





































































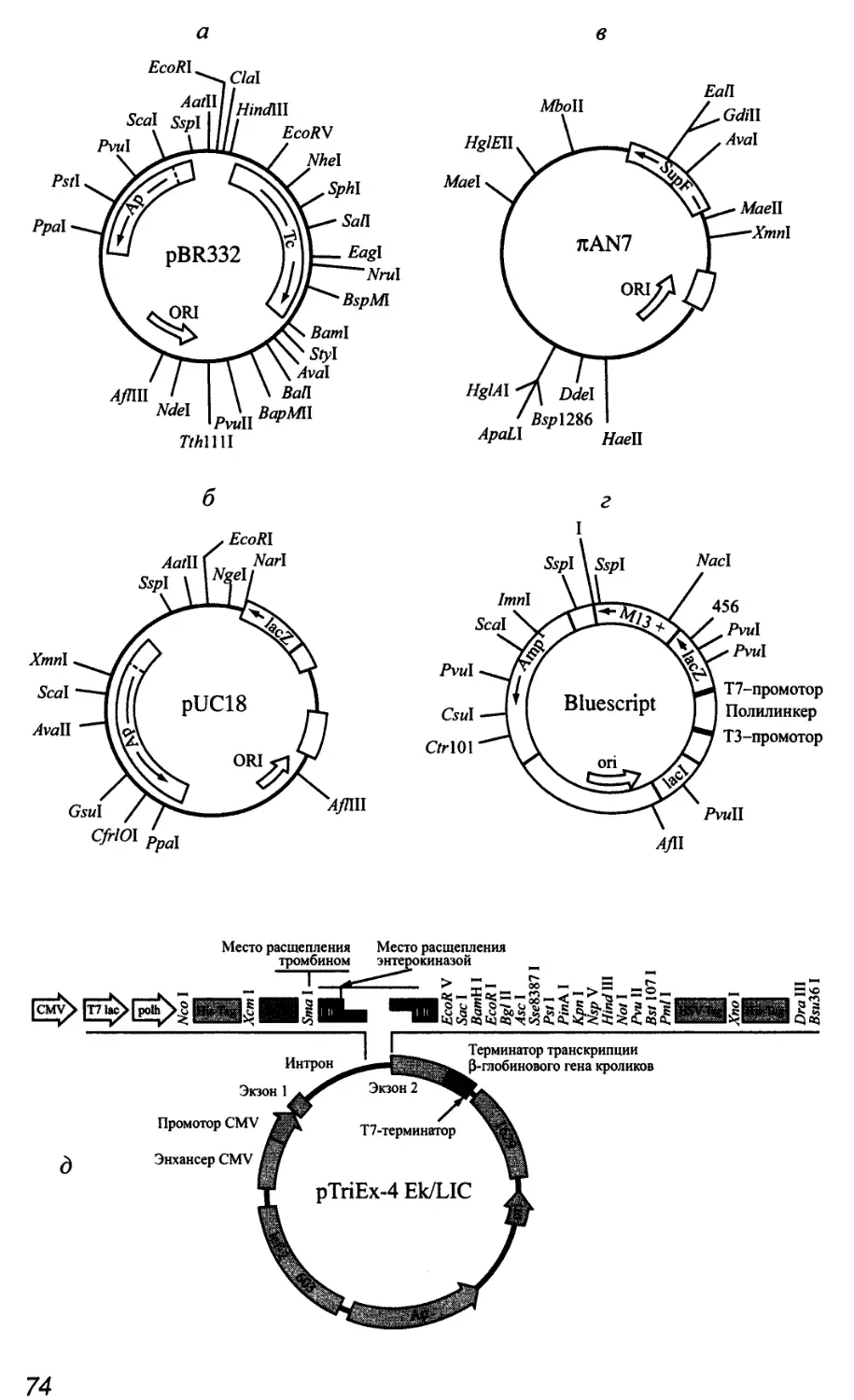









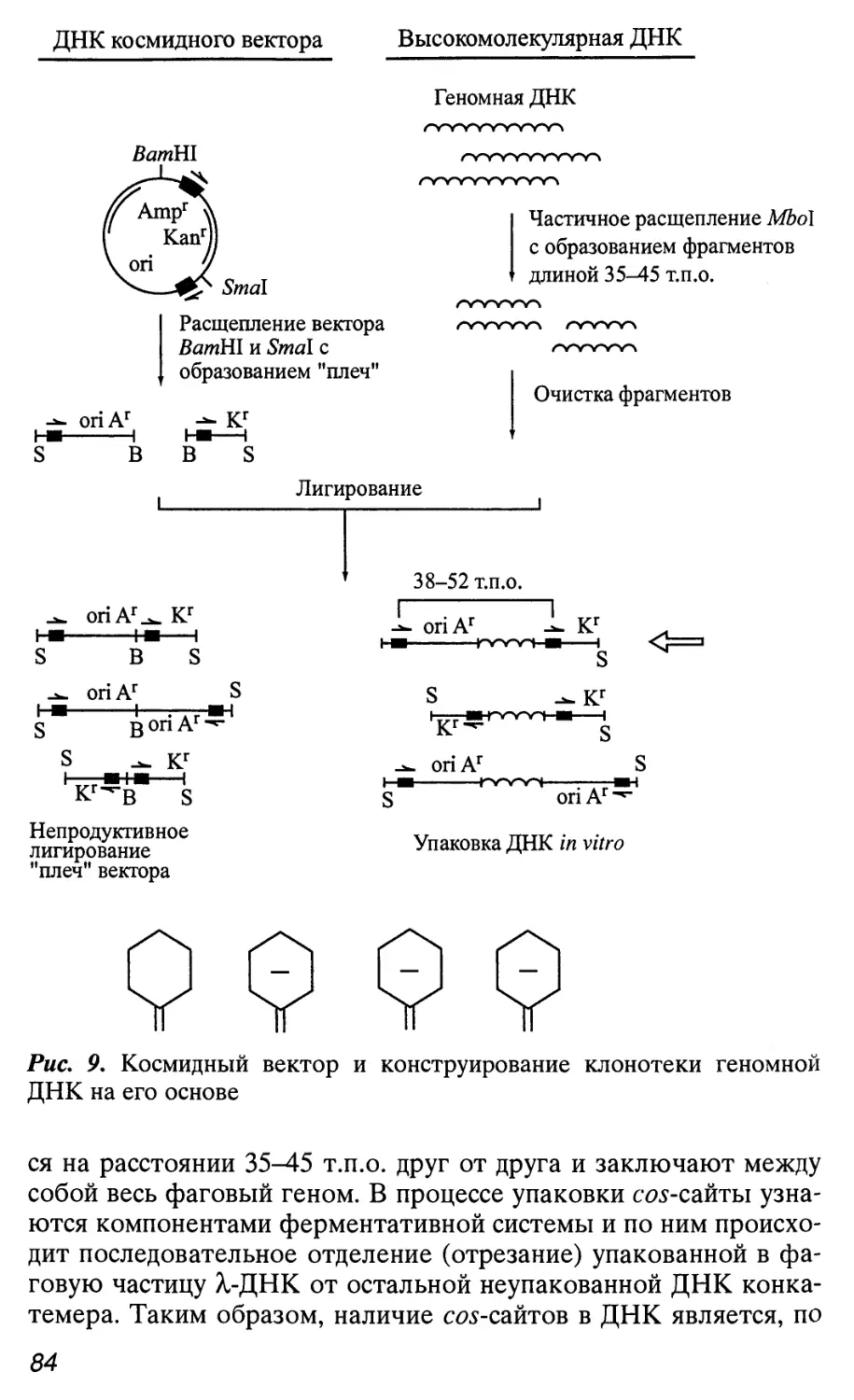




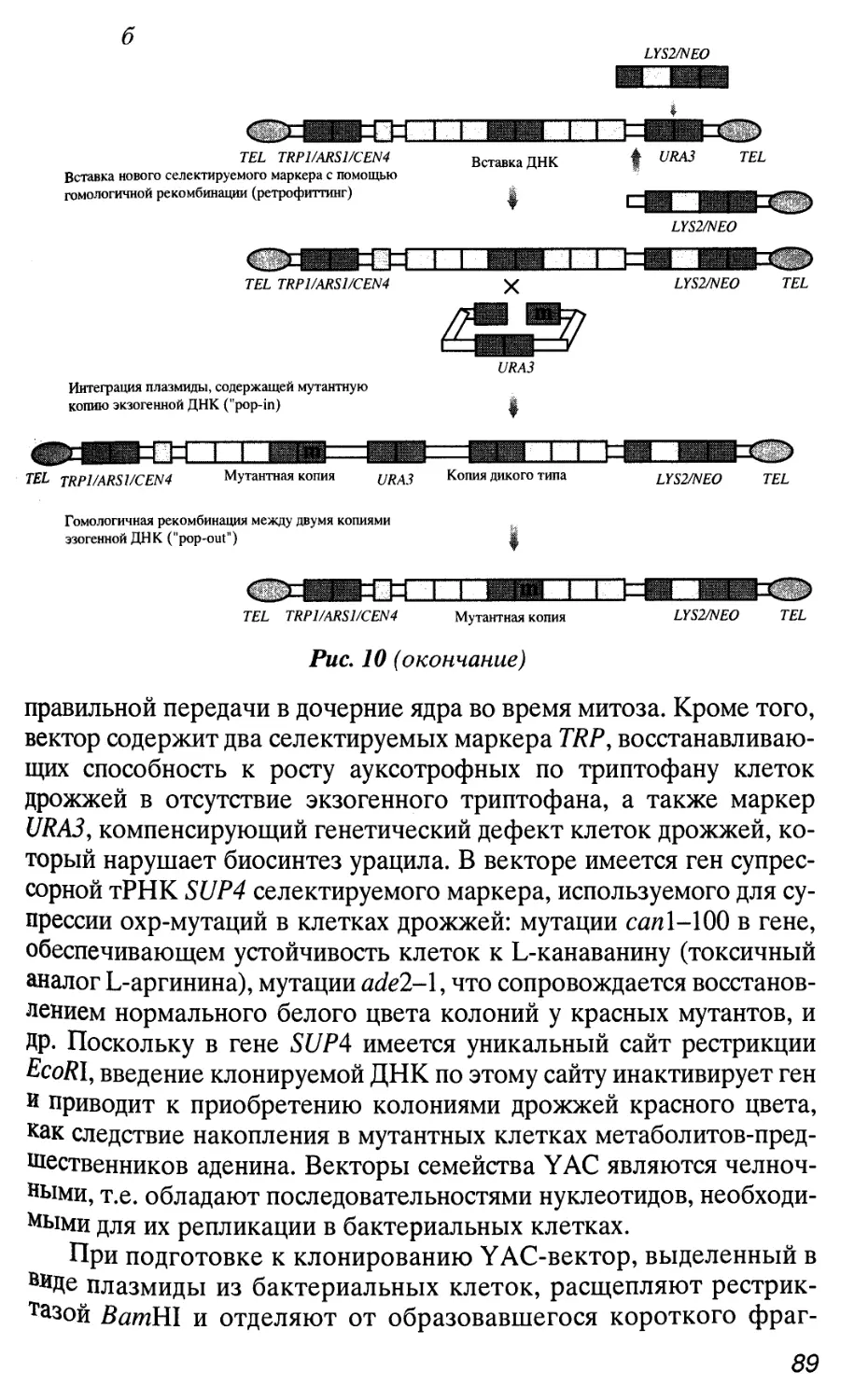












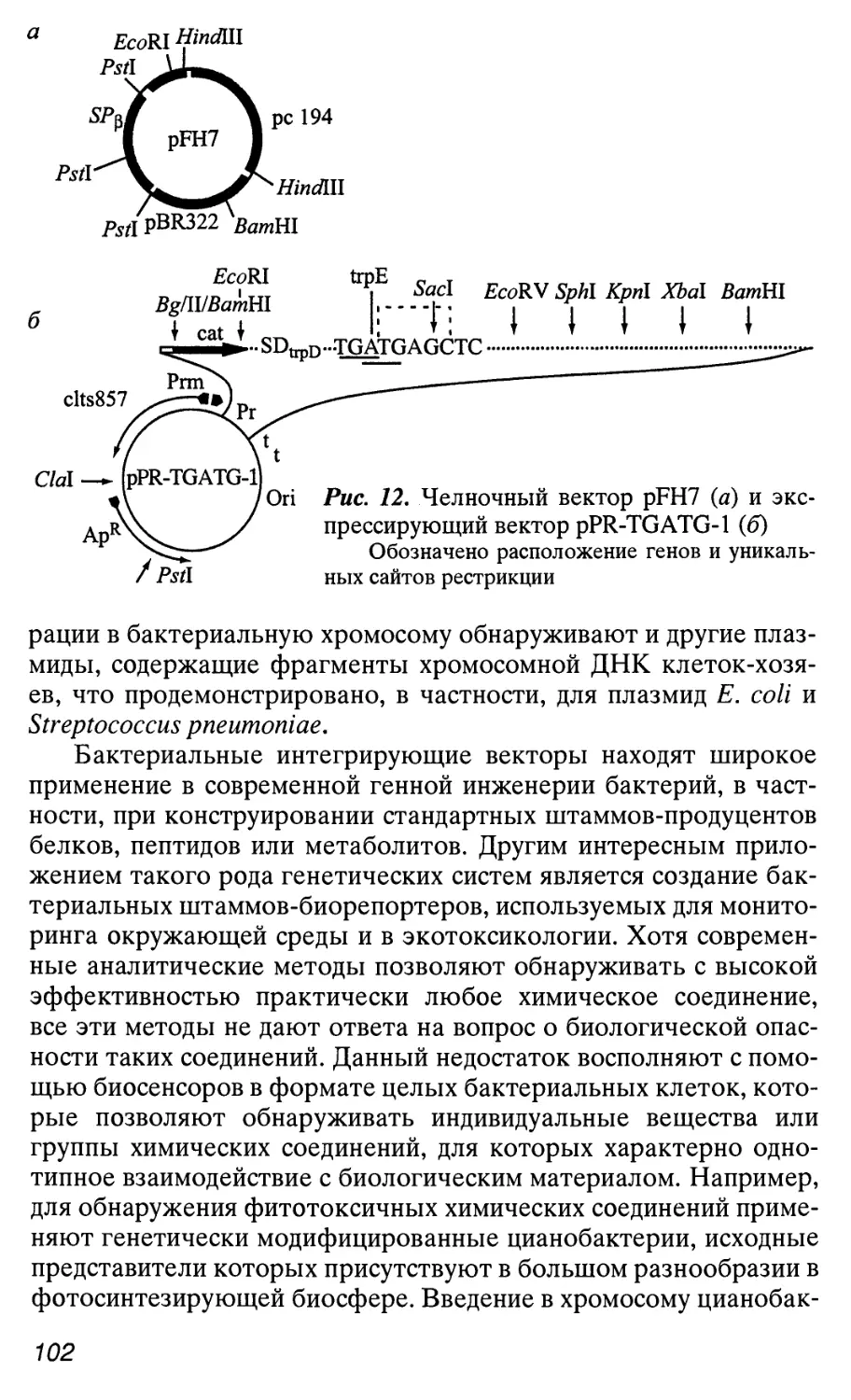












































































































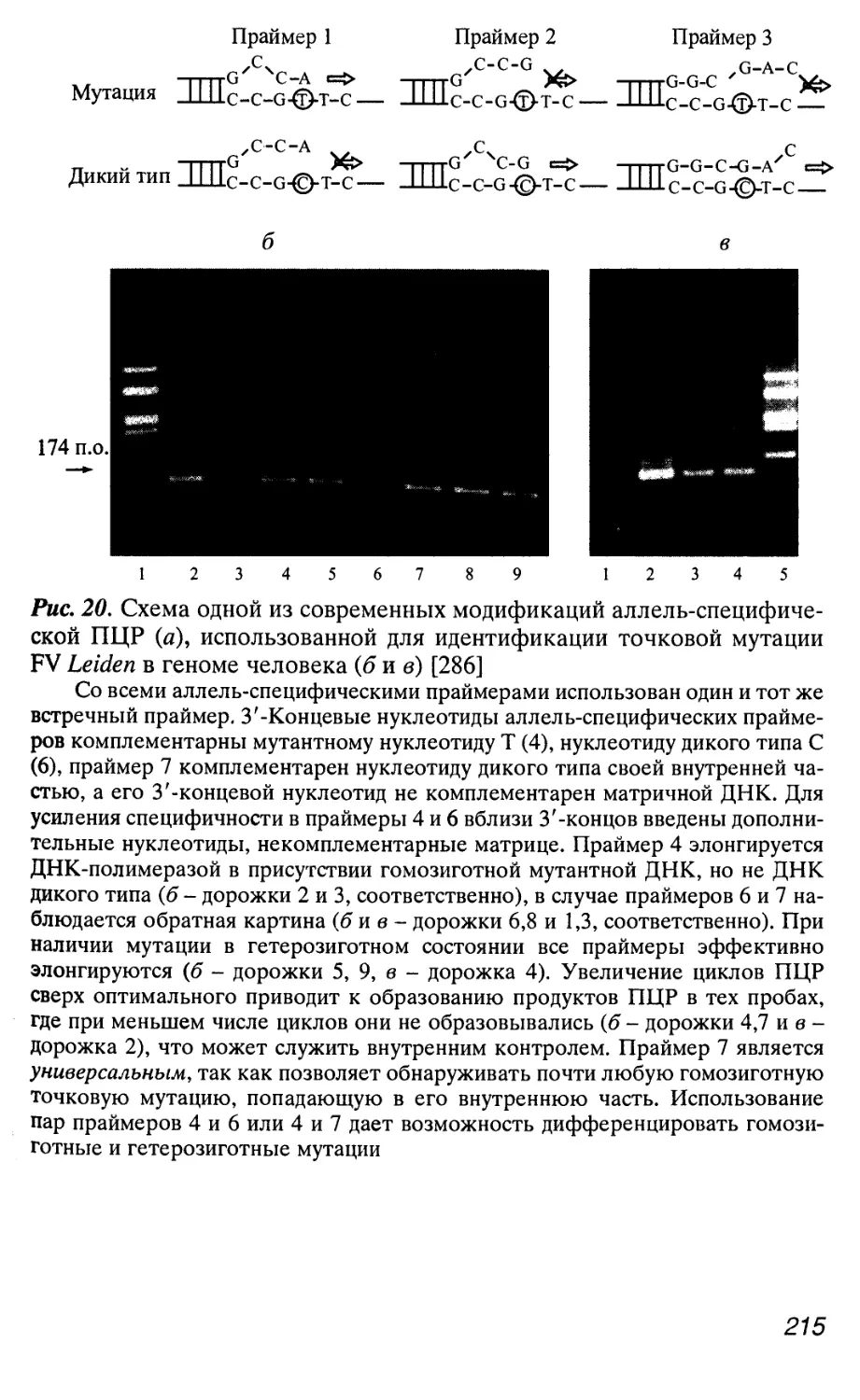



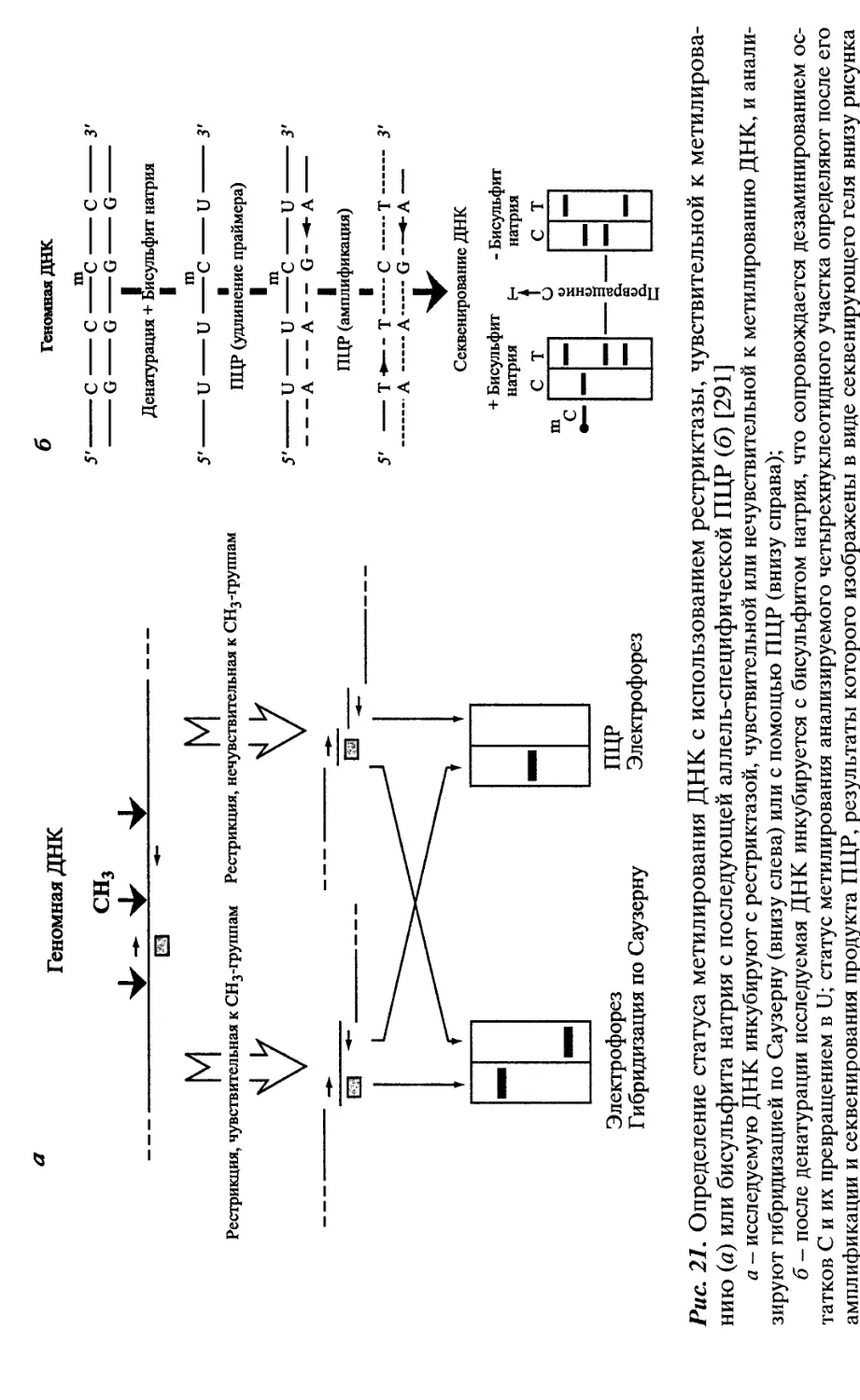


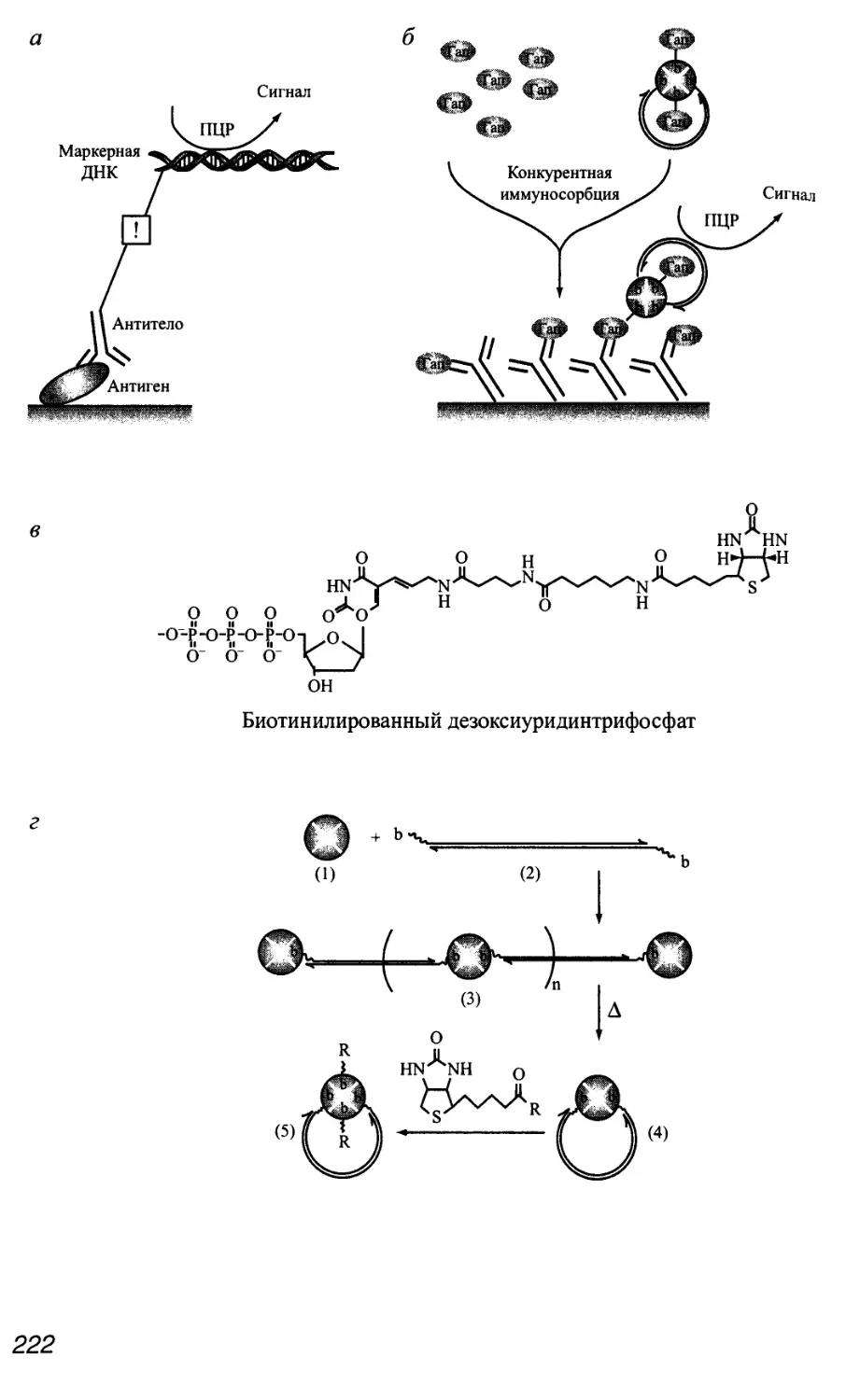


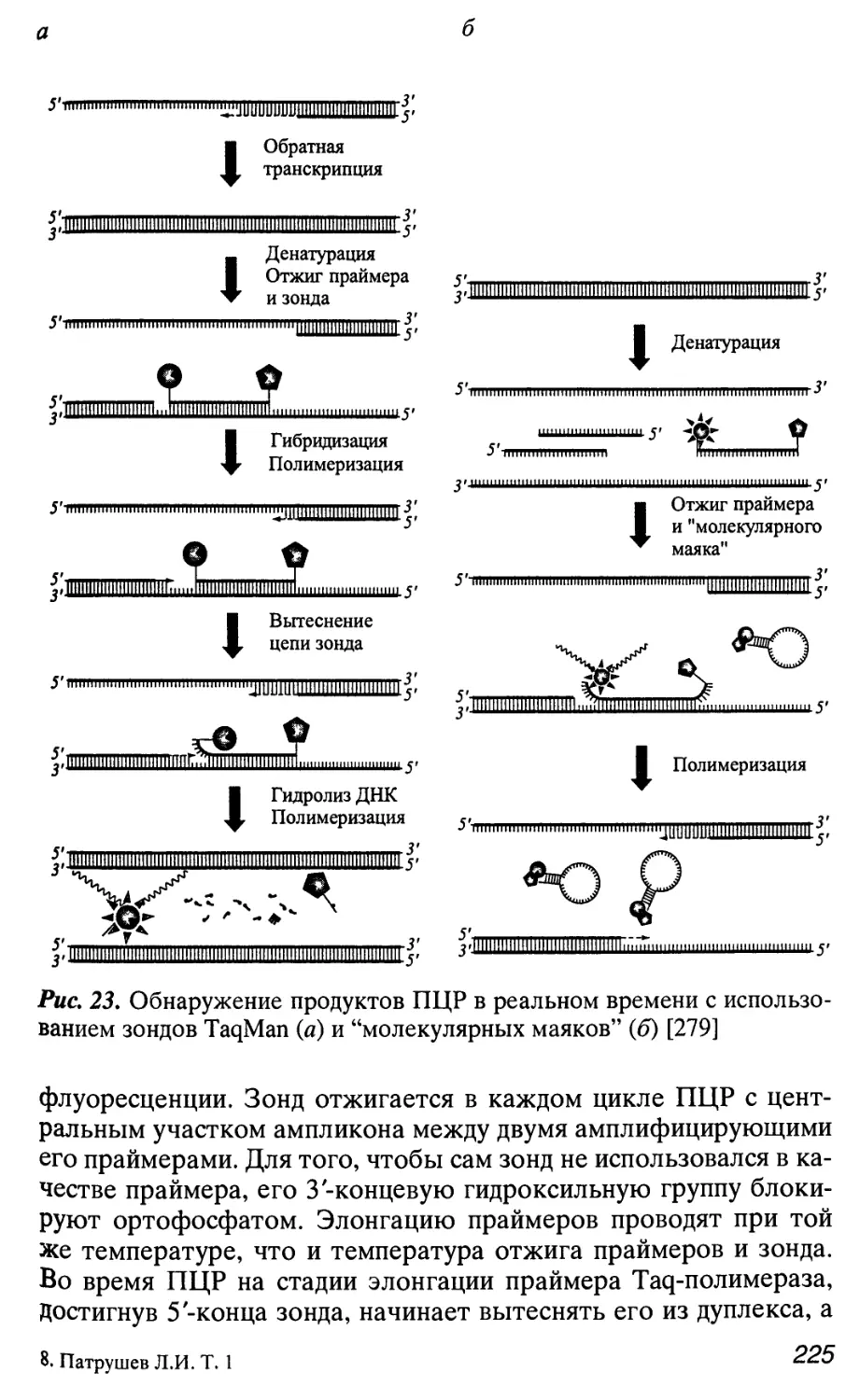






































































































































































































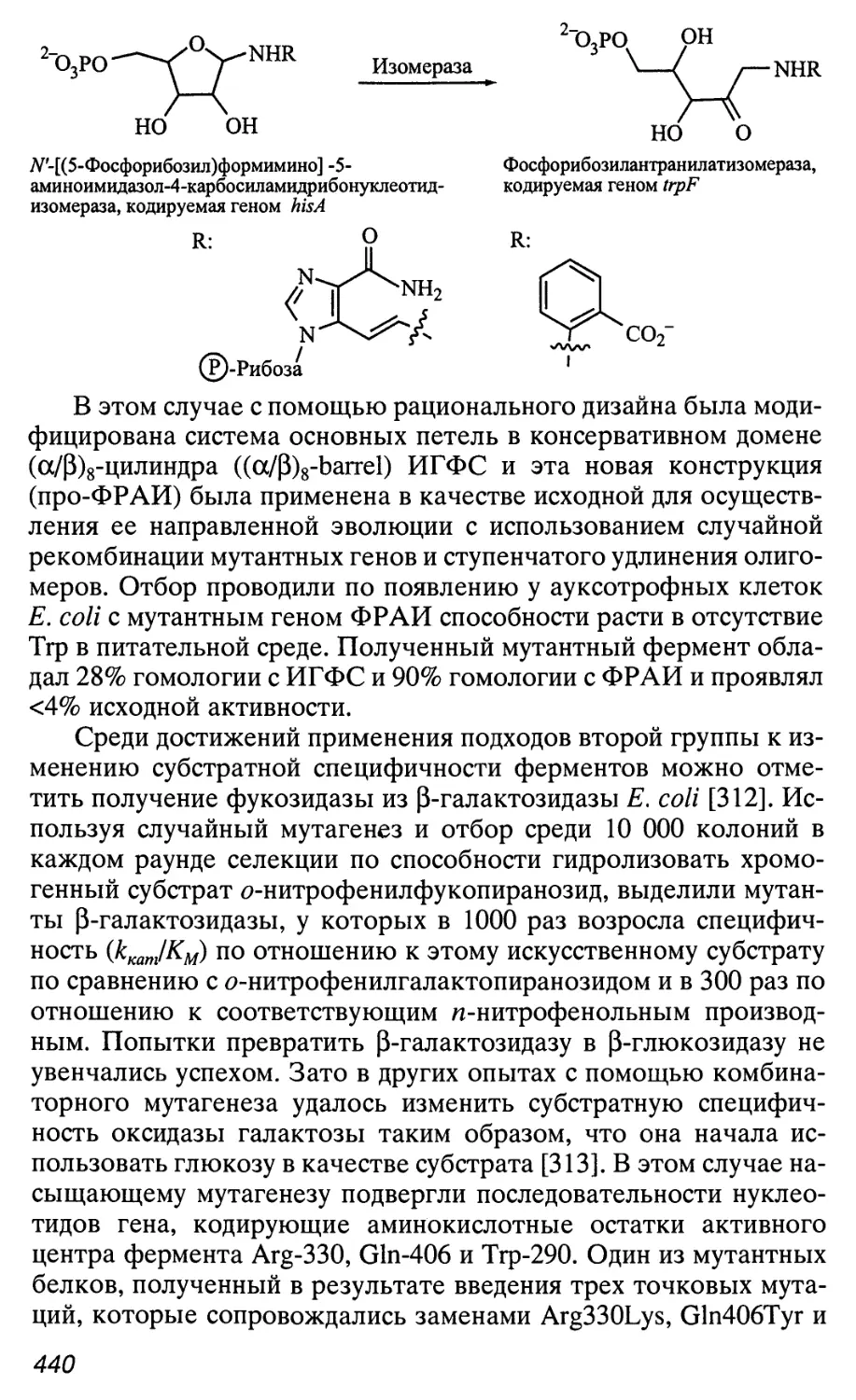



















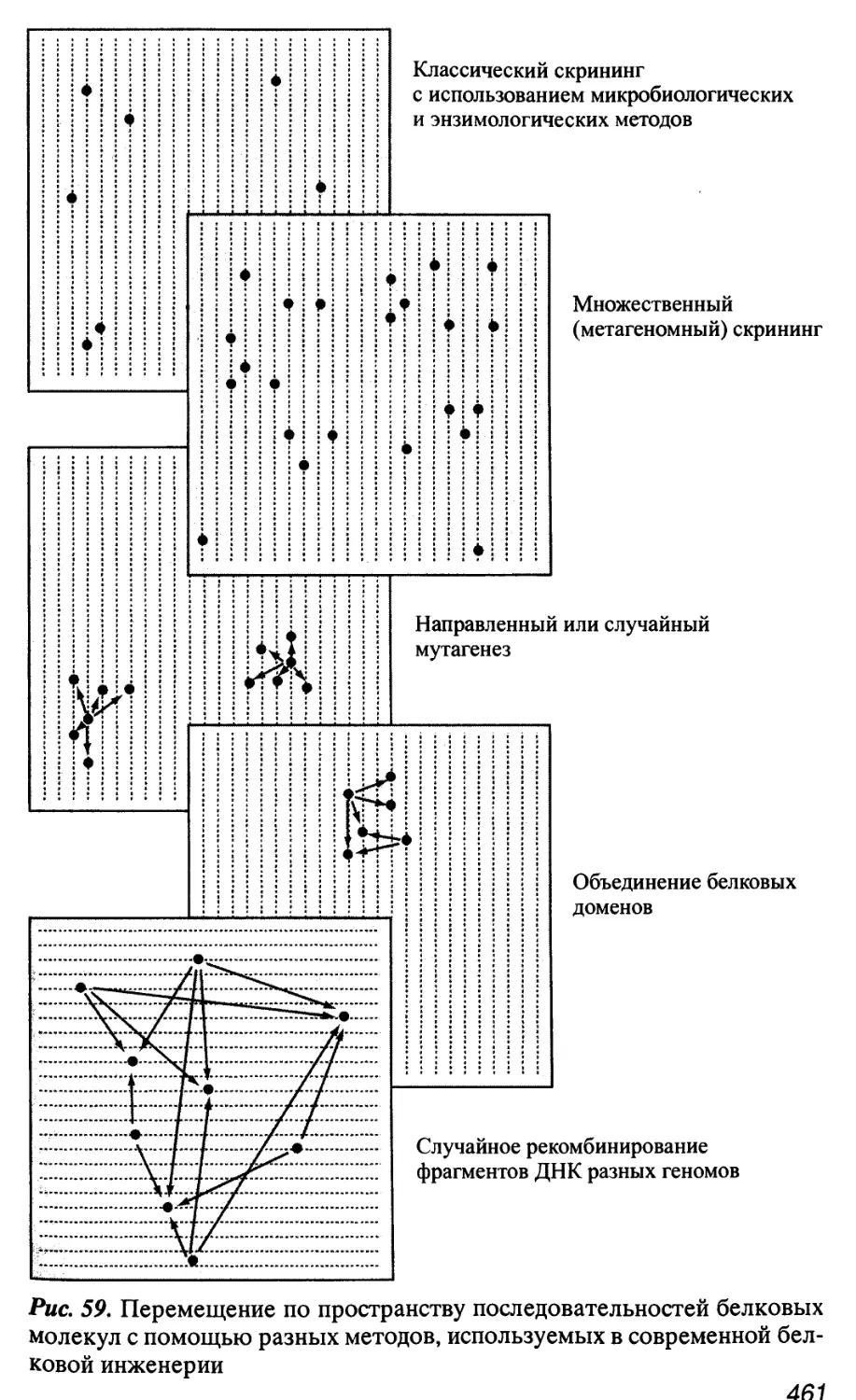




































































Автор: Патрушев Л.И.
Теги: общая генетика общая цитогенетика иммуногенетика эволюционное учение видообразование филогенез генная инженерия белковая инженерия
ISBN: 5-02-033278-X
Год: 2004




Л. И. Патрушев
генетические
системы
Генная и белковая инженерия
НАУКА
РОССИЙСКАЯ АКАДЕМИЯ НАУК
ИНСТИТУТ БИООРГАНИЧЕСКОЙ ХИМИИ
м. М.М.ШЕМЯКИНА И Ю.А.ОВЧИННИКОВА
RUSSIAN ACADEMY OF SCIENCES
SHEMYAKIN AND OVCHINNIKOV
INSTITUTE OF BIOORGANICAL CHEMISTRY
U. Pafrushev
rtificial
genetic
systems
Vol. ?
Genefic and profein engineering
MOSCOW NAUKA 2004
Л.И. Патрушев
скусственные
генетические
системы
Том 1
Генная и белковая инженерия
МОСКВА НАУКА 2004
УДК 575
ББК 28.04
П20
Ответственный редактор
академик А.И. МИРОШНИКОВ
Рецензенты:
член-корреспондент РАН ВТ. ДЕБА БОВ,
доктор биологических наук Г.В. ШПАКОВСКИЙ
На обложке гравюра М.К. Эшера "Спираль"
"В пространстве парит женский образ, напоминающий о спиралях срезае-
срезаемой фруктовой кожуры или полой, разбитой на куски скульптуры. Ощущение
глубины подчеркнуто грядой облаков, уменьшающихся и рассасывающихся по
мере приближения к горизонту"
(Комментарий М.К. Эшера)
Патрушев Л.И.
Искусственные генетические системы / Л.И. Патрушев; Ин-т био-
биоорганической химии им. М.М. Шемякина и Ю.А. Овчинникова РАН. -
М.: Наука. - ISBN 5-02-033278-Х
Т. 1: Генная и белковая инженерия / Отв. ред. А.И. Мирошников. -
М.: Наука, 2004. - 526 с. - ISBN 5-02-032893-6
В первом томе монографии рассмотрены современные методические подходы, ис-
используемые в генной и белковой инженерии для создания рекомбинантных ДНК и бел-
белков. Вначале обсуждаются основные принципы и методы генной инженерии, включая
клонирование ДНК, создание клонотек нуклеотидных последовательностей и систем их
экспрессии. Отдельная глава посвящена ПЦР и альтернативным способам амплификации
ДНК. Во второй части описаны принципы методов, используемых при реализации двух
основных направлений белковой инженерии: рационального дизайна и направленной эво-
эволюции белковых молекул, в том числе, направленного и случайного мутагенеза, лигиро-
вания синтезированных белков, фагового и рибосомного дисплеев и т.п.
Для научных работников, аспирантов и студентов, специализирующихся в области
химии и биологии.
ТП 2004-1-117
ISBN 5-02-033278-Х (общ.) © Л.И. Патрушев
ISBN 5-02-032893-6 (том 1) © Российская академия наук, 2004
© Оформление. Издательство "Наука",
2004
Предисловие
ответственного редактора
По мнению ведущих мировых экспертов, генная инженерия
будет широко использоваться человечеством для решения своих
глобальных проблем в XXI веке, и по масштабам применения бу-
будет сравнима с информационными технологиями, а также техно-
технологиями, применяемыми для производства новых материалов и
энергии. Внедрение новых методов, основанных на генной инже-
инженерии, несомненно, окажет большое влияние на многие сферы
деятельности человеческого общества, включая медицину, сель-
сельское хозяйство, нанотехнологии и промышленное производство.
В соответствии с прогнозами американских экономистов, только
генетические подходы обеспечат экономике США к 2025 г. 20%
прирост валового национального продукта, что составит около 2
триллионов долларов чистой прибыли.
Уже в настоящее время с помощью генно-инженерных мето-
методов удается получать трансгенные животные и растения, кото-
которые объединяют в своем геноме гены двух или нескольких видов
организмов. Это позволяет контролировать рост таких организ-
организмов, период их полового созревания и пищевую ценность, а
также использовать в качестве биореакторов для производства
важных биотехнологических продуктов. Создаются трансгенные
растения, успешно противостоящие насекомым-вредителям, ви-
вирусным инфекциям и гербицидам. Постепенно вводятся в сель-
сельскохозяйственную практику морозоустойчивые и засухоустойчи-
засухоустойчивые трансгенные растения. Все эти достижения и открывающие-
открывающиеся перспективы являются прочной основой для второй Зеленой
революции в мировом сельском хозяйстве.
Расширяются сферы использования рекомбинантных мик-
микроорганизмов, которые сегодня включают получение сыров и
вин в пищевой промышленности, а также тканей и бумаги. Ми-
Микроорганизмы, созданные руками генных инженеров, будут на-
находить все большее применение в производстве лекарственных
препаратов, для очистки окружающей среды от токсичных со-
соединений, добычи полезных ископаемых, в том числе меди, син-
теза веществ сложной структуры. Действительно, химическая
инженерия сегодняшнего дня претерпевает "биологизацию".
Все чаще в крупномасштабном химическом синтезе использу-
используются ферменты, модифицированные методами белковой инже-
инженерии, а принципы самосборки биологических макромолекул
успешно применяют для объединения химических веществ в
упорядоченные комплексы.
Современная генетика не обходит стороной и информацион-
информационные технологии. Благодаря ее успехам возникла новая научная
дисциплина - биоинформатика, которая помогает использовать и
интерпретировать обширную информацию, получаемую в моле-
кулярно-генетических исследованиях, особенно при анализе пер-
первичной структуры целых геномов высших организмов, а также
глобальных профилей экспрессии генов с применением биочи-
биочипов. Более того, формируется новое направление исследований
по созданию биокомпьютеров, которые в будущем могут соста-
составить сильную конкуренцию современным кремниевым техноло-
технологиям. Так, будущие биофотонные компьютеры на основе фотон-
фотонных процессоров, построенных с использованием биомолекул, по
своему быстродействию могут оставить далеко позади вычисли-
вычислительную технику сегодняшнего дня.
Современная молекулярная генетика вносит существенный
вклад в диагностику, предупреждение и лечение более 4000 за-
заболеваний человека. Хотя генотерапия, основанная на замене
поврежденных генов пациента на полноценные, в наши дни де-
делает еще свои первые шаги, есть все основания считать, что в
нынешнем веке этот подход станет одним из доминирующих в
восстановлении здоровья человека и предупреждении его забо-
заболеваний. Не исключено, что с помощью генно-инженерных ме-
методов в конце концов удастся существенно увеличить продол-
продолжительность жизни людей и повысить ее качество в преклон-
преклонном возрасте.
Несмотря на такие захватывающие перспективы, необходи-
необходимо помнить, что меч, вложенный в руки современного человека
развитием генной инженерии, является обоюдоострым. Необос-
Необоснованное использование генно-инженерных методов для изме-
изменения существующих биологических видов может нарушить
сложившийся экологический баланс в биосфере и иметь для че-
человечества непредсказуемые последствия. Чтобы избежать та-
такого рода неприятностей и даже экологической катастрофы
при конструировании генетически модифицированных видов
животных и растений, необходимо ясно представлять себе мо-
молекулярные механизмы, лежащие в основе биологических явле-
ний, сопровождающих формирование организмов и отдельных
генетических признаков. Это, в свою очередь, требует проведе-
проведения глубоких фундаментальных исследований, а также крити-
критического осмысления научной информации, полученной к насто-
настоящему времени, и объем которой продолжает непрерывно уве-
увеличиваться.
Решению этой задачи должна способствовать предлагаемая
вниманию читателей книга Л.И. Патрушева "Искусственные ге-
генетические системы", первый том которой посвящен методоло-
методологическим и методическим вопросам генной и белковой инжене-
инженерии. Внимательное прочтение книги несомненно даст заинтере-
заинтересованному читателю представление о современном состоянии
этих ключевых направлений молекулярной генетики и должно
помочь найти свой путь в море научной литературы, которое не-
неизбежно приходится осваивать перед проведением собственных
экспериментальных исследований.
Хотелось бы подчеркнуть, что научная работа Л.И. Патру-
Патрушева не ограничивается теоретическими изысканиями и литера-
литературной деятельностью. Эту сторону научной работы ему удается
плодотворно совмещать с экспериментальными исследованиями
генетических явлений. Вместе со своими сотрудниками автор мо-
монографии внес заметный вклад в биотехнологию, получив одним
из первых в России высокопродуктивный бактериальный штамм-
продуцент термостабильной ДНК-полимеразы Thermus aquaticus
и разработав эффективные методы ее очистки. В настоящее вре-
время этот фермент находит широкое применение во многих лабо-
лабораториях у нас в стране и за рубежом. Являясь членом Междуна-
Международного общества по исследованию тромбозов и гемостаза
(ISTH), Л.И. Патрушев разработал универсальную систему ал-
аллель-специфической ПЦР, которая, в частности, позволяет диа-
диагностировать мутации, ассоциированные с тромбофилиями, и с
ее помощью впервые осуществил анализ генетической структу-
структуры российской популяции в отношении распространенности дан-
данных генетических маркеров. В этом направлении на стыке гене-
генетики и медицины он продолжает активно работать и в настоящее
время. То, что Л.И. Патрушев знает о генетических методах не
понаслышке, придает его книге особую ценность, так как чита-
читатель может найти в ней много советов и рекомендаций (см., на-
например, раздел о полимеразной цепной реакции в первой части
книги), которые могут оказать практическую помощь при прове-
проведении его собственных исследований.
Выход в свет первого тома монографии Л.И. Патрушева "Ис-
"Искусственные генетические системы" - заметное явление в
7
современной русскоязычной генетической литературе. Книга
должна способствовать распространению современных генети-
генетических знаний в России и воспитанию нового поколения россий-
российских генетиков.
Л.И. Мирошников
Апрель 2004 г.
Будущим создателям
искусственных генетических систем
Предисловие автора
Жизнь подобна карточной игре,
в которую ты играешь, не зная правил.
ПЛ. Капица
Книга, предлагаемая вниманию читателей, основана на мате-
материале, который используется мной при чтении спецкурса по ис-
искусственным генетическим системам для студентов четвертого
года обучения кафедр биоорганической химии МГУ и физико-
химической биологии и биотехнологии Московского физико-
технического института. Как и следует из названия, монография,
в основном, посвящена изложению методических, а скорее даже
методологических достижений современной молекулярной гене-
генетики. При этом я умышленно сузил понятие термина "генная ин-
инженерия", сохранив за ним разделы по конструированию генов и
систем их экспрессии как таковых в рамках центрального посту-
постулата молекулярной биологии (ДНК<->РНК—>белок). Методичес-
Методические разделы таких направлений исследований, как клонирование
млекопитающих, трансгенез, антисмысловые и биочиповые тех-
технологии, ДНК-диагностика, генотерапия и тому подобные науко-
наукоемкие приложения генно-инженерных подходов, которые в
большей степени направлены на системное познание генетичес-
генетических явлений, предполагается объединить в дальнейшем под на-
названием "Нуклеиновые кислоты как объект и инструмент иссле-
исследований" во втором томе "Искусственных генетических систем".
К сожалению, правда жизни такова, что из-за беспрецедент-
беспрецедентной сложности генетических систем, занимаясь их исследованием
и конструированием, мы слабо представляем себе, что же проис-
происходит в таких системах на самом деле. Даже "простейшая" поли-
меразная цепная реакция, в которой одновременно участвуют
три миллиарда пар нуклеотидов ДНК человека, на мой взгляд,
является довольно темным ящиком, который будет еще долго
удивлять вдумчивого экспериментатора. Что же говорить о кло-
клонировании многоклеточных организмов, вокруг которого сего-
сегодня так много шумных дискуссий и больше поражений, чем по-
9
бед? Играя в эти опасные игры, мы напоминаем специалистов по
компьютерам, которых так много на Савеловском и Митинском
радиорынках г. Москвы. Они вполне успешно собирают работа-
работающую машину, смутно представляя себе, как устроены и функ-
функционируют ее отдельные составные части, в том числе, и цент-
центральный процессор. При этом, по большому счету, рискуют та-
такие специалисты значительно меньше, чем генетики. Успех же
коммерческой деятельности подобных фирм определен одно-
однозначным соответствием отдельных блоков компьютеров друг-
другу, что определяется международными стандартами, которым
строго следуют при конструировании отдельных плат расшире-
расширения, а также открытым характером архитектуры вычислитель-
вычислительных машин. Последнее свойство современных компьютеров,
предусмотренное пионерами их разработки, позволяет с помо-
помощью относительно независимых электронных модулей легко из-
изменять технические возможности базовых конструкций.
Несмотря на то, что все известные живые системы объедине-
объединены общими законами функционирования на молекулярном уров-
уровне, в отличие от компьютеров, им не свойственна открытость.
Перенос генов между организмами разных таксономических
групп в природе резко ограничен. А успешный обмен комплекса-
комплексами дифференцированных геномов в составе клеток, тканей или
органов еще более проблематичен. Конкретные живые системы
создаются и поддерживаются уникальными системами генов, ко-
которые составляют единое целое с контролируемым этими генами
фенотипами. Относится это не только к организмам разных ви-
видов, но и отдельным особям многоклеточных организмов одного
вида, неповторимый фенотип которых определяется многочис-
многочисленными аллельными вариантами генов популяции и генетичес-
генетическими особенностями онтогенеза. Поэтому наши попытки замены
одних генов на другие в геномах живых существ (а именно эти по-
попытки одухотворяют генную инженерию в широком смысле это-
этого термина) неизбежно нарушают систему гомеостаза трансген-
трансгенного организма, ослабляют его жизненные силы. С учетом таких
соображений я весьма скептически отношусь к возможности осу-
осуществления скачкообразной генно-инженерной эволюции суще-
существующего биологического вида многоклеточных организмов,
конечным результатом которой, на мой взгляд, может быть лишь
создание курьезных "домашних" животных и растений, нежизне-
нежизнеспособных в природных условиях. Давайте вспомним, что из-за
несопоставимой выживаемости в экстремальных условиях для ко-
космического полета всегда отбирают дворовых собак, а не породи-
породистых красавцев с искусно подогнанными аллелями.
10
Программой-максимумом генной инженерии является созда-
создание полностью искусственного организма на основе проектов,
разработанных в лаборатории, конструирование жизнеспособно-
жизнеспособного организма de novo. Но, как видно из второй части этой книги,
рациональный дизайн даже одной белковой молекулы с заранее
заданными свойствами сегодня является неразрешимой задачей.
Более плодотворными оказываются подходы, основанные на на-
направленной эволюции белков, когда среди огромного числа слу-
случайных вариантов полипептидных цепей с помощью изощрен-
изощренных методов отбора удается находить полипептиды с требуемой
активностью. Однако крупномасштабное использование таких
методов ограничивается комбинаторной проблемой белковой
инженерии: из-за чисто физических препятствий мы можем ра-
работать лишь в весьма ограниченном пространстве аминокислот-
аминокислотных (и нуклеотидных) последовательностей. Прорыв в этой об-
области можно ожидать в результате качественного изменения воз-
возможностей вычислительной техники, новым поколениям кото-
которой окажется по силам моделировать виртуальные (потенциаль-
(потенциально жизнеспособные) клетки, их ансамбли и многоклеточные ор-
организмы. Но не стоит забывать и о том, что в этом подходе нель-
нельзя полностью полагаться на аксиому молекулярного детерминиз-
детерминизма: даже после синтеза правильных молекул, их будет не просто
однозначно уложить в результате самосборки в задуманные
структуры. В сложных биологических системах всегда будут су-
существовать альтернативные варианты реализации потенциаль-
потенциальных возможностей объединения молекул, окончательный выбор
между которыми еще (очень) долго будем определять не мы.
Наглядным подтверждением правдоподобия вышеизложен-
вышеизложенных рассуждений является, на мой взгляд, то, что даже при нали-
наличии большого желания мне не удалось состыковать главы, пара-
параграфы, абзацы, предложения и слова этой книги именно так, как
этого хотелось изначально, в соответствии с четким планом, в
лучших традициях школьных сочинений. Идеи, воплощенные в
генной и белковой инженерии, проявили упорное стремление са-
самоорганизовываться, и стоило большого труда хотя бы отчасти
удержать их в задуманных рамках. Ведь автор и его произведение
являются неразрывной искусственной генетической системой, а
книга - это виртуальный плод функционирования макромолекул
в соответствии с законами молекулярной биологии и генетики,
далекими от наивного детерминизма. Поэтому, выставляя на суд
читателей своего искусственного, хотя и старательно причесан-
причесанного монстра, хочется рассчитывать на снисхождение строгих
критиков - в конце концов (и как всегда) во всем виноваты идеи.
11
Вот уже более десяти лет моим родным домом является Ин-
Институт биоорганической химии им. академиков М.М. Шемякина и
Ю.А. Овчинникова РАН и лаборатория биотехнологии ИБХ,
возглавляемая А.И. Мирошниковым, который взял на себя не-
нелегкий труд по научному редактированию рукописи. Хотелось
бы выразить большую благодарность за поддержку, оказанную
изданию книги, дирекции и Ученому совету ИБХ, а также лично
А.И. Мирошникову, которым в наше непростое время удается
создавать и сохранять творческую обстановку в Институте и ла-
лаборатории.
Как всегда, моим пастырем при подготовке рукописи к печа-
печати была заведующая научно-информационным отделом Инсти-
Института Т.И. Соркина, без реальной помощи и советов которой эта
книга никогда не увидела бы свет в конце тоннеля бесконечных
сомнений и дефицита времени. Кроме того, Т.И. Яковлева силь-
сильно облегчила мне жизнь при подготовке иллюстративного мате-
материала, а Г.А. Черняев работу с литературой. Им я хочу выразить
отдельную искреннюю признательность.
Глубокоуважаемые мной рецензенты В.Г. Дебабов и
Г.В. Шпаковский своим высокопрофессиональным анализом по-
помогли освободить рукопись от многих ошибок и неточностей, а
B.C. Микоян сумел сделать читаемым мой далекий от совершен-
совершенства английский язык. Не меньшую помощь в этом нелегком де-
деле усовершенствования рукописи оказала мне и редактор изда-
издания Н.М. Александрова. За это я чувствую себя перед ними в не-
неоплатном долгу.
Постоянные встречи со студентами на лекциях и семинарах
оставляют заметный след в моей жизни. Их свежий взгляд на ге-
генетику и неожиданные вопросы являются дополнительным силь-
сильным стимулом к продолжению самообразования. Хотелось бы
поблагодарить своих молодых коллег за внимательное отноше-
отношение к проблемам молекулярной генетики и пожелать им светло-
светлого будущего.
Отдельно хочется поблагодарить всех зарубежных коллег,
регулярно обеспечивавших меня научной литературой по обсуж-
обсуждаемым вопросам. Их поддержка помогла и постоянно помогает
не отставать от жизни.
Несмотря на неоценимую помощь большого коллектива про-
профессионалов, вся ответственность за фактическое содержание
книги лежит только на мне, и я заранее благодарен читателям за
конструктивную критику.
Часть I
Принципы генной инженерии
Само название раздела молекулярной генетики, именуемого ген-
генной инженерией, указывает на то, что в результате такого рода
исследований создаются искусственные генетические конструк-
конструкции, в которых отдельные части генов или гены целиком объеди-
объединяются в требуемой последовательности руками эксперимента-
экспериментатора - генного инженера. Это позволяет определять их взаимное
влияние и функциональное значение, а также проводить экспрес-
экспрессию генов в новом генетическом окружении. Таким образом, в
экспериментальных условиях имеет место обмен генетической
информацией как между отдельными генами организмов одного
и того же вида, так и между организмами разных таксономичес-
таксономических групп, что не происходит в природе из-за непреодолимых ба-
барьеров репродуктивной изоляции [1]. Тем не менее, практически
во всех методах современной генной инженерии фрагментарно (в
адаптированном виде) используются элементы природных моле-
кулярно-генетических механизмов.
Глава 1
Введение. Природные системы генов,
их организация и экспрессия
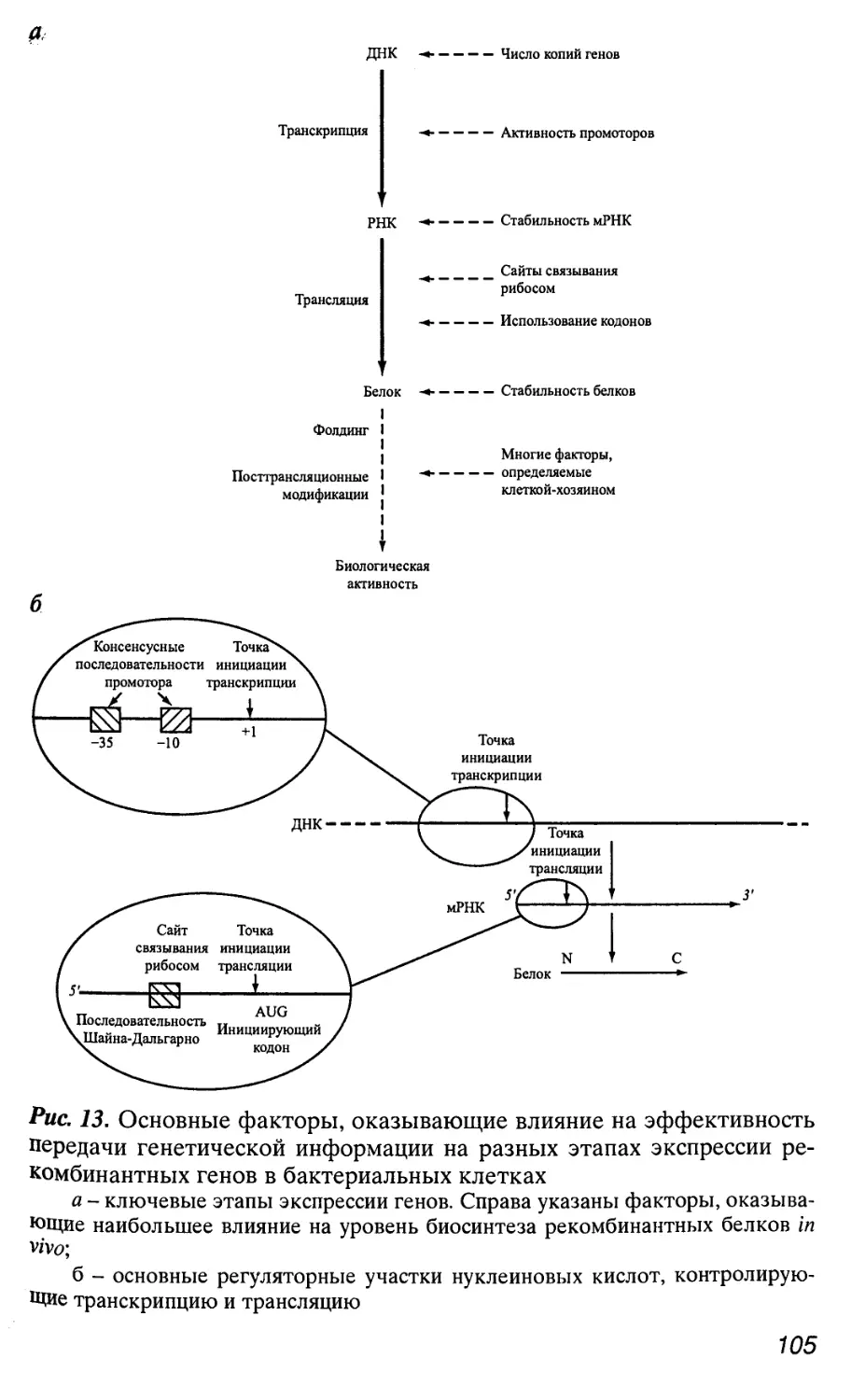
1.1. Гены и хромосомы
Генетическая информация о структуре отдельных белков и
нуклеиновых кислот у всех организмов заключена в молекулах
ДНК или РНК в виде последовательностей нуклеотидов, называ-
называемых генами [2-5]. Однако одной информации о структуре мак-
макромолекул, кодируемых генами, недостаточно для их функцио-
функционирования. Координированная работа (экспрессия) большого
числа генов возможна лишь благодаря наличию тонких регуля-
торных механизмов, определяющих место, время и уровень экс-
экспрессии конкретного гена или группы генов. Для того чтобы экс-
экспрессия гена была регулируемой, он должен содержать индиви-
индивидуальную (регуляторную) метку, по которой регуляторные ком-
13
поненты генетической системы клетки или организма могли бы
безошибочно оказать на ген необходимое воздействие. В соот-
соответствии с этим любой ген состоит из двух основных функцио-
функциональных частей (последовательностей нуклеотидов) - регуля-
торной и структурной. Регуляторная часть обеспечивает пер-
первые этапы реализации генетической информации, заключенной
в структурной части гена, которая, в свою очередь, содержит ин-
информацию о структуре конкретных белков или нуклеиновых
кислот. Поэтому размер гена складывается из размеров его
структурной и регуляторной частей. Однако определить протя-
протяженность гена не так просто, особенно в случае генов эукариот.
Отдельные элементы регуляторной области генов, например,
энхансеры, могут располагаться на значительном (>60 т.п.о.) рас-
расстоянии от структурной части гена - как перед ней, так и позади
нее или даже в ней самой. В самой же структурной части боль-
большинства эукариотических генов кодирующие последовательнос-
последовательности нуклеотидов (экзоны) перемежаются протяженными некоди-
рующими последовательностями (нитронами). Суммарный раз-
размер интронов у высших организмов как правило, многократно
превышает суммарный размер экзонов конкретных генов [6-8].
Уже исходя из этого факта, можно сделать вывод о том, что ге-
геном любого эукариотического организма содержит не только по-
последовательность нуклеотидов с генетической информацией о
белках и нуклеиновых кислотах, но и большое количество после-
последовательностей нуклеотидов, не несущих такой информации.
Однако помимо интронов в геноме эукариот есть и другие не-
кодирующие последовательности нуклеотидов, главным обра-
образом, различные повторяющиеся последовательности [9-10]. По-
Поэтому общая длина некодирующих последовательностей нуклео-
нуклеотидов в геномах большинства эукариот в десятки раз превышает
длину кодирующих последовательностей [11]. Не до конца выяс-
выясненные и очень большие размеры генов эукариот, к тому же рас-
расположенных в геноме среди многочисленных некодирующих по-
последовательностей нуклеотидов, создают значительные труднос-
трудности для изучения их структуры и функционирования in vivo.
Как у прокариотических, так и у эукариотических организ-
организмов все гены располагаются группами на отдельных молекулах
ДНК, которые при участии белков и других макромолекул кле-
клеток организуются в хромосомы. Зрелые клетки зародышевой ли-
линии (гаметы - яйцеклетки, спермин) многоклеточных организ-
организмов содержат по одному (гаплоидному) набору хромосом орга-
организма. У диплоидных (полиплоидных) организмов, клетки кото-
которых содержат по одному (несколько) набору хромосом каждого
14
из родителей, одинаковые хромосомы получили название гомо-
гомологичных хромосом, или гомологов. Гомологичными являются и
одинаковые хромосомы разных организмов одного биологичес-
биологического вида. Гены и некодирующие последовательности нуклеоти-
дов, заключенные в хромосомах ядер клеток, представляют
большую (основную) часть генома организма. Кроме того, в ге-
геном организма входят и внехромосомные генетические элемен-
элементы, которые во время митотического цикла воспроизводятся не-
независимо от хромосом ядер [12-14]. Так, митохондрии грибов и
млекопитающих содержат менее 1 % всей ДНК, тогда как у поч-
почкующихся дрожжей Saccharomyces cerevisiae количество суммар-
суммарной митохондриальной ДНК составляет до 20% всей ДНК клет-
клетки. ДНК пластид растений составляет от 1 до 10% суммарного
количества их ДНК.
Поскольку гены, входящие в состав отдельных хромосом, на-
находятся в одной молекуле ДНК, они образуют отдельную генети-
генетическую группу сцепления и в отсутствие рекомбинации совмест-
совместно передаются от родительских клеток дочерним. Остаются до
конца не понятыми физиологическое значение распределения ге-
генов по отдельным хромосомам и природа факторов, определяю-
определяющих число хромосом в геномах различных эукариот. Например,
невозможно объяснить эволюционные механизмы появления
большого числа хромосом у конкретных организмов только ог-
ограничениями, накладываемыми на максимальный размер моле-
молекул ДНК, входящих в состав этих хромосом. Так, геном амери-
американской амфибии Amphiuma содержит в ~30 раз больше ДНК,
чем геном человека, и вся ДНК заключена только в 28 хромосо-
хромосомах, что вполне сопоставимо с кариотипом человека D6 хромо-
хромосом). И даже самая маленькая из этих хромосом больше самых
крупных хромосом человека. Остаются неизвестными и факто-
факторы, ограничивающие верхний предел числа хромосом у эукариот.
Например, у бабочки Lysandra nivescens диплоидный набор со-
составляет 380-382 хромосомы, и нет основания считать, что это
значение является максимально возможным.
По-видимому, большинство особенностей структурной и функ-
функциональной организации генома должны обеспечивать надежность
его функционирования, т.е. точность передачи генетической ин-
информации от родительских клеток дочерним на протяжении мно-
многих клеточных генераций и правильную работу генов. Поэтому
можно предполагать, что эти особенности имеют, прежде всего,
отношение к обеспечению надежности передачи и внутриклеточ-
внутриклеточной реализации генетической информации, проявляющейся в упо-
упорядоченной во времени и безошибочной экспрессии генов [15].
15
1.2. Геном
Термином "геном" описывают полную совокупность генов и
некодирующих последовательностей живого организма, отдель-
отдельной клетки, клеточной органеллы или вируса. Такое определе-
определение указывает на то, что геном живого организма составляет вся
его ДНК. Структурная организация генома является фундамен-
фундаментальным таксономическим признаком, лежащим в основе совре-
современной систематики живых существ. Несмотря на то, что эволю-
эволюционные отношения между группами организмов до конца не вы-
выяснены и являются предметом оживленных дебатов, в соответст-
соответствии со структурной организацией генома все живые организмы в
настоящее время разделяют на три домена: Archaea (археи, архе-
бактерии), Bacteria (эубактерий) и Еикагуа (эукариоты) [16]. Ге-
Геном архей и эубактерий не заключен в ядро, ограниченное ядер-
ядерной мембраной, и его воспроизведение не сопровождается мито-
митозом. Клетки эукариот содержат оформленное ядро, и митоз за-
завершает редупликацию их генома. Археи представляют третью
крупную ветвь эволюционного древа и занимают промежуточ-
промежуточное положение между эукариотами и эубактериями, в том числе,
по структурно-функциональной организации их генома и, в част-
частности, первичной структуре генов рРНК. Характерная структура
генома архей, эубактерий и эукариот накладывает отпечаток на
морфологические, физиологические, биохимические и генетиче-
генетические особенности этих организмов, которые в конечном счете
определяются генетической информацией, заключенной в их ге-
геномах и реализующейся через экспрессию соответствующих ге-
генов. Таким образом, можно без преувеличения сказать, что
структура генома лежит в основе всех тех внутренних и внешних
проявлений жизнедеятельности любого организма, которые оп-
определяют его положение в иерархии живых существ, населяю-
населяющих нашу планету.
Изучение генома направлено, прежде всего, на познание за-
законов его функционирования. Время, в котором мы живем, рас-
рассматривается в молекулярной биологии как постгеномная эра.
Действительно, исследования последних десяти лет принесли зна-
знания о полной первичной структуре геномов многих бактерий,
дрожжей, круглого червя нематоды, дрозофилы, арабидопсиса.
Приближается к окончательному завершению расшифровка по-
последовательности нуклеотидов генома человека [17, 18], а также
одного из важнейших объектов его хозяйственной деятельнос-
деятельности - риса [19, 20]. Близки к полному завершению и другие не ме-
менее глобальные проекты исследования генома эукариотических
16
организмов, в том числе геномов африканской шпорцевой ля-
лягушки Xenopus leavis и мыши. На фоне этих блестящих достиже-
достижений ясно обозначился разрыв между формальными знаниями
первичной структуры генома эукариот и полным отсутствием ка-
какой-либо информации о функциях большинства его генов. Ост-
Острое ощущение этого пробела в знаниях привело к созданию но-
нового направления в молекулярно-генетических исследованиях -
геномики. В отличие от классической и молекулярной генетики,
которые в основном заняты изучением отдельных генов, геноми-
геномика пытается одновременно рассматривать все гены организма в
качестве единой динамической системы, компоненты которой,
формируя генные сети, взаимодействуют друг с другом в прост-
пространстве и во времени. При этом функциональная геномика зани-
занимается поиском биологических функций последовательностей
нуклеиновых кислот, которые в результате компьютерного ана-
анализа отнесены к категории генов, исследованием функциональ-
функциональных взаимодействий между генами (в том числе, продуктами их
экспрессии) в норме и при патологических состояниях организма.
Функциональная геномика в широком смысле включает в себя
многие традиционные и новые молекулярно-генетические дис-
дисциплины, и в своих исследованиях использует большое количест-
количество экспериментальных приемов. В настоящее время интенсивно
развиваются методы анализа транскриптома и протеома (соот-
(соответственно совокупностей всех видов РНК или белков клетки
или организма), основанные на технологиях биологических мик-
микрочипов, генных нокаутов, антисмысловых РНК, рибозимов и
других современных подходах, принципы которых будут рассма-
рассматриваться во втором томе монографии. По-видимому, програм-
программой-максимумом функциональной геномики можно считать ус-
установление функциональных отношений между всеми взаимо-
взаимодействующими генами организма, познание фундаментальных
отношений между генотипом и фенотипом.
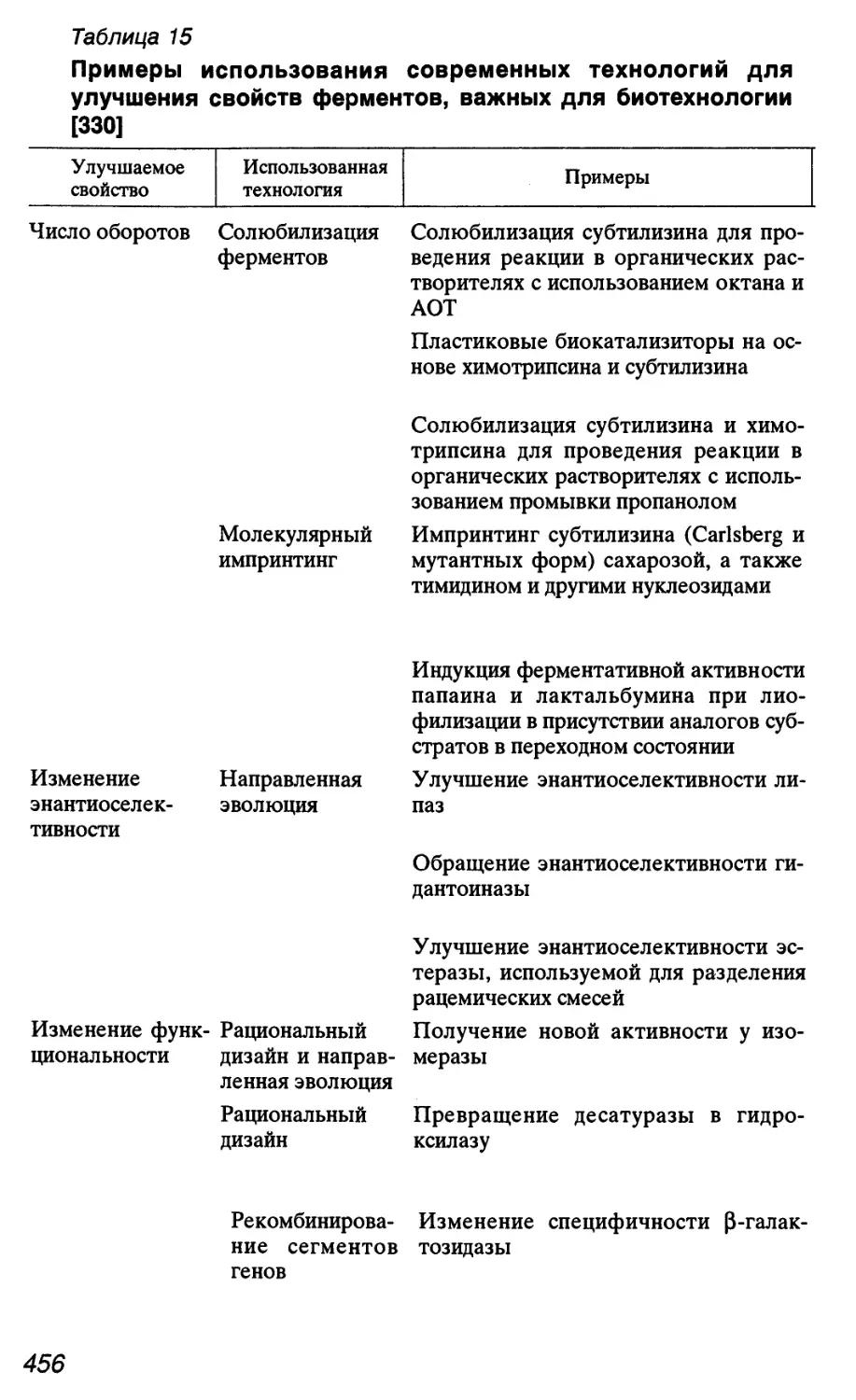
1.2.1. Размеры геномов
Как видно из табл. 1, размеры геномов организмов разных
видов значительно отличаются друг от друга. При этом часто не
наблюдается корреляции между уровнем эволюционной сложно-
сложности биологического вида и размером его генома.
Суммарное количество ДНК в гаплоидном геноме принято
обозначать латинским символом С. В 1978 г. Т. Кавалье-Смит
описал в качестве парадокса тот факт, что у эукариот транскри-
транскрибируется лишь незначительная часть последовательностей
17
Таблица 1
Размер гаплоидного генома у некоторых групп организмов и их
отдельных представителей
Группа или представитель организмов
Мелкие вирусы
Микоплазмы
Mycoplasma genitalium G-37
Mycoplasma mycoides subsp. mycoides SC PG1
Эубактерии
Bacillus subtilis 168
Escherichia coll K-12
Nostoc punctiforme ATCC 29133
Археи
Methanococcus jannaschii
Halobacterium salinarum
Грибы
почкующиеся дрожжи Saccharomyces cerevisiae
пекарские дрожжи Schizosaccharomyces pombe
Насекомые
Drosophila melanogaster
Моллюски
Костистые рыбы
Амфибии
бесхвостые
шпорцевая лягушка Xenopus leavis
хвостатые
Рептилии
Птицы
Млекопитающие
человек Homo sapiens
мышь Mus musculus
Растения
голосеменные
покрытосеменные
рис Oryza sativa
лилия Lilium longiflorum
Простейшие
Amoeba dubia
Примечание. * Для групп организмов указан средний
Размер генома, п.о.*
1,0-104
1,6-106
0,58- 106
1,28-106
2,0 • 106
4,2 • 106
4,6 • 106
1,0-107
2,1 • 106
1,7 • 106
4,0 • 106
4,7 • 107
1,2 -107
1,4 • 107
2,3 • 109
1,8 • 108
1,6 109
1,4 109
2,7 • 109
3,1 • 109
1 Г\
3,6 • 10ю
1,5 • 109
1,2-109
2,6 • 109
3,0 • 109
3,5 • 109
1,6-1010
2,7 • 1010
4,7 • 108
1,8- 10"
6,7-1011
размер генома.
18
нуклеотидов генома (~3% генома человека). В соответствии с
этим необъясненный до настоящего времени феномен значи-
значительной избыточности генома эукариот в отношении некодиру-
ющих последовательностей нуклеотидов известен в генетике под
названием "парадокса С" (C-value paradox) [10,21, 22]. По мнению
автора книги, эволюционное включение избыточных последова-
последовательностей нуклеотидов в исходный геном-предшественник ста-
стабилизировало заключенную в нем генетическую информацию,
что, в свою очередь, создало необходимые условия для возникно-
возникновения многоклеточности в природе [23].
1.2.2. Геном вирусов
По определению X. Френкель-Конрата, «вирусы - это части-
частицы, состоящие из одной или нескольких молекул ДНК или РНК,
обычно (но не всегда) окруженных белковой оболочкой; вирусы
способны передавать свои нуклеиновые кислоты от одной клет-
клетки-хозяина к другой и использовать ее ферментативный аппарат
для осуществления своей внутриклеточной репликации путем на-
наложения собственной информации на информацию клетки-хозя-
клетки-хозяина; иногда вирусы могут обратимо включать свой геном в геном
хозяина (интеграция), и тогда они либо ведут "скрытое существо-
существование", либо так или иначе трансформируют свойства клетки-хо-
клетки-хозяина» [24]. В приведенном определении отмечены характерные
особенности жизненного цикла вирусов, которые находят отра-
отражение в организации их генома. Вирусы являются внутриклеточ-
внутриклеточными паразитами и используют для своего размножения
белоксинтезирующий аппарат клетки-хозяина. Жизненный цикл
вируса начинается с проникновения внутрь клетки. Для этого он
связывается со специфическими рецепторами на ее поверхности
и либо вводит свою нуклеиновую кислоту внутрь клетки, остав-
оставляя белки вириона на ее поверхности, либо проникает целиком в
результате эндоцитоза. В последнем случае после проникнове-
проникновения вируса внутрь клетки следует его "раздевание" - освобожде-
освобождение геномных нуклеиновых кислот от белков оболочки, что де-
делает вирусный геном доступным для ферментных систем клетки,
обеспечивающих экспрессию генов вируса.
После проникновения вируса в клетку может происходить его
размножение, часто сопровождаемое гибелью самой клетки (виру-
(вирулентный путь развития), либо вирус может длительное время су-
существовать внутри клетки, внешне себя не проявляя (латентная
инфекция). В этом случае его геном встраивается в геном клетки-
хозяина и реплицируется вместе с ним или находится во внехромо-
19
сомном состоянии. После проникновения вирусной нуклеиновой
кислоты в клетку, заключенная в ней генетическая информация
должна быть расшифрована (прочитана) генетическими система-
системами клетки-хозяина и использована для синтеза компонентов вирус-
вирусных частиц. Поскольку для своего размножения вирусы использу-
используют главным образом, ферментные системы клетки-хозяина, их ге-
геном характеризуется относительно малыми размерами и кодирует
структурные белки вирионов, а также белки и ферменты, которые
перестраивает метаболизм клетки для нужд размножения вируса,
делая процесс репликации вирусов максимально эффективным.
Геном вирусов может быть представлен одноцепочечными или
двухцепочечными ДНК или РНК. Кроме того, все гены вирусов
могут быть заключены в одной молекуле нуклеиновой кислоты
или разделены на несколько блоков (вирусных хромосом). Напри-
Например, у реовирусов геном представлен двухцепочечной РНК и со-
состоит из десяти сегментов. Геномы вирусов, содержащих одноце-
почечную РНК, также могут быть либо цельными (например, у
ретровирусов), либо сегментированными (например, у ортомиксо-
вирусов или аренавирусов). Геном РНК-содержащих вирусов пред-
представлен только линейными молекулами РНК.
Все известные ДНК-содержащие вирусы позвоночных имеют
геном, заключенной в одной "хромосоме", линейной или кольце-
кольцевой, одно- или двухцепочечной молекуле ДНК. У некоторых виру-
вирусов, например, вируса гепатита В, геном представлен кольцевой
ковалентно замкнутой молекулой двухцепочечной ДНК, в обеих
цепях которой в разных местах обнаружены одноцепочечные уча-
участки. У аденоассоциированных вирусов комплементарные цепи
ДНК находятся в различных вирусных частицах.
Между понятиями "вирус" и "отдельная вирусная частица"
или "вирион" имеются существенные различия. Вириону прису-
присущи конкретные физические и биохимические свойства, которые
составляют фенотипические признаки, определяемые последова-
последовательностью нуклеотидов генома, заключенного в данной вирус-
вирусной частице, его генотипом. В отличие от этого, вирус обладает
целым рядом дополнительных свойств (например, особенностя-
особенностями цикла размножения), которые проявляются лишь после того,
как он образует неразрывное целое с клеткой-хозяином. Поэто-
Поэтому понятие вируса нельзя редуцировать до отдельной вирусной
частицы. Оно должно включать в себя и биологические особен-
особенности существования вируса в биосфере. В соответствии с этим
геном вируса (как это и следует из определения генома), не мо-
может рассматриваться как последовательность нуклеотидов гене-
генетического материала, заключенного в индивидуальном вирионе.
20
1.2.3. Геном эубактерий
Электронно-микроскопическое изучение срезов бактериаль-
бактериальных клеток в разных условиях и более ранние исследования бак-
бактерий с помощью светового микроскопа продемонстрировали
компактное распределение ДНК в бактериальной клетке. По-
Поскольку такие структуры отдаленно напоминали ядра эукариот,
они получили название нуклеоидов, или ДНК-плазмы [25]. Эти
термины подчеркивают генетические функции нуклеоида, но в
то же время и существенные морфологические отличия от обыч-
обычных интерфазных ядер эукариот, прежде всего, отсутствие ядер-
ядерной оболочки, которая бы отделяла гены бактерии от окружаю-
окружающей их цитоплазмы. Исследование бактериальных клеток с по-
помощью электронной микроскопии в мягких условиях без предва-
предварительной химической фиксации показало, что нуклеоиды пред-
представлены в виде диффузно окрашенных областей, свободных от
рибосом. При этом вытянутые участки ДНК на внешней части
нуклеоидов направлены в окружающую цитоплазму. С помощью
специфических антител установлено, что молекулы РНК-поли-
меразы, ДНК-топоизомеразы I и гистоноподобного белка HU ас-
ассоциированы с нуклеоидами. Вытянутые участки ДНК по пери-
периферии нуклеоидов обычно интерпретируют как сегменты бакте-
бактериальной хромосомы, вовлеченные в транскрипцию. Полагают,
что эти участки состоят из петель ДНК бактериальной хромосо-
хромосомы, которые в зависимости от физиологического состояния
клетки находятся в транскрипционно-активном состоянии или
втягиваются внутрь нуклеоидов при подавлении транскрипции.
Таким образом, нуклеоид бактериальных клеток не является
статическим внутриклеточным образованием или компартмен-
том, который можно четко определять морфологически. Напро-
Напротив, во время различных фаз роста бактериальных клеток нукле-
нуклеоид непрерывно меняет форму, что, по-видимому, сопряжено с
транскрипционной активностью определенных бактериальных
генов. Так же, как и в хромосомах эукариот, ДНК нуклеоида ас-
ассоциирована со многими ДНК-связывающими белками, в частно-
частности, с гистоноподобными белками HU, H-NS и IHF, а также топо-
изомеразами, которые оказывают большое влияние на функцио-
функционирование бактериальных хромосом и их внутриклеточную ком-
пактизацию. Однако детальные молекулярные механизмы кон-
конденсации бактериальной ДНК с образованием лабильных "ком-
пактосом" (по аналогии со стабильными нуклеосомами эукари-
эукариот) пока неизвестны. В последнее время возрастает интерес к
бактериальному так называемому LP-хроматину (low protein
21
chromatin), для которого характерно относительно низкое содер-
содержание белкового компонента. Аналогичный LP-хроматин обна-
обнаруживают у вирусов, в митохондриях, пластидах и у динофлагел-
лят (жгутиконосцев). Следовательно, этот тип структурной орга-
организации генетического материала претендует на универсаль-
универсальность и ассоциирован с определенными формами регуляции экс-
экспрессии генов, свойственными прокариотическим организмам.
В последние годы наблюдается значительный прогресс в иссле-
исследовании первичной структуры бактериальных хромосом. Опреде-
Определена полная последовательность нуклеотидов хромосом паразити-
паразитических бактерий: микоплазмы Mycoplasma genitalium, M. pneumonia,
и Haemophilus influenzae. В 1997 г. усилиями интернациональных
коллективов ученых была определена полная первичная структура
хромосом Escherichia coli и Bacillus subtilis длиной соответственно в
~4,6 и ~4,2 млн п.о. соответственно [26, 27]. Общее количество бак-
бактериальных геномов, секвенированных к настоящему времени,
приближается к 100. Большинство этих геномов содержит ~1000
(±15%) открытых рамок считывания (ОРС) на 1 млн п.о. бактери-
бактериальной ДНК. При этом обнаружены консерватизм в порядке рас-
расположения генов рибосомных белков и РНК, а также примеры го-
горизонтального переноса генов в процессе эволюции. Вне зависимо-
зависимости от занимаемой экологической ниши и филогенетического по-
положения представителей бактерий, значительная часть их генома
C0-50%) представлена генами, уникальными для конкретного ми-
микроорганизма, или генами с неизвестными функциями. При этом
разнообразие генов выше у бактерий, которые существуют в изме-
изменяющихся условиях окружающей среды.
1.2.4. Геном архей
Домен (надцарство Archaea) состоит их трех царств -
Euryarchaeota, которое составляют метаногены, экстремальные
галофилы и некоторые гипертермофилы, Crenarchaeota, вклю-
включающего гипертермофилов и нетермофильные микроорганиз-
микроорганизмы, а также Korarchaeota (слабо изученная группа микроорганиз-
микроорганизмов, обитающих в термальных источниках, выделенная на осно-
основании особенностей первичной структуры 16S РНК). Геном ар-
архей представлен кольцевыми ковалентно замкнутыми молекула-
молекулами ДНК, размер которых приближается к таковому эубактерий
[28]. У архей обнаружены плазмиды, в том числе, и мегаплазми-
ды, размер которых может превышать 700 т.п.о., причем мега-
плазмиды могут содержать жизненно важные гены.
Геномы содержат три типа генов: 1) типично бактериальные
22
гены, 2) гены, подобные эукариотическим, и 3) гены, специфич-
специфичные для этих микроорганизмов. К первой группе относятся гены,
обеспечивающие метаболизм, биосинтез небольших молекул,
транспорт и регуляторные процессы. Гены, которые более похо-
похожи на эукариотические, вовлечены в транскрипцию, трансляцию
и метаболизм ДНК. Гены, типичные для архей, контролируют
биосинтез жгутиков и уникальных липидов клеточных стенок, а
также метаногенез. Недавние исследования показали, что между
бактериями и археями имеет место горизонтальный перенос ге-
генов в обоих направлениях.
У архей, по аналогии с эубактериями, многие гены организо-
организованы в кластеры (опероны), транскрибируемые с одного промо-
промотора, которые транслируются в виде длинных некэпированных
мРНК, содержащих короткие поли(А)-последовательности эу-
бактериального типа. Однако структура самих промоторов резко
отличается от эубактериальной и напоминает таковую промото-
промоторов РНК-полимеразы I эукариот [29]. В отличие от эубактерии и
эукариот архей содержат, как правило, одну копию гена рРНК.
Интроны у архебактерий, в том числе и мобильные интроны, об-
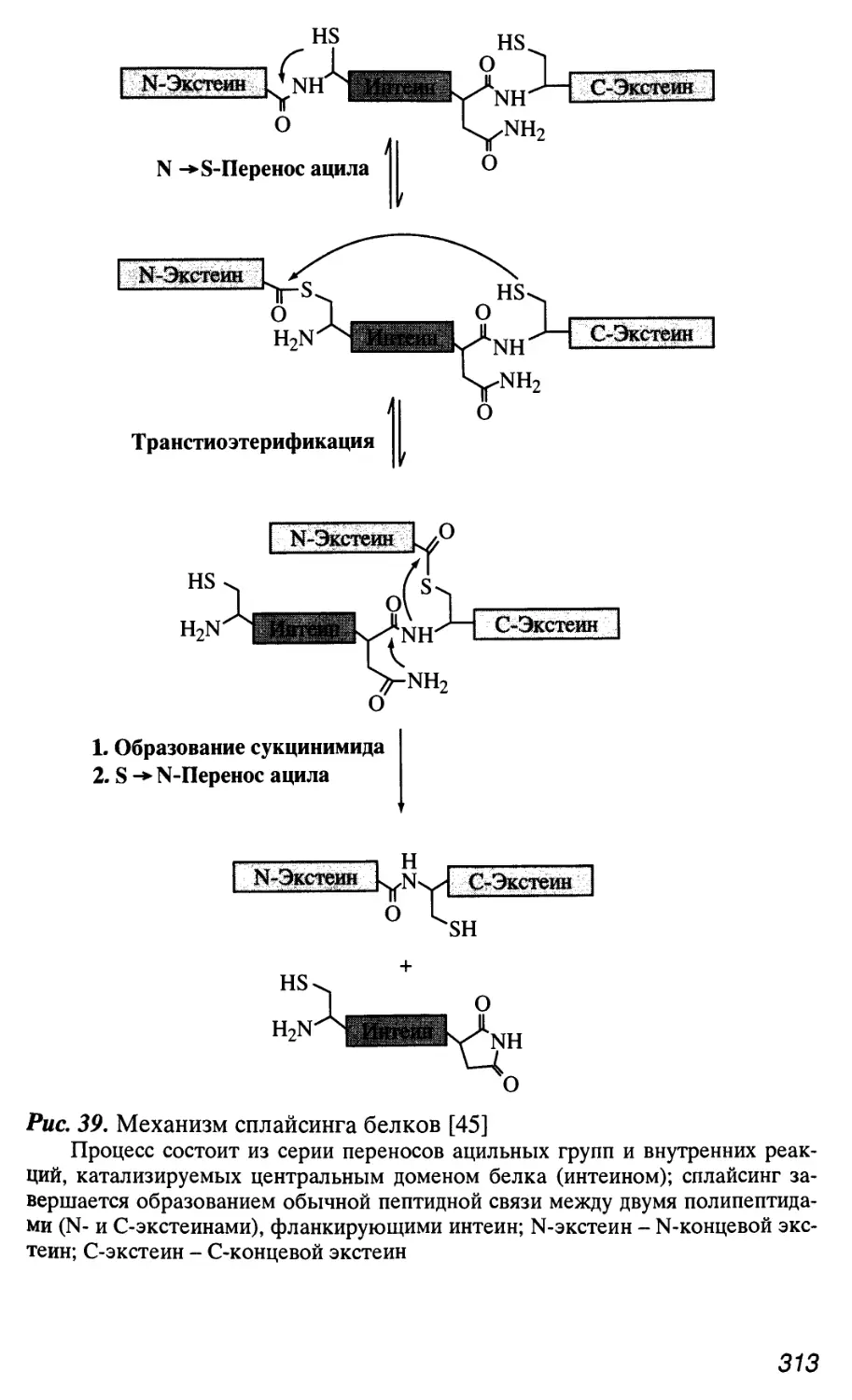
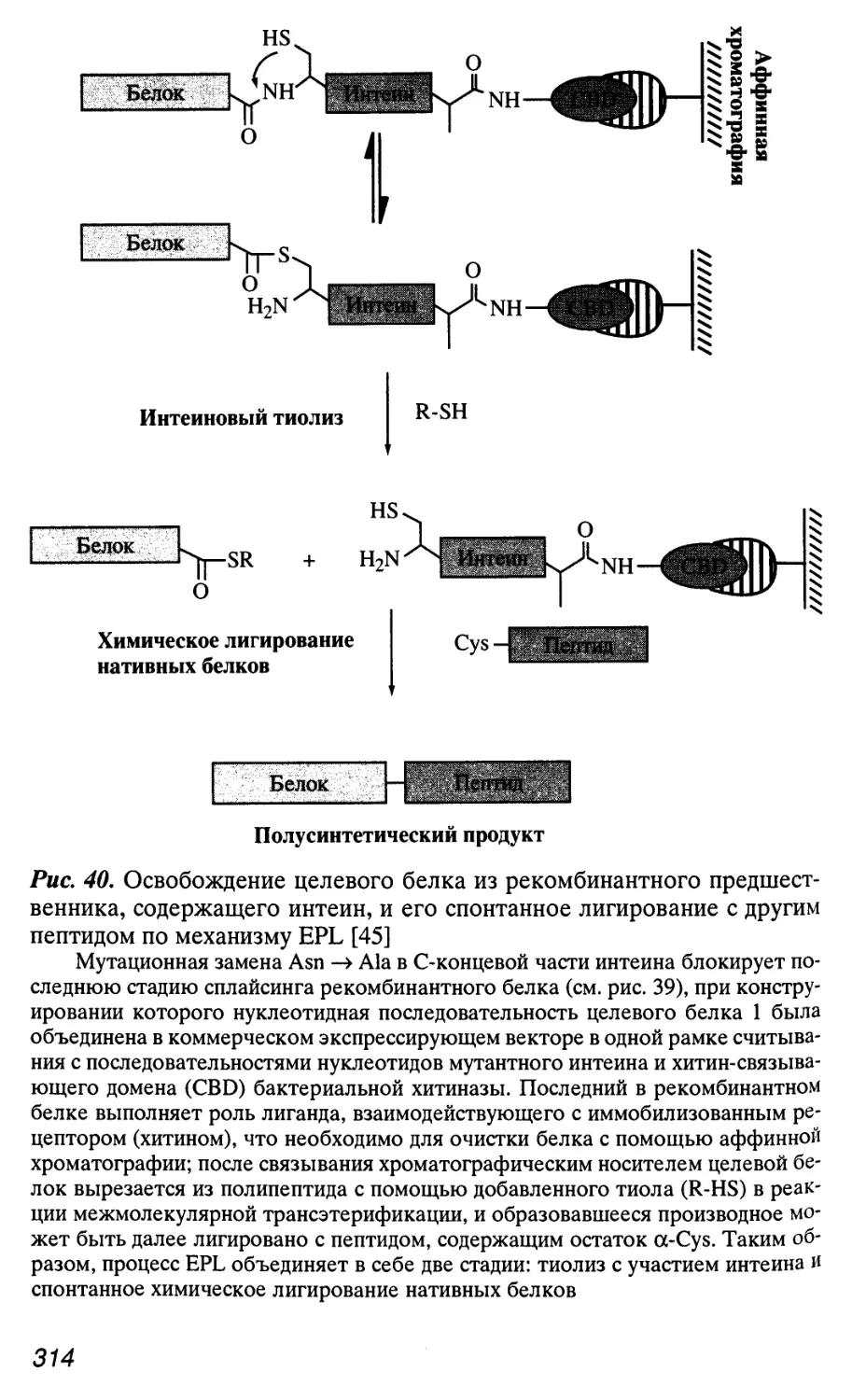
обнаружены только в генах стабильных РНК (рРНК и тРНК), тог-
тогда как присутствие интеинов в белках (до трех в одном полипеп-
полипептиде) является обычным явлением.
1.2.5. Геном эукариот
В отличие от прокариот основная часть генома эукариот на-
находится в специальном клеточном компартменте (органелле), по-
получившем название ядра, а значительно меньшая часть - в мито-
митохондриях, хлоропластах и других пластидах. Так же, как и у про-
прокариот, информационной макромолекулой генома эукариот яв-
является ДНК, которая неравномерно распределена по хромосо-
хромосомам в виде комплексов с многочисленными белками. Эти ДНК-
белковые комплексы эукариот получили название хроматина.
На протяжении клеточного цикла хроматин претерпевает высо-
коупорядоченные структурные преобразования в виде последо-
последовательных конденсаций-деконденсаций. В соматических клетках
при максимальной конденсации в метафазе митоза эти преобра-
преобразования сопровождаются формированием видимых в микроскопе
метафазных хромосом. Как морфология метафазных хромосом,
так и их число, являются уникальными характеристиками вида.
Совокупность внешних признаков хромосомного набора эукари-
эукариот получила название кариотипа. Эти признаки широко исполь-
используются в биологической систематике.
23
Последовательности нуклеотидов эукариотического генома.
Геном эукариот составляют уникальные и повторяющиеся по-
последовательности нуклеотидов. Содержание уникальных после-
последовательностей в геноме, определенное на основании кинетики
реассоциации фрагментированной ДНК, варьирует у разных ор-
организмов, и их доля составляет 15-98% от всей ДНК. Несмотря
на то, что во фракцию уникальных последовательностей попада-
попадают многие структурные гены, большая часть этих последова-
последовательностей является некодирующей и обычно не заключает в се-
себе генетической информации в общепринятом значении этого
термина: не кодирует функционально значимые полипептидные
цепи или РНК. Примером таких уникальных последовательнос-
последовательностей являются интроны, появление которых в геноме эукариот
пока не нашло своего объяснения [6].
Не менее загадочным с эволюционной точки зрения остается
и феномен появления в геноме многоклеточных организмов
большого количества некодирующих повторяющихся последова-
последовательностей. Такие повторы представлены в гаплоидном геноме
эукариот множественными копиями. В современной классифика-
классификации повторов принято различать часто повторяющиеся последо-
последовательности, число которых превышает 105 на гаплоидный ге-
геном, и умеренно повторяющиеся, представленные 10-Ю4 копия-
копиями. Представителем первых является сателлитная ДНК, кото-
которая состоит из коротких тандемных повторов длиной 1-20 п.о.,
организованных в длинные блоки [30]. Содержание сателлитной
ДНК в геноме эукариот может достигать 5-50% от суммарного
количества ДНК. Микро- (от 1 до 4 п.о. в основном повторяю-
повторяющемся блоке) и минисателлитные (с большим числом п.о. в ин-
индивидуальном повторе) ДНК характеризуются высокой вариа-
вариабельностью по числу копий в геномах организмов даже одного
вида и в ряде случаев обладают генетической нестабильностью
как в норме, так и при некоторых патологических состояниях ор-
организмов. Благодаря этому свойству мини- и микросателлиты ча-
часто называют тандемными повторами с изменяющимся числом
копий VNTR (variable number of tandem repeats).
Другой тип повторов - диспергированные повторяющиеся по-
последовательности ДНК, не организованные в крупные блоки, а
рассеянные по геному [31, 32]. Повторы этого типа, иначе назы-
называемые умеренно повторяющимися последовательностями
(medium reiterated frequency repeats - MERs), разделяют на два об-
обширных класса: SINE (short interspersed elements) - короткие и
LINE (long interspersed elements) - длинные диспергированные эле-
элементы. Длина SINE-элементов составляет 90-400 п.о., тогда как
24
длина LINE-последовательностей может достигать 7 т.п.о. [31].
Хорошо изученными повторами класса SINE в геноме человека и
некоторых приматов являются так называемые Alu-повторы,
длина повторяющейся единицы которых составляет ~300 п.о. Alu-
повторы представлены в геноме человека ~106 копиями и в сред-
среднем встречаются через каждые 4 т.п.о., составляя ~5% от суммар-
суммарного количества ДНК. Аналогичные в структурном отношении
повторы, названные В1, обнаружены в геноме мышей, а под дру-
другими названиями они описаны также у многих млекопитающих.
Хотя LINE-последовательности заключают в себе гены об-
обратных транскриптаз, что является признаком ретротранспозо-
нов (мобильных генетических элементов животных, обладаю-
обладающих структурным сходством с геномом ретровирусов), для них
характерно отсутствие последовательностей длинных концевых
повторов (long terminal repeats - LTR), типичных для ретротранс-
позонов. В качестве примера LINE-последовательности можно
упомянуть LINE-1-повтор, широко распространенный в геноме
животных. LINE-1-элемент мышей содержит две открытые рам-
рамки считывания ORF-1 и ORF-2, вторая из которых кодирует бе-
белок, гомологичный обратной транскриптазе. ОРС фланкирова-
фланкированы короткими нетранслируемыми последовательностями, а сами
LINE-1 - короткими прямыми повторами (SDR - short direct
repeats). 5'-Концевые последовательности повтора функциониру-
функционируют в качестве промоторов транскрипции. Этот участок LINE-1
грызунов (но не человека) построен из коротких тандемных по-
повторов двух типов, А и F, называемых мономерами. Длина моно-
мономеров у крыс составляет 600 п.о. При этом А- (но не F-) мономе-
мономеры обладают активностью промоторов.
Так же, как и сателлитные ДНК, SINE- и LINE-повторы харак-
характеризуются генетической нестабильностью. Их общими чертами
являются транскрибируемость и способность к транспозициям.
Последовательности РНК, транскрибированные с умеренных
повторов, обнаруживают среди гетерогенных ядерных РНК, где их
доля достигает 20-30%. Имеются экспериментальные свидетельст-
свидетельства того, что новые копии повторяющихся элементов обоих ти-
типов возникают в геноме в результате функционирования меха-
механизма, названного ретротранспозицией, или ретропозицией.
При участии подобного механизма под действием обрат-
обратной транскриптазы сначала образуется кДНК на матрице
РНК-транскрипта соответствующего повтора, которая далее интег-
интегрируется в новый локус генома, как это имеет место у ретровирусов.
Такой механизм дает возможность локально изменять число копий
определенных последовательностей нуклеотидов в эукариотичес-
25
ком геноме. Тем не менее, большая часть LINE-последовательностей
неспособна к транспозициям, и их ОРС, по-видимому, могут быть от-
отнесены к псевдогенам - неэкспрессирующимся последовательнос-
последовательностям, гомологичным последовательностям истинных генов.
Вообще, транспозоны (transposable elements) составляют по
крайней мере 45% всего генома человека [32]. Транспозоны мле-
млекопитающих разделяют на две группы: ДНК-транспозоны и рет-
ротранспозоны. По своей структуре ДНК-транспозоны живот-
животных напоминают бактериальные транспозоны и перемещаются в
геноме по механизму вырезания и вставки ("cut and paste" mecha-
mechanism) с использованием транспозазы (см. раздел 3.1.5). ДНК-
транспозоны составляют ~3% генома человека и в большинстве
своем неактивны [17]. Ретротранспозоны, составляющие ~42%
генома человека, перемещаются по геному с использованием ме-
механизма копирования и вставки ("copy and paste" mechanism), в ко-
котором участвует обратная транскриптаза и РНК-матрица [33].
Ретротранспозоны животных разделяют на автономные и неав-
неавтономные. Автономные транспозоны кодируют ферменты, не-
необходимые для их перемещения. Однако их автономность не яв-
является полной, поскольку перемещение требует участия и фер-
ферментов клетки, в частности, используемых в репарации ДНК.
Автономные транспозоны, в свою очередь, разделяют на два
класса: содержащие и не содержащие LTR. LTR-содержащие ретро-
ретротранспозоны животных по своей структуре напоминают ретровиру-
сы, однако у них отсутствует ген белка оболочки env.
К ним относятся интрацистернальные А-частицы мышей (intracister-
nal A-particles - IAPs) [34], эндогенные ретровирусы человека
(human endogenous retroviruses - HERVs) [32] и MaLR-повторы (mam-
(mammalian LTR-retrotransposons) длиной в 2-3 т.п.о. До 8% генома чело-
человека составлено дефектными эндогенными ретровирусами и оди-
одиночными LTR, которые возникают вследствие рекомбинации меж-
между 51- и З'-LTR обладающих ими ретротранспозонов. К ретротранс-
позонам, не содержащим LTR, относятся уже обсуждавшиеся LINE-
последовательности, которые, в свою очередь, включают неактив-
неактивные Ь2-элементы человека и активные в отношении транспозиций
элементы L1 человека и мышей [35]. Автономные ретротранспозо-
ретротранспозоны, не содержащие LTR, составляют ~21% генома человека.
Кроме автономных ретротранспозонов геномы млекопитаю-
млекопитающих содержат большое количество неавтономных ретротранспо-
ретротранспозонов, которые для транспозиции нуждаются в ферментах, коди-
кодируемых автономными транспозонами. К таким ретротранспозо-
нам относятся вышеупомянутые Alu-элементы человека и гомо-
гомологичные им В1-последовательности мышей.
26
Хроматин. Хроматином называют сложную смесь веществ, из
которых построены хромосомы эукариот. Основными компонен-
компонентами хроматина являются ДНК, гистоны и негистоновые белки,
образующие высокоупорядоченные в пространстве структуры
[2]. Соотношение ДНК и белка в хроматине составляет ~1 : 1, а
основная масса белка хроматина представлена гистонами. Гисто-
Гистоны образуют семейство высококонсервативных основных белков,
которые разделяются на пять больших классов, названных HI,
Н2А, Н2В, НЗ и Н4. Размер полипептидных цепей гистонов лежит
в пределах ~220 (HI) и 102 (Н4) а. о. Гистон HI сильно обогащен
остатками Lys, для гистонов Н2А и Н2В характерно умеренное со-
содержание Lys, полипептидные цепи гистонов НЗ и Н4 богаты Arg.
Внутри каждого класса гистонов (за исключением Н4) на основа-
основании аминокислотных последовательностей различают несколько
субтипов этих белков. Такая множественность особенно харак-
характерна для гистонов класса HI млекопитающих. В этом случае раз-
различают семь субтипов, названных Н1.1-Н1.5, Н1° и Hit.
Важным результатом взаимодействия ДНК с белками в со-
составе хроматина является ее компактизация. Суммарная длина
ДНК, заключенной в ядре клеток человека, приближается к 1 м,
тогда как средний диаметр ядра составляет ~10 мкм. Длина моле-
молекулы ДНК, заключенной в одной хромосоме человека, в среднем
равняется ~4 см. В то же время длина метафазной хромосомы со-
составляет ~4 мкм. Следовательно, ДНК метафазных хромосом че-
человека компактизована по длине, по крайней мере, в ~104 раз.
Степень компактизации ДНК в интерфазных ядрах значительно
ниже и неравномерна в отдельных генетических локусах.
С функциональной точки зрения различают эухроматин и гете-
рохроматин [36-39]. Эухроматин характеризуется меньшей по
сравнению с гетерохроматином компактизацией ДНК, и в нем
главным образом локализуются активно экспрессирующиеся ге-
гены. В настоящее время все еще широко распространено мнение
о генетической инертности гетерохроматина. Поскольку его ис-
истинные функции сегодня нельзя считать установленными, эта
точка зрения по мере накопления знаний о гетерохроматине мо-
может измениться. Уже сейчас в нем находят активно экспрессиру-
экспрессирующиеся гены.
Гетерохроматизация определенных участков хромосом часто
сопровождается подавлением транскрипции расположенных в
них генов [36]. В процесс гетерохроматизации могут быть вовле-
вовлечены протяженные участки хромосом и даже целые хромосомы.
В соответствии с этим считается, что регуляция транскрипции ге-
генов эукариот в основном происходит на двух уровнях. На первом
27
из них компактизация или декомпактизация ДНК в хроматине
может приводить к длительной инактивации или активации про-
протяженных участков хромосом или даже целых хромосом в онто-
онтогенезе организма. Более тонкая регуляция транскрипции активи-
активированных участков хромосом достигается на втором уровне при
участии негистоновых белков, включающих многочисленные
факторы транскрипции.
1.3. Экспрессия генов
Конечным результатом экспрессии генов, кодирующих белки
или нуклеиновые кислоты, должно быть образование этих пол-
полноценных в функциональном отношении макромолекул, сопро-
сопровождаемое формированием определенного фенотипа организма.
В соответствии с основным постулатом молекулярной биологии
генетическая информация в процессе ее реализации передается
однонаправленно от нуклеиновых кислот к белкам. При этом ре-
реализуется следующая обобщенная схема: ДНК <-> РНК -» белок,
которая подчеркивает, что в ряде специальных случаев возмож-
возможна передача генетической информации от РНК к ДНК с исполь-
использованием механизма обратной транскрипции. До сих пор не об-
обнаружена передача генетической информации от белков к нукле-
нуклеиновым кислотам. На первом этапе экспрессии генов происходит
переписывание генетической информации, заключенной в генах,
на матричные (информационные) РНК (мРНК - messenger RNA,
mRNA), которые являются местом промежуточного хранения
этой информации при ее реализации. В некоторых случаях уже
сами РНК являются конечным результатом экспрессии генов, и
после ряда ферментативных модификаций они непосредственно
используются в клеточных процессах. Это относится, прежде
всего, к рибосомным и транспортным РНК (рРНК и тРНК), ко-
которые вместе составляют основную часть суммарной РНК клет-
клетки. Кроме того, к таким РНК принадлежат малые ядерные РНК
(мяРНК), участвующие в процессинге предшественников мРНК
эукариот, а также РНК, входящие в состав некоторых фермен-
ферментов, и природные антисмысловые РНК.
Синтез РНК происходит в результате сложной последова-
последовательности биохимических реакций и называется транскрипцией.
Появление русифицированного термина "мРНК" связано с тем,
что на втором этапе реализации генетической информации, на-
называемом трансляцией, последовательность нуклеотидов мРНК
согласно генетическому коду однозначно определяет последова-
28
тельность аминокислотных остатков синтезируемых белков, т.е.
проявляет некоторые свойства матрицы, в соответствии с после-
последовательностями нуклеотидов которой происходит соединение
аминокислотных остатков друг с другом в полипептидных цепях
белков во время их биосинтеза. Таким образом, экспрессию ге-
генов определяют два глобальных молекулярно-генетических ме-
механизма: транскрипция генов и трансляция синтезированных
мРНК рибосомами, которая завершается образованием полипеп-
полипептидных цепей, кодируемых генами. Однако процесс экспрессии
генов не ограничивается только транскрипцией и последующей
трансляцией синтезированной мРНК. Существенными момента-
моментами экспрессии генов являются посттранскрипционные и пост-
посттрансляционные модификации мРНК и белков, которые вклю-
включают процессинг их предшественников (удаление избыточных
последовательностей и другие ковалентные модификации после-
последовательностей РНК и белков). Посттранскрипционные модифи-
модификации предшественников мРНК обеспечивают подготовку мРНК
к эффективной трансляции рибосомами и определяют продол-
продолжительность ее существования в цитоплазме. Посттрансляцион-
Посттрансляционные модификации белков также необходимы для их полноценно-
полноценного функционирования.
1.3.1. Транскрипция
В процессе транскрипции генов происходит биосинтез моле-
молекул РНК, комплементарных одной из цепей матричной ДНК,
сопровождаемый полимеризацией четырех рибонуклеозидтри-
фосфатов (ATP, GTP, СТР и UTP) с образованием 3'-5' - фосфо-
диэфирных связей и освобождением неорганического пирофос-
фата. Основными ферментами, осуществляющими транскрип-
транскрипцию, являются ДНК-зависимые РНК-полимер азы, которые
функционируют с участием многочисленных факторов транс-
транскрипции - регуляторных белков, осуществляющих высокоспе-
высокоспецифические белок-белковые и белково-нуклеиновые взаимо-
взаимодействия [40]. Взаимодействия факторов транскрипции с регу-
ляторными нуклеотидными последовательностями генов, друг с
Другом и с молекулами РНК-полимеразы необходимы для пра-
правильного узнавания транскрипционным комплексом регулятор-
регуляторных последовательностей в составе генов и приводят к повыше-
повышению или понижению уровня транскрипции соответствующих
последовательностей при ответе клеток на внешние или внут-
внутренние регуляторные сигналы. Благодаря факторам транскрип-
транскрипции и регуляторным последовательностям генов становится
29
возможным синтез специфических РНК в определенный мо-
момент времени и осуществляется регуляция экспрессии генов на
уровне транскрипции.
ДНК-зависимые РНК-полимеразы. РНК-полимеразы под-
подразделяются на две группы. К первой группе относятся фермен-
ферменты, состоящие только из одной субъединицы, среди них -
РНК-полимеразы митохондрий и небольших бактериофагов,
например SP6 и Т7. Эти РНК-полимеразы транскрибируют не-
небольшое число генов простых геномов и для их функционирова-
функционирования не требуется сложных регуляторных воздействий. Вторую
группу составляют сложно устроенные РНК-полимеразы бак-
бактерий и эукариот, которые представляют собой многосубъеди-
ничные белковые комплексы, транскрибирующие сотни и ты-
тысячи различных генов. Такие ферменты во время своего функ-
функционирования реагируют на многочисленные регуляторные
сигналы, поступающие от регуляторных последовательностей
нуклеотидов и белковых факторов. У живых организмов, начи-
начиная с бактерий, возникает потребность в РНК-полимеразах
сложной структуры, способных осуществлять обширную про-
программу реализации генетической информации. Вероятно, по-
поэтому наблюдается иерархия в степени сложности строения
указанных ферментов, которая достигает верхнего предела в
случае РНК-полимераз эукариот.
Единицы транскрипции (транскриптоны). Синтез РНК моле-
молекулами РНК-полимераз in vivo начинается в определенных мес-
местах ДНК, называемых промоторами, и завершается на особых
регуляторных последовательностях - терминаторах. Последова-
Последовательности нуклеотидов ДНК, заключенные между промоторами
и терминаторами, называют транскрипционными единицами,
или транскриптонами. В пределах каждого транскриптона
транскрибируется только одна цепь ДНК, которая получила на-
название значащей или матричной. Термины "транскрипционная
единица" или "транскриптон" по смыслу близки термину "ген",
но они не всегда совпадают. Так, транскрипционные единицы
прокариот, как правило, заключают в себе генетическую инфор-
информацию нескольких генов и называются оперонами. Продуктами
транскрипции оперонов являются полицистронные мРНК, при
трансляции которых рибосомами образуется несколько белков.
Белки, кодируемые полицистронными мРНК, обычно функцио-
функционально связаны друг с другом и обеспечивают протекание како-
какого-либо метаболического процесса, например, биосинтеза опре-
определенной аминокислоты или утилизацию углеводов в качестве
источника углерода. Организация генов в виде оперонов облег-
30
чает координированную регуляцию их экспрессии на уровне
транскрипции. Согласованная регуляция транскрипции (и других
этапов экспрессии) многих генов, не образующих одного оперо-
на, чаще всего осуществляется специфическими белками-регуля-
белками-регуляторами, которые взаимодействуют с гомологичными регулятор-
ными нуклеотидными последовательностями, характерными для
генов данной группы.
Этапы транскрипции. Процесс транскрипции в настоящее
время принято подразделять на четыре основные стадии: 1) свя-
связывание молекул РНК-полимеразы с ДНК и распознавание про-
промотора; 2) инициация; 3) элонгация; 4) терминация [41]. Три по-
последних этапа характерны для биосинтеза большинства других
макромолекул клетки, особенно для тех из них, синтез которых
является матричным, в частности белков. После связывания с
ДНК молекулы РНК-полимеразы осуществляют поиск промо-
промоторов, на которых происходит формирование инициационных
комплексов. Начальная стадия инициации транскрипции завер-
завершается образованием нескольких первых фосфодиэфирных
связей в молекуле вновь синтезируемой РНК, после чего транс-
транскрипция переходит в стадию элонгации - последовательного уд-
удлинения синтезируемых молекул РНК. Стадия элонгации закан-
заканчивается по достижении молекулами РНК-полимераз специаль-
специальных регуляторных последовательностей ДНК, называемых
терминаторами транскрипции, после чего происходит осво-
освобождение синтезированных молекул РНК и РНК-полимераз из
транскрипционных комплексов. Освободившиеся молекулы
РНК-полимераз приобретают способность вступать в новый
цикл транскрипции. Следует помнить, что четкого разделения
единого процесса транскрипции на отдельные стадии в реаль-
реальной жизни не существует; оно используется главным образом
для удобства описания механизмов биосинтеза РНК и является
упрощением.
В обычных условиях РНК-полимеразы эубактерий для ини-
инициации транскрипции не требуют дополнительных факторов. В
отличие от этого для точной инициации транскрипции РНК-по-
лимеразой II требуется наличие, кроме ее субъединиц, еще и ос-
основных факторов транскрипции. Синтез РНК, который не зави-
зависит от присутствия регуляторных молекул, получил название ба-
залъной транскрипции. Транскрипция в клетках является регу-
регулируемым процессом, который требует участия белков-актива-
белков-активаторов или репрессоров. Белок-активатор (в том числе тканеспе-
Цифический фактор транскрипции) взаимодействует с регуля-
торными последовательностями ДНК и активирует синтез РНК.
31
Такая транскрипция получила название индуцированной, или ак-
активированной. Следовательно, базальная транскрипция не мо-
может происходить in vivo, и этот термин используется только при
описании результатов исследований синтеза РНК in vitro, в бес-
бесклеточных системах транскрипции.
1.3.2. Котранскрипционные и посттранскрипционные
модификации РНК
Транскрипция у любого организма является первым этапом
реализации генетической информации - экспрессии генов. Одна-
Однако первичные транскрипты, как правило, представляют собой
лишь предшественники зрелых мРНК - пре-мРНК, которые пе-
перед выполнением своих функций должны претерпеть многочис-
многочисленные изменения, называемые посттранскрипционными моди-
модификациями. Кроме того, у эукариот зрелые мРНК должны быть
доставлены от места их биосинтеза (ядра) к месту трансляции
(цитоплазматическим рибосомам), т.е. экспортироваться из ядра
в цитоплазму.
Одной из наиболее удивительных посттранскрипционных мо-
модификаций пре-мРНК является редактирование (editing) их пер-
первичной структуры. В результате посттранскрипционно изменяет-
изменяется смысл генетической информации, заключенной в соответству-
соответствующем гене [42-45].
Посттранскрипционные модификации РНК особенно харак-
характерны для эукариотических организмов, у которых в силу моза-
мозаичной интрон-экзонной структуры их генов первичные транс-
транскрипты представлены гигантскими предшественниками, включа-
включающими в себя последовательности как экзонов, так и интронов.
5'-Конец предшественника мРНК чаще всего подвергается ко-
транскрипционным модификациям, в результате которых к его
5'-концевому нуклеотиду особым образом присоединяется оста-
остаток гуанозина с образованием "шапочки" - кэпа [46]. Эта котран-
скрипционная модификация создает условия для прохождения
следующего этапа процессинга мРНК - сплайсинга, в том числе,
и альтернативного сплайсинга, сопровождающегося вырезани-
вырезанием последовательностей интронов и объединением экзонов с об-
образованием непрерывной кодирующей последовательности
мРНК [47]. Альтернативный сплайсинг, объединяя экзоны одно-
одного и того же гена в разных сочетаниях, является точно регулиру-
регулируемым процессом, создающим дополнительное разнообразие бел-
белковых продуктов, кодируемых одним геном, что увеличивает ко-
кодирующий потенциал генома без физического увеличения его
32
размера. Одновременно со сплайсингом от З'-конца предшествен-
предшественника мРНК (пре-мРНК) путем эндонуклеазного расщепления от-
отделяется избыточный фрагмент РНК, и к оставшейся части при-
присоединяется поли(А)-последовательность. Эта совокупность ре-
реакций получила название полиаденилирования мРНК. После та-
таких котранскрипционных и посттранскрипционных модифика-
модификаций пре-мРНК образовавшаяся зрелая, стабилизированная
мРНК переносится из ядра в цитоплазму, часто к специфическо-
специфическому месту своей внутриклеточной локализации, где может быть
депонирована или эффективно транслироваться рибосомами.
Каждый из этапов посттранскрипционных модификаций может
использоваться для регуляции уровня экспрессии соответствую-
соответствующих генов.
1.3.3. Трансляция
В процесс биосинтеза белка, называемого трансляцией, во-
вовлечено множество макромолекул и макромолекулярных ком-
комплексов [48, 49]. На этом этапе реализации генетической инфор-
информации происходит считывание генетической информации, за-
заключенной в мРНК, рибосомами и ее передача полипептидным
цепям белков, т.е. биосинтез полипептидных цепей, последова-
последовательность аминокислот в которых, как правило, однозначно оп-
определена последовательностью нуклеотидов в транслируемых
мРНК в соответствии с генетическим кодом. Свободные амино-
аминокислоты не узнаются рибосомами. Чтобы это произошло, амино-
аминокислоты должны поступать в рибосомы в виде конъюгатов с
тРНК (аминоацилированных тРНК), последовательности нукле-
нуклеотидов которых распознаются аппаратом трансляции. В каждой
молекуле тРНК имеется участок из трех нуклеотидов, компле-
комплементарный кодону мРНК. Именно эта последовательность, на-
называемая антикодоном, в основном определяет положение той
или иной аминокислоты в полипептидной цепи. В ходе каждого
индивидуального акта трансляции рибосома распознает кодон
мРНК и в соответствии с ним выбирает аминоацилированную
тРНК, антикодон которой соответствует транслируемому кодо-
кодону. После этого происходит соединение посредством пептидной
связи очередной аминокислоты с С-концевой аминокислотой
растущей цепи полипептида.
Таким образом, во время трансляции рибосома после связы-
связывания мРНК начинает последовательно, кодон за кодоном, пере-
перемещаться вдоль матрицы, выбирая из окружающей среды моле-
молекулы аминоацилированных тРНК. При этом каждый индивиду-
2. Патрушев Л.И. Т. 1 33
альный акт трансляции завершается присоединением выбранной
молекулы аминокислоты к С-концевой аминокислоте синтезиру-
синтезируемой цепи белка посредством пептидной связи.
Процесс биосинтеза белка как и биосинтез любой другой ма-
макромолекулы клетки, условно разделяют на три основных этапа:
инициацию, элонгацию и терминацию. Во время инициации
трансляции происходит сборка нативной 70S или 80S рибосомы на
транслируемой мРНК и подготовка к образованию пептидной
связи между первыми двумя N-концевыми аминокислотными ос-
остатками синтезируемого полипептида. При элонгации наблюда-
наблюдается последовательное удлинение растущей цепи полипептида
аминокислотными остатками, а терминация трансляции сопро-
сопровождается прекращением синтеза полипептида и его высвобожде-
высвобождением из трансляционного комплекса. При этом наблюдается так-
также разделение рибосомы и мРНК, после чего они вступают в но-
новый цикл трансляции. В ходе трансляции рибосома последова-
последовательно перемещается вдоль транслируемой молекулы мРНК, счи-
считывая заключенную в ней в виде триплетного генетического кода
генетическую информацию. Трансляция начинается с 5'-концевой
части мРНК, а завершается в ее З'-концевой части. Биосинтез по-
полипептида начинается с его N-концевой аминокислоты.
1.3.4. Системы надзора за мРНК и белками
Консервативная у всех эукариот система надзора за мРНК
является частью общей системы организмов, обеспечивающей
точность передачи генетической информации от генов к фено-
типическим признакам. Специальный белковый комплекс учи-
учитывает расстояние между терминирующим кодоном мРНК и
последовательностью, расположенной ближе к ее З'-концу. По-
После терминации трансляции такой комплекс собирается на
мРНК и сканирует ее на определенное расстояние в направле-
направлении 3' —» 5', пытаясь обнаружить особенности структуры, харак-
характерные для нормальных мРНК. Если искомая информация ком-
комплексом не обнаруживается, он направляет мРНК на путь де-
деградации. Согласно альтернативной точке зрения на механизм
надзора, для комплекса важна информация, которая располага-
располагается непосредственно после кодона терминации трансляции. В
любом случае эта система обеспечивает деградацию мРНК, в
которой присутствуют бессмысленные кодоны, образовавшие-
образовавшиеся в результате мутаций [50].
Функциональным аналогом этой системы у бактерий являет-
является механизм деградации недостроенных пептидов, синтезирован-
34
ных на фрагментах мРНК неполной длины. В том случае, если
транслируемая бактериальная мРНК укорочена в своей У - кон-
концевой части и рибосома не находит в соответствующей рамке
считывания терминирующий кодон, она не может закончить
трансляцию с помощью стандартного механизма терминации.
Столкнувшись с такой ситуацией, рибосома останавливается на
конце мРНК и с ней взаимодействует lOSa РНК, кодируемая у
Е. coli геном ssrA, которая запускает процесс деградации частич-
частично синтезированного полипептида. Эта необычная РНК, амино-
ацилированная остатком Ala на своем 3'-конце, подобном стан-
стандартной акцепторной последовательности тРНК, сочетает в себе
свойства тРНК и мРНК. После взаимодействия с рибосомой эта
РНК вступает в стандартный цикл трансляции: ее остаток Ala
включается в недостроенную цепь полипептида. Вслед за этим
рибосома транслирует короткую последовательность нуклеоти-
дов lOSa РНК, которая теперь уже функционирует в качестве ма-
матрицы, добавляя десять аминокислот - ANDENYALAA - в С-ко-
нец укороченной полипептидной цепи, и терминирует трансля-
трансляцию на UAA-кодоне lOSa РНК. С-Концевая олигопептидная по-
последовательность освободившегося полипептида далее распозна-
распознается специфической протеиназой, которая расщепляет этот по-
полипептид, обеспечивая его утилизацию бактериальной клеткой в
процессе катаболизма [51].
1.3.5. Котрансляционные и посттрансляционные
модификации белков
Синтезом полноценного полипептида в результате трансля-
трансляции кодирующей его мРНК рибосомами обычно завершается
процесс передачи генетической информации от генов к белкам
как у бактерий, так и у высших организмов. Однако в большин-
большинстве случаев при синтезе конечного белкового продукта эукари-
отическими клетками производятся различные его модифика-
модификации, в результате которых полипептид и приобретает требуемые
свойства. Особой посттрансляционной модификацией является
сплайсинг белков, механизм которого будет рассмотрен во вто-
второй части книги (раздел 1.3.2).
Кроме того, необходимо иметь в виду, что конечный уровень
содержания конкретных белков в клетке зависит не только от
скорости их биосинтеза, но и от скорости внутриклеточной де-
деградации [52]. Поэтому дифференциальная регуляция стабильно-
стабильности белков является важнейшим механизмом, регулирующим
экспрессию генов у любого организма.
2* 35
Многие белки и секретируемые пептиды претерпевают раз-
различные структурные изменения в результате котрансляционных и
посттрансляционных модификаций, т.е. во время или после завер-
завершения их синтеза рибосомами. В настоящее время известно более
500 различных модификаций белковых молекул. Подобные моди-
модификации заметно влияют на функциональную активность белков
и пептидов и вносят существенный и дополнительный вклад в ре-
результаты экспрессии генов, кодирующих эти молекулы [53, 54].
Одна из таких модификаций, а именно фосфорилирование рецеп-
рецепторов, факторов транскрипции, гистонов и ассоциированных с ни-
ними белков, является одним из ключевых механизмов регуляции
экспрессии генов у эукариот [55, 56]. Кроме того, посттрансляци-
посттрансляционные модификации включают в себя гликозилирование остатков
Asn в последовательностях Asn-X-[SerThr], N-концевое ацилирова-
ние, циклизацию N-концевого остатка Glu с образованием пиро-
глутаминовой кислоты, С-концевое амидирование последователь-
последовательностей освобождающихся пептидов, гидроксилирование остатков
Lys и Pro, метилирование различных остатков аминокислот и т.д.
[57]. Среди ковалентных посттрансляционных модификаций пеп-
пептидов, синтезируемых рибосомами, особое место занимает эпиме-
ризация L-аминокислотных остатков с образованием D-энантио-
меров, присутствие которых оказывает принципиальное влияние
на биологическую активность пептидов [58].
Много этапов различных преобразований информационных
макромолекул и белков отделяют генетическую информацию,
заключенную в конкретных генах, от ее реализации в соответст-
соответствующих фенотипических признаках клеток и организма, кото-
которые обладают этой информацией. Такая реализация требует точ-
точной координации в функционировании многих генетических сис-
систем организма на всех основных уровнях экспрессии генов. Мно-
Многоступенчатый и сложный характер регуляции функционирова-
функционирования генов, особенно у высших организмов, накладывает большие
ограничения на возможности получения полноценной экспрессии
рекомбинантных генов в гетерологичных искусственных генети-
генетических системах.
1.4. Использование генной инженерии для создания
искусственных генетических систем
Обмен генетической информацией между носителями этой
информации - живыми организмами, а также составляющими их
соматическими и половыми клетками является фундаменталь-
36
ным принципом существования всего живого. Половой процесс
освобождает организмы от груза необратимых изменений в виде
соматических мутаций и других многочисленных модификаций
макромолекул, которые нарушают его нормальное функциони-
функционирование в старости. Кроме того, в результате полового процесса
геном нового организма воссоздается в новом сочетании алле-
аллелей, что, как правило, сопровождается расширением его адаптив-
адаптивных возможностей. Этот широко распространенный способ об-
обмена генами между организмами с их передачей по вертикали,
т.е. от поколения к поколению, не исчерпывает всего феномена
обмена генами, непрерывно происходящего в биосфере.
Известна обширная группа генетических явлений, связанная с
горизонтальной передачей генов в пределах одного поколения
организмов, а также между клетками одного и того же многокле-
многоклеточного организма. Характерными примерами такого рода явля-
являются специфическая и неспецифическая трансдукции, осуществ-
осуществляемые бактериофагами, в результате чего происходит перенос
небольших частей генома микроорганизмов. Большое значение в
эволюции бактерий играет и обмен генами с помощью конъюга-
тивных плазмид и транспозонов, в частности, распространение
генов устойчивости к различным химическим веществам как в
популяциях родственных бактерий, так и между представителями
таксономически удаленных друг от друга групп [59]. Ретровиру-
сы, по-видимому, и в природных условиях способны осуществ-
осуществлять горизонтальный перенос генов у млекопитающих, а с помо-
помощью Ti-плазмид происходит горизонтальный обмен генами и у
растений. Перемещение генетической информации и изменение
характера ее экспрессии возможны и в пределах геномов самих
одноклеточных и многоклеточных организмов под действием
разнообразных мобильных генетических элементов, а также при
воздействии мутагенных факторов окружающей среды [60].
Приведенные примеры показывают, что в природе обмен
блоками генов, отдельными генами и их фрагментами как в пре-
пределах геномов, так и между различными геномами и организма-
организмами - обычное явление. В результате этих событий переносимые
гены не только сохраняют свою способность к экспрессии в но-
новом генетическом окружении, но и могут значительно менять ее
Уровень, что часто сопровождается характерным изменением
фенотипа организмов.
Образование новых сочетаний генов в природных условиях,
по-видимому, не носит целенаправленного характера, и лишь же-
жесткая проверка естественным отбором дает возможность оце-
оценить жизненную значимость таких преобразований геномов.
37
Однако, сознательное использование в лабораторных условиях
генетических принципов, лежащих в основе природных переме-
перемещений генов, позволило разработать более эффективные систе-
системы передачи генетической информации между организмами и
приступить к беспрецедентным по информативности исследова-
исследованиям генетических явлений на молекулярном уровне. У нас на
глазах произошло рождение нового направления в молекулярной
биологии - генной инженерии, значение которого не ограничива-
ограничивается результатами конкретных фундаментальных и прикладных
исследований. По-видимому, именно с этого момента начался но-
новый этап эволюции биосферы Земли, все последствия которого
мы в настоящее время не в состоянии предвидеть.
Необходимость манипулирования генами диктуется конкрет-
конкретными задачами фундаментальных и прикладных исследований.
Для понимания молекулярных механизмов функционирования от-
отдельных генов и взаимосвязанных генетических систем большое
значение имеет работа с изолированными генами. Такие исследо-
исследования позволяют определять границы генов, выделять их в чис-
чистом виде и идентифицировать элементы структуры, существен-
существенные для функционирования. Доказательством функциональной
значимости выделенного участка генома может быть только его
нормальная экспрессия в модельной генетической системе. По-
Поэтому следующим этапом изучения выделенного гена всегда явля-
является перемещение его в такую генетическую систему, где экс-
экспрессия гена легко обнаруживается. Результаты экспрессии оце-
оценивают либо по появлению белкового продукта, кодируемого ис-
исследуемым геном, либо по изменению функций биологической
системы вследствие появления в ней новой ферментативной или
другой активности, например, по компенсации присутствующей в
этой системе мутации. Таким образом, в результате исследования
структуры конкретного гена и моделирования его экспрессии в
искусственной генетической системе можно понять особенности
его функционирования в живом организме. Подобный подход ус-
успешно применяют как к известным генам, которые выделяются
целенаправленно, так и к неидентифицированным ранее последо-
последовательностям нуклеотидов, функциональную значимость кото-
которых определяют лишь после выделения их в чистом виде. Послед-
Последний подход реализуется в так называемой обратной генетике.
В настоящее время с помощью методов генной инженерии по-
получены данные о структуре и функционировании генов разнооб-
разнообразных организмов, что позволило перейти на качественно новый
уровень генетических исследований. Это, во-первых, возможность
переноса гена в новое для него генетическое окружение с дальней-
38
шей его экспрессией, что ведет к изменению свойств организма, в
геном которого вводится ген (например, создание продуцентов би-
биологически активных веществ или трансгенных животных), а так-
также осуществление генотерапии наследственных и приобретенных
заболеваний путем искусственного замещения мутантных алле-
аллелей. Во-вторых, стало реальным конструирование новых генов пу-
путем объединения in vitro как известных, так и новых, искусственно
синтезированных последовательностей нуклеотидов. Этот подход
используется в белковой инженерии для исследования функцио-
функциональной значимости отдельных аминокислот и доменов в полипеп-
полипептидных цепях ферментов, а также для создания новых белков.
В-третьих, в современной биотехнологии появилась возможность
применять изолированные гены в составе генно-инженерных кон-
конструкций для получения пищевых продуктов и биологически ак-
активных веществ белковой природы.
Поскольку в экспериментальных условиях невозможно рабо-
работать с одной копией гена, создание необходимого числа идентич-
идентичных копий гена (или его частей) является первой и одной из ос-
основных задач генной инженерии. Для ее решения используют ме-
метод молекулярного клонирования [61]. Сущность метода заклю-
заключается в том, что нуклеотидная последовательность, которую не-
необходимо выделить или размножить, ковалентно встраивается в
самореплицирующиеся молекулы нуклеиновой кислоты, называ-
называемые векторами. Далее такая последовательность нуклеотидов в
составе вектора вводится в клетки про- или эукариотического
организма, и эти гибридные клетки в селективных условиях,
обеспечивающих сохранение вектора внутри клеток, выращива-
выращивают на питательной среде. В результате образуется клон клеток,
теоретически содержащих идентичные векторные молекулы с
одной и той же вставкой чужеродной последовательности нукле-
нуклеотидов. Объединение молекул клонируемой последовательности
нуклеотидов и вектора является не чем иным как рекомбинацией
in vitro, поэтому такие гибридные молекулы называют рекомби-
нантными молекулами.
В настоящее время разработаны многочисленные методы,
позволяющие выделять определенные последовательности нук-
нуклеотидов из сложной смеси фрагментов хромосомной ДНК, а
также осуществлять обмен между строго определенными фраг-
фрагментами генов и другими последовательностями нуклеиновых
кислот. Во всех этих реакциях, как правило, используются высо-
коочищенные препараты нуклеиновых кислот и ферментов нук-
нуклеинового обмена, особенности использования которых будут
кратко рассмотрены ниже.
39
Глава 2
Выделение нуклеиновых кислот и
ферменты, используемые для работы с ними
Большинство ферментов, применяемых для молекулярного
клонирования нуклеиновых кислот, участвует в метаболизме нук-
нуклеиновых кислот in vivo. Это означает, что генная инженерия в
своем развитии опирается на достижения исследований фермент-
ферментных систем метаболизма нуклеиновых кислот и существует бла-
благодаря возможности получения таких ферментов в высокоочи-
щенном состоянии. В то же время сам процесс клонирования и ис-
исследования клонированных последовательностей нуклеотидов
сводится к последовательному проведению in vitro определенных
ферментативных реакций с использованием очищенных фермен-
ферментов и их субстратов - нуклеиновых кислот.
2.1. Основные приемы очистки нуклеиновых кислот
Генно-инженерные работы, как правило, начинаются с полу-
получения препаратов высокоочищенных нуклеиновых кислот. Кри-
Критериями качества используемого метода очистки ДНК и РНК,
как, впрочем, и других макромолекул клетки, является чистота
конечного препарата, уровень фрагментации выделяемых моле-
молекул, общий выход, а также время, требуемое для проведения вы-
выделения нуклеиновых кислот.
2.1.1. Традиционные методы
При очистке ДНК и РНК в основном используются одни и те
же приемы [62]. Различия в методах их фракционирования опре-
определяются значительно большей чувствительностью молекул
РНК к гидролитическому расщеплению из-за присутствия у ри-
бонуклеотидов, входящих в состав РНК, свободных 2'-ОН групп,
их более низкой молекулярной массой и наличием у одноцепо-
чечных молекул характерной (компактной) пространственной
структуры. Все современные методы выделения нуклеиновых
кислот состоят из трех этапов: разрушения клеток, инактивации
нуклеаз и собственно очистки. Для выделения нуклеиновых кис-
кислот в нативном состоянии необходимо соблюдать, по крайней ме-
мере, две предосторожности: следует использовать мягкие условия
разрушения тканей биологического объекта и инактивировать
гидролитические ферменты (ДНКазы или РНКазы) до того, как
40
они успеют гидролизовать молекулы нуклеиновых кислот. С уче-
учетом этого ингибиторы нуклеаз вводятся в буферные растворы до
разрушения клеток или тканей. В качестве ингибиторов нуклеаз
используют как неспецифические денатурирующие агенты (ион-
(ионные детергенты, гидроокись ртути, гуанидинхлорид, гуанидин-
изотиоцианат, фенол, хлороформ и т.п.), так и специфические
ингибиторы, например, ингибитор РНКазы из плаценты челове-
человека или ванадиевые комплексы рибонуклеозидов.
Работу с геномной двухцепочечной ДНК (дцДНК) (особенно
с ДНК эукариотических клеток) сильно усложняют значитель-
значительная молекулярная масса и высокая жесткость выделяемых моле-
молекул. Следствием этого являются большая вязкость растворов вы-
высокомолекулярной ДНК и легкость ее фрагментации в таких рас-
растворах. В связи с этим, при выделении высокомолекулярной
ДНК допускается лишь мягкое перемешивание ее раствора в
процессе депротеинизации (освобождения от белков). Для этого
на первых этапах очистки обычно применяют протеолитические
ферменты (протеиназу К из Trirachium album или проназу из
Streptomyces griseus) [63], фенол и хлороформ [64]. Поскольку
при освобождении от белков происходит одновременная очистка
ДНК и РНК, для отделения примесей РНК используют высоко-
очищенные препараты РНКаз без примесей ДНКаз (чаще всего
панкреатическую РНКазу), а в случае выделения РНК - препара-
препараты ДНКаз, не загрязненных РНКазами.
При использовании большинства методов выделения ДНК из
тканей животных происходит одновременное выделение геном-
геномной и митохондриальной ДНК. Последняя, как правило, присут-
присутствует в клетках в виде множественных копий, что приводит к
высокой представленности митохондриальных генов в получен-
полученных препаратах ДНК и в клонотеках, создаваемых на основе
этих препаратов. В случае необходимости, для освобождения от
митохондриальной ДНК на первых стадиях очистки геномной
ДНК получают ядра клеток анализируемых тканей. Альтерна-
Альтернативно, при необходимости выделения чистой митохондриальной
ДНК вначале получают эти органеллы [65].
Электрофоретическое и хроматографическое разделение
нуклеиновых кислот. Для дополнительной очистки плазмидной
ДНК иногда применяют гель-фильтрацию на Сефакриле S1000
(крупнопористый синтетический аналог сефадекса) [66], а также
электрофорез в агарозном геле. Следует отметить, что электро-
электрофорез в агарозном, полиакриламидном или смешанном агарозно-
полиакриламидном гелях используют для определения размеров
и выделения небольших фрагментов дцДНК, образующихся при
41
действии эндонуклеаз рестрикции или во время секвенирования
ДНК. Проявление фрагментов в этом случае проводят с помо-
помощью бромистого этидия, интеркалирующего между основаниями
ДНК и флуоресцирующего в ультрафиолетовом свете. Более
чувствительными методами обнаружения фрагментов ДНК в ге-
гелях являются окраска солями серебра, авторадиография и введе-
введение флуоресцентной метки.
Отдельно следует упомянуть метод аффинной хромато-
хроматографии на олиго(с1Т)-целлюлозе, используемый для специфиче-
специфической очистки эукариотических мРНК, большинство из кото-
которых содержит на З'-конце поли(А)-последовательность. В оли-
го(а?7>целлюлозе последовательности олиго(а?ГI2_18 ковалент-
но присоединены к молекулам целлюлозы своими 5'-концевы-
ми фосфатными группами. При использовании этого метода
фракцию суммарной РНК наносят на колонку с олиго(я?Г)-цел-
люлозой в условиях, при которых происходит гибридизация по-
ли(А)-последовательностей мРНК с комплементарными им по-
последовательностями олиго(я?Г)-целлюлозы. После отмывки не-
связавшихся со смолой молекул РНК, большинство из которых
являются рРНК и тРНК, фракцию поли(Л)-РНК элюируют,
прогревая колонку до температуры плавления поли(Л)жоли-
го(а?7>гибридов или разрушая гибриды понижением ионной си-
силы элюирующего раствора.
Перечисленные выше способы очистки и фракционирования
нуклеиновых кислот позволяют получать лишь относительно ко-
короткие последовательности нуклеотидов. Однако в настоящее
время в арсенале молекулярной биологии имеются методы, кото-
которые дают возможность выделять в гомогенном состоянии после-
последовательности нуклеотидов, эквивалентные целым хромосомам.
Одним из таких методов является электрофорез в импульсном
электрическом поле (pulsed field gel electrophoresis - PFGE), с по-
помощью которого удается препаративно разделять, например,
ДНК целых хромосом дрожжей [68, 69]. Другой эффективный
метод, с помощью которого можно фракционировать метафаз-
ные хромосомы млекопитающих - это сортировка хромосом с
помощью проточной цитофлуориметрии [70-72]. Метод позво-
позволяет получать индивидуальные метафазные хромосомы в коли-
количестве, достаточном для конструирования клонотек генов инди-
индивидуальных хромосом.
42
2.1.2. Выделение метафазных хромосом
с помощью проточной цитометрии
По не вполне понятным причинам природа позаботилась о
том, чтобы большие геномы эукариот были разделены на строго
определенные сегменты ДНК, расположенные в отдельных хро-
хромосомах. Трудно предположить, что при этом учитывались инте-
интересы генетиков, хотя такая организация генома, несомненно, об-
облегчает его исследование. Действительно, полное секвенирова-
ние и генетическое картирование генома всегда начинается с изу-
изучения отдельных хромосом. Ниже будут рассматриваться клоно-
теки геномной ДНК, в том числе, и соответствующие отдельным
хромосомам исследуемого биологического вида. Для создания та-
таких клонотек, без которых не обойтись при систематическом
изучении индивидуальных генов, прежде всего, необходимо полу-
получить препараты метафазных хромосом исследуемого биологиче-
биологического вида. Для решения этой непростой задачи в настоящее вре-
время успешно используют проточную цитометрию, группу инст-
инструментальных методов, специально разработанную для сорти-
сортировки микроскопических частиц [72]. Для обозначения проточ-
проточной сортировки клеток и хромосом часто используется аббреви-
аббревиатура FACS (fluorescence-activated cell (chromosome) sorting).
Принцип действия проточных цитофлуориметров-сортиров-
щиков. Для фракционирования микроскопических объектов в
проточных сортировщиках суспензию частиц метят флуорес-
флуоресцентным красителем и пропускают в струе жидкости через луч
лазера. Жидкость, содержащая флуоресцентно меченые части-
частицы, при прохождении через сопло, в результате вибрации его
разбивается на капли, содержащие отдельные микрочастицы.
Каждая из капель приобретает электростатический заряд. Капли
пролетают между двумя электродами, на которые под управле-
управлением компьютера подается напряжение, создающее электричес-
электрическое поле определенной направленности, что приводит к отклоне-
отклонению траектории падения частиц (рис. 1, a-в). Буферный раствор
подается из резервуара в проточную кювету, в которой происхо-
происходит ее смешивание с анализируемыми микрочастицами (до
Ю5 частиц на 1 мл суспензии), после чего через сопло диаметром
50-250 мкм суспензия впрыскивается в центральную стационар-
стационарную часть струи жидкости, называемую коаксиальным потоком,
Диаметр которой составляет 5-20 мкм. Частицы захватываются
потоком в результате гидродинамического фокусирования и пе-
перемещаются вместе с жидкостью. Движение частиц в коаксиаль-
коаксиальном потоке позволяет им точно попадать далее в сфокусирован-
43
Давление воздуха
Воздух
Буферный раствор
Буферный резервуар
Проточная кювета
Пробирка с
образцом
Буферный
раствор
1
Суспензия
хромосом
Создание электрического
заряда на каплях
Зеленый
фильтр
ЭВМ
Рис. 1. Сортировка микрочастиц в проточном цитометре [70]
а - подача анализируемого образца в проточную кювету цитометра. Под
давлением, создаваемым сжатым воздухом, буферный раствор и суспензия ана-
анализируемых микрочастиц подаются в проточную кювету цитометра; б - основ-
основные компоненты сортирующей и оптической анализирующей систем четырех-
параметрического проточного цитометра; в - формирование капель в процессе
сортировки микрочастиц. Между моментом анализа частицы и приданием кап-
капле, ее содержащей, электрического заряда проходит время, достаточное для то-
того, чтобы только одна (или три) капли приобрели заряд и изменили траекторию
полета в электрическом поле, создаваемом отклоняющими пластинами; X - рас-
расстояние между центрами соседних капель после их выхода из сопла; г - разде-
разделение метафазных хромосом человека с помощью проточной цитометрии [71]
44
в
. Точка анализа
хромосом
Задержка в создании
заряда = ЮЛ.
Отклоняющие
электроды
&
Y
8
о
о
Точка отрыва
Ъ,
00
«Л
m
m
?
.X
u
о
u
I
Область
сортировки
Интенсивность флуоресценции (Хромомицин A3)
4,
ный лазерный луч и оказаться равномерно освещенными, что не-
необходимо для их предсказуемой флуоресценции. При этом анали-
анализируемые частицы следуют в потоке на некотором расстоянии
друг от друга, которое зависит в том числе от их концентрации в
анализируемой суспензии. В проточной камере или сразу после
выхода из нее микрочастицы облучаются сфокусированным ла-
лазерным лучом, и жидкость разбивается на капли, которые приоб-
приобретают электрический заряд.
Место, где лазерный луч пересекает поток жидкости, называ-
называют точкой наблюдения или точкой анализа. Это место окружено
линзами, которые собирают свет, испускаемый частицей в ре-
результате флуоресценции и составляющий анализируемый сиг-
сигнал. Световые сигналы попадают на фотодетекторы (фотодиоды
или фотоумножители). Фотодетекторы, улавливающие рассеян-
рассеянный свет под прямым углом к лазерному лучу и по его ходу, поз-
позволяют оценивать форму и размеры анализируемых частиц, а
также их состав (по коэффициенту преломления их внутренней
среды) и, как следствие, метаболическое состояние клеток. Дли-
Длина волны такого рассеянного света соответствует длине волны
света, испускаемого лазером. Кроме того, в проточных цитофлу-
ориметрах имеется несколько других фотодетекторов, направ-
направленных на точку анализа, которые расположены под разными
фиксированными углами по отношению к лазерному лучу и снаб-
снабжены различными оптическими фильтрами. Это позволяет раз-
разделять и количественно оценивать излучение, возникающее в ре-
результате флуоресценции разных флуорохромов, которыми окра-
окрашивают анализируемые микрообъекты. От количества фотоде-
фотодетекторов, окружающих точку анализа, зависит число парамет-
параметров, которые могут фиксироваться и анализироваться прибором.
В соответствии с этим сами приборы могут быть многопарамет-
многопараметрическими (обычно пяти-, шестипараметрическими). В фотоде-
фотодетекторах световые сигналы преобразуются в электрические, ко-
которые далее обрабатываются с помощью вычислительной техни-
техники. Получив группу сигналов от фото детекторов компьютер в
соответствии с заданной программой дает команду о создании
электрического потенциала определенной полярности на элект-
электродах, которые отклоняют траекторию свободного падения кап-
капли жидкости, содержащей микрочастицу, в ту или другую сторо-
сторону. В результате капля попадает в нужную пробирку, то есть про-
происходит сортировка частиц в соответствии с их свойствами.
Сортировка метафазных хромосом. Один из результатов раз-
разделения метафазных хромосом человека с помощью проточной
цитометрии представлен на рис. 1, г [71]. В этом опыте суспен-
46
Таблица 2
Количество индивидуальных хромосом человека, требуемых
для реализации разных молекулярно-генетических проектов [72]
Область применения Количество
требуемых хромосом
ПЦР» цитогенетическое окрашивание хромосом 3,0 х 102
Клонотеки на основе фаговых векторов 1,6 х 106
(размер вставки ~17 т.п.о.)
Клонирование в космидах (размер вставки ~37 т.п.о.) 6,4 х 106
Клонирование в YAC-векторах (размер вставки 6,4 х 107
-100 т.п.о.)
зию метафазных хромосом метили двумя красителями: Хёхстом
33258 и хромомицином A3, первый из которых взаимодействует
преимущественно с АТ-парами ДНК, а второй - GC-парами. Ок-
Окрашенные таким образом хромосомы, содержание ДНК в кото-
которых может различаться на 50%, разделяли с помощью двухлазер-
ного проточного цитометра. Видно, что в результате сортировки
получают фракции всех хромосом за исключением хромосом, от-
относящихся к группе 9-12. В этом конкретном опыте на основании
различий в содержании ДНК удалось разделить родительские го-
гомологичные хромосомы 19 и 21 (отмечены стрелками). Хромосо-
Хромосомы 9-12, которые не различаются сортировщиком на основании
их флуоресценции с вышеупомянутыми красителями, выделяют
путем сортировки суспензии метафазных хромосом гибридных
клеток хомячок-человек, в которых остается только одна иссле-
исследуемая хромосома человека (см. ниже).
Узким местом, ограничивающим применение проточных
цитометров для наработки метафазных хромосом в препаратив-
препаративном количестве, достаточном для их использования в молеку-
молекулярно-генетических исследованиях, является скорость сорти-
сортировки, а следовательно, и время, которое необходимо затратить
для реализации соответствующих проектов. В табл. 2 приведе-
приведены количества метафазных хромосом, которые требуются для
проведения ряда генно-инженерных экспериментов. При ис-
использовании обычных сортировщиков, которые анализируют
До 1000 частиц/сек, индивидуальные хромосомы человека мож-
можно накапливать лишь со скоростью ~40 хромосом/сек A000/23).
Ясно, что в этом случае 106 хромосом теоретически могут быть
получены только за семь часов непрерывной работы прибора, а
следовательно, для наработки 6,4 х 107 хромосом, требуемых
47
для получения одной клонотеки на основе искусственных хро-
хромосом дрожжей (YAC), потребуется по крайней мере 21 день.
Естественно, решение этой проблемы лежит на пути совершен-
совершенствования техники сортировки. Высокоскоростные сортиров-
сортировщики позволяют получать в сутки до 1,5 х 107 индивидуальных
хромосом человека. Еще большей производительности удается
достигнуть с применением оптической сортировки хромосом.
Этот подход позволяет нарабатывать в сутки до 7,5 х 107 от-
отдельных хромосом человека.
Все упомянутые выше методы очистки нуклеиновых кислот
при использовании в качестве исходного материала клетки бак-
бактерий, животных или растений дают в руки исследователей слож-
сложную смесь генов и некодирующих нуклеотидных последователь-
последовательностей. Для выделения конкретных последовательностей нукле-
отидов и работы с ними необходимо использовать многочислен-
многочисленные ферменты. Свойства некоторых ферментов, широко исполь-
используемых в генной инженерии, кратко рассмотрены ниже.
2.2. Ферменты рестрикции и модификации нуклеиновых кислот
Среди ферментов, используемых в генной инженерии для
клонирования, большое значение имеют эндонуклеазы рестрик-
рестрикции - рестриктазы. Эти ферменты, впервые открытые как
часть системы рестрикции-модификации ДНК у бактерий, специ-
специфически гидролизуют молекулы двухцепочечных ДНК при нали-
наличии в них определенных последовательностей нуклеотидов, на-
называемых сайтами рестрикции. Названия рестриктаз складыва-
складываются из первых букв видовых названий бактерий, в которых они
были обнаружены, например, Есо - Е. coli. В том случае, когда
различные по специфичности действия рестриктазы присутству-
присутствуют в клетках разных штаммов одного вида бактерий, в название
рестриктазы вводят дополнительную букву, например, рестрик-
рестриктазы Hinc и Hind выделены из бактериальных клеток
Haemophilus influenzae, штаммы с и d. Цифры, следующие за бук-
буквенными обозначениями, отражают последовательность откры-
открытия соответствующих рестриктаз в клетках бактерий одного ви-
вида, например, Hael, Haell и НаеШ - из Н. aegipticus.
2.2.1. Классификация систем рестрикции-модификации
По механизму действия и молекулярной структуре различа-
различают пять типов систем рестрикции-модификации и соответствен-
соответственно рестриктаз, но нет основания считать, что эта классификация
48
Тип
системы
II
IIS
III
IV
Субъединичный
состав
Примеры Субстрат Метали-
Метались расщепление) рование
TGAN8TGCT
V318NV3OV
AACN6GTGC (N6mA)
11О9МЭУЭО
CGATN7GTTA
EcoRl GAAATTC (N6mA)
Hhal GCGAC
PvuW
CAGACTG
GGATGN9A (N6mA)
331V3N13A (N6mA)
GGTGAN8A (N6mA)
GAAGAN8A (N6mA)
Eco?l AGACCN?A
Eco?\5 I CAGCAGN25A (N6mA)
StyLTl CAGAGN?
Eco51\ CTGAAGN16A
Gsul CTGGAGN16A
Mmel TCCRACN20
5с^-подобная
^ 5cgl
¦^ 5p/I
5ael
AN10CGAN6TGCN12A
AN8GAGN5CTCN13A
AN10ACN4GTAYCN12
- Распознающий домен
- Модифицирующая метилаза
-ДНК-геликаза
- Эндонуклеаза рестрикции
Рис. 2. Типы бактериальных систем рестрикции-модификации [76]
Подчеркнуты нуклеотиды сайтов рестрикции, подвергающиеся метилиро-
метилированию гомологичными системами модификации по указанному положению
AQ
исчерпывает все классы природных ферментов (рис. 2). Рестрик-
тазы типа I представляют собой сложные мультимерные ком-
комплексы, которые входят в состав системы рестрикции-модифика-
рестрикции-модификации, построенной из пяти субъединиц, две из которых (HsdM -
host specificity determinant) обладают ДНК-метилазной активнос-
активностью, для двух других (HsdR) характерна эндонуклеазная и ДНК-
хеликазная активности, и одна (HsdS) обеспечивает специфич-
специфичность взаимодействия комплекса с ДНК [73]. Тримерный ком-
комплекс M2S метилирует ДНК, однако рестрикцию может осуще-
осуществлять только гетеропентамер M2SR2- Рестриктазы типа I для
проявления своей активности требуют присутствия АТР, S-адено-
зилметионина и ионов Mg2+. Они распознают асимметричные, со-
состоящие из двух частей последовательности нуклеотидов, и в том
случае, если сайт неметилирован, прочно связываются с ним, что
приводит к АТР-зависимой активации хеликазы. В результате
комплекс начинает перемещаться вдоль ДНК, расплетая ее и ос-
оставляя по бокам обе цепи ДНК в виде петель. Расщепление ДНК
происходит после остановки комплекса в том случае, если он
встречает непреодолимое препятствие на своем пути, например в
виде второго такого же комплекса или структуры Холидея, обра-
образовавшейся в ходе рекомбинации. Линейные молекулы ДНК, со-
содержащие один сайт связывания, являются плохими субстратами
для этих ферментов. В результате расщепление ДНК происходит
за тысячи п.о. от сайта узнавания с образованием длинных 3'-вы-
3'-выступающих одноцепочечных концов. С учётом гомологии амино-
аминокислотных последовательностей рестриктазы типа I подразделя-
подразделяют на четыре семейства (IA-ID). В силу всех вышеупомянутых
особенностей эти интересные ферменты не находят широкого
применения в генной инженерии.
Системы рестрикции-модификации типа II устроены наибо-
наиболее просто [74, 75]. В таких системах модифицирующая ДНК-ме-
тилтрансфераза функционирует в виде свободного мономера, а
эндонуклеаза рестрикции - в виде димера, причем эти два фер-
фермента не зависят друг от друга. Рестриктазы типа II узнают спе-
специфические последовательности нуклеотидов в точке расщепле-
расщепления ДНК или непосредственной близости от нее, они активны в
присутствии ионов Mg2+ без каких-либо дополнительных кофак-
кофакторов и чаще всего используются при молекулярном клонирова-
клонировании, а также в ДНК-диагностике.
В системах рестрикции-модификации типа III субъединица
ДНК-метилтрансферазы (Mod) распознает асимметричный сайт
рестрикции (рис. 2) [76]. Эндонуклеаза (Res) может выполнять
свои функции только в комплексе с Mod-субъединицей, причем
50
рестрикция стимулируется S-аденозилметионином. Res-субъеди-
ница обладает активностью АТР-зависимой ДНК-хеликазы, не-
необходимой для транслокации комплекса вдоль ДНК, которая
прекращается после встречи комплекса с препятствием. Однако,
в отличие от рестриктаз типа I, связавшийся с ДНК репрессор
может блокировать транслокацию, но разрыв происходит не в
месте остановки комплекса. Для эффективного расщепления
ДНК требуется наличие двух расположенных по соседству ком-
комплексов, взаимодействующих с неметилированными сайтами уз-
узнавания, которые расположены в обратной ориентации по отно-
отношению друг к другу. Например, хромосома фага Т7 содержит
36 сайтов узнавания рестриктазой ЕсоР151, но в одной ориента-
ориентации, и эта ДНК полностью устойчива к действию данной эндо-
нуклеазы. Ясно, что такая ориентация сайтов не могла возник-
возникнуть случайно (вероятность 1/236 или 1/7 х 1010) и является резуль-
результатом естественного отбора. Механизм запуска расщепления
ДНК в этой системе пока неизвестен.
Системы рестрикции-модификации типа IV распознают
асимметричные последовательности, отступя от которых на оп-
определенные расстояния, делают разрезы ДНК, что сближает их с
системой IIS (см. ниже и рис. 2) [77]. Однако, в отличие от IIS, в
системе типа IV эндонуклеазная и ДНК-метилтрансферазная ак-
активности находятся в составе одной полипептидной цепи, а эндо-
эндонуклеазная активность стимулируется S-аденозилметионином.
Эта метилтрансфераза метилирует лишь одну цепь ДНК, чего
недостаточно для подавления рестрикции в данном сайте. В сис-
системе типа IV обнаружена вторая метилтрансфераза, которая ме-
метилирует обе цепи ДНК, что делает сайт устойчивым к соответ-
соответствующей эндонуклеазе.
Zteg-подобная система рестрикции-модификации названа так
по своему ферменту-прототипу Bcgl, выделенному из Bacillus
coagulans [78]. Так же, как и в предыдущем случае, в этой систе-
системе эндонуклеазная и ДНК-метилтрансферазная активности объ-
объединены в одной полипептидной цепи субъединицы А или а, с ни-
ними не ассоциирована ДНК-хеликазная активность, действие эн-
донуклеазы стимулируется S-аденозилметионином (который
также является донором метальных групп для метилтрансфера-
зы) и не требует АТР. В отличие от системы IV, Zteg-подобная си-
система обладает второй субъединицей В (или Р), которая обеспе-
обеспечивает специфичность действия ферментов. Аналогично систе-
системе I, эта система распознает двойной сайт, разделенный случай-
случайной последовательностью, однако вносит два двухцепочечных
разрыва по обе стороны от сайта (см. рис. 2).
51
2.2.2. Рестриктазы типа II -
основной инструмент генной инженерии
Большинство рестриктаз типа II специфически узнают на
ДНК тетра- и гексануклеотидные последовательности, и, по
крайней мере, три из них - октануклеотиды. При этом некото-
некоторые ферменты узнают частично вырожденные последовательно-
последовательности (например, Hindi - GTYRAC, где Y и R, соответственно, пи-
пиримидин и пурин), другие - симметричные последовательности,
разделенные случайной последовательностью нуклеотидов
(Bgll - GCCN5GGC), сайты рестрикции третьих -квазисимметрич-
ны {Btrl - САС A GTC, где А обозначает точку расщепления),
из-за чего их иногда называют ферментами muna UQ. Ферменты
типа US (от англ. shift) вносят разрывы в ДНК, которые смеще-
смещены на несколько нуклеотидов относительно сайта рестрикции
(см. рис. 2).
Чем короче олигонуклеотидная последовательность сайта
рестрикции, узнаваемого рестриктазой, тем чаще он встречается
в случайной последовательности нуклеотидов, в которой каждый
из четырех нуклеотидов представлен с одинаковой частотой
E0% А-Т-пар и 50% G-C-nap). Так, случайная тетрануклеотид-
ная последовательность встречается в среднем через каждые
256 п.о. D4), а гексануклеотидная - через каждые 4096 п.о. D6).
Однако, в природных ДНК распределение нуклеотидов может
заметно отличаться от случайного. Например, для эукариотичес-
ких ДНК характерна низкая частота встречаемости динуклеоти-
да CpG и, соответственно, сайтов рестрикции, содержащих эти
динуклеотиды (рестриктазы Hha\, Hpall, Taql, Thai, Aval, Haell,
HindU, Sail, Smal, Xhol, Xmal). Существенное отклонение частоты
встречаемости сайтов рестрикции от ожидаемого при случайном
их распределении вдоль ДНК имеет место и в хромосомах термо-
термофильных бактерий, которым, напротив, свойственно (хотя и не
во всех случаях) обогащение по G-C-парам. Для большинства
сайтов, узнаваемых рестриктазами типа II, характерно наличие в
них симметрии второго порядка, т.е. узнаваемые ими последова-
последовательности представляют собой палиндромы, например, у рест-
рестриктазы EcoRl - 5'-GAATTC-3'. Это означает, что нуклеотиды,
расположенные в каждой из цепей на равном расстоянии от оси
симметрии, комплементарны друг другу. Если точки расщепле-
расщепления противоположных цепей ДНК смещены друг относительно
друга в сайте рестрикции, то образующиеся в результате рест-
рестрикции концы ДНК содержат выступающие одноцепочечные
участки. Поскольку такие участки комплементарны сами себе и
52
I
5'— GGA I ТСС
iii ! iii
3' — CCT I AGG— 5'
1
t
5' — G
i
3'— CCTAG— 5'
5'— GATCC — 5'
Q—yBamHl
5'
3'
— GGT I ACC —
III III I
— CCA ! TGG—
3' 5'— GGTA — 3' CC — 3'
__ II II
5' 3'—CC 3'— ATGG — 5' P"
t -
5'
3'
{
¦CAG ! CTG-
III | III
¦GTC i GAC-
1
5'
3'
5'— CAG
Ml
3'— GTC
CTG — 3'
Ml
GAC—5'
Mbol
5' —
3' —
G
1 +
CCTAG —5'
5' —
GATCT — 3'
1
A—5'
5'
*" 3'
— G
1
— С
GATC
MM
CTAG
T — 3'
A—5'
Рис. З. Формы разрывов двухцепочечных ДНК, образующихся под дей-
действием рестриктаз
а - 5'-выступающие A), 3'-выступающие "липкие" B) и "тупые" C) концы
ДНК, образующиеся под действием рестриктаз BamHl, Kpnl и Pvull, соответст-
соответственно. Стрелками обозначены места разрывов цепей ДНК, пунктирной лини-
линией - ось симметрии сайтов рестрикции; б - лигирование с потерей сайта рест-
рестрикции
друг другу и могут между собой взаимодействовать, их часто на-
называют "липкими" концами. В липких концах выступающим од-
ноцепочечным участком может быть как 5'-, так и 3'-конец
(рис. 3, а). Формальным признаком образования 5'- или 3'-высту-
3'-выступающих липких концов в сайтах рестрикции является расположе-
расположение точки расщепления цепей ДНК в последовательности, ис-
используемой для обозначения сайта рестрикции, слева или справа
53
от оси симметрии, соответственно. У некоторых рестриктаз точ-
точки расщепления обеих цепей ДНК расположены непосредствен-
непосредственно друг под другом в сайте рестрикции. В этом случае после рас-
расщепления ДНК липких концов не образуется, а получаются так
называемые "тупые" концы, в которых нет выступающих одно-
цепочечных участков ДНК (см. рис. 3, а). Имеется одно принци-
принципиальное функциональное различие между 5'- и 3'-выступающи-
3'-выступающими липкими концами - последние невозможно пометить путем их
достройки ДНК-полимеразой. Эту особенность следует учиты-
учитывать при выборе рестриктаз для получения рестрикционных
фрагментов ДНК, которые предполагается использовать в каче-
качестве зондов.
При конструировании рекомбинантных молекул полезно по-
помнить, что, хотя рестриктазы BamHl, Bell, Bglll и Xholl узнают
разные сайты рестрикции, они образуют одни и те же липкие
концы, GATC. То же характерно и для группы рестриктаз SalGl,
Xhol и Aval (NCGA). При лигировании (см. ниже) фрагментов
ДНК, образованных рестриктазами одной из таких групп, проис-
происходит их объединение, но при этом исходные сайты рестрикции
теряются, так как в результате образуется новая непрерывная
последовательность нуклеотидов (см. рис. 3, б).
Последовательности нуклеотидов липких концов, образую-
образующихся под действием рестриктаз типа IIS, являются уникальными
для каждого такого сайта рестрикции. Вследствие этого рестрик-
ционные фрагменты ДНК, образовавшиеся под действием дан-
данной рестриктазы, в смеси соединяются друг с другом лишь в стро-
строго определенной исходной последовательности, которая задается
уникальными последовательностями нуклеотидов в липких кон-
концах рестрикционных фрагментов ДНК. Например, при расщеп-
расщеплении рестриктазой типа IIS репликативной формы ДНК фага
фХ174 образуется 14 фрагментов, которые in vitro объединяются
в правильную последовательность с образованием инфекцион-
инфекционной фХ 174-ДНК.
2.2.3. Универсальные рестриктазы для одноцепочечных ДНК
На основе рестриктаз, узнающих асимметричные последова-
последовательности нуклеотидов, разработана система, позволяющая рас-
расщеплять молекулы одноцепочечных ДНК (оцДНК) в любой задан-
заданной точке. С этой целью синтезируется олигонуклеотид, 5'-конце-
5'-концевая часть которого содержит сайт, узнаваемый такой рестрикта-
рестриктазой, а последовательность нуклеотидов 3'-концевой части компле-
комплементарна участку ДНК, в который необходимо внести эндонукле-
54
Участок I
А
5'
5'
Рис. 4. Расщепление одноцепочечной ДНК универсальной рестрикта-
зой
а - синтетический олигонуклеотид; б - одноцепочечная ДНК. После обра-
образования шпильки и гибридизации с одноцепочечной ДНК-мишенью олигонук-
олигонуклеотид образует сайт связывания рестриктазы (участок I) и сайт расщепления
ДНК-ДНК-гибрида (участок II). Стрелки указывают места эндонуклеазного
расщепления ДНК и олигонуклеотида
азный разрыв. В результате гибридизации олигонуклеотида с
оцДНК образуется структура, изображенная на рис. 4. При этом
фермент взаимодействует с сайтом узнавания (участок I), а разры-
разрывы вносятся по местам, обозначенным стрелками. Таким образом,
положение места эндонуклеазного расщепления будет целиком за-
зависеть от последовательности нуклеотидов участка II синтетичес-
синтетического олигонуклеотида, комплементарного ДНК-субстрату.
2.2.4. Изошизомеры
В клетках разных видов бактерий могут содержаться рест-
рестриктазы, узнающие одни и те же сайты рестрикции. Они получи-
получили название изошизомеры. Эти ферменты с успехом используют-
используются для обнаружения метилированных участков ДНК в геноме.
Так, рестриктазы Hhal и Hpall расщепляют неметилированные
последовательности GCGC и CCGG, соответственно, и утрачива-
утрачивают способность к расщеплению, если хотя бы один из остатков
цитозина в этих сайтах метилирован. В то же время фермент
Mspl (изошизомер Hpall) расщепляет последовательность CCGG
независимо от того, метилированы или неметилированы остатки
Цитозина в таком сайте. N-Метилирование остатков аденозина в
ДНК можно обнаружить с помощью изошизомеров Sau3A (рас-
(расщепляет как метилированные, так и неметилированные последо-
последовательности GATC), Dpnl (расщепляет только метилированные
последовательности GMeATC) и Mbol (расщепляет только неме-
неметилированные последовательности).
Среди изошизомеров есть ферменты, которые узнают одни и
те же последовательности, но разрезают их по-разному. Такие ре-
рестриктазы иногда называют гетерошизомерами. Например, рест-
55
риктазы Ассв5\ и Kpnl узнают сайт GGTACC, разрывы в ДНК
вносят следующим способом: G-lGTACC и GGTAC-lC, соответст-
соответственно, а, следовательно, создают разные липкие концы. Гетеро-
шизомеры Smal и Xmal расщепляют ДНК с образованием тупых
концов (CCCiGGG) и липких - (CiCCGGG), соответственно, что
позволяет по-разному клонировать одинаковые по длине фраг-
фрагменты ДНК, образовавшиеся под действием этих ферментов.
2.2.5. Изменение специфичности действия рестриктаз
Влияние неоптимальных условий. Рестриктазы являются вы-
высокоспецифическими ферментами. Однако для поддержания
этой специфичности in vitro необходимо соблюдать в реакцион-
реакционной смеси оптимальные условия для действия ферментов. При
нарушении таких условий у некоторых рестриктаз начинает про-
проявляться вторичная (так называемая штриховая) активность. В
частности, рестриктаза EcoRl расщепляет последовательность
GAATTC при рН 7,3, 100 мМ NaCl в присутствии 5 мМ MgCl2, од-
однако при изменении значений рН, понижении концентрации NaCl
или замене ионов Mg2+ на Мп2+, а также в присутствии органиче-
органических растворителей у фермента появляется тенденция к расщеп-
расщеплению более короткой последовательности ААТТ (так называе-
называемая активность EcoRI* "со звездой")- К рестриктазам, обладаю-
обладающим подобными свойствами, относятся также эндонуклеазы
BamHl, Bstl, Bsul, Ddel, Hha\, Pstl, Sail, Sstl, Xbal.
Действие рестриктаз на необычные субстраты. Помимо двух-
цепочечных ДНК многие рестриктазы способны использовать
ДНК-РНК-гибриды в качестве субстрата. Это относится, в част-
частности, к рестриктазам EcoRl, Hindll, Sail, Mspl, Hhal, Alul, Taql и
Haelll. Некоторые рестриктазы, например, Haelll, Hhal и Sfal,
способны расщеплять оцДНК фага фХ174, хотя и со значительно
меньшей скоростью, чем соответствующую двухцепочечную
RF-форму. Такая способность была продемонстрирована для не-
некоторых других рестриктаз, а также ДНК-субстратов. Остается
неясным, узнают ли эти рестриктазы истинные одноцепочечные
сайты или же последовательности нуклеотидов, заключенные в
элементы вторичной структуры.
С развитием метода полимеразной цепной реакции (ПЦР)
(см. ниже гл. 6) часто возникает необходимость расщепления ре-
стриктазами амплифицированных олигонуклеотидов недалеко
от их концов, т.е. в условиях, когда сайт рестрикции фланкирован
с одного из своих концов одним или несколькими нуклеотидами.
В этом случае установлена четкая зависимость способности
56
Таблица 3
Эффективность расщепления коротких последовательностей
ДНК некоторыми распространенными рестриктазами [79]
Рестриктаза
BamHl
Bgtll
Clal
EcoRl
Hindlll
Nhel
Pstl
Pvul
Sad
Sacll
Smal
Spel
Последовательность
олигонуклеотидов
в окрестностях сайта
рестрикции
CGGATCCG
CGGGATCCCG
CGCGGATCCCGC
CAGATCTG
GAAGATCTTC
GGAAGATCTTCC
CATCGATT
GATCGATC
CCATCGATGG
CCCATCGATGGG
GGAATTCC
CGGAATTCCG
CCGGAATTCCGG
CAAGCTTG
CCAAGCTTGG
CCCAAGCTTGGG
GGCTAGCC
CGGCTAGCCG
CTAGCTAGCTAG
GCTGCAGC
TGCACTGCAGTGCA
CCGATCGG
ATCGATCGAT
TCGCGATCGCGA
CGAGCTCG
GCCGCGGC
TCCCCGCGGGGA
CCCGGG
CCCCGGGG
CCCCCGGGGG
TCCCCCGGGGGA
GACTAGTC
GGACTAGTCC
CGGACTAGTCCG
CTAGACTAGTCTAG
Длина
цепи,нт
8
10
12
8
10
12
8
8
10
12
8
10
12
8
10
12
8
10
12
8
14
8
10
12
8
8
12
6
8
10
12
8
10
12
14
Процент расщепления
олигонуклеотида после
инкубации в течение
2ч
10
>90
>90
0
75
25
0
0
>90
50
>90
>90
>90
0
0
10
0
10
10
0
10
0
10
0
10
0
50
10
10
0
0
10
10
0
0
20 ч
25
>90
>90
0
>90
>90
0
0
>90
50
>90
>90
>90
0
0
75
0
25
50
0
10
0
25
10
10
0
>90
10
10
50
>90
>90
>90
50
50
57
Таблица 3 (окончание)
Рестриктаза
Последовательность
олигонуклеотидов
в окрестностях сайта
рестрикции
Длина
цепи,нт
Процент расщепления
олигонуклеотида после
инкубации в течение
2ч
20 ч
Sphl
Xbal
Xhol
GGCATGCC
CATGCATGCATG
ACATGCATGCATGT
CTCTAGAG
GCTCTAGAGC
TGCTCTAGAGCA
CTAGTCTAGACTAG
CCTCGAGG
CCCTCGAGGG
CCGCTCGAGCGG
Примечание. Жирным курсивом
рестрикции.
8
12
14
8
10
12
14
8
10
12
выделены
0
0
10
0
>90
75
75
0
10
10
0
25
50
0
>90
>90
>90
0
25
75
последовательности сайтов
определенных рестриктаз расщеплять сайты рестрикции от ко-
количества фланкирующих сайт нуклеотидов. Данное свойство ре-
рестриктаз объясняют, в частности, тем, что на самих концах моле-
молекулы дцДНК происходит локальное плавление двойной спирали
ДНК с образованием коротких одноцепочечных участков, захва-
захватывающих сайт узнавания рестриктазами. Частично избежать
локальное плавление можно понижением температуры реакци-
реакционной смеси во время проведения рестрикции таких олигонукле-
олигонуклеотидов. Поскольку эти данные имеют большое значение для
практической генной инженерии, они суммированы в табл. 3.
Проблема возможности изменения субстратной специфично-
специфичности рестриктаз методами белковой инженерии обсуждается в
разделе 3.6.2. второй части книги.
2.2.6. Изменение концов рестрикционных фрагментов ДНК
Часто из-за особенностей первичной структуры в клонируе-
клонируемой ДНК или векторе отсутствуют сайты рестрикции, необходи-
необходимые для их соединения. Для преодоления этих затруднений, свя-
связанных с этим обстоятельством, разработан ряд методов, позволя-
позволяющих изменять последовательности нуклеотидов на концах фраг-
фрагментов ДНК, образованных эндонуклеазами рестрикции. Прежде
всего, можно "затупить" липкие концы путем заполнения их нук-
леотидами с помощью ДНК-полимеразы в том случае, если одно-
цепочечный участок является 5'-выступающим. При наличии
58
3'-выступающих концов их "затупляют" с помощью нуклеаз, спе-
специфичных в отношении оцДНК, а также ДНК-полимераз, облада-
обладающих 3' —> 5'-корректирующей экзонуклеазной активностью (см.
ниже).
К тупым концам фрагментов ДНК можно присоединять ко-
короткие двухцепочечные олигонуклеотиды, называемые линкера-
линкерами, которые содержат требуемый сайт рестрикции. По заверше-
завершении лигирования по тупым концам линкера и фрагмента ДНК
для получения липких концов образовавшиеся гибридные фраг-
фрагменты инкубируют с рестриктазой, специфичной в отношении
сайта рестрикции линкера.
Для замены одного липкого конца на другой используют двух-
двухцепочечные олигонуклеотиды, называемые адаптерами, состоя-
состоящими из двух частично перекрывающихся олигонуклеотидов, ко-
которые по обеим сторонам образуют одноцепочечные липкие кон-
концы. Один из них соответствует липкому концу фрагмента ДНК, а
другой обладает требуемой последовательностью нуклеотидов.
2.2.7. ДНК-метилазы и урацил-ДНК-гликозилазы
Большинство штаммов Е. coli содержит два типа ферментов,
метилирующих ДНК независимо от систем рестрикции-модифи-
рестрикции-модификации: Dam- и "Dcm-метилазы, которые контролируют уровень
экспрессии некоторых генов и репликацию бактериальной ДНК,
а также метят родительскую цепь ДНК, что необходимо для пра-
правильной коррекции ошибочно спаренных нуклеотидов во время
репарации ДНК [80]. Dtfm-метилазы осуществляют перенос ме-
тильных групп в Ыб-положение аденина (N6mA) в последователь-
последовательности GATC. В таком случае многие рестриктазы (например,
ВсП, Mbol или Clal), в состав сайтов рестрикции которых входит
данная метилированная последовательность, перестают расщеп-
расщеплять ДНК по этим сайтам. Аналогичное действие на некоторые
рестриктазы, например EcoRll, оказывают и Dcm-метилазы, осу-
осуществляющие метилирование остатков цитозина в положении
С5 EтС) в последовательностях CMeCAGG и CMeCTGG. Для того
чтобы избежать нежелательного влияния этих метилаз на клони-
клонируемые ДНК, в качестве хозяев используют мутантные штаммы
E.coli: dam- и dcmr. ДНК-метилазы применяют для блокирования
in vitro соответствующих сайтов рестрикции на исследуемых
фрагментах ДНК с целью получения под действием гомологич-
гомологичных рестриктаз фрагментов больших размеров.
Для ДНК-метилтрансфераз эукариот характерна только спе-
специфичность 5шС. Метилирование ДНК происходит редко у био-
59
логических видов, размер генома которых < 108 п.о., и оно повсе-
повсеместно распространено у организмов таксономических групп с
большим размером генома. Метилтрансфераза животных Dnmtl
(DNA methyltransferase 1) относится к так называемым поддержи-
поддерживающим метилтрансферазам, которые обладают слабой актив-
активностью на полностью неметилированной ДНК, и высокой - если
в сайте метилирована одна цепь. В этом случае метилирование
ДНК ассоциировано с хроматином, неактивным в отношении
транскрипции. Также, как и системы рестрикции-модификации у
бактерий, метилтрансферазы животных и растений, по-видимо-
по-видимому, выполняют защитные функции - инактивируют последова-
последовательности, транскрипция которых нежелательна, например мно-
многочисленных ретроэлементов. По непонятным причинам систе-
системы рестрикции-модификации ДНК у эукариот отсутствуют.
Единственным повреждением ДНК, которое является непо-
непосредственным следствием ее ферментативного метилирования,
является окислительное дезаминирование 5тС с образованием
урацила. Такие повреждения репарируются урацил-ДНК-глико-
зилазой, которая сканирует ДНК и удаляет остатки U. Этот фер-
фермент активно используется при проведении направленного мута-
мутагенеза с целью удаления родительской цепи ДНК, в которую
вместо Т исходно вводят остатки U (см. раздел 1.2.2 части II).
2.3. ДНК- и РНК-лигазы
ДНК-лигазы являются важнейшими ферментами метаболиз-
метаболизма ДНК в живых клетках, участвующими в процессах реплика-
репликации ДНК, репарации ее повреждений и рекомбинации [81-83].
Создание фосфодиэфирных связей в одноцепочечных разрывах
дц ДНК с помощью ДНК-лигаз является наряду с рестрикцией
одним из важнейших этапов получения рекомбинантных ДНК
in vitro. Реакция лигирования протекает в три этапа (рис. 5). Вна-
Вначале (этап 1) образуется промежуточный конъюгат фермента с
AMP, в котором остаток AMP соединяется фосфороамидной
связью с остатком Lys в последовательности KXDG полипептид-
полипептидной цепи фермента (однобуквенное обозначение, в котором X -
любой аминокислотный остаток). В зависимости от происхож-
происхождения фермента донорами AMP могут быть NAD+ или АТР. По-
После этого остаток AMP переносится на 5'-фосфатную группу
концевого нуклеотида в точке разрыва ДНК (этап 2) с образова-
образованием промежуточного соединения аденилат-ДНК. (АрррДНК).
Возникшая фосфодиэфирная связь гидролизуется во время нук-
леофильной атаки З'-ОН группы соседнего нуклеотида, что при-
60
Е+АТР
" E-AMP
E-AMP
ОН 9
3'
о"
5'
Р ^О
О"
•О о
ф
i ^
н
о
о
О
AMP
Аденозин
О
II
LO Р O-J
о
Рис. 5. Механизм лигирования ДНК Т4-ДНК-лигазой
7 - образование промежуточного комплекса фермент Е-AMP; 2 - образо-
образование фосфодиэфирной связи
водит к созданию новой фосфодиэфирной связи, восстанавлива-
восстанавливающей целостность сахаро-фосфатного остова и освобождением
AMP (этап 3).
ДНК-лигазы разделяют на два семейства в зависимости от ис-
используемого ими кофактора в качестве донора AMP [84]. АТР-за-
висимые лигазы обнаруживают у бактериальных и эукариотиче-
ских вирусов, архей, дрожжей, млекопитающих и эубактерий.
КАО+-зависимые ДНК-лигазы имеются почти исключительно у
эубактерий. Единственное известное исключение в этом отноше-
отношении составляют энтомопоксвирусы насекомых Melanoplus san-
guinipes и Amsacta moorei. Наибольшее применение в генно-инже-
генно-инженерных исследованиях сегодняшнего дня находит АТР-зависимая
ДНК-лигаза бактериофага Т4. Т4-ДНК-лигаза осуществляет со-
соединение фрагментов дц ДНК, обладающих комплементарными
61
липкими или тупыми концами. Как следует из механизма реак-
реакции, необходимым условием протекания лигирования является
наличие 5'-концевого фосфата и 3'-концевого гидроксила в точ-
точках разрыва цепей ДНК. При этом эффективность соединения
фрагментов ДНК по тупым концам Т4-ДНК-лигазой возрастает
в присутствии Т4-РНК-лигазы, которая осуществляет ковалент-
ное соединение 5'-фосфорилированных концов оцДНК или РНК
с З'-ОН группами одноцепочечных нуклеиновых кислот.
Использование ДНК-лигаз для проведения лигазной цепной
реакции рассматривается в разделе 6.6.1 этой части книги [85].
2.4. Ферменты матричного синтеза ДНК и РНК
К ферментам матричного синтеза нуклеиновых кислот отно-
относятся многочисленные ДНК- и РНК-зависимые ДНК- и РНК-по-
лимеразы, осуществляющие зависимый от матричных ДНК или
РНК синтез нуклеиновых кислот. Эти ферменты обычно исполь-
используются в генной инженерии для получения двухцепочечных мо-
молекул ДНК из одноцепочечных, а также для обратной транс-
транскрипции, т.е. синтеза дцДНК, комплементарных мРНК, которые
называют комплементарными ДНК (кДНК).
2.4.1. ДНК-зависимые ДНК-полимеразы
ДНК-зависимые ДНК-полимеразы осуществляют синтез
ДНК на ДНК-матрицах в процессе репликации вирусных, бакте-
бактериальных и эукариотических, митохондриальных и хлоропласт-
ных ДНК, а также во время их репарации и рекомбинации in vivo
[86-88]. Среди ДНК-зависимых ДНК-полимераз наибольшее
применение в генной инженерии находят ДНК-полимераза I
Е. coli и ее большой фрагмент (фрагмент Клёнова [89]), Т4-ДНК-
полимераза и в последнее время термостабильные ДНК-полиме-
ДНК-полимеразы, особенно ДНК-полимераза Thermus aquaticus (Гяд-полиме-
раза). Все эти ферменты в присутствии ионов Mg2+ из четырех
дезоксирибонуклеозидтрифосфатов (dATP, dCTP, dGTP и ТТР)
осуществляют синтез ДНК, комплементарной матричной ДНК, и
для своего функционирования требуют наличия затравки на мат-
матричной оцДНК, т.е. олиго- или полидезоксирибонуклеотида со
свободным З'-ОН-концом, комплементарного матричной ДНК.
ДНК-полимераза IE. coli состоит из одной полипептидной цепи с
молекулярной массой около 109 к Да и обладает тремя активнос-
активностями: полимеризующей в направлении 5' -> 3', 5' -> З'-экзонук-
леазной и 3' —> 5'-экзонуклеазной. Кроме того, в присутствии из-
62
бытка неорганического пирофосфата в реакционной смеси ДНК-
полимераза I может осуществлять пирофосфоролиз ДНК, т.е.
реакцию, обратную полимеризации нуклеотидов. Большой фраг-
фрагмент ДНК-полимеразы IE. coli (фрагмент Клёнова) является ча-
частью полипептидной цепи ДНК-полимеразы I с молекулярной
массой около 76 кДа, у которой отсутствует домен, соответству-
соответствующий 5' —> З'-экзонуклеазе.
Как ДНК-полимераза I, так и ее фрагмент применяется для
введения радиоактивно меченых дезоксирибонуклеотидов в син-
синтезируемые цепи ДНК путем ник-трансляции, т.е. перемещения
одноцепочечного разрыва вдоль молекулы дцДНК, в котором
З'-ОН-конец используется в качестве затравки для ферментов
[90]. При этом ДНК-полимераза I прокладывает себе путь с по-
помощью 5' —> З'-экзонуклеазы, а фрагмент Клёнова вытесняет
цепь ДНК с 5'-конца. Кроме того, фрагмент Клёнова использу-
используют для синтеза второй цепи кДНК, секвенирования ДНК по ме-
методу Сэнгера, заполнения 5'-выступающих липких концов ДНК с
образованием тупых концов, введения концевой радиоактивной
метки, а также для удаления 3'-выступающих концов рестрикци-
онных фрагментов ДНК 3' -> 5'-экзонуклеазой этого фермента.
Как и ДНК-полимераза I E. coli, Т4-ДНК-полимераза обладает
3' —> 5'- (но не 5' —> 3'-) экзонуклеазной активностью, которая у
последней, по крайней мере, в 200 раз выше. Это позволяет ис-
использовать Т4-ДНК-полимеразу, в частности, для введения ра-
радиоактивной метки путем реакции обмена немеченного 3'-конце-
3'-концевого нуклеотида на меченный радиоактивным изотопом. Кроме
того, это свойство фермента используется для получения векто-
векторов для клонирования без использования ДНК-лигазы (см. ниже
раздел 3.1).
Термостабильная Гяд-ДНК-полимераза в настоящее время
широко применяется для проведения ПЦР (см. главу 6.1), а ее мо-
модифицированный (мутантный) аналог, для которого характерно
многократное повышение сродства к терминирующим синтез
ДНК дидезоксирибонуклеозидтрифосфатам, - для секвенирова-
секвенирования ДНК по методу Сэнгера.
2.4.2. РНК-зависимые ДНК-полимеразы
Обратные транскриптазы, иногда называемые ревертазами,
способны осуществлять синтез ДНК на матрице РНК, полимери-
зуя четыре дезоксирибонуклеозидтрифосфата, как это имеет ме-
место в случае ДНК-зависимых ДНК-полимераз. Обратные транс-
транскриптазы, так же, как и ДНК-полимеразы, функционируют
63
только при наличии затравки. Широко используемая в генной ин-
инженерии обратная транскриптаза вируса миелобластоза птиц,
помимо полимеризующей активности, обладает 5' —> 3'- и
3' —> 5'-экзорибонуклеазными активностями и расщепляет РНК
в ДНК-РНК-гибридах по процессивному механизму, т.е. облада-
обладает активностью РНКазы Н. Кроме данного фермента для синте-
синтеза ДНК на матрице РНК часто используют обратную транскрип-
тазу вируса лейкоза мышей Молони (MMLV). Обратные транс-
криптазы находят применение в синтезе дцДНК, комплементар-
комплементарных РНК (особенно мРНК), для последующего ее клонирования
в плазмидных векторах при получении клонотек кДНК (см. ниже
раздел 4.1.2). Кроме того, эти ферменты, подобно ДНК-полиме-
разам, могут быть использованы для введения радиоактивной
или флуоресцентной метки в ДНК-зонды в составе соответству-
соответствующим образом меченых дезоксирибонуклеозидтрифосфатов.
Недавно способность синтезировать ДНК на матрице РНК в
определенных условиях была продемонстрирована для термоста-
термостабильных ДНК-полимераз Thermus thermophilus и Т. flavus. Это
позволяет использовать их для прямого обнаружения специфиче-
специфических РНК в биологических образцах методом ПЦР. Современ-
Современные модификации такого подхода дают возможность в одной ре-
реакционной смеси (и пробирке) синтезировать в реакции обратной
транскрипции небольшое число копий амплифицируемого фраг-
фрагмента ДНК на матрице РНК, которые сразу же используются
тем же ферментом в качестве матрицы в обычной ПЦР (см. ни-
ниже главу 1.6).
2.5. Другие ферменты, используемые в генной инженерии
Среди других многочисленных ферментов, используемых в
генной инженерии, прежде всего следует упомянуть полинуклео-
тидкиназы, которые осуществляют перенос у-фосфатных групп
АТР на 5'-ОН группы ДНК или РНК. Полинуклеотидкиназа бак-
бактериофага Т4 широко используется для введения радиоактивной
метки в ДНК или РНК с целью получения радиоактивно мече-
меченых зондов или секвенирования нуклеиновых кислот. Два ти-
типа реакций используется для введения концевой метки в ДНК с
помощью полинуклеотидкиназы: прямая реакция, когда у-фос-
фатная группа переносится ферментом непосредственно к дефо-
сфорилированному 5'-концу ДНК, и реакция обмена. В послед-
последнем случае присутствие в реакционной смеси избытка ADP вы-
вынуждает полинуклеотидкиназу осуществлять перенос фосфатной
64
группы с 5'-конца ДНК на ADP с образованием АТР, а освобо-
освободившаяся ОН-группа далее фосфорилируется у-фосфатом по
обычному механизму.
Терминальная трансфераза из тимуса теленка (терминальная
дезоксирибонуклеотидилтрансфераза), осуществляющая после-
последовательное присоединение дезоксирибонуклеозидмонофосфа-
тов из пула дезоксирибонуклеозидтрифосфатов к 3'-ОН-группам
молекул ДНК, используется для введения радиоактивной метки в
составе меченых нуклеотидов в 3'-концы ДНК, а также присое-
присоединения к 3'-концам фрагментов ДНК (особенно кДНК) протя-
протяженных гомополимерных последовательностей нуклеотидов
{коннекторов) для последующего их клонирования.
Щелочные фосфатазы бактерий (ВАР) и кишечника теленка
(CIAP) катализируют удаление 5'-фосфатных групп ДНК или
РНК, а также расщепление макроэргических связей рибо- и дез-
оксирибонуклеозидтрифосфатов. Их используют при подготовке
фрагментов нуклеиновых кислот к введению 5'-концевой радио-
радиоактивной метки 32Р, а также для предотвращения лигирования
векторных молекул ДНК самих на себя. Как уже упоминалось
выше, ДНК-лигаза способна образовывать фосфодиэфирные
связи в одноцепочечных разрывах лишь при наличии в них
5'-концевого фосфата. Удаление 5'-концевых фосфатных групп
молекул вектора, линеаризованных с помощью одной рестрикта-
зы во время подготовки его для клонирования соответствующе-
соответствующего фрагмента ДНК, предотвращает образование кольцевых мо-
молекул вектора (без вставки), а также его олигомеров во время ли-
лигирования с клонируемой последовательностью. Необходимые
для лигирования со вставкой фосфатные группы содержатся в са-
самих клонируемых фрагментах ДНК, а остающиеся в результате
неполного лигирования два одноцепочечных разрыва репариру-
ются in vivo после введения вектора со вставкой в бактериальные
клетки. Гены секретируемых щелочных фосфатаз находят при-
применение и в качестве высокочувствительных генов-репортеров
при исследовании функционирования регуляторных элементов
различных генов в прокариотических и дрожжевых системах экс-
экспрессии, а также в клетках млекопитающих [91].
В заключение следует рассмотреть некоторые нуклеазы, ча-
часто используемые в генной инженерии. Нуклеазы - это гидроли-
гидролитические ферменты, расщепляющие фосфодиэфирные связи в
нуклеиновых кислотах. По механизму расщепления субстратов
нуклеазы разделяют на экзо- и эндонуклеазы. Экзонуклеазы осу-
осуществляют последовательное отщепление мононуклеотидов или
небольших олигонуклеотидов с концов молекул ДНК или РНК,
3. Патрушев Л.И. Т. 1 65
тогда как эндонуклеазы вносят в молекулы нуклеиновых кислот
внутренние разрывы. В частности, к эндонуклеазам относятся
многочисленные рестриктазы, рассмотренные выше. Некоторые
нуклеазы обладают более широкой специфичностью и могут ги-
дролизовать как РНК, так и ДНК, а также одновременно прояв-
проявлять эндо- и экзонуклеазную активности. Нуклеаза Ва1Ъ\ из
Alteromonus espejina последовательно отщепляет отдельные мо-
но- и олигонуклеотиды с 5'- и 3'-концов дцДНК, укорачивая обе
цепи ДНК приблизительно с равной скоростью. При этом нукле-
нуклеаза Ва1Ъ\ гидролизует и оцДНК.
Экзонуклеаза III E. coli катализирует последовательное от-
отщепление 5'-нуклеотидов из дцДНК в направлении 3' —> 5'. Фер-
Фермент обладает эндонуклеазной активностью по отношению к
апуринизированной ДНК, активностью РНКазы Н (гидролиз
РНК в РНК-ДНК-гибридах) и З'-фосфатазной активностью.
Экзонуклеаза фага X. катализирует последовательное отщеп-
отщепление 5'-мононуклеотидов в дцДНК при наличии в них 5'-конце-
5'-концевых фосфатных групп. Ее используют для получения молекул
оцДНК с целью их последующего секвенирования.
Нуклеаза S1 из Aspergillus orizae C5 кДа, 267 а.о.), в присут-
присутствии ионов Zn2+ специфически расщепляет молекулы оцДНК
и РНК с образованием 5'-фосфорилированных моно- и олиго-
нуклеотидов. Фермент специфически распознает и расщепляет
одноцепочечные участки в одноцепочечных разрывах, брешах
и петлях двухцепочечных ДНК, но не в одиночных ошибочно
спаренных нуклеотидах. Те же свойства присущи и нуклеазе зо-
золотистой фасоли (mung bean), а также нуклеазе Р1 из Penicillium
citrinum.
Панкреатическая рибонуклеаза А (РНКаза А) является не-
небольшим белком A24 а.о.), который обладает активностью эндо-
рибонуклеазы, специфически расщепляющей фосфодиэфирные
связи, образованные пиримидиновыми нуклеотидами [92]. Про-
Продуктами гидролиза являются З'-фосфорилированные пиримиди-
новые мононуклеотиды и олигонуклеотиды, содержащие конце-
концевые пиримидин-3'-монофосфаты. Во многих тканях всех млеко-
млекопитающих обнаружен специфический белковый ингибитор RI
D50-460 а.о.), обладающий высоким сродством к РНКазе А [93].
Панкреатическая дезоксирибонуклеаза I (ДНКаза I) пред-
представляет собой эндонуклеазу, гидролизующую как одно-, так и
дцДНК с образованием сложной смеси моно- и олигонуклеоти-
дов, содержащих 5'-фосфатные группы. В присутствии ионов
Mg2+ ДНКаза I независимо атакует каждую цепь ДНК, при этом
места разрывов располагаются статистически вдоль молекулы, а
66
в присутствии ионов Мп2+ расщепляет обе цепи ДНК приблизи-
приблизительно напротив друг друга.
Рассмотренные ферменты не исчерпывают всего списка, ис-
используемого в генной инженерии. Особенности применения мно-
многих из этих ферментов для решения конкретных генно-инженер-
генно-инженерных задач будут еще не раз обсуждаться при дальнейшем изложе-
изложении принципов генной инженерии.
з* 67
Глава 3
Векторы
Ферменты, описанные в разделе 1.2, позволяют работать как
с протяженными молекулами ДНК, так и с их фрагментами. Од-
Однако с использованием только этих ферментов еще нельзя ре-
решить одну из основных методических задач молекулярной гене-
генетики - выделение любой требуемой нуклеотидной последова-
последовательности в чистом виде и в количестве, достаточном для иссле-
исследования этих последовательностей биохимическими методами.
Исключение составляет метод ПЦР, однако его применение ог-
ограничивается короткими последовательностями нуклеотидов.
Основная идея, позволяющая решать эту задачу, заключает-
заключается в том, чтобы присоединить исследуемые фрагменты ДНК к
молекуле-переносчику, которая могла бы автономно существо-
существовать внутри бактериальных или эукариотических клеток в виде
одной или нескольких копий и передаваться вместе со встроен-
встроенным в нее фрагментом ДНК от одной клетки к другой. Такие мо-
молекулы-переносчики фрагментов нуклеиновых кислот были со-
созданы, их называют векторами, и они являются одним из важ-
важнейших инструментов генной инженерии.
Идеальная векторная молекула должна обладать нескольки-
несколькими обязательными свойствами. Во-первых, любой вектор дол-
должен длительное время существовать в популяции клеток-хозяев,
т.е. реплицироваться автономно или вместе с хромосомами кле-
клеток. Во-вторых, в любом векторе должны быть биохимические
или генетические маркеры, которые позволяли бы обнаружи-
обнаруживать его присутствие в клетках. В-третьих, структура векторной
молекулы должна допускать встраивание в нее чужеродной по-
последовательности нуклеотидов без нарушения ее функциональ-
функциональной целостности. Для конструирования векторов в генной инже-
инженерии используют небольшие молекулы нуклеиновых кислот,
способные к автономной репликации в бактериальных и эука-
эукариотических клетках - плазмиды и эписомы, хромосомы виру-
вирусов, а также фрагменты хромосом эукариотических клеток.
3.1. Плазмидные векторы
Первые эффективные векторы для клонирования фрагмен-
фрагментов чужеродной ДНК, не утратившие своего значения и поныне,
были получены с использованием бактериальных плазмид. И се-
68
годня векторы на основе плазмид чаще всего используются в мо-
лекулярно-генетических исследованиях. Целый ряд уникальных
биологических свойств плазмид способствовали их широкому
применению в качестве векторных молекул.
3.1.1. Биологические свойства бактериальных плазмид
Плазмиды являются молекулярными эндосимбионтами кле-
клеток, в которых они постоянно существуют [94]. Гены природных
плазмид часто берут на себя выполнение функций, которые да-
дают преимущество клеткам-хозяевам в борьбе за существование
в изменяющейся окружающей среде. Так, одна группа генов
обеспечивает клеткам устойчивость к токсическим веществам
(антибиотикам, ионам тяжелых металлов и т.п.), другие плаз-
мидные гены кодируют белки (например, колицины), токсичные
для клеток, не обладающих такими плазмидами, третьи обеспе-
обеспечивают прикрепление бактериальных клеток к субстрату, чет-
четвертые помогают осваивать клеткам-хозяевам новые источники
углерода. В определенных условиях практически любой ген бак-
бактериальной хромосомы может быть перенесен с помощью ре-
рекомбинации в плазмиды и далее с их помощью распространить-
распространиться в популяции микроорганизмов. Существование плазмид в
клетках-хозяевах во многом зависит от них, однако способность
к автономной репликации дает плазмидам определенную свобо-
свободу от конкретного хозяина.
Таким образом, способность к автономной репликации явля-
является важнейшим биологическим свойством любой плазмиды,
обеспечивающим ее независимое (в определенных пределах) су-
существование [95]. Автономная репликация обеспечивается нали-
наличием области начала репликации, на котором происходит сбор-
сборка макромолекулярного комплекса, осуществляющего инициа-
инициацию и продолжение синтеза плазмидной ДНК. Для своей репли-
репликации различные плазмиды имеют разную потребность в фер-
ферментах клетки-хозяина. Так, плазмиды ColEl, используемые в ка-
качестве основы при конструировании многих векторных плазмид,
для синтеза своей ДНК используют исключительно белки клет-
клетки-хозяина, в том числе ДНК-полимеразу I E. coli на ранних ста-
стадиях репликации и ДНК-полимеразу III - на заключительных.
Синтез ДНК на этих плазмидах можно моделировать с использо-
использованием бесклеточных экстрактов бесплазмидных штаммов бак-
бактериальных клеток.
Существование плазмид, не зависимое от хромосомы клетки-
хозяина, невозможно без осуществления контроля числа копий
69
плазмидной ДНК в клетке, осуществляемого самими плазмидами
[96]. Все плазмиды осуществляют негативный контроль синте-
синтеза ДНК с использованием специфических ингибиторов реплика-
репликации. Одним из распространенных механизмов негативного кон-
контроля является синтез на матрице плазмидной ДНК антисмысло-
вой РНК (контртранскрипта), который может быть комплемен-
комплементарен или мРНК белка-инициатора репликации Rep, или РНК-
праймеру (в том числе, его предшественнику), необходимому для
инициации синтеза плазмидной ДНК. Такой механизм функцио-
функционирует у небольших плазмид типа ColEl, а также контролирует
репликацию низкокопийных больших плазмид наподобие поло-
полового F-фактора [97]. Другой тип негативной регуляции реплика-
репликации, обнаруженный, в частности, у плазмиды pSClOl, использует
специальные регуляторные последовательности - итероны, ко-
которые представляют собой прямые повторы, расположенные
вблизи области начала репликации и гена rep, кодирующего по-
позитивный транс-действующий регулятор репликации плазмидной
ДНК [98]. Итероны конкурируют с областью начала репликации
за связывание Rep-белка, присутствующего в клетке в неболь-
небольшом количестве, и связывают его полностью при достижении
числа копий плазмиды определенного уровня, что полностью
блокирует инициацию репликации. Наличие уникальных меха-
механизмов контроля числа копий плазмид в клетке обеспечивает
поддержание соответствующих плазмид в клетке на уровне од-
ной-двух копий (низкокопийные плазмиды) или 10-100 (плазми-
(плазмиды с умеренной копийностью и высококопийные плазмиды).
С учетом вышеперечисленных механизмов удалось получить
мутантные миниплазмидные производные плазмиды R1 с нару-
нарушенным контролем числа копий. Такие миниплазмиды обладают
температурочувствительными репликонами, которые при 30°С
обеспечивают их существование в виде 20-50 копий на клетку, а
при повышении температуры сверх пермиссивной и инкубации
клеток на питательной среде в течение 2-3 ч количество плаз-
плазмидной ДНК достигает 70% от суммарной клеточной ДНК [99].
Векторы, сконструированные на основе таких плазмид, способ-
способны обеспечивать очень высокий уровень экспрессии рекомби-
нантных белков, гены которых клонированы с их помощью. По-
Повышение числа внутриклеточных копий плазмид в ряде случаев
может быть достигнуто и другим распространенным способом.
Как уже упоминалось, репликация многих плазмид, в том числе
ColEl, не требует синтеза белков, кодируемых их генами, тогда
как контроль числа копий зависит от синтеза плазмидных макро-
макромолекул. Инкубация клеток, содержащих такие плазмиды, с
70
хлорамфениколом блокирует репликацию ДНК клетки-хозяина,
но не плазмидной ДНК. Это приводит к резкому (~50-кратному)
увеличению числа молекул плазмид по отношению к хромосом-
хромосомной ДНК, синтез которых обеспечивается предсуществующими
белками бактериальной клетки.
Еще одним важным для генной инженерии свойством плаз-
плазмид является консервативность их размера. Минимальный раз-
размер плазмиды диктуется необходимостью расположения в ее мо-
молекуле всех генов и регуляторных последовательностей, необхо-
необходимых для поддержания ее внутриклеточной автономии. Другое
требование, которое необходимо соблюдать при конструирова-
конструировании искусственных плазмид - это близкое друг к другу располо-
расположение взаимозависимых регуляторных участков молекулы. Это
необходимо для обеспечения высокой вероятности их совмест-
совместной передачи в дочерние клетки. Повышение размера плазмиды
сверх оптимального будет отрицательно сказываться на ее ста-
стабильности. Случайно возникающие в популяции бактерий му-
тантные формы плазмиды меньшего размера будут реплициро-
реплицироваться быстрее, и постепенно бактериальные клетки с мутантны-
ми плазмидами вытеснят клетки с исходными молекулами.
И, наконец, в завершение этого раздела, необходимо вспом-
вспомнить о совместимости разных плазмид в бактериальных клетках.
Различные близкородственные плазмиды, как правило, не могут
длительное время сосуществовать друг с другом в клетках потом-
потомства исходной их содержащей бактериальной клетки [100]. В ре-
результате после некоторого количества клеточных делений в до-
дочерних клетках остается только один вид плазмид из нескольких
первоначально в них присутствующих. Это свойство плазмид на-
называется несовместимостью. Считается, что две плазмиды от-
относятся к разным группам несовместимости, если они способны
стабильно сосуществовать в одной бактериальной клетке. При-
Причина несовместимости близкородственных плазмид в бактери-
бактериальных клетках проста: все они обладают одним и тем же или
очень похожим механизмом контроля числа их копий. Если ре-
прессор одной плазмиды будет подавлять репликацию другой и
наоборот, решение о том, какая плазмида будет реплицировать-
реплицироваться в данное время, будет приниматься случайно, что с большой
вероятностью будет приводить к изменению их соотношения.
В том случае, если это соотношение изменится, например, до
3 : 1, а сами плазмиды окажутся неразличимыми по поведению
при сегрегации в дочерние клетки, они будут утрачиваться слу-
случайным образом, независимо друг от друга, и одна из плазмид мо-
может быть полностью потеряна во время следующего клеточного
71
деления. Тот же механизм будет функционировать и при распре-
распределении многокопийных плазмид, таких как pBR322, между до-
дочерними клетками, поскольку их передача в дочерние клетки
осуществляется случайным образом. Эти особенности биологии
плазмид объясняют, почему, несмотря на наличие возможности
одновременного проникновения в бактериальные клетки во вре-
время их трансформации нескольких рекомбинантных плазмид, от-
отдельные рекомбинантные клетки содержат плазмиды только од-
одного вида. Одновременно все это еще раз указывает на необходи-
необходимость соблюдения в своей экспериментальной деятельности зо-
золотого правила микробиологии - всегда начинать работу с бакте-
бактериальными штаммами после их рассева до отдельных колоний.
Плазмиды серии pBR как основа для конструирования плаз-
плазмидных векторов. Большая серия векторных плазмид, обозна-
обозначенных символом pBR, создана на основе репликона природной
плазмиды ColEI, придающей клеткам Е. coli устойчивость к коли-
цину путем его объединения с генами устойчивости к антибиоти-
антибиотикам. Таким образом, бактериальные клетки, несущие подобные
комбинированные плазмиды, приобретают устойчивость к соот-
соответствующим антибиотикам, и их было легко отличить от бес-
плазмидных клеток путем простого посева на питательную среду
с антибиотиками. Генетическая карта одного широко распрост-
распространенного вектора этой серии - pBR322 изображена на рис. 6, а.
Такая плазмида представляет собой кольцевую ковалентно замк-
замкнутую молекулу ДНК длиной 4363 п.о. Последовательность нук-
леотидов pBR322 полностью известна. Плазмида содержит гены
устойчивости к тетрациклину (Тс) и ампициллину (Ар), которые
были перенесены в плазмиду pBR322 из плазмиды pSClOl и
транспозона ТпЗ, соответственно. Оба этих гена являются селек-
селектируемыми генетическими маркерами плазмиды, т.е. позволяют
проводить отбор бактериальных клеток с плазмидой pBR322 по
их способности к росту на питательных средах в присутствии те-
тетрациклина и(или) ампициллина. Плазмида pBR322 содержит
также обеспечивающий ее стабильную репликацию в клетках
Е. coli участок ДНК, который включает область начала реплика-
репликации (ori). Характерной чертой плазмиды pBR322, как и любого
современного вектора, является наличие в ней нескольких уни-
уникальных сайтов рестрикции, обозначенных на генетической кар-
карте. Следует иметь в виду, что встраивание в плазмиду клонируе-
клонируемых чужеродных фрагментов ДНК по сайтам рестрикции, распо-
расположенным в генах Ampr или Тсг (например, Pstl или ВатШ), бу-
будет нарушать целостность этих генов и их функциональную ак-
активность. В результате происходит утрата бактериальными клет-
72
ками, содержащими рекомбинантные плазмиды, устойчивости к
соответствующим антибиотикам. По такому признаку легко раз-
различить бактериальные клетки, не содержащие плазмиды (не рас-
растут в присутствии ампициллина и тетрациклина), клетки с плаз-
мидой, не содержащей вставки клонируемой ДНК (растут в при-
присутствии обоих антибиотиков), и клетки с рекомбинантными
плазмидами (в зависимости от локализации вставки могут расти
на среде только с одним из двух вышеупомянутых антибиотиков).
Следовательно, наличие в векторных молекулах селектируемых
маркеров резко повышает эффективность клонирования из-за
возможности проведения быстрого отбора рекомбинантных
плазмид на селективных питательных средах.
Помимо генов устойчивости к антибиотикам в качестве се-
селектируемых маркеров используют и другие гены или их фраг-
фрагменты. В частности, для этих целей часто применяются гены раз-
различных ферментов, присутствие которых в клетках в составе
плазмиды обнаруживают по появлению соответствующей фер-
ферментативной активности. В часто используемых векторах серии
pUC таким селектируемым маркером является 5'-концевая часть
гена C-галактозидазы Е. сой - lacZ' (см. рис. 6, б). Эта часть гена,
находящаяся под контролем lac-промотора, кодирует N-конце-
вую часть р-галактозидазы (так называемый а-пептид), которая
путем объединения с недостающей С-концевой частью полипеп-
полипептида без образования пептидной связи восстанавливает фермен-
ферментативную активность р-галактозидазы. р-Галактозидаза облада-
обладает способностью расщеплять искусственный субстрат Xgal
E-бром-4-хлор-3-индолил-/)-галактопиранозид) с образованием
окрашенного в голубой цвет продукта реакции. В том случае,
когда в бактериальных клетках, которые содержат в хромосоме
недостающую экспрессирующуюся 3'-концевую часть гена lacZ и
выращиваются на среде с Xgal, в результате комплементации
(Х-пептидом вектора появляется активность р-галактозидазы, об-
образованные этими клетками бактериальные колонии окрашива-
окрашиваются в голубой цвет. Уникальные сайты рестрикции для клони-
клонирования ДНК локализованы в начале гена р-галактозидазы в со-
составе полилинкера (синтетической последовательности нуклео-
тидов, содержащей несколько перекрывающихся уникальных
сайтов рестрикции), который не нарушает функциональной це-
целостности последовательности нуклеотидов ос-пептида. Вставка
рекомбинантной ДНК в эти векторы разрывает структурную
часть гена р-галактозидазы и инактивирует его, в связи с чем ко-
колонии бактерий с подобными рекомбинантными плазмидами, вы-
выросшие на питательной среде с Xgal, не окрашены. Поскольку
73
а
в
EcoRl
Pstl
Ppal
Clal
Pvul
Mboll
AfllU
HglAl
Ddel
Bspl2S6
ApaLl Haell
Maell
Xmnl
EcoR\
Nar\
Nad
Afllll
456
Pvul
Pvul
Т7-промотор
Полилинкер
ТЗ-промотор
Pvull
Место расщепления Место расщепления
тромбином энтерокиназой
Экзон 1
Промотор CMV
Энхансер CMV
00 х
Терминатор транскрипции
C-глобинового гена кроликов
II!
74
векторы серии pUC одновременно содержат и ген устойчивости к
ампициллину, отбор бактерий, несущих рекомбинантные плаз-
*ШДЫ> можно проводить одновременно по этим двум маркерам.
На питательной среде с ампициллином и Xgal вырастают только
бактерии, устойчивые к антибиотику, т.е. содержащие плазмиду
pUC, а среди выросших колоний лишь неокрашенные содержат
вставку чужеродной ДНК. В качестве селектируемых маркеров в
векторных молекулах часто используются гены, присутствие ко-
которых может быть обнаружено косвенно по комплементации ге-
генетических дефектов бактериальных клеток-хозяев, что делает
их жизнеспособными в определенных селективных условиях. По-
Подробнее об одной из таких систем см. в разделе 7.3.3.
Плазмиды серии Bluescript. За короткий период развития
генной инженерии было сконструировано труднообозримое ко-
количество векторных плазмид, обеспечивающих конкретные по-
потребности исследователей. Одной из вершин генно-инженерного
искусства, прекрасно иллюстрирующей возможности генной ин-
инженерии, в настоящее время являются полифункциональные
векторы серии Bluescript, полученные фирмой "Stratagene"
(США) (см. рис. 6, г). Вектор Bluescript Ml3+ представляет собой
кольцевую ковалентно замкнутую молекулу ДНК длиной около
3 т.п.о. Он включает в себя ген устойчивости к ампициллину
Атрт, ген C-галактозидазы lacZ, в N-концевую часть которого
встроен полилинкер, содержащий уникальные сайты рестрикции
для 21 рестриктазы, промоторно-операторную область lacZ, a
также ген lac-репрессора lad. В результате встраивания клониру-
клонируемого фрагмента ДНК в полилинкер происходят разрыв кодиру-
Рис. 6. Различные плазмидные векторы для клонирования ДНК и их ре-
стрикционные карты
Обозначены положения уникальных сайтов рестрикции, а также функцио-
функционально значимых генов
а - векторная плазмида pBR322; б - экспрессирующая векторная плазмида
pUC18; в - векторная плазмида 7CAN7, предназначенная для отбора рекомби-
нантных клонов с использованием гомологичной рекомбинации; г - много-
многофункциональная векторная плазмида Bluescript; д - вектор pTriEx-4 Ek/LIC
(Novagen, США), используемый для клонирования ДНК без применения ДНК-
лигазы с последующей кратковременной экспрессией клонированных генов в
клетках животных. LIC - участки вектора, содержащие липкие концы; S- HSV
и His-tags - последовательности, кодирующие аффинные пептидные метки (см.
табл. 5); CMV и Л lac и polh - промоторы цитомегаловируса, Т7-РНК-полиме-
разы (гибрид с /ас-промотором) и гена полигедрина бакуловирусов для транс-
транскрипции клонированных генов в клетках млекопитающих, бактерий или насе-
насекомых, соответственно; цифры внутри ОРС соответствуют нумерации нуклео-
тидов в молекуле вектора
75
ющей части гена lacZ и инактивация C-галактозидазы, что, как и
в случае вектора pUC18, можно обнаружить по исчезновению ок-
окраски колоний бактерий, содержащих этот вектор со вставкой
клонированной ДНК. Кроме того, встроенный в полилинкер
фрагмент ДНК попадает под контроль промоторно-операторной
регуляторной последовательности гена lacZ и в присутствии ин-
индуктора IPTG может быть экспрессирован в клетках Е. coli. В до-
дополнение к этому полилинкер в векторной плазмиде содержит на
одном конце промотор для Т7-, а на другом - для ТЗ-РНК-поли-
мераз, которые ориентированы навстречу друг другу, что позво-
позволяет транскрибировать любую из цепей клонированного фраг-
фрагмента ДНК in vitro с помощью той или другой РНК-полимеразы
и получать препаративные количества мРНК или же комплемен-
комплементарной ей антисмысловой РНК. Кроме того, вектор Bluescript
М13+ обладает межгенной областью (IG) фага fl, родственного
фагу М13. Эта область детерминирует все цмс-действующие
функциональные последовательности нуклеотидов фага, необхо-
необходимые для репликации его хромосомы и упаковки ее в фаговые
частицы. В присутствии фага-помощника М13 происходит пре-
преимущественная упаковка образовавшейся в результате реплика-
репликации одноцепочечной плазмиды в фаговые частицы М13. ОцДНК
Bluescript M13+ после очистки может быть использована непо-
непосредственно для секвенирования клонированной ДНК или прове-
проведения сайт-специфического мутагенеза. Векторы типа Bluescnpt
М13+, способные существовать либо в виде плазмиды, либо в со-
составе фаговых частиц нитевидных бактериофагов, называют
фагмидами.
3.1.2. Векторы для клонирования
без применения ДНК-лигазы
Клонирование без лигирования вектора и вставки (ligation-
independent cloning - LIC) становится возможным в том случае, ес-
если комплементарные одноцепочечные участки (липкие концы)
вектора и клонируемого фрагмента ДНК после отжига друг на
друга остаются стабильными без их предварительного ковалент-
ного соединения в условиях трансформации компетентных бак-
бактериальных клеток. Такие гибриды становятся достаточно проч-
прочными после того, как гибридизуемые друг с другом последова-
последовательности достигают определенной длины, обычно —12—15 нук-
нуклеотидов.
Векторы с такими свойствами получают путем инкубации ли-
линеаризованных молекул с Т4-ДНК-полимеразой в присутствии
76
только одного дезоксирибонуклеозидтрифосфата, например dT,
поскольку Т4-ДНК-полимераза обладает 3' —> 5'-экзонуклеазной
активностью, она начинает гидролизовать цепи ДНК в указан-
указанном направлении до тех пор, пока не встретит нуклеотид, трифо-
сфат которого присутствует в реакционной смеси (в данном при-
примере остаток Т). После этого реакция приходит в динамическое
равновесие: отщепление остатка Т сопровождается его повтор-
повторным включением в вектор. Такая процедура, проведение которой
планируется с учетом информации о первичной структуре векто-
вектора, сопровождается образованием двух специфических, неком-
некомплементарных друг другу одноцепочечных концов молекулы не-
необходимой длины [101, 102].
Клонируемый фрагмент ДНК получают с помощью ПЦР в
присутствии праймеров, содержащих на 5'-концах последова-
последовательности, комплементарные одноцепочечным участкам подго-
подготовленного вектора. Образовавшийся продукт ПЦР очищают от
dNTP и плазмидной ДНК, если она была использована в качест-
качестве матрицы, и также инкубируют с Т4-ДНК-полимеразой в реак-
реакционной смеси, содержащей один дезоксирибонуклеозидтрифос-
фат, комплементарный вышеупомянутому (в нашем примере
dATP). Вектор и подготовленный фрагмент ДНК смешивают
друг с другом и после инкубации в течение 5 мин смесь использу-
используют для трансформации компетентных клеток. Лигирование од-
одноцепочечных разрывов происходит непосредственно в бактери-
бактериальных клетках, после чего образуются кольцевые ковалентно
замкнутые молекулы рекомбинантной плазмиды, пригодные для
непосредственного применения в системах экспрессии и других
приложениях генной и белковой инженерии. Готовые наборы
для создания обсуждаемого семейства векторов имеются в прода-
продаже, и их можно приобрести, например, на фирме Novagen (США).
Детальную схему одного из представителей данного семейства
векторов, используемых для экспрессии клонированных генов в
клетках животных, можно видеть на рис. 6, д.
3.1.3. Векторы для прямого клонирования продуктов ПЦР
Термостабильные ДНК-полимеразы, у которых отсутствует
3' —> 5'-экзонуклеазная активность, включают в оба конца про-
продукта ПЦР независимо от матрицы 3'-выступающий остаток dA
[103]. Это обстоятельство препятствует непосредственному кло-
клонированию продуктов ПЦР по тупым концам линеаризованного
вектора и требует предварительного удаления выступающих ос-
остатков с помощью ферментов, обладающих экзонуклеазной ак-
77
тивностью, которые используют при проведении ПЦР в качестве
добавок к основному полимеризующему ферменту. Несмотря на
некоторое неудобство, доставляемое генным инженерам 3'-вы-
3'-выступающими одноцепочечными нуклеотидами ПЦР-продуктов,
это свойство ДНК-полимераз используется при создании плаз-
мидных векторов для непосредственного клонирования синтези-
синтезированных продуктов ПЦР без предварительного "затупления" их
концов. Такие векторы представляют собой линейные молекулы
ДНК, у которых на концах имеются 3'-выступающие остатки dU,
которые образуют комплементарные пары с 3'-выступающими
остатками dA ПЦР-продуктов. Инкубация таких векторов с про-
продуктами ПЦР в присутствии ДНК-лигазы при обычной темпера-
температуре A6 °С) в течение 0,5-2 ч сопровождается их эффективным
лигированием. При этом для лигирования можно брать продукты
ПЦР без предварительной очистки после экстракции реакцион-
реакционной смеси хлороформом для инактивации 7ад-ДНК-полимеразы.
В качестве примера вектора для непосредственного клонирова-
клонирования продуктов ПЦР можно упомянуть имеющийся в продаже мно-
многофункциональный вектор pETBlue-1 AccepTor фирмы Novagen
(США).
3.1.4. Использование транспозонов для клонирования ДНК
Транспозоны, повсеместно распространенные в живой при-
природе, являются мобильными генетическими элементами, облада-
обладающими способностью перемещаться из одной части генома в
другую путем вырезания и последующей интеграции по механиз-
механизму, независимому от гомологичной рекомбинации. Отдельные
представители этого обширного семейства мобильных генетиче-
генетических элементов бактерий сильно различаются по своей структу-
структуре и механизмам внутригеномного перемещения. В настоящее
время известно, по крайней мере, четыре механизма транспози-
транспозиции [104]. При консервативной транспозиции транспозон выре-
вырезается из репликона-донора и в неизменном виде интегрируется в
репликон-реципиент. Образовавшаяся в исходном репликоне
брешь восстанавливается в результате репарации. Репликатив-
ная транспозиция характеризуется тем, что транспозон не выре-
вырезается из локуса, в котором он расположен. Вместо этого репли-
кон-донор и репликон-реципиент образуют коинтеграт (объе-
(объединяются друг с другом), в процессе формирования которого воз-
возникает вторая копия транспозона. Следующее за этим разреше-
разрешение коинтеграта приводит к разделению репликонов, каждый из
которых становится обладателем копии исходного транспозона.
78
Процесс разделения коинтеграта осуществляется с помощью го-
гомологичной рекомбинации или при участии сайт-специфической
рекомбиназы (резолъвазы), которая кодируется транспозоном и
распознает специфические последовательности (res). В процессе
эксцизионной транспозиции, сайт-специфическая эндонуклеаза
(интеграза) при участии других белковых факторов вырезает
транспозон из репликона-донора, восстанавливая кольцевую
структуру репликона. При этом одновременно образуется коль-
кольцевой ковалентно замкнутый мобильный элемент в качестве
промежуточного соединения, который встраивается в репликон-
реципиент. Наконец, в процессе ретротранспозиции имеет мес-
место транскрипция ретротранспозона с образованием РНК, содер-
содержащей интрон группы II, который с участием матуразы удаляет-
удаляется из исходной РНК и интегрируется в безинтронную РНК, не
обязательно того же вида, что и РНК-донор (обратный сплай-
сплайсинг). Далее РНК-реципиент с помощью обратной транскрипции
превращается в кДНК и интегрируется в репликон-реципиент по
механизму RecA-зависимой гомологичной рекомбинации. Аль-
Альтернативно, в редких случаях, интрон-содержащий транскрипт
ретротранспозона, может с помощью обратного сплайсинга
встраиваться в одну из цепей расплетенной двухцепочечной
ДНК-реципиента. После внесения во вторую цепь ДНК одноце-
почечного разрыва с помощью эндонуклеазы, образовавшийся
3'-конец ДНК служит местом инициации обратной транскрипции
на матрице интегрированной РНК, что приводит в конечном сче-
счете к образованию двухцепочечной ДНК со вставкой последова-
последовательности ретротранспозона. Всеми тремя активностями: обрат-
обратной транскриптазы, матуразы и эндонуклеазы обладает белок,
кодируемый геном iep (intron encoded protein), локализованный в
интроне ретротранспозона.
По выбору мест интеграции в репликон-реципиент транспо-
зоны разделяют на три группы. Некоторые из них, наподобие
Тп5 и бактериофага Ми, интегрируют в геном случайным обра-
образом, другие встраиваются в ограниченное число участков ДНК,
для третьих характерна сайт-специфическая интеграция. Именно
способность ряда транспозонов встраиваться случайным образом
в геном клетки-хозяина часто используется в современной моле-
молекулярной генетике для клонирования неизвестных последова-
последовательностей и картирования генома.
Встраивание транспозона в кодирующую часть гена в резуль-
результате транспозиции, как правило, его инактивирует, что сопровож-
сопровождается развитием соответствующего фенотипа. Фрагмент ДНК с
транспозоном может быть выделен из клонотеки последователь-
79
ностей с помощью зондов, для создания которых используются
последовательности транспозона. Поскольку последовательности
транспозонов, используемых в такой работе, известны, не пред-
представляет труда с помощью подходящих рестриктаз выделить
транспозон вместе с фланкирующими его неизвестными последо-
последовательностями нуклеотидов. С помощью набора рестриктаз мож-
можно построить детальную физическую карту неизвестных последо-
последовательностей, прилегающих к интегрированному транспозону, и
осуществить дальнейшее продвижение в сторону неизвестных по-
последовательностей в обе стороны от транспозона. Такой подход
включает в себя элементы позиционного клонирования, принци-
принципы которого будут рассмотрены ниже в разделе 4.2.
3.2. Векторы на основе хромосомы фага X
Основным недостатком плазмидных векторов для клониро-
клонирования является их малая емкость в отношении клонируемых
фрагментов ДНК. Размер вставок клонируемой ДНК в плазмид-
плазмидных векторах, которые способны стабильно в них существовать,
как правило, не превышает нескольких тысяч пар оснований.
Большие вставки ДНК в векторных плазмидах нестабильны, и их
размеры постепенно уменьшаются по мере увеличения числа ра-
раундов репликации таких рекомбинантных плазмид in vivo. Выра-
Выраженное делетирование чужеродной ДНК в плазмидах большого
размера связано с тем, что в бактериальных клетках селективное
преимущество получают те плазмиды, время репликации кото-
которых минимально. Поэтому нуклеотидные последовательности
ДНК, не участвующие в репликации векторных плазмид, посте-
постепенно элиминируются посредством делеций при длительном
культивировании рекомбинантных бактерий.
Емкость клонирующих векторов была значительно повыше-
повышена с появлением векторов, сконструированных на основе хромо-
хромосомы бактериофага X. Получившие широкое распространение
векторы серий Charon, Xgtll и XEMBL обладают, по крайней ме-
мере, двумя существенными преимуществами перед плазмидными
векторами. Во-первых, векторы на основе ДНК фага X обладают
значительно большей емкостью, в них можно клонировать фраг-
фрагменты ДНК длиной от 5 до 25 т.п.о. Во-вторых, фаговые частицы,
содержащие упакованную ДНК, способны проходить литический
цикл развития внутри бактериальных клеток и поэтому образо-
образовывать стерильные пятна (бляшки) на газоне бактерий. Такие
бляшки содержат в концентрированном виде как сами фаговые
80
рис. 7. Упаковка рекомбинант-
ной фаговой ДНК в фаговые
частицы in vitro
рис. 8. Генетическая карта хро-
лосомы бактериофага А, -
EMBL3
а - расположение генов на хро-
хромосоме; 6 - шкала длины хромосом-
хромосомной ДНК в процентах от длины X
ДНК и т.п.о.; в - участок генома, за-
замещаемый на клонируемый фраг-
фрагмент ДНК соответствующего разме-
размера. S, В и R - сайты рестрикции SalGl,
BamHl и EcoRl, соответственно
Штамм 1
Штамм 2
Упаковочный
экстракт
Вектор со вставкой
Упаковка
Рекомбинантный фаг
a AW В С EF UVCTHMLKI J 1"Я IpyNrex trpE NS^OPQ SR
I H I III И И II HI II I CD ШШ DC
? ОПШШ Гены
в
О
10
20
30
40
50
60
70
80
1 1 1
10
1 1 1
20
SBR
\/У///////
1
30
RBS
|
40
i
i
т.п.о.
EMBL
частицы с упакованными в них рекомбинантными молекулами
ДНК, так и все продукты, появляющиеся в результате экспрессии
клонированных бактериальных генов. Каждая бляшка возникает
вследствие развития индивидуальной фаговой частицы, содержа-
содержащей рекомбинантную ДНК только одного типа, а, следовательно,
все фаговые частицы одной бляшки (~1010) представляют собой,
как правило, клон идентичных фаговых частиц (они могут разли-
различаться в редких случаях за счет мутационных изменений их гено-
генома, произошедших в процессе жизненного цикла фага, либо в том
случае, если одна бактериальная клетка заражается несколькими
фаговыми частицами одновременно). Все это позволяет легко об-
обнаруживать в фаговых бляшках искомые ферментативные актив-
активности или последовательности нуклеотидов и идентифицировать
клонированные последовательности ДНК. Основой конструиро-
конструирования фаговых векторов служат несколько простых принципов
(рис. 7). В середине молекулы Х-ДНК длиной ~45 т.п.о. располо-
расположен участок хромосомы (-15 т.п.о.), который не является необхо-
необходимым для литического развития бактериофага. Поэтому, в
81
принципе, его можно заменить на любой фрагмент ДНК анало-
аналогичного размера и осуществить клонирование фрагмента путем
размножения рекомбинантного бактериофага. Поскольку меха-
механизм упаковки хромосомной ДНК в фаговые частицы основан на
включении ДНК строго определенного размера, рекомбинантные
ДНК, содержащие фрагменты клонируемой ДНК, которые не со-
соответствуют оптимальному размеру, не упаковываются и не кло-
клонируются. Это позволяет легко освобождаться от фаговых час-
частиц, не содержащих вставки клонируемой ДНК, и оптимизиро-
оптимизировать процесс клонирования путем снижения в упаковочных экс-
экстрактах доли нежизнеспособных фаговых частиц.
Процесс упаковки фаговой ДНК в зрелые фаговые частицы
осуществляется в смеси бесклеточных экстрактов двух штаммов
Е. coli, лизогенных по дефектным бактериофагам X. Обычно в
одном штамме амбер-мутацией инактивирован один из белков
фагового капсида (продукт гена Е), а в другом - ген А, продукт
которого необходим для включения фаговой ДНК в головку бак-
бактериофага. Имеются и другие пары лизогенных штаммов Е. coli,
позволяющие производить упаковку ДНК в фаговые частицы с
использованием тех же общих принципов. Объединение бескле-
бесклеточных лизатов обоих штаммов Е. coli приводит к взаимной ком-
комплементации недостающих функций с помощью соответствую-
соответствующих белков дикого типа. Таким образом, в объединенных экс-
экстрактах имеются все компоненты, необходимые для сборки зре-
зрелых инфекционных фаговых частиц, в них происходит упаковка
рекомбинантной ДНК с эффективностью образования
104— 105 фаговых частиц на 1 мкг упаковываемой ДНК. Помимо
вышеупомянутых мутаций ДНК Х-лизогенов содержат темпера-
турочувствительную мутацию в репрессоре cl, который инакти-
вируется после переноса лизогенных клеток Е. coli на непермис-
сивную температуру D2 °С), что сопровождается индукцией про-
фага X и накоплением внутри бактериальных клеток белковых
продуктов, необходимых для упаковки ДНК. ДНК профагов так-
также содержит делецию Ы, элиминирующую сайт att, требуемый
для интеграции фаговой ДНК в бактериальную хромосому. Это
предотвращает выход ДНК профага из бактериальной хромосо-
хромосомы, а следовательно, и ее упаковку in vitro. Кроме того, в хромо-
хромосоме профага имеется мутация, инактивирующая ген S, кодирую-
кодирующий лизоцим, что препятствует преждевременному лизису бакте-
бактериальных клеток после индукции профага и позволяет сконцент-
сконцентрировать бактериальные клетки перед получением упаковочных
экстрактов. И, наконец, бактериальные лизогенные клетки со-
содержат мутацию reck, которая предотвращает гомологичную ре-
82
комбинацию между ДНК профага и рекомбинантными ДНК,
упаковываемыми в фаговые частицы.
В качестве примера рассмотрим генетическую карту век-
тррной ДНК бактериофага X-EMBL и кратко обсудим возмож-
возможности этого вектора (рис. 8). Векторы серии EMBL являются
производными ДНК бактериофага XI059. Их хромосомная
ДНК длиной в 42364 п.о. содержит центральный сегмент ДНК
длиной ~15 т.п.о., который замещается на клонируемый фраг-
фрагмент ДНК соответствующего размера. При этом в фаговые ча-
частицы может быть упакована рекомбинантная ДНК общей дли-
длиной в 9-23 т.п.о. Замещаемый фрагмент фаговой хромосомы
фланкирован с обоих концов последовательностями полилинке-
полилинкера, содержащего рестриктазные сайты EcoRl, BamHl и SalGl, по
которым встраивают клонируемые фрагменты ДНК. При этом
во время подготовки вектора к работе нет необходимости отде-
отделять "плечи" вектора от центрального фрагмента. Сначала цен-
центральный фрагмент ДНК выщепляется рестриктазой по одному
из сайтов полилинкера, а затем смесь образовавшихся фрагмен-
фрагментов обрабатывается другой рестриктазой, сайт для которой на-
находится в полилинкере. Возникающие при этом олигонуклео-
тидные фрагменты полилинкера удаляются при переосаждении
ДНК спиртом, а липкие концы "плеч" вектора и центральной
последовательности получаются некомплементарными друг
другу и не могут объединяться в процессе лигирования с обра-
образованием исходной формы ДНК вектора.
3.3. Космиды и фазмиды
Как уже упоминалось выше, фаговые векторы позволяют
клонировать фрагменты ДНК длиной 15-25 т.п.о. Однако этого
явно недостаточно, чтобы клонировать целиком многие гены
животных и растений, длина которых зачастую превышает
35-40 т.п.о. Требуемой емкостью обладают векторные молеку-
молекулы, называемые космидами (рис. 9). Космиды представляют со-
собой небольшие плазмиды, в которые in vitro введены cos-сайты
ДНК фага X. Отсюда происходит название всего типа данных
векторов (cosmid). В ДНК нормальных фаговых частиц cos-сай-
ТЬ1 расположены на концах молекул, они разделяют мономеры
фаговой ДНК в конкатемерах, объединяющих несколько соеди-
соединенных "голова к хвосту" мономеров, которые являются предше-
предшественниками зрелых фаговых ДНК перед упаковкой в фаговые
Частицы. В таких конкатемерах соседние cos-сайты располагают-
83
ДНК космидного вектора
Высокомолекулярная ДНК
Геномная ДНК
ВатШ
Smal
Расщепление вектора
ВатШ и Smal с
образованием "плеч"
Частичное расщепление Mbol
с образованием фрагментов
длиной 35-45 т.п.о.
oriAr
в в
кг
Кг
н
В
опАг
s
h-
гН
; ori Аг ¦
_ Kr
i 1
Непродуктивное
лигирование
"плеч" вектора
Очистка фрагментов
Лигирование
38-52 т.п.о.
oriAr
^ Кг
Kr'
_*. oriAr
S ori Ar "^
Упаковка ДНК in vitro
Рис. 9. Космидный вектор и конструирование клонотеки геномной
ДНК на его основе
ся на расстоянии 35^5 т.п.о. друг от друга и заключают между
собой весь фаговый геном. В процессе упаковки сш-сайты узна-
узнаются компонентами ферментативной системы и по ним происхо-
происходит последовательное отделение (отрезание) упакованной в фа-
фаговую частицу А,-ДНК от остальной неупакованной ДНК конка-
темера. Таким образом, наличие cos-сайтов в ДНК является, по
84
существу, единственным необходимым условием упаковываемо-
Ьти ДНК в фаговые частицы. Это означает, что последователь-
последовательность нуклеотидов А,-ДНК, расположенная между двумя cos-сш-
^ами, которая заключает в себе весь фаговый геном
C5-45 т.п.о.), может быть замещена in vitro на аналогичный по
длине C8-52 т.п.о.) фрагмент чужеродной ДНК и эффективно
упакована в фаговые частицы (такова максимальная емкость го-
головки фага). Естественно, что такая искусственная фаговая час-
частица оказывается нежизнеспособной.
Однако после адсорбции химерной фаговой частицы на по-
поверхности бактериальной клетки заключенная в ней ДНК прони-
проникает (вводится фаговой частицей) внутрь бактерии и начинает
автономно реплицироваться как плазмида, размер которой со-
составляет 30-40 т.п.о. Поскольку такая плазмида (космида) содер-
содержит в своем составе селектируемые маркеры в виде генов устой-
устойчивости к антибиотикам, ее поддерживают в бактериальных
клетках путем выращивания бактерий на среде с соответствую-
соответствующими антибиотиками. Несмотря на то, что емкость космидных
векторов значительно выше фаговых, эффективность клониро-
клонирования в космидах ниже, хотя и достигает в ряде случаев
105-106 колоний на 1 мкг клонируемой ДНК. При такой эффек-
эффективности упаковки требуется всего лишь 2-4 мкг клонируемой
ДНК для получения полной клонотеки большинства эукариоти-
ческих геномов.
Стадия упаковки ДНК космид в фаговые частицы использу-
используется лишь для облегчения процесса введения рекомбинантных
ДНК большого размера внутрь бактериальных клеток. Такой
процесс имитирует проникновение фаговой хромосомы в бакте-
бактерии во время фаговой инфекции. В случае космид сходство
между их проникновением в бактериальные клетки и фаговой
инфекцией на этом заканчивается. Однако сходство является
более глубоким в случае векторов, называемых фазмидами.
Фазмиды представляют собой векторные молекулы ДНК, кото-
которые содержат в себе генетические элементы плазмид и хромо-
хромосом бактериофагов. Они могут обладать емкостью в отношении
клонируемой ДНК, характерной для А,-векторов, и существо-
существовать в определенных условиях в бактериальных клетках в виде
плазмиды или же упаковываться в фаговые частицы in vivo при
изменении этих условий.
85
3.4. Искусственные хромосомы
Хромосомы высших организмов содержат в своем составе про-
протяженные молекулы ДНК. Например, длина ДНК одной из типич-
типичных хромосом человека составляет 100-200 млн п.о. Исследование
генов в хромосомах высших растений, животных и человека, в том
числе и полное секвенирование их геномов, потребовало создания
векторов для клонирования фрагментов ДНК длиной в несколько
сотен тысяч пар оснований. Этой задаче отвечают недавно создан-
созданные системы для клонирования сверхдлинных молекул ДНК на ос-
основе искусственно полученных мини-хромосом дрожжей (YAC -
yeast artificial chromosome), бактерий (ВАС - bacterial artificial chro-
chromosome) и бактериофагов (РАС - PI phage-derived artificial chromo-
chromosome). Разработка таких векторов позволила не только сделать ре-
реальным определение полной первичной структуры больших гено-
геномов, но и открыла новые возможности для получения трансгенных
соматических клеток человека, животных и растений. Создание
этих технологий, в свою очередь, оказало глубокое влияние на при-
приложения биотехнологии в молекулярной биологии и генетике (ис-
(исследование экспрессии и функций индивидуальных генов), медици-
медицине (генотерапия) и сельском хозяйстве (трансгенные животные и
растения с новыми потребительскими свойствами).
Дальнейшее развитие этого направления исследований при-
привело к созданию искусственных хромосом животных (MAC -
mammalian artificial chromosome) и человека (НАС - human artifi-
artificial chromosome) с помощью которых оказалось возможным вве-
введение протяженных фрагментов ДНК в дифференцированные и
эмбриональные стволовые клетки.
3.4.1. Сверхъемкие векторы YAC, ВАС и РАС
Плазмиды дрожжей. В ядрах клеток большинства штаммов
дрожжей S. cerevisiae присутствует плазмидная ДНК в виде двухце-
почечных кольцевых ковалентно замкнутых молекул в количест-
количестве 30-100 копий на клетку, что может составлять до 5% от суммар-
суммарной ядерной ДНК. Плазмиды получили название 2|im. Клетки, со-
содержащие такие плазмиды, обозначают cir+, а бесплазмидные
клетки - cir°. Единственное фенотипическое различие между клет-
клетками этих двух типов заключается в том, что скорость деления
сп^-клеток незначительно меньше, чем cir°. В этой связи плазми-
плазмиды 2цт рассматривают как внутриклеточные паразиты, и эти
свойства отличают их от плазмид других грибов, которые, как
правило, кодируют белки, необходимые для жизнедеятельности
86
Микроорганизмов-хозяев. Стабильное существование плазмид
в клетках, как и в случае бактериальных плазмид, требует функ-
функционирования механизмов их автономной репликации, контроля
числа копий и распределения между дочерними клетками.
Присутствующие в плазмидах последовательности ARS
(autonomously replicating sequence) необходимы для инициации
репликации плазмидной ДНК, которая осуществляется с участи-
участием белков клетки-хозяина. Однако таких последовательностей
самих по себе недостаточно для стабильного существования
плазмиды в клетках дрожжей, и для этого требуется функциони-
функционирование четырех дополнительных генов. Один из них, FLP, коди-
кодирует рекомбиназу, которая при некоторых обстоятельствах мо-
может увеличивать число копий плазмид внутри клеток, хотя здесь,
как обычно, сохраняется только одна инициация репликации
плазмиды в каждой S-фазе клеточного цикла. Два других гена
REP1 и REP2 кодируют белки-репрессоры гена FLP, а ген RAF
кодирует антирепрессор, и его активность негативно регулирует-
регулируется комплексом белков Replp/Rep2p.
Векторы для клонирования ДНК в клетках дрожжей. Выше-
Вышеперечисленные особенности биологии плазмид 2цт учитывают-
учитываются при конструировании соответствующих векторов и искусст-
искусственных хромосом YAC. Современные дрожжевые векторы пред-
представлены несколькими семействами.
Плазмидные интегрирующие векторы дрожжей YIP (yeast
integrative plasmids) предназначены для встраивания клонирован-
клонированных фрагментов ДНК в клетки дрожжей. Эти векторы не репли-
реплицируются автономно в клетках дрожжей, но обладают селекти-
селектируемыми маркерами, обеспечивающими рост клеток в селекти-
селектирующих условиях. Они способны реплицироваться в клетках
Е. coli. Остальные нижеперечисленные векторы относятся к чел-
челночным векторам, т.е. способны реплицироваться в бактериаль-
бактериальных и дрожжевых клетках. Дрожжевые реплицирующиеся плаз-
плазмиды YRP (yeast replicating plasmids) содержат область начала
репликации ARS, в отсутствие селектирующего агента существу-
существуют в дрожжевых клетках крайне нестабильно. В отсутствие се-
селекции в каждой генерации утрачивается, как правило, > 1/10 всех
Молекул плазмид. Дрожжевые эписомные плазмиды YEP (yeast
episomal plasmids) сконструированы на основе природной плаз-
плазмиды 2 цт. Они присутствуют в клетках в большом числе ко-
копий и наследуются значительно более стабильно, чем плазми-
ДЫ семейства YRP. Дрожжевые плазмиды, содержащие центро-
центромеры YCP (yeast centromeric plasmids) содержат последовательности
ARS и CEN. Последнее обстоятельство делает их в отсутствие спе-
87
CEN4
Bam
Bam
1 Расщепление ВатШ
2 Расщепление EcoRl
TEL TRP
CEN4
ни—а
TEL
ARS1
3 Лигирование с крупными
EcoRl - фрагментами ДНК
URA3
Искусственная хромосома дрожжей
со вставкой 100-700 т.п.о.
Рис. 10. Схема клонирования а
сверхдлинных молекул ДНК с
использованием вектора YAC
(а) и их изменение с помощью
ретрофиттинга (б) [105 а]
а - клонирование ДНК в дан-
данном YAC-векторе включает три
основных этапа: / - линеаризацию
ДНК вектора рестриктазой BamHl;
2 - расщепление линеаризованной
ДНК вектора рестриктазой EcoRl с
образованием "плеч"; 3 - введение
в вектор клонируемого EcoRl-
фрагмента ДНК;
б - введение мутаций в ген,
клонированный в YAC-векторе, ме-
методом ретрофиттинга. Вначале в
YAC-векторе со вставкой инакти-
вируют его исходный селектируе-
селектируемый маркер URA3 путем введения в
этот ген последовательностей но-
новых селектируемых маркеров LYS2
и Neo посредством гомологичной
рекомбинации между YAC-векто-
ром и соответствующей плазмидой; на втором этапе с помощью линеаризован-
линеаризованной дрожжевой интегрирующей плазмиды во вставку YAC-вектора вместе с мар-
маркером URA3 вводят мутантную последовательность (т); после интеграции плаз-
плазмиды (pop-in) образуется тандемная дупликация модифицируемого гена, в кото-
которой присутствуют исходный и мутантный гены; при выращивании клеток дрож-
дрожжей без селекции по маркеру URA3 одна из копий гена спонтанно и равновероят-
равновероятно утрачивается с низкой частотой вместе с маркером в результате гомологичной
рекомбинации (pop-out)
циальных селективных условий в 10-100 раз более стабильными по
сравнению с изогенными (во всем остальном идентичными) плаз-
мидами без С?М-последовательностей. Векторы этой серии были
использованы для создания искусственных хромосом дрожжей.
Искусственные хромосомы дрожжей YAC. Первые минихро-
мосомы дрожжей были получены А. Мюрреем и Дж. Шостаком в
1983 г. Мини-хромосомы дрожжей YAC представляют собой коль-
кольцевые молекулы ДНК, содержащие большинство вышеупомянутых
генетических элементов, которые позволяют им стабильно сущест-
существовать во внехромосомном состоянии в клетках дрожжей (рис. 10).
Типичный вектор включает в себя две теломерные последователь-
последовательности нуклеотидов TEL (Tetrahymena), необходимые для репликации
концов мини-хромосомы, и область начала репликации ARS1, соеди-
соединенную с последовательностью центромеры (CEN). Все эти функци-
функциональные элементы требуются для репликации YAC-вектора и его
88
б
LYS2/NE0
TEL TRP1/ARS1/CEN4
Вставка нового селектируемого маркера с помощью
гомологичной рекомбинации (ретрофиттинг)
TEL TRP1/ARS1/CEN4
Интеграция плазмиды, содержащей мутантную
копию экзогенной ДНК ("pop-in)
TEL TRP1/ARS1/CEN4
Мутантная копия
URA3
Гомологичная рекомбинация между двумя копиями
эзогенной ДНК ("pop-out")
LYS2/NEO
LYS2/NEO TEL
URA3
Копия дикого типа
LYS2/NEO
TEL
TEL TRP1/ARS1/CEN4
Мутантная копия
LYS2/NEO
TEL
Рис. 10 (окончание)
правильной передачи в дочерние ядра во время митоза. Кроме того,
вектор содержит два селектируемых маркера TRP, восстанавливаю-
восстанавливающих способность к росту ауксотрофных по триптофану клеток
дрожжей в отсутствие экзогенного триптофана, а также маркер
URA3, компенсирующий генетический дефект клеток дрожжей, ко-
который нарушает биосинтез урацила. В векторе имеется ген супрес-
сорной тРНК SUP4 селектируемого маркера, используемого для су-
супрессии охр-мутаций в клетках дрожжей: мутации сап\-\00 в гене,
обеспечивающем устойчивость клеток к L-канаванину (токсичный
аналог L-аргинина), мутации ade2-\,4TO сопровождается восстанов-
восстановлением нормального белого цвета колоний у красных мутантов, и
Др. Поскольку в гене SUP4 имеется уникальный сайт рестрикции
EcoRl, введение клонируемой ДНК по этому сайту инактивирует ген
и приводит к приобретению колониями дрожжей красного цвета,
как следствие накопления в мутантных клетках метаболитов-пред-
метаболитов-предшественников аденина. Векторы семейства YAC являются челноч-
челночными, т.е. обладают последовательностями нуклеотидов, необходи-
необходимыми для их репликации в бактериальных клетках.
При подготовке к клонированию YAC-вектор, выделенный в
ВиДе плазмиды из бактериальных клеток, расщепляют рестрик-
тазой ВатШ и отделяют от образовавшегося короткого фраг-
89
мента ДНК, который не требуется для репликации YAC-вектора
в дрожжах (этап 1, рис. 10, а). После этого проводят второе рас-
расщепление вектора рестриктазой ЕсоШ, сопровождающееся обра-
образованием двух его "плеч", каждое из которых на одном из концов
содержит теломерные последовательности хромосомы дрожжей
(этап 2). На заключительном этапе C) полученные "плечи" лиги-
руют с крупными ?соШ-фрагментами клонируемой ДНК, кото-
которые получают путем частичного расщепления высокомолеку-
высокомолекулярной хромосомной ДНК, предназначенной для клонирования.
Полученными таким образом рекомбинантными ДНК трансфор-
трансформируют протопласты клеток дрожжей, и образовавшиеся транс-
трансформанты отбирают на селективной твердой питательной среде
по комплементации мутаций trpl и игаЗ, что свидетельствует о
присутствии в рекомбинантной конструкции обоих плеч вектора.
Для стабильного существования в клетках дрожжей и правиль-
правильной передачи дочерним клеткам существенным оказывается раз-
размер искусственных хромосом. В таких векторах удается осуще-
осуществлять клонирование фрагментов ДНК длиной до 2 млн п.о., что
значительно превышает емкость космид E0 т.п.о.).
Векторы YAC, содержащие клонируемые последовательнос-
последовательности, существуют в среднем в виде одной копии на клетку, и даже в
отсутствие селектирующих условий утрачиваются с очень низ-
низкой частотой A0-3-10-5 за клеточную генерацию). При увеличе-
увеличении общего размера вектора до 140 т.п.о. и выше частота потери
его молекул не превышает таковую, характерную для обычных
хромосом дрожжей.
Клоны, полученные с использованием векторов YAC, могут
быть модифицированы в дрожжевых клетках с помощью гомоло-
гомологичной рекомбинации методом так называемого "ретрофиттин-
га" (retrofitting), что создает условия для изменения далее самой
клонированной последовательности (рис. 10, б) [105а,б]. Для прове-
проведения ретрофиттинга клетки, содержащие YAC с клонированной
последовательностью, трансформируют последовательностью
ДНК (кассетой генов), содержащей подходящий генетический мар-
маркер, например ген LYS2 или Neo, фланкированный короткими по-
последовательностями, идентичными участкам ДНК одного из мар-
маркеров, например, URA3, присутствующих в искусственной хромосо-
хромосоме. Ген Neo, обеспечивающий устойчивость клеток к антибиотику
неомицину, часто используется при отборе трансформантов клеток
животных. Гомологичная рекомбинация между соответствующими
последовательностями кассеты генов с новыми генетическими
маркерами и URA3 приводит к интеграции новых маркеров в ген
URA3 и инактивации последнего. В результате появляется возмож-
90
цость его повторного введения в клонированную последователь-
последовательность YAC в качестве селектируемого маркера вместе с новыми
последовательностями, изменяющими исходную. Ретрофиттинг
позволяет преодолевать технические сложности, которые возника-
возникают при необходимости проведения генно-инженерных манипуля-
манипуляций с протяженными последовательностями нуклеотидов, клони-
клонированными в YAC-векторах, т.к. исключает необходимость исполь-
использования рестриктаз и лигирования, обычно применяемых для этих
целей при работе с более короткими последовательностями.
Использование YAC для получения клонотек нуклеотидных
последовательностей. При создании искусственных хромосом
дрожжей in vitro в среднем удается клонировать фрагменты ДНК
длиной ~300 т.п.о. Однако с помощью гомологичной рекомбина-
рекомбинации, проводимой непосредственно в клетках дрожжей, можно по-
получать вышеупомянутые вставки в несколько млн п.о. Для реали-
реализации полной емкости вектора используют предварительно полу-
полученные YAC-конструкции, в которых клонированы частично пере-
перекрывающиеся последовательности. Для обнаружения перекрыва-
перекрывающихся клонов используют рестрикционное картирование с гибри-
гибридизацией по Саузерну, метод "прогулки по хромосоме" и ряд других
стандартных методов исследования генома, которые будут рассмо-
рассмотрены во втором томе этой книги. После обнаружения таких пере-
перекрывающихся клонов проводят скрещивание гаплоидных клеток,
содержащих требуемые YAC, в полученных диплоидных штаммах
индуцируют мейоз, при котором с высокой частотой возникают
требуемые рекомбинантные YAC с протяженными непрерывными
последовательностями - контигами - исследуемого генома. Для
предотвращения образования в результате рекомбинации дицент-
рических и ацентрических YAC, которые нестабильны, объединяе-
объединяемые вставки должны быть клонированы в одной и той же
5' —» 3'-ориентации по отношению к маркерам вектора. После за-
завершения клетками мейотических делений и споруляции в спорах
обнаруживают требуемые рекомбинанты, конечная длина кото-
которых после проведения серии последовательных скрещиваний мо-
может превышать 2 млн п.о. [105, в].
Получение трансгенных животных с использованием YAC.
После проведения вышеупомянутого ретрофиттинга для введения
селектируемых маркеров, применяемых при отборе трансформи-
трансформированных клеток животных, модифицированные таким образом
YAC со вставками исследуемых последовательностей могут быть
использованы для получения трансгенных животных. Очищенные
Молекулы рекомбинантных YAC можно вводить в пронуклеус
Яйцеклеток с помощью микроинъекции или липофекции.
При всех своих достоинствах системы клонирования, осно-
основанные на векторах семейства YAC, обладают рядом существен-
существенных недостатков. На стабильность YAC-векторов в дрожжевых
клетках оказывает влияние их генотип. Так, потеря вектора про-
происходит с высокой частотой в клетках, содержащих мутантные
гены BUB1, BUB2 и BUB3, MAD1, MAD2 и MADS, CDC6, CDC20,
PDS1 и др. Кроме того, стабильная сегрегация вектора в дочер-
дочерние клетки происходит лишь в том случае, если вставки клониру-
клонируемой ДНК не содержат последовательностей, гомологичных по-
последовательности ARS, чего нельзя полностью исключить при
клонировании больших фрагментов эукариотической ДНК с не-
неизвестной первичной структурой.
В рекомбинантных ДНК, поддерживаемых в таких системах,
часто возникают внутренние делеции некоторых, например, по-
повторяющихся последовательностей. Кроме того, при введении
рекомбинантных ДНК в клетки дрожжей иногда имеет место
проникновение в одну клетку нескольких молекул вектора со
вставками. В итоге отдельные клоны дрожжевых клеток могут
содержать несколько несцепленных друг с другом молекул ре-
рекомбинантных ДНК, а рекомбинация между ними вообще может
приводить к образованию химерных молекул, которые являются
одной из главных проблем, возникающих при использовании
YAC: 40-60% клонов могут содержать химерные молекулы. Все
это очень затрудняет физическое картирование генов в хромосо-
хромосомах исследуемых объектов.
Искусственные хромосомы бактерий. Для преодоления труд-
трудностей, возникающих при использовании искусственных хромосом
дрожжей, были сконструированы альтернативные векторные сис-
системы, среди которых наиболее популярными в настоящее время
являются системы, основанные на искусственных хромосомах
бактерий - ВАС (bacterial artificial chromosome) [106]. В векторных
системах ВАС используется ДНК полового фактора
(F-фактора) Е. coli - гигантской плазмиды мужских бактериаль-
бактериальных клеток, которые являются донорами бактериальной ДНК при
конъюгации с женскими клетками (рис. 11). Типичный F-фактор
содержит гены oriS, repE, par А и рагВ, регулирующие собственную
репликацию и контролирующие число копий в бактериальных
клетках. В частности, гены oriS и герЕ обеспечивают однонаправ-
однонаправленную репликацию F-фактора, а гены par А и рагВ поддерживают
число его копий на уровне одной-двух на бактериальную клетку.
Классический вектор ВАС (pBAC108L) включает в себя все эти
гены и ген устойчивости к хлорамфениколу, используемый в каче-
качестве селектируемого маркера. Вектор содержит также фрагмент
92
рис. И- Представители се-
семейств ВАС- (а) и РАС- (б) век-
векторов
а
Hindm ВагпШ
NotVEagI
LoxP
XmaV Smal
NotVBgH/Sfil
Sail
б
Плазмидный
репликон
rep
Полилинкер pUC19
loxP
Репликон
литического PI
rep lyt
Плазмидный
репликон
rep
kanR
ДНК, по которому производится клонирование. В этом фрагмен-
фрагменте имеются типичный полилинкер, а также два уникальных сайта
рестрикции ШпсШ и BamHl, фланкированные промоторами Т7- и
§|N-РНК-полимераз. Такие промоторы могут быть использованы
Для получения РНК-зондов, необходимых для осуществления
Прогулок по хромосомам", а также прямого секвенирования кло-
^рованной ДНК в месте стыковки с вектором. Кроме того, во
фрагменте имеется сайт cosN, обеспечивающий расщепление век-
вектора со вставкой в уникальном месте с помощью терминазы фа-
Ш А, без применения ферментов рестрикции. Этот фермент ис-
Црльзуется бактериофагом для специфического разрезания конка-
?|Меров своей хромосомы при упаковке в фаговые частицы. Для
т®й же цели может быть применен и сайт loxP бактериофага Р1,
93
который является мишенью для фаговой эндонуклеазы Сге. Эти
сайты используются для получения специфических концов клони-
клонированной ДНК с целью ее дальнейшего рестрикционного картиро-
картирования путем введения концевой метки с последующим неполным
расщеплением рестриктазами и электрофоретическим разделени-
разделением образовавшихся фрагментов.
Современные ВАС-векторы позволяют клонировать фрагмен-
фрагменты ДНК длиной до 300 т.п.о. и выше. Рекомбинантные молекулы
вводятся в клетки Е. coli с помощью электропорации (см. раз-
раздел 3.8), причем эффективность образования трансформантов в
10-100 раз выше, чем при обычной трансформации сферопластов
дрожжей векторами семейства YAC. Это позволяет уменьшить ис-
исходное количество ДНК, необходимое для конструирования ре-
репрезентативных клонотек генов (см. гл. 4). При скрининге таких
клонотек используются традиционные методы работы с бактери-
бактериальными колониями. В отличие от YAC-ДНК, которая находится
в клетках дрожжей в линейной форме, ВАС-векторы со вставка-
вставками, как и традиционные F'-факторы, существуют в бактериальных
клетках в виде кольцевых суперскрученных молекул. Это облег-
облегчает их выделение и последующую работу с рекомбинантными
молекулами ДНК в растворе, а кроме того, допускает повторное
введение в бактериальные клетки этих ДНК, выделенных мини-
препаративными методами. Поскольку рекомбинантные ВАС-
векторы существуют в бактериальных клетках в виде одной ко-
копии, исключаются совместное клонирование в одной клетке раз-
разных фрагментов ДНК и образование химерных молекул, что
очень важно для физического картирования больших геномов ме-
методами "снизу вверх". Весьма существенным свойством системы
клонирования, основанной на векторах семейства ВАС, является
ее генетическая стабильность. Исходная структура клонирован-
клонированных фрагментов ДНК в пределах точности использованных мето-
методов сохраняется в таких векторах даже после 100 серийных пере-
пересевов бактериальных клеток, содержащих рекомбинантные моле-
молекулы ДНК. Все вышеперечисленные свойства переводят векторы
ВАС в разряд сверхъемких векторов нового поколения.
Искусственные хромосомы PL В заключение следует упомя-
упомянуть о семействе векторов РАС (PI-derived artificial chromosome),
также часто используемых в современных исследованиях. Векто-
Векторы этой серии содержат гены умеренного бактериофага Р1, обес-
обеспечивающие репликацию фаговой хромосомы в зараженных
бактериальных клетках. Рекомбинантные ДНК на их основе
(размер вставки 150-200 т.п.о.) также вводятся в бактериальные
клетки с помощью электропорации.
94
Бактериофаг PI, заражающий клетки Е. coli так же, как и ко-
дифаг X, относится к категории умеренных, т.е. способен длитель-
длительное время существовать в зараженных клетках в скрытом виде как
йрофаг. Однако, в отличие от бактериофага X, который во время
скрытого (лизогенного) состояния встраивает свою хромосому в
хромосому бактерии-хозяина, фаг Р1 поддерживает хромосому в
цитоплазме бактериальных клеток в виде кольцевой ковалентно
замкнутой молекулы, напоминающей плазмиду, размер которой
составляет 100 т.п.о. Размер репликона, который способен обеспе-
обеспечивать репликацию хромосомы Р1 в лизогенном состоянии, состав-
составляет всего 1,5 т.п.о. Этот сегмент ДНК содержит область начала
репликации oriR длиной в 245 п.о., ген герА, кодирующий белок-
инициатор репликации B86 а.о.), а также последовательность incA
длиной в 285 п.о., которая осуществляет контроль репликации.
Структура одного из классических векторов семейства РАС
(pNS582) представлена на рис. 11, б [107]. Вектор состоит из двух
доменов, фланкированных последовательностями сайт-специфи-
сайт-специфической рекомбинации фага PI loxP, ориентированными в одном и
том же направлении. Рекомбинация между ними с участием Р1-
Cre-рекомбиназы, которую кодирует бактериальная клетка-хозя-
клетка-хозяин, приводит к разделению доменов с образованием двух кольце-
кольцевых молекул. Один из доменов содержит ген устойчивости к ампи-
ампициллину атрг, многокопийный репликон плазмиды pBR322 и рас-
сайт бактериофага, обеспечивающий упаковку ДНК в его голо-
головку. Второй домен вектора обладает плазмидным Р1-репликоном,
функционирующим во время лизогенного состояния бактериофа-
бактериофага, литическим Р1-репликоном, который находится под контролем
/ас-промотора, геном устойчивости к канамицину капг, кодирую-
кодирующим аминогликозид-З'-фосфотрансферазу, и полилинкером. Ис-
Использование двух репликонов фага Р1 позволяет поддерживать
вектор со вставкой в виде 1-2 копий при конструировании клоно-
1$к последовательностей, а после индукции /яс-промотора IPTG
Повышать количество копий вектора в ~25 раз как следствие
функционирования литического репликона, обычно используемо-
используемого фагом для интенсивной репликации хромосомы во время лити-
Ческого цикла развития при образовании фаговых частиц.
Упаковка ДНК в головки бактериофага Р1, как и ДНК фага X,
Происходит по заполняющему (headful) механизму (см. раздел 3.3).
Процесс инициируется комплексом фаговых белков, получившим
йазвание наказы (pacase), который специфически расщепляет
йРедшественник ДНК по рас-сшту (аналогу уже обсуждавшегося
fcos-сайта бактериофага X), состоящий из восьми гексануклеотид-
** повторов 5'-TGATCA(G), симметрично расположенных отно-
95
сительно точки разрыва ДНК. Начиная с этого конца, ДНК посту-
поступает в предшественник головки, после заполнения которого ДНК
разрезается второй раз (headful cleavage), что приводит к отделе-
отделению упакованной ДНК от ее внешней части. Второй разрыв вно-
вносится независимо от последовательности нуклеотидов гидролизуе-
мого участка, поэтому, в отличие от ДНК фага X, последователь-
последовательности двух концов упакованной хромосомы Р1 не идентичны.
Клонирование последовательностей ДНК проводят по уникаль-
уникальным сайтам рестрикции полилинкера (pcs). Если размер клониро-
клонированной ДНК соответствует емкости головки бактериофага, внутрь
головки попадают обе последовательности сайт-специфической.ре-
сайт-специфической.рекомбинации loxP, что позволяет рекомбинантным молекулам после
перехода в бактериальные клетки из фаговых частиц замыкаться в
кольцо и реплицироваться по одному из вышеупомянутых механиз-
механизмов. Превышение лигированной вставкой допустимого размера
приводит в процессе упаковки ДНК в фаговые частицы к удалению
одного из /ахР-сайтов, что делает невозможным образование внут-
внутриклеточных кольцевых молекул, и они утрачиваются.
Дальнейшее развитие векторных РАС-систем привело к созда-
созданию семейства векторов нового поколения, pCYPAC с полилинке-
полилинкером, внедренным в ген sacBII, кодирующий токсичный для клеток
фермент, который участвует в метаболизме сахарозы (рис. 11,6,
справа) [108]. Это позволяет проводить позитивный отбор клеток,
содержащих вектор со вставкой, поскольку только такие клетки
выживают в селективных условиях. Вектор не требует упаковки в
фаговые частицы и вводится в бактериальные клетки с помощью
электропорации, содержит те же два уже рассмотренных реплико-
на, один /ахР-сайт, а также репликон плазмиды pUC19, разрываю-
разрывающий ген sacBII. Это понижает токсичность гена во время наработ-
наработки самого вектора в препаративном количестве, а при клонирова-
клонировании исследуемых фрагментов ДНК репликон удаляется. Дополни-
Дополнительный отбор векторных молекул осуществляют с использовани-
использованием гена устойчивости к канамицину. Вставки рекомбинантной
ДНК в таких векторных системах стабильны, и образование химер-
химерных молекул, которые являются бичом векторных YAC-систем, не
отмечено, что делает весьма удобным использование РАС-векто-
ров для конструирования геномных клонотек крупных геномов жи-
животных и растений с последующим их анализом.
3.4.2. Искусственные хромосомы животных и человека
Использование высокоемких векторов YAC, ВАС и РАС ока-
оказало и продолжает оказывать неоценимую пользу при реализа-
96
ции крупных научных проектов по секвенированию больших ге-
геномов, включая геном человека. Тем не менее все вышеперечис-
вышеперечисленные векторные системы не удовлетворяют полностью запро-
запросов современной молекулярной генетики, особенно в областях
получения трансгенных животных и генотерапии. Для осуществ-
осуществления экспрессии генов в трансгенных организмах в настоящее
время чаще всего применяют кДНК и искусственные промото-
промоторы, что исключает осуществление полноценного контроля
транскрипции рекомбинантных генов. В этой связи возникает не-
необходимость введения в клетки протяженных геномных последо-
последовательностей с полноценной интрон-экзонной структурой и соб-
собственными регуляторными участками, а также целых областей
хромосом высших организмов. Осуществлять такие генно-инже-
генно-инженерные манипуляции позволяют искусственные хромосомы жи-
животных (mammalian artificial chromosomes - MAC) и одна из их
разновидностей - искусственные хромосомы человека (human
artificial chromosome - НАС), которые, благодаря собственной си-
системе, обеспечивающей правильную сегрегацию в митозе, дают
возможность не прибегать к введению рекомбинантных молекул
в геном модифицируемых клеток, а следовательно, избегать не-
нежелательных последствий его модификации, сопровождаемой
эффектами положения, инсерционным мутагенезом и тому по-
подобными неприятными явлениями. При конструировании MAC и
НАС, в основном, используют две стратегии [109-112].
Конструирование MAC методом "сверху вниз". Данная стра-
стратегия основана на последовательном укорачивании природных
хромосом с сохранением их элементов, обеспечивающих реплика-
репликацию и митотическую сегрегацию. Эта группа методов известна
как фрагментация хромосом с использованием теломерных по-
последовательностей (telomere-associated chromosome fragmen-
fragmentation - TACF) или укорачивание с помощью теломер (telomere
directed truncation - TDT). Метод основан на гомологичной реком-
рекомбинации между вектором (YAC) и укорачиваемой хромосомой, в
результате которой происходит замена большей части последова-
последовательностей плеч хромосомы на последовательности вектора. Та-
Такой вектор исходно содержит последовательности, гомологичные
таковым изменяемой хромосомы, по которым происходит крос-
синговер, селектируемый маркер (обычно ген устойчивости к ан-
антибиотику пео или gpt, кодирующий гуанинфосфорибозилтранс-
феразу) и теломерные последовательности. Гомологичную ре-
рекомбинацию при модификации хромосом человека проводят по
последовательностям альфоидной ДНК центромер или уникаль-
уникальным последовательностям, отбирая рекомбинантов на селектив-
4. Патрушев Л.И. Т. 1
ной среде с антибиотиком G418 или на среде HAT. При получении
удачных конструкций мини-МАС, содержащие область центроме-
центромеры, две теломерные последовательности и области начала репли-
репликации, не утрачиваются культивируемыми клетками на протяже-
протяжении 60 генераций, даже в том случае, если клетки выращивают не
в селективных условиях. Сначала MAC и НАС получали в клетках
человека или соматических гибридах человек-хомячок, однако,
из-за низкой частоты гомологичной рекомбинации в таких клет-
клетках в последнее время стали использовать клетки цыплят линии
DT 40, предшественников В-клеток, трансформированных виру-
вирусом лейкоза, для которых характерна высокая частота кроссинго-
вера. Полученные в них MAC далее переносят в клетки человека
для дальнейшего изучения. С помощью данного метода было ус-
установлено, что размер Х-хромосом человека может быть умень-
уменьшен до 500 т.п.о. без потери ими способности к автономному ста-
стабильному существованию внутри клеток [113].
Получение искусственных хромосом с использованием са-
теллитной ДНК (satellite DNA-based artificial chromosome -
SATAC) является другим распространенным методом "сверху
вниз", основанном на использовании горячих точек нестабиль-
нестабильности генома в амплифицируемых перицентромерных областях
акроцентрических хромосом [114]. В этом случае плазмидную
ДНК, содержащую селектируемый маркер, вводят с помощью
гомологичной рекомбинации в перицентромерную область ге-
терохроматина хромосом мышей или человека. Клетки, содер-
содержащие такие хромосомы, далее культивируют на селективной
среде, что сопровождается амплификацией маркерной последо-
последовательности плазмиды и окружающих перицентромерных по-
последовательностей (подробнее о механизме такой амплифика-
амплификации см. раздел 5.2.1). В итоге формируется дицентрическая (со-
(содержащая две центромеры) хромосома, что сопровождается ее
разрывом во время митотических делений с образованием до-
дополнительных искусственных хромосом, размер которых мо-
может быть в пределах 10-360 млн п.о. Эти хромосомы, состоящие
из последовательностей перицентромерного гетерохроматина,
которые перемежаются введенной рекомбинантной ДНК, далее
могут быть перенесены в клетки животных других видов и че-
человека. Такие искусственные хромосомы правильно сегрегиру-
сегрегируют в митозе в дочерние клетки и содержат большое количество
специфических повторяющихся последовательностей, что поз-
позволяет легко выделять их с помощью проточной цитометрии
(раздел 2.1.2). Кроме того, показано, что такие хромосомы мо-
могут быть использованы для создания стандартными методами
98
трансгенных мышей, которые содержат искусственные хромо-
хромосомы во всех соматических клетках [115].
Конструирование MAC методом "снизу вверх". При этом
подходе искусственную минихромосому собирают из отдельных
последовательностей, соответствующих теломерам, центроме-
центромерам и областям начала репликации природных хромосом, с кото-
которыми объединяют требуемую рекомбинантную ДНК. Теломер-
ные последовательности животных представляют собой тандем-
но повторяющиеся последовательности вида (TTAGGG)n, кото-
которые, будучи объединенными в повторы длиной ~1 т.п.о., эффек-
эффективно функционируют в клетках человека. В качестве областей
начала репликации могут быть использованы различные после-
последовательности, среди которых наиболее изучены соответствую-
соответствующие последовательности C-глобинового гена человека.
Последовательности, составляющие центромеры, исследова-
исследованы в меньшей степени, и с ними сложнее работать генно-инженер-
генно-инженерными методами. Основным компонентом центромер человека яв-
является а-сателлитная (алъфоидная) ДНК, которая сформирова-
сформирована семейством повторяющихся последовательностей, специфич-
специфичных для центромер приматов. Альфоидные мономеры (-170 п.о.)
в центромерах человека в свою очередь составляют протяженные
последовательности длиной в несколько млн п.о., организованные
в повторы более высокого порядка (higher order repeats - HOR). Го-
Гомология между мономерами альфоидной ДНК отдельных хромо-
хромосом составляет 60-80%, тогда как организация тандемно повторя-
повторяющихся HOR близка к идентичной. С учетом структуры альфоид-
ных мономеров и HOR хромосомы человека разделяют на не-
несколько семейств [116]. Кроме обычных центромер, построенных
на основе альфоидной ДНК, в маркерных хромосомах человека,
иначе называемых также небольшими добавочными хромосомами
(small accessory chromosomes - SACs), обнаружены неоцентроме-
неоцентромеры, не содержащие ос-сателлитной ДНК. Неоцентромеры также
используются при конструировании MAC методом "снизу вверх".
Котрансфекция (липофекция) культивируемых клеток чело-
человека смесью синтетической альфоидной ДНК, теломерных после-
последовательностей, геномной ДНК человека и маркерного гена со-
сопровождается образованием нескольких минихромосом, как след-
следствие укорачивания эндогенных хромосом клетки. Кроме того, в
таких клетках имело место образование искусственных минихро-
минихромосом de novo. Более эффективным способом создания MAC и
НАС является их генно-инженерное конструирование на основе
стандартных векторов YAC, ВАС и РАС путем введения в них аль-
альфоидной ДНК и теломерных последовательностей. В этом слу-
4* 99
Таблица 4
Сравнительные характеристики различных векторов
для клонирования ДНК
Вектор
Плазмиды
Хромосома
фага А,
Космиды
ВАС
РАС
YAC
MAC
Хозяин
Е. coli
Е. coli
Е. coli
Е. coli
Е. coli
S. cerevisiae
Клетки животных
Структура
Кольцевая
Линейная
Кольцевая
Кольцевая
Кольцевая
Линейная хромосома
Линейная или кольцевая
Размер
вставки
(т.п.о.)
0,1-10
5-25
35-45
До 300
100-300
100-2000
41000 (?)
чае, несмотря на наличие теломерных последовательностей, ис-
искусственные хромосомы обнаруживали в клетках в кольцевой
форме в виде молекул, амплифицированных до размера 1-10
млн п.о. Кольцевые НАС могут быть получены и вообще без те-
теломерных последовательностей при наличии в РАС-векторе по-
последовательности альфоидной ДНК длиной в 70 т.п.о., наличие
которой совершенно необходимо для функционирования сегреги-
сегрегируемых искусственных хромосом. Искусственные хромосомы
MAC и НАС дают возможность вводить в них целые генетические
локусы и получать экспрессию соответствующих генов в культи-
культивируемых клетках или трансгенных животных. Основная пробле-
проблема, с которой приходится сталкиваться при использовании данных
искусственных генетических систем, это неконтролируемая экс-
экспрессия рекомбинантных генов, которые часто присутствуют в
таких хромосомах в большом числе копий. Технологии MAC и
НАС в настоящее время находятся в самом начале своего разви-
развития, и можно надеяться, что в недалеком будущем эти векторные
системы станут гораздо более совершенными. Работы в этом на-
направлении не прекращаются, ведь основная идея, лежащая в осно-
основе данных векторов, а именно, возможно более полная функцио-
функциональная имитация природных эукариотических хромосом, выгля-
выглядит весьма привлекательно, и, после ее полноценной реализации,
позволит генным инженерам получать полноценную экспрессию
гигантских генов высших эукариотических организмов.
В заключение этого раздела приводятся сравнительные ха-
характеристики рассмотренных основных векторных систем, кото-
100
рые используются в настоящее время для клонирования фраг-
фрагментов ДНК (табл. 4).
3.5. Интегрирующие и челночные (бинарные) векторы
Векторы, пригодные для клонирования ДНК в бактериях, от-
отличающихся от Е. coli, должны обладать всеми характерными
чертами, которые были отмечены выше для плазмидных векто-
векторов. От рассмотренных они отличаются главным образом тем,
что содержат природные или искусственные генетические эле-
элементы, функционирующие в новых клетках-хозяевах.
3.5.1. Интегрирующие векторы грамположительной бактерии
Bacillus subtilis
В. subtilis, как и Е. coli, является излюбленным объектом ген-
генной инженерии. Это связано с тем, что В. subtilis - непатогенный
микроорганизм, многие штаммы которого широко используются
в микробиологической промышленности для производства био-
биологически активных соединений и пищевых веществ. В. subtilis в
отличие от Е. coli способна секретировать белки и пептиды, что
облегчает их очистку и дальнейшее использование. Большинство
векторов для клонирования ДНК в клетках В. subtilis создано на
основе плазмид других видов бацилл, а также Staphylococcus
aureus и бактерий рода Streptococcus.
Основным свойством интегрирующих векторов является их спо-
способность стабильно встраиваться в геном клетки-хозяина. Это ста-
становится возможным благодаря наличию в таких векторах последо-
последовательностей нуклеотидов, гомологичных последовательностям ге-
геномной ДНК. В результате функционирования общей системы ре-
рекомбинации происходит объединение хромосомной и плазмидной
ДНК интегрирующего вектора и стабильное включение всей век-
векторной плазмиды в хромосому. Примером интегрирующего векто-
вектора служит плазмид a pFH7 В. subtilis (рис. 12, а). Векторная плазмид а
содержит фрагмент ДНК умеренного бактериофага SPC и после по-
попадания в клетки В. subtilis, лизогенные по данному бактериофагу,
эффективно интегрируется в профаг. Поскольку плазмида содер-
содержит ген устойчивости к хлорамфениколу cat, клетки приобретают
этот признак. Индукция профага приводит к образованию фаговых
частиц, трансдуцирующих такую плазмиду и ассоциированный с ней
признак устойчивости к хлорамфениколу. Интеграция плазмиды
SPC в бактериальную хромосому происходит по механизму гомоло-
гомологичной рекомбинации с участием гена гесЕ. Способность к интег-
101
а
Pstl
б
'Bf 1 pc 194
' pFH7
JL
Hindlll
Pst{pBR322 'BamKl
EcoRl trpE
Bg/ll/BamHl
cat
Sad EcoRV Sphl Kpnl Xbal BamKl
"fi \ \ I I I
clts857
Clal
/ Pstl
'Ori Рис. 12. Челночный вектор pFH7 (а) и экс-
прессирующий вектор pPR-TGATG-1 (б)
Обозначено расположение генов и уникаль-
уникальных сайтов рестрикции
рации в бактериальную хромосому обнаруживают и другие плаз-
миды, содержащие фрагменты хромосомной ДНК клеток-хозя-
клеток-хозяев, что продемонстрировано, в частности, для плазмид Е. coli и
Streptococcus pneumoniae.
Бактериальные интегрирующие векторы находят широкое
применение в современной генной инженерии бактерий, в част-
частности, при конструировании стандартных штаммов-продуцентов
белков, пептидов или метаболитов. Другим интересным прило-
приложением такого рода генетических систем является создание бак-
бактериальных штаммов-биорепортеров, используемых для монито-
мониторинга окружающей среды и в экотоксикологии. Хотя современ-
современные аналитические методы позволяют обнаруживать с высокой
эффективностью практически любое химическое соединение,
все эти методы не дают ответа на вопрос о биологической опас-
опасности таких соединений. Данный недостаток восполняют с помо-
помощью биосенсоров в формате целых бактериальных клеток, кото-
которые позволяют обнаруживать индивидуальные вещества или
группы химических соединений, для которых характерно одно-
однотипное взаимодействие с биологическим материалом. Например,
для обнаружения фитотоксичных химических соединений приме-
применяют генетически модифицированные цианобактерии, исходные
представители которых присутствуют в большом разнообразии в
фотосинтезирующей биосфере. Введение в хромосому цианобак-
102
терии Synechocystis РСС 6803 гена люциферазы светлячков (luc)
под контролем tac-промотора превращает его в стабильного про-
продуцента этого фермента. Функционирование люциферазы, в
свою очередь, зависит от концентрации АТР в клетках (см. раз-
раздел П.3.2.2), которая отражает их метаболическую активность.
По подавлению биолюминесценции судят о токсическом дейст-
действии веществ на метаболизм этой бактерии.
Интегрирующие векторные системы, в которых использует-
используется тот же принцип гомологичной рекомбинации, разработаны и
для эукариотических клеток, включая клетки животных и расте-
растений. В конце концов, такие работы привели к развитию целого
направления исследований по созданию трансгенных животных и
растений, стабильно наследующих и экспрессирующих гены, ис-
искусственно введенные в их геном.
Наличие гомологичной рекомбинации между хромосомной
ДНК клетки-хозяина и векторной ДНК, содержащей гомологич-
гомологичные хромосомной ДНК последовательности нуклеотидов, прихо-
приходится учитывать при получении соответствующих генно-инженер-
генно-инженерных конструкций. Такая неконтролируемая рекомбинация в боль-
большинстве случаев нежелательна, так как может приводить к потере
или структурным перестройкам клонированных фрагментов ДНК.
Для того, чтобы свести последствия этого явления к минимуму, ис-
используют специальные штаммы клеток-хозяев, в которых общая
система рекомбинации не функционирует, например, вследствие
мутационной инактивации гена reck E. coli или гесЕ В. subtilis.
3.5.2. Челночные векторы
Интегрирующая плазмида pFH7 (рис. 12, а) дает возможность
проиллюстрировать еще один важный принцип, широко исполь-
используемый при конструировании векторных систем в генной инже-
инженерии. Эта плазмида получена путем объединения двух реплико-
нов, один из которых берет начало от плазмиды рС194 В. subtilis,
а другой - от плазмиды pBR322 E. coli, что дает возможность век-
вектору существовать и стабильно реплицироваться как в клетках
Е. coli, так и В. subtilis. Такие векторы, способные реплициро-
реплицироваться в клетках-хозяевах разных биологических видов, называ-
называют челночными, или бинарными векторами.
Принципы конструирования и функционирования челночных
векторов одинаковы, они должны включать в себя репликоны
тех генетических систем, в которых будет происходить реплика-
репликация челночного вектора. При этом используются области начала
репликации генетических элементов, которые автономно суще-
103
ствуют во внехромосомном состоянии в природных условиях.
Так, интегрирующий вектор pFH7 В. subtilis обладает свойствами
челночного вектора, поскольку для его конструирования приме-
применены репликоны двух видов бактерий. Более эффектными при-
примерами челночных векторов являются плазмидные ДНК, способ-
способные реплицироваться в клетках высших (животных и растений) и
низших организмов. Необходимость использования челночных
векторов в генной инженерии связана с тем, что наработку в пре-
препаративном количестве векторной ДНК для проведения генно-
инженерных манипуляций удобнее проводить в бактериальных
клетках, тогда как получение биологически активных продуктов
клонированных генов высших организмов во многих случаях воз-
возможно только в клетках своего или близкого вида, в которых эти
гены экспрессируются в природных условиях, т.е. в своем обыч-
обычном генетическом окружении (подробнее см. раздел 5.2).
3.6. Конструирование экспрессирующих векторов
и их функционирование
Интенсивные исследования последних лет принесли и про-
продолжают приносить новые знания о механизмах, обеспечиваю-
обеспечивающих высокоэффективную и высокоспецифическую экспрессию
генов [15]. Эту информацию успешно используют для эффектив-
эффективной экспрессии рекомбинантных генов в гомологичном или гете-
рологичном генетическом окружении [117]. После рассмотрения
основных принципов конструирования векторов для клонирова-
клонирования ДНК можно перейти к обсуждению проблемы экспрессии
клонированных генов в искусственных генетических системах.
Именно экспрессия клонированных генов является одной из ос-
основных задач генной инженерии и биотехнологии. Действитель-
Действительно, функциональную роль отдельных генов и их частей в живом
организме можно понять и оценить лишь на основании экспрес-
экспрессии этих последовательностей, т.е. по фенотипическому проявле-
проявлению их потенциальных биологических возможностей. Кроме то-
того, крупномасштабная наработка биотехнологических продуктов
требует осуществления эффективной экспрессии конкретных ге-
генов в искусственно созданных условиях. Для получения полно-
полноценной экспрессии клонированных генов используют экспресси-
рующие векторные системы, принципы конструирования кото-
которых в настоящее время хорошо разработаны.
Напомним, что экспрессия генетической информации, заклю-
заключенной в генах живых организмов, в соответствии с основным по-
104
а
ДНК -* Число копий генов
Транскрипция
Активность промоторов
РНК "• Стабильность мРНК
Трансляция
_^ Сайты связывания
рибосом
-* Использование кодонов
Белок -* Стабильность белков
Фолдинг
Посттрансляционные
модификации
Многие факторы,
определяемые
клеткой-хозяином
Биологическая
активность
Консенсусные Точка
последовательности инициации
промотора транскрипции
ч
Точка
инициации
транскрипции
Точка
инициации
трансляции
Сайт Точка
связывания инициации
рибосом трансляции
N
Белок
Последовательность
Шайна-Дальгарно
Инициирующий
Рис. 13. Основные факторы, оказывающие влияние на эффективность
передачи генетической информации на разных этапах экспрессии ре-
рекомбинантных генов в бактериальных клетках
а - ключевые этапы экспрессии генов. Справа указаны факторы, оказыва-
оказывающие наибольшее влияние на уровень биосинтеза рекомбинантных белков in
vivo;
б - основные регуляторные участки нуклеиновых кислот, контролирую-
контролирующие транскрипцию и трансляцию
105
стулатом молекулярной биологии осуществляется путем ее пере-
переноса от нуклеиновых кислот к белкам по схеме: ДНК <-» РНК —»бе-
—»белок, и эффективность прохождения любого из этих, а также неко-
некоторых промежуточных этапов влияет на общий выход зрелого бел-
белкового продукта (рис. 13). Полноценный в функциональном отно-
отношении ген, в том числе и рекомбинантный, состоит из двух основ-
основных частей - регуляторной и структурной (рис. 13, б). Генетичес-
Генетический код, с помощью которого последовательность нуклеотидов в
кодирующей структурной части гена определяет последователь-
последовательность аминокислот в кодируемом этим геном белке, за немногими
исключениями, универсален для всех живых организмов. Это дает
принципиальную возможность получения полноценной экспрессии
самых разнообразных осмысленных последовательностей нуклео-
нуклеотидов, в том числе и искусственно синтезированных, в любом при-
природном генетическом окружении. Таким образом, универсаль-
универсальность генетического кода является основой для создания функци-
функционально активных генно-инженерных конструкций.
Регуляторные части генов, а также продукты их экспрессии,
мРНК и белки, распознаются соответствующими ферментными
системами организма и обеспечивают упорядоченную экспрессию
структурной части гена. При этом регуляторные участки генов и
промежуточных продуктов их экспрессии, как правило, высоко-
высокоспецифичны в отношении своих природных генетических эффек-
эффекторов (РНК-полимераз, рибосом, факторов транскрипции и
трансляции, белковых факторов сплайсинга, ферментов, осуще-
осуществляющих посттрансляционные модификации полипептидов, и
т.п.), и чаще всего они не могут эффективно функционировать в
гетерологичном генетическом окружении. Очевидно, что при
конструировании высокоэффективных экспрессирующих векто-
векторов необходимо, прежде всего, учитывать особенности структуры
регуляторной части рекомбинантного гена, исходя из того, в ка-
каких генетических условиях клонированный ген будет экспресси-
роваться. Однако не только регуляторные последовательности
генов являются препятствием для высокоэффективной экспрес-
экспрессии чужеродных рекомбинантных генов. Как уже было отмечено,
структурные части генов про- и эукариот фундаментально отли-
отличаются друг от друга по наличию у последних внутри генов интро-
нов. Следовательно, гены эукариот не могут эффективно экс-
прессироваться в бактериальных клетках, поскольку у прокариот
отсутствуют соответствующие системы сплайсинга. Кроме того,
у предшественников эукариотических мРНК не может осущест-
осуществиться в бактериальных клетках и правильный процессинг 3'- и
5'-концевых некодирующих последовательностей. Даже такой
106
беглый взгляд на проблему экспрессии рекомбинантных генов в
чужеродном генетическом окружении позволяет увидеть много-
многочисленные препятствия, лежащие на пути решения этой пробле-
проблемы. Ниже будут рассмотрены факторы, которые необходимо
учитывать при конструировании экспрессирующих векторов.
3.6.1. Факторы, оказывающие влияние
на эффективность экспрессии рекомбинантных генов
в бактериальных клетках: транскрипция
Для экспрессии клонированного гена в клетках Е. coli или
любого другого организма необходимо, чтобы этот ген был
транскрибирован РНК-полимеразой, а образовавшаяся РНК
транслирована рибосомами с образованием нативного белкового
продукта. Таким образом, для эффективной экспрессии в клет-
клетках Е. coli любой ген должен находиться под контролем регуля-
торных элементов данного микроорганизма, основным из кото-
которых является промотор [118].
Базальный уровень транскрипции рекомбинантных генов обес-
обеспечивается небольшим набором упорядоченных регуляторных по-
последовательностей. Например, у Е. coli это происходит при наличии
перед структурной частью клонированного гена последовательно-
последовательностей в окрестностях нуклеотидов +1, -10 и -35 (где нуклеотид в по-
положении +1 указывает на точку инициации транскрипции).
-35 -10 +1
...TTGACA ТАТААТ cat - консенсус
...ТТГАСА ТАТА7Т cat - /ас-промотор
...TTGACA ТТ4АСТ cat - frp-промотор
...TTGACA GATACT cat - /^-промотор фага X
В представленных последовательностях промоторов Е. coli, на-
наиболее часто используемых в генно-инженерных конструкциях для
экспрессии в этих клетках, отмечены различия (выделены курси-
курсивом), которые оказывают влияние на эффективность использова-
использования промотора РНК-полимеразой ("силу" промотора).
В конечном счете, сила промотора определяется легкостью обра-
образования так называемого открытого комплекса между РНК-по-
РНК-полимеразой и промотором, в котором происходит стабильное ло-
локальное расплетение цепей ДНК, что необходимо для инициации
транскрипции. Естественно, легкость такой ферментативной дена-
денатурации ДНК зависит от первичной структуры окружающих этот
участок последовательностей нуклеотидов. Консенсусная последо-
107
вательность является усредненной последовательностью, которая
чаще всего встречается в генах Е. coli, а /^-промотор фага X - од-
одним из наиболее сильных природных промоторов, функционирую-
функционирующих в этой бактерии. Из приведенных последовательностей видно,
что ни одна из критических последовательностей природных про-
промоторов не идентична консенсусной последовательности, что ука-
указывает на возможность изменения последовательности нуклеоти-
дов промоторов для дальнейшего повышения эффективности их
функционирования. Действительно, знание основных принципов
строения и функционирования промоторов позволяет конструиро-
конструировать новые промоторы, которые по своей эффективности могут
превосходить природные. Примером такого промотора, используе-
используемого в некоторых экспрессирующих векторах, является промотор
Taq, который в своем составе объединяет последовательности про-
промоторов лактозного и триптофанового оперонов Е. coli.
В клетках Е. coli (и других бактерий) транскрипция большин-
большинства генов носит регулируемый характер, что обеспечивается си-
системой белков-репрессоров и активаторов, а также низкомолеку-
низкомолекулярных эффекторов, которые главным образом влияют на взаи-
взаимодействие РНК-полимеразы с транскрибируемыми генами
[119-121]. Необходимость регулирования экспрессии генов в ген-
генной инженерии диктуется прежде всего тем, что сверхэкспрессия
клонированных генов в клетках-продуцентах рекомбинантных
белков при использовании сильных конститутивных промоторов
может отрицательно сказываться на их жизнеспособности, по-
поскольку промежуточные и конечные продукты экспрессии часто
обладают цитотоксическим действием [122]. В этом случае сверх-
сверхэкспрессия клонированных генов сопровождается снижением
скорости или даже полной остановкой роста бактериальных или
иных клеток. Для предотвращения этих эффектов используются
генно-инженерные конструкции, в которых экспрессия клониро-
клонированных генов в значительной степени подавлена (репрессирована)
на ранних фазах роста культуры рекомбинантных клеток и мо-
может быть дерепрессирована в любой нужный момент времени.
В качестве примера рассмотрим три системы регулируемой
экспрессии рекомбинантных генов в клетках Е. coli, которые полу-
получили широкое распространение. В одном случае для контроля экс-
экспрессии клонированных генов используют хорошо изученную сис-
систему регуляторных элементов лактозного оперона [123], в дру-
другом - систему промотор-репрессор-оператор фага X [124], в треть-
третьем - промоторы и РНК-полимеразы бактериофагов [125]. Прин-
Принцип регуляции экспрессии рекомбинантных генов в первых двух
случаях один и тот же. В векторные молекулы вводятся регуля-
108
дррные последовательности фага X или /ас-оперона, за которыми
следуют последовательность нуклеотидов полилинкера с несколь-
несколькими уникальными сайтами рестрикции, по которой проводится
зонирование гена. Одновременно в тот же вектор вводится ген-
регулятор, кодирующий белок-репрессор. В отсутствие индуциру-
индуцирующего воздействия молекулы репрессора, ген которого экспрес-
сируется конститутивно, связываются с оператором, тем самым
препятствуя взаимодействию РНК-полимеразы с промотором, под
контролем которого находится клонированный рекомбинантный
ген. Это приводит к резкому снижению уровня транскрипции кло-
клонированного гена, низкому внутриклеточному содержанию соот-
соответствующей мРНК и белкового продукта ее трансляции.
Существенным различием двух систем регулируемой экспрес-
экспрессии является способ их индукции. В случае регуляторной системы
/ас-оперона чаще всего используют синтетический аналог при-
природного индуктора (ялло-лактозы) - изопропил-р-О-тиогалакто-
пиранозид (IPTG). Индуктор связывается с молекулой репрессора
и нарушает его взаимодействие с операторным участком ДНК.
Область /ас-промотора становится доступной для РНК-полимера-
РНК-полимеразы, которая с высокой эффективностью начинает транскрибиро-
транскрибировать гены, расположенные вслед за промотором. В отличие от
этого в системе репрессии-дерепрессии фага X используют ген ре-
репрессора с1857, содержащий температурочувствительную мута-
мутацию. Наличие такой мутации приводит к тому, что мутантный бе-
белок-репрессор сохраняет свою нативную конформацию, а следо-
следовательно, и способность подавлять синтез РНК только при пер-
миссивной температуре (как правило, 28-30°С). При повышении
температуры окружающей среды до 42°С происходят инактива-
инактивация белка-репрессора и дерепрессия генов, расположенных под
контролем промоторов PL или PR фага X.
Использование системы репрессии-дерепрессии, чувствитель-
чувствительной к температуре, в ряде случаев более технологично по сравне-
сравнению с химической индукцией по двум обстоятельствам:
во-первых, это менее дорогой способ индукции; во-вторых, для
системы фага X характерна более высокая степень репрессии ге-
генов, находящихся под ее контролем, и, соответственно, более низ-
низкий базальный уровень их экспрессии. Такой фактор становится
решающим при клонировании и получении экспрессии генов, бел-
белковые продукты которых токсичны для клетки-хозяина. Однако,
несмотря на технологичность системы репрессии-дерепрессии,
чувствительной к температуре, сверхпродукция рекомбинантных
белков, особенно эукариотических, часто сопровождается внут-
внутриклеточным образованием их нерастворимых агрегатов, что в
109
ряде случаев полностью отсутствует при использовании для выра-
выращивания рекомбинантных клеток температур ниже 30°С (подроб-
(подробнее см. ниже). Эти эффекты накладывают ограничения на ис-
использование векторов с температурочувствительным контролем
экспрессии для продукции эукариотических и в меньшей степени
прокариотических белков в бактериальных клетках.
Самый сильный уровень репрессии рекомбинантного гена в
бактериальных клетках обеспечивает система транскрипции с учас-
участием фагоспецифических РНК-полимераз Т7, ТЗ и SP6 (TV-подоб-
(TV-подобный фаг Salmonella), что особенно важно при клонировании генов
белков, токсичных для клеток-хозяев, таких как колицины, эндо- и
экзонуклеазы, протеиназы и т.п. Нечетные Т-фаги и SP6 кодируют
просто устроенные (односубъединичные) РНК-полимеразы, кото-
которые осуществляют транскрипцию большей части поздних генов
этих вирусов. При этом фаговые ферменты очень эффективно ис-
используют собственные промоторы, которые совершенно не распоз-
распознаются РНК-полимеразой Е. coli из-за особенностей первичной
структуры. Поэтому в отсутствие фаговых РНК-полимераз транс-
транскрипция генов, клонированных под контролем фаговых промото-
промоторов, полностью блокирована. В таких системах индукцию рекомби-
рекомбинантных генов осуществляют двумя способами. В одном случае ре-
комбинантными плазмидами трансформируют штамм Е. coli, лизо-
генный (т.е. содержащий в своей хромосоме интегрированную фа-
фаговую хромосому) по фагу DE3, производному X, обладающему об-
областью иммунности фага 21, геном Т7-РНК-полимеразы под кон-
контролем /ас(УК5-промотора и геном /ас-репрессора lacl [126]. Индук-
Индукция транскрипции гена Т7-РНК-полимеразы осуществляется искус-
искусственным индуктором /ас-оперона IPTG. В присутствии IPTG в
клетках нарабатывается Т7-РНК-полимераза, которая с высокой
эффективностью начинает транскрибировать рекомбинантный ген.
Уровень репрессии гена Т7-РНК-полимеразы в системе, рас-
рассмотренной выше, бывает недостаточным для предотвращения ле-
летального действия очень токсичных рекомбинантных белков на
клетку-хозяина. В этом случае для индукции рекомбинантного ге-
гена может быть использовано заражение клеток, содержащих экс-
прессирующий вектор, фагом СЕ6, который содержит в своей хро-
хромосоме ген Т7-РНК-полимеразы, под контролем ^-промоторов PL
и PR, регулируемых температурочувствительным репрессором
с1857. После заражения таким фагом рекомбинантных клеток и ин-
индукции гена Т7-РНК-полимеразы транскрипция рекомбинантных
генов происходит настолько эффективно, что это предотвращает
нормальное развитие самого фага. Оба типа систем имеются в про-
продаже и распространяются фирмой Novagene (США).
110
Для обеспечения регулируемой тканеспецифической экс-
экспрессии рекомбинантных генов в соматических клетках живот-
животных и растений в составе векторов используют энхансеры, кото-
которые избирательно стимулируют транскрипцию в соответствую-
соответствующих тканях и не оказывают такого действия на гены в тканях,
клетки которых не экспрессируют необходимые регуляторные
белки. Кроме того, популярным становится введение в экспрес-
сирующие эукариотические векторы пограничных последова-
последовательностей нуклеотидов, фланкирующих клонируемые гены, ко-
которые помогают обеспечивать экспрессию рекомбинантных ге-
генов, сводя к минимуму эффект их положения в хромосомах сома-
соматических клеток.
Наряду с промоторами векторы, экспрессирующие рекомби-
нантные гены в клетках Е. coli, должны содержать и гомологич-
гомологичные терминаторы транскрипции. Необходимость в этом вызвана
тем, что даже нетранслируемые избыточные 3'-концевые после-
последовательности нуклеотидов мРНК, транскрибированной с реком-
бинантного гена, отрицательно сказываются на эффективности
ее трансляции, что, по-видимому, связано с участием таких по-
последовательностей в образовании сложной третичной структуры
рекомбинантных мРНК.
3.6.2. Факторы, влияющие на эффективность
экспрессии рекомбинантных генов
в бактериальных клетках: трансляция
Как следует из изложенного выше, уровень экспрессии кло-
клонированных генов в бактериальных и эукариотических клетках
во многом зависит от эффективности их транскрипции, т.е. от
уровня внутриклеточного содержания соответствующих мРНК.
Однако экспрессия рекомбинантных генов не в меньшей степени
зависит и от эффективности трансляции таких мРНК рибосома-
рибосомами, поскольку процесс передачи генетической информации при-
природных генов и на этом уровне является объектом многочислен-
многочисленных регуляторных воздействий [127]. При работе с рекомбинант-
ными ДНК чаще всего приходится иметь дело с гетерологичны-
ми системами, в которых клонированные экспрессируемые ДНК
чужеродны по отношению к генетическому окружению и фер-
ферментативным системам бактериальных или эукариотических
клеток-хозяев. Поэтому всегда необходимо учитывать уровень
Соответствия регуляторных факторов клеток-хозяев регулятор-
ным последовательностям мРНК, которые являются транскрип-
транскриптами рекомбинантных генов. В частности, на эффективность
Трансляции чужеродных мРНК в бактериальных клетках оказы-
111
вает сильное влияние первичная структура небольшого участка
5'-концевой части мРНК перед инициирующим AUG-кодоном,
частично комплементарная 3'-концевой части 16S рРНК. Этот
участок мРНК называют последовательностью Шайна-Далъ-
гарно, или SD-последователъностъю [128, 129].
SD-последовательность мРНК служит сигналом для взаимо-
взаимодействия рибосом с мРНК и обеспечивает инициацию трансляции
с правильного AUG-кодона. Она представляет собой короткую по-
последовательность нуклеотидов, обогащенную пуриновыми осно-
основаниями, и расположена за 4-9 нуклеотидов перед AUG-кодоном:
16S рРНК hoAUUCCUCCACUAGG 5'
lacZ-мРН К иисАС дс AGG AAAC AGCU auqacc aug auu • • • • 3'
(в приведенном примере прописными буквами у /acZ-мРНК изоб-
изображена SD-последовательность нуклеотидов, комплементарная
соответствующей пиримидиновой последовательности 16S рРНК,
и подчеркнут инициирующий кодон). Различия в эффективности
распознавания такого сайта на мРНК рибосомами приводят к раз-
различиям в уровнях экспрессии соответствующих генов. Достаточ-
Достаточно наличия в SD-сайте трех нуклеотидов, комплементарных соот-
соответствующему участку 16S РНК, чтобы рибосомы приобретали
способность связывать эту мРНК. Помимо SD-последовательнос-
ти в мРНК, по-видимому, имеются и другие участки, необходимые
для инициации трансляции, так как на основании только первич-
первичной структуры мРНК в настоящее время невозможно предсказать
эффективность сайта связывания рибосом. Более того, сайты
инициации трансляции на мРНК, обладающие протяженной гомо-
гомологией с 16S рРНК, функционируют плохо, возможно, из-за
слишком прочного связывания рибосом с мРНК.
Существенным фактором, оказывающим влияние на уровень
трансляции мРНК рибосомами, может быть вторичная структура
мРНК в окрестностях инициирующего ко дона [130]. Таким обра-
образом, из всего вышеизложенного следует, что структура некоди-
рующей 5'-концевой части мРНК играет важную роль в трансля-
трансляции мРНК, и, по крайней мере, эти особенности синтеза белка не-
необходимо учитывать при конструировании векторов для экспрес-
экспрессии рекомбинантных генов в клетках Е. coli. Современные экс-
прессирующие векторы построены таким образом, что промо-
промотор, с которого начинается транскрипция клонированного гена, а
также следующая за ним последовательность нуклеотидов,
включающая инициирующий ATG-кодон, являются частью ге-
генов, эффективно экспрессирующихся в клетках Е. coli. Сразу
112
фр за ATG-кодоном вектора располагают уникальные сайты
рестрикции, по которым проводят встраивание кодирующей час-
f# клонируемого гена, экспрессию которого хотят получить. В
гом случае, если сайт рестрикции, по которому производится
клонирование, расположен в начале кодирующей части природ-
природного гена, в результате транскрипции такой рекомбинантной мо-
молекулы образуется химерная мРНК. 5'-Концевая часть этой
мРНК от промотора до сайта рестрикции для клонирования соот-
соответствует последовательности нуклеотидов вектора и кодирует
несколько аминокислот природного гена, регуляторные последо-
последовательности которого использовали для конструирования экс-
прессирующего вектора. Протяженная же 3'-концевая часть та-
такой мРНК будет соответствовать клонированной последователь-
последовательности нуклеотидов. Если соединение этих двух частей мРНК про-
произошло в одной рамке считывания, то в результате ее трансляции
будет образовываться химерный полипептид, N-концевая часть
которого кодируется вектором, а С-концевая - клонированным
фрагментом ДНК. Примерами таких экспрессирующих векторов
могут быть плазмиды pUC18 и pUC19 (см. рис. 6, б), продукты
экспрессии которых представляют собой химерные белки, а
N-концевая часть содержит N-концевые аминокислотные остат-
остатки р-галактозидазы Е. coli.
Превосходным вектором, хорошо зарекомендовавшим себя в
работе, является экспрессирующий вектор pPR-TGATG-1, скон-
сконструированный в лаборатории СВ. Машко (ГосНИИ Генетики и
селекции промышленных микроорганизмов, г. Москва). Вектор
функционирует по тому же самому принципу. В нем структурная
часть экспрессируемого гена соединяется через уникальный сайт
рестрикции Sad с инициирующим кодоном ATG (см. рис. 12, б).
В результате образуется гибридный оперон, транскрибируемый с
промотора X-PR, дистальный цистрон которого представлен кло-
клонируемым геном, а цистрон, проксимальный промотору PR, со-
содержит гибридный ген cro'-cat'-trpE, состоящий из слитных час-
частей генов сто фага X, хлорамфениколацетилтрансферазы cat и
trpEE. coli. 3'-Концевая часть цистрона содержит область реини-
циации трансляции, SD-последовательность гена trpD E. coli и пе-
перекрывающиеся кодоны терминации и инициации трансляции
(TGATG). В этой последовательности 3'-концевой нуклеотид G
является первым нуклеотидом уникального сайта рестрикции
Sad (GAGCTC). В процессе синтеза белка, направляемого векто-
вектором, образуется короткий гибридный пептид Cro'-Cat'-TrpE дли-
Ной ~120 аминокислот. Синтез пептида высокоэффективен, по-
поскольку все регуляторные элементы вектора оптимизированы
113
для его образования. Сразу же после терминации синтеза пепти-
пептида происходит реинициация синтеза полипептида, кодируемого
клонированным геном, на перекрывающемся с кодоном TGA
ATG-кодоне. Поскольку при этом не происходит освобождения
рибосом из транслирующего комплекса, стадия реинициации
трансляции протекает с высокой скоростью. Промотор PR в этом
векторе контролируется температурочувствительным репрессо-
ром фага X clts857, поэтому экспрессия клонированного гена ин-
индуцируется повышением температуры окружающей среды до
42°С и сопровождается инактивацией репрессора.
Приведенные примеры четко демонстрируют роль двух ос-
основных факторов, оказывающих влияние на уровень экспрессии
чужеродных клонированных генов, находящихся в составе экс-
прессирующих векторов в бактериальных клетках. Этими фак-
факторами являются, во-первых, оптимальная транскрипция клони-
клонированных генов и, во-вторых, эффективная трансляция транс-
транскрибированной мРНК. В обоих случаях наилучшие результаты
могут быть получены при объединении в составе вектора струк-
структурной части клонированного чужеродного гена с генетическими
элементами, регулирующими транскрипцию и трансляцию (про-
(промоторы, SD-последовательности и т.п.) бактериальных клеток-
хозяев или их вирусов. Знание главных принципов, лежащих в ос-
основе регуляции транскрипции и трансляции, позволяет конструи-
конструировать новые регуляторные последовательности (например, про-
промотор Taq) путем объединения известных регуляторных элемен-
элементов или использования искусственно синтезированных последо-
последовательностей нуклеотидов, представляющих собой усредненные
канонические регуляторные последовательности, учитывающие
особенности большого числа природных регуляторных элемен-
элементов. Однако этими важными факторами не исчерпывается воз-
возможность оптимизации экспрессии рекомбинантных генов в чу-
чужеродном генетическом окружении.
Среди других факторов, оказывающих влияние на эффектив-
эффективность трансляции, следует упомянуть частоту использования
кодонов при кодировании белков в структурных частях разных
генов [131]. В настоящее время установлено, что использование
синонимических кодонов (кодирующих одну и ту же аминокисло-
аминокислоту) вырожденного генетического кода не случайно и отражает
количественную представленность отдельных изоакцепторных
тРНК в клетках организма. С другой стороны, частота использо-
использования кодонов в разных генах одного и того же организма явля-
является эффективным фактором, регулирующим уровень экспрес-
экспрессии этих генов в процессе трансляции. Чем реже тот или иной ко-
114
«он используется для кодирования аминокислоты в данном орга-
организме, тем с меньшей скоростью он будет транслироваться рибо-
рибосомами вследствие низкой внутриклеточной концентрации
тРНК, узнающей такой кодон. Так, в группе белков дрожжей с
высоким уровнем экспрессии 96% аминокислот кодируются
только 25 из 61 возможного кодона. Замена этих 25 предпочти-
предпочтительных ко донов в 5'-концевых частях генов белков с высоким
уровнем экспрессии на редко встречающиеся кодоны снижает
уровень синтеза как белков, так и их мРНК. Трансляция чуже-
чужеродной мРНК, содержащей редко встречающиеся кодоны, со-
сопровождается временной задержкой синтеза белка, преждевре-
преждевременной терминацией трансляции, сдвигом рамок считывания и
ошибочным включением аминокислотных остатков. При этом
отрицательный эффект редко встречающихся кодонов проявля-
проявляется сильнее всего, если они локализованы в 5'-концевой части
мРНК, кодирующей N-концевые аминокислотные остатки ре-
комбинантного белка.
Кроме того, на эффективность трансляции кодона оказывает
влияние и контекст кодона, т.е. соседние, окружающие кодон по-
последовательности нуклеотидов [132-134]. Этот феномен предпо-
предпочтительного использования одних кодонов перед другими необ-
необходимо учитывать в опытах по оптимизации экспрессии чуже-
чужеродного генетического материала в гетерологичном генетичес-
генетическом окружении, поскольку несоответствие в частотах использо-
использования кодонов у организмов разных видов может служить серьез-
серьезным препятствием для получения эффективной экспрессии кло-
клонируемых генов. Снижение уровня экспрессии под влиянием по-
подобных факторов, в частности, наблюдали при попытках экс-
экспрессии генов растений в клетках дрожжей и других гетероло-
гичных системах. Таким образом, эффекты, возникающие в свя-
связи с различной частотой использования кодонов, необходимо
учитывать при подборе генетических систем для эффективной
экспрессии клонированных чужеродных генов. Этот фактор мо-
может оказаться одним из решающих при получении полностью
синтетических генов, например генов пептидных гормонов, для
использования их в биотехнологии, а также при планировании
мутационных замен нуклеотидов в белковой инженерии. Для
обеспечения сверхэкспрессии эукариотических рекомбинантных
белков, мРНК которых содержит редкие для Е. coli кодоны,
сконструированы штаммы, в которых происходит синтез недо-
недостающих изоакцепторных тРНК. Так, в штаммах Rosetta™
(Novagene) экспрессируются на отдельной плазмиде, относящей-
относящейся к одной группе совместимости с векторной плазмидой, гены
115
тРНК, распознающих кодоны AUA, AGG, AGA, CUA, ССС и
GGA. В таких штаммах происходит резкое возрастание синтеза
рекомбинантных белков, для которых данный фактор является
лимитирующим [52, 53].
3.6.3. Факторы, влияющие на эффективность
экспрессии рекомбинантных генов:
стабильность рекомбинантных РНК и белков
Важным фактором, определяющим уровень экспрессии ре-
рекомбинантных генов в реципиентных клетках, является стабиль-
стабильность кодируемых этими генами мРНК и белков. Во введении
упоминалось значение таких факторов для регуляции экспрессии
генов. Не менее важную роль эти феномены играют и в контро-
контроле уровня экспрессии рекомбинантных генов в искусственных ге-
генетических системах. Замены ко донов в мРНК дрожжей на сино-
синонимические приводят не только к снижению уровня трансляции
измененных мРНК в клетках дрожжей, но и сопровождаются
уменьшением стабильности самих мРНК [137,138]. Достаточный
уровень стабильности мРНК является одним из факторов, опре-
определяющих давление отбора на использование конкретных сино-
синонимических кодонов для кодирования белков. Важную роль во
внутриклеточной стабильности мРНК играют элементы ее вто-
вторичной структуры. Так, образование шпилек на 3'-конце мРНК
препятствует ее деградации под действием бактериальных экзо-
нуклеаз, РНКазы II и полинуклеотидфосфорилазы. Поскольку
изменение первичной структуры с учетом возможной вторичной
структуры без изменения функциональных свойств представля-
представляется в настоящее время сложной задачей, проблема внутрикле-
внутриклеточной стабильности рекомбинантных РНК решается, главным
образом, путем введения мутаций в гены экзо- и эндонуклеаз
бактериальной клетки-хозяина. Так называемые безРНКазные
штаммы Е. coli находят применение в качестве реципиентов ре-
рекомбинантных плазмид в генной инженерии. При этом решается
двойная задача: с одной стороны, в таких клетках повышается
стабильность транскриптов рекомбинантных генов, а с другой -
пониженный внутриклеточный уровень определенных РНКаз
способствует более эффективной очистке рекомбинантных бел-
белков, так как для многих препаратов ферментов даже незначи-
незначительные примеси нуклеазной активности бывают нежелательны.
Правило N-концевой последовательности (N-end rule). На
стабильность рекомбинантного белка оказывает заметное влия-
влияние природа аминокислотного остатка, непосредственно следую-
116
|цего за инициаторным N-концевым метионином. Этот амино-
аминокислотный остаток определяет эффективность удаления остатка
формилметионина (fMet), с которого всегда начинается синтез
долипептидных цепей в бактериях. Такой N-концевой процес-
синг полипептида осуществляется метиониламинопептидазой,
которая удаляет fMet, и с увеличением размера аминокислотного
остатка, непосредственно следующего за fMet, уменьшается эф-
эффективность этого процесса [139]. Резкое снижение или полное
прекращение процессинга имеет место в том случае, если в этом
положении полипептидной цепи находятся остатки His, Gin, Glu,
phe, Met, Lys, Туг, Тгр или Arg. При наличии в этом положении
любого из оставшихся аминокислотных остатков от 16% до 97%
полипептидов подвергаются процессингу.
Зависимость между природой N-концевых остатков в белках
и их стабильностью в бактериальных клетках определяется пра-
правилом N-концевой последовательности [140]. В том случае, если
на N-конце полипептида после удаления fMet оказываются Arg,
Lys, Phe, Leu, Trp и Туг, время полужизни белка составляет
< 2 мин, и оно становится > 10 мин при наличии в этом положе-
положении всех остальных аминокислотных остатков. Соответствую-
Соответствующие аминокислотные остатки являются сигналом для распозна-
распознавания белков убиквитин-зависимой системой протеолиза.
Проблема внутриклеточной стабильности рекомбинантных
белков больше связана с деградацией небольших пептидов, по-
поскольку крупные нативные белки более стабильны в бактериаль-
бактериальных клетках. Один из подходов, позволяющих стабилизировать
короткие чужеродные пептиды в клетках Е. coli, - включение тре-
требуемого пептида в состав гибридного белка. В этом случае после-
последовательность нуклеотидов, кодирующую гибридный белок, со-
соединяют в составе экспрессирующего вектора в одной рамке счи-
считывания с бактериальным геном, кодирующим белок (например,
геном р-галактозидазы). Образующийся в результате экспрессии
такого рекомбинантного гена гибридный белок в своем составе
содержит в N- или С-концевой части требуемый пептид, защищен-
защищенный основным белком-носителем от протеолитической деграда-
деградации. Для отделения пептида от белка-носителя их соединяют друг
с другом последовательностью аминокислот, по которой можно
провести специфическое расщепление полипептидной цепи. В том
случае, если пептиды не содержат метионина, соединение белка-
носителя и пептида осуществляют через эту аминокислоту, и от-
Щепление пептида производят с помощью бромистого циана. Та-
Такой подход был использован, например для получения рекомби-
рекомбинантного соматостатина, а также А- и В-цепей инсулина.
117
Для разделения рекомбинантного пептида и белка-носителя
можно использовать и протеолитические ферменты. Для этого
требуемый пептид соединяют с белком-носителем с помощью
последовательности аминокислот, специфически расщепляемой
протеолитическим ферментом, например пептидазой клостридий
(клострипаином) в случае получения кальцитонина или трипси-
трипсином для получения р-эндорфина.
Второй подход, позволяющий защищать рекомбинантные
белки от внутриклеточного протеолиза, - выведение их в пери-
плазматическое пространство или окружающую среду после за-
завершения биосинтеза. При таком подходе рекомбинантные поли-
полипептиды соединяют с сигнальными последовательностями ами-
аминокислот белков (р-лактамаза, OmpA, OmpF, PhoA), секретируе-
мых во внешнюю среду и периплазматическое пространство. По-
Подобным путем были получены, например проинсулин, гормон
роста человека и C-эндорфин.
3.6.4. Другие причины низкой эффективности
экспрессии рекомбинантных генов в бактериях
Вторичные сайты инициации трансляции. Иногда в результа-
результате экспрессии рекомбинантного гена в бактериальных клетках
образуется укороченная форма рекомбинантного белка, что свя-
связывают обычно с протеолитической деградацией исходной поли-
полипептидной цепи. Однако наличие сайта вторичной инициации
трансляции в мРНК также может быть причиной этого явления
[141]. Последовательность внутри мРНК, напоминающая сайт
связывания рибосом AAGGAGG, которая расположена за
5-13 нуклеотидов перед AUG-кодоном, может стать причиной та-
такой ошибочной инициации синтеза белка.
Вторичная структура транскрипта рекомбинантного гена.
Неблагоприятная вторичная структура гибридных рекомбинант-
рекомбинантных генов, особенно в окрестностях сайта инициации трансляции,
также может приводить к неэффективной трансляции соответст-
соответствующих мРНК [130]. Поэтому компьютерный поиск последова-
последовательностей, комплементарных последовательностям, находя-
находящимся вблизи сайта связывания рибосом транслируемой мРНК с
последующим дизайном рекомбинантного гена может решить
эту проблему. В ходе дизайна самокомплементарность внутри
мРНК можно преодолеть путем введения синонимических замен
нуклеотидов, которые не изменяя смысла кодонов окажут влия-
влияние на образование неблагоприятной вторичной структуры
мРНК.
118
Неожиданное возникновение кодонов терминации трансля-
трансляции. Возникновение при экспрессии укороченного, легко дегра-
йфуемого рекомбинантного белка может быть следствием появ-
появления в результате мутации в кодирующей части рекомбинантно-
рекомбинантного гена нового терминирующего кодона. Наличие такого арте-
артефакта обычно обнаруживается при секвенировании гена или его
экспрессии в бесклеточных системах трансляции, сопряженной с
Транскрипцией.
5'-концевой блок трансляции E'-translational blockage). Введе-
Введение редко используемых кодонов в мРНК вблизи сайта инициа-
й(ии трансляции часто сопровождается подавлением трансляции
мРНК гена-репортера. Ингибирования не происходит, если тот
же самый кодон расположить ниже в рамке считывания. Данный
Эффект является следствием первичной структуры мРНК, а не
особенностей ее пространственной структуры, которая могла бы
формироваться при участии этого кодона, и связан с недостатком
соответствующей тРНК.
Все эти и еще многие другие особенности регуляции трансля-
трансляции необходимо учитывать при конструировании искусственных
генетических систем экспрессии исследуемых генов [142, 143].
Влияние терминатора транскрипции вектора. Наличие дейст-
действенного терминатора транскрипции позади экспрессируемого ре-
рекомбинантного гена является еще одним условием эффективной
экспрессии рекомбинантного белка. В том случае, если РНК-по-
лимераза не прекращает синтеза РНК на границе рекомбинант-
рекомбинантного гена, может образоваться полицистронный транскрипт, в
котором сайты инициации трансляции векторной плазмиды бу-
будут конкурировать за соответствующие участки целевой реком-
бинантной мРНК. В частности, этот эффект может иметь место
при включении в полицистронную мРНК последовательности ге-
гена Ыа, определяющего устойчивость клеток к ампициллину.
Все эти и еще многие другие особенности регуляции трансля-
трансляции необходимо учитывать при конструировании искусственных
генетических систем экспрессии исследуемых генов [142, 143].
3.6.5. Особенности биосинтеза эукариотических рекомбинантных
белков в бактериальных клетках: тельца включения
Экспрессия эукариотических генов в бактериальных клетках
связана еще с одной проблемой: большая часть рекомбинантных
эукариотических белков, а также некоторые прокариотические
белки при очень высоком уровне биосинтеза переходят в нерас-
нерастворимое состояние, образуя в клетках так называемые тельца
119
включения. Последние представляют собой частицы, состоящие
главным образом из агрегатов рекомбинантного белка и ряда бак-
бактериальных белков. Рекомбинантные белки в тельцах включения
находятся, как правило, в денатурированном неактивном состоя-
состоянии, и для их получения в растворимом виде приходится применять
мощные денатурирующие агенты, такие как мочевина, гуанидинх-
лорид и детергенты [144-146]. Солюбилизированный в этих жест-
жестких условиях рекомбинантный белок для восстановления натив-
ной конформации требует ренатурации, в процессе обработки де-
денатурирующими агентами белки могут изменять свою структуру,
а при концентрировании из разбавленных растворов, в которых
проводится ренатурация, часто вновь выпадают в осадок.
В чем же причина такого аномального поведения рекомби-
нантных эукариотических белков в бактериальных клетках? Для
поддержания белков в растворимом состоянии в клетках эукари-
от реализуются три основных механизма: компартментализация
продуктов трансляции, белок-белковые взаимодействия и пост-
посттрансляционные модификации. Образующиеся в эндоплазмати-
ческом ретикулуме полипептидные цепи не распределяются хао-
хаотически в цитоплазме эукариотических клеток, но последова-
последовательно переходят через ряд компартментов, где претерпевают
посттрансляционные модификации. При этом внутренние усло-
условия отдельных компартментов могут существенно различаться.
Так, молекулы инсулина накапливаются in vivo в секреторных
гранулах, рН в которых составляет 4,5-5,5, в то время как рН ци-
цитоплазмы бактериальных клеток приближается к 7,8. В этих ус-
условиях инсулин лишь слабо растворим. Многие гидрофобные эу-
кариотические белки не существуют в растворимой форме в от-
отсутствие фосфолипидов мембран. Белки молока являются интег-
интегральной частью мицелл, стабилизированных ионами Са2+.
Посттрансляционные модификации белков, в частности, фо-
сфорилирование, гидроксилирование, гликозилирование и огра-
ограниченный протеолиз, происходящие в определенных компарт-
ментах эукариотических клеток, также оказывают большое вли-
влияние на растворимость белков, их стабильность и биологические
функции. Это особенно относится к секретируемым белкам эука-
риот. Поскольку в бактериальных клетках соответствующие си-
системы посттрансляционных модификаций белков отсутствуют, в
них не могут быть получены биологически полноценные реком-
рекомбинантные эукариотические полипептиды, что накладывает ог-
ограничения на использование бактерий в биотехнологии.
Наконец, третьим ключевым фактором, оказывающим влия-
влияние на растворимость белков, является их нативная конформация
120
g растворе. Правильное сворачивание (фолдинг) полипептидных
кепей некоторых белков в клетках эукариот обеспечивается спе-
специфическими белками, называемыми шаперонами, которые не-
необходимы для эффективного формирования третичной структу-
структуры полипептидных цепей других белков, но не входят в состав ко-
конечной белковой структуры. Шапероны оказывают влияние лишь
1Ш конформацию полипептидных цепей и не действуют на кова-
яентные связи белковых молекул. К этому классу полипептидов
относятся цитозольные белки теплового шока Hsp70 и Hsp60, a
5»акже гомологичные им белки, например белок, связывающийся
е тяжелой цепью иммуноглобулинов (BiP). Шапероном является
также ядерный нуклеоплазмин, обеспечивающий сборку нуклео-
сом. У Е. сой соответствующие функции выполняют белки SecB,
триггерный фактор, а также GroEL и GroES, обеспечивающие
экспорт полипептидов из цитоплазмы и участвующие в морфоге-
морфогенезе бактериофагов. Два последних белка - GroEL и GroES, изве-
известные также как СрпбО и СрпЮ, кодируются генами оперона groE,
регулируемого тепловым шоком. Белки, эволюционно родствен-
родственные белку GroEL, получили название шаперонинов.
Из других белков, участвующих в сворачивании полипептид-
полипептидных цепей, следует упомянуть т.н. фолдазы и изомеразы. Наибо-
Наиболее хорошо изученными ферментами этой группы являются тио-
редоксин и глутаредоксин, которые наряду с глутатионзависи-
мым превращением рибонуклеотидов в дезоксирибонуклеотиды
способны восстанавливать дисульфидные связи в белках, способ-
способствуя формированию у них правильной третичной структуры. Те
же функции выполняет и дисульфидизомераза белков, осуществ-
осуществляя образование дисульфидных связей в секретируемых белках
после их переноса через мембраны эукариотических клеток. Ли-
Лимитирующей стадией в образовании правильной третичной
структуры полипептидных цепей является цис-транс-изомериза-
ция остатков пролина в полипептидных цепях. Этот процесс в
клетках эукариот осуществляется с участием пролш-цис-транс-
изомеразы. Поскольку все вышеперечисленные условия, необхо-
необходимые для поддержания эукариотических рекомбинантных бел-
белков в растворимой форме, не могут быть реализованы в бактери-
бактериальных клетках, в них происходит агрегация рекомбинантных
белков с образованием телец включения. В настоящее время не
существует универсального метода, который бы позволял полу-
получать рекомбинантные эукариотические белки в бактериальных
^летках в нативном виде. В некоторых случаях понижение тем-
температуры выращивания рекомбинантных бактерий до 30 °С при-
приводит к образованию полностью растворимых рекомбинантных
121
белков [147]. Это наблюдали в случае рекомбинантного у-интер-
ферона, А-цепи рицина, щелочного фактора роста фибробластов
и в ряде других случаев.
Более универсальным подходом к получению рекомбинант-
ных белков в растворимой форме является обеспечение секрети-
руемого характера их экспрессии, т.е. секреции синтезирующих-
синтезирующихся рекомбинантных белков в питательную среду [148]. Системы
секреции у грамположительных и грамотрицательных бактерий
интенсивно изучаются. Получение рекомбинантных эукариоти-
ческих белков в секретируемой форме высокотехнологично, по-
поскольку значительно облегчает их очистку. Кроме того, у секре-
тируемых белков больше шансов приобрести нативную конфор-
мацию, так как сворачивание полипептидных цепей происходит в
большом объеме, а следовательно, и более низкой их концентра-
концентрации, что предотвращает агрегацию полипептидных цепей, не
полностью сформировавших свою третичную структуру. Приме-
Примером эффективного использования секреторной системы Е. coli
является получение рекомбинантного инсулиноподобного фак-
фактора роста I (IGF-I) в секретируемой форме [149]. В этой системе
удавалось нарабатывать до 1 г рекомбинантного белка в 1 л бак-
бактериальной культуры. Секретируемая система экспрессии на ос-
основе грамотрицательной бактерии Pseudomonas aeruginosa будет
кратко рассмотрена ниже в разделе 5.1.1.
Еще одним возможным, хотя и более труднореализуемым
подходом к эффективному синтезу растворимых рекомбинант-
рекомбинантных белков в бактериальных клетках является введение экспрес-
сирующихся генов шаперонов в клетки бактерий-хозяев [150,
151]. Так, сверхэкспрессия гомологичного gro-оперона в клетках
Е. coli, содержащих экспрессирующиеся гены большой и малой
субъединиц Rubisco цианобактерии Anacistis nidulans, приводила
к 5-10-кратному возрастанию внутриклеточного содержания на-
тивного рекомбинантного белка Rubisco. В некоторых случаях
внутриклеточную агрегацию белков сверхэкспрессирующихся
генов удается предотвратить искусственной заменой отдельных
аминокислот в полипептидных цепях методами белковой инже-
инженерии. Например, удаление семи аминокислот перед спиралью G
в N-концевой части полипептидной цепи фрагмента Клёнова
ДНК-полимеразы IE. coli, а также замена нескольких гидрофоб-
гидрофобных аминокислот, боковые цепи которых экспонированы на по-
поверхности спирали G, на остатки аспарагина полностью предот-
предотвращали внутриклеточную агрегацию фрагмента и образование
агрегатов в процессе его очистки. В нативной молекуле ДНК-по-
ДНК-полимеразы I участок полипептидной цепи, в котором проведены
122
замены, закрыт доменом 5' —> З'-экзонуклеазы, отсутствующим
у фрагмента Кленова, и не оказывает влияния на агрегационные
свойства фермента.
Правильный выбор вектора и штамма-хозяина помогает ре-
решать вышеперечисленные проблемы, связанные с низкой эффек-
эффективностью экспрессии рекомбинантных белков в гетерологичных
системах. С помощью современных векторов можно повысить
растворимость и эффективность фолдинга рекомбинантных бел-
белков, по крайней мере, тремя путями. Во-первых, экспрессировать
рекомбинантный белок в составе гибридного полипептида, дру-
другой компонент которого сам по себе обладает высокой раствори-
растворимостью. Это относится, например, к глютатион-Б-трансферазе,
тиоредоксину, бактериальному белку NusA [152,153]. Во-вторых,
обеспечить слияние рекомбинантного белка с ферментом, участ-
участвующим в образовании дисульфидных связей, например, тиоре-
доксином, или белками DsbA и DsbC. В-третьих, снабдить реком-
рекомбинантный белок сигнальной последовательностью, которая бы
обеспечивала его экспорт в периплазматическое пространство.
Фолдинг рекомбинантных белков в цитоплазме может быть
улучшен при использовании для их экспрессии бактериальных
штаммов, содержащих мутации в генах тиоредоксинредуктазы
trxB и глютатионредуктазы gor [154]. Это связано с тем, что у
Е. coli образование дисульфидных связей в белках происходит
преимущественно во время их экспорта в периплазматическое
пространство, поскольку цитоплазма обладает высоким восста-
восстанавливающим потенциалом [155]. У двойных мутантов по этим
генам имеет место, по крайней мере, 10-кратное повышение
уровня образования нативных белков, для которых существен-
существенным является правильное формирование дисульфидных связей.
Многие вышеупомянутые векторы и бактериальные штаммы в
настоящее время можно приобрести у некоторых фирм, напри-
например, на фирме Novagene (США).
Следует отметить также, что образование нерастворимых те-
телец включения в бактериальных клетках в ряде случаев (напри-
(например, при крупномасштабной наработке инсулина человека) явля-
является преимуществом, а не недостатком, поскольку тельца могут
облегчать очистку рекомбинантных белков и защищать их от
нротеолитической деградации.
Несмотря на определенные успехи в получении нативных ре-
рекомбинантных эукариотических белков в бактериальных клет-
клетках, эти системы экспрессии эукариотических генов в настоящее
Бремя еще далеки от совершенства. По-прежнему остается от-
открытым вопрос о посттрансляционных модификациях рекомби-
123
нантных белков в бактериях, а для многих рекомбинантных бел-
белков не решена даже проблема их внутриклеточной растворимос-
растворимости. С этой точки зрения идеальными системами экспрессии ре-
рекомбинантных эукариотических генов являются гомологичные
генетические системы, т.е. сами эукариотические клетки живот-
животных или растений. Действительно, такие системы экспрессии, хо-
хотя и более дорогие, с успехом используются в биотехнологии.
При этом одним из ключевых факторов, обеспечивающих эф-
эффективное функционирование подобных систем, могут быть эу-
эукариотические экспрессирующие векторы, основные особеннос-
особенности которых будут рассмотрены ниже в разделе 3.7.
3.6.6. Очистка рекомбинантных белков с использованием
аффинных аминокислотных последовательностей
Быстрое, и даже бурное развитие протеомики в последние
несколько лет определяет интенсивное использование в исследо-
исследовательской работе рекомбинантных белков. Естественно, работа
с индивидуальными белками требует их эффективной очистки.
Как всегда, любая задача может быть решена разными способа-
способами. Использование для быстрого выделения индивидуальных
белков аффинной метки (affinity tag) в виде специфической ами-
аминокислотной последовательности, присоединенной к одному из
концов исследуемого полипептида, позволяет просто и эффек-
эффективно решать эту задачу [156]. Присоединение аффинных после-
последовательностей проводят методами генной инженерии, объеди-
объединяя в составе экспрессирующего вектора кодирующую часть ис-
исследуемого гена с последовательностью нуклеотидов того или
иного пептида или полипептида, обладающего избирательным
сродством к лиганду (табл. 5). В целом, системы очистки реком-
рекомбинантных белков, основанные на аффинных аминокислотных
последовательностях, обладают целым рядом уникальных
свойств. Так, они позволяют проводить выделение любого гиб-
гибридного белка в один этап путем его адсорбции на соответствую-
соответствующем носителе. Сама аффинная метка оказывает минимальное
влияние на третичную структуру основного белка и может быть
легко удалена из гибридного полипептида. Кроме того, она допу-
допускает проведение быстрой и точной количественной оценки со-
содержания белка в искусственных системах экспрессии рекомби-
рекомбинантных генов. Тем не менее использование каждой аффинной
метки требует соблюдения конкретных условий, которые не яв-
являются универсальными и могут оказывать сильное влияние на
биохимические свойства выделяемого белка.
124
Таблица 5
Свойства аффинных аминокислотных последовательностей,
используемых в современной генной инженерии [156]
Дффинная
последова-
последовательность
Поли(А^)
II(wra(His)
FLAG
3 х FLAG
Strep-tag II
c-myc
S-Tag
HAT (при-
(природная Не-
последова-
последовательность)
Кальмоду-
лин-связы-
вающий
Пептид
Сорбент
Катионо-
обменные
смолы
Ni2+-NTA,
Со2+-СМА
(Talon)
Монокло-
нальные
антитела
анти-FLAG
Модифици-
Модифицированный
стрептави-
дин Strep-
Tactin
Монокло-
нальные
антитела
S-фрагмент
РНКазы А
Со2+-СМА
(Talon)
Кальмоду-
лин
Условия
элюирования
Линейный
градиент
NaCl @-
400 мМ) при
щелочных
значениях
рН > 8,0
Имидазол,
20-250 мМ
или низкие
значения рН
рН 3,0 или 2-
5 мМ ЭДТА
2,5 мМ
дестиобио-
тин
Низкие
значения рН
Низкие
значения рН
Имидазол
150 мМ или
низкие зна-
значения рН
ЭГТА или
ЭГТАсШ
NaCl
Количе-
Количество
остатков
5-6
(обыч-
(обычно 5)
2-10
(обыч-
(обычно 6)
8
22
8
11
15
19
26
Моле-
куляр-
кулярная
масса,
кДа
0,80
0,84
1,01
2,73
1,06
1,20
1,75
2,31
2,96
Первичная
структура
RRRRR
НННННН
DYKDDDDK
DYKDHDGD-
YKDHD1DYK-
DDDDK
WSHPQFEK
EQKLISEEDL
KETAAAKFER-
QHMDS
KDHLIHNVH-
KEFHAHAHNK
KRRWKKNFI-
AVSAANRFK-
KISSSGAL
125
Таблица 5 (окончание)
Аффинная
последова-
последовательность
Сорбент
Условия
элюирования
Количе-
Количество
остатков
Моле-
куляр-
кулярная
масса,
кДа
Первичная
структура
Домен,
связываю-
связывающий
целлюлозу
SBP
Целлюлоза
Стрептави-
дин
Семейство I:
хлористый
гуанидин
или моче-
мочевина >4 М.
Семейство
11/Ш: эти-
ленгликоль
Биотин,
2мМ
27-189
38
3,0-
20,00
4,03
Домен,
связываю-
связывающий хитин
Глюта-
тион-S-
трансфе-
раза
Белок,
связываю-
связывающий
мальтозу
Хитин
Глютатион
Амилоза
с попереч-
поперечными
сшивками
Объедине-
Объединение с после-
дователь-
довательностью ин-
теина: ди-
тиотреитол,
30-50 мМ
Восстанов-
Восстановленный
глютатион,
5-10 мМ
Мальтоза,
10 мМ
51
211
396
5,59
Домены
MDEKTTGW-
RGGHVVEGL-
AGELEQLRA-
RLEHHPQGQ-
REP
TNPGVSAWQ-
VNTAYTAGQ-
LVTYNGKTY-
KCLQPHTSL-
AGWEPSNVP-
ALWQLQ
26,00 Полипептид
40,00 Полипептид
Все аффинные метки, составленные из аминокислотных по-
последовательностей, можно разделить на две группы. К первой
группе относятся небольшие пептиды, которые оказывают ми-
минимальное влияние на основной белок. Такие метки не иммуно-
генны, и во многих случаях не возникает необходимости их уда-
удаления из гибридных полипептидов. Влияние коротких последова-
последовательностей на свойства основных полипептидов зависит от пер-
первичной структуры метки и положения в полипептидной цепи ре-
комбинантных белков [157]. Вторая группа аффинных меток
включает большие пептиды или белки. Их использование спо-
способствует повышению растворимости основного белка, однако
126
часто возникает необходимость удаления метки перед дальней-
дальнейшей работой с рекомбинантными белками, например, при прове-
проведении кристаллографических исследований или получении анти-
антител. В целом, трудно a priori принять решение об использовании
конкретной метки, которое будет зависеть от свойств изучаемо-
изучаемого белка (его стабильности, гидрофобности и т.п.), а также пред-
предполагаемого дальнейшего применения. В табл. 5 приведены све-
сведения о наиболее часто используемых аффинных аминокислот-
аминокислотных последовательностях.
Полиаргининовая система. Полиаргининовые аффинные
метки обычно содержат 5-6 остатков Arg и добавляются к С-кон-
цам белков, экспрессирующихся в бактериальных клетках. В со-
сочетании с хроматографией на SP-сефадексе такие последова-
последовательности позволяют достигать 95% чистоты рекомбинантного
белка и 44% выхода [158]. Метка может быть удалена с помощью
карбоксипептидазы В. Система используется для иммобилизации
функциональных белков на плоских поверхностях с целью изуче-
изучения их взаимодействия с лигандами, а также на слюдяных под-
подложках, обычно применяемых при электронной сканирующей
микроскопии. В настоящее время применяется не очень часто.
Полигистидиновая система широко применяется для аффин-
аффинной хроматографии белков на сорбентах с иммобилизованными
ионами переходных металлов, включая Со2+, Ni2+, Cu2+, и Zn2+
(immobilized metal-affinity chromatography - IMAC). Функциональ-
Функциональные группы имидазольного кольца His образуют с ионами проч-
прочные координационные связи. На таких колонках ионы Ni2+ удер-
удерживаются с помощью хелатного агента, например, иммобилизо-
иммобилизованной нитрилотриуксусной кислоты (NTА), которая обладает
четырьмя валентностями, взаимодействующими с четырьмя из
шести валентностей ионов металла. Оставшиеся две свободные
валентности каждого иона взаимодействуют с биополимерами.
При этом прочность связи зависит от длины поли(Н18)-последо-
вательности в рекомбинантном белке. Элюирование связавших-
связавшихся белков можно осуществлять с помощью имидазола как в мяг-
мягких неденатурирующих условиях, так и в присутствии 6 М гуани-
Дина или 8 М мочевины.
Другим распространенным носителем, разработанным для
очистки белков, обладающих полигистидиновой меткой, являет-
является Talon, в состав которого входит Со2+-карбоксиметиласпартат
(Со2+-СМА), иммобилизованный на твердом носителе [159]. Talon
Позволяет выделять белки в более мягких условиях, чем те, кото-
которые допускает Ni2+-NTA и для этой хроматографической систе-
, по-видимому, характерно более низкое неспецифическое
127
связывание белков. Ее использование позволило разработать но-
новую аффинную метку на основе 19-звенной природной поли(Н1з)-
последовательности (HAT), которая позволяет производить очи-
очистку рекомбинантных белков в мягких условиях. В целом, систе-
система с полигистидиновой меткой позволяет производить очистку
рекомбинантных белков из всех основных систем экспрессии,
включая бактерии, клетки дрожжей, млекопитающих и насеко-
насекомых. Благодаря небольшому размеру и низкому электрическому
заряду метка оказывает незначительное влияние на активность
рекомбинантного белка. В том случае, если это все-таки проис-
происходит, перенос аффинной последовательности с одного конца ре-
рекомбинантного полипептида на другой, как правило, решает про-
проблему [160]. Использование системы не рекомендуется для очи-
очистки ферментов, содержащих ионы металлов в активном центре
из за их возможного взаимодействия с NTA, а также в анаэроб-
анаэробных условиях, которые уменьшают эффективность взаимодейст-
взаимодействия аффинной метки с №2+-1ЧТА-носителями.
Система FLAG. В этой системе используется короткая
8-звенная аминокислотная последовательность. Пептид FLAG
специфически взаимодействует с антителами подклассов Ml, M2
и М5. При этом остается непонятным является ли это взаимодей-
взаимодействие Са2+-зависимым [161, 162]. Условия очистки белков в такой
системе неденатурирующие, однако их взаимодействие с носите-
носителями на основе моноклональных антител менее прочное, чем в
вышеописанных системах с ионами металлов. Система на основе
трехкратного повтора последовательности FLAG C х FLAG)
позволяет обнаруживать до 10 фмолей синтезированного ре-
рекомбинантного белка. Последовательность FLAG может быть
удалена энтерокиназой, которая специфична в отношении
5-звенной С-концевой аминокислотной последовательности
(AspAspAspAspLysi) аффинной метки.
Система Strept-tag. Данная аффинная метка представляет со-
собой семейство искусственных пептидов, разработанных для очист-
очистки белков на колонках с иммобилизованным стрептавидином (о
стрептавидине подробнее см. в разделе 1.6.3.11) [163]. Мутантная
форма стрептавидина, названная стрептактином (Strep-Tactin), с
заменами аминокислотных остатков в положениях 44, 45 и 47 об-
обладает большим сродством к пептиду Srept II, чем к исходному (по-
(последовательность см. в табл. 5). Рекомбинантные белки, несущие
эту аффинную метку, связываются в мягких условиях с иммобили-
иммобилизованным биотином и могут быть элюированы производными би-
биотина, например, дестиобиотином. Биотинилированные белки
также могут связываться со стрептактином, однако такое взаимо-
128
действие можно заблокировать авидином. Такую систему реко-
рекомендуется использовать для очистки рекомбинантных белков в
анаэробных условиях, а также для ферментов, содержащих в сво-
своем составе ионы металлов. При этом можно проводить совмест-
совместную очистку субъединиц мембранных белков, не содержащих аф-
аффинной метки. Данная аффинная метка, кроме того, находит при-
применение в эукариотическом клеточном дисплее (см. раз-
раздел П.2.3.2), и в последнее время становится все более популярной,
в том числе, в исследованиях белок-белковых взаимодействий
[164] и в приложениях с использованием ЯМР-спектроскопии.
Системы на основе c-myc-последовательностей. Мышиные
моноклональные антитела 9Е10 к белку с-тус широко использу-
используются в качестве иммунохимического реагента в клеточной биоло-
биологии и белковой инженерии [165]. Эпитоп, распознаваемый антите-
антителами, который представляет собой последовательность из 11 ами-
аминокислотных остатков, может быть экспрессирован в составе раз-
различных белков и остается распознаваемым антителами. Эта аф-
аффинная метка используется в Западном блоттинге, для иммуно-
преципитации белков, в проточной цитометрии, а также для мони-
мониторинга экспрессии генов на уровне трансляции в бактериальных
и эукариотических системах, в том числе, и для быстрой очистки
рекомбинантных белков, которые могут быть закристаллизованы
[166, 167]. Система часто используется для обнаружения рекомби-
рекомбинантных белков, но редко - для их выделения в чистом виде.
Система S-Tag. Работа системы основана на взаимодействии
15-звенного S-пептида рибонуклеазы А со 103-звенной последо-
последовательностью того же фермента, которые образуют специфиче-
специфический очень прочный комплекс (Kd~ 10"9 М) [168]. Последователь-
Последовательность S-Tag присоединяют генно-инженерными методами к
N-концевой части рекомбинантного белка в составе экспрессиру-
ющего вектора и по завершении экспрессии гибридный белок
выделяют с помощью аффинной хроматографии на колонке с
протеин S-агарозой. Элюирование целевого белка можно осуще-
осуществлять с помощью энтерокиназы, распознаваемую последова-
последовательность которой вводят в виде линкера между целевым реком-
бинантным белком и последовательностью S-Tag. Для тех же це-
целей может быть использован и тромбин в сочетании с расщепля-
расщепляемой этой протеиназой аминокислотной последовательностью. В
Данном случае с колонки освобождается нативный рекомбинант-
ный белок с правильной N-концевой последовательностью. Аль-
Альтернативно, элюирование гибридного белка с колонки можно
проводить в денатурирующих условиях (ЗМ гуанидинтиоциана-
том, 0,2 М цитратом, рН 2, или 3 М MgCl2). В этом случае маркер-
5. Патрушев Л.И. Т. 1 129
ная последовательность в составе гибридного белка сохраняется,
а колонка автоматически регенерируется. Если же рекомбинант-
ный белок образует в бактериальных клетках тельца включения,
его солюбилизируют 6 М мочевиной, наносят на колонку в 2 М
мочевине, а элюирование производят либо энтерокиназой непо-
непосредственно в мочевине, либо в вышеупомянутых денатурирую-
денатурирующих условиях. Один мл аффинного сорбента может связывать до
500 мкг рекомбинантного белка.
Ни пептид S-Tag, ни 103-звенный полипептид, используемый
в качестве аффинного сорбента, не обладают ферментативной
активностью, однако после образования комплекса в последнем
индуцируется рибонуклеазная активность. Это позволяет коли-
количественно оценивать эффективность исследуемых систем экс-
экспрессии. Однако еще большей чувствительностью и удобством в
применении обладают системы, в которых S-белок ковалентно
связан с флуоресцентными красителями или ферментами (напри-
(например, с классической пероксидазой хрена). Такую систему можно
эффективно использовать для идентификации рекомбинантного
белка с помощью вестерн-блоттинга и ее часто применяют одно-
одновременно со второй аффинной меткой.
Кальмодулин-связывающий пептид. Очистка гибридных бел-
белков, содержащих кальмодулин-связывающий пептид, была впер-
впервые описана в 1992 г. [169]. В ней используется 26-звенный пептид,
заключенный в С-концевой последовательности киназы легкой
цепи миозина скелетных мышц. Кальмодулин в присутствии ионов
Са2+ обладает высоким сродством к этому пептиду. Прочность
комплекса допускает промывку колонки при аффинной хромато-
хроматографии в жестких условиях, что позволяет достигать высокой сте-
степени очистки целевого полипептида. Система особенно пригодна
для очистки рекомбинантных белков из клеток Е. coli, поскольку
их собственные белки не взаимодействуют с кальмодулином. При
этом хроматографию можно проводить в присутствии восстанав-
восстанавливающих агентов и детергентов в концентрациях до 0,1%. Эта
аффинная последовательность позволяет вводить радиоактивную
метку с помощью у-[32Р] и протеинкиназы А, что используется для
исследования белок-белковых взаимодействий [170]. Данную аф-
аффинную последовательность предпочтительнее вводить в С-конец
исследуемого полипептида, поскольку его N-концевая локализа-
локализация часто приводит к ингибированию трансляции.
Домены, связывающие целлюлозу. Известно более 13 се-
семейств белков, обладающих доменами, связывающими целлюло-
целлюлозу (cellulose-binding domains - CBDs), размеры которых варьиру-
варьируют в пределах 4-20 кДа. Некоторые CBD необратимо взаимодей-
130
ствуют с целлюлозой и могут быть использованы для иммобили-
иммобилизации ферментов на соответствующем носителе [171]. Связь дру-
других CBD с целлюлозой менее прочная, и они находят применение
для очистки и разделения гибридных белков, обладающих такой
аффинной меткой. В частности, CBD класса I, обратимо связыва-
связывающиеся с кристаллической целлюлозой, особенно пригодны для
аффинной хроматографии [172]. Преимущества целлюлозы пе-
перед другими носителями заключаются в ее инертности, низком
неспецифическом взаимодействии с биологическими макромоле-
макромолекулами, доступности в виде разнообразных форм и приложимос-
приложимости к созданию фармакологических препаратов. Обычно связь
CBD с целлюлозой настолько прочная, что гибридный белок
можно отделить от носителя только в присутствии денатурирую-
денатурирующих агентов, что делает необходимым проведение рефординга
элюированных белков для восстановления их биологической ак-
активности. В отличие от этого, рекомбинантные белки, содержа-
содержащие CDB классов II и III, элюируются в мягких условиях этилен-
гликолем, который, в свою очередь, может быть удален с помо-
помощью диализа [173]. Эффективная экспрессия гибридных белков,
меченных CDB-доменами, происходит в прокариотических и эу-
кариотических системах всех типов.
Стрептавидин-связывающий пептид SBP. Недавно получен-
полученный стрептавидин-связывающий пептид (streptavidin-binding pep-
tide - SBP) образует прочный комплекс со стрептавидином с
Kd ~2,5 нМ [174]. Рекомбинантные белки, содержащие такую
С-концевую метку, эффективно экспрессируются в бактериаль-
бактериальных клетках и могут быть очищены с помощью аффинной хро-
хроматографии из бесклеточных экстрактов [175].
Хитин-связывающий домен. Аффинная метка на основе хи-
тин-связывающего домена Bacillus circulans [175] широко исполь-
используется в сочетании с интеинами, что позволяет освобождать иммо-
иммобилизованный на хитиновом носителе рекомбинантный белок в
результате белкового аутосплайсинга (подробнее об интеинах см.
раздел П. 1.3.2, а о применении хитиновых доменов в разделе
Н.3.2.1). Рекомбинантные белки, содержащие N-концевые или
С-концевые хитиновые домены, ассоциированные с интеинами,
эффективно синтезируются в бактериальных клетках [176, 177].
Глютатион-Э-трансфераза в качестве аффинной метки. Ги-
Гибридные белки, содержащие в качестве аффинной метки после-
последовательность глютатион-Б-трансферазы (GST), могут быть
очищены хроматографией на колонках с иммобилизованным
глютатионом [178]. Гибридные белки в большинстве случаев
растворимы в водных растворах, существуют в них в виде диме-
5* 131
ров и легко обнаруживаются по активности GST или иммуноло-
иммунологическими методами. Последовательность GST обычно удаля-
удаляют из гибридного полипептида с помощью специфических про-
теиназ: тромбина или фактора Ха. Гибридные белки на основе
GST стали одним из основных инструментов молекулярной
биологии. Они используются для изучения белок-белковых и
белково-нуклеиновых взаимодействий [179, 180].
Белок, связывающий мальтозу (maltose-binding protein -
МВР) с молекулярной массой 40 кДа, кодируется геном malE
Е. coli К12. Гибридные белки, содержащие в своем составе МВР,
могут быть очищены в один этап с помощью аффинной хрома-
хроматографии на амилозе, стабилизированной поперечными сшив-
сшивками [181]. Kd этого комплекса находится в микромолярном ди-
диапазоне. Объединение МВР с основным (особенно эукариотиче-
ским) рекомбинантным белком в одной полипептидной цепи по-
повышает растворимость последнего, что в ряде случаев предот-
предотвращает его агрегацию при сверхэкспрессии в бактериальных
клетках. Система МВР широко используется для очистки ре-
комбинантных белков [182].
В целом, введение аффинных меток в виде обсуждавшихся
выше пептидных и полипептидных последовательностей сущест-
существенно облегчает очистку меченых таким образом рекомбинант-
ных белков, а также предоставляет дополнительные возможнос-
возможности наблюдать за эффективностью экспрессии рекомбинантных
генов. Выбор для этих целей системы очистки зависит от кон-
конкретного белка, который предстоит получать в очищенном со-
состоянии, а также от целей его дальнейшего использования. В ря-
ряде современных систем в рекомбинантные белки вводится двой-
двойная аффинная метка, одна из которых служит для очистки белка,
а другая - для мониторинга за его биосинтезом. Также с помо-
помощью двойной метки может быть достигнута большая степень
очистки рекомбинантных белков. Разработаны множественные
аффинные аминокислотные последовательности, которые вклю-
включают в себя кальмодулин-связывающий пептид, специфический
сайт расщепления протеиназой и белок А для иммобилизации
всей рекомбинантной конструкции. С помощью этих систем уда-
удается изучать белок-белковые взаимодействия путем выделения
соответствующих белковых комплексов при исследованиях про-
теома [183, 184]. Методы иммобилизации белков с помощью аф-
аффинных последовательностей используются при создании пеп-
пептидных и белковых микрочипов, клонотек на их основе, для ад-
адресной доставки белков и во многих других приложениях. О не-
некоторых из них будет дополнительно рассказано в разделе П.3.2.
132
3.7. Векторы для переноса ДНК в клетки животных и растений
Все основные принципы, используемые при конструировании
бактериальных векторов, применимы и для получения векторов
эукариотических клеток. Как и в случае бактерий, эукариотиче-
ский вектор представляет собой небольшую молекулу ДНК, спо-
способную автономно реплицироваться в клетках животных или
растений. Помимо последовательностей нуклеотидов, обеспечи-
обеспечивающих репликацию, эукариотические векторы могут содер-
содержать гены, используемые в качестве селектируемых маркеров, а
также один или несколько уникальных сайтов рестрикции, по ко-
которым производится встраивание клонируемых последователь-
последовательностей нуклеотидов ДНК. Поскольку непосредственное клони-
клонирование рекомбинантных ДНК в клетках животных или расте-
растений было бы дорогостоящей и малоэффективной процедурой,
эукариотические векторы используют, как правило, для получе-
получения экспрессии уже клонированных последовательностей нукле-
нуклеотидов в клетках высших эукариот, а сам процесс клонирования
проводят в бактериях. Следовательно, эукариотические векто-
векторы, помимо всего прочего, должны быть челночными вектора-
векторами. Для экспрессии в клетках рекомбинантные ДНК помещают
под контроль регуляторных элементов, узнаваемых и используе-
используемых ферментативными системами эукариотических клеток.
В отличие от бактерий в клетках эукариот внехромосомные,
автономно реплицирующиеся генетические элементы типа бак-
бактериальных плазмид встречаются редко. Поэтому при конструи-
конструировании векторов, способных реплицироваться и осуществлять
экспрессию в клетках животных, чаще всего используют регуля-
торные генетические элементы вирусов животных или, в зависи-
зависимости от задач, эукариотических генов домашнего хозяйства, а
также генов, для которых характерна тканеспецифическая экс-
экспрессия.
3.7.1. Экспрессирующий вектор pTriEx
Структура двух современных векторов, предназначенных для
обеспечения экспрессии клонированных генов в клетках живот-
животных, представлена на рис. 14 (Novagen, США).
Вектор pTriEx™ "широкого спектра действия" обеспечивает
кратковременную экспрессию рекомбинантных генов в большом
числе клеток позвоночных животных. С использованием поли-
полилинкера экспрессируемый ген помещают под контроль промото-
Ров актинового гена цыплят или промотора цитомегаловируса
(CMV), эффективное функционирование которых обеспечивает-
133
а
Промотор CMV
Энхансер CMV Промотор (J-активного
гена цыплят
Терминатор транскрипции
Р-глобинового гена кроликов
Т7-терминатор
Энхансер CMV
Промотор й-активного гена циплят
Экзон 1
Интрон
Т7-терминатор
Терминатор транскрипции
Р-глобинового гена кроликов
Рис. 14. Представители векторов, обеспечивающих экспрессию реком-
бинантных генов в клетках животных
а - вектор рТпЕх™ широкого спектра действия, обеспечивающий кратко-
кратковременную экспрессию генов; б - вектор того же семейства (рТпЕх), использу-
используемый для стабильной экспрессии рекомбинантных генов (Novagen, США)
ся энхансером CMV (рис. 14, а). Вектор содержит сайт полиаде-
нилирования C-глобинового гена кроликов. В РНК, синтезируе-
синтезируемую с этих промоторов, включается последовательность интро-
на, что необходимо для правильного процессинга и экспорта
РНК. Вектор обладает также промотором Т71ас (гибридный про-
промотор, содержащий последовательность /ас-оператора, что обес-
обеспечивает дополнительный контроль транскрипции) терминато-
терминатором Т7-РНК-полимеразы для осуществления при необходимости
транскрипции клонированного гена в бактериальных клетках и
промотором рЮ бакуловирусов, который обеспечивает высокий
уровень экспрессии гена в клетках насекомых, зараженных баку-
ловирусами (см. раздел 6.2.5). Сигналы инициации трансляции
вектора включают сайт связывания бактериальных рибосом,
вслед за которым расположена консенсусная последователь-
последовательность, обеспечивающая инициацию трансляции в клетках млеко-
млекопитающих. Следует отметить наличие в обоих концах полилин-
полилинкера сайтов для рестриктаз, расщепляющих ДНК с образованием
тупых концов, которые позволяют при необходимости клониро-
клонировать в любой из трех возможных рамок считывания ген устойчи-
устойчивости к ампициллину в качестве селектируемого маркера, об-
области начала репликации плазмид семейства pUC, а также мар-
маркерную последовательность, кодирующую пептид His6_10 для очи-
134
стки рекомбинантного белка на никелевой колонке. Кроме того,
по этим сайтам можно вводить маркерную последовательность
(например, кодирующую стандартно используемую в качестве
метки последовательность GlnProGluLeuAlaProGluAspProGluAsp
вируса простого герпеса), обеспечивающую обнаружение реком-
рекомбинантного белка с помощью моноклональных антител, а также
последовательности, кодирующие сайты действия протеолитиче-
ских ферментов для удаления вышеупомянутых маркерных по-
последовательностей из рекомбинантных белков.
Для получения стабильной экспрессии рекомбинантных ге-
генов в клетках животных можно использовать другую разновид-
разновидность вектора pTriEx (рис. 14, б). Принцип функционирования та-
такого вектора заключается в том, что рекомбинантный белок экс-
прессируется в виде гибридной молекулы, С-концевая часть ко-
которой содержит маркерный полипептид (неомицин- или гигроми-
цин В-фосфотрансферазу), обеспечивающий клеткам устойчи-
устойчивость к антибиотикам неомицину или гигромицину, соответст-
соответственно. В таких системах клетки, устойчивые к антибиотику, га-
гарантированно экспрессируют и рекомбинантный белок, что лег-
легко контролируется путем выращивания клеток на соответствую-
соответствующих селективных питательных средах. Трансляцию гибридной
мРНК обеспечивает кэп-независимый энхансер трансляции CITE
(cap-independent translation enhancer) вируса энцефаломиокардита
(EMCV) или внутренний сайт связывания рибосом IRES (internal
ribosome entry site).
3.7.2. Векторы растений
Необходимость и последствия использования векторов рас-
растений. Разработка методов введения новых или измененных ге-
генов в геном растений [185] позволяет получать трансгенные ге-
генетически модифицированные (genetic modified - GM) растения,
эффективно экспрессирующие эти гены, что привело к зеленой
революции в сельском хозяйстве, которая и сегодня далека от
своего завершения. Значительная часть сельскохозяйственных
угодий, особенно в США, занята генетически измененными рас-
растениями, в частности, хлопком, кукурузой и соей. Для большин-
большинства генетически модифицированных растений характерна высо-
высокая урожайность, что достигается, в том числе, приданием расте-
растениям устойчивости к насекомым и гербицидам. Введение транс-
трансгенов позволяет повышать качественные характеристики транс-
трансгенных растений. Например, методами генной инженерии удает-
удается изменять форму и расцветку цветов, а также увеличивать про-
135
должительность сохранения ими товарного вида в срезанном со-
состоянии. Созданы сорта картофеля со сбалансированным содер-
содержанием крахмала в клубнях, а также сорта риса, содержащие в
зернах провитамин А, что позволяет избегать авитаминоза при
преимущественном использовании риса для питания. На основе
трансгенных растений получены эффективные вакцины, реали-
реализующие свое действие после использования таких растений в пи-
пищу. Воплощение в жизнь масштабных проектов, связанных с ге-
генетически модифицированными растениями, потребовала разра-
разработки новых векторных систем, а также методов введения векто-
векторов в клетки растений [186].
Возможность получения трансгенных растений основана на
тотипотентности их некоторых клеток, т.е способности в оп-
определенных условиях под действием фитогормонов дифференци-
дифференцироваться в разных направлениях с образованием всех тканей ор-
организма растения и самого полноценного растения. Для создания
трансгенного растения необходимо трансформировать культиви-
культивируемые клетки, их протопласты или клетки в составе органов и
тканей подходящим вектором, содержащим требуемый клониро-
клонированный ген, отделить от ^трансформированных и получить из
них целые трансгенные растения.
Селектируемые маркеры и гены-репортеры, используемые
при трансформации клеток растений. Клетки растений чувстви-
чувствительны к некоторым антибиотикам, особенно тем из них, кото-
которые подавляют биосинтез белка в хлоропластах, в том числе ка-
намицину и гигромицину. Поэтому в состав векторов вводят
бактериальные гены устойчивости к таким антибиотикам под
контролем генетических регуляторных элементов растений
(табл. 6). В качестве других селектируемых маркеров в векто-
векторах растений используют гены устойчивости к гербицидам. Не-
Недавно в качестве селектируемого маркера стали применять ген
маннозо-6-фосфат изомеразы (pmi): экспрессирующие его клет-
клетки приобретают способность использовать маннозу в качестве
единственного источника углерода, а следовательно, расти на
среде с этим углеводом. Современные технологии позволяют
вести наблюдение за экспрессией трансгенов в растениях и с по-
помощью визуальных методов. В частности, экспрессия гена
C-глюкуронидазы (C-glucuronidase - GUS) придает трансформи-
трансформированным клеткам способность расщеплять 5-бром-4-хлор-3-
индолил-C-О-глюкуроновую кислоту (X-Gluc) и 4-метилумбел-
лиферил-C-О-глюкуроновую кислоту (MUG) [187]. Специфиче-
Специфическое расщепление X-Gluc сопровождается освобождением со-
соединения, которое под действием кислорода воздуха приобрета-
136
Таблица 6
Маркерные гены и гены-репортеры, используемые
в современной генной инженерии микроводорослей [189]
Ген
Белковый продукт, функция
Источник гена
Применение
AphVIII Аминогликозид-З'-фосфо-
трансфераза (устойчивость
к паромомицину, канами-
цину и неомицину)
Ble Белок устойчивости к блео-
мицину (устойчивость
к таллизомицину и род-
родственным антибиотикам)
Nptll Неомицинфосфотрансфера
за II (устойчивость к G418)
Als Ацетолактатсинтаза
(устойчивость к гербици-
гербицидам, производным сульфа-
нилмочевины)
Cryl-1 Рибосомный белок S14
Оее-1 Белок-энхансер выделения
кислорода (oxygen evolving
enhancer protein)
Cat Хлорамфениколацетил-
трансфераза (устойчивость
к хлорамфениколу)
Hpt Гигромицин В-фосфотранс-
фераза
AadA Аденилилтрансфераза
(устойчивость к спектино-
мицину)
Nat Устойчивость к ноурсео-
трицину
Sat-1 To же
EGFP Модифицированный зеле-
зеленый флуоресцирующий
белок
ChGFP To же
Streptomyces M
rimosus
Streptoalloteichus M
hindustanus
Транспозон Tn5 M
Е. coli
Chlamydomonas M
То же
Тоже
Транспозон Тп9
Е. coli
Эубактерии
Streptomyces
noursei
Е. coli
Искусственный
(оптимизирован-
(оптимизированный по использо-
использованию кодонов)
Тоже
М
М
М
М
М
М
М
Р
Р
13/
Таблица 6 (окончание)
Ген
Gus
Glutl
Hupl
e-frustulin
Ars
Luc
Примечание
Белковый продукт, функция
Р-Глюкуронидаза
Переносчик глюкозы
Переносчик гексоз
Кальций-связывающий
гликопротеин
Арилсульфатаза
Люцифераза
Источник гена
Е. coli
Человек
Chlorella kessler
Navicula
pelliculosa
Chlamydomonas
Horatia parvula
. М - маркерный ген, Р - ген-репортер.
Применение
P
МилиР
M илиР
P
P
P
ет голубую окраску, а ткани, экспрессирующие трансген, при-
приобретают соответствующий цвет. В процессе метаболизма
MUG с участием гена-репортера освобождается флуоресцирую-
флуоресцирующее соединение. При этом количественное определение флуо-
флуоресценции позволяет охарактеризовать уровень экспрессии ге-
гена gus, а следовательно, и само трансгенное растение с точки
зрения экспрессируемости трансгена. В последнее время все
большую популярность приобретает использование в качестве
репортера гена зеленого флуоресцирующего белка (GFP) [188].
Дополнительные сведения о маркерных генах и генах-репорте-
генах-репортерах, применяемых в современной генной инженерии растений,
можно найти в табл. 6.
Два типа векторных систем, используемых для трансформа-
трансформации клеток растений. На основании способа доставки трансгенов
в клетки различают два типа векторных систем. В одной из них
осуществляют так называемое прямое введение трансгена в
клетки или протопласты, лишенные клеточной стенки, одно-
однодольных и двудольных растений с использованием группы стан-
стандартных методов, включающих Са2+-зависимую трансформацию
в присутствии полиэтиленгликоля, электропорацию, бомбарди-
бомбардировку клеток микрочастицами, нагруженными ДНК, и т.п. (см.
разделы 3.8.3. и 3.8.4.). Эта группа подходов к трансформации
клеток растений известна также под названием "перенос генов,
опосредованный ДНК" (DNA-mediated gene transfer - DMGT). Си-
Системы второго типа основаны на применении ферментативного
аппарата доставки ДНК бактерий Agrobacterium tumefaciens или
A. rhizogenes, которые обладают способностью трансформиро-
трансформировать клетки растений в природных условиях.
Векторы для прямой трансформации клеток. Специфика
векторов этого типа обусловлена, главным образом, регуля-
138
торными последовательностями нуклеотидов, которые обес-
обеспечивают эффективную транскрипцию генов в растительных
тканях. Например, в известном векторе pCaMVCAT использу-
используются промотор и сайт полиаденилирования гена 55 РНК виру-
вируса мозаики цветной капусты, под контролем которых находит-
находится ген бактериальной хлорамфениколацетилтрансферазы {cat)
[190]. Этот ген-репортер применяется для оптимизации усло-
условий введения векторной ДНК в клетки растений (поскольку
имеются очень чувствительные методы обнаружения активно-
активности этого фермента) и может быть заменен на любой другой
ген. Вектор pCaMVCAT является челночным, т.е. может реп-
реплицироваться как в клетках Е. coli, так и растений. В первом
случае селектируемым маркером служит, как обычно, ген ус-
устойчивости к ампициллину. Векторы этого типа с нерегулиру-
нерегулируемым промотором используются с целью получения времен-
временной экспрессии рекомбинантных генов и проведения фунда-
фундаментальных исследований функциональной организации гено-
генома растений. При конструировании векторов для постоянной
экспрессии генов в клетках растений ген cat обычно заменяют
на доминантный селектируемый маркер, например, ген устой-
устойчивости к канамицину [189].
Векторы на основе Ti-плазмид. Бактерию Agrobacterium
tumefaciens иногда называют природным генным инженером за
ее способность переносить свою ДНК в клетки зараженных
растений, интегрировать ее в геном организма-хозяина и вызы-
вызывать стабильную трансформацию этих клеток введенными ге-
генами, т.е. индуцировать рост опухолей. Вирулентные штаммы
агробактерий содержат Ti-плазмиды, размер которых составля-
составляет 200-250 т.п.о. Во время инфекции определенная последова-
последовательность Ti-плазмиды, получившая название Т-ДНК (trans-
(transferred), переносится в клетки и случайным образом интегриру-
интегрируется в геном и ее гены экспрессируются в зараженных клетках.
Т-ДНК содержит гены, обеспечивающие биосинтез фитогормо-
нов ауксина и цитокинина, которые, в свою очередь, приводят к
независимой от гормонов пролиферации клеток и росту опухо-
опухолей. Т-Область плазмиды не содержит генов, которые обеспе-
обеспечивают их мобилизацию, а соответствующие функции выпол-
выполняют ~20 генов Vir-области плазмиды, организованных в опе-
рон. Экспрессия генов vir индуцируется в условиях, имитирую-
имитирующих таковые сока растений, т.е. при низких значениях рН и в
присутствии специфических фенольных соединений типа пред-
предшественника лигнина и продуктов его деградации. Индукция
опосредована рецептором этих соединений VirA и активатором
139
транскрипции VirG. Белок VirA в комплексе с лигандом фосфо-
рилирует VirG, который после этого приобретает способность
взаимодействовать с регуляторной последовательностью vir-
оперона и активировать его транскрипцию.
Активация v/r-генов приводит к образованию одноцепочеч-
ной копии Т-области Ti-плазмиды. Этот процесс происходит при
участии белков VirDj и VirD2, которые вносят одноцепочечные
разрывы по обеим сторонам Т-области, фланкированной 24-нук-
леотидными повторами. Белок VirD2 остается ковалентно свя-
связанным с цепью Т-ДНК, и этот нуклеопротеиновый Т-комплекс
переносится в клетки [191,192]. Транспорт обеспечивают 11 virB-
генов, продукты которых формируют структуры, напоминаю-
напоминающие половые ворсинки Е. coli. Кроме того, белок VirE2 покрыва-
покрывает оболочкой переносимую одноцепочечную ДНК, предохраняя
ее от деградации и также, как и VirD2, содержит сигнал ядерной
локализации (NLS), который обеспечивает перенос Т-комплекса
из цитоплазмы в ядро.
Введение генов непосредственно с помощью Ti-плазмид не
используется, поскольку приводит к образованию опухолевых
клеток, из которых невозможно получить целое растение. Для
этих целей применяют векторные молекулы на основе Ti-плаз-
Ti-плазмид с удаленными онкогенами. Вместо онкогенов в вектор мож-
можно встраивать стандартными методами по сайтам рестрикции по-
последовательности клонируемой ДНК [193, 194]. В таких челноч-
челночных векторных плазмидах сайты рестрикции, по которым произ-
производят клонирование, расположены в искусственной Т-области,
фланкированной вышеупомянутыми повторами. В качестве се-
селектируемых маркеров в этих плазмидах используют располо-
расположенные в Т-области гены устойчивости к антибиотикам или гер-
гербицидам, которые позволяют отбирать трансформированные
клетки растений. После введения клонируемой последовательно-
последовательности рекомбинантным вектором трансформируют клетки агро-
бактерий и с их помощью при участии v/r-генов осуществляют
перенос всей генно-инженерной конструкции в клетки растений.
Гены вирулентности могут находиться как в составе самого век-
вектора, так и в Гга/15-положении на плазмиде-помощнике. Для полу-
получения генетически модифицированных растений суспензию агро-
бактерий можно инкубировать с протопластами, интактными
клетками, тканями, листьями или даже целыми растениями. Во
всех случаях достигается высокоэффективный перенос рекомби-
нантной ДНК в геном трансформируемых клеток. Интеграция
рекомбинантной ДНК происходит случайным образом с неболь-
небольшим предпочтением участков активно транскрибируемых генов.
140
Для введения больших фрагментов ДНК в клетки растений в по-
последнее время были получены соответствующие космидные век-
векторы [195], а также векторы на основе искусственных бактери-
бактериальных хромосом (ВАС), получившие название ТАС-векторов
(transformation-competent bacterial artificial chromosomes) [196].
ТАС-векторы могут быть реплицированы как в клетках Е. coli,
так и агробактерий, что позволяет с помощью этих микроорга-
микроорганизмов вводить в клетки растений фрагменты ДНК длиной более
150 т.п.о.
Векторы для переноса рекомбинантных генов в хлоропласты
высших растений. Получение трансгенных растений, содержа-
содержащих генетически измененные хлоропласты, является одним из
быстро развивающихся направлений современной генной инже-
инженерии растений [197]. Интерес к этой области исследований оп-
определяется большей безопасностью таких трансгенных растений
в отношении загрязнения окружающей среды. Поскольку хлоро-
хлоропласты не переносятся с пыльцой, значительно уменьшается
опасность распространения трансгенов среди перекрестно опы-
опыляемых растений близких видов. Кроме того, сама пыльца транс-
трансгенных растений менее токсична для насекомых, которые не яв-
являются мишенью ее токсического воздействия. Для генома хло-
хлоропластов (пластома) характерен высокий уровень полиплоидии
A03-105 копий на клетку), поэтому отдельная клетка с трансфор-
трансформированными хлоропластами содержит тысячи копий трансгена,
что позволяет получать очень высокий уровень экспрессии соот-
соответствующих рекомбинантных белков (до 25% от суммарного
растворимого клеточного белка).
Векторы для трансформации хлоропластов представляют со-
собой кольцевые молекулы ДНК, содержащие маркерный ген для
отбора трансформантов, а также два участка длиной 1-2 т.п.о.
каждый, гомологичные последовательностям пластома, в кото-
которые происходит интеграция вектора с помощью гомологичной
рекомбинации [198]. В геноме хлоропластов описано, по крайней
мере, 16 сайтов, по которым удалось получить продуктивную ин-
интеграцию вектора. Гомологичные последовательности, получив-
получившие название LTR- (left) и RTR-последовательностей (right target-
targeting regions), фланкируют полилинкер, по которому производят
клонирование исследуемых фрагментов ДНК. В зависимости от
размера пластома данная векторная система позволяет вводить в
геном хлоропластов фрагменты ДНК длиной до 50 т.п.о., содер-
содержащие от 20 до 30 генов.
Для получения эффективной экспрессии клонированных
генов их вводят между двумя кассетными последовательностя-
141
ми вектора. 5'-Концевая PL-кассета (promoter and leader) содер-
содержит промотор и регуляторные последовательности, обеспечи-
обеспечивающие трансляцию синтезируемой с этого промотора мРНК,
которые могут быть либо 5'-концевой нетранслируемой обла-
областью E' UTR), либо 5'-концевым участком, контролирующим
трансляцию E' TCR), включающим, в свою очередь, 5' UTR и
участок, кодирующий N-концевую часть лидерного пептида.
Т-кассета содержит З-иТК-последовательность, которая стаби-
стабилизирует мРНК, что является одним из условий достижения
высокого уровня экспрессии трансгена в хлоропластах. Для по-
получения эффективной транскрипции клонированного гена час-
часто используют сильный Prrn-промотор оперона рРНК (rrri). В
современных векторах присутствуют системы последователь-
последовательностей, позволяющие убирать из стабильных трансгенных рас-
растений маркерные последовательности для предотвращения
распространения генов устойчивости к антибиотикам в окру-
окружающей среде, а также для более устойчивой экспрессии самих
трансгенов.
Введение рекомбинантных ДНК в составе векторньгх моле-
молекул в хлоропласты высших растений в основном осуществляют
биолистическими (biolistic) методами путем бомбардировки
клеток микрочастицами с сорбированными на них молекулами
ДНК [199]. Принцип таких методов будет кратко рассмотрен в
разделе 3.8.3. По-прежнему весьма популярным остается метод
трансформации хлоропластов с использованием полиэтилен-
гликоля [200].
3.8. Введение рекомбинантных ДНК в клетки
После завершения лигирования векторных молекул с клони-
клонируемыми фрагментами ДНК образовавшиеся рекомбинантные
молекулы необходимо вводить в подходящие клетки, где бы они
могли длительное время существовать и стабильно передаваться
дочерним клеткам в процессе клеточных делений. По способу
введения рекомбинантных ДНК в клетки все существующие ме-
методы разделяют на четыре большие группы: биологические, хи-
химические, физические и механические. Биологические методы
основываются на переносе генов в процессе вирусной инфекции
и использовании клеточных рецепторов для захвата чужеродной
ДНК, в том числе и бактериальными клетками с природной ком-
компетентностью. Группа химических методов включает в себя ис-
использование ионов металлов (Са2+, Rb+), ДЭАЭ-декстрана, липо-
142
сом (липофекция) и т.п. химических реагентов. В соответствии со
своим названием в физических методах применяют электриче-
электрические поля высокой напряженности (электропорация), а также ла-
лазерное излучение для создания пор в мембранах клеток-реципи-
клеток-реципиентов рекомбинантных ДНК. Наконец, при механическом введе-
введении ДНК имеет место грубое нарушение целостности мембран
клеток, например, при микроинъекции или бомбардировке кле-
клеток микрочастицами, на которых сорбированы рекомбинантные
молекулы.
Наиболее часто используемый в генно-инженерных исследо-
исследованиях процесс поглощения экзогенной ДНК бактериальными
клетками, сопровождающийся приобретением ими новых гене-
генетических маркеров, называют генетической трансформацией
[201]. В том случае, если бактериальные клетки захватывают
ДНК бактериофагов и в результате происходит образование зре-
зрелых фаговых частиц, говорят о трансфекции фаговой ДНК. По-
Поглощение экзогенной ДНК в процессе трансформации и транс-
трансфекции может осуществляться лишь компетентными клетка-
клетками. Под компетентностью понимается такое состояние бактери-
бактериальных клеток, при котором они способны сорбировать экзоген-
экзогенную ДНК на своей поверхности и поглощать ее, после чего экзо-
экзогенная ДНК может быть интегрирована в бактериальную хромо-
хромосому или существовать в виде внехромосомного элемента (эписо-
мы, плазмиды).
3.8.1. Природная компетентность бактериальных клеток
Для многих грамположительных бактерий компетентность
является естественным свойством, которое становится макси-
максимально выраженным на определенных этапах роста культуры.
В природных условиях данный процесс является одним из про-
проявлений коллективного поведения микроорганизмов в популя-
популяции, сопровождающегося саморегулированием ее численности
и синхронным изменением характера экспрессии генов в от-
отдельных клетках [202, 203]. В то же время, у наиболее важных
Для генной инженерии грамотрицательных клеток Е. coli есте-
естественная компетентность вообще отсутствует и клетки приоб-
приобретают ее лишь в искусственных условиях. За исключением
Neisseria и родственных им бактерий, для которых характерна
непрерывная компетентность, остальные часто используемые
бактерии, обладающие природной компетентностью, генетиче-
генетически регулируют ее регулоном компетентности. Компетент-
Компетентность для таких бактерий является нормальной физиологиче-
143
ской стадией развития. Грамположительные бактерии регули-
регулируют свою компетентность секретируемыми внешними фак-
факторами компетентности или фермонами (phermones), а также
экспрессией определенных генов, которая сопровождается син-
синтезом внутренних и внешних белков, участвующих в развитии
компетентности. Грамотрицательные бактерии такие, как
Haemophilus influenzae, контролируют компетентность преиму-
преимущественно внутренними механизмами.
Биологический смысл природной компетентности бактерий
не вполне понятен. Процесс трансформации бактериальных кле-
клеток в природных условиях обеспечивает поддержание жизненно
важного динамического состояния генома бактериальных кле-
клеток. Развитие компетентности тесно сопряжено с рекомбинацией
и репарацией бактериальных хромосом и является одним из мо-
молекулярных механизмов, обеспечивающих горизонтальный пе-
перенос генов у микроорганизмов [204]. В настоящее время имеют-
имеются указания на то, что донорная ДНК, которая захватывается
бактериальными клетками в природных популяциях микроорга-
микроорганизмов, появляется не только из-за случайной гибели клеток.
Развитие компетентности, по крайней мере у Streptococcus pneu-
moniae, индуцирует лизис части клеток этой популяции и осво-
освобождение геномной ДНК, а следовательно, процессы освобожде-
освобождения ДНК и ее захвата бактериальными клетками в таких систе-
системах координированы [205]. Суммируя данные о биологическом
значении природной компетентности бактериальных клеток,
можно заключить, что при участии этого процесса происходит
обмен генетической информацией в популяциях микроорганиз-
микроорганизмов, что необходимо для поддержания генетического разнообра-
разнообразия вида и распространения генов, важных для выживания бакте-
бактерий в изменяющихся условиях окружающей среды. Кроме того,
трансформирующая ДНК может участвовать в репарации по-
повреждений бактериальных хромосом после генотоксических воз-
воздействий [206].
У наиболее важной для генной инженерии грамположитель-
ной бактерии В. subtilis компетентность в искусственных услови-
условиях можно развить, выращивая клетки в специальных минималь-
минимальных питательных средах, содержащих глюкозу в качестве источ-
источника углерода, а также ионы Mg2+ и Са2+ [207]. Ионы Mg2+ необ-
необходимы для активации специфических нуклеаз, участвующих в
захвате экзогенной ДНК. Одним из практических способов до-
достижения компетентности является двухступенчатое понижение
скорости роста клеток в культуре. Биосинтетические процессы в
компетентных клетках замедлены, что может проявляться, в ча-
144
стности, в их устойчивости к антибиотикам (ампициллину). Био-
Биосинтез макромолекул ассоциирован, главным образом, с индук-
индукцией системы SOB-репарации (аналог SOS-системы), что являет-
является следствием накопления брешей в хромосомной ДНК бактерий
из-за нарушений нормальной репликации при замедлении скоро-
скорости роста.
Молекулярные события, связанные с развитием компетент-
компетентности у В. subtilis, схематически изображены на рис. 15, д. В про-
процессе становления компетентности клетки экскретируют два
пептида: 9-10-звенный модифицированный фермой СотХ и фак-
фактор, стимулирующий компетентность (competence stimulating fac-
factor - CSF). Покинув клетку, оба фактора взаимодействуют как с
ней самой, так и соседями, создавая состояние бактериальной
культуры, называемое восприятием кворума (quorum sensing),
одного из способов межклеточного общения грамположитель-
ных и грамотрицательных бактерий, при котором низкомолеку-
низкомолекулярные эффекторы (аутоиндукторы), синтезируемые одним
организмом, индуцируют экспрессию генов в других. У грамот-
грамотрицательных бактерий в качестве аутоиндукторов часто высту-
выступают ацетилированные лактоны гомосерина. СотХ далее взаи-
взаимодействует с мембранной протеинкиназой СотР, которая фос-
форилирует саму себя и белок Com А. Фактор CFS, синтезируе-
синтезируемый в то же самое время, воспринимается мембранной олигопеп-
тидпермеазой (ОРР), которая импортирует его внутрь клетки, где
фактор выполняет роль ингибитора дефосфорилирования
СотА. Фосфорилированный СотА активирует яг/-оперон, состо-
состоящий из четырех генов биосинтеза антибиотика сурфактина.
Среди них ген comS кодирует белок, который активирует белок
СотК путем освобождения его из комплекса. Во время логариф-
логарифмической фазы роста бактериальных клеток СотК находится в
комплексе с белком МесА, направляющим СотК на путь про-
теолитической деградации протеиназой ClpC. Активированный
белок СотК стимулирует свой собственный биосинтез и транс-
транскрипцию поздних генов компетентности.
Компетентные клетки В. subtilis имеют 20-50 мест связыва-
связывания дцДНК [208]. ОцДНК, РНК или ДНК-РНК-гибриды пере-
переносятся в клетку крайне неэффективно. Ключевую роль в про-
процессе связывания играет трансмембранный белок ComEA, ко-
который связывает преимущественно дцДНК, а также белки
группы ComG (ComGA-ComGG) (рис. 15, б). Последние обеспе-
обеспечивают формирование канала в плотном слое пептидогликана.
После взаимодействия с ComEA дцДНК быстро становится од-
Ноцепочечной под действием ассоциированной с ComEA экзо-
145
нуклеазы (N). Длина фрагментов оцДНК составляет 7-15 т.п.о.
Белок СотЕС формирует трансмембранный водный канал, че-
через который ДНК может быть перенесена в обоих направлени-
направлениях: как 5'> 3', так и 3'> 5'. Белок ComFA обладает активностью
геликазы и за счет гидролиза АТР, по-видимому, поставляет
энергию, необходимую для переноса ДНК через мембрану.
ОцДНК, проникшая в клетку, далее ассоциируется с белками
RecA и SSB, что необходимо для ее интеграции в бактериаль-
бактериальную хромосому. С момента добавления ДНК к компетентным
клеткам до появления трансформантов проходит не менее двух
мин. Такого рода эксперименты, особенно опыты по котранс-
формации удаленных маркеров, позволили определить ско-
скорость переноса ДНК в клетку как постоянную на уровне
180нт/секпри28°С.
Процесс интеграции ДНК в процессе трансформации грампо-
ложительных бактерий разделяют на пять стадий: 1) интернали-
зация ДНК и ее расположение в непосредственной близости от
хромосомы; 2) образование нестабильного донорно-реципиент-
ного комплекса; 3) образование стабильного нековалентного
комплекса; 4) образование ковалентного комплекса между до-
норной и реципиентной ДНК; 5) разрешение ковалентного гете-
родуплекса, содержащего ошибочно спаренные нуклеотиды. По-
Подробное рассмотрение всех этих вопросов выходит за рамки кру-
круга обсуждаемых нами проблем. Замечу только, что переход меж-
между стадиями 2 и 3 блокируется каумермицином, ингибитором
ДНК-гиразы, что указывает на важную роль пространственной
структуры ДНК в рассмотренном процессе рекомбинации между
донорной ДНК и реципиентной ДНК бактериальной хромосомы.
После установления компетентности клетки В. subtilis остаются в
таком состоянии в течение нескольких часов.
Рис. 15. Развитие природной компетентности (а) и проникновение ДНК
в клетку (б) у В. subtilis [20]
Становление компетентности инициируется фермоном СотХ и фактором,
стимулирующим компетентность CSF; СотХ воспринимается гистидиновой
протеинкиназой СотР, которая после аутофосфорилирования, фосфорилирует
белок СотА, активирующий srf-оперон, связанный в другой рамке считывания
с геном белка ComS; CSF является ингибитором дефосфорилирования ComA, a
также активирует собственный синтез через свой ген phrC; ComS освобождает
из предсуществующего комплекса СотК, который стимулирует транскрипцию
как своего собственного гена, так и поздних генов компетентности; белок YlbF
повышает активность ComS, а также участвует в спорообразовании.
Р - в генах обозначает промотор, а рядом с белками - ортофосфатную
группу; ОРР - олигопептидпермеаза
146
ComK MecA
ClpC
ClpP
Пептидогаикан
Мембрана
Споруляция
Поздние гены
споруляции
ДНК
Пептидогликан
Мембрана
RecA <Y SSB
147
3.8.2. Искусственная компетентность Е. coli
Клетки Е. coli не обладают природной компетентностью. Од-
Однако ввиду важности этого организма для молекулярной генети-
генетики, были разработаны методы, позволяющие придавать Е. coli
искусственную компетентность. При искусственной компетент-
компетентности трансформирующие плазмиды взаимодействуют со специ-
специфическими каналами на внешней поверхности клеток. Каждая
клетка обладает 10-200 каналами, обеспечивающими транспорт
питательных веществ и других ингредиентов, необходимых для
роста и размножения бактерий. В опытах по конкуренции транс-
трансформирующей плазмиды pBR322 и ее производной pXf 1 было ус-
установлено, что когда их смешивают в отношении 1:1000 (в сумме
10 молекул на клетку), это никак не сказывается на эффективно-
эффективности трансформации pBR322. Уменьшение этой пропорции до
1:10 000 или 1:100 000 A00 молекул на клетку) понижало эффек-
эффективность трансформации лишь на 60 и 80%, соответственно. Это
показывает, что имеется много каналов для проникновения
ДНК, а сами клетки должны конкурировать друг с другом за ее
захват.
В настоящее время установлено, что при низких температу-
температурах и в присутствии двухвалентных катионов клетки Е. coli и эк-
экзогенная ДНК продуктивно взаимодействуют друг с другом и эк-
экзогенная ДНК может с высокой эффективностью проникать
внутрь бактериальных клеток. Частоту трансформации и транс-
фекции клеток Е. coli можно повысить разными способами. Сре-
Среди них наиболее популярными являются тепловой шок, введение
в инкубационную смесь одновалентных катионов (Rb+), хлорида
гексаминкобальта (III) или выращивание бактериальных клеток
на среде с повышенным содержанием ионов Mg2+ A0-20 мМ).
Предполагается, что в присутствии двухвалентных катионов
или диметилсульфоксида при низкой температуре или в резуль-
результате действия других вышеупомянутых агентов нарушается це-
целостность и структура липополисахаридного слоя бактериаль-
бактериальных клеток, что может сопровождаться демаскировкой или по-
повышением доступности каналов захвата, которые могут быть
ассоциированы с местами взаимодействия внешней и внутрен-
внутренней мембран (т.н. мостики Байера). Хлорид гексаминкобаль-
гексаминкобальта (III) возможно специфически усиливает взаимодействие ДНК
с каналами.
Современные методики трансформации бактериальных кле-
клеток позволяют получать до 107— 108 трансформантов или транс-
фектантов на 1 мкг суперскрученной плазмидной ДНК. Посколь-
148
ку трансформация и трансфекция проводятся при низких концен-
концентрациях экзогенной ДНК, а для появления у трансформирован-
трансформированной бактерии устойчивости к антибиотикам или развития фаго-
фаговой бляшки достаточно проникновения внутрь бактериальной
клетки одной молекулы рекомбинантной ДНК, образовавшиеся
колонии или бляшки трансформантов и трансфектантов содер-
содержат, как правило, только один тип рекомбинантных молекул
ДНК с идентичными вставками клонируемых последовательнос-
последовательностей нуклеотидов. В случае же проникновения в бактериальные
клетки рекомбинантных плазмид с разными вставками клониру-
клонируемой ДНК в процессе клеточных делений происходит их быстрая
сегрегация в разные клетки, поскольку они относятся к одной
группе совместимости (см. раздел З.1.1.).
Еще более высокой эффективности трансформации (до
109-Ю10 трансформантов на 1 мкг ДНК) достигают при использо-
использовании электроперации [209]. В этом случае клетки вместе с плаз-
мидной или фаговой ДНК помещают в небольшую ячейку между
двумя электродами и подвергают кратковременному C-5 мс) воз-
воздействию электрического поля высокой напряженности. Быстрая
поляризация клеточных мембран приводит к их обратимому по-
повреждению (образованию пор), сопровождаемому повышением
их проводимости и проницаемости для макромолекул. В это вре-
время происходит эффективный перенос экзогенных молекул ДНК
внутрь клеток, подвергнутых электрическому шоку.
После того как рекомбинантные молекулы ДНК введены в
бактериальные клетки и получены бактериальные колонии или
фаговые бляшки, каждая из которых содержит рекомбинантную
ДНК только одного типа, возникает важная задача отбора тех
колоний или бляшек, которые заключают в себе искомые после-
последовательности нуклеотидов рекомбинантных ДНК.
3.8.3. Способы и последствия введения ДНК
в культивируемые клетки животных
Современные технологии позволяют вводить рекомбинант-
рекомбинантные ДНК в клетки любых типов и исследовать последствия экс-
экспрессии в них соответствующих генов. Этот процесс переноса ге-
генов в клетки высших организмов также, как и упомянутое выше
введение ДНК бактериофагов в бактериальные клетки, называ-
называют трансфекцией. Трансфекция эукариотических клеток, как
правило, включает два основных этапа: перенос ДНК через кле-
клеточные мембраны в цитоплазму клеток и последующий их транс-
транспорт в ядра. Часто используемые приемы преодоления барьера
149
клеточных мембран будут кратко рассмотрены ниже. Сам же ме-
механизм попадания экзогенной ДНК в ядра пока еще мало изучен.
По-видимому, в этом случае имеет место пассивное перемещение
линейных молекул через ядерный поровый комплекс без участия
системы транспорта ядерных белков [211]. На основании биоло-
биологических последствий, сопровождающих возникновение клеток-
трансфектантов, различают стабильную трансфекцию, когда
рекомбинантные ДНК интегрируются в хромосомы клеток-ре-
клеток-реципиентов и становятся их неотъемлемой частью, а также вре-
временную трансфекцию (transient transfection), при которой молеку-
молекулы рекомбинантной ДНК существуют и транскрибируются в яд-
ядрах во внехромосомном состоянии непродолжительное время.
Перенос генов с помощью вирусов основан на природном
процессе вирусной инфекции. Эффективные векторы созданы на
основе хромосомной ДНК ретровирусов, вирусов SV40, аденови-
аденовирусов, аденоассоциированных вирусов, вируса вакцины, вируса
герпеса и др. Ретровирусные векторы используются особенно ча-
часто для переноса генов в клетки животных [212].
Перенос генов, опосредованный клеточными рецепторами,
основан на интернализации рецепторов в процессе эндоцитоза.
В этом случае природный лиганд рецептора используют для ад-
адресной доставки молекул ДНК к клеткам, обладающим такими
рецепторами. В одной из таких систем в качестве переносчика
рекомбинантной ДНК применяется конъюгат трансферрина с
полилизином. Последний, являясь поликатионом, прочно взаимо-
взаимодействует с ДНК и осуществляет ее конденсацию с образованием
плотных комплексов [213].
Электропорация. Метод может быть использован для введе-
введения ДНК в клетки любых типов. Кратковременное A0 мксек-
100 мсек) воздействие на суспензию клеток с экзогенной ДНК
электрическим полем высокой напряженности E0-1500 V/см) со-
сопровождается локальным нарушением целостности мембран с
образованием микропор (диаметр 20-120 нм), через которые за-
заряженные молекулы, в том числе и ДНК, как линейная, так и су-
перскрученная, проникают внутрь клеток [214]. Поры закрыва-
закрываются спустя несколько мсек после прекращения действия им-
импульса. Тем не менее даже в оптимальных условиях в результате
этого шока приблизительно половина клеток погибает. Электро-
порированные клетки далее инкубируют в течение 48 час на не-
неселективной питательной среде, после чего производят отбор
трансфектантов по селектируемому маркеру, например, на среде
с антибиотиком неомицином. Наличие свободных концов в ли-
линейных векторах способствует их интеграции в хромосомы кле-
150
ток-реципиентов и образованию стабильных трансфектантов.
Метод электропорации особенно эффективен для переноса
внутрь клеток небольших молекул ДНК, включая бактериаль-
бактериальные плазмиды, и его эффективность быстро уменьшается с уве-
увеличением размера фрагментов. Тем не менее, эффективность
электропорации еще остается на достаточно высоком уровне,
позволяющем получать высококачественные клонотеки с ис-
использованием искусственных хромосом ВАС и РАС (размер кло-
клонированных фрагментов - 100-300 т.п.о.). Трансфекция клеток
животных методом электропорации гораздо менее эффективна
по сравнению с бактериальными, так как исходное количество
клеток в суспензии в этом случае значительно ниже, а ДНК, по-
попадая в цитоплазму, подвергается быстрой деградации. В этой
связи повышение эффективности метода может быть достигнуто
путем обеспечения защиты ДНК от нуклеазного гидролиза и
снабжением молекул сигнальными последовательностями, кото-
которые после объединения с соответствующими белками обеспечи-
обеспечивали бы направленный перенос молекул в ядра трансфецируе-
мых клеток.
Создание микроотверстий в клеточных мембранах с помо-
помощью лазера. Кратковременное облучение суспензии клеток с по-
помощью лазера обратимо создает микроотверстия в клеточных
мембранах, через которые экзогенная ДНК может проникать
внутрь клеток [215].
Микроинъекции. Введение ДНК внутрь клеток с помощью
этого метода осуществляют с использованием микроманипулято-
микроманипуляторов, оснащенных стеклянными микрокапиллярами, которыми
прокалывают мембраны клеток. Метод особенно интенсивно ис-
используется при создании трансгенных животных. В этом случае
большие фрагменты ДНК из векторов YAC, ВАС или РАС вво-
вводят непосредственно в ядра индивидуальных клеток. Из всех ис-
используемых методов только микроинъекция позволяет вводить в
клетки строго дозированное количество молекул ДНК.
Бомбардировка клеток микрочастицами. Эта группа мето-
методов, иначе называемых баллистической (biolistic) инъекцией,
первоначально была разработана для введения рекомбинантных
ДНК в клетки растений и в настоящее время приспособлена так-
также для введения нуклеиновых кислот в клетки микроорганизмов,
животных и человека как in vitro, так и in vivo [210, 216]. При
этом подходе микрочастицы, содержащие ДНК, с помощью по-
порохового заряда, сжатого гелия или электрического поля разго-
разгоняют до высоких скоростей (-1000 м/сек) и поток частиц кратко-
кратковременно направляют на поверхность клеток. Некоторые из ча-
151
стиц после проникновения внутрь клеток вместе со связанной с
ними ДНК достигают ядер, что может сопровождаться стабиль-
стабильной трансфекцией клеток-мишеней. В качестве носителей ДНК
используют микрочастицы вольфрама, золота или льда. Баллис-
Баллистическая инъекция позволяет вводить нуклеиновые кислоты не
только в ядра клеток, но и находящиеся непосредственно в клет-
клетках органеллы: митохондрии и хлоропласты [217]. В настоящее
время данные подходы широко используются в генотерапии, для
получения трансгенных животных и растений, индукции сайлен-
синга генов небольшими интерферирующими РНК (siPHK), a
также для прямой иммунизации организмов с помощью ДНК-
вакцин в виде плазмидных ДНК, действие которых особенно эф-
эффективно против вирусных инфекций [218].
Трансфекция клеток, опосредованная фосфатом кальция
(calcium phosphate coprecipitation method), является одним из наи-
наиболее широко используемых и наименее воспроизводимых по
эффективности методов ведения рекомбинантных ДНК [219].
Раствор СаС12 с помещенной в него ДНК медленно смешивает-
смешивается с фосфатным буфером, что сопровождается образованием
нерастворимого фосфата кальция и одновременным осаждени-
осаждением ДНК. Суспензия вносится в чашки Петри с культивируемы-
культивируемыми клетками. Через некоторое время частицы фосфата каль-
кальция, содержащие ДНК, сорбируется на поверхности клеток и
проникают внутрь в процессе эндоцитоза. Далее небольшая
фракция рекомбинантной ДНК оказывается в ядре, где может
быть интегрирована в хромосому или некоторое время сущест-
существовать во внехромосомном состоянии. Метод прост в своей реа-
реализации, однако высоко чувствителен к условиям проведения
эксперимента (концентрациям ДНК и СаС12, значениям рН, про-
продолжительности трансфекции) и поэтому трудно воспроизво-
воспроизводим. При слишком длительной инкубации клеток с фосфатом
кальция проявляется его цитотоксическое действие. С учетом
этого, для каждой линии клеток требуется проведение тщатель-
тщательной оптимизации условий.
Метод трансфекции клеток, основанный на использовании
ДЭАЭ-декстрана, является альтернативой рассмотренному выше
кальций-фосфатному методу [220]. Этот подход широко исполь-
используется для получения клеточных линий, временно экспрессирую-
щих рекомбинантные гены. ДЭАЭ-декстран смешивают с ДНК в
питательной среде с низким содержанием сыворотки, и смесь на
некоторое время добавляют к культивируемым клеткам. Ком-
Комплексы ДНК с ДЭАЭ-декстраном сорбируются на поверхности
клеток и поступают внутрь с помощью эндоцитоза. Доставку
152
комплексов внутрь клеток можно стимулировать, повысив про-
проницаемость клеточных мембран диметилсульфоксидом. К недо-
недостаткам метода следует отнести подавление роста клеток во вре-
^я трансфекции, а также появление в самих клетках различных
морфологических изменений. При этом стабильные трансфек-
танты возникают редко. Трансфекция с применением ДЭАЭ-дек-
страна является одним из примеров возможности использования
катионных полимеров для осуществления этого процесса. Для
эффективной трансфекции по тому же принципу можно исполь-
использовать дендримеры (сильно разветвленные молекулы полиме-
полимеров), полилизин, протамины и полиэтиленимин [221]. Вторая
группа родственных методов основана на применении для тех же
целей катионных липидов.
Липосомы в трансфекции. Использование липосом является
еще одним популярным, эффективным и универсальным мето-
методом введения рекомбинантных ДНК в клетки животных и чело-
человека [222]. Для создания липосом в этом случае чаще всего ис-
используют катионные липиды, которые являются амфифильны-
ми самоассоциируемыми молекулами, которые в присутствии
ДНК, благодаря электростатическим и другим взаимодействиям,
конденсируются в частицы, получившие название липоплексов
(lipoplexes). Липоплексы, несущие суммарный положительный
заряд, взаимодействуют с сиаловыми кислотами клеточных мем-
мембран и обеспечивают эффективное проникновение ДНК внутрь
клеток в процессе эндоцитоза [223]. Хотя большинство ДНК по-
попадает в лизосомы и деградирует, значительная их фракция до-
доставляется в ядро трансфецируемых клеток.
Для получения липоплексов катионные липиды и ДНК сме-
смешивают в бессывороточной питательной среде, поскольку сыво-
сыворотка препятствует их объединению, и культивируемые клетки
кратковременно инкубируют в их присутствии. Метод позволяет
осуществлять как временную, так и постоянную трансфекцию с
образованием стабильных клеточных линий. По сравнению с
ДЭАЭ-декстраном эффективность этих методов, по крайней ме-
мере, в 5-100 раз выше при получении временной трансфекции и в
3-20 раз при создании стабильных трансфектантов. При этом с
помощью липосом удается осуществлять трансфекцию в слож-
сложных случаях тех клеточных линий, которые не поддаются введе-
введению рекомбинантных ДНК другими способами. Более того, дан-
данная группа методов допускает проведение трансфекции в суспен-
суспензионных культурах, не требует митотического деления клеток в
Момент проведения эксперимента и, как правило, высоковоспро-
изводима.
153
Конъюгативный перенос бактериальных генов в клетки жи-
животных. Перенос генов во время конъюгации бактериальных
клеток, когда мужские и женские клетки вступают в контакт
друг с другом через объединяющий их цитоплазматический мос-
мостик, является широко распространенным и хорошо изученным
генетическим явлением [224, 225]. Недавно была продемонстри-
продемонстрирована возможность конъюгативного переноса ДНК из бактери-
бактериальных клеток в культивируемые клетки животных [226]. В этой
серии экспериментов В.Л. Ватерсу удалось показать, что гены ус-
устойчивости к антибиотикам, находящиеся в составе конъюгатив-
ной плазмиды Е. coli, переносятся с низкой частотой в клетки
яичников китайских хомячков СНО К1 из бактериальных клеток,
давая возможность клеткам-реципиентам выживать на селектив-
селективной среде в присутствии соответствующих антибиотиков. При
этом не происходило поглощения бактериальных клеток клетка-
клетками животных посредством эндоцитоза, и перенос имел место в
присутствии ДНКазы в питательной среде, что исключало непо-
непосредственный захват ДНК клетками из культуральной жидкости.
Чужеродная ДНК реплицировалась в клетках животных, а экс-
экспрессия генов генетических маркеров происходила лишь в том
случае, если гены находились под контролем эукариотических
промоторов. Хотя конъюгативный перенос генов бактерий в
клетки дрожжей, а также растений (Ti-плазмиды) известен дав-
давно, обсуждаемая работа впервые продемонстрировала возмож-
возможность непосредственного обмена генами между бактериями и
клетками высших животных. В том случае, если данный процесс
удастся оптимизировать, у генной инженерии появится дополни-
дополнительная возможность введения очень больших молекул ДНК в
клетки животных, в том числе и в целях генотерапии.
154
Глава 4
Клонотеки генов
Любой индивидуальный ген занимает лишь небольшую часть
генома живого организма. В то же время размер генома даже
наиболее просто организованных бактерий в среднем составляет
2-Ю6 п.о., а суммарный размер молекул ДНК, составляющих гап-
гаплоидный геном человека и высших животных, по крайней мере,
на три порядка больше. Из этого следует, что уникальные гены,
представленные в гаплоидном геноме только одной копией, зате-
затеряны среди других последовательностей генома, и для работы с
индивидуальными рекомбинантными ДНК требуется их очистка
от ненужного генетического материала. Такая задача в генной
инженерии решается через создание репрезентативных (пред-
(представительных) клонотек последовательностей нуклеотидов
ДНК или, иначе говоря, клонотек генов.
4.1. Получение клонотек генов
Клонотека генов представляет собой набор разных последо-
последовательностей нуклеотидов ДНК, клонированных в составе век-
векторных молекул, которые в сумме составляют весь геном иссле-
исследуемого организма или какую-либо известную его часть. При
этом репрезентативная клонотека должна заключать в себе с вы-
высокой долей вероятности любую последовательность нуклеоти-
нуклеотидов изучаемого генома. Аналогичный по смыслу и широко рас-
распространившийся в русскоязычной литературе термин "библио-
"библиотека" генов или "библиотека" последовательностей, который
представляет собой буквальный перевод английского термина
"library", нельзя считать удачным, поскольку набор нуклеотид-
ных или аминокислотных последовательностей не имеет никако-
никакого отношения к книгохранилищу.
Как уже подробно обсуждалось выше, для большинства ге-
генов эукариот характерна интрон-экзонная структура, а интроны,
присутствующие в первичных транскриптах таких генов, выреза-
вырезаются в процессе сплайсинга с образованием зрелых молекул
мРНК. Для получения экспрессии эукариотических генов в клет-
клетках прокариот, а также изучения тканеспецифической экспрес-
экспрессии генов возникает необходимость в получении кодирующих по-
последовательностей эукариотических генов без интронов. В этом
случае задача решается через создание репрезентативных клоно-
155
тек кДНК, в которых с высокой вероятностью представлены по-
последовательности нуклеотидов мРНК, синтезирующихся в тка-
тканях, культурах тканей или отдельных соматических клетках.
На рис. 9 представлена схема конструирования клонотеки ге-
геномной ДНК с использованием космидного вектора. Следует
иметь в виду, что общие принципы получения клонотек генов
приложимы и к любым другим векторам. На первом этапе кон-
конструирования осуществляют подготовку вектора и клонируемой
ДНК. Космидный вектор расщепляют рестриктазами ВатШ и
Smal, причем рестриктаза Smal расщепляет ДНК с образованием
тупых концов. В результате образуются два "плеча" космидного
вектора, содержащих область начала репликации ori, используе-
используемую системой репликации бактериальных клеток, и два селекти-
селектируемых маркера, которые представляют собой гены устойчивос-
устойчивости к антибиотикам - ампициллину (Атрг) и канамицину (Kanr), a
также два сш-сайта хромосомы бактериофага X. Параллельно со
всеми необходимыми предосторожностями получают препараты
высокомолекулярной ДНК, которую необходимо клонировать.
Геномную ДНК подвергают частичному гидролизу мелко-
щепящей рестриктазой Mbol (узнает последовательность из че-
четырех нуклеотидов GATC, которые комплементарны липким
концам, образуемым рестриктазой ВатШ). Время рестрикции и
концентрацию фермента подбирают таким образом, чтобы
средняя длина образующихся фрагментов ДНК составляла
35-45 т.п.о. Обогащенную фракцию полученных фрагментов
ДНК получают центрифугированием смеси рестрикционных
фрагментов в градиенте концентрации сахарозы. Далее эти
фрагменты лигируют с приготовленными "плечами" вектора в
условиях, при которых образование сшивок по тупым концам
минимально. При этом, помимо требуемых молекул рекомби-
нантных ДНК, могут образовываться и артефактные молеку-
молекулы, которые не будут упаковываться в фаговые частицы либо
из-за их неоптимального размера, либо вследствие неправиль-
неправильной ориентации сш-сайтов.
Полученную после лигирования сложную смесь молекул ре-
комбинантных ДНК упаковывают в фаговые частицы стан-
стандартным способом, и образовавшимися фаговыми частицами
заражают подходящие бактериальные клетки. В итоге колонии
бактериальных клеток, которые выросли в присутствии анти-
антибиотиков, должны заключать в себе различные последователь-
последовательности нуклеотидов клонируемой ДНК приблизительно одина-
одинаковой длины.
156
4.1.1. Клонотеки геномной ДНК
На вышеприведенном частном примере был рассмотрен один
из подходов к конструированию клонотеки геномной ДНК с ис-
использованием космидного вектора, который иллюстрирует все
основные этапы получения клонотек генов с применением и дру-
других векторов, плазмидных или фаговых [227, 228]. Во всех случа-
случаях для получения репрезентативных клонотек генов следует по-
полнить о необходимости случайной фрагментации клонируемой
высокомолекулярной ДНК, чтобы все участки генома в образу-
образующейся смеси фрагментов были представлены равновероятно.
Кроме того, размеры образуемых фрагментов ДНК должны со-
соответствовать емкости вектора, используемого для их клониро-
клонирования. Так, для клонирования в космидах необходимо получать
фрагменты ДНК длиной 35-45 т.п.о., тогда как для клонирования
в фаговых векторах типа Charon и плазмидах эти размеры долж-
должны составлять соответственно 20-24 и 1,5-3,0 т.п.о.
Чем короче фрагменты, используемые для получения клоно-
клонотек генов, и чем сложнее исследуемый геном, тем большее число
клонов необходимо получить, чтобы клонотека была полной.
Например, для того чтобы создать геномную клонотеку бакте-
бактерий из фрагментов ДНК длиной в 20 т.п.о., в которой любая по-
последовательность была бы представлена с вероятностью 99%, не-
необходимо иметь 460 клонов, тогда как для млекопитающих это
число возрастает до 600 000, а для покрытосеменных растений
оно еще в ~10 раз больше. Отсюда следует, что в случае получе-
получения репрезентативных клонотек генов бактерий для решения та-
такой задачи вполне достаточно плазмидных векторов, несмотря
на их небольшую емкость. В то же время использование плазмид
для создания полных клонотек генов млекопитающих или расте-
растений совершенно бесперспективно, так как потребовало бы под-
Держания в клонотеке огромного количества клонов, многие из
которых с большой вероятностью могут быть утрачены.
Размер клонотеки, которая будет представлять с требуемой
вероятностью любую последовательность исследуемого генома,
можно рассчитать по формуле:
- Р)
N =
- /)
N - требуемое число клонов, Р - вероятность того, что кло-
йотека содержит требуемую последовательность ДНК, / - доля
г©нома, в среднем, представленная в каждом клоне, которая вы-
выделяется путем деления среднего размера вставки на размер ис-
157
следуемого генома. Эта формула позволяет произвести расчет
числа независимых клонов, т.е. числа рекомбинантных бактерий
или фаговых частиц, которые необходимо получить непосредст-
непосредственно после трансформации или трансфекции компетентных
клеток. После ресуспендирования колоний или фаговых бляшек
происходит амплификация клонотеки, в которой каждый клон
уже будет представлен тысячами бактерий или фаговых частиц.
Оценку качества клонотеки осуществляют путем определе-
определения среднего размера вставок в случайно выбранных клонах.
Клонотеки, полученные с использованием бактериальных
клеток, обычно хранят при -70°С в виде суспензии в 30% глице-
глицерине, а суспензии фаговых частиц - в диметилсульфоксиде, что
предохраняет клетки и вирусные частицы от гибели при замора-
замораживании и оттаивании. Этой процедуры, впрочем, рекомендует-
рекомендуется избегать для большей сохранности клонотеки и отбирать бак-
бактерии или фаговые частицы без оттаивания основной суспензии.
В этой связи бывает полезно исходную клонотеку хранить от-
отдельно, а на ее основе получить вторичную клонотеку, которую,
разделив на несколько частей, использовать в повседневной ра-
работе, оттаивая каждую часть ограниченное число раз.
4.1.2. Клонотеки кДНК
При конструировании клонотек кДНК прежде всего прово-
проводят очистку мРНК хроматографией на олиго-сГГ-целлюлозе, ко-
которую используют в качестве матрицы при обратной транскрип-
транскрипции (рис. 16) [228, 229]. Обратная транскриптаза (как и ДНК-за-
ДНК-зависимая ДНК-полимераза) для своего функционирования требу-
требует затравки в виде олигонуклеотида, как правило олиго(сГГ), ко-
который отжигают с 3'-концевой поли(А)-последовательностью
мРНК [230]. Элонгируя этот праймер, обратная транскриптаза
синтезирует первую цепь кДНК. Удаление РНК-матрицы из гиб-
гибрида проводят с помощью РНКазы Н или путем щелочного гид-
гидролиза мРНК. Образовавшиеся молекулы оцДНК имеют тенден-
тенденцию самопроизвольно формировать вторичную структуру, что
приводит к образованию структуры типа "стебель-петля" на 3-
конце оцДНК. Этот конец, в свою очередь, используется в каче-
качестве затравки при синтезе второй цепи кДНК ДНК-полимеразой
I. В итоге образуется дцДНК, обе цепи которой на одном конце
соединены между собой петлей из оцДНК. Петлю удаляют инку-
инкубацией с S1 -нуклеазой, специфически гидролизующей одноцепо-
чечные нуклеиновые кислоты. Альтернативно в цепи мРНК, ис-
использованной в качестве матрицы при синтезе первой цепи
158
¦АААААЗ' мРНК
г
3'
Олиго(сПГ)-праймер
Праймер
Обратная транскриптаза
¦ААААА 3' мРНК
ТТТТТ 5'
¦ААААА
щщ ^т^ч ^Т^^ ^Т^^ ^Т^^ ^Т^^
Удаление мРНК
(гидролиз щелочью
или РНКазой Н)
^т^ч ^Т^^ ^Т^^ ^Т^^ ^Т^^
Образование шпильки
^в*^ ^Т^^ ^Т^^ ^Т^^ ^Т^^
Синтез ДНК
ДНК-полимеразой
гв^ гв^ гв^ гв^ гв^
мРНК
ДНК
Удаление шпильки
Достройка концов
ДНК-полимеразой
Двухцепочечная
кДНК
Рис. 16. Синтез кДНК на матрице мРНК обратной транскриптазой [273]
кДНК, с помощью РНКазы Н делают одноцепочечные разрывы
[231]. Образовавшиеся в итоге фрагменты мРНК далее использу-
используются ДНК-полимеразой IE. coli в качестве затравок при синтезе
второй цепи кДНК. Повторная инкубация кДНК с ДНК-полиме-
ДНК-полимеразой приводит к окончательному "затуплению" ее концов, кон-
концы кДНК фосфорилируют с помощью полинуклеотидкиназы,
что необходимо для проведения последующего клонирования
этих макромолекул.
Для клонирования к концам кДНК присоединяют с помощью
ДНК-лигазы олигонуклеотидные адаптеры, содержащие сайты
рестрикции, необходимые для соединения с линеаризованным
вектором. После этого кДНК фракционируют по размерам, очи-
Щают от оставшихся молекул адаптера и клонируют по липким
159
концам стандартными методами. Поскольку длина кДНК редко
превышает несколько т.п.о., в качестве вектора обычно исполь-
используют плазмиды или инсерционные векторы на основе фага X (на-
(например, gtlO или gtll).
Необходимо иметь в виду, что синтезированные таким спосо-
способом молекулы кДНК редко заключают в себе всю последователь-
последовательность мРНК-матрицы. Обратная транскриптаза обычно не транс-
транскрибирует цепь мРНК до конца, кроме того потеря информации
происходит при гидролизе одноцепочечного участка. Для преодо-
преодоления этих затруднений в качестве затравок при синтезе первой
цепи кДНК часто используют вместо олиго(с1Т)-праймеров слу-
случайные праимеры - короткие олигонуклеотиды, которые могут
отжигаться с матрицей, в том числе и ближе к ее 5'-концу. Кроме
данного преимущества, использование случайных праймеров поз-
позволяет получать кДНК для бактериальных РНК и геномных РНК
вирусов, которые, как правило, не содержат протяженных 3'-кон-
3'-концевых поли(А)-последовательностей. Системы синтеза кДНК, ос-
основанные на использовании случайных праймеров, в настоящее
время продаются многими фирмами. Использование терминаль-
терминальной трансферазы для присоединения гомополимера к 3'-концу
первой цепи кДНК позволяет избежать необходимости использо-
использования 3'-концевой петли для синтеза второй цепи, так как в этом
случае в качестве затравки уже можно использовать праймер,
комплементарный гомополимерному участку. Получить допол-
дополнительную информацию о 5'-концевых последовательностях
мРНК можно с помощью эффективного метода амплификации
5'-концов кДНК E'-RACE), уже рассмотренного в разделе, посвя-
посвященном полимеразной цепной реакции, принцип которого пред-
предполагается рассмотреть во втором томе монографии.
4.1.3. Случайные и упорядоченные клонотеки
В рассмотренных выше случаях клонотеки последовательно-
последовательностей представляли собой суспензии рекомбинантных бактериаль-
бактериальных клеток или фаговых частиц, в которых отдельные клоны,
представленные многими тысячами индивидуальных частиц, ни-
никак не были упорядочены в пространстве друг относительно дру-
друга. Такие клонотеки отвечают многим генно-инженерным зада-
задачам, например, могут быть эффективно использованы для отбо-
отбора клонов методом гибридизации колоний (см. ниже). Однако в
ряде случаев, например, когда процесс отбора занимает длитель-
длительное время и требует анализа каждого индивидуального клона, не-
невозможно одновременно исследовать все клоны клонотеки. По-
160
этому после анализа небольшого числа клонов снова приходится
обращаться к клонотеке за новой аликвотой бактериальных кле-
клеток, но в результате в анализ со значительной вероятностью мо-
могут попадать клоны, которые уже были изучены. Для того, что-
чтобы избежать такого повторения, после первого рассева транс-
трансформированных клеток выросшие колонии, перед тем как на-
начать с ними работать, упорядочивают, например, путем перека-
перекалывания в виде матрицы на чистую чашку Петри с присвоением
каждому клону определенного номера или подращивания инди-
индивидуальных клонов в отдельных лунках планшетов для микроти-
микротитрования. Это позволяет после обнаружения рекомбинантной
ДНК, обладающей требуемыми свойствами, легко вернуться к
соответствующему исходному клону и далее работать только с
ним, автоматизировать процесс скрининга и получать идентич-
идентичные копии клонотеки. В результате таких простых манипуляций
клонотека последовательностей поддерживается в виде матрицы
(arrayed (gridded) library), в которой несмотря на пространствен-
пространственную упорядоченность отсутствует какая-либо внутренняя логи-
логика, которая бы связывала друг с другом соседние клоны [232].
Такая связь достигается в упорядоченных клонотеках в ко-
которых клоны, располагающиеся по соседству, содержат пере-
перекрывающиеся последовательности, а весь их ряд представляет
непрерывную последовательность генома [233, 234]. Естествен-
Естественно, упорядочивание таких клонов возможно только после секве-
нирования клонированных последовательностей ДНК или опре-
определения особенностей их первичной структуры другими способа-
способами. Упорядоченные клонотеки содержат гораздо меньшее число
клонов по сравнению с неупорядоченными, поскольку в послед-
последних каждый клон многократно дублирован. С такими клонотека-
ми проще работать, но их значительно труднее создавать. Упоря-
Упорядоченные клонотеки ВАС-клонов ДНК человека были использо-
использованы международным консорциумом при выполнении програм-
программы "Геном человека". В отличие от этого, американская фирма
Selera при решении той же задачи пошла по другому пути, ис-
используя случайные клонотеки и метод дробовика (shot-gun), в
котором осуществляли тотальное секвенирование потенциально
перекрывающихся случайно выбираемых из клонотеки клонов,
которые объединяли в непрерывную последовательность с помо-
помощью вычислительной техники.
Выше были рассмотрены условия, которые необходимо со-
соблюдать для получения репрезентативных клонотек геномной
ДНК. Однако во многих случаях не возникает необходимости ра-
работать с полными клонотеками генов, и исследователи ограничи-
6. Патрушев Л.И. Т. 1
ваются неполными клонотеками, обогащенными отдельными
участками генома. Так, клонотеки кДНК заключают в себе по-
последовательности нуклеотидов, которые в виде зрелых мРНК
специфически экспрессируются в различных клетках или тканях.
То же можно сказать и о клонотеках повторяющихся последова-
последовательностей нуклеотидов, отобранных из препарата суммарной
ДНК на основании кинетики их реассоциации.
Сведения о приемах работы с клонотеками случайных нукле-
отидных последовательностей, которые используются при кон-
конструировании белков методами направленной эволюции макро-
макромолекул, можно найти в разделе П.2.1 этой книги.
4.2. Поиск последовательностей в клонотеках генов
Способы получения требуемых последовательностей нуклео-
нуклеотидов из клонотек генов можно разделить на три группы. При ис-
использовании первой группы методов рекомбинантные бактерии
или фаговые частицы исследуют на присутствие в них искомых по-
последовательностей нуклеотидов путем последовательного перебо-
перебора случайных клонов. При таком подходе, получившем название
скрининга, творческие усилия исследователя направлены только
на облегчение самого процесса анализа клонов, например, на его
автоматизацию. Во втором случае, присутствие нужных последо-
последовательностей обнаруживают косвенно, по появлению в бактери-
бактериальных клетках или фаговых лизатах бляшек продуктов экспрес-
экспрессии искомых генов - РНК, белков или ферментативной активнос-
активности, т.е. определенного фенотипа, который отличает такие клоны
от соседних, не содержащих соответствующих последовательнос-
последовательностей. В этом случае исследователь среди большого количества сум-
суммарных клонов осуществляет выбор тех, которые резко отлича-
отличаются от соседних по своему фенотипу, например, цвету колоний.
При таком подходе производится выбор требуемого фенотипа
среди большого числа других фенотипов. Реализация третьего
подхода требует создания селективных условий, при которых пре-
преимущество в размножении получают те клоны, которые отвечают
требованиям отбора, например, приобрели способность к росту на
селективных питательных средах в присутствии антибиотика или
в отсутствие аминокислоты в случае исходно ауксотрофного
штамма. Последний подход, кроме своего необыкновенного изя-
изящества в замысле, демонстрирует самую высокую эффективность,
так как позволяет "в одно касание" освободиться от всех нежела-
нежелательных примесей в виде ненужных клонов.
162
4.2.1. Использование меченых зондов
Для выявления конкретных последовательностей нуклеоти-
дов в клонотеках генов рекомбинантные бактерии или фаги рас-
севают на чашках Петри и образовавшиеся колонии или бляшки
переносят на нитроцеллюлозные или нейлоновые фильтры [235,
236]. Для этого стерильные фильтры накладывают на поверх-
поверхность чашек, в результате чего часть бактериальных клеток или
фаговых частиц прочно связывается с поверхностью фильтров.
Расположение их на фильтрах и поверхности чашек Петри точно
совпадает. Бактериальные клетки или фаговые частицы, сорби-
сорбированные на фильтрах, лизируют щелочью, что сопровождается
одновременной денатурацией заключенной в них ДНК. Освобо-
Освободившуюся ДНК фиксируют на фильтрах и гибридизуют с мече-
меченым олигонуклеотидным зондом, последовательность нуклеоти-
дов которого точно соответствует последовательности нуклеоти-
дов искомой ДНК. В результате гибридизации происходит специ-
специфическое прочное связывание зонда с идентичными и/или гомо-
гомологичными последовательностями нуклеотидов в ДНК, содержа-
содержащейся в отдельных бактериальных колониях или бляшках.
После отмывки фильтров от несвязавшегося радиоактивного
зонда их высушивают и анализируют с помощью авторадиогра-
авторадиографии с использованием чувствительной к радиоактивному излуче-
излучению рентгеновской пленки. Образование специфических гибри-
гибридов обнаруживают после проявления рентгеновской пленки по
появлению на ней четких гибридизационных сигналов в виде тем-
темных пятен, положение которых точно соответствует таковому
определенных колоний или бляшек на исходной чашке Петри. В
результате проведения исследования идентифицируются бакте-
бактериальные колонии или фаговые бляшки, содержащие искомые
рекомбинантные ДНК. С этого момента с помощью обычных
микробиологических и биохимических методов можно получать
неограниченное количество идентичных копий клонированной
последовательности нуклеотидов ДНК для дальнейших исследо-
исследований. В ряде случаев, когда нет необходимости в скрининге
большого числа клонов, гибридизацию с зондами можно заме-
заменить ПЦР. При таком подходе в качестве источника матричной
ДНК используют суспензию бактериальных клеток или фаговых
частиц отдельных клонов без специальной очистки матричных
нуклеиновых кислот.
Гомологичные зонды. Использование гомологичных зон-
зондов, т.е. зондов, последовательность которых полностью соот-
соответствует исследуемому гену, становится возможным в том слу-
6* 163
чае, если известна (например, из литературы или базы данных)
первичная структура последовательности, которую необходимо
обнаружить в клонотеке. Такая задача часто возникает, когда
определенный ген или кДНК хотят использовать в прикладных
целях, например, в биотехнологических разработках или для ге-
нотерапии наследственных заболеваний. Та же самая задача мо-
может возникнуть и при необходимости получения последователь-
последовательности целого гена, если предварительно клонирована его часть.
Подход с использованием гомологичных зондов может быть
применен и при исследовании популяционного полиморфизма
исследуемых последовательностей. Сложнее обстоит дело в том
случае, если на момент клонирования последовательность нук-
леотидов исследуемого участка хромосомы совершенно неизве-
неизвестна. Такие задачи иногда могут быть решены с использовани-
использованием гетерологичных зондов.
Гетерологичные зонды. Гетерологичные олигонуклеотид-
ные зонды получают на основании предположения о наличии го-
гомологии в последовательностях у генов организмов разных био-
биологических видов, которые выполняют аналогичные функции.
При гибридизации гетерологичных зондов с ДНК исследуемой
клонотеки используют более низкие температуры их отжига для
того, чтобы даже при наличии нескольких неправильно спарен-
спаренных оснований в гибриде он оказался достаточно стабильным для
обнаружения. Понижение температуры отжига зонда значитель-
значительно ниже его температуры плавления будет почти неизбежно со-
сопровождаться получением ложноположительных результатов,
т.е. гибридизацией зондов с неродственными последовательнос-
последовательностями, которые обладают определенной гомологией с искомой.
Во многих случаях этот результат не опасен, поскольку после та-
такого предварительного отбора, круг поиска нужной последова-
последовательности существенно сужается. Проведение повторной гибри-
гибридизации в других условиях среди предварительно отобранных
клонов может привести к положительному результату. Ситуация
становится угрожающей в том случае, если число ложноположи-
ложноположительных клонов, выявленных на первом этапе отбора, становит-
становится слишком большим. В этом случае цена повторного скрининга
может оказаться слишком высокой.
Обратная трансляция. Выбрать последовательность зонда и
праймеров для ПЦР к неизвестному гену иногда оказывается воз-
возможным путем очистки небольшого количества белка, кодируе-
кодируемого клонируемым геном, и определения последовательности
его нескольких N-концевых или С-концевых аминокислотных ос-
остатков [237, 238]. На основании этих данных с учетом вырожден-
164
ности генетического кода и частоты встречаемости аминокис-
аминокислотных остатков у исследуемого организма синтезируют олиго-
нуклеотидные зонды, которые используют для гибридизации in
situ, аналогично тому как было описано выше. Такой подход к
определению последовательности нуклеотидов гена на основа-
основании последовательности аминокислот белка, кодируемого этим
геном, получил название обратной трансляции (back (reverse)
translation). При решении данной задачи, вырожденность генети-
генетического кода можно преодолеть путем синтеза вырожденных
олигонуклеотидов посредством введения в каждое положение
синтезируемого олигонуклеотида нескольких требуемых генети-
генетическим кодом нуклеотидов. Однако сознательное повышение
вырожденности зонда уменьшает его специфичность и повыша-
повышает вероятность получения ложноположительных результатов.
Ситуация может быть существенно улучшена путем использова-
использования при отборе нескольких вырожденных зондов, соответствую-
соответствующих разным участкам клонируемого гена.
Обратная трансляция аминокислотной последовательности
Met-Phe-Asn-Cys-Trp указывает на необходимость синтеза
15-звенного вырожденного олигонуклеотида со следующей пер-
первичной структурой: ATGTT(T/C)AA(T/C)TG(T/C)TGG, в которой
нуклеотиды в скобках отмечают места с неоднозначной последо-
последовательностью. Теоретически олигонуклеотид длиной в
14-15 нуклеотидов, полученный путем обратной трансляции по-
последовательности из пяти аминокислотных остатков должен
быть уникальным для геномов такого размера, как геном челове-
человека. Впрочем, это утверждение верно лишь в том случае, если ге-
геном представлен случайной последовательностью. На практике
это не так. В любом большом геноме встречаются мультигенные
семейства с близкой первичной структурой, и наблюдается гомо-
гомология между многими последовательностями, которая отражает
их зависимое друг от друга происхождение. Поэтому 15-звенный
олигонуклеотид при исследовании генома человека скорее всего
не окажется геноспецифическим. Однако извлечь специфичес-
специфическую геномную последовательность с помощью таких олигонук-
олигонуклеотидов можно при использовании их в качестве праимеров для
ПЦР на матрице кДНК соответствующей клонотеки. В этом слу-
случае неспецифические олигонуклеотиды скорее всего не будут
участвовать в образовании продукта ПЦР.
Современные методы позволяют определять аминокислотную
последовательность полипептида, выделенного из пятна, которое
выявляется после проведения двумерного электрофореза, а также
во фракциях, получаемых с помощью аналитической ВЭЖХ или
165
капиллярного электрофореза. Для этих целей в настоящее время
широко используется MALDI-TOF-масс-спектрометрия, основан-
основанная на лазерной десорбции и ионизации макромолекул, опосредо-
опосредованной матриксом (matrix assisted laser desorption ionization -
MALDI) и детекторами ионов, которые позволяют определять
время их полета (time of flight - TOF). В этой группе методов им-
импульсное облучение сорбента, с которым связаны анализируемые
макромолекулы приводит к их высокоэффективной фрагмента-
фрагментации и десорбции в вакуум благодаря избирательному поглощению
сорбентом энергии света. При этом время полета образовавшихся
ионов в анализаторе прямо пропорционально их массе.
Использование антител для отбора клонов в экспрессирую-
щих клонотеках. Получение клонотеки генов или кДНК с ис-
использованием экспрессирующих векторов дает ряд преимуществ
в их последующем отборе. В этом случае, если 5'-концевая часть
клонируемого гена находится близко от промотора вектора или
его кодирующая часть соединена в одной рамке считывания с
инициирующим ATG-кодоном через последовательность, коди-
кодирующую часть другого белка, например, C-галактозидазы, то в
бактериальных клетках или фаговых лизатах можно обнаружить
появление специфического белкового продукта или фермента-
ферментативной активности. Если же рекомбинантный белок неактивен и
представляет собой лишь часть полипептидной цепи нативного
фермента, его обнаруживают иммунохимическими методами.
Отбор нужных клонов может проводиться и с помощью чисто ге-
генетических методов с использованием специально сконструиро-
сконструированных векторных плазмид.
Позиционное клонирование. Метод позиционного клониро-
клонирования часто используется для выделения неизвестных генов с не-
неизвестным положением на генетической карте, присутствие ко-
которых можно определить только на основании характерного фе-
фенотипа организма, например, симптомов заболевания [239-241].
Для клонирования таких генов в больших выборках определяют
связь между передачей определенного молекулярного маркера
потомству и исследуемого фенотипа. В качестве таких молеку-
молекулярных маркеров часто используют характерный полиморфизм
длин рестрикционных фрагментов ДНК (ПДРФ-маркеры - мар-
маркеры полиморфизма по длинам рестрикционных фрагментов
ДНК - RFLP - restriction fragment length polymorphism) или длину
полиморфных микросателлитных локусов. Анализ проводится в
большом количестве семей, у которых наблюдается передача ис-
исследуемого фенотипа от родителей потомству. Если в ходе про-
проведения таких исследований достоверно устанавливается, что фе-
166
нотип с высокой частотой передается потомству вместе с кон-
конкретным молекулярным маркером, то делается вывод о располо-
расположении маркера и искомого гена, определяющего исследуемый
фенотипический признак, на одной хромосоме недалеко друг от
друга. Это дает возможность провести отбор клонов в клонотеке
последовательностей, составленной из крупных фрагментов
ДНК, с помощью зонда на выявленный полиморфный маркер. В
составе обнаруженного таким образом фрагмента с определен-
определенной вероятностью может оказаться и ген, определяющий появле-
появление исследуемого фенотипического признака (симптомов забо-
заболевания). Поскольку данный метод отбора основан исключи-
исключительно на определении положения искомого гена относительно
полиморфного маркера (в том числе и другого гена), он получил
название позиционного клонирования.
4.2.2. Повторный скрининг
Часто при первичном отборе клоны на чашках Петри вырас-
вырастают настолько плотно, что после гибридизации с зондом быва-
бывает трудно выделить с первого раза бактерии, относящиеся к од-
одному клону или фаговые частицы одной бляшки, которые дали
положительный сигнал во время проведения первого раунда от-
отбора. В этом случае бактериальные клетки или фаговые части-
частицы, выделенные из зоны сигнала, рассевают до отдельных коло-
колоний или бляшек и используют во втором раунде отбора. Как пра-
правило, с помощью такого простого подхода удается "расчистить"
нужные клоны и выделить их в гомогенном состоянии.
4.2.3. Субклонирование
При субклонировании вставку рекомбинантной ДНК, выде-
выделенную из вектора изолированного клона, переносят в новый
вектор, который дает возможность ее исследования в более про-
простых условиях и с затратой меньших усилий. Если вставка была
получена в фаговом, космидном векторе или с использованием
искусственной хромосомы, что предполагает ее большой исход-
исходный размер, то она может быть переклонирована по частям в
плазмидном векторе, и это может значительно облегчить ее
дальнейшее исследование, например, с помощью секвенирова-
ния.
167
Глава 5
Системы экспрессии рекомбинантных генов
Выделение любого нового рекомбинантного гена, как правило,
заканчивается попытками получения его полноценной экспрессии
в искусственных генетических системах, т.е. наработки в препара-
препаративном количестве полноценного в функциональном отношении
рекомбинантного белка. В настоящее время имеется множество си-
систем экспрессии рекомбинантных генов, среди которых наиболь-
наибольшее применение в биотехнологии находят модифицированные раз-
различным образом микроорганизмы. Поскольку такие системы не
позволяют решить многих проблем, связанных с экспрессией эука-
риотических рекомбинантных генов, в том числе, осуществления
посттрансляционных модификаций рекомбинантных белков, в ря-
ряде случаев для решения этих проблем используют культивируемые
клетки животных и растений. Для изучения механизмов экспрессии
генов в лабораторных условиях большую пользу приносят так на-
называемые пермеабилизованные культивируемые клетки с полу-
полупроницаемыми мембранами, что можно сделать в определенных
условиях. Кроме того, незаменимыми помощниками молекуляр-
молекулярных биологов и генетиков остаются бесклеточные белоксинтези-
рующие системы, один из вариантов которых, а именно проточ-
проточные системы, находит применение и в промышленном производст-
производстве пептидов. Принципы, лежащие в основе использования таких си-
систем, будут кратко рассмотрены ниже.
5.1. Экспрессирующие системы микроорганизмов
Приемы конструирования векторов, экспрессирующих ре-
комбинантные гены в бактериальных клетках, а также основные
проблемы, возникающие при попытках экспрессии чужеродных
генов в бактериях, были уже кратко рассмотрены в разделе 3.6.
Здесь же мы проиллюстрируем этот материал некоторыми со-
современными достижениями в разработке бактериальных систем
экспрессии.
5.7.7. Грамотрицательные бактерии
Escherichia coli. Кишечная палочка Е. coli, несомненно, явля-
является одним из наиболее изученных биологических объектов, ис-
используемых в биотехнологии для получения рекомбинантных
белков. В том случае, если для конкретного белка удается подо-
168
Таблица 7
Примеры использования клеток Е. coli для промышленного про-
производства рекомбинантных белков
Область применения
Примеры использования
рекомбинантных белков
Преимущества систем
экспрессии и очистки
Антигены
Антитела и их
фрагменты
Ферменты, в том
числе, с улучшен-
улучшенными свойствами
Структурные ис-
исследования белков
Лекарственные
средства
Получение поликлональных ан-
антител [242]
Получение рекомбинантных
одноцепочечных антител и их
фрагментов (см. П.3.4.2)
Получение катехолдиоксигеназ
для устранения хлорсодержа-
щих загрязнений окружающей
среды [243], разнообразные
ферменты для научных иссле-
исследований
Получение меченых белков для
ЯМР-спектроскопии [244]
Производство рекомбинантного
инсулина, интерферона и т.д.
[245]
Возможность масш-
масштабирования, отсутст-
отсутствие эндотоксинов
Возможность
масштабирования
Правильный фол-
динг, возможность
масштабирования
Правильный фолдинг
Высокая степень
очистки, отсутствие
эндотоксинов
брать адекватную систему вектор-хозяин, то его получение в
препаративном количестве, в том числе, и крупномасштабное
производство становится недорогим, быстрым и легко масштаби-
масштабируемым процессом. В табл. 7 приведены некоторые примеры ис-
использования Е. coli в современной биотехнологии для производ-
производства рекомбинантных белков.
Pseudomonas aeruginosa. Как уже упоминалось, экспрессия
чужеродных белков в стандартном хозяине, Е. coli, чаще всего
сопровождается образованием телец включения, из которых
выделение нативного белка может представлять серьезную
проблему. Данную проблему позволяют решать системы экс-
экспрессии рекомбинантных генов, разработанные на основе гра-
мотрицательной бактерии P. aeruginosa, обеспечивающих сек-
секрецию синтезирующихся гетерологичных белков в питатель-
питательную среду [246]. Эта бактерия осуществляет биосинтез и секре-
тирует большое количество внеклеточных ферментов, в том
числе, и различные гидролазы, которые представляют интерес
для биотехнологии. Для фолдинга и секреции таких ферментов
у P. aeruginosa имеются высокоэффективные, специфические
169
механизмы. У этой бактерии известно, по крайней мере, пять
путей секреции, в реализации которых задействовано от трех до
двенадцати различных белков [247]. Кроме того, несколько спе-
специальных дисульфидоксидоредуктаз и протеиназ участвуют в
фолдинге секретируемых белков в периплазматическом прост-
пространстве [248, 249]. Все эти особенности P. aeruginosa были ис-
использованы для создания высокоэффективных штаммов-проду-
штаммов-продуцентов гетерологичных рекомбинантных белков.
Штамм PABST7.1 P. aeruginosa эффективно используется для
сверхэкспрессии рекомбинантных генов, транскрипция которых
осуществляется с сильного Т7-промотора, успешно используемо-
используемого для тех же целей в клетках Е. coli. Ген Т7-РНК-полимеразы,
осуществляющей транскрипцию с этого промотора, был клони-
клонирован под индуцируемым промотором, который контролируется
Lac-репрессором, кодируемым геном lacl% расположенным сразу
за геном Т7-РНК-полимеразы. Оба этих гена стабильно интегри-
интегрировали в хромосому штамма P. aeruginosa PABS1, дефектного по
биосинтезу липазы, превратив бактериальные клетки в сверхпро-
сверхпродуцента Т7-РНК-полимеразы. Одновременно был получен штамм
P. aeruginosa PABST7.7, у которого ген Т7-РНК-полимеразы был
интегрирован непосредственно в структурную часть гена липазы
Ир А. Оба штамма оказались пригодными для сверхэкспрессии му-
тантных генов липазы. Такой подход позволяет внедрять ген Т7-
РНК-полимеразы в любой хромосомный ген P. aeruginosa, что со-
сопровождается его инактивацией на фоне сверхэкспрессии Т7-
РНК-полимеразы, и позволяет следить за появлением активности
клонируемого гомологичного рекомбинантного гена.
Вторую часть обсуждаемой системы экспрессии составляет
плазмида pUCPL6AN, в которой липазный оперон UpAIH клони-
клонирован под контролем Т7-промотора. Плазмида содержит уни-
уникальные сайты рестрикции Nhel и Apal, локализованные в 5'- и 3'-
концевых частях гена НрА, что позволяет заменять этот ген на
любой клонируемый. При этом не затрагиваются сигнальная по-
последовательность и 3'-концевые регуляторные последовательно-
последовательности исходного гена, которые необходимы для специфического
распознавания белка фолдазой, специфичной в отношении липа-
липазы. Такая система позволяет осуществлять сверхэкспрессию и
скрининг больших клонотек мутантных генов в направленной
эволюции белковых молекул непосредственно на основании фер-
ферментативной активности секретируемых мутантных белков, а
также нарабатывать нативные рекомбинантные белки в препа-
препаративном количестве.
170
5.1.2. Клетки дрожжей как экспрессирующие системы
Наряду с бактериальными системами, клетки дрожжей
Saccharomyces cerevisiae или Schizosaccharomyces pombe широко ис-
используются для экспрессии рекомбинантных генов и позволяют по-
получать большое количество рекомбинантных белков с высоким
выходом. Однако, в отличие от бактериальных клеток, клеточные
стенки которых содержат опасные для здоровья токсичные и пиро-
генные компоненты, дрожжи лишены такого недостатка. Это де-
делает дрожжевые системы экспрессии предпочтительными при про-
производстве лекарственных и пищевых рекомбинантных продуктов.
Кроме того, дрожжи, как эукариоты, обладают развитой системой
эндоплазматического ретикулума и аппарата Гольджи, а, следова-
следовательно, способны к осуществлению посттрансляционных модифи-
модификаций рекомбинантных белков. Как и в случае любых других сис-
систем экспрессии рекомбинантных генов, для того чтобы дрожжевая
система функционировала, необходимо ввести рекомбинантные ге-
гены в клетки в составе стабильного экспрессирующего вектора, и
обеспечить эффективную транскрипцию и трансляцию рекомби-
рекомбинантных нуклеотидных последовательностей.
Типы дрожжевых векторов и их характерные особенности.
При создании систем экспрессии рекомбинантных генов на осно-
основе дрожжей в настоящее время используются четыре типа векто-
векторов, которые различаются по способу, которым они обеспечива-
обеспечивают существование чужеродной ДНК в дрожжевых клетках [250,
251]. Векторы первого типа, к которым относятся, в частности,
векторы на основе дрожжевой интегрирующейся плазмиды Yip
(yeast integrating plasmid), используются для встраивания рекомби-
нантной ДНК в хромосомы дрожжей. У этих векторов отсутству-
отсутствует область начала репликации и они не способны самовоспроиз-
самовоспроизводиться в трансформированных клетках. Поэтому единствен-
единственным способом сохранения их последовательностей в клетках
дрожжей является совместная репликация в составе хромосом.
Второй тип векторов принадлежит к семейству дрожжевых
реплицирующихся плазмид YRp (yeast replicating plasmid). Эти
плазмиды способны к автономной репликации в клетках дрож-
дрожжей благодаря наличию у них ARS-последовательностей
(autonomously replicating sequences), о которых уже упоминалось в
связи с искусственными хромосомами дрожжей (YAC). ARS-no-
следовательности, по-видимому, представляют собой области на-
начала репликации на хромосомах дрожжей. Векторы третьего
шипа образуют так называемое семейство дрожжевых центро-
мерных плазмид YCp (yeast centromere plasmid), которые, как и
171
следует из их обозначения, содержат в своем составе цетромер-
ные последовательности хромосом дрожжей и обладают способ-
способностью не только к автономной репликации, но и более стабиль-
стабильному существованию в клетках дрожжей в виде внехромосомных
элементов по сравнению с векторами предыдущего семейства.
Наконец, векторы семейства дрожжевых эписомных плазмид
YEp (yeast episomal plasmid) получены из природной многокопий-
ной кольцевой плазмиды дрожжей B }im), и объединяют в себе
большинство положительных качеств вышеупомянутых векто-
векторов. Они существуют в клетках дрожжей во внехромосомном со-
состоянии в большом числе копий, автономно реплицируются и
стабильно сегрегируют между делящимися клетками [252].
Все перечисленные векторы являются челночными, т.е. допус-
допускают проведение этапов субклонирования фрагментов ДНК и на-
наработки их в большом количестве с использованием клеток Е. coli.
Это становится возможным благодаря наличию у них области на-
начала репликации плазмиды pBR322, а также селектируемых марке-
маркеров в виде генов Ыа и ш, придающих бактериальным клеткам ус-
устойчивость к ампициллину и тетрациклину, соответственно. Селек-
Селективный характер роста клеток дрожжей при наличии у них вектор-
векторных молекул также обеспечивают селектируемые маркеры век-
векторных плазмид. К ним относятся гены, дающие возможность де-
деления ауксотрофных штаммов дрожжей на средах без соответству-
соответствующих пищевых добавок: URA3, LEY2, HIS3, TRP1 или LYS2, кото-
которые восстанавливают способность ауксотрофов синтезировать, со-
соответственно, урацил, а также аминокислоты Leu, His, Trp или Lys.
Использование гена URA3 в качестве контр-селективного маркера
рассмотрено в разделе о дрожжевых дигибридных системах оценки
белок-белковых взаимодействий во второй части книги.
Эффективную транскрипцию рекомбинантных генов в клет-
клетках дрожжей обеспечивают специфические регуляторные элемен-
элементы, промоторы и терминаторы транскрипции. Высокий уровень
синтеза РНК на клонированных последовательностях в клетках
S. cerevisiae может иметь место при использовании промоторных
участков генов гликолиза: GALA, ADH, PGK или GAPDH. При этом
для исключения отрицательного влияния рекомбинантных белков
на дрожжевые клетки промоторы должны быть индуцируемыми.
Фрагменты ДНК, выполняющие роль терминаторов транскрип-
транскрипции часто берут начало от 3' -концевых последовательностей ге-
генов PGK, PHO5 или TRP1. При этом последовательности 3'-конце-
3'-концевых нетранслируемых участков генов могут оказывать большое
влияние на эффективность транскрипции и трансляции клониро-
клонированных рекомбинантных последовательностей.
172
Соблюдение условий, необходимых для транскрипции и транс-
трансляции клонированных генов, как и в случае бактериальных систем
экспрессии, еще не гарантирует высокий уровень синтеза кодируе-
кодируемого этим геном белкового продукта. Многие белки (и мРНК) не-
нестабильны в клетках дрожжей. Так, проинсулин человека крайне
нестабилен в дрожжевых системах экспрессии (это не относится к
рекомбинантному интерферону а2). На эффективность экспрессии
рекомбинантных генов в клетках дрожжей оказывает влияние
множество до конца не изученных факторов, и оптимизация этого
процесса при решении конкретных биотехнологических задач в
каждом случае требует проведения специальных исследований.
Введение векторной ДНК в клетки дрожжей может быть осу-
осуществлено несколькими способами: в сферопласты в присутст-
присутствии ионов Са2+ и полиэтиленгликоля, которые, в свою очередь,
получают разрушением клеточной стенки с помощью фермен-
ферментов. Кроме того, используются методы, основанные на примене-
применении ацетата лития или электропорации [253, 254].
Использование дрожжевых систем экспрессии в биотехнологии
и научных исследованиях. Крупномасштабный синтез рекомби-
рекомбинантных белков является одной из основных целей, для достижения
которой конструируются системы экспрессии рекомбинантных ге-
генов в клетках дрожжей [255]. Синтезируемые рекомбинантные
белки могут накапливаться в цитоплазме клеток, после чего их вы-
выделяют по традиционной схеме с использованием многостадийной
очистки из бесклеточных экстрактов, получаемых после разруше-
разрушения клеток. Популярными также являются системы, обеспечиваю-
обеспечивающие секрецию рекомбинантных белков в культуральную среду. В
этом случае секрецию можно обеспечить, например, включением в
рекомбинантный белок аминокислотной лидерной последователь-
последовательности препро-а-фактора S. cerevisiae, которая направляет синтези-
синтезируемые молекулы в секреторные гранулы [256].
Метилотрофные дрожжи Pichiapastoris. С 1984 г., когда ме-
тилотрофные дрожжи впервые начали использовать в биотехно-
биотехнологии, с помощью этих микроорганизмов было получено более
300 рекомбинантных белков. Популярность этого вида дрожжей
связана прежде всего с тем, что в их системах экспрессии можно
использовать один из самых сильных регулируемых промоторов
гена алкогольоксидазы (AOXI), что обеспечивает высокий уро-
уровень экспрессии рекомбинантных генов. Промотор репрессиру-
репрессируется глюкозой или другими источниками углерода и активирует-
активируется в 1000 раз после переноса клеток на среду с метанолом в каче-
качестве единственного источника углерода. Кроме того, имеются
конститутивный промотор гена GAP, а также промотор гена
173
FLDI, кодирующего глютатион-зависимую формальдегиддегид-
рогеназу, который индуцируется метанолом и метиламином (не-
(нетоксичным источником азота). Кроме того, векторные плазмиды
этого микроорганизма допускают стабильную интеграцию ре-
рекомбинантных генов в строго определенные участки дрожжево-
дрожжевого генома в виде одной или нескольких копий. При этом сами
клетки могут культивироваться в ферментерах с высоким выхо-
выходом. Немаловажным обстоятельством является и доступность
дрожжевых штаммов в виде наборов для клонирования, которые
продаются фирмой Invitrogen (США).
На основе Saccharomyces cerevisiae созданы высокоэффектив-
высокоэффективные системы экспрессии рекомбинантных генов и крупнотон-
крупнотоннажного синтеза рекомбинантных белков человека, животных и
растений, в том числе и рекомбинантных антител (см. раз-
раздел И.3.4) [257]. Кроме того, экспрессия рекомбинантных белков
в этих клетках активно используется для улучшения свойств бел-
белков методами направленной эволюции с использованием клеточ-
клеточного дисплея (раздел П.2.3.2), изучения белок-белковых взаимо-
взаимодействий в дрожжевых л-гибридных системах (раздел П.2.3.4) и
для множества других не менее важных целей.
5.2. Системы экспрессии, основанные
на культуре клеток животных
Только на первый взгляд экспрессия рекомбинантных генов в
бактериальных и дрожжевых клетках под контролем хорошо
изученных регуляторных элементов представляется простой за-
задачей. На практике, как уже обсуждалось ранее, экспрессия эука-
риотических генов в бактериях происходит неэффективно из-за
образования нерастворимых телец включения и отсутствия необ-
необходимых посттрансляционных модификаций рекомбинантных
полипептидных цепей. Для преодоления этих и некоторых других
затруднений в последнее время широко используются культиви-
культивируемые клетки животных и растений.
Эффективный синтез рекомбинантных белков зависит не
только от используемых клеточных линий. Основное влияние на
этот процесс оказывают конструкция экспрессирующего векто-
вектора, а также метод введения рекомбинантных ДНК в эукариоти-
ческие клетки. В последнем случае низкая эффективность до-
доставки рекомбинантных молекул в клетки и неоптимальная ло-
локализация сайтов их интеграции в геном могут свести на нет все
достоинства клеточных линий и экспрессирующих векторов. Для
повышения уровня экспрессии рекомбинантных генов, стабиль-
174
но интегрированных в геном культивируемых клеток, разработа-
разработаны методы их селективной амплификации. С использованием од-
одного из них получены мутантные сублинии клеток яичников ки-
китайских хомячков СНО, в которых амплификация происходит в
присутствии селектирующего агента, например, метотрексата.
Другие системы амплификации основаны на подавлении экспрес-
экспрессии жизненно важных ферментов, например, глутаминсинтетазы
или аденозиндезаминазы, под действием специфических ингиби-
ингибиторов: метионинсульфоксимина или 2'-дезоксикоформицина, со-
соответственно. Клеточные линии и клоны, стабильно продуциру-
продуцирующие рекомбинантные белки, как правило, получают путем от-
отбора из пула гетерогенных клеток. И, наконец, еще одним суще-
существенным условием успешной экспрессии рекомбинантных генов
является оптимизация самого процесса культивирования клеток,
включая тщательный подбор культуральных сред.
Для получения небольшого количества рекомбинантных бел-
белков и контроля функциональной целостности генно-инженерных
конструкций кроме вышеупомянутого подхода часто применяют
временно экспрессирующиеся векторы, которые обладают способ-
способностью к репликации в культивируемых клетках COS во внехромо-
сомном состоянии. В этом случае источником рекомбинантных
белков являются супернатанты клеток. При другом подходе реком-
рекомбинантные белки синтезируют в культивируемых клетках насеко-
насекомых после их заражения векторами на основе генома бакуловиру-
сов. Последний подход становится все более популярным, так как
допускает стабильную экспрессию многих рекомбинантных генов,
продукты которых могут находиться как внутри клеток, так и в ас-
ассоциированном с поверхностью клеток состоянии, или они могут
секретироваться в культуральную жидкость.
Недавно швейцарскими исследователями было проведено
сравнительное изучение эффективности экспрессии гена цитоки-
на человека - фактора, игнибирующего леккозы (leukemia
inhibitory factor - huLIF) в разных эукариотических системах
[257а]. Фактор huLIF представляет собой белок среднего разме-
размера, полипептидная цепь которого содержит несколько потенци-
потенциальных сайтов N-гликозилирования и в процессе фолдинга обра-
образует три дисульфидные связи. Поскольку полученные результа-
результаты представляют большой практический интерес для генной ин-
инженерии и биотехнологии, так как иллюстрируют многие основ-
основные принципы функционирования рекомбинантных генов в гете-
рологичных системах экспрессии, они будут кратко рассмотрены
ниже.
175
5.2.1. Клетки яичников китайских хомячков (линия СНО)
Эта линия клеток и ее многочисленные производные часто
используются для синтеза рекомбинантных белков после пред-
предварительной эндогенной амплификации соответствующих ре-
рекомбинантных генов, введенных в клетки с помощью трансфек-
трансфекции. Спонтанная амплификация генов в нормальных клетках
животных является редким событием, а в трансформированных
(опухолевых) клетках она происходит с частотой Ю-4-Ю-6 за
клеточную генерацию. Частота амплификации определенных
генов может быть повышена в результате некоторых экзоген-
экзогенных воздействий, включая инкубацию клеток с гидроксимоче-
виной, афидиколином, канцерогенами, а также под действием
ультрафиолетового или у-излучения и в условиях гипоксии.
Амплификация гена обычно начинается с его вырезания из хро-
хромосомы в составе протяженного сегмента ДНК, сопровождае-
сопровождаемого образованием кольцевой структуры, которая продолжает
существовать в виде внехромосомного элемента (двойной мини-
хромосомы (double-minutes - DM)), способного к автономной
репликации. Предполагают, что DM является промежуточным
продуктом амплификации, образующимся в условиях низкого
давления селектирующего фактора окружающей среды. На бо-
более поздних стадиях амплифицирующаяся ДНК после повтор-
повторной интеграции в геном обнаруживается преимущественно в со-
составе удлиненного участка хромосомы (extended chromosomal
region - ECR), иначе называемого гомогенно окрашивающейся
областью (homogeneously staining region - HSR). На этой стадии
амплифицированный участок генома клеток, а следовательно, и
их фенотип становятся более стабильными.
Для биотехнологических целей в системе амплификации ре-
рекомбинантных генов в качестве селектируемого маркера часто ис-
используют ген дигидрофолатредуктазы (ДГФР), интегрированный
в экспрессирующий вектор. ДГФР катализирует превращение фо-
лиевой кислоты в тетрагидрофолат, необходимый для синтеза эу-
кариотическими клетками глицина, тимидинмонофосфата и пури-
новых оснований. В этой связи клетки СНО, в которых ген ДГФР
инактивирован под действием мутации, не растут на средах без
нуклеозидов и приобретают эту способность после трансфекции
геном ДГФР. Растущие трансфектанты далее отбираются по при-
признаку амплификации гена ДГФР на фоне увеличивающихся кон-
концентраций метотрексата - ингибитора ДГФР в питательной среде,
так как увеличение числа копий гена будет придавать клеткам ус-
устойчивость к ингибитору в больших концентрациях. После прове-
176
SV40/AdMLP
huLIF
ДГФР pA VA
amp
IgL huLIF
IgH
pA
neo
amp
в
1
huLIF
pA amp
Рис. 17. Кассеты генов в экспрессирующих векторах, использованные для
получения фактора, ингибирующего лейкозы, в культурах клеток [257а]
а - кДНК huLIF (черные стрелки), включающая лидерные последователь-
последовательности (незаштрихованные прямоугольники), была интегрирована в экспресси-
экспрессирующии вектор рХМТЗ под контроль гибридного промотора, расположенного
между точкой начала репликации вируса SV40 и основным поздним промото-
промотором аденовируса (SV40/AdMLP), ДГФР, amp, neo - гены дигидрофолатредукта-
зы и устойчивости к ампициллину и неомицину соответственно, VA - трансля-
трансляционный энхансер; рА - сайт полиаденилирования;
б - вектор pSV2neo-Ig-huLIF, сконструированный на основе транскрипци-
транскрипционных и трансляционных регуляторных элементов генов тяжелой (IgH) и лег-
легкой (IgL) цепей иммуноглобулинов. Транскрипция кассеты контролируется мы-
мышиным промотором гена IgL к и энхансером гена IgH. Транскрипт, содержащий
последовательность huLIF, в результате сплайсинга объединяется с экзоном 2
гена IgL, кодирующего лидерную последовательность, которая обеспечивает
секрецию зрелого huLIF клетками;
в - вектор pGES-LIF; прямоугольники 1-4 - регуляторная область кластера
глобиновых генов, короткая черная стрелка - промотор C-глобинового гена,
ломаной линией отмечены последовательности, удаляемые сплайсингом. Во
всех случаях стрелки указывают направление транскрипции
дения множественных раундов селекции получают популяцию
клеток, содержащих до нескольких сотен копий гена ДГФР.
Размер амплифицированного геномного локуса значительно
больше размера селектируемого гена и может составлять не-
несколько сотен тысяч пар оснований. Трансфекция клеток геном
ДГФР и исследуемым рекомбинантным геном, заключенными в
один экспрессирующии вектор, сопровождаемая их интеграцией в
177
геном, будет приводить к их совместной амплификации на фоне
метотрексата и усилению синтеза требуемого рекомбинантного
белка, как следствие увеличения дозы его гена. Данные о возмож-
возможности сохранения высокого уровня экспрессии гена в отсутствие
селектирующего агента, т.е. генетической стабильности проду-
продуцентов, противоречивы. Результат зависит от генотипа конкрет-
конкретных клонов клеток, в том числе и локализации сайта интеграции
экспрессирующего вектора в геном клеток СНО. Эффективным
экспрессирующим вектором, используемым в описываемой систе-
системе, является двухцистронная молекула ДНК, в которой исследуе-
исследуемый рекомбинантный ген находится под контролем сильного про-
промотора перед геном ДГФР, что обеспечивает высокое сопряже-
сопряжение синтеза рекомбинантного белка с уровнем устойчивости кле-
клеток к метотрексату (рис. 17, а).
5.2.2. Клетки мышиной миеломы (линия SP2/0)
Клетки миеломы находят широкое применение для получения
гибридом - линий клеток, производящих моноклональные антите-
антитела определенной специфичности. Поскольку эти клетки осуществ-
осуществляют эффективную секрецию рекомбинантных белков, они хоро-
хорошо изучены в отношении экспрессии в них соответствующих ре-
рекомбинантных генов, введенных с помощью трансфекции. Кроме
того, такие клетки способны расти в суспензионной культуре (без
прикрепления к поверхности субстрата), что облегчает их культи-
культивирование при необходимости крупномасштабной наработки.
Линия клеток мышиной миеломы Sp2/0 Agl4, утративших
способность синтезировать или секретировать иммуноглобулины,
часто используется для производства рекомбинантных белков.
Клетки этой линии легко трансфецируемы рекомбинантными
ДНК и могут выращиваться в больших количествах на питатель-
питательной среде без дорогостоящей сыворотки. Клетки Sp2/0 экспресси-
руют эндогенный ген ДГФР, что делает невозможным их прямое
применение для амплификации рекомбинантных ДНК с исполь-
использованием метотрексата. Это затруднение обычно преодолевают
путем введения в экспрессирующий вектор мутантного гена
ДГФР, в котором точковая мутация, сопровождаемая заменой
Т —> G, приводит к замещению Leu —> Arg в положении 22 поли-
полипептидной цепи ДГФР, следствием чего является понижение срод-
сродства фермента к метотрексату в 270 раз. Благодаря этому после
трансфекции может быть проведен отбор клеток, содержащих
мутантный ген ДГФР вместо нормального, путем постепенного
увеличения концентрации метотрексата в среде. При умеренных
178
концентрациях ингибитора эндогенный ген ДГФР полностью
инактивируется, тогда как мутантный ген остается активным и
может быть амплифицирован в результате описанного выше ме-
механизма после повышения концентрации метотрексата в среде.
Однако уровень амплификации гена ДГФР и ассоциированного с
ним рекомбинантного гена в такой системе, как правило, ниже,
чем в обычной, так как меньшее число внутриклеточных копий
мутантного гена требуется для обеспечения необходимого уровня
устойчивости клеток к ингибитору. Кроме того, было показано,
что высокий уровень внутриклеточной экспрессии рекомбинант-
ных генов, введенных в клетки Sp2/0 с помощью трансфекции,
может обеспечиваться после инкубации с метотрексатом за счет
увеличения скорости их транскрипции, а не в результате их амп-
амплификации. Такой результат может быть получен в результате
введения множественных копий гена ДГФР при трансфекции.
Структура одного из векторов, пригодного для использования с
миеломными клетками Sp2/0, представлена на рис. 17, б. Транс-
Транскрипция трансгенов в этом векторе обеспечивается промоторно-
энхансерными элементами гена иммуноглобулина с участием эн-
эндогенных регуляторных факторов клеток миеломы.
5.2.3. Клетки селезенки мышей (линия MEL)
Обе системы экспрессии, описанные выше, базируются на
амплификации трансгенов, обеспечивающей высокий уровень
внутриклеточного синтеза кодируемых ими рекомбинантных
белков в отобранных клонах клеток. Тем не менее, в природе
уникальные гены (т.е. присутствующие в виде одной копии на
гаплоидный геном) могут экспрессироваться с очень высокой
эффективностью. Примером этого являются, например, имму-
иммуноглобулины, секретируемые плазматическими клетками, или
глобины, синтезируемые в больших количествах клетками эри-
троидного ряда. Высокий уровень транскрипции глобинового
локуса становится возможным не только благодаря функциони-
функционированию эффективных промотора и энхансера, но и в результа-
результате присутствия так называемой доминантной регуляторной
области (dominant control region - DCR), изменяющей простран-
пространственную структуру хроматина всего локуса. DCR расположена
за ~50 т.п.о. перед кластером глобиновых генов и вначале была
идентифицирована благодаря присутствию в ней нескольких
сайтов, гиперчувствительных к ДНКазе. Путем комбинирова-
комбинирования последовательностей нуклеотидов, окружающих сайты,
удалось сконструировать небольшую искусственную DCR, об-
179
ладающую всеми основными свойствами исходной. Рекомби-
нантные гены, объединенные в векторе с такой DCR, экспрес-
сировались с высокой эффективностью в клетках эритроидно-
го ряда независимо от места их интеграции в геном и не требо-
требовали для этого предварительной амплификации. Таким обра-
образом, рассматриваемая система позволяет избежать отрицатель-
отрицательного влияния эффекта положения у рекомбинантных генов, что
избавляет от необходимости скрининга большого числа транс-
фецированных клеток для получения высокопродуктивных
продуцентов рекомбинантных белков.
Для биотехнологических целей в настоящее время часто ис-
используется линия клеток MEL-E9, которая является производной
линии клеток MEL-NP5, полученных из селезенки мышей NFS, за-
зараженных дефектным по репликации вирусом, образующим фоку-
фокусы селезенки (spleen focus forming virus - SSFV). Эти клетки не об-
образуют вирусных частиц, но вызывают возникновение опухолей
после инъекции сингенным мышам. Они не претерпевают спон-
спонтанной дифференцировки, однако, под действием экзогенных
факторов (диметилсульфоксида, гемина или гексаметиленбисаце-
тамида) дифференцируются в клетки, содержащие гемоглобин.
На рис. 17, в изображена схема экспрессирующего вектора,
применяемого с этой линией клеток. В нем область DCR соеди-
соединена с промотором, экзоном 2, интроном 2 и экзоном 3 C-глоби-
нового гена, поскольку показано, что присутствие последова-
последовательностей нуклеотидов интрона 2 абсолютно необходимо для
эффективной экспрессии генов, направляемой этим промотором.
Рекомбинантный ген может быть встроен справа или слева от
интрона 2. После успешной трансфекции этот экспрессирующий
вектор обеспечивает независимую от эффекта положения и чис-
числа копий тканеспецифическую экспрессию рекомбинантных ге-
генов в клетках эритроидного ряда.
5.2.4. Клетки африканской зеленой мартышки (линия COS)
Получение временной экспрессии генов в клетках COS часто
используется для быстрой наработки рекомбинантных белков и
ДНК. При конструировании клеток COS клетки зеленой мартыш-
мартышки CV-1 были трансформированы вирусом SV40 с дефектной об-
областью начала репликации. В результате были получены три ли-
линии клеток (COS-1, 2 и 7), конститутивно экспрессирующих боль-
большой Т-антиген вируса. После проведения трансфекции клеток экс-
прессирующими плазмидами, содержащими область начала репли-
репликации вируса SV40, последняя эффективно взаимодействует с эн-
180
догенным Т-антигеном, что сопровождается внехромосомной реп-
репликацией плазмиды с образованием большого числа ее копий в ци-
цитоплазме клеток. Если рекомбинантный ген в такой плазмиде на-
находится под контролем подходящего промотора, то это приводит к
внутриклеточному образованию рекомбинантных белков в боль-
больших количествах.
Число копий плазмид, введенных в клетки COS, достигает
максимума через ~48 ч после трансфекции. Далее внутриклеточ-
внутриклеточный уровень плазмидной ДНК начинает постепенно снижаться
из-за ее цитопатического действия и гибели клеток. В результа-
результате системы, основанные на клетках COS, не могут быть исполь-
использованы для крупномасштабной наработки рекомбинантных бел-
белков в течение длительного времени. Максимальное внутрикле-
внутриклеточное содержание рекомбинантных белков в этих системах на-
наблюдают через 72 ч после трансфекции, и их синтез продолжает-
продолжается в течение последующих 5-10 дней на фоне медленного сниже-
снижения количества клеток в культуре, что позволяет все же исполь-
использовать клетки COS для препаративного синтеза рекомбинантных
белков. Для этого одновременно трансфецируют до 108 клеток,
выращивают их на роллерах или микроносителях, проводя мно-
многократный сбор культуральной жидкости. Такой подход позволя-
позволяет получать до нескольких миллиграммов рекомбинантного бел-
белка из вышеописанного пула трансформированных клеток.
Эффективность системы экспрессии, основанной на клетках
COS, зависит, в первую очередь, от природы рекомбинантного
белка, которая определяет его чувствительность к внутрикле-
внутриклеточным протеиназам, а также метода трансфекции, соотношения
числа клеток и молекул рекомбинантных плазмид, используемых
для трансфекции, и состава питательной среды. Пример экспрес-
сирующей кассеты генов, используемой в этой системе, представ-
представлен на рис. 17, а. Такой фрагмент ДНК может быть интегрирован
в подходящую коммерческую плазмиду, в частности, в экспресси-
рующий вектор рХМТЗ.
5.2.5. Клетки насекомых, зараженные бакуловирусами
Многочисленное семейство бакуловирусов, размножающих-
размножающихся в клетках беспозвоночных, обладает геномом в виде двухцепо-
чечной кольцевой ковалентно замкнутой ДНК длиной в
80-220 т.п.о. Круг хозяев бакуловирусов ограничен, главным об-
образом, членистоногими. Клетки млекопитающих могут быть за-
заражены бакуловирусами только при высокой множественности
инфекции. При этом вирус проникает в клетки, но в них не вос-
181
производится, не экспрессирует рекомбинантные гены с промо-
промоторов насекомых и не способен находиться (персистировать) в
клетках животных длительное время.
Интерес к бакуловирусам как потенциальным переносчикам
рекомбинантных генов и системам, обеспечивающим их экспрес-
экспрессию, возник после обнаружения их способности к синтезу боль-
большого количества вирусных белков на поздних стадиях инфекции.
В частности, это касается вирусных полиэдрина и белка рЮ, ге-
гены которых находятся под контролем сильных поздних промото-
промоторов и которые могут быть удалены без ущерба для размножения
вирусов. Поэтому замещение указанных генов рекомбинантны-
ми сопровождается усиленной экспрессией последних.
Первоначально использование бакуловирусов с культурами
клеток вызывало большие затруднения из-за экзотичности объ-
объекта исследования. Однако с ростом интереса к этим системам
экспрессии были разработаны многочисленные варианты реком-
рекомбинантных вирусов и векторов на их основе, включая челночные
векторы для клеток дрожжей и бактерий. Кроме того, успех этой
системы связан с получением удобных линий клеток насекомых.
В настоящее время наиболее часто используемыми клеточными
линиями остаются IPLB-Sf-21-AE (получена из Spodoptera
frugiperda), ее клональная производная Sf9, а также BTI-TN-
5В1-4 и TN368 из Trichopsula ni. Современные клеточные линии
насекомых выращивают в виде суспензионных культур на ролле-
роллерах и в ферментерах на питательных средах, не содержащих фе-
тальной сыворотки.
Для проявления в полной мере биологической активности
многих рекомбинантных белков требуются посттрансляционные
модификации их полипептидных цепей. Оказалось, что клетки
насекомых обладают способностью производить большое число
таких модификаций, включая гликозилирование, фосфорилиро-
вание, ацилирование остатками жирных кислот и амидирование.
Рекомбинантные белки могут претерпевать в них адекватный
протеолитический процессинг путем удаления сигнальных после-
последовательностей аминокислот и переноситься в соответствующие
клеточные компартменты. Были проведены интенсивные иссле-
исследования механизмов N-гликозилирования в клетках насекомых,
которые показали, что хотя оно и происходит, но не соответству-
соответствует в полной мере таковому клеток млекопитающих.
182
5.25. Сравнение эффективности
рассмотренных систем экспрессии
Проведено сравнение эффективности рассмотренных выше
систем экспрессии эукариотических рекомбинантных генов с
использованием гена huLIF в качестве модели. In vivo этот бе-
белок с молекулярной массой 32-67 кДа в зависимости от источ-
источника получения синтезируется под контролем уникального ге-
гена, содержащего три экзона. Фрагмент кДНК, кодирующий
зрелый процессированный huLIF, клонировали в составе трех
векторов, структура которых изображена на рис. 17, a-e. Для
трансфекции клеток COS и СНО использовали одну и ту же кон-
конструкцию (см. рис. 17, а). Оказалось, что все пять исследован-
исследованных линий клеток обладали способностью к синтезу рекомби-
нантного huLIF, обладающего биологической активностью. В
клетках линий СНО, Sp2/0 и MEL после проведения электрофо-
электрофореза продукт обнаруживали в виде отчетливой, хотя и широкой
полосы. В то же время рекомбинантный белок в клетках, зара-
зараженных бакуловирусами, был представлен дискретными фрак-
фракциями меньшей молекулярной массы, что указывало на его не-
неполное гликозилирование. Рекомбинантный материал в клет-
клетках COS был представлен дисперсной фракцией с молекулярной
массой 20—40 кДа, что интерпретировалось в пользу ограничен-
ограниченной способности этих клеток осуществлять процессинг сильно
гликозилированных полипептидных цепей.
Стабильные клетки-продуценты, созданные на основе ампли-
амплификации рекомбинантных генов, позволяют получать рекомби-
нантные продукты в более высоких титрах по сравнению с клет-
клетками, адаптированными к "быстрой" кратковременной экспрес-
экспрессии рекомбинантных белков (клетки MEL или COS). Для получе-
получения высокого уровня экспрессии рекомбинантных белков во всех
обсуждаемых системах необходимо, прежде всего, учитывать
особенности структуры белка, который должен быть синтезиро-
синтезирован, и конструировать экспрессирующие векторы в соответствии
с требованиями, предъявляемыми теми или иными клетками. В
ряде случаев предварительные исследования такого рода бывает
удобнее провести в бесклеточных белоксинтезирующих систе-
системах, которые позволяют иногда получать даже препаративные
количества рекомбинантных белков.
183
5.3. Клетки с нарушенной проницаемостью
внешних мембран
Группа методов, основанная на использовании клеток с ис-
искусственно нарушенной проницаемостью внешних мембран для
биосинтеза белковых молекул (semi-permeabilised (SP) cell sys-
systems), по механизму действия и возможностям, которые они пре-
предоставляют исследователям, занимает промежуточное положе-
положение между системами трансляции живых клеток и бесклеточны-
бесклеточными белоксинтезирующими системами [258, 259]. Такие системы
позволяют реконструировать начальные этапы сборки и пост-
посттрансляционных модификаций белковых молекул, сопряженные
с их биосинтезом, а также активно влиять на эти процессы в ус-
условиях эксперимента. SP-Клетки также дают возможность ак-
активно исследовать секреторные пути белков в условиях, прибли-
приближающихся к таковым живых клеток.
Для получения полупроницаемых мембран клетки животных
выращивают в культуре и обрабатывают детергентом дигитони-
ном, повреждающим внешнюю мембрану в мягких условиях, что,
в конечном счете, позволяет выделять клетки, свободные от
компонентов цитозоля, но сохраняющие основные черты своей
внутренней архитектуры. В последующих опытах SP-клетки
можно добавлять в бесклеточные системы трансляции мРНК
(см. ниже) и наблюдать за прохождением синтезирующегося бел-
белка через внутриклеточные компартменты. Для получения SP-
клеток могут быть использованы любые клеточные линии, одна-
однако, каждая из них для достижения наилучших результатов требу-
требует проведения специальных этапов оптимизации. С использова-
использованием SP-клеток так же, как и бесклеточных систем, можно легко
исследовать влияние отдельных компонентов на прохождение
всего моделируемого внутриклеточного процесса в условиях,
максимально приближенных к нативным. Кроме того, добавле-
добавление к SP-клеткам реагентов, осуществляющих поперечные сшив-
сшивки, позволяет фиксировать синтезирующиеся белки в просветах
эндоплазматического ретикулума, что дает новые возможности
для локализации систем фолдинга и внутриклеточного транспор-
транспорта белков. Поскольку получение SP-клеток допускает использо-
использование любых клеточных линий, в том числе и мутантных, с их по-
помощью можно исследовать роль генетических факторов во всех
вышеупомянутых процессах.
184
5.4. Бесклеточные белоксинтезирующие системы
Среди искусственных систем биосинтеза белка важное место
занимают бесклеточные системы [260]. Любая бесклеточная сис-
система создается, прежде всего, для моделирования конкретных би-
биохимических процессов, происходящих в живом организме, и во
время функционирования воспроизводит некоторые существен-
существенные особенности жизнедеятельности клетки. В генной инженерии
бесклеточные белоксинтезирующие системы часто используются
для исследования кодирующего потенциала и механизмов экспрес-
экспрессии клонированных генов in vitro, а также на промежуточных эта-
этапах конструирования рекомбинантных генов для идентификации
мРНК или фрагментов ДНК по кодируемым белкам.
Большим преимуществом бесклеточных систем перед целыми
клетками является доступность их отдельных компонентов для
экспериментальных воздействий. Такие системы позволяют ис-
исследовать влияние различных экзогенных факторов на их функци-
функционирование (ионные условия, рН, ингибиторы и активаторы и
т.п.). Кроме того, в бесклеточной системе можно легко заменять
отдельные компоненты или непосредственно воздействовать на
них в изолированном состоянии и затем по реакции системы по-
познавать их функциональную значимость. Большинство результа-
результатов, полученных с помощью бесклеточных систем, невозможно
было бы иметь при использовании живых клеток, так как послед-
последние при нарушении гомеостаза нередко гибнут. При этом бывает
трудно определить, какой же компонент оказался критическим. К
сожалению, перечисленные достоинства бесклеточных систем од-
одновременно являются их слабым местом, поскольку после разру-
разрушения клеток безвозвратно исчезают те многочисленные взаимо-
взаимодействия между их компонентами, благодаря которым можно без
труда отличить живую клетку от бесклеточного экстракта.
Бесклеточные белоксинтезирующие системы представляют
собой одни из самых сложных и многокомпонентных систем in
vitro, используемых в биохимии. Это связано с необходимостью
воспроизведения в пробирке всех этапов биосинтеза белка,
включая транскрипцию, аминоацилирование тРНК и трансля-
трансляцию мРНК рибосомами. Тем не менее, уже более 30 лет удается
успешно осуществлять процесс биосинтеза белка in vitro и ис-
использовать такие системы по трем основным направлениям: для
анализа кодирующего потенциала нуклеиновых кислот, исследо-
исследования механизмов биосинтеза белка и препаративной наработки
некоторых рекомбинантных белков и пептидов.
По природе компонентов, которые определяют способность
185
бесклеточных систем осуществлять трансляцию определенных
мРНК, принято различать прокариотические и эукариотические
системы. Естественно, что наиболее эффективная трансляция
мРНК происходит в гомологичных бесклеточных системах. Одна-
Однако под контролем гомологичных регуляторных элементов в бес-
бесклеточных системах могут быть успешно транслированы и чуже-
чужеродные мРНК - транскрипты рекомбинантных генов.
В некоторых бесклеточных системах транслируют предвари-
предварительно очищенную мРНК или используют эндогенную мРНК,
присутствующую в полисомах. В других белоксинтезирующих си-
системах - системах сопряженной транскрипции и трансляции, одно-
одновременно происходят синтез мРНК и ее трансляция рибосомами.
Из-за высокой стоимости очищенных компонентов в подав-
подавляющем большинстве случаев бесклеточные системы биосинте-
биосинтеза белка используют в аналитическом варианте, когда объем
пробы не превышает 100 мкл. Однако в последнее время разра-
разработаны проточные белоксинтезирующие системы, в которых
процесс трансляции удается поддерживать длительное время, не-
непрерывно подводя извне расходуемые вещества, с одновремен-
одновременным удалением синтезируемых белков и продуктов деградации
компонентов системы. Основные принципы получения и функ-
функционирования всех разновидностей белоксинтезирующих систем
будут рассмотрены ниже.
5.4.1. Прокариотические системы
Среди прокариотических бесклеточных белоксинтезирую-
белоксинтезирующих систем наибольшее распространение получили системы на
основе экстрактов клеток Е. coli, хотя основные принципы, ис-
используемые для их получения, могут быть применены и для дру-
других бактериальных клеток. Прокариотическая, как и любая дру-
другая бесклеточная система биосинтеза белка, должна содержать
несколько обязательных компонентов: 1) рибосомы и белковые
факторы трансляции; 2) матричный полирибонуклеотид (мРНК
или синтетические полинуклеотиды); 3) аминоацилированные
тРНК; 4) GTP в качестве источника энергии; 5) одновалентные
(К+ или NH4 ) и двухвалентные (Mg2+ или Са2+) катионы, а также
буферные вещества для поддержания рН системы в физиологи-
физиологических пределах.
Одним из наиболее важных условий функционирования бес-
бесклеточных систем биосинтеза белка является нативное состоя-
состояние рибосом и факторов трансляции. В качестве источников этих
компонентов в бактериальных бесклеточных системах чаще все-
186
го используют экстракты соответствующих клеток. Бактериаль-
Бактериальные клетки выращивают на богатой питательной среде, суспен-
суспендируют в буфере и разрушают тем или иным способом. Лизат
центрифугируют при 30 000 g. При этом в супернатанте (так на-
называемом БЗО-экстракте) остаются рибосомы и остальные бел-
белковые факторы трансляции, необходимые для функционирова-
функционирования системы. Кроме того, в нем содержатся бактериальные ДНК
и мРНК, которые, если от них не освободиться, приводят к неспе-
неспецифическому синтезу белка в бесклеточной системе.
В том случае, если разрушение бактериальных клеток прово-
проводят жесткими методами с применением ультразвука, растирания
со стеклянными бусами и т.п., освобождение от эндогенных нук-
нуклеиновых кислот становится сложной задачей и требует фракци-
фракционирования экстрактов с помощью ионообменной хроматогра-
хроматографии. При такой хроматографической очистке фракции S100 из
нее удаляются все аминоацилированные и свободные молекулы
тРНК. Поэтому при реконструировании бесклеточной белоксин-
тезирующей системы к объединенным фракциям рибосом и S100-
экстракта добавляют препарат очищенной суммарной тРНК.
Экстракты бактериальных клеток S30 и S100, кроме всех не-
необходимых факторов трансляции, содержат и основные компо-
компоненты системы транскрипции, включая РНК-полимеразу, фак-
фактор терминации транскрипции р, CRP-белок и т.п. Поэтому для
создания ДНК-зависимой системы сопряженной транскрипции и
трансляции, в которой происходит полная экспрессия генов, на-
находящихся под контролем бактериальных регуляторных элемен-
элементов, как правило, не требуется введения в нее дополнительных
белков. Достаточно внесения в систему экзогенной ДНК-матри-
ДНК-матрицы, транскрибируемой бактериальной РНК-полимеразой, а так-
также четырех рибонуклеозидтрифосфатов, чтобы в ней начала ак-
активно синтезироваться мРНК и одновременно транслироваться
рибосомами. В итоге в таких бесклеточных системах в ряде слу-
случаев удается синтезировать высокомолекулярные белки, облада-
обладающие ферментативной активностью.
Экстракты бактериальных клеток содержат многочислен-
многочисленные нуклеазы и протеолитические ферменты, которые понижа-
понижают эффективность синтеза белков in vitro, разрушая мРНК и об-
образуемые полипептиды. Для преодоления этих затруднений в
бесклеточных системах часто используют экстракты мутантных
бактериальных клеток, дефектных по РНКазам и полинуклео-
тидфосфорилазе, а в сами бесклеточные системы при необходи-
необходимости вводят ингибитор РНКазы из плаценты человека или ин-
ингибиторы протеиназ: лейпептин, пепстатин, химостатин и т.п.
187
Не существует больших ограничений на первичную структу-
структуру мРНК, транслируемых в бактериальных бесклеточных систе-
системах. Единственным необходимым условием их эффективной
трансляции является отсутствие совершенной вторичной струк-
структуры или каких-либо особо прочных спиральных участков, ста-
стабилизированных кооперативными взаимодействиями. Вторич-
Вторичную структуру транслируемых мРНК обычно разрушают крат-
кратковременным прогреванием с последующим быстрым охлажде-
охлаждением непосредственно перед внесением ее в пробы.
GTP относится к обязательным компонентам бесклеточной
белоксинтезирующей системы. Он не может быть заменен на
любой другой из известных рибонуклеозидтрифосфатов и необ-
необходим для мРНК-зависимого связывания аминоацил-тРНК с ри-
рибосомами и транслокации. Поскольку при трансляции происхо-
происходит непрерывное расходование GTP, а образующийся при этом
GDP является ингибитором трансляции, в процессе функциони-
функционирования бесклеточной системы осуществляют постоянную реге-
регенерацию GTP. Для этого применяют АТР в качестве донора фо-
фосфатных групп, которая, в свою очередь, регенерируется путем
переноса фосфатной группы вводимого в бесклеточную систему
фосфоэнолпирувата с помощью пируватфосфокиназы. Энергия
макроэргических связей АТР используется также при аминоаци-
лировании тРНК аминоацил-тРНК-синтетазами. В том случае,
если создается бесклеточная система сопряженной транскрипции
и трансляции, для осуществления синтеза РНК в нее вводятся
еще два недостающих рибонуклеозидтрифосфата: UTP и СТР.
Для функционирования бесклеточных белоксинтезирующих
систем необходимо обеспечивать в них определенные ионные ус-
условия. Наиболее существенным фактором в этом случае является
концентрация ионов Mg2+. Достаточно изменения оптимальной
концентрации Mg2+ в системе на 1-2 мМ, чтобы в ней перестали
синтезироваться полипептиды, обладающие ферментативной ак-
активностью. При этом влияние ионов Mg2+ на суммарное включе-
включение аминокислот в синтезируемые полипептидные цепи проявля-
проявляется в меньшей степени, что, по-видимому, объясняется нарушени-
нарушением точности включения аминокислот в белки при неоптимальных
концентрациях ионов Mg2+. В системах in vitro ионы Mg2+ могут
быть заменены на ионы Са2+ и даже Мп2+, а также частично заме-
замещены полиаминами: спермидином или спермином, которые благо-
благоприятно влияют на трансляцию и образование нативных белков.
Биосинтез белка в бесклеточных системах происходит также
в присутствии ионов К+ или NH4 , оптимальные концентрации
188
которых составляют ~100 мМ. Ионы Na+ ингибируют трансля-
трансляцию, а ацетат-анионы в используемых солях предпочтительнее
анионов СГ. Ионы Mg2+, K+ и NH4 необходимы для ассоциации
субчастиц рибосом и поддержания их в компактной форме, а так-
также обеспечения других их функций.
Несмотря на то, что в бесклеточной трансляции чаще всего
находят применение системы с использованием S30- и SlOO-экс-
трактов с добавленными рибосомами, современный уровень зна-
знаний молекулярных механизмов трансляции позволяет получать
бесклеточные белоксинтезирующие системы из полностью очи-
очищенных компонентов. Однако создание таких систем является
трудоемким процессом, поэтому их применяют только в аналити-
аналитическом варианте для решения специальных задач.
5.4.2. Эукариотические системы
Несмотря на относительную простоту получения бактери-
бактериальных белоксинтезирующих систем, их использование ограни-
ограничивается трансляцией бактериальных и фаговых мРНК или ре-
комбинантных последовательностей нуклеотидов, находящихся
под контролем генетических регуляторных элементов бактерий
или бактериофагов. В этой связи возникла необходимость в раз-
разработке бесклеточных систем биосинтеза белка, пригодных для
непосредственной трансляции мРНК высших организмов.
В настоящее время наиболее часто применяемыми системами
такого рода являются белоксинтезирующие системы из ретику-
лоцитов кроликов, зародышей пшеницы и культивируемых сома-
соматических клеток различного происхождения. При этом системы
всех трех типов могут быть использованы почти с одинаковым
успехом для трансляции разнообразных мРНК и, как правило, та-
такие системы не обнаруживают видоспецифичности. Основные
принципы конструирования прокариотических белоксинтезиру-
белоксинтезирующих систем применяют и для получения систем биосинтеза бел-
белков высших организмов. Специфика последних связана, прежде
всего, с биологическими особенностями объектов, которые слу-
служат источником белковых компонентов систем, а также с разли-
различиями механизмов биосинтеза белка у прокариот и эукариот.
Ретикулоциты - безъядерные предшественники эритроцитов
человека и животных, осуществляют активный синтез гемогло-
гемоглобина и в связи с этим обладают всеми необходимыми компонен-
компонентами белкового синтеза. В то же время отсутствие ядер в таких
клетках дает возможность получать бесклеточные экстракты,
189
минимально загрязненные геномной ДНК. Эти особенности ре-
тикулоцитов животных делают их излюбленным источником
бесклеточных экстрактов, способных осуществлять эффектив-
эффективную трансляцию разнообразных экзогенных мРНК.
Все основные компоненты, необходимые для функциониро-
функционирования бактериальных бесклеточных систем биосинтеза белка,
должны присутствовать и в бесклеточной белоксинтезирующей
системе из ретикулоцитов кроликов. Однако необходимо отме-
отметить две существенные особенности такой системы. Во-первых,
для регенерации АТР в качестве доноров фосфатных групп ис-
используют креатинфосфат - универсальный аккумулятор энергии
в макроэргических связях у большинства позвоночных, а сам пе-
перенос групп осуществляется вводимой в систему креатинфосфо-
киназой. Во-вторых, для предотвращения ингибирования транс-
трансляции в процессе синтеза белка в систему вводят гемин.
Функционирование системы из зародышей пшеницы и ее со-
состав существенно не отличаются от таковых ретикулоцитарной си-
системы биосинтеза белка. Различия заключаются, главным обра-
образом, в методах получения бесклеточных экстрактов. Зародыши
обычно выделяют из сухих зерен озимой пшеницы. Преимущества-
Преимуществами этих бесклеточных белоксинтезирующих систем перед другими
являются возможность длительного хранения и доступность исход-
исходного биологического материала (зерен сортовой пшеницы), что
обеспечивает воспроизводимость получаемых результатов и не
требует проведения экспериментов с лабораторными животными.
Несмотря на то, что разные эукариотические бесклеточные
белоксинтезирующие системы активно транслируют гетероло-
гичные мРНК, гомологичные матрицы, как правило, транслиру-
транслируются более эффективно. Например, глобиновые мРНК исполь-
используются в синтезе белка в семь-восемь раз лучше лизатами рети-
ретикулоцитов, чем экстрактами культивируемых клеток яичников
китайских хомячков. Кроме того, в клетках разных типов диф-
дифференцированных тканей могут присутствовать специфические
белковые ингибиторы трансляции определенных мРНК. Факты
такого рода необходимо учитывать при выборе бесклеточной си-
системы для решения конкретных экспериментальных задач.
5.4.3. Проточные системы
Бесклеточные системы биосинтеза белка позволили генной
инженерии получать экспрессию изолированных генов, не при-
прибегая к помощи живых клеток. До недавнего времени все обсуж-
обсуждавшиеся выше бесклеточные системы существовали только в
190
аналитическом варианте и были функционально активны на про-
протяжении короткого времени D0-60 мин). Для всех этих систем
характерен малый выход синтезируемого белка (не более 1 мкг
на 1 мл экстракта), и на одну молекулу мРНК в них синтезирует-
синтезируется лишь одна-две молекулы полипептида.
Значительный прогресс в этой области исследований был до-
достигнут в лаборатории А.С. Спирина в 1988 г. С помощью про-
простых усовершенствований удалось получить эффективную бес-
бесклеточную белоксинтезирующую систему. Бесклеточные экс-
экстракты бактериальных или эукариотических клеток помещают в
ячейку, закрытую с двух сторон полупроницаемыми мембрана-
мембранами. Размер пор позволяет проходить через мембраны вместе с
током жидкости низкомолекулярным химическим веществам и
небольшим белкам. Содержимое ячейки, в которой имеются все
компоненты, необходимые для бесклеточной трансляции, инку-
инкубируют при обычной температуре. При этом с одной стороны в
такую ячейку-реактор со скоростью ~1 мл/ч непрерывно посту-
поступают ингредиенты, расходуемые в процессе биосинтеза белка
(аминокислоты, ATP, GTP), а с другой - из нее выходят синтези-
синтезированные белковые продукты (если их молекулярная масса и от-
отсутствие способности к агрегации позволяют пройти через поры
мембраны).
Такое простое усовершенствование бесклеточной системы
сильно изменило характер ее функционирования. Биосинтез бел-
белка в ней может продолжаться непрерывно в течение нескольких
десятков часов, причем на одну молекулу транслируемой мРНК
синтезируются сотни копий полипептидных цепей белков, сум-
суммарный выход которых может достигать 200 мкг и более на 1 мл
бесклеточного экстракта.
С разработкой проточной бесклеточной белоксинтезирую-
щей системы стало возможным проводить биосинтез рекомби-
нантных белков в препаративных количествах in vitro. Такая сис-
система позволяет синтезировать в больших количествах полипеп-
полипептиды, подверженные быстрой внутриклеточной деградации про-
теиназами или образующие тельца включения в живых клетках.
Проточная система может быть полезна также для получения
белков, обладающих цитотоксической активностью, или других
рекомбинантных полипептидов, для которых нежелательны ар-
тефактные посттрансляционные модификации, происходящие in
vivo. Недавно в таких системах удалось получить препаративные
Количества функционально активного интерлейкина 6 человека.
191
Глава 6
Полимеразная цепная реакция
и другие способы
амплификации ДНК и сигналов
В 1985 г. К.Б. Мюллис и др. опубликовали и запатентовали
метод полимеразной цепной реакции (ПЦР), которому суждено
было оказать глубочайшее влияние на все области исследования
и прикладного использования нуклеиновых кислот [261,262].
Значение этого метода для молекулярной биологии и генетики
оказалось столь велико и очевидно, что уже через семь лет авто-
автору была присуждена Нобелевская премия по химии. Возмож-
Возможность амплификации любого сегмента ДНК, последовательность
нуклеотидов которого известна, и получение его по завершении
ПЦР в гомогенном виде и препаративном количестве делают
ПЦР альтернативным методом молекулярного клонирования ко-
коротких фрагментов ДНК. При этом не возникает необходимости
в применении сложных методических приемов, которые исполь-
используют в генной инженерии при обычном клонировании. Разработ-
Разработка метода ПЦР во многом расширила методические возможнос-
возможности молекулярной генетики, и, в частности, генной инженерии,
причем настолько, что это кардинально изменило и усилило на-
научный потенциал многих ее направлений. Реалии исследователь-
исследовательской жизни указывают на то, что этому методу нужно уделить в
нашем рассказе более пристальное внимание.
6.1. Проведение ПЦР
6.1.1. Общая схема ПЦР
Принцип ПЦР заключается в следующем (рис. 18). Реакцион-
Реакционная смесь обычно содержит матричную ДНК, четыре дезоксири-
бонуклеозидтрифосфата dATP, dCTP, dGTP и ТТР, Taq-полиме-
разу и два синтетических олигодезоксирибонуклеотидных прай-
мера длиной 15-30 нуклеотидов, комплементарных участкам
противоположных цепей ДНК, фланкирующих исследуемый
участок ДНК-матрицы. ПЦР начинается с кратковременного
A-2 мин) прогревания реакционной смеси при ~95°С. При этом
происходит плавление цепей ДНК и инактивация примесных бел-
белков, которые могут присутствовать в реакционной смеси, если
используется недостаточно очищенная ДНК, например, из кли-
192
Циклы
Олигонуклеотидные
праймеры
3'
5'
\
5'
\v Цепи
,хднк
5'
3'
5'
Продукты
ПЦР
3'
23 продуктов ПЦР
п
2й продуктов ПЦР
Рис. 18. Полимеразная цепная реакция (ПЦР)
нических образцов. Далее реакционную смесь охлаждают до тем-
температуры отжига праймеров с матричной ДНК C7-65°С). В ре-
результате праймеры связываются с комплементарными местами
на матрице и с этого момента начинают служить затравками для
синтеза Taq-полимеразой новых цепей ДНК, комплементарных
каждой из цепей денатурированной матричной ДНК. Для прове-
проведения синтеза ДНК в оптимальных для Taq-полимеразы услови-
условиях температуру реакционной смеси поднимают до 72°С и при та-
такой температуре проводят элонгацию цепей вновь синтезирую-
7. Патрушев Л.И. Т. 1 193
щейся ДНК в течение 0,5-2 мин. По окончании стадии синтеза
ДНК заканчивается первый цикл ПЦР, после чего проводятся та-
такие же второй, третий и последующие циклы в автоматическом
режиме. После завершения третьего цикла уже образуется дис-
дискретный продукт ПЦР - фрагмент дцДНК, содержащий на своих
5'-концах последовательности нуклеотидов праймеров. Количе-
Количество продукта ПЦР в реакционной смеси после завершения каж-
каждого цикла удваивается и, следовательно, по мере прохождения
реакции экспоненциально возрастает, достигая нескольких мик-
микрограммов в реакционной смеси объемом 25-50 мкл. В финале
ПЦР содержимое реакционной смеси анализируют с помощью
электрофореза в агарозном геле в присутствии бромистого эти-
дия, где продукт реакции выявляется в УФ-свете. Продукт ПЦР
можно выделить из агарозного геля в чистом виде и работать с
ним как с обычным фрагментом дцДНК, т.е. гидролизовать рес-
триктазами, клонировать в плазмидных векторах по липким и ту-
тупым концам, секвенировать или использовать в качестве зондов
при гибридизации.
С помощью ПЦР можно амплифицировать in vitro сегменты
ДНК длиной от 0,1 до 5-7 т.п.о. и более, а для получения положи-
положительного результата достаточно присутствия в реакционной сме-
смеси одной-двух копий амплифицируемой последовательности нук-
нуклеотидов (например, геномной ДНК, содержащейся в одной-двух
соматических клетках). При этом теоретически нет необходи-
необходимости в тщательной очистке матрицы, так как большинство на-
находящихся в реакционной смеси белков и ферментов инактивиру-
ется в первых же циклах ПЦР и не оказывает влияния на проте-
протекание реакции при высоких температурах.
6.1.2. Критические компоненты реакции
При практическом использовании ПЦР, по крайней мере, три
характеристики этого метода наиболее важны для исследовате-
исследователя - это специфичность реакции, точность синтеза ДНК и его
эффективность [263]. При абсолютной специфичности ПЦР
продукт должен быть копией именно того локуса, который амп-
лифицировали, а другие амплифицированные последовательнос-
последовательности нуклеотидов в продукте ПЦР не должны присутствовать. При
этом, как правило, не обращают внимания на то, имеются ли в
продукте ПЦР ошибочно включенные нуклеотиды, которые не-
некомплементарны амплифицируемому участку ДНК и появляю-
появляющиеся благодаря сбоям в работе ДНК-пол имеразы. Такой подход
имеет место, например, при ДНК-диагностике инфекционных за-
194
болеваний, когда определяют наличие в исследуемом образце
нуклеиновых кислот возбудителя. В этом случае вполне доста-
достаточно, чтобы специфический продукт ПЦР был представлен эле-
ктрофоретически гомогенным набором молекул, соответствую-
соответствующих амплифицируемому участку матрицы.
Требования к точности амплификации резко повышаются в
том случае, если последовательность нуклеотидов продукта ПЦР
критична для проводимого исследования. Ошибки в амплифика-
амплификации недопустимы при определении первичной структуры ампли-
фицируемого локуса, или при использовании продукта ПЦР для
синтеза кодируемого им белка в генно-инженерных системах экс-
экспрессии.
Эффективность ПЦР количественно оценивается по выходу
продукта ПЦР после завершения реакции. Хотя теоретически ко-
количество продукта в реакционной смеси после каждого цикла
ПЦР удваивается, на практике этого никогда не происходит. Как и
в любой другой химической реакции выход продукта ПЦР можно
увеличить путем оптимизации критических параметров реакции.
Однако действие многих компонентов в ПЦР взаимозависимо, по-
поэтому изменение свойств одного из компонентов, как правило,
влечет за собой необходимость в оптимизации свойств и других.
Одними из наиболее критических компонентов для специ-
специфичности ПЦР и ее эффективности являются олигонуклеотид-
ные праймеры, поэтому наш краткий обзор компонентов реак-
реакции, которые оказывают существенное влияние на ее протека-
протекание, мы начнем именно с дизайна праймеров.
6.1.3. Конструирование праймеров
Определившись с локусом в ДНК, который в зависимости от
цели исследования необходимо амплифицировать, переходят к
разработке праймеров. В большинстве случаев длина праймеров
должна быть в пределах 18-30 нт, однако при проведении направ-
направленного мутагенеза могут быть использованы праймеры боль-
большей длины, в том числе и мегапраймеры (см. раздел 1.2.4, час-
части II).
GC-Состав праймеров, который определяет температуру
плавления (Тт) гибрида, образованного праймерами и матрицей,
должен находиться в пределах 35-65% (в идеале 45-55%) и по
возможности быть одинаковым у обоих праймеров. Конечно,
состав праймеров полностью определяется первичной структу-
структурой амплифицируемого участка матрицы. Но в том случае, ес-
если состав оказывается сильно смещенным в сторону AT или GC,
7* 195
допускается добавление на 5'-концы праймеров нескольких ос-
остатков G и/или С, и, соответственно, А и/или Т, некомплемен-
некомплементарных матрице. Имеется еще один прием, который позволяет
повышать Тт гибридов без изменения первичной структуры
праймеров, что бывает необходимо для амплификации участков
ДНК с высоким GC-составом и при наличии в них самокомпле-
самокомплементарных повторяющихся последовательностей. В этом слу-
случае при синтезе праймеров применяют аналоги нуклеотидов,
которые образуют более прочные связи со своими комплемен-
комплементарными партнерами [263].
Во время амплификации обычно используют температуру
отжига (Та) праймеров на 5 °С ниже их расчетной (Тт), которую
обычно определяют с помощью олигонуклеотидных кальку-
калькуляторов (см., например, http://www.nwfsc.noaa.gov/protocols/
oligoTMcalc.html). Различия между Тт у пары праймеров в 4-6 °С
обычно не сильно оказывают влияние на эффективность ПЦР, од-
однако лучше если их Тт совпадают. Если различия между праймера-
ми слишком велики, то можно изменить их GC-состав, удлинив
один из праймеров в соответствии с амплифицируемой последова-
последовательностью с 3 - или 5-конца (в первом случае не происходит изме-
изменения размера образующегося продукта ПЦР). Для приблизитель-
приблизительного расчета Та вручную можно воспользоваться формулами [263]:
[4(G + С) + 2(А + Т)] - 5°С при длине праймера < = 20 нт, или
62,3°С + 0,41 °С (%G-C) - 500/длина праймера - 5°С, при дли-
длине праймера >20 нт.
Кроме вышеупомянутых особенностей дизайна праймеров, при
их конструировании полезно иметь в виду следующее:
- при выборе мест отжига праймеров на матрице необходимо из-
избегать участков, которые в одноцепочечной форме образуют
вторичную структуру, например, палиндромных последова-
последовательностей, особенно в том случае, если Тт этих участков боль-
больше Та праймеров;
- по мере необходимости допускается присоединение к 5-концам
праймеров некомплементарных матрице последовательности,
которые могут заключать в себе требуемые сайты рестрикции,
промоторные участки, кодоны инициации и терминации транс-
трансляции и т.п.;
- для эффективной ПЦР не требуется полной комплементарнос-
ти праймеров матрице, хотя полное соответствие желательно;
- для исключения отжига праймеров в неспецифических местах
анализируемых последовательностей, необходимо подтвер-
196
дить с помощью компьютерного анализа отсутствие в них до-
дополнительных мест посадки. Это особенно важно при анализе
таких больших геномов как геном человека, животных и расте-
растений. Практически подобный анализ проводят с использованием
баз данных последовательностей исследуемого организма, до-
доступных через Интернет. Необходимо изменять последователь-
последовательность праймеров в том случае, если их гомология с возможны-
возможными вторичными сайтами посадки превышает 70%. При этом ис-
использование интронных последовательностей в качестве мест
посадки дает хороший результат, поскольку они, как правило,
дивергированы даже в мультигенных семействах;
полезно начинать и заканчивать последовательность праймера
с одного-двух пуриновых нуклеотидов, однако следует избегать
включения остатков G или С на его 3'-конец, так как это повы-
повышает вероятность неспецифического отжига праймеров из-за
высокой прочности соответствующих водородных связей;
недопустима самокомплементарность праймеров, особенно в их
3'-концевых областях. Ее наличие способствует амплификации
димеров праймеров, а протяженная самокомплементарность
делает праймеры нефункциональными;
• при амплификации последовательностей в мультигенных се-
семействах, когда не удается сделать праймеры полностью специ-
специфичными для одного локуса, необходимо приложить усилия,
чтобы по крайней мере два (чем больше, тем лучше) последних
3'-концевых нуклеотида были полностью специфичны в отно-
отношении анализируемого участка ДНК. Таким простым приемом
часто удается предотвратить появление неспецифических про-
продуктов ПЦР в этих сложных специальных случаях;
¦ ионные условия при проведении ПЦР и количество циклов
должны определяться индивидуально для каждой новой пары
праймеров;
• стандартные концентрации праймеров в реакционной смеси со-
составляют 0,1-1,0 мкМ, хотя в большинстве случаев хорошие ре-
результаты получаются при их концентрациях 0,2-0,5 мкМ. При
этом необходимо иметь в виду, что чем больше концентрация
праймеров в реакционной смеси, тем выше вероятность образо-
образования неспецифических продуктов ПЦР и тем дороже обходится
каждая проба. В этой связи минимальные концентрации прайме-
праймеров для конкретных условий проведения реакции должны быть
определены эмпирически. Однако для повышения эффективно-
эффективности ПЦР, когда вышеупомянутый критерий не является критиче-
критическим (например, при амплификации плазмидной ДНК), необхо-
необходимо увеличивать соотношение праймер/матрица.
197
Осуществление дизайна праймеров в настоящее время сильно
облегчается наличием высокоэффективных пакетов программ-
программного обеспечения, которые созданы специально для этой цели.
Но ни одна из современных компьютерных программ не осво-
освобождает исследователя от необходимости затраты интеллекту-
интеллектуальных усилий при попытке решения собственных проблем.
6.1.4. Термостабильные ДНК-зависимые ДНК-полимеразы
Другим важнейшим компонентом реакционной смеси при
проведении ПЦР является термостабильная ДНК-полимераза.
В настоящее время, благодаря наличию высокоразвитой дилер-
дилерской сети, имеется много источников разных термостабильных
ДНК-полимераз, хотя, как это обычно бывает, первичных произ-
производителей этих ферментов намного меньше. Поэтому разнообра-
разнообразие доступных препаратов ферментов не так велико, как кажет-
кажется на первый взгляд.
Выбор конкретной ДНК-полимеразы зависит от целей экспе-
эксперимента. Ферменты, обладающие более высокой точностью син-
синтеза ДНК, и меньшей частотой включения в продукт ПЦР оши-
ошибочных (некомплементарных матрице) нуклеотидов, как прави-
правило, обладают 3' -» 5'-экзонуклеазной (корректирующей) актив-
активностью. В том случае, если на 3'-конец строящейся цепи ДНК
включается некомплементарный матрице нуклеотид, он удаляет-
удаляется из цепи с участием этой активности ДНК-полимеразы. К сожа-
сожалению, наиболее широко распространенная в настоящее время
Taq-ДНК-полимераза, такой активностью не обладает, а следо-
следовательно, точность синтеза ДНК этим ферментом невелика по
сравнению с ДНК-полимеразами, способными корректировать
собственные ошибки. К последним ферментам относятся, напри-
например, Pfu-, Vent- и Deep Vent-ДНК-полимеразы.
Несмотря на более высокую точность действия этих фермен-
ферментов, только с ними не работают по ряду причин. Основная из
них - неспецифический гидролиз праймеров, который происхо-
происходит под действием корректирующей экзонуклеазы. При этом,
чем длиннее праймер, тем выше начальная скорость его деграда-
деградации. Кроме того, точности работы Taq-ДНК-полимеразы оказы-
оказывается вполне достаточно для большей части экспериментов, в
которых она используется в настоящее время. Тем не менее, точ-
точность Taq-полимеразы можно повысить, если ее применять в со-
составе смесей с более корректно работающими ферментами
(-50:1), что позволяет уменьшить скорость деградации прайме-
праймеров и значительно повысить точность синтеза ДНК во всей сис-
198
Таблица 8
Частота ошибок при синтезе ДНК, осуществляемом
термостабильными ДНК-полимеразами при проведении ПЦР
в оптимальных условиях
п Частота мутаций
ДНК-полимераза (на { нуклеотид/1 раунд репликации)
Pfu
Deep Vent
Vent
Taq
exo(-)Pfu и UITma
Смесь Taq/Pfu или Klentaq/Pfu
1,3 • Ю-6
2,7 • Ю-6
2,8 • Ю-6
8,0 • Ю-6
5,0 • 10
-5,0 • Ю-6
теме. Частота ошибочно включенных нуклеотидов в результате
одновременного функционирования двух разных ДНК-полиме-
раз в смесях, применяемых для амплификации длинных сегмен-
сегментов ДНК (long PCR), оказывается ниже, чем при использовании
Taq-полимеразы, но выше, чем для очищенной ДНК-полимеразы
Pfu (табл. 8).
Мутации, которые возникают при использовании Taq-ДНК-
полимеразы, как правило, представляют собой транзиции
AT -» GC. В том случае, если матрица обладает тенденцией к об-
образованию элементов вторичной структуры, возникновение де-
леций также становится весьма вероятным [263]. Klentaq, мутант
Taq-ДНК-полимеразы, у которого отсутствует 5' -» З'-экзонук-
леазная активность из-за делеции N-концевой части полипептид-
полипептидной цепи, получил свое название по аналогии с фрагментом Клё-
нова ДНК-полимеразы IE. coli, у которого по той же причине от-
отсутствует эта активность, присущая ферменту дикого типа. В от-
отличие от Taq-ДНК-полимеразы Klentaq обладает более широким
оптимумом в отношении концентрации ионов Mg2+, что позволя-
позволяет осуществлять эффективный поиск оптимальных условий про-
проведения реакции, при изменении других компонентов реакцион-
реакционной смеси. В смеси с ферментами, обладающими корректирую-
корректирующей экзонуклеазной активностью, его часто используют для
амплификации протяженных участков ДНК. Приблизительно
теми же свойствами обладает фрагмент Штоффеля Taq-ДНК-
полимеразы, который представляет собой фрагмент С-концевой
части ее полипептидной цепи длиной в 544 а.о.
Tth-ДНК-полимераза, выделенная из термофильной бакте-
бактерии Thermus thermophilus, обладает в присутствии ионов Мп2+ ак-
активностью РНК-зависимой ДНК-полимеразы, а в присутствии
199
ионов Mg2+ активностью обычной ДНК-полимеразы, сравнимой
с таковой у Taq-ДНК-полимеразы. Благодаря этим свойствам
фермент используется для одновременного проведения обратной
транскрипции и амплификации транскрибированных последова-
последовательностей в одной пробирке.
Pfu-ДНК-полимераза из Pyrococcus furiosus обладает высокой
термостабильностью, 3' -» 5'-корректирующей экзонуклеазной
активностью, и, как следствие, высокой точностью синтеза ДНК.
При использовании в смеси Taq-ДНК-полимеразы она обеспечи-
обеспечивает образование продуктов ПЦР с тупыми концами, поскольку
3'-выступающий А отщепляется экзонуклеазой Pfu-ДНК-поли-
меразы.
Vent-ДНК-полимераза из Thermococcus litoralis по многим
свойствам напоминает Pfu-полимеразу, однако, в отличие от нее,
этот фермент неспособен амплифицировать матрицы, если в них
присутствуют остатки I или dU.
Deep Vent-ДНК-полимераза из Pyrococcus species GB-D в от-
отличие от только что рассмотренного фермента более устойчива
к присутствию формамида и диметилсульфоксида в реакционной
смеси, которые используются для понижения температуры плав-
плавления амплифицируемых участков ДНК, обладающих высоким
GC-составом и сложной вторичной структурой. С помощью это-
этого фермента удается амплифицировать участки ДНК по крайней
мере до 14 т.п.о.
UlTma-ДНК-полимераза из Thermotoga maritima применяет-
применяется для амплификации участков ДНК, длина которых не превы-
превышает 3 т.п.о. Как и в случае всех остальных ДНК-полимераз, об-
обладающих 3'—>5'-корректирующей активностью, под действием
UlTma-ДНК-полимеразы образуются продукты ПЦР, не содер-
содержащие 3'-выступающих А, и, следовательно, эти продукты мо-
могут быть непосредственно использованы для клонирования по
тупым концам. Все приведенные здесь данные взяты из руко-
руководства [263].
Прежде чем закончить раздел о ДНК-полимеразах, применя-
применяемых в ПЦР, хочется дать небольшой комментарий по поводу
концентраций ферментов в пробах, которые целесообразно
применять при проведении реакции. В рутинных экспериментах
в пробы объемом 25-100 мкл обычно добавляют 0,5-0,25 ед.
фермента. Вообще, чем выше концентрация фермента в пробе,
тем больше вероятность образования неспецифических продук-
продуктов. В то же время необходимо помнить, что амплификация, осу-
осуществляемая, в частности, Taq-ДНК-полимеразой, происходит по
дистрибутивному механизму, т.е. в процессе синтеза особенно
200
длинных цепей ДНК фермент, начавший синтез, редко его закан-
заканчивает. Происходит диссоциация комплекса фермент-(про-
дукт-матрица), и синтез продолжает новая молекула фермента.
Исходя из этого, очевидно, что для получения продуктов ПЦР
большой длины необходимо повышать концентрацию фермента
в пробе вплоть до 10 ед. активности на 50 мкл реакционной сме-
смеси. В эффективности такого незамысловатого приема приходи-
приходилось неоднократно убеждаться на практике.
6.1.5. Другие критические компоненты и параметры ПЦР
Матричные нуклеиновые кислоты. В обычных опытах в про-
пробы добавляют 102— 105 копий матрицы. При уменьшении концен-
концентрации матрицы до уровня ниже оптимального возрастает веро-
вероятность образования неспецифических продуктов ПЦР. Пониже-
Понижением отношения праймер/матрица добиваются увеличения спе-
специфичности реакции, в то же время с увеличением этого отноше-
отношения, как правило, происходит возрастание количества образовав-
образовавшегося продукта.
Ионы двухвалентных металлов. Концентрация ионов Mg2+
должна превышать концентрацию дезоксирибонуклеозидтрифо-
сфатов (dNTPs) в реакционной смеси на 0,5-3,0 мМ. Такой избы-
избыток необходим потому, что основным источником фосфатных
групп во время ПЦР являются dNTPs, а ионы Mg2+ образуют с ни-
ними растворимый комплекс. В то же время ионы взаимодействуют
еще и с матричной ДНК, праймерами, самой ДНК-полимеразой и
являются абсолютно необходимыми для функционирования лю-
любых ферментов матричного синтеза нуклеиновых кислот. Их
концентрация влияет на процесс отжига праймеров, температуру
плавления двойной спирали нуклеиновых кислот, активность и
точность функционирования ДНК-полимеразы. В этой связи,
требуется точное определение оптимальной концентрации ионов
Mg2+ в реакционной смеси для каждого нового сочетания прайме-
праймеров и матрицы. В общем, повышение концентрации ионов Mg2+
выше оптимальной уменьшает специфичность ПЦР, что часто
сопровождается образованием "шмира".
Температурный режим. Первому циклу ПЦР обычно должна
предшествовать преинкубация реакционной смеси при 92-96°С в
течение 0,5-10 мин, что сопровождается инактивацией примес-
примесных белков, если они там присутствуют, а также способствует
более полной первоначальной денатурации матричной ДНК.
Введение этого этапа повышает эффективность ПЦР. При за-
завершении последнего цикла пробы обычно выдерживают в тече-
201
ние 5-10 мин при 72°С для полного завершения синтеза начавших
синтезироваться цепей в ПЦР-продукте. Необходимость такой
задержки именно на последнем этапе синтеза ДНК связана с тем,
что к этому моменту накапливается большое количество продук-
продукта ПЦР, и незавершенность синтеза его цепей особенно сильно
проявится в гетерогенности состава молекул реакционной смеси.
В то же время недостроенные молекулы, остающиеся в проме-
промежуточных циклах, достраиваются сами собой и не оказывают
влияния на общий результат реакции.
Время элонгации праймеров. Скорость элонгации праймеров
при ПЦР обычно приближается к 50 нт/сек при 72°С. Из этого
следует, что для амплификации участка матрицы длиной < 400 нт
вполне достаточно 15 сек. Это время можно сократить еще боль-
больше, поскольку процесс элонгации праймеров начинается уже в
фазе их отжига. В случае амплификации очень длинных последо-
последовательностей время элонгации следует увеличивать в ходе ПЦР с
целью компенсации ингибирующего влияния возрастающей вяз-
вязкости реакционной смеси.
Циклы. Количество циклов, которое необходимо провести
для амплификации требуемой последовательности, обратно про-
пропорционально количеству копий матрицы в реакционной смеси.
Обычно используют такое число копий, чтобы видимый продукт
образовывался после проведения 25-50 циклов. Во время ПЦР
наблюдается экспоненциальная фаза увеличения числа синтези-
синтезированных молекул дцДНК, которая продолжается до тех пор, по-
пока это число не достигнет —1012. Затем скорость накопления
продукта резко уменьшается. Существует несколько основных
факторов, которые препятствуют экспоненциальному накопле-
накоплению продукта, как это должно происходить теоретически. Из них
следует упомянуть истощение субстратов реакции (праймеров
или dNTPs), стабильность компонентов реакционной смеси, инги-
бирование ПЦР конечным продуктом реакции, конкуренцию за
субстраты неспецифическими продуктами ПЦР или димерами
праймеров, повторный отжиг продуктов ПЦР, препятствующий
элонгации праймеров, неполную денатурацию в присутствии
большого количества продукта ПЦР и ряд других. С учетом этих
факторов из-за "эффекта плато" увеличение числа циклов само
по себе не приводит к возрастанию специфичности ПЦР или на-
накоплению специфического продукта. Для повышения специфич-
специфичности ПЦР необходимо понижать число циклов и уменьшать
продолжительность их отдельных сегментов (время денатурации,
отжига и элонгации праймеров). Однако для повышения эффек-
эффективности ПЦР при амплификации больших участков матрицы
202
(>1 т.п.о.) продолжительность сегментов ПЦР в каждом цикле
нужно увеличивать. На продолжительность отдельных сегмен-
сегментов циклов оказывают влияние форма капли реакционной смеси,
толщина стенок пробирок, а также особенности конструкции на-
нагревающего блока амплификатора (термоциклера).
Объем реакционной смеси. Стандартный объем пробы, в
которой проводят ПЦР, обычно составляет 20-100 мкл, тем не
менее, при использовании современных амплификаторов для
аналитических целей может вполне подойти объем в 5 мкл.
Пробы большого объема неэффективно прогреваются и ох-
охлаждаются, в маленьких объемах синтезируется небольшое ко-
количество ПЦР-продукта.
6.2. Проблемы, возникающие при постановке ПЦР
Весь мой опыт биохимической работы показывает, что ка-
каким бы авторитетным источником данных ты ни пользовался
при налаживании искусственной генетической системы, для то-
того, чтобы она хорошо работала, конкретные условия необходи-
необходимо подбирать самому. Это особенно относится к сложным мно-
многокомпонентным системам. Этап оптимизации системы в экспе-
экспериментальной работе является очень полезным не только из-за
чисто утилитарных соображений. В ходе предварительных опы-
опытов убеждаешься, что у тебя наладился диалог с системой и она
отвечает на твои вопросы. Любая система на адекватно (разум-
(разумно) поставленный вопрос дает исчерпывающий ответ, но
нашего интеллекта, как правило, не хватает, чтобы сразу ин-
интерпретировать полученный результат правильно. Только в хо-
ходе налаженного диалога с системой можно постепенно прибли-
приблизиться к истине.
6.2.1. Появление неспецифических продуктов ПЦР
Одним из самых неприятных артефактов, с которым прихо-
приходится сталкиваться при постановке ПЦР, это появление продук-
продукта ПЦР в обязательном отрицательном контрольном образце,
т.е. в отсутствие матрицы. Такие артефакты относятся к катего-
категории ложноположительных результатов. Причиной, чаще всего,
является загрязнение компонентов реакции или лабораторного
пластика продуктами ПЦР, что может легко произойти в процес-
процессе постановки массовых ПЦР, например, в ДНК-диагностике при
недостаточно чистой работе. При появлении этого грозного при-
признака неаккуратной работы приходится менять загрязненный
203
компонент, на практике же, в целях экономии времени, - все
компоненты реакционной смеси. Поэтому, по возможности, не-
необходимо работать с небольшими аликвотами компонентов, а их
основные запасы хранить отдельно, подальше от аппаратов для
электрофореза и открывать в редких случаях. Кроме того, гото-
готовить образцы для ПЦР и проводить электрофоретический ана-
анализ продуктов ПЦР следует в разных помещениях.
Из предлагаемых способов борьбы с кроссконтаминацией об-
образцов продуктами ПЦР предыдущих опытов можно отметить об-
облучение лабораторного пластика ультрафиолетовым светом [264]
или инкубацию с гипохлоритом натрия [265], а также использова-
использование фотохимических и ферментативных методов [266-269].
Дополнительные неспецифические продукты могут образовы-
образовываться и при безукоризненном отрицательном контроле. В этом
случае причиной их появления могут быть слишком высокая кон-
концентрация праймеров или их чересчур большая вырожденность.
В ряде случаев приходится менять праймеры, так как условия ис-
использования некоторых из них нельзя оптимизировать. Весьма ча-
часто избавиться от неспецифических продуктов в виде "шмира" или
дискретных полос можно с помощью "горячего старта" и/или
"гнездовой" ПЦР, но у последнего подхода есть свои недостатки
(см. ниже). Плохая денатурация матрицы также может приводить
к появлению неспецифических продуктов. К тому же результату
может приводить и высокая концентрация ионов Mg2+, dNTPs или
ДНК-полимеразы. В случае с ионами Mg2+, их концентрацию оп-
оптимизируют путем постепенного уменьшения с интервалом
0,2-0,5 мМ. Присутствие ингибиторов ПЦР в пробе часто отрица-
отрицательно сказывается на специфичности реакции.
Слишком низкая температура отжига праймеров приводит к
их взаимодействию с матрицей в неспецифических местах и также
может быть причиной образования неспецифических продуктов
ПЦР в конкретном случае, что может быть и следствием чрезмер-
чрезмерной продолжительности циклов, излишнего числа циклов для дан-
данного количества копий матрицы в пробе или медленной скорости
перехода между отдельными сегментами циклов. Скорость пере-
перехода от сегмента к сегменту в цикле ПЦР называют рэмпом. В со-
современных амплификаторах этот параметр можно изменять, и он
также требует оптимизации в сложных случаях.
Димеры праймеров. При наличии основного продукта ПЦР
образование димеров праймеров, которые иногда появляются в
виде полосы, идущей при электрофорезе впереди бромфеноло-
вого синего, как правило, можно игнорировать, хотя это и не
радует глаз. Однако, если данный процесс становится интенсив-
204
ным, то происходит конкуренция за субстраты ПЦР со стороны
димеров. В редких случаях образуются исключительно димеры
и тогда приходится с ними бороться. Причинами образования
димеров могут быть: комплементарность праимеров в их 3'-
концевых частях, слишком короткие праймеры, их чрезмерно
высокая концентрация или же низкая концентрация матрицы.
Димеры праимеров могут образовываться и при наличии у них
гомологии с повторяющимися последовательностями матрич-
матричной ДНК, а также при "переамплификации" с избыточным чис-
числом циклов и использовании субоптимальных температур от-
отжига праимеров,
Появление продуктов ПЦР, не соответствующих по разме-
размерам ожидаемым. Такой результат ПЦР чаще всего указывает на
артефакт, но иногда может быть и открытием нового полимор-
полиморфизма в матричной ДНК, возникшего в результате делеции или
вставки. По крайней мере, подобный результат должен настора-
настораживать. Система амплификации на основе ПЦР, как и любая дру-
другая искусственная генетическая система, является "вещью в се-
себе", законы существования которой не вполне понятны. Это осо-
особенно относится к тем сложным случаям амплификации, когда в
качестве матрицы используется ДНК целого большого генома,
например, генома человека. В данном случае единственным до-
доказательством того, что амплифицирован нужный локус, будет
подтверждение происхождения продукта на уровне его первич-
первичной структуры. Быстрее всего убедиться в адекватности продук-
продукта можно путем исследования сайтов рестрикции. Однако, даже
наличие предсказанных сайтов из-за сложности исследуемого
объекта не дает полной гарантии того, что артефакта удалось из-
избежать. Окончательным доказательством амплификации нужно-
нужного генетического локуса может служить только полное секвени-
рование продукта ПЦР.
Дискретные "неспецифические" продукты. Исследование
этого феномена в каждом случае требует индивидуального под-
подхода. Например, по нашему опыту амплификация генетических
локусов, содержащих повторяющиеся последовательности, мо-
может сопровождаться образованием нескольких воспроизводимых
дискретных полос, которые не отражают гетерогенности длин
исследуемого участка ДНК в геноме. Амплификация индивиду-
индивидуального клонированного продукта ПЦР в данном случае приво-
приводит к образованию точно тех же дискретных продуктов. Это ука-
указывает на то, что сам продукт ПЦР, образовавшийся в первых
циклах, формирует гетерогенную матрицу. Конечно, причиной
появления дискретных неспецифических полос могут быть и бо-
205
лее тривиальные факторы, связанные с неоптимальными услови-
условиями проведения ПЦР.
Высокомолекулярный "шмир" на фоне дискретных про-
продуктов ПЦР или при полном их отсутствии. "Шмир" очень час-
часто возникает при использовании плохо очищенных матриц.
При появлении "шмира" светится вся дорожка в агарозном ге-
геле после проведения электрофореза и окрашивания его броми-
бромистым этидием. В этом случае имеют место множественные не-
неспецифические инициации синтеза ДНК из-за присутствия в ре-
реакционной смеси низкомолекулярных затравок в виде неспеци-
неспецифических фрагментов ДНК или РНК, а также повторяющихся
последовательностей в матрице, которые "залипают" друг на
друга. Денатурированные белки, неспецифически удерживая
концы фрагментов ДНК вблизи матрицы, могут создавать ме-
места множественной инициации синтеза ДНК и т.п. Имеются, по
крайней мере, две возможности преодоления такого эффекта:
дополнительная очистка матрицы, например, с помощью фе-
нольной депротеинизации или с использованием ионообменной
смолы Chelex-100 [270], а также, если по каким-то причинам
хорошо очищенные матрицы недоступны, применение моди-
модифицированных ДНК-полимераз в ПЦР с "горячим стартом"
(см. ниже).
6.2.2. Низкий выход или полное отсутствие продуктов ПЦР
В сильно неоптимальных условиях ПЦР не происходит. При
наличии такого результата необходимо убедиться в том, что амп-
лификатор работает правильно, проверить (путем последова-
последовательной замены на работающие) все компоненты реакционной
смеси, попытаться резко понизить температуру отжига прайме-
ров или снова выделить ДНК.
Однако, в том случае, если для выделения нуклеиновых
кислот используются упрощенные и ускоренные методы, что
неизбежно при проведении массовых анализов в клинических
лабораториях, оценке качества пищевых продуктов или мони-
мониторинге окружающей среды, причиной отсутствия продуктов
ПЦР в реакционной смеси могут быть многочисленные инги-
ингибиторы реакции. Подробные данные о таких ингибиторах,
а также стимуляторах ПЦР, можно найти в специальном обзо-
обзоре [271].
206
6.3. Методы ПЦР
6.3.1. Стандартная ПЦР
В простых случаях подходы, основанные на протоколах стан-
стандартной ПЦР, разработанные К.Б. Мюллисом и др., как правило,
дают хорошие результаты при амплификации последовательнос-
последовательностей, длина которых не превышает 3 т.п.о. Такой метод наиболее
применим для амплификации коротких последовательностей,
любых последовательностей в составе гомогенных векторных
молекул на основе плазмид и хромосом бактериофагов, а также
при реамплификации продуктов ПЦР. В этом случае точное сле-
следование прописи является гарантией получения нужного ПЦР-
продукта.
6.3.2. Множественная ПЦР
Присутствие в реакционной смеси нескольких пар прайме-
ров, специфичных в отношении разных генетических локусов, в
ряде случаев позволяет проводить одновременную амплифика-
амплификацию соответствующих участков ДНК-матрицы. Такой вариант
системы ПЦР получил название множественной ПЦР (мПЦР).
(multiplex PCR, mPCR). В простом случае это становится возмож-
возможным, если оптимальные условия проведения реакции близки для
всех пар используемых праймеров. Если мПЦР проводят в фор-
формате аллель-специфической ПЦР (см. раздел 6.3.11 этой главы),
и целью эксперимента является одновременная идентификация
двух аллелей одного гена, которые различаются по одному нук-
леотиду, то аллель-специфические праймеры делают разной
длины, что позволяет по размеру продуктов ПЦР, разделяемых
с помощью капиллярного электрофореза, идентифицировать
исследуемые аллели.
6.3.3. Асимметричная ПЦР
Асимметричная ПЦР (asymmetrical PCR), иначе называе-
называемая однонаправленной ПЦР (single-sided PCR), используется
Для наработки одноцепочечных фрагментов ДНК, соответст-
соответствующих амплифицируемому участку ДНК-матрицы. В этом
случае при постановке ПЦР один из праймеров находится в
низкой концентрации, поэтому в процессе ПЦР он быстро ис-
истощается, и синтез ДНК с этого момента продолжается с одно-
одного праймера, исходно присутствовавшего в избытке, а накопле-
накопление ПЦР-продукта уже происходит не экспоненциально, а ли-
207
нейно. В итоге продукт ПЦР оказывается представленным, в
основном, одноцепочечными молекулами ДНК. Кроме выше-
вышеупомянутого случая с множественной ПЦР, асимметричную
ПЦР используют также для проведения непосредственного
секвенирования продукта ПЦР [272], для наработки одноцепо-
чечных ДНК при создании олигонуклеотидных микрочипов
[273] и в других приложениях (например, при исследовании по-
полиморфизма одноцепочечных фрагментов ДНК (single-strand-
(single-stranded conformation polymorphism - SSCP) в ДНК-диагностике, тре-
требующих для своей реализации использования оцДНК.
6.3.4. Гнездовая ПЦР
Этот метод часто выручает, когда другими способами не
удается освободиться от неспецифических продуктов ПЦР и
"шмира". Принцип метода гнездовой ПЦР (nested PCR) заклю-
заключается в последовательном применении двух пар праймеров,
специфичных в отношении исследуемого локуса (рис. 19). Вна-
Вначале проводят ПЦР с внешними праймерами. В результате по
разным причинам синтезируется некий гетерогенный продукт,
в котором нужный фрагмент может визуально не наблюдаться
из-за его низкой концентрации. Небольшое количество обра-
образовавшегося продукта ПЦР без какой-либо очистки далее ис-
используют в качестве матрицы в новом раунде ПЦР, которую
осуществляют с помощью новых {внутренних) праймеров,
комплементарных внутренним последовательностям локуса,
амплифицированного в первом раунде ПЦР. Поскольку только
у специфического продукта ПЦР будут присутствовать места
посадки для внутренних праймеров, амплификация всех осталь-
остальных продуктов ПЦР, представляющих "шмир" и полосы непо-
непонятного происхождения, с внутренними праймерами происхо-
происходить не будет. К одному из недостатков метода, который одно-
одновременно является и его достоинством, следует отнести его
чрезвычайную чувствительность, что при неаккуратной работе
может стать легким источником ложноположительных резуль-
результатов из-за загрязнения специфическим продуктом ПЦР ком-
компонентов реакционной смеси.
6.3.5. ПЦР с "горячим стартом"
Хотя оптимальной температурой для синтеза ДНК термоста-
термостабильными ДНК-полимеразами является ~72°С, они активны и
при комнатной температуре. В то же время при низкой темпера-
температуре создаются идеальные условия для неспецифического взаи-
208
ДНК-матрица
Отжиг внешних праймеров
5'
3'
Праймер 2
Праймер 1
3'
5'
Первая амплификация
Первый
ПЦР-продукт
Отжиг внутренних праймеров
Праймер 4
Праймер 3
Вторая амплификация
Конечный продукт ПЦР
Рис. 19. Гнездовая ПЦР [273]
С помощью праймеров 1 и 2 синтезируют первый продукт ПЦР, который
служит матрицей во втором раунде ПЦР с праймерами 3 и 4
модействия праймеров и матрицы в случае их неполной компле-
ментарности в соответствующих участках. В такой ситуации 3'-
концы праймеров, доступные ДНК-полимеразе при комнатной
температуре и выше вплоть до температуры денатурации ком-
комплексов праймер-матрица, удлиняются, а образующиеся при
этом полностью комплементарные матрице участки служат эф-
эффективными праймерами в следующих циклах ПЦР. В итоге об-
образующиеся по данному механизму продукты ПЦР не имеют ни-
ничего общего с последовательностью нуклеотидов, которую было
209
необходимо амплифицировать. Для преодоления этого затрудне-
затруднения необходимо создать условия, при которых удлинение прайме-
ров начиналось бы только после их отжига с соответствующими
последовательностями матрицы, при понижении температуры
после стадии денатурации в первом цикле ПЦР.
Имеется несколько подходов, получивших название ПЦР
с "горячим стартом" (hot start PCR), которые позволяют до-
достичь желаемого результата. В самых первых системах ДНК-по-
лимеразу и остальную часть реакционной смеси разделяли с по-
помощью легкоплавкого физического барьера (воска) [274]. При
таком подходе все компоненты реакционной смеси, кроме ДНК-
полимеразы и матрицы, вносят в нижнюю часть пробирки, на
смесь наносят легкоплавкий полимер и пробирки прогревают с
тем, чтобы он расплавился, и затем охлаждают до его застыва-
застывания. Далее сверху наслаивают масло, а под масло вносят недоста-
недостающие компоненты. Плавление барьера происходит в первом
цикле ПЦР после прогревания пробирок до ~55°С, что сопровож-
сопровождается объединением всех компонентов реакционной смеси, и
ПЦР начинается.
В альтернативных подходах объектом воздействия является
непосредственно ДНК-полимераза. В одном случае образуют
комплекс моноклональных антител с ферментом, в котором ан-
антитела инактивируются при прогревании в первом цикле ПЦР
[275], в другом случае фермент модифицируют по некоторым
аминокислотным остаткам термолабильными химическими
группами, что делает его неактивным при комнатной температу-
температуре. Активация Taq-ДНК-полимеразы происходит после ее про-
прогрева в течение 2-4 мин при 95°С [276]. Недостатком этого мето-
метода является снижение удельной активности Taq-ДНК-полимера-
Taq-ДНК-полимеразы, а, следовательно, и эффективности ПЦР в целом. Олигонук-
леотидные аптамеры, специфически взаимодействующие с Taq-
ДНК-полимеразой, также могут быть использованы для горяче-
горячего старта [277]. Однако, по своей природе этот метод не является
универсальным, так как в ряде случаев невозможно исключить
взаимодействие аптамеров с праймерами или матричной ДНК.
Наконец, в последнее время разработан вариант ПЦР с горя-
горячим стартом, в котором из реакции при низкой температуре вы-
выводятся праймеры, образующие характерную вторичную струк-
структуру, разрушающуюся при нагревании [278]. Для реализации это-
этого подхода к 5'-концам праймеров присоединяют одинаковые по-
последовательности, которые отсутствуют в матричной ДНК. ПЦР
. проводят в присутствии трех праймеров: двух, специфичных в от-
отношении амплифицируемого участка ДНК, и одного, комплемен-
210
тарного избыточной 5'-концевой последовательности прайме-
ров. Первые циклы ПЦР проводят при температуре отжига
праймеров, которая обеспечивает амплификацию соответствую-
соответствующего участка матрицы. Затем температуру отжига повышают,
что дает возможность лишь третьему праймеру взаимодейство-
взаимодействовать с дополнительной последовательностью на концах первых
двух. В результате образовавшиеся в предыдущих циклах про-
продукты ПЦР продолжают амплифицироваться только с помощью
этого одного праймера. С помощью такого подхода удается по-
повысить специфичность ПЦР и избежать образования димеров
праймеров, поскольку короткие продукты ПЦР из-за присутст-
присутствия на их концах самокомплементарных последовательностей
образуют структуры типа "ручки сковородки" (шпилька-одноце-
почечная петля). ДНК с подобными структурами являются при
проведении ПЦР плохими матрицами.
6.3.6. ПЦР, сопряженная с обратной транскрипцией
Метод ПЦР, сопряженной с обратной транскрипций
(ОТ-ПЦР) (RT PCR, one tube PCR), позволяет обнаруживать в об-
образцах последовательности РНК и количественно определять их
содержание, что широко используется для оценки дифференци-
дифференциальной экспрессии генов на уровне транскрипции и в ДНК-диа-
ДНК-диагностике возбудителей инфекционных заболеваний, например,
для обнаружения вируса гепатита С или ВИЧ [279]. Во время
проведения ОТ-ПЦР на матрице РНК с помощью обратной
транскриптазы синтезируют кДНК, последовательности кото-
которой далее, в свою очередь, амплифицируют в обычной ПЦР.
В современных вариантах ОТ-ПЦР (обратную транскрипцию и
ПЦР) проводят в одной пробирке, что объясняет одно из распро-
распространенных названий этого метода [263].
Для проведения обратной транскрипции используется не-
несколько ферментов. Наиболее эффективной (но и дорогостоя-
дорогостоящей) является обратная транскриптаза вируса миелобластоза
птиц (AMV), с помощью которой удается синтезировать кДНК
длиной до 14 т.п.о., вируса лейкоза мышей Молони (MMLV)
A0 т.п.о.), а также ДНК-полимеразы термофильных бактерий
Thermus thermophilus (Tth-ДНК-полимераза), Т. flavus и
Carboxydothermus hydrogenoformans (С. therm.), с помощью кото-
которых удается амплифицировать последовательности меньшей дли-
длины (до 3 т.п.о.). Бактериальные ДНК-полимеразы обнаруживают
способность использовать РНК в качестве матриц только в при-
присутствии ионов Мп2+ (кроме С. therm., которая активна в присутст-
211
вии ионов Mg2+) при повышенной температуре E0-70°С), что осо-
особенно удобно в случае амплификации последовательностей с вы-
высоким GC-составом и сложной вторичной структурой. Вариант
метода с раздельной обратной транскрипцией и ПЦР подходит
для исследования сложных смесей РНК или при необходимости
транскрибирования очень длинных последовательностей. В дан-
данном случае обратную транскрипцию проводят в условиях, полно-
полностью оптимизированных для этих ферментов, чего не удается сде-
сделать при проведении обеих стадий реакции в одной пробирке.
Ввиду чрезвычайно высокой чувствительности метода ис-
источником ложноположительных результатов могут быть даже
небольшие примеси ДНК в анализируемых образцах. Проблему
решают путем подбора праймеров таким образом, чтобы они
были комплементарны последовательностям соседних экзонов.
В этом случае продукт ПЦР не образуется на матрице ДНК из-
за большого размера интрона, если же он все-таки синтезирует-
синтезируется, его можно будет легко отличить по размерам от ожидаемо-
ожидаемого фрагмента ДНК, который должен синтезироваться на безин-
тронной матрице кДНК. Альтернативным способом борьбы с
такими артефактами является обработка анализируемых образ-
образцов ДНКазой.
6.3.7. Амплификация больших участков ДНК
с высокой точностью
Имеется несколько факторов, ограничивающих в обычных
условиях проведение амплификации протяженных последова-
последовательностей ДНК с высокой точностью (long PCR, long and accu-
accurate (LA) PCR). Среди них следует упомянуть депуринизацию
ДНК при высоких температурах, используемых для денатурации
ДНК, подавление элонгации растущих цепей ДНК стабильными
элементами вторичной структуры амплифицируемых одноцепо-
чечных участков матрицы, слишком короткое время элонгации
праймеров в соответствующих сегментах ПЦР или фрагмента-
фрагментация матричной ДНК. Первое затруднение удается частично пре-
преодолевать путем повышения значений рН реакционной смеси при
проведении ПЦР, что подавляет протонирование пуриновых ос-
оснований, которое само по себе ускоряет процесс депуринизации.
Проблема вторичной структуры матрицы может быть решена
включением в состав реакционной смеси денатурирующих аген-
агентов типа диметилсульфоксида, а третье затруднение - увеличени-
увеличением продолжительности элонгации праймеров при температуре
72°С. Образование двухцепочечных разрывов матрицы можно
212
предотвратить, выделяя ДНК в более мягких условиях, в том чис-
числе и электрофорезом в импульсном электрическом поле. Однако
наиболее серьезным фактором, ограничивающим возможность
амплификации протяженных участков ДНК-матрицы, является
низкая точность синтеза ДНК очищенными термостабильными
ДНК-полимеразами, у которых, как уже упоминалось, отсутству-
отсутствует 3' —> 5'-экзонуклеазная корректирующая активность. Нуклео-
тиды, ошибочно включенные в продукт ПЦР, резко увеличива-
увеличивают вероятность терминации синтеза ДНК в этой точке и обрыва
строящейся цепи ДНК. Такая преждевременная терминация син-
синтеза ДНК полностью предотвращает амплификации участков
ДНК, длина которых превышает 5-10 т.п.о.
Для преодоления затруднения, связанного с низкой точнос-
точностью синтеза ДНК Taq-полимеразой разработаны варианты ПЦР,
которые позволяют амплифицировать протяженные (до
40 т.п.о.) участки ДНК с меньшей частотой включения ошибоч-
ошибочных нуклеотидов по сравнению с обычными методами [280].
В таких системах обычно используют смесь двух ДНК-полиме-
раз, содержащую укороченный вариант Taq-ДНК-полимеразы
(Klentaq), не обладающей 5'—>3'-экзонуклеазной активностью, к
которому добавлена 1/160 или 1/500 часть (по единицам активно-
активности) Pfu- или Deep Vent-ДНК-полимеразы, соответственно. Два
последних фермента обладают 3'—>5 '-корректирующей активно-
активностью, что позволяет им удалять нуклеотиды, ошибочно включен-
включенные ДНК-полимеразой Klentaq в 3'-конец строящейся цепи ДНК.
А это не только способствует более точному синтезу ДНК, но и
образованию более длинных продуктов ПЦР.
Метод LA-PCR в настоящее время находит применение в
ДНК-диагностике для обнаружения крупных перестроек геном-
геномной ДНК, сопровождающихся экспансией тринуклеотидных по-
повторов [281], выявления протяженных транслокаций, а также для
амплификации целых генов, например, гена ретинобластомы
[282] или вирусных геномов [283].
6.3.8. ПЦР in situ
Этот вариант ПЦР разработан для амплификации последова-
последовательностей ДНК или РНК непосредственно на фиксированных
препаратах тканей, клеток или хромосом [284, 285]. Если с помо-
помощью обычной гибридизации in situ с использованием меченой
ДНК в качестве зонда можно обнаруживать до 10 копий анализи-
анализируемых последовательностей на клетку, то чувствительность
ПЦР in situ в -10 раз выше, т.е. позволяет обнаруживать одну ко-
213
пию ДНК на фоне 1 мкг неспецифической ДНК или (с помощью
ОТ-ПЦР) небольшое количество РНК, образовавшейся в резуль-
результате транскрипции уникального гена. Поскольку метод не требу-
требует полного разрушения клеток или тканей, что необходимо для
проведения гибридизации по Саузерну или Северного (Northern)
блоттинга, с помощью ПЦР in situ удается идентифицировать и
локализовать клетки, обладающие исследуемыми последова-
последовательностями, и определить их количество.
Постановка ПЦР in situ включает в себя четыре этапа: приго-
приготовление и фиксацию препаратов клеток или тканей, получение
полупроницаемых клеточных мембран, амплификацию последо-
последовательностей с помощью ПЦР и обнаружение ПЦР-продукта.
Фиксацию клеток и тканей обычно производят 10%-ным форма-
формалином. Далее ткани заключают в парафиновые блоки, а клетки
промывают и суспендируют в воде, содержащей диэтилпирокар-
бонат, для инактивации ДНКаз и РНКаз. После этого среды тка-
тканей и клетки переносят на предметные стекла, обработанные си-
ланом, и ткани дополнительно промывают ксиленом и этанолом
для удаления парафина. Мембраны клеток делают полупроница-
полупроницаемыми с помощью протеолитических ферментов: пепсина, трип-
трипсина или протеиназы К, причем эффективность этой стадии зави-
зависит от продолжительности фиксации препаратов формалином.
Использование первых двух ферментов предпочтительнее, по-
поскольку они менее стабильны, чем протеиназа К, и легко инакти-
вируются во время инкубации препаратов при высоких значени-
значениях рН. Окончательную инактивацию протеиназы проводят с по-
помощью диэтилпирокарбоната и промыванием препаратов абсо-
абсолютным этанолом. Далее с помощью амплификатора, снабжен-
снабженного специальным блоком, в капле на препарате проводят ПЦР с
горячим стартом, используя dUTP, меченный стероидом диокси-
генином, или биотином, и по завершению этого этапа препараты
инкубируют с антителами к диоксигенину или стрептавидином,
находящимися в комплексе с хромогеном, для обнаружения соот-
соответствующего комплекса.
6.3.9. Аллель-специфическая ПЦР
Известно, что для эффективного осуществления ПЦР
3'-концевой нуклеотид праймера должен быть комплементарен
соответствующему нуклеотиду матричной ДНК. В противном
случае эффективность удлинения праймера во время ПЦР резко
снижается и при определенных сочетаниях ошибочно спаренных
нуклеотидов не происходит. Именно эта особенность ПЦР и ле-
214
Праймер 1
Мутация И Мс_
Праймер 2
x-c-G
C-G-Q>T-C
-LLu-C-C-G-Cr>T-C
Дикий тип
,C-C-A ,CN
ЖС ~^ I I IIG C"G ^ I I 11G-G-
c-c-g-©-t-c— _LLLLc-c-g-©-t-c— I4IC c ,
Праймер 3
,G-A-C
C-C-G-0-T-C
-G-G-C-G-Ax c=>
в
174 П.О.
Рис. 20. Схема одной из современных модификаций аллель-специфиче-
аллель-специфической ПЦР (а), использованной для идентификации точковой мутации
FV Leiden в геноме человека {б и в) [286]
Со всеми аллель-специфическими праймерами использован один и тот же
встречный праймер. 3'-Концевые нуклеотиды аллель-специфических прайме-
ров комплементарны мутантному нуклеотиду Т D), нуклеотиду дикого типа С
F), праймер 7 комплементарен нуклеотиду дикого типа своей внутренней ча-
частью, а его 3'-концевой нуклеотид не комплементарен матричной ДНК. Для
усиления специфичности в праймеры 4 и 6 вблизи 3'-концов введены дополни-
дополнительные нуклеотиды, некомплементарные матрице. Праймер 4 элонгируется
ДНК-полимеразой в присутствии гомозиготной мутантной ДНК, но не ДНК
дикого типа (б- дорожки 2 и 3, соответственно), в случае праймеров 6 и 7 на-
наблюдается обратная картина (б и в - дорожки 6,8 и 1,3, соответственно). При
наличии мутации в гетерозиготном состоянии все праймеры эффективно
элонгируются {б - дорожки 5, 9, в - дорожка 4). Увеличение циклов ПЦР
сверх оптимального приводит к образованию продуктов ПЦР в тех пробах,
где при меньшем числе циклов они не образовывались (б - дорожки 4,7 и в -
дорожка 2), что может служить внутренним контролем. Праймер 7 является
универсальным, так как позволяет обнаруживать почти любую гомозиготную
точковую мутацию, попадающую в его внутреннюю часть. Использование
пар праймеров 4 и 6 или 4 и 7 дает возможность дифференцировать гомози-
гомозиготные и гетерозиготные мутации
215
жит в основе метода обнаружения мутаций с помощью аллель-
специфической ПЦР, иногда называемой аллель-специфической
элонгацией праймера (allele specific primer extension). При иссле-
исследовании способности Taq-ДНК-полимеразы инициировать синтез
ДНК на праймерах, 3'-концевой нуклеотид которых некомпле-
некомплементарен матричной ДНК, было установлено, что не все пары
ошибочно спаренных нуклеотидов праймера и ДНК-матрицы
одинаково эффективно предотвращают синтез ДНК. По способ-
способности направлять синтез ДНК все такие пары 3'-концевого нук-
леотида праймера и соответствующего нуклеотида матричной
ДНК располагаются в следующий ряд: С-С, A-G, G-A,
G-G < А-А < Т-С, С-Т, Т-Т « А-С, С-А « G-T, T-G (данные
фирмы "Perkin-Elmer Cetus"). При этом наименее благоприятны-
благоприятными для продолжения синтеза ДНК являются пары нуклеотидов
пурин-пурин и пиримидин-пиримидин.
На рис. 20 представлены схема высокоэффективного варианта
аллель-специфической ПЦР, разработанного нами и использован-
использованного для диагностики мутации FV Leiden при тромбофилиях, а так-
также полученные с помощью данного метода результаты [286]. Эта
мутация локализована в экзоне 10 гена фактора V системы свер-
свертывания крови человека и часто ассоциирована с синдромом ее по-
повышенной свертываемости. В соответствии с последовательнос-
последовательностью нуклеотидов анализируемого участка генома синтезируют два
праймера, 3'-концевой нуклеотид одного из которых комплемен-
комплементарен мутантному нуклеотиду матричной ДНК, а у другого - нук-
леотиду дикого типа (см. рис. 20 а, б). Для усиления специфичнос-
специфичности действия праймеров вблизи их 3'-концов были введены неком-
некомплементарные матрице нуклеотиды. Для исключения ложноотри-
цательных результатов в пробах, где продукт ПЦР отсутствует,
повышали число циклов ПЦР сверх оптимального, после чего, ес-
если система работает нормально, продукт ПЦР появляется и в этих
пробах, что может служить дополнительным внутренним контро-
контролем. Другой тип разработанных нами универсальных аллель-спе-
аллель-специфических праймеров содержит 3'-концевой нуклеотид, всегда
некомплементарный матрице, а мутантный нуклеотид матрицы
попадает в его внутреннюю часть (рис. 20 а, в). В этом случае про-
продукты ПЦР отсутствуют, если в гибриде во внутреннюю часть
праймера попадает любой некомплементарный мутантный нукле-
нуклеотид матричной ДНК вне зависимости от его точной локализации.
Такие праймеры позволяют обнаруживать любые точковые мута-
мутации в гомозиготном состоянии и у гаплоидных микроорганизмов.
Для выявления гетерозиготного состояния анализируемого
гена иногда используют мутантный и нормальный праймеры раз-
216
ных размеров, которые различаются по длине их 5'-концевых по-
последовательностей, полностью комплементарных анализируе-
анализируемой ДНК. В этом случае в процессе ПЦР в реакционную смесь
можно одновременно добавлять оба аллель-специфических
праймера: мутантный и нормальный вместе с общим для обоих
праймеров - встречным. Образовавшиеся продукты реакции, со-
соответствующие мутантному аллелю и аллелю дикого типа, далее
разделяют электрофоретически в гелях, применяемых для секве-
нирования нуклеиновых кислот.
При использовании аллель-специфических праймеров с це-
целью обнаружения мутаций необходимо иметь в виду, что не все
сочетания ошибочно спаренных с матрицей 3'-концевых нуклео-
тидов одинаково эффективны в блокировании ПЦР. Наимень-
Наименьшей способностью к элонгации праймеров с ошибочно спарен-
спаренными 3'-концевыми нуклеотидами обладает так называемый
фрагмент Шоффлера Taq-полимеразы, который представляет
собой молекулу ДНК-полимеразы Т. aquaticus, укороченную с
N-конца на несколько десятков аминокислотных остатков.
В то же время в этом методе не применимы термостабильные
ДНК-полимеразы, обладающие 3'—>5 '-корректирующей актив-
активностью, например, Vent-полимераза.
ПЦР с аллель-специфическими праймерами является про-
простым и эффективным методом обнаружения мутаций в геном-
геномной ДНК обследуемых индивидуумов. В отличие от всех рас-
рассмотренных выше методов аллель-специфическая ПЦР в такой
постановке позволяет находить небольшое количество мутант-
ных ДНК на фоне большого числа молекул ДНК дикого типа.
Аналогичная генетическая ситуация может иметь место в том
случае, если соматические мутации возникают в процессе онто-
онтогенетического развития организмов, и лишь небольшая часть
соматических клеток (клон соматических клеток) таких орга-
низмов-мозаиков содержит анализируемые мутации. В этом
случае, например, при онкологических заболеваниях, мутант-
ные ДНК в препаратах суммарной ДНК сильно разбавлены со-
соответствующими последовательностями дикого типа и их труд-
трудно обнаружить другими методами. Аллель-специфические
праймеры, полностью комплементарные лишь мутантным по-
последовательностям анализируемой ДНК, вовлекаются в ампли-
амплификацию таких мутантных последовательностей, а последова-
последовательности нуклеотидов ДНК дикого типа не амплифицируются.
Подобный подход позволяет обнаруживать несколько десятков
или сотен молекул мутантной ДНК на фоне десятков тысяч мо-
молекул ДНК дикого типа.
217
Зонды типа "висячий замок" (padlock probes) не имеют пря-
прямого отношения к ПЦР. Но они также, как и аллель-специфиче-
аллель-специфическая ПЦР, позволяют обнаруживать точковые мутации и нахо-
находят применение в анализе неамплифицированной геномной ДНК,
в том числе ос-сателлитных последовательностей в центромер-
ных участках метафазных хромосом человека [287]. Поэтому о
них целесообразно упомянуть в этом разделе. Такие зонды пред-
представляют собой линейные олигонуклеотиды, концы которых, со-
соединенные случайной последовательностью нуклеотидов, ком-
комплементарны анализируемому участку ДНК и после гибридиза-
гибридизации с исследуемой ДНК сближаются настолько, что между ними
остается только одноцепочечный разрыв, который может быть
ликвидирован с помощью ДНК-лигазы [288]. После этого исход-
исходный олигонуклеотид превращается в кольцевую ковалентно
замкнутую молекулу, через которую проходит цепь анализируе-
анализируемой ДНК. Циркуляризованные таким образом олигонуклеотиды
далее могут быть использованы в качестве матриц в амплифика-
амплификации по типу катящегося кольца (rolling circle amplification - RCA)
(см. раздел 6.6.3 этой главы), что позволяет увеличить количест-
количество молекул зонда до уровня, допускающего осуществлять анализ
отдельных уникальных последовательностей нуклеотидов [289].
Дальнейшее внедрение экспоненциальной амплификации лиги-
рованных зондов с помощью разветвленной амплификации по
типу катящегося кольца (branched RCA, bRCA) позволило прово-
проводить анализ замен отдельных нуклеотидов в плашках для микро-
микротитрования в гомогенной реакционной смеси при постоянной
температуре [290].
6.3.10. ПЦР, чувствительная к метилированию матрицы
По крайней мере, семь интенсивных ковалентных модифика-
модификаций азотистых оснований геномной ДНК обнаружены у живых
организмов, относящихся к различным таксономическим груп-
группам, и биологические последствия этих явлений составляют
предмет интенсивных исследований [291]. Одноклеточные эука-
риоты содержат Ы6-метиладенин (т6А) и 5-гидроксиметилурацил
(hm5U). В клетках человека и насекомых были обнаружены т6А
и №-метилгуанин (m7G). У прокариот наиболее часто встречают-
встречаются т6А и 5-метилцитозин (т5С), а также, в меньшей степени,
№-метилцитозин (т4С). Все модификации связаны с системами
рестрикции и модификации ДНК, а ш6А, кроме того, участвует в
контроле репликации и репарации ДНК у этих организмов.
У бактериофага Т4 все остатки С представлены 5-гидроксиме-
218
Геномная ДНК
сн3
б
Геномная ДНК
С
G
С
G
m
[ + bi
¦С
¦G
С
G-
3'
Денатурация + Бисульфит натрия
м м
Рестрикция, чувствительная к СН3-группам Рестрикция, нечувствительная к СН3-группам
I
У-
и
•и
m
U
3'
ПЦР (удлинение праймера)
А А G--«-A
I
ПЦР (амплификация)
I
У Т+— -Т С Т 3'
А А G -—«-а —
Секвенирование ДНК
+ Бисульфит
натрия н
С Т t
- Бисульфит
натрия
С Т
m
Электрофорез
Гибридизация по Саузерну
ПЦР
Электрофорез
и
S
S
и
в
ев ^~~
О,
са
и
О.
С
Рис. 21. Определение статуса метилирования ДНК с использованием рестриктазы, чувствительной к метилирова-
метилированию (а) или бисульфита натрия с последующей аллель-специфической ПЦР (б) [291]
а - исследуемую ДНК инкубируют с рестриктазой, чувствительной или нечувствительной к метилированию ДНК, и анали-
анализируют гибридизацией по Саузерну (внизу слева) или с помощью ПЦР (внизу справа);
б - после денатурации исследуемая ДНК инкубируется с бисульфитом натрия, что сопровождается дезаминированием ос-
остатков С и их превращением в U; статус метилирования анализируемого четырехнуклеотидного участка определяют после его
амплификации и секвенирования продукта ПЦР, результаты которого изображены в виде секвенирующего геля внизу рисунка
тилцитозином (hm5C) или гликозилированными hm5C, а у расте-
растений до 50% остатков С метилированы (т5С). Вообще, последняя
модификация наиболее часто встречается у эукариот с размером
генома > 108 п.о. (т.е. у животных и растений), но редка у организ-
организмов с меньшим геномом (в том числе дрожжей, двукрылых насе-
насекомых и нематод). Хотя в клетках млекопитающих только 3-10%
остатков С метилированы, данная модификация играет большую
роль в функционировании их генома. У этих организмов образо-
образование т5С ассоциировано с регуляцией транскрипции, в том чис-
числе инактивацией Х-хромосом, импринтингом, дифференциров-
кой клеток и различными патологическими процессами, вклю-
включая канцерогенез.
Как уже упоминалось, одним из простых и эффективных ме-
методов обнаружения метилированных остатков С и А в ДНК явля-
является использование эндонуклеаз рестрикции, чувствительных к
метилированию (ЭРЧМ) (см. раздел 2.2) (рис. 21, а). В этом случае
инкубация изучаемой ДНК с ЭРЧМ в зависимости от статуса ме-
метилирования исследуемого участка может приводить или не при-
приводить к разрушению ампликона и подавлению его амплифика-
амплификации в ПЦР. Однако, поскольку не всегда в нужных местах генома
присутствуют соответствующие сайты рестрикции, применение
таких ферментов имеет ограничения. При другом распространен-
распространенном подходе, лишенном этого недостатка, используют спо-
способность бисульфита (HSO3) превращать в оц ДНК остатки С
(но не т5С) в U. В этом случае праймеры в ПЦР могут быть по-
подобраны таким образом, что их элонгация будет происходить при
наличии соответствующих модификаций в анализируемом участ-
участке ДНК, однако, еще более информативным окажется секвени-
рование продуктов ПЦР, в результате которого легко обнаружи-
обнаруживаются замены С —> U (рис. 21, б). Данный метод обладает высо-
высокой чувствительностью (требуется не более 10 нг ДНК) и позво-
позволяет количественно оценивать наличие т5С в исследуемых об-
образцах, обнаруживая до 5% метилированных остатков С в анали-
анализируемом ампликоне.
6.3.11. Иммуно-ПЦР
Иммуно-ПЦР (immuno-PCR - IPCR) является сверхчувстви-
сверхчувствительным методом обнаружения белков и других антигенов
[292-294]. Разработанный впервые Т. Сано и др. в 1992 г. метод
иммуно-ПЦР обеспечивает ~ 1000-кратное увеличение чувстви-
чувствительности при детекции антигенов по сравнению с обычными им-
муноферментными методами анализа (enzyme-linked immunosor-
220
bent assay - ELISA). В этой группе методов используют ковалент-
ные конъюгаты фрагментов одноцепочечной или двухцепочеч-
ной ДНК с антителами или их генно-инженерными производны-
производными в качестве зондов к исследуемым антигенам (рис. 22, я). В та-
таком варианте антиген, иммобилизованный на твердом носителе,
инкубируют с антителами в виде конъюгатов с маркерным фраг-
фрагментом ДНК. После отмывания избытка несвязавшегося конъю-
гата оставшийся ампликон обнаруживают с помощью ПЦР. Во
второй модификации метода (рис. 22, б) иммуно-ПЦР использу-
используют для конкурентного обнаружения низкомолекулярных антиге-
антигенов. В одном из таких случаев, иллюстрирующем принцип данно-
данного метода, свободный анализируемый гаптен конкурирует с
конъюгатом гаптен-стрептавидин за сайты связывания в молеку-
молекулах антител, иммобилизованных на твердой подложке. Конъю-
гат, оставшийся в связанном состоянии, определяется по присут-
присутствию маркерных фрагментов ДНК с помощью ПЦР.
Системы биотин-стрептавидин или биотин-авидин часто ис-
используются для формирования различных межмолекулярных
комплексов. Авидин - белок куриного яйца, и его функциональ-
функциональный аналог, бактериальный белок стрептавидин из Streptomyces
avidinii, образуют чрезвычайно прочные комплексы с авидином
(формула изображена на рис. 22, в) с константой ассоциации
~1015. Использование стрептавидина в различных приложениях с
нуклеиновыми кислотами предпочтительнее, чем авидина, по-
поскольку его изоэлектрическая точка (pi) лежит вблизи 7,0, а сле-
следовательно при физиологических значениях рН неспецифичес-
неспецифическое взаимодействие этого белка с ДНК менее вероятно. Кроме
того, молекулы стрептавидина не гликозилированы, что опять-
таки предотвращает образование неспецифических комплексов.
Молекулы биотина могут быть присоединены ковалентными
связями к 3'- и 5'-концевым нуклеотидам нуклеиновых кислот, а
также включены во внутренние части макромолекул в составе
различных конъюгатов с дезоксирибонуклеозидтрифосфатами,
один из которых изображен на рис. 22, в.
Стрептавидин является гомотетрамерным белком, содержа-
содержащим четыре сайта связывания молекул биотина, по одному на
каждой цепи. На рис. 22, г представлена схема самосборки олиго-
мерных конъюгатов стрептавидин-ДНК из биотинилированных
фрагментов ДНК и молекул стрептавидина. В этом примере
фрагменты ДНК биотинилированы по обоим 5'-концам и, взаи-
взаимодействуя с четырехвалентными молекулами стрептавидина,
образуют трехмерные межмолекулярные сети. Анализ с помо-
помощью электрофореза и сканирующей силовой микроскопии пока-
221
а
Сигнал
Маркерная
ДНК
Конкурентная
иммуносорбция
Сигнал
в
о
HN
0 0 0
-°--р-о-р-о-р-о-|^о
О" О" О"
он
н
О
я
HN HN
Биотинилированный дезоксиуридинтрифосфат
A)
B)
C)
О
D)
рис. 22. Использование ДНК-белковых конъюгатов в иммуно-ПЦР
[294]
а - обнаружение высокомолекулярных антигенов. Антиген, иммобилизо-
иммобилизованный на твердом носителе, взаимодействует с конъюгатом антитело-ДНК,
после чего наличие маркерной ДНК в комплексе выявляют с помощью ПЦР;
прямоугольник с восклицательным знаком указывает на важность этапа синте-
синтеза конъюгата для создания эффективной системы;
б - обнаружение низкомолекулярных гаптенов с помощью конкурентной
иммуно-ПЦР. Анализируемый свободный гаптен конкурирует за иммобилизо-
иммобилизованные антитела с конъюгатом гаптен-стрептавидин-ДНК; по окончанию инку-
инкубации и отмывки несвязавшихся молекул маркерную ДНК обнаруживают с по-
помощью ПЦР; b - сайты связывания биотина на гомотетрамерной молекуле
стрептавидина, Гап - анализируемый гаптен, дуга изображает маркерную ДНК,
меченную по обоим 5'-концам биотином, стрелки находятся на 3'-концах;
в - биотинилированный дезоксиуридинтрифосфат, используемый для вве-
введения биотиновой метки в молекулы ДНК в процессе их биосинтеза;
г - схема самосборки конъюгатов гаптен-стрептавидин-ДНК; 1 - стрепта-
видин; 2 - фрагмент ДНК, меченный по 5'-концам биотином; 3 - олигомерный
конъюгат стрептавидин-ДНК; 4 - наночастицы стрептавидин-ДНК, преимуще-
преимущественно образующиеся после термального разрушения комплексов 3; 5 - ком-
комплекс наночастицы с конъюгатом биотин-гаптен; R - молекула гаптена; стрел-
стрелки обозначают 3'-концы ДНК
зал, что после термального разрушения таких сетей и повторной
сборки комплексов в них начинают преобладать двухвалентные
молекулы стрептавидина, соединяющие соседние молекулы
ДНК, получивших название ДНК-содержащих наночастиц
(nanocircles). Как следствие, олигомерные конъюгаты стрептави-
стрептавидина с ДНК еще сохраняют значительную способность взаимо-
взаимодействовать с биотином, в том числе и в иммуно-ПЦР-тестах, на-
направленных на конкурентное обнаружение низкомолекулярных
гаптенов (рис. 22, б). В целом, фрагменты ДНК соединяют с ан-
антителами с помощью химических поперечных сшивок [293], хи-
химерных белков с двойной специфичностью [292], путем взаимо-
взаимодействия между стрептавидином и биотином [295], а также с ис-
использованием только что рассмотренных олигомерных самосо-
самособирающихся конъюгатов стрептавидин-ДНК [296].
6.3.12. Количественная ПЦР (ПЦР в реальном времени)
При исследовании нуклеиновых кислот методом ПЦР часто
возникает задача определения числа копий матричной ДНК или
РНК в пробе. Сравнение интенсивности свечения продуктов
ПЦР, полученных обычными методами, после их разделения в
агарозном геле в ряде случаев может дать какое-то представле-
представление о содержании матрицы, однако такой метод оценки не явля-
223
ется количественным. Между тем, имеется острая необходимость
в точном определении различий в содержании матричной ДНК в
1,5 или 2 раза (например, для идентификации трисомии по хромо-
хромосоме 21 при синдроме Дауна или гетерозиготных мутаций, соот-
соответственно). Не менее важной задачей является и определение
числа копий мРНК в клетке для количественной оценки уровня
экспрессии конкретных генов.
Теоретически количество ПЦР-продукта в пробе после за-
завершения каждого цикла ПЦР должно удваиваться. Однако это-
этого не происходит, поскольку в реальных условиях ни одна из ста-
стадий реакции не заканчивается со 100%-ным выходом. На эффек-
эффективность прохождения отдельных циклов оказывают большое
влияние различные ингибиторы ПЦР, причем наименее эффек-
эффективными оказываются завершающие циклы. Поэтому предска-
предсказать количество продукта, которое должно образоваться по за-
завершению ПЦР в каждом конкретном случае не представляется
возможным. Этот клубок труднопреодолимых проблем частично
удается распутать с помощью группы современных методов, по-
получивших название ПЦР в реальном времени (real-time PCR).
В основе таких методов лежит количественное определение со-
содержания продукта ПЦР в реакционной смеси в каждом цикле
реакции. Для этой цели разработано несколько подходов, в кото-
которых используются флуоресцентно меченные олигонуклеотиды.
Система TaqMan. В 1991 г. П. Холланд с соавт. предложили
использовать 5'—>3'-экзонуклеазную активность Taq-полимеразы
для контроля за эффективностью прохождения циклов ПЦР по
освобождению в реакционную смесь флуоресцентной метки, ас-
ассоциированной с 5'-концевым нуклеотидом зонда [297]. Именно
этот подход лег в основу одного из наиболее широко распростра-
распространенных современных методов количественной ПЦР в реальном
времени. В разработке фирм Perkin-Elmer-Applied Biosystems дан-
данный метод получил название "TaqMan". При реализации этого
подхода обычно используют Taq- или Tth-полимеразы, однако, в
принципе, те же функции может выполнять любая другая ДНК-
полимераза, обладающая 5'—>3'-экзонуклеазной активностью,
например, ДНК-полимераза Tfl [298].
На рис. 23, а представлена схема использования метода
TaqMan для количественного определения мРНК. После синтеза
кДНК стандартным методом с помощью обратной транскрипта-
зы ее используют в качестве матрицы в ПЦР. В реакционную
смесь, кроме двух обычных праймеров, добавляют олигонуклео-
тидный зонд, меченный по 5-концу флуорофором, а также со-
содержащий в своей центральной или 3-концевой части тушитель
224
а б
5'111" l"Z'M
¦ Обратная
1 транскрипция
у """»" tlHIIIIIHIIIinilllllll 1 1111 Illllllllllllllll Зу
щ Денатурация
1 Отжиг праймера у v
Ў и зонда з'111111111111111111111111111111111111111Н11111111Н11111111Н111111111Н111111ШШ11-5>
5>" ' ' ' NIIIIHIIIIIIIIIIIIIIIII j, ¦
¦ Денатурация
j'"'"""llll 1ИП...|||1111 Illllllllllllll , , с. ^
Гибридизация у J ^ j
Полимеризация
3'"'"""""""" iiiiiimiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiiij'
5'||||||||1ни11111111П11111 пинии iiiiiiiiiiiiiiiiiiiiiiiiiiiii-?' | Отжиг праймера
1 и "молекулярного
~ маяка"
jllllllllllllllllllllllllllll llHIIIIIIIIIIIIIIIIIIHIll <> У Illllllllllllllllllllllllly
Вытеснение
цепи зонда
5>1 ' ' IIIIIIIIIIIIIIIIIIIMIIIIIIIII л. s, ^ <^
у I'^.Zi I I
j'llllllllllllllllllllllllllllllHiiiilllllllilllillllllllllliiiiiiiiiiiiiiiiiiiiiiniiiij' ¦ Полимеризация
¦ Гидролиз ДНК
Ж. Полимеризация с. у
^г •' niiiiiiiiiiiiniiitiiMHiiiiiiiiiuimnHn \\\\\|Ц|||Ц||MIIHlllllllllliHIII
у " 5'
g-111111111111 1 Illtllininillliffll
5 7ТТ1ТТПТЛТТГПШТТШТТПТ1ТТТ1ТГПТ1ШТТТТТТТТТТТТТТТТПТТТТТТТТТТПТТЛТТТПТТТТ'3 ?г1ШИ|И1|1ШШШШММ111И11Димт1тпт^т^тш^Ш^Ш^^^^. ?'
j'lllllllllllllllllllllllllllllllllllllllllllllllllllllllllllllllllllllllllllllllffllEj' J
Рис. 23. Обнаружение продуктов ПЦР в реальном времени с использо-
использованием зондов TaqMan (а) и "молекулярных маяков" (б) [279]
флуоресценции. Зонд отжигается в каждом цикле ПЦР с цент-
центральным участком ампликона между двумя амплифицирующими
его праймерами. Для того, чтобы сам зонд не использовался в ка-
качестве праймера, его 3'-концевую гидроксильную группу блоки-
блокируют ортофосфатом. Элонгацию праймеров проводят при той
Же температуре, что и температура отжига праймеров и зонда.
Во время ПЦР на стадии элонгации праймера Taq-полимераза,
Достигнув 5'-конца зонда, начинает вытеснять его из дуплекса, а
8. Патрушев Л.И. Т. 1
затем отщепляет 5'-концевой нуклеотид зонда, меченный флуо-
рофором, в точке раздвоения цепей ДНК и зонда [299]. Посколь-
Поскольку экзонуклеазная активность Taq-полимеразы специфична в от-
отношении двухцепочечных ДНК [300], зонд, находящийся в реак-
реакционной смеси в свободном состоянии, остается интактным. По
мере углубления ПЦР интенсивность флуоресценции реакцион-
реакционной смеси увеличивается, и это возрастание происходит ступен-
ступенчато по завершению стадии элонгации праймера в каждом цикле
ПЦР. Свободный зонд, находясь в растворе в контакте с тушите-
тушителем флуоресценции, создает небольшой базальный уровень флу-
флуоресценции, который преодолевается по мере накопления сво-
свободного флуорофора в реакционной смеси. С этого момента, оп-
определяемого числом прошедших циклов ПЦР, которое называют
порогом числа циклов (Ct - threshold cycle), появляется возмож-
возможность наблюдать в реальном времени за кинетикой накопления
флуорофора в реакционной смеси. Значение Ct обратно пропор-
пропорционально количеству амплифицируемой матрицы в пробе. Чем
выше концентрация амплифицируемой последовательности в ре-
реакционной смеси, тем меньшее количество циклов требуется для
того, чтобы интенсивность флуоресценции освободившегося
флуорофора в пробе превысила базальный уровень.
Физическое разделение флуоресцентных красителей - репор-
репортера и тушителя флуоресценции - в процессе гидролитического
разрушения зонда снимает подавление флуоресценции в анализи-
анализируемой части спектра. Максимальный сигнал флуоресценции по-
получают в том случае, если в зонде оба красителя максимально
удалены друг от друга, т.е. находятся на 5'- и 3'-концах олигонук-
леотида. Это вызвано, по-видимому, более эффективным рас-
расщеплением зонда ДНК-полимеразой [301]. Поскольку ДНК-по-
ДНК-полимераза может гидролизовать зонд лишь в составе гибрида с
ДНК-матрицей, температура стадии элонгации праймера не
должна превышать температуру плавления (Тм) гибрида. Боль-
Большинство используемых зондов обладают Тм ~70°С, поэтому на
практике при проведении ПЦР в реальном времени в системе
TaqMan стадии отжига зонда и праймеров, а также элонгации
объединяют и проводят при 60-62°С. Это приводит к тому, что
ДНК-полимераза функционирует в субоптимальных условиях и
делает всю систему TaqMan менее эффективной и гибкой по
сравнению с системами ПЦР в реальном времени, основанными
на других принципах (см. ниже).
На рис. 24 представлены результаты использования ПЦР в
реальном времени в формате TaqMan для обнаружения гетерози-
гетерозиготной делеции, приводящей к развитию ос-талассемии у челове-
226
i'HVR
в
Зонд TaqMan
SI S2
S3
Делеция
SEA
9 11 13 15 17 19 21 23 25 27 29 31 33 35 37 39
Число циклов
y = 30,54-2,87x
Коэффициент корреляции = -0,9965
0,28
1,4 2,8 7 14 28
ДНК (нг)
Рис. 24. Обнаружение делеции SEA в гетерозиготном состоянии у паци-
пациентов с а-талассемией с помощью ПЦР в реальном времени в формате
TaqMan [302]
а - схема расположения кластера а-глобиновых генов человека и делеции
SEA; HVR - гипервариабельный участок, S1-S3 - праймеры, использованные в
ПЦР, стрелка указывает на флуоресцентно меченный зонд;
б - кинетика накопления свободного флуорофора при анализе ДНК паци-
пациента, содержащего гетерозиготную делецию SEA; ПЦР проводили в разных
пробирках: 1 - с праймерами S1 и S2; 2 - с праймерами S1 и S3; 3 - базовая ли-
линия, полученная с помощью контрольного образца, в котором отсутствует мат-
матрица;
в - определение чувствительности метода с праймерами S1 и S2, Ct - порог
числа циклов
8* 227
ка [302]. ос-Талассемия представляет собой генетическое заболе-
заболевание, сопровождающееся резким снижением или полным пре-
прекращением биосинтеза ос-глобина. В Юго-Восточной Азии
60-90% потерь плода, вызванных водянкой, связано с гомозигот-
гомозиготной делецией SEA длиной ~20,5 т.п.о., которая удаляет оба ос-гло-
биновых гена, локализованных на хромосоме 16, но оставляет
интактным ген эмбрионального ^-глобина (рис. 24, а). В этой свя-
связи определение носительства делеции среди населения Юго-Вос-
Юго-Восточной Азии является весьма актуальным.
Праймер S1 и зонд гибридизуются с последовательностью,
расположенной непосредственно перед делецией, и эта мутация
не оказывает влияния на их взаимодействие с матричной ДНК.
Место посадки праймера S2 удаляется делецией, и в этом случае
соответствующий ПЦР-продукт не образуется в присутствии
праймеров S1 и S2 в реакционной смеси, хотя реакция на мутант-
ной матрице пройдет в присутствии праймеров S1 и S3, которые,
в свою очередь, не функционируют на ДНК без делеции из-за
слишком большого расстояния между этими праймерами. Таким
образом, образование продукта ПЦР как в присутствии прайме-
праймеров SI + S2, так и S1 + S3 в реакционной смеси свидетельствует о
наличии гетерозиготной делеции SEA в ДНК, как это представ-
представлено на рис. 24, б. Хотя теоретически эффективность образова-
образования продуктов ПЦР в присутствии обеих пар праймеров при на-
наличии гетерозиготной делеции SEA должна быть идентичной,
выявляемые с помощью ПЦР в реальном времени различия мо-
могут быть связаны с длинами ампликонов, поскольку все осталь-
остальные условия проведения ПЦР в этих опытах были одними и теми
же. С помощью серийных разведений образцов ДНК определили
чувствительность системы ПЦР в реальном времени, которая в
обсуждаемом примере позволяла обнаруживать 0,28 нг (~20 ко-
копий) ДНК в пробе (рис. 24, в).
Прогресс количественной ПЦР в формате TaqMan во многом
обязан разработке зондов, которые позволяют эффективно об-
обнаруживать 5'—>3'-экзонуклеазную активность термостабильных
ДНК-полимераз в реальном времени. В качестве флуорофора
часто используют 6-карбоксифлуоресцеин (FAM), а тушителя
флуоресценции - 6-карбокситетраметилродамин (TAMRA) [303].
В качестве тушителя флуоресценции часто применяют Dabcyl D-
D'-dimethylaminophenylazo)benzoic acid). В паре с ним TAMRA уже
выступает в роли флуорофора. Оптимальная длина ампликона
для ПЦР в реальном времени, как правило, меньше 100 п.о.
F0-80 п.о.). ПЦР-продукты, полученные с использованием амп-
ампликонов такой длины, более эффективно денатурируют, а время
228
фазы элонгации может быть сокращено до 10-15 сек, что замет-
заметно ускоряет весь процесс амплификации.
Система FRET. Оценку скорости образования продуктов
ПЦР в реальном времени осуществляют и с использованием дру-
других физических принципов. В частности, функционирование зон-
зондов второго типа основано па резонансном переносе энергии флу-
флуоресценции (fluorescent resonance energy transfer - FRET) [304].
В этом случае два олигонуклеотида, специфичные в отношении
продукта ПЦР, тандемно гибридизуют с продуктом в каждой фа-
фазе отжига праймеров. Выше расположенный зонд содержит на
своем З'-конце возбуждающий краситель (в частности, FITC), а
следующий за ним олигонуклеотид мечен по 5'-концу красите-
красителем-репортером (например, RED640). Таким образом, в этой сис-
системе два вышеупомянутых красителя оказываются пространст-
пространственно сближенными друг с другом после гибридизации с продук-
продуктом ПЦР. После этого возбуждение FITC сопровождается пере-
переносом электронов на акцепторную молекулу RED640, которая
испускает кванты света определенной энергии в более длинно-
длинноволновой части спектра. Интенсивность флуоресценции четко
коррелирует с количеством связавшихся зондов, а, следователь-
следовательно, и с количеством образовавшегося продукта ПЦР.
"Молекулярные маяки". Третий тип зондов, используемых
для обнаружения специфических продуктов ПЦР в реальном вре-
времени, получил название "молекулярных маяков" (molecular bea-
beacons) [305, 306] (рис. 23, б). Такие олигонуклеотиды обладают тре-
тремя характерными чертами. К 5'- и З'-концам "молекулярного ма-
маяка" присоединены, соответственно тушитель флуоресценции и
флуорохром. Последовательности, непосредственно примыкаю-
примыкающие к этим красителям, самокомплементарны, поэтому в реакци-
реакционной смеси такие зонды приобретают структуру типа "стебель-
петля", в которой тушитель и флуорохром пространственно сбли-
сближены, и флуоресценция флуорохрома-репортера подавлена. В ка-
качестве тушителей флуоресценции в этих системах часто использу-
используют нефлуоресцирующие хромофоры, которые, получив энергию
от флуорофора, рассеивают ее в виде тепла. Область петли зонда
содержит последовательность, комплементарную анализируемо-
анализируемому продукту ПЦР. При температурах более низких, чем темпера-
температура отжига зонда на продукт ПЦР, структура стебля сохраняет-
сохраняется и "молекулярный маяк" находится в выключенном состоянии.
После образования гибрида между зондом и продуктом ПЦР или
Денатурации зонда тушитель и флуорохром пространственно раз-
разделяются, флуорохром начинает флуоресцировать, то есть "ма-
"маяк" зажигается и начинает функционировать. Возбуждение элек-
229
тронов красителей осуществляют аргоновым лазером, а с помо-
помощью оптико-волоконного кабеля испускаемый свет выводится в
прибор с зарядовой связью, данные от которого обсчитываются
компьютером с помощью специальных алгоритмов.
Системы ПЦР в реальном времени, основанные на "молеку-
"молекулярных маяках", обладают гораздо большей специфичностью,
чем система TaqMan, и позволяют обнаруживать одиночные за-
замены нуклеотидов в ДНК. Кроме того, в отличие от последней
системы, они пригодны для количественной оценки содержания
продукта ПЦР в реакционной смеси по завершении реакции с по-
помощью флуориметра. "Молекулярные маяки" могут быть ис-
использованы и для обнаружения продуктов реакции в системах
изотермической амплификации, которые будут подробнее рас-
рассмотрены ниже [307].
К недостаткам этой группы методов следует отнести необхо-
необходимость проведения очень тщательного дизайна зондов, для каж-
каждого из которых требуется проводить определение индивидуаль-
индивидуальных профилей плавления. Многие зонды такого типа имеют тен-
тенденцию к образованию в процессе рефолдинга после тепловой
денатурации альтернативных пространственных структур, что
сопровождается резким усилением фоновой флуоресценции, со
всеми вытекающими отсюда негативными последствиями для
чувствительности системы.
Флуоресцентные красители, связывающиеся с ДНК. Четвер-
Четвертый тип систем обнаружения продуктов ПЦР в реальном време-
времени основан на непосредственном взаимодействии флуоресцент-
флуоресцентных красителей (например, SYBR Green) с дц ДНК [308]. Краси-
Краситель, не связавшийся с ДНК, обнаруживает слабую флуоресцен-
флуоресценцию в растворе, которая возрастает в результате связывания кра-
красителя дц ДНК по мере элонгации праймеров в цикле ПЦР. На
стадии денатурации интенсивность флуоресценции снова умень-
уменьшается. В соответствии с этим интенсивность флуоресценции ре-
реакционной смеси измеряют в конце этапа элонгации каждого
цикла. Хотя метод исключает необходимость в дополнительных
флуоресцентных зондах, его специфичность низка и целиком оп-
определяется специфичностью взаимодействия праймеров с ДНК-
матрицей. Тем не менее, эффективность такой системы можно
повысить, фиксируя интенсивность флуоресценции суммарного
продукта ПЦР как функцию от температуры плавления (Тм) амп-
ликона. Для этого температуру реакционной смеси медленно по-
повышают сверх значений Тм ампликона, фиксируя при этом флу-
флуоресценцию, что в ряде случаев позволяет идентифицировать
сигнал, исходящий от требуемого продукта ПЦР в виде достаточ-
230
но узкого пика на фоне широких пиков, появляющихся при дру-
других температурах, которые относятся к артефактам ПЦР. Вто-
Вторым недостатком данного метода является зависимость интен-
интенсивности флуоресценции от размера продукта ПЦР. Чем больше
длина синтезированной ДНК, тем большее количество молекул
красителя с ней соединяется.
Праймеры типа "Скорпион". В одной из последних разрабо-
разработок по обнаружению продуктов ПЦР в реальном времени ис-
используются праймеры, которые сами заключают в своей после-
последовательности флуоресцентно меченные зонды [309]. Такие
праймеры выполняют двойную функцию: обычную, обеспечивая
синтез цепей ДНК, и функцию зонда, которая реализуется после
включения праймера в продукт ПЦР. Принцип действия прайме-
ров "Скорпион" представлен на рис. 25, из которого также видно,
почему этот класс олигонуклеотидов получил такое экзотичес-
экзотическое название. Действительно, графическое изображение прайме-
праймера в составе продукта ПЦР отчасти напоминает профиль этого
отшельника пустыни.
З'-Концевая часть олигонуклеотида "Скорпион" заключает в
себе последовательность, комплементарную анализируемому
участку ДНК, и в ПЦР выполняет непосредственную роль прай-
праймера. 5'-Концевая часть олигонуклеотида сконструирована та-
таким образом, что образует структуру типа "стебель-петля". За
счет этого происходит сближение флуорофора, присоединенного
непосредственно к 5'-концу "Скорпиона" с тушителем флуорес-
флуоресценции, расположенным в центральной части олигонуклеотида
на границе последовательности праймера. В этом отношении
пространственная структура олигонуклеотидов "Скорпион" на-
напоминает таковую "молекулярных маяков" и выполняет ту же
функцию - обеспечивает тушение флуоресценции флуорофора
до тех пор, пока олигонуклеотиды находятся в свободном состо-
состоянии. Однако, в отличие от "молекулярных маяков", участок
ДНК, находящийся в петле "Скорпиона", комплементарен после-
последовательности нуклеотидов амплифицируемой последовательно-
последовательности ДНК (ампликона), что и обеспечивает ему уникальные свой-
свойства. Кроме того, непосредственно за тушителем флуоресценции
в цепь ДНК вводится мономер гексэтиленгликоля (HEG), кото-
который блокирует прохождение ДНК-полимеразы вдоль матрицы
до 5'-конца, т.е. прекращает амплификацию в этой точке, и по-
последовательность, образующая структуру типа "стебель-петля",
остается неамплифицированной.
Во время амплификации праймеры "Скорпион", как обычно,
включаются в продукт ПЦР (рис. 25, а). При прохождении стадий
231
3' 5'
а
5'-
Обратная транскри1щия
б
5'™
Денатурация
Отжиг
Полимеризация
5'
•5'
¦5'
Тепловая денатурация
,-3'
Охлаждение
Измерение флуоресценции
Тушитель флуоресценции
Флуорофор
ПЦР-праймер
Блокатор
Рис. 25. Праймеры типа "Скорпион", используемые для обнаружения
продуктов ПЦР в реальном времени [279]
Олигонуклеотиды этого типа объединяют в одной (а) или двух (б) молеку-
молекулах специфический праймер для ПЦР, расположенный в их 3'-концевой части,
флуорофор на 5'-конце, тушитель флуоресценции, находящийся вне праимера
ближе к его 5'-концу, и ингибитор (блокатор) синтеза ДНК, препятствующий
использованию 5'-концевой части олигонуклеотида во время ПЦР в качестве
матрицы. Кроме того, центральная часть олигонуклеотида комплементарна 5'-
концевому участку амплифицируемого продукта ПЦР, что удерживает флуоро-
флуорофор вдали от тушителя флуоресценции после образования петли ДНК в фазе
отжига праймеров
отжига праймеров и их удлинения последовательность петли
"Скорпиона" внутримолекулярно гибридизуется с продуктом
ПЦР, что приводит к разделению флуорофора и тушителя и со-
сопровождается появлением сигнала флуоресценции. Последова-
Последовательность ампликона, с которой гибридизуется петля, обычно
начинается через три нуклеотида от З'-конца праимера "Скорпи-
"Скорпион". В этих условиях внутримолекулярная гибридизация стано-
становится кинетически более предпочтительной, поскольку она не за-
зависит от вероятности встречи зонда, который присутствует в ре-
реакционной смеси в низкой концентрации, с другим продуктом
ПЦР. Это позволяет проводить ПЦР в условиях более коротких
232
циклов, а возникающий при этом сигнал обладает значительно
большей интенсивностью, чем сигналы, генерируемые зондами
TaqMan и "молекулярными маяками" [310].
При использовании стандартных праймеров "Скорпион" име-
имеет место некоторое тушение флуоресценции даже после их гиб-
гибридизации с ампликоном, из-за пространственной близости флу-
орофора и тушителя флуоресценции. Дальнейшее усиление ин-
интенсивности сигнала в этой системе достигается путем отделения
флуорофора и тушителя друг от друга, которые связывают с от-
отдельными олигонуклеотидами, комплементарными друг другу
(рис. 25, б).
6.4. Амплификация последовательностей
с неизвестной первичной структурой
Для амплификации последовательностей с использованием
обычной ПЦР необходимо знание первичной структуры обеих
последовательностей, фланкирующих анализируемый ампликон,
что является существенным ограничением использования метода
в анализе последовательностей неизвестной структуры. В иссле-
исследовательской работе часто встречаются ситуации, когда известна
последовательность, расположенная с одной стороны ампликона,
а другую требуется определить, то есть совершить переход от из-
известной последовательности генома к соседней неизвестной. Для
решения этой задачи создано несколько методов, объединенных
под общим названием односторонней ПЦР (single-sided PCR).
6.4.1. Использование ПЦР для продвижения вдоль ДНК
от участка с известной первичной структурой
в сторону неизвестной последовательности
Теоретически наиболее просто проблема амплификации не-
неизвестной последовательности решается в том случае, если она
входит в состав рестрикционного фрагмента ДНК, длина которо-
которого допускает его использование для ПЦР, и к ней прилегает по-
последовательность с известной первичной структурой (изображе-
(изображена пунктирной линией на рис. 26, а) [311]. К обоим концам фраг-
фрагмента по открытым сайтам рестрикции с липкими концами при-
присоединяют олигонуклеотидные адаптеры, иногда называемые
шплинтами (splints), что позволяет в дальнейшем использовать
последовательности адаптеров в качестве мест посадки прайме-
праймеров при ПЦР. В удачно складывающихся обстоятельствах при ис-
использовании двух праймеров, комплементарных известному уча-
233
Цитирование
адаптеров
а
5'ОН
-|—|-
З'ОН
tZD *-
ОН Р ...
он - р
Лигирование
I Денатурация
1 ДНК-полимераза I
Jnup
Добавление I
праймеров \
СИ]
—rzu-
ПЦР I
—•сиз
—•сиз +
си—
ПЦР
в
Очистка со
стрептавидином
CD-
Лигирование
| Денатурация
1 Отжиг праймера
.п.9_
I Продолжение ПЦР
"ПС
стку матрицы и направленных в противоположные стороны по
направлению к неизвестным последовательностям, а также одно-
одного праймера, соответствующего использованному адаптеру, уда-
удается получить продукты ПЦР, большая часть которых составле-
составлена неизвестными последовательностями (рис. 26, б). Однако ча-
чаще всего реакционная смесь после проведения ПЦР в таких усло-
условиях оказывается сильно загрязнена неспецифическими продук-
продуктами, которые берут начало от других неспецифических фраг-
фрагментов ДНК без известной последовательности, фланкирован-
фланкированных теми же адаптерами (рис. 26, в).
Повышение специфичности ПЦР в этом случае может быть
достигнуто несколькими путями, в том числе, с помощью очист-
очистки промежуточных продуктов ПЦР (метод аффинной ПЦР -
capture PCR). Для этого праймеры к известной последовательно-
последовательности можно пометить биотином, и меченные таким образом про-
продукты ПЦР отделить от остальных продуктов реакции с помо-
помощью аффинной хроматографии на колонках с иммобилизован-
иммобилизованным стрептавидином. Альтернативно можно не допустить амп-
Рис. 26. Использование ПЦР для продвижения вдоль ДНК от участка с
известной первичной структурой в сторону неизвестной последователь-
последовательности [311]
а - молекула ДНК, содержащая в центральной части известную последова-
последовательность (пунктирные линии), фланкированную с обеих сторон неизвестными
последовательностями (сплошные линии);
б - успешная амплификация неизвестных последовательностей, располо-
расположенных рядом с известной;
в - артефакт: амплифицирована неизвестная последовательность, не свя-
связанная с известным участком ДНК;
г - амплификация неизвестных последовательностей с использованием
очистки промежуточных продуктов ПЦР с помощью биотинилированных прай-
меров, специфичных в отношении известной последовательности; после синте-
синтеза цепей ДНК в первом цикле ПЦР с биотинилированных праймеров, специ-
специфичных в отношении известной последовательности, цепи ДНК выделяют с по-
помощью аффинной хроматографии на иммобилизованном стрептавидине и ис-
используют в качестве матрицы в ПЦР с праймерами, специфичными в отноше-
отношении адаптера и известной последовательности. Черные прямоугольники - адап-
адаптеры и соответствующие им праймеры, белые прямоугольники - праймеры,
комплементарные известным последовательностям;
д - достижение специфичности в амплификации неизвестной последова-
последовательности с использованием специального адаптера, который становится до-
доступным праймеру после достройки комплементарной цепи;
е - тот же эффект, достигаемый с использованием "пузырьковой" (bubble)-
ПЦР, где а и b - некомплементарные последовательности равной длины, при
этом последовательность b идентична таковой далее используемого праймера,
Место посадки для которого образуется после синтеза комплементарной цепи,
замещающей последовательность а
235
лификации неспецифических последовательностей, проведя не-
несколько первых циклов ПЦР только в присутствии специфичес-
специфических праймеров, меченных биотином (рис. 26, г). Далее одноцепо-
чечные продукты реакции необходимо очистить с помощью аф-
аффинной хроматографии и использовать в качестве матрицы при
достройке второй цепи с помощью праймера к адаптеру, а обра-
образовавшийся двухцепочечный продукт реакции амплифицировать
с использованием праймеров к специфической последовательно-
последовательности и адаптеру.
Еще один вариант решения задачи освобождения от неспеци-
неспецифических продуктов ПЦР предполагает создание матричной по-
последовательности, комплементарной адаптеру, на цепи ДНК, ко-
которая синтезируется с праймера, комплементарного известной
последовательности (рис. 26, д). При реализации этого принципа
концы фрагментов ДНК лигируют с дефосфорилированным
адаптером, что предотвращает объединение молекул адаптера
друг с другом. Кроме того, адаптер содержит короткий двухцепо-
двухцепочечный АТ-богатый участок. По завершении лигирования обра-
образовавшиеся молекулы гибридной двухцепочечной ДНК денату-
денатурируют и отжигают с праймером, комплементарным известной
последовательности анализируемой ДНК, после чего этот прай-
мер используют в качестве затравки для синтеза комплементар-
комплементарной цепи ДНК, на которой создается сайт посадки для встречно-
встречного универсального праймера, расположенный в адаптере. Далее,
в присутствии универсального и специфического праймеров про-
проводят амплификацию исследуемого участка ДНК. По-другому
решается та же самая задача методом "пузырьковой" ПЦР (bub-
(bubble PCR) (рис. 26, ё) [312]. Здесь анализируемые фрагменты ДНК
лигируют с адаптером, содержащим в центральной части неком-
некомплементарные друг другу последовательности а и Ь, из которых Ъ
идентична последовательности универсального праймера. Свое
название метод получил из-за того, что некомплементарные од-
ноцепочечные участки формируют структуру, напоминающую
вздутие или пузырек. После лигирования, денатурации и отжига
специфического праймера с достраивают комплементарную
цепь, которая содержит сайт посадки универсального праймера,
после чего амплифицируют анализируемый участок ДНК в при-
присутствии двух праймеров Ъ и с.
В заключение этого раздела следует упомянуть еще об одном
простом и эффективном способе исследования участков ДНК с не-
неизвестной первичной структурой, расположенных в окрестностях
короткой известной последовательности, с помощью обратной
или инвертированной ПЦР (inverse PCR) [313] (рис. 27). В этом
236
Рестрикция
Лигирование при низкой
концентрации ДНК
Отжиг праймеров
Амплификация
рис. 27. Обратная (инвертиро-
(инвертированная) ПЦР [311]
Пунктирная линия обознача-
обозначает известную последовательность,
прямоугольники - праймеры, а
стрелки рядом с ними - направле-
направление синтеза ДНК
случае исследуемый препа-
препарат инкубируют с рестрикта-
зой для получения компле-
комплементарных друг другу лип-
липких концов, образовавшиеся
фрагменты разбавляют до
очень низкой концентрации
и лигируют. В таких услови-
условиях фрагментны преимущест-
преимущественно замыкаются в кольцо,
а не объединяются в линей-
линейные молекулы. Только коль-
кольцевые молекулы далее на-
направляют образование про-
продукта ПЦР при использовании неочищенного продукта лигирова-
ния в качестве матрицы при проведении ПЦР в присутствии прай-
праймеров, комплементарных известной последовательности и направ-
направленных в противоположные стороны. По завершении амплифика-
амплификации образуется продукт ПЦР в виде линейной двухцепочечной
ДНК, содержащий объединенную по сайту рестрикции неизвест-
неизвестную последовательность, фланкированную с двух сторон последо-
последовательностями известной первичной структуры. Эффективность
амплификации можно повысить, если перед проведением ПЦР
кольцевые молекулы линеаризовать по сайту рестрикции, искус-
искусственно созданному между сайтами посадки двух праймеров.
6.4.2. Понижение сложности амплифицируемой ДНК
с помощью ПЦР (метод RAPD)
Термин "сложность генома" используют для описания всевоз-
всевозможных присутствующих в нем последовательностей ДНК. Для
генома, состоящего из повторяющихся и уникальных последова-
последовательностей, сложность генома определяется в виде нормирован-
нормированной суммы всех последовательностей по следующей формуле:
О—
¦П
237
где mi обозначает число повторов в геноме /-той последователь-
последовательности, состоящей из N{ п.о. [314]. Сложность генома определяет
скорость гибридизации уникальных последовательностей с пре-
препаратами ДНК. В этой связи специфическое селективное пони-
понижение сложности препарата ДНК могло бы ускорить и облег-
облегчить проведение его анализа с помощью гибридизации. Решение
этой проблемы затрудняется тем, что исследователь редко рас-
располагает достаточным количеством информации о последова-
последовательностях ДНК анализируемого генома с неизвестной первич-
первичной структурой для осуществления выбора его представительной
части с целью дальнейшего использования при амплификации с
помощью ПЦР.
Несмотря на нетривиальный характер задачи, в настоящее
время имеется методический подход, позволяющий с помощью
ПЦР понижать сложность образца ДНК без каких-либо предва-
предварительных знаний о его первичной структуре. Метод получил на-
название случайной амплификации полиморфных последователь-
последовательностей ДНК (random amplified polymorphic DNA-RAPD). Метод
основан на использовании в ПЦР коротких праймеров со случай-
случайной последовательностью нуклеотидов. При этом в конкретной
ПЦР чаще всего используется только один праймер. В этом слу-
случае продукт ПЦР будет образовываться, если инвертированные
повторяющиеся последовательности, которые содержат в себе
сайт посадки такого праймера, будут располагаться в анализируе-
анализируемом геноме достаточно близко друг от друга, на расстоянии, допу-
допускающем эффективное прохождение ПЦР. Приняв такое рассто-
расстояние равным 2 т.п.о., и исходя из статистических расчетов вероят-
вероятности встречаемости в геноме размером 3 х 109 п.о. двух одинако-
одинаковых случайных последовательностей конкретной длины, можно
теоретически определить число соответствующих ампликонов в
таком геноме. Результаты таких расчетов представлены в табл. 9.
Из результатов, представленных в таблице, становится яс-
ясным, что, используя простейший прием включения в реакцион-
реакционную смесь одного короткого праймера со случайной последова-
последовательностью, можно с помощью ПЦР амплифицировать конкрет-
конкретную (хотя заранее не определенную) часть генома с неизвестной
первичной структурой. При этом сложность образующихся про-
продуктов ПЦР можно контролировать с помощью длины олигонук-
леотида, используемого в качестве праймера. Этот метод успеш-
успешно используется для поиска информативных полиморфных мар-
маркеров, пригодных для физического картирования геномов жи-
животных и растений, а также идентификации организмов путем
получения фингерпринтов их ДНК [315, 316].
238
Таблица 9
Расчетный результат ПЦР-амплификации методом RAPD
с помощью одного нуклеотида указанной длины [314]
Длина олигонуклеотида-
праймера(нт)
Число ампликонов
Суммарная длина амплифици-
рованной ДНК (п.о.)
8 1500 1,5 х 106
9 100 1,0 х 105
Ю 6 6,0 х 103
Примечание. Суммарная длина амплифицируемого генома принимается равной
Зх109п.о.
6.5. Одновременная амплификация
последовательностей целого генома
Одним из ограничений проведения генетических исследо-
исследований in vitro является малая доступность биологического ма-
материала. В частности, клетки дифференцированных тканей
часто невозможно культивировать без утраты ими своего ис-
исходного фенотипа. Многие типы клеток, например спермато-
сперматозоиды, вообще невозможно культивировать. Хотя с помощью
известных праймеров можно амплифицировать отдельные
участки генома, после проведения ПЦР остальной неамплифи-
цированный генетический материал оказывается потерянным.
В этой связи предпринимаются попытки разработки методов
амплификации значительной части генетического материала
на первом этапе его изучения с целью резервирования для
дальнейшей работы.
Метод преамплификации с удлинением праймеров (primer
extension preamplification - PEP) позволяет частично решать вы-
вышеупомянутую проблему [317,318]. При реализации этого подхо-
подхода в ПЦР используют смесь вырожденных коротких (часто 15-
нуклеотидных) праймеров, которые представлены всевозможны-
всевозможными D15) последовательностями. Хотя концентрация каждого от-
отдельного праймера в такой смеси мала, ее оказывается достаточ-
достаточно для амплификации с некоторой эффективностью каждого со-
соответствующего сегмента анализируемой ДНК с образованием
~30 копий продуктов ПЦР после проведения 50 циклов. Ампли-
фицированные участки могут представлять до 80% последова-
последовательностей всего генома. Практическая реализация обсуждаемо-
обсуждаемого подхода сопровождается артефактами, связанными с димери-
239
зацией праймеров. Действительно, поскольку популяция прайме-
ров включает все возможные последовательности, в ней находят-
находятся пары праймеров, комплементарные друг другу, что является
предпосылкой для образования димеров, т.е. происходит незави-
независимая от матрицы неспецифическая амплификация последова-
последовательностей.
Проблему образования димеров в РЕР-ПЦР пытаются ре-
решать с помощью варианта метода, получившего название ПЦР
со случайными мечеными праймерами (tagged random primer
PCR - T-PCR) [319]. В этом случае при амплификации использо-
использовали смесь всевозможных 9-звенных олигонуклеотидных прай-
праймеров, каждый из которых содержал 5'-концевую метку в виде
постоянной 17-нуклеотидной последовательности:
GTTTTCCCAGTCACGACN9,
где N - один из четырех возможных нуклеотидов. После прове-
проведения нескольких раундов ПЦР праймеры и их димеры отделя-
отделяли от продуктов реакции с помощью гель-фильтрации, а сами
продукты использовали в качестве матрицы в ПЦР с 17-звен-
ным праймером, соответствующим 5'-концевым последовате-
последовательностям матричных продуктов реакции. В итоговой смеси
продуктов ПЦР последовательности нуклеотидов исследуемой
хромосомы I дрожжей S. ротЬе были равномерно и специфиче-
специфически представлены. Дальнейшее усовершенствование этого под-
подхода привело к созданию праймеров следующей похожей
структуры:
AAGTCGCGGCCGCN6ATG,
в которых случайная 6-звенная последовательность была флан-
фланкирована постоянными последовательностями 13-нуклеотидной
5'-концевой маркерной и З'-концевым тринуклеотидом. Послед-
Последний тринуклеотид позволял праймерам инициировать синтез
ДНК со специфических фиксированных точек анализируемых
последовательностей хромосом, случайная 6-нуклеотидная по-
последовательность служила для стабилизации праймеров в гибри-
гибридах, поскольку в сложной смеси исходных праймеров всегда нахо-
находился один, З'-концевая 9-звенная последовательность нуклеоти-
нуклеотидов которого была полностью комплементарна соответствую-
соответствующей последовательности ампликона. Длину постоянной З'-конце-
вой последовательности праймеров в этих исследованиях изменя-
изменяли в пределах трех-шести нуклеотидов.
Разработка систем амплификации целых геномов особенно
актуальна для проведения молекулярно-генетических исследо-
240
ваний на уровне отдельных клеток, в том числе для осуществ-
осуществления пренатальной диагностики на стадии предимплантации
зародыша [320]. Информацию о других применениях ПЦР с ис-
использованием нуклеиновых кислот индивидуальных клеток в
качестве матриц (single-cell PCR) можно найти в недавнем об-
обзоре [321].
6.6. Альтернативные способы амплификации
нуклеиновых кислот in vitro
Рассмотренные выше многочисленные приложения ПЦР к
генетическим и молекулярно-биологическим исследованиям на-
наглядно иллюстрируют неисчерпаемые возможности этой группы
методов, которые во многих случаях способны заменить клони-
клонирование ДНК классическими генно-инженерными способами.
В настоящее время трудно представить себе, что в ближайшем
будущем ПЦР может быть вытеснен каким-либо альтернатив-
альтернативным методом исследования нуклеиновых кислот. Тем не менее,
поиск новых методов амплификации нуклеотидных последова-
последовательностей, менее требовательных к строгому контролю за тем-
температурными режимами проведения реакции, продолжается и по
сей день. Работы в этом направлении стимулируются еще и тем,
что исследователей не оставляет надежда преодоления патент-
патентных ограничений, связанных с методами, в основе которых ле-
лежит ПЦР. На этом фронте молекулярно-генетических исследо-
исследований уже имеются несомненные достижения.
6.6.1. Лигазная цепная реакция
Способность ДНК-лигаз восстанавливать фосфодиэфир-
ные связи в одноцепочечных разрывах двухцепочечных моле-
молекул ДНК используется при проведении лигазной цепной реак-
реакции - ЛЦР (ligase chain reaction - LCR) [85, 322, 323]. Для этого
синтезируют два олигонуклеотида, комплементарные непре-
непрерывной последовательности нуклеотидов ДНК, которые по-
после гибридизации с такой последовательностью примыкают
друг к другу и разделены лишь одноцепочечным разрывом.
При этом З'-конец предшествующего нуклеотида должен со-
содержать свободную З'-ОН-группу, а в 5'-положении 5'-конца
олигонуклеотида, следующего за ним, должна находиться фо-
фосфатная группа.
В первом цикле ЛЦР два синтезированных олигонуклеотида
отжигаются с исследуемой денатурированной ДНК (как и при
241
ПЦР) в присутствии термостабильной ДНК-лигазы, а также
всех остальных необходимых кофакторов и ионов. Реакцион-
Реакционную смесь прогревают до температуры, оптимальной для лиги-
рования, после чего между отожженными олигонуклеотидами
образуется фосфодиэфирная связь, соединяющая оба олигонук-
леотида друг с другом. Затем температуру реакционной смеси
повышают до температуры плавления гибрида, образованного
анализируемой ДНК и лигированными олигонуклеотидами, ко-
которые освобождаются в реакционную смесь в составе единого
фрагмента. Во время второго цикла температуру реакционной
смеси вновь понижают до температуры отжига олигонуклеоти-
олигонуклеотидов, которые, находясь в молярном избытке по отношению к
освободившимся продуктам реакции, преимущественно гибри-
дизуются с ДНК. Далее, связавшиеся олигонуклеотиды снова
лигируют, и образовавшийся продукт реакции освобождают из
гибрида повышением температуры. При этом содержание про-
продукта в реакционной смеси удваивается. После проведения п
циклов ЛЦР в реакционной смеси теоретически может оказать-
оказаться пх молекул продукта реакции, где х - число пар олигонукле-
олигонуклеотидов, вступающих в реакцию в каждом цикле, теоретически
равное числу копий анализируемой последовательности нукле-
отидов в реакционной смеси.
ЛЦР не происходит в том случае, если З'-концевой нуклеотид
первого олигонуклеотида или 5'-концевой нуклеотид второго не-
некомплементарны соответствующим нуклеотидам ДНК. Поэтому
при наличии мутации в тех сайтах ДНК, где олигонуклеотиды
стыкуются друг с другом, в реакцию со своим партнером вступа-
вступает только измененный олигонуклеотид, полностью комплемен-
комплементарный мутантной ДНК, т.е. продукт реакции образуется лишь
при наличии в реакционной смеси "мутантного" олигонуклеоти-
олигонуклеотида, но не олигонуклеотида дикого типа. Таким образом, принци-
принципы, лежащие в основе метода определения мутаций с помощью
ЛЦР, в ряде существенных моментов идентичны принципам ал-
аллель-специфической ПЦР.
Для количественной оценки ЛЦР один из олигонуклеотидов
обычно метят биотином, а другой (соответствующий мутантному
аллелю) - с помощью радиоактивной или флуоресцентной мет-
метки. По завершении ЛЦР олигонуклеотиды, меченные биотином,
связывают с мембранными фильтрами с иммобилизованным
стрептавидином, а количество лигированных олигонуклеотидов
определяют по количеству связавшейся с фильтрами радиоак-
радиоактивной или флуоресцентной метки после отмывки непрореагиро-
вавших олигонуклеотидов.
242
6.6.2. Изотермические системы
амплификации нуклеиновых кислот,
основанные на транскрипции
Изотермическая самоподдерживающаяся репликация по-
последовательностей (isothermal self-sustained sequence replication -
3SR) [324]. При использовании этого метода амплификации
нуклеотидных последовательностей РНК является наиболее
предпочтительной исходной матрицей, хотя молекулы ДНК
также могут выполнять эту функцию. В последнем случае мо-
молекулы ДНК копируются с помощью праймеров, содержащих
промотор для РНК-полимеразы, например, Т7-РНК-полимера-
зы, которая далее используется для получения копии РНК с
ДНК-матрицы. Комплементарная цепь ДНК амплифицируемой
РНК синтезируется в процессе обратной транскрипции с учас-
участием РНК-зависимой ДНК-полимеразы вируса миелобластоза
птиц (AMV), которая начинает синтез с праймера, содержащего
промотор для Т7-РНК-полимеразы. Обратная транскриптаза
AMV обладает активностью РНКазы Н, которая специфически
удаляет цепь РНК из образовавшегося дуплекса ДНК-РНК.
В результате образуется одноцепочечная ДНК, комплементар-
комплементарная исходной РНК. После этого в дело идет второй праймер,
комплементарный этой ДНК, который используется для синте-
синтеза второй цепи ДНК обратной транскриптазой (рис. 28, а). По
завершению синтеза второй цени ДНК образовавшаяся молеку-
молекула уже содержит промотор для Т7-РНК-полимеразы, так что
этот фермент может быть использован далее для синтеза мно-
множества копий РНК с этой матрицы, которые комплементарны
исходной РНК (рис. 28, б).
Начиная с этого момента, обратная транскриптаза начинает
циклически синтезировать цепи ДНК на РНК-матрицах, расщеп-
расщеплять молекулы РНК в гибридах и превращать их в двухцепочеч-
ные молекулы, которые, в свою очередь, начинают служить ма-
матрицами для Т7-РНК-полимеразы для синтеза новых копий одно-
цепочечных РНК. Замечательным свойством данной системы яв-
является возможность осуществления всех этих реакций одновре-
одновременно и при одной постоянной температуре. В итоге имеет место
самоподдерживающаяся амплификация исходной последователь-
последовательности РНК, специфичность которой задается первичной структу-
структурой используемых праймеров.
Амплификация с замещением цепи ДНК (strand displacement
amplification - SDA). По своей идеологии амплификация с заме-
замещением цепи очень близка к только что рассмотренному методу
243
а
5' ДНК
ДНК
5'
5'CZZl-
В
Амплифицируемая РНК
5' ДНК
Обратная транскрипция/РНКаза Н
Амплифицируемая РНК
15' ДНК
15' ДНК
I Обратная транскрипция
(Обратнаятранскрипция/РНКаза Н
I Синтез второй цепи ДНК
5'
15' ДНК
ДНК
ДНК
5' ДНК
ДНК
5'
Рис. 28. Изотермическая самоподдерживающаяся репликация последо-
последовательностей [311]
а - реакцию начинают с синтеза двухцепочечной ДНК на матрице ампли-
фицируемой РНК с помощью обратной транскриптазы; для синтеза первой це-
цепи ДНК используют праймер, специфичный в отношении 3'-конца анализируе-
анализируемой РНК, содержащий промотор для Т7-РНК-полимеразы (прямоугольник,
правая часть которого окрашена), вторую цепь синтезируют тем же ферментом
с праймера В;
б - в фазе амплификации синтезированную двухцепочечную ДНК транс-
транскрибируют Т7-РНК-полимеразой, а образующиеся РНК циклически превраща-
превращают в двухцепочечную ДНК той же обратной транскриптазой с помощью выше-
вышеупомянутых праймеров; эта ДНК вновь транскрибируется Т7-РНК-полимера-
зой и т.д.
ЗБЯ-амплификации, хотя и отличается от него рядом деталей
(рис. 29) [325-327]. В данном методе используются особые свой-
свойства рестриктазы BsoB 1, которая распознает гексануклеотидный
сайт рестрикции и расщепляет его по следующему механизму:
CiTCGGG. Показано, что введение в сайт рестрикции ос-тио-
групп вместо соответствующего фосфата помощью a-S-dCTP не
остается незамеченным ферментом и он перестает расщеплять
тиоэфирную связь. Аналогичными свойствами обладает рест-
риктаза Hindi (GTTnLGAC). Однако, в этом случае, если включе-
включение производного происходит в верхнюю цепь ДНК в сайте рест-
244
Подготовка анализируемой ДНК
Денатурация (95°С)
I Отжиг праймеров E2°С)
E)
Комплементарные
праймеры
ДНК-полимераза
Амплифицируемая
область ДНК
IdUTP, dATP
thiodCTP, dGTP
I ДНК-полимераза
D) Sl- Рис. 29. Амплификация с замещением
цепи [325]
При подготовке матрицы (верхняя
часть рисунка) амплифицируемую ДНК G)
отжигают с двумя праймерами В] (bumper) и
S ], содержащим сайт рестрикции BsoBl, кото-
которые далее элонгируют ДНК-полимеразой в
присутствии четырех dNTPs, в том числе тио-
лированного dCTP B); цепь, инициированная
с праймера В], вытесняется из дуплекса C, 4)
и может быть использована в качестве мат-
матрицы во время синтеза ДНК с новых прайме-
праймеров В2 и S2 E); образовавшаяся в итоге двух-
цепочечная ДНК F) в отличие от исходной
содержит сайт рестрикции BsoBl.
В экспоненциальной фазе реакции
(нижняя часть рисунка) рестриктаза BsoBl
вносит одноцепочечный разрыв (обозна-
(обозначен треугольником) в верхнюю цепь ДНК,
синтез которой был инициирован с прайме-
праймера S], но не в нижнюю цепь, поскольку по-
последняя содержит в сайте рестрикции мо-
s, cucggg 0 дифицированный остаток dCMP G); нали-
налицо)———— gagccc чие одноцепочечного разрыва позволяет
ДНК-полимеразе (обозначена звездочкой)
инициировать синтез ДНК в этом месте и построить новую цепь ДНК, вытеснив дочернюю из дуплекса, при этом проис-
происходит восстановление немодифицированного сайта рестрикции, который по-прежнему остается измененным в нижней це-
цепи (8-Ю), что дает возможность образованному дуплексу вступить в новый цикл синтеза ДНК, а вытесненная цепь также
используется в качестве матрицы вначале с праймерами В2 и S2, а затем В] и S], что обеспечивает экспоненциальный ха-
характер амплификации
Экспоненциальная амплификация
участка ДНК
(б).
CTCGGG
•GAGCCC'
(в)
BsoBl\
¦ cVtcggg-
G)
¦GAGCCC¦
| ДНК-полимераза
X. новый цикл
С*
GAGCCC
CUCGGG
GAGCCC
рикции, модифицированный сайт расщепляется эндонуклеазой
как обычно. Если же изменяется нижняя цепь сайта, то фермент
перестает его расщеплять. Как это свойство рестриктазы BsoB 1
используется для осуществления изотермической амплификации
ДНК иллюстрирует нижняя часть рис. 29.
Для синтеза ДНК при SDA-амплификации используют тер-
термостабильную Bst-ДНК-полимеразу, у которой отсутствует
5'-»3'-экзонуклеазная активность. Такой фермент осуществля-
осуществляет вытеснение цепи ДНК из дуплекса при замене ее вновь син-
синтезируемой в результате полимеризации четырех дезоксирибо-
нуклеозидтрифосфатов. После денатурации ДНК с ней отжига-
отжигают два прайм ера, один из которых (S,) содержит на 5'-конце
сайт BsoBl, некомплементарный матрице, а другой (В,), полно-
полностью комплементарный матрице, выполняет роль "буфера",
вытесняя 5'-конец праймера S! (этап 1). Праймеры Bj и S, начи-
начинают одновременно удлиняться ДНК-полимеразой в присутст-
присутствии oc-S-dCTP и остальных дезоксирибонуклеозидтрифосфатов,
что приводит к синтезу двух почти идентичных комплементар-
комплементарных цепей ДНК, из которых одна без сайта рестрикции остает-
остается в дуплексе, а другая, содержащая новый сайт рестрикции
BsoRl, введенный с помощью праймера Sb вытесняется в реак-
реакционную смесь (этапы 3 и 4). Вытесненная цепь может далее ги-
бридизоваться с комплементарными ей праймерами S2 и В2
(этап 5), элонгация которых приводит к образованию двухцепо-
чечного продукта реакции (б), участвующего далее в фазе экс-
экспоненциальной амплификации. Механизм амплификации схе-
схематически представлен в нижней части рисунка.
В фазе экспоненциальной амплификации рестриктаза
BsoBl вносит одноцепочечный разрыв в верхнюю цепь ДНК, в
которой сайт рестрикции был создан праймером Sj и не содер-
Рис. 30. Амплификация последовательностей геномной РНК вируса
иммунодефицита человека [311] Qp-репликазой [162]
РНК анализируемого образца гибридизуют с зондами двух типов: полири-
бонуклеотидами MDV и олигодезоксирибонуклеотидами, биотинилированны-
ми по своим концам; все зонды специфичны в отношении анализируемой РНК;
после встречи с ВИЧ-РНК зонды образуют с ней специфические гибриды, ко-
которые отделяют от остальных компонентов смеси аффинной хроматографией
на носителе с иммобилизованным стрептавидином; связавшиеся с носителем
комплексы элюируют РНКазой Н, которая гидролизует РНК только в ДНК-
РНК-гибридах, и инкубируют с Т4-ДНК-лигазой, что приводит к объединению
двух MDV-зондов, после чего они могут быть транскрибированы QP-реплика-
зой; каждый из зондов по отдельности не может быть использован этим фер-
ферментом в качестве матрицы
246
I I I I I I Г—\
Зонд MDV
РНКаза Н
1 1 1
ВИЧ-РНК
Иммобилизующий
зонд
Зонд MDV
Гибридизация
Иммобилизация
Промывание
РНКазаН
1 1 1
Т4-ДНК-лигаза
-I 1 I II ' ' I I
Qp-Репликаза
Амплифицированная РНК
247
жит модифицированного нуклеотида. Зато тиолированный
dCMP присутствует в нижней цепи, и она по этой причине оста-
остается интактной (этап 7). ДНК-полимераза связывается с одно-
цепочечным разрывом и начинает синтез новой цепи ДНК, вы-
вытесняя старую из дуплекса (этапы 8-10), что приводит к воспро-
воспроизведению конструкции G) и весь процесс повторяется. Вытес-
Вытесненные цепи связываются с комплементарными им праймерами
S2 и В2, выполняя роль матриц в синтезе ДНК, что обеспечива-
обеспечивает экспоненциальный характер амплификации анализируемой
последовательности.
Современные варианты SDA-амплификации могут быть реа-
реализованы в формате реального времени, а также на микроэлек-
микроэлектронных чипах [238] и активно используются для проведения
ДНК-диагностики возбудителей инфекционных заболеваний та-
таких, как Chlamydia trachomatis и Neisseria gonorrhoeae.
Использование Qp-репликазы для амплификации нуклеотид-
ных последовательностей. Этот способ амплификации осуществ-
осуществляется на уровне РНК с использованием РНК-репликазы, кото-
которая обычно участвует в репликации геномной РНК колифага QC
[329]. Соответствующие РНК-матрицы могут быть получены
субклонированием требуемых последовательностей в виде фраг-
фрагментов ДНК, внедренных в последовательности Qp-ДНК под
контроль промотора Т7-РНК-полимеразы в подходящем векто-
векторе. С помощью Т7-РНК-полимеразы на ДНК-матрице такого
плазмидного вектора можно получать копии РНК, которые бу-
будут служить матрицами при синтезе РНК Qp-репликазой in vitro.
При более общем подходе к реализации этих идей получают два
РНК-зонда, которые гибридизуются с соседними последователь-
последовательностями анализируемой РНК таким образом, что между ними ос-
остается одноцепочечный разрыв, который может быть ликвиди-
ликвидирован с помощью Т4-ДНК-лигазы. Оба зонда по отдельности не
могут реплицироваться Qp-репликазой, однако, после ковалент-
ного соединения в составе объединенной молекулы становятся
для нее хорошей матрицей (рис. 30).
Процесс репликации QJ3-PHK характеризуется рядом уни-
уникальных черт. Для него не требуются праймеры, и во время
синтеза новых цепей РНК не образуется промежуточных двух-
цепочечных молекул. Фермент распознает на реплицируемои
РНК специфические последовательности и элементы вторич-
вторичной структуры, после чего делает копии соответствующих
участков РНК, комплементарные исходной матрице, которые
отделяются от матрицы в процессе их синтеза, формируя свою
собственную стабильную пространственную структуру. Эти
248
Таблица 10
Сравнительные характеристики основных методов
амплификации нуклеиновых кислот in vitro
Метод
Амплифици-
руемая после-
последовательность
Используемый темпера-
температурный режим (°С)
Требуемое ко-
количество зон-
зондов
Уровень ам-
амплификации
PCR
QJ3R
LCR
3SR
SDA
Матрица
Зонд
Зонд
Матрица
Матрица
50-98, циклически
37, изотермально
50-98, циклически
42, изотермально
37, изотермально
2 или более
1
4
2
4
10
ш9
105
10
107
12
10
Примечание. QPR-QP-репликация, LCR - лигазная цепная реакция, 3SR
изотермическая самоподдерживающаяся репликация последовательностей, SDA
амплификация с замещением цепи ДНК
элементы структуры также обеспечивают репликацию вновь
синтезированной РНК Qp-репликазой. Таким образом, фер-
фермент одновременно реплицирует родительские и дочерние це-
цепи РНК, что приводит к амплификации соответствующих по-
последовательностей.
Синтез РНК, осуществляемый Qp-репликазой, характеризу-
характеризуется очень высокой эффективностью. В общем случае удается
получать 107-108 копий исходной последовательности. При
этом репликация может начинаться с одной молекулы. Однако
реализация этого метода сталкивается с рядом технических
трудностей. В частности, сама QfJ-репликаза является сложно-
устроенным четырехсубъединичным ферментом и не всегда до-
доступна. Кроме того, имеется целый ряд ограничений, наклады-
накладываемых на последовательности анализируемой РНК, что вызва-
вызвано соблюдением структурных особенностей молекул, распозна-
распознаваемых репликазой. Все это ограничивает широкое распростра-
распространение системы в анализе ДНК. Тем не менее, этот подход в бу-
будущем может оказаться весьма полезным в тех случаях, где тре-
требуется очень высокий уровень амплификации последовательно-
последовательностей при постоянной температуре, а в настоящее время он ус-
успешно используется для обнаружения РНК вируса иммунодефи-
иммунодефицита человека.
Основные характеристики всех рассмотренных выше отлича-
отличающихся от ПЦР методов амплификации нуклеиновых кислот
представлены в табл. 10 [330].
249
6.6.3. Амплификация по типу катящегося кольца
В заключение этой главы кратко рассмотрим уже упомяну-
упомянутый метод изотермической амплификации нуклеиновых кислот,
осуществляемой на кольцевых ковалентно замкнутых молекулах
ДНК по типу катящегося кольца (rolling circle amplification -
RCA) [331]. В методе линейной RCA используется один праймер,
комплементарный кольцевой одноцепочечной ДНК, которая мо-
может быть получена денатурацией исходных двухцепочечных мо-
молекул. Синтезировав комплементарную цепь, ДНК полимераза
начинает вытеснять 5'-конец синтезированной цепи из гибрида,
причем процесс продолжается непрерывно на протяжении всей
реакции, во время которой фермент совершает множество оборо-
оборотов вокруг амплифицируемой кольцевой матрицы. Продуктом
RCA является длинная одноцепочечная ДНК, в которой мономе-
мономеры, комплементарные исходной кольцевой молекуле, следуют
друг за другом в виде конкатемера. Поскольку в этом случае ги-
гигантская молекула ассоциирована с единственным праймером, ме-
метод RCA с успехом используется для амплификации сигнала на
микроматрицах [331]. При этом на двумерных и трехмерных мик-
микроматрицах имеет место возрастание сигнала в 10 000 раз по срав-
сравнению с сигналом, получаемым от отдельного зонда, меченного
флуоресцентным красителем. Ковалентное связывание 5'-концов
праймеров для RCA с антителами позволяет обнаруживать белки
в концентрации до 0,1 пг/мл анализируемой жидкости [332, 333].
Добавление в реакционную смесь с кольцевой ДНК в качест-
качестве матрицы второго встречного праймера делает RCA-амплифи-
кацию экспоненциальной в изотермических условиях [331]. Экс-
Экспоненциальная RCA была использована для обнаружения линей-
линейных молекул ДНК с использованием зондов типа "висячий за-
замок", принцип действия которых уже рассматривался выше [335].
Как видно из всего представленного в этой главе материала, в
последние несколько лет было разработано много новых подходов
амплификации последовательностей нуклеиновых кислот как с
помощью различных вариантов ПЦР, так и альтернативных мето-
методов, включая изотермическую амплификацию. Методы амплифи-
амплификации, с помощью которых в ряде случаев можно заменять тради-
традиционное клонирование и осуществлять детекцию специфических
последовательностей, становятся все менее трудоемкими, более
быстрыми и чувствительными. Прослеживается тенденция к авто-
автоматизации всех указанных процессов. Метод ПЦР стал незамени-
незаменимым в молекулярно-генетических исследованиях, и пока не видно
предела его дальнейшему совершенствованию и развитию.
250
Заключение. Стратегия выделения нового гена
После обсуждения основных экспериментальных приемов,
используемых в современной генной инженерии, становится яс-
ясно, каким образом можно решить одну из основных методичес-
методических задач молекулярной генетики, а именно: выделить требуе-
требуемый ген и заставить его работать в новых для него (искусствен-
(искусственных) генетических условиях. Такая задача наиболее просто ре-
решается в случае бактериальных и вирусных генов, поскольку ге-
геном этих микроорганизмов невелик. Кроме того, бактериальные
гены, как правило, не содержат интронов и даже гетерологичные
бактериальные гены хорошо экспрессируются в клетках Е. coli.
Но и клонирование эукариотических генов в настоящее время
представляется вполне посильной задачей.
Стандартный подход
Приняв решение о клонировании какого-либо эукариотиче-
ского гена, прежде всего необходимо ответить на вопрос: нуж-
нужна ли экспрессия рекомбинантного гена в клетках микроорга-
микроорганизмов или же можно ограничиться исследованием его структу-
структуры? Очевидно, что в первом случае нужно думать о создании
клонотеки безинтронных кДНК, а во втором - клонотеки ге-
геномной ДНК исследуемого объекта, что в ряде случаев техни-
технически менее сложно. Задача становится более трудной, если от-
отсутствуют сведения о первичной структуре клонируемого гена.
Тогда единственным источником получения частичной инфор-
информации о последовательности его нуклеотидов может быть коди-
кодируемый этим геном белок.
Определив последовательность нескольких N-концевых ами-
аминокислот белка, можно в соответствии с генетическим кодом син-
синтезировать ряд олигонуклеотидных зондов и использовать их для
скрининга клонотеки генов. В этом случае удобнее использовать
экспрессирующую клонотеку кДНК, так как для получения необ-
необходимой информации нужно будет исследовать меньшее количе-
количество клонов. Действительно, в клонотеках кДНК отсутствует
большинство некодирующих последовательностей нуклеотидов,
суммарная длина которых может значительно (на два-три поряд-
порядка) превышать таковую значимых последовательностей. Однако
при использовании таких клонотек особенно остро встает вопрос
об их репрезентативности, поскольку внутриклеточное содержа-
содержание индивидуальных мРНК сильно различается в клетках разных
тканей. После выделения рекомбинантной ДНК, гибридизующей-
ся с упомянутыми выше олигонуклеотидными зондами, можно
251
идентифицировать кодируемый кДНК белок после ее экспрессии
с использованием специфических антител. Альтернативно: коди-
кодирующий потенциал клонированной кДНК определяют после ее
гибридизации с суммарной мРНК, выделяя фракцию мРНК, за-
задерживаемой иммобилизованной кДНК на носителе, с последую-
последующей трансляцией мРНК в бесклеточной белоксинтезирующей си-
системе. Кроме того, если клонированная кДНК кодирует мРНК ис-
исследуемого белка, то они и в растворе образуют специфический
ДНК-РНК-гибрид, и мРНК перестает участвовать в трансляции в
бесклеточной системе (метод прерванной трансляции). Наличие
или отсутствие белкового продукта бесклеточной трансляции
легко обнаружить с помощью специфических антител. После
проведения таких исследований можно точно идентифицировать
клонированную кДНК и использовать ее в качестве зонда для вы-
выделения целого гена из клонотеки геномной ДНК. Одновременно
возможна экспрессия клонированной к ДНК в клетках микроорга-
микроорганизмов или в гомологичных эукариотических клетках.
Клонирование in silico
Попытки клонирования кДНК редко завершаются выделени-
выделением клона, который бы заключал в себе полную последователь-
последовательность мРНК, послужившей матрицей при синтезе этой макромо-
макромолекулы с помощью обратной транскрипции. В ряде случаев иден-
идентифицировать новый клон и выяснить его полноразмерную по-
последовательность можно с помощью методов современной био-
биоинформатики, не прибегая к дальнейшим генно-инженерным ма-
манипуляциям [335]. В частности, можно провести компьютерный
поиск гомологичной последовательности в базах данных EST-no-
следовательностей, доступных через Интернет.
Маркеры экспрессирующихся последовательностей - EST
(expressed sequence tags) - представляют собой последовательнос-
последовательности нуклеотидов, полученные при секвенировании 5'- и З'-кон-
цевых последовательностей кДНК случайно выбранных клонов,
длина которых обычно не превышает 1,5 т.п.о. Такие клоны
можно получить, например, с использованием быстрой амплифи-
амплификации концов к ДНК (rapid amplification of cDNA ends - RACE). Ба-
Базы данных, насчитывающие в настоящее время миллионы этих
последовательностей, каждой из которых присвоен уникальный
идентификационный номер, являются мощным инструментом
идентификации и поиска новых генов, особенно в сложноустро-
енных геномах эукариот.
При клонировании in silico на основании гомологии с полу-
252
ченной последовательностью необходимо прежде всего найти в
базе данных подходящий EST-маркер. Это осуществляется с по-
помощью определенного алгоритма, например BLAST, с исполь-
использованием различных интерфейсов, среди которых в настоящее
время популярен, находящийся на сайте Центра биотехнологи-
биотехнологической информации (Center for Biotechnology Information -
NCBI http://www.ncbi.nih.gov/BLAST). Сделав запрос в форме
нуклеотидной или аминокислотной последовательности, убеж-
убеждаются в наличии или отсутствии в базе данных идентичной или
гомологичной последовательности EST. Путем организации та-
таких запросов можно выяснить, экспрессируется ли обнаружен-
обнаруженная экспериментально последовательность в клетках, и в случае
положительного ответа на вопрос использовать эти данные для
позиционного клонирования целого гена (см. ниже). После вы-
выявления в базе данных EST, гомологичного исследуемой после-
последовательности, необходимо убедиться в наличии или отсутствии
данных о последовательности нуклеотидов другого конца выяв-
выявленной мРНК, а также о перекрывающихся с ними последова-
последовательностях. Перекрывающиеся последовательности в базах
данных кластеризованы, и к ним имеется быстрый доступ. При
удачном стечении обстоятельств экспериментально получен-
полученную последовательность, полученную путем объединения пе-
перекрывающихся последовательностей, удается поместить в
контиг, который заключает в себе всю последовательность
мРНК, т.е. завершить виртуальное клонирование целой кДНК.
Позиционное клонирование
По-прежнему уникальной задачей, которая по плечу лишь
большим международным коллективам, остается клонирование
неизвестных генов животных и растений, функционирование ко-
которых можно заметить по сложным фенотипическим признакам,
например, симптомам системного наследственного заболевания.
При выделении таких генов из генома человека работа начинает-
начинается с проведения скрупулезного анализа сцепления исследуемого
фенотипического признака и какого-либо полиморфного моле-
молекулярного маркера, например, редкого аллеля HLA или миниса-
теллитного локуса всех членов семьи больного. После обнаруже-
обнаружения сцепленных маркеров задача сводится к клонированию круп-
крупных фрагментов ДНК, включающих эти маркеры, их секвениро-
ванию, выявлению открытых рамок считывания, в которых пы-
пытаются обнаружить мутации, отсутствующие у здоровых индиви-
индивидуумов. В случае удачи дальнейшая работа может проводиться
253
по одной из вышеприведенных схем и должна закончиться иден-
идентификацией гена и его белкового продукта.
Примерно такой, но на самом деле намного более сложный,
путь исследований в генной инженерии привел за последние
30 лет к клонированию множества генов, в том числе и неизвест-
неизвестных ранее, определению их структуры и особенностей функцио-
функционирования. Аналогичные подходы легли в основу многих новых
направлений молекулярной биологии и генетики. Об использова-
использовании этих методов при целенаправленном конструировании ре-
комбинантных белков речь пойдет во второй части книги.
Литература
1. Matic /., Taddei F., Radman М. Genetic barriers among bacteria // Trends
Microbiol. 1996. Vol. 4. P. 69-72.
2. Георгиев Г.П. Гены высших организмов и их экспрессия. М.: Наука,
1989. 254 с.
3. Лъюин Б. Гены. М.: Мир, 1987. 544 с.
4. Жиму лев И.Ф. Общая и молекулярная генетика. Новосибирск: Си-
Сибирское университетское издательство, 2003. 480 с.
5. УотсонДж. Молекулярная биология гена. М.: Мир, 1967. 461 с.
6. Duret L. Why do genes have introns? Recombination might add a new piece
to the puzzle // Trends Genet. 2001. Vol. 17. P. 172-175.
7. Logsdon J.M., Jr. The recent origins of spliceosomal introns revisited //
Curr. Opin. Gen. Develop. 1998. Vol. 8. P. 501-508.
8. Sharp P.A. Split genes and RNA splicing // Cell. 1994. Vol. 77. P. 805-815.
9. SmitA.F.A. The origin of interspersed repeats in the human genome // Curr.
Opin. Genet. Develop. 1996. Vol. 6. P. 743-748.
10. Britten RJ., Graham D.E., Neufeld B.R. Analysis of repeating DNA
sequences by reassociation // Meth. Enzymol. 1974. Vol. 29. P. 363-418.
11. Petrov D.A. Evolution of genome size: New approaches to an old problem
// Trends Genet. 2001. Vol.17. P. 23-28.
12. Gray M.W. Evolution of organellar genomes // Curr. Opin. Genet. Develop.
1999. Vol. 9. P. 678-687.
13. Martin W., Stobe В., Goremykin V. et al. Gene transfer to the nucleus and
the evolution of chloroplasts // Nature. 1998. Vol. 393. P. 162-165.
14. Lang B.F., Gray M.W., Burger G. Mitochondrial DNA evolution and the
origin of eukaryotes // Annu. Rev. Genet. 1999. Vol. 33. P. 351-397.
15. Патрушев JIM. Экспрессия генов. М.: Наука, 2000. 528 с.
16. Woose C.R., Kandler О., Wheelis M. Towards a natural system of organ-
organisms: Proposal for the domains Archaea, Bacteria, and Eucarya // Proc. Nat.
Acad. Sci. USA. 1990. Vol. 87. P. 4576-4579.
17. Lander E.S., Linton L.M., Birren B. et al. Initial sequencing and analysis of
the human genome // Nature. 2001. Vol. 409. P. 860-921.
18. Venter J.C., Adams M.D., Myers E.W. et al. The sequence of the human
genome // Science. 2001. Vol. 291. P. 1304-1351.
254
19. Yu J., Ни S., Wang J. et al. A draft sequence of the rice genome {Oryza sati-
va L. ssp. indica) // Science. 2002. Vol. 296. P. 79-91.
20. Gojf S.A., Ricke D., Lan Т.Н. et al. A draft sequence of the rice genome
(Oryza sativa L. ssp. japonica) II Ibid. 2002. Vol. 296. P. 92-100.
21. Gall J.G. Chromosome structure and the C-value paradox // J. Cell Biol.
1981. Vol. 91.3-14.
22. Hartl D.L. Molecular melodies in high and low С // Nature Rev. Genet.
2000. Vol. l.P. 145-149.
23. Patrushev L.I. Altruistic DNA. About protective functions of the abundant
DNA in the eukaryotic genome and its role in stabilizing genetic informa-
information // Biochem. Mol. Biol. Intern. 1997. Vol. 41. P. 851-860.
24. Френкелъ-Конрат X. Химия и биология вирусов. М.: Мир, 1972.
С. 15.
25. Sharpe M.E. Upheaval in the bacterial nucleoid: An active chromosome seg-
segregation mechanism // Trends Genet. 1999. Vol. 15. P. 70-74.
26. Blattner F.R., Plunkett G., Ill, Block C.A. et al. The complete genome
sequence of Escherichia coll K-12 // Science. 1997. Vol. 277. P. 1453-1474.
27. Kunst F., Ogasawara N., Moszer I. et al. The complete genome sequence of
the gram-positive bacterium Bacillus subtilis I I Nature. 1997. Vol. 390.
P. 249-256.
28. Klenk H.-P., Clayton R.A., TombJ.F. et al. The complete genome sequence
of the hyperthermophilic, sulphate-reducing archaeon, Archaeoglobus
fulgidus II Nature. 1997. Vol. 390. P. 364-370.
29. Soppa J. Transcriptional initiation of Archaea: Facts, factors and future
aspects // Mol. Microbiol. 1999. Vol. 31. P. 1295-1305.
30. Csink A.K., HenikoffS. Something from nothing: The evolution and utility
of satellite repeat // Trends Genet. 1998. Vol. 14. P. 200-204.
31. WeinerAM. SINEs and LINEs: The art of biting the hand that feeds you //
Curr. Opin. Cell Biol. 2002. Vol. 14. P. 343-350.
32. Smit A.F.A. Interspersed repeats and other mementos of transposable ele-
elements in mammalian genomes // Curr. Opin. Genet. Develop. 1999. Vol. 9.
P. 657-663.
33. Sassaman DM., Dombroski В A., Moran J.V. et al. Many human LI ele-
elements are capable of retrotransposition // Nature Genet. 1997. Vol. 16.
P. 37-43.
34. KuffEL., Lueders K.K. The intracisternal A-particle gene family: Structure
and functional aspects // Adv. Cancer Res. 1988. Vol. 51. P. 183-276.
35. Ostertag EM., Kazazian H.H. Jr. Biology of mammalian LI retrotrans-
posons. Annu. Rev. Genet. 2001. Vol. 35. P. 501-538.
36. Жиму лев И.Ф. Гетерохроматин и эффект положения гена. Новоси-
Новосибирск: Наука, 1993. 491 с.
37. Гвоздев В.А., Алаторцев В.В., Аравин А.А. и др. Гетерохроматин:
Молекулярная эволюция и эффекты положения генов у Drosophila
melanogaster II Молекуляр. биология. 1999. Т. 33. С. 14-25.
38. Гвоздев В.А. Гетерохроматин (структура, молекулярная эволюция и
регуляторные взаимодействия) // Проблемы и перспективы молеку-
молекулярной генетики. Т. 1. Москва. Наука, 2003. С. 15-27.
255
39. HenikojfS. Heterochromatin function in complex genomes // Biochim. bio-
phys. acta. 2000. Vol. 1470. P. 1-8.
40. Lee T.I. Young R.A. Transcription of eukaryotic protein-coding genes //
Annual Rev. Genet. 2000. Vol. 34. P. 77-137.
41. Smale ST., Kadonaga J.T. The RNA polymerase II core promoter // Annu
Rev. Biochem. 2003. Vol. 72. P. 449-479.
42. Samuel C.E. RNA editing minireview series // J. Biol. Chem. 2003
Vol. 278. P. 1389-1390.
43. Smith H.C., GottJM., Hanson M.R. A guide to RNA editing // RNA. 1997
Vol. 3. P. 1105-1123.
44. Smith H.C., Sowden M.P. Base modification RNA editing: The good, the
bad and the unregulated // Trends Genet. 1996. Vol. 12. P. 418-424.
45. Bass B.L. RNA editing by adenosine deaminases that act on RNA // Annu.
Rev. Biochem. 2002. Vol. 71. P. 817-846.
46. Shatkin AJ., Manley J.L. The ends of the affair: Capping and polyadenyla-
tion // Nature Struct. Biol. 2000. Vol. 7. P. 838-842.
47. Smith C.W.J., Valcarcel J. Alternative pre-mRNA splicing: The logic
of combinatorial control // Trends Biochem. Sci. 2000. Vol. 25. P. 381-
388.
48. Spirin A.S. Ribosomes. Ed. P. Siekevitz. Norwell (MA): Kluwer, 1999.
49. Translational control of gene expression. Ed. N. Sonenberg,
J.W.B. Hershey, M.B. Mathews. Cold Spring Harbor (NY): Cold Spring
Harbor, 2000. 1020 p.
50. Hilleren P., Parker R. Mechanisms of mRNA surveillance in eukaryotes //
Annu. Rev. Genet. 1999. Vol. 33. P. 229-260.
51. Muto A., Ushida C, Himeno H. A bacterial RNA that functions as both a
tRNA and an mRNA // Trends Biochem. Sci. 1998. Vol. 23. P. 25-29.
52. Kirschner M. Intracellular proteolysis // Ibid. Vol. 24. P. M42-M45.
53. Bradshaw R.A., Stewart A.E. Analysis of protein modifications: Recent
advances in detection, characterization and mapping // Curr. Opin.
Biotechnol. 1994. Vol. 5. P. 85-93.
54. Han K.K., Martinage A. Post-translational chemical modification(s) of pro-
proteins // Intern. J. Biochem. 1992. Vol. 24. P. 19-28.
55. Cohen P. Signal integration at the level of protein kinases, protein phos-
phatases and their substrates // Trends Biochem. Sci. 1992. Vol. 17.
P. 408-413.
56. Stroud R.M. Mechanisms of biological control by phosphorylation // Curr.
Opin. Struct. Biol. 1991. Vol. 1. P. 826-835.
57. Kraft R., Wittmann-Liebold B. Proteins: Postsynthetic modifications //
Encyclopedia of life sciences. 2001. Nature Publishing Group.
58. Мог A., Amiche M., Nicolas P. Enter a new post-translational modification:
D-amino acids in gene-encoded peptides // Trends Biochem. Sci. 1992.
Vol. 17. P. 481-485.
59. Миндлин С.З., Басе И.А., Богданова Е.С. и др. Горизонтальный пе-
перенос генов в природных популяциях бактерий. Гены и транспозоны
устойчивости к соединениям ртути // Проблемы и перспективы мо-
молекулярной генетики. М.: Наука. 2003. С. 124-146.
256
60. The horizontal gene pool // Ed. CM. Thomas. Harwood: Acad. publ. 2000.
419 p.
61. Maniatis Т., Fritsch E.F., Sambrook J. Molecular cloning: A laboratory
manual. Edn. 2 // Cold Spring Harbor Laboratory Press, Cold Spring
Harbor, NY; 1989.
62. Molecular cloning. A laboratory manual on the WEB. Chapter 6:
Preparation and analysis of eukaryotic genomic DNA. Chapter 7 Extraction,
purification, and analysis of mRNA from eukaryotic cells. Cold Spring
Harbor Laboratory, http://www.molecularcloning.com.
63. Gross-Bellard M., Oudet P., Chambon P. Isolation of high molecular-
weight DNA from mammalian cells // Europ. J. Biochem. 1973. Vol. 36.
P. 32-38.
64. Blin N., Stafford D.W. A general method for isolation of high molecular
weight DNA from eukaryotes // Nucl. Acids Res. 1976. Vol. 3.
P. 2303-2308.
65. Bignell G.R., Miller A.R., Evans I.H. Isolation of mitochondrial DNA //
Meth. Mol. Biol. 1996. Vol. 53. P. 109-116.
66. Gomez-Marquez J., Freire M., Segade F. A simple procedure for the large-
scale purification of plasmid DNA // Gene. 1987. Vol. 54. P. 255-259.
67. Celano P., Vertino P.M., Casero R.A. Jr. Isolation of polyadenylated RNA
from cultured cells and intact tissues // Biotechniques. 1993. Vol. 15. P. 26-28.
68. Anand R. Pulsed field gel electrophoresis: A technique for fractionating
large DNA molecules // Trends Genet. 1986. Vol. 2. P. 278-283.
69. Schwartz D.C., Cantor C.R. Separation of yeast chromosome-sized DNAs
by pulsed field gradient gel electrophoresis // Cell. 1984. Vol. 37. P. 67-75.
70. Givan A.L. Flow cytometry: First principles. 2nd ed. NY: Wiley-Liss, 2001.
256 p.
71. Trask BJ. Human cytogenetics: 46 Chromosomes, 46 years and counting //
Nature Rev. Genet. 2002. Vol. 3. P. 769-778.
72. Roslaniec M.C., Bell-Prince C.S., Crissman H.A. et al. New flow cytomet-
ric technologies for the 21st century // Hum. Cell. 1997. Vol. 10. P. 3-10.
73. Rao D.N., Saha S., Krishnamurthy V. ATP-dependent restriction enzymes //
Progr. Nucl. Acids Res. 2000. Vol. 64. P. 1-63.
74. PingoudA., Jeltsch A. Recognition and cleavage of DNA by type-II restric-
restriction endonucleases // Europ. J. Biochem. 1997. Vol. 246. P. 1-22.
75. Kovall R.A., Matthews B.W. Type II restriction endonucleases: Structural,
functional and evolutionary relationships // Curr. Opin. Chem. Biol. 1999.
Vol. 3. P. 578-583.
76. Blumenthal R.M., Cheng X. Restriction-modification systems // Modern
microbial genetics. Ed. U.N. Streips, R.E. Yasbin. 2rd ed. N.Y.: Wiley,
2002. P. 177-225.
77. Janulaitis A., Petrusayte M., Maneliene Z. et al. Purification and properties
of the Eco51\ restriction endonucleases and methylase - Prototypes of the
new class (type IV) // Nucl. Acids Res. 1992. Vol. 20. P. 6043-6049.
78. Kong H., Roemer S.E., Waite-Rees PA. et al. Characterization of Bcgl, a
new kind of restriction-modification system. J. Biol. Chem. 1994. Vol. 269.
P. 683-690.
9. Патрушев Л.И. Т. 1
79. New England Biolabs Technical Literature, http://www.neb.com/
neb/frame_teoh.html
80. Messer W., Noyer-Weindner M. Timing and targeting of biological func-
functions of Dam methylation in E. coli I I Cell. 1998. Vol. 54. P. 735-737.
81. Wilkinson A., Day J., Bowater R. Bacterial DNA ligases // Mol. Microbiol.
2001. Vol. 40. P. 1241-1248.
82. Doherty, AJ., Suh S.W. Structural and mechanistic conservation in DNA
ligases // Nucl. Acids Res. 2000. Vol. 28. P. 4051-4058.
83. Timson DJ. et al. DNA ligases in the repair and replication of DNA //
Mutat. Res. 2000. Vol. 460. P. 301-318.
84. Cao W. DNA ligases: Structure, function and mechanism // Curr. Organ.
Chem. 2002. Vol. 6. P. 827-839.
85. Cao W. Recent developments in ligase-mediated amplification and detection
// Trends Biotechnol. 2004. Vol. 22. P. 38-44.
86. Hubscher U., Spadari S. Eukaryotic DNA polymerases // Annu. Rev.
Biochem. 2002. Vol. 71. P. 133-163.
87. Steitz T.A. DNA polymerases: Structural diversity and common mechanisms
//J. Biol. Chem. 1999. Vol. 274. P. 17395-17398.
88. Rattray AJ., Strathern J.N. Error-prone DNA polymerases: When making a
mistake is the only way to get ahead // Annu. Rev. Genet. 2003. Vol. 37.
P. 31-66.
89. Beese L.S., Derbyshire V., Steitz T.A. Structure of DNA polymerase I
Klenow fragment bound to duplex DNA // Science. 1993. Vol. 260.
P. 352-355.
90. Rigby P.W.J., Dieckmann M., Rhodes C, Berg P. Labeling deoxyribonucle-
ic acid to high specific activity in vitro by nick translation with DNA poly-
polymerase I // J. Mol. Biol. 1977. Vol. 113. P. 237-251.
91. Yang T.T., Sinai P., Kitts P.A., Kain S.R. Quantification of gene expression
with a secreted alkaline phosphatase reporter system // Biotechniques. 1997.
Vol. 23. P. 1110-1114.
92. Raines R.T. Ribonuclease A // Chem. Rev. 1998. Vol. 98. P. 1045-1065.
93. Hofsteenge J. Ribonuclease inhibitor // Ed. G. D'Alessio, J.F. Riordan.
Ribonucleases - structures and functions. San Diego: Academic Press,
1997. P. 621-658.
94. Perlin M.H. The subcellular entities a.k.a. plasmids // Modern microbial
genetics. Ed. U.N. Streips, R.E. Yasbin. 2nd ed. N.Y.: Wiley, 2002.
P. 507-560.
95. DelSolar G., Giraldo R., Ruiz-Echevarria MJ. et al. Replication and control
of circular bacterial plasmids // Microbiol. Mol. Biol. Rev. 1998. Vol. 62.
P. 434-464.
96. DelSolar G., Espinosa M. Plasmid copy number control: An ever-growing
story // Mol. Microbiol. 2000. Vol. 37. P. 492-500.
97. Tomizawa J., Itoh T. Plasmid ColEl incompatibility determined by interac-
interaction of RNA1 with primer transcript // Proc. Nat. Acad. Sci. USA. 1981.
Vol. 78. P. 6096-6100.
98. Nordstrom K. Control of plasmid replication - how do DNA iterons set the
replication frequency? // Cell. 1990. Vol. 63. P. 1121-1124.
258
99. Bittner M., Vapnek D. Versatile cloning vectors derived from runaway-
replication plasmid pKN402 // Gene. 1981. Vol. 15. P. 319-329.
100. Novick R.P. Plasmid incompatibility // Microbiol. Rev. 1987. Vol. 51.
P. 381-395.
101. Aslanidis C, de Jong PJ. Ligation-independent cloning of PCR products
(LIC-PCR) // Nucl. Acids Res. 1990. Vol. 18. P. 6069-6074.
102. Haun R.S., Serventi I.M., Moss J. Rapid, reliable ligation-independent
cloning of PCR products using modified plasmid vectors // Biotechniques.
1992. Vol. 13. P. 515-518.
103. Clark JM. Novel non-templated nucleotide addition reactions catalyzed by
prokaryotic and eukaryotic DNA polymerases // Nucl. Acids Res. 1988.
Vol. 16. P. 9677-9686.
104. Hayes F. Transposon-based strategies for microbial functional genomics
and proteomics // Annu. Rev. Genet. 2003. Vol. 37. P. 3-29.
105. (a) Bruschi C.V., Gjuracic K. Yeast artificial chromosomes // Encyclopedia
of Life Sciences. Nature Publ. Group. 2002. http://www.els.net;
F) Adam C, Mullen J.A., Kindle K. Retrofitting YACs for direct DNA
transfer into plant cells // Plant J. 1997. Vol. 11. P. 1349-1358; (e) Den
Dunnen J.T., Grootscholten P.M., Dauwerse J.D. et al. Reconstruction of
the 2.4 Mb human DMD-gene by homologous YAC recombination //
Hum. Mol. Genet. 1992. Vol. 1. P. 19-28.
106. Monaco A.P., Larin Z. YACs, BACs, PACs and MACs: Artificial chro-
chromosomes as research tools // Trends Biotechnol. 1994. Vol. 12.
P. 280-286.
107. Sternberg N. Bacteriophage PI cloning system for the isolation, amplifica-
amplification, and recovery of DNA fragments as large as 100 kilobase pairs // Proc.
Nat. Acad. Sci. USA. 1990. Vol. 87. P. 103-107.
108. Ioannou P.A., Amemiya C.T., Games J. et al. A new bacteriophage Pi-
derived vector for the propagation of large human DNA fragments //
Nature Genetics. 1994. Vol. 6. P. 84-89.
109. Murray A.W., SzostakJ.W. Construction of artificial chromosomes in yeast
//Nature. 1983. Vol. 305. P. 189-193.
110. Brown W.R.A., Mee PJ., Shen M.H. Artificial chromosomes: Ideal vec-
vectors? // Trends Biotechnol. Sci. 2000. Vol. 18. P. 218-223.
111. Vos J.-M.H. Mammalian artificial chromosomes as tools for gene therapy
// Curr. Opin. Gen. Develop.1998. Vol. 8. P. 351-359.
112. Larin Z., Mejia J.E. Advances in human artificial chromosome technology
//Trends Genet. 2002. Vol.18. P. 313-319.
113. Mills W., Critcher R., Lee C, Farr CJ. Generation of an approximately
2.4Mb human X centromere-based minichromosomes by targeted telom-
ere-associated chromosome fragmentation in DT40. // Hum. Mol. Genet.
1999. Vol. 8. P. 751-761.
114. Kereso J., Praznovsky Т., Cserpan I. et al. De novo chromosome forma-
formations by large-scale amplification of the centromeric region of mouse chro-
chromosomes // Chromosome Res. 1996. Vol. 4. P. 1-14.
115. Co D.O., Borowski A.H., Leung J.D. et al. Generation of transgenic mice
and germline transmission of a mammalian artificial chromosome intro-
9* 259
duced into embryos by pronuclear microinjection. // Chromosome Res.
2000. Vol. 8. P. 183-191.
116. Lee C, Wevrick R., Fisher R.B., Ferguson-Smith MA. et al. Human cen-
tromeric DNAs // Hum. Genet. 1997. Vol. 100. P. 291-304.
117. Makrides S. Strategies for achieving high-level expression of genes in
Escherichia coli // Microbiol. Rev. 1996. Vol. 60. P. 512-538.
118. Busby S., EbrightR.H. Promoter structure, promoter recognition, and tran-
transcription activation in prokaryotes // Cell. 1994. Vol. 79. P. 743-746.
119. Struhl K. Fundamentally different logic of gene expression in eukaryotes
in prokaryotes // Cell. 1999. Vol. 98. P. 1-4.
120. Rojo F. Repression of transcription initiation in bacteria // J. Bacteriol.
1999. Vol. 181. P. 2987-2991.
121. Rhodius V.A., Busby SJ. Positive activation of gene expression // Curr.
Opin. Microbiol. 1998. Vol. 1. P. 152-159.
122. Ricci J.C.D. et al. Plasmid effects on Escherichia coli metabolism // Crit.
Rev. Biotechnol. 2000. Vol. 20. P. 79-108.
123. Matthews K.S., Nichols J.C. Lactose repressor protein: Functional proper-
properties and structure // Prog. Nucl. Acid Res. Mol. Biol. 1998. Vol. 58.
P. 127-164.
124. Ptashne M., Jeffrey A., Johnson A.D. et al. 1980. How the X repressor and
Cro work. // Cell. Vol. 19. P. 1-11.
125. DubendorffJ.W., Studier F.W. Controlling in an inducible T7 expression
system by blocking the target T7 promoter with lac repressor // J. Mol.
Biol. 1991. Vol. 219. P. 45-59.
126. Studier F.W., Moffatt В A. Use of bacteriophage T7 RNA polymerase to
direct selective high-level expression of cloned genes //J. Mol. Biol. 1986.
Vol. 189. P. 113-130.
127. McCarthy J.E.G. Posttranscriptional control of gene expression in yeast //
Microbiol. Mol. Biol. Rev. 1998. Vol. 62. P. 1492-1553.
128. Shine J., Dalgarno L. The З'-terminal sequence of Escherichia coli 16S
ribosomal RNA: Complementarity to nonsense triplets and ribosome bind-
binding sites // Proc. Nat. Acad. Sci. U.S.A. 1974. Vol. 71. P. 1342-1346.
129. McCarthy J.E.G., Brimacombe R. Prokaryotic translation: The interactive
pathway leading to initiation // Trends Genet. 1994. Vol. 10. P. 402-407.
130. De SmitM.H., van DuinJ. Control of prokaryotic translational initiation by
mRNA secondary structure // Prog. Nucl. Acid Res. Mol. Biol. 1990.
Vol. 38. P. 1-35.
131. Grantham R., Gautier C, Gouy M. et al. Codon catalog usage is a genome
strategy modulated for gene expressivity. Nucl. Acids Res. 1981. Vol. 9.
P. 43-74.
132. Xia X. How optimized is the translational machinery in Escherichia coli,
Salmonella typhimurium and Saccharomyces cerevisiael // Genetics. 1998.
Vol. 149. P. 37-44.
133. Mottagui-Tabar S., Isaksson LA. The influence of the 5' codon context on
translation termination in Bacillus subtilis and Escherichia coli is similar
but different from Salmonella typhimurium I I Gene. 1998. Vol. 212.
P. 189-196.
260
134. Moszer I., Rocha E.P.C., Danchin A. Codon usage and lateral gene trans-
transfer in Bacillus subtilis // Curr. Opin. Microbiol. 1999. Vol. 2. P. 524-528.
135. Rosenberg A.H., Goldman E., Dunn, JJ. et al. // J. Bacteriol. 1993.
Vol. 175. P. 716-722.
136. Del Tito BJ. Jr., Ward J.M., Hodgson J. et al // Ibid. 1995. Vol. 177.
P. 7086-7091.
137. Caponigro G., Parker R. Mechanisms and control of mRNA turnover in
Saccharomyces cerevisiae II Microbiol. Rev. 1996. Vol. 60. P. 233-
249.
138. Jacobson A., Peltz S.W. Interrelationships of the pathways of mRNA decay
and translation in eukaryotic cells // Annu. Rev. Biochem. 1996. Vol. 65.
P. 693-739.
139. Ralph A., Bradshaw, William W. et al. N-Terminal processing: The
methionine aminopeptidase and Na-acetyl transferase families // Trends
Biochem. Sci. 1998. Vol. 23. P. 263-267.
140. Varshavsky A. The N-end rule: Functions, mysteries, uses // Proc. Nat.
Acad. Sci. USA. 1996. Vol. 93. P. 12142-12149.
141. Preibisch G., Ishihara H., Tripier D., Leineweber M. I I Gene. 1988.
Vol. 72. P. 179-186.
142. Lindahl L., Hinnebusch A. Diversity of mechanisms in the regulation of
translation in prokaryotes and lower eukaryotes // Curr. Opin. Genet.
Develop. 1992. Vol. 2. P. 720-726.
143. McCarthy J.E., Gualerzi C. Translational control of prokaryotic gene
expression // Trends Genet. 1990. Vol. 6. P. 78-85.
144. Lilie H., Schwarz E., Rudolph R. Advances in refolding of proteins pro-
produced in E. coli II Curr. Opin. Biotechnol. 1998. Vol. 9. P. 497-501.
145. Panda AX. Bioprocessing of therapeutic proteins from the inclusion bod-
bodies of Escherichia coli I I Adv. Biochem. Engin. Biotechnol. 2003. Vol. 85.
P. 43-93.
146. De Bernardez Clark E. Refolding of recombinant proteins // Curr. Opin.
Biotechnol. 1998. Vol. 9. P. 157-163.
147. Schein C.H., Noteborn МММ. Formation of soluble recombinant proteins
in Escherichia coli is favored by lower growth temperature //
BioTechnology. 1988. Vol. 6. P. 291-294.
148. Braun P., Gerritse G., van DijlJ.-M., Quax W.J. Improving protein secre-
secretion by engineering components of the bacterial translocation machinery //
Curr. Opin. Biotechnol. 1999. Vol. 10. P. 376-381.
149. Joly J.C., Leung W.S., Swartz J.R. Overexpression of Escherichia coli oxi-
doreductases increases recombinant insulin-like growth factor-I accumula-
accumulation // Proc. Nat. Acad. Sci. USA. 1998. Vol. 95. P. 2773-2777.
150. GoloubinoffP., Gatenby A.A., Lorimer G.H. GroE heat shock proteins pro-
promote assembly of foreign prokaryotic ribulose bisphosphate carboxylase
oligomers in Escherichia coli // Nature. 1989. Vol. 337. P. 44-47.
151. Thomas J.G., Ayling A., Baneyx F. Molecular chaperones, folding cata-
catalysts, and the recovery of active recombinant proteins from E. coli. Appl.
Biochem. Biotechnol. 1997. Vol. 66. P. 197-238.
152. Hlavac F., Rouer E. Expression of the protein-tyrosine kinase p561ck by
261
the pTRX vector yields a highly soluble protein recovered by mild sonica-
tion //Protein Expr. Purif.1997. Vol. 11. P. 227-232.
153. Oswald Т., Wende W., Pingoud A., Rinas U. Comparison of N-terminal
affinity fusion domains: Effect on expression level and product hetero-
heterogeneity of recombinant restriction endonuclease EcoRV // Appl.
Microbiol. Biotechnol. 1994. Vol. 42. P. 73-77.
154. Prinz W.A., Aslund F., Holmgren A., Beckwith J. The role of the thiore-
doxin and glutaredoxin pathways in reducing protein disulfide bonds in the
Escherichia coll cytoplasm. // J. Biol. Chem. 1997. Vol. 272.
P. 15661-15667.
155. Kadokura H., Katzen F., Beckwith J. Protein disulfide bond formation in
prokaryotes // Annu. Rev. Biochem. 2003. Vol. 72. P. 111-135.
156. Terpe K. Overview of tag protein fusions: From molecular and biochemi-
biochemical fundamentals to commercial systems // Appl. Microbiol. Biotechnol.
2003. Vol. 60. P. 523-533.
157. Bucher M.H., Evdokimov A.G., Waugh D.S. Differential effects of short
affinity tags on the crystallization of Pyrococcus furiosus maltodextrin-
binding protein // Biol. Cryst. 2002. Vol. 58. P. 392-397.
158. Sassenfeld H.M., Brewer S.J. A polypeptide fusion designed for purifi-
purification of recombinant proteins // Bio/Technology. 1984. Vol. 2.
P. 76-81.
159. Chaga G., Hopp J., Nelson P. Immobilized metal ion affinity chromatog-
raphy on Co2+-carboxymethylaspartate-agarose superflow, as demon-
demonstrated by one-step purification of lactate dehydrogenase from chicken
breast // Biotechnol. Appl. Biochem. 1999. Vol. 29. P. 19-24.
160. Halliwell СМ., Morgan G., Ou СР., Cass A.E. Introduction of a (poly)his-
tidine tag in L-lactate dehydrogenase produces a mixture of active and
inactive molecules // Anal. Biochem. 2001. Vol. 295. P. 257-261.
161. Hope T.P., Gallis В., Prikett K.S. Metal-binding properties of a calcium
dependent monoclonal antibody // Mol. Immunol. 1996. Vol. 33.
P. 601-608.
162. Einhauer A., Schuster M., Wasserbauer E., Jungbauer A. Expression and
purification of homogenous proteins in Saccharomyces cerevisiae based
on ubiquitin-FLAG fusion // Protein Expr. Purif. 2002. Vol. 24.
P. 497-504.
163. Schmidt T.G.M., Koepke J., Frank R., Skerra A. Molecular interaction
between the Strep-tag affinity peptide and its cognate target, Streptavidin
// J. Mol. Biol. 1996. Vol. 255. P. 753-766.
164. Stray J.E., Lindsley J.E. Biochemical analysis of the yeast condensin
Smc2/4 complex. An ATPase that promotes knotting of circular DNA // J.
Biol. Chem. 2003. Vol. 278. P. 26238-26248.
165. Evan G.I., Lewis G.K., Ramsay G., Bishop J.M. Isolation of monoclonal
antibodies specific for human c-myc proto-oncogene product // Mol. Cell
Biol. 1985. Vol. 5. P. 3610-3616.
166. Ferrando A., Koncz-Lalman Z., Farras R. et al. Detection of in vivo pro-
protein interactions between Snfl-related kinase subunits with intron-tagged
262
epitope-labeling in plant cells // Nucl. Acids Res. 2001. Vol. 29.
P. 3685-3693.
167. McKern N.M., Lou M., Frenkel M.J. et al. Crystallization of the first three
domain of the human insuline-like growth factor-1 receptor // Prot. Sci.
1997. Vol. 6. P. 2663-2666.
168. Karpeisky M.Y., Senchenko V.N., Dianova M.V., Kanevsky V. Formation
and properties of S-protein complex with S-peptidecontaining fusion pro-
protein // FEBS Lett. 1994. Vol. 339. Vol. 209-212.
169. Stofko-Hahn R.E., Carr D.W., Scott J.D. A single step purification for
recombinant proteins. Ibid. 1992. Vol. 302. P. 274-278.
170. Vaillancourt P., Zheng C.-F., Hoang D.Q., BreisterL. Affinity purification
of recombinant proteins fused to calmodulin or calmodulin-binding pep-
tides // Meth. Enzymol. 2000. Vol. 326. P. 340-362.
171. Xu M.-Q., Paulus #., Chong S. Fusions to self-splicing inteins for protein
purification. Ibid. P. 376-418.
172. Tomme P., Boraston A., McLean B. et al. Characterization and affinity
applications of cellulose-binding domains // J. Chromatogr. B. 1998.
Vol. 715. Vol. 283-296.
173. McCormick M., Berg J. Purification and S Tag detection of CBD fusion
proteins // in Novations. 1997. Vol. 7. P. 12-15.
174. Keefe A.D., Wilson D.S., Seelig В., Szostak J.W. One-step purification of
recombinant proteins using a nanomolar-affinity streptavidin-binding pep-
tide, the SBP-Tag // Prot. Expr. Purif. 2001. Vol. 23. P. 440-446.
175. Watanabe Т., Ito Y., Yamada T. et al. The role of the C-terminal domain
and type III domains of chitinase Al from bacillus circulans WL-12 in
chitin degradation //J. Bacteriol. 1994. Vol. 176. P. 4465^472.
176. Cantor EJ., Chong S. Intein-mediated rapid purification of Cre recombi-
nase // Prot. Expr. Purif. 2001. Vol. 22. P. 135-140.
177. Sweda P., PladzykR., KotlowskiR., KurJ. Cloning, expression, and purifi-
purification of the Staphylococcus simulans lysostaphin using the intein-chitin-
binding domain (CBD) system // Prot. Expr. Purif. 2001. Vol. 22.
P. 467^71.
178. Smith D.B., Johnson K. Single-step purification of polypeptides expressed
in Escherichia coli as fusions with glutathione S-transferase // Gene. 1988.
Vol. 67. P. 31^0.
179. Beekman J.M., Cooney AJ., Elliston J.F. et al. A rapid one-step method
to purify baculovirus-expressed human estrogen receptor to be used in
the analysis of the oxytocin promoter // Gene. 1994. Vol. 146.
P. 285-289.
180. Ron D., Dressier H. pGSTag - a versatile bacterial expression plasmid for
enzymatic labelling of recombinant proteins // Biotechniques. 1992.
Vol. 13. P. 866-869.
181. Di Guan C, Li P., Riggs P.D., Inouye H. Vectors that facilitate the expres-
expression and purification of foreign peptides in Escherichia coli by fusion to
maltose-binding protein //Gene. 1988. Vol. 67. P. 21-30.
182. Podmore A.H., Reynolds P.E. Purification and characterization of
VanXYC, a d,d-dipeptidase/d,d-carboxypeptidase in vancomycin-resistant
263
Enterococcus gallinarum BM4174 // Europ. J. Biochem. 2002. Vol. 269.
P. 2740-2746.
183. Gavin A.C., Bosche M., Krause R. et. al. Functional organization of the
yeast proteome by systematic analysis of protein complexes // Nature.
2002. Vol. 415. P. 141-147.
184. Braun P., LaBaerJ. High throughput protein production for functional pro-
teomics//Trends Biotechnol. 2003. Vol. 21 P. 383-388.
185. Генная инженерия растений. Лабораторное руководство. Ред.
Дж. Дрейнер, Р. Скотт, Ф. Армитидж, Р. Уолден. М., Мир. 1991.
407 с.
186. Нarisen G., Wright M.S. Recent advances in the transformation of plants //
Trends Plant Sci. 1999. Vol. 4. P. 226-231.
187. Jefferson R.A., Kavanagh T.A., Bevan M.W. GUS fusions: C-
Glucuronidase as a sensitive and versatile gene fusion marker in higher
plants // EMBO J. 1987. Vol.6. P. 3901-3907.
188. HaselojfJ., Siemering K.R., Prasher D.C., Hodge S. Removal of a cryptic
intron and subcellular localization of green fluorescent protein are required
to mark transgenic Arabidopsis plants brightly // Proc. Nat. Acad. Sci.
USA. 1997. Vol. 94. P. 2122-2127.
\%9.Leon-Banares R., Gonzalez-Ballester D., Galvan A., Fernandez E.
Transgenic microalgae as green cell-factories // Trends Biotechnol. 2004.
Vol. 22. P. 45-52.
190. Fromm M.E., Taylor L.P., Walbot W. Expression of genes transferred into
monocot and dicot plant cells by electroporation // Proc. Nat. Acad. Sci.
USA. 1985. Vol. 82. P. 5824-5828.
191. Zupan J., Zambryski P. The Agrobacterium DNA transfer complex // Crit.
Rev. Plant Sci. 1997. Vol. 16. P. 279-295.
192. Tzfira Т., Citovsky V. Partners-in-infection: Host proteins involved in the
transformation of plant cells by Agrobacterium // Trends Cell Biol. 2002.
Vol.12. P. 121-129.
193. Hellens R., Mullineaux P. A guide to Agrobacterium binary Ti vectors //
Trends Plant Sci. 2000. Vol. 5. P. 446^51.
194. Karimi M., Inze D., Depicker A. GATEWAY™ vectors for
Agrobacterium-m&diat&d plant transformation // Ibid. 2002. Vol. 7.
P. 193-195.
195. Lazo G.R., Stein P.A., Ludwig R.A. et al. A DNA transformation-compe-
transformation-competent Arabidopsis genomic library in Agrobacterium I I Bio/Technology.
1991. Vol. 9. P. 963-967.
196. Shibata D., Liu Y.-G. Agrobacterium-medi&ted plant transformation with
large DNA fragments // Trends Plant Sci. 2000. Vol. 5. P. 354-357.
197. Daniell H., Khan M.S., Allison L. Milestones in chloroplast genetic engi-
engineering: An environmentally friendly era in biotechnology // Ibid. 2002.
Vol. 7. P. 84-91.
198. Maliga P. Progress towards commercialization of plastid transformation
technology // Trends Biotechnol. 2003. Vol.21. P. 20-28.
199. Svab Z. et al. Stable transformation of plastids in higher plants // Proc. Nat.
Acad. Sci. USA. 1990. Vol. 87. P. 8526-8530.
264
200. Golds T. et al. Stable plastid transformation in PEG-treated protoplasts of
Nicotiana tabacum // Biotechnology. 1993. Vol. 11. P. 95-97.
201. Streips U.N. 2002. Transformation, in: Modern Microbial Genetics,
Second Edition. Edited by Streips U.N., Yasbin R.E., Wiley-Liss, Inc.
P. 429-461.
202. Прозоров АЛ. Трансформация у бактерий. М: Наука, 1988. 255 с.
203. Fuqua С, Parsek M.R., Greenberg Е.Р. Regulation of gene expression by
cell-to-cell communication: Acyl-homoserine lactone quorum sensing //
Annu. Rev. Genet. 2001. Vol. 35. P. 439^68.
204. Tonjum Т., Havarstein L.S., Koomey M., Seeberg E. Transformation and
DNA repair: Linkage by DNA recombination // Trends Microbiol. 2004.
Vol.12. P. 1-4.
205. Steinmoen H., Knutsen E., Havar stein L.S. Induction of competence in
Streptococcus pneumoniae triggers lysis and DNA release from a subfrac-
tion of the cell population // Proc. Nat. Acad. Sci. USA. 2002. Vol. 99.
P. 7681-7686.
206. Bjedov I., Tenaillon O., Gerard B. et al. Stress-induced mutagenesis in
bacteria // Science. 2003. Vol. 300. P. 1404-1409.
207. Bott K.F., Wilson G.A. Development of competence in the Bacillus subtilis
transformation system //J. Bacteriol. 1967. Vol. 94. P. 562-570.
208. Dubnau D. DNA uptake in bacteria // Ann. Rev. Microbiol. 1999. Vol. 53.
P. 217-244.
209. Wirth R., Friesinger A., Fiedler S. Transformation of various species of
gram-negative bacteria belonging to eleven different genera by electropo-
ration // Mol. Gen. Genet. 1989. Vol. 216. P. 175-179.
210. SanfordJ.C, Smith F.D., Russell JA. Optimizing the biolistic process for
different biological applications // Meth. Enzymol. 1993. Vol. 217.
P. 483-509.
211. Salman H., Zbaida D., Rabin Y., Chatenay D., Elbaum M. Kinetics and
mechanism of DNA uptake into the cell nucleus // Proc. Nat. Acad. Sci.
USA. 2001. Vol. 98. P. 7247-7252.
212. Peng K.-W., Russell S.J. Viral vector targeting // Curr. Opin. Biotechnol.
1999. Vol. 10. P. 454-457.
213. Wagner E., Zenke M., Cotten M., Beug H., Birnstiel ML. Transferrin-poly-
cation conjugates as carriers for DNA uptake into cells // Proc. Nat. Acad.
Sci. USA. 1990. Vol. 87. P. 3410-3414.
214. Neumann E., Kakorin S., Toensing K. Fundamentals of electroporative deliv-
delivery of drugs and genes // Bioelectrochem. Bioenerg. 1999. Vol. 48. P. 3-16.
215. Tao W., Wilkinson J., Stanbridge E.J., Berns M.W. Direct gene transfer
into human cultured cells facilitated by laser micropuncture of the cell
membrane//Proc. Nat. Acad. Sci. USA. 1987. Vol. 84. P. 4180-4185.
216. Klein T.M., Arentzen R., Lewis PA., Fitzpatrick-McElligott S.
Transformation of microbes, plants and animals by particle bombardment
// Bio/Technology. 1992. Vol. 10. P. 286-291.
217. Finer J.J., Vain P., Jones M.W., McMullen M.D I I Development of the par-
particle inflow gun for DNA delivery to plant cells // Plant Cell Rep. 1992.
Vol. 11. P. 323-328.
265
218. Waterhouse P.M., Helliwell C.A. Exploring plant genomes by RNA-
induced gene silencing // Nature Rev. Genet. 2002. Vol. 4. P. 29-38.
219. Jordan M., Schallhorn A., Wurm F.M. Transfecting mammalian cells:
Optimization of critical parameters affecting calcium-phosphate precipi-
precipitate formation //Nucl. Acids Res. 1996. Vol. 24. P. 596-601.
220. Lopata M.A., Cleveland D.W., Sollner-Webb B. High-level expression of a
chloramphenicol acetyl-transferase gene by DEAE dextran-mediated DNA
transfection coupled with a dimethylsulfoxide or glycerol shock treatment
//Ibid. 1984. Vol. 12. P. 5707-5717.
221. BoussifO., LezoualchF., Zanta M.A. et al. A versatile vector for gene and
oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine
//Proc. Nat. Acad. Sci. USA. 1995. Vol. 92. P. 7297-7301.
222. Scherman D., Bessodes M., Cameron B. Application of lipids and plasmid
design for gene delivery to mammalian cells // Curr. Opin. Biotechnol.
1998. Vol. 9. P. 480-485.
223. Friend D.S., Papahadjopoulos D., Debs RJ. Endocytosis and intracellular
processing accompanying transfection mediated by cationic liposomes //
Biochem. biophys. acta. 1996. Vol. 1278. P. 41-50.
224. Жакоб Ф., Волъман Э. Пол и генетика бактерий. М: Изд. Иностр.
лит. 1962. 476 с.
225. Porter R.D. Conjugation // Modern microbial genetics. Ed. U.N. Streips,
R.E. Yasbin. 2rd ed. N.Y.: Wiley, 2002. P. 463-506.
226. Waters V.L. Conjugation between bacterial and mammalian cells // Nature
Genet. 2001. Vol. 29. P. 375-376.
227. Evans G.A., Lewis K., Rothenberg B.E. High efficiency vectors for cosmid
microcloning and genomic analysis //Gene. 1989. Vol. 79. P. 9-20.
228. Molecular cloning. A Laboratory manual on the WEB. Cold Spring Harbor
Laboratory Press. 2002. http://www.molecularcloning.com
229. Gubler U., Hoffman BJ. A simple and very efficient method for generat-
generating cDNA libraries // Gene. 1983. Vol. 25. P. 263-269.
230. Gerard G.F., D'Alessio JM. Reverse transcriptase (EC2.7.7.49): The use
of cloned Moloney murine leukemia virus reverse transcriptase to synthe-
synthesize DNA from RNA // Meth. Mol. Biol. 1993. Vol. 16. P. 73-94.
231. Gubler U. Second-strand cDNA synthesis: mRNA fragments as primers //
Meth. Enzymol. 1987. Vol. 152. P. 330-335.
232. Borodin A., Kopatnzev E., Wagner L. et al. An arrayed library enriched in
hncDNA corresponding to transcribed sequences of human chromosome
19: Preparation and analysis // Genetic Analysis. 1995. Vol. 12. P. 23-31.
233. Grothues D., Cantor C.R., Smith C.L. Top-down construction of an ordered
Schizosaccharomyces pombe cosmid library // Proc. Nat. Acad. Sci. USA.
1994. Vol. 91. P. 4461-4465.
234. Branscomb E., Slezak Т., Рае R. et al. Optimizing restriction fragment fin-
fingerprinting methods for ordering large genomic libraries // Genomics.
1990. Vol. 8. P. 351-366.
235. Grunstein M., Hogness D.S. Colony hybridization: A method for the isola-
isolation of cloned DNAs that contain a specific gene // Proc. Nat. Acad. Sci.
USA. 1975. Vol. 72. P. 3961-3965.
266
236. Hanahan D., Meselson M. Plasmid screening at high colony density //
Gene. 1980. Vol. 10. P. 63-67.
237. Lee C.C., Wu X., Gibbs R.A. et al. Generation of cDNA probes directed by
amino acid sequence: Cloning of urate oxidase // Science. 1988. Vol. 239.
P. 1288-1291.
238. Rose T.M., Schultz E.R., HenikoffJ.G. et al. Consensus-degenerate hybrid
oligonucleotide primers for amplification of distantly related sequences //
Nucl. Acids Res. 1998. Vol. 26. P. 1628-1635.
239. Parrish J.E., Nelson D.L. Methods for finding genes: A major rate-limiting
step in positional cloning. Gene Anal. Tech. Appl. 1993. Vol. 10. P. 29-41.
240. Oh J., Bailin Т., Fukai K. et al. Positional cloning of a gene for Hermansky-
Pudlak syndrome, a disorder of cytoplasmic organelles // Nature Genet.
1996. Vol. 14. P. 300-306.
241. Wang Q., Curran M.E., Splawski I. et al. Positional cloning of a novel
potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias.
Nature Genet. 1996. Vol. 12. P. 17-23.
242. Kawakami Y., Battles J.K., Kobayashi T. et al. Production of recombinant
MART-1 proteins and specific antiMART-1 polyclonal and monoclonal
antibodies: Use in the characterization of the human melanoma antigen
MART-1 //J. Immunol. Meth. 1997. Vol. 202. P. 13-25.
243. Broderick J.B. Catechol dioxygenases // Essays Biochem. 1999. Vol. 34.
P. 173-189.
244. Gardner K.H., Kay L.E. The use of 2H, 13C, 15N multidimensional NMR to
study the structure and dynamics of proteins // Annual Rev. Biophys.
Biomol. Struct. 1998. Vol. 27. P. 357^06.
245. Schmidt M., Babu K.R., Khanna N. et al. Temperature-induced production
of recombinant human insulin in high-cell density cultures of recombinant
Escherichia coli II J. Biotechnol. 1999. Vol. 68. P. 71-83.
246. Jaeger K.-E. Microbial lipases form versatile tools for biotechnology //
Trends Biotechnol. 1998. Vol. 16. P. 396-403.
247. Rosenau F., J eager J.-E. Bacterial lipases from Pseudomonas: regulation
of gene expression and mechanisms of secretion // Biochemie. 2000.
Vol. 82. P. 1023-1032.
248. Urban A., Leipelt M., Eggert T. et al. DsbA and DsbC affect extracellular
enzyme formation in Pseudomonas aeruginosa I I J. Bacteriol. 2001.
Vol.183. P. 587-596.
249. Liebeton K., Zacharias A., Jeager K.-E. Disulfide bond in Pseudomonas
aeruginosa lipase stabilizes the structure but is not required for interaction
with its foldase // Ibid. P. 597-603.
250. Emr S.D. Heterologous gene expression in yeast // Meth. Enzymol. 1990.
Vol. 185. P. 231-233.
251. Stearns Т., Ma H., Botstein D. Manipulating yeast genome using plasmid
vectors // Ibid. P. 280-297.
252. Rose A.B., Broach J.R. Propagation and expression of cloned genes in
yeast: 2-Microns circle-based vectors // Ibid. P. 234-279.
. Ho H., Fukuda Y., Murata K. Kimura A. Transformation of intact yeast
cells treated with alkali cations //J. Bacteriol. 1983. Vol. 153. P. 163-168.
267
254. Becker DM., Guarente L. High-efficiency transformation of yeast by elec-
troporation//Meth. Enzymol. 1991. Vol. 194. P. 182-187.
255. Cereghino G.P.L, Cregg J.M. Applications of yeast in biotechnology:
Protein production and genetic analysis // Curr. Opin. Biotechnol. 1999.
Vol. 10. P. 422^27.
256. Zsebo KM., Lu H.S., Fieschko J.C. et al. Protein secretion from
Saccharomyces cerevisiae directed by the prepro-alpha-factor leader
region // J. Biol. Chem. 1986. Vol. 261. P. 5858-5865.
257. Romanos M.A., Scorer C.A., Clare JJ. Foreign gene expression in yeast:
A review // Yeast. 1992. Vol. 8. P. 423^88; (a) Giesse S., Gram H.,
Kleuser B. et al. Eukaryotic expression systems: A comparison // Protein
Expression and Purification. 1996. Vol. 8. P. 271-282.
258. Wilson R., Oliver J., Brookman J.L. et al. Development of a semi-perme-
abilised cell system to study the translocation, folding, assembly and trans-
transport of secretory proteins // Biochem. J. 1995. Vol. 307. P. 679-687.
259. Farmery M.R., Allen S., Allen A.J., BulleidNJ. The role of ERp57 in disul-
fide bond formation during the assembly of MHC class I in a synchronized
semi-permeabilised cell translation system // J. Biol. Chem. 2000.
Vol. 275. P. 14933-14938.
260. Jermutus L., Ryabova L.A., Pliickthun A. Recent advances in producing
and selecting functional proteins by using cell-free translation // Curr.
Opin. Biotechnol. 1998. Vol. 9. P. 534-548.
261. Saiki R.K., ScharfS., Faloona F. et al. Enzymatic amplification of C-glo-
bin genomic sequences and restriction site analysis for diagnosis of sickle
cell anemia // Science. 1985. Vol. 230. P. 1350-1354.
262. Mullis KB, Faloona F. Specific synthesis of DNA in vitro via a polymerase
catalyzed chain reaction // Meth. Enzymol. 1987. Vol. 155. P. 335-350.
263. Alkami Quick Guide™ for PCR. A laboratory reference for the polymerase
chain reaction. 1999. http://www.alkami.com
264. Sarkar G., Sommer 5.5. Shedding light on PCR contamination // Nature.
1990. Vol. 343. P. 27.
265. Prince AM., Andrus L. PCR: How to kill unwanted DNA // Biotechniques.
1992. Vol. 12. P. 358-360.
266. De Filippes FM. Decontaminating the polymerase chain reaction // Ibid.
1991. Vol. 10. P. 26-30.
267. Espy MJ., Smith T.F., Per sing DM. Dependence of polymerase chain
reaction product inactivation protocols on amplicon length and sequence
composition //J. Clin. Microbiol. 1993. Vol. 31. 2361-2365.
268. Furrer В., Candrian U., Wieland P., Luthy J. Improving PCR efficiency //
Nature. 1990. Vol. 346. P.324.
269. Meier A., Persing D.H., Finken M., Bottger E.C. Elimination of contami-
contaminating DNA within polymerase chain reaction reagents: Implications for a
general approach to detection of uncultured pathogens // J. Clin. Microbiol.
1993. Vol. 31.646-652.
270. Walsh P.S., Metzger D.A., Higuchi R. Chelex-100 as a medium for simple
extraction of DNA for PCR-based typing from forensic material //
Biotechniques. 1991. Vol. 10. P. 506-513.
268
271. Wilson l.G. Inhibition and facilitation of nucleic acid amplification // Appl.
Envir. Microbiol. 1997. Vol. 63. P. 3741-3751.
272. Nichols R.C., Raben N. Hints for direct sequencing of PCR-generated sin-
single-stranded DNA // Biotechniques. 1994. Vol. 17. P. 412-414.
273. Dale J.W., von Schantz M. From genes to genomes. Concepts and applica-
applications of DNA technology. N.Y.: Wiley, 2002.
274. Horton R.M., Hoppe B.L., Conti-Tronconi B.M. AmpliGrease: "Hot
Start" PCR Using Petroleum Jelly // Biotechniques. 1994. Vol. 16.
P. 42-43.
275. Kellogg D.E., Rybalkin I., Chen S. et al. TaqStartTM Antibody: "Hot
Start" PCR Facilitated by a Neutralizing Monoclonal Antibody Directed
Against Taq DNA Polymerase // Biotechniques. 1994. Vol. 16.
1134-1137.
276. Birch D.E. Simplified hot start PCR // Nature. 1996. Vol. 381. P. 445^46.
277. Dang C, Jayasena S.D. Oligonucleotide inhibitors of Taq DNA poly-
polymerase facilitate detection of low copy number targets by PCR // J. Mol.
Biol. 1996. Vol. 264. P. 268-278.
278. Brownie J., Shawcross S., Theaker J. et al. The elimination of primer-
dimer accumulation in PCR // Nucl. Acids Res. 1997. Vol. 25.
P. 3235-3241.
279. Bustin S.A. Absolute quantification of mRNA using real-time reverse
transcription polymerase chain reaction assays //J. Mol. Endocrinol. 2000.
Vol. 25. P. 169-193.
280. Barnes W. PCR Amplification of up to 35-kb DNA with high fidelity and
high yield from bacteriophage templates // Proc. Nat. Acad.Sci. USA.
1994. Vol. 91. P. 2216-2220.
281. Lamont PJ., Davis M.B., WoodN.W. Identification and sizing of the GAA
trinucleotide repeat expansion of Friedreich's ataxia in 56 patients - clini-
clinical and genetic correlates // Brain. Vol. 1997, 120. P. 673-680.
282. Dittmar M.T., Simmons G., Donaldson Y. et al. Biological characterization
of human immunodeficiency virus type 1 clones derived from different
organs of an AIDS patient by long-range PCR // J. Virol. 1997. Vol. 71.
P. 5140-5147.
283. Li D., Vijg J. Multiplex co-amplification of 24 retinoblastoma gene exons
after pre-amplification by long-distance PCR // Nucl. Acids Res. 1996.
Vol. 24. P. 538-539.
284. Gosden J., Hanratty D. PCR in situ: A rapid alternative to in situ hybridiza-
hybridization for mapping short, low copy number sequences without isotopes //
Biotechniques. 1993. Vol. 15. P. 78-80.
285. PCR in situ hybridization. 3rd ed., Philadelphia: Lippincott-Raven, 1998.
286. Patrushev L.I., Zykova E.S., Kayushin A.L. et al. New DNA diagnostic sys-
system for detection of Factor V Leiden // Thrombosis Res. 1998. Vol. 92.
P. 251-259.
287. Nils son M., Krejci K., Koch J. et al. Padlock probes reveal single-
nucleotide differences, parent of origin and in situ distribution of cen-
tromeric sequences in human chromosomes 13 and 21 // Nature Genet.
1997. Vol. 16. P. 252-255.
269
288. Nilsson M., Malmgren H., Samiotaki M. et al. Padlock probes:
Circularizing oligonucleotides for localized DNA detection // Science.
1994. Vol. 265. P. 2085-2088.
289. Barter J., Nilsson M., Mendel-Hartvig M., Landegren U. Signal amplifica-
amplification of padlock probes by rolling circle replication // Nucl. Acids Res.
1998. Vol. 26. P. 5073-5078.
290. Faruqi A.F. et al. High-throughput genotyping of single nucleotide poly-
polymorphisms with rolling circle amplification // BMC Genomics. 2001.
Vol. 2. P. 4.
291. Rein Т., DePamphilis ML., Zorbas H. Identifying 5-methylcytosine and
related modifications in DNA genomes // Nucl. Acids Res. 1998. Vol. 26.
P. 2255-2264.
292. Sano T. Smith C.L., Cantor C.R. Immuno-PCR: Very sensitive antigen
detection by means of specific antibody-DNA conjugates // Science. 1992.
Vol. 258. P. 120-122.
293. Hendrickson E.R., Hafield-Truby T.M., JoergerR.D. et al. High sensitiv-
sensitivity multianalyte immunoassay using covalent DNA labeled antibodies and
polymerase chain reaction // Nucl. Acids Res. 1995. Vol. 23. P. 522-
529.
294. Niemeyer CM. The developments of semisynthetic DNA-protein conju-
conjugates // Trends Biotechnol // 2002. Vol. 20. P. 395^01.
295. Niemeyer СМ., Adler M., Blohm D. Fluorometric polymerase chain reac-
reaction (PCR) enzyme-linked immunosorbent assay for quantification of
immuno-PCR products in microplates // Anal. Biochem. 1997. Vol. 246.
P. 140-145.
296. Niemeyer CM. et al. Self-assembly of DNA-streptavidin nanostructures
and their use as reagents in immuno-PCR // Nucl. Acids Res. 1999.
Vol. 27. P. 4553-4561.
297. Holland P., Abramson R.D., Watson R., Gelfand D.H. Detection of specif-
specific polymerase chain reaction product by utilizing the 5/->3/ exonuclease
activity of Thermus aquaticus DNA polymerase // Proc. Nat. Acad. Sci.
USA. 1991. Vol. 88. P. 7276-7280.
298. Gut M., Leutenegger СМ., Huder J.B. et al. One-tube fluorogenic reverse
transcription-polymerase chain reaction for the quantitation of feline coro-
naviruses // J. Virol. Meth. 1999. Vol. 77. P. 37^6.
299. Lyamichev V., Brow M.A., Dahlberg J.E. Structure specific endonucle-
olytic cleavage of nucleic acids by eubacterial DNA polymerases //
Science. Vol. 260. 1993. P. 778-783.
300. Heid C.A., Stevens J., Livak K.J., Williams P.M. Real time quantitative
PCR // Genome Research. 1996. Vol. 6. P. 986-994.
301. Livak KJ., Flood S.J., Marmaro J. et al. Oligonucleotides with fluorescent
dyes at opposite ends provide a quenched probe system useful for detect-
detecting PCR product and nucleic acid hybridization // PCR Methods and
Applications. 1995. Vol. 4. P. 357-362.
302. Sun C-F., Lee C.-H., Cheng S.-W. et al. Real-time quantitative PCR analy-
analysis for oc-thalassemia-1 of Southeast Asian type deletion in Taiwan. Clin.
Genet. 2001. Vol. 60. P. 305-309.
270
303. Glazer A.N., Mathies RA. Energy-transfer fluorescent reagents for DNA
analyses // Curr. Opin. Biotechnol. 1997. Vol. 8. P. 94-102.
304. Wittwer C.T., Herrmann M.G., Moss A A., Rasmussen R.P. Continuous flu-
fluorescence monitoring of rapid cycle DNA amplification // Biotechniques.
1997. Vol. 22. P. 130-131, 134-138.
305. Tyagi S., Bratu D.P., Kramer F.R. Multicolor molecular beacons for allele
discrimination // Nature Biotechnol. 1998. Vol. 16. P. 49-53.
306. Antony Т., Subramaniam V. Molecular beacons: Nucleic acid hybridization
and emerging applications // J. Biomol. Struct. Dynamics. 2001. Vol. 19.
P. 497-504.
307. Tyagi S., Landegren U., Tazi M. et. al. Extremely sensitive, background-
free gene detection using binary probes and beta replicase // Proc. Nat.
Acad. Sci. USA. 1996. Vol. 93. P. 5395-5400.
308. Morrison T.B., Weis J.J., Wittwer C.T. Quantification of low-copy tran-
transcripts by continuous SYBR Green I monitoring during amplification.
Biotechniques // 1998. Vol. 24. P. 954-962.
309. Whitcombe ?>., TheakerJ., Guy S.P. et al. Detection of PCR products using
self-probing amplicons and fluorescence // Nature Biotechnology. 1999.
Vol. 17. P. 804-807.
310. Thelwell N., Millington S., Solinas A. et al. Mode of action and application
of Scorpion primers to mutation detection // Nucl. Acids Res. 2000.
Vol. 28. P. 3752-3761.
311. Cantor C.R., Smith C.L. I I Genomics: The science and technology behind
the human genome project. N.Y. Wiley: 1999. P. 98-130.
312. Riley J., Butler R., Olgivie D. et al. A novel, rapid method for the isolation
of terminal sequences from yeast artificial chromosome (YAC) clones //
Nucl. Acids Res. 1990. Vol. 18. P. 2887-2890.
313. Trig На Т., Peterson M.G., Kemp DJ. A procedure for in vivo amplification
of DNA segments that lie outside the boundaries of known sequences //
Ibid. 1988. Vol. 16. P. 8186.
314. Cantor C.R., Smith C.L. II Genomics: The science and technology behind
the human genome project. N.Y. Wiley: 1999. P. 112-113.
315. Mudge J., Anderson W.R., Kehrer R.L., Fairbanks DJ. A RAPD genetic
map of Saccharum officinarum II Crop Sci. 1996. Vol. 36. P. 1362-1366
316. Zarlenga D.S., Higgins J. PCR as a diagnostic and quantitative technique in
veterinary parasitology //Veterinary Parasitol. 2001. Vol. 101. P. 215-230.
317. Zhang L., Cui X., Schmitt K. et al. Whole genome amplification from a sin-
single cell: Implications for genetic analysis // Proc. Nat. Acad. Sci. USA.
1992. Vol. 89. P. 5847-5851.
318. Cheung V.G., Nelson S.F. Whole genome amplification using a degenerate
oligonucleotide primer allows hundreds of genotypes to be performed on
less than one nanogram of genomic DNA // Ibid. 1996. Vol. 93.
Vol. 14676-14679.
319. Grothues D., Cantor C.R., Smith C.L. PCR amplification of megabase
DNA with tagged random primers (T-PCR) // Nucl. Acids Res. 1993.
Vol. 21. P. 1321-1322.
320. Samura O., Pertl В., Sohda S. et al. Female fetal cells in maternal blood:
271
Use of DNA polymorphisms to prove origin // Hum. Genet. 2000.
Vol. 107. P. 28-32.
321. Hahn S., Zhong X.Y., Troeger C. et al. Current applications of single-cell
PCR // Cell. Mol. Life Sci. 2000. Vol. 57. P. 96-10.
322. Barany F. Genetic disease detection and DNA amplification using cloned
thermostable ligase // Proc. Nat. Acad. Sci. USA. 1991. Vol. 98.
P. 189-193.
323. Jarvius J. et al. Oligonucleotide ligation assay // Meth. Mol. Biol. 2003.
Vol. 212. P. 215-228.
324. Fahy E., Kwoh D.Y., Gingeras T.R. Self-sustained sequence replication
CSR): An isothermal transcription-based amplification system alternative
to PCR // PCR Meth. and Appl. 1991. Vol. 1. P. 25-33.
325. Schweitzer В., Kingsmore S. Combining nucleic acid amplification and
detection // Curr. Opin. Biotechnol. 2001. Vol. 12. P. 21-27.
326. Walker G.T., Little M., Nadeau J.G., Shank D.D. Isothermal in vitro ampli-
amplification of DNA by a restriction enzyme/DNA polymerase system // Proc.
Nat. Acad. Sci. USA. 1992. Vol. 89. P. 392-396.
327. Little M., Andrews J., Moore R. et al. Strand displacement amplification
and homogeneous real-time detection incorporated in a second-generation
DNA probe system, BDProbeTecET // Clin. Chem. 1999. Vol. 45.
P. 777-784.
328. Westin L., Xu X., Miller C. et al. Anchored multiplex amplification on a
microelectronic chip array // Nature Biotechnol. 2000. Vol. 18.
P. 199-204.
329. Tyagi S., Landedren U., Tazi M. et al. Extremely sensitive, background-
free gene detection using binary probes and Q|3 replicase // Proc. Nat.
Acad. Sci. USA. 1996. Vol. 93. P. 5395-5400.
330. Abramson R.D., Myers T.W. Nucleic acid amplification technologies //
Current Biol. 1993. Vol. 4. P. 41-^7.
331. Lizardi P., Huang X., Zhu Z. et al. Mutation detection and single molecule
counting using isothermal rolling circle amplification // Nature Genet.
1998. Vol. 19. P. 225-232.
332. Schweitzer В., Wiltshire S., Lambert J. et al. Immunoassays with rolling
circle DNA amplification: A versatile platform for ultrasensitive antigen
detection//Proc. Nat. Acad. Sci. USA. 2000. Vol. 97. P. 10113-10119.
333. Wiltshire S., O'Malley S., Lambert J. et al. Detection of multiple allergen-
specific IgE on microarrays by immunoassay with rolling circle amplifica-
amplification // Clin. Chem. 2000. Vol. 46. P. 1990-1993.
334. Thomas D., Nardone G., Randall S. Amplification of padlock probes for
DNA diagnostics by cascade rolling circle amplification or the polymerase
chain reaction //Arch. Pathol. Lab. Med. 1999. Vol. 123. P. 1170-1176.
335. Banfi S., Guffanti A., Borsani G. How to get the best of dbEST // Trends
Genet. 1998. Vol. 14. P. 80-81.
Часть II
Белковая инженерия
Введение
Ферменты, повсеместно распространенные в живой природе,
обладают целым рядом свойств, которые делают их весьма при-
привлекательными объектами исследований в биотехнологии [1, 2].
Они осуществляют ферментативные реакции в мягких условиях
с кажущимися константами специфичности (ккат/км) в пределах
106— 108 М-1 • сек-1. При этом имеет место ускорение химических
реакций (&ка1ДНекат) Д° Ю17 раз. Уникальная геометрическая орга-
организация активных центров ферментов, обеспечивающая одно-
однозначное расположение в них субстратов реакции, делает фермен-
ферменты высокоспецифическими катализаторами, действие которых
не сопровождается образованием побочных продуктов и не тре-
требует высоких затрат энергии. Эти свойства ферментов имеют
большое значение для охраны окружающей среды. В то же вре-
время большая специфичность ферментов имеет для биотехнологии
и свою оборотную сторону: с помощью природных ферментов
можно осуществлять лишь химические реакции, происходящие в
организме. Из этого весьма существенного ограничения возник-
возникла важная задача: создание ферментов с новыми свойствами, ко-
которые бы осуществляли новые химические реакции.
Интерес к направленному изменению свойств ферментов не
ограничивается запросами современной индустрии и медицины.
Исследование механизмов биокатализа и особенностей структур-
структурно-функциональных взаимоотношений в белках имеет большое
фундаментальное значение, поскольку все эти явления лежат в
основе жизни. А говорить о понимании природы ферментативно-
ферментативного катализа можно только после искусственного создания поли-
полипептидов с заранее заданными признаками. Получением белков и
ферментов с новыми свойствами занимается одно из наиболее
активно развивающихся направлений современной молекуляр-
молекулярной биологии - белковая инженерия.
273
Два направления исследований в белковой инженерии.
В современной белковой инженерии активно развиваются два
тесно связанных друг с другом подхода: рациональный дизайн
(rational design) белковых молекул и их направленная эволюция
(directed evolution) [3]. Как следует из самого названия научного на-
направления, исследователи, работающие в области рационального
дизайна, предпринимают попытки создания новых белков, исходя
исключительно из теоретических представлений о будущей конст-
конструкции [3,4]. В этом случае на чертеже располагают остатки ами-
аминокислот конструируемого белка таким образом, чтобы синтези-
синтезированная по данному плану полипептидная цепь после приобрете-
приобретения ею пространственной структуры в результате фолдинга оказа-
оказалась способной катализировать требуемую химическую реакцию.
Естественно, реализация таких проектов требует глубоких знаний
законов, определяющих соответствие между первичной и третич-
третичной структурами белков, которые, несмотря на интенсивные ис-
исследования, на сегодняшний день не вполне понятны. Поскольку
формирование пространственной структуры белков определяет
большое количество внутримолекулярных и межмолекулярных
взаимодействий, конструирование элементов вторичной и третич-
третичной структур белков не всегда удается даже в простейших случаях.
Мощностей современных компьютеров не хватает для проведения
расчетов таких взаимодействий с использованием имеющихся ал-
алгоритмов даже в коротких полипептидных цепях. Однако, несмот-
несмотря на эти ограничения, которые основываются исключительно на
нашей неполной осведомленности в области физики и химии бел-
белка, а также недостаточно высоком уровне развития современной
вычислительной техники, за рациональным дизайном, несомнен-
несомненно, большое будущее. Можно сказать, что реализация в полной
мере подходов на основе рационального дизайна является про-
граммой-максимум белковой инженерии, которая когда-нибудь
будет выполнена. И тогда перед человечеством откроются неогра-
неограниченные возможности создания искусственных генетических сис-
систем, в том числе и новых форм жизни.
Подход, получивший название направленной эволюции бел-
белковых молекул, не требует досконального знания особенностей
строения и функционирования создаваемых белковых молекул
[5-7]. В основе этого экспериментального направления исследо-
исследований лежат идеи, хорошо известные в эволюционной биологии.
Для осуществления направленной эволюции белков в условиях
лаборатории создают большие клонотеки случайных аминокис-
аминокислотных последовательностей, из которых вначале тем или иным
274
методом отбирают полипептидные цепи, обладающие хотя бы в
небольшой степени требуемыми свойствами. Далее на основе
найденных последовательностей с использованием случайного
мутагенеза получают новые клонотеки белков, которые приме-
применяют в следующем раунде селекции, и так далее. При осуществ-
осуществлении направленной эволюции используют генно-инженерные
конструкции, экспрессирующие исследуемые белки, которые ос-
остаются физически связанными с отбираемыми белками, напри-
например, в составе фаговой частицы или в комплексе с кодирующей
белок мРНК. Следовательно, в таких искусственных генетичес-
генетических системах генотип оказывается однозначно связанным с фе-
фенотипом, что позволяет через генотип, изменяемый направлен-
направленным или случайным мутагенезом, оказывать влияние на фенотип
белковой молекулы и отбирать полипептиды, обладающие необ-
необходимыми свойствами. Использование методов направленной
эволюции позволило добиться выдающихся результатов в белко-
белковой инженерии, часть из которых будет рассмотрена ниже.
Исследователи, работающие в области направленной эволю-
эволюции, изначально используют клонотеки последовательностей,
которые, несмотря на случайный характер их создания, охваты-
охватывают лишь небольшую часть теоретически возможного прост-
пространства последовательностей, представляющего набор моле-
молекул с аминокислотными остатками (или нуклеотидами) во все-
всевозможных сочетаниях. Поскольку такие клонотеки получают
руками экспериментатора, то с их помощью можно конструиро-
конструировать макромолекулы, не встречающиеся в природных условиях.
Однако размер полного пространства последовательностей
слишком велик, чтобы с ним можно было работать эксперимен-
экспериментально. Поэтому в белковой инженерии на практике часто реа-
реализуют компромиссный вариант подхода, в котором в качестве
исходной последовательности при конструировании нового фер-
фермента используют известную последовательность с исследован-
исследованной пространственной структурой, функциональность которой
уже была апробирована в природе. В этом случае на основе име-
имеющихся знаний о структуре и механизмах функционирования ак-
активного центра исходного фермента с помощью направленного
мутагенеза заменяют конкретные аминокислотные остатки в на-
надежде получить фермент с требуемыми свойствами. Такой под-
подход получил название рационального редизайна. Одним из плодо-
плодотворных подходов в этом направлении исследований является ин-
инженерия белковых поверхностей (patch engineering), при котором
с помощью мутаций изменяют участки полипептидной цепи в ок-
окрестностях аминокислотных остатков, сближенных на поверхно-
275
сти белковой глобулы, но находящихся в полипептидной цепи на
значительном расстоянии друг от друга.
Поскольку большинство изменений аминокислотных остат-
остатков в составе полипептидных цепей проводится в искусственных
генетических системах с использованием направленного или слу-
случайного мутагенеза, целесообразно в конце этого краткого вве-
введения вспомнить типы мутаций и их основные характеристики.
Мутации, мутагенез и их последствия
для белковых молекул.
Мутации - это наследуемые изменения структуры генома, а
следовательно и генов, представляющих его существенную часть.
Поскольку основу любого генома составляют нуклеиновые кисло-
кислоты - ДНК или РНК, то под действием мутаций происходит, преж-
прежде всего, изменение структуры геномных нуклеиновых кислот.
Процесс возникновения мутаций, основанный на различных меха-
механизмах, называют мутагенезом. В зависимости от факторов, вы-
вызывающих мутации, последние принято разделять на спонтанные
и индуцированные. Считается, что спонтанные мутации возника-
возникают самопроизвольно на протяжении всей жизни организма в нор-
нормальных для него условиях окружающей среды. При этом широ-
широкое распространение получило мнение о том, что спонтанные му-
мутации в эукариотических клетках возникают с частотой 10-9-10-12
на нуклеотид за клеточную генерацию. Теперь, однако, становит-
становится ясно, что такие цифры не отражают реальности. Они не учиты-
учитывают того факта, что частоты спонтанных мутаций могут сущест-
существенно (на несколько порядков) изменяться от локуса к локусу и,
скорее всего, указывают на нижний предел частоты мутаций в от-
отдельных наиболее стабильных участках генома.
Индуцированными мутациями называют наследуемые изме-
изменения генома, возникающие в результате тех или иных мутаген-
мутагенных воздействий в искусственных (экспериментальных) услови-
условиях или при неблагоприятных воздействиях окружающей среды.
Среди важнейших мутагенных факторов, прежде всего, необхо-
необходимо отметить химические мутагены - органические и неоргани-
неорганические вещества, вызывающие мутации, а также ионизирующее
излучение. При детальном рассмотрении спонтанных и индуциро-
индуцированных мутаций становится ясно, что между этими двумя типами
нет существенных различий. Действительно, большинство спон-
спонтанных мутаций возникает в результате мутагенного воздейст-
воздействия, которое их индуцирует, но не регистрируется эксперимента-
экспериментатором. На более прочном фундаменте находится классификация
276
мутаций, в которой учитываются молекулярные процессы, лежа-
лежащие в основе их возникновения.
В классификации, базирующейся на размерах сегментов ге-
генома, подвергающихся преобразованиям, мутации разделяют на
геномные, хромосомные и генные. При геномных мутациях у ор-
организма-мутанта происходит внезапное изменение числа хромо-
хромосом, кратное целому геному. Если через 2п обозначить число
хромосом в исходном диплоидном геноме, то в результате геном-
геномной мутации, называемой полиплоидизацией, происходит образо-
образование полиплоидных организмов, геном которых представлен An,
вп и т.д. хромосомами. В зависимости от происхождения хромо-
хромосом в полиплоидах различают аллополиплоидию, в результате
которой происходит объединение при гибридизации целых не-
неродственных геномов, и аутополиплоидию, для которой харак-
характерно адекватное увеличение числа хромосом собственного гено-
генома, кратное 2п. При хромосомных мутациях происходят как изме-
изменение числа отдельных хромосом в геноме (анеуплоидия), так и
крупные перестройки структуры отдельных хромосом. Послед-
Последние получили название хромосомных аберраций. В этом случае
наблюдаются потеря (делеции) или удвоение части (дупликации)
генетического материала одной или нескольких хромосом, изме-
изменение ориентации сегментов хромосом в отдельных хромосомах
(инверсии), а также перенос части генетического материала с од-
одной хромосомы на другую (транслокации) (крайний случай -
объединение целых хромосом).
На генном уровне изменения первичной структуры ДНК под
действием мутаций менее значительны, чем при хромосомных
мутациях, однако, генные мутации встречаются более часто.
В результате генных мутаций происходят замены, делеции и
вставки одного или нескольких нуклеотидов, транслокации, дуп-
дупликации и инверсии различных частей гена. В том случае, когда
под действием мутации изменяется лишь один нуклеотид, гово-
говорят о точковых мутациях. Поскольку в состав ДНК входят азо-
азотистые основания только двух типов - пурины и пиримидины, все
точковые мутации с заменой оснований разделяют на два класса:
транзиции (замена пурина на пурин или пиримидина на пирими-
пиримидин) и трансверсии (замена пурина на пиримидин или наоборот).
Из-за вырожденности генетического кода могут быть три гене-
генетических последствия точковых мутаций: сохранение смысла ко-
дона (синонимическая замена нуклеотида), изменение смысла
кодона, приводящее к замене аминокислоты в соответствующем
месте полипептидной цепи (миссенс-мутация) или образование
бессмысленного кодона с преждевременной терминацией (нон-
277
сенс-мутация). В генетическом коде имеются три бессмыслен-
бессмысленных кодона: амбер - UAG, охр - UAA и опал - UGA. В соответст-
соответствии с этим получают название и мутации, приводящие к образо-
образованию бессмысленных триплетов (например, амбер-мутация).
По влиянию на экспрессию генов мутации разделяют на две
категории: мутации типа замен пар оснований и типа сдвига рам-
рамки считывания (frameshift). Последние представляют собой де-
леции или вставки нуклеотидов, число которых не кратно трем,
что связано с триплетностью генетического кода. Первичную
мутацию иногда называют прямой мутацией, а мутацию, вос-
восстанавливающую исходную структуру гена, - обратной мута-
мутацией, или реверсией. Возврат к исходному фенотипу у мутантно-
мутантного организма вследствие восстановления функции мутантного
гена нередко происходит не за счет истинной реверсии, а вслед-
вследствие мутации в другой части того же самого гена или даже дру-
другого неаллельного гена. В этом случае возвратную мутацию на-
называют супрессорной. Молекулярно-генетические механизмы,
благодаря которым происходит супрессия мутантного фенотипа,
весьма разнообразны.
Умение индуцировать мутации в клонированных генах с це-
целью получения мутантных белков лежит в основе белковой ин-
инженерии. При этом используют две группы методов, приводящих
к разным последствиям на молекулярном уровне. Первая группа
основана на случайном мутагенезе, т.е. введении в мутагенизиру-
емый участок гена многих мутаций, положение каждой из кото-
которых не контролируется исследователем, а ограничивается лишь
размером фрагмента нуклеиновой кислоты, в который эти мута-
мутации вводятся. Более или менее случайный мутагенез имеет мес-
место, в частности, при инкубации нуклеиновых кислот с химически-
химическими мутагенами или осуществлении их синтеза с помощью ДНК-
полимераз с ослабленной специфичностью в отношении субстра-
субстратов-предшественников. Совокупность этих подходов активно ис-
используется при проведении направленной эволюции белковых
молекул. Вторая группа методов, получившая название направ-
направленного или сайт-специфического мутагенеза, обеспечивает
введение мутаций в строго определенные участки нуклеиновых
кислот, что позволяет заменять отдельные аминокислотные ос-
остатки в кодируемых этими молекулами белках и ферментах. На-
Направленный мутагенез является сердцем (но не мозгом) рацио-
рационального дизайна и редизайна белковых молекул, к рассмотре-
рассмотрению которого мы сейчас переходим.
275
Глава 1
Рациональный дизайн и редизайн
белковых молекул
Рациональный дизайн белков основывается на сознательном
использовании законов формирования пространственной струк-
структуры белков и механизмов ферментативного катализа для созда-
создания новых макромолекул, обладающих требуемыми свойствами.
Конструирование белковой молекулы с чистого листа de novo в
наиболее сложной и интригующей форме предполагает получе-
получение последовательности, не встречающейся в природе, которая
претерпевает фолдинг с образованием белковой глобулы с пред-
предсказанной в проекте третичной структурой и ферментативной
активностью. Термин "дизайн de novo" также часто используют
для описания процесса конструирования белковой молекулы с за-
заложенными исследователем особенностями пространственной
структуры ее скелета без учета данных о точных координатах
атомов макромолекулы. Кроме того, все вышеупомянутые воз-
возможности успешно применяются для перестройки известных
белков с целью направленного изменения их свойств и биологи-
биологической активности. В этом случае в процессе редизайна пересма-
пересматриваются проекты основного дизайнера белков - природы, как
правило, в двух основных направлениях: для создания мини-бел-
мини-белков, более короткие полипептидные цепи которых сохраняют
важные свойства исходных природных макромолекул, а также
для получения ферментов с измененной субстратной специфич-
специфичностью или обладающих большей стабильностью в экстремаль-
экстремальных условиях их использования (экстремозимов).
1.1. Проектирование новых белков и ферментов
1.1.1. Энергетические требования,
предъявляемые к новой белковой конструкции
В процессе биосинтеза или по его завершению линейная мо-
молекула полипептида должна свернуться правильным образом с
образованием нативной пространственной структуры, необходи-
необходимой для ее функционирования. Процесс сворачивания (фолдинга)
полипептидной цепи может происходить как самопроизвольно,
так и с участием белков-помощников - шаперонов. В соответст-
соответствии с современными моделями, высококооперативный процесс
фолдинга полипептидных цепей происходит по механизму нукле-
279
ации, т.е. начинается с формирования ядер фолдинга, в котором
участвует небольшое количество аминокислотных остатков, во-
вокруг которых начинают формироваться другие внутримолеку-
внутримолекулярные взаимодействия [8].
Теоретически разработанная полипептидная цепь будет при-
приобретать в процессе фолдинга уникальную правильную прост-
пространственную структуру, если она будет удовлетворять, по край-
крайней мере, двум требованиям [9]. Во-первых, цепь должна содер-
содержать позитивные элементы, стабилизирующие ее правильную
третичную структуру. Различия в свободной энергии поли-
полипептидных цепей природных белков в правильной конформа-
ции и денатурированном состоянии достигают значений
4-10 ккал/моль. Во-вторых, полипептидные цепи должны за-
заключать в себе элементы негативного дизайна для создания
большого энергетического разрыва между нативным состояни-
состоянием молекулы и другой возможной конформацией. В противном
случае, в процессе сворачивания полипептидной цепи белок вме-
вместо единственной правильной конформации будет предпочти-
предпочтительно формировать альтернативные элементы вторичной
структуры, организованные в глобулы. Таким образом, для ус-
успешного дизайна белка требуются не только стабилизация его
пространственной структуры в функциональном состоянии, но и
дестабилизация всех возможных альтернативных элементов
вторичной структуры, которые иначе будут служить кинетичес-
кинетическими и энергетическими ловушками на пути реализации пра-
правильного фолдинга. Действительно, уже в ранних эксперимен-
экспериментах по белковому дизайну de novo было установлено, что для ус-
успешного завершения работы необходимо предвидеть и с помо-
помощью негативного дизайна нейтрализовать как можно больше
альтернативных внутримолекулярных структур, обладающих
низкой свободной энергией.
Информация, требуемая для правильного фолдинга полипеп-
полипептидной цепи, распределена вдоль нее в последовательности ами-
аминокислотных остатков и реализуется в сети многочисленных вну-
внутримолекулярных полярных и гидрофобных взаимодействий,
каждое из которых вносит в данный процесс свой энергетичес-
энергетический вклад. При этом наиболее важное влияние оказывают ами-
аминокислотные остатки, расположенные внутри белковой глобу-
глобулы, и бывает трудно оценить роль отдельных аминокислотных
остатков в определении пространственной структуры белка. В
идеальном случае рациональный дизайн белков должен учиты-
учитывать совокупность всех этих взаимодействий.
280
1.1.2. Выбор пространственной структуры
При реализации структурного подхода в дизайне белков рабо-
работа начинается с построения трехмерной модели полипептидного
скелета макромолекулы. При дизайне de novo такие координаты
атомов должны быть получены путем математических расчетов.
Компьютерный дизайн совершенно новых белков начинают с оп-
определения координат скелета полипептидной цепи и построения
схемы силовых полей, взаимодействующих в макромолекуле.
Альтернативно скелет белка может быть построен с использова-
использованием клонотеки известных элементов вторичных структур. В том
случае, если целью исследования является редизайн природного
белка, при построении модели используют координаты атомов ис-
исходной макромолекулы, предварительно полученные с помощью
рентгеноструктурного анализа или ЯМР-спектроскопии.
1.1.3. Комбинаторная проблема обратного дизайна белков
После выбора конформации скелета создаваемого белка пе-
переходят к следующему этапу конструирования - так называемому
обратному дизайну: определению последовательности аминокис-
аминокислотных остатков, которая может свернуться в требуемую прост-
пространственную структуру. Полный набор случайных последова-
последовательностей, который можно получить для 100-звенного полипеп-
полипептида из 20 разных аминокислотных остатков, представлен 10130
последовательностями. Учитывая большое влияние на фолдинг
гидрофобных и полярных аминокислотных остатков, прежде все-
всего занимаются их распределением вдоль полипептидной цепи.
Введение только пяти таких остатков позволяет ограничить про-
пространство последовательностей в 1060 раз. Но даже в этом случае
число оставшихся вариантов является слишком большим A070).
Имеется несколько путей снижения остроты этой проблемы.
Во-первых, известно, что большое число последовательностей
может приобретать одну и ту же пространственную структуру,
что дает возможность рассматривать не все возможные последо-
последовательности, а лишь одну из многих. Во-вторых, при конструиро-
конструировании последовательности можно использовать не все 20 амино-
аминокислот, а значительно меньшее количество. Водородные связи и
электростатические взаимодействия между боковыми цепями
аминокислотных остатков, погруженных в белковую глобулу, иг-
играют большую роль в формировании пространственной структу-
структуры белка. Поэтому расчет возможных взаимодействий внутри
белковой глобулы с использованием специальных алгоритмов по-
помогает осуществить выбор оптимальной последовательности из
281
многочисленных возможных, а одной из основных задач является
разработка эффективных алгоритмов, позволяющих находить
оптимальные решения данной проблемы.
1.1.4. Дизайн поверхности белковой глобулы
Менее четверти боковых цепей аминокислотных остатков на-
находится внутри белковой глобулы вне контакта с растворителем.
Остальные 75% остатков помогают стабилизировать основную
пространственную структуру белка и дестабилизировать возмож-
возможные альтернативные состояния. В настоящее время создано про-
программное обеспечение, позволяющее автоматизировать процесс
разработки участков полипептидных цепей, которые взаимодей-
взаимодействуют с растворителем. В результате его использования удается
предсказывать стабильность ос-спиральных участков белка, кон-
контактирующих с водной фазой, и характер взаимодействия сосед-
соседних групп в этих последовательностях.
7.7.5. Примеры использования методов рационального дизайна
С помощью дизайна de novo пока удается конструировать
элементы вторичной структуры белков или их отдельные доме-
домены [9]. Гораздо более успешными оказываются попытки осуще-
осуществлять рациональный редизайн известных белков, используя в
качестве исходных молекул белки и ферменты, функциональ-
функциональность которых уже была апробирована самой природой в живых
организмах. Одним из подходов в этом случае является создание
новых активных центров в белках, пространственная структура
которых хорошо изучена.
Создание нового активного центра в молекуле тиоредоксина.
Недавно при использовании в качестве исходной молекулы
инертного белка тиоредоксина после проведения предваритель-
предварительного компьютерного дизайна удалось внедрить в его полипептид-
полипептидную цепь новый активный центр, способный осуществлять гид-
гидролиз активированных эфиров [10]. В этом случае для решения
проблемы были использованы знания, накопленные при исследо-
исследовании механизмов функционирования каталитических антител
(абзимов). Была создана серия виртуальных модифицированных
молекул тиоредоксина с минимальной общей свободной энерги-
энергией. Требуемое высокоэнергетическое состояние конкретных мо-
молекулярных участников реакции моделировали с помощью боко-
боковых цепей аминокислотных остатков, ориентированных в прост-
пространстве с целью создания условий, необходимых для осуществле-
осуществления катализа (реакция а). На рисунке справа (б) изображена схе-
282
a Q
+ His
о в о
Г Я А
> R!-0 0-R2 Ri-0 I O-Ri
Rl-0^4R2 I °"
His О
1 "
o-
ма той же самой реакции, включающая гипотетическое проме-
промежуточное соединение в переходном состоянии, аналог которого
(в) был использован для иммунизации животных при получении
абзимов (об абзимах подробнее см. ниже раздел 3.4.3).
Был получен набор (клонотека) вариантов, по-разному ориен-
ориентированных в пространстве боковых цепей (ротамеров), находя-
находящихся в высокоэнергетическом состоянии, которые размещали в
различных участках полипептидной цепи тиоредоксина таким об-
образом, чтобы они сохраняли способность взаимодействовать с суб-
субстратом. (Ротамерами называют статистически значимые конфор-
мации боковых цепей аминокислотных остатков в составе поли-
полипептидной цепи). Путем теоретической оценки способности пред-
предполагаемых активных центров, сформированных в разных участ-
участках полипептидной цепи, взаимодействовать с субстратом удалось
предсказать несколько "протозимов", потенциально способных
осуществлять соответствующую реакцию. Экспериментальное ис-
исследование показало, что, действительно, такие белки гидролизо-
вали эфир по механизму нуклеофильного катализа с участием ос-
остатка His. Хотя удельная активность подобных искусственных фер-
ферментов была умеренной (на уровне абзимов первого поколения),
сам подход представляется весьма перспективным, так как его реа-
реализация может способствовать созданию искусственных фермен-
ферментов, катализирующих реакции, которые не происходят в природе.
Конструирование новых металлоферментов. В настоящее
время достаточно хорошо изучены механизмы реакций, осуще-
осуществляемых ферментами, которые содержат в своем активном
Центре ионы металлов (металлоферментами). Перед началом
работ по конструированию искусственных металлоферментов
253
уже имелся метод компьютерного предсказания участков поли-
полипептидных цепей, способных образовывать комплексы с ионами
металлов [11]. На основе этого подхода опять-таки в молекуле
тиоредоксина были смоделированы центры связывания ионов
металлов в комплексе с молекулой кислорода [12]. Многие из та-
таких искусственно созданных белков обладали способностью свя-
связывать ионы металлов и осуществлять реакции с участием моле-
молекулярного кислорода.
1.1.6. Ландшафт функциональности среди пространства
последовательностей белковых молекул
Переходя к обсуждению результатов по созданию белков и
ферментов с новыми функциями, которые удается получать ме-
методами рационального дизайна в белковой инженерии, необходи-
необходимо сформулировать два важных понятия, которые часто исполь-
используются в молекулярной генетике. Совокупность всех возможных
последовательностей аминокислотных остатков полипептидной
цепи (или полинуклеотида) определенной длины составляет ее
полное пространство последовательностей (sequence space). На-
Например, полное пространство последовательностей 100-звенной
полипептидной цепи, построенной из 20 природных аминокислот-
аминокислотных остатков, будет представлено 20100 (~10130) последовательнос-
последовательностями. Хотя полное пространство последовательностей является п-
мерным и его невозможно изобразить графически, для удобства
обсуждения его часто изображают в виде двумерной плоскости,
где каждая точка плоскости соответствует индивидуальной после-
последовательности этого пространства (рис. 31).
По оси z на рисунке отражен уровень функциональности кон-
конкретных молекул пространства последовательностей, выражен-
выраженный в произвольных единицах, который в случае ферментов коли-
количественно характеризует их способность осуществлять химичес-
химическую реакцию (например, число оборотов kmT или коэффициент
специфичности ктт/Км данного фермента). Введение третьего из-
измерения позволяет получать т.н. ландшафты функциональности
(fitness landscapes), на которых холмы, в том числе и их склоны,
графически представляют функциональности отдельных макро-
макромолекул, причем отдельные холмы соответствуют разным функ-
функциям белков анализируемого пространства последовательностей.
Вершины холмов занимают макромолекулы с максимальной
функциональностью (в случае ферментов - удельной активнос-
активностью, специфичностью, стабильностью и тому подобными свойст-
свойствами, важными для их функционирования в определенных услови-
284
p.
3
20
40
20
0
Индивидуальные последовательности
Рис. 31. Ландшафт функциональности среди пространства последова-
последовательностей белковых молекул [13]
Каждая точка на плоскости отражает индивидуальную последовательность
полипептидной цепи заданной длины, а все вместе - всевозможные последова-
последовательности, собранные из данного количества аминокислотных остатков, кото-
которые составляют полное пространство последовательностей для полипептида
определенного размера; функциональность - количественно выраженная (в
произвольных единицах) способность конкретного белка из данного простран-
пространства последовательностей выполнять конкретную функцию, например, с неко-
некоторым сродством связывать лиганд
ях). Природные последовательности, обнаруженные в живых ор-
организмах, составляют отдельную немногочисленную группу обще-
общего пространства, тогда как его оставшаяся часть относится к дру-
другим теоретически возможным последовательностям. Белковая ин-
инженерия позволяет создавать и исследовать последовательности
белков, не встречающиеся в природе и обладающие совершенно
новыми биохимическими свойствами, что неуклонно приводит к
изменению ландшафта, характерного для конкретного простран-
пространства последовательностей. При этом необходимо иметь в виду, что
такие статические ландшафты лишь частично отражают свойства
пространства последовательностей, поскольку и само пространст-
пространство, и уровни функциональности составляющих его членов могут
Непрерывно изменяться, особенно в ходе направленной эволюции
285
белков, не говоря уже о сложных, эволюционирующих биологиче-
биологических системах [14].
Создание методами рационального редизайна искусственных
белков, обладающих совершенно новыми функциями, на основе по-
полипептидного скелета биологически инертных макромолекул, как
правило, требует введения нескольких мутантных аминокислотных
остатков в разные участки их полипептидных цепей, так как оди-
одиночные мутации не приводят к желаемому результату. Говоря о воз-
возможностях метода в терминах ландшафтов функциональности,
можно предположить, что введение множественных мутаций позво-
позволяет пересекать биологически инертные пространства последова-
последовательностей и попадать в зоны новых холмов, на их склоны. Это от-
открывает путь для дальнейшего совершенствования функционально-
функциональности нового белка, полученного методами белковой инженерии, и
может приводить к изменению вида самого ландшафта.
Для реализации замысла по созданию новой белковой моле-
молекулы имеется несколько экспериментальных путей, в том числе
и химический синтез полипептидных цепей. Однако наиболее по-
популярным подходом к получению совершенно новых, а также из-
измененных природных белков, является их биосинтез in vivo или in
vitro. В этом случае создают ген исследуемого белка, клонируют
его в экспрессирующем векторе и далее осуществляют транс-
транскрипцию гена с последующей трансляцией образовавшихся
мРНК. Замены аминокислотных остатков в исследуемом белке
проводят целенаправленно, создавая мутации в определенных
участках рекомбинантного гена, т.е. осуществляя процесс на-
направленного мутагенеза.
1.2. Методы направленного мутагенеза
Развитие генной инженерии облегчило процесс получения
мутаций в конкретных участках генома и анализ последствий
этих мутаций на молекулярном уровне. Совокупность методов
получения конкретных мутаций на основе генно-инженерных
подходов называют направленным, или сайт-специфическим му-
мутагенезом. Прообразом направленного мутагенеза является про-
простая инкубация клонированного гена с химическими мутагенами
in vitro, которая приводит к четкой локализации возникающих
мутаций в пределах этого гена. В настоящее время разработано
много эффективных методов сайт-специфического мутагенеза,
позволяющих сознательно производить мутационные замены
конкретных нуклеотидов. Получение заранее определенных му*
таций в сегментах рекомбинантных генов, введение мутантных
286
генов в организм и исследование влияния полученных мутаций на
функционирование гена - этот подход прямо противоположен
классическим методам получения мутаций путем отбора спон-
спонтанно возникающих мутантов, а также обработкой целых орга-
организмов или их репродуктивных органов мутагенами. Сумма по-
подобных подходов к исследованию функций генов получила назва-
название обратной генетики.
1.2.1. Получение делеций и вставок
Делецией называют потерю части нуклеотидов в геноме орга-
организма. Такой вид мутаций удобнее всего использовать для локали-
локализации (картирования) функционально значимых участков в генах
и кодируемых этими генами белках. Действительно, последова-
последовательное удаление все новых и новых участков ДНК на границах
генов с помощью делеций оказалось исключительно плодотвор-
плодотворным в обнаружении регуляторных элементов генов, исследовании
их структурно-функциональных особенностей, взаимного распо-
расположения и влияния друг на друга. В то же время укорочение поли-
полипептидных цепей белков путем введения делеций в их гены дает
возможность получать мини-белки и мини-ферменты для струк-
структурно-функциональных исследований и биотехнологии. Простой и
эффективный метод создания делеций любого размера разрабо-
разработан с использованием экзонуклеазы ВаВ\ [15]. Метод позволяет
удалять нуклеотиды в окрестностях сайтов рестрикции (рис. 32).
Ген, клонированный в плазмиде, расщепляют по уникально-
уникальному сайту рестрикции и образовавшиеся линейные молекулы
ДНК инкубируют в присутствии экзонуклеазы Bal3\. При этом
экзонуклеаза последовательно удаляет нуклеотиды с обоих кон-
концов ДНК, причем количество удаляемых нуклеотидов прямо про-
пропорционально времени инкубации ДНК с нуклеазой, а также за-
зависит от температуры инкубации и концентрации фермента. В
результате подобного действия образуется набор фрагментов
ДНК разной длины, содержащих делеций различных размеров по
обе стороны выбранного сайта рестрикции. К "тупым" концам
Таких молекул ДНК с помощью ДНК-лигазы присоединяют
Двухцепочечные олигонуклеотидные линкеры, содержащие уни-
уникальный (часто исходный) сайт рестрикции, обрабатывают соот-
соответствующей рестриктазой, замыкают молекулы в кольцо по-
посредством лигирования и затем вводят их в бактериальные клет-
клетки. Точное картирование концов делеций осуществляют секвени-
рованием соответствующих участков ДНК мутантных плазмид.
В результате получают набор делеций разного размера, положе-
257
EcoRl
ВатШ
Расщепление с помощью
_ _т ВатШ D UT
EcoRl ВатШ
Экзонуклеаза Ва131
C' - и 5' - экзонуклеазы)
V//////////A
V//////A
У///Л
Увеличение
времени
расщепления
BamHI-линкеры
ВатШ + ДНК-лигаза
замыкание в кольцо
EcoRl
Клоны
с делециями
ВатШ
ВатШ
ВатШ
ВатШ
Рис. 32. Получение делеций с помощью нуклеазы ВаВ 1
ние которых в исследуемом фрагменте ДНК строго локализова-
локализовано. Нуклеаза ВаВ\ является сложноустроенным ферментом и
имеет склонность к асинхронному расщеплению двухцепочечных
молекул ДНК, что сопровождается образованием делеций разно-
разного размера. В отличие от этого процессивно функционирующая
экзонуклеаза III дает возможность получать более гомогенный
набор делеций.
Экзонуклеаза III позволяет не только создавать двунаправ-
двунаправленные делеций, но и однонаправленно удалять последователь-
последовательности ДНК по соседству с выбранным сайтом рестрикции. В по-
последнем случае участок ДНК, в котором необходимо получить
делецию, расщепляют двумя рестриктазами, одна из которых со-
создает "тупой" или 5'-выступающий конец вблизи делетируемого
сегмента ДНК, а другая - четырехнуклеотидный 3'-выступаю-
3'-выступающий участок. Однонаправленность действия экзонуклеазы III
обеспечивается ее специфичностью, поскольку она может начи-
начинать гидролиз ДНК только с "тупого" или 3'-утопленного конца.
Возникающие в итоге одноцепочечные участки далее удаляются
нуклеазами 51 (или mung bean), а в случае необходимости к обра-
образовавшимся "тупым" концам присоединяют линкеры для даль-
дальнейшего клонирования модифицированного фрагмента.
Небольшие делеций в окрестностях сайтов рестрикции можно
получать быстрее удалением "липких" концов линеаризованной
плазмидной ДНК после рестрикции с последующим замыканием
линейной ДНК в кольцо лигированием по образовавшимся "ту-
"тупым" концам. В этом случае размер делеций соответствует разме-
размеру одноцепочечных "липких" концов в сайтах рестрикции. Кроме
того, такой метод допускает простую проверку наличия мутаций
в требуемом участке ДНК, так как в результате мутации происхо-
происходит потеря уникального сайта рестрикции. Для получения вставок
коротких или протяженных последовательностей нуклеотидов в
исследуемые участки клонируемых генов также разработаны эф-
эффективные и надежные методы. В простейшем случае такая зада-
задача решается путем расщепления исследуемого гена рестриктазой
по уникальному сайту рестрикции и встраивания по этому сайту
фрагмента природной ДНК или синтетического двухцепочечного
олигонуклеотида, который фланкирован соответствующими
"липкими" концами. Лигирование может проводиться и по "ту-
"тупым" концам. В таком случае последовательности нуклеотидов
на концах вставки уже не имеют существенного значения для
встраивания этого фрагмента по сайту рестрикции.
Для создания множественных вставок коротких или протя-
протяженных последовательностей нуклеотидов в исследуемых участ-
Ю. Патрушев Л.И. Т. 1 289
ках ДНК в основном используют два подхода. В первом случае с
помощью панкреатической ДНКазы в низких концентрациях в
присутствии ионов Мп2+ вносят случайным образом один двухце-
почечный разрыв в каждую векторную плазмиду, содержащую
клонированный ген. К концам образовавшихся линейных моле-
молекул ДНК присоединяют с помощью ДНК-лигазы синтетические
олигонуклеотидные линкеры, содержащие сайт рестрикции, ко-
который отсутствует в исследуемой плазмиде. Образовавшиеся ли-
линейные молекулы ДНК с линкерами обрабатывают рестрикта-
зой, узнающей сайт рестрикции линкера, что приводит к образо-
образованию липких концов, и замыкают в кольцо с помощью ДНК-ли-
ДНК-лигазы. В итоге кольцевые молекулы ДНК содержат исследуемый
клонированный ген, в котором имеется по одной вставке локали-
локализованных случайным образом (в соответствии с расположением
исходных двухцепочечных разрывов) олигонуклеотидных линке-
линкеров. Во втором случае статистические разрывы в двухцепочеч-
ной ДНК получают путем частичного (неполного) гидролиза
мелкощепящими рестриктазами, которые узнают сайт рестрик-
рестрикции длиной в четыре нуклеотида. Метод получения вставок с ис-
использованием синтетических олигонуклеотидных линкеров по-
получил название сканирования линкером.
1.2.2. Мутагенез с использованием олигонуклеотидов
Благодаря своей высокой эффективности и простоте направ-
направленное введение мутаций с использованием олигонуклеотидов по
модифицированной методике Т.А. Кункеля до сих пор находит
широкое применение [16, 17]. В этом подходе для направленного
введения мутаций используют 20-25-звенный олигонуклеотид,
комплементарный мутагенизируемому участку гена, который со-
содержит все требуемые замены нуклеотидов. Мутагенизируемыи
ген или его участок клонируют в фагмидном векторе, в котором
далее стандартными методами заменяют ряд остатков тимина на
остатки уридина и переводят в одноцепочечную форму. Олиго-
Олигонуклеотид гибридизуют с такой кольцевой одноцепочечной ДНК
и используют в качестве праймера для модифицированной
Т7-ДНК-полимеразы (секвеназы), с помощью которой в присут-
присутствии обычных четырех дезоксирибонуклеозидтрифосфатов
синтезируют комплементарную цепь ДНК (рис. 33).
Остающийся одноцепочечный разрыв лигируют и образовав-
образовавшиеся двухцепочечные кольцевые ковалентно замкнутые моле-
молекулы ДНК инкубируют с урацил-ДНК-гликозилазой. При этом
удаляются остатки урацила из матричной цепи ДНК с образова-
образовало
оцДНК-матрица, содержащая урацил
U
U
1. Отжиг мутантного I Мутантный праймер
праймера
2. Синтез ДНК и
лигирование
U
U
U
5. Репликация
4. Трансформация
Е. coli ung*
3. Инкубация с UDG
in vitro
Рис. 33. Схема направленного мутагенеза по методу Т.А. Кункеля [17].
Праймер, содержащий мутантный нуклеотид (помечен крестиком), отжи-
отжигают с одноцепочечной ДНК (/), в которой большая часть остатков Т была
предварительно заменена на U; после синтеза второй цепи ДНК-полимеразой и
лигирования одноцепочечного разрыва B) остатки U в дочерней цепи удаляют
с помощью урацил-ДНК-гликозилазы (UDG) C) и модифицированную ДНК
вводят в клетки Е. coli, содержащие нативный ген UDG - ung+ D), в которых
происходит деградация дочерней цепи и построение новой цепи E), содержащей
мутантный нуклеотид в требуемом положении. оцДНК - одноцепочечная ДНК
нием апиримидиновых сайтов, в которых спонтанно в условиях
щелочных значений рН возникают одноцепочечные разрывы
ДНК. Это сопровождается эффективной элиминацией исходной
цепи ДНК, не содержащей мутаций, а следовательно, предотвра-
предотвращает возможность репарации введенных мутаций и восстановле-
восстановления исходной первичной структуры мутагенизируемого участка.
Такими синтезированными in vitro молекулами ДНК далее транс-
трансформируют компетентные клетки Е. coli, содержащие функцио-
функциональный ген урацил-ДНК-гликозилазы (ung), что способствует
дальнейшему удалению остатков урацила в родительской цепи
ДНК и повышению эффективности мутагенеза. На этом этапе
также происходят достройка второй цепи плазмидной ДНК и
фиксация мутаций, вводимых с помощью мутантного олигонук-
леотида. К достоинствам метода следует отнести его простоту:
все основные реакции по подготовке ДНК к трансформации про-
проводят в одной пробирке, использование секвеназы позволяет бы-
быстро и эффективно достраивать цепь ДНК. Эффективность ме-
ю*
291
тода достигает 80%, т.е. только один из пяти полученных клонов
не содержит требуемых мутаций.
Имеются варианты метода, которые позволяют избегать ис-
использования ДНК с модифицированными остатками урацила и
всех последующих манипуляций, связанных с отбором мутант-
ных плазмид [18, 19]. В этом случае после денатурации двухцепо-
чечной плазмидной ДНК щелочью мутагенизируемую цепь гиб-
ридизуют одновременно с двумя олигонуклеотидами, из которых
первый содержит требуемые мутации, а второй вносит мутации в
ген C-лактамазы, используемой в качестве селектируемого мар-
маркера. Получаемая в результате мутантная C-лактамаза, обеспе-
обеспечивает устойчивость трансформированных клеток к смеси анти-
антибиотиков: ампициллина, цефотаксима и цефтриаксона, чего не
происходит при наличии в клетках исходного вектора. Таким об-
образом, приобретение клетками устойчивости к смеси антибиоти-
антибиотиков с высокой вероятностью свидетельствует об одновременном
присутствии в векторе с клонированным мутагенизируемым ге-
геном требуемых мутаций.
Еще одним широко распространенным способом отбора мо-
молекул ДНК - потомков мутантной цепи плазмидной ДНК являет-
является расщепление двухцепочечных молекул плазмидной ДНК, по-
полученных на матрице родительской немутантной цепи, по уни-
уникальному сайту рестрикции, который элиминируется в мутант-
мутантной цепи вторым олигонуклеотидом [20]. Этапы реализации это-
этого подхода в общих чертах совпадают с вышеописанными для
C-лактамазы.
Эффективным способом освобождения от исходных нему-
тантных молекул ДНК может быть использование рестриктаз,
действие которых зависит от метилирования сайтов рестрикции
[21, 22]. При таком подходе работу по введению мутаций с помо-
помощью олигонуклеотидов начинают на плазмидной ДНК, сайты ре-
рестрикции на которой для этих рестриктаз, в частности Dpnl, пол-
полностью метилированы Dam-метилтрансферазой in vitro. После
Рис. 34. Схема направленного мутагенеза с использованием ПЦР и рес-
триктазы Dpnl [22]
Мутагенизируемую ДНК полностью метилируют Dam-метилазой и ис-
используют в качестве матрицы в ПЦР с праймерами, содержащими требуемую
мутацию (помечена); по завершению ПЦР, в которой образуется неметилиро-
ванная ДНК, родительскую ДНК убирают с помощью рестриктазы (/), а про-
продукт ПЦР очищают электрофорезом B); после фосфорилирования концов
ДНК в одноцепочечных разрывах и их лигирования, метилирования C) и осво-
освобождения от неполностью метилированных молекул с помощью рестриктазы
D) проводят ПЦР с новыми мутагенизирующими праймерами
292
Me
Me
1. Инкубация с DprA
2. Очистка в геле
3. Лигирование
Фосфорилирование
Dam-метилирование
4. Очистка ДНК
Me
Me
Следующий цикл ПЦР-мутагенеза
Me
М
Me
достройки дочерних цепей ДНК-полимеразой с использованием
мутагенизирующих олигонуклеотидов в качестве праймеров, ро-
родительские полностью метилированные молекулы избирательно
разрушают рестриктазой Dpnl в условиях высокой ионной силы,
благодаря которой метилированные наполовину дочерние ДНК
остаются интактными. Дальнейшее повышение эффективности
этого метода достигается сочетанием метилирования исходных
ДНК и введения урацила вместо тимина или использованием вто-
второго олигонуклеотида, изменяющего селектируемый сайт рест-
рестрикции, как было описано выше.
В сочетании с ПЦР метод элиминации метилированных моле-
молекул ДНК рестриктазой Dpnl позволяет направленно вводить мно-
множественные мутации, избегая необходимости трансформации
бактериальных клеток и выделения плазмидной ДНК на проме-
промежуточных стадиях. При таком подходе исходную мутагенизируе-
мую плазмидную ДНК C-5 т.п.о.) полностью метилируют Dam-
метилтрансферазой in vitro и используют в качестве матрицы при
ПЦР с двумя перекрывающимися мутантными праймерами
(рис. 34). После завершения ПЦР, в результате которой амплифи-
цируется вся молекула исходной плазмидной ДНК, продукт ПЦР
полностью неметилирован. Это позволяет удалить исходную (не-
мутантную) метилированную матричную ДНК с помощью рест-
риктазы Dpnl, и после ликвидации одноцепочечных разрывов с
помощью ДНК-лигазы полученный продукт ПЦР, содержащий
первую мутацию, после электрофоретической очистки использу-
используют в качестве матрицы в новом раунде ПЦР со второй парой му-
тантных праймеров для введения другой мутации по той же схеме.
Общее количество таких циклов мутагенеза in vitro определяется
количеством мутаций, которые необходимо ввести в исходную
молекулу плазмидной ДНК. На заключительном этапе сконстру-
сконструированную таким образом мутантную ДНК вводят с помощью
трансформации в компетентные бактериальные клетки и мутант-
мутантную плазмидную ДНК получают в препаративном количестве.
Эти принципы легли в основу коммерчески доступной и одной из
наиболее широко используемых в настоящее время систем на-
направленного мутагенеза "QuikChange™ Site-Directed Mutagenesis
System (QCM)", разработанной фирмой Stratagene (США).
1.2.3. ПЦР с перекрывающимися праймерами
Разработка метода полимеразной цепной реакции принципи-
принципиально изменила ситуацию в исследованиях по направленному му-
мутагенезу. Использование ПЦР для направленного мутагенеза ос-
294
новано на применении в качестве праймеров олигонуклеотидов,
не полностью комплементарных матричной ДНК. Повышенные
требования к комплементарности накладываются лишь на по-
последний, 3'-концевой, нуклеотид праймера, но, даже в случае
3'-концевой некомплементарности, праймеры часто продолжа-
продолжают функционировать в системе ПЦР, хотя и с разной эффектив-
эффективностью. В то же время некоторая способность 17-нуклеотидного
праймера инициировать ПЦР проявляется даже при наличии все-
всего восьми нуклеотидов, комплементарных матрице, три из кото-
которых расположены на его 3'-конце. Таким образом, простейшим
способом введения сайт-специфических мутаций в амплифициру-
емый фрагмент ДНК является использование праймеров, частич-
частично комплементарных матрице, т.е. содержащих необходимые му-
тантные нуклеотиды. Этот способ применяется для создания в
амплифицируемом продукте новых сайтов рестрикции с целью
его последующего клонирования. Такой подход удобен для
встраивания амплифицируемого продукта в экспрессирующий
вектор в одной открытой рамке считывания (ОРС) с инициирую-
инициирующим ATG-кодоном вектора. Действительно, как уже обсужда-
обсуждалось в первой части книги, экспрессирующие векторы часто со-
содержат 5'-концевую часть будущего рекомбинантного гена,
включая регуляторные последовательности, промотор, ATG-ko-
дон и иногда дополнительные кодоны нескольких последующих
аминокислот. При конструировании таких векторов в последова-
последовательности, следующие за ATG-кодоном, вводят сайты рестрик-
рестрикции, по которым и встраивают фрагмент экспрессируемого гена,
кодоны которого (для полноценной экспрессии гена) должны на-
находиться в одной ОРС с ATG-кодоном вектора. Положение при-
природных сайтов рестрикции в клонируемом гене редко отвечает
требованиям ОРС вектора. Поэтому введение искусственных
сайтов рестрикции в клонируемый фрагмент с помощью прайме-
праймеров, не полностью комплементарных матрице, является хорошим
решением проблемы.
Вырожденные праймеры, которые представляют собой
сложную смесь олигонуклеотидов, содержащих многие точко-
вые мутации, также используют для сканирования определен-
определенных участков ДНК [23]. В таком классическом варианте поста-
постановки ПЦР, кроме точковых мутаций, можно легко получать
делеции и вставки. Дальнейшим усовершенствованием метода
направленного мутагенеза с помощью ПЦР явилась разработка
подхода к созданию гибридных генов с помощью перекрываю-
перекрывающихся праймеров [24] (рис. 35). Этот метод позволяет не только
целенаправленно получать точковые мутации, делеции и встав-
295
ки, но и гибридные молекулы ДНК без применения ДНК-лига-
зы. Для получения точковых мутаций используют пару прайме-
ров, в которых изменены соответствующий нуклеотид или не-
небольшая группа нуклеотидов (мутантный сайт обозначен на
рис. 35,7 прямоугольниками внутри праймеров). При этом внут-
внутренние праймеры b и с комплементарны друг другу. В первых
ПЦР в отдельных пробирках амплифицируют правую и левую
части мутагенизируемого сегмента ДНК с использованием пар
праймеров а + b и с + d, что приводит к образованию двух фраг-
фрагментов ДНК, перекрывающихся на участках, которые содер-
содержат включенные праймеры (этапы 1 и 2). Фрагменты очищают
от праймеров, смешивают друг с другом в эквимолярном соот-
соотношении и после цикла тепловой денатурации и ренатурации
используют в качестве матрицы в другой ПЦР с внешними
праймерами and (этап 3). На этом этапе в первом цикле ПЦР
происходит достройка цепей перекрывающихся фрагментов
ДНК (пунктирные линии на рис. 35,7), и образовавшийся фраг-
фрагмент двухцепочечной ДНК, содержащий требуемую мутацию,
далее служит матрицей для амплификации и по завершении
ПЦР присутствует в реакционной смеси в препаративном коли-
количестве.
Тот же принцип объединения двух фрагментов ДНК, пере-
перекрывающихся за счет праймеров, используется при получении
делеций и вставок. Для создания делеции (на рис. 35,2 обозначе-
обозначена прямоугольниками на цепях ДНК) берут внутренние прайме-
праймеры b и с, 5'-концы которых комплементарны матрице по одну
сторону делетируемой последовательности нуклеотидов, а
3'-концы - последовательности нуклеотидов, фланкирующей
другой ее конец. Образующиеся при этом продукты ПЦР АВ и
CD перекрываются в точке делеции и далее используются как и
в предыдущем случае. При получении вставок (см. рис. 35,3) при-
применяют внутренние праймеры b и с, комплементарные друг дру-
другу своими 5'-концевыми частями, которые соответствуют обра-
образуемой олигонуклеотидной вставке во фрагмент матричной
ДНК. 3'-Концы этих праймеров комплементарны участкам
Рис. 35. Использование ПЦР для получения мутаций
Получение точковых мутаций (/), делеций B) и вставок C): and- внеш-
внешние праймеры, b и с - внутренние праймеры. АВ и CD - фрагменты ДНК, об-
образовавшиеся в результате ПЦР с использованием праймеров а и Ь, а также с
и d, соответственно. После гибридизации друг с другом объединяемых фраг-
фрагментов ДНК сформировавшимися комплементарными последовательностями
нуклеотидов одноцепочечные участки достраиваются ДНК-полимеразой в
процессе ПЦР
296
Точковые мутации
а с
Делеции
I
b N-
АВ
Вставки
а
I
CD
АВ
CD
ДНК-матрицы, непосредственно примыкающим к сайту, в кото-
который производится вставка олигонуклеотида.
С помощью метода ПЦР удается легко получать и множест-
множественные случайные мутации в конкретных участках ДНК. В этом
случае в реакционной смеси искусственно создают условия, не-
неоптимальные для точности функционирования термостабильной
ДНК-полимеразы (подробнее см. раздел 2.2.2.).
1.2.4. Мегапраймеры в направленном мутагенезе
Недавно разработанные системы направленного мутагенеза
с использованием ПЦР и мегапраймеров позволяют существенно
облегчить процесс получения мутаций, что достигается отказом
от использования перекрывающихся мутантных праймеров для
синтеза мутантных фрагментов ДНК [25, 26]. Принцип этого ме-
метода заключается в следующем (рис. 36,я). В первом раунде ПЦР
синтезируют мутантный фрагмент ДНК, в который вводят мута-
мутации с помощью одного из праймеров (праймер М). Поскольку об-
образующийся продукт ПЦР обладает значительными размерами
(его допустимые размеры 70-800 п.о.) и используется в качестве
праимера в следующем раунде ПЦР, он получил название мега-
праймера. Фрагмент очищают электрофорезом в агарозном геле
и используют в качестве праимера во втором раунде репликации
со встречным праймером А. Праймеры А и В могут быть универ-
универсальными и содержат на своих 5'-концах сайты рестрикции для
дальнейшего клонирования фрагмента. Конечный продукт ПЦР,
образовавшийся после второго раунда амплификации, длина ко-
которого может быть в пределах 400-2500 п.о., после очистки и ин-
инкубации с соответствующими рестриктазами клонируют в подго-
подготовленном векторе, заменяя исходный фрагмент ДНК на мутант-
мутантный, полученный с использованием мегапраймера.
Имеется модифицированная методика, при которой во время
ПЦР с мегапраймером не используется встречный праймер А, а в
качестве праймеров функционируют обе цепи двухцепочечного
ПЦР-продукта-мегапраймера (рис. 36,6) [27]. Метод позволяет по-
получать как точковые мутации, так и делеции, вставки или замеще-
замещения длиной до 25 п.о. В рассмотренном варианте метод с мегапрай-
мерами предусматривает очистку промежуточных продуктов амп-
амплификации и последующие манипуляции с ними. Однако имеются
его современные модификации, включающие ряд уже обсуждав-
обсуждавшихся выше подходов, в частности, использование метилирования
для освобождения от родительской ДНК, которые в комбинации
позволяют получать мутантный ген в одной пробирке [27].
298
б
Матрица
\
\
1. ПЦР с праймерами В и М
2. Очищенный продукт
Мегапраймер
ПЦР с праймером А и
мегапраймером
Мегапраймер
\
Рис. 36. Направленный мутагенез с использованием мегапраймеров
[26 (а) и 27 (б)]
а - многоступенчатый вариант метода. Первый раунд ПЦР проводят на плаз-
миде с мутагенизирующим праймером М и встречным - В; после очистки продукт
ПЦР, содержащий требуемые мутации, в свою очередь, используют в качестве
праймера (мегапраймера) со встречным праймером А; итоговый продукт ПЦР
содержит на своих концах уникальные сайты рестрикции, введенные с помощью
праймеров А и В, что позволяет далее клонировать всю конструкцию;
б - мутагенез в одной пробирке. В первой фазе реакции (А-В) в стандарт-
стандартных условиях на матрице плазмидной ДНК, полностью метилированной по сай-
сайту Dpnl, синтезируют короткий продукт ПЦР с использованием двух мутагени-
зирующих праймеров A и 2) или пары, включающей мутагенизирующий B) и
встречный универсальный C) праймеры; во второй фазе мутагенеза температу-
температуру отжига в циклах повышают, что исключает участие в ПЦР коротких прай-
праймеров A-3), а их роль берут на себя обе цепи мегапраймера (С-Е); образовав-
образовавшийся продукт ПЦР инкубируют с рестриктазой Dpnl с целью удаления роди-
родительской ДНК и реакционной смесью без очистки трансформируют клетки
В. coli; для замещения участка гомологичной последовательности в исследуе-
Mqm родственном гене на последовательность мегапраймера последний после
синтеза (этапы А-В) очищают и используют в ПЦР с плазмидным вектором,
содержащим соответствующий рекомбинантный ген (этапы G-I); точки обо-
обозначают мутации; выделены модифицируемые участки плазмид
299
Получение нескольких мутаций в последовательных раун-
раундах ПЦР. При необходимости получения нескольких (до шес-
шести) сайт-специфических мутаций, удаленных друг от друга на-
настолько, что их невозможно включить в один или два мутагени-
зирующих праймера, можно использовать множественные ра-
раунды ПЦР в соответствии с недавно разработанной методикой
[28, 29] (рис. 37). При таком подходе применяют один универ-
универсальный праймер и несколько мутагенизирующих, последова-
последовательности которых содержат требуемые мутации. В первом ра-
раунде ПЦР при введении в продукт ПЦР первой мутации с помо-
помощью мутантного праймера 1 в качестве матрицы используют
ДНК, содержащую как мутагенизируемую последователь-
последовательность, так и последовательность, комплементарную универ-
универсальному праймеру. Образовавшийся в итоге продукт ПЦР от-
отделяют от матричной ДНК электрофорезом в полиакриламид-
ном геле и, в свою очередь, применяют в качестве матрицы во
втором раунде вместе с матричной ДНК, содержащей мутаге-
мутагенизируемую последовательность, но у которой отсутствует по-
последовательность, комплементарная универсальному прайме-
праймеру. Поскольку обе эти матрицы перекрываются, то в первом
цикле второго раунда амплификации, происходящем в присут-
присутствии универсального праймера и встречного праймера, содер-
содержащего вторую мутацию, происходит достройка одной из пере-
перекрывающихся цепей с участием первого продукта ПЦР в каче-
качестве праймера с образованием двухцепочечной ДНК, содержа-
содержащей в достроенной цепи первую мутацию. Далее образовав-
образовавшийся гетеродуплекс используется в качестве матрицы в по-
последующих циклах. В итоге продукт ПЦР после завершения
второго раунда уже содержит две требуемых мутации. При по-
повторении аналогичных раундов с использованием новых мута-
мутагенизирующих праймеров и универсального праймера происхо-
происходит удлинение продукта ПЦР и введение в него новых требуе-
требуемых мутации. Конечный продукт ПЦР, содержащий множест-
множественные мутации, может быть клонирован в соответствующем
векторе стандартными методами, например, путем замещения
исходного участка гена дикого типа.
Рис. 37. Схема введения нескольких сайт-специфических мутаций в по-
последовательных раундах ПЦР [28]
S1 и S2 - боковые праймеры; Р1-Р6 - мутантные продукты ПЦР; ml-тб -
мутагенизирующие праймеры; узкие черные полосы обозначают мутации; ши-
широкая черная полоса слева - место посадки праймера S1, штриховка справа -
место посадки праймера S2
300
Матрица А (содержит сайт посадки праймера S1)
1
1.ПЦР
2. Очистка продукта Р1 в геле
(для удаления матрицы А)
Матрица Б (не содержит сайт посадки праймера S1)
т2
ПЦР
тЗ
S1
5 циклов ПЦР с неочищенным
продуктом Р2 и матрицей Б
с последующими 25 циклами
после добавления праймеров
SI hS2
ml
m2 m3 m4 m5
тб
5 циклов ПЦР с неочищенным
продуктом Р6 и матрицей Б
с последующими 25 циклами
после добавления праймеров
S1hS2
S2
301
1.2.5. Мутагенез с использованием нонсенс-сулрессоров
Генетический код, определенный в лабораториях Г. Кораны
и М. Ниренберга в начале 1960-х гг., является основой для осуще-
осуществления белковой инженерии. За немногими исключениями ге-
генетический код универсален для всех живых организмов, что поз-
позволяет планировать конкретные замены аминокислот в белках и
проектировать целые белковые молекулы путем создания струк-
структурных частей соответствующих генов в виде конкретных после-
последовательностей нуклеотидов. В то же время универсальность ге-
генетического кода оказывает отрицательное влияние на творчес-
творческий потенциал белковых инженеров, так как не позволяет ис-
использовать аналоги аминокислот при проектировании новых
белковых молекул и ограничивает их возможности 20-тью при-
природными аминокислотными остатками. Такая ситуация стимули-
стимулировала разработку методов, которые позволяют обходить выше-
вышеупомянутое затруднение. Группа методов, получивших название
белковой инженерии, основанной на mPHK (tRNA-mediated pro-
protein engineering (TRAMPE)), позволяет расширить возможности
использования генетического кода и обойти запреты, накладыва-
накладываемые системами трансляции на применение аналогов аминокис-
аминокислот при построении полипептидных цепей [30-32].
В ранних работах такого рода использовали химические мо-
модификации аминокислотных остатков непосредственно в составе
аминоацилированных тРНК. В частности TPHKLys, аминоацили-
рованную остатком Lys, после модификации агентом, осуществ-
осуществляющим поперечные сшивки после активации видимым светом,
удалось превратить в Ы-е-E-азидо-2-нитробензоил)-Ьу8-тРНКьУ5,
которая направляла включение этого аналога в белки в бескле-
бесклеточной системе трансляции. Синтезированные таким образом
модифицированные белки далее использовали для исследования
их участия в белок-белковых взаимодействиях путем образова-
образования поперечных сшивок с соседними белками [33].
При другом подходе, в одной из разновидностей направленно-
направленного мутагенеза, получившей название сайт-специфического заме-
замещения неприродными аминокислотами (site-directed non-native
amino acid replacement - SNAAR), использовали аминоацилиро-
ванные аналоги тРНК, специфически распознающие бессмыс-
бессмысленные кодоны UAG (amber), UAA (ochre) или UGA (opal) (т.е. су-
прессорные тРНК), для специфического включения соответству-
соответствующих аналогов аминокислот в участки полипептидных цепей,
определяемые нонсенс-кодонами. В этом случае стоп-кодоны
вводили в кодирующие участки анализируемых генов стандарт-
302
ными генно-инженерными методами, а соответствующим обра-
образом аминоацилированные супрессорные тРНК обеспечивали
считывание таких кодонов во время трансляции мРНК.
В настоящее время существуют, по крайней мере, три мето-
метода, которые позволяют осуществлять аминоацилирование тРНК
неприродными остатками аминокислот. Во-первых, возможно
проводить химическое аминоацилирование супрессорных тРНК
соответствующими аналогами аминокислот. В этом случае тре-
требуемым аналогом ацилируют олигонуклеотид pdCpA, который
затем лигируют с укороченной соответствующим образом тРНК.
Во-вторых, как уже упоминалось выше, существуют подходы,
позволяющие модифицировать аминокислотные остатки непо-
непосредственно в составе аминоацилированных тРНК [34, 35]. В-
третьих, ошибочное аминоацилирование аналогами можно осу-
осуществлять и с помощью самих нативных аминоацил-тРНК-синте-
таз, не оставляя им выбора в подходящей аминокислоте. К сожа-
сожалению, этот метод ограничен лишь небольшим числом аналогов
аминокислот, которые аминоацил-тРНК-синтетазы способны ис-
использовать в качестве субстратов. К таким аналогам, в частно-
частности, относятся фтортирозин [36] и тиопролин [37]. В то же время
химическое аминоацилирование тРНК является универсальным
средством решения данной проблемы.
Использование избыточных кодонов для осуществления
сайт-специфического замещения неприродными аминокислота-
аминокислотами. Основная трудность, с которой столкнулись исследователи
при использовании бессмысленных кодонов для включения ана-
аналогов аминокислот в белки, была преждевременная термина-
ция трансляции, происходящая из-за частого гидролиза пепти-
дилтРНК релизинг-факторами трансляции, распознающими
стоп-кодоны. Это сопровождалось освобождением недостроен-
недостроенного полипептида после включения аналога в строящуюся поли-
полипептидную цепь. Для решения такой проблемы были разработа-
разработаны системы трансляции из Micrococcus luteus. У этого микроорга-
микроорганизма имеются шесть кодонов, совершенно не задействованных в
синтезе белка, и именно их использовали для включения непри-
неприродных аминокислотных остатков в полипептидные цепи [38]. В
частности, избыточный кодон AGA, обычно кодирующий Arg,
был включен в последовательности генов, и такая ДНК была
применена в качестве матрицы в бесклеточной системе трансля-
трансляции, сопряженной с транскрипцией. При наличии соответствую-
соответствующей аминоацил-тРНК в реакционной смеси происходит включе-
включение аминокислотного остатка в полипептидную цепь, а в ее от-
отсутствие - терминация трансляции. Это явилось основой для со-
303
здания систем SNAAR, не зависимых от наличия в транслируе-
транслируемых мРНК стоп-кодонов и супрессорных тРНК.
При другом подходе повышение эффективности супрессии
бессмысленных кодонов достигается применением специфичес-
специфических природных супрессорных тРНК [39]. Дрожжевая тРНК^,
чаще всего используемая для супрессии амбер-кодонов, обеспе-
обеспечивает низкую эффективность супрессии (особенно путем вклю-
включения в полипептидные цепи полярных аминокислот), что приво-
приводит к очень низкому выходу соответствующего белкового про-
продукта. Более высокой эффективностью in vitro обладают супрес-
сорные тРНК?иРА и tPHK^a E. coli, причем последняя оказа-
оказалась наиболее эффективной, так как с одинаковым успехом на-
направляет включение в белки как полярных, так и неполярных
аминокислотных остатков.
Большинство разновидностей методов SNAAR реализуется с
использованием бесклеточных систем трансляции, сопряженных
с транскрипцией (см. раздел 6.3 в первой части книги). В оптими-
оптимизированных бесклеточных системах удается синтезировать на
матрице рекомбинантной ДНК до 0,4 мг белка, содержащего из-
измененные аминокислотные остатки, в 1 мл бесклеточного экс-
экстракта. Кроме того разработаны соответствующие системы
SNAAR, функционирующие in vivo, в частности в ооцитах X. lae-
vis. В этом случае модифицированные аминоацилированные
тРНК вводят в ооциты с помощью микроинъекций [40]. Хотя та-
такая система непригодна для крупномасштабной наработки моди-
модифицированных белков, в ней удается получать белки, прошедшие
посттрансляционные модификации, что невозможно осущест-
осуществить в аналогичных бесклеточных системах.
Альтернативой микроинъекциям при создании модифици-
модифицированных белков методом SNAAR in vivo является разработка
специфических пар тРНК : аминоацил-тРНК-синтетаза, в кото-
которых фермент проявляет высокую специфичность к конкретно-
конкретному аналогу аминокислоты и лишь его использует для аминоаци-
лирования тРНК [41]. Для получения соответствующих фер-
ферментов может быть использован весь арсенал методов белко-
белковой инженерии.
В некоторых особых случаях удается включать неприродные
аминокислотные остатки в белки in vivo, не прибегая к генно-ин-
генно-инженерным манипуляциям. В частности, можно встраивать
и-фторфенилаланин (p-F-Phe) в специфическое положение поли-
полипептидной цепи дигидрофолатредуктазы в клетках специального
штамма Е. coli, содержащего мутантную аминоацил-тРНК-синте-
304
тазу для тРНКРЬе, которая утратила способность взаимодейство-
взаимодействовать активным центром с p-F-Phe, токсичным для клеток. Для
направляемого амбер-кодоном включения p-F-Phe в соответст-
соответствующее место полипептидной цепи дигидрофолатредуктазы в
бактериальные клетки вводили генно-инженерную конструк-
конструкцию, обеспечивающую экспрессию дрожжевой супрессорной
тРНК?иеА и гомологичной аминоацил-тРНК-синтетазы, равно
как и саму дигидрофолатредуктазу, ген которой содержал требу-
требуемую амбер-мутацию. При культивировании таких клеток в сре-
среде, где содержание p-F-Phe было значительно больше, чем Phe,
дрожжевая синтетаза аминоацилировала гомологичную
тРНК^а » что обеспечивало эффективную супрессию амбер-ко-
дона. Выход модифицированного белка в такой системе состав-
составлял 8-12 мг/л бактериальной культуры [42]. Хотя такой метод
получения модифицированных белков не является универсаль-
универсальным, с его помощью можно вводить стабильную изотопную мет-
метку в виде 19F-Phe в белки для их дальнейшего исследования с по-
помощью ЯМР-спектроскопии.
Несмотря на большой потенциал, обсуждаемая группа мето-
методов TRAMPE из-за ряда недостатков пока еще не получила широ-
широкого распространения в современной белковой инженерии. Для
дальнейшего внедрения этих методов требуются усовершенство-
усовершенствования в дизайне тРНК, подвергающейся аминоацилированию,
разработка более эффективных химических методов аминоаци-
лирования тРНК неприродными аминокислотами, а также повы-
повышение производительности самих систем биосинтеза модифици-
модифицированных белков. Работы в данном направлении не прекращают-
прекращаются, а, следовательно, и прогресс в этой области исследований не-
неизбежен.
Химическая модификация мутантных белков, полученных
сайт-специфическим мутагенезом. Комбинированное использова-
использование методов направленного мутагенеза и химической модификации
боковых цепей мутантных белков, полученных первым методом,
дает возможность включать в полипептидные цепи неприродные
аминокислотные остатки, что не может быть реализовано каждым
из этих методов в отдельности. При таком подходе с помощью на-
направленного мутагенеза в исследуемый участок полипептидной це-
цепи вводят остаток природной аминокислоты, содержащий легко"
модифицируемую функциональную группу (например, остаток
Cys), которую далее изменяют химическими методами.
Остаток Cys в составе белковой молекулы можно тиоалкили-
ровать с помощью метантиосульфоната (CH3SO2S-R), который
305
специфически и количественно реагирует с сульфгидрильными
группами. Этот метод, особенно пригодный для белков, не содер-
содержащих природных SH-групп, был успешно применен для моди-
модификации субтилизина [43]. Кроме того, остатки Cys, образовав-
образовавшие дисульфидные связи и расположенные внутри белковых гло-
глобул, после таких манипуляций остаются интактными. Замена ос-
остатков Cys на Ser в том случае, если первые не входят в состав ак-
активного центра, как правило, является нейтральной по отноше-
отношению к ферментативной активности мутагенизируемого белка.
Это дает дополнительную возможность специфической модифи-
модификации конкретных остатков Cys после предварительной замены
части лишних остатков направленным мутагенезом.
Обсуждаемый комбинированный подход к модификации бел-
белков может быть использован для изменения окружения активно-
активного центра ферментов, исследования топологических особеннос-
особенностей белковых молекул и их стабилизации. В частности, с помо-
помощью этой группы методов удается манипулировать активностью,
специфичностью и рН-оптимумом субтилизина, а также констру-
конструировать комбинаторные клонотеки белков, в которых исследуе-
исследуемая полипептидная цепь целенаправленно сканируется остатком
Cys [44].
1.3. Химико-ферментативный синтез
в создании полусинтетических полипептидов:
лигирование синтезированных белков
Разработанный в 1998 г. подход к получению модифициро-
модифицированных белков, получивший название "лигирование синтезиро-
синтезированных белков" (expressed protein ligation, EPL), оказал и продол-
продолжает оказывать большое влияние на развитие белковой инжене-
инженерии [45]. Так же, как и рассмотренный выше метод нонсенс-су-
прессирующего мутагенеза, EPL позволяет вводить в белки не-
неприродные аминокислотные остатки и получать модифициро-
модифицированные полипептидные цепи, которые трудно, а порой и невоз-
невозможно, создать другими имеющимися методами. Метод EPL ос-
основан на ковалентном соединении друг с другом полипептидов,
полученных химическим синтезом или с использованием техно-
технологий рекомбинантных ДНК, что сопровождается образованием
требуемого модифицированного белкового продукта. Возмож-
Возможность такой сборки достигается введением на соответствующие
концы объединяемых полипептидов реакционноспособных
групп, обеспечивающих образование ковалентных связей в вод-
водных растворах при физиологических значениях рН.
306
1.3.1. Полусинтетические белковые молекулы
в создании новых полипептидов
Метод EPL является одной из разновидностей подхода, полу-
получившего название полусинтеза (semisynthesis) белков. Он вклю-
включает в себя группу методов, позволяющих осуществлять сайт-
специфические модификации белковых и пептидных молекул,
включая объединение фрагментов полипептидных цепей, полу-
полученных протеолитическим или химическим гидролизом исход-
исходных белковых молекул. В одном из простых вариантов полусин-
полусинтеза методом направленного мутагенеза в требуемый участок по-
полипептидной цепи вводят остаток Cys, который далее модифици-
модифицируют химическим зондом, специфически взаимодействующим с
сульфгидрильной группой этого аминокислотного остатка. С по-
помощью таких подходов удается вводить в исследуемые участки
полипептидных цепей фотоактивируемые химические группы,
осуществляющие поперечные сшивки полипептидных цепей [46],
флуорофоры [47], а также создавать фотоловушки для фермен-
ферментов, т.е. получать белки, сайт-специфически меченные флуорес-
флуоресцентными зондами [48].
Меньшие значения рКа ос-аминогрупп в белках по сравнению
с е-аминогруппами остатков Lys позволяют осуществлять специ-
специфическое аминоацилирование N-концов полипептидов при уме-
умеренно кислых значениях рН реакционной смеси [49]. С использо-
использованием другого химико-ферментативного подхода также удается
вводить в белки сайт-специфическую метку [50]. В этом случае
на первом этапе с помощью протеинкиназы и аналога [ATP]-y-S
осуществляют специфическое присоединение к полипептидной
цепи тиофосфатной группы, которую далее S-алкилируют флуо-
флуоресцирующим зондом. Таким способом была получена, в частно-
частности, меченая протеинкиназа А [51].
При другом общем подходе полусинтеза белков используют
протеолитические ферменты для лигирования пептидов. В этом
случае для обращения протеолитической реакции условия реак-
реакции изменяют таким образом, что аминолиз промежуточного
пептидил-ферментного комплекса становится предпочтительнее
его гидролитического расщепления. Необходимые условия со-
создаются путем включения в реакционную смесь органических
растворителей, таких как глицерин, диметилформамид или аце-
тонитрил. При наличии в реакционной смеси второго пептида в
процессе аминолиза происходит его объединение с пептидом из
комплекса посредством амидной связи [52,53]. Обратный проте-
олиз был успешно использован для лигирования пептидов с папа-
ином, трипсином и субтилизином. С использованием такого рода
307
подходов путем последовательного ферментативного лигирова-
ния олигосахаридов и полипептидов удается получать специфи-
специфически гликозилированные производные белков, например, гли-
козилированную рибонуклеазу В [54].
Путем изменения активных центров протеолитических фер-
ферментов с помощью генно-инженерных методов удалось значи-
значительно улучшить их способность лигировать пептиды. В частно-
частности, Д. Джексон с соавт. в 1994 г. получили двойной мутант суб-
тилизина, названный субтилигазой, обладающий высокой проте-
инлигазной активностью [55]. Замена Ser —> Cys в активном цен-
центре фермента придавала ему способность ацилироваться пепти-
пептидами, этерифицированными по С-концу гликолат-фенилаланил-
амидной группой. Вторая замена Pro —> Ala в том же активном
центре компенсировала негативные стерические эффекты пер-
первой мутации и усиливала аминолиз промежуточного тиоэфирно-
го комплекса фермент-пептид N-концевой аминогруппой второ-
второго пептида, сопровождаемый объединением этих двух пептидов с
помощью пептидной связи. Субтилигаза была успешно использо-
использована для полного синтеза нескольких белков и получения цикли-
циклических пептидов. В частности, с ее помощью удалось осущест-
осуществить синтез нативной рибонуклеазы, что будет подробнее рас-
рассмотрено ниже [55].
Д. Джексоном с соавтор, была разработана стратегия синте-
синтеза крупных полипептидных цепей с использованием субтилига-
зы, реализованная в синтезе модифицированной рибонуклеазы
А, обладающей ферментативной активностью [55]. На первом
этапе синтеза был получен полностью незащищенный пептид,
представляющий собой С-концевой фрагмент РНКазы А, кото-
который был использован в дальнейшем в качестве акцепторной мо-
молекулы (см. схему).
Следующий N-концевой донорный фрагмент полипептидной
цепи этерифицирован по С-концу гликолат-фенилаланиламидом
(glc-F-NH2). Полученный эфир эффективно ацилируется субти-
субтилигазой, что отражает особенности ее субстратной специфично-
специфичности: фермент предпочитает использовать в качестве субстрата
эфиры, в которых освобождается группа glc-F-NH2. В отличие от
этого донорный пептид содержит на N-конце дополнительную
изоникотиноильную защитную группу, что предотвращает его
лигирование самого на себя. Такая группа вводится на последней
стадии твердофазного синтеза пептидов и проявляет устойчи-
устойчивость к разбавленной плавиковой кислоте (HF), которая исполь-
использовалась для снятия защиты с боковых цепей пептидов и отщеп-
отщепления самих пептидов от твердого носителя. После каждого ли-
308
гирования пептидов изоникотиноильную группу удаляли в усло-
условиях мягкого восстановления (в присутствии цинка и уксусной
кислоты) для освобождения ЫН2-группы - необходимой участни-
участницы очередного цикла лигирования.
Исходя из субстратной специфичности субтилигазы (фер-
(фермент эффективно использует крупные гидрофобные донорные
субстраты и малоэффективен с отрицательно заряженными
R-NH-пептид y-CO-R' + Н2М-пептид2-СООН
\ 1. Субтилигаза
Я-Ш-пептиду-СО-Ш-пептидг-СООН
2. Zn/CH3COOH
H2N-nenTHflY-CO-NH-nenTHflZ-COOH
R-NH-neirnmx-CO-R т 3. Повторение этапов 1 и 2
Н2Н-пептидх-СО-ЫН-пептиду-СО-ЫН-пептид2-СООН
R =
Изоникотиноил Гликолат-фенилаланиламид
X, Y, Z - различные пептиды
остатками аминокислот или остатками Pro, попадающими в его
активный центр), карту полипептидной цепи РНКазы А разде-
разделили на шесть фрагментов. Эти фрагменты были синтезирова-
синтезированы и лигированы друг с другом в соответствии с вышеприведен-
вышеприведенной схемой с выходом, достигающим 70% на каждом этапе. В
309
результате была получена полноразмерная полипептидная цепь
РНКазы А A24 аминокислотных остатка), которая после спон-
спонтанного фолдинга приобрела искомую ферментативную актив-
активность. В ходе синтеза отдельных пептидов исследователи заме-
заменили два остатка His, находящихся в активном центре фермента
(положения 12 и 119), на остатки 4-фторгистидина. Поскольку
это производное His обладаетр?а = 3,5 (рКа His = 6,8), у искусст-
искусственной РНКазы А наблюдался резкий сдвиг в оптимуме рН ре-
реакции гидролиза субстрата без заметных изменений &кат, что бы-
было особенно неожиданным результатом, позволившим сделать
интересные выводы о механизме ферментативной реакции, осу-
осуществляемой РНКазой А. Позднее субтилигаза была успешно
использована для синтеза модифицированного гормона роста
человека.
Дальнейшее усовершенствование стратегии полусинтеза бел-
белков было сделано Р. Оффордом и К. Роузом [56], которые впер-
впервые использовали химию гидразонов и оксимов для лигирования
синтетических и рекомбинантных пептидов. При полусинтезе
(комбинированном синтезе) модифицированного колоний-стиму-
лирующего фактора гранулоцитов (G-CSF) вначале с помощью
специфического протеолиза по связям Lys-Ser рекомбинантного
фактора были получены фрагменты его полипептидной цепи. С
помощью периодатного окисления на N-концах пептидов были
получены альдегидные группы, а на С-концах с использованием
обратного протеолиза - гидразиды. Такие производные легко ли-
гируются друг с другом с образованием исходного полипептида
длиной в 174 а.о., обладающего биологической активностью.
При этом ряд исходных пептидных фрагментов можно было за-
заменять на искусственно полученные химическим синтезом и со-
содержащие модифицированные аминокислотные остатки. Сила
подхода, основанного на химии гидразонов и оксимов, заключа-
заключается в его универсальности. Вышеупомянутые модификации N- и
С-концов пептидов могут проводиться в присутствии любых бо-
боковых групп природных белков и не требуют их специальной за-
защиты. Именно это существенно облегчило химический синтез
белков и легло в основу группы современных методов, получив-
получивших название лигирования синтезированных белков, которые бу-
будут подробнее рассмотрены ниже.
Еще один подход, позволяющий осуществлять лигирование
полностью незащищенных пептидов в водных растворах при ней-
нейтральных значениях рН, был разработан в лаборатории С. Кента
в 1994 г. [57]. В этом случае на первом этапе проводят химиосе-
лективную реакцию между пептидом, содержащим N-концевой
310
NH3 NH OH
* i i О
NH3 NH OH
Подшпептщ 1
e
Обратимая транстиоэтерификация
-R-S"
h3n-|
NH, NH OH
^ Г i
H,N,
T
CONH
Спонтанный S -*¦ N-перенос ацила
h3n4
NH3
NH
OH
Полййептид 1
CO
e
>e
о ^-sh
/ [
I\H CONH
Рис. 38. Лигирование нативных белков [45]
Оба полностью незащищенных пептида реагируют друг с другом при ней-
нейтральных значениях рН; пептид справа содержит N-концевой остаток Cys, пеп-
пептид слева - С-концевую тиоэфирную группу; изображены незащищенные боко-
боковые функциональные группы аминокислотных остатков; в квадратных скобках
показано промежуточное соединение, спонтанно претерпевающее перестрой-
перестройку; один из объединяемых пептидов может быть синтетическим, другой - при-
природным
остаток Cys (oc-Cys), и вторым пептидом, содержащим ос-тиоэ-
фирную группу на С-конце. Первоначальная реакция транстиоэ-
терификации (рис. 38) сопровождается спонтанной внутримоле-
внутримолекулярной перестройкой (ацильное смещение S —> N) с образова-
образованием амидной связи. Методики, основанные на этой реакции, в
последнее время претерпели множество усовершенствований,
что сделало данный подход еще более эффектным и эффектив-
эффективным [58, 59].
311
1.3.2. Сплайсинг и транс-сплайсинг белков
в лигировании пептидов
Белковый сплайсинг представляет собой природный постран-
сляционный процесс, в котором белок-предшественник претер-
претерпевает спонтанную внутримолекулярную перестройку, в резуль-
результате которой происходит точное удаление внутреннего сегмента
полипептидной цепи (интеина) и лигирование внешних пептидов
(экстеинов) друг с другом [60]. Как правило, сплайсинг белков не
требует присутствия специфических последовательностей ами-
аминокислот в экстеинах. В отличие от этого, интеины характеризу-
характеризуются рядом консервативных последовательностей, которые об-
образуют семейство доменов, насчитывающее более 100 членов
(см. базу данных в Интернете: www.neb.com/neb/inteins.html). Во
время реализации стандартного механизма белкового сплайсинга
на первом этапе происходит ацильный перенос N —> S или N —> О,
при котором N-концевой экстеин переносится на SH- или
ОН-группу боковых цепей остатков Cys/Ser интеина, располо-
расположенных вблизи его N-конца (рис. 39). Образовавшийся в итоге
разветвленный интермедиат далее разрешается в реакции цикли-
циклизации с участием остатка Asn в С-концевой части интеина, кото-
который таким образом вырезается в виде С-концевого сукцинимид-
ного производного. Эта реакция частично катализируется интеи-
ном, обладающим специфической пространственной структурой,
благодаря которой расщепляемая амидная связь приобретает
конформацию с высокой энергией. На заключительном этапе
сплайсинга в результате S —> N или О —> N переноса ацила обра-
образуется амидная связь между двумя экстеинами процессируемой
полипептидной цепи, что напоминает механизм химического ли-
гирования незащищенных пептидов, рассмотренный выше.
Несмотря на то, что биологическое значение сплайсинга бел-
белков до сих пор остается невыясненным, сам он уже находит при-
применение в биотехнологии. Имеются, по крайней мере, две облас-
области применения интеинов. При одном подходе используют мутант-
ные формы интеинов, у которых произведена замена Asn —> Ala
в С-концевой части, что делает возможным прохождение только
первого этапа сплайсинга. В таких системах рекомбинантные
белки, синтезируемые в виде гибридных полипептидов с модифи-
модифицированным интеином, отщепляются с участием тиольной груп-
группы в результате внутримолекулярной реакции трансэтерифика-
ции (рис. 40). В итоге происходит высокоспецифическое протео-
литическое освобождение рекомбинантного белка от предшест-
предшественника, что облегчает его последующую очистку. Причем в
процессе такого освобождения могут быть получены ос-тиоэфир-
312
N -^S-Перенос ацила
Н-Экстаин
Транстиоэтерификация
1. Образование сукцинимида
2. S -*• N-Перенос ацила
N-Экстеин
0
-Экстеин
О
NH
Рис. 39. Механизм сплайсинга белков [45]
Процесс состоит из серии переносов ацильных групп и внутренних реак-
реакций, катализируемых центральным доменом белка (интеином); сплайсинг за-
завершается образованием обычной пептидной связи между двумя полипептида-
полипептидами (N- и С-экстеинами), фланкирующими интеин; N-экстеин - N-концевой экс-
теин; С-экстеин - С-концевой экстеин
313
¦Бёяйг
Белок
Интеиновый тиолиз
-ш-
I
R-SH
Бедок
о
+ H2N
Химическое лигирование
нативных белков
Белок
Полусинтетический продукт
Рис. 40. Освобождение целевого белка из рекомбинантного предшест-
предшественника, содержащего интеин, и его спонтанное лигирование с другим
пептидом по механизму EPL [45]
Мутационная замена Asn —> Ala в С-концевой части интеина блокирует по-
последнюю стадию сплайсинга рекомбинантного белка (см. рис. 39), при констру-
конструировании которого нуклеотидная последовательность целевого белка 1 была
объединена в коммерческом экспрессирующем векторе в одной рамке считыва-
считывания с последовательностями нуклеотидов мутантного интеина и хитин-связыва-
ющего домена (CBD) бактериальной хитиназы. Последний в рекомбинантном
белке выполняет роль лиганда, взаимодействующего с иммобилизованным ре-
рецептором (хитином), что необходимо для очистки белка с помощью аффинной
хроматографии; после связывания хроматографическим носителем целевой бе-
белок вырезается из полипептида с помощью добавленного тиола (R-HS) в реак-
реакции межмолекулярной трансэтерификации, и образовавшееся производное мо-
может быть далее лигировано с пептидом, содержащим остаток oc-Cys. Таким об-
образом, процесс EPL объединяет в себе две стадии: тиолиз с участием интеина я
спонтанное химическое лигирование нативных белков
314
о
NH
С-Эвсстеин
N-Зкстеин
О
JL
NH
Объединение
фрагментов
NH
С-Экстеин
Сплайсинг белка
NH
С-Экстеин
Рис. 41. Транс-сплайсинг белка с использованием разделенного интеина
[45]
Полипептидную цепь интеина можно разделить на две части, N-концевую
(IN) и С-концевую (Iе), которые сами по себе не обладают способностью осуще-
осуществлять белковый сплайсинг, но после нековалентного объединения активиру-
активируются
ные производные белков, которые являются ключевыми компо-
компонентами реакции нативного химического лигирования и далее
могут быть непосредственно использованы в этой реакции для
получения полусинтетических белковых продуктов [61-63].
Недавно несколькими независимыми группами исследовате-
исследователей была продемонстрирована возможность разделения интеи-
нов на две части, каждая из которых сама по себе не обладает
ферментативной активностью, однако после нековалентного их
объединения комплекс активируется [64-67]. На основе этого от-
открытия были разработаны высокоэффективные системы, осу-
осуществляющие транс-сплайсинг белков (рис. 41). При одновре-
315
менной экспрессии обеих частей интеина в одной клетке проис-
происходит спонтанный транс-сплайсинг белков, однако в бесклеточ-
ных системах для его прохождения необходимо проводить после-
последовательно денатурацию и ренатурацию рекомбинантных бел-
белков. Недавно, у Synechocytis (Ssp) в белке DnaE был обнаружен
составной интеин, который не требует для своей активации про-
проведения цикла денатурации-ренатрурации [66]. Не исключено,
что с его помощью удастся решить эту биотехнологическую про-
проблему. С использованием транс-сплайсинга белков возможно
получение полусинтетических белковых продуктов, объединяю-
объединяющих в себе рекомбинантный белок с полностью синтетическим
пептидом, в который введена изотопная метка. Кроме того, не-
недавно такой подход был применен для синтеза циклических пеп-
пептидов [69], изучения белок-белковых взаимодействий [70] и осу-
осуществления контроля за функциями белков [71], что позволяет
угадывать за ним большое будущее.
В заключение этого раздела следует упомянуть о возможно-
возможности использования транс-сплайсинга белков для исследования
белок-белковых взаимодействий в цитозоле прокариотических и
эукариотических клеток [72, 73]. При таком подходе получают
гибридные рекомбинантные белки путем слияния половинок зе-
зеленого флуоресцирующего белка (GFP) или люциферазы с двумя
фрагментами расщепленного интеина. Другой конец одного из
фрагментов интеина соединяют с белком-рецептором (приман-
(приманкой), как это делается в классических системах дигибридного
скрещивания у дрожжей, а у другого фрагмента - с большим ко-
количеством потенциальных белков-лигандов. В том случае, если
между рецептором и лигандом происходит взаимодействие, два
фрагмента интеинов объединяются друг с другом (см. выше,
рис. 41) и осуществляют транс-сплайсинг, завершающийся обра-
образованием полноценного флуоресцирующего белка или люцифе-
люциферазы, что легко обнаруживается по изменению уровня флуорес-
флуоресценции инкубационной смеси.
1.3.3. Метод EPL в белковой инженерии
Как можно видеть из вышеизложенного, метод EPL обладает
большими преимуществами перед подходами, основанными на
использовании только химических методов синтеза полипепти-
полипептидов. При использовании лигирования синтезированных белков
большая часть белковой молекулы может быть синтезирована
рибосомами, и полученные фрагменты полипептидных цепей да-
далее легко объединяются с пептидами, синтезированными хими-
316
чески. Это позволяет проводить почти любые направленные из-
изменения небольших участков полипептидных цепей сложных
белковых молекул и изучать их влияние на функциональность
исследуемых белков. В участки полипептидных цепей, получен-
полученных химическим синтезом, можно вводить любые функциональ-
функциональные группы, в том числе и аналоги аминокислотных остатков,
которые невозможно включить в состав полипептидных цепей с
использованием аппарата трансляции. С учетом таких возможно-
возможностей, метод EPL был немедленно использован для сайт-специфи-
сайт-специфического введения в белки разнообразных молекулярных зондов,
включая флуорохромы и стабильные изотопы, что позволило
эффективно исследовать структурно-функциональные отноше-
отношения в сложных белках и макромолекулярных комплексах. Метод
EPL дает возможность получать производные белков чрезвычай-
чрезвычайно высокой молекулярной массы, что в настоящее время невоз-
невозможно сделать никакими другими методами. В частности, с помо-
помощью этого метода были получены модифицированные производ-
производные р'-субъединицы РНК-полимеразы Е. coli, длина полипептид-
полипептидной цепи которой составляет 1407 а.о. [47].
Методом EPL в настоящее время уже исследованы десятки
белков прокариотических и эукариотических организмов. В этот
список входят киназы, фосфатазы, факторы транскрипции,
РНК-полимеразы, белки, участвующие в цитоплазматической
передаче сигналов, ионные каналы, рецепторы и антитела. Ниже
мы кратко рассмотрим некоторые из наиболее важных приложе-
приложений метода EPL в белковой инженерии.
Имитация посттрансляционных модификаций белков. Пост-
Посттрансляционные модификации белков являются одним из важ-
важнейших этапов экспрессии генов. Большинство эукариотических
белков становятся функционально-активными лишь после кова-
лентного присоединения к их функциональным группам модифи-
модифицирующих молекул. Кроме того, посттрансляционные модифи-
модификации, например, фосфорилирование или ацетилирование белко-
белковых молекул, являются важным механизмом регуляции их биоло-
биологической активности. Исследование молекулярных механизмов
регуляции активности белков под действием их ковалентных мо-
модификаций сдерживается отсутствием адекватных модельных си-
систем. Например, стандартным методом получения специфически
фосфорилированных белков является их инкубация с соответст-
соответствующей протеинкиназой в присутствии субстратов. Однако
обычно не удается контролировать уровень фосфорилирования
белка, а также специфичность самой реакции. Использование
метода EPL позволяет решить эту проблему путем объединения
317
рекомбинантных немодифицируемых фрагментов исследуемого
белка с требуемым модифицированным фрагментом, синтезиро-
синтезированным химическими методами. В частности, методом EPL уда-
удалось получить фосфорилированный по четырем критическим
положениям рецептор трансформирующего фактора роста р ти-
типа I (TpR-I), объединяя синтетический модифицированный фраг-
фрагмент полипептидной цепи с соответствующим рекомбинантным
фрагментом фактора. Оказалось, что фосфорилирование, при-
приводящее к активации TpR-I, увеличивает его сродство к природ-
природному субстрату - фактору транскрипции Smad2 и уменьшает к
белку-ингибитору FKBP12 [74]. Данные такого рода позволили
построить новую модель механизма активации рецептора TpR-I.
В аналогичных опытах с белком Smad2 было установлено, что
его фосфорилирование сопровождается образованием тримера,
диссоциацией его комплекса с рецептором и гетероолигомериза-
цией с регуляторным белком Smad4 [75]. С помощью аналогич-
аналогичного подхода начато исследование молекулярных последствий
гликозилирования и липидирования белков. Последняя посттран-
посттрансляционная модификация включает N-концевое меристоилиро-
вание, присоединение остатков холестерина к С-концам, S-npe-
нилирование (присоединение фарнезильных и геронилгерониль-
ных групп) остатков Cys вблизи С-концов белковых молекул, а
также S-пальмитоилирование остатков Cys вдоль полипептид-
полипептидных цепей [76]. Липидирование белков повышает их сродство к
мембранам, что оказывает влияние на внутриклеточную локали-
локализацию и функции образующихся липопротеинов.
Приведенные примеры показывают, что метод EPL позволя-
позволяет легко объединять белки как друг с другом, так и со многими
химическими соединениями другой природы.
318
Глава 2
Направленная эволюция белков
Основные идеи, лежащие в основе обсуждаемого подхода к
получению новых белковых молекул с требуемой биологической
активностью, заимствованы из дарвиновской теории биологиче-
биологической эволюции, объясняющей причины разнообразия жизни на
Земле. В соответствии с концепциями неодарвинистов, внутриви-
внутривидовое генетическое разнообразие, которое является следствием
глобального мутационного процесса, рано или поздно находит
свое фенотипическое проявление. Фенотипически различающие-
различающиеся организмы конкурируют друг с другом за жизненное прост-
пространство и ресурсы, что сопровождается борьбой за существова-
существование и естественным отбором, в результате которых выживают те
мутантные особи, фенотип которых наиболее приспособлен к
данным экологическим условиям.
При искусственном отборе, осуществляемом человеком,
проводится направленная селекция индивидуумов, фенотип ко-
которых отвечает требованиям селекционера. В этом случае ли-
лимитирующим фактором становится количество фенотипов, до-
доступных экспериментатору. По мере углубления понимания
молекулярных механизмов изменчивости живых организмов
появилась возможность искусственно влиять на нее посредст-
посредством усиления процесса мутагенеза с помощью тех или иных му-
мутагенов.
С учетом этих простых и "очевидных" соображений легко
сделать следующий шаг от живых организмов к молекулам с тем,
чтобы применить вышеупомянутые принципы к молекулярным
объектам для получения молекул с нужными свойствами. Но по-
поскольку "легко срубить только упавшее дерево", такой шаг суж-
суждено было сделать отнюдь не нам, а Солу Спигельману с соавт. в
1965 г. [77].
Опыты С. Спигельмана. В классических опытах С. Спигель-
мана была использована система репликации геномной РНК бак-
бактериофага Q|3 in vitro, в которой синтез РНК осуществлялся
Qp-репликазой. Помимо фермента и матрицы исходная смесь со-
содержала все четыре рибонуклеозидтрифосфата, т.е. предшест-
предшественники мономеров реплицируемых макромолекул, необходи-
необходимые для построения комплементарных цепей РНК. После инку-
инкубации в течение определенного времени, достаточного для синте-
синтеза полноразмерных молекул РНК, аликвоту реакционной смеси
Переносили в новую пробирку, содержащую все исходные компо-
319
0,015
03
ю
о
I
S
I
S
о
S
Я
0,005 -
100
200
Время (мин)
300
400
Рис. 42. Изменение скорости синтеза Qp-PHK в опытах С. Спигельма-
на [78]
Скорость синтеза РНК измеряли по включению радиоактивного GTP; по-
после проведения определенного количества циклов отбора имело место скачко-
скачкообразное возрастание скорости синтеза РНК
ненты кроме РНК, и инкубацию продолжали в течение того же
времени. Поскольку точность репликации РНК Qp-репликазой
невысока, ожидалось, что в результате серийных переносов бу-
будут постепенно отбираться мутантные молекулы матрицы с бо-
более высокой скоростью репликации, которые, в конце концов,
вытеснят исходные молекулы.
Ожидания авторов полностью подтвердились. После 70 пас-
пассажей скорость репликации QJ3-PHK возросла в ~10 раз. Конеч-
Конечную популяцию составляли укороченные мутантные РНК, более
эффективно используемые в репликации. Уже после первых пас-
пассажей имело место трехкратное повышение скорости реплика-
репликации (рис. 42), переход к более высоким скоростям носил ступен-
ступенчатый характер. На первых переносах наблюдалось уменьшение
скорости репликации, по мнению авторов, связанное с появлени-
появлением (из-за накопления мутаций) неблагоприятных для репликации
вариантов макромолекул.
320
По современным представлениям, в том случае, когда накоп-
накопление мутаций в популяции эволюционирующих молекул не ней-
нейтрально по отношению к их "фенотипу", в ней имеется преобла-
преобладающая последовательность (master sequence), которая в наи-
наибольшей степени приспособлена к условиям воспроизведения in
vitro [78]. Кроме того, если мутации возникают достаточно часто,
ведущая последовательность окружена облаком мутантов, ко-
которые также обладают приспособленностью, достаточной для их
сохранения в условиях эксперимента. При постоянных условиях
ведущая последовательность и облако мутантов находятся в рав-
равновесии, и такой набор эволюционирующих макромолекул, для
которого характерно стационарное распределение генотипов,
получил название квази-вида. Это понятие эффективно исполь-
используется для описания эволюции вирусов, хотя в данном случае рав-
равновесное состояние генотипов не является обязательным требо-
требованием к возможности применения такого понятия [79].
Идеи С. Спигельмана оказались исключительно продуктив-
продуктивными для последующего развития молекулярной биологии и лег-
легли в основу одного из ключевых подходов к получению новых
белков и ферментов с использованием методов направленной
эволюции макромолекул. Однако, в отличие от обсуждавшихся
выше классических опытов, разнообразие последовательностей
нуклеиновых кислот, которые кодируют белки, эволюциониру-
эволюционирующие in vitro в такой системе, возникает не спонтанно, а создает-
создается исследователем с помощью случайного мутагенеза перед каж-
каждым новым раундом отбора белков с требуемыми свойствами.
Поэтому эксперименты по направленной эволюции белков начи-
начинаются с формирования комбинаторных клонотек случайных по-
последовательностей, которые охватывают определенную часть
исследуемого пространства последовательностей.
2.1. Комбинаторные клонотеки
последовательностей нуклеотидов
Как следует из предшествующего изложения, для реализации
концепции направленной эволюции белков требуется прежде
всего иметь в распоряжении большой набор (выбор) макромоле-
макромолекул, среди которых можно было бы ожидать наличие белков с
требуемыми свойствами. Если бы исходный пул макромолекул
включал все возможные комбинации аминокислотных последо-
последовательностей, и желаемое свойство было бы присуще хотя бы од-
одной из них, то получение нужного белка или фермента в требуе-
11. Патрушев Л.И. Т. 1 321
мых количествах было бы делом грамотного применения мето-
методов отбора и генно-инженерной техники. Следовательно, созда-
создание представительного пула (клонотеки) нуклеотидных и, как
следствие, аминокислотных последовательностей является одной
из первых задач белковой инженерии, основанной на применении
принципов направленной эволюции. Однако, несмотря на значи-
значительный прогресс, на пути создания таких клонотек лежат значи-
значительные трудности.
Само по себе создание представительных клонотек и тести-
тестирование свойств даже небольших белков со всеми возможными
комбинациями аминокислотных остатков является в настоящее
время неразрешимой задачей. Исчерпывающая клонотека долж-
должна включать полное пространство последовательностей. Как уже
упоминалось выше, пространство последовательностей полипеп-
полипептидной цепи определенной длины представляет собой сеть всех
теоретически возможных взаимосвязанных аминокислотных по-
последовательностей, в которой каждый узел представляет собой
уникальную последовательность аминокислот. Пространство по-
последовательностей для белка в п аминокислотных остатков (а.о.)
будет содержать ~20" членов, т.е. в пространстве последователь-
последовательностей даже небольшого белка длиной в 200 а.о. будет заключе-
заключено ~20200 разных макромолекул, а это значение превышает число
атомов в видимой части вселенной, которое оценивается ~1079.
Большой по объему задачей представляется даже исследо-
исследование влияния одиночных замен аминокислотных остатков по-
полипептидной цепи известного белка длиной в 200 а.о., так как
здесь количество возможных вариантов составит 3800, а при не-
необходимости изучения в белке еще и всех двойных замен -
~7,3 • 106. Однако, как уже упоминалось, для радикального изме-
изменения каталитических свойств ферментов, как правило, необхо-
необходимы множественные замены аминокислотных остатков их по-
полипептидных цепей.
В настоящее время неизвестны частоты встречаемости бел-
белков с заданной вторичной, а тем более третичной структурами,
среди набора полипептидов со случайными последовательностя-
последовательностями аминокислотных остатков. Поэтому для поиска белковых мо-
молекул, обладающих требуемыми свойствами, необходимо ис-
использовать клонотеки очень большого размера, в идеале содер-
содержащие максимально возможное количество компонентов, с ко-
которым еще можно работать экспериментальными методами. Раз-
Разнообразие в клонотеках нуклеотидных последовательностей, ко-
кодирующих исследуемые белки, создается введением в гены слу-
случайных мутаций.
322
2.2. Методы введения случайных мутаций
Рассмотренные подходы к получению сайт-специфических
мутаций с помощью олигонуклеотидов позволяют с высокой
точностью и эффективностью производить замены отдельных
нуклеотидов в строго определенных локусах. Однако чаще всего
невозможно предсказать фенотипические последствия мутацион-
мутационных замен отдельных аминокислот в полипептидных цепях бел-
белков, и для получения необходимых фенотипических изменений
требуется внесение множественных мутаций в определенные
участки гена с последующим отбором мутантов требуемого фе-
фенотипа. Для решения подобных задач был разработан и эффек-
эффективно используется метод кассетного мутагенеза.
При реализации такого подхода из гена, клонированного в
составе векторной плазмиды, по двум близко расположенным
уникальным сайтам рестрикции вырезается фрагмент ДНК, в
который требуется внести мутации, и на его место встраивается
синтетический двухцепочечный олигонуклеотид, содержащий
необходимые замены нуклеотидов (кассету мутаций). В этом
случае, если в окрестностях мутагенизируемого локуса гена от-
отсутствуют подходящие природные сайты рестрикции, их вводят
с помощью направленного мутагенеза. Разработка автоматиче-
автоматических синтезаторов ДНК сделала синтез олигодезоксирибонук-
леотидов простой и даже рутинной процедурой. Более того, ис-
использование на некоторых этапах синтеза вместо одного нукле-
отида смеси из двух, трех или даже всех четырех дезоксирибо-
нуклеозидтрифосфатов позволяет получать за один прием
сложную смесь олигонуклеотидов, которые могут содержать в
определенных сайтах наборы кодонов для многих или даже всех
20 природных аминокислот. Это дает возможность осуществ-
осуществлять одновременный скрининг по искомому мутантному фено-
фенотипу большого числа разных мутантных клонов, полученных в
одном цикле клонирования. С помощью кассетного мутагенеза
можно определять функциональную роль отдельных сайтов и
целых доменов в полипептидных цепях конкретных белков и
создавать рекомбинантные белки с новыми, подчас неожидан-
неожиданными свойствами.
Одной из разновидностей кассетного мутагенеза является
способ введения мутаций, основанный на ПЦР с вырожденными
праймерами. В этом случае при проведении ПЦР используют
праймеры, которые получают введением в определенные поло-
положения при их синтезе разных нуклеотидов, что достигается при-
применением на конкретных этапах синтеза не отдельных предшест-
п* 323
венников, а их смеси, что приводит к одновременному образова-
образованию набора олигонуклеотидов.
Внесение множественных мутаций в разные участки поли-
полипептидных цепей позволяет при наличии соответствующих эф-
эффективных методов отбора получать белки с требуемыми свой-
свойствами и уже после этого определять, какие именно аминокисло-
аминокислоты придают эти свойства белку. Альтернативным методом опре-
определения функциональной значимости отдельных аминокислот в
белках является их целенаправленная замена на нейтральную
аминокислоту, например, аланин. Такая последовательная заме-
замена аминокислот в полипептидных цепях на остатки Ala получила
название сканирование аланином. Введение аланина в полипеп-
полипептидные цепи не изменяет их общей конформации, как это имеет
место, например, в случае замены на глицин или пролин, и не со-
сопровождается ярко выраженными электростатическими или сте-
рическими эффектами. Кроме того, аланин часто встречается в
полипептидных цепях и с одинаковой частотой представлен как
на внутренних, так и на внешних участках полипептидных цепей
белковых глобул. С помощью сканирования аланином можно ло-
локализовать аминокислоты, образующие активный центр фер-
ферментов, исследовать участки полипептидных цепей, существен-
существенные для взаимодействия белков с другими макромолекулами и
низкомолекулярными лигандами, изучать структуру эпитопов
полипептидных цепей, а также ряд других структурных и функ-
функциональных особенностей белков.
2.2.1. Химический мутагенез
Делеции и вставки, создаваемые в структурных частях генов,
как правило, их инактивируют, особенно в тех случаях, когда эти
мутации приводят к сдвигу открытых рамок считывания. Поэто-
Поэтому делеции и вставки in vitro используют главным образом для
поиска и изучения регуляторных элементов генов, влияющих на
эффективность их экспрессии. Большое значение для исследова-
исследования функционирования белков имеют методы мутагенеза in vitro,
направленные на получение точковых мутаций, следствием кото-
которых являются одиночные замены аминокислот в полипептидных
цепях. Распространенным методом введения большого числа
точковых мутаций разной локализации в исследуемые части ге-
генов in vitro является химический мутагенез одноцепочечных уча-
участков рекомбинантных ДНК. Принцип подобных методов заклю-
заключается в том, что некоторые химические мутагены, такие как би-
бисульфит натрия, гидроксиламин или метоксиламин, действуют
324
только на одноцепочечные участки ДНК. Следовательно, полу-
получив молекулы ДНК, содержащие одноцепочечные бреши в ис-
исследуемых участках генов, можно с помощью бисульфита натрия
дезаминировать остатки цитозина в этих участках, т.е. превра-
превратить их в остатки урацила. После достройки цепи такой мутаге-
низированной молекулы ДНК с помощью фрагмента Клёнова
ДНК-полимеразы I Е. сой происходит замена исходных G-C-nap
на T-U. Затем мутагенизированные молекулы ДНК с помощью
трансформации вводят в бактериальные клетки, где по заверше-
завершении первого раунда репликации в молекуле осуществляются за-
замены остатков U на Т и полная замена G-C-пары на А-Т, т.е.
имеет место обычная транзиция.
Одноцепочечные мутагенизированные участки ДНК удобно
получать путем гибридизации одноцепочечнои и двухцепочечнои
ДНК одного и того же вектора, содержащего клонированный ген
или его участок, который необходимо мутагенизировать. В этом
случае в образующемся гибриде-гетеродуплексе, одна цепь кото-
которого принадлежит вектору без вставки, а другая - вектору со
вставкой, происходит выпетливание последовательности вставки
в виде одноцепочечного участка ДНК. Обсуждаемый подход к
получению статистического набора точковых мутаций с исполь-
использованием химических мутагенов позволяет легко создавать боль-
большое число мутантных молекул ДНК, содержащих одну или не-
несколько мутаций в разных сочетаниях. Последующий отбор му-
мутантов на основе новых биохимических или иных параметров му-
мутантных белков (исчезновение, ослабление или усиление фер-
ферментативной активности, появление новой активности или новых
иммунологических свойств и т.п.) дает возможность идентифици-
идентифицировать остатки аминокислот в исследуемых белках, отвечающие
за эти изменения.
Несмотря на удобство введения такого рода мутаций в ДНК
in vitro, химический мутагенез накладывает ограничения на
спектр возникающих мутаций, так как лишь определенные ос-
остатки нуклеотидов ДНК претерпевают обязательные измене-
изменения. Поэтому многие мутации никогда не могут быть получены
с помощью химических мутагенов. Проблему можно частично
решить, используя для репарации одноцепочечных брешей
ДНК аналоги нуклеотидов, например, N-гидроксицитозинтри-
фосфат, который в составе ДНК одинаково хорошо спаривает-
спаривается с А и G, или создавая такие условия, при которых репариру-
ющая ДНК-полимераза начинает ошибочно включать в синте-
синтезируемую цепь ДНК некомплементарные матрице нуклеотиды.
Все перечисленные выше методы локального мутагенеза, осу-
325
ществляемого in vitro, позволяют, в конечном счете, получать
набор случайных мутаций, локализованных на определенном
исследуемом участке ДНК. Мутагенизированные молекулы
ДНК из одной реакционной пробирки представляют собой
сложную смесь, в которой каждая молекула несет несколько
независимо возникших мутаций. Для введения мутаций в опре-
определенный локус исследуемого гена необходимо проводить тру-
трудоемкую процедуру отбора, сопряженную с анализом большого
числа мутантов. Подлинную революцию в направленном мута-
мутагенезе произвела разработка методов с использованием ПЦР и
синтетических олигонуклеотидов.
2.2.2. Синтез ДНК с ошибками
Искусственное увеличение частоты включения в продукт
ПЦР некомплементарных матрице (ошибочных) нуклеотидов во
время проведения реакции является простым и широко использу-
используемым методом получения последовательностей нуклеотидов, из-
измененных случайным образом. Имеется несколько способов
уменьшения точности синтеза ДНК во время ПЦР, среди кото-
которых следует отметить создание специальных (неоптимальных)
условий проведения реакции и использование специальных
ДНК-полимераз.
Как уже упоминалось в разделе 3.1.5 первой части книги, по-
повышение концентрации дезоксирибонуклеозидтрифосфатов и
ионов Mg2+ в реакционной смеси приводит к возрастанию числа
ошибок амплификации ДНК во время ПЦР. К тем же последст-
последствиям ведет замена ионов Mg2+ на ионы Мп2+ или их совместное
использование [80]. Для достижения максимального мутагенного
эффекта необходимо соблюдать определенное соотношение
между концентрациями всех четырех дезоксирибонуклеозидтри-
фосфатов в инкубационной смеси [81]. К сожалению, даже в оп-
оптимальных условиях замены нуклеотидов в системах такого рода
не происходят случайным образом: транзиции (замены пурина на
пурин и пиримидина на пиримидин) превалируют над трансверси-
трансверсиями (замены пурина на пиримидин и наоборот). Благодаря этому,
в результате такого мутагенеза в среднем было обнаружено по-
появление в конкретном участке мутантной полипептидной цепи
лишь 5,7 а.о. из 20 теоретически возможных [82]. Следовательно,
данный подход в его обычном воплощении не пригоден для со-
создания репрезентативных клонотек нуклеотидных последова-
последовательностей, кодирующих все возможные варианты полипептид-
полипептидных цепей, исследуемых методом направленной эволюции.
326
В последнее время была разработана основанная на ПЦР си-
система случайного мутагенеза, которая позволяет преодолевать
описанное выше затруднение [83, 84]. Мутагенез проводят в две
стадии. На первом этапе ПЦР осуществляют с помощью Taq-
ДНК-полимеразы в присутствии ионов Мп2+, что сопровождается
ошибочным включением нуклеотидов в синтезируемую ДНК с
преобладанием транзиций. Образовавшийся продукт используют
в качестве матрицы во второй ПЦР, при проведении которой в
реакционную смесь добавляют дезоксиинозинтрифосфат (dITP),
который, как известно, образует комплементарные пары с пири-
мидинами С и Т матрицы. В итоге происходит коррекция спектра
мутаций, образовавшихся в первом раунде, в сторону образова-
образования трансверсий. Если при использовании обычных подходов
имеет место образование 74,8% транзиций и только 25,2% транс-
трансверсий, то в случае системы Mn2+-dITP доля трансверсий возрас-
возрастала до 41,7%, что позволяло повысить число возможных замен
а.о. в каждом участке мутагенизируемых полипептидных цепей
более, чем в два раза.
2.2.3. Случайное объединение гомологичных
и негомологичных участков генов
Случайное объединение гомологичных мутантных фрагмен-
фрагментов ДНК, в сумме составляющих всю структурную часть гена
(DNA shuffling), с последующей экспрессией собранных таким пу-
путем генно-инженерных конструкций является еще одним распро-
распространенным и эффективным способом повышения разнообразия
пула белковых молекул, используемых в направленной эволю-
эволюции [85]. Реализацию этого подхода осуществляют в два этапа.
Прежде всего, структурную часть гена в составе экспрессирую-
щего вектора подвергают мутагенному воздействию, получая по-
популяцию молекул ДНК, содержащих случайные мутации. Му-
Мутантные белки (следовательно, и кодирующие их гены) подверга-
подвергают отбору на наличие в них определенных свойств (см. ниже). На
втором этапе отобранные мутантные гены, которые кодируют
требуемые белки, фрагментируют ДНКазой I и объединяют друг
с другом in vitro с помощью ПЦР в отсутствие праймеров
(рис. 43, а). Вместо инкубации с ДНКазой I для фрагментации ге-
генов также применяют ПЦР со случайным праймированием, осу-
осуществляемым короткими праймерами, которые обладают не-
несколькими местами посадки в гене (рис. 43, б).
Возможность правильной сборки гена из таких фрагментов
ДНК определяется тем, что в процессе фрагментации образуют-
327
Повторение
операции
5'—*¦
3' * х
5'
* 5'
3'
Случайный мутагенез,
скрининг
Отобранные клоны
Й й =
х-
х-
-х*-
-х-
-X-
-X-
-X-
-X-
-X-
-X-
| Фрагментация ДНКазой I
х ¦<—х-
х-
х
х-
| Сборка полноразмерных
t молекул и амплификация
3' 5'-
-х-
х-
-х-
-х-
I Ююнирование и скрининг
Отобранные клоны
Синтез со случайно {
отжигающейся затравкой
5'-.
3'
5'
3'
- 3'
5'
| Удаление
| матричной ДНК
ся перекрывающиеся молекулы ДНК, в сумме представляющие
всю исходную молекулу, которые во время ПЦР на стадии отжи-
отжига находят комплементарные гомологи. При этом из-за случай-
случайного характера проведенного мутагенеза на данной стадии с вы-
высокой вероятностью образуются гетеродуплексы. Мутации, при-
присутствующие в гетеродуплексах, фиксируются при достройке вы-
выступающих одноцепочечных участков ДНК ДНК-полимеразой, а
также при последующих циклах ГЩР. После такой перетасовки
мутантных частей и экспрессии гибридных генов среди новых му-
тантных ферментов проводят второй цикл отбора белков с тре-
требуемыми свойствами, что, как правило, позволяет улучшить па-
параметры исходных белков.
При случайном объединении ДНК фрагментированных ге-
генов одного семейства (family shuffling) фрагментации и после-
последующему объединению подвергают последовательности гомо-
гомологичных генов, принадлежащих одному семейству. При этом
подходе случайному объединению в одной молекуле ДНК под-
подвергаются фрагменты ДНК, последовательности которых ди-
вергировали в процессе филогенетического развития организ-
организмов и, судя по значительной гомологии, происходят от общего
гена-предшественника. Технически процесс объединения фраг-
фрагментов ДНК в данном случае не отличается от только что опи-
описанного.
Основной проблемой, с которой приходится сталкиваться при
объединении фрагментов ДНК разных генов одного семейства,
является низкий выход рекомбинантных фрагментов ДНК: из
двух генов, гомология между которыми составляет 80%, образу-
образуется менее 1% рекомбинантных молекул. Возможное объясне-
объяснение этого явления заключается в том, что в данной системе обра-
образование гомодуплексов при первом отжиге фрагментов ДНК
Рис. 43. Случайное объединение гомологичных мутантных фрагментов
ДНК (DNA shuffling) [85]
Получение клонотек случайных последовательностей и их направлен-
направленную эволюцию проводят в два этапа: 1) исследуемую последовательность
нуклеотидов подвергают случайному мутагенезу и отбору на основании
свойств кодируемого этой последовательностью белка, 2) популяцию отоб-
отобранных последовательностей фрагментируют случайным образом с помо-
помощью ДНКазы I (а) или методом ПЦР с короткими случайно отжигающими-
отжигающимися праймерами (б) и перекрывающиеся фрагменты объединяют друг с дру-
другом с помощью ПЦР без праймеров; образовавшимися в итоге полнораз-
мерными молекулами ДНК трансформируют компетентные клетки, среди
которых проводят отбор по фенотипу на основании свойств исследуемого
рекомбинантного белка
329
друг на друга происходит предпочтительнее, чем соответствую-
соответствующих гетеродуплексов. Проблему решают применением в реком-
бинировании предварительно полученных фрагментов одноцепо-
чечной ДНК разных цепей объединяемых генов. При другом спо-
способе решения этой проблемы в объединении используют фраг-
фрагменты ДНК, созданные не случайным расщеплением ДНКазой I,
а с помощью рестриктаз, причем ДНК разных генов инкубируют
с различными рестриктазами. В таком случае гомодуплексы из
фрагментов одного и того же гена образуются с той же высокой
частотой, однако элонгируются только гетеродуплексы с 5'-вы-
5'-выступающими одноцепочечными участками при несовпадении рас-
расположения сайтов рестрикции на двух объединяемых последова-
последовательностях ДНК.
Процесс ступенчатого удлинения олигомеров (staggered
extension process (StEP)). Этот простой и эффективный метод ча-
часто используют для осуществления рекомбинации между двумя
или несколькими ДНК-матрицами in vitro с помощью модифи-
модифицированной ПЦР [86]. Две или несколько ДНК-матриц отжига-
отжигают с набором коротких олигонуклеотидных праймеров в одной
реакционной смеси, которые взаимодействуют с ДНК в случай-
случайных местах. За этим следуют повторяющиеся циклы денатура-
денатурации, очень непродолжительного отжига праймеров и их элонга-
элонгации ДНК-полимеразой. Процесс продолжается до тех пор, пока
в реакционной смеси не образуется ПЦР-продукт необходимой
длины. Переключение синтеза ДНК с одних матриц на другие,
которое происходит в каждом новом цикле ПЦР, в конце кон-
концов приводит к образованию набора полноразмерных генов, в
которых случайным образом объединены участки исходных ге-
генов-матриц.
Рис. 44. Рекомбинирование негомологичных белков [87]
Исходный димер объединяемых генов состоит из генов 1 и 2, объединен-
объединенных линкером, содержащим подходящие сайты рестрикции; (/) димер фраг-
ментируют ДНКазой I и образовавшиеся концы "затупляют" нуклеазой S1; B)
отбирают фрагменты, размер которых соответствует длине одного гена плюс
длина линкера, C) переводят их в кольцевую форму лигированием по "тупым"
концам и D) линеаризуют с помощью рестриктазы по сайту, который нахо-
находится в линкере; образующаяся в итоге библиотека генов содержит гибрид-
гибридные молекулы ДНК, кодирующие N-концевую часть белка 2 и С-концевую
часть белка 1; E) химерные гены клонируют в экспрессирующем векторе сра-
сразу или после амплификации с помощью ПЦР с использованием по одному из
праймеров, комплементарных концам объединяемых генов, которые соседст-
соседствуют с линкером, и после экспрессии химерных генов получают клонотеку хи-
химерных белков
330
Ген1
(I)
Ген 2
B)
C)
Клонотека гибридных генов
D)
E)
Клонотека гибридных белков
331
Рекомбинирование фрагментов генов и белков, не зависимое
от гомологии. Во всех подходах, рассмотренных выше, необходи-
необходимым условием обмена последовательностями двух рекомбиниру-
емых генов является наличие между ними протяженных участков
гомологии. Для преодоления этого ограничения можно исполь-
использовать специальный метод, который позволяет получать из двух
сильно дивергировавших кодирующих последовательностей кло-
нотеки гибридных генов, объединяющих в себе последователь-
последовательности исходных генов в разных пропорциях [87].
Работа по созданию таких клонотек начинается с объедине-
объединения двух генов в виде димера "голова к хвосту", между которыми
находится короткая линкерная последовательность, содержащая
нужные сайты рестрикции (рис. 44, стадия 1). Димеры фрагмен-
тируют случайным образом, инкубируя их с ДНКазой I, образо-
образовавшиеся концы молекул "затупляют" нуклеазой S1, а сами
фрагменты ДНК фракционируют по размерам, отбирая молеку-
молекулы, длина которых соответствует размерам одного из исходных
генов (стадия 2). Далее осуществляют внутримолекулярное лиги-
рование этих фрагментов по тупым концам (стадия 3), что сопро-
сопровождается образованием кольцевых молекул ДНК, которые да-
далее линеаризуют с помощью рестриктаз по последовательности
линкера (стадия 4). Итоговые гибридные молекулы ДНК кодиру-
кодируют N-концевую часть белка 2 и С-концевую часть белка 1, кото-
которые могут быть синтезированы в виде единой полипептидной це-
цепи в результате экспрессии рекомбинантных генов после их кло-
клонирования в составе экспрессирующего плазмидного вектора.
При этом, соотношение последовательностей двух объединяе-
объединяемых белков в составе гибридных молекул варьирует в широких
пределах.
2.3. Скрининг и отбор белков с требуемыми свойствами
Создание репрезентативных клонотек нуклеотидных и коди-
кодируемых ими белковых последовательностей является первым
этапом, который необходимо преодолеть при конструировании
белков методом направленной эволюции. Следующий важный
шаг на этом пути - обнаружение белка с требуемыми свойствами
среди большого числа макромолекул, составляющих получен-
полученную клонотеку. Имеются три принципиально различающихся
стратегии поиска нужного молекулярного объекта: случайный
скрининг, улучшенный (facilitated) скрининг и отбор. Подходы,
основанные на скрининге, предполагают в оптимальном вариан-
варианте изучение всех объектов клонотеки. При случайном скрининге,
332
который является наиболее трудоемким и наименее интеллекту-
интеллектуальным подходом, каждый молекулярный объект (в виде клонов)
один за другим исследуется на наличие у него требуемых свойств,
при этом его выбор из клонотеки происходит "вслепую", ничем
не ограничивается и является случайным перебором.
Улучшенный скрининг становится возможным в том случае,
если объекты, составляющие клонотеку, различаются феноти-
пически, например, по наличию у них потенциальной фермента-
ферментативной активности. Примером улучшенного скрининга является
обнаружение окрашенных бактериальных колоний, что может
быть следствием экспрессии в бактериальных клетках искомого
фермента. Хотя при таком качественном тесте все равно прихо-
приходится анализировать все объекты клонотеки, которые будут за-
затеряны среди ненужных представителей (в нашем примере -
большого числа неокрашенных бактериальных клеток), улуч-
улучшенный скрининг существенно облегчает процесс поиска требу-
требуемых молекулярных объектов. Тем не менее, эта группа мето-
методов, как правило, характеризуется низкими значениями отноше-
отношения сигнала к шуму, а также имеет место конкуренция (в том чис-
числе и неспецифическая) за ресурсы (в частности, компоненты пи-
питательной среды или субстрат реакции) со стороны ненужных
компонентов клонотеки, что может блокировать проявление фе-
нотипического признака.
В системах, основанных на истинном отборе или селекции,
создаются условия, обеспечивающие избирательное сохранение
компонентов клонотеки, которые обладают определенными
свойствами. Например, при отборе конкретных представителей
клонотек на основе микроорганизмов в систему можно ввести се-
селектирующий агент, например антибиотик, в присутствии кото-
которого будут выживать лишь те объекты, которые экспрессируют
ген устойчивости к этому антибиотику. Поскольку системы се-
селекции обладают наибольшей разрешающей способностью, да-
далее будут подробнее рассмотрены принципы действия некоторых
современных вариантов таких систем, которые эффективно ис-
используются для получения белковых молекул методом направ-
направленной эволюции.
2.3.1. Фаговый дисплей
Метод фагового дисплея, разработанный Г.П. Смитом в
1985 г., представляет собой простую и эффективную систему от-
отбора белков и пептидов с требуемыми свойствами, экспрессиро-
ванных на поверхности частиц нитевидных бактериофагов [88].
Возможность эффективного использования фагового дисплея
333
для проведения направленной эволюции белковых молекул опре-
определяется наличием в системе прямой связи между аминокислот-
аминокислотной последовательностью исследуемого белка и кодирующей ее
последовательностью нуклеотидов генома бактериофага, т.е.
между генотипом фаговой частицы и ее фенотипом [89]. Это до-
достигается клонированием целевой нуклеотидной последователь-
последовательности в составе гибридной последовательности, которая, кроме
нее, включает ген одного из белков фаговой оболочки. Во время
фаговой инфекции в результате экспрессии гибридного гена про-
происходит образование химерного белка, который встраивается в
оболочку бактериофага, а исследуемая полипептидная цепь экс-
экспонируется наружу фаговой частицы и может быть изучена на
предмет наличия у нее определенных свойств. Использование
при клонировании различных (например, измененных под дейст-
действием мутаций) нуклеотидных последовательностей приводит к
созданию клонотеки таких последовательностей в составе фаго-
фаговых частиц и одновременно клонотеки полипептидов, в которой
каждый гибридный полипептид не теряет связи с кодирующим
его геном. Это дает возможность при необходимости продолжать
работу с такой искусственной генетической системой, как на ген-
генном уровне, так и с самим очищенным белком, после заражения
бактериальных клеток отдельной фаговой частицей и очистки
бактериофага.
Процедура отбора требуемой фаговой частицы из клонотеки
проста. В том случае, если ведется отбор на способность гибрид-
гибридного белка взаимодействовать с конкретным иммобилизованным
лигандом, после инкубации суспензии фаговых частиц с сорбен-
сорбентом несвязавшиеся частицы отмывают, а оставшиеся в комплек-
комплексе с лигандом бактериофаги после элюирования используют для
заражения бактериальных клеток. Амплифицированные таким
образом гибридные гены являются исходным материалом в но-
новом раунде мутагенеза и селекции. Эта и некоторые другие воз-
возможности отбора будут подробнее рассмотрены ниже.
Бактериофаги. При конструировании клонотек нуклеотид-
нуклеотидных последовательностей для фагового дисплея чаще всего ис-
используют хромосомы нитевидных колифагов М13, fl и fd, а так-
также фагмиды, полученные на их основе. Эти фаги заражают муж-
мужские клетки Е. coli через половые ворсинки (F-пили), которые со-
содержат белковые рецепторы, обеспечивающие их сорбцию. Хро-
Хромосома фагов представляет собой одноцепочечную, кольцевую,
ковалентно замкнутую ДНК, которая заключена внутри цилинд-
цилиндрической белковой оболочки, обеспечивающей выживание виру-
вирусов в экстремальных условиях: при значениях рН 2,5-11 и темпе-
334
ратурах до 80°С. Оболочка построена из белков нескольких ти-
типов. Основной белок оболочки g8p E,2 кДа), представленный
~2700 копиями, формирует цилиндр вокруг молекулы ДНК.
Верхняя часть цилиндра закрыта белками g7p и g9p C,6 кДа),
присутствующими в пяти копиях каждый. Противоположный ко-
конец закрывается пятью молекулами белков g6p A2,3 кДа) и g3p
D2,5 кДа), которые обеспечивают адсорбцию фагов на половых
ворсинках бактериальных клеток. Полипептидная цепь белка
?3р, часто используемого в качестве носителя при построении
клонотек на основе фагового дисплея, состоит из трех доменов,
разделенных повторяющимися последовательностями, обога-
обогащенными остатками Gly. N-Концевой домен (остатки 1-67) и
центральный домен (остатки 87-217), пространственная структу-
структура которых определена, обеспечивают инфекционность фаговых
частиц [90, 91]. С-Концевой домен (остатки 257-406) удерживает
белок в составе оболочки [92].
Нитевидные бактериофаги не вызывают лизиса бактериаль-
бактериальных клеток во время инфекции, но постоянно выходят из них че-
через пору диаметром ~8 нм во внешней мембране клетки. Размер
поры обеспечивает прохождение одной фаговой частицы. Во
время переноса через внутреннюю бактериальную мембрану од-
ноцепочечная геномная ДНК покрывается белками оболочки,
которые локализованы во внутренней мембране. При этом их
N-концы экспонированы в периплазматическое пространство, а
С-концы контактируют с цитоплазмой. Белки g8p и g3p первона-
первоначально синтезируются в виде предшественников, содержащих
N-концевую сигнальную последовательность, которая удаляется
после их внедрения в мембрану.
Хотя все пять белков оболочки уже были использованы для
получения гибридных белков фагового дисплея, g3p и g8p оста-
остаются наиболее популярными объектами таких исследований.
При этом g8p чаще всего применяются для экспонирования ко-
коротких пептидов на поверхности бактериофага, тогда как g3p
объединяют, в том числе, и с крупными глобулярными белками.
Выбор определяется тем, что включение больших полипептид-
полипептидных цепей в состав g8p увеличивает диаметр фаговой частицы,
что препятствует их прохождению через поры в бактериальной
мембране.
Когда полная замена исходного белка оболочки на гибрид-
гибридный летальна для бактериофага, гибридный белок нарабатыва-
нарабатывают в бактериальных клетках под контролем фагмиды, а белок
дикого типа обеспечивают заражением бактериальных клеток,
содержащих фагмиду, фагом-помощником. В такой системе со-
335
держание гибридного белка g3p в фаговых частицах поддержива-
поддерживается на уровне одной копии (вместо трех-пяти, как обычно), что
бывает необходимым для отбора рецепторов, обладающих осо-
особенно высоким сродством к лиганду. Иногда для преодоления
токсического действия гибридных белков их экспрессию осуще-
осуществляют под контролем промоторов фагового шока, которые ак-
активируются только после заражения бактериальных клеток фа-
фагом-помощником [93].
В фагмидных экспрессирующих векторах на месте стыковки
гена белка оболочки и клонируемого гена часто помещают ко-
дон терминации трансляции. При таком устройстве вектора сис-
система дисплея начинает функционировать только в супрессорных
штаммах Е. coli, а в отсутствие супрессии в бактериальных клет-
клетках синтезируется исследуемый белок клонотеки. Этот подход
исключает необходимость переклонирования изучаемой после-
последовательности нуклеотидов после отбора белка с нужными
функциями с помощью фагового дисплея. Однако ограниченная
эффективность супрессии может сделать уровень экспрессии ги-
гибридного белка на поверхности фаговых частиц очень низкой.
Кроме обсуждавшихся выше нитевидных бактериофагов в
фаговом дисплее могут быть использованы и другие колифаги, в
том числе X и Т4 [94, 95]. Но из-за более низкой эффективности
экспрессии гибридных белков оболочки на поверхности капсидов
этих фагов, а также трудностей, связанных с получением репре-
репрезентативных клонотек нуклеотидных последовательностей на
основе их хромосом, такие системы пока не находят широкого
распространения.
Фаговый дисплей в исследовании коротких пептидов и эпито-
пов. В живом организме большинство биологических процессов
управляется посредством специфических белок-белковых или
белково-нуклеиновых взаимодействий. К таким процессам отно-
относятся, например, регуляция транскрипции генов под действием
различных белковых факторов, взаимодействие белковых лиган-
дов с рецепторами на поверхности клеток, а также специфичес-
специфическое связывание антигенов соответствующими антителами. Пони-
Понимание молекулярных механизмов взаимодействия белковых ли-
гандов с рецепторами имеет большое фундаментальное и при-
прикладное значение. В частности, разработка новых лекарствен-
лекарственных препаратов белковой природы обычно начинается с иденти-
идентификации исходной последовательности аминокислот, обладаю-
обладающей требуемой биологической активностью (так называемая
"основная" (lead) последовательность). Однако пептиды с основ-
основной последовательностью аминокислот могут обладать и неже-
336
дательными биологическими свойствами: низкой активностью,
токсичностью, малой стабильностью в организме и т.п.
До появления клонотек пептидов улучшение их биологичес-
биологических свойств осуществляли путем последовательного синтеза
большого числа аналогов и проверкой их биологической актив-
активности, что требовало больших затрат времени и средств. В по-
последние годы появилась возможность с помощью автоматичес-
автоматических синтезаторов создавать за короткое время тысячи различных
пептидов. Разработанные методы направленного мутагенеза так-
также позволили резко расширить число белков, получаемых одно-
одновременно и последовательно тестируемых на биологическую ак-
активность. Однако только недавно разработанные подходы к со-
созданию клонотек пептидов привели к получению миллионов по-
последовательностей аминокислот, требуемых для проведения эф-
эффективного скрининга с целью выявления пептидов, максималь-
максимально удовлетворяющих предъявляемым критериям. Такие клоно-
теки используются для исследования взаимодействия антител с
антигенами, получения новых ингибиторов ферментов и антими-
антимикробных агентов, конструирования молекул, обладающих требу-
требуемой биологической активностью, или придания новых свойств
белкам, например, антителам.
По способам получения клонотеки пептидов разделяются на
три группы [95а]. К первой группе можно отнести клонотеки хи-
химически синтезированных пептидов, в которых индивидуальные
пептиды иммобилизованы на микроносителях. При таком подхо-
подходе после присоединения очередных аминокислот в индивидуаль-
индивидуальных реакционных смесях к пептидам, иммобилизованным на ми-
микроносителях, содержимое всех реакционных смесей объединя-
объединяют и разделяют на новые порции, которые применяют на следу-
следующей стадии присоединения новых аминокислотных остатков.
После проведения ряда таких этапов оказываются синтезирован-
синтезированными пептиды, содержащие последовательности использован-
использованных в синтезе аминокислот во всевозможных случайных сочета-
сочетаниях. Клонотеки пептидов, иммобилизованных на микроносите-
микроносителях, обладают существенным недостатком: они требуют при
скрининге очищенных препаратов рецепторов, находящихся в
растворимой форме. В то же время в большинстве случаев при
биологических испытаниях, проводящихся для фундаментальных
и фармакологических исследований, чаще всего применяют ре-
рецепторы, ассоциированные с мембранами.
Кроме того клонотеки пептидов могут быть получены с по-
помощью твердофазного синтеза пептидов, при котором на каждой
стадии химического присоединения очередной аминокислоты к
337
растущим пептидным цепям используют эквимолярные смеси
всех или некоторых аминокислот-предшественников. На конеч-
конечной стадии синтеза проводят отделение пептидов от носителя,
т.е. переводят их в растворимую форму. Третий подход к конст-
конструированию клонотек пептидов стал реальным именно благода-
благодаря развитию методов генной инженерии. Он прекрасно иллюст-
иллюстрирует возможности таких методов и, несомненно, является
крупным достижением в их применении. В этой связи рассмот-
рассмотрим более подробно результаты использования клонотек пепти-
пептидов в исследовании эпитопов (антигенных детерминант) белков.
Генно-инженерная технология получения гибридных белков
позволила разработать эффективный метод наработки коротких
пептидов для анализа их биологической активности. Как и в слу-
случае клонотек генов, клонотеки пептидов, полученные генно-ин-
генно-инженерными методами, представляют собой большой (часто ис-
исчерпывающий) набор коротких пептидов. Недавно сделанные
наблюдения позволяют рассматривать клонотеку пептидов одно-
одновременно и в качестве клонотеки эпитопов белков. Во-первых,
короткие пептиды могут включать все основные остатки амино-
аминокислот, играющие главную роль во взаимодействии с антитела-
антителами, и они в состоянии имитировать крупные антигенные детер-
детерминанты белков. Во-вторых, в большинстве случаев нековалент-
ные связи, образующиеся между немногими наиболее важными
остатками аминокислот белковых лигандов и их рецепторами,
вносят основной вклад в общую энергию взаимодействия лиганд-
рецептор. С учетом этого любой пептид можно рассматривать
как потенциальный лиганд, гаптен или часть антигенной детер-
детерминанты более крупных полипептидов, а любую клонотеку пеп-
пептидов - как клонотеку эпитопов белков или потенциальных ли-
лигандов для соответствующих белковых рецепторов.
Клонотека пептидов, полученная в результате реализации
третьего подхода, в современном виде представляет собой набор
десятков или даже сотен миллионов коротких различающихся
последовательностей аминокислот, которые экспрессированы на
поверхности вирионов бактериофагов в составе их собственных
структурных белков. Это становится возможным благодаря вве-
введению в геном бактериофагов гибридных рекомбинантных ге-
генов, кодирующих измененные структурные белки его вирионов.
В результате экспрессии таких генов образуются гибридные бел-
белки, на N- или С-концах которых (см. ниже) присутствуют допол-
дополнительные последовательности аминокислот. В наиболее хоро-
хорошо разработанной системе, позволяющей конструировать клоно-
клонотеки пептидов генно-инженерными методами, используют не-
338
Олигонуклеотидная
вставка
pVIII-Эпитоп
Олигонуклеотидная
вставка
#
Л
\J
рШ-Эпитоп
Рис. 45. Экспрессия пептидных эпитопов на поверхности оболочки ни-
нитевидных колифагов [95а]
Пептидные эпитопы находятся в составе гибридных полипептидных цепей
минорного белка рШ (а) или основного белка pVIII вирусной оболочки (б).
Стрелки указывают положение кодирующих эпитопы олигонуклеотидных
фрагментов в геноме бактериофага, а также локализацию самих эпитопов. В
составе полипептида рШ (а) показана только одна копия эпитопа (на самом де-
деле их число достигает 4-5)
большой нитевидный колифаг f 1 и два его белка: основной и ми-
минорный белки оболочки pVIII и pill [95a]. In vivo оба белка синте-
синтезируются в виде полипептидных цепей с короткими N-концевы-
ми сигнальными последовательностями, которые отщепляются
сигнальной пептидазои во время их созревания после переноса к
внутренней части бактериальной мембраны. Зрелые белки
встраиваются в оболочку бактериофага в процессе ее сборки.
При этом белок pVIII образует основную оболочку бактериофа-
339
га, тогда как четыре или пять молекул рШ ассоциированы с кон-
концевой частью вириона и обеспечивают взаимодействие вирусных
частиц с половыми ворсинками клеток Е. coli (рис. 45). Генно-ин-
Генно-инженерными методами пептиды соединяют с белками - непосред-
непосредственно с их N-концевыми последовательностями или на неболь-
небольшом от них расстоянии. Концевые последовательности большин-
большинства белков являются более гибкими и, как правило, экспониру-
экспонируются на поверхности глобулы, что позволяет получать гибрид-
гибридные рекомбинантные белки без существенного нарушения их ос-
основных свойств, а также делает интегрируемые пептиды доступ-
доступными для распознавания извне. Кроме того, в таком положении
и пространственная структура самих пептидов испытывает мень-
меньшее влияние белка-носителя. В ходе экспериментов было уста-
установлено, что введение чужеродных пептидов в N-концевую часть
белка рШ не оказывает существенного влияния на жизнеспособ-
жизнеспособность и инфекционность фаговых частиц, тогда как соединение
пептидов длиной >5 аминокислотных остатков с N-концевой час-
частью белка pVIII нарушает сборку вирионов. Последнее затрудне-
затруднение можно преодолеть доставкой к месту сборки вирионов моле-
молекул белка pVIII дикого типа, синтез которых направляется соот-
соответствующим геном вируса-помощника. В этом случае оболочка
бактериофага будет содержать как измененные белки pVIII, так
и полипептиды дикого типа от вируса-помощника.
При конструировании клонотеки пептидов прежде всего син-
синтезируют два комплементарных друг другу олигонуклеотида, об-
образующих после отжига двухцепочечную молекулу, центральная
часть которой кодирует собственно пептиды (рис. 46, я), а высту-
выступающие по концам одноцепочечные участки комплементарны
"липким" концам вектора, получающимся под действием соот-
соответствующей рестриктазы (рис. 46, б).
Для кодирования аминокислот пептидов используют вырож-
вырожденные кодоны вида NNK или NNS, которые включают все че-
четыре нуклеотида (N) в первом и втором положениях, G или Т(К),
а также G или C(S) в третьем положении. При таком подходе ин-
информация о всех 20 аминокислотах и одном стоп-кодоне заклю-
заключена в 32 различных кодонах NNK и NNS, а не в 64, как это име-
имеет место в случае природного генетического кода.
В процессе синтеза вырожденных олигонуклеотидов, кодиру-
кодирующих исследуемые пептиды, на каждой стадии используют инди-
индивидуальные нуклеотиды для кодонов инвариантных аминокис-
аминокислот, фланкирующих вариабельный участок пептида, а также эк-
вимолярные смеси нуклеотидов для участков, кодирующих слу-
случайные последовательности. Образовавшийся в итоге набор вы-
340
Вырожденный GGGCT (NNK) 6GGGGCCGCTG
олигонуклеотид tgccccga (nnm) 6ccccggc
Вектор S*l+ W
— CTATTCTCACTCGGCCGACG TGGCCTGGCC^
— GATAAGAGTGAGCCGGC TGGACCGGACCGGJ
CTCGGGCCGAAACTGTTGAA —
.GACCCCGGCTTTGACAACTT —
в Вектор
со вставкой
— CTATTCTCACTCGGCCGACGCfGGCT (NNK) 9GGGGCCGCI?tGGCCGAAACTGTTGAA
— GATAAGAGTGAGCCGGCjTGCCCCGA (NNK) 9CCCCGGCGACCCCGGCTTTGACAACTT —
I
Гибридный
pill NH2-A DGAX6 GAAGA
ETVE-
Отсутствует у pill дикого типа
Рис. 46. Создание рекомбинантного вирусного генома, содержащего
вставки вырожденных олигонуклеотидов, для получения клонотеки
эпитопов [95а]
Двухцепочечный олигонуклеотид (а), содержащий вырожденные кодоны
NNK и те же самые сайты рестрикции в составе линкеров, лигируют с ДНК век-
вектора Fuse5 (б), расщепленного рестриктазой Sfil, с образованием рекомбинантно-
рекомбинантного генома (в), который направляет синтез гибридного рекомбинантного белка (г),
содержащего на N-конце указанную аминокислотную последовательность
рожденных олигонуклеотидов далее клонируют в виде одноцепо-
чечных фрагментов в соответствующих сайтах гена белка обо-
оболочки бактериофага в составе фагового вектора или фазмиды.
Альтернативно для такого набора олигонуклеотидов (химически
или с помощью ПЦР) синтезируют комплементарные цепи с
включением инозина в вариабельные участки, поскольку его ос-
остатки, как известно, спариваются с основаниями С и Т матрицы,
что облегчает образование правильных дуплексов между соот-
соответствующими олигонуклеотидами. Образующиеся двухцепо-
чечные олигонуклеотиды в случае необходимости обрабатыва-
обрабатывают требуемыми рестриктазами и клонируют в фаговом векторе.
Итоговые рекомбинантные молекулы (см. рис. 46, в) ДНК вводят
в бактериальные клетки, получая ~109 трансформантов на 1 мг
рекомбинантной ДНК, образовавшиеся фаговые частицы раз-
размножают в бактериях и после очистки анализируют на присутст-
присутствие рекомбинантных пептидов (см. рис. 46, г), способных взаимо-
взаимодействовать с исследуемыми рецепторами в белках их вирионов.
Число индивидуальных фаговых клонов в клонотеке является
определяющим для ее использования. Например, клонотека,
включающая все возможные гексапептиды, должна содержать
64 млн B06) разных шестичленных аминокислотных последова-
341
тельностей, кодируемых ~1 млрд C26) различных гексакодонов
C2 - число кодонов, с помощью которых можно закодировать лю-
любую из 20 аминокислот с использованием кодонов NNK или NNS).
Для решения такой задачи необходимо получать очень большие
клонотеки, содержащие, по крайней мере, 2 • 108- 3 • 108 индивиду-
индивидуальных независимых клонов, а величина 109 в настоящее время яв-
является верхним пределом для числа индивидуальных клонов кло-
клонотеки, которую еще можно практически использовать.
Исходя из этого, можно заключить, что максимальная длина
пептидов, включающих в себя все возможные сочетания 20 ами-
аминокислот, с которыми возможно работать с помощью клонотек
пептидов, составляет шесть аминокислотных остатков. Тем не
менее, следует иметь в виду, что клонотека 15-членных пептидов
того же размера B-3 • 108 клонов) будет содержать больше раз-
разнообразных гексапептидов, чем рассмотренная выше клонотека
6-членных пептидов. Кроме того, поскольку лишь ограниченное
число аминокислотных остатков в пептиде действительно опре-
определяет его биологическую активность, клонотека 15-членных
пептидов может оказаться представительнее клонотеки более
коротких пептидов с тем же числом клонов.
Для того, чтобы выделить из клонотеки пептиды с искомой
биологической активностью, применяют различные методы
скрининга. В частности, для выделения пептидов, имитирующих
определенные эпитопы, используют биотинилированные моно-
клональные антитела (mAb) соответствующей специфичности,
которые иммобилизуют на твердой подложке с помощью стреп-
тавидина (рис. 47). Фаговые частицы, экспрессирующие на своей
поверхности соответствующие эпитопы, взаимодействуют с ан-
антителами и задерживаются подложкой, тогда как другие реком-
бинантные фаговые частицы удаляются в процессе промывания.
Задержанные на подложке фаговые частицы элюируют буфе-
буфером с низкими значениями рН, индивидуальные клоны дополни-
дополнительно размножают в бактериальных клетках, и экспрессирован-
ные на них эпитопы исследуют по различным критериям. Нали-
Наличие идентичных или сходных последовательностей нуклеотидов
среди клонированных последовательностей свидетельствует о
специфичности процесса очистки. Индивидуальные клоны затем
охарактеризовывают другими, в частности иммуноферментными
методами. На заключительной стадии осуществляют синтез вы-
выделенных пептидов и их всестороннее изучение в очищенном со-
состоянии.
Результаты, полученные при скрининге пептидных клонотек
на основе фагового дисплея, можно разделить на две группы [96].
342
Олигонуклеотидная
вставка,
кодирующая
эпитоп
Эпитоп
Биотинилированное
Чашка Петри, покрытая
стрептавидином
Рис. 47. Схема отбора фаговых частиц, обладающих требуемыми эгти-
топами
Показаны три рекомбинантных фаговых частицы, экспрессирующие раз-
разные эпитопы в составе белка рШ. Только эпитоп центральной фаговой части-
частицы распознается молекулой биотинилированного антитела, иммобилизованно-
иммобилизованного на чашке Петри с помощью стрептавидина и использованного для скринин-
скрининга клонотеки
Прежде всего были получены многочисленные пептиды, имити-
имитирующие линейные эпитопы белковых молекул, т.е. соответству-
соответствующие группе аминокислотных остатков, расположенных подряд
друг за другом в полипептидной цепи молекулы антигена. В од-
одной из таких работ из клонотеки были выделены пептиды, после-
последовательность аминокислот которых резко отличалась от после-
последовательности аминокислот истинного эпитопа исследуемого ан-
антигена. Тем не менее, такой пептид прочно связывался со специ-
специфическими антителами и конкурировал за связывание с природ-
природным антигеном. Это позволило сделать вывод о возможности су-
343
ществования мимотопов - коротких пептидов, имитирующих
природные эпитопы, последовательности аминокислот которых
существенно различаются между собой. Удалось установить ка-
канонические последовательности аминокислот пептидов, имити-
имитирующих эпитопы природных белков, а среди них идентифициро-
идентифицировать аминокислотные остатки, играющие ключевую роль во вза-
взаимодействии антиген-антитело.
Одним из многообещающих приложений клонотек пептидов
является идентификация пептидных лигандов, имитирующих
"структурные" эпитопы, образующиеся на поверхности белко-
белковых глобул в результате сворачивания полипептидных цепей, что
сопровождается пространственным сближением аминокислот-
аминокислотных остатков, расположенных в полипептидной цепи на значи-
значительном расстоянии друг от друга. С помощью пептидных клоно-
клонотек возможна идентификация пептидных аналогов различных
эпитопов небелковой природы. По-видимому, в ближайшем бу-
будущем возможно использование пептидных клонотек для полу-
получения новых лекарственных препаратов, создания диагностичес-
диагностических средств и производства эффективных вакцин. В области кон-
конструирования новых лекарственных препаратов усилия исследо-
исследователей могли бы быть направлены на создание пептидных ли-
лигандов, специфически взаимодействующих с рецепторами, пред-
представляющими медико-биологический интерес. Знание структуры
таких лигандов позволило бы упростить получение на этой осно-
основе лекарственных препаратов небелковой природы.
Клонотеки пептидов и эпитопов найдут свое применение и в
исследованиях механизмов гуморального иммунного ответа, а
также заболеваний иммунной системы. В частности, большинст-
большинство аутоиммунных заболеваний сопровождается образованием ау-
аутоантител против антигенов собственного организма. Эти анти-
антитела во многих случаях служат специфическими маркерами того
или иного аутоиммунного заболевания. С использованием клоно-
клонотеки эпитопов, в принципе, можно получать пептидные маркеры,
с помощью которых было бы возможно следить за специфично-
специфичностью аутоантител во время развития патологического процесса
как в индивидуальном организме, так и в группе пациентов и,
кроме того, определять специфичность аутоантител при заболе-
заболеваниях неизвестной этиологии.
Клонотеки пептидов и эпитопов потенциально могут быть
использованы также для скрининга иммунных сывороток для вы-
выявления пептидов, специфически взаимодействующих с защит-
защитными антителами. Такие пептиды будут имитировать антиген-
антигенные детерминанты патогенных организмов и служить мишенями
344
для защитных антител организма. Это позволит использовать по-
подобные пептиды для вакцинации пациентов, у которых отсутст-
отсутствуют антитела против соответствующих патогенов. Изучение
эпитопов с помощью клонотек пептидов является частным слу-
случаем одного из многочисленных направлений их использования в
прикладных и фундаментальных исследованиях взаимодействия
лигандов и рецепторов. Дальнейшее усовершенствование этого
подхода должно способствовать созданию новых лекарственных
препаратов на основе коротких пептидов и быть полезным в ис-
исследованиях механизмов белок-белковых взаимодействий.
Фаговый дисплей в исследовании ферментов. С 1991 г. с по-
помощью фагового дисплея было исследовано более 30 ферментов
[97]. Поскольку перед встраиванием в фаговую оболочку гибрид-
гибридные белки должны быть как минимум перенесены в периплазма-
тическое пространство бактериальных клеток, наибольшей эф-
эффективности фагового дисплея удается достичь с секретируемы-
ми белками: протеиназами, бактериальными секретируемыми и
периплазматическими ферментами, антителами и др. Помимо
этого ограничения имеется немало и других узких мест, которые
мешают эффективному использованию системы для изучения
полноразмерных полипептидных цепей ферментов.
Прежде всего, это необходимость правильного фолдинга ис-
исследуемого полипептида в составе гибридной молекулы, что
особенно проблематично в том случае, когда белок имеет не-
небактериальное происхождение. Кроме того, рекомбинантный
белок, будучи экспонированным на поверхности фаговых час-
частиц, должен обладать достаточной стабильностью. Эти и мно-
многие другие проблемы, связанные с недостаточной эффективно-
эффективностью экспрессии гибридных генов в системе фагового дисплея,
должны решаться с использованием общих подходов к оптими-
оптимизации экспрессии рекомбинантных генов в бактериальных сис-
системах, рассмотренных в первой части книги. С помощью подхо-
подходящих бактериальных штаммов и условий культивирования
можно решать многие из этих проблем. Тем не менее, при отбо-
отборе ферментов с требуемыми свойствами чаще всего приходится
пользоваться оригинальными подходами, к счастью опирающи-
опирающимися в своей основе на общие принципы, которые будут кратко
перечислены ниже.
Отбор по связыванию с лигандом. Анализ популяции фа-
фаговых частиц на наличие на их поверхности белков, взаимодей-
взаимодействующих с ингибитором исходного фермента или монокло-
нальными антителами (mAb) соответствующей специфичности,
может быть как самостоятельной задачей, так и хорошим мето-
345
дом исследования качества самой фаговой клонотеки. Фаги ин-
инкубируют с иммобилизованным ингибитором или mAb и элюи-
руют тем же ингибитором в условиях пониженного его сродст-
сродства к ферменту, например, в результате изменения значений рН.
Отношение числа связавшихся фаговых частиц из клонотеки к
количеству связавшихся частиц из популяции немодифициро-
ванных фагов рассматривают как меру правильности фолдинга
гибридного фермента, а также качества фагового дисплея вооб-
вообще. Отбор по степени связывания лиганда оказывается полез-
полезным при попытках получения методами белковой инженерии
дополнительных сайтов связывания в рекомбинантном фермен-
ферменте с целью осуществления аллостерической регуляции его ак-
активности. Такие модифицированные молекулы могут использо-
использоваться в качестве биосенсоров для количественной идентифика-
идентификации конкретных лигандов.
Отбор на основании ферментативной активности. Отбор
частиц фагового дисплея на основании ферментативной активно-
активности исследуемых гибридных белков требует от экспериментато-
экспериментатора большего искусства по сравнению с только что рассмотрен-
рассмотренным. В этом случае необходимо найти способ, который бы учи-
учитывал способность фермента метаболизировать субстрат и осо-
особенности его выделения из сложной смеси фаговых частиц.
В простом случае, когда объектом поиска является мутант-
ный фермент с измененной субстратной специфичностью, его
можно отделить от остальных производных на колонке с иммо-
иммобилизованным аналогом субстрата, который должен специфи-
специфически распознаваться этим, но не исходным ферментом. Отбор с
использованием ингибиторов, полученных на основе аналогов
субстратов, был успешно применен, в частности, для мутантных
производных нуклеазы стафилококков с целью изменения ее
специфичности. Нуклеаза стафилококков является Са2+-зависи-
мой фосфодиэстеразой, гидролизующей ДНК с небольшим пред-
предпочтением к остаткам тимидина на 5'-конце расщепляемой связи.
Из клонотеки, содержащей мутагенизированный с помощью
ПЦР ген нуклеазы, выделяли ферменты с повышенным сродст-
сродством к иммобилизованным олигонуклеотидам, построенным из
фосфоротиоатных аналогов тимидина или гуанидина. Такие про-
производные олигонуклеотидов устойчивы к действию данной нук-
нуклеазы. Некоторые производные нуклеазы, выделенные с помо-
помощью этого метода на тимидиновом субстрате, обладали анало-
аналогичной удельной активностью, что и фермент дикого типа, тогда
как удельная активность мутантных ферментов с повышенным
сродством к олиго(С) была в 10 раз ниже. Одновременно проис-
346
ходило ожидаемое изменение специфичности производных фер-
фермента [98].
Поскольку многие ферменты обладают большим сродством
к субстратам в переходном состоянии, именно иммобилизован-
иммобилизованный аналог субстрата, имитирующий его переходное состояние,
используют в качестве лиганда при аффинной хроматографии. В
основе этой концепции лежит одно из главных положений тео-
теории ферментативного катализа, впервые выдвинутое Л. Полин-
гом, в соответствии с которым активный центр фермента должен
быть в большей степени комплементарен форме субстрата, нахо-
находящейся в переходном состоянии, т.е. претерпевшей предвари-
предварительные структурные перестройки на пути превращения в про-
продукт реакции, но не в основном исходном состоянии [99].
Третьим широко используемым способом отбора является
введение аффинной метки в активный центр фермента в со-
составе фаговой частицы с последующим отбором меченых частиц.
Разновидностью этого подхода является использование субстра-
субстрата, метаболизируя который, фермент совершает самоубийство,
так как в результате ферментативной активации субстрат нео-
необратимо модифицирует активный центр фермента.
Отбор in vivo. Все рассмотренные выше подходы к отбору
белков и пептидов, экспонированных на поверхности фаговых
частиц, были основаны на применении селектирующих лигандов
вне организма, т.е. in vitro. Несмотря на большой, во многом уже
реализованный потенциал этой группы методов, их использова-
использование имеет весьма существенные ограничения. Например, для ле-
лечения многих сердечно-сосудистых заболеваний, включая тром-
тромбозы, атеросклероз, воспаление при аутоиммунных реакциях ор-
организма, а также процессы атерогенеза в опухолях, необходима
адресная доставка лекарственных средств к клеткам эндотелия
кровеносных сосудов. Как и во всех других подобных случаях, из-
избирательное воздействие на эндотелий невозможно без исполь-
использования макромолекулярных мишеней, специфичных для такой
ткани. При этом характер экспрессии генов в клетках эндотелия
зависит от их локализации в организме, т.е. органной принадлеж-
принадлежности. Поиск молекулярных мишеней эндотелия затруднен не-
невозможностью сохранения специфической экспрессии его генов
в условиях культивирования in vitro. В культуре клеток или тка-
ткани эндотелий утрачивает свои специфические свойства. Все это
относится не только к эндотелию, но и ко многим другим специ-
специализированным (высокодифференцированным) тканям организ-
организма. Такое состояние дел стимулировало поиск маркеров непо-
непосредственно в живом организме.
347
Одним из таких подходов, имеющих прямое отношение к об-
обсуждаемой теме, является отбор белковых лигандов фагового
дисплея непосредственно в живом организме [100, 101]. В этом
случае суспензию фаговых частиц дисплея вводят в кровоток
экспериментального животного, через некоторое время живот-
животное забивают и из исследуемого органа выделяют находящиеся
там фаговые частицы. Выделенные фаги размножают в бактери-
бактериальных клетках и проводят новый раунд отбора. После несколь-
нескольких раундов удается выделять фаговые частицы, специфически
задерживаемые данным органом, а следовательно, экспрессиру-
ющие на своей поверхности лиганды, специфичные в отношении
рецепторов органа (или ткани). При этом остальные органы и
ткани организма осуществляют отрицательный отбор тех фаго-
фаговых частиц, которые экспрессируют лиганды универсальной спе-
специфичности и взаимодействуют со всеми органами и тканями ор-
организма. С использованием такой системы отбора пептидов фа-
фагового дисплея удалось получить пептидные аптамеры, специфи-
специфически взаимодействующие с многими тканями эксперименталь-
экспериментальных животных [102-105], в том числе с клетками гладких мышц
мышей в области рестеноза [106], а также эпителием атероскле-
ротических сосудов [107].
Антитела, полученные с помощью фагового дисплея на чис-
чистых мышиных моделях, часто реагируют с теми же антигенами
грызунов других видов и человека. Это редко удается наблюдать
при создании моноклональных антител (mAb) на основе гибри-
домной технологии, поскольку организм иммунизируемого жи-
животного должен быть толерантен к эпитопам с подобной эволю-
эволюционной консервативностью. Сама по себе консервативность та-
таких аминокислотных последовательностей может указывать на
их важное функциональное значение. Тем не менее, не всегда
специфичность создаваемых антител является универсальной и
распространяется на соответствующие антигены человека.
Одним из путей решения этой проблемы является использо-
использование мышей линий nude, SCID или SCID/NOD, которым транс-
трансплантируют ткани человека. Поскольку у мышей nude отсутству-
отсутствует тимус, мыши SCID не содержат Т- и В-лимфоцитов, а мыши
SCID/NOD дефицитны в отношении нормальных киллеров, реак-
реакции отторжения трансплантата у них резко снижены. Хотя кро-
кровеносные сосуды мышей прорастают в трансплантат, его сосуди-
сосудистая система не является полностью мышиной и сохраняет гене-
генетические маркеры организма-донора.
В одном из экспериментов мышам линии SCID была переса-
пересажена синовиальная оболочка человека от пациентов с ревмато-
348
лдным артритом или остеоартритом, а другим мышам - ткань ко-
кожи в качестве контроля. Мышам вводили внутривенно суспен-
суспензию фаговых частиц из клонотеки фагового дисплея, экспресси-
рующего семизвенные пептиды со случайной последовательнос-
последовательностью [108]. Через 15 мин после инъекции трансплантаты изолиро-
изолировали, гомогенизировали и использовали для выделения фаговых
частиц, которые далее размножали в бактериальных клетках и
подвергали еще двум дополнительным циклам отбора по той же
схеме. В результате были получены пептиды, специфически вза-
взаимодействующие с сосудистой системой синовиума, но не кожи,
которые рассматриваются в качестве перспективных лигандов
для адресной доставки лекарственных препаратов к пораженным
тканям. Отбор in vivo пептидов и белков, экспрессирующихся в
фаговом дисплее, может быть применен и для разработки новых
лигандов белковой природы, специфичных в отношении других
тканей, в том числе и опухолевых, что может быть использовано
для лечения соответствующих заболеваний.
2.3.2. Клеточный дисплей
Использование поверхности живых клеток в качестве дис-
дисплея для белков, подвергаемых отбору в ходе направленной эво-
эволюции, обладает рядом преимуществ перед применением для тех
же целей классического фагового дисплея [109,110]. Прежде все-
всего, клеточный дисплей дает возможность анализировать белки
значительно большего размера. Введение флуоресцентной мет-
метки в лиганды к рецепторам, экспрессирующимся на поверхности
клеток, позволяет эффективно использовать проточную цитоме-
трию для отбора соответствующих рецепторов. С помощью со-
современных цитометров можно анализировать и сортировать до
50 000 клеток/сек и, следовательно, проводить быстрый скрининг
больших комбинаторных клонотек белков и пептидов. При этом,
В отличие от фагового дисплея, на поверхности клеток может
акспрессироваться множество (до 104) молекул анализируемого
белка, что понижает отношение интенсивности сигнала к шуму и
Дает возможность более эффективно исключать ложноположи-
тельные результаты. Такие параметры систем на основе клеточ-
клеточного дисплея позволяют in situ количественно определять кон-
константы связывания и скорости диссоциации комплексов, форми-
формирующихся на поверхности клеток.
Все вышеперечисленные особенности делают клеточный
дисплей весьма привлекательным для белковой инженерии и не
только. Системы клеточного дисплея используют для производ-
349
ства живых вакцин, создания биоадсорбентов с целью удаления
из окружающей среды токсических веществ и ионов тяжелых ме-
металлов, получения биокатализаторов в виде целых клеток с им-
иммобилизованными на их поверхности ферментами, биосенсоров
для диагностики и мониторинга окружающей среды. В качестве
исходных клеток при создании клеточного дисплея применяют
клетки микроорганизмов (грамотрицательных и грамположи-
тельных бактерий, а также дрожжей) и животных.
Принцип действия клеточного дисплея заключается в экс-
экспрессии на поверхности клеток гетерологичных белков (белков-
пассажиров), которые отсутствуют у данного организма, объеди-
объединенных в составе гибридной молекулы с помощью пептидного
спейсера с полипептидной цепью белка-носителя, обеспечиваю-
обеспечивающего заякоривание всей конструкции в мембране клеток. При
этом используют гибридные белки трех типов, в которых белок-
пассажир находится на N-конце, С-конце или во внутренней час-
части белка-носителя в виде сэндвича. Для успешного выполнения
своих функций белки-носители должны отвечать, по крайней ме-
мере, четырем требованиям: 1) обладать эффективной сигнальной
или транспортной последовательностью, обеспечивающей про-
прохождение гибридного белка через внутренние мембраны клеток;
2) проявлять сильные якорные свойства для прочного удержива-
удерживания белка-пассажира на поверхности клеток; 3) должны быть
совместимыми с белками-пассажирами, т.е. не дестабилизиро-
дестабилизироваться после объединения с ними; 4) демонстрировать устойчи-
устойчивость к протеолитическим ферментам, присутствующим в пери-
плазматическом пространстве или культуральной жидкости. В
качестве векторов для генов гибридных белков используют экс-
прессирующие плазмиды или хромосомы вирусов.
Природа белка-пассажира также имеет большое значение
для эффективности функционирования клеточного дисплея и во
многих случаях является определяющей. Эти белки оказывают
сильное влияние на транслокацию (перенос) гибридных белков
через мембрану, их фолдинг и стабильность.
Функциональность клеточного дисплея в большой степени
зависит и от самих клеток-хозяев. Так, грамотрицательные бак-
бактерии из-за хрупкости внешней мембраны сильно затрудняют по-
получение клеточного дисплея. Тем не менее, клетки Е. coli исполь-
используются для этих целей благодаря наличию хорошо разработанно-
разработанного молекулярно-генетического инструментария, в том числе эф-
эффективных методик трансформации, а также охарактеризован-
охарактеризованных мутантов, которые облегчают применение Е. coli. Грамполо-
жительные бактерии, особенно В. subtilis и бактерии рода
350
Staphylococcus, особенно пригодны для создания биокатализато-
биокатализаторов и адсорбентов, так как обладают мембранами, достаточно ус-
устойчивыми к внешним воздействиям. Однако эти бактерии избе-
избегают использовать для скрининга клонотек белков и пептидов.
Для этой цели обычно берут клетки дрожжей S. cerevisiae, по-
поскольку в них системы фолдинга и секреции белков близки тако-
таковым клеток животных, они считаются безопасными для челове-
человека, условия их культивирования хорошо изучены и, кроме того,
заякоривание гибридных белков на мембранах в данном случае
можно осуществлять с помощью остатка гликозилфосфатидили-
нозитола и дисульфидных связей. Основываясь на этих принци-
принципах, клетки дрожжей используют для создания оральных вакцин
и других биотехнологических целей.
В целом, метод отбора эволюционирующих белков с помо-
помощью клеточного дисплея удобен, прежде всего, своей прозрачно-
прозрачностью, так как позволяет непосредственно in situ следить за кине-
кинетическими параметрами отбираемых макромолекул [111], что,
как правило, невозможно осуществить с помощью дисплеев, ос-
основанных на использовании субмикроскопических частиц - бак-
бактериофагов и рибосом. В этой связи данная область исследова-
исследований в настоящее время активно развивается.
2.3.3. Рибосомный дисплей и мРНК-дисплей
Одним из основных параметров, обеспечивающих успех ис-
использования комбинаторных клонотек нуклеотидных последова-
последовательностей в отборе тех из них, которые кодируют белки или
пептиды с требуемыми свойствами, является представительность
набора последовательностей в клонотеке. Трудно поймать чер-
черную кошку в темной комнате, особенно, если ее там нет. Найти
последовательность в клонотеке даже самыми изощренными ме-
методами можно лишь в том случае, если она в ней присутствует.
Репрезентативность клонотек, основанных на использовании
бактериальных, дрожжевых клеток или вирусов, зависит от эф-
эффективности трансформации или трансфекции клеток, верхний
предел которых ограничен 10ю и 107 последовательностями для
бактерий и дрожжей соответственно. Преодолеть ограничение
можно путем отказа от попыток введения рекомбинантных мо-
молекул ДНК внутрь клеток и перехода к работе непосредственно
с рекомбинантными последовательностями нуклеотидов в бес-
бесклеточных системах, что позволяет создавать клонотеки, содер-
содержащие 1012- 1015 разных последовательностей и эффективно ра-
работать с ними. В этом случае верхний предел размера и молеку-
351
лярного разнообразия клонотеки определяет количество моле-
молекул матричной ДНК, которую можно амплифицировать in vitro
(обычно 5-50 нмолей) в разумных объемах реакционных смесей
для проведения ПЦР @,1-1,0 л).
В настоящее время разработано два типа систем отбора нук-
леотидных последовательностей комбинаторных клонотек, ко-
которые основаны на их транскрипции, с последующей трансляци-
трансляцией образовавшихся мРНК в бесклеточной системе. В одной из си-
систем отбора первого поколения транскрибируемые последова-
последовательности были сконструированы таким образом, что у кодируе-
кодируемых ими мРНК отсутствовали кодоны терминации трансляции.
После завершения трансляции мРНК тройной комплекс мРНК-
рибосома-растущая полипептидная цепь не распадается, и каж-
каждый синтезированный таким образом полипептид остается в кон-
контакте с кодирующей его последовательностью нуклеотидов. Фе-
Фенотип оказывается прямо связанным с породившим его геноти-
генотипом. Поэтому нужные последовательности нуклеотидов можно
обнаруживать и выделять на основании свойств взаимодействую-
взаимодействующих с ними полипептидов, например, их хроматографических па-
параметров. Задача еще более упрощается при использовании бес-
бесклеточных систем второго поколения, в которых связь между
РНК и кодируемым ею полипептидом становится ковалентной, а
следовательно, повышается надежность связи между генотипом
и фенотипом эволюционирующих in vitro макромолекул.
Рибосомный дисплей. В системах с использованием рибосом-
ного дисплея цикл направленной эволюции белков выглядит сле-
следующим образом (рис. 48, а) [112-114]. Двухцепочечную ДНК ис-
исходной клонотеки нуклеотидных последовательностей, кодирую-
кодирующих различные мутантные формы исследуемого белка, транс-
транскрибируют in vitro, например, с помощью очищенной Т7-РНК-
полимеразы. Образовавшуюся мРНК, которая на 3'-конце не со-
содержит терминирующего кодона, транслируют в бесклеточной
белоксинтезирующей системе из ретикулоцитов кроликов. По-
После того, как работающие рибосомы достигают 3'-концов транс-
транслируемых мРНК, синтез белка прекращается без распада трой-
тройных комплексов мРНК-рибосома-синтезированный белок. Ком-
Комплексы дополнительно стабилизируют добавлением в реакцион-
реакционную смесь ионов Mg2+ и фракционируют на основании свойств
синтезированных полипептидов, например, с помощью аффин-
аффинной хроматографии. После чего очищенную мРНК используют в
качестве матрицы для обратной транскрипции с целью получе-
получения двухцепочечной кДНК, которую, в свою очередь, амплифи-
цируют с помощью ПЦР. Поскольку один из праймеров содер-
352
а
Рибосомный дисплей
Транскрипция in vitro
мРНК-дисплей
Транскрипция in vitro
мРНК
мРНК
, Трансляция in vitro
ДНК
Лигирование с линкером
J'
3' Нативный белок,
связанный с
рибосомой
Аффинная хроматография
ПЦР
Мутагенез
с помощью
ПЦР
I Элюирование мРНК
~ 3'
Обратная транскрипция
ПЦР
Мутагенез
с помощью
ПЦР
Трансляция in vitro i
5'
J' Гибридная ,,
мРНК
Пептидил-
трансферная
реакция
Очистка конъюгата i
мРНК с белком т
5'
Обратная транскрипция I
Аффинная хроматография
Элюирование кДНК
из комплекса
©0 ©
Рис. 48. Рибосомный дисплей и мРНК-дисплей [115]
а - в рибосомном дисплее связь между генотипом и отбираемым феноти-
фенотипом (между мРНК и кодируемым полипептидом) осуществляется нековалентно
непосредственно через рибосомные частицы;
б - в мРНК-дисплее мРНК и кодируемый полипептид связаны ковалентно
через молекулу пуромицина (Р), что значительно повышает эффективность си-
системы
12. Патрушев Л.И. Т. 1
353
жит последовательность промотора Т7-РНК-полимеразы, про-
продукт амплификации может быть снова транскрибирован in vitro,
а полученная мРНК задействована в новом цикле отбора по рас-
рассмотренной выше схеме.
мРНК-дисплей (дисплей на основе РНК-белковых конъюга-
тов). В 1997 г. Р. Робертсу и Дж. Шостаку удалось существенно
повысить эффективность рибосомного дисплея с помощью од-
одной замечательной модификации исходного метода [114—116]. В
их системе окончание трансляции мРНК in vitro сопровождалось
образованием ковалентной связи между С-концевым аминокис-
аминокислотным остатком синтезированного полипептида и 3'-концом
мРНК, в результате трансляции которой этот полипептид обра-
образовался. Связь между мРНК и пептидом оказывалась более проч-
прочной, чем в предыдущем случае, что облегчало их совместную
очистку и использование в направленной эволюции белковых
молекул по схеме, основные этапы которой совпадают с уже рас-
рассмотренными выше. При таком подходе к 3'-концам молекул
мРНК, синтезированным in vitro на первом этапе (рис. 48, б), при-
присоединяли короткий A9-30 нт) олигодезоксирибонуклеотидный
линкер, ковалентно связанный своим 3'-концом с молекулой пу-
пуромицина. Стандартный 30-звенный линкер -это 5'-(dAJ7dCdCP,
где Р - остаток пуромицина. Молекула пуромицина, классическо-
классического ингибитора биосинтеза белка, по своей структуре имитирует
аминоацилированную тРНК и после попадания в А-участок рибо-
рибосомы присоединяется рибосомой к НООС-группе С-концевого
аминокислотного остатка растущей цепи полипептида амидной
связью. Это сопровождается прекращением трансляции и осво-
освобождением недостроенной полипептидной цепи. Наличие в лин-
линкере олиго(ёА)-последовательности позволяет производить пол-
полное отделение свободной мРНК, оставшейся после завершения
трансляции, на олиго(ёТ)-колонках от конъюгатов для исключе-
исключения участия данных молекул в последующих раундах отбора.
Модифицированные мРНК нормально транслируются рибо-
рибосомами до тех пор, пока рибосомы не достигают границы РНК-
ДНК, где трансляция заканчивается распознаванием предпослед-
предпоследнего 3'-концевого кодона мРНК перед последовательностью
ДНК-линкера [114]. Остаток пуромицина, который находится на
3'-конце линкера, попадает в А-участок рибосомы и присоединя-
присоединяется к С-концевому аминокислотному остатку растущего поли-
полипептида по вышеописанной схеме. Это сопровождается образо-
образованием конъюгата синтезированного белка с кодирующей его
мРНК и освобождением конъюгата в реакционную смесь. Фено-
типический признак в виде структурных и функциональных осо-
354
бенностей синтезированного белка оказывается связанным с ге-
генотипом (информацией, заключенной в первичной структуре)
кодирующей его мРНК, поскольку в такой системе конъюгат
мРНК с пуромицином выполняет двойную функцию: матрицы и
акцептора строящегося полипептида. После выделения конъю-
гатов их можно использовать в последовательных циклах обога-
обогащения по исследуемому свойству полипептида, амплификации
матрицы в виде кДНК, ее транскрипции и трансляции в бескле-
бесклеточной системе. При этом этап отбора полипептида может быть
проведен как до синтеза первой цепи ДНК обратной транскрип-
тазой, так и после осуществления ее синтеза.
2.3.4. N-гибридные системы в исследовании белков
Специфические белок-белковые и белково-нуклеиновые
взаимодействия лежат в основе функционирования всех без ис-
исключения генетических систем. Не менее важную роль в дан-
данных процессах играют и взаимодействия между белками и низ-
низкомолекулярными соединениями. Поэтому исследование меха-
механизмов таких взаимодействий, а также идентификация молеку-
молекулярных партнеров, участвующих в формировании функциони-
функционирующих макромолекулярных комплексов, является важнейшей
задачей молекулярной биологии. Грандиозность задачи при-
привлекла в эту область большое количество исследователей и по-
породила обилие разработок, направленных на ее решение. Од-
Одним из методических прорывов последнего времени в изучении
белок-белковых взаимодействий было создание дрожжевой ди-
гибридной системы, которая с тех пор широко используется для
решения многих аналогичных проблем, в том числе и для отбо-
отбора рекомбинантных белков, полученных методами белковой
инженерии [117, 118].
Дрожжевая дигибридная система оценки белок-белковых
взаимодействий (Y2H). Система Y2H (yeast two-hybrid assay) была
разработана С. Филдсом и О. Сонгом в 1989 г. для исследования
белок-белковых взаимодействий в клетках дрожжей [119]. В по-
последующие годы система получила широкое распространение и
претерпела множество изменений и усовершенствований благо-
благодаря трем несомненным достоинствам, изначально присутствую-
присутствующим в этом подходе. Во-первых, метод позволяет прямо и непо-
непосредственно обнаруживать взаимодействие между двумя белками
в живой клетке. Во-вторых, функционирование системы не тре-
требует предварительных знаний о природе пары белков, которые в
ней исследуются, следовательно, система может быть использо-
12*
355
вана для изучения взаимодействий между любыми, в том числе и
ранее неизвестными белками. Наконец, данные об аминокислот-
аминокислотных последовательностях анализируемых белков можно легко
извлечь из нуклеотидных последовательностей рекомбинантных
плазмид, которые их кодируют, что существенно облегчает иден-
идентификацию белковых молекул.
Принцип действия системы Y2H основан на наблюдении, что
для восстановления активности эукариотических активаторов
транскрипции, два домена которых - активирующий (activation
domain, AD) и ДНК-связывающий (DNA binding domain, DBD) -
физически разделены путем разрыва полипептидной цепи, не
требуется восстановления разрыва. Активность белков в ряде
случаев можно восстановить просто смешивая домены друг с дру-
другом в растворе, в том числе и в комбинации с доменами других ак-
активаторов транскрипции [120,121].
Система Y2H включает в себя два гибридных белка: "приман-
"приманку" (bait) - X-AD и "хищника" (prey) - Y-DBD, каждый из них, в
свою очередь, построен из двух половин: белков X или Y, взаимо-
взаимодействие между которыми исследуется, а также одного из двух
доменов: активатора транскрипции DBD или AD (рис. 49, а). Кро-
Кроме того, интегральной частью системы Y2H является ген-репор-
ген-репортер, находящийся под контролем промотора, активируемого
фактором транскрипции с доменами AD и DBD. Если белки X и
Y взаимодействуют между собой, то их димеризация будет сопро-
сопровождаться сближением ассоциированных с ними доменов, что
приведет к образованию гетеродимера с активностью исходного
фактора транскрипции и активацией гена-репортера, экспрессию
которого легко обнаружить фенотипически, например, по фер-
ферментативной активности кодируемого этим геном белка. Плаз-
миду-"приманку" и плазмиду-"хищника" вводят в гаплоидные
клетки дрожжей, относящиеся к разным типам спаривания, а и а,
скрещивание между которыми приводит к образованию диплоид-
диплоидных штаммов, содержащих обе плазмиды.
Первоначально работоспособность системы Y2H была про-
продемонстрирована с использованием двух дрожжевых белков, фи-
физически взаимодействующих друг с другом in vivo. Были сконст-
сконструированы две плазмиды, каждая из которых кодировала гибрид-
гибридный белок, в одном случае, серин-треониновую протеинкиназу
SNF1, объединенную в одной полипептидной цепи с DBD-доме-
ном фактора транскрипции Gal4, а в другом - белок-активатор
протеинкиназы SNF4 с AD-доменом того же фактора. Последо-
Последовательность нуклеотидов, с которой специфически взаимодейст-
взаимодействует Gal4, поместили перед геном C-галактозидазы, а сам реком-
356
а
AD
б
Сайт, связывающий DBD Ген-репортер
Сайт, связывающий DBD Ген-репортер
Сайт, связывающий DBD
Ген-репортер
Рис. 49. Варианты «-гибридных дрожжевых систем, разработанные для
исследования взаимодействия белков друг с другом и различными ли-
гандами [118]
а) дигибридная система Y2H, б) моногибридная система Y1H; в и г - вари-
варианты тригибридной системы Y3H. AD - активирующий домен фактора транс-
транскрипции; DBD - ДНК-связывающий домен; X и Y - белки, между которыми ис-
исследуется взаимодействие; MS2 - белок оболочки фага MS2. В системе (в), раз-
разработанной для исследования взаимодействий белков с РНК, изучают гибрид-
гибридную РНК, у которой последовательность R всегда взаимодействует с белком
MS2, а под вопросом взаимодействие последовательности X с белком Y. В сис-
системе (г) использованы два лиганда, соединенных линкером; молекула слева яв-
является специфическим лигандом для белка X, а исследуется взаимодействие мо-
молекулы, расположенной справа, с белком Y
бинантный ген интегрировали в локус URA3 хромосомы дрож-
дрожжей. Обе плазмиды были введены в клетки данного штамма
дрожжей, после чего в них определяли активность C-галактози-
дазы стандартным калориметрическим методом. В контрольных
экспериментах было показано, что ни домены сами по себе, ни
каждый из химерных белков по отдельности не активировали
транскрипцию гена-репортера. Однако одновременное присутст-
присутствие в клетках дрожжей двух плазмид сопровождалось 200-крат-
200-кратным возрастанием внутриклеточной активности C-галактозида-
357
зы. Нативный фактор транскрипции Gal4 стимулировал экспрес-
экспрессию этого гена в 4000 раз.
Плазмиды, использованные в экспериментах, содержали по-
последовательности нуклеотидов, кодирующие домены AD и DBD,
были фланкированы последовательностями полилинкера и нахо-
находились под контролем промотора гена алкогольдегидрогеназы
ADH1, конститутивного промотора промежуточной силы. Обе
плазмиды относятся к семейству челночных векторов, которые
способны реплицироваться как в клетках дрожжей, так и Е. coli.
Репликацию в бактериальных клетках обеспечивает область на-
начала репликации ori pBR, а ген устойчивости к ампициллину Ыа
выполняет функции селектируемого маркера. В дрожжевых
клетках функционирует область начала репликации CEN4, обес-
обеспечивая поддержание плазмиды в клетках в виде 1-5 копий. В
этом случае в качестве селектируемых маркеров используются
гены TRP1 и LEU2, которые дают возможность ауксотрофным
мутантам дрожжевых клеток расти без добавления аминокислот
Тгр и Leu в питательную среду.
Во время функционирования системы Y2H в клетках дрож-
дрожжей одновременно присутствуют три плазмиды, включая две,
рассмотренные выше, и плазмиду, содержащую ген-репортер. В
этой связи возникает проблема очистки одной из трех плазмид,
кодирующих исследуемый белок для ее дальнейшего использова-
использования в секвенировании данного гена, а также в новых раундах му-
мутагенеза и отбора мутантных белков. Для решения этой задачи
во все плазмиды, кроме отбираемой, вводят контр-селективные
маркеры, например, ген URA3, кодирующий оротидин-5'-фос-
фат-декарбоксилазу [122]. Этот жизненно важный фермент уча-
участвует в биосинтезе урацила, но, кроме того, он обладает способ-
способностью превращать нетоксичную 5-фтороротовую кислоту
E-FOA) в токсичный 5-фторурацил. Таким образом, экспрессия
гена URA3 летальна для клеток дрожжей, выращиваемых в при-
присутствии 5-FOA. Альтернативно плазмидами, выделенными из
дрожжевых клеток, трансформируют ауксотрофные клетки
Е. coli trpl~, Ieu2~, и трансформанты высевают на селективную
среду, в которой отсутствуют соответствующие аминокислоты.
В результате вырастают клетки, содержащие плазмиду с генами
TRP1 и LEU2. Кроме того, в отбираемую плазмиду могут быть
введены и другие селектируемые маркеры, которых нет в других
плазмидах, например, гены устойчивости к антибиотикам, в част-
частности, к зеоцину {zeor).
В дополнение к системе Y2H, основанной на DBD- и AD-до-
менах белка Gal4, разработано несколько систем, использующих
358
другие активирующие и ДНК-связывающие домены. Среди них
наиболее известны системы на основе DBD LexA-penpeccopa
Е. coli и AD белка VP16 вируса простого герпеса [123], а также
DBD репрессора cl фага X [124] и эстрогенового рецептора чело-
человека [125].
Гены-репортеры в системе Y2H. Кроме приложений в стан-
стандартных исследованиях белок-белковых взаимодействий, систе-
система Y2H может быть эффективно использована в направленной
эволюции белков для отбора мутантов, взаимодействующих с
изучаемыми лигандами. В этом случае, удобство системы заклю-
заключается в том, что интенсивность отбора эволюционирующих бел-
белков может быть регулируема. Так, на ранних этапах отбора, ког-
когда необходимо обнаруживать белки, обладающие небольшой ак-
активностью, используются высоко чувствительные системы ге-
генов-репортеров. Для выявления более активных мутантов следу-
следующего поколения силу отбора постепенно понижают, применяя
менее селективные гены.
Современные системы Y2H обеспечивают три уровня кон-
контроля отбора благодаря последовательностям нуклеотидов трех
типов: вышерасположенных активирующих последовательнос-
последовательностей UAS (upstream activating sequences), промотора и самого гена-
репортера, которые в разных комбинациях присутствуют в соот-
соответствующих плазмидах. Первый уровень контроля достигается
с помощью UAS. Такие последовательности специфически рас-
распознаются DBD-доменами белков-активаторов транскрипции и
служат местом взаимодействия комплекса DBD-AD с ДНК в ок-
окрестностях гена-репортера. При этом уровень активации транс-
транскрипции коррелирует с числом DBD-связывающих сайтов, распо-
расположенных перед промотором, количество которых в генно-ин-
генно-инженерных конструкциях может варьировать от одного до четы-
четырех [126]. Например, DBD LexA202 взаимодействует с соответст-
соответствующей UAS в виде гомодимера, и наиболее чувствительные ген-
генно-инженерные конструкции содержат восемь тандемно распо-
расположенных последовательностей операторов LexA. Конечно, не-
необходимо иметь в виду, что чем чувствительнее система с геном-
репортером, тем большее количество ложноположительных ре-
результатов будет в ней фиксироваться.
Промотор, под контролем которого происходит транскрип-
транскрипция гена-репортера, обеспечивает второй уровень контроля чув-
чувствительности системы отбора. Как правило, промотор обеспе-
обеспечивает некий базальныи уровень транскрипции независимо от
наличия белка-активатора. При этом, чем ниже такой уровень,
тем сильнее ген-репортер реагирует на действие белка-активато-
359
pa, и тем чувствительнее система отбора. Для понижения базаль-
ного уровня транскрипции в ряде случаев приходится вводить в
конструкцию перед промотором цмс-действующую репрессирую-
репрессирующую последовательность URS (upstream repressing sequence) [127].
Наконец, третий уровень чувствительности системы отбора
обеспечивает ген-репортер как таковой. В рассматриваемой сис-
системе Y2H применяют гены, обеспечивающие калориметричес-
калориметрические способы обнаружения их активности, а также способность
роста клеток в определенных условиях культивирования. Ген
lacZ, использованный в системе Y2H первого поколения в каче-
качестве репортера, кодирует C-галактозидазу Е. coli. Этот фермент
обладает широкой субстратной специфичностью и может рас-
расщеплять ряд хромогенных субстратов, включая 5-бром-4-хлор-3-
индолил-C-О-галактопиранозид (X-gal) и о-нитрофенил-p-D-ra-
лактопиранозид (ONPG), продукты расщепления которых могут
быть обнаружены как на твердой питательной среде (по цвету
колоний), так и в жидких средах. Кроме того, широкое распрост-
распространение получили гены MEL1 и gusA, кодирующие, соответствен-
соответственно, ос-галактозидазу и C-глюкуронидазу, активность которых об-
обнаруживают с использованием хромогенных субстратов 5-бром-
4-хлор-3-индолил-а-О-галактопиранозида (X-oc-D-gal) и 5-бром-
4-хлор-3-индолил-C-О-глюкуроновой кислоты (X-gluc) [124].
Применение в качестве репортера зеленого флуоресцирующего
белка (green fluorescent protein - GFP) или его производных дела-
делают систему детекции еще более простой и эффективной, так как
в этом случае для обнаружения экспрессии его гена не требуется
никаких дополнительных условий, кроме наличия биосинтеза са-
самого белка и, конечно, соответствующего оборудования. (О флу-
флуоресцирующих белках подробнее см. ниже раздел 3.2.2.)
Введение в число генов-репортеров ауксотрофных маркеров
значительно повысило эффективность отбора взаимодействую-
взаимодействующих макромолекул в системе Y2H, так как позволило одновре-
одновременно следить за взаимодействием до 106 разных белков. Для
этой цели в дрожжевых системах наиболее часто используют ге-
гены HIS3, LEU2, ADE2 и URA3, кодирующие жизненно важные
ферменты биосинтеза аминокислот или нуклеотидов. Это обес-
обеспечивает клеткам способность роста без соответствующего до-
добавления аминокислот или нуклеотидов в питательные среды. В
частности, для гена HIS3 характерен высокий базальный уровень
активности, которая может регулироваться 3-амино-1,2,4-триа-
золом C-АТ), конкурентным ингибитором белкового продукта
этого гена. Эффективность отбора с использованием генов LEU2
и ADE2 контролируется изменением концентраций, соответст-
360
а
Пептидное
ингибирование
димеризации
белков
I Сайт, связывающий PBPJ | URA3 ген-репортер |
|Сайт,связьшающийРВР| \URA3 ген-репортер |
Белковое
ингибиро-
ингибирование диме-
[Сайт, связывающий РВР| | tet | ризации | Сайт, связывающий DBD | | tet \
белков
TetR
| Сайт, связывающий TetR| | HIS3 ген-репортер]
| Сайт, связывающий TetR | | HIS3 ген-репортер|
в
| Сайт, связывающий РВР| [С//?АЗ ген-репортер | |Сайт, связывающий РВР|
| URA3 ген-репортер|
Рис. 50. Варианты дигибридной дрожжевой системы, разработанные
для отбора ингибиторов белок-белковых взаимодействий (а, б) или бел-
белков, взаимодействующих между собой (в) [118]
а - обратная система Y2H. Взаимодействие белков X и Y активирует ген-
репортер URA3, что при наличии 5-фтороротовой кислоты E-FOA) в питатель-
питательной среде приводит к гибели клеток; подавление взаимодействия исследуемым
белком делает клетки жизнеспособными в этих условиях;
б - разделенная гибридная система. Взаимодействие между белками X и Y
активирует ген tet, кодирующий репрессор TetR, который, в свою очередь, по-
подавляет транскрипцию гена-репортера HIS3, необходимую для роста клеток на
среде без гистидина; нарушение взаимодействия X и Y активирует ген ///55, что
делает клетки жизнеспособными;
в - система с репрессируемым трансактиватором. Активирующий белок X
(АХ) обладает способностью активировать ген URA3, что в присутствии 5-FOA
приводит к летальным последствиям для клетки; его взаимодействие через ис-
исследуемый домен X с доменом Y гибридного белка репрессора, объединенного
в одной молекуле с репрессирующим доменом (RD), подавляет транскрипцию
гена URA3
361
венно, Leu и аденина в питательной среде. Ген URA3, как уже упо-
упоминалось выше, обеспечивает позитивный и негативный отбор.
Кроме того, для негативного отбора применяют ген CYH2, по-
поскольку его экспрессия делает клетки дрожжей чувствительны-
чувствительными к циклогексимиду, ингибитору элонгации полипептидных це-
цепей у эукариот [127].
Варианты системы Y2H. Разработан ряд вариантов основной
дрожжевой дигибридной системы, использующихся для отбора
мутантных белков, взаимодействие между которыми нарушается
в результате мутаций. В системе, получившей название обрат-
обратной Y2H, для отбора белков с нарушенным взаимодействием при-
применяют Gal4 DBD и AD, а также ген-репортер URA3 в качестве
контр-селектируемого маркера (рис. 50, а). В такой системе вна-
вначале подтверждают взаимодействие между исследуемыми белка-
белками, выращивая клетки в отсутствие урацила, а затем отбор ве-
веществ, нарушающих взаимодействие между белками, проводят в
присутствии 5-FOA. Клетки выживают лишь в том случае, если
исследуемые белки в результате внешнего воздействия переста-
перестают взаимодействовать друг с другом, и экспрессия гена URA3
прекращается.
В разделенной гибридной системе (split-hybrid system) обна-
обнаружение белок-белковых взаимодействий проводят с использо-
использованием двух генов-репортеров (рис. 50, б) [128]. В этой системе
взаимодействие белков друг с другом активирует транскрипцию
гена tet E. coli, который кодирует репрессор TetR, взаимодейству-
взаимодействующий с ДНК. Экспрессия гена HIS3 находится под контролем
двух тандемных операторов. Первоначально исследуемое белок-
белковое взаимодействие активирует ген tet, а образующийся ре-
репрессор TetR блокирует транскрипцию гена H1S3, что делает не-
невозможным рост клеток в отсутствие His в питательной среде.
Нарушение белок-белкового взаимодействия приводит к обрат-
обратному эффекту, что позволяет проводить отбор соответствующих
ингибиторов по росту клеток в отсутствие аминокислоты.
Система с репрессируемым трансактиватором (repressed
transactivator system - RTS) является еще одной разновидностью
системы Gal4 Y2H, в которой AD-домен Gal4 заменен на N-koh-
цевой репрессорный домен RD дрожжевого белка-репрессора
TUP1 [129,130] (рис. 50, в). В такой системе ген-репортер URA3
находится под контролем промотора гена GALL В отсутствие
взаимодействия между исследуемыми белками ген URA3 активи-
активирован, и, следовательно, дрожжевые клетки погибают в присут-
присутствии 5-FOA. Если же домены Gal4-DBD и TUP1-RD, находящие-
находящиеся в составе гибридных белков, сближаются друг с другом благо-
362
даря взаимодействию исследуемых участков их полипептидных
цепей, то RD-домен подавляет транскрипцию гена URA3 с промо-
промотора GAL1, и клетки становятся устойчивыми к 5-FOA.
Системы Y2H, не требующие активации или подавления
транскрипции. Использование синтеза РНК для обнаружения
взаимодействия белков друг с другом является с биохимической
точки зрения очень сложным подходом. Его реализация требует
переноса гибридных белков в ядро с последующим влиянием на
аппарат транскрипции, что открывает большой простор для ар-
артефактов, часть которых была отмечена выше. В этой связи, уси-
усилия исследователей направлены на создание дигибридных сис-
систем, не зависящих от транскрипции генов-репортеров. Некото-
Некоторые из таких систем основаны на появлении новой ферментатив-
ферментативной активности после объединения двух гибридных белков, каж-
каждый из которых сам по себе ею не обладает. Один из вариантов
дигибридных систем основан на обнаружении активности дигид-
рофолатредуктазы, которая появляется в результате объедине-
объединения двух фрагментов полипептидной цепи данного фермента, что
является одной из разновидностей генетической комплементации
[131]. У дигидрофолатредуктазы N-концевой и С-концевой доме-
домены A-105 и 106-186 а.о., соответственно) не приобретают пра-
правильной пространственной конформации, не объединившись
друг с другом. На этом основании их ассоциацию обнаруживают
по появлению ферментативной активности или способности
взаимодействовать с конъюгатом ингибитора метотрексата с
флуоресцеином. Описаны системы и на основе зеленого флуо-
флуоресцирующего белка (GFP) [70].
Проблема ложноположителъных и ложноотрщательных
результатов при использовании системы Y2H. Ложноположи-
тельные результаты, в которых желаемое выступает под видом
действительного, являются неизменными спутниками любой на-
научно-исследовательской работы, в том числе, и систем отбора
белков in vivo. По механизму возникновения ложноположитель-
ные результаты можно разделить на две категории: технические
и биологические [132]. К первой группе относятся артефакты, не
связанные со взаимодействием двух гибридных белков как тако-
таковым. В этом случае экспрессия некоторых гибридных белков со-
сопровождается необъяснимой индукцией гена-репортера. Такие
технические проблемы можно минимизировать, используя, на-
например, несколько респонсивных промоторов для гена-репорте-
гена-репортера [133]. Ложноположительные результаты биологической при-
природы являются следствием взаимодействия между исследуемыми
белками, которого не происходит в клетке. Доля таких взаимо-
363
действий, которую можно оценить путем анализа данных, полу-
полученных с известными белками или по глобальным профилям экс-
экспрессии двух генов, кодирующих анализируемые белки, состав-
составляет 40-50% [133, 134].
Возникновение ложноположительных клонов особенно ве-
вероятно в первых раундах отбора белков, эволюционирующих in
vitro, когда их активность еще слабо выражена, и для ее обнару-
обнаружения приходится использовать высокочувствительные гены-ре-
гены-репортеры. Часто клетки, выжившие во время такого отбора, не
содержат белков, взаимодействующих друг с другом, а являются
мутантами, которые обладают искомым фенотипом. Помимо
этого, найдется множество других причин, по которым могут вы-
вырасти фоновые колонии клеток дрожжей. Подробное описание
подходов и конкретных методов, с помощью которых можно
снизить влияние ложноположительных клонов на общий исход
эксперимента, можно найти в специальных руководствах [132].
Здесь же отметим, что для успешного решения задач по отбору
белков в ходе направленной эволюции, необходимо на основании
данных литературы выбрать формат оптимальной системы Y2H,
в котором соблюден баланс между чувствительностью системы и
ее устойчивостью к посторонним шумам. Выводы о взаимодейст-
взаимодействии белков друг с другом можно делать лишь по степени воспро-
воспроизводимости результатов с несколькими генами-репортерами.
Положительные клоны, полученные при первичном скрининге,
должны исследоваться в новом раунде отбора. При этом во вто-
вторичном скрининге часто используют систему lacZ с геном C-га-
лактозидазы в качестве репортера. Само собой разумеется, что
на всех этапах проведения отбора необходимо ставить контроль-
контрольные пробы, так как весь опыт экспериментальной работы пока-
показывает, что лучше поставить десять лишних контролей, чем не
поставить одного нужного. Ведь в случае ошибки потери време-
времени будут несопоставимы со временем, затраченным на постанов-
постановку контрольных экспериментов.
Гораздо менее понятным феноменом являются ложноотрица-
тельные результаты отбора в системе Y2H, которые проявляются
в том, что взаимодействующие друг с другом белки не оказывают
влияния на транскрипцию гена-репортера. Хотя такие артефакты
не представляют серьезной проблемы в опытах по направленной
эволюции белков, основной задачей которой является отбор бел-
белков, эволюционирующих к нужным свойствам, к такой возможно-
возможности необходимо относиться очень серьезно при использовании раз-
разных систем Y2H в крупномасштабных исследованиях. Например,
попытки воспроизвести результаты работы по изучению интер-
364
актома дрожжей (совокупности всех белок-белковых взаимодей-
взаимодействий клетки), полученные с помощью одной из таких систем, в
другой системе позволили выявить только -25% обнаруженных
ранее взаимодействий [135]. Подобные проблемы вероятно будут
отступать параллельно с ростом понимания механизмов белок-
белковых взаимодействий и активации транскрипции.
Бактериальные дигибридные системы (В2Н). Одним из пре-
преимуществ системы В2Н перед Y2H при осуществлении отбора во
время направленной эволюции белков является более высокая
эффективность трансформации бактериальных клеток, что поз-
позволяет исследовать одновременно до 108 белков, а это на два по-
порядка превышает возможности дрожжевой системы. Высокая
скорость деления бактериальных клеток сокращает время прове-
проведения экспериментов, кроме того клетки Е. coli являются этало-
эталоном в опытах по генной и белковой инженерии. В связи с этим си-
системы В2Н становятся все более популярными в исследованиях
белок-белковых взаимодействий.
В одной группе систем В2Н используют свойства бактериаль-
бактериальных репрессоров и активаторов транскрипции, которые часто яв-
являются димерами со структурно различающимися доменами
DBD и AD. На рис. 51 схематически изображены функциональ-
функциональные взаимодействия в трех В2Н-системах. В одной из первых раз-
разработанных систем С-концевой домен АД34-репрессора, обеспе-
обеспечивающий его димеризацию, что необходимо для проявления его
активности, был заменен на функционально эквивалентный до-
домен "лейциновая застежка" дрожжевого белка-активатора
транскрипции GCN4 (рис. 51, а) [136]. Химерный белок оказался
стабильным и хорошо выполнял функции белка-репрессора in
vivo. Одновременно такая замена предотвращала образование ге-
теродимеров между ХАЪА- и А,-репрессорами, которые распозна-
распознают разные последовательности операторов. Исследуемый белок
X объединяли с С-концом модифицированного А434-репрессора,
который взаимодействует с оператором ХАЪА. Другой анализиру-
анализируемый белок Y соединяли с С-концом А,-репрессора, который сам
по себе не может эффективно взаимодействовать с мутантным
^-оператором. Его взаимодействие происходит лишь в том слу-
случае, если белки X и Y образуют комплекс. При этом А,-репрессор
подавляет транскрипцию гена-репортера lacZ, предотвращая вза-
взаимодействие РНК-полимеразы с промотором. Такая система
обеспечивала лишь 10-кратное изменение уровня транскрипции
гена-репортера на фоне продуктивного взаимодействия между
исследуемыми белками и, следовательно, была мало пригодна
для изучения слабых белок-белковых взаимодействий.
365
I araBAD I Оператор LexA
1 . ' 1___<Оч/Т\—
Оператор АгаС
Оператор LexA
axzBAD
в
Оператору | | Ген-репортер |
Рис. 51. Варианты бактериальных дигибридных систем В2Н [115]
а - система, основанная на природных бактериальных белках-репрессорах.
С-концевая часть репрессора Фага X 434 соединена через домен "лещиновая за-
застежка" (L), обеспечивающий димеризацию молекул, с исследуемым белком X.
Взаимодействие таких гибридных молекул с оператором X 434 не влияет на
транскрипцию гена-репортера, однако если белки X и Y образуют комплекс,
домены ?1-репрессора приобретают способность связываться с гомологичным
низкоаффинным оператором, конкурируя с РНК-полимеразой за промотор ге-
гена-репортера;
б - подавление транскрипции изгибанием ДНК. Гибридный белок АгаС-Х
активирует ген-репортер araBAD; взаимодействие АгаС-Х с гибридным белком
LexA-Y (L-Y) сопровождается связыванием LexA (L) оператором и подавлени-
подавлением транскрипции;
в - ?1-Репрессор, объединенный с белком X, может активировать РНК-по-
лимеразу, если белок X взаимодействует с белком Y, объединенным в одной
полипептидной цепи с N-концевым доменом ос-субъединицы РНК-полиме-
разы
Дальнейшее 10-кратное повышение чувствительности было
достигнуто в системах второго типа при использовании белков
AraC-LexA (рис. 51, б) [137]. В этом случае три тандемных копии
оператора LexA вводят перед сайтом инициации транскрипции ге-
гена lacZ, который находится под контролем промотора гена
araBAD. Белок АгаС, как сам по себе, так и в виде гибрида с ис-
исследуемым белком, активирует транскрипцию гена lacZ. Если ги-
366
бридные белки X-LexA и Y-AraC взаимодействуют друг с другом,
то происходит изгибание ДНК в области промотора, что приво-
приводит к предотвращению транскрипции гена-репортера по неясно-
неясному до конца механизму и может быть следствием инактивации
РНК-полимеразы в этой области или предотвращения ее взаимо-
взаимодействия с промотором.
В системе третьего типа последовательность ^-оператора по-
помещают перед промотором гена-репортера таким образом, что
он сам по себе не влияет на транскрипцию даже в присутствии со-
соответствующего репрессора (рис. 51, в) [138]. В этой системе ис-
используют гибридный белок Х-А,-репрессор, а также гибридную,
измененную в N-концевой части а-субъединицу РНК-полимера-
РНК-полимеразы Y-oc, входящую в состав холофермента. Такая модифициро-
модифицированная РНК-полимераза самостоятельно не может эффективно
взаимодействовать с промотором, однако, если между исследуе-
исследуемыми белками X и Y имеет место продуктивное взаимодействие,
фермент стабилизируется на промоторе, и происходит активация
транскрипции гена-репортера lacZ. В этой системе взаимодейст-
взаимодействие гибридных белков Gal4 и Gall 1 между собой с KD = 1О7 М ак-
активирует транскрипцию гена в 45 раз. На подобном принципе бы-
была разработана В2Н-система с геном-репортером ЯШ, позволя-
позволяющим проводить контр-селекцию в присутствии 5-FOA [139], а
кроме того и система Y2H [140]. Так же, как и в случае Y2H-chc-
тем, у систем В2Н имеются варианты, основанные не на модуля-
модуляции транскрипции, а на восстановлении ферментативной актив-
активности при сближении двух белковых фрагментов, например, ак-
активности аденилатциклазы [141].
Сегодня В2Н-системы еще находятся в начальной стадии сво-
своего развития. Несмотря на ряд отмеченных выше преимуществ
таких систем, они обладают и существенными недостатками. В
частности, в бактериальных клетках труднее, чем у дрожжей, до-
добиться сосуществования нескольких плазмид. Кроме того, про-
процесс интеграции генов в хромосому бактерий менее эффективен,
чем у дрожжей. Тем не менее, поскольку интерес к системам В2Н
сохраняется, преодоление технических затруднений является во-
вопросом времени.
Моногибридные (Y1H) и тригибридные (Y3H) дрожжевые
системы. Моногибридная система (one hybrid system), основанная
на той же концепции, что и Y2H, является ее упрощенным вари-
вариантом. Как следует из названия, основным действующим нача-
началом моногибридной системы является один гибридный белок, в
котором AD-домен соединен с гетерологичным DBD-доменом
(см. рис. 49, б). Система позволяет осуществлять поиск белков,
367
взаимодействующих со специфическими последовательностями
ДНК. В том случае, если исследуемый полипептид обладает ис-
искомой DBD-активностью, AD-домен активирует транскрипцию
гена-репортера. Альтернативно с помощью системы Y1H можно
проводить in vivo поиск последовательности нуклеотидов, с кото-
которой взаимодействует конкретный DBD в клонотеке таких после-
последовательностей, созданной на индивидуальных плазмидах. Оба
этих подхода плодотворно используются в современных исследо-
исследованиях, в том числе для идентификации новых факторов транс-
транскрипции (см. например [142-144]).
Тригибридная дрожжевая система (Y3H) является дальней-
дальнейшим развитием системы Y2H [118,142]. Этот вариант чаще всего
используется с целью исследования РНК-белковых взаимодейст-
взаимодействий. Для активации (или в другой модификации ингибирования)
транскрипции гена-репортера в ней задействованы три гибрид-
гибридных макромолекулы: два гибридных белка, один из которых со-
содержит DBD-домен, соединенный с универсальным РНК-связы-
вающим доменом (в данном примере с белком оболочки фага
MS2), и AD-домен, объединенный с белком Y, взаимодействие
которого с конкретной последовательностью РНК анализирует-
анализируется (рис. 49, в). Третьим компонентом является гибридная РНК,
содержащая универсальную якорную последовательность, взаи-
взаимодействующую с белком MS2, и последовательность X, способ-
способность которой взаимодействовать с белком Y исследуется. В том
случае, когда имеет место РНК-белковое взаимодействие, фор-
формируется тройной комплекс, активирующий промотор гена-ре-
гена-репортера. В другом варианте система Y3H может быть использо-
использована для изучения взаимодействий белков-рецепторов с низкомо-
низкомолекулярными лигандами, получившими название химических ин-
индукторов димеризации (chemical inducers of dimerization - CID)
(рис. 49, г). В этом случае в качестве третьего компонента систе-
системы используют двойной лиганд L1-L2, у которого часть L1 взаи-
взаимодействует с белком X, а взаимодействие L2 с белком Y иссле-
исследуется. (Возможен и обратный вариант, когда изучается взаимо-
взаимодействие L1 с белком X.) При наличии взаимодействия происхо-
происходит активация (или в других конструкциях подавление) активнос-
активности гена-репортера.
368
Глава 3
Достижения белковой инженерии
У белковой инженерии большое будущее. Но даже достиже-
достижения сегодняшнего дня в данной области весьма значительны.
Мы являемся свидетелями активного использования белков и
ферментов для крупномасштабного тонкого химического син-
синтеза, эффективного разделения рацемических смесей энантио-
меров, в качестве биосенсоров, лекарственных средств при за-
заместительной терапии многих заболеваний человека, для полу-
получения пищевых продуктов, детергентов и эффективных мою-
моющих средств. Все это требует проведения поиска еще более эф-
эффективных биокатализаторов, в том числе и создания искусст-
искусственных измененных ферментов методами белковой инженерии.
Наш краткий обзор не ставит задачи дать исчерпывающую кар-
картину полученных результатов, и основной акцент делается на
использовании генно-инженерных методов при конструирова-
конструировании белков с новыми свойствами. Однако было бы несправед-
несправедливым умолчать о химических подходах получения биокатали-
биокатализаторов с новыми свойствами, активно применяемых в совре-
современной биотехнологии.
3.1. Химические модификации белков
Использование химических методов для направленного изме-
изменения свойств ферментов, по-видимому, началось в 1966 г. с ра-
работ Л. Полгара и М. Бендера [145], а также К. Ниита и Д. Кош-
ланда [146], которые применили своеобразный "химический му-
мутагенез" на уровне белковой молекулы для превращения остатка
Ser в Cys в каталитической триаде активного центра субтилизи-
на. Интерес к этому направлению исследований был стимулиро-
стимулирован во второй половине 1980-х гг. опытами Е. Кайзера, в частно-
частности, присоединившего флавиновый кофермент к папаину, что
привело к превращению протеиназы в оксидоредуктазу [147]. Се-
Сегодня ковалентные и нековалентные химические модификации
белковых молекул являются широко распространенным подхо-
подходом к изменению их свойств [148]. Ниже будут кратко рассмотре-
рассмотрены три современных подхода к улучшению свойств ферментов
разными химическими методами.
369
3.1.1. Искусственные поперечные сшивки в белках
Введение молекулярных поперечных сшивок в белки широко
применяется в настоящее время для их стабилизации. Для этих
целей создают внутримолекулярные и межмолекулярные попе-
поперечные сшивки с использованием различных бифункциональных
и полифункциональных реагентов. В частности, хорошие резуль-
результаты были получены на основе глутарового альдегида, который
вначале был применен для стабилизации кристаллов карбокси-
пептидазы с целью их дальнейшего исследования с помощью
рентгеноструктурного анализа.
О
Уже в этих первых опытах было установлено, что фермент в
виде кристаллов, содержащих поперечные сшивки, сохранял до
5% своей первоначальной активности. Вскоре последовало даль-
дальнейшее исследование возможности достижения большей эффек-
эффективности в сохранении активности кристаллических ферментов,
поскольку повышение их стабильности в такой форме было оче-
очевидным. Итогом явилось получение термолизина в виде нерас-
нерастворимых кристаллов фермента, содержащих поперечные
сшивки (crosslinked enzyme crystal - CLEC), и этот термин дал на-
название всей группе подобных методов химической модификации
белковых молекул [149]. Модифицированный термолизин CLEC
сохранял значительную часть своей исходной ферментативной
активности, а полученный таким же образом субтилизин обнару-
обнаруживал высокую стабильность как в органических, так и водных
средах и обладал способностью катализировать синтез пептидов
в ацетонитриле [150]. Во всех первых опытах была отмечена низ-
низкая активность ферментов CLEC, особенно в органических сре-
средах, на фоне их высокой стабильности. Низкую эффективность
катализа связывали с пониженной скоростью переноса субстрата
и продукта реакции в кристаллах в органических средах. Для пре-
преодоления этого недостатка CLEC-ферментов была предложена
предобработка кристаллов краун-эфиром 18-краун-6 с последую-
последующей лиофилизацией кристаллов. В итоге способность CLEC-суб-
тилизина Carlsberg образовывать пептидные связи в органичес-
370
ких средах была повышена в 13 раз, что связывали с улучшением
обмена молекулами воды между активным центром фермента и
окружающей средой с участием краун-эфира (см. сводную
табл. 15 в конце книги) [151].
Ковалентная модификация ферментов монофункциональ-
монофункциональными реагентами используется для введения в белки новых
функциональных групп, повышения их растворимости в орга-
органических средах и других изменений биохимических свойств.
Особенной популярностью пользуется модификация фермен-
ферментов полиэтиленгликолем (ПЭГ), которая приводит не только к
стабилизации в органических растворителях, но и сопровожда-
сопровождается повышением временем их полужизни в биологических
жидкостях, а также снижением антигенности макромолекул,
что важно при терапевтическом применении ферментов [152].
Кроме того, для тех же целей используют ковалентные моди-
модификации белков амфифильным (содержащим гидрофобные и
гидрофильные группы) детергентом полиоксиэтиленлауроило-
вым эфиром (Brij 35).
N\ / C1 С12Н25(ОСН2СН2J3Оч C12H25(OCH2CH2J3Ov
с12н25(осн2сн2J3он _9 ^ Ч Vе1 ЕШг ¦ N\ V-NH~E
С12Н25(ОСН2СН2J3О / С12Н25(ОСН2СН2J3О '
В этом случае наблюдается повышение растворимости ката-
лазы в абсолютном толуоле и 1,1,1-трихлорэтане, причем в по-
последнем растворителе активность модифицированного фермента
была в 200 раз выше, чем у исходного белка, а его активность в
водных средах была выше в 15-20 раз [153]. Другие многочислен-
многочисленные примеры ковалентных модификаций ферментов с целью
введения в них функциональных групп небольшого размера (в
том числе и коферментов), а также замещения отдельных атомов
можно найти в обзоре [154].
В целом, химические модификации представляют собой груп-
группу быстро реализуемых и недорогих методов стабилизации фер-
ферментов и изменения их субстратной специфичности путем введе-
введения небольших функциональных групп, что невозможно сделать
с помощью направленного и случайного мутагенеза. К недостат-
недостаткам этих методов следует отнести невозможность осуществления
контроля за полнотой прохождения реакций и изменения кон-
конкретных участков полипептидных цепей. Решение проблемы ле-
лежит на пути комбинированного применения направленного мута-
мутагенеза с последующей химической модификацией боковых цепей
371
специально введенных аминокислотных остатков, что, как уже
рассматривалось выше, в ряде случаев позволяет преодолевать
все упомянутые затруднения.
3.1.2. Оптимизация ферментов для функционирования
в неводных средах
Использование органических растворителей в качестве сре-
среды для проведения химических реакций на основе биокатализа
имеет много преимуществ перед применением водных сред для
осуществления аналогичных реакций [154]. В частности, к несо-
несомненным достоинствам данной группы методов относятся повы-
повышение растворимости гидрофобных субстратов и сопутствую-
сопутствующий этому сдвиг равновесия химической реакции в сторону обра-
образования продуктов реакции.
При исследовании структуры ферментов в органических сре-
средах было установлено, что кристаллические или лиофильно вы-
высушенные полипептиды сохраняют свои основные характерные
особенности. Так, с помощью рентгеноструктурного анализа
кристаллов субтилизина, содержащих небольшое количество по-
поперечных сшивок, было продемонстрировано, что фермент в та-
таких условиях не изменяет свою общую структуру, а структура ак-
активного центра практически не отличается от таковой в водных
растворах и других органических растворителях [155]. Похожие
данные были получены и с помощью инфракрасной спектроско-
спектроскопии с преобразованием Фурье (FTIR spectroscopy) [156]. На осно-
основании таких результатов были сделаны выводы о независимости
свойств ферментов как биокатализаторов от природы органиче-
органических растворителей. Несмотря на то, что способность ферментов
к катализу зависит от состава среды, эту зависимость нельзя объ-
объяснить изменениями их структуры. В отличие от кристалличес-
кристаллических и лиофильно высушенных форм, у ферментов, находящихся
в растворе, наблюдалось изменение вторичной структуры, кото-
которое зависело от природы использованного органического соеди-
соединения и коррелировало с изменениями уровней ферментативной
активности растворенных белков. Особенно сильная потеря ак-
активности имеет место при значительном отклонении структуры
ос-спиральных участков и C-слоев ферментов от таковой, наблю-
наблюдаемой в водных растворах.
Ферменты, солюбилизированные в органических раствори-
растворителях, содержащих небольшое количество воды или анионных
детергентов, способны совершать правильный фолдинг. Следо-
Следовательно, вода не является единственной уникальной средой, ко-
372
торая обеспечивает внутримолекулярные взаимодействия, необ-
необходимые для приобретения ферментом пространственной струк-
структуры, нужной для появления его каталитической активности.
Уровень протонирования молекул фермента в органических рас-
растворителях обычно задается значениями рН водного раствора, из
которого производится их высушивание. Этот феномен получил
название рН-памяти белков. Вообще лиофилизация белков мало
сказывается на ионизации их заряженных функциональных
групп, которая изначально имела место в водных растворах.
Как следует из вышеизложенного, для проявления фермента-
ферментами максимальной активности в органических средах, требуется
проведение предварительной подготовки их препаратов, в част-
частности кристаллизации или высушивания. Другим распространен-
распространенным приемом является иммобилизация белков, т.е. ковалентное
или нековалентное их присоединение к растворимым или нерас-
нерастворимым носителям. Интересной новой разработкой в этом на-
направлении было создание так называемых пластиковых биока-
биокатализаторов (biocatalytic plastics) [157,158]. Пластиковые биока-
биокатализаторы получают непосредственно в неводной среде путем
сополимеризации виниловых мономеров с химически модифици-
модифицированными ферментами, растворимыми в органических раство-
растворителях, и имеющими на своей поверхности ковалентно присое-
присоединенные функциональные группы с полимеризующимися двой-
двойными связями. При использовании субтилизина Carlsberg и ос-хи-
мотрипсин в качестве модельных объектов, было показано, что
ферменты полностью сохраняют свою исходную активность в
гексане в течение, по крайней мере, трех недель. Технологии на
основе биоинкапсуляции в синтетические полимеры, по-видимо-
по-видимому, найдут широкое применение в органической химии и биотех-
биотехнологии [159]. Необходимость дегидратации ферментов перед
использованием в безводных средах часто является причиной их
низкой активности. Предварительная же иммобилизация фер-
ферментов, например, на силикагеле, с последующей дегидратацией
абсолютным н-пропанолом позволяет в несколько десятков раз
повысить их активность в органических растворителях [160].
Многие ферменты быстро инактивируются при высоких кон-
концентрациях органических растворителей, которые требуются
для проведения соответствующих реакций. Одним из способов,
успешно применяемых для решения этой проблемы, является по-
получение комплексов ферментов с липидами или детергентами,
что делает их растворимыми и стабилизирует в неводных средах.
Например, различные C-галактозидазы в комплексе с липидами
приобретают способность катализировать реакции трансглико-
373
зилирования с разными гидрофобными спиртами в безводном ди-
изопропиловом эфире [161]. Аналогичный подход был успешно
применен и к другим гликозидазам [162]. Еще одним современ-
современным методом стабилизации ферментов в органических раствори-
растворителях является молекулярный импринтинг. Поскольку примене-
применение этого подхода не ограничивается решением задач такого ро-
рода и распространяется на контроль субстратной специфичности
белков и ферментов, а также активно используется в других при-
приложениях белковой инженерии и биотехнологии, представляется
целесообразным рассмотреть его более подробно.
3.1.3. Молекулярный импринтинг
Феномен молекулярного импринтинга был впервые обнару-
обнаружен в 1972 г. Для его реализации в водном растворе получают ма-
кромолекулярные комплексы низкомолекулярных лигандов с
полимерами, которые далее высушивают и промывают раство-
растворителем, избирательно освобождающим комплексы от лиганда,
но не растворяющим макромолекулы [163]. Поскольку подвиж-
подвижность макромолекул в твердой фазе ограничена, они сохраняют
конформацию, которая была индуцирована в них соответствую-
соответствующим лигандом, даже после его удаления из комплекса. В итоге
образуется новый класс искусственных материалов, обладающих
свойствами специфических рецепторов, поскольку заключают в
себе отпечаток пространственной структуры лиганда-матрицы.
Такие материалы обладают высоким сродством и избирательно-
избирательностью по отношению к лигандам, уникальной стабильностью, зна-
значительно превышающей таковую природных биоматериалов, и
их довольно просто получать в большом количестве. Они актив-
активно внедряются в практику для синтеза, разделения, идентифика-
идентификации и связывания матричных лигандов и их производных, а так-
также создания биосенсоров. Лигандами же могут служить микроор-
микроорганизмы, белки, нуклеиновые кислоты, аминокислоты, сахара,
алкалоиды, стероидные соединения, токсины, гербициды, арома-
ароматические и гетероциклические химические соединения, ионы ме-
металлов и вещества в газообразной фазе.
Работы по применению молекулярного импринтинга в белко-
белковой инженерии начались после того, как двумя группами исследо-
исследователей в Японии и Великобритании была продемонстрирована
возможность с помощью аналогов субстратов в переходном со-
состоянии придать ферментативную активность белкам, у которых
она отсутствует - альбумину и лактоглобулину [164,165]. Вскоре
молекулярный импринтинг был использован и для контроля суб-
374
стратной специфичности субтилизина в отношении ацилирова-
ния нуклеозидов в органических средах [166]. Лиофилизация суб-
субтилизина в присутствии нуклеофильных субстратов стимулиро-
стимулировала способность фермента ацилировать нуклеозиды и углеводы
в органических растворителях. В том случае, если импринтинг
осуществляли в присутствии сахарозы, имела место 40-кратная
стимуляция способности ацилировать сахарозу по сравнению с
исходным ферментом, а скорость ацилирования тимидина возра-
возрастала только в четыре раза. Обратную картину наблюдали, если
для импринтинга использовали тимидин: происходила девяти-
девятикратная стимуляция ацилирования сахарозы и 52-кратное увели-
увеличение скорости реакции при применении тимидина в качестве
субстрата. Другие примеры использования всех вышеперечис-
вышеперечисленных подходов приведены в табл. 15 (см. далее). Этими приме-
примерами мы заканчиваем далеко не исчерпывающее рассмотрение
приложения химических методов к белковой инженерии и пере-
переходим к обсуждению недавних достижений в конструировании
белков и ферментов с помощью подходов, целиком основанных
на методах молекулярной генетики.
3.2. Гибридные белки
Гибридные (химерные) белки представляют собой полипеп-
полипептидные цепи, искусственно составленные из фрагментов природ-
природных белков разного происхождения. Современные методы ген-
генной и белковой инженерии позволяют вводить в полипептидные
цепи элементы вторичной структуры других белков, встраивать
новые функциональные домены, заменять одни функциональ-
функциональные домены на другие, а также объединять целые белковые мо-
молекулы в одной полипептидной цепи. Из этого следует, что со-
создание гибридных белков является одной из разновидностей ра-
рационального редизайна белковых молекул.
Целью конструирования гибридных белков является улучше-
улучшение свойств, имеющихся у исходных макромолекул, для более
производительного их использования в биотехнологии, а также
создание белков и ферментов с новыми свойствами. Работы в об-
области гибридных белков, с одной стороны, направлены на изме-
изменение кинетических параметров ферментов, их субстратной спе-
специфичности, оптимума рН, термостабильности и других биотех-
биотехнологических параметров. При другом подходе усилия исследо-
исследователей сосредоточены на создании бифункциональных и много-
многофункциональных белковых молекул, объединяющих в одной по-
полипептидной цепи несколько функционирующих доменов.
375
3.2.1. Гибридные ферменты
В условиях живой клетки ферменты не существуют сами по
себе в виде отдельных молекул.
С помощью современных систем исследования белок-белко-
белок-белковых взаимодействий in vivo даже у одноклеточных организмов вы-
выявлены гигантские сети взаимодействующих белков, в организа-
организацию которых вовлечены тысячи макромолекул [167]. Организа-
Организация цитоплазмы на молекулярном уровне создает условия для
строгого упорядочивания метаболических процессов и их глобаль-
глобальной регуляции. Наиболее сильные взаимодействия такого рода
удается наблюдать путем экспериментальной очистки и изучения
многофункциональных ферментных комплексов и белковых сис-
систем. В подобных комплексах имеет место строгая пространствен-
пространственная упорядоченность каталитических субъединиц, которая обеспе-
обеспечивает определенную последовательность протекания фермента-
ферментативных реакций путем канализированной передачи промежуточ-
промежуточных продуктов метаболизма от одного активного центра к друго-
другому, что значительно повышает эффективность этих процессов.
Одними из наиболее впечатляющих примеров мультифер-
ментных комплексов являются природные системы синтеза поли-
кетидов и нерибосомного синтеза пептидов. Во всех этих случаях
синтез происходит путем последовательного добавления к продук-
продуктам реакции химических элементов с помощью пространственно
упорядоченных ферментных модулей [168, 169]. Каталитические
модули, в свою очередь, являются частью многомодульных поли-
полипептидов, которые образуют гигантские синтазные комплексы,
осуществляющие полный синтез поликетидов и пептидов. Таким
образом, природа и пространственная организация модулей полно-
полностью определяют структуру конечного продукта, синтезируемого
с их участием. Продемонстрирована возможность искусственного
изменения структуры конечных продуктов реакции путем генно-
инженерных модификаций, влияющих на структуру и взаимное
расположение каталитических модулей, и это направление являет-
является предметом интенсивных исследований [170, 171].
Искусственные бифункциональные и полифункциональные
ферменты. Потенциальная возможность создания бифункцио-
бифункциональных ферментов на основе гибридных белков была реализо-
реализована в конце 1980-х гг. с целью осуществления сопряженных
ферментативных реакций. Были получены гибридные белки,
объединяющие в одной полипептидной цепи C-галактозидазу и
галактозодегидрогеназу [172], галактозодегидрогеназу и бакте-
бактериальную люциферазу [173], а также малатдегидрогеназу и цит-
ратсинтазу [174]. Суммарная активность таких систем в стацио-
376
нарном состоянии была в два-три раза выше, чем активность сме-
смесей отдельных ферментов, а лаг-период перед выходом систем в
стационарное состояние был в четыре-шесть раз короче. Макси-
Максимальное увеличение эффективности в сопряженных системах на-
наблюдали в условиях, когда прохождение первой стадии реакции
лимитировало весь процесс, например, в неоптимальных для пер-
первого фермента значениях рН или при дефиците его субстрата.
Ожидается, что в будущем точное позиционирование активных
центров каталитических доменов в гибридных бифункциональ-
бифункциональных ферментах друг относительно друга должно значительно по-
повысить эффективность таких систем.
Гибридные мультиферментные комплексы нескольких ката-
каталитических доменов могут быть получены и без синтеза длинных
химерных полипептидных цепей, содержащих эти домены. Раз-
Разработка таких альтернативных систем была начата с использова-
использования целлюлосомы Clostridium thermocellum, которая обеспечива-
обеспечивает гидролиз целлюлозы, служащей источником углерода для
этой бактерии. Целлюлосома представляет собой высокоупоря-
доченный комплекс более 25 ферментов, осуществляющих гид-
гидролиз целлюлозы и полисахаридов, ассоциированных с клеточ-
клеточной стенкой клеток растений. Каждая из каталитических субъе-
субъединиц этого комплекса заякорена на определенных участках по-
полипептидной цепи белка СурА с помощью докериновых доменов
(dockerin domains) без привлечения ковалентных связей. В свою
очередь, белок СурА, выполняющий скелетные функции, содер-
содержит девять высокогомологичных рецепторных последовательно-
последовательностей, названных когезиновыми доменами (cohesin domains), кото-
которые связывают каталитические субъединицы целлюлосомы, вза-
взаимодействуя с их докериновыми доменами [175].
Докериновые домены могут быть объединены с другими
ферментами в составе гибридных полипептидных цепей, что бу-
будет обеспечивать взаимодействие таких рекомбинантных белков
со скелетной последовательностью СурА. Более того, хотя коге-
зиновые домены обладают высокой гомологией, они не полно-
полностью идентичны и предпочтительно взаимодействуют с докери-
докериновыми доменами определенной первичной структуры. Это со-
создает условия для упорядоченной сборки каталитических субъ-
субъединиц, вводимых в состав мультиферментного комплекса, на
скелетной последовательности по воле исследователя путем ли-
линейного упорядочивания когезиновых доменов вдоль полипеп-
полипептидной цепи СурА методами белковой инженерии [176]. Первые
результаты использования такого подхода на практике уже полу-
получены [177, 178].
377
Получение ферментов с новыми свойствами путем объеди-
объединения гомологичных белковых доменов. Случайное объедине-
объединение участков полипептидных цепей двух близкородственных
ферментов, методическое обеспечение которого рассматрива-
рассматривалось выше, находит широкое применение в направленной эво-
эволюции белков. Однако такой подход порождает большое коли-
количество химерных молекул, полностью утративших биологичес-
биологическую активность, так как в ходе подобных манипуляций с боль-
большой вероятностью происходит нарушение тонких внутримоле-
внутримолекулярных взаимодействий, обеспечивавших функциональность
исходных белков.
В отличие от этого продуманный обмен конкретными струк-
структурными субдоменами белков, функции которых известны, мо-
может приводить к более предсказуемым результатам. Например,
сериновые протеиназы семейства S1 состоят из двух гомологич-
гомологичных субдоменов типа C-баррел, интерфейс которых формирует
активный центр типа "клешни". Гидрофобные коровью структу-
структуры и триада каталитических остатков аминокислот консерватив-
консервативны у этой группы ферментов. Однако пептидные петли на по-
поверхности ферментов, окружающие активный центр, заметно ва-
варьируют по своей первичной структуре, что коррелирует с уни-
уникальными субстратными специфичностями ферментов и особен-
особенностями их регуляции. С учетом этих предпосылок была получе-
получена гибридная протеиназа fXYa с объединенными N-концевым
субдоменом активированного фактора свертывания крови
Ха (fXa) и С-концевым субдоменом трипсина [179]. Анализ трех-
трехмерной структуры гибридного белка показал, что расположение
консервативных гидрофобных участков в окрестностях активно-
активного центра в основном было сохранено, однако пространственная
структура элементов поверхности белковой глобулы заметно из-
изменилась. Исследование ферментативной активности гибридно-
гибридного белка с помощью искусственных субстратов обнаружило на-
наличие у него амидолитической активности. При этом гибридная
протеиназа проявляла менее выраженную субстратную специ-
специфичность, чем исходные ферменты, сохраняя высокую способ-
способность использовать субстраты, характерную для фактора Ха.
Адресная доставка гибридных белков. Адресные домены
(tagging domains) и сигнальные последовательности белков широ-
широко используются в белковой инженерии для направленного изме-
изменения внутриклеточной и внеклеточной локализации гибридных
полипептидов. Разработано большое количество векторов, в том
числе и коммерческих, которые позволяют с помощью простых
генно-инженерных манипуляций получать гибридные белковые
378
молекулы, у которых основной функциональный участок объе-
объединен с тем или иным адресным доменом или сигнальной после-
последовательностью. Такие модификации могут обеспечивать гиб-
гибридной молекуле способность к специфической секреции разны-
разными бактериальными хозяевами. Функциональность подобным ги-
гибридным белкам придает способность адресных доменов претер-
претерпевать автономный фолдинг в процессе биосинтеза и, следова-
следовательно, оказывать минимальное воздействие на пространствен-
пространственную структуру основной части гибридного белка.
К той же категории относятся и векторы, в которых клони-
клонируемые гены объединяют с последовательностями, кодирующи-
кодирующими белковые домены со свойствами специфических лигандов или
рецепторов. В качестве адресных доменов в настоящее время ис-
используют белок, связывающий мальтозу, глютатион-8-трансфе-
разу, домены, взаимодействующие с целлюлозой или хитином,
гексагистидиновую последовательность и ряд других. Такие мо-
модификации значительно облегчают очистку рекомбинантных
белков с помощью аффинной хроматографии. Интересной раз-
разработкой в данной области является коммерчески доступный
вектор, в котором последовательность хитин-связывающего (ад-
(адресного) домена объединена с последовательностью нуклеоти-
дов интеина дрожжей S. cerevisiae [61]. Во время экспрессии кло-
клонированного с помощью этого вектора гена происходит образо-
образование гибридного белка, содержащего на С-конце основного ре-
комбинантного белка интеин и адресный домен. Как уже рассма-
рассматривалось в разделе 1.3.2 этой части книги, интеины обеспечива-
обеспечивают прохождение аутосплайсинга белков, их содержащих. Ис-
Использованный в данной конструкции интеин был изменен таким
образом, что активировался в присутствии реагентов, восстанав-
восстанавливающих сульфгидрильные группы. Поэтому после аффинной
сорбции гибридного белка на колонке с иммобилизованным хи-
хитином происходит освобождение основного рекомбинантного
белка из гибридной молекулы в присутствии дитиотриэтола или
C-меркаптоэтанола как следствие расщепления пептидной связи
интеином.
Экспрессия гибридных белков на поверхности бактериаль-
бактериальных клеток. Еще одним интенсивно развивающимся направлени-
направлением белковой инженерии, которое использует адресные домены и
сигнальные последовательности, является экспонирование ре-
рекомбинантных белков на поверхности бактериальных клеток для
создания бактериального дисплея, живых вакцин, а также иммо-
иммобилизации самих клеток в процессе культивирования. Примене-
Применение бактериального дисплея для направленной эволюции белков
379
было подробно рассмотрено выше (раздел 2.3.2) [109]. В основ-
основном те же самые принципы используются и для экспрессии на
клеточной поверхности антигенных эпитопов, обеспечивающих
проведение вакцинации.
Адресная доставка гибридных белков к специфическим по-
последовательностям нуклеиновых кислот. Технологии гибрид-
гибридных белков могут быть использованы для доставки полипепти-
полипептидов к специфическим последовательностям нуклеиновых кис-
кислот. В частности, путем объединения доменов типа "цинковые
пальцы", участвующих в распознавании последовательностей
ДНК факторами транскрипции, с каталитическим доменом рес-
триктазы Fokl удается изменить специфичность действия этой
эндонуклеазы [180].
С помощью аналогичного подхода можно изменять специ-
специфичность и самих факторов транскрипции, что удалось проде-
продемонстрировать с помощью гибридных а-субъединиц РНК-поли-
меразы Е. coli. В этих опытах объединяли N-концевую часть ос-
основной а70-субъединицы с С-концевой последовательностью а32-
субъединицы, которая обеспечивает распознавание РНК-поли-
меразой Е. coli промоторов генов теплового шока. Полученный
гибридный белок после объединения с минимальным ферментом
РНК-полимеразы, не содержащим а-субъединицу, придавал ему
способность инициировать транскрипцию на гибридных промо-
промоторах, составленных из консенсусных (-35)-последовательности
а32-зависимых промоторов и (-Ю)-последовательности промото-
промоторов а70 [181]. Путем замены в молекуле РНК-полимеразы Е. coli
ос-субъединицы дикого типа на гибридную а-субъединицу, кото-
которая содержала С-концевой домен ос-субъединицы В. subtilis полу-
получали фермент, регулируемый активатором транскрипции этой
бактерии [182].
Реконструкция факторов транскрипции из отдельных химер-
химерных белков широко применяется в последнее время в дигибридных
системах, используемых для исследования белок-белковых взаи-
взаимодействий. Принцип действия таких систем и особенности их
функционирования уже подробно рассматривались в разделе 2.3.4.
Проектирование аллостерических взаимодействий в много-
многофункциональных белках с искусственно объединенными домена-
доменами. У большинства гибридных бифункциональных и многофунк-
многофункциональных белков, полученных по обсуждаемым выше техно-
технологиям, каталитические домены соседствуют друг с другом в по-
полипептидной цепи и, по-видимому, приобретая свою пространст-
пространственную структуру в результате независимого фолдинга, облада-
обладают значительной конформационной свободой. Это повышает ве-
380
роятность сохранения функциональной активности доменов в но-
новом для них окружении, но в то же время приводит к отсутствию
оптимальной взаимной ориентации каталитических центров до-
доменов, а их относительная независимость затрудняет осуществ-
осуществление координированных регуляторных воздействий на актив-
активность всего гибридного белка. В этой связи, одним из направле-
направлений исследований в данной области белковой инженерии являет-
является разработка подходов к обеспечению аллостерических взаимо-
взаимодействий между отдельными активными центрами многофункци-
многофункциональных гибридных белков и остальными частями молекул. Ре-
Реальным путем функционального сближения независимых ката-
каталитических доменов может быть их физическое сближение, в
том числе размещение одного домена внутри другого.
Возможность реализации такой стратегии на практике была
продемонстрирована для гибридных белков, объединяющих в од-
одной полипептидной цепи бактериальные |3-лактамазу и мальто-
декстрин-связывающий белок MalE [183]. Были получены генно-
инженерные конструкции, в которых последовательность C-лак-
тамазы соединяли либо непосредственно с С-концом белка MalE,
либо встраивали внутрь его полипептидной цепи. Оказалось, что
в том случае, если полипептидную цепь C-лактамазы встраивали
после а.о. 133 или 303 MalE, секретируемый в периплазматичес-
кое пространство гибридный полипептид сохранял активность
обоих ферментов. Эти участки полипептидной цепи MalE были
выбраны потому, что локализованные здесь короткие делеции и
вставки не оказывали влияния на активность белка. С-Концевые
модификации MalE приводили к образованию гибридного белка
с теми же активностями, и который кроме того был менее чувст-
чувствителен к деградации эндогенными протеиназами, а в присутст-
присутствии мальтозы C-лактамазный домен становился более устойчи-
устойчивым к денатурации мочевиной. Таким образом, в гибридной по-
полипептидной цепи имели место аллостерические взаимодействия
между двумя каталитическими доменами.
Рекомбинантные ферменты, обладающие аллостерическими
свойствами, могут быть использованы для создания новых био-
биосенсоров. В частности, попытки проведения модификаций тако-
такого рода были предприняты в отношении щелочной фосфатазы
[184]. Пептидные эпитопы, специфичные для ВИЧ типа I и виру-
вируса гепатита С, были встроены в полипептидную цепь щелочной
фосфатазы, после чего исследовали влияние антител к этим эпи-
Топам на активность рекомбинантных ферментов. Установлено,
что антитела снижали на 20% активность гибридных ферментов,
Полученных на основе щелочной фосфатазы дикого типа, однако
381
стимулировали ее в два-три раза у гибридных ферментов, создан-
созданных на основе температуро-чувствительных мутантов.
Методами белковой инженерии можно оказывать влияние и
на аллостерическую регуляцию в сложных мутильтимерных
комплексах. Известно, что с помощью замены единственного а.о.
в полипептидной цепи L-лактатдегидрогеназы (ЛДГ) можно из-
изменить ее субстратную специфичность, превратив в малатдегид-
рогеназу (МДГ) [185]. Оба фермента являются тетрамерами, по-
построенными из идентичных субъединиц. При этом полученная
направленным мутагенезом МДГ в отличие от исходной ЛДГ ут-
утрачивает способность взаимодействовать с оксаматом (неметабо-
лизируемым аналогом пировиноградной кислоты). Оказалось,
что оксамат стимулирует активность МДГ в гибридных гетеро-
тетрамерах, составленных из субъединиц ЛДГ и МДГ, взаимодей-
взаимодействуя с субъединицами ЛДГ. Следовательно в такой системе ок-
оксамат ведет себя как настоящий аллостерический эффектор.
Приведенные примеры иллюстрируют первые шаги, сделан-
сделанные на пути конструирования ферментов с аллостерическими
свойствами. Полученные результаты показывают потенциаль-
потенциальную плодотворность данного направления исследований. Однако
для наиболее полного воплощения этих идей в жизнь при конст-
конструировании таких полипептидов необходимо учитывать прост-
пространственные особенности взаимодействия объединяемых ката-
каталитических доменов в гибридных белках, а это невозможно без
понимания связей между первичной и пространственной структу-
структурами изучаемых полипептидов, что, впрочем, относится и ко всей
области рационального дизайна белковых молекул. Интенсив-
Интенсивные структурные исследования белков с высоким разрешением и
молекулярное моделирование в конце концов позволят решать
такого рода проблемы.
3.2.2. Зеленый флуоресцирующий белок (GFP) и его аналоги
в качестве белков-репортеров
В исследовательской работе часто требуется оценивать ско-
скорость биосинтеза и содержание конкретных белковых молекул в
живой клетке, следить за их перемещением во внутриклеточном
пространстве или выделять труднодоступные белки в индивиду-
индивидуальном виде из сложной смеси. В этом случае белки-репортеры
оказываются незаменимыми. Например, присоединив к С-концу
анализируемого белка в качестве белка-репортера C-галактози-
дазу, можно производить очистку рекомбинантного белка по ак-
активности C-галактозидазы или отслеживая ее антигенные детер-
382
минанты иммунохимическими методами. Соединив фрагменты
ДНК, содержащие открытые рамки считывания (ОРС), с генами
белков-репортеров, получают возможность очистить такие гиб-
гибридные белки по активности белка-репортера и использовать их
для иммунизации лабораторных животных. Антитела далее мож-
можно применить для очистки нативного белка, в состав которого
входит кодируемый ОРС, рекомбинантный полипептид, и тем са-
самым идентифицировать клонированный фрагмент гена.
Кроме активного использования в изучении экспрессии ге-
генов, белки-репортеры оказываются незаменимыми помощни-
помощниками при исследовании, в частности, факторов транскрипции и
механизмов передачи сигналов, при поиске новых лекарствен-
лекарственных препаратов и конструировании биосенсоров на основе кле-
клеток. В принципе, многие ферменты могут быть использованы в
качестве репортеров, и их выбор определяется конкретными
задачами и возможностями исследователя. Основными крите-
критериями выбора являются удобство идентификации белка-репор-
белка-репортера в составе молекулярной химеры, его стабильность и другие
важные качества. К таким свойствам можно также отнести
безопасность в отношении окружающей среды, простоту ис-
использования, высокую чувствительность при обнаружении и
низкую фоновую активность.
Кроме вышеупомянутой C-галактозидазы в качестве белков-
репортеров в настоящее время интенсивно используются и дру-
другие природные или генетически модифицированные белки,
включая хлорамфениколацетилтрансферазу, различные флуо-
флуоресцирующие белки, например, люциферазу светлячков, обелин,
экворин, а также зеленый флуоресцирующий белок (GFP) и его
аналоги. Флуоресцирующие белки, т.е. белки, способные испус-
испускать свет в результате контакта с электромагнитным излучени-
излучением, в последнее время получили очень широкое распространение.
Поэтому некоторые их свойства и области их применения будут
кратко рассмотрены ниже.
Биолюминесценция. Некоторые живые организмы обладают
способностью светиться, испуская электромагнитное излучение
в видимой части спектра. Биолюминесценцией называют образо-
образование видимого света в результате химической реакции, протека-
протекающей в живом организме. Этот важный биологический феномен
используется организмами для защиты от внешних врагов, при
половом размножении и в целях маскировки. В настоящее время
описано более 30 различных биолюминесцентных систем у бак-
бактерий, грибов, червей, ракообразных, насекомых и рыб [186]. Во
всех этих системах, как и в случае с рассмотренным экворином,
383
Таблица 11
Примеры биолюминесцентных систем различных организмов
Организм (таксон)
Бактерии (фотобактерии;
Vibrio)
Жгутиконосцы (Gonylaulax;
Pyrocystis)
Стрекающие (Aequorea; Obelia)
Аннелиды (Diplocardia)
Моллюски (Latia)
Ракообразные (Vargula;
Cypridina)
Насекомые (Photinus; Photuris)
Люциферин и другие кофакторы
FMNH2; RCHO
Тетрапиррол; Н+
Колентеразин; Са2+
N-Изовалерил-З-аминопропа-
нал; Н2О2
Енолформиат; терпен
ароматического альдегида
Ядро имидазопиразина
(Бензо)тиазол; ATP; Mg2+
^макс нм
495-500
475
460-490
500
500
560
465
происходит ферментативная реакция окисления субстратов, из-
известных под общим названием люциферинов, сопровождаемая
испусканием видимого света. В таких процессах участвуют окси-
геназы, получившие название люцифераз, которые, как правило,
используют молекулярный кислород для окисления связанных с
ними люциферинов, переводя эти молекулы в возбужденное со-
состояние по следующему механизму (звездочка обозначает воз-
возбужденное состояние молекулы):
Люциферин + О2 (или Н2О2) —>
—> Оксилюциферин* —> Оксилюциферин + ho
Субстраты в различных реакциях биолюминесценции существен-
существенно различаются по своей структуре и принадлежат к разным
классам химических соединений. Примеры светящихся организ-
организмов и их субстратов приведены в табл. 11 [186].
Сам процесс свечения живых организмов также может быть
различным. Одни организмы обладают способностью испускать
свет непрерывно, тогда как другие - короткими вспышками, ко-
которые продолжаются менее 10 сек. В последнем случае в начале
вспышки происходит быстрый запуск химической реакции с по-
последующим истощением субстрата и его мобилизацией к момен-
моменту следующей вспышки. Запуск реакции в этом случае может
быть результатом быстрого изменения значений рН в светящей-
светящейся органелле, доступности молекул кислорода или ионов кальция
[187]. В настоящее время охарактеризованы, по крайней мере,
384
семь фотопротеинов: талассиколин (thalassicolin), экворин
(aequorin), митрокомин (mitrocomin), также известный как халис-
таурин (halistaurin), клитин (clytin), известный и под именем фиа-
лидина (phialidin), обелин (obelin), мнемиопсин (mnemiopsin) и бе-
ворин (bevorin). Среди Са2+-зависимых фотопротеинов экворин
был выделен и охарактеризован одним из первых в 1962 г. и в по-
последующие годы вместе с ассоциированным с ним зеленым флу-
флуоресцирующим белком получил широкое распространение в ка-
качестве белка-репортера в искусственных генетических системах.
Система экворина и зеленого флуоресцирующего белка
(GFP). Зеленый флуоресцирующий белок (green fluorescent pro-
protein - GFP) вызывает характерное свечение тела медузы Aequoria
victoria, которая имеет форму зонтика, диаметром 7-10 см и оби-
обитает в северо-западной части Тихого океана. Световой орган ме-
медузы представлен мелкими гранулами, распределенными по
краю зонтика. О. Шимомуре с соавт. удалось показать, что пре-
препараты медузы в присутствии КС1 светятся зеленым светом, а по-
после лизиса клеток - голубым [188]. Позднее было установлено,
что голубое свечение обусловлено Са2+-связывающим фотобел-
фотобелком экворином, а зеленое - GFP, возбужденным в результате пе-
переноса энергии без излучения от экворина. Этот феномен био-
биолюминесценции редко встречается в живой природе и характе-
характерен, главным образом, для морских организмов. Экворин облада-
обладает широким спектром люминесценции с максимумом при 469 нм,
тогда как спектр флуоресценции GFP узок, и его максимум нахо-
находится при 508 нм [189].
Экворин. Фотобелок экворин состоит из трех компонентов:
апоэкворина, колентеразинового хромофора и молекулярного
кислорода [190]. Полипептидная цепь экворина, построенная из
189 а.о., содержит три Са2+-связывающих сайта. После связыва-
связывания ионов Са2+ экворин претерпевает конформационную перест-
перестройку, сопровождаемую окислением связанного с ним коленте-
колентеразина с образованием нестабильного производного диоксетано-
на, что приводит к освобождению молекулы СО2 и кванта света
(рис. 52, а). При этом люминесценцию экворина индуцируют ио-
ионы Са2+ в пределах концентраций 0,1-10 мкМ. Регенерацию экво-
экворина проводят, диализуя препарат белка против ЭДТА для удале-
удаления ионов Са2+, с последующим добавлением колентеразина. По
данным рентгеноструктурного анализа молекула экворина со-
содержит гидрофобную центральную часть, с которой ассоцииро-
ассоциирован колентеразин, находящийся в форме перекиси. Испускание
света экворином зависит от концентрации ионов Са2+, что позво-
позволяет его использовать для количественного определения данных
13. Патрушев Л.И. Т. 1
а
но'
Колентеразин
0-0,
Производное диоксетанона
но
'ОН
о
v N"
hv D69 нм)
+0,
но'
-он
Колентерамид
Вид сбоку
Вид сверху
6
О
1. Циклизация
2. Окисление
Рис. 52. Структурные и функциональные особенности системы эквори-
на и GFP [193]
а - предполагаемый механизм реакции хемилюминесценции, осуществляе-
осуществляемой экворином;
б-пространственная структура GFP. Молекула содержит 11 |3-слоев, окру-
окружающих центральный ос-спиральный участок;
в - механизм образования хромофора в GFP из трех аминокислотных ос-
остатков его полипептидной цепи; Р - продолжение полипептидной цепи
386
ионов в различных компартментах живой клетки. Экворин так-
также применяется в качестве эффективного репортера, так как его
наличие уже в аттомолярных концентрациях можно легко обна-
обнаруживать при низких значениях сигнала фоновой биолюминес-
биолюминесценции.
Экворин отвечает большинству требований, предъявляемых
к белкам-репортерам. Тем не менее, широкое его использование
в молекулярно-биологических исследованиях, по-видимому,
сдерживается зависимостью от внешнего субстрата, колентера-
зина, а также характером люминесценции, которая происходит в
виде вспышки с продолжительностью ~5 сек, что создает опреде-
определенные затруднения в детекции белка в динамических опытах.
Однако благодаря чрезвычайно высокой чувствительности сис-
систем, созданных на основе экворина, он постепенно внедряется в
практику, хотя несоизмеримо более низкими темпами, чем его
природный партнер - GFP.
Зеленый флуоресцирующий белок и его производные. Перво-
Первоначально обнаруженный в медузе GFP синтезируется и другими
морскими организмами. У медузы этот белок осуществляет сме-
смещение спектра биолюминесценции из голубой области в зеле-
зеленую, что сопровождается повышением квантового выхода,
уменьшением светорассеяния и распространением света на боль-
большие расстояния. Полипептидная цепь GFP построена из 238 а.о.
молекулярной массы 27 кДа и организована в 11-цепочечный
C-ствол, внутри которого находится ос-спиральный участок
(рис. 52, б). Хромофор GFP представляет собой 4-(и-гидроксибен-
зилиден)-имидазолидин-5-он, образуемый в результате посттран-
посттрансляционной модификации полипептидной цепи, в которой участ-
участвуют аминокислотные остатки Ser-65, Tyr-66 и Gly-67. Образова-
Образование хромофора происходит в два этапа: реакции циклизации
между этими аминокислотными остатками и последующего
окисления с участием молекулярного кислорода. Путем укорачи-
укорачивания полипептидной цепи было показано, что, за небольшим ис-
исключением, для функционирования белка важны все его амино-
аминокислотные остатки. Изменение полипептидной цепи GFP с помо-
помощью мутаций позволяет повышать или понижать стабильность
белка, увеличивать эффективность образования его хромофора
и получать молекулы с новыми спектральными характеристика-
характеристиками, представленными в табл. 12.
GFP дикого типа обладает двумя максимумами поглощения
при 395 и 470 нм и одним максимумом испускания при 509 нм.
Наличие двух максимумов поглощения обусловлено присутст-
присутствием двух химически различающихся форм хромофора - анион-
13* 387
Таблица 12
Флуоресцентные свойства GFP и его мутантных производных
Мутант
Мутации
Возбуждение
макс, (нм)
Испускание
макс, (нм)
Квантовый
выход
Дикий тип
EGFP S65T
BFP Y66H, Y145F
YFP S65G, V68L,
S72A, T203Y
CFP T66W
Примечание. В скобках отмечены
395 D70)
488
380
513D98)
433 D53)
дополнительные
509
509
440
527
475 E01)
максимумы.
0,77
0,70
0,20
0,63
0,24
ной и нейтральной. Замена Ser-65 —> Thr приводит к образова-
образованию мутанта, получившего название улучшенного GFP
(enhanced GFP-EGFP) с единственным максимумом возбуждения
при 488 нм. Этот аналог, в отличие от белка дикого типа, может
претерпевать эффективный фолдинг при 37 °С, и интенсив-
интенсивность его флуоресценции в живых клетках значительно выше,
чем у исходного белка. Двойной мутант Туг-66 —> His, Туг-145 —>
—> Phe, получивший название голубого флуоресцирующего бел-
белка (BFP), после возбуждения при 381 нм испускает свет с длиной
волны 445 нм. Голубое излучение BFP может возбуждать EGFP,
в связи с чем одновременное использование пары этих белков в
качестве репортеров позволило разработать популярную груп-
группу методов на основе резонансного переноса энергии при флуо-
флуоресценции (fluorescence resonance energy transfer - FRET). Два
других мутанта - циановый флуоресцирующий белок (CFP) и
желтый флуоресцирующий белок (YFP) также применяются во
FRET-системах.
Замечательным свойством GFP является его полная функци-
функциональная независимость от каких-либо дополнительных кофак-
кофакторов. Кроме того, белок мало токсичен для клеток, легко обна-
обнаруживается и его внутриклеточное содержание можно оценивать
количественно с высокой точностью. GFP стабилен при значени-
значениях рН 5-12, устойчив к действию протеиназ и высоких темпера-
температур (вплоть до 65 °С), не инактивируется в присутствии детерген-
детергентов A% SDS), 6M хлористого гуанидина, а также в препаратах,
фиксированных формальдегидом. При этом флуорофор практи-
практически не обесцвечивается в процессе флуоресцентной микроско-
микроскопии. Из-за небольшого размера GFP оказывает минимальное
влияние на подвижность и локализацию гибридного белка-парт-
белка-партнера, что делает особенно удобным его использование в генно-
388
инженерных конструкциях, применяемых для исследования экс-
экспрессии генов в живых тканях in vivo.
GFP-подобные белки. В последнее время начинают полу-
получать широкое применение GFP-подобные белки кораллов клас-
класса Anthozoa, гены которых впервые были клонированы и экс-
прессированы в группе С.А. Лукьянова в Институте биооргани-
биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинни-
Овчинникова РАН (Москва) [194]. Такие GFP-подобные белки образуют
семейство полипептидов с молекулярной массой 25-30 кДа, флу-
флуоресцирующих в зеленой (GFP), желтой (YFP) и красной (RFP)
областях спектра, а также не обладающих способностью к флу-
флуоресценции (nonfluorescent chromoproteins - СР). Введение в прак-
практику GFP-подобных белков и мутантных форм GFP позволяет
использовать в исследованиях феномен биофлуоресценции бел-
белков-репортеров в широкой области спектра - 442-645 нм. GFP-
подобные белки так же, как и GFP, формируют хромофорную
группу из аминокислотных остатков своих полипептидных цепей
и не требуют для этого никаких дополнительных кофакторов
кроме молекулярного кислорода. В общей сложности у флуо-
флуоресцирующих белков в настоящее время известно, по крайней
мере, четыре типа хромофорных групп, различающихся по сво-
своей структуре, что, наряду с эффектами разных внутримолеку-
внутримолекулярных взаимодействий хромофоров и окружающих аминокис-
аминокислотных остатков, объясняет спектральное разнообразие свойств
этих макромолекул.
Введение мутаций в гены GFP-подобных белков позволяет
изменять максимумы испускания и квантовый выход исходных
макромолекул, в том числе, создавать двухцветные белки с двумя
максимумами испускания. Кроме того, удается получать из не-
флуоресцирующего СР полипептиды, которые испускают кван-
кванты света в дальней красной области спектра, что само по себе,
по-видимому, не является пределом возможностей подходов, ос-
основанных на мутагенезе генов GFP-подобных белков. Одним из
особо примечательных мутантов красного флуоресцирующего
белка RFP, известного под коммерческим названием dsRed, явля-
является белок dsRed-E5, который изменяет свой цвет с зеленого на
красный во времени, что отражает процесс формирования его
хромофора и происходит в результате резонансного переноса
энергии флуоресценции (FRET) между рано созревающим E-6 ч
после индукции экспрессии) и поздно созревающим (> 9 ч) моно-
мономерами в тетрамере dsRed-E5. Это позволяет использовать му-
мутант в качестве своеобразного флуоресцентного хронометра
(fluorescent timer) в клеточной биологии.
389
Одной из серьезных проблем, ограничивающих применение
GFP-подобных белков Anthozoa является олигмеризация в
физиологических условиях. Это не мешает их использованию в
качестве простых репортеров экспрессии генов, но превращает-
превращается в серьезное ограничение при получении гибридных белков
особенно в том случае, когда исследуемый белок сам существует
в виде олигомера. В этом случае производные dsRed, как прави-
правило, образуют внутриклеточные агрегаты. Решение этой пробле-
проблемы может лежать на пути создания мутантов dsRed с нарушенной
олигомеризацией. К преимуществам белков этого семейства сле-
следует отнести низкую аутофлуоресценцию в красной области спе-
спектра, и возможность раздельной детекции их флуоресценции и
флуоресценции GFP (в том числе его производных), что допуска-
допускает многоцветное окрашивание биологических объектов.
Использование экворина, GFP и GFP-подобных белков в ис-
искусственных генетических системах. Благодаря своим уникаль-
уникальным свойствам экворин активно используется в качестве флуо-
флуоресцентной метки в иммуноферментных методах, в зондах на ос-
основе нуклеиновых кислот, а также в высокочувствительных сис-
системах количественного определения содержания универсального
вторичного мессенджера Са2+ внутри клеток. Изменение концен-
концентрации ионов Са2+ в цитоплазме и ее микрокомпартментах часто
сопровождает стимуляцию внеклеточных рецепторов, принадле-
принадлежащих к обширному классу рецепторов, сопряженных с G-белка-
ми. Эти рецепторы активируются в ответ на многочисленные
внеклеточные стимулы, включая воздействия светом, нейроме-
диаторами, гормонами и веществами, имеющими запах. В по-
последнее время в исследованиях по функциональной геномике от-
открывается большое количество новых рецепторов, для которых,
как правило, неизвестны эндогенные лиганды [191]. Кроме того,
оценивать уровень активации этих рецепторов необходимо при
разработке новых лекарственных препаратов, специфически вза-
взаимодействующих с ними. Для решения таких задач были разра-
разработаны автоматизированные высокочувствительные методы
оценки активности рецепторов с использованием экворина в ка-
качестве белка-репортера [192].
На основе экворина были созданы клеточные микросенсоры
для быстрого обнаружения патогенных вирусов и бактерий. Для
этой цели генно-инженерными методами были получены В-кле-
В-клеточные линии, экспрессирующие на своей поверхности молеку-
молекулы разных иммуноглобулинов, специфичных в отношении кон-
конкретных возбудителей, а также апоэкворин. Взаимодействие им-
иммуноглобулинов с лигандами запускает внутриклеточный каскад
390
передачи сигналов, приводящих к освобождению ионов Са2+, и,
как следствие, люминесценции экворина. Клетки иммобилизова-
иммобилизовали на стеклянной подложке фотолитографическим методом, ко-
которую помещали в небольшую проточную камеру, содержащую
питательную среду. Процесс обнаружения возбудителей в такой
системе занимает менее одной минуты.
Зеленый флуоресцирующий белок, благодаря своей способ-
способности к аутофлуоресценции, активно используется в качестве ре-
репортера для исследования в реальном времени молекулярных
процессов, происходящих в живых клетках. Одно из ограничений
его применения связано с наличием в клетках других флуоресци-
флуоресцирующих молекул, которые в ряде случаев снижают чувствитель-
чувствительность обнаружения GFP. Кроме того, значительная внутрикле-
внутриклеточная стабильность этого белка (у GFP дикого типа время полу-
полужизни более 24 ч) затрудняет его использование для оценки
транскрипционной активности генов, так как его внутриклеточ-
внутриклеточное содержание уже может не коррелировать с наличием соот-
соответствующих мРНК. В таком случае используют менее стабиль-
стабильный мутантный аналог EGFP, так называемый дестабилизиро-
дестабилизированный EGFP (d2EGFP), время полужизни которого в клетке со-
составляет ~2 ч. d2EGFP оказывается пригодным для исследования
изменения уровней экспрессии генов в процессе передачи сигна-
сигналов, в том числе при внешних воздействиях на клетку, что успеш-
успешно применяется фармацевтическими фирмами для скрининга но-
новых лекарственных препаратов [190].
GFP и его аналоги представляют собой исключительно удоб-
удобные белки-репортеры для анализа экспрессии генов [193]. В ча-
частности, добавление к любому из концов его полипептидной це-
цепи сигнальных последовательностей не нарушает способность
GFP к флуоресценции, однако позволяет контролировать его
внутриклеточную локализацию в исследуемых компартментах,
например, в ядре или митохондриях. Объединение GFP в одной
гибридной полипептидной цепи с белковыми рецепторами (в ча-
частности, глюкокортикоидным) или лигандами, также часто не
нарушает функциональных свойств ни одной из макромолекул
гибрида. С помощью такой системы можно следить за перемеще-
перемещением соответствующих белков и их комплексов в живых клетках
и тканях как при проведении фундаментальных исследований,
так и прикладных, например при разработке систем для геноте-
рапии.
GFP-подобные белки Anthozoa в настоящее время эффек-
эффективно используются в качестве пассивных белков-репортеров
при исследовании регуляторных элементов генов бактерий, жи-
391
вотных и растений. Кроме того их применяют в качестве репор-
репортеров активации генов в биосенсорах, основанных на бактери-
бактериальных клетках, при исследовании механизмов дифференци-
ровки клеток, для слежения за динамикой внутриклеточных пе-
перемещений белков, органелл и вирусных частиц, при исследова-
исследовании механизмов фагоцитоза. Интересные перспективы в приме-
применении этих белков открываются в связи с недавним обнаруже-
обнаружением способности исходно нефлуоресцирующих белков asulCP
приобретать эту способность после облучения зеленым светом.
Через некоторое время белок возвращается в свое первона-
первоначальное состояние или может быть искусственно "потушен" об-
облучением голубым светом. В результате мутагенеза генов бел-
белков этого семейства в группе К.А. Лукьянова (ИБХ, Москва)
были получены так называемые зажигающиеся флуоресцирую-
флуоресцирующие белки (kindling fluorescent proteins - KFPs), которые под дей-
действием света определенной длины волны могут быть превраще-
превращены из СР-белков во флуоресцирующие FP. Более продолжи-
продолжительное облучение исходных молекул может переводить их в
необратимое "зажженное" состояние. Активированные таким
образом KFP начинают флуоресцировать в красной области
спектра, что может быть использовано для слежения за переме-
перемещением клеток в тканях или внутриклеточным движением ор-
органелл. Кратковременное обратимое "зажигание" таких белков
позволяет последовательно получать картины флуоресцирую-
флуоресцирующих биообъектов в разные промежутки времени эксперимента.
Более того, высушенные KFP сохраняют способность претерпе-
претерпевать аналогичные превращения под действием света, следова-
следовательно могут быть использованы для получения биосенсоров, а
также в современных нанотехнологиях для хранения информа-
информации и других не менее важных целей.
3.2.3. Гибридные токсины
Концепция "магической пули", которая бы избирательно по-
поражала больные (в том числе раковые) клетки и оставляла не-
невредимыми здоровые, до сих пор остается востребованной. Иде-
Идеальное лекарственное средство строго специфического (избира-
(избирательного) действия должно обладать, по крайней мере, следую-
следующими структурно-функциональными особенностями (рис. 53, а).
Препарат должен заключать в себе действующее начало для до-
достижения физиологического эффекта и лиганд, распознающий
рецептор на поверхности клеток-мишеней. Кроме того, в нем
должны быть структурные элементы, узнаваемые системой
392
NH2
NH2
а
Лиганд
Переносчик
Спейсер
Действующее начало
Узнавание
мишени
б
Защита,
транспорт
Отщепление
лекарства
Физиологический
эффект
la
II
Ib
III
соон
252
365 404
613
в
Лиганд
II
Транслокация
III
Токсичность
NH2
vL
II
III
Рис. 53. Лекарственные препараты направленного действия на основе
гибридных токсинов
а - обобщенная схема структуры лекарственного препарата направленно-
направленного действия;
б - строение псевдомонадного токсина (цифрами обозначено положение
аминокислотных остатков);
в - строение гибридного токсина;
г - гибридный токсин на основе моноклональных миниантител
транспорта организма, для доставки лекарства к клеткам-мише-
клеткам-мишеням, а также спейсерный участок, необходимый для отделения
действующего начала от остальных функциональных частей пре-
препарата после его доставки по адресу. Именно такая идеальная
схема реализуется в природном экзотоксине Pseudomonas aerugi-
nosa. Исследование возможности его использования в качестве
действующего начала в гибридных токсинах было проведено од-
одним из первых И. Пастаном [195].
Экзотоксин А Р. aeruginosa представляет собой белок, состо-
состоящий из одной полипептидной цепи длиной в 613 аминокислот,
которая организована в три функциональных домена (см.
рис. 53, б). N-Концевой домен 1а (аминокислотные остатки 1-252)
необходим для взаимодействия с рецепторами ос2-макроглобули-
на на поверхности клеток-мишеней (прототип лиганда идеально-
идеального лекарства направленного действия) [196]. Функции домена Ib
(аминокислотные остатки 365-404) неизвестны, и он может быть
удален из полипептидной цепи токсина без потери активности.
Домен II (аминокислотные остатки 253-364) обеспечивает эф-
эффективный перенос токсина в цитозоль клеток (система транс-
393
порта лекарства), а домен III (аминокислотные остатки 405-613)
осуществляет ADP-рибозилирование фактора элонгации транс-
трансляции EF2, что приводит к подавлению трансляции и гибели кле-
клеток-мишеней. Эффективность действия псевдомонадного токси-
токсина чрезвычайно высока. Его внутривенное введение мышам в ко-
количестве 0,3 мкг приводит к смерти животных в течение суток. А
гибель культивируемых клеток вызывает еще меньшее количе-
количество молекул токсина.
Таким образом, для оказания цитотоксического действия эк-
экзотоксину А необходимо с помощью домена 1а распознать рецеп-
рецепторы на поверхности клеток, проникнуть в клетку с помощью эн-
доцитоза, опосредованного рецепторами, и быть транслоциро-
ванным через внутреннюю мембрану в цитозоль, где локализует-
локализуется фактор EF2 [196]. Основная идея в создании токсинов направ-
направленного действия заключалась в том, чтобы заменить домен 1а на
какой-либо иной пептидный лиганд, взаимодействующий с дру-
другой группой рецепторов на поверхности клеток, и тем самым из-
изменить специфичность действия токсина в отношении самих кле-
клеток (рис. 53, в).
Было установлено, что удаление домена 1а генно-инженер-
генно-инженерными методами резко (в сотни и тысячи раз) снижает токсич-
токсичность такого укороченного белка как в отношении клеток раз-
различных линий, так и in vivo. Присоединение к С-концевой части
укороченного полипептида молекулы интерлейкина 2 человека
осуществляли путем объединения структурных частей соответст-
соответствующих генов в экспрессирующем векторе. Очищенный гибрид-
гибридный токсин оказался чрезвычайно токсичным в отношении кле-
клеток, несущих на своей поверхности рецепторы интерлейкина 2, и
не действовал на клетки, у которых эти рецепторы отсутствова-
отсутствовали и которые погибали под действием природного токсина. Ин-
тернализация (транслокация внутрь клеток) гибридного токсина
была опосредована субъединицами р55 и р70 рецептора интер-
интерлейкина 2. Таким образом, в результате действия гибридного
токсина на популяцию клеток, часть из которых экспрессирует
на своей поверхности рецепторы интерлейкина 2, происходит из-
избирательная гибель именно этих клеток.
В организме большинство покоящихся Т-клеток и Т-клеток
памяти не экспрессируют на своей поверхности высокоаффин-
высокоаффинных рецепторов интерлейкина 2, тогда как Т-клетки, стимулиро-
стимулированные аллоантигенами, содержат такие рецепторы. Поэтому
внутрибрюшинное введение гибридного токсина крысам с экспе-
экспериментальным артритом - заболеванием, обусловленным пато-
патологической активацией Т-клеток, снижало симптомы заболева-
394
ния. Гибридный токсин существенно уменьшал у мышей и реак-
реакции отторжения трансплантата.
Вслед за этими пионерскими работами последовала целая се-
серия исследований, направленных на создание аналогичных сис-
систем адресной доставки различных цитотоксических полипепти-
полипептидов. В процессе дальнейшего усовершенствования системы ад-
адресной доставки псевдомонадного токсина с использованием ин-
терлейкина 2 в качестве лиганда исследователи отказались от
полного удаления адресного домена токсина и ограничились его
инактивацией путем введения в ген токсина четырех сайт-специ-
сайт-специфических мутаций. Молекулы такого гибридного токсина оказа-
оказались в 10-100 раз более эффективными цитотоксическими аген-
агентами против клеток человека и обезьян, экспрессирующих на
своей поверхности рецепторы для интерлейкина 2, а также обла-
обладали значительно большим временем полужизни в крови мышей
in vivo по сравнению с ранее полученной конструкцией.
На основе псевдомонадного токсина были созданы гибрид-
гибридные токсины, содержащие в качестве лигандов полипептидные
цепи интерлейкинов 4 и 6, трансформирующего фактора роста
типа а и инсулиноподобного фактора роста I. Для всех этих гиб-
гибридных белков была показана высокоспецифическая цитоток-
сичность в отношении опухолевых клеток (включая клетки мие-
ломы человека), обладающих соответствующими рецепторами.
Использование в гибридном токсине в качестве лиганда части по-
полипептидной цепи CD4 - гликопротеина поверхности Т-клеток,
который является рецептором вируса ВИЧ и взаимодействует с
его антирецептором, гликопротеином gpl20, позволило избира-
избирательно поражать Т-клетки, зараженные вирусом ВИЧ и экспрес-
сирующие на своей поверхности вирусный белок gpl20.
Тот же принцип подавления инфекции, вызванной вирусами
ВИЧ, растворимыми рецепторами CD4 был использован при
конструировании гибридных белков, объединяющих части поли-
полипептидных цепей CD4 с константными частями тяжелых или лег1
ких цепей иммуноглобулинов человека. При этом в процессе
объединения генов были удалены последовательности нуклеоти-
дов, кодирующие трансмембранный и цитоплазматический доме-
домены CD4, а также вариабельную часть полипептидных цепей им-
иммуноглобулинов. Образующиеся гибридные молекулы, назван-
названные иммуноадгезинами, за счет константной части молекулы им-
иммуноглобулина приобретали повышенную стабильность в орга-
организме и, кроме того, сохраняли специфические свойства, опосре-
опосредуемые константными частями иммуноглобулинов: связывание
Fc-рецептора и белка А, способность к фиксации комплемента и
395
перенос через плацентарный барьер. Совокупность всех этих
свойств давала возможность иммуноадгезинам прерывать инфек-
инфекцию Т-клеток вирусом ВИЧ-I, блокируя как сам вирус, так и за-
зараженные им клетки, экспрессирующие на своей поверхности ви-
вирусный антиген gpl20.
Дальнейшее усовершенствование генно-инженерных конст-
конструкций на основе псевдомонадного экзотоксина А произошло по-
после того, как в качестве адресной части гибридного токсина ста-
стали использовать вариабельные домены шАЬ к компоненту р55
рецептора интерлейкина 2 человека. В этом рекомбинантном
белке с помощью 15-звенного пептидного линкера аминокислот
соединяли вариабельный домен тяжелой цепи иммуноглобулина
с вариабельным доменом его легкой цепи, а С-конец легкой це-
цепи - с N-концом укороченного псевдомонадного токсина
(рис. 53, г). Такие молекулы гибридного токсина также оказались
высокоспецифичными цитотоксическими агентами по отноше-
отношению к лейкозным клеткам человека, экспрессирующим на своей
поверхности рецепторы интерлейкина 2.
Разработанный подход показал возможность использования
специфических антител в качестве адресных частей гибридных
токсинов. Это дает в руки исследователей универсальный спо-
способ адресной доставки токсинов, который в будущем позволит
оказывать цитотоксическое действие на любые группы клеток,
экспрессирующих на своей поверхности специфические антиге-
антигены, т.е. значительно расширить количество мишеней для хи-
миотерапевтических воздействий с помощью рекомбинантных
белков.
Помимо псевдомонадного экзотоксина А в качестве действу-
действующего начала в гибридных токсинах успешно применяли дифте-
дифтерийный токсин, фактор некроза опухолей и А-цепь рицина. По-
Поскольку А-белок избирательно взаимодействует с константными
(Fc) частями иммуноглобулинов класса G многих млекопитаю-
млекопитающих, такой гибридный токсин в паре с иммуноглобулином, полу-
полученным против какого-либо антигена на поверхности клеток, из-
избирательно связывается с этими клетками и убивает их. Иммуно-
токсины являются еще одним потенциальным противоопухоле-
противоопухолевым агентом и могут быть использованы против клеток, экспрес-
экспрессирующих на своей поверхности специфические антигены. В на-
настоящее время некоторые иммунотоксины уже прошли клиниче-
клинические испытания и активно применяются в клинической практике
США и Западной Европы для лечения онкологических и аутоим-
аутоиммунных заболеваний [197-199].
396
3.3. Экстремозимы
Современные методы генной инженерии являются основой
получения любого фермента в чистом виде и, практически, в не-
неограниченном количестве. Это позволяет использовать фермен-
ферменты в биотехнологической промышленности для крупномасштаб-
крупномасштабного проведения химических реакций с эффективностью, кото-
которая, как правило, недоступна методам органической химии. Су-
Существенным ограничением широкого применения ферментов в
промышленности является низкая стабильность, поскольку их
первичным источником чаще всего являются мезофильные орга-
организмы, т.е. организмы, существующие в условиях обычной тем-
температуры, давления и других параметров окружающей среды.
Тем не менее имеется обширная группа микроорганизмов, полу-
получивших название экстремофилов, обитающих в экстремальных
условиях окружающей среды: температуре в пределах -2°-
-15 °С и 60°-110 °С, высокой ионной силе B-5М NaCl), или зна-
значениях рН <4 и >9. Большинство экстремофилов относится к
Archaea, одному из трех филогенетических доменов жизни (эука-
риоты, эубактерии и археи), различаемых на основании первич-
первичной структуры 16S рРНК, однако экстремофилы встречаются и
среди бактерий [200].
3.3.1. Экстремофилы и их ферменты
Экстремофилы обладают ферментами, для которых харак-
характерна чрезвычайно высокая стабильность в экстремальных усло-
условиях проведения ферментативных реакций. Такие ферменты по-
получили название экстремозимов [201]. Последние весьма привле-
привлекательны для биотехнологии, так как обладают свойствами, ко-
которые ранее считались несовместимыми с биологическими объ-
объектами. Более того, сам факт наличия экстремозимов указывает
на принципиальную возможность изменения обычных фермен-
ферментов методами белковой инженерии для увеличения их стабильно-
стабильности. При этом экстремозимы, по-видимому, задают верхнюю
планку параметров ферментов, к которым белковые инженеры,
работающие в соответствующей области биотехнологии, могут
и, возможно, должны стремиться.
Считается, что ферменты максимально адаптированы к
функционированию в условиях, оптимальных для роста организ-
организма-хозяина. Классическим путем получения бактериальных экс-
экстремозимов для биотехнологических целей является выделение
микроорганизма в виде чистой культуры с последующей очист-
очисткой фермента из биомассы, которую обычно выращивают в фер-
397
ментерах. Альтернативно, из ДНК клеток чистой культуры вы-
выделяют ген требуемого фермента и с использованием экспресси-
рующего вектора клонируют его в бактериальных клетках, удоб-
удобных для крупномасштабного культивирования, чаще всего в
клетках Е. coli. К сожалению, в настоящее время лишь 0,1-1,0%
микроорганизмов окружающей среды могут быть выделены и
культивированы в чистом виде. Для использования ферментатив-
ферментативных ресурсов этих организмов применяют другие подходы. В ча-
частности, ДНК, выделяемую прямо из почвы или других объектов
окружающей среды, используют для получения так называемых
клонотек ДНК окружающей среды (environmental libraries), кото-
которые являются неисчерпаемым источником новых генов.
Альтернативно с помощью методов белковой инженерии можно
модифицировать известные мезофильные ферменты для прида-
придания им новых свойств, которые бы позволили ферментам функ-
функционировать в экстремальных условиях. Реализация этого подхо-
подхода требует знаний механизмов, обеспечивающих природным экс-
тремозимам их исключительные качества.
Исследование первичной и пространственной структуры экс-
тремозинов выявило ряд существенных особенностей их строе-
строения, отличающих эти ферменты от мезофильных гомологов. В
частности, для ферментов термофилов и гипертермофилов ха-
характерно наличие развитой сети внутримолекулярных ионных
взаимодействий, в том числе и между субъединицами [202]. Это
приводит к формированию у таких ферментов более компактной
пространственной структуры. Тем не менее, данный механизм
стабилизации термофильных ферментов не является универсаль-
универсальным, и у гомологов различных видов термофилов могут быть за-
задействованы разные механизмы, например, дополнительные гид-
гидрофобные взаимодействия вместо ионных. У р-гликозидазы
Sulfolobus solfataricus развитая сеть ионных взаимодействий на
поверхности молекулы сочетается с наличием гидрофильных ка-
каналов, проникающих глубоко в белковую глобулу, что также
считается фактором, способствующим стабилизации этого фер-
фермента [203]. В целом, в настоящее время еще не установлены
универсальные закономерности, определяющие высокую термо-
термостабильность ферментов, что само по себе может оказаться об-
общим правилом, указывающим на уникальность каждого стабили-
стабилизирующего механизма.
В отличие от гипертермофилов ферменты психрофильных
микроорганизмов функционируют в условиях низких температур
и быстро инактивируются уже при умеренных температурах.
Кристаллографическое изучение этих ферментов показывает,
398
что для них характерна более высокая гибкость полипептидных
цепей по сравнению с соответствующими функциональными ана-
аналогами мезофильных организмов [204]. Кроме того, защита та-
таких ферментов от денатурации в условиях низких температур ча-
частично достигается ослаблением гидрофобных внутримолеку-
внутримолекулярных взаимодействий в этих условиях.
Экстремозимы галофилов адаптированы к функционирова-
функционированию при высокой ионной силе, поскольку ионные условия внут-
внутри клеток данных микроорганизмов изотоничны по отношению
к окружающей среде. Например, внутри клеток археи
Methanopyrus kandleri, размножающейся при 98 °С, концентрация
фосфата достигает 1,5 М [205, 206]. Белки галофильных бакте-
бактерий являются значительно более кислыми по сравнению с анало-
аналогами мезофилов и содержат существенно большее количество
отрицательно заряженных аминокислотных остатков на своей
поверхности. Полагают, что эти остатки удерживают гидратиру-
ющие ионы у поверхности белковой глобулы, а также, будучи ги-
гидрофильными, препятствуют агрегации молекул в условиях вы-
высокой ионной силы за счет гидрофобных межмолекулярных вза-
взаимодействий. Сети внутримолекулярных взаимодействий также
вносят вклад в поддержание особых свойств ферментов этой
группы.
У ацидофильных и алкалифилъных экстремофилов, которые
существуют при сильно кислых и сильнощелочных значениях рН
окружающей среды, условия внутри клеток близки к нейтраль-
нейтральным. Тем не менее, внеклеточные ферменты этих микроорганиз-
микроорганизмов вынуждены функционировать именно в таких условиях и об-
обладают повышенной стабильностью.
3.3.2. Достижения белковой инженерии
в повышении стабильности ферментов
Осмысленное следование законам природы является основой
успеха конструирования искусственных генетических систем во-
вообще и новых белков в частности. Попытки использовать знания
о механизмах функционирования природных экстремозимов при
конструировании белков с помощью рационального редизайна
уже приносят определенные плоды. Так, введение в белки допол-
дополнительных внутримолекулярных ионных связей путем добавле-
добавления в полипептидную цепь новых аминокислотных остатков при-
приводит к их стабилизации, хотя часто ценой уменьшения удельной
активности [201]. Элиминация же таких связей дестабилизирует
ферменты. Конформационная гибкость ферментов тесно связа-
399
на с их каталитическими функциями. Методами белковой инже-
инженерии было установлено, что термостабильность фермента и его
способность катализировать химическую реакцию, по крайней
мере, частично не зависят друг от друга, при этом мутации, кото-
которые усиливали бы данные свойства, встречаются редко.
Распространенным способом получения экстремозимов яв-
является их биосинтез в мезофильных микроорганизмах. В этой
связи, уместно вспомнить о нашей работе, завершившейся со-
созданием высокопродуктивного штамма продуцента термоста-
термостабильной ДНК-полимеразы Thermus aquaticus YT1 [207]. Ген
фермента был клонирован в плазмидном экспрессирующем
векторе (см. раздел 4.6 первой части книги) под контролем тер-
моиндуцируемого промотора и введен в клетки Е. coli. Первич-
Первичная структура гена была адаптирована для его эффективной
экспрессии в данной искусственной генетической системе, что
потребовало замены двух аминокислотных остатков в N-конце-
вой части рекомбинантного фермента. В итоге из 100 г биомас-
биомассы удается получать до 2 млн единиц активности высокоочи-
щенного фермента, который в настоящее время используется
многими лабораториями для проведения ПЦР. Благодаря вы-
вышеупомянутым заменам, улучшившим потребительские свойст-
свойства белка, наш фермент содержит еще и фирменную N-конце-
вую метку из уникальных аминокислотных остатков, что позво-
позволяет при желании легко идентифицировать его среди фермен-
ферментов других производителей.
Плодотворным подходом в белковой инженерии этого на-
направления является искусственный перенос особенностей пер-
первичной и пространственной структур экстремофилов к мезо-
фильным аналогам, т.е. использование методов рационального
дизайна и редизайна. В частности, введение нескольких мутант-
ных аминокислотных остатков в полипептидную цепь термоли-
зиноподобной протеиназы умеренного термофила Bacillus
stearothermophilus позволило в 340 раз повысить стабильность
фермента при 100 °С по сравнению с исходным белком со време-
временем полужизни в таких условиях - 170 мин и сохранить неизмен-
неизменной удельную активность фермента [208]. Не удивительно, что
этот фермент стал гидролизовать субстраты, устойчивые к дей-
действию исходной протеиназы. Как полагают, такие мутации, при-
приводящие к заменам Gly —> Ala и Ala —> Pro, а также образованию
новой дисульфидной связи, сопровождаются понижением значе-
значений энтропии для денатурированной формы фермента, которая
становится из энергетических соображений менее предпочти-
предпочтительной по сравнению с нативной конформацией.
400
а
Исходная плашка
OSQOOOOOOOOO
ОООООООООООО
Gooooooooeoo
оооооооооооо
оооооооосооо
йааооооооооа
RT
НТ
о оо в оо о о о а о<
оооооооооооо
оовооооо»ооо
00080,0000.000
А;
оввооооооосоХ
OQCOOOODOO00
оосооооооооо
OtJCOOOOOOOOO
оооаооооаооо
Аг
о
О
I
с;
к
ю
ез
(-
1,0
0,8
0,6
0,4
0,2
0,0
Активность (А;)
1000
с* 100
со
S
S
о
О.
10
3-2G7
_/Я
Насыщающий /
мутагенез ^'
/^/ Рекомбинация
V
Дикий тип
Ход эволюции
о
о
as
аз
S
сз
№
сз
5
5
О 10 20 30 40 50 60 70 80 90
Температура (°С)
Рис. 54. Получение термостабильного производного субтилизина из
психрофильного гомолога S41 антарктической бактерии рода Bacillus
методом направленной эволюции белковых молекул [213]
а - отбор термостабильных производных фермента из библиотеки случай-
случайных мутантов проводили путем параллельного переноса мутантов в новые
плашки с последующей их инкубацией при комнатной (RT) и повышенной (НТ)
температурах и определением оставшейся ферментативной активности мутан-
мутантов. Aj и Аг - удельные активности ферментов при нормальной и повышенной
температурах соответственно;
б- термостабильность индивидуальных мутантов и их удельная активность
при обычной температуре. Эллипсом обведены точки, характеризующие по ис-
исследуемым параметрам разные клоны фермента дикого типа, что позволяет
оценить воспроизводимость опытов. Пунктирные линии отмечают усреднен-
усредненные характеристики фермента дикого типа;
в - схема, отражающая термостабильность производных субтилизина, по-
полученных на отдельных этапах направленной эволюции. Применение на заклю-
заключительном этапе отбора методики случайного объединения сегментов генов
разных термостабильных аналогов позволяет повысить их термостабильность
еще по крайней мере в 10 раз;
г - зависимость удельной активности от температуры у исходного, мезо-
фильного и мутантного термостабильного субтилизина пятого поколения
401
Методы направленной эволюции белков активно используют-
используются для улучшения свойств ферментов, важных для биотехнологии.
В качестве еще одного примера изменения термостабильности
ферментов с помощью направленной эволюции белковых моле-
молекул рассмотрим недавно опубликованный метод получения тер-
термостабильного аналога субтилизина из гомолога этого фермента
(субтилизина S41) психрофильной (предпочитающей понижен-
пониженную температуру окружающей среды) антарктической бактерии
Bacillus TA41 [209, 210]. В этом случае клонированный в экспрес-
сирующем векторе ген психрофильного субтилизина мутагенизи-
ровали с помощью ПЦР, получая клонотеку последовательнос-
последовательностей, содержащих случайные мутации. Отбор термостабильных
вариантов клонов проводили при повышенной температуре, до-
достаточной для того, чтобы полностью инактивировать исходный
фермент (рис. 54,а). Отношение удельной активности мутантных
ферментов в условиях высокой температуры к активности при
обычной температуре (A^/A,) использовали в качестве меры для
оценки термостабильности мутантов. Одновременно осуществля-
осуществляли мониторинг за термостабильностью отдельных мутантов и их
удельной активностью при обычных температурах. Контрольные
эксперименты с независимыми клонами фермента дикого типа
показали, что ошибка в оценке этих ключевых параметров актив-
активности ферментов не слишком высока (рис. 54,6). Один из мутан-
мутантов с повышенной термостабильностью, полученный в первом ра-
раунде отбора, далее модифицировали с помощью насыщающего
мутагенеза с последующей оценкой термостабильности новых
мутантов (рис. 54,в - отмечены эллипсом). Это позволило увели-
увеличить термостабильность исходного мутанта в ~10 раз, а дальней-
дальнейшее 10-кратное повышение стабильности было достигнуто путем
случайного обмена фрагментами мутантных генов ферментов
первого поколения. Общий результат направленной эволюции
субтилизина представлен на рис. 54,г. Точно так же путем введе-
введения случайных мутаций и отбора in vitro удалось существенно по-
повысить термостабильность и удельную активность 3-изопропил-
малатдегидрогеназы В. subtilis, которую пришлось, однако, экс-
прессировать в клетках Thermus thermophilus [211]. Число таких
примеров можно без труда увеличить [211].
В целом, подходы рационального дизайна и направленной эво-
эволюции белков оказываются весьма плодотворными в преодолении
пропасти между жесткими требованиями, предъявляемыми к про-
промышленным ферментам, и их реальными биохимическими свойст-
свойствами. Несомненно, активные исследования в этой области сделают
нас в ближайшие годы свидетелями новых достижений.
402
3.4. Белковая инженерия антител
Антитела, благодаря своей высокой специфичности взаимо-
взаимодействия с высокомолекулярными и низкомолекулярными лиган-
дами различной природы, находят широкое применение в различ-
различных областях биологии и медицины. Уникальные свойства анти-
антител позволяют использовать их для введения специфических ме-
меток и обнаружения макромолекул как in vivo во внутриклеточном
пространстве, так и на фиксированных цитологических препара-
препаратах, и делают незаменимыми в медицинской диагностике. В виде
конъюгатов с токсинами и радионуклидами они находят примене-
применение в качестве средства адресной доставки этих соединений к клет-
клеткам-мишеням для их избирательной элиминации в терапевтичес-
терапевтических целях. Антитела, обладающие ферментативной активностью,
обнаружены при некоторых заболеваниях человека, а также явля-
являются предметом интенсивных исследований в биотехнологии.
Разработка методов получения в неограниченных количест-
количествах mAb заданной специфичности еще больше расширила об-
область применения этих белков. В свою очередь, само появление
моноклональных антител (mAb) стимулировало исследователей
к применению методов белковой инженерии для направленного
изменения исходных макромолекул, что привело к созданию це-
целой группы белков с совершенно новыми свойствами. Получение
абзимов, т.е. антител, обладающих ферментативной активнос-
активностью, явилось одним из самых неожиданных открытий в этой об-
области исследований. Достижения в конструировании антител и их
производных хорошо иллюстрируют возможности современной
белковой инженерии.
3.4.1. Моноклональные антитела
Поликлональная антисыворотка, полученная в результате
иммунизации животного индивидуальным антигеном, содержит
целый набор антител, принадлежащих к различным классам им-
иммуноглобулинов, которые взаимодействуют с разными эпитопа-
ми антигена, обладают разной аффинностью и присутствуют в
неодинаковых пропорциях у отдельных животных, а также в раз-
разные периоды жизни индивидуального организма. Эпитопом (ан-
(антигенной детерминантой) называют часть молекулы антигена,
распознаваемую молекулой антитела или рецептора, специфич-
специфичного в отношении данного антигена, и взаимодействующую с
ним. Как следует из названия, моноклоналъные антитела (mAb)
представляют собой иммуноглобулины, продуцируемые одним
клоном иммортализованных В-лимфоцитов, и представляют со-
403
бой гомогенную популяцию идентичных белковых молекул од-
одной специфичности [212]. Современные методы получения mAb
позволяют целенаправленно нарабатывать белки заданной спе-
специфичности, взаимодействующие с конкретными эпитопами мо-
молекул-мишеней или определенной группой эпитопов.
Получение mAb. Ключевая идея, лежащая в основе получе-
получения mAb (Г. Колер и С. Милыитейн, 1975 г.), как и всё гениаль-
гениальное, очень проста: сделать бессмертным В-лимфоцит, который
исходно синтезирует антитела одного вида и специфичности, но
сам по себе может совершать в культуре лишь ограниченное чис-
число делений в течение 10-14 дней культивирования. Практически
это достигается путем слияния первичных В-лимфоцитов, секре-
тирующих антитела, с опухолевыми миеломными клетками, ко-
которые данной способностью не обладают, но могут неограничен-
неограниченно долго делиться в культуре. В результате образуются гибрид-
гибридные клетки (гибридомы), которые приобретают от одной из ис-
исходных клеток способность продуцировать антитела требуемой
специфичности, а от другой - способность к продолжительному
росту и делению в культуре. Реализация данной схемы начинает-
начинается с иммунизации экспериментальных животных, как правило
мышей, антигенами, против которых предполагается получать
антитела. Иммуногенами могут быть препараты очищенных
макромолекул, их различные смеси, а также целые клетки или
субклеточные структуры. После достижения антителами в сыво-
сыворотке крови мышей необходимого титра селезенку иммунизиро-
иммунизированных животных используют в качестве источника В-клеток,
продуцирующих искомые антитела.
Для создания перевиваемых линий клеток свежевыделенные
лимфоциты селезенки сливают в присутствии фьюзогена (поли-
этиленгликоля) с опухолевыми (плазмацитомными или миелом-
миеломными) лимфоцитами животного в идеале того же вида, которые
могут делиться неограниченно долго. Такие иммортализованные
клетки должны обладать развитым эндоплазматическим ретику-
лумом и не синтезировать иммуноглобулины, кодируемые их
собственными генами. Кроме того, эти клетки должны обладать
чувствительностью к селектирующему агенту (аминоптерину
или азасерину). Гибридные клетки (гибридомы) отделяют от ис-
исходных опухолевых клеток на селективной питательной среде
HAT (hypoxanthine-aminopterin-thymidine), содержащей указанные
соединения, включая ингибитор биосинтеза нуклеотидов ами-
ноптерин, в которой исходные миеломные клетки не могут де-
делиться и погибают. Это происходит потому, что миеломные
клетки не содержат одного из ферментов: гипоксантин-фосфо-
404
рибозилтрансферазы (HPRT) или тимидинкиназы (ТК), необхо-
необходимых для роста клеток на селективной среде. Гены данных фер-
ферментов клетки получают в результате слияния от В-лимфоцитов
селезенки. Сами же клетки селезенки сохраняют свою жизнеспо-
жизнеспособность в культуре в течение 10-14 дней, после чего спонтанно
погибают. Суспензию гибридных клеток распределяют по лун-
лункам 96-луночных плашек, содержащих в виде монослоя питаю-
питающие клетки, которые обеспечивают гибридомы факторами рос-
роста, необходимыми для их эффективного размножения. В совре-
современном варианте культивирования питающие клетки могут быть
заменены полноценными питательными средами.
Выращенные таким образом в небольшом количестве куль-
культуры гибридных клеток проверяют на способность секретиро-
вать в питательную среду специфических антител и далее рассе-
вают до отдельных клеток методом последовательных разведе-
разведений. В результате, каждая клетка потенциально может стать ро-
родоначальницей клона, продуцирующего mAb требуемой специ-
специфичности. Поскольку синтез антител в ряде случаев может быть
токсичным для клетки-продуцента, вместо культивирования in
vitro на этой стадии параллельно используют культивирование in
vivo в виде асцитных опухолей у мышей. Отмечено, что непро-
непродолжительное культивирование in vivo в дальнейшем способству-
способствует стабилизации клеточных линий, продуцирующих mAb. Полу-
Полученные таким образом, охарактеризованные клоны гибридом яв-
являются источником неограниченного количества mAb данной
специфичности, которые можно получать, культивируя гибридо-
гибридомы в искусственной питательной среде или асцитной жидкости
организма мышей.
Для культивирования гибридом in vitro в основном использу-
используют два подхода. При одном из них гибридомы выращивают в те-
течение ~10 дней в виде суспензионных клеточных культур на ис-
искусственных питательных средах в присутствии фетальной теля-
телячьей сыворотки в качестве источника факторов роста. В ряде
случаев гибридомы удается адаптировать к росту в бессыворо-
бессывороточных средах. Концентрация mAb в таких средах к моменту за-
завершения культивирования достигает ~10 мг/мл. mAb можно
сконцентрировать и очистить стандартными хроматографичес-
кими методами, включая аффинную хроматографию на колон-
колонках с иммобилизованными протеином А или протеином G, одна-
однако некоторые антитела не переносят таких манипуляций и дена-
денатурируют, утрачивая свою активность.
Попытки получать mAb in vitro в более высоких концентра-
концентрациях, не прибегая к специальному концентрированию, привели к
405
разработке систем культивирования клеток с использованием
полупроницаемых мембран для отделения клеток и синтезируе-
синтезируемых mAb от питающего раствора. В этом случае культура клеток
находится в сосуде объемом ~5 мл, отделенном от сосуда с пита-
питательной средой большего объема мембраной, поры которой поз-
позволяют диффундировать веществам с молекулярной массой
1-30 к Да. Такая система позволяет работать с клетками и mAb
независимо от питательной среды, а также легко менять ее со-
состав. При этом концентрация антител, синтезируемых в подоб-
подобных системах, сопоставима с их концентрацией в асцитной жид-
жидкости. Особенно высокую плотность клеток в культуре удается
получать с использованием биореакторов, в которых питатель-
питательную среду и газы прокачивают через полупроницаемые капилля-
капилляры (hollow-fiber bioreactor).
Хотя в клеточных культурах in vitro удается синтезировать
более 90% mAb, в ряде сложных случаев невозможно обойтись
без культивирования гибридом в асцитных опухолях. Это осо-
особенно относится к трансфектомам - клеточным линиям, полу-
полученным в результате трансфекции миеломных клеток вектор-
векторными молекулами ДНК, экспрессирующими мутантные гены
иммуноглобулинов, полученные генно-инженерными метода-
методами. Асцитный метод становится незаменимым и в тех случаях,
когда необходимо получать в небольших количествах разнооб-
разнообразные концентрированные препараты mAb, например во вре-
время скрининга антител определенной специфичности. Этот ме-
метод часто используют для получения mAb изотипов М и G3 (IgM
и IgG3), которые легко денатурируют в процессе очистки, что
сопровождается потерей комплементсвязывающей активности,
а также IgA-антител. Иногда в клеточных культурах образуют-
образуются неправильно гликозилированные mAb, что отрицательно
сказывается на их антигенсвязывающей активности и других
биологических свойствах.
Эти и некоторые другие ограничения использования для про-
производства mAb культивируемых гибридом не позволяют полно-
полностью отказаться от асцитной жидкости как источника таких бел-
белков. Поэтому, несмотря на введение ограничений на вивисекцию
экспериментальных животных, культивирование гибридомных
клеток in vivo и по сей день остается популярным и в ряде случа-
случаев единственным способом, позволяющим добиваться приемле-
приемлемых результатов в данной области биотехнологии.
Свойства mAb. Важнейшими свойствами антител являются
их специфичность и аффинность. Аффинность (КА) является
термодинамическим параметром, количественно описывающим
406
силу взаимодействия антигена и антитела в растворе, и определя-
определяется по закону действующих масс как отношение концентрации
комплекса антиген-антитело к произведению концентраций ком-
компонентов. Значения аффинности для mAb лежат в пределах
105-1012 М-1 для высокоаффинных и низкоаффинных соответст-
соответственно. Общая стабильность комплекса обозначается термином
авидность, которая определяется аффинностью, валентностью
антигена и антитела, а также особенностями пространственной
структуры антигена, создающими стерические препятствия для
образование комплекса.
У mAb известны два механизма перекрестных иммунологиче-
иммунологических реакций с разными антигенами. В одном случае, причина
кроется в антигене, и это происходит, когда разные антигены об-
обладают одинаковыми эпитопами (например, лактатдегидрогена-
за и изоцитратдегидрогеназа в области NAD-связывающего сай-
сайта). В другом случае, mAb (обычно класса IgM) сами по себе мо-
могут взаимодействовать с альтернативными антигенными детер-
детерминантами, сформированными разными остатками аминокислот.
Последнее свойство иногда сильно ограничивает применение
конкретных mAb для диагностики и терапии. Полиспецифич-
Полиспецифичность, как правило, является свойством mAb, обладающих низ-
низкой аффинностью.
Области применения mAb. Моноклональные антитела и про-
производные на их основе, о которых речь пойдет ниже, находят ши-
широкое применение во многих областях современной биологии и
медицины. Оборот mAb на мировом рынке в 1998 г. превышал
три млрд долларов. Практическое использование mAb продол-
продолжает расширяться, хотя структура рынка непрерывно меняется,
что связано с постоянным внедрением новых технологий, в част-
частности, ПЦР, которая при той же информативности является во
многих тестах гораздо более чувствительной, чем иммунологиче-
иммунологические методы.
Прежде всего, mAb являются прекрасным инструментом ко-
количественного и качественного иммуноферментного (ИФА) и
радиоиммунологического (РИА) анализа антигенов любого про-
происхождения. Особо следует отметить их незаменимость в иссле-
исследовании системы маркеров дифференцировки клеток (CD-мар-
(CD-маркеров - cluster of differentiation (CD) system) в иммунологии. В на-
настоящее время известно более 200 таких маркеров, и по их при-
присутствию на поверхности клеток в разных сочетаниях делают
обоснованные выводы о состоянии иммунной системы организ-
организма: стадиях дифференцировки клеток иммунной системы, уровне
их активации и т.п. С помощью проточной цитометрии и флуо-
407
ресцентно-меченых mAb удается наблюдать за динамикой раз-
различных субпопуляций лимфоцитов во время иммунного ответа
организма, определять иммунодефицитные состояния, а также
диагностировать онкологические заболевания и следить за эф-
эффективностью терапии.
Для обнаружения комплекса антиген-антитело в ИФА, как
правило, используют цветные реакции, однако данный подход
может быть легко адаптирован к детекторам флуоресценции или
биоэлектронным приборам (биосенсорам). Биосенсоры, создан-
созданные на основе mAb, успешно применяются для непрерывного мо-
мониторинга окружающей среды, биотехнологических процессов, а
также внутренней среды организма человека и животных.
Отдельная большая область применении mAb - это их ис-
использование в качестве лекарственных средств. Первыми mAb,
которые были допущены в США к применению в клинике, были
ОКТЗ. Эти мышиные mAb блокируют все Т-клетки, экспрессиру-
ющие на своей поверхности комплекс CD3. Они успешно исполь-
используются для подавления острой реакции отторжения транспланта-
трансплантата при пересадке органов. Поскольку их действие основано на
иммуносупрессии, они не вызывают иммунного ответа в организ-
организме человека. Антитела к антигенам CD2, CD5, CD7 и CDw52, a
также к цитокинам и их рецепторам проходят клинические испы-
испытания для лечения аутоиммунных заболеваний. Целью примене-
применения данных mAb является специфическое удаление конкретных
субпопуляций клеток иммунной системы таким образом, чтобы
обновленный организм не утрачивал способности противостоять
обычным инфекциям.
Использование mAb в качестве антибактериальных агентов и
средств, направленных против бактериальных эндотоксинов,
сдерживается их высокой стоимостью, поэтому во многих случа-
случаях проблема решается применением высокоэффективных и де-
дешевых антибиотиков. Однако последние не могут быть исполь-
использованы при развитии у бактерий лекарственной устойчивости. В
этой связи, перспективным лекарственным средством может
оказаться IgG человека, специфичный в отношении липида А, од-
одного из основных компонентов липополисахаридных эндотокси-
эндотоксинов бактерий. Такие mAb успешно применяют при сепсисе ново-
новорожденных, а также при сепсисе, возникающем у пациентов по-
после оперативного вмешательства.
Мечтам о возможности использования mAb в качестве эф-
эффективных противоопухолевых агентов пока, к сожалению, не
дано воплотиться в жизнь в полной мере. Одной из причин отсут-
отсутствия выраженного прогресса в этой области является то, что
408
большинство опухолей человека, развивающихся в результате
малигнизации клеток эпителия кишечника, молочных желез,
легких или простаты, появляется вследствие активации внутри-
внутриклеточных онкогенов, которые недоступны для mAb в качестве
мишеней. (Косвенным подтверждением этому механизму являет-
является тот факт, что иммуносупрессия у человека не сопровождается
возрастанием частоты возникновения опухолей данного типа.)
Тем не менее, исследования в области получения противоопухо-
противоопухолевых иммунотоксинов, направленных против клеток иммунной
и кроветворной систем, интенсивно продолжаются и в настоящее
время (см. ниже).
В заключение далеко не исчерпывающего рассмотрения во-
вопроса о сферах применения mAb следует упомянуть и о недавно
открытых каталитических антителах (абзимах), обсуждению ко-
которых будет посвящен специальный раздел этой главы.
3.4.2. Рекомбинантные антитела
Хотя гибридомные технологии еще продолжают достаточно
активно использоваться, с появлением новых эффективных ме-
методов белковой инженерии, в том числе систем отбора белков на
основе разнообразных дисплеев и репрезентативных клонотек
случайных белковых последовательностей, mAb начинают по-
постепенно сдавать свои позиции. Новые технологии позволяют от-
отбирать антитела требуемой специфичности непосредственно из
суспензии фаговых частиц без иммунизации лабораторных жи-
животных и при этом получать белки с совершенно новой специ-
специфичностью к антигенам, которые неиммуногенны in vivo. Новые
подходы дают возможность снять ограничения, накладываемые
на производство антител особенностями иммунного ответа живо-
живого организма. В последние годы удалось получить большое коли-
количество рекомбинантных антител с новыми свойствами: значи-
значительно уменьшить размер их молекул, а также объединить анти-
антитела в поливалентные гибридные комплексы, сильно повысив
при этом их авидность. Генно-инженерными методами удалось
объединить фрагменты антител с разнообразными аминокислот-
аминокислотными последовательностями для обеспечения адресной доставки
макромолекул. Такие гибридные молекулы, кроме антител,
включают ферменты для активации предшественников цитоток-
сичных лекарственных препаратов, токсины, белки вирусных ча-
частиц, используемые в генотерапии, и сами могут быть включены
в липосомы для повышения эффективности химиотерапии. Ре-
Рекомбинантные антитела применяют для получения биосенсоров,
используемых при мониторинге исследуемых молекул в реаль-
409
ном времени, в том числе опасных для здоровья веществ, загряз-
загрязняющих окружающую среду [213]. Фрагменты рекомбинантных
антител находят применение, по крайней мере, в 30% систем со-
современной иммунодиагностики. Ниже будут кратко рассмотрены
эти и некоторые другие приложения технологии рекомбинант-
рекомбинантных антител к современным фундаментальным и прикладным
исследованиям.
Снижение иммуногенности антител. Аминокислотные после-
последовательности полипептидных цепей mAb первого поколения це-
целиком кодировались геномом тех животных, которых использо-
использовали в качестве источника лимфоцитов при получении гибридом.
Хотя такое строение антител не вызывало неудовольствия у ис-
исследователей, антитела животного происхождения не находили
широкого применения в клинике по двум обстоятельствам. Во-
первых, введение чужеродных белков в организм человека часто
вызывало у пациентов сильный иммунный ответ, получивший
название синдрома антимышиных антител человека (human anti-
mouse antibody - НАМА). Это делало невозможным проведение
длительной терапии мышиными антителами. Во-вторых, все изо-
типы мышиных и крысиных иммуноглобулинов класса IgG (за
исключением т2а) не выполняли функций эффекторов иммунно-
иммунного ответа у человека.
Решение этой проблемы было начато с создания методами
генной инженерии химерных антител, построенных из гибрид-
гибридных полипептидных цепей, у которых вариабельные участки
имели мышиное происхождение, а константные целиком пред-
представляли собой последовательности иммуноглобулинов человека
[214] (рис. 55,а). Дальнейшим развитием этого направления ис-
исследований было получение так называемых "очеловеченных"
(humanized) или перестроенных (reshaped) антител, в которых
только антигенсвязывающие петли CDR имели мышиное проис-
происхождение [215]. Наконец, недавно с помощью двух альтернатив-
альтернативных методов были получены рекомбинантные антитела, содер-
содержащие только последовательности иммуноглобулинов человека.
В частности, проблема была решена созданием трансгенных
мышей (XenoMouse), экспрессирующих после иммунизации толь-
только гены иммуноглобулинов человека [216,217]. Для этого на пер-
первом этапе были получены трансгенные мыши, у которых с помо-
помощью технологии генного нокаута были удалены обеспечиваю-
обеспечивающие рекомбинацию участки локусов тяжелых цепей и легких
к-цепей иммуноглобулинов. Поскольку рекомбинация является
абсолютно необходимым этапом сборки генов иммуноглобули-
иммуноглобулинов в эмбриогенезе и во время иммунного ответа, нокаутирован-
410
Моноклональные
антитела грызунов
Химерные антитела
"Очеловеченные" или
перестроенные антитела
Антитела человека
Fab
scFv
Fv
VH
Fab'2
Моноклональные
шпйпсеяа гршунбв
Двухвалентные димерные
шниантитеяа
Тримерные мшшантйтела
в
bisAb
Fab'2
Димерные
миниантитела
cFv
Антитела двойной специфичности и их фрагменты
Рис. 55. Стратегии снижения иммуногенности антител, типы рекомби-
нантных антител и их фрагментов
а - этапы снижения иммуногенности антител. Путем постепенной замены
участков полипептидных цепей антител грызунов соответствующими последо-
последовательностями человека удалось в организме грызунов получить нативные ан-
антитела человека;
б-типы фрагментов антител, которые обладают антигенсвязывающей ак-
активностью
в - антитела и их фрагменты, обладающие двойной специфичностью
ные мыши полностью утратили способность синтезировать соб-
собственные антитела. На следующем этапе работы в геном нокау-
нокаутированных мышей были введены гомологичные гены тяжелых
цепей и легких к-цепей иммуноглобулинов человека. Для этого с
помощью искусственной хромосомы дрожжей (YAC) был клони-
клонирован фрагмент геномной ДНК человека длиной в 1 м.п.о., содер-
содержащий указанный локус в эмбриональной конфигурации (т.е. не
411
претерпевший перестроек в результате рекомбинации в процес-
процессе эмбриогенеза). Эта рекомбинантная ДНК была использована
далее для получения трансгенных мышей стандартным методом.
Анализ таких трансгенных мышей показал, что их геном содер-
содержит 34 функционально активных V-гена тяжелых цепей, все их
D- и J-гены, а также участок геномной ДНК в эмбриональной
конфигурации, фланкированный последовательностями, кодиру-
кодирующими константные области тяжелых цепей Cji и С5. Трансген-
Трансгенный локус легких к-цепей включал 18 функционально активных
V-генов, все J-гены и константную область гена Ск.
Основной характеристикой рекомбинантных антител являет-
является их специфичность в отношении конкретных антигенов. Одна-
Однако, кроме этого, они должны обладать способностью выполнять
эффекторные функции, обеспечиваемые их константными обла-
областями. В классе IgG иммуноглобулинов человека имеются четыре
подкласса (изотипа) IgGl-IgG4, которые различаются по способ-
способности взаимодействовать с Fc-рецепторами на поверхности эф-
фекторных клеток (макрофагов и нормальных киллеров), а так-
также по способности фиксировать комплемент. В зависимости от
задач, которые предполагается решать с помощью рекомбинант-
рекомбинантных антител, предпочтительными оказываются антитела опреде-
определенного изотипа. Так, если с помощью антител предполагается
непосредственно элиминировать клетки-мишени (например, опу-
опухолевые клетки), то предпочтительными оказываются антитела
изотипа IgGl, так как они эффективно взаимодействуют с Fc-ре-
Fc-рецепторами и фиксируют комплемент, что необходимо для моби-
мобилизации вышеупомянутых клеток-киллеров. Если же антитела
будут использованы только для предотвращения взаимодействия
лигандов с рецепторами, то антитела должны принадлежать к
изотипам IgG4 или IgG2. С учетом этих соображений было полу-
получено несколько линий трансгенных мышей, каждая из которых
продуцировала антитела человека определенного изотипа.
Ферментативная система мышей, обеспечивающая рекомби-
рекомбинацию частей генов иммуноглобулинов в эмбриогенезе и в про-
процессе иммунного ответа, оказалась полностью совместимой с ге-
генами иммунной системы человека. У трансгенных мышей проис-
происходило переключение классов иммуноглобулинов человека, а
также имел место соматический гипермутагенез. Поскольку
единственными белками человека, которые синтезировались в
организме трансгенных мышей, были рекомбинантные иммуно-
иммуноглобулины, их организм сохранял способность образовывать ан-
антитела на любой антиген человека. Такие трансгенные мыши
могли быть использованы для получения гибридом и mAb по
412
стандартной технологии, рассмотренной выше. В течение
двух-четырех месяцев у исследователей была возможность со-
создавать панель mAb требуемой специфичности со сродством к
антигенам в пределах 10—1000 пМ. Первыми по этой технологии
были получены mAb к интерлейкину 8, которые были использо-
использованы в клинических испытаниях для лечения псориаза. Время по-
полужизни антител в кровотоке пациентов составило ~3 недели,
что соответствовало стабильности обычных антител человека.
Кроме того, ни у одного больного не наблюдали образования ан-
антител к использованному иммуноглобулину. Также успешно про-
проходят клинические испытания антител к рецепторам эпидермаль-
ного фактора роста, сверхэкспрессия которого имеет место при
многих видах онкологических заболеваний.
Рекомбинантные многовалентные антитела. Природные ан-
антитела являются многовалентными молекулами, обладающими
несколькими идентичными участками связывания антигена. Та-
Такое строение антител значительно повышает их функциональ-
функциональную аффинность (авидность), так как позволяет одновременно
связываться несколькими участками с одной или многими моле-
молекулами антигена и затрудняет диссоциацию иммунных комплек-
комплексов. Иммуноглобулины класса IgG являются двухвалентными мо-
молекулами с двумя антигенсвязывающими Fab-участками поли-
полипептидных цепей. В то же время молекулы IgM в физиологичес-
физиологических условиях являются пентамерами, обладают 10 Fab-участка-
Fab-участками, и им свойственна более высокая авидность, чем антителам
класса IgG. Кроме того, несмотря на конформационную гибкость
молекул IgG, они часто бывают не в состоянии взаимодейство-
взаимодействовать с соседними рецепторами на поверхности клетки, что, как
правило, необходимо для специфической активации внутрикле-
внутриклеточной передачи сигналов и апоптоза. Этого недостатка лишены
полученные с использованием поперечных сшивок многовалент-
многовалентные антитела [218]. Однако такие многовалентные молекулы
иммуноглобулинов обладают слишком большой молекулярной
массой (>300 кДа), что затрудняет их проникновение к клеткам-
мишеням, например, опухолевым клеткам. Следуя данной логи-
логике, можно сформулировать задачу для белковой инженерии: не-
необходимо повысить авидность антигенсвязывающей молекулы
на основе иммуноглобулинов за счет увеличения ее валентности,
т.е. числа антигенсвязывающих модулей, на фоне одновременно-
одновременного уменьшения молекулярной массы [219]. Исследования, прове-
проведенные в этом направлении, не замедлили принести свои плоды.
Антигенсвязывающие модули полипептидных цепей имму-
иммуноглобулинов Fab и Fv моновалентны. Модуль Fv построен из
413
расположенных рядом друг с другом вариабельных частей тяже-
тяжелой и легкой цепей иммуноглобулина VH и VL, наименьших фраг-
фрагментов молекулы иммуноглобулина, которые еще могут полно-
полностью сохранять антигенсвязывающие свойства исходной молеку-
молекулы. Исходя из этого, прежде всего, были получены химическими
методами двухвалентные и трехвалентные Fab-фрагменты имму-
иммуноглобулинов, молекулярная масса которых F0-120 кДа) соот-
соответствовала задачам их доставки к клеткам-мишеням [220, 221]
(рис. 55,6).
Рекомбинантные Fv-фрагменты антител, которые были на-
названы одноцепочечными Fv (single-chain Fv - scFv), представляют
собой объединенные в одну полипептидную цепь VH- и Уь-участ-
ки полипептидных цепей иммуноглобулинов, соединенные ко-
коротким пептидным линкером (рис. 55,6). Недостатками таких мо-
молекул являются одновалентность, сопровождаемая низкой аф-
аффинностью, и низкая молекулярная масса (~30 кДа), что приво-
приводит к их быстрой протеолитической деградации в кровотоке. Для
решения этих проблем рекомбинантные scFv-модули объединя-
объединяли с помощью пептидных линкеров или адгезивных белковых до-
доменов в мультимерные агрегаты. Простейшим вариантом реком-
бинантных мультимеров являются димеры с молекулярной мас-
массой ~60 кДа, получившие название димерных миниантител (dia-
body) [222]. В такой конструкции домены VH и VL в каждом моду-
модуле scFv соединены коротким пятизвенным пептидным линкером,
что препятствует внутримолекулярному объединению V-доме-
нов и способствует агрегации scFv-модулей друг с другом
(рис. 55,6). Димерные миниантитела обладают двумя функцио-
функциональными антигенсвязывающими сайтами. Неожиданно оказа-
оказалось, что уменьшение длины линкера до трех остатков и менее,
подавляет образование димерных миниантител и вместо этого
три молекулы scFv объединяются в тример (тримерные миниан-
миниантитела - triabody) с тремя антигенсвязывающими сайтами и мо-
молекулярной массой ~90 к Да [222] (рис. 55,6). Также были получе-
получены четырехвалентные тетрамеры в результате объединения че-
четырех молекул scFv, названные тетрамерными миниантитела-
ми (tetrabody) [223].
Рекомбинантные антитела с несколькими специфичности-
ми. Во всех случаях, рассмотренных выше, возможно объедине-
объединение в единый комплекс scFv-модулей с разными антигенсвязыва-
антигенсвязывающими сайтами, что приводит к образованию антигенсвязываю-
щих белков с несколькими специфичностями. Простейшая мак-
макромолекула, которая обладает двумя специфичностями, должна
взаимодействовать с двумя разными эпитопами, расположенны-
414
ми на одной молекуле антигена (рис. 55,в). Это повышает ее
авидность по отношению к антигену-мишени. Кроме того, моле-
молекула антитела с двумя специфичностями может быть сконструи-
сконструирована таким образом, что будет в состоянии одновременно рас-
распознавать разные антигены. Подобные белковые конструкции
привлекательны для применения в терапии онкологических и
других заболеваний, при которых требуется элиминация сомати-
соматических клеток.
Стратегия использования антител с двумя специфичностями
для этих целей такова: один антигенсвязывающий участок взаи-
взаимодействует с антигеном-маркером клетки-мишени, а другой
специфически распознает эпитопы на поверхности цитотоксиче-
ских Т-лимфоцитов, макрофагов или цитолитического вируса,
облегчая их контакт с клеткой, которую необходимо элиминиро-
элиминировать [224,225]. Аналогичный подход может быть применен и для
радиотерапии: здесь антитела с двумя специфичностями исполь-
используют для адресной доставки к опухолевым клеткам радиолиган-
да, поражающего клетки-мишени [226]. Кроме того, антитела с
несколькими специфичностями могут быть получены с помощью
уже упомянутых выше химических методов путем образования
поперечных сшивок между остатками cys в шарнирных областях
Fab-фрагментов [227]. Альтернативно антитела с двумя специ-
специфичностями могут быть синтезированы с помощью гибридных
гибридом, иначе называемых квадромами (quadroma). Квадромы
являются продуктом слияния клеток двух разных клеточных ли-
линий гибридом и во время культивирования секретируют в виде
единого гибридного белка антитела с двумя специфичностями, в
котором тяжелые и легкие цепи, различающиеся антигенсвязы-
вающими участками, объединяются случайным образом [228]. К
сожалению, случайное объединение тяжелых и легких цепей в
таких гибридных клетках приводит к образованию до десяти раз-
различных вариантов антител, из которых только один обладает ис-
истинной двойной специфичностью. К тому же сам процесс произ-
производства антител двумя вышеупомянутыми методами является
весьма трудоемким и дорогостоящим. Эти и ряд других недостат-
недостатков обсуждавшихся методов получения антител с двумя специ-
специфичностями стимулировали исследования, направленные на их
получение генно-инженерными методами.
Все рассмотренные выше подходы, используемые для полу-
получения рекомбинантных поливалентных антител, могут быть при-
применены и для конструирования макромолекул с несколькими
специфичностями. В этом случае, димеризуя scFv-модули разны-
разными методами, получают антитела двойной специфичности трех
415
типов: миниантитела (minibody), одноцепочечные антитела
двойной специфичности (sc-bsAb) и димерные антитела. Для ге-
теродимеризации scFv-модулей при получении миниантител ис-
используют домены типа "лейциновая застежка" факторов транс-
транскрипции Fos и Jun, которые встраивают генно-инженерными ме-
методами в полипептидные цепи разной специфичности [229]. Та-
Такой подход обеспечивает при сборке миниантител образование
исключительно гетеродимеров. Объединение двух scFv-модулей
разной специфичности в одной полипептидной цепи приводит к
образованию sc-bsAb-антител. Такие рекомбинантные белки об-
обладают достаточно высокой стабильностью in vivo и могут быть
использованы в терапевтических целях [230]. Наконец, при кон-
конструировании димерных антител используют два рекомбинант-
ных модуля разной специфичности, в которых VL- и Ун-последо-
Ун-последовательности соединены очень короткими пептидными линкера-
линкерами, что исключает их внутримолекулярную димеризацию и обес-
обеспечивает образование гетеродимеров, составленных из разных
молекул.
Оригинальный, высокоэффективный метод получения ди-
димерных и тримерных миниантител с несколькими специфичнос-
тями был недавно разработан группой СМ. Деева (Институт
биоорганической химии им. академиков М.М. Шемякина и
Ю.А. Овчинникова, РАН) [231]. В этой работе авторы использо-
использовали свойство секретируемой рибонуклеазы (барназы) Bacillus
amyloliquefaciens образовывать очень прочный комплекс
(KD ~10-14 M) со своим белковым ингибитором барстаром. Благо-
Благодаря небольшим размерам (длина полипептидных цепей барназы
и барстара составляет 110 и 89 а.о., соответственно), их можно
легко объединять стандартными генно-инженерными методами
в одной полипептидной цепи с scFv-фрагментами антител, нара-
нарабатывать в большом количестве в бактериальных клетках и по-
после очистки таких гибридных белков соединять друг с другом в
разных комбинациях через образование комплекса барназа-бар-
стар. Введение в гибридную полипептидную цепь тандемно двух
молекул барназы позволяет присоединять к ней две гибридные
молекулы миниантител, содержащих в своем составе молекулу
барстара, что может приводить к созданию трехвалентных мини-
миниантител, в том числе и обладающих тройной специфичностью.
Благодаря простоте и высокой эффективности данный элегант-
элегантный метод может найти в будущем большое применение в раз-
разных областях молекулярной биологии и медицины, хотя пока не
ясно, какова иммуногенность комплекса барназа-барстар для ор-
организма человека.
416
Димерные миниантитела двойной специфичности и scFv-ди-
меры также обладают и двумя существенными преимуществами
перед аналогичными полноразмерными антителами. Поскольку
у них отсутствуют Fc-домены, то обладая соответствующими
специфичностями, они способны лишь активировать Т-клетки
после их сближения с клетками-мишенями, но не повреждать их
через систему комплемента (подробнее см. ниже). По тем же
причинам, а также вследствие их малого размера эти молекулы
обладают значительно меньшей иммуногенностью.
Молекулярные мишени эффекторных и атакуемых клеток
для антител двойной специфичности. Наиболее изученными
рецепторами, активирующими цитотоксическое действие эф-
эффекторных клеток, являются Т-клеточные рецепторы (TCR) и
рецепторы Fcy (FCyR). Все зрелые Т-лимфоциты экспрессируют
на своей поверхности TCR, которые могут быть универсальны-
универсальными мишенями для bsAb, одна из специфичностей которых на-
направлена против константных участков полипептидных цепей
TCR-комплекса, например, CD3. Однако ответ лимфоцитов на
взаимодействие TCR с антителами зависит от стадии дифферен-
цировки данных клеток. Например, исходные СО8+-Т-лимфоци-
ты не способны лизировать клетки-мишени и для приобретения
ими этой способности необходимо, чтобы активация TCR проис-
происходила одновременно с воздействием на них интерлейкина 2. В
отсутствие цитокинов стимуляция TCR индуцирует апоптоз, что
завершается гибелью Т-клеток [232]. Такого рода эффекты яв-
являются серьезным затруднением на пути направленного исполь-
использования цитотоксических Т-лимфоцитов (CTL) против опухоле-
опухолевых клеток in vivo.
Специфичные в отношении TCR bsAb могут быть использо-
использованы и для активации СБ4+-Т-хелперов, хотя и в этом случае,
так же как и в случае CD8+-CTL, последствия стимуляции TCR за-
зависят от присутствия цитокинов. В такой ситуации цитокины вос-
воспаления вызывают дифференцировку СО4+-Т-клеток с образова-
образованием клеток ТЫ, тогда как цитокины IL-4 и IL-10 приводят к об-
образованию клеток Th2. Какой тип клеток предпочтительнее, бу-
будет зависеть от вида и локализации опухоли, против которой
предполагается их использовать. Все это указывает на то, что ис-
использование bsAb для лечения онкологических заболеваний в
большой степени зависит от одновременного грамотного приме-
применения цитокинов и наверняка требует учета других факторов, ко-
которые еще предстоит обнаружить и исследовать.
Положительный эффект от применения bsAb можно полу-
получить и в случае их направленности против каждого из четырех
14. Патрушев Л .И. Т. 1 417
FcR, экспрессирующихся на поверхности лейкоцитов человека:
FCyRI (CD64), FCyRII (CD32), Fc^III (CD16) и FcaRI (CD89) [233,
234]. В этом случае использование bsAb вместо обычных mAb,
прямо направленных против антигенов опухолевых клеток, дает
преимущество, поскольку IgG другой специфичности, присутст-
присутствующие в кровотоке в высокой концентрации, препятствует вза-
взаимодействию комплекса опухолевых клеток и IgG с рецепторами
FCyR на поверхности эффекторных клеток. Антитела двойной
специфичности, обладающие более высоким сродством к FCyR
или взаимодействующие с эпитопами, локализованными вне сай-
сайта связывания Fc-домена антител, позволяют избегать ингибиру-
ющего влияния эндогенных IgG. Большинство клеточных медиа-
медиаторов врожденного иммунитета (нормальные киллеры, моноци-
моноциты, макрофаги и гранулоциты), в которых bsAb стимулируют ци-
тотоксическое действие, фагоцитоз и воспалительные реакции,
экспрессируют на своей поверхности FcR. Для этих клеток харак-
характерны тенденции к существованию в конститутивно активиро-
активированном состоянии, что требует меньшей костимуляции цитокина-
ми по сравнению с покоящимися Т-клетками для проявления ци-
тотоксического эффекта после заякоривания с помощью bsAb на
поверхности клеток-мишеней как in vivo, так и in vitro.
Кроме FcR, мишенями для запуска цитотоксического ответа
у NK-клеток, макрофагов и гранулоцитов могут быть использо-
использованы адгезивные рецепторы CD2, CD38, CD44, CD69 [235, 236].
Кроме того, в стимуляции цитотоксического ответа NK-клеток
принимают участие рецепторы NKRP-1, NK-TR, Ly-49D и KIR,
которые также могут быть объектом воздействия bsAb [237,
238]. Рецепторы, связывающие маннозу, "рецепторы-чистиль-
"рецепторы-чистильщики" и Toll-подобные рецепторы (TLR) макрофагов и моноци-
моноцитов рассматриваются в качестве перспективных объектов воз-
воздействия bsAb.
В приведенных примерах мишенями антител, полученных ме-
методами белковой инженерии, были белки, экспонированные на
поверхности клеток. Однако не меньшего биологического эф-
эффекта удается достичь с помощью рекомбинантных антител, дей-
действие которых направлено против внутриклеточных лигандов.
Внутриклеточные антитела. К рассмотренным выше уни-
уникальным свойствам миниантител для полноты картины необхо-
необходимо добавить еще одно - возможность их эффективного био-
биосинтеза непосредственно внутри соматических клеток высших
животных и растений [239, 240]. Такая аберрантная {эктопичес-
{эктопическая) экспрессия генов миниантител сопровождается образовани-
образованием внутриклеточных антител (intrabodies), которые, сохраняя
418
а
в
Эндоплазматический ретикулум
Рецептор, фермент,
свободный или
ассоциированный с
мембранами, функцио-
функциональный белок
Специфичес-
Специфический фрагмент
scFv
Субстрат,
1 вторичный
метаболит,
гормон
Эндоплаз-
матичес-
матический рети-
ретикулум
Взаимодей-
Взаимодействующие
белки
Рис. 56. Молекулярные механизмы иммуномодулирующего действия
внутриклеточных антител [242]
Антитела, связываясь с белком-мишенью (а, б, г-ж), а также низкомоле-
низкомолекулярным субстратом или гормоном (в), блокируют их взаимодействие друг с
другом, конкурируя за сайт связывания (а), или изменяя конформацию белка
(б), или делая недоступным субстрат, гормон, аллостерический эффектор (в),
нарушают фолдинг белка (г), его взаимодействие с регуляторными участками
нуклеиновых кислот (е) или сборку многосубъединичного комплекса, а также
могут блокировать транспорт белков в органеллы или микрокомпартменты
клетки (ж)
способность к специфическому взаимодействию с соответствую-
соответствующими высоко- и низкомолекулярными мишенями, могут оказы-
оказывать существенное влияние на внутриклеточные процессы и ста-
статус многоклеточного организма в целом. Поскольку в силу сво-
своей конструкции эти антитела не секретируются клетками, они
накапливаются в цитоплазме и, в частности, в их эндоплазмати-
ческом ретикулуме. Группа методов, использующих селектив-
селективное влияние внутриклеточных антител на биохимические про-
процессы многоклеточного организма, получила название иммуно-
модуляции.
Конкретные механизмы, посредством которых внутрикле-
внутриклеточные антитела могут оказывать свое иммуномодулирующее
действие, весьма разнообразны (рис. 56). Прежде всего антитела,
14* 419
специфически взаимодействуя с функциональными доменами
белков-мишеней, могут конкурировать за соответствующие уча-
участки связывания с субстратами, вторичными метаболитами или
гормонами или нарушать такие взаимодействия путем измене-
изменения пространственной структуры молекулы-мишени, а также
сборки многосубъединичных макромолекулярных комплексов
(рис. 56, а,б,ё). Кроме того, к аналогичным биологическим по-
последствиям может приводить непосредственное взаимодействие
антител с вышеупомянутыми низкомолекулярными соединения-
соединениями (рис. 56,в). В результате образования комплексов со строящи-
строящимися цепями вновь синтезируемых белков может предотвращать-
предотвращаться формирование активных макромолекул, как следствие нару-
нарушения их правильного фолдинга (рис. 56,г). Нейтрализация с по-
помощью антител ДНК-связывающих участков факторов транс-
транскрипции и других регуляторных белков, специфически взаимо-
взаимодействующих с ДНК, может изнутри клетки модулировать экс-
экспрессию генов на уровне транскрипции (рис. 56,д). Отдельно сле-
следует упомянуть возможность использования внутриклеточ-
внутриклеточных антител для изменения направлений адресной доставки мак-
макромолекул и низкомолекулярных эффекторов внутри клеток,
экспрессирующих соответствующие рекомбинантные гены
(рис. 56, ж,з). Специфическая доставка этих соединений в различ-
различные компартменты клетки достигается с помощью сигнальных
аминокислотных последовательностей, присоединяемых к scFv-
фрагментам антител или транзитных пептидов, направляющих
перенос белков в митохондрии и хлоропласты растений [241].
Исследование многих из вышеперечисленных механизмов
уже осуществляется на практике. Так, внутриклеточные реком-
рекомбинантные антитела, полученные к хемокиновому рецептору ма-
макрофагов человека CCR5, который используется вирусом ВИЧ
для входа в макрофаги, предотвращают экспрессию этого рецеп-
рецептора на поверхности клеток и соответственно их заражение виру-
вирусом [242]. Внутриклеточная экспрессия миниантител позволяет
сгладить симптомы нейродегенеративной болезни Хантингтона,
которые являются следствием экспансии CAG-повтора внутри
гена хантингтина, что приводит к образованию повторов поли-
глутаминовой кислоты в полипептидной цепи этого белка, со-
сопровождаемому формированием внутриклеточных белковых аг-
агрегатов [243]. Показано, что внутриклеточные антитела к хан-
тингтину эффективно предотвращают образование таких агрега-
агрегатов. Полагают, что данный подход может быть применен и для
лечения других нейродегенеративных заболеваний, развиваю-
развивающихся по похожему механизму: болезней Альцгеймера, Паркин-
420
сона, прионовых заболеваний и спиноцеребральной атаксии.
Приложения внутриклеточных антител к миру растений рассмо-
рассмотрены в обзоре [240].
Доменные антитела. В конце 1980-х гг. группой Г. Винтера
(Великобритания) из клонотеки Ун-фрагментов мышиных анти-
антител, экспрессирующихся в клетках Е. coli, были выделены поли-
полипептиды, специфически взаимодействующие с лизоцимом [244].
Такие фрагменты антител (молекулярная масса 15 кДа) были
вдвое короче обсуждавшихся выше одноцепочечных scFv-фраг-
ментов, и они являются наименьшими фрагментами антител, ко-
которые еще сохраняют свою специфичность при взаимодействии с
молекулами антигена (рис. 55,6). Поскольку эти фрагменты
включают в себя вариабельные домены тяжелых цепей иммуно-
иммуноглобулинов, они получили название доменных антител (domain
antibodies - dAb) [245]. Аналогичными свойствами обладают ва-
вариабельные домены легких цепей иммуноглобулинов. Каждая
молекула dAb обладает тремя из шести природных CDR-участ-
ков вариабельных частей антител, определяющих специфич-
специфичность этих макромолекул. Природным прототипом таких моле-
молекул являются антитела верблюдов, которые представлены толь-
только тяжелыми цепями. Антигенсвязывающий участок полипеп-
полипептидных цепей этих антител представлен одним неспаренным ва-
вариабельным доменом, обозначаемым VHH.
Высокая специфичность и малый размер dAb делают их осо-
особенно удобными для получения путем отбора из комбинатор-
комбинаторных клонотек последовательностей, а также для осуществления
аффинного созревания и наработки в большом количестве с по-
помощью генно-инженерных методов. Кроме того, небольшие
молекулы легче доставляются к мишеням внутри организма
при терапевтических воздействиях. В то же время короткие по-
полипептиды значительно менее стабильны в биологических жид-
жидкостях по сравнению с полноразмерными антителами, что огра-
ограничивает их клиническое применение. Для преодоления этого
затруднения проводятся работы по созданию конъюгатов dAb с
полиэтиленгликолем, сывороточным альбумином, а также Fc-
фрагментами или целыми константными частями молекул ан-
антител. По предварительным данным каждый из таких методов
существенно улучшает свойства dAb как потенциальных лекар-
лекарственных препаратов. Так же, как и в случае scFv-фрагментов
антител, dAb удобно димеризовать для повышения авидности и
создания миниантител двойной специфичности. Альтернативно,
гетеродимеризацию можно проводить, используя дисульфид-
ную связь между доменами СН1 и CL, которая приводит к обра-
421
зованию Fab-подобной молекулы, обладающей двойной специ-
специфичностью, обеспечиваемой одним антигенсвязывающим сай-
сайтом. Уникальные свойства dAb позволяют рассматривать их в
качестве удобной основы для создания полифункциональных
антител нового поколения.
Несомненно важнейшим свойством как исходных антител,
так и их рекомбинантных производных является их специфич-
специфичность, что должно проявляться в отсутствии перекрестных имму-
иммунологических реакций с макромолекулами, обладающими общи-
общими свойствами с антигенами-мишенями. С помощью обсуждав-
обсуждавшихся методик получения антител, в конечном счете опираю-
опирающихся на иммунизацию экспериментальных животных, очень
трудно быстро и эффективно решить эту задачу. Как и во многих
других случаях на помощь пришли методы генной и белковой ин-
инженерии, которые обеспечили значительный прогресс в данном
направлении исследований.
Клонотеки фрагментов антител. Разнообразие природных
антител возникает, прежде всего, в процессе рекомбинации шес-
шести CDR-областей генов тяжелых и легких цепей иммуноглобули-
иммуноглобулинов и соматического гипермутагенеза. Количество молекул им-
иммуноглобулинов, различающихся по своей первичной структуре,
которые теоретически могут возникнуть в отдельном организме,
оценивается в 1012-1014. В реальной жизни это число ограничива-
ограничивается размером популяции В-лимфоцитов организма и принимает-
принимается равным ~108. Данные значения определяют верхний предел ре-
репрезентативности клонотек, при конструировании которых мо-
могут быть использованы два фундаментально различающихся пер-
первичных источника последовательностей нуклеотидов: 1) природ-
природные V-гены, полученные из ДНК доноров, 2) синтетические V-
гены, полученные in vitro.
Природные источники V-генов иммуноглобулинов. При ис-
использовании природных источников последовательностей V-ге-
V-генов имеется в виду, что сама иммунная система животных или че-
человека создает требуемое разнообразие этих последовательнос-
последовательностей. При таком подходе мРНК В-лимфоцитов доноров использу-
используют для получения кДНК с последующей амплификацией с помо-
помощью ПЦР соответствующих последовательностей. При этом как
у человека, так и экспериментальных животных доноры могут
быть двух типов: предварительно иммунизированные (инфици-
(инфицированные) [246, 247] и не имевшие контакта с конкретным анти-
антигеном [248], Современные базы данных предоставляют полную
информацию о всех последовательностях генов клеток зароды-
зародышевой линии человека VH, VK, Vy, а также J- и D-сегментов
422
(http://www.mrc-cpe.cam.ac.uk/imt-doc/vbase-homepage.html), что
позволяет амплифицировать с помощью ПЦР любую из этих по-
последовательностей и использовать ее для конструирования кло-
нотек генов.
Клонотеки, полученные на основе генетического материала
доноров с выраженным иммунным ответом (иммунные клоноте-
клонотеки), являются источником получения высококачественных анти-
антител, которые могут быть использованы для профилактики забо-
заболеваний, иммунотерапии, а также исследования особенностей гу-
гуморального ответа пациентов. В этом случае донорами являются
пациенты с инфекционными [249], онкологическими [250] и ауто-
аутоиммунными [251] заболеваниями. В таких клонотеках последова-
последовательности, кодирующие антитела с требуемой специфичностью,
составляют ощутимую фракцию, а следовательно требуемые ан-
антитела с высокой аффинностью могут быть получены уже из
клонотек, содержащих ~105 клонов. Кроме того, многие гены
этих клонотек кодируют зрелые (affinity matured) антитела, опти-
оптимизированные в отношении аффинности, тогда как для получе-
получения аффинно-зрелых антител из синтетических клонотек требу-
требуется проведение специальной стадии для осуществления их "со-
"созревания" (affinity maturation) (см. ниже).
К недостаткам иммунных клонотек следует отнести невоз-
невозможность осуществления контроля за иммунным ответом, что в
ряде случаев исключает получение антител с требуемыми харак-
характеристиками. Механизмы природной толерантности, как прави-
правило, не допускают получения антител к антигенам организма-до-
организма-донора. Помимо всего прочего, токсичность некоторых антигенов
(и антител) может стать причиной смерти иммунизируемого ор-
организма. При использовании в качестве доноров ДНК пациентов
с иммунным ответом обычно в дело идут только мало представ-
представленные плазматические и В-клетки, а другие лимфоидные ткани,
которые могли бы быть более богатым источником исходного
биоматериала, недоступны. Кроме того, для получения антител к
каждому новому антигену требуется создание новой клонотеки
последовательностей.
Клонотеки, получаемые на основе ДНК неиммунизирован-
ных доноров (неиммунные клонотеки), имеют целый ряд пре-
преимуществ перед только что рассмотренными. В этом случае
клонотеки получаются значительно более представительными,
так как теоретически в них можно обнаружить все возможные
варианты антител данной специфичности, и для отбора требуе-
требуемых клонов можно продуктивно использовать существенно
большее количество антигенов, в том числе и аутоантигены.
423
Однако, как это часто бывает в жизни, упомянутое достоинство
при рассмотрении под другим углом зрения становится недо-
недостатком таких систем. Из-за больших размеров клонотеки за-
заметно усложняется сам процесс скрининга, а представленность
последовательностей в клонотеке становится неконтролируе-
неконтролируемой и непредсказуемой.
В качестве источников ДНК при конструировании неиммун-
неиммунных клонотек применяют В-клетки различных лимфоидных тка-
тканей: периферической крови, костного мозга, тонзилл [252]. При
этом для синтеза кДНК, содержащих Ун-последовательности, ис-
используют IgM-мРНК или суммарную мРНК соответствующих
тканей. Представленность последовательностей в клонотеках,
полученных на основе IgG-мРНК, сильно смещена в сторону ан-
антител неизвестного происхождения, как результат неконтроли-
неконтролируемого иммунного ответа на неизвестные антигены, и такие
клонотеки рассматриваются как нерепрезентативные [248]. Из
общих соображений следует, что более представительные клоно-
клонотеки позволяют получать к конкретному антигену более разно-
разнообразные антитела с большей аффинностью. Так, из клонотеки,
содержащей 1010 клонов, удается получать антитела со сродством
К)-8 - 0,3- К)-9 М, те же параметры для наилучших антител из
клонотек, насчитывающих 3-107 клонов, составили только
10-5-10-7 м [248, 252].
Клонотеки на основе синтетических последовательностей
\-генов. В отличие от клонотек, составленных на основе природ-
природных последовательностей, "синтетические" клонотеки позволя-
позволяют использовать весь потенциал генной и белковой инженерии
при их конструировании и дальнейшем применении. Первые кло-
клонотеки такого рода были построены на основе 49 Ун-сегментов
генома человека клеток зародышевой линии, которые собирали
случайным образом в полные V-гены с помощью ПЦР, что ими-
имитировало V-D-J-рекомбинацию, происходящую in vivo в процессе
созревания генов иммуноглобулинов. Это приводило к появле-
появлению Ун-генов, кодирующих иммуноглобулины со случайной или
частично случайной последовательностью из 4-15 а.о. в CDR3-
области. Такие Ун-гены объединяли в одну последовательность с
одной из семи Уь-последовательностей, претерпевших перест-
перестройки в природных условиях. В подобных клонотеках удалось
обнаружить последовательности, специфичные в отношении
многих антигенов, но соответствующие антитела обладали низ-
низкой (микромолярной) аффинностью [253-256].
В клонотеках большего размера F,5 • 1010 клонов), при полу-
получении которых VH- и Уь-последовательности объединяли в век-
424
торе с помощью lox-Сге-рекомбиназы in vitro, удавалось обнару-
обнаруживать гены, кодирующие антитела с аффинностью до 1нМ, что
соответствует характеристикам антител, образующихся у мышей
во время вторичного иммунного ответа [257]. В целом, использо-
использование ряда последовательностей иммуноглобулинов в качестве
скелетных, в которых определенные участки систематически из-
изменяют с помощью мутагенеза in vitro, позволяют получать но-
новые антитела с хорошими характеристиками [258, 259].
Вторичные клонотеки, используемые для осуществления
"аффинного созревания" рекомбинантных антител. Имеются
задачи, для решения которых аффинность рекомбинантных ан-
антител, получаемых с помощью описанных выше первичных кло-
нотек, является слишком низкой. С целью дальнейшего улучше-
улучшения их характеристик проводят процедуру, получившую название
"аффинного созревания". Для этого новые раунды отбора осу-
осуществляют среди последовательностей новой, целиком синтети-
синтетической клонотеки, полученной на основе последовательностей
антител с лучшими характеристиками, которые были выявлены
в результате селекции первичной клонотеки.
Для реализации этого подхода прежде всего следует выбрать
молекулу рекомбинантного иммуноглобулина, характеристики
которого необходимо улучшить. Далее конкретные участки по-
последовательности нуклеотидов, кодирующей выбранное антите-
антитело, изменяют с помощью насыщающего мутагенеза, а молекулы
антител, которые образуются в результате экспрессии мутант-
ных последовательностей, отбирают по выбранным критериям.
Антитела с наивысшей аффинностью (KD 10-100 рМ) были полу-
получены из клонотек мутантов по области CDR3 тяжелых и легких
цепей молекулы иммуноглобулина, а также других CDR-последо-
вательностей, которые формируют центр связывания антигенов
[260, 261]. В ряде случаев удается значительно улучшить аффин-
аффинные характеристики антител, изменяя с помощью случайных му-
мутаций всю V-область исходного фрагмента иммуноглобулина,
что напоминает механизм соматического гипермутагенеза при
созревании В-клеток. При таком подходе фаги, полученные в
каждом цикле отбора, размножали в клетках мутаторного штам-
штамма Е. coli mutD5, что позволяло работать с клонотеками очень
большого размера (~10и фаговых частиц) [262]. В результате аф-
аффинность исходных антител к гаптену удавалось повысить более
чем в 100 раз.
Фаговый дисплей. Отбор рекомбинантных антител на основе
фагового дисплея является одним из наиболее популярных мето-
методов получения антител требуемой специфичности. В этом случае
425
нуклеотидные последовательности, кодирующие антитела, чаще
всего, в формате scFv, объединяют с 5'-концевой частью гена 3
белка оболочки бактериофага fl в составе фаговой хромосомы
[263] или фагмиды [260, 264-267]. Существует также клонотека
более протяженных последовательностей, кодирующих Fab-
фрагменты антител [266]. Источниками последовательностей
служили как ДНК периферической крови человека, так и реком-
бинантные гены, в которых разнообразие создавалось с помо-
помощью вырожденных синтетических нуклеотидов стандартными
генно-инженерными методами. Фаговые частицы таких клоно-
тек экспрессировали на поверхности вириона разные аминокис-
аминокислотные последовательности иммуноглобулинов, объединенные с
белком оболочки в составе гибридной молекулы. Теоретически
подобные клонотеки могут состоять из 1011 разных фаговых час-
частиц, что определяется верхним пределом эффективности транс-
фекции бактериальных клеток векторной ДНК. Однако возмож-
возможность осуществления случайной рекомбинации последовательно-
последовательностей первичной клонотеки между собой с помощью сге-рекомби-
назы или ПЦР позволяет значительно расширить представитель-
представительность итоговой клонотеки [268].
Отбор фаговых частиц, экспрессирующих антитела требуе-
требуемой специфичности, как правило осуществляется с помощью от-
отдельных антигенов, иммобилизованных на твердом носителе с
использованием 96-луночных плашек для микротитрования и
стандартного иммуноферментного метода [269]. Разработаны и
высокопроизводительные методы скрининга с применением ми-
микроматриц антигенов, иммобилизованных на мембранах [269].
Сам отбор проводят в виде двух-трех последовательных раундов,
каждый из которых включает сорбцию фаговых частиц, экспрес-
экспрессирующих соответствующие антитела, на носителях с иммобили-
иммобилизованным антигеном, отмывку несвязавшихся частиц, элюирова-
ние оставшихся связанными и размножение полученных реком-
бинантных бактериофагов.
Несмотря на простоту идей, лежащих в основе получения ре-
комбинантных антител с использованием фагового дисплея, его
реализация на практике (как впрочем и любой другой искусст-
искусственной генетической системы) требует учета массы весьма суще-
существенных деталей. В частности, антигены, применяемые в такой
системе, должны быть высокоочищенными; полученные с помо-
помощью фагового дисплея антитела часто не взаимодействуют с на-
тивными белками; эффекты авидности могут затруднять отбор
высокоаффинных клонов; разные клоны с одинаковой аффинно-
аффинностью трудно дифференцировать друг от друга и т.д. [270-272].
426
3.4.3. Антитела, обладающие ферментативной активностью:
абзимы
В 1986 г. двумя группами исследователей, руководимых
Р.А. Лернером и П.Г. Шультцем, было показано, что молекулы
антител могут обладать ферментативной активностью [273, 274].
С этого времени берет начало новое направление белковой ин-
инженерии по искусственному конструированию ферментов на ос-
основе полипептидных цепей иммуноглобулинов, которые получи-
получили название каталитических антител или абзимов. Более того,
вскоре после этого открытия в организме человека были обнару-
обнаружены абзимы, появление которых ассоциировано с некоторыми
распространенными заболеваниями. Результаты данных исследо-
исследований еще раз подчеркивают плодотворность рационального
подхода к решению проблем белковой инженерии, который по
мере углубления знаний о механизмах функционирования белко-
белковых молекул, несомненно, станет доминирующим.
Принципы получения каталитических антител и их фермен-
ферментативная активность. Современные представления о механизмах
ферментативного катализа основываются на теории переходно-
переходного состояния, главные положения которой были сформулирова-
сформулированы Л. Полингом [99], а в термодинамических терминах - М. По-
лани в 1920-х гг. В соответствии с этой концепцией образование
фермент-субстратного комплекса сопровождается конформаци-
онными и структурными перестройками субстрата, значительно
понижающими энергетический барьер, который необходимо
преодолеть для превращения субстрата в продукт реакции. Такое
временное нестабильное состояние субстрата в фермент-субст-
фермент-субстратном комплексе получило название переходного состояния.
Исходя из этих представлений, можно заключить, что активные
центры ферментов в ходе эволюции были сформированы таким
образом, чтобы обеспечивать не только связывание субстратов,
но и их фиксацию в промежуточном активированном - переход-
переходном - состоянии. В настоящее время установлена структура мно-
многих субстратов в переходном состоянии и осуществлен химичес-
химический синтез их стабильных аналогов (transition state analogue -
TSA), которые активно используются для получения эффектив-
эффективных ингибиторов ферментов.
Молекулы антител напоминают ферменты по их способнос-
способности образовывать с высокой специфичностью прочные комплек-
комплексы с низкомолекулярными гаптенами и антигенными детерми-
детерминантами больших макромолекул. Однако, поскольку в отличие
от ферментов в функции антител не входит дальнейший метабо-
метаболизм антигена (аналога ферментного субстрата), его перестрой-
427
"•'•"'•••¦ :"''BlitCk»3
Дизайн и синтез аналога субстрата в переходном состоянии (TSA)
Экс]
менте
прессия генов фраг-
)в антител с помощью
фагового дисплея
Фрагменты антител
scFv, Fab
Получение конъюгата TSA с
белком-носителем
Спленэктомия
Получение
гибридом
Моноклональные
антитела
Отбор по
связыванию с
TSA
Иммунизация
мышей
Очис
сывор
¦
тка
отки
¦
Поликлональные
антитела
О (бор антител-каталшаторов исследуемой реакции
Рис. 57. Основные этапы получения каталитических антител (абзи-
мов) [279]
ка с образованием переходного состояния не происходит. Исходя
из этих данных, Р.А Лернер и П.Г. Шультц решили получить ан-
антитела путем иммунизации экспериментальных животных ста-
стабильными аналогами субстратов в переходном состоянии. При
этом предполагалось, что сайты связывания антител, оптималь-
оптимально соответствующие структуре субстрата в переходном состоя-
состоянии, будут взаимодействовать с самим субстратом, фиксировать,
как и фермент, субстрат в переходном состоянии и тем самым ус-
ускорять (катализировать) протекание химической реакции в нуж-
нужном направлении, то есть выполнять ферментативные функции.
428
Современная схема получения абзимов на основе аналогов
субстратов в переходном состоянии (TSA) представлена на
рис. 57 [275]. Работа начинается с выбора реакции, которую не-
необходимо осуществить с помощью каталитических антител, да-
далее следует разработка структуры стабильных TSA и их химиче-
химический синтез. Для повышения иммуногенности низкомолекуляр-
низкомолекулярных TSA получают их конъюгаты с высокомолекулярным носи-
носителем, например БСА, которыми иммунизируют лабораторных
животных. Селезенку иммунизированных животных после спле-
нэктомии можно затем использовать для получения поликло-
нальных и/или mAb к TSA по гибридомной технологии и/или для
выделения TSA-связывающих фрагментов антител с применени-
применением методов фагового или рибосомного дисплеев. На заключи-
заключительном этапе среди представителей антител, взаимодействую-
взаимодействующих с TS А, отбирают макромолекулы, катализирующие исследу-
исследуемую ферментативную реакцию.
Такая схема, получившая название стабилизации переходно-
переходного состояния, оказалась очень продуктивной. Вначале усилия ис-
исследователей были сконцентрированы на хорошо изученных ре-
реакциях, в том числе переносе ацильных групп, и была продемон-
продемонстрирована возможность осуществления таких химических пре-
превращений абзимами [276]. При этом типичное ускорение реакции
достигало значений в 103-106. Вскоре были получены каталити-
каталитические антитела, катализирующие химические реакции, трудно-
трудноосуществимые с помощью обычных химических методов, в част-
частности, анти-Болдуиновская циклизация [277], и гидролиз ВАС2-
карбоматного эфира [278]. Кроме того, абзимы, полученные ме-
методами белковой инженерии, могут осуществлять реакции, кото-
которые не катализируются природными ферментами, что было ис-
использовано для избирательной активации предшественников
токсических веществ при химиотерапии онкологических заболе-
заболеваний [279, 280]. Продемонстрирована возможность применения
каталитических антител для проведения реакций, сопровождаю-
сопровождающихся образованием высокореакционноспособных промежуточ-
промежуточных соединений: карбокатионов [281], 1,3-диполей [282] и трип-
летных бирадикалов [283]. С помощью абзимов удается осуще-
осуществлять контроль хиральности химических соединений, участву-
участвующих в реакциях, при проведении которых такие искусственные
ферменты демонстрируют высокую энантио- и диастереоспеци-
фичность. Недавно продемонстрирована возможность получения
абзимов, гидролизующих токсичные фосфорорганические со-
соединения, в связи с чем обсуждается возможность их использова-
использования в качестве антидотов для ликвидации последствий примене-
429
ния химического оружия [284]. Такого рода примеры указывают
на чрезвычайно широкий потенциал каталитических антител как
искусственных биокатализаторов химических реакций.
Как следует из вышеприведенной схемы получения абзимов,
иммунизация экспериментальных животных является основным
этапом, определяющим их специфичность. В настоящее время
разработаны методы иммунизации, которые позволяют полу-
получать каталитические антитела, реализующие механизмы кислот-
кислотного, основного или ковалентного катализа. Так, использование
для иммунизации гаптенов, содержащих положительно заряжен-
заряженные группы, например четвертичные амины, как правило, сопро-
сопровождается включением в активный центр абзима отрицательно
заряженных аминокислотных остатков (Glu, Asp), а для гаптенов
с отрицательно заряженными группами наблюдается обратная
картина [285]. Иммунизация гаптенами, которые ковалентно вза-
взаимодействуют с аминокислотными остатками центра связывания
антител, приводит к образованию каталитических антител, со-
содержащих в активном центре нуклеофильные аминокислотные
остатки (Lys, Ser), которые участвуют в реакциях ковалентного
катализа [286]. Такая схема иммунизации позволяет получать аб-
абзимы, осуществляющие специфический гидролиз ароматических
эфиров [287].
С помощью клонотек последовательностей, кодирующих Fab-
фрагменты или scFv-фрагменты антител, и всего арсенала мето-
методов направленной эволюции белков удается осуществлять продук-
продуктивный поиск каталитических антител требуемой специфичности
(величины ктт/Км) с последующим повышением эффективности
катализа для производных этих абзимов в последовательных цик-
циклах отбора, имитирующих аффинное созревание антител [288].
Как уже отмечалось выше, использование фаговых и рибосомных
дисплеев в направленной эволюции белков дает ряд преимуществ
по сравнению с классической гибридомной технологией. Во-пер-
Во-первых, эта группа методов дает возможность работать с репертуа-
репертуаром иммунной системы человека и получать абзимы на основе его
иммуноглобулинов, что облегчает их последующее внедрение в
клиническую практику. Во-вторых, использование клонотек боль-
большого размера в принципе позволяет вовлекать в циклы отбора по-
последовательности, не существующие в природе. Наконец, эти под-
подходы исключают необходимость иммунизации животных, а следо-
следовательно с их помощью конечный результат может быть получен
быстрее и с меньшими затратами.
Рациональный дизайн также способен приносить плоды при
конструировании абзимов с заданными свойствами [289]. Была
430
поставлена задача методами белковой инженерии на основе им-
иммуноглобулинов, специфически взаимодействующих с динитро-
фенильными группами антигенов, получить абзимы, гидролизу-
ющие эфиры динитрофенола следующей структуры:
Исходя из того, что имидазол действует в качестве нуклео-
фильного катализатора при гидролизе карбоксиэфиров в водных
растворах, предполагали, что введение остатка гистидина в центр
связывания динитрофенильных лигандов иммуноглобулина мо-
может придать ему способность осуществлять гидролиз сложных
эфиров динитрофенола. В качестве исходного белка при конст-
конструировании абзима использовали VL- и Ун-фрагменты тяжелой и
легкой цепей иммуноглобулина, специфически взаимодействую-
взаимодействующего с динитрофенильными группами антигенов. Фрагмент
ДНК, кодирующий Уь-область легкой цепи, синтезировали in
vitro и экспрессировали в клетках Е. coli. При этом в процессе
синтеза кодон, кодирующий Туг-34, был изменен на кодон His,
так как было известно, что именно Туг-34 контактирует с моле-
молекулами динитрофенола в комплексах антиген-антитело. Очи-
Очищенный рекомбинантный Уь-фрагмент объединяли с Ун-фраг-
ментом тяжелой цепи природного иммуноглобулина, взаимодей-
взаимодействующего с динитрофенолом, что приводило к образованию Fv-
фрагмента.
Оказалось, что мутантный Fv-фрагмент способен катализи-
катализировать гидролиз кумаринового эфира 5-B,4-динитрофенил)ами-
нопентаноевой кислоты (п = 4) в 90 000 раз более эффективно,
чем 4-метилимидазол, и начальная скорость гидролиза была в
45 раз выше, чем у Fv-фрагмента дикого типа. При этом мутант-
мутантный Fv-фрагмент был неактивен в отношении аминопропаноево-
го (п = 2) и аминогексаноевого {п = 5) аналогов субстрата. Таким
образом, с помощью замены единственной аминокислоты в анти-
генсвязывающем центре иммуноглобулина удалось создать на
его основе высокоспецифичный гидролитический фермент, а сам
процесс конструирования был основан на знании молекулярных
механизмов катализа и взаимодействия антигенов с антителами.
Природные каталитические антитела. В настоящее время не-
неясно, имеет ли гигантский каталитический потенциал антител ка-
431
кое-либо физиологическое значение для организма животных.
Недавно у всех исследованных молекул иммуноглобулинов неза-
независимо от их происхождения и специфичности в отношении анти-
антигенов была обнаружена способность синтезировать перекись во-
водорода из синглетного кислорода ( O2J [290]. Авторы предпола-
предполагают, что эта ферментативная активность антител может иметь
непосредственное отношение к их основной защитной функции
[275]. Известно, что антимикробное действие полиморфноядер-
ных лейкоцитов сопряжено с обоазованием активных форм кис-
кислорода - супероксид аниона [О2 ), перекиси водорода, ! О2, ра-
радикалов гидроксила (НО») и других сильных окислителей, син-
синтез которых происходит в ответ на интернализацию микроорга-
микроорганизма, покрытого антителами, и его поглощение фагосомами
лейкоцитов. В этой связи полагают, что антитела после попада-
попадания в фагосомы могут принимать участие в синтезе Н2О2 и, соот-
соответственно, в разрушении патогенного микроорганизма. Антите-
Антитела оказались уникальными в том отношении, что способны син-
синтезировать до 500 ^юлярных эквивалентов Н2О2 из 102 без поте-
потери активности, тогда как другие исследованные белки синтезиру-
синтезируют в тех же условиях <10 молярных эквивалентов или не образу-
образуют перекиси водорода вообще [291]. Таким образом, природные
антитела, по-видимому, сами по себе обладают всеми свойствами
"магической пули", так как сочетают в себе способность к специ-
специфическому распознаванию объекта воздействия со способностью
генерировать токсическое вещество, разрушающее этот объект.
Другие природные абзимы, обладающие ДНК-никазной и
РНКазной активностью, обнаружены в сыворотке крови челове-
человека при аутоиммунных заболеваниях (системной красной волчан-
волчанке) и инфекциях вирусом гепатита В [292, 293]. С использовани-
использованием разных тРНК в качестве субстратов было установлено, что
такие абзимы не обладают специфичностью в отношении после-
последовательностей нуклеотидов, но распознают особенности прост-
пространственной структуры этих макромолекул. По-видимому, ДНК-
никазные антитела по своей природе являются антиидиотипиче-
скими и структура их активного центра имитирует активный
центр соответствующих нуклеаз [294, 295]. В организме больных
астмой были выявлены аутоиммунные антитела, специфически
расщепляющие эндогенно образующийся пептидный гормон -
вазоактивный кишечный пептид (VIP), и действие этого абзима,
вероятно, вносит существенный вклад в развитие патологическо-
патологического процесса [296]. Тяжелые формы гемофилии могут возникать
432
вследствие образования аутоиммунных каталитических антител,
расщепляющих фактор VIII [297].
Все приведенные примеры каталитических антител, как при-
природных, так и получаемых с помощью белковой инженерии, не
являются исчерпывающими и лишь иллюстрируют эту активно
развивающуюся область исследований. Полную информацию по
данному кругу вопросов можно, в частности, получить из обзо-
обзоров, недавно опубликованных в ноябрьском номере J. Immunol.
Methods за 2002 г., который целиком посвящен абзимам. Сколь-
Сколько же потенциальных ферментативных активностей заключено
во всей популяции антител одного индивидуума, репертуар кото-
которых практически безграничен благодаря запрограммированному
процессу соматического мутагенеза? Насколько общим явлени-
явлением могут быть побочные ферментативные активности белков,
возникающие в результате соматических мутаций в живом орга-
организме? Попытка дать ответ на эти вопросы будет предпринята
ниже в разделе о ксенобиозе.
3.5. Пептидные аптамеры
Пептидные аптамеры представляют собой искусственно по-
полученные короткие 15-30-звенные аминокислотные последова-
последовательности, встроенные генно-инженерными методами в полипеп-
полипептидную цепь белка-носителя, которые обладают способностью
специфически распознавать и взаимодействовать с высоко- или
низкомолекулярными соединениями. В качестве белка-носителя,
чаще всего, используют бактериальный тиоредоксин [298], одна-
однако в последнее время разработаны аналогичные системы на ос-
основе зеленого флуоресцирующего белка (GFP) [299] и нуклеазы
стафилококков [300]. Таким образом, пептидные аптамеры пред-
представляют собой гибридные белки, в которых полипептидная цепь
белка-носителя выполняет скелетную функцию, обеспечивая
правильное пространственное расположение пептида-рецептора
на поверхности белковой глобулы носителя, а также стабиль-
стабильность пептида и высокий уровень его экспрессии во внутрикле-
внутриклеточном окружении.
Пептидным аптамерам присущи некоторые черты антител:
пептидная часть гибридного белка является аналогом вариабель-
вариабельного участка полипептидных цепей иммуноглобулинов, кроме
того, похожей является и функциональная нагрузка на эти два
класса макромолекул - и те и другие обладают свойствами высо-
высокоспецифических белков-рецепторов с константами диссоциации
комплексов с лигандами (Kd) в пределах 10-М О-8 М. Пептидные
433
аптамеры получают путем скрининга комбинаторных пептидных
клонотек как in vitro с помощью рибосомного и мРНК-дисплея,
так и непосредственно в живой клетке в дигибридных системах.
Отобранные по способности взаимодействовать с иммобилизо-
иммобилизованными молекулами, пептидные аптамеры могут специфически
распознавать не только белки, относящиеся к одному семейству,
но и менее значительно различающиеся аллельные варианты по-
полипептидов.
Экспрессия пептидных аптамеров в клетках позволяет специ-
специфически подавлять функционирование белков-мишеней. Такой
подход является альтернативой генному нокауту и антисмысло-
вым олигонуклеотидам в исследовании функций белков in vivo.
Кроме того, с помощью пептидных аптамеров удается блокиро-
блокировать пролиферацию эукариотических клеток, а также прекра-
прекращать бактериальную и вирусную инфекции. Например, пептид-
пептидные аптамеры к циклин-зависимой киназе Cdk2 предотвращают
ее взаимодействие со своими субстратами и после их синтеза в
клетках человека, препятствуют прохождению клеток по кле-
клеточному циклу [301]. Имеется достаточно примеров, указываю-
указывающих на то, что внутриклеточная экспрессия пептидных аптаме-
аптамеров специфически нарушает белок-белковые взаимодействия и
может изменять внутриклеточную локализацию белков, что час-
часто сопровождается доминантным фенотипическим эффектом
[302]. Такого рода соображения стимулировали исследования с
использованием пептидных аптамеров для контроля передачи
сигналов в клетках человека и животных и разработку на основе
аптамеров лекарственных препаратов, обладающих противоопу-
противоопухолевой активностью [303].
Созданы системы отбора пептидных аптамеров, обладаю-
обладающих бактерицидным действием, которые основаны на экспрес-
экспрессии последовательностей пептидных клонотек в бактериях.
В этом случае отбираются пептиды с бактерицидным действием
только в присутствии индуктора транскрипции гена пептидного
аптамера в векторе, который находится непосредственно в бак-
бактериальной клетке [304]. Исследование механизмов бактерицид-
бактерицидного действия аптамеров, полученных в результате такого скри-
скрининга, показало, что их мишенью могут быть конкретные фер-
ферменты (тимидилатсинтаза), в том числе участвующие во внутри-
внутриклеточных процессах, связанных с ростом клеток. В последнем
случае пептидные аптамеры служат инструментом поиска новых
мишеней, воздействие на которые останавливает рост бактери-
бактериальных клеток и вызывает их гибель. Пептидные аптамеры, вза-
взаимодействующие с онкобелком Е6 вируса папилломы человека,
434
который, по-видимому, блокирует апоптоз зараженных клеток,
вызывают апоптотическую гибель клеток и прекращение вирус-
вирусной инфекции. При этом незараженные клетки, экспрессирую-
щие данный пептидный аптамер, не погибают [305]. Пептидные
аптамеры могут вызывать защиту клеток растений от вирусной
инфекции, что крайне важно для успешного продолжения "зеле-
"зеленой революции" в сельском хозяйстве. В частности, недавно по-
получены трансгенные растения табака, экспрессирующие 29-
звенный пептидный аптамер, взаимодействующий с N-белками
нуклеокапсида различных тосповирусов. Такие растения оказа-
оказались устойчивыми к заражению несколькими представителями
этого семейства [306].
Все это показывает, что пептидные аптамеры могут быть
эффективным инструментом контроля бактериальной и вирус-
вирусной инфекций высших животных и растений, а наличие высоко-
высокоэффективных систем отбора делает пептидные аптамеры весьма
привлекательными объектами исследований по разработке бел-
белковых препаратов, влияющих на экспрессию генов. Кроме того
эти рекомбинантные белки пригодны для эффективного изуче-
изучения механизмов белок-белковых и белково-нуклеиновых взаимо-
взаимодействий.
3.6. Изменение специфичности ферментов
Мир ферментов бесконечно разнообразен. Мало того, что в
соответствии с современной классификацией ферментов, осно-
основанной на их субстратной специфичности, число объектов в этом
мире превышает 3000 [307]. В настоящее время имеются все ос-
основания считать, что значительную часть ферментов еще только
предстоит обнаружить и исследовать. Основным источником но-
новых ферментов несомненно является биосфера Земли, в том чис-
числе и микроорганизмы (табл. 13).
В зависимости от происхождения пробы (почва, вода, воздух
и т.п.) количество известных микроорганизмов в ней оценивает-
оценивается в пределах 0,1-1,0% от общего числа присутствующих видов.
Но даже среди представителей обширных коллекций микробов
детально с биохимической точки зрения исследована только их
небольшая часть.
Как уже упоминалось во введении к этой части книги, фер-
ферменты, в силу их уникальных свойств, являются чрезвычайно
привлекательными объектами биотехнологии. Они использу-
используются в органической химии для синтеза чистых в отношении
435
Таблица 13
Количество биологических видов, населяющих современную
Землю [308]
Группа организмов
Общее количество
видов
Количество извест-
известных видов(%)
Животные (млекопитающие, птицы,
рыбы)
Членистоногие/беспозвоночные
Нематоды
Высшие растения
Водоросли
Мохообразные
Грибы
Бактерии
Археи
Вирусы
3,5 • 104
106-107
5- 105
2,7 • 105
104-105
2,5 • 104
1,5 • 106
104-105
105-106
105-106
Примечание. В квадратные скобки заключены приблизительные
>90
10
3
>90
[70]
70
[5]
[1-10%]
[0,1-1%]
[4]
значения.
оптической активности соединений, детергентов и эмульгато-
эмульгаторов, входят в состав компонентов для стирки белья, активно ис-
используются в пищевой промышленности. Поскольку многие
реакции органического синтеза не происходят в природных ус-
условиях, предпринимаются попытки изменения субстратной
специфичности ферментов и переориентации их на осуществ-
осуществление таких химических превращений. Этими сугубо приклад-
прикладными направлениями не ограничивается круг интересов биохи-
биохимиков. Еще более захватывающими оказываются результаты
фундаментальных исследований структурно-функциональных
отношений в белках, в которых обнаруживаются удивитель-
удивительные по красоте решения проблем химического катализа при-
природными ферментами. В этом случае попытки изменения суб-
субстратной специфичности ферментов методами белковой инже-
инженерии предпринимаются для подтверждения правильности кон-
концепций, лежащих в основе современного понимания механиз-
механизмов ферментативных реакций. И вряд ли стоит осуждать иссле-
исследователя, если он работает в данной области просто из любви к
искусству.
436
3.6.1. Проблема изменения специфичности ферментов:
желаемое и действительное
Несмотря на большое разнообразие известных ферментатив-
ферментативных активностей, сами ферменты построены из ограниченного
числа блоков - функциональных доменов, в состав которых, в
свою очередь, входит вполне определенное количество элемен-
элементов вторичных структур. Все это проявляется в наличии у фер-
ферментов гомологичных участков аминокислотных последователь-
последовательностей разной протяженности и модулей, с похожей пространст-
пространственной укладкой полипептидных цепей. Такое строение фермен-
ферментов a priori накладывает ограничения на ферментативные актив-
активности, которые они потенциально могут приобрести в результа-
результате замены нескольких аминокислотных остатков. В этой связи
возможность изменения субстратной специфичности одного фер-
фермента с появлением у него новой специфичности, которой исход-
исходно обладает другой фермент, более вероятна в том случае, если
имеется сходство в строении двух ферментов, имеющее отноше-
отношение к их активности [309]. Например, поскольку в превращении
субстрата у липаз и амидаз принимает участие триада аминокис-
аминокислотных остатков Ser-His-Asp, она не должна изменяться при по-
попытках превращения амидазы в липазу и наоборот, а само нали-
наличие сходства между ферментами делает их взаимопревращение, в
соответствии с данной концепцией, теоретически возможным. В
то же время превращение эстераз в оксигеназы, которые резко
отличаются от первой группы ферментов по своей пространст-
пространственной структуре, а также используют другие субстраты и ко-
кофакторы, кажется невозможным, или же реализация такого про-
проекта столкнется со значительно большими трудностями.
Правильность такого рода соображений, а может быть про-
просто их сильное влияние на психологию исследователей, которые
отказываются подниматься на теоретически недостижимые вер-
вершины, подтверждается результатами, полученными в этой обла-
области. Все результаты по взаимопревращению или созданию но-
новых ферментов условно разделяют на три группы [309]. К пер-
первой группе относят изменения ферментов, приводящие к тому,
что они начинают метаболизировать субстраты, использованию
которых природным аналогом мешают стерические препятст-
препятствия на пути нового субстрата к активному центру, нетипичные
взаимодействия электронов и т.д. Во вторую группу попадают
результаты, в которых измененный исходный фермент начина-
начинает катализировать новую реакцию, известную для другого фер-
фермента, и между обоими ферментами имеет место высокая гомо-
437
о
сЛ
/яре/я-Бутилацетат
Эстераза
ОН
О
/ире/я-Бутанол Уксусная кислота
он]
Глюкозид
O-R
Галактозидаза
+ ROH
он
Глюкоза Спирт
I2
о
о
Фосфолипид
Фосфолипаза А]
(быстро)
Липаза
(медленно)
О
II
,о-р-х
Оэ
1 -Лизофосфолипид
RjCOOH
Жирная
кислота
Рис. 58. Ферментативные реакции, иллюстрирующие три подхода к из-
изменению субстратной специфичности ферментов методами белковой
инженерии
1 - устранение стерических препятствий: эстеразы не действуют на третич-
третичные спирты;
2 - создание новой активности: галактозидазы могут использовать в каче-
качестве субстрата галактозиды, но не глюкозиды;
3 - усиление имеющейся небольшой активности: по сравнению с фосфоли-
пазой А1, липазы некоторых микроорганизмов, например, Bacillus sp. или
Staphylococcus sp., обладают слабой активностью в отношении snl -групп фос-
фолипидов
логия. Изменения, при которых исходный фермент начинает ис-
использовать с высокой эффективностью субстрат, в отношении
которого была отмечена лишь слабая исходная активность, со-
составляют третью группу достижений в этой области. Химичес-
Химические реакции, иллюстрирующие три вышеперечисленных подхо-
подхода к изменению субстратной специфичности ферментов, пред-
представлены на рис. 58.
Первое сообщение, в котором описывалось изменение фер-
фермента методом направленной эволюции таким образом, что он
начинал метаболизировать субстрат, не распознаваемый фер-
ферментом дикого типа, появилось в 1998 г. [310]. В качестве ис-
исходного соединения был взят эфир, содержащий гидроксиль-
ную группу в положении 3, напоминавший по своему строению
один из строительных блоков, участвующих в биосинтезе эпо-
438
тилонов (недавно выделенных из миксобактерий макроцик-
лических поликетидов, обладающих противоопухолевой актив-
активностью).
Мутант
эстеразы
О ОН
1а
+ Этанол
—+¦ + Глицерин
a:R = H3CCH2~ Сдвиг рН => Изменение окраски 16
б: R = НОСН2СН(ОН)СН2-
Источник углерода
=> Ускорение роста
Такое соединение не могло быть использовано в качестве
субстрата 20-ю исследованными гидролазами A8 липазами и
двумя эстеразами). В то же время близкородственные 3-гидро-
ксиэфиры стереоспецифически гидролизовались этими фермен-
ферментами. Получение специфических продуктов гидролиза помимо
чисто теоретического имело практическое значение, так как
они могли быть использованы в синтезе эпотилонов. С помо-
помощью мутаторного бактериального штамма Epicurian coli XL1-
Red (Stratagene) была получена клонотека мутантов эстеразы.
Отбор вариантов эстеразы, которые в результате мутагенеза
приобрели способность гидролизовать это соединение, проводи-
проводили на основе изменения цвета колоний, как следствие изменения
значений рН, происходившего под действием продукта гидроли-
гидролиза этилового эфира \а. Обнаруженные таким образом варианты
далее дополнительно исследовали на среде, содержащей произ-
производное глицерина 1 б в качестве дополнительного источника уг-
углерода. В том случае, если мутантный фермент приобретал спо-
способность гидролизовать это соединение, освобождавшийся гли-
глицерин использовался клетками, что проявлялось в ускоренном
их росте и образовании более крупных бактериальных колоний.
В результате были получены двойные мутанты исходной эсте-
эстеразы, которые приобрели требуемую активность, характеризу-
характеризующуюся умеренной стереоспецифичностью. Использование на-
насыщающего мутагенеза в окрестностях обнаруженных мутаций
позволило отобрать новые мутанты со значительно более высо-
высокой стереоспецифичностью.
Другим примером такого рода явилось создание на основе
полипептидной цепи индол-3-глицеролфосфатсинтазы
(ИГФС), участвующей в биосинтезе гистидина, нового фермен-
фермента, обладающего активностью фосфорибозилантранилатизо-
меразы (ФРАИ), которая участвует в биосинтезе триптофана
[311].
439
2-,
NHR
Изомераза
^У'-[E-Фосфорибозил)формимино] -5-
аминоимидазол-4-карбосиламидрибонуклеотид-
изомераза, кодируемая геном hisA
R:
"O3PQ
ОН
NHR
НО
О
Фосфорибозилантранилатизомераза,
кодируемая геном trpF
R:
JO-Рибоза
В этом случае с помощью рационального дизайна была моди-
модифицирована система основных петель в консервативном домене
(ос/р)8-цилиндра ((oc/p)8-barrel) ИГФС и эта новая конструкция
(про-ФРАИ) была применена в качестве исходной для осуществ-
осуществления ее направленной эволюции с использованием случайной
рекомбинации мутантных генов и ступенчатого удлинения олиго-
меров. Отбор проводили по появлению у ауксотрофных клеток
Е. coli с мутантным геном ФРАИ способности расти в отсутствие
Тгр в питательной среде. Полученный мутантный фермент обла-
обладал 28% гомологии с ИГФС и 90% гомологии с ФРАИ и проявлял
<4% исходной активности.
Среди достижений применения подходов второй группы к из-
изменению субстратной специфичности ферментов можно отме-
отметить получение фукозидазы из р-галактозидазы Е. coli [312]. Ис-
Используя случайный мутагенез и отбор среди 10 000 колоний в
каждом раунде селекции по способности гидролизовать хромо-
генный субстрат онитрофенилфукопиранозид, выделили мутан-
мутанты р-галактозидазы, у которых в 1000 раз возросла специфич-
специфичность (ккат/Км) по отношению к этому искусственному субстрату
по сравнению с онитрофенилгалактопиранозидом и в 300 раз по
отношению к соответствующим я-нитрофенольным производ-
производным. Попытки превратить р-галактозидазу в р-глюкозидазу не
увенчались успехом. Зато в других опытах с помощью комбина-
комбинаторного мутагенеза удалось изменить субстратную специфич-
специфичность оксидазы галактозы таким образом, что она начала ис-
использовать глюкозу в качестве субстрата [313]. В этом случае на-
насыщающему мутагенезу подвергли последовательности нуклео-
тидов гена, кодирующие аминокислотные остатки активного
центра фермента Arg-330, Gln-406 и Тгр-290. Один из мутантных
белков, полученный в результате введения трех точковых мута-
мутаций, которые сопровождались заменами Arg330Lys, Gln406Tyr и
440
Trp290Phe, эффективно окислял D-глюкозу, а также некоторые
другие первичные спирты и один вторичный спирт.
С помощью случайного мутагенеза и отбора удалось изме-
изменить субстратную специфичность Р450-монооксигеназы В. mega-
terium (P450 ВЗ) таким образом, что образующиеся продукты под
действием кислорода воздуха спонтанно превращались в индиго и
индирубин, которые удавалось нарабатывать в миллиграммовых
количествах [314]. Всего три замены аминокислотных остатков
обеспечивали мутантному ферменту изменения его каталитичес-
каталитических свойств.
К достижениям использования подходов третьей группы
можно отнести возможность резкого (~20-кратного) усиления
активности Р450-монооксигеназы P. putida (P450cam) в отношении
гидроксилирования нафталина, хотя ее наиболее эффективным
субстратом является камфора [315]. Данный эффект достигался
небольшим числом замен аминокислотных остатков в исходном
ферменте. Среди таких мутантов были обнаружены и ферменты
с изменившейся региоспецифичностью гидроксилирования. С по-
помощью направленной эволюции удается превращать липазу
S. aureus в фосфолипазу, что сопровождается ~ 17-кратным уси-
усилением исходной (очень низкой) фосфолипазной активности
[316]. С помощью того же подхода были получены 70-кратное
усиление активности альдолазы Е. coli по отношению к нефосфо-
рилированному D-2-кето-З-дезоксиглюконату и ослабление -
в отношении его фосфорилированного аналога, а также новая
активность в отношении L-глицеральдегида [317]. Количество
аналогичных примеров можно увеличить.
Подходы, основанные на химической белковой инженерии,
также дают возможность оказывать весьма существенное влия-
влияние на субстратную специфичность ферментов. В результате ре-
реализации таких подходов удается сочетать высокую избиратель-
избирательность катализа, которую обеспечивает полипептидная цепь, с ка-
каталитической универсальностью синтетических активных цент-
центров. Эффективность применения данной группы методов иллюс-
иллюстрируют результаты, полученные с классическим объектом бел-
белковой инженерии - сериновой протеиназой субтилизином [318].
Фермент был закристаллизован и непосредственно в кристалли-
кристаллическом состоянии стабилизирован поперечными сшивками с по-
помощью глутарового альдегида. В таком состоянии субтилизин
оказался нерастворимым в воде и органических растворителях, и
демонстрировал очень высокий уровень стабильности. Далее ос-
остаток Ser-221, находящийся в активном центре субтилизина, был
превращен химическими методами в производное селеноцистеи-
441
на. В результате протеолитический фермент приобрел новую ак-
активность пероксидазы и избирательно катализировал восстанов-
восстановление гидроперекисей. Более того, поскольку полу синтетическая
пероксидаза и исходный протеолитический фермент обладали
идентичными сайтами связывания субстрата, а субстратная спе-
специфичность субтилизина хорошо изучена, избирательность селе-
носубтилизина в отношении субстратов оказалась предсказуе-
предсказуемой. Это позволило осуществлять рациональный подбор субст-
субстратов для исследования новой пероксидазной активности полу-
полусинтетического фермента.
3.6.2. Рестриктазы
Эндонуклеазы рестрикции класса II, которые обладают уни-
уникальной способностью расщеплять ДНК по специфическим сай-
сайтам рестрикции, широко используются в генной инженерии (см.
раздел 2.1 в первой части книги). Кроме того эти ферменты яв-
являются излюбленным объектом фундаментальных исследова-
исследований, в ходе которых предпринимаются попытки объяснения их
высокой специфичности. В настоящее время имеются данные
рентгеноструктурного анализа, по крайней мере, для 12 рестрик-
таз, в том числе свободных ферментов, а также ферментов в
комплексе с ДНК, содержащими соответствующие сайты рест-
рестрикции, и неправильными ДНК-субстратами. Среди этих фер-
ферментов эндонуклеаза EcoRV наиболее хорошо изучена и чаще
всего используется в белковой инженерии [319].
Помимо чисто научного, у белковых инженеров имеется к ре-
стриктазам и утилитарный интерес, как незаменимому инстру-
инструменту молекулярной генетики. Действительно, если среди фер-
ферментов, узнающих четырех- и шестинуклеотидные сайты рест-
рестрикции, имеются представители, которые расщепляют все теоре-
теоретически возможные палиндромные сайты такой длины, то круп-
нощепящих рестриктаз, действующих на 8-звенные, а тем более
10-звенные сайты рестрикции, известно очень мало. Скорее все-
всего, список природных крупнощепящих рестриктаз не будет силь-
сильно расширен, поскольку их присутствие в бактериальной клетке
не дает ей больших селективных преимуществ. Действительно,
из статистических соображений 8-звенная уникальная последова-
последовательность встречается в случайной последовательности нуклео-
тидов не чаще, чем через каждые ~66 000 п.о., и система рестрик-
рестрикции-модификации, построенная с использованием таких фермен-
ферментов не сможет эффективно защитить бактерию от вирусной ин-
инфекции. Тем не менее крупнощепящие рестриктазы остро вос-
442
требованы в современных исследованиях для получения и клони-
клонирования больших фрагментов ДНК. В этой связи предпринима-
предпринимаются попытки искусственного создания таких ферментов.
Использование рационального редизайна полипептидных це-
цепей рестриктаз EcoRl и EcoRW с целью изменения их субстратной
специфичности пока не завершилось успехом [320]. Замены ами-
аминокислотных остатков, вовлеченных в процесс распознавания
субстрата, неизменно сопровождались снижением специфичнос-
специфичности фермента и уменьшением его удельной активности. Предпо-
Предполагается, что сложная сеть ДНК-белковых взаимодействий при
взаимодействии рестриктазы с ДНК формируется кооперативно,
и любые вмешательства в эту сеть приводят к отрицательным
последствиям. В настоящее время остаются в значительной сте-
степени непонятными энергетические характеристики динамичес-
динамических структур, формирующихся в комплексах фермент-ДНК, а
также, что еще более важно, нет возможности количественной
оценки влияния каждого контакта в таких комплексах на все дру-
другие контакты, то есть на кооперативность взаимодействий. Кро-
Кроме того, на сегодняшний день отсутствует понимание структур-
структурных и термодинамических особенностей, а также функциональ-
функциональной роли альтернативных конформаций ферментов, которые он
принимает в комплексах с частично измененными сайтами рест-
рестрикции и которые не может расщеплять. Однако без таких зна-
знаний трудно рационально воздействовать на специфичность дейст-
действия рестриктазы.
Пока с помощью рационального редизайна удается изменить
специфичность рестриктаз только в отношении их действия на
модифицированные субстраты. С использованием такого подхо-
подхода был получен мутант EcoRl, который с разной эффективнос-
эффективностью расщеплял сайты рестрикции, содержащие в положении 4
остатки урацила или тимина (GAAUTC и GAATTC) [321]. Также
были получены мутанты EcoRV, которые проявляли 220-кратное
предпочтение в отношении сайта GATAUC перед сайтом
G AT АТС [322]; другой мутант этого фермента предпочтительно
(в ~103 раз более эффективно) расщеплял сайт, где одна из фос-
фосфатных групп была заменена метилфосфонатом (GATATgC)
(подчеркнута), хотя фермент дикого типа действует на обе после-
последовательности одинаково [323].
Полипептидная цепь рестриктаз класса II состоит из двух до-
доменов - распознающего ДНК и каталитического. Исходя из это-
этого, были получены химерные белки, содержащие домены разных
ферментов (не обязательно рестриктаз), и исследована их специ-
специфичность. Наибольшей функциональностью обладал гибридный
443
белок, полученный из каталитического домена рестриктазы
Fokl, объединенного с ДНК-связывающим доменом, который со-
состоит из трех "цинковых пальцев", а также аналогичных доменов
факторов транскрипции. Поскольку каждый из "цинковых паль-
пальцев" взаимодействует с тремя основаниями ДНК, то путем изме-
изменения их количества в гибридном белке, а также аминокислот-
аминокислотных остатков, из которых они построены, удавалось менять спе-
специфичность действия такой химерной рестриктазы [324]. Объе-
Объединение того же каталитического домена в одной полипептидной
цепи с ДНК-распознающими доменами факторов транскрипции,
относящихся к гомеодоменному типу, а также типу Gal4, также
приводило к созданию активной эндонуклеазы конкретной спе-
специфичности [325, 326]. В этом случае эффективное расщепление
двухцепочечной ДНК наблюдали только при наличии в полипеп-
полипептиде двух близко расположенных копий ДНК-связывающего до-
домена, что соответствует механизму действия димерной рестрик-
рестриктазы Fokl. В отличие от нативной рестриктазы, которая разреза-
разрезает ДНК, отступя 9 и 13 п.о. от сайта узнавания, т.е. создает одно-
цепочечные липкие концы длиной в 4 нт, гибридные рестрикта-
рестриктазы создают липкие концы другой длины. Химерные рестриктазы
потенциально могут использоваться для стимуляции гомологич-
гомологичной рекомбинации в клетках in vivo.
3.6.3. Изменение стереоспецифичности
Стереоспецифический синтез хиральных химических соеди-
соединений привлекает большое внимание исследователей, работаю-
работающих в области фундаментальной химии и промышленного орга-
органического синтеза. Вспомним, что хиралъностъю называют
свойство объектов, обладающих жесткой пространственной
структурой (в том числе геометрического места точек или ато-
атомов молекулы), быть несовместимыми со своими зеркальными
отображениями. По определению такие объекты не имеют эле-
элементов симметрии второго порядка. В химии каждого из пары
хиральных соединений, являющихся несовместимым зеркальным
отображением один другого, называют энантиомером. Проведе-
Проведение стереоспецифического синтеза в лаборатории или промыш-
промышленных масштабах требует большого количества очищенных оп-
оптических изомеров соединений-предшественников и является
весьма дорогостоящей процедурой. В этой связи не прекращает-
прекращается поиск эффективных энантиоселективных катализаторов, сре-
среди которых все большее внимание привлекают ферменты и ката-
каталитические антитела. Хотя в ряде случаев стереоспецифичность
444
ферментов оставляет желать лучшего, ее можно существенно
улучшить с помощью направленного мутагенеза, изменяя после-
последовательность аминокислотных остатков вблизи активного цен-
центра. Однако одно из радикальных решений проблемы улучшения
энантиоселективности ферментов дает недавно примененный
для этой цели подход, основанный на направленной эволюции
белковых молекул [327].
Методы отбора энантиоселективных мутантных ферментов.
Традиционные методы, основанные на анализе продуктов реак-
реакции методом ВЭЖХ, газовой хроматографии в хиральной фазе
или капиллярного электрофореза, в силу своей трудоемкости и
низкой производительности, не позволяют проводить крупномас-
крупномасштабный отбор мутантов из больших клонотек. С использовани-
использованием этих методов удается проанализировать в день не более не-
нескольких десятков образцов, тогда как потребности белковой ин-
инженерии в таком анализе на несколько порядков выше. Одним из
наиболее перспективных современных методов, используемых
для решения этой задачи является масс-спектрометрия с иони-
ионизацией распылением в электрическом поле (electrospray ionization
mass spectrometry - ESI-MS) [328]. (R)- и E)-энантиомеры облада-
обладают одинаковыми масс-спектрами, и их невозможно определить
количественно без предварительного хроматографического раз-
разделения. Однако, если один из энантиомеров пометить тяжелым
изотопом (дейтерием), смесь меченого и немеченого энантиоме-
энантиомеров уже можно эффективно анализировать по масс-спектру и ко-
количественно определять избыток конкретного энантиомера
(enantiomeric excess - ее) в смеси, а следовательно и фактор селек-
селективности фермента (Е) в отношении конкретных энантиомеров.
Два других подхода, активно используемых для количествен-
количественного определения энантиомеров в смеси, включают капилляр-
капиллярный матричный электрофорез (capillary array electrophoresis -
САЕ) [318] и капиллярный электрофорез на микрочипах (capil-
(capillary electrophoresis on microchips - САЕ on chips) [313]. Оба метода
кроме того применяются при секвенировании ДНК, анализе оли-
гонуклеотидов и продуктов ПЦР и позволяют исследовать сотни
тысяч образцов в день. В случае САЕ прибор для капиллярного
электрофореза, оснащенный 96 капиллярами длиной в 50 см, поз-
позволяет одновременно анализировать соответствующее количест-
количество образцов из 96-луночных плашек.
С использованием недавно разработанного подхода, кото-
который был назван дерацемизацией, удается осуществлять биосин-
биосинтез оптически чистых аминов [329]. В этом случае аминооксида-
зе Aspergillus niger, которая исходно обладает способностью
445
Таблица 14
Промышленное использование модифицированных ферментов
для стереоспецифического синтеза химических соединений в
Германии [330]
Процесс
Разделение
Разделение
Кинетическое
разделение
Аминирова-
ние
Субстрат
Рацемиче-
Рацемические спирты
Рацемиче-
Рацемические амины
Рацемиче-
Рацемические амиды
аминокислот
Фумаровая
кислота
Продукт
Энантио-
меры
спиртов
R- и S-ами-
ДЫ
L-амино-
кислоты
L-acnapa-
гиновая
кислота
Фермент
Липаза
Липаза
Амидаза
Аспартат-
аммиак-
лиаза
Масштаб
производ-
производства (т/год)
1000
100
100
100
Фирма
BASF
BASF
DSM
DSM
окислять бензиламин, с помощью направленной эволюции при-
придали способность использовать в качестве субстрата стереоизо-
мер (?)-ос-метилбензиламин. Полученный таким образом му-
тантный фермент (Asn336Ser) оказался в 50 раз более активным
в отношении (?)-ос-метилбензиламина, чем исходный, и в семь
раз превышал его энантиоселективность. Использование этого
фермента в сочетании с восстанавливающим агентом позволи-
позволило дерацемизировать рацемическую смесь ос-метилбензиламина
с выходом 73% (Я)-энантиомера и 93%-ным избытком энантио-
мера (ее).
В последнее время некоторые мутантные аналоги фермен-
ферментов, обладающих повышенной стереоспецифичностью, активно
внедряются в промышленное производство (табл. 14). В качестве
заключительного примера, которым далеко не исчерпывается
весь список современных достижений в этой области, можно упо-
упомянуть получение из природной D-специфической гидантоиназы
с помощью мутагенеза и последующего отбора L-специфическо-
го фермента, в настоящее время используемого для биосинтеза
L-метионина [331].
446
Глава 4
Комбинаторная белковая инженерия
в природе: ксенобиоз?
Успехи белковой инженерии, демонстрирующие возмож-
возможность изменения субстратной специфичности ферментов путем
замены одной или нескольких аминокислот с помощью рацио-
рационального редизайна или эволюционных подходов, наводят на
многочисленные размышления. В частности, возникает вопрос:
почему изменения субстратной специфичности ферментов явля-
являются редким событием, а мутации в конкретном гене, как прави-
правило, сопровождаются ослаблением или потерей ферментативной
активности вообще? В силу ограниченности наших знаний о
структурно-функциональных взаимоотношениях в белках ответ
на такой вопрос кажется очевидным: одиночные замены амино-
аминокислот в активном центре фермента или его окрестностях приво-
приводят к конформационным или иным изменениям полипептидной
цепи, делающим активный центр нефункциональным в отноше-
отношении своего природного субстрата.
Вероятно, на самом деле последствия таких мутационных за-
замен аминокислот могут быть более значительными. Начнем с то-
того, что внимание исследователей, изучающих свойства мутант-
ных ферментов, как правило, целиком направлено на изучение
влияния мутаций на его основную активность. При этом не учи-
учитывается возможность появления новой ферментативной актив-
активности, которая остается незамеченной просто потому, что исход-
исходно неизвестно, появления какой активности следует ожидать.
А между тем при ближайшем рассмотрении такие последствия
почти любой миссенс-мутации (точковой мутации, которая при-
приводит к замене одного осмысленного код он а на другой) в струк-
структурной части гена, кодирующего полипептидную цепь, кажутся
весьма вероятными. Действительно, в природе, по-видимому, не
существует ферментов с абсолютной субстратной специфичнос-
специфичностью, и полифункциональность является изначальным и фунда-
фундаментальным свойством сложных полипептидов. Какой бы узкой
ни была субстратная специфичность ферментов по отношению к
природным субстратам, для них всегда можно синтезировать ис-
искусственные субстраты, расширяющие их субстратную специ-
специфичность. Полифункциональность описана для многих природ-
природных белков. Например, кристаллины (одни из основных белков
хрусталика глаза позвоночных) у млекопитающих являются од-
одновременно малым белком теплового шока неизвестной функ-
447
ции, у птиц и рептилий обладают активностью аргининосукци-
натлиазы, другая же форма (8) у рыб и рептилий обладает лак-
татдегидрогеназной активностью и является ДНК-связывающим
белком. Изоформа кристаллинов т, характерная для рыб, птиц и
рептилий, является одновременно ос-энолазой и белком теплово-
теплового шока. У гемоглобина - основного переносчика кислорода и уг-
углекислого газа животных - обнаружены анилингидроксилазная,
каталазная и пероксидазная активности. Все вышеперечислен-
вышеперечисленные активности гемоглобина, по-видимому, не имеют физиоло-
физиологического значения и могли возникнуть именно как побочные ак-
активности в процессе эволюционирования основной функции это-
этого белка.
Еще более яркий пример такого рода демонстрируют абзимы
(антитела, обладающие ферментативной активностью). Помимо
хорошо известной гидролитической активности абзимов в отно-
отношении различных эфиров и кокаина описана их способность осу-
осуществлять синтез амидов, реакции стереоспецифической цикли-
циклизации, перегруппировку Клайзена, приводящую к образованию
префеновой кислоты из хоризмовой, а также ряд других реакций,
которые уже были рассмотрены подробнее в разделе об абзимах
(раздел 3.4.3).
Исходя из этого можно ожидать появления в организме чело-
человека и животных антител, обладающих самыми неожиданными
ферментативными активностями, гены большей части которых
элиминируются в процессе эмбрионального развития. Подтверж-
Подтверждением данного предположения служит открытие в организме
больных астмой аутоиммунных антител, специфически расщеп-
расщепляющих эндогенно образующийся пептидный гормон - вазоак-
тивный кишечный пептид (VIP), а также аутоиммунного расщеп-
расщепления фактора VIII свертывания крови у больных гемофилией.
По-видимому, по мере накопления экспериментальных данных
число таких примеров будет увеличиваться. На скрытую потен-
потенциальную полифункциональность полипептидных цепей указы-
указывает также наличие у многих ферментов аллостерической регу-
регуляции, проявляющейся в различных формах, разная субстратная
специфичность аллельных вариантов различных ферментов, а
также изменение субстратной специфичности некоторых фер-
ферментов при изменении условий окружающей среды.
Таким образом, при ближайшем рассмотрении традиционное
представление о ферментах как о высокоспецифических биоло-
биологических катализаторах кажется упрощенным. Высокая специ-
специфичность ферментов проявляется лишь в определенных услови-
условиях внутренней среды организма, как одна из возможностей, де-
448
терминированных конкретной первичной структурой данного
полипептида. Незначительные изменения первичной структуры
таких полипептидных цепей под действием точковых мутаций,
по-видимому, могли бы способствовать проявлению скрытых
возможностей полипептидных цепей, вызывая появление у них
новой субстратной специфичности. Реализация некоторых вновь
возникших субстратных специфичностей мутантных ферментов
может происходить в клетках в том случае, если для них найдет-
найдется соответствующий субстрат среди многочисленных нормаль-
нормальных метаболитов и не менее многочисленных макромолекул (что
имеет место, в частности, в случае аутоиммунных антител, рас-
расщепляющих VIP).
Таким образом, вырисовывается следующая гипотетическая
цепь событий, приводящих к образованию ферментов с новой
субстратной специфичностью: соматическая мутация изменяет
первичную структуру полипептидной цепи какого-либо фермен-
фермента таким образом, что он начинает распознавать эндогенный ме-
метаболит в качестве нового субстрата и метаболизировать его с
образованием нового химического соединения, не свойственного
данному организму. Эндогенно синтезированному химическому
соединению можно было бы присвоить парадоксальное назва-
название эндогенного ксенобиотика. Естественно, что мутационное
изменение субстратной специфичности ферментов должно быть
редким событием. Однако с учетом громадного числа соматиче-
соматических клеток организмов животных и человека, а также вполне
значимой частоты спонтанных соматических мутаций AО8-1О9
и выше на клеточную генерацию) этот механизм может реали-
зовываться.
Возможность такого события должна существенно возрас-
возрастать из-за увеличения частоты соматических мутаций под дей-
действием неблагоприятных экологических факторов. Более того,
вышеупомянутые низкие частоты соматических мутаций мож-
можно рассматривать только как средние. В разделе 5.3 книги
"Экспрессия генов [I. 15] мной была обоснована дифференци-
дифференциальная защищенность отдельных генетических локусов от хи-
химических мутагенов, которая обусловливается особенностями
их пространственной структуры в составе хроматина.
Если предположение об образовании эндогенных ксеноби-
ксенобиотиков в онтогенезе верно, то подобный процесс должен быть
свойствен большинству многоклеточных организмов и носить
глобальный характер. В соответствии с этой гипотезой процесс
образования эндогенных ксенобиотиков в многоклеточном ор-
организме ферментами, субстратная специфичность которых из-
15. Патрушев Л.И. Т. 1 449
менена под действием соматических мутаций, можно было бы
назвать ксенобиозом. Какими же могут быть последствия ксе-
нобиоза для многоклеточного организма? В качестве единст-
единственного примера рассмотрим возможное влияние соматичес-
соматических мутаций на систему цитохрома Р-450 в организме человека
и животных.
Цитохромы группы Р-450 являются одними из основных ком-
компонентов обширной системы детоксикации чужеродных химиче-
химических соединений (ксенобиотиков), поступающих в организм че-
человека и животных из окружающей среды (подробнее см. раз-
раздел 5.1.1). Отличительной особенностью системы цитохромов
Р-450 является их способность распознавать в качестве субстра-
субстратов и метаболизировать множество экзогенных химических со-
соединений, что, с одной стороны, объясняется наличием большо-
большого числа изоформ цитохромов Р-450, а с другой - их широкой суб-
субстратной специфичностью.
Среди цитохромов Р-450 встречаются изоформы и со строгой
субстратной специфичностью. Эти ферменты участвуют в мета-
метаболизме эндогенных химических соединений, в частности в био-
биосинтезе стероидных гормонов. Таким образом, налицо противо-
противоречивый характер действия цитохромов группы Р-450. С одной
стороны, они распознают и метаболизируют миллионы ксеноби-
ксенобиотиков, а с другой - как правило, не метаболизируют сотни ты-
тысяч эндогенных метаболитов. Следовательно, система цитохрома
Р-450 толерантна в отношении эндогенных метаболитов собст-
собственного организма, многие из которых обладают большим
структурным сходством с экзогенными ксенобиотиками. Каким
же образом можно представить себе механизм потери толерант-
толерантности системы цитохрома Р-450 по отношению к обычным мета-
метаболитам соматических клеток и каковы возможные последствия
этого явления? Метаболизм ксенобиотиков цитохромами группы
Р-450 сопровождается двойственным физиологическим эффек-
эффектом. Во-первых, как уже упоминалось (см. раздел 5.1.1), происхо-
происходит детоксикация ксенобиотиков, а во-вторых, их метаболичес-
метаболическая активация - образование нестабильных, реакционноспособ-
ных промежуточных химических соединений, обладающих мута-
мутагенной и канцерогенной активностью. Этот механизм является
одним из основных в активации химических проканцерогенов в
канцерогены, особенно в случае полициклических ароматичес-
ароматических углеводородов.
При этом можно говорить о возможности нарушения толе-
толерантности системы цитохрома Р-450 в результате каких-либо
патологических процессов, как это имеет место в случае иммун-
450
ной системы при развитии аутоиммунных заболеваний. С уче-
учетом приведенных фактов можно предполагать, что под действи-
действием соматических мутаций может произойти изменение активно-
активного центра цитохрома Р-450 таким образом, что он начнет рас-
распознавать в качестве субстрата эндогенный метаболит организ-
организма, например ароматическую аминокислоту или стероидный
гормон, и метаболизировать его по эпоксид-диольному пути с
образованием промежуточного эпоксида, обладающего мута-
мутагенной активностью (см. рис. 54). Образовавшийся эндогенный
ксенобиотик в том случае, если он не окажется особенно ток-
токсичным, будет непрерывно мутагенизировать геном как своей
собственной клетки, так и соседних клеток до тех пор, пока не
произойдут мутации, нарушающие контроль клеточной проли-
пролиферации (например, в онкогенах или генах-супрессорах опухо-
опухолей), что будет сопровождаться малигнизацией клеток, в том
числе их неконтролируемым ростом, т.е. развитием злокачест-
злокачественного новообразования.
В том случае, если будет малигнизироваться клетка, содержа-
содержащая мутантный ген цитохрома Р-450, то все клетки образующей-
образующейся опухоли будут синтезировать эндогенный ксенобиотик и опу-
опухоль будет оказывать сильный токсический эффект на целый ор-
организм. Этого не произойдет, если будут малигнизироваться
клетки, расположенные по соседству с мутантной. В настоящее
время уже описаны эксперименты, в которых с помощью направ-
направленного введения точковых мутаций удалось изменить специ-
специфичность цитохрома Р-450. Кроме того, как было упомянуто вы-
выше, изменение субстратной специфичности под действием точко-
точковых мутаций описано и для других ферментов. Все это указывает
на возможность развития событий по предлагаемому гипотети-
гипотетическому сценарию.
Появление мутаторного фенотипа у клеток злокачественных
новообразований описано в настоящее время для многих форм
рака. Основной причиной мутаторного фенотипа раковых кле-
клеток считают мутационные нарушения ферментных систем репа-
репарации, что имитирует внутриклеточное возрастание концентра-
концентрации мутагенов. Предлагаемый механизм указывает на еще один
возможный путь малигнизации нормальных клеток через обра-
образование эндогенных ксенобиотиков - эндогенных мутагенов.
При развитии такой цепи событий повреждение систем репара-
репарации будет вторичным по отношению к первичному синтезу эндо-
эндогенного мутагена.
Помимо онкологических заболеваний, эндогенные ксенобио-
ксенобиотики могли бы вызывать некоторые аутоиммунные заболевания
15* 451
в ответ на образование аддуктов с макромолекулами организма,
что придавало бы модифицированным макромолекулам иммуно-
генность и нарушало иммунологическую толерантность организ-
организма. Эндогенные ксенобиотики могли бы быть также одной из по-
постоянно действующих причин старения организма вследствие его
постепенного отравления эндогенными ксенобиотиками по мере
накопления с возрастом в клетках мутантных ферментов, обла-
обладающих измененной субстратной специфичностью.
Процесс спонтанного и индуцированного соматического му-
мутагенеза происходит непрерывно на протяжении всей жизни мно-
многоклеточного организма. Следовательно, в организме постоянно
образуются новые мутантные белки, которые исходно не были
закодированы его геномом. Мутантные белки могут контактиро-
контактировать в клетках с большим количеством эндогенных метаболитов
и при наличии структурного соответствия между метаболитом и
новым активным центром мутантного белка метаболизировать
его с образованием эндогенного ксенобиотика, например, про-
продуктов расщепления упомянутого выше пептидного гормона VIP,
наличие которых в организме больных доказано. Многоклеточ-
Многоклеточный организм является своеобразным полигоном, на котором
ежеминутно на протяжении всей жизни происходят испытания
вновь образующихся белков на способность метаболизировать
эндогенные химические соединения, с которыми они находятся в
контакте. Таким образом, ксенобиоз в том случае, если он рас-
распространен так широко, как я думаю, должен быть обычным фе-
феноменом, присущим всем многоклеточным организмам.
Если предположение о ксенобиозе верно, то организм дол-
должен обладать эффективными системами защиты от этого неиз-
неизбежного явления. Помимо высокоэффективных репаративных
систем, понижающих частоту спонтанного мутагенеза в сомати-
соматических клетках, защиту от эндогенных ксенобиотиков отчасти
могла бы осуществлять сама иммунная система. В таких случаях
с участием иммунной системы элиминировались бы те мутант-
мутантные соматические клетки, которые экспонируют мутантные бел-
белки на своей поверхности и которые в этом случае распознавались
бы как чужеродные антигены. Однако в большинстве случаев,
по-видимому, этого не должно происходить, так как мутантные
белки будут локализованы внутри клеток.
Другим средством защиты от образующихся ферментов с но-
новой субстратной специфичностью (и ксенобиоза) у многоклеточ-
многоклеточных организмов могут быть убиквитин- и АТР-зависймые систе-
системы внутриклеточного протеолиза аномальных белков. Однако
эти системы должны срабатывать в первую очередь в отношении
452
мутантных белков, у которых резко нарушена пространственная
структура, что при одиночных заменах аминокислот, по-видимо-
по-видимому, происходит редко. В пользу такого предположения свиде-
свидетельствуют все те многочисленные мутантные соматические
клетки, которые описаны в литературе и из которых можно вы-
выделять мутантные белки.
Вероятно, основную защиту от эндогенных ксенобиотиков
могла бы обеспечивать обсуждавшаяся выше система цитохрома
Р-450. Действительно, широкая совокупная субстратная специ-
специфичность системы цитохрома Р-450 позволяет ей метаболизиро-
вать и в конечном счете обезвреживать химические соединения.
По аналогии с иммунной системой, которая помимо защиты ор-
организма от экзогенных инфекционных агентов выполняет функ-
функции иммунного надзора за антигенным состоянием соматических
клеток, основной функцией системы цитохрома Р-450 может
быть освобождение организма от эндогенных ксенобиотиков.
В соответствии с этим система цитохрома Р-450 эволюционно
могла возникнуть для поддержания, прежде всего, метаболичес-
метаболического гомеостаза многоклеточных организмов в условиях непре-
непрерывно образующихся эндогенных ксенобиотиков, а уже затем ее
функции распространились и на экзогенные химические соедине-
соединения, вредные для организма.
453
Глава 5
Белковая инженерия в поисках полипептидов,
максимально отвечающих требованиям
современной биотехнологии
Белковая инженерия, как и все остальные научные направле-
направления, имеет перед собой две цели: фундаментальную, направлен-
направленную на познание окружающего мира, и прикладную, призванную
улучшить жизнь человека отныне и навсегда. Биотехнологичес-
Биотехнологическая промышленность является одним из крупнейших потребите-
потребителей фундаментальных и прикладных достижений белковой инже-
инженерии, которая оказывает на развитие современной индустрии
глубокое влияние.
5.1. Изменение парадигмы оптимизации
биотехнологических процессов
При разработке биотехнологического процесса в современ-
современных условиях приходится учитывать не только основные издерж-
издержки на его проведение, но и необходимость осуществления процес-
процесса в жестких временных рамках, что накладывает особые требо-
требования на свойства биокатализаторов [330]. Необходимость ин-
интенсификации ферментативных процессов особенно актуальна
для фармацевтической промышленности, поставленной в труд-
трудные условия из-за ограничений срока действия патентов. При
обычной разработке стадий биосинтеза в конкретном биопроцес-
биопроцессе исходят из свойств имеющихся ферментов, с помощью кото-
которых предполагается осуществлять требуемые химические реак-
реакции в промышленном масштабе. Свойства фермента определяют
особенности дизайна биопроцесса в целом. Это определяется
тем, что для оптимального функционирования конкретного фер-
фермента необходимо создавать строго контролируемые условия
проведения реакции, а стоимость биокатализатора часто состав-
составляет основную часть издержек производства.
С увеличением стоимости исходных субстратов реакции и
расширением возможностей белковой инженерии в отношении
изменения свойств ферментов в нужную сторону структура из-
издержек производства начинает меняться. При проектировании
современного биотехнологического процесса все чаще становит-
становится возможным брать за основу свойства реакций как таковых и
технические возможности их проведения, а биокатализатор со-
454
здавать, исходя из предварительно сформулированных требова-
требований. При таком подходе современная идеология разработки био-
биотехнологического процесса меняется на противоположную по
сравнению с обычно применяемым для данной цели подходом.
В этом случае основной упор делается на поиск "идеального"
биокатализатора, который бы максимально соответствовал тре-
требованиям спланированного процесса биосинтеза требуемого хи-
химического соединения.
Тем не менее, в обстановке изменяющейся парадигмы дизай-
дизайна биотехнологического процесса возможности поиска новых
биокатализаторов ограничиваются независимыми от фермента
свойствами и условиями проведения реакций, такими как термо-
термодинамика реакции, а также свойствами исходных реагентов и об-
образующихся продуктов. Например, растворимость и стабиль-
стабильность субстратов и продуктов реакции определяет состав, рН и
температуру среды, в которой данная реакция должна прово-
проводиться, а равновесие реакции должно быть сдвинуто в сторону
образования продуктов в высокой концентрации. Методы и тех-
технологии белковой и генной инженерии сегодняшнего дня, рас-
рассмотренные в этой книге, позволяют эффективно менять ключе-
ключевые свойства исходных ферментов, существенные для проведе-
проведения биотехнологических процессов.
5.2. Современные технологии,
позволяющие улучшать свойства ферментов
Некоторые из впечатляющих достижений белковой инжене-
инженерии были рассмотрены в гл. 3. Табл. 15 суммирует и дополняет
эту информацию.
Интенсивные исследования стабильности ферментов, прове-
проведенные на протяжении двух последних десятилетий, привели к
обнаружению новых молекулярных механизмов стабилизации
белковых молекул, в том числе сетей внутримолекулярных ион-
ионных взаимодействий [343], которые позволили сознательно улуч-
улучшать соответствующие свойства ферментов. Особенно продук-
продуктивными в этом отношении оказались методы направленной эво-
эволюции белковых молекул, применение которых позволяет повы-
повышать температуру денатурации белковых глобул на 10-15 °С без
ущерба для их ферментативной активности. Особенно впечатля-
впечатляющим достижением здесь является 340-кратное повышение вре-
времени полужизни протеиназы Bacillus stearothermophilus при
100 °С без ухудшения ее каталитических свойств [211]. Такого
455
Таблица 15
Примеры использования современных технологий для
улучшения свойств ферментов, важных для биотехнологии
[330]
Улучшаемое
свойство
Использованная
технология
Примеры
Число оборотов
Солюбилизация
ферментов
Молекулярный
импринтинг
Солюбилизация субтилизина для про-
проведения реакции в органических рас-
растворителях с использованием октана и
АОТ
Пластиковые биокатализиторы на ос-
основе химотрипсина и субтилизина
Солюбилизация субтилизина и химо-
химотрипсина для проведения реакции в
органических растворителях с исполь-
использованием промывки пропанолом
Импринтинг субтилизина (Carlsberg и
мутантных форм) сахарозой, а также
тимидином и другими нуклеозидами
Изменение
энантиоселек-
тивности
Направленная
эволюция
Индукция ферментативной активности
папаина и лактальбумина при лио-
филизации в присутствии аналогов суб-
субстратов в переходном состоянии
Улучшение энантиоселективности ли-
липаз
Обращение энантиоселективности ги-
дантоиназы
Изменение функ- Рациональный
циональности дизайн и направ-
направленная эволюция
Рациональный
дизайн
Улучшение энантиоселективности эс-
теразы, используемой для разделения
рацемических смесей
Получение новой активности у изо-
меразы
Превращение десатуразы в гидро-
ксилазу
Рекомбинирова- Изменение специфичности |3-галак-
ние сегментов тозидазы
генов
456
Полученный результат
Константа специфичности ккат/Км субтилизина, солюбилизированного в окта-
октане, возрастает в ~103 раз по сравнению с суспендированным ферментом и в
10 раз ниже, чем в водных растворах; стабильность - в 103 раз выше, чем в
водных растворах [332]
Включение ферментов в синтетические полимеры повышает скорость реакции
в 104 раз и увеличивает их стабильность в полярных растворителях, что зна-
значительно облегчает проведение синтеза пептидов [158]
Активность ферментов в 103 раз выше, чем у лиофилизированных препаратов
и сопоставима с таковой препаратов CLEC [161]
Скорость ацилирования тимидина субтилизином в THF возрастает в ~50 раз по
сравнению с ^модифицированным ферментом; при использовании для им-
принтинга альтернативных нуклеофилов соответствующим образом изменяет-
изменяется субстратная специфичность субтилизина в отношении других нуклеозидов, а
скорость реакции возрастает в 50-180 раз [167]
Трехкратное возрастание скорости реакции ^-элиминации по сравнению с
белками, не подвергнутыми импринтингу [166]
У мутантных липаз Pseudomonas aeruginosa четвертого поколения значение ее в
отношении S-энантиомеров возрастает с 2% до 81% [333]
Изменение селективности с D- на L- и пятикратное повышение удельной
активности у гидантоиназы Arthrobacter sp., используемой при производстве
L-метионина; природные L-специфические гидантоиназы неизвестны [334]
Мутанты эстеразы Pseudomonas fluorescens, экспрессирующейся вЕ. coli, с воз-
возросшими до 21% значениями ее в отношении смеси 3-гидроксиэфиров [313]
Превращение индол-3-глицеролфосфатсинтазы Е. coli в фосфорибозилантра-
нилатизомеразу, сопровождаемое уменьшением в два раза значений ккат и в
15 раз Км по сравнению с исходным ферментом [314, 334]
Изменение способности олеатдесатуразы вводить в субстрат двойные связи на
способность осуществлять его гидроксилирование на основании сравнения пер-
первичной структуры исходного фермента и неродственных гидроксилаз; потребо-
потребовало замены с помощью направленного мутагенеза семи а.о. [335]
Десятикратное возрастание активности фукозидазы и 40-кратное уменьшение
активности Р-галактозидазы в результате изменения шести а.о. [315]
457
Таблица 15 (окончание)
Улучшаемое
свойство
Использованная
технология
Примеры
Использование
неприродных
субстратов
Направленная
эволюция
Получение диоксигеназы из би-
фенилоксигеназ
Повышение ста-
стабильности фер-
ферментов
Направленная
эволюция
Рациональный
дизайн и случайный
мутагенез
Получение термостабильной эсте-
эстеразы
Повышение термостабильности
протеиназы
Повышение ста-
стабильности и ак-
активности фер-
ферментов
Мутагенез с от-
отбором в клеточном
дисплее
Улучшение свойств левансукразы
Химические мо- Ковалентная им-
дификации для мобилизация по
повышения ста- многим участкам
бильности фер- полипептидной
ментов цепи
Химические по-
поперечные сшивки
Метод CLEC
Сверхстабилизация
термостабильной эстеразы
Использование полимерного
диальдегиддекстрана для создания
поперечных сшивок в белке
Создание поперечных сшивок и хи-
химические модификации субтили-
зина
Создание для фер- Использование гликозидгидролаз в
ментов липидной органических средах
оболочки
Химические мо- Модификация
дификации фер- монофункциональ-
ментов с целью ными агентами
повышения их
активности
Изменение реак- Направленная
ционной среды эволюция
Модификация каталазы детерген-
детергентом Brij 35
Повышение активности эстеразы в
водных средах, содержащих органи-
органические растворители
458
Полученный результат
Обмен сегментами генов бифенилоксигеназ Pseudomonas pseudoalcaligenes и
Burkholderia cepacia, обладающих разными субстратными специфичностями,
привел к созданию диоксигеназы, эффективно метаболизирующей полихло-
рированные бифенилы [336]
После шести циклов отбора произошло повышение Тц на 14 °С без потери
активности; результат указывает на то, что эти два свойства ферментов не
исключают друг друга [337]
Замена восьми а.о. в термолизиноподобной протеиназе Bacillus stearothermo-
philus увеличивала время полужизни фермента в присутствии денатурирующих
агентов до 170 мин при 100 °С (у исходного фермента —1 мин), что не сопро-
сопровождалось понижением удельной активности [211]
Коэкспрессия на поверхности клеток Е. coli левансукразы Zymomonas mobilis и
фермента, вызывающего нуклеацию льда Pseudomonas syringae, повышала ско-
скорость превращения сахарозы в глюкозу и увеличивала стабильность фермента
[338]
Повышение в 3 • 105 раз термостабильности эстеразы Bacillus stearothermophilus
после иммобилизации по аминогруппам на глиоксил-агарозе [339]
Девятикратное повышение времени полужизни ацилазы пенициллина G при
55 °С без уменьшения значений Утах [340]
Проведение химических модификаций и введение поперечных сшивок в крис-
кристаллический полусинтетический селеносубтилизин сопровождались появле-
появлением пероксидазной активности, десятикратным увеличением стабильности
фермента без потери энантиоселективности [341]
Создание липидной оболочки вокруг маннозидазы, глюкозидазы и глюкоза-
минидазы делает поверхность ферментов гидрофобной и в семь раз повышает
их способность к трансгликозилированию в среде изопропилового эфира [163]
По сравнению с исходным ферментом активность модифицированной ката-
лазы в трихлороэтилене возрастает в 200 раз, а в водных растворах - в 15 раз
[154]
50-кратное повышение n-нитробензилэстеразной активности субтилизина в
30% диметилформамиде [343]
459
рода результаты привели к необходимости пересмотра широко
распространенного мнения, согласно которому активность и ста-
стабильность ферментов связаны друг с другом реципрокными от-
отношениями благодаря глобальной конформационной гибкости
полипептидных цепей, задействованной в ферментативном ката-
катализе. Постепенно складывается впечатление, что процесс дена-
денатурации запускается общими инициаторами и/или механизмами,
которые индуцируются высокой температурой, неоптимальными
значениями рН, органическими растворителями или детергента-
детергентами. Как следствие этих наблюдений появилась возможность пу-
путем улучшения термостабильности ферментов одновременно по-
повышать их устойчивость и к другим вредным экстремальным ус-
условиям. Хотя среди современных методов стабилизации белков
преобладают химические (иммобилизация и введение химичес-
химических поперечных сшивок), разработка новых подходов, в которых
используется экспрессия белков на поверхности клеток [338],
многоточечное ковалентное сшивание белков с носителями
[344], введение поперечных сшивок в кристаллические фермен-
ферменты [341], а также рассмотренные выше методы направленной
эволюции белков и, возможно, рационального дизайна будут
превалировать в создании новых биокатализаторов для нужд
промышленности будущего.
Конструирование ферментов методами белковой инженерии
сопровождается постепенным освоением новых областей иссле-
исследуемого пространства последовательностей и на этом пути про-
продвижения в неведомое наблюдается значительный прогресс
(рис. 59). Методы классического скрининга белков с новыми
свойствами, основанные на последовательном переборе отдель-
отдельных различающихся последовательностей, дают возможность ис-
исследовать лишь очень ограниченное количество не связанных
друг с другом представителей из теоретически доступного прост-
пространства последовательностей в разных его областях. Картина из-
изменяется лишь незначительно при анализе большого количества
геномов разных видов в процессе метагеномного скрининга. Ме-
Методы направленного и случайного мутагенеза позволяют незна-
незначительно отклониться от исходной последовательности и иссле-
исследовать пространство последовательностей в ее окрестностях.
Случайное объединение доменов разных белков и, особенно,
комбинаторная перетасовка нуклеотидных последовательностей
разных геномов дают возможность обследовать более удаленные
друг от друга области пространства последовательностей. Тем не
менее, наши современные возможности пока не позволяют по-
подойти к решению этой проблемы более широко, и в исследуемом
460
Классический скрининг
с использованием микробиологических
и энзимологических методов
\\f
V
Множественный
(метагеномный) скрининг
Направленный или случайный
мутагенез
Объединение белковых
доменов
Случайное рекомбинирование
фрагментов ДНК разных геномов
Рис. 59. Перемещение по пространству последовательностей белковых
молекул с помощью разных методов, используемых в современной бел-
белковой инженерии
4G1
пространстве последовательностей еще долгое время будут оста-
оставаться обширные белые пятна.
Вне зависимости от того, какая из двух обсуждавшихся выше
концепций дизайна биотехнологических процессов будет преоб-
преобладать в будущем, одним из основных вопросов белкой инжене-
инженерии, по-видимому, останется вопрос о пределе совершенства иде-
идеального биокатализатора. Прежде всего необходимо выяснить,
какие факторы лимитируют функции известной белковой моле-
молекулы, а поняв это потребуется найти способы преодоления таких
ограничений. Рациональный дизайн белков развивается медлен-
медленно из-за сложности объекта исследований. Пока не удалось даже
установить основные закономерности, определяющие термоста-
термостабильность белковых молекул для того, чтобы сознательно изме-
изменять это свойство в нужном направлении. В то же время методы
направленной эволюции белков в последнее время быстро рас-
распространяются и находят все большее применение, поскольку по
своей сути не требуют от исследователя предварительных знаний
о структурно-функциональных отношениях в эволюционирую-
эволюционирующих макромолекулах. Прогресс в понимании структурно-функ-
структурно-функциональных взаимосвязей в белковых молекулах определит по-
популярность рационального дизайна белков. А поскольку откры-
открытия в науке предсказать невозможно, делать прогнозы о ближай-
ближайшем будущем развитии таких мощных направлений молекуляр-
молекулярной биологии и генетики как генная и белковая инженерия - де-
дело неблагодарное.
Литература
1. Radzicka A., Wolfenden R. A proficient enzyme // Science. 1995. Vol. 267.
P. 90-93.
2. Schmid A., Dordick J.S., Hauer B. et al. Industrial biocatalysis today and
tomorrow // Nature. 2001. Vol. 409. P. 258-268.
3. Solon D.N., Voigt C.A., Mayot S.L. De novo design of biocatalysts // Curr.
Opin. Chem. Biol. 2002. Vol. 6. P. 125-129.
4. Cedrone F., Menez A., Quemeneur E. Tailoring new enzyme functions by
rational redesign // Curr. Opin. Struct. Biol. 2000. Vol. 10: P. 405^10.
5. Directed evolution of proteins, or how to improve enzymes for biocatalysis /
Ed. S. Brakmann, K. Johnsson. N.Y.: Wiley, 2002.
6. Petsko G.A. Enzyme evolution: Design by necessity // Nature. 2000.
Vol. 403. P. 606-607.
7. Griffiths A.D., Tawfikt D.S. Man-made enzymes - from design to in vitro
compartmentalization // Curr. Opin. Biotechnol. 2000. Vol. 11. P. 338-353.
8. Mirny L., Shakhnovich E. Protein folding theory: From lattice to all-atom
models //Annu. Rev. Biophys. Biomol. Struct. 2001. Vol. 30. P. 361-396.
462
9. De Grado W.F., Summa CM. De novo design and structural characteriza-
characterization of proteins and metalloproteins // Annu. Rev. Biochem. 1999. Vol. 68.
P. 779-819.
10. Bolon D.N., Mayo S.L. Enzyme-like proteins by computational protein
design // Proc. Nat. Acad. Sci. USA. 2001. Vol. 98. P. 14274-14279.
11. Hellinga H.W., Richards F.M. Construction of new ligand binding sites in
proteins of known structure. 1. Computer-aided modeling of sites with pre-
predefined geometry // J. Mol Biol. 1991. Vol. 222. P. 763-785.
12. Benson D.E., Wisz M.S., Hellinga H.W. Rational design of nascent metal-
loenzymes // Proc. Nat. Acad. Sci. USA. 2000. Vol. 97. P. 6292-6297.
13. Sole R.S., Manrubia S.C, Benton M. et al. Criticality and scaling in evolu-
evolutionary ecology //Trends Ecol. Evol. 1999. Vol. 14. P. 156-160.
14. Kauffman S., Ellington A.D. Thinking combinatorial^ // Curr. Opin. Chem.
Biol. 1999. Vol. 3. P. 256-259.
15. Molecular cloning: A laboratory manual on the Web. Chap. 13. Cold Spring
Harbor; Laboratory press, 2001. http://www.molecularcloning.com/
index.html
16. Kunkel ТА. Rapid and efficient site-specific mutagenesis without pheno-
typic selection // Proc. Nat. Acad. Sci. USA. 1985. Vol. 82. P. 488-492.
17. Handa P., Thanedar S., Varshney U. Rapid and reliable site-directed muta-
mutagenesis using Kunkel's approach // In vitro mutagenesis protocols. 2nd ed.
Totowa (N.J.). Humana press, 2002. P. 1-6. (Meth. in Mol. Biol.; Vol. 182).
18. Andrews С A., Lesley SA. Selection strategy for site-directed mutagenesis
based on altered p-lactamase specificity // Biotechniques. 1998. Vol. 24.
P. 972-977.
19. Andrews C.A., Lesley SA. Site-directed mutagenesis using altered p-lacta-
p-lactamase specificity // In vitro mutagenesis protocols. 2nd ed. Totowa (N.J.).
Humana press, 2002. P. 7-17. (Meth. in Mol. Biol.; Vol. 182).
20. Deng W.P., NickoloffJA. Site-directed mutagenesis of virtually any plas-
mid by eliminating a unique site // Anal. Biochem. 1992. Vol. 200.
P. 81-88.
21. Li S., Wilkinson M.F. Site-directed mutagenesis: A two-step method using
PCR and Dpnl // Biotechniques. 1997. Vol. 23. P. 588-590.
22. Li F., MullinsJJ. Site-directed mutagenesis facilitated by Dpnl selection on
hemimethylated DNA // In vitro mutagenesis protocols. 2nd ed. Totowa
(N.J.). Humana press, 2002. P. 19-28. (Meth. in Mol. Biol.; Vol. 182).
23. Gamper M., Hilvert D., Kast P. Probing the role of the C-terminus of
Bacillus subtilis chorismate mutase by a novel random protein termination
strategy // Biochemistry. 2000. Vol. 39. P. 1407-1408.
24. Ho SJV., Hunt H.D., Horton R.M. et al. Site-directed mutagenesis by over-
overlap extension using the polymerase chain reaction // Gene. 1998. Vol. 77.
P. 51-59.
25. Br0ns-Poulsen J., Peter sen N.E., Horder M., Kristiansen K. An improved
PCR-based method for site directed mutagenesis using megaprimers // Mol.
Cell. Probes. 1998. Vol. 12. P. 345-348.
26. Br0ns-Poulsen J., N0hrJ., Larsen L.K. Megaprirner method for polymerase
chain reaction-mediated generation of specific mutations in DNA // In vitro
463
mutagenesis protocols. 2nd ed. Totowa (N.J.). Humana press, 2002.
P. 71-76. (Meth. in Mol. Biol.; Vol. 182).
27. Kirsch R.D., Joly E. An improved PCR-mutagenesis strategy for two-site
mutagenesis or sequence swapping between related genes // Nucl. Acids
Res. 1998. Vol. 26. P. 1848-1850.
28. MeeteiA.R., Rao M.R.S. Generation of multiple site-specific mutations in a
single polymerase chain reaction product // Anal. Biochem. 1998. Vol. 264.
P. 288-291.
29. Meetei A.R., Rao M.R. Generation of multiple site-specific mutations by
polymerase chain reaction // Meth. Mol. Biol. 2002. Vol. 182. P. 95-102.
30. Wang L., Brock A., Herberich В., Schultz P.G. Expanding the genetic code
of Escherichia coli I I Science. 2001. Vol. 292, N 5516. P. 498-500.
31. Rothschild KJ., Gite S. tRNA-mediated protein engineering // Curr. Opin.
Biotechnol. 1999. Vol. 10. P. 64-70.
32. SaksM.E. Making sense out of nonsense //Proc. Nat. Acad. Sci. USA. 2001.
Vol. 98. P. 2125-2127.
33. Krieg U.C., Walter P., Johnson A.E. Photocrosslinking of the signal
sequence of nascent preprolactin to the 54-kilodalton polypeptide of the sig-
signal recognition particle // Ibid. 1986. Vol. 83. P. 8604-8608.
34. Noren CJ., Anthony-Cahill SJ., Griffith M.C., Schultz P.G. A general
method for site-specific incorporation of unnatural amino acids into pro-
proteins // Science. 1989. Vol. 244. P. 182-188.
35. Anthony-Cahill SJ., Griffith M.C., Noren CJ. et al. Site-specific mutagen-
mutagenesis with unnatural amino acids // Trends Biochem. Sci. 1990. Vol. 14.
P. 400-403.
36. Xiao G., Parsons J.F., Tesh K. et al. Conformational changes in the crys-
crystal structure of rat glutathione transferase Ml-1 with global substitution
of 3-fluorotyrosine for tyrosine // J. Mol. Biol. 1998. Vol. 281.
P. 323-339.
37. Budisa N., Minks C, Medrano FJ. et al. Residue specific bioincorpora-
tion of non-natural, biologically active amino acids into proteins as
possible drug carriers: Structure and stability of the per-thiaproline
mutant of annexin V // Proc. Nat. Acad. Sci. USA. 1998. Vol. 95.
P. 455-459.
38. Kowal A.K., Oliver J.S. Exploiting unassigned codons in Micrococcus
luteus for tRNA-based amino acid mutagenesis // Nucl. Acids Res. 1997.
Vol. 25. P. 4685-4689.
39. Chad S.T., Liu D.R., Froland WA., Schultz P.G. Development of improved
tRNAs for in vitro biosynthesis of proteins containing unnatural amino
acids // Chem. Biol. 1996. Vol. 3. P. 1033-1038.
40. Nowak M.W., Gallivan J.P., Silverman S.K. et al. In vivo incorporation of
unnatural amino acids into ion channels in Xenopus oocyte expression sys-
system // Meth. Enzymol. 1998. Vol. 293. P. 504-529.
41. Liu D.R., Magliery TJ., Pastrnak M., Schultz P.G. Engineering a tRNA and
aminoacyl-tRNA synthetase for the site-specific incorporation of unnatural
amino acids into proteins in vivo // Proc. Nat. Acad. Sci. USA. 1997.
Vol. 94. P. 10092-10097.
464
42. Furter R. Expansion of the genetic code: Site-directed p-fluoro phenylala-
nine incorporation in Escherichia coli // Protein Sci. 1998. Vol. 7.
P. 419-426.
43. De Santis G., Jones J.B. Combining site-specific chemical modification
with site-directed mutagenesis. Versatile strategy to move beyond structur-
structural limitations of 20 natural amino acids side chains in protein engineering //
P. 55-56.
44. Plettner E., Khumtaveepom K., Shang X., Jones J. A combinatorial
approach to chemical modification of subtilisin Bacillus lentus // Bioorg.
Med. Chem. Lett. 1998. Vol. 8. P. 2291-2296.
45. Muir T.W. Semisynthesis of proteins by expressed protein ligation // Annu.
Rev. Biochem. 2003. Vol. 72. P. 249-289.
46. Chen Y., Ebright Y.W., Ebright R.H. Identification of the target of a tran-
transcription activator protein by protein-protein photocrosslinking // Science.
1994. Vol. 265, N 5168. P. 90-92.
47. Mukhopadhyay J., Kapanidis A.N., Mekler V. et al. Translocation of
sigmaG0) with RNA polymerase during transcription: Fluorescence reso-
resonance energy transfer assay for movement relative to DNA // Cell. 2001.
Vol. 106, N 4. P. 453^63.
48. Curley K., Lawrence D.S. Photoactivation of a signal transduction pathway
in living cells // J. Amer. Chem. Soc. 1998. Vol. 120, N 33. P. 8573-
8574.
49. Wetzel R., Halualani R., Stults J.T., Quan C. A general method for highly
selective cross-linking of unprotected polypeptides via pH-controlled mod-
modification of N-terminal alpha-amino groups // Bioconjug. Chem. 1990.
Vol. 1,N2.P. 114-122.
50. Zou K., Miller W.T., GivensRS., Bayley H. Caged thiophosphotyrosine pep-
tides // Angew. Chem. Intern. Ed. 2001. Vol. 40, N 16. P. 3049-3051.
51. Zou K., Cheley S., Givens R.S., Bayley H. Catalytic subunit of protein
kinase: A caged at the activating phosphothreonine // J. Amer. Chem. Soc.
2002. Vol. 124, N 28. P. 8220-8229.
52. Homandberg G.A., Laskowski M. Enzymatic resynthesis of the hydrolyzed
peptide bond(s) in ribonuclease S // Biochemistry. 1979. Vol. 18, N 4.
P. 586-592.
53. Vogel K., Chmielewski J. Rapid and efficient resynthesis of proteolyzed
triose phosphate isomerase // J. Amer. Chem. Soc. 1994. Vol. 116, N 24.
P. 11163-11164.
54. Witte K., Sears P., Martin R., Wong C.-H. Glycopeptide condensation and
glycosylation // Ibid. 1997. Vol. 119, N 9. P. 2114-2118.
55. Jackson D.Y., Burnier J., Quan С et al. A designed peptide ligase for total
synthesis of ribonuclease. A with unnatural catalytic residues // Science.
1994. Vol. 266, N 5183. P. 243-247.
56. Gaertner H.F., Rose K., Cotton R. et al. Construction of protein analogues
by site-specific condensation of unprotected fragments // Bioconjug. Chem.
1992. Vol. 3, N 3. P. 262-268.
57. Dawson P.E., Muir T.W., Clark-Lewis I., Kent S.B. Synthesis of proteins by
native chemical ligation // Science. 1994. Vol. 266, N 5186. P. 776-779.
16. Патрушев Л.И. Т. 1 465
58. Dawson P.E., Kent S.B. Synthesis of native proteins by chemical ligation //
Annu. Rev. Biochem. 2000. Vol. 69. P. 923-960.
59. Hofmann R.M., Muir T.W. Recent advances in the application of expressed
protein ligation to protein engineering // Curr. Opin. Biotechnol. 2002.
Vol. 13, N 4. P. 297-303.
60. Paulus H. Protein splicing and related forms of protein autoprocessing //
Annu. Rev. Biochem. 2000. Vol. 69. P. 447^96.
61. Chong S., Mersha F.B., Comb D.G. et al. Single-column purification of free
recombinant proteins using a self-cleavable affinity tag derived from a pro-
protein-splicing element // Gene. 1997. Vol. 192. P. 278-281.
62. Evans T.C., BennerJ., Xu M.Q. The in vitro ligation of bacterially expressed
proteins using an intein from Methanobacterium thermoautotrophicum I IJ.
Biol. Chem. 1999. Vol. 274, N 7. P. 3923-3926.
63. Mathys S., Evans T.C., Jr., Chute C.I. et al. Characterization of a self-splic-
self-splicing mini-intein and its conversion into autocatalytic N- and C-terminal
cleavage elements: Facile production of protein building blocks for protein
ligation//Gene. 1999. Vol. 231, N 1/2. P. 1-13.
64. Southworth M.W., Adam E., Panne D. et al. Control of protein splicing by
intein fragment reassembly // EMBO J. 1998. Vol. 17, N 4. P. 918-926.
65. Mills K.V., Lew B.M., Jiang S.-Q., Paulus H. Protein splicing in trans by puri-
purified N- and C-terminal fragments of the Mycobacterium tuberculosis RecA
intein // Proc. Nat. Acad. Sci. USA. 1998. Vol. 95, N 7. P. 3543-3548.
66. Wu H., Ни Z., Liu X.Q. Protein trans-splicing by a split intein encoded in a
split DnaE gene of Synechocystis sp. PCC6803 // Ibid. N 16. P. 9226-9231.
67. Yamazaki Т., Otomo Т., О da N. et al. Segmental isotope labeling for protein
NMR using peptide splicing // J. Amer. Chem. Soc. 1998. Vol. 120, N 22.
P. 5591-5592.
68. Shingledecker K., Jiang S.-Q., Paulus H. Molecular dissection of the
Mycobacterium tuberculosis RecA intein: Design of a minimal intein and of
a trans-splicing system involving two intein fragments // Gene. 1998.
Vol. 207, N 2. P. 187-195.
69. Scott СР., Abel-Santos E., Wall M. et al. Production of cyclic peptides and
proteins in vivo II Proc. Nat. Acad. Sci. USA. 1999. Vol. 96, N 24.
P. 13638-13643.
70. Ozawa Т., Nogami S., Sato M. et al. A fluorescent indicator for detecting
protein-protein interactions in vivo based on protein splicing // Anal. Chem.
2000. Vol. 72, N 21. P. 5151-5157.
71. Mootz H.D., Muir T.W. Protein splicing triggered by a small molecule // J.
Amer. Chem. Soc. 2002. Vol. 124, N 31. P. 9044-9045.
72. PelletierJ., Sidhut S. Mapping protein-protein interactions with combinato-
combinatorial biology methods // Curr. Opin. Biotechnol. 2001. Vol. 12. P. 340-347.
73. Ozawa Т., Kaihara A., Sato M. et al. Split luciferase as an optical probe for
detecting protein-protein interactions in mammalian cells based on protein
splicing // Anal. Chem. 2001. Vol. 73, N 11. P. 2516-2521.
74. Huse M., Muir T.W., Xu L. et al. The TGF-beta receptor activation process:
An inhibitor- to substrate-binding switch // Mol. Cell. 2001. Vol. 6, N 3.
P. 671-682.
466
75. Wu J., Ни M., Chai J. et al. Crystal structure of a phosphorylated Smad2.
Recognition of phosphoserine by the MH2 domain and insights on
Smad function in TGF-beta signaling // Ibid. Vol. 8, N 6. P. 1277-
1289.
76. Peters C, Wagner M., VolkertM., Waldmann H. Bridging the gap between
cell biology and organic chemistry: Chemical synthesis and biological
application of lipidated peptides and proteins // Naturwissenschaften. 2002.
Vol. 89. P. 381-390.
77. Spiegelman S. An approach to the experimental analysis of precellular evo-
evolution // Quart. Rev. Biophys. 1971. Vol. 4, N 2. P. 213-253.
78. Shuster P. Evolutionary biotechnology - from ideas and concepts to exper-
experiments and computer simulations // Directed molecular evolution of pro-
proteins: or how to improve enzymes for biocatalysis / Ed. S. Brackmann,
K. Johnsson. N.Y.: Wiley, 2002. P. 5-28.
79. Domingo E., Holland JJ. RNA virus mutations and fitness for survival //
Annu. Rev. Microbiol. 1997. Vol. 51. P. 151-178.
80. Lin-Goerke J.L., Robbins D.J., BurczakJ.D. PCR-based random mutagene-
sis using manganese and reduced dNTP concentration // Biotechniques.
1997. Vol. 23. P. 409-412.
81. Fromant M., Blanquet S., Plateau P. Direct random mutagenesis of gene-
sized DNA fragments using polymerase chain reaction // Anal. Biochem.
1995. Vol. 224. P. 347-353.
82. Miyazaki K., Arnold F.H. Exploring nonnatural evolutionary pathways by
saturation mutagenesis: Rapid improvement of protein function // J. Mol.
Evol. 1999. Vol. 49. P. 716-720.
83. Xu H., Petersen E.I., Petersen S.B., El-Gewely M.R. Random mutagenesis
libraries: Optimization and simplification by PCR // Biotechniques. 1999.
Vol. 27. P. 1102-1104, 1106, 1108.
84. Fenton C, Xu H., Petersen E.I. et al. Random mutagenesis for protein
breeding // Meth. Mol. Biol. 2002. Vol. 182. P. 231-241.
85. Kikuchi M., Harayama S. DNA shuffling and family shuffling for in vitro
gene evolution // In vitro mutagenesis protocols / Ed. J. Braman. Totowa
(N.J.). Humana press, 2002. P. 243-257.
86. Zhao H., Giver L., Shao Z. et al. Molecular evolution by staggered extension
process (StEP) in vitro recombination // Nature Biotechnol. 1998. Vol. 16.
P. 258-261.
87. Sieber V., Martinez C.A., Arnold F.H. Libraries of hybrid proteins from dis-
distantly related sequences // Ibid. 2001. Vol. 19. P. 456^60.
88. Smith G.P. Filamentous fusion phage: Novel expression vectors that display
cloned antigens on the virion surface // Science. 1985. Vol. 228, N 4705.
P. 1315-1317.
89. Fernandez-Gacio A., Uguen M., Fastrez J. Phage display as a tool for the
directed evolution of enzymes // Trends Biotechnol. 2003. Vol. 21.
p. 408^14.
90. Riechmann L., Holliger P. The C-terminal domain of TolA is the corecep-
tor for filamentous phage infection of E. coli // Cell. 1997. Vol. 90, N 2.
P. 351-360.
16* 467
91. Holliger P., Riechmann L., Williams R.L. Crystal structure of the two N-ter-
minal domains of g3p from filamentous phage fd at 1.9 angstrom: Evidence
for conformational lability // J. Mol. Biol. 1999. Vol. 288, N 4. P. 649-657.
92. Rakonjac J., Feng J.N., Model P. Filamentous phage are released from the
bacterial membrane by a two-step mechanism involving a short C-terminal
fragment of pill // Ibid. 1999. Vol. 289, N 5. P. 1253-1265.
93. BeekwilderJ., Rakonjac J., Jongsma M., Bosch D. A phagemid vector using
the E. coli phage shock promoter facilitates phage display of toxic pro-
proteins // Gene. 1999. Vol. 228, N 1/2. P. 23-31.
94. Dunn I.S. Assembly of functional bacteriophage-lambda virions incorporat-
incorporating C-terminal peptide or protein fusions with the major tail protein // J.
Mol. Biol. 1995, Vol. 248, N 3. P. 497-506.
95. Efimov V.P., Nepluev I.V., Mesyanzhinov V.V. Bacteriophage-T4 as a sur-
surface display vector // Virus Genes. 1995. Vol. 10, N 2. P. 173-177.
{a) Scott J.K. Discovering peptide ligands using epitope libraries // Trends
Biochem. Sci. 1992. Vol. 17. P. 241-245.
96. Roberts R.W., Ja W.W. In vitro selection of nucleic acids and proteins: What
are we learning? // Curr. Opin. Struct. Biol. 1999. Vol. 9. P. 521-529.
97. Soumillion P., FastrezJ. Investigation of phage display for the directed evo-
evolution of enzymes // Directed evolution of proteins, or how to improve
enzymes for biocatalysis / Ed. S. Brakmann, K. Johnsson. N.Y.: Wiley,
2002. P. 79-110.
98. Light J., Lerner RA. Random mutagenesis of staphylococcal nuclease and
phage display selection // Bioorg. Med. Chem. 1995. Vol. 3, N 7.
P. 955-967.
99. Pauling L. Chemical achievement and hope for the future // Amer. Sci.
1948. Vol. 36. P. 51-58.
100. Pasqualini R., Ruoslahti E. Organ targeting in vivo using phage display
peptide libraries // Nature. 1996. Vol. 380, N 3572. P. 364-366.
101. George A.J.T., Lee L., Pitzalis С Isolating ligands specific for.human vas-
culature using in vivo phage selection // Trends Biotechnol. 2003. Vol. 21.
P. 199-203.
102. Samoylova T.I., Smith B.F. Elucidation of muscle-binding peptides by
phage display screening // Muscle Nerve. 1999. Vol. 22. P. 460-466.
103. Arap W., Haedicke W., Bernasconi M. et al. Targeting the prostate for
destruction through a vascular address // Proc. Nat. Acad. Sci. USA. 2002.
Vol. 99, N3. P. 1527-1531.
104. Essler M., Ruoslahti E. Molecular specialization of breast vasculature:
A breast-homing phage-displayed peptide binds to aminopeptidase P in
breast vasculature // Ibid. P. 2252-2257.
105. Trepel M., Arap W., Pasqualini R. et al. Modulation of the immune
response by systemic targeting of antigens to lymph nodes // Cancer Res.
2001. Vol. 61, N 22. P. 8110-8112.
106. Michon I.N., Hauer A.D., Von der Thusen J.H. et al. Targeting of peptides
to restenotic vascular smooth muscle cells using phage display in vitro and
in vivo II Biochim. biophys. acta. 2002. Vol. 1591, N 1/3. P. 87-97.
107. Houston P., Goodman J., Lewis A. et al. Homing markers for atheroscle-
468
rosis: Applications for drag delivery, gene delivery and vascular imaging //
FEBS Lett. 2001. Vol. 492, N 1/2. P. 73-77.
108. Lee L., Buckley C, Blades M.C. et al. Identification of synovium-specific
homing peptides by in vivo phage display selection // Arthritis Rheum.
2002. Vol. 46, N 8. P. 2109-2120.
109. Lee S.Y., Choi J.H., Xu Z. Microbial cell-surface display // Trends
Biotechnol. 2003. Vol. 21. P. 45-51.
110. Wittrup K.D. Protein engineering by cell-surface display // Curr. Opin.
Biotechnol. 2001. Vol. 12. P. 395-399.
111. Wittrup K.D. Directed evolution of binding proteins by cell surface dis-
display: Analysis of the screening process // Directed evolution of proteins, or
how to improve enzymes for biocatalysis / Ed. S. Brakmann, K. Johnsson.
N.Y.: Wiley, 2002. P. 111-126.
112. Mattheakis L.C., BhattR.R., Dower W.J. An in vitro polysome display sys-
system for identifying ligands from very large peptide libraries // Proc. Nat.
Acad. Sci. USA. 1994. Vol. 91, N 19. P. 9022-9026.
113. Hanes J., Pliickthun A. In vitro selection and evolution of functional pro-
proteins by using ribosome display // Ibid. 1997. Vol. 94. P. 4937^942.
114. Roberts R.W., SzostakJ.W. RNA-peptide fusions for the in vitro selection
peptides and proteins // Ibid. 1997. Vol. 94. P. 12297-12302.
115. Schaffitzel C, Pliickthun A. Protein-fold evolution in the test tube // Trends
Biochem. Sci. 2001. Vol. 26. P. 577-579.
116. Takahashi T.T., Austin R.J., Roberts R.W. mRNA display: Ligand discov-
discovery, interaction analysis and beyond // Ibid. 2003. Vol. 28. P. 159-165.
117'. McDonald P.N. Two-hybrid systems: Methods and protocols:
Introduction // Meth. Mol. Biol. 2001. Vol. 177. P. v-viii.
118. Carter B.T., Lin H., Cornish V.W. Yeast n-hybrid systems for molecular
evolution // Directed evolution of proteins, or how to improve enzymes for
biocatalysis /Ed. S. Brakmann, K. Johnsson. N.Y.: Wiley, 2002.
P. 127-158.
119. Fields S., Song O. A novel genetic system to detect protein-protein inter-
interactions // Nature. 1989. Vol. 340, N 6230. P. 245-246.
120. Brent R., Ptashne M. A eukaryotic transcriptional activator bearing the
DNA specificity of a prokaryotic repressor // Cell. 1985. Vol. 43, N 2/3.
P. 729-736.
121. Hope I.A., Struhl K. Functional dissection of a eukaryotic transcriptional
activator protein, GCN4 of yeast // Cell. 1986. Vol. 46, N 6. P. 885-894.
122. Boeke J.D., La Croute F., Fink G.R. A positive selection for mutants lack-
lacking orotidine-5'-phosphate decarboxylase activity in yeast: 5-fluoro-orotic
acid resistance // Mol. Gen. Genet. 1984. Vol. 197, N 2. P. 345-346.
123. Vojtek А.В., Hollenberg S.M., Cooper J.A. Mammalian Ras interacts
directly with the serine/threonine kinase Raf// Cell. 1993. Vol. 74, N 1.
P. 205-214.
124. Serebriiskii I., Khazak V., Golemis E. A two-hybrid dual bait system to dis-
discriminate specificity of protein interactions // J. Biol. Chem. 1999.
Vol. 274, N 24. P. 17080-17087.
125. Le Douarin В., Pierrat В., Von Ваш E. et al. A new version of the two-
469
hybrid assay for detection of protein-protein interactions // Nucl. Acids
Res. 1995. Vol. 23, N 5. P. 876-878.
126. Ptashne M. A genetic switch. 2nd ed. Cambridge: Blackwell: Cell press,
1992.
127. Vidal M., Brachmann R.K., Fattaey A. et al. Reverse two-hybrid and one-
hybrid systems to detect dissociation of protein-protein and DNA-protein
interactions // Proc. Nat. Acad. Sci. USA. 1996. Vol. 93, N 19.
P. 10315-10320.
128. Shih H.M., Goldman P.S., De Maggio AJ. et al. A positive genetic selec-
selection for disrupting protein-protein interactions: Identification of CREB
mutations that prevent association with the coactivator СВР // Ibid. N 24.
P. 13896-13901.
129. Hirst M., Ho C, Sabourin L. et al. A two-hybrid system for transactivator
bait proteins // Ibid. 2001. Vol. 98, N 15. P. 8726-8731.
130. Tzamarias D., StruhlK. Functional dissection of the yeast Cyc8-Tupl tran-
scriptional co-repressor complex // Nature. 1994. Vol. 369, N 6483.
P. 758-761.
131. MichnikS.W. Exploring protein interactions by interaction-induced folding
of proteins from complementary peptide fragments // Curr. Opin. Struct.
Biol. 2001. Vol. 11, N 4. P. All-All.
132. Но Т., Ota K., Kubota H. et al. Roles for the two-hybrid system in explo-
exploration of the yeast protein interactome // Mol. Cell. Proteomics. 2002.
Vol. 1. P. 561-566.
133. Ito Т., Chiba Т., Ozawa R. et al. A comprehensive two-hybrid analysis to
explore the yeast protein interactome // Proc. Nat. Acad. Sci. USA. 2001.
Vol. 98. P. 4569^574.
134. Deane СМ., Salwinski L., Xenarios I., Eisenberg D. Protein interactions:
Two methods for assessment of the reliability of high throughput observa-
observations // Mol. Cell. Proteomics. 2002. Vol. 1. P. 349-356.
135. Matthews L.R., Vaglio P., Reboul J. et al. Identification of potential inter-
interaction networks using sequence-based searches for conserved protein-pro-
protein-protein interactions or "interologs" // Genome Res. 2001. Vol. 11.
P. 1771-1775.
136. Ни J.C., О'Shea EX., Kim P.S., Sauer R.T. Sequence requirements for
coiled-coils: analysis with lambda repressor-GCN4 leucine zipper
fusions // Science. 1990. Vol. 250, N 4986. P. 1400-1403.
137. Kornacker M.G., Remsburg В., Menzel R. Gene activation by the AraC
protein can be inhibited by DNA looping between AraC and a LexA
repressor that interacts with AraC: possible applications as a two-hybrid
system // Mol. Microbiol. 1998. Vol. 30, N 3. P. 615-624.
138. Whipple F.W. Genetic analysis of prokaryotic and eukaryotic DNA-bind-
ing proteins in Escherichia coli II Nucl. Acids Res. 1998. Vol. 26, N 16.
P. 3700-3706.
139. Joung J.K., Ramm E.I., Pabo CO. A bacterial two-hybrid selection system
for studying protein-DNA and protein-protein interactions // Proc. Nat.
Acad. Sci. USA. 2000. Vol. 97, N 13. P. 7382-7387.
140. Marsolier M.C, Prioleau M.N., Sentenac A. A RNA polymerase Ill-based
470
two-hybrid system to study RNA polymerase II transcriptional regulators //
J. Mol. Biol. 1997. Vol. 268, N 2. P. 243-249.
141. Karimova G., Ullmann A., Ladant D. A bacterial two-hybrid system that
exploits a cAMP signaling cascade in Escherichia coli II Meth. Enzymol.
2000. Vol. 328. P. 59-73.
142. Vidal M., Legrain P. Yeast forward and reverse "n"-hybrid systems //
Nucl. Acids Res. 1999. Vol. 27, N 4. P. 919-929.
143. Salero E., Perez-Sen R., Aruga J. et al. Transcription factors Zicl and Zic2
bind and transactivate the a Apolipoprotein E gene promoter // J. Biol.
Chem. 2001. Vol. 276. P. 1881-1888.
144. Hock M.B., Brown M.A. Nuclear factor of activated T cells 2 transactiva-
tion in mast cells: A novel isoform-specific transactivation domain confers
unique FceRI responsiveness // Ibid. 2003. Vol. 278, N 29.
P. 26695-26703.
145. Polgar L., Bender ML. A new enzyme containing a synthetically formed
active site: Thiolsubtilisin // J. Amer. Chem. Soc. 1966. Vol. 88.
P. 3153-3154.
146. NeetK.E., KoshlandD.E., Jr. The conversion of serine at the active site of
subtilisin to cysteine: A 'chemical mutation' // Proc. Nat. Acad. Sci. USA.
1966. Vol. 56. P. 1606-1611.
147. Kaiser E.T. Catalytic activity of enzymes altered at their active sites //
Angew. Chem. Intern. Ed. 1988. Vol. 27. P. 913-922.
148. De Santis G., Jones J.B. Chemical modification of enzymes for enhanced
functionality // Curr. Opin. Biotechnol. 1999. Vol. 10. P. 324-330.
149. St. Clair N.L., Navia M.A. Cross-linked enzyme crystals as robust biocata-
lysts // J. Amer. Chem. Soc. 1992. Vol. 114. P. 7314-7316.
150. Wang Y.-F., Yakovlevsky K., Zhang В., Margolin AL. Cross-linked
enzyme crystals (CLEC) of subtilisin: A versatile catalyst for organic syn-
synthesis // J. Org. Chem. 1997. Vol. 32. P. 3488-3495.
151. Van Unen D.-J., Sakodinskayal.K., EngbersenJ.FJ., Reinhoudt D.N'. Crown
ether activation of cross-linked subtilisin Carlsberg crystals in organic sol-
solvents // J. Chem. Soc. Perkin Trans. 1998. Vol. 20. P. 3341-3343.
152. Lundbland R.L., Bradshaw R.A. Applications of site-specific chemical
modification in the manufacture of biopharmaceuticals. 1. An overview //
Biotechnol. Appl. Biochem. 1997. Vol. 26. P. 143-151.
153. Jene Q., Pearson J.C., Lowe C.R. Surfactant modified enzymes: Solubility
and activity of surfactant-modified catalase in organic solvents // Enzyme.
Microb. Technol. 1997. Vol. 20. P. 69-74.
154. Khmelnitsky Y.L., Rich J.O. Biocatalysis in nonaqueous solvents // Curr.
Opin. Chem. Biol. 1999. Vol. 3. P. 47-53.
155. Schmitke J.L., Stern L.J., Klibanov A.M. The crystal structure of subtilisin
Carlsberg in anhydrous dioxane and its comparison with those in water and
acetonitrile // Proc. Nat. Acad. Sci. USA. 1997. Vol. 94. P. 4250-4255.
156. Griebenow K., Klibanov A.M. Can conformational changes be responsible
for solvent and excipient effects on the catalytic behavior of subtilisin
Carlsberg in organic solvents? // Biotechnol. Bioeng. 1997. Vol. 53.
P. 351-362.
471
157. Wang P., Sergeeva M.V., him L., Dor dick J.S. Biocatalytic plastics as
active and stable materials for biotransformations // Nature Biotechnol.
1997. Vol. 15. P. 789-793.
158. Dordick J.S., Novick SJ., Sergeeva M.V. Biocatalytic plastics // Chem.
Industry. 1998. P. 17-20.
159. Gill I., Ballesteros A. Bioencapsulation within synthetic polymers. (Part 2).
Non-sol-gel protein-polymer biocomposites // Trends Biotechnol. 2000.
Vol. 18. P. 469-Ф79.
160. Partridge J., Hailing P.J., Moore B.D. Practical route to high activity
enzyme preparations for synthesis in organic media // Chem. Commun.
1998. Vol. 7. P. 841-842.
161. Okahata Y., Mori T. Effective transgalactosylation catalysed by a lipid-
coated P-D-galactosidase in organic solvents // J. Chem. Soc. Perkin Trans.
Part I. 1996. Vol. 23. P. 2861-2866.
162. Mori Т., Okahata Y. A variety of lipid-coated glycoside hydrolases as
effective glycosyl transfer catalysts in homogeneous organic solvents //
Tetrahedron Lett. 1997. Vol. 38. P. 1971-1974.
163. Piletsky S.A., Alcock S., Turner A.P.F. Molecular imprinting: At the edge
of the third millennium // Trends Biotechnol. 2001. Vol. 19. P. 9-12.
164. Ohya Y., Miyaoka J., Ouchi T. Recruitment of enzyme activity in albumin
by molecular imprinting // Macromol. Rapid Commun. 1996. Vol. 17.
P. 871-874.
165. Slade CJ., Vulfson E.N. Induction of catalytic activity in proteins by
lyophilization in the presence of a transition state analogue // Biotechnol.
Bioeng. 1998. Vol. 57. P. 211-215.
166. Rich J.O., Dordick J.S. Controlling subtilisin activity and selectivity in
organic media by imprinting with nucleophilic substrates // J. Amer.
Chem. Soc. 1997. Vol. 119. P. 3245-3252.
167. Tucker CL., Gera J.F., Uetz P. Towards an understanding of complex pro-
protein networks // Trends Cell Biol. 2001. Vol.11, N 3. P. 102-106.
168. Carreras C.W., Santi D.V. Engineering of modular polyketide synthases to
produce novel polyketides // Curr. Opin. Biotechnol. 1998. Vol. 9.
P. 403^11.
169. Mootz H.D., Marahiel M.A. Design and application of multimodular pep-
tide synthetases // Ibid. 1999. Vol. 10. P. 341-348.
170. Xu Z., Bae W., Mulchandani A. et al. Heavy metal removal by novel CBD-
EC20 sorbents immobilized on cellulose // Biomacromolecules. 2002.
Vol. 3. P. 462-465.
171. Crane D.E., Walsh СТ., Khosla C. Harnessing the biosynthetic code:
Combinations permutations, and mutations // Science. 1998. Vol. 282.
P. 63-67.
172. Ljungcrantz P., Carls son H., Mansson M.-O. et al. Construction of an arti-
artificial bifunctional enzyme, oc-galactosidase/glactose dehydrogenase,
exhibiting efficient galactose channeling // Biochemistry. 1989. Vol. 28.
P. 8786-8792.
173. Lindbladh C, Persson M., Bulow L., Mosbach K. Characterization of
recombinant galactose dehydrogenaseAuciferase, displaying an improved
472
bioluminescence in a three-enzyme system // Europ. J. Biochem. 1991.
Vol. 204. P. 241-247.
174. Lindbladh C, Rault M., Hagglund C. et al. Preparation and kinetic charac-
characterization of a fusion protein of yeast mitochondrial citrate synthase and
malate dehydrogenase // Biochemistry. 1994. Vol. 33. P. 11692-11698.
175. Beguin P., Lemaire M. The cellulosome: An exocellular, multiprotein
complex specialized in cellulose degradation // Crit. Rev. Biochem. Mol.
Biol. 1996. Vol. 31. P. 201-236.
176. Bayer E.A., Morag E., Lamed R. The cellulosome - a treasure-trove for
biotechnology //Trends Biotechnol. 1994. Vol. 12. P. 379-386.
177. Leibovitz E., Beguin P. Comparison of two scaffolding polypeptides for
the integration of different proteins in synthetic complexes derived from
the Clostridium thermocellum cellulosome // Enzyme Microb. Technol.
1998. Vol. 22. P. 588-593.
178. Pages S., Bela'ich A., Belaich J.-P. et al. Species specificity of the cohesin-
dockerin interaction between Clostridium thermocellum and Clostridium
cellulolyticum - prediction of specificity determinants of the dockerin
domain // Proteins. 1997. Vol. 29. P. 517-527.
179. Hopfner K.-P., Kopetzki E., Krefie G.-B. et al. New enzyme lineages by
subdomain shuffling // Proc. Nat. Acad. Sci. USA. 1998. Vol. 95.
P. 9813-9818.
180. Kim Y.-G., С ha J., Chandrasegaran S. Hybrid restriction enzymes: Zinc
finger fusions to Fokl cleavage domain // Proc. Nat. Acad. Sci. USA. 1996.
Vol. 93. P. 1156-1160.
181. Kumar A., Grimes В., Logan M. et al. A hybrid sigma subunit directs RNA
polymerase to a hybrid promoter in Escherichia coli II J. Mol. Biol. 1995.
Vol. 246. P. 563-571.
182. Mencia M., Monsalve M., Rojo F., Salas M. Substitution of the C-terminal
domain of the Escherichia coli RNA polymerase a subunit by that from
Bacillus subtilis makes the enzyme responsive to a Bacillus subtilis tran-
scriptional activator// Ibid. 1998. Vol. 275. P. 177-185.
183. Betton J.M., Jacob J.P., Hofnung M., Broome-Smith J.K. Creating a
bifunctional protein by insertion of a C-lactamase into maltodextrin-bind-
ing protein // Nature Biotechnol. 1997. Vol. 15. P. 1276-1279.
184. Brennan C.A., Christianson K., La Fleur M.A., Mandecki W. A molecular
sensor system based on genetically engineered alkaline phosphatase //
Proc. Nat. Acad. Sci. USA. 1995. Vol. 92. P. 5783-5787.
185. Fushinobu S., Ohta Т., Matsuzawa H. Homotropic activation via the sub-
unit interaction and allosteric symmetry revealed on analysis of hybrid
enzymes of L-lactate dehydrogenase // J. Biol. Chem. 1998. Vol. 273.
P. 2971-2976.
186. Hastings J.W. Chemistries and colors of bioluminescent reactions:
A review // Gene. 1996. Vol. 173, N 1: Special number. P. 5-11.
187. Lewis J.C., Daunert S. Photoproteins as luminescent labels in binding
assays // Fresenius J. Anal. Chem. 2000. Vol. 366. P. 760-768.
188. Shimomura O. A short story of aequorin // Biol. Bull. 1995. Vol. 189, N 1.
P. 1-5.
473
189. Johnson F.H., Shimomura 0., Saiga Y. et al. // J. Cell. Сотр. Physiol.
1962. Vol. 60. P. 85.
190. Deo S.K., Daunert S. Luminescent proteins from Aequorea victoria'.
Applications in drug discovery and in high throughput analysis // Fresenius
J. Anal. Chem. 2001. Vol. 369. P. 258-266.
191. Pausch M.H. G-protein-coupled receptors in Saccharomyces cerevisiae:
High-throughput screening assays for drug discovery // Trends Biotechnol.
1997. Vol. 15, N 12. P. 487^94.
192. Ungrin M.D., Singh L.M., Stocco R. et al. An automated aequorin lumi-
luminescence-based functional calcium assay for G-protein-coupled recep-
receptors // Anal. Biochem. 1999. Vol. 272, N 1. P. 34-42.
193. WahlforsJ., Loimas S., Pasanen Т., Hakkarainen T. Green fluorescent pro-
protein (GFP) fusion constructs in gene therapy research // Histochem. Cell.
Biol. 2001. Vol. 115. P. 59-65.
194. Verkhusha V.V., Lulcyanov К A. The molecular properties and applications
of Anthozoa fluorescent proteins and chromoproteins // Nature Biotechnol.
2004. Vol. 22. P. 289-296.
195. Reiter Y., Pastan 1. Recombinant Fv immunotoxins and Fv fragments as
novel agents for cancer therapy and diagnosis // Trends Biotechnol. 1998.
Vol. 16. P. 513-520.
196. Falnes P.0., Sandvig K. Penetration of protein toxins into cells // Curr.
Opin. Cell Biol. 2000. Vol. 12. P. 407^13.
197. Kreitman RJ. Immunotoxins in cancer therapy // Curr. Opin. Immunol.
1999. Vol. 11. P. 570-578.
198. White С A., Weaver R.L., Grillo-Lopez A.I. Antibody-targeted
immunotherapy for treatment of malignancy // Annu. Rev. Med. 2001.
Vol. 52. P. 125-145.
199. Weinberg A.D. OX40: Targeted immunotherapy - implications for tem-
tempering autoimmunity and enhancing vaccines // Trends Immunol. 2002.
Vol. 23. P. 102-108.
200. Brown J.R., Doolittle W.F. Archaea and the prokaryote-to-eukaryote tran-
transition//Microbiol. Mol. Biol. Rev. 1997. Vol. 61. P. 456-502.
201. Hough D.W., Danson M.J. Extremozymes // Curr. Opin. Chem. Biol. 1999.
Vol. 3. P. 39^6.
202. Yip K.S.P., Britton K.L., Stillman T.J. et al. Insights into the molecular
basis of thermal stability from the analysis of ion-pair networks in the glu-
tamate dehydrogenase family // Europ. J. Biochem. 1998. Vol. 255.
P. 336-346.
203. Aguilar C.F., Sanderson I., Moracci M. et al. Crystal structure of the C-gly-
cosidase from the hyperthermophilic Archaeon Sulfolobus solfataricus:
resilience as a key factor in thermostability // J. Mol. Biol. 1997. Vol. 271.
P. 789-802.
204. Russell RJ.M., Gerike U., Danson M.J. et al. Structural adaptations of the
cold-active citrate synthase from an Antarctic bacterium // Structure. 1998.
Vol. 6. P. 351-361.
205. Ermler U., Merckel M.C., Thauer R.K., Shima S. Formylmethanofuran:
Tetrahydromethanopterin formyltransferase from Methanopyrus
474
kandleri - new insights into salt-dependence and thermostability //
Structure. 1997. Vol. 5. P. 635-646.
206. Danson M.J., Hough D.W. The structural basis of protein halophilicity //
Сотр. Biochem. Physiol. A. 1997. Vol. 117. P. 307-312.
207. Патрушев Л.И., Валяев А.С., Головченко П.А. и др. Клонирование
гена термостабильной ДНК-полимеразы Thermus aquaticus YT1 и
его экспрессия в клетках Escherichia coli // Молекуляр. биология.
1993. Т. 27. С 1100-1112.
208. Van den Burg В., Vriend G., Veltman O.R. et al. Engineering an enzyme
to resist boiling // Proc. Nat. Acad. Sci. USA. 1998. Vol. 95.
P. 2056-2060.
209. Miyazaki K., Wintrode PL., Grayling R.A. et al. Directed evolution study
of temperature adaptation in a psychrophilic enzyme // J. Mol. Biol. 2000.
Vol. 297. P. 1015-1026.
210. Arnold F.H., Wintrode P.L., Miyazaki K., Gershenson A. How enzymes
adapt: Lessons from directed evolution // Trends Biochem. Sci. 2001.
Vol. 26. P. 100-106.
211. Akanuma S. et al. Further improvements of the thermal stability of a par-
partially stabilized Bacillus subtilis 3-isopropylmalate dehydrogenase variant
by random and site-directed mutagenesis // Europ. J. Biochem. 1999.
Vol. 260. P. 499-504.
212. Winter G., Milstein C. Man-made antibodies // Nature. 1991. Vol. 349.
P. 293-299.
213. Kramer K., Hock В. Recombinant antibodies for environmental analysis //
Anal. Bioanal. Chem. 2003. Vol. 377. P. 417-426.
214. Dall'Acqua W., Carter P. Antibody engineering //Curr. Opin. Struct. Biol.
1998. Vol. 8. P. 443-450.
215. Riechmann L., Clark M., Waldmann H., Winter G. Reshaping human anti-
antibodies for therapy // Nature. 1988. Vol. 332. P. 323-327.
216. Green L.L., Jakobovits A. Regulation of В cell development by variable
gene complexity in mice reconstituted with human immunoglobulin yeast
artificial chromosomes // J. Exp. Med. 1998. Vol. 188. P. 483-495.
217. Davis C.G., Gallo ML., Corvalan J.R.F. Transgenic mice as a source of
fully human antibodies for the treatment of cancer // Cancer and Metastasis
Rev. 1999. Vol. 18. P. 421-425.
218. Colcher D., Pavlinkova G., Beresford G. et al. Pharmacokinetics and
biodistribution of genetically engineered antibodies // Quart. J. Nucl. Med.
1998. Vol. 42. P. 225-241.
219. Muller K.M., Arndt K.M., Pluckthun A. Model and simulation of multiva-
lent binding to fixed ligands // Anal. Biochem. 1998. Vol. 261. P. 149-157.
220. Casey J.L., Pedley R.B., King DJ. et al. Dosimetric evaluation and
radioimmunotherapy of anti-tumour multivalent Fab fragments // Brit. J.
Cancer. 1999. Vol. 81, N 6. P. 972-980.
221. Humphreys D.P., Vetterlein O.M., Chapman A.P. et al. F(ab'J molecules
made from Escherichia coli produced Fab' with hinge sequences confer-
conferring increased serum survival in an animal model // J. Immunol. Meth.
1998. Vol. 217. P. 1-10.
475
222. AtwellJ., Breheney KA., Lawrence LJ. et al. ScFv multimers: Length of
the linker between VH and VL domains dictates precisely the transition
between diabodies and triabodies // Protein Eng. 1999. Vol. 12.
P. 597-604.
223. Le Gall F., Kipriyanov S.M., Moldenhauer G., Little M. Di-, tri- and
tetrameric single chain Fv antibody fragments against human CD 19: Effect
of valency on binding // FEBS Lett. 1999. Vol. 453. P. 164-168.
224. Bauer S., Renner C, Juwana J.P. et al. Immunotherapy of human tumors
with T-cell activating bispecific antibodies: Stimulation of cytotoxic path-
pathways in vivo II Cancer Res. 1999. Vol. 59. P. 1961-1965.
225. BartlettJS., KleinschmidtJ., Boucher R.C., Samulski RJ. Targeted adeno-
associated virus vector transduction of nonpermissive cells mediated by a
bispecific F(ab'gammaJ antibody // Nature Biotechnol. 1999. Vol. 17.
P. 181-186.
226. Kraeber-Bodere F., Faibre-Chauvet A., Sai-Maurel C. et al. Bispecific
antibody and bivalent hapten radioimmunotherapy in CEA-producing
medullary thyroid cancer xenograft // J. Nucl. Med. 1999. Vol. 40.
P. 198-204.
227. Glennie MJ., McBride H.M., Worth A.T., Stevenson G.T. Preparation and
performance of bispecific F(ab)'2 antibody containing thioetherlinked
Fab' gamma fragments //J. Immunol. 1987. Vol. 139. P. 2367-2375.
228. Milstein C, Cuello A.C. Hybrid hybridomas and their use in immunohis-
tochemistry // Nature. 1983. Vol. 305. P. 537-540.
229. Pack P., Pluckthun A. Miniantibodies: Use of amphipathic helices to pro-
produce functional, flexibly linked dimeric Fv fragments with high avidity in
Escherichia coli II Biochemistry. 1992. Vol. 31. P. 1579-1584.
230. De Jonge J., Heirman C, De Veerman M. et al. In vivo retargeting of T cell
effector function by recombinant bispecific single chain Fv (anti-
CD3 x antiidiotype) induces long-term survival in the murine BCL1 lym-
phoma model//J. Immunol. 1998. Vol. 161. P. 1454-1461.
231. Deev S.M., Waibel R., Lebedenko E.N. et al. Design of multivalent com-
complexes using the barnase. barstar module // Nature Biotechnol. 2003.
Vol. 21. P. 1486-1492.
232. Noel PJ., Boise L.H., Thompson C.B. Regulation of T cell activation by
CD28 and CTLA4 // Adv. Exp. Med. Biol. 1996. Vol. 406. P. 209-
217.
233. Deo Y.M., Sundarapandiyan K., Kelleher K. et al. Bispecific molecules
directed to the Fc receptor for IgA (FcaRI, CD89) and tumor antigens effi-
efficiently promote cell-mediated cytotoxicity of tumor targets in whole
blood//J. Immunol. 1998. Vol. 160. P. 1677-1686.
234. Van Spriel A.B., Van Den Herik-Oudijk I.E., Van Sorge N.M. et al.
Effective phagocytosis and killing of Candida albicans via targeting FCyRI
(CD64) or FcaRI (CD89) on neutrophils // J. Infect. Diseas. 1999.
Vol. 179. P. 661-669.
235. Segal DM., Vance B.A., Sconocchia G. Bispecific antibodies // Antibodies
in diagnosis and therapy. Technologies, mechanisms and clinical data / Ed.
S. Matzku, R.A. Stahel. Amsterdam: Harwood, 1999. P. 49-80.
476
236. Cesano A., Visonneau S., Deaglio S. et al. Role of CD38 and its ligand in
the regulation of MHC-nonrestricted cytotoxic T cells // J. Immunol. 1998.
Vol. 160. P. 1106-1115.
237. Lanier L.L. NK cell receptors // Annu. Rev. Immunol. 1998. Vol. 16.
P. 359-393.
238. Yokoyama W.M. Natural killer cell receptors // Curr. Opin. Immunol. 1998.
Vol. 10. P. 298-305.
239. Marasco W.A. Intracellular antibodies (intrabodies) as research reagents
and therapeutic molecules for gene therapy // Immunotechnology. 1995.
Vol. 1. P. 1-19.
240. Conrad U., Manteuffel R. Immunomodulation of phytohormones and
functional proteins in plant cells // Trends Plant Sci. 2001. Vol. 6.
P. 399-402.
241. Conrad U., Fiedler U. Compartmentspecific accumulation of recombinant
immunoglobulins in plant cells: An essential tool for antibody production
and immunomodulation of physiological functions and pathogen activity //
Plant Mol. Biol. 1998. Vol. 38. P. 101-109.
242. Steinberger P., Andris-Widhopf J., Buhler B. et al. Functional deletion of
the CCR5 receptor by intracellular immunization produces cells that are
refractory to CCR5-dependent HIV-1 infection and cell fusion //Proc. Nat.
Acad. Sci. USA. 2000. Vol. 97. P. 805-810.
243. LecerfJ.-M., Shirley T.L., Zhu Q. et al. Human single-chain Fv intrabodies
counteract in situ huntingtin aggregation in cellular models of
Huntington's disease // Ibid. 2001. Vol. 98. P. 4764-4769.
244. Ward E.S., Gussow D., Griffiths A.D. et al. Binding activities of a reper-
repertoire of single immunoglobulin variable domains secreted from
Escherichia coli II Nature. 1989. Vol. 341. P. 544-546.
245. Holt L.J., Herring C, Jespers L.S., Woolven B.P. Domain antibodies:
Proteins for therapy // Trends Biotechnol. 2003. Vol. 21. P. 484-490.
246. Clackson Т., Hoogenboom H.R., Griffiths A.D., Winter G. Making anti-
antibody fragments using phage display libraries // Nature. 1991. Vol. 352.
P. 624-628.
247. Burton D.R., Barbas C, Persson M.A. et al. A large array of human mon-
monoclonal antibodies to type 1 human immunodeficiency virus from combi-
combinatorial libraries of asymptomatic seropositive individuals // Proc. Nat.
Acad. Sci. USA. 1991. Vol. 88. P. 10134-10137.
248. Marks J.D., Hoogenboom H.R., Bonnert T.P. et al. By-passing immuniza-
immunization: Human antibodies from V-gene libraries displayed on phage // J. Mol.
Biol. 1991. Vol. 222. P. 581-597.
249. Ohlin M., Borrebaeck C.A. Characteristics of human antibody repertoires
following active immune responses in vivo II Mol. Immunol. 1996.
Vol. 33. P. 583-592.
250. Pereira S., Van B.P., Elder D. et al. Combinatorial antibodies against
human malignant melanoma // Hybridoma. 1997. Vol. 16. P. 11-16.
251. Roben P., Barbas S.M., Sandoval L. et al. Repertoire cloning of lupus
anti-DNA autoantibodies // J. Clin. Invest. 1996. Vol. 98. P. 2827-
2837.
477
252. Vaughan T.J., Williams A.J., Pritchard K. et al. Human antibodies with
sub-nanomolar affinities isolated from a large non-immunized phage dis-
display library // Nature Biotechnol. 1996. Vol. 14. P. 309-314.
253. Hoogenboom H.R., Winter G. By-passing immunization: Human antibod-
antibodies from synthetic repertoires of germline VH gene segments rearranged in
vitro IIJ. Mol. Biol. 1992. Vol. 227. P. 381-388.
254. Nissim A., Hoogenboom H.R., Tomlinson 1.М. et al. Antibody fragments
from a 'single pot' phage display library as immunochemical reagents //
EMBO J. 1994. Vol. 13. P. 692-698.
255. De KruifJ., Boel E., Logtenberg T. Selection and application of human sin-
single chain Fv antibody fragments from a semisynthetic phage antibody dis-
display library with designed CDR3 regions // J. Mol. Biol. 1995. Vol. 248.
P. 97-105.
256. De KruifJ., Terstappen L., Boel E., Logtenberg T. Rapid selection of cell
subpopulation-specific human monoclonal antibodies from a synthetic
phage antibody library // Proc. Nat. Acad. Sci. USA. 1995. Vol. 92.
P. 3938-3942.
257. Griffiths A.D., Williams S.C., Hartley O. et al. Isolation of high affinity
human antibodies directly from large synthetic repertoires // EMBO J.
1994. Vol. 13. P. 3245-3260.
258. KnappikA., Ge L., Hanegger A. et al. Fully synthetic human combinatori-
combinatorial antibody libraries (HuCAL) based on modular consensus frameworks
and CDRs randomized with trinucleotides // J. Mol. Biol. 2000. Vol. 296.
P. 57-86.
259. Desiderio A., Franconi R., Lopez M. et al. A semi-synthetic repertoire of
intrinsically stable antibody fragments derived from a single-framework
scaffold // Ibid. 2001. Vol. 310, N 3. P. 603-615.
260. SchierR., McCall A., Adams G.P. et al. Isolation of picomolar affinity anti-
c-erbB-2 single-chain Fv by molecular evolution of the complementarity'
determining regions in the center of the antibody binding site // Ibid. 1996.
Vol. 263. P. 551-567.
261. Yang W.P., Green K., Pinz S.S. et al. CDR walking mutagenesis for the
affinity maturation of a potent human anti-HIV-1 antibody into the pico-
picomolar range // Ibid. 1995. Vol. 254. P. 392-403.
262. Low N.M., Holliger P.H., Winter G. Mimicking somatic hypermutation:
Affinity maturation of antibodies displayed on bacteriophage using a bac-
bacterial mutator strain // Ibid. 1996. Vol. 260. P. 359-368.
263. Huie M.A., Cheung M.C., Muench И.О. et al. Antibodies to human fetal
erythroid cells from nonimmune phage antibody library // Proc. Nat. Acad.
Sci. USA. 2001. Vol. 98, N 5. P. 2682-2687.
264. Vaughan TJ., Williams A.J., Pritchard K. et al. Human antibodies with
sub-nanomolar affinities isolated from a large non-immunized phage dis-
display library // Nature Biotechnol. 1996. Vol. 14, N 3. P. 309-314.
265. Sblattero D., Bradbury A. Exploiting recombination in single bacteria to
make large phage antibody libraries // Ibid. 2000. Vol. 18. P. 75-80.
266. De Haard H.J., Van Neer N., Reurs A. et al. A large non-immunized
human Fab fragment phage library that permits rapid isolation and kinetic
478
analysis of high affinity antibodies //J. Biol. Chem. 1999. Vol. 274, N 26.
P. 18218-18230.
267. Sheets M.D., Amersdorfer P., Finnern R. et al. Efficient construction of a
large nonimmune phage antibody library; the production of panels of high
affinity human single-chain antibodies to protein antigens // Proc. Nat.
Acad. Sci. USA. 1998. Vol. 95, N 2. P. 6157-6162.
268. Sblattero D., Lou J., Marzari R., Bradbury A. In vivo recombination as a
tool to generate molecular diversity in phage antibody libraries // J.
Biotechnol. 2001. Vol. 74, N 4. P. 303-315.
269. De Wildt R.M., Mundy C.R., Gorick B.D., Tomlinson l.M. Antibody arrays
for high-throughput screening of antibody-antigen interactions // Nature
Biotechnol. 2000. Vol. 18, N 9. P. 989-994.
270. Griffiths A.D., Duncan A.R. Strategies for selection of antibodies by phage
display //Curr. Opin. Biotechnol. 1998. Vol. 9. P. 102-108.
271. Bradbury A., Velappan N., Verzillo V. et al. Antibodies in proteomics 1:
Generating antibodies // Trends Biotechnol. 2003. Vol. 21. P. 275-281.
272. Bradbury A., Velappan N., Verzillo V. et al. Antibodies in proteomics II:
Screening, high-throughput characterization and downstream
applications // Ibid. P. 312-317.
273. Tramontano A., Janda K.D., Lerner R.A. Catalytic antibodies // Science.
1986. Vol. 234. P. 1566-1570.
274. Pollack SJ., Jacobs J.W., Schultz P.G. Selective chemical catalysis by an
antibody // Ibid. P. 1570-1573.
275. Wentworth P. Antibody design by man and nature // Ibid. 2002. Vol. 296.
P. 2247-2249.
276. Wentworth P., Janda K.D. Catalytic antibodies // Curr. Opin. Chem. Biol.
1998. Vol. 2, N1. P. 138-144.
277. Janda K.D., Shevlin C.G., Lerner R.A. Antibody catalysis of a disfa-
disfavored chemical transformation // Science. 1993. Vol. 259, N 5094.
P. 490-493.
278. Wentworth P., Jr., Datta A., Smith S. et al. Antibody catalysis of BAc2 aryl
carbamate ester hydrolysis: A highly disfavored chemical process // J.
Amer. Chem. Soc. 1997. Vol. 119, N 9. P. 2315-2316.
279. Wentworth P., Datta A., Blakey D. et al. Toward antibody-directed
"abzyme" prodrug therapy, ADAPT: Carbamate prodrug activation by a
catalytic antibody and its in vitro application to human tumor cell killing //
Proc. Nat. Acad. Sci. USA. 1996. Vol. 93, N 2. P. 799-803.
280. Shabat D., Lode H.N., Peril U. et al. In vivo activity in a catalytic antibody-
prodrug system: Antibody catalyzed etoposide prodrug activation for
selective chemotherapy // Ibid. 2001. Vol. 98. P. 7528-7533.
281. Li Т., Janda K.D., Ashley J.A., Lerner R.A. Antibody catalyzed cationic
cyclization // Science. 1994. Vol. 264, N 5163. P. 1289-1293.
282. Toker J.D., Wentworth P., Jr., Ни Y. et al. Antibody-catalysis of a bimol-
ecular asymmetric 1,3-dipolar cycloaddition reaction // J. Amer. Chem.
Soc. 2000. Vol. 122, N 13. P. 3244-3245.
283. Jones L.H., Harwig C.W., Wentworth P., Jr. et al. Conversion of enediynes
into quinones by antibody catalysis and in aqueous buffers: Implications
479
for an alternative enediyne therapeutic mechanism // Ibid. 2001. Vol. 123,
N 15. P. 3607-3608.
284. Vayron P., Renard P.-Y., Frederic T. et al. Toward antibody-catalyzed
hydrolysis of organophosphorus poisons // Proc. Nat. Acad. Sci. USA.
2000. Vol. 97. P. 7058-7063.
285. Wentworth P., Jr., Liu Y., Wentworth A.D. et al. A bait and switch hapten
strategy generates catalytic antibodies for phosphodiester hydrolysis //
Ibid. 1998. Vol. 95, N 11. P. 5971-5975.
286. Wirsching P., Ashley JA., Lo C.H. et al. Reactive immunization // Science.
1995. Vol. 270, N 5243. P. 1775-1782.
287. Lo C.-H.L., Wentworth P., Jr., Jung K.W. et al. Reactive immunization
strategy generates antibodies with high catalytic proficiencies // J. Amer.
Chem. Soc. 1997. Vol. 119, N 42. P. 10251-10252.
288. Takahashi N., Kakinumal H., Liu L. et al. In vitro abzyme evolution to
optimize antibody recognition for catalysis // Nature Biotechnol. 2001.
Vol. 19. P. 563-567.
289. BanerJ., Nilsson M., Mendel-Hartvig M., Landegren U. Signal amplifica-
amplification of padlock probes by rolling circle replication // Nucl. Acids Res.
1998. Vol. 26. P. 5073-5078.
290. Wentworth A.D., Jones L.H., Wentworth P., Jr. et al. Antibodies have the
intrinsic capacity to destroy antigens // Proc. Nat. Acad. Sci. USA. 2000.
Vol. 97, N 20. P. 10930-10935.
291. Wentworth P., Jr., Jones L.H., Wentworth A.D. et al. Antibody catalysis
of the oxidation of water // Science. 2001. Vol. 293, N 5536.
P. 1806-1811.
292. Shuster AM., Gololobov G.V., Kvashuk O.A. et al. DNA hydrolyzing
autoantibodies // Ibid. 1992. Vol. 256. P. 665-667.
293. Nevinsky G.A., Buneva V.N. Human catalytic RNA- and DNA-hydrolyzing
antibodies // J. Immunol. Meth. 2002. Vol. 269. P. 235-249.
294. Gololobov G.V., Chernova E.A., Schourov D.V. et al. Cleavage of super-
coiled plasmid DNA by autoantibody Fab fragment: Application of the
flow linear dichroism technique // Proc. Nat. Acad. Sci. USA. 1995.
Vol. 92, N1. P. 254-257.
295. Gololobov G.V., Rumbley C.A., Rumbley /JV. et al. DNA hydrolysis by
monoclonal anti-ssDNA autoantibody BV 04-01: Origins of catalytic
activity // Mol. Immunol. 1997. Vol. 34, N 5. P. 1083-1093.
296. Paul S., Voile DJ., Beach CM. et al. Catalytic hydrolysis of vasoactive
intestinal peptide by human autoantibody // Science. 1989. Vol. 244,
N4909. P. 1158-1162.
297. Bayry J., Lacroix-Desmazes S., Pashov A. et al. Autoantibodies to factor
VIII with catalytic activity // Autoimmun Rev. 2003. Vol. 2. P. 30-35.
298. Colas P., Cohen В., Jessen T. et al. Genetic selection of peptide aptamers
that recognize and inhibit cyclin dependent kinase 2 // Nature. 1996.
Vol. 380. P. 548-550.
299. Abedi M.R., Caponigro G., Kamb A. Green fluorescent protein as a scaf-
scaffold for intracellular presentation of peptides // Nucl. Acids Res. 1998.
Vol. 26. P. 623-630.
480
300. Norman T.C., Smith D.L., Sorger P.K. et al. Genetic selection of peptide
inhibitors of biological pathways // Science. 1999. Vol. 285. P. 591-595.
301. Cohen B.A., Colas P., Brent R. An artificial cell-cycle inhibitor isolated
from a,combinatorial library // Proc. Nat. Acad. Sci. USA. 1998. Vol. 95,
N 24. P. 14272-14277.
302. Colas P., Cohen В., Ferrigno P.K. et al. Targeted modification and trans-
transportation of cellular proteins // Ibid. 2000. Vol. 97. P. 13720-13725.
303. Buerger C, Nagel-Wolfrum K., Kunz C. et al. Sequence-specific peptide
aptamers, interacting with the intracellular domain of the epidermal growth
factor receptor, interfere with Stat3 activation and inhibit the growth of
tumor cells //J. Biol. Chem. 2003. Vol. 278. P. 37610-37621.
304. Blum J.H., Dove S.L., Hochschild A., Mekalanos JJ. Isolation of peptide
aptamers that inhibit intracellular processes // Proc. Nat. Acad. Sci. USA.
2000. Vol. 97. P. 2241-2246.
305. Butz K., Denk C, Ullmann A. et al. Induction of apoptosis in human papil-
lomavirus-positive cancer cells by peptide aptamers targeting the viral E6
oncoprotein // Ibid. P. 6693-6697.
306. Rudolph C, Schreier P.H., Uhrig J.F. Peptide-mediated broad-spectrum
plant resistance to tospoviruses // Ibid. 2003. Vol. 100. P. 4429-4434.
307. Webb E.C. Enzyme nomenclature. San Diego: Acad. press, 1992.
308. Morrison T.B., Weis JJ., Wittwer C.T. Quantification of low-copy tran-
transcripts by continuous SYBR Green I monitoring during amplification.
Biotechniques // 1998. Vol. 24. P. 954-962.
309. Bornscheuer U.T. Evolutionary generation of enzymes with novel sub-
substrate specificities // Directed evolution of proteins, or how to improve
enzymes for biocatalysis / Ed. S. Brakmann, K. Johnsson. N.Y.: Wiley,
2002. P. 329-341.
310. Bornscheuer U.T., Altenbuchner J., Meyer H.H. Directed evolution of an
esterase for the stereoselective resolution of a key intermediate in the syn-
synthesis of epothilones //Biotechnol. Bioeng. 1998. Vol. 58. P. 554-559.
311. Altamirano MM., Blackburn J.M., Aguayo C, Fersht A.R. Directed evo-
evolution of new catalytic activity using the alpha/beta-barrel scaffold //
Nature. 2000. Vol. 403, N 6770. P. 617-622.
312. Zhang J.H., Dawes G., Stemmer W.P. Directed evolution of a fucosidase
from a galactosidase by DNA shuffling and screening // Proc. Nat. Acad.
Sci. USA. 1997. Vol. 94, N 9. P. 4504-4509.
313. Sun L., Baiter Т., Alcade M. et al. Modification of galactose oxidase to
introduce glucose-6-oxidase activity // Chem. Biochem. 2002. Vol. 3.
P. 781-783.
314. Li Q.-S., Schwaneberg U., Fischer P., SchmidRD. Directed evolution of
the fatty acid hydroxylase P-450 BM3 into the indole-hydroxylating cata-
catalyst // Chemistry. 2000. Vol. 6, N 9. P. 1531-1536.
315. Joo #., Lin Z., Arnold F.H. Laboratory evolution of peroxide-mediated
cytochrome P450 hydroxylation // Nature. 1999. Vol. 399, N 6737.
P. 670-673.
316. Kauffmann I., Schmidt-Dannert C. Conversion of Bacillus thermocatenu-
latus lipase into an efficient phospholipase with increased activity towards
481
long-chain fatty acyl substrates by directed evolution and rational design //
Protein Eng. 2001. Vol. 14, N 11. P. 919-928.
317. Fong S., Machajewski T.D., Мак С.С., Wong C. Directed evolution of D-
2-keto-3-deoxy-6-phosphogluconate aldolase to new variants for the effi-
efficient synthesis of D- and L-sugars // Chem. Biol. 2000. Vol. 7, N 11.
P. 873-883.
318. Haring D., Schreier P. Chemical engineering of enzymes: Altered catalyt-
catalytic activity, predictable selectivity and exceptional stability of the semisyn-
thetic peroxidase seleno-subtilisin // Naturwissenschaften. 1999. Vol. 86.
P. 307-312.
319. Lanio Т., Jeltsch A., Pingoud A. Evolutionary generation versus rational
design of restriction endonucleases with novel specificity // Directed evo-
evolution of proteins, or how to improve enzymes for biocatalysis / Ed.
S. Brakmann, K. Johnsson. N.Y.: Wiley, 2002. P. 309-328.
320. Jeltsch A., Wenz C, Wende W. et al. Engineering novel restriction endonu-
endonucleases: Principles and applications // Trends Biotechnol. 1996. Vol. 14,
N 7. P. 235-238.
321. Jeltsch A., Alves J., Oelgeschlager T. et al. Mutational analysis of the func-
function of Glnll5 in the EcoRI restriction endonuclease, a critical amino acid
for recognition of the inner thymidine residue in the sequence -GAATTC-
and for coupling specific DNA binding to catalysis // J. Mol. Biol. 1993.
Vol. 229, N1. P. 221-234.
322. Wenz C, Selent U., Wende W. et al. Protein engineering of the restric-
restriction endonuclease EcoRV: Replacement of an amino acid residue in the
DNA binding site leads to an altered selectivity towards unmodified and
modified substrates // Biochim. biophys. acta. 1994. Vol. 1219, N 1.
P. 73-80.
323. Lanio Т., Selent U., Wenz C. et al. EcoRV-T94V: A mutant restriction
endonuclease with an altered substrate specificity towards modified^
oligodeoxynucleotides//Protein Eng. 1996. Vol. 9, N 11. P. 1005-1010.
324. Smith J., Bibikova M., Whitby F.G. et al. Requirements for double-strand
cleavage by chimeric restriction enzymes with zinc finger DNA-recogni-
tion domains // Nucl. Acids Res. 2000. Vol. 28, N 17. P. 3361-3369.
325. Kim Y.G., Chandrasegaran S. Chimeric restriction endonuclease // Proc.
Nat. Acad. Sci. USA. 1994. Vol. 91, N 3. P. 883-887.
326. Kim Y.G., Smith J., Durgesha M., Chandrasegaran S. Chimeric restriction
enzyme: Gal4 fusion to Fokl cleavage domain // Biol. Chem. 1998.
Vol. 379, N 4/5. P. 489-495.
327. Reetz M.T., Jaeger K.-E. Directed evolution as a means to create enantios-
elective enzymes for use in organic chemistry // Directed evolution of pro-
proteins, or how to improve enzymes for biocatalysis / Ed. S. Brakmann,
KJohnsson. N.Y.: Willey. 2002. P. 245-279.
328. Swali V., Langley J., Bradley M. Mass spectrometric analysis in combina-
combinatorial chemistry // Curr. Opin. Chem. Biol. 1999. Vol. 3. P. 337-341.
329. Alexeeva M. Enright A., Daw son MJ. et al. Deracemisation of oc-methyl-
benzylamine using an enzyme obtained by in vitro evolution // Angew.
Chem. Intern. Ed. 2002. Vol. 41. P. 3177-3180.
482
330. Burton S.G., Cowan D.A., Woodley J.M. The search for the ideal biocata-
lyst // Nature Biotechnol. 2002. Vol. 20. P. 37-45.
331. May O., Nguyen P.Т., Arnold F.H. Inverting enantioselectivity and
increasing total activity of a key enzyme in a multi-enzyme synthesis cre-
creates a viable process for production of L-methionine // Ibid. 2000. Vol. 18.
P. 317-320.
332. Wangikar P.P., Michels P.C., Clark D.S., DordickJS. Structure and func-
function of subtilisin BPN solubilized in organic solvents // J. Amer. Chem.
Soc. 1997. Vol. 119. P. 70-76.
333. Reetz M.T., Zonta A., Schimossek K. et al. Creation of enantioselective bio-
catalysts for organic chemistry by in vitro evolution // Angew. Chem.
Intern. Ed. 1997. Vol. 36. P. 2830-2833.
334. Sutherland J.D. Evolutionary optimization of enzymes // Curr. Opin.
Chem. Biol. 2000. Vol. 4. P. 263-269.
335. Broun P., Shanklin J., Whittle E., Somerville C. Catalytic plasticity of fatty
acid modification enzymes underlying chemical diversity of plant lipids //
Science. 1998. Vol. 282. P. 1315-1317.
336. Kumamaru Т., Suenaga H., Watanabe Т., Furukawa K. Enhanced degra-
degradation of polychlorinated biphenyls by directed evolution of biphenyl
dioxygenase//Nature Biotechnol. 1998. Vol. 16. P. 663-666.
337. Giver L., GershensonA., FreskardP.O., Arnold F.H. Directed evolution of
a thermostable esterase // Proc. Nat. Acad. Sci. USA. 1998. Vol. 95.
P. 12809-12813.
338. Jung H.C., LebeaultJM., Pan J.G. Surface display of Zymomonas mobilis
levansucrase by using ice-nucleation protein of Pseudomonas syringae I I
Nature Biotechnol. 1998. Vol. 16. P. 576-580.
339. Fernandez-Lafuente R., Cowan D.A., WoodA.N.P. Hyperstabilization of a
thermophilic esterase by multipoint covalent attachment // Enzymol.
Microb. Technol. 1995. Vol. 17. P. 366-372.
340. Kazan D., Ertan H., Eraslan A. Stabilization of Escherichia coli penicillin
G acylase against thermal inactivation by cross-linking with dextran
dialdehyde polymers // Appl. Microbiol. Biotechnol. 1997. Vol. 48.
P. 191-197.
341. Haring D., Schreier P. Novel biocatalysts by chemical modification of
known enzymes: Cross-linked microcrystals of the semi-synthetic peroxi-
dase seleno-subtilisin // Agnew. Chem. Intern. Ed. 1998. Vol. 37.
P. 2471-2473.
342. Moore J.C., Arnold F.H. Directed evolution of a para-nitrobenzyl esterase
for aqueous-organic solvents // Nature Biotechnol. 1996. Vol. 14.
P. 458-462.
343. Daniel R.M., Cowan D.A. Biomolecular stability and life at high tempera-
temperatures // Cell. Mol. Life Sci. 2000. Vol. 57. P. 250-264.
344. Blanco R.M., Guisan J.M. Stabilization of enzymes by multipoint covalent
attachment to agarose aldehyde gels - borohydride reduction of trypsin
agarose derivatives // Enzymol. Microbiol. Technol. 1989. Vol. 11.
P. 360-366.
Предметный указатель
MUG. См. Метилумбеллиферил-
р-глюкуроновая кислота
ONPG, 360
S-Аденозилметионин, 50, 51
Talon, 127
Xgal, 73
X-gluc, 136, 360
X-oc-gal, 360
YAC. См. Искусственные хромо-
хромосомы дрожжей
Аберрации хромосомные, 277
Абзимы, 282, 403, 409, 427-433,
448
определение термина, 427
природные, 431
рациональный дизайн, 430
схема получения, 429
ферментативные активности,
429
Авидин, 221
Авидность антител, 407
Авитаминоз, 136
Авторадиография, 42, 163
Адаптеры, 59, 159, 233
Аденилилтрансфераза, 137
Аденовирусы, 150
Аденозиндезаминаза, амплифи-
амплификация гена, 175
Азасерин, 404
Алкогольоксидаза, промотор ге-
гена дрожжей, 173
Аллополиплоидия, 277
Аллостерические ферменты,
конструирование, 380
Альдолаза, 441
Альцгеймера болезнь, 420
Амидирование, 36
484
Амилоза, 126, 132
Аминоацил-тРНК-синтетазы,
188
ошибки аминоацилирования,
303
специфичность в отношении
аналогов аминокислот, 304
Аминогликозид-З'-фосфотранс-
фераза, 95, 137
Аминооксидаза, 445
Аминоптерин, 404
Ампликон, 220,221,225,228,230,
231,238,239
Амплификация
генов в клетках животных, 176
нуклеиновых кислот, изотер-
изотермическая
с замещением цепи ДНК,
243
с помощью Qp-репликазы,
248
самоподдерживающаяся
репликация последователь-
последовательностей, 243
по типу катящегося кольца,
218
разветвленная, 218
Анеуплоидия, 277
Анилингидроксилаза, 448
Антикодон, 33
Антимышиных антител челове-
человека синдром, 410
Антирепрессор, 87
Антирецептор, 395
Антисмысловые
олигонуклеотиды, 434
РНК, 9, 17, 28
регуляция репликации
плазмид, 70
синтез, 76
Антитела
антиидиотипические, 432
аффинное созревание, 421,
423, 424, 430
in vitro, 425
определение термина, 425
внутриклеточные, 418
двойной специфичности, одно-
цепочечные, 416
димерные, 416
доменные, 421
каталитические. См. Абзимы
клонотеки фрагментов, 422
моноклональные
авидность, 407
аффинность, 406
в растениях, 421
снижение иммуногенности,
410
специфичность, 422
Аптамеры
олигонуклеотидные, 210
пептидные, 348, 433-435
практическое применение,
434
Арабидопсис, первичная струк-
структура генома, 16
Аргинин, 89
в качестве аффинной метки,
127
Аргининосукцинатлиаза, 448
Аренавирусы, 20
Арилсульфатаза, 138
Археи (архебактерии), 16,18,436
ДНК-лигазы, 61
систематика, 22
структура генома, 23
экстремофилы, 397, 399
Асцитная жидкость, 405, 406
Ауксин, 139
Аутоиндукторы, 145
Аутополиплоидия, 277
Аутосплайсинг белковый, 131
Афидиколин, 176
Аффинность антител, 406
Аффинные пептидные метки,
124-132
белок, связывающий мальто-
мальтозу, 132
глутатион-Б-трансфераза, 131
двойные, 132
домены, связывающие целлю-
целлюлозу, 130
кальмодулин-связывающий
пептид, 130
на основе с-тус, 129
полигистидин, 127
система Flag, 128
система S-Tag, 129
система Strept-tag, 128
стрептавидин-связывающий
пептид, 131
хитин-связывающий домен,
131
Ацетолактатсинтаза, 137
Ацетонитрил, 307
Байера мостики, 148
Бактерии
Agrobacterium rhizogenes, 138
Agrobacterium tumefaciens, 138,
139
Arthrobacter sp., 457
Aspergillus niger, 445
Bacillus amyloliquefaciens, 416
Bacillus circulans, 131
Bacillus megatehum, 441
Bacillus stearothermophilus, 400,
455, 459
Bacillus subtilis, 22, 144, 350,
380, 402
Bacillus TA4\, 402
Burkholderia cepacia, 459
Carboxydothermus hydrogeno-
formans, 211
Chlamydia trachomatis, 248
Clostridium thermocellum, 377
Epicurian coli, 439
Haemophilus influenzae, 22, 144
Methanopyrus kandleri, 399
Micrococcus luteus, 303
485
Neisseria, 143, 248
Nostoc punctiforme, 18
Pseudomonas aeruginosa, 393,
457
экспрессия рекомбинант-
ных генов, 169
Pseudomonas fluorescens, 457
Pseudomonas pseudoalcaligenes,
459
Pseudomonas putida, 441
Pseudomonas syringae, 459
Pyrococcus furiosus, 200
Pyrococcus species GB-D, 200
Staphylococcus aureus, 441
Staphylococcus sp., 351
Streptococcus pneumoniae, 144
Streptomyces avidinii, 221
Sulfolobus solfataricus, 398
Thermococcus litoralis, 200
Thermotoga maritima, 200
Thermus aquaticus, 1
Thermusflavus, 211
Thermus thermophilus, 199, 211,
402
Zymomonas mobilis, 459
агробактерии, 141
Бактериофаги
фХ174
расщепление оцДНК рест-
риктазами, 56
репликативная форма
ДНК, 54
СЕ6, система индукции реком-
бинантных генов, ПО
П
межгенная область IG,
76
фаговый дисплей, 334, 339,
426
fd, фаговый дисплей, 334
М13
фаговый дисплей, 334
фаг-помощник, 76
MS2, белок оболочки, 368
Ми, интеграция хромосомы в
ДНК, 79
Р1
486
ряс-сайт, 95
биология, 95
репликоны, 95
упаковка ДНК в фаговые
частицы, 95
эндонуклеаза Сге и loxP-
сайт, 94, 95
Qp, направленная эволюция
генома, 319
SPp, конструирование векто-
векторов, 101
SP6, РНК-полимераза, ПО
Т4
модификации ДНК, 218
фаговый дисплей, 336
Т7, сайты рестрикции хромо-
хромосомы, 51
X
1059, строение хромосомы,
83
cos-сайты, 83, 93, 95
дефектные, 82
емкость головки фага, 85
лизогения, 82, 95
литический цикл развития,
80
промотор PL, свойства,
107
профаги, 82
температурочувствитель-
ный репрессор с1857, 109
упаковка ДНК в фаговые
частицы, 82, 95, 96
фаговый дисплей, 336
химерные фаговые части-
частицы, 85
морфогенез, 121
нитевидные
выход из клетки, 335
фаги-помощники, 336
фагмиды, 76
Бакуловирусы,134,175, 181
Барназа, 416
Барстар, 416
Белки
GFP-подобные
dsRed, 389, 390
зажигающиеся флуоресци-
флуоресцирующие, 392
нефлуоресцирующие хро-
мопротеины (СР), 389
флуоресцентный хроно-
хронометр, 389
рН-память, 373
денатурация, 460
иммобилизация, 373
негистоновые, 27, 28
рибосомные
S14, 137
расположение генов, 22
связывающие мальтозу,
очистка рекомбинантных
белков, 379
стабильность, 35
теплового шока, 121, 447
химико-ферментативный син-
синтез, 306
экспорт из цитоплазмы, 121
Белоксинтезирующие системы
бесклеточные
прокариотические, 186-189
проточные, 190-191
эукариотические, 189-190
Бензотиазол, 384
Библиотеки. См. Клонотеки
Биоадсорбенты, 350
Биоинформатика, 6, 252
Биокатализаторы пластиковые,
373
Биолюминесценция, 383, 385
Биореакторы
мембранные, 406
трансгенные организмы, 5
Биосенсоры на основе
аллостерических ферментов,
381
белков, 369
клеток, 350, 383, 392
молекулярного импринтинга,
374
моноклональных антител, 408
флуоресцирующих белков, 392
Биотин, 126, 128, 214, 221, 235,
242, 342
Биочипы, 6, 9
Бисульфит ион
механизм действия, 324
превращение С в U, 220
Блеомицин, 137
Блоттинг
Западный, 129, 130
Северный, 214
Бляшки фаговые, 81
Бромфеноловый синий, 204
Бычий сывороточный альбумин,
429
Вакцины, 136, 344
ДНК, 152
живые, 350
оральные, 351
пептидные, 345
Ванадиевые комплексы рибонук-
леозидов, 41
Векторы, 68-155
YAC. См. Искусственные хро-
хромосомы дрожжей
космиды и фазмиды, 83-85
на основе ДНК фага X, 80-83
Charon, ^gtll и EMBL, 80
общие особенности строения,
68
определение термина, 39, 68
плазмидные, 80
Bluescript, 75
pBR322, 72
pPR-TGATG-1, 113
pUC18 и pUC19, 73, 96, 113,
134
pUCPL6AN, 170
для прямого клонирования
продуктов ПЦР, 77
клонирование без исполь-
использования ДНК-лигазы, 76
сравнительные характеристи-
характеристики, 100
фаговые, 47
Вивисекция, 406
Вирион, 19, 20, 338, 340, 426
Вирусная инфекция, лечение
487
аптамерами, 434
ДНК-вакцинами, 152
Вирусные частицы
отличие от понятия вирус, 20
стабилизация глицерином, 158
Вирусы
SV40, 150, 180
аденоассоциированные, 20,
150
бактериальные. См. Бактерио-
Бактериофаги
в качестве векторов, 142
вакцины, 150
вирулентный путь развития,
19
гепатита
B, 20, 432
C, 211,381
жизненный цикл, 19
иммунодефицита человека
(ВИЧ), 211, 249, 381, 395,
420
латентная инфекция, 19
лейкоза, 98
лейкоза мышей Молони, 64,
211
миелобластоза птиц, 64, 211,
243
мозаики цветной капусты,
промотор, 139
образующие фокусы в селе-
селезенке (SSFV), 180
определение термина, 19
папилломы человека, 434
патогенные, детекция, 390
простого герпеса, 135, 150, 359
разнообразие, 436
регуляторные элементы гено-
генома, 133
энцефаломиокардита, энхан-
сер, 135
ВИЧ. См. Вирусы иммунодефи-
иммунодефицита человека
Внехромосомные генетические
элементы, 15
Водорода перекись, 432
биосинтез антителами, 432
488
как кофактор биолюминес-
биолюминесценции, 384
Вольфрама микрочастицы, 152
Галактозидаза
ос-пептид, 73
в дигибридной системе, 356,
360, 364
ген в плазмидном векторе,
113, 117, 166
гибрид с галактозодегидроге-
назой, 376
изменение субстратной специ-
специфичности, 373
репортер при очистке реком-
бинантных белков, 382
Галактозодегидрогеназа, 376
Гаметы, 14
Гаплоидный
геном, 17, 24, 155, 179
набор хромосом, 14
организм, 14, 91, 216, 356
Гексаметилен-бис-ацетамид как
фактор дифференцировки
клеток, 180
Гексаминкобальта(Ш) хлорид,
148
Гель
агарозно-полиакриламидйый,
41
агарозный, 41, 194, 206, 223,
298
полиакриламидный, 41, 301
Гемин, 180, 190
Гемоглобин, 180
побочные активности, 448
синтез в ретикулоцитах, 189
Гемофилия аутоиммунная, 432
Генетически модифицированные
(GM) растения. См. Трансген-
Трансгенные растения
Геном
архей, 22-23
вирусов, 19-21
как таксономический признак,
16
определение, 16
размеры, 17-19
африканская шпорцевая ля-
лягушка, 17, 18
вирусы, 18
голосеменные растения, 18
дрожжи Saccharomyces cere-
visiae, 18
дрожжи Schizosaccharomy-
cespombe, 18
дрозофила, 18
лилия Lilium longiflorum,
18
микоплазмы, 18
простейшие, 18
птицы, 18
рептилии, 18
рис, 18
сложность, 237
эубактерий, 21-22
эукариот, 23-28
Геномика функциональная, 17,
390
Генотоксичность, 144
Гены
горизонтальный перенос, 22,
23, 37, 144
домашнего хозяйства, 133
общая структура, 14
поздние, ПО
Гербициды, 5, 135, 136, 137, 140,
374
Геронилгеронильные группы.
См. Пренилирование белков
Гетерохроматизация, 27
Гетерохроматин, 27
перицентромерная область, 98
Гетерошизомеры, 55
Гибридизация
in situ, 165, 213
колоний, 160
мРНК с иммобилизованной
кДНК, 252
неродственных геномов, 277
оценка кинетической сложно-
сложности генома, 238
с гетерологичными зондами,
164
Гибридомы, 412, 415, 430
замена технологиями in vitro,
409
замена фаговым дисплеем, 348
культивирование, 405
получение, 404
получение из клеток миело-
мы, 178
проблема иммуногенности ан-
антител, 410
Гигромицин, 135, 136
Гигромицин р-фосфотрансфера-
за, 137
Гидантоиназа, изменение стерео-
специфичности, 446
Гидроксила радикалы, 432
Гидроксилирование Lys и Pro,
36
Гидроксиметилурацил (hm5U),
218
Гидроксиметилцитозин (hm5C),
220
Гидроксимочевина, 176
Гидроксицитозинтрифосфат как
мутаген, 325
Гипоксантин-фосфорибозил-
трансфераза, 405
Гипоксия, стимуляция амплифи-
амплификации генов in vivo, 176
Гираза, 146
Гистидин
в активном центре
абзимов, 431
модифицированного тиоре-
доксина, 283
в качестве аффинной метки,
125, 127, 134, 379
индол-3-глицеролфосфатсин-
таза в белковой инженерии,
439
как селектируемый маркер,
172
фторпроизводные, 310
Гистоноподобные белки HU, Н-
NS и IHF, 21
Гистоны, 27
HI, субтипы, 27
489
посттрансляционные модифи-
модификации, 36
Гликозилирование, 36, 318
5-гидроксиметилцитозина,
220 in trans, 374, 459
белков субтилизином, 308
ошибочное антител, 406
Гликозилфосфатидилинозитол в
заякоривании белков на мемб-
мембранах, 351
Гликопротеины
CD4, 395
gpl20 как антирецептор CD4,
395
кальций-связывающие, 138
Глиоксил-агароза, 459
Глутаминовая кислота
полимеры при болезни Хан-
Хантингтона, 420
циклизация на N-концах,
36
Глутаминсинтетаза, амплифика-
амплификация гена, 175
Глутаредоксин, 121
Глукозаминидаза, 459
Глукозидаза, 459
Глукуронидаза р, 136, 138,
360
Глутатион иммобилизованный,
131
Глутатион-Б-трансфераза, 123
как аффинная метка, 126,
131
очистка рекомбинантных бел-
белков, 379
Глутатионредуктаза, 123
Гольджи аппарат, 171
Гомогенно окрашивающаяся об-
область (хромосомы), 176
Гомосерин, ацетилированые лак-
тоны, 145
Гранулоциты, 418
Гуанидинизотиоцианат, 41
Гуанидинтиоцианат, 129
Гуанидинхлорид, 41, 120, 126,
127, 388
490
Гуанинфосфорибозилтрансфера-
за, 97
Дезаминирование
окислительное 5-метилцито-
зина, 60
цитозина бисульфитом на-
натрия, 325
Дезоксикоформицин, 175
Дендримеры, 153
Депротеинизация нуклеиновых
кислот, 41, 206
Дерацемизация, 445
Дерепрессия, 108, 109
Дестиобиотин, 125, 128
Диастереоспецифичность, 429
Дигидрофолатредуктаза
восстановление активности из
двух фрагментов, 363
как селектируемый маркер,
176
модификации аналогами ами-
аминокислот, 304
Диизопропиловый эфир, 374
Диметилсульфоксид
как фактор дифференцировки
клеток, 180
понижение температуры плав-
плавления ДНК, 200, 212
стабилизация фаговых частиц,
158
трансфекция клеток, 153
трансформация бактерий, 148
Диметилформамид
в полусинтезе белков, 307
неводная среда в фементатив-
ном катализе, 459
Динитрофенола эфиры, 431
Диоксигенин, 214
Диплоидные организмы, 14, 91,
356
Диплоидный геном, 15, 277
Дисплей, 409
клеточный, 174, 349-351
бактериальный, 379
модификация ферментов,
458
эукариотический, 129
мРНК, 355
рибосомный, 351-355
получение пептидных апта-
меров, 434
фаговый, 333-349
используемые бактериофа-
бактериофаги, 334
исследование коротких пеп-
пептидов и эпитопов, 336
методы отбора белков, 346
определение термина, 333
отбор in vivo, 347
отбор рекомбинантных ан-
антител, 425
получение абзимов, 429,
430
Дистрибутивный механизм элон-
элонгации праймеров, 200
Дисульфидизомераза, 121
Диэтилпирокарбонат, 214
ДНК
альфоидная, 97, 99
комплементарная (кДНК)
определение термина, 62
получение, 158
сателлитная, 24
ДНКаза I, 66, 290, 327
ДНК-диагностика, 6
SSCP, 208
гепатит С, 211
инфекционные заболевания,
194, 248
использование рестриктаз, 50
поиск мутаций, 7
поиск перестроек генома, 213
пренатальная, 241
проблема контаминации, 203
тромбофилии, 216
ДНК-зависимые ДНК-полимера-
зы
IЕ. coli, 62
бактериофага Т4, 62, 76
термостабильные. См. Поли-
меразная цепная реакция,
ДНК-зависимые ДНК-по-
ДНК-полимер азы
ДНК-лигазы
АТР-зависимые, 61
NAD+-3aBHCHMbie, 61
бактериофага Т4, 61
два семейства, 61
ликвидация одноцепочечных
разрывов ДНК, 218, 248,
294
получение кольцевых ДНК,
290
присоединение
адаптеров к кДНК, 159
линкеров, 287, 290
ДНК-плазма. См. Нуклеоид
ДНК-топоизомераза, 21
Домены
адресные, 378
активаторов транскрипции,
356
белка gp3 нитевидных бакте-
бактериофагов, 335
белковые, конструирование de
novo, 282
в систематике организмов, 16
в создании гибридных белков,
375
гомологичные, объединение в
белковой инженерии, 378
докериновые, 377
каталитические, 377
когезиновые, 377
лейциновая застежка, 365
связывающие хитин, 131,
379
связывающие целлюлозу, 130,
379
со свойствами специфических
лигандов или рецепторов, 379
цинковые пальцы, изменение
специфичности рестриктазы
Fok I, 380
Доминантная регуляторная об-
область DCR, 179
Дробовика метод, 161
Дрожжи
векторы
плазмидные интегративные
(YIP), 87, 171
491
плазмиды, содержащие
центромеры (YCP), 87,171
реплицирующиеся плазми-
плазмиды (YRP), 87, 171
эписомные плазмиды
(YEP), 87, 172
метилотрофные Pichia pastoris
использование в биотехно-
биотехнологии, 173
пекарские S. pombe
использование в биотехно-
биотехнологии, 171
ПЦР-амплификация гено-
генома, 240
почкующиеся S. cerevisiae
интеины, 379
использование в биотехно-
биотехнологии, 171, 174
плазмиды, 86
фолдинг белков, 351
Дрозофила, первичная структура
генома, 16
ДЭАЭ-декстран
трансфекция, 152, 153
трансформация бактерий, 142
Енолформат, 384
Заместительная терапия, 369
Зеленый флуоресцирующий бе-
белок и его производные, 363,
382-92
Зеоцин,358
Золота микрочастицы, 152
Зонды
вырожденные, 165
гетерологичные, 164
гомологичные, 163
кДНК, 252
конъюгаты фрагментов ДНК
с антителами,221
лигированные, 218
получение из продуктов ПЦР,
194
радиоактивно меченные, 163
рестрикционные фрагменты
ДНК, 54
492
РНК, 93
скрининг клонотек, 251
стабильные изотопы, 317
типа висячий замок, 218
флуоресцентные для белков,
307
химические, модификация
SH-rpynn, 307
Изовалерил-3-аминопропанол,
384
Изоляция репродуктивная, 13
Изомеразы, 121
Изопропил-р-?)-тиогалактопира-
нозид (IPTG), 76, 95, 109,
ПО
Изопропилмалатдегидрогеназа,
повышение термостабильнос-
термостабильности, 402
Изопропиловый эфир, 459
Изошизомеры, 55
Имидазол, 127
Имидазопиразин, 384
Иммунодиагностика, 410
Иммуномодуляция, 419
Иммуноферментный анализ
(ИФА), 220, 342, 390, 407, 408,
426
Импринтинг
генетический, 220
молекулярный, 374, 456
Индол-3-глицеролфосфатсинта-
за, превращение в фосфорибо-
зилантранилатизомеразу, 439
Инженерия
белковая
белковых поверхностей,
275
в природе, 447
глобальные цели, 454
комбинаторная проблема,
281
направленная эволюция мо-
молекул, 274
обратный дизайн, 281
определение термина, 273
основанная на тРНК, 302
рациональный дизайн и ре-
дизайн, 274
генная
методические основы, 40
опасность применения, 6
определение термина, 13
основная задача, 39
перспективы использова-
использования, 5
химическая, биологизация, 6
Инициаторы репликации, 70, 95
Инсулин
биосинтез in vivo, 120
рекомбинантный, 117, 118,
123, 169, 173
Инсулиноподобный фактор рос-
роста
I, рекомбинантный, 122
в составе гибридных токсинов,
395
Интеграза, 79
Интеины, 23, 131, 312, 316, 379
Интерактом, 365
Интерлейкин
2
как цитокин, 417
рецепторы, 394
6, биосинтез в проточной сис-
системе, 191
8, моноклональные антитела
к, 413
в составе гибридных токсинов
2,394
4,395
6,395
Интерферон, 169
<х2, 173
Y, 122
Интрацистернальные А-частицы
(IAPs), 26
Интроны
в экспрессирующих векторах,
134, 180
влияние на экспрессию реком-
бинантных генов, 106, 155, 251
группы II, 79
использование в ПЦР, 197,212
мобильные, 23, 79
определение термина, 14
у архей, 23
Искусственные хромосомы
Р1, 94-96
РАС pNS582, 95
pCYPAC,96, 151
конструирование НАС, 100
получение геномных кло-
нотек, 96
электропорация, 151
бактерий,92-94
pBAC108L, 92
конструирование геномных
клонотек, 151
получение MAC и НАС, 99
секвенирование генома че-
человека, 161
создание ТАС-векторов,
141
дрожжей YAC, 88-92, 94
ацентрические, 91
дицентрические, 91
нестабильность, 96
получение MAC и НАС, 97,
99
получение клонотек нукле-
отидных последовательнос-
последовательностей, 91
получение трансгенных жи-
животных, 91, 411
трансфекция, 151
животных, 86
человека, 86
Итероны, 70
Кальмодулин, 130
Кальмодулин-связывающий пеп-
пептид, 125, 130, 132
Кальцитонин, 118
Кальция ионы
как вторичный мессенджер,
390
как кофактор биолюминес-
биолюминесценции, 384
определение внутриклеточно-
внутриклеточного содержания, 390
493
получение компетентных кле-
клеток, 144
стабилизация белков молока,
120
трансфекция клеток
дрожжей, 173
животных, 152
растений, 138
цитотоксическое действие,
152
трансформация
бактериальных клеток, 142
Камфора, 441
Канаванин, 89
Канамицин, 95, 96, 136, 137, 139,
156
Канцерогенез, 220
Канцерогены
амплификация генов, 176
образование из проканцероге-
нов, 450
Капсид, 82, 336
Карбоксиметиласпартат кобаль-
кобальта, 127
Кариотип, 15, 23
Каумермицин, 146
Квадромы, 415
Кворума восприятие, 145
кДНК. См. ДНК комплементар-
комплементарная
Киллеры природные, 348, 412,
418
Кислород
в биолюминесценции, 384
как участник ферментатив-
ферментативных реакций,284
синглетный, 432
Кленова фрагмент, 63, 122, 123,
199, 325
Клетки
африканской зеленой мар-
мартышки (COS), 180-181
бактериальные, 21
гибридные, 39
животных как системы экс-
экспрессии генов, 174-183
зародышевой линии, 14
494
мышиной миеломы Sp2/0,
178-179
насекомых, зараженные баку-
ловирусами, 181-183
пермеабилизованные, 184
питающие, 405
половые, 36
селезенки мышей MEL, 179—
180
соматические, 23
хозяева для вирусов, 19
эукариотические, 16
яичников китайских хомячков
(СНО), 176-178
Клонирование
in silico, 252
многоклеточных организмов, 9
молекулярное
без использования ДНК-ли-
газы, 63
определение термина, 39
по частям, 167
стандартный подход, 251
позиционное, 80, 167
Клонотеки, 155-168
Ун-фрагментов антител, 421
геномной ДНК, 43 '
амплификация, 158
особенности хранения, 158
геномные, 96
ДНК окружающей среды,
398
индивидуальных хромосом, 42
кДНК, 64, 158
комбинаторные
белков и пептидов, 349, 351,
409
максимальный размер, 351
нуклеотидных последова-
последовательностей, 351
методы поиска последова-
последовательностей, 162
последовательностей пепти-
пептидов и белков отбор пептид-
пептидных аптамеров, 434
последовательностей фраг-
фрагментов антител, 422
вторичные, 425
иммунные, 423
неиммунные, 423
поиск абзимов, 430
синтетические, 424
репрезентативность, 155,351
расчет числа клонов, 157
скрининг
высокопроизводительный,
445 повторный, 167
случайные, 160
упорядоченные, 161
Клостропаин, 118
Кобальт-ионы в аффинной хро-
хроматографии, 127
Когезиновые домены, 377
Код генетический, 28
вырожденность, 114, 165, 277
Кодоны
избыточные, 303
инициирующие, 112, 113, 166,
295
синонимические, 114, 118, 277
терминации и инициации
трансляции перекрывающие-
перекрывающиеся, 113
частота использования, 114
Коинтеграты, 78
Колентеразин, 384
Колифаги, 95, 248, 334, 336, 339
Колицины, 69, 72, ПО
Компактосомы, 21
Компетентность бактерий
искусственная, 148-149
В. subtilis, 144
определение термина, 143
природная, 143-148
факторы, 144
Конкатемеры ДНК
Р1,93
X, 83, 84
при амплификации, 250
Коннекторы, 65
Консенсус. См. Последователь-
Последовательности консенсусные
Контиги
определение термина, 253
получение с использованием
YAC, 91
Контртранскрипт, 70
Кораллы Anthozoa, 389
Космиды, 47, 83, 85, 90, 100, 141,
156, 157, 167
Краун-эфиры, 370
Крахмал, 136
Кристаллины, 447
Ксенобиоз, 433
определение термина, 450
Ксенобиотики эндогенные, 449
Ксилен,214
Кукуруза, 135
Культура клеток суспензионная,
178
Кэп, 23, 32, 135, 6
Лазеры
аргоновые, 230
в проточной цитометрии, 43
десорбция и ионизация макро-
макромолекул, 166
создание микроотверстий в
клеточной мембране, 143,
151
ЛактамазаC, 118,292,381
Лактатдегидрогеназа
активность кристаллинов, 448
превращение в малатдегидро-
геназу, 382
эпитопы, 407
Лактоза алло-, 109
Ландшафт функциональности,
284
Левансукраза, 459
Лейкоциты
как мишени антител, 418
полиморфноядерные, 432
Лейпептин, 187
Лигазная цепная реакция, 62,
241-242, 249
Лигирование ДНК, механизм ре-
реакции, 60
Лигнин, 139
Лизогения, 82, 95, 101, ПО
495
Лизоцим
фаговый, инактивация, 82
эпитопы, 421
Линкеры, 59, 289
олигонуклеотидные, 287, 290,
354
пептидные, 129, 396, 414,
416
сканирование, 290
Липазы, 439
в стереоспецифическом синте-
синтезе, 446,456
мутанты P. aeruginosa, 170
превращение в фосфолипазы,
441
Липидирование белков, 318
Липиды катионные, 153
Липкие концы ДНК, 53, 58
Липоплексы, 153
Липопротеины, 318
Липосомы, 153
в химиотерапии, 409
введение ДНК в клетки, 143
Липофекция, 91,99, 143
Ложноположительные результа-
результаты, 363
Люцифераза, 138, 316, 376, 383,
384
Люциферин, 384
Магическая пуля, 392
Магния ионы
как кофактор биолюминес-
биолюминесценции, 384
получение компетентных кле-
клеток, 144
Макрофаги, 418
взаимодействие с антителами,
412
Малатдегидрогеназа, 376
получение из лактатдегид-
рогеназы, 382
Мальтодекстрин-связывающий
белок, 381
Мальтоза, 379, 381
Манноза, 136,418
Маннозидаза, 459
Маннозо-6-фосфатизомераза, 136
Марганца ионы
77Л-ДНК-полимераза, стиму-
стимуляция активности обратной
транскриптазы, 199
влияние на ДНКазу I, 67
изменение специфичности ре-
стриктаз, 56
ошибки синтеза ДНК, 326
эндонуклеазные разрывы
ДНК, 290
Маркеры
генетические, котрансформа-
ция, 146
контр-селектируемые
HIS3, 367
URA3, 172, 358, 362
селектируемые
ген sacBII, 96
доминантные, 139
устойчивость к гербицидам,
140
экспрессирующихся последо-
последовательностей (EST), 252, 253
Масс-спектрометрия
MALDI-TOF, 166
с ионизацией распылением в
электрическом поле (ESI-MS),
445
Матураза, 79
Мегаплазмиды, 22
Мегапраймеры, 195, 298
Меристоилирование белков,
318
Металлоферменты, рациональ-
рациональный дизайн,283
Метанол и метиламин как индук-
индукторы, 174
Метиладенин (т6А), 218
Метилаза, ДНК-метилаза. См.
Метилтрансферазы
Метилгуанин (m7G), 218
Метилимидазол, 431
Метилирование
азотистых оснований ДНК, 59
218
496
аминокислотных остатков, 36
определение статуса с помо-
помощью ПЦР, 218-220
Метилтрансферазы
Dam, Dem, 59
Mod, 50
модифицирующие, 50
поддерживающие (Dnmtl), 60
эукариот, 59
Метилумбеллиферил-ос-О-глю-
куроновая кислота (MUG), 136
Метилцитозин (т4С), 218
Метиониламинопептидаза, 117
Метионин, 117, 446, 457
Метионинсульфоксимин, 175
Метотрексат, 175, 176, 178, 179,
363
Миелома, 178, 404
Микоплазмы, 22
Микросателлиты, 24
Микросенсоры клеточные, 390
Микроскопия
оптическая, 21
флуоресцентная, 388
электронная, 21
сканирующая, 127, 221
Миксобактерии, 439
Миниантитела, 416
димерные, 414
тетрамерные, 414
тримерные, 414
Минисателлиты, 24
Минихромосомы, 88, 99
двойные, 176
Миозин, киназа легкой цепи,
130
Митохондрии
LP-хроматин, 22
выделение ДНК, 41
геном, 15,23
РНК-полимеразы, 30
суммарное содержание ДНК,
15
трансфекция, 152
Модификации
посттранскрипционные РНК,
29, 32-33
17. Патрушев Л.И. Т. 1
посттрансляционные белков,
29, 35-36
в гетерологичных системах
экспрессии рекомби-
нантных генов, 120, 124,
168
в клетках дрожжей, 171,
174
в клетках насекомых, 182
в ооцитах, 304
в пермеабилизованных клет-
клетках, 184
в проточных бесклеточных
системах, 191
имитация методом EPL, 317
Мозаицизм, 217
Мониторинг
внутренней среды организма,
408
мутантов микроорганизмов,
402
окружающей среды, 206, 350,
408
синтеза молекул в реальном
времени, 132,409
экспрессии генов, 129
Моноциты, 418
Мочевина, 127
мРНК. См. РНК матричная
Мутагенез
кассетный, 323
с вырожденными праймера-
ми, 323
направленный, 286-306
введение множественных
мутаций, 294
делеции и вставки, 287
мегапраймеры, 298
метод Т.А. Кункеля, 290
нонсенс-супрессоры, 302
ПЦР с перекрывающимися
праймерами, 295
сайт-специфическое заме-
замещение неприродными
аминокислотами, 302
сайт-специфический. См. Му-
Мутагенез направленный
497
сканирование аланином, 324
случайный, 323
объединение гомологич-
гомологичных и негомологичных
участков генов (DNA
shuffling), 327
синтез ДНК с ошибками,
326
ступенчатое удлинение оли-
гомеров, 330
химический, 324-326
бисульфит натрия, 324
гидроксиламин, 324
метоксиламин, 324
Мутации
геномные, хромосомные, ген-
генные, 277
делеции,277
дупликации,277
инверсии, 277
миссенс-, 277
нонсенс-
амбер, 82, 277, 304
охр и опал, 277
прямые и обратные, 278
реверсии, 278
точковые, 277
транзиции и трансверсии,
277
сдвиг в частотах возникно-
возникновения, 326
транслокации, 277
Наночастицы, ДНК-содержа-
щие, 223
Насекомые
Spodoptera frugiperda, 182
Trichopsula ni, 182
Нейлоновые фильтры, 163
Нейромедиаторы, 390
Нематоды
геном, 16
количество видов, 436
метилирование ДНК, 220
Неомицин, 90, 135, 137, 150
Неомицин C-фосфотрансфераза,
135
498
Неомицинфосфотрансфераза II,
137
Неоцентромеры, 99
Нетранслируемая область
З'-концевая C'UTR), 111, 172
5'-концевая хлоропластов
E'UTR), 142
Ник-трансляция, 63
Нитрилуксусная кислота, 127
Нитроцеллюлозные фильтры,
163
Ноурсеотрицин, 137
Нуклеазы
Ва131,66
PI Penicillium citrinum, 66
SI, 66, 289
золотистой фасоли (mung
bean), 66, 289
ингибиторы, гидроокись рту-
ртути, 41
экзонуклеазы, 65
III E. coli, 66, 289
фага X, 66
эндонуклеазы, 66
Нуклеоид, 21
Нуклеокапсид, 435
Нуклеоплазмин, 121
Нуклеосомы, 21
сборка, 121
Обелин, 383
Область начала репликации
бактериальных плазмид, 69,
70, 72, 103, 134, 172, 358
вируса SV40, дефектная, 180
дрожжевых векторов, 171, 358
дрожжевых плазмид, 87
искусственных хромосом жи-
животных, 98, 99
космид, 156
фага Р 1,95
Обратная транскриптаза
активность у термостабиль-
термостабильных ДНК-полимераз, 64, 211
в мРНК-дисплее, 355
вируса лейкоза мышей Моло-
hh(MMLV), 64,211
вируса миелобластоза птиц,
64,211
изотермическая амплифи-
амплификация ДНК, 243
использование в ОТ-ПЦР,
211
количественное определение
мРНК, 224
ретротранспозонов, 25,79
синтез кДНК, 158
Оксамат как аллостерический
эффектор, 382
Оксилюциферин, 384
Олигопептидпермеаза, 145
Олигосахариды, лигирование,
308
Онкогены, 140,409,451
Оперон, 23
srfB. subtilis, участие в транс-
трансформации, 145
Ti-плазмид, vir, 139
гибридный, 113
лактозный, регуляция, 108
липазный НрА, 170
определение термина, 30
рРНК, промотор, 142
теплового шока, 121
триптофановый и лактозный,
промоторы, 108
Оротидин-5'-фосфат-декарбок-
силаза, 358
Ортомиксовирусы, 20
Открытые рамки считывания
(ОРС), 22, 25, 35, 113, 117, 134,
166, 253, 295, 383
сдвиг, 115, 278,324
Паказа, 95
Пальмитоилирование белков,
318
Парадокс С, 19
Парафин, удаление из препара-
препаратов, 214
Паркинсона болезнь, 421
Паромомицин, 137
Пепсин, 214
лигирование пептидов, 307
17*
Пепстатин, 187
Пептидил-тРНК, 303
Пептиды
лигирование, 307
нерибосомный синтез, 376
Переклонирование, 336
Переносчики
гексоз, 138
глюкозы, 138
кислорода, 448
рекомбинантных ДНК, 68,
150, 182
углекислого газа, 448
Переходное состояние субстрата
аналоги, 427,429
стабилизация, 429
Периплазматическое простран-
пространство, 118, 123, 170, 335, 345,
350, 381
Перицентромерные области хро-
хромосом, 98
Пероксидаза
активность, 448
получение, 442, 459
хрена, 130
Пили F-, 334
Пирогенные загрязнения, 171
Пироглутаминовая кислота, 36
Пирофосфат неорганический,
29,63
Пирофосфоролиз ДНК, 63
Плазмиды
Ti
векторы на основе, 139
горизонтальный перенос
генов, 37, 154
бактериальные
Со1Е1,69,70,72
pSC101,72
группы совместимости, 71
консервативность размера,
71
контроль числа копий, 70
способность к автономной
репликации, 69
температурочувствитель-
ная репликация, 70
499
дрожжевые, 2jim, 86
конъюгативные, горизонталь-
горизонтальный перенос генов, 37
Пластиды, 22, 23
Пластом, 141
Полиаденилирование
мРНК, 33
сайты в векторах, 134, 139
Поликетиды, 376, 439
Полилизин, 150, 153
Полилинкеры, 73, 75, 83, 93, 95,
96, 108, 133, 141, 358
Полимеразная цепная реакция,
192-250
ДНК-зависимые ДНК-поли-
меразы, 198
Bst, 246
Deep Vent, 200, 213
Klentaq, 213
Pfu,200, 213
Tfl, 224
Tth, 199
UITma, 200
Vent, 200
фрагмент Штоффеля, 199
затруднения при постановке
димеры праймеров, 204
дискретные неспецифичес-
неспецифические продукты, 205
дополнительные неспеци-
неспецифические продукты, 204
низкий выход продуктов,
206
образование продуктов в
отсутствие матрицы, 203
появление шмира, 204, 206
размеры продуктов не соот-
соответствуют ожидаемым,
205
зонды
TaqMan, 224
молекулярные маяки, 229
критические компоненты
ионы двухвалентных метал-
металлов, 201
конструирование прайме-
праймеров, 195
500
концентрация фермента в
пробе, 200
подготовка матрицы
Chelex-100, 206
требования к матрице, 201
общие параметры
время элонгации прайме-
праймеров, 202
выход продукта, 194
кинетика накопления про-
продуктов, 202
объем реакционной смеси,
203
размер продукта, 194
температурный режим, 201
точность, 195
циклы, 202
общие принципы, 192
праймеры
вырожденные, 239
Скорпион, 231
разновидности
in situ, 213
аллель-специфическая, 207,
216
амплификация протяжен-
протяженных ДНК с высокой точ-
точностью, 212
асимметричная (однона-
(однонаправленная), 207
аффинная, 235
гнездовая, 208
иммуно, 220
количественная, в реаль-
реальном времени, 223
множественная, 207
обратная (инвертирован-
(инвертированная), 236
односторонняя, 233
преамплификации с удлине-
удлинением праймеров, 239
пузырьковая, 236
с горячим стартом, 208, 214
с ДНК индивидуальных
клеток, 241
случайная амплификация
полиморфных последо-
вательностей (RAPD),
238
со случайными мечеными
праймерами, 240
сопряженная с обратной
транскрипцией, 211
стандартная, 207
чувствительная к метили-
метилированию матрицы, 218
рэмп, 204
Полиморфизм
длин рестрикционных фраг-
фрагментов ДНК (ПДРФ), 166
маркеры для картирования ге-
генома, 238, 253
одноцепочечных фрагментов
ДНК, 208
популяционный, 164, 205
Полинуклеотидкиназа, 64, 159
Полинуклеотидфосфорилаза,
116,187
Полиплоидия, 14, 141, 277
Полиэтиленгликоль
использование для трансфор-
трансформации
дрожжей, 173
клеток растений, 138
хлоропластов, 142
конъюгаты с антителами, 421
слияние клеток, 404
стабилизация ферментов, 371
Полиэтил енимин, 153
Половой фактор Е. coli, 92
Половые ворсинки (F-пили), 140,
334
Поперечные сшивки
в SP-клетках, 184
в амилозе, 126, 132
в кристаллах белков, 370
в тРНК, 302
индукция светом, 307
стабилизация белков, 458
стабилизация субтилизина,
441
Порог числа циклов, 226
Последовательности аминокис-
аминокислотные
аффинные, 124
лидерные, 142, 173
сигнальные, 118, 123, 170, 182,
335,339,350,379,391,420
транспортные, 350
Последовательности нуклеотид-
ные
LTR и RTR, 141
URS, 360
VNTR, 24
ос-сателлитные, 218
гомополимерные, 160
избыточные, 29
кодирующие, 14
консенсусные, 107, 134, 380
некодирующие, 14
повторяющиеся, 14, 24
высокого порядка, 99
пограничные, 111
поли(А)
бактериальные и вирусные,
160
при очистке мРНК, 42
синтез кДНК, 158
регуляторные, 29, 31
сигнальные, 151
транскрибируемые, 19
умеренно повторяющиеся
(MER), 24
Alu, 25
В1,25
LINE, 24
SINE, 24
короткие прямые повторы,
SDR, 25
мономеры А и F, 25
уникальные, 24
Постгеномная эра, 16
Праймеры
для обратной транскриптазы,
158
к гомополимерам, 160
олиго(сГГ), 160
РНК в репликации плазмид, 70
случайные, 160
универсальные, 301
Пренилирование белков, 318
501
Прионы, 421
Провитамин А, 136
Прогулки по хромосомам, 93
Пролил-цмс-трянс-изомераза,
121
Промоторы
lac, 73
рЮ бакуловирусов, 134
актинового гена цыплят,
133
вируса мозаики цветной капу-
капусты, 139
гена рРНК, 142
гибридные
Т71ас, 134
Го?, 108, 114
нерегулируемые, 139
определение термина, 30
распознавание РНК-полиме-
разой, 31
фага X,?R, 114
хлоропластов, 142
цитомегаловируса, 133
Проназа, 41
Пространство последовательнос-
последовательностей
аминокислотных, 11, 284, 322,
460
нуклеотидных, 11
Протамины, 153
Протеиназы
К, 41, 214
процессинг рекомбинантных
белков, 129
Протеинкиназы
А, 130, 307
SNF1 в дигибридной системе,
356
в химико-ферментативном
синтезе белков, 307
мембранная СотР, 145
модификация рекомбинант-
рекомбинантных белков, 317
Протеинлигаза, 308
Протеолиз
внутриклеточный, 118
защита от, 123, 187
обратный, 307
502
ограниченный in vivo, 120
при трансформации бактерий,
145
специфический, 135, 182, 307
Протеом, 17, 132
Протеомика, 124
Протозимы, 283
Профаг, 101
Псевдогены, 26
Псориаз, лечение антителами,
413
Пуромицин в мРНК-дисплее, 354
ПЦР. См. Полимеразная цепная
реакция
Ревертаза. См. Обратная транс-
криптаза
Регулон, 143
Резольваза, 79
Резонансный перенос энергии
флуоресценции (FRET), 229
Рекомбиназы
Сге, 95,425, 426
дрожжевая, FLP, 87
при транспозиции, 79
Рекомбинантные молекулы, 39
Рекомбинация
in vitro, 39
V-D-J, 424
гомологичная
RecA-зависимая, 79
RecE-зависимая, 101
в компетентных бактери-
бактериальных клетках, 144, 146
влияние на сцепление гене-
генетических маркеров, 15
инактивация в иммунной
системе, 410
интегрирующие векторы,
101, 141
использование ПЦР, 330
клонирование длинных по-
последовательностей, 91
между LTR, 26
образование химерных
ДНК, 92
перенос хромосомных ге-
генов в плазмиды, 69
предотвращение, 83, 91, ЮЗ
разделение коинтегратов,
79
ретрофиттинг, 90
роль ДНК- и РНК-лигаз, 60
создание искусственных
хромосом, 97
стимуляция химерными ре-
стриктазами, 444
участие ДНК-полимераз, 62
сайт-специфическая, ис-
использование сге-рекомби-
назы, 426
система Р 1,95, 96
случайная, 440
Реовирусы, 20
Репарация ДНК
SOS-система, 145
брешей аналогами нуклеоти-
дов при химическом мутагене-
мутагенезе, 325
брешей при репликативной
транспозиции, 78
в компетентных бактериаль-
бактериальных клетках, 144
контроль т6А у прокариот,
218 .
нарушение при мутаторном
фенотипе, 451
одноцепочечных разрывов в
векторе in vivo, 65
предотвращение при направ-
направленном мутагенезе, 291
при перемещении автономных
транспозонов, 26
ферменты
ДНК-зависимые ДНК-по-
лимеразы, 62
ДНК-лигазы, 60
ДНК-метилазы, 59
Репрессия, 108, ПО
глюкозой у дрожжей, 173
последовательностью URS,
360
трансактиватора в дигибрид-
ных системах, 362
Репрессор
cl фага X, 359
Lac, 75, ПО, 170
LexA, 359
REP, 87
TetR, 362
TUP1 дрожжей, 362
в группах совместимости плаз-
мид, 71
действие на рестриктазы типа
1,51
как регулятор транскрипции,
31
температурочувствительный,
с1857, 82, ПО, 114
у бактерий, 108, 365
фага Х434, 365
Рестриктазы
вторичная активность, 56
действие на
короткие последовательно-
последовательности, 56
одноцепочечные ДНК, 56
значение для генной инжене-
инженерии, 48
изменение специфичности
в неоптимальных условиях,
56
действие на модифициро-
модифицированные ДНК, 443
методами белковой инже-
инженерии, 443
с помощью доменов факто-
факторов транскрипции,444
крупнощепящие, 442
номенклатура, 48
рентгеноструктурный анализ,
442
типы
1,50
11,50
III, 50
IIS,51,52
IV, 51
универсальные, 54
503
химерные полипептиды, 444
чувствительные к метилиро-
метилированию ДНК, 55, 220
Ретикулоциты, бесклеточный
синтез белка, 189
Ретинобластомы ген, 213
Ретровирусы, 20
векторы на их основе, 150
геном, 25
горизонтальный перенос ге-
генов, 37
эндогенные, 26
Ретротранспозиция, ретропози-
ция, 25, 79
Ретрофиттинг, 90
Ретроэлементы, подавление
транскрипции метилировани-
метилированием, 60
Рефолдинг полипептидных це-
цепей, 230
Рецепторы
сопряженные с G-белками,
390
хемокиновые макрофагов, 420
Рибозимы, 17
Рибонуклеаза
В,гликозилирование, 308
Рицин,122, 396
РНК
lOSa, 35
информационные. См. РНК
матричные, 28
малые ядерные, 28
матричные
модификации котранскрип-
ционные, 32
процессинг, 32
редактирование, 32
система надзора, 34-35
стабильность, 116
транспорт из ядра в цито-
цитоплазму, 33
небольшие интерферирую-
интерферирующие (siPHK), 152
рибосомные, 28
транспортные
аминоацилирование непри-
504
родными аналогами ами-
аминокислот, 303
аминоацилированные, 33,
188
биосинтез, 28
в бесклеточных белоксин-
тезирующих системах,
185,186
введение в ооциты, 304
гены супрессорных в векто-
векторе, 89
изоакцепторные, 114
интроны в генах, 23
использование в белковой
инженерии, 302
коррекция редко встречаю-
встречающихся кодонов, 116, 119
при очистке мРНК, 42
расщепление абзимами, 432
супрессорные, 302, 304
РНКаза
А. См. РНКаза панкреатичес-
панкреатическая
Н
активность у экзонуклеазы
III, 66
в синтезе кДНК, 158
в составе обратной транс-
криптазы,64, 243
II
участие в деградации
мРНК, 116
активность абзимов, 432
дефектные штаммы Е. coli,
116,187
ингибиторы
ванадиевые комплексы ри-
бонуклеозидов, 41
диэтилпирокарбонат, 214
из плаценты человека, 41,
187
панкреатическая
S-фрагмент в качестве аф-
аффинной метки,125, 129
ингибитор RI, 66
использование для осво-
освобождения от ДНК, 41
описание фермента, 66
синтез с использованием
субтилигазы, 308
РНК-полимеразы ДНК-зависи-
ДНК-зависимые, 62, 106, 107
бактериальные, 21, 31
гибридные с-субъединицы,
380
минимальный (кор) фер-
фермент, 380
модификация ос-субъедини-
цы, 367, 380
модификация методом EPL,
317
в бесклеточных белоксинтези-
рующих системах, 187
взаимодействие с промотором,
107
митохондриальные, 30
номенклатура, 30
фаговые, 108
SP6,30,93, ПО
ТЗ, 76, 110
Т7, 30, 76, 93, ПО, 134, 170,
243, 248, 352, 354
эукариотические, 29, 30
1,23
11,31
Ротамеры, 283
Ртути гидроокись, 41
Рубидия ионы, 148
Рэмп, 204
Сайленсинг, 152
Сайты рестрикции
вырожденные, 52
квазисимметричные, 52
определение, 48
потеря при лигировании, 54
природные, упорядоченные,
51
симметричные, 52
частота встречаемости, 52
эффективность использова-
использования на концах ДНК, 56
Самосборка макромолекул, 6, 11
Саузерн-гибридизация, 91, 214
Сверхэкспрессия клонированных
генов, 108
Секвеназа, 290, 291
Секреторные гранулы, 120
Секреция рекомбинантных бел-
белков, 122
Селективные условия, 39, 162
Селеносубтилизин, 442, 458
Селеноцистеин в активном цент-
центре субтилизина, 442
Сепсис, лечение с использовани-
использованием mAb, 408
Серебро, окраска ДНК, 42
СефадексБР, 127
СефакрилБЮОО, 41
Силан, обработка предметных
стекол,214
Синонимические замены нуклео-
тидов. См. Кодоны синоними-
синонимические
Соматостатин рекомбинантный,
117
Спектиномицин, 137
Сплайсинг, 32
альтернативный, 32
белковый, 35, 312
in trans, 315
в белковой инженерии,
312-316
очистка рекомбинантных
белков, 379
обратный, 79
препятствие для экспрессии
генов в бактериях, 106, 155
факторы, 106
Спленэктомия, 429
Среда питательная, селективная,
HAT, 404
Стрептавидин, 125, 126, 128, 235,
242, 342
происхождение, 221
самосборка комплексов с био-
тинилированной ДНК, 221
Стрептавидин-связывающий пеп-
пептид SBP, 131
Стрептактин,128
505
Субдомены белков гомологич-
гомологичные, случайное объединение,
378
Субклонирование рекомбинант-
нойДНК, 167, 172,248
Субтилигаза, 308, 309, 310
Субтилизин
Carlsberg, 370, 373
ацилирование нуклеозидов и
углеводов, 375
в неводных средах, 372,456
изменение активного центра,
369
как протеинлигаза, 308
лигирование пептидов, 307
молекулярный импринтинг,
375,456
нитробензилэстеразная актив-
активность, 459
превращение в пероксидазу,
441
стабилизация поперечными
сшивками, 370
термостабильные аналоги, 402
тиоалкилированные произ-
производные, 306
Сульфанилмочевина, 137
Супероксид анион, 432
Сурфактин, 145
Сферопласты, 94,173
Сцепление
анализ при позиционном кло-
клонировании, 253
генетическая группа, 15
Талассемия а, 226
Таллизомицин, 137
Тельца включения, 120, 121, 123,
130, 169, 174,191
Терминаза фага X, 93
Терминальная трансфераза, 65,
160
Терминатор транскрипции, 30,
31,111,119,134,172
Терпены, 384
Тетрапиррол,384
Тимидилатсинтаза, 434
506
Тимидинкиназа, 405
Тиопролин, 303
Тиоредоксин, 121, 123, 283
в качестве белка-носителя,
433
создание активного центра,
282
создание центров связывания
ионов металлов, 284
Тиоредоксинредуктаза, 123
Топоизомераза. См. ДНК-топо-
изомераза
Тосповирусы, 435
Тотипотентность, 136
Трансгенез, 9
Трансгенные животные, 39, 86,
97, 103
XenoMouse, 410
использование YAC-векторов,
91
с искусственными хромосома-
хромосомами, 99
Трансгенные растения, 86, 103,
135,136,435
освобождение от селектируе-
селектируемых маркеров, 142
устойчивость к патогенам, 5
Трансгены, 135
наблюдение за экспрессией,
136
прямое введение в клетки рас-
растений, 138
экспрессия, 179
Трансгликозилирование. См.
Гликозилирование in trans
Трансдукция, 101
специфическая и неспецифи-
неспецифическая, 37
Транскриптом, 17
Транскриптон, 30
Транскрипционные единицы, 30
Транскрипция, 29-32
базальная, 31
влияние на экспрессию реком-
бинантных генов, 107-111
индуцированная (активиро-
(активированная), 32
обратная, 28, 62, 64, 79
определение термина, 28
основные этапы, 31, 107
у бактерий, 108
Трансляция
влияние на экспрессию реком-
бинантных генов
5'-концевой блок, 119
вторичная структура
мРНК, 118
вторичные сайты инициа-
инициации трансляции,118
неожиданное возникнове-
возникновение терминирующих кодо-
нов, 119
правило N-концевой после-
последовательности, 116
терминатор транскрипции
вектора, 119
обратная, 164, 165
определение термина, 33
реинициация на перекрываю-
перекрывающихся кодонах, 114
терминация в системах надзо-
надзора, 34
этапы, 34
Трансплантат
отбор в фаговом дисплее, 349
отторжение
подавление антителами,
408
подавление токсинами, 395
у мышей nude, 348
Транспозиция
консервативная, 78
репликативная, 78
эксцизионная, 79
Транспозоны
ТпЗ, 72
автономные
LTR-содержащие, 26
не содержащие LTR, 26
горизонтальный перенос ге-
генов, 37
ДНК-транспозоны, 26
использование для клонирова-
клонирования генов, 78-80
неавтономные, 26
ретротранспозоны, 26
Трансфектанты, отбор, 150
Трансфектома, 406
Трансфекция
временная, 150
конъюгативный перенос бак-
бактериальных генов в клетки
животных, 154
копреципитация ДНК фосфа-
фосфатом кальция, 152
определение термина, 143, 149
с помощью
баллистической (биолисти-
ческой) инъекции, 151
дендримеров, 153
диметилсульфоксида, 153
ДЭАЭ-декстрана, 152
катионных липидов, 153
клеточных рецепторов, 150
липосом, 153
микроинъекций, 151
электропорации, 150
стабильная, 152
Трансферрин, 150
Трансформация генетическая,
143
Трансформирующий фактор
роста а в составе гибридных
токсинов,395
Триггер ный фактор, 121
Трипсин, 118
гибридный, 378
лигирование пептидов, 307
пермеабилизация клеток,
214
Тромбин, 129, 132
Тупые концы ДНК, 54, 58
Тяжелых металлов ионы
удаление из окружающей сре-
среды, 350
устойчивость бактериальных
клеток, 69
Убиквитин, 117,452
Удлиненный участок хромосо-
хромосомы, 176
507
Урацил,362
биосинтез, 358
в направленном мутагенезе,
292
в сайтах рестрикции, 443
в случайном мутагенезе, 325
образование из 5тС, 60
Урацил-ДНК-гликозилаза, 60,
290
Фаги. См. Бактериофаги
Фагмиды, 76, 290, 334, 335, 336,
426
Фагосомы, 432
Фагоцитоз, исследование, 392
Фазмиды, 85
Фактор свертывания крови Ха
создание гибридной протеина-
зы, 378
удаление избыточных после-
последовательностей рекомбинант-
ных белков, 132
Факторы роста, 122, 405
Фарнезильные группы. См. Пре-
нилирование белков
Фенол, 41,206
Фингерпринты ДНК, 238
Фитогормоны, 136, 139
Флуоресцеин, 228, 363
Флуоресценция
базальный уровень, 226
красители
SYBR Green, 230
тушители, 226
DABCYL, 228
TAMRA, 228
флуорофоры
FAM, 228
FITC, 229
RED640, 229
TAMRA, 228
Флуорохромы. См. Флуоресцен-
Флуоресценция, флуорофоры
Фолдаза, 121, 170
Фолдинг полипептидных цепей,
121,123,169,175,184,230,274,
508
279,345,350, 372,379, 380, 388,
420
Формалин, влияние на ПЦР in
situ, 214
Формальдегиддегидрогеназа, глу-
татион-зависимая, 174
Формилметионин, 117
Фософодиэстераза, Са2+-зависи-
мая, 346
Фосфатаза
синтез методом EPL, 317
щелочная
бактерий и кишечника те-
теленка, 65
создание аллостерического
центра, 381
Фосфолипазы, получение из ли-
липаз, 441
Фосфорибозилантранилатизоме-
раза, получение из индол-3-
глицеролфосфатсинтазы, 439
Фосфоротиоатные аналоги оли-
гонуклеотидов, иммобилизо-
иммобилизованные, 346
Фотонные процессоры, 6
Фотопротеины
беворин,385
клитин, 385
митрокомин, 385
мнемиопсин, 385
обелин, 385
талассиколин, 385
фиалидин, 385
халистаурин, 385
экворин, 385
Фторгистидин, 310
Фтороротовая кислота, 358
Фтортирозин, 303
Фторурацил, 358
Фторфенилаланин, 304
Фьюзоген, 404
Хантингтин, 420
Хантингтона болезнь, 420
Хёхст 33258, 47
Химостатин, 187
Химотрипсин
в неводных средах, 373
как пластиковый биокатали-
биокатализатор, 456
Хиральность, 444
Хитин-связывающий домен, 126,
131,379
Хлопок, 135
Хлорамфеникол, 92, 101, 137
амплификация плазмидной
ДНК, 71
Хлорамфениколацетилтрансфе-
раза, 113, 137,139,383
Хлоропласты
биосинтез белков, 136
геном, 23
суммарное содержание ДНК,
15
трансгенные, 141
транспорт антител, 420
трансфекция, 152
Хлороформ, 41,78
Холидея структуры, влияние на
рестрикцию, 50
Хроматин
бактериальный LP, 21
защитная функция, 449
компактизация ДНК, 27
определение термина, 23
состав, 27
Хроматогр афия
аффинная, 42, 127, 129, 347,
352, 379
ионообменная, 187
Хромомицин A3, 47
Хромосомы
X человека, 98
акроцентрические, 98
гомологичные, 15
дицентрические, 98
искусственные, 86-100
маркерные, 99
метафазные
выделение с помощью про-
проточной цитометрии, 43-
48
как таксономический при-
признак, 23
оптическая сортировка, 48
небольшие добавочные, 99
определение, 14
проблема числа, 15
термофильных бактерий, 52
электрофоретическое выде-
выделение, 42
Хромофор GFP, образование из
аминокислотных остатков,
387
Целлюлоза
гидролиз целлюлосомой,
377
домен, связывающий, 126,130,
379
олиго(AТ), 42, 158
Целлюлосома, 377
Цефлотаксим, 292
Цефтриаксон, 292
Циан бромистый, 117
Циануринхлорид, 371
Цитозин,дезаминирование, 325
Цитокинин,139
Цитомегаловирусы, 133
Цитометрия проточная, 42,
43-48,98,129,349,407
FACS, 43
принцип метода, 46
Цитохром Р450, 450, 451, 453
Цитратсинтаза, 376
Шайна-Дальгарно последова-
последовательность, 112
Шаперонины, 121
Шапероны, 121, 122,279
Шок
тепловой, 148
промоторы генов, 380
фаговый, 336
электрический,149, 150
Шоффлера фрагмент Taq-ДНК-
полимеразы, 217
Шплинты, 233
ЭГТА, 125
Экворин, 383, 385, 390
509
Экзоны
в предшественниках мРНК, 32
в экспрессирующих векторах,
180, 183
использование в ОТ-ПЦР, 212
определение, 14
суммарный размер, 14
Экспрессия генов, 28-29
Экстеины, 312
Экстремофилы ацидофильные и
алкалифильные, 399
Электропорация клеток, 143
бактерий, 94, 96, 149
дрожжей, 173
животных, 138, 150
Электрофорез
в импульсном электрическом
поле, 42, 213
двумерный, 165
капиллярный, 166, 207, 445
матричный, 445
на микрочипах, 445
Энантиомеры
D-аминокислот, 36
определение термина, 444
Энантиоселективность
катализаторов, 444
ферментов, 446,457
Эндонуклеазы рестрикции. См.
Рестриктазы
Эндорфин р-, 118
Эндоцитоз, 19, 150, 152, 154, 394
Энолаза а, 448
Энтерокиназа, 128, 129
Энтомопоксвирусы, 61
Энхансеры, 14
в составе векторов, 111, 134,
179
трансляции, 135
Эписомы, 68,143
дрожжей, 87, 172
Эпитопы, 129, 324, 336, 338, 342,
343, 348, 380, 381,403, 414, 418
одинаковые у разных антиге-
антигенов, 407
Эпотилоны, 439
Эстеразы, изменение стереоспе-
цифичности действия, 439
Этидий бромистый, 42, 194, 206
Эубактерии, 16
Эукариоты, 16
Эухроматин, 27
Ядерный поровый комплекс, 150
Ядро
интерфазное, 21
мембрана, 16
перенос ДНК, 152, 153
сигнал ядерной локализации
(NLS), 140
ЯМР-спектроскопия, 129, 169,
281,305
Англо-русский словарь использованных
словосочетаний
5' translational blockage: 5'-концевой блок трансляции
activation domain - AD: активирующий домен
affinity maturation: созревание аффинности
affinity tag: аффинная метка
allele specific primer extension: аллель-специфическая элонгация прай-
мера
arrayed (gridded) library: клонотека, упорядоченная в виде матрицы
asymmetrical PCR, single-sided PCR: асимметричная ПЦР, однонаправ-
однонаправленная ПЦР
autonomously replicating sequence - ARS: автономно реплицирующаяся
последовательность
back (reverse) translation: обратная трансляция
bacterial artificial chromosome - ВАС: искусственная хромосома бактерий
biocatalytic plastics: пластиковые биокатализаторы
bispecific antibody - sc-bsAb: антитело двойной специфичности
blue fluorescent protein - BFP: голубой флуоресцирующий белок
branched rolling circle amplification, branched RCA, bRCA: разветвленная
амплификация по типу катящегося кольца
bubble PCR: пузырьковая ПЦР
calcium phosphate coprecipitation method: трансфекция, опосредованная
фосфатом кальция
capillary array electrophoresis - САЕ: капиллярный матричный электро-
электрофорез
capillary electrophoresis on microchips - САЕ on chips: капиллярный элек-
электрофорез на микрочипах
cap-independent translation enhancer - CITE: кэп-независимый энхансер
трансляции
capture PCR: аффинная ПЦР
cellulose-binding domain - CBD: домен, связывающий целлюлозу
Center for Biotechnology Information - NCBI: Центр биотехнологической
информации
chemical inducers of dimerization - CID: химические индукторы димериза-
ции
cluster of differentiation (CD) system: система кластера дифференцировки
(лимфоцитов)
511
cohesin domain: когезиновый домен
crosslinked enzyme crystal - CLEC: кристаллы фермента, содержащие по-
поперечные сшивки
cyan fluorescent protein - CFP: циановый флуоресцирующий белок
diabodies - димерные миниантитела
directed evolution: направленная эволюция
DNA binding domain - DBD: ДНК-связывающий домен
DNA shuffling: случайное объединение фрагментов ДНК
DNA-mediated gene transfer - DMGT: перенос генов, опосредованный
ДНК
dockerin domain: докериновый домен
domain antibodies - dAb: доменные антитела
dominant control region - DCR: доминантная регуляторная область
double-minutes - DM: двойные минихромосомы
electrospray ionization mass spectrometry - ESI-MS: масс-спектрометрия с
ионизацией распылением в электрическом поле
enantiomeric excess: избыток энантиомера
enhanced GFP - EGFP: улучшенный зеленый флуоресцирующий белок
environmental library: клонотека ДНК окружающей среды
enzyme-linked immunosorbent assay - ELISA: иммуноферментный метод
expressed protein ligation, EPL: лигирование синтезированных белков
expressed sequence tags - EST: маркеры экспрессирующихся последова-
последовательностей
extended chromosomal region - ECR: удлиненный участок хромосомы
family shuffling: случайное объединение последовательностей одного се-
семейства
fitness landscape: ландшафт функциональности
fluorescence-activated cell (chromosome) sorting -FACS: сортировка флуо-
флуоресцентно меченых клеток (хромосом)
fluorescent resonance energy transfer - FRET: резонансный перенос энер-
энергии флуоресценции
fluorescent timer: флуоресцентный хронометр
Fourier-transform infrared (FTIR) spectroscopy: инфракрасная спектроско-
спектроскопия с преобразованием Фурье
genetic modified - GM: генетически модифицированный
green fluorescent protein - GFP: зеленый флуоресцирующий белок
headful cleavage - разрезание (ДНК) после заполнения головки (бакте-
(бактериофага)
higher order repeats - HOR: повторы высокого порядка
hollow-fiber bioreactor: мембранный биореактор
homogeneously staining region - HSR: гомогенно окрашивающаяся об-
область (хромосомы)
host specificity determinant - HsdM(S): детерминанта специфичности
клетки-хозяина
hot start PCR: ПЦР с "горячим стартом"
human anti-mouse antibody - HAMA: синдром антимышиных антител че-
человека
512
human artificial chromosome - НАС: искусственная хромосома человека
humanized antibodies: "очеловеченные" антитела
immobilized metal-affinity chromatography - IMAC: аффинная хроматогра-
хроматография на иммобилизованных ионах металлов
immuno-PCR - IPCR: иммуно-ПЦР
internal ribosome entry site - IRES: внутренний сайт связывания рибо-
рибосом
intrabodies: внутриклеточные антитела
intron encoded protein - iep: белок, кодируемый интроном
inverse PCR: инвертированная (обратная) ПЦР
isothermal self-sustained sequence replication - 3SR: изотермическая само-
самоподдерживающаяся репликация последовательностей
kindling fluorescent proteins - KFPs: зажигающиеся флуоресцирующие
белки
leukemia inhibitory factor - huLIF: фактор, ингибирующий лейкозы
ligase chain reaction - LCR: лигазная цепная реакция (ЛЦР)
ligation-independent cloning - LIC: клонрование без применения ДНК-ли-
газы
long PCR, long and accurate (LA) PCR: амплификация протяженных по-
последовательностей ДНК с высокой точностью
maltose-binding protein - МВР: белок, связывающий мальтозу
mammalian artificial chromosome - MAC: искусственная хромосома мле-
млекопитающих
master sequence: преобладающая последовательность
matrix assisted laser desorption ionization - MALDI: лазерная десорбция и
ионизация (макромолекул), опосредованная матриксом
molecular beacons: молекулярные маяки
multiplex PCR, mPCR: множественная ПЦР
nanocircles: наночастицы
N-end rule: правило N-концевой последовательности
nested PCR: гнездовая ПЦР
nonfluorescent chromoproteins - СР: нефлуоресцирующие хромопротеины
nuclear localization signal - NLS: сигнал ядерной локализации
one-hybrid system: моногибридная система
ordered library: упорядоченная клонотека
overlap extension PCR: амплификация перекрывающихся последователь-
последовательностей
oxygen evolving enhancer protein - Oee: белок-энхансер выделения кисло-
кислорода
PI phage-derived artificial chromosome - РАС: искусственная хромосома
бактериофага PI
padlock probes: зонды типа "висячий замок"
patch engineering: инженерия белковых поверхностей
primer extension preamplification - PEP: преамплификация с удлинением
праймеров
promoter and leader - PL: PL-кассета (содержащая промоторную и лидер-
ную последовательности)
513
pulsed field gel electrophoresis - PFGE: электрофорез в импульсном элек-
электрическом поле
QuikChange™ Site-Directed Mutagenesis System - QCM: система направ-
направленного мутагенеза QuikChange™
quorum sensing: восприятие кворума
random amplified polymorphic DNA - RAPD: случайная амплификация по-
полиморфных последовательностей
rapid amplification of cDNA ends - RACE: быстрая амплификация концов
кДНК
rational design: рациональный дизайн
real-time PCR: ПЦР в реальном времени
red fluorescent protein - RFP: красный GFP-подобный белок
repressed transactivator system - RTS: система с репрессируемым трансак-
трансактиватором
reshaped antibodies: перестроенные антитела
restriction fragment length polymorphism - RFLP: полиморфизм по длинам
рестрикционных фрагментов ДНК - ПДРФ
right (left) targeting regions - RTR (LTR): RTR- AЛТ*-)последовательности
rolling circle amplification - RCA: амплификация по типу катящегося
кольца
RT PCR, one tube PCR: ПЦР, сопряженная с обратной транскрипцией
(ОТ-ПЦР)
satellite DNA-based artificial chromosome - SAT AC: искусственная хромо-
хромосома на основе сателлитной ДНК
semi-permeabilised (SP) cell systems: клеточные системы с полупроницае-
полупроницаемыми мембранами
sequence space: пространство последовательностей
shot-gun method: метод дробовика
single-cell PCR: ПЦР с ДНК индивидуальных клеток
single-chain bispecific antibody - sc-bsAb: одноцепочечное антитело двой-
двойной специфичности
single-chain Fv - scFv: одноцепочечный Fv-фрагмент (антител)
single-stranded conformation polymorphism - SSCP: полиморфизм одноце-
почечных фрагментов ДНК
site-directed non-native amino acid replacement (SNAAR): сайт-специфиче-
сайт-специфическое замещение неприродными аминокислотами
small accessory chromosome - SAC: небольшая добавочная хромосома
spleen focus forming virus - SSFV: вирус, образующий фокусы селезенки
split-hybrid system: разделенная гибридная система
staggered extension process - StEP: процесс ступенчатого удлинения оли-
гомеров
strand displacement amplification - SDA: амплификация с замещением це-
цепи ДНК
streptavidin-binding peptide - SBP: стрептавидин-связывающий пептид
tagged random primer PCR - T-PCR: ПЦР со случайными мечеными прай-
мерами
tagging domain: адресный домен
514
telomere directed truncation - TDT: укорачивание с помощью теломер
telomere-associated chromosome fragmentation - TACF: фрагментация хро-
хромосом, с использованием теломерных последовательностей
tetrabodies: тетерамерные миниантитела
threshold cycle, Ct: порог числа циклов
transformation-competent bacterial artificial chromosomes - TAC: трансфор-
трансформируемые искусственные хромосомы бактерий
transient transfection: временная трансфекция
transition state analogue - TSA: аналог (субстрата) в переходном состоя-
состоянии
triabodies: тримерные миниантитела
tRNA-mediated protein engineering - TRAMPE: белковая инженерия, осно-
основанная на тРНК
untranslated region - UTR: нетранслируемая область (мРНК)
upstream activating sequence - UAS: вышерасположенная активирующая
последовательность
upstream repressing sequence - URS: вышерасположенная репрессирую-
репрессирующая последовательность
vasoactive intestinal peptide - VIP: вазоактивный кишечный пептид
yeast artificial chromosome - YAC: искусственная хромосома дрожжей
yeast centromeric plasmids - YCP: дрожжевые плазмиды, содержащие
центромеры
yeast episomal plasmids - YEP: эписомные плазмиды дрожжей
yeast integrative plasmid - YIP: интегрирующие плазмиды дрожжей
yeast replicating plasmids - YRP: реплицирующиеся плазмиды дрожжей
yeast two-hybrid assay - Y2H: дрожжевая дигибридная система
yellow fluorescent protein - YFP: желтый флуоресцирующий белок
Оглавление
Предисловие ответственного редактора 5
Предисловие автора 9
Часть I
ПРИНЦИПЫ ГЕННОЙ ИНЖЕНЕРИИ 13
Глава 1. Введение. Природные системы генов, их организация и
экспрессия 13
1.1. Гены и хромосомы 13
1.2. Геном 16
1.2.1. Размеры геномов 17
1.2.2. Геном вирусов 19
1.2.3. Геном эубактерий 21
1.2.4. Геном архей 22
1.2.5. Геном эукариот 23
1.3. Экспрессия генов 28
1.3.1. Транскрипция 29
1.3.2. Котранскрипционные и посттранскрипционные
модификации РНК 32
1.3.3. Трансляция 33
1.3.4. Системы надзора за мРНК и белками 34
1.3.5. Котрансляционные и посттрансляционные моди-
модификации белков 35
1.4. Использование генной инженерии для создания искусст-
искусственных генетических систем 36
Глава 2. Выделение нуклеиновых кислот и ферменты, использу-
используемые для работы с ними 40
2.1. Основные приемы очистки нуклеиновых кислот 40
2.1.1.Традиционные методы 40
2.1.2. Выделение метафазных хромосом с помощью
проточной цитометрии 43
2.2. Ферменты рестрикции и модификации нуклеиновых
кислот 48
2.2.1. Классификация систем рестрикции-модифика-
рестрикции-модификации 48
2.2.2. Рестриктазы типа II - основной инструмент генной
инженерии 52
516
2.2.3. Универсальные рестриктазы для одноцепочечных
ДНК 54
2.2.4. Изошизомеры 55
2.2.5. Изменение специфичности действия рестриктаз .... 56
2.2.6. Изменение концов рестрикционных фрагментов
ДНК 58
2.2.7. ДНК-метилазы и урацил-ДНК-гликозилазы 59
2.3. ДНК- и РНК-лигазы 60
2.4. Ферменты матричного синтеза ДНК и РНК 62
2.4.1. ДНК-зависимые ДНК-полимеразы 62
2.4.2. РНК-зависимые ДНК-полимеразы 63
2.5. Другие ферменты, используемые в генной инженерии 64
Глава 3. Векторы 68
3.1. Плазмидные векторы 68
3.1.1. Биологические свойства бактериальных плазмид 69
3.1.2. Векторы для клонирования без применения ДНК-
лигазы 76
3.1.3. Векторы для прямого клонирования продуктов
ПЦР 77
3.1.4. Использование транспозонов для клонирования
ДНК 78
3.2. Векторы на основе хромосомы фага X 80
3.3. Космиды и фазмиды 83
3.4. Искусственные хромосомы 86
3.4.1. Сверхъемкие векторы YAC, ВАС и РАС 86
3.4.2. Искусственные хромосомы животных и человека 97
3.5. Интегрирующие и челночные (бинарные) векторы 101
3.5.1. Интегрирующие векторы грамположительной бак-
бактерии Bacillus subtilis 101
3.5.2. Челночные векторы 103
3.6. Конструирование экспрессирующих векторов и их
функционирование 104
3.6.1. Факторы, оказывающие влияние на эффективность
экспрессии рекомбинантных генов в бактери-
бактериальных клетках: транскрипция 107
3.6.2. Факторы, влияющие на эффективность экспрес-
экспрессии рекомбинантных генов в бактериальных клет-
клетках: трансляция 111
3.6.3. Факторы, влияющие на эффективность экспрес-
экспрессии рекомбинантных генов: стабильность реком-
рекомбинантных РНК и белков 116
3.6.4. Другие причины низкой эффективности экспрес-
экспрессии рекомбинантных генов в бактериях 118
3.6.5. Особенности биосинтеза эукариотических ре-
рекомбинантных белков в бактериальных клетках:
тельца включения 119
517
3.6.6. Очистка рекомбинантных белков с использовани-
использованием аффинных аминокислотных последовательнос-
последовательностей 124
3.7. Векторы для переноса ДНК в клетки животных и расте-
растений 133
3.7.1. Экспрессирующий вектор рТпЕх 133
3.7.2. Векторы растений 135
3.8. Введение рекомбинантных ДНК в клетки 142
3.8.1. Природная компетентность бактериальных кле-
клеток 143
3.8.2. Искусственная компетентность Е. coli 148
3.8.3. Способы и последствия введения ДНК в культиви-
культивируемые клетки животных 149
Глава 4. Клонотеки генов 155
4.1. Получение клонотек генов 155
4.1.1. Клонотеки геномной ДНК 157
4.1.2. Клонотеки кДНК 158
4.1.3. Случайные и упорядоченные клонотеки ..'. 160
4.2. Поиск последовательностей в клонотеках генов 162
4.2.1. Использование меченых зондов 163
4.2.2. Повторный скрининг 167
4.2.3. Субклонирование 167
Глава 5. Системы экспрессии рекомбинантных генов 168
5.1. Экспрессирующие системы микроорганизмов 168
5.1.1. Грамотрицательные бактерии 168
5.1.2. Клетки дрожжей как экспрессирующие сис-
системы 171
5.2. Системы экспрессии, основанные на культуре клеток
животных 174
5.2.1. Клетки яичников китайских хомячков (линия
СНО) 176
5.2.2. Клетки мышиной миеломы (линия SP2/0) 178
5.2.3. Клетки селезенки мышей (линия MEL) 179
5.2.4. Клетки африканской зеленой мартышки (линия
COS) 180
5.2.5. Клетки насекомых, зараженных бакуловиру-
сами 181
5.2.6. Сравнение эффективности рассмотренных систем
экспрессии 183
5.3. Клетки с нарушенной проницаемостью внешних мем-
мембран 184
5.4. Бесклеточные белоксинтезирующие системы 185
5.4.1. Прокариотические системы 186
5.4.2. Эукариотические системы 189
5.4.3. Проточные системы 190
518
Глава 6. Полимеразная цепная реакция и другие способы ампли-
амплификации ДНК и сигналов 192
6.1. Проведение ПЦР 192
6.1.1. Общая схема ПЦР 192
6.1.2. Критические компоненты реакции 194
6.1.3. Конструирование праймеров 195
6.1.4. Термостабильные ДНК-зависимые ДНК-полиме-
разы 198
6.1.5. Другие критические компоненты и параметры
ПЦР 201
6.2. Проблемы, возникающие при постановке ПЦР 203
6.2.1. Появление неспецифических продуктов ПЦР 203
6.2.2. Низкий выход или полное отсутствие продуктов
ПЦР 206
6.3. Методы ПЦР 207
6.3.1. Стандартная ПЦР 207
6.3.2. Множественная ПЦР 207
6.3.3. Асимметричная ПЦР 207
6.3.4. Гнездовая ПЦР 208
6.3.5. ПЦР с "горячим стартом" 208
6.3.6. ПЦР, сопряженная с обратной транскрипцией 211
6.3.7. Амплификация больших участков ДНК с высокой
точностью 212
6.3.8. ПЦР in situ 213
6.3.9. Аллель-специфическая ПЦР 214
6.3.10. ПЦР, чувствительная к метилированию матрицы ... 218
6.3.11. Иммуно-ПЦР 220
6.3.12. Количественная ПЦР (ПЦР в реальном вре-
времени) 223
6.4. Амплификация последовательностей с неизвестной
первичной структурой 233
6.4.1. Использование ПЦР для продвижения вдоль ДНК
от участка с известной первичной структурой в
сторону неизвестной последовательности 233
6.4.2. Понижение сложности амплифицируемой ДНК с
помощью ПЦР (метод RAPD) 237
6.5. Одновременная амплификация последовательностей
целого генома 239
6.6. Альтернативные способы амплификации нуклеиновых
кислот in vitro 241
6.6.1. Лигазная цепная реакция 241
6.6.2. Изотермические системы амплификации нуклеи-
нуклеиновых кислот, основанные на транскрипции 243
6.6.3. Амплификация по типу катящегося кольца 250
Заключение. Стратегия выделения нового гена 251
Литература 254
519
Часть II
БЕЛКОВАЯ ИНЖЕНЕРИЯ
Введение 273
Глава 1. Рациональный дизайн и редизайн белковых молекул 279
1.1. Проектирование новых белков и ферментов 279
1.1.1. Энергетические требования, предъявляемые к но-
новой белковой конструкции 279
1.1.2. Выбор пространственной структуры 281
1.1.3. Комбинаторная проблема обратного дизайна бел-
белков 281
1.1.4. Дизайн поверхности белковой глобулы 282
1.1.5. Примеры использования методов рационального
дизайна 282
1.1.6. Ландшафт функциональности среди пространства
последовательностей белковых молекул 284
1.2. Методы направленного мутагенеза 286
1.2.1. Получение делеций и вставок .' 287
1.2.2. Мутагенез с использованием олигонуклеотидов .... 290
1.2.3. ПЦР с перекрывающимися праймерами 294
1.2.4. Мегапраймеры в направленном мутагенезе 298
1.2.5. Мутагенез с использованием нонсенс-супрессоров ... 302
1.3. Химико-ферментативный синтез в создании полусинте-
полусинтетических полипептидов: лигирование синтезированных
белков 306
1.3.1. Полусинтетические белковые молекулы в созда-
создании новых полипептидов 307
1.3.2. Сплайсинг и транс-сплайсинг белков в лигировании
пептидов 312
1.3.3. Метод EPL в белковой инженерии 316
Глава 2. Направленная эволюция белков 319
2.1. Комбинаторные клонотеки последовательностей нуклео*
тидов 321
2.2. Методы введения случайных мутаций 323
2.2.1. Химический мутагенез 324
2.2.2. Синтез ДНК с ошибками 326
2.2.3. Случайное объединение гомологичных и негомо-
негомологичных участков генов 327
2.3. Скрининг и отбор белков с требуемыми свойствами 332
2.3.1. Фаговый дисплей 333
2.3.2. Клеточный дисплей 349
2.3.3. Рибосомный дисплей и мРНК-дисплей 351
2.3.4. N-гибридные системы в исследовании белков 355
520
Глава 3. Достижения белковой инженерии 369
3.1. Химические модификации белков 369
3.1.1. Искусственные поперечные сшивки в белках 370
3.1.2. Оптимизация ферментов для функционирования
в неводных средах 372
3.1.3. Молекулярный импринтинг 374
3.2. Гибридные белки 375
3.2.1. Гибридные ферменты 376
3.2.2. Зеленый флуоресцирующий белок (GFP) и его
аналоги в качестве белков-репортеров 382
3.2.3. Гибридные токсины 392
3.3. Экстремозимы 397
3.3.1. Экстремофилы и их ферменты 397
3.3.2. Достижения белковой инженерии в повышении
стабильности ферментов 399
3.4. Белковая инженерия антител 403
3.4.1. Моноклональные антитела 403
3.4.2. Рекомбинантные антитела 409
3.4.3. Антитела, обладающие ферментативной активнос-
активностью: абзимы 427
3.5. Пептидные аптамеры 433
3.6. Изменение специфичности ферментов 435
3.6.1. Проблема изменения специфичности ферментов:
желаемое и действительное 437
3.6.2. Рестриктазы 442
3.6.3. Изменение стереоспецифичности 444
Глава 4. Комбинаторная белковая инженерия в природе: ксено-
биоз? 447
Глава 5. Белковая инженерия в поисках полипептидов, макси-
максимально отвечающих требованиям современной биотех-
биотехнологии 454
5.1. Изменение парадигмы оптимизации биотехнологичес-
биотехнологических процессов 454
5.2. Современные технологии, позволяющие улучшать свой-
свойства ферментов 455
Литература 462
Предметный указатель 484
Англо-русский словарь использованных словосочетаний 511
Contents
Editor-in-chief preface 5
Author preface 9
Parti
PRINCIPLES OF GENETIC ENGINEERING 13
Chapter 1. Introduction. Natural gene systems, their organization
and expression 13
1.1. Genes and chromosomes 13
1.2. Genome 16
1.2.1. Genome sizes 17
1.2.2. Viral genome 19
1.2.3. Eubacterial genome 21
1.2.4. Archaeal genome 22
1.2.5. Eukaryotic genome 23
1.3. Gene expression 28
1.3.1. Transcription 29
1.3.2. Cotranscriptional and posttranscriptional RNA modifi-
modifications 32
1.3.3. Translation 33
1.3.4. mRNA and protein surveillance systems 34
1.3.5. Cotranslational and posttranslational protein modifica-
modifications 35
1.4. Use of genetic engineering for construction of artificial
genetic systems 36
Chapter 2. Nucleic acid purification and the enzymes commonly used
for their manipulation 40
2.1. Basic methods for nucleic acid purification 40
2.1.1. Conventional methods 40
2.1.2. Metaphase chromosome isolation by flow cytometry 43
2.2. Nucleic acid restriction and modification enzymes 48
2.2.1. Classification of restriction-modification systems 48
2.2.2. Type II restriction endonucleases as essential tools of
genetic engineering 52
2.2.3. Universal restriction endonucleases for single-stranded
DNA 54
522
2.2.4. Isoschizomers 55
2.2.5. Changing the sequence specificity of restriction
enzymes 56
2.2.6. Changing the form of DNA fragment ends 58
2.2.7. DNA methyltransferases and uracil DNA glycosylases 59
2.3. DNA and RNA ligases 60
2.4. The enzymes of DNA and RNA template synthesis 62
2.4.1. DNA-dependent DNA polymerases 62
2.4.2. RNA-dependent DNA polymerases 63
2.5. Other enzymes used in genetic engineering 64
Chapter 3. Vectors 68
3.1. Plasmid vectors 68
3.1.1. Biological properties of bacterial plasmids 69
3.1.2. Vectors for ligation-independent cloning 76
3.1.3. Vectors for direct cloning of PCR products 77
3.1.4. Use of transposons for DNA cloning 78
3.2. Vectors based on phage X chromosome 80
3.3. Cosmids and phasmids 83
3.4. Artificial chromosomes 86
3.4.1. Supercapacity vectors YAC, ВАС and РАС 86
3.4.2. Human and animal artificial chromosomes 97
3.5. Integrative and shuttle (binary) vectors 101
3.5.1. Integrative vectors of gram-positive bacteria Bacillus
subtilis 101
3.5.2. Shuttle vectors 103
3.6. Engineering of expression vectors and their functioning 104
3.6.1. Factors which affect recombinant gene expression
levels in bacterial cells: transcription 107
3.6.2. Factors which affect recombinant gene expression
levels in bacterial cells: translation Ill
3.6.3. Factors which affect recombinant gene expression
levels in bacterial cells: stability of recombinant RNA
and proteins 116
3.6.4. Other reasons for low recombinant gene expression in
bacteria 118
3.6.5. The peculiarity of the eukaryotic recombinant protein
biosynthesis in bacterial cells: inclusion bodies 119
3.6.6. Purification of recombinant proteins by affinity tag
sequences 124
3.7. Vectors for DNA transfer in animal and plant cells 133
3.7.1. Expression vector pTriEx 133
3.7.2. Plant vectors I35
3.8. Recombinant DNA uptake into cells I42
3.8.1. Natural competence of bacterial cells 143
л л о
3.8.2. Induction of competence in E. coli 14°
3.8.3. Methods and consequences of DNA uptake into cul-
cultured animal cells
523
Chapter 4. Gene libraries 155
4.1. Construction of gene libraries 155
4.1.1. Genomic libraries 157
4.1.2. cDNA libraries 158
4.1.3. Random, arrayed and ordered libraries 160
4.2. Search of right sequences in gene libraries 162
4.2.1. Use of the labeled probes 163
4.2.2. Rescreening 167
4.2.3. Subcloning 167
Chapter 5. Recombinant gene expression systems 168
5.1. Microorganisms as expression systems 168
5.1.1. Gram-negative bacteria 168
5.1.2. Yeast cells as expression systems 171
5.2. Cultured animal cells as expression systems 174
5.2.1. Chinese hamster ovary cells (CHO line) 176
5.2.2. Mouse myeloma cells (SP2/0 line) 178
5.2.3. Mouse spleen cells (MEL line) , 179
5.2.4. African green monkey kidney cells (COS lines) 180
5.2.5. Baculovirus-infected insect cell systems 181
5.2.6. Comparing efficacy of the expression systems 183
5.3. Permeabilised cells 184
5.4. Cell-free protein synthesizing systems 185
5.4.1. Prokaryotic systems 186
5.4.2. Eukaryotic systems 189
5.4.3. Continuous-flow cell-free expression systems 190
Chapter 6. Polymerase chain reaction and other methods for DNA and
signal amplification 192
6.1. PCR in practice 192
6.1.1. Basic principles of the PCR reaction 192
6.1.2. Most important components of the PCR reaction 194
6.1.3. Designing the primers 195
6.1.4. Thermostable DNA-dependent DNA polymerases 198
6.1.5. Other critical components and parameters of PCR
reaction 201
6.2. PCR troubleshooting 203
6.2.1. Non-specific product yields 203
6.2.2. Little or no product yield 206
6.3. PCR methods 207
6.3.1. Standard PCR 207
6.3.2. Multiplex PCR 207
6.3.3. Asymmetrical PCR 207
6.3.4. Nested PCR 208
6.3.5. Hot start PCR 208
6.3.6. Reverse-transcriptase (RT) PCR 211
6.3.7. Long and accurate (LA) PCR 212
6.3.8. In situ PCR 213
524
6.3.9. Allele-specific PCR 214
6.3.10. Methylation-specific PCR 218
6.3.11. Immuno-PCR 220
6.3.12. Quantitative (real-time) PCR 223
6.4. Amplification of sequences with unknown primary structure 233
6.4.1. Use of PCR for walking from a known region of the
genome into flanking unknown sequences (single-
sided PCR) 233
6.4.2. Reducing DNA complexity with PCR (RAPD method) 237
6.5. Total genome amplification methods 239
6.6. Alternative methods for nucleic acid amplification in vitro 241
6.6.1. Ligase chain reaction 241
6.6.2. Transcription-based isothermal amplification of
nucleic acids 243
6.6.3. Rolling circle amplification 250
Summary. The strategy for new gene isolation 251
Literature 254
Part II
PROTEIN ENGINEERING
Introduction 273
Chapter 1. Rational protein design and redesign 279
1.1. Designing proteins and enzymes 279
1.1.1. The energy requirements for new protein construction 279
1.1.2. Choosing a three-dimensional structure 281
1.1.3. Combinatorial problem of inverse protein design 281
1.1.4. Designing protein surface 282
1.1.5. Applications of rational design methods 282
1.1.6. Molecular fitness landscape in protein sequence space 284
1.2. Methods for site-directed mutagenesis 286
1.2.1. Generation of deletions and insertions 287
1.2.2. Oligonucleotide-based mutagenesis 290
1.2.3. Overlap extension PCR 294
1.2.4. Megaprimers in site-directed mutagenesis 298
1.2.5. Nonsense suppressors in site-directed mutagenesis 302
1.3. Chemo-enzymatic synthesis in establishing semisyn-
thetic polypeptides: expressed protein ligation 306
1.3.1. Combining chemical synthesis and enzymatic semisyn-
thesis in designing new polypeptides 307
1.3.2. Protein splicing and trans-splicing in peptide ligation 312
1.3.3. Applications of expressed protein ligation methods for
protein engineering 316
Chapter 2. Directed protein evolution 319
2.1. Combinatorial libraries of polynucleotide sequences 321
2.2. Random mutagenesis methods 323
525
2.2.1. Chemical mutagenesis 324
2.2.2. Error-prone DNA synthesis 326
2.2.3. Random shuffling of homologous and nonhomologous
gene regions 327
2.3. Screening and selection of proteins with required pro-
properties 332
2.3.1. Phage display 333
2.3.2. Cell-surface display 349
2.3.3. Ribosomal display and mRNA display 351
2.3.4. N-hybrid systems as a tools for protein research 355
Chapter 3. Advances in protein engineering 369
3.1. Chemical modifications of proteins 369
3.1.1. Artificial protein crosslinking 370
3.1.2. Optimization of enzymes for functioning in nonaqueous
solvents 372
3.1.3. Molecular imprinting 374
3.2. Hybrid proteins „ 375
3.2.1. Hybrid enzymes 376
3.2.2. Green fluorescent protein (GFP) and its analogs as a
reporters 382
3.2.3. Hybrid toxins 392
3.3. Extremozymes 397
3.3.1. Extremophiles and their enzymes 397
3.3.2. Advances of protein engineering in enzyme stability
enhancement 399
3.4. Antibody protein engineering 403
3.4.1. Monoclonal antibodies 403
3.4.2. Recombinant antibodies 409
3.4.3. Antibodies with enzymatic activity: abzymes 427
3.5. Peptide aptamers 433
3.6. Changing the enzyme substrate specificity 435
3.6.1. The problem of changing enzyme substrate specificity:
the wishes and reality 437
3.6.2. Restriction endonucleases 442
3.6.3. Changing the stereospecificity 444
Chapter 4. Combinatorial protein engineering in nature: xenobiosis? 447
Chapter 5. Protein engineering in search for polypeptides which
maximally meet the requirements of modern biotechnology 454
5.1. The optimization of biotechnological processes: the
paradigm shift 454
5.2. Modern technologies available for improving enzyme
characteristics 455
Literature 462
Object's index 484
English-Russian dictionary of used combination of words 511
Научное издание
Патрушев Лев Иванович
Искусственные
генетические
системы
Том 1
Генная и белковая
инженерия
Утверждено к печати
Ученым советом Института
биоорганической химии
им. академиков М.М. Шемякина и Ю.А. Овчинникова
Российской академии наук
Зав. редакцией Н.А. Степанова
Редактор Н.М. Александрова
Художник Е.А. Шевейко
Художественный редактор Ю.И. Духовская
Технический редактор О.В. Аредова
Корректоры А.Б. Васильев,
Р.В. Молоканова, ЕЛ. Сысоева
Подписано к печати 08.07.2004
Формат 60 х 90 l/i6- Гарнитура Тайме
Печать офсетная
Усл.печ.л. 33,0. Усл.кр.-отт. 33,3. Уч.-изд.л. 31,3
Тираж 1000 экз. Тип. зак. 3496
Издательство "Наука"
117997, Москва, Профсоюзная ул., 90
E-mail: secret@naukaran.ru
Internet: www.naukaran.ru
Отпечатано с готовых диапозитивов
в ГУП "Типография "Наука"
199034, Санкт-Петербург, 9 линия, 12
Patrushev L.I.
Artificial Genetic Systems / Shemyakin-Ovchinnikov Institute of
Bioorganic Chemistry Russian Academy of Science - Moscow: Nauka -
ISBN 5-02-033278-X
Vol. 1: Genetic and Protein Engineering / L.I. Patrushev; Ed. by
A.I. Miroshnikov. - Moscow: Nauka, 2004. - 526 p. - ISBN 5-02-032893-6
The first volume of monograph presents a comprehensive view of the modern techniques
employed in the genetic and protein engineering for construction of recombinant DN A and proteins.
In the beginning, basic principles and methods of genetic engineering are discussed, including DNA
cloning, preparation of genomic and cDNA libraries, as well as systems for their expression. The
second part of the monograph deals with principles of the techniques employed in the realization of
two modern approaches in protein engineering: the rational design and directed evolution of pro-
proteins. It embraces the key aspects of directed and random mutagenesis, expressed protein ligation,
cell-surface, phage and ribosomal displays, and so on.
For specialists in genetics, molecular biology and chemistry, postgraduates, and students.
LI. Pafrushev
Сознательное использование принципов, лежащих в основе природно-
природного переноса генов, позволило генным инженерам разработать аналогичные
более эффективные искусственные системы и приступить к беспрецедентным
по информативности исследованиям генетических явлений на молекулярном
уровне. По-видимому, 1970-е годы можно считать началом нового этапа эво-
эволюции биосферы Земли, все последствия которого мы в настоящее время
не в состоянии предвидеть.
Интерес к направленному изменению свойств ферментов не ограничи-
ограничивается запросами современной индустрии и медицины. Исследование
механизмов биокатализа и особенностей структурно-функциональных взаи-
взаимоотношений в белках имеет большое фундаментальное значение, поскольку
все это лежит в основе жизни. Говорить же о понимании природы фермента-
ферментативного катализа можно только после искусственного создания методами
белковой инженерии ферментов с заранее заданными свойствами.
ISBN 5-02-032893-6
9 785020 328938"