактерный цеолит с очень пористым связующим NaA II,
то отклонение получается для NaA I 30%, а для СаА ГКЗ 4%.
Так как этот метод не претендует на высокую точность, то и само опре-
деление удельной поверхности связующего может проводиться упрощенно.
•120
Для этого вполне достаточно измерить адсорбцию азота при —195° в од-
ной точке — при относительном давлении около 0.14-0.15, т. е. при
~ 100 мм рт. ст. Это можно делать на том основании, что для подавляю-
щего большинства непористых или крупнопористых адсорбентов точка ат,
соответствующая заполненному мономолекулярному слою, лежит на поло-
гом участке изотермы и не выходит по относительному давлению за пре-
делы 0.1 4-0.17. При таком методе точность определения константы урав-
нения БЭТ ат не меньше 10—15%. Аппаратурное оформление такого
метода будет также очень просто: потребуется наличие жидкого азота,
форвакуумный насос, ртутный манометр и газовая бюретка.
ЭЛЕКТРОННО-МИКРОСКОПИЧЕСКОЕ ИССЛЕДОВАНИЕ
СИНТЕТИЧЕСКИХ ЦЕОЛИТОВ МЕТОДОМ РЕПЛИК
В. М. Лукьянович, Э. И. Евко, В. В. Чураков
Введение
Целью данного сообщения является краткое подведение итогов и не-
которая оценка перспективы применения электронной микроскопии в об-
ласти исследования синтетических цеолитов.
Достигнутые здесь до последнего времени результаты были весьма
скромны. Обычно при помощи электронного микроскопа определяли
размер и форму кристаллов цеолитов в порошкообразных препаратах
и в растворах после кристаллизации [1, 2]. Применение с этой целы»
электронной микроскопии является целесообразным: кристаллы нередка
имеют размеры порядка единиц и десятых долей микрона, так что воз-
можности световой микроскопии в определении формы и размеров кри-
сталлов цеолитов весьма невелики.
Однако прямое исследование в электронном микроскопе кристаллов
цеолитов дает лишь ограниченную информацию об их морфологии и раз-
мерах, так как удается наблюдать лишь силуэты частиц. Поэтому мелкие
кристаллы, прилипшие к более крупным и находящиеся выше или ниже
последних, вообще могут быть не замечены на микрофотографиях при
подсчете частиц и кривые распределения кристаллов по размерам, полу-
ченные на основе таких данных, будут искажены — сдвинуты в сторону
больших размеров. Очевидно также, что в случае неправильной формы
кристаллов (например, при значительном отклонении их формы от наи-
более распространенной — кубической) и произвольной ориентации кри-
сталлов на пленке-подложке судить об их форме на основании наблюдаемых
силуэтов весьма затруднительно.
По-видимому, эти обстоятельства были причиной столь ограниченного
применения электронной микроскопии для исследования цеолитов. Зна-
чительно расширяет возможности электронной микроскопии в этом отно-
шении метод реплик. Изучение в микроскопе не самих кристаллов, но
тонких пленок-отпечатков с них не только позволяет исключить указан-
ные выше недостатки, но и дает возможность включить в круг объектов
исследования более массивные образцы — гранулированные цеолиты.
Методика
Известно немало различных вариантов изготовления реплик [3].
Нами были отработаны следующие способы получения наиболее распро-
страненных углеродных реплик.
121
1. Получение реплик с порошкообразных ^препаратов.
На стеклянную пластинку наносят каплю 1 %-го раствора коллодия в амил-
ацетате. Наклоном пластинки капле дают стечь и высохнуть следу. На
это место помещают каплю водной суспензии цеолита такой концентрации,
чтобы после испарения воды коллодиевая пленка была покрыта плотным
слоем частиц. Затем кристаллы покрывают слоем распыленного в вакууме
углерода толщиной 200—300 А. Поверх всего наносят несколько капель
нагретого концентрированного раствора желатины. После застывания
раствора желатиновую пленку вместе с захваченным ею препаратом от-
деляют от стекла, желатину растворяют в горячей воде, коллодиево-угле-
родную пленку промывают и переносят в смесь концентрированных соля-
ной и плавиковой кислот (1 : 1) для растворения цеолитов. Промытые
в воде пленки вылавливают на сетку, высушивают, растворяют коллодие-
вую пленку в ацетоне и оставшуюся углеродную реплику оттеняют хро-
мом или платиной. Успех всей этой довольно сложной операции в значи-
тельной степени обусловлен тем, что почти во все время препарирования
хрупкая углеродная реплика укреплена коллодиевой пленкой.
2. Получение реплик с гранулированных цеолитов.
Гранулу раскалывают, и реплику получают одним из следующих двух
способов:
а) На скол гранулы в вакууме напыляют слой углерода, укрепляют
его парафином, и гранулу помещают в смесь кислот до растворения цео-
лита, на что, в зависимости от свойств гранулы, требуется от 3 часов до
1 суток. Отделившуюся углеродную пленку с парафиновым слоем промы-
вают в воде, вылавливают на сетку так, чтобы парафиновый слой был
внизу, высушивают, и растворяют парафин в толуоле. После оттенения
металлом препарат готов для просмотра в микроскопе.
б) На скол гранулы под углом 30° напыляют тонкий слой платины,
затем углерода (предварительно оттененная реплика). Гранулу полно-
стью растворяют в смеси кислот, реплику переносят в подкисленную со-
ляной кислотой воду и промывают затем в чистой воде. Реплику вылав-
ливают на сетку и, не давая ей высохнуть, быстро промывают в спирте и
эфире. Последняя операция требуется для замены воды на жидкость с не-
большим поверхностным натяжением, иначе реплика при высыхании
будет разорвана силами поверхностного натяжения.
Из двух приведенных вариантов, являющихся в общем вполне удо-
влетворительными, предпочтение следует отдать первому: работа с укреп-
ленными репликами дает почти 100 %-й выход годной продукции, тогда
как препарирование по второму способу нередко осложняется разрывом
.реплик.
Полученные результаты и их обсуждение
Необходимо подчеркнуть, что из нескольких сотен полученных нами
микрофотографий ниже приведены лишь типичные, которые следует
считать скорее иллюстрациями, чем доказательством. Выводы делались
нами на основании совокупности материала с широким использованием
стереоскопических снимков.
На рис. 1 показана углеродная реплика с кристаллов цеолита NaX.1
Углеродные реплики-чехлы хорошо воспроизводят форму кристаллов.
В то же время они достаточно прозрачны для того, чтобы отчетливо раз-
1 В виде исключения зта реплика получена в самом электронном микроскопе.
Известно, что при просмотре в микроскопе препараты покрываются углеродистым слоем
благодаря крекингу паров углеводородов, имеющихся в приборе. Покрытый таким
слоем после получасового просмотра препарат был обработан кислотами для раство-
рения цеолита, затем снова помещен в микроскоп и сфотографирован.
122
дичать мелкие кристаллы на фоне более крупных. На некоторых снимках
удается видеть ступени роста, параллельные ребрам кристаллов. Преиму-
Рис. 1. Углеродная реплика с кристаллов цеолита.
тцество метода реплик перед непосредственным исследованием кристаллов
убедительно доказывается подобными снимками. Изучение стереоснимков
Рис. 2. Предварительно оттененная платиной углеродная репли-
ка с глины, применяемой в качестве связующего при формиро-
вании гранул цеолитов.
реплик, полученных различными способами, убеждает в том, что углерод-
ные чехлы со всех сторон воспроизводят поверхность кристаллов. Это
123
свидетельствует, по всей видимости, о значительной миграции наносимых
атомор углерода по поверхности кристаллов.
На рис. 2 приведена микрофотография реплики с поверхности куска
глины, применяемой в качестве связующего при получении гранул цео-
Рис. 3. Предварительно оттененная платиной углеродная
реплика с поверхности скола гранулы цеолита.
а — с малым увеличением; б —с большим увеличением.
литов. Наблюдаемая картина довольно типична для глин: плоские час
тицы местами образуют сравнительно гладкие участки, местами сцеплены
в агрегаты.
На рис. 3, а, б при разных увеличениях показаны реплики с поверх-
ности скола гранул цеолита NaA. На снимке с малым увеличением (рис. 3, а)
видно множество беспорядочно расположенных одиночных кристаллов
124
приблизительно кубической формы с преобладающим размером 2—4 мк.
На снимке с большим увеличением (рис. 3, б) хорошо заметно, что кри-
Рис. 4. Предварительно оттененная платиной углеродная репли-
ка с гранулы цеолита, полученной вибросмешением.
сталлы равномерно покрыты какой-то пленкой, сглаживающей промежутки
между кристаллами и содержащей образования пластинчатой формы.
Рис. 5. Предварительно оттененная платиной углеродная реп-
лика с гранулы французского цеолита К-10 фирмы СЕСА.
Поскольку ничего подобного не наблюдается на репликах с чистых кри-
сталлов, имеются все основания считать эту пленку за слой глины, вве-
денной при формовании цеолитов. Примерно такая же картина наблю-
125
дается и в том случае, если гранулы с такими же кристаллами NaA и
такой же глиной получают при помощи вибросмешивания (рис. 4).2 Оче-
видно, при вибросмешении не происходит сколько-нибудь заметного дроб-
ления кристаллов, не заметно отличия и в распределении глины по срав-
нению с обычным способом приготовления гранул.
Нами было изучено также несколько образцов зарубежных гранули-
рованных цеолитов. На рис. 5 показана реплика с французского цеолита,
изготовленного по патенту Линде и характеризующегося повышенной
механической прочностью. По размеру кристаллов этот образец не отли-
Рис. 6. Углеродная реплика со скола гранулы цеолита США
4A-XW фирмы Линде. Оттенена платиной.
чается от рассмотренных выше. Поверхность кристаллов не гладкая7и,
по-видимому, покрыта слоем какого-то вещества, по виду не похожего
на слой глины, вводимой в отечественные цеолиты. Интересным оказался
американский цеолит 4A-XW фирмы Линде (рис. 6), гранулы которого
характеризуются высокой прочностью и небольшим объемом вторичной
пористости (0.16 см3/г). Прежде всего обращает на себя внимание тог
факт, что кристаллы здесь примерно на порядок меньше по размерам,
чем в ранее рассмотренных образцах, и в то же время отличаются широким
распределением по размерам. Кристаллы имеют далеко не завершенную
кубическую форму, содержат’много фигур и ступеней роста. Слоя связую-
щего материала на кристаллах не заметно. Местами встречаются значи-
тельные по площади гладкие участки, причина появления которых оста-
ется неясной.
Характерную столбчатую или пластинчатую форму имеют кристаллы
морденита — одного из самых богатых кремнеземом алюмосиликатов
(рис. 7, 8).3 Реплики хорошо воспроизводят целые колонии этих кристал-
лов. Такие же столбчатые структуры были нами обнаружены в гранулах
молекулярного сита «цеолон», изготовленного на основе синтетического
морденита фирмой США Нортон (рис. 9).
2 Этот образец гранулированного цеолита бы получен А. И. Сараховым.
3 Образец морденита приготовлен Э. Э. Сендеровым [4].
126
В заключение уместно сказать несколько слов о перспективах приме-
нения электронной микроскопии для исследования цеолитов.
Метод реплик позволяет надежно определять форму и размеры кри-
сталлов в порошкообразных препаратах и на сколах гранул. Следова-
Рис. 7, 8. Углеродная реплика с кристаллов морденита. От-
тенена платиной.
тельно, теперь появилась возможность получать достоверные кривые рас-
пределения кристаллов по размерам, что открывает пути проведения
серьезных электронно-микроскопических исследований. Можно ожидать,
например, что тщательное определение дисперсности частиц на последо-
вательных стадиях процесса кристаллизации позволит получить ценные
127'
сведения о кинетике и механике роста кристаллов и тем самым поможет
сознательно управлять процессом кристаллизации [3].
Более сложная и отдаленная проблема — визуализация отверстий пор
и самих адсорбирующих пор в цеолитах. Хотя разрешающая способность
Рис. 9. Углеродная реплика со скола гранулы цеолита «цеолон»,
фирма Нортон, США.
современных микроскопов доходит до 5 А, вряд ли целесообразно пытаться
непосредственно увидеть эти поры. Вероятно, более эффективен здесь
будет метод декорирования — «проявление» пор посредством осаждения
на них частиц тяжелых металлов. Этот метод позволяет обнаруживать на
обычных кристаллах дефекты атомного масштаба, и следует ожидать,
что применение его к исследованию цеолитов будет оправдано.
Выводы
1. Отработана методика получения углеродных реплик с порошкооб-
разных и гранулированных синтетических цеолитов.
2. При помощи метода реплик проведено исследование ряда отече-
ственных и зарубежных цеолитов и отмечены особенности их строения.
ЛИТЕРАТУРА
1. М.М. Дубинин, Е. Д. 3 а в е п и н а, В. М. Лукьянович, Н. П. Хар-
ла м о в, Изв. АН СССР, ОХН, 1961, 8, 1380.
2. С. А. Л евин а, Н. И. П л ющевский, Н. Ф. Ермоленко, ДАН БССР,
1962, 6, 8.
.3. В. М. Лукьянович. Электронная микроскопия в физико-химических иссле-
дованиях. Изд. АН СССР, 1960, 90, 133.
-4. Э. Э. С е н д е р о в, Геохимия, 1963, 9, 820.
ПОЛУЧЕНИЕ ЦЕОЛИТОВ
ХИМИЧЕСКИЕ АСПЕКТЫ
ПРОЦЕССОВ КРИСТАЛЛИЗАЦИИ СИНТЕТИЧЕСКИХ ЦЕОЛИТОВ
С. П. Жданов, Н. Н. Самулевич, Е. Н. Егорова
Со времени выхода первой работы Бэррера по гидротермальному син-
тезу цеолитов [1] прошло уже более 15 лет. С тех пор были получены и
описаны многие типы синтетических цеолитов [2—11], некоторые из ко-
торых обладают рядом ценных свойств и нашли уже широкое практиче-
ское применение. Количество публикаций, посвященных исследованиям
структуры и свойств синтетических цеолитов и вопросам их практического
использования, и интерес к этим материалам возрастают с каждым годом.
Но нельзя не обратить внимания на крайне ограниченное количество ра-
бот, посвященных вопросам синтеза цеолитов, физико-химическим и хими-
ческим аспектам процессов кристаллизации цеолитов. В настоящей ра-
боте делается попытка обобщения результатов исследований условий кри-
сталлизации цеолитов в системе Na20—А12О3—SiO2—Н2О, ведущихся
в лаборатории силикатных сорбентов Института химии силикатов
им. И. В. Гребенщикова АН СССР, и их анализ с точки зрения химии
процессов кристаллизации. Вопросы термодинамики процессов кристал-
лизации цеолитов должны быть предметом специальных исследований и
не рассматриваются в этой работе. В работе приводятся также некоторые
результаты исследований процессов кристаллизации калиевых и сме-
шанных цеолитов, выполйенных в нашей лаборатории.
Поля кристаллизации натриевых цеолитов на диаграммах
составов Исходных гелей
Синтетические цеолиты чаще всего получаются путем кристаллизации
щелочных силикаалюмогелей при определенных температурах. При этом
в зависимости от составов исходных гелей из них преимущественно кри-
сталлизуются цеолиты того или иного типа. Для того чтобы выяснить,
как влияет состав исходных гелей на направление процесса кристал-
лизации, необходимо исследовать поля кристаллизации цеолитов раз-
ных типов на диаграммах составов. Для четырехкомпонентной системы
Na2O—А12О3—SiO2—Н2О удобнее такие исследования проводить при по-
стоянной температуре. Полное выражение состава такой системы может
быть сделано лишь на пространственных диаграммах, например при исполь-
зовании для этого тетраэдра. Однако графическое изображение областей
кристаллизации на таких пространственных диаграммах затруднительно.
Значительно более простыми, а вместе с тем и вполне корректными и на-
глядными являются изображения полей кристаллизации на треуголь-
9 Цеолиты
129
Рис.
Поля кристаллизации цеолитов в
Na2O—А12О3—SiO2—Н2О на тре-
ных сечениях тетраэдра плоскостями, параллельными одной из его гр а-
ней (рис. 1). Удобно пользоваться диаграммами составов, отвечающими
постоянным содержаниям НгО в этой системе [12]. Каждая точка внутри
треугольника будет точно характеризовать содержание трех других ком-
понентов системы.
На рис. 2 и 3 приведены два
таких треугольника составов на-
триевых силикаалюмогелей с на-
несенными на них полями кристал-
лизации цеолитов разных типов,
получающихся из этих гелей
при 90°.
Для построения таких полей
были использованы результаты
многочисленных синтезов цеоли-
тов из натриевых силикаалюмо-
гелей во всей области составов,
где реально осуществимо их по-
лучение из смесей щелочных рас-
творов силиката и алюмината на-
трия.1 Каждое из полей ограни-
. „ чивает область составов гелей, в
1. Схема выражения составов четы- - _
рехкомпонентной системы. пределах которой данный тип цео-
лита кристаллизуется либо в чи-
стом виде, либо преимущественно
с незначительными примесями кристаллов других цеолитов. Наличие дру-
гих фаз в продуктах кристаллизации в значительных количествах наблю-
дается обычно лишь в областях составов, прилегающих к границам полей.
Существенно, что каждый из ти-
пов цеолитов в пределах своего
поля кристаллизуется вполне
устойчиво и воспроизводимо
и что чаще всего выдерживание
кристаллов в контакте с маточ-
ным раствором не приводит
к фазовым превращениям. Эти
обстоятельства свидетельствуют
о том, что получающиеся кри-
сталлы могут длительно суще-
ствовать в контакте с раство-
ром при данной температуре
в пределах своего поля кристал-
лизации и могут быть выде-
лены из растворов, но, вообще
говоря, они не всегда являются
истинно равновесными фазами.
Случаи образования метаста-
бильных цеолитных фаз и их
перекристаллизации при дли-
тельном выдерживании были
ранее отмечены [4, 13, 14].
На диаграммах (рис. 2 и 3) по-
казаны поля кристаллизации первичных фаз. Условия образования рав-
Рис. 2.
системе
угольной диаграмме при постоянном содержа-
нии Н2О, равном 80%.
Прерывистой линией показана граница гелеобразо-
вании из растворов алюмината и силиката. Ж —
цеолит типа содалита; А — неолит типа А: X — цео-
лит типа фожазита; Е — шабазит; В — филлипсит.
1 Всего в системе Na2O—А12О3—SiO2—Н2О было выполнено несколько сотен
синтезов. Принадлежность продуктов кристаллизации к той или иной фазе подтвер-
ждалась на основании рентгеновского и микроскопического анализов.
130
Рис. 3. Поля кристаллизации цеолитов
в системе Na2O—Al203—Si02—Н20 на
треугольной диаграмме при постоян-
ном содержании Н20, равном 90%.
Обозначения те же, что на рис. 2.
новесных и метастабильных фаз и фазовые превращения в этих системах
должны быть предметом специальных исследований.
Как видно из диаграмм рис. 2 и 3, каждый из типов цеолитов кристал-
лизуется из гелей, составы которых изменяются в определенных пределах.
При этом не только положение полей кристаллизации и их границ, но
и сама возможность кристаллизации цеолитов того или иного типа опре-
деляются не только содержанием окислов, образующих скелет геля и
кристаллическую решетку цеолита
(SiO2, А12О3, Na2O), но и содержа-
нием воды в геле. Из рис. 2 и 3 видно,
как сильно отразилось на положении
полей кристаллизации цеолитов из-
менение содержания воды в исход-
ных силикаалюмогелях всего на 10 %
(от 80 до 90%). Влияние изменения
содержания воды в составе силика-
алюмогелей на положение полей кри-
сталлизации прослеживается и по
другим разрезам тетраэдра, отвечаю-
щим постоянным содержаниям воды,
которые не приведены в этой ра-
боте. Исследования полей кристал-
лизации цеолитов в системе Na2O—
А12О3—SiO2—Н2О полностью еще не
закончены и будут предметом спе-
циальной публикации.
Поля кристаллизации калиевых
цеолитов на треугольных диаграммах
составов, построенных аналогичным
образом, как это видно из данных работы [15], также хорошо разгра-
ничиваются, если содержание всех четырех компонентов геля строго
учитывается. Следует обратить внимание на то, что из калиевых сили-
каалюмогелей соответствующего состава не образуется калиевых анало-
гов цеолитов А и X.
Из приведенных исследований и из результатов, представленных на
рис. 2 и 3, следует, что при данной температуре возможность кристалли-
зации цеолита того или иного типа из щелочных силикаалюмогелей пред-
определена химическим составом гелей.
О химической структуре щелочных силикаалюмогелей
В принципе для получения цеолитов в щелочных алюмосиликатных
системах необязательно исходить из щелочных силикаалюмогелей. Цео-
литы могут кристаллизоваться как из вполне гомогенных смесей щелочных
растворов силикатов и алюминатов, так и из гетерогенных суспензий,
содержащих частицы нерастворенного SiO2. Однако в широкой области
составов при смешении щелочных растворов силиката и алюмината обра-
зуются щелочные силикаалюмогелй, которые и являются исходными при
синтезе цеолитов. Поэтому вопросы химического строения таких силика-
алюмогелей имеют непосредственное отношение к химии процессов кри-
сталлизации цеолитов.
Несмотря на чрезвычайно большое количество работ, посвященных
исследованиям структуры силикаалюмогелей [16], не существует единой
точки зрения относительно механизма образования их кремнеалюмокис-
лородного скелета, его химической и атомной структуры и состояния AI
в скелете. Общепризнанными являются, по-видимому, лишь представ-
ления о корпускулярном строении скелета силикаалюмогелей,. основан-г
9* 131
ные на непосредственных электронномикроскопических наблюдениях [17—
19]. Из этих представлений следует, что такие смешанные гидрогели
должны состоять из скелета, образованного контактирующими глобулами
(мицеллами) и интермицеллярной жидкости, заполняющей пространства
между частицами. При ис-
Таблица 1 следовании процессов кри-
Образец Соотношение окислов в исходных гелях Соотношение окислов в ске- лете геля И ей о ь- о сталлизации цеолитов из таких гелей важно знать, как изменяется состав ске-
SiO, Na,O NasO SiO, Na, О лета и интермицеллярной
А1,О3 А1,О3 SiO, A12O3 А1,О3 Ои д й" жидкости у гелей разных
составов. Для этих целей
Д 462-1 . . 1.0 3.0 3.0 2.4 0.73 1 мы исследовали химиче-
Д 464-1 . . Д 467 .. . 1.0 2.0 3.0 3.0 3.0 1.5 2.5 2.8 0.75 0.89 1 1 ский состав скелета раз-
Г 471 . . . 5.0 4.0 0.8 3.4 0.99 — ных по составу гелей, от-
Г 196-1 . . 5.0 4.0 0.8 3.5 0.97 1.5 мытых от интермицелляр-
Г 196-11 . . 5.0 4.0 0.8 3.6 0.88 1.5 ной жидкости дистилли-
Г 662 . . . Г 660 . . . 50.8 36.0 31.2 17.2 0.61 0.48 4.1 6.6 0.97 0.95 2.05 2.55 рованной водой.
Исследования кине-
тики процесса вымывания
щелочи из силикаалю-
могелей показывают, что существуют две стадии этого процесса. Стадия
быстрой отмывки, в течение которой из геля сравнительно легко удаляется
основная масса щелочи и pH
промывных вод понижается
приблизительно до 10.5, и ста-
дия медленной отмывки, кото-
рая характеризуется крайне
малым переходом оставшейся в
геле щелочи в промывные воды
даже при использовании во
много раз больших количеств
воды в этой стадии промывания
(рис. 4).
Существенно, что первая бы-
страя стадия заканчивается тог-
да, когда отношение Na2O :
А12О3 в скелете отмытого геля
оказывается близким к единице
(на рис. 4 это состояние отвечает
примерно пересечению обеих
кривых). Следовательно, как и
в кристаллических цеолитах,
щелочь, содержащаяся в геле
в количествах, эквивалентных
содержанию AI, прочно связа-
на со скелетом геля. Это нашло
подтверждение при отмывке си-
Рис. 4. Изменение pH промывных вод и со-
держания остаточной щелочи в силикаалю-
могеле в зависимости от количества воды, ис-
пользованной для промывания геля.
Различные обозначения точек на кривой pH относят-
ся к трем параллельным опытам.
ликаалюмогелей самых различ-
ных составов. Соответствующие
аналитические данные приведе-
ны на рис. 5 и в табл. 1. Соста-
вы исходных гелей и составы их
скелетов после отмывки от ин-
термицеллярной жидкости даны на рис. бив табл. 1 без учета содержа-
ния воды. В последнем столбце таблицы приведены отношения Si : Al
1?2
в решетке кристаллов цеолитов, полученных из соответствующих
гелей.
Как видно из этих данных, при самых различных отношениях SiOa:
А12О3=п (50.8 п 1) и Na20 : А12О3 (от 3 до 31.2) в исходных
гелях, отношение Na2O : А12О3 в скелете всех гелей оказывается близким
к 1, а величина п всегда больше 2 и изменяется в значительно более узких
пределах, чем в исходных гелях (6.6 п 2.4).
Следовательно, как и в кристаллических алюмосиликатах [20], со-
держание А1 в кремнеалюмокислсродной сетке щелочных силикаалюмо-
6
Рис. 5. Изменение составов щелочных силикаалюмогелей при отмыв-
ке их от интермицеллярной жидкости.
Черные кружочки — исходные щелочные силикаалюмогели, светлые — сили-
каалюмогели, отмытые от интермицеллярной жидкости (моль. %).
гелей никогда не превышает содержания кремния и каждый атом А1
в сетке гелей, как и в каркасах кристаллических алюмосиликатов, напри-
мер цеолитов, удерживает по одному одновалентному катиону. Но эти
обстоятельства указывают на то, что А1 в кремнеалюмокислородной сетке
щелочных силикаалюмогелей, как и в кристаллических алюмосиликатах,-
находится в тетраэдрах, несущих избыточный отрицательный заряд, и
щелочные катионы присутствуют в геле для его компенсации. Так как
время до гелеобразования в смесях щелочных растворов силиката и
алюмината в некоторых случаях очень мало и измеряется секундами и
долями секунд, то тетраэдрические структурные элементы, из которых
строится сетка гелей, должны существовать уже в исходных смешиваемых
растворах. Ионы SiO3/z и АЮ2' из-за нарушения координационных прин-
ципов и невозможности образования двойных связей атомами Si и А1
с кислородом не могут реально существовать в растворах; наиболее ве-.
роятны их гидратные формы
133
_ ОН --2
I
О—Si—О и
I
ОН _
он
I
НО—А1—ОН
I
ОН
Эти тетраэдрические гидратные ионы могут играть роль элементарных
звеньев, из которых строится по конденсационному механизму общая
кремнеалюмокислородная сетка силикаалюмогелей.
О - -2 ОН “О ОН - -3
1 НО-Si—ОН 1 1 -|- НО-А1—ОН —> 1 1 1 НО—Si—О—AI—ОН 1 1 + н2о
О он -о ок-
~ он -3 он он ОН - -2
1 О—Si—О । 1 + НО—А1—ОН —> 1 1 1 О—Si—О—AI—ОН 1 1 + ОН-
II О он 1 1 О он
1 НО—AI—ОН 1 1 НО-AI—ОН
он 1 он
При использовании всех возможностей конденсации и полимеризации
в сетке геля окажутся заряженными лишь Al-тетраэдры, заряд которых,
как и в кристаллических алюмосиликатах, компенсируется щелочными
катионами. Относительное содержание Si-тетраэдров в сетке гелей, как
видно из данных табл. 1, растет с увеличением общего содержания SiO2
в гелях, но оно зависит также от содержания избыточной (по отношению
к А12О3) щелочи в геле. Чем больше содержание избыточной щелочи в геле,
тем больше SiQ2 вместе с ней находится в интермицеллярной жидкости.
Отношение Si : Al в сетке гелей разных составов близко к пределам изме-
нения этого отношения в кристаллах получающихся из них цеолитов.
Таким образом, кремнеалюмокислородная сетка скелета силикаалюмо-
гелей, так же как и каркас цеолитов, должна быть построена из чередую-
щихся Si- и Al-тетраэдров, образующих в отличие от кристаллов про-
странственно неупорядоченную структуру.
Существует точка зрения, согласно которой щелочные силикаалюмо-
гели, получающиеся при смешении растворов силиката и алюмината нат-
рия, представляют собой смесь соосаждающихся гидроокисей кремния
и алюминия 121]. Эти представления малоправдоподобны, поскольку
в сильно щелочных средах как алюмогели, так и силикагели не могут быть
достаточно стабильными из-за их растворимости в щелочах.
> .Изменение состава кристаллов цеолитов одного и того же типа
с изменением состава гелей
Некоторые типы синтетических цеолитов отличаются постоянством
отношения Si : А1=а: их кремнеалюмокислородного каркаса, у других
типов цеолитов это отношение непостоянно и зависит от состава исходных
гелей. К первой группе цеолитов принадлежат натриевые цеолиты Ж
и А и калиевые F и К-1 [151. Ко второй группе относятся синтетические
фожазиты, натриевые и калиевые шабазиты и филлипситы и, вероятно,
также высококремнеземистые цеолиты, кристаллизующиеся из смешанных
натриево-калиевых силикаалюмогелей. У цеолитов первой группы отно-
шение х равно 1, и они получаются либо из сильно щелочных, либо из
сильно алюминатных гелей. У цеолитов второй группы значение х может
изменяться от 1 до 2.5 и более, в зависимости от содержания SiO2 в исход-
ных гелях. Зависимость величин х кристаллов фожазита от состава гелей,
из которых они образовались, показана на рис. 6. Как видно из этого ри-
134
сунка, отношение Si : Al в решетке фожазита увеличивается с увеличением
содержания SiO2 в исходных гелях. Подобным же образом зависит от
состава гелей отношение Si : Al у калиевых шабазитов, что видно из сле-
дующих данных:
№ образца ..............................
Содержание SiO2 в безводных гелях, % . .
n = SiO2: А1.2О3 в гелях................
х = Si: Al в цеолитах...................
13
18.6
2
1.0
22
24.0
3
1.6
57
33.5
6
2.1
Бэррер [4] связывал рост относительного содержания SiO2 в кристал-
лах цеолитов с увеличением отношения п в гелях. Однако из данных
табл. 1 и рис. 6 можно видеть, что
Как было показано в [12], при
одинаковых значениях п исходных
гелей величины х кристаллов фо-
жазита, получающихся из этих
гелей, при постоянной концентра-
ции в них Na2 О зависят от концен-
трации SiO2, увеличиваясь с ро-
стом последней.
Роль интермицеллярной жидкости
в процессах кристаллизации
цеолитов
Благодаря ограничению макси-
мально возможного содержания
Al-тетраэдров в кремнеалюмокис-
лородной сетке гелей состав ин-
термицеллярной жидкости должен
быть существенно различным у ге-
лей с различным значением п.
У гелей с п < 2 интермицелляр-
ная жидкость должна быть обога-
щена А1203, а у гелей с п > 2 — SiO2.
Из данных, приведенных выш
величины х зависят не только от п.
Рис. 6. Изменение отношения Si : Al в кри-
сталлической решетке синтетических фо-
жазитов в зависимости от состава исход-
ных щелочных силикаалюмогелей.
Заштриховано — поле кристаллизации цеолитов
с решеткой фожазита; кривые с цифрами — линии
равных отношений Si : Al в решетке.
I, следует, что с ростом х = Si : Al
в кремнеалюмокислородной сетке скелета гелей растут и величины х
в крист аллах получающихся из них цеолитов. Это может указывать на то,
чт о элементы структуры решетки цеолитов — различным образом организо-
ванные группы сочетающихся Si- и А1-тетраэдров — формируются уже
в структуре сетки гелей. Однако процесс кристаллизации цеолитов из
гелей не сводится лишь к упорядочению структуры этой сетки. Из данных
табл. 1 видно, что во всех без исключения случаях кристаллизация со-
провождается выходом части SiO2 из сетки скелета геля. И этот процесс
можно рассматривать лишь как результат химического воздействия
щелочных растворов интермицеллярной жидкости на кремпеалюмокис-
лородную сетку геля, сопровождающийся в условиях повышенных тем-
ператур разрывами Si—О—Si связей и перестройкой структуры сетки.
Насколько велика роль интермицеллярноп жидкости в процессах
кристаллизации цеолитов из силикаалюмогелей, видно из результатов
опытов по управляемой кристаллизации гелей путем изменения состава
интермицеллярноп жидкости. В этих опытах исходные образцы гелей, из
которых нормально получались цеолиты А и X, отмывались от интерми-
целлярной жидкости, а затем к скелету геля А была добавлена интерми-
целлярная жидкость, переводящая гель А в область составов гелей X,
а к скелету геля X добавлена жидкость, переводящая его в область ге-
лей А. В результате из модифицированного геля А был получен после его
135
Таблица 2
Изменение направления процесса кристаллизации силикаалюмогелей путем
изменения состава интермицеллярной жидкости
Силикаалюмогель А Силикаалюмогель X
Состав исходного геля . . 3Na2O • AI2O3- SiOo 4Na2O • A12O3 • 5SiO2
Состав цеолита, нормаль- но получаемого из это- го геля Na2O • Al2O3-2SiO2 Na2O • AI2O3-3SiO2
• Тип цеолита NaA NaX
Состав геля после отмыв- ки (состав скелета геля) 0.75Na2O • А1яОа • 2.54SiO9 0.88Na2O • A12O3 • 3.63SiO2
Состав добавленной интер- мицеллярной жидкости Раствор 1 н. NaOH Щелочной раствор алюми-
Общий состав модифици- рованного геля 2Na2O • AI2O3 • 2.54SiO2 ната с отношением Na2O: А12О3 = 2.42 1.68Na2O • А12О3- 1.74SiO2
Состав кристаллов полу- ченного цеолита .... 0.95Na2O - A12O3 • 2.23SiO2 0.98Na2O • А12ОЧ • 2SiO,
Тип цеолита NaX NaA
Примечание. Составы гелей и кристаллов во всех случаях приведены без учета содер-
жания воды.
выдерживания при 90° цеолит типа X, а из геля X — цеолит типа А. Со-
ответствующие данные по составам гелей и кристаллов приведены в таб-
лице 2.
Таким образом, направление процесса кристаллизации — образование
кристаллов цеолитов того или иного типа—не задается составом и струк-
турой кремнеалюмокислородной сетки первичного геля, а регулируется
составом интермицеллярной жидкости. Иначе говоря, тип цеолита, ко-
торый будет кристаллизоваться из данного геля, при данной температуре
определяется составом геля в целом. Результат кристаллизации геля не
зависит от пути, каким образом гель был получен, а лишь от его состава.
Эти результаты подтверждают ранее сделанные выводы о том, что возмож-
ности кристаллизации цеолитов того или иного типа в конечном счете
определяются химическим составом гелей.
Общие закономерности в расположении полей кристаллизации
цеолитов на диаграммах составов
В результате исследования продуктов кристаллизации большого
числа силикаалюмогелей .самого .различного состава (несколько сотеи
составов) при 90° нам удалось выявить границы полей кристаллизации
натриевых цеолитов на треугольных диаграммах составов силикаалюмо-
гелей по разрезам тетраэдров, отвечающих постоянным содержаниям
Н2О в них 70, 75, 80, 85, 90 и 95%. Эти диаграммы в деталях еще подле-
жат уточнениям, но общие закономерности в расположении полей кри-
сталлизации цеолитов разных типов на диаграммах составов могут быть
выявлены уже теперь. Эти закономерности следующие.
1. Положение поля кристаллизации цеолита данного типа на треуголь-
ных диаграммах составов, относящихся к постоянной температуре, опре-
деляется химическим составом кристаллов цеолита. Но, так как отноше-
ние Na : Al в решетках всех цеолитов постоянно и равно 1, то положение
поля кристаллизации зависит от отношения Si : Al в кремнеалюмокисло-
родном каркасе цеолита или содержанием SiO2 в составе цеолита. Чем
больше содержание SiO2 в цеолите (чем больше отношение Si : Al в ре-
136
тетке), тем из более кремнеземных гелей и смесей кристаллизуется
цеолит.
Это находит подтверждение на схеме расположения полей кристалли-
зации натриевых цеолитов на диаграмме рис. 7.2 Как видно из этой схемы,
морденит с наибольшим отношением Si : А1=5, известным для цеолитов,
кристаллизуется в области составов, наиболее богатых SiO2; цеолит Ж
с наименьшим отношением Si : AL
неземной области составов. Между
по величинам a:=Si: Al типов цео-
литов располагаются поля кри-
сталлизации цеолитов с промежу-
точными значениями а: от 1.1 до 2.6
(фожазиты, филлипситы и шаба-
зиты). В натриевой системе не по-
лучены цеолиты с х между 2.6 и
5.0, но из калиево-натриевых сили-
каалюмогелей в нашей лаборато-
рии получены цеолиты со значе-
ниями х, лежащими в пределах
3.0 <С х <Z 3.5, и поля кристалли-
зации этих цеолитов лежат в более
кремнеземной области составов,
между полями шабазита (а:=2—
2.5) и морденита (х=5).
2. Из силикаалюмогелей с от-
ношением SiO2 : А12О3 2 кри-
сталлизуются только цеолиты, в
решетке которых отношение Si :
А1=1. Это обстоятельство нахо-
дится в связи с правилом Ле-
венгатейна, которое, несмотря на
наличие большого избытка А12О3
в гелях, ограничивает возможность
использования алюмокислородных
тетраэдров в построении каркаса
цеолитов в большем количестве,
= 1 кристаллизуется в наименее крем-
полями кристаллизации этих крайних
Рис. 7. Схема расположения областей кри-
сталлизации синтетических цеолитов на
треугольной диаграмме составов (схема не
учитывает содержания Н2О и относится
к различным температурам).
Ж — цеолит с решеткой содалита; А — цеолит
типа А (х=1); X — фожазит («=1.1 ч-1.5);
Y — фожазит (х=1.5 ч- 2.55); В — филлипсит
(х= 1.43 ч- 1.76); Е — шабазит (х=2 ч- 2.43);
Г — смешанные K.Na-цеолиты (ж=3 ч- 3.5);
М — морденит (х=5).
чем кремнекислородных тетраэдров.
Поэтому цеолиты с Si : А1=1, получающиеся из гелей, относительно бога-
тых А1203, представляют собой предельно обогащенную этим компонен-
том фазу.
3. Положение полей кристаллизации цеолитов разных типов на
диаграмме составов зависит также от устойчивости их кремнеалюмо-
кис л сродных каркасов к щелочным средам, что связано с особенностями
структуры этих каркасов и их химическим составом. Связи А1—О—А1,
присутствующие, например, в алюминатах [22], не устойчивы к действию
щелочных растворов, так же как и связи Si—О—Si в кремнеземе. Из самого
факта образования цеолитов в сильно щелочных средах следует, что связи
А1—О—Si в структурах цеолитов, образованных чередующимися тетра-
эдрами кремния и алюминия, стабильны к действию щелочных растворов.
При одинаковом химическом составе цеолитов Ж и A (Na2O • А12О3 •
2SiO2) их поля кристаллизации лежат в разных областях составов гелей
(рис. 7). Цеолит А, решетка которого построена из, сдвоенных четырех-
членных колец тетраэдров (рис. 8, 2), кристаллизуется из менее щелочных
2 Эта принципиальная схематическая диаграмма не относится к постоянной тем-
пературе. Поле кристаллизации морденита приближенно определено для 260°, а поля
кристаллизации Na.K-цеолитов — для 100°. Расположение полей остальных цеоли-
тов относится к 90°.
137
гелей. Это указывает на меныпую стабильность сдвоенных четверных
колец с равным содержанием Si- и Al-тетраэдров к щелочным растворам
по сравнению с простыми кольцами (рис. 8, 7), из которых построена
решетка цеолита Ж. Действительно, обработка растворами NaOH кри-
сталлов цеолита А приводит к его перекристаллизации в цеолит Ж, что
отмечалось и ранее 14, 5|.
Решетка филлипсита также построена из сдвоенных четырехчленных
колец, устойчивость которых к щелочным растворам должна быть пони-
жена еще и из-за того, что содержание Al-тетраэдров в этих кольцах
Рис. 8. Структурные группы из Si- и Al-тетраэдров, уча-
ствующие в построении каркасов различных типов цеолитов.
1 — четверные одинарные кольца в цеолите Ж (решетка содалита);
2 — четверные сдвоенные кольца в неолите А и филлипсите; 3, 4 —
шестичленные сдвоенные кольца в цеолите X и шабазите; 5 — цепоч-
ки в решетке морденита (заштрихованы А1О4-тетраэдры).
в случае филлипсита меньше содержания Si-тетраэдров. Поэтому филлип-
сит кристаллизуется также из менее щелочных гелей, чем цеолит Ж и
даже цеолит А, в пределах полей кристаллизации шабазита и фожа-
зита.
Решетки шабазита и фожазита можно рассматривать построенными из
шестичленных сдвоенных колец Si- и Al-тетраэдров (рис. 8, 3 и 4) с раз-
ным, но всегда меньшим относительным содержанием последних. Устой-
чивость таких колец к щелочным растворам должна возрастать с увели-
чением в них содержания Al-тетраэдров. Наблюдающаяся зависимость
состава кристаллов фожазита от состава гелей (рис. 6) может быть свя-
зана с различной устойчивостью этих шестичленных колец с разными от-
ношениями Si : Al к щелочным растворам. В шабазитах отношение Si : Al
велико (2—2.5), и поля их кристаллизации лежат в областях сравнительно
малых содержаний Na2O в гелях (рис. 2 и 3).
Наконец, решетка морденита построена из цепочек, в которых на
5 кремнекислородных тетраэдров приходится только 1 алюмокислородный
[23] (рве. 8, 5). В этих цепочках, следовательно, преобладают связи
Si—О—Si, и поэтому кристаллы морденита должны быть неустойчивыми
к щелочным растворам. Действительно, уже при обработке 0.5 н. NaOH
кристаллы морденита разрушаются, и он может кристаллизоваться
поэтому только в области малощелочных составов смесей Na2O, А12О3,
SiO2 и Н2О.
138
Выводы
1. При постоянной температуре поля кристаллизации цеолитов раз-
ных типов, кристаллизующихся в системах Na20(K20)—А1203—Si02—Н2О,
хорошо разграничиваются, если их представлять на треугольных диаграм-
мах, отвечающих сечениям тетраэдра составов по постоянным содержа-
ниям Н2О.
2. При заданной температуре из силикаалюмогелей, содержание всех
четырех компонентов которых строго определено, кристаллизуются устой-
чиво и воспроизводимо цеолиты определенного типа.
3. В кремнеалюмокислородиой сетке скелета гелей, получаемых при
взаимодействии щелочных растворов силикатов и алюминатов, как и
в каркасах кристаллических цеолитов и щелочных алюмосиликатов вообще,
содержание А! никогда не превышает содержания Si, а содержание щелоч-
ного катиона эквивалентно содержанию А1. Это свидетельствует об общ-
ности принципов построения кремнеалюмокислородных каркасов в кри-
сталлах цеолитов и силикаалюмогелях и одинаковом координационном
состоянии А1 в них.
4. Направление процесса кристаллизации силикаалюмогелей не опре-
деляется только составом их скелета, но может быть изменено путем из-
менения состава интермицеллярной жидкости. Природа образующихся
из геля кристаллов не зависит от пути, по которому был получен гель,
а лишь от его конечного состава.
5. При переменном химическом составе кристаллов цеолита одного
и того же типа отношение Si : Al в их решетке изменяется в зависимости
от изменения состава исходных силикаалюмогелей. Это отношение растет
с ростом содержания Si02 в гелях.
6. Положение полей кристаллизации цеолитов разных типов на тре-
угольных диаграммах составов определяется их химическим составом и
различной устойчивостью их решетки к действию щелочных растворов.
ЛИТЕРАТУРА
1. R. М. В а г г е г, J. Chem. Soc., 1948, 2158.
2. R. М. В а г г е г, Е. A. W h i t е, J. Chem. Soc., 1951, 1167.
3. R. M. В а г г e г, J. W. В a у n h a m, J. Chem. Soc., 1956, 2882.
4. R. M. В а ггег, J. W. Baynham et al., J. Chem. Soc., 1959, 195.
5. D. W. В г e c k, W. E v e г s о 1 e, R. M i 11 о n, T. R e e d, T. T h о m a s.
J. Am. Chem. Soc., 1956, 78, 5963.
6. T. B. R e e d, D. W. В reck, J. Am. Chem. Soc., 1956, 78, 5972.
7. D. W. В r e c k, J. V. Smith, Scient. American, 1959, 1, 85.
8. L. Broussard, D. Shoemaker, J. Am. Chem. Soc., 1961, 83, 4675.
9. C h e m. a. Engng. News, 1962, 40, 12, 52.
10. G. T. К e r r, G. Koko tailo, J. Am. Chem. Soc., 1961, 83, 4675.
И. С. П. Ж д а н о в, H. H. Б унта рь, ДАН СССР, 1962, 147, 1118.
12. С. П. Ж д а и о в, Н. Н. Бунта р ь, Е. Н. Е г о р о в а, Изв. АН СССР,
сер. хим., 1963, 11, 2061.
13. С. П. Жданов, Н. Н. Бунтарь, в сб.: Синтетические цеолиты, Изд.
АН СССР, 1962, 105.
14. Э. Э. С е н д е р о в, Геохимия, 1963, 9, 820.
15. М. Е. О в с е п я н, С. П. Ж д а н о в, Изв. АН СССР, сер. хим., 1965, 1, 11.
16. В. К. С к а р ч е н к о. Алюмосиликатные катализаторы. Изд. АН УССР, Киев,
1963.
17. С. R. Adams, Н. Н. V о g е, J. Phys. Chem., 1957, 61, 722.
18. Я. В. Мирский, Колл, ж., 1957, 19, 696.
19. А. В. К и с е л е в, В. И. Л ы г и н, И. Е. Н е й м а р к, И. В. С л и н я к о в а,
Чень Вень-хан, Колл, ж., 1958, 20, 52.
20. W. Loewenstein, Am. Mineral., 1954, 39, 92.
21. Л. И. П и г у з о в а, А. В. Агафонов, в сб.: Синтетические цеолиты, Изд.
АН СССР, 1962, 152.
22. F. W. В а г th, J. Chem. Phys., 1935, 3, 323.
23. W. М. М е i е г, Z. KristalL, 1961, 115, 439.
139
ПОЛУЧЕНИЕ КАЛИЕВЫХ
И СМЕШАННЫХ НАТРИЕВО-КАЛИЕВЫХ ЦЕОЛИТОВ
М. Е. Овсепян, С. П. Жданов
Условия кристаллизации синтетических калиевых цеолитов и условия
кристаллизации цеолитов из смешанных натриево-калиевых силикаалю-
могелей мало изучены. По синтезу калиевых цеолитов известна лишь
одна публикация [1], в которой исследована сравнительно узкая область
составов системы К2О—А12О3—SiO2—Н2О. По синтезу цеолитов из сме-
шанных гелей также опубликованы лишь весьма ограниченные данные [2].
Нами была исследована почти вся область возможного образования
щелочных калиевых силикаалюмогелей в четырехкомпонентной системе
К2О—А12О3—SiO2—Н2О с содержанием НЛО в гелях от 70 до 95%. Кри-
сталлизация гелей производилась при 90° в закрытых стеклянных кол-
бах. Опыты по кристаллизации заканчивались после того, как структура
первоначального геля полностью разрушалась и на дне колбы осаждались
кристаллы цеолитов с прозрачным маточным раствором над ними. Кри-
сталлизация гелей продолжалась от нескольких дней до 2—3 недель.
Еще более длительное выдерживание образовавшихся кристаллов в кон-
такте с маточным раствором не приводило к перекристаллизации образо-
вавшихся фаз, что свидетельствует о достаточно стабильном состоянии
полученной гетерогенной системы. Принадлежность кристаллов к тем
или иным продуктам определялась на основании микроскопических
исследований и данных рентгеновского фазового анализа. При темпера-
туре 90° и указанных содержаниях Н2О в гелях из калиевых силикаалю-
могелей устойчиво и воспроизводимо кристаллизуются 4 синтетических
цеолита: калиевый шабазит, калиевый филлипсит, цеолит K-F и новый
цеолит К-I, не имеющий природных аналогов и ранее не описанный в ли-
тературе. Образование других кристаллических калиевых алюмосилика-
тов в этих условиях не наблюдалось. Составы синтетических калиевых
цеолитов приведены в табл. 1.
Таблица I
Типы цеолитов
Калиевые шабазиты........
Калиевые филлипситы . . .
K-F......................
К-1......................
Химический состав
Покава-
тели
прелом-
ления
К2о • А12О3 • (2.0—4.2)SiO2 • (3.8—6.2)Н2О
К2О • А12О3 - (3.5—4.4)SiO2 • (3.8—4.4)Н2О
К2О • А12О3 • 2SiO2 • 3.4Н2О
К2О • А12О8 • 2SiO8 • 3.8Н9О
1.464-1.491
1.486-1.489
~ 1.470
~ 1.475
На рис. 1—4 показаны поля кристаллизации калиевых цеолитов
при 900.1 Эти поля нанесены на треугольных диаграммах, представляющих
собой сечения тетраэдра составов для четырехкомпонентной системы
К2О—А12О3—SiO2—Н2О плоскостями, отвечающими постоянным содер-
жаниям воды 70, 80, 90 и 95% соответственно, как это делалось в [3—5].
Каждая точка треугольника полностью определяет состав геля и отвечает
только одному точно определенному соотношению всех четырех его ком-
понентов. В пределах каждого поля кристаллизуется исключительно или
преимущественно цеолит данного типа. Присутствие примесей других
кристаллических фаз не исключено, но это имеет место обычно в обла-
1 Прерывистая линия ограничивает область возможного получения однородных
силикаалюмогелей.
140
стях составов, прилегающих к границам полей. Как видно из рис. 1—4,
поля кристаллизации разных калиевых цеолитов хорошо разграничи-
ваются, и это указывает на то,
что при постоянной температу-
ре (и давлении) образование цео-
литов того или иного типа пред-
определяется составом системы.
Закономерности в расположе-
нии полей кристаллизации ка-
лиевых цеолитов на диаграм-
мах состава такие же, как и
натриевых [5]. Цеолиты с мень-
шим и постоянным отношением
Si/А1=1 в решетке (F, К-I) кри-
сталлизуются из более щелоч-
ных и менее кремнеземных ге-
лей, а из более кремнеземных и
менее щелочных гелей кристал-
лизуются цеолиты с Si /Al > 1—
филлипситы и шабазиты. При
этом содержание SiO2 в составе
кристаллов данного типа возра-
стает с ростом его содержания
в гелях, из которых кристал-
лы образовались.
Как следует из сравнения
полей кристаллизации цеоли-
Рис. 1. Поля кристаллизации синтетических
цеолитов в четырехкомпонентной системе-
К2О—А12О3—SiO2—Н2О при 90°, изображен-
ные на треугольном сечении тетраэдра со-
ставов (вес. %) при содержании воды 70%.
G — синтетический калиевый шабазит; М — синте-
тический калиевый филлипсит; F — цеолит типа
K-F; К-1 — новый синтетический калиевый цеолит.
тов в калиевой и натриевой системах [4, 5], в области кристаллизации
натриевых цеолитов Ж, А и X в калиевой системе лежат поля кристал-
Рис. 2. Поля кристаллизации синтетических
цеолитов в четырехкомпонентной системе К2О—
А12О3—SiO2—Н3О при 90°, изображенные на
треугольном сечении тетраэдра составов
(вес. %) при содержании воды 80%.
Обозначения те же, что на рис. 1.
лизацци цеолитов других струк-
турных типов. Калиевые анало-
ги цеолитов типа Ж, А и X пря-
мым синтезом из калиевых си-
ликаалюмогелей не получаются..
Это обстоятельство, вероятно,
находится в связи с различием
в размерах ионов Na+ и К+
(rNa=0.98 А; гк=1.33 А), кото-
рые не могут вследствие этого
занимать одни и те же положе-
ния в решетке упомянутых цео-
литов. Действительно, как струк-
тура кристаллов цеолита Ж,
так и структура кристаллов
цеолитов А и X строится из
кубооктаэдрических блоков, со-
стоящих из 24 (Si—А1)-тетраэд-
ров, образующих так называе-
мые малые полости с ведущими
в них окнами из шестичленных
кислородных колец. Диаметры
этих окон 2.2 к [6], и большая
часть Ионов Na+ в таких струк-
турах располагается как раз в центрах этих колец, но эти положения не
могут занять ионы К+, так как их диаметр (2.66 А) оказывается больше
диаметра шестичленных колец. Ионным обменом ионы К+ могут в этих
141
кристаллизации синтетических
четырехкомпонентной системе
Рис. 3. Поля
цеолитов в
К2О—А12О3—SiO2—Н2О при 90°, изображен-
ные на треугольном сечении тетраэдра со-
ставов (вес. %) при содержании воды 90% .
Обозначения те же, что на рис. 1.
уже сформировавшихся, жестких каркасах из (Si—А1)-тетраэдров заме-
щать ионы Na+, но, так же как и ионы Са++, К+-ионы в таких структурах,
по-видимому, не занимают тех
же положений, которые зани-
мали в них ионы Na+. Об этом
можно, например, заключить по
тому, что уменьшение адсорб-
ционного пространства, доступ-
ного для паров воды, в кристал-
лах цеолита А в результате
полной замены Na+на К + (около
0.08 см3/г) оказывается гораздо
большим, чем разница в сум-
марных объемах ионов Na+n К +
(0.02 см3/г). Значительное умень-
шение адсорбционного объема
даже для таких малых моле-
кул, как молекула воды, по-
следовавшее в результате заме-
ны Na+ на К+, указывает на не-
доступность некоторой доли
объема полостей в кристаллах
К-формы цеолита вследствие их
блокировки более крупными ио-
нами калия. Такая блокировка
не могла бы иметь места, если
бы ионы К+ занимали те же позиции, что и ионы Na+ до обмена.
Количество синтетических калиевых
в системе К2О—А12О3—SiO2—Н2О,
четырьмя типами. Эти четыре
типа получаются только из ге-
лей с содержанием воды от 70
до 95 %. Из менее водных гелей
могут быть получены другие
калиевые цеолиты, но эта об-
ласть составов пока еще по-
дробно нами не исследована. Тем
не менее из сильно щелочных
гелей, содержащих Н2О меньше
70%, нами получен еще один
новый калиевый цеолит К-П.
Этот цеолит кристаллизуется в
виде очень тонкодисперсных
кристалликов размером около
1—2 мк с показателем прелом-
ления —1.50. Состав кристал-
лов отвечает формуле 0.87К2О •
А12О3 • 2.03SiO2 • 1.71Н2О. Этот
цеолит в отличие от аналогич-
ного по составу цеолита К-I,'раз-
рушающегося уже при 200° [7],
является вполне термостойким
вплоть до 600° С, и даже после нагревания при 800° цеолит теряет только
около 25% своей адсорбционной способности по воде.
Термостойкость шабазитов и филлипситов различна у разных образцов
и зависит, по-видимому, от содержания кремнезема в составе кристаллов.
цеолитов, кристаллизующихся
ограничивается упомянутыми
не
Рис. 4. Поля кристаллизации синтетических
цеолитов в четырехкомпонентной системе
К2О—А12О3—SiO2—Н2О при 90°, изобра-
женные на треугольном сечении тетраэдра
составов (вес. %) при содержании воды 95%.
Обозначения те же, что на рис. 1.
142
На рис. 5 приведены изотермы адсорбции воды на всех пяти типах
синтезированных калиевых цеолитов. Из рисунка видно, что синтетиче-
ские калиевые цеолиты, так же как и натрие-
вые, являются весьма эффективными осуши-
телями, так как при самых малых относи-
тельных давлениях они поглощают от 8 до
20% воды по отношению к собственному весу.
Подъем изотерм в области plps=O.S—1.0 обя-
зан, по-видимому, капиллярной конденсации
во вторичной пористой структуре, образован-
ной контактирующими кристалликами цео-
литов [8].
Результаты исследования кристаллиза-
ции цеолитов из смешанных калиево-натрие-
вых гелей с постоянным отношением SiO2/
А12О3, равным 6 и 10, приведены на диа-
граммах рис. 6 и 7. На этих диаграммах
поля кристаллизации цеолитов разных типов
разграничиваются вполне четко; если же
учитывать К2О и Na2O в виде суммы ще-
лочей, объединяя эти компоненты, то в од-
них и тех же точках, отвечающих одинаковым
суммарным содержаниям щелочей, но разному
их соотношению, кристаллизуются разные
цеолиты. Это указывает на то, что направле-
ние процесса кристаллизации цеолитов из сме-
шанных гелей определяется при равном содер-
жании других компонентов не только общей
раздельным содержанием каждого из окислов
Рис. 5. Изотермы адсорбции
паров воды на различных ти-
пах синтетических калиевых
цеолитов при 18°.
1 — шабазит; 2 — K-F; 3 — К-1;
4 — филлипсит; 5 — K-II.
щелочностью гелей, но и
и их соотношением. Нане-
сенные на диаграммах гра-
ницы полей кристаллиза-
ции из-за сравнительной
ограниченности количе-
ства синтезов следует счи-
тать приближенными, но
закономерности в располо-
жении полей кристаллиза-
ции разных типов цеоли-
тов определенным образом
проявились и здесь. Цео-
лит F с малым содержа-
нием SiO2 в решетке кри-
сталлизуется в обоих слу-
чаях пз гелей с наимень-
шим содержанием SiO2.
Шабазит, так же как и из
чисто калиевых гелей, кри-
сталлизуется из гелей с
малым содержанием SiO2,
а филлипсит — из гелей с
большим его содержанием.
Пожалуй, наиболее ин-
Рис. 6. Поля кристаллизации синтетических смешан-
ных Na.K-цеолитов в пятикомпонентной системе
Na2O—К2О—А12О3—SiO2—Н2О при постоянном со-
держании воды 85% и мольном отношении
SiO2/Al2O3=6.
тересным результатом является получение цеолита Na,K-F из гелей с от-
ношением Na2O : К2О=8 : 1. В чисто натриевой системе этот цеолит не
кристаллизуется 12, 9 5]. Интересно также получение Ма,К-цеолитов
типа X, которые, по [2], не кристаллизуются из смешанных Na.K-гелей.
143
Таблица 2
Химический состав исходных гелей и полученных из них цеолитов
№ образца Состав исходных смешанных гелей Отношение Na,0 : К2О Состав полученных цеолитов 4 % Тип цеолита
гель цеолит
869 90Na2O • 7.45К2О • А12О3 • 10SiO2 • 330Н2О 12:1 3.6:1 0.72Na20 • 0.20K2G Л12О3 • 2S1О2 Na, K-F
835 60Na20 • 4.95К2О • А12О3 • 6SiO2 • 218Н2О '12:1 3.7:1 0.75Na2O • 0.21К20 А12О3 • 2SiO2 Na, K-F
810 4.0Na2O • 1,0К20 А12О3 2S Ю2 • 165Н2О 4:1 4.5 : 1 0.77Na2O 0.17К2О • А12О3 2SiO2 Na, К-A
801 2.5Na2O • 2.5К2О А12О3 • 2S Ю2 • 79Н2О 1 : 1 1 : 1 0.47Na20 • 0.47К20 • А12О3 • 2SЮ2 Na, K-G
803 2.5Na2O • 2.5К2О А12О3 2 Si О2 • 120Н20 1 : 1 1 : 1 0.36Na2O 0.38К20 • А12О3 • 2SiO2 Na, K-G
814 1.0Na2O • 4.0К2О • А12О3 • 2S1О2 168Н2О 1:4 1 :3 O.24N32O • 0.71К.20 • AI2O3 * 2SiO2 Na, K-G
870 14Na2O • 1.87К2О А12О3 • 10SiO2 82Н2О 7.6:1 3:1 0.63Na20 0.21К2О А12О3 • 2.64S1O. Na, K-X
Цеолиты типа X, как видно из рис. 6 и 7, кристаллизуются из гелей, от-
носительно богатых Na2O. В составе кристаллов X, образующихся из
смешанных гелей, присут-
ствуют оба щелочных окис-
ла. По-видимому, как ио-
ны Na+, так и К+, содер-
жащиеся в цеолитах, не
образующихся в соответ-
ствующей гомощелочной
системе, являются вторич-
ными и проникают в ре-
шетку кристаллов в ре-
зультате ионного обмена.
Это находит подтвержде-
ние в химическом составе
кристаллов таких смешан-
ных цеолитов. Отношение
Na : К в таких кристаллах
зависит не от общего соста-
ва Ма,К-гелей, а, скорее,
только от изменения от-
ношения Na : К в них
(табл. 2).
Рис. 7. Поля кристаллизации синтетических сме-
шанных Na, К-цеолитов в пятикомпонентной си-
стеме Na20—К2О—А12О3—SiO2—Н2О при постоян-
ном содержании воды 85% и мольном отношении
SiO2/Al2O3=10.
Из смешанных гелей с
сталлизуются только такие
могут получаться из гомощелочных гелей.
постоянным отношением SiO2/Al2O3=2 кри-
типы цеолитов, которые в близких условиях
Таблица 3
Влияние природы щелочного катиона в общей щелочности смешанных
Ка,кК-силикаалюмогелей на направление процесса кристаллизации цеолитов
(во всех гелях SiO2 •• А12О3 = 2)
н2о, % Общая кон- центрация щелочи, моль/л Продукты кристаллизации
Na-гели К-гели смешанные Na, К-гели при соотношении щелочных окислов
4Na2O • К2О 2.5Na2O - 2.5К2О Na2O • 4K2O
70 3.0 Ж G + К-1 Na, К-А Na,K-G Na,K-G + Na,K-I
74 2.5 Ж G + К-1 Na, К-А Na,K-G Na,K-G + Na,K-I
78 2.0 Ж G Na, К-А Na,K-G + Na, К-A Na,K-G + Na,K-I + Na,K-A
82 1.5 А G Na,K-A Na,K-G + Na,K-A Na,K-G + Na,K-A
87 . 1.0 А G Na,K-A Na,K-G + Na,K-A Na,K-G + Na,K-A
Ю Цеолиты
145
Из данных табл. 3 видно, что природа кристаллов цеолитов, образую-
щихся из таких бищелочных Na.K-гелей, при постоянном суммарном
содержании щелочных окислов в них определяется как общей концентра-
цией обеих щелочей, так и отдельно концентрацией каждого из щелочных
окислов. Закономерности, управляющие процессами кристаллизации
цеолитов из бищелочных Ка,К-силикаалюмогелей, во многих отношениях
еще не раскрыты, и нужны дополнительные исследования полей кристал-
лизации цеолитов в таких сложных пятикомпонентных системах, для того
чтобы разобраться в этих закономерностях. Эти исследования представ-
ляют не только чисто научный, но и практический интерес, так как в та-
ких пятикомпонентных системах могут быть получены высококремнезе-
мистые цеолиты, не получающиеся ни в чисто натриевой, ни в чисто ка-
лиевой системе. Такие цеолиты уже получены в нашей лаборатории, но
область их кристаллизации подробно еще не исследована. В дальнейших
исследованиях могут быть получены новые интересные данные и новые
типы цеолитов.
ЛИТЕРАТУРА
1. R. М. В а г г е г, J. W. Baynham, J. Chem. Soc., 1956, 2882.
2. R. М. В а г г е г, J. W. В а у n h a m, F. W. Bultitude, W. М. Meier,
J. Chem. Soc., 1959, 195.
3. С. П. Ж да нов, И. Н. Б у и т а р ь, Е. Н. Е г о р о в а, Изв. АН СССР, сер.
хим., 1963, 11, 2061.
4. М. Е. О всепян, С. П. Ж д а н о в, Изв. АН СССР, сер. хим., 1965, 1, 11.
5. С. П. Ж д а н о в, Н. Н. С а м у л е в и ч, Е. Н. Е г о р о в а, настоящий сборник,
стр. 129.
6. D. W. В г е с k, W. G. Е v е г s о 1 е, R. М. М i 11 о n, Т. R е е d, Т. Т h о m a s,
J. Am. Chem. Soc., 1956, 78, 5963.
7. С. П. Ж д а н о в, М. Е. О в с е п я н, ДАН СССР, 1964, 157, 913.
8. М. М._ Дубинин, М. М. В и ш н я к о в а, Е. Д. 3 а в е р и н а, Е. Г. Ж у-
ковская, Е. А. Леонтьев, В. М. Лукьянович, А. И. Сар а-
хов, Изв. АН СССР, ОХН, 1960, 396.
9. С. П. Ж д а н о в, Н. Н. Бунтарь, в сб.: Синтетические цеолиты, Изд.
АН СССР, 1962, 105.
СИНТЕЗ И ОСНОВНЫЕ СВОЙСТВА РАЗЛИЧНЫХ ИОНООБМЕННЫХ
И ИЗОМОРФНОЗАМЕЩЕННЫХ ФОРМ ЦЕОЛИТОВ
И. Е. Неймарк, М. А. Пионтковская, А. И. Растрененко, Г. С. Шамеко,
В. Г. Ильин, А. М. Еременко, С. Н. Антоновская
С развитием новых отраслей химической и нефтеобрабатывающей
промышленности требования к адсорбентам, в том числе и к синтетиче-
ским цеолитам, резко повышаются. В связи с этим работы по изменению
избирательной способности синтетических цеолитов приобретают особую
актуальность.
В настоящей работе изложены результаты исследований по получению
новых цеолитных материалов, изготовляемых как прямым синтезом из
щелочных алюмосиликатных систем, так путем ионного обмена и изо-
морфного замещения кремния в тетраэдрах SiO4 на элементы с коорди-
национным числом четыре.
Влияние постепенного замещения натрия другими катионами
на сорбционные свойства цеолитов типа А и X
Эффективные размеры «окон» больших полостей, их проходимость
в значительной степени зависят от размеров обменных катионов, разме-
щенных в непосредственной близости к этим «окнам» и без которых отри-
146
цательно заряженная кристаллическая решетка цеолита существовать
не может. Путем обмена ионов натрия в цеолитах А и X на катионы иных
размеров можно влиять на молекулярно-ситовые свойства этих адсорбен-
тов, которые определяются прежде всего соотношением размеров «окон»
и эффективным диаметром молекул поглощаемого вещества.
Известно, что величина адсорбции на цеолитах, в особенности молекул
полярных веществ, в значительной мере определяется ион-дипольным
взаимодействием молекул адсорбируемого вещества с катионами [1, 2],
и поэтому, кроме величины ионного радиуса катиона, важное значение
для адсорбции будут иметь заряд и природа катиона, его деформируемость
и положение в анионном каркасе.
Для приготовления образцов цеолитов с разной степенью замещения
натрия другими катионами исходный образец цеолита многократно
обрабатывали слабыми растворами соответствующих солей. Степень
обмена натрия зависела от числа обработок. Применяемая методика обес-
печивала наиболее мягкие условия для реакций обмена и сводила к мини-
муму разрушающее действие на структуру цеолита гидролитической кис-
лотности некоторых растворов.
Для получения образцов с высоким содержанием соответствующего
обменного катиона вместо натриевого цеолита применялся аммонийный
цеолит, который готовился из натриевой формы обработкой его смесью
NH4OH и NH4C1 (pH смеси не превышал 10).
Степень обмена натрия т-». , в цеолите
(Me) + (Na)
определяли по резуль-
татам анализа равновесных растворов при помощи пламенного фото-
метра (натрий), фотоколориметра (кобальт, никель) и весовым методом
(кальций, магний).
Нами были синтезированы ионообменные производные цеолитов типов
А и X с различной степенью обмена на катионы щелочных, щелочнозе-
мельных и некоторых тяжелых металлов.
Таблица 1
Ряды полученных ионообменных форм цеолита NaA
0.15 KNaA 0.16 LiNaA 0.16 RbNaA
0.33 KNaA 0.30 "LiNaA 0.40 RbNaA
0.47 KNaA 0.42 LiNaA 0.49 RbNaA
0.63 KNaA 0.66 LiNaA 0.55 RbNaA
0.79 KNaA 0.81 LiNaA 0.64 RbNaA
0.92 KNaA 0.87 LiNaA 0.74 RbNaA
0.95 KNaA 0.90 LiNaA 0.84 RbNaA
0.15 CaNaA 0.32 MgNaA 0.17 CoNaA
0.19 CaNaA 0.43 MgNaA 0.33 CoNaA
0.23 CaNaA 0.49 MgNaA 0.44 CoNaA
0.29 CaNaA 0.52 MgNaA 0.55 CoNaA
0.35 CaNaA 0.57 MgNaA 0.69 CoNaA
0.50 CaNaA 0.64 MgNaA 0.73 CoNaA
0.64 CaNaA 0.81 CaNaA 0.96 CaNaA 0.90 MgNaA 0.78 CoNaA
0.16 NiNaA 0.15 CeNaA
0.24 NiNaA 0.19 CeNaA
0.39 NiNaA 0.27 CeNaA
0.43 NiNaA
0.59 NiNaA
0.66 NiNaA
В табл. 1 и 2 даны ряды полученных ионообменных форм цеолита.
Числа указывают на степень обмена натрия на соответствующий катион
металла.
10*
147
Таблица 2
Ряды полученных ионообменных форм цеолита NaX
0.15 KNaX 0.10 LiNaX 0.17 (NH4)NaX
0.27 KNaX 0.13 LiNaX 0.27 (NHJNaX
0.33 KNaX 0.15 LiNaX 0.34 (NH4)NaX
0.46 KNaX 0.19 LiNaX 0.42 (NHJNaX
0.24 LiNaX 0.57 (NHJNaX
0.35 LiNaX 0.62 (NHJNaX 0.73 (NH4)NaX 0.78 (NHJNaX
0.20 CaNaX 0.08 MgNaX 0.05 CoNaX
0.43 CaNaX 0.12 MgNaX 0.12 CoNaX
0.59 CaNaX 0.14 MgNaX 0.15 CoNaX
0.71 CaNaX 0.19 MgNaX 0.18 CoNaX
0.77 CaNaX 0.25 MgNaX 0.21 CoNaX
0.82 CaNaX 0.30 MgNaX 0.26 CoNaX
0.90 CaNaX 0.40 MgNaX
0.93 CaNaX 0.11 CeNaX 0.47 MgNaX 0.35 (NH4)NaY
0.36 CeNaX 0.50 (NHJNaY
0.66 CeNaX 0.63 (NHJNaY 0.80 (NHJNaY
Характеристики полученных цеолитов изучены различными методами:
адсорбционным, хроматографическим, химическим, и некоторые образ-
цы — методом ядерного магнитного резонанса. Исследовались также
Рис. 1. Изобары адсорбции паров
воды, метанола и С02 цеолитом
типа А, содержащим патрий и ка-
лий в различных соотношениях.
р/р„ : Н,0 — 0.14, СНзОН — 0.24. р для
СО, — 20 мм.
теплоты смачивания водой.
На ионообменных формах цеолитов в
высоковакуумной адсорбционной установ-
ке .исследованы изотермы адсорбции па-
ров воды, спиртов, некоторых углеводо-
родов, углекислого газа и других адсор-
батов. Теплоты смачивания определялись
в. адиабатическом калориметре.
На рис. 1—4 представлены изобары и
изотермы сорбции различных адсорбатов
и теплоты смачивания водой в зависи-
мости ют степени обмена натрия на дру-
гие катионы в цеолитах типа А и X.
Как видно из этих рисунков, по мере
замещения натрия в цеолитах типа NaA
на калий адсорбция паров воды снижает-
ся (рис. 1), снижаются и величины теплот
смачивания (рис. 2). Аналогичная картина
наблюдалась и для натрий-рубидиевых
цеолитов. По-иному ведут себя образцы,
содержащие ионы лития, кальция, ко-
бальта, никеля и магния. По мере заме-
щения натрия в цеолите на эти катионы
адсорбция паров воды растет, увеличи-
ваются также и теплоты смачивания во-
дой (рис. 2 и 3).
Особенно сильный рост адсорбции во-
ды характерен для магнийз вмещенных
образцов цеолита. На рис. 4 приведены
изотермы сорбции паров на порошках пористых кристаллов NaA и маг-
нийзамещенных образцах. Как видно из рис. 4, изотермы сорбции паров
воды на магнийз вмещенных обрвзцах рвсполвгаются знвчительно выше
изотермы сорбции пиров воды ни натриевой форме цеолита. Чем более
полно замещен натрий на ион магния, тем выше изотерма сорбции. Теп-
148
лота смачивания водой магниевого цеолита типа А достигает при заме-
щении натрия приблизительно на 50% 150 кал., вместо 110 кал. на на-
триевом цеолите (рис. 2).
Изменение адсорбционных свойств цеолита в результате замены натрия
на другие катионы может являться результатом сужения или расшире-
ния «окон» полостей цеолита, увеличения или уменьшения объема внутри-
кристаллического пространства за счет изменения объема, занимаемого
самими катионами, изменения энергии связи молекул адсорбируемого
вещества с поверхностью цеолита, изменения плотности адсорбированного
вещества.
Уменьшение адсорбции паров воды на
калиевых и рубидиевых цеолитах нельзя
связывать с проявлением ситового дей-
ствия, так как размеры «окон» больших
полостей после обмена все же остаются
доступными для молекул воды. Уменьше-
ние адсорбционного пространства Аош
в результате замещения натрия (г=0.98 А)
на катионы больших размеров калия
(г=1.33 А) или рубидия (г=1.47 А) на-
много меньше наблюдаемого уменьшения
объема сорбированной воды Да. Отноше-
ние-^-^>10. Нет соответствия между
увеличением объема адсорбированной
воды и увеличением объема внутрикри-
сталлического пространства за счет уве-
личения объема в результате замены
натрия на катионы лития, кальция, ни-
келя, кобальта.
Анализируя полученные данные, легко
заметить симбатность между гидратацион-
ной способностью катионов и адсорбируе-
мостью паров воды на соответствующих
ионных производных цеолитов. Такая же
симбатность наблюдается между величи-
Рис. 2. Зависимость теплоты сма-
чивания цеолита типа А от сте-
пени обмена натрия на ионы К+,
Li+, Rb+, Mg2+, Са2+, Со2+,
Ni2+.
нами теплот смачивания и гидратацион-
ной способностью катиона, компенсирующего зардд анионной решетки
(табл. 3).
Таким образом, величина адсорбции полярных веществ на цеолитах
в значительной степени определяется взаимодействием молекулы адсор-
бируемого вещества с катионами.
При помощи ЯМР [3] было показано, что на магнитный резонанс про-
тонного поглощения существенное влияние оказывают обменные катионы
цеолита, которые по силе влияния на величину протонного поглощения
располагаются в ряд: магний > кальций > натрий. Данный порядок
расположения катионов соответствует последовательности этих катионов
в отношении их чисел гидратации в водных растворах [4].
Вероятно, имеются различия в размещении катионов в цеолитах
типа А и X, о чем свидетельствует хотя бы тот факт, что замещение нат-
рия на кальций в цеолите А приводит к увеличению эффективного диа-
метра «окон», а в цеолите X аналогичное замещение вызывает сужение
«окон». Несмотря на это, различия изобары на цеолитах типа X с разной
степенью замещения натрия на другие катионы имеют тот же характер,
что и на цеолите типа А. По мере замещения натрия на калий адсорбция
воды понижается. Замена катиона натрия на катионы с большой гидрата-
149
Таблица 3
Зависимость между теплотами смачивания и гидратацией на ионных
производных цеолита
Rb+ к+ Na+ L1+ Са!+ Со’+ NP+ Mg2+
Теплота смачивания, кал./г, 6 = 0.4 30 73 108 110 112 115 120 145
Энергия гидратации 74 80 98 121 375 470 485 470
ционной способностью (кальций, кобальт, никель) приводит к повышению
адсорбции воды (рис. 5).
На основании наших данных и работ других исследователей [3, 5]
можно считать установленным, что при заполнении первичных пор цео-
литов молекулами воды и другими полярными молекулами последние
в первую очередь располагаются у катионов, компенсирующих заряд ре-
шетки; они являются активными центрами полярных веществ.
Рис. 3. Изобары адсорбции паров воды
на цеолите типа А, содержащем на-
трий и кальций, натрий и кобальт, на-
трий и литий, натрий и никель, натрий
и калий в разных соотношениях.
p/pg : Са — 0.23, Li — 0.17, К — 0.14, N1 — 0.02,
Со — 0.02.
Рис. 4. Изотермы сорбции паров
воды на синтетических цеолитах
типа А, содержащих катионы
натрия и магния в различных
соотношениях.
7.05% [MgO (7); 6.4% MgO (2); 4.62%
MgO (3) и NaA (7).
Были также изучены свойства металлзамещенных цеолитов, катионы
которых образуют с аммиаком комплексы. К ним относятся цеолиты, со-
держащие катионы цинка, меди, кобальта, никеля и кадмия. Обмен нат-
рия.:, на эти катионы производился через аммонийную форму цеолита.
Для этих образцов характерна повышенная активность по отношению
к аммиаку (рис. 6). Изотермы сорбции аммиака на NiA, ZnA, СоА, СпА
лежат выше изотермы сорбции аммиака на NaA. Эти результаты также
указывают на дополнительное электростатическое взаимодействие амми-
ака с обменными катионами.
Была исследована адсорбция пропана, н-гексана, н-гептана и н-де-
кана на образцах цеолита типа А, содержащих катионы натрия, кальция,
кобальта и никеля в различных соотношениях. На рис. 7 представлены
изобары адсорбции разных углеводородов на цеолитах с разной степенью
замещения натрия на кальций; на рис. 8 — изобары этих углеводородов
156
на цеолитах с разной степенью замещения натрия на катионы кобальта
и никеля. На всех изобарах наблюдено резкое возрастание адсорбции
°РИ (Ме)+%а) =°-2~0-3- Адсорбция
углеводородов на цеолите NaA
практически не происходит, и только при степени замещения натрия
на кальций, никель или кобальт выше 20°/о наблюдается резкое воз-
растание адсорбции углеводородов С3—С10.
Тот факт, что только при достижении степени обмена натрия в цеолите
NaA на кальций, кобальт, никель, близкой к 0.3, внутреннее сорбционное
пространство цеолита становится до-
ступным молекулам углеводородов,
говорит о существовании в цеолите
определенной группы катионов, отли-
чающихся от остальных своим положе-
нием по отношению к решетке. Очевид-
Рис. 5. Изобары сорбции паров воды
на цеолите типа X, содержащем натрий
и калий, натрий и литий, натрий и
кальций, натрий и магний, натрий и
кобальт, натрий и церий в различ-
ных соотношениях.
ViPs : КХ — 0.28, СоХ — 0.52, СеХ — 0.4,
СаХ —0.17, ЫХ—0.07, MgX — 0.17.
Рис. 6. Изотермы адсорбции аммиака при
20° С на цеолитах типа А, в которых натрий
замещен катионами, способными к ком-
плексообразованию.
но, именно эти, >обменивающиеся в пер-
вую очередь катионы и определяют
доступность молекул углеводородов в
полости цеолита.
Согласно рентгеноструктурным исследованиям Броуссарда и Шо-
мекера [6], из 12 ионов натрия, содержащихся в элементарной ячейке
цеолита NaA, восемь катионов находятся вблизи шестичленных «окон»
малых полостей, а четыре катиона статистически распределены по объему
больших полостей. Весьма вероятно, что замещение именно этих четырех
катионов приводит к образованию в цеолите типа А более открытой пори-
стой структуры.
Из рис. 7 также следует, что поглощение углеводородов с разной дли-
ной цепи начинается при разных степенях замещения натрия на кальций.
Длина молекулы вдоль оси составляет примерно 5, 10, 11, 15 А соответ-
ственно для пропана, гексана, гептана и декана. Молекулы длиной около
5 А могут легко обойти случайные препятствия в «окне» цеолита, изменив
направление движения в ближайшей свободной полости, и проникнуть
дальше в глубь решетки. Более длинные молекулы не могут так изменять
направление движения, так как это связано со значительной деформа-
цией углеводородной цепочки. Поэтому адсорбция углеводородов от С5
и выше происходит на цеолитах при более высокой степени обмена натрия
151
на кальций, когда все ионы натрия, могущие затруднить диффузию мо-
лекул адсорбата, будут полностью удалены. Следовательно, пропан можно
выделять из смеси углеводородов с более длинной цепью, применив в ка-
честве молекулярного сита цеолит типа А с соответствующей степенью
обмена натрия на кальций.
Интерес представляет выяснение поведения металламмиачных ком-
плексов тяжелых металлов в ионном обмене на синтетических цеолитах
и влияния этих комплексов на изменение селективных свойств пористых
Рис. 7. Адсорбция углеводородов с разной
длиной цепи на цеолите типа А, содержа-
щем натрий и кальций в разных соотно-
шениях.
Рис. 8. Адсорбция С3Н8 (7),
С8Пи (2) и С7НИ (3) при
р=30 мм цеолитами с раз-
ным содержанием никеля
(светлые точки) и кобальта
(черные точки).
кристаллов. Нами были получены образцы ряда металламмиачнозамещен-
ных цеолитов на основе образцов NaA и NaX путем многократной обра-
ботки исходного цеолита растворами комплексных аммиакатов:
[Cu(NH3)4]SO4; [Ni(NH3)6]Cl2.
[Co(NH3)6] Cl3.
Обмен проводили при комнатной температуре. После обмена цеолит
отмывался от солей спиртово-аммиачным раствором во избежание разру-
шения комплексных групп в результате гидролиза. В табл. 4 указаны
синтезированные нами металламмиачные цеолиты и степень обмена натрия
на комплексные катионы. Меньшая степень обмена наблюдается у цеолита
типа А, что вызвано стерическими затруднениями. В таблице также приве-
дены данные о термической устойчивости металламмиачных цеолитов. По
мере их нагрева выше 120° уменьшается количество аммиака в цеолите,
т. е. комплексы разрушаются. Однако даже при 300° в цеолите еще остается
аммиак. Следовательно, до 300° в цеолите наряду с катионом-комплексо-
образователем находятся также и комплексные катионы. Такие полика-
тионные цеолиты должны обладать другими молекулярно-ситовыми
152
Таблица 4
Металламмиачнозамещенные цеолиты, степень замещения и термостабильность
Цеолит Степень замеще- ния. 70 Содержание аммиака (в %) в зависимости от температуры прокалки
20° 170° 200° 300°
|Co(NH3)6JNaX 15.50 9.09 2.79 ‘ 1.60 0.53
[Ni(NH3)6jNaX 53.90 3.40 1.33 0.62 0.22
[Cu(NH3)4]NaX 73.23 4.91 . 3.40 — 0.30
Таблица 5
Предельные сорбционные объемы при p/ps — 0.5 металламмиачных
цеолитов в смЗ/г
Цеолит Предельно-сорбционный объем, см3/г
CHSOH с2н5он сдан С4Н5ОН
NaX . . .• 0.238 0.233 0.210
[CO(NH3)6]NaX 0.278 0.261 0.239 0.084
fNi(NH3)6]NaX 0.247 0.270 0.243 0.064
[Cu(NH3)4]NaX 0.243 0.268 0.260 0.070
[Co(NH3)6]NaA 0.210 0.242 0.175 0.002
Cu(Nn3)J6NaA 0.235 0.203 0.198 0.009
Ni(NH3)6]NaA 0.218 0.191 0.194 0.007
NaA 0.200 0.167 0.080 —
свойствами, чем исходные цеолиты. Действительно, на поликатионных
цеолитах наблюдается повышенная адсорбция спиртов низшего гомологи-
ческого ряда — метилового, этилового, пропилового по сравнению с цео-
литами NaA и NaX. Бутиловый спирт слабо адсорбируется даже металл-
аммиачным цеолитом типа X (табл. 5). Этим обстоятельством можно вос-
пользоваться, например, для обезвоживания бутанола и т. д.
Получение цеолитов путем изоморфного замещения кремния
в кремнекислородных тетраэдрах на бериллий и магний
Одним из методов получения новых цеолитных материалов является
метод изоморфного замещения кремния в кремнекислородных тетраэдрах
на элементы с координационным числом четыре.
Известно, что координационное число двухвалентного иона бериллия
по отношению к кислороду всегда равно 4 [7]. Это связано с небольшим
размером иона бериллия: его радиус равен 0.34 А, т. е. приближается
к размеру иона кремния (0.39 А). Таким образом, бериллий и кремний
изоструктурны, и следует ожидать большого сходства у этих элементов
при построении кристаллов. Действительно, в минералах ионы бериллия
не встречаются вне кремнекислородпой решетки, так как они заменяют
кремний в тетраэдрах SiO4 [7].
С целью получения цеолитов, в которых кремний в кислородных
тетраэдрах замещен бериллием, были изучены условия гелеобразования
и кристаллизации силикатов и алюмосиликатов бериллия. Было установ-
лено, что гели силикатов бериллия при нагреве до 100° в течение 6 часов
не кристаллизуются.
153
Уэллс [8], исследуя осаждение солей металлов силикатом натрия,
пришел к выводу, что при применении двух солей с разными катионами
происходит более полное осаждение силикатов. Вероятно, это положение
может быть распространено на процесс получения сложных бериллий-
алтомосиликатов.
У гидроокиси бериллия и алюминия наблюдается сходство основных
свойств. Хотя радиусы и заряды ионов бериллия и алюминия отличаются
друг от друга, однако потенциалы ионизации довольно сходны (Ф^ = 5.9;
Рис. 9. Изотермы сорбции паров
воды (2), метанола (2), бензола
(5), н-гексана (4) на бериллий-
алюмосиликатном цеолите.
Фд^ = 5.3), вследствие чего эти ме-
таллы следуют один за другим в про-
цессе гидролитического разложения
солей [9J.
Нами были исследованы факторы,
влияющие на процессы получения
и кристаллизации бериллийалюмоси-
ликатных гелей. Было изучено влия-
ние на процессы кристаллизации pH
среды, условий смешения, состава
Рис. 10. Спектрограмма, снятая
с бериллийалюмосиликата.
геля, температуры и продолжительности кристаллизации. В результате
получения сотен образцов разработаны методы получения бериллийалюмо-
силикатных пористых кристаллов. Синтезированы образцы с разным со-
держанием окиси бериллия. В зависимости от количества внедренного
бериллия и условий кристаллизации получены кристаллы разных типов.
Бериллийалюмосиликаты с содержанием окиси бериллия до 13%
являются хорошо окристаллизованными порошками. Их кристаллическая
структура совпадает со структурой цеолита типа А (табл. 6). По адсорб-
ционным и молекулярно-ситовым свойствам они также идентичны цео-
литу NaA. При изменении условий кристаллизации и внедрения большого
количества ВеО в цеолит (образец 817, табл. 6) межплоскостные расстоя-
ния у такого образца уже не соответствуют кристаллической структуре
шабазита. Он состоит из отличных друг от друга по структуре фаз: 'одна —
с кубической решеткой, идентичной решетке цеолита Курнакова (гидро-
содалита), но с более высоким, чем у последнего, периодом идентичности
элементарной ячейки, а=9.03 (у гидросодалита а=8.88), другая — с ге-
ксагональной решеткой типа берилла. Обе фазы хорошо окристаллизованы.
В образцах не обнаружено сколько-нибудь заметных количеств аморфной
фазы.
154
Таблица б
Межплоскостные расстояния (d) и интенсивности соответствующих линий (1) некоторых кристаллов
817 Гидросодалит Берилл 833 1023 NaX
I dA hkl I dA hkil I dA I dA I dA hkl I dA
20 8.8 — — — — — — 74 14.7 100 14.7 111 ОС 14.5
48 6.31 310 71 6.28 — — — 62 8.8 21 8.8 220 cpc 8.8
22 4.56 — — — 0002 8 4.57 29 7.20 23 7.3 311 cpc 7.6
40 3.85 — — — 2020 8 3.97 37 5.78 36 5.80 331 c 5.74
100 3.70 211 100 3.62 — — — 30 5.52 — — — — —-
14 3.29 — — — 1122 10 3.24 27 4.91 32 4.81 511 cp 4.81
15 2.95 — — — 2022 8 3.00 27 4.45 15 4.21 440 cpc 4.42
22 2.85 310 43 2.81 2131 10 2.87 63 3.85 30 3.88 533 cpc 3.81
40 2.59 222 91 2.59 2132 8 2.51 34 3.35 20 3.45 642 cp 3.32
60 2.39 — — — 2240 7 2.29 33 2.97 — — 660 cp 2.94
13 2.26 — — — 1014 5 2.21 31 2.90 — — 555 c 2.87
14 2.19 — — — 2133 8 2.14 24 2.79 12 2.70 480 cp 2.79
40 2.11 — — — 2242 4 2.05 20 2.63 10 2.58 931 срсл 2.62
17 1.967 — — — 2024 8 1.989 43 2.39 8 2.31 1022 cp 2.40
8 1.830 — — — 3143 7 1.793 19 2.20 — — — — —
17 1.757 — — — 3034 8 1.737 — — — — — — —
7 1.637 — — — 2244 8 1.626 — — — — — — —
13 1.585 — — — 3253 5 1.578 — — — — — — —
15 1.537 — — — 0006 4 1.532 — — — — — — —
Примечание, ос — очень сильная; с — сильная; срс — среднесильная; ср — средняя; срсл — среднеслабая; сл — слабая; осл — очень слабая.
Рентгенографическое исследование образцов 817, гидросодалита 833, 1023 выполнено Е. Г. Куковским, за что авторы выражают ему благодарность.
Образец 833 с содержанием ВеО 21.4% относится по структуре
к фожазиту (табл. 6), по адсорбционным свойствам близок к свойствам
цеолита типа X. Он адсорбирует бензол, н-гексан. Однако адсорбционная
емкость бериллийалюмосиликатного цеолита несколько ниже по парам
воды, криптону, бензолу и н-гексану (рис. 9).
Для доказательства того, что бериллийалюмосиликатный цеолит пред-
ставляет собой продукт изоморфного замещения ионов кремния в тетра-
эдрах SiO4 на бериллий, была изучена реакция обмена полученных кри-
сталлов, содержащих разное количество бериллия, с раствором хлори-
стого натрия. Было показано, что содержание бериллия в образце до и
после обмена остается практически постоянным. Кристаллическая струк-
тура и адсорбционные свойства при такой обработке не меняются.
Методом ИКС установлено, что тетраэдры ВеО4 являются составной
частью структуры синтезированных цеолитов. На рис. 10, наряду с по-
лосой поглощения, отвечающей кремнекислородным и алюмокислород-
ным тетраэдрам, появляется полоса поглощения, отвечающая колебаниям
тетраэдров ВеО4. Таким образом, приведенные результаты указывают на
то, что синтезированные нами бериллийалюмосиликатные пористые кри-
сталлы являются продуктами изоморфного замещения ионов кремния
в тетраэдрах SiO4 на ионы бериллия.
Кроме бериллийалюмосиликатных пористых кристаллов, в аналогич-
ных условиях были получены алюмомагниевые силикаты. Согласно
рентгенограмме (табл. 6), синтезированный образец 1023 имеет кристалли-
ческую структуру, идентичную цеолиту NaX. Адсорбционные исследова-
ния указывают на то, что эти .кристаллы являются пористыми и обладают
молекулярно-ситовыми свойствами.
Синтез цеолитов с повышенным содержанием кремнезема
Начиная с 1961 г. после опубликования патента Брэка [10] внимание
исследователей привлек цеолит типа Y, который является синтетическим
аналогом природного цеолита фожазита. Кристаллическая структура
и адсорбционные свойства цеолита Y мало чем отличаются от молекуляр-
ного сита типа X, однако повышенное содержание кремнезема придает
ему некоторые новые свойства. Он обладает устойчивостью по отношению
к воздействию органических кислот, большей термостойкостью, чем NaX,
и высокой каталитической активностью в некоторых реакциях углево-
дородов. Расстояния между катионными участками — тетраэдрами
А1О4“® — достигают 7 А, и это, видимо, является важнейшей особенностью
нового цеолита. По данным Брэка [10], цеолиты с соотношением
SiO2/Al2O3 > 4 можно получить, только применяя в качестве одного из
основных компонентов высококонцентрированный золь кремневой кис-
лоты. Мы поставили перед собой задачу выяснить возможность получения
цеолита типа Y силикатным методом. В результате многочисленных
опытов нами разработаны методы получения цеолитов химического со-
става 0.8—0.9 Na2O • А12О3 • (3.5—5.1)SiO2 • (6—8)Н2О. Полученные об-
разцы подвергались рентгеноструктурному исследованию, изучению ад-
сорбционных свойств, устойчивости к воздействию уксусной кислоты
и термоустойчивости.
Параметр элементарной кубической ячейки кристаллов цеолитов с со-
отношением SiO2/Al2O3 > 4 а°=24.72—24.68 А. Адсорбционная емкость
по парам воды при p/ps=0.14 (20°) достигает 0.29 см3/г и парам бензола —
0.28 см8/г.
Синтезированные образцы не растворяются в 3 н. уксусной кислоте,
в отличие от цеолитов типов А и X, и сохраняют адсорбционную емкость
156
ио парам воды после обработки кислотой в течение 30 мин. при 95—98°,
равной 0.2 г/г. На рис. 11 представлена изотерма сорбции по парам воды
до и после обработки его кислотой.
Установлено, что термообработка до 800° не приводит к разрушению
кристаллической решетки.
Синтезированные цеолиты типа Y могут быть декатионированы мето-
дом ионного обмена катиона натрия на аммонийный ион с последующим
Рис. 11. Изотерма сорбции паров
воды на цеолите типа Y до (7) и
после (2) его обработки уксусной
кислотой.
Рис. 12. Изотермы сорбции паров
воды на цеолите типа Y до (7) и
после (2) декатионирования.
прокаливанием или методом электродиализа. Получен декатионированный
цеолит типа Y с соотношением SiO2/Al2O3=4.7 со степенью декатионирова-
ния 94%, на рентгенограмме которого интенсивность дифракционных ли-
ний практически не изменилась по сравнению с исходным цеолитом. На
изотерме сорбции по парам воды уменьшается несколько крутизна восхо-
дящей ветви изотермы (рис. 12). Декатионированный в таких же условиях
цеолит NaX полностью разрушается, на рентгенограмме не обнаружено
дифракционных линий.
Таким образом, путем постепенного замещения в цеолитах ионов нат-
рия на другие катионы, изоморфного замещения кремния в кремнекисло-
родных тетраэдрах на элементы с координационным числом четыре и
изменения соотношения SiO2/AlaO3 в цеолитах можно в широких преде-
лах менять избирательные свойства пористых кристаллов.
ЛИТЕРАТУРА
1. О. М. Д иигит, А. В. К и с е л е в, Г. Г. М уттик, Колл, ж., 1963, 25, 34.
2. Д. П. Тимофеев. Кинетика адсорбции. Изд. АН СССР, 1962.
3. И. В. М ат яш, М. А. П и он тк о в ск а я, Л. М. Т а р а с е н к о, Р. С. Тю-
тю н н и к, Ж. структ. химии, 1962, 3, 214; 1963, 4, 106.
4. Н. А. Измайлов. Электрохимия, Изд. ХГУ, 1959.
5. О. М. Джигит, С. П. Ж д а н о в, А. В. К и с е л е в, Г. Г. М у т т и к,
ЖФХ, 1962, 36, 919.
6. L. Broussard, D. Р. S h о е m а к е г, J. Am. Chem. Soc., 1960, 82, 1041.
7. К. Ранкама и др., в сб. переводных статей «Бериллий», ИЛ, 1955, 7.
8. Р. А й л е р. Коллоидная химия кремнеземов и силикатов. Госстройиздат, М.,
1959.
9. В. Э й т е л ь. Физическая химия силикатов. ИЛ, 1962.
10. Д. Брэк, Пат. ФРГ, 1098929, 1961.
157
Образец 833 с содержанием ВеО 21.4% относится по структуре
к фожазиту (табл. 6), по адсорбционным свойствам близок к свойствам
цеолита типа X. Он адсорбирует бензол, н-гексан. Однако адсорбционная
емкость бериллийалюмосиликатного цеолита несколько ниже по парам
воды, криптону, бензолу и н-гексану (рис. 9).
Для доказательства того, что бериллийалюмосиликатный цеолит пред-
ставляет собой продукт изоморфного замещения ионов кремния в тетра-
эдрах SiO4 на бериллий, была изучена реакция обмена полученных кри-
сталлов, содержащих разное количество бериллия, с раствором хлори-
стого натрия. Было показано, что содержание бериллия в образце до и
после обмена остается практически постоянным. Кристаллическая струк-
тура и адсорбционные свойства при такой обработке не меняются.
Методом ИКС установлено, что тетраэдры ВеО4 являются составной
частью структуры синтезированных цеолитов. На рис. 10, наряду с по-
лосой поглощения, отвечающей кремнекислородным и алюмокислсрод-
ным тетраэдрам, появляется полоса поглощения, отвечающая колебаниям
тетраэдров ВеО4. Таким образом, приведенные результаты указывают на
то, что синтезированные нами бериллийалюмосиликатные пористые кри-
сталлы являются продуктами изоморфного замещения ионов кремния
в тетраэдрах SiO4 на ионы бериллия.
Кроме бериллийалюмосиликатных пористых кристаллов, в аналогич-
ных условиях были получены алюмомагниевые силикаты. Согласно
рентгенограмме (табл. 6), синтезированный образец 1023 имеет.кристалли-
ческую структуру, идентичную цеолиту NaX. Адсорбционные исследова-
ния указывают на то, что эти ,кристаллы являются пористыми и обладают
молекулярно-ситовыми свойствами.
Синтез цеолитов с повышенным содержанием кремнезема
Начиная с 1961 г. после опубликования патента Брэка [10] внимание
исследователей привлек цеолит типа Y, который является синтетическим
аналогом природного цеолита фожазита. Кристаллическая структура
и адсорбционные свойства цеолита Y мало чем отличаются от молекуляр-
ного сита типа X, однако повышенное содержание кремнезема придает
ему некоторые новые свойства. Он обладает устойчивостью по отношению
к воздействию органических кислот, большей термостойкостью, чем NaX,
и высокой каталитической активностью в некоторых реакциях углево-
дородов. Расстояния между катионными участками — тетраэдрами
А1О4~6 — достигают 7 А, и это, видимо, является важнейшей особенностью
нового цеолита. По данным Брэка [10], цеолиты с соотношением
SiO2/Al2O3 > 4 можно получить, только применяя в качестве одного из
основных компонентов высококонцентрированный золь кремневой кис-
лоты. Мы поставили перед собой задачу выяснить возможность получения
цеолита типа Y силикатным методом. В результате многочисленных
опытов нами разработаны методы получения цеолитов химического со-
става 0.8—0.9 Na2O • А12О3 • (3.5—5.1)SiO2 • (6—8)Н2О. Полученные об-
разцы подвергались рентгеноструктурному исследованию, изучению ад-
сорбционных свойств, устойчивости к воздействию уксусной кислоты
и термоустойчивости.
Параметр элементарной кубической ячейки кристаллов цеолитов с со-
отношением SiO2/Al2O3 > 4 а°=24.72—24.68 А. Адсорбционная емкость
по парам воды при р/рв=0.14 (20°) достигает 0.29 см3/г и парам бензола —
0.28 см®/г.
Синтезированные образцы не растворяются в 3 н. уксусной кислоте,
в отличие от цеолитов типов А и X, и сохраняют адсорбционную емкость
156
по парам воды после обработки кислотой в течение 30 мин. при 95—98°,
равной 0.2 г/г. На рис. 11 представлена изотерма сорбции по парам воды
до и после обработки его кислотой.
Установлено, что термообработка до 800° не приводит к разрушению
кристаллической решетки.
Синтезированные цеолиты типа Y могут быть декатионированы мето-
дом ионного обмена катиона натрия на аммонийный ион с последующим
0 0.2 ОД 0.6 0.8 1.0p/ps
Рис. 11. Изотерма сорбции паров
воды на цеолите типа Y до (7) и
после (2) его обработки уксусной
кислотой.
10.0'---1----1---1----1----L-
О 0.2 ОД 0.6 0.6 1.0 p/ps
Рис. 12. Изотермы сорбции паров
воды на цеолите типа Y до (7) и
после (2) декатионирования.
прокаливанием или методом электродиализа. Получен декатионированный
цеолит типа Y с соотношением SiO2/Al2O3=4.7 со степенью декатионирова-
ния 94%, на рентгенограмме которого интенсивность дифракционных ли-
ний практически не изменилась по сравнению с исходным цеолитом. На
изотерме сорбции по парам воды уменьшается несколько крутизна восхо-
дящей ветви изотермы (рис. 12). Декатионированный в таких же условиях
цеолит NaX полностью разрушается, на рентгенограмме не обнаружено
дифракционных линий.
Таким образом, путем постепенного замещения в цеолитах ионов нат-
рия на другие катионы, изоморфного замещения кремния в кремнекисло-
родных тетраэдрах на элементы с координационным числом четыре и
изменения соотношения SiO2/Al2Oa в цеолитах можно в широких преде-
лах менять избирательные свойства пористых кристаллов.
ЛИТЕРАТУРА
1. О. М. Джигит, А. В. К и с е л е в, Г. Г. М уттик, Колл, ж., 1963, 25, 34.
2. Д. П. Тимофеев. Кинетика адсорбции. Изд. АН СССР, 1962.
3. И. В. М а т я ш, М. А. Пионтковская, Л. М. Та ра сенко, Р. С. Тю-
тю н н и к, Ж. структ. химии, 1962, 3, 214; 1963, 4, 106.
4. Н. А. Измайлов. Электрохимия, Изд. ХГУ, 1959.
5. О. М. Джигит, С. П. Ж д а и о в, А. В. К и с е л е в, Г. Г. Муттик,
ЖФХ, 1962, 36, 919.
6. L. Broussard, D. Р. Shoemaker, J. Am. Chem. Soc., 1960, 82, 1041.
7. К. P а в к а м а и др., в сб. переводных статей «Бериллий», ИЛ, 1955, 7.
8. Р. А й л е р. Коллоидная химия кремнеземов и силикатов. Госстройиздат, М.,
1959.
9. В. Э й т е л ь. Физическая химия силикатов. ИЛ, 1962.
10. Д. Брэк, Пат. ФРГ, 1098929, 1961.
157
РАВНОВЕСИЕ ИОННОГО ОБМЕНА НА ЦЕОЛИТЕ ТИПА А
А. С. Плачинда
С целью получения цеолитов типа А с переменным составом обменных
катионов исследовалось равновесие ионного обмена для следующих пар
катионов: Na+—Li+, Na+—K+,Na+—Rb+, Na+—Ag+, Na+—Ca2+, Na+—Co2+,
Na+—Ni2+, Ca2+—Co2+, Ca2+—Ni2+.
Навески цеолита, синтезированного в нашей лаборатории, заливались
водными растворами хлоридов металлов (в случае Rb и Ag брались ни-
траты). Анализировался равновесный раствор и в ряде случаев — обра-
зец. Так как применялись растворы концентраций главным образом 0.01—
0.1 и.' можно было вычислить активности ионов в растворе, исходя из пра-
Рис. 1. Зависимость логарифма коэффициента
равновесия от соотношения обмениваемых ка-
тионов для разных пар.
1 — Na+—Ni!+; 2 — Na—Со2+; 3 — Na+—Са2+.
вила ионнои силы и предполо-
жения, что активности ионов
К+ и С!" в растворах КС1
равны.
Было показано, что в слу-
чае обмена Na+ на однозаряд-
ные катионы в основном соблю-
дается закон действующих масс.
Наоборот, при обмене Na+ на
двухзарядные 'катионы коэф-
фициент равновесия К'А оказы-
вается непостоянным:
меняется в 100 раз, A'JJa—
в 1000 раз и К'со—почти
в 10000 раз (рис. 1).
Здесь
К'А == NZBMZAfZAl NZaMZ^4Z1s
л В — ;vA УИВ <В I JVB тА ‘А .
где N. = ----эквивалентная доля катиона в цеолите, С{ — кон-
А са + св
центрация катионов в цеолите в мэкв/г анионного каркаса цеолита,
тА
Zf — заряд катионов, МА~ 10Ц)----------------мольная доля катиона
~18~ + тА + гов + п
в растворе, т( и п — моляльные концентрации катионов и аниона
в растворе, yf— коэффициенты активности катионов в растворе.
Равновесия Са2+—Ni2+, Са2+—Со2+ исследовались лишь в небольшом
интервале замещений, однако и в этом случае было обнаружено изменение
коэффициента равновесия в 10 раз.
При обмене Na+ на двухзарядные катионы для большей части интер-
вала замещений соблюдается уравнение
СМе1СЯя — К (аМе1аТЯа)Р>
(1)
где а, — активности катионов в растворе (гэкв/л).
Эмпирические константы Аир этого уравнения, значения которых
приведены в табл. 1, не зависят от концентрации раствора (в изученных
пределах).
Из полученных данных можно вычислить значения коэффициентов
активности обменных катионов в твердой фазе (рис. 2) и значения термо-
динамической константы ионообменного равновесия (табл. 2), восполь-
158
Таблица 1 Таблица 2
Эмпирические константы Значения термодинамической
уравнения (1) константы ионообменного равновесия
Me к P K-Na KCa к-Na KCo JTNa KNi
N12+ 0.046 0.32 — 20 — 80 — 1400
Co2+ 0.17 0.22
Ca2+ 0.23 0.52
зовавшись для этого соответствующими выражениями, полученными
Гейнсом и Томасом [1]. Пренебрегая набуханием цеолита и адсорбцией
электролита, получаем
, loir f-
b/AB=-(ZA-ZB)^B-7VBln^A+ ‘
АВ
+ J (2)
О
bl /вА= (^А - zb) л'а + Л'а In -
1
- flntf'A.d2VB, (3)
*в
1
= ^в-za + J 0)
О
К,А/гв
где/£А=—---------константа рав-
^в
новесия, — коэффициенты ак-
тивности катионов в твердой
фазе (/А = 1 при 2Va = 1, /в = 1
Рис. 2. Зависимость логарифма коэффи-
циента активности катионов в твердой
фазе цеолита от их соотношения.
1 и Г — Na+ и Са2+ соответственно; 2 и 2' — Na+
и Nl2+; 3 и 3' — Na+ и Со2+.
при 7VB = 1).
Как следует из рис. 2, величина коэффициента активности данного
обменного катиона в цеолите сильно зависит от природы второго катиона.
Автор выражает благодарность проф. И. Е. Неймарку за интерес
к работе и обсуждение ее результатов.
ЛИТЕРАТУРА
1. G. L. G a i n е s, Н. С. Tho mas, J. Chem. Phys., 1953, 21, 714.
159
ОСОБЕННОСТИ СИНТЕЗА ЦЕОЛИТА СТРУКТУРЫ ТИПА ФОЖАЗИТА
В УСЛОВИЯХ ЛАБОРАТОРИИ И ОПЫТНОЙ УСТАНОВКИ
Л. И. Пигузова, А. С. Витухина, В. Ф. Дмитриева
Высококремнеземнистым синтетическим цеолитам со структурой и
отношением SiO2/Al2O3, отвечающим природному фожазиту, в патент-
ной литературе присвоено обозначение «У» [1, 2]. В последнее время
этот цеолит привлекает к себе внимание в связи с появлением в литературе
работ по его применению в катализе [3—5].
Цеолит Y, так же как и цеолит X, имеет кубическую решетку, отли-
чаемую параметром а, равным 24.65 А, вместо 24.9 А — для сита типа X.
Диаметр входного канала их также одинаков и равен 8—9 А [2, 6].
Мольное отношение SiO2/Al2O3 в цеолите Y изменяется от 3.1 до 6.0
против 2.4—2.8 для цеолита типа X [2, 5, 7, 8]. В элементарной ячейке
цеолита Y содержится вдвое меньше тетраэдров А1О4 и катионов по срав-
нению с цеолитом X, в чем, по-видимому, и кроется основная причина
отличий в свойствах двух цеолитов, а именно более высокая термическая
стабильность Na и особенно декатионированной формы цеолита Y, повы-
шенная устойчивость к кислотам, а также более высокая активность Са
и декатионированной формы в каталитических реакциях: крекинга, изо-
меризации, алкилирования и др.
Целью нашей работы было нахождение способа синтеза цеолита типа Y,
разработка метода получения активного и стабильного золя кремневой
кислоты, а также изучение основных свойств — термической и химиче-
ской стабильности.
Проведенное исследование адсорбционных свойств цеолита Y по воде,
СО2, бензолу, метанолу, 1,3,5-триэтилбензолу и др. показало, что по этим
свойствам фожазит близок к цеолиту X.
Согласно теории академика Д. С. Коржинского [9], активность Si02
зависит от щелочности среды кристаллизации: чем ниже щелочность,
тем выше активность Si02 и богаче кремнеземом минерал. Руковод-
ствуясь этим в синтезе фожазита, мы пользовались уменьшением щелоч-
ности при одновременном пересыщении кремнеземом реакционной среды,
которое достигалось практически при использовании золя кремневой кис-
лоты. В связи с этим возникли и два различных метода синтеза, условно
названные нами силикатным (при использовании силиката натрия) и
кремнезольным (при синтезе из золя кремневой кислоты).
Экспериментальная часть
В силикатном методе синтеза в лабораторных условиях исследовался
широкий диапазон изменения мольного отношения компонентов SiO2/Al2O3
от 7 до 16 и Na2O/SiO2 от 0.70 до 0.95. Однако отношение SiO2/Al2O3
в цеолите изменялось в большинстве опытов в пределах 3.1—3.6. В табл. 1
представлены оптимальные условия получения высококремнистого цео-
лита силикатным методом в лабораторных условиях (образцы 1 и 2) и
в условиях опытной установки (образцы 3 и 4).
Получение цеолита, более богатого кремнеземом, близкого по составу
к фожазиту, оказалось возможным при использовании золя кремневой
кислоты, позволяющего создать большие избытки SiO2 при малой щелоч-
ности и вводить к реакционную среду SiO2 в наиболее активной форме.
Из имеющихся литературных сведений нами был выбран распростра-
ненный способ получения золя кремневой кислоты с использованием иони-
тов [10, 11]. Разработанный вариант этого способа, применительно
160
упаривание до концентрации
35
Рис. 1. Влияние pH золя кремневой
с концентрацией SiO2 107 г/л на его
ность.
О 5 t0 15 20 25 30
Время, сутки
к синтезу цеолита типа фожазита, заключается в сочетании двух про-
цессов: обесщелачивание разбавленного раствора силиката натрия на
катионите марки КУ-2 и последующее его
150—250 г/л SiO2.
Было проведено исследо-
вание влияния pH золя крем-
невой кислоты различной
концентрации на его стабиль-
ность. При содержании
100 г/л SiO2 наиболее ста-
бильными оказались золи с
величинами pH 1.1—2.0 и
8.85—10.9 (рис. 1). При уве-
личении концентрации золя
до 250 г/л SiO2 предел вели-
чины pH, в котором золи
оставались стабильными, зна-
чительно сузился. Наиболее
стабильным оказался золь
с pH ~ 9 (рис. 2), в отличие
от золей с величиной pH
6-7.
Величины pH приготов-
ленных партий концентриро-
ванного золя были равны 8.5—9.0, а вязкость изменялась в пределах
1.5—4.0 сст и выше. Такой Золь был достаточно стабильным и активным
в синтезе высококремнистого цеолита.
Как уже отмечалось выше, характерной особенностью синтеза вы-
сококремнистого цеолита является
кислоты
стабиль-
время, сутки.
кислоты
стабиль-
Рис. 2. Влияние pH золя кремневой
с концентрацией SiO2 250 г/л на его
ность.
пониженная величина отношения
Na2O : SiO2, близкая к 0.4—
0.6, при высокой величине
отношения SiO2 : А12О8.
В табл. 1 представлены
наиболее характерные по хи-
мическому составу лабора-
торные образцы, полученные
кремнезольным способом.
Как видно из данных таб-
лицы, уменьшение количе-
ства щелочи в среде кристал-
лизации по отношению
Na2O : SiO2 с 0.60 до 0.47
приводит к постепенному
увеличению содержания SiO2
с 4.0 до 4.6 в готовом цео-
лите (обр. 5—8). Дальнейшее
уменьшение содержания ще-
лочи вызывало резкое увели-
чение продолжительности'
кристаллизации и загрязне-
ние целевого продукта не-
Некоторое повышение содер-
и путем увеличения концентра-
ции реагирующих веществ, и относительного избытка двуокиси кремния
в реагирующей массе (образец 9). Те же закономерности подтвердились
на образцах, полученных в укрупненном масштабе (образцы 10—12).
закристаллизовавшейся аморфной SiO2.
жания SiO2 в готовом цеолите достигалось
Ц Цеолиты
161
Таблица 1
Зависимость химического состава и адсорбционной емкости по парам воды
цеолитов от условий их кристаллизации
№ об- разца Условия кристаллизации Мольное отношение SiO2/Al2O3 в готовом цеолите Влагоемкость в г/г сорбента, прокаленного при 600° С, при относительной влажности воздуха, %
концентрации мольные отношения
SiO,, г/л А12О3, г/л S iO 2/ AljOg Na,O/SiO, ЩО/Na.O
100 20 <0.1
И
Силикатный метод
1 75 14.0 9.0 0.80 50 3.3 0.32 0.24 0.24
2 85 9.6 15.0 0.90 50 3.5 0.31 0.29 0.27
3 85.5 10.0 15.5 0.93 40 3.3 0.29 0.25 0.23
4 85.5 10.0 15.5 0.93 40 3.4 0.33 0.29 0.27
Кремнезольный метод
5 6 7 8 . 100 8.5 20 0.58 0.55 0.50 0.47 34 40 55 67 4.0 4.2 4.4 4.6 0.31 0.32 0.36 0.34 0.27 0.29 0.30 0.29 0.27 0.27 0.29 0.29
9 130 8.2 27 0.47 67 5.2 0.27 0.23 0.22
10 0.52 50 4.0 0.33 0.26 0.25
11 100 8.5 20 0.50 55 4.2 0.31 0.26 0.21
12 0.47 65 5.2 0.26 0.21 0.20
Таблица 2
Сопоставление рентгеноструктурных данных, полученных на образцах цеолита
типа фожазита различного химического состава
образ- ца Название образца и тип Химический состав, моль Константы решетки а, А
Na,O СаО А12О, S1O,
1 Природный фожазит [12] 0.43 0.54 1.0 5.0 24.74—24.65
Кремнезольный метод
2 NaY (по Бреку) [1] . . 0.94 — 1.0 4.8 24.60
3 NaY-54 0.93 .— 1.0 5.7 24.63
4 NaY-68 0.85 -— 1.0 4.5 24.67
5 NaY-14 0.85 — 1.0 3.8 24.70
Силикатный метод
6 NaY (по Бреку) [1] 0.92 — 1.0 3.29 24.87—24.77
7 NaY-20-МОБ 1.0 ‘ •— 1.0 3.35 24.75
8 NaX 1.0 — 1.0 2.63 24.90
Проведенное исследование позволило представить зависимость со-
держания SiO2 в готовом цеолите от содержания щелочи в среде кристал-
лизации в виде примерной кривой, показанной на рис. 3.
На рис. 4 представлены поля кристаллизации цеолитов структуры X
и Y, силикатного и кремнезольного методов получения. Участок, лежащий
выше линии, проходящей через деление, составляет поле кристаллизации
высококремнистого цеолита — аналога фожазита.
Результаты рентгеноструктурного анализа, выполненного Н. А. Ши-
шаковым, представлены в табл. 2. Показано постепенное увеличение па-
162
раметра а с уменьшением содержания SiO2 в цеолите. Кроме того, с по-
мощью рентгеноструктурного анализа выявлялось присутствие в цеолите
некоторого количества паразитирующего минерала типа NaP и аморфной
SiO2. Получение чистой фазы фожазита составляло основную тонкость
Na.zO/StOz В мелях 6реляционной среде
Рис. 3. Изменение химического состава готового цеолита в за-
висимости от щелочности реакционной среды в процессе
кристаллизации.
синтеза и являлось наиболее трудным при переходе от лабораторных
условий (реактор емкостью 5 л) к масштабам пилотной установки (реактор
емкостью 80 л).
Было применено подкисление золя соляной кислотой, которое способ-
ствовало некоторой деполимеризации коллоидных частиц и увеличению
активности SiO2 ИЗ].
Рис. 4. Поля кристаллизации цеолитов структуры Y и X.
Из данных, представленных в табл. 3, видно, что подкисление щелоч-
ного золя с малой вязкостью и, следовательно, малой степенью полимери-
зации не приводило к существенному увеличению активности SiO2, но
увеличивало содержание SiO2 в цеолите с 3.7 до 4.1 молей (образцы 1—3).
Увеличение активности SiO2 в золе становится заметным при подкисле-
нии более вязких золей, которые до этого в синтезе цеолита были неак-
тивны (образцы 4, 5). Та же закономерность проверялась в оптимальных
11*
163.
Таблица 3
Влияние величины pH исходного золя кремневой кислоты на качество
получаемого сорбента типа фожазита
Условия кристаллизации
X арактеристика
золя
концентра- мольные отноше-
нии ния
Адсорбционная
емкость сорбента,
прокаленного
при 600° С, г/г
Химический со-
став цеолита, моль
Относительная
влажность, %
1
2
3
4
5
6
7
8
8.7
2.0
1.5
8.0
1.75
8.4
1.75
0.75
100
1
ИЗО
0.60 25 0.34 0.30
20 0.50 55 0.33 0.0 0.33
27 0.47 65 0.25 0.24
0.05
20 <0.1 О £ О 5
0.31 0.29 0.85 1.0
0.26 0.25 1.1 1.0
0.30 0.27 0.90 1.0
0.0 0.0 — —
0.29 0.25 1.0 1.0
0.21 0.20 1.0 1.0
0.20 0.19 0.80 1.0
0.02 0.01 0.81 1.0
6
те
3.7
4.1
4.1
4.4
4.6
5.0
5.3
о
о
условиях осаждения, при соотношении Na2O : SiO2, равном 0.47 (образцы
6—8). Наилучшие результаты дало подкисление золя до pH 2.0—1.5.
Изучение зависимости термической стабильности образцов от хими-
ческого состава как для Na-формы, так и декатионированной позволило
сделать вывод, что цеолиты с мольным отношением SiO2 : А12О3=3.4—6.0
стабильны до 800° С (табл. 4).
Процесс декатионирования (до 80%) приводит к повышению порога
термической стабильности до 900° С только для образцов цеолита, по хи-
мическому составу близких к фожазиту (образцы 9, И). Высококремнистые
Таблица 4
Зависимость термической и химической устойчивости цеолита структуры
Y и X от химического состава
№ об- разца Название образцов, тип цеолита Мольное отношение SiO2/Al2O8 в готовом . сорбенте Величина адсорбции сорбента, прокаленного при различных температурах, при относительной влажности воздуха <0.1 г/г Степень рас- творения цеолита (в пересчете на А12О3), вес. %
600° 800° 900° после об- работки Зн. СНзСООН, 98° С (600°)
1 NaX ВНИИНП } 2-5 { 0.25 0.0 0.0 0.0 100
2 NallX ВНИИНП 0.16 0.0 0.0 0.0 100
3 NaY-20-МОБ } м ( 0.27 0.16 0.0 0.0 100
4 CaY-20-МОБ 0.26 0.23 0.0 0.0 100
5 NaY-39-МОБ } 4.0 { 0.25 0.21 0.0 0.09 60
6 NaHY-39-МОБ 0.26 0.20 0.0 0.07 70
7 NaY-48 ' 4.7 0.16 0.16 0.0 0.14 2.0
8 NaY.-41-МОБ } 5-2 { 0.20 0.17 0.0 0.16 2.0
9 HNaY-41-МОБ 0.20 0.18 0.17 0.14 3.0
10 NaY-54 1 5-’ { 0.19 0.18 0.0 0.17 2.0
И HNaY-54 0.20 0.18 0.16 0.16 1.0
164
образцы, содержащие около 5 молекул SiO2, оказались практически нерас-
творимыми в уксусной кислоте в отличие от образцов цеолита типа X и Y
силикатного метода приготовления.
Обработка 3 н. НС1 приводила к разрушению цеолита типа фожазита
и снижению адсорбционной емкости по воде.
Выводы
1. Разработана в лаборатории и опробована в условиях опытной уста-
новки пропись приготовления цеолита типа фожазита различного хими-
ческого состава с воспроизводимыми свойствами на базе использования
силиката натрия и золя кремневой кислоты.
2. Разработан метод приготовления концентрированного (до 250 г/л
SiO2) стабильного щелочного и кислого золя кремневой кислоты, пригод-
ного для получения высококремнистого цеолита типа фожазита. Показана
возможность повышения активности золя при подкислении его с рН=
=8—9 до рН=1.5—2.0.
3. Показана повышенная устойчивость структуры типа Y к кислотам
при температуре 95—98° С и продолжительности обработки 1 час по срав-
нению со структурой X, разрушающейся в этих условиях.
ЛИТЕРАТУРА
1. R. М. Milton, D. W. В reck, US Pat., 2988503, 1961.
2. D. W. В reck, FDR Pat., 1098929, 1961.
3. P. A. Howell, J. Phys. Chem., 1960, 3, 64, 364.
4. J. Rah о, P. Picker t, D. Stamires, Actes Congr. Internet. Catalyse,
Paris, 1960, 2, 2055.
5. T. L. Thomas. Доклад на VI Мировом нефтяном конгрессе в 1963 г. в ФРГ.
6. R. М. М i 11 о n, US Pat., 2882244, 1959.
7. D. F г е е m a n et al., J. Chem. Phys., 1961, 35, 3, 799.
8. Л. И. П и г у з о в а, А. С. Витухина и др., в сб.: Синтетические цеолиты,
Изд. АН СССР, 1962, 152.
9. Д. С. К о р ж и н с к и й, Геохимия, 1956, 7, 3.
10. Р. К. А й л е р. Коллоидная химия кремнезема и силикатов. Гос. изд. лит.
по строит., архитектуре и строит, материалам, М., 1959.
11. Ch. Kimber lin et al., US Pat., 2798857, 1957.
12. R. M. Barter, W. В u s e r et al., Chimia, 1955, 9, 118.
13. K. J о t o, J. Phys. Chem., 1956, 60, 7, 1007.
О ЗАВИСИМОСТИ ОТНОШЕНИЙ SiO2/Al2O;i В ФАЗАХ
ПЕРЕМЕННОГО СОСТАВА ОТ ЩЕЛОЧНОСТИ
МИНЕРАЛООБРАЗУЮЩЕГО РАСТВОРА
Э. Э, Сендеров
Для целей практического получения кислотостойких цеолитов важно
выяснение условий, в которых образуются обогащенные кремнеземом
фазы. Именно такие цеолиты оказываются устойчивыми в кислых средах.
Ранее на примере изучения кристаллизации в системе морде-
нит—Na2O—Н2О [1] нами было показано, что поле кристаллизации обо-
гащенной кремнеземом фазы — морденита сужается при увеличении
щелочности минералообразующего раствора, и эта фаза может быть
вытеснена более бедными SiO2 анальцимом или (при более низких тем-
165
пературах) шабазитом. Объяснение этому явлению может быть дано
на основе представлений о зависимости активности кислотных и основных
компонентов в растворе от условий кислотности или щелочности [2].
При постоянной суммарной концентрации данного кислотного компо-
нента его активность с увеличением щелочности падает, а с уменьшением
ее, т. е. с увеличением кислотности, возрастает. Для более сильных кис-
лот изменение активности значительнее. Качественно это явление можно
объяснить тем, что в более щелочном растворе бблыпая доля кислотного
компонента связана тем или иным способом с каким-либо основанием,
в то время как активность этого компонента пропорциональна количе-
ству несвязанных и неионизированных его частиц.
Поскольку SiO2 является в растворе слабой кислотой, возрастание
щелочности раствора вследствие увеличения количества избыточной NaOH
в смеси приводит к уменьшению активности кремнезема. Поэтому равно-
весие фаз, содержащих различные его количества, сдвигается в сторону
более бедных SiO2 разностей [1J.
Однако для цеолитов характерно в той или иной степени образование
фаз переменного состава с различным отношением SiO2/Al2O3. Они пред-
ставляют как бы твердые растворы двух конечных членов интервала сме-
симости: наиболее бедной и наиболее богатой кремнеземом разности.
В этом случае распределение кремнезема между раствором и кристаллом
должно подчиняться закону распределения, согласно которому отношение
активностей SiO2 в растворе и кристалле равно постоянной величине —
коэффициенту распределения. Поэтому уменьшение щелочности раствора,
в котором происходит кристаллизация, вызывая возрастание активности
кремнезема, должно привести к образованию разности, обогащенной
двуокисью кремния.
Это подтверждается данными по изменению состава анальцима при
кристаллизации его из смесей, содержащих различные количества избы-
точной гидроокиси натрия. Было показано, что анальцим обладает пере-
менным составом по содержанию SiO2 и А12О3, причем величина а0 — ребра
элементарной кубической ячейки анальцима—линейно уменьшается с уве-
личением отношения SiO2/Al2O3 в составе кристаллов [3, 4], что вызвано
меньшим размером тетраэдра SiO4 по сравнению с АЮ4. Эти данные были
использованы нами для определения составов анальцимов, полученных
в условиях различной щелочности.
Было приготовлено несколько серий смесей с различными соотноше-
ниями SiO2 и А12О3 (n=SiO2/Al2O3), указанными в таблице. Смеси готови-
лись из свежеосажденного силикагеля и алюмината натрия. Сверх того
количества Na2O, которое нужно для построения решетки натриевого
алюмосиликата, в смесь добавлялся избыток Na2O в виде гидроокиси
натрия. Избыточная щелочность регулировалась или добавлением раз-
ных количеств NaOH, или нейтрализацией избытка NaOH соляной кис-
лотой.
Смеси помещались в автоклав вместе с количеством воды, рассчи-
танным таким образом, чтобы отношение SiO2 к общему количеству Н2О
(вода смеси+добавленная вода) в каждой серии опытов (n=const) оста-
валось одним и тем же. Отношения количества Na в смеси к общему коли-
честву воды во всех опытах с исходным Na2O/Al2O3=4 было также по-
стоянным и равнялось 30 мг/мл. Только в серии опытов, где щелочность
регулировалась уменьшением количества первоначально добавленной
NaOH, эта концентрация последовательно уменьшалась.
Смеси подвергались кристаллизации при 250° С и давлении, равном
давлению насыщенного пара раствора, в течение двух—четырех суток.
Единственным продуктом оказался анальцим. Для него были сняты рент-
генограммы на приборе УРС-55 в камере с диаметром 57.3 мм. Использо-
166
валось фильтрованное железное излучение. По рентгенограммам был рас-
считан параметр а0 анальцима по отражениям от плоскостей с Л2+й2-Н2,
равным 180, 182, 186, 190, 194, 196 и 198. По а0 с помощью данных Саха [3,
4] было определено отноше-
ние SiO2/Al2O3 в кристаллах
анальцима.
Эти результаты представ-
лены в таблице и показаны
на рисунке. Они показывают
возрастание отношения
SiO2/Al2O3 в анальцимах,
кристаллизующихся из менее
щелочных растворов, т. е, из
растворов, в которых актив-
ность SiO2 должна быть выше.
Поскольку анальцим явля-
ется единственным продуктом
кристаллизации, такая зави-
симость и ожидалась. Отно-
шение SiO2/Al2O3 зависит
только от концентрации
NaOH; на него мало влияет
С ЫагОЧйг03'гМОлгНС1 Б ЗБа.г0-А1гО3-Б.8510г
В Шг0-А1г03-п$10+ЫС1 В гНагО-А1г03-Б.88:Ог
Г Ыа30-А1г03-п310£бНС1 Г Na.zO-Al303^.8SiOe
присутствие или отсутствие
в растворе дополнительных
солей. Это подтверждается
сходным изменением состава
анальцима в серии опытов,
проведенных в присутствии
и отсутствии NaCl (см. ри-
сунок, а, б), Образующегося
Зависимость отношения SiO2/Al2O3 в анальциме
от количества избыточной щелочи в исходной
смеси (по отношению к содержанию Na2O в аналь-
циме).
Исходные смеси: а -» с добавкой HC1; б — без добавки
НС1. п — отношение SiO2/Al2O3 в исходных смесях.
при нейтрализации гидроокиси натрия соляной кислотой.
По-видимому, зависимость содержания SiO2 в кристаллах и, следова-
тельно, активности SiO2 в растворе от концентрации NaOH является
линейной.
Значения а0 для анальцимов, кристаллизовавшихся при 250° С из смесей
с различным избытком NaOH, и соответствующие величины отношения
SiO2/Al2O3 в них
Соотношения компонентов в исходных смесях a0 в кристаллах, к SiO2/Al2O3 в кристаллах
4Na2O • А12О3 • 4.2SiO2+2HCl 13.724+0.003 3.9+0.1
4NaaO • А12О3 • 4.2SiO2+4HCl 13.719+0.004 4.1+0.2
’ 4Na2O • А12О3 • 4.2SiO2+6HCl 13.698 +0.004 4.8+0.2
4Na2O • A12O3 • 4.8SiO2 13.729 + 0.004 3.7+0.2
4Na2O • A12O3 • 4.8SiO2+2HCl 13.717+0.003 4.1+0.1
4Na2O • A12O3 • 4.8SiOa+4HCl 13.697+0.005 4.8+0.2
4Na2O • A12O3 • 4.8SiO2+6HCl 13.683+0.004 5.4+0.2
4Na2O • A12O3 • 6.4SiO2+2HCl 13.692+0.004 5.1+0.2
4Na2O • A12O3 • 6.4SiO2+4HCl 13.672+0.002 5.7+0.1
4Na2O • A12O3 • 6.4SiO2+6HCl 13.655+0.002 6.3+0.1
4Na2O • A12O3 • 4.8SiO2 13.729 +0.004 3.7+0.2
3Na2O • A12O3 • 4.8SiO2 13.719+0.002 4.0+0.1
2Na2O • A12O3 • 4.8SiO2 13.690 +0.004 5.1+0.2
lNa2O • A12O3 • 4.8SiO2 13.684 +0.004 5.3±0.2
Примечание. Отношение S1O2/A12O3 в кристаллах. Рассчитано с помощью данных
Саха [*].
167
Интересно отметить, что при отсутствии избыточной щелочи или при
полной ее нейтрализации отношение Si02/Al203 в анальцимах может быть
несколько выше, чем в исходной смеси, что свидетельствует об обогащении
кристаллов кремнеземом в этих условиях по сравнению с минералообра-
зующим раствором, если, конечно, данные Саха [4] точны.
Таким образом, в случае кристаллизации фаз с переменным отношением
SiO2/Al2O3 в их составе понижение щелочности раствора способствует
образованию обогащенных кремнеземом разностей. Это обстоятельство
важно при получении, например, обогащенных двуокисью кремния раз-
ностей фожазита [5].
ЛИТЕРАТУРА
1. Э. Э. С е н д е р о в, Геохимия, 1963, 9, 820.
2. Д. С. Коркинский, Геохимия, 1956, 7, 3.
3. Р. Saha, Amer. Mineral., 1959, 44, 300.
4. Р. Saha, Amer. Mineral., 1961, 46, 859.
5. С. П. Жданов, H. H. Бунта рь, Изв. АН СССР, сер. хим., 1963, 11, 2061.
СИНТЕЗ И ИССЛЕДОВАНИЕ СВОЙСТВ
ВОДОРОДНЫХ ФОРМ ЦЕОЛИТОВ
Г. В. Цицишвили, Г. Д. Багратишвили, К. А. Бежашвили,
К. Е. Авалиани, Ц. А. Геджадзе, М. С. Шуакришвили, Ц. М. Окропиридзе„
М. Г. Адолашвили
Исследование свойств и возможностей применения водородных ионо-
обменных форм цеолитов привлекает значительный интерес исследовате-
лей. Этот интерес обусловлен рядом специфических свойств водородной
формы цеолитов (высокая каталитическая активность, адсорбционная
способность, кислотостойкость и др.).
Ранее спектроскопическим и химическим методами нами [1] было
показано, что при термической обработке аммониевых цеолитов происхо-
дит разложение иона аммония и таким путем можно получать водородные
формы цеолитов
NH4A = HA+ nh3,
где А — анионная часть алюмосиликатного скелета цеолита.
Термическая обработка аммониевых цеолитов типа А и X проводилась
нами при температуре около 300° С в течение трех часов. При этом для
сравнения проводились опыты по термической обработке аммониевых
цеолитов на воздухе и в вакууме. Изучалось также влияние содержания
иона аммония в исходных аммониевых цеолитах на свойства полученных
водородных форм. Дебаеграммы полученных образцов водородных форм
цеолитов показывали структуру, характерную для цеолитов данного типа.
Исследование на микровесовой установке адсорбционных свойств во-
дородных форм цеолитов типа А и X, полученных термообработкой аммо-
ниевых форм в вакууме, показывает, что с увеличением степени замещения
Na+ на NH4+ в цеолите наблюдается вначале некоторое повышение адсорб-
ционной способности по парам воды, которая затем уменьшается (рис. 1).
Уменьшение адсорбционной способности водородных форм цеолитов по
парам воды, по-видимому, частично обязано их деструкции. Кривые
зависимости величины адсорбции паров воды водородными формами цео-
168
литов от содержания водорода 1 в цеолитах типа А и X приведены на
рис. 2. Как видно из рисунка, адсорбционная способность водородных
форм цеолитов зависит от степени замещения ионов натрия на ионы
аммония (водорода). Верхней границей, соответствующей стабильности
водородных форм цеолитов типа А, является степень замещения около
20%, а в цеолите типа X степень замещения значительно выше (рис. 2).
Из сопоставления изотерм адсорбции паров воды на водородных фор-
мах цеолитов, полученных термическим разложением аммониевых цео-
литов в условиях вакуума
или на воздухе (рис. 3),
вытекает важный вывод о луч-
шем сохранении цеолитной
структуры водородных форм
молекулярных сит в процессе
получения в условиях ваку-
ума. При этом различие в ад-
сорбционных свойствах по
воде водородных форм цеоли-
тов, полученных в вакууме
или на воздухе, больше для
цеолитов с высокой'степенью
замещения. Эта разность
особенно наглядно проявля-
ется при исследовании низко-
температурной (—195.8° С)
адсорбции паров азота на
водородных формах цеолитов
(рис. 4). Образец водород-
ного цеолита, полученного
на воздухе, примерно на 70%
теряет способность адсорбции
азота по сравнению с образ-
цом водородной формы цео-
лита того же состава, полу-
ченного в вакууме. Однако
Рис. 1. Изотермы адсорбции паров воды при
20° С на натриевых и водородных цеолитах.
1 — NaA; 2 — 0.15Н, 0.85NaA; 3 — 0.2Н, 0.8 NaA;
4 — 0.38Н, 0.62NaA; 5 — 0.51H, 0.49 NaA; 6— 0.7 H,
0.3NaA; 7 — NaX; 8 — 0.55H, 0.45 NaX; 9 —0.63H,
0.37 NaX и 10 — 0.74 H, 0.26 NaX.
возможно, что причина этого
явления кроется не в агрессивном действии кислорода воздуха на
цеолит водородной формы вообще, а в действии воздуха во время процесса
его получения. В самом деле, если водородной формы цеолит, получен-
ный вакуумной термообработкой, нагреть повторно при доступе воздуха
в тех же условиях, как и при получении цеолита на воздухе, то его ад-
сорбционная способность по азоту снижается всего лишь на 10% (рис. 4).
Водородная форма цеолитов обладает одним важным свойством, ко-
торое привлекает особое внимание. Как известно, преобладающее боль-
шинство природных и искусственных цеолитов сравнительно легко раз-
лагается кислотами, т. е. они нестойки в кислой среде, что значительно
сужает область их практического применения. Кислотостойкость цеолитов
существенно зависит от отношения кремния к алюминию. В самом деле,
отношение Si02/Al203 для кислотостойких форм цеолитов — «Зеолона»
доходит до 5, а в мордените оно достигает 10.
Нами замечено, что полученные водородные формы цеолитов типа А
и X с отношением SiO2/Al2O3 1.95 и 2.5 обладают значительно повышен-
1 Содержание водородных ионов в водородных цеолитах принято условно.
Для упрощения обозначений допускается сохранность количества ионов водорода,,
образующихся при термической деструкции ионов аммония.
169
а, ммоль/г
5
а, ммоль/г
О 0.2 0.4 0.6 0.8 1.0 р/р
Рис. 3. Изотермы адсорб-^jg
ции паров воды при 20°С
на водородных формах
цеолитов, полученных
путем термической об-
работки аммониевых
цеолитов состава 0.15 Н,
0.85 NaA в вакууме (7)
и’на воздухе (2) и соста-
ва 0.70 Н, 0.30 NaA в ва-
кууме (2) и на воздухе (4).
I I -»---—i—'
0 40 so
Рис. 2. Зависимость
адсорбции паров воды
при p/ps=0A на во-
дородных цеолитах
типа А и X, полу-
ченных в вакууме,
от степени замещения
Na+na Н+ (NH4+) в
цеолите.
h, ммоль/г
Рис. 4. Изотермы адсорб-
ции паров азота при
—195.8°С на цеолите NaA (4)
и на образцах водородных
цеолитов типа А, получен-
ных путем термической об-
работки аммониевого цео-
лита в вакууме (7), на воз-
духе (2) и полученном в
вакууме и затем обработан-
ном на воздухе аналогично
предыдущему образцу (2).
а, ммоль/г
0 0.2 0.4 0.6 0.8 1.0 p/ps
Рис. 5. Изотермы адсорбции
паров воды при температуре
20°С на NaA-цеолите (2), на
водородном цеолите состава
0.2 Н, 0.8 NaA (7) и на образ-
цах водородных цеолитов пре-
дыдущего состава, обработан-
ных соляной кислотой концен-
трации 0.1 и. НС1 в течение
6 часов (2), 0.3 и. НС1 в тече-
ние 6 часов (4) и 0.3 н. НС1
в течение 30 часов (5).
ной стойкостью к кислотам по сравнению с натриевыми и другими катион-
ными формами (рис. 5).
Следует отметить, что натриевые цеолиты, полученные обратным ион-
ным обменом из водородной формы цеолита, теряют характерную для во-
дородной формы кислотостойкость. Таким образом, по-видимому, ки-
слотостойкость водородных цеолитов обусловлена именно существованием
в них водорода, а не структурными изменениями, которые можно было
допустить при термообработке аммониевых цеолитов в процессе полу-
чения водородных форм.
ЛИТЕРАТУРА
1. Г. В. Цицишвили, Г. Д. Багратишвили, К. А. Б еж аш в и л и,
Д. И. Б ар и а б ишвил и, М. С. Ш уак ришви ли, ДАН, 1 936, 152,
1136.
ИССЛЕДОВАНИЕ ВОЗМОЖНОСТИ ПОЛУЧЕНИЯ ЦЕОЛИТОВ
С ИЗОМОРФНЫМ ЗАМЕЩЕНИЕМ ХРОМОМ
Н. Ф. Ермоленко, Л. В. Пансевич-Коляда
Использование цеолитов в различных областях производства выдвигает
насущную потребность в получении цеолитов, обладающих самыми раз-
ными свойствами. В связи с этим путем варьирования условий в настоя-
щее время получено большое количество разных типов цеолитов. Широко
ведутся исследования по получению новых форм цеолитов путем катионо-
обмена. Особый интерес представляет получение новых форм цеолитов
путем изоморфного замещения как кремния, так и алюминия в кристал-
ле молекулярного сита. Последнее модифицирование было осуществлено
Бэррером и др., получившими цеолиты с замещением кремния на герма-
ний, а алюминия на галлий [1, 2].
В настоящей работе мы попытались получить цеолиты, в которых
алюминий изоморфно замещался бы на хром. Подобный выбор был про-
диктован, с одной стороны, тем, что хром и алюминий изоморфны, оба
являются трехвалентными металлами, способными в щелочной среде
давать однотипные растворимые соединения, с другой стороны, получение
цеолита, содержащего хром в изоморфно-замещенном виде, представляет
интерес с точки зрения изучения возможности использования его как ка-
тализатора. Указания на применение цеолитов, содержащих хром в ка-
честве катализаторов, имеются в некоторых патентах и работах (3].
Нами был получен ряд образцов цеолитов, содержащих в своем составе
хром в неионообменной форме. В настоящей работе приведены данные по по-
лучению цеолита типа NaA, в состав которого входит хром в различных
соотношениях, изучены его структура, адсорбционные и некоторые ка-
талитические свойства.
Нами получено шесть образцов цеолитов типа NaA, содержащих хром
в неионообменной форме. Детали метода будут рассмотрены в другой ра-
боте. В данной статье мы остановимся на характеристике полученных
образцов.
Кроме исследованных цеолитов, для получения сравнительных дан-
ных по каталитическим свойствам образцов нами были приготовлены
также катализаторы; содержащие хром: 1) обработкой готового цеолита
типа NaA промышленного изготовления раствором хромата с последую-
щим выдерживанием смеси в течение 6 часов при температуре 110°, с даль-
171
нейшим фильтрованием, отмыванием до pH 9 и высушиванием при 6U—
80°; 2) путем нанесения свежеприготовленного, отмытой) от щелочи
гидрата окиси хрома в определенном соотношении на промышленный-
цеолит NaA, с последующей сушкой и измельчением.
Образцы синтезированных цеолитов подвергались химическому и рент-
геноструктурному анализу. Рентгеноструктурный анализ проводили в ап-
парате УРО 50-И. Условия съемки: напряжение 32 кв, сила тока 10 мка,
ширина щели расходимости В=2 мм, ширина
щели счетчика а=0.25 мм, скорость счетчика
0.5 град/мин. Образец неподвижен, Сп-излу-
чение, Ni-фильтр. Регистрация излучения про-
водилась счетчиком Гейгера—Мюллера МСТР-4.
Рентгенографические исследования осуществле-
ны А. А. Прокоповичем.
Идентичность полученных нами образцов
с цеолитом NaA подтверждена данными рент-
геноструктурного анализа. Адсорбционную ем-
кость по воде проверяли в вакуумной установке
с пружинными кварцевыми весами; температура
прокаливания образцов в установке 265°.
Каталитические свойства изучались в про-
точной установке обычного типа по реакции
дегидратации этилового спирта.
В табл. 1 представлен состав полученных
цеолитов по данным химического анализа.
Рис. 1. Образец 9. Как вытекает из табл. 1, увеличение содер-
жания SiO2 и Сг2О3 в исходных растворах ведет
к увеличению процентного содержания этих компонентов в конечном
пРоДУкте. Увеличение содержания Сг2О3 происходит за счет уменьше-
ния частично Si<J2, в основном же за счет уменьшения А12О3 в кристал-
лическом веществе.
Электронномикроскопическое исследование образцов показало, что
при меньшем содержании Сг2О3 кристаллы получаются на снимке более
четкими. Образец 9 содержит всего 3.5% Сг2О3 (рис. 1). Для препаратов 37,
39, 40, содержащих от 8 до 1196 Сг2О3 (рис. 2—4), характерны довольно
хорошо оформленные кристаллы, но появляется некоторая «лохматость»
поверхности, вероятно благодаря присутствию небольшого количества
аморфной фазы. У препаратов 83, 84, в состав которых входит 12.5—
26.596 Сг2О3 (рис. 5, 6), тоже имеются хорошо оформленные кристаллы,
Таблица 1
Состав синтезированных цеолитов типа 4А, изоморфно замещенных хромом
№ образ- ца Состав конечного кристаллического продукта, % Мольный состав кристаллической фазы
Na2O SiO2 AI2Os Cr2O3
NaA 14 35.6 31 0.75Na2O • А12О3 • 1.9SiO2
37 12.9 25.9 16.3 8.1 1.3Na2O • Al2O3 • О.ЗЗСг203 - 2.78Юг
39 14.8 28.8 . 20 10.9 1.2Na2O • А12О3 - 0.34Сг2О3 • 2.4SiO2
40 16.6 25.1 17.4 11.1 1.6Na2O • А12О3 • 0.43Сг2О3 • 2.4SiOa
9 16 33 25.3 3.5 Ша20 • А12О3 • 0.09Сг203 • 2.2S1O,
83 11 24.5 20.4 26.5 0.88Na2O • А12О3 • 0.87Сг2О3 • 2Si62
84 13 32.9 22.4 12.4 0.93Na2O • А12О3 • 0.37Сг2О3 • 2.5SiO2
51 13.9 29.8 — 24.9
172
Рис. 3. Образец 39.
Рис. 2. Образец 37.
Рис. 4. Образец 40.
Рис. 5. Образец 83.
7Д
Рис. 6. Образец 84.
Рис. 7. Образец 51.
но аморфная фаза дает похожую картину. При большом количестве Сг
он, очевидно, весь не входит в решетку и кристаллы кажутся «лохматыми».
В случае образца 51 или хромосиликата, хотя под микроскопом были
видны достаточно четкие частички, напоминающие цеолит NaA, оказа-
лось, что он является рентгеноаморфным (рис. 7). В отношении остальных
образцов рентгеноструктурный анализ подтвердил наличие кристалли-
ческой структуры типа NaA (табл. 2).
Таблица 2
Данные рентгеноструктурного анализа образцов синтезированных цеолитов
NaA наши данные Номер обраеца
37 39 40 83 84
I d I d Г d I d I d I d
93 12.36 61 12.47 100 12.47 50 12.53 75 12.34 92 12.3
— —— — — — — 10 10.75 —— — — —
49 8.74 33 8.73 21 8.73 15 8.51 25 8.74 43 8.73
-—. — ——• — — — — — —— — —
30 7.12 45 7.15 42 7.13 35 7.12 25 7.11 19 7.23
— — 45 6.39 50 6.39 — — 46 6.49 — —
-—. — — — — — .— .— -— — — —
— — — — — — — •— — — — —
— , . — —— — — ——
25 5.51 — — — — 25 5.43 — — 36 5.66
— — — — 16 5.01 — — — — 10 5.17
20 4.35 22 4.50 — — 15 4.36 — — 12 4.77
— — —— .—• —— — — — , — —
65 4.10 56 4.10 100 4.10 57 4.10 — — 54 4.11
84 3.71 100 3.70 67 3.70 100 3.72 71 3.71 68 3.71
— — 55 3.63
18 3.41 40 3.40 16 3.39 28 3.41 —— — — —
86 3.29 90 3.28 80 3.28 60 3.27 58 3.29 64 3.30
— — —— — 33 3.16 -—. — — — — •—
— — — — — — —— — — —
100 2.98 84 2.97 92 2.97 80 2.98 100 2.98 100 2.98
—. — 67 2.94 32 2.94 80 2.94 — -—. -—- —
12 2.89 16 2.89 — — 43 2.88 — — — —
— — 25 2.80 — —— — —— —— ——
18 2.75 — — 42 2.74 15 2.74 — — — —
7 2.69 — — 16 2.68 20 2.68 — — — —
84 2.62 60 2.61 75 2.61 70 2.62 67 2.63 70 2.63
Как можно видеть из указанной таблицы, для всех образцов [сохра-
няются в основном все линии, наиболее характерные для цеолита типа
NaA. Это линии большой интенсивности и характеризующие их меж-
плоскостные расстояния: 12,36; 8.74; 4.10 и 3.29.
Однако для синтезированных хромовых цеолитов появляются и но-
вые линии, довольно сильные по интенсивности — 7=45—50 и 7=67—80,
с межплоскостными расстояниями d=6.39 и d=2.94.
Для характеристики адсорбционных свойств синтезированных нами
образцов была исследована адсорбция паров воды. По данным адсорб-
ционных измерений построены изотермы адсорбции воды (рис. 8).
Как видно из приведенных изотерм, активность образцов различна.
Наиболее активным оказался 84, а 83 и 39 — почти одинаковы. Несколько
ниже их по активности образцы 37 и 40.
174
Каталитические свойства синтезированных цеолитов
За последнее время в литературе все чаще затрагивается вопрос о ка-
талитических свойствах цеолитов, о внутрикристаллическом катализе
на молекулярных ситах [5—10]. В этих работах изучался катализ как
н-алканов, так и некоторых спиртов [1], отмечался внутрикрпсталли-
ческий, селективный по отношению к соединениям изо-строения и арома-
тическим характер каталитического превращения, а также была сделана
попытка изучения кинетики и механизма этого процесса [8—10]. Необхо-
свойства молекулярных сит еще да-
димо отметить, что каталитические
леко не изучены.
Нами для оценки каталитиче-
ской активности образцов была
выбрана классическая реакция
дегидратации этилового спирта на
твердом катализаторе. Этиловый
спирт для катализа был выбран
потому, что его молекулы явля-
ются по размеру достаточно малы-
ми, чтобы имелась возможность
проникать в поры цеолита 4А.
Процесс проводился в обычной
проточной установке со скоростью
подачи 5 мл/час во всех опытах.
Реакционная трубка заполнялась
насадкой и катализатором. Образ-
цы катализатора предварительно
таблетировались под давлением
в 100 ат, измельчались, и отби-
ралась фракция с сита № 3 в объ-
еме 5 мл и весом от 3 до 4 г. Ката-
лиз проводился с этиловым спир-
том-ректификатом. d42O=0.809 см3/г
и 93% по объему.
Время контактирования колеба-
лось от 20 мин. до 1—1.5 часов, в за-
Рис. 8. Изотермы адсорбции паров води
на хромовых цеолитах.
висимости от активности образца. Продукты реакции были газообразные,
а также имелся катализат. Газ собирался в газовой бюретке над насыщен-
ным раствором NaCl и анализировался сразу после реакции на газоана-
лизаторе ВТИ-2. Однако ВТИ-2 не позволяет установить, какой непре-
дельный углеводород получается в результате реакции, а дает общий
объемный процент ненасыщенных соединений в газе. Мы предполагали,
что должен получаться этилен. Для установления состава газовой фазы
газообразные продукты катализа на NaA пропускались в бромную воду,
которая постепенно обесцвечивалась. Продукт реакции высушивался и
перегонялся на воздухе. Выделенное вещество кипело при £КЩ1=129—
130°; df= 1.5379; n2j?=2.1736; МКнайд=27.03; МКВЫЧ=26.97.
Полученные данные совпадают с литературными для дибромэтана:
п™=2.1804, d42O=1.5379.
Анализ свидетельствует о том, что в газообразных продуктах реакции
присутствует из непредельных соединений только этилен. Кроме водо-
рода и этилена, в газовой фазе обнаруживалось от 1 до 5% СО и предель-
ных углеводородов, С02 — в количестве от 4 до 8%.
Катализ этилового спирта на цеолите NaA показал, что при увеличе-
нии температуры увеличиваются скорость реакции и количество выделяю-
175.
щегося газа. Причем, после 4 часов работы катализатора наблюдается
падение его активности.
При катализе на образцах с хромом, полученных различными путями,
реакция шла по-другому. Для образцов, различных по способу приготов-
ления, она имела свои особенности, о которых будет сказано ниже, но
во всех случаях в начале реакции при прибавлении первых капель эти-
лового спирта наблюдается экзотермический эффект. Температура повы-
шается сразу на 50—100°, что является характерным для Сг2О3 [11].
При дальнейшем добавлении спирта с определенной скоростью наблю-
дается постепенное восстановление Сг+6 до Сг+3, который частично
окислился на воздухе при разогревании трубки и установлении температур-
ного режима [12, 13]. Для того чтобы сохранить постоянной каталити-
ческую активность образцов, дальнейшие опыты проводились при нагре-
вании в среде этилового спирта как восстановителя.
В случае хромовых цеолитов в катализате, кроме спирта, было уста-
новлено наличие уксусного альдегида по его аддукту с димедоном. Тем-
пература плавления аддукта 139—140°. Проба смешения плавилась при
той же температуре. Данные анализа газовой фазы приведены в табл. 3.
В зависимости от способа получения образцы делятся на две группы:
группу, проявляющую при катализе преимущественно дегидратирующую
способность, при этом большую часть газа составляет зтилен, и группу
образцов, проявляющих преимущественно дегидрогенизационную спо-
собность. Образец CrNaAIII, полученный путем нанесения хромата на
промышленный цеолит NaA, дает преимущественно этилен в газовой
фазе, несмотря на содержание Сг2О3 13.5%. Так же ведет себя и образец
•Cr(OH)34~NaA, полученный путем смешения промышленного цеолита
NaA и свежеприготовленного, отмытого от щелочи гидрата окиси хрома,
хотя содержит 15.7% Сг2О3.
В отличие от указанных препаратов хромовые цеолиты (37, 39, 40)
дают в газовой фазе преимущественно водород, т. е. реакция идет в на-
правлении дегидрогенизации. Аморфный хромосиликат 51, полученный
нами из силиката и уксуснокислой соли хрома с добавлением щелочи,
Таблица 3
Содержание Н2 и С2Н4 в газовой фазе в результате
каталитического превращения этилового спирта на цеолитах
NaA, содержащих хром
№ образца i, °C % с.;п. % н2
NaA 350 42.3
370 80.1 0.5
Ст-4АШ (NaA+хромат) 350 64.4 17.2
Cr(OH)3+NaA 350 79.5 12.9
380 56.0 22.4
37 (тип NaA) 320 10.5 48.0
380 9.7 62.7
39 (тип NaA) 350 11.4 68.0
380 13.6 74.2
40 (тип NaA) 350 6.5 73.5
380 5.3 61.6
51 (аморфный) 350 6.8 73.8
380 5.7 61.5
84 (тип NaA) 350 40.0 37.5
380 39.0 41.3
83 (тип NaA) ..... 350 26.1 54.6
380 36.0 49.5
476
является катализатором дегидрогенизации и содержит 24.9% окиси
хрома.
Интересно отметить особую группу кристаллических образцов. Цео-
литы 83, 84 близки по содержанию SiO2 и общему содержанию А12О3 и
Сг2О3 к NaA. Особенно это относится к хромовому цеолиту 84. В отноше-
нии каталитической активности эти препараты являются как бы проме-
жуточными между первой группой и NaA. У них содержание водорода
и этилена в газе приблизительно одинаковое.
Таким образом, качественное сравнение катализа этилового спирта
на различных образцах показывает, что в случае чистого цеолита идет
исключительно дегидратация этилового спирта с образованием этилена;
в случае механической смеси Cr(OH)3 с NaA и нанесения хромата на цео-
лит тоже преобладает реакция дегидратации; в случае синтезированных
кристаллических цеолитов в основном идет дегидрогенизация этилового
спирта; в случае аморфного образца 51 без алюминия тоже преимуще-
ственно происходит дегидрогенизация.
Специфичность каталитических реакций для синтезированных кри-
сталлических образцов по сравнению со смешанными может служить,
как нам кажется, подтверждением того, что хром частично заместил алю-
миний и кремний в решетке цеолита. Об этом говорят также результаты
химического и рентгеноструктурного анализов. Кроме того, наши
данные позволяют сделать вывод о том, что полученные образцы, как и
сам цеолит NaA, обладают каталитической активностью и что в зависи-
мости от метода получения можно синтезировать хромовые цеолитные ка-
тализаторы, имеющие способность либо дегидрировать этиловый спирт,
либо проявлять себя как катализатор смешанного действия.
Выводы
1. Получены кристаллические цеолиты, содержащие в своем составе
хром.
2. Рентгеноструктурным анализом установлено, что полученные цео-
литы по типу соответствуют NaA, но имеют и некоторое отклонение
в структуре от цеолита NaA.
3. Адсорбционная емкость синтезированных цеолитов по воде соот-
ветствует NaA.
4. Образец NaA проявляет дегидратирующую способность, получен-
ные по первому методу образцы — дегидрирующую, а по второму — ведут
себя как катализаторы смешанного типа.
5. В отличие от синтезированных цеолитов обработанный хрома-
том NaA и смешанный с гидроокисью катализатор, несмотря на
высокий процент окиси хрома, оказывают в основном дегидратирующее
действие.
6. Появление новых, довольно сильных линий по рентгеноструктур-
ным данным, результаты химического анализа и специфичность катали-
тического действия синтезированных нами образцов дают основание пред-
полагать о возможности изоморфного замещения алюминия и кремния
хромом в кристалле цеолита.
ЛИТЕРАТУРА
1. . J. S е 1 b u n, R. В. Mason, J. Inorg. a. Nucl. Chem., 1961, 20, 3—4, 222.
2. R. М. В а г г е г, J. W. В а у nh а ш, F. W. В u 1 t i t и d е, W. М. М е i е г,
J. Chem. Soc. 1959 195.
3. J. Johnson, Brit. Pat. 8462, 1915; A. O. J a e g n e r, J. А. В о r t s c h,
US Pat., 1696546, 1928.
4. P. B. W e i s с, V. A. F r i 11 e 11 e, J. Phys. Chem., 1960, 64, 3, 382.
12 Цеолиты
177
5. A. N. К е о gh, L. В. S a n d, J. Am. Chem. Soc., 1961, 83, 16, 3536.
6. В. Я. Николина, И. E. Неймар к, M. А. Пионтковская, Усн.
химии, 1962, 29, 9, 1089.
7. П. Н. Галич, А. А. Г у т ы р я, В. С. Г у т ы р я, И. Е. Н е й м а р к, ДАН
СССР, 1962, 144, 1.
8. К. В. Топчиева, Б. В. Романовен ий, X о Ши Тхоанг, ДАН
СССР, 1963, 149, 5, 644.
9. Г. В. Цицишвили, 1П. И. Сидамонидзе, Ш. А. Зедгенидзе,
ДАН СССР, 1963, 153, 6.
10. Т. В. Роде. Кислородные соединения хрома и хромовые катализаторы. Изд.
АН СССР, 1962, 96, 182.
11. А. М. Рубинштейн, Усн. химии, 1952, И, 1315.
12. 3. Г. Зульфугаров, Г. И. Алимарданов, 3. С. А г ами р з о е в а,
Азёрб. Хим. журн., 1962, 3, 75.
ВЛИЯНИЕ СТЕПЕНИ КАТИОННОГО ОБМЕНА
В СИНТЕТИЧЕСКОМ ФОЖАЗИТЕ НА АДСОРБЦИЮ ИЗ РАСТВОРОВ
С. П. Жданов, В. Н. Новикова, Л. Ф. Павлова, Ю. А. Элътеков
Ранее [1, 2] было показано, что частичное (на 50—70%) замещение
ионов Na+ на ионы Са++ в синтетических цеолитах типа фожазита увели-
чивает избирательность адсорбции бензола и тиофена из н-алканов.
В настоящей работе было проведено систематическое изучение влия-
ния степени обмена Na+ на Са++ в синтетическом фожазите на адсорбцию
из растворов. Для этого был синтезирован Na-фожазит (Г-196 VII п)
и обработкой растворами Ca(NO3)2 в различных условиях получены об-
разцы фожазита с различным содержанием Са++ (см. таблицу).
Характеристики образцов
м образца Состав образца
Na+ Са++ аю2- SiO2
1 1 1 1.55
2 0.9 0.05 1 1.55
3 0.75 0.125 1 1.55
4 0.5 0.25 1 1.55
5 0.09 0.44 1 1.55
Перед опытами образцы прокаливались при 350—400° в течение 10—
12 часов. Методика измерения описана в [1, 2].
На рисунке представлены изотермы адсорбции тиофена из растворов
в н-гептане и в бензоле и бензола из растворов в н-гексане. Избирательность
адсорбции бензола и тиофена из н-алканов велика, и при весьма малых
концентрациях достигается предельная адсорбция. Избирательность ад-
сорбции из смесей тиофен+бензол чрезвычайно мала, что указывает на
близость адсорбционных свойств цеолита в отношении этих двух веществ.
Сопоставление изотерм адсорбции бензола и тиофена из растворов в н-ал-
канах на образцах фожазита с различным содержанием иона Са++ пока-
зывает, что введение небольшого количества ионов Са++ заметно увеличи-
вает избирательность адсорбции из этих растворов. Увеличение степени
замещения ионов Na+ на Са++ от 10 до 50% практически не влияет на
избирательность адсорбции. Дальнейшее же уменьшение ионов Na+
178
в результате ионного обмена резко снижает адсорбцию бензола и тиофена
примерно на 20% по отношению к образцам 2—4 и на 10—15% по сравне-
нию с исходным Na-фожазитом.
Изотермы адсорбции тиофена (а) из растворов в и-гептапе (верхние кривые), в бен-
золе (нижняя кривая) и бензола (б) из растворов в и-гексане.
Цифры в обозначениях’ соответствуют номерам образцов таблицы.
0.2
Таким образом, введение даже небольшого количества ионов Са++
в Na-фожазит заметно увеличивает адсорбцию бензола и тиофена из рас-
творов, а максимальное замещение ионов Na+ на Са++ резко снижает ад-
сорбцию этих веществ по сравнению с исходным Ка+-фожазитом.
ЛИТЕРАТУРА
1. С. П. Ж д а п о в, А. В. К и с е л е в, Л. Ф. П а в л о в а, Кинетика и катализ,
1962, 3, 445.
2. А. В. Киселев, В. Н. Новикова, Ю. А. Эльтеков. Нефтехимия,
1965, 5, 136.
ВЛИЯНИЕ ПРИРОДЫ И СТЕПЕНИ ЗАМЕЩЕНИЯ НЕКОТОРЫХ
КАТИОНОВ В ЦЕОЛИТАХ ТИПА X НА ИХ ХРОМАТОГРАФИЧЕСКИЕ
СВОЙСТВА
Т. Г. Андроникашвили, Г. В. Цицишвили, Ш. Д. Сабелашвили,
Т. А. Чумбуридзе
В газо-адсорбционной хроматографии наряду с активированным углем,
силикагелем и окисью алюминия в настоящее время большое распростра-
нение получают синтетические цеолиты, или так называемые молекуляр-
ные сита.
В основном в практике газовой хроматографии используются про-
мышленные образцы сит, известные под маркой СаА, а также и натрие-
вая форма цеолита типа X — NaX [1].
Из литературных источников известно, что адсорбционная способность
цеолитов зависит от природы и степени замещения катионов, входящих
в состав кристаллической решетки молекулярных сит [2, 3]. Это, по-ви-
димому, связано с некоторым изменением в строении цеолита, в частности
12*
179
с сужением или расширением входных окон полостей, а также с химиче-
ской природой катиона. Так как в основе процесса хроматографического
разделения лежит адсорбция, то нарушение соотношения между состав-
ляющими межмолекулярных сил приводит к изменению характера хро-
матограмм. Несомненно, что введение и замена в цеолитах одних катионов
другими приводят не только к геометрической модификации каналов цео-
литов, но также и к изменению межмолекулярных взаимодействий раз-
деляемых компонентов и адсорбента, что в свою очередь оказывает боль-
шое влияние на хроматографический процесс.
В нашем исследовании за исходную форму был взят натриевый цео-
лит типа X (продукт Горьковской опытной базы ВНИИНП, партия
Ц-202-98), на основе которого по ранее разработанной методике [4] был
приготовлен ряд катионообменных форм с различной степенью замеще-
ния катиона натрия на ионы кальция, бария, калия и серебра.
В табл. 1 приведены химические формулы исходного безводного нат-
риевого цеолита и его катионопроизводных.
Таблица 1
.№ образца
Химический состав
1
2
3
4
5
6
7
8
9
0.93 Na2O • А12 03 • 2.50SiO2
0.25 СаО • 0.67 Na2O • А12О3 • 2.66
0.57 СаО • 0.50 Na2O - А12О3 • 2.51
0.83 СаО • 0.09 Na2O • А12О3 • 2.45
0.25 ВаО - 0.71 Na2O • А12О3 • 2.50
0.35 К2О • 0.57 Na2O • А12О3 • 2.48
0.22 Ag2O • 0.69 Na2O • А1,О3 • 2.52
0.39 Ад2О • 0.55 Na2O • АГ2О3 • 2.60
0.85 Ag20 • 0.10 Na2O • А12О3 2.76
SiO2
SiO2
SiO2
SiO2
SiO2
SiO2
SiO2
SiO2
Сохранность цеолитной кристаллической структуры синтезированных
образцов была установлена снятием дебаеграмм. Хроматографические ис-
следования проводились на хроматографе ХТ-2М в колонке длиной 3.5 м.
Колонка заполнялась молекулярными ситами зернением 15—30 меш
(0.5—1 мм). Сорбент для загрузки в хроматографические колонки под-
готовлялся следующим образом. Из порошка цеолита приготовлялись
таблетки без добавки связующего, затем эти таблетки измельчались,
просеивались через сито и для испытания отбиралась вышеуказанная
фракция. Использование цеолитов без связующего (каолина) исключает
возможность возникновения вторичных эффектов, влияющих на хромато-
графические процессы, вызванных присутствием глины в качестве свя-
зующего.
Все образцы подвергались активации нагреванием при 450° в течение
4—5 часов (исключение составляла серебряная форма, где максимальная
температура активации не превышала 250° С). После загрузки в хромато-
графические колонки цеолиты перед началом работы прогревались при
температуре 300° в течение получаса в потоке газа-носителя (воздуха)
для устранения возможно попавшей влаги. Необходимость такой тща-
тельной активации объясняется тем, что влага, содержащаяся в цеолите,
может значительно изменить хроматографические свойства молекулярных
сит [4].
Хроматографические свойства всех изготовленных образцов изучались
на модельной смеси углеводородных газов Сх—С4, а также на окиси угле-
рода и водороде. В качестве газа-носителя использовался воздух, скорость
которого менялась в пределах 30—100 мл/мин. Температура нагрева
180
80 120 160 200 260 280 t°C
Рис. 1. Зависимость коэффициен-
тов разделения в системе этилен—
пропан от температуры нагрева
колонки, заполненной цеолитом
NaX.
хроматографической колонки поддерживалась в интервале от комнатной
температуры до 300° С. В некоторых случаях для выявления каталитиче-
ской активности цеолитов температура нагрева колонки поднималась
до 400° С.
Вначале нами исследовалась натрие-
вая форма цеолита типа X, и получи-
лась такая же картина разделения, как
в случае использования цеолита со свя-
зующим [5]. В этом случае мы обнару-
жили инверсию последовательности вымы-
вания этилена — пропана в зависимости от
температуры нагрева колонки, которая не
была замечена в предыдущей работе [5],
где исследовался сравнительно узкий ин-
тервал температур от 120 до 200° С. При тем-
пературе нагрева колонки около 80° спер-
ва вымывается пропан, а затем этилен.
Дальнейшее повышение температуры на-
грева колонки ухудшает разделение этих
компонентов, доводит до минимума, и при-
близительно с 120° С начинается инверсия
порядка вымывания этих компонентов
(рис. 1). Дальнейшее повышение температуры до 240° С приводит к улуч-
шению разделения этих компонентов.
Подобные исследования проводились нами также с кальцийсодержа-
щими цеолитами с различной степенью замещения натрия на кальций,
Рис. 2. Зависимость коэффициентов раз-
деления в системе этилен—пропан от тем-
пературы нагрева колонки, заполнен-
ной кальцийсодержащим цеолитом.
а именно с малым, средним и почти
полным замещением (образцы 2—4).
На цеолитах с малым и средним со-
держанием кальция (образцы 2 и 3)
наблюдается, как и на исходной на-
триевой форме, изменение последова-
тельности вымывания этилена—про-
пана в зависимости от температуры на-
грева колонки. Однако характер этой
зависимости также в большой мере
определяется содержанием ионов
кальция в цеолите. На рис. 2 пред-
ставлена кривая зависимости коэф-
фициентов разделения этих компонен-
тов, по Жуховицкому Кг [6] для слу-
чаев полного разделения и по Струппе
б [7] для случаев неполного разделе-
ния, от температуры нагрева колонки
на кальцийсодержащих цеолитах.
Как видно из рис. 2, на образце 2
(форма с малым содержанием кальция) имеющая место инверсия последо-
вательности вымывания этилена—пропана по своему характеру очень
близка к инверсии этих же компонентов на натриевой форме. На образце 3
(среднее замещение) при нагревании колонки до температуры 120° сперва
вымывается пропан, а затем этилен, при 140—180° их разделения не
происходит. Дальнейший подъем температуры до 240° приводит к инвер-
сии последовательности вымывания этих компонентов и к улучшению
разделения этой пары. И, наконец, на образце 4 почти с полным замеще-
нием натрия на кальций на всем исследованном температурном интервале
не происходит изменения последовательности вымывания этих компонен-
181
тов. Такое влияние содержания кальция в цеолите на порядок вымывания
предельных и непредельных соединений, по всей вероятности, можно
объяснить более сильными поляризационными свойствами иона кальция
по сравнению с натрием [8]. Также замечено, что с увеличением степени
Рис. 3. Влияние скорости газа-носителя на ка-
талитическое превращение пропилена. Адсор-
бент — кальциевая форма цеолита (образец 4).
Температура нагрева хроматографической ко-
лонки 300°.
Скорость газа-носителя: 1 — 30, 2 — 50, з — 70 и 4 —
100 мл/мин.
v,m6
7
пилена. Уменьшение скорости носителя от
замещения натрия на каль-
ций коэффициенты разделе-
ния этилена и пропана уве-
личиваются.
При определенных темпе-
ратурах нагрева колонки у
этих форм цеолитов прояв-
ляются каталитические свой-
ства относительно ненасыщен-
ных углеводородов С3—С4.
Так, при хроматографирова-
нии пропилена в колонке,
нагретой до 280—300°, и ско-
рости газа-носителя 100 мл/
мин. пропилен начинает под-
вергаться каталитическому
воздействию сорбента, и вме-
сто одного пика, характери-
зующего вымывание пропи-
лена, на хроматограмме по-
являются два пика, причем
пик, соответствующий обра-
зовавшемуся веществу, по-
является раньше пика про-
100 до 30 мл/мин. приводит
к исчезновению пика пропилена, с сохранением пика, соответствующего
образовавшемуся веществу (рис. 3). Можно предположить, что исчезно-
вение пика пропилена на хроматограмме связано с полимеризацией про-
пилена на сорбенте, а появление
пика нового вещества вызвано или
изомеризацией пропилена в цикло-
пропан, или самогидрогенизацией
в пропан, так как удерживаемые
объемы пропана и циклопропана
на цеолитах близки и в некоторых
случаях их отдельная идентифи-
кация несколько затруднена. Ана-
логичное каталитическое воздей-
ствие оказывает кальциевая фор-
Рис. 4. Зависимость коэффициентов разде-
ления в системе этилен—пропан от темпера-
туры нагрева колонки, заполненной ба-
риевым цеолитом (образец 5).
ма и на бутилен, только при бо-
лее низких температурах, чем на
пропилен.
Для бариевой формы цеолита
(малое замещение натрия на барий),
так же как и для кальциевой формы (образец 2), свойственна инверсия
последовательности вымывания этилена—пропана в зависимости от тем-
пературы нагрева колонки. На рис. 4 представлена кривая зависимости
коэффициентов разделения от температуры нагрева колонки, заполненной
бариевым цеолитом. Как видно из рисунка, характер инверсии на этой
форме очень близок к результатам, полученным на образце 2. Подобная
инверсия имеет место и для смеси бутана и пропилена, но при высоких
температурах не происходит разделения этих компонентов, хотя время
182
вымывания пропилена меньше, чем время элюирования бутана, т. е. про-
исходит изменение порядка вымывания. Однако разница во времени их
вымывания недостаточна для разделения этой пары. Следует отметить,
что удерживаемые объемы всех исследованных компонентов на бариевой
форме превышают удерживаемые объемы соответствующих соединений на
кальциевой форме (образец 2).
Калийзамещенная форма цеолита (образец 6) по своим хроматографи-
ческим свойствам несколько отличается от остальных исследованных
образцов молекулярных сит. Во-первых, на этом сорбенте с данной сте-
пенью замещения натрия на калий не удается разделить окись углерода
и метан, и даже удерживаемый объем метана немного больше удерживае-
мого объема окиси углерода, что обычно не свойственно цеолитам. Эта
Рис. 5. Хроматограмма разделения искусственной смеси газов, состоящей из
водорода, метана, окиси углерода, этана, этилена, пропана, пропилена, н-бу-
тапа, транс-бутилена и цис-бутилена.
Сорбент — калийзамещенная форма цеолита, температура нагрева колонки 200° С, ско-
рость газа-носителя 100 мл/мин.
разность невелика и недостаточна для разделения указанных веществ.
Во-вторых, на этом сорбенте не наблюдается инверсии последовательности
вымывания этилена и пропана в зависимости от температуры нагрева ко-
лонки в отличие от остальных изученных образцов. На хорошо высушен-
ной калийзамещенной форме при всех температурах нагрева колонки
после каждого насыщенного углеводорода вымывается ненасыщенный
углеводород с таким же числом атомов углерода (рис. 5).
На хроматографической колонке, заполненной калиевым цеолитом,
при температуре нагрева колонки 200° С и скорости газа-носителя
100 мл/мин. удалось разделить транс- и цис-бутилены (коэффициент
разделения по Струппе 6=0.59), а также Р-бутилен и изо-бутилен, 6=0.67.
И, наконец, были изучены хроматографические свойства трех образ-
цов [7—9] серебряных цеолитов с различной степенью замещения натрия
на серебро. На всех образцах ни при высоких, ни при низких температурах
нагрева колонки водород, окись углерода, а также ненасыщенные угле-
водороды Са—С4 не элюируются потоком газа-носителя; что касается на-
сыщенных соединений, то они разделяются и вымываются из хроматогра-
фической колонки. Замечено, что с увеличением степени замещения иона
натрия на серебро растут удерживаемые объемы предельных углеводо-
родов.
Для всех изученных образцов молекулярных сит характерны некото-
рые общие свойства. Коэффициенты разделения К для большинства слу-
чаев с увеличением температуры нагрева колонки уменьшаются, а
в пределе изменения скорости газа-носителя от 30 до 100 мл/мин. практи-
183
чески остаются постоянными. Для изученных насыщенных и ненасыщен-
ных углеводородов состава Q—С4 наблюдается увеличение коэффициентов
разделения с ростом числа углеродных атомов. На основании газо-хро-
матографических данных [9] были рассчитаны теплоты адсорбции иссле-
Таблица 2
Теплоты адсорбции (ккал./мол.)
Компонент № образца
1 2 3 4 5 6 7 8 9
сн4 3.7 (4.5) 4.7 4.2 4.1 4.6 4.3 3.6 3.6 3.5
со 5.5 6.5 7.0 7.9 6.1 — — — —
с2нп 6.2 6.6 6.3 6.2 6.8 6.2 5.5 5.5 6.0
С2Н4 8.3 8.5 8.8 10.9 9.1 7.3 — — —
с3н8 8.0 7.3 7.3 7.2 8.0 8.0 7.8 7.4 8.2
с3не 9.9 10.8 11.2 13.9 11.9 9.3 — — —
С4Н10 9.8 9.9 9.5 9.2 10.0 9.9 9.4 9.2 —
дуемых соединений на ряде образцов цеолитов (табл. 2). При расчете
теплот адсорбции этих компонентов методом газовой хроматографии были
выбраны такие температурные интервалы, при которых пики соответствую-
щих веществ на хроматограммах имеют наиболее симметричную форму.
Значения теплот адсорбции компонентов на NaX получены более высо-
кие, чем на тех же образцах со связующим [10]. Это в большой степени
вызвано тем, что в предыдущей работе не было обращено должного вни-
мания на возможность увлажнения сорбента во время эксперимента. Для
образцов 2—4 с увеличением степени замещения катионов натрия на ион
кальция характерен рост теплот адсорбции ненасыщенных соединений
и окиси углерода, в то время как теплоты адсорбции предельных соеди-
нений практически мало изменяются. На калиевой форме цеолита значе-
ния теплот адсорбций предельных углеводородов по сравнению с натрие-
вой формой цеолита мало изменяются; что касается величин теплот адсорб-
ций ненасыщенных соединений, то они уменьшаются. Величины теплот
адсорбции исследованных соединений на трех образцах серебряных цео-
литов по предварительным данным не подчиняются каким-либо опреде-
ленным закономерностям.
ЛИТЕРАТУРА
1. Т. Г. Андроникашвили, Г. В. Цицишвили, в сб.: Поверхностные
явления на алюмосиликатах, Изд. АН ГрузССР, 1965.
2. Г. В. Цицишвили, Т. Г. Андроникашвили, в сб.: Синтетические
цеолиты, Изд. АН СССР, 1962, 112.
3. D. W. В г е с k, W. G. Е v е г s о 1 е, R. М. М i 11 о n, Т. В. R е е d, Т. L. Т h о-
m a s, J. Am. Chem. Soc., 1956, 78, 23, 5963.
4. J. I а пак, М. К г е j с i, Е. D u b s к у, Annals N. Y. Academy of Sci., 1959,
72, 13, 731.
5/ T. Г. Андроникашвили, Ш. Д. Сабелашвили, Г. В. Цици-
швили, Нефтехимия, 1962, 2, 248.
6. Н. М. Туркельтауб, А. А. Жуховицкий. Газовая хроматография.
Изд. АН СССР, 1960, 144.
7. Н. G. S t г u р р е, I Symp. Gas Chromatogr. DDR Akademie-Verlag, Berlin,
1959 28.
8. R. M. В arrer, D. A. L a n d 1 e y, J. Chem. Soc., 1959, 2838.
9. S. A. G r e e n e, H. P u s t, J. Phys. Chem., 1958, 62, 1, 55.
10. Г. В. Цицишвили, T. Г. Андроникашвили, Ш. Д. Сабела-
швили, T. А. Чумбуридзе, Сообщ. АН ГрузССР, 1962, 30, 6, 731.
184
ВАКУУМ-ТЕРМИЧЕСКАЯ АКТИВАЦИЯ
И АДСОРБЦИОННЫЕ СВОЙСТВА АММОНИЕВЫХ ФОРМ ЦЕОЛИТОВ
В. Г. Ильин, И. Е. Неймарк, А. И. Растрененко
Существуют цеолиты (тип Y), допускающие декатиониройание [1].
Аммониевые формы этих цеолитов после термообработки имеют кристал-
лическую структуру и адсорбционные свойства, характерные для натрие-
вых форм. Известно также, что процесс декатионирования приводит к раз-
рушению структуры синтетических цеолитов типов А и X [2]. Очевидно,
что возможность декатионирования связана со структурными особенно-
стями и химическим составом цеолитов.
В настоящей работе изучались адсорбционные свойства аммониевых
форм цеолитов А, X и Y, подвергавшихся вакуум-термической активации
при разных температурах. В качестве исходных были взяты цеолиты,
синтез которых осуществлялся силикатным способом. Аммониевые формы
цеолитов получались методом ионного обмена. Данные о химическом со-
ставе и структуре исследованных цеолитов приведены в таблице. Рент-
геноструктурный анализ подтвердил сохранность структуры цеолитов
после замещения Na+ на NH4+ (в цеолите 9 обнаружена примесь других
фаз).
Характеристика исследованных цеолитов
№
образца
Исходные цеолиты и их NHj-произ-
водные
Степень обмена Na+
на NH.+, %
1
2
3
4
5
6
7
8
9
10
11
12
13
NaA «0= 12.20 А
0.81 Na2O • А12О3 • 1.82 SiO2 • 4.1 Н2О
NH4NaA
NH4NaA
NH4NaA
NaX a0=24.84 A
0.86 Na2O • Ala03 • 3.0 SiO2 • 5.0 H2O
NH4NaX
NH4NaX
NH4NaX
NaY a0=24.68A
1.1 Na2O • Ala03 • 5.0 SiO2 • 6.5 H2O
NH4NaY
NH4NaY
NaY a0=24.67A
0.75 Na2O • A12O3 • 4.3 SiO2 • 7.0 H2O
NH4NaY
50
71
80
62
73
78
66
78
74
На рис. 1 представлена зависимость адсорбции паров воды от условий
вакуум-термической активации цеолитов.1
Активность всех цеолитов по парам воды растет при температурах
активации до 100—150° С. Дальнейшее повышение температуры до 400° С
вызывает значительное уменьшение активности NH4 — производных
цеолитов типов А и X, причем наиболее резко — у образцов с высокой
степенью замещения. При термической обработке МН4-форм цеолитов
имеет место, вероятно, образование структурных гидроксильных групп,,
что ведет к разрушению кристаллической структуры [3].
На кривых рис. 1 для цеолитов типа Y наблюдается максимум, сви-
детельствующий о значительном увеличении адсорбционной активности
1 Последовательно проводилась вакуум-термическая активация в интервале тем-
ператур 50—400° С.
185-
при температуре, близкой к 300° С. Более высокая термостойкость NH4-
лроизводных цеолитов типа Y в этом интервале температур объясняется
Рис. 1. Изменение адсорбционной активности цеолитов А, X
и Y по отношению к парам воды (20° С, р/рв=0.3) в зависи-
мости от температуры вакуум-термической активации (10~3 мм
рт. ст., 5 часов).
Номера кривых па всех рисунках настоящей статьи соответствуют
номерам образцов в таблице.
«большим отношением SiO2/Al2O3 и, следовательно, большими расстояниями
между катионными позициями, в результате чего локальные перестройки
Рис. 2. Изотермы адсорбции при 20° С паров СН8ОН, СОг и NHS
цеолитами типа А после вакуум-термической активации при 350° С
(дли СН8ОН — после повторной активации при 350° С).
в этих цеолитах не влияют на сохранность алюмокремнекислородпого
•остова в целом.
Из исследованных нами цеолитов типа Y наиболее стойким оказался
•образец 13, о чем свидетельствуют адсорбционные измерения и рентгено-
структурный анализ. После прокаливания в течение 5 часов при темпе-
ратуре 400° С на дебаеграммах не обнаружено уменьшения-интенсивности
186
дифракционных линий, столь значительного для Ш14-форм цеолитов
А и X. При высоких температурах разрушению подвергался также цео-
лит 9.
Данные об исследовании адсорбционных свойств цеолитов по парам
СН3ОН, С02. NH3 и С6Н6 приведены на рис. 2 и 3. ГШ4-формы цеолита А
Рис. 3. Изотермы адсорбции при 20° С паров СвН6
и NHS цеолитами X и Y после вакуум-термиче-
ской активации при 350 ° С.
•оказались наименее устойчивыми к воздействию температуры и значи-
тельно снижали адсорбционную активность. Активность КН4-производ-
ных цеолита X по отношению к парам С6Нв и NH3 и образца 13 (NH4NaY)
по отношению к парам СвНв после вакуум-термической активации при
350° С повышалась по сравнению с исходными цеолитами (рис. 3).
ЛИТЕРАТУРА
1. J. R а b о, Р. Р i с k е г t, D. S tamires, J. Е. Boyle, Actes Congr. Inter-
net. Catalyse, Paris, 1960, 2, 2055.
2. D. W. В r e c k, W. G. E v e r s о 1 e, R. M. Mi 1 ton, T. B. R e e d, T. L. T h o-
m a s, J. Am. Chem. Soc., 1956, 78, 5963.
3. С. П. Ж д a h о в, А. В. К и с e л e в, В. И. Л ы г и п, Т. И. Т и т о в а, ДАН
СССР, 1963, 150, 3, 584.
ВОПРОСЫ СИНТЕЗА
И ТЕХНОЛОГИИ ПРОИЗВОДСТВА ЦЕОЛИТОВ
А. В. Агафонов, С. М. Вайнштейн, И. Э. Гельме, Б. А. Липкинд,
Л. И. Пигузова
Основные принципы синтеза различных цеолитов
Принципиальная схема синтеза основных цеолитов, предназначен-
ных для промышленного применения, в настоящее время общеизвестна
и используется в различном технологическом или аппаратурном оформле-
нии многими авторами и держателями патентов как в Советском Союзе,
так и в зарубежных странах. Основные операции синтеза и получаемые
•промежуточные продукты представлены в нижеследующем перечне.
187
Операции
Продукты и полупро-
дукты
1. Подготовка сырья.
2. Приготовление и дозировка
исходных растворов.
3. Смешение и подогрев растворов,
осаждение гидрогелей.
4. Кристаллизация.
5. Промывка кристаллов и про-
ведение обмена катионов.
6. Грануляция с добавкой свя-
зующего.
7. Дегидратация.
1. Исходное промышленное сырье?
силикат глыба, гидрат окиси алю-
миния, каустическая сода.
2. Рабочие растворы силиката на-
трия и алюмината натрия.
3. Смесь гидрогелей окисей кремния
и алюминия.
4. Кристаллы щелочного (натрие-
вого) цеолита.
5. Кристаллы цеолита с обменным
катионом.
6. Гранулированный необезвожен-
ный цеолит.
7. Готовый адсорбент.
Приведенная схема включает в себя то общее, что свойственно синтезу
большинства известных цеолитов.
Особенности при синтезе разновидностей цеолитов по химическому
составу и структуре состоят главным образом в подборе необходимого
состава реакционной массы и режима кристаллизации, т. е. в создании
условий реакции, определяемых так называемым полем кристаллизации.
Следует отметить, что синтез основной группы цеолитов возможен
в сравнительно широком диапазоне физико-химических, режимных и
технологических условий.
Это и создало предпосылки для разностороннего варьирования тех-
нологического и аппаратурного оформления в их промышленном произ-
водстве, а как следствие — и многообразие патентов, выданных на раз-
личные способы получения однотипных синтетических цеолитов, выраба-
тываемых под своеобразными торговыми наименованиями во многих
странах.
Можно, однако, добавить, что свобода варьирования условий при син-
тезе различных цеолитов, если иметь в виду их высокое качество, убывает
от цеолитов группы АкХ, от X к Y и далее, к некоторым другим разно-
видностям цеолитов.
Далее, некоторые особенности в синтезе и технологии производства
возникают в случаях различного предназначения однотипных по своей
кристаллической форме цеолитов. Примером может служить глубина
катионного обмена в цеолитах СаА или СаХ, оптимальный размер кри-
сталлов, различный подбор связующих материалов, используемых для
грануляции, методы грануляции и форма гранул.
Важнейшим элементом условий синтеза любого из цеолитов является
соотношение реагирующих масс гидрогелей кремния и
алюминия, а также щелочи и воды при кристаллизации.
Так, для кристаллизации цеолита типа NaA постоянного состава
успешно могут использоваться гидрогели в широком диапазоне мольного
отношения SiO2/Al2O3 (0.5—2.0) при соответствующих колебаниях также
и отношения Na2O/SiO2 (например, 1 и 4 в крайних точках).
Соотношение SiO2/Al2O3 в геле, подвергаемом кристаллизации, может
быть равным или ниже, чем в кристалле цеолита NaA. В последнем слу-
чае раствор алюмината натрия в реагирующей массе находится в из-
бытке.
Практический выбор дозировки у границы верхних значений отноше-
ния SiO2/Al2O3 продиктован как оптимумом по чистоте фазы, так и тех-
188
нико-экономическими соображениями. Сближение составов реагирующей
массы и кристалла увеличивает выход цеолита, сокращает расходы ре-
агентов и увеличивает производительность аппаратуры равной емкости.
При повышении отношения SiO2/Al2O3 более 2 надежность в воспроиз-
водимости качества этого цеолита резко снижается.
Для цеолита NaX характерна кристаллизация из реакционной массы,
имеющей избыток SiO2, т. е. при отношении SiO2/Al2O3, превышающем
~2.5, величину, соответствующую готовому кристаллу.
Увеличению отношения SiO2/Al2O3 обычно сопутствует уменьшение
величины отношения Na2O/SiO2. Цеолит типа NaX требует более строгого
соблюдения режима кристаллизации, чем цеолит NaA, поэтому для его
надежного получения предпочтительнее вести синтез не у границ, а в сред-
ней зоне поля кристаллизации.
Экономические соображения, требующие сближения составов среды
и кристаллов, в данном случае отодвинуты на второй план.
Для цеолита NaY характерна растянутость поля кристаллизации
в широком диапазоне отношений SiO2/Al2O3 более 5, но имеется и строгая
ограниченность поля по ординате, зависящей от отношения Na2O/SiO2,
т. е. высокая чувствительность кристаллизации к изменению щелочно-
сти среды.
Практические затруднения при осуществлении этого синтеза в про-
мышленном масштабе состоят в том, что во избежание сложного оборота
большого количества маточных растворов вопреки оптимальности для
кристаллизации высоких значений отношений SiO2/Al2O3 условия син-
теза приходится несколько смещать в сторону меньших отношений
SiO2/Al2O3. Из этого вытекает необходимость вести синтез в условиях
большой точности.
Ограничения по содержанию щелочи и то, что щелочь, вводимая с си-
ликатом, при больших отношениях SiO2/Al2O3 начинает преобладать над
алюминатной, заставляют решать задачу этого синтеза путем применения
в качестве источника нещелочных силикатов, а именно специально при-
готовленных золей кремнекислоты, свободных от ионов Na.
Вторым по важности фактором для синтеза является концентрация
взятых для реакции растворов, обобщенно характеризуемая
содержанием суммы SiO2-|-Al2O3 в среде, подвергаемой кристаллизации.
Наименее требователен к соблюдению точной концентрации среды
цеолит NaA; для него не было выявлено ярко выраженного оптимума по
содержанию SiO2-|-Al2O3 в процессе кристаллизации.
Этот цеолит может быть получен из растворов достаточно высоких
концентраций, успех синтеза при этом зависит от достигнутого качества
смешения растворов перед осаждением и от гомогенизации смеси гидро-
гелей до кристаллизации. С повышением концентраций гомогенное сме-
шение затрудняется.
С другой стороны, допуская на данном этапе развития промышленного
производства цеолитов осуществление кристаллизации в периодически
действующем оборудовании, мы не считаем задачу повышения концен-
трации реагирующих растворов достаточно острой. Поэтому для синтеза
цеолита NaA нами выбраны умеренно средние концентрации 80—85 г
SiO2 или А12О3 на литр в каждом из соответствующих растворов.
Иначе обстоит дело с синтезом цеолита NaX. В этом случае было най-
дено, что хорошая воспроизводимость качества цеолита достигается в огра-
ниченных пределах концентраций реагирующих растворов, которые
в сумме находятся в пределах 5—8 вес. % в пересчете на сухие SiO2
и А12О3.
Эти результаты заставили твердо ограничить верхний предел концен-
трации растворов, используемых для синтеза цеолита NaX, не считаясь
189
с некоторым снижением потенциала производительности кристаллиза-
ционного оборудования по сравнению с производством цеолита NaA.
Условия смешения растворов и нагревания
смеси имеют довольно существенное значение для цеолита.
Способы достаточно хорошего смешения были отработаны примени-
тельно к различным вариантам смешения в потоке или при приливании
одного раствора к другому.
Несколько принципиальнее оказался вопрос о нагревании смеси.
Следует отметить влияние условий нагревания реагирующих веществ на
размер кристаллов цеолита. Было найдено, что цеолиты, получаемые при
смешении растворов, нагретых при температуре кристаллизации, имеют
преобладающий размер кристаллов 5—7 мк, тогда как при подогревании
смеси после смешения растворов получаются кристаллы размером в ос-
новном 2—3 мк.
Гранулированные цеолиты той или другой формации были тщательно
изучены, особенно в тех процессах, где динамика адсорбции играет ре-
шающую роль. Было, однако, найдено, что адсорбционные и механиче-
ские свойства гранулированных цеолитов независимо от размера исполь-
зованных кристаллов в пределах точности методов контроля равноценны.
В тех случаях, когда нагреваются гидрогели, особенно после появле-
ния зародышей кристаллизации, важен выбор способа нагрева. Осуще-
ствление режимов смешения и нагрева геля, не соответствующих требо-
ваниям, приводит к снижению качества цеолитов особенно NaX и NaY.
Процесс кристаллизации, будучи периодическим и требуя
для получения большинства цеолитов покоя кристаллизуемой смеси,
в значительной мере определяет компановку всей схемы технологического
процесса их производства. Важнейшими и взаимосвязанными факторами
при кристаллизации остаются температура и время. Известно, что для
получения большинства цеолитов промышленного типа кристаллизацию
удается осуществлять под атмосферным давлением и при температурах,
ограниченных интервалом 90—100° С. Применение гидротермальных спо-
собов кристаллизации под давлением ведет к осложнениям технологии,
причем не столько в кристаллизационной аппаратуре, сколько в более
громоздкой схеме приготовления исходных гелей, подвергаемых кри-
сталлизации.
Оптимальное время кристаллизации различных цеолитов тщательно
изучалось. Найдено, что кристаллизация цеолита типа NaA может быть
проведена достаточно быстро, в основном за 4—6 часов, однако за это
время процесс кристаллизации не заканчивается и продолжаются еще
некоторые процессы перекристаллизации, способствующие повышению
чистоты фазы. При недостаточной чистоте фазового состава цеолитных
порошков имеет место и некоторое снижение динамической активности
адсорбента. Наряду с этим у цеолитов NaA обнаружено закономерное
снижение прочности по мере увеличения длительности их кристаллизации.
Продолжительность кристаллизации цеолита NaX больше, нежели
цеолита NaA, общий и местный перегрев при кристаллизации свыше
100° С ведет к ухудшению их адсорбционных свойств.
Катионный обмен с учетом многообразия ионообменных форм
цеолитов является достаточно большим и самостоятельным вопросом их
синтеза. Различная активность катионов и неравные условия их распо-
ложения в кристаллической решетке цеолита, естественно, влияют на
кинетику и равновесие катионного обмена в каждом частном случае.
Из основных итогов работы ВНИИНП в этой области для наиболее
распространенных типов цеолитов (СаА, СаХ, NaA, AgA) представляется
интересным упомянуть о роли предварительной промывки цеолита и о срав-
нении степени обмена катионов, осуществляемого в порошке и гранулах.
190
Требования к промывке кристаллов перед проведением катионного
обмена, естественно, зависят от относительной активности обмениваемых
катионов. Поскольку это касается обмена Na' на Са", т. е. производства
основных промышленных цеолитов, то при высокой относительной ак-
тивности Са, а также существенной растворимости образующегося ги-
драта окиси кальция промывка кристаллов перед катионным обменом
смогла быть значительно сокращена.
Качество цеолитов общего значения, где степень катионного обмена
Na' на Са " ограничивается 70—75%, в результате сокращения промывки
ни по одному из показателей ТУ затронуто не было.
Случаи, когда те же цеолиты должны готовиться с полным обменом
(например, СаХ для использования в катализе или для молекулярно-
ситового разделения узких фракций углеводородов), являются исключе-
нием.
Не все катионы, введенные в состав цеолита, в равной степени противо-
стоят гидролизу при последующей его отмывке от солей, являющихся
продуктами ионного обмена. Такое положение характерно, например,
для калиевых цеолитов. Качество калиевых цеолитов (с достаточно пол-
ным обменом) оказалось весьма чувствительным к отклонениям от регла-
ментированного режима промывки.
При получении серебряного цеолита в связи с его низкой термостой-
костью и ограничением температуры прокаливания возникли трудности
с приданием ему необходимой прочности. Эти обстоятельства заставили
нас заняться изучением катионного обмена в гранулах. ‘
Было показано, что катионный обмен в прокаленных гранулах может
быть проведен практически с той же полнотой, что и при обработке по-
рошкообразного гидратированного цеолита. Различия в случаях обмена
катионов в порошке или готовых гранулах наблюдаются, естественно,,
в кинетике и динамических показателях этого процесса. Для эффективного
проведения катионного обмена в гранулах необходимо увеличивать тем-
пературу и продолжительность процесса. Существенной неоднородности
степени обмена по глубине гранулы в этом случае замечено не было.
Вопросы грануляции цеолитов и выбора связующего материала имеют
большое значение для качества получаемых товарных адсорбентов. Свя-
зующее и метод грануляции больше, нежели другие факторы, влияют на
динамическую активность цеолитов, на возможное развитие паразитиче-
ской каталитической активности или на нарушение селективности в ад-
сорбционных процессах. Вместе с тем в этой операции решается вопрос
оптимального сочетания двух основных параметров качества адсорбента:
прочности и динамической активности.
Общие вопросы в технологии и аппаратурном оформлении промышленного
производства синтетических цеолитов
Аппаратурное оформление промышленного процесса производства
цеолитов в большой степени влияет на экономику производства. Тип
выбранного оборудования определяет поточность и производительность
линии, реализуемую степень механизации и автоматизации технологи-
ческого процесса, маневренность установки при выпуске адсорбентов раз-
личного типа. Наиболее принципиальными технологическими узлами
в части аппаратурного оформления промышленного производства яв-
ляются узлы кристаллизации, промывки и грануляции. ВНИИНП про-
ведены значительные работы по моделированию и испытанию соответ-
ствующего оборудования различных типов.
До тех пор пока процесс кристаллизации остается периодическим, мы
пришли к выводу о целесообразности рассматривать кристаллизатор как
191
аппарат комплексного назначения, в котором последовательно проводится
ряд технологических операций периодического характера: от осаждения
геля до катионного обмена включительно. При этом поточность процесса
обеспечивается регламентированным графиком работы батареи однотип-
ных аппаратов. Основными критериями работоспособности такого аппа-
рата являются интенсивность перемешивания и время, необходимые для
взмучивания отстоявшихся осадков. Большое значение имеет также тер-
мостатирование аппарата в период длительной кристаллизации, осуще-
ствляемой в покое. Конструкция кристаллизатора, снабженного пропел-
лерной мешалкой и термостатированного водяной рубашкой, была нами
отработана на моделях и опытно-промышленных аппаратах емкостью
.до 15 м3.
Для выбора оборудования узла промывки проведены опыты фильтро-
вания и промывки на нутч-фильтрах, фильтрпрессах, барабанных вакуум-
фильтрах непрерывного действия, центрифугах. В результате этих опы-
тов в качестве фильтрующего оборудования однозначно выбраны бара-
банные вакуум-фильтры, которые позволяют при фильтровании обеспе-
чить съем осадка до 3 т/м2 в сутки в пересчете на сухой продукт, а по
фильтрату соответственно 12—15 м3/м2 в сутки. Центрифуги в силу
специфических условий съема осадка и значительного уноса твердой фазы
на больших производительностях оказались менее пригодными.
Были испытаны различные способы грануляции цеолитов в основном
на базе применения глиняных связующих, в том числе на машинах, ра-
ботающих по принципу вмазывания массы в формовочные ячейки, «экстру-
зии» в щнек-прессах, закатки на вращающихся тарельчатых гранулято-
рах. Были разработаны также приемы, позволяющие формовать таблетки,
спрессованные из сухих порошков. Следует отметить, что грануляция еще
остается слабым звеном в технологии производства цеолитов, и прежде
всего потому, что промышленность не располагает проверенными моделями
машин большей производительности.
Регламенты технологического процесса производства и показатели
работы оборудования, выявленные на полузаводской установке, под-
тверждены опытами, проведенными в заводских условиях, и легли в ос-
нову проекта более крупной промышленной установки по производству
синтетических цеолитов, разработанного Государственным институтом
прикладной химии.
По этому проекту весь технологический процесс производства цеоли-
тов различных марок после кристаллизации удалось уложить в одну
компактную непрерывно-поточную линию.
СИНТЕЗ И ПРОИЗВОДСТВО ЦЕОЛИТОВ ТИПОВ А и X
Я. В. Мирский, М. Г. Митрофанов, Б. М. Попков, Л. Т. Болотов,
Л. Ф. Ручко
Синтетические цеолиты являются новыми высокоэффективными ад-
сорбентами, применение которых в различных областях адсорбционной
техники позволяет повысить эффективность процессов тонкой осушки и
очистки веществ и осуществить новые процессы разделения смесей.
Работы, связанные с получением и применением синтетических цео-
литов, обладающих молекулярно-ситовыми свойствами, стали развиваться
сравнительно недавно [1—6]. Первая работа в этой области, выполненная
Р. М. Бэррером, относится к 1948 г. В 1956 г. были опубликованы сообще-
192
ния о свойствах синтетических цеолитов, обладающих высокой адсорб-
ционной емкостью и хорошей селективностью, которые были синтезиро-
ваны исследователями фирмы Линде (США) и получили наименование
цеолитов типа А [7, 8]. Способ и условия получения этих цеолитов в то
время не были опубликованы в литературе. Первые зарубежные сообще-
ния о технологии получения синтетических цеолитов и патентные описа-
ния появились в 1959 г. [9].
В Советском Союзе способ и технология получения синтетических
цеолитов были разработаны в 1959 г. [10, 11].
Некоторые вопросы синтеза цеолитов типов А и X
Как было ранее установлено, наиболее удобным в технологическом
отношении способом получения гидрогелей для синтеза цеолитов типов А
и X является смешение растворов алюмината натрия и жидкого стекла [15—
18]. Скорость образования цеолитов при кристаллизации гидрогелей
довольно значительна: длительность кристаллизации 6—12 часов до-
статочна для получения цеолитов обоих типов с высокой адсорбционной
емкостью.
Та блица 1
Адсорбционная емкость цеолитов типа А, полученных при различной длитель-
ности кристаллизации при 90°
Состав гидрогеля, моли на 1 моль А12О3 Продолжи- тельность кристаллиза- ции, часы Адсорбционная емкость (см3/г) по парам воды при относительном давлении
Na2O SiO2 н2о 0.1 0.5 0.9
2.02 0.30 50.0 1 0.22 0.23 0.25
2.08 0.50 55.0 1 0.24 0.26 0.28
2.08 0.50 55.0 3 0.24 0.25 0.26
2.26 1.00 65.0 1 0.24 0.25 0.28
2.26 1.00 65.0 3 0.25 0.26 0.28
2.42 1.40 70.0 1 0.25 0.26 0.28
2.42 1.40 70.0 3 0.26 0.27 0.28
2.48 1.70 45.0 1 0.24 0.26 0.27
2.78 1.70 50 1 0.23 0.26 0.27
2.78 1.70 65 6 0.23 0.25 0.25
2.48 1.70 70 1 0.11 0.12 0.15
2.35 1.70 70 2 0.22 0.24 0.25
2.48 1.70 70 6 0.23 0.25 0.26
2.78 1.70 75 6 0.23 0.25 0.26
2.78 1.70 100 6 0.24 0.27 0.28
2.64 2.00 60 1 0.25 0.27 0.29
2.64 2.00 80 3 0.25 0.26 0.28
2.34 2.00 70 6 0.24 0.25 0.28
2.64 2.00 80 12 0.25 0.27 0.29
2.34 2.00 70 24 0.23 0.25 0.27
2.34 2.00 70 48 0.22 0.24 0.26
2.71 2.2 85 1 0.06 0.08 0.10
2.71 2.2 85 3 0.25 0.27 0.29
2.71 2.2 85 12 0.24 0.25 0.28
2.71 2.2 85 24 0.26 0.28 0.30
В табл. 1 приведены данные об адсорбционной емкости цеолитов, по-
лученных при температуре кристаллизации 90° С из гидрогелей различ-
ного состава.
При получении гидрогелей во всех случаях применялся 2 н. (по NaOH)
раствор жидкого стекла с содержанием SiO2 180—190 г/л. Используемые
растворы алюмината натрия содержали от 80 до 250 г/л А12О3 и имели кау-
13 Цеолиты
193
стический модуль 1.7—1.9. Приготовление гидрогеля осуществлялось
путем одновременного сливания гелеообразующих растворов, при ком-
натной температуре, с интенсивным перемешиванием образующегося
золя. Перемешивание производилось и после коагуляции золя, что при-
водило к образованию текучего гидрогеля, в котором частицы собственно
гидрогеля находятся во взвешенном состоянии в маточном растворе.
Установлено, что предварительное выдерживание гидрогеля в течение
нескольких часов при комнатной температуре перед нагреванием и кри-
сталлизацией не оказывает влияния на адсорбционную емкость образую-
щихся при кристаллизации цеолитов.
Образцы, данные для которых приведены в табл. 1, обладали адсорб-
ционными свойствами и кристаллической структурой, характерными для
цеолитов типа А. Из таблицы видно, что чем меньше содержание SiO2
в гидрогеле и чем ниже содержание воды, тем быстрее происходит кри-
сталлизация.
Цеолиты типа X, так же как и цеолиты типа А, могут быть получены
из гидрогелей, приготовленных совершенно различными способами.
Во всех случаях, если только конечный состав гидрогелей отвечает тре-
буемому, образуется цеолит типа X.
С целью определения необходимой продолжительности кристаллиза-
ции для получения цеолитов типа X с высокой адсорбционной емкостью
были синтезированы образцы цеолитов при времени кристаллизации
гидрогелей от 1 до 48 часов. Полученные данные (табл. 2) показали, что
Таблица 2
Адсорбционная емкость цеолитов типа X, полученных при различной
продолжительности кристаллизации
Состав гидрогеля, моли на 1 моль Д12О3 Условия кристалли- зации Адсорбат Адсорбционная емкость (см3/г) при относительном давлении
Na2O SiO2 н2о темпера- тура, °C продол- житель- ность, часы 0.1 0.5 0.9
3.90 3.00 175 95—100 6—24 M-CyHjg 0.28—0.30 0.29—0.31 0.30—0.33
4.55 3.50 205 95—100 6—48 н-С7Н16 0.27—0.30 0.28—0.31 0.28—0.32
5.20 4.00 230 95—100 6—48 н-С7Н16 0.28—0.29 0.29—0.30 0.30—0.31
минимально необходимой продолжительностью кристаллизации является
5—6 часов, 12 часов кристаллизации обеспечивает во всех случаях полу-
чение цеолитов с высокими адсорбционными свойствами. Дальнейшее
увеличение продолжительности кристаллизации до 48 часов не приводит
к изменению адсорбционных свойств цеолита.
Кристаллизация цеолитов происходит в коллоидной системе, пред-
ставляющей собой гидрогель, состоящий из дисперсной фазы — водного
щелочного алюмокремнегеля и дисперсионной среды — раствора, со-
держащего те же компоненты. В связи с этим представляет интерес вы-
яснение роли отдельных фаз этой системы в процессе синтеза цеолитов.
С этой целью было изучено распределение компонентов между отдельными
фазами в гидрогеле и проведены опыты по кристаллизации гидрогелей,
из которых предварительно отделялся фильтрат, т. е. часть дисперсион-
ной среды.
Отделение фильтрата от исходного гидрогеля производилось путем
вакуумного фильтрования. Полученный при этом отжатый гидрогель
подвергался кристаллизации обычным образом, но в отсутствии филь-
194
Таблица 3
Состав отжатых гидрогелей и адсорбционная емкость полученных из них
цеолитов
Тип цео- лита Содержание компонентов в исходном гидрогеле, моли на 1 моль А12О3 Объем выделен- ного филь- трата, В°/о к объему исходно- го гидро- геля Содержание компонентов в фильтрате, моли на 1 моль А12О3 Содержание компонентов в отжатом ги- дрогеле, моли на 1 моль А12О3 Адсорбат Адсорбционная ем- кость цеолитов (см3/г), полученных при кри- сталлизации отжа- тых гидрогелей при относительном давлении
Na.O S1O, Na2O S1O2 Na,O S1O2 0.1 0.5 0.9
А 2.48 1.70 48.1 3.93 0.07 2.10 2.20 н2о 0.24 0.26 0.23
X 5.20 4.00 49.8 36.00 11.60 3.73 3.65 н-С8Н20 0.28 0.29 0.29
трата. Данные, характеризующие состав фаз гидрогелей и адсорбционную
емкость цеолитов, полученных кристаллизацией отжатых гидрогелей,
представлены в табл. 3.
Отделение от гидрогеля фильтрата, составляющего почти половину
объема гидрогеля, привело к некоторому изменению состава отжатого
гидрогеля в связи с переходом части компонентов в фильтрат. Так, со-
держание Na2O в гидрогеле в обоих случаях заметно уменьшилось, со-
держание SiO2 при получении цеолита А возросло от 1.70 до 2.20, а при
получении цецлита X уменьшилось от 4.00 до 3.65. В результате отделе-
ния фильтрата содержание воды в гидрогеле уменьшилось почти вдвое.
В то же время кристаллизация гидрогелей, из которых выделен фильтрат,
приводит к образованию цеолитов с высокими адсорбционными свой-
ствами. Следовательно, образование цеолита происходит лишь в дисперс-
ной фазе. Компоненты дисперсионной фазы (фильтрата) непосредственно
в синтезе не участвуют. В то же время распределение компонентов между
фазами, определяя состав собственно гидрогеля, в котором происходит
кристаллизация, оказывает существенное влияние на процесс синтеза
цеолита.
Вышеприведенный вывод о роли фаз гидрогеля в образовании цеоли-
тов подтверждается результатами опытов по синтезу цеолитов, проведен-
ных следующим образом. Гидрогели готовились путем смешения креп-
кого раствора алюмината натрия (А12О3 240—250 г/л) и жидкого стекла
(SiO2 180—190 г/л). Перемешивание золя производилось вначале энер-
гично, а перед началом коагуляции прекращалось. В результате коагу-
ляции золя образовывался сплошной, весьма плотный гель, который
разрезался на кусочки и затем погружался на несколько часов в воду в слу-
чае цеолита Айв щелочной раствор в случае цеолита X. При этом в рас-
твор переходила часть щелочи и глинозема в случае цеолита А и кремне-
зема в случае цеолита X. Исходный же концентрированный алюмоси-
ликатный гель при этом не разрушался, сохраняя форму и размеры, что
обеспечивало четкое разделение фаз гидрогеля в процессе кристалли-
зации.
Объем воды, в которую погружался гидрогель при получении цеолита А,
в 1.5—4 раза превышал объем гидрогеля, а объем щелочного раствора,
в который погружался гидрогель при получении цеолита X, в 2 раза
превышал объем гидрогеля. Затем гели кристаллизовались обычным об-
разом. Полученные при этом цеолиты обладали высокими адсорбционными
свойствами (табл. 4).
13*
195
Таблица 4
Адсорбционные свойства цеолитов
(температура кристаллизации 90° С)
Тип об- разца Состав исходного гидрогеля, моли на 1 моль А12О3 Состав конечного гидрогеля, моли на 1 моль А12О3 Концентра- ция Na2O, моли на 1 л геля Время кристал- лизации, часы Адсорбат Адсорбцион- ная емкость (см3/г) при 20° С и значе- ниях р/р8
Na2O SiO2 н2о Na,O SiO2 Н2О исход- ная конеч- ная 0.1 0.5
2.48 1.7 49 2.7 1 СН3ОН 0.22 0.23
2.48 1.7 49 2.48 1.7 122 2.7 1.16 6 » 0.24 0.26
2.48 1.7 49 2.48 1.7 122 2.7 1.16 12 » 0.24 0.25
2.48 1.7 49 2.48 1.7 122 2.7 1.16 24 » 0.23 0.25
2.34 1.7 48 2.34 1.7 155 2.5 0.82 6 » 0.24 0.25
in ал 2.48 1.7 49 2.48 1.7 171 2.7 0.84 12 » 0.24 0.26
2.48 1.7 49 2.48 1.7 171 2.7 0.84 24 » 0.25 0.27
2.48 1.7 49 2.48 1.7 171 2.7 0.84 6 » 0.23 0.24
2.48 1.7 49 2.48 1.7 171 2.7 0.84 12 » 0.24 0.25
2.53 1.7 47 2.53 1.7 169 2.72 0.83 6 Н2о 0.25 0.26
Al' "V 1 3.04 3.5 71 4.55 3.5 205 2.21 1.20 12 н-гептан 0.29 0.29
in ал ; 3.04 3.5 71 4.55 3.5 205 2.21 1.20 12 » 0.30 0.31
Технология производства синтетических цеолитов
Сырьем для получения цеолитов является силикат-глыба, гидроокись
алюминия, едкий натр и хлористый кальций. Процесс получения цео-
литов включает следующие основные стадии: а) приготовление раствора
алюмината натрия путем растворения гидроокиси алюминия в кипящем
растворе едкого натра; б) приготовление раствора жидкого стекла пу-
тем разварки силикат-глыбы; в) смешение растворов в пропорциях, необхо-
димых для получения гидрогеля заданного состава; г) кристаллизация
гидрогеля; д) промывка кристаллов образовавшегося цеолита для удале-
ния избытка щелочи; е) обработка кристаллов цеолита раствором хлори-
стого кальция (в случае получения цеолита в кальциевой форме) и про-
мывка кристаллов для удаления соли; ж) смешение влажной массы кри-
сталлов цеолита с порошком глины; з) гранулирование; и) сушка гранул;
к) прокалка гранул.
Принципиальная схема получения цеолитов показана на рис. 1.
Силикат-глыба разваривается острым паром в автоклаве (1), крепкий
раствор жидкого стекла выдавливается из автоклава в емкость (2), где
разбавляется водой и несколько охлаждается. Охлажденный и несколько
разбавленный раствор откачивается в другую емкость (3), где разбав-
ляется до получения рабочего раствора заданной концентрации.
В емкости (5) раствор отстаивается для отделения от него шлама, ко-
торый спускается в канализацию. Осветленный раствор жидкого стекла
откачивается в емкость рабочего раствора (4), а затем в напорный бачок (5),
откуда он расходуется при получении гидрогеля.
Крепкий раствор щелочи (едкого натра) из емкости (6) поступает
в реактор для получения раствора алюмината натрия (7), оборудованный
лопастной мешалкой и змеевиком для нагрева глухим паром. В реактор
также подается вода для разбавления крепкой щелочи. После измерения
уровня раствора щелочи и проверки содержания в нем NaOH раствор
нагревается до температуры кипения и в него загружается отмеренное
количество гидроокиси алюминия. После растворения гидроокиси алюми-
ния в щелочи нагревание раствора прекращается и он направляется в ем-
196
Рис. 1. Схема получения синтетических цеолитов.
кость (S), служащую для хранения крепкого раствора алюмината. При-
готовление рабочего раствора алюмината натрия осуществляется в ем-
кости (Р) путем разбавления крепкого раствора алюмината. Дальнейшие
операции при получении цеолита А производятся следующим образом.
Готовый раствор алюмината натрия из емкости (Р) насосом откачивается
в напорный бачок раствора алюмината (10), откуда через ротаметр (11)
выдавливается в струйный смеситель (13). Туда же из напорного бачка (5)
через ротаметр (12) поступает раствор жидкого стекла. Смесь растворов
направляется в емкость для получения геля (11), оборудованную мешалкой,
где при непрерывной работе мешалки происходит образование гидро-
геля и затем его размешивание до образования жидкой подвижной пульпы.
Эта пульпа непрерывно стекает в кристаллизатор (15). После заполнения
кристаллизатора содержимое его нагревается при перемешивании до
Рис. 2. Узел промывки цеолитов методом противото-
ка с использованием отстойных центрифуг.
температуры кристаллизации и выдерживается при этой температуре за-
данное время. Нагревание гидрогеля до температуры кристаллизации
может производиться как глухим паром, так и острым при перемешивании.
При большом объеме кристаллизатора более удобен нагрев острым па-
ром, подаваемым в кристаллизатор через распределительное устройство,
так как такой способ нагрева позволяет сравнительно быстро нагреть
большую массу гидрогеля при умеренном механическом перемешивании.
По окончании кристаллизации содержимое кристаллизатора (суспензия,
цеолита) откачивается в декантатор (16), где осуществляется промывка
цеолита декантацией. После нескольких декантаций кристаллы цеолита
подаются на фильтрпресс (20), где окончательно промываются от избытка
щелочи.
Лепешка с фильтрпресса направляется в бегуны (25) для приготовле-
ния массы для таблетирования. Масса для таблетирования готовится
путем смешения цеолита с порошком глины, являющимся связующим
материалом. Глинопорошок в бегуны поступает из бункера (27). Приго-
товленная в бегунах масса направляется на перетирочные вальцы (26),
а затем на гранулятор (28)\ полученные на нем гранулы поступают в лен-
точную сушилку (29). Сухие гранулы отсеиваются от крошки на барабан-
ном сите (30), после чего направляются в прокалочпую печь (31), а крошка
возвращается в бегуны (25) для повторного использования.
При получении кальциевой формы цеолита лепешка с фильтрпресса
загружается в емкость (17), куда подается раствор хлористого кальция.
Необходимая степень обмена может быть достигнута при однократной
обработке, однако при крупномасштабном производстве целесообразно про-
водить двухкратную обработку, первый раз — отработанным раствором
хлористого кальция, второй раз — свежим. При таком способе проведе-
ния ионного обмена достигается весьма высокая степень использования
198
хлористого кальция, что позволяет расходовать лишь минимально необ-
ходимое количество этого реагента. Для приготовления раствора хлори-
стого кальция служит емкость (18), для хранения частично использован-
ного раствора — емкость (19).
При получении микросферического цеолита производится распыли-
тельная сушка подготовленной соответствующим образом цеолитной
пульпы. Эта пульпа закачивается в бункер, откуда постепенно через
пневматический распылитель поступает в сушильную колонну. При рас-
пылении пульпа образует сферические капли, которые, высыхая в сушиль-
ной колонне в потоке горячих дымовых газов, превращаются в порошок
цеолита, состоящий из микросферических частиц. Дымовые газы, покидая
сушильную колонну, проходят через циклоны, где из них выделяется
микросферический цеолитный порошок, который поступает на прокалку.
Прокаленный цеолит затаривается в мешки или железные бочки.
В описанной технологической схеме одной из важных операций яв-
ляется промывка суспензий. Способ промывки кристаллов цеолитов с при-
менением отстоя пульпы в декантаторах пригоден для небольшой по про-
изводительности установки. Крупномасштабные установки требуют ис-
пользования более производительных методов промывки. К числу таких
методов относится, в частности, промывка с помощью отстойных центри-
фуг (рис. 2).
В зтом случае промывка производится следующим образом. По окон-
чании кристаллизации маточный раствор откачивается из кристаллиза-
тора, а суспензия цеолита разбавляется водой (или раствором, поступаю-
щим со второй ступени промывки). Разбавленная суспензия направляется
насосом в отстойную центрифугу первой ступени промывки (1). Промывка
осуществляется ступенчатым противотоком с использованием четырех
ступеней. Каждая ступень включает смесительный бачок (6—8) и центри-
фугу. В первой ступени промывки роль смесительного бачка выполняет
кристаллизатор (9). Чистая вода из бачка (5) в четвертую ступень про-
мывки движется противоточно по отношению к кристаллической пульпе.
Раствор из первой ступени промывки сбрасывается в канализацию.
В первых трех ступенях промывки отжатая масса выгружается шнеком
из центрифуги, смешивается с раствором, поступающим из центрифуги
следующей ступени промывки и направляется в смесительный бачок, где
разбавляется тем же раствором в 6—10 раз. Из центрифуги последней
ступени промывки (4) пульпа поступает на фильтрпресс, где продувкой
теплого воздуха подсушивается до образования плотной массы, поддаю-
щейся транспортировке на ленточном транспортере. Эта масса поступает
в бегуны для смешения со связующим веществом и затем подвергается
гранулированию.
Применение ступенчато-противоточной промывки позволяет за счет
многократного использования воды резко сократить расход ее на про-
мывку цеолита, что является существенно важным при крупномасштабном
получении цеолитов. Кроме того, приме ле противоточной ступенчатой
промывки способствует сокращению потерь цеолита с промывными водами.
Для разделения водных суспензий цеолитов в процессе промывки
разработана специальная центрифуга НОГШ-500 (конструктор Н. П. До-
дин). Эта центрифуга имеет следующие основные технические характери-
стики:
Производительность:
по суспензии.......................15 м3/час
по осадку........................1.2 т/час
Диаметр ротора..................... 500 мм
Число оборотов ротора.............. 2650 об/мин.
Мощность электродвигателя..........28 квт
Вес центрифуги...................... 2400 кг
199
Приготовление цеолитов типа X производится в принципе по такой же
схеме, как и цеолитов типа А. Имеются лишь некоторые отличия, связан-
ные с тем, что при получении цеолитов типа X необходимо приготовление
гидрогелей с относительно высоким содержанием окиси кремния (SiO2
от 2.8 до 4.0 молей) и щелочи (от 2.8 до 5.2 молей Na2O). Другим важным
элементом в технологии приготовления цеолитов типа X является исклю-
чение перемешивания гидрогеля после нагревания его до температуры
кристаллизации.
В 1960 г. было начато опытное производство таблетированных цеолитов
в промышленных условиях по вышеописанной технологии [12—14].
Получение гидрогелей производилось при одновременной подаче
растворов в смеситель. Как показал длительный опыт работы, способ
осаждения гидрогелей при одновременном смешении растворов практи-
чески весьма удобен, позволяет стабильно получать весьма однородные
по составу гидрогели. Образующийся в смесителе золь поступал в емкость
с мешалкой, где он коагулировал, затем, подвергаясь энергичному раз-
мешиванию, приобретал хорошую текучесть и самотеком поступал
в кристаллизатор. После заполнения кристаллизатора, в случае получе-
ния цеолитов типа А, производился нагрев гидрогеля при механическом
перемешивании либо глухим паром, через змеевик, находящийся в кри-
сталлизаторе, либо острым паром, подаваемым через распределитель,
расположенный в нижней части кристаллизатора.
По завершении кристаллизации маточный раствор сливался из кри-
сталлизатора, а кристаллический осадок размешивался с водой и отка-
чивался в декантатор. Кристаллы цеолитов при отстаивании образуют
чрезвычайно плотные осадки. Размешивание этих осадков с водой возможно
при использовании переносного воздушного барбатера или путем подачи
в слой осадка струи воды с давлением в несколько атмосфер. Образовав-
шаяся после размешивания кристаллического порошка цеолита с водой
жидкая суспензия легко перекачивается центробежным насосом. По
окончании перекачки трубопроводы промывались водой для удаления
остатков суспензии во избежание отложения плотных осадков в трубо-
проводах. При этом использовалась обычная запорная арматура.
Как показал опыт производства цеолитов, жидкая суспензия цеолит-
ного порошка в воде обладает хорошей прокачиваемостью по трубопро-
водам и, при соблюдении указанного условия, отложения осадков в трубо-
проводах не наблюдается.
Промывка цеолитов после кристаллизации во всех случаях осуще-
ствлялась вначале декантацией, затем на фильтрпрессе. По окончании
промывки производилась подсушка лепешки на фильтрпрессе продувкой
воздуха, что улучшает транспортабельность цеолитной массы после вы-
грузки ее с фильтрпресса. Выгруженная из фильтрпресса масса содер-
жала 40—42% влаги и в таком виде поступала в бегуны для смешения
с порошком глины. Количество добавляемой в цеолит глины составляло
15—20%. Масса, подготовленная для таблетирования, таблетировалась
с использованием барабанной таблеточной машины. Сушка свежесформо-
ванных таблеток производилась в камерной сушилке на сетчатых стелла-
жах при циркуляции через слой таблеток воздуха, нагретого до 120° С.
Высушенные таблетки хранились в железных бочках до момента загрузки
их в прокалочную печь.
Прокалка цеолита осуществлялась в шахтной печи при движении
таблеток сплошным слоем сверху вниз и дымовых газов снизу вверх (за
счет работы дымососного вентилятора). Температура дымовых газов перед
поступлением их в печь строго контролировалась. За весь период прокалки
эта температура не превышала 600° С. Прокаливание адсорбента произ-
водилось при температуре 575—600° С. Длительность пребывания адсор-
200
бейта в зоне прокаливания, составляла примерно 10 часов. Циркуляция
адсорбента через печь после вывода ее на режим продолжалась до тех пор,
пока выходящий из печи цеолит не приобретал механической прочности,
характерной для прокаленных таблеток, и содержание влаги в нем было
не выше 1—1.5%. Затем выгруженный из печи адсорбент затаривался
в железные бочки, а печь догружалась свежими сухими таблетками.
Полученные цеолиты обладали четко выраженными молекулярно-
ситовыми свойствами, характерной кристаллической структурой и ка-
тионно-обменными свойствами, присущими цеолитам.
ЛИТЕРАТУРА
1. R. М. В а г г е г, J. Chem. Soc., 1948, 127.
2. R. М. Barr er et al., J. Chem. Soc., 1952, 1561.
3. R. M. Barr er et al., J. Chem. Soc., 1953, 1466.
4. R. M. В а г г e r et al., J. Chem. Soc., 1956, 2992.
5. R. M. Barter et al., J. Chem.-Soc., 1959, 195.
6. R. M. Barter, Brit. Chem. Engng., 1959, 1.
7. D. W. В reck et al., J. Am. Chem. Soc., 1956, 23, 5963.
8. R. S с a r r, Chem. Age India, 1957, 8, 1, 81.
9. E. L. L a b i n e, Chem. Engng., 1959, 66, 16, 104.
10. Я. В. Мирский, Авторское свидетельство СССР, 127997.
11. Я. В. Мирский, ДАН СССР, 1960, 130, 1, 116.
12. Я. В. М и р с к и й, М. Г. Митрофане в, Хим. и технол. топлив и масел.
1960, 10, 16.
13. Я. В. М и р с к и й, М. Г. Митрофанов, Новости нефт. техн., сер. пере-
работка, 1960, 10, 8.
14. Я. В. М и р с к и й, М. Г. Митрофан ов, Л. Т. Б о л о т о в, А. И. Меж-
ду м о в а, К. Ф. Бунин, В. Н. Д у л ь с к а я, А. Н. Мельник,
Новости нефт. техн., сер. переработка, 1962, 2, 13.
15. И. В. Неймарк и др., Колл, ж., 1961, 23, 4, 115.
16. М. С. М и с и н, Л. М. М а к с и м о в а и др., в сб.: Синтетические цеолиты,
Изд. АН СССР, 1962, стр. 135.
17. Л. И. П и г у з о в а, Хим. и технол. топлив и масел, 1961, 6, 9.
18. С. П. Ж д а н о в, Н. Н. Б у н т а р ь, ДАН СССР, 1961, 138, 119.
ВЛИЯНИЕ УСЛОВИИ ГРАНУЛЯЦИИ И ХРАНЕНИЯ ЦЕОЛИТОВ
НА ИХ МЕХАНИЧЕСКИЕ СВОЙСТВА
А. Т. Слепнева, Б. А. Липкинд, В. А. Бурылов,
С. В. Капацинский
Применяющиеся в настоящее время способы грануляции синтетиче-
ских цеолитов и виды связующих материалов крайне разнообразны. Од-
нако в промышленной практике наиболее распространена грануляция
цеолитиых порошков с глиняными связующими. Для этой цели были пред-
ложены глины, существенно отличающиеся друг от друга по минералоги-
ческому и химическому составам. Представлялось интересным исследо-
вать свойства основных из предложенных образцов глин как связующих.
При изучении механических и адсорбционных свойств гранулированных
цеолитов применялись методы анализа, предусмотренные временными
техническими условиями.
В задачу настоящего сообщения не входит обсуждение связи этих
свойств с физико-химическими свойствами изученных глин. Можно
только отметить, что свойства глин как связующих для цеолитов не на-
ходятся в прямой зависимости ни от минералогического, ни от химиче-
ского составов глин.
201
Для каждого из пяти изученных образцов глин при грануляции цео-
литов четырех форм (NaA, СаА, NaX, СаХ) подбирались минимально
допустимые количества связующего, обеспечивающие получение гранули-
рованных цеолитов с прочностью на раздавливание и истирание, соответ-
ствующей требованиям ВТУ. Полученные при этом результаты приве-
дены в табл. 1. Как следует из приведенных в таблице данных, изученные
Таблица 1
Количество глины, необходимое для получения
показателей механической прочности, соответствующих
ВТУ
Месторождение глины Количество глины, необходимое для полу- чения показателей механической прочности, соответствующих ВТУ, в % по отношению к кристаллиту
форма цеолита
NaA СаА NaX СаХ
Глуховское 15 18 15 18—20
Латненское 22 25—26 22 22—27.5
Курцовское .... Смесь: огланлинское 9—10 15 9—10 16—18
(1 часть) и глухо- вецкое (4 части) . > 35 > 35 > 35 > 35
Харьковское .... 10—12 > 25 10—12 > 25
образцы глин по своим связующим свойствам располагаются в следующем
порядке: курцовская, глуховская, латненская, смесь огланлинской и
глуховецкой глин. Последняя оказалась совсем непластичной, и даже при
добавке 35% этой смеси глин не удалось получить гранулированный цео-
лит, который по своим механическим свойствам соответствовал бы тре-
бованиям ВТУ.
Особое место занимает харьковская глина, которая оказалась высоко-
пластичной при грануляции натриевых форм цеолитов, но имела очень
низкие связующие свойства при грануляции цеолитов в кальциевой форме.
Известно, что для окончательного выбора глины совершенно недостаточно
оценки только лишь связующих ее свойств, так как некоторые высоко-
пластичные глины могут значительно снижать показатели динамической
адсорбционной активности гранулированного цеолита. В связи с этим
образцы гранулированных цеолитов, приготовленные с добавкой указан-
ных в табл. 1 количеств глины, оценивались на адсорбционную активность
в динамических условиях по парам воды, пентану и тиофену в соответ-
ствии с требованиями ВТУ. Результаты этих определений (средние из
Таблица 2
Динамическая активность образцов, сгранулированных на минимальном
количестве связующего, еще обеспечивающего соответствующую ВТУ прочность
Месторождение глины Динамическая активность для цеолитов разных форм, вес. %
NaA СаА NaX
Н2О Н2О пентан Н..О тиофен
Глуховское 14—15 12—13 6—7 16—17 8.5—10
Латненское 14—15 12—13 6—7 16—17 8.5—10
Курцовское 12—14 11—12 0 16—17 2—3
Харьковское 9—10 12—13 6—7 — 3—4
202
ряда параллельных опытов) приведены в табл. 2 и показывают, что наи-
более пластичные из изученных глин, курцовская и харьковская, могут
быть использованы только лишь для приготовления осушителей. Так,
КоличестВо глухоВской глины, % Количество глухоВекой глины, %
Рис. 1. Влияние количества глуховской глины на механическую прочность
гранул цеолита типа NaA (а) и СаА (б).
1 — прочность на истирание (%); г — прочность на раздавливание (кг/мм1).
если' даже при адсорбции воды гранулированными цеолитами, приго-
товленными со сравнительно небольшими количествами курцовской глины,
заметно некоторое снижение динамической активности цеолита, то при
Рис. 2. Влияние количества латненской глины на механическую прочность
гранул цеолита типа NaA (а) и СаА (б).
1 — прочность на истирание (%); 2 — прочность на раздавливание (кг/мм2).
адсорбции более трудно сорбирующихся молекул динамическая актив-
ность таких сорбентов падает очень резко, снижаясь в отдельных случаях
до нуля (например, адсорбция пентана из смеси его с изопентаном).
Более высокую динамическую активность при адсорбции всех изучен-
ных веществ имели цеолиты, сформованные с глуховской и латненской
глинами. Именно поэтому обе эти глины нашли в настоящее время наиболее
широкое применение в качестве связующего для цеолитов. При условии
2ЭЗ
равных адсорбционных свойств гранулированных цеолитов предпочтение
должно быть отдано наиболее пластичной глине, обеспечивающей полу-
чение механически прочных гранул при меньшей добавке — глине глу-
ховского месторождения. Преимущество ее связующих свойств перед
связующими свойствами латненской глины особенно наглядно видно
из рис. 1 и 2.
Таким образом, лучшим из изученных глиняных связующих по сово-
купности всех свойств формованных цеолитов является глина глухов-
ского месторождения. Поэтому на этой глине проводилось изучение влия-
ния режима грануляции на механические свойства формованных цеолитов.
Основными параметрами, определяющими, при прочих равных усло-
виях, механические свойства формованных цеолитов, являются: тонина
помола глины, характер смешения и пластификации массы из глины и
кристаллита, влажность формовочной массы и вид оборудования для гра-
нуляции. Ранее было показано решающее влияние влажности формовоч-
ной массы на механические свойства цеолита [1], остальные параметры
процесса грануляции изучались в настоящей работе. В табл. 3 приведены
данные по влиянию тонины помола глшы на механические свойства фор-
мованных цеолитов. Из приведенных в этой таблице данных видно, что
тонина помола глины оказывает заметное влияние на механические свой-
ства формованных цеолитов.
Таблица 3
Влияние тонины помола глины на механическую прочность цеолитных fpanyn
Наименование образца Тонина помола глины, мк Тип гранулятора Насып- ной вес, Г;СМ3 Механическая проч- ность
на раз- давлива- ние, кг/мм2 на исти- рание, %
NaA-202-362-935 420 Лабораторная шнековая 0.71 0.4 38.3
NaA-202-362-936 315 машина То же 0.72 0.6 50.0
NaA-202-362-938 150 » 0.73 0.8 58.5
NaA-202-362-939 < 105 » 0.73 0.9 68.0
NaA-27-951 260 » 0.66 0.4 53.0
NaA-27-951 A 190 » 0.68 0.6 60.0
NaA-27-951 Б 95 » 0.69 0.9 69.0
NaA-202-394-1 > 315 Вмазывающая таблеточ- 0.70 0.6 36.9
NaA-202-394-2 200 ная машина ТМ-1 То же 0.72 0.7 43.3
NaA-202-394-3 < 150 » 0.73 1.1 50.0
Измельчение глины в изученных пределах приводило к повышению
механической прочности цеолитов как на раздавливание, так и на исти-
рание. Проведенные совместно с А. И. Сараховым опыты по дальнейшему
измельчению глины методом вибропомола с последующей грануляцией
цеолита с этой глиной показали, что при еще более тонком помоле глины
может быть достигнуто некоторое увеличение механической прочности
сформованных гранул при равном количестве связующего: 1.5 кг/мм2 и
81.5% вместо 1.4 кг/мм2 и 74.5% для исходного образца. Изменение
дисперсности второго компонента гранулируемой массы кристаллов
цеолитов в пределах от 2 до 8 мк не оказывало влияния на механические
свойства гранулированного цеолита.
Исходные компоненты, глина и кристаллы, могут смешиваться по
двум разным схемам. По первой схеме кристаллы подсушиваются до по-
лучения хорошо сыпучего материала и смешение производится в порош-
204
ках. По второй схеме отжатая и немного подсушенная лепешка смеши-
вается с глиной в пасте. Ниже (табл. 4 и 5) приведены данные, показываю-
щие влияние способа смешения на механическую прочность «цеолитных
гранул.
Таблица 4
Влияние способа смешения на уровень механической прочности формованных
гранул
№ серии № опыта Наименование образца Способ смешения Длитель- ность смеше- ния. часы На- сып- ной вес, г/см3 Механическая прочность
на раз- давлива- ние, кг/мм2 на ис- тира- ние, %
1 NaA-925 Сухое смешение. 0.5 0.75 0.7 67.1
1 1 2 NaA-926 Смешение в пасте. 0.5 0.73 0.5 69.3
п / 3 Проба 1 Сухое смешение. 0.5 0.73 0.8 52.0
111 4 Проба 2 Смешение в насте. 0.5 0.74 0.9 54.0
пт / 5 Проба 3 Сухое смешение. 2.0 0.76 1.0 67.0
111 1 6 Проба 4 Смешение в пасте. 2.0 0.75 1.1 65.0
Сухое смешение. 0.74 0.8 62.0
средние показатели | Смешение в пасте. — 0.74 0.8 62.0
Результаты опытов, приведенные в табл. 4, показывают, что при до-
статочно длительном смешении (не менее 0.5 часа) средний уровень ме-
ханической прочности гранулированных цеолитов не зависит от способа
смешения компонентов. Для оценки однородности механических свойств
цеолитов в зависимости от способа смешения образцы формовочных масс
гранулировались в равных условиях на лабораторном шнековом грану-
ляторе, а затем отдельные таблетки подвергались испытанию на раздав-
ливание (табл. 5). Как следует из приведенных в табл. 5 данных, оба спо-
Таблица 5
Влияния способа смешения на механическую прочность отдельных гранул
(шнековый гранулятор)
Наименование образца Способ смешения Длитель- ность сме- шения, часы Механическая прочность на раздавливание, кг/мм2
№ опыта
1 2 3 4 5 6 7
NaA-925 Сухое смеше- ние. 0.5 0.7 0.7 0.8 0.7 0.7 0.7 0.7
NaA-928 Смешение в пас- те. 0.5 0.6 0.6 0.7 0.7 0.7 0.6 0.7
соба смешения обеспечивают примерно равную и достаточно высокую
однородность механической прочности гранул цеолитов. Следовательно,
обе схемы смешения дают примерно равные показатели по механическим
свойствам цеолитов и выбор того или иного способа смешения должен осу-
ществляться на основании чисто технологических соображений. Преиму-
ществом схемы смешения в порошках является возможность использова-
ния малогабаритного, высокопроизводительного, непрерывнодействую-
щего оборудования и лучшие возможности для автоматизации процесса
205
и точной дозировки компонентов. Вторая схема, смешение в пасте, более
выгодна по энергетическим соображениям.
Тщательно смешанная и пластифицированная масса поступает на
грануляцию. Наибольшее распространение в настоящее время для гра-
нуляции цеолитов получили шнековые и барабанные вмазывающие гра-
нуляторы. Были сопоставлены данные по механической прочности гра-
нул, полученных на этих типах грануляторов в полузаводских и про-
мышленных условиях. На основании изучения большого количества об-
разцов и получения средних данных было показано, что уровень механи-
ческой прочности на раздавливание примерно одинаков, а уровень меха-
нической прочности на истирание заметно более высок при использовании
шнекового гранулятора. Это преимущество особенно сильно проявляется
при более низком содержании связующего в цеолите. Недостатком вма-
зывающего гранулятора является также разнородность механических
Таблица 6
Характеристика однородности механической прочности таблеток,
сформованных на вмазывающем барабанном грануляторе ТМ-1
№ опыта Прочность на раздавливание (кг/мм2) отдельных таблеток образца
1 2 3 4 5 6 7 8
1 0.5 0.9 0.7 0.9 0.6 1.0 0.5 0.9
2 0.9 1.1 0.8 0.2 1.5 0.4 1.5 1.2
3 0.6 1.1 0.4 0.8 0.6 1.8 0.8 0.9
свойств формуемых таблеток. В табл. 6 приведены в качестве примера
данные, характеризующие прочность на раздавливание отдельных таб-
леток, отбиравшихся при грануляции на барабанном вмазывающем гра-
нуляторе производительностью около 0.5 т/сутки.
Таблица 7
Влияние условий и длительности хранения формованных цеолитов
на их свойства
Ы» серии Условия хранения № опыта Длительность хра- пения, годы с лэлси’ииидер/пании после хранения, % Насыпной вес, г/см3 Активность по парам воды, % Мех аническая прочность
в статиче- ских усло- виях в динамиче- ских усло- виях С5 С В п ff те Г гг те Р ние, кг/мм2 истирание, %
В открытом сосуде 1 0 18.0 0.75 20.0 14.5 1.3 65
т в помещении. 2 1 20.0 0.76 20.2 13.9 1.2 68
3 2 19.6 0.76 22.1 14.5 1.1 69
4 3 19.5 0.77 20.1 14.3 1.3 69
( В открытом сосуде 5 0 33.0 0.75 22.1 14.3 0.9 66
II ) под водой в поме- 6 1 32.0 0.76 22.2 13.9 0.7 45
11 < щении. 7 2 32.0 0.76 20.6 14.0 0.7 51
1 8 2.5 33.0 0.77 21.0 14.1 0.7 50
В открытом сосуде 9 0 20.0 0.74 21.0 14.8 1.0 66
111 1 на улице в зимних 10 2 мес. 20.0 0.73 20.6 —— 0.8 62
условиях. 11 3 » 20.3 0.74 21.0 14.5 1.0 66
206
Причиной такой неоднородности служит неравномерное вмазывание
массы в центре и на периферии барабана, что было подтверждено* спе-
циальными опытами.
В заключение приводятся имеющие определенный практический ин-
терес данные о влиянии условий длительного хранения формованных
цеолитов на их механические свойства. Образцы цеолитов хранились в те-
чение 3 лет в герметичной и открытой таре в помещении, в открытом сосуде
под водой и в течение 3 зимних месяцев в открытом сосуде на улице. Ре-
зультаты некоторых из опытов приведены в табл. 7.
Во всех случаях, кроме длительного хранения цеолита в воде, механи-
ческие свойства его остались на первоначальном уровне. Длительное
пребывание цеолита в воде сперва привело к снижению его механической
прочности, которая потом длительное время не изменялась и оставалась
на уровне, близком к требованиям ВТУ. Как и следовало ожидать, ад-
сорбционные свойства цеолитов не изменялись во всех изученных случаях.
ЛИТЕРАТУРА
1. Б. А. Л и п к и в д, В. А. Б у р ы л о в и др., в сб.: Синтетические цеолиты,
Изд. АН СССР, 1962, 191.
СОВЕРШЕНСТВОВАНИЕ МЕТОДОВ
И ПОКАЗАТЕЛЕЙ ВРЕМЕННЫХ ТЕХНИЧЕСКИХ УСЛОВИЙ
НА ФОРМОВАННЫЕ СИНТЕТИЧЕСКИЕ ЦЕОЛИТЫ
С. М. Вайнштейн, Е. Я. Гиенко, А. Т. Слепнева, Б. А. Липкинд,
. В. А. Бурылов
Введение временных технических условий на формованные синтети-
ческие цеолиты типов А и X в значительной мере содействовало улучше-
нию адсорбционных и механических свойств цеолитных адсорбентов,
изготовляемых на опытных и промышленных установках Советского Союза.
Накопленный к настоящему времени опыт применения ВТУ показал, что
принятые методы оценки позволяют надежно контролировать качество
цеолитов, а показатели, принятые в ВТУ, предъявляют к ним достаточно
высокие требования. Хотя действующие временные технические условия
в общем регламентируют выпуск качественных цеолитов, накапливающийся
опыт производства и применения цеолитных адсорбентов требует дальней-
шего совершенствования методов оценки и показателей качества. Этому
вопросу и посвящено данное сообщение.
Одним из основных показателей, характеризующих адсорбционные
свойства цеолитов, является их динамическая активность по парам воды.
При оценке динамической активности цеолитов по парам воды для сокра-
щения времени проведения опыта принято высокое влагосодержание осу-
шаемого воздуха (ф=70—75%, что соответствует точке росы 4-15°).
Осушка потоков с таким высоким влагосодержанием не характерна для
цеолитов, которые применяются в основном для глубокой доосушки
газов или жидкостей. Поэтому возникало некоторое сомнение в том, ха-
рактеризует ли показатель динамической активности, полученный по
режиму, принятому в ВТУ, активность цеолитов при глубокой осушке
продуктов с относительно низким исходным влагосодержанием. Для уточ-
нения этого вопроса были использованы два образца цеолитов СаА, один
из которых был некондиционным по показателю динамической активности
207
по парам воды,
по парам
ноет!
Для обоих образцов была изучена динамическая актив-
воды при режимах и
на
20
Относительное Влагосодерэюание Воздуха,%
* > 6
- г
аппаратуре, предусмотренных
ВТУ, но с изменением от-
носительного влагосодержа-
ния осушаемого воздуха от
<р=75% до <р=0.3% (точка
росы от -j-15° до —47°). Ре-
зультаты этого опыта приве-
дены на рис. 1 и показывают,
что изменение влагосодержа-
ния осушаемого воздуха в
пределах от <р=75% до <р=
=0.3 % не привело к сколько-
нибудь существенному изме-
нению отношения динамиче-
ских активностей двух изу-
чавшихся цеолитов. Во всем
—।—।—।—।—।___________।___।__। ।
0 20 4Z7 80 80
Относительное Влигоеодерзюание Воздуха, %
Рис. 1. Влияние влагосодержания осушаемого
воздуха на динамическую активность цеолитов.
а — относительное влагосодержание воздуха — от 0.3 до
2%, б — от 2 до 75%. 1 —кондиционный цеолит; 2 —-
некондиционный цеолит.
изученном интервале величи-
на этого отношения сохраня-
лась на уровне — 130%. Ана-
логичные результаты были
получены для цеолитов NaA
и NaX в интервале влагосо-
держаний осушаемого возду-
ха 2—75%, с той лишь раз-
ницей, что у цеолитов типа X
активность не оставалась в этом интервале концентраций постоянной, а
уменьшалась на 15—20%. Таким образом, принятый в ВТУ показа-
тель достаточно надежно характе-
ризует динамическую активность гра-
нулированного цеолита по парам во-
ды в широком интервале влагосодер-
жания осушаемого потока.
Метод оценки динамической актив-
ности по парам воды, регламенти-
рованный ВТУ, предусматривает до-
статочно жесткие режимы осушки:
сравнительно небольшая высота слоя
адсорбента и высокие скорости осу-
шаемого потока. В таких условиях
величина динамической активности
должна зависеть от размеров гра-
нул формованных цеолитов. Однако,
хотя ВТУ предусматривает выпуск
цеолитов в виде таблеток диаметром
и высотой 2.0 и 4.0 мм, показатели
динамической активности в зависимо-
сти от размера гранул не дифференци-
рованы. В связи с этим была изучена
зависимость динамической активности
от размера гранул для всех форм
Рис. 2. Увеличение динамической ак-
тивности с уменьшением размера гра-
нул от 4.0 до 2.0 мм для разных форм
цеолитов.
цеолитов. На рис. 2 показано увеличение динамической активности цео-
литов при уменьшении размеров таблеток от 4.0 до 2.0 мм. Приведенные
данные являются средними из большого числа опытов, в которых все
условия приготовления образцов, кроме диаметра отверстия матриц при
грануляции, были равными. Из графика, приведенного на рис. 2, видно,
208
что, как и следовало ожидать, эффект увеличения динамической актив-
ности цеолитов с уменьшением их гранул увеличивается по мере уменьше-
ния размера входных окон в полости цеолита. На основании полученных
данных рекомендуется ввести в ВТУ дополнительные повышенные пока-
затели для 2.0-миллиметровых таблеток.
В настоящее время формованные цеолиты в отдельных случаях вы-
пускаются не только в виде таблеток, как это предусмотрено ВТУ, но
и в виде гранул диаметром 4 и 2 мм различной длины. Поэтому было про-
верено влияние длины гранул на их динамическую активность при оценке
в стандартных условиях. В опытах, результаты которых представлены
в табл. 1, оценивалась динамическая активность гранул длиной 15—20 мм
и 0 4 мм и приготовленных из них таблеток с длиной, равной диаметру.
Таблица 1
Влияние длины гранул на показатель динамической активности по парам воды
№ опыта № образца Длина гра- нулы, мм Насыпной вес, г/см3 Адсорбционная активность
в статических условиях при <Р = 0.03% в динамических условиях
вес. % МГ/СМ3
1 4 0.72 20.9 12.6 90.7
1 | 2 15 0.64 21.5 10.6 67.2
3 4 0.73 20.7 14.1 103.5
2 { 4 20 0.67 21.2 12.5 83.7
3 ! 5 4 0.72 20.0 16.1 116.0
1 6 20 0.68 20.5 13.0 89.0
Из приведенных данных видно, что оценка динамической активности
цеолитов в виде гранул длиной 15—20 мм приводит к снижению показа-
теля активности, причем в связи с одновременным уменьшением насып-
ного веса особенно резко уменьшается величина, характеризующая ак-
тивность единицы объема (мг/см3). В связи с этим рекомендуется в ВТУ
уточнить гранулометрический состав гранулированного цеолита при его
испытании на динамическую активность.
Механические свойства формованных цеолитов в соответствии с ВТУ
оцениваются их прочностью по отношению к раздавливанию и истиранию.
Причем оба эти показателя определяются для цеолитов в дегидратирован-
ном состоянии (остаточное влагосодержание не выше 2.0%). Известно,
что прочность цеолитов зависит от их влагосодержания (непрерывно умень-
шается с увеличением степени гидратации). В связи с этим возникает
вопрос: характеризует ли в достаточной мере оценка механической проч-
ности дегидратированного цеолита, предусмотренная ВТУ, его прочность
в процессе эксплуатации? Ответ на этот вопрос может быть дан на основа-
нии анализа отношения прочности цеолитов в дегидратированном и гидра-
тированном состояниях. Если величина этого отношения сохраняет при-
мерно постоянное значение вне зависимости от изменения режимов
основных операций технологического процесса производства цеолитов, то
предусмотренная ВТУ оценка прочности является достаточной для харак-
теристики их механических свойств. Проведенный анализ большого количе-
ства образцов различных форм цеолитов, приготовленных на опытной
установке, показал, что отношение прочностей определенной формы цео-
лита в дегидратированном и гидратированном состояниях изменяется
сравнительно в небольших пределах. Средние показатели, характеризую-
щие величину этих отношений, приведены в табл. 2. Особенный интерес
14 Цеолиты
209
Таблица 2
Характеристика прочности цеолитов в дегидратированном
и гидратированном состояниях
Тип цеолита Прочность цеолитов в дегидратированном состоянии Прочность цеолитов в гидратированном состоянии Отношение прочности цеолита дегидратированного к гидратированному
равна влива- ние, кг-мм2 истирание, % раздавлива- ние, КГ/ММ2 истирание, % раздавлива- ние истирание
А 0.6—1.5 60—75 0.4—0.6 20—30 1.5—2.5 2.0—3.5
X 0.6—0.9 60—70 0.3—0.4 2.0—5.0 2.0—3.0 ~20
представляло сопоставление отношения прочностей цеолитов, изготовлен-
ных различными организациями. Эти данные приведены в табл. 3. Об-
разцы цеолитов формы NaA или сходных с ней форм, представленные
в этой таблице, отличались друг от друга регламентом приготовления и
типом связующего.
Таблица 3
Характеристика механической прочности цеолитов, приготовленных
в различных организациях
Наименование цеолита Характер свя- вующего Содержа- ние влаги перед определе- нием проч- ности, % Механическая прочность Отношение прочности цеолита дегидрати- рованного к гидра- тированному
раздавли- вание, кг мм2 истира- ние, % раздавли- вание истира- ние
Образец авторов. Глина. { 1.8 21.0 1.1 0.6 78.5 35.0 1.8 2.2
Образец Я. В. Мир- ского NaA—10 Гкз. Глина. | 1.9 19.5 1.0 0.5 73.0 28.0 2.0 2.6
Образец В. Я. Ни- колиной Ц. Синтети- ( ческое. | 1.7 19.0 0.3 0.1 16.0 5.0 3.0 3.2
Образец фирмы «Линде» 4AXW. — { 1.6 20.0 1.8 0.6 58.0 28.0 3.0 2.0
Из данных, приведённых в табл. 3, видно, что для всех изученных
образцов, вне зависимости от условий их приготовления, отношение проч-
ностей дегидратированных и гидратированных цеолитов оставалось при-
мерно на одном уровне.
Были проведены специальные опыты, в которых изучалось влияние
типа и количества глиняного связующего, характера подготовки массы
к грануляции и режима термообработки формованного цеолита на величину
отношения прочности цеолитов в дегидратированном и гидратированном
состояниях. Пока не удалось разработать режимов, которые позволяли бы
заметно уменьшить величину указанного отношения прочностей цеоли-
тов без заметного ухудшения его адсорбционных свойств. Полученные
результаты показывают, что в настоящее время нет необходимости вво-
дить в ВТУ дополнительно оценку механических свойств формованных
цеолитов в гидратированном состоянии. Однако в исследовательских и
опытных работах целесообразно оценивать механические свойства цеоли-
тов при различной степени их гидратации.
210
Накопленный опыт производства и применения цеолитных адсорбен-
тов показал, что в ВТУ не включены показатели, характеризующие не-
которые важные эксплуатационные свойства формованных цеолитов,
а именно: водостойкость и пылевыделение. В связи с этим разрабатывались
методики для определения этих характеристик. Способ определения водо-
стойкости был предложен лабораторией И. Э. Гельмса и заключается
в следующем. Навеска адсорбента, отсеянного на сите 1.5 мм и предвари-
тельно прокаленного при 350° С 4 часа, помещается в стакан на 250 см®
и заливается водой. Адсорбент выдерживается в воде в течение 24 часов,
затем вода декантацией отделяется, а гранулы высушиваются, отсеваются
на сите 1.5 мм и прокаливаются при
350° 4 часа. По разности весов до и
после опыта определяется водостойкость
цеолитов. При оценке образцов цеоли-
тов, приготовленных различными орга-
низациями, были получены показатели
водостойкости от 0 до 100%. Низкая
водостойкость формованных цеолитов
объясняется неудачным выбором свя-
зующего или недостаточно эффективным
режимом термообработки сформованного
цеолита. Поскольку разработаны ва-
рианты технологии, позволяющие гаран-
тированно получать высокую водостой-
кость формованных цеолитов без ухуд-
шения других их абсорбционных и ме-
ханических свойств, рекомендуется
ввести в ВТУ показатель водостойкости
Влажность цеолита, °/о
Рис. 3. Влияние степени гидратации
цеолита на механическую прочность
формированных гранул.
1 — цеолит NaA; 2 — цеолит СаА.
для всех форм цеолитов пе менее 98 %.
Для оценки содержания пыли на фор-
мованных цеолитах были разработаны
два метода: метод мокрого рассева и ме-
тод отдувки пыли при вибрации цеолита.
Метод мокрого рассева. По этому методу навеска (25—30 г)
предварительно прокаленных при 650° в течение 4 часов гранул цеолита
загружается в колбу и заливается 100 см3 воды. После 5—10 мин. стоя-
ния содержимое колбы переносится на сито с размером ячейки 1 мм и на
сите дополнительно промывается водой (50 мл). Вода с содержащейся
в ней пылью собирается в фарфоровую чашку и выпаривается. Получен-
ный осадок прокаливается при 650° 2 часа и взвешивается. Вес осадка,
отнесенный к первоначальной навеске и выраженный в процентах, дает
содержание пыли на цеолите. Сходимость параллельных определений до
0.1 абс.% — при оценке образцов с содержанием пыли менее 1.0% и
0.15—0.20 абс. % — при содержании пыли на образце выше 1.0%.
Метод отдувки пыли при вибрации цеолита. На рис. 4
приведена схема прибора для определения пыли методом вибрации. Реак-
тор (5) с загруженной навеской полностью гидратированного цеолита уста-
навливается на вибраторе (1) типа ЭЛ-1. Амплитуда колебаний вибратора
регулируется латром (2), напряжение около 100 в. Для отдувки пыли
в реактор через реометр (4) подается воздух со скоростью 1 м/сек. Дли-
тельность определения составляет 12 мин., после чего навеска цеолита
взвешивается и по разности весов определяется содержание пыли на цео-
лите. На рис. 5 приведены графики зависимости уменьшения веса испытуе-
мого образца от длительности испытания. Как видно из данных, приве-
денных на рис. 5, графики износа отчетливо разделяются на два участка.
На первом участке величина износа достаточно резко уменьшается по
14*
211
мере увеличения длительности испытания; второй — участок постоянного
износа цеолита при испытании. Такой характер износа позволяет исполь-
зовать первый период испытания (дли-
,-^-быброс тельность вибрации 12 мин.) для
Рис. 4. Схема прибора для определения
содержания пыли на формованных цео-
литах методом вибрации.
1 — вибратор; 2 — латр; 3 — реактор; 4 —
реометр.
оценки содержания пыли на поверх-
ности цеолита. Определенное методом
вибрации содержание пыли на об-
разцах цеолитов, приготовленных по
различным вариантам технологии и
в различных организациях ко-
лебалась в пределах 0.2—3.0%.
Сходимость параллельных определе-
ний— 0.1 абс. %.
Формованные цеолиты не долж-
ны содержать более чем 0.4—0.5%
пыли при определении тем или дру-
гим методом. Окончательный выбор
метода определения содержания пыли
и показателя кондиционности будут
произведены после накопления экспериментального материала на основе
оценки цеолитов промышленного производства.
Рис. 5. Дифференциальные и интегральные кривые
убыли веса цеолита при определении содержания пыли
методом вибрации.
1, 2 из —дифференциальные кривые: 1’, 2' и 3' —интеграль-
ные кривые. Содержание пыли на образцах: 1 и 1Г — 1.2%;
2 и 2’ — 1.0%; з и з' — 0.5%.
Все предлагаемые дополнения к ВТУ должны способствовать улучше-
нию эксплуатационных характеристик формованных цеолитов, выпускае-
мых нашей промышленностью.
212
ВЛИЯНИЕ ТИПА СВЯЗУЮЩЕГО
И НЕКОТОРЫХ УСЛОВИЙ ПРИГОТОВЛЕНИЯ ФОРМУЕМЫХ МАСС
НА СВОЙСТВА ГРАНУЛИРОВАННЫХ ЦЕОЛИТОВ
Г. М. Белоцерковский, Т. Г. Плаченов, К. Г. Ионе, Ю. В. Ежов,
В. Ф. Карельская, Е. Н. Долгова
Цель настоящего исследования заключалась в выяснении влияния
природы связующего и некоторых условий приготовления формуемой
массы на вторичную пористую структуру, динамическую активность и
механическую прочность полученных гранул.
При формовании гранул цеолитов были применены в качестве связую-
щего глины трех различных минералогических типов: монтмориллонито-
вого (бентониты), каолинитового и палыгорскитового. Глины этих типов
отличаются друг от друга пластичностью, набухаемостью, пептизируе-
мостью в воде и катионным составом’И—3, 6]. Для формования гранул
было также применено синтетическое связующее — 5,6 оксинитрат алю-
миния.
Приготовление формуемой массы проводили различными способами:
в бегунах, в малаксёре и в малаксёре с предварительной виброобработкой
смеси твердых компонентов в течение 1 мин. и 60 мин. Образцы, приго-
товленные одним и тем же способом из кристаллов одной и той же партии,
объединены в отдельные серии. Содержание связующего во всех образцах
составляло 15% по отношению к весу дегидратированных гранул.
Формуемая масса образцов I серии готовилась смешением компонентов
в малаксёре в течение 30 мин. Для формования применялся шнек-грану-
лятор ЛТИ.1 . .*-•
Образцы II серии получены от К. Т. Подорвана.. Глины предвари-
тельно обрабатывали на коллоидной ,мельнице, смесь ее с цеолитом три
раза пропускали через сито с отверстиями 2 мм. Массу, затворенную водой,
перемешивали ручным способом в течение часа и формовали. на шнеко-
вом грануляторе.
При приготовлении образцов III и IV серий для разрущрния ком-
ков кристаллов цеолита и получения более однородной смеси твердых
компонентов последние подвергались виброобработке 2 в течение 1 мин.
(III серия и один образец IV серии) .или 60 мин. (IV серия). Воздущ-
носухая смесь твердых компонентов переносилась в малаксёр, куда
дозировали со скоростью 100 мл/мин. минимальное количество воды, необ-
ходимое для получения пластичной хорошо формуемой массы; полученную
массу с содержанием 29—32% воды перемешивали 30 мин. и формовали на
опытном шнек-грануляторе. Термическая обработка гранул проводилась
при следующих условиях: 3 часа при 150—180° и 6 часов при 575+25° С.
В IV серию включены образцы, исходная смесь для которых готови-
лась различными способами: в бегунах и виброобработкой в течение 1 мин.
и 60 мин. В бегунах проводили смешение твердых компонентов в течение
5 мин., затворяли смесь водой, полученную массу перемешивали в течение
20 мин. и формовали ее на шнек-грануляторе.
Вторичная пористая структура гранулированных цеолитов исследова-
лась с помощью метода ртутной порометрии |4, 5] и на основании опреде-
ления кажущихся (б) и истинных (d) удельных весов. Механическая проч-
ность гранул характеризовалась средней величиной нагрузки на единицу
поперечного сечения гранулы (кг/мм2), при которой происходило ее раз-
рушение. Динамическая активность (ад, мг/см3) гранулированных цеолй-
1 Образцы были получены В. Н. Мазиным и И. В. Сучковым.
2 Вибромельница — модель № 7 М-ОСЗ. Число колебаний -- 3000 кол./мин.
Емкость — 10 дм3.
213
тов определялась в стандартной установке при удельной скорости паро-
воздушного потока 1>=1.б л/мин.см2.
Рис. 1. Распределение объема вторичных пор
в гранулированных цеолитах по эквивалент-
ным радиусам в зависимости от способа приго-
товления смеси исходных компонентов.
1 — в бегунках; 2 — виброобработкой в течение
1 мин.; з — виброобработкой в течение 60 мин.
Результаты определения вторичной пористой структуры гранулиро-
ванных цеолитов приведены в таблице, из которой видно, что суммарная
пористость образцов I, И, III
серий, полученных гранулиро-
ванием с помощью глин раз-
личного минералогического со-
става, колеблется всего в ин-
тервале 0.380—0.410 см3/см3.
Преобладающие эквивалентные
радиусы пор, составляющие 60—
70% от всего объема вторичных
пор, в образцах всех трех серий
находятся в одном и том же диа-
пазоне 6900—1500 А. Несмотря
на такую идентичность вторич-
ной пористой структуры образ-
цов I, II и III серий, они суще-
ственно отличаются между со-
бой по динамической активности
по парам воды (значения од1
и и механической прочности,
преследовалась цель выяснить
При получении IV серии образцов
возможность увеличения динамической активности и механической проч-
ности гранул за счет применения виброобработки смеси кристаллов цео-
лита и глины перед малоксажем. Из данных, приведенных в таблице, видно,
что при переходе от образца, получен-
ного в бегунах, к образцам, для ко-
торых смешение сухих компонентов
перед малоксажем производилось виб-
рообработкой в течение 1 и 60 мин.:
а) кажущийся удельный вес (б) воз-
растает с 0.926 до 1.217 см3/см3;
б) суммарный объем пор (i>6) умень-
шается с 0.461 до 0.299 см3/см3; в) пре-
обладающий эквивалентный радиус
пор сдвигается в сторону меньших
значений (рис. 1); г) механическая
прочность и динамическая активность
возрастают.
Наблюдаемое изменение вторичной
пористой структуры гранул и их
свойств при возрастании интенсив-
ности и длительности смешения твер-
дых компонентов смеси можно объяс-
нить тем, что при виброобработке
происходит более гомогенное распре-
деление связующего между кристал-
Рис. 2. Динамическая активность по
парам воды гранулированных цеоли-
тов, полученных в ГО Б ВНИИНП NaA
Ц-202-174 (7) и в ЛТИ — из смеси исход-
ных компонентов, обработанной на виб-
ромельнице в течение 1 мин. (2) и 60
мин. (3).
лами цеолита, что, по-видимому, приводит к увеличению числа контактов
кристаллов цеолита друг с другом и кристаллов с вяжущей добавкой.
Все это способствует более плотной упаковке кристаллов в гранулах,
уменьшению объема вторичных пор и их преобладающего эквивалентного
радиуса, увеличению содержания собственно кристаллов цеолита в единице
объема гранул, что и приводит к существенному увеличению динамической
активности слоя. Из таблицы и рис. 2 видно, что по динамической активно-
го
Вторичная Пористая структура, динамическая активность й механическая прочность Гранулированных цеолитов
Связующее Механическая прочность на раздавливание, кг-мм2 8, г, см’ d, Г/СМ5 W|, см’/см’ V пор, см3/см3 Объем пор (см’/см3) при значениях гэкв мг/см’ В, мин. при кон- центрации 13.5 мг/Л
дегидра- тация 2 часа при 400е гидрата- ция при = = U. 75 31-2.9 А 1500-31 А 6900—1500А >6900 А
I серия кристалл «средни й»
Глуховская глина . — 0.68 1.048 1.696 0.381 0.367 0.014 0.068 0.290 0.007 98 138
Оглан.тинекий бентонит ...... — 0.77 1.027 1.714 0.401 0.407 0.000 0.080 0.281 0.046 78 109
Крымский кил __ 1.0 1.055 1.716 0.390 0.352 0.038 0.047 0.282 0.023 54 75
Аскангель — 1.12 1.056 1.713 0.383 0.366 0.017 0.047 0.301 0.018 76 107
II серия, кристалл парти и № 15
Харьковский бентонит 2.24 1.15 1.099 1.768 0.379 0.372 0.007 0.068 0.283 0.021 71 100
Черкасский » 2.06 1.09 1.037 1.718 0.396 0.388 0.008 0.078 0.282 0.028 54 76
Аттанул ьгит 0.95 0.91 1.015 1.717 0.409 0.390 0.019 0.107 0.291 0.002 104 146
III серия— образцы ЛТИ, крист алл Л 2 21, В 1 т б р о о б р а б о тка смеси твердых компонентов
на вибром е л ь н и це в течение 1 мин.
Глуховская глина 1.30 0.70 1.052 1.718 0.388 0.368 0.020 0.082 0.279 0.007 116 163
Латненская глина 0.98 0.56 1.077 1.787 0.398 0.408 0.000 0.114 0.293 0.001 116 163
5,6 оксинитрат алюминия 1.55 0.95 1.112 1.833 0.392 0.361 0.031 0.037 0.204 0.002 125 176
IV серия — об р а а ц ы ЛТИ, крист алл I) [-202-268 , связующее — глуховская глина
Способ смешения компонентов: 0.135 81 114
в бегунах 1.1 0.66 0.926 1.717 0.461 0.444 0.017 0.074 0.205
виброобработкой в течение 1 мин. виброобработкой в течение 60 мин. 1.24 0.77 1.076 1.731 0.379 0.363 0.016 0.083 0.266 0.009 116 163
1.36 0.70 1.217 1.735 0.299 0.313 0.000 0.168 0.145 0.000 128 180
Примечание: Значение аД1 соответствует средней минимальной точке росы —72±2°.
сти по парам воды образец, полученный виброобработкой смеси кристал-
лов цеолитов и глины в течение 60 мин., превосходит другие исследованные
опытные и опытно-промышленные образцы гранулированных цеоли-
тов.
Выше было показано, что минералогический тип глины при одинаковом
способе получения гранул из одной и той же партии кристаллов мало
влияет на суммарный объем вторичных пор и их распределение по экви-
валентным радиусам, но существенно сказывается на их механической
прочности, динамической активности по парам воды и водоустойчивости.
Незначительное различие во вторичной пористой структуре гранулиро-
ванных цеолитов I, II, III серий, полученных из кристаллов одной и той же
Рис. 3. Влияние удельной скорости
паровоздушного потока на динами-
ческую активность гранулированных
цеолитов, приготовленных с помощью
различных связующих.
партии с помощью различных глин,
можно, по-видимому, объяснить прак-
тически одинаковой упаковкой (коор-
динацией) в гранулах кристаллов, а
также кристаллов и частиц связующего
друг относительно друга; этому способ-
ствует одинаковая дисперсность кри-
сталлов цеолитов, приблизительно оди-
наковая дисперсность связующего и
один и тот же способ формования и
гранулирования массы. Различная же
динамическая активность гранулирован-
ных цеолитов, имеющих практически
одинаковую вторичную пористую струк-
туру, может явиться следствием частич-
ной блокировки связующим окон, ве-
дущих в полости кристаллов. Под «бло-
кировкой» мы понимаем не «замазыва-
Свявующие: 1 — 5,6 оксинитрат алюми- НИ6» ОКОН СВЯЗуЮЩИМ, а ЭКраНирОВа-
ния: глимГ7- лЬатненс1Дя-глина™СКаЯ ние им части окон кристаллов, способ-
ствующее созданию дополнительного
диффузионного сопротивления для молекул адсорбата. Блокировка,
по-видимому, будет определяться некоторыми физико-химическими свой-
ствами глины, в частности ее термоустойчивостью и пептизируемостью;
эти свойства влияют и на механическую прочность гранул. Можно пред-
положить, что пониженная термостойкость глины и повышенная пепти-
зируемость ее в воде будут способствовать блокировке окон кристаллов
связующим (а следовательно, понижению динамической активности) и
увеличению механической прочности. Высказанные предположения на-
ходятся в соответствии со свойствами гранулированных цеолитов, полу-
ченных при применении в качестве связующих бентонитовых, коалинито-
вых глин и А12О3 — связующего, образующегося при термическом разло-
жении 5,6 оксинитрата алюминия (таблица).
Можно предполагать, что одним из свойств связующего, которое
может снизить эффект блокировки, является сквознопористость его час-
тиц. На это свойство связующего ранее не обращалось внимания. Между
тем характер пористости частиц самого вяжущего компонента может ска-
заться на кинетике адсорбции готовыми гранулами.
Для того чтобы выяснить влияние характера пористости частиц свя-
зующего на кинетику сорбции гранул, была исследована при различных
удельных скоростях паровоздушного потока динамическая активность
по парам воды гранулированных цеолитов, полученных с помощью глу-
ховской и латненской глин и связующих, заведомо отличающихся от них
большей степенью сквознопористости: А12О3 (из 5,6 оксинитрат алюминия)
и аттапульгита [6].
216
При изменении удельной скорости с 1.6 до 3 л/мин.см2 динямичестуя
активность гранул (ад мг/см3), полученных с помощью аттапульгита и
А12О3 — падает на 14—15% (рис. 3), а гранул, полученных с помощью
глуховской и латненской глин — на 30—35%. Такое различное умень-
шение динамической активности с увеличением удельной скорости паро-
воздушного потока для исследованных образцов свидетельствует о неоди-
наковой скорости адсорбции паров воды. Так как образцы несущественно
отличаются по своей вторичной структуре, то можно предположить, что
причиной различной кинетики адсорбции являются особенности пористой
структуры связующего, а именно его сквознопористость.
По-видимому, для получения гранулированных цеолитов, обладающих
более высокой степенью использования адсорбционного объема при боль-
ших удельных скоростях паровоздушного потока (3 л/мин.см2), пред-
почтение следует отдать связующему, частицы которого после термиче-
ской обработки сами обладают относительно развитой сквознопористой
структурой. А12О3 — связующее, образующееся при термической обра-
ботке 5,6 оксинитрата алюминия, и аттапульгит, цо-видимому, удовлетво-
ряют этому условию в большей степени, чем глуховская и латненская
глины.
Выводы
1. Минералогический тип глин при одинаковом способе получения
гранул из одной и той же партии кристаллов мало влияет на объем вто-
ричных пор и их распределение по эквивалентным радиусам, но суще-
ственно сказывается на механической прочности, динамической актив-
ности по парам воды и водостойкости гранул.
2. Гранулы цеолитов, полученные с помощью бентонитовых глин
(черкасской, харьковской, крымского кила и аскангеля) по сравнению
с образцами, гранулированными при применении Каолинитовых глин
(глуховской и латненской) обладают большей механической прочностью;
но уступают последним по динамической активности и водостойкости-.
3. Виброобработка воздушносухой смеси кристаллов цеолитов и
глины в течение 60 мин. приводит к получению наиболее плотного грану-
лированного цеолита, превосходящего по динамической активности по
парам воды другие исследованные опытные и опытно-промышленные
образцы.
4. Применение относительно сквознопористого связующего (атта-
пульгит, А12О3) способствует получению гранул цеолита, обладающих
большей скоростью адсорбции в условиях динамического опыта, чем об-
разцы, гранулированные с помощью каолинитовых глин.
ЛИТЕРАТУРА
1. Ф. Д. Овчаренко. Гидрофильность глин и глинистых минералов. Изд. АН
УССР, Киев, 1961.
2. П. А. Земятченский, Глины СССР. Изд. АН СССР, 1935.
3. Ф. Д. Овчаренко, И. Г. Кириченко, Д. И. Коваленко и
А. И. Растрененко. Украинские бентониты. Изд. АН УССР, Киев, 1958.
4. Т. Г. И л а ч е н о в, ЖПХ, 1955, 28, 3.
5. Т. Г. И л а ч е н о в. Ртутная порометрическая установка. Изд. ЛТИ им. Ленсо-
вета, 1961.
6. Ф. Д. Овчаренко, Е. Г. Куковский, С. И. Ничипоренко,
Н. В. Вдовенко, В. Ю. Третинник, Н. Н. Круглицкий,
А. А. П а н а с е в и ч. Коллоидная химия палыгорскита. Изд. АН УССР,
Киев, 1963.
217
ИССЛЕДОВАНИЕ ВЛИЯНИЯ ПРИРОДЫ СВЯЗУЮЩЕГО
И УСЛОВИЙ ФОРМОВАНИЯ НА СОРБЦИОННЫЕ
И МЕХАНИЧЕСКИЕ СВОЙСТВА ГРАНУЛИРОВАННЫХ ЦЕОЛИТОВ
В. Н. Мазин, И. В. Сучков]
Синтетические цеолиты, полученные путем кристаллизации из алюмо-
силикатных гелей, представляют собой порошки кристаллов различной
формы и размеров |1|. Цеолиты в виде гранул, таблеток и сфер могут быть
получены путем формования достаточно пластичной массы, состоящей из
кристаллов цеолита и какого-либо связующего материала. В качестве
последнего в настоящее время широко используются глины различных
типов. Нами изучалось влияние природы глин на механическую проч-
ность и динамическую активность по парам воды формованных цеоли-
тов NaA.
В качестве связующего использовались бентонитовые глины кил-
аскангель и огланлинский белый бентонит, а также глуховская глина
каолинитового типа. Результаты химического анализа использованных
в работе образцов глин представлены в табл. 1.
Таблица 1
Химический состав глин
Образец Содержание, %
S1O, А1,О, t Fe,O3 СаО MgO к,о Na,0
Кил 49.9 18.3 3.1 5.6 4.6 Нет. 0.3
Аскангсль 55.2 19.2 2.9 1.9 4.8 » 1.75
Огланлинский бентонит 71.6 16.2 2.1 2.3 3.6 0.4 0.7
Глуховская глина . . . 54.1 36.9 7.76 Нет. Нет. Нет. Не определяли.
Цеолит NaA применялся в виде кристаллических порошков трех раз-
личных степеней дисперсности: фракции с преобладающим размером
частиц 10, 2—3 и менее 1 мк.
Формование цеолитов с применением указанных связующих проводи-
лось на лабораторном шпек-прессе, сконструированном и изготовленном
в лаборатории Т. Г. Плаченова. Масса для грануляции готовилась путем
перемешивания порошков цеолита и глины в течение 0.5 часа в лаборатор-
ном смесителе-малаксёре с добавлением необходимого количества воды.
Количество связующего во всех опытах составляло 15%. В табл. 2 пред-
ставлены данные о влажности формуемой массы в зависимости от вида
глины и степени дисперсности порошка цеолита. Формуемость характери-
вуется величиной интервала влажностей массы, при котором грануляция
на прессе данной конструкции проходила нормально, т. е. без налипания
массы на шнек, вытекания массы через фильеры и т. д. Формуемость массы
определяется как степенью дисперсности порошка цеолита, так и приро-
дой связующего. В случае применения глин как бентонитового, так и као-
линитового типов наиболее технологичным является применение кри-
сталлов средней дисперсности.
Гранулы цеолитов, при получепии которых в качестве связующего
использована глина, становятся прочными и неразмокаемыми в воде
после их прокалки. Это происходит, очевидно, за счет необратимой де-
гидратации связующего, которая протекает при определенной температуре,
характерной для каждого типа глины.
218
Таблица 2
Влажность формовочной массы при нормальном режиме прессования
Связующее
Дисперсность порошка
цеолита, мк
Влажность, %
Кил.
Огланлинский бентонит.
Аскангель.
Глуховская глина.
10
2—3
Менее 1
10
2-3
Менее 1
10
2—3
Менее 1
10
2—3
Менее 1
28—30
22—28
27—29
28—29
23—24
26
Масса не прессуется.
23-24
25-26
Масса не прессуется.
23
Масса не прессуется.
На рис. 1 представлены термограммы примененных нами образцов
глин, полученные А. И. Батраковой. Как известно 12], удаление кри-
Рис. 1. Кривые нагревания различных глин.
А — аскангель; Б — кил; В — огланлинский бентонит;
Г — глуховская глина.
сталлизационной воды из глины, ведущее к ее практически необратимой
дегидратации, происходит при температуре, соответствующей второму
эндотермическому пику на кривой нагревания. В исследуемом случае,
219
как это видно из рис. 1, такими температурами являются для глины кил—
670°, для аскангеля — 680°, для огланлинского бентонита — 690° и для
глуховской глины — 571° С.
Таблица 3
Зависимость прочности формованных цеолитов от вида
связующего и температуры прокалки
Свявующее Температура Прокалки. °C Прочность на раздавливание, кг/гранула
500 10.2
Кил. | 600 12.1
670 13.2
500 9.3
Бентонит огланлинский. | 600 11.4
690 14.6
500 10.3
Аскангель. | 600 11.3
680 12.4
500 10.5
Глуховская глина. | 600 11.4
571 11.0
В табл. 3 приведены величины механической прочности гранул, сфор-
мованных на шнек-прессе из массы, содержащей 15% связующего, и по-
рошка цеолита средней степени дисперсности. Исследовались образцы,
прокаленные при температуре 500, 600° С и температуре, соответствую-
щей второму эндотермическому пику для каждого образца глины. Опре-
деление прочности проводилось по методике, приведенной в ВТУ на цео-
литы типа А и X [3].
Как следует из табл. 3, максимальную прочность в исследованном ин-
тервале температур имеют образцы, прокаленные при температуре, от-
вечающей необратимой Дегидратации связующего. Бентонитовые глины,
использованные в качестве связующего, обеспечивают несколько более
высокую механическую прочность формованных цеолитов по сравнению
с каолинитовой глуховской глиной.
Сорбционные свойства формованных цеолитов исследовались путем
снятия изотерм адсорбции паров воды на весах Мак-Бена при 20° С,
а также определением динамической активности по парам воды по стан-
дартной методике [3]. В табл. 4 приведены значения динамической ем-
кости образцов формованных цеолитов, содержащих 15% связующего
и прокаленных при 600° С.
Таблица 4
Динамическая активность формованных цеолитов
по парам воды
Обравец
Динамическая
активность
а_,о, мг/см3
NaA — кил............................
NaA — аскангель......................
NaA — огланлинский бентонит..........
NaA — глуховская глина...............
28.5
36.5
59
103
Равновесные величины адсорбции паров воды для всех указанных
образцов (табл. 4) практически совпадают, как это видно из рис. 2.
220
В связи с этим можно предположить, что различная динамическая ак-
тивность образцов связана с различием кинетики сорбции паров воды на
формованных цеолитах, полученных с применением различных связующих.
Таблица 5
Динамическая активность по парам воды цеолитов,
полученных методом полусухого прессования
Образец Содержа- ние свя- зующего, % Насыпной вес, г/см3 Динамическая активность а_70, мг/см3
2 0.75 120
NaA — кил? j 5 0.75 117.5
15 0.76 102
2 0.74 121
NaA — глуховская гли- 1 5 0.76 116.3
на. | 15 0.76 109
В табл. 5 представлены значения динамической активности цеолитов
сформованных методом полусухого прессования. Основная особенность
Рис. 2. Изотермы адсорбции паров воды при 20° С на
формованных цеолитах.
1 — NaA (огланлинский бентонит); 2 —Na (глуховская глина);
5 —Na (кил); 4— Na (аскангель).
этого метода состоит в том, что формование проводится путем прессования
тонких пластинок цеолита из массы, содержащей лишь 10—12% влаги.
Полученные пластинки после сушки и прокалки дробятся и рассеива-
ются на требуемые фракции.
221
Как следует из табл. 5, динамическая активность цеолитов, получен-
ных методом полусухого прессования, практически не зависит от природы
связующего.
При этом наибольшее различие динамической активности в зависимо-
сти от метода формования проявляется у образцов, полученных с исполь-
зованием высокопластичных бентонитовых глин, в то время как в случае
глуховской глины этот эффект проявляется незначительно. Вероятно,
указанное явление может быть объяснено тем, что в зависимости от содер-
жания влаги в формуемой массе в различной степени протекает набухание
бентонитов, что и отражается на степени «замазывания» окон, ведущих
в полости цеолита.
ЛИТЕРАТУРА
1. В. Я. Николина, И. Е. Неймар к, М. А. Пионтковская, Уеп.
химии, 1960, 29, 9, 1088..
2. О. П. Мчедлов-Петрбсян, веб.: Физико-химические основы керамики,
Промстройиздат, 1956, 56.
3. Временные технические условия на цеолиты А и X. М., 1962.
ПРИМЕНЕНИЕ ОСНОВНЫХ СОЛЕЙ АЛЮМИНИЯ
ДЛЯ ГРАНУЛЯЦИИ ЦЕОЛИТОВ
И ИССЛЕДОВАНИЕ СВОЙСТВ ГРАНУЛИРОВАННЫХ ОБРАЗЦОВ
К. Г. Ионе, Г. М. Белоцерковский, Т. Г. Плаченов
В настоящее время при промышленном изготовлении гранулированных
цеолитов в качестве связующего применяют глины |1, 2|. Глины как
связующее для цеолитов в силу своей физико-химической природы не-
полностью удовлетворяют требованиям, сформулированным ранее [3].
Известный интерес как связующее для цеолитов представляют ос-
новные соли алюминия. Они могут быть добавлены в кристаллы в виде
раствора или золя, способны к полимеризации [4, 51.
Первые попытки применить раствор 5,6 оксинитрата алюминия для
связывания кристаллов цеолитов в гранулы дали положительные резуль-
таты: были получены механически прочные зерна с удовлетворительной
величиной равновесной адсорбции по парам воды (31.
В настоящей работе изучалась пористая структура, динамическая
активность по парам воды, механическая прочность и термостойкость
цеолитов, гранулированных с помощью солей алюминия различной основ-
ности. Выяснялось воздействие основных солей алюминия на кристалли-
ческую структуру цеолитов.
Гранулировались кристаллы цеолитов, полученные от Б. А. Лип-
кинда (Ц-202-125 и др.), от В. Я. Николиной (№№ 12, 21, 25), от В. Н. Ма-
зина.
Основные соли алюминия состава AI(OH)2NO3, Al2(OH)aNO3,
AI3(OH)SNO3 и Al^OHJjjNOs1 готовились растворением свежеосажденной
гидроокиси алюминия 16J в стехиометрическом количестве азотной кис-
лоты.
При концентрации 150—250 г А12О3 на 1 л соли с соотношением
AI/NO3=1 и 2 представляли собой опалесцирующие растворы (золи),
соли с соотношением A1/NO3 >2 — студни.
1 Приведенные формулы не отражают полного состава молекул основных солей
алюминия, так как не включают аква-, гидрооксо-, оксо- и других групп, характер-
ных для этих соединений 14,5J.
222
Рис. 1. Распределение объема пор по эквпвалепт-
ным радиусам в гранулах, получеппых формова-
нием цеолитов с помощью оксинитратов алюминия
различной основности.
1 — AI2(OH),NO,; 2 — AI.fOHjsNO,; S — AI.lOHh.NO,.
Методика формования гранул заключалась в следующем. Воздушно-
сухие кристаллы цеолита в течение 3 мин. перетирались в бегунах. Затем
к порошку добавляли водный раствор 5,6 оксинитрата алюминия с таким
содержанием А1гО3, чтобы его вес составлял 13—14% от веса сухой массы
смеси, и с таким количест-
вом воды, которое должно
придать готовой массе
влажность 40—41 %. При
такой влажности она ста-
новится пластичной и хо-
рошо формуется.
После добавления свя-
зующего масса перемеши-
валась в бегунах еще 10—
12 мин., а затем формова-
лась в шнековом грануля-
торе.
Сырые гранулы под-
вергались термообработ-
ке. Выбранный режим тер-
мообработки основан на
данных термогравиметри-
ческого анализа основных
солей алюминия (в интер-
вале 200—600°) и гранули-
рованных с их помощью
цеолитов. За оптимальный
был принят режим, при
котором температура гра-
нул от 20 до 450° С поднималась за 8 часов с двухчасовыми выдержками
в интервалах 60—120, 150—200, 260—300, 400—450° С; затем гранулы
прокаливались 4 часа при 600+25°.
Механическая прочность готовых гранул характеризовалась сопро-
тивлением на раздавливание (кг/мм2).
Таблица 1
Сравнительная характеристика цеолитов, гранулированных с помощью солей
алюминия различной основности
Кристаллы цеолита NaA Ц-202-30 ГОБ
Связующее Механи- ческая прочность воздушно- сухих гранул, кг, мм2 Кажущая- ся плот- ность, г/см’ Суммар- ная пори стость, ”2, см’/см3 Равновес- ная вели- чина адсорбции паров воды, Яр. % при = 0.1 Динамическая актив- ность по ларам воды. L = 1Ь см, Пуд=1.0 ЛМИН.СМ’
минималь- ная точка росы, °C Сд,3 мг/см3
AMOH)r,NO3 1.24 1.210 0.339 21.4 —76 98.0
A13(U II )3NO3 0.62 1.045 0.435 21.0 —76 83.0
A14(OH)uNO3 0.40 0.958 0.479 21.0 —77 73.5
Объем вторичных пор и распределение их по эквивалентным радиусам
определялись методом ртутной порометрии |8|, динамическая активность
образцов по парам воды — в стандартной установке. Была приготовлена
223
серия образцов цеолитов, гранулированных с помощью оксинитратов
алюминия, в которых отношение A1/NO3 менялось от 1 до 4.
Установлено, что с увеличением основности связующего при прочих
неизменных условиях получения суммарный объем вторичных пор в гра-
нулах увеличивается, а механическая прочность падает (табл. 1);
Рис. 2. Динамическая активность гранулирован-
ных цеолитов но парам воды при Куд = 1.6 л/мин. см2.
1 — Линде 4А; 2 — опытно-промышленный образец Кв 1; 3 —
опытно-промышленный образец Кв 2; 4 — ЛТИ крист. Ц-202-
125 ГОБ, 14% АЦО,; 5 — ЛТИ крист. .№ 25, 14% А12О„.
при этом заметно уменьшается и динамическая активность. В то же время
основность связующего не влияет на равновесную величину адсорбции
паров воды (ар мг/г). Падение динамической активности (ат_, мг/см3)
связано с более развитым объемом вторичных пор в образцах, полученных
с помощью A13(OH)8NO3 и A14(OH)uNO3 и, следовательно, с меньшим
содержанием собственно цеолита в единице объема слоя. Наилучшими
из этой серии образцов являются цеолиты, гранулированные с помощью
5,6 оксинитрата алюминия: по механической прочности (табл. 2) и дина-
Таблица 2
Механическая прочность образцов цеолитов NaA
(на раздавливание, кг/мм2)
Механическая прочность,
кг/мм2
Наименование образца
дегидратиро-
ваны
2 часа при
400° С
гидратиро-
ваны при
Р/Р« = 0-75
Связующее — 5,6- оксинитрат алюминия
Кристалл средний, 13% А12О3. зернение 3 мм 1.15 0.79
Кристалл Ц-202-125 ГОБ, 13% А12О3, зернение 3 мм . . . 1.09 0.98
Кристалл № 25, 14% А12О3, зернение 3 мм 1.83 - 1.26
Кристалл № 25, 14% А12О3, зернение 2 мм 2.26 1.12
Опытно-промышленные образцы
NaA Ц-202-174 ГОБ.....................................
Линде 4А..............................................
0.72
0.66
0.28
0.33
224
мической активности (табл. 1 и рис. 2) они превосходят как образцы,
гранулированные солями более высокой основности, так и цеолиты опыт-
но-промышленных партий — Линде 4А и ГОБ.
Цеолиты, гранулированные с помощью бентонитовых глйн и прока-
ленные при температуре 500—550° С, разваливаются в воде частично или
полностью. Дополнительная их термообработка при 600+25° С в течение
4—6 часов повышает стойкость к жидкой воде, но заметно снижает ди-
намическую активность (табл. 3).
Таблица 3
Влияние температуры термообработки на осушающую способность
гранулированных цеолитов NaA по парам воды в динамических условиях
Условия опыта: Z = 30 см; Гуд. = 1.6 л/мин.см2
Образец Конечная температура термообработки
500° 600°
минимальная точка росы, °C мг/см3 минимальная точка росы, °C ад, мг/см3
Связующее — глины
Кристалл крупный, связующее — крымский кил Кристалл № 14, харьковский бен- —72 52 —73 53.5
тонит 15% —73 109 —73 82
Кристалл средний, 15% аскангель —73 76 —74 40
Связующее — 5,6 оксин итрат алюминия
Кристалл № 12, 13% А12О3 . . . — — —79 126
Ц-202-125 ГОБ, 13% А12О3 . . . — —78 114
Кристалл средний, 13% А12О3 — — —75 103
Гранулы, в которых кристаллы цеолитов связаны А12О3 с помощью
5,6 оксинитрата алюминия, приобретают стойкость к жидкой воде уже
после прокаливания при 450—550° С. После термообработки при 600 +25°С
они сохраняют высокую динамическую активность (табл. 3). Повышение
температуры прокаливания гранул до 660—670° С не вызывает понижения
их равновесной величины сорбции по парам воды (табл. 4).
Таблица 4
Изменение равновесной величины сорбции паров воды на гранулированных
цеолитах в зависимости от конечной температуры термообработки
Образец NaA P/Ps = 0.05 р/р« = 0.1 P/Ps = 0.5
Температура термообработки
600° 670° 600° 670° 600° 670°
Кристалл ГОБ Ц-202-125, 13% А12О3 Кристалл № 12, 13% А12О3 .... 17.8 20.1 18.4 20.3 21.8 23.6 21.8 24.1 25.5 25.6 25.3 26.1
С помощью рентгеноструктурного анализа было установлено, что
продукт термической обработки основных солей при температурах свыше
350° представляет собой у—А12О3. По-видимому, эта модификация А12О3
15 Цеолиты
225
Рис. 3. Влияние удельной скорости
паровоздушного потока на динами-
ческую активность цеолитов, формо-
ванных с помощью различных свя-
зующих.
Связующие: 1 — раствор 5,6 оксинитрата
алюминия; 2 — глуховская глина; 3 —
латненская глина.
и является цементирующим компонентом в гранулах и придает им выше-
указанные свойства — термо- и водостойкость.
Этот вяжущий компонент вводится в массу кристаллов цеолита в вы-
сокодиспер^ированном виде в составе раствора основной соли. Благодаря
этому в гранулах образуется гораздо
большее число контактов — цеолит-
связующее, чем при применении вяжу-
щего компонента в виде макрочастиц,
что, по-видимому, является одним из
условий, способствующих получению
механически прочных гранул.
Для более полной оценки адсорбента
важно изучить его динамическую актив-
ность при различных удельных скоро-
стях паровоздушного потока.
Для того чтобы сравнить влияние
А12О3-связующего и глин-связующих
на кинетические свойства полученных
гранул цеолита с помощью раствора
5,6 оксинитрата алюминия, глуховской
и латненской глин, были приготовлены
образцы гранулированных цеолитов.
Благодаря строго одинаковым условиям
приготовления они практически не
отличались по суммарному объему вто-
ричных пор и распределению их по эк-
вивалентным радиусам [9], а также по величине равновесной емкости
по парам воды.
Изучение динамической активности образцов при различных удельных
скоростях паровоздушного потока показало, что для цеолитов, гранули-
Рис. 4. Выходные кривые для гранулированных цеолитов по
С02, полученных с помощью различных связующих.
Связующие: 1 —5,6 оксинитрат алюминия (15% А12О3); 2— глуховская
глина (15%); 3 — латненская глина (15%); 4—образец ГОБ СаА
(Ц-202-117).
рованных с помощью раствора 5,6 оксинитрата алюминия, падение вели-
чины ад с увеличением нуд от 1.6 до 3.0 л/мин. см2 (рис. 3) было наимень-
226
шим. Причина, по-видимому, заключается в том, что глины частично
экранируют окна, ведущие внутрь адсорбционных полостей кристаллов
цеолита. Возможно также, что определенную роль в кинетике сорбции
гранул цеолита при поглощении слоем из потока играет и проницаемость
частиц связующего. В этом отношении у—А12О3, внесенная в цеолит в со-
ставе раствора основной соли, должна намного превосходить глины, так как
она сама по себе является высокопористым продуктом.
Более высокие кинетические свойства цеолитов, сформованных в гра-
нулы с помощью 5,6 оксинитрата алюминия, по сравнению с гранулами,
Рис. 5. Рентгенограммы исходного кристалла цеолита NaA Ц-202-125 ГОБ (7)
и растертых в порошок гранул, полученных с помощью 5, 6 оксинитрата алюминия (2)
и 5, 6 оксихлорида алюминия (3). .
в которых вяжущим компонентом являются глины, подтверждаются опы-
тами Г. А. Мусакина и Н. И. Козловой при исследовании ими динамики
сорбции СО2 на цеолитах (рис. 4).
С помощью рентгеноструктурного анализа и инфракрасной спектро-
скопии была сделана попытка установить характер воздействия кислого
связующего (каким являются основные соли алюминия) на кристаллы
цеолитов.
Для исследования с помощью рентгеноструктурного анализа и ИК
спектроскопии готовые гранулы растирались в агатовой ступке. Рент-
генограммы снимались ионизационным методом на Cu-катоде; спектры —
на КВг-подложке в области 6—12 мк, характерной для алюмосилика-
тов [7].1
Рентгенограммы порошков, полученных истиранием гранул, не от-
личаются от рентгенограмм исходных кристаллов (рис. 5). По-видимому,
кристаллическая решетка цеолита при грануляции его с помощью окси-
нитратов алюминия не претерпевает каких-либо заметных изменений.
На рентгенограмме образца, полученного с помощью 5,6 оксихлорида
алюминия, обнаружены добавочные линии (отмечены крестиком), указы-
вающие на присутствие в образце NaCl. По-видимому, этим объясняется
пониженная равновесная емкость цеолитов, гранулированных с помощью
5,6 оксихлорида алюминия: при pip, =0.05, 0.1 и 0.6 они поглощают
16, 18 и 21 % воды, в то время как цеолиты, связанные 5,6 оксинитратом
алюминия, поглощают соответственно 18, 22 и 26%.
С помощью инфракрасной спектроскопии был исследован алюмоси-
ликатный каркас кристаллов цеолитов, гранулированных с помощью
1 Спектры поглощения инфракрасных лучей были сняты А. Шевяковым.
15*
227
1200 1000 900 800 700 СМ
тНМ—А-т-Н—г1—
в 10 12 19
Рис. 6. ИК спектры образцов.
1 — воздушносухая соль 5, 6
оксинитрата алюминия; 2 — про-
дукт термической обработки соли
при 450° С; 3 — цеолит, гранули-
рованный 5, 6 оксинитратом алю-
миния (до термической обработ-
ки); 4 — то же самое после тер-
мической обработки при 450”; 5 —
исходные кристаллы цеолита ГОБ
NaA Ц-202-125.
3. Г. М. Б е л о ц е р к о в с к и й,
5,6 оксинитрата алюминия (рис. 6). Рассмотрение спектров исходных
кристаллов и гранулированного цеолита позволяет заключить следую-
щее.
Спектр поглощения гранул можно рас-
сматривать как сумму спектров исходных
кристаллов и связующего.
В спектре порошка, полученного рас-
тиранием термически обработанных гра-
нул, основной пик поглощения находится
у 1000 см-1, а в спектре исходных кристал-
лов — у 1010 см-1; подобное смещение
слишном невелико, чтобы свидетельство-
вать о некотором изменении кремнекисло-
родной сетки.
Отсутствуют полосы поглощения, ко-
торые могли бы указывать на появление
в гранулированном цеолите свободных
групп SiO2 и алюминия в шестичленной
координации и тем самым служить при-
знаком частичной аморфизации кристал-
лов [7].
Выводы
1. Гранулы цеолита, полученные при
применении в качестве связующего рас-
твора 5,6 оксинитрата алюминия, содер-
жащие 13—14% А12О3-связующего, пре-
восходят по динамической активности по
парам воды и углекислоте, механической
прочности, устойчивости к жидкой воде
опытные и опытно-промышленные образцы
гранулированных цеолитов.
2. Методами рентгеноструктурного
анализа и инфракрасной спектроскопии
не обнаружено изменений в кристалличе-
ской решетке цеолитов при их грануля-
ции растворами основных солей алюминия
с соотношением A1/NO3=2 — 4.
ЛИТЕРАТУРА
1. U S Pat., 2973327, 1961.
2. Синтетические цеолиты. Изд. АН
СССР, 1962.
К. Г. И о и е, Т. Г. П л а ченов, в сб.: Син-
тетические цеолиты, Изд. АН СССР, 1962, стр. 174.
4. Химия комплексных соединений. Под ред. Дж. Бейлара. ИЛ, М., 1960.
5. R. Р. Grata m, J. Am. Chem. Soc., 1947, 69, 816.
6. Э. А. Левицкий, Изв. ВУЗов, Цветная металлургия, 1961, 2, 71.
7. В. А. К о л е с о в а, Е. Ф. Г р о с с, ЖФХ, 1952, 26, 1637.
8. Т. Г. П л а ч е н о в. Ртутная порометрическая установка. Изд. ЛТИ
им. Ленсовета, 1961.
9. Г. М. Белоцерковский, Т. Г. Плаченов, К. Г. И о н е, Ю. В. Е ж о в,
В. Ф. Карельская, Е. Н. Долгова, настоящий сборник, стр. 213.
228
ПУТИ ПОЛУЧЕНИЯ ПОРИСТЫХ СТЕКОЛ — МОЛЕКУЛЯРНЫХ сит
С ЗАДАННЫМИ ХАРАКТЕРИСТИКАМИ НА ОСНОВЕ СТЕКОЛ
КАЛИЕВОАЛЮМОСИЛИКАТНОЙ СИСТЕМЫ
Л. С. Ястребова, А. А. Бессонов, С. С. Хвощев
В последние годы работами [1—2] было показано, что путем обра-
ботки некоторых щелочносиликатных стекол в кислотах удается полу-
чать пористые продукты с выраженными молекулярно-ситовыми свой-
ствами по отношению к простым и малым молекулам. Хотя пористые
стекла этого типа значительно уступают синтетическим цеолитам по об-
щей сорбционной емкости, однако они обладают и одним очень ценным
преимуществом — чрезвычайно высокой кислотоустойчивостыо. Послед-
нее позволяет говорить о перспективах их использования в качестве мо-
лекулярных сит для тех процессов химической и нефтехимической про-
мышленности, которые протекают в кислых средах.
Однако, прежде чем ставить вопрос о практическом использовании
пористых стекол — молекулярных сит для какого-либо реального про-
цесса, следовало более обстоятельно изучить, в каких пределах и
какими путями можно изменять их основные сорбционные характеристики:
размеры пор и предельные объемы сорбционного пространства. Нам было
известно, что для двухкомпонентных щелочносиликатных стекол в ка-
честве таких путей могут быть использованы изменения количества и
природы щелочного катиона, содержащегося в исходном стекле [1—3],
а также его термическая обработка [3—4]. Так, по мере увеличения со-
держания щелочного окисла в стекле-основе возрастают общие объемы
и размеры пор в выщелоченных (пористых) стеклах. При одинаковых
молярных составах из калиевосиликатного стекла получаются пористые
стекла, обладающие большей сорбционной емкостью и большими разме-
рами пор, чем из натриевых. И, наконец, закаленное стекло данного со-
става характеризуется большим предельным объемом сорбционного про-
странства и большим размером пор, чем отожженное. Как видно, во всех
перечисленных случаях увеличение сорбционной емкости пористых стекол,
вызываемое изменением их исходного состава или термической обработ-
кой, сопровождается увеличением размеров пор, причем возможность
варьирования последних довольно ограничена.
В поисках путей для более тонкого и разностороннего регулирования
структуры пористых стекол — молекулярных сит мы обратились к нашим
данным о влиянии небольших добавок различных окислов на пористую
структуру поверхностных пленок высококремнеземистых натриевосили-
катных стекол. Согласно этим данным [5], наибольший интерес представ-
ляет введение в щелочноеиликатное стекло окиси алюминия, небольшие
добавки которой (3—5 моль. %) приводили к заметному увеличению об-
щего объема пор пленки при одновременном повышении химической устой-
чивости стекла. Последнее обстоятельство имело большое значение и при-
менительно к рассматриваемому случаю — получению пористых стекол —
молекулярных сит, так как наиболее интересные с этой точки зрения
калиевосиликатные стекла с высоким (30—35 моль. %) содержанием
К2О отличаются крайне низкой химической стойкостью, что усложняет
работу с ними до начала их-выщелачивания. Уже первые опыты по вве-
дению в калиевосиликатные стекла такого типа небольших (1—4 моль.%)
добавок окиси алюминия [3] показали перспективность этого направле-
ния работы, ибо оказалось, что наряду с увеличением предельного объема
сорбционного пространства в выщелоченном стекле в данном случае имеет
место уменьшение размеров пор. Одновременно наблюдалось заметное
понижение гигроскопичности исходного стекла.
229
Для детального изучения влияния различных (но всегда небольших)
добавок окиси алюминия к калиевосиликатному стеклу на сорбционные
свойства получаемых после выщелачивания пористых стекол были сва-
рены две серии стекол, в которых к стеклу-основе состава SiO2 — 65,
К2О — 35 моль. % (серия I) и SiO2 — 70, К2О — 30 моль. % (серия II)
добавлялось сверх 100% от 1 до 11 вес. % А1203. Стекла варились в квар-
цевых тиглях в пламенной печи при температурах 1500—1550° С, после
чего отливались в железные формы и подвергались грубому отжигу в ка-
ленице от температуры около 600° С. Для получения закаленных образ-
цов кусочки стекла после выдерживания их в специальной печи при
~ 620° С выбрасывались в четыреххлористый углерод. Готовые стекла
Рис. 1. Зависимость сорбционных свойств пористых стекол от со-
держания окиси алюминия в стекле-основе.
дробились в порошок и выщелачивались в 2 н. соляной кислоте до момента
полного извлечения из них ионов калия, о чем судили по результатам
химического анализа выщелоченных стекол. Одновременно с анализом
на К + ионы, полученные пористые стекла анализировались на содержание
в них окиси алюминия. Сорбционное исследование пористых стекол про-
водилось в вакуумной объемной установке по парам воды, метанола и эта-
нола. Результаты, полученные для обеих серий стекол, оказались одно-
типными, вследствие чего ниже будут приведены данные только для сте-
кол серии I.
На рис. 1 представлены изотермы, снятые на образцах, приготовленных
из отожженных стекол, выщелачивавшихся при 50° С в течение несколь-
ких недель.1 Здесь и дальше в числителе дробных условных обозначений
стекол стоит моль. % К2О в стекле-основе, в знаменателе — вес. % до-
бавленной к нему окиси алюминия. Совершенно несомненно существова-
ние четкой, но довольно сложной зависимости сорбционных свойств по-
ристых стекол от количества А1203, введенной в исходное калиевосиликат-
ное стекло. Первые ее добавки вызывают увеличение общего объема пор.
1 То обстоятельство, что здесь и на следующих рисунках изотермы адсорбции
спиртов располагаются в ряде случаев над изотермами адсорбции воды обусловлено,
по-видимому, особенностями адсорбции этих веществ в порах, соизмеримых с ними по
размерам.
230
при уменьшении их размеров, однако последующие ведут не только к умень-
шению размеров пор, но и к уменьшению общей сорбционной емкости
пористого стекла. Последнее связано, по-видимому, с постепенным уплот-
нением скелета стекла, в результате чего часть пор становится недоступ-
ной даже для молекул воды. Начиная со стекла 35/7 объемы более крупных
пор, доступных для спиртов, а также предельные объемы сорбционного
пространства по воде вновь начинают возрастать. При этом различия
в размерах пор между соседними стеклами ряда не превышают нескольких
десятых долей ангстрема.
В поисках причин столь своеобразного влияния окиси алюминия на
структуру получаемых при выщелачивании пористых стекол была изу-
чена начальная стадия процесса разрушения кислотами исходных стекол
ряда. Не имея возможности в рамках настоящей статьи подробно останав-
ливаться на этой части работы, ограничимся лишь упоминанием о неко-
торых ее результатах. Было установлено, что из большинства исследован-
ных стекол весь калий удаляется (в условиях опыта) в течение первых
четырех часов, причем по мере увеличения содержания алюминия в стекле
скорость извлечения калия в самый начальный период выщелачивания
несколько затормаживается. Алюминий, при содержании А1203 до
~ 3 моль. % (стекла 35/1—35/5), практически не удаляется из стекла
в первые часы выщелачивания, однако при больших ее содержаниях
(стекла 35/6—35/11) начинает интенсивно переходить в раствор уже после
непродолжительного воздействия кислоты на стекло. По-видимому,
именно с этим связано увеличение объемов и размеров пор в последних
двух стеклах рассматриваемого ряда (рис. 1), ибо процесс извлечения
алюминия должен сопровождаться разрушением алюмокислородных тет-
раэдров стекла, за счет чего и появляются дополнительные объемы пус-
тот в выщелоченном стекле. Прямым подтверждением этому являются
данные о содержании А12О3 в исследованных пористых стеклах. Из табл. 1
видно, что с увеличением содержания окиси алюминия в исходных стек-
лах относительно все большие ее количества переходят в раствор в про-
цессе кислотной обработки стекла.
Таблица 1
Зависимость количеств окиси алюминия, переходящих
в раствор, от содержания ее в исходном стекле
Условное обозначение стекла. Содержание окиси алюминия в пористых стеклах (вес. 7о), Количество А12О3, остающейся в пори- стом стекле, в % к ее первоначаль- ному содержанию
принимая, что алюминий при выщелачивании не извлекается из стекла (рас- четные данные), А фактически
35/1 1.8 1.8 100
35/3 5.2 3.9 75
35/5 8.4 4.5 54
35/7 11.4 3.5 31
35/9 14.1 2.1 15
Для того чтобы установить, для каких составов и в какой мере можно
увеличить предельный объем сорбционного пространства пористых сте-
кол за счет извлечения из стекла алюминия, были проведены следующие
опыты. Порошки стекол от 35/1 до 35/11 выщелачивались в двунормальной
соляной кислоте при комнатной температуре в течение двух сроков:
231
пяти дней и одного месяца, после чего анализировались ца содержание
калия и алюминия и подвергались сорбционному исследованию. Резуль-
таты представлены на рис. 2. Из рисунка видно, что для стекол 35/1—35/5
увеличение продолжительности выщелачивания с пяти дней до одного
месяца не приводит к извлечению дополнительных количеств алюминия,
а вместе с тем и не меняет сорбционных свойств пористых стекол. Для
стекол 35/7—35/11, напротив, с увеличением срока выщелачивания все
большие количества алюминия переходят из стекла в раствор и вместе
с тем возрастает сорбционная емкость соответствующих пористых стекол,
а также доля в них более крупных пор, доступных для молекул метанола
см3/г
35/1
ВыщелачиВание 5ди. ВыщелачиВание 1мес.
А1г03~1.61% А1г03-1Л2%
СВ30Н CHSOH
ч.и >-1 1 ? w>h я—.—г-®-;—г~®
35 П
ВыщелачиВание 5Он. ВыщелачиВание 1мес.
А1303- 7.00 % А1г03-5.91°/о
Не0
Вг0
' ОН^ОН
f ОН.ОН
(° с^н.он i fill
35/5
Выщелачибание 58н. А1г03-Ш% ВыщелачиВание 1мес. А1г03~9.60°/о
Вг0 Н30
ен3он f сн3он -> А 4 А
35/11
ВыщелачиВание 5дн. А1г03~5.52°/о ВыщелачиВание 1мес. А1г03~2.92%
нго L сн3оц %0 1 1 1 1
СгВ5ОН , >1 ill!
О 0.2 0.4 0.6 0.8 1.0 0.2 0.4 0.6 0.8 1.0 0.2 0.4 0.6 0.8 1.0 0.2 0.9 0.6 0.8 1.0 p/ps
Рис. 2. Влияние продолжительности выщелачивания на структуру пористых стекол
в зависимости от содержания окиси алюминия в стекле-основе.
и этанола. Практическая значимость результатов этой серии опытов не-
сомненна, так как, опираясь на них, можно наметить оптимальные ре-
жимы получения пористых стекол для каждого из исследованных соста-
вов. Так, если считать, что весь калий извлекается из стекла за короткое
время, продолжительность кислотной обработки стекол первой группы
может составлять всего несколько часов. Стекла второй группы должны
выщелачиваться значительно дольше; однако следует иметь в виду, что
путем повышения температуры выщелачивания время, необходимое для
получения из них пористых стекол с заданными характеристиками,
удается и в этом случае значительно сократить.
Задачей дальнейшего исследования было выяснение влияния терми-
ческой обработки калиевоалюмосиликатных стекол на сорбционные
свойства продуктов их выщелачивания. Опыты проводились на стеклах
35/1—35/7, отожженные и закаленные образцы которых выщелачивались
в строго одинаковых условиях. Данные о содержании окиси алюминия
в полученных пористых стеклах представлены в табл. 2, а на рис. 3 при-
ведены результаты их сорбционного исследования.
Сопоставление данных рисунка и таблицы позволяет заключить, что
для стекол 35/1—35/3 увеличение сорбционной емкости и размеров пор
232
Таблица 2
Влияние термической обработки исходного стекла на содержание
окиси алюминии в пористых стеклах
Условное обозначение стекла Содержание окиси алюминия в пористых стеклах (вес. %), в % к А
принимая, что алюми- фактически в отожжен- ном образце в закаленном образце
не извлекается из стекла (расчетные данные), А в отожжен- ном образце в закаленном образце
35/1 1.8 1.42 1.50 80 83
35/3 5.2 3.12 3.53 61 67
35/5 8.4 4.60 3.37 55 40
35/7 11.4 5.91 1.14 52 10
закаленных образцов по сравнению с отожженными обусловлено только
процессом собственно закалки, которая приводит к увеличению межатом-
ных расстояний в стекле, т. е. к разрыхлению его скелета. Для стекол
см3/г
0.16
0.08 У
О
0.&-
0.16 Ь
0.08
о о.г оя о.б о.в to о.г оя о.б о.в ю 0.2 оя о.б о.о 1.0 0.2 оя 0.6 о.о top/ps
Рис. 3. Влияние термической обработки калиевоалюмосиликатных стекол на струк-
туру получаемых из них пористых стекол.
35/5—35/7 (а также для следующих стекол ряда) имеет значение еще и
второй фактор — увеличение степени извлечения из стекла алюминия,
чему несомненно способствует вызываемое закалкой разрыхление сетки
стекла.
Изложенный экспериментальный материал свидетельствует о том, что
исследованные стекла калиевоалюмосиликатной системы чрезвычайно
чувствительны к действию целого ряда факторов, влияющих на структуру
образующихся при их выщелачивании пористых продуктов. Важнейшими
233
из таких факторов являются: содержание окиси алюминия в стекле,
термическая обработка стекла и условия его выщелачивания.
Влияние содержания окиси алюминия связано как с изменением
структуры скелета стекла по мере введения алюминия, так и с возмож-
ностью извлечения последнего из стекла в ходе его обработки кислотой,
в результате чего происходит образование дополнительного объема пустот
в пористом стекле.
Термической обработкой стекла можно или уменьшить размеры и
объемы пор, или, напротив, увеличить их. Первое достигается путем тща-
тельного отжига стекла, второе — его закалкой. Влияние закалки об-
условлено увеличением межатомных расстояний в стекле, что в свою
очередь способствует увеличению степени извлечения из него алюминия.
Условия выщелачивания — его продолжительность и температура —
имеют значение в тех случаях, когда речь идет о составах, отдающих
в раствор алюминий. Они определяют степень извлечения алюминия из
стекла и вместе с тем — структуру получаемого пористого стекла.
Зная направление влияния каждого из перечисленных факторов,
можно (путем соответствующего их подбора) регулировать процесс полу-
чения пористых стекол — молекулярных сит в желаемом направлении.
В настоящее время на основе стекол калиевоалюмосиликатной системы
мы получаем пористые стекла — молекулярные сита с диаметром пор
от ~ 3 до 5—7 А при различиях в диаметрах пор между соседними стек-
лами на десятые доли ангстрема. Предельный объем сорбционного про-
странства (по воде) у разных стекол ряда составляет от 0.08 до 0.14 см3/г.
ЛИТЕРАТУРА
1. С. П. Ж данов, Е. В. Коромальди, ДАН СССР, 1961, 138, 870.
2. С. П. Жданов, Л. С. Ястребова, Е. В. Коромальди, в сб.: Син-
тетические цеолиты, Изд. АН СССР, 1962, 68.
3. Л. С. Я с т р е б о в а, в сб.: Стеклообразное состояние. 1964, 3, 4, 147.
4. Л. С. Ястребова, ЖПХ, 1963, 8, 1858.
5. Л. С. Ястребова, ЖПХ, 1961, 34, 448.
НАБОР ПОРИСТЫХ СТЕКОЛ — МОЛЕКУЛЯРНЫХ СИТ И ЕГО
ХАРАКТЕРИСТИКИ ПО АДСОРБЦИОННЫМ ДАННЫМ
Е. В. Коромальди, С. С. Хвощев, С. П. Жданов
Если расположить широко известные синтетические цеолиты по прин-
ципу возрастания размеров каналов, ведущих в полости их кристалли-
ческих решеток, то получится ряд КА—NaA—СаА—СаХ—NaX. Весь
этот ряд построен на основе цеолитов только двух типов — А и X, у ко-
торых диаметры «окон», ведущих в большие полости для Na-форм состав-
ляют соответственно ~ 4.2 и ~ 7.5 А [1, 2]. Эти размеры определяются
особенностями строения кремнеалюмокислородного каркаса цеолитов
данных типов и являются для них достаточно стабильными. Стабильность
размеров «окон» в каркасах цеолитов, с одной стороны, является благо-
приятным фактором для селективности адсорбции на цеолитах веществ,
отличающихся по размерам молекул, но, с другой стороны, эта же ста-
бильность не благоприятствует широкому модифицированию молеку-
лярных сит — пористых кристаллов по размерам входных «окон». Моди-
фицирование, осуществляемое с помощью замены одних катионов на дру-
234
Рис. 1. Селективность адсорбции на наборе пористых стекол-молекулярных сит.
Крестики — десорбция.
гие, не может привести к изменению этих размеров в достаточно широ-
ких пределах.
В приведенном ряду катионным обменом не удается получить пористые
кристаллы с размерами каналов, лежащими между их размерами у СаА
и СаХ, и для заполнения существующего в этом ряду пробела, по-види-
мому, будут нужны новые типы синтетических цеолитов, возможности
получения которых подлежат еще выяснению. Для специфических слу-
чаев разделения смесей и для других специальных целей могут потребо-
ваться молекулярные сита с меньшими диаметрами каналов, чем у КА,
и с большими, чем у NaX. Но возможности получения пористых кристал-
лов с размерами каналов, изменяющимися в широких пределах путем
прямого синтеза, ограничены особенностями строения кремнеалюмокис-
лородных каркасов в цеолитах.
В связи с этим, как мы уже отмечали ранее [3], создание набора пори-
стых стекол — молекулярных сит могло бы привести не только к попол-
нению, но и к расширению набора молекулярных сит — пористых кри-
сталлов, как в сторону меньших, так и в сторону больших размеров пор.
Целесообразность исследований в этом направлении теперь подтверждается
еще и тем, что в последнее время в работах, выполненных Г. М. Бело-
церковским, выявились реальные возможности получения пористых
стекол в виде прочных гранул. В настоящем сообщении приводятся ре-
зультаты исследования адсорбции разных веществ на созданном в нашей
лаборатории наборе пористых стекол — молекулярных сит с постепенно
возрастающими размерами пор.
Для того чтобы определить в каком отношении каждый из образцов
пористых стекол этого ряда находится к упомянутому цеолитному ряду
молекулярных сит, нужно сопоставить данные по адсорбции одних и
тех же веществ на образцах пористых стекол этого ряда и на цеолитах.
В таблице приведены данные по адсорбции паров воды, спиртов, углево-
дородов и азота на цеолитах типов КА, NaA и СаА, а на рис. 1 — изо-
термы адсорбции этих же веществ на наборе пористых стекол, располо-
женных в порядке возрастания размеров пор.
Из сравнения данных таблицы и рисунка видно, что этот набор пори-
стых стекол — молекулярных сит по размерам пор перекрывает предел
изменения размеров пор в упомянутом выше наборе молекулярных сит —
пористых кристаллов.
Равновесные величины адсорбции некоторых паров на цеолитах
Адсорбат КА NaA СаА
Линде, гранулиро- ванные К-83 Линде 1 Na-83 Линде 1 Са-152
ео Р, см’/г Р/Рв см3/г Р/Рв о ео Р, Р, § ес Р. Р см/®г Р/Рв см’/г
Н2О 0.5 0.196 0.5 0.258 1.0 0.289 0.5 0.260 1.0 0.305 0.5 0.323
СН3ОН .... 0.5 0.083 0.5 0.184 1.0 0.238 — — 1.0 0.232 — —.
С2Н&ОН . . . 0.5 0.038 0.5 0.043 1.0 0.196 0.5 0.243 — — — —
С4Н8ОН . . . — — — — — — 0.5 0.02 — — 0.5 0.316
/-С4Н10. . . . — — —. — 0.3 0.006 2 — .—. 0.3 0.005 2 — —
i-CgHjg .... 0.5 0.003
Ns — —. — — 0.9 0.0075 0.5 0.015 0.9 0.266 0.5 0.11
c6He — —• — — 1.0 0.03 — — 1.0 0.02 — —•
1 По данным Брека [1|.
2 Адсорбция в г/г.
236
кинетикой, а для молекул СаН6ОН поры
изменений состава не-
малых
Пористое стекло 1 является несомненно более тонкопористым, чем
цеолит КА, что следует из сопоставления объемов пор, доступных для ад-
сорбции молекул СНдОН у сравниваемых молекулярных сит. С другой
стороны, последние члены ряда (пористые стекла 7 и 8) являются, по-ви-
димому, более крупнопористыми, чем цеолит NaX, так как на изотермах
адсорбции воды на этих пористых стеклах уже имеется заметный гисте-
резис, отсутствующий у цеолита NaX.
Пористое стекло 2 по размерам пор близко к цеолиту КА. Метиловый
спирт, хотя и адсорбируется на этом стекле в относительно больших ко-
личествах, но с замедленной
этого образца уже совсем
недоступны (цеолит КА ад-
сорбирует при р/р„=0.5
около 0.04 см3/г С2Н5ОН).
Стекло 3 из набора по раз-
мерам пор, вероятно, близко
к цеолиту NaA, что следует
из его адсорбционных
свойств. Пористые стекла 4
и 5 близки к цеолиту СаА,
что следует из сравнения ве-
личин адсорбции этанола,
бутанола, а также изобута-
на, изобутанола и изооктана
на этих пористых стеклах и
цеолитах. Однако они адсор-
бируют несколько большие
количества бензола, но мень-
шие, чем СаА, количества бу-
танола (табл. 1 и рис. 1).
Стекла 6 и 7 по размерам
пор должны находиться в об-
ласти изменения размеров
каналов цеолитов СаХ и
NaX, что следует, например,
из данных по адсорбции 1,3,5-
триэтилбензола. Стекло 6,
так же как и цеолит СаХ,
время как стекло 7 поглощает, как и цеолит NaX, 1,3,5-триэтилбензол.
Несоответствие объемов пор, доступных для адсорбции молекул воды
и для более крупных молекул 1,3,5-триэтилбензола и изооктана на по-
ристых стеклах 7 и 8, может быть связано как с наличием в скелете стекла
некоторого количества тонких пор, через которые молекулы 1,3,5-три-
этилбензола и изооктана не могут проникнуть в полости, так и с вероят-
ным отклонением плотности упаковки молекул адсорбированных веществ
в порах от нормальной плотности жидкости, используемой в расчетах
адсорбционного объема. И в случае адсорбции на цеолите NaX 1,3,5-три-
этилбензола объем адсорбированной жидкости при допущении нормаль-
ной ее плотности в каналах цеолита составляет только около 2/3 адсорб-
ционного объема, определенного по адсорбции воды.
Исследованный нами набор пористых стекол — молекулярных сит
получен на основе щелочносиликатных стекол, за исключением стекла
8, полученного из натриевоборосиликатного стекла. Возможности ис-
пользования двухкомпонентных стекол этих систем, так же как и щелочно-
силикатных стекол с малыми добавками А12О3, для получения молеку-
лярных сит были установлены в [5]. Впервые селективность адсорбции
Рис. 2. Влияние
триевоборосиликатных стекол на адсорбцию па-
ров воды получающимися из них пористыми
стеклами.
Состав исходных стекол (моль. %): 1 — SiO2 — 60,
В2О3 —40; 2 —SiO2 —60, В2О3 — 38, Na20 — 2; 3 —
SiO2 — 60, В2О3 — 36, Na2O — 2, А12О3 — 2. Черные
значки — десорбция.
не адсорбирует 1,3,5-триэтилбензол, в то
237
на пористых стеклах, полученных из натриевоборосиликатных стекол,
была отмечена в [4].
Для управления структурой пористых стекол мы использовали вы-
явленные ранее [3—6] возможности варьирования размеров пор пори-
стых стекол, полученных из щелочносиликатных и щелочноборосиликат-
ных стекол, путем изменения состава исходных стекол, условий их тер-
мической обработки и выщелачивания. Яркой иллюстрацией влияния
малых изменений состава исходных боросиликатных стекол на структуру
получающихся из них пористых стекол являются изотермы адсорбции
паров воды (рис. 2). Изотерма 1 относится к пористому стеклу, получен-
ному из бесщелочного боросиликатного стекла, изотерма 2 — к пори-
стому стеклу, полученному из этого же исходного стекла, но с добавкой
2 моль. % Na2O вместо В2О3, а изотерма 3— к стеклу с добавкой еще 2моль. %
А12О3 также за счет В2О3. Форма изотерм свидетельствует о том, что
малые добавки Na2O к исходному бесщелочному стеклу приводят к обра-
зованию в пористом стекле неоднородных пор бутылкообразной формы,
а последующая добавка к этому стеклу 2 моль. % А12О3 приводит к полу-
чению снова однороднотонкопористого стекла. Эти изменения размеров
и формы пор в пористых стеклах, происходящие в результате малых доба-
вок Na2O и А12О3 в исходное стекло, находятся в связи с изменениями
размеров областей химической неоднородности в исходных стеклах в за-
висимости от того, находится ли Na2O в стекле в связи с В2О3 или с А12О3.
В первом случае Na2O взаимодействует с В2О3 с образованием заряжен-
ных тетраэдрических групп [BO«/J-, которые ассоциируются с незаря-
женными ВОз/,-группами, образуя обособленные боратные области
в стекле, что делает стекла химически неоднородными. В том случае,
когда в стекле присутствуют одновременно В2О3 и А12О3, Na2O преиму-
щественно связывается с последним с образованием тетраэдрических
групп [AlOi/J-, которые образуют общую сетку с 8Ю*/2-тетраэдрами,
и такое стекло становится более гомогенным, как и стекло В2О3—SiO2,
не содержащее Na2O. Структура пористых стекол, полученных из щелоч-
носиликатных стекол в значительно меныпей степени чувствительна к из-
менению состава и условий обработки исходных стекол, но все же удается
таким путем существенно изменять структуру и адсорбционные свойства
пористых стекол.
При точном соблюдении условий получения пористых стекол их струк-
тура вполне воспроизводима. Казалось бы, что при беспорядочном строе-
нии скелета гидратированного кремнезема в пористых стеклах размеры
каналов, образующихся в зтом скелете в результате извлечения из исход-
ного, также аморфного стекла отдельных его компонентов (атомов или
ионов), не могут быть сколько-нибудь постоянными, как постоянны раз-
меры «окон», ведущих в полости кристаллической решетки цеолитов.
Однако из изотерм рис. 1 видно, что адсорбция на пористых стеклах ха-
рактеризуется большой селективностью.
Результаты исследования адсорбции различных веществ на цеолитах
не всегда могут быть объяснены, исходя из представлений о полной регу-
лярности решетки цеолитов. Нельзя, например, объяснить молекулярно-
ситовым эффектом ограниченность адсорбции метилового и зтилового спир-
тов на цеолите КА, не сделав заключения о недоступности для моле-
кул СН3ОН и С2Н6ОН значительной части объема полостей в цеолите,
доступных для адсорбции молекул воды, т. е. о структурной неравно-
ценности разных частей зтих объемов или, в противном случае, о значи-
тельных различиях плотности упаковки молекул в каналах цеолитов от
их упаковки в нормальных жидкостях. Известно также, что синтетиче-
ские мордениты, получаемые разными авторами при идентичности их со-
става и близости структуры, определяемой из рентгеновских данных,
238
также могут весьма существенно различаться по их селективности при
адсорбции одних и тех же веществ. Равным образом и у других цеолитов
одного и того же типа, получаемых в разных условиях, наблюдаются
некоторые различия в их селективности.
Существенно не отличаясь по селективности адсорбции от цеолитов,
пористые стекла — молекулярные сита обладают несколько меньшей,
чем цеолиты, адсорбционной емкостью и меньшей адсорбционной способ-
ностью по парам воды при малых давлениях. Последнее связано с различ-
ной природой адсорбционных центров (ионы у цеолитов и гидроксилы
у пористых стекол), а не с различием в размерах пор пористых кристал-
лов и пористых стекол. Но в ряду пористых стекол крутизна изотермы
адсорбции воды, как это видно на рис. 1, заметно возрастает с уменьше-
нием размеров пор (в направлении от образца 8 к образцу 1). Специфиче-
ской особенностью пористых стекол, выгодно отличающей их от цеолитов,
является их абсолютная устойчивость к кислым средам. Эта их особенность
и выявившиеся возможности получения пористых стекол в виде механи-
чески прочных гранул, делают пористые стекла новыми перспективными
адсорбентами. Ближайшей задачей исследователей и практиков является
выявление конкретных возможностей практического использования этих
адсорбентов.
ЛИТЕРАТУРА
1. D. W. В г е с k, W. G. Е v е г s о I е, R. М. М i 11 о n, Т. В. R е е d, Т. L. Т h о-
m a s, J. Am. Chem. Soc., 1956, 78, 5963.
2. L. В г о u s s а г d, D. Р. S h о е m а к е г, J. Am. Chem. Soc., 1961, 82, 1041.
3. С. П. Ж д а н о в, Е. В. Корома льди, ДАН СССР, 1961, 138, 870.
4. С. П. Жданов, Е. В. Коромальди, Изв. АН СССР, ОХН, 1959, 4, 5.
5. С. П. Ж д а н о в, С. К. Д у б р о в о, Ю. А. Ш м и д т, Е. В. Ко рома л ь ди,
Описание изобретения к авторскому свидетельству № 139783. Бюлл. изобретений,
1961,14.
6. С. П. Жданов, Л. С. Ястребова, Е. В. Коромальди, в сб.: Синте-
тические цеолиты. Изд. АН СССР, 1962, 68.
ПРИМЕНЕНИЕ ЦЕОЛИТОВ
ТОНКАЯ ОЧИСТКА ГАЗОВ ОТ ДВУОКИСИ УГЛЕРОДА
С ОДНОВРЕМЕННЫМ УДАЛЕНИЕМ ПАРОВ ВОДЫ — КАК ПЕРВАЯ
ФАЗА ПРОЦЕССА РАЗДЕЛЕНИЯ И ПЕРЕРАБОТКИ ГАЗОВ
Н. С. Торочешников, А. И. Сидоров, Н. В. Вельцев
Современная химическая промышленность и другие отрасли народ-
ного хозяйства во все возрастающем объеме используют в качестве сырья
водород и углеводородсодержащие газы и атмосферный воздух. Во всех
агрегатах разделения газов удаляют вредные примеси двуокиси углерода
и пары воды. Как правило, эта операция осуществляется многоступен-
чато с применением главным образом жидких поглотителей. Для дости-
жения большей степени очистки газов от двуокиси углерода применяют
растворы щелочей, а для осушки газов — твердые поглотители, силика-
гель или активную окись алюминия. В связи с большой сложностью
применяемых методов процесса осушки и очистки газов в настоящее время
изыскиваются более рациональные методы решения указанной задачи.
В частности, в проблемной лаборатории по разделению газов МХТИ
им. Д. И. Менделеева проводятся работы по разработке процесса тонкой
очистки газов от двуокиси углерода с одновременным удалением паров
воды адсорбционным способом, с применением синтетических цеолитов.
Эти работы, помимо изучения общих закономерностей процесса адсорб-
ции на цеолитах, имеют целью получение данных для создания укрупнен-
ных опытно-промышленных установок для конкретных технологических
процессов, как например очистки и осушки воздуха высокого давления
перед низкотемпературной ректификацией, создания защитных атмосфер
и др.
Исследование процесса тонкой очистки газов от двуокиси углерода
в присутствии паров воды охватывает изучение: 1) адсорбционных
свойств цеолитов в статических условиях; 2) адсорбционных свойств
цеолитов в динамических условиях стационарного слоя, при барометри-
ческих и повышенных давлениях; 3) различных методов регенерации
цеолитов.
Исследование адсорбционных свойств искусственных цеолитов по двуокиси
углерода в статических условиях
Процесс совместного удаления паров воды и двуокиси углерода в ос-
новном определяется адсорбцией двуокиси углерода. Поэтому основной
характеристикой цеолитов для данного случая является их адсорбционная
способность по последней. На вакуум-адсорбционной установке с квар-
цевыми весами по известной методике были получены изотермы адсорбции
двуокиси углерода в широком диапазоне парциальных давлений и тем-
240
ператур на большом количестве различных партий отечественных и зару-
бежных цеолитов типов А, X, Y. В табл. 1 представлены данные по стати-
ческой активности ряда исследованных цеолитов при 20° С.
Таблица 1
Изотермы адсорбции двуокиси углерода на цеолитах
Парци- альное давление СО, мм рт. ст. Статическая активность, г/100 г
5А США СаА Ц-202-117 СаА Ц-202-20П СаА ЧССР 13Х США NaX Ц-202-216- 218 NaX ГрозНИИ NaX Ц-202-115
2 5.3 4.2 2.6 3.8 1.9
4 7.3 6.0 3.6 6.3 5.2 5.1 5.3 2.6
6 8.6 7.4 4.4 7.0 5.8 5.6 5.8 3.4
12 9.9 9.6 5.8 9.4 7.6 7.1 7.7 4.5
25 11.8 11.8 8.4 12.0 9.8 9.2 10.0 6.2
30 12.5 12.3 9.0 12.8 10.3 9.8 10.5 6.7
50 13.9 13.6 10.5 14.0 12.0 12.0 12.1 8.0
70 14.7 14.5 11.2 14.6 13.2 13.0 13.0 9.3
100 16.3 16.1 16.0 15.8 15.0 14.8 12.5
Цеолиты типа СаА (Ц-202-117) и NaX (Ц-202-216-218) производства
ГОБ ВНИИНП по своим адсорбционным свойствам аналогичны цеоли-
там фирмы Линде, а в пределе^малых парциальных давлений (до 6 мм рт. ст.)
их статическая активность по двуокиси углерода несколько ниже. Другие
партии цеолитов ГОБ ВНИИНП, как например СаА (Ц-202-20П) и NaX
(Ц-202-115), обладают худшей адсорбционной способностью во всем
диапазоне давлений и особенно в области низких концентраций двуокиси
углерода. Исследованные нами образцы цеолитов ГрозНИИ, в частности
цеолит типа NaX, как видно из табл. 1, адсорбируют двуокись углерода
несколько лучше, чем цеолит NaX Ц-202-216-218. Однако он уступает ука-
занному цеолиту по механической прочности.
Исследования динамики адсорбции двуокиси углерода и паров воды
в стационарном слое цеолитов при атмосферном давлении
Основным узлом установки, на которой проводились динамические
исследования, являлся стеклянный адсорбер с внутренним диаметром
32 мм, имеющий по высоте штуцера для отвода газа на анализ и ввода
термопар. Для нагрева адсорбента в процессе регенерации адсорбер
имеет нихромовую обмотку. В опытах применялись адсорберы высотой
600 и 1000 мм. В установке имеются специальные узлы для предваритель-
ной осушки или увлажнения газа. Анализ газа на двуокись углерода
в процессе адсорбции и регенерации проводился с помощью газоанализа-
торов типов ГЭУК-21 — на большие содержания СО2 и JRGA (инфракрас-
ный газоанализатор) — на малые содержания СО2. Влагосодержание газа
определялось с помощью прибора по точке росы.
На экспериментальной установке исследовалось влияние различных
факторов на адсорбцию двуокиси углерода из газа-носителя (концентра-
ции паров воды, двуокиси углерода, температуры, скорости газового
потока). Опыты проводились с цеолитами типа СаА и NaX отечествен-
ного и зарубежного производства со средним зернением от 1 до 3 мм.
Опыты показали-, что в динамических условиях цеолиты обладают
высокой скоростью адсорбции паров воды, значительно превосходящей
скорость адсорбции других газов и паров, особенно при скоростях газо-
вого потока свыше 1.5 л/мин. см2. Это обстоятельство позволяет прово-
16 Цеолиты
241
дить на цеолитах процесс одновременной осушки и очистки газов, в том
числе газов, содержащих значительное количество двуокиси углерода.
Это было подтверждено при исследовании процесса получения защитной
атмосферы [1]. В указанном исследовании было показано, что вместе
с очисткой газа от двуокиси углерода происходила его осушка, в частно-
сти остаточная влажность при адсорбции на отечественном цеолите NaA
(52 ГОБ) составляла —50° С по точке росы, а на цеолите 4А США —63° С
(при начальном содержа-
а,г/100г
нии паров воды порядка
9 г/м3 и концентрации
двуокиси углерода 10.8%).
В опытах по получению
защитных атмосфер на
цеолитах типа СаА были
получены результаты по
осушке газа (по точке росы)
на цеолите СаА Ц-202-
117 — 61—62° С, а на цео-
лите 5А США —64° С.
Изменение влагосодержа-
ния в исходном газе в пре-
делах 0.4—4 г/м3 практи-
, . , , , , , , , , , чески не отражалось на
0 г 4 6 8 10 °1о С0г адсорбции СО2, априуве-
Рис. 1. Зависимость динамической активности син-
тетических цеолитов от концентрации двуокиси
углерода в газовой смеси.
1 — СаА Ц-202-117, 5А США; 2 — NaX Ц-202-216, 13Х
США; 3 — 5А США; 4 — NaX Ц-202-216-218; 5 — СаА
Ц-202-117.
личении влажности до
25 г/м3 адсорбция СО2
уменьшилась на 12—15%.
В защитной атмосфере со-
держание СО2 не превы-
шало 5 см3/м3.
При небольших кон-
центрациях двуокиси угле-
рода, если процесс очистки проводится под атмосферным давлением,
влияние исходной влажности газа оказывается в большей степени.
В табл. 2 представлены данные по одновременному поглощению паров
воды на цеолите NaX и двуокиси углерода при содержании в газе по-
следней ~0.5 %.
Из табл. 2 видно, что увеличение влажности газа от 0.12 до 11.20 г/м3
вызывает падение динамической активности цеолита по двуокиси углерода
более чем на 30%. Следует отметить, что влияние паров воды на адсорб-
цию СО2 начинает заметно сказываться при влажности газа свыше 1.0 г/м3.
При наличии влаги свыше 1.0 г/м3 целесообразно проводить предваритель-
ную осушку газа.
Таблица 2
Динамическая активность цеолита NaX по углекислому газу
в присутствии паров воды
Влагосодер- жание газа. г/м3 Адсорбировано СО8, г вСО2, г/100 г Адсорбировано Н2О, г Адсорбировано СО2 + Н2О, г
0.12 8.75 3.65 0.116 . 8.86
0.85 8.55 3.57 0.76 9.31
4.80 6.50 2.70 3.12 9.62
11.20 5.90 2.47 6.70 12.60
242
В процессе поглощения двуокиси углерода из газовой смеси проис-
ходит заметный разогрев цеолитов. При концентрации СО2 в газе ~ 10%
температура слоя повышается на 40—50° С. Если процесс идет в адиаба-
тических условиях, что чаще всего имеет место на практике, то в резуль-
тате разогрева адсорбента его адсорбционная способность сильно пони-
жается. На рис. 1 показана зависимость динамической активности цео-
литов по двуокиси углерода от ее концентрации в поступающем газе при
высоте слоя адсорбента 1000 мм и скорости газового потока 0.6 л/мин.см2.
Верхние кривые представляют собой изотермы адсорбции двуокиси угле-
рода цеолитов СаА и NaX при 20° С.
Как видно из рисунка, динамическая
активность цеолита 5А США при
данных условиях составляет около
а^, г/100г
Рис. 2. Влияние температуры
термостатирующей жидкости на
динамическую активность цеоли-
тов по двуокиси углерода.
Т— 5А США 10% СО2, w =
0.6 л/мин.см8; 2 — СаА Ц-202-117
10% СО2, г^=0.6 л/мин.см2; 3— NaX
Ц-202-216 0.5% СО£, гс==1.3 л/мин.см2.
Рис. 3. Зависимость динами-
ческой активности цеолитов
по двуокиси углерода от ско-
рости газового потока.
1 — 5А США, с отводом части теп-
ла адсорбции; 2 — 5А США, адиа-
батические; 3 — СаА, Ц-202-117,
ади абатические.
8.2 г/100 г, а отечественного цеолита СаА Ц-202-117 — примерно на 15%
ниже, хотя его адсорбционная способность в статических условиях та-
кая же, как и у американского образца. При проведении опытов с цйрку-
ляцией термостатирующей жидкости с температурой 20° С разогрев ад-
сорбента был в среднем на 15° ниже, чем в адиабатических условиях,
а динамическая активность увеличилась почти на 20%. На рис. 2 пока-
зано влияние температуры термостатирующей жидкости на динамическую
активность цеолитов по двуокиси углерода.
В динамических опытах важное значение имеет изучение характера
влияния скорости газового потока на процесс адсорбции. Обычно по ха-
рактеру этого влияния судят о кинетике процесса и тех факторах, кото-
рые ее определяют (внешняя или внутренняя диффузия). Непременным
условием в данных опытах является изотермичность процесса. В опытах
с большими концентрациями двуокиси углерода это условие не соблюда-
лось, так как не удавалось при имеющейся конструкции адсорбера быстро
отводить тепло адсорбции. На рис. 3 показано влияние скорости газового
потока на динамическую активность цеолитов СаА по двуокиси углерода
при ее концентрации в газе — 10%. Приведенные данные позволяют
судить о влиянии скорости потока и определить оптимальную скорость-
16*
243
Рис. 4. Зависимость высоты рабо-
тающего слоя (зоны массопере-
дачи) цеолитов по двуокиси угле-
рода от скорости газового потока.
w,ji/muh смг
газового потока для данных условий адсорбции двуокиси углерода. Влия-
ние скорости газа в большей степени сказывалось при адсорбции двуокиси
углерода на отечественном цеолите СаА, чем на цеолите 5А США. Это
явление, как и меньшая величина динамической активности при равных
условиях процесса, указывает на замедленную скорость адсорбции дву-
окиси углерода цеолитом СаА Ц-202-117. Аналогичные опыты были про-
ведены с концентрациями двуокиси углерода 0.5 и 0.03%. Полная изо-
термичность процесса достигалась при адсорбции двуокиси углерода из
атмосферного воздуха. По выходным кри-
вым с помощью уравнения
L (**р тпр)
£° = — (1 — /) Bp — -Sip)
(L — общая высота слоя, тп — время до
наступления равновесия, тлр — время до
проскока, / — доля адсорбента, не насы-
щенная адсорбатом к моменту проскока)
определялись высоты работающего слоя—
зоны массопередачи [2]. На рис. 4 пока-
зана зависимость высоты работающего
слоя Lo от скорости газового потока для
цеолитов типа СаА и NaX. Характер кри-
вых позволяет предположить различие
в адсорбции двуокиси углерода на этих
цеолитах. В отношении цеолита NaX для
всех исследованных скоростей потока,
вероятно, большее значение имеет внеш-
СаА при скоростях выше 1 л/мин. см2 —
няя диффузия, а для цеолита
внутренняя диффузия. Наши наблюдения носят предварительный харак-
тер и в дальнейшем будут уточнены в специальных опытах для выясне-
ния характера зависимости адсорбции от скорости потока.
Следует отметить, что различные партии отечественных цеолитов од-
ного и того же типа при равных условиях процесса адсорбции в динами-
ческих опытах показывали значительные расхождения в скорости погло-
щения. Наряду с советскими и американскими цеолитами были проведены
также предварительные опыты с цеолитами чехословацкого производства
типов СаА (Calsit-5) и NaX (Nalsit-13), гранулированных в виде шариков.
Эти цеолиты в статических условиях обладают хорошими адсорбционными
свойствами по двуокиси углерода. В динамических опытах они показали
худшие результаты, чем советские цеолиты. Можно предположить, что
причина этого — условия грануляции цеолитов.
Динамика адсорбции двуокиси углерода при высоких давлениях
В большинстве случаев очистку от двуокиси углерода и других при-
месей, а также осушку газов проводят при повышенном давлении. Так,
например, на некоторых установках по разделению воздуха рабочее дав-
ление последнего — 200 атм. Процесс осушки проводится обычно при
этом давлении с помощью силикагеля или активной окиси алюминия,
а очистка от двуокиси углерода — химическим методом (растворами щело-
чей) при давлении 6—10 атм. В последнее время был поставлен ряд ис-
следований [3—5] с целью замены щелочной очистки на адсорбционную
с применением таких поглотителей, как силикагель, окись алюминия и
активированные угли. При этом выяснилось, что названные адсорбенты
достаточно хорошо поглощают двуокись углерода только при низких
244
температурах. Это обстоятельство создает большие трудности при осуще-
ствлении процесса в заводских условиях. В связи с этим в МХТИ
им. Д. И. Менделеева был изучен процесс адсорбции двуокиси углерода
из воздуха с помощью цеолитов при плюсовых температурах. При адсорб-
ции двуокиси углерода из воздуха
при атмосферном давлении и положи-
тельных температурах (20—25° С) на
цеолитах типов СаА и NaX (высота
слоя 600 мм и скорость газа
0.6 л/мин. см2) динамическая актив-
ность составила 0.6—0.7 г/100 г.
С повышением давления воздуха ад-
сорбционная способность цеолитов
по двуокиси углерода несколько уве-
личивалась. Опыты по адсорбции
двуокиси углерода (в присутствии
паров воды) при высоких давлениях
с помощью цеолитов показали, что
имеет место то же явление, что и при
атмосферном давлении — большая
скорость адсорбции паров воды по
сравнению со скоростью адсорбции
двуокиси углерода. В связи с этим
обстоятельством адсорбция паров
Рис. 5. Зависимость динамической ак-
тивности цеолита СаА от давления при
постоянной линейной скорости сжатого
воздуха.
воды происходит в основном в первых
по ходу газа слоях адсорбента. Коли-
чество адсорбированной влаги определяется временем адсорбции двуокиси
углерода. Опыты показывают, что при давлении — 200 ати и темпера-
туре +20° С количество цеолитов, сорбирующих только влагу, состав-
ляет — 5—7%.
Адсорбция двуокиси углерода с повышением давления (от атмосфер-
ного) сначала увеличивается, а затем падает. На рис. 5 показано уменьше-
Рис. 6. Зависимость динамической активности
цеолита СаА от давления при постоянном ко-
личестве очищаемого воздуха в единицу вре-
мени (считая на расширенный). Высота слоя
адсорбента — 920 мм.
ние динамической активности
СаА по адсорбции двуокиси
углерода из воздуха при повы-
шенных давлениях свыше 50 атм.
для двух линейных скоростей.
Подобное явление наблюдалось
при адсорбции двуокиси угле-
рода из воздуха высокого дав-
ления на силикагеле и активи-
рованном угле при низких тем-
пературах [4, 5]. На рис. 6 по-
казано влияние повышения дав-
ления на динамическую актив-
ность цеолитов по двуокиси
углерода при двух постоянных
количествах пропускаемого че-
рез адсорбер сжатого воздуха
(в пересчете на дросселирован-
ный воздух). Из данных рис. 6
видно, что для цеолита СаА в районе 100 атм. имеется максимум динами-
ческой активности. Для цеолита NaA этот максимум при тех же условиях
скоростей имеет место при несколько большем давлении.
Понижение степени адсорбции двуокиси углерода с повышением
давления в определенных пределах может объясняться влиянием адсорб-
245
60 80 100 120 /40 t,MUH
Рис. 7. Выходные кривые адсорбции двуокиси
углерода из воздуха высокого давления.
р=200 атм.; и—25 нл/мин.см2.
худшие адсороционные свойства по дву-
Рис. 8. Зависимость высоты работающего
слоя цеолитов по двуокиси углерода от ско-
рости газового потока при адсорбции из воз-
духа высокого давления.
р=200 кг/см2; 4=18° С; высота слоя 93 см.
ции других компонентов воздуха (главным образом азота). Это наблюде-
ние совпадает с данными некоторых исследователей [6], считающих, что
количество адсорбированного азота при давлении воздуха в 200 атм.
составляет — 0.5 вес. %. Хотя эта величина и невелика, однако очевидно,
что цеолиты очень быстро насы-
щаются азотом и адсорбция
двуокиси углерода происходит
в условиях предадсорбции азо-
та. К этому следует добавить,
что с повышением давления
ухудшаются и внешнедиффу-
зионные условия — затрудня-
ется подвод молекул двуокиси
углерода к поверхности зерен
цеолита.
На рис. 7 показаны выход-
ные кривые адсорбции двуокиси
углерода из воздуха при давле-
нии — 200 атм. и скорости газо-
вого потока 25 л/мин. см2. Они
показывают, что скорость ад-
сорбции двуокиси углерода на цеолите NaX выше, чем на цеолите СаА.
На рис. 7 видно, что время защитного действия цеолита NaX несколько
меньше. Однако, как указывалось выше, образец цеолита NaX Ц-202-115,
использованный в опытах, имел
окиси углерода по сравнению
с американским цеолитом 13Х.
При высоких давлениях из
- конструктивных соображений
более выгодно выбрать такую
скорость воздуха, которая обе-
спечивала бы высокую произво-
дительность процесса при наи-
меньшем диаметре адсорбера.
Продолжительность адсорбции
и степень использования адсор-
бента будут определяться при
этом высотой слоя. Гидравличе-
ское сопротивление слоя при
высоких давлениях не имеет
существенного значения. Как
показали наши расчеты, для
установок производительностью
3000 нм3/час воздуха ско-
рость должна быть не менее
25—30 нл/мин. см2
.030 м/сек. на полное
сечение адсорбера при рабочих
условиях).
На рис. 8 показана зависимость высоты работающего слоя (зоны массо-
передачи) от скорости воздуха. Данные рис. 8 показывают, что в диапазоне
скоростей 25—30 нл/мин. см2 высота работающего слоя цеолита NaX
в два с лишним раза меньше, чем цеолита СаА. При дальнейшем повыше-
нии скорости потока газа высота работающего слоя цеолита СаА резко
возрастает. Учитывая свойства цеолита NaX при адсорбции двуокиси
углерода и, как будет показано ниже, лучшие свойства при регенерации,
порядка
(0.025-0
246
можно считать, что этот цеолит при условии повышения адсорбционной
характеристики будет наиболее пригодным для применения в процессе
адсорбционной очистки воздуха высокого давления от двуокиси углерода.
При адсорбционной осушке и очистке воздуха на синтетических цео-
литах наблюдается стабильная во времени и высокая степень осушки и
очистки, не достигаемая при существующих методах осушки и очистки
газов под давлением. •
Остаточное содержание двуокиси углерода не превышает 2—3 см3/нм3,
а влажность осушенного воздуха — ниже —76° С по точке росы (менее
1 ppm). При этом происходит удаление из воздуха примесей углеводоро-
дов (ацетилена, паров масла и продуктов его пиролиза, имеющего место
в ряде случаев).
Лабораторные исследования и предварительные опыты в заводских
условиях позволили получить материал для выдачи рекомендаций пред-
приятиям и для создания опытно-промышленных установок адсорбцион-
ной осушки и очистки воздуха высокого давления. На этих установках
будут отработаны оптимальные варианты осуществления процесса тонкой
осушки и очистки сжатых газов, в частности воздуха.
Регенерация цеолитов по шарам воды и двуокиси углерода
Если в процессе одновременного поглощения паров воды и двуокиси
углерода в целом процесс определяется адсорбцией углекислого газа,
то при регенерации цеолитов приходится ориентироваться на десорбцию
влаги, хотя она и адсорбиро-
вана в малом количестве. К со-
жалению, зто в значительной
мере усложняет процесс регене-
рации, требуя большего расхода
тепла. Сама по себе десорбция
двуокиси углерода из цеолитов
протекает довольно легко, осо-
бенно на цеолитах типа NaX.
На рис. 9 показана десорбция
двуокиси углерода из зерен цео-
литов СаА и NaX при различ-
ных температурах и вакуумиро-
вании (р=10-2 мм рт. ст.). Из
рис. 9 видно, что при темпера-
туре 0° С за 15 мин. из цеолита
NaX десорбируется 88% от об-
щего количества адсорбирован-
ной двуокиси углерода, а из
цеолита СаА — 72%. Скорость
десорбции двуокиси углерода
из цеолита NaX выше, чем из
Рис. 9. Кинетика десорбции двуокиси угле-
рода из зерен цеолитов типа СаА и NaX.
1 — СаА Ц-202-100; 2 —NaX Ц 202-216; 3 —СаА
Ц-202-100; 4 — NaX Ц 202-216.
цеолита СаА. С увеличением
температуры скорость десорб-
ции возрастает. Так, при 53° С
за указанное время из цеолита
NaX десорбируется 97% СО2.
Исследования десорбции двуокиси углерода из слоя цеолитов с по-
мощью вакуумирования показали, что при начальной температуре 20° С
десорбция происходит медленно как на цеолитах СаА, так и NaX. В про-
цессе десорбции температура слоя понижается, что замедляет скорость
десорбции.
247
На практике регенерация адсорбентов обычно осуществляется про-
дувкой каким-либо инертным газом, нагретым до определенной темпера-
туры. Остаточное содержание десорбируемого газа в адсорбенте опре-
деляется температурой и концентрацией вещества в отдувочном газе.
Если имеется сухой отдувочный газ, не содержащий адсорбированного
компонента, то десорбция многих газов и паров из цеолитов может быть
осуществлена в изотермических условиях, т. е. при той же температуре,
при которой происходила адсорбция. Однако скорость десорбции будет
довольно медленной, особенно в последней стадии. Нами проведены опыты
по десорбции двуокиси углерода с продувкой слоя цеолитов NaX возду-
хом, осушенным и очищенным от двуокиси углерода с помощью цеолитов.
Предварительно исследуемый цеолит после тщательной регенерации
при высокой температуре (300—350°) и вакуумировании насыщался дву-
окисью углерода при концентрации 0.03% и температуре 20° С до равно-
весия (W=0.7 л/мин. см2). Затем при той же температуре и скорости по-
давался продувочный газ противотоком процессу адсорбции. Время от-
дувки принималось равным времени адсорбции. Опыты показали, что
при данном процессе регенерации десорбируется более 50% от адсорби-
рованного ранее количества двуокиси углерода, причем лобовой слой
цеолита (при адсорбции этот слой является последним) регенерируется
практически полностью. Установлено также, что время практически
полной десорбции двуокиси углерода (95%) почти в 10 раз больше,
чем время защитного действия слоя при поглощении (для данных
условий).
Повышение температуры слоя при регенерации увеличивает скорость
десорбции двуокиси углерода как в случае вакуумирования, так и в слу-
чае применения отдувочного газа. На основании анализа опытных дан-
ных нами установлено, что при вакуум-термической регенерации для цео-
литов типа NaX оптимальной температурой, обеспечивающей полную
десорбцию двуокиси углерода при достаточной скорости ее (при времени,
не превосходящем цикла адсорбции и охлаждения), является температура
65—80° С. Для цеолитов типа СаА эта температура несколько выше и
составляет 100—120° С. Такой же порядок температур регенерации
остается и в случае применения сухого и свободного от двуокиси углерода
продувочного газа. Здесь могут, применяться два способа нагрева адсор-
бента. Первый, когда подвод тепла осуществляется через стенки адсор-
бера, на практике применяется редко, так как в промышленных условиях
диаметр адсорбера довольно большой, а цеолиты обладают низкой тепло-
проводностью; второй, когда подвод тепла осуществляется с помощью
продувочного газа, нагретого до определенной температуры, применяется
чаще. В этом случае скорость десорбции будет определяться скоростью
подвода тепла, т. е. в конечном счете зависеть от количества продувочного
газа и его температуры. В промышленных условиях довольно редко
имеется в достаточном количестве сухой (точка росы порядка —60° С)
и не содержащий двуокиси углерода газ. Наиболее часто для целей реге-
нерации используют природный газ или атмосферный воздух. Влажность
воздуха меняется в более широких пределах, чем у природного газа. В этом
случае для удаления двуокиси углерода из цеолитов типа NaX темпера-
ТУРУ в период десорбции следует иметь 100—120° С, а для цеолита СаА —
150—180° С. Однако, поскольку требуется удаление не только двуокиси
углерода, но и паров воды, температура регенерации должна быть выше.
В процессе получения защитных атмосфер, если влажность отдувочного
газа составляет 5—7 г/м3, нагрев цеолита СаА должен быть проведен до
230—250° С. При этом остаточное содержание паров воды в цеолите будет
составлять 1.0 4- 1.5 вес. %, что практически не отразится на дальнейшем
процессе.
248
При адсорбционной осушке и очистке воздуха высокого давления в про-
цессе его низкотемпературного разделения в достаточном количестве
имеется сухой и свободный от двуокиси углерода (содержание СО2 3—
’5 см3/нм3) отходящий азот. Десорбция двуокиси углерода из цеолита
NaX при использовании этого азота в качестве отдувочного газа будет
происходить достаточно быстро и нацело при температуре порядка 80° С.
Однако, учитывая, что необходимо десорбировать также поглощенную
влагу, температуру следует поднять до 150—180° С. Энергетические за-
траты на регенерацию цеолитов в данном процессе могут быть снижены.
Считая, что почти вся влага адсорбируется в первых по ходу газа слоях
адсорбента, некоторое количество его (примерно 10%) следует поместить
в отдельный адсорбер примерно такого же сечения, но соответственно
меньшей высоты. Тогда регенерация цеолитов в этом адсорбере может
осуществляться при высоких температурах (порядка 200° С), а регенера-
ция основной массы — при температуре — 80° С.
Выводы
1. Исследование процесса адсорбции двуокиси углерода в присутствии
паров воды на цеолитах показало целесообразность его осуществления
в промышленных условиях для осушки и очистки газов перед низкотем-
пературным разделением (воздуха, углеводородных газов) и для получе-
ния защитных атмосфер.
2. В работе получены данные, характеризующие динамику процесса
адсорбции двуокиси углерода в присутствии паров воды в зависимости
от различных факторов, и определены зоны массопередачи для указан-
ного процесса.
3. На основании полученных экспериментальных данных разрабо-
таны рекомендации для создания опытно-промышленных установок для
тонкой осушки и очистки сжатых газов, в частности воздуха.
ЛИТЕРАТУРА
1. С. Е. Б а р к, Н. В. К е л ь ц е в, И. П. О г л о бин а и др., в сб.: Синтети-
ческие цеолиты. Изд. АН СССР, 1962, 276.
2. Р. Е. Т г е у b а 1. Mass Transfer Operation. N. Y., 1955, 503.
3. E. К a rw a t, Chem. Engng. Progress, 1959, 55, 5, 79.
4. И. П. Ишкин, H. Ф. Катина, ЖПХ, 1960, 12, 2623.
5. Я. Д. Зельвенский, Е. Н. Харьковская, Хим. пром., 1960, 4.
6. N. Kase, Chem. Ind. (Japan), 1959, 10, 6, 520; 8, 729.
ГЛУБОКАЯ ОСУШКА ГАЗОВ В СТАЦИОНАРНОМ ДВИЖУЩЕМСЯ
И ПСЕВДООЖИЖЕННОМ СЛОЯХ ЦЕОЛИТА
Т. Г. Плаченов, А. Н, Ширяев
Глубокая осушка газов является в настоящее время необходимой и
важной стадией ряда технологических .процессов при получении газов
высокой степени чистоты, переработке и транспортировке природного и
промышленного газов.
В течение длительного времени в промышленности для осушки газов
пользовались гигроскопическими жидкостями. Одной из первых жидко-
стей, примененных для осушки городского газа, был глицерин. Для осушки
природного газа в начале 30-х годов был применен раствор хлористого
кальция, а с 1936 г. — диэтиленгликоль.
249
В 1957 г. в США и Канаде осушка природных газов этиленгликолем
проводилась не менее чем на 5000 установках [1, 2]. За последние
25 лет для удаления паров воды из газов широкое распространение полу-
чил адсорбционный метод. В 1957 г. в США для осушки газов было ис-
пользовано около 9000 т адсорбента [3]. В качестве адсорбентов для целей
осушки газов нашли применение силикагель, активная окись алюминия и
активированный боксит [4—15].
В настоящее время каждая индустриальная страна производит ад-
сорбенты-осушители и применяет их для осушки газов.
За последнее десятилетие во многих странах мира разработана про-
мышленная технология получения новых алюмосиликатных адсорбен-
тов — осушителей синтетических цеолитов.
Синтетические цеолиты имеют значительные преимущества перед дру-
гими осушителями как по равновесной адсорбционной емкости при малых
концентрациях паров воды, так и по динамической активности во всем
J диапазоне величин начальной влажности газового потока.
В адиабатических условиях сорбции паров воды динамическая емкость
цеолита NaA более чем в два раза превышает емкость силикагеля.
В отличие от силикагеля и активной окиси алюминия синтетические
цеолиты NaA и NaX обладают высокой динамической адсорбционной
емкостью по парам воды при больших скоростях газового потока и пре-
восходят их по глубине осушки.
Высокое сродство цеолитов к воде определяет преимущественную
сорбцию паров воды из влажного газа, содержащего другие примеси,
даже в случае цеолитов, структура которых обеспечивает проникновение
Голекул этих примесей в объемы полостей кристаллов.
При осушке многокомпонентного газа в первых участках слоя цеолита
всегда адсорбируются водяные пары. По мере насыщения цеолита зона
адсорбции паров воды перемещается и вытесняет при этом другие адсор-
бированные вещества. Степень осушки газов цеолитами зависит от глу-
бины их регенерации и температуры работающего слоя цеолита. Для
'^количественной характеристики процесса глубокой осушки газов и уста-
новления параметров адсорбента, обеспечивающих одновременно осушку
и очистку их от примесей, в настоящее время требуются эксперименталь-
ные данные, характеризующие динамическую адсорбционную активность
цеолитов по парам воды и ее изменение в зависимости от условий экс-
плуатации адсорбционных установок.
С целью выяснения наиболее эффективных условий применения син-
тетических цеолитов для глубокой осушки газов нами была изучена
осушка воздуха в стационарном, движущемся и псевдоожиженном слоях
цеолита NaA отечественного производства.
Изучение процесса адсорбции водяных паров из потока воздуха про-
водилось на установке, схема которой приведена на рис. 1.
При изучении процесса осушки воздуха в стационарном и движущемся
слоях центральные переточные трубки в адсорбционной колонне 1 уда-
лялись, а распределительные поддерживающие решетки заменялись
кольцами.
Для регенерации цеолита и подготовки адсорбционной колонны к опыту
часть воздуха от очистительной системы 3 направлялась на блок осушки 9.
Регенерация цеолита проводилась в аппарате цилиндрической
формы с загрузочной емкостью 3.5 кг. Диаметр аппарата — 150 мм. Тем-
пература в регенераторе поднималась до 400° С в течение одного часа,
фиксировалась хромель-копелевой термопарой и поддерживалась с по-
мощью терморегулятора марки ЭРМ-47. В процессе регенерации пода-
вался сухой, подогретый до 350—400° С воздух со скоростью
0.15 л/мин. см2.
250
Перед началом проведения опытов система продувалась воздухом
с точкой росы 60 ~т- 70° С. После просушки системы ток сухого воздуха
не прекращался и в колонну 1 загружался адсорбент из регенератора 18.
Перед началом опытов в непрерывном псевдоожиженном и движу-
щемся слоях устанавливалась заданная скорость движения адсорбента
с помощью автоматического дозатора 21. Началом проведения опыта
считался момент включения автоматического дозатора адсорбента и
пуск влажного воздуха в адсорбционную колонну.
В процессе проведения опыта через каждые 15 мин. прибором по точке
росы 12 определялась проскоковая концентрация паров воды на одной из
Рис. 1. Схема установки.
1 — адсорбционная колонна; 2 — компрессор; з — очистительная система; i и 6 —
диафрагменный ротаметр; S — увлажнительная емкость; 7 — смеситель; 8 — психро-
метр мокрый; 9 — блок осушки; 10 — кран-переключатель; 11 и 16 — ротаметр;
12— прибор для определения точки росы; 13—термопары; 14—переключатель;
15—потенциометр; 17 — калорифер; 18 — регенератор; 19—ватный фильтр;
20 — образцовый манометр; 21 —• автоматический дозатор; [22 — емкость для отра-
ботанного цеолита.
полок. Одновременно с помощью термопар 13 и потенциометра 15 заме-
рялась температура слоя в адсорбционной колонне; психрометром 8 и
прибором по определению точки росы контролировалась влажность паро-
воздушного потока, подаваемого в адсорбционную колонну.
В процессе проведения эксперимента образцовым манометром 20
фиксировалось сопротивление адсорбционной колонны.
При непрерывном псевдоожиженном и движущемся слоях адсорб-
ционная динамическая емкость осушителей определялась в процессе
опыта с помощью .торзионных весов путем десорбции при температуре
400° С.
Первая серия опытов по изучению процесса осушки воздуха цеолитов
NaA показала, что при многоцикловой работе имеет место значительное
падение равновесной адсорбционной емкости цеолита по парам воды.
На 28-м цикле (табл. 1) наблюдалось снижение этой величины с 23.5 до 9%.
Для выяснения причин такого снижения адсорбционной емкости цео-
литов нами было проведено по тридцать циклов регенерация—насыщение
на торзионных весах при температуре регенерации 400 и 650° для цео-
лита NaA.
Величина равновесной статической емкости изменялась за 30 циклов
при температуре регенерации 400° С на 9 % и при температуре 650° С
на 12% от начальной адсорбционной равновесной емкости. Несовпадение
251
Таблица 1
Равновесная адсорбционная емкость, кажущаяся и истинная
плотности цеолитов
Наименование образца 8, г/см3 d, г/см3 Число циклов Равновесная статическая емкость при P/Ps = 0.75, %
NaA-173-0.5 1.18 1.82 23.5
1.42 2.04 12 10.5
NaA-173-0.8 1.18 1.81 —. 23.4
1.42 2.11 28 9.0
NaA-174-0.5 1.13 1.84 — 23.5
1.38 2.02 10 12.0
NaA-174-0.8 1.13 1.80 — 23.6
1.22 1.96 8 15.4
результатов в опытах цикловой работы цеолитов на установке и на тор-
зионных весах могло явиться следствием различия в условиях регене-
рации.
Количество цеолита, применяемое при работе на адсорбционной уста-
новке, составляло 3.5 кг за один цикл, тогда как навеска, используемая
в опытах на торзионных весах, не превышала 0.2 г. Изменение адсорб-
ционных свойств, истинной и кажущейся плотностей гранул цеолита
в процессе цикловой работы при относительно больших его количествах
могло быть обусловлено воздействием насыщенного водяного пара, обра-
зующегося при регенерации.
С целью выяснения влияния водяного пара при повышенных темпера-
турах на структуру и сорбционные свойства цеолита NaA последний в ко-
личестве 28 г подвергался действию водяного пара при температурах
100, 200, 300, 400 и 500° С в течение 40 часов для каждого температур-
ного режима. Экспериментальные данные этой серии опытов показали,
что цеолит NaA устойчив при воздействии водяного пара до 200° С. Од-
нако повышение температуры до 300° С уже приводит к снижению равно-
весной адсорбционной емкости по парам воды до 10.5%, а при обработке
цеолита водяным паром в течение 40 часов при температуре 500° С его
адсорбционная емкость по парам воды снизилась до 4.0%.
Исследование гранул цеолита, обработанных водяным паром при 500° С,
показало увеличение истинной плотности, определенной с помощью
бензола, до 2.18 при весьма малом изменении пористой структуры единицы
объема. Полученные результаты свидетельствовали о разрушении кри-
сталлов цеолита NaA.
При изучении процесса регенерации цеолита выяснялись условия,
обеспечивающие устойчивость кристаллов и сохранение адсорбционной
емкости при многократной регенерации.
Результаты опытов показали, что степень падения адсорбционной
емкости цеолита NaA по парам воды в процессе цикловой его работы за-
висит от скорости повышения температуры при регенерации и количества
регенерируемого цеолита.
В наших условиях регенерации уменьшение скорости подъема тем-
пературы до 150° в час обеспечило устойчивую работу цеолита в стацио-
нарном, движущемся и псевдоожиженном слоях.
Сравнительное изучение динамики адсорбции стационарным и псевдо-
ожиженным слоями цеолита NaA проводилось с навеской, равной 515 см3;
при этом опыты с кипящим слоем ставились в колонке с пятью полками
без передвижения цеолита с полки на полку.
252
В опытах со стационарным слоем поток воздуха подавался сверху
вниз для того, чтобы слой цеолита был прижат к решетке колонки. В табл. 2
приведены величины динамической адсорбционной емкости при постоян-
ной проскоковой концентрации паров воды (Лд) и до точки росы —60°
(Ад1) для стационарных и псевдоожиженных слоев цеолита.
Таблица 2
Динамическая адсорбционная емкость слоя цеолита NaA, по парам воды
Слой цеолита Ад, г Ад ДО 60», Г г?, л/мин. • см2 t росы, °C Размер гранул, мм Ар, ММ рт. ст.
Стационарный 70.5 72.0 1.5 —70 0.4—0.6 28
Псевдоожиженный .... 69.5 70.5 1.5 —68 0.4—0.6 15
Стационарный 68.0 69.0 2.0 —70 0.6—1.0 46
Псевдоожиженный .... 67.0 68.0 2.0 —68 0.6—1.0 17
Из приведенных в таблице данных следует, что динамическая адсорб-
ционная емкость псевдоожиженного слоя по парам воды не превышает
динамической активности стационарного поджатого слоя.
Степень осушки воздуха стационарным слоем даже несколько больше,
чем в случае псевдоожиженного слоя; последнее, очевидно, можно объяс-
нить большим сопротивлением поджатого слоя.
Результаты изучения процесса глубокой осушки воздуха непрерывно
движущимся слоем цеолита приведены в табл. 3. В этой таблице показано
изменение динамической адсорбционной емкости (ал) по парам воды в за-
висимости от скорости поступательного движения цеолита и распределе-
ния адсорбированной воды по полкам колонки.
Таблица 3
Динамическая адсорбционная емкость цеолита NaA
Скорость движе- ния цеолита, см/час Од, мг/г Распределение адсорбированной воды, %
I полка II полка III—V полки проскок
6.0 210 94.18 5.87 0.027 0.023
8.3 160 96.28 3.70 0.027 0.023
11.0 121 99.15 0.90 0.027 0.023
12.9 103 99.45 0.50 0.027 0.023
Из приведенных в таблице данных следует, что процесс адсорбции
паров воды псевдоожиженным непрерывным потоком цеолита завершается
на первых двух полках адсорбционной колонки, при этом основная доля
поступающих в колонку паров воды адсорбируется первой лобовой пол-
кой. Увеличение скорости поступательного движения цеолита в колонке
сопровождается снижением его динамической адсорбционной емкости.
При минимальной скорости степень использования статической адсорб-
ционной емкости цеолита достигает единицы.
Исследование процесса адсорбции паров воды стационарным слоем
цеолита NaA при разных скоростях паровоздушного потока показало,
что динамика сорбции хорошо описывается уравнением Шилова
6 = K(L — h),
где 6 — время защитного действия слоя длиной L,
253
h==~K
т = KL2 — в2.
Полученные экспериментальные динамические характеристики слоя
цеолита приведены в табл. 4. Выходные кривые и кривые изменения тем-
Рис. 2. Выходные и температурные
кривые при скорости воздуха
2.5 л/мии. см2.
пературы стационарного слоя пока-
заны на рис. 2—4.
Символами К, т, h, Лд, A, v в
таблице обозначены соответственно:
коэффициент защитного действия,
потеря времени защитного действия,
длина неиспользованного слоя, ди-
намическая адсорбционная емкость
слоя, равновесная статическая ад-
сорбционная емкость и скорость
паровоздушного потока.
Лд = с0г>5б, Л = аоДв£,
где «0— равновесная статическая
адсорбционная емкость (мг/г), А —
гравиметрическая плотность адсор-
бента, s —поперечное сечение слоя
шихты, L — длина слоя.
В табл. 5 приведены результаты
расчета зоны массопередачи в не-
подвижном слое адсорбента по фор-
муле, предложенной Михаэлисом—
Трейболом [16—18]:
^нас ®ир
La = L eHaf — (i — /) (онас — епо)
ИЛИ
^нас ™пр
^нас /) (^яас ^пр)
На оси ординат: слева — температура росы,
справа — температура слоя цеолита. 1 — ло-
бовой слой на высоте 8 см; 2 — средний слой
на высоте 15 см; з — вамыкающий слой на
высоте 26 см.
L - (/) (£0)
а= L '
где Lo—длина зоны массопередачи
(длина работающего слоя адсор-
бента), 0ВВС— время до равновесного насыщенного слоя, 6пр— время
защитного действия при постоянной проскоковой концентрации, / — не-
использованная часть адсорбента в зоне массопередачи, твас— общее
количество сухого воздуха за время 6нас, тщ—общее количество воз-
Таблица 4
Динамические характеристики слоя цеолита NaA, С0 = 7.5 мг/л
К мин./см т, мин. h, см Т y/~V Kv h V V я > 1 м о D. л/мин.-см2
11.9 61.0 5.4 88 22.6 3.83 0.87 2.0
9.0 56.5 6.3 89 22.4 3.97 0.83 2.5
7.7 53.5 7.0 92 22.8 4.04 0.80 3.0
254
Рис. 3. Выходные и температурные кри-
вые при скорости воздуха 2 л/мин. см2.
Обозначения те же, что на рис. 2.
Рис. 4. Выходные и температурные кри-
вые при скорости воздуха 3 л/мин. см2.
Обозначения те же, что на рис. 2.
Рис. 5. Выходные и температурные кри-
вые для движущегося слоя при скорости
воздуха 2 л/мин. см2.
Обозначения те же, что на рис. 2.
Таблица 5
Изменение длины зоны массопередачи в зависимости от длины
слоя адсорбента и скорости паровоздушного потока
L, см V. л/мин.* см2 L„, см а А
10 2.0 8.2 0.57 0.54
10 2.5 9.2 0.54 0.41
10 3.0 12.5 0.37 0.26
15 2.0 7.5 0.75 0.71
15 2.5 9.3 0.69 0.64
15 3.0 11.1 0.63 0.57
26 2.0 7.2 0.86 0.87
26 2.5 9.1 0.83 0.83
26 3.0 10.8 0.79 0.80
духа за время 01Г[), а — степе
емкости.
Из данных, приведенных в
дачи изменяется с ростом дл1
Рис. 6. Выходные и температурные к]
вые для движущегося слоя при ско]
сти воздуха 3 л/мин. см2.
Обозначения те же, что на рис. 2.
концентрации паров воды на
и 15 см увеличиваются, как это
чих равных условий работы дви
потока воздуха.
использования равновесной статической
,бл. 5, следует, что длина зоны массопере-
а слоя адсорбента, а для малых слоев и
скорости потока воздуха, равной
3 л/мин. см2, она превышает длину
слоя адсорбента.
С помощью уравнения Трейбола
можно с известным приближением
рассчитывать длину зоны массопере-
дачи для слоя цеолита больше 20 см.
Результаты изучения процесса
адсорбции водяных паров из воздуш-
ного потока движущимся слоем цео-
лита приведены на рис. 5—7.
t°C На рис. 5 показаны постоянные
проскоковые концентрации паров
воды на участках движущегося слоя
“° 8, 15 и 26 см и соответственно ртим
участкам — температуры слоя цео-
5V лита.
Начальный участок верхней кри-
вой характеризует нарастание кон-
центрации паров воды за слоем 8 см
до момента выхода адсорбционной
колонки на режим; последующий
слой длиной 7 см понижает проскоко-
вую концентрацию паров воды пер-
вого слоя до концентрации, отве-
чающей точке росы —70° С; замыкаю-
щий слой 11 см практически не
jqZ адсорбирует пары воды и только
снижает температуру потока воздуха
на 5—6° С.
При увеличении скорости потока
воздуха постоянные проскоковые
участках колонки движущегося слоя 8
видно на рис. 6, где при сохранении про-
«ущегося слоя увеличена только скорость
256
Влияние скорости движения цеолита в адсорбционной колонке и ско-
рости паровоздушного потока на динамическую активность цеолита по
парам воды показано на рис. 7, где на оси абсцисс отложены скорость дви-
жения цеолита в колонке, а на оси ординат — динамическая адсорбцион-
ная емкость в процентах.
Для сравнения на этом же рисунке показано изменение динамической
адсорбционной емкости по парам воды в зависимости от скорости потока
воздуха для стационарного слоя цеолита.
Из приведенных графиков следует, что динамическая адсорбционная
емкость цеолита по парам воды с увеличением скорости потока воздуха
и уменьшением скорости движения цеолита в колонке возрастает. При
минимальной скорости движений цеолита в колонке его динамическая
адсорбционная емкость достигает равновесной емкости.
«д-,%
ZZ-
В\---------1--------1---------1--------1--------1----
О 100 гор 300 ООО 500 w, см/чае
1.75 г.00 г.25 2.50 г.75 3.00 VtA/MUH-CM
Рис. 7. Динамическая адсорбционная активность по парам воды
цеолита NaA (в %).
На оси абсцисс: w — скорость движения цеолита, v — скорость потока
воздуха.
На основании уравнения материального баланса представляется воз-
можным определить минимальную скорость движения цеолита в колонке.
Wap=(C0 — Cnp)svt,
где W — количество выгруженного цеолита из колонны за время t,
av—равновесная адсорбционная емкость, Со — начальная концен-
трация водяных паров в потоке воздуха, Спр— постоянная проско-
ковая концентрация, s — поперечное сечение слоя цеолита в колонне,
V.— фиктивная скорость потока воздуха.
Постоянная проскоковая концентрация паров воды для цеолита со-
ставляет от 0.025 до 0.008% от начальной концентрации при ее изменении
от 20 до 7.5 г/м3. Поэтому в уравнении материального баланса постоянной
проскоковой концентрацией можно пренебречь.
или
Wav = Со svt.
W Ср ,
= = м/час.
Для расчета адсорбционной колонны необходимо знать только падение
равновесной адсорбционной емкости в процессе цикловой работы цеолита.
17 Цеолиты
257
При глубокой осушке газов, содержащих компоненты, понижающие
адсорбционную активность цеолита, необходимо экспериментально уста-
навливать падение равновесной емкости ускоренным методом для каждого
частного случая.
Результаты изучения процесса глубокой осушки воздуха движущимся
и псевдоожиженным слоями цеолита показали, что наиболее эффективным
процессом является осушка газов движущимся слоем.
Производительность единицы объема адсорбционной колонны с дви-
жущимся слоем превышает производительность единицы объема адсорб-
ционной колонны с псевдоожиженным слоем в 3—3.5 раза.
Адсорбционные колонны с движущимся слоем по конструкции и экс-
плуатационным качествам имеют значительные преимущества по сравне-
нию с адсорбционными колоннами с псевдоожиженным слоем.
Адсорбционные колонны с движущимся слоем позволяют вести про-
цесс осушки, очистки и разделения газов с переменной скоростью газового
потока.
Выводы
1. Изучен процесс глубокой осушки воздуха стационарным, движу-
щимся и псевдоожиженным слоями цеолита и показано, что: динамика
сорбции паров воды из потока воздуха стационарным слоем цеолита опи-
сывается уравнением Шилова; производительность единицы объёма ад-
сорбционной колонны с движущимся слоем превышает производитель-
ность колонны с псевдоожиженным слоем в 3—3.5 раза.
2. Из материального баланса процесса глубокой осушки воздуха
получено уравнение для расчета минимальной скорости движения цео-
лита в адсорбционных колоннах с движущимся и псевдоожиженным
слоями.
ЛИТЕ РАТУРА
1. А. Л. Коул ь, Ф. С. Ризенфельд. Очистка газов. Перевод с англ. ГНТ,
изд. «Нефтяная и горно-топливная литература», М., 1962.
2. W. S w е г d 1 о f f, Oil and Gas J., 1957, 55, 17, 122.
3. H. M. Barry, Chem. Engng., 1960, 67, 3, 105.
4. С. H. Никитин. Силикагель и его применение в черной металлургии. Мет.
изд., М.—Л., 1941.
5. В. L. R a t m е 11, Р. Bateman, J. Institution of Heating and Ventilating
Engineers, 1952, 19, 198 , 471.
6. Modern Practice in Gas Dehydration. Oil in Canada, 1953, 5, 28, 24, 26.
7. W. L. Ross, Heat Piping and Air Condit., 1957, 29, 3, 161.
8. L. D. P о 1 d e r m a n, Oil and Gas J., 1957, 55, 38, 107.
9. R. W. Parker, Petrol. Engrs., 1943, 14, 4, 110.
10. W. A. La Lande, W. McCarter, J. B. Sanborn, Ind. Engng. Chem.,
1944, 36, 2, 99.
11. R. C. A m e г o, J. W. Moore, R. G. C a p e 1 1, Chem. Engng. Progress, 1947,
43, 7, 349.
12. R. S. Capell, E. G. H a m m e r s c h m i d t, N. N. G e s c h n e r, Ind.
Engng. Chem., 1944, 36, 9, 779.
13. J. W. Carte r, Brit. Chem. Engng., 1960, 7, 472.
14. J. W. Carter, Brit. Chem. Engng., 1960, 8, 552.
15. J. W. Carter. Brit. Chem. Engng., 1960, 8, 625.
16. J. Nutter, J. Burnet. A. I. Ch. E. Journal, 1963, 9, 2, 202.
17. A. Michaels, Ind. Engng. Chem., 1952, 44, 1922.
18. R. T г e у b a 1. Mass Transfer Operations. McGrow Hill, N. Y., 1956.
258
РАЗДЕЛЕНИЕ И ТОНКАЯ ОЧИСТКА
СИНТЕТИЧЕСКИМИ ЦЕОЛИТАМИ УГЛЕВОДОРОДОВ,
ПРИМЕНЯЕМЫХ В ПРОИЗВОДСТВЕ ВЫСОКОПОЛИМЕРОВ
Таблица 1
Примерное содержание некоторых
примесей в дивиниле, применяемом
при производстве СКВ из спирта,
и в мономере для получения
Ч'мс-1,4-полибутадиена
Компонент Содержание, вес. %
для СКВ для,поли- бута- диена
С4Н10 0.65 0.8
С4Н8 6.1 ——
Эфиры 0.25 0.001
С6 0.05 0.001
Ацетиленовые —. 0.005
Вода 0.03 0.001
неизмеримо возросли требования
В. С. Виноградова, Л. С. Кофман
Наиболее важные с практической точки зрения свойства полимеров,
вся совокупность их так называемых технических качеств в значительной
степени зависят от чистоты исходных мономеров и веществ, применяемых
как растворители при полимеризации. Весьма небольшое количество
примесей влияет не только на кинетику процесса полимеризации, но и на
структуру полимера. Между тем
если, например, в цепи молекул поли-
бутадиена или полиизопрена содер-
жится лишь несколько звеньев, при-
соединенных в положении 1,2 или же
транс-звеньев, эти полимеры суще-
ственно уступают по качеству при-
родному каучуку.
Одним из наиболее наглядных
показателей научно-технического
прогресса промышленности тяжелого
органического синтеза является круп-
нотоннажное производство индиви-
дуальных углеводородов такой чис-
тоты, которая совсем недавно была
недостижима даже для исследова-
тельских лабораторий. С развитием
знаний в области получения ме-
таллорганических и комплексных
катализаторов и их применения для
синтеза стереорегулярных полимеров
к чистоте углеводородов. Некоторое представление об этом можно полу-
чить из табл. 1—3, причем следует учесть, что при совместном присутствии
нескольких из перечисленных примесей содержание каждой из них
должно быть еще меньшим.
Не менее жесткие требования предъявляют к чистоте растворителей,
содержание которых в направляемой на полимеризацию шихте в несколько
раз больше, чем самого мономера. Весьма ограничено присутствие при-
месей и в растворителях каталитических систем. Так, при производстве
Таблица 2
Примерное содержание некоторых примесей
в техническом изопрене и в мономере для получения
цис-1,4~полиизопрена
Содержание, вес. %
Компонент технический продукт мономер
Меркаптановая сера ...... Ацетиленовые Пиперилен Циклопентадиен Вода до 0.001 до 0.06 13.0 до 1.0 0.003 0.0002 0.0004 0.06- 0.0005 г',.:-;! 0.001
17* 259
Таблица 3
Желательный состав пропилена и этилена, применяемых
в качестве мономеров
Компонент Содержание
Ацетиленовые 0.001 объема. %
Бутадиен 0.001 »
СО 0.0005 »
H2S 6.10-5 вес.%
Сернистые в пересчете на серу .... 0.0002 »
Н2О 0.005 объемн.%
С02 0.001 »
о2 0.0001 »
полиэтилена низкого давления требуется циклогексан, содержащий не
более 0.01% ароматических и менее 0.001% воды.
Большую часть углеводородов для производства высокополимеров
выделяют из продуктов термической и каталитической переработки
нефти. Количество примесей в этих продуктах зависит не только от усло-
вий переработки, но и от чистоты исходного сырья. Например, если в изо-
пентане содержится н-пентан, то при дегидрировании в катализате об-
наруживают, помимо изопентенов, также и увеличенное содержание пен-
тенов нормального строения. При наличии в перерабатываемом сырье
соединений, содержащих серу или азот, соответствующие примеси будут
содержаться и в целевых продуктах. Между тем многие каталитические
системы очень чувствительны к сернистым соединениям; в ряде случаев
число звеньев диена-мономера, присоединенных в положении 1,2, резко
возрастает в присутствии всего лишь нескольких молекул сернистых
соединений на миллион молекул углеводорода.
С другой стороны, чистота целевых углеводородов зависит от способа
их выделения, так как некоторые примеси могут быть внесены вслед-
ствие контакта с применяемыми разделяющими агентами. При разделе-
нии диенов от алкенов экстрактивной ректификацией углеводороды за-
грязняют следами растворителя и продуктов его гидролитического и
термического распада, а при хемосорбции — растворами солей закиси
меди, аммиаком. Оба эти метода, так же как и экстрактивная кристалли-
зация с мочевиной, непригодны к тому же для разделения легких угле-
водородов, отличающихся по строению скелета молекулы, а хемосорб-
цией нельзя разделить и алкан-алкеновые смеси. При наиболее распро-
страненном способе извлечения изобутилена из содержащих его фракций
продуктов нефтепереработки обводненной серной кислотой в целевой
продукт переходит некоторое количество метилмеркаптана, образую-
щего азеотропные смеси с углеводородами С4 и поэтому трудноотделимого.
Поскольку абсолютное содержание примесей, подлежащих удалению
из углеводородов, как правило, невелико, а степень очистки должна быть
очень высокой, весьма перспективно для целей тонкой очистки примене-
ние адсорбционной техники. Однако наиболее распространенные сор-
бенты: силикагель, активная окись алюминия и активированные угли —
не обладают требуемым для тонкой очистки углеводородов комплексом
свойств. В виде примера можно указать, что сорбцией окисью алюминия
практически невозможно достигнуть глубокой осушки даже алканов,
а при контакте с непредельными и без того малая (3 г/100 г) динамическая
активность по воде становится еще меньшей, особенно при температурах
выше 15°. Молекулы непредельных, например изопрена, свободно про-
260
никая в пористую структуру алюмогеля, образуют олигомеры, не удаляе-
мые десорбцией в токе инертного газа. При термическом воздействии
олигомеры превращаются в коксообразные продукты, выжиг которых
сопровождается рекристаллизацией окиси алюминия в неактивную
форму И].
Иная картина наблюдается при сорбции синтетическими цеолитами,
промышленное получение которых недавно освоено в СССР. Для каждого
из легких углеводородов, в том числе и непредельных, может быть по-
добрана такая форма цеолитов, в полости кри-
сталлов которых не проникает данный угле-
водород. Это резко снижает возможность его
полимеризации и позволяет достигнуть очень
глубокой осушки и высокой динамической ак-
тивности сорбента даже при большой скорости
осушаемого в жидкой фазе потока (табл. 4).
На основе изучения изотерм адсорбции ин-
дивидуальных углеводородов может быть сде-
лан только предварительный выбор марки цео-
литов. Так, из рис. 1 следует, что для осушки
изопентана и изопрена возможно применение
любой формы цеолитов типа А, при осушке
пиперилена нельзя пользоваться цеолитами
СаА, а для осушки дивинила и винилацети-
лена применимы только цеолиты КА. Однако
достоверно установить пригодность конкретных
типов и партий цеолитов можно лишь на ос-
нове опытов, проведенных в динамических
условиях при циклической работе.
В докладе на 1-й конференции по цеолитам
мы приводили [2] данные о том, что при оценке
пригодности данного образца формованных цео-
Рис. 1. Изотермы адсорб-
ции непредельных углеводо-
родов формованными цеоли-
тами А при 20° С.
1 — пиперилен (СаА); 2 — изо-
прен (СаА); 3 — дивинил (NaA);
1 — винилацетилен (NaA); б —
дивинил (КА).
литов для различных случаев разделения нельзя ограничиться изучением
адсорбции компонентов в статических условиях. Молекулярно-ситовые
свойства цеолитов, внутренняя поверхность пор которых имеет отрица-
тельный заряд, не могут быть полностью сопоставлены и по сорбции та-
кого полярного вещества, как вода. Поэтому при составлении обязатель-
ных методик оценки молекулярно-ситовых свойств формованных цеолитов
СаА были учтены данные разделения алканов С4—Св изо- и нормального
строения |3—5].
Еще в большей мере приведенные соображения важны при переходе
к промышленному применению цеолитов. Для проектирования крупных
адсорбционных установок необходимо знать, как будет изменяться дина-
Таблица 4
Основные показатели процесса глубокой осушки изопрена в жидкой фазе
цеолитами NaA (£ = 23 см) при проскоковой концентрации воды 1-10-4
вес. % в динамических условиях
Исходное содержание воды, вес. % № цикла сорбции ш “д ^0
л/л час м/сек. г/см2 г/100 г г/см2 см
0.03 1 10 7- КГ4 3.1 18.5 0.14 3.4
0.03 3 20 14 •10-4 3.5 21.0 0.16 2.0
0.003 3 10 7 • 10~4 2.2 13.5 0.00 1.8
0.003 6 10 7 • 10"4 2.3 14.0 0.10 1.8
261
мическая активность сорбента и длина зоны массопередачи («работаю-
щего слоя») при разных составах разделяемой смеси и при различных
условиях сорбции в длительной циклической работе, какой метод десорб-
ции следует применить, располагать сведениями о теплотах сорбции, ки-
нетических показателях при сорбции и десорбции и т. п. Если исходить
только из эмпирических данных, потребовалось бы выполнить неоправ-
данно большой объем экспериментальных работ при разработке каждого
промышленного процесса.
В трудах М. М. Дубинина и его учеников [6—8] содержатся убеди-
тельные доказательства применимости уравнений теории объемного за-
а, г/ЮОг
р,мм ртст
Рис. 2. Изотермы адсорб-
ции цеолитами СаА инди-
видуальных н-бутенов ,при
100°.
1 — wc-2-бутен; 2 — 1-бутен;
3 щранс-Ё-бутен; 4 — 1-бутен
(равновесная смесь). Покривей
4 ' крестики — вычисленные
точки;; кружочки — эксперимен-
тальные.
полнения пор для корреляции в широком диапа-
зоне состава и температуры данных исследова-
ния изотерм сорбции аполярных веществ цео-
литами. Однако попытки описания указанными
уравнениями изотерм адсорбции полярных и
поляризуемых соединений, в том числе непре-
дельных углеводородов, не привели к полностью
сопоставимым результатам, что объяснялось
повышенным «сродством» цеолитов к данным
веществам. Поскольку производство большин-
ства важнейших полимеров основано на приме-
нении непредельных углеводородов, данные
о сорбции которых различными цеолитами
в литературе почти не отражены, нами были
предприняты исследования в этой области.
Основные результаты разработки процессов
с применением формованных цеолитов отече-
ственного производства для глубокой осушки
и разделения непредельных углеводородов,
топкой очистки от некоторых полярных соеди-
нений и т. п. изложены в других статьях [9—
11, 14]. В данном сообщении приводятся пре-
имущественно положения, обобщающие зти
работы.
. При контакте с формованными цеолитами
в определенных условиях наблюдаются превра-
J ’ щепия непредельных углеводородов: полимери-
зация, изомеризация и даже крекинг. Экспериментально установлено на
прймёрё изобутилена, что образование олигомеров легко полимеризующихся
Углеводородов, имеющих весьма ограниченный доступ в пористую струк-
туру кристаллов цеолита, обусловлено природой связующего и актива-
цией его поверхности кислородом при повышенной температуре. Поли-
мер’йНуйщую паразитарную каталитическую активность цеолитов можно
предЪт'враТйть путей исключения их контакта с кислородом, подбором
связующего," ""а также модифицированием предадсорбцией некоторых
веществ [11], Крекинг углеводородов происходит лишь в тех случаях,
когда десорбцию их из цеолитов осуществляют при высокой температуре.
Что касается изомеризации проникающих в поры цеолитов непредельных
углеводородов — с перемещением кратной связи и пространственной
перегруппировкой атомов в более устойчивое положение, то она проте-
кает, в полостях кристаллов сорбента и тем в большей степени, чем выше
температура. Полностью предотвратить ее, по-видимому, невозможно;
в литературе’ содержатся указания лишь о частичном подавлении изоме-
ризующего влияния алюмосиликатов при уменьшении относительного
содержания А12О3. На чистом силикагеле изомеризации н-бутенов заме-
чено не было [12]. Между тем именно изомеризацией могут быть объяс-
пены некоторые из отклонений изотерм адсорбции непредельных от урав-
нений теории объемного заполнения пор.
Так (рис. 2), изотермы адсорбции 1-бутена на цеолите СаА, экспери-
ментальная и рассчитанная [7] по уравнению
lga = lg^-0.434-~ - (1)
не совпадают. Если же учесть фактический состав адсорбата, в котором
вследствие изомеризации содержатся цис- и шранс-2-бутены, то точки
изотермы, рассчитанные по аддитивности, укладываются непосредственно
на экспериментальной кривой (рис. 2). При расчете по приведенному
уравнению на основе данных о сорбции каждого из изомеров 2-бутена
оказалось, что значения предельного объема адсорбционного простран-
ства (И70) во всех случаях весьма близки, а коэффициенты аффинности (р)
резко отличаются (табл. 4). Теплоты сорбции индивидуальных алкенов при
равной степени заполнения пор тем больше, чем больше значение р.
Следовательно, уравнения теории объемного заполнения пор могут
быть, очевидно, применены и для описания сорбции непредельных угле-
водородов и их смесей, если учитывать изменение состава адсорбата вслед-
ствие изомеризации.
Таблица 5
Значения коэффициентов аффинности (р) и предельных объемов адсорбционного
пространства (7F0) по сорбции некоторых индивидуальных веществ
формованными цеолитами СаА и NaX
Наименование вешества Тип цеолита Wo. см5,'г эксп теор ^эксп/^теор
1-Бутен СаА 0.197 3.78 2.38 1.68
транс-2-Буген . . . » 0.188 2.84 2.38 1.22
цис-2-Бутен .... » 0.190 4.40 2.38 1.88
Этилмеркаптан . . . » 0.170 2.34 2.11 1.11
Изопентан NaX 0.227 2.95 3.16 0.94
Циклогексан .... » 0.158 3.40 3.28 1.04
Бензол » 0.211 3.76 2.96 1.3
Тиофен » 0.193 4.58 2.65 1.7
•Этилмеркаптан . . . » 0.239 3.80 2.11 1.8
«Сероуглерод .... » 0.160 1.07 1.62 0.7
Из данных табл. 5, относящихся к цеолитам NaX и СаА, видно, что
значения Wo, вычисленные по данным для полярных, поляризуемых и
аполярных соединений, в ряде случаев совпадают. В то же время наблю-
дались расхождения в значениях fV0 и для веществ с одинаковой степенью
отклонения Р8КСП от оцененного по парохорам, а значения Р8КСП тем больше
отличаются от расчетных, чем более отчетливо выражены полярные свой-
ства данного соединения. На основе теории объемного заполнения пор
можно с достаточным для технических целей приближением скоррели-
ровать сравнительно ограниченные экспериментальные данные и приме-
нительно к процессам тонкой очистки углеводородов от примеси полярных
веществ.
Средние значения Wo как для цеолитов СаА, так и NaX мало отли-
чаются от приведенных в литературе [6]; отклонения значений Wo для
циклогексана и сероуглерода подлежат уточнению. Однако пониженное
значение коэффициента аффинности сероуглерода согласуется с экспери-
ментально установленными фактами о крайне незначительной сорбции
его цеолитами NaX при 20° в области малых концентраций (рис. 3) и
о невозможности в этих условиях полного удаления его из алканов ад-
сорбцией.
263
Рис. 3. Изотермы адсорбции
цеолитами NaX при 20° С.
1 — этилмеркаптан; 2 — сероуг-
лерод.
При рассмотрении кривых адсорбционного равновесия изомеров ал-
кенов С4 и С6 в паровой фазе на цеолитах СаЛ было обнаружено обращение
относительной адсорбируемости (а) некоторых разделяемых пар, в част-
ности стереоизомеров 2-алкенов. Это явление выражено наиболее отчет-
ливо для смесей стереоизомеров 2-пенте-
на [14], которые невозможно разделить при
составе, близком к эквимолекулярному
(рис. 4). Выделение чистого унс-2-бутена
принципиально возможно даже в случаях
преобладающего содержания нгранс-изомера
в разделяемой смеси [11]. Значение а пары
1-бутен—н-бутан возрастает при уменьше-
нии содержания 1-бутена.
Изучена также соадсорбция цеолитами
NaX смесей сернистых соединений с изопен-
таном или с циклогексаном. При этом уста-
новлено, что статическая активность сорбен-
та по тиофену и этилмеркаптану из соответ-
ствующих жидких углеводородов несколько
ниже, чем при сорбции паров чистых ве-
ществ (рис. 5), однако в области низких
концентраций Сернистых значений а дохо-
дит до 1500, что позволяет оценивать формо-
ванные цеолиты NaX по разделению стандартной смеси циклогексан-тио-
фен в динамических условиях [3].
Экспериментально установлена возможность тонкой очистки алканов
и циклоалканов от бензола, тиофена и этилмеркаптана; селективность их
адсорбции цеолитами NaX убывает по
мере уменьшения отношения Рэксп к Ртеор.
Однако при малых концентрациях тиофе-
на полной очистки от него бензола дости-
гнуть не удается. Следует учесть, что по-
ры цеолитов NaX доступны для всех ком-
понентов перечисленных смесей. Поэтому
селективность сорбции в данном случае
определяется не основанным на различиях
в размерах молекул молекулярно-сито-
вым эффектом (как это происходит, на-
пример, при разделении изо- и и-алка-
нов цеолитами СаА), а отличиями в кине-
тике сорбции и взаимодействии молекул
разной полярности и поляризуемости с
отрицательно заряженной поверхностью
пор цеолита.
При корреляции данных о разделении
в динамических условиях рассчитывали
длину зоны массопередачи (£0) при данной длине L слоя сорбента
по уравнению [12], в котором fm — отношение площади над выходной
кривой (рис. 6) на участке нарастания концентрации (от проскоковой
до начальной) к общей площади этого участка; тБ—_тЕ— тА — время,
необходимое для исчерпывания динамической активности сорбента с мо-
мента проскока.
Рис. 4. Кривая адсорбционного
равновесия системы гщс-2-пентен,
траис-2-пентен на цеолитах_СаА.
т т ________________
(2)
264
Слева на рис. 6 показано изменение концентрации сорбируемой при-
меси по длине слоя сорбента. На концевом участке слоя, соответствующем
зоне массопередачи, концентрация примеси возрастает от нуля до исход-
ной. Это отвечает моменту проскока; незаштрихованная часть слоя харак-
теризует неиспользованную долю его актив-
ности. При дальнейшем пропускании разде-
ляемой смеси происходит полное насыщение
сорбента и концентрация примеси в рафи-
нате увеличивается, что отражено формой
выходной кривой в правой части рисунка,
симметричной левой кривой. Для техниче-
ских расчетов успешно пользуются урав-
нением Шилова
тд = Х (£-*), (3)
где h пропорциональна неиспользованной
доле статической активности слоя. При изу-
чении адсорбционной очистки промышлен-
ных потоков более уместно основываться на
количестве (кд) очищенного продукта (ра-
фината), чем на времени защитного дей-
ствия тА, являющегося основным показате-
лем при процессах, аналогичных проти-
вогазовой технике. Заменив т на v и учи-
тывая, что h = Lo, находим:
гА=ХД£-/т£0); К' =
I
VA
— L-fm.L0 мл/см’ &
а.г/100г
гОг
__________I_________।_________।
О 0.1 0.2 0.3
Концентрация S, бес. %
Рис. 5. Адсорбция этилмер-
каптана цеолитами NaX при
20° С.
1 — пары чистого этилмеркаптана;
2 — из раствора в изопентане.
При совместном решении уравнений (2) и (4) приходим к следующим
выражениям:
£ = £0
К'=
(4а).
При этом вместо коэффициента защитного действия К введена по-
стоянная К1, выражающая количество очищенного в зоне массопере-
Рис. 6. Схемы к расчету Lo по уравнению Трейбола.
дачи рафината, отнесенное к единице длины слоя (мл/см или г/см).
Подставляя значение гд из (4) в уравнение (5) динамической актив-
ности до проскока
^дРСо
“д- 1-С0
(5).
265-
где Со — весовая доля сорбируемой примеси, ар—ее плотность, после
шреобразований приходим к выражению:
“д К' • ₽ - Со
-L-Ь^Ц = ~Т—Со = г/™3- (6)
Величина ае характеризует полную (т. е. до выравнивания составов
-очищаемой смеси и рафината) динамическую активность сорбента. Как
это видно из ее размерности (г/см3 сорбента), а, в отличие от ад постоянна
при заданных параметрах (температура, скорость) для любого слоя сор-
бента, что облегчает расчеты при переходе от лабораторных к промышлен-
ным адсорберам. Практическая применимость уравнения (6) подтверди-
лась при сопоставлении с экспериментальными данными разделения стан-
дартной (3) смеси различными образцами цеолитов СаА (1)—(4):
Образцы цеолитов
12 3 4
ав по уравнению (6).................
ае фактически.......................
7.95 8.97 8.46 • 9.12
8.00 8.65 8.40 9.45
Экспериментальные данные по измерению длины зоны массопередачи
(Ln) в зависимости от линейной скорости хорошо согласуются, даже при
сорбции из жидких потоков, с эмпирическим уравнением
Lo = В' d • (о , (7)
поскольку при разных скоростях значение В' оставалось практически
постоянным. Поэтому для приближенного расчета Lo при изменении
линейной скорости от со, до <в2 или при переходе к иным размерам (</)
гранул пользовались уравнением
Из результатов изучения стабильности молекулярно-ситовых свойств
цеолитов после длительного нагрева при умеренно высоких температу-
рах [10] вытекало, что следует стремиться к возможному ограничению
предельного нагрева цеолитов при их регенерации. Вместе с тем очевидно,
что от полноты удаления сорбированных веществ зависят все показатели
последующего адсорбционного цикла. В связи с этим условия десорб-
ции—регенерации цеолитов разрабатывали применительно к каждому
данному процессу. В качестве элюента применяли вещества, сорбируемые
цеолитами (полярные и аполярные) и несорбируемые ими при температуре
регенерации. При регенерации цеолитов водяным паром в вакууме [13]
собственно десорбция полностью заканчивается в момент заполнения пор
сорбента молекулами воды. Однако восстановление сорбционных свойств
цеолитов зависит от условий последующей активации. Влияние содержа-
ния остаточной влаги на показатели последующей сорбции зависит не
столько от природы сорбируемого вещества, сколько от типа цеолита,
уменьшаясь по мере увеличения размера «окон» (табл. 6). Следовательно,
активация цеолитов NaX возможна при более низкой температуре и боль-
шем остаточном давлении, чем цеолитов типа А. ?. .
При изучении некоторых кинетических показателей десорбции в токе
азота или паров углеводорода строили выходные кривые в координатах:
концентрация адсорбированного вещества в выходящих из адсорбата
парах как функция удельного (л/л сорбента) расхода газа-вытеснителя
266
Таблица 6
Влияние повышения содержания остаточной влаги цеолитов с 0.5 до 1.5°/0
на динамические показатели сорбции различных веществ (относит. %)
Тип цеолита Адсорбат “в ь0
СаА н-бутены .... 40 35 55
NaX этилмеркаптан 15-20 10 10—25
(рис. 7) при данной скорости потока. Для ускорения опытов заданную
температуру поддерживали только до прекращения существенного из-
менения состава паров, после чего быстро повышали температуру сор-
бента до 350—370° с целью полного удаления из цеолитов сорбированного
ими компонента. Общее количество десорбата и объем газа-вытеснителя,
требуемый для полного завершения
интегрированием площади под выход-
ной кривой с учетом асимптотического
приближения ее к оси абсцисс.
В результате проведенных работ
установлено, что удельное количество
газа-вытеснителя (у л/л сорбента) для
каждой данной температуры почти
не менялось при увеличении скорости
газа в два раза. Таким образом, с
достаточным для технических целей
приближением можно принять
v = о - т, (9)
тде т — продолжительность десорб-
ции единицы объема слоя сорбента
в часах, <в — объемная скорость
элюента в л/л сорбента час.-1. С дру-
гой стороны, в полулогарифмических
координатах зависимость удельного
расхода газа-вытеснителя от темпера-
туры, десорбции может быть описана
линейным уравнением
lg f = At-\-B, (10)
десорбции, находили графическим
Рис. 7. Выходные кривые десорбции из
цеолитов.
1 —н-бутен (СаА); 2 — этилмеркаптан (NaX).
что позволяет найти расчетным путем наиболее экономически выгодные
условия регенерации цеолитов. Минимально допустимую температуру,
при которой еще достигается достаточно полное удаление сорбированных
веществ, определяли, сопоставляя высоту концентрационных пиков вы-
ходных кривых при различных температурных условиях.
Характерно, что значения коэффициентов уравнения (10) в большей
мере зависят от типа цеолита, чем от природы сорбата. Из данных табл. 7
видно, что при io=const для десорбции из цеолитов NaX полярного этил-
меркаптана требуется не намного больше времени,, чем для удаления 1-бу-
тена из цеолитов СаА. Это согласуется с изложенными выше соображе-
ниями о влиянии предадсорбции равного количества воды на активность
цеолитов типа А и X.
Удаление сорбированных цеолитами примесей достигается при лю-
бом из указанных вариантов десорбции. Вытеснение сорбата водяным
267
Таблица 7
Сопоставление расхода элюента на десорбцию и продолжительность
регенерации различных типов и форм цеолитов при 250°, <7 = 1.5 мм
и и = 100 л/л час
Тип цеолита и сорбированное вещество —А • 103 В Л/Л сорбента Часы
NaX, этилмеркаптан 4.6 3.65 . 320 3.2
СаА, 1-бутен 6.5 4.00 240 2.4
NaA, ацетонитрил 4.6 4.50 2200 22
паром в вакууме с наибольшим успехом может быть применено при та-
ких процессах, как тонкая очистка углеводородов от легкополимеризую-
щихся или термически нестабильных в присутствии следов кислорода
примесей, например при десорбции меркаптанов из цеолитов NaX. Этот
метод полезен и как предшествующий регулируемой окислительной реге-
нерации в тех случаях, когда ее невозможно избежать, так как при этом
уменьшается вероятность частичного разрушения цеолитов вследствие
местного перегрева. Если при этом не ставится задача рекуперации сор-
бата, вместо вакуума можно применить разбавление водяного пара инерт-
ным газом, а иногда и воздухом.
Изложенные методы эксперимента и обработки опытных данных
успешно применены при разработке ряда процессов, предназначенных
для промышленного внедрения.
ЛИТЕРАТУРА
1. I. D е i t е г, Chem. Engng., 1960, 67, 22, 115.
2. В. С. Виноградова, Л. С. Кофман, в сб.: Синтетические цеолиты,
Изд. АН СССР, 1962, 99.
3. В Т У на синтетические цеолиты А и X, № МРТУ-6-01-567-63.
4. Т р у д ы по химии и химической технологии. НИИ химии Горьковского гос.
унив. 1962, 2, 268.
5. В. С. В и н о г р а д о в а, Л. С. К о ф м а и, в сб.: Синтетические цеолиты,
Изд. АН СССР, 1962, 245.
6. М. М. Д у бинин и др., в сб.: Синтетические цеолиты, Изд. АН СССР, 1962,
7 и 18.
7. Г. В. Цицишвили, Т. Г. Андроникашвили, в сб.: Синтетические
цеолиты, Изд. АН СССР, 1962, 127.
8. Е. П. Жуковская, Диссертация, М., 1963.
9. Г. В. У р б а н, В. С. Виноградова и др., настоящий сборник, стр. 268.
10. В. С. Виноградова, Л. С. К о ф м а н и др., настоящий сборник, стр. 285.
11. В. С. Виноградова, Т. А. Марковаи др., настоящий сборник, стр. 283.
12. L. L i 111 е et al., Canad. J. Chem., 1961, 39, 1, 42.
13. Л. С. Кофман, В. С. Виноградова, И. П. Потепалова, Автор-
ское свидетельство № 157966.
14. Н. В. Захарова, Л. С. Кофман и др., настоящий сборник, стр. 274.
ТОНКАЯ ОЧИСТКА АЛКАНОВ С4—С6 ОТ СЕРНИСТЫХ СОЕДИНЕНИЙ
Г. В. Урбан, В. С. Виноградова, В. Н. Комарова, Л. С. Кофман
В присутствии некоторых сероорганических соединений, в частности
меркаптанов, происходит либо полное отравление большинства катали-
тических систем, применяемых при полимеризации непредельных угле-
водородов, либо образование полимеров нежелательной структуры, тех-
268
нические свойства которых при этом резко ухудшаются. Поэтому в угле-
водородах-мономерах и растворителях содержание подобных соединений
должно быть сведено к минимуму и
во всяком случае не превышать
2 • 10~4 вес. % в пересчете на серу.
Поскольку в ближайшие годы раз-
витие нефтехимической промышлен-
ности планируется в основном на ба-
зе сернистых нефтей, вопросы тонкой
очистки легких углеводородов от сер-
нистых соединений приобрели осо-
бую актуальность. В углеводородных
фракциях С4, С5 и Св, выделенных
из продуктов добычи и переработки
нефтей некоторых месторождений,
содержится ~ 0.02 % серы. Приме-
нявшиеся до последнего времени ме-
тоды сероочистки дороги, громоздки
и не могут рассматриваться как до-
статочно надежное средство уменьше-
ния количества сернистых примесей
в углеводородах по крайней мере на
2 порядка.
Целью нашего исследования яв-
лялось изучение возможности исполь-
зования уникальных сорбционных
свойств синтетических цеолитов для разработки промышленного процесса
тонкой очистки от сернистых соединений алканов С4—Св и циклоалка-
Рис. 2. Изотермы адсорбции на формован-
ных цеолитах в логарифмической шкале.
NaX : 1 — этилмеркаптан, 3 — сероуглерод, 4 —
изопентан, б — циклогексан, 6 — тиофен; СаА:
2 — этилмеркаптан.
достигнуто не было.
Исследование изотерм адсорбции производили на вакуумной адсорб-
ционной установке с весами Мак-Бена. Исследуемые формованные цео-
литы активировали при 350° и давлении 10~2 мм рт. ст. При корреляции
экспериментальных данных по уравнению теории объемного заполнения
пор [1] получили достаточно херошее приближение точек изотерм к пря-
NaX: 1 — этилмеркаптан, 3 — сероуглерод,
4 — изопентан; 6 — циклогексан, 7 — тио-
фен; СаА: 2 — этилмеркаптан; NaA: 5 — этил-
меркаптан; прерывистой линией обозначены
изотермы адсорбции, рассчитанные по урав-
нению теории «объемного заполнения пор».
нов, применяемых при производ-
стве высокополимеров в качестве
исходных материалов и раствори-
телей. Особое внимание было уде-
лено удалению наиболее вредных
примесей, таких, как меркаптаны
и тиофен. Попутно решались во-
просы глубокой осушки углево-
дородов.
В результате изучения изотерм
адсорбции индивидуальных ’ве-
ществ, углеводородов и сернистых
соединений, различными типами
цеолитов (рис. 1) было предвари-
тельно установлено, что наиболее
эффективно применять для наме-
ченной цели цеолиты NaX. Низ-
шие меркаптаны (например, этил-
меркаптан) частично сорбируются
и цеолитами СаА, но из смесей с
изопентаном в динамических усло-
виях должной глубины очистки
269
мой в интервале двух—трех порядков равновесных относительных дав-
лений.
В результате графической обработки изотерм адсорбции (рис. 2) вы-
числили 12] предельный адсорбционный объем (РГ0) и коэффициент аф-
Рис. 3. Изотермы адсорбции, рассчитан-
ные по уравнению (1) для малых пар-
циальных давлений на цеолитах.
NaX : 1— тиофен, 2 — этилмеркаптан, 3 — се-
роуглерод; СаА: 4 — этилмеркаптан.
фена и сероуглерода при 20° С
финности (р). В качестве стандарт-
ного вещества был выбран азот; в со-
ответствии с данными 13] значение В
принято для цеолитов СаА — 4.95 • 10-6
и для NaX — 6.55 • 10-6.
При адсорбции изопентана, бен-
зола, тиофена и этилмеркаптана зна-
чения Wo близки к найденным дру-
гими исследователями [3] при сорбции
на цеолитах NaX. Отклонения между
экспериментальными и расчетными
значениями р аполярных соедине-
ний (изопентана и циклогексана) не
превышают погрешностей опыта. Что
касается коэффициента аффинности
исследованных примесей сернистых
соединений, то их значения тем больше
вычисленныхпоинкрементам, чем луч-
ше сорбируется данное вещество [2].
По экспериментальным значениям
Wo и Р вычислены координаты изо-
терм адсорбции этилмеркаптана, тио-
для равновесных давлений, соответствую-
щих предполагаемому содержанию сернистых соединений в очищаемых
углеводородах. Из приведенных на рис. 3 изотерм адсорбции видно, что
в области низких концентраций се-
роуглерод (в отличие от этилмер-
каптана и тиофена) практически не
сорбируется цеолитами NaX из па-
ровой фазы и поэтому не может
быть полностью удален из угле-
водородов. Впрочем, в этом нет
особой необходимости, так как
допустимое содержание сероугле-
рода и сероокиси углерода в мо-
номерах при стереонаправленной
полимеризации на 2—3 порядка
больше, чем меркаптанов [2].
Кроме изотерм адсорбции, по
которым можно судить об адсорб-
ционной емкости цеолитов, для
решения вопроса об эффектив-
ности разделения необходимо рас-
полагать данными о специфич-
ности адсорбции из реальных си-
стем. Для этого исследовали изо-
термы соадсорбции зтилмеркап-
а,,г1К0г
Рис. 4. Изотермы адсорбции цеолитами
NaX.
1 — пары чистого этилмеркаптана; 2 — этилмер-
каптан из раствора в изопентане; 3 — сероуглерод
из раствора в изопентане.
тана и сероуглерода из изопентана в жидкой фазе в области концентра-
ций от 2 • 10-4 до 1 • 10~2 вес. % (в пересчете на серу). При этом установ-
лено (рис. 4), что статическая активность цеолитов NaX по этилмеркап-
тану при сорбции из жидкой фазы в присутствии избытка изопентана не-
сколько меньше, чем из паров чистого вещества. Тем не менее эффективная
270
очистка цеолитами NaX изопентана и циклогексана вполне осуществима
даже при малом содержании соответственно этилмеркаптана и тиофена
в очищаемой фракции. Кривая фактического фазового равновесия (рис. 5)
системы этилмеркаптан—изопентан—цеолит NaX, построенная по экс-
Рис. 5. Кривая сорбционного равновесия
системы этилмеркаптан—изопентан—цео-
лит NaX при малых концентрациях серы.
X — мольная доля изопентана в адсорбате; У —
мольная доля изопентана в рафинате.
Рис. 6. Схема лабораторной уста-
новки тонкой очистки алканов
С4—Св от сернистых соединений.
1 — дозатор с рубашкой; 2 — адсор-
бер с цеолитом NaA; 3 — адсорбер с
цеолитом NaX; 4 — приемник; 5 —
сосуд Дьюара.
Объем р/иримта
периментальным данным при сорбции из жидкой фазы, в области малых
концентраций приближается к оси абсцисс.
Динамические и некоторые кинетические показатели процесса очистки
были определены на лабораторной установке (рис. 6), состоящей из не-
скольких адсорберов, первый из ко-
торых загружали цеолитами NaA
(для осушки), а последующие — цео-
литами NaX.
Стадию адсорбции проводили при
комнатной температуре, пропуская
очищаемый поток с заданной ско-
ростью через слой сорбента снизу
вверх до выравнивания концентрации
серы в исходной смеси и в рафинате.
В регулярно отбираемых пробах ра-
фината определяли содержание серы
методом гидрирования с никелем Ре-
нея с точностью до 1 • 10~5%. Про-
скоковой концентрацией примеси счи-
тали 2 • 10“4%, в пересчете на серу.
Затем строили выходные кривые
(рис. 7), по которым вычисляли [2]
основные динамические показатели:
длину зоны массопередачи, динами-
ческую активность до «проскока» (ад)
и полную (ая).
Основные результаты тонкой очистки изопентана, в том числе содер-
жащего циклопентадиен (ЦПД), в динамических условиях представлены
в таблице. Полученные результаты отображены графически в коорди-
натах: ae^f(C0), ад=ф((о) и £о=ф((о), где Со — начальная концентрация
сернистых в очищаемой смеси, <о — линейная скорость потока, Lo — длина
зоны массопередачи в стационарном слое сорбента. .
Рис. 7. Пример выходной кривой.
’’Б и VE — объемы рафината: до «проскок а»-
и до «исчерпывания» соответственно; Со — ис-
ходная концентрация адсорбируемого компо-
нента.
271
Динамические показатели процесса очистки изопентана от сернистых
соединений цеолитами NaX (при длине слоя сорбента 33.5 см)
№ цикла Исходная смесь Скорость потока Ьо> см а Д °F
концен- трация серы, вес. °/0 содержа- ние ЦПД. вес. % л/л час М/сек. 10‘ г/см2 г/100г Г/СМ3 г/100г Вз
2 0.016 0.022 3.0 2.32 14.0 0.43 2.1 0.015 2.4 6100
11 0.027 0.022 9.2 8.5 25.0 0.36 1.7 0.017 2.7 5700
12 0.012 0.022 9.5 8.8 26.0 0.23 0.9 0.012 1.8 5850
В среднем . . 5880
6 0.249 0.022 1.0 0.93 9.7 1.10 5.22 0.038 6.10 6750
7 0.249 0.022 3.0 2.78 15.6 1.00 4.74 0.037 5.83 6250
8 0.249 0.022 4.9 4.55 22.4 0.88 4.20 0.038 6.10 7000
9 0.249 0.022 7.7 6.87 23.7 0.83 3.96 0.038 6.10 6050
10 0.249 0.022 10.2 9.30 30.5 0.75 3.61 0.037 5.83 6620
В среднем. . 6530
15 0.102 0.000 3.0 2.32 10.2 1.00 4.74 0.035 5.55 4500
13 . 0.010 0.000 3.0 2.32 9.2 0.82 3.90 0.028 4.44 4080
Изменение динамической активности сорбента зависит от концентрации
<серы и других сорбируемых примесей. Зависимость длины зоны массо-
передачи от скорости потока (рис, 8) при постоянной концентрации серы
Рис. 8. Сопоставление уравнения (2)
с экспериментальными данными.
в очищаемом потоке удовлетворительно
описывается эмпирическим уравнением
Lq = - d • by •
Средние значения коэффициента В3,
характеризующего особенности данной
системы, вычисленные по эксперимен-
тальным данным, в присутствии цикло-
пентадиена снизились с 6530 до 5880 при
уменьшении концентрации серы на один
порядок (с 0.25 до 0.015—0.027 вес. %).
В отсутствие ЦПД при прочих равных
условиях значения В3 значительно
меньше (~ на 30%) вследствие улучше-
ния кинетики сорбции основной приме-
си. Пользуясь найденными в лабора-
торных условиях значениями В3 и а8.
можно вычислить зависимость ал от
линейной скорости очищаемого потока и в условиях работы промыш-
ленных адсорберов.
Циклопентадиен (ЦПД), содержащийся в некоторых промышленных
потоках изопентана наряду с серой, также избирательно сорбируется
цеолитами NaX. При соадсорбции ЦПД динамические показатели про-
цесса очистки от сернистых соединений значительно ухудшаются (рис. 9).
Так, в присутствии 0.02% ЦПД динамическая активность сорбента умень-
шается тем больше, чем меньше относительное содержание серы и ЦПД
(рис. 9, 2). Кроме того, вследствие блокировки пористой структуры сор-
бента образующимся дициклопентадиеном возникает необходимость в ре-
гулируемой окислительной регенерации.
272
Десорбцию цеолитов NaX после их насыщения сернистыми соедине-
ниями осуществляли в отсутствие кислорода в десорбирующем агенте
двумя путями:
а) пропусканием перегретого водя-
ного пара через нагретый до 120—150°
сорбент при давлении 5—100 мм рт. ст.
с последующей активацией цеолитов
при 200—350°. С третьего до двадцать
второго цикла сорбции при десорбции
в токе перегретого водяного пара актив-
ность цеолитов сохранялась на одном и
том же уровне при условии постоянства
остаточного содержания влаги в цеоли-
тах. Кинетика десорбции в данном слу-
чае определяется скоростью адсорбции
Рис. 9. Зависимость полной динамиче-
ской активности (а,) цеолитов NaX по
этилмеркаптану от исходной концентра-
ции S.
1 — в отсутствие ЦПД; 2 — в присутствии 0.022
вес. % ЦПД.
Рис. 10. Десорбция этилмеркап-
тана из цеолитов NaX в токе
паров изопентана.
а — выходные кривые десорбции;
б — кривые подъема температуры;
1 — при 200® и скорости гава-вытес-
нителя 160 л/л сорб. час; 2 — при
150® и скорости гава-вытеснителя
80 л/л сорб. час.
сорбированные цеолитами сернистые соединения;
очищенного углеводорода, например, изопентана.
воды, вытесняющей
б) в токе паров
Кинетику десорбции парами изопентана
250° С при разных скоростях газа-вытесни-
теля. По аналитическим данным о содержа-
нии серы в десорбате строили выходные
кривые десорбции в координатах С"=/(р),
где С' — концентрация серы в десорбате,
v — количество углеводорода, затрачен-
ное на десорбцию (рис. 10). По выход-
ным кривым вычисляли зависимость рас-
хода газа-вытеснителя от температуры при
разных скоростях, отображая это графи-
чески (рис. И) в координатах где
v — удельный расход газа (л/л сорбента),
t — температура. В полулогарифмических
исследовали в области 100—
Рис. 11. Зависимость от темпера-
туры удельного (ъ>хл/л) расхода
газа-вытеснителя на десорбцию
сернистых соединений (этилмер-
каптана) из цеолита NaX.
координатах эта зависимость описывается
линейным уравнением
lg V = —0.0045/ + 3.72.
18 Цеолиты
273
Из сопоставления кривых подъема температуры с выходными кривыми
десорбции следует, что для практически полной десорбции сернистых
соединений достаточна температура порядка 200—240°.
Выводы
1. Экспериментально установлена перспективность одновременного
осуществления глубокой осушки и тонкой очистки от сернистых соеди-
нений легкокипящих алканов и циклогексана путем последовательной
адсорбции цеолитами NaA и NaX.
2. Изучены изотермы адсорбции цеолитами СаА и NaX каждого
из индивидуальных углеводородов, подлежащих очистке, а также серни-
стых соединений, подлежащих удалению.
3. По уравнению теории объемного заполнения пор рассчитаны зна-
чения коэффициентов аффинности и других констант, необходимых для
корреляции изотерм адсорбции цеолитами NaX.
4. На примере зтилмеркаптана изучена соадсорбция цеолитами NaX
сернистых соединений с изопентаном из жидкой фазы и определена их
относительная сорбируемость.
5. В динамических условиях изучена соадсорбция этилмеркаптана и
циклопентадиена.
6. Разработаны условия регенерации цеолитов после улавливания ими
сернистых соединений и выведено уравнение для расчета удельного рас-
хода вытеснителя при разных параметрах.
ЛИТЕР А}Т УРА]
1. М. М. Дубинин и др., ДАН СССР. 1961, 138, 4, 866; Изв. АН СССР.^ОХН,
1961, 7, 1183; 8, 1962; 5, 760; 6, 960.
2. В. С. Виноградова, Л. С. Кофман, настоящий сборник, стр. 259.
3. М. М. Д у би нин, 3. А. Жуков а, Н. В. К е л ь ц е в, в сб.: Синтетические
цеолиты, Изд. АН СССР, 1962.]
РАЗДЕЛЕНИЕ АМИЛЕНОВ ИЗО- И НОРМАЛЬНОГО СТРОЕНИЯ
НА СИНТЕТИЧЕСКИХЩЕОЛИТАХ
Н. В. Захарова, В. С. Виноградова, Л. С. Кофман
Разделение углеводородов С5 с одинаковым числом атомов углерода
и равным числом кратных связей, например изо- и нормального строения
или с различным положением кратной связи в молекуле, невозможно
осуществить обычными методами разделения — ректификацией (простой
или с разделяющими агентами), хемосорбцией, а также экстрактивной
кристаллизацией с мочевиной.
При некоторых нефтехимических синтезах, в частности при получении
изопрена, исходят из пентенов изостроения. При этом н-пентены рас-
сматривают, в лучшем случае, как балласт.
В связи с зтим особый интерес вызывает применение синтетических
цеолитов для разделения изомеров алкенов С5.
Для выяснения возможности разделения изомеров амиленов синтети-
ческими цеолитами типа СаА посредством прибора, изображенного на
рис. 1, определили относительную сорбируемость каждой из пар алкенов С5.
274
По экспериментальным данным строили кривые фактического адсорб-
ционного равновесия (рис. 2 и 3) и рассчитывали относительную сорби-
руемость по уравнению
_ У1 • ж2
ар~ у2- х1'
Как видно из приведенных данных, синтетические цеолиты могут быть
применены для разделения не только алкенов С5 изо- и нормального строе-
К термостату
Рис. 1. Схема прибора для изу-
чения относительной сорби-
руемости разделяемых пар
компонентов.
1 — уравнительная склянка со
ртутью; 2 — газовая бюретка;
3 — сосуд с адсорбентом.
Рис. 2. Кривая адсорбцион-
ного равновесия 2-метил-1-
бутен: пентен-1 при 70° С.
ния, но и стереоизомеров 2-пентена. Кривая фазового равновесия стерео-
изомеров 2-пентена пересекает диагональ в точке, соответствующей —50%
yizc-2-пентена, т. е. при пропускании смеси стереоизомеров 2-пентена
Рис. 3. Кривая адсорбционного
равновесия тронс-2-пентен: цис-
2-пентен при 70° С (цеолиты
СаА).
Рис. 4. Зависимость динамической актив-
ности формованных цеолитов СаА до
«проскока» н-пентенов от линейной ско-
рости потока.
через цеолиты СаА рафинат обогащается либо транс-, либо цис-изоме-
ром, в зависимости от состава исходной смеси. Это обстоятельство позво-
ляет понять несоответствие между данными работ [1] и [2].
При контакте алкенов С5 с цеолитами происходит изомеризация ал-
кенов с перемещением двойной связи. Степень изомеризации 1-пентена
в 2-пентены при 70° составляла 26%, при 350° степень изомеризации до-
18*
275.
стигала 69%. Изомеризации З-метил-1-бутена, связанной с более корен-
ной перестройкой структуры, не наблюдалось. При 70° С 2-метил-1-бутен
изомеризуется в 2-метил-2-бутен на 87%; в этих же условиях 2-метил-
2-бутен изомеризуется в 2-метил-1-бутен на 13%.
Эффективность разделения алкенов С5 на синтетических цеолитах
проверяли в динамических условиях (диаметр адсорбера 13 мм, длина
слоя сорбента 270 мм). Для каждого опыта на основании данных хромато-
графического анализа проб рафината строили выходные кривые, по ко-
торым рассчитывали длину зоны массопередачи и динамическую актив-
Рис. 5. Зависимость длины зо-
ны массопередачи от линей-
ной скорости потока.
Рис. 6. Зависимость 1g расхода газа на де-
сорбцию от температуры.
ность сорбента до «проскока» и до «исчерпывания» (в г/100 г и г/см2). За
проскоковую концентрацию принимали содержание н-амиленов в рафи-
нате 0.05%. Длину зоны массопередачи рассчитывали по уравнению,
приведенному в литературе [3].
При 70° и содержании н-амиленов в исходной смеси 5—6% динамиче-
ская активность цеолита до «проскока» составляла 6.2 г/100 г (1.33 г/см2),
при 200° активность снизилась до 3 г/100 (0.64 г/см2). Зависимость динями-
ческой активности цеолитов от линейной скорости потока показана на
рис. 4, а на рис. 5 — зависимость длины зоны массопередачи от линейной
скорости потока.
Регенерацию цеолитов проводили при 350°, в условиях, исключающих
попадание воздуха в систему; газом-элюентом служил бутан или природ-
ный газ. Из рис. 6 видно, что зависимость расхода газа на десорбцию от
температуры явлйется практически линейной.
Пользуясь приведенными данными, можно рассчитать размеры ад-
сорберов при заданных линейных скоростях и динамической активности
цеолитов.
При циклической работе цеолита в течение 22 циклов динамическая
активность до «проскока» при 70° стабильно сохранялась на уровне
5.6 г/100 г. Количество образовавшихся димеров изоамиленов не превы-
шало 0.02%.
ЛИТЕ РАТУРА
1. В. С. В в но г р а до в а, Л. С. К о ф м а н, в сб.: Синтетические цеолиты, Изд.
АН СССР, 1962, 245.
2. USA Pat. 2850549, 1958.
3. Н. Barry, Chem. Engng., 1960, 3, 105.
ПРОЦЕСС ОЧИСТКИ ИЗОБУТИЛЕНА АДСОРБЦИЕЙ
СИНТЕТИЧЕСКИМИ ЦЕОЛИТАМИ
В. С. Виноградова, Л. О. Димент, В. Н. Комарова, Т. А. Маркова,
Л. С. Кофман
Изобутилен относится к числу углеводородов, широко применяемых
в производстве высокополимеров, особенно бутилкаучука, обладающего
весьма ценными техническими свойст-
вами: химической инертностью, высокой
термостабильностью, близкой к нулю
электропроводностью, способностью к са-
мозатягиванию порезов и т. п. Не слу-
чайно выпуск бутилкаучука за рубежом
неуклонно возрастает: в Канаде выработ-
ка его увеличилась с 1950 по 1960 г. в
2.5 раза [1], а в США составила в 1963 г.
120 тыс. т, что на 30% больше, чем в
1962 г. [2].
Однако указанные специфические свой-
ства бутилкаучука достигаются лишь при
отсутствии в изобутилене-мономере при-
месей бутенов нормального строения, осо-
бенно 1-бутена. В то же время экстракция
50—60-процентной серной кислотой, яв-
ляющаяся фактически единственным мето-
дом извлечения изобутилена из промыш-
ленных фракций С4, не обеспечивает долж-
ную чистоту мономера, в котором остает-
ся некоторое количество нормальных бу-
Рис. 1. Изотермы адсорбции ал-
кенов С4 на цеолитах СаА при
20° С.
1 — цис-2-бут&г, 2 — 1-бутен; 3 —
трсшс-2-бутен.
тенов. Даже при больших энергетических
затратах возможно очистить изобутилен четкой ректификацией только
от изомеров 2-бутена, а 1-бутен, относительная летучесть которого близ-
[TlP Ps/p]2'10'"
Рис. 2. Изотермы адсорбции ал-
кенов С4 на цеолитах СаА в ко-
ординатах уравнения
lg a — [TlgPg/p]2.
1 — uuc-2-бутен; 2 — 1-бутен; 3 —
тпранс-2-бутен.
ка к единице, переходит в дистиллят
вместе с целевым продуктом.
В связи с изложенным нами были про-
ведены исследования по тонкой очистке
технического изобутилена формованиями
цеолитами СаА.
Предварительно на вакуумной адсорб-
ционной установке исследовали изотермы
адсорбции (рис. 1) каждого из индиви-
дуальных алкенов С4 промышленными 13]
образцами формованных цеолитов СаА,
активированными при 350° С и остаточ-
ном давлении 10-3 мм рт. ст. Полученные
данные коррелировали по уравнению для
адсорбентов 1-го структурного типа [4].
Экспериментальные точки изотерм адсорб-
ции каждого из изомеров н-бутенов груп-
пировались около соответствующих пря-
мых (рис. 2), что подтверждает принци-
пиальную возможность применения урав-
нений теории объемного заполнения пор
для описания изотерм адсорбции цеолитами СаА и непредельных угле-
водородов.
277
Таблица 1
Значение констант уравнения адсорбции цеолитами СаА бутенов
нормального строения
Наименование углеводорода Wo ₽эксп ₽ = Р/Ро Степень отклонения
1-Бутен 197 3.78 2.33 1.68
транс-2-Бутен 188 2.84 2.33 1.22
qwc-2-Бутен 190 4.40 2.33 1.88
По построенным в координатах 1g а—(lg pjp)2 изотермам определили
(табл. 1) предельные адсорбционные объемы (РЙ0) и коэффициенты аффин-
ности (Р). В качестве стандартного вещества приняли азот, при значении
константы В, равном 4.95 • 10-6 [4]. Значения Wo, рассчитанные по изо-
термам сорбции н-бутенов и
Рис. 3. Изотермы адсорбции цео-
литами СаА при 100° С.
1 — расчетная изотерма ituc-2-бутена;
2 — расчетная изотерма 1-бутена;
3 — расчетная изотерма тпранс-2-бутена;
4 — изотерма смеси н-бутенов. По кри-
вой 4 крестики — точки, рассчитан-
ные по аддитивности с учетом процен-
та изомеризации, треугольники —
экспериментальные точки.
определенные другими исследователями по
изотермам аполярных веществ [5], прак-
тически совпадают.
Полученные экспериментальные дан-
ные были распространены в соответствии
с теорией объемного заполнения пор на
область более высоких температур (до 100—
150°), при которых применение сорбента
в промышленности более рационально.
Сорбция н-бутенов при повышенной тем-
пературе (100° и выше) сопровождается
изомеризацией (перемещением кратной
связи) бутенов. Так, при 100° после кон-
такта с цеолитами 1-бутена в сорбате со-
держится— 14.5% транс-2-бутена и 1.5%
i/.uc-2-бутена. Поэтому экспериментальная
изотерма отвечает фактическому составу
сорбата, а не собственно ‘1-бутена. Это
подтверждается совпадением эксперимен-
тальных точек изотермы 1-бутена при 100°
с точками, рассчитанными по аддитив-
ности с учетом изменения состава сор-
бированной фазы (рис. 3, 4).
Все изомеры н-бутена хорошо, хотя и
в различной степени, сорбируются цео-
литами СаА, что создает возможность отделения их от изобутилена,
используя молекулярно-ситовой эффект. Различия в адсорбции инди-
видуальных изомеров н-бутена могут быть использованы для разделения
их между собой.
Так, при изучении относительной сорбируемости стереоизомеров най-
дено, что более сорбируемый в чистом виде ywc-изомер становится менее
сорбируемым из смеси в довольно широком интервале концентраций.
Относительная сорбируемость шранс-2-бутен : г/ис-2-бутен в изученной
области состава практически постоянна и равна ~ 3.5. По аналогии
с расчетом числа теоретических тарелок (Т. Т.) при ректификации про-
веден графоаналитический расчет числа теоретических ступеней адсорбции
(Т. С. А.). При содержании в исходной смеси 10% транс-изомера выде-
ление унс-2-бутена концентрацией 99.9% может быть достигнуто в ад-
сорбере непрерывного действия эффективностью три Т. С. А. Возможность
278
выделения чистого унс-2-бутена из смеси стереоизомеров была подтвер-
ждена на лабораторной установке в стационарном слое формованных
цеолитов СаА в динамических условиях.
Динамические показатели процесса очистки технического изобутилена
от бутенов нормального строения были подробно изучены на лаборатор-
ных установках (рис. 4). Изобутилен в паровой или жидкой фазе пропу-
Рис. 4. Схема лаборатррной установки для адсорбцион-
ной очистки изобутилена.
1 — баллон; 2 — ротаметр; з — форсорбер; 4 — адсорбер; 5 —
приемник полимеров; 6 — реометр; 7 — градуированные прием-
ники.
скали со скоростью, устанавливаемой по реометру, последовательно через
форсорбер с цеолитом NaA и адсорбер с цеолитом СаА, снизу вверх. Хро-
матографическим анализом проб рафината, т. е. очищенного изобутилена,
отбираемых через каждые 10 мин., определяли содержание н-бутенов
с точностью до 0.05 вес. %. При этом было установлено, что изобутилен
липть незначительно (0.5 г/100 г при 600 мм и 20° С) сорбируется формован-
ными с глуховской глиной цеолитами СаА, но при контакте с последними
частично полимеризуется, образуя олигомеры, преимущественно димеры.
Наибольшая паразитарная каталитическая активность была обнару-
жена (табл. 2) у цеолитов, сформованных посредством латненской и .глу-
ховской глин. При контакте с кристаллитами СаА, спресованными в гра-
Таблица 2
Выход полимеров изобутилена на различных образцах цеолитов СаА
Наименование образца Выход полимеров изобутилена в вес. % к пропущенному изобутилену при циклах
1 2 3
СаА ГрозНИИ с латненской глиной СаА-171 ГОБ ВНИИ НН с 18% Глуховской 58.0 55.0 —
Г JIИ НЬТ СаА тамбовский с 15% оглонлинского бенто- 37.0 10.2 5.5
нита 10.9 2.6 0.9
СаА тамбовский с 15% нросяновского каолина СаА харьковский с 14% А12О3 в виде оксини- 0.1 1.0 —
трата 0.0 0.0 0.0
СаА ГОБ без связующего 0.0 0.0 0.0
NaA ГОБ с 18% глуховской глины 0.0 0.0 0.0
279
пулы без применения связующего и посредством оксинитрата алюминия
или с просяновским каолином, а также при контакте с цеолитами NaA,
сформованными с применением любой из исследованных глин, полимери-
зация изобутилена практически не происходила. Образования димеров
не наблюдалось и после частичной дезактивности цеолитов СаА, сформо-
ванных с глуховской глиной, если прини-
Рис.' 5. Дифференциальная кри-
вая образования полимеров в те-
чение одного цикла сорбции.
мались меры, полностью предотвращаю-
щие контакт нагретых гранул цеолитов
с кислородом.
Таким образом, можно полагать, что
димеризация изобутилена протекает при
многократном контакте со связующим
наиболее активных молекул его, проникаю-
щих в пористую структуру цеолитов и
тут же вытесняемых из нее молекулами
н-бутенов. Паразитарная каталитическая
активность связующего определяется его
природой и, очевидно, стимулируется хемо-
сорбцией кислорода активированной по-
верхностью глины. Каталитическая актив-
ность глуховской глины быстро умень-
шается и в течение одного и того же цикла
адсорбции изобутилена (рис. 5), что можно
объяснить блокированием активных цен-
тров связующего образовавшимися олиго-
мерами.
На дезактивированных одним из способов цеолитах СаА были иссле-
дованы основные параметры циклического процесса сорбции и десорбции
при изменении концентрации н-бутенов от 0.5 до 3% в интервале давления
до 5—6 атм. и скоростей потока изобутилена 0.01—0.13 м/сек. По выход-
ным кривым вычислили значения длины зоны массопередачи [6]. Полу-
ченные результаты изменения длины зоны массопередачи в зависимости
от динамической активности и температуры представлены в табл. 3, а на
Таблица 3
Влияние температуры адсорбции на динамическую активность цеолитов СаА
по «-бутенам (при длине слоя адсорбента 27 см)
№ цикла Состав исход- ной смеси, вес. 7о О о иас- м Динамическая активность А ° S со ее м г. Динамический коэф- фициент, М = — а
скорость потока газа, м/сек.
до «проскока» (Од) полная (а,)
Т5 а- с § Длина зоны : s" -б а о итатичеикан ТИВИОСТЬ по I терме, г/100 I
1-бу- тен изобу- тилен г/см’ г/100 т/см3 г/100 г
3 2.2 97.8 !0 0.018 10.4 1.27 6.72 0.058 8.28 10.2 0.81
5 2.3 97.7 20 0.017 10.6 1.26 6.67 0.058 8.28 10.2 0.81
6 2.3 97.7 !0 0.015 12.5 1.23 6.52 0.061 8.70 10.2 0.85
В среднем • • • 1.25 6.62 0.059 8.40 10.2 0.82
2 22 97.8 100 0.023 10.9 0.89 4.71 0.041 5.85 7.1 0.82
3 . 1.1 98.9 100 0.023 9.6 0.80 4.23 0.040 5.72 6.8 0.84
В среднем • - • 0.85 4.50 0.040 5.78 — 0.83
280
рис. 6 показана зависимость динамической активности до «проскока»
н-бутена (проскоковая концентрация 0.05%) от линейной скорости по-
тока. Зависимости длины зоны массопередачи от линейной скорости
потока скоррелированы по эмпири-
ческому уравнению
Lq Ва • d • Via,
применяемому для расчета длины
работающего слоя активированных
углей. Значения коэффициента Ба,
вычисленные по экспериментальным
данным для различных скоростей
очищаемого потока, отличаются от
среднего значения не более чем на
+5—7% (табл. 4), что позволяет рас-
считать динамические показатели
сорбции на цеолитах при переходе
от адсорберов лабораторных разме-
ров к промышленным. Это было под-
тверждено при опытных работах на
Рис. 6. Зависимость динамической
активности до «проскока» от линейной
скорости потока (для слоя сорбента
£=50 см).
пилотной установке с адсорберами
полезной емкостью 5 л сорбента. Сте-
пень использования статической ак-
тивности (М), т. е. отношение aja
(табл. 3), равна ~ 0.83, независимо от
температуры сорбции и линейной скорости. Отличие между а и as, по-
видимому, обусловлено соадсорбцией незначительных количеств изобу-
тилена, сернистых соединений и других неидентифицированных сорби-
руемых’ примесей.
Как указывалось выше, при регенерации цеолитов и десорбции из
них бутенов необходимо избегать присутствия в газе-вытеснителе даже
небольших 0.2% примесей кислорода. Нами было изучено несколько
вариантов регенерации цеолитов.
ь Таблица4
Влияние линейной скорости разделяемого потока на динамические
показатели процесса при 20° С
Скорость очищаемого потока Длина слоя сорбента, см Динамическая активность до «проскока» (ад) Длина работаю- щего СЛОЯ, см Полная дина- мическая актив- ность (Cg) Значение констан- ты В3
л/л час (по жид- ким угле- водородам) м/сек. (по парам) г/см1 г/100 г г/см’ г/100 г
0.5 0.010 25 1.27 7.25 9.6 0.059 8.40 640
1.2 0.024 25 1.03 5.90 15.8 0.060 8.56 660
2.0 0.040 25 0.85 4.85 21.6 0.058 8.28 721
3.0 0.060 25 0.73 4.17 26.0 0.060 8.56 693
5.0 6.4 0.105 0.128 25 25 0.30 0.17 1.72 0.97 1 Не определены.
1.5 0.060 50 2.22 6.35 27.1 0.061 8.70 720
2.5 0.105 50 1.98 5.65 31.5 0.059 8.40 730
3.2 0.128 50 1.89 5.40 37.2 0.060 8.56 677
В среднем . . . 0.059 8.40 691
281
При регенерации цеолитов перегретым до 120—150° водяным паром
в вакууме после собственно десорбции осуществляли активацию сорбента
при 250—350° С при том же давлении. При увеличении остаточного со-
держания воды в цеолитах на 1 % активность сорбента по н-бутенам сни-
жается более чем на 3 г/100 г, т. е. почти вдвое. Одновременно увеличи-
вается и длина зоны массопередачи. Поэтому при активации цеолитов
после десорбции водяным паром температура должна быть не ниже 300°.
В результате изучения кинетики процесса десорбции в токе инертного
газа установлено, что полная десорбция 1-бутена из цеолитов СаА может
быть достигнута практически при лю-
бой температуре. Кинетику десорбции
исследовали в области температур
20—300° при разных объемных скоро-
стях потока газа-вытеснителя. По ана-
литическим данным строили выход-
v, л/л сорбента
Рис. 8. Зависимость удельного расхода
газа на десорбцию и-бутеновой темпе-
ратуры десорбции.
Рис. 7. Выходные кривые < десорбции
в токе пропана.
1 — 160»; 2 — 200°; 3 — 260°; 4, 5, в — кри-
вые подъема температуры.
ные кривые десорбции в координатах С'1=/(у), где v — удельный расход
газа (л/л сорбента), С1 — концентрация бутенов в газе (рис. 7), и вычис-
ляли графическим интегрированием приближенный расход газа-вытесни-
теля на десорбцию при заданной температуре.
В результате проведенных работ показано, что зависимость удельного
расхода газа (г, л/л) и его скорости (со, л/л • час) от температуры (<) де-
сорбции может быть выражена (рис. 8) в координатах lg(p)=<p(O линей-
ными уравнениями
lg v = —0.006624-|-3.98, v =
где т — продолжительность десорбции.
Максимумы концентрационных пиков соответствуют температуре
240—260°, которую и следует считать максимально необходимой для за-
вершения десорбции с наименьшим расходом газа-вытеснителя.
Совокупность полученных данных позволяет осуществить проекти-
рование опытных установок тонкой очистки изобутилена адсорбцией
формованными цеолитами СаА [7].
Выводы
1. Разработан процесс тонкой очистки изобутилена от примесей бу-
тиленов нормального строения, основанный на молекулярно-ситовом эф-
фекте синтетических цеолитов СаА.
2. На примере алкенов С4 показано, что адсорбция непредельных
углеводородов цеолитами СаА может быть описана уравнениями теории
282
объемного заполнения пор при условии учета изменения состава вслед-
ствие изомеризации.
3. Определены значения предельного адсорбционного объема и коэф-
фициентов аффинности цеолитов СаА каждого из изомеров н-бутена.
4. Показана возможность разделения стереоизомеров 2-бутена по-
средством цеолитов СаА.
5. Определены причины образования олигомеров изобутилена при
контакте с формованными цеолитами СаА и обсуждены пути подавления
паразитарной каталитической активности сорбента.
6. Найдены оптимальные условия десорбции и-бутенов из цеолитов
и регенерации сорбента и выведено уравнение для расчета удельного
расхода газа-вытеснителя при разных условиях.
ЛИТЕРАТУРА
1. Химическаяи резиновая промышленность в капиталистических странах.
НИИТЭХИМ, 1962.
2. Ch е m. a. Engng. News, 1963, 41, 44, 25.
3. В Т У на синтетические цеолиты типа А и X, № МРТУ-6-01-567-63.
4. М. М. Д у би н и н, 3. А. Ж у к о в а, Н. В. К е л ь ц е в, в сб.: Синтетические
цеолиты, Изд. АН СССР, 1962, 7.
5. 3. А. Жукова, Н. В. К е л ь ц е в. Разделение и анализ углеводородных
газов. М., 1963, 134.
6. В. С. Виноградова, Л. С. К о ф м а и, настоящий сборник, стр. 259.
7. Авторское свидетельство 150108, 1961.
ПРИМЕНЕНИЕ СИНТЕТИЧЕСКИХ ЦЕОЛИТОВ
ДЛЯ ИЗВЛЕЧЕНИЯ АЦЕТОНИТРИЛА ИЗ ЕГО СМЕСЕЙ
С УГЛЕВОДОРОДАМИ С4
Т. А. Маркова, В. С. Виноградова, Л. С. Кофман
При разделении продуктов дегидрирования алканов экстрактивной
ректификацией с ацетонитрилом (АН) с дистиллятами уносится некоторое
количество ацетонитрила. Так как дистилляты (например, бутан) возвра-
щают на дегидрирование, ацетонитрил необходимо рекуперировать. ,
На адсорбционной вакуумной установке были определены изотермы
адсорбции АН формованными цеолитами NaA-124 в интервале температур
20—200° и двумя образцами цеолита КА при 20° (рис. 1).
Очистку бутана от АН цеолитами NaA в динамических условиях
осуществляли при 20° в жидкой фазе и давлении 4—5 атм. Для каждого
опыта на основании данных хроматографического анализа проб рафината
строили выходные кривые (рис. 2), по которым рассчитывали длину зоны
массопередачи [1] и динамическую активность до «проскока» и до «исчер-
пывания» [2]. За проскоковую концентрацию принимали содержание АН
в рафинате 0.05 вес. %. При содержании АН в исходной смеси 2.0—
3.5 вес. % и скорости жидких углеводородов 0.02 см/сек. динамическая
активность цеолита NaA составляла: до «проскока» (пд) 10.0—12.0 г/100 г,
полная (а8) — 0.152—0.170 г/см3 (21.8—24.2 г/100 г) при регенерации цео-
литов в токе инертного газа в условиях циклической работы.
Кинетические показатели десорбции АН при разных объемных скоро-
стях потока осушенного газа (азот, бутан) изучали в области температур
100—300°. По данным хроматографического анализа газов десорбции
строили кривые (рис. 3) зависимости концентрации АН в газе десорбции
283
от объема пропущенного газа. Графическим интегрированием находили
количество газа, необходимое для регенерации цеолитов при заданной
температуре. Зависимость логарифма
расхода газа (и) на десорбцию АН от
температуры (t) выражается прямой ли-
нией (рис. 4), описываемой уравнением
1g р = —0.0462*+ 4.5.
Рис. 2. Выходная кривая при очистке
бутана от АН цеолитом NaA.
Линейная скорость жидкости—0.02см/сек.,
давление — 5 атм., температура — 20° С.
На оси ор&инат — концентрация АН в бу-
тане; на оси абсцисс — количество жидко-
сти, пропущенной через слой цеолита (мл).
Со — начальная концентрация АН.
р,мм рт.ст.
Рис. 1. Изотермы адсорбции
ацетонитрила формованными
цеолитами.
1, 2, 3 — NaA при 20, 100 и 250°С,
соответственно; 4, 5 — КА (226 и
548) при 20° С.
После десорбции парами бутан-бутиленовой смеси с 2—3% дивинила,
полимеризующегося в порах сорбента, активность формованных цеолитов
Рис. 3. Десорбция АН из цеолитов
NaA в токе бутана.
1 — выходная кривая десорбции; 2 — кри-
вая подъема температуры..
NaA снизилась за 7 циклов сорб-
ции—десорбции с 16.5 до 3.5%.
При регенерации цеолитов в токе
перегретого до 120—150° водяного
пара в сочетании с вакуумом или па-
ровоздушной смесью было рекупери-
ровано — 80% от адсорбированного
количества ацетонитрила в виде
Рис. 4. Зависимость удельного (i>, л/л)
расхода газа-вытеснителя на десорб-
цию АН из цеолита NaA от темпера-
туры.
75 %-го водного раствора. В последних двух случаях после собственно
десорбции осуществляли активацию сорбента при 350° и давлении
284
5 мм рт. ст. Полная динамическая активность по парам АН при последую-
щих циклах сорбции устойчиво сохранялась на уровне 15—17 г/100 г.
Наиболее полная рекуперация АН была достигнута при десорбции
в парах углеводородов с температурой кипения выше 125°. Так, при де-
сорбции в токе паров ксилола практически полная рекуперация АН до-
стигается при удельном расходе ксилола 7.0—17.0 г/г сорбента при тем-
пературе 300—200° соответственно; удельный расход ксилола при реку-
перации 95% АН составляет 3.0—7.0 г/г сорбента. Последующее выделе-
ние АН из его 2—3%-го раствора в ксилоле может быть осуществлено
без особых затруднений обычной ректификацией, так как относительная
летучесть пары АН—л-ксилол приближается к десяти.
ЛИТЕРАТУРА
1. R. Treybal. Mass Transfer Operations. McGrow Hill, N. Y., 1955.
2. В. С. Виноградова, Л. С. Кофман, настоящий сборник, стр. 259.
ИССЛЕДОВАНИЕ ТЕРМОСТАБИЛЬНОСТИ СИНТЕТИЧЕСКИХ
ЦЕОЛИТОВ
В. С. Виноградова, Л. С. Кофман, Б. А. Липкинд, Л. М. Медведева,
А. Т. Слепнева
Обычно сорбционные процессы осуществляют циклически, причем
на стадии десорбции-регенерации прибегают к повышению температуры,
как правило, тем большему, чем селективнее действие сорбента.
Синтетические цеолиты отличаются от других мелкопористых сорбен-
тов исключительно высокой сорбционной способностью при малых кон-
центрациях сорбируемых веществ. Это свойство, являясь весьма положи-
тельным фактором на стадии адсорбции, затрудняет десорбцию, для пол-
ного осуществления которой обычно требуется высокая температура.
При контакте формованных цеолитов с углеводородами, в которых
присутствуют, на первый взгляд, незначительные количества легко поли-
меризирующихся веществ, в условиях циклической работы постепенно на-
капливаются отложения полимеров. Вследствие этого постепенно ухуд-
шаются кинетические показатели сорбции и активность цеолитов р'езко
снижается. В таких случаях неизбежна окислительная регенерация при
температуре порядка 400—500°; в результате местных перегревов возмо-
жен подъем температуры и выше 500°.
Таким образом, вопрос о стабильности свойств цеолитов при длитель-
ном воздействии умеренно высокой температуры весьма актуален, по-
скольку срок службы цеолитов в условиях циклической работы является
одним из решающих при оценке экономичности сорбционных процессов.
Между тем в литературе содержатся лишь общие указания о температур-
ном пределе устойчивости цеолитов — либо при весьма ограниченной про-
должительности нагрева, либо вообще без указаний о продолжительности.
Например, сообщалось, что при 800° кристаллическая структура цеоли-
тов NaA и СаА разрушается менее чем за 2 часа, а при 700° не изменяется
в течение 6 часов [1].
По данным [2], температура 600—650° выдерживается цеолитом, од-
нако если регенерацию цеолитов осуществлять при этой температуре,
их адсорбционные свойства ухудшаются через относительно небольшое
число циклов. В то же время, если осуществляют регенерацию при тём-
285
пературе не выше 315° С, срок службы их превышает 2 тыс. циклов, при-
чем уменьшение активности (на 30%) наблюдается только в течение пер-
вых 200 циклов.
В связи с этим при разработке ряда процессов очистки и разделения
углеводородов цеолитами СаА особое внимание было обращено на их тер-
мическую стабильность при ци-
клической работе. При этом
было замечено, что активность
некоторых образцов за первые
10—20 циклов уменьшалась бо-
лее чем в 2 раза, хотя в тех же
условиях активность ряда дру-
гих образцов цеолитов того же
типа сохранялась практически
на одном уровне.
На рис. 1—3 сопоставлены
20\______।______।_______।______।------1— изменения активности в резуль-
0 200 600 В00 800 1000 тате нагрева при 500° 4 образцов
Продолжительность термообработки,часы цеолитов СаА, два из которых
Рис. 1. Изменение динамической активности
до «проскока» в зависимости от продолжи-
• тельности термообработки.
Образцы: 1 — 171, 2 117, 3 — Линде 5А Кг 1, 4 —
Линде 5А Кг 2.
получены в опытно-промышлен-
ных условиях и два — образцы
фирмы Линде.
Динамическая активность
образца 171 после 1000-часовой
обработки снизилась на 20%.
Образец 117 после 200 часов
нагрева потерял около 55% первоначальной активности до «проскока»,
а полная динамическая активность упала более чем на 75% от исходной.
Изученные образцы цеолитов СаА фирмы Линде занимали промежуточное
положение, несколько различаясь между собой по термической стабиль-
ности.
В связи с вышеизложенным
представлялось целесообразным
изучить влияние на термиче-
скую стабильность режимов всех
стадий приготовления формо-
ванных цеолитов СаА.
В настоящем сообщении при-
водятся результаты исследова-
ния влияния некоторых усло-
вий процесса производства цео-
литов СаА на их термическую
стабильность, в частности изу-
чали влияние: продолжитель-
ности кристаллизации, степени
катионного обмена, глубины
отмывки натриевого кристал-
лита, температуры и продолжи-
и^г[100г
10.0 F
4^1--------1------1--------1-------1-------1—
0 200 W0 Б00 800 1000
Продолжительность термообработки, часы
Рис. 2. Изменение полной динамической ак-
тивности от продолжительности термообра-
ботки.
Образцы: 1 —-171, 2 — 117.
тельности прокалки.
Активность цеолитов оценивали в соответствии со стандартной мето-
дикой [3], основанной на определении динамических показателей процесса
разделения смеси изопентан-н,-пентан в паровой фазе. На основе построен-
ных по данным анализа рафината выходных кривых рассчитывали дина-
мическую активность данного слоя L сорбента до «проскока» (ад г/см2 и
г/100 г) и длину зоны массопередачи.
286
Для расчета значений Lo воспользовались известным уравнением [4],
подставив взамен времени защитного действия соответствующие ему
Рис.
от
Рис. 4. Выходная 'кривая при’ра-
боте сорбента на исчерпывание.
3. Изменение длины зоны массопередачи
продолжительности термообработки,
1 — 171; 2 — 117, 3 — Линде 5А К» 1 и Ks 2.
объемы рафината, собранного до «проскока» (гБ) и до «исчерпывания» (fE):
РЕ РБ
L°~L "е -(l-Д (^-"н) ’ (П
где / — доля площади под выходной
кривой на участке нарастания концен-
трации, отнесенная ко всей площади
этого участка (рис. 4).
Поскольку все опыты осуществляли
в стандартных условиях, найденные
значения ал и Lo вполне сопоставимы.
Кроме того, определяли полную дина-
мическую активность (а„ г/см3 или
г/100 г), значение которой постоянно
практически при любой длине слоя сор-
бента и мало зависит от скорости по-
тока.
Расчет значений а, производили по
уравнению (2):
“л
“» = £ — £0/ ’ <2)
соответствие которого эксперименталь-
ным данным было нами подтверждено.
Для определения термостабильно-
сти цеолитов применили ускоренную
Рис. 5. Зависимость динамической
активности цеолитов СаА от содержа-
ния в них СаО.
До «проскока»: 1 — исходный цеолит,
2 — после 168-часового нагрева; полная:
3 — исходный пеолит; 4 — после 168-ча-
методику, заключающуюся В продол- сового нагрева.
жительном нагреве образцов сорбента
при 500° в муфельной печи с автоматической регулировкой и непрерывной
регистрацией температуры посредством прибора ЭПД-12. Через опреде-
ленные промежутки времени отбирали пробы испытуемых образцов
цеолитов и определяли их молекулярно-ситовые свойства, как указано
выше.
287
Из рассмотрения фактического материала, приведенного в табл. 1—4
и на рис. 5 и 6, вытекает следующее.
1. Продолжительность кристаллизации 12 часов вполне достаточна
для сохранения стабильности цеолитов. Дальнейшее увеличение продол-
жительности кристаллизации не изменяет качества цеолита (табл. 1).
2. Степень катионного обмена является одним из факторов, резко
сказывающихся на термостабильности синтетических цеолитов СаА
Рис. 6. IЗависимость динамической активно-
сти .до «проскока» от продолжительности
термообработки.
Образцы после первичного прокаливания при 600° Cs
1 — 4 часа; 2 — 24 часа.
Цеолиты, содержащие от 10.5 до 13%
бильными; длина зоны массопередачи и
(табл. 2). Цеолиты, содержащие
8% СаО, характеризуются в три
раза большей, чем стандартные
цеолиты, длиной зоны массо-
передачи и пониженным значе-
нием полной динамической ак-
тивности сорбента. Уже после
168-часового нагрева при 500°
полная динамическая актив-
ность уменьшается на 20—25 %.
То же относится и к образ-
цам с высоким содержанием
СаО, которые уже в начальный
момент характеризуются не-
сколько пониженным значением
активности до «проскока», а в
результате 168-часового нагрева
при 500° теряют 35% первона-
чальной активности.
СаО, являются вполне термоста-
t наименьшая и практически не
изменяется, так же как и динамическая активность до «проскока» и полная,
даже после более чем 500-часовой выдержки при 500° (табл. 2 и 4).
3. Глубина отмывки кристаллита в широком интервале расхода про-
мывной воды (4—100 т/т) не оказывает влияния ни на исходную динами-
ческую активность цеолита, ни на его термическую стабильность. Только
при очень низком расходе промывной воды (1 т/т) отмечено некоторое
уменьшение термической стабильности цеолита (табл. 3).
4. Увеличение длительности первичного прокаливания при температуре
600° С с 6 до 24 часов не оказывает влияния на термическую стабильность
цеолита (табл. 4 и рис. 6).
На основании результатов проведенного исследования были подобраны
оптимальные режимы на всех стадиях процесса приготовления формован-
Таблица 1
Влияние продолжительности кристаллизации на стабильность цеолита
(содержание СаО —12.0%)
Продолжи- тельность кристаллиза- ции, часы Продолжи- тельность нагрева, часы см а по н-пентану А а8 по н-пентану а по парам А ВОДЫ, Г/100 г
г/см’ г/100 г г/см3 г/100 г
12 9.0 1.25 7.1 0.053 8.5 14.9
12 168 8.8 1.29 7.2 0.054 8.6 12.4
24 — 7.0 1.15 6.4 0.048 7.3 15.0
24 168 6.4 1.20 6.7 0.045 7.2 13.1
48 — 9.2 1.18 6.3 0.051 7.5 15.1
48 168 6.2 1.40 6.9 0.056 8.2 9.3
288
Таблица 2
Влияние степени ионного обмена на термостабильность цеолита
№ об- разца Содер- жание СаО, вес. % Продол- житель- ность нагрева, часы LOi см по м-пентану а8 по н-пентану °д по парам воды, г/100 г
г/см2 г/100 г г/см3 г/100 г
788 | 8.2 32.6 0.57 3.2 0.046 7.3 15.3
8.2 168 31.0 0.43 2.5 0.036 5.7 14.0
791 | 10.9 9.4 1.17 6.9 0.049 7.8 13.8
10.9 168 9.4 1.16 6.7 0.048 7.7 14.8
792 | 13.1 12.5 1.27 6.9 0.058 8.6 13.0
13.1 168 7.2 1.37 7.3 0.054 8.2 12.0
793 { 17.6 . . 16.4 1.15 6.2 0.056 8.9 10.2
17.6 168 17.3 0.79 4.2 0.040 5.9 10.7
Таблица 3
Влияние глубины отмывки на термическую стабильность цеолита
(содержание СаО = 12.0%)
№ об- разца Расход воды, т/т Продол- житель- ность нагрева, часы I-а, см ад по м-пентану а8 по н-пентану а по Д парам воды, г/100 г
г/см2 г/100 г г/см3 г/100 г
788А | 1 9.6 1.16 6.2 0.049 7.0 14.1
1 168 11.5 1.08 5.9 0.047 7.2 13.9
788Б | 4 7.8 1.28 7.1 0.052 7.8 15.0
4 168 7.4 1.32 7.4 0.052 8.1 15.4
625 | 8 10.4 1.16 6.1 0.050 7.5 —
8 168 11.6 1.25 6.3 0.058 8.4 —
100 13.0 1.18 6.1 0.054 7.9 —
627 | 100 168 11.0 1.32 7.2 0.058 8.4 —
Таблица 4
Влияние температуры и длительности первичного прокаливания
на термостабильность цеолита (содержание СаО = 11.7%)
ЛЬ об- t разца Условия прокаливания LOt см Продол- житель- ность нагрева, часы по м-пентану а9 по м-пен- тану д по парам воды, г/100 г
t, °C продол- житель- ность, часы г/см2 г/100 г г/см3 г/100 г
13.8 1.45 7.7 0.063 9.3 14.1
13.6 168 1.37 7.6 0.063 9.0 13.3
634 600 6 12.0 340 1.29 7.1 0.056 9.0 —
10.9 484 1.38 7.5 0.059 8.9 —
23.0 680 0.87 4.6 0.053 8.1 —
11.4 — 1.35 7.0 0.058 8.7 11.7
10.1 168 _ 1.16 5.9 0.050 7.3 11.6
635 600 24 13.6 340 1.13 6.0 0.052 7.5 —
14.8 536 1.12 6.0 0.049 7.3 —
32.0 880 0.061 3.2 0.047 6.5 —
19 Цеолиты
289
ного цеолита СаА. Приготовленные при этих условиях образцы имеют
достаточно высокие и хорошо воспроизводимые показатели по первона-
чальной адсорбционной активности и термической стабильности.
ЛИТЕРАТУРА
1. D. W. В г е с k, W. G. Eversole, R. М. М i 11 о n, J. Am. Chem. Soc., 1956,
78, 23, 5963.
2. Ch. К. Hersh. Molecular Sieves. Reinhold Publ. Co., N. Y., London, 1961.
3. В T У на синтетические цеолиты типа А и X, № МРТУ-6-01-567-63.
4. R. Т г е у b а 1. Mass Transfer Operations. McGrow Hill, N. Y., 1955.
5. В. С. В и н о г p а д о в а, Л. С. Кофман, настоящий сборник, стр. 259.
РАЗДЕЛЕНИЕ УГЛЕВОДОРОДОВ В ПСЕВДООЖИЖЕННОМ СЛОЕ
ЦЕОЛИТА СаА
Н. С. Корнилова, И. Н. Морина, Н. А. Нечаева
С целью разработки метода очистки изобутилена от примесей н-бути-
ленов с применением псевдоожиженного слоя мелкодисперсного цеолита
была создана опытная установка, схема которой приведена на рис. 1.
Адсорбер представляет собой полый цилиндрический аппарат диаметром
Рис. 1. Схема опытной установки для очистки изобутилена в псевдоожи-
женном слое цеолита СаА (стадия адсорбции).
1 — баллон с изобутиленом; 2 — испаритель; 3 — ротаметр; 4 — форсорбер; в — гильза
для термометра; в — бункер; 7 — адсорбер; 8 — циклон; 9 — конденсатор; 10 — сбор-
ник; 11 — пробоотборник.
70 мм, высотой 1.5 м, с расширителем в верхней части и распределитель-
ной решеткой — в нижней. Десорбция н-бутиленов проводилась азотом
при температуре 350° С в аппарате, аналогичном адсорберу.
Исходное сырье содержало 2.5—^2.7 вес. % а-бутилена, 0.4—0.5%
лгранс-Р-бутилена и в некоторых случаях до 0.1% цис-fi бутилена. Ад-
сорбция проводилась при температуре 30—50° С,: весовой скорости по-
дачи сырья 0.8—0.9 час.-1, линейной скорости сырья в свободном сечении
290
адсорбера 0.05—0.07 м/сек. и высоте псевдоожиженного слоя цеолита
около 1 м. Опыты проводились на дробленом цеолите СаА [1]. Использо-
валась фракция цеолита 0.16—0.63 мм с эквивалентным диаметром час-
тиц 0.418 мм. Было проведено 12 циклов адсорбции.
Рассчитывалась полная динамическая активность цеолита и активность
его до проскока в рафинат 0.2 вес. % н-бутиленов. Десорбция цеолита
проводилась азотом, содержащим заметные количества кислорода, и азо-
том, очищенным от кислорода. Было установлено, что при десорбции
н-бутиленов азотом, содержащим 0.5—1.0 объемн. % кислорода, актив-
Рис. 2. Выходные кривые адсорбционной очистки изобутилена
цеолитом СаА в псевдоожиженном слое.
Температура 30—50° С; в. с. 0.8—0.9 час.-*1; скорость гава 0.05—
0.07 м/сек. Цифры у кривых — номера циклов.
ность цеолита до проскока составляла 2.5—3.6 вес. %, или 1.3—1.9 г/см2,
полная динамическая активность равнялась 3.5—5.0 вес. %. Одновре-
менно наблюдалось образование димеров изобутилена в количестве др
40 вес. % от пропущенного сырья в первом цикле с постепенным пониже-' '
нием димеризации до 10% (в пятом цикле). При проведении десорбции
азотом, содержащим 0.02—0.04 объемн. % кислорода, активность цер- '
лита до проскока повысилась до 4.0—5.0 вес. %, или 2.1—2.5 г/См2/
полная динамическая активность увеличилась до 5.3—7.0 вес. %. Обра-
зования димеров при этом не наблюдалось. Длина зоны массопередачи, .
подсчитанная на основе выходных кривых (рис. 2), колебалась от 70 до '.'
120 см. :
Проведенные исследования показали возможность осуществления
процесса разделения углеводородов изо- и нормального строения на псев-
доожиженном слое мелкодисперсного цеолита СаА. Для более широкого
исследования процесса адсорбции мелкодисперсным цеолитом на уста-
новке проводятся опыты по уточнению влияния гидродинамических фак- ,
торов на процесс.
ЛИТЕРАТУРА
1. Л. С. К о ф м а н, В. С. Виноградова, В. М. Комарова, авторское
свидетельство № 150108, 1961.
АДСОРБЦИЯ ИЗ СМЕСИ БЕНЗОЛА И ТИОФЕНА
СИНТЕТИЧЕСКИМИ ЦЕОЛИТАМИ ТИПА X
А. Н. Салюкое, Л. Ф. Кулиш, В. Н. Мазин, Л. Е. Фенелоноеа
Очистка бензола от сернистых соединений, в частности от тиофена,
представляет одну из важных проблем. Однако в ряде работ (1,2] пока-
зано, что из растворов бензол 4-н-гексан и тиофен 4-н-гептан на цеолите
13Х преимущественно адсорбируются бензол и тиофен, что определяется
природой этих систем (наличие ионов натрия на поверхности цеолита и
л-связей в молекулах тиофена и бензола).
Для отделения бензола от тиофена нами использовалась возможность
химического модифицирования поверхности цеолита типа X путем ион-
ного обмена. Разделение проводилось в жидкой фазе в динамических
условиях при длине слоя сорбента 20 см, зернении 0.5—1.5 мм, удельной
скорости потока 0.45 мл/мин.см2 и исходной концентрации тиофена в бен-
золе 0.011%. Результаты опытов приведены в табл. 1.
Таблица 1
Степень очистки бензола
и динамическая емкость
по тиофену различных цеолитов
Образец Максималь- ная степень очистки бензола (содержание тиофена, %) Динамиче- ская емкость по тиофену, мг/г
NaX 0.0071 0.027
СаХ 0.0076 0.01
NiNaX 0.0004 0.08
NiCaX 0.0 0.54
Таблица 2
Динамическая емкость цеолита
NiCaX по тиофену в зависимости
от содержания Ni в цеолите
Содержание Ni в цеолите NiCaX, г на 100 г цеолита Динамическая емкость по тиофену, мг/г
2.80 0.32
3.14 0.37
4.09 0.54
Как следует из таблицы, цеолиты NaX и СаХ практически не разделяют
«смесь тиофен—бензол. Путем замены в цеолитах СаХ и NaX части катио-
нов на ионы никеля удается получить сорбенты с повышенным сродством
к тиофену. Никелевый цеолит, полученный путем катионного обмена из
деолита СаХ (NiCaX), обладает более выраженной разделяющей способ-
ностью, нежели цеолит, полученный из NaX (NiNaX).
Были проведены опыты по изучению влияния содержания Ni в цео-
лите NiCaX на динамическую емкость по тиофену. Результаты опытов
приведены в табл. 2.
Как видно из таблицы, с увеличением содержания Ni в цеолите NiCaX
динамическая емкость цеолита по тиофену повышается.
ЛИТЕРАТУРА
1. С. П. Ж д а н о в, А. В. К и с е л е в, Л. Ф. П а в л о в а, Кинетика и катализ,
1962, 3, 451.
2. А. В. К и с е л е в, В. Н. С е м е н ов а, Ю. А. Э л ь т е к о в, Кинетика и ка-
тализ, 1962, 6, 937.
ВЫДЕЛЕНИЕ ЭТИЛЕНОВЫХ УГЛЕВОДОРОДОВ ИЗ КОКСОВОГО
ГАЗА С ПРИМЕНЕНИЕМ СИНТЕТИЧЕСКИХ ЦЕОЛИТОВ
М. П. Корш, Е. И. Казаков, Н. А. Кунакова, Н. Ф. Карпова
Разработка эффективных способов извлечения этилена из различных
содержащих его газов представляет исключительный интерес и имеет
большое практическое значение.
Научно-исследовательских ра-
бот по извлечению этилена из га-
зов с помощью цеолитов почти не
проводилось.
В нашей работе адсорбция
этилена из коксового газа изуча-
лась на стационарном слое сор-
бента в динамических условиях.
Исследовались цеолиты типа NaA,
СаХ и NaX при различных ско-
ростях газового потока. В каче-
стве исходного был взят газ Мо-
сковского коксогазового завода.
В результате проведенной работы
было исследовано влияние объем-
ной скорости газа на время защит-
ного действия цеолитов и скорость
процесса адсорбции.
Результаты опытов представлены на рис. 1.
Десорбция адсорбированных этиленовых углеводородов производи-
лась путем ступенчатого повышения температуры цеолита до 300—350° С.
Кроме этиленовых углеводоро-
дов, все цеолиты адсорбируют
небольшие количества других
компонентов коксового газа.
В процессе десорбции послед-
ние десорбируются в первую
очередь до 100—150° С. На ос-
новании этого мы использовали
ступенчатый способ термической
десорбции, позволяющий выде-
лить из десорбата фракцию до
150° С с большим содержанием
СО, Н2 и СН4 и тем самым уве-
личить в остальном десорбате
содержание этиленовых угле-
водородов до 90—92%. Метод
термической ступенчатой десорбции адсорбированных цеолитом газов
может быть рекомендован как один из простых способов разделения сор-
бата на различные фракции.
Результаты опытов по десорбции газов, адсорбированных на цеолите
NaA, представлены на рис. 2.
Выводы
Рис. 1. Выходные кривые адсорбции эти-
лена из коксового газа на цеолитах NaA
w'°
wo - '
80 -
60 -
W -
20 -
О
0.026 м/сек
0.055м/сек\
0.069м/сек.
100
200
300 t°c
Рис. 2. Содержание СтН„ в десорбате при
различных температурах десорбции.
температурах десорбции.
В результате проведенной работы по выделению этилена из коксового
газа с помощью молекулярных сит установлено следующее.
1. С помощью цеолита типа NaA можно получить концентрат с содер-
жанием 90—92% непредельных углеводородов, где 97.7—95.0% этилена.
293
2. Метод термической, ступенчатой десорбции адсорбированных цео-
литами газов может быть рекомендован как один из простых способов
разделения сорбата на различные фракции.
ОБЛАГОРАЖИВАНИЕ ТУРКМЕНСКОГО БЕНЗИНА ПРИ ПОМОЩИ
СИНТЕТИЧЕСКОГО ЦЕОЛИТА СаА
X. И. Арешидзе, Г. О. Чивадзе
Применение высокооктановых бензинов представляет большой народно-
хозяйственный интерес, так как повышает коэффициент полезного дей-
ствия двигателей внутреннего сгорания и удлиняет срок их службы.
Известно, что алканы нормального строения обладают низкими окта-
новыми числами, поэтому их присутствие в бензинах снижает октановую
характеристику. Таким образом, алканы нормального строения являются
нежелательными компонентами бензинов. Для того чтобы повысить окта-
новое число бензинов за счет алканов нормального строения, надо или
изменить их природу, или удалить их из бензинов. До открытия замеча-
тельных свойств цеолитов избирательно адсорбировать н-алканы из угле-
водо’родпых смесей не было возможности удалять н-алканы из бензи-
нов [1].
В данной работе ставилась задача выделить н-алканы с помощью
синтетического цеолита кальциевой формы типа А из низкооктанового
туркменского бензина и проследить, как повлияет этот процесс на по-
вышение антидетонационных свойств бензина.
Удаление н-алканов из бензина дает возможность уточнить, какое ко-
личество алканов из общего количества падает на алканы нормального
. и изостроения. Знание подобного состава, помимо теоретического, имеет
и практическое значение. Впервые синтетические цеолиты для подобного
анализа бензинов были применены Шварцем [2].
Зная групповой состав бензина и количественное распределение в нем
алканов нормального и изостроения, можно предусмотреть значение его
октановой характеристики. В результате проведенного исследования нами
показано, что из общего количества алканов туркменского бензина на
н-алканы падает 20.2%, а на изоалканы — 33.0%. Таким образом, уда-
ление н-алканов из бензина имеет и аналитическое значение.
Показано, что синтетические цеолиты, хотя и в малом количестве,
но все же удаляют и алканы изостроения. Из-за малых количеств выделен-
ных изоалканов их структура не установлена. Надо полагать, что эти изо-
алканы должны быть мало разветвленными. Так, например, если имеем
изоалкан строения СН3—СИ—СН2—СН2—СН2—СН2—СН3, у которого
I
сн3
метильная группа находится у второго углеродного атома, он своей
прямой цепочкой может войти в окно синтетического цеолита, т. е. эта
часть молекулы углеводорода может себя вести как алкан нормального
строения.
На майской сессии Комиссии по цеолитам 9 мая 1962 г. в Тбилиси
одним из авторов настоящей статьи были доложены результаты работ,
касающихся изменения октановой характеристики мирзаанского бензина
в результате удаления н-алканов синтетическим цеолитом СаА. Было
показано, что октановое число мирзаанского бензина повышается на
14 пунктов.
294
Бензины отличаются количественным содержанием н-алканов, и
поэтому при удалении их эффект улучшения топливных свойств бензи-
нов будет разным. В данной работе мы поставили цель проследить, как
изменится октановое число туркменского бензина в результате удаления
н-алканов.
Браун, Райтмар и Стреккер [3] при помощи синтетического цеолита
СаА удалили н-алканы из бензинов с целью повышения их октановой
характеристики.
Я. В. Мирский и М. Г. Митрофанов [4] изучали разделяющую спо-
собность синтетического цеолита СаА на смесях н-гептана с толуолом и
метилциклогексаном. Разделение осуществлялось как в жидкой, так и
в паровой фазах.
С. Н. Павлова, 3. В. Дриацкая, М. А. Мхчиян [5] определили содер-
жание алканов нормального строения с помощью синтетических цеоли-
тов в петролейном эфире, бензольных, толуольных и ксилольных фрак-
циях туймазинской, майкопской, могутовской и провернской нефтей.
Показано, что содержание алканов нормального строения во всех иссле-
дуемых фракциях с повышением температуры кипения фракций умень-
шается. Это изменение особенно заметно на примере исследуемых образ-
цов могутовской нефти.
Л. Н. Квитковский и Е. В. Грушецкая [6] определили н-алканы в ис-
кусственных смесях углеводородов и в нефтяных фракциях. Показано,
что расхождение параллельных опытов в случае искусственных смесей
не превышает 0.5%, в то время как для нефтяных фракций оно колеблется
от 0.87 до 4.76%. Авторами [6] показано, что крекинг при регенерации
не должен иметь места, так как отложения кокса или смол в кристалле
цеолита не было.
Л. Ф. Павлова [7] изучила адсорбцию н-гексана из смеси н-гексан-
бензол порошкообразным синтетическим цеолитом СаА и синтетическим
таблетированным цеолитом Линде 5А. Исследование показало, что таб-
летированный синтетический цеолит 5А полностью извлекает н-гексан.
М. Б. Вольф и Р. В. Алексеева [8] исследовали изменение октановой
характеристики бензина в результате удаления н-алканов. Ими показано,
что бензин с температурой кипения 70—150° С, содержащий в своем со-
ставе н-гексан, н-гептан, н-октан и н-нонан, после депарафинизации ха-
рактеризуется октановым числом, которое выше начального на 17.5 пунк-
тов. В случае бензиновой фракции с температурой кипения 60—94° С
тем же способом была повышена октановая характеристика на 21.5 пунк-
тов.
М. Г. Митрофанов и Я. В. Мирский [9] извлекали н-алканы из бензи-
новых фракций с применением синтетического цеолита типа СаА. Опыты
проводились в лабораторных условиях и на укрупненной лабораторной
установке. В зависимости от образца бензина изменение октановой ха-
рактеристики имело место от 11.2 до 20.3.
Н. В. Кельцев, А. Ф. Старовойтова и Н. С. Торочешников [10] отде-
лили н-пентан от изопентана с помощью синтетического цеолита СаА.
В. С. Виноградова и Л. С. Кофман [11] разделили изомерные алканы
и алкены состава С4—С8 при помощи синтетического цеолита СаА. Ав-
торы предполагают, что возможно практически выделить 100% изопен-
тана из смеси с н-пентаном или 100% изобутана из смеси с н-бутаном.
Они отделили изобутилен от цис- и транс-бутен-2. Поданным авторов [11],
синтетический цеолит СаА может быть применен также для разделения
стереоизомерных алкенов состава С4—С6.
И. Н. Самсонова и С. П. Жданов [12] с сотрудниками использовали
цеолиты 5А и пористое стекло № 10 для количественного определения
содержания н-алканов в искусственных углеводородных смесях и бен-
295
зиновых фракциях. В работе использовался проточный и весовой методы.
В результате проведенных исследований показано, что стекло № 10
с успехом можно использовать для количественного определения н-ал-
канов в деароматизированных бензинах.
М. Г. Митрофанов, Я. В. Мирский, А. 3. Дорогочинский с сотрудни-
ками [13] выделили н-алканы из бензинов грозненской, озексуатской,
ромашкинской нефтей, а также из дистиллата бензина «Галоша» и из
фракции экстракционного бензина при помощи синтетического цеолита
типа СаА. В результате удаления
Схема установки для получения ден-
парафинироваппого бензина.
1 — капельная воронка; 2 — трубка с адсор-
бентом; 3 — электропечь; 4 — холодильник;
5 — приемник; 6 — лопушка; 7 — манометр;
8 — термопара; 9 — вакуум-насос.
н-алканов октановое число используе-
мых образцов повысилось на 11.2—
18.2 пунктов.
Первая промышленная установка
[14] по извлечению н-алканов ме-
тодом, разработанным компанией
«Линде», была построена в Силебии
(США, штат Техас).
Бензин из туркменской нефти
(продукция Батумского нефтеперера-
батывающего завода) имеет столь
низкое октановое число (55), что без
его повышения он не находит при-
менения в народном хозяйстве. Низ-
кое октановое число данного бензина
обусловлено присутствием алканов
нормального строения, поэтому их
удаление должно повышать антиде-
тонационные свойства бензина. Объ-
ектом исследования в данной работе
был бензин из туркменской нефти
с температурой кипения 37—158° С,
а в качестве адсорбента н-алканов
применяли синтетический цеолит СаА
в виде гранул — образец Горьковской
опытной базы ВН1ДИНП Ц-202-238.
Л. И. Пигузова [15] показала, что искусственный цеолит СаА полу-
заводской установки Горьковской опытной базы термостабилен при
450—750°. Повышенная термостабильность синтетического цеолита СаА
объясняется автором [15] большим зарядом иона кальция по сравнению
с ионом натрия.
Синтетический цеолит помещали в стеклянную трубку высотой 1000 мм,
диаметром 22 мм, насыпной объем 300 мл; поверхность синтетического
цеолита была покрыта битым стеклом для предварительного испарения
бензина. Трубку с адсорбентом переносили в вертикально установленную
трубчатую электропечь. Цеолит сушился постепенным повышением тем-
пературы до 400° С в течение 3 часов под вакуумом 5 мм рт. ст. Адсорб-
цию н-алканов проводили при 180° С и давлении 400 мм рт. ст. с разными
объемными скоростями подачи бензина в адсорбер. Для установления
влияния скорости подачи бензина на полноту выделения н-алканов она
менялась от 0.15 до 1.0 час.-1. Экспериментально было найдено, что ско-
рость 0.15 час.-1 является более приемлемой, поэтому в дальнейшем мы
придерживались скорости 0.15 час.-1.
Денпарафинированпый бензин собирали в приемник и ловушку, охла-
ждаемые жидким азотом. После окончания процесса адсорбции денпара-
финированный бензин, находящийся на поверхности адсорбента, удалялся
при 120° в токе азота. Схема установки дана на рисунке. Десорбцию
296
н-алканов проводили при 340—350° С и давлении 5 мм рт. ст. с помощью
азота. Образование кокса не имело места.
Многими исследователями было показано, что оптимальной темпера-
турой для полной десорбции н-алканов состава С6—С8 является 350° С
при давлении 3 мм рт. ст.
На примере десорбции н-алканов из фракции 60—85° С [13] было
показано, что при 250° С десорбируется 50% адсорбированных н-алка-
нов, а при 350° С — 100%.
Полнота выделения н-алканов контролировалась повторным пропу-
сканием одного и того же образца бензина. Выделение н-алканов счи-
талось законченным тогда, когда при повторном пропускании бензина
с последующей десорбцией выделение н-алканов не имело места.
Для того чтобы убедиться, что прекращение выделения н-алканов вы-
звано отсутствием их в исследуемом образце, а не падением адсорбцион-
ной способности синтетического цеолита, последняя проверялась на све-
жем образце бензина, и она оказалась почти прежней. Опыты по выделению
н-алканов одним и тем же образцом синтетического цеолита СаА проводи-
лись в продолжении 200 часов. За это время работы его адсорбционная
способность оставалась постоянной.
Выделенные н-алканы были исследованы методом газо-жидкостной
хроматографии.1 Исследование показало, что смесь н-алканов содержит
12.1% н-пентана, 27.8% н-гексана, 39.1% н-гептана, 12.7% н-октана,
0.7% изогексана, 1.8% изогептана и 3°/0 изооктана. 2.8% соответствует
остальным углеводородам. Физические свойства бензина до и после уда-
ления н-алканов даны в таблице.
Результаты опытов по выделению н-алканов из бензина
Свойства бензина Исходный бензин Денпарафи- нированный бензин Смесь н-алканов
Выход, вес. % 76.8 20.2
Удельный вес d420 0.7229 0.7301 0.6792
Показатель преломления пд20 Групповой углеводородный состав, вес.% 1.4109 1.4193 1.3840
парафиновые 53.2 41.2 91.7
нафтеновые 35.7 44.6 —
ароматические 11.1 14.0 —
Максимальная анилиновая точка 56.8 50.4 69.5
Октановое число Фракционный состав, р С: 55 72.7 — .
начало кипения 37 43 37
выкипает 10% 77 79 78
» 50% НО 115 103
» 90% 145 147 130
конец кипения 158 158 151
Как видно из табличных данных, после денпарафинизации уменьшение
максимальной анилиновой точки, увеличение удельного веса, показателя
преломления и октанового числа указывают на то, что из бензина удали-
лись те компоненты, которые имеют анилиновую точку выше, а показа-
тель преломления, удельный вес и октановое число ниже, чем у нафтено-
ароматической части бензина.
1 Эта часть работы выполнена в Институте органической химии имени Н. Д. Зе-
линского АН СССР ст. научи, сотрудником М. И. Розенгартом, за что ему выражаем
глубокую благодарность.
297
Проведенным исследованием показано, что в результате удаления
н-алканов из туркменского бензина его октановое число повышается на
17.7 пунктов.
Определение октанового числа производилось моторным методом на
Батумском нефтеперерабатывающем заводе.
ЛИТЕРАТУРА
1. В. М. Barrec, J. Soc. Chem. Ind., 1945, 64, 5.
2. R. D. Schwartz, D. J. Brasseaux, Anal. Chem., 1957, 29, 1026.
3. G. R. Bro wn, R. A. R i g h t m i г e, H. A. Strecker, Oil a. Gas J., 1959,
57, 189.
4. Я. В. Мирский, M. Г. Митрофанов, ЖФХ, 1961, 35, 460.
5. С. H. П а в л о в a, 3. В. Д р и а ц к а я, М. А. М х ч и я и, Хим. и технол.
топлив и масел, 1962, 3, 58.
•6. Л. Н. К в и т к о в с к и й, Е. В. Груш ед к а я, Хим. и технол. топлив и
масел, 1962, 3, 61.
7. Л. Ф. Павлова, в сб.: Синтетические цеолиты, Изд. АН СССР, 1962, 225.
8. М. Б. В о л ьф, Р. В. А л е к с е е в а, в сб.: Синтетические цеолиты, Изд. АН
СССР, 1962, 233.
fl. М. Г. М и т р о ф а н о в, Я. В. М и р с к и й, в сб.: Синтетические цеолиты, Изд.
АН СССР, 1962, 236.
10. Н. В. Кельцев, А. Ф. Старовойтова, Н. С. Торочешников,
в сб.: Синтетические цеолиты, Изд. АН СССР, 1962, 239.
11. В. С. В и н о г р а д о в а, Л. С. Ко фм ан, в сб.: Синтетические цеолиты,
. Изд. АН СССР, 1962, 245.
12. Й. Н. С а м с о н о в а, С. П. Ж д а н о в, Н. Н. Б у н т а р ь, Е. В. К о р о-
мальди, В. А. Голубева, ЖПХ, 1963, 36, 2502.
13. М. Г. Митрофанов, Я. В. Мирский, А. 3. Дорогочинский,
А. П. Д р о н и н, С. В. М а к а р ь е в, Б. И. Л у г о в о й. Тр. ГрозНИИ,
Гостоптехиздат, М., 1963, 15, 84.
14. Chem. Engng., 1962, 69, 58.
15. Л. И. П и г у з о в а, Хим. и технол. топлив и масел, 1961, 4, 9.
ОСУШКА ДИВИНИЛА
Л. О. Дымент, В. С. Виноградова, Л. С. Кофман
Во избежание разрушения комплексных каталитических систем, при-
меняемых при стереонаправленной полимеризации 1,3-бутадиена(диви-
нила), содержание в нем влаги должно быть минимальным — порядка
10 ~4 и менее процентов. Путем азеотропной ректификации относительно
нетрудно достигнуть снижения концентрации воды до 0.005%. Более глу-
бокая осушка дивинила, например адсорбцией силикагелем или алюмо-
гелем или обработкой щелочными металлами, осложнена и связана с по-
терями продукта. Не может быть рекомендовано для этой цели и примене-
ние цеолитов NaA, в полости которых дивинил частично проникает
(рис. 1), полимеризуясь в них. Нами экспериментально установлено до-
вольно быстрое снижение активности цеолитов NaA по воде после пропу-
скания дивинила.
На лабораторной вакуумной адсорбционной установке были изучены
изотермы сорбции паров воды и некоторых углеводородов С4 на нескольких
образцах формованных цеолитов NaA и КА (рис. 2). Образец 253 сформо-
ван посредством глины кил, остальные — с глуховской глиной. Стати-
ческая активность по воде калиевой формы цеолитов А приблизительно
на 40 отн. % ниже, чем формы NaA. Тем не менее для осушки дивинила
применение цеолитов КА полностью оправдано, поскольку исключается
возможность полимеризации несорбируемого ими дивинила.
298
Исследование процесса осушки дивинила в динамических условиях
осуществляли на лабораторной установке (рис. 3) при объемной скорости
1—5 л/л (в пересчете на жидкий углеводород), что соответствовало ли-
нейной скорости паров 0.01—0.05 м/сек.
Рис. 1. Изотермы 'адсорбции
1,3-бутадиена.
1 — на цеолитах КА-526; 2 — на
цеолитах NaA.
Рис/ 2. Изотермы адсорбции воды
на цеолитах.
1 — КА-253; 2 — КА-253 после 13
циклов; 3 — КА-226-526 после 10 цик-
лов сорбции—десорбции; 4 — КА-226-
526 после 48 часов нагрева при 500°С.
Основные результаты опытов представлены в виде выходных кривых
(рис. 4), из которых видно, что остаточное содержание влаги в дивиниле
не превышало 0.0001 вес. % (точка помутнения < —70° С). Динамиче-
ские показатели процесса глубокой осушки дивинила приведены в таб-
Рис. 3. Схема установки для осуш-
ки дивинила.
1 — баллон с дивинилом; 2 — реометр;
з — адсорбер с цеолитами; 4 — пробоот-
борная ампула; б — баллон для осушен-
ного дивинила.
Рис. 4. Выходные кривые при
осушке дивинила в динамических
условиях.
1,2,3 — цеолитами КА-226-526 (цик-
лы 1, 2, 4); 4,5,6 — цеолитом КА-253
(циклы 6, 9, И).
лице для проскоковой концентрации воды 7 • 10-4% (температура по-
мутнения —60° С).
Кинетические показатели осушки дивинила в динамических условиях
цеолитами КА вполне удовлетворительны. Длина работающего слоя
нала даже при больших скоростях потока, вследствие чего динамическая
активность до проскока (ад) и полная (а„) практически совпадают. Наблю-
даемое постепенное уменьшение активности цеолитов КА при цикличе-
299
Осушка дивинила формованными цеолитами КА в паровой фазе при скорости
0.05 м/сек.
Н аименование образца № цикла Содержание влаги в осу- шаемом дивиниле, моль % £о> см а д Содержание влаги в осушенном дивиниле, вес. %
г/см2 г/100г г/см3 г/100г
2 2.4 3.43 16.89 0.136 17.67 < 0.0001
КА-202-226-526 | 4 — 2.3 2.98 14.65 0.118 15.34 < 0.0001
7 — — 1.58 7.76 — — sg 0.0001
2 1.78 4.5 3.05 14.12 0.122 15.70 < 0.0001
3 —. -—. 2.89 13.42 0.107 13.80 < 0.0001
5 2.00 — 2.63 12.22 0.098 12.70 < 0.0001
6 0.85 2.2 2.17 10.04 0.081 10.40 —
7 0.78 2.0 2.03 9.42 0.076 9.80 —
КА-202-253 8 0.75 2.0 1.77 8.22 0.066 8.50 —.
10 0.62 1.8 1.89 8.77 0.070 9.00 —
11 0.62 1.8 1.59 7.37 0.059 7.60 -—.
12 0.62 .—. 1.50 6.97 0.056 7.20 < 0.0001
14 0.75 — 1.30 6.05 0.048 6.20 < 0.0001
16 — —_ 1.15 5.32 —— — —
Примечание. Цикл № 14 — после окислительной регенерации.
ской работе вызвано не полимеризацией дивинила, а пониженной термо
стабильностью данных образцов цеолитов КА. Это видно из изотермы
адсорбции паров воды образцом цеолита КА после нагрева в течение
48 часов при 500° С (рис. 2). Следует учесть, что при регенерации цеолитов
их нагревали до 350—400° после каждого цикла сорбции.
Применение цеолитов КА для осушки легкополимеризующихся угле-
водородов (диенов, ацетиленовых) перспективно, но требуется создать
условия получения термостабильных сорбентов этого типа, а также уточ-
нить оптимальный режим их регенерации. Поскольку изотермы адсорбции
паров воды на калиевой форме цеолита А более пологи в области малых
концентраций, чем на NaA, следует полагать, что и температура десорб-
ции воды может быть соответственно понижена. Исследования в указан-
ной области продолжаются.
ГЛУБОКАЯ ОСУШКА ПРОПИЛЕНА
С ПОМОЩЬЮ СИНТЕТИЧЕСКИХ ЦЕОЛИТОВ ТИПА NaA и КА
Р. П. Кирсанова, С. Ш. Бык
В связи с освоением в СССР производства полипропилена проблема
глубокой осушки исходного мономера (пропилен) приобретает значитель-
ный практический и теоретический интерес. Наиболее перспективным и
эффективным методом осушки пропилена без сомнения является метод
адсорбционной осушки с помощью синтетических цеолитов.
Опыты по осушке пропан-пропиленовой фракции (пропилена —
77.93 объемн. %, пропана — 22.06 объемн. %, этилена — 0.00085 объемн. %,
бутилена — 0.00015 объемн.%) проводились на лабораторном стенде
(рис. 1) при следующих условиях: /=20° С, р=9—10 ати, относительная
влажность подаваемого на осушку газа <р=0.9, количество адсорбента
300
5—6 г, время контакта 5 сек., температура процесса регенерации 350° С,
точка росы регенерационного газа (азота) равняется —60° С. Влагосо-
держание осушенной пропан-пропиленовой фракции определялось влаго-
мером «Shaw», позволяющим определять влажность любого газа до точки
росы —100° С. В качестве осушителей были выбраны цеолиты NaA и
Рис. 1. Схема установки.
1 — баллоны с С,Н«; 2 — термостат; з — увлажнитель; 4 — каплеотбой-
ник; 5 — колонка с адсорбентом; е — манометр образцовый; 7 — влагомер
фирмы Шоу; 8 — газовые часы; 9 — колонка с осушителем.
КА, гранулированные с применением в качестве связующего глины —
крымского кила. Полученные экспериментальные данные представлены
в табл. 1.
Таблица 1
Динамическая активность цеолита NaA по парам воды
Осушаемый газ
Влагосодержание осу-
шенного газа,
отвечающее точке
росы, °C
Динамиче-
ская актив-
ность, %
Пропан-пропиленовая
фракция ............
Азот................
—72+1
—72 ±1
14.5
19.0
В табл. 1 приведены усредненные данные большого количества опытов;
данные, характеризующие осушку азота, были получены при тех же
условиях, что и для пропан-пропиленовой фракции. Как это следует из
табл. 1, пропилен в заметной степени ( « на 25%) подавляет адсорбцию
паров воды на цеолитах NaA.
Значительный практический интерес представляло также исследование
характера изменения адсорбционной активности цеолита в результате
длительного воздействия пропилена. С этой целью на описанном выше
лабораторном стенде было проведено 50 циклов осушка—регенерация.
Было показано, что после 50 циклов точка росы осушенного газа и, что
самое существенное, динамическая адсорбционная емкость цеолита оста-
ются в пределе ошибок опыта неизменными (—72° С и 14.5%).
30.1
Для оценки эффективности цеолита NaA (рекомендованного нами для
технологической схемы осушки пропилена) в области низких исходных
влагосодержаний и при умеренно повышенных температурах представ-
ляло интерес определить изотермы адсорбции паров воды на цеолите
NaA при 20 и 60° С. Это исследование проводилось динамическим методом,.
Рис. 2. Изотерма адсорбции паров
воды на цеолите NaA при 20° С.
Рис. 3. Изотерма адсорбции паров
воды на цеолите NaA при 60° С.
адсорбционная активность цеолита определялась с помощью выходных
кривых, а также весовым методом, влагосодержание осушенного (при
р=1 атм.) азота (точка росы —72° С) определялась с помощью кулоно-
метрического влагомера «Beckman». Из рис. 2, 3 видно, что даже при
<р=0.05 и f=60° С исследованные образцы цеолита характеризуются
достаточно высокой адсорбционной активностью ( да 15 вес.%).
Кроме детального исследования процесса осушки пропилена на цео-
литах NaA, нами по описанной выше методике было проведено несколько
циклов осушка—регенерация с опытными образцами цеолитов КА. По-
лученные результаты представлены в табл. 2.
Таблица 2
Динамическая активность цеолита КА по парам воды
Осушаемый гав Влагосодержание осу- шенного гава, отве- . чающее точке росы, °C Динамиче- ская актив- ность, %
Пролан-цропиленовая фракция —49 14
Азот —73 19
Для сравнения на изотермической лабораторной установке при р=
=1 атм. и i=20° С были определены также осушающие свойства импорт-
ных (английских) цеолитов типа КА: точка росы осушенного азота —73° С,
динамическая адсорбционная емкость 17%.
Выводы
1. Исследован процесс осушки пропан-пропиленовой фракции на цео-
литах NaA со связующим материалом — глиной крымский кил; показано,
что после 50 циклов осушка—регенерация адсорбционная активность
цеолита практически не снижается.
302
2. Проведены опыты по осушке пропан-пропиленовой фракции оте-
чественными опытными образцами цеолитов типа КА. Определены осу-
шающие свойства импортных (английских) цеолитов КА.
РЕЗУЛЬТАТЫ ИСПЫТАНИЙ ОСУШКИ ПРОПАНА ЦЕОЛИТАМИ
И ДРУГИМИ СОРБЕНТАМИ
Г. И. Вялкина, Н. В. Жданова, Н. В. Вельцев, Г. С. Фролов
В сжиженных углеводородных газах (пропане и бутане) растворимость,
воды при понижении температуры уменьшается, что ведет к выделению
капельной влаги и, как следствие этого, в определенных рабочих усло-
виях — к образованию твердых кристаллогидратов углеводородов. Чтобы
избежать возможности выделения кристаллогидратов при транспортировке,
хранении и переработке пропана и бутана, предъявляются жесткие
требования к их влагосодержанию. Наиболее эффективными промышлен-
ными осушителями являются твердые адсорбенты: силикагель, окись алю-
миния, синтетические цеолиты. Высокие адсорбционные показатели
цеолитов как осушителей стимулировали их промышленное применение
для сушки промышленных газов [1, 2], адсорбционного масла на газо-
бензиновых заводах [3], олефинового и парафинового сырья в процессах
алкилирования [4, 5] и других органических жидкостей [6].
Сравнительная оценка осушающих свойств окиси алюминия была
получена на лабораторной установке, основным элементом которой слу-
жил адсорбер с загрузкой 60 г адсорбента — активированная окись алю-
миния или цеолит NaA; размеры частиц составляли соответственно 2—
5 мм, d=A=4.5 мм, высота слоя — 8 см. Опыты проводились с техниче-
ским пропаном при температуре 20° С и давлении 7—8 атм. Каждому
опыту предшествовала регенерация адсорбента в вакууме при 300° С
и остаточном давлении 150 мм рт. ст. Линейные скорости жидкого про-
пана составляли 0.003—0.004 м/сек. Исходная влажность пропана соот-
ветствовала насыщению пропана водой при 22° С — 0.3 г/м3.
В результате испытаний было установлено, что как в случае окиси алю-
миния, так и в случае цеолитов остаточное содержание влаги в пропане-
составляет менее 0.01 г/м3, однако как равновесная (при насыщении),
так и динамическая активность до проскока цеолитов в 4—5 раз превос-
ходит динамическую активность окиси алюминия. Так, динамическая
активность окиси алюминия составляет 3—4 г Н2О/Ю0 г, цеолита — 12—
15 г Н2О/100 г. Защитное время действия слоя цеолита высотой 8 см при
указанных выше скоростях потока составляет около 24 часов, окиси алю-
миния — 6 часов.
Аналогичные результаты были получены при испытании процесса
осушки цеолитами и окисью алюминия на опытной установке газобен-
зинового завода с двумя попеременно работающими адсорберами, высота
слоя промышленных адсорбентов в которых составляла 1 м, а диаметр
5 см; регенерация производилась горячим воздухом при температуре
200-250° С.
При осушке цеолитами NaA, имеющими входные окна в адсорбцион-
ные полости меньших размеров (4А), чем критический диаметр молекул-
пропана (4.9 А), отсутствует одновременное поглощение пропана и выс-
ших углеводородов, характерное для процесса осушки окисью алюминия.
Высокие адсорбционные свойства цеолитов по растворенной в жидком
пропане влаге, глубина осушКи, исключающая возможность образования
303-
кристаллогидратов при любом охлаждении, позволяют рекомендовать
цеолиты для широкого использования в качестве осушителя сжиженных
газов.
ЛИТЕРАТУРА
1. 3. А. Ж у к о в а, Н. В. К ел ьцев, И. П. О г л о б и н а, Н. С. Т о р о чет-
ников, Хим. промышленность, 1962, 2, 100.
2. G. S. Cochrane, Chem. Engng., 1959, 66, 17, 129.
3. J. J. С о 11 i n s, Oil a. Gas J., 1962, 10, 100.
4. J. J. Collins. Oil a. Gas J., 1962, 60, 27, 97.
5. Chem and Engng. News, 1962, 40, 16, 66.
6. В. А. С о к о л о в, Н. С. Т о р очешников, Н. В. К е л ь ц е в. Молекуляр-
[ные сита и их применение. Изд. «Недра», 1964.
АДСОРБЦИЯ ВОДЫ, РАСТВОРЕННОЙ В СПИРТАХ Q—С4,
СИНТЕТИЧЕСКИМИ ЦЕОЛИТАМИ NaA
М. Е. Селин, Д. С. Лаврухин, Л. Б. Кулемина
Важной и перспективной областью применения синтетических цео-
литов является осушка жидкостей. Однако до настоящего времени прак-
тически отсутствовали данные, количественно характеризующие свой-
ства цеолитов при адсорбции воды, растворенной в органических жид-
костях. Исключением является работа [1], в которой приведены изотермы
адсорбции воды из ее растворов в фурфуриловом спирте и пиридине цео-
литами СаА. Исследовав область высоких равновесных концентраций
воды, авторы экстраполировали полученные изотермы в область низких
концентраций, что, по нашему мнению, не вполне правомерно. По этой же
причине нельзя согласиться с теоретическими выводами авторов.
Изучение адсорбции из растворов в области малых концентраций ад-
сорбата представляет не только теоретический, но и большой практиче-
ский интерес. В частности, во многих случаях лабораторной практики
требуются растворители (например, спирты) с минимальным содержанием
воды; с помощью цеолитов эту задачу можно решить весьма успешно.
Применение цеолитов в качестве осушителей требует ответа на вопросы:
а) какой тип цеолита наиболее эффективен в том или ином конкретном
случае и б) какова адсорбционная емкость цеолитов при осушке до опре-
деленного минимального содержания воды.
Настоящая работа посвящена исследованию адсорбции воды, раство-
ренной в спиртах Сг—С4, синтетическими цеолитами NaA. Ограничение
области исследования цеолитами только одного типа было обусловлено
тем, что предварительные опыты показали меньшую эффективность цео-
литов СаА и NaX при осушке спиртов.
Методика исследования
Адсорбент — таблетированный синтетический цеолит NaA — прокали-
вался при 350° С и давлении 10 ~4 мм рт. ст. Спирты очищались обычными
методами [2], дополнительно осушались цеолитом NaA и ректифицирова-
лись на колонке с 12 теоретическими тарелками. Показатели преломления
соответствовали литературным данным 12]. Вода — бидистиллировацная,
п2>20=1.3333. Приготовленные растворы контактировались с адсорбен-
том в запаянных ампулах в течение 3 суток, при периодическом встряхи-
вании. Концентрации и объемы исходных растворов и навески адсорбента
304
брались с расчетом перекрытия интервала равновесных концентраций
от 0.0001 до 0.99 весовых долей воды при обеспечении наибольшего коли-
чества экспериментальных точек в области малых концентраций. Содер-
жание воды в равновесных растворах определялось электрометрическим
титрованием реактивом Фишера [3], что давало более высокую точность
измерения, чем другие возможные способы. В частности, считающийся
весьма точным интерферометрический способ определения концентраций
растворов в данном случае оказался малопригодным, ввиду погрешностей
за счет ионного обмена цеолит—вода.
Относительная ошибка метода анализа составляла 0.001 в области
малых концентраций воды. Величины адсорбции X и полного содержания
воды в адсорбционном объеме а рассчитывались по известным формулам [4]
(в граммах на грамм адсорбента):
(1)
а = X vi — при адсорбции из растворов воды в метаноле и этаноле,
X
а = — при адсорбции из растворов воды в пропиловых и бутиловых спйр-
тах,
где go — вес раствора, у0 и у — весовые доли воды в исходном и равновес-
ном растворах, т — навеска адсорбента, v — адсорбционный объем.
Оцененная по изотерме адсорбции паров воды величина адсорбцион-
ного объема составляла 0.215 см3/г. Для ограниченно растворимых бути-
ловых спиртов изотермы определялись в интервалах концентраций, со-
ставлявших 70—80% от пределов растворимости.
Результаты опытов и их обсуждение
Изотермы адсорбции воды при 20° С в интервале^ от 0 до 1 даны на рис. 1.
Изотермы полного содержания воды в адсорбционном объеме показаны
на рис. 2. По виду изотерм (и по характеру процесса) адсорбции воды из
растворов цеолитами следует различать два основных случая:
1) когда компоненты смеси имеют такие размеры молекул, что один
из компонентов способен проникать в поры, а второй может адсорбиро-
ваться только на внешней поверхности кристаллов;
2) когда оба компонента способны проникать внутрь кристалла и
конкурировать друг с другом в адсорбционном объеме.
К первому случаю и относится адсорбция воды из растворов в пропи-
ловых и бутиловых спиртах. Изотерма адсорбции круто поднимается
в области малых концентраций, достигает максимума при у=0.007—
0.025 и линейно падает до нуля при у=1. Экстраполирование прямоли-
нейного участка дает при пересечении с осью ординат величину адсорб-
ционного объема. Однако было бы ошибочным считать, что вся изотерма
должна быть прямой линией, как это утверждается в работе [1]. Авторы
этой работы, обнаружив максимум на изотерме адсорбции в системе пи-
ридин—вода на цеолите 5А, приписывают появление максимума каким-то
аномалиям в свойствах системы. Одновременно, не исследовав области
малых концентраций в системе фурфуриловый спирт—вода, авторы делают
вывод о прямолинейности всей изотермы. Критические замечания по ра-
боте [1] были сделаны А. В. Киселевым и Л. Ф. Павловой [5]. Можно
только добавить, что измеренные нами изотермы адсорбции воды из рас-
творов в пропиловых и бутиловых спиртах подтверждают вывод о распо-
ложении максимума в области отличных от нуля концентраций.
Смещение максимума в сторону возрастания концентрации, как это
было в системе пиридин—вода—цеолит 5А, объясняется более низкой
величиной энергии адсорбции воды в этой системе по сравнению с системой
20 Цеолиты
305
спирт—вода. Если считать энергию адсорбции воды из раствора в первом
приближении равной разности теплоты адсорбции чистой воды и теплоты
Рис. 1. Изотермы адсорбции воды, растворенной в спиртах.
1 — ивобутиловый; 2 — н-бутиловый; 3 — изопропиловый; 4 — н-про-
пиловый; 5 — этиловый; в — метиловый.
растворения воды в данном растворителе, то чем больше теплота растворе-
ния воды, тем меньше будет энергия адсорбции и тем больше удаление
Рис. 2. Изотермы полного содержания ад-
сорбированной цеолитами NaA воды, рас-
творенной в спиртах.
1 — ивобутиловый; 2 — изопропиловый; 3 — «-бу-
тиловый; 4 — и-пропиловый; б — этиловый; в — ме-
тиловый.
максимума от оси ординат (у=0).
Так, теплота растворения во-
ды в пиридине (А=2.324 ккал./
моль [6], а в пропиловом спир-
те— ^2=1.520 ккал./моль, т. е.
Qt Q2 и, следовательно,
т, _ > г9 , что и под-
37 — ^макс 37 ~ а?м акс
тверждается экспериментом.
Адсорбция воды из раство-
ров в этиловом и метиловом
спиртах относится ко второму
случаю, когда оба компонента
проникают в адсорбционный
объем. Полученные изотермы
показывают, что адсорбция во-
ды положительна во всей об-
ласти концентраций. Изотерма
адсорбции воды из раствора в
этиловом спирте близка к ли-
нейной в интервале у=0.5—1,
что говорит о более или менее
полном вытеснении спирта из
адсорбционного объема при высокой концентрации воды.
Изотерма адсорбции воды из раствора в метиловом спирте не имеет
линейного участка; следовательно, метиловый спирт не вытесняется пол-
ностью из адсорбционного объема во всем интервале концентраций. Пол-
306
Рис. 3. Начальные участки изотерм ад-
сорбции воды, растворенной в спиртах.
1 — ивобутиловый; 2 — ивопропиловый; 3 —
н-бутиловый; 4 —- н-пропиловый; 5 — этиловый;
6 — метиловый.
ное содержание воды в адсорбционном объеме достигает предельного зна-
чения в области низких концентраций воды в системах, содержащих
пропиловые и бутиловые спирты, тогда как в системах, содержащих эти-
ловый и метиловый спирты, заполнение адсорбционного объема водой
имеет место при у=0.6 и у=1 соответственно. Для всех исследованных
систем величина адсорбционного объема составляет около 0.21 см3/г,
что находится в хорошем соответствии со значением этой величины, полу-
ченной из изотермы адсорбции па-
ров воды.
Из рассмотрения начальных
участков изотерм адсорбции (рис. 3)
видно, что максимумы на изотер-
мах адсорбции воды из спиртов
С3—С4 не совпадают, значение
Та:=хМакс возрастает в ряду: изо-
бутанол, изопропанол, н-бутанол,
н-пропанол. В таком же порядке
будет уменьшаться эффективность
адсорбента при осушке спиртов.
Начальные участки изотерм (рис. 3)
позволяют количественно оценить
емкость адсорбента при осушке
спирта до любого заданного содер-
жания воды. Так, например, при
конечном содержании воды, рав-
ном 0.1% (вес.), емкость адсорбен-
та составляет: при осушке изобу-
тилового спирта — 13.8%, изопропилового — 13.0%, н-бутилового —
12.0%, н-пропилового — 10.5%, этилового — 4.0% и метилового — 0.7%.
Следовательно, применение цеолитов NaA будет сравнительно мало-
эффективным при абсолютировании этилового спирта и совершенно неце-
лесообразным при осушке метилового спирта, тогда как для остальных
спиртов цеолиты являются хорошими осушителями.
Расчетно-графическая обработка экспериментальных данных показала,
что изотермы адсорбции воды из растворов в спиртах С3—С4 хорошо ап-
проксимируются уравнением, типа уравнения Лэнгмюра:
ОрС (1 — с)
д-|-с
(2)
Приводим значения констант уравнения (2), определенных по двум-
трем сериям опытов, для расчета величин адсорбции воды цеолитами:
Растворитель «о ь
н-Пропанол 0.20 0.00090
Изопронанол .... 0.21 0.00062
н-Бутанол 0.202 0.00068
Изобутанол 0.210 0.00051
С помощью формул (1) и (2) и табличных данных можно рассчитать
потребное количество тг цеолита для осушки gr спирта от начальной кон-
центрации у0 до любой заданной концентрации у.
ЛИТЕРАТУРА
1. 1.1. К i р 1 i n g, Е. М. W г i g h t, Trans. Faraday Soc., 1959, 55, 1185.
2. А. Вайсбергер и др. Органические растворители. ИЛ, 1958.
3. Дж. Митчел, Д. С м и т. Акваметрия. ИЛ, 1952, 101.
20*
307
4. А. В. Киселев, Усн. химии, 1956, 25, 705.
5. А. В. К и с е л е в, Л. Ф. П а в л о в а, Кинетика и катализ, 1961, 2, 599.
6. L. Socconi, J. Am. Chem. Soc., 1960, 82, 3829.
ПРИМЕНЕНИЕ СИНТЕТИЧЕСКИХ ЦЕОЛИТОВ ДЛЯ ОСУШКИ
АЦЕТОНА В ПРОЦЕССЕ ПРОМЫШЛЕННОГО ПОЛУЧЕНИЯ ДИКЕТЕНА
А. Н. Салю ков, П. Н. Эндюсъкин, В. Н. Мазин, В. Г. Широков
В настоящее время осушку ацетона проводят с помощью ректификации,
что позволяет снизить содержание воды от 1.0—1.5% до 0.5—0.7%.
Для более глубокой осушки ацетона нами был предложен адсорбционный
метод с применением в качестве сорбента-осушителя синтетического цео-
лита NaA. В связи с этим было необходимо предварительное изучение не-
которых закономерностей процесса адсорбции воды цеолитом NaA из
жидких растворов воды в ацетоне.
Для определения зависимости величины адсорбции воды от равновес-
ной концентрации в ацетоне была снята изотерма адсорбции из раствора.
Статическая активность определялась по разнице исходной и равновесной
концентраций. Результаты опытов приведены в таблице.
Зависимость величины адсорбции воды из ацетона от равновесной
концентрации на цеолите NaA
Равновесная концентрация, С Величина ад- сорбции» а, мг/г Равновесная концен- трация, С Величина адсорбции а. мг/г
% мг/г % мг/г
0.10 1.0 85 1.0 10 205
0.16 1.6 110 1.2 12 208
0.21 2.1 112 1.5 15 210
0.31 3.1 146 2.1 21 210
0.65 6.5 190 2.6 26 211
Зависимость величины адсорбции от равновесной концентрации удо-
влетворительно описывается уравнением Лэнгмюра [1]. Были проведены
опыты по изучению скорости адсорбции воды из ацетона слоем формован-
ного цеолита NaA в 1 см в зависимости от зернения сорбента, природы
связующего и скорости потока раствора. Установлено, что кинетика ад-
сорбции удовлетворительно описывается уравнением
da
— (1)
где а — величина адсорбции за время, с0 — исходная концентрация,
у — равновесная концентрация, р — кинетический коэффициент.
Скорость адсорбции не зависит от вида связующего. Величина коэф-
фициента р связана со скоростью и зернением сорбента зависимостью
w0.53
₽ = 0.64 да, (2)
где со — скорость потока, d — зернение.
[ В наших исследованиях опыты проводились при концентрациях воды
в ацетоне, соответствующих второй области изотермы адсорбции. Из-
308
вестно (2], что для данной области концентраций при внешнедиффузион-
ном процессе уравнение динамики имеет вид
(3)
где т — время защитного действия слоя сорбента до проскока концен-
трации с, а0-—величина динамической емкости, со — скорость потока,
с0 — исходная концентрация сорбата, L — длина слоя сорбента, р— кине-
Продолжительность опыта., t,Mu.n.
Рис. 1. Расчетные (обозначены прерывис-
тыми линиями) и опытные выходные кри-
вые осушки ацетона при длине слоя сор-
бента 20 см.
1—при ш=0.85 см/мин. и Со=1.5%; 2— при
ш = 0.45 см/мин. и Со=1.5%; 3— при а>=
= 0.45 см/мин. и Со=1.0%.
Рис. 2. Схема установки для осушки
ацетона с помощью цеолита.
1 — напорный бак; 2 — ротаметр; 3 — адсор-
беры; 4 — сборник; 5 — емкость; 6 — элек-
тронагреватель; 7 — холодильник.
тический коэффициент, р = -^~, где у1 — равновесная концентрация при
а =г/2о0.
На рис. 1 приведены рассчитанные по уравненйю (3) и эксперимен-
тальные выходные кривые для слоя сорбента длиной 20 см при различных
скоростях потока и исходных концентрациях воды в ацетоне. Как видно
из рис. 1, динамика осушки ацетона удовлетворительно описывается урав-
нением (3).
Уравнение (3) было использовано нами с целью расчетов адсорберов
опытной установки для осушки ацетона. На рис. 2 показана схема уста-
новки для осушки ацетона с помощью цеолита NaA.
Ацетон из напорного бака (7) самотеком поступал в один из адсорберов
(5) и проходил через слой снизу вверх со скоростью 0.7 см/мин. Ско-
рость потока регулировалась ротаметром (2). Осушенный ацетон поступал
в сборник (7). Через каждые два часа определялась концентрация воды
в ацетоне за слоем. Проскоковая концентрация в первый момент времени
соответствовала содержанию воды в ацетоне 0.02—0.03%. Время защит-
ного действия до проскока воды в ацетоне 0.05% составляло 45—50 часов.
Регенерация слоя сорбента проводилась продувкой адсорбера подо-
гретым до температуры 300° азотом. Предварительно слой продувался
в течение 2 часов без подогрева. Общее время регенерации составило
26 часов. Более глубокая осушка ацетона позволила увеличить выход
дикетена на 6.5%.
ЛИТЕРАТУРА
1. С. Б р у и а у э р. Адсорбция газов и паров. ИЛ, 1948.
2. Е. И. Серпионова. Промышленная адсорбция газов и паров. Госхимиздат,
1956.
309
ЖИДКОФАЗНАЯ АДСОРБЦИЯ ВОДЫ ЦЕОЛИТАМИ ИЗ
ОЛЕФИНО-АРОМАТИЧЕСКИХ УГЛЕВОДОРОДНЫХ СМЕСЕЙ
Г. Я. Александрова, Г. С. Гольдин, Г. С. Шор
Настоящее сообщение посвящено исследованию жидкофазной ад-
сорбции воды из олефино-ароматических углеводородных смесей. В со-
став исследуемой смеси входят преимущественно олефиновые и олефино-
ароматические углеводороды с содержанием углерода в молекуле от 6
до 10 атомов. Предельное содержание воды в смеси составляет 0.1—
Рис. 1. Изотермы адсорбции воды на гранулирован-
ных и кристаллических цеолитах при температуре
20° С.
1 — NaA; 2 — СаА; 3 — СаХ; 4 — NaX. Светлые круж-
ки — кристаллы, темные — гранулы.
Рис. 2. Выходные кривые адсорбции во-
ды при различных скоростях потока.
1 — 0.72, 2 — 0.64, 3 — 0.48, 4 — 0.36; 5 —
0.20 л/час/см2.
0.15 вес.%. Для исследования применены образцы цеолитов А и X, как
гранулированные, так и мелкодисперсные кристаллические порошки.
Содержание воды в исходных и в осушенных углеводородных смесях
определялось гидридкальциевым методом. Снятие изотерм адсорбции
воды из смеси олефино-ароматических углеводородов проводилось по
общепринятой методике.
На рис. 1 приведены изотермы
адсорбции воды гранулированными и
кристаллическими цеолитами. Из ри-
сунка видно, что изотермы адсорбции
имеют крутой подъем в области ма-
лых равновесных концентраций и при
равновесной концентрации воды, рав-
ной 0.04%, практически наступает
насыщение. Из исследуемых образцов
(NaX, NaA, СаА, СаХ) наилучшим
осушителем углеводородной смеси
является цеолит NaA. Повышенная
эффективность действия цеолитов
NaA объясняется сочетанием двух
факторов: полярности адсорбируе-
мого вещества и наличием «отсеивающего эффекта».
Исследования в динамических условиях проводились на адсорбцион-
ной колонке следующих размеров: внутренний диаметр 18 мм, высота
800 мм. Чтобы добиться равномерной отработки сорбента, смесь подава-
лась снизу вверх: скорость потока регулировалась и в пределах одного
опыта поддерживалась постоянной.
310
Выходные кривые адсорбции воды их углеводородной смеси при раз-
личных скоростях потока приведены на рис. 2, из которого видно, что
первые порции смеси имеют различную глубину осушки в зависимости
от скорости потрка.
В течение некоторого промежутка времени степень осушки при дан-
ной скорости остается постоянной, а затем содержание влаги в смеси на-
чинает возрастать и постепенно достигает исход-
ного значения. На основании опытных данных бы-
ло рассчитано количество влаги, адсорбированной
цеолитом при различных скоростях потока угле-
водородной смеси. Зависимость динамической ак-
тивности цеолита от скорости потока углеводо-
родной смеси представлена на рис. 3, откуда вид-
но, что с увеличением скорости подачи углеводо-
родной смеси динамическая активность по воде
резко снижается.
В исследуемых пределах скоростей потока за-
висимость динамической активности от скорости
линейна. Эффективная осушка осуществляется
при скоростях потока не более 0.5 л/час/см2.
На рис. 4 приведены выходные кривые адсорб-
Рис. 3. Зависимость ди-
намической активности
цеолитов от скорости
потока (длина слоя
70 см).
ции воды из углеводородной смеси при скоростях
потока 0.36 л/час/см2.
Обработка опытных данных показала, что на-
блюдается линейная зависимость объема осушен-
ного раствора и динамической активности от дли-
ны слоя сорбента (рис. 5).
Дегидратации цеолитов предшествовала промывка сорбента легко-
кипящим растворителем, например бензином Б-70, с последующим отду-
вом паров углеводорода азотом, воздухом или вакуумированием. После
Рис. 4. Выходные кривые адсорбции
воды при скорости потока 0.36 л/час/
см2, температуре 20°, исходной кон-
центрации влаги 0.1 вес. % при раз-
личной длине слоя (см).
2— 20, 2 — 35, 3 — 50, 4 — 70 см.
осушенного раствора и дина-
мической активности цеолитов
от длины слоя сорбента (ско-
рость потока 0.36 л/час/см2).
1 — динамическая активность; 2 —
объем осушенной смеси.
85 циклов сорбции—десорбции влаги снижения адсорбционной емкости
цеолита не отмечено.
Лабораторные исследования подтверждены на пилотной установке
(внутренний диаметр адсорбера 51 мм, длина слой сорбента 2000 мм).
Проведенные исследования показали, что применение цеолитов марки
NaA дает возможность осуществить глубокую осушку смеси олефино-аро-
матических углеводородов без изменения ее химического состава.
311
ОСУШКА НИЗКОМОЛЕКУЛЯРНЫХ ЖИРНЫХ кислот
С ПОМОЩЬЮ ПОРИСТЫХ СТЕКОЛ — МОЛЕКУЛЯРНЫХ сит
И СИНТЕТИЧЕСКОГО МОРДЕНИТА
Т. М. Буркат, Д. П. Добычин, А. С. Мошкевич, Т. А. Ушакова
Разрабатывая и исследуя пористые стекла — молекулярные сита [1 —
4], мы обратились к осушке низкомолекулярных жирных кислот как
к специфически подходящему примеру возможного применения этих но-
вых сорбентов в области тонкой очистки и разделения смесей. Основани
ями к тому были высокая химическая стойкость пористых стекол к кисло-
там, а также меньшая, чем у цеолитов, полярность, что позволяло рас-
считывать на более легкую регенерируемость, чем у цеолитов.
Результаты наших первых работ показали [2—4], что пористые стекла —
молекулярные сита действительно обладают способностью к глубокой
осушке уксусной и пропионовой кислот, протекающей по молекулярно-
ситовому механизму. Лучшие результаты достигались на стеклах, близ-
ких по размерам «окон» пор к цеолитам типа NaA, причем осушка про-
пионовой кислоты протекала эффективнее, чем уксусной.
Цеолиты NaA, напротив, оказались совершенно непригодными для
подобных целей в силу полного их разрушения в уксусной кислоте [2, 3].
Как теоретический, так и практический интерес представлял вопрос
о способности пористых стекол к молекулярно-ситовой осушке муравьи-
ной кислоты и ее смесей с другими жирными кислотами. Нами были про-
ведены опыты по осушке в динамических условиях смеси муравьиной и
уксусной кислот (1 : 2) с 2.5% воды. В этих опытах были использованы
пористые стекла, близкие по размерам пор к цеолитам типа NaA и более
крупнопористые, а также к цеолитам типа КА (сорбирующим воду, но
мало или практически совсем не сорбирующим метиловый спирт). Такие
пористые стекла были получены нами из натриевоборосиликатных стекол,
а также предоставлены нам Л. С. Ястребовой, которая получила их из
калиевоалюмосиликатных стекол.
Опыты проводились на колонке длиной 50 см на пористых стеклах
фракции 0.1—0.25 и 0.25—0.5 мм при скоростях потока от 0.3 до 2.3 см8/см2
мин. Определение содержания воды производилось по методу Фишера.
Стекла типа NaA и более крупнопористые оказались неспособными к от-
делению воды от муравьиной кислоты. Так, на образце 40, имеющем поры,
близкие к таковым у цеолитов типа А, а также и более крупные, муравьи-
ная кислота проникает в поры сорбента, подавляя сорбцию воды. Пони-
жение содержания воды в смеси кислот сопровождалось на этом образце
понижением содержания в первых пробах элюата и муравьиной кислоты
(20.8% вместо 33% в исходной смеси). Заполнение пространства пор во-
дой при полной отработке слоя составляет 0.010 г/г, тогда как запол-
нение муравьиной кислотой равно 0.039 г/г и соответственно общее за-
полнение сорбционного пространства составляет 0.049 г/г (около 25%
общего объема пор).
Максимальная глубина осушки смеси муравьиной и уксусной кислот
была достигнута на пористом стекле типа КА (образец 22). Остаточная
концентрация воды в смеси кислот составляла 0.15—0.25% при исходном
содержании около 2.5%. Общее заполнение сорбционного пространства
составляло лишь 0.026 г/г (около 13—15% общего объема пор). Однако
количество сорбированной воды в этом случае составляло 0.021 г/г, и
только 0.005 г/г приходилось на муравьиную кислоту.
Достигнутый выход при высоте слоя 500 мм и скорости потока около
0.4 см®/см2мин. не превышал 0.75 г/г сорбента. Повышение температуры
от 20 до 40° С на стекле № 21 (КА) увеличивало заполнение пор водой от
312
Осушка уксусной кислоты (начальное содержание воды ~~2%)
на пористых стеклах — молекулярных ситах
Опыты проводились с пористым стеклом фракции 0.1—0.25 мм на колонке диаметром 5.5 мм
длиной 42.5 см, за исключением опыта № 48 на стекле № 41, где длина колонки составляла 30 см
№ опыта I Образец С адсорбции воды «s„ о см3/г Отношение величин адсорбции спиртов и воды при р/р9 = = 0.6—0.7 Скорость подачи, и, см’/см’мин. Выход осушенной кислоты, г/г Динамиче- ская актив- ность по воде ад, г/г Использо- вание объема пор % | Длина зоны массопе- | редачи, Lq, г мм |
д о к р С к р я о и О к р до про- скока до насы- щения до про- скока до насы- щения до про- скока до насы- щения
38 30/2 ГОИ 0.130 1 с 0.6 2.7 3.1 0.057 0.06 43.8 46 65
39 30/4 ГОИ 0.096 0.27 0.5 0.47 5.6 0.009 0.039 9.3 41 >1000
37 30/6 ГОИ 0.120 0.33 0 0.48 1.0 5.1 0.02 0.05 16.7 41.6 470
36 № 1 IIХС 0.150 0.74 6 0.039® 0.54 2.9 3.2 0.062 0.063 41.3 42.0 26.5
40 № 40 0.213 1 1 0.44 1.0 1.4 0.019 0.025 8.9 11.7 149
48 № 41 0.196 0.51 0.078 0.53 2.05 3.5 0.042 0.05 21.4 25.6 172
а Изотерма адсорбции спирта идет несколько выше изотермы адсорбции воды,
является, очевидно, увеличение плотности спирта в адсорбированном состоянии.
6 При р/Рз = 0.3.
в Приведено соотношение величин адсорбции азота и воды.
г Величина Lo вычислялась обычным методом, согласно [®].
причиной чему
0.016 до 0.019 г/г. Уменьшение скорости потока и размера зерна (от
0.25—0.5 до 0.1—0.25 мм) также несколько улучшало результаты. Сле-
довательно, определяющей стадией процесса является диффузия внутри
пор сорбента. Поэтому возможности интенсификации процесса лежат
прежде всего на пути уменьшения внутридиффузионного сопротивления,
а также повышения полезной емкости сорбента посредством улучшения
его пористой структуры.
При изучении осушки уксусной кислоты, кроме упоминавшихся
выше образцов пористых стекол, был использован также представлен-
ный С. П. Ждановым образец 1 пористого стекла — молекулярного си-
та [5, 3]. Опыты велись в условиях, сходных с описанными выше, при
содержаниях воды в осушаемой уксусной кислоте около 0.2 и 2—2.5 % и при
скоростях подачи от 0.2 до 2 г/см2мин.
В табл. 1 приведены результаты ряда проведенных экспериментов.
На рис. 1 изображены выходные кривые наиболее характерных опытов,
а на рис. 2 — изотермы адсорбции воды, метилового и этилового спиртов
на наших образцах.
Глубина осушки уксусной кислоты доходила до 0.015—0.02%. Мак-
симальный выход осушенной уксусной кислоты при Со=2% Н2О дости-
гал 2—3 г/г, а в случае ледяной (0.4% Н2О) — до 6.4 г/г. Судя по соот-
ношениям сорбции воды, метанола и веществ с более крупными молеку-
лами, пористая структура наиболее активных образцов 1 и 30/2 близка
к NaA, но размеры пор у образца 30/2 несколько больше, чем у 1 (см. таб-
лицу). Несколько более узки «окна» пор у примыкающего к ним образца 41.
Сорбция метилового спирта на нем почти вдвое меньше сорбции воды.
Функция распределения объема пор по радиусу у этого образца шире,
чем у 30/2: адсорбция этанола составляет 15—20% от адсорбции метанола.
Эти отличия в пористой структуре и понижают эффективность процесса
обезвоживания уксусной кислоты на образце 41 по сравнению с 1 и 30/2.
Хотя диффузия в порах сорбента остается лимитирующей стадией про-
цесса и в случае осушки уксусной кислоты, внутридиффузное сопротив-
313.
.ление на ряде образцов незначительно. Это проявляется в сокращении
длины’зоны массопередачи у лучших образцов до 2.5—6.5 см. Однако у бо-
лее мелкопористых стекол (например, 30/4 и 30/6), приближающихся
к КА, внутридиффузионное сопротивление велико, зона массопередачи
растянута и эффективность их действия понижена. Не исключено, что сни-
Рис. 1. Выходные кривые опытов по осушке уксусной кислоты на по-
ристых стеклах — молекулярных ситах (условия опытов см. в табл. 1).
* 1 — образец 1 ИХС АН СССР; 2 — образец 30/2 ГОИ; 3 — образец 41; 4 —< об-
разец 40; 5 — образец 30/6 ГОИ.
жение тем или иным путем (уменьшение размера зерна, повышение тем-
пературы и т. п.) внутридиффузионного сопротивления на стеклах таких
типов повысит эффективность их действия и позволит иначе подойти
к оценке их пригодности для практических целей.
Рис. 2. Изотермы адсорбции паров воды, метилового и этилового спиртов на
образцах пористых стекол № 40 (а), № 41 (б) и синтетическом мордените (в).
1 — вода (адсорбция); 2 — вода (десорбция); 3 — метиловый спирт; 4 — этиловый спирт.
Малоэффективны также и стекла с чрезмерно крупными размерами
пор (образец 40), причем на заметное повышение эффективности их дей-
ствия за счет изменения кинетических условий процесса, естественно,
рассчитывать не приходится. Тот факт, что в случае более крупнопори-
стого стекла (40) длина зоны массопередачи в несколько раз больше,
чем у более мелкопористых образцов 1 и 30/2, вызывается, очевидно, его
неоднороднопористой структурой и наличием весьма мелких пор.
С увеличением размера зерен пористого стекла зона массопередачи
растягивается. Так, у образца 40, обладающего и мелкими, и крупными
314
{типа А и крупнее) порами, при переходе от зерен размером 0.1—0.25 мм
к 0.25—0.5 мм длина зоны массопередачи
(от 23—25 до 45 см при скоростях подачи
К увеличению длины зоны массопередачи (Lo)
на фракции 0.1—0.25 мм этого стекла от 15
до 23—25 см приводит и увеличение скорости
потока (U) от 0.44 см3/см2 • мин. до 1.25 см3/см2 •
мин., что соответствует известной обратной
пропорциональности зависимости Lo oi\/U-
Были проведены опыты по осушке уксус-
ной кислоты на гранулированном пористом
•стекле типа КА. Гранулы были изготовлены
Г. М. Белоцерковским и В. И. Захаровым из
пористого стекла 30/6, измельченного до раз-
мера — 1 мк. Опыты показали, что при оди-
наковом зернении длина зоны массопередачи
•сокращается от > 75 до 35—40 см (рис. 3).
В ряде опытов после проскока наблюда-
лось превышение концентрации воды в элюа-
те ее содержания в исходной кислоте. Этот
эффект может быть объяснен только тем,
что вследствие большей скорости диффузии
вода первой проникает в поры, а более круп-
ные молекулы кислоты входят в поры с не-
которым запаздыванием и вытесняют при этом
находящуюся там воду. Вследствие этого
увеличивается почти вдвое
порядка 1.5 см3/см2 - мин.).
2 злюата/г сорбента
Рис. 3. Выходные кривые опы-
тов по осушке уксусной кисло-
ты на зерненом и гранулиро-
ванном пористом стекле типа
КА (образец 30/6 ГОИ). Зер-
нение 0.1—0.25 мм, длина
слоя 42.5 см.
1 — зерненое пористое стекло; 2 —
гранулированное пористое стекло.
•содержание воды в элюате возрастает до
значений, превосходящих исходную концентрацию. Такого рода ано-
мальные эффекты, возможные только в случаях молекулярйо-ситового
Рис. 4. Кинетика сорбции образцом
40 из уксусной кислоты, содержащей
25% воды.
1 —концентрация воды (%) в сорбированной
фазе; 2 — общая величина адсорбции (г/г);
з — содержание воды в порах (мг/г).
разделения по диффузионному ме-
ханизму, уже отмечались [2, 7].
Исследование состава сорбирован-
ной фазы 1 дало прямые эксперимен-
тальные доказательства справедли-
вости изложенной картины. При ад-
сорбции в статических условиях из
уксусной кислоты с 2% воды сорби-
рованная фаза содержит 15—20% во-
ды. По мере того, как общая величина
адсорбции (воды и кислоты) в процес-
се опыта растет, концентрация воды
в сорбированной фазе падает, а абсо-
лютное количество сорбированной
воды проходит через максимум
(рис. 4).
Среди данных табл. 1 представ-
ляет особый интерес связь между эф-
фективностью осушки уксусной кис-
лоты на данном стекле и соотношение м
сорбции на нем метилового и этило-
вого спиртов и воды. Полученные результаты указывают на то, что для
данной системы оптимальным является такой размер «окон» пор, при ко-
тором сорбция метилового спирта близка к сорбции воды, а сорбция эти-
лового — подавлена, т. е. размер, близкий к NaA.
1 Экспериментальная часть проведена практикантом ЛГУ Р. А. Абакумовой.
315
Как и ожидалось, относительно небольшая полярность пористых сте-
кол (по сравнению с цеолитами) действительно облегчает их регенерацию.
Многократное восстановление сорбционных свойств пористых стекол
в циклических опытах обеспечивалось их прогревом при температурах
до 120—150° С в токе осушенного воздуха.
Изложенные результаты могут служить для оценки показателей про-
цесса осушки уксусной кислоты, а также дают возможность научно обо-
снованных поисков пористых стекол — молекулярных сит с повышенной
емкостью и оптимальными для данной системы размерами пор.
В литературе имелись указания на возможность осушки кислых сред
с помощью синтетических цеолитов с повышенным содержанием кремне-
зема [8, 9]. В 1963 г. такого типа цеолит (морденит) был синтезирован
Рис. 5. Выходные кривые опытов по осушке уксус-
ной кислоты на синтетическом мордените (морденит
предварительно пропитан осушенной кислотой).
1 — опыт 30 : II = 10 см; U = 0.5 см3 * * * 7/см2 мин.
2 — опыт 32 : Н = 45.5 см; U = 1.96 см3/см2 мин.
3 — опыт 35 ; Н = 30 см; U = 2.04 см3/см2 мин.
4 — опыт 41 : Н = 30 см; U = 1.8 см3/см2 мин.
Э. Э. Сендеровым [10], и нам были предоставлены для испытаний его
образцы. Они представляли собой тонкие порошки, структура которыхт
согласно рентгеноструктурным определениям, проведенным Н. В. Ивано-
вой, отвечала данным Сендерова [10], а кристаллы имели размеры по-
рядка десятых долей микрона. Для проведения динамических опытов
исходный порошок был спрессован и раздроблен до получения фракции
0.25—0.5 мм. Рентгеноструктурные и адсорбционные определения сви-
детельствовали об идентичности структуры исходного порошка и прессо-
ванных зерен.
Как видно на рис. 5, выходные кривые после проскока поднимались
круто. Зона массопередачи была коротка и изменялась от 17—18 до 75—
80 мм при увеличении скорости подачи от 0.5 до 3 см3/см2 • мин. На мор
дените, как и на пористых стеклах, наблюдался аномальный эффект'пре-
вышения в элюате после проскока начального содержания воды (рис. 5,
7, 2, 3). Обсуждавшаяся выше диффузионная природа этого явления была
доказана на примере морденита прямым экспериментом. При осушке
уксусной кислоты с 2% воды на слое морденита, выдержанного в осушен-
ной кислоте в течение 12 часов, было обнаружено (рис. 5, 4) полное ис-
чезновение этого эффекта. Тем самым было доказано, что упомянутый
аномальный эффект порождается конкурентной диффузией воды и кис-
лоты в порах сорбента.
Кроме изложенного, нами исследовалась осушка на мордените и по-
ристом стекле 41 смесей винилацетата с уксусной кислотой, получаемых
в НИИПП при синтезе винилацетата. При начальных содержаниях воды
0.1—0.7% на обоих сорбентах достигалась глубина осушки 0.01% и
ниже. При начальных содержаниях воды 0.15 и 0.7% полная емкость.
316
пористого стекла и морденита соответственно составляла 0.024 и 0.078 г/г.
Полученные результаты свидетельствуют о возможности успешного ис-
пользования морденита и пористых стекол и в этом направлении.
ЛИТЕРАТУРА
1. Д. П. До бычин, ЖПХ, 1962, 35, 51.
2. Д. П. Добычин, Т. М. Буркат и Н. Н. К и. с е л е в а, в сб.: Синтети-
ческие цеолиты, Изд. АН СССР, 1962, 75.
3. Т. М. Б у р к а т, Д. П. Д о б ы ч и н и С. П. Ж д а н о в, ДАН СССР, 1963,
150 1293.
4. Т. М.’б у р'к а т, Д. И. Д о б ы ч и н, ЖПХ, 1965, 38, 9, 1994.
5. С. П. Жданов и Е. В. К о р о м а л ь ди, ДАН СССР, 1961, 138, 870.
6. R. Т г е у b а 1. Mass Transfer Operation. Me Grow Hill, N. Y., 1955, 504.
7. Л. Г. Тонконог и К. В. Ч мутов, в сб.: Синтетические цеолиты, Изд.
АН СССР, 1962, 230.
8. P.-W. Lowenberg, J. Appl. Chem., 1911, 9, 8, 417.
9. Chem. and Engng. News, 1962, 40, 12, 52, 54.
10. Э. Э. С e н д e p о в, Геохимия, 1963, 9, 820.
ОЧИСТКА ВОЗДУХА ОТ СЛЕДОВ АЦЕТИЛЕНА ЦЕОЛИТАМИ СаА
НА ПИЛОТНОЙ УСТАНОВКЕ
Н. Г. Сихарулидзе, Г. В. Цицишвили, Ц. В. Маградзе,
Т. Г. Андроникашвили
Известно [1, 2], что одним из важнейших условий безопасной работы
блоков разделения воздуха является полная очистка воздуха от примесей
ацетилена.
Между тем в последнее время появилось сообщение [3] о том, что при-
менением синтетических цеолитов 5А на кислородных установках обеспе-
Рис. 1. Схема очистки воздуха от следов ацети-
лена на пилотной установке.
1 — компрессор 039-А; 2 — маслоотделитель; 3 — адсор-
бер с силикагелем; 4 — адсорбер с цеолитом СаА; 5 —
электроподогреватели; 6 — расходомер; 7 — пробоотборные
точки, I—IV — слои сорбента.
чивается гарантийная очистка воздуха от ацетилена при р =20 ати и
/=4° С.
Ранее нами [4] в лабораторных условиях изучалась возможность пол-
ной очистки воздуха от ацетилена при нормальном давлении и темпера-
туре в пределах 20—70° С. На основании лабораторных данных была смон-
тирована пилотная установка (рис. 1).
317
Испытания проводились при линейных скоростях 0.27, 0.36 и 0.42 м/сек-
при содержании ацетилена 2.4 мл/м3, т. е. примерно в 80 раз больше воз-
можного содержания его в воздухе (0.03 мл/м3).
При каждой линейной скорости исследовалось влияние высоты слоя
сорбента на эффективную емкость цеолита СаА, по отношению к ацети-
лену. Изучалась также динамика насыщения ацетиленом отдельных слоев
Рис. 2. Влияние высоты слоя сор-
бентов на эффективную емкость цео-
литов СаА по отношению к ацети-
лену.
1 — 10=0.27, 2 — 10=0.36, 3 — 10=0.42
м/сек.
цеолитов.
Результаты опытов по изучению влия-
ния высоты слоя сорбентов на эффек
тивную емкость цеолитов представлены
на рис. 2.
Из рисунка видно, что общая эффек-
тивная емкость цеолитов не соответст-
вует обычному нарастанию емкостей,
полученных при лабораторных испыта-
ниях [4]. Причиной этого является, по-
видимому, неудовлетворительная реге-
нерация верхнего и нижнего слоев в
адсорбере, что подтверждается данными
изучения работы отдельных слоев при
общей высоте слоя 40 см (см. таблицу).
Причиной ненормальной работы слоев
I и IV является, по-видимому, попада-
ние влаги в процессе активации и ре-
генерации цеолитов воздухом, предва-
рительно осушенным в силикагелевом
адсорбере. Опыт работы показывает,
что силикагель при низких давлениях
и температуре 25 4- 35° С не произво-
дит эффективной осушки воздуха. На-
блюдается повышение температуры воз-
духа на выходе из цеолитного адсорбе-
ра на 15 4- 20° С, причиной чего, по всей вероятности, является теп-
лота адсорбции паров воды.
Нами изучалась также десорбция ацетилена из цеолитов в процессе
очистки воздуха на пилотной установке (рис. 3) для разработки оптималь-
ного режима работы более крупных адсорберов.
Как видно из рис. 3, десорбция ацетилена наиболее быстро протекает
на I и IV слоях, причем IV (нижний) слой при наступлении проскока
почти мгновенно теряет поглотительную способность.
На основании проведенных опытов на пилотной установке можно
сделать следующие выводы:
1. Изучена динамическая емкость цеолитов СаА по отношению к аце-
тилену на пилотной установке, которая может служить моделью для про-
мышленных установок.
Эффективная емкость отдельных слоев
цеолита (мг/100 г)
№ слоя сорбента Скорость воздуха, м/сек.
0.27 0.36 0.42
I 6.0 5.0 3.50
II 22.0 18.60 15.90
III 34.2 31.60 28.30
IV 18.8 16.70 13.90
318
2. Наиболее активными, по отношению к поглощению ацетилена, яв-
ляются средние слои сорбента, емкость которых близка к 20—30?мг/100 г.
Следовательно, при определении полезной высоты адсорбера в промышлен-
_i_
1401,мин.
Рис. 3. Десорбция ацетилена по слоям цеолита СаА (Z—IV)
при общей высоте слоя 40 см.
ных установках следует учитывать возможность образования малоэффек-
тивных слоев адсорбента.
3. Учитывая влияние влаги на процесс сорбции ацетилена, следует
предусмотреть двухступенчатую очистку воздуха от ацетилена цеолитами
типа СаА, первая ступень которой будет работать в основном в качестве
осушителя воздуха.
ЛИТЕРАТУРА
1. Кислород, 1958, 5, 65.
2. Chem. Engng. Progress, 1960, 6, 68.
3. С a n a d. Chem. Processing, 1963, 3, 47.
4. Н.Г.С ихарулидзе, Г. В. Цицишвили, Т. Г. Андроникашвили,
ЖПХ, 1965, 38, 1536.
РАЗДЕЛЕНИЕ ИЗОТОПОВ ВОДОРОДА ПРИ АДСОРБЦИИ НА
СИНТЕТИЧЕСКИХ ЦЕОЛИТАХ
В. Е. Кочурихин, Я. Д. Зелъвенский
Использование адсорбции как метода разделения изотопов представ-
ляет значительный интерес, так как в системе газ—твердое тело возможны
высокие значения коэффициентов разделения. При этом в качестве адсор-
бентов наибольшее внимание привлекают синтетические цеолиты [1J.
С другой стороны, смеси изотопов являются удобными модельными сме-
сями для изучения адсорбционных свойств и разделяющей способности
синтетических цеолитов.
В связи с этим нами были проведены измерения коэффициента разде-
ления смеси Н2—D2 при адсорбции на различных синтетических цеолитах.
Эксперименты проводили статическим методом на вакуумной циркуля-
ционной установке. Анализ на содержание D2 проводили методом тепло-
проводности. В работе использовали гранулированные синтетические цео-
литы NaX, NaA, СаА и СаХ.
На рис. 1 изображена зависимость коэффициента разделения а смеси
Н2—D2 на цеолитах NaA и NaX от давления в интервале давлений 40—
319'
600 мм рт. ст. Из графика видно, что коэффициент разделения падает
по мере роста давления в интервале 40—200 мм рт. ст. и остается постоян-
Рис. 1. Зависимость коэффициента разделе-
ния смеси Н2—Юг на цеолитах NaA и NaX
от давления.
J — цеолит NaA, Т = 78°К; 2 — цеолит NaA, Т =
= 82°К; 3 — цеолит NaX, Т = 79° К; 4 — цеолит NaX,
Т = 84° К.
ным при дальнейшем увеличе-
нии давления.
Зависимость коэффициента
разделения смеси Н2—D2 от
температуры изучена при дав-
лениях 60 и 200 мм рт. ст. в
интервале температур 77—90° К
на цеолитах NaA и NaX. По-
лученные результаты представ-
лены на рис. 2 в координатах
1g а—ЦТ. Как видно из этого
графика, точки удовлетворитель-
но укладываются на прямые.
Коэффициент разделения при
равных внешних условиях на
цеолите NaA выше, чем на цео-
лите NaX. Кроме того, из гра-
фика видно, что точки, полу-
ченные при р=200 мм рт. ст.
для гранулированного и порошкообразного цеолитов типа NaA, хорошо
укладываются на одну прямую. Это говорит о том, что связующая добавка
не влияет на коэффициент разделения (хотя снижает величину адсорбции
водорода и дейтерия).
Мы исследовали также влияние за-
мены катиона Na+ цеолитов типа А и X
другими катионами на коэффициент
разделения. Для этой цели путем ион-
ного обмена были получены цеолиты
LiA, SrA, MnA, СоА, NiA, SrX и ВаХ.
На этих цеолитах измерены коэффи-
циенты разделения при адсорбции сме-
си Н2—D2 при давлении р=200 мм рт.
ст. в интервале температур 77—90° К.
Результаты представлены на рис. 3 и
4 в координатах 1g а—ЦТ.
Во всех изученных случаях точки
удовлетворительно укладываются на
прямые в этих координатах. Коэффи-
циенты разделения при р=200 мм рт. ст.
и Т= 77.3° К на этих цеолитах распола-
гаются в следующих последовательно-
стях: NaA > LiA > СаА > SrA >
> NiA > СоА > MnA и SrX > СаХ >
> NaX > ВаХ.
Тангенс угла наклона прямых на
Рис. 2. Зависимость коэффициента
разделения смеси Н2—D2 от темпе-
ратуры в координатах 1g а — 102/71
на цеолитах NaA и NaX.
1 — цеолит NaA, р = 60 мм рт. ст.; 2 — цео-
лит NaA, р = 200 мм рт. ст. (черные круж-
ки — гранулированный цеолит, светлые —
порошкообразный); 3 — цеолит NaX, р =
=60 мм рт. ст.; 4 — цеолит NaX, р = 200 мм
рт. ст.
рис. 2—4 определяет разность нуле-
вых энергий АЕ0, близкую к разности теплот адсорбции D2 и Н2. Так как
отношение теплот адсорбции D2 и Н2 можно считать постоянным, то тан-
генс угла наклона этих прямых пропорционален теплоте адсорбции водо-
рода (или дейтерия).
Из представленных данных видно, что замена катиона Na+ на другие
катионы меняет энергию взаимодействия молекулы водорода с поверх-
ностью цеолита. При замене Na+ на Li+, когда радиус «окон», ведущих
в большие полости цеолитов, увеличивается от 4.2 А для NaA до 4.4 А
32(1
для Li А [2], энергия взаимодействия молекулы с цеолитом типа А умень-
шается.
Рис. 3. Зависимость коэффициента раз- Рис. 4. Зависимость коэффициента раз-
деления смеси Н2—D2 от температуры деления смеси Н2—D2 от температуры
на различных цеолитах типа X в коор- на различных цеолитах типа А в ко-
динатах 1g а — 102/Т. ординатах 1g а — 102/Г.
1 — SrX; 2 — NaX; 3 — СаХ; 4 — ВаХ. 1 — NaA: 2 — L1A; 3 — СаА; 4 — SrA; 5 — NiA;
6 —СоА; 7 — МпА.
Однако при переходе от СаА к SrA и от СаХ к ВаХ, когда радиус
«окон» убывает, энергия взаимодействия молекулы Н2 с цеолитом в этом
ряду также убывает.
Из изложенного выше следует, что простой и однозначной связи между
размером пор цеолитов и их разделяющей способностью нет и что причи-
ной изменения адсорбционных свойств цеолитов в этом случае является
изменение химического состава.
ЛИТЕРАТУРА
1. D. Basmadijan, Canad. J. Chem., 1960, 38, 141.
2. И. E. H ейма p к, М. А. Пионтковская, А. Е. Л у к а ш, Р. С. Тю-
тю н н и к, в сб.: Синтетические цеолиты, Изд. АН СССР, 1962, 49.
ПРИМЕНЕНИЕ СИНТЕТИЧЕСКИХ ЦЕОЛИТОВ
ДЛЯ АДСОРБЦИОННО-ТЕРМИЧЕСКОЙ ОЧИСТКИ АРГОНА
ОТ КИСЛОРОДА
Г. А. Головко
Получение чистого аргона непосредственно из современного блока
разделения воздуха практически невозможно. Объясняется это трудно-
стями ректификации смеси аргон—кислород, так как температуры кипе-
ния этих газов отличаются всего на 3°, а летучести — на 1296.
В связи с этим на воздухоразделительных установках производится
так называемый «сырой аргон», содержащий от 2 до 10% кислорода и не-
сколько процентов азота (хотя последний можно легко отделить методом
ректификации). Сырой аргон очищается от кислорода путем связывания
21 Цеолиты
321
последнего кислородоактивными металлами, либо методом каталитиче-
ского гидрирования с помощью водорода. Метод каталитического гидри-
рования, являющийся сейчас основным, позволяет получать аргон высо-
Рис. 1. Принципиальная схема установки для
адсорбционно-термического разделения смеси ар-
гон—кислород.
1 — баллон с чистым кислородом; 2 — баллон с чистым
аргоном: з — емкость для смеси; 4, 9 — фильтр; 5 — теп-
лообменник-вымор аживатель; 6 — динамическая трубка;
7 — сосуд Дьюара; 8 — электропечь; 10 — баллон с ре-
генерирующим газом; 11 — газгольдер.
кой чистоты, однако необходимость использования дорогостоящего палла-
диевого катализатора и водорода повышает стоимость установки, делает
ее взрывоопасной и значительно усложняет технологическую схему.
Рис. 3. Выходная
кривая по кислороду
(содержание кислоро-
да в очищенном арго-
не) при температуре
—180 ° С.
Рис. 2. Ход процесса реге-
нерации и охлаждения цео-
лита во времени.
Участок 1—2 — откачка до 1—
10 мм рт. ст.; 2—з — охлажде-
ние за счет холодного воздуха;
3—4 — охлаждение за счет ис-
парения жидкого кислорода;
4—5 — рабочий режим.
Именно несовершенство методов очистки определяет высокую стоимость
чистого аргона.
В настоящее время имеется возможность [1—3] организовать очистку
аргона от кислорода на новой взрывобезопасной основе — методом низко-
температурной адсорбции с помощью синтетических цеолитов.
Для исследования этого вопроса созданаукрупненная установка, осна-
щенная современными измерительными приборами. Принципиальная
322
схема установки ясна из приведенного рис. 1. В динамическую трубку
(изготовленную из латунной трубы диаметром 51X3.5 мм) загружено
1050 г цеолита NaA.
С целью изучения свойств цеолита при повышенных термических на-
грузках проведено более 20 циклов в соответствии с режимом регенерации
и охлаждения, представленным на рис. 2. Существенного ухудшения
адсорбционных свойств цеолита не наблюдалось.
Селективное разделение смеси аргон—кислорода (95 4-98% и 5 4“ 2%
О2) достигается при температуре, близкой к температуре кипения кисло-
рода и скорости потока до 1.2 л/мин. см2. Количество окклюдированного
из смеси кислорода составляет несколько процентов к весу дегидратиро-
ванного цеолита. Характер выходных кривых (рис. 3) указывает на хоро-
шие адсорбционные свойства исследуемого цеолита. Остаточное содержа-
ние кислорода в очищенном аргоне менее 0.005% по объему.
Таким образом, результаты проведенных опытов позволяют сделать
вывод о перспективности применения отечественных синтетических цео-
литов для очистки аргона от кислорода.
ЛИТЕРАТУРА
1. R. М. В а г г е г, Brennstoffchemie, 1954, 35, 21/22.
2. R. A. Jouns, В. М. Milton, US Pat., 2810454, 1957.
3. J. W. Armon d, Brit. Pat., Spec. Cl. 90, К 1A, 809168, 1959.
МОЛЕКУЛЯРНЫЕ СИТА ДЛЯ ПОЛИМЕРОВ
Е. К. Богачева, Ю. А. Элыпеков
Применение цеолитов в качестве молекулярных сит ограничивается
низкомолекулярными веществами. Принцип молекулярно-ситового дей-
ствия может быть распространен на разделение и исследование высоко-
молекулярных соединений, в том числе и растворов полимеров. Для
определения доступности внутренней поверхности пористых адсорбен-
тов для макромолекул изучена адсорбция полистирола (ПС) и полиди-
метилсилоксана (ПДМС) из разбавленных растворов на пористых и непо-
ристых адсорбентах. На рис. 1 приведены изотермы гиббсовой адсорбции
полистирола с молекулярным весом 717=290 000 из разбавленных раство-
ров с СС14 на непористом аэросиле (с удельной поверхностью по азоту
s=170 м2/г) и широкопористых силикагелях С-41 (s=40 м2/г, преимуще-
ственный диаметр пор d=750 А), СГ-6 (s=120 м2/г, (2=280 А) и ШСК
(s=340 м2/г, d=120 А). Аэросил и силикагели С-41 и СГ-6 хорошо адсор-
бируют полистирол, адсорбция же его на образце ШСК ничтожно мала
вследствие того, что размеры макромолекул значительно больше размеров
устьев пор. Из сопоставления величин адсорбции полистирола, отнесен-
ных к единице поверхности адсорбента, видно, что макромолекулы поли-
стирола не могут проникнуть в наиболее узкие поры образца СГ-6 и по-
этому величины адсорбции на этом образце меньше соответствующих
величин для образца С-41. Можно предполагать, что образец СГ-6 не ад-
сорбирует те макромолекулы, у которых молекулярный вес больше сред-
нечислового и из-за стерических затруднений (размер макромолекул
соизмерим с поперечником пор) весьма медленно достигается истинное
равновесие. На рис. 2 приведены кинетические данные по адсорбции поли-
стирола на образцах С-41 и СГ-6. В случае образца С-41 равновесие практи-
21*
323
чески достигается за трое суток. Скорость адсорбции полистирола на сили-
кагеле СГ-6 сильно зависит от количества адсорбента: для 1 г адсорбента
количество полистирола, адсорбированного за 1 месяц, составляет 50%
Рис. 1. Изотермы адсорбции полистирола из растворов CCI4.
1 — аэросил; 2 — силикагель С-41; з — силикагель СГ-6; 4 — силика-
гель ШСК. Изотермы получены после шестимесячного перемешивании
суспензий. Справа — величины адсорбции в мг/м*.
равновесного, для меньшей навески — 0.25 г, равновесие также еще не до-
стигается за 1 месяц, но величина адсорбции отличается от равновесной
только на 10%. Следует подчеркнуть характерную особенность для адсорб-
ции полистирола на образце СГ-6: вначале адсорбированная фаза оказы-
Рис. 2. Зависимость адсорбции полистирола от времени
контакта с силикагелями.
1 — С-41; 2 — СГ-6 для количества адсорбента гп=1 г; 3 — СГ-6
для количества адсорбента т—0.25 г. Черные кружки обозначают
вторую серию опыта.
вается обогащенной молекулами растворителя, и в первые минуты наблю-
дается отрицательная адсорбция полистирола (рис. 2).
Результаты показывают, что в среднем размер глобул полистирола
(2I/-29O ООО) менее 280, но больше 120 А. Для получения более четких
результатов нужны более узкие фракции полистирола и адсорбенты с уз-
ким распределением числа пор по размерам. Близкие результаты были
324
получены при изучении адсорбции ПДМС с М-350 ООО из растворов в н-
гексане на аэросиле, силикагелях С-41 и ШСК, окиси алюминия и цеолите
13Х из ВНИИНП (см. таблицу). Эти результаты показывают, что раз-
мер макромолекул ПДМС меньше 120, но больше 80 А.
Адсорбция ПДМС в мг/м2 при равновесных концентрациях
Образец 8, М2/Г d, А 7, мг/г
1 5 10 20
Ааросил . . . 170 0.60 0.70 0.80 0.80
С-41 40 750 0.40 0.5 0.60 0.63
ШСК .... 340 120 0.70 0.80 0.80 0.80
A12Os .... 250 80 0.04 0.06 0.07 0.07
13 X (600) 10 0.00 0.02 0.02 0.03
Таким образом, однороднопористые силикагели могут служить моле-
кулярными ситами для больших молекул полимеров и использоваться
при их фракционировании.
Авторы благодарят профессора А. В. Киселева за постоянный интерес
к этой работе, Ю. С. Никитина и Л. И. Пигузову за предоставление об-
разцов и В. Н. Новикову за участие в работе.
ПРИМЕНЕНИЕ НАПОЛНЕННЫХ ЦЕОЛИТОВ В ШИННОЙ
ПРОМЫШЛЕННОСТИ
X. Н. Бородушкина, Г. А. Блох, Д. Б. Богуславский, И. Е. Неймарк,
М. Б. Зеликин, Т. Р. Гендлер, Г. М. Левит, А. Ф. Чернухина,
В. Я. Николина, К. Т. Подорван
В ранее опубликованных работах были изложены результаты исследо-
ваний по изучению возможности применения синтетических цеолитов
как носителей эффективных ускорителей вулканизации, использование
которых в обычных условиях практически невозможно. Введенные в цео-
литы активные соединения (газообразные амины, этаноламины), будучи
адсорбционно связанными, не проявляют своего действия при температу-
рах обработки смесей и легко десорбируются при достижении температуры
вулканизации. Было показано, что применение синтетических цеолитов
типа NaX, наполненных газообразными аминами и этаноламинами, в ка-
честве вторичных ускорителей вулканизации в стандартных и шинных
смесях из натурального и бутадиенстирольного маслонаполненного кау-
чуков позволяют существенно ускорить процесс вулканизации, при со-
хранении устойчивости сырых смесей к подвулканизации и высокого
уровня физико-механических свойств вулканизатов.
В настоящем сообщении рассматриваются а) результаты производ-
ственных испытаний синтетических цеолитов, наполненных аммиаком,
для вулканизации протекторных и брекерных смесей; б) дальнейшие ис-
следования по введению в резиновые смеси н-бутиламина, этилендиамина
и пиперидина на цеолитах для ускорения вулканизации.
325
Производственные испытания цеолитов, наполненных аммиаком
Цеолиты типа NaX, предварительно дегидратированные при темпера-
туре 400° С в течение 3 часов, заполняли аммиаком из баллона до постоян-
ного веса. Определяли допустимое время хранения наполненных цеоли-
тов при различных условиях. Наполненные цеолиты до введения в рези-
новые смеси хранились в закрытых сосудах, полиэтиленовой упаковке
и непродолжительное время без упаковки.
Таблица 1
.Изменение содержания аммиака на цеолите в зависимости от условий
и сроков хранения
Вид упаковки Содержание аммиака, %
Сроки хранения
0 7 суток 1 месяц
Полиэтиленовая пленка, К • 0.06 мм 100 91.9
Полиэтиленовая пленка, К 0.22 мм 100 98.4 —
Стеклянная закрытая тара 100 100 100
Примечание. Содержание аммиака на цеолите определялось по количеству азота.
В" табл. 1 приведены данные, характеризующие изменение содержания
аммиака на цеолите в зависимости от условий его хранения.
Из приведенных данных видно, что синтетические цеолиты, наполнен-
ные аммиаком, могут храниться длительное время в закрытых сосудах
и в полиэтиленовой упаковке с толщиной пленки 0.22 мм. Для изучения
кинетики десорбции аммиака в зависимости от влажности воздуха и вре-
мени экспозиции синтетический цеолит NaX, наполненный аммиаком,
выдерживался при температуре 20° С в течение 0.5—48 часов в эксика-
торе при различной влажности. Содержание аммиака в навеске цеолита
до и после экспозиции определялось химически по количеству азота.
Экспериментальные данные о влиянии влажности воздуха на кинетику
десорбции аммиака из цеолита представлены в табл. 2. Как и следовало
Таблица 2
Влияние влажности воздуха на кинетику десорбции аммиака
Относи- тельная влажность воздуха. % Соль, поддержи- вающая постоянную влажность Содержание аммиака, % к первоначальному содержанию
Продолжительность экспозиции, часы
0.5 1 2 3 5 24 48
45 К2СО3-2Н2О 96.0 94.5 85.0 82.9 75.0 29.5 0
58 NaBr-2H2O 93.5 91.0 83.5 80.0 73.0 20.5 0
80 NH4C1 88.0 85.5 79.0 78.0 73.0 15.7 0
98 Pb(NO3)2 87.0 83.0 80.1 77.3 70.2 7.8 0
ожидать, с повышением влажности воздуха процесс вытеснения аммиака
из цеолита водой ускоряется. Хранение наполненных аммиаком цеоли-
тов в открытом состоянии длительное время невозможно. Если цеолиты,
наполненные аммиаком, при влажности воздуха 58% оставить без упа-
ковки на 30 мин., то потери аммиака незначительны.
Наполненные цеолиты использовали для вулканизации протекторной
смеси из бутадиенстирольного маслонаполненного каучука и брекерной
326
смеси из НК. В качестве первичного ускорителя вулканизации в этих
смесях применяли сульфенамид Ц.
Изготовление смесей производили в скоростных резиносмесителях
по принятым в производстве режимам. Технологические свойства протек-
торных и брекерных резин на всех стадиях производства были удовлетво-
рительными.
Опытные смеси с аммиаком характеризуются более высокой скоростью
вулканизации и имеют показатели сопротивления подвулканизации, рав-
ноценные соответствующим серийным смесям, в которых в качестве
вторичного ускорителя для протекторных резин применялся дифенил-
гуанидин, для брекерных — альтакс (табл. 3).
Таблица 3
Результаты испытаний смесей и готовых покрышек размером 6 • 70—15
Свойства смесей и резин
Муни
Свойства сырых смесей
Сопротивление подвулканизации по Муни при I—
=120° С протекторных смесей, мин. .
Сопротивление подвулкапизации по
при «=110° С брекерных смесей, мин.
Физике-механические свойства
резин из покрышек
Протекторные
Модуль при 300% удлинении, кгс/см2..........
Сопротивление разрыву, кгс/см2..............
Относительное удлинение, %..................
Остаточное удлинение, %.....................
Сопротивление раздиру, кгс/см...............
Твердость по Шору, ед.......................
Эластичность по отскоку, %, н/у.............
70° С....................................
100° С....................................
Потери при истирании, см3/квт-час...........
Содержание свободной серы, %................
Брекерные
Модуль при 300% удлинении, кгс/см2..........
Сопротивление разрыву, кгс/см2..............
Относительное удлинение, %..................
Остаточное удлинение, %.....................
Содержание свободной серы, %................
Прочность связи, кгс:
протектор-брекер ...........................
брекер-брекер.............................
брекер-каркас.............................
Пробег шин на стенде, км....................
Испытания
опытные серийные
26 22
37 33
93 87
183 185
553 545
22 21
51 52
61 60
42 —
60 —
63 —
174 178
0.07 —
100 88
278 279 *
593 572
29 27
0.1 0.15
11.8 10.2
12.1 —
15.0 12.0
3665 3800
С применением исследуемых смесей была изготовлена опытная партия
легковых шин размера 6.70—15. Вулканизацию шин производили по
режимам, сокращенным на 13%, по сравнению с действующими в произ-
водстве.
Результаты физико-механических и стендовых испытаний покрышек
приведены в табл. 3.
Из приведенных данных видно, что по всему комплексу физико-
механических свойств протекторная и брекерная резины из опытных
покрышек равноценны соответствующим резинам из серийных шин.
Опытные шины, изготовленные с применением в протекторе и брекере
резин, содержащих в своем составе цеолиты, наполненные аммиаком,
характеризуются высокими показателями прочности связи между эле-
ментами покрышки и по уровню пробега на стенде не уступают серийным
шинам.
Предварительные результаты производственных испытаний подтвер-
дили ранее сделанные выводы о возможности использования синтетиче-
Рис. 1. Кинетика вулка-
низации протекторных
резин из БСК в присут-
ствии цеолита, напол-
ненного н-бутиламином.
Первичный ускоритель —
сульфонамид Ц 0.7 вес. ч.
Сопротивление
разрыву: 1 — ДФГ
0.6 вес. ч. (контрольная
резина); 2 — н-бутиламин
0.4 вес. ч.; з — и-бутиламин
0.8 вес- ч. Модуль при
300 %-м удлинении:
4 — ДФГ 0.6 вес. ч.; 5 —
н-бутиламин 0.4 вес. ч.;
в — н-бутиламин 0.8 вес. ч.
ских цеолитов для интенсификации процесса вулканизации при сохра-
нении стойкости резиновых смесей к подвулканизации.
Испытания в резиновых смесях синтетических цеолитов, наполненных
активными аминами
Синтетические цеолиты типа NaX были
(^кип—78° С), этилендиамином (/кил=118с С) и
наполнены н-бутиламином
пиперидином (£К1Ш=106° С).
Рис. 2. Кинетика вулка-
низации брекерных ре-
зин из НК в присутст-
вии цеолитов, наполнен-
ных н-бутиламином.
Первичный ускоритель —
альтакс 0.6 вес. ч.
Сопротивление
разрыву: 1 — ДФГ 0.4
вес. ч. (контрольная рези-
на); 2 — н-бутиламин 0.2
вес. ч.; з — н-бутиламин
0.4 вес. ч. Модуль при
300 %-м удлинении:
4 — ДФГ 0.4 вес. ч.; 5 —
н-бутиламин 0.2 вес. ч.;
6 — н-бутиламин 0.4 вес. ч
Исследовали ускоряющее действие адсорбированных аминов в брекер-
ных смесях из натурального каучука в сочетании с первичным ускори-
телем альтаксом и в протекторных смесях из бутадиенстирольного масло-
328
наполненного каучука в комбинации с ускорителем сульфенамидом Ц.
Изучение влияния указанных аминов на изменение вязко-текучих свойств-
сырых смесей и физико-механических показателей вулканизатов произ-
Продолжительность прогреби, при. Р=11О°С,мин.
Рис. 3. Сопротивление подвулканизации по Муни при 2=110° брекерных резин
из НК, содержащих различные цеолиты, наполненные н-бутиламином. Дозировка
к-бутиламина — 0.2 вес. ч.
1 — м-бутиламин на сите типа NaX; 2 — м-бутиламин на сите типа MgA; 3 — нгбутиламин на
сите типа СаА.
водили в сравнении со смесями аналогичного состава, содержащими в ка-
честве вторичного ускорителя дифенилгуанидин.
.//-бутиламин, введенный в смеси на сите, ускоряет процесс вулкани-
зации (рис. 1—2).
Брекерная и протекторная
смеси с 0.2—0.4 вес. ч. н-бу-
тиламина по скорости и степени
вулканизации равноценны ре-
зинам с дифенилгуанидином.
С повышением концентрации
н-бутиламина в смесях до
0.4—0.8 вес. ч. время вулкани-
зации, необходимое для дости-
жения оптимальных свойств
резин, сокращается соответ-
ственно до 20—30 мин. против
40—50 мин. для контрольной
смеси.
Продолжительность Вулканизации
при t=138°C'MUH
При использовании в каче-
стве вторичного ускорителя
н-бутиламина для обеспечения
высокой стойкости смесей к
преждевременной вулканизации
большое значение имеет пра-
вильный выбор типа цеолита.
Синтетический цеолит NaX хо-
рошо заполняется н-бутилами-
ном, но ввиду несоизмеримости
Рис. 4. Кинетика вулканизации брекерных
резин из НК в присутствии цеолита, напол-
ненного этилендиамином. Первичный уско-
ритель — альтакс 0.6 вес. ч.
Со противление разрыву: 1 — ДФГ
0.4 вес. ч. (контрольная резина); 2 — этилендиамин
0.3 вес. ч.; з — этилендиамин 0.5 вес. ч. Модуль
при 300 %-м удлинении: 4 — ДФГ 0.4 вес. ч.;
5 — этилендиамин 0.3 вес. ч.; 6 — этилендиамин
0.5 вес. ч.
размеров его молекул с разме-
рами входных «окон» и большой летучести он плохо удерживается ситом
NaX и выделяется при изготовлении смесей, что вызывает снижение стой-
кости их к подвулканизации.
329
5.
Продолжительность Вулканизации
при t=143°C, мил.
Кинетика вулканизации протек-
Сйятетические цеолиты СаА, MgA хорошо адсорбируют и удерживают
н-бутиламин при повышенных температурах. Применение указанных
цеолитов для введения н-бутиламина в резиновые смеси значительно
повышает их сопротивление подвулканизации (рис. 3).
Аналогичное действие на процесс вулканизации протекторных и бре-
керных смесей оказывают синтетические цеолиты, наполненные этилен-
диамином. Последний, являясь активным ускорителем процесса вулкани-
зации, дает возможность получить резиновые смеси с большей скоростью
вулканизации (рис. 4). По основным физико-механическим свойствам
вулканизаты с н-бутиламином и
этилендиамином равноценны ре-
зинам с дифенилгуанидином
(рис. 1, 2, 4).
Устойчивость резиновых смесей
с этилендиамином к преждевремен-
ной вулканизации также опреде-
ляется типом синтетического цео-
лита.
По сравнению с н-бутиламином
и этилендиамином пиперидин явля-
ется более активным ускорителем.
Протекторные смеси, содержа-
щие в своем составе всего 0.15
вес. ч. пиперидина, адсорбирован-
ного на сите NaX, по скорости
изменения физико-механических
свойств идентичны смесям с 0.6
вес. ч. дифенилгуанидина. Повы-
шение содержания пиперидина
в смеси не оказывает существен-
ного влияния на изменение вязко-
текучих свойств сырых смесей
при температуре 120° С. Время же,
необходимое для достижения опти-
мальных свойств резин, в этом
случае сокращается до 20 мин.
против 40—50 мин. для контроль-
ной смеси. Например, после вулка-
низации при температуре 143° С в течение 40—50 мин. смесь с дифенил-
гуанидином имеет показатели: модуль при 300%-м удлинении 80 кгс/см2,
сопротивление разрыву 204 кгс/см2, относительное удлинение 577%. Для
смесей с пиперидином в количестве 0.4 вес. ч. на 100 вес. ч. каучука такой же
уровень физико-механических показателей достигается за 20 мин. (рис. 5).
Время подвулканизации указанных смесей по Муни, характеризующее
сохранение вязкости при температуре 120° С, составляет 22 мин. для
смесей с 0.4 вес. ч. пиперидина, для смесей с 0.2 вес. ч. — 24 мин., для
контрольной смеси это время равно 26 мин.
Аналогичные закономерности получены при изучении ускоряющего
действия газообразных и жидких аминов, адсорбированных синтетиче-
скими цеолитами, в рецептуре шинных резин из стереорегулярных поли-
меров (СКИ-3, СКД).
На основании проведенных исследований установлена возможность
практического применения синтетических цеолитов, наполненных актив-
ными ускорителями, в резиновой промышленности для интенсификации
процесса вулканизации многослойных изделий — шин, при сохранении
стойкости резиновых смесей к преждевременной вулканизации.
Рис.
торных резин из БСК в присутствии цео-
лита, наполненного пиперидином. Первич-
ный ускоритель—сульфенамидЦ 0.7 вес. ч.
Сопротивление разрыву: 4 —
0.6 вес. ч.; 6 — пиперидин 0.15 вес. ч.; 6 — пи-
перидин 0.4 вес. ч. Модуль при 300%-м
удлинении: 1 — ДФГ 0.6 вес. ч. (контроль-
ная резина): 2 — пиперидин 0.15 вес. ч.; з —
пиперидин 0.4 вес. ч.
330
ЛИТЕРАТУРА
1. Rubber World, 1961, 143, 4, 60.
2. M. M. Дубинин, ДАН СССР, 1961, 138, 866.
3. М. М. Дубинин и др., Изв. АН СССР, ОХН, 1961, 7, 8, 1183, 1380.
4. В. Я. Н и к о л и н а, И. Е. Неймар к, М. А. Пионтковская, Усп.
химии, 1960, 29, 9.
5. Г. А. Б л о х, И. Е. Неймар к, Ю. Г. Шевченко, Д. Б. Богу-
славский, X. Н. Беродушкина, А. Д. Чугай, Тез. докл. IV
научно-техн. конф, по вопросам хим. и техн, каучука и резины, Ярославль,
1962, 36.
6. Г. А. Б л о х, X. Н. Б о р о д у ш к и н а, Ю. Г. Ш е в ч е н к о, Д. Б. Бо-
гу с л а в с к и й, И. Е. Н е й м а р к, Изв. ВУЗов, Легкая промышлен-
ность, 1963, 3, 24.
7. X. Н. Б о р о д у ш к и н а, Г. А. Б л о х, Т. Р. Г е н д л е р, И. Е. Ней-
мар к, Д. Б. Богуславский, М. Б. Зелики н, Тез. докл. II
научно-техн, совещ. по пробл. сырья для шинной промышл. и его качества,
Днепропетровск, 1963, 52.
•8. X. Н. Б о р о д у ш к и н а, Г. А. Блох, Д. Б. Богуславский,
Т. Р. Г е н д л е р, И. Е. Н е й м а р к, Каучук и резина, 1964, 2, 1.
ИССЛЕДОВАНИЕ ЭФФЕКТИВНОСТИ ПРИМЕНЕНИЯ
ВЫСОКОАКТИВНЫХ ВУЛКАНИЗУЮЩИХ СИСТЕМ
В ШИННЫХ РЕЗИНАХ С ПОМОЩЬЮ МОЛЕКУЛЯРНЫХ СИТ
ТИПА X
Ю. И. Ерофеева, Т. Н. Бухмшп, М. С. Фелъдштейн,
3. В. Скородумова
В последние годы в связи с внедрением в промышленность высокоско-
ростных режимов изготовления и обработки резиновых смесей, исполь-
зованием жестких каучуков и введением в них больших количеств напол-
нителей особенно острой становится проблема предотвращения прежде-
временной вулканизации. Для решений этой проблемы используются
различные технологические приемы: применение агентов и ускорителей
вулканизации, характеризующихся замедленным действием в начальном
периоде [1], введение в резиновые смеси замедлителей подвулкани-
зации и бинарных систем ускорителей [2]. Весьма перспективным' яв-
ляется метод предотвращения подвулканизации резиновых смесей вве-
дением в них вулканизующих систем с помощью молекулярных сит [3—8].
Настоящая работа посвящена получению химически наполненных
молекулярных сит различными агентами и ускорителями вулканизации
и определению эффективности их действия в резиновых смесях на основе
различных каучуков.
Проводились работы по наполнению молекулярных сит 13Х различ-
ными аминами: пиперидином, морфолином, моно- и диэтаноламином. Эти
вещества сильно ускоряют процесс вулканизации, однако их использова-
ние в свободном виде не представляется возможным из-за летучести при
введении в резиновые смеси, что приводит к получению неоднородных
вулканизатов, а также вследствие вызываемой ими преждевременной вул-
канизации. Введение указанных аминов в резиновые смеси с помощью
молекулярных сит позволяет устранить эти недостатки.
Большая летучесть перечисленных аминов позволила вести наполне-
ние ими молекулярных сит. из паров. Были использованы два метода:
статический и динамический.
331
Наполнение пиперидином и морфолином проводилось при комнатной
температуре, моно- и диэтаноламином — при 50—55° С.
Полученные данные показали, что количество адсорбированного ве-
щества, а также скорость наполнения зависят от строения и температуры
Рис*. 1. Изотерма адсорбции
паров пиперидина на моле-
кулярном сите 13Х.
Черные кружки — адсорбция, бе-
лые — десорбция.
кипения аминов. Так, для пиперидина и мор-
фолина, температура кипения которых 106 и
130° С соответственно, количество адсорби-
рованного вещества составляет 18—20 г на
100 г молекулярных сит, для моно- и ди-
этаноламина (температура кипения 171 и
270° С) — 15 г.
Н. В. Ковалевой были сняты изотермы
адсорбции и десорбции пиперидина на мо-
лекулярном сите 13Х (рис. 1). Изотерма ад-
сорбции пиперидина на молекулярном сите
13Х имеет вид, характерный для адсорбции
на таком типе адсорбентов — высокая ад-
сорбционная емкость при малых давлениях.
Десорбция же пиперидина очень затруд-
нена. При 100° С имеет место лишь незна-
чительное удаление пиперидина; при тем-
пературе 160° С достигается практически
полная десорбция. Таким образом, полу-
ченные данные говорят о том, что при тем-
пературах, соответствующих температурам
технологической обработки резиновых сме-
сей, десорбция пиперидина практически не
идет, тогда как при температурах вулканизации достигается почти пол-
ное удаление адсорбированного вещества.
Наполненные аминами молекуляр-
ные сита 13Х были применены в ка-
честве вторичных ускорителей в сме-
сях на основе бутадиен-стирольного,
полибутадиенового и полиизопрено-
вого каучуков. Основным ускорите-
лем в этих смесях является ускори-
тель сульфенамидного типа N-цик-
логексил-2-бензтиазолсульфе н амид
(сульфенамид Ц). В качестве напол-
нителя использовалась сажа типа
ХАФ.
Как видно из рис. 2, введение
вторичных ускорителей: дифенилгуа-
нидина (ДФГ) и тетраметилтиурам-
моносульфида ’ (ТМТМ) — приводит
к резкому возрастанию скорости и
повышению степени вулканизации.
Такой же эффект достигается при
применении молекулярных сит, на-
полненных пиперидином (13ХП). Од-
нако, если в первом случае, т. е. при
применении ДФГ и ТМТМ, повышение
Рис. 2. Влияние наполненных пипери-
дином молекулярных сит на модуль
вулканизатов из бутадиен-стирольного
каучука (СКМС—ЗОАРКМ)
1 — сульфенамид Ц 0.8 вес. ч.; 2 — то же+-
+ДФГ 0.3 вес. ч.; 3 — то же+ТМТМ 0.1 вес. ч.;
4 —то же+13ХП 0.48 вес. ч.; б —то же+
+13ХП 0.96 вес. ч. Светлая часть столбиков —
продолжительность вулканизации 30 мин., за-
штрихованная — 20 мин.
скорости вулканизации сопровождается уменьшением стойкости резино-
вых смесей к под вулканизации, то в случае применения пиперидина на
молекулярных ситах склонность смесей к преждевременной вулканизации
не увеличивается (рис. 3). При этом следует отметить, что введение мо-
332
лекулярных сит, наполненных пиперидином, существенно не изменяет
сопротивления разрыву, относительного и остаточного удлинения вулкани-
затов (табл. 1).
Рис. 3. Влияние наполненных пиперидином молеку-
лярных сит на изменение пласто-эластических свойств
резиновых смесей из бутадиен-стирольного каучука
(СКМС—ЗОАРКМ)
1 —сульфонамид Ц 0.8 вес. ч.; 2 —то же+ДФГ 0.3 вес. ч.;
3— то же4-ТМТМ 0.1 вес. ч.; 4—то же+13ХП 0.48 вес. ч.
Гранулированные полиакрилнитрилом молекулярные сита по адсорб-
ционной емкости к пиперидину и по своему действию в резиновых сме-
сях идентичны порошкообразным образцам.
Исследование действия наполненных пиперидином молекулярных сит
в смесях на основе полибутадиенового (СКД) каучука показало, что их
применение дает примерно такой же
эффект повышения интенсивности вул-
канизации, что и ТМТМ (рис. 4), но
резко снижает склонность смесей
к преждевременной вулканизации
(рис. 5). На этом же рисунке показано
действие пиперидина на молекулярных
ситах по сравнению с действием пипе-
ридина, введенного в резиновую смесь
в свободном виде.
Аналогичные действия оказывают
наполненные пиперидином молекуляр-
ные сита и в смесях на основе поли-
изопренового каучука — СКИ-3.
Молекулярные сита, наполненные
моноэтаноламином [9, 10], были опро-
бованы в смесях из натурального и
бутадиенстирольного каучуков. Показа-
но, что введение наполненных моноэта-
ноламином молекулярных сит не приво-
Рис. 4. Влияние наполненных пи-
перидином молекулярных сит на
модуль вулканизатов из полибута-
диенового каучука (СКД).
1 —сульфенамид Ц 0.7 Овес, ч.; 2 — то
же+ДФГ 0.3 вес. ч.; 3 — то же+ТМТМ
0.1 вес. ч.; i —то же+ 13ХП 0.45 вес. ч.;
5 — то же+13ХП 0.S0 вес. ч.
Продолжительность вулканизации 50 мин.
дит к существенному увеличению скоро-
сти и повышению степени вулканизации
(табл. 2), но значительно улучшает пласто-эластические свойства смесей
(рис. 6).
Представлялось интересным выяснить возможность наполнения мо-
лекулярных сит 13Х высокоактивными ускорителями вулканизации,
333
Рис. 5. Влияние наполненных пиперидином молекулярных
сит на изменение пласто-эластических свойств резиновых сме-
сей из полибутадиенового каучука (СКД).
1 — сульфенамид ц 0.7 вес. ч.; 2 — то же+ДФГ 0.3 вес. ч.; 3 —• то
же+ТМТМ 0.1 вес. ч.; 4—то же+13ХП 0.48 вес. ч.; 5 — то же+
+пиперидин 0.2 вес. ч.
Рис. 6. Влияние наполненных моноэтаноламином молеку-
лярных сит на изменение пласто-эластических свойств ре-
зиновых смесей из натурального каучука.
1 — сульфенамид Ц 0.6 вес. ч.; 2 — то же+ДФГ 0.3 вес. ч.; 3 — то
же+моноэтаноламин 0.3 вес. ч.; 4—то же+13ХМЭ 2.0 вес. ч.
Таблица 1
Влияние состава вулканизующих групп на физико-механические свойства
вулканизатов из бутадиенстирольного каучука (СКМС—ЗОАРКМ)
(температура вулканизации 153° С, продолжительность — 30 мин.)
Показатель Сульфен- амид Ц, 0.8 вес. ч. Сульфен- амид Ц + ДФГ Сульфен- амид Ц -j- + ТМТМ Сульфен- амид Ц + + 0.48 13ХП Сульфен- амид ц + + 0.97 13ХП Сульфен- амид Ц 4- 4-0.97 13ХП гранул
Сопротивление разрыву, кгс/см2 225 201 224 228 230 205
Относительное удлинение, % 564 464 476 492 464 444
Остаточное удлинение, % . 20 17 13 15 16 14
Примечание. 13ХП — наполненные пиперидином молекулярные сита 13Х.
Таблица 2
Влияние состава вулканизующих групп на модуль вулканизатов
из натурального каучука
(температура вулканизации 143° С)
Показатель Время нагрева- ния, мин. Сульфен- амид ц, 0.6 вес. ч. Сульфен- амид. Щ-ДФГ Сульфен- амид Ц 4~ моно- этанол- амин Сульфен- амид Ц 4- 13ХМЭ
( 5 78 103 87 76
10 117 148 128 127
Модуль при 300%-м удлинении, 1 15 149 160 155 149
кгс/см2 20 150 165 160 158
1 30 149 159 157 155
Примечание: 13ХМЭ — наполненные моноэтанол амином молекулярные сита 13 X.
применяемыми в промышленности. Большинство таких ускорителей вул-
канизации, известных в качестве ультраускорителей, представляет
собой кристаллические вещества.
Наполнение молекулярных сит этими веществами можно осуществить
адсорбцией из растворов. Но, благодаря тому что ускорители вулкани-
зации главным образом растворимы в растворителях, молекулы которых
гораздо меньше молекул веществ, предназначенных к наполнению, то
их адсорбция на молекулярных ситах из растворов связана с большими
трудностями.
Были сделаны попытки наполнения молекулярных сит диэтилтиомо-
чевиной, тетраметилтиурамдисульфидом, диэтилдитиокарбаматом цинка
и 2-меркаптобензтиазолом из их растворов в бензоле. Полученные данные
показали, что адсорбция этих веществ на молекулярном сите 13Х при
комнатной температуре очень незначительна и составляет для диэтил-
тиомочевины — 3 г на 100 г молекулярных сит, для 2-меркаптобенз-
тиазола — не более 4 г; тетраметилтиурамдисульфид и диэтилдитиокар-
бамат цинка на молекулярных ситах из раствора бензола в указанных
условиях не адсорбировались.
Выводы
Результаты проведенного исследования показали возможность полу-
чения химически наполненных молекулярных сит различными летучими
ускорителями вулканизации из паров.
335
Наполненные молекулярные сита содержали 15—20% адсорбирован-
ного вещества.
По данным исследования адсорбции и десорбции пиперидина на моле-
кулярных ситах установлено, что при температурах, соответствующих
температурам технологической обработки резиновых смесей, пиперидин
практически не удаляется, тогда как при температурах вулканизации
достигается почти полная его десорбция.
Применение молекулярных сит, наполненных пиперидином, в резино-
вых смесях на основе бутадиен-стирольного, полибутадиенового и поли-
изопренового каучуков позволяет существенно увеличить скорость и по-
высить степень вулканизации, сохраняя высокую стойкость смесей к пре-
ждевременной вулканизации.
ЛИТЕ РАТУ РА
1. М. С. Ф е л ь д ш т е й н, Каучук и резина, 1962, 3.
2. Б. А. Д о г а д к и н, М. С. Фельдштейн, Э. Н. Беляева, Высоко-
мол. соединения, 1959, 1, 2, 254; 1960, 2, 2, 247.
3. F. О’ Connor, Т. L. Thomas, M.L. Dunham, Ind. Engng Chem.,
1959, 51, 4, 531.
4. F. O’ Connor, R. L. M ays, J. Chem. Eng. Data, 1960, 5, 4, CA 53,13641 g.
5. Rubber World, 1958, 138, 1, 128.
6. Rubber World, 1958, 138, 3, 424.
7. Rubber World, 1961, 143, 4, 60.
8. Rubber Age, 1958, 83, 3, 482.
9. Г. А. Б л о x, И. E. Неймар к, X. H. Б о p о д у ш к и н а, Д. Б. Б о-
гуславский, Ю. Г. Шевченко, Изв. ВУЗов, Технология легкой
промышленности, 1963, 4.
10. X. Н. Бородушкина, Г. А. Блох, Д. Б. Богуславский,
Т. Р. Г е н д л е р, И. Е. Н е й м а р к, Каучук и резина, 1964, 2.
О НАПОЛНЕНИИ ЦЕОЛИТОВ СЕРОВОДОРОДОМ,
СЕРНИСТЫМ ГАЗОМ И ОРГАНИЧЕСКИМИ ПЕРЕКИСЯМИ
А. Ф. Носников, С. Е. Рапчинская, Г. А. Блох,
И. Е. Неймарк
Цеолиты открывают новые перспективы для осуществления вулкани-
зации массивных резиновых изделий с помощью газообразных или легко
летучих вулканизующих систем. Мы изучали возможность вулканизации
каучука с помощью сероводорода и сернистого газа и органических ле-
тучих перекисей, вводимых в состав резиновых смесей на цеолитах. Как
известно, обычными приемами осуществить вулканизацию массивных
изделий с помощью газообразных вулканизующих систем не представ-
ляется возможным. Были изучены процессы сорбции и десорбции на цео-
литах различных типов: NaX, NaA, СаХ, СаА, MgA. Установлено, что
при температуре 350—400° С за 9(5—100 мин., а при 450—500° С за 15—
30 мин. достигается практически полное удаление сорбированной воды из
цеолитов.
Данные, характеризующие максимальное заполнение цеолитов при
комнатной температуре, приводятся в таблице.
При наполнении цеолита типа NaX сероводородом происходит час-
тичное его разложение с выделением свободной серы, которая легко окис-
ляется кислородом (при свободном доступе воздуха происходит самовоз-
горание наполненного сероводородом цеолита). Была изучена десорбция
сероводорода, сернистого газа и перекиси дитретичного бутила (пере-
336
Тип цеолита % максимального впачения
пары перекиси ДТБ so2 H„S
NaX (порошок) 20—25 28—32 19—21
NaA » Не адсорбируется. 26—31 10—14
NaA (гранулы) » » 22—23 —
СаА » —~ 22—23 10
MgA » — 12-13 12.5
NaA » 10—13 — —'
киси ДТБ) в интервале температур от 20 до 180° С. Десорбция серни-
стого газа из цеолита типа NaX при 60° С составляет 11—12 %, при 100° С —
до 24%, при 143—170° С — 45—49% за время 30 мин. Десорбция серо-
водорода из цеолита типа СаА (гранулы) при 115—180° С составляет
7.0—16%, из цеолита типа MgA (гранулы) при 143° С — 44%, из NaA
(порошок) при 143° С — 80—82 %. Десорбция перекиси ди-третичного бутила
из цеолита NaX (порошок) начинается только при 120° С и за 15 мин.
составляет 4—6%, при 130—180° С — 50—80%. Более длительные ре-
жимы десорбции (60—120 мин.) на величину десорбции практически не
влияют.
Была осуществлена вулканизация натрийбутадиенового каучука как
с помощью сероводорода и сернистого газа, так и с помощью сернистого
газа, введенного на цеолите, и 2-меркаптобензтиазола, введенного в кау-
чук обычным путем. В первом случае резина имела сопротивление раз-
рыву 47—49 кгс/см2, во втором — 39—40 кгс/см^, в то время как кон-
трольная стандартная ненаполненная резина на СКБ имела всего лишь
12—15 кг/см2. Таким образом, здесь доказаны не только практическая
возможность осуществления вулканизации массивных изделий газообраз-
ной вулканизующей системой, но и радикальный характер этих реакций.
Установлено, что перекись ди-третичного бутила, вводимая в полиэти-
лен-пропиленовый каучук (СКЭП) обычным путем, позволяет получать
вулканизаты с пределом прочности 180—220 кгс/см2. Однако по мере вы-
леживания сырых смесей с ДТБ происходит улетучивание перекиси из
смеси, что резко сказывается на физико-механических показателях резин,
приготовленных из этих смесей. Прочность вулканизатов, полученных из
резиновых смесей после двухчасовой вылежки, падает на 10—20%,
после 24 часов — на 30—50%, через 48 часов — на 50—70%, а через
96 часов — на 70—100%, т. е. практически получаются сырые, невулкани-
зованные смеси. Резиновые смеси (СКЭП), в которых перекись ДТБ
введена на цеолитах, независимо от времени их лежки перед вулканиза-
цией, не утрачивают перекиси ДТБ, и получаемые резины обладают
неизменно высокими физико-механическими свойствами.
22 Цеолиты
337
ИССЛЕДОВАНИЕ СКОРОСТИ
СТАЦИОНАРНОЙ ДИФФУЗИИ ОРГАНИЧЕСКИХ ПАРОВ В СТЕРЖНЯХ
ИЗ СПРЕССОВАННОГО ЦЕОЛИТА NaX
А. С. Пономарев, Д. П. Тимофеев
Наличие бидисперсной структуры в синтетических цеолитах приводит
к тому, что при адсорбции из газовой фазы перенос вещества внутри гра-
нулы цеолита осуществляется двумя существенно отличающимися по
своей природе диффузионными потоками: один поток — во вторичных
порах, образованных между элементарными кристаллами, и второй —
непосредственно в кристаллах. В процессе заполнения сорбционной ем-
кости диффузионное сопротивление кристаллов может меняться, в то
время как диффузионное сопротивление во вторичных порах, если не
Рис. 1. Зависимость коэффициента диффузии паров
П30-С5Н12, Я-С5Н12, CgHi^, С7Н16, CgHg, СН3ОН
от температуры в цеолите NaX.
происходит капиллярной конденсации, остается постоянным. Возникает
вопрос, какая доля общего диффузионного потока через гранулу на пути
от поверхности к центральным частям приходится на кристаллы и какая
доля — на вторичные поры.
В ряде работ [1—3] отмечались довольно низкие значения величин
коэффициентов диффузии в первичных порах. Однако непосредственного
сопоставления диффузионного потока во вторичных порах с общим по-
током, включающим также диффузию в кристаллах, сделано не было.
Если принять, как в работах [4—7], что перенос несорбирующегося газа
осуществляется объемной диффузией, не связанной с наличием адсорб-
ционного поля адсорбента, то, сопоставляя приведенные диффузионные
потоки несорбирующегося и хорошо адсорбирующегося газа, можно при-
ближенно оценить влияние адсорбции на общий перенос, или, точнее,
величину составляющей, общего диффузионного потока в адсорбционной
фазе, т. е. в кристаллах.
Для измерения диффузионных потоков по сорбирующемуся и несор-
бирующемуся газу был использован описанный ранее метод измерения
диффузионных потоков в стационарных условиях [8]. В качестве объекта
исследования был выбран кристаллический цеолит типа NaX, спрессо-
ванный в стержень (длина L=2.1 мм, диаметр d=4.4 мм) до пористости
е=0.46. Измерения проводились по парам н-С5Н12, н-С6Н14, н-С7Н1в,
СвНв, СН3ОН и нзо-СБН12. В качестве неадсорбпрующегося газа, или,
точнее, малоадсорбирующегося в условиях опыта, был выбран азот.
338
Измерения велись в атмосфере гелия, который являлся газом-носителем,
при общем давлении газовой смеси 1 атм. и в интервале температур от
50 до 300° С. Вычисление коэффициентов диффузии проводилось по урав-
нению
V • L • С2
D = F • Сг
где и — расход гелия на тыльной
стороне зерна (см8/сек.), F и L — се-
чение и длина гранулы; G и Са —
концентрации на торцах зерна.
Условие С2 Сг, при котором
справедливо уравнение (1), выполня-
лось. Экспериментально найденная
температурная зависимость коэффи-
циента диффузии азота в спрессо-
ванных зернах составила D — 711-75,
что указывает на отсутствие пере-
носа в адсорбционной фазе и на то,
что перенос во вторичных порах про-
исходил путем обычной диффузии.
Расчет коэффициента диффузии орга-
нического пара из данных, получен-
а,мл/г
Рис. 2. Изотермы адсорбции паров бен-
зола (7 и 2) на цеолите NaX.
у, и р2 — давление паров бензола на входе
и на выходе из зерна.
ных с азотом, проводился по уравнению
где о12=^у-?, о1>3-
1
т3
~Г
т2
°1, 2
^Орг- пара ==
°1, 3
—- mr, т2, т3, dt, d2, d3—массы и газокинетические
(2)
диаметры соответственно молекул гелия,
азота и данного углево-
дорода, и т. д. Таким обра-
зом, коэффициенты диффу-
зии органических паров,
рассчитанные по формуле
(2), представляют собой
те значения, которые были
бы, если данная пористая
среда была бы по отноше-
нию к рассматриваемому
органическому пару инерт-
ной. Экспериментальные
значения коэффициентов
диффузии в виде кривой
зависимости D—Т приве-
дены на рис. 1. На этом
Рис. 3. Зависимость коэффициента диффузии от же РисУнке приведены ве-
температуры при разных парциальных давлениях личины коэффициента диф-
паров бензола в газовой смеси. фузии, рассчитанные по
Цеолит NaX (е=0.46); р,=5, р2=11, р,=27 мм рт. ст. формуле (2) (пОКЭЗЭНЫ
прерывистой линией). Как
видно из рис. 1, рассчитанные по азоту значения коэффициентов диф-
фузии мало отличаются от экспериментальных величин и, следовательно,
общий диффузионный поток органического пара определяется в основ-
ном составляющей потока в газовой фазе, т. е. во вторичной пористости.
22*
339
Измеренные коэффициенты диффузии получены при довольно больших
(в среднем 0.6 4" 0.9) относительных заполнениях, соответствующих
выбранным парциальным давлениям н-С8Н12, нзо-С8Н12, н-СвН14, С7Н1в,
СвНв и СН3ОН в 110, 170, 24, 34, 27, 29.6 мм рт. ст. соответственно. По-
этому сделанный вывод, вообще говоря, может и не распространяться на
малые заполнения. Достигнуть малых заполнений на цеолите NaX по
использованным парам при низких температурах весьма сложно по при-
чине значительной крутизны изотермы и возникающих в связи с этим труд-
ностей подвода вещества, создания и контроля низких парциальных
давлений. Более простым оказалось измерение коэффициента диффузии
при малых заполнениях в области температур 200—300° С, где изотерма
адсорбции СвНв на NaX1 более пологая (рис. 2). Можно видеть, что при
температурах 250 и 300° С в адсорбционной фазе создаются значительные
градиенты при относительных заполнениях 6=0.2 -4- 0.4. Полученные
экспериментальные и рассчитанные по азоту коэффициенты диффузии
паров бензола для этих температур (рис. 3) и в этом случае весьма близки
по величине. Одновременно можно видеть, что общий коэффициент не
зависит от величины заполнения.
Таким образом, скорость стационарной диффузии исследованных ор-
ганических паров как при малых, так и при больших заполнениях опре-
деляется скоростью переноса во вторичных порах.
ЛИТЕРАТУРА
1. R. М. В а г г е г, D. W. В г о о с k, Trans. Faraday Soc., 1953, 49, 1049.
2. Д. П. Тимофеев, И. Т. Е р а ш к о, Изв. АН СССР, ОХН, 1961, 1192.
3. Д. И. Тимофеев, О. Н. К а б а и о в а, И. Т. Е р а ш к о, А. С. По-
номарев, в сб.: Синтетические цеолиты, Изд. АН СССР, 1962, 24.
4. Р. Clausing, Ann. der Physik, 1930, 7, 489.
5. G. D a m k 6 h 1 e г, Z. Phys. Chem., 1935, A 174, 222.
6. R. M. C arman, F. A. R a a 1, Proc. Roy. Soc., 1951, 209, 38, 1096.
7. R. M. В arrer, E. S t г a c h m a n, Proc. Roy. Soc., 1951, A 209, 38.
8. А. С. Пономарев, Д. П. T и м о ф е е в, ДАН СССР, 1963, 150, 1081.
9. Я. В. Ми реки й, М. Т. Ми трофанов, в сб.: Синтетические цеолиты,
Изд. АН СССР, 1962, 95.
ИССЛЕДОВАНИЕ КИНЕТИКИ СОРБЦИИ и-ПЕНТАНА
И ДИЭТИЛОВОГО ЭФИРА НА ЦЕОЛИТАХ СаА и NaX
И. Т. Ерашко
Одним из главных направлений применения синтетических цеолитов
является процесс осушки. Поэтому наши предыдущие работы в области
изучения кинетических свойств цеолитов были посвящены именно иссле-
дованию кинетики сорбции паров воды цеолитами NaA и СаА [1—2].
Было установлено, что коэффициент диффузии паров Н2О 'зависит от
концентрации воды в адсорбированной фазе. Изучив несколько образцов-
с различающейся вторичной пористостью, мы пришли к выводу, что, по
всей видимости, основным является перенос воды внутри самих кристал-
ликов цеолита, а характер вторичной пористости не оказывает заметного
влияния на скорость процесса. В силу малых размеров молекулы воды
размер входного отверстия, разный у NaA и СаА, заметного влияния на
скорость процесса не оказывает.
1 По данным Я. В. Мирского и М. Г. Митрофанова [9].
340
Представлялось интересным продолжить исследование с такими веще-
ствами, диаметр молекул которых близок к диаметру входного отверстия
цеолита. Такими веществами были взяты н-пентан и диэтиловый эфир
(их диаметр близок к 5 А, т. е. к диаметру входного окна СаА). Эти ве-
щества, обладая одинаковыми размерами молекул, вместе с тем различны
по своему взаимодействию с решеткой цеолитов.
Исследования кинетики сорбции паров диэтилового эфира и н-пентана
проводились методом нестационарной диффузии. Была использована ва-
куумная установка с кварцевыми весами [3]. Давление во время опытов
поддерживалось приблизи-
тельно постоянным. И?
Найденное из опыта
время т05, т. е. время,
за которое достигается ве-
личина адсорбции при по-
стоянном давлении, равное
половине равновесной ад-
сорбции при этом давле-
нии, связано с коэффици-
ентом диффузии соотно-
шением
/?2
D K——. i)
1^0.5 '
Исследованные грану-
лы формованных цеолитов
имели форму цилиндра рис. Зависимость коэффициента диффузии диэти-
С d—l. Для этого случая лового эфира от величины адсорбции.
Пе=0.318 (2). Так как
Л Т0.5
коэффициент диффузии может зависеть от заполнения, изотерма адсорбции
разбивается на участки, для которых можно считать, что£>е постоянно. Для
первого впуска (давление паров составляет сотые доли миллиметра рт. ст.),
где адсорбция протекает на чистом образце, предварительно эвакуиро-
ванном при 350° в течение 3—5 часов, коэффициент диффузии нами не
рассчитывался. В этой области не выполняется условие постоянства давле-
ния. После достижения равновесной адсорбции кран на адсорбционную
трубку перекрывался и в приборе создавалось нужное давление. Зачтем
кран открывался вновь и измерялся прирост адсорбции во времени, откуда
находилось т0 5 для расчета D„ по вышеприведенной формуле (2). Далее
процедура повторялась прп другом давлении.
Кинетика сорбции диэтилового эфира и н-пентана была изучена
в области давлений, не превышавших 30 мм рт. ст. В этой области выпол-
няется кнудсеновский режим, и перенос вещества не зависит от давления
(в отличие от переходной и вязкой области,, которая достигается при дав-
лениях свыше 30 мм рт. ст.).
С целью исследования температурной зависимости коэффициента диф-
фузии исследуемых паров проводились опыты при £=50, 100, 150°
и некоторые опыты при 75° С. От температур, меньше 50°, пришлось от-
казаться в силу того, что процесс сорбции пентана (и особенно эфира) на
образцах СаА идет настолько медленно, что равновесие устанавливается
сутками. Нет уверенности в том, что при 20° вообще возможно в течение
нескольких суток получить равновесную изотерму для эфира. Сказанное
не относится к образцам NaX, на которых равновесие устанавливается
гораздо быстрее.
341
На рис. 1 и 2 представлены кинетические кривые, полученные для
цеолита СаА-I, т. е. зависимость De от величины адсорбции при разных
температурах для эфира и пентана на этом образце. Как видно из рисун-
ков, для
Рис. 2. Зависимость коэффициента диф-
фузии н-пентана от величины адсорбции.
всех температур коэффициенты диффузии н-пентана намного
превышают коэффициенты диффу-
зии диэтилового эфира (разни-
ца при 50 и 100° почти на один
порядок). Это значит, что скорость
адсорбции н-пентана намного вы-
ше, чем диэтилового эфира. На
рис. 3 и 4 приведены изотермы ад-
сорбции этих же паров при 50,
100 и 150° на образце СаА-I. Далее
был проведен опыт при £=100°
с диэтиловым эфиром на образце
СаА-П, резко отличающемся раз-
мерами вторичных пор от первого
образца. Характеристика образцов
приведена в таблице.
На рис. 2 крестиками обозна-
чены коэффициенты диффузии эфи-
ра на втором образце СаА. Как
видно, эти результаты хорошо со-
впадают с полученными для об-
разца I. Заметного влияния вто-
ричная пористость на скорость
процесса, видимо, не оказывает.
Интересно, что сорбция паров
эфира и пентана на цеолите NaX
протекает очень быстро. К сожа-
лению, это не позволило подроб-
нее исследовать кинетику процесса
в данном случае, т. е. надежно
найти Dе в широкой области заполнения. В силу того что т0 s зачастую
было меньше половины минуты, для каждой температуры удалось полу-
при 50° и средней величине адсорб-
1.1 ммоль/г для пентана, De соот-
чить не более двух значений De. Так,
ции, равной 1.2 ммоль/г для эфира и
ветственно равны 1 • 10-6 и 2х
Х10-всм2/сек.; при 100°иа=0.8 ммоль/г
для эфира и 0.7 ммоль/г для пентана
De соответственно равны 3 • 10“® и
5 • 10“® см2/сек. Скорость сорбции
н-пентана здесь также выше, чем
эфира, но эта разница меньше, чем
в первом случае. Изотермы адсорб-
ции н-пентана и эфира приведены
для образца NaX на рис. 5 и 6.
Из полученных результатов уда-
лось оценить энергию активации
диффузионного процесса для некоторых величин адсорбции. Эта
для диэтилового эфира в интервале от 1.1 до 1.4 ммоль/г составляет
10—11 ккал./моль (при 100—150°). Для н-пентана она составляет 12—
13 ккал./моль в интервале от 0.5 до 0.6 ммоль/г. Если вспомнить, что
по уравнению активированной диффузии D связано с Е и Д S
И AS
D = Do е RTe R ,
Наиболее вероятный радиус
вторичных пор цеолитов
Марка цеолита Диаметр гранулы, см Радиус пор, А
СаА-I Линде . 0.17 2700
СаА-П . . . 0.23 8900
NaX Линде . 0.18 2500
величина
342
то значительное различие в коэффициентах диффузии и довольно близкие
значения энергии активации говорят о различии энтропийного фактора
активации. Это означает, что молекулы диэтилового эфира и н-пентана,
адсорбируясь, могут по-разному взаимодействовать с катионным составом
Рис. 3. Изотермы адсорбции эфира на СаА-1.
решетки. К аналогичным выводам приводит сравнение теплот адсорб-
ции эфира и пентана, которое было произведено в работах А. В. Киселева
и др.^[4—6]. Найдено, что эти два, близких по размеру, вещества имеют
разную теплоту адсорбции на цеолитах типа А и X. Во всех исследованных
авторами случаях теплота адсорбции диэтилового эфира больше, чем
н-пентана. Очевидно, что в случае эфира имеется еще, кроме дисперсион-
ного, электростатическое взаимодействие с катионами решетки. Авторы
приходят также к выводу, что взаимное расположение этих молекул
внутри полостей может быть различно.
343
а, ммоль/г
Рис. 6. Изотермы адсорбции и-пентана на NaX.
Общий коэффициент диффузии в формованных цеолитах является
функцией частных значений коэффициентов Бг, Бгг, DIU, обусловленных
соответственно скоростью переноса в сорбционной полости элементарных
ячеек, скоростью переноса во вторичных порах и скоростью прохожде-
ния через входное отверстие в сорбционную полость. Третий фактор не
имел практического значения в случае сорбции воды (ее молекулы доста-
точно малы). Повышение скорости сорбции диэтилового эфира и н-пен-
тана на цеолите NaX говорит о заметном влиянии фактора проходимости
окна в случае сорбции углеводородов. Скорость прохождения молекулы
через входное отверстие в сорбционную полость обычно связывают с раз-
мерами молекулы и окна. Разница между коэффициентами диффузии н-пен-
тана и диэтилового эфира, более ощутимая при сорбции этих веществ на
цеолите СаА, чем NaX, показывает, что не только чисто геометрические
соотношения играют существенную роль для скорости процесса. Очень
важны также энергетические взаимодействия между молекулой, прохо-
дящей в полость, и окном. Молекулы диэтилового эфира, испытывая до-
полнительное действие ионов решетки, медленнее проникают внутрь
полостей, где происходит основной процесс адсорбции.
ЛИТЕРАТУРА
1. Д. П. Тимофеев, И. Т. Е р а ш к о, Изв. АН СССР, ОХН, 1961, 7, 1192.
2. Д. П. Тимофеев, И. Т. Е р а ш к о, Изв. АН СССР, сер. хим., 1964,
10, 1761.
3. Д. П. Т и м о ф е е в, И. Т. Е р а ш к о, ДАН СССР, 1960, 132, 5, 1144.
4. О. М. Джигит, А. В. Киселев, Г. Г. Му ттик, Колл, ж., 1963,
25, 34.
5. О. М. Джигит, А. В. Киселев, Г. Г. М у ттик, J. Phys. Chem.,
1962, 66, 2127.
6. О. М. Д ж и г и т, А. В. К и с е л е в, Г. Г. М у т тик, ЖФХ, 1962, 36, 919.
О КИНЕТИКЕ И ДИНАМИКЕ СОРБЦИИ ГАЗОВ И ПАРОВ
НА СИНТЕТИЧЕСКИХ ЦЕОЛИТАХ
В. Б. Фенелонов, Л. Е. Фенелонова, А. Н. Салюков,
В. Н. Мазин
В связи с широким применением синтетических цеолитов для очистки
и осушки газов и паров необходима разработка методов расчета подоб-
ных процессов.
В большинстве случаев расчет сорбционных процессов сводится к уста-
новлению зависимости между временем защитного действия или количе-
ством поглощенного вещества и длиной слоя, удельной скоростью потока,
зернением сорбента, температурой и т. д.
Если для сорбционных процессов на углях, силикагелях и других
сорбентах эти зависимости широко известны, то методы расчета сорбции
на цеолитах изучены значительно меньше.
Нами исследовались кинетика и динамика сорбции СО2 и Н2О на син-
тетических цеолитах.
Было показано, что кинетика сорбции удовлетворительно описывается
уравнением вида:
345
где а — количество вещества, поглощенного за время т; а0 — предельная
величина сорбции при данных условиях; w — начальная скорость сорб-
ции, причем гло=аор, где 0 — кинетический коэффициент.
Уравнение (1) предложено Трепнелом [1] для случая, когда при сорб-
ции молекула занимает одновременно два центра.
В случае сорбции паров воды цеолитом весьма вероятно, что молекула
Н2О взаимодействует одновременно с двумя кислородными атомами с об-
разованием водородных связей [2].
На рис. 1 представлен график некоторых экспериментальных дан-
ных в координатах уравнения (1). Из графика видно, что кинетика
сорбции СО2 на синтетических цеолитах удовлетворительно описывается
Рис. 1. Кинетика сорбции СО2 и паров воды на
синтетических цеолитах (гранулы NaA ГОБ
ВНИИНП).
1. 2 -СО, 10%, 1 = 20° С; 3, 4-Н2О, СН.о - 7.3 мг/л,
1 = 20° С.
уравнением (1).
После интегрирования
при условиях
i = const, а = 0 при т = 0,
Со = const, а — а0 при т = t
уравнение (1) приобретает
вид
или в линейной форме
у = аор — а,В, (3)
где величина кинетического
коэффициента 0 при концен-
трации адсорбата Со в по-
токе и зернении сорбента d
определяется из
Со
(4)
где К — константа уравне-
ния.
Для вывода уравнения динамики сорбции было использовано урав-
цение материального баланса в форме, предложенной В. Л. Анохиным [3]:
а0 dt
dv = dm -f- иа0 da.
(5)
вещества, прошедшего за слой за время т; а0 —
сорбента в данных условиях; Со — исходная кон-
га— вес сорбента; и — объемный расход смеси;
где v — количество
предельная емкость
центрация сорбата;
С •
а—отношение =-, где С—концентрация сорбата за слоем в момент
со
времени т.
Уравнение кинетики сорбции (1) в динамических условиях при из-
меняющейся по ширине адсорбционного фронта концентрации С с учетом
условия (4) преобразуется в
da
-^-==,80(1-0)2, (6)
С- и
где с = —- = -----необходимое условие стационарного режима,
о о л0
346
Совместное решение уравнений (5) и (6) приводит к уравнению дина-
мики сорбции:
где А — насыпной вес сорбента, v — удельная скорость потока, L —
длина слоя сорбента, 0 — кинетический коэффициент.
Уравнение (7) удовлетворительно описывает динамику сорбции СО2
и Н2О на синтетических цеолитах в диапазоне относительных концентра-
ций за слоем 0.05 о 0.75. о
На рис. 2 представлены
экспериментальные выходные
•кривые сорбции СО2 и Н2О,
прерывистой линией обозначен
ход кривых по уравнению (7).
Кривая 2 была рассчитана по
уравнению (7) с использованием
величин Од и Р, полученных из
обработки кривой 1, а затем
подтверждена эксперименталь-
но.
На рис. 3 те же выходные
кривые представлены в коор-
динатах
Ь1 — о а , .
-------з---Г т
а 1—а 1
уравнения (7).
Из угла наклона прямой
•определяется величина динами-
ческого коэффициента.
Значение динамической ем-
Рис. 2. Экспериментальные и расчетные (пре-
рывистая линия) выходные кривые сорбции
СО2 и паров Н2О на синтетических цеолитах
(таблетки ГОБ ВНИИНП).
1 — NaX 3% СО2, Ь=5 см, и=0.5 л/мин. см2; 2 —
тоже, £=10 см; 3— NaX 0.25% СО2, £=10 см,
г=0.5 л/мин.см2; 4 = СаА 14.7 мг/л Н2О, £=15 см,
v=1.7 л/мин.см2; 5 — СаА 7 мг/л, £=15 см,
т= 1.7 л/мин.см2.
кости ад рассчитывается из
Q
уравнения (7), когда относительная концентрация за слоем — с — 0.5
и уравнение(7) имеет вид
«од г
то-в= Cov L'
откуда
Сог
“0— Д£ т0.5
(9)
Таблица 1
• Параметры опыта и расчетные значения «0 и (3
Образец Концентрация сорбата Насыпной вес Д. г/см3 Удельная скорость V, л/мин. • см2 Длина слоя 1, см ®0» г/см3 1/мин.
3% со2 0.65 0.5 5 78 0.80
NaX 3% со2 0.65 0.5 10 78 0.80
0.25% СО2 0.65 0.5 10 40 0.134
14.7 мг/л Н2О 0.75 1.70 15 218 0.130'
СаА 7 мг/л Н2О 0.75 1.70 15 202 0.070
Примечание. Все образцы изготовлены в ГОБ ВНИИНП,
347
В табл. 1 приведены опытные и расчетные величины для опытов, пред-
ставленных на рис. 2 и 3.
Уравнение динамики (7) можно использовать для пересчета данных
при изменении длины слоя и концентрации. В последнем случае можно-
пользоваться достаточно хорошо выполняющимся соотношением
-уг^- = const. (10)
С помощью уравнения (7) может быть рассчитана средняя динамиче-
ская активность сорбента до проскока о.
Пусть проскоку о,- соответствует время тогда по графику сорбции
может быть рассчитана динамическая активность из выражения
CovS
а*~ &LS
vS Г*
-aZsJ (ii).
о
Ввиду трансцендент-
ности уравнения (7) функ-
ция с(т) в явном виде не
выражается, но
ч
vCo f , . , сой
о
—(12).
Рис. 3. Выходные кривые сорбции в координатах
уравнения (7).
1 — NaX 3% СО2, Ь=5 см, »=0.5 л/мин.см2; 2 — NaX 3%
СО2, L=10 см, с=0.5 л/мин.см2; 3—NaX 0.25% СО2,
L=10 см, 15=0.5 л/мин.см2; 4 — СаА 14.7 мг/л Н2О,
Ь=15 см, ®=1.7 л/мин.см2; 5 — СаА 7 мг/л Н2О, L=15 см,
15=1.7 л/мин.см2.
После решения урав-
нение (11) принимает вид
Сог Г
°д = й0° + -д^1 В — °) ’ —
Совместное решение (7) и (13) приводит
расчета динамической активности:
к простому уравнению для
Сор Г, 1 — о "|
йД = °0— Д£.р [_1п - +1J-
(14)
Уравнение (13) несколько лучше описывает экспериментальные дан-
ные, чем уравнение (14). Возможно, это вызвано наложением погреш-
ностей уравнений (7) и (13). Поэтому расчет динамической активности
можно производить по полуэмпирическому уравнению
C(ir Г 1 — о 1
(15)
В табл. 2 показана применимость приведенных уравнений динами-
ческой активности для расчета. Для сравнения приведены данные по
динамической активности, рассчитанные графически.
Приведенные уравнения динамики и кинетики сорбции позволяют
вести расчеты сорбционных процессов с достаточной степенью точ-
ности.
348
Таблица 2
Применение уравнений (12)—(15) для расчета величины динамической активности
•z, мин. 3 Динамическая активность а, мг/г Погрешность kaia П определения
графиче- ская по (13) по (14) по (15) по (13) ПО (14) по (15)
51 0.10 39 39.8 43.2 39.8 + 2.0 +10.7 +2.0
60 0.20 46.4 45.8 50.0 45.8 — 2.6 + 7.7 —1.3
68 0.30 51.1 51.0 54.6 50.3 — 1.2 + 5.8 —3.2
76 0.40 55.8 55.6 59.2 54.0 - 0.3 + 6.1 —3.2
84 0.50 60.2 59.4 61.6 57.4 — 1.3 + 2.3 —4.7
93.5 0.60 63.0 63.2 65.2 60.8 + 0.6 + 5.1 —4.45
104 0.70 66.2 66.8 68.8 64.6 + 0.9 + 3.9 —2.4
122 0.80 68.6 71.8 70.4 69.0 + 4.7 + 2.6 +0.61
149 0.90 69.6 79.4 80.8 70.2 +14.0 +29 +0.8
Условия опыта: концентрация СО, в потоке Г7с, удельная скорость потока 0.5 л/мин.см2,
.длина слоя сорбента 20 см.
ЛИТЕРАТУРА
1. Б. Т р е и н е л. Хемосорбция. ИЛ, 1958.
2. Г. В. Цицишвили, Г. Д. Багратишвили, в сб.: Синтетические
цеолиты, Изд. АН СССР, 1962, 38.
3. В. Л. Анохин, ЖФХ, 1957, 31, 5.
МЕТОДЫ РАСЧЕТА АДСОРБЕРОВ С МОЛЕКУЛЯРНЫМИ СИТАМИ
П. Пиру, М. Ралек, О. Грубнер
Введение
Одним из самых распространенных способов практического использо-
вания молекулярных сит является сорбционная сушка газов. Несмотря
на это, в литературе до сих пор отсутствуют данные о расчете адсорбе-
ров, наполненных молекулярным ситом в качестве сорбента. Внимание
было уделено расчету адсорберов [1], работающих на основе обычных
адсорбентов, какими являются, например, силикагель, окись алюминия
и активные угли. Предметом настоящего сообщения является более де-
тальная оценка возможности применения некоторых методов для описа-
ния хода процесса сушки в слое молекулярного сита.
Для описания процесса сушки в адсорбере необходимо получить
прежде всего ответ на следующие вопросы: 1) какое количество молеку-
лярного сита является необходимым для того, чтобы в течение сорбцион-
ного цикла концентрация удаляемого вещества на выходе из адсорбера
не превышала заданной концентрации; 2) в продолжении какого времени
адсорбер с данной навеской адсорбента будет работать так, чтобы концен-
трация сорбата на выходе не превышала заранее заданной концентрации.
Главное внимание поэтому в отдельных методах расчета уделяется
выходным кривым, т. е. кривым, описывающим зависимость концентрации
сорбата на выходе из адсорбера от времени. Выходные кривые необходимо
теоретически рассчитать, поскольку это возможно, на основе доступных
349
физических и физико-химических данных. Для расчета выходных кривых
можно воспользоваться методами Розена [2, 3], Гоугена и Маршалла [4],
Мичелса [5] или Иглтона и Блисса [6]. В случае адсорберов с молеку-
лярными ситами, которые по своим сорбционным и структурным свой-
ствам значительно отличаются от до сих пор применяющихся сорбентов,
необходимо подобрать такой метод, который содержит как можно меньше
упрощающих допущений и который наиболее точно описывает сложный
сорбционный процесс, протекающий в слое молекулярного сита. С этой
точки зрения самыми выгодными методами являются методы Розена |2, 3]
и Гоугена и Маршалла [4]. Остальные методы не оправдали себя даже для
расчета адсорберов с обычными сорбентами (силикагель), не говоря уже
о расчете выходных v кривых для адсорберов с молекулярными ситами,
так как в них отсутствуют некоторые данные о переносе вещества. Вслед-
ствие этого мы провели анализ только первых двух методов.
Методы расчета
При теоретическом расчете выходных кривых по Розену [2, 3] учиты-
ваются скорость переноса сорбата к поверхности зерен сорбента и скорость
переноса сорбата во внутрь зерен сорбента. При этом пренебрегают влия-
нием скорости собственной сорбции и влиянием продольной диффузии
в слое сорбента. Далее предполагается, что равновесная сорбция водяного
пара удовлетворяет линейной изотерме. Применение линейной изотермы
адсорбции при расчете является допустимым упрощением. На практике
адсорберы работают обычно при условиях, когда концентрация сорбата,
например водяного пара, на выходе незначительна, т. е. в крутой или
практически линейной области изотермы адсорбции. Кривая выхода
по Розену дана в интегральном виде теоретическим соотношением
С_
у + — J е sin (4р — xh^v) g idg,
о
(1)
где С и Со — концентрация водяного пара на выходе и входе в адсорбер;
v, х и у — безразмерные параметры, характеризующие перенос вещества,
длину слоя и время контакта; hv и h2 — функции; р — переменная ин-
тегрирования.
Метод Гоугена и Маршалла [4] еще далее упрощает представления
Розена, пренебрегая влиянием внутренней диффузии и учитывая только
перенос сорбата к поверхности сорбента. Выходная кривая по Гоугену
и Маршаллу дана в интегральном виде теоретическим соотношением
Яг
-g- = 1 — ео/ j (2i Vot • gz) d (gz),
о
(2>
где at и gz — безразмерные параметры, характеризующие время контакта
и перенос вещества; Jo — функция Бесселя первого рода и нулевого по-
рядка.
Проверка методов расчета
На рис. 1 и 2 приведены результаты расчета выходной кривой по обоим
методам (кривые — по уравнениям (1), (2), точки — по эксперименталь-
ным данным [7]). На оси у отложено значение С/Со, а на оси х — время
в минутах от начала сорбционного процесса. Использован был лаборатор-
ный адсорбер, работающий изотермически при 20° С с навеской 9.4 г
350
молекулярного сита типа Линде 4А с эффективным диаметром зерен 1.3 мм.
Длина слоя 7.4 см, диаметр слоя 1.5 см. Содержание водяного пара в воз-
духе б.ыло 0.014 г/г. Скорость
тока влажного воздуха в слое
молекулярного сита была
4.5 г/см2 • час. При расчете
для константы Генри было
использовано значение
0.152 • 106, а для коэффици-
ента диффузии водяного пара
в воздухе в зернах молеку-
лярного сита по Д. П. Тимо-
фееву [8] — значение 0.5Х
ХЮ-6 см2/сек.
Из сравнения теоретиче-
ской выходной кривой с экспе-
риментальными данными вы-
текает, что процесс сорбции
описан достаточно точно бо-
Рис. 1 ."Выходная кривая, рассчитанная ’ по
уравненшо Гоугена и Маршалла.
лее общим методом Розена.
Таким образом, этот метод
может быть использован для
описания процесса сушки на
молекулярных ситах. Инте-
ресно, что при помощи этого
метода можно хорошо опи-
сать процесс сорбции даже
при относительно высоких
концентрациях водяного пара
на входе в адсорбер. Это
обстоятельство расширяет
границы применимости мето-
да Розена. Было бы поэтому
интересно проверить метод
Розена с целью расчета вы-
ходных кривых для промыш-
ленных адсорберов с моле-
кулярными ситами и далее
распространить этот метод
(поскольку доступны значе-
ния коэффициентов внутрен-
ней диффузии) на расчет
сорбции других сорбатов
на молекулярных ситах. Ме-
тод Гоугена и Маршалла не
является подходящим для
Кружками отмечены экспериментальные данные.
Рис. 2. Выходная кривая, рассчитанная по
уравнению Розена.;
Кружками отмечены экспериментальные данные.
описания процесса сушки на молекулярных ситах. Сдвиг теоретической
выходной кривой к существенно более высоким значениям времени по
отношению к экспериментальным данным, вероятно, вызван тем обстоя-
тельством, что здесь играет роль также и диффузия водяного пара в по-
рах зерен молекулярного сита, которую этот метод при расчете не учиты
вает.
ЛИТЕРАТУРА
1. J. W. Carter, Brit. Chem. Engng., 1960, 5, 472, 625; 1961, 6, 308.
2. J. В. Roz en, J. Chem. Phys., 1952, 20, 387.
3. J. B. R о z e n, Ind. Engng. Chem., 1954, 46, 1590.
351
4. О. A. Н о u g e n, W. R. Marshall, Chem. Engng. Progress, 1947, 49, 197.
5. R. F. T г e у b a 1, Mass Transfer Operations. McGraw-Hill, N. Y., 1955.
6. L.C. E aglet on, H. В 1 i s s, Chem. Engng. Progress, 1953, 49, 543.
7. O. Gru bn er, P. J i r u, M. R a 1 e k. Molekulova sita. SNTL, Praha, 1964.
8. Д. П. Тимофеев, О. H. К а б а н о в а, Ю. Т. Е р а ш к о, А. С. П о но-
ма р е в, в сб.: Синтетические цеолиты, Изд. АН СССР, 1962, 24.
КИНЕТИКА ВАКУУМНОЙ ДЕСОРБЦИИ
НОРМАЛЬНЫХ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ ИЗ ЗЕРНА
ЦЕОЛИТОВ
Ю. М. Афанасьев, 3. А. Жукова, Н. В. Вельцев
Одна из основных стадий всякого адсорбционного процесса — десорб-
ция поглощенных веществ — может осуществляться различными методами:
путем повышения температуры слоя адсорбента, снижения давления в си-
стеме, отдувки адсорбата в токе газа-носителя, применением вакуума
и т. п., однако во всех случаях необходимым условием успешного осуще-
ствления десорбционной стадии является наличие достаточно высоких
коэффициентов диффузии адсорбата внутри зерна' адсорбента. Величина
коэффициента диффузии адсорбата внутри зерна должна во многом опре-
делять выбор температурного режима десорбции. Ранее нами было пока-
зано, что для эффективного осуществления процесса десорбции коэффи-
циенты внутренней диффузии должны быть не менее 20 • 10“7 см2/сек.
Для случая десорбции воды из цеолитов такие значения коэффициентов
достигаются при температурах выше 150° С, для случая десорбции дву-
окиси углерода — выше 0° С [1].
В настоящей работе была исследована кинетика десорбции нормальных
парафиновых углеводородов С4—С9 из гранул синтетических цеолитов.
Выбор адсорбата был продиктован прикладным значением работы, ши-
роким использованием цеолитов типа СаА для извлечения нормальных
парафинов из различного нефтяного сырья [2—4].
Изучение кинетики десорбции проводилось на вакуумной адсорб-
ционной установке с кварцевыми весами. Каждому опыту предшествовало-
изучение изотермы адсорбции при той же температуре. Данные адсорб-
ционного равновесия углеводородов, полученные нами на советских цео-
литах, были в хорошем соответствии с величинами адсорбции н-парафинов
на образцах фирмы Линде [5]. Подготовка образцов заключалась в от-
качке при 350° С до постоянного веса при остаточном давлении 10 ~5 мм рт. ст.
Насыщенный при заданной температуре образец соединяли с вакуумной
системой, в которой поддерживалось -ззление 10~2 мм рт. ст. Насыщение
образца углеводородом соответствовало относительному давлению угле-
водорода р/рв=0.95 при 20° С. Изменение веса гранулы цеолита фикси-
ровалось через каждые 10 мин. катетометром, имеющим чувствительность
1 • 10 ~3 мм. В основном опыты проводились с образцами гранули-
рованных цеолитов типа NaX (Ц-202-216-218) и СаА (Ц-202-100), у ко-
торых диаметр гранул был равен высоте и составлял 4.3 мм.
На рис. 1 приведены кинетические кривые десорбции углеводородов
Св—Св из цеолитов NaX и СаА при 150° в координатах у—t, где y=atlaco —
степень насыщения, t — время (мин.) at — адсорбировано вещества при
времени t, ат — адсорбировано вещества при равновесии перед началом
десорбции. Сравнение данных, полученных на образцах цеолитов типов
NaX и СаА, показывает, что скорость десорбции нормальных углеводоро-
352
дов из цеолита СаА благодаря диффузионному торможению кристалли-
ческой решетки, имеющей размер входных окон в адсорбционные полости,
близкий к критическому диаметру молекул нормальных парафинов
(4.9 А), значительно ниже, чем у цеолитов типа NaX. Продолжительность
экспозиции для достижения равной степени десорбции из цеолита СаА
в 12—30 раз ниже, чем из цеолита NaX, причем разность в скоростях
десорбции возрастает с увеличением цепи углеводорода. Скорость отвода
вещества из гранул в процессе вакуумной десорбции характеризуется
коэффициентом диффузии, который численно соответствует количеству
вещества, диффундирующего через единицу поверхности при градиенте
Рис. 1. Десорбционные кривые нормальных парафинов при 150° С из
цеолитов NaX (а) и СаА (б).
концентрации, равном единице. Коэффициенты диффузии для цилиндра
конечных размеров выражаются уравнениями диффузии
СО со •
а 32 VV 1 Г. / р» .
p2(2m-l)eXPL1-\^2
я=1 т=1 Гп ' '
(2т— 1)2 Tt2
£2
(1)
(2)
где р„ — корни функции Бесселя J0 (ря); п=1, 2, 3. . .; тга=1, 2, 3 . . .
Решение уравнения (1) для цилиндрических гранул с длиной гра-
нулы L, равной ее диаметру d, приводит к простому уравнению для опре-
деления коэффициента диффузии при величине степени насыщения у=0.5:
0;3187?2
0 = —2---
где т0 5 — время, при котором достигается степень заполнения адсорбента,
•у=0.5 [6, 7]; R — радиус гранулы.
Величины коэффициентов диффузии, вычисленные из эксперименталь-
ных данных по адсорбции нормальных парафинов, приведены в табл. 1,
данные которой показывают, что основным фактором, определяющим
скорость десорбции, является температура. С повышением температуры
отмечается резкое возрастание коэффициента диффузии. Температурный
режим десорбции должен выбираться в зависимости от требуемой степени
десорбции, продолжительности экспозиции, энергозатрат на нагрев ад-
сорбента и т. п.
Для каждого углеводорода существует температурный порог, при ко-
тором коэффициент диффузии достигает величины D > 20 • 10~7 см2/сек.,
открывающей путь к практическому применению. Эти пороговые темпе-
23 Цеолиты
353
Таблица 1
Коэффициенты диффузии нормальных парафиновых углеводородов при
десорбции из гранулированных цеолитов
з г, °с Цеолит с. с5 с6 с. С8 с„
/• г NaX • 11.1 - - —_
СаА 1.75 — — — — —
, 50 | NaX 32.0 6.1 — • — — —
, . СаА 6.65 0.3 . — — — . —
100 | NaX — 120 22.3 1.6 <0.1 <0.1
СаА — 25 1.65 0.08 <0.1 <0.1
150 | NaX >200 — 120 • 45 10 1.4
СаА >200 — 18.8 2.58 0.4 0.05
200 NaX >200 — >200 160 50 22.3
СаА >200 — — 10 3.1 2.6
250 | NaX >200 >200 >200 >200 >200 >200
СаА >200 >200 >200 — 41.0 12.2
Рис. 2. Температуры, при которых дости-
гаются коэффициенты диффузии D > 20Х
ХЮ~7 см2/сек. — t' (сплошные линии) и ос-
таточное содержание углеводородов
а > 1 г/100 г в течение 30 мин. — t" (пре-
рывистые линии).
ратуры, как показывает табл. 2, возрастают по мере увеличения молеку-
лярного веса углеводорода и температуры его кипения. На рис. 2 порого-
вые температуры выражены как функция чи ;ла атомов углерода в моле-
куле углеводорода. Для цеолита NaX разница между температурой, при
которой величина D достигает ве-
личины 20 • 10-7 см2/сек., и тем-
пературой кипения углеводорода
колеблется от 40° С для к-С4Н10,.
до 60° С для н-С9Н22.
Диффузионное торможение кри-
сталлической решетки цеолита СаА
при десорбции нормальных пара-
финовых углеводородов значи-
тельно более резко выражено, чем
при вакуумной десорбции воды из
цеолита КА [1]. Одинаковые вели-
чины коэффициентов диффузии
достигаются, например, если цео-
лит СаА при десорбции н-бутана
перегреть по сравнению с цеоли-
том NaA на 30° С. С удлинением
углеводородной цепи в молекуле
диффузионное торможение решет-
ки повышается, в связи с чем раз-
ница в температурах равных
коэффициентов диффузии увели-
чивается, достигая для н-нонана
70° С.
Остаточное содержание углеводорода в гранулах цеолита зависит от
температуры и цродолжительности экспозиции с вакуумом. На рис. 3
представлены кривые остаточного содержания нормальных парафиновых
углеводородов в цеолите NaX в зависимости от температуры при времени
экспозиции 30 мин.
Экспериментальные кривые позволяют установить минимальные тем-
пературы, которые требуется применить, чтобы достичь необходимой сте-
пени удаления каждого углеводорода. Так, для того чтобы остаточное-
354
Таблица 2
Характерные температуры для процесса десорбции нормальных парафиновых
углеводородов
с. с5 С7 ^8 Сд
Цеолит температура кипения, °C
0 36 69 98 126 150
Температура, при которой / D =20 • 10~7 см2/сек. 1 NaX СаА 40 76 100 100 160 140 210 170 240 210 280
Температура, при которой в те- ( чение 30 мин. достигается оста- 1 точное содержание углеводе- j рода < 1 г/100 г. 1 NaX СаА 65 100 140 185 185 240 245 280 285 310
содержание нормального парафина в цеолите NaX не превышало 1 г/100 г,
требуются температуры: н-С4Н10 — 70° С, н-С5Н12 — 100° С, н-С8Н14 —
140° С, н-С7Н1в — 190° С, н-С8Н18 - 240° С, н-С9Н20 — 280° С. Анало-
Рис. 3. Остаточное содержание углеводородов при
десорбции из цеолита NaX в течение 30 мин.
Г — С4Н10, S —С5Н„, 3 — С«Н>(, 4 — С,Н,,. 5 — С8Н,8.
6 С8Н80.
гичные температуры для цеолита СаА, показанные на рис. 3 прерывистой
линией, приблизительно на 40° С выше.
Таким образом, изученные кривые десорбции нормальных парафино-
вых углеводородов из цеолитов и вычисленные коэффициенты диффузии
являются исходными для расчета технологических десорбционных про-
цессов и выбора температурного режима десорбции в процессах депарафи-
низации нефтяных продуктов.
ЛИТЕРАТУРА
1. Н. В. К е л ь ц е в, Газ. промышленность, 1964, 4, 51.
2. G. J. Griesmer, Н. В. Rhodes, К. Kigonaga, Petrol. Refiner,
1960, 39, 6, 125.
3. Н. В. К е л ь ц е в, А. Ф. Старовойтова, Газ. промышленность, 1961,
8, 34.
4. М. Б. Вольф, Р. В. Алексеева, в сб.: Синтетические цеолиты,
Изд. АН СССР, 1962, 233.
5. D. L. Р е t е г s о п, О. R е d 1 i с h, J. Chem. and Engng. Data, 1962, 7, 4, 570.
6. Д. И. T и м о ф e e в, И. T. E p а ш к о, ДАН СССР, 1960, 132, 5, 1144.
7. Д. П. Тимофеев, И. Т. Е р а ш к о, Изв. АН СССР, ОХН, 1961, 7,1192.
23*
355
ИЗОТЕРМИЧЕСКАЯ ДЕСОРБЦИЯ ЭТИЛЕНА ИЗ СЛОЯ ЦЕОЛИТА NaX
Н. В. Келъцее, Н. С. Торочешников, Ю. И. Шумяцкий
Одним из путей решения проблемы улавливания и концентрирования
непредельных углеводородов, в том числе этилена, из различных про-
мышленных газов является применение новых высокоактивных промыш-
ленных сорбентов с большой удельной поверхностью. К таким сорбентам
относятся синтетические цеолиты. Высокая адсорбционная способность
цеолитов по этилену, даже в области низких парциальных давлений его
в исходном газе, селективность адсорбции при выделении из смесей с дру-
гими (парафиновыми) углеводородами, достаточная механическая проч-
ность позволили поставить вопрос о применении цеолитов для получения
этиленового концентрата из газов [1].
Эффективность всякого адсорбционного процесса разделения газовых
смесей во многом определяется условиями десорбции поглощенных ве-
ществ, устанавливаемыми обычно опытным путем.
В литературе отсутствуют подробные данные по десорбции олефиновых
углеводородов из цеолитов, и в связи с этим нами было проведено исследо-
вание процесса десорбции этилена газообразным азотом из слоя гранули-
рованного цеолита NaX, размельченного до размера частиц 0.5—1.0 мм.
Изучение изотермической десорбции этилена проводилось в интервале
температур 25—75° С в лабораторном адсорбере диаметром 9.5 мм, с вы-
сотой слоя цеолита 46 см; загрузка цеолита составляла 16.4 г. Адсорбер
был снабжен термостатирующей рубашкой, через которую циркулиро-
вала вода с заданной температурой, подаваемая из ультратермостата.
Перед началом каждого опыта цеолит подвергали регенерации, нагревая
адсорбер извне электрическим током до 340—360° С с одновременной
продувкой азотом в течение 2.5 часа. В этих условиях достигалось прак-
тически полное обезвоживание цеолита.
После регенерации адсорбер охлаждали до температуры опыта, а затем
через него пропускали смесь этилена с азотом до тех пор, пока не уста-
навливалось адсорбционное равновесие, о наступлении которого судили
по идентичности составов входящего и выходящего газов.
Десорбция этилена осуществлялась продувкой азотом. Температура
при десорбции поддерживалась постоянной и была равна той температуре,
при которой проводилась адсорбция. Расход отдувочного газа — азота
при десорбции устанавливался реометром. Выходящая из адсорбера смесь
этилена с азотом непрерывно анализировалась на хроматографе ГСТЛ
с алюмогелем в качестве неподвижной фазы и воздухом в качестве газа-
носителя.
Применявшийся в опытах этилена по данным хроматографического
и химического анализов имел чистоту выше 99%.
На рис. 1 приведено изменение во времени концентрации этилена
в выходящем газе при скорости продувочного газа (азота) 0.5 л/см2 мин.
Предварительное насыщение цеолита этиленом было различным и отве-
чало условиям равновесия с газом, в котором концентрация этилена из-
менялась от 0.8 до 14%.
График показывает, что в любом случае на выходной кривой десорб-
ции можно отметить два характерных участка. На первом горизонтальном
участке имеет место постоянство содержания этилена в газе десорбции,
и это содержание соответствует концентрации в газе предшествующей ста-
дии, т. е. при насыщении. Наличие такого участка свидетельствует о том,
что между газовой фазой и определенными слоями сорбента, включая
последние по ходу движения газового потока, успевает установиться
состояние, близкое к равновесному. Второй участок характеризуется за-
356
кономерным падением содержания этилена в газе десорбции. Десорбцион-
ные кривые, полученные при разных исходных степенях заполнения цео-
лита этиленом и отвечающие второму периоду десорбции, достаточно
точно ложатся на единую десорбционную кривую. Это означает, что время
отдувки до некоторой заданной величины остаточной адсорбции не за-
висит от исходной степени насыщения цеолита. Продолжительность пер-
вого периода десорбции определяется графически как горизонтальный
отрезок, проведенный из точки концентрации на оси абсцисс, соответствую-
/4
12
10
8
А 63
о 89
е Mi
08
1.5
г.5
90
В
г
А &-А-А
Расход отдувочного газа
(л/смг-мин.)
• 198
х 1.03
А 0.50
е 0.25
150 180 б.мин
-о-» х, о
О 3 9
Количество пропущенного отдувочного
газа (л/г)
Содержание этилена в смеси,
подаваемой, на адсорбцию (°/о)
о
Рис. 1. Десорбция этилена азотом при температуре 25° С, расходе азота 0.5 л/мин. см2
(я) и при разных расходах азота (б).
щей равновесной степени насыщения, до пересечения с единой десорб-
ционной кривой второго периода. Длина горизонтального участка умень-
шается с увеличением степени насыщения цеолита этиленом.
Опыты, проведенные с разными скоростями продувочного газа в ин-
тервале от 0.5 до 2.0 л/см2мин., показали, что степень десорбции этилена
зависит только от количества пропущенного газа. Кривые, построенные
в координатах «содержание этилена в газе десорбции», «количество
пропущенного азота на единицу веса адсорбента», при разных скоростях
потока совпадают. Эта закономерность сохраняется как в случае высокой,
так и в случае низкой исходной степени заполнения цеолита этиленом
(рис. 1).
По кривым, представленным на рис. 1, и значениям равновесной ста-
тической активности, согласно изотермам адсорбции этилена, была рассчи-
тана зависимость количества недесорбированного этилена в процессе
отдувки при разном удельном расходе азота.
Эти данные для трех температур приведены на рис. 2. Прерывистые
линии, соответствующие первому участку выходной кривой, являются
357
касательными к единой десорбционной кривой, они пересекают ось орди-
нат в точке, которая соответствует
Рис. 2. Зависимость количества неде-
сорбированного этилена от объема про-
* пущенного аэота.
А — остаточное количество этилена, исм’/г
сорбента; Б — количество пропущенного от-
дувочного газа, л/г.
в газе, / (с) — изотерма адсорбции,
заполнению сорбента этиленом перед
началом десорбции.
Наличие достаточно размытого
фронта десорбции этилена из цеоли-
тов, характерное для крутых вы-
пуклых изотерм адсорбции, позво-
ляет применить последние для вычис-
ления выходных десорбционных кри-
вых [2, 3].
Если предположить, что при
исследованных скоростях газового
потока равновесие между газовой и
твердой фазами устанавливается до-
статочно быстро, тогда десорбцион-
ные кривые, отвечающие второму
периоду, могут быть рассчитаны из
изотерм адсорбции по уравнению [4]
V
— =МГ(с), (1)
где v — объем пропущенного отду-
вочного газа, х — длина адсорбера,
М — вес сорбента на единицу длины
колонки, с — концентрация этилена
f(c) — производная изотермы адсорб-
ции по концентрации.
Для больших концентраций, кроме того, учитывалось изменение
объема газовой смеси согласно уравнению Викке [5]:
V / С \2
- = с) - (2)
где С — суммарная концен-
трация газа в объеме адсор-
бера, равная сумме концен-
. траций сорбированного газа
(С2Н4) и газа-носителя (N2).
Изотермы адсорбции эти-
лена, полученные на уста-
новке с кварцевыми весами
после эвакуирования образца
цеолита при 350° С до оста-
точного давления 10 ~5 мм
рт. ст., приведены на рис. 3.
На рис. 4 представлен
пример расчета общей десорб-
ционной кривой этилена из
цеолита NaX по изотерме ад-
сорбции при 25° С. Графиче-
Рис. 3. Изотермы адсорбции (1—5) С2Н4 на
гранулированном цеолите NaX.
А — активность сорбента, исм3/г; Б — концентрация
этилена, объемн. %; В—давление этилена, мм рт. ст.
ское дифференцирование изотермы для расчета по уравнению (1) выполнено
по методике М. И. Яновского [6]. Изотерма этилена была разбита на ряд
участков, например 0—0.5,0.5—1,1—2, 2—3, 3—4,4—6 и т. п. Через точки,
отвечающие местам пересечения изотермы с линиями, параллельными
оси ординат, проведены касательные к изотерме. На оси концентраций
358
влево от точки О отложен отрезок ОР, равный MX (в нашем случае 16.4 г).
Из Р исходят лучи, параллельные касательным к изотерме. Через каждую
точку пересечения луча с осью ординат Б проведена параллельная оси
абсцисс прямая до пересечения с соответствующей вертикальной гранич-
ной линией. Точки, образующиеся при последнем пересечений, по смыслу
построения отвечают значениям v уравнения (1) для соответствующих кон-
центраций. Пунктирная линия получена с учетом поправочного множи-
теля (1 — с/С)2. Экспериментальные точки достаточно точно наклады-
ваются на теоретически полученную кривую, что позволяет рекомендовать
метод графического дифференцирования для широкого использования при
Рис. 4. Построение общей десорбционной кривой из изотермы адсорб-
ции этилена при 25° С.
А — концентрация этилена, объемн. %; В — активность сорбента, исм’/г; В —
количество пропущенного отдувочного гаэа, л/г. Черные треугольнички — выбороч-
ные экспериментальные точки.
расчете десорбции углеводородов из цеолитов. Расчеты десорбционных
кривых этилена, выполненные на основании изотерм адсорбции при тем-
пературах 25, 50, 75, 200 и 300° С, позволили сделать вывод, что доста-
точно быстрая десорбция этилена может быть достигнута уже при 75° С;
однако большой удельный расход отдувочного газа ( ~ 1 л/г цеолита)
исключает возможность получения в данном случае концентрированного
этилена в газах десорбции. Если принять согласно изотермам, что в про-
цессе адсорбции этилена при 25° С из смеси, содержащей 3% его, 1 г
цеолитов поглощает 27 нем3, а при десорбции выделяется 90% поглощен-
ного этилена, то в газе десорбции при 75° С будет содержаться лишь около
4.3% этилена. С повышением температуры расход, отдувочного газа резко
снижается, в результате чего, например при 200°, концентрация этилена
в газах десорбции может быть повышена до 30%, а* при 300° — до 50%;
Результаты работы позволяют выбрать оптимальные условия десорб-
ции этилена и других углеводородов из цеолитов, применяя метод графи-
ческого дифференцирования изотерм адсорбции. 1
359
ЛИТЕРАТУРА
1. Н. В. Кельцев, Газ. промышленность, 1960, 9, 49.
2. Г. Ш а й. Теоретические основы хроматографии газов. ИЛ, 1963.
3. С. Е. Р о г и н с к и й, М. И. Я н о в с к и й, ЖФХ, 1950, 24, 137.
4. J. W. W i 1 s о n, J. Am. Chem. Soc., 1940, 62, 1583.
5. Е. W i с k е, Angew. Chemie, 1947$ 13, 15.
6. М. И. Яновский, ЖПХ, 1951, 24,6,661.
ИЗУЧЕНИЕ АДСОРБЦИИ ПАРОВ ВОДЫ
ПРИРОДНЫМИ ЦЕОЛИТАМИ
В. Т. Быков, А. Н. Виргинцев, А. В. Лукьянов, Н. Е. Щербатюк
Адсорбция на природных цеолитах изучалась в основном Р. М. Бэр-
рером. Было показано, что анальцим, гармотом, филлипсит, цеолиты
семейства натролита, стильбита и гейландита обычно сорбируют только
небольшие полярные молекулы, такие, как вода или аммиак [1—3].
Среди алюмосиликатных кристаллов, которые характеризуются как сор-
бенты поглощением газов, в работе [1, 4] изучался морденит. Предложены
структуры для анальцима [1, 5], волокнистых цеолитов семейства натро-
лита [1, 6], фожазита, эрионита, шабазита, гмелинита и цеолита типа
гармотом [1].
Дана классификация некоторых молекулярных сит, в которой выделено
пять типов цеолитов. За основу принят предельный размер сечения моле-
кул сорбатов, которые способны проникать в полости цеолитов. Согласно
этой классификации, изучаемый в данной работе Na-морденит относится
к 4-му типу молекулярных сит и может поглощать молекулы газов и па-
ров с критическим диаметром не более 4 А. Вообще природные цеолиты
характеризуются меньшим диапазоном свободных диаметров «окон» по
сравнению с искусственными цеолитами. Например, известно, что фожа-
зит является цеолитом с наиболее открытой решеткой. Диаметр его «окон»
около 8 А, второй послео фожазита цеолит со сравнительно большими се-
чениями «окон» — 4—5 А — шабазит. Свободные диаметры «окон» других
природных цеолитов меньше, чем у шабазита. Кроме работ Бэррера по
изучению адсорбции на природных цеолитах, имеются данные по адсорб-
ции паров воды на десмине и натролите в работе [7].
В настоящей работе ставилась цель — изучение адсорбции водяных
паров некоторыми природными цеолитами в широких интервалах равно-
весных относительных давлений.
Объектами исследования являлись три образца природного морденита
из района Нидым, притока Нижней Тунгуски, образец ломонтита из того же
района, образец натролита из Хибин и десмина из района пос. Тетюхе
Приморского края.
Измерение изотерм адсорбции паров воды производилось изопиести-
ческим методом в установке, описанной в работе [8], в лаборатории зон-
ной плавки Института неорганической химии СО Ан СССР.
Образцы морденита Т, ломонтита, натролита были любезно предостав-
лены Г. В. Букиным. Образцы цеолитов использовались очищенными и
измельченными. Брались навески образцов по 1 г, а в случае мордени-
тов — по 0.2—0.4 г, находящихся в равновесии с влагой помещения. Под-
готовка образцов к эксперименту сводилась к термической дегидратации.
Установление оптимального режима обезвоживания цеолитов приобре-
360
Рис. 1. Изотермы адсорбции па-
ров воды на природных цеолитах.
1 — десмин; 2 — натролит; з — мор-
денит; 4 — ломонтит.
тает решающее значение для их практического применения. Режим де-
гидратации выбирался на основании изучения дифференциальных кривых
нагревания и кривых обезвоживания исследуемых цеолитов с учетом того,
чтобы при наиболее полном удалении полостной воды сохранилась обра-
тимость поглощения.
Образцы цеолитов прокаливались в течение 4—6 часов при следующих
температурах: морденит — 400° С [9], десмин — 300° С [10], натролит —
440° [11], ломонтит — 280° С [10].
Чашечки с прокаленными цеолитами помещались в прибор для изо-
пиестирования. Давление водяных паров над цеолитами создавалось при-
сутствием в приборе бюкса со стандартным раствором, в качестве которого»
служил водный раствор серной кислоты,
а в отдельных случаях — водный рас-
твор хлористого натрия.
Равновесное давление водяных паров
в приборе определяется легко, так как
равновесная концентрация серной кислоты
в стандартном растворе известна. Поэтому,
используя справочные данные по зависи-
мости давления паров воды от концентра-
ции кислоты (или соли) [12], путем линей-
ной интерполяции можно определить и
равновесные давления паров воды. Пред-
варительными опытами было показано,
что для достижения равновесия необхо-
димо 48—72 часа. Все опыты проводились
при 25° С. Точность поддерживания тем-
пературы +0.01° С. Используемая уста-
новка работала круглосуточно.
Поглощающая способность цеолитов
связана с химическими особенностями
адсорбируемых молекул: цеолиты наибо-
лее эффективны в отношении полярных
молекул и ненасыщенных углеводородов.
С другой стороны, цеолиты — эффек-
тивные адсорбенты для веществ, молеку-
лы которых способны проникать через
«окна» в полости дегидратированных кри-
сталлов, т. е. в их первичную пористую
структуру. Полярные молекулы воды с
лекулы, равным 2.6 А, свободно проникают через «окна» в полости изу-
чаемых цеолитов.
На рис. 1 приведены изотермы адсорбции паров воды на десмине (7),
натролите (2), мордените (3) и ломонтите (4).
Десмин по строению алюмосиликатного каркаса относится к цеоли-
там второй группы по классификации Тейлора [13]. Структура цеолитов
этого класса до сих пор окончательно не выяснена.
Предполагают [13], что структура их образована бесконечной дву-
мерной связью из тетраэдров (Al, Si)04. Слои связаны между собой более
слабыми силами, чем силы связи между тетраэдрами внутри слоя. Опыты
по дегидратации этих цеолитов подтверждают эту общую картину. При
обезвоживании десмин сокращается в направлении, перпендикулярном
к слоям. Это естественно, если справедливо предположение о том, что мо-
лекулы воды расположены между плоскостями. Удаление этой воды при-
ведет к сближению между слоями и одномерному сокращению элементар-
ной ячейки решетки.
«критическим диаметром» мо-
361.
Кривая изотермы адсорбции десмина поднимается полого по мере воз-
растания величины относительного давления пара, что говорит о посте-
пенном заполнении пустот между слоями. При давлении р/рк=О.О1 ве
личина адсорбции составляет 68% от предельной при p/pt=OAO.
Натролит — представитель волокнистых цеолитов, алюмосиликат-
ный каркас которых образован идущими параллельно рядами тетраэдров
(Al, Si)04 (наподобие волокон) [13]. «Волокна» образуются цепочкой из
звеньев тетраэдров состава Al2Si3O10. При различном соединении этих
звеньев между собой получаются отдельные представители класса волок-
нистых цеолитов. Каналы в промежутках между волокнами заполняются
катионами и молекулами воды. В полостях структуры натролита может
находиться лишь по одной молекуле воды, что хорошо иллюстрируется
кривой изотермы адсорбции. При давлении р/ре=6.01 величина адсорб-
ции составляет 96% от предельной при plps=QAQ.
Морденит — тоже относится к классу волокнистых цеолитов.
Каналы в промежутках между волокнами заполнены катионами натрия
и молекулами воды.
Свободный диаметр канала 6.0 А, по [15], и 6.6 А, по [14], диаметр вось-
мерных «окон» — 4 А. Несмотря на широкий просвет (6 А) основных ка-
налов в мордените, он может поглощать и обменивать молекулы с диамет-
ром, лишь немного больше 4 А.
Изотермы адсорбции паров воды на всех трех образцах морденитов
подобны друг другу по форме, величины адсорбции при р//\=0.01 соот-
ветственно равны: MI — 59%, МП — 66%, МТ — 70% от предельной
при р/рв=0.40.
Образец МТ был наиболее чистым и однородным, поэтому, сопоставляя
полученные величины, считаем, что заполнение 70% от предельного для
морденита Т самое точное.
Ломонтит — это кальциевый цеолит. А. Е. Ферсман [16] отметил,
что ломонтит легко теряет половину молекулы воды и переходит во вто-
ричный леонгардит, обладающий характерным свойством — способно-
стью к распылению и рассыпанию.
Дифференциальные кривые нагревания ломонтита показывают, что
выделение воды из ломонтита происходит в два этапа [10]: первая порция
воды (7.5%), или 2Н2О выделяется в интервале температур 240—280° С;
выделение оставшихся двух молекул воды задерживается до температуры
400—420° С. В нашем опыте ломонтит, прокаленный при температуре
400° С, терял обратимость поглощения. Можно предположить, что при
такой обработке разрушается кристаллическая решетка этого цеолита.
Подобное явление уже отмечалось в работе [7]. Поэтому ломонтит прока-
Таблица 1
Величины сорбции при различных равновесных давлениях (ммоль/г)
Цеолит Величины сорбции паров Н2О при 25° С и различных значениях j>/pg
т о с-01 • 24 1 • io-3 — О сч © ° О О СО о О со © © © ©
Морденит I 1.97 3.12 3.55 5.55 6.05 6.30 6.50 6.70 8.09
Морденит II 2.10 3.09 3.68 5.25 5.57 5.75 5.85 5.95 7.03
Морденит Т 2.20 3.61 4.16 5.65 5.90 6.00 6.00 6.10 6.55
Десмин — 3.48 4.84 6.75 7.30 7.50 7.80 7.95 8.26
Натролит 1.18 2.77 5.75 5.95 6.00 6.05 6.35 6.85 10.31
Ломонтит — 1.41 1.94 2.70 3.20 3.35 4.10 4.70 5.40
.362
ливался на воздухе только до 280° С, т. е. до потери половинного содер-
жания воды. При этом обратимость поглощения сохранялась.
Кривая изотермы адсорбции ломонтита (вернее, леонгардита) подни-
мается по мере возрастания величины р/ре полого, что связано с постепен-
ным заполнением каналов ломонтита.
При значении р/р8=0.01 величина адсорбции составляет 48% от пре-
дельной.
Целесообразно предельные величины адсорбции для воды оценивать
при давлении p/pg=0.40 при 20° С, так как при более высоких относи-
тельных давлениях пара возможна капиллярная конденсация рассматрива-
емых паров в зазорах между контактирующими кристаллами цеолитов [17].
Результаты опытов приведены
в табл. 1, где даны величины ад-
сорбции для различных относи-
тельных давлений.
Предельные адсорбционные
объемы были оценены нами как
по графикам изотерм адсорбции
при р/ре=0А0, так и расчетным
путем по уравнению адсорбции
для адсорбентов 1-го структур-
ного типа, к которым относятся
цеолиты [18, 19].
Для вычисления предельных
адсорбционных объемов дегидра-
тированных цеолитов было исполь-
зовано уравнение изотермы ад-
сорбции для адсорбентов 1-го
структурного типа при Т <Z Ткр
Рис. 2. Изотермы адсорбции паров воды
на природных цеолитах в линейной форме
уравнения (1).
Значения кривых те же, что на рис. 1.
тде а — величины адсорбции (ммоль/г) для равновесных относительных
давлений p/ps и абсолютных температур Т; Wo — предельный объем
адсорбционного пространства (см3/г); В — константа, зависящая от раз-
меров микропор адсорбента; и — объем 1 ммоля вещества в адсорби-
рованном состоянии (в виде жидкости с плотностью, соответствующей
объемной жидкой фазе при температуре опыта), v = 0.01807 см3/ммоль;
р — коэффициент аффинности характеристических кривых, зависящий
от природы адсорбируемого вещества. Для установления соответствия
экспериментальных данных уравнениям адсорбции обычно пользуются
линейной формой уравнения (1) или
lg а = 1g -0.434^-(f 1g-^)2; (2)
1g а = С - D (1g )2 , (3)
где
C=lg-^ = lga0 (4)
и
D = 0.434В (j-J . (5)
363
Построены изотермы адсорбции паров воды при 25° С в линейной форме
уравнения (2).
На рис. 2 изображены в линейной форме изотермы адсорбции паров
воды. Экспериментальные точки хорошо укладываются на прямые линии
в интервале равновесных относительных давлений от р/р ==5.7 • 10~4
до 0.23. В табл. 2 приведены константы С и D уравнения в форме (3)и ука-
Таблица 2
Константы уравнения изотермы адсорбции в форме (2)
Цеолит С D • 10’ Интервал применимости
P/Ps е
Морденит I 0.760 4.516 6.7- 10-* —2.34-10-1 0.31—0.91
Морденит II 0.748 4.110 5.7 • IO'4 — 2.34 -10-1 0.31—0.95
Морденит Т 0.775 4.148 5.7 -10-4 _ 2.34 • 10-1 0.30—0.88
Десмин 0.865 7.849 9 • Ю-з — 2.34 • 10-1 0.48—0.94
Натролит 0.790 6.870 5.7 • IO”4 — 2.34 10-1 0.20—0.82
Ломонтит 0.490 8.167 9-10-3 — 2.34-10-1 0.45—0.91
заны интервалы равновесных относительных давлений и заполнений О
предельного объема адсорбционного пространства, для которых экспери-
ментальные данные находятся в соответствии с уравнением (3). Как видно
из таблицы, константы С для всех трех образцов морденитов близки по
значениям. В табл. 3 приведены величины констант уравнения (1): пре-
Таблица 3
Константы уравнения изотермы адсорбции в форме (1)
Цеолит Расчетные значения по уравнению (1) Определенные по изотерме адсорбции при р/р8 = 0.40
а0, ммоль/г Wo, см3/г В/₽2-10" Go, ММОЛЬ/Г TVo, см3/г
Морденит I 5.754 0.1040 1.172 5.95 0.1075
Морденит II ... . 5.598 0.1012 1.066 5.60 0.1012
Морденит Т . . . . 5.957 0.1076 1.076 5.95 0.1075
Десмин 7.328 0.1324 2.037 7.25 0.1310
Натролит 6.166 0.1114 1.782 6.0 0.1084
Ломонтит 3.090 0.0558 3.15 3.15 0.0569
дельные объемы адсорбционного пространства Wo и отвечающие им пре-
дельные величины адсорбции а0 и константы В1$2, характеризующие
кривизну изотерм адсорбции.
Величины Wo отнесены к единице массы полностью дегидратированных
цеолитов.1 Предельные адсорбционные объемы для всех образцов мордени-
тов, определенные из уравнения (1) и по изотермам адсорбции при р/р„='
=0.40, близки по значениям. Более точны значения Wo, определенные
из уравнения (1). В наших опытах предельный объем адсорбционного про-
1 Исключение составляют десмин и ломонтит.
364
•странства для десмина 0.13 см3/г, т. е. немного меньше по сравнению с ве-
личиной 0.18 см3/г для десмина в работе 17], а для натролита в нашем опыте
величина Wo=OAl см3/г, т. е. в два раза больше, чем у образца
натролита в работе [7].
В заключение следует отметить, что изотермы адсорбции паров воды
на натролите, десмине и мордените являются типичными для мелкопори-
стых адсорбентов и уже при малых относительных давлениях достигается
почти полное заполнение адсорбционного пространства. Ломонтит дает
меньшее заполнение в этих же условиях.
Подтверждена применимость уравнения изотермы адсорбции для ад-
сорбентов 1-го структурного типа по Дубинину к исследованным цеоли-
там.
Представляет интерес уточнить полученные константы по другому
адсорбату, например азоту.
ЛИТЕРАТУРА
1. R. М. В а г г е г, D. Sc. (N. L.), Brit. Chem. Engng., 1959, 5, 4.
2. В. M. В а г г е г, Proc. Roy. Soc., 1938, А 167, 392.
3. R. М. В а г г е г, (unpublished).
4. R. М. Barre г, Trans. Faraday Soc., 1944, 40, 374, 555.
5. R. M. Barre r, J. Soc. Chem. Ind., 1945, 44, 130.
6. В. P., 548, 905; US Pat., 2306, 610.
7. С. П. Ж д а н о в, H.H. Бунтарь, в сб.: Синтетические цеолиты, Изд.
АН СССР, 1962, 105.
8. А. Н. К и р г и и ц е в, А. В. Лукьянов, ЖФХ, 1963, 37, 1.
9. В. П. III а ш к и и а, Минерал, сб. Львовск. гос. унив., 1958, 12, 170.
10. Э. А. Лазаренко, М. М. С л и в к о, Минерал, сб. Львовск. гос. унив.,
1961, 15, 235.
11. А. Д. Федосеев, Э. Н. Корыткова, Зап. Всес. минерал, общ., 1963,
сер. II, ХСП, 4, 305.
12. R. A. R obinso n, R. Н. S t о k е. Electrolite Solution. London, 1959.
13. В. Т е й л о р, в сб.: Основные идеи геохимии, ОНТИ, Химтеорет, 1937.
14. W. М. Meier, Z. Kristallogr., 1961, 115, 5—6, 439.
15. Е. А. И о б е д и м с к а я, Н. В. Белов, Кристаллография, 1963, 8, 6.
16. А. Е. Ферсман, Тр. Геол, и минерал, музея АН СССР, 1902, 2, 6.
17. М. М. Дубинин, Е. Г. Жуковская, К. О. М у р д м а а,
Изв. АН СССР, ОХН, 1962, 6, 960.
18. М. М. Д у б и н и н, Е. Д. 3 а в е р и н а, ЖФХ, 1949, 23, 1129.
19. М. М. Дубинин, 3. А. Ж у к о в а, Н. В. К е л ь ц е в, в сб.: Синтети-
ческие цеолиты, Изд. АН СССР, 1962, 7.
СОРБЦИОННЫЕ СВОЙСТВА
НЕКОТОРЫХ СОЛЕЙ ГЕТЕРОПОЛИКИСЛОТ
П. К. Мигалъ, М. А. Кердиваренко, Г. А. Кренис
Важным вопросом является изыскание новых веществ, обладающих
свойствами молекулярных сит и устойчивых в кислых средах.
В этой связи нами проведено исследование сорбционных свойств не-
которых труднорастворимых солей гетерополикислот. Гетерополикислоты
и их соли являются высокогидратированными кристаллическими соеди-
нениями, причем процесс удаления воды обратим и не ведет к разложе-
нию [1, 2]. Роль воды, находящейся в гетерополисоединениях, остается
пока не совсем ясной, несмотря на важность этого вопроса для
изучения их строения [3, 4]. Часть воды заполняет пустоты в кристал-
365
лах и носит цеолитный характер [5]. Адсорбционные свойства микро-
кристаллического фосфоромолибдата аммония замечены Турне и Деви-
ном [6].
Нами исследовались сорбционные свойства аммониевых и цезиевых
солей кремнемолибденовой, кремневольфрамовой, кремнемолибдоволь-
фрамовой и фосфорномолибденовой кислот. Соли получали осаждением
хлоридами аммония и цезия из синтезированных [7] или промышленных
кислот. Кремнемолибдат и кремневольфрамат аммония получали при
двух условиях: из концентрированных растворов кислот при быстром
добавлении осадителя и из разбавленных растворов при медленном до-
бавлении осадителя. В первом случае получался мелкокристаллический
осадок с кристаллами величиной порядка микрона, во втором случае
осадок состоял из больших прозрачных шестигранных кристаллов вели-
чиной порядка 10 мк. Сорбцион-
ные свойства мы исследовали для
мелкокристаллического осадка.
Исследование проводили в ва-
куумной адсорбционной установке
а.мг/г
, ммоль/г
1
15
200
f-0 P/Ps
100 -
Рис. 1. Изотермы сорбции паров воды и
бензола при 20° С на солях гетерополикис-
лот и цеолите NaX (15% глины).
Г —NaX, вода; 2 —фосфорномолибдат аммония,
вода; з — кремнемолибдат аммония, вода; 4 —
кремнемолибдовольфрамат цезия, вода; 5 — крем-
немолибдат цезия, вода; 6 — кремневольфрамат
аммония, Г вода; 7 — кремнемолибдат аммония,
бензол.
Рис. 2. Изотермы сорбции паров уксус-
ной кислоты при 20° С на солях гетеро-
поликислот и цеолите NaX (15% глины).
1 — NaX; 2— кремнемолибдовольфрамат це-
зия; 3 — кремнемолибдат цезия; 4 — кремне-
вольфрамат аммония; S — фосфорномолибдат
аммония; в — кремнемолибдат аммония. •
с кварцевыми весами. Навески дегазировались непосредственное установке
в течение 18—20 часов с одновременным нагреванием до 150—300° С до
постоянного веса и остаточного давления 5 • 10-s мм рт. ст.
Нами изучалась сорбция паров воды, метилового спирта, бензола и
уксусной кислоты. Параллельно адсорбция велась также и на цеолите NaX.
На рис. 1 приведены изотермы адсорбции паров воды при 20° С на
цеолите NaX (7), фосфорномолибдате аммония (2), кремнемолибдате
аммония (5), кремнемолибдовольфрамате цезия (4), кремнемолибдате
цезия (5), кремневольфрамате аммония (6) и паров бензола на кремнемо-
либдате аммония (7). Как видно из рис. 1, изотермы сорбции на солях
гетерополикислот имеют вид, характерный для цеолитов. Величины сорб-
366
ции паров воды на кремнемолибдате и фосфорномолибдате аммония близки
к адсорбционной емкости цеолита NaX. Незначительная сорбция бензола
кремнемолибдатом свидетельствует о молекулярно-ситовом характере
этой соли. *
В качестве кислого адсорбата нами использовалась уксусная кислота.
Изотермы сорбции СН3СООН при 20° С приведены на рис. 2: (2) — на цео-
лите NaX, (2) — на кремнемолибдовольфрамате цезия, (5) — на кремне-
молибдате цезия, (4) — на кремневольфрамате аммония, (5) — на фосфорно-
молибдате аммония и (6) — на кремнемолибдате аммония. Максимальную
сорбционную емкость по кислоте обнаруживают цезиевые соли. Значи-
тельное снижение величины сорбции СН3СООН на кремнемолибдате и
фосфоромолибдате аммония по сравнению с водой подтверждает молеку-
лярно-ситовой характер этих солей. Кислотостойкость всех исследованных
соединений следует из обратимости сорбции, выражающейся в отсутствии
гистерезиса, который на цеолите NaX выражен весьма сильно.
ЛИТЕ РАТУ РА
1. Е. Я. Р о д е, М. М. И ванова, ЖНХ, 1958, 3, 2323.
2. Е. Я. Роде, Н. А. Кротов, ЖНХ, 1959, 4, 1782.
3. А. А. Б а б а д - 3 а х р я п и н, Усп. химии, 1956, 25, 1373.
4. A. Bradley, I. Illingworth, Proc. Roy. Soc., 1936, A 157, 113.
5. S. W e s t, L. Audrieth, J. Phys. Chem., 1955, 59, 1069.
6. C. Tourneux, C. Devin, C. r., 1951, 232, 2430; 1951, 233, 45.
7. Неорганические синтезы, I. ИЛ, M., 1951.
КАТАЛИТИЧЕСКАЯ АКТИВНОСТЬ СИНТЕТИЧЕСКИХ ЦЕОЛИТОВ
В РЕАКЦИИ КРЕКИНГА КУМОЛА
Е. Г. Борескова, К.' В. Топчиева, Л. И. Пигу зова
Синтетические цеолиты в настоящее время приобретают большое зна-
чение не только как адсорбенты, но и как катализаторы разнообразных
реакций. Возможность химического модифицирования поверхности цео-
литов путем ионного обмена делает их перспективными для осуществления
ряда каталитических процессов. Молекулярно-ситовые свойства цеолитов
позволяют надеяться получить катализаторы со значительно большей се-
лективностью, чем используемые в настоящее время.
В изучении молекулярных сит как катализаторов наметилось два
основных направления: подход с точки зрения геометрической доступно-
сти внутренней поверхности цеолитов для реагирующих молекул и удале-
ния продуктов реакции из пор [1J, с другой стороны — изучение харак-
тера промежуточного взаимодействия, зависящего от химических свойств
и электронной структуры катализатора [2].
Настоящее исследование касается второго из указанных направлений
и посвящено изучению каталитической активности синтетических цеоли-
тов типа X и Y, структура которых исключает влияние размера пор на
селективность реакции. Химический состав образцов варьировался в ши-
роких пределах как в отношении природы и степени обмена замещающего
катиона, так и в соотношении SiO2/Al2Og в структуре.
Каталитическая активность цеолитов определялась по модельной
реакции крекинга кумола, кинетический механизм которой был изучен
ранее на аморфных алюмосиликатных катализаторах.
367
Таблица 1
Химический состав образцов молекулярных сит
Тип цеолита SiO,/AI..O3 CaO Na2O % обмена
КаХЦ-202-127 2.4 17.8 0
'СаХ-1.0 экв 15.8 3.3 82.3
СаХ-0.75 экв 2.4 12.6 6.1 66.6
СаХ-0.5 экв —. 9.4 9.08 49.7
CaY-20-MOE-l.O экв — 14.1 1.1 93
CaY-20-MOE-0.75 экв 3.4 10.4 4.06 78
NaNH4-20-MOE-2 — — 7.7 23
NaNII4-20-MOE-l 3.4 — 9.0 50
Как известно, каталитически активные центры этой реакции в алюмо-
силикатах связывались с наличием протонизированных мест на их по-
верхности. С этой точки зрения катионные формы цеолитов (Na и Са) не
должны были бы проявлять значительной активности в этой реакции.
Поскольку реакция крекинга кумола все же протекает на этих образцах,
вероятно, природа активных центров цеолитов иная, так как, исходя из
•структурных данных и щелочной реакции цеолитов в воде, трудно пред-
положить наличие протонных мест на катионированной поверхности
цеолитов.
Химический состав изученных образцов приведен в табл. 1. Эти об-
разцы были получены на укрупненной установке МОБ.
Подбор образцов позволял проследить, с одной стороны, влияние на
каталитическую активность природы и степени обмена ионов, с другой —
изменение химического состава решетки цеолита и влияние декатиони-
рования на изменение ак-
Рис. 1. Схема установки для проведения реакции
в хроматографическом режиме.
Д— баллон с гелием; 2 — вентиль тонкой регулировки;
з — система очистки газа-носителя; 4 — детектор; 5 — реак-
тор; 6 — ловушка; 7 — аналитическая колонка; 8 — кон-
вертор; р — пенный измеритель скорости.
тивности.
Все исходные образцы
цеолитов предварительно
прокаливались при 500° С
в течение 6 часов и допол-
нительно прогревались в
реакторе перед каждым
опытом 1 час в токе инерт-
ного газа при той же тем-
пературе. Опыты по кине-
тике крекинга кумола про-
водились как в реакторе
проточного типа, так и
импульсным хроматогра-
фическим методом [4, 5].
Хроматографи ч е с к и й
метод в последнее время
с успехом применяется
не только для опреде-
ления теплот адсорбции, но и для изучения каталитических процессов.
Он позволяет быстро и с достаточной точностью получить кинети-
ческие характеристики реакции. Схема установки приведена на
рис. 1.
В поток газа-носителя (гелия), проходящего через реактор с катали-
затором (5), вводилась небольшая порция кумола (4 • 10-3 г). Кумол,
проходя с током гелия через колонку, превращался последовательно
368
по всему сдою катализатора. Продукты реакции и ненрореагировавший
кумол вымораживались жидким азотом в ловушке (6), а затем вводились
в аналитическую колонку (7). Введение аналитической колонки исклю-
чало перекрывание пиков реагентов, которое наблюдалось из-за непол-
ного разделения их в реакторе. В разделительной колонке в качестве
неподвижной фазы использовалась полярная жидкость ОП-7 на диатоми-
товом кирпиче. После аналитической колонки все компоненты конверти-
ровались до СО2 и детектировались. Степень превращения кумола опре-
делялась как отношение веса пика бензола к сумме весов пиков бензола
и кумола. Реакция проводилась в температурном интервале 400—525° С.
Следует отметить высокую избирательность реакции, т. е. в продуктах
реакции обнаруживали только бензол и пропилен. Лишь при температурах
выше 525° С наблюдалось образование небольших количеств побочных
продуктов. Экспериментальные данные (рис. 2) обрабатывались по урав-
нению, выведенному для реакции 1-го порядка Бассетом, Рогинским [4, 5]:
F 1
А К = R2T6-m ’ lg Г^х ’
In t/1-Х
Рис. 2. Зависимость активности
цеолитов от времени контакта.
где к — константа скорости реакции, К — константа адсорбционного
равновесия, F — скорость газа-носителя, т — навеска катализатора,
х — степень превращения.
Как видно из рис. 2, экспериментальные точки в координатах уравне-
ния (1) хорошо ложатся на прямые 1—6. Из уравнения (1) следует, что
тангенс угла наклона прямой (рис. 2) пропорционален произведению кон-
стант скорости реакции и адсорбционного равновесия; константа адсорб-
ционного равновесия вычислялась из величин удерживаемых объемов,
а затем из величин их произведения рассчитывалась константа скорости.
Положение прямых показывает соот-
ношение констант скоростей. Исходный
Ыа+-образец обладает ничтожной актив-
ностью. Замещение на ионы Са2+ заметно
увеличивает активность, причем возраста-
ние активности пропорционально степени
обмена. Увеличение отношения SiO2/Al2O3
в структуре вызывает заметное увеличе-
ние активности. Максимальная величина
константы скорости была получена для
декатионированных образцов.
В координатах In In 1_х от ЦТ при
постоянной скорости газа-носителя угол
наклона прямой дает величину кажущейся
энергии активации.
На рис. 3 показано изменение значе-
ний энергии активации для различных
цеолитов. Следует отметить, что для каж-
дого типа цеолитов наклоны прямых оди-
наковы для различных степеней обмена
катиона. Для определения величин истин-
ных энергий активации необходимо было
определить теплоты адсорбции кумола на этих катализаторах. Нами были
определены теплоты адсорбции всех компонентов реакции в предкатали-
тической области температур (290—450° С).
На рис. 4 дана графическая зависимость величин удерживаемых
объемов в зависимости от ЦТ для бензола и кумола на цеолитах разных
типов.
1/4 24 Цеолиты
369
Таблица 2
Теплоты адсорбции кумола и бензола на молекулярных ситах
Тип цеолита
Н аимеяование сорбтива NaX СаХ-0.5 СаХ-0.75 СаХ-10 CaY-0. а CaY-1.<i NaNH,-l см и й £ SR 10 X- Линде
Бензол 20 + 1 19 24 27 18 21 15 15 18 21
Кумол 27 + 1 26 25 26 23 24 21 21 25 28
(18 ккал./моль) хорошо
величина
реакции крекинга ку-
разных типов.
Рис. 3. Энергии активации
мола на цеолитах
Интересно отметить, что полученная
согласуется с величинами теплот адсорбции бензола на цеолите 13Х,
полученными Бэррером [8] — 17 ккал./моль при 25—70° С — и Эбер-
ли [9] — 15.5 ккал./моль при 260—482° С. По-видимому, независимость
теплоты адсорбции от температуры распространяется на достаточно
широкий интервал температур.
Как видно из рис. 4, время удерживания кумола для всех образцов
больше, чем бензола. У цеолитов типа X теплота адсорбции бензола воз-
растает с увеличением со-
держания кальция. Тепло-
ты адсорбции кумола при-
мерно постоянны (табл. 2).
Интересно, что мини-
мальные времена удержи-
вания как для бензола, так
и для кумола обнаружены
у декатионированных мо-
лекулярных сит типа Y.
На этих образцах наблюда-
лись и минимальные тепло-
ты адсорбции бензола. Теп-
лоты адсорбции кумола на
декатионированных образ-
цах близки к величинам
теплот на катионных фор-
мах.
В работе были опреде-
лены теплоты адсорбции
участков реакции и на
каталитически неактивных цеолитах NaX (ВНИИНП и фирмы Линде).
Полученные значения оказались близкими к величинам теплот адсорб-
ции для активных образцов; по-видимому, адсорбция происходит не только
на специфических каталитических центрах.
В условиях проведения реакции в хроматографическом режиме не
было обнаружено заметной адсорбции пропилена, что свидетельствует
об отсутствии тормозящего действия его на протекание реакции кре-
кинга кумола на синтетических цеолитах. Этот факт объясняет разли-
чие в кинетическом механизме реакции при ее протекании на аморф-
ных алюмосиликатах и синтетических цеолитах в хроматографическом
режиме. Для последних наблюдается преобладание адсорбции исходного
вещества по сравнению с адсорбцией продуктов реакции; на аморфном
алюмосиликате наблюдается обратное явление.
370
Таблица 3
Энергии активации и константы скоростей реакции крекинга кумола
на синтетических цеолитах
Тип цеолита
Энергия активации и константы NaX СаХ-0.5 СаХ-0.75 СаХ-10 CaY-0.75 CaY-10 NaNH,-1 см и § 10 X- Линде
Е, ккал./моль . . . 47 40 38 38 29 31 30 32 39
Л, сек.-1 | 0.5 10-з 0.90 10-2 1.25 10-2 1.5 10-2 4.4 10-2 2.8 10-2 0.15 0.25 0.8-10-2
К, моль • ат м--1 • г-1 | 6.0 W-3 1.5 IO-3 2.2 Ю-з 3.5 Ю-з 0.62 Ю-з 1.4 Ю-4 4.1 10-4 4.1 1.2- Ю-з
Величины истинных энергий активации, констант скоростей и кон-
стант адсорбционного равновесия для всех изученных образцов приве-
дены в табл. 3.
+2 -
Рис. 4. Теплоты адсорбции бензола, кумола и пропиле-
на на различных молекулярных ситах.
Из этих данных видно, что с увеличением степени замещения натрия
на кальций и при декатионировании цеолитов константа скорости реакции
заметно растет. Так, при переходе к декатионированным образцам кон-
станта скорости реакции увеличивается более чем на порядок.
В полученных величинах энергий активации не обнаруживается столь
ясной закономерности. Для неактивного натриевого образца величина
1/2 24 Цеолиты
371
энергии активации высока — 47 ккал./моль. Введение в цеолит ионов
кальция заметно снижает энергию активации — до 38 ккал./моль. Однако
в зависимости от степени обмена величина энергии активации не меняется,
ее значение примерно одинаково 40 38 ккал./моль для всех степеней
обмена. Как видно из полученных нами данных, наблюдается резкое раз-
личие в величинах энергий активации для цеолитов типа X и Y при близ-
ких степенях обмена. Цеолит типа Y имеет значительно меньшую вели-
чину энергии активации, чем цеолит типа X (38 и 29 ккал./моль соответ-
ственно). Декатионированные образцы типа Y, обладая значительно
большей активностью, нежели катионные формы типа X и Y, дают зна-
чения энергии активации, близкие к образцу CaY (30—32 ккал./моль по
сравнению с 29—31 ккал./моль для CaY).
В табл. 3 для сравнения приведены данные, полученные нами по ка-
талитической активности цеолита 10Х фирмы Линде. Как видно из таб-
реакции близка к величине, получен-
ной для цеолита СаХ, с близкой
степенью обмена.
Исходя из теоретических предпо-
сылок [2, 7, 3], на поверхности моле-
кулярных сит у алюмокислородных
тетраэдров можно предположить су-
ществование избыточных отрицатель-
ных зарядов, компенсируемых вклю-
чением соответствующих катионов.
Одновалентные катионы локализуют-
лицы, величина энергии активации
Рис. 5. Изменение каталитической ак-
тивности декатионированного цеолита в
зависимости от количества адсорбиро-
ванного хинолина.
ся у электроотрицательных центров
поверхности и полностью компен-
сируют отрицательные заряды, ли-
шая цеолит каталитической актив-
на дивалентные катионы каждый
ности, как это, по-видимому, имеет
место в Na-образце. При замещении
двухвалентный катион должен компен-
сировать отрицательные заряды двух алюмокислородных тетраэдров.
При этом энергетически более выгодным является асимметричное поло-
жение катиона, при котором полностью компенсируется отрицательный
заряд одного тетраэдра, а другой остается неполностью компенсирован-
ным [2, 7]. С этой точки зрения может быть понятным возрастание ката-
литической активности цеолита при увеличении содержания в нем Са
и при увеличении отношения SiO2/Al2O3 в структуре. В первом случае,
т. е. при увеличении степени замещения, происходит увеличение числа
некомпенсированных электроотрицательных центров, а во вотором при
увеличении отношения SiO2/Al2O3 увеличивается степень некомпенсиро-
ванности отрицательного заряда за счет увеличения расстояния между
соседними алюмокис дородными тетраэдрами.
Особый интерес представляют декатионированные формы цеолитов.
Они занимают несколько обособленное место, так как при разложении
аммонийной формы и удалении протонов происходят небольшие, но опре-
деленные изменения в решетке, связанные с потерей атомов кислорода
структуры. Как указывается в работе Рабо, на поверхности декатиони-
рованных цеолитов образуются атомы алюминия в трехвалентном состоя-
нии (центры кислоты Льюиса). Но наряду с ними сохраняются отрица-
тельно заряженные алюмокислородные тетраэдры, аналогичные тетра-
эдрам кальциевой формы цеолитов. Однако оставалось неясным, какие
из этих типов центров являются каталитически активными. В работе,
доложенной на Парижском конгрессе по катализу, на основании близкой
активности катионных и декатионированных образцов в реакции изоме-
372
ризации утверждается, что активным центром для этой реакции является
отрицательно заряженный алюмокислородный тетраэдр с неполностью
компенсированным зарядом. Более точно ответить на этот вопрос можно
было бы, изучив адсорбцию на цеолитах органических оснований, которые
являются ядами для всех кислотных центров, в том числе и центров типа
Льюиса.
С этой целью нами было изучено отравляющее действие хинолина на
каталитическую активность кальциевых и декатионированных образцов.
Был выбран хинолин, так как он устойчив при температуре опыта 420° С.
В описанной выше хроматографической установке измерялась актив-
ность исходного цеолита, а затем в каталитическую колонку попеременно
вводились определенные порции хинолина и кумола.
На рис. 5 показано падение каталитической активности декатиониро-
ванного цеолита в зависимости от количества адсорбированного хинолина.
Как видно из рисунка, первоначально активность образца сильно сни-
жается пропорционально количеству введенного хинолина, достигая
небольшого, но постоянного значения. Минимум активности достигается
при введении 0.09 ммоль/г. Подобная картина наблюдалась при отрав-
лении алюмосиликатных катализаторов органическими основаниями и
ионами щелочных и щелочноземельных металлов [10]. Полученная за-
висимость падения активности декатионированных цеолитов при отрав-
лении их хинолином подтверждает существование на поверхности цеоли-
тов по крайней мере двух видов центров (отрицательно заряженных алю-
мокислородных тетраэдров и образующихся при удалении протонов ато-
мов алюминия в трехвалентном состоянии — центры типа кислоты
Льюиса). Вероятно, при отравлении этих образцов хинолин адсорбируется
именно на апротонных центрах поверхности, которые определяют ка-
талитическую активность, так как для катионных форм (СаХ и CaY),
обладающих высокой активностью, приписываемой А104 тетраэдрам,
нами не было обнаружено заметного снижения их активности при адсорб-
ции хинолина.
Таким образом, отравляющее действие хинолина на цеолиты подтвер-
ждает наличие кислотных центров на поверхности декатионированных
цеолитов, .но не обнаруживает таковых в катионных формах цеолитов.
С целью подтверждения наличия кислотных центров на поверхности
декатионированных цеолитов были исследованы ПК спектры поглощения
исходных цеолитов и цеолитов после высокотемпературной адсорбции
хинолина.1 Близость спектров адсорбированного хинолина на декатиони-
рованном цеолите типа Y и окиси алюминия, обладающей сильными элек-
троноакцепторными центрами, свидетельствует о существовании анало-
гичных центров на декатионированном цеолите.
На основании полученных результатов можно считать, что активными
центрами декатионированных цеолитов являются атомы алюминия в трех-
координационном положении, у катионных форм активность, по-види-
мому, связана с неполностью компенсированными алюмокислородными
тетраэдрами.
ЛИТЕ РАТУ РА
1. U. J. Frilette, Р. В. Weisz, R. L. Golden, J. of Catalysis, 1962, 1,4, 3.
2. S. R a b о, P. P i c k e г t, D. N. S t a m i г e s, Actes 2 Congr. internal.
Catalyse, Paris, 1960; T. 2, Paris, 1961, 2055.
3. T. B. Reed, D. W. В г e c k, J. Am. Chem. Soc., 1956, 78, 5972.
4. С. 3. Рогинский, M. И. Яновский, Г. А. Газиев, ДАН СССР,
1961, 140, 5, 1125.
1 Работа по высокотемпературной адсорбции хинолина и и зучению спектров
поглощения хинолина находится в печати.
24*
373
5. D. В. В asset, Н. W. Н a b g о о d, J. Phys. Chem., 1960, 64, 6, 769.
6. Г. А. Г а з и e в, В. Ф. Ф и л и н о в с к и й, М. И. Я н овский, Кинетика
и катализ, 1963, 4, 5, 688.
7. G. М. S chwa b, R. Lieb, Z. f. Naturforschung, 1963, 18а, 164.
8. R. М. Ваггег, F. М. В u 11 i t u d e, J. W. Sutherland, Trans. Fa-
raday Soc., 1957, 1111.
9. P. E. E berly, J. Phys. Chem., 1962, 66, 812.
10. К. В. Топчиева, И. Ф. M о с к о в с к а я, ДАН СССР, 1958, 123, 5, 891.
КАТАЛИТИЧЕСКИЕ РЕАКЦИИ УГЛЕВОДОРОДОВ
И КИСЛОРОДСОДЕРЖАЩИХ СОЕДИНЕНИЙ НА ЦЕОЛИТНЫХ
КАТАЛИЗАТОРАХ СО СТРУКТУРОЙ Y
X. М. Миначев, В. И. Гаранин, Я. И. Исаков, Л. И. Пигузова,
А. С. Витухина
Синтетические цеолиты начинают находить все большее применение
как катализаторы органических и неорганических реакций. Круг этих
реакций расширяется. Несмотря на то что исследования в зтом направле-
ний начаты совсем недавно (1959—1960 гг.), уже опубликованы сообщения
об использовании молекулярных сит как катализаторов в крекинге [1—5],
для полимеризации олефинов [6], дегидратации спиртов [1, 3, 6, 7], изо-
меризации углеводородов [8, 9] и др. Имеются патентные указания на
возможность применения цеолитов в реакциях окисления [10], дегидри-
рования [11], синтеза метанола [12] и других.
На основании специфических свойств цеолитов можно ожидать, что это
новое направление их использования (в катализе) приведет к результатам,
имеющим большое научное и практическое значение. Кроме того, иссле-
дования в этом направлении помогут выработать рекомендации по по-
давлению так называемой паразитной каталитической активности цео-
литов, проявляющейся при осушке и очистке мономеров (полимеризация,
изомеризация и др.).
В опубликованных работах исследовались каталитические свойства
различных катионных форм синтетических цеолитов типа А и X, и лишь
в работах Рабо с сотрудниками [8, 9] использовались катализаторы на
основе цеолитов типа Y, заряженных благородными металлами.
В настоящем сообщении приводятся результаты исследований катали-
заторов на основе отечественных цеолитов типа Y в реакциях изомери-
зации н-пентана, н-гексана [13], н-гептана, циклогексана, гидрирования
и гидроизомеризации бензола [14], дегидратации спиртов и алкилирова-
ния бензола олефинами.
Экспериментальная часть
В работе использовались цеолиты типа Y в различных катионных
формах, синтезированные во ВНИИНП Л. И. Пигузовой.
Катализаторы готовились введением Pd, Pt, Bh и Jr в соответствую-
щие цеолиты в количестве 0.5% (вес.), таблетировались и восстанавлива-
лись водородом при 380°. Для реакций алкилирования бензола олефинами
и дегидратации спиртов использовали обезвоженные чистые цеолиты.
В качестве исходных веществ использовались углеводороды и спирты
ч. д. а., за исключением этилового спирта, который применялся в виде
ректификата, и пропан-пропиленовая фракция с содержанием про-
374
пилена 75% (объемн.). Опыты по изомеризации н-алканов, циклогек-
сана и гидрированию и гидроизомеризации бензола проводили в проточ-
ной установке высокого давления, а алкилирование бензола и дегидрата-
цию спиртов — в кварцевом реакторе
при атмосферном давлении. Во всех
случаях в реактор загружалось 10 мл
катализатора. Анализ продуктов реак-
ций проводили хроматографически.
Изомеризация н-алканов и
циклогексана
Рис. 1. Зависимость глубины изомери-
зации н-гексана от концентрации пал-
ладия.
1 — 0.25% Pd/CaY; 2 — 0.5% Pd/CaY; 3 — 1%
Pd/CaY.
Изомеризацию н-гексана прово-
дили на платиновом, палладиевом,
родиевом и иридиевом цеолитных ка-
тализаторах на основе натриевой и
кальциевой форм цеолитов с различ-
ным отношением SiO2/Al2O3 (табл. 1).
Катализаторы на основе натриевой формы цеолитов в реакциях изомери-
зации оказались неактивными.
Таблица 1
Изомеризация н-гексана на Ме-цеолитных катализаторах
(р = 30 атм.; о.с. =1 час.-1; Н2/СвН14 = 3.2)
Катализатор SiO,/AI2O3 t, °C Состав катализата, моль. %
2,2-диме- тилбутан 3-метил- пентан 2.3-диметил- бутан -4-2-ме- тилпентан н-гексан
0.5% Pt/CaY 3.4 400 7.5 19.2 32.7 40.6
3.4 400 7.3 19.4 31.3 42.0
0.5% Pd/Cai . . . . < 4.5 350 11.0 22.9 36.4 29.7
Как видно из табл. 1, в которой приведены данные, полученные при
оптимальной температуре,’ платиновый и палладиевый катализаторы по
изомеризующим свойствам практически между собой не отличаются. При
400° на обоих катализаторах суммарный выход изомерных гексанов со-
ставляет около 60%. Увеличение отношения SiO2/Al2O3 в цеолите дозво-
ляет значительно повысить выходы изомерных гексанов (в том числе
2,2-диметилбутана) и снизить температуру реакции. В случае палладие-
вого катализатора на цеолите с отношением SiO2/Al2O3=4.5 удалось до-
стичь приближения к равновесию в отношении всех изомеров на 88%,
а в отношении неогексана — на 55 %. Родиевый и иридиевый катализа-
торы (в табл. 1 не показано) наряду с реакцией изомеризации в значи-
тельной степени проводят гидрокрекинг исходного углеводорода. Так,
на родиевом катализаторе при 390° С количество изомерных гексанов и
продуктов гидрокрекинга составляет 25.8 и 21.9% соответственно, а на
иридиевом при 310° — 6.3 и 26.2%.
На рис. 1 представлена зависимость глубины изомеризации н-гек-
сана от концентрации палладия, введенного в цеолит CaY с отношением
SiO2/Al2O3=3.4. Как видно, изменение концентрации металла от 0.25
до 1% (вес.), т. е. в 4 раза, в общем мало сказывается на активности ката-
лизатора.
Кроме изомеризации н-гексана, на цеолите структуры Y с отношением
SiO2/Al2O3=3.4, содержащем палладий, исследована также изомеризация
н-пентана и н-гептана. Экспериментальные данные приведены в табл. 2.
375
Таблица 2
Изомеризация w-пентана и и-гептана
(р = 30 атм.; о. с. = 1 час.-1; Н2/СН=3.2)
Исходный углеводород °C Состав продукта, вес. %
изомеры продукты крекинга
н-Пентан { 400 420 49.5 52.8 2.5 3.7
/(-Гептан 360 47.2 10.3
Из приведенных данных следует, что и в этих реакциях катализатор
проявляет высокую активность и селективность. Так, в изомеризации
н-пентана приближение к равновесию составляет выше 80%.
Изомеризация циклогексана была исследована впервые. Полученные
данные представлены в табл. 3.
Таблица 3
Изомеризация циклогексана на Ме-цеолитных катализаторах
. (р = 30 атм.; о. с. = 1 час.-1, Н2/С6Н12 = 3.2)
Катализатор SiO2/Al,O3 I, °C Состав катализата, моль. %
метилцик- лопентан цикло- гексан
0.5% Pt/CaY .... 3.4 370 47.0 51.2
3.4 370 43.0 57.0
05% Pd/CaY 4.5 4.5 300 320 45.7 51.5 54.3 48.5
4.5 330 57.0 43.0
Примечания. Для 0.5% Pt/CaY содержание бензола в катализате 1.8%. При 1=330° для
0.5% Pd/CaY продукты крекинга составляют 7.9%.
Данные табл. 3 показывают, что платиновый и палладиевый катали-
заторы по изомеризующей активности отличаются незначительно. Повы-
шение отношения SiO2/Al2O8 в цеолите приводит, как и в случае н-гексана,
к значительному возрастанию активности катализатора и снижению темпе-
ратуры, при которой достигается максимальный выход метилциклопен-
тана. На иридиевом катализаторе (в табл. 3 не указано) циклогексан уже
при 330° крекируется на 52 %, а выход метилциклопентана составляет
всего 4%.
Гидрирование и гидроизомеризация бензола
В табл. 4 представлены результаты, полученные при гидрировании
бензола.
Приведенные данные показывают, что по гидрирующей активности
исследованные катализаторы располагаются в ряд: Rh > Jr > Pd.
Как и следовало ожидать, увеличение отношения SiO2/Al2O3 в цеолите
не сказывается на активности катализаторов в гидрировании. Так же мало
влияет и природа катиона в цеолите.
На указанных катализаторах по мере роста температуры, кроме гид-
рирования бензола в циклогексан, начинает протекать гидроизомериза-
376
Таблица 4
Гидрирование бензола
(р = 30 атм.; о. с. =0.5 час.-1; Н2/СвНв = 5)
Катализатор SiO2/Al2Oa t, °C Состав катализата, вес. %
цикло- гексан бензол
0.5% Rh/CaY . . . 3.4 200 97.8 2.2
0.5%Ir/CaY 3.4 200 91.0 9.0
3.4 200 82.5 17.5
0.5% Pd/CaY • • | 4.5 200 80.2 19.8
4.5 200 87.6 12.4
ция, причем, как видно из табл. 5, удельный вес реакции зависит от отно-
шения SiO2/Al2O3 в цеолите.
Наиболее активным в гидроизомеризации является палладиевый ка-
тализатор, на котором при 320° выход метилциклопентана достигает
Таблица 5
Гидроизомеризация бензола на палладий-цеолитном катализаторе
(р = 30 атм.; о.с. =0.5 час.-1; Н2/С6Нв = 5)
Катализатор S1O2/AI2Os t, °C Состав катализата, вес %
метилцикло- пентан циклогексан бензол
3.4 320 20.1 63.5 16.4
0.5% Pd/CaY .... | 3.4 370 51.8 33.3 14.9
4.5 320 67.3 28.4 4.3
Примечание. При S1O2/AI2Os = 4.5 и
= 320° С продукты крекинга составляют 5.7°/0.
67.3%. Увеличение отношения SiO2/Al2O3 в палладий-цеолитном катали-
заторе с 3.4 до 4.5 почти в 3.5 раза увеличивает глубину гидроизомериза-
ции бензола. Иридиевый катализатор оказался гидрокрекирующим; так,
при 320° выход продуктов гидрокрекинга составил 60%. В тех же усло-
виях на родиевом катализаторе глубина гидрокрекинга достигала
16.9%. Доля реакции гидроизомеризации на этих катализаторах при 320°
составила 5.8 и 12% соответственно.
Дегидратация спиртов
Опыты проводили на цеолите с отношением SiO2/Al2O3=3.4, получен-
ные результаты представлены в табл. 6.
Как видно из табл. 6, при 250° все нормальные первичные спирты почти
нацело дегидратируются до соответствующих олефинов. Более реак-
ционноспособный изопропиловый спирт селективно дегидратируется в про-
пилен при еще более низкой температуре (230°). Гладко и селективно про-
текает дегидратация циклогексанола. Уже при 150° этот спирт на 68%
превращается в циклогексен, а при 170° дегидратация проходит полно-
стью и катализат состоит из чистого циклогексена.
Провести селективную дегидратацию н- и изобутилового спиртов не
удалось, так как в условиях опытов имела место интенсивная полимери-
зация соответствующих олефинов. Так, в случае изобутилового спирта
377
Таблица 6
Дегидратация спиртов на цеолите CaY
(ск. подачи = 3.87 г-м/л-час.)
Спирт t, °C Продукт реакции Степень превращения
Этиловый 250 Этилен. —100
и-Пропиловый f 250 Пропилен. 90
1 275 » 100
Изопропиловый 230 » 100
и-Бутиловый 250 Бутилен. 100
Изобутиловый .... 250 Изобутилен. 100
150 Циклогексан. 68
Циклогексиловый | 160 » 95—98
170 » 100
при 250° в виде газа выделялось только 65 % (от теоретического) изобути-
лена, остальное его количество полимеризовалось.
Следует отметить, что в аналогичных условиях на промышленном
алюмосиликатном катализаторе (Уфимский) полная дегидратация эти-
лового спирта в этилен наблюдалась лишь при 300°, т. е. при температуре
на 50° выше, чем на цеолите.
Алкилирование бензола олефинами
Данные по использованию цеолитов в качестве катализаторов алкили-
рования бензола в научной литературе отсутствуют. Имеется лишь один
патент [15], в котором сообщается об алкилировании ароматических угле-
водородов олефинами на цеолите структуры X с отношением SiO2/Al2O3=
=2.5 и размером пор — 13 А. При алкилировании бензола пропиленом на
цеолите (форма не указана) при 205° С, отношении бензол/пропилен=
=1.5/1 и атмосферном давлении получен алкилат с выходом
~ 9.0 объемн. %, который на 83 объемн. % состоял из изопропилбен-
зола, остальные 17 объемн. % составляли полиалкилбензолы.
Мы проводили алкилирование бензола олефинами (этиленом, пропи-
леном) в паровой фазе, при атмосферном давлении на цеолитах с различ-
ным отношением SiO2/Al2O3 и размером пор 10 А в различных катионных
формах.
При определенных условиях были получены изопропилбензол и этил-
бензол с выходами соответственно 83 и 45% от теоретического за один
проход. Данные по алкилированию бензола пропан-пропиленовой фрак-
цией приведены в табл. 7.
Из табл. 7 видно, что, как и в реакциях изомеризации углеводородов,
натриевая форма цеолитов неактивна. При замене Na+ на NH4+ и после-
дующем декатионировании активность цеолитов резко возрастает. Вы-
сокой активностью обладают также двухвалентные катионные формы.
Для кальциевой формы оптимальная температура находится в области
250-300°.
При увеличении объемной скорости по бензолу с 0.3 час. 1 до 1.2 час.-1
выход изопропилбензола постепенно падает. При изменении молярного
отношения бензол : олефин с 2 : 1 до 6 : 1 выход алкилбензолов от тео-
ретического растет, при этом образуется главным образом моноалкилбен-
зол. С увеличением отношения SiO2/Al2O3 в цеолите активность его
растет.
378
Таблица 7
Алкилирование бензола пропан-пропиленовой фракцией на цеолитах
Катализатор t, °C o. c. no C6He Мольное отноше- ние СвН«/пропан- пропилен Выход изопро- пиленбензола в % от теорети- ческого
CaY-I NaY-II NaY-П «D» CaY-II «D» ( CaY-II | 300 300 300 250 300 300 300 300 0.3 0.3 0.3 0.3 0.3 0.6 0.6 0.6 2:1 2:1 2:1 2:1 2:1 2:1 4:1 6:1 64 0 50 66 70 55 75 83
Следует отметить, что в продуктах реакции наряду с непрореагиро-
вавшим бензолом и соответствующим алкилбензолом были обнаружены
также углеводороды С6 (до 3 вес. %) и полиалкилбензолы. Однако при
соответствующих условиях удалось достичь селективности в отношении
образования моноалкилбензола, равной 95—100%.
Изучение активности декатионированных цеолитов, полученных из
натриевой и кальциевой форм, показало, что катализатор, полученный из
кальциевой формы, является более активным. Этот факт можно объяснить
тем, что в результате операции NaY -> CaY -> DY удаляется больше
ионов Na+, чем в случае NaY -> DY; кроме того, в результате неполного
обмена Са++ на NH4+ имеет место как бы совместное действие декатиони-
рованной и кальциевой форм, за счет чего повышается активность катали-
затора.
Полученные экспериментальные данные показывают, что исследован-
ные цеолитные катализаторы обладают рядом серьезных преимуществ перед
известными катализаторами типа Ме/А12О3 • SiO2, А12О3 • SiO2, Ме/А12О3,
Me/SiO2, Н3РО4/кизельгур и др.
Определяющую роль в активности цеолитных катализаторов играет
природа катиона. Так, натриевые формы цеолитов в условиях, оптималь-
ных для кальциевых форм, в реакциях изомеризации и алкилирования уг-
леводородов оказались неактивными (вне зависимости от состава цеолита).
Увеличение отношения SiO2/Al2O3 в цеолитах структуры Y с од-
ним и тем же катионом приводит к росту активности катализаторов и
позволяет снижать температуру реакций (приблизительно на 40—50°
при увеличении отношения с 3.4 до 4.5).
Важными являются и вопрос о влиянии степени катионного обмена,
специально не изученный в этой работе, а также селективность в отноше-
нии размеров молекул и возможность протекания реакций при определен-
ных условиях и на внешней поверхности цеолитов, когда размеры их
каналов меньше размеров реагентов.
ЛИТЕРАТУРА
1.
2.
3.
4.
Н. К е о g h, L. В. Sand, J. Am. Chem. Soc., 1961, 83, 3536.
V. J. F r i 1 e t t e, P. B. Wei s.z, R. L. Golden, J. Catalysis, 1962,
1, 301.
P. B. Weis z, V. J. F r i 1 e 11 e, R. W. M a a t m a n, E. В. M о wer,
J. Catalysis, 1962, 1, 307.
К. В. Топчиева, В. В. Романовский, X о - Ш и Тхоанг,
ДАН СССР, 1963, 149, 5, 644.
379
5. .П. И. Г1 и г у з о в а, Л. С. В и т у х и н а, Химия и технология топлив п
масел, 1963, 6, 17.
6. С. J. Norton, Chem. and Ind., 1962, 6, 268.
7. Г. В. Цицишвили, Ш. И. Сидамбнидзе, Ш. А. Зедгеяидзе,
ДАН СССР, 1963, 156, 6, 1395.
8. J. A. R аЬо, Р. Е. Р i с к е г t, R. L. М ays, Ind. Engng. Chem., 1961, 53,
9, 733.
9. J. A. R abo, P. E. P i с к e r t, D. S t a m i r e s, J. В о у 1 e, Congr. in-
ternal. Catalyse, Paris, 1960, 2, 104.
10. D. W. В reck, R.M. Milton, US Pat., 3013985, 1961.
11. S. W. В u к a t a, C. R. Castor, R. M. Milton, US Pat., 3013988, 1961.
12. D. W. В r e c k, US Pat., 3013984, 1961.
13. X. M. Миначев, В. И. Г аранин, JI. И. П и г у з о в а, А. С. В и т у-
хина, Изв. АН СССР, ОХН, 1965, 7, 1169.
14. X. М. М и я а ч е в, В. И. Г а р а я и я, Л. И. П и г у з о в а, А. С. В и т у-
хина, Изв. АН СССР, ОХН, 1965, 6, 999.
15. Wm. J. Mattox, Wm. F. A г e у. US Pat., 2904607, 1959.
ИССЛЕДОВАНИЕ СВОЙСТВ НИКЕЛЬЦЕОЛИТНЫХ КАТАЛИЗАТОРОВ
Н. В. Боруноёа, Л. X. Фрейдлин, И. Е. Неймарк,
В. Г. Ильин, Л. И. Гвинтер
Цеолиты, обладающие широкоразвитой однороднопористой структу-
рой (размер пор 4—5 или 10—13 А), представляют безусловный интерес
как катализаторы. Можно ожидать, что такие катализаторы будут про-
являть высокую селективность действия в реакциях гидрирования и де-
гидрирования углеводородов, молекулы которых обладают различными
размерами.
В работе [1] установлено, что на Pt/CaA-цеолите нормальные углево-
дороды селективно гидрируются в присутствии изоолефинов: степень
насыщения бутилена достигала 70%, а изобутилена — только 2%.
Галичем, Гутыря и одним из авторов [2] показано, что на СаА-цеолите
в отличие от алюмосиликатных и алюмохромовых катализаторов н-ал-
каны дегидрируются в линейные олефины без циклизации и изомеризации
скелета. Этот-факт был объяснен тем, что образующиеся при дегидроцикли-
зации молекулы не имеют выхода из внутренних полостей через проход-
ные каналы сечением 4—5 А [3]. Аналогичные результаты получены на
СаА-цеолите с добавками Pt и Ni.
Согласно японскому патенту [4], на Ni-цеолите, полученном осажде-
нием, проводилось гидрирование масел при 180° С и повышенном давле-
нии. В нескольких патентах описываются методы приготовления Ni-цео-
литных катализаторов без указания их применения.
Настоящее исследование посвящено изучению Ni-цеолитных катали-
заторов, полученных катионным обменом или осаждением, в реакциях
гидрирования углеводородов.
Катализаторы 1 и 2 были приготовлены методом ионного обмена.
Катализаторы 3 и 4 были получены путем смешения водной суспензии
цеолита NaA с нейтральным карбонатом никеля.
Ниже представлены данные о химическом составе использованных
катализаторов.
№ катали-
затора
1
2
3
4
5
Химический состав
0.22NiO • 0.495Na2O • А12О3 • 1.89SiO2 • 4.1Н2О
0.53NiO • 0.405Na20 • А12О3 • 2.14SiO2 • 5.6Н2О
0.218NiO • 0.810Na20 • A12O3 • 1.82SiO2 • 4.1H2O
0.355NiO • 0.810Na20 • A12O3 • 1.82SiOa • 4.1H2O
— 0.810Na20 • A12O3 - 1.82SiO2 • 4.1H9O
Содержание
никеля, вес.%
3.84
7.87
3.65
5.75
Примечания. Коэффициенты показывают, сколько молей N1O, Na2O, S1O2 и Н2О прихо-
дится на 1 моль А12О3. Катализатор 5 — молекулярное сито NaA.
380
На рис. 1 и 2 приведены изотермы адсорбции паров воды молекуляр-
ными ситами NaA и NaX, а также полученными на их основе Ni-цеолит-
ными катализаторами до и после их восстановления водородом. Гидри-
Рис. 1. Изотермы адсорбции
паров воды при 20° С.
1 — молекулярное сито типа NaA;
2 — никельцеолитный катализатор
Ка 1 до восстановления; з — ни-
кельцеолитный катализатор Ка 1
после восстановления.
Рис. 2. Изотермы адсорб-
ции паров воды при 20° С.
т — молекулярное сито типа
NaX; 2 — никельцеолитный ка-
тализатор № 2 до восстановле-
ния; 3 — никельцеолитный ка-
тализатор № 2 после восста-
новления.
рование проводилось в проточной системе при обычном давлении. Условия
опытов и полученные результаты представлены в таблице и на рисунках.
Катализаты анализировали рефрактометрически [5], бромировани-
ем [6] и методом газо-жидкостной хроматографии.
Результаты и обсуждение
Из таблицы следует, что Ni-цеолит с размером входного канала 4 А
(катализатор 1) проявляет высокую избирательность: степень гидриро-
вания н-октена достигла 81.5%, тогда как изоолефины и бензол, которые
не могут проникнуть во внутреннюю полость к никелю, практически не
гидрируются.
На Ni-цеолите NaX с размером входного канала 10 А (катализатор 2)
октен гидрировался почти нацело, а бензол — лишь на 5%.
Известно, что на Ni-катализаторах на носителях, содержащих 4%
и более Ni, степень гидрирования бензола достигает 100% [7]. Низкую
активность катализатора 2 нельзя объяснить также малым размером
«окна» цеолита, размеры которого 10 А доступны для бензола.
Из сравнения изотерм адсорбции паров воды на катализаторах 1 и 2
до и после их обработки водородом видно, что в процессе восстановления
нарушается их макроструктура, особенно заметно у катализатора 2.
Возможно, что этим объясняется низкая активность последнего в реак-
ции гидрирования бензола.
Интересно отметить большую роль активации катализаторов прокали-
ванием в токе сухого воздуха при 350° в течение 6 часов, с последующим
восстановлением при этой же температуре. Из сравнения опытов 5 и 7
25 Цеолиты
381
следует, что после такой обработки активность катализатора в реакции
гидрирования н-октена возросла почти в 5 раз.
Таким образом, на Ni-цеолитных катализаторах, полученных методом
катионного обмена и отличающихся размером входных каналов, можно
осуществить селективное гидрирование углеводородов, отличающихся
размерами и строением молекул (н-олефины и изоолефины; «-олефины и
бензол, и т. д.).
Исследование поведения катализаторов, приготовленных методом на-
несения, показало, что на контакте 3, с меньшим количеством никеля
(3.5%), степень гидрирования бензола низка — 5%, тогда как на катали-
заторе 4 с 5.8% Ni она достигла 66.6% (опыты 9 и 13). На обоих катали-
заторах октен гидрировался нацело.
Циклогексен на катализаторе 3 при 180° С не гидрировался, но пре-
терпевал необратимый катализ (87%). На одном цеолите NaA (катализа-
тор 5) циклогексен при этой температуре в токе водорода не претерпевал
изменений.
На катализаторе 4 степень гидрирования алкилбепзолов (30%) была
значительно ниже степени гидрирования незамещенного бензола (66%).
Выводы
.1. Изучались свойства Ni-цеолитных катализаторов.
2. Установлено, что Ni/NaA-цеолитный катализатор, полученный ме-
тодом катионного обмена, проявляет высокую избирательность: степень
гидрирования н-октана — 81.5%, а изоолефины практически не гидри-
руются.
3. Ni/NaX-цеолит (методом катионного обмена), содержащий 7.8%
Ni, также проявляет селективность: катализирует полное гидрирование
н-октена, а бензола — только на 5%.
4. На катализаторе, полученном нанесением 5.8% Ni на NaA, степень
гидрирования бензола — 66.6%, а алкилбензолов значительно ниже
Степень гидрирования углеводородов различными цеолитными
катализаторами
№ опыта Метод приготовле- ния катали- затора Размеры входного канала цеолита, А Гидрируемое соединение Температура гидрирования, °C Степень гидрирования, вес. %
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 Катион- ный обмен. Нанесе- ние. 4 10 { 4 Октен. Ивоамилен. Диизоамилен. Бензол. Октен. Бензол. Октен. Октен. Бензол. Циклогексан. Циклогексен. Октен. Бензол. Бутилбензол. Вторичный бутил- бензол. 130 50 160 180 130 180 130 130 180 300 180 130 180 180 180 81.5 0.0 0.0 0.0 90.1 5.0 20.0 100.0 5.0 15.0 0.0(87) 100.0 66.6 38.6 30.0
Примечания. В опыте № 7 гидрирование проводилось на неактивированном катализаторе.
В опыте Me 10 дегидрирование циклогексана при 300°. Для циклогексена в скобках приведена сте-
пень превращения его в реакции диспропорционирования.
382
(30—36%). На аналогичным образом приготовленном катализаторе с 3.65%
Ni циклогексен не гидрировался, но претерпевал необратимый катализ
(87%).
ЛИТЕРАТУРА
1. Р. В. W е i s г, V. G. F г i 1 е 11 е, R. W. Maut man, Е. В. М over, J.of
Catalysis, 1962, 1, 307.
2. Н. П. Г а л и ч, А. А. Гутыря, В. С. Г у т ы р я, И. Е. Неймарк,
ДАН СССР, 1962, 144, 1, 147.
3. В. Я. Н и к о л и н а, И. Е. Неймарк, М. А. Пионтковская, Усп.
химии, 1960, 29, 1088.
4. Японский патент, 336, 1955.
5. Н. Д. 3 е л и н с к и й, Г. С. П а в л о в, ЖРХО, 1926, 58, 1309.
6. Т. Д. Гальперин, Тр. Инет, нефти АН СССР, Изд. АН СССР, 4, 116, 140.
7. С. С. Н о в и к о в, X. М. Миначев, И. Д. Р о ж д е с т в е н с к а я,
ДАН СССР, 1950, 72, 911.
ПРИМЕНЕНИЕ МОЛЕКУЛЯРНЫХ СИТ В КАТАЛИЗЕ
Л. И. Пигу зова
За последние три года за рубежом и в Советском Союзе проводятся
широкие исследования в области применения синтетических цеолитов
в самых различных каталитических реакциях. Благодаря своим струк-
турным особенностям, высокой термической и химической стабильности
цеолиты непосредственно или в качестве носителей для катализаторов на-
чали проникать в различные области катализа, как-то: крекинг, изомери-
зация, полимеризация, дегидратация спиртов, синтез спиртов и эфиров
и др. Рассмотрение данных как литературных, так и патентных позволило
разделить цеолитные катализаторы на несколько групп, положив в основу
такого деления характер их каталитических свойств.
Работы по выявлению крекирующих свойств синтетических цеолитов
начали публиковаться с 1960 г. [1]. Наиболее широкие исследования от-
носятся к использованию цеолитов в реакции крекинга [1 —10, 13—21,
32—33], часть из них проведена советскими исследователями [15—21,
28, 35, 36, 41]. В 1961—1962 гг. появились патенты на крекинг легкого
и тяжелого газойля [4] нефтяных углеводородов, содержащих азотистые
соединения [13], опубликованы результаты исследования крекинга н-де-
кана и керосино-газойлевой фракции на цеолитах X [15], превраще-
ния додекана на цеолите СаА [17], кумола и псевдокумола на цеолите
X [8, 16, 20]. В [1, 9] отмечается, что при 470° более высокая актив-
ность цеолита X в Na- и Са-формах при крекинге н-декана по сравнению
со стандартным алюмосиликатным катализатором. Замещение в цеолите X
катиона Na на Са приводит к повышению активности.
Продукты, получающиеся на цеолите СаХ, сходны с продуктами,
образующимися в присутствии алюмосиликатного катализатора. В отли-
чие от них продукты, образующиеся в присутствии NaX, не содержат
изосоединений и характеризуются в два раза большим образованием не-
предельных соединений.
В 1962—1963 гг. Л. И. Пигузовой и А. С. Витухиной проводились ис-
следования по крекингу н-декана и керосино-газойлевой фракции на цео-
литах типа X и Y [14, 15, 19].
Наши исследования крекинга керосино-газойлевой фракции показали,
что с повышением степени обмена катиона Na на Са (до 100%) в цеолите X
индекс активности повышался с 33.8 до 43 вес. % при объемной ско-
25*
383.
рости подачи сырья, равной 1 час."1. Н- и Са-формы цеолита Y при рав-
ных выходах показали производительность в два раза выше, чем у цео-
лита СаХ. В равных условиях подобной производительности не давал
ни один из ранее известных Al-Si-катализаторов.
Смешанные катализаторы, полученные наполнением гидроокиси крем-
ния или шарикового алюмокремнегеля кристаллическими цеолитами
типа СаХ или CaY, взятыми в количестве не менее 25%, имели понижен-
ный индекс активности (31—38 вес. %). Однако после обработки таких
катализаторов водяным паром (100%, 650°, 20 часов) активность их вновь
можно довести уже до очень высо-
Изменение энергии активации реакции
крекинга кумола на цеолите типа X в
зависимости от природы замещающего
катиона и степени замещения.
кого уровня.
Полученный крекинг-бензин со-
стоял из ароматических и парафино-
нафтеновых соединений при малом
содержании непредельных углеводо-
родов (йодное число 7—9). Крекинг-
газ был богат изосоединениями
(S4—С4 и i—С6), выход которых до-
ходил до 48—55 вес. %.
Активность цеолитов X и Y в
Са-форме (близкой к 100 %-й степени
обмена) и цеолита Y в глубоко дека-
тионированной Н-форме не снижа-
лась как при проведении 4—5-цик-
ловых, так и 12-цикловых опытов.
Коксовые отложенияна катализаторе,
при содержании 4% и выше, выжи-
гались без затруднения в течение
1.5—2.5 часов.
Ряд новых промышленных катали-
заторов повышенной активности типа
«Дюрабед 5», «XZ-15», выпускаемых
фирмами США, основан на применении
кристаллических алюмосиликатов типа молекулярных сит [10—12, 38].
Фрилет с соавторами [7] нашел, что при крекинге кумола активность
у цеолита СаХ в два раза больше, чем у цеолита NaX. Эта активность
характеризовалась выходом бензола, равным 49.1%, и пропилена —
38.2%.
В работе [8] изучалось изменение энергии активации реакции крекинга
кумола и р-кумола (псевдокумола) при 500° на цеолите типа X в зависи-
мости от природы замещающего катиона и степени замещения им иона Na.
Из рисунка видно, что замещение в цеолите NaX катиона Na на двухва-
лентные катионы (Mg и Са) почти вдвое снижает энергию активации реак-
ции, замещение иона Na на Zn, Мп снижает энергию активации при тех же
степенях замещения заметно меньше, введение Cd, наоборот, увеличивает
энергию активации. Значения энергии активации проходят через минимум.
Авторы объясняют это тем, что при больших степенях обмена замещение
происходит в более глубоких слоях твердой фазы, недоступных для ку-
мола (см. рисунок).
К. В. Топчиева с соавторами [16] также изучала модельную реакцию
крекинга кумола, проводя параллельные опыты на молекулярном сите
10Х США (партия 106) и рентгеноаморфном Al-Si-катализаторе. В ре-
зультате исследования показано близкое направление процесса крекинга
на двух катализаторах. Однако обнаружено, что цеолит 10Х фирмы Линде
обладает значительно меньшей активностью, чем алюмосиликатный ка-
тализатор типа Гудри.
384
Исследование кинетики реакции разложения кумола на цеолите диф-
ференциальным методом позволило обнаружить большую величину
энергии активациц и протекания на нем (до 528° С) реакции крекинга
кумола в кинетической области. Для реакции крекинга кумола на цео-
лите энергия активации равна 35 ккал./моль. В том же направлении изу-
чалась реакция крекинга кумола на отечественных цеолитах ВНИИНП
структуры X и Y [20].
Г. В. Цицишвили с соавторами исследовал ту же реакцию крекинга
кумола на цеолитах NaX, СаА и НХ [21]. Показано, что значительная
активность в крекинге достигалась при температуре выше 400°.
В работе Вольфа [3] изучалась связь между коэффициентом распреде-
ления газ—жидкость и крекирующей активностью цеолитов. Для изуче-
ния влияния структуры полярных носителей на процесс распределения
при газо-жидкостной хроматографии были использованы наиболее харак-
терные Н-цеолиты с различным содержанием окиси алюминия (от 20 до
80%). Наблюдалась симбатность между величинами кислотности и актив-
ности цеолитов.
Исследования Галич, Гутыря и др. [18] природы углистых отложений,
образующихся на молекулярном сите СаА при крекинге н-алканов, по-
казали, что в полостях этого цеолита все же протекают реакции дегидро-
циклизации и изомеризации, т. е. реакции, типичные для А1—Si-катали-
заторов, но ввиду ограниченности размеров «окон» у 5А образовавшиеся
углеводороды, не имея выхода, подвергаются уплотнению с образованием
кокса (8—10%), который удалось извлечь только путем разрушения
структуры цеолита.
На необычайно высокую каталитическую активность синтетического
морденита в Na- и Н-формах при относительно низких температурах
(350° и ниже), даже в условиях дезактивации коксом, впервые указали
американские исследователи [2]. Кислая форма морденита, стабильная
до 800°, характеризуется адсорбционными свойствами, промежуточными
между цеолитами типа А и фожазитом.
Расщепляющие свойства кислого морденита в крекинге н-декана
подобны свойствам цеолита типа 10Х [1] и отличаются по характеру
действия от А1—Si-катализатора большей гидрирующей активностью, вы-
ражающейся более высокой величиной отношения парафин/олефин в про-
дуктах крекинга (соответственно 4.6 против 3.3). Расщепляющую способ-
ность проявлял и Mg-морденит. Близкие данные получены нами на син-
тетическом Н-мордените.
Таким образом, многие разрозненные исследования достаточно четко
выявили интересные возможности синтетических цеолитов для использо-
вания их в качестве катализаторов крекинга.
Об изомеризации н-бутана в i-бутан на катализаторе А1С13 с цеоли-
тами в качестве носителя имеется первое указание в работе Б. Н. Дол-
гова [28].
Первые из полученных нами декатионированных образцов цеолитов
структуры X и Y, имевшие соотношения SiO2/Al2O3 около 2.5—3.5, ис-
пытывались в начале 1962 г. во ВНИИ Нефтохима в качестве основы пла-
тинированного катализатора применительно к процессу изомеризации
н-пентана. Более детальные исследования нами были продолжены совме-
стно с Институтом органической химии АН СССР.
Об изомеризующих свойствах катализаторов на основе цеолита типа Y
впервые стало известно в 1961 г. [29, 30]. В этих работах исследовались
различные молекулярные сита, использованные в качестве носителя Pt-
катализатора (0.5% Pt), активность их определялась по изомеризации
нормальных парафинов С6—Св.
385
Как и следовало ожидать, все исследованные цеолитные Pt-катализа-
торы в Са-форме были активными в отличие от неактивной Na-формы.
Газообразные продукты реакции состояли преимущественно из метана,
в них отсутствовал пропан, что свидетельствует, по мнению авторов,
о протекании реакции через образование радикалов, а не через образова-
ние ионов карбонил. Относительно длительное время контактирования
продуктов реакции, удерживаемых в порах, с поверхностью катализа-
тора обусловливает на цеолитах высокое газообразование. Декатиони-
рованный аморфный алюмосиликат по своей активности как носитель
занимает промежуточное положение между декатионированными цеоли-
тами типа X и Y. В работах указывается, что молекулярные сита типа Y
после обмена в них Na на Са, Мп или Се являются прекрасной основой
катализаторов изомеризации. Однако для катализаторов, содержащих
трех- и многовалентные катионы, отмечается меньшая селективность дей-
ствия при малой стабильности активности.
В патенте [31] указано на возможность проведения реакции изомери-
зации н-олефинов С4—С6 или СБ—С6 с разветвленной цепью в паровой
фазе при температуре 80—260° (с образованием в основном цис- и транс-
изомеров) в присутствии цеолита 5А с примесью: Fe, Mg, РЬ и В. Допу-
скаются только следы Na или К.
В работе [41] по разделению изопентан-пентановых и изобутен-буте-
новцх смесей на цеолите СаА показано его паразитное, в данном случае
изомеризующее действие (при температуре 350—480°) по отношению
к алкенам С4, выражающееся в перемещении двойной связи и простран-
ственной изомерии с образованием смеси 2-бутена, содержащей цис-
изомер (25%) и транс-изомер (54%).
В 1962 г. появились сообщения фирмы Нортона о возможности про-
ведения изомеризации на новом высококремнистом цеолите зеолоне [6,
22, 25, 27]. Кроме того, имеются указания на реакции изомеризации,
риформинга и др., протекающие на цеолитах, в работе [34].
Процесс гидрокрекинга высокоароматизированного сырья проводился
на катализаторе типа цеолитов 13Х и 10Х с добавлением гидрирующих
компонентов [37], в качестве которых использовались добавки МоО3
(9 вес. %) и СоО (3 вес. %) или их сульфиды. Сырьем служила газой-
левая фракция, выкипающая в интервале 204—427° С. В случае сырья,
состоящего из парафиновых углеводородов, более благоприятным для
проведения процесса является интервал температур 399—510°. По мне-
нию авторов, эти катализаторы целесообразно применять и в процессах
типа изокрекинга и изомакса.
В патенте [39] сообщается о способе приготовления катализаторов,
содержащих металлы или окислы Сг, Мо и W во внутренней адсорбцион-
ной полости природного цеолита-фожазита, синтетических молекулярных
сит типа X, Y и L. Для этого использовался комплексный, растворимый
в воде металл-аминный катион (органический и неорганический), который
вводился в адсорбционную полость цеолита путем замены им цеолитпосвя-
занного натрия и затем в струе инертного газа при температуре около
500° восстанавливался до элементарного металла.
Подобными способами заряжались металлами и нецеолитные носители:
А12О3, SiO2 и А12О3—SiO2, однако паилучшими катализаторами оказались
молекулярные сита, так как осажденные на них металлы или их окислы
принимали более активную форму. Катализаторы рекомендуются для ис-
пользования в реакциях дегидрогенизации пропана, полимеризации эти-
лена, гидросульфирования, риформинга, гидрирования олефинов.
В патенте [40] показана, в отличие от обычного гидрообессеривающего
А1—Со—Мо-катализатора, возможность приготовления Со—Мо-катали-
затора гидроочистки на основе синтетического молекулярного сита с боль-
386
шим диаметром пор — 13А. С этой целью Na-форма цеолита 13А прев-
ращалась в Со-форму, которая затем смешивалась с соответствующим
количеством окисла молибдена. Авторы указывают и способ приготовле-
ния сита 13А. Приготовленные таким образом катализаторы, содержащие
примерно 3% СоО и 10% МоО3, при переработке западно-техасского тяже-
лого лигроинового сырья (0.3% серы) в стандартных условиях процесса
гидроочистки оказались высокоактивными. При объемной скорости по-
дачи сырья 1.0 и 1.8 час.-1 в жидких продуктах содержание серы снижа-
лось с 0.3% соответственно до 0.0015—0.034%.
Сообщения о полимеризации на дегидратированных цеолитах типа А
и X были опубликованы в рекламных материалах фирмы Нортона в 1961—
1962 гг. [25]. Указывалось, что ответственными за ведение реакции поли-
меризации являются в цеолите участки, состоящие из «твердой кислоты»
[22, 24].
Кастер в одном из своих патентов [42] предложил принципиально
новый способ приготовления цеолитных катализаторов, заключающийся
в нанесении на цеолит щелочных металлов 1-й группы периодической си-
стемы. Для этой цели автор применяет обработку цеолита парами щелоч-
ного металла в инертной атмосфере. Заряженный цеолит используется
в качестве катализатора для селективной полимеризации смеси олефинов.
В работах [6, 23, 26] имеются указания о возможности проведения на цео-
лите типа зеолона реакции алкилирования олефинами, в частности то-
луола и кумола.
В более раннем патенте [43] в качестве катализатора для алкилирова-
ния бензола пропиленом было предложено молекулярное сито структуры X
с размером пор 13А. Алкилирование проводилось при мольном отношении
углеводородов 1.5 : 1 при температуре 204° С и атмосферном давлении.
Получаемые при этом с выходом 9.0 объемн. % продукты алкилирования
содержали 83 объемн. % мопоизопропилбензола, остальные 17 объемн. %
составляли полиалкилбензолы. На молекулярном сите 4А такой продукт
получен не был. Из различных катионообменных форм цеолита X наиболее
подходящей оказалась магниевая форма. Чтобы получить такой же вы-
ход алкилбензола с катализатором, представляющим собой фосфорную
кислоту, нанесенную на кизельгур при давлении 28 атм. и 268° С, должно
было быть примерно более высокое мольное отношение С6Н6/С3Нв, равное
5—10 : 1, и соответственно высокий рисайкл. В работе [44] также ука-
зывается на полимеризующие и алкилирующие свойства цеолитов.
В работе [9] сообщается, что на Pt-катализаторе на основе цеолита
типа А можно осуществить процесс гидрирования смеси 1-бутена и 2-ме-
тилпропена, взятых в равных объемах при температуре 25°, времени
контакта т=0.1 сек. При этом только 1-бутен превращается на 50%
в бутан. В то же время Pt-катализатор на основе А12О3 способствует
превращению почти равных количеств обоих олефинов.
В патентной литературе в 1961 г. появились указания на применение
многофункциональных катализаторов на основе молекулярных сит типа
5А и 13Х [45]. Они отличаются тем, что обладают одновременно как сорб-
ционными, так и каталитическими свойствами. Метод приготовления та-
ких катализаторов состоит в обработке предварительно прокаленного
цеолита типа 5А раствором той или иной соли металлов: Cr, Pt, Ni и Мо.
В патенте [46 [ предлагается готовить активные и стабильные катали-
заторы для процессов гидрирования, дегидрирования, дегидратации спир-
тов, синтеза NH3 и синтеза по Фишеру—Тропшу. Для этого цеолит типа
NaX заряжают одним или несколькими металлами подгруппы Fe, (Fe,
Со, Ni) ионным обменом цеолита с раствором соли указанного металла,
раствором аминокомплекса или в инертной атмосфере жидким соедине-
нием металла (карбонил, гидрокарбонил или ацетилацетонат). Затем ме-
387
талл окисляют воздухом или О2 и используют цеолиты, содержащие СоО
в процессе гидрообессеривания, a Fe2O3 — для очистки Н2 от СО.
В [47] сообщается о возможности использования цеолитов A, D,
L, R, S, Т, X, Y, а также природных цеолитов в качестве основы для
приготовления катализатора в целях окисления органических соедине-
ний, в частности этилена в окись этилена.
В работе [48] указывается на возможность получения ряда сложных
эфиров непосредственным взаимодействием карбоновых кислот со спир-
тами в присутствии катализаторов — молекулярных сит.
В [49] приводится способ зарядки металлом Zn или его окислом мо-
лекулярных сит типа A, D, L, R, S, Т, X, Y и ряда природных мине-
ралов с целью получения катализатора для синтеза метилового спирта,
а также для очистки кислорода и удаления вредных газов.
Г. В. Цицишвили с соавторами [21] изучал дегидратацию изопро-
пилового спирта на цеолитах типа NaX, СаА и НХ. Дегидратирую-
щие свойства их расположены в следующий ряд: НХ > NaX > СаА.
На цеолите NaX в интервале температур 240—270° энергия дегидратации
равна 7.4 ккал./моль и для цеолита НХ — 27 ккал./моль.
В патенте [50] описывается способ приготовления Cd-катализатора
на основе цеолита типа X путем зарядки металлом Cd. В случае окис-
ления Cd в CdO цеолит можно использовать в качестве катализатора
разложения этилового спирта на этилен и уксусный ангидрид, а также
для дегидрирования первичных спиртов. Другие способы «зарядки»
описаны в [51—55].
Вейс и др. [1] с 1960 г. начали развивать теоретические представления
о каталитических реакциях, протекающих в сильно развитых полостях
кристаллического тела. В отличие от поверхностного типа катализа
на обычных катализаторах они назвали такой тип селективного катализа
«вну трикриста ллическим». Например, Са-форма цеолита А ускоряет
реакцию дегидратации только 1-бутанола в присутствии изобутилового
спирта, в то время как в присутствии стандартного катализатора прохо-
дит дегидратация обоих спиртов. Са-форма цеолита структуры А в отли-
чие от цеолита структуры X ведет реакцию крекинга исключительно
н-парафинов, т. е. селективно, а образующиеся продукты имеют только
н-строение. Однако Вейс на основании опытов утверждает, что не всякая
каталитическая активность на цеолитах имеет внутрикристаллическую при-
роду. В качестве доказательства приводится пример, когда на цеолите 4А
несмотря на малую внешнюю поверхность — 10 м2/г против 700 м2/г
для общей поверхности при реакции гидратации окиси этилена имело
место образование этилен-гликоля и полигликолей, размер молекул ко-
торых не соответствовал размерам пор. По мнению Вейса, понятие о по-
верхности, несущей активные центры, принятое для обычных катали-
заторов с трехмерной структурой, утрачивает свое значение в случае
цеолитных катализаторов, в которых реагирующие молекулы могут на-
ходиться под действием необычных для катализа кулоновских сил,
играющих основную роль при взаимодействии между ионами.
Особенностями внутрикристаллического катализа можно объяснить
и поведение цеолитных катализаторов в процессе крекинга. Так, меньшую
устойчивость активности цеолита НХ, декатионированного больше чем
на 40% по сравнению с цеолитом HY, декатионированного даже на 30—
90%, как нам представляется, можно объяснить геометрическим фактором,
т. е. более близким расположением в нем тетраздров А104 (расстояние
2.6 А), которых в цеолите X больше в два раза, чем в цеолите типа фожазита
Y (расстояние 5.2 А).
Процесс глубокого декатионирования цеолита, связанный с дегидра-
тацией, приводит к ослаблению связей А1—Si каркаса и в конечном итоге
388
ведет к разрушению последнего. В декатионированном цеолите Y струк-
тура элементарной ячейки мало изменяется, хотя в ней также один тетра-
эдр алюминия (А104) теряет один электрон и одновременно другой тетра-
эдр алюминия переходит в А12О3 типа Льюиса, обусловливая тем самым
появление у цеолита кислых свойств.
Что касается высокой активности в реакциях крекинга цеолита Y
в Са-форме в отличие от Na-формы, то можно согласиться с мнением Рабо
о природе каталитической реакции изомеризации [29]. Причины этого*
различия прежде всего лежат в удлинении (до 7А) и ослаблении связей
у иона с тетраэдрами в результате уменьшения катионной плотности,
а затем уже в самой природе катионов.
Относительно кислые свойства цеолитных катализаторов, содержащих
большое количество щелочных металлов, не позволяют, однако, говорить
по аналогии со свойствами Al-Si-катализатора [56—61] о кислой при-
роде цеолитов в Na-форме.
Предварительно результаты качественной оценки кислых свойсти
отечественных цеолитов (без связующего), полученные Е. Н. Россолов-
ской (МГУ), по реакции их с индикатором р-диметил аминоазобензол ом
указывают на основные свойства Na-формы и кислые свойства декатиони-
рованной и Са-формы цеолитов типа X и Y. Кислотность измеряется вели-
чинами от 0.1 до 0.9 мзкв./г, близкими к Al-Si-катализатору.
Шваб объясняет природу каталитической активности цеолитов в реак-
ции крекинга кумола [8] ненасыщенностью электроотрицательных (анион-
ных) центров, возникающих при замещении одновалентного Na на дву-
валентный катион. Повышение энергии активации при переходе от Mg
к Zn автор объясняет более сильным поляризующим действием иона.
Отравляющее действие катиона Cd предположительно объясняется уси-
лением ковалентного характера связи (цвет окисла). Авторы считают,
что, кроме кислотных центров, для крекинга необходимо второе условие —
наличие на поверхности отрицательно заряженных центров — оснований
Льюиса. Подтверждение этого авторы видят в более легком превращении
р-кумола по сравнению с кумолом.
В работе [29] экспериментально показано, что с повышением степени
декатионирования цеолита NaY увеличивается его каталитическая ак-
тивность. Са-форма (80% обмен. Са) ведет себя подобно декатионирован-
ной форме катализатора (80% обмен, катиона Na на NH4), как по актив-
ности, так и по стабильности. Это указывает на одинаковый характер ак-
тивных участков. Подтверждением этой точки зрения являются опыты
Е. Н. Россоловской.
По мнению Рабо, каталитический эффект декатионированного цео-
лита типа Y можно объяснить высокой концентрацией непарных электро-
нов у А1, находящегося в четверной координации, и асимметричным поло-
жением в случае цеолита в Са-форме. В этой связи встает вопрос, какое же
влияние оказывает сам катион.
Полное отсутствие каталитической активности у одновалентно- и
многовалентно-катионных цеолитов с низким отношением Si/Al свиде-
тельствует, что эти катионы сами по себе почти или совсем не оказывают
влияния на каталитическую активность. Активность и стабильность цео-
литов с высоким отношением Si/Al обязаны образованию прочной кри-
сталлической тетраэдрической структуры А104—Si04 [29].
Из аналогии с аморфным алюмосиликатным катализатором, для ко-
торого принят протонный механизм действия, возникает и второй вопрос:
что представляют собой активные участки, например цеолитного Pt-ка-
тализатора, — протонные кислоты или акцепторы пары электронов?
Рабо, признавая, что каталитическое действие цеолитных катализа-
торов в реакциях углеводородов протекает через промежуточное образо-
38»
вание ионов карбония, считает, что не обязательно чтобы в любом слу-
чае эти активные участки представляли собой протонные или апротонные
кислоты. В случае изомеризации парафинов, по его мнению, наиболее
эффективными участками должны явиться тетраэдры А1О4, которые имеют
очень мало катионов (Н-форма) или только слабо связаны с катионами
(Са-форма).
ЛИТЕРАТУРА
1. Р. В. W е i s z, V. J. F г i 1 е 11 е, J. Phys. Chem., 1960, 3, 44, 382.
2. А. Н. К е о v g h, L. В. Sand, J. Amer. Chem. Soc., 1961, 16, 83, 3536.
3. F. W о 1 f, A. Losse, Chem. Ing. Techn., 1961, 33, 6, 442.
4. Американский патент, 1961, 2916437; 1961, 2916437.
5. E. A. S о 1 о m о и, P. A. L e f r a n s с о i s, Rev. Recent. USA Pat., 1962,
33, 1, 42.
6. Petr. Refiner, 1962, 5, 284.
7. V. J. F r i 1 e t t e, P. B. W e i s z, R. L. Golden, J. Catalysis, 1962,
1, 4, 301.
8. I. M. Schwab, R. S i e b, Z. Naturforsch., 1963, 18a, 164.
9. P. B. W e i s z, V. J. F r i 1 e 11 e, R. W. M aa t man, E. B. Mower,
J. Catalysis, 1962, 1, 307.
10. Французский патент, 1962, 1310520.
11. Oil a. Gas J., 1962, 10, 14, 21, 23, 96—144.
12. Che m. Eng. News, 1962, 17, 40, 25.
13. Американский патент, 1961, 2962435.
14. M_e т о д ы анализов нефтей и нефтепродуктов, Сборник ВНИИ НИ, ГТТП, 1955.
15. Л. И. Пи г уз ов а и А. С. В и т у х и и а, Хим. и технол. топлив и масел,
1963, 6, 17.
16. К. В. Топчиева, В. В. Романовский и др., ДАН СССР, 1963, 149,
3, 644.
17. И. Н. Галич, И. Т. Голубенко и др., в сб.: Синтетические цеолиты,
изд. АН СССР, 1962, 260.
18. П. Н. Г а л и ч и др., Нефтехимия АН СССР, 1962, 2, 193.
19. Л. И. И и г у з о в а и др., Авторская заявка № 845219/23 от 4 июля 1964 г.
20. К. В. Топчиева, Е. Г. Борескова, Л. И. Пигузова, настоя-
щий сборник, стр. 367.
21. Г. В. Ц и ц и ш в и л и и др., ДАН СССР, 1963, 153, 6, 1395.
22. С. J. Norton, Chem. Ind., 1962, 6, 258; 10, 24—25.
23. Che m. Engng., 1962, 69, 9, 36.
24. Chem. a. Engng. News, 1962, 14, 40, 52.
25. Che m. Age, 1962, 87, 2235, 785.
26. Che m. a. Engng. News, 1962, 11, 40, 52.
27. A n g e w. Chem., 1962, 9, 129.
28. В. H. Долгов. Катализ в органической химии. Госхимиздат, Л., 1959, 554.
29. J. A. R а Ь о, R. Е. Р i с h е г t, D. Stamires, J. Boyle, Докл. на
конгрессе по катализу, состоявшемся в Париже в 1960 г., 2, 2055.
30. J. A. R а Ъ о, Р. Е. Р i с h е г t, R. L. М а у s, Ind. Engng. Chem., 1961, 53,
9 733.
31. Английский патент, 1962, 886716.
32. Американский патент, 1961, 2971904.
33. Oil a. Gas J., 1959, 57, 27, 54.
34. Y. J. Yriesmer, Н. В. Rhodes, К. Kiyonada, Petrol. Refiner,
1960, 39, 6, 125.
35. X. М. М и н а ч е в., В. И. Г а р а н и н, Л. И. П и г у з о в а, А. С. В и т у-
хина, Изв. АН СССР, ОХН, 1965, 7, 1116.
36. X. М. Миначев, В. И. Г а р а н и н, Я. И. И с а к о в, настоящий сбор-
ник, стр. 374.
37. Американский патент, 1961, 2983670.
38. D. Н. Stormont, Oil a. Gas J., 1963, 10, 61, 64—66.
39. Американский патент, 1961, 3013988.
40. Американский патент, 1961, 2967159.
41. В. С. Виноградова, Диссертация на соискание ученой степени канди-
дата наук, Гипрокаучук, М., 1964.
42. Американский патент, 1961, 3013986.
43. Американский патент, 1957, 2904607.
44. Р. Sherwood, Techn. Petr., 1963, 11, 18, 204, 23.
45. Американский патент, 1961, 2970968.
46. Американский патент, 1961, 3013990.
47. Американский патент, 1961, 3013985.
390
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
€0.
61.
J. Chem. Soc. Japan, Pure Chem. Sec. 1962, 83, 2, 195.
Американский патент, 1961, 3013984.
Американский патент, 1961, 3013983.
Американский патент, 1961, 3013982.
Американский патент, 1961, 3013987.
Американский патент, 1962, 3033642.
Американский патент, 1961, 3013989.
Американский патент, 1961, 3006863.
Ю. А. Б е т е п а ж, ЖОХ, 1947, 17, 199.
Е. О b 1 a d. Advances in Catalysis, III. Academic Press, N. Y., 1951, 199.
J. D. D a n f о r t h. A. D. F. M artin, J. Phys. Chem., 1956, 60, 4, 422.
К. В. Топчиева, И. Ф. Московская, ДАН СССР, 1958, 123, 5, 891.
Y. J. Т г a m b о u z е et al., Actes du douzieme Congres internationale de Cata-
lyse, 1961, 1, 1313.
H. В e n e s i, J. Phys. Chem., 1957, 61, 970.
СОДЕРЖАНИЕ
Стр.
Предисловие.......................................................... 3.
Теоретические вопросы адсорбции на цеолитах и исследование их свойств
М. М. Дубинин. Особенности адсорбции паров различных веществ на
цеолитах как на микропористых адсорбентах............................. 5
А. В. Киселев. Природа адсорбции цеолитами.......................... 13.
Л. В. Радушкевич. К вопросу о теории уравнения изотермы сорбции
паров на пористых сорбентах так называемого первого структурного
типа............................................................ 25
Б. П. Беринг, В. В. Серпинский, С. И. Суринова. Особен-
ности адсорбции смесей паров на микропористых адсорбентах .... 30
П. Бройер, А. А. Лопаткин, С. Шпигиль. Расчет энергии взаи-
модействия простых молекул с различными катионными формами цео-
лита типа X .................................................... 36
В. И. К в л и в и д з е. Исследование спектров ЯМР протонов в цеолитах
при низких температурах.............................................. 42
О. М. Д ж и г и т, С. П. Ж д а н о в, К. Н. М и к о с. Изотермы и диффе-
ренциальные теплоты адсорбции веществ с различной электронной струк-
турой молекул разными катионными формами цеолита типа фожа-
зит ................................................................. 46
С. П. Жданов, В. И. Л ы г и н, Т. И. Титова. Инфракрасные спектры
цеолитов различной структуры......................................... 53
В. И. Л ы г и н. Инфракрасные спектры адсорбированных цеолитами моле-
кул ................................................................ 58.
Г. Д. Багратишвили, Г. В. Цицишвили, М. Л. Канта-
рия, Н. И. Ониашвили. Исследование адсорбционного взаи-
модействия веществ с синтетическими цеолитами методом инфракрасных
спектров поглощения.................................................. 65
С. П. Г а б у д а. Подвижность и форма связи молекул воды в цеолитах ... 68
Н. Н. А в г у л ь, Г. Г. Муттик. Теплота адсорбции полярных моле-
кул цеолитом NaX................................................ 75
М. В. С е р д о б о в. Влияние заместителей на теплоту адсорбции неполяр-
ных производных насыщенных углеводородов графитированной сажей
и синтетическим Na-фожазитом.................................... 78
Т. Н. Шишакова, М. М. Дубинин. Термическая обработка гранул
и адсорбционные свойства кристаллических и формованных синтетиче-
ских цеолитов NaX и СаА ........................................ 79
Э. Н. Корыткова, А. Д. Федосеев. О структурных изменениях
некоторых природных цеолитов при нагревании..................... 81
Б. А. Г л а з у н, М. М. Дубинин, И. В. Ж и л е н к о в, В. М. Фе-
до р о в. Исследование диэлектрической релаксации цеолита NaA при
адсорбции воды.................................................. 83
Г. В. Цицишвили. Физико-химические свойства новых ионообменных
форм цеолитов................................................... 87
М. А. Пионтковская, И. Е. Неймар к, Г. С. Шамеко,
Г. И. Денисенко, И. Н. Червецов а. О некоторых свойствах
различных ионообменных форм цеолитов типа А..................... 97
В. Босачек. Адсорбция благородных газов на цеолитах типа X, моди-
фицированных ионным обменом.................................... 103
Е. Ф. П о л с т я н о в, М. М. Дубинин. Адсорбция паров циклогексана,
бензола и н-пентана на цеолитах NaX и СаХ...................... 109
О. К а д л е ц. Адсорбция паров бензола на цеолите NaX.............. 114
392
А. И. Capa хов, M. M. Дубинин, Ю. Ф. Березкина. Удель-
ная поверхность вторичных пор синтетических цеолитов.......... 117
В. М. Лукьянович, Э. И. Е в к о, В. В. Чураков, Электропно-
микроскопическое исследование синтетических цеолитов методом реплик 121
Получение цеолитов
С. П. Ж д ан ов, Н. Н. С а м у л е в и ч, Е. Н. Е г о р о в а. Хими-
ческие аспекты процессов кристаллизации синтетических цеолитов 129
М. Е. О в с епян, С. П. Ж д а и о в. Получение калиевых и смешанных
натриево-калиевых цеолитов.................................... 140
И. Е. Неймарк, М. А. Пионтковская, А. И. Растрененк о,
Г. С. Ш а м е к о, В. Г. И л ь и н, А. М. Еременко, С. Н. Анто-
новская. Синтез и основные свойства различных ионообменных и
изоморфнозамещенных форм цеолитов............................. 146
А. С. П л а ч и н д а. Равновесие ионного обмена на цеолите типа А . . . 158
Л. И. П и г у з о в а, А. С. В и т у х и н а, В. Ф. Дмитриева. Осо-
бенности цеолита структуры типа фожазита в условиях лаборатории и
опытной установки........................................ 160
3. Э. С е н д е р о в. О зависимости отношений SiO2/Al2O3 в фазах перемен-
ного состава от щелочности минералообразующего раствора.. 165
Г. В. Цицишвили, Г. Д. Баграгишвили, К. А. Бежашвили,
К. Е. А в а л и а н и, Ц. А. Г е д ж а д з е, М. С. Ш у а к р и-
швили, Ц. М. Окропиридзе, М. Г. А д о л а ш в и л и. Син-
тез и исследование свойств водородных форм цеолитов........... 168
Н. Ф. Ермоленко, Л. В. Пансевич-Коляда. Исследование
возможности получения цеолитов с изоморфным замещением хро-
мом .......................................................... 171
С. П. Ж д а н о в, В. Н. Новикова, Л. Ф. П а в л о в а, Ю.А.Эль-
т е к о в. Влияние степени катионного обмена в синтетическом фожазите
на адсорбцию из растворов..................................... 178
Т. Г. Андроникашвили, Г. В. Цицишвили, Ш. Д. С а бе-
ла ш в и л и, Т. А. Чумбуридзе. Влияние природы и степени
замещения некоторых катионов в цеолитах типа X на их хроматографи-
ческие свойства .............................................. 179
В. Г. Ильин, И. Е. Неймарк, А. И. Р а с т р е н е н к о. Вакуум-
термическая активация и адсорбционные свойства аммониевых форм
цеолитов ................................................ 185
А. В. А г а ф о н о в, С. М. Вайнштейн, И. Э. Г е л ь м с, Б. А. Л и п-
к и н д, Л. И. Пигу зов а. Вопросы синтеза и технологии произ-
водства цеолитов.............................................. 187
Я. В. Мирский, М. Г. Митрофанов, Б. М. П о п к о в, Л. Т. Бо-
лотов, Л. Ф. Р у ч к о. Синтез и производство цеолитов типов
А и X......................................................... 192
А. Т. С л е п н е в а, Б. А. Л и п к и и д, В. А. Б у р ы л о в, С. В. К а-
пацинский. Влияние условий грануляции и хранения цеолитов на •
их механические свойства ................................ 201
С. М. Вайнштейн, Е. Я. Г и е н к о, А. Т. Слепнева, Б. А. Л и п-
к и н д, В. А. Б у р ы л о в. Совершенствование методов и показате-
лей временных технических условий на формованные синтетические
цеолиты....................................................... 207
Г. М. Белоцерковский, Т. Г. Плаченов, К. Г. Ионе,
Ю. В. Ежов, В. Ф. Карельская, Е. Н. Долгова
Влияние типа связующего и некоторых условий приготовления форму-
емых масс на свойства гранулированных цеолитов................ 213
В. Н. Ма зин, И. В. С у ч к о в. Исследование влияния природы связую-
щего и условий формования на сорбционные и механические свойства
гранулированных цеолитов...................................... 218
* К. Г. Ионе, Г. М. Белоцерковский, Т. Г. Плаченов. При-
менение основных солей алюминия для грануляции цеолитов и иссле-
дование свойств гранулированных образцов........................ 222
Л. С. Я с т р е б о в а, А. А. Бессонов, С. С. X в о щ е в. Пути полу-
чения пористых стекол — молекулярных сит с заданными характеристи-
ками на основе стекол калиевоалюмосиликатной системы.......... 229
Е. В. К о р о м а л ь д и, С. С. X в о щ е в, С. П. Жданов. Набор
пористых стекол — молекулярных сит и его характеристики по адсорб-
ционным данным .......................................... 234
393
Применение цеолитов
Н. С. Т о р о ч е ш и и к о в, А. И. Си доров, Н. В. К е л ь ц е в. Тон-
кая очистка газов от двуокиси углерода с одновременным удалением
паров воды — как первая фаза процесса разделения и переработки
газов ........................................................... 240
Т. Г. П л а ч е и о в, А. Н. Ш и р я е в. Глубокая осушка газов в стационар-
ном, движущемся и псевдоожиженном слоях цеолита ................. 249
В. С. В и н о г р а д о в а, Л. С. Кофман. Разделение и тонкая очистка
синтетическими цеолитами углеводородов, применяемых в производстве
высокополимеров.................................................. 259
Г. В. Урбан, В. С. Виноградова, В. Н. Комарова, Л. С. К о ф-
м а н. Тонкая очистка алканов С4—Св от сернистых соединений . . . 268
Н. В. 3 а х а р о в а, В. С. Виноградова, Л. С. Кофман. Разде-
ление амиленов изо- и нормального строения на синтетических цеолитах 274
В. С. Виноградова, Л. О. Д ы м е н т, В. Н. Комарова,
Т. А. М а р к о в а, Л. С. Кофман. Процесс очистки изобутилена
адсорбцией синтетическими цеолитами ....................... 277
Т. А. Маркова, В. С. Виноградова, Л. С. Кофман. Приме-
нение синтетических цеолитов для извлечения ацетонитрила из его сме-
сей с углеводородами С4 ......................................... 283
В. С. Виноградова, Л. С. Кофман, Б. А. Л и п к и н д,
Л. М. Медведева, А. Т. Слепнева. Исследование термо-
стабильности синтетических цеолитов.............................. 285
Н. С. К о р н и л о в а, И. Н. Морина, Н. А. Нечаева. Разделение
углеводородов в псевдоожиженном слое цеолита СаА............ 290
А. Н. С а л ю к о в, Л. Ф. Кулиш, В. Н. Мазин, Л. Е. Ф е не-
ясно в а. Адсорбция из смеси бензола и тиофена синтетическими
• цеолитами типа X -......................................... 292
М. П. К о р ш, Е. И. Казаков, Н. А. К у н а к о в а, Н. Ф. Кар-
пова. Выделение этиленовых углеводородов из коксового газа с при-
менением синтетических цеолитов............................. 293
X. И. А р е ш и д з е, Г. О. Ч и в а д з е. Облагораживание туркменского
бензина при помощи синтетического цеолита СаА............... 294
Л. О. Д ы м е н т, В. С. Виноградова, Л. С. Кофман. Осушка
дивинила ................................................... 298
Р. П. Кирсанова, С. Ш. Бык. Глубокая осушка пропилена с по-
мощью синтетических цеолитов типа NaA и КА.................. 300
Г. И. В я л к и н а, Н. В. Жданова, Н. В. К е л ь ц е в, Г. С. Ф р о-
л о в. Результаты испытаний осушки пропана цеолитами и другими
сорбентами ................................................. 303
М. Е. С е л и н, Д. С. Л а в р у х и н, Л. Б. Кулемина. Адсорб-
ция воды, растворенной в спиртах Сг—С4, синтетическими цеолитами NaA 304
А. Н. С а л ю к о в, П. Н. Эндюськин, В. Н. М а з и и, В. Г. Ши-
роков. Применение синтетических цеолитов для осушки ацетона
в процессе промышленного получения дикетена ..................... 308
Г- Я. Александрова, Г. С. Гольдин, Г. С. Шор. Жидкофаз-
ная адсорбция воды цеолитами из олефино-ароматических углеводород-
ных смесей.................................................. 310
Т. М. Б у р к а т, Д. П. Д о б ы ч и н, А. С. М о ш к е в и ч, Т. А. У ш а-
к о в а. Осушка низкомолекулярных жирных кислот с помощью по-
ристых стекол — молекулярных сит и синтетического морденита ... 312
Н. Г. Сихарулидзе, Г. В. Цицишвили, Ц. В. Маградзе,
Т. Г. Андроиикашвили. Очистка воздуха от следов ацетилена
цеолитами СаА на пилотной установке........................ 317
В. Е. Кочурихин, Я. Д. Зельвенский. Разделение изотопов водо-
рода при адсорбции на сйнтетических цеолитах..................... 319
Г. А. Головко. Применение синтетических цеолитов для адсорбционно-
термической очистки аргона от кислорода..................... 321
Е. К. Богачева, Ю. А. Эльтеков. Молекулярные сита для поли-
меров ...................................................... 323
X. Н. Бородушкина, Г. А. Блох, Д. Б. Богуславский,
И. Е. Н е й м а р к, М. Б. 3 е л и к и и, Т. Р. Г е и д л е р, Г. М. Л е-
в и т, А. Ф. Чернухина, В. Я. Николина, К. Т. no-
fl о р в а н. Применение наполненных цеолитов в шинной промышлен-
ности ..................................................... 325
10. И. Ерофеева, Т. Н. Б у х м и т, М. С. Ф е л ь д ш т е й н,
3. В. Скородумова. Исследование эффективности применения
высокоактивных вулканизующих систем в шинных резинах с помощью
молекулярных сит типа X.................................... 331
394
А. Ф. Н о сни к о в, С. Е. Рапчинская, Г. А. Б л о х, И. Е. Н е й-
м а р к. О наполнении цеолитов сероводородом, сернистым газом и
органическими перекисями............................................... 336 -
А. С. Пономарев, Д. П. Тимофеев. Исследование скорости стацио-
нарной диффузии органических паров в стержнях из спрессованного
цеолита NaX .................................................... 338
И. Т. Е р а ш к о. Исследование кинетики сорбции н-пентана и диэтилового
эфира на цеолитах СаА и NaX..................................... 340
В. Б. Фенелонов, Л. Е. Фенелонова, А. Н. Салюков,
В. Н. Мазин. О кинетике и динамике сорбции газов и паров на син-
тетических цеолитах............................................. 345
П. Й и р у, М. Р а л е к, О. Г р у б и е р. Методы расчета адсорберов
с молекулярными ситами.......................................... 349
Ю. М. А ф а и а с ь е в, 3. А. Ж у к о в а, Н. В. К е л ь ц е в. Кинетика
вакуумной десорбции нормальных парафиновых углеводородов из зерна
цеолитов ....................................................... 352
Н. В. К е л ь ц е в, Н. С. Т о р о ч е ш и и к о в, Ю. И. Ш у м я ц к и й.
Изотермическая десорбция этилена из слоя цеолита NaX............ 356
В. Т. Б ы к о в, А. Н. К и р г и и ц е в, А. В. Лукьянов, Н. Е. Щ ер-
батю к. Изучение адсорбции паров воды природными цеолитами . . . 360
П. К. М и г а л ь, М. А. К е р д и в а р е н к о, Г. А. К р е н и с. Сорб-
ционные свойства некоторых солей гетерополикислот.............. 365-
Е. Г. Борескова, К. В. Топчиева, Л. И. Пигузова. Ка-
талитическая активность синтетических цеолитов в реакции крекинга
кумола ............................................................... 367
X. М. Миначев, В. И. Г а р а н и н, Я. И. И с а к о в, Л. И. П и г у-
з о в а, А. С. В и т у х и н а. Каталитические реакции углеводородов и
кислородсодержащих соединений на цеолитных катализаторах со струк-
турой Y............................................................... 374
Н. В. Бо рунов а, Л. X. Ф р е й д л и и, И. Е. Н е й м а р к,
В. Г. И л ь и и, Л. И. Г в ин тер. Исследование свойств никельцео-
литных катализаторов................................................. 380'
Л. И. Пигузова. Применение молекулярных сит в катализе.......... 383,
ЦЕОЛИТЫ, ИХ СИНТЕЗ, СВОЙСТВА И ПРИМЕНЕНИЕ
(Материалы II всесоюзного совещания по цеолитам)
Утверждено к печати
Научным советом по адсорбентам АН СССР
Редактор издательства А. А. Борисов
Художник В. В. Грибакин
Технический редактор Р. А. Замараева
Корректоры Л. М. Бова и Н. 3. Петрова
Сдано в набор 26/VI 1965 г. Подписано к печати
29/Х 1965 г. РИСО АН СССР № 43-80В. Формат
бумаги 70Х1081/,,. Бум. л. 123/8. Печ. л. 243/,=
=33.90 усл. печ. л. Уч.-ивд. л. 33.52. Изд. № 2614.
Тип. зак. № 351. М-27117. Тираж 2500.
Доп. ТП 1965 г. 201. Цена 2 р. бб к.
Ленинградское отделение издательства «Наука»
Ленинград, В-164, Менделеевская лин., д. 1
1-я тип. издательства «Наука», Ленинград, В-34,
9 линия, д. 12
ИСПРАВЛЕНИЯ И ОПЕЧАТКИ
Стра- ница Строка Напечатано Должно быть
29 Табл., графа 1 4 сверху н-С-Н14 н-СвН14
153 Табл. 5, графа 5 С4НБОН С4Н8ОН
153 Табл. 5, графа 1 2 сверху CO(NH3)e Co(NH3)e
153 Табл. 5, графа 1 3 снизу [Cu(NHs)]e [Cu(NHs)4]
211 26 сверху абсорбционных адсорбционных
211 Подпись к рис. 3, 3 сверху формированных формованных
218 14 сверху ГЛИНЫ кил- глины: кил»
222 23 снизу способны способных
282 Подпись к рис. 8, 2 сверху н-бутеновой н-бутенов от
382 23 сверху н-октана н-октена
393 16 сверху цеолита синтеза цеолита
Цеолиты.