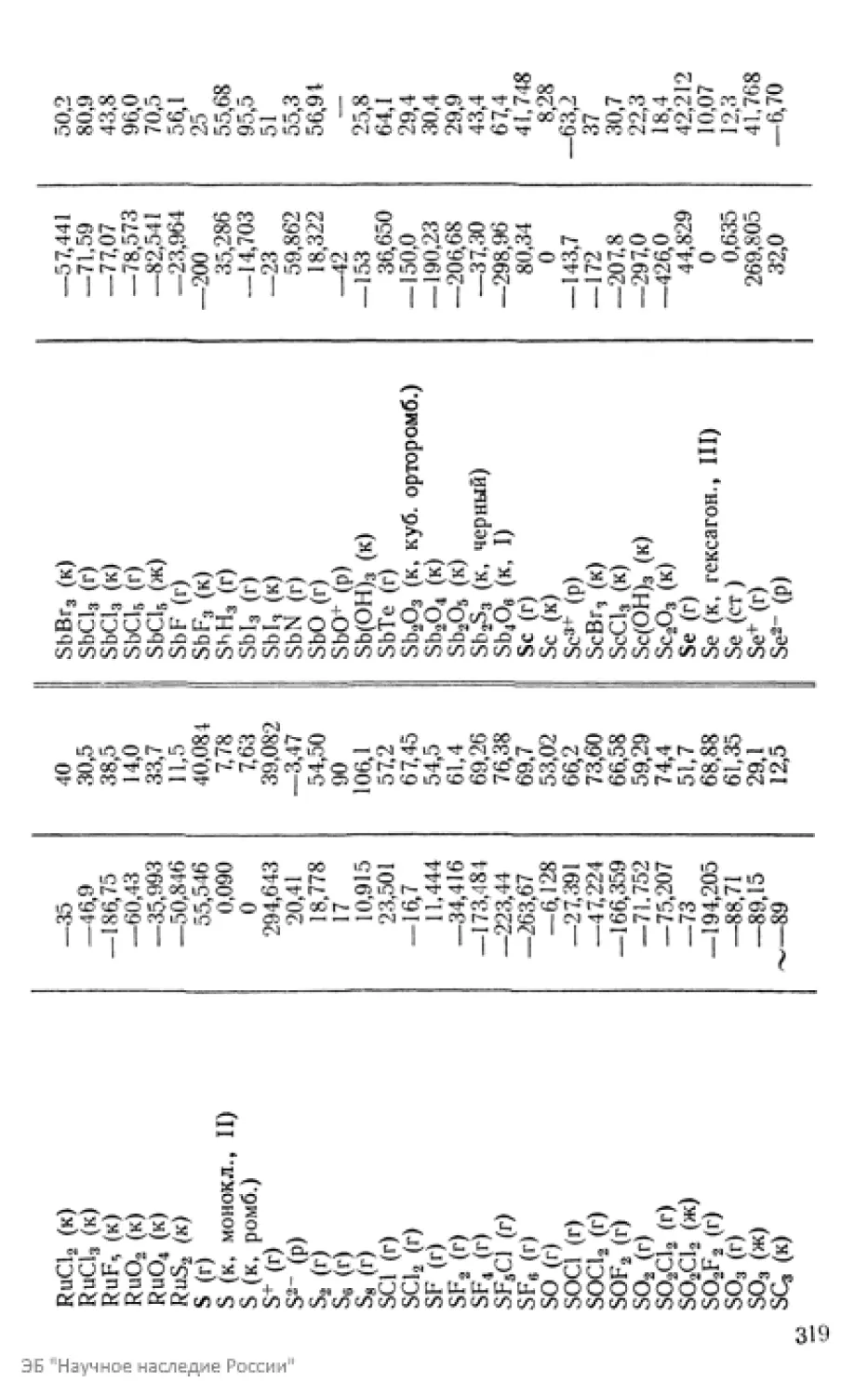
/

















































































































































































































































































































































Текст
М X • КАГАЛ ЕТЬЯ И Ц
ПЕРИОДИЧЕСКАЯ СИСТЕМ \ ЭЛЕМЕНТОВ Д.И. МЕНДЕЛЕЕВА
Периоды РЯДи Г РУППЕ’ Э Л Е М F. И То»
I 1 II 1 Ш_1 [V 1 V J {У 1 VII VIII
I 1 1 LOOTS? Ц 1 Водород «wig Н е 1ешк
II 2 J и1 6^4В Li Литий ».* 4 9Л>1?11 Ве 1*ри МИЙ t , 5 24*»/ ЮЛ В Lop »а*2ра * иг.ои С УглерМ Sa*}*1 7 ил»? N А Я 2^Р4 11^4 ° Киг.юро 1 9 2*3рдв 19.99140 Г Фтор ,.*м ” ».i7t N е Неон
III 3 И it1 n.wfJ Nji Натрий 12 3** М30У М в Магний . , ,J 34*ар’ 2ДОГ154 Al Алюминий 14 Э«»9ра 2МИЙ SI Кремний IS1 3*аЭ₽* 30.973?* !* ♦Мир 1* Зва0|>* Сера 17 За*Эр6 35.413 С1 Хлор 18 3a!Jp* J9.P48 А г Арсам
[V 4 19 4|1 лк К Кх.тий ... " Wjt С а К1ъикй 21 3d*4aa S С 44.955* (кли-хий 22 ad W Т 1 4Т.90 Титан В V Ю .04 ! 4 Вана дни 24 1 3d54*’ С Г 51491, Л’*” 25 М*4р» М П Ч.93Ю Мартане и 2« mW ге 5544, 27 МТ4а» С» М.9332 Кайвлат 28 3**41* N1 м.то Нике*
5 29 Си «5*в Мем Я М*4аа Zn *vt Цинк 4IJ4 ' 31 69.72 G а 1 а.ыик Л 71J, G с ['срчаиий 3J1 4аЛ4*> 74.9116 АД MlHnihAK я 4aJ4p* 7I.9a S е Сс.агк T9,904 В Г Бром iw Кг Криптон
V 6 У7 •t1 «.46/, К Ь |>енлмй -38 S Г Строений 3* 4rf яа2 Y Н.909 ; Игтрии 40 4d*9*a Z Г 9М1 Цирконий 41 4<АЛ Nb 92.МИЛ] Нмйин 42 ad’s*1 М о M<vm6 МП 43 4tf*M3 Тс 97 9062 Техткшт 44 ftf’sa 1 R U |ф|.0? Рутений * <А..| й Ь Ю2.9О55 . Родни 1 М Р d 106.4 | [111П ТНН
7 47 Л й »•Л6* Серебро 41 4d*S»? С d 112.40 Кадмий 49 So’Sp* 114 Д2 1П Индий 50 М»5р» S1 О.ЮМ ( я] •«V 121,7 j S Ь Сурьмя 52 ♦а1**4 d?j0 Те Темур » s и UejKH5 1 И«л | V 54 баЧр» кЛ.м X е Ксенон
VI 8 55 «а1 ш«я С« Цемй 54 84 s 137 В Я Бярнй 57 5<W 1 L а ндеоз^ Й Л а нта и - 72 5d*6a» ?! Hf 144, f Гафнии 73 3 J id в* * Та siomiJ Tamil 74 54*в«Л W 11М4 Вольфрам 75 Й4*Йа* R е ckjot Ретаи 76 Os ИОЛ Осмий 77 44*01* 1т 191Л2 Иридий 78 мЧт' Pl 195Д, Ила сииа
9 7» (id41)**' А и !«.«.« Ъ.ТйТй »| 541 веа3 Н S 1W0 Ргуть а , Я1 в!2^1 ИМЗт TI Та_гтмй S2 6вЛ0р3 м?,1 РЬ Синжи 8J ва*»?1 204.9904 В I 01К мм1 84 Ь»год* 4о9] Ро По I'jhhm. 85 '.’а,9 put A t Астат 86 •а*8р* 13221 ЙП Ратон
VII 10 «7 т*1 дея Г г Франции м 7а» 27£,ОН4 Ra Галий м 1 . W’ra’ А С (2П| j Актниий * ? 104 Г Ud’fi’) К U [2611 Курчатовий
Се Церий Min w Pr Ира илл им 140.90?? м л4в*а Nd Неодим M4J, *1 arW Pm Прометий 11*51 ** 4t4»s Sm Самарий 150.4 43 4/»а* Ен Lapoirtin HLW М 4ГТы'б»| Gd Гадали*^ К7Д, 4Г*41* |ь да.*: $* « .r’i,’ Dy Днспрогий М1Д( 67 4f"fla* Но Гольмий I64.9M4 и «’»,» Er M.i l»TJ. *’ 4/W Tn Тулий | 161.934 2 7® (4Гм)ва* Yb НТТГртНл I73J»4 Scr’e»* Lu Лю гений 174,9?
“А К Т И Ml Н О И Д Ы
w ad*?!*] Th 1оркй 3C.‘Hay4H(j 81 ^Чятв* Ра Протактинии е ^жУпед^е Рос ,2-^э«<гУ»* и > рл» СИИНЗд ^,3 Нептуний 2JT/HI2 М%а» Ри 11.1)1 опий |244| *5 Ат Ачерминй 12*31 « «Чг' j С m Кюрий П47[ ! В к |верк.1 им !bv 98 ($F«w4a’J Cf Калнфиримн 1«И м Ь”?.о Es JwHinribimi ?М| 18ft (b,’W) F» Фермии 12171 (w’b.’) M d M ttiie легший I2M] lW(4,“n»l (No! (Нобелий) 12Ж (Лоуренсии! 125*8
М. X. КАРАПЕТЬЯНЦ
ВВЕДЕНИЕ
В ТЕОРИЮ
ХИМИЧЕСКИХ
ПРОЦЕССОВ
Издание третье,
лерсработалное
л доноллепное
Допущено
Министерством высшего л среднего
спыщальвого обрйэлйазлзя СССР
и качиство учебного пособия
для студентов
к н мпко-те г но лопгч ескп я
слецна.пкяостей
высших учебных заведений
ф
МОСКВА «высшая школа* ia&f
ЭБ 'Научное наследие России"
ББК 24.1
К 21
УДК 54 + 546
Рецензент; Лроф. Н. н, Павлов
(Московский текстильный н петиту т)
Каря Ететья нц М, X.
К21 Введение в теорию химических процессов: Учеб,
пособие для вузов. — З-екэд., псрераб. и доп, — М.;
Высш, школа, 1981. — 333 с., ил.
В пер. 50 к.
В кяигЁ- излагаются обшне зэкпномср(гоет.н прогскапия х и м нч еск ИХ
реакций н с&П|^ьождающнк их процессор чц eprei ц к? Штцсгссв,
уч<цне л химическом сродстве. элементы учения о скорости и ныл-
ННЗ-МС реакций,. LJk?fiCT&;l pj цтНСцюв, Прльед^ни НрИмСрЫ и pH bl Г11£|| ня
оснив гермидшгзмнкч к химии ?лгч с-нтсп
Третье издание переработано и ДрООЛИСНа НОВЫМ матери алом
(2-е изла^не яыилр м 1975 r.J.
для er^tffKT-QO Гл Г4(#С1Г] &-
ц 6^№KNJr К hCijm tu2n! frHOTTt’ji.
, 20502—172 , A 540
К------------46—SJ iB02000000 celt oj i
00l(0])-8| ББК 24Л
© Издательство «Висшая шкйлй*> 1975
© Издательство * Вы сизая Еийола>4 ШI, с нэ.ч внемля мя
ЭБ ''Научное наследие России"
ПРЕДИСЛОВИЕ
Преподавание химии в вузе должно отличаться от школь-
ного не только широтой охвата материала, нои глубиной его
рассмотрения; вузовский курс должен быть курсом высшей
химии, читаемым на научной основе. Поэтому предшество-
вать курсу неорганической химии в вузе должны основопо-
лагающие сведения и прежде всего современные представ-
ления о строении вещества и главных закономерностях про-
цессов. Основное внимание при этом следует уделить во-
просам, позволяющим обобщить и объяснять материал по
составу и свойствам веществ; в то же время следует разумно
разгрузить описательную часть Курса.
Физико-химическое введение делает курс неор ганичсской
химии научно целенаправленным, дает возможность опи-
раться в изучении фактического материала на теоретические
обобщения, приблизить изложение предмета к современному
состоянию химической науки.
В природе и в искусственных условиях приходится
постоянно сталкиваться с самыми разнообразными процес-
сами, такими, как синтез, диссоциация, всевозможные
химические реакции, с процессами, в которых переплета-
ются признаки физических и химических явлении (напри-
мер, растворение) с физическими изменениями (например,
с плавлением веществ). Если к тому же учесть сложность
многих из них и разнообразие условий их протекания—
температуры, давления, концентрации веществ, то станет
очевидным, сколь непроста задача, даже хотя бы в общих
чертах, разобраться во всем этом многообразии, наметить
некоторые, пускай и приближенные, обобщения, охватываю-
щие тот или иной круг явлений.
Без знания строения атомов и молекул, природы химиче-
ской связи и межмолекулярного взаимодействия сделать
это невозможно. Однако эти сведения лишь необходимы,
ноне достаточны. Ведь свойства веществ познаются прежде
всего во взаимодействии с другими веществами. Поэтому,
приступая к изучению химии, нужно знать общие законо-
мерности протекания химических реакций и сопровождаю-
щих их процессов.
Одна из попыток решить первую из поставленных задач
была осуществлена изданием книги М. X. Карапетьянца и
1* 3
ЭБ ,:НауччоЕ наследие России11
С. И. Др а к it ня «Строение вещества* (3-е изд. М., Высшая
школа, 1978).
Б настоящем пособии сделана попытка наметить одно из
мыслимых решений второй задачи — изложить основные
закономерности химических превращений и сопутствующих
им процессов.
В первой его части рассмотрен Круг вопросов, связан-
ных с энергетикой процессов; во второй изложено учение
о химическом сродстве. Третья часть (одержит элементы
учения о скорости и механизме реакций. Свойствам раство-
ров посвящена четвертая часть, В пятой части приведены
некоторые примеры применения материала второй части
к химии элементов, имея в виду, что эти сведения могут
быть использованы и в описательной части курса неоргани-
ческой химии. Таким образом, круг рассмотренных вопро-
сов не совсем традиционен. Отличаются от общепринятых
и характер, и последовательность изложения материала.
Так, например, наряду с обычными примерами примене-
ния закона Гесса (часть первая) рассмотрено его использо-
вание в различных термохимических циклах, включающих
такие величины, как энергия ионизации, электронное срод-
ство, энергия решетки, теплота гидратации. Это позволяет
продемонстрировать студентам универсальность простого
метода расчета и уже с самого начала связать излагаемый
материал с вопросами строения вещества.
Во второй и третьей частях главное внимание уделено
химическому сродству. Разумеется, вопросы кинетики не
менее (а зачастую даже более) важны, чем вопросы статики
процессов. Однако если принять во внимание специфику
и большое разнообразие кинетических факторов, а также
огромную сложность учета их влияния на реакционную
способность веществ, изменение представлений о механизме
протекания процессов по мере углубления наших знаний к,
наконец, то обстоятельство, что большинство подлежащих
рассмотрению вопросов связано со статикой различных про-
цессов, то этот выбор вряд ли можно счесть спорным. Дей-
ствительно, и закон действующих масс, и принцип Ле Ша-
телье, и многие свойства растворов (в их числе раствори-
мость, температуры отвердевания и кипения, давление пара),
и процессы в них (диссоциация, нейтрализация, сольвата-
ция, комплексообразование, гидролиз и т. д.) — это прежде
всего проблемы равновесия. Вместе с тем надо отчетливо
показать, что вопросы статики к кинетики — это проблемы
возможности и действительности и что значение эцергетиче-
ЭЕ ' t-йуччов наследие России"
с кого (термодинамического) м кинетического факторов
неодинаково для различных типов процессов: для реакций
в растворах электролитов (например, при нейтрализации),
для высокотемпературных реакция и других «быстрых»
процессов кинетические соотношения несущественны; на-
оборот, для медленных реакций и таких, продукты которых
гораздо устойчивее исходных веществ (например, при горе-
нии), не и грают ощутимой роли равновесные соотношения.
Мы сочли необходимым ввести в курс понятия об энтро-
пии S н ее изменении AS и об изменении энергии Гиббса АО,
так как твердо уверены в том, что нельзя излагать химию
в вузе, опираясь только на понятие о тепловых эффектах
ДН. С другой стороны, мы отдавали себе отчет в том, что
на первом курсе информация о величинах AG и AS не мо-
жет быть ни полной, ни строгой; она в доступной форме
должна передавать лишь главное, давая общую ориенти-
ровку. Приучить студентов с первого курса пользоваться
энтальпийными и энтропийными характеристиками — это
означает не только привить нм навыки изучения с общих по-
зиций самых различных процессов (химическое взаимодей-
ствие, растворение и т, д.), но и подготовить их к постоян-
ному применению этих фундаментальных характеристик —
вначале на материале неорганической, а затем аналитиче-
ской к органической химии. В курсе физической химии эти
представления лат учат дальнейшее развитие, уточнение,
детализацию, будут поставлены на прочный математический
фундамент, Поэтому, в частности, при рассмотрении окис-
лительно-восстановительных реакций уделено внимание не
только составлению уравнений, т, е. чисто формальной сто-
роне, но и решению вопроса о направлении этих процессов,
о глубине их протекания,
В настоящее время все чаще приходится иметь делос про-
цессами, происходящими как в водных, так и в неводных
растворах, при низких и при повышенных температурах,
кроме того, изучать все возрастающее число реакций, про-
текающих при высоких и сверхвысоких температурах и дав-
лениях, в расплавах, газах. Поэтому ограничиваться рас-
смотрением электродных потенциалов £°, а иа их основа-
нии и э. д, с. £ означало бы искусственно сузить круг изу-
чаемых явлений совершенно частным (хотя н практически
важным) примером очень разбавленных раство-
ров в одном растворителе при одной температуре
(25 *С) иодном давлении. В связи с этим мы уделили
внимание н неводным растворам, и высокотемпературным
5
ЭБ ' На^ччоЕ наследие России"
процессам и сочли целесообразным прийти к величине Е
(£°) через Л<?.
В пособии скорость химических процессов рассматри-
вается после учения о химическом сродстве. Это отвечает
и логике исследования: пока не выяснен вопрос о принци-
пиальной осуществимости процесса, бессмысленно решать
кинетические задачи.
Вопросы строения вещества нашли отражение не только
при описании энергетики процессов, т. е. в первой части
пособия, но и в других его частях. Уделено большое внима-
ние и пер поди чес кой системе элементов Д. И. Менделеева,
К ней автор обращается и при рассмотрении тепловых
Эффектов процессов, и при исследовании реакционной спо-
собности веществ, и при анализе свойств растворов, и при
изложении некоторых вопросов, связанных с химией эле-
ментов (часть V*). Он старался показать, что периодическая
система важна не только для систематизации материала
химии, но и для плодотворного использования на ее основе
различных закономерностей н, в первую очередь, методов
сравнительного расчета.
Таковы основные принципы построения курса.
Настоящее пособие написано на основе многолетнего
опыта чтения лекций по курсу общей и неорганической хи-
мии в МХТИ им. Д. И, Менделеева и отражает стремление
к усилению в нем элементов термодинамики. Можно с удов-
летворением отметить, что идея тер моди нами зацнн курса
химия нашла поддержку как н практике преподавай ня в дру-
гих вузах, так и в ряде книг, вышедших в последние годы.
Автор полагает, что эта книга будет полезна и для сту-
дентов тех вузов, где читается лишь курс общей химии.
Физико-химическое введение в этот курс будет не только
содействовать повышению теоретическою уровня его из-
ложения, но и усилению связи с профилирующими дисцип-
линами.
Гл. V четвертого раздела этого пособия написана проф.
С, И. Дракиным; в составлении этой главы принял участие
докт. хим, наук В. А, Дроздов,
Автор выражает благодарность канд. техн, наук, доц.
М. Л. Каралетьяни за большую помощь в работе.
Автор
* См. также М. X. Кдрапетьнкц. Конспект лекций по неоргани-
ческий химии. Выгт. I, II, IJ1. М-, изд. МХТИ им. Д. И. Менделеева,
1973. 1974. J977.
ЭБ гНауччоЕ наследие России"
РАЗДЕЛ Т
ЭНЕРГЕТИКА ПРОЦЕССОВ
вгзгдкнгге
В любом процессе соблюдается закон сохранения энер-
гии', ему отвечает равенство
Q-АУ + Д (1.1)
которое означает, что если к системе (веществу или совокуп-
ности веществ) подводится теплота Q, то в общем случае
она расходуется на изменение ее внутренней энергии &U
и на совершение работы Д. Под внутренней энергией си-
стемы U подразумевается общий ее запас, включая энергию
поступательного и вращательного движения молекул, энер-
гию внутримолекулярных колебаний атомов и атомных
групп, энергию движения электронов в атомах, внутриядер-
ную э!1ергню и т. д. — словом, все виды энергии, кроме
кинетической и потенциальной энергии системы в целом.
Под величиной А имеют в виду работу против всех сил,
действующих на систему (внешнее давление, электриче-
ское поле, магнитное поле и т. д.).
Работу Д при переходе системы из состояния J в состо-
яние 2 удобно представить в виде суммы
г
А=*Д'+|МРь (1.2)
первый член которой Д' равен работе против всех сил, дей-
ствующих на систему, кроме внешнего давления р.
Если изучаемая система находится только под действием
последнего, то равенство (1<£) примет вид
а
A^jpdV. (1,3)
В частности» для изобарного процесса (р = const)
(L4)
где ДР — изменение объема. Для химической реакции вели-
чина ДР будет равна сумме объемов продуктов реакции за
ЗБ ’Научное наследие России"
вычетом суммы объемов исходных веществ- Так, для про-
цесса
о А ]-&В -р. l. e^a'D +гЕ . ч (I)
где VAl VB........ Уд, Ve ... — мольные объемы веществ
А, В...... D, Е ... .
В случае р const равенство (1.1) примет вид
<?P=^1+pV1}-(C/J + P^1)-
(f.5)
Рис. I. К пояснению
физического смысла
siiwuriMH (Н = U +
+ РИ
Обозначим сумму U + pV, стоящую в скобках, буквой
Н. Величина Н называется энтальпией (от греч. entab
реуа — нагреваться). Таким образом,
энтальпия определяется уравнением
n=~u^pV. (1.6)
Она подобно объему, давлению, тем*
пер а туре it внутренней энергии яв-
ляется свойством вещества, Смысл
этого свойства .можно пояснить сле-
дующим образом. Если газ находит-
ся в цилиндре, где он «заперт» не-
весомым поршнем (рис. I), то энер-
гия газа и поршня с грузом будет
равна и + psh, где s — сечение пор-
шня, а потенциальная энер-
гия поршня с грузом. Но psh = pV‘t
это дает основание рассматривать энтальпию как энергию
расширенной системы.
В соответствия с (1.6) для изобарного процесса имеем
(?р=ДЯ = Е^|1_£/Уис1. (].7)
Для изохорного процесса (Li) трансформируется в
О)
Г21Л1ГП I
ЭНЕРГЕТИЧЕСКИЕ ЭФФЕКТЫ
Химические процессы протекают либо с выделен if ем,
либо с поглощением теплоты: первые называются экзотер-
мическими (от грсч. ехо — вне, снаружи), вторые — эндо-
термическими (от греч. entlon— внутри). Так, реакция
Н-Н+С1—С1= Я—С! + Н—Cl (II)
6
ЭБ ,:Научнов наследие России"
является процессом экзотермическим: выигрыш энер-
гии» обусловленный образованием двух связей Н—Cl
(103,3 к к ал /моль), перекрывает расход энергии на разрыв
связей Н—Н (104,2 ккал/моль) к С1—0 (58,0 к кал/моль).
Действительно, 2< 103,3 >(104,2 4- 58,0).
Количество выделенной (или поглощенной) теплоты на-
зывают тепловым эффектом процесса *. Чтобы этой вели-
чине придать полную определенность, надо условиться об
ее знаке, выбрать единицы намерения, установить, к ка-
кому количеству вещества ее следует относяТь, и выбрать
режим протекания процесса Примем положительным теп-
ловой эффект эндотермических процессов; условимся выра-
жать его в килокалориях (ккал).
Энтальпия ЭЛТЯДЬГТИА
f ^пал
, * <1 '
Рис 2Г Эитальпийнаи диаграмма (схема):
4 — »ндйт&ринческнЯ процесс (Atf > fl); d —
}н?йТ£ряи чески fl прсдесс (Л Я < □)
Почти все процессы протекают или при постоянном объ-
еме, т. е. в закрытом сосуде (например, в автоклаве), или
при постоянном давлении, т, е. в открытом сосуде (напри-
мер, в колбе), причем подавляющее их большинство прово-
дится при р = const. ИзобарЕшй режим (как правило,
р = J атм) является типичным для лабораторного и про-
мышленного осуществления всевозможных процессов. Что
касается температуры, то предполагается,что она одина-
кова для всех реагентов (обычно при 25-С).
Таким образом, есть все основания остановить СВОЙ вы-
бор ня тепловом эффекте при р, Т = const, т. е, на разно-
сти энтальпий Д/У, характеризующей «теплоспособность»
системы (рис. 2). Вит почему принято говорить об измене-
* В дальнейшем ради краткости наряду с термином леллкзэой
эффект» (процесса) будет употребляться и термин (теплота» (процесса),
О
ЭБ ,:Научнов наследие России"
пии энтальпии при реакции (кратко — об энтальпии реак-
ции) как о синониме теплового эффекта.
Запись химической реакции k сопровождаемую указа-
нием теплового эффекта+ принято называть /гсермокияtwe*
ckxlm домдеядел'.
Сказанное целесообразно распространить па любые
процессы, т. е. не ограничиваться только химическими реак-
циями (см. с, 19 и ел.). Остается добавить одно— необхо-
димо сопровождать символы реагентов указанием их состо-
яния.
За п з юь пр оцесса (II), отвечающа я тер мох ими чес кому
уравнению
(г) + (г)» НС1 (г), dJ/ — ЙЙ кхал/ноль,
означает, что превращение
Рис. 3. Зависимость эепзлытии
fl г — ff0 (ыкал/молъ) свинца от
температуры 71 (К) при р =
= 1 атх:
&АПЛ “ темсгга (Ш1,1синч;
лЯпяр. — чплут* парчобрлм^.
кил; Гпл — vtune^typa
н н я। Гннл — Ttujf натура к и пенни
0Ф5 моль газообразного водорода
иОг5моль газообразного хло-
ра в I моль хлористого водо-
рода сопровождается выделе-
нием 22 иная теплоты. В со-
ответствии с законом сохра-
нения энергии тепловой эф-
фект этого процесса может
быть вычислен н так:
йН=я (^Н-н4"еС]-С1)я
(1.9)
т. е. Art равна алгебраиче-
ской сумме энергии образо-
вания соответствующих свя-
зей. Действительно, подста-
вив в (1.9) значения энергий
связи Н—Ci, IL и С1г, полу-
чим ДД —22 ккал/моль.
Следует иметь в виду, что
символом ДЯ принято обозна-
чать не только те г моты различных изобарно-изотермиче-
ских процессов (химических реакций Д/Л.р, фазовых пре-
вращений ДЯфл), но н изменение энтальпии в результате
изменения состояния данного объекта вслед-
ствие изменения какого-либо параметра — температуры,
давления, концентрации (например, в результате нагрева-
ния вещества). Так, из рис. 3 следует, что помимо ДЯфп
при данных р И Т, тг е. &Нр.т (ДЯП,„ А^п,р), приходится
иметь дело и с величинами ДЯ₽Г которые можно рассчи-
ЗБ 'IftyHHBB наследие России"
тать по уравнению
HTi-HT = f Vr- (1Л0)
г
где Ср—теплоемкость при постоянном давлении (изобар-
ная теплоемкость) *. Если же температурный интерна л
охватывает и изобарно-изотермический процесс, то (]. 10)
превращается в многочлен, нечетные члены которого отве-
чают изменению температуры h а четные — изменению состо-
яния. В примере, представленном па рис. 3^ получим
мю>&
^Г-500” j (Cp)pb <ю ^Г + (4Ч1л)рЪ +
5СЧ?
2024 2"ЮП
+ £ + )pb+ f (Ср)рЬ(г^Гь
ВДШ.5 2Й31
Раздел химии, в котором изучаются тепловые эффекты
различных процессов (образования веществ, их сгорания,
взаимодействия, растворения, плавления и т. д.). назы-
вается
Измерение тепловых эффектов производится и кдлеуш-
В простейшем случае калориметр представляет со-
бой сосуд, содержащий вещество с известной теплоемкостью
и ок ружейный оболочкой из плохо проводящего тепло мате-
рика. Если допустить отсутствие теплообмена между кало-
риметром и окружающей средой (для этого изучаемый про-
цесс нужно проводить как можно быстрее), то
ДА
где с — теплоемкость всех составных частей калориметра;
Д# — изменение темлературы.
Главв 11
ЗАКОН ГЕССА
Основной закон термохимии был сформулирован русским
ученым Г- И. Гессом (1840): если из данных исходных ве-
ществ можно различными способами получить заданные но-
* По определению, Сл = Интеграл (1.10} можно найтн
графически иЕ1тегрнроваи1^м зависимости Ср — f (fy
Что касается вел «чипы, отложенной по оси ординат рис. 3 (а также
рис, 4. 6—1 l)i то ее выбор обусловлен тем, что ^7_9к т* 0 (суще-
ствует T9 к ЕЕазывлемая гцмшя зчераыя, отличающаяся от Нуля); отне-
сение А к эЕЕтальпим при абсолютном нуле (Яв) не отражается на резуль-
тату выч!1сленняь так как начало отсчета при вы Чита и и ее исключается:
так, - Ац) - (ЯГ1 - = АГ( -
ЭБ ,:Научнов наследие России"
Л
печные продукты, то независимо от путей получения (напри-
мер, от вида промежуточных продуктов) суммарный тепло-
вой эффект будет одним н тем же, иными словами, таимой
эффект зависит только от вида и состояния исходных ее-
имеете и конечных продуктов, но не зависит от пути пере-
хода. Смысл этого положения покажем на простом примере
получения двуокиси углерода из графита:
С (графит)-|-7/4 (г) «СО (г), Д/Л
CO(r)+J/A(H-COi(r), ЛЯ»
С (графитЦ- О* (г)" СО2 (г), Л/7
Представим эти реакции следующей схемой:
С (графит)—-------—'^1 (О
ЛЯ| -СО (г)
Очевидна,
ДН =1 ДЯ^ДЯ,
или
ДЯ1+ДЯЖ-АЯ)-0. (1.11)
Это означает, что если все три процесса удовлетворяют тре-
бованию Г1|С( = 7’К<И1 ирт( = рКОи1 то независимо от того,
сгорает графит сразу в СО„ или сначала а СО, а затем СО
в COj, Тепловой эффект будет одним и тем же. Из (Т.Н),
в частности, следует, что
Л/Л=Л//—Л/?г.
Измерив А/7 и Л//4т можно рассчитать величину А//|}
которую опытным путем найти сложно (при сгорании С
в общем случае получается смесь СО н СОа, и каким именно
образом наблюдаемое выделение теплоты распределяется
между СО и СО,, решить трудно). Обобщая этот пример,
получаем
Е Afit = 0 = А1Ц + ЛЯ, + ДЯ5 4-...+ (-ЛЯ) I
или } (1.!2)
&tf-AHl+Ani+&H3+„, 1
Другим примерам использования закона Гесса может
служить равенство (1,9).
Из закона Гесса, который является одним из следствий
закона сохранения энергии, вытекает ряд выводов, Наибо-
лее важны из них два: 1) тепловой эффект реакции равен
сумме теплот образования (AH0(Sp) продуктов реакции за
12
ЭБ ,:Научнов наследие России"
вычетом суммы тбПАот образования исходных вешрсте\
2) тепловой эффект реакции равен сумме те плот сгорания
(Atfe ) исходных веществ за вычетом суммы теп лот ссора-
пая продуктов реакции. При алгебраическом суммировании
следует учитывать стехиометрические коэффициенты.
Так, для реакции (i)
(L13J
или
^=Р(л^₽)а + &Р^^)в+-]-
-Щ^горЬ+Чл^в+Ч. (TJ4>
Первое следствие имеет общее значение, второе важно
для органических соединений.
Закон Гесса и его следствия не могут быть использованы,
если мы не условимся, какой смысл вкладывать в понятия
теплота образования и теплота сгорания вещества.
Под теплотой образования обычно понимают тепловой
эффект образования I моля вещества из простых веществ,
устойчивых при 25 °C н I атм (графит, ромбическая сера,
белый фосфор, жидкий бром, белое олово, Кристаллический
йод и т. д.), Под теплотой сгорания обычно- подразумевают
тепловой зф(])ект сгорания 1 моля вещества до СО4 (г) и
НаО (ж); для остальных элементов в каждом случае ука-
зываются продукты окисления.
Таким образом, если в реакции (1) участвуют простые
вещества, то в уравнение ([.13) не войдут теплоты их обра-
зования, так как они по определению равны пулю; если же
реагентами служат негорючие вещества, то в уравнении
(1.14) не будут содержаться их теплоты сгорания, которые
по определению равны нулю. Так, для реакции
CHjCOOH -1. Ct НЬОН = CHjCOOC, H4 + H(O (111)
в соответствии с (1.13) и (1.14) имеем
= [(Л//ов p)ci1iCOOCfHl+ (Д^овр)н,о]~
- [(д^р)сн,соои + <й^6р)^^ОН]
или
Л//= (Д^егоР)сН,СООН + (Л^Р£ф)с,Н,ОЦ “ (^^crepJcH.COO^Hр
поскольку (АЯСГ0р)н,о = О,
Стандартные тепловые эффекты. Значения АН зависят
и от природы веществ, и от условий их существования, я
13
ЭБ ,:Научнов наследие России"
от характера рассматриваемого процесса. Поэтому их при-
нято унифицировать, относя к так называемому стандарт'
ному состоянию. Под последним при данной температуре
(чаще всего при 25 еС = 298,16 К) подразумевают: для кри-
сталлических и жидких веществ (конденсированные фазы)
их устойчивое состояние при р = 1 атм, а для газов их со-
стояние под атмосферным давлением и при условии, что они
обладают свойствами, присущими им при очень низких дав-
лениях, т, е. являются идеальными газами. Последнее уточ-
нение необходимо, так как при р = 1 атм каждый газ в той
или иной — различной! — степени отклоняется от уравне-
ния Клапейрона—.Менделеева, а это отклонение может ока-
заться существенным (особенно для тех веществ, которые
при данной температуре и атмосферном давлении близки
к конденсации). Для вещества в растворе принимают его
концентрацию С, = I моль на литр, причем предполагается,
что раствор ведет себя, как и при весьма небольших (точнее
при бесконечно малых) концентрациях, т, е. является иде-
альным. Отнесение &Н при р = 1 (С, = I) к идеально-
газовому или бесконечному разбавленному состоянию при-
водит к единообразной системе величин и тем самым к воз-
можности их сочетания. Значения Д Н при указанных
условиях получили название стандартных тепловых эффек-
тов. Их принято обозначать как &Н'г. Наиболее употреби-
тельной является характеристика при комнатной темпера-
туре, т. е. if/,,,, (нижний индекс отвечает округленному
значению Т = 298,16 К). Унификация важна и потому,
что разница в значениях i//j- (ДЯ^,) образования двух
веществ отражает лишь различия в их природе.
Значения стандартных тепловых эффектов приведены
в табл 1,2, 3, 4, а также их можно найти в различных спра-
вочниках, где они Объединены по виду веществ и характери-
стикам процессов. Такие таблицы упрощают расчеты, так
как путем комбинирования нескольких сотен известных
величин можею получить значения ДЯ0 для десятков тысяч
реакции. К сожалению, эти данные скудны, анх достовер-
ность подчас сом еги тел в ня. Так, если для единичных ве-
ществ (к их числу относятся, например, Н2О и СОг) точ-
ность значеЕЕий ДЯ«Н отвечает погрею пости порядка ±0,1
и меньше, то для большинства изученных объектов она со-
ставляет ±1, достигая ДЛЯ многих ±10 ккал/моль и даже
более значительных величин. Еще более скудны н менее
достоверны данные по высокотемпературным стандартным
системам,
ЭЕ тйучшэв наследие России"
Значения H'itt (ДНт) представляют собой систему
взаимно согласованных величин. Поэтому замена в справоч-
ных таблицах ранее известных значении вновь найденными
для единичных веществ в принципе ошибочна, даже если
последние гораздо точнее первых. Такая замена допустима
лишь при условии, что одновременно пересматриваются
значения H‘iVI (Д/fr) для всех веществ, связанных с тем
веществом, для которого эта замена произведена. Так,
уточнен fie стандартной энталь-
пия образования а-кварца по-
требовало пересчета Л//^ для
образования всех силикатов, си-
лицидов, кремни йор панических
соединений и других веществ,
содержащих кремний. Естествен-
но, что расхождение (A/7i»0siO,,
скажем, в 10 ккал .'моль может
привести для сложных сили-
катов к погрешности порядка
100 ккал/моль и более.
Влияние на тепл овин эффект
температуры к давления. Как
быть в тех случаях, когда надо
знать тепловой эффект процесса
при иных температурах и дав-
лениях? Н первом приближении
KOiri; Г= W» К
к
ЙО
15
10
5
jNHrHOyri;r=jO0QK
О
Рис. 4. Энтальпийная диа-
грамма для расчета дли ян» я
температуры на тепловой
эффект реакции
можно принять, что изменения VaNB (0+7/3^ (r)—NO(r)
температуры и давления мало
отражаются на величине Д/Д Небольшую чувствитель-
ность А// к изменен ню Г (обусловленную возрастанием
энтальпии всех реагентов с нагреванием) можно показать
на примере реакции
С (графи г) 4- СОг (г) =. 2СО (г) {IV)
для которой — 41,5, а Д/fJw = 39,5 ккалт т. е. изме-
нение температуры Еза 1000 °C изменяет тепловой эффект
всего на 2 ккал, 11а рис. 4 приведен другой пример.
Разница энтальпий исходных веществ при 298 и 4000 К
(1,6 ккал) почтя не отличается от этой величины для продук-
тов реакции (1,1 ккал).
Следует, однако, иметь в виду, что в общем случае при
большом перепаде температур нельзя принять, что ДЯ
= const; в ряде процессов (в частности, для реакций в расъ
ворах) изменение А// с температурой может бить значитель-
15
ЭБ ' Изучим наследие России"
ним и в узком интервале температур. В общем случае оно
будет тем больше, чем больше сумма теплоемкостей продук-
тов реакции отличается от Суммы теплоемкостей исходных
веществ (см, также с. 19).
Действительно, строго говоря, так как
в соответствии с (1.10)
Г Г Г j
ЛЯГ1- &nTt - f 2 Г S <9««4Г= f iCpdT
Vs *E *J
или, в частности+
т
&H'r-Wnt+( iCpdT.
где ДСР (АСр) — алгебраическая сумма теплоемкостей
(стандартных теплоемкостей) реагентов, взятая с учетом стег
хнеметрических коэффициентов. Так, для реакции (1)
^“H(^D + e(C₽}E + ...]-[e(Cp)A+ft(Cp)0+...]
Однако уменьшаемое и вычитаемое в последнем равенстве
часто мало отличаются друг от друга, т. е.
Еще меньше влияние давления; к тому же па практике
возможный интервал давлений обычно сравнительно неве-
лик. Так, для реакции
1/tN1(r)+V1H1(r)-NHt(r) (V)
различие между A//P=|fl™ и ДЯр=#о«атч составляет всего
окало 5 %.
Гл-qiea Ш
ТЕПЛОВЫЕ ЭФФЕКТЫ РАЗЛИЧНЫХ ПРОЦЕССОВ
1. Химические реляции
Тепловые эффекты, как правило, велики и колеблются
в широких пределах. Теплоты образования веществ обычно
составляют величины порядка 10—100 к кал/моль, сравни-
тельно редко снижаясь до 1 и достигая 1000 ккал и более.
* Допущение Att* ~ const == 0) тем Сол ее спрэ&еинво,
чем ближе к нулю нэяетне числа мелей газообразных реагентов
(Дл£ р). Так, Для М различных случайно взятых двух-г трех- и четырех-
атомных веществ- изменение их стандартных тел лот образован ня к нН’
тервалеЭДЙ—10OQK составляет в среднем 0,35 ккал/моль [Д =
t= 0J7 ккал/моль|,
16
ЭБ ,:Научнов наследие России"
Теплоты сгорания, как правило, больше те плот образова-
ния и обычно превышают 100 ккал. Если теплоты сгорания
всех веществ отрицательны, то теплоты образования отри-
цательны лишь для большинства соединений. Известны
вещества, образование которых связано с поглощением тел-
лоты.
Эндотерм ич ними сред 1 г неорганических соединений яв-
ляются гидриды неметаллов (СилаВы, бораны и др.), оксиды
азота н хлора, нитриды, карбиды, цианиды, соединения
золота н некоторые другие вещества; средн органических
соединений —это многие углеводороды-
Т е плоты р еа к ци й сои змер и мы с те пл отам и обр азова ни я *.
Термохимические уравнения
As (к) -р */jClj (г) ж* АзС!□ (ж) „ A ff Jn #« — 753 ккал;
С3Н4 (г) + 3V А (Г) = 200f frj + ЗНЯО {ж)> ₽ —372,80 к^л;
1/А (г) + V А (г)»NO(r), =21 г 57 ккал
н
SOi (г) + 2HjS ft) = 35 (ронб) + 2Н±О (ж). Д = -55.67 к кал
подтверждают изложенное
В табл. 1 приведены зЕтачения стандартных те пл от обра-
зования, а в табл. 2 — стандартных тепло? сгорания неко-
торых веществ.
С помощью подобных таблиц можно осуществлять разно-
образные расчеты. Так, из тзбл, 1-находим разность теплот
образования Н2О (г) к HtO (ж); она отвечает теплоте паро-
образования воды Д//°гн = 10,519 ккал/моль,
К чисто химическим реакциям можно отнести и процесс
распада ионных кристаллов на газообразные ионы, напри-
мер;
GF (к) »Cs+ {г) + F’ (г), АП = 180,5 ккал/«ол^
Тепловой эффект такого рода процессов, как иэвсстею. назы-
вается й ррше/ти. Приходится изу-
чать и обратные процессы, а также реакции диссоциации мо-
лекул (радикалов) на атомы или ионы, К последним принад-
лежат процессы тала
Nj (г) = 2N (г). Д/У = —224.8 ккал/маль.
• Поэтому п тех случаях, когда для расчета искомого теплового
аффекта можно поспальзовап>ся обоими следствиями нт закона Гесса
(сми с. 13). предпочтительное пользоваться первым из них; это приво-
Дит к кеиыией логрешЕюсти шчислений.
17
ЭБ ,:Научнов наследие России'1
Таблица t
Стандартные теплоты образования Л7Л,Ч (нивл/моль)
некоторых веществ и нонен
Пещ^сгмз ДЯт R^mtCTBO
АйВг (к) —24,06 HjO (г) -57.7Й6
AfeCO, (К) —120,96 И Jo (ж) Н£ (г) —6 Ь.315
А1Д (к, а) —400,5 —5,0
AsCb (ж) BaSO, (я) —75,4 HgO (к, красная) -21.72
-ЗЗДЙ L1+ (г) 163J0
С (алмаз) 0,437 LiCl (к) —97,о8
С (графит} 0 N (г) Н2Д«4
СН* (г} —17.88 NO (г} 21,5?
00 (Г) Na (г) 25J4
СОо (г) -44,052 Na4- (г) 144,45
сЛ (Г) СйО, (к) 54,02 NaBr (к) —Ш>Л8
—312н42 NaCl (к) -S8.26
CdO (к} -41,9 NaF (К) —137, J
CI (г) 28Ж1 Na! {к> - 69,4#
01- (г) —54337 О (г) 59,556
Cs+ (г) О, (г) 34,0
GsF (К1 ^132н7 он - и —32,154
CuSO4 (р) -20L73 SO, (г) SrSO, (к) -70,96
F (г) 19,003 —350,82
F" (г) -К2.066 TiN (к) -S1.1
FeA (к) -196,5 XcF4 (к) -60
Н (г) 52.098 ZnO (х) —83,80
Н- (г) 367. (63 ZnSOj (р) —254,45
HCI (г) -22а06
Таблица S
Стандартные теплоты сгорания Л7/^ва (ккал/моль}
некоторых веществ ’
13 сЩсСгво АН BcUicCtbo дн;?4
СН| (г) —212,80 сна, (ж) —10^31
GHn (г) -372,80 С13ЯОН (ж) —173,45
С,Н, {г) —530,ЙО НСООН (ж) - 60.6?
w<JIin (г} —687,55 СН/2ОСН1 (ж) -427.8
изг1‘С1Н|0 (г) C,HS (г) -685,03 CJIbCOOH (к) -772,2
—310,44 C]qHs (к) -123L8
QH, (г) —337,27 C|jHinO|i (к) COS (г) -1318,90
QA (ж) —780,98 -154,76
18
ЭБ 'Научное наследие России"
К простейшим химическим процессам можно отнести и
отрыв электрона от атома, например
Na (г) = Мл" (г) 4-е", I [8.7 ккал/иоль,
и его присоединение к атому, в частности,
F(r)-|-c“ = F“ <г). &f! = —&!,[ ккал/моль
Они отвечают соответственно энергии ионизации (первому
потенциалу ионизации натрия Inb) и энергии электронного
сродства (фтора £F)-
2. Фазовые превращения
Тепловые эффекты перехода из одного агрегатного состо-
яния в Другое Д/Уфр обычно значительно меньше таковых
для химических процессов В частности, теплоты парообра-
зования ДЯ|1Л> (прн I атм) составляют величины порядка
10 (реже нескольких десятков) к кал/моль, теплоты плавле-
ния AtfnT, перехода из ^морфного состояния в кристалли-
ческое и Преврасцелпя одном модификации в другую — по-
рядка 1—5 ккал/моль (см. рис. 3). Эти величины для ряда
веществ приведены в табл. 3. Из нее видно, что лишь тогда,
когда температура фазового превращения под атмосфер-
ным давлением сильно отличается от комнатной (например,
для Ag), различием в теплотах парообразования, а поэтому
и сублимации, т. е. непосредственного перехода из кри-
сталлического Состояния в газообразное, минуя жидкую
фазу, нельзя пренебречь. Велики они и для тугоплавких
(бысококипящнх) веществ. Так, для W (р = 1 атм)
184 ккал/моль.
3. Процессы о растворах
Рассмотрим взаимодействие водных растворов КВ г и
Ag\O3. Его можно свести к процессу:
(р) 4 Вт (р) — Ag Вт (к), Л // = —20,27 кх ал/молъ
и тем самым считать, что тепловой эффект реакции не за-
висит от природы аннона соли серебра н катиона бромида.
Это допущение справедливо при условии, ЧТО оба реагента
пол]।остью диссоциированы; последнее обеспечивается до
статочным разбавлением растворов (символ «р» и означает
весьма большое разбавление *). Подобным образом теплота
* Часто прнмеЕ1яют сиииол «aqi.
19
ЭБ ,:Научнов наследие России"
нейтрализации сильной кислоты сильным основанием не за-
висит ОТ их природы — в Любом случае взаимодействие сво-
дится к реакции
Е Г (р)4-он- (р) = Н,0 (ж), Дй;„ = —13.335 ккшфпль» (VI)
Иное дело — слабая кислота или (и) слабое основание.
В этом случае теплота нейтрализации меньше, так как иони-
зация кислоты (основания) требует Затраты энергии *.
Таблица 3
Стандартные теплоты фазовых превращений
(А#*3,)ф. 1Г (ккал/мпль) некоторых веществ*
Внцество
(iJJ ГТ-
(АЯин^флг
Вещество
Парообразование (АЯП4ф)
Ай
AsFa
Вга
60.1 (2410 К)
В,4В
I по
1 г-й (эз1 к)
10Л2
9J7 (373 К}
нр (
Плавление (ЛЯ^)
2JI (37DJ5 К)
ОЛЗ (145 К)
5,59
АШг,
BF&
so3
Сублимация (АЯсуй^)
BECI,
т!
27?6&
ЧЯ2
43Д1
Переход из аморфного
состояния н кристаллическое (Л//кр)
Ву,Оз
HaSeO4 НаО
Sb
Те
-6,0
-43
- 2Л1
-2Р7
Полиморфное превращение
ASjOi (октаэдр }^
-* мпиокд.
С (графит-к алмаз)
Sr (серое -н белое)
14
0,437
ОрО
* 3 скобках >'явз-&ны вмя чан к н т&илер агурц ? тех елу чаях н К-ОГда ли*
АТлнчввтся от 25* Сг
Из этих примеров ясно, что термах ими чес к не расчеты
для реакций в растворах целесообразно проводить не по
Теплотам образования нейтральных молекул, а по те,мотам
образования ионов. Измерить их не представляется возмож-
ным, так как нонам одного знака всегда сопутствуют НОИЫ
противоположного знака. Однако подобно тому как для
всех элементарных веществ величина теплоты образования
условно считается равной нулю (с. 13), так и дли ио-
• Запись Н* (pj является условной; см. с. 177*
ЭБ "Н4^нм наследие России"
нов можно ввести начало отсчета, приняв ДЯо5р одного из
них за нуль. Нулевой условились считать стандартную теп-
лоту образования нона Н+ (р). Это допущение, не отражаясь
на правильности результатов вычисления (так как во все
расчеты входят разности величин), вместе с тем поз-
ВОляет создать систему значений ДЯовр ионов. Так, сочетая
уравнения (VI) и
Hs (г) 4-1/4О (г) = Н 8О (Ж), = — 68,32 X мл/ыод ь,
находим
Н1 (г) + VA (г) — Н+ (р) 4- ОН - (р), длг;а ™ —54,98 кхад/мозъ,
но согласно условию
поэтому
VsHa (г) -f-coH,O = Н+ (р) 4- с, ЛH‘ni=0.
W I (г) 4- ’/А (О + ®Н:О(ж) . ОН- (р);
AWa« = —54,98 кхал/моль.
Это н будет &Но6р нона OFI- (р). Подобным образом можно
найти ДЯо|(р других ионов. Для некоторых водных ионов
эти величины приведены в табл, 4.
Таблица 4
Стандартные теплоты обрааоаанил (ккал,'моль)
некоторых ВОДНЫХ ИОНОЙ
WflH [|<JH AA/10S
Ag* ЗД23 LI* —456^5
Ег- 2S.O2 М£=+ —110Л6
ОО*‘ -I6L84 Ка+ —5X46
С[- —39,91 -19r57
Cj‘* 16,00 он- —54^8
F- —79,79 SOJ- —2i 7J3
Н+ (W Zn*+ -36J2
К+ —Й0^9
Теплоты растворения ДЯ», сравнительно невелики и
обычно составляют величину порядка 10 ккал. Они менее
чувствительны к природе веществ, чем теплоты химических
процессов. В табл. 5 приведены значения АЯр;к„ некото-
рых веществ. Эти величи ееы отвечают процессу растворения
I моля данного вещества в определенном количестве раство-
рителя {п моль), указанном в табл, 5.
ЭБ ,:Н-аучнов наследие России"
21
В свяли с этим следует обратить внимание Е<а зависимость
Affpann: от концентрации раствора. Так, если
HjSQt (ж) 4- 6Н2О (ж) = HjSQ* 6HtO, ЛН;^=—14,51 к кал/коль,
то
И jSOj (ж)+coHsP (ж) =« HjSOi (р), АН = — 23,15 кк а л/но ль.
Влияние концентрации на теплоту растворения серной кис-
лоты иллюстрируется рис, 5. Об энергетических эффектах
,ик*Л
^2343____
Рне. S. Зависимость теплоты раство-
рения H^SOj (ж) в Н2О (ж) при 25 °C
от числа молен воды лИ|О
в растворах см, также с. 144 и ел,, 149 н ел., 155 и сл.ф
180 н сл.
Таблица 5
Теплити растворения ЛЯрпети (кнал.'мадь)
некоторые веществ в воде при 25е С
Л + н НЯО (ж)ыД в я 11^0 (ж)
Вещество п iWJHCTe Веществ п Л Триста
енрн (Ж) С(Х (г) СаСЪ (к) CaCL (1Й2О (х) FeCl, (г) НВг (г) НС1 [г) QP 3600 [00 400 1000 со OD —0.37 —4,72 -17,00 4,311 -31,64 —20,35 ~-17рвЙ ИГ (г) НиВО3 (к) NEI, (г) NiltCl (к) NaCI (к) NaOH (к) N^SOi 10Н2О (к) 8 88 8Ц1 -14,5 3,21 -11,15 3.53 1.21 — 10J0 Ь6 76
Глйнв IV
ПРИМЕРЫ ПРИМЕНЕНИЯ ЗАКОНА ГЕССА
Располагая значениями тепловых эффектов образова-
ния. сгорания, растворения, парообразования и т. д.,
можно с помощью закона Гесса рассчитать теплоты самых
ЭЕ Н Яннов наследие России"
разнообразных процессов, в частности таких, эксперимен-
тальное изучение которых затруднительно ил it вообще не-
возможно ь Несколько иллюстраций было при неделе ранее.
Ниже рассмотрен еще ряд примеров. Ради единообразия
и наглядности все они представлены в виде далпфн/шйншг
Ладгролс — «энергетических лестниц* (см, рис. 2)т причем
с соблюпением маешта-
If яд,тир, атоцъ!
ба (см. рис, 4). Преж-
де чем обратиться к
нимФ приведем в схема-
тическом виде диаграм-
му уровней энтальпий
в некоторых из рассмот-
ренных соуояний (рис.
6; он отвечает экзотер-
мичности как процесса
образования соединения
из простых веществ, так
и процесса его раство-
рения).
Энергия связи. Для
расчета энергии связи
Е надо знать теплоту
образования газообраз-
ного соединения из га-
зообразных атомов. Эта
величина называется
ЛЯдЛ1;с
и
Продукты сгорания
Ртгс\ 6
Энтальпийная диаграмма Для
личных состояний fvXd-Мй)
аяю.илряой тяшоята? образования (A//aTJ- Для ее опреде-
ления нужно знать теплоты диссоциации простых веществ
и теплоту образования соединения. В рассматриваемом слу-
чае (рис. 7) в соответствии с уравнением (1.12) получаем
+ -{AHodp)Hl0(r) =
±= ([04 ,1 96 -|- оа5 119. i 12) - (-57,796) = 23 L 5*8 ккал/имь,
откуда
*0-н “G’S 07?4г)]— 110,8 кмал.
Теплота гидратации. Расчет теплоты гидратации может
быть осуществлен на основании термохимических циклов,
изображенных на рис 8 л 9; для металлов (рис. 8) надо знать
теплоты сублимации ионизации / = Д//^, и гид-
ратации A/Zjiup газообразных катионов; для неметаллов
(рис. 9) — теплоту диссоциации ДЯШС, электронное срод-
стио Е == АЕВ>1 и теплоту гидратации А//г,Г1р газообразных
анионов.
23
ЭБ ,:Научнов наследие России"
Различие результатов расчета теплот гидратации для Zn
и Си (рис. 8) объясняется следующим. При примерно одина-
ковых энергиях ионизации и энергиях гидратации катионов
(что обусловлено близостью их радиусов) медь — битее туго-
плавкий металл, чем цинк, поэтому
(4йсУ«а)си > (Д^<у«л)гп*
В результате если процесс перехода кристаллического цинка
в состояние иона в водном растворе является экзотермиче-
ским (ДЯда = —36,72 ккал), то аналогичный процесс
для меди эндотерм и чен
(А//™ = 16,00 ккал).
Теплоты гидратации F,
и С12 одинаковы по знаку,
но первая по абсолютной
величине больше второй
(рис. 9), так как при близ-
ких значениях сродства к
электрону теплота гидра-
тации нона F" значительно
больше, чем иона Ci- (ска-
зывается различие в их
радиусах). Расхождение
было бы меньше, если бы
переход от хлора к фтору
(подобно переходу от иода
к брому и от брома к хло-
ру) сопровождался ростом
теплоты диссоциации. Как известно, это не имеет места:
в то время как в молекуле С12 кратная связь вследствие
образования двух дополнительных донорно-акцепторных
связей Зр — 3d, в молекуле Fj связь одинарная (у атома
фтора нет валентных d-орбиталей).
Расчет теплоты гидратации стехиометрической смеси
ионов может быть произведен по энергии кристаллической
решетки U и теплоте растворения соединения. Так, для слу-
чая, представленного на рис. 10, а,
- (iffrHjpJuci" 205 -(—8) = 213 ккал.
УровеЕгь энергии для гидратированных ионов может лежать
и выше первоначального; не исключена возможность и почти
полного совпадения с ним (например, для случая NaCl).
Энергия ионной решетки. Для расчета энергии крЕгстал-
лической решетки часто пользуются термохимическим цнк-
ЭБ кучнее наследие России"
ff-Яо. хма
200Н
100
о
2Н(т)+О<г)
ffp ксс )о, ।
-1 гмц&т.
| Н;<г)т|о3(г]
_ j HjOTri
-100 ; —
Рис. 7. Энтальпийная диаграмма
для расчета атомарной теплоты об-
разования HSO
a
fl
Рис. 8, Энтальпий Era я диаграмма ллл распе-
та теплоты процесса
Ме₽ кЕаО« Мел+ (р) -р лй“
<i — u,hilxf й — кедъ (п “
Рнс+ 5+ ЭЕЕта.тьгшЙная диаграмма для рае*
чета теплоты процесса
VaXj (г) +®НВО + ле" = X*- (р)
а — ЧлОр; б -- фтор (п = I)
ЭБ ' Научное наследие России"
лом, известным как цикл Борна — Габера. На рнс, 10, б
этот цикл приведен для NaCI, т. е. представлена сле-
дующая совокупность процессов: переход Na из кри-
сталлического состояния в состояние одноатомного
газа (35,74 ккял /моль), диссоциация молекул Cij на
свободные атомы (0,5 х 57,986 ккал/моль), иониза-
ция атомов Na (118,71 к кал/моль), переход атомов СI
в ионы СГ (—84,83 ккал/моль), соединение ИОНОВ Na*
и Cl' с образованием кристаллов NaCI (—189 ккал/моль).
200
кийц
юс-
5<?
МвЧгНСНт}
Хь*. г; ч-СГ i= I
Z |Nakij-air;.
". - — I^ChfrF
- I -(* 1 Nta
-50 - tf**!--1 TJ*C1
-waE.-----------------
4
!£=r(i₽M )qi
о
Pur, 10. Энтальпийная диаграмма ддя расчета:
4з — те л л 01 ы гидратации £Д!Ггид- сто ли о нетрн чеекоД емвен конов;: б
энергия нри ста л л ил d-с кой решитки С-1 х.порлдэ л а трин (цикл Еорлэ — Гябгря)
Последняя величина равна и противоположна по знаку
величине энергии кристаллической решетки NaCI, По
табл, 1 сразу находим ОмаС1 = 144,45 + (—54,837) —
— (—98,26) = 187,9 ккал/моль.
Обобщив этот цикл на нонепяс кристаллы типа МеХ,
получим
U Me X = “ 0flobp) М« X + (ДУСMe + 'А Идиес) X, +
+ (йЯМЯ1ИЯЫе-0*„)х.
Естественно, что в каждом из рассмотренных примеров
соответствующими термохимическими циклами можно вос-
пользоваться для нахождения любой величины, если из-
вестны остальные (а не только тех величин, которые отве-
чают подряду ночным подписям и отмечены жирными стрел-
ЭЕ Йтучнее наследие России"
ками). Так, по те платам раствор etui я и гидратации можно
вычислить энергию кристаллической решетки U веще-
ства (рис. 10, я), никл Борна — Габера (рис. 10, й) исполь-
зовать для вычисления электронного сродства и т. д.
Глава V
НЕКОТОРЫЕ ЗЛКОНОМЕРНОСТИ
1. Теплоты сходных процессов
В ряду сходных веществ величины Л/У меняются законо-
мерно (см., например, табл. 1 и 2). Поэтому закономерно
изменяются и тепловые эффекты аналогичных процессов.
Так, представление о закономерном изменении тепл от
образования и сгораЕзия можно получить, проследив его,
например, для гомологических рядов органических соеди-
нении (СН*, ОН4, СаНл, CjHl0, ,..) или в ряду соединений,
сгруппированных по какому-либо другому признаку. В част-
ности, можно установить, что (Дпсйр) в гомологиче-
ском ряду для высших членов ряда растет линейно с увели-
чением молекулярной массы соединения.
Тепловые эффекты н периодический закон. Для неорга-
нических соединений тепловые эффекты однотипных про-
цессов являются периодической функцией порядкового но-
мера соответствующего элемента. Для физических превра-
щений это показано на рис. 11, для химических — на рис. 12.
Рис. 12 как бы распадается на несколько областей: зона
острых и абсолютных пиков (хлориды s-элементов), область
сглажеЕ1НЫХ и меньших максимумов (хлориды р* и d-элемен-
тов) н область сравнительно небольшого изменения значе-
ний (хлориды /-элементов). Господствующие пики
занимают хлориды щелочных металлов, меньшие пики —
ZnCij и CdCI*. Надо иметь в виду, что для многих соединений
значения Л/?ы>р неизвестны или определены лишь для газо-
образЕгого и жидкого состояний. Кроме того, не всегда из-
вестны значения соединений, в которых элемент
находится в степени окисления, отвечающей номеру группы
или близкой к нему. Увеличение степени окисления приво-
дит к уменьшению грамм-эквивалентной AHo6fi (это видно
на примере UO„). Наконец, надо учитывать и различия
в характере связи, координации и т, д. Тем не менее, рнс. 12
не только дает общую картину периодичности, но и свиде-
тельствует об определенных закономерностях в изменении
хлоридов. В частности, мысленно соединив точки для
27
ЭБ 'Науччое наследие России"
ЭБ ,:Научнов наследие России1
ЗБ "Научное наследие России1
Рис. 12. Зависимость стандартной теплоты образования (ккал/моль) хлоридов от порядкового номера
Элемента Z
хлорилов элементов III группы, мы убедились бы в более
закономерном изменении ’значений Д//пСр в ряду ВС1я“
—A!CIS—ScCI3— YCI4— LaCIs—АсС1э по сравнению с рядом
BCIS—AlCl^GaCI^-hiCls—Т1С1а и тем самым в проявле-
нии в последнем вторичной периодичности. Подобного рода
графики можно построить и для других веществ (бромиды,
оксиды, сульфиды н т. д.).
Рис. 13- Взаимосвязь между стандартными теплотамее реакций
обратна км я:
а — ялорндоя элемента я нгг тертого (1>, пятого ([[) периодов (Ккэл/моль)
н tf — тс плотями реЛкцнй й//’иН (ккдл/моль)
Э (ОН), (1С)+СО, (г) =ЭСО, (К) + HjO (ж) (Г)
и
2ЭОН (к) + СО, (г}= Э^СОя (к)+ Нр (ж) (I I)
для элементов гтсд групп йеряялня я лития
Пример закономерности. Количественная закономер-
ность прослеживается в коде изменения теплот образова-
ния в двух рядах 1 и Н подобных соединений. Аналогичный
ход значений &.И в fuix часто приводит к линейным соотно-
шениям вида
Л\Н, + В, (1,15)
соответствующим одному из методов сравнительного рас-
чета— сопоставлению при данных условиях значений од-
ного свойства в двух рядах сходных веществ (процессов).
Соотношение (1.15) иллюстрируется рис. 13, а. Оно справед-
ливо и для более сложных процессов, например для диссо-
циации карбонатов н титанатов элементов подгруппы берил-
лия, и подтверждено данным!! (рис. 13, 6), свидетельствую-
щими о сходстве элементов подгрупп бериллия и лития.
чное наследие России"
ЭЕ
2. Теплоты образования it структура
Сопостав-геиие экспериментальных и теоретических зна-
чений те пл от образования веществ в ряде случаев позволяет
сделать tie которые выводы об их структуре. Рассмотрим не-
сколько примеров.
По теплоте образования (или сгорания) С4Н]Й можно
решить, является ли изучаемый препарат нормальным
бутаном или изобутаном (см. табл. 2).
Приняв для молекулы озона треугольное строение
,ОХ
Oz X). по величине энергии связи £0-0 = 35 ккал полу-
чаем теплоту атомизации ДЯ„ОМ1|| s + 105 ккал; но опыт
дает для озона Д//обр = 34,0 к к ал /моль, чему отвечает значе-
ние AtftTOlini =* +145 ккал. Следовательно, молекуле озона
нельзя приписать треугольную структуру. Ее геометрия
такова;
О
0/Х()
Это отвечает и другим экспериментальным фактам, в част-
ности и тому, что дипольный момент молекулы О., не равен
нулю.
Сравнения вычисленных значений цикланов СЛН^
(л 3) с экспериментальными показывают, что, лишь на-
чиная с циклопентана (« =5), значения практически сов-
падают друг с другом; для п = 3 и 4 экспериментальные
величины больше теоретических. Из этого следует
два вывода: I) молекулы циклопропана я циклобута па на-
ходятся в напряженном состоянии и поэтому неустойчивы
(углы С—С—С, не отвечающие тетраэдру, равны соответ-
ственно &0 н 90е}; 2) у высших цикланов (л 6) напряжение
снимается, так как за счет ненлоского строения сохраняются
углы 109*.
И еще один пример. Наряду с соединениями постоянного
состава (характеризующимися целочисленным!г стехиомет-
рическими коэффициентами), ДЛЯ которых справедливы за-
коны постоянства состава н Кратных отношений, сущест-
вуют соединен и Я переменного состава (многие оксиды, суль-
фиды, карбиды, нитриды я т. д ). Так, карбид циркония
имеет состав не ZrC (в соответствии с местом элементов-
партнеров в периодической системе элементов), a ZrCj-j,
где аг н границах области непрерывного изменения состава
меняется в широких пределах. К подобным выводам можно
31
ЭБ ,:Научнов наследие России"
прийти не только на основании изучения структуры, но и
в результате термохимических исследований, так как в со
ответствии с непрерывным изменением состава будет непре-
рывно меняться и теплота образования таких веществ.
Риг, Ы. Зввисммть стан-
дартной теплоты образова-
нии (ккал/моль) нит-
рида титана Т1Г4Л от вели-
чины х
На примере Д//обр нитрида титана это иллюстрирует
рис. 14. Из него непосредственно следует, что в интервале
однородности (л = 0,45 + 0,98; опытные дан!1ые)
—4 Я, ~ 17,84-59.4*
и что Д //tin = —80 ккал /моль, Разложение нитрида ти-
тана должно описываться не уравнением
TiN=T| + VtN1
а уравнением
Т1 K =
ЭБ 'Научное наследие России"
РАЗДЕЛ ГТ
ХИМИЧЕСКОЕ СРОДСТВО
ВНЕДЕ1ШЕ
Химика интересует не только энергетика процессов,
в части ости теплота образования (разложения) данного ве-
щества или теплота его взаимодействия с другими вещест-
вами, но и реакционная способность различных веществ.
Например, карбид алюминия не распадается, но не терпит
соприкосновения с водой, образуя метан СН4 и гидроксид
алюминия А1 (ОН)Я. Почему это так?
Для решения вопроса о возможности протекания реак-
ции недостаточно обладать «химической интуицией», не-
обходим количественный критерии п р и и ц и п и а л ь-
ной осуществимости процесса, С его помощью можно
определить, насколько далеко и л ст процесс; нельзя ли до-
биться (и как это сделать) увеличения степени превращения;
если данное вещество пе реакционноспособно, то можно ли
создать условия, при которых оно может взаимодействовать
с другими веществами; как влияют на течение процесса тем-
пература, давление, разбавление инертным газом, варьиро-
вание концентрации реагентов; можно ли заставить изучае-
мую реакцию протекать в обратном направлении и т. д„
и т, п,
Глава г
НЕОБРАТИМЫЕ П ОБРАТИМЫЕ ПРОЦЕССЫ
Изучение химических реакции Приводит к следующему
выводу: наряду с процессами, которые, начиная протекать
в одном направлении, затем идут в обоих направлениях
(за счет взаимодействия продуктов реакции), т. е. являются
двусторонними, встречаются и такие, которые протекают
практически односторонне, до полного исчерпания исход-
ных веществ. Первые процессы — К НИМ относится подав-
ляющее большинство реакций — принято называть химиче-
ски обратимыми, вторые—их меньше — химически необ-
ратимыми.
2 М- X Клраи гт ьянц 33
ЭБ ,:Научнов наследие России"
Хотя теоретически необратимый процесс можно пред-
ставить протекающим В определенных условиях обратимо,
Т. е. в принципе можно считать вес реакции обратимыми.
однако химику нередко приходится встречаться с процес-
сами, в которых преобладает реакция, идущая в одном на-
правленны. Эго бывает и в тех случаях, когда продукты
взаимодействия удаляются из сферы реакции (выпадение
осадка, выделение газа, образование, например, в случае
ионных реакций практически неднссоцнированпых продук-
тов) или же когда за счет огромного избытка исходных ве-
ществ противоположный процесс практически подавляется.
Таким образом, естественное или искусственное исключен не
Рнс. 15. Механическая
модель — аналог химиче-
ского равиоиссия
возможности обратной реакции по-
зволяет довести процесс практиче-
ски ДО конца.
Примерами могут служить: вза-
имодействие растворов хлорида
бария с сульфатом натрия, броми-
да меди с а мм и а Ком, нейтрализа-
ция сапяной кислоты раствором
едкого натра. Это все примеры
лишь практически необра-
тимых процессов, так как я BaSO4
несколько растворим, и комплекс-
ный катион [Cu(NHa)4P не абсолютно устойчив, и Н2О
немного диссоциирует. К совершенно необратимым про-
цессам относятся реакции
КСЮа = KCI + 1,50fe w_p|j = Pb 4-3Nt
В обычных условиях невозможно осуществить образование
бертолетовой соли иэ хлорида калия и кислорода, а также
синтез азида свинца из свинца и азота.
Равновесие. В результате обратимого процесса насту-
пает истинное (устойчивое) равновесие. Оно характери-
зуется следующими признаками;
а) при отсутствии внешних воздействий состояние си-
стемы остается неизменным во времени;
б) система следует за изменением внешних воздействий,
сколь малы бы они ни были. Перемена направления воздей-
ствия вызывает перемену направления изменения в системе.
Если внешнее воздействие снимается, система вновь возвра-
щается в исходное состояние (подробнее см, с. 78 и сл.).
Механическая модель на рис, 15 иллюстрирует аналогич-
ное равновесие; небачыное усилие, приложенное к ни-
зе 'нЗ^нов наследие России"
ли imp у, сместит его из изображенного положения (где его
энергия минимальна). Степень смещения определяется
величиной силы, а направление смещения — направлением
ее действия. Если воздействие прекратится, цилиндр вер-
нется в псрвоЕшчалыюе положение * *. Таким образом,
между действием и его результатом существует кол и ч е-
стае иная связь;
в) состояние системы будет одинаковым независимо
от того, с какой стороны она подходит к равновесию (в ме-
ханической системе независимо от того, слева или справа
скатывается цилиндр).
Примером системы, находящейся в истинном химическом
равновесии, может служить эквимолекулярная смесь COiR
СО и Н.О над катализатором при 810 еС. Это соотноше-
ние между реагентами сохраняется сколь угодно долго.
Повышение температуры сместит равновесие реакции
CO-f-HjpaCQi+Ht (I)
влево; охлаждение вызовет сдвиг равновесия вправо. (Эти
см ei цен и я окажутся тем значительнее, чем больше будет
отличаться температура от 810 *С.) ** Если же восстано-
вить первоначальную температуру, то система вернется в ис-
ходное состояние.
От истинного следует отличать кажущееся равновесие,
которое называют также заторможенны.» или ложным.
В сущности это состоя и нс нельзя называть равновесным,
так как оно только по одному признаку тождественно истин-
ному равновесию—по неизменности состояния во вре-
мени.
Тан, может существовать сколь угодно долго (& отсут-
ствие возмущающих факторов) и гремучая смесь (Ht с О2).
и термит (смесь Fe,Oac Al) н условиях, когда каждая из этих
пар веществ реакционноспособна. Взаимодействия не про-
исходит потому, что на пути процесса существуют апреЕтят-
ствегя» (см. часть III). Но достаточно в гремучую смесь вве-
сти платинированный асбест, а термит поджечь, как нач-
нется энергичное взаимодействие. Происходят односторон-
ние процессы
(г)+VA М - И (И)
и
F?A (к) -f 2А ((к) = АIА (к) 4- ЕГе (к)
• Аналогия был» бы полной, сели бы скатывающийся цилиндр
□становился сразу.
*• О влиянии температуры на я ими чесное равновесие см, £> 79.
2* 35
ЭБ ,:Научнов наследие России"
сопровождающиеся большим выдел с ни ем теплоты (соответ-
ственно 68,32 п 204,0 ккал). Катализатор в первом случае
и небольшой подвод теплоты — во втором приводят к ко-
лоссальному эффекту; инициаторы процесса как бы про-
буждают дремлющие силы.
Эти н нм подобные примеры приводят к общим выводам:
а) неизменность системы (вещества) во времени явля-
ется недостаточным условием для суждения о том, нахо-
дится лп система в истинном или кажущемся равновесии;
б) при заторможенном равновесии нет количественного
соответствия между величиной действия, вызывающего про-
цесс, и его результатом;
в) в результате «размораживания» заторможенного рав-
новесия в системе чаще всего происходит односторонний
(необратимый) процесс.
Представление о ложном равновесии, возможно, и ле за-
служивало бы того, чтобы на нем останавливаться, если бы
нс одно обстоятельство. Мы живем в основном в мире таких
равновесий; ведь нас окружают главным образом твердые
вещества, течение процессов в которых резко затормо-
жено медленностью диффузии в них.
Глава 1Т
ЭНТРОПИЯ
Состояние любой совокупности частиei можно охаракте-
ризовать двояко: 1) указать значения непосредственно изме-
ряемых свойств вещества, таких, например, как его темпе-
ратура к давление; это характеристика макросостояния
вещества; 2) указать мгновенные характеристики каждой
частицы вещества — ее положения п пространстве, скорости
и направления перемещения; это характеристика микросо-
стояния вещества. Так как тела состоят из огромного числа
частиц, то данному макросостоянию отвечает колоссальное
число различных мпкросостояпнГц при неизменном состоя-
нии вещества (например, его температуры) и положение
частиц, и скорость их движения в результате их перемеще-
ния претерпевают непрерывные изменения.
Число микросостояний, с помощью которых осуществля-
ется данное макросостояние вещества, называется вероят-
ностью его состояния (<и). Таким образом, величина ю есть
число различных способов реализации данного состояния
вещества.
36
ЭБ ,:Научнов наследие России"
Введем еще одно свойство вещества — его энтропию S
(от [ реч trope — превращение).
Эта величина определяется формулой Больцмана:
S = -j^-]n<u, (II.I)
где R— универсальная газовая постоянная, A'rt— число
Авогадро Учитывая, что R = 1,987 кал/моль-К, и умно-
жая обе части (11.1) на Afe, т. е. относя величину 5 (как мы
делали Для V, А// и Других величин) К I НОЛЬ вещества
(а не к | молекуле), находим, что энтропия выражается
в кал/моль'К; часто пользуются и условным названием
энтропийная единица (э, е.): 1 э. е. = 1 кал/моль-К,
Для оценки порядка величины S надо знать, чему равны
пели чины т. Последние колоссальны. Даже для совокупно’
ста из 10 частиц га порядка 10*. Но нам приходится иметь
дело с обьектами, содержащими 10й частиц (ведь 1010 ато-
мов железа весят всего 10-12 г); для них « огромны. Однако
если согласна (11.1) произвести лога рифм нрава fine, то полу-
чатся сравнительно небольшие величины — порядка десят-
ков (реже сотен) энтропийных единиц.
1. Энтропия — мера неупорядоченности
Пусть какое-либо твердое вещество (например, кри-
сталл CsF) находится при данных температуре и давлении.
Ионы Cs* и Р, расположенные в узлах кристаллической ре-
шетки, совершают колебания около равновесных положе-
ний. Представим себе, что мы могли бы так быстро «фотогра-
фировать» этот кристалл, что фиксировали бы мгновенные
положения колеблющихся ионов Тогда количество неодина-
ковых фотоснимков равнялось бы а. В условиях более низ-
кой температуры амплитуда колебаний этих частиц умень-
шится, сократится и число несовпадающих фотоснимков;
величина <t> станет меньше. Допустим, что наш кристалл
взят при Т — 10 К; тогда ш станет совсем незначительной.
Наконец, если бы мы достигли Т = 0 К, то обнаружили бы,
что ионы авмерзлн» в узлы решетки, г. е. вариации в осу-
ществлении данного состояния были бы исключены, иными
словами, ш — I, В соответствии с уравнением (II I) это
значит, что при Т = 0 К и S = 0. Мысленно осуществив
обратный процесс, приходим к выводу, что нагревание вы-
зовет возрастание энтропии.
37
ЭБ ,:Научнов наследие России"
Для веществ, структура которых не характеризуется
дальним порядком (аморфные тела, твердые растворы),
очевидно, 5лЛ >0. При Т = 0 К энтропию, ОТЛиЧнуЮ от
нуля, имеют и И.,, СО, Н4О, NSO и некоторые другие веще-
ства, что вызвано различными причинами. Так, для СО
это объясняется псу порядочен [{ОСТЬЮ кристаллической ре-
шетки, обусловленной случайным расположением концов
соседних молекул. Возможны два варианта:
СО ОС СО ОС и СО СО СО СО
Если бы аба он и были равновероятны, то в соответствии
с уравнением (II. 1) Sr,0 = 7? In 2 = 1,38 э. е. В действи-
тельности Sr-# = 1,12 э, е. Расхождение обусловлено тем,
что молекулы несколько чаще располагаются первым спо-
собом. Незначительность этого расхождения свидетель-
ствует и О близости свойств углеродного и кислородного
концов молекул СО — результат сходства этих атомов (в со-
ответствии с расположением этих элементов в периодиче-
ской системе элементов Д. И. Менделеева).
Изложенное означает, что энтропия — мера неупорядо-
ченности состояния системы. Действительно, опа растет
нс только с повышением температуры, но и при плавлении
(и сублимации) твердого вещества, при кипении жидкости,
словом, при переходе вещества из состояния с меньшей
энергией в состояние с большей энергией (рис. 16). Сопро-
вождаются ростом энтропии, и процессы расширения (на-
пример, газа) и растворения кристаллов, и химическое
взаимодействие, протекающее с увеличением объема (напри-
мер, диссоциация соединения)} когда вследствие ргхтга
числа частиц неупорядоченность возрастает. Наоборот, все
процессы, связанные с увеличением упорядоченности,—
охлаждение, олвердеванне, конденсация, сжатие, кристал-
лизация из растворов, химическая реакция, протекающая
с уменьшением объема (например, полимеризация), — со-
провождаются уменьшением энтропии.
Возрастание энтропии вещества при повышении темпе-
ратуры иллюстрируется рис. 16 (на том же примере, что и
рис. 3).
Влияние давления на энтропию можно показать на та-
ком примере: если ггры Т = 500 К и р — I атм Snhj =
= 50,7 э. е., то при Т = 500 К и р = 300 атм Snh, =
— 35.0 э. е.
Порядок величины Д5 часто можно ориентировочно оце-
нить по знаку и величине изменения объема Д1 в процессе.
3$
ЭБ ,:Научнов наследие Рогсии"
Последнюю же легко определить по знаку изменения числа
молен газообразных реагентов Ан’, так как в первом при-
ближении объемом негазообразных реагентов по сравнению
с объемом газообразных реагентов можно пренебречь.
Действительно, если, на пример, 1 моль твердого вещества
занимает незначительный, I
обычно несколько больший
объем, то объем ! моля газа
примерно в тысячу раз боль-
ше. Надо также учесть, что
при высоких температурах
объемы твердых и жидких
тел возрастают незначитель-
но, тогда как газообразных
веществ — очень сильно. Та-
ким образом несоизмеримость
объемов газов и твердых
(жидких) веществ очевидна.
Поэтому при реакциях и кон-
денсированных системах (т.
с, не содержащих газообраз-
ных реагентов), например с
процессе
ЗМп (к) + С (грлфйт)= Мл/! (к)
можно считать AS 0 (дейст-
вительно, AS,”ino = “0,9 э, е.), Тот же результат получится
и когда Ацг = 0, например для реакции
С(графвт)+0^(г) = СО»(г)
(ASinon = 0,3 э. е.) *,
Иное дело— процесс растворения. Хотя при растворении
твердых веществ (и жидкостей) в жидкостях суммарный
объем растворяемого вещества и растворителя почти не от-
личается от объема раствора, т. е. хотя в этом процессе
А V 0, однако в нем AS =/= 0 Действительно, процесс рас-
творения твердого вещества означает распространение его
молекул в объеме, который в десятки, сотни, а то и в тысячи
раз больше его собственного объема. Поэтому процесс рас-
творения В соответствии с уравнением (11,2) может сопро-
вождаться значительным изменением энтропии (подробнее
см. с. 146 н сл.).
* Даже в гея случаях, когда ДЗ 0 (хотя. Д V — 0}, энгролиниый
вклад в величину Лб часги не является решающим.
Рис, 16. Зависимость энтропии
S (з. с.) свинца аг температуры
Т (К) при р = 1 атм.
Д^гл — нзменемня энтропии при
плавленым (2.0 э с L —
изменение ?итраП]И1 njnk п-нрообр?-
зсваинн |?1.1 э с). Г|РЯ течпе-
ритурв плз fwit-н н и №40,5 К.), /кнгт—
темпервтура К ИпеняЯ (3021
39
ЭБ ,:Научнов наследие России"
Если исходные реагенты негазообразны, а продукты
процесса содержат газы, то AV >0 и AS >0. Примером
служат реакции диссоциации различных твердых веществ
(кар Со на то а, сульфидов и т- Д-). Действительно, для реак-
ции СаСО3 = СаО + СО, (Анг = i) ASw = 88,47 э, е.
В процессах, я которых 0, очевидно, н AS 0, Это
утверждение относится, например, к реакции
СН4 (г) 4-со, (Г) = 2СО (г) + 211, (Г)
для которой при Т = 1300 К величина AS3 = 68 э, е.
Наоборот, если АУ< 0, то, как правило, л Д$ < 0;
например в процессе
С (графит}-И 2НЯ (г}₽= СН( (г)
в котором Ап1 — —1, ASjuu = “25,3 э. е.
Однако было бы неправильным полагать, что для любой
реакции изменение энтропии определяется только величи-
ной изменения объема. Это справедливо лишь тогда, когда
реагенты близки по структуре, и поэтому не может сказаться
чувствительность энтропии к особенностям строения. Так,
в процессе изомеризации, например при превращении бу-
тана в изобутан, A V = 0 (Длг = 0), однако AS Q. Воз-
растание «упорядоченности:» в этом процессе (изобутан —
более симметричная молекула, чемм-бутан) приводит к тому,
что в данном случае AS < 0.
Стандартные эн троп it it. Чтобы энтропии веществ были
сравнимы (для сопоставления и определения изменения
энтропии в различных процессах, в том числе и химических
реакциях), нх, Как и Тепловые .эффекты, принято относить
к определенным условиям. Чаще всего значения S берут
при t = 25 X и р " I атм; при этом газы считают идеаль-
ными, а для растворов (и ионов в растворах} принимают их
состояние при концентрации, равной единице, предполагая,
что раствор обладает свойствами бесконечно разбавленного
раствора, Э1 [Троп if я при этих условиях обозначается Si,s
и называется стандартной энтропией (см, табл, 6 и прило-
жение I).
Наряду со значениями Sgw пользуются и величинами Sy.
Их тоже называют стандартными, однако высокотемпера-
турные значения известны для сравнительно небольшого
числа веществ.
Отрицательные значения некоторых ионов не должны
смущать читателей; это означает лишь, что их энтропия
меньше (5мь)ц*1р(, принятой равной! нулю.
40
ЭБ ,:Научнов наследие России"
Таблица 6
Сгамдаруиы? энтропии (?. ej некоторых
веществ is минор
Вещество ! Вещество Sd*B
Ай1 (Р) 1736 Ft (Г) 48,45
Ар-СОя (к; 40.0 Н- (р) 0
Efer. (х> -5G И, (г) 31J9S
сн4 (Г) шз Hto (г) <106
СО’ • (?} -И.Д95 H,Q (ж) IRL75
Catlj (г) <00 он (Р) —2.60
QHufr), 1 .генсек ш,Аа I РЬ (г) <889
Q11,5 (г); ц>1к,югсксан 71-28 РЪ (к) 15,49
СеИц(г). и-гексан 02ЛЗ РЪ (ж) 17,14
Ct (р) 13Д1 WCIa (к) ^31,1
С12 (г) 53^293 WCI* (к) -Л&.4
С«! (к) 7,923 WClfl IK) «0,70
Си4' (Р) -23J65 Zu {К, р} 9,95
Г <Р> —3X1 ZtP- (pl —26,434
Zr (0 <315
Хотя, как видно из табл. 6 и приложения [, энтропии
различных веществ меняются в широких пределах, однако
в первом приближении можно принять, что это двузнач-
ные величины. Оценим п связи с этим ориентировочно мас-
штаб и. Осуществим это на примере водорода. В Соответ-
ствии с уравнением (11.1) имеем
« = ^/* = 10
31,195-6,023 I02’ 1ПН
1,937 2,303 = ,v *
Согласитесь с тем, что это умопомрачительная величина.
2. Энтропия и природа вещестна
Так же, как и в случае ДЯг (ЛЛ/ИН), различие в значе-
ниях Si (Зи») отражает лишь различие в свойствах веществ.
Это позволяет привести ряд примеров, свидетельствующих
о связи энтропия с природой вещества.
Усложнение молекулы приводит к возрастанию энтро-
пии. Таи, = 38,467; (S;9S)O, = 49,005; (S»i)o, =
= 57,08 э. е.; если у атомарного кислорода возможно
только поступательное движение частиц, то у молекул кис-
лорода — и поступательное, и вращательное, и колебатель-
ное движение, а у угловых молекул озона набор вращатель-
ных и колебательных движений увеличивается; это оз на-
41
ЭБ 'Научное наследие России"
чает, что «о, а поэтому и So, > So* > So.
Л на,ior и чп о имеем: (Si«)so* (61,35) > (S1,„)SO1 (59,29) >
(53,02). Этот вывод справедлив для жидких и
кристаллических веществ:
WCIt (к) WClj (к) WCIj (к)
s;„, э. е......... ~3i,i — 49,4 60,70
Чем тверже вещество, тем меньше его энтропия. Если при
Т - 500 К S₽b = 21,9 э. е. (см. рис. !6h то Sfr = 11,1.
Sec атм aj = 2 э. е. То же относится и к соединениям: так,
если (Ss»)cdt. = 56. то (SjW)cb* — 6,48 э. е
Наименьшей энтропией обладает алмаз (S^ = 0,566
э, е.), что естественно, так как это самое твердое простое
вещество.
В аморфном и стеклообразном состояниях энтропия
больше, чем в ирис галл и чес ком. Так. S$rt для МайВ*О?(к)
равна 45,3, а для Ka3S.jO, (ст) 48,3 э. е. Другой пример:
(S«ih)a!{Oh^{bm) = 19,8, а ^ц^дцонык* = 16,75 э. е.
Энтропия чувствительна и к степени дисперсности веще-
ства. Например, для кристаллов Ni (ОН)4 имеем;
Раз мор. гм . . ....t<H J(b* 10*
Э-С..............19,10 19,2 19.5
Вещество в растворе, как правило, обладает большей
энтропией, чем в чистом состоя ими, так как при растворении
обычно преобладают процессы разу поря дочивання:
ZnCIg (к) ZrC12 (Щ
s;„. ?. е. , . 26,64 эш
Обратимся теперь к органическим соединениям.
В пределах данного гомологического ряда S^ возра-
стает, причем, начиная примерное четверто го-пятого члена,
ее увеличение становится практически неизменным:
j (г) ! 2 .3 4 5 б 7
э. е. 4-1,53 54,85 64,51 74,12 83,40 92.83 102,77
ю5Г" "адз-'
Повышение кратности связи приводит к падению 5»я.
Так, переход в последовательности метановые — этил ено
вые — ацетиленовые углеводороды сопровождается умень-
шением S«k:
4-C,Htn (г) 1-СлНя {г} 1'С*НМ (г)
3;в„ э. е........ 74,12 ж 69л I
ЭЕ й^уччее наследие России"
Сказывается упорядочивание структуры при переходе от
соединения с одинарной связью к соединениям с двойной и
тройной связью *.
Бросается в глаза значительное падение 5дИ при пере-
ходе от 1-пентина к спиро пента ну;
CH—С-С Н 2- СНй-СНа (г)
I-лентни
s:u, 9. е. ..,78,82
Cnilpurifll’TSII
67,45
Здесь проявляется жесткость молекул второго вещества.
По этой же причине цикланы (циклоалканы) имеют мень-
шую энтропию, чем соответствующие им по массе алкены;
СИ*
СНи-СН-(СНЛ-СН,(г)
э_е...,71,28 9(,93
Энтропия и периодический закон. Сходство природы и
структуры соединений rrp if водит к за кономерному измене-
нию их энтропий. Например:
(г) С!, (г) В г, (г) 1а (г)
. . 48,45 5ф&3 58.G45 62,284
S пределах подгруппы энтропия растет, однако не по-
тому, что она является одвоЗ!гачЕгой функцией массы. В по-
следнем легко убедиться, рас-
смотрев ход энтропии !Го пе-
риоду. Так, ХОТЯ в ряду
Na — Аг атомная масса уве-
личивается, однако Зда пре-
терпевает сложное изменение
(рнс. 17), Переход от мягкого
натрия к твердому кремнию
Рис. 17. Зависимость стаидарт1;ой
Энтропии St\, (5. е.) от порядкового
когьера элементов Z для элементов
третьего периода
* В какой-то степени на падение SJ„ в этом ряду ил и нет и нско
TOjioe уменьшение массы молекулы.
43
ЭБ ,:Научнов наследие России"
ЭБ ,:Научнов наследие России1
Рис- 18и Зависимость стандартной энтропии SJB4Ai (кнл/моль-К) кристаллических хло-
ридов от порядкового ЕЮМОра ЫО№НТЭ Z
сопровождается уменьшением энтропии; затем она несколько
возрастает н в конце периода достигает весьма большого
значения — сказывается газообразность хлор а а аргона.
Хотя В ряду Li — Be — В — С атомная масса увеличи-
вается, однако s:st падает — сказывается увеличение твер-
дости.
Другой пример: (5ж)яь (1Й,22 э. е.) почти вдвое больше
(10,17 э. е.), хотя моль первого на 20 % легче моля
второго.
Рис. 19. Взаимосвязь между стаклартЕгьг&ги энтрОЕпглми
(з. ej:
л — tacMtUTOT ncflrhynii ли тиа 11) и бериллия (([) и fl " гаэмвисшы*
гатДО * ло-р кляв ir тстраерая идо и Клементон подгруппы- угле[Н1Ы
Более полной будет картина при рассмотрении хода эн-
тропии для совокупности подобных веществ по всей перио-
дической системе Такого рода зависимость на примере од-
ной группы соединений изображена на рис. 18. На нем не
представлены жидкие и газообразные соединения, так как
для ннх (особенно для последних) ЗЕгачеПЕтн S выпадают из
общей закономерности — сказывается чувствительность эн-
тропии к агрегатному состоянию вещества. Кроме того, на
графике изображена не мольная, а гр амм-э к ви валентная
энтропия, что почти устраняет влияние на значение
увеличения числа атомов в молекуле соединения (например,
рост в р яду R ЬСI—SrC li— ¥ С18—2гС 14— ...). Пери оди чность
величин S"i'a4hi была бы более отчетливой, если бы мы рас-
полагали значениями энтропий соединений, в которых
входящие в них элементы находились бы в соотпетствующих
степенях окисления (см., например, положение точек на
рис. 18 для FeC)t, FeClj, TICi, AuCiJ.
ЗБ гНауччов наследие России" ®
Некоторые закономерности. Если нэять два ряда сход-
ных веществ, то будет наблюдаться параллелизм измене-
ния энтропии ц них. Нередко он отвечает приближенному
линейному соотношению (рис. 19):
$п=А5г + 0> (II. з>
аналогичному (1.15). Мыслимо и сопоставление рассматри-
ваемого свойства в ряду сходезых веществ с другими их ха-
Рнс. 20. Взс<иА|О<-&язь между
платностью р(г/сми) и стзггдарт-
LLQbl 9НТрОЕ[МСЙ (э. С.) РАЗ-
ЛИЧНЫХ ыаднфикзцнА двуокиси
кремния:
/ а-грнд]|мнт: 2 — й-кри стеба-
лип J — aj кварц; 4 — kgsceih 5 —
<ТЯШ1>В1ГТ
рактер в стиками. В ряде случаев оно также отвечает линей-
ной зависимости (рис. 20).
3. Изменение энтропии
Л соответствии с (11.1) при переходе вещества из исход-
ного состояния в конечное мольное изменение энтропии вы*
лажается уравнением
Рассмотрим в качестве примера плавление кристаллов.
Мы знаем, что если частицы, образующие кристаллы, распо-
ложены вполне определенным образом (в узлах кристалл и*
ческой решетки}, то в жидкости они располагаются менее
упорядоченно (ближний порядок). Это означает, что со* >
>ши. Поэтому в процессе кристаллизации, например
НйО (к, р = 1 агм, Т = 273,16 К} -+ НгО (ж, р = 1 атм,
Т = 273,16 К), происходит увеличение энтропии на вели-
чину
S“-5l,₽J?ln-^-1 т. е. S"-SK^ASnrl >0;
4G
ЭБ ,:Научнов наследие России1
очевидно,
5к-Хж--А51ГрЯв1<0.
причем ASK|l,rfT и ASnjI будучи противоположными по знаку,
равны по величине.
Аналогично ©г > «". Поэтому а процессе парообразова-
шгя, в частности Н,О (ж, р = ] атм, Т = 373,16 К) ->
-*- НпО (г, р = 1 атм, Т = 373,16 К)Р происходит возраста-
ние мольной энтропии на величину
Sr-S*=J?lri^
й
т. е.
5г-Зи-Д£П1(>>0;
очевидно^
— Sf = Л5К0цяецС С О,
Так как о/ больше отличается от t»“, чем <о* от и*, то
ASjr^.
К подобному выводу приводит н рис. 16: разность энтропий
в точках d и с больше таковой в-точках b и д.
Оба примера относились к процессам фазовых превраще-
ний ('кристаллизация — плавление, парообразование—кон-
денсация, Сублимация—десублннация, полиморфные из-
менения), А ОЕ1и Характеризуются тем, что обе фазы могут
сосуществовать, т. е находиться в равновесии. Это значит,
что путем сколь угодно малого изменения температуры п
(или) давления можно осуществить соответствующий сдвиг
равновесия. Так, подвод небольшого количества теплоты
к системе, состоящей из кипящей воды и сухого и асы пол-
ного пара, приводит к смещению равновесия в процессе па-
рообразования в одну сторону, небольшое сжатие — в про-
тивоположную. А£фП колеблются в до ват ь но широких
пределах — от небольших величин (порядка 0,1 э. е.) для
превращения веществ из аморфного состояния в кристалли-
ческое до десятков единиц для сублимации, причем оче-
видно:
Л5сув1 '^лпр = ;^ ям — к
Представление о порядке величин AS*n мют значения
этой характеристики для различных процессов (табл. 7).
Следует обратить внимание на то, что стандартная энтро-
пия фазового перехода (Л5й»)ф.п в общем случае отличается
47
ЭБ ,:Научнов наследие России"
or А£ф Эго различие будет 1ем значнгель нее, чем больше
несовпадение Тф „ с 298,16 К. Так, если для Н2О (ASfjiJnnp =
== 28,36 э. е. и (Д$зпд)плр = 26,04 э. е., то, например, для
меди (ASJwKyu., = 31,82 э. е., в то время как (AS.**^) >
> 75 э. е.
Таблица 7
Значение величин Д$ф п для некоторых процессы
Екщостпф Beiutcrns а е (4як)* и * е
В (к) 1ГЮ в (ам) 1,М 0,16
С (алмаз) I С (графит) 1,372 0.816
HzO (к) ^11,5 Н/> (ж) 16,75 -5,25
HNQ, (л) Л. 20 } Н\О,(г) 6Э.7& 28^9
NjCX (к) NA(r) 42,4
AgNO, (к) 33 AgNOjtp) ИЛ1 18^86
Изменение энтропии в химических реакциях вычисля-
ется подобным путем; так, для процесса
nA + tB + „.™<ilD+₽E 4*,,. (Ill)
AS«(rfSD+eSE+,)-(аЗл + б$в+ ), (IT.3)
причем энтропии всех веществ берутся при условиях про-
текания реакции. Таким образом, расчет Л5 осуществля-
ется приемом, аналогичным вычислению A/f по закону Гесса
(см. с. II). Казалось бы, надо брать, по аналогии с сто при-
менением, алгебраическую сумму энтропий образования
AS всех реагентов, но, очевидно, величина
[rf ( 4So6p) D + е PS0«p) F + ] ' [Я <Л5о6Г) А + 4й Sofi],) В + ' ]
будет равна AS (П.З). Поэтому, например, для процесса
С (графит)-(-004 (г) = 2СО (г); = 25^ -j-
в частности, при Т — 1500 К
Д31ч0 = 2 59,5 - (8,0 4-65,8) = 41,2 э.е.,
что мало отличается от величины 5«( = 41,986 э. е.
Обобщая этот пример на другие химические реакции,
можно сделать вдже1ыГ[ вывод1 хотя S (S’) каждого вещества
возрастает с температурой (и в данном интервале Т тем
значительнее, чем больше его теплоемкость), но AS (AS*)
с температурой меняется мало (в рассмотренном примере
48
ЭБ ,:Научнов наследие России"
изменение температуры на 1200 “С привело к изменению AS
лишь на 0,8 э, е.).
Изменение энтропии и периодический закон- Общую
картину изменения энтропии при фазовых переходах
(ЛЗф. „) ласт обращение к пер и одическому законуД. И. Мен-
делеева. На одном примере это показано на рис. 21 при
р = I а™. Если ДЗпд показывает явную периодичность, то
Рнс. 21, За&и сините изменения энтропии хлоридов при их плавлении
iSnj (э. е.) и парообразования Л5ппр (э, е.) от порядкового номера
элемента
ДЛЯ AS„ap периодичность выражена менее отчетливо. Дей-
ствительно, если н вменен не Д5пд для хлорилов охватывает
интервал от~3 до ^2! э. е., то амплитуда колебаний Д5|1а|,
для тех же веществ лежит в пределах лишь ~20 * 27 з. е.
Что касается изменений энтропий в химической реакции
(Д5±р), то эта величина не является периодической функцией
порядкового номера элемента. Так, хотя для реакции
УчЭ(к) + ^С!иИ-^ЭС!л (к)
А5 = Vn-Sgcp, — 'л5, ™ */*SCjt(
ЭБ ,:Научнов наследие России"
49
значения l/nSacin и Sa для различных Э неодинаковы,
однако разность между ними по сравнению со значением
1/2 Sci, от элемента к элементу меняется очень мало. Поэ-
тому в первом приближении AS равна —J/2Sci,. Особенно
близки значения AS для аналогов по подгруппе, на пример.
AS в реакциях
ЭО (к)+00g (г) ₽ ЭСО., (л)
для всех двухвалентных элементов близки, а для элементов
подгруппы бериллия почти неотличимы,
4. Энтропийный и энтальпийный факторы процесса
Если величина АН отражает в ОСНОВНОМ взаимное ат ля-
ни е атомов в молекуле, стремление к объединению частиц
в более крупные сочетания, т. е. способность их к агрега-
ции, то величина AS отражает противоположную тенден-
цию — стремление к беспорядочному расположению частиц,
к их дезагрегации. Система переходит в состояние с мини-
мальной энергией лишь тогда, когда AS = 0; если же
АН — 0, то система переходит в наиболее неупорядоченное
состояние.
С одной стороны, частицы вещества па пути к минималь-
ной энергии стремятся сблизиться, взаимодействуя друг
с другом, и дать прочные агрегаты, заняв при этом мини-
мальный объем; с другой стороны, тепловое движение раз-
брасывает Частины, распространяя их па возможно боль-
ший объем (в частности, при растворении приводит к вырав-
ниванию концентраций). Словом, поведение реагентов напо-
минает поведение частиц воздуха; последние и не падают на
землю, и не разлетаются. На каждую из этих противополож-
ных тенденций, выражаемых величинами АН и AS, влияют
природа вещества и условия протекания процесса (тем-
пература, давление, соотношение между реагентами и
т. д.).
Для количественного сопоставления этих противодей-
ствующих факторов необходимо выразить олицетворяющие
их величины в одинаковых единицах. Это можно осущест-
вить двояко —• либо разделив АН на Т, либо умножив AS
па Т (переведя предварительно АН в кал/моль или AS
в ккал/К-мать). Остановимся на втором из этих вариан-
тов, мотивируя свой выбор тем, что оба сомножителя в ве-
личине TAS выражают стремление к беспорядку,
50
ЭБ ,:НаучноЕ наследие России"
Peiccmotpим случай, когда обе тенденции одинаковы, т. е.
энтальпийный (ДЙ) л энтропийный (TAS) факторы компен-
сируют друг друга. Равенство
ДЯ = ТД$, (ПЛ)
од и ] I е тез а р я ющее «у р а в и ове j и и ва и и с», у п и вс р са л ы ю. Оно
относится и к равновесию между кипящей жидкостью и
ее насыщенны А! паром (например^ к воде при / = |ООйС
и = I атм\ п к равновесию между плавящимися кристал-
лами и отвердевающей жидкостью (примером может служить
бензол при / = 5,6X ир = 1 атм), и к другим видам фазо-
вы х пр ев р аще ни й и иди виду ал ьн ы х веществ (су бл и м а и и я,
полиморфные превращения). Это равенство справедливо
и для фазовых превращений с участием растворов, в частно-
сти для равновсси я между насыщенным раствором я кристал-
лами растворенного вещества (например, между сахаром н
его водным раствором, содержащим при 20 °C 200 г CflH|3Ofl
на 100гНйО). Для химического процесса это равенство отве-
чает равновесию между исходными веществами н продук
гамн взаимодействия (например, для реакции между СОн
и HSi с одной стороны, н СО и Н3О — с другой].
Другими словами, уравнение (П.4)г являясь условием
равновесия, Характеризует такое состояние данной системы,
когда скорости' протекающих в ней противоположных процес-
сов/ч а нрпмер, испарения и кон де Etc а г щи. прямой я обратной
химической реакция) становятся равными
По пут ею отметим, что с помощью равенства (1L4) можно
рассчитать изменение эЕггропии в данном равновесном про-
цессе из непосредственно измеримых нелнчпн, так как это
равенство означает, что
Тк е,, разделив теплоту процесса на абсолютную темпера*
туру, мы получаем искомую величину. Таи, вновь возвра-
щаясь к рис. 16а мы могли бы сказать, что отрезок пй равеЕ!
отношению теплоты плавления к температуре плавления
свинца, а отрезок cd — отношению его теплоты парообразен
вапия к температуре кипения. Из уравнения (Н.4) следует,
в частности, что
^ПЛ ==
Поэтому, например, полимеры (если они ise обладают гиб-
кими цепями) должны иметь высокие «точки плавления»:
в Е1их сочетается значительное меж цеп ное взаимодействие
51
ЭБ ,:Научнов наследие России"
(большие АЯпя) с незначительной гибкостью целей (неболь-
шие Д£й.,),
В дальнейшем мы продолжим рассмотрение вопросов,
затронутых в настоящем разделе, а сейчас познакомимся
еще с одной важной величиной,
Г-Швл HI
ЭНЕРГИЯ ГИББСА
(, Критерий направлении процесса
В механике большое значение имеет принцип стремле-
ния к минимуму потенциальной энергии. Тенденция тела
к перемещению сверху вниз определяется разностью уров-
ней в его начальном и конечном положениях независимо от
траектории падения. Движение прекращается, когда гра-
витационный потенциал достигает минимума. Убыль гра-
витационного потенциала (отнесенная к единице массы)
равна работе падения тела, величина которой от пути пере-
мещения не зависит.
В химических процессах тоже есть свой потенциал. По-
добно механическому (гравитационному) потенциалу он
убывает в самопроизвольно протекающих процессах. Ис-
черпание движущей силы взаимодействия знаменуется до-
стижением минимума этого потенциала. Так как мы услови-
лись рассматривать процессы при р, Т = const, то потен-
циал , являющийся движущей силой химических процес-
сов, целесообразно назвать изобарн о- изотерм ическим по-
тенциалом или, ради краткости, изобарным потенциалом.
Его обозначают буквой 6 в честь американского ученого
Гиббса, которому принадлежит разработка этого вопроса,
В его же честь эту величину теперь принято называть
энергией Гиббса. В дальнейшем мы будем пользоваться по-
следним термином *. Убыль этого потенциала тоже не за-
висит от пути процесса, в частности от его «химической
траектории», и равна максимальной работе реакции за вы-
четом работы против ВЕзешнсго Давления (см с. 7);
(П&)
это работа перехода от данного состояния к рев новее пому.
* в литературе подчас применяются термяни-сшгонимы свобод-
на я энтальлня, с&ойвднэя энергия при постоянном ддплеини, фуЕсн-
пня (лотенцнал) Гиббса. Наряду с <? иногда используются и другие
обй.^!#11&||цп? □ частности Z.
52
ЭБ ,:Научнов наследие России"
Условием и р и и ц и и и а л ь н о ii осуществ к-
мости процесса, т. е. возможности протекания ре-
акции о прямом Направлении без Затраты работы, является
неравенство
Чем больше химическое сродство реагентов, другими
словами, чем более отдалена совокупность данных веществ
от состояния равновесия (для химически обратимых процес-
сов), гем сильнее стремление к протеканию процесса, тем
больше будет убыль G.
Если рассматриваемая реакция осуществима в прямом
направлении, то в обратном направлении она при данных
р и Т невозможна, так как для противоположного процесса
в этом случае
т>°- {II.7)
Это неравенство является индикатором, принципи-
альной невозможности процесса.
Л юбая реакция может протекать самопроизвольно тол ько
в [^правлении, приближающем систему к состоянию рав-
новесия. Если в системе пасту пи л о истинное химическое
равновесие, то дальнейшее изменение энергии Гиббса про-
исходить не будет, те,
iCP г = ° (rfG₽.r= °). (НЛ)
Итак, критерий течения процесса найден — мерой хими-
ческого сродства является убыль (?, т. е. — AG. Таким обра-
зом, для совокупности веществ при данных температуре и
давлении (концентрации) мерой химического сродства бу-
дет величина ЛСр.г (в дальнейшем индекс р, Т опускаем),
Это движущая сила процесса. Чем Л(7 меньше, тем дальше
система от состояния химического равновесии и тем более
она реакционноспособна,
Эти соображения в схематической форме иллюстрирует
рис. 22. По оси абсцисс отложен состав реакционной смеси;
левая ордината соответствует веществам, вступающим во
взаимодействие, правая — получающимся веществам. Чем
ближе точки к первой, тем больше в смеси исходных веществ;
так, точка А отвечает реагирующей смеси, содержащей
примерно 80 % исходных веществ и 20 % продуктов реак-
ции. По оси ординат отложена энергия Гиббса. Точки А и
В отвечают ее Значению соответственно для исходных и по-
S3
ЭБ ,:Научнов наследие России"
лученных пещсств. Рис. 22, и иллюстрирует принципиаль-
ную возможность полного перехода исходных веществ в про
дукты реакции, рис, 22,6—невозможности взаимодей-
ствия исходных веществ (мыслим лишь обратный процесс),
рис. 22, а, аналогичный рис. 151 —осуществимость двусто-
МСЮДНШЙ | ЛСиГОчИЬгЮ
pfl RHQPEC Hfcl е
Рис. 22. Изменение энергии
Гиббса ио мерс лротекамия
процесса (схема) г
ы — процесс DoJbiOHitiH G — н-t-
пуэыожеий f — осуществив п
Обо к t ни лра яле IIII ях
ровинх изменений, на котором
кривая АСВ характеризует за-
висимость Сг от состава смеси.
Положение кривой АН опре-
деляется I гр и род ой процесса, а
также условиями его протека-
ния. Для данного процесса и
данных р и Т оно фиксировано.
Точка С в соответствии с усло-
вием (П.В) отвечает равновесию,
а се абсцисса — равновесному
составу. Чем больше величина
Од отличается от величины 6а,
тем ближе к одной из границ
диаграммы положение равнове-
сия. Сближение <м и Св смеща-
ет положение равновесия к се-
редине диаграммы. В любом со-
стоянии левее С (т. е. для сме-
сей, более богатых исходными
веществами по сравнению с рав-
новесной) или правее С (снесл
богаче продуктами взаимодейст-
вия, чем равновесная смесь) си-
стема реакционноспособна, так
как перемещен не по кривой в
сторону точки С будет сопро-
вождаться падением С, что от-
вечает условию осуществимости
процесса fl 1.6).
Для вычисления значения
ДО реакции применим метод
расчета, аналогичный способу вычисления Д/У (с. J2) и
Д$ (с. 48) реакции. Так, для процесса (111) при данных р
и Т по аналогии с (1.13) и (Н.З) имеем
дб=[rf (Дбойр)с + f (Лбцйр)г 4-..,] -
(]].9)
ЭБ ,:Научнов наследие России1
причем, как н а случае значения AGorp простых ве-
ществ принимаются равными нулю. Так, применив (П-9)
к процессу перехода вещества из аморфного состояния в кри-
сталлическое, мы бы получили G< 0 (в частности, для
SiOa~—б ккал/моль). Этот результат отвечает большей
реакционной способности вещества в аморфном состоянии
по сравнению с кристаллическим.
В безошибочности результатов анализа процессов на ос-
новании определения знака величины AG мы убедимся
в дальнейшем неоднократно. Сейчас же важно еще раз под-
черкнуть, что прогноз совершенно ле Зависит от механизма
реакции, от того пути, по которому фактически протекает
процесс, ибо величина АО равна разности значений G про-
дуктов реакции и начальных веществ, т. е. подобно А// (и
AS) не зависит от пути процесса. Однако это означает в то
же время, что никакой информации о самом процессе и,
в частности, о его скорости, которая весьма чувствительна
именно к пути реакции, извлечь из АО невозможно. Следо-
вательно, эти вопросы надо анализировать с лени алы го.
Они будут обсуждены ниже (часть HI). Здесь подчерняем
лишь, что между пршищпиальиой осуществимостью про-
цесса и его практической реализацией подчас дистанция
огромного размера; вопреки (11.6) нередко реакцию прове-
сти не удается, так как реакционноспособная система нахо-
дится п состоянии заторможенного равновесия.
Затруднения на пути мыслимого процесса приводят к
к тому, что существует немало веществ, для распада которых
AG< О (т. е. для цх образования AG >0); тем не менее они
могут существовать сколь угодно долго. Примером первого
случая могут служить реакции горения различных органи-
ческих соединений: хотя для всех этих процессов уже при
комнатной температуре AG О, эти вещества горят только
при высокой температуре. Примером случая, когда AG 0,
являются ацетиленовые углеводороды": они неустойчивы
к разложению на углерод и водород, причем их неустой-
чивость с ростом молекулярной массы возрастает, однако
только при высокой температуре скорость их распада ста-
новится ощутимой. Эти обстоятельства позволяют сделать
важный вывод: если для образования данного вещества из
элементарных веществ AG > 0, то его можно получить толь-
ко косвенным путем. Действительно, все оксиды хлора н
азота (кроме NO) н многие другие соединения (в частности,
BSH4, SiHj, НгТе) не могут быть получены прямым синте-
зом.
55
ЭБ ,:Научнов наследие России"
Величина AG связана с величинами Л2У и AS важным
соотношением:
Дб^ЛН-ТДЗ, (II .10)
ему отвечает уравнение
G — H-TS. (ПН)
являющееся о пределен и ем функции G.
Действительно, применив (II.И) для всех веществ,
участвующих □ данном процессе, при р, Т = const полу-
чаем (11,10).
В соответствии с (11.10) величину Л6, выражающую
«работоспособность» Совокупности реагирующих веществ,
измеряют в тех же единицах, что и величину АН, выражаю-
щую их «теплое пособи ость», г. е. в килокалориях.
2. Энтальпийный и энтропийный факторы и направление
процесса
Из соотношения (11.10) в соответствии с неравенством
(II.6) следует, что тенденция к реакции тем значительнее,
чем отрицательное А Я п положительнее AS; реакции, для
которых А//>*0, a AS<0, в принципе невозможны
(AG >0), т. е. мыслимы лишь в противоположном направ-
лении. Таким образом, различные злаки АТ/ л AS сви-
детельствуют о возможности двустороннего течения про-
цесса (при условии соизмеримости AW и TAS); очевидно,
одинаковые знаки АН и AS указывают на возможность те-
чения лишь одностороннего процесса.
Схематически эти обобщения иллюстрируются рис. 23-
Верхняя часть поля чертежа — это область недозволенных
процессов, нижняя часть — область разрешенных процес-
сов. На рис. 87 эти же результаты представлены в виде диа-
грамм физико-химического анализа— в координатах свой-
ство—состав.
В предельных случаях, когда AS = 0 или ЛИ = 0, не-
обходимым условием протекания процессов будет соответ-
ственно выполнение неравенств АТУ <; 0 и AS > 0; иными
словами, изоэнтропийные процессы могут быть только эк-
зотермическими, а изоэнтальпийные всегда должны проте-
кать с ростом энтропии. (Очевидно, что экзотермическими
были бы любые процессы при Т -+0К.)
Следовательно, знак и величина AG определяются конку-
ренцией энтальпийного (АН) и энтропийного (ГАЗ) фа кто-
ЭБ Ц^ччсе наследие России"
AG
рае, причем роль последнего при прочих равных условиях
тем значительнее, чем выше температура, — вывод, отве-
чающий тому, что первая величина олицетворяет стремле-
ние к упорядоченности (уменьшение энергии в результате
агрегации), а вторая — к беспорядку (увеличение энтро-
пии в результате дезагрегации), Единство и борьба этих
противоположностей и разрешается в соответствии с (11,10)
в величине АО, являющейся своеобразной равнодействую-
щей этих противоборствую-
щих тенденций, Поэтому кри-
терием равновесия (лротеи а -
ния *10 у сторон них процессов)
и является равенство &Н =
= ТДЗ, т. е. (П 8).
С прохождением величины
ДО через нуль связано тече-
ние двусторонних (обрати-
мых) процессов. Так, хотя при
низкой температуре водород
не диссоциирует, т. е. равно-
весие реакции
Ht(r)-2H (г)
практически нацело смещено
в одну сторону, однако в со-
ответствии с уравнением
= Ifl - FAS t04,20 — 0.02359Т
Рке. 23, Различные саршпиы
соотношении пел и ч ни i/Л iS
и AG на графике- Лб = / (Г);
точки С отвечают состшиним
ГН1ПЕ[ОП£СИЛ
при Т 4400 К повышение ил if понижение температуры
смещает равновесие в соответствующем направлении.
Влияние температуры. Как мы уже отмечали, в первом
приближении можно пренебречь влиянием температуры на
А// (с. 15) и на АЗ (с. 48). Это означает, что часто изменения
А// и АЗ невелики по сравнению с изменением Г, т. е. что
(П-Ю) можно считать уравнением прямой, нэклоег которой
будет определяться знаком АЗ.
Важной то, что при неизменности агрегатл ого состояния
реагентов уравнение (II. 10) может оказаться справедливым
нвсравнительвотнрокоминтервалетемпсратуршо-первых,
в тех случаях, когда нельзя пренебрегать изменен пом ДЯ
и 43 с Т, следует учесть, что знаки d (SH)idT и d (&S}/dT
противоположны; во-вторых, первая из этих величин мень-
ше второй в Т раз. Это приводит к тому, что изменение Д//
67
ЭБ ,:Научнов наследие России"
ii AS с T мало отражается па величине AG, гораздо меньше,
чем изменение Т.
В соответствии с (П.10) роль энтропийного фактора тем
значительнее, чем больше абсолютное значение AS. Это
иллюстрируется рис 24. Из него следует, и частности, что
ASiv и AS\ ] положительны, причем ASvi > ASiv, что Д5п
и ASvil отрицательны, причем | ASvii I > | ASij |, что
ASi о, a ASv = 0; что при достаточно высоких темпера-
турах в реакциях (I), (II) и (VII) должны образоваться ве-
щества, записанные в левой части равенства, в реакциях
-100
{J $00 КИЮ 1500 2«Ю 2500 3000
2
Рнс. 24 Записи мости (ккал) исколорык
реакций от температуры Т (К)
СО(ГННР(|}-СОИГНМНО (I)
IL(')-h’/A(f)-H20(>K) (II)
С (Графит) + COt (г) = 12 СО (г) < i II)
С (графит) -Ь ОНО - СА (Г) (IV)
Cii* (Г)т COj (г) - ЙСО (г) + 2Н> (г) (V)
С (графит) + 2Нг (rj = СН, (г) (VI)
(IV) и (VI) — вещества, записанные в правой части равен-
ства; что же касается реакции (V), то опа при любых тем-
пературах в принципе должна протекать слева направо.
При мер н а я л и ней ность огр а н нчен а точка м и фазового п ре-
вращения; так, если, например, один из реагентов при не-
которой тем (Герату ре из твердого переходит в жидкое состо-
яние, то при этой температуре на ASX р наложится AS^ П1
что приведет к изменению наклона прямой (увеличению или
уменьшению в зависимости от того, произойдет ли фазовое
превращение с одним из продуктов реакции пли с одним из
исходных веществ).
Увеличение роли энтропийного фактора с ростом темпе-
ратуры можно проиллюстрировать на примере возрастания
относительной устойчивости нормального угле-
водорода с повышением температуры. Действительно, при
ЭЕ ^й^ччее наследие России"
низких температурах в равно веси ой смеси бутана и изобу -
тана преобладает язобутаи, а при высоких температурах —
« бутан. В качестве другого примера укажем на процессы
изомеризации «-Октана. Этот углеводород имеет 17 изоме-
ров. Наибольшей теплотой образования обладает гексаме-
тил этап. Казалось бы, он должен быть и наиболее стабиль-
ным. В действительности это самый неустойчивый изомер —
сказывается его высокая симметрия. В реакциях гидриро-
вания, например в процессах
CtH.t(r) + H2(r) = CflII1Hr}
1’ftKCCIP Н. ССКСЛН
и
(г) (г) — C^Hj^ (г)
цнчлогекеая >оГ*кСйн
изменение объема одно и то же (Дпг = —1), однако неоди-
наковы не только AH]» (соответственно —30,0 и —10,53
ккал), но и Так как для 1-гексена S«H — 91,93,
а для циклогексана = 71,28, то Д5^( этих двух реак-
ций будут отличаться друг от друга более чем на 20 э. е.
Вооружившись уравнением (11.10), мы можем решить
еще один важный вопрос.
Выло время, когда полагали, что мерой химического
сродства является тепловой :и/хрс’кш реакции (принцип
Бсртло). Этот критерий (ДА/) заманчив, так как тепловой
эффект легко намерить и осмыслить, и, на первый взгляд,
правдоподобен. В самом деле, чем больше выделится теп-
лоты при реакции, тем, казалось бы, «охотнее» вещества
вступают в нее. тем прочнее образующиеся продукты и
полнее протекает процесс (см. с. 36).
Справедливость этих рассуждений можно подтвердить
реакциями (IV) и (V) (рис. 24) и многими другими, в част-
ности процессами:
Ка (к)^-'/EPs (rJ^NsF (к), At Г,,™—137 J ККЯЛ/ЧОЛЬ
Na (к) 4- '//11 а (г) = NaCl (к), = —38,26 хкал/цоль
Na (к)-р/гВга (ж) = NaBr (к). АН’т = —85,38 ккал/мояь
и
Na (х) -|- a/sIа (к) = Na I (к), = —69.46 хкял/моль
ЗЕ1ачигельное выделение теплоты (Д//ЙА *С 0) при обра-
зовании галогенидов натрия можно рассматривать как
критерий их устойчивости относительно простых веществ,
а возрастание "этой величины в ряду Nal — NaBr — NaCl —
S9
ЭБ ,:Научнов наследие России"
— NaF — как свидетельство увеличения химического срод-
ства галогенов к натрию с уменьшением их порядкового
номера (различие в агрегатном состоянии галогенов, разу*
местся, не отражается на ходе значений Оба вы-
вода отвечают действительности,
К этим примерам можно было бы прибавить н многие
другие. Более того, если бы мы ограничились рассмотре-
нием только низкотемпературных процессов, то убедились
бы в том, что все они, в соответствии с принципом Бертло,
являются экзотермическими. Однако чем выше темпера-
тура, тем все чаше встречаются самопроизвольные про-
цессы, сопровождающиеся, вопреки принципу Бертл о,
ire выделением, а поглощением теплоты. Так, реакция (VI)
при Т = 1300 К протекает слева направо (рис, 24), хотя
для нее при этой температуре тспловое"! эффект И положи-
телен, и велик (ДЯ = 62,0 ккал).
Есть и другое противоречие. Процесс, идущий в дан-
ных условиях (например, при данной температуре) с выде-
лением теплоты, в иных условиях (например, при другой
температуре) протекает в обратном направлении, т. е. с по-
глощением теплоты.
Таким образом, существование самопроизвольно про-
текающих эндотермических реакций и химическая обра-
тимость мезогих процессов ~ вот факты, свидетельствую-
щие о том, что в общем случае тепловой эффеЕ(Г реакции
fie яЕзляется мерой химического сродства.
Обращение к уравнению <11.10) снимает эти противоре-
чия: в общем случае не вся энергия процесса ЛЯ может
быть превращена в работу (—ДО); часть ее («связанная»
энергия ГД5) не используется.
Повышение температуры препятствует силам межатом-
ного (межмолекулярного) притяжения, способствующим
упорядочению системы, и усиливает хаотическое движение,
г. е. дезагрегацию частиц. Поэтому при очень низких
температурах преобладает первая тенденция, при высоких—
вторая. Действительно, если процессы ассоциации моле-
кул и синтеза веществ для своего осуществления, как пра-
вило, требуют низкотемпературного режима, то реак-
ции разложения обычно протекают при высоких темпера-
турах.
Соотношение между «порядком» и «беспорядком» и опре-
деляет направление реакции.
Так как значение второго члена правой части урав-
нения (И. 10) при прочих равных условиях тем меньше,
ЭЕ^Й-зучноЕ наследие России"
чем Ei иже температура, то ясно, что при достаточно низ-
ких температурах можно им пренебречь, т. е. судить о на-
правлении процесса непосредственно по его тепловому
эффекту. Это и означает, что пользоваться принципом
Бертл о независимо от природы процесса с уверенностью
можно только для процессов, идущих при низких темпе-
ратурах, Для процессов, протекающих при высоких тем-
пературах, второй член может стать настолько значитель-
ным, что АС и АИ могут отличаться не только по вели-
чине, но н по знаку, А в двух случаях будет наблюдаться
противоречие принципу Бертл о: а) для экзотермического
процесса (ДЯ<0), когда 7 > О и AS <0 и поэтому
Т'ДД’^О и ДБ >-0; б) для эндотермического процесса
(i\H >-0) при Т 0 и AS "> 0, когда ДБ< 0.
При достаточно высоких температурах все химические
соединения разлагаются, хотя процессы их распада эндо-
терыичны (ДА/>0); уже из этого примера видна роль
энтропийного фактора.
3, Стандартные изменения энергии Гиббса
Чтобы можно было производить алгебраические дей-
ствия с ДБ; отдельных веществ (для определения ДБ про-
цесса), необходимо эти величины отнести к одинаковым
условиям- По аналогии с лА/^й и ДА/j (с. 14) и и
Д5) (с. 4!) пользуются системой стандартных энергий
Гиббса А6||и (Лйт) образования данного вещества. Для
элементарных веществ, устойчивых в стандартном состоя-
нии, принимается ЛБ«Л = 0. Для химического процесса
величина ДС^ (ДБг) отвечает условию, когда парциаль-
ные давлений (концентрации) всех реагентов в течение
всего процесса осгзеотся неизменными и равными единице.
ЕстёственнО, что это условие подразумевает неизменный
состав реакционной смеси, т, е. отвечает допущению, что
количества всех веществ в реакционной зоне несоизмеримо
больше количества прореагировавших it образовавшихся
веществ по уравнению реакции, что и обеспечивает по-
стоянство р (С) в течение всего процесса. Так, для уже упо-
минавшейся реакции
СО (г) + Н,о (г) = 60s (гЦ- н4 (г)
значение = —6,843 ккал/моль означает следующее:
если бы в бесконечно большом количестве смеси СО, Н2О,
COi и Н4, взятой при 25 “С, в которой = рн^о =
61
ЭБ ,:Научнов наследие России"
= Рсо, — рн^ — I атм, г. e, рол, = 4 атм (все газы иде-
альны), прореагировало бы по 1 молю СО и НаО, То в ре-
зультате энергия Гиббса уменьшалась бы на 6,843 ккал/
Удобство оперирования с величинами AGJ,H (AG г),
различие которых для разных веществ выражает лишь
несходство этих веществ, очевидно. Важно и то, что стан-
дартные энергии Гиббса по определению не зависят ин от
давлений, нн от концентраций, а лишь от температуры.
Если знак AG позволяет судить о направлении процесса,
то знак AGC в общем случае этой информации не песет;
ведь последняя величина отвечает гипотетическому про-
цессу^ Но можно считать, что при ДСр 0 процесс осуще-
ствим не только в стандартных, во и в любых реальных
условиях; наоборот, неравенство AGr > 0 отвечает прин-
ципиальной осуществимости обратного ему процесса как
в стандартных, так п в каких угодно условиях. К сожале-
нию, не существует определенных значений AG?, за пре-
делами которых процесс можно считать в принципе реали-
зуемым в прямом (AG< 0) или в обратном (AG >0)
направлениях. Лишь в качестве очень грубого при-
ближения этот порог можно считать равным при-
мерно ± 10 ккал/моль; его ориентировокНОСТЬ связана
с тем, что он зависит от значений стехиометрических коэф-
фициентов в уравнении реакции (см. ниже). Важна и дру-
гая сторона вопроса: если величина AGj- по абсолютному
значению невелика, то путем соответствующего изменения
условий процесс можно сделать протекающим в том или
другом направлении. Ясно также и то, что условие ЛС) = О
в отличие от (11,10) не является критерием равновесия.
Значения AG^ для ряда веществ приведены в табл, 8
и в приложении 1. В дальнейшем мы везде пользуемся
значениями AGf (в основном AGiHl)*.
Рассмотрим несколько примеров. Величина AGiss =
= —76,64 ккал/моль для реакции
Zn (к)-Н/А (r) = ZnO (к)
свидетельствует о том, что цинк может окисляться не только
при ро, = I атм, но и при любом сколь угодно малом дав-
лен ни кислорода.
Так как для реакции
Ns (г) 4- EHtO (ж)« NH+ Ср) 4- NO; <р}
• Большинство приведенных ранее значений изменений функций
Гиббса также были йО;и,
62
ЭБ ,:Научнов наследие России"
AGm = 85,55 ккал, то этот весьма заманчивый способ
фиксации азота неосуществим: сколь велико бы Hit было рм,,
□ воде не появятся ноны азотистокислого аммония-
Таблица 8
Стандартные изменения функции Гиббса (ккад.'модь)
образования некогарык веществ н нож»
В^ЦсСгвп ОЙ ЛСгд^н
ВЩОН^ {р) —Ж2 MriClj (к) ^-i03
CBtj (к) «,в Мп/Л (к)
CHjCOOH {PJ —9^5 Мо1| (к}
а, (и ~Э0 NaNllj (к) -HJ
Crh (И - 55 Na3Sb (к) —50
CuFs (к) -11 еда SbOCl (К) ^—8t>
CuSO. (р) —16ВД $ЩОН)3 (к) -v—153
HCN (В) 2G.8 WJj (к) —3
НдВД» (Р) -178.0 2п^ (р) —33.173
HfN (к) -81Л (Zn(CN)J=- (р) 103Д
Из того, что л л я процесса образования аммиака из азота
и водорода Абдо = 5,8 к кал/моль, можно лишь сделать
вывод, что синтез аммиака при 400 LC неосуществим, если
каждый из реагентов находится под атмосферным давле-
нием, Но считать, что этот процесс при 400 ®С вообще невоз-
можен, было бы ошибочным. Действительно, при ВЫСОКИХ
давлениях синтез аммиака становится возможным.
4. Природа вещества п изменение анергии Гиббса
Химическое соединение тем устойчивее, чем отрицатель-
нее его Д(зф||р (ДДобр) (это обстоятельство, в частности,
определяет последовательность вытеснения элементарных
веществ из их соединений другими элементарными веще-
ствами). Так, химическая инертность перфтора л капов,
а также многих Других соединений, в том числе SIF* н
SF(, обусловлена большими отрицательными значениями
AGpe их образования (для CFJT S1F4 it SF* AGjSJj равны соот-
ветственно —375.8; —212,3 и —263,7 ккал/моль). На этой
же особенности MgO и А1,Оз основаны методы получения
металлов — магний- и алюмотермия. Обилие комплексных
соединений обусловлено тем, что они прочнее простых:
сказывается большая прочность комплексных ионов
[A! lOH)i {р) (AGjSS^—312,3 ккал/моль), SiF| (р) (—510),
ЭБ ' Науч ное наследие России" v
uor (p) (—246,4), SbO4 (p) (—42)1. Наоборот, чем поло-
жительней dGrfp. тем менее устойчиво данное вещество.
Взрывоопасность NCJ, (ж) отвечает AGiw £= 70 ккал/моль.
Надо помнить, что для твердых веществ Дб;,н зависит
не только от природы вещества, но и от модификации, сте-
пени дисперсности и других факторов. Так, для Zn (0Н)г(к)
п Zn ЮН)г (ам) Аби» соответственно равны —132,5 и
— 131,5 ккал/моль; для модификаций A^SiOji кнДНнта,
андалузита и силлиманита — они составляют —584,16;
—584,33 и —583,74 ккал/моль; для свежсосаждснпого
Сс(ОН). н этого же вещества после старения — соответ-
ственно —108,2 н —109,3 ккал/моль.
Обобщая данные гго значениям AG«h диссоциации СаСО3
(31,22) и KjCOj (83,19 ккал/моль), приходим к выводу,
что карбонаты щелочных металлов устойчивее карбонатов
элементов подгруппы бериллия. Действительно, при той
температуре, когда рсо, Для СаСОа становится равным I атм,
рсо, для KjCOj достигает давления, лишь несколько пре-
восходящего 1 мм рт. ст. Поэтому если ЭСО3 разлагаются
па воздухе при нагревании, то ЭаС03 плавятся без разло-
жения. Анализ значений AG’i^ позволяет заключить также,
что карбонат данного элемента стабильнее сто бикарбоната:
2К HCOj (к) = КзСОа (к) - С£\ (г) -j-Н/3 (ж), AG^„S = 8,92 лка.1/ыоль
Ks<.'Oj {к) = К (к) 4- СО2 (г), AGJ „ = ЙЗ, 19 ККал/МОЛЬ
В ряду алкан — алкен—алкин реакционная способ-
ность возрастает; это непосредственно видно из сопоставле-
ния значений AGin (ккал/моль) в ряду газообразных
О,НЧ (—5,61), С3Н, (И,99) и С3Н, (46.47).
Значения изменения энергии Гиббса родственных про-
цессов (например, образования сходных веществ), как
и значения &Н и AS для них, представляют собой систему
величин (рис. 25). Из него вытекает ряд выводов, в част-
ности, о большей химической активности элементов под-
групп лития к бериллия но сравнению с элементами под-
групп меди и цинка (подробнее см. часть V).
Обращает на себя внимание сходство рис. 25 с рис. 12.
Оно обусловлено тем, что разница в значениях Дб; и Д//°
в соответствии с уравнением (11.10) равна /AS, а при
Т = const н AS const (см. с. 50). Поэтому точки на
рис. 25 смещены по вертикали но сравнению с соответствую-
щими точками на рис. 12 примерно на одну и ту же вели-
чину.
ЗБ "1^4чое наследие России"
ЭБ ,:Научнов наследие России"
Рис, 25. Згьнснмоеть изменен еея стандартной энергии Гиббса образов ееи я ДСг^/л (хкал/швдь) хлоридов от
порядкового номера элемента 2
Некоторые закономерности. Здесь так же, как и в слу-
чаях Д77 (см. ряс. i3), S (см, рис, 19) и AS, можно говорить
и о количественных закономерностях. Но одном при-
мере это демонстрируется на рис, 26, отвечающем прибли-
женной зависимости
ДС|1^ЛДО1+А, (11.12)
аналогичной (IJ5); в ней сравниваются значения AG в двух
рядах 1 и II сходных процессов. По рис. 26 легко оценить
величину Образования нитрата бериллия из оксидов,
а по лей — и стандартное изменение энергии Гиббса при
образовании Be (NOs)t из простых веществ.
-гьо -ИЮ -1W -|Й) -SO
-Рис. 27. Взаимосвязь между
Знамениямл А Н у {ккая/мйпь)
И Абу (ккал/моль) в реак-
циях
Э (к)+ 20, (г) =30,00
где Э = Si, Ge, Sr, РЬ при
298” (/} И НЮОК (2)
Рис, 26. Взаимосвязь между
аца чеки я мн AGj, t (к кал/молъ)
в реакциях
МеО (к) -f-SO3 (г) ™ MeSOj (к)
(I)
и
McO(K) + NA(r) =
= Me(NOi)l(K) (II)
Можно также воспользоваться приближенным линей-
ным соотношением
iG^A&fi+S, (II. 13)
являющимся примером другого метода сравнительного рас-
чета (сопоставление значений двух свойств в одном ряду
сходных веществ (процессов)}, В нем при одинаковых усло-
виях сравниваются тепловые эффекты и изменения энер-
гии Гиббса в ряду сходных реакций. Это соотношение
ЭБ 'Научное наследие России"
иллюстрируется рис. 27, нарушение последовательности
в ряду SiOj—РЬО2 обусловлено вторичной периодич-
иостью. Точка для СО, не ляжет на прямую — скажется
отличие в значении Д5 для этого оксида от других. Так как
ЛН мало изменяется с Т, то абсциссы точен на низно-
н высокотемпературных прямых почти совпадают; это
позволяет оценить значение з по нему и зна-
чение ASftOip
5. Определение изменения энергия Гиббел процесса
Как же найти величину Д(7? Обратимся к одному из воз-
можных путей решения этой задачи. Осуществим это на
Рис. 58. Проведение реакции
Zn (к} -pCuSOi (р)= Си (н) + ZnSO4 (р)
при 25 X и I эти (схема):
л — в тсpuflcri тс: б — гм mi# м и tес КФм йл^мс-Нте
примере реакции вытеснения шиком меди из раствора мед-
кого купороса (рис, 28, и):
Zn (ж>-FCuSO* (p)-ZnSO, (р)Ч- Си (к).
Протекание этого процесса в термостате, т. е. при постоян-
ных давлении и температуре (р — 1 атм, t = 25’С), со-
гласно закону Гесса, будет сопровождаться тепловым
эффектом:
Д// = (ДЯс$р)гп$о. (р)— (AffoeP)ciiso, (pj ="
= (— 254,45) —(201,73) = — 52.72 ккэл/моль.
Проведем теперь эту же реакцию в гальваническом элементе
(рис. 2S, б). Для этого погрузим пластинку цинка (один
электрод) в раствор сульфата цинка^ а пластинку меди
ЭБ "НаучАе наследие России"
(другой электрод)—в раствор сульфата меди, Если сое-
динить оба полуэлемента п-образной трубкой, заполненной
токопроводящим раствором, то мы создадим гальванический
элемент — источник электродвижущей силы (э. д. с.). Это
элемент Даниэля — Якоби. В первом пол у элементе будет
происходить растворение цинка с превращением его ато-
мов в ионы, т, е, процесс
Zn«=Zii»h4- 2г
во втором — разрядка ионов, сопровождающаяся осажде-
нием меди:
Си** + 2е“ =<11°
а суммарно—токообразующая реакция:
zu*+Cu*',=zn#''+ai* (viii)
Схема этого гальванического элемента может быть
изображена так:
2г
ZtPIZn^BGiQjC^
{5бЭ*=
(нзбыточные сульфат-ноны перемещаются в направлении,
противоположном на пр а вл он ню движения электронов во
внешней цепи).
Если замкнуть внешнюю цепь на какое-либо омическое
сопротивление, то будет происходить лишь выделение
«бесполезной! джоуле вой теплоты, не сопровождающееся
полезной работой 4' (см. с. 7 и 53). Соединив же элемент
с электромотором, якорь которого будет нраищться с такой
скоростью, что развиваемая им обратная электродвижу-
щая сила практически уравновесит э. д. с. элемента, полу-
чим иной результат: тепловые потери станут минимальными,
а работа, наоборот, достигнет предельного значения 4тязе
К тому же результату можно прийти, скомпенсировав
э. д. с, данного элемента протнвоэлектродеи-
ж у щ е й силой другого элемента.
Для понимания разницы в результатах этих двух опы-
тов можно провести такую аналогию. Если газ, заключен-
ный в цилиндр, снабженный крышкой-поршнем, расширя-
ется, то он производит работу (1.3)
.'1=^ pdV,
где р — внешнее давление; V, в Vs — его начальный и
конечный объемы. Чем меньше pf тем меньше работа рас-
63
ЭБ ,:Научнов наследие России"
ширення. В предельном (идеализированном) случае от-
сутствия противодавления (невесомый поршень) А — О
(гак как р = 0). Наоборот, чем больше р, тем больше
п А. Работа будет максимальной, когда внешнее давление
станет равным давлению газа (практически меньше его
на очень небольшую — математик скажет — на бесконечно
малую величину) •* *
Химику приходится решать сходную задачу при взве-
шивании. Ведь чтобы определить массу, ее надо сном пен*
сировать равной массой.
В рассматриваемом случае при коротком замыкании
цепи (отсутствие п роти воэлектродвижу щей с пл и) «химнче-
скне силы» остаются нсуравновсшсннЕями и работа тока Л'
равна нулю. Наоборот, если э. д. с. гальванического эле-
мента полностью скомпенсирована, то будет совершаться
максимальная работа. Вог тогда мы получим возможность
вычислить AG токообразующего процесса, так как в этом
случае будет справедливо равенство (J1.5), которое для
работы тока примет вид
—A<j = nFE. (11.14)
где п—количество грамм-эквивалентов вещества (заряд
нона); F — число Фарадея (величина nF равна количеству
прошедшегоэлектрячества); Е = э, д. с. элемента **.
Изложен нос озн а ч а ет, что и з.ме не н и е G л р и о кисл ительно-
восстановительных процессах может служить источником
э. д. с. гальванических элементов.
В связи с содержанием настоящего раздела хотелось бы
обратить внимание на реакцию; С(гр) 4- О, = С<Л (г).
Так как для нее Д5 О, ТО—ДЯ —AG. Следовательно,
если бы мы сумели сделать эту реакцию токообразующим
процессом, то могли бы практически полностью перевести
энергию горения угля в электрическую энергию (а в цели
топка — паровой котел — турбина — генератор коэффи-
циент полезного действия достигает лишь 20—31) %).
6. Стандартные электродные потенциал гл
Для химической реакции, протекающей в гальваниче-
ском элементе в стандартных условиях, уравнение (11.12)
примет вид
- iG’=flFEe, (ПЛ5)
* Дикисйшсе nnobinicitHc давления оы34екп изменение знака
работы.
** — Ай (кал) = 23 0Ы пЕ tB),
ЭБ ,:Научнов наследие России"
69
ЭБ ,:НаучноЕ наследие России1
рис. 29. Зависимость стандартного электродною потенциала в йодном растворе
от /ЕОрядкояого номера элемерЕтя Л 0j4+ = I; все ионы гидратированы)
где Е° — электродвижущая сила при условии, что все реа-
генты находятся в стандартном состоянии. Подобно тому
как величину Лб’ реакции можно вычислить путем алге-
браического суммирования значений AG° образования реа-
гентов, воспользовавшись для этого уравнением вида (II. 9),
так и величину £’ можно рассчитать по разности стан-
дартных электродных (оки^ттелино-восстановительных)
потенциалов Еа{. Так, для реакции (VIIJ) £° = £си — Е%п.
Абсолютную величину Е, измерить невозможно, так как
в любом гальваническом элементе протекают две электрод-
ные реакции, и его напряжение равно разности электрод-
ных потенциалов. Поэтому приходится пользоваться отно-
сительными электродными потенциалами. Условно при-
нимают равной нулю величину Е* водородного электрода
(платиновый стержень в растворе кислоты, насыщенной
водородом) при I = 25 X, концентрацию ионов водорода
в водном растворе — равной единице и давлении водо-
рода— 1 атм. Разумеется, такая условность фиксирован-
ного начала отсчета не сказывается на получаемых резуль-
татах, так как приходится иметь дело не с абсолютными
величинами, ас разностью двух величин.
Значения стандартных (нормальных) электродных потен-
циалов Езда по отношению к водородному электроду для
некоторых «полуреакцнй» приведены в табл. 9.
Здесь они расположены по убывающей величине отри-
цательного потенциала, чему отвечает падение восстанови-
тельной и рост окислительной активности. В соответ-
ствии со сказанным ла с. 61 эти величины относятся к р ®=
= 1 атм для газов и С = I Л! для растворов.
В значениях стандартных электродный потенциалов
проявляется периодичность свойств элементов (рис. 29),
Следует подчеркнуть, что можно говорить лишь о сход-
стве графиков ЛСДк = / (Z) (см. рис. 25) и = ф (Z)
(рис. 29), а не об их подобии, так как первый отвечает
кристаллическим веществам, а второй — гидратированным
ионам.
Значения EJSi могут быть использованы при решении
ряда задач.
Рассмотрим один пример. Расположив металлы по вели-
чине их £'ijS, получим ряд напряжений металлов. В нем
каждый предыдущий металл активнее последующего и
поэтому может его вытеснять из раствора. Так, цинк стоит
выше меди; поэтому и происходит реакция (VIII). Дей-
ствительно, для нее £iSS = (finfcu == + 0,34 — (—0,76) =
ЭБ ,:Научнов наследие России"
71
T a fl л и ц а 9
Стандартные электродные потеЕкциали (В) н водных
рйстаорйх (Crirj]L = 1 М; все коны гидратированы)*
Элеитрiu.ii пя реждип
Окислен we L i * + е = L i Восста нов ленное —3,01
Ceci оя к и с К Ь+ -f- е = R Ь елето ы ине Очель слабые K+^f”K Очень сильные 1 -2,98 —2,93
= ок нсл ме ли Ва3 н + 2е и В а восста новин Cai4--|r2e=Ca Nfl* + s = Na Асаг -j- Зе= Ас La4 н + Зе ™ L а CeJi’4’3ff=Cc ME»++2s=.4g ¥^!Ч-Зе = Yb Sft+3e=Sc Th*v-|-4e^Tl! Ве?нЧ-2е=Ве AI^+3e-A| ZnO’ - + Й HtO -f 2е = 2 n + 4ОН“ Те+2₽=Тй»- ЗОГ+НР+2е=SOJ+2OH- 2Н/)4-^=Н£+ 2ОН- И13ОИ iL 1 1 1 1 1 11 II 1 1 1 Н -— ~ Г* “ “ J w ’ -- '< N bi KJ р — К c -1У >, (u Jj > i к j( Ь С H- J
Й Q g -0ДО
Я x —0,K3
я Zn*4-2e=4n я
м 4s+3Hi-|-3e = AsH! >я mi <11 CJ1 О» ft -4S?^
о 2SO=’ + 3HjO + 4e= S/Й - + GOHJ S|O{-4 4! 4^2e —2H,S<X я
S + 2e = S*’ 5
5 FtfH-f-2e=Fe = Ш 1 —0^4
д Cr»'-|-e=Cra+ £ —0,41
5 ti--h=ti ' Я
= Со=Ц-2е=Сй s
м о Ni* h-j-2e^ Ni о р —0,25
CJ S Sn*’’-f'&”Sn CfOf ’MH10-|-3e—одень (PJ + 5OH- 1 —0J4 -41. L3
д Pbs4'S₽=Pb —0.ГЗ
и MnO, +2H UO+ 2e= Мпдень (p}+ 2OH’ tj —0,05
L=k Hg|?+H-2e = Hg + iI' Q. —0,04
СП D [П £ 0.00
iXi NO; 4- H.O+2C = NO; + 2OH" ЛйВ’г4-е=Ди4-Вг- Srponj / 4- 2Hr 4- 2e — HsS (p) Cn’h-|-e»Cuh Sn'+ + 2fr-Sn*+ AjjCJ -h?= Ag -f'Cl’ PbO, 4-H/J +2f = Р1Ю + 2OH’ CJO, +Нде + 2« = СЮ; +2OH- Ca=i--|-2f=Cti GO; 4-Нде+2p «CIO; 4-2011" SO;-4-3lJ++fie^S-]-4l!BO 0,01 0,07 0J4 0,15 0J5 43,22 0/25 0,33 OpH 0.35 0.35
72
ЭБ 'Научное наследие России1
реакция
Лроболлишда fFUfri. 5
₽1И
I
я
S
н
ГС
Оа Ч-2Н-0-к 4е =ЧОН-
HjSOj Ч-4Н* Ч-4еS 4- ЗНгО
Cui+-pe=Cu
1.(к)+а?=й1-
МпОг4-е=МпО1"
MnO; -1- ЙН jQ -|-Зй « МпО* + 4ОН-
BrO; +ЗНХ)+бе« Вг~+6ОН*
С1О,- + HjO + 2е ~ СЮ"+2ОН-
Qa 4-2H++2# ^HjO,
Fe*++t=Fe**
Ag' -M= Ай
S4g»*+2c=[Hg1l»*
NOr + 4 H1 4- 3t = NO 4.2 H ,0
HNQ»+ »++r ₽ NO 4- H j5
Brs (я0+2е^2Вг"
J О,- + 6H ¥+5f=vf 1 i (к)+3H.C
01-MH - -F-4e=2H1O
MiiOs-<-4 H *-|-2e == M rt?f + 2 H ,0
Q, (r) + H»0+& = <>! + 20H“
TP +2f — T[+
2HN0. ([>) + 1H -Ц-4e -N,O (r)-|-3H4O
СггОГ + HH +te = 2Сг’ь-|- 7H .0
CI.+£e=X|-
Au,1r-|-3? = Ati
PbO, -MH ► -I- 2e PIP*- 4-2H1O
МпО; + Я H x+5e == Mn*h 4- 4H E0
ЕЗгОгЧ-СН1 +&±-V,Br, (jkJJMHjO
O-bFLO + Ze^aOH-
НС1ОН-Н*-М=Н/}+С1,
MnO; + 4 H+ J- 3e _ M nOs + 2 H s0
Au+-M= Au
HA+2H+4-2#=aH1p
Xt-0,4- 6H+ = Xc + 3HSO
Co»*-H=Ctf*
JC
;=
ь
й>
C2l
0’ifMib Ft4-2s = £F-
сильные Н|Хе£Ц+2Н‘ Ч--₽=
окислители ^ХсОдЧ-ЗНйО
FIKH++2e=2HFl?)
Окисленное
состояние
Очень
слабые
еосстйко-
Ештелм
Восстанов-
ленное
состояние
0,40
0,45
ОД*
0Л4
0Д4
0^30
0,61
0.66
0.6Я
0,77
0,80
0.92
0.96
1,00
1,07
1J9
К 23
1,23
L2I
1.25
1,29
!Р36
1J2
1,46
1.5J
1.52
1.59
1,63
1,70
1г?0
1J8
1,8
1,81
2,07
2,86
3.00
З.об
*Длв угрАщеиия bjttheb опуЩЕнО лг^етлнсн! СостояI'Kti в в символе f—
Днзк минус.
73
ЭБ ' Науччое наследие России1
— 1,10 В, т. е. > О и в соответствии с (П. 15) < 0.
Очевидно, при контактировании двух металлов в растворе
первым будет растворяться тот, который расположен выше
в ряду напряжений. Лишь после его полного растворения
начнется растворе si не второго, более благородного металла.
Так, из табл. 9 непосредственно следует, что оцинкованное
железо не будет корродировать до растворения всего цинка.
Иное дело луженое железо; достаточно обнажиться неболь-
шому участку железа, как оно начнет растворяться.
Для выяснения причин различия в электродных потенция лак,
я поэтому и в значениик AG каждого элемента надо оценить энта.ль-
пиннъей и энтропийный вклады ц величину AG. Например, процессу
Me(K) = Me*i-(p)+2e
для 2п отвечает АН^ц = — 36J2, а для Си -Н6ХЮ ккал (табл. 4).
Следовательно, Д7/ в рассмотренssoft на с. 67 реакции (—52,^0) почти
не отличается от AG для кее- К тем же результатам можно прийти
и ня ос нова ин и данных табл. 6- для Zsi полупим = —36,384 эг е,р
а для Си —30,088 эг ег Следовательно, различие в величинах Д$ столь
мало (*~6,3 5, с.), что членом TAS можно пренебречь. Иначе говоря,
в данном случае рсакциоЕЕную способность определяет тепловой эффект,
хотя, как уже отмечалось ранее (с. 26), лсекость отрыва электронов
от атомов ни и кз и меди примерно одинакова. Подобным же образом,
несмотря на то, что JLi > /N,. a литий и кальций нмоют
более отрицательный электродный потенциал — сказываются малый
размер иона лития и большой заряд иона кальция (по сравнению с но
ком натрия).
Хотя и процессе
//23,(r)+e^3’(pJ
для фтора ASJOt = —27,58 э. е., а для хлора —33,14 з. е. (см. табл. 6),
т, е. Д (ASJhJ = 14 s. а поэтому .разница в значениях TAS для них
составляет^ 4,3 икал, однако значения Д/Z {соответственно —79,79
и —39,94 ккал; ем. табл. 4) отличаются на вынчину (-*- 40 ккал),
превышающую предыдущую на порядок. Поэтому и в данном случае
можно пренебречь энтропийной составляющей и объяснить большее
значение Ай (£) для фтора по сравнению с хлором энергетикой с<ют-
ветствующего процесса (хоти сродство к электрону у них почти нс от-
личается) *.
Конечно, все это не озЕшчасг, чти можно пренебрегать энтропий-
ной составляющей величины Аб во веек случаях.
Не следует преувеличивать возможностей использова-
ния значений Ej14 (н+ в частности, роль энергии гидрата-
дни). Ведь они относятся к частному случаю
разбавленных (строго говоря, бесконечно разбавленных)
водных растворов при t — 25 X ч р = 1 атм. Поэтому
они не позволяют судить □ направлении реакций при иных
* См. также с. 23 и ел,
ЭЕ Й4-."нов наследие России"
температурах, очень высоких давлениях и значительных
концентрациях, а также в неводных растворах *. Правда,
в большинстве случаев можно считать, что давление не ока*
аывает существенного влияния на £, а отличие концентра'
ций от единицы можно учесть; однако последнее приводит,
как правило, лишь к приближенным результатам, Основное
же заключается в том, что вне количественной оценки
остается множество самых разнообразных процессов. В этом
отношении значения Е уступают универсальному Крите’
рию Д<1
7, Константа химического равновесия
С момента наступления равновесия в реагирующей си-
стеме концентрации (парциальные давления) веществ не из-
меняются. Поэтому для процесса (I), протекающего при
р, Т — const, будут неизменными и отношения (стр. 108
и сл.)
Кс=^~ ("16>
G д1. D ..,
И
где Ci и pt — соответственно равновесные кон-
центрации и парциальные давления. Величины /<с и
называются хилач^ского равновесия. Их чис-
ловое значение при прочих равных условиях зависит
от характера процесса и глубины его протеканий. Так,
для реакции СН* (г) + CQs (г) 7 2СО (г) -j- 2Нг (г)
rt--rt--1
Это выражение легко конкретизировать. Действительно,
полагая, что справедлив закон Дальтона
Pi = Робщ^Ь (H-1S)
где — мольная доля, и в соответствии с тем, что по опре-
делению (см. с. 140)
(П. 19)
• Данные л о э на пеня ям для неводных растворов весьма
ограниченны, £ ыиогне йз ныеющнхея противоречивы,
ЭБ ,:Научнов наследие России"
75
получим
к f_________V
* F?CHj'*COi ^rtCo+rtH1+flCFlb + rtCO1
где nf — число молей компонента i и смеси, р — общее
давление.
Рассмотрим процесс, в котором наряду с газообразными
реагентами участвуют твердые или жидкие вещества (не об-
разующие друг с другом растворов). При данной темпера-
туре парциальные давления реагентов, находящихся и кон-
денсированном состоянии, постоянны л не зависят от ко-
личества реагентов (как и давление насыщенного пара
какого-либо вещества, например воды, при данной темпе-
ратуре). Поэтому эти парциальные давления независимо
оттого, пренебрежимо малы они или существенны, из вес тезы
или неизвестны, можно включить в константу равновесия.
Следовательно, в подобных случаях в выражение (ПЛ7)
следует ввести только равновесные парциальные даЕзле-
ння газообразных реагентов. Так. для реакции CaCO;t (к) =
СаО (к) 4- СО2 (г) КР = Рсйг
Константа химического равновесия зависит от пр tip оды
реагентов и температуры; tin от давления (при не очень
высоких давлениях), ни от концентраций, наличия или
отсутствия примесей она не зависит.
Очевидно, чем значительнее убыль энергии Гиббса,
тем больше константа равновесия. Математически эта связь
выражается простой зависимостью:
й<Гг ₽ Д/Гг -Т AS}. - — In Я - -4,576Т In X, (I Г.20)
позволяющей по величине Дб’ (или AH’ п AS’) вычис-
лить К, а затем, пользуясь (JI.16) или (11.17), и равна-
весные концентра ним (парциальные давления) реагентов.
Следует подчеркнуть, что соотношение между стандарт-
ным изменением энергии Гиббса процесса и константой
его равновесия является универсальным. Оно применимо
к любому равновесию — н к диссоциации электролита
в растворе (см. раздел IV, гл. IV), и к равновесию между
кипящей жидкостью и Сухим насыщенным пэром (% —
давление пара при данной температуре), и к равновесию
растворенное вещество — насыщенный раствор (К = Смм).
Сочетай tie уравнений (11.20) и (11,15) позволяет найти
константу равновесия и я электрохимическом процессе.
Таким образом, целесообразность в веде Finn величины AG*
подкрепляется еще одним весьма существенным обстоя тел ь-
ЗБ ,:|ТДчнов наследие России"
с твои. Из (И. ЙО) непосредственно следует, что большим
отрицательным значениям ДО’ отвечает большое значение А',
т. е. преобладание в равновесной смеси продуктов реакции.
Наоборот, если Абг 0, то в равновесной смеси будут
преобладать исходные вещества. Следовательно, в первом
случае можно пренебречь наличием исходных веществ,
во втором—продуктов реакции. Если же Лбэ = 0, то
К — I и в соответствии с уравнением (11.20) ^0,
что подтверждает сказанное на с. 62,
Так, для реакции
2CHjr)=cgHt (4+зн, (г)
ЛЯ!„ = 54,02 - 2 (— 17,88) -89,78 ккал (табл. I);
Д$;„ =48,«Н 3-31,195-2-44,53=52,725 э. <?. (табл. 6),
откуда Лбт (кал) = 89 780 — 52,725 Г и, в частности,
Д0й» = 74,060 ккал (последняя величина совладает с най-
денной по Приложению I: 49,827 4 2 (—12,146) =
= 74,09 ккал). Следовательно, в соответствии с (11.20)
74 060= — 4,576 238,2 1g Кр,
откуда
(А = 5,35- ГО"Й,
^сн,
т. е. равновесие смещено влево. Для оценки масштаба этой
величины укажем, что если (рр>|Ш)сн, = (^Jh, = 1„-
то мы получили бы концентрацию С:Нй, равную 1 моле-
куле в—' 10®* кубических километрах. Однако Д5 0;
поэтому при высоких температурах равновесие смещается
вправо.
Уравнению (11.20) можно придать следующий вид:
_ (15*
К=С 7ГГе"1Г, (Ji.21)
где е — основание натуральных логарифмов. Это уравне-
ние, как I! (11,10). отражает противоположное влияние
на равновесие энтальпийного и энтропийного факторов.
Первый показател в степени сил ьно эавнсит от температуры,
второй можно считать постоя иным: при низких темпера-
турах решающее значение приобретает АЯ^, при высо-
ких — AS° Вид уравнения (11.21) свидетельствует о боль-
шой чувствительности, величины К к значениям А//’
77
ЭБ ,:Научнов наследие России'1
и Д5в; даже небольшое изменен не последняя приводит
к значительному изменению К н в соответствии с (11.16)
к существенному изменению состава равновесной смеси.
Вифиъ следует подчеркнуть, что урявненне (П.20) пркуеннмл
к любому равновесию (при р, Г ™ соо$1) кипящая жидкость — насы-
щенный парь насыщенный раствор — избыток растворенного вещества
и ъ д.
В заключение отмстим, что величины Лб и Ди связаны с уравне-
ниями
cict...
iO = AGeH-J?7’— (И.22)
cjcU.
и
ЛG = iCF + RT in -?-=-, (11.23)
pVb
из которых в соответствии e on pc делением величины d(j° следует coot-
ношение Cl-E.’M). Для злсктрокимЕсческнл реакций в соответствии
с {11.14) и (II.1&) имеем
ЯГ
----|л
nF
(11.24)
Ура вне неен (11.221 л (11.23) называются
я уравнение (П^4)—^нан^ниед tffpHfffia.
Главе IV
ВЛИЯНИЕ ИЗМЕНЕНИЯ УСЛОВИЯ НА ХИМИЧЕСКОЕ
РАВНОВЕСИЕ
L Принцип Ле Шателье
Влияние изменения условий на положение равновесия
определяется правилом, которое называется принципом
Ле Шателье (1834) или принципом подвижного равно-
весия-, если на систему, находящуюся в истинном равнове-
сии, оказывают воздействие извне путем изменения какого-
либо из условий, определяющих положение равновесия,
то оно смещается в направлении того процесса, протекание
которого ослабляет эффект произведенного воздействия.
Противодействуя произведенному изменен ню, система пере-
ходит из одного состояния равновесия в другое, отвечаю*
7В
ЭБ ,:Научнов наследие России"
шее новым условиям. Это связано с тем, что внешнее воз-
действие в разной стегте!1и изменяет скорость двух взаимно
противоположных процессов.
Принцип Ле Шателье справедлив м в отношении равно-
весных систем, несвязанных с химическими превращениями
(кипение, кристаллизация, растворение и т. д ; см. часть IV).
Он не применим к системам, находящимся в кажущемся
равновесии, ибо выход из такого равновесия означает
течение одностороннего процесса.
Рассмотрим влияние различных факторов на химиче-
ское равновесие.
2. Влияние температуры
В соответствии с принципом Ле Шателье нагревание
вызывает смещение равновесия в сторону того из двух
встречных процессов, протекание которого сопровожда-
ется поглощением теплоты, иначе говоря, повышение тем-
пературы вызывает возрастание константы равновесия
эндотермического процесса. При этом подразумевается,
что константа равновесия отвечает выражениям {II. 16)
или (11.17), т. е. d их числителе стоят концентрации (пар-
циальные давления) продуктов реакции. Естественно, что
понижение температуры приводит к противоположному
результату: равновесие смещается в сторону того процесса,
протекание которого сопровождается выделением теп-
лоты, иначе говоря, охлаждение благоприятствует экзо-
термическому процессу и вызывает рост константы его
равновесия.
Из изложенного следует, что для суждения о влиянии
температуры надо звать тепловой эффект изучаемого про-
цесса. Его можно найти или ОПЫТНЫМ путем, или С помощью
Закона Гесса.
Выводы, к которым мы пришли, можно показать на гра-
фике (рис. 30). Рис. 30, л отвечает эндотермическому про-
цессу (при повышении температуры равновесие смещается
в сторону продуктов реакции); рис. 30, б соответствует
экзотермической реакции (нагревание приводит к обрат-
ному результату).
Направление смещения равновесия в резуль-
тате изменения температуры определяется знаком теп-
лового эффекта. Степень смещения равновесия опре-
деляется величиной теплового эффекта: чем больше
ЭБ 'Научное наследие России"
79
&fi, тем значительнее влияние температуры; наоборот,
если Д// близко к нулю, то температура практически
не влияет на равновесие (см. также с. 82 и ел.). Хотя, как
уже отмечалось, тепловые Эффекты с Температурой ме-
няются незначительно, но для химического равновесия
в очень широком интервале температур следует принимать
Рис. 3D, Влияние температуры на химическое
Р = const (схема):
равновесие при
d — sii д (>т(!ры й ч ка л реакции; £ — экзотермическая реакция
во вЕпзмание возможное изменение Д7/, Если по абсолют-
ной величине значение Д/У невелико, то может произойти
и перемена знака A/Y. Это будет означать что изменение
температуры вызовет изменение знака температурного
коэффициента константы равновесия.
Все эти выводы непосредственно следуют из уравне-
ния (IL2I), с помощью которого можно количественно
определить влияние температуры на равновесие.
Процессы разложения химических соединений обычно
протекают с поглощением теплоты, а процессы синтеза —
с выделенЕтем (см,р однако. с 31). Поэтомут например,
равновесие реакции
Нд=2Н (IX)
при повышении температуры смещается вправо, а при
понижении температуры влево; етными словами, нагрева-
икс вызывает увеличение в равновесной смеси концентра-
ции атомарного водорода, а охлаждение — молекулярного
водорода. Соответствующим образом меняется и константа
равновесия этой реакции. Для этой реакции в согласии
ЭБ ' 1-Й?ччсе наследие России"
с уравнениями (ПЛ7) и (II. 18) степень диссоциации се
равна * *
«=]- (№5)
Так как изобарное повышение температуры вызывает
рост Kpt то происходит увеличение я. На рис. 31 зависи-
мость « от Т показана при разных давлениях (линии ad
Чем Прочнее
тем сильнее
на подобного
о; fl
Цб
04
0.2
о
300Q 3000 4М0 5000
Г
Рис. 31. Влияние темпе-
ратуры Т (К) и давления
р (етм) На степень диссп-
11ИЯЦМН а водорода
и сс; р — I атм),
молекулы вещества,
влияет температура
рода равновесия.
Другим примером может слу-
жить равновесие реакции 2NO, =
NiO4. Легко догадаться, чти на-
грева н не стимулирует диссоциа-
цию молекул М/)ъ охлаждение —
ассоциацию молекул NOS. Этот
пример является хорошим демон-
страционным экспериментом. Дело
в том, что диоксид азота окрашен
в бурый цвет, а четырехокись азота
бес цветя а. Возьмите П-образную
трубку, нижние концы которой
раздуты в форме шаров, и запол-
ните ее диоксидом азота. Затем
опустите один из шаров в горячую
воду, а другой — в холодную или
тающий лед.
Изменение окраски препарата
свидетельствовать о влиянии температуры на рассматри-
ваемое равновесие.
будет непосредственно
Н1=2Н
* Исходное состояние системы , + J моль 0 моль
Равновесное состоя н tie систем и . , (I—я) моль 2a мол!>
Общее количество рслгеггтса . . + d +*) мъть
Пэ ринал ь н ые дав л tun я ре а та- ] tt 2a
той д непкнт равновесия .... р /j
В соответствии с (IIJ7)
2а у
tf ^11 ... У Г IJ-,
4л‘
₽T^ai'
Следовательно^ JCP — w2(/fp4- 1/?} или (11.25).
fl[
ЭБ ,:Научнов наследие России1
Выясним теперь, как минет температура на направле-
ние смещения равновесия в реакциях:
СОг(г)+ниг)«СО(г)+НаО(г)
С (гр) + со»(г)“SCO(Г)
Определим вначале, пользуясь законом Гесса, тепловой
эффект первого процесса. Для этого можно поступить
двояко: или от суммы те плот образования СО и HtO отнять
теплоту образования СО», или из теплоты сгорания Нг
вычесть теплоту сгорания СО. В результате получим вели-
чину ЛИ — 9,84 к кал/моль. Следовательно, нагревание
будет смещать равновесие влево, охлаждение — вправо
(см. с. 35); иными словами, с повышением температуры
в смеси будет расти содержанке оксида углерода и водя-
ного пара, причем, так как ЛН не очень велико, этот рост
не будет значительным.
Судить о влиянии температуры на втором процесс можно
по такому признаку: на стенках камер и дымовых труб,
через которые проходят продукты этой реакции, оседает
сажа. Объяснение очевидно: так как смесь СО н СО»,
удаляясь из зоны взаимодействия, охлаждается и при этом
равновесие смещается влево, то выделение углерода явля-
ется Процессом экзотермическим, с решением этого во-
проса можно связать и некоторые другие. Например, горя-
щий уголь при интенсивном дутье дает большую темпера-
туру, чем при медленной подаче воздуха: образующийся
при окислении угля диоксид углерода при интенсивном
дутье не успевает восстановиться до СО; значит, э ц дотер -
мическая реакция, течение которой в условиях затруд-
ненного теплообмена (уголь — плохой проводник тепла)
вызывает понижение температуры, не происходит.
Характер влияния температуры на равновесие можно
было бы рассмотреть и на основании уравнения (П.10),
исходя из знака н абсолютного значения AS. Ведь при
равновесии ДО = 0, поэтому при Д5 <; 0 н Д£/ с 0, а при
AS >0 и А// >0.
Это соображение существенно, так как в связи со ска-
занным на с. 38 знак AS и порядок этой величины легко
определить.
Высокотемпературные процессы. Исследуя злнянне тем перату ры
на ленный процесс, следует иметь н виду не только возрастающее
с повышением температуры действие энтропийного фактора, но и воз-
можность резкого илменонвя устойчивости различных неществ.
Повышение температуры может вызнать и течение новых про-
цессов. Вот два примера. Равновесие реакции ,/s.N'a -|- J/s О» — NO
ЭБ кучнее наследие России"
с ловышечЕЕем темпер-этуры смещается дЕврэво (для ее» АН > 0), т. е.
пасревание вызывает увеличенне содержатся окиси азота в равновес-
ной сМеСн (в)роцесс проводит в щхчътовоЛ дуге) Однако если бы мы
попытал весь оиущестьнть згу реакцЕвю при еще более выгонон темпера-
туре, то выход КО, пройдя через махсикЕум при — 3500 К (— 8 К NO),
с дпльнейшнм повышенном температуры начал бы падать н при Т =
— БОСО К снизился бы до 4 %
(□место того чтобы подняться до
20 ?4) (рле 32). Это лишь кажу-
щем а противоречие принципу Ле
Шательс При очень высоких тем-
пературах усЕмивается диссоциация
молекул и прежде всего молекул
кислорода: анергия связи для О»
меньше, чем для Ns. Действитель-
но, при 3000 К кислород днссосш-
ирует на 10%, а азот всего на
0.1 %. Диссоциация приводит к
Реес, 32, Зависимость выхода
NO в реакции
V^+^-NO
от температуры Г (К) и давле-
ния j? (атм)
расходу кислорода чше ло л аз Есаче -
Е1ию*, а поэтому и к уменьшению
выхода NO (рис 32),
Обсуждая осуществимость син-
теза аммиака при очень высоких
температурах, надо учесть плия-
ние на равновесЕЕе деессоцеейцеен
водорода и взаимодействия атомарного водорода с азотом: это
взаимодействие приведет к росту выхода аммиака с тем перату рой .
N При температурах в тысячи и
Рис. 33- Зависимость равееоецк-
ногосостава воздуха (смссее ада-
та и кислорода) в мочь ныл до-
лях от температуры Г (К)
десятки тысяч градусов Скорость про-
текания процессов становится столь
BHaHEET&ibHOkl, что торможения прак-
тически не существуем т. е, проблемы
скорости отходят на задний план; н
ЭТИХ у СЛ0ВЕЕ Л к рСЗ КЦИ0И n а я способ-
ность веществ определяется л основной
энергетикой процессов. Однако это
вовсе ле означает упрощения хими-
ческого поведения веществ. Достаточно
указать еез то, что процессы парооб-
разовании и сублимации при высоких
температурах сопровождаются хими-
ческими лревращеЕЕнямн. Наряду с
усилением диссоциации и упрощением
частиц (и чаел1остгг1 образованием раз-
л ичн ы х двух атом ны к части ц — Cj,
ОН, CaCI, SiO. АЮ, M<*N, SO ее мио
гих других) происходит форм ирон 3 НЕЕС
более сложееых частиц (например, за
счет димеризации), прячем нередко их
концентрация растет е температурой;
да и многие р^дьскады прнобрстаЕст
свойства молекул. Так, в высокотен-
пературиых парах алюмофторидд лиги я
наряду с молекулами LiAIFj содержатся частицы LiF, (L]F)j, AIF3,
В парах карбида крен tin я при высоких температурах наряду с иоде-
ез
ЭБ ,:Научнов наследие России"
кулэми SiC зарегистрированы чзгтниы Sis, Si3, SiCa, SisC SijCj. Si/^;
в пэрах VnOr, — частицу VjOm. V.A, VbO]n VhO(i, V/?j и т. д. Чем
выше температура, тем больше содержание частил, в которых зле-
менты ггахолятся а несвойственегых нм степенях окисления. Таким
образом, представлен не о ларе как о среде, бедной процесса ми > уста-
рело. Особенно велико разнообразие частиц в ноппэнровамнои
паре.
Причинами существования и высокотемпературных парах укруп-
ненных чао гни квляются участие is их формировании валентно ненасы-
щенныч а гомон (или радикалов), образование продуктов меж ди лол ь-
пого взаимодействия [например. (LiF)a]. образование частиц а воз-
бужденном состояния (Mgt) и т. д,
Сложность и своеобразие изменения состоя определяются соот-
ношением значений тегглот диссоциации, парообразования, сублима-
ции, димеризация и других параметров процесса.
Возьмем в качестве примера такие сравнительно простые частицы,
как молекулы На<Х В водяном паре уже при температурах порядка
2000 °C можем обнаружить частицы Н,, Otl ОН, Н и О, я при больших
температурах — многочислениые продукты ионизации ОН, Н и О.
Эти явления характерны я для других систем. Так. из мыслимого на-
бора частиц СаС2 — СаС1+ — — Са* — Са'1+ по мере повышения
температуры постепенно исчезают стоящие слева.
На рис. 33 представлены результаты расчета термической диссо-
циации воздуха пол атмосферным давлением. Подобная картина полу-
чился и для водяного пара. Так, при Г> 3600 К копией грация & рав-
новесной смеси нестабильных при обычных температурах частиц ОН
оказывается больше концентрации стабильных при обычных усло-
виях молекул воды. т. е. при таких температурах эти группы устойчи-
вее водяного пара.
3. Влияние да пл они fi
Давление не изменяет константы равновесия, так как
в согласии с (11.20) она зависит только от температуры;
следовательно, с давлением меняется Д6 (а не ДСС).
В соответствии с принципом Ле Шателье сжатие сме-
щает химическое равпоосспе в направлении процесса,
сопровождающегося уменьшением объема, а понижение
давления вызывает сдвиг равновесия в противоположную
сторону. Таким образом, направление смещения
равновесия определится знаком AV. Так, повышение
давления будет смещать равновесие реакций (IV) и (VI)
влево, а реакций (II) и (VII) — вправо (см. рис, 24 и 34).
Очевидно^ при вычислении ДУ можно пренебречь объемом
нс га зообразн ы х реа ге нтов.
Для определения степени смещения равновесия
надо знать абсолютную величину Так, если на реак-
ции
Ия-кЗНг-2КНл (X)
и
СО + 2Н^СНЭОН 1X1)
ЗБ 1-Й^чсе наследие России"
повышение давления влияет почти одинаково, то для реак-
ции 2SOt 4* Ог = 2SOa его действие окажется менее эффек-
тивным.
В предыдущую фразу слово «почти» вставлено не слу-
чайно. При обычных давлениях 1 моль любого газа при дан-
ной температуре занимает практически один и тот же
объем. Поэтому если бы реакции (X) к (XI) осуществлялись
Иетспди не Продукты Исходные Продукты
pt агенты реакции реагенты g реакция
Рис. 34. Влияние давления на химическое рзвновссне
при Г = consl (схема):
4 — реакция, сопровождающаяся уменьшением объема;
£ — рз-экцр! к, сопрйважля юща яс я у lie л нче jhibm объема
при невысоких давлениях, то они сопровождались бы
одинаковым изменением объема — в обоих процессах коли-
чество продуктов реакции на 2 моля меньше, чем исходных
веществ. Но при высоких давлениях — порядка несколь-
ких сот атмосфер [целесообразность такого режима выте-
кает из принципа Ле Шателье) — сказывается индивиду-
альность каждого газа, различие в отклонении его ОТ
свойств идеального газа, в частности различие в сжимае-
мости; поэтому при очень высоких давлениях AV в реак-
циях (X) и (XJ) будут не совсем одинаковыми (хотя Аиг
равны).
Повышение давления уменьшает степень диссоциации
газов (ДУ< 0). Так, сжатие в соответствии с уравне-
нием (1L22) смещает равновесие реакции (IX) влево,
понижение давления— вправо (линии о!? и on на рис. 31).
В подобных случаях, когда повышение давления и тем-
пературы приводит к противоположным результатам, для
Достижения одного и того же состояния требуется одно-
«б
ЭБ ,:Научнов наследие России"
стороннее изменение давления или температуры. Этот
вывод следует и из рис. 31 (сдвиги о/ и ое}.
Чем меньше абсолютное значение А V, тем меньше влия-
ние давления на равновесие. Поэтому на равновесие реак-
ции (I —рис. 24) давление не влияет. Однако, строго
говоря, это справедливо лишь при нс очень больших дав-
лениях. Если бы нас заинтересовал вопрос, будет ли влиять
давление на это равновесие при р — 500 атм. то следовало
бы ответить утвердительно. На первый взгляд, это легко
объяснить так: при р — 500 атм водяной пар сконденси-
руется, а объемом жидкости можно пренебречь; следова-
тельно, течение реакции вправо будет сопровождаться
значительным уменьшением объема. Но вспомним режим
процесса: ведь он осуществляется при температурах, пре-
вышающих критическую температуру воды! Равновесие
здесь сместится вправо, но незначительно, и по другой
причине. Степень отклонения неполярных молекул СО2
и Нг и полярных молекул СО и 112О от свойств идеальных
газов неодинакова; поэтому реакция будет сопровождаться
некоторым изменением (н данном случае уменьшением)
объема. Это означает, что при высоких давлениях повы-
шение давления вызовет сдвиг равновесия вправо.
Мы остановились на этих примерах потому, что все
шире и шире используются высокие и сверхвысокие дав-
ления. В этих условиях нельзя считать, что признаком
неизменности объема при процессе служит равенство
Ддг = 0. Ведь тождественность критериев A V = Он Ап = 0,
строго говоря, справедлива только для идеальных газов.
И еще одпЕ1 пример. Как будет влиять и а диссоциацию
карбонатов, оксидов, сульфидов и других соединений соз-
дание вакуума? Так как для процессов, подобных СаСОэ —
= СаО 4- СО2 н ЙА&О = 4Ag + <Л. АИ >0, то пониже-
ние давления будет фактором благоприятным.
Мы обращали внимание на то, что объемом конденси-
рованной фазы можно пренебречь. Однако надо учиты-
вать, что при сверхвысоких давлениях объем газов ста-
новится соизмеримым с объемом твердых тел и жидкостей
(см. с, 153). Не приходится говорить о случаях, когда среди
реагентов нет газообразных веществ; примером подобного
рода могут служить процессы модификационных превра-
щений, например С (графит) — С (алмаз). Кстати говоря,
так как в данном случае AV ничтожно мало, то даже для
незначительного сдвига равновесия вправо требуется весьма
значительное изменение (повышение) давления — так как
ЭЕ йтуччее наследие России"
плотность алмаза (3,5 г/см3) ЛИШЬ немного превыпгает
плотность графита (2,2 г/см1).
Для точного учета пл няни я давления на равновесие
реакций, особенно при высоких давлениях и в широком
интервале давлений, следует принимать во внимание изме-
нение величины AV с р; так, если по мере повышения дав-
ления абсолютное значение AV уменьшается, то будет
уменьшаться н эффект действия давления.
Остается добавить немногое; сочетание высоких темпе-
ратур с высоким давлением открывает новые горизонты.
Подтверждением этому служит искусственное получение
многочисленных веществ и синтез новых модификаций
различных соединений, в частности BN и SiO,.
4, Влияние концентрации
В соответствии с принципом Ле Шателье введение в рав-
новесную систему дополнительных количеств какого-либо
реагента вызывает сдвиг равгговссия в том направлении,
при котором его концентрация уменьшается. Поэтому
избыток исходного вещества (исходных веществ) вызывает
смещение равновесия вправо, увеличивая степень превра-
щения других реагентов' добавление продукта (продуктов)
реакции вызывает смещение равновесия влево, т. е. умень-
шение степени полноты ее протекания. Так, избыток кис-
лорода увеличивает степень превращения SOs в SQa при
контактном получении три оксида серы — возрастание кон-
центрации молекул веществ ускоряет ту реакцию, которая
ИХ расходует, причем константа равновесия не изменится,
так как она зависит для данной реакции только от темпе-
ратуры. Если направление смещения равновесия
в процессе определяется тем, какой из реагентов взят
в избытке, ТО степень Смещения равновесия при Дан-
ном количестве реагента определится величиной стехио-
метрических коэффициентов.
Рассмотрим протекающую в водном растворе реакцию
RCls4-3KSCN =t Ft; (SCN),-|-3KC1
Прибавление к равновесной смеси FeCls или KSCN вызовет
смещение равновесия вправо, что легко обнаружить по уси-
лению интенсивности окраски раствора (вследствие уве-
личения количества окрашенных молекул роданида же-
леза.) Наоборот, введение в раствор КО сдвигает равно-
весие в сторону исходных веществ — окраска становится
ЭБ ,:Научнов наследие России"
87
менее интенсивной. В соответствии с уравнением (fl. 16)
константа равновесии этой реакции
г
дг LFe(SCNhLKC£
С CFeCUCft5CN
Вид этого выражения показывает не только направление,
но и степень смещения равновесия- Изменение концентра-
ции КС! или KSCN вызовет более значительный сдвиг
положения равновесия, чем изменеЕтие концентрации
Fe (SCN)5 или FeCls; стехиометрические коэффициенты
в уравнении реакции для KSCN и КО втрое больше, чем
для FeCI3 н Fe(SCN)3.
Во многих случаях смещение равновесия в сторону
продуктов взаимодействия можно осуществить и их уда-
лением из реакционной эоны, связывая их в малодиссо-
циирующие, трудно раствор и мыс или нелетучие вещества.
Так, введение в равновесную систему
сн/)н 1 ci Tycoon — CH^OOCf 1э+НаО
водоотнимающих веществ (например, 1LSO4) позволяет
сместить эту реакцию вправо.
S. Влияние инертного газа
При введении в равновесную систему (при = const)
инертного газа концентрации (парциальные давления)
реагентов уменьшаются. Если течение процесса связано
е уменьшением объема (например, в случае синтеза амми-
ака). то равновесие сместится в сторону исходных веществ;
наоборот, для реакций, которые сопровождаются возраста-
нием объема, разбавление инертным газом будет вызывать
увеличение полноты реакции. Если же ДУ = 0, то си-
стема будет нечувствительна к пр1Есутствию инертного
газа. Эти выводы непосредственно следуют и егз закона
Дал ьто на. Дейс тли тел ь но, из уравнения (IL18) вид но,
что при данном давлении эффект разбавления (уменьше-
ние A/J подобен эффекту уменьшения общего давления ^6и[
в системе при данном составе.
6. Оптимальные условия осуществлепия процесса
Анализ влияния температуры, давления, соотноЕпенпя
между реагентами позволяет для каждого процесса выбрать
наилучшнй режим,
ЭБ Й^учнсе наследие России"
Так, если реакция является экзотермической и про-
текает с уменьшеЕ^ием объема, то для получения макси-
мального выхода продуктов реакции необходимо сочета-
ние низких температур и высоких давлении {рис. 35).
Говоря об оптимальном соотношении между реагентами,
необходимо обратить внимание на следующее: если рас-
ход одного из них по тем
или иным причинам лими-
тирован, то увеличение
концентрации сореатентов
позволит увеличить сте-
пень его использования;
она будет тем большей, чем
больше введено другого
реагента. При этом надо
иметь в виду не только за-
труднения! связанные с
удалением из равновесной
смеси избытка нелрсреа-
гировавшего реагента, по
и то обстоятельство, что
для получения максималь-
ного выхода продуктов
реакции нужна стехиоме-
трическая смесь.
Так, если синтез оксида
Рис. 35- Зависимость выхода амми-
ака в реакции 7sNj (г) + Я/,Н₽ (г) —
= ^Нэ (г) ст температуры и дав’
ЛСЕЕ11Н
азота осуществлять не из
стехиометрической смеси азота и кислорода (Ма : Оа = 1 : I),
а из воздуха, то выход NO снизится, причем величина умень-
шения выхода будет одинаковой как для воздуха (Nt : Ой =
= 4 : i), так и для смеси, обогащенной кислородом, если
в последней N3 : О3 = 1 : 4.
Окончательный выбор условий требует учета их влия-
ния на скорость процесса (см, часть III)- В связи с этим
разл₽1чают к с/лглеяи яол-
рдогцци. Первая отвечает установлен и ео в системе
равновесия (абсциссы минимумов на кривых рис. 22, 30
и 34), т. е. степени преврэщежЕя, которую легко опреде-
лить на основдеши выражения для константы равновесия
данного процесса. Однако далеко не всегда реакция дохо-
дит до равновесия; мешает этому медленность многих
процессов. Правда, с помощью катализаторов реакции
можно ускорить, однако и в этих случаях их часто нс до-
водят до равновесия, В связи с этим надо иметь в виду
S'J
ЭБ ,:Научнов наследие России"
возможность противоположного влияния данного фактора
if а статику н кинетику процесса.
Противоположное влияние повышения температуры на
ранповесне эндотерм и четкого и экзотермического процес-
сов, с одной стороны, н, как правило, благоприятное
влияние повышен и я температуры на скорость процесса —
с другой, приводят к выводу, что в первом случае опти-
мальным будет высокотемпературный режим. Во втором же
случае надо взвесить эффективность противоположных
влияний; однако часто введен ее с активного катализатора
позволяет без ущерба для скорости перейти на низкотем-
пературны и режим: примером могут служить экзотермиче-
ские каталитические процессы {X) фис. 35) и (XI),
Обсуждая влияние повышения давления и введения
инертного газа, следует помнить не только о разном на-
правлении смещения равновесия под действием этих фак-
торов, но я о том, что первый из них обычно ускоряет
процесс, а второй, наоборот, замедляет.
Гл а на V
ОКИС ЛИТЕ Л ЬНО-Ы)ССТАНЦВ ИТЕЛЬНЫЕ РЕ АКЦИИ
Все реакции можно разделять па две группы: в одних
степень окисления атомов остается постоянной, в других
она меняется. К первым относятся обменные реакции,
некоторые процессы синтеза и распада веществ, В отличие
от них протекание окислительно-восстановительных реак-
ции связано со сдвигом или-полным переходом электронов
от одних атомов (ионов) к другим — от восстановителя
к окислителю. Примером первых могут служить процессы
AgNQ, + N aCl = AgCl + N а NO,
BaO+HiO=Da(OH)1 и CaCOa = CaO+COt
примером вторых — реакция
SnCI,+2FeC!j = SnC!, + SFeCl,
В первых трех процессах как в исходные вещества, так и
в продукты входят одни и те же частицы *, в четвертом
набор частиц меняется, гак как течение процесса слева
направо сопри же ею с увеличением степей к Окисления олова
и с уменьшением степени окисления железа: произошло
* Об обменwx реакциях в расглоре см. с, 202 и сл,
ЭБ ’кучнее наследие России"
ок и еле] । we Sn1+ в Sn4f и одновременно восстановление Fe3+
в Fe2*.
Можно было бы привести примеры различных ок цели-
тельно- восстановительних реакций и не только ионных,
но и многих других. Все они сопровождаются изменением
степени окисления—ее увеличение является процессом
окисления, уменьшение—процессом восстановления, К про*
стен шей окислительно-восстановительной системе относится
электролизер: катод служит восстановителем, анод — окис-
лителем (электролиз —- универсальный и наиболее мощ-
ный окислительно-восстановительный метод).
Окислительно-восстановительные реакции чрезвычайно
важны. Это и Процессы горения, к получение различных
веществ (в частности, металлов и кислот), это и электро-
химические процессы, дыхание, фотосинтез, работа нерв-
ной системы и т, д,
Об окислительно-восстановительных свойствах элемен-
тов и соединений можно судить, руководствуясь периоди-
ческом системой элементов Д. И, Менделеева.
Т и пи ч я ы Мн ОКи ели тел ям] i я вл я юте я: а) п р осТЫС вещества,
атомы которых обладают большой электроотрицательностью
(элементы VIA- и VПЛ-групп); из них наиболее активен
фтор, а также кислород и хлор: б) ионы с дефицитом элек-
тронов: простые катионы с высшей или большой степенью
+4 +1 +5 +1
окисления, например Pb, Fe, TJ. Се, и сложные анноны,
в которых более электроположительный элемент имеет
высшую илн значительную степень пкцсленця, например
Л + ► -I ? J e I 1
(сад-, (СгАГ, (N<v. (Mnoj-, (SO.r, (doj-.tcioj-,
(BiOsy. (PbO3)4-, (CIO)-, (BrOi)-.
Растворы кислот—более сильные окислители, чем
растворы их солей, причем окислительная активность пер-
вых тем значительнее, чем выше их концентрация. Так,
КМОц (р) почти не проявляет окислительных свойств
(необходим очень сильный восстановитель), разбавленный
раствор HNQj является слабым окислителем, а концентри-
рованная азотная кислота — один лэ наиболее энергичных
окислителей. К окислителям относятся также пероксиды
металлов. Рост степени окисления не всегда означает
усиление окислительной способности. Так, в водных раст-
ворах НС [о — более сильный окислитель, чем HCLOj.
Типичные восстановители—это: а) элементы, атомы
которых обладают наименьшей электроотрицательностью
91
ЭБ ,:Научнов наследие России"
(элементы основных подгрупп I и 11 группы); б) анионы,
как простые, например Cl", S1", так и сложные, в которых
более электроотрицательный элемент ле имеет предельной
степени окисления, например (SQj)4", (NOs)r; в) катионы,
у которых степень окисления может возрасти, например
Ge4*, Мп**, Cr**", Ti®*; г) некоторые вещества при высоких
температурах, например С, СО, На.
В качестве восстановителей применяют также нагретые
магний, алюминий, цинк, железо н некоторые другие ме-
таллы. Восстановительная активность металла тем больше,
чем меньше их потенциал ионизации.
Вещества, содержащие элементы в максимальной и ми-
нимальной степенях окисления, могут быть соответственно
только окислителями или только восстановителями; ве-
щества, содержащие элементы в промежуточной степени
окисления, могут быть как окислителями (под влиянием
более актирного, чем они, восстановителя), так и восстано-
вителями (под влиянием более энергичного, чем они, окис-
лителя). К первым относятся высшие кислородные соеди-
нения, например РЬОг, HsSO4, HN03, КгСгаО7; ко вторым —
H;S и HNOe; к последним — HaSO3 и HNOS. Действительно,
например,
SO’" (Р) 4- 25 (Р)+6Н* (р)₽ 35е (ромб) + 3HjO (ж),
Дб«1 —— 94,59 ккал;
но
CIS (г) 4-5ОГ (?) 4- HaQ (ж) = SOf- (р)+2С1- (р) -J- 2Н* (р),
AGjjt = — 67,92 ккал.
Слабый окислитель проявляет окислительную активность
только иод влиянием энергичного восстановителя; мало-
активный восстановитель вступает в окнелительнО’Висста-
повительные реакции лишь под действием сильного окис-
лителя.
При записи окислительно-восстановительных реакций
обычно показывают, сколько электронов отдано окисли-
телем и сколько приобретено восстановителем. Условно
принято окисление отождествлять с отдачей электро*
нов, а восстановление — с приобретением элек-
тронов, т. е. не принимаются во внимание строение частиц,
природа химической связи в них и механизм протекающего
процесса. Ради упрощения записи обычно указывают
степени окисления лишь тех атомов, у которых она меня-
ется. Условным является и приписывание окислительно-
92
ЭБ ,:НауччоЕ наследие России"
восставовительных свойств молекулам вещества, В дей-
ствительности ими обладают, как правило, атомы (ионы).
Так, принято говорить, что энергичным окислителем слу-
жит перманганат (KMnOj, хотя речь идет об ионе (МпО4)-.
Большей частью трактовка окислнтельно-восстанови-
тельяого процесса как простой передачи электронов — лишь
удобный прием, облегчающий написание уравнения реак-
ции и подбор коэффициентов. Так, лишь формально можно
считать, что в реакции
2г-
I !
г и +! -»
Zn -р VjO^ = ZiiO
происходит потеря атомом пинка двух электронов к при-
обретение нх атомом кислорода. Ведь многозарядных
атомарных конов, в частности ионов 0s-, вообще не может
быть, тем более по соседству с катионом. В действитель-
ности связь между цинком и кислородом в молекуле ZnO
пол ярка: по экспериментальным данным в этой молекуле
эффективный заряд на цинке равен т. е. почти
вдвое меньше заряда +2. (На основе этих и других данных
можно заключить, что Эффективные заряды, как правило,
значительно меньше степеней ок келен к я,)
В молекулах CH,, СНдОН, НСОН, НСООН и С02 угле-
род четырех валентен, и отличия в типе его связи, разуме-
ется, не столь велики, чтобы допустить, что набор степеней
окисления в них (от—4 до +4) отражает истинное строение
его атома.
], Составление у рак нений реакций
Ради удобства и единообразия при записи окнелнтельно-
восстановительных процессов целесообразно применять оп-
ределенный порядок: для исходных веществ сначала будем
записывать восстановитель (окисляющееся вещество), затем
окислитель (восстанавливающееся вещество), затем там,
где это необходимо, среду; для продуктов реакции сначала
будем писать продукт окисления восстановителя, затем
продукт восстановления окислителя, затем и другие ве-
щества.
В процессе составления уравнения выясняется, вхо-
дит ли в реакцию вода или получается в результате ее
протекания; будем записывать ее в конце. В связи с этим
отметим, что вода часто расходуется на реакцию
о=--1-н1о^гон-
АЗ
ЭБ ,:Научнов наследие России"
или naif у чаете я в результате реакции
д+2Н< 4-Н,С
2-
(так как Частины О не могут появляться в свободном виде).
В подавляющем большинстве случаев можно считать, что
степень окисления водорода и кислорода в молекуле волы
ле меняется. К редким исключениям относятся лишь те
процессы, в которых вода подвергается действию или очень
сильного окислителя (например, фтора), или очень энер-
гичного восстановителя (например, ионов Н*).
Для составления уравнения окислительно-восстанови-
тельной реакции надо знать химические формулы реаген-
тов и продуктов реакции (они часто определяются на осно-
вании опыта). Сначала производят подбор коэффициентов
для веществ, атомы которых меняют степень окисления;
при этом исходят из того, что число электронов, отданных
восстановителем, должно быть равно числу электронов,
полученных окислителем.
Составление уравнения окислительно-восстановительной
реакции рассмотрим на примере окисления сероводорода
перманганатом калия в кислой среде.
L В результате проведения этого процесса малиновый
раствор обесцвечивается (вследствие перехода марганца
из состояния со степенью окисления ф-7 в состояние со сте-
пенью окисления +2) и появляется муть (выпадение серы).
Следовательно, реагенты и их степени окисления отвечают
записи
H,S*+ к MnO* + i! jSOA * s*+ AnSOj
ясс: г окне л и- сгеда
era- толь
ИйЕН-
тсль
2. Степень окисления серы повышается на 2 единицы,
а марганца уменьшается на 5 единиц. Следовательно,
баланс «электронного обмена» гакон:
окисление
восстановление
-J *
S—3ff- — S
j$n+fkn—Mn+B
множитель
5
2
С помощью найденных множителей можно уравнять числа
электронов^ отданных восстановителем и приобретенных
ЭБ Научнее наследие России"
окислителем, и найти основные коэффициенты в уравнении
реакции:
51 ij S -f- 2КМлО| + HjSO4 -* 5$+ ZMnSO,
[If III
3. Внесем коррективы, учитывая число атомов каждого
элемента в обеих частях уравнении (пока без кислорода
и водорода):
SHgS-l-aKMrA-i-S^SOi -+ 5S4-2MHSO* + KsSO|
4. Допишем уравнение реакции, проверяя число ато-
мов водорода в исходных веществах и в продуктах реак-
ции к определяя попутно число молекул воды:
5Н»5 + 2К MnOt -J- 3H,SO* =-as+2MOSO,+КзЗО4+8Н tO {X11)
5. Наконец, проверяем правильность записи по кисло-
роду (слева и справа по 20 атомов).
Последние две операции МОЖНО объединить, а при из-
вестном навыке записывать процесс сразу. Разумеется,
независимо от имеющегося опыта рассмотрение операции
целесообразно осуществлять без м[югократного перелнсыва-
ния уравнения реакции.
Разобранный процесс (XII) можно было бы предста-
вить к в иной (ионной) форме:
5S^ -+2 (М nO*r + 16Н+- 5S+2Мп«-+8Н^О (XI Га)
полагая, что ни источник кислой среды, ни природа катиона
перманганата не отражаются на результатах.
Часто (особенно для процессов с участием ионов) при-
меняется несколько иной варЕзант записи — составление
«пол у реакций» с последующим их суммированием. Рас-
смотрим этот прием на примере того же процесса. Ему
отвечают:
а) окисление и он а-восстанови тел я:
S (а)
вмета- <жие-
но ел tn- лей-
ка я ня я
ферма фор згв
б) восстановление иона-оккслнтеляЕ
(MiA)~+8H+4-5e-- 1й -НН>0 (б)
окислен^ яосета^
пая новлен-
фории нл я
форма
ЭБ ,:Научнов наследие России'1
95
(ионы H+ необходимы для связывания кислорода в моле*
кулы НаО, а электроны для баланса зарядов). Умножая
равенство (а) на 5, а (б) на 2. после суммирования получаем
уравнение (ХНа).
Это пример наиболее распространённого вада Записи
окислительно-восстановительных реакций (см. с. 97).
Если средой, и которой протекает реакция, является
окислитель или восстановитель, то ради наглядности сум-
марное количество этого вещества целесообразно разбить
на две части.
Проиллюстрируем этот прием на примере растворе-
ния меди в концентрированной азотной кислоте:
fit-
в + ч*
Си 4 2Н NOa + 2HNQ, = Си (NO,), -f- 2NO, + 2НаО (XIJI)
flKUC.lIl- со-лсоС-
TtJIb разова-
тсль
н в разбавленной азотной кислоте:
&-
1 1 4» 4
aril+2НЫО^+6Н NQ,» 3Cu (NOi); + 2ТО + 4HiO (X [V)
оннсли- сслеоб-
тгль рэзовл-
тель
(Эти реакции, как и многие другие, рассмотренные в на-
стоящей главе, воспроизводят лишь преобладаю-
щее направление процессов. Так, при растворении Си
в HNCX, выделяется несколько различных оксидов азота.)
Примером процесса, в котором необходим дополнитель-
ный расход восстановителя, является реакция
_r J d п
2НЬ 4“ Я л О, + 2НС1 = 1, 4- м’пС!» + 2Н,0 (XV)
яссста- седе-
нови- tfrps-
тсль эовятгль
Этот прием записи целесообразно распространять и
на процессы, в которых происходит изменение степени
окисления различных атомов в. одной и той же молекуле,
ЭЕ 'Й9учнов наследие России"
например на реакции
J 1
4FcSi -h Q, + 100а = 2&0,+
* 1
4-JU++)f-
3$Sf-
+я "t +» +з^ . _ . +s «1* н й
3 A^Sa + J H NOa + 24 H iTOj+4H SO = 6H, AsO, + 23NO+9НЭ SO,
J________________t
3-3ti+6)«“
3fc- fc-
i
04 !
rt
В a (IO5), + 3Ba (Yo^г + В а (1ОДЙ = В a5 (16B)B + 4?2 + Д (X VIJ
I t
a-Sc-
Составление подобных и других сложных (по величине
коэффициентов) реакций не может вызвать особых затруд-
нений. Так, суммируя вспомогательные уравнения
+ * +а 1
2As — 4е' = 2As
3if-21ir = 3S’
я N.|-3e-=Jf
получаем костяк предпоследнего процесса.
Читателям предлагается самим в приведенных ниже
реакциях, подобных рассмотренным, расставить коэффи-
циенты и показать направление перехода электронов::
Д] -р Fcj|O[ —Р" Fg
2л + HNO3 -> Zn (NQ,)B + NHjKOij h Ha0
H
Co (NQj)j CoaOij-4" NQi H Oj
В тех случаях, когда трудно решить вопрос о степени
окисления атомов (например, в реакциях с участием орга-
нических соединений), основные коэффициенты можно
находить по числу атомов кислорода, полагая, что каж-
дому вошедшему в реакцию атому кислорода отвечает
переход двух электронов. Так, в реакции
SCfHuO^ ^-24 KMnO44-3GH2SO4-
111
= 30OCi -I- JMMnSO* + J 2K sS Oj 4- 66HaO
4 M- X. К.Л p-а nerbail □,
ЭБ ,:Научнов наследие России"
iга окисление молекулы глюкозы надо затратить 12 атомов
кислорода, что отвечает отдаче 24 электронов.
В обиход химиков, изучающих рассматриваемые про-
цессы, наряду с химическим эквивалентом вошли так назы-
ваемые окислительный и восстановительный эквиваленты.
Эго частное от деления молекулярной массы на число
приобретаемых (теряемых) электронов. Так, в реакции
(XII) для КМпО* первый равен 158,15/5, а для сероводо-
рода второй — < ютов । г нс его молекулярной массы
2. Типы окислительно-восстановительных реакций
Меж молекулярные реакции. Он it протекают с измене-
нием Степени окисления атомов в разных молекулах. Эти
процессы составляют наиболее обширную группу окисли-
тельно-восстановительных реакций. Вот несколько при-
меров.
Синтез:
Й О +J -*
Mg+Vjpj^Meo
Вытеснение:
2J?1 + FcA-Й А+ 2Fe
Другие процессы:
10HCI4- 2КМгО4 -f-6HCI -= 5СЫ-2МлС184-2КС14 8! !2О
A Mg 4- 2H+foj 4- 8Н NCj = 4Mg 4- NjO 4- SH,O
2 An (NQ,), -f-5 +вН,0 =
« 2H MnO4 4- 5 (N HJt Sb* 4- 5H aSO* 4- 4 H NQs
3M nSO, 4- 2KM11O* -p 2HtO = 5M nO4 4- 2KHSO* 4- HS$O* (X VI I)
К']О»4-5К1* +ЗН#5О*=« 3^4-3^30*4-3HSO (XVIII)
Внутримолекулярные реакции. В них происходит изме-
нение степени окисления разных атомов в одной и той же
молекуле. Обычно это реакции термического разложения
веществ (AgNO3, ZrT*, КС10а н др.). Например:
HgO - 1/Д4- Не
и
= Na 4-Сг*Од 4" 4Н-.0
Реакции самоокисления — самовосстановления (реакции
диспропорционирования, дисмутацнн). Их протекание со-
провождается одвоврсменным уменьшением и увеличением
ЭЕ "Й&ччое наследие России"
степе»if окисления атомов одного и того же элемента. По-
этому эти реакции принципиально осуществимы лишь для
тех веществ, в молекулах которых есть атомы со степенью
окисления, промежуточной между минимально и макси-
мально возможной. Легкость их течения при прочих рав-
ных условиях связана с близостью энергетических уров-
ней электронов в состояниях атомов, отвечающих разным
степеням окисления.
При мерой реакции самоокисления — самовосстановле^
ния служат процессы
2AuF + AuF ₽ AuFj+SAu; V Д -f- >/Д+ Н,О= HCIO -f- Hcl
NO* 4-tlOi + H,0=H SfOi + H NOi
Сюда же относятся реакции диспропорционирования S,
СО, Н2Ог, Cui, GcBrt1 SeCl,, TiCl3 u др. * Укажем также
на процессы
Н +2Н ЙЬ4=нйГр, + амЬ + Н,0
N аЙО -|- 2NaClO = N аЙОа+2NaCl
зкс!О? а- ксюэ - зкЗ о*+кс1
SNajSbb + Na^ =3Na*sb4 4- NaaS*
И
»/Д + ’/Д+ 3NaOH+3HtO= SNaHjPOj 4-PHj
Мыслимы и реакции диспропорционирования, в которых
при изменении степени окисления элемента его валент-
ность остается постоянной. К ним относятся многие про-
цессы с участием органических соединений, например
реакции
ИСОН -f- НСОН 4- HjO = НОСЮН4- сЦоН
и
CHjCOH 4-CHjCOH 4- КОН=СН^СООК 4- СНзСНвОН
Если рассмотренные в этом разделе реакции проте-
кали бы справа налево, то мы имели бы дело с процессами,
описанными в начале параграфа. Реакциями днелропорцио-
нировання были бы процессы, противоположные (XVII)
и (XVIII).
" ** +1 I 41/4.i
• Диспропорционирование вида STiOCf? ™ ТЮЯ + ТLC-Ij !с« яалл-
ется окнелнтельно-восстановитыьлым орошссо»г, Ряим рассмотрен^
пая реакция (XVI) 5Вл (EOS) — Вя4 (lOfl)a 41я + служит при-
мерм сочетания диспропорциицровання с внутримолекулярным
окнелнтсл ьно- восста новител ь н ым л роиессом.
ЭБ,:Нау> ДА наследие России"
99
Реакции самоокисления — самовосстановления обычно
записывают в более лаконичной форме:
3AuF=AtjFI4-2Au; «К^Юь^ЗКСЮ*-!-КС’1 н г, д.
При затруднениях в подборе коэффициентов для реак-
ций диспропорционирования эти процессы следует считать
протекающими как бы в обратном направлении, г. е. начи-
нать расстановку коэффициентов для веществ с различными
степенями окисления элементов (тогда задача сводится
к уже рассмотренной на с. 94 и сл.).
Внутримолекулярные реакции окисления — восстанов-
ления, в которых происходит выравнивание степеней окис-
ления атомов одного и того же элемента, т. е, обратные
ранее рассмотрен ним, являются процессами кои тр диспро-
порционирования (конмутации). Вот два примерз:
NH(Nbj = ^,4-2HsO И NH^bb-fiiO-f-SHjO
3- Вл и я пне среды па характер реакций
Явится ли данное вещество окислителем или восстано-
вителем, нередко зависит от среды. В зависимости от нее
может меняться и характер протекания процесса между
одними и теми же реагентами, Вот лишь одни пример:
как известно, перманганат — сильный окислитель. Но
наибольшую окислительную активность ионы (MnOj про-
являют в сильнокислой среде, восстанавливаясь до ио-
нов Мп2*, меньшую — в нейтральной, а также в слабокис-
лой и слабощелочной средах, где они восстанавливаются
-rt + b
до Мп (МлОа), и минимальную — в енльнощелочнон среде,
+4 44
восстанавливаясь в ней лишь до Мп !иоегя (MnOJ4-].
объясняется темФ что новы водорода внедряются в анноны
+1
(МпОд)', вызывая ослабление связи между марганцем н
кислородом, тем самым облегчая действие восстановителя.
+7
В нейтральной среде деформация анионов (MnOJ значительно
меньше, так как поляризующее действие молекул воды
гораздо слабее влияния ионов Я*. Гидроксид-ноны, нао-
борот, даже несколько упрочняют связь Мп —О,
Итак, в силънскнслсй среде, где концентрация ионов Н*
велика, связываются все атомы О иона MnQf, в среде,
близкой к нейтральной, где концентрация Ц+ незначи-
тельна, связывается лишь половина этих атомов; наконец,
ЗБ ' Л^чс-а наследие России"
б склькощелочной среде, где концентрация Н* исчезающе
мала, эти ионы не могут связывать нн одного атома кисло-
рода. Сказанное иллюстрируется реакциями
SNaesbe+2K.MnO* -К 3! IaSOa — bNa^j +2MnSO4 -J- K*SO4 4-3HsO
3N л ;SQ, + 2K MnO4 -I- HSO = 3N as§ba -f 2AtaOs + 2K.OII
И
+ 2КМпО<+2KOH— \Ta3sbt + ЙК,Уг1О4 -J- НЭО
Отсюда становится понятной необходимость при реак-
циях в нейтральной среде прибегать к активным окислите-
лям (например, содержащим непрочные ионы), а при реак-
циях в щелочной среде применять нагревание и энергичные
восстановители.
Для создания кислой среды обычно пользуются раз-
бавленном серной кислотой, так как азотная кислота про-
являет окислительные, а соляная кислота—восстано-
вительные свойства. Для создания щелочной среды при-
нято прибавлять \гаОН (р) или КОН (р).
Существуют процессы, в которых роль среды выполняет
окислитель [см., например, реакции (ХШ) и (XlV)J или
восстановитель [реакция (XV)).
Интересны примеры влияния среды на Течение процесса
с участием перекиси водорода:
2И’/ + нД-1±+2нЗ
Nis?+1 гД+гСНоРООН s+ Sl-lfl О н- N i (СНзСОО)а
Р b $ + 411Д = РЪ5О4+4На О
2&С1,+ ЗН Д + lONaOH = 2Ыаа&О4-|- Шаб’ 4-6NaCl
5Н Д + 2КМпОл+3HiSO* = 5Oi 4- SMnSOj + K*SO* Д 8Н,О
H Д+PbOt + SCHjOOOH = 2НИО + 6* 4-pii (CH3COO)t
5H Д 4- 2i lTon = ’г4- 5&J H-GHaCf
H Д + aXgOH = t$s -|-2НгЙ?4- 2^g
В этих процессах H2O2 выступает то как окислитель (пер-
вне четыре реакции^ то как восстановитель. Кромв того+
известна и реакция диспропорционирования чистой пере-
киси водорода
нД=на'д+‘/Д
Таков «причудливый» характер этого соединения.
101
ЭБ ,:Научнов наследие России"
Влияние среды сказывается и в тех случаях, когда
степень окисления в продуктах реакция меняться не может,
но данное вещество образует амфотерное (с. 190) соедине-
нна Так, если в кислой среде происходит процесс
AI-&r = Al»i-
то в щелочной идет процесс
А! 4- лОН - - Зг = (Л1 (ОН)Я | *'»*
Приходится сталкиваться и с реакциями типа
(СгАР -|-11Нн -J- 6s- = 2Сг -|- ТН/)
(СтО4)2 - 4- SHsO 4- 3^ = (CrOj)-+4ОН-
Первая протекает в кислой среде, вторая — в щелочной
(между бихромат-хромат-ионами существует равновесие
До?)«-+11,0*- 2 (СлО,)а- 4- 21Г
смещающееся в кислой среде влево, а в щелочной —
вправо).
Сопоставляя даЕоаые по окислитель но-восстановитель-
ным реакциям, можно сделать некоторые обобщения,
В тех случаях, когда у и он а-восстановителя происхо-
дит увеличение числа атомов О, оно достигается за счет
молекул воды (в нейтральной или кислой среде) либо
ионов ОН- в щелочной среде). В первом случае появля-
ются ионы FT, во втором эти ионы связываются нонами
ОН' в Н,О.
Связывание же атомов О нона-окисли тел я осуществля-
ется молекулами воды (в нейтральной и щелочной среде),
либо ионами Н+ (в кислой среде). В первом случае обра-
зуются НОНЫ ОН", во втором — молекулы Н2О-
Если в результате реакции возрастает число атомов О,
связанных с атомом-восстановителем (анион бескислородной
кислоты превращается в аннон кислородной кислоты,
анион с меньшим числом атомов О превращается в анион
с большим числом атомов О, катион с небольшим зарядом
превращается в анион и т. д.), то среда влияет на восста-
новительные свойства, В подобных случаях процесс облег-
чается в щелочной среде, так как поставщиком атомов О
являются ионы ОН-.
Наоборот, характерный для атома-о кисли тел я переход,
свяэаи!1ый с уменьшением числа присоединенных атомов О
(кислородсодержащий анион трансформируется в бескяс-
ЗБ нУЙчсе наследие России"
дородный катион, анион с большим количеством атомов О
переходит в аннон с меньшим количеством атомов О и т. д.),
облегчается в кислой среде.
Кислая среда способе! в у ет процессам, в которых рас-
ход ионов И’ на восстановление больше расходов ионов ОН'
на окисление. 13 противоположных случаях целесообразно
применение щелочной среды.
Если же расход Н+- и ОН'-ионов одинаков, то реакцию
можно проводить и в нейтральной среде.
На течение процессов может оказать влияние не только
вещество среды, но и его концентрация. Иногда это влия-
ние усложняется действием температуры. Так, реакция
диспропорционирования хлора в щелочном растворе в за-
висимости от условий протекает по-разному:
24 Д+1/Д +6NaOH « NaClO, + 4- 3H,0
гор Я Ч
кшгце игр.
к
Д+‘/Zb+2NaOH = nZiO 4- N аС1 + НаО
ХФЛ-,
p J li Л.
t_E
GcO — энергичны!! восстановитель в щелочной среде,
так как основной процесс сопровождается образованием
HSO и, следовательно, убылью а нерп г и Гиббса. Наоборот,
РЬОа проявляет большую окислительную активность в кис-
лой среде, так как кислород связывается ионами Н+ опять-
таки в воду, а (Лб^ч)н,о гж> — —56,703 ккал /мол ь.
В ряде случаев за счет среды можно даже изменить
направление процесса. Так, реакция
31,4-3!1аО = 11?Оа + 51 ff
в щелочной среде идет слева направо, а в кислой — справа
налево. Примером процесса, еще более чувствительного
к среде, может служить взаимодействие HuQs с н HJOa:
в сильно кислой среде пероксид водорода окисляет иод
по уравнению
1,-|-шД-2Н Го, 4-11-1,0
а в несколько менее кислой среде — восстанавливает йодно-
ватую кислоту по уравнению
511^0,4- 2НIО j = 5Ы j {3 4-1 з + ьО;г
103
ЭБ ,:Научнов наследие России"
fi. Направление реакций
Било бы неверным ограничиваться умением записы-
вать различные окислитель но-восстанови тел иные реак-
ции, так как такая задача требует нс столько знания хи-
мии, сколько умения составлять пропорции. И хотя с уве-
личением числа записанных реакций автоматически растет
и «химический багаж», однако не менее важно умение опре-
делять направление данного процесса.
Если взаимодействует сильный окислитель с сильным
восстановителем, то происходит односторонний процесс.
Он практически протекает до конца, так как его продукты—
обычЕ<о вещества со слабо выраженными окислительно-
восстановительными свойствами. При малом отличии окис-
ли тел ынивосстанови тельной активности исходных веществ
и продуктов реакции процесс является двусторонним.
Для количественной оценки направленности процессов
пользуются значениями ДО? (Д^й«) образования реагентов,
а для весьма частного (хотя и практически важного) слу-
чая — разбавленных холодных несжатых водных раство-
ров — значениями нормальных электродных потенциа-
лов
Расчет будем производить по значениям Аб«Нз кото-
рые записаны под формулами реагентов.
На примере процессов диспропорционирования рассмот-
рим две окислительно-восстановительные реакции:
3AuF (Kj = AuFa (к) <Х1Х)
3( —14) — 71 2-D
И
ЗПС1 (М = 2Т] (к) + Tl1CI, (к) (XX)
3[-U,2DS) 2 0 —гО
Из приведенных данных следует, что (Дв^Ьтх —
= —29,0 и (ДС^)хх = 62,624 ккал, т. е. первая реакция
будет протекать вправо, а вторая влево. Обобщая этот ре-
зультат, приходим к выводу, что для золота типично трех-
валентное состояние, а для таллин одновалентное.
В заключение на двух примерах рассмотрим примене-
ние таблиц стандартных электродных потенциалов для
определения направления реакций (в разбавленных вод-
ных растворах при 25 °C и I атм). Использование этих
таблиц основано на возможности разделения ок и ели тел ьно-
восста лови тельной реакции на две «пол у реакции*, каждая
из которых включает окислительно-восстанови тельную пару
ЭЕ "ьННчое наследие России"
вида, приведенного в тайл. 9, Сочетая различные полу-
реакции, можно получить самые разнообразные продукта.
J. В какую сторону будут протекать процессы
2КГ- (р)4-2FcJ• С]э (р) =Г!4- 2KCI (р) + 2Fe-*CI4 (р)
(Г—F, а, вг, п?
Из табл, 9 выписываем значения Е’П11 для пяти электродных
реакций: F^+ (р) 4- е- = Fe« >• fp), = + 0,77 В Fs (г) + & = 2F- (р), £J(I = + 2,86 В Cls(г)4- &-= 2С] (р), £„, = + t ,3f> В Вг4(>к)4-2₽_=2Вг (р), ],07 В I* (к) 4- аг=аг (р), е;„~+о,54 в
Представив заданную реакцию в ионной форме:
2Г- (р)+2Fe*<- (р)= FS 4- 2Fe*h (р)
получаем: для KF ELe = 0,77-2,86 = — ВДВ; для KCI е;18 = о,77 - ] ,36 = - о,5<3 В; для КВт E;Ba=D,77—1,07 =—0,30 В; ДЛЯ KI Е;„—0,77-0^4-0,23 В.
Полученные результаты означают, что только иодил калия
будет восстанавливать FeCla.
2. Можно ЛИ Окислить НВг (р) с помощью растворов
перманганата и бихромата?
Из табл, 9 имеем:
{Мп(\)- (р) 4- 8Н11 (Р) н- &-=Мтг+ (р) 4- « if о. £:»»= 1ЛI В
(Сг2О,)г (р) + ! 4 Н '• (р) 4- бе - - 20* '• (р)+7 i i го, е;„ = 1,33 в
&,(ж)4-2с’«2Вг- (р), £^₽1,07 В
Следовательно, оба раствора будут окислять бромисто-
водородную кислоту. (В растворе же НС! окислителем
был бы лишь перманганат-и он.)
ЭБ 'Научное наследие России"
РАЗДЕЛ Ш
СКОРОСТЬ И МЕХАНИЗМ
ХИМИЧЕСКИХ РЕАКЦИИ
ВВЕДЕНИЕ
Материал, содержащийся во второй части, позволяет
устанавливать принципиальную осуществимость процесса,
решать вопрос о предельной его полноте н об условиях
равновесия. Теперь следует научить скорости процесса,
решить вопрос как быстро можно достичь равновесия, Ведь
важно знать не только, какому соотношению реагентов
отвечает состояние равновесия, но и насколько быстро
оно достигается. В общем случае нельзя считать, что чем
дальше реагенты находятся от состояния равновесия, т, е.
чем отрицатель Flee А<7, тем быстрее оно будет достигнуто:
между скоростью достижения равновесия и его положением
никакой зависимости ист. Так, хотя для реакции NO (г) +
+ VA (г) = NC\ (г) ДСий = —8,37. а для реакции На (г)+
4- (0 — Н2О (г) = —54,638 ккал, первая в от-
личие от второй протекает очень быстро при обычной
температуре.
Если расчет свидетельствует о невозможности проте-
кания реакции в данных условиях, т, е. если 4<jJ>CL
то, разумеется, бессмысленно пытаться ее реализовать.
Но и в том случае, когда согласно расчету процесс прин-
ципиально осуществим (Д<3< 0}, он может не идти из-за
каких-либо препятствий на его пути. И таких случаев
не мало. Так, например, хотя многие углеводороды неустой-
чивы в отношении разложения на углерод и водород (см.
Приложение i) и их неустойчивость в каждом гомологи-
ческом ряду возрастает с увеличением молекулярной массы
и с повышением температуры, однако только при высоких
температурах скорость их распада становится ощути-
мой *. Для процессов горения AGr< 0- Это значит, что
все органические вещества должны окисляться кислородом
воздуха- Однако и растения, и животные, и угольт и нефть
* Например, разложения с^тапляюишк углевдоролог. является
причиной посте пен егого отложен нм крхса при крекинге м«фти.н
ЭБ WS.'hge наследие России"
не подвергаются заметному окислению. Хотя (Езд)м« <
< (£йв)гп, магний не вытесняет цинк. Часто мыслимо
полное превращение исходных веществ, а процесс вовсе
не идет.
Что делать в подобных, весьма многочисленных слу-
чаях? Как поступить, когда вопреки условию Д0< О
попытки осуществления процесса оказываются безуспеш-
ными или недостаточно результат в ними? Ответ один — все
усилия надо направить на решение проблемы скорости,
на преодоление кинетических трудностей реализации про-
цесса. Надо при этом иметь в виду, что взаимодействие
органических соединений обычно протекает медленнее вза-
имодействия неорганических веществ и редко доходит
до состояния равновесия *.
Таким образом нельзя, не предварив опыт расчетом,
бороться с инертностью вещества с помощью катализатора,
а кажущуюся вялость процесса (при весьма неблагоприят-
ном положении равновесия) пытаться преодолеть введением
более активного катализатора.
В истории химии и химической технологии известно
немало примеров, когда пренебрежение необходим остью
предварительного выяснения вопросов об осуществимости
процесса и о равновесном соотношении приводило к непро-
изводительной затрате труда, времени и материальных
средств. Однако отсутствие сведений о численных значе-
ниях свойств соответствующих веществ, с одной стороны,
и насущная необходимость быстрого решения вопроса
о рсализацнЕ1 данного процесса, с другой стороны, приво-
дят к тому, что опыты по его изучению, к сожалению, часто
Приходится ставить без предварительной оценки значе-
ний AG.
Если, исходя из «энергоспособносгна (А//) и фзбото-
способности» (&<?), любой процесс можно представить
записью
исходные реагенты -> продукты реакции (1)
т. е, рассчитать его (см. части I и II), то куда сложнее
обстоит дело с точки зрения течения его во времени, когда
Приходится расшифровывать запись
исходные реагенты -+ промежуточные вещества -► продукты реакции (II)
1№рехсд1и» состояние
* Разумеется, мадлесгиоегь процесса или его отсутствие зача-
стую является фактором пйложительнын, Как правило, мы эн интерн
сопаны в том, чтобы органические вещества не окислялись, металлы
“е ржавели, краски не выцветали.
ЭБ ,:НауччоЁ наследие России"
107
Выявление н учет среднего звена, раскрывающего меха-
низм процесса и не представляющего никакого интереса
с точки зрения вычисления значений A//, AS л ДО, — в об-
щем случае чрезвычайно сложная проблема, так как в от-
личие от исходных реагентов и продуктов реакции про-
межуточные продукты выделить и изучить удается очень
редко. В большинстве случаев механизм процесса и осо-
бенности промежуточного состояния являются плодом кос-
венных соображений, т, е, лишь более или менее правдо-
подобной гипотезой.
Нужно помнить также и о том, что любая реакция идет
в энергетически наиболее выгодном направлении (конечно,
простота се написания не означает легкости ее протека-
ния).
Гаана [
ОСНОВНЫЕ ГТОПЯТПЯ
Исследование течения реакции во времени составляет
содержание химической кинетики. Под кинетикой в широ-
ком смысле слова понимают учение о скоростях различных
процессов — химических реакций, растворения, кристал-
лизации, парообразования и т, д. Мы будем рассматри-
вать лишь химические процессы.
Химику приходится иметь делос реакциями, протекаю-
щими со всевозможными скоростями — от исчезающе ма-
лых (охватывающих геологические периоды) до колоссаль-
ных (взрывные реакции). Процессы инициируются под
влиянием повышения температуры, действия света (фото-
химия), механического усилия (механохимня), излучения
большой энергии (в частности, ионизирующего излучения,
вызывающего радиолиз воды и другие процессы, рассма-
триваемые в радиохимии) и т. д.
Изучение кинетики реакций позволяет выявить их меха-
низм и решить задачу интенсификации процесса, т. е. имеет
и теоретическое, и практическое значение.
Реакция может протекать в объеме фазы (гомогенно)
или на границе раздела фаз (гетерогенно). К гомогенным
реакциям относятся, например, процессы в растворах,
к гетерогенным — реакции на границе газ — твердое ве-
щество.
Скорость реакции равна числу ее актов в единицу вре-
мени: для гомогенных реакций — в единице объема, для
ЗБ наследие России"
гетерогенных — на единице поверхности разделафаз. Можно
пользоваться и пропорциональными им величинами, ха-
рактеризуя, например, скорость изменения во времени
концентраций реагирующих веществ (концентрацию обычно
выражают в моль/л, а время — в мяв). Так как в общем
случае концентрации реагирующих веществ непрерывно
изменяются, то следует говорить о мгновенной скорости
реакции и, т. е. о ее скорости в данный момент времени т.
В общем виде она выражает изменение количества реаги-
рующих веществ и единицу времени в единице реакцион-
ного пространства, т, е.
р= + *»1,
Для гомогенных реакций d большинстве случаев
(П1.1)
(рис. 36; tf — tga). Здесь С—концентрация любого реа-
гента (так как все они связаны стехиометрическими коэф-
фициентами), Следует только
исходных веществ убывает
(dC < 0), а продуктов ре-
акции возрастает''(4(7 >0).
Поэтому для первых эту
производную егядо брать С
минусом^С 0, ((т >0),
для вторых — с плюсом
учесть, что концентрация
Рнс, 36. Изменение ко времени т
концентрации С разлагающегося ве-
щества;
J — медлешия РевКЧЙЯ", ? — «ИСфИйДЬ-
нлн* рс-имцичг J — GtitTpafl реакций
В подавляющем боль-
шинстве случаев запись
процесса не отражает ис-
тинного пути реакции. Это
относится даже К тем ре-
акциям, которым отвечают
сравнительно простые сте-
хиометрические уравне-
ния. Почти все они явля-
ются сложными многостадийными процессами,т.е. осущест-
вляются в результате одновременного (или последователь-
ного) протекания нескольких простых реакций; при этом про-
дукты промежуточных стадий обычно быстро расходуются
и поэтому присутствуют в небольших количествах. Стадиями
реакций могут быть не только химические процессы, но и,
например, переход вещества из объема фазы к ее границе,
на которой протекает реакция, или перенос продуктов
109
ЭБ "Научнов наследие России"
взаимодействия от этой поверхности В объем. Скорость
подобных процессов определяется темпом диффузии.
Скорость реакции зависит от многих причин- На нее
влияют: природа и концентрация реагентов, давление (для
реакций с участием газов}, температура, катализатор, при-
меси н их концентрации, степень измельчения (в реакциях
с участием твердых веществ), среда (для реакций в раство-
рах), форма сосуда (в цепных реакциях (см. с, 115 и сл,)),
интенсивность света (в фотохимических реакциях), потен-
циал электродов (в электрохимических реакциях), мощ-
ность дозы излучения (в радиационно-химических про-
цессах), Таким образом, лишь некоторые из факторов, дей-
ствующих па скорость реакции, одновременно оказывают
влияние на химическое равновесие. В связи с этим надо
отметить огромную трудность учета действия различных
факторов Eia скорость реакции и тем более количественной
их оценки.
Скорость процессов контролируется по изменен ню кон-
центрации реагентов. За их темпом можно следить и по
изменению какого-либо свойства (показатели преломле-
ния, электропроводности, спектра и т. д.), не нарушая
течения данной реакции. Если процесс протекает медленно,
то через определенные промежутки времени отбирают
пробы и определяют в них содержание данного вещества
(например, титрованием). Для реакций, протекающих
с большой скоростью, пользуются резким охлаждением,
останавливая их. Учитывая то обстоятельство, что ско-
рость реакции очень чувствительна к изменению темпера-
туры (см. с. 119), реагирующую смесь обычно помещают
в термостат.
Основными параметрами, которые приходится учиты-
вать почти во всех процессах, являются концентрации
(давления) реагентов, температура и действие катализа-
тора, Поэтому главной пашей задачей будет рассмотрение
их влияния на скорость процесса и решение вопроса, на-
сколько быстро с их помощью можно достичь равновесия
в реагирующей системе.
Глава ][
ВЛИЯНИЕ КОНЦЕНТРАЦИИ
1, Гомогенные и гетерогенные реакция
Как мы уже знаем, гомогенные процессы характери-
зуются взаимодействием вещества в одезой фазе. В гете-
рогенных реакциях наряду с химическими превращениями
110
ЭБ ' Научнов наследие России"
имеются стадии переноса веществ. Их влияние егя процесс
в целом зависит от условий его протекания. Если наиболее
медленной стадией является химическая реакция, то гово-
рят, что процесс протекает в кинетической области, если
же, наоборот, звеном, тормозящим процесс в целом, служит
перенос веществ, то говорят о диффузионной области. Что
является лимитирующей стадией: взаимодействие ве-
ществ — можно установить по температурной зависимости
скорости реакции; а первом случае скорость гораздо чув-
ствительнее к температуре, чем во втором,
2. Простые и сложные реакции
Если процесс протекает в одну стадию в соответствии
со стехиометрическим уравнением, т, е, отвечает одному
этапу, его называют простым. Сложные реакции — это
совокупность простых; поэтому их кинетические уравне-
ния содержат несколько констант скорости. К сложным
реакциям относятся обратимые, параллельные, последо-
вательные и др. Они различаются по характеру связи
между отдельными этапами взаимодействия. Для каждой
стадии применимы уравнения простых реакций, Если
темп протекания отдельных стадии сильно различается,
то суммарная скорость процесса определяется (лимитиру-
ется) темпом самой медленной стадии и может быть описана
кинетическим уравнением этой простой реакции.
При исследовании сложных реакций пользуются прин-
ципом независимости, считая, что каждая из проходящих
реакций описывается основным законом и протекает неза-
висимо от других, т. е. полное изменение системы пред-
ставляет собой их сумму.
Обратимые реакции. Рассмотрим какую-либо односта-
дийную обратимую реакцию, протекающую в газообраз-
ной фазе, например
H^O-l-Wz±HI(rH-HI(r) (til)
Пусть в закрытом сосуде приведены в соприкосновение
водород и иод. Если это произошло при такой температуре,
при которой их смесь будет реакционноспособна (например,
при 450 X), то начнется взаимодействие. Его скорость будет
пропорциональна концентрациям реагентов, т. е. выразится
соотношением
?=Л?НС11( (1112)
m
ЭБ ,:Научнов наследие России'1
где k —константа скорости прямой реакции. Эго выра-
жение называете я законом действующих, масс (Гульдбсрг,
Вааге, 1864—1867 ). Как только образуются молекулы Н1,
начинается обратная реакция со скоростью
Р (111.3)
здесь k — константа скорости обратной реакции. Кинети-
ческие уравнения (II 1.2) и (IIL3) реакции (III) основы-
ваются на том факте, что чем выше концентрация реаген-
тов, тем больше вероятность их встречи, а поэтому и вза-
имодействия. В связи с уменьшением концентрации Нг
и fa и с увеличением концентрации HI скорость прямой
реакции о будет падать, а скорость обратной реакции v воз-
растать. Спустя некоторое время скорости встречных
процессов сравняются:
w-Г- (II1.4)
В системе наступает химическое равновесие. Начиная
с этого момента число образовавшихся и распавшихся
молекул Hl будет одинаковым: в Системе наступит под-
вижное (динамическое) равновесие. Макроскопические из-
менения прекращаются, микроскопические сохраняются,
В соответствии с (II 1.2) и (II1-3)
(11.1-5)
или
X/A^CЙ[/(C’н^Cl1)• (ПГ.6)
В уравнения (II 1.5) и (ПГ.6) в отличие от уравнений (II 1.2)
п (III .3) подставляют не текущие, т. е- переменные (мгно-
венные), а равновесные концентрации.
Константа скорости данной реакции при фикс про ват-
ной температуре постоянна; поэтому будет постоянным
и отношение
^=T/Л. (Ш.7)
Опа называется константой химического равновесия.
Таким образом, мы пол учили уравнение
= ("Г.8)
являющееся примером соотношения (И. 16). Введя вместо
концентраций парциальные давления р, компонентов, мы
получили бы уравнение
^р=Рш'(Рн,Р1,)1 (1Л.9)
являющееся примером соотношения (11.17),
ЭЕ чсе наследие России"
Так как положение равновесия не зависит от состава
исходной смеси (см. с. 35 и 54). то к результату, представ-
ленному уравнением (Ш.6), привело бы и помещение
в сосуд только молекул HI; разница заключалась бы лишь
в том, что вначале v = и. Можно было бы взять и про-
извольную смесь Н4, 14 и HI; в этом случае лишь быстрее
было бы достигнуто равновесие, но его положение ока-
залось бы прежним (рис. 37, а
также см. рис. 22, a). % я/
Представим теперь, что нас
интересует химически обрати-
мая реакция
йА | ЛН -f-,., 71 г/О ~₽Е "4"... (IV)
Для нее, аналогично процес-
су (HI).
сг = йСдф.. и « = R£)Ce ...,
откуда следует соотношение
с£съ...
Кс=^—’ (41.10)
тождественное (11.16).
Хотя уравнение (III.8) мож-
но трактовать как частный слу-
чай соотношения (III. 10), одна-
ко и это надо подчеркнуть,
последнее в общем случае нс
может быть обосновано так, как
это было сделано для уравнения
(JII.B), т. е. на основании
выражений, аналогичных (III.2)
н (111,3). Дело d том, что
Рис. 37. Ход реакции обра-
зования (/) и распада (5) HI
при Т“ 44В "С (пллюстра-
ння двустороннего лрибли -
женин к равновесию)
механизм реакции не задается ее стехиометрическим
уравнением. Поэтому в подавляющем большинстве
случаев (л во всех, когда коэффициенты а, 5,... велики) надо
также учесть, что часто се кинетические законы, т. е. связь
между скоростью и концентрацией, сложны или неизвестны.
Однако важно другое; соотношение (111.10) справедливо
в любом случае (если давление не очень велико); оно может
быть выведено совершенно строго методами, изложение
которых выходит за рамки данного курса.
113
ЭБ 'Научное наследие России"
Параллельные реакции. К ним относятся реакции, одно-
временно протекающие в нескольких направлен лях типа
fr,,B
(VI)
Примерами (VI) могут служить: разложение при умерен-
ном нагревании бертолетовой соли
д^йка-т
6KCIQZ
^KCIOj + KCI
нитрование фенола С^НГ,ОН (в результате чего получаются
сразу три изомера — орто-, мета- л лйра-интрофенолы) к
распад некоторых радиоактивных элементов.
Естественно, что параллельные реакции — они особенно
часты в органической химии — осуществляются лишь тогда,
когда сети термодинамически возможны. Однако преоблада-
ние той или иной из них, а поэтому и относительные коли-
чества получаемых продуктов однозначно определяются
соотношением их скоростей.
Последовательные реакции. Последовательные (или кон-
секутлвяые от лат. consequent— последовательный} реак-
ции—это процессы, । г роте кающие через промежуточные
стадии, когда одна реакция идет вслед за другой, В про-
стейшем случае им отвечает схема
А — В—С (VIJ)
Эти процессы очень важны, так как большинство химиче-
ских реакций протекает потому или иному механизму через
ряд последовательных стадий. В Одних случаях промежу-
точными являются валентно-насыщенные молекулы, в дру-
гих— атомы или радикалы. Точное решение кинетических
уравнений для последовательных реакций возможно лишь
для сравнительно простых процессов.
Сопряженные реакции. Эго процессы, которые проис-
ходят согласно схеме
A-^B->D (VIII); A-f-C-t-E (IX)
Протекание одной из них обусловливает протекание другой:
первичный процесс (VII1) идет самопроизвольно, вторич-
ный процесс (JX) —только при течении (VI И), иначе
Е14
ЭБ ,:Научнов наследие России"
говоря, од!>а реакция возбуждает (инициирует) вторую.
Общее для обеих реакций вещество А называют актором (от
лаг. actor — Действующий); вещество С — акцептором (от
лат. acceptor — принимающий); В — индуктором (от лат.
inductor — побудитель). В качестве примера укажем па
реакцию окисления FeSO< перекисью водорода, которая воз-
буждает (индуктор — ион Fe*+) взаимодействие НгОг (ак-
тор) с HI (акцептор). Рассматриваемые реакции Важны
в биохимии: они протекают в клетках, причем энергию,
необходимую для течения реакции с Д(?] >0, доставляет
реакция, для которой &G2<i 0, причем [ Д(34 | > | Дб1 |,
т. е. процесс в целом, естествен;to, удовлетворяет усло-
вию (II.6).
Механизм сопряженных реакций исследован еще недо-
статочно. Чаще всего он заключается в возникновении ак-
тивных промежуточных продуктов, участвующих в обоих
процессах.
Явление химической индукции впервые было исследовано
Н. Л, Шиловым (1905), который положил начало разработке
его теории.
Цепные реакции. Эти реакции вызываются свободными
радикалами — активными частицами, за счет которых про-
исходит превращение неактивных молекул в активные в ре-
зультате возникновения реакционноспособной частицы в
каждом акте процесса.
Существуют два типа цепных реакций — с неризветелеН'
ними и разверченными цепями.
К первому типу относится фотохимический синтез хло-
ристого водорода. Возникновению цепи отвечает образо-
вание атомов-ради калов *:
C!,+Av«.2a
развитию цели — чередующиеся процессы:
Вг +С12₽НС1 + И- '
H'-f-C]l=--l1CI+CI'
Нг +СГ = НСЦ-1Г
H’ + Qt-HCl-l-Cl-
(X)
(Xi)
(ХН)
обрыву цепи—реакции рскомбиЕЩЦНк:
НЧ-.Н'^Н,)
СГ+СГ -С12 ]
* Под действием кванта света диссоциируют молекулы хлора,
а не водорода, так как £^1—01 = 58,0, 0 ю время как =
— 104,2 ккал/моль.
115
ЭБ ,:Научнов наследие России"
Обобщим этот пример. Пусть за счет внешнего источника
энергии (свет, электроразряд, нагревание, а-, р- или у-излу-
чение, электронный удар) образуются свободные радикалы
или атомы, обладающие ненасыщенными валентностями.
Они взаимодействуют с ИСХОДНЫМИ молекулами, причем
в каждом звене цепи вновь образуется новая активная
частица. Путем попеременного повторения одних и тех же
элементарных процессов происходит распространен не реак-
ционной цепи. Ее длина может быть очень большой (в рас-
сматриваемом примере на каждый поглощенный квант обра-
зуется до 100 000 молекул НО). Столкновение двух одина-
ковых радикалов при условии, что выделяющаяся при ЭТОМ
энергия может быть отдана третьему телу, приводит к об-
рыву цепи. Причиной обрыва может служить не только
рекомбинация свободных радикалов (XII), но и их захват
стенкой реакционного сосуда, взаимодействие радикала
с примесями (если они не служат источником свободных
радикалов), а также образование малоактивного радикала
(обрыв в объеме) Вот почему скорость цепной реакции
очень чувствительна к наличию посторонних частиц и
к форме сосуда. Так, содержание в хлороводородной смеси
долей процента кислорода в сотни раз уменьшает длину це-
пей, а поэтому и скорость синтеза: атом Н, легко реагируя
с О:, образует малоактивным радикал НО,, нс способный
вступать в реакцию с Ht
НО, (г) + Нг (Г) - н А (г) + Н(Г), (= 15,398),
приводящую к регенерации атома Н; а радикалы НО2 могут
вступать во взai[модеметипе между собой:
' НО, (г) + НОИг) - НА (г) + О» (г), (ДО™=- 41J 45).
Примерами процессов с неразветвленными цепями слу-
жат также реакции хлорирования углеводородов (в част-
ности, метана), разложения органических соединений (на-
пример, CHjCOQH), полимеризации (хлоропрена) и др.
Процессы с разветвленными цепями отличаются от рас-
смотренных тем, что в них единичная реакция одного сво-
бодного радикала приводит к возникновению более чем
одного нового свобошгого радикала. Один из них как бы
продолжает цепь, а другой (другие) — начинает новую
(новые). К этому типу процессов относится окислен не во-
зе (Ц^исе наследие России"
дорода, которое при определенных условиях протекает так:
н-ьа*«он*4-о (хш)
СЬГ + На = Н3С4-Н (XIV)
О + Нг=ОН-+Н... (XV)
Образующиеся а реакциях (XIII) и (XIV) радикалы обеспе-
чивают развитие иеразветвленной цепи, а атом кислорода,
обладающий двумя свободными валентностями, входя в ре-
акцию (XV), образует два добавочных радикала, начинаю-
щих разветвление. Так возникает огромное количество сво-
бодных радикалов. Этны рассматриваемые реакции отли-
чаются от процессов первого типа, в которых концентрации
радикалов невелики. * Размножение* радикалов приводит
к лавинообразному течению процесса, которое может вы-
звать взрыв. Однако и в этих процессах происходят обрывы
цепей. Причем лишь в том случае, когда темп разветвления
опережает темп обрыва, происходит бурное увеличение ско-
рости процесса.
Темп разветвления определяется числом встреч реак-
ционноспособных частиц с молекулами исходного вещества,
т. е. пропорционален концентрации вещества и объему
сосуда. Скорость обрыва складывается из скорости этого про-
цесса на частицах примесей (она пропорциональна концент-
рации последних) к скорости обрыва на стенках (пропор-
циональна поверхности сосуда). Из этого следует существо-
вание некоторых предельных (критических) условий. Изме-
нение давления, температуры и т. п. вызывает внезапный
переход от медленного течения процесса к взрыву. Чем
меньше скорость разветвления, тем менее резок этот пе-
реход.
Тетраэтилсвинец, иод и некоторые другие вещества, до-
бавляемые в горючее, являются антидетонаторами. Они
способствуют взаимодействию с активными центрами (ато-
мами и радикалами), в результате чего образуются устой-
чивые соединения.
Теория цепных реакций создана трудами акад. Н. Н. Се-
менова, X кишельвуда (Англия) и других ученых.
3. Молекулярность и порядок реакции
По числу молекул, принимающих участие в элементар-
ном акте химического Превращения, различают реакции од-
номолекулярные, двухмолекулярпые и т, д. Вероятность
одновременного соударения многих частиц очень мала; по-
117
ЭБ ' Науччое наследие России"
атому трехмолекулярные реакции редки, а четырех молеку-
лярные неизвестны,
Сумма показателей степеней в уравнениях вида (Ш-2)+
относящихся к изменяющимся концентрациям, называется
общим (суммарЕ1Ы№) /крлщри; таким образом, он
определяет характер зависимости скорости от коннеитрз’
цин.
В тех весьма многочисленных случаях, когда процесс
многостадиен и поэтому ^брутто^эались fl) лишь фиксирует
исходное и конечное состояния системы, совершенно не
раскрывая механизма процесса, а также тогда, когда по
условиям эксперимента различие концентраций реагентов
весьма велико, порядок реакции не совпадает с ее модны/-
лясностью. Примерами несовпадения могут служить про-
цессы
2NA^4N0,4-O3 (XVI)
и
Cj Jf«O|i+ HjQ =CflEiiaQi-j- G(H|/3e (XVII)
трклшкф- МЮКМ1 ФРУКТОЛ
Blflft <аХПр
являющиеся реакциями первого порядка. Первый из них,
вероятно, протекает в две стадии:
К'20л — N2Qj -| О2
(XVIII)
(Х]Х)
причем Vxvni<^x[x. В реакции (XVII) избыток воды
(в сочетании с ее относительно незначительной молекуляр-
ной массой} позволяет пренебречь уменьшением ее концент-
рации, т. е. считать в уравнении
tfjybL ^СпНлОц с'н,о
Сн.о = const.
Существуют реакции нулевого порядка (например, раз-
ложение некоторых соединений на поверхности различных
веществ, когда скорость распада веществ не зависит от их
концентрании в объеме) и реакции дробного порядка (на-
пример, При многостадийных процессах, если самые мед-
ленные стадии имеют разный порядок, но скорости их соиз-
меримы), Разумеется, ни нулевой, ни дробной молекуляр-
ности быть не может, так как она относится к механизму
реакции, а не К описывающему ее уравнению.
118
ЭБ ,:Научнов наследие России"
Глчпя Ш
ПЛПЯНПЕ ТЕМПЕРАТУРЫ
Скорость химических реакций я подавляющем большин-
стве случаев с нагреванием возрастает. Она очень чувстви-
тельна к изменению температуры. Так, если осуществить
синтез Н/Эпри 20 *Сдаже на 15 % практически невозможно
(на это потребовалось бы 54 млрд, лет), то при 500 сС для
этого необходимо всего 50 мин, а при 700 ГС реакция про-
исходит молниеносно.
Почему нагревание вызывает столь значительное ускоре-
ние процесса? Так как скорость реакции пропорциональна
частоте столкновений между молекулами, то, на первый
взгляд, это легко объяснить учащением соударений реагиру-
ющих частиц. Однако это предположение нс подтверждает-
ся— скорость движения частиц при нагревании па 10=
увеличивается всего лишь на I—2 %. Кроме того, если бы
необходимым и достаточным условием протекания реакций
являлось Лишь соударение частиц, то нельзя было бы объяс-
нить различие в скоростях процессов при одинаковых кон-
центрациях реагентов; было бы непонятным и действие ка-
тализатора, и его специфичность, и мцогое другое. Да и
если бы каждое столкновение оканчивалось актом взаимо-
действия, го все реакции протекали бы со скоростью взрыва:
ведь молекулы, содержащиеся в 1 см’ газа, испытывают
ежесекундно такое колоссальное число со ударен и Гт, что ему
отвечают скорости, превышающие экспериментальные в сот-
ни миллиардов раз; так. при р = I атм и 7’ = 500 °C еже-
секундное количество столкновений и I см3 газа равно
лЛО29. Последнее соображение не перечеркивает обоснова-
ния уравнения вида (I П.2), так как число столкновений, при-
водящих к реакции, пропорционально общему их числу.
В 1879 г. голландским ученый Вант-Гофф обнаружил,
что лошм/емне темлератрры на каждые 10 ’С увеличивает
скорость гомогенных реакций в 2—4 раза (правило Вант-
Г оффа)>
Точнее зависимость скорости реакции от температуры
передает соотношение
1пЛ₽—(111.11}
полученное первоначально опытным путем шведским уче-
ным Аррениусом (1889). В (Ш.11) а и b — независимые от
температуры коэффициенты, определяемые природой реа-
119
ЭБ ,:Научн(ЭЕ наследие России"
гейтов, т. е. постоянные для данного процесса. Их легко
Найти, располагая значениями константы скорости (&) изу-
чаемой реакции при двух температурах (решив два уравне-
ния с двумя неизвестными).
Предположив, что реагировать могут только молекулы,
находящиеся в особой — активной — модификации (кото-
рые образуются из обычЕ1ых молекул эндотермически), до-
пустив при этом, что образование этой моде [фи к а щит яв-
ляется обратимым процессом и что ее концентрация ни-
чтожно мала, Аррениус применил к равновесию между
активными и неактивными молекулами уравнение зависи-
мости константа химического равновесия от температуры.
Это позволило ему обосновать свое уравнение, получив его
в виде
Е -/А
1п*=--ег+с нли k=Ae RT *
где предэкспоиеицналъный множитель А — коэффициент
пропорциональности (In Л — С) и Ея ( = «/?) — энергия пе-
ревода в активное состояние (ее обычно называют опытной
энергией активации). Так, для разложения Hl эксперимен-
тальные данные приводят к зависимости
4J дао
k= 1,2 IOue (си8ноль'"1-С”1}.
Для процессов между валентно-Насыщенными молеку-
лами £л порядка десятков килокалорий етэ моль, для реак-
ций С участием Свободных атомов и радикалов — 1—
10 к кал/моль, а для взаимодействия нопов близка к нулю,
В соответствии с (Ш.12) при данной температуре скорость
реакции тем больше, чем ^теньзгге Ея: поэтому многие про-
цессы протекают по механизму, включающему участие
в них свободных атомов и радикалов.
ЭЕтергии активации некоторых реакций приведены в
Табл. 10,
Выяснив характер связи £я с k и установив ее масштаб,
мы в согласии с уравнением (111.12) вновь дол ж егы отметить
сильное влияние температуры на скорость процесса (изме-
нение температуры в арифметической прогрессии вызывает
изменение константы скорости в геометрической прогрессии).
Важно при этом подчеркнуть, что хотя скорость реакции
сильно меняется с температурой, однако с увеличением и
растет и и; поэтому константа равновесия К в отличие от
ЭБ ^$<чсе наследие России"
констант скоростей k и k [см. уравнение (Ш.7)| меняется
сравнительно мало. Колебание величины Е„ для различных
реакций в не очень белый и л пределах означает и сравни-
тельно небольшое варьирование температурного коэффи-
циента скоростей различных реакции, что отвечает правилу
Бант-Гоффа. Из уравнения (111.12) следует также, что чем
больше энергия активации, тем значительнее влияние тем-
пературы на скорость реакции.
Таблица 10
Энергии активации Ея (ккал) некоторых реакций
Реакция Р£?КЦИП
СНя -|- Hj-^з CHj ] [ 10,0 2Н[ = Нг-|-Г4 44,6
CHj ae*C--f- SD.O ЙКН, = Ы,+ЗН1 78,0
CH30H = CH.4C0 45,5 2N<X«2N0+Q| 265
СяН5Вг«аН4 4 2НВг 53J& 2КгО=2К44-Оа 5У,5
2С1яО=2С1г+0, 21,0 2501+04=250, 60,0
.Масштабы величины Е„ приводят, кроме того, к другому
важному вы золу. Рассматривая предэк^поненту А (в J 11.12)
как экстраполяционное значение константы скорости Ат„
при Еа = О, т, е, полагая, что она отвечает предельному
случаю (реакционноелособмы вес частицы), можно уравне-
нию (LIJ.I2) придать следующий вид:
, £й
tiu.ia)
1. Теория химической кинетики
Исходя из представлений об активных частицах в хими-
ческой кинетике, используют два метода, представляющие
собой различные подходы к теоретическому обоснованию
одного и того же явления, — теорию активных соударений
и теорию активированного комплекса.
Теория активных соударений. В ее основе лежит пред-
ставление о том, что для осуществления реакции достаточно
тесного сближения частиц; поэтому скорость взаимодейст-
вия должна быть пропорциональна числу их стол к но пен и й
за данный промежуток времени. Расчеты, проведенные для
бимолекулярных реакции, показали, что доля реагирующих
молекул по отношению к сталкивающимся ничтожно мала:
121
ЭБ ,:Научнов наследие России"
она порядка 10*n + 10"*®. Эта колоссальная диспропорция
объясняется несколькими причинами*
Столкнувшиеся молекулы могут не прореагировать, если
они должным образом не сориентированы в пространстве,
т. е. нс образуют конфигурации, способствующей разрыву
одних и формированию Других связей* Взаимодействию
благоприятствует столкновение молекул в положениях,
когда соприкасаются их реакционноспособные участки. Не-
обходимость определенной конфигурации резко уменьшает
число актов взаимодействия при столкновении активных
молекул; особенно важен этот эффект для сложных молекул.
Так, если бензойная кислота —СООН лег ко взаи-
модействует со спиртом ROH. то соединение типа
/—хХ
СООН (где X = F, С1, В г) медленно реагирует
с ним, так как близко расположенные атомы галогенов X
экранируют реакционный центр. Долю столкновений с над-
лежащей ориентацией оценивают с помощью крадтмюл-
ноао или стерического (от греч. stcrios — пространственный)
фактора — множителя Р. Он гораздо меньше единицы и тем
меньше, чем сложнее форма молекул реагентов. Его опре-
деляют сравнением рассчитанной скорости с найденной опыт-
ным путем. Возможны и другие затруднения. Например,
если в результате взаимодействия получаются возбужден-
ные молекулы, то ДЛЯ их стабилизации надо «сбросить? из-
быточную энергию; в противном случае сформировавшаяся
молекула распадается. Для этого необходимо «третье тело»
(которым иногда может быть и стенка сосуда). Мыслимы и
квантовые препятствия, когда течение процесса связано
с изменением электронного состояния; вероятность же не-
которых электронных переходов невелика, что приводит
к замедлению реакции.
Наконец, и это главное (так как перечисленные причины
лишь в отдельных случаях могут уменьшить число резуль-
тативных соударений), для эффективности столкновений не-
обходимо, чтобы они происходили между частицами, обла-
дающими определенной энергией, избыточной по сравнению
со средней.
Из молекул яри о-кинетической теории столкновений вы-
текает, что
£_
к = 2 К Те , (111.14)
ЭБ 'НЛ'яноё наследие России"
где Z' — козыри Циен г. не Зависящий от температуры и оп-
ределяемый молекулярностью процесса; для бимолекуляр-
ных реакций
, . Г мд+Л1в
Г = (rt+fl)= \/ , (ELIЛб)
। А В
где /\ и га— радиусы молекул; МА и ЛГВ — молекулярные
мясе и реагентов. В (I1L14) f—истинная энергия акти-
вации.
rfw
du
Рис, 38. Распределение молекул:
д — Пй склрпсгян Hfi прянем NrOj. (гИ V — Н рецепт йыекм. силроегь кото-
рые Л^ЖИТ К^ЭКДУ cJ Id [J. —|— 0.<Н муе> б — 1ЬГ» 441ECTH4lStKuft >и«ргим при рМ-
лнчпык тем пратура л (?лем*)2 Т5 > Гг > Г1
Следует подчеркнуть, что хотя (II1.14) передает темпе-
ратурную зависимость k, однако, как правило, йТ(,ор > й,ип
(подчас на несколько порядков). Эго н означает, что необ-
ходимо ввести упомянутый выше множитель Р, который
в соответствии со сказанным может достигать значений
16"э—10"4, а для медленных реакций — даже 10'1'— IO_f.
Для объяснения природы активированных молекул
Д, А. Алексеев (J9J5—1924) воспользовался законом Мак-
свелла—Больцмана. Ему отвечает кривая, выражающая
при данной температуре распределение молекул по скоро-
стям, На рнс. 38, а по оси абсцисс отложена скорость моле-
кул, а по оси ординат“ процент молекул, движущихся
в определенном интервале скоростей. Каждая из этих изо-
терм, круто поднявшись и пройдя через максимум (он отве-
чает наиболее вероятной скорости при данной температуре),
медленно опускается, асимптотически приближаясь к осн
абсцисс. Так как при больших значениях ц изотермы прак-
тически сливаются с осью абсцисс, то для и >1000 м/с
кривые даны в огромном увеличении (по оси ординат —
123
ЭБ ,:Научнов наследие России"
правая часть рис. 38, п). Площадь под каждой кривой про-
порциональна общему числу частиц, скорость которых лежит
в пределах от 1350 до 1500 м/с).
Энергия молекул определяется скоростью их движения.
Поэтому их энергетическим «спектр» будет подобен рас-
смотренному. Схематически он показан на рис. 38, 6. В от-
личие от рис. 38, а площадь под изотермой рис. 38, б в со-
ответствии с тем, что
ей
Г J JU
I —т— N.
,1 du
о
будет равна общему числу частиц. Реакционноспособными
будут лишь те частицы, энергия которых отвечает некото-
рому порогу ц£. Соответствующая величине иЕ энергия
(по сравнению со средней энергией молекул) и есть энергия
активации. Число реакционноспособных частиц равно
□&
г J и
J
"Е
И графически выражается заштрихованной на рис. 38, б
площадью. Отношение ее ко всей площади под изотермой
равно доле активных молекул. В соответствии с законом
Максвелла — Больцмана последняя равна:
м -
-^=* ЯГ- (Ш.16)
Облик этого уравнения свидетельствует о том, что доля
реакционноспособных частиц ничтожно мала.
Итак, результативными будут л ишь стол к новен и я между
частицами, обладающими (в расчете на 1 моль получающе-
гося вещества) достаточным избытком энергии — энергией
активации Е. Она определяется природой реагирующих
веществ н отвечает необходимости ослабления связей в тех
молекулах, которые вступают во взаимодействие. Рис. 39
поясняет эти представления. Здесь по горизонтали отложен
ход процесса, а по вертикали энергия рассматриваемой
совокупности веществ. Если прямая реакция эндотермиче-
ская, то диаграмма уравнения энергии молекул реагирую-
щих веществ, продуктов реакции и активных молекул будет
представлена граф! г ком типа рис. 39, а; для экзотермиче-
ских реакций — типа рис. 39, б. На них Е„ и Ё„ — энергии
активации прямой и обратной резкий и. В соответствия с за-
ЭБ Ж 41-ide наследие России"
коном сохранения энергии разность между ними равна теп-
ловому эффекту реакции,
Чем меньше энергетический барьер (или чем левее пунк-
тирные вертикали на рис. 38, а и вертикали иР. на рис. 38, б),
тем легче осуществляется процесс, т. е, тем больше его ско-
рость: кол нчественно этот вывод отвечает уравнению ([ 11.14).
С ростом температуры распределение молекул по энер-
гиям меняется: кривая несколько смещается вправо, ее
Реакционный.. путь
Рис, 39, Изменение энергии реагирующей системы (схема):
4а — црдотгрйнчсскл я реакция: й — зкэот&риячтск* я реакция
верхняя часть становится белее низкой и пологой (см.
рис. 38, б) — количество частиц с энергией больше Et
резко возрастает. Об этом можно судить, сравнивая пло-
щади под участками кривых, расположенных правее пунк-
тирной вертикали. Таким образом, повышение температуры
вызывает резкий прирост числа активных частиц и, следо-
вательно, значительное ускорение реакции.
Теория активированного комплекса. Эта теория осно-
вана на предположении, что в ходе взаимодействия началь-
ная конфигурация частиц реагентов переходит в конечную
в результате непрерывного изменения межатомных расстоя-
ний. Продукты реакции могут получаться лишь при усло-
вии образования промежуточной конфигурации, называе-
мой переходным состоянием или активированным комплек-
сом. (Понятие о нем ввели Эйрнпг и Полдни в 1935 г.). Эта
конфигурация состоит из атомов реагентов, во в ней ста-
рые связи еще не целиком разрушились, а новые нс успели
полностью образоваться. Ее образование связано с затратой
энергии; эта затрата и есть энергия активации, Необходи-
мость предварительных энергетических затрат при сближе-
]25
ЭБ ,:Нзучнов наследие России"
нии частиц вызвана их взаимным отталкиванием, обуслов-
ленным тождественностью электронов (обменный Эффект),
Поскольку при образовании переходного состояния старые
связи еще не разрушились, то энергия активации сущест-
вен но меньше энер гк и диссоциаци н. Конфи гур а цня а ктн вн-
рованного комплекса и его свойства для подавляющего боль-
шинства реакции неизвестны. Несмотря на это с помощью
приближенных квантовомеханичееккх расчетов можно по-
лучить качественные результаты, помогающие понять за-
кономерности протекания процессов. В их основе лежит
анализ изменения потенциальной энергии системы при
взаимодействии одного атома (С) с двухатомной молекулой
(АВ) по реакции
АВ .(-С —А+ВС (XX)
Сближение С с АВ требует меньших энергетических затрат,
если происходит вдоль линии связи А—В, т. е. следующим
образом:
А-В~С
По мере сближения атома С с молекулой АВ связь В—С уси-
ливается, а А—В ослабляется. На некотором расстоянии
возникает активированный комплекс А.,,В,.,С, который
представляет собой неустойчивое образование; распадаясь,
он дзет А и ВС. Изменение потенциальной энергии системы
из трех атомов В Процессе взаимодействия С С АВ можно
охарактеризовать с помощью диаграммы на примере про-
цесса
Н4-Н,^
rfE ifj
Это трехмерная поверхность (ряс. 40, о), представляющая
собой две глубокие «йОжбкны», разделенные сравнительно
«невысокиms перевалом. Проекция изоэнергетически х раз-
резов этой поверхности изображена на рис. 40, б. Путь
процесса показан жирной крЕзвой. Плато отвечает трем ато-
мам, разделенным достаточно большим расстоянием. Энер-
гетически наиболее выгоден путь взаимодействия по дну
одной ложбины, затем через перевальную точку, отвечаю-
щую активированному комплексу, и затем скатывание по
дну другой ложбины. Если по этому наиболее вероятному
пути реакции сделать вертикальный разрез, перпендику-
лярный плоскости чертежа, и развернуть его в линию езз
одну плоскость, то получится профиль реакционного пути,
характеризующий изменение энергии вдоль лутц реакции.
Эта кривая будет подобна изображению на рис. 39, а. Ось
ЗБ ' Шюв наследие России"
абсцисс (координата реакции) на этом графике отразит из-
менения в положении ядер и распределении электронов по
мере прохождений каждого акта реакции г Максимум будет
отвечать активированному комплексу- Разность энтальпий
переходного состояния н исходных реагентов — это энер-
гия активации прямой реакции; аналогичная разность для
Рнс. ^0. Ход реакции:
tr - п простри ястве: б — и Проекции (сясма)ь клжДля «рлядя отясч«тт <и?рв-
ДСЛЫШОНу уровне BFICpTnic: CTptAKufl ЗЮК41МИ ПУТЬ IКОДРДНN-IIГЛ} I ЗРкШССС-Н й
□влиrult контур (tirrpcDiлЫМИ тонкл*) огвечпет 41 ктИвлровлн!iочу комплексу
продуктов реакции — это энергия активации обратной ре-
акции. Теоретический анализ приводит к выводу, что,
строго говоря, в данном случае энергия активации Е„ (А Я*)
представляет собой разницу между соответствующими нуле-
выми энергетическими уровнями.
Таким образом, построение поверхности потенциальной
Энергии позволяет судить и о природе активировдЕЗЕЗого
комплекса, и о межатомных расстояниях, и о величине
энергии активации, т, е. вооружает сведениями, необходи-
мыми для теоретического расчета скорости реакции на ос-
новании свойств исходных молекул. Вот почему рассмат-
риваемую теорзио иногда называют теорией абсолютных
скоростей, 11равда, математические трудности не позво-
ляют осуществить строгий расчет. Лишь в простых системах,
например ДЛЯ реакции
D-|-Ha = HD-|-H
удалось это сделать, да и то приближенно. Однако эти труд-
ности непринципиальны, И можно полагать, что в дальней-
шем они будут преодолены.
12?
ЭБ ,:Научнов наследие России"
Теория переходного состояния дает следующее выра-
жение для константы скорости:
ат лн'ц xsj
к=^^~~Йт1ПГг (Ш. 17)
где х'— так называемый трансмиссионный (от лат, trans-
miss io— передача) коэффициент, учитывающий скорость
прохождения активировали ого комплекса через переваль-
ную точку, т, е. вероятность его перехода в продукты взаи-
модействия; ДЯа и AS,— соответственно энтальпия (энер-
гия активации £\) и энтропия образования этого комплекса;
Л — постоянная Планка.
Обращает на себя внимание то, что в соответствии с
(111.17) скорость реакции определяется не Д/7д активации,
a AGI активации.
Сопоставление (II 1.16) с уравнением
_Е»
ft^Pc (1П.18)
отвечающим фантам, изложенным на с. 122, свидетельст-
вует о том, что множитель Р отражает влияние изменения
энтропии в процессе активации. Для взаимодействия про-
стых молекул A Sa очень мало, для сложных может быть
значительным. Для очень быстрых реакций (Р сравнительно
велико) ASj 0, для весьма медленных (Р очень мало)
ASj < 0. Возможны случаи, когда большому значению AS1
отвечает совсем незначительная величина Дб^ это будет
наблюдаться тогда, когда и iWi велцка.
Примером подобных процессов служит денатурация бел-
ков. В этой реакции вследствие разрыхления структуры
белка и разрыва при активации части солевых мостиков
между кислотными и основными группами энтропия при
активации возрастает (AS# >0), н хотя энергия активации
велика, Абы мало, а поэтому скорость реакции значительна.
Таким образом, уравнения (Ш.14) и (111.17) являются
уравнениями «аррениусова* типа, т. е. типа (III. 12). Можно
пользоваться всеми тремя, но чаще всего применяют (111.17)
как теоретически наиболее обоснованное.
Успешное описание зависимости k = f{T'} возможно и
с помощью теории соударений, и с помощью теории пере-
ходного состояния. Отклонения становятся существенными
при (E/RT) < ^5, т. е. при высоких температурах и для
быстрых реакций.
128
ЭБ ,:Научнов наследие России"
Энергетические Константы уравнений (111.12), (III.И) и
(111.17) связаны (см. также с. 120, 122—128) простым соот-
ношением
Et = E-J- 1/2/?Г = ДЯа -F- 4 (III Л 9)
позволяющим найти непосредственно неопределяемые вс-
лнчины по опытной энергий активации £а. Целесообразно
Ре виц и oh Н н Й путь РеаЛ Ц >0 и и ы Й путь Реакционный путь
а 6 4
Рис. 41. Реакционный путь процессов
СНИО+Г (г)-СНпг)+НГ(г}
где Г — F (а); С1 (6); Вт (й) (схема)
в связи с этим заметить, что во всех трех теориях энергети-
ческие параметры активации пр и мер ею одинаковы. Вспом-
ним, ведь £t, как правило,
колеблется в пределах ~10—
60 ккал/моль, а величина RT
при обычных температурах
составляет всего 0,6ккал,что
часто соизмеримо с погреш-
ностью определения £,.
В заключение укажем на
два обстоятельства.
В сходных процессах про-
исходит закономерное изме-
нение £.- Так, уменьшение
реакционной способности в
ряду F+—С1+—Вг4 приводит к
увеличению энергии актива-
ции £ в процессе
СН4 (г) + Р (г)-СН* (г)+НГ (г)
Рис, 42. Вэанмосаяэъ между
5П ер гней актива ци и Е (ккал/мел ь)
н тепловым эффекты А//
(нмл/моль) в реакциях
СИ,4 (г) -Ь RH (г)-СН4(г) + R (г)
где R = CH, (J); (3); их =
CJI, (J): изо= CjHuWi их =
= C,HLq (5); (CHsh С (О
с возрастанием порядкового номера галогена, а уменьше-
ние прочности связи в ряду Н—F, H—CI, И—Вг— к пере-
к 2^
3 МР X, Кй ПС1ЬЯJLL1,
ЭБ гНаучнов наследие России"
Ходу or экэптермнческнх процессов (а) и (б) к sei дотер мп ве-
скому процессу (tf) (рис. 41).
Хотя между тепловым эффектом и энергией активации
в общем случае связи нет (первая величина определяется
исходным и конечным состояниями реагирующей системы,
а вторая — путем превращения), однако для сходных про-
цессов наблюдается параллелизм в изменении Л?/ и £
(рис. 42).
Г-тйнй IV
ВЛИЯНИЕ КАТАЛИЗАТОРА
1, Общие сведения
Катализатор * ~ это вещество, которое или резко ме-
няет скорость реакции, или вызывает ее, если она не идет,
ио принципиально осуществима h т. е. Дб< 0. Так, смесь
алюминия и иода при комештнон температуре вс обнару-
живает заметных признаков взаимодействия, во достаточно
капли воды, чтобы вызвать бурную реакцию 1(АС^)д]]3 =
= —72,671 ккал/моль]» Реже приходится иметь дело с яв-
лением датокй/тмиггза, когда катализатором служит один ла
продуктов реакции. Огромное практическое значение ката-
лизаторов обусловлено возможностью быстро, без затраты
энергии получать в больших количествах самые разнооб-
разные вещества.
Хотя каталитические процессы были известны очень
давно (приготовление спиртов, эфиров, уксуса, вина и т, д ).
однако лишь а XIX в, было обращено внимание на их ха-
рактерные особенности.
В зависимости от того, находится ли катализатор □ той
же фазе, что и реагирующие вещества, будучи равномерно
распределен d реакционном объеме, или образует самостоя-
тельную фазу, говорят о или ол« лата-
лазе. В последнем случае ускорение процесса обычно свя-
зано с каталитическим действием поверхности твердого тела
(катализатора^ В гетерогенном катализе применяются пе-
реходные металлы, их оксиды, сульфиды и другие соедине-
ния. Гомогенными катализаторами обычно служат растворы
* Катализ — от греч. возбуждение, лрекраш&ннй. разложение,
разрутонне. Впервые этот термин ввел немецкий химик Берцелиус
(1036).
130
ЭБ ,:Научнов наследие России"
кислот, оснований, солей и прежде всего солей d-элементов
(Сг« Мп, Fe, Со. NI, Си и др.),
Между количествами реагентов и катализаторов сущест-
вует огромная диспропорция (хотя действие последнего
примерно пропорционально его количеству). Так, одна
массовая часть катализатора вызывает превращение мил-
лиона массовых частей аммиака при его окислении в азот-
ную кислоту, Эта диспропорция особенно велика для таких
активных катализаторов, как ферменты, являющиеся уско-
рителями биологических процессов: одна молекула фер-
мента каталазы за 1 с способна разложить i 00 000 молекул
НД.
В ходе каталитической реакции катализатор остается
химически неизменным, а его количество постоя иным (если
не учитывать механического уноса и возможности протека-
ния побочных процессов, в которых катализатор участвует
как реагент).
Катализаторы отличаются избирательностью (селектив-
ностью) действия. Так, на окиси алюминия при 350—360 “С
происходит дегидратация этанола
QisOl 1=₽СЛ*+Н*О
а в присутствии меди при 200—250 "С — его дегидрирова-
ние
с2н4он^сн3сно+Н:.
В отсутствие катализатора обе реакции протекают парал-
лельно. Специфичность действия особенно велика у био-
катализаторов. Избирательность зависит не только от при-
роды катализатора, но и от условий его применения. В при-
сутствии катализатора удается реализовать такие прин-
ципиально возможные процессы, которые в отсутствие ката-
лизатора не протекают (к ним относятся, в частности, мно-
гие реакции биосинтеза сложных молекул).
Катализатор не влияет на истинное равновесие, т, е, не
меняет константу равновесия и равновесные концентрации.
Он в равной степени ускоряет и прямую, и обратную реак-
ции, Если повышение температуры не только убыстряет
процесс, но и смещает равновесие, то катализатор лишь
изменяет время его достижения; оно тем меньше, чем актив-
нее катализатор. Вводя катализатор в реакционную зону
экзотермических процессов, можно осуществить снижение
температуры, ле проигрывая вскорости процесса — обстоя-
тельство, имеющее первостепенное значение, так как многие
Б* 33!
ЭБ ' Научное наследие России"
промышленно важные реакции протекают с выделением
теплоты.
Присутствие в зоне реакции посторонних веществ ока-
зывает различное влияние на катализатор: одни нейтральны,
другие усиливают действие катализатора, третьи его ослаб-
ляют или прекращают. Ускорители каталитических про-
цессов называются промоторами или активаторами. Так,
небольшая добавка щелочных сульфатов в сотни раз повы-
шает активность VjOj — катализатора окисления SCt в SO3.
Например, кислород и его соединена я являются каталити*
ческами ядами, вызывающими обратимое отравление желез-
ного катализатора; в процессе синтеза NHS снимает отра-
вление этого катализатора тщательно очищенная свежая
азотоводород нал смесь. Присутствие серы и се соединений
в этом же процессе вызывает необратимое отравление ката-
лизатора: восстановить его активность действием свежей
смеси не удается.
Велико значение методики приготовления и обработки
катализатора. Чем более рыхлой, неустойчивой и значи-
тельной является его поверхность, тем больше его актив-
ность. Низкотемпературный режим приготовления катали-
затора не позволяет его частицам перегруппировываться
в более устойчивые формы.
Таковы основные черты каталитических процессов.
2, Элементы теории катализа
Действие катализатора связано с тем, что он вступает
в промежуточное взаимодействие с реагирующими вещест-
вами, направляя процесс на новый реакционный путь.
Сложные каталитические процессы протекают по несколь-
ким направлениям с образованием различных иродуктон,
но □ любом случае должно соблюдаться условие AG<t О
(конечно, не обязательно ускоряется именно тот процесс,
для которого величина ДО наиболее отрицательна). О про-
межуточных соединениях обычно судят по косвенным при-
знакам; Лишь в редких случаях удается зафиксировать их
существование (например, путем спектрального анализа).
Если исключить сравнительно немногочисленные слу-
чаи ускорения за счет возникновения цепной реакции,
когда появляются богатые энергией частицы (см. с. 115),
то действие положительного катализатора в основном объяс-
няется тем, что он, меняя механизм процесса, снижает энер-
гию активацци. (Из изложенного ясно, что Ё и t пони-
ЗБ (-/ауччее наследие России"
жаются на одну я ту же величину, т. е. встречные реакции
ускоряются одинаково.)
На рис. 43 и 44 схематически представлено изменение
энергии реакционной системы вдоль бес каталитического
(тонкие кривые) и каталитического (жирные кривые) путей
реакции — снижение
энергии активации в
присутствии катализа-
торов Д£к>1 (подробнее
см. стр. 134)— будет тем
значительнее, чем акти-
внее катализатор. Так,
энергия активации реак-
ции 2НТ = Н, + ^сни-
жается в присутствии Au
с 44 до 25, а в присутст-
вии Pt — до 14 ккал.
Действие катализаторов
можно проиллюстриро-
вать и рис, 38, б, сме-
щение вертикали и£
влево приводит к воз-
растанию долл реакци-
онноспособных частиц.
Рис. 43. Реакционный путь процесса
при lOMorcHHOM катализе (кривая
/-s'j-a"-2) (схема):
ifкат — умел injitii |аг нергнн лктчгШин
иод влиянием kiiTJi^itjatfipai J _ ИСХОД-
НЫ* вгшгет*я: ? — килечные продуши:
J — (промежуточное соединение (крпгля
Ьй-3 — &сСкаталп1Нчеекнй путь
Для оценки порядка величины ускоряющего действия
катализатора обратимся к уравнению (II 1.12). Так как ве-
личина £ стоит в показателе степени, то даже сравнительно
небольшое ее уменьшение под действием катализатора
должно привести к значительному увеличению константы
скорости. Действительно, снижение £ с 50 до 30 ккал/моль
вызовет (при Т = 500 К) ускорение реакции в
деки 20(КЮ
е RT «i2'500 =fM д= 4,85 10й раз.
А ведь на практике приходится иметь дело с процессами,
в которых £ уменьшается в 2—3 и в большее число раз.
Так, Pt вызывает уменьшение £ в реакции
ИОь (r) + Oi (r)=2SQi (г)
с 60 до 15 К кал/моль. Энергия активации гомогенных ката-
литических процессов обычно больше энергии активации
гетерогенных каталитических процессов. Если в последних,
где достигается ускорение в 10й -s- IO1® раз, £ составляет
ЭБ ,:Научнов наследие России"
Ш
примерно 15-5-30 ккал/моль, то в ферментативных про-
цессах— всего 8-5-12 к кал/моль.
Так обстоит дело с формально-энергетической стороиоГг
каталитических процессов *.
Каков же механизм действия катализаторов?
Для гомогенных реакции считают, что катализатор обра-
зует з той же фазе промежуточные реакционноспособные
продукты: медленно про-
текающий процесс, на-
пример реакция
Л-|-В=АВ
в присутствии катализа-
тора К I гр он с ходит в две
стадии?
Релкцпшжый путь
А+К = АК
и
АК + 9 = АВ+К,
Рлс, 44, Реакционный путь процесса
при гетерогенном катализе (кривая
/й'-З-а" — 4-ч'" - 2) (схема):
йЕкйт “ уменьшение №?Гнн ант ie бал. ни
п<?д влиянием к*тйлннто|м: у —н-сход.
rNc веществе; ? — Конечные продукты;
j — л дсо(| бяро rtf ini ис и с иодные вещесгм:'
<а" — лдсогбиропаннМЛ -НИ ГИВИ рол* IIII ий
комплекс! 4 — д д серб кровв л и ые продукты
рез к ци к (к рн р? я У -и-J — беек в тэ .1 куч чсСн и 21
путь процесса ]
т. е. образуются частицы
промежуточного соеди-
нения А К (затем актив-
ный комплекс ЛВК) я
конечные продукты с ре-
генерацией катализато-
ра, Примером может
служить реакция
сна-
для которой Е = 45,5 к кал/моль. Проведение ее в присут-
ствии паров иода вызывает течегзие двухсталийного про
цесса с A£kn = 13 ккал/моль (ускорение в |(? раз).
Здесь величина (см. рис, 43) — это разность между
энтальпией образования активного комплекса, содержа-
щего катализатор (точка а'), я энтальпией образования
• Расчеты, подобные гтркмщешгому, не всегда дакгг удовлетворл-
тельное согласие с опытом. Это объясняется тем, что катализатор
может ле только уменьшить £, но н увеличить энтропию актнвацнн
(см. ниже). Тогда в ссютвстстиин с уравнен кем (111.17} при вычислен ин
<AS »> к ,T-'A / 1,>И
надо принять во кинмэкие в множителье * “Jt fe “ l!tKnl . Этот
множитель Fte следует учитывать, если можно предположить, что кон-
фигурация активного комплекса относительно исходных молекул
одинакова как без катализатора, так и с ним,
ЭЕ Шуччсе наследие России"
активного комплекса, в состав которого входят только ис-
ходные вещества (точка д). Если даже суммарная разность
уровнен 2 — а* н 3 —д' будет больше разности уровней
/ — u\ Ti С» еСЛИ (El “h Да) ^*1иск-*!
£1|СНТ1 то и в этом случае скорость реакции иоз-
растет.
Характер промежуточный соединений с катализаторам различен.
Для кислогна-основных реакнищ когда электронные пары переме>
иупотсч без разобщеннsi электронов (гетеролитнчехкнй разрыв аалепр
них связей! — это комплексы типа солей; для окислительно ьосстапо-
внтельиых реакций^ когда э.тсктрозшыс пары раздел я кхпея (гомолити-
ческие или радикальные реакции}! это. как правило, комплексы с уча-
стием молекул или ионов, содержащих металлы переменной салеит-
ности К первой группе относятся процессы, в которых катализато-
ром служат кислоты или основания; это реакции присохли пен ня (err
щемления) полярных молекул. Ко второй группе о гное як я процессы,
в которых катализаторами служат ноны ^элементов пли образованные
ими комплексы (в частностиi реакции с участием атомов Н плн Ок
В последних перенос электрона осуществляется за счет попеременного
окисления н восстановление я иояаг содержащего металл. М а пример.
Мл3*+(COO)J" М п {СОТ)J
катали- ут
элтФр Ьродунт
Мп(СОО)Г +сог+ ООСГ
coo’+at^o^+ci+ci-
н
(!|4-Мп^—Мл^+СГ
пли суммарно
* - МлЧ-
CI + (СОО)=- 2CO,+ 2С1-.
Хотя промежуточные продукты являются весьма реак-
ЦП он нос пос об}! ымн (это очень нестойкие соединения с ма-
лым временем жизни), однако для ряда процессов их уда-
лось выделить, доказав тем самым их существование,
Более сложен механизм гетерогенного катализа. Однако
бесспорно, что в этом случае существенную роль играет
поглощение поверхностью катализатора реагирующих ча-
стиц. Процесс также протекает в нес коль ко стадий, но здесь
их больше и они иные: за счет днффуз и и частицы исходных
реагентов подводятся к катализатору я его поверхность
поглощает их (активированная адсорбция). Этот процесс
сопровождается сближением молекул и повышением — под
влиянием силового поля поверхностных атомов катализа-
тора — их химической активности; изменяется структура
электронных оболочек молекул и, как следствие, пони-
жается активационный барьер. В результате на катализа-
ЭБ ,:Научнов наследие России1
-гире происходит реакция. Затем продукты взаимодействия
покидают катализатор и, наконец, за счет диффузии пере-
ходят в объем. Таким образом, промежуточными в гетеро
генном Катализе являются поверхностные соединения.
Всегда (по крайней мере в первом приближении) можно
считать, что одна из пяти стадий — наиболее медленная —
определяет скорость процесса в целом.
Энергетика второй, третьей и четвертой стадий, совокуп-
ность которых и составляет собственно гетерогенный ката-
литический процесс, схема-
тически отражена на рис, 44.
Адсорбция исходных веществ
Рис. 45. Зависимость катали-
тической активности {k) неко-
торых элементов (в отношении
разложения аммиака i:j>h i =
800 “С и р = I атм} ОТ их ло-
ридкового Etohccjiai Z
сопряжена с энергией акти-
вации, отвечающей возрас-
танию энтальпии На участ-
ке 1 — а'. Она приводит к
энергетическому уровню 3.
Возникает активный ком-
плекс (д'), адсорбирующийся
на катализаторе (процесс
3 — а"). Затем образуются
адсорбированные на катали-
заторе продукты реакции
(а’ — 4), после чего послед-
ние покидают поверхность ка-
тализатора (десорбции отве-
чает участок кривой 4— а").
Таким образом, и в гетеро-
генном катализе ускоряющее
действие катализатора связа-
ло с тем, что реагирующие вещества образуют промежуточ-
ные соединения .что приводиткснижению энергии активаЦПи,
Процесс формирования промежуточных поверхностных
соединений происходит на активных участках (активные
центры) катализатора. Силовые поля активных центров
ослабляют связи между атомами адсорбированных молекул,
что л приводит к возрастанию реакционной способности.
Важно подчеркнуть, что активные центры составляют весьма
небольшую долю поверхности. Это подтверждается дейст-
вием каталитических ядов, блокирующих активные центры;
ничтожное количество их уменьшает активность катализа-
тора пли выводит из строя большие его массы. Путем дози-
ровки отравления можно даже определить число активных
центров на поверхности катализатора,
136
ЭБ ‘Научное наследие России"
Для увеличения попер хи ости и, тем самым, количества
активных центров катализатора его нужно тонко измель-
чить. Чтобы он не уносился и не создавал сопротивления
току газа, его наносят (осаждают) на инертный носитель
С развитой поверхностью (силикагель, асбест, пемза и т. д.),
Представляется существенным не только наличие актив-
ных центров, но и число их и характер расположения. Чем
больше геометрическое соответствие между расположением
атомов в адсорбируемых молекулах исходных веществ и
расположением активных центров на поверхности катали-
затора, тем активнее последний. Если число активных цент-
ров или же их расположение (по их рисунку или по рас-
стояниям между ними) не соответствует структуре молекул
реагирующих веществ, процесс ускоряться не будет. Из
этого следует существенное значение энтропийного фактора
в гетерогс н н ом катализе, Рол ь п ромоторов с вод и тс я гл явным
образом к {Доукомплектованию» активных центров.
Вопрос о природе (строении) активных нейтрон нахо-
дится в стадии изучения н является предметом научных
дискуссий. Вследствие этого единой теории действия, а по-
этому и подбора катализаторов нс существует. Можно лишь
говорить об общих соображениях. Таковыми являются:
I) катализатор должен быть способен к химическому взаи-
модействию хотя бы с одним реагентом; 2) изменение энер-
гии Гиббса взаимодействия катализатора с реагентами
должно быть менее отрицательным, чем его изменение и ка-
тализируемой реакции. Однако в последние годы достиг-
нуты большие успехи в представлениях о механизме ката-
лиза. позволившие выдвинуть некоторые общие принципы
выбора катализаторов для различных типов реакций, Так,
во многих случаях определяющим фактором в подборе ка-
тализаторов является положение элементов в периодиче-
ской системе Д, И. Менделеева, На рис. 45 представлены
результаты изучения каталитической активности металлов V
и VI периодов в реакции разложения аммиака. Налицо
периодичность изменения каталитических свойств с макси-
мумами активности у железа и его аналогов — рутения и
осмия.
ЭБ 'Научное наследие России"
РАЗДЕЛ IV
РАСТВОРЫ
ПП ЕДЕНИЕ
Если привести в соприкосновение два или несколько вс*
ществ, то можно получить либо новые соединения, либо
неоднородную смесь (которую можно вновь разделить на
составные части с помощью механических или простых фи-
зических методов), либо, наконец, однородную систему.
В первом случае протекает кнк-тческая реакция, во вто*
ром — механический процесс (в результате которого полу*
чается смесь, причем ее неоднородность будет определяться
лишь усилиями, приложенными при перемешивании). Тре-
тий же случай — процесс образования раствора — проме-
жуточный между химическим и механическим процессами,
Состав растворов в некотором интервале концентрации, тем-
ператур и давлений может меняться непрерывно. Отсутст-
вием у них постоянства состава и неприменимостью к ним
закона кратных отношений и закона эквивалентов растворы
приближаются к механическим смесям. С химическими сое-
динениями их роднит однородность (часть тождественна
целому); другим общим признаком являются довольно зна-
чительные объемные и энергетические эффекты, сопровож-
дающие процесс растворения-многих веществ.
Первые обширные работы по свойствам растворов при*
надлежит М. В. Ломоносову. Им же была намечена програм-
ма исследования растворов, не утратившая актуальность
и до настоящего времени.
Растворы играют важную роль в живой и неживой при-
роде, а также в науке я технике. ФЕГзиологические процессы
в организмах животных н в растениях, всевозможные про-
мышленные процессы (например, в производстве щелочей,
солей), образование осадочных пород н другие в большин-
стве своем протекают в растворах. Повсеместность растворов
объясЕзяется, в частности, и тем, что процесс растворения
самопроизволен, т. е. сопровождается убылью энергии
Гнббса, Поэтому найти чистые вещества ез естественных
условиях или приготовить их в лаборатории чрезвычайно
трудно,
ЗБ ' наследие России"
Абсолютно нерастворимых веществ FfCT, В сущности н
серебро, и золото растворимы в воде, хотя их раствори-
мость крайне незначительней
Различают и рислмары. В первых вещества
рас11а даются tta частицы., ранные размерам молекул нлн ионов (КГ1 —
— 10Г* см). Коллоидные системы лежат асжду истинными растворами
ее механической. смесью: это мп1фпгстсрогснлые высоко дисперсные *
системы (радиус чвстеец Ю"1 — KTZ cuj ее грубоднсперслые сеестсмы
(радиус частиц 10"э—10"* см). Они ззрегативно Егеустойчивы, так
как без специальной (пополнительной) стабилизации коллоидные
части цы объединяются ее оседают,
ГрубоДЕЕСПерСНЫС системы обычно бывают либо я виде C^TtWrJUn —
твердые частицы В ЖИДКОСТЯХ, либо В виде ЬСН£ — ЖНДКЕЕе частицы
одного вещества в другом жидком веществе (при сох ранен ни гетеро-
генности).
Примерами коллоидных систем служат растворы клея и желатины
(коллоидные растворы или эолн: от греч. со На— клей) . туман и
дым (аэрозоля — твердые нлн жидкие чистины в газе), не которые
цветные стекла. Типичной эмульсией является молоко, в котором мел-
кие Енарини масла плаядЕот в жидкость!. Суспензии получаются, напри-
мер, при взбалтывании гл илы в воде. В последнем случае твердые час-
тицы со временем Оседают и а Дно сосуда.
Коллоидные растворы отличаются ог истин пых тем, что их чи*
стицы сильно рассеивают проходящий через них спет н делают заметь
ным путь про» уте иного светового луча
Мы сстэЕЕоппмея только на чети иных растворах. Учение о кол-
лоидных расгпорах обычно рзссматрияастся отдельею,
Растворы бывают газообразные (газовые смеси), жидкие
и твердые. К газообразным растворам относится, налрн-
мер, воздух, Морская вода — наиболее распроетраЕ1енныЙ
жидкий раствор различЕзых солей ее газов в воде. Твердыми
являются растворы водорода в палладии, воды и других
яшдкостей в цеолитах и т. д. Многие металлические сплавы
из-за нх однородности относят к твердям растворам.
Большое значение имеют жидкие растворы. Поэтому на
них мы сосредоточим основное внимание. В этих растворах
обычно протекает большинство реакций, так как имеются
весьма благоприятные условЕзя как для перемещения мо-
лекул, так и для тесного их сближения, необходимого для
химического взанмодейстния,
Наибольшее значение имеют водные растворы, так как
вода — самый распространенный и универсальный раство-
ритель. Она играет очёезь важную роль в нашей жизни и
обладает рядом особенностей, обусловлеЕтых наличием
различных структур, отличающихся как энергетически, так
• Слово «дисперсный* происходит от лат. dkper^are— рассеиьвть,
раздроблять.
ЭБ ,:Научнов наследие России1
139
if химически. Важно также следующее: так как водные
растворы обычно изучаются при температурах, близких
к температуре замерзания воды, то ее структурность выра-
жена весьма чет ко.
Растворитель и растворенные вещества. Эти понятия
в известной степени условны. Какое вещество, например,
считать растворителем в смеси, состоящей из равных коли-
честв воды и спирта? Иногда за растворитель принимают
то вещество, которое при охлаждении раствора кристалл и-
зустоя первым. Так, из водно-спиртового раствора сначала
выделяется вода; ее можно считать здесь растворителем. Но,
например, из многих водно-солевых растворов кристалли-
зуется сначала соль, а иногда одновременно и соль, и лед.
Можно принять за растворитель и то вещество, которое
находится в преобладающем количестве, по при этом надо
иметь в виду, что нередко оно при охлаждении раствора
отвердевает не первым. За растворитель принимают веще-
ство. добавление которого не изменяет агрегатного состоя-
ния раствора, Если одним из составляющих раствор ве-
ществ является жидкость, а другими — газы пли твердые
вещества, то растворителем обычно считают жидкость.
Обозначения свойств составных частей при fin то сопро-
вождать индексами 1, 2. 3, ..., причем для растворителя
обычно используют индекс 1.
Концентрация. Наряду с температурой и давлением
основным параметром состояния раствора считается его
концентрация.
Существует много способов выражения концентрации.
Их можно разделить на две группы: способы, в которых
количество данного компонента н количество всей смеси
выражаются в одинаковых единицах, и способы, в которых
они выражаются в различных единицах. Наиболее употре-
бительны следующие варианты.
Процентная концентрация по массе
(массовые проценты)-— число массовых частей
растворенного вещества в 100 массовых частях раствора.
Мольная доля (Д7() — отЕгошение числа молей
данного компонента i к сумме чисел молей всех компонентов
раствора. Для бинарного раствора
11 л I , ,
ЛГ( = -—:-- И /У4=-----п J
где rtj н — числа молей растворителя и растворенного
вещества .
ЭБ 'М-Ячое наследие Рогсии"
Аналогично определяются массовая и объемная доли,
Моляльность (/я) — число молей растворенного
вещества, содержащихся в 1000 г растворителя.
Молярность (или мольност ъ) (Л!) — число
молей растворенного вещества в [ л раствора.
Нормальность (и.) — Число Грамм-эквивалентов
растворенного вещества в 1 л раствора.
Титр (Г) — число граммов растворенного вещества
в [ МЛ раствора.
Часто концентрацию выражают через плотность ра-
створа.
Для пересчета объемных единиц концентрации в весо-
вые или в мольные и обратно нужно знать плотности ра-
створов и молекулярные массы растворенных веществ.
В научных исследованиях часто пользуются мольными
долями, мол я л ьн остью или массовыми процентами (они не
зависят от температуры). При проведении реакций между
растворенными веществами удобно пользоваться раство-
рами, концентрация которых выражена через нормаль-
ность.
Необходимо помнить, что различные способы выражения
концентрации дают разные величины (табл. И),
Таблица 11
Растворимость сери в бензоле И толуоле при 25° С
Раегворямость 17л МолЬн % Мас %
Eelmcji 18,5 0,60 2
Толуол И,К 0,74 2
Глава I
О ПР А ЗОЕ А11IIЕ FACT Л О РОЛ
При образовании раствора в общем случае происходит
изменение свойств и растворителя, и растворенного веще-
ства (растворенных веществ). Эго обусловлено тем, что
в растворе действуют силы, вызывающие и межмолекул яр-
кое взаимодействие (электростатическое, ван-дер-ваалъсовы
силы), к ионно-дипольное взаимодействие, проявляющиеся
па сравнительно значительных расстояниях, и специфиче-
ское взаимодействие (донорно-акцепторное, водородная
1-1J
ЭБ 'Научное наследие России"
связь), сказывающееся на сравнительно небольших рас-
стояниях, Первое является общим для всех веществ; оно
связано с совокупностью физических процессов, Второе
связано с перестройкой электронных оболочек молекул,
атомов и ионов: оир обусловлено химическими изменениями.
Сторонники «физической» теории растворов трактовали
образование раствора как суммарный результат молеку-
лярного движения и взаимного сцепления частиц, т. е. по-
лагали, что при растворении доминируют физические про-
цессы смешения веществ друг с другом. Наоборот, привер-
женцы «химической» теории подчеркивали преобладающую
роль взаимодействия между различными частицами в ра-
створе, полагая, что силы, действующие в растворах, чисто
химические, только менее интенсивные. Эти крайние точки
зрения дополняют друг друга. Поэтому правильнее было
бы не противопоставлять их, а объединять, подчеркивая
при этом, что в за вне и мости от природы компонентов раство-
ров и условий их образования (соотношение между вещест-
вами, температура, давление) влияние «физических» и «хи-
мических» факторов может быть различным. Основу сов-
ременной теории растворов и составляет синтез этих точек
зрения, восходящих к представлению о растворах, которое
было дано Д. И. Менделеевым. Рассматривая растворы как
смеси непрочных химических соединений определенного со-
става, находящихся в состоянии частичной диссоциации, он
подчеркивал необходимость создания общей теории ра-
створов, способной объяснить с едином точки зрения всо
наблюдаемые факты.
Совокупность всех процессов, возникающих в результате
появления в растворителе растворенного вещества, назы-
вается сольватацией (от лат. solvere — растворять). Для
частного случая водных растворов говорят о гидратации
(от греч. hydor— вода). Понятие о сольватации было вве-
дено И. А. Каблуковым (1891).
В результате различных взаимодействий в растворе мо-
гут возникнуть соединения переменного состава между ча-
стицами растворенного вещества к растворителя. Такие
соединения называются «шмалшш (для водных раство-
ров — гидратами).
Нельзя понять сложных явлений в растворе и дать вер-
ную его характеристику, не учитывая всех факторов, а при-
нимая во внимание лишь некоторые из них и тем более один
(например, влияние водородной связи), даже если он пре-
обладает. Необходимо учитывать все типы взаимодействия
14Й
ЭБ ,:Научнов наследие России"
между всеми видами частиц, включая те из них, которые воз-
никли при образовании раствора (они могут быть связаны
не только с формированием новых частиц, но и с разруше-
нием существовавших в индивидуальных веществах), Воз-
никновение и распад любых агрегатов описываются зако-
ном действующих масс, так как в растворе устанавливается
динамическое равновесие между всеми входящими в пего
частицами. Это позволяет охарактеризовать раствор как
равновесную однородную систему, которая достигла мини-
мума энергии Гиббса в результате взаимодействия всех ее
частиц за счет всех возможных типов взаимодействия между
ними *.
По мере повышения концентрации раствора, как ггра-
вило, усиливается интенсивность взаимодействия между со-
держащими его частицами и усложняется его структура;
наоборот, по мере уменьшения доли растворенного вещества
строение раствора упрощается и взаимодействие между ча-
стицами ослабевает. Все это делает понятной огромную слож-
ность развития количественной теории концентрированных
растворов. До сих пор не удалось полностью выяснить пи
состав, ни пространственную конфигурацию продуктов взаи-
модействия, природу связи в них, взаимодействия между
частицами растворенного вещества. Сложность взаимодей-
ствия в растворах усугубляется отсутствием математиче-
ской теорий строения жидкостей. Поэтому разработка тео-
рии растворов высокой концентрации — дело неблизкого
будущего. Гораздо проще обстоит дело с очень разбавлен-
ними растворами, количественная теория которых создана
уже давно (см. гл. II данного раздела).
Прибавление к жидкости второго вещества в зависимо-
сти от природы компонентов раствора может вызвать как
стабилизацию ее структуры, так и ее частичное (или полное)
разрушение. Первый тип взаимодействия обычно связан
с образованием растворов посредством внедрения частиц
растворенного вещества в пустоты растворителя. Примером
могут служить атомы благородных газов, входящие при
растворении в пустоты воды. В подобных случаях введение
молекул растворенного вещества тормозит движение окру-
жающих его молекул растворителя. Вода вокруг таких ча-
стиц становится более связанной. Ко второму типу отио-
* В твердых одиородных системах (см. с, ООО и ся.) очень часто
ссылается заторможенное состояние (ложное равновесие}. Несмотря
на это, их также принято относить к растворам,
143
ЭБ ':НаучноЕ наследие России"
<щтся раствор четырех хлористого углерода в этиловом
спирте. Появление молекул СС14 вызывает диссоциацию
ассоциированных комплексов растворителя.
Влияние природы растворителя можно продемонстриро-
вать и на следующем примере. Стремление к упорядочению
в NHa {ж) меньше, чем в НгО (ж), так как в отличие от
молекулы воды в молекуле аммиака лишь одна не поделен-
ная пара электронов; кроме того, аммиак не может образо-
вать трехмерные агрегаты, да и водородные связи и нем
слабее, чем в воде.
1, Изменение свойств при образовании растворов
В результате изменения структуры при переходе на
индивидуального состояния в раствор, а также в резуль-
тате происходящих при этом взаимодействий изменяются
свойства системы. На это указывает, в частности, наличие
тепловых (АН) и объемных (AV) эффектов при растворении,
Так, при смешении I л этилового спирта с I л воды объем
образующегося раствора оказывается равным не2л, а 1,93л
(25 X). В данном случае уменьшение объема (на 3,5 %)
обусловлено в основном образованием водородных связей
между гидроксильными группами спирта и воды, а также
разрушением ЛЪДОПОДОбных структур последней,
Если раствор образуется с поглощением: теплоты (АН >
>0), то в соответствии с общим уравнением Д6 =
= Л//~ TAS (II.10) условием растворения служит не-
равенство AS >0. Эго означает, что энтропия раствора
будет больше суммы энтропий' растворителя п растворен-
ного вещества. Если же растворение является процессом
экзотермическим (Л/У < 0), то в соответствии с упомянутым
уравнением возможны два случая: AS >-0 и AS С 0.
Первый вероятнее, fiO чаще встречается второй, так как,
в силу специфичности взаимодействия в растворе, чем
больше &Н, тем более должна быть упорядочена структура
раствора.
Часто процесс растворения сопровождается изменением
агрегатного состояния (например, при растворении кри-
сталлических и газообразных веществ в жидкостях). Тогда
теплоту растворения можно рассматривать как сумму теп-
лоты соответствующего фазового перехода (ф, п.) и теплоты
сольватации:
П-f-^Ясольи- (1V-I)
144
ЭБ "Научим наследие России11
Аналогично для энтропии имеем
и = А 5ф_ |Е Л ^сольи' (IV. 2)
Для частного случая растворения кристаллических ве-
ществ в жидкостях первое слагаемое в них соответствует
разрушению кристаллической решетки; поэтому АЛЛ, п >0
и А51кп >0.
Сольватация — процесс экзотермический (A0),
поэтому теплота растворения может иметь различный знак.
Сольватация означает упорядочение состояния системы
(происходит уменьшение числа частиц). Следователию,
Л3смь»< 0; однако ло абсолютной величи ее е последнее
слагаемое обычно невелико, поэтому растворение сопро-
вождается возраста fin ем э ее тропи и.
Для растворения газа в жидкости первое слагаемое
в этих уравнениях отвечает конденсации, т. в. А//(,„< 0 и
Д5Ф.,.< 0.
Для опенки степени чувствительности энтропии раст-
ворения к структуре укажем, что для растворения ар гое; а
в воде Л&ольв = —22 э. е.. а эффект ограничения неупоря-
доченности вследствие уменьшения объема в результате
растворения газа в жидкости состаЕзляет 12 э, е. Следова-
тельно, разница между этими величинами (—10 э, е.) при-
ходится на упорядочение структуры растворителя.
Идеальные растворы. При смешении жидкостей, моле-
кулы которых не полярин и сходны между собой по струк-
туре н по природе химической связи, тепловые и объемные
изменения очень малы. Например, при сливании толуола
с бензолом и Af/^Ои Дк ^0. Если смешение двух жид-
костей сопровождается лишь хаотическим распределением
частиц без изменения межч а стачного взаимодействия, то
теплота смешения patnia нулю, а энтропия меняется лишь
d результате изменения концентрации.
Смеси, в процессе образования которых тепловые и
объемные эффекты отсутствуют, т, е. АН = 0 и A V = О,
называются идеальными (совершенными) растворами.
Иногда эту особенность идеалье;ых растворов выражают
другими словами — говорят, что для них энтальпия и объем
аддитивны.
Свойства идеальных растворов, подобно свойствам раз-
реженных газовых смесей, не зависят от природы раство-
ренного вещества, а определяются лишь их концеЕпрацией.
Следовательно, единственной причиной их образования (как
и образования газовых смесей) является возрастание энт-
L15
ЭБ ,:Научнов наследие России"
решим при смешении. Для идеального раствора ,£ О
(так как растворение связано с изменен нем величины ®
для любого раствора). Однако Д$гвеш в этом случае не будет
зависеть от природы компонентов, а однозначно опреде-
лится ия соотношением (мольными долями).
Близки к идеальным крайне разбавленные растворы раз-
личных веществ (см. гл, II); в них можно пренебречь взаи-
модействием между частицами растворенного вещества из-за
их разобщенности. Однако уподобить поведение растворен-
ного вещества в разведенных растворах поведению разре-
женного газа (а растворителя — вакууму) и основывать на
этой аналогии все рассуждения и выкладки было бы опро-
метчиво. Такая аналогия носят скорее формальный харак-
тер. Ведь строение жидких и твердых растворов отвечает
структуре вещества не в газообразном, а в конденсирован-
ном состоянии (см. с. 165).
Если агрегатное состояние компонентов и их смеси не-
одинаково, то процесс образования идеального раствора
уже не будет ни изоэнтальпийным, ни изохорным. Очевидно,
в этом случае
А^мсгь и л н ДУ рдет* в Уфь л*
Так, при растворен]!]! твердого нафталина в жидком бен-
золе
" й^ = (д^Ьн,.
т. е, должны наблюдаться эффекты, отвечающие переходу
нафталина в состояние жидкости [переохлажденной, так
как Т< (TnJcfH.L
2. Растворимость
Процесс растворения связан с диффузней, т. е. с само-
произвольным распространением частиц одного вещества
между частицами другого. Так, например, при внесении
куска сахара в стакан с водой сахар растворяется, т. е.
раздробляется до молекул и равномерно распределяется по
всему объему раствора. Суммарный объем сахара и воды
почти не меняется, но объем, в котором находились частицы
сахара до растворения, значительно возрастает после раст-
ворения. Происходит изменение агрегатного состояния,
«расширение» сахара и разу порядочен не движения его мо-
лекул-
ЗБ 1^Ьчсе наследие России"
Процесс растворения идет самопроизвольно (AG С 0) н
раствор остается ненасыщенным. Когда энтальпийный и
энтропийный факторы в уравнении (11.10) станут од Ивано-
выми, т. е. Лб = 0, система окажется в состоянии нстин-
н о по р а вновес и я. Раствор ста ко в нтся насыщен яым > В та ком
растворе неопределенно долго могут сосуществовать без
каких-либо изменений раствор и избыток растворяемого ве-
щества. Так как скорость, с которой молекулы, отрываясь
от поверхности твердого тела (при наличии его избытка),
переходят в раствор, равна скорости осаждения молекул
растворенного вещества па той же поверхности; равновес-
ное состояние может быть нарушено только в результате
изменения температуры, давления млн введения других
веществ (см. с. ] 49 и ел.). Из изложенного следует, что
растворимости Твердых веществ способствует склонность
к возрастанию неупорядоченности, а их кристаллизации —
энергетический фактор, т. е. склонность к понижению по-
тенциальной энергии. Равновесие соответствует концентра-
ции, отвечающей уравновешиванию обоих процессов. На-
оборот, растворимости газообразных веществ благоприятст-
вует тенденция к уменьшению неупорядоченности.
Можно получить я пересыпанные растворы, T. е. такие,
концентрации которых выше концентрации насыщенного
раствора (при данных температуре и давлении). Пересыщен-
ные растворы можно приготовить осторожным и медленным
охлаждением насыщенных при высокой температуре раство-
ров (например, NasSaO3 и CH3COONa). Они представляют
собой систему, находящуюся в кажущемся равновесии
(AG >0). Встряхивание или внесение н них кристаллов того
же вещества, которое находится в растворе, или другого,
например изоморфного с ним, вызывает кристаллизацию;
происходит выделение избытка растворенного вещества и
раствор становится насыщенным. Система таким образом
переходит в состояние истинного равновесия, что сопрово-
ждается убылью энергии Гиббса. Здесь, как н в случае
химической заторможенной системы, ничтожный нм пул вс
может вызвать огромный эффект (см. с. 35).
Итак, растворимость данного eeutpemea отвечает вели-
чине его концентрации в нааицрнном растворе. Она опре-
деляется энтальпийным и энтропийным факторами. Для
разных веществ состояние истинного равновесия в растворах
может наступить при различных соотношениях между ними.
Это связано с различной природой растворенного вещества
и растворителя, с неодинаковой интенсивностью взаимолей-
L47
ЭБ ,:Научнов наследие России"
ствня их частиц и др. Поэтому концентрация насыщенных
растворов различных веществ имеет разную величину.
Растворимость — наиболее изученное свойство раство-
ров. Для различных веществ она колеблется в значитель-
ных пределах. В табл. 12 приводится растворимость неко-
торых [semccTB в одном растворителе, а в табл. 13 — раство-
римость одного вещества в различных растворителях.
Таблица 12
Растворимость некоторых веществ и воде
(в г на ЮТ г HjO) при 20’ С
! Расгворияосгь Вещество Растворимость
€.Н,А NaCI HjBQ, 1 200 36 3 ^астворимисть кг в различу ОСО, Agt >днда калин (мае. IX растворителях 0.0013 0.00000013 Таблица 13 96»
Растворитель Растер и мОсгь Рлстиоранль P?CThQP И
Н2О NH, CHXJH сн/осна 593 Й4,5 33,01 1-1ДЭ7 1,302 OOgOu г'Ях: -е* -1- hJL, □ ч 0,307 0,20 0,05 0,00016 0,00012
Разумеется, понятия ^(асыщеинын» и «концентрирован-
ный* не тождественны. Настрнмер, 35%-ный водный раствор
КВт является концентрированным, но он ненасытен, так
как при 20 аС в 100 г воды растворяется около 65,8 г КВг.
Наоборот, насыщенный раствор сульфата кальция прн
20 *С на каждые 100 г воды содержит всего 0J? г Са5Ол;
сказывается малая растворимость этого вещества; раствор
будет насыщенным, но весьма разбавленным. Так как насы-
щенные растворы различных веществ охватывают концент-
рации от ничтожно малых до весьма значительных, то они
подобны лишь с формальной стороны. Их структура может
быть самой разнообразно ft — от почти неискаженного строе-
148
ЭБ 'Научнов наследие России"
ния чистого растворителя до структуры, близкой к кристал-
лической решетке растворенного вещества.
Рассмотрим более сложный случай. Если ввести в си-
стему, состоящую из двух несмешнвающихся жидкостей,
растворимое в них вещество, то оно распределится между
этими жидкостями Так, что при данных температуре и дав-
лении отношение его концентраций в этих растворителях
остается ней змеиным (независимо от общего количества
распределенного вещества), т. е.
Ct/Ci = К, (1V.3)
где константа равновесия К называется коэффицамтом рас-
пределения. Уравнение (1V-3) называется законом распре-
деления. Его можно рассматривать как пример примене-
ния уравнения (11.16).
Растворимость определяется тремя факторами: а) при-
родой вещества; б) агрегатным состоянием и в) вегйшними
условиями (температурой, давлением и др.).
3. Влияние па растворимость природы компонентов
К сожалению, до настоящего времени нет теории, с по-
мощью которой можно было бы предсказать и вычислить
растворимость. Обусловлено это уже упоминавшимися при-
чинами — сложностью взаимодействия в растворе н отсут-
ствием общей теории жидкого состояния. Исключениями
являются лишь идеальные растворы и растворы очень плохо
растворимых веществ *. Тем не менее многие Еэонросы, свя-
занные с растворимостью, можно объяснить, исходя из
общих закономерностей ее строения компонентов раствора.
Известно, что силы, действующие между ковалентными
молекулами, обычно не специфичны и для мало поляр вых
молекул невелики.
Если в ковалентную жидкость В в вест ее однотипные мо-
лекулы А, то энергия взаимодействия U частиц А и В не
будет существенно отличаться от энергии взаимодействия
между частицами А и А или частицами В и В. Поэтому
подобно тому как смешиваются лЕобые количества одного
и того же вещества, будут неограниченно растворяться
друг в друге также и различные ковалеЕгГНЫе жидкости.
По этой причине и молекулярные кристаллы обычно хорошо
• Казалось бы, для идеального расюора не может идти речь
о растворимости. Однако здесь имеются п сяду случаи, когда темпе-
ратура олыт<< нюкс точки кристалл и га ци и растворителя.
119
ЭБ ,:Научнов наследие России"
растворяются в ковалентных жидкостях. Например, раст-
воримость толуола а бензоле не ограничена, а кристал-
лический нафталин хорошо растворам а неполярных
жидкостях.
Из сказанного становится понятной справедливость ста-
ри н fi о го, найденного из многовекового опыта правила;
«подобное растворяется д подобном».
Наоборот, если энергия взаимодействия молекул Л н А
или В и В больше, чем А и В, то одинаковые молекулы
каждого компонента предпочтительно будут связываться
между собой и растворимость понизится. Это часто наблю-
дается при значительной полярности одного из компонен-
тов раствора. Этим, например, можно объяснить плохую
растворимость НС! в бензоле; небольшую растворимость Е1е-
л ал яр них и малаполярных веществ в полярном раствори-
теле, например в воде, Как известно, положительный заряд
в молекулах воды сосредоточен на атомах водорода, а отри-
цательный — я а орбиталях двух электронных пар атома
кислорода; это приводит к значительному разделению заря-
дов, что вызывает интенсивную ассоциацию воды (за счет
водородных связей). Поэтому притяжение неполярных Мо-
лекул неэлектролита к молекулам воды будет меньше, чем
притяжение молекул воды друг к другу.
Если взаимодействия между соответствующими части-
цами гаковы, что имеет место соотношение
^а-в > у (уа-л+
то оба компонента склонны к образованию друг с другом
сольватов или химических соединений разной степени
прочности, 0 этом случае наблюдается повышенная раст-
воримость вещества, а между растворителем к растворен-
ным веществом часто образуются водородные или до нор ио-
акцепторные связи. Примером большой растворимости с
образованием водородных связей является раствор этило-
вого спирта в воде, а с образованием донор но акцепторной
связи — раствор аммиака в воде. Растворимость спирта не
ограничена, а аммиак в воде растворяется в соотношении
приблизительно 700 : I (по объему).
Растворимость солей в воде в значительной степени за-
висит от их природы. Так, почти все соли щелочных ме-
таллов н аммония хорошо растворимы в воде [за исключе-
нием Li^POj., LijCOa, NaSb(OI ])$, K;sCo(XOj)g, KaP^CIj,
(NHtJiPtCle, (NHJ/Zo(NO«)«J. Хорошо растворимы нитраты,
150
ЭБ ,:Научнов наследие России"
галиды (кроме галидов серебра, ртути, свинца и таллия)
и сульфаты (кроме сульфатов щелочноземельных металлов
и свинца). Для переходных металлов характерна неболь-
!й£
шая растворимость их
сульфидов, фосфатов, кар-
бонатов н некоторых дру-
гих солей, а также их
ГИДрОКСПДОВн
Чем эндотермичЕгсе рас-
творение, тем меньше рас-
творимость. Так, в ряду
SO3—NaCI—NaHCOjt—PbL
растворимость в воле па-
да ет. Дл я с ход ны х вещее тв
это падение можно
выразить к ол и ч сс твен ио
(рис* 46), Некоторые пра-
вильности удается устано-
вить и в связи с изменением
1
Рнс. 46. В за иное в я зь между
тсллот&н растворения
(икзл/мОль) н растворимостью га-
зов С и CJTAOH при /= 25 °C:
J — Не. ? - N« J — Nl * — СО.
5 — Лг: - сот
степени окисления, а также с положением элементов в перио-
дической системе. Так t в ряду НСЮ—HClOj—HCIC^—HCIOj
растворимость растет. В соответствии с возрастагшем проч-
ности ‘ веществ в ряду
CaS04— SrSOi“BaSO.j их
Nt CjHjN
Рис. 17. Зависимость расТДО[>11м<№тЕ1
(\j) jd-jiJOKcuCpji^&fla (резорцина)
пр и 30 *С от дн электрической upo-
Нашаемости « растворителя
растворимость надает.
СвЗО4(Ю+л$-СааЧр) +
+ (ад-{Р)<
= —S.1 кал/мсль.
SrSOj fK)-|-l?i7 [= Sra ? (р) +
+ (SQi)s^(pX ля^-
= ОД кал/мол bh
BaSCIj (к)Д-й<7 = Ва111' (р)-J-
+ (soji-(p)1 дя;<к=
= G,3 кал/дюль.
Установлен и ряд зако-
номерностей, характера
зующнй влияние природы
растворителя на раствори-
мость. К их числу отно-
сится (дмгеячэдкв.' potrnaopjMfocim л>
npojcodwm через я ряду рйсп?аорнте-
лей, располо^секк^л: ™ сазрастпакчцему з/*йч^нию жж-
люлекулярного s ныл+ Максимум отвечает
I51
ЭБ ,:Научнов наследие России"
тому растворителю, молекулярное поле которого близко
к молекулярному полю растворенного вещества (рис. 47).
В ряде случаев создается впечатление несправедливости
правила Семенченко (когда удается реализовать только
ветвь кривой). Вместе с тем надо подчеркнуть, что это пра-
вило не является строгим. Так, при образовании концент-
рированных твердых растворов и при химическом взаимо-
действии между компонентами растворимость резко увели-
чивается — точки оказываются над кривой, отвечающей
правилу Семенченко.
Хотя накоплен огромный материал по растворимости
разнообразных веществ в различных растворителях, од-
нако, как мы уже отмечали, количественная теория раст-
воримости еще не разработана. На пути ее создания воз-
никают большие трудности, так как объектами служат
главным образом концентрированные растворы.
ft. Виннице на растворимость внешних условий
Растворимость характеризуется истинным равновесием.
Поэтому для определения влияния температуры и давления
Рис. 4Я. Закнсчмостъ раствори-
мости С (л/ЮО г 11/)) некото-
рых галев а воде при р= I атм
от температуры t (°C)
на растворимость можно вос-
пользоваться принципом Ле
Шателье: характер дей-
ствия Тир будет определя-
ться соответственно знаком
теплоты (энтропии) растворе-
ния в насыщенном растворе
ДЯрв„л (Д5ра„я) и изменением
объема А/рястр в этом рас-
творе, а степень их влияния
— их абсолютным значением.
Растворимость газов в
жидкостях. Растворение га-
зов почти всегда сопровож-
дается выделением теплоты
(сольватации их молекул).
Поэтому, согласно принципу
ЛеШателье, повышение тем-
пературы понижает раствори-
мость газов (рис. 48). Примером служит процесс образования
пузырьков газа при нагревании водопроводной или речной
воды. Однако известны случаи, когда нагревание вызывает
ЭЕ Н}53нсе наследие России"
рост растворимости газов (растворение благородных газов
в некоторых органических растворителях).
Так как при растворении газообразных веществ в жид-
кости Л^ра-твС 0, то давление способствует росту раство-
римости газов. Ога зависимость для малорастворимых ве-
ществ выражается законом Генри (1802): растворимость
газа при постоя»пой температуре пропорциональна его
давлению.
Рис. 49. Зависимость рас-
твОрн мости GOj С (л/лН^)
при 15 вС от давления
р (атм)
i — по икону Гснря: г —
экспериментальные данные
Рнс. 50. Зависимость раствори-
мости С (мае, %) азота в жил-
кой двуокиси серы от давления
р (кг/см1) при 25 (,С
При растворении смеси газов растворимость каждого из
них пропорциональна его парциальному давлению. Так,
например, вода при соприкосновении с воздухом раство-
ряет столько же кислорода, сколько она растворила бы его,
соприкасаясь с чистым кислородом, находящимся пол дав-
лением I /5 атм (парциальное давление кислорода в воз-
духе).
Закон Генри справедлив лишь для сравнительно раз-
бавленных растворов, при невысоких давлениях и отсутст-
вии химического взаимодействия между молекулами раст-
воряемого газа и растворителем, В области низких и уме-
ренных давлений растворимость газов всегда растет с по-
вышением давления (рнс. 49). При очень высоких давлениях
растворимость может достигнуть максимума (рнс. 50).
Обусловливается это тем, что при очень высоких давле-
ниях изменение объема жидкости в результате растворения
153
ЭБ ,:НаучноЕ наследие России"
Рнс. 51. Зависимость
взаимной растворимости
С (мае. %) анилина н
воды аг-температуры i (’С):
в ней газа становится соизмеримым с объемом растворен-
ного газа.
Для небольших давлений закон Генри можно формули-
ровать и так: объем растворенного газа не зависит twn дав-
ления (это непосредственно следует из закона Бойля —
Мариотта).
Растворимость жидкостей в жидкостях. Разделяются
или смешиваются две жидкости, зависит от соотношения
между величинами Д//ржС!В и ГЛ5„а„и. Для идеальных раст-
воров Д$Сие1и определяется только
концентрацией молекул. Поэтому
при смешении данных весовых ко-
личеств веществ с ростом их моле-
кулярной массы АЬ'смш будет
уменьшаться (при неизменном зна-
чении Д//С.еш = 0). Отсюда можно
было бы заключить, что, напри-
мер, Д5ритв полимерных веществ
в низкомолекулярной жидкости
будет зависеть только от энерге-
тического эффекта, т. е. процесс
растворения всегда окажется экзо-
термическим. Однако если учесть
мыслимые варианты размеров,
формы и степени гибкости поли-
мерных молекул, то станет понят-
ным, что этот вывод будет нахо-
диться в соответствии с эксперимен-
том лишь для некоторых высоконо-
л и мер ны х веществ с вес ьма жестки м и
молекулами. Если молекулы очень гибкие, то при переходе
в раствор они перестают мешать друг Другу. I [оявляется
множество новых комбинаций в их расположении, что мо-
жет привести и значительному увеличению энтропии. Энт-
ропийный эффект может оказаться столь большим, что роль
взаимодействия н тем самым величины и знака ока-
жутся несущественными.
При смешивании жидкостей, между молекулами кото-
рых проявляются различной силы взаимодействия, воз-
можны три случая: а) неограниченная растворимость;
б) ограниченная растворимость и в) полная нерастворимость.
Ограниченная растворимость жидкостей наблюдается,
например, при смешивании воды и анилина. На рис. 51
приведена их взаимная растворимость в зависимости от гем-
151
ЭБ "Нлучное наследие России"
точки (nd отиечакэт ссют-
лстствгнно концентрациям
насыщенного рнфриора ани-
лин в л ноле и 4и?ды л ани-
лине при t160 йС
пературы. Кривая разделяет области существования гомо-
генных и гетерогенный систем. Заштрихованная на диа-
грамме площадь — это область расслаивания жидкостей,
Так, 50%-пая смесь при t = 160 °C расслаивается лэ два
взаимно насыщенных
раствора (точки с и d).
Температура, соответст-
вующая точке /С, — кри-
тическая температура
растюрения, т. е. та
температура, начиная с
которой имеет место не-
ограниченная взаимная
смешиваемость обоих
компонентов. Рост вза-
имной растворимости с
температурой в данном
случае обусловлен эп-
дотермпчиостью процес-
Рис, S2. Зависимость раствор [[мости в
ваде С (мае. %) некоторых солей От
температуры I (“Q
са растворения.
Демонстрировать ограниченную растворимость можно
на следующем опыте. В делительной воронке взбалтывается
эфир с водой. После отстаивания отдельно сливают водный
и эфирный слон. При поднесений горящей лучинки водный
слой воспламеняется, что свидетельствует о наличии в нем
горючего эфира. В эфирном слое бесцветная сернокислая
медь синеет, так как за счет имеющейся в нем воды обра-
зуется кристаллогидрат синего цвета.
К практически нерастворимым жидкостям относится
смесь масла с водой.
На растворимость жидкостей в жидкостях давление
почти не влияет, поскольку при растворении отсутствуют
заметные объемные эффекты.
Растворимость твердых тел в жидкостях, П отличие от
газов ла растворимости твердых тел давление значительно
не сказывается (так как ДУра„ = 0), Его влияние стано-
вится ощутимым лишь при очень высоких давлениях. Так,
например, лишь при давлениях порядка десяти тысяч атмо-
сфер растворимость fin трата аммония в воде падает >
>0) почти вдвое.
Изменение растворимости с температурой определяется
знаком и величиной теплового эффекта растворения. Тем-
пературную зависимость растворимости твердых веществ
часто выражают графически, в виде кривых растворимости
tss
ЭБ 'Научное наследие России'1
(рис. 52). Растворимость нитрата рубидия и хлората калия
при нагревании от 0 до 100 увеличивается в несколько
раз. Подобные изменения растворимости в соответствии
с принципом Ле и1ательс характерны для веществ, процесс
растворения которых протекает с поглощением теплоты.
Для сульфата иттербия теплота гидратации преобладает
над теплотой разрушения кристаллической решетки; сто
растворение экзотермично, поэтому растворимость с ростом
температуры падает. Растворимость хлорида иттрия прак-
тически не зависит от температуры, что связано с почти
нулевым тепловым эффектом его растворения. Излом кри-
вой для сульфата натрия обусловлен разрушением кристал-
логидрата при 32,4 °C До этой температуры насыщенный
раствор находится в равновесии с осадком кристаллогидрата
NatSOv IOHjO, выше ее — с безводным Ма2ЭОг Резкое
изменение хода кривой обусловлено значительной эндотер-
мичностью процесса растворения кристаллогидрата и экзо-
терм ич нос тыо растворения безводной соли.
5. Влияние на растворимость посторонних веществ
В присутствии примесей растворимость данного веще-
ства обычно уменьшается. Так, растворимость газов часто
сильно уменьшается при внесении в воду солей или других
растворимых н ней веществ. Например, в I г воды при 20 °C
растворяется около 3 см* хлора, а и I г насыщенного раст-
вора NaCl растворяется всего 0,3 см1 хлора (поэтому хлор
хранят над водным раствором соли).
Подобно газам, растворимость многих жидкостей пони-
жается в присутствии солей. Растворимость фенола, напри-
мер, в чистой воде больше, чем в солевом растворе.
Понижение растворимости в присутствии солей назы-
вается высаливанием. Одной из се причин может быть соль-
ватация солен, ведущая к уменьшению числа свободных мо-
лекул растворителя, а с ним и к понижению растворяющей
способности жидкости.
6. Некоторые эакономервеетн
При изучении свойств растворов нередко прослежи-
ваются проявления периодического закона. Покажем это
на примере тепловых эффектов. Из таблиц, в которых соб-
раны значения Д/?.]и образования различных ионов, выте-
кает закономерный ход этих величин в ряду сходных частиц
ЗБ ' J^Hce наследие России"
ЭБ ,:НзуччоЕ наследие России1
Рнс. БХ Зависимость стандартной тс плети растворы! и я (АЯж/лЦмп (ккал/г-экв) ллоридон от порядко-
вого помора элемента Z (л[!О = щ!
(натфЕшер. F“—Cl"—Вт—Г). Рассматривая н одинаковых
условиях совокупность значения теплот растворения родст-
венных соединений» легко обнаружить периодичность в
Рнс. 64l Взаимосвязь между
стяЕгдартггыш! тё л логами раствсь
рения (Д//^н)]ьй±тв (кмд/моль)
хлоридов и бромидов щелочных
металлов
ее изменении. Даже если
учесть неполноту данных,
представленных па рис< 53,
и невысокую точность значе^
ннй тепл от растворения не-
которых хлоридов, все же
можно прийти к выводу о
су щес твова и и и о л ре делен ной
закономерности в ходе этих
кара ктер и стик р аствор ов.
В подобных закономерностях
содержатся и кол нчествен-
ные соотношения. Один из
мыс л нмы х пр и меров п ред ’
ставлен на рис. 54, на кото-
ром сопоставлены теплоты
растворения хлоридов и бро-
мидов щелочных металлов
(пн.о = оо).
Глава П
РАЗБАВЛЕННЫЕ РАСТВОРЫ НЕЭЛЕКТРОЛИТОВ
Если в результате смешивания различных веществ полу-
чается новая однородная система — раствор, то его свой-
ства отличаются от свойств каждого из компонентов а от-
дельности. Это измен с иле свойств связано, с одной стороны,
с характером взаимодействия между молекулами компонен-
тов и вновь образованными продуктами и, с другой стороны,
с уменьшением концентрации свободных молекул каждого
из веществ при распределении в нем другого вещества.
Влияние всех этих факторов усиливается с ростом кон цент-
рани и, а их количественный учет представляет очень труд-
ную за чач у. Поэтому заслуж и ва Ют в ни и a 11 и я к р а й не разба в-
ленные растворы,для образования которых Af=0.
В таком растворе частицы растворенного вещества нахо-
дятся на столь большом расстоянии друг от друга, что их
взаимное влияние можно исключить, а растворитель прак-
тически не меняет своих свойств; растворы приближаются
ЗБ ’^уччеа наследие России'1
к идеальным л изучение их упрощается; становятся и более
простыми уравнения, описывающие свойства таких раство-
ров.
Ниже рассмотрены четыре свойства разбавленных раст-
воров,
В уд ем считать, что из раствора при соответствующем из-
менении температуры будет выделяться только один из
компонентов (растворитель) и что растворенное вещество
tie является электролитом.
1, Давление пира
В результате естественного процесса псп а реп и Я лад
жидкостью образуется пар, давление которого можно из-
мерить с помощью манометра, предварительно удалив из
Рис. 55, Равновесие жид-
кость — пар (схема)
Рис. 56. Г Ее ренос раствори-
теля:
а — сосуд С р-НС ГИОРКТГЛСНг
5 —- сосуд е растБсрои
сосуда воздух (рис. 55). Эндотермический процесс испаре-
ния обратим: одновременно с ним протекает экзотермиче-
ский процесс конденсации. При определенных условиях
ус Tafia вливается равновесие (Л(7 = 0). РаВН овес нос состоя-
ние при данной температуре характеризуется давлением
насыщенного пара.
Представим себе, что в равновесную систему «жидкость —
пар» введено нелетучее вещество *. Его переход в паровую
* Нелетучим можно считать псщсстпо, у которого т&мпоратура
кнпення примерно па 150 °C отличается от температуры кипения раст-
ворителя.
159
ЭБ 'Научное наследие России"
фазу исключен; оно попадает только о жидкость. Но прев-
ращен ле растворителя в раствор будет означать нарушение
равновесия (/Vj-C Д'J). В соответствии с принципом Ле
I Пателы? возникает процесс, стремящийся ослабить эффект
воздействия, т. е. конденсация. Это означает падение дав-
ления пара. Таким образом, мы пришли к выводу, что
давление пара пйд раствором (рт) меньше, чем над чистым
раствор 11 тел см {pt).
Иллюстрацией сказанному может служить такой легко
осуществимый опыт (рнс. 56). Под стеклянный колпак ста’
вят стакан а с чистым растворителем и другой стакан б
с таким же количеством раствора. Спустя некоторое время
уровень жидкости в стакане а понижается, а уровень раст-
вора повышается, что обусловлено более низким давлением
лара над раствором. В данном случае происходит перенос
растворителя.
Теперь перейдем к количественной стороне явления.
Очевидно, понижение давления будет тем значительнее, чем
больше введено растворенного вещества, иными словами,
давление пара над раствором pj будет пропорционально кон-
центрации растворителя Л\:
Pi — Kfli-
Для раскрытия смысла коэффициента пропорционально-
сти АГ| в этом уравнении применим его для все белее н более
разбавленных растворов. В пределе, когда Л/, = I, полу-
чим ру = pl. Следовательно, это уравнение может быть
записано так:
/>1=А <TV ,4J
т. е. давление нашивного пара над растворам равно его
давлению над чисапм» растворителем, умнолсенному на
мольную долю растворителя >
Уравнению (IV.4) можно придать и несколько иной вид.
В соответствии с (IV. 1) = 1 — Л/,. Отсюда Л = р}Х
X (I — Л\) = р\ — p"tNs или р] — р = p\Ns. Разделив по-
следнее равенство на pj, получим
Следовательно, относительное понижение давления м«еы-
щенноеи чара растворители над раствором равно малиной
доле рмтворенкиео вещества в растворе,
ию
ЭБ ,:НаучноЕ наследие России"
Мы приведи здесь два варианта формулировки первого
закона Рауля (188G). В случае идеального раствора он спра-
ведлив для всех веществ
2, Температуры ни пенил и отпер девали л
В прямой зависимости от давления насыщенного пара
раствора нелетучего вещества находится температура ки-
пения раствора.
Температурой кипения жидкости Является температура,
при которой давление ее паров становится равным внеш-
нему давлению (при р = I атм 100 °C дня воды, 80 °C для
бензола и т. д ). Следует обратить внимай не на важное
отличие раствора от чистого вещества. Температура кипе-
ния раствора отвечает его равновесию с первым пузырьком
пара (начало кипения). Действительно, в силу нелету-
чести растворенного вещества утрата раствором даже ни
чтожно малого количества испарившейся жидкости при-
водит к увеличению концентрации раствора. Она будет
отличаться от первоначальной и поэтому свойства раствора
и, в частности, его температура кипения станут иными.
Вернемся к рис. 55. Понижение давления пара в ре-
зультате растворения означает необходимость повышения
температуры раствора для восстановления нарушенного
равновесия. Тогда давление насыщенного лара будет до-
ведено до первоначального (панример, атмосферного). Таким
образом, температура кипения раствора выше температуры
кипения растворител я.
С понижением давления пара над раствором связано по-
нижение температуры отвердевания последнего по сравне-
нию с чистым растворителем (температура отвердевания —
это температура, при которой
давление пара над жидкостью
становится равным давлению па-
ра над твердой фазой). В связи с
этим рассмотрим теперь систему
при более низкой температуре,
когда в равновесии находятся
твердая и жидкая фазы (например,
лед и вода или кристаллический и
жидкий бензол) (рис. 57). Если в
такую систему внести растворимое
Рис, 57. Равновесие кри
сталлы — жиДкОсть(|;Хема)
вещество, не переходящее в твердую
фазу (например, сахар в первом случае и нафталин — ио
втором), то равновесие нарушится. Это нарушение связано
& MX Клр-опстьяпц
ЭБ гНаучнов наследие России"
с конденсацией избытка парообразной фазы, протекающей
с выделением тепла. Чтобы восстановить равновесное со-
стояние, необходимо а соответствии с принципом Ле IIla-
тел не понизить температуру. Следовательно, температура
отвердевания раствора ниже температуры отвердевания
ч истого растворителя.
Здесь также надо обратить внимание иа важное в данном
случае отличие раствора от чистою растворителя Темпе-
ратура отвердевания раствора отвечает выделению из него
первого кристаллика твердой фазы (начало отвердева-
ния), так как выпадение из нею даже ничтожно малого
количества растворителя приведет к изменению (увеличе-
нию) концентрации раствора
Так как для разбавленных растворов понижение давле-
ния пара пропорционально концентрации, то и повышение
температуры кипения, и понижение температуры отверде-
вания разбавленных рас-
творов пропорционально
их концентрации. В дан-
ном случае (в отличие от
Закона Рауля) концент-
рацию принято выра-
жать моляльностью. Та-
ким образом,
Лип “
G
цац
Т
Рис. 58. Зависимость анергии Гиббса G.
От температуры Т над чистым раство-
рителем в различных его агрегатных
состояниях п вад раствором (схема),
кривые rfc. ею л ай отлечают чнетоыу
растворителю соответственна в кристалл и
ческой, жидком и газообразном состоя ни я х.
рфыная Jib отвечает раствору» АТ^ — ло
и и жен не тем нерв тури отвердела и и я ч
— повышение тем л ара туры ннле-
пня раствора (по сравнению с чистые
растворителем}
физический смысл константы
не представляется возможным. Действительно,
математически Е = ДГИНЙ и К = при m = 1, однако
концентрация in = 1 столь далека от большого разбавле-
ния (342 г тростникового сахара на i л воды!), что уравне-
ния (IV.6) и (IV.7) становятся несправедливыми. Чтобы
(IV6)
И
(IV.7)
пропор-
называ-
ЛТ’вщ t= Km
Коэффициенты
циональностн
ются соответственно
вбуллиоскопичес-коИ Е
(лат. ebbul io~ выкипаю)
и криоскопической К
(греч «крнос»— холод)
константами.
Воспользоваться тем
же приемом, с помощью
которого мы выяснили
уравнения (IV.4), здесь
хотя
ЭЕ ' 4^1 нов наследив России"
найти численные значения Е или /С, надо результаты изме^
рения для очень разбавленных растворов пересчи-
тать на I моль растворе Fin ого вещества. Таким образом^
Е н являются экстраполяционными по-
стоя иными,
Обо они ле зависят от природы растворенного
вещества, а характеризуют лишь растворитель
Их значения для некоторых растворителей приведены
в табл. 14.
Т а бл и ца 14
Эйулл^освдпичгскнс £ к криоскопические £ коесстзнгы некоторых растворителей (град/киль)
Рлс1Вц1:нТел1> £ Л
Вода 0,53 6 1,86
Бен эод 2^7 5,] 2
Уксусная кислота 3J 3,9
1 ItfTpufciLKi.l 5,27 6^9
Поскольку ДТК|ГИ и Л7^тв изменяются пропорционально
числу грамм-молекул, а каждая грамм-молекула содержит
одинаковое число молекул (6,024 IO3*), то эти характер и С’
тики раствора зависят только от числа частиц растворен-
ного вещества. Отсюда опорой зпкйк Родля: лоэышешде
рштгмярся я/юло/?цдоядл&«« чнеш/ чдтиц /юсядео/мкмоео
одцдопвд w зпйпедт от гао
Связь между повышением температуры кипения и по-
НИ жен нем температуры отвердевания можно показать с по-
мощью рис. 58. На нем представлены четыре кривые,
изображающие температурную зависимость энергии Гибб-
са четырех фаз — твердого, жидкого и газообразного
растворителя и раствора- Точка й отвечает кипению, тоЧ’
* Можно локаут, что
Ё Д^и)! н
1 ото j ооо
.VI j лИ i
где /? — универсальная газовая постоянная. Таким odpaiQMj в эти
уравнения входят только характеристики растворителя!
6* 163
ЭБ ,:Научнов наследие России"
к а о — отвердеванию растворителя; точки hid — соответ-
ственно кнпению и отвердеванию раствора. В каждой из
этих точек в соответствии с общими критериями равнове-
сия соблюдается равенство энергий Гиббса сосуществующих
фаз.
3. Осмотическое давление
Представим себе, что в сосуд л /рис. 59) с чистым раство-
рителем опущен цилиндр б, нижняя половина которого
изготовлена из материала, свободно пропускающего моле-
Рнс. 59. Схема Опыта,
пояс някпце! о осмотичес-
кое давление:
и — сосуд с р««эоркгея*и:
б — сосуд с распаром;
а — поршень; г — ppyj
перехода растворителя
кули растворителя, ио непроходи-
мого для частиц растворенного ве-
щества (пол у п р oi 11 [ [ [эсм а я перего-
родка) *. В цилиндре свободно
может передвигаться запирающий
раствор поршень а. Это неравно-
весная система, так как если в рас-
творителе A?j = lf то в растворе
Л\< L Поэтому в системе нач-
нете я самопроизвмьный проiiecc
выравнивания концентраций (он
обусло вл сн то к же причиной, что
и процесс растворен нм; см. с. 146
и ел). При этом молекулы рас-
творителя будут переходить в ци-
линдр с раствором (обратный пере-
ход растворенного вещества по усло-
вию опыта исключен). Это явление
напоминает перенос растворители
в опыте, описанном нас. 160„ где
рол ь с воеобраз ной пол ул р он и цае-
мон перегородки играла газовая
фаза. Процесс самопрoi[звольного
в раствор через полупроницаемую
перегородку называется
В результате осмоса объем жидкости в цилиндре уве-
личится и поршень поднимется. Чтобы предотвратить
* В качестве гкхлу проницаемых перегородок дли нудных растось
рол применяются пленки рэститьтьного я ли животного происхожде-
ния, а также полученные искусственным путем (пергамент, целлофан).
Для Е1зготовлепия прочных сосудов с полупроницаемыми стеихдои
примени ют керамические изделия, поры которых заполняются желези-
стое и и? роди стой медью Сщ [Fe (CN)J.
164
ЭБ 'Научнов наследие России"
возрастание объема с разбавлением раствора и остановить
осмос, необходимо извне создать давление на раствор.
Для этого на поршень устанавливаются грузи ни г. Внешнее
давление вызывает обратный процесс — выход раствори-
теля из раствора. При определенной нагрузке наступит
динамическое равновесно между скоростями ВХОДЯЩИХ
в цилиндр и выходящих из пего частиц растворителя.
Поршень при этом займет первоначальное положение.
Давлен tie, которое нужно приложить к раствору, чтобы
осмос прекратился, называется осмотическим давлением *.
Изучение осмотического давления для разбавленных
растворов показало, что оно не зависит от природы компо-
нентов и возрастает пропорционально концентрации раство-
ренного вещества и абсолютной температуре, причем ко-
эффициент пропорциональности оказался универсальной
константой, численно равной газовой постоянной R, Та-
ким образом,
Рогя=£ЛГ. (IV.8)
Выражая концентрацию числом молей растворенного
вещества в единице объема, вместо (IV. 8) получим
P^V-^RT. (IV.9)
Последнее по форме совладает с уравнением состояния
идеального газа, что позволило голландскому фнзико-
химику Венгт-Гоффу (1887), обратившему на это внимание,
прийти к выводу, что осмотическое давление равно тому
давлению, которое оказывало бы растворенное вещество,
если бы оно, находясь в газообразном состоянии при той же
температуре, занимало тот же объем, который занимает
раствор (принцип Вант-Гоффа).
Необходимо вновь подчеркнуть, что аналогия (установ-
ленная Вант-Гоффом) между газообразным и растворенным
состоянием вещества является формальной, так как меха-
низм газового и осмотического давления совершенно раз-
личен. Ведь движение молекул в газах хаотическое, в то
* Выло бы не п ранил ьным в ан и св ином опыте снять п рати вала о-
ленне (пызгзя, что в результате пОЛьема уровня раствора сйадэетгя
столб жидкости, вес которого и будет определять осмотическое дав-
ление). Bc.If. переход растворителя в цилиндр понизит коицеЕпрации?
раствора, т е. нес столба будет соответствовать осмотическому дав-
лению не первоначально взятого растворе.
165
ЗБ ,:Н-аучнов наследие России"
□ремя хак в растворах, подобно жидкостям, существует
ближний порядок, и их структура при низких темпера-
турах ближе к строению жидких и твердых тел, чем к
«бесструктурному* газовому состоянию.
h. Определение молекулярной массы
растворен него вещества
Уравнения, приведенные в трех предыдущих парагра-
фах, позволяют рекомендовать четыре метода определения
молекулярной массы.
Придав уравнению (IV,5) вид
(IV. 10)
можно определить молекулярную массу растворенного ве-
щества ЛПТ зная его навеску git навеску растворителя
и молекулярную массу растворителя ЛГ j и измерив при
данной температуре рт и pj (последнюю величину можно
заимствовать из справочника).
Введя согласно определению в (IV .6) и (IV.7) величину
т~Мл1 1000 *
после преобразования получим
_.£^1000
A'J’kh 1Г.Й\
. Х&1000
ДТ отв^1
(IV.II)
(IV. 12)
Эти уравнения позволяют найти М2 подстановкой значений
соответствующих величин.
Заменив в (IV.9) величину ла отношением g2/Mit после
преобразования находим уравнение
М
&RT
Росм^
(IV. 13)
позволяющее определить Л12 по навеске glt зная темпера-
туру и объем раствора и измерив его осмотическое давление,
366
ЭБ ,:НаучноЕ наследие России"
5Г Некоторые выводы
Естественно» что существует связь не только между
ЛТ'кил и АЛни {рнс- 58), но ir связь между последними и
Др (рис. 60). Кривая кипения (конденсации) СМ. вдоль ко-
торой сосуществуют кипящая жидкость и сухой насыщен-
ный пар н в каждой точке которой <J“ = бг выражает
взаимосвязь между температурой кипения и давлением
(пара) над чистым жидким растворителем. Кривая субли-
мации £Ю, вдоль которой сосуществуют сублимирующийся
Ряс. 60. Взаимосвязь ггежду температурой
Т и Да ваенмСМ р при рай новее кН различных
фаз (схема)г
□Я — pSBIIOBeCla* SfctiflKCKTb — PSp (D КйжДфС|
тэт ке ** CrJ: ОД — pfieHOPtctat KpiFtrfirtrt —
ЫФ {G* • Gr)S OG — p±i я иvprCHt к^истМл —
ЖНДКМТЬ (бК " б* К (fb — pJlEli&»rCHC pa Ст*
вор — IFDP (C|S *= 0r)i C> — ДОИ1РОЕГС-НЧ KpWtTJiffdl
— жидкость — Jisp (G* " C* “ Gr)C d — рав-
новесие КрнСПЛЛ — FflCTMip — FIJI p (fiK «= C'p =
= Grk Л7ОТВ,— Лиикжгяне TcifncpBTVPtf отлерде-
ьакня; drKi|[f — повышенно тс^йг pa ту p ы кипе-
ния; Лр — Пфии*С|1НГ JWBrtrilHrt [|АрЙ PSCTFOPS
(по сравнению с -чистии растворителей!
твердый растворитель и сухой насыщенный лар (G“ = Gr),
выражает взаимосвязь между температурой сублимации
и давлением пара над чистым твердым растворителем. Эти
кривые смыкаются В тройной точке О, где в равновесии
находятся все три фазы — твердая, жидкая и парообраз-
ная (Gr = G* = Gr)- Из точки О вверх (почти вертикально)
идет кривая плавления ОС, т. е. сосуществования твердой
и жидкий фаз (6К = G“). Она выражает взаимосвязь между
167
ЭБ ,:НаучноЕ наследие России"
температурой и давлением при плавлении. Кривая Bd от-
носи тс я к р аст вор у; вдел ы гее cot у ществ у ют ки п я щи й раствор
и сухой насыщенный лар. Она выражает температурную
зависимость давления пара над раствором (G1’ = G'). Эта
кривая в соответствии с первым законом Рауля (с. 160)
располагается ниже кривой 0/1. Внизу кривая dB закан-
чивается в новой тройной точке d (GK = G1' = G') сосущест-
вования раствора с твердым и парообразным растворите-
лем.
Если данному (например, атмосферному) давлению от-
вечает горизонталь mn, то абсцисса точки а отвечает темпе-
ратуре кипения растворителя, а абсцисса точки b—тем-
пературе кипения раствора, т. е. АТК1|П = I ab |. Опустив
из точки а перпендикуляр на кривую </В, получим точ-
ку Ь'; ее ордината равна давлению пара над растворим,
т. е, понижение давления пара над раствором Ар = | а!)' |.
Наконец, расстояние между абсциссами точек О и d равно
понижению температуры отвердевания ДГв|В. Из рис. 60
видно, что чем концентрированнее раствор, т. е. чем ниже
будет пунктирная кривая, тем больше будут значения Д/7,
ДГ,„П и ДГМ(.
Графика, иллюстрирующего одновременно все четыре
рассмотренных свойства, нс может бытьн так как последнее
(осмотическое давление) в отличие от остальных трех от-
носится к неравновесной системе.
Изумшнгё с(юйст0 разйавлемных рлстйирос представляет не только
теоретический, ito я практический жстерёс. Доснточно напомнить
о возможности определен и я молекулярной мдссы и о том. что нередко
приходится иметь дела с растворами плохо растворимых веществ,
которые сама природа сделала гсчеЕ[ъ разбавлеггиымн. Кроме того,
котя уравнен к я (lv.4)> (IV. 6), (IV.7) и (IV. 9), отражающие предел ь-
ные за коим ( как к закон Генри, относятся к разведенным (строго го-
воря, к бесконечно разведенным) растеорзм, однако с некотором по-
грешностью ими можно пользовать я н при умерешеом рялбавчтенни.
Для концентрирован них же растворов, где пни совершенно непри-
менимы. ими можно воспользоваться для грубой ориентировки, что
тоже небесполезно.
Рассмотрим несколько примеров, иллюстрирующих последнее
соображение.
Урльиенне (IV.41 гюзвюллет понять, почему гигроскопичные ве-
щества (например, .MgCy па воздухе ряепд^ются. а некоторые,
наоборот, выветриваются.
На ос нова неги уравнения ([ V.6) легко ориентироваться в выборе
нешеств для получения бани с возможно большей температурой кипе-
ния, Ладо Подобрать мщсствОь максимально растворимое в раствори-
теле, который, в свою очередь, гуттал бы двум требованиям обладал
бы наибольшей температурой кнленни и нацменыпей. удельной теплей
тай парообразования (см. с. 1бЦ)г
168
ЭБ ,:Научнов наследие Рогсии"
Ур-апнепнс {IV.9) делает попятным все ecue существующий раз-
нобой в значениях точек плавления некоторых веществ1 ничтожная
примесь сильно уменьшает температуру плавления веществ (особенно
тугоплавких) Оно также позволяет осмыслить следующее обстоя-
телаччио вопреки пому, что температура окружающей среды ниже тем-
пературы отвердевания «ччиестпя, оно нередко остается жидким. Этот
парадоксальный факт объясняется гигроскопичностью- Например,
уксусная кислота 16,7 42) и глицерин (tnjl = 19 °C) не отверде-
вают лаже при сравнительно низкой температуре'в помещении. На сни-
жении температур отвердевания раствора по сравнению с чистым раст-
верителем основа ио действие соли, которой посыпают лед и снег {при
небольшом мерою) для их тляннп.
Очевидно, с уравЕ1ением (IV. 7) можно связать и получение лег-
коплавких сплавов, и действие флюсов.
Явление Осмоса играет важную роль в жизнедеятельности живот-
ных и растений, Оболочки клеток представляют собой перепонки.
легко лроииЕЕдсмыс для воды, по почти нои ран н наем ые для веществ,
растворенных л клеточном соке. Поэтому пресноьодиые рыбы нс мо-
гут j-кссть в соленой воде (где рпги = Й8 атм). а морские рыбы — в прес-
пой. Этим же обтреплется и то, что когда мы ныряем в реке, открыть
глаза больно, н то время как н море, где концентрация солей выше
и приближается к концентрации солей □ клетках роговицы, зга боль
ощущается гораздо слабее Физиологический раствор (0,9 % -лый
водный раствор NaCI} Еса человека и тесмокровных животных ока-
Аыяаст благотворЕЕое действие, так как его осмотическое Давление
7 атм) близки к осмотическому давлению плазмы крови.
Для очистки вещегти весьма перспективным является так назы-
ваемый обратный осмо? — разделение раствора за счет приложении
к нему давления, превышающего ое.мопгчсскос. Расчеты показывают,
что лтог метод значительно
дешенчТе традиционных спс-
собаи разделения (например,
ныыоражн в а пнем, дистнлл я
цисн н др.). Его примене-
ние ос обе него целесообразно
для смесей сходных всшеств
(например, изомеров).
Вновь подчеркнем,
что все рассмотренные
уравнения справедливы
лишь для разбавленных
растворов. Поэтому, на-
пример, прямые, отве-
чающие уравнению
(IV.7), в общем случае
будут распространяться
на очень небольшую
концентрационную зону
(рис. 61). Она может
Рнс. 61. ПФннткекие температуры ot-
вердевання одного вещества » резуль-
тате растворения в нем другого ве>
щества (схема}:
/ — u ps<TTitbpeiro в Л; 2 — А рл с творен*
я В Л В принят тя 100 %); верти-
кали Д к В отвечу юг чисты к компонента kJ
□казаться м значительной. Но за-
тем эти линии искривляются.
169
ЭБ ,:НаучноЕ наследие России1
Глена ITT
РАСТВОРЫ ЭЛЕКТРОЛИТОВ
Описанные в гл. И закономерности и, в частности, урав-
нения ([V.4), (IV.6), (IV.7) л (IV.9) справедлины лишь для
очень разбавлен!,их растворов нелетучих веществ при
условии, что размеры их частиц в чистом состоянии и в
растворе одинаковы (неэлектролиты).
Сохранив первые два условия, рассмотрим теперь
растворы электролитов,
Электропроводность металлов в твердом и в расплав-
ленном состоянии обусловливается тем, что между их час*
типами осуществляется металлическая связь, за счет сво-
бодно перемещающихся электронов электронного газа (про-
водники первого рода).
Почти все чистые жидкости, газы и большинство твер-
дых неметаллических геЛ электрический ТОк тэе проводят
(непроводники). Но в растворенном или расплавленном
состоянии многие неметаллические вещества тоже проводят
электрический ток. Их проводимость существенно отли-
чается от проводимости металлических проводников: про-
хождение тока через растпоры и расплавы сопровождается
разложением вещества — электролизом. Вещества, раство-
ры и расплавы которых проводят электрический ток при
одновременном протекании химического процесса, назы-
ваются электролитами (проводники второго рода).
1. Электропроводность
Электропроводность растворов электролитов значитель-
но ниже электропроводности металлов и зависит от кон-
центр эпи и раствора. Опытным путем обычно определяют
не электропроводность, а сопротивление прохождению тока.
Электропроводность раствора — величина, обратная сопро-
тивлению:
<?=— Ом"1,
г
Электропроводность столбика раствора между двумя
отстоящими друг от друга на J см электродами, каждый
из которых имеет площадь I см4, называется удельной элек-
тропроводностью х. С разбавлением раствора она умень-
шается, так как уменьшается относительное количество
электролита. Электропроводность раствора, содержащего
1 г-экв растворенного вещества, весь объем которого за-
I70
ЭБ 'Научнов наследие России"
ключей между двумя параллельно расположенными злею
тродами, отстоящими друг от друга на расстоянии 1см, на-
зывается эквивалентной электропроводностью)*. Очевидно,
1-ХУ, (IV. 14)
где V — объем (см8), занимаемый грамм-эквивалентом рас-
творенного вещества.
В отличие от удельной эквивалентная электропровод-
ность растет с увеличением разбавления, стремясь к неко-
торому пределу (рис. 62).
Таковы факты. Прежде чем обратиться к их толкованию
и количественным выводам, рассмотрим основные положе-
ния теории электролитической диссоциации.
2. Оеновы теории электролитической диссоциации
Еще в начале прошлого века, излагая свою теорию элек
трота, проф. Юрьевского (ныне Тартуского) уни вереи
тета Ф. И. Гротгус (1805) выска-
зал мнение, что в растворе под
действием тока растворенное
вещество распадается на про-
тивоположно заряжен ные части-
цы, которые нейтрализуются на
электродах- Позже Фарадей
(1833) назвал подобные заря-
женные частицы нонами (от
греч. «ион» — идущий), В 1878 г.
петербургский проф. Р, Э. Ленц,
исследуя электропроводность
раствора, высказал предполо-
жение, что молекулы веществ
уже при растворении могут рас-
падаться па ионы. Все эти и
подобные высказывания оста-
Рис. 62. Зависимость экви-
взле 11тной электрон роводн о-
сти X (сы-/Ом) ат разйннле-
иил Р (л/г-эШ для ВОДНЫХ
растаскав некоторых з-.тек-
тралнтов прл /= 25 °C
вались предположениями И тог-
да, когда шведский ученый
Аррениус (1887) занимался не-
ся едова ни я м и электропроводпо-
сти растворов. Закономерности
в изменениях эквивалентной
электропроводности с кон цент-
рациси указывали на то, что в растворе молекулы элект-
ролита, очевидно, распадаются на ионы. Из хода кривых
L7I
ЭБ 'Научное наследие России"
на рнс, 6S вытекает, что сначала распадается часть элек-
тролита, а по мере разбавления раствора диссоциация
увеличивается, что н ведет к росту L
К этому времени В ант* Гофф (1887) установил, что в
растворах солей, кислот и оснований изменение осмоти-
ческого давления превышает вычисленное по уравнению
(IV.9). Подобные отклонения измеренных величин от вы-
численных по соответствующим уравнениям наблюдаются
в сторону повышения для температуры кипе fin я it в стороны
понижения для температуры отвердевания этих растворов.
Так, например, молекулярная масса газообразного NaCl
равна 58,5, а на основании криоскопических измерений
(с. 166) она оказалась равной примерно 30.
Не зная, чем можно объяснить эти отклонения, по стре-
мясь сделать соответствующие уравнения пригодными
для этих растворов, Вант-Гофф ввел в них поправочный
множитель /, названный изотоническим коэффициентом *.
Подстановка коэффициента Вант-Гоффа г в уравнение ос-
мотического давления и в уравнения закона Рауля сделали
их пригодными для разбавленных растворов всех веществ,
в том числе и для растворов солей, кислот и оснований.
Коэффициент i вводится в соответствующие уравнения
как сомножитель. Для растворов электролитов вместо
(IV.4), (IV,6), (IV.7) и (IV,9) получим соответственно
Лр— jC, КИЛ = if (IV.I5) (IV. 16) (IV.I7) (IV.J8)
где i >1+ Таким образов
(IV-] 9)
/ = А Г |( ИII /Л 7 ц и П h (IV.20)
f =^= ДГоти/АТ отв. (IV.21)
i = £Vi-- н/ргр- цн (IV. 22}
где 4/?', ДТ'хнп, ЛТртъ и — измеренные величины пони-
жения давления пара, повышения температуры кипения,
понижения температуры отвердевания и осмотического
Давления, а Др, и рог„— те же величины, вы-
• Растворы с одинаковым <кмощческчм давлением нааыиаюгс.ч
иютоиическиыи,
ЭЕ Научнее наследие России"
численные соответственно по уравнениям (IVЛ), (IV-6),
(1V.7) и (IV.9),
Аррениус нашел, что соли, кислоты, основания и дру-
гие вещества, для которых i > I, являются электролитами,
Он установил, что подобно изменению эквивалентной элек-
трон роводности для растворов этих веществ коэффициент I
растет с разбавлением (для неэлектролитов i = I).
Эти отклонения нельзя было объяснить ничем иным,
кроме как увеличением числа растворенных частиц, т, е,
диссоциацией молекул электролита в растворе на более
мелкие частицы.
Установив, что величины коэффициентов I, полученные
измерением понижения точки отвердевания, совпадают
с подсчитанными им самим па основании его данных из
электропроводности, т. е. что растворы электролитов не*
дут себя аналогично и при пропускании электрического
тока, и в его отсутствие, Аррениус окончательно убедился,
что диссоциация молекул растворенных электролитов на
ионы происходит уже при самом растворении независимо
от того, пропускают через раствор электрический ток или
нет. Такой распад электролитов в а ионы под действием
растворителя называется электролитической диссоциа-
цией , Благодаря этому процессу в растворе увеличивается
число частиц, что н ведет к повышению осмотического дав-
лен ня и к значению i > ]. Отсюда на рис. 58 и 60 кривые
растворов при одной и той же концентрации расположатся
для электролитов ниже, чем для неэлектролитов.
Тщательное изучение коэффициента i показало, что
лишь в некоторых случаях он имеет близкое к целому числу
значение. Так, например, если в разбавленном растворе
КС] величина i 1,85, а для SrClt i 2,5, то для уксус*
ной кислоты CHjCOOH 1,05. Из этого можно заклю-
чить, что не все электролиты полностью диссоциируют
в растворе на ионы.
О неполной диссоциации на ионы многих электролитов
в растворе говорит также и возрастание эквивалентной
электропроводности с разбавлением. При повышении кон-
центрации, наоборот, эквивалентная электропроводность
уменьшается. Поэтому процесс электролитической диссо-
циации можно считать обратимым.
В растворе СНаСООН часть молекул остается н ед ассо-
циирован ной на ионы. Для количественной характерис-
тики соотношения диссоциированных и недассоцииро-
ванных молекул электролита при данных условиях пиль-
J73
ЭБ ,:Научнов наследие России"
зуютея понятием степени диссоциации. Степень электроли-
тической диссоциации а равна отношению числа молекул,
распавшихся на ионы, к обш/ему числу растворенных молекул.
(Подобной величиной мы пользовались и ранее; см. с. S1.)
Можно предположить, что эквивалентная электропро-
водность растворов электролитов пропорциональна сте-
пени их электролитической диссоциации а в растворах:
(IV.23)
Из рис, 62 следует, что при очень большом разбавлении
электролиты полностью распадаются на ионы, т. е. в этих
условиях я = 1 (см. также с. 185). Таким образом, восполь-
зовавшись приемом* аналогичным приему нахождения кон-
станты уравнения (IV,4), приходим к выводу, что
где а» — эквивалентная электропроводность при очень
большом (бесконечном) разбавлении. Подставляя это зна-
чение К. в (IV.23), получаем
А и
откуда
«=АД₽. (IV. 24)
т. е. степень электролитической диссоциации равна отно-
шению эквивалентной электропроводности при данном
разбавлении к эквивалентной электропроводности при бес-
конечном разбавлении, Удобно определять а также с по-
мощью коэффициента I, которым сравнительно легко на-
ходится эбуллио- или криоскопическим методом.
Рассмотрим раствор, содержащий, например* I моль
электролита, т. е. Л’в молекул. Если каждая из них распа-
дается на т ионов, то при степени диссоциации а в растворе
образуется Лг4*та ионов. Число недиссоцпированных моле-
кул составит Vo— JV^cc = Na (1 — а). Общее число частиц
(молекул и ионов) пекле растворения будет равно N^na 4-
+ No (1 — а). Отношение этого числа к числу растворен-
ных молекул и даст изотонический коэффициент:
_ Урта + УоО-а) !)«+!] _, _ ,, , ,
Л'ч Но ( J
откуда
(«ад
ЭБ Рз^ччсе наследие России"
В водном растворе к сильным электролитам относятся
почти все соли, многие неорганические кислоты {HNO3,
H4SO4, HCIO4, НС1, НВт, Ш и др.) и гидроксиды щелочных
и щелочноземельных металлов. Электролитами средней
Силы являются водные растворы некоторых органических
и неорганических кислот (щавелевой муравьиной
ИСООН, сернистой HjSO#, фосфорной НЛРО4 и др.),К сла-
бым электролитам принадлежат такие кислоты, как раст-
поры HjS, HCN, H2SiOa, HSBO3, гидроксиды многих d-эле*
ментов [Си (ОН)2, Ст (ОН )8 и др.], а также водные растворы
некоторых солей [HgCI*, Mg(CN)tt CdClt, Fe(SCN)s и др. I.
Все вышеизложенное и составило основу теории элек-
тролитической диссоциации Аррениуса. В ней не учиты-
вается взаимодействие между коками я растворе и между
ионами растворенного вещества и растворителем. Поэтому
в том виде, в котором излагал ее Аррениус, эта теория
охватывала лить крайне разбавленные растворы слабых
электролитов
3. Образование растворов
Сказанное на с. 141 и сл. относилось к растворам вообще,
а поэтому в частности и к растворам электролитов. Теперь
мы остановимся на последних подробнее, по-прежнему
имея в виду главным образом водные растворы (о неводных
растворах см. гл. V).
Ос обе ез и ость растворов электролитов обусловлена, во-
первых, тем, что d них находятся заряженные частицы,
во-вторых, что это частицы имеют заряд разного знака.
Основная составляющая взаимодействий в таких раство-
рах — это взаимодействие между ионами и молекулами
растворителя. Ион оказывает значительное поляризующее
влияние — индуцируемый им дипольный момент в моле*
кулак растворителя соизмерим с дипольным моментом мо-
лекул даже такого полярного растворителя, каким яв-
ляется вода (поэтому введен не в воду первых порций элек-
тролита вызыва ет особенна бол ьшое возму[ пд ющее де йствие).
Однако было бы неправильным считать, что все сво-
дится к взаимодействию заряженных частиц со средой,
к действию электростатических сил. Так, может происхо-
дить частичный или полный перенос электронов от ионов
к молекулам растворителя, приводящий к перераспределе-
нию заряда между ионом и его сольватной оболочкой. При
больших концентрациях растворенного вещества, а для
17S
ЭБ ,:Научнов наследие России"
растворителя с низкой диэлектрической проницаемостью
и при сравнительно небольших его концентрациях в ре-
зультате усиления плияНИ я заряженных частиц друг на
друга могут образоваться ионные пары и более сложные
группировки, содержащие как ионы, так и молекулы.
Характер в интенсивность взаимодействия между всеми
этими частицами определяются природой компонентов.
Так, в водных растворах она различна для ИОНОВ Н+, ионов
с в-оболочкой, с незавершенной d-оболочкой и т. д.
Ион в растворе резко отличается по свойствам от нейт-
рального атома этого же элемента. Объясняется это тем,
что химические свойства атома обусловливаются способ-
ностью нейтральной частицы отдавать или принимать элек-
троны с образованием химической связи. Например, атом
калия вступает в химическую реакцию с водой, отдавая ей
свой электрон. Ион калия валентного электрона не имеет
и поэтому с водой химически не взаимодействует, Атом
хлора способен при и it мать электрон, в то время как ион
хлора этой способностью не обладает. Поэтому связь иона
с молекулами растворителя может осуществляться разно-
образными силами, начиная со слабых дисперсионных,
водородных, электростатических и кончая донор но-акцеп-
торными. Чисто ковалентная и ионная связи обычно не
реализуются. Поэтому-то растворы нельзя назвать истин-
ными химическими соединениями. Однако прочность связи
между компонентами раствора иногда достигает больших
величин. Ионы всегда сольватированы. Так, и воде нет
ионов Си4*, а существуют комплексные ионы [CiHfLO^F*.
Об этом можно Судить по тому, что безводная соль CuSO4
бесцветна, в то время как ее раствор окрашен в голубой
цвет. Аналогично образуются гидратированные ионы
и др. Л1ногне из них довольно прочны и при
испарении воды выделяются в виде кристаллогидратов,
например MgCL-6HsO и AICln-6HjO.
Особый характер носит гидратация попа водорода. Сна-
чала протон Н* вступает в химическую реакцию с одной
1 олеку л ой воды, образуя ион гидр оксо Е1и я:
Н+(г}+Н,О{ж)=НгОЧр).
затем последний (в котором все протоны равноценны) может
гидратироваться с некоторым числом молекул волы:
HjO' (p)+rtHiO (ж) - НаО* лН,0 (р)
ЗБ Т-)?$чсе наследие России"
наиболее устойчивой является конфигурация H»Ot(HjO*x
x3Ht0) *. Такая схема обосновывается термохимичес-
кими расчетами теплоты гидратации иона водорода -
Разложение воды на свободные газообразные ионы про-
исходит по реакции
11,0 (ж) = Нг (гЦ-ОН- (г); /V/J,,» + 403,324 к кал/моль.
Диссоциации воды в растворе с образованием гидратиро-
ванных ионов соответствует уравнение
Нг0 (ж) + а(|=Н‘ (р); 13,335 ккал/мояь-
Теплота гидратации ионов волы будет равна
Д//Г111р = ] 3,335 — 403,324=5— 390,0 кклл/моль-
Следовательно, теплоты гидратации ионов очень велики
и имеют порядок величины теплот химических реакций.
Хотя последний из рассмотренных процессов протекает
с возрастанием числа частиц, однако для него Л£< О
(—19,35 э. е.). Это объясняется значительным упорядочи-
вающим влиянием молекул воды в результате сольватации
ими конов.
Ввиду того, что нсгндратиронанные ионы в растворе
отсутствуют, свойства растворов определяются составом
гидратированных иОНОв. Так, свойства водных растворов
Сг’* обусловлены свойствами аква комплекса |CrfH1O)JS4’,
а кислая реакция ОС13 ф) — его диссоциацией, например
(Сг(Н40)в1э* = [Сг(Н,О)5(ОН)Р+ + н+.
Под влиянием поля ионов меняется и строение раство-
рителя,
В непосредственной близости от иона располагается
структурированный растворитель. Число его частиц, нахо-
дящихся в этом сольватном слое, называется координаци-
онным числом емматации. Для большинства катионов
оно равно 4, 6 или 8. При записи различных уравнений
(электролитической диссоциации, взаимодействия между
ионами) обычно координационная сфера ионов не указы-
вается.
Гидратированный ион не представляет собой заряжен-
ный Шарик, облепленный некоторым количеством молекул
волы. Он входит в структуру жидкого раствора как полно-
правный ее участник. Известно, что вода обладает ближним
порядком, т. е. представляет собой как бы размытую легко
* Поэтому запись Н+ (р) яплвдтгя условной,
177
ЭБ ,:Научнов наследие России"
тырьмя соседними, образуя
I)
и
структуры
Рнс. 63. Элемент
БОДЫ
деформируемую кристаллическую структуру, напоминаю-
щую структуру льда. Каждая молекула ее связана с че-
тетр аэ д ри ч ес к у ю я не й к у. Rtгут-
ря этой ячейки имеется пусто-
та, размер которой превышает
размер молекулы воды (рис.
63). Попадая в такую струк-
туру, простейший небольшой
ион благородпогазового типа
может занять место любой
из молекул воды. При этом
силами электростатического
притяжениям л ндр у того вида
взаимодействия он плотно
свяжется с ближайшим окру-
жением из молекул воды. В
простейшем случае при этом
наблюдается выделение пер-
вичной теплоты растворения.
Более сложные ионы могут во-
влечь в сферу своего действия
и отдаленные с л ои молекул воды. В этом случае и само вза-
имодействие усложняется. Выделяемая при этом энергия
называется вторичной теплотой гидратации Крупные ионы
(например, С’ОГ и др.) попадают в тетраэдрические пустоты
и связываются с большим числом молекул воды. В этом слу-
чае благодаря большому размеру иона может произойти
значительное нарушение структуры воды н растворенное
вещество резко изменит сцойства растворителя.
Ионы в растворе могут, меняясь местами с окружающими
молекулами воды, перемещаться из одного тетраэдра в дру-
гой (тр алел я ни он ное движение). Этим обусловливаются и
электропроводность, и теплопроводность растворов, а так-
же и многие другие их свойства.
Как мы уже отмечали, ориентирующее влияние ионов
на молекулы воды очень велико. Поэтому оно, по-видимому,
распространяется за пределы первой гидратной сферы,
т. е. между упомянутым слоем и молекулами воды, харак-
теризующимися собственной (неискаженной) структурой
растворителя, располагается промежуточная сфера «воз-
бужденных» молекул воды. Все три структурные зоны рас-
творителя находятся в равновесии между собой. Таким
образом, эффект внедрения иона в растворитель можно
расчленить на две составляющие — непосредственное с вя-
зе ручное наследие России"
зыванне ионом молекул растворителя и его воздействие
на первичную структуру последнего. Благодаря уплотне-
нию молекул растворителя вокруг иона процесс сольвата-
цин обычно сопровождается сжатием раствора.
С возрастанием концентрации электролита зона неиска-
женной и частично искаженной структуры воды сокра-
щается. При Концентрации, отвечающей связыванию всех
молекул растворителя в первичные сольватные оболочки,
достигается граница полной сольватации. Понятие об этой
границе было введено К. П. Мищенко ir А. М. Сухотиным.
Ей отвечает такая конце]!грация раствора, которой соот-
ветствует сумма координационных чисел катионов и анио-
нов, т. е. отсутствие свободного растворителя и наибольшая
Рнс. 64. Механизм про-
цесса дкс«оциаинн. крис-
таллического aemeetaa на
ионы (схема)
удаленность ионов друг от друга. При дальней! и ем увели-
чении содержания растворенного вещества начинается
борьба за молекулы растворителя. В этой ко и курен цин
одерживают верх ноны, обладающие меньшим размером
и большим Зарядом, В результате происходит Либо обра-
зование Кристал л осоль вата, либо, например в случае вод-
ного раствора хлористого водорода, формирование недис-
социнрующнх летучих молекул растворенного вещества.
Сольватация существенным образом меняет и свойства
иона, так как диполи молекул воды оказывают большое
деформирующее действие на ион; это приводит к образова-
нию новой электронной системы с собственным уровнем
энергии, резко отличающимся от уровня энергии «цевоэ-
мущенного» иона.
Все это приводит к выводу, что общепринятая нагляд-
ная трактовка процесса растворения — она приведена
ниже— представляет собой лишь весьма упрощенную схему.
К ионам на поверхностном слое кристалла притягиваются
Противоположно заряженными концами полярные моле-
кулы растворителя (рис. 64). При сближении молекул с ио-
нами потенциальная энергия уменьшается и ионы передо-
|7й
Э Б ' Науч ное наследие Росси и" ' *
дят в раствор. По мере накопления ионов в растворе
усиливается обратный процесс— кристаллизация. При вы-
равимванин их скоростей наступает равновесие: раствор
становится насыщенным.
Диссоциация и ионизации полярных молекул протекают
аналогично растворению ионных кристаллов по схеме,
приведенной на рис. 65,
Вещества, в молекулах которых частицы связаны раз-
личными видами химической связи, распадаются на ионы
сначала по ионным свя-
____0 '<>,0 о^>
О0 0CD
0
Рис. 65- Механизм диссоциации поляр-
ной молекулы в растворе на ионы
(схема)
зям, а затем по поляр-
ным, Так, например,
бисульфат калия
К-СК ZO
/Sv легко дис-
Н—Oz "О
социнрует по ионной
связи К—О, труднее по
полярной СВЯЗИ 1’1—О
и практически не распадается на ионы по малололяр-
ным связям серы с кислородом.
Велики трудности создания математически разработан-
ной теории растворов электролитов. Было бы очень просто,
если бы можно было рассматривать такую систему, как
совокупность заряженных шар и ков-ионов в растворителе,
представляющем собой непрерывную среду с диэлектри-
ческой проницаемостью е. Такая модель не может дать
согласия с опытом. Ведь надо учесть совокупное действие
ряда факторов: изменение е растворителя в зависимости
□г природы ионов и их концентрации, влияние собственного
объема ионов, влияние концентрации несвязанного раство-
рителя, возможность формирования сложных (тройных
и др.) частиц, изменение энергии сольватации нойон с кон-
центрацией раствора, неполноту диссоциация электролита,
изменение структуры раствора с его концентрацией. Оби-
лие этих факторов и различный их вклад (в зависимости
от природы компонентов раствора, его концентрации и
температуры) делают невозможным их строгий количест-
венный учет во всей совокупности. Современный уровень
квантовомеханичсского и электростатического подходов
совершенно недостаточен для этого.
Учитывая интенсивное взаимодействие даже в разбав-
ленных растворах электролитов, уравнение
180
ЭБ ,:НауччоЕ наследие России"
предусматривающее суммирование изменения энтальпии
при взаимном удалении попои от расстояния в кристалл и-
ческой решетке до бесконечности [например, К*А" =
= К+ (г) 4- А" (г)] с изменением энтальпии в результате
сольватации катионов и анионов (например'. К’ (г) +
4- д- (г) К* (р) + А_(р)|, следует считать справедливым
лишь для очень разбавленных (теоретически бесконечно
разбавленных) растворов.
Первое слагаемое в (IV.26) представляет собой энталь-
пию разрушения кристаллической решетки. Эго большая
эндотермическая величина. Второе слагаемое — это при-
мерно равновеликая ей экзотермическая величина. Так,
для MgClj энергия кристаллической решетки равна 595,
а теплота растворения — 37 к к ал/моль, Следовательно,
по уравнению (IV. 26) получаем Д/Гсольь = —632 к к ал/моль,
т. е. величину порядка тепловых эффектов химических
процессов. Поэтому ДИ|)лети по абсолютной величине обычно
невелико {от 20 до —40 ккал) и нередко близко к нулю (см.
табл. 5). Следовательно, в подобных случаях небольшая
погрешность в определении одного из двух слагаемых мо-
жет привести к качественно неверным результатам (относи-
тельно знака величины Л.Н^Я1 а поэтому и характера
влияния температуры на растворимость •). Для других
концентраций Д#реш должна отвечать удалению ионов
до среднего расстояния между ними в растворе, а Д^сольв
должна учитывать и образование ионных пар, it нсдиссо-
цнировапных молекул, и новых продуктов их взаимодей-
ствия с растворителем и т. д.
Теплоты растворения электролитов в органических
жидкостях более экзотермичны, чем в воде. Кроме того,
э изотерм и чн ость ДЯрае„ с температурой в неводных раство-
рах падает; в водных же растворах наблюдается обратное
явление—нагревание приводит к более отрицательным
значениям А/Ураств.
Уравнение
•^SpatTB ₽ ДЗреш + А5^вг,ьа, (IV.2?}
аналогично? (IV.26), справедливо н для энтропии растворе-
ния.
Чтобы найти теплоту гидратации индивидуальных ио-
нов, надо
^соль» “ (Д-^со ЛЫ1) к ! + (Д л- {1V. 28)
* Имен и вяду Анри» в насыщенном раствора.
18 J
ЭБ ,:Научнов наследие России"
Рнс. 66. Зависинмть теплоты
гидратации ДЯГин>. (ккал/ммь)
некоторых одновалентных ка-
тионов от их радиуса /? (А)
при t - 23
разделить на составляющие для кати Он Ов и анионов. Для
этой цели необходимо знать теплоту гидратации хотя бы
одного нона. Эта задача может быть решена дол у щей кем
равенства теплот гидратации иэдадектронных ионов цезия
и иода. Разделнв суммарную величину теплоты гидратации
CsI, определяют ее значение для ионов Cs* и [', которые
используют затем для вы-
числения ионных тел л от ги-
дратации остальных копов.
Величины вычисленных та-
ким образом тепл от гидрата-
ции отдельных полов близки
к тепловым эффектам хими-
ческих реакций (ем. также
с. 177). Например, Д/7а- =
= —8J ккал/моль, ДНма+ =
— —101 ккал/моль, &Hst- =
= — 330 ккал/моль, ДНс«н- =
— —386 ккал/моль. Зная
теплоты гидратации отдель-
ных ионов и энергии крис-
таллических решеток элек-
тролитов, можно производить
вычисления теплот раство-
рения и, наоборот, л о теп-
лота м растворения и теплотам гидратации ионов опреде-
лять энергию кристаллических решеток (см. рис. 10).
Изучение ИОННЫХ теплот гидратации показало, что их
величины зависят от радиусов понов (рис. 66). Теплота
гидратации с ростом радиусов уменьшается; уменьшается
и число молекул растворителя, связанных с попом. Этим
объясняется наблюдаемое уменьшеЕше электропроводности
растворов от Cs4- к L? и, наоборот, рост ее в этом порядке
в расплавах их солен, где сольватация отсутствует.
Теплоты гидратации ионов возрастают примерно про-
порционально квадрату заряда. Приблизительно так же
меняются и энтропии гидратации; так, для Э* и 3м послед-
ние равны соответственно ~ —15 и —55 э. е. Из этих дан-
ных следует, что для малозарядных катионов, учитывая
к тому же небольшие, как правило, значения температур,
величинами ТД£растк можно пренебречь.
Интенсивность взаимодействия растворителя с ионом
зависит от целого ряда факторов: к ним относится и энер-
гия ионизации / атомов-иоиоабразователей. Поэтому с /
ЭБ 1-1^$чсе наследие России"
связаны 1! теплоты, и энтропии сольватации. В растворах
м но гоза рядных ионов структурирование растворителя ста-
новится столь значительным, что они приобретают свой-
ства, присущие кристаллосольватам, Наоборот, ионы, об-
ладающие деструктурнрующнм действием (К*, NOj и др.),
оказывают разрыхляющее влияние на прппонпую зону
сольватов, В последнем случае обычно А Я >0 и 45 >0,
Мыслимы El случаи, когда б результате двустороннего пе-
реноса заряда междз1 ионами н соприкасающимися с ними
молекулами раствор if тел и происходит полный переход
электронов, т. е. ок и с л и тел ыю- восстанови тел ы гы н процесс,
приводящий к коренным изменениям природы раствора,
Последнее делает понятным отсутствие высокозарядных
ионов в водных растворах. Так, растворение солей CoJ’ не
Приводит к образованию Со^(р) — происходит окислитель-
но-восстал они тел ьгичй процесс, сопровождающийся выде-
лением кислорода.
Располагая значениями к Д5млы, легко вы-
числить Дбсольц, Для этого можно также воспользоваться
уравнением Борна
(IV 23}
где в — диэлектрическая проницаемость растворителя*.
Это уравнение дает грубо приближенный результат (его
вывод не учитывает ни изменения в вокруг ионов, ни их
структуру). Однако для больших ионов с небольшими за-
рядами погрешность вычисления не очень велика, В то же
время (IV.29) отвечает примерной пропорциональности
АСсаям) квадрату Заряда ионов (см. выше), Не пр и ходится
говорить о качественном согласии с экспериментом: чем
большее, тем значительнее AGcn4t.тем лучше растворяется
ионное соединен не
Большое значение е имеют растворители с сильно по-
лярными молекулами. Поэтому электролиты хорошо рас-
творяются (н диссоциируют) в Н^О (в = 81), СНаСООН
(57), хуже в С2НЬОН (21), плохо в бензоле (2,5) и в других
неполярных жидкостях.
• БьгнОД [IV.29) можно найти в пособии М. К. Карапетьяица
и С. И. Дрюпч! «^троение пешлетвзк 3-е изд. М-, Высшая школа, 1973.
183
ЭБ ,:Научнов наследие России"
Гллдл TV
ЛОННЫЕ РАВНОВЕСИЯ If ОБМЕННЫЕ РЕАКЦИИ
В РАСТВОРАХ ЭЛЕКТРОЛИТОВ
Как указывалось выше, электролитическая диссоциа-
ция — обратимый процесс. Поэтому для диссоциации рас-
творенных веществ на яоегы справедливы общие законы
равновесия. Так, для процесса
К„лл “лК++^А-
величинэ константы равновесия связана со стандартны ми
изменениями энергии Гиббса, энтальпии и энтропии при
реакции равенством (1L20), н уравнение, выражающее кон-
станту равновесия. Запишется так:
к JK'FlA-r rI™
*—1ВДГ' <IV3”
Ионную концентрацию находят умножением мольной
концентрат г и электролита ня степень диссоциации а и if а
число пт показывающее, сколько ионов данного вида обра-
зуется при распаде одной молекулы.
Закон действующих масс в виде (IV. 30} справедлив
лишь для слабых электролитов них разбавленных раство-
рах. При его применении к концентрированным растворам
и к растворам сильных электролитов необходимо учиты-
вать поправочные коэффициенты (СМ. С. 186 и СЛ.).
Ниже основное внимание уделяется применению урав-
нений (11.20) и (IV,30),k раХ1пчлы.м типам ионных равно-
весийдиссоциации слабых электролитов (в том числе
воды и плохо растворимых солей, комплексных ионов)
и гидролизу. Во всех случаях, если это не оговорено спе-
циально, подразумевается, что константа равновесия отно-
сится к 25 СС.
1. Равновесие в растворах слабых электролитов
При растворении слабого бинарного электролита КА
в растворе установится равновесие
КА = к*ч-А-
Применяя к нему закон действующих масс, можно написать:
*= ГК(КА] “ (IV'31)
184
ЭБ ,:Научнов наследие России"
Если растворен I моль электролита, у которого степень
электролитической диссоциации равна а, то
[КА]=ТгД( [К>“ н |А->~,
где V — объем р а створ и. Полета ил я я эти величины в ура в-
наше f IV.31). получим
. а-3 as х
Й “ 1—а *
(IV. 32]
Где С — мольная концентрация. Это уравнение называется
заяонсм* рдзбааления {Оствальд, 1888). Оно выражает не
изменение К в зависимости
от С (для данного электролита
пр» постоя незой температуре
К const), а зависимость а
от С
Чем больше ft, тем значи-
тельнее диссоциация электро-
лита. Так, увеличение Я в
ряду кислот СНдСООН —
CHjCJCOOH—СНС1ЙСООН —
CCl^COOH, обусловленное от-
та л ки ва и и ем э л е ктронов от
связи ОН электроотрицатель-
ным атомом хлора, означает
вместе с тем и роста в этом
Рис. 67. Изменение степени
алскт^йлити’сескоч диссоциации
« с ]]аяб4МСЕ1Иси и (схема}
ряду.
Со глас но (J V. 32) с тепен ь электр ал ига 1чес кой дне соц и з цн и
слабого электролита с разбавлением раствора растетч
Так, в растворе, содержащем I ^оль СНдСООН в 1 л НоО^
а — 0,0038 (0ъ38%)т возрастая при стократном разбавлении
до 0,041 (4,1 %), а при десятнтысячекратпом — до 0,306
(30,6 %).
Схематически эта зависимость представлена на рис. 67.
При очень большом разбавлении а достигает предела-
Естественна, что рис. 67 подобен рис. 62. Иногда уравнению
(IV.32) придают иной вид. Подставляя а пего значение а
из уравнения (1V.24), получим
X-
(^м
(IV.33)
С помощью этого уравнения можно вычислить константу
Электр литической диссоциации ва основании юмере] i и и
ЭЛеКТрО! Ер О ВОД! [ОСТН.
1Й5
ЭБ ,:Научнов наследие России"
Константа элсктрвднтнчсской диссоциации Л' является
характерной величиной для данного электролита и раство-
рителя и зависит лишь от температуры. Повышение тем-
пературы оказывает различное влияние на К (рис. 68).
Для многих веществ К проходит через максимум. В соответ-
ствии с принципом Jit Шателье это объясняется переменой
знака А//рмгк> связанЕюй с различным влиянием темпера-
туры на электролитическую диссоциацию молекул и на
гидратацию ионов.
2. Растворы сильных электролитов
Строго говоря, закон действующих масс применим лишь
к идеальным газам и идеальным растворам, поскольку при
его выводе не учитываются силовые поля частиц. Опыт по-
1,101—J--1—I—I—j_
ю го м w so
t
Рис. 68. Зависимость к&ц.
сганти диссоциации К не-
которых слабых электроли-
тов в водны?; растворах от
температуры I ft)
казывает, что он применим и
к разбавленным растворам сла-
бых электролитов и неэлектро-
литов. Так, например, кон-
станта электролитической дис-
социации уксусной кислоты с
изменением концентрации ос-
тается величи ной. постоя иной.
Но даже для очень разбавлен-
ных растворов сильных элек-
тролитов уравнение (IV.30.) не-
применимо вопреки сравнению
(11.20)
Л «/(С).
Можно предположить, что
сильные электролиты в раство-
рах полностью распадаются на
ноны. Спектральные и оптичес-
кие исследован и я । юдтвер жд ают
отсутствие в растворе сильных
электролитов нейтральных моле-
кул, в то время как в растворах слабых электролитов не-
диссоциированные молекулы обнаруживаются. Но тогда
следовало бы ожидать, что для раствора NaCl i=2. Однако
ЭТОГО lie наблюдается. Только в предельно разбавленных
растворах поваренной солн значение i приближается к
двум.
186
ЭБ ''Научное наследие России1
ЭкВНВалеНТНЗЯ ЭЛСКТрОПрОВОДНОСТЬ раСТВОрОВ СИЛЬНЫ»
электролитов не остается постоянной при изменении кон-
центрации, как этого можно ожидать при полной диссо-
циации, з увеличивается с ростом разбавления. П то время
как подобные факты для растворов слабых электролитов
хорошо объяснимы с помощью представления о степени
диссоциации, для растворов сильных электролитов многое
остается непонятным. Таким образом, нельзя отрицать
факт полной диссоциации в растворах сильного электро-
лита ла ионы п в то же время трудно просто объяснить
многие их свойства. Причина i.e котор ого сходства свойств
растворов сильных электролитов со слабыми заключается,
очевидно, в следующем Ионы сильного электролита в раст-
воре взаимодействуют друг с другом благодаря наличию
значительных электростатических сил, действующих меж-
ду ними. Это взаимодействие усиливается с ростом кон-
центрации, т. с. с уменьшением расстояния между ионами.
При этом каждый нон в растворе окружается ближайшими
противоположно заряженными ионами, в то время как
ионы одноименного знака располагаются дальше. Так
каждый ион окружается роем других ионов, В системе
создаются условия для равномерного размещения ионов
подобно их положению в кристаллах, но Era более удален-
ном друг от друга расстоянии. При этом ноны сольвати-
руются, что Также отражается на их свойствах и свойствах
растворителя.
Такое взаЕ)модействие ионов друг с другом и раствори-
телем влияет на скорость их движения в электрическом паче,
что ведет к понижению электропроводности с концентра-
цией.
Кроме описанных явлений, в растворах сильных элек-
тролитов при повышенных концентрациях может происхо-
дить ассоциация ионов. Так, в водных растворах, например,
установлено образование ионов ВаС1+, AgC3;, LiCli и др.
Прзг разбавлении эти частицы диссоциируют. Поэтому
с геоны шепнем концентрации сильных электролитов даже
при полной их диссоциации происходят изменения свойств
раствора, аналогичные тому, как если бы при этом уменfa-
шал ась степень диссоциации электролита. Ясно, что эти
изменения не связаны с изменением истинной степени дне-
соцнащш, как у слабых электролЕЕтов, а обусловливаются
проявлением кажущейся степени диссоциации. Пе>
следЕ1яя, в свою очередь, не отражается концентрацией
раствора, чем и обусловливается неприменимость закона
187
ЭБ ,:Научнов наследие России"
действующих масс при подстановке в него нс тин пых кон-
центра! И г Л растворов электролитов.
Формальный выход из положен к я был предложен аме-
риканским ученым Льюисом {1907). Он ввел понятие о ка-
жущейся концентрации, которую назвал активностью (а).
Активность — это величина, подстановка которой вместо
концентраций в уравнение закона действующих масс делает
его справедливым (для всех электролитов « неялектролатив)
при любых концентрациях. Она выражает активную кон-
центрацию вещества н, не представлял какого-либо реаль-
ного его свойства, отражает суммарно вес возможные про-
цессы в растворе. Поэтому введение активности нс раскры-
вает механизма процессов взаимодействия ионов и раство-
рителя. Это лишь удобный прием, позволяющий находить
свойства любых растворов. Активность связана с концен-
трацией уравнением
в=уС, (IV.34)
где у — коэффициент активности. Последний характери-
зует отличие свойств растворов сильных электролитов и
вообще реальных растворов от идеальных растворов.
Для предельно разбавленных растворов, где отсутствуют
силы взаимодействия ионов из-за их отдаленности друг
от друга. а=С, т. е. у=1. Активность ионов, так же как
и концентрация, выражается в моль/л и является как бы
их эффективней концентрацией.
А к these юсть и коэффициент актив еюсти можно определить
на «Х'новании экспериментальных данных (по повытенпЕО
точек кипения и понижению точек отвердевания растворов,
а также по давлению пара над раствором и другими мето-
дами).
3- Диссоциация кислот, ос пинаний и солей в воде
Кислоты в воде диссоциируют на ионы водорода (гнд-
роксоння) и на анион *. В табл. 15 приведены значения
констант диссоциации некоторых электролитов в воде.
Максимальное число ионов водорода, образуеоецихся
из одной молекулы кислоты, определяет ее основность,
В зависимости от разбавления и других условий многоос-
HOEEiue кислоты могут диссоцинроЕзать ступенчато, после-
* Более общее определение понятий «кислота» и «основание»
см. в гл. V.
18S
ЭБ ,:Научнов наследие России"
дователыю отщепляя один ион водорода за другим. Так,
для ортофосфор ной кислоты константы диссоциации каждой
ступени при 25 °C равны:
К; =
|Н||НгР0.1
HW4
Ю-з;
К8 =
_ |Н J|po;-|
* [НРО- ] ‘
ГН*]|НРО»-]
[М.РО(]
-3,6-10-м,
= L9 |0-«1
Подобная картина характерна и для других mi югоосновных
кислот: первый ион водорода отрывается от молекулы
легче, последующие все труднее, так как возрастает отри-
цательный Заряд кислотного остатка. Поэтому обычно
в растворе фосфорной кислоты ионов POJ очень мало.
Таблица 15
Константы hohtimipih К нскиторык веществ
и ионов в воде при I *= 25“ С
Формула к Формулл л
СНтрООН ],я- Ю-5 нхо,, 3,6 - i0"T
HCN 4Д». 10’» HflOj 2,4 - 10-и
НСО7 4,7- 10” НгРОГ i.fl io-*
HF ’ (5.S 10 н RbS 1,1 - 10-’
нрог Зр- 10'1Л ад io-*
HS- КЗ-10 « H^POj 7,11 10 я
HSOf 1.0 10 1 |,а - io-»
Способность многоосное пых к н с лот диссоциировать
ступенчато объясняет их склонность к образованию кис-
лых солей.
Как мы уже отмечали, константа диссоциации в отли-
чие от степени диссоциации нс меняется с разбавлением
раствора. Поэтому она является важной характеристикой
слабого электролита.
Если из силу бескислородных кислот НЧЭ влияет при-
рода 9, то сила кислородсодержащих кислот Эг*,+яО1Гг ЮН)Л
зависит как от природы Э, так и от значений т и п. Правда,
ввиду большой электроотрицательности атома кислорода
Он оттягивает электроны от связей О—Н; поэтому влияние
ft не существенно. О сказанном можно судить но значениям
первой константы диссоциации кислородсодержащих кис-
лот в водных растворах: С1(ОН), В(ОН)Э, Si(OH)*, Te(OH)fl
189
ЭБ ‘Научное наследие России"
(f/i=O) являются слабыми кислотами (для них .^< 10"*);
NO(OH), SO(OH)4, IO(OH)S (m = I) — значительно силь-
нее (для них Ki = 10"Б -5- 10"1); КОз(ОН), SOs(OH)s On =
= 2)— сильные кислоты.
11одобно много основным кислотам, основан!)я .чного-
зарядных катионов могут диссоциировать ступенчато. На-
пример:
Fe (ОН )г Fe (ОН)++ОН-; Fe (ОН Fe (ОНГ+ 4- ОН-
Fe(OH)*+^ Fe”4-OH“
Этцм объясняется способность основания многовалентных
металлов образовывать основные соли, например Fe(OH)CI?i
Cus(OH)sCOs и др.
Гидроксиды многих металлов в водных растворах могут
диссоциировать и по кислотному, и по основному типам.
Например, гидроксид цинка диссоциирует:
(ZntHaOJd«* 2П(ОНЬ (H/J),[Zn(OH)*p-
Прн этом в кислой среде равновесие сдвигает влево, в ще-
лочной — вправо. Это находится в соответствии с законом
действующих масс. Оба процесса приводят к образованию
Очень слабого электролита Н.гО.
Соединения, которые в зависимости от условий прояв-
ляют как кислотные, так и основные свойства, называются
амфотерными или амфолитами (от греч. «амфотерос» —
обоюдный, в смысле — двусторонний). К амфотерным элек-
тролитам относятся Zn(OH)2, А1(ОН)а , Ве(ОН),. Ga(OH)3t
Cr(OH)s, Gc(OH)f1 Sn(OH)41 Pb(OH)j и многие другие.
Амфотерность проявляется и у соединений, содержащих
большие атомы неметаллов, например у HOI. Борная кис-
лота НЭВО$ при взаимодействии с более сильной фосфорной
кислотой НаРО4 проявляет основные свойства, давая сое-
динение BPOj.
Солями называются электролиты, которые при электро-
литической диссоциации образуют катионы металлов или
сложных групп, и одноатомные или многоатомные анионы.
Соли бывают средние (нейтральные) Iнапример, Саа(РО*)г],
кислые (СаНРО4), основные iZn(0H)Ol, смешанные
[CaCl(OO))t комплексные lK4Fe(CN)»h двойные (KMgFj).
Двойными называются комплексные соли, которые в раство-
ре почти полностью распадаются на ионы солей, из которых
они образовались.
]0О
ЭБ ,:НаучноЕ наследие России"
Кислые солк диссоциируют ступенчато, легко отщепляя
вначале ионы металлов, а затем ИОНЫ водорода. Например,
NsHaPO4 — Na" + H-PQ;; I^PO,- “ Н* -|- Н РО»-
н
HPOJ-=H*-|-POr
В последних двух ступенях равновесие сдвинуто влево;
поэтому ионов Нт в растворе очень мало.
Основные соли также диссоциируют ступенчато, легко
отделяя кислотный остаток, а затем гидр оксид-нон ОН',
например:
Fe {О Н)г С] Fe (ОН) Л- СГ; Fe (ОН J (u “ Fe {ОН + ОН -
И
Fe(OH)i+ = Fe1L-f-OH-
Некоторые гидроксосолн, легко отщепляя воду, neper
ходят а оксОСОЛн. Вот ОДИН Пример:
SMOH^a-SbOCH-HtO
Оксосоли диссоциируют на оксопневый катион и аннон
кислоты. В указанном примере
SbOCl- (SbOf+Cl-
(см- также с. 209—210).
А. Влияние од поименных ионов на диссоциацию
слабых электролитов
Мы установили (см. с. 185), что на состояние динами-
ческого равновесия электролитической дпссоцнаили слабого
электролита влияет разбавление раствора. Сместить рав-
новесие можно также изменением концентрации одного из
находящихся в растворе ионов. Так, для раствора уксусной
кислоты
СН^ООН Z2 СН^СОО-Ч- ьг
и в соответствии с уравнением
|С11аС(ХУ|| Н-]
- [СНаСООН|
(IV.35)
прибавление хорошо диссоциирующей ее соли (например,
СН3СООМа) увеличит концентрацию ионов СН3СОО‘, что
поведет к значительному сдвигу равновесия влево к к по-
нижению кислотности среды.
19!
ЭБ ,:Научнов наследие России"
ПодоСетым же образом уменьшается концентрация конов
011 при прибавлении к водному раствору аммиака какой-
нибудь аммонийной соли Следовательно, введение в раствор
слабого электролита одноименных ионов уменьшает его
диссоциацию
5, Диссоциация коды
Применим закон действующих масс и к воде Судя по ее
электропроводности, она хотя и весьма незначительно,
но все же диссоциирует на ноны
гн2О — Н/У+ОН-
Для простоты вместо иона тндроксолня по-прежнем у будем
писать иегидратнрованный ион Н+, тем более, что это не
отразится на выводах * Итак,
Н,о- Н' + ОН-
Следователъно, вода — самый типичный амфотерный элек-
тролит, т. е она может действовать в равной степени и как
кислота, и как основание
Для диссоциации воды на ионы АД]и== 13,335 ккал/моль
и (15^ =—19,35 э е (см с 177) Поэтому в соответствии
с уравнением (II 20)
— 4,576 298,21£ К «13 335+J9,35
откуда
11 lEU|
Из значения ЭТОЙ величины видно, что вода относится к очень
слабым электролитам Так как ее степень диссоциации
чрезвычайно мала, то пренебрежимо мала и концентрация
распавшихся молекул по сравнению с концентрацией не-
диссоциироваялых молекул, поэтому последнюю можно
считать величиной постоянной и равной
I Н>°) = = S5-56 ыоль^л
10| U10
Подстановка этой величины в выражение константы дис-
социации Н4О дает:
[Н+] JOH-] = 1,8- ИГ3* 55,56 ^1,0-10-“
* Эта замена совершенно условна, так как протонов к воде не мо-
жет бить (ио расчетам сущкпомине одною протока требует объема
10-* л), см с, I7G.
J92
ЗБ ,:Н-аучноЕ наследие России"
Произведение концеЕгграций водородные и гидр оксид-ионов,
являющееся при данной температуре постоя иной величи-
ной, называется ионным произведением воды Kw Итак,
ирп 35 “С
Это означает, что увеличение кои центр а шеи водородных
ионов вызывает соответствующее уменьшение концентра-
ции гидр о кс ид-ионов, и наоборот. Равновесие между иона-
ми FT и 01Г существует не только в воде, но и во всех вод-
ных растворах. Поэтому указанное соотношение может
характеризовать кислотность и
основность различных сред.
Для процесса
НЧр) + 01Г (р)₽н.р (ж)
= —13,335 к кал/моль,
т. е. диссоциация воды связана
с большой затратой энергии.
Отсюда в соответствии с прин-
ципом Ле Шателье температура
будет оказывать значительное
влияние sra (рис. 69).
Так как Ktr то не мо-
жет быть водного раствора, в
котором величина (НЧйлн (ОН-)
равнялась бы нулю. Следова-
тельно, в любом водном рас-
творе всегда присутствуют сов-
местно ионы [Н*] и [ОН-]. Для
Рне. 69. Зависимость кон-
станты диссоциации воды
Kw от тем пер а тури i (°C)
нейтральной среды 1Н*) = [ОН- J == V10“'* == 10-7 моль/л.
В кислой среде [Н*1 > Ю*’ и [ОН_!< 10”т; в щелочной
среде, наоборот, |H+J< 10’7 и ЮН.-! > Ю-т. ,Можно
пользоваться любой из этих величин, так как
(О-1-* Е0'и
« l^-b-iHTp
Обычно кислотность и щелочность среды принято характе-
ризовать концентрацией водородных ионов.
Строго говоря, постоянство конного произведения
воды справедливо лишь в том случае, если истинные кон-
центрации ионов заменены их активностями. Это особенно
важно для водных растворов сильных кислот и оснований,
так как для малых 1Н*] и (ОГГI значения концентрации и
активности практически совпадают.
7 М- X. Kiijhiutibfl иц
ЭБ ,:Научнов наследие России"
1УЗ
Для удобства при опенке кислотности и щелочности
среды пользуются не концентрацией водородных ионов,
а величиной водородного показателя pH; он ранен отрица-
тельному десятичному логарифму концентрации водород-
ных ионов:
PH = -)glH'J. (IV.36)
Если, например, [ЯЧ = 10"5, то pH => —lg I0’6 = 5. В ней-
тральной среде, где [HI = 10“’, pH = 7; в кислой среде,
где [Н*1 > 1СН, рН< 7; в щелочной среде, где 1Н*1< 10"т,
pH >7. Чем выше концентрация водородных ионов, тем
меньше pH. В качестве примера рассчитаем pH раствора,
в котором [НЧ = 2-101*1, В соответствии с (IV,36) получаем
pH = —1g 2 + 7 = 6.7, Водородный показатель может
быть больше 14, а также может принимать отрицательные
значения (в очень кислой среде).
Водородный показатель измеряется различными мето-
дами, Сравнительно грубое, по быстрое определение pH
можно произвести с помощью специальных реактивов —
индикаторов, окраска которых меняется в зависимости
от концентрации водородных ионов. Свойства некоторых
индикаторов приведены в табл. 16.
Таблица J6
Свойства некоторых индикаторов
Инт^рпял pH П<-рС V О-Д.9 «краени Окрвскч
Индикатор в кислой среде п щелочкой *ред*
Метил в ио лет .Метялевый оранжевый Лакмус Фенолфталеин Индигокармин 0—3 3,1— 4,4 5—8 8,3—10,0 1'2—14 Желто-зеле- ный Красны1! Бес лиг 11 е J й Голубей Фнолетопо- голубой Желтый Синий К рас EELEhl Желтый
Окраска индикатора в области перехода меняется посте-
пенно, Поэтому с помощью различных индикаторов можно-
определить значение pH. Так, если исследуемый раствор
краснеет при прибавлении лакмуса и желтеет от метилового
оранжевого, то pH его лежит в пределах от 4,4 до 5,0
(табл, 16). Если далее один из этих индикаторов прибавить
к одинаковым количествам испытуемого раствора л эта*
194
ЭБ ' Науччое наследие России"
лонного, koi г центра ни я водородных конов которого из вест-
на, ТО путем сравнения окраски можно установить до воль нс
точное значение pH (колориметрический метод).
6. Буферные растворы
Получить очень разбавленные растворы кислот или ще-
лочен с постоянным: значением pH путем разбавления силь-
ных КИСЛОТ или оснований невозможно, так как незначи-
тельные количества СОг воздуха, щелочей из стекла или
загрязнений в дистиллированной воде могут заметно из-
менить реакцию таких растворов. Тем не менее, в лабо-
раторной практике часто требуется иметь раствор с вполне
определенным и устойчивым значением pH. Такие растворы
готовят смешиванием слабых кислот или слабых оснований
С ИХ СОЛЯМИ.
Рассмотрим в качестве примера раствор» содержащий
уксусную кислоту и ацетат натрия. Из уравнения (IV.35)
следует, что
ГН + 1 К iCbljCOOH] /IV 471
|Н 1=*1сП£ба=Г (IV'37)
Пр л введении в раствор хорошо диссоциирующего ацетата
натрия диссоциация кислоты подавляется. В результате
этого кон центра ц и я недиссоци л рова и i гы х и ел е к ул СНаСОО Н
становится почти равной общей концентрации КИСЛОТЫ, а
концентрация CHSCOO — общей концентрации соли. Тог-
да (IV.37) можно записать так:
[ И+ Н X ^лслотз^ (ГV.38)
[соль]
Так как Д' — постоянная величина, то концентрация во-
дородных ионов в таком растворе будет определяться от-
ношением концентрации кислоты к концентрации соли.
Разбавление этого раствора практически не изменит его pH.
Даже добавление некоторого количества сильной кислоты
не повлияет заметно на pH такого раствора, так как вве-
денные ионы IP свяжутся с избытком анионов и иеднссоции-
рующие молекулы слабой кислоты. Почти не изменится
pH и при добавлении щелочи. Растворы слабой кислоты
и ее соли (или слабого основания и его соли), концентрация
водородных ионов в которых почти нс меняется при введе-
нии в них сильной кислоты или сильного основания, на-
зываются буфершлми растоирчми. Они играют большую
Z* 193
ЭБ ,:Науч1НОЕ наследие России"
роль в регулировании жизненных процессов в организмах
животных и растений.
Легко прийти к аналогичным выводам, рассмотрев
раствор, содержащий слабое основание и его соль.
7. Произведение растворимости
Применим закон действующих масс к равновесной сис-
теме, состоящей из кристаллов плохо растворимого вещества
и его ИОНОВ в насыщенном растворе (рис. 70):
КлА|м " лК++мА"
При установившемся равновесии в единицу времени в раст-
вор переходит столько
ионов, сколько их вновь возвра-
щается в осадок (вследствие
ничтожно малой растворимости
считаем, что ot = I*), Для этой
системы, согласно сказанному
на с. 188,
Sr«SoKP-pSJJ:™): Для трудкорастворимого элек-
а - насыщен^ р«»ор: ТрОЛИТЗ АКТИВНОСТЬ ИОНОВ равна
б-осадок их концентрации, поскольку
раствор получается очень раз-
бавленным. Следовательно, это уравнение можно заменить
(IV. 31); ио знаменатель правой части последнего есть ве-
личина постоянная. Поэтому
[КЦ"[А-^=Х|КЧЛЙГ|-const.
Следовательно, в насыщенном растворе труднорастворимого
электролита произведение концентраций его ионов в степени
стехиометрических коэффициентов при данной температуре
есть величина постоянная. Она называется произведением
растворимости
ПР = [К+р[А-]'4. (IV. 39}
Для расчета ПР можно воспользоваться уравнением
(11.20). Покажем это на примере А&СОз;
ЛЙ:СО3 (кнч = 2Ag+ (р)-НСРГ) (Р)
Полная Д13СГОИИЯЦЕ1Я плохо растворимого вещества вытекает
из у равней нн (1УД2Х тян как 4? очевь мало1 я К сравнительно велико,
19&
ЭБ ,:Научнов наследие России"
Из табл. I it 4 = 9,58 ккал/моль; из табл. 6 ASJ« “
= —19,0 э. е. Следовательно,
— ЦТ In dgiCcoj- « ДЙТ = 9580 + J9,ОТ (кал/моль),
откуда при 25 °C
КС=ПР^6,71 io-ч
Чем меньше величина ПР, тем труднее в соответствии
с уравнением (11.20) осуществляется переход веществ
в раствор.
Произведения растворимости некоторых солей приве-
дены в табл. 17. (Справочник ХнмиКа/Под ред. Николь-
ского Л. М, 3-е изд., перераб, и доп. —М.: Химия, 1954).
Таблица 17
Произведение растворимости ПР некоторых малцраетдорняыл
в воде веществ при
вещество ПР Fst-mtcTBO ПР
АдВг 5,0 JO 1й CdS 7J Ft)
AgSCN Ll(i.|0-« CoS 1,9- 10
AgCl l,fi. Ю-i® CuS 3,5- F0 <2
Agl 3,1 • 10» ClLfS 2,0 10 «
AgaS 4,0 !0 « HgS 3,(b 10 41
BaCOj 4,9 . IO-» 1,08 tO-i» NtS IJ 10 «
BaSO, ZnS io-^
OSO, 6,3. io-* мЁ(ОН)а 5,3.10-й
Даже из этих отрывочных данных можно сделать ряд вы-
водов, в частности, о падении растворимости в рядах
AgCl—AfiBr—Agl, CaSOj—SrSOq—BaSOj, ZnS—Cd^HgS,
CuS—Cu2S, о близости свойств однотипных соединений
кобальта и никеля, о ничтожно малой растворимости HgS
и г. д.
Так, падению растворимости в ряду ZnS~€dS— HgS
отвечает то, что в реакциях
3S (к)+2НС1 (р) = эаа (pHHsS (г)
Д б*а! = 4,71 (Хл), 9,96 (Cd} и 43,37 (Нд) ккм/м*ль.
Располагая величинами ПР, можно вычислить кон-
центрацию ионон к соли в насыщенном растворе. Напри-
мер, для системы
AgSCN (р) AgT (p) + SCN- (р)
197
ЭБ ':На\ччоЕ наследие России1
где
i Ag+1 = ISCN-], nPAj(SCN = [ Ag 4 [SCN-] = 1,16- I(H«,
пт к уда
(Ari = [SCN-J^/Ti3“i0-i»=l,08.10-» ыоль/л;
следовательно, [AgSCN] —Ш [0-в моль/л.
Зная произведение растворимости, можно предвидеть,
выпадет в ли не выпадет да иная соль в осадок. Так, в милли-
мольном растворе CaSO4 осадок не выпадет: величина
ICes+J [SOj I = 0,001-0,00] = I0-* меньше ПР^о. =
= 63‘]0’* (табл, 17).Следовательно,этот раствор будет не-
насыщенным. Поэтому его соприкосновение с кристаллами
соли вызовет их растворение. Если же 1К+? 1АЧ'1’ > ПР,
то раствор окажется пересыщенным и из него будет выпа-
дать осадок.
Так как ПР = const, то введение одноименного иона
будет смещать равновесие в сторону образования осадка.
Так, при прибавлении хорошо растворимого ВаС14 я насы-
щенному раствору BaSO4 концентрация ионов Ва1+ возрас-
тает, Это приведет к связыванию ионов SO*-, т. е, к обра-
зованию осадка (произведение концентрации ионов Ва*+
н SO* должно остаться неизменным). Чем больше ПР, тем
легче добиться растворения осадка. Если Ag! (ПР =
= 8,1-КГ17) нерастворим в NH#1 то AgCl (ПР =
растворим в МН,. При этом образуется аммиачный комплекс
(см. $ 8).
8. Диссоциация комплексных попов
Теперь применим закон действующих масс к диссоциа-
ции комплексов.
В растворе комплексные ноны всегда несколько диссо-
ииируют; так,
ЮЧЫНзЫ* (p)-Qi" (р)Ч-вмн, (р)
Обычно такое равновесие сильно сдвинуто влево. Действи-
тельно, для данного процесса
К имт
(Gp^KNH^
[CofNlU,]^
7,8 - J0-a-
Поэтому и здесь, как и при диссоциации слабого электро-
лита (§2), воды (§ 6) и плохо растворимого вещества (jj 7),
концентрация ионов d растворе очень мала. Эти и позво-
ляет н ЭТИХ случаях Применить Закон действующих масс
198
ЭБ ,:Научнов наследие России"
в виде однотипных простых выражений, содержащих кон-
центрации соответствующих частиц.
Величина К,„ет называется кояс/лдк/пой нестойкое/ли *
Чем больше она, тем легче распадается комплексный ион
Ла составные части и, следователь!)о, тем менее он устойчив.
В зависимости от прочности связи между центральным
ионом и лигандами значения констант нестойкости для раз-
ных ионов различны, iio в Аолырвнетве случаев невелики
(табл. 18).
Таблице S8
Константы нестойкость некоторых водных комплексных моею а
прн 25 С
Ф&рх у ла 1 । ФорыуЛ! । «И
lAtfCNjJ- s-jo^ i IWiahl** 5,2 JO *»
AupJjJ- 5- 10 a* FeSCN]»* 5.10 a
lCd(CNh]=- м jo 17 Wr^P- 7,8 -10 M
GofNHjJep* ^8 -10 * 1,8 Г0 s
Cu(O}.|- J JO * Zn(CN)ds i,3- to «:
(Hg(CNhr 4- JO « [ZntNfW* ЗЛ !0"1Q
Большая устойчивость цианида цинка по сравнению с его
аммиакатом обусловлена более прочной связью между комп-
лексообразавателем и лигандами.
Комплексные ионы, как правило, диссоциируют с ту ле л
чат, например:
(AgfSAhP- (Р) - И«(«А)Г (PJ+SjPi- (р)
(AfiiSiQjJr (Р)~ Agr (р)4-3/3>- (р)
Каждая ступень характеризуется своей константой. В дал-
ном случае (K„r(1)i = 2.3-10"* л (/С,|ссп)4 = 1,5-КГ’.
При многоступенчатой диссоциации часто пользуются
общей константой нестойкости, которая равна произвелс
нию констант нестойкости по всем ступеням диссоциации-
l^neei)i (^иегт)а
причем (К„кт)| >(# 11^1)4 > ” - -
К диссоциации комплексного иона применимо правило
сдвига равновесия с изменением концентрации одноимен-
ного иона (см. § 4). Так, для диссоциации нова (Ag (SjO^fl4"
_ l,V ] [З^СЧ-р
"е" _ ЙЖЕЬГ
* Величина, обратная константе вестийксктн, называется кон-
ста и той уггойчивоети комплекса,
199
ЭБ ' Научное наследие России"
Если увеличить концентрацию попон 5,ОГ (например,
введеннем в раствор тиосульфата натрия NajS^Oj), то рав-
новесие сместится влево, так как при возрастании числи-
теля дроби должен возрасти и знаменатель (условие по-
стоянства значения Х„сС1). Следовательно, должна увели-
читься и концентрация не распавшегося комплексного нона.
Наоборот, удаление тиосульфатного иона из раствора вы-
зывает разложите комплекса.
Комплексные соединения с большим значением кон-
станты нестойкости относят к двойным солям (см. с. 190).
Например, кали ево-алюми пневые к другие квасцы в раство-
ре почти полностью диссоциируют на составляющие их
ионы и воду по реакции
КIАЦЗО^] U2HjP - к+ 4-АР++2S0J- 4- 12HSO
К ним, как и к растворам других сильных электролитов,
закон действующих масс в его обычной форме неприменим.
Образование комплексного соединения из простых ве-
ществ обусловлено уменьшением энергии Гиббса в процесса
комплексообразования - Следовательно, константа нестойко-
сти также связана с энергией Гиббса, т. е. с изменением и
энтальпийного (Д//), и энтропийного (7Д$) факторов.
По количеству теплоты, выделившемуся при реакции комп-
лексообразования, и по изменению энтропии при этом опре-
деляется устойчивость комплексного соединения.
Энтальпийный фактор обусловлен величиной энергии
связи. Если связь между лигандами и комплексообразова-
телем близка к чисто электростатической, то ее энергия
растет с увеличением заряда и с уменьшением радиусов
взаимодействующих частиц. В этом направлении растет
и прочность комплексного соединения. Например, иои Al**
образует более устойчивый комплекс с небольшими Района-
ми, чемсбольшимиСГ-нонамн: (К1|Кт)[д1р*)-< (KukJiaici*)--
Большой однозарядный ион СЮ* проявляет очень малую
склонность к комплексообразованию с металлами.
Если связь между центральным атомом металла и ли-
гандами преимуществен и о ковалентная, то прочность ее
растет с ростом способности лиганда принимать электроны
от металла, т. е. при соединении с лигандами имеющими
свободные d-орбитали, или с лигандами, с л особ н ими отда-
вать электронные пары iia свободные d-орбитали металлов.
Так, прочные комплексы образуют переходные металлы VII,
VIII и I групп, т. с. d-элементы середины больших пери-
200
ЭБ ,:НаучноЕ наследие России"
одов, и анноны или полярные молекулы, склонные к обра-
зованию донорно-акцепторной связи, В ряду
f AgFa]"” I ArCI3 J" - f AgB г,Г - [Agl ,Г
прочность комплешжого лона повышается за счет увели-
чения степени ковалентности связи Ав — Г при переходе
от F- к [“. Легко объяснить и понижение устойчивости комп-
лекса в ряду (Au (CN)SF — [Ag (CN)J' — (Си (CN)J*
(табл. 18).
Энтропийный фактор также играет большую роль в ус-
тойчивости комплексного соединения. С ростом энтропии
протекает процесс комплексообразования, в котором поло-
жительный ион, взаимодействуя с отрицательными лиган-
дами, образует комплекс с более низким зарядом. Напри-
мер, в водном растворе может идти реакция
[Cr(HtO), Р++2С1- = [Ст(Н4О)4 C1J++2Н.Р
Сольватация с понижением заряда ослабляется; в данном
случае изменение энтропии происходит благодаря усилению
неупорядоченности в слабее сольватированной системе.
Энтропийный эффект проявляется также ив случае
замены лигандов, занимающих по одному координационно-
му месту во внутренней сфере, па лиганды, каждый из кото-
рых способен занимать по два и больше мест (хелаты).
Аммиак NHa, например, занимает одно координационное
место в комплексе, а этнленднамнн (en) КНпСНХН^КН. —
два- Несмотря на то что по количеству теплоты, выделяю-
щейся при реакциях комплексообразования, две молекулы
NH3 эквивалентны одной молекуле еп, комплексы этилен-
ди амина значительно устойчивее аналогичных комплексов
аммиака. Так, если
(ст)[NJ IN mid" ₽ 1 ,Я ’ 1 ТО (КMMT)[N1 (en),jrk ”7>8 Iо***.
Это объясняется тем, что процесс
INI 4-Эсп = [Mi (enM^+GNl <а
протекает с увеличением числа частиц, а это ведет к росту
энтропии.
В еще большей степени возрастает прочность комплекса
при замене обычных лигандов на этилен ди аминтетр а аце-
татный ион (ЭДТА), занимающий три или шесть координа-
ционных мест. Это используется в аналитической химии
201
ЭБ ,:Научнов наследие России"
для получения очень прочных комплексных соединений
щелочноземельных, редких и других металлов по реакции
(ЭД Т Ар - + [ Me (н гО)« 1» - - [ МеЭДТЛ |а' -МН20
9. Обменные реакции
Реакции обмена между нонами протекают обычно с боль-
шой скоростью и большей частью без разрушения образо-
вавшихся при растворении ионов.
Несмотря на разнойменноеть зарядов, ионы одного к
того же электролита, благодаря их сольватации, лишь
в некоторой степени снова соединяются между собой. Со-
единившиеся ионы могут вновь распадаться. В равновесной
системе число соединяющихся ионов равно числу образую-
щихся за то же время ионов.
Если прибавляемый к раствору электролит образует
ионы, резко отличающиеся по свойствам от присутствую-
щих, тогда между противоположно заряженными нонами
различных электролитов может произойти взаимодействие.
В этом случае пр отекает химическая реакция и появляется
новое соединение за счет образования прочной связи между
нонами. Оно отличается от исходных тем, что либо является
менее растворимым и удаляется из раствора в виде осадка
или газа, либо в меньшей степени диссоциирует на состав-
ляющие его ионы. Все эти процессы сопровождаются убылью
энергии Гиббса.
В соответствии со сказанным рассмотрим четыре типич-
ных случая обменных реакции в растворах электролитов,
равновесие которых сильно смещено в сторону образования
продуктов реакции.
Образование осадка. При сливании, например, раство-
ров хлорида бария и сульфата натрия образуется осадок
сульфата бария, Молекулярное уравнение этой реакции
запишется так:
BaClg (р)+ Na25O4 (р) =Ва$94 (к)( -f-2NaCI (р)
Сущность этого процесса выражает ионное уравнение
Ва^ (p)+SOj-(p) = BaSQ1 (х)|
(Плохо растворимое вещество, будучи весьма скудным
поставщиком ионов, может быть изображено в молекуляр-
ном виде). Уравнение показывает, что ноны взаимодей-
ствуют между собой как неделимые частицы и что только
202
ЭБ ^Нз^ччое часледие России"
эпг два пола в данной системе образутот молекулы нового
вещества. Поскольку последнее выпадает в осадок и не
участвует в обратной реакции, то и равновесие рассматри-
ваемого процесса сильно смещено вправо, т. е. реакция
практически идет до конца (AGjw == —14,32 ккал/моль).
Образование газообразных (легко летуч их) веществ. При
взаимодействии концентрированных растворов хлорида нат-
рия и серной кислоты выделяется газообразный хлористый
водород:
NaCl-f-H»SO4 = HCIf + NaHSO4 ИЛИ FP+СГ-НСН
Равновесие этой реакции Смещено вправо. Путем Етагрева-
ння можно почти полностью удалить НС) из раствора,
добиваясь значнтелы!ого смещения равновесия. Действи-
тельно, так как в подобного рода процессе AS >0, то в со-
ответствии с уравнением (НЛО) повышение температуры
приведет к еще более отрицательным значениям ДО.
Образование слабых электролитов. Если к раствору
цианида кальция прибавить, например, раствор серной
кислоты, то произойдет обменная реакция
3KCN (pJ + H.50, (p)-2HCN (р)+K>S04 (р)
иди
CN- (р)-ЬЬГ (Р>НСК (р)
Ц.ф] о ».чз*
В результате получается малодиссоцинрующее соеди-
нение (ITCN). Поэтому равновесие сместится почти пол-
ностью вправо (Абия = —12,58 ккал /моль), т. е. реакция
практически дойдет до конца.
Обобщая этот пример, можно сделать вывод, что силь-
ные кислоты вытесняют слабые из растворов их солей
(аналогично сильные оснований вытесняют слабые основа-
ния).
К обменным ионным процессам относятся также реак-
ции нейтрализации, в результате которых образуется сла-
бый электролит—вода. Так, например,
HCI-J-KOHi=HiO+KCl или Н++ОН-<=Н4О
Реакции нейтрализации любых других сильных кислот и
оснований протекают аналогично. Поскольку соль пол-
ностью диссоциирует на ионы, реакция в любом случае
протекает лишь между ионами Н’ и ОН”, т. е. процесс ней-
* Под формулами вещиитв здесь в далее указами значения
ЭБ "Научное наследие России"
тра-тизации и общем виде выражается одним и тем же урзн^
неняем:
Н+ (р)+ОН" {р) = НцО (ж); Д<4Й1—— 39J03 ккал/наль
Образование комплексных волов. Часто ноны различных
электролитов, соединяясь между собой или с молекулами
растворителя, образуют новые, более сложные комплекс-
ные ноны. Например:
1 *еа т (р) + 4CN- (р) = I Hg(CK}4 |i- (р)1 AG;M => — 53t 16 ккал/моль
или
Zn2h (р) + 4\На (rJ^[Zn(NI JjhP*" (p)i —2116® ХКйл/мойь
Комплексные ионы обычно слабо диссоциируют и поэтому
реакция протекает практически необратимо’ в сторону их
образования (см. также § 8).
Из рассмотренных примеров можно заключить, что
обменные реакции в растворах электролитов являются
обычно реакциями между попами. Реакции обмена в раство^
рах электролитов практически идут необратимо и до конца
r тех случаях, когда в качестве продуктов получаются либо
малорастворимые вещества (осадки и газы), либо мал од нс-
сониирующие соединения (слабые электролиты или комп-
лексные ионы) (правило SepmowX
Часто приходится сталкнвлтъся с процесса ин. d которых реали-
зуется не одни из четырех рзссмотреннУХ типов обмен elms реакций
а та или иная комби ня ни я их. Так, многие обменное реакции в раство-
рах электролитов сопронождяются одновременным образованием и
малорястворнмых веществ, и слабых электролитов. Если в процессе
NajS (p) + HCf{p)₽2NaCl (рЯ-На5 (г)]
образуется мал «раствор имев газообразное мщество (H^S), являю-
щееся однойремеиио и очень слабым электролитом (Хн с = ЦОЛО"1),
то в реакции
Ba(OH)s (р}4 HgSOj (р) = BaSOd (k) + 2HsO (ж)
ИЛИ
Ва=+ -I-ЗОН - + 2Н "+5О*- « BaSOj [ + 2Н4О
одиодрсмспЕю образуются и трудиорастЕорииыс, и малсщнссоцпирую-
щие вещества.
В леки гор их реакциях все иоееы участвуют в образовании различ-
ных осадков:
CuSOj (p)+BaS (p)=±CuS (k)J +BaSOj (k)J
Cu^+SO|~ + Ba^+S’" = CleS J + BaSOj |
Во всех подобных случаях равновесие сильно cmlweeo в сторону нрн-
яой реакции. Действительно* в первом из этих примеров ==
=" —52,526 ккал, а во втором Лб^>$ ™ — 63,2 ккал.
ЭБ ' Е^^чсе наследие России"
Некоторые практически необратимые ионные реакции
сопровождаются образованием непрочных соединений, рас-
падающихся па малодиссоцинрующие или мало раствори-
мые вещества, пли на те и другие одновременно, например
.SO, (г)
Ka^SOj (р)-Ь2НС1 (p) = 2NaCl (р) 4- HsSOa (р)<
*Н4О (ж)
или
SO? (Р)+2Н+ (p) = SOs ф+Н^О (ж)
В данном случае образуются очень слабый электролит и
газ. Оба продукта смещают равновесие вправо (Дб»в =
= —J2JJ5 ккал). Повышение температуры будет способ'
ствовать течению этой реакции (Д5 >0).
Часто встречаются процессы, при которых слабые элект-
ролиты или малорастворимые соединении имеются и в числе
исходных веществ, и в числе конечных продуктов реакции.
Например, в процессах
HCN’ (р)+СНаСОО- (р)^СН^ООН (p)+CN- (р)
и
КН4ОН (р)+Н+ (р)^Н/> W + NH+ (р)
слабые электролиты есть и в левой, и в правой частях урав-
нений реакций.
Равновесие обратимого процесса в этих случаях сме-
щается в сторону образования вещества, обладающего
меньшей константой диссоциации. В первой реакции рав-
новесие смещено влево (Кисы = 4,9* ИГ1’; Кен,союн =
= 1,8-1(Г4), во второй сильно сдвинуто вправо (Кн^о =
— 1т8*10гМ; Knh.oh = 1,8-КГ*). Это’ отвечает значениям
AGjas = -”10,29 н ^—20 ккал соответственно для первой
и второй реакций.
Примерами процессов, в которых слева и справа имеются
труднорастворимые вещества, могут служить реакции
AgCl (к)1 H-Nal (р) Agi (к)Ц-МаС1 (р)
И
BaCOj (к)| -|-N3jSO4 (р) д: BaSO4 (кД -t-Na/X^
Равновесие в таких системах смещается в сторону образо-
вания менее растворимого соединения. В первой реакции
оно смещено вправо, так как иРдР[<С ПРд^; здесь
= —13.7 ккал. Во втором процессе равновесие лишь
несколько сдвинуто d сторону образования BaSOi
205
ЭБ ,:Научнов наследие России"
(ПРваса, ™4,9- IO"’; np^so, =1,08- КГ1»); здесь
Нередко встречаются процессы, в уравнениях которых
с одной стороны равенства стоит труднор ас твори мое соеди-
нение, а с другой — слабый электролит. Так, равновесие
в системе
AgCN (к)| +НЧ?) - HCN (Р) (Р)
значительно смещено вправо, поскольку ион CN' более
прочно связывается в молекулу очень слабого электролита,
чем в молекулу труднорастворимого вещества, Поэтому
□садок цианида серебра растворяется прп добавлении азот-
ной кислоты,
Аналогичной причиной обусловливается растворение
гидроксидов многих металлов я присутствии кислот. Так,
например, гидроксид магния полностью растворяется при
прибавлении к раствору азотной кислоты:
MgfOHh (х)| (р}-2Н,0 (ж) +Mg«- (р)
Здесь ионы ОН- связываются в малодиссоциирующне мо-
лекулы волы полнее, чем в гидроксид магния/Это видно
из сопоставления следующих величин:
пр5ионь=5>5 ^№ = ю-ч
Действительно, в данном случае Аб^ч = —22,966 ккал.
Подоб! 1ы ми же п р ич и ням11 обусл о вл е но р аствор е ни е
амфотерных гидроксидов в растворах щелочей. Например,
в процессе
Вс (ОН)В [ + ЮН- = [ Be з-
введение ионов ОН" среды способствует образованию ма-
л одиссоци и р у юти х гид роксоко м гтл ексо в; р з вн овес не с м е-
щается вправо.
Вышеприведенные примеры объясняют причину раство-
рения в кислотах и щелочах многих вещесгвт которые в воде
практически нерастворимы- Они объясняют также влияние
среды на направление процесса.
Некоторые труднорастворимые соли многоосновных кис-
лот растворяются в присутствии избытка этой же кислоты
благодаря образованию малодиссоциирующсго кислого а НИ’
она, например:
СаСО3 (к)| + Н* (р}=Ся^ (р)+НСО, (р)
Эта реакция (AG^ = —2,58 ккал) обусловливает наличие
временной жесткости в природных водах.
ЗБ Ж™ наследие России*1
Многие хорошо растворимые комплексные соединения
можно разрушить действием других электролитов или
растворителя, если в результате этого образуются мало-
растворимые осадки или новые комплексные ионы, диссо-
циация которых меньше диссоциации комплексного ионе
исходного вещества. Например, прибавлением к раствору
lAg(CN)ar сульфида натрия можно полностью разрушить
комплексное соединение, осаждая нон серебра в виде труд
норастворимоги осадка Ag4S. Эго происходят благодаря
тому, что произведение растворимости Ag2S значительно
меньше константы нестойкости комплексного иона (ПРДй5 =
= 4,0-КГи, (Klltf'r)[AS(CNi,]- — 8-10“!Э*). При образовании
осадка ионы серебра более полно удаляются из раствора,
чем при связывании их в комплексный ион.
Действием сильных кислот можно разрушить аммиакаты,
так как молекулы аммиака связываются водородными
ионами в прочный нон аммония. Например, HNO., разру-
шает комплексное соединение (Ag(NH9)zl Cl, связывая ам-
миак по реакции
NHHpH Н+ (p)=nii; Ср)
(AGJ^ = —12,6 ккал/модь); этому способствует также од
повременное осаждение ионов серебра в виде AgCl.
Разрушение комплекса может произойти н в том слу-
чае, если к раствору комплексного соединения прибавить
вещество, которое с комплексообразователем образует более
прочный коп. Например:
12п(МН5)(]^(р>-|-4СМ- (p)-[Zn(CN)4p- (PH4NH, (г)|
5,1 ккал)
Образующийся цнаянд-нон значительно прочнее аммиа-
ката, так как сила поля лиганда, а поэтому и энергия рас-
щепления для CN’ больше, чем для МНЭ; этому отвечает н
различие в значениях констант нестойкости:
(^netr)[Zn(M H,)JI =М - 10 (^„KT)[ZnfCN)d«- " 1.3 - 10
Аналогичное превращение одного комплексного лона в дру-
гой происходит и при попадании комплексного соединения
в растворитель, молекулы которого образуют с комплексо-
оСразователем более прочную связь, чем связь лигандов
• Только при большой диспропорция значений IIP и можно
сделать бесспорное заключение, так как это — раз личные лара мери-
стики и поэтому неерланкные мньду собой.
207
ЭБ ,:Н-аучноЕ наследие России"
с центральным ИОНОМ растворенного комплекса. Так, при
растворении Си [(HjO)J SOj-HjO в жидком аммиаке обра-
зуется ион (Си (NHs)4|!*.
10. Гидролиз
Обменные реакции между растворителем и растворен-
ным веществом, которые обычно протекают с образованием
слабо диссоциирующих или труднорастворимых веществ,
представляют особый интерес. Поэтому мы посвятим им
отдельный раздел. Реакции взаимодействия между состав-
ными частями растворителя и растворенного вещества
называются сольволизом (для воды — гидролизом) *.
Гидролизу могут подвергаться химические соединена я
различных классов: соли, углеводы, белки, эфиры, жиры
нт, д, В неорганической химии чаще всего приходится иметь
дело с гидролизом солей, т, е. с обменным взаимодействием
ионов соли с молекулами воды, в результате которого сме-
щается равновесие электролитической диссоциации воды.
Г ।! д р ол 1! з — р ез у л ьта т поляризационного вз а имод е ЙСТ-
вия ионов соли с их гидратной оболочкой. Чем значитель-
нее это взаимодействие, тем интенсивнее протекает гидро-
лиз. Для ориентировки можно воспользоваться следующим
упрощенным ходом рассуждений.
Катионы связаны с молекулами воды донор ио-акцеп-
тор ной связью; донором являются атомы кислорода, имею-
щие две свободные электронные пары, акцептором — ка-
тионы, имеющие свободные электронные ячейки. Чем боль-
ше заряд иона и чем меньше его размер, тем значительнее
будет «катионная доля» поляризующего действия Кл*
па На0. Аииое1Ы связаны с молекулами воды водородной
связью. Сильное влияние может привести к полному отрыву
прото на — водородная связь становится ковалентной. До-
норная активность Ал' будет тем значительнее, чем больше п
и меньше /?д-, В зависимости пт силы поляризующего
влияния К'1* и Ал’ па молекулы Н30 будут получаться раз-
личные результаты. Так, катионы элементов побочных
подгрупп и непосредственно следующих за ними элементов
подвергаются более интенсивному гидролизу, чем другие
ноны одинаковых С ними Заряда и радиуса, так как ядра
первых менее эффективно экранируются rf-электронамн.
Обычно различают четыре случая действия воды па соли.
• о №П1>велнзе см. гл, V.
ЗЕ ручное наследие России"
i. Если катионы и анионы имеют небольшие заряды и
значительные размеры, то ИХ поляризующее влияние па
молекулы воды невелико, Т. е. взаимодействия соли с Н/)
практически не происходит, Это относите я к таким катио-
нам, как К+ и Са,+, и к таким аннонам, какС!’ и NOS. Иными
словами, соли сильного основания it сильной кислоты
гидролизу не подвергаются. В этом случае равновесие дис-
социации воды в присутствии ионов соли почти не смещается.
Поэтому pH =7, т. е. растворы таких солей практически
нейтральны.
2. Если катионы являются слабыми акцепторами элек-
тронов, но анионами служат части цы, обладающие значи-
тельной донорной активностью (S*~. СОТ, SOj ), то проис-
ходит гидролиз по аниону. Примером будет процесс взаимо-
действия NaiCOj с Н2О:
СОГ+НОН=НСО;Ч-ОН-
сопровождающийся образованием слабоднссоциирующего
бикарбонат-нона. Обобщая этот пример на подобные ему,
приходим к выводу, что гидролизу по аннону подвержены
соли сильного основания и слабой кислоты (СНаСООК.
KCN, NaaPO, и т. д.). Так как в растворе образуется избы-
ток гидроксид-попов, то раствор будет иметь щелочную
реакцию (pH > 7). Чем больше поляризующее влияние
анионов, тем сильнее гидролиз. В соответствии с законом
действующих масс это означает, что гидролиз будет тем
интенсивнее, чем слабее кислота.
3. Если, наоборот, анионы — слабые доноры электро-
нов, а катионы обдадаЕот значительным поляризующим
действием (Zn11’, Cu2+ и др.), то взаимодействие обусловлено
ьтияннем на молекулы НгО катионов, т, е. происходит
гидролиз по катиону. Примером служит процесс
Си** + НОН - [Си (ОН)]*+11+
Гидролнз обусловлен образованием плохо диссоциирующих
частиц СиОН*. В результате равновесие электролитической
диссоциации воды смещается и в растворе появляется из-
быток водородных ионов; поэтому реакция среды кислая
(pH с 7). Очевидно, чем слабее основание, тем полнее
будет гидролиз, т. е. гидролизу по катиону подвержена
соль слабого основании и сильной кислоты (NH4Br, ZnCl2
и т. д.).
В ряде случаев при гидролизе солей, образованных
слабым основанием и сильной кислотой, получаются оксо-
£09
ЭБ ,:Научнов наследие России"
соли, например, для Э = Sb, Bi:
ЭСЬ + Нго «ЭОСЦ- 2НС1
В результате гидролиза образуются плохо растворимые
в воде хлориды антимонила (SbO’) и висмутила (BiO+); так,
для SbCla вместо
SbClj (K)-f-3H/> (3K)=Sb (ОН)Я (к) + ЗНС1 (р)
протекает реакция
St>CI, (к)+ HtO (ж)=5ЬОС1 (к) + 2НС1 (р)
Действительно, если для первого процесса =
= 1,7 ккал, то для второго — 9,0 ккал сказывается проч-
ность ионов SbO‘: (4GiW)sbo^P) — —42 ккал/моль. Обра-
зование оксосолей при гидролизе, обусловленное прочно-
стью связи Э—О, характерно также для элементов подгруп-
пы титана,
4, Если и катионы, и анионы обладают умеренным по-
ляризующим действием, то в процессе обменного разло-
жен ня воды участвуют п те и другие. Происходит гидролиз
по катиону и аниону. Примером служит процесс
NHt-+CHjCOO-4- НОН = NH(OH 4- СН/ХЮН
Б зависимости от соотношения констант диссоциации обра-
зующихся при гидролизе кислоты и основания растворы
солей этого типа могут иметь слабо кислую или слабощелоч-
ную реакцию, т. с. pH близко к 7. Этот тип гидролиза ха-
рактерен для солей, абразива иных слабым основанием и
слабой кислотой.
5. Если взаимодействуют с водой вещества типа SiCI«,
CSj, РВг6 и т. д., т. е. отличающиеся тем, что молекулы
содержат частицы с большим поляризующим влиянием
J 1 +1
(Si „ Р и т. д.), то в отличие от всех ранее рас€.мотрс1шых
случаев при гидролизе образуются две кислоты — это
л р । [ зн ан гидр (Xi । [ за га логе в а н г и др и д ов и те i оз н г и др и д ов.
Само название последних свидетельствует об особенностях
процесса и его продуктах, Вот три примера;
j
ВCl?-] ЗНаО= HyBOg + ЗНС1; CSg -^3^0 = J- 2HjS
и
WCU-I 4НИО = Н^ТОН’6НС!
210
ЭБ ,:НаучноЕ наследие России"
К рассмотренным процессам можно было бы присоеди-
нить реакции вида
Ci» + HtO = нбю+нб и X eFeH- нЗЬ— X е ч- VA -f- н F
отличающиеся от предыдущих тем, что их протекание со-
провождаегся изменением степени окисления.
Следует подчеркнуть, что, связывая гидролиз с поляри-
зующим влиянием ионов сми^ с акцепторной способно-
стью катионов и донорной способностью анионов. мы имеем
в негду лишь качественную сторону явления. К количеств
венным или полу количественным результатам такой под-
ход принести пока Fte может Инос дело рассмотрение гид-
рсмтцта с энергетических позиций. Так, для реакций
и
РС1а (жЬНЗН^О (ъ) = НзРОл (р)-?-ЗНС1 (р)
АчС13 (ж) + ЗН2О (ж)-НаЛ&О3 (р) -р ЗНС1 fp}
получаем А(7^Р1 равньге соответственно—66,5 и —1217 ккал,
что отвечает большей необратимости первой из этих про-
цессов. Другими словами, гидролиз в ряду PCL-AsCl*—
—SbCl3 будет ослабе-
вать; то же заключение
относится^ например, и
к ряду GeClj-—SnCIB—
- РЬС1г
Закономерность в
интенсивности гидроли-
за и положении элемен-
тов в периодической си-
стеме видна 1тз следую-
щего npirMCpa: для
процессов
Рнс. 7L Зависимость pH водный
растворов перхлоратов элементов
II группы от порядкового намеря
элемента ? при m « I и I — 25 aL
Г4 + НаО (*) = НГО (р) +
+ НГ (р)
AGjM -6,16 (С1Д 13.15
(Br,) и 20,63 ккал/моль
(1Дт.е. равновесие взаимодействияГав рядуCL—Вг2—1а с
водой все более смещается влево, И еще один пример. Как
видно из рис. 71, гидролиз в ряду Be(C10q)i—В а (С Ю.|)2 ослаб-
ляется, а длч перхлоратов элементов подгруппы цинка
он не только сильнее^ но и меняется немонотонно. Анало-
гичный ход по подгруппам и мест сумма двух первых иониза-
ционетых потенциалов; следователыю+ здесь проявляется
211
ЭБ ,:Научнов наследие России"
вторичная периодичность, Чем больше энергия взаимодей-
ствия между гидратируемым ионом и молекулами воды,
тем больше деформация электронных, оболочек молекул
воды, что создает больше возможностей увеличения кис-
лотности раствора.
Ступенчатый гидролиз. Если соль содержит однозаряд-
ный катион (анион) и многозарядный анион (катион), то
возможен ступенчатый гидролиз. Так, образующиеся в ре-
зультате первой ступени гидролиза по аниону кислые соли
могут подвергаться дальнейшему взаимодействию с водой.
Однако вторая и последующие ступени гидролиза выражены
менее сильно. Это обусловлено уменьшением константы
диссоциации при переходе от К] к К2, от К, к Кз и т. д.
(см. с. 189). Так, поскольку ион НСОЛ ди ссоцип руст слабее,
чем Н?СОз (К'г< Ки см. табл. 15), то он и образуется в пер-
вую очередь. В качестве другого примера укажем на гидро-
лиз фосфата натрия:
NajPO* (р)+Н,О (») = Na1HPO4 (p)-f-NaOE-i (р)
РОЛ (р)+ НОН (ж)=11РОГ (р)+ОН - (р): К1 - 2,9 IQ-»
н РОГ (р) + нон (ж) = HJ4) Г (р) -|- ОН- (р); кг - Ь.З I о *
HjPO; (р) + НОН (ж) = Hi?04 (р) 4- ОН- (р); К, ~ 1,4 !©-»•«
где Ki, Кг и «з — константы гидролиза. Таким образом,
гидролизом солен слабых многоосновных кислот по второй
и третьей ступеням можно пренебречь
Процесс ступенчатого гидролиза соли слабого основания
и слабой кислоты, образованной многозарядным катионом
и однозарядным анионом, может иллюстрироваться реак-
циями
AI (СНзСОО)з + Нг0 - А1 (ОН) (CH/lOO)-. 4-СНаСООЦ
или
А1 (ОН) (СНяСОО).; + Н£О — А1 (О! 1)3 C! iaOOO + СН^СООД
Степень гидролиза. Количественно гидролиз характери-
зуется степенью гидролиза а, н константой гидролиза Кс.
Первая величина представляет собой отношение числа мо-
лекул, подвергшихся гидролизу, к общему числу молекул,
т. е.
вг^Сх/С, (IV. 40)
где Сг — концентрация гид рол изованной части вещества;
С—общая ковце[1трааия растворенного вещества.
В растворах умеренных концентраций степень гидро-
лиза при комнаткой температуре невелика; для солей, обра-
зованных сильным основанием и сильной кислотой, она
212
ЭБ ,:Научнов наследие России"
практически равна нулю; для солей, образованных
слабым основанкем и сильной кислотой или сильным
основанием я слабой кислотой, опа составляет около I %.
Для солей, образованных слабым основанием и слабой
кислотой, степень гидролиза может приближаться к J00 %,
а для галогенангмдр идо в достигает 100 %, Так, дли NH4C1
при концентрации в 0,01 М<хг = 0,01 %; для 0,1 и. раствора
CH3COONI1| ~ 0,5 %; для растворов и (NH^S
она равна соответствен!го 77 и 99 %.
Если в результате гидролиза образуется осадок или га-
зообразный продукт, т, е< одно из веществ удаляется из
сферы реакции, гидролиз может происходить практически
необратимо. Поэтому при взаимодействии солей Al3*, Сга*
и Рея* с растворами сульфидов и карбонатов в осадок вы-
падают не сульфиды и карбонаты этих катионов, а соот-
ветствующие гидроксиды:
2А1С1а (K)+3Na,S (x} + 6HsO (ж) =
« 2АI (ОН)Я (к) | + SHjS {ф -К 6NaCI (к)
2CiCla (к) + ЗМааСОа (кЦ-ЗН3О (ж}—
= 2Сг(ОН), (к)Ц-ЗСОа (r)jH-GNaCJ {к)
Константа гидролиза. Реакцию гидролиза для соли КА
в общем виде МОЖНО представить уравнением
KA + HsO—KOH-|-liA
Константа равновесия этой реакции
(КОН] [НА!
л [КАцН^Г
Так хак [Н/Э) в разбавленных растворах можно считать
величиной постоянной, то и произведение К (Н4ОI также
будет постоянным. Эту величину называют аонстантаИ
гидролиза. Таким Образом,
. [КОН] |JJA]
В случае гидролиза по аннону (например, для KCN) это
уравнение примет вид
[OH-JIHA] (НА]К„ ка,
к'”—С*Т— "л" ^’Тл-ТНТ = зд
обобщая, подучаем
— Лц.//(ннслг
(1V.41J
а 13
ЭБ ,:Научнов наследие России"
Аналогично для гидролиза по катиону (например, для
МВД)
(I V42)
Для гидролиза к по катиону, и по аниону, очевидно,
^Сг—^wWkhcji^wu)- (FV.43)
Эти уравнения служат количественным подтверждением
сделанного ранее вывода: чем слабее кислота (основание),
Рис. 72, Зависимость степени гид-
ролиза ctr водного раствора соды
От разбавления V (л/моль) при
/ = 20 X
жден нем этого обстоятельства
сол ь к оторой (которого)
по двор гаетс я гидр од i t зу,
тем полнее он протекает.
Влияние условий на
степень полноты гидролиза.
С уменьшением концсЕгтрЭ’
и и и раство р ов гид рол и з
усиливается. Рост степени
гидролиза с разбавлением
на примере процесса
Na±COj(p) + НОН (ж} =i
= NaHO0rfp) + NaOH{p)
нлл юстрируется рис. 72. Эк-
с и ер нм ен тал ы еы м подтвер-
может служить помутнение
растворов некоторых солей (например, нитрата висмута) ио
мере их разведения. Прибавление воды приводит к пере-
ходу в плохо растворимый нитрат висмутнла.
Поскольку при обратимом гидролизе устанавливается
динамическое равновесие, то в соответствии с законом дей-
ствующих масс можно сместить это равновесие в ту или
иную сторону за счет введения в раствор кислоты или
основан я. Этим часто пользуются для усиления или по-
давления процесса гидролиза/в частности, с целью поддер-
жания постоя Е]ной концентрации ионов водорода в растворе.
Прибавление кислоты вызывает усиление гидролиза по анио-
ну (смещение равЕ]овеспя вправо в результате связывания
ионов в воду); гидролиз по катиону усиливается в прп*
сутстшЕн щелочи. Так, если к раствору буры NaMB4O, до-
бавить кислоту, т. е. связать гидроксид-ионы, то гидролиз
Na, В A 4-7HaO^4H9BOs+2NaOH
усилится; если же добавить основание, то равновесие вдви-
нется влево — гидролиз будет подамеи*
214
ЭБ 'Научнов наследие России"
Константа диссоциации воды увеличивается с темпера-
турой е большей степени, чем константы диссоциации про-
дуктов гидролиза слабых кислот н оснований. Поэтому при
нагрева инн степень гид-
ролиза возрастает. К
этому выводу легко
прийти и иначе: так как
реакция нейтрализации
экзотермична, то гидро-
лиз, будучи противопо-
ложным ей процессом,
э н дотер мочен; поэтом у
в соответствии с прин-
ципом Ле Шателье на-
гревание вызывает уси-
ление гидролиза. На
примере процесса
Рис. 73. Зя и ценность степени гидроли-
3aaL водного рй атвора [Сг (НлО\]С1д о г
температуры i (°C)
forfH/Jh |CWp ) -f- нон (ж> - (OHJ JCI/P)+на (р)
влияние температуры на гидролиз иллюстрируется рис. 73.
Нагревание вызывает уменьшение влияния комплексо-
образования на гидролиз, так как константа нестойкости
комплексов с повышен и ем температуры увеличивается.
Процесс гидролиза солей в ряде случаев может быть
очень СЛОЖНЫМ. Поэтому простые уравнения реакции гид-
ролиза в общепринятой записи являются в общем случае
условными. Решить вопрос о продуктах гидролиза можно
лишь на основании их аналитического исследования. Уста-
новлено, что очень часто в тех случаях, когда гидролизу
подвергаются комплексные соединения (или гидратирован-
ные ноны), продуктами гидролиза могут быть многоядер -
ные комплексы. Так, если в растворах Hgs* есть только
одноядерные комплексы, то в растворах FeV помимо комп-
лексов |FeQH]2+ н [Fc(OH)xl* содержится двухъядерный
комплекс (Fe^OHjJ1*; в растворах BeSr в ос нов ei ом обра-
зуются мЕюгоядерпые комплексы состава [ Ве,(ОН)3 F*; в
растворах Sn1+ образуются комплексные ноны [Sn1(OH)JFr,
[Snt(OH)sF*, SnOH+; в растворах BiJ* наряду с 1ВЮН12+
находятся комплексные ионы состава [В!((ОЙ)1г1с+.
Рёзищнн гидролиза h привюДнщне к обрз зойй илю полияд^риых
«омплексов, е общем виде можно преДстлэлть урлвнениел1
тМ? > Ч-пНр=“+лН+
213
ЭБ ,:Научнов наследие России"
где кя колеблется от ! да 9Р а п может принимать значения от I до 15.
Проникание такого рода реакций а настоящее дрен л установлено бтсс
чем дитя 3D катионов Чем сильнее гидролиз тем нероятнес образова-
ние поли ядерник гидрааксоком^лснсон Было найдено, что каждому
Заряду а большинетие случаев отвечает олредс псинаяформа комплекса.
Тдх, для ионол Me-* характерна форма димеров [Мсг (01Т)!^. для ионов
Me**—IMeJOHJJ" а для - форма |.Мев (ОН)9|Ь+ и более слож-
ные, напрнмёр [Zrj (OH)Jb+,
При высоки к температура к и больших значениях pli образуются
и оксокомплекси
2МеОН = МсОМй+НйО
или
Н Н
ч /\ Z(\ у
>Ме<^Ме/ ^-Ме-О-Ме-О-Ме— + пНаО
Н И
Структурно полнядерныс комплексы можно ирелегапнть в виде октз-
эдрак, сосд и пенным между собой по вершине (fr = ^)г ребру (3) нлн
грани (4) посредством различных мостиков (О. ОН н др.)
Сложений состав имеют и продукты гидролиза карбона-
тов ряда металлов. Так, прзк взаимодействии растворимых
солей jMg2"» CuE+i Znz\ РЬ,+ и т, д. с раствором соды обра-
зуются ire средине карбонаты, а соответствующие гид-
р оксо карбонату например Mg^tOH^COj, Ct^OH^CQg,
Zns(OH)s(CO3hP РЬэ(ОН)2(СО3)й В частности, можно назвать
реакцию
2-VigSOj + 2Na?C0^ + Н20 = М^ОН^СО, -f- 2NajSO4 + CO.f
Наиболее распространенным случаем гидролиза но ка-
тиону является гидролиз'солей, образованных многозаряд-
ным катионом и оДЕЮэарядиым анионом, например А1С13.
В растворах этих солей наименее диссоциированное сое-
дннеяие образуется за счет присоединения первого гидрою
сид-нона к иону металла. Необходимо учитывать, что ион
А1Эн в растворе гидратирован. Поэтому первая стадия гид-
ролиза выражается уравнением
f АI (НаО)аГн + НОН == (AI(HsO)sOH j«+ + НаСГ
При обычной температуре гидролиз этих солей практически
ограничивается этой стадиен. При нагревании, а также при
разбавлении раствора происходит гидролиз по второй сту-
пени с образованием одиозамещенных основных солей:
[ АЦНьОЪОН ] = •*+ На0= [ АКН^ЫОНЬГ +11дО+
2J6
ЭБ 'Научнов наследие России"
Такны образом, кислая реакция водного раствора соли
объясняется тем, что гидратированный катион теряет про-
тон и акватрупла превращается в гидроксогруппу. В рас-
смотрен еюм случае могут образоваться и более сложные
комплексы н например [AijOH)jb)l5*+ а также комплексные
ионы BJt.ua [А1О(ОН)ЛР" и [AiOa(OH)J®'1 что связано с боль-
шим сродством алюминия к кислороду.
В случае гидролиза аммиакатов кислотность среды можно
объяснить течением процесса
(Co(NHs)Bp'+НиО-= [Co(Nf UNHtp+ -f- Н/Г
Глава V
ТЕОРИЯ КИСЛОТ И ОСНОВАВ Ш)
Теория кислот и оснований уже в течение [{ескольких
столетий является одним из важЕюГииих разделов химии.
Существовавшие в этой области представления за послед-
ние десятилетия подверглись основательному пересмотру
в связи с успехами многостороннего исследования неводных
растворов и развитием химии комплексных соединений^
Здесь возникли новые понятия и теории, которые в настоя-
щее время стали неотъемлемой частью химической науки.
L Краткая история вопроса
Первой стала известна людям уксусная кислота, которая обра-
зуется при скисании вина. Этот процесс был им знаком Задолго до
начала нашей Эры. Более тысячи лет винный уксус Оставался един-
ственной известной кислотой. Эго получило Отражен не в терминоло-
гии — 11о латыни acehim — уксус, acitiiLm — кислота.
Также и глубине веков теряется открытие иешестр, имеющих
в растн&ре щелочную реакцию, — соаы NajCQj и ногата К/ЗОа. Сода
иногда встречается а природе (содешые озера), поташ содержится и золе
растений.
Серную, соляную и азопгую кислоты получили алхимики прибли-
зительно в VII в. нашей эры: точную дату получения этнк псгцеста
устаЕговить не удается. Арайс кие алхимнкп научились повышать ед-
кость иоташа н седы, добавляя к их растворам известь:
+ Са(ОН)4 = СаСО^ I + ЙМеО1- [
Так были приготовлены шелочщ вернее их растворы. В твердом виде
NaOlI и КОН были выделены только в 1792 г. петербургские акад.
Т. Ловицем. Алхиинкн ввели понятие <солъ>, которое играло важ-
ную роль я их представлениях. Oleh называли солью всякое твердое
г растворимое вещество.
Представления о взаимных отношениях кислот, оснований и солей
сформировались н XVH —XVIII вв.
21?
ЭБ ,:Научнов наследие России1
В середине XVII в, ЙлЛль отмстил, что Асе кислоты имеют ряд
общих свойств, в том числе способность энергично растворить раз-
личные естества н изменять цпет не которых растите,!ьпых красок.
Органические соединения, нзыспянщие спою окраску от действия ямс-
лог el ос копаний h применяются и в настоящее время в качестве инди-
Каторон (см с. 194—IU5).
Вполне четкая формулировка понятий кислота^ основание*
и <солъ» была лака в работах франиузского химика Румя (1714). Ок
предложил называть солью любое вещество, сбра довейте соединен) ем
минеральной нли oprai । и час кой кислоты с bcvlcvtbom, которое служит
основанием (base) и сообщает соли твердость. Рузль ввел понятия
передняя соль» и «кислая соль».
Пример его с этого времени теория кислот м оси&наций ста копится
одним из кардинальных вопросов хи при ческой науки.
В (778 г. Лавуазю была выдвинута лиглорейяпл пто/?ия лшыот.
Установив, что в состав серной, фосфорной> азотной, угольной
м других имел от входит кислород. Лавуазье стал считать, что кислород
является обязательной составной частью мсек кислот, что общие сной-
ства кислот обусловлены наличием в них кислорода (отсюда кислород
в получил спи илэлангге).
Лавуазье геол а га л, что все Кислоты состоят из двух простых, дэлге
неразложимых веществ — кислорода и кислотного ради калл ь Так.
на призер, сер нал кислота, согласно его взглядам, состоит из кисло-
рода и серы (в то время не было еще установлено различие между кис-
лотой н ее аигидридом) •.
Теория Лавуазье объяснила большое число реакций образования
кислот и солей. 'Она получила широкую поддержку со сторо?гы имин-
ков того времени н интенсивно разрабатывалась в течение нескольких
Десятилетий.
Одни ко эта теории сразу же встретилась с затруднениями. Три,
в соляной кислоте нс удалось' обнаружить кислород. Лавуазье счи-
тал, что со временем это будет сделано. Он предположил, что соля-
ная кислота является кислородным соединением некоторого ради-
кала. названного им му рисы (mnria — старинное латинское название
поваренной соли). Тем не менее тщательные исследования состава
синильной н сероводородном кислот, выполненные Бертолле. и даль*
ней шее исследование состава _ соляной кислоты, проведенное Гей*
Люссакон и Тенарсм (Франни*) п Дяш (Англия), понтли, что кис*
лорол в этих веществах не содержится. То же самсе было усынов-
лен о Л>'|я фтор иодородпой, иодводородилй я бромиодородпой кислот.
Эти факты Eta холились d непреодолимом противоречии с кнелородиой
теорией Лавуазье, Кроне того, зга теория не объясняла, почему оксиды
металлов, которые тоже содержат кислород, обладают не' кис л or
ееыыи4 а основными свойствами.
В 1814 г. Дэви обнаружил! что соединение исцп с кислородом
(теперь эти соединение называется йодноватый ангидрид) проявляет
кислотные свойств? только при присоединении: к нему полы. При заме-
щении и образовавшемся емдннеиин водорода на металл получались
ооли. Так впервые была установлена разница между ангидридом и кис-
лотой. На основании результатов этого исследования Дэви л ре д поло-
* Отголоском этих представлений является термин «углекислота»,
который и сейчас иногда употребляется для обозначения двуокиси
углерода СОд.
218
ЭБ ,:Научнов наследие России"
}нилг что носителем кислотны к соойстп является тге кислород! а водо-
род тЬог вам яд был риэвпт н обоснован на большом Э№юрндентя ль-
ном материале- Дюлочгом и осилен ио Лнбвдим Кислородную теорию
яамегЕнла теория жаглолг
Либиху принадлежит определение кислотыh которое и сейчас
иожш) встретить в учебниках: «kfimots — водородное соединение,,
а котором водород может быть snIkhcii на металл* Это определение
нс отряжает наиболее существенною свойства кислот, благодаря кото-
рому эти ишдестяа были пи делены в особый класс соединений, — спо-
собности вступать в реакцию нейтрализации с основаниями. Кроме
того, согласно этому определению, к кислотам следует при числить
такие веыестед, кяк уммияк> ко горы» легко реагирует со щеточными
в иьмочиоземсл иными металлам щ ла при мер:
2Na (k)-|-2NH4 (r) = £NaNH3 (кЦ-На (г)
ЯМ11Д
lijiTphtf
(д,пя этой реакции dGJ(( = —20,2 ккал),
В 1887 г. Аррениусом была предложена теория электро*
литической диссоциации (см. гл. Ill, §2), которая по-новому
решила вопрос о Природе кислот и оснований. Согласно этой
теории кислота — это вещестио, дпссиипнруютее в раст-
воре с образованием копив Н\ Все общие свойства кислот —
кислый вкус, действие на металлы, индикаторы и т, и, яв-
ляются свойствами ионов водорода. Основание — это веще-
ство, диссоциирующее с образованием ионов ОН". Реакция
нейтрализации сводится к взаимодействию водородных и
гидроксид-ионов, приводящему К образованию недиссоции-
ровалпых молекул воды.
Однако несмотря на то что теория электролитической дис-
социапин полностью подтвердилась, представления о кисло-
тах н основаниях, возникшие в начале развития этой теории,
не удовлетворяют современному состоянию химической па-
уки по следующим соображениям:
1, Кислоты и основания могут реагировать между собой -
и не будучи диссоциированы на иолы. Например, газообраЭ’
Ныл хлористый водород легко реагируете твердой щелочью.
То же относится к изменению окраски индикаторов. Напри-
мер, хлористый водород, растворенный в бензоле, совер-
шенно не диссоциирует на ионы (для бензола в = 2,3);
однако если к такому раствору добавить индикатор, он пока-
жет кислую реакцию.
2. Известно много реакций образования солей, анало-
гичных реакции нейтрализации, однако Происходящих без
участия ионов Н* или ОН" Простейшим примером является
NHa (r)-|-HCl (*). AG';d( = —2!,И кмл/чоль
219
ЭБ ,:Научнов наследие России"
Аммиак, играющий в этой реакции роль основания, не со-
держит группы ОН*.
Аналогично аммиаку реагирует его органическое произ-
водное триметилам и н:
(CH,bN + НС1 - [ (€ H3)3N Н ]С1
хяорнд
трагмети лэм-
чонни
Триметнламкн может реагировать не только с хлористым
водородом, но и с хлористым метилом:
(CHjJjN+CHeCl ₽ [ (СИЛ N ]С1
хлорид
тгтрзмстил им-
МфНИЙ
Получающийся хлорид тетраметил аммония обладает всеми
свойствам и соли — это кристаллическое вещество, хорошо
растворимое в воде и в растворе полностью диссоциирующее
на коны:
I(CH,hN)Cl-[(CHshN|++a-
В данном пр is мерс соль образуется из веществ, не содержа-
щих ни ионов Н+, ни ионов ОН".
3, Как уже указывалось, нейтрализация, согласно тео-
рии Аррениуса, сводится к образованию молекул воды из
ионов Н* к ОН*, Однако возникает вопрос, какие осЕгованпя
имеются для того, чтобы одно вещество — воду — выделить
средн всех существующих в природе соединений и вещества,
диссоциирующие tia ионы, из которых образуется вода
(Н+ и ОН"), отнести к особым классам — кислотам и основа-
ниям. Исключительное значение воды связано только с тем,
что ее на Земле очень много,
1 Ьдводя итог, можно сказать, что теория кислот и ос-
нований Аррениуса полностью применима лишь при усло-
вии, что вещества реагируют в водном растворе. Поэтому
детальное изучение процессов, протекающих без участия
растворителя, а также реакций в неводных средах потре-
бовало существенного дополнения и обобщения этой теории.
Естественно, что любая более общая теория кислот и осно-
ваний должна включать теорию Аррениуса как частный
случай.
' В настоящее время используются и разрабатываются
несколько обобщенных теорий кислот и оснований. Наиболее
широко применяются три — теория сольвосистем, начало
которой положили работы американских химиков Кэди и
220
ЭБ гНзуччсэЕ наследие России"
Франклина, опубликованные в 1396 — 1905 гг.; л/юлмная
теория кислот и оснований, выдвинутая в 1923 г. незави-
симо датским уценим Брепстедом и английским ученым
Лоури; электронная теория, предложенная а 1923 г. амери-
канским фнзнко-химнком Льюисом. Хотя эти теории исхо-
дят из разных предпосылок, они не противоречат друг другу.
В них рассматривают проблему с различных точек зрения;
каждая из ЭТИХ теорий анализирует те особен пости кислотно-
основного взаимодействия, которых другая теория не ка-
сается Это три различных инструмента познания природы.
Вопрос о том, какую из перечисленных теорий использо-
вать, следует решать, исходя из конкретных условий поста е*
ленной научной или технической задачи.
В разработке современных представлений о кислотах л
основаниях 311Д Ч ительная роль пр и н а дл ежи т со ветскн м уче-
ным— А, Н. Сахарову, В. А. Плотникову, Н, А, Измай-
лову, А. И. Шатен штейн у, М. И. Усановнчу н др.
2, Теория сольвосистем
Вспомним реакцию диссоциации воды:
Htp^H+4-OH- .
Иопы Н* и ОН'как и все прочие коны, в водном растворе
гидратируются. Гидратация койа водорода отличается от
гидратации других ионов. Имеется много фактов, свидетель-
ствующих о том, что нон водорода интенсивно взаимодей-
ствует с одной молекулой воды:
Н*Н-Н8О==Н^0*
Соединения, содержащие нон гндроксонпя Н/У, могут
быть выделены в кристаллическом состоянии. Например,
рентгеноструктурное исследование гидрата хлорной кис-
лоты НСЮ4-Н4О показало, что это вещество представляет
собой перхлорат гндроксонпя [Ha0J*(C104)‘. Таким образом,
собственная диссоциация воды может быть выражена урав-
нением
2HsO^HaO*+OH-
В 1905 г. появилась работа Франклина, который детально
исследовал реакции различных веществ в среде жидкого
аммиака. (Аммиак превращается в жидкость при —33 :С.)
Жидкий NH,, растворяет многие неор ганическис соединения;
эти растворы хорошо провидят ток, что свидетельствует об
221
ЭБ ,:Научнов наследие России"
электролитической диссоциации В исследованиях Фран-
клина, которые затем были продолжены многими другими
учеными, была обнаружена глубокая аналогия между реак-
циями электролитов в водкой среде н в среде жидкого
аммиака.
Выяснилось, что аммиак, хотя и в ничтожной степени,
Диссоциирует на ионы:
NH,;* H++NHj
Ион водорода в растворе сольватирован; очевидно, наиболее
интенсивно он будет взаимодействовать с одной молекулой
растворителя:
H+4-NHs=NH+
В результате получится ион аммония — аналог пока гид*
роксоимя — прочный комплекс, который давно известен
химикам. Таким образом, собственная диссоциация жидкого
аммиака запишется совершенно аналогично диссоциации
воды:
2NHa^ NHf + NH]
Как известно, а жидком аммиаке хорошо растворяются
щелочные и щелочноземельные металлы; при этом образу-
ются растворы синего цвета, обладающие металлической
проводимостью. Однако эти растворы неустойчивы, в них
постепенно происходит реакция
2Ме + 2N Н,=2MeN Н* + Н J
ВЫМД
металл*
Если принять во внимание реакцию собственной диссоциа-
ции аммиака, то станет ясно, что взаимодействие щелочного
металла с аммиаком аналогично его взаимодействию с во-
дой:
2Мс 1 211,0= 2МйОН 4-Н»|
Г|ЕЛр0К{11Д
Ж-/ЛЛЛ а
Получающиеся в результате реакции с аммиаком амиды ме-
таллов содержат отрицательный ион NHu, образующийся
при собственной диссоциации аммиака. Эго аналог иона
ОН”, получающегося при собственной диссоциации воды.
Таким образом, амиды металлов являются аналогами гид-
роксидов. Эта аналогия усиливается тем, что ионы ОЦ-
и КН;, а также молекулы Н20 и NHe изоэлектронны.
222
ЭБ гНауччоЕ наследие России"
Указанная аналогия нс является формальной, она про-
является во множестве свойств Подобно водным растворам
щелочей, аммиачные растворы амидов хорошо проводят
электрический то в, что обусловлено диссоциацией
MeNllj^Me'H NIL
Фенолфталеин в этих растворах окрашивается в красный
цвет, при прибавлении кислот происходит их нейтрализа-
ция Даже растворимость амидов изменяется в той же после-
довательности, что н растворимость гидроксидов Li КН,—
нерастворим, N а К Н, — трудно растворим, KN Н 4, RbN112
и CsNH4— хорошо растворимы
На основании изложенного можно сделать вывод, что
амиды металлов в среде жидкого аммиака ведут себя как
сильные основания, хотя эти вещества не содержат груп-
пу ОН-.
Рассмотрим поведение кислот в растворе жидкого ам-
миака.
При добавлении к аммиаку какой-либо кислоты, напри-
мер хлорной, сразу произойдет реакция образования аммо-
нийной соли:
NHj + HClOj-dNEJJCLO*
перхлорат
;1ЧНО1ЕЕ1Я
Аналогичный процесс имеет место и в водном растворе:
Н/>+НСЮ*=1 IHjOJCIO,
п tpj.idjinT
г ЯДррИ Corl kl Й
Очевидно, если растворить в жидком NHa соль аммония,
то получится тот же результат, что и при растворении соот-
ветствующей кислоты— и в том и в другом случае обра-
зуется раствор аммонийной соли.
Франклин установил, что соли аммония, растворенные
в жидком аммиаке, реагируют с амидами металлов как ти-
пичные кислоты:
KNH, 4- ЫЦ.СЮ4 - КСЮ4 Ц- 2NH, НЛн SNjNH^fNHJiSO* =
= K’BjS0,4-4NH,
Сравните:
КОН + НСЮ* - КСЮ4 4- HjO или КОН 4- (Н«Р)СЮ^КСЮ4 4*гН^О
Активные металлы вытесняют водород из растворов аммо-
нийных солей в жидком аммиаке*
Мй 4- 2NH4N0, = Mg[NOa)j - г 2NHa4- H,t
£23
ЭБ ,:Научнов наследие России"
Таким образом, в растворе жидкого аммиака кислотами яв-
ляются аммонийные соли. Реакция нейтрализации в жидком
аммиаке сводится к взаимодействию
NHJ4-NH;-»2KHS
приводящему к образованию недиссоцнЯрова иных молекул
растворителя,
Рассмотренные закономерности были обобщены В новой
теории кислот и оснований — теории сольвосистем.
Согласно зтой теории кислотой является веилретво, даю-
tape в растворе те положительные ионы, которые образу-
ются при собственной диссоциации растворителя. Основа-
нием теория смьвоеисгнем называет seipecmeo, дающее » ра-
створе те отрицательные ионы, которые образуются при
собственной диссоциации растворителя.
Теория сольвОСИстем в некоторых чертах сходна с кис-
лородной теорией Лавуазье, в частности, наличие водорода
в составе молекулы не считается необходимым признаком
кислоты.
Разберем теорию сольвосистем сначала на примере реак-
ций в жидком аммиаке, которые в настоящее время изучены
очень подробно, а затем кратко рассмотрим некоторые дру-
гие неводные растворители. Везде, где это возможно, будем
указывать на аналогию с реакциями в водных растворах.
Жидкий аммиак. Как известно, при действии щелочей
на соли тяжелых металлов в водном растворе выпадают
осадки нерастворимых гидроксидов, на пример:
CuSO4+2КОН -Cu(OHU + KjSO!
В жидком аммиаке происходит выпадение в осадок нераст-
воримых амидов (растворимы в жидком NH^ только амиды К,
Rb и Cs), например:
Zn( NOb)a+2 KN 11* = Zn(N НI + 2К КО,
В водном растворе в ряде случаев выпадают не гидроксиды,
а оксиды металлов, например;
Hg(NQJ3d-2KOH = HgO| Ч-2ККО, +1 JaO
Аналогичное явление имеет место и в жидком аммиаке —
в осадок выпадают имиды или нитриды:
3HgCl,+6KNHt=HgiNil -Г 6КС14-4NH,
4П1Т|1ИД
рту Г И
ЭБ' |Шчое наследие России"
Ряд гидроксидов в водном растворе проявляет амфо-
терные свойства; они растворяются в избытке щелочи с об-
разованием Г|гдроксоком[1лексО0. Например:
Z п(ОН)а + 2КОН - K2l Zn{OI IJ« |: РЬО+ЖОН = KjPtA + Hsp
Аналогичное растворение с образованием амидных комплек-
сов наблюдается для соответствующих соединений в жидком
аммиаке:
Zn(NHsh +2KNHS = Ks|Zn(NHJj I
PbNH 4- 2KNHi= K,l>b(N'H)t 4- NHj
Амфотерность среди амидов гораздо более распространена»
чем среди гидроксидов. Например, амфотерны Sr(NHj2 и
AgNHs—аналоги неамфотерных Sr(()Hj2 и А&О. Даже
NaNH< — аналог NaOH — проявляет признаки амфотер-
ности ,
Гидролизу в водных растворах соответствует происхо-
дящий в среде жидкого аммиака ал,монолит
HgClj-b нон = HgOHCI н-на
и
I IgClj + HNH, = HgNH£l + HCI(NH4CI)
или
ticia-i- знон = В(он)4+эна
ВО,+3 HN Hs = В( NH jbH- 3HCI(NHjCl)
Реакции, аналогичные гидролизу, происходят и в других
неводи гы л растворителях; для их обозначения употребляется
общее название — сольволиз.
Поскольку NHa диссоциирует на ионы в значительно
меньшей степени, чем FLO, многие соединения, гидролизуе-
мые водой, не подвергаются сольволизу в присутствии амми-
ака. &гу особенность NI4:t используют в тех случаях, когда
необходимо избежать сольволиза. Так, Например, чистый
безводный KsS получают взаимодействием калия с серой
в среде жидкого NHj, после чего удаляют аммиак нагрева-
нием, Выделяемы Ге из водных растворов кристаллогидрат
KiS*5HsO обезводить нагреванпем не удастся, гак как при
этом происходит гидролиз и получаемый продукт загрязнен
значительным количеством КОН.
Жидкий фгорводород. Собствен пая диссоциация раство-
рителя:
2HF 1LF+-I-F-
Молекула HF изоэлектрон на НгО, а ион F" — иону ОН’.
Согласно теории соль вое истем основаниями в безводном HF
„ nu 8 м. х, Кмррлис^янч 225
ЭБ Науччое наследие Россинг
я вл я ЕОТСЯ фториды щелочных металлов: при их диссоциации
увеличивается концентрация ионов К. Кислотами являются
вещества, образующие прочные комплексы с ионами фтора —
BF„ SbFi и др.; при их растворении в HF возрастает кон-
центрация ионов HjR:
или
BF#-f-HF=H|BFt]
!l[Brd] + HF^HsF+-h[BFd-
SbFu+HF—HJSbF»]
H|SbFt| -f HF H,F+ + |SLFn|-
Имеются и амфотерные соединения в среде жидкого HF.
К их числу относятся фториды алюминия н хрома. Напри-
мер,
Q-Fj+SNaF^NaHQF,]
сравните эту реакцию с идущей в водном растворе:
Сг(ОН)а -|- 3NaOH = Naj[Cr(OH),]
С Другой стороны,
Naa|CrFd 4-ЗН[ВГ(| -* CrF,|-j-3Na| BFJ + 3HF
Эта реакция аналогична процессу, проходящему в водной
среде:
N аа[Сг(О Н ]* | + ЗЬ1С! * Сг (О] 1)а| -|- 3 NaCI 4- 31130
В среде жидкого HF кислота I tlSbFj растворяет многие
металлы, например медь.
Интересно, что сила кислот в ряду H[PF(|—H[AsF(] —
H(SbF5| в безводной HF возрастает, тогда как в соответ-
ствующем ряду кислородсодержащих кислот в водных раст-
ворах она падает.
Расплавленный бромид сурьмы. Собственная диссоциация рзствсь
ритсля агл = 97 *С}
SbBr^SbBri^ Вг
Ocj 10ван]| я пси в этом растворителе являются бром иди ш-ла'шых метал-
лов и NI!jBrr так как при их дЕЕссоццацЕси образуются иоцы брома г
КВг^К+ + Вг*
А1Вгэ будет KELCJlDTOEh потом у что
SbBra + Д1Вгя = |5ЬВг?Г | [АШцГ
Реакция нейтралам ними;
К+1 Бг- + |SbBrJ+ | [AlBfJ' - К+1 + SbBr3
В резудьтити образуются псдиссаци кровя иные молекулы растворителя,
22li
ЭБ ,:Научнов наследие России"
Жидкая четы решись азота. Это соединенно превращается в жид-
кость при 21,15 42. Ее собственная диссоциация такова:
NA~ NO* + NO;
Нптро- тгтрлт-
ЗНЛ-ИОЫ ион
Этот растворитель реагирует со многими металлами, реакция идет
по сломе
Ме+ NА -► MefNQjb -|- NO
Сравните;
Me+НОН -* Мс(ОН)л+Иа
Реакция металлов с безводной NaOa широко используется в настоя-
щее время для получен и я безводны к нитратов, которые приготовить
другими методами не удается. Так получают без води ие нитраты цинка,,
меди, бериллия и др.
Реакция металла с NA часто сильно тормоаптся образующейся
на поверхности пленкой нитрата — соли трудно растворимы в NA»
так как Этот растворитель имеет низкое знамен не в. Для ускорения
реакций разбавляют NSO, растворителями с высоким значением * —
нитрометаном или этилаиетатсм — или борут для резкими не металл,
а его карбонил, например.
№1(С0)м+4N А 2Mn(M0a)f -I-4NO-I- (ОСО
В настоящее время детально изучено много растворите-
лей, в которых протекают реакции, описываемые теорией
сольвосистем. Исследование этих р'еакций выявило огром-
ное количество новых соединений, подчас весьма не похо-
жих на ^обычные» вещества, с которыми привыкли иметь
дело химики„ Таким образом, если неорганическая химия
XIX в. была в основном химией водных растворов, то теперь
мы уже имеем множество различных «химий», каждая из
которых обещает выявить по крайней мере такое же много-
образие соединений, как то, с которым оперировала неорга-
ническая химия прошлого. Эти результаты имеют важное
значение как для развития теоретических представлений,
так и для решения многих прикладных задач; об этом под-
робнее будет сказано в конце данной главы.
Хотя теория ссль&осистем дает единую схему для боль-
шого числа реакций в неводных растворах, она не охваты-
вает всех особенностей кислотно-основного Взаимодействия.
Ограниченность этой теории сводится в основном к следую-
щему,
]. Теория сольвоспстем подразделяет вещества на кис-
лоты и основания, исходя из собственной диссоциации раст-
ворителя. Однако для реакции кислоты с основанием во
многих случаях не требуется какой-либо среды. Так, на-
пример, аммиак реагирует с хлористым водородом как
3" 227
ЭБ ,:Научнов наследие России"
в газовом фазе, так к в среде неполярных растворителей
(бензол и др,), где не Происходит распада веществ на ионы и
образования соль вое пстемы с растворителем.
2. Поскольку в теории сольвослетом всякая реакция
нейтрализации сводится к образованию недисСОЦнирОвая-
ных молекул растворителя, важнейший результат взаимо-
действия кислоты с основанием — получение соли — рас-
сматривается как какой-то побочный процесс.
От первого из указанных недостатков свободна протон-
ная теория кислот и оснований.
3. П|Югйнная теория
Выше указывалось, что нет никаких причин считать йод у
совершенно и с ключи тельным веществом средн других хими-
ческих соединений, Однако один из ионов, входящий в со-
став ВОДЫ,— ион водорода — может быть выделен средн
всех остальных: это единственный ион, не имеющий элек-
тронных оболочек. Протон примерно в 10 000 рзз меньше
остальных ионов. Он обладает целым рядом совершенно
особых свойств — исключительной подвижностью, способ-
ностью глубоко внедряться в электронные оболочки других
ионов и молекул, осуществлять химическую связь (водород-
ная связь) н т. д.
Поскольку выделены в особый класс реакции, в которых
осуществитяетс я Iюреход электроиов, — реакции окисле-
ния— восстановления (см. р. II, гл. V), целесообразно
также отнестн в отдельный класс процессы, происходящие
с участием другой элементарной частицы—протона.
Эти обстоятельства йызвали появление протонной тео-
рии кислот и оснований.
Согласно этой теории кислотой является всякая частица
(молекула иди ион), отдающая протон. Основанием явдя-
е/пся частица (молекула или ион), присоединяющая протон.
Данные определения могут показаться несколько непри-
вычными, поскольку в число кислот и оснований включаются
не только нейтральные молекулы, но и ионы. Например,
согласно протонной теории, основанием является карбо-
нат-ион, так как в водном растворе легко происходит реак-
ция
СО'" -f- Н+ X HCOj; N asCOa + НОН ~ N аИ СОЛ + N аОН
Однако вспомним, что соду и гидроксид натрия химики нау-
чились различать сравнительно недавно и этот факт нало-
ЭБ Т-Г§?$чсе наследие России"
жил отпечаток на терминологию: Ма2СОа — сода, NaOH —
каустическая сода.
Согласно протонной теории кислоты могут быть трех ти-
пов:
I) нейтральные кислоты — нейтральные молекулы, на-
пример;
НС1^ННЧ-С|- п HjSOi* H++HSOJ
2) анионные кислоты — отрицательные ионы, например:
ИЗО;?:Н+4-SOf и НРОГ^Н+ + РОГ
3) катионные кислоты — положительные ноны, напри-
мер:
+ И Н£*7±НаР + Н+
К числу катионных кислот относятся также гидратиро-
ванные ионы многовалентных металлов, например гидрати-
рованный ион алюминия» Как мы знаем (с. 216), соли элю-
ми пи я имеют в растворе кислую реакцию; это обусловлено
процессом
[А|(Н,О)„|«*- ;± |Д|(НаО)*ОН ]«*+ Н+ (Ки = ], 12. 10«)
К числу кислот относятся многие комплексы, например
| PHNHJjHsO)" г jPt(NHahOH]3*+ H*
ИЛИ
[Р1СЦ1 ldO)a]* Г IPtCLfOHk)*- -f-2H+
Аналогичная классификация может быть дана и для осно-
ваний: нейтральные (Н4О, NHa, C*H4NHS), анионные
(Cl-, ОН", NOj), катионные (HaN—NHj), Анионы многоос-
новных кислот амфотерны.
Растворители типа воды п жидкого аммиака, согласно
протонной теории, также относятся к амфотерным вещест-
вам. Например:
НгО^Н+4-ОН-
В этом процессе водя отщепляет протон, следовательно,
является КИСЛОТОЙ. Однако в реакции
НаР-НГ# Н30+
молекула воды присоединяет протон; следовательно, ее
можно считать основанием.
Любая реакция отщепления протона, согласно протон-
ной теории, выражается схемой
кислота основание -J- Н+
22^
ЭБ ,:Научнов наследие России"
Основание и кислота, участвующие в таком процессе, назы-
ваются сопряженными. Так, например, ион HSO* —осно-
вание, сопряженное кислоте H;SO4, я ион НаО* — кислота,
сопряженная основанию Н2О.
Протонная теория объясняет основной характер таких
органических соединений, как амины, эфиры, кетоны и тио-
эфиры. Присоединяя при помощи донорно-акцепторной
связи протон, перечисленные соединения образуют так на-
зываемые оний-катионы, являющиеся сопряженными кис-
лотами:
^Nt + H+jt^NsH >С-О:+Н+?:^С-О:Н
^От + Н*гЪ6:Н
Растворители примято делить на и
Такая классификация основывается ен характере их участил а кислот-
EL0-OCHO3JIOM равновесии.
Протолитические растворители — кислые {НСООН. СНлСООН>
HsSO4 и др.). В результате собственной ионизации сеш образуют лио-
ний-воны вида &Н+Р где S — молекула растворителя; например,
асндсоон ендооог +снасоо
Лрот&лнтнческпе растворнтели могут ггрисоедцнт протоны, по спо-
собность отдавать прогтоиы выражена у них значительно сильнее;
сравним взаимодействие уксусной кислоты с и НО:
СН3СООН-ркН3-ь кн^+сн3соо-
СН^ООН + НС1 х CHjCOOH • + Cl-
Первая из этих реакций протекает до конца, вторая — обратима^
Ап ротон ггые растворители — растворители игЕсртлые (бензол, толу-
ол, CClj. дихлорэтан и др.). Способность присоединять или отщеплять
протоны у них выражена очень слабо.
Отметим факторы, влияющие на кислотно-основные свой-
ства веществ.
Сила кислоты характеризуется констаЕг™! ее диссоциа-
цни- Обычно приводят ис величины J(3I а их логарифмы^
взятые с обратным знаком, которые обозначают рА'а (индекс
«а» от слова aciduin— кислота).
Сила кислот зависит от природы растворителя. Напри-
мер, хлористый водородh растворенный в бензоле, почти не
проводит электрического тока, в то время как водный раст-
вор НС1 — сильная кислота. Вода в отличие от бензола
ИЕГгенспвно ионизирует молекулы хлористого водорода.
Доказано, что ионизации в растворе подвергаются не сами
кислоты, а продукты нх взаимодеЕ“1СТВпя с растворите л емФ
например:
НС1 + Н/Э -+ Нзр > HCI -г Н5ОТ -j-Q-
ЭБ Научнее наследие России"
Вопрос о влиянии растворителя на силу кислот и оснований
будет рассмотрен ниже (с. 1236 н сл.).
Кислотно-основные свойства веществ зависят также от
структуры молекулы и взаимного влияния атамов и групп
атомов в молекуле.
Взаимное &,*!нннstc атомов п групп, атмоя и молекулу выаыгдю-
шсс перераспределение электронной плотности, Егазывают
ндш (обозначается ±г 7-эф<[»екг). Заместители, облнтдающпс
+ /-эффектом, отталкивают связывающую их электронную пару к во-
дороду, что понижает способность иона ГГ к отщеплению. Так, для
карбиновых кислот смещение электронксай плотности под гочдсйствием
заместителя X, обладающего-;- /-эффектом, можно представить записью
где стрелки показывают смещение электронам. + / -эфс[лк г лроян.тчют
атомы К О, S. При этом наибольший Н-Лэффект проявляет N. нан-
меяьший — S.
Чем больше метильных групп у атома углерода^ тем сильнее за-
меститель проявляет + /-эффект, он увеличивается и ряду “СН31
—CJkCH31—CH (CHj)ai —С (СГ]а)я. Эго можно проследить на при-
мере кнсЛОт (ь скобках у Казаны рХА а, водном растворе}; Н “
-СОСИ п,7), СН3-С00Н (4.7) CE-ljCIFa— СООН (4,9), (С1Ц£-
- СООН ЯО)
Заместители, облАдакицие — /-эффектом, оттягшиют электронные
облака других атомов, что облегчает отщепление иона водорода;
Заместители X тем сильное притягивают электроны, чем выше нл
электроотрицательность. В частности, галоген уксусные кислоты зна-
чительно сильнее, чем уксусная кислота (в скобках — рЯ" в водном
растворе): СНХООН (4.7) FCHE-COOH (2 >7}. С1СНЙ — COQH (2,9),
ВгСН2— СООН (2,9), 1СНЙ—СООН (J,0}.
Затухание в.тиянпя индукционного эф|)скта при отдален ни атома
галогена от карбоксильной группы можно проследить при сравнении
силы хлормасляных кислот;
масляная кислота CHj—СНИ-СНЙ—СООН. рК ;=4,8
a-juiopv асл я нал к и слога CI 1$—С113—СЬ [ — ССО J, р К = 2,В
i
ci
В-хлормасляная кислота CHj—СН—СИ,— СООН, p/(=4?Q
c!j
у-дсл ормас ля н ая к ксяога СЯС JI и —□ L иСН s- СООН, рХ = 4Р5
ЭБ ,:Научнов наследие России1
231
В водном растворе сила основания определяется
степенью сдвига слева направо равновесия
R»N: + НаО q± RjfliH q-НСг
ЕЦел очные свойства раствора основания обусловлены обра-
зующимися НО’-ионами. Для приведенной реакции можно
записать выражение константы равновесия (или константы
основности):
_ [R3NH||HO-1
[RiN]
(индекс «в» от слова base — основание; в формулу не вклю-
чена равновесная концентрация воды, поскольку НгО при-
сутствует в большом количестве л концентрация ее посто-
янна).
На практике обычно пользуются не величинами
основании, а значениями рКв, которые численно равны
— Jg Кл. Основание тем сильнее, чем меньше значение рЛ\.
Силу основания можно оценивать и величиной p/fai
рассматривая при этом равновесие отщепления протона от
атома, который его удерживает, например:
R^H+HiOstHjCr+RjN
Здесь К._1И£«1
|R,NH|
Для водных растворов величины р/(4 и р/<и связаны с вели-
чиной ионного произведения воды (pKs = 14,00 при 25 °C)
уравнением
Р^+рЛв-1Ш-
Аналогичные соотношения справедливы и для неводных
растворов,
Осноьлые свойства амилов зависят от того, накую орбиталь за и и*
мает нйгЕЦД^енная пэра электронов, образующая донор но-акцептор-
ную связь. Ирм одинаковой пространственной Доступности кеподелен-
ной пары электронов енла основании уменьшаете и в ряду
^N: ($рэ-орбнталь)1 ^N: (зр^орбиталь)ь К: (^орбиталь}.
В среде нейтрального растворителя, например хлороформа, метил-
амины но своей силе как основал ня располагаются в ряд:
СН3ч СН3\
Nil., CH,^NH., )NH, CH.-TSN
СИ/ сн/
(слайде (сильная
ФСНОЫг основа-
нне) пне]
232
ЭБ 'Научное наследие России"
Стрелками обоя кам еи индукционный эффект метильной группы. Ука-
занный порядок может измениться, когда пропс ходит интенсивное
взаимодействие растворителя с веществом. Так, в водном растворе
мал скул и воды сильно взаимодействуют с атамами но лоро ла дм и но-
группы, образуя водородные связи. Это приводит к изменению ос ион-
ности перечисленных аминов (в скобках рЛ.,) МН^ (4,75), СН.ЫНЛЗДв),
<СНЛ)3 NH (3,23) и (СНА N (4,20).
Важное значение в протонной теории имеет собственная
диссоциация растворителя, происходящая с отщеплением
протона. Запишем эту реакцию для нескольких веществ,
учитывая, что образующийся ион водорода — протон —
ввиду своих чрезвычайно малых размеров будет сразу же
внедряться а электронные оболочки молекулы раствори-
теля, давая ион типа гпдроксония — нон лиония; образую-
щимся отрицательный ион называется лиата.
Вода:
2НЛ^НэОк-|-ОН-
Жндкин аммиак:
2NHjXNHH-NHj
Жидкий фтор водород:
2HF^H3F++F"
(с этими реакциями мы уже встречались при рассмотрении
теории соль вое метем).
Уксусная кислота:
2СН ,000f I * СН/2ООН1 -J- сы/:оо -
Серная кислота:
SHjSOiTt H,SO; + Н8О,-
Количественной мерой собственной диссоциации (иони-
зации) * растворителя является константа автопротолиза
(другое название — ионное произведение автопротолиза).
Как из вес лето, для воды произведение концентраций ионов
водорода и гидр оксид-ионов при данной температуре яв-
ляется величиной постоянной — это ионное произведение
ВОДЫ (см. с. 193). Учитывая, что ион водорода в водном
растворе существует в виде иона Hs0\ формулу ионного
произведения воды следует записывать так:
(H/>+][CH-] = Kw
• <Иош)звция» — более строгий термин, поскольку диссоциация
может быть не электролитической,
233
ЭБ ,:Научнов наследие России"
Естественно, что аналогичное соотношение, вытекающее из
закона действующих масс, будет соблюдаться И для всех
других раствори телей:
(,1 ио ни ft Ц л и ата | ₽ К /
Ks — коне та и та автопротолиза раствор нтсл я.
Величина Кл может быть найдена из измерения электро-
проводности растворителя. Очевидно, Чём Сильнее собствен-
ная ионизация растворителя, тем выше его кислотность.
Так же, как в случае констант диссоциации, для констант
автопротолиза /<Л обычно указывают их логарифмы, взятые
с обратным знаком, которые обозначают р/С. В табл. I9
приведены значения рК4 для некоторых растворителей.
Таблица I9
pKs некоторых растворителей ирл 25 X
Вещество PA# B-BUIfrrta PK#
нр I ЛОО Ci Г-iOl-i 1П.7
NFL (при —33.1’С) 22 сАон 19Д
СНЯОЮН I2.fi HF IL,7
КСООН 6Р2 ! CH^N I0h5
HjSO* 3
Как известно, собственная ионизация воды является
причиной гидролиза веществ в водном растворе (с. 208
и ел,). Например:
NaCM^COO + НОН ~ Cl Т3ССЮН 4- Na+ + ОН'
Аналогично этому собствен! га я ионизация неводных раство-
рителей вызывает сольволиз растворенных веществ. На-
пример;
N aCHj^COO + С.ЩОН S CH jCOOH 4- №*+CJUO'
Согласно изложенным вы trie представлениям гидролиз иона
аммония протекает по уравнению
NHj-|-HilO^Hs0*+NHs
Аналогично происходит его сольволиз в спиртовой среде;
N Н i+С»ЩОН NHS+С»М^ОН;
ион
этаксонни
Очевидно, чем сильнее собственная ионизация раствори-
теля— больше константа автопротолиза, тем в большей
234
ЭБ гНа\ччоЕ наследие России"
степени происходит сольволиз. Например, и водном раст-
воре соли азотной кислоты не подвергаются гидролизу,
в растворе же безводной уксусной кислоты идет интенсив-
ный сольволиз:
МО; + СНаСОО11 Г HNQ, 4-СН/Х)О-
или
К коа -1- СИаСООН 7± н N оа + к ’• + сн/тоо -
Еще интенсивнее протекает сольволиз солей, растворенных
d безводной серной кислоте:
KNQ, + HjSO* XT HNOa + К+ -М ISO;
В этой среде все без исключения соли подвергаются далеко
идущему с ол ьвол । [ зу.
Другой важной характеристикой растворителя в теории
Брсистеда является сродство к протону. Оно определяется
энергией, которая выделяется при сольватации протона
в данном растворителе. Чем больше протонное сродство,
тем сильнее выражены основные свойства растворителя.
В настоящее время точные значения энергий сольвата-
ции протона неизвестны ни для одного растворителя. По-
скольку протон наиболее прочно связывается с одной моле-
кулой растворителя, образуя ион лиония, очевидно, что
наибольшая доля энергии сольватации падает гза этот про-
цесс. Определение этой доли энергии—менее сложная
задача, чем нахождение полной энергии сольватации про-
тона. Для ряда молекул она известна. Так, для реакций
HSO (г) Ч-Н' (г) = Н/)' (г), ДС’ = —162,7 ккал
NH, (г)4-Н- (г)=» NH; (г), 10*=— I89J ккал
Как видно, молекула аммиака гораздо энергичней взаимо-
действует с протоном, чем молекула воды.
Несмотря на то что абсолютные величины протонного
сродства пока неизвестны, порядок их изменения при пере-
ходе от одного растворителя к другому установлен довольно
точно. Ниже приводится ряд растворителей, расположен-
ных в последовательности, соответствующей уменьшению
их сродства к протону: NHS, N..H]. Н?О, CjHjOH, HCN,
HjS, CHjCOOH, НСООН, HF, HjSOi, HNO3, HCIO4.
Следовательно, протонное сродство изменяется прибли-
зительно в порядке, обратном изменению констант автопро-
толиза. Однако это правило весьма приблизительно. Напри-
мер, для бензола С«Нв и других углеводородов ионное про-
235
ЭБ ' Изучим наследие России"
из ведение ничтожно мало — эти вещества не диссоциируют
на ноны, ио протонное сродство также невелико.
Вещества, легко присоединяющие протоны, называют
протофильными (NH3, МгН4 и др.), легко их отщепляю-
щие— протогенными (HF, HNOj и др.). Соединения, спо-
собные как присоединять, так и отщеплять протоны, назы-
вают амфипротонными (Н?О, СНзОН и др.).
Приведенный ряд изменения протонного сродства поз-
воляет сделать важные выводы о проявлении кислотных
и основных свойств в различных растворителях.
Наибольшим сродством к протону обладает жидкий
аммиак. Поэтому лрн растворении в нем любого вещества,
которое может отдавать протоны, произойдет ил присоеди-
нение к молекуле NHa:
т. е. любое вещество, способное отдавать протоны, будет
реагировать в жидком аммиаке как кислота. Например,
если растворить в нем воду, то произойдет реакция
NHj-f-HdO-^NHl + OH-
Вода, растворенная в жидком аммиаке, будет вести себя
как довольно сильная кислота: ее легко можно оттитровать
раствором основания. Вспомним, что основаниями в раст-
воре жидкого NHS являются амиды щелочных металлов.
Вещества, которые в водном растворе мы знаем как сла-
бые кислоты, в жидком аммиаке станут сильными кисло-
тами, например;
HCN-bNHs-»-NHt+CK-
В нем сильными кислотами будутСН3СООН, HtS, HF ит. д.
Более того, в среде жидкого NH3 будут вести себя как
кислоты такие вещества, которые в водном растворе не
проявляют кислотных свойств, например спирты:
qH^OH + NHj^NHf+CjHjP’
Раствор этилового спирта в жидком аммиаке можно титро-
в |ть как кислоту амидом калия. Даже углеводороды, раст-
воренные в жидком МН3, проявляют слабые кислотные
свойства:
C^+NH^NHt+CiHj
EnT61f
CJ1, -J-NHjSNHf+qH;
Э7ИЛС!11
бензол
ЭБ ' наследие России"
Углеводороды можно титровать как кислоты в растворе
жидкого аммиака раствором металлического калия; при этом
идет реакция
2NHJ- Ч- 2К = 2К4’ + 2NH,+НЛ f
Таким образом, в среде жидкого аммиака кислотные
свойства веществ проявляются сильнее, чем в воде. Кислоты,
сильные в водном растворе (H,$OjT HCl, HNO3 и т, п,),
останутся сильными и в жидком аммиаке (сила этих кислот
при переходе из водной среды в аммиачную даже несколько
уменьшится ввиду того, что диэлектрическая проницае-
мость аммиака меньше, чем воды, и это затрудняет диссоциа-
цию на ноны). С другой стороны, слабые в водном растворе
кислоты HjjS, HCN и другие в среде жидкого аммиака ста-
нут приблизительно такими же по силе, как FLSO0, НС1,
HNO5 и т. п. Таким образом, в растворе жидкого аммиака
различия в силе кислот сглаживаются; это обусловлено
большим сродством аммиака к протону.
Вода также обладает большим сродством к протону (хотя
и меньшим, чем NHS), поэтому в водном растворе тоже сгла-
живаются различия между кислотами, правда, в меньшей
степени, чем в среде жидкого аммиака. В водном растворе
на, НВг, HI, H4SO4, HNO3, НСЮ^ и т. д, — почти одина-
ково сильные кислоты: степень их диссоциации в разбав-
ленных растворах практически равна 100%.
Растворители, в которых сглаживаются различия в силе
кислот (или оснований), называются нивелирующими.
Нивелирующими растворителями для кислот будут веще-
ства с большим сродством к протону — аммиак и его орга-
нические производные, гидразин, вода.
Теперь рассмотрим, что будет происходить с кислотами
и основаниями в среде с гораздо меньшим сродством к про-
тону, чем у жидкого аммиака и воды, например в безводной
уксусной кислоте.
Таблица 30
Константы диссоциации некоторых кислот в СН^СООН
прн 20 СС
КнслСТ< Кислоте
HClOj НВт на ]г6 Ш-й 4- 10 * 1,4 10 * HnSO, HNO3 в - то-* 4,2-10-1"
Й37
ЭБ ':НауччоЕ наследие России"
В уксусной кислоте протон сольватируется очень слабо,
поэтом у д иссоц и аци я кислот в ЭТОМ раствор и теле затруднена.
Кислоты, сильные в водном растворе, будут слабыми
в уксусной кислоте. В табл. 20 приведены константы диссо-
циации некоторых кислот в среде безводной СН<£ООН.
Все эти кислоты являются слабыми в среде уксусной
кислоты, но неодинаково слабыми. В водном растворе НСЮ4
н HNO,, — одинаково сильные кислоты, а в безводной
СН/2ООН азотная кислота приблизительно в 4000 раз сла-
бее хлорной. Таким образом, в данной среде становятся
более заметными различия в силе кислот.
Растворители, в которых в большой степени проявляются
различия в силе кислот (или оснований), называются диф-
ференцирующим и.
Проявление дифференцирующего действия имеет боль-
шое практическое значение. Оно позволяет, подобрав соот-
ветствующий неводный растворитель, проводить анализ и
разделение таких веществ, которые в водном растворе ведут
себя практически одинаково.
Дифференцирующими растворителями для кислот яв-
ляются СН9СООН. CsHj.OH, ацетон CHj—СО—СНд и дру-
гие органические растворители, обладающие меньшим срод-
ством к протону, чем вода.
Разберем, как будут вести себя основания в среде без-
водной уксусной кислоты.
Вследствие болы ной величины константы автопротолиза
этого растворителя все соединения, способные присоеди-
нять протон, будут вступать в эту реакцию. Поэтому все
основания в безводной уксусной кислоте станут одинаково
сильными (хотя и не очень сильными ввиду малой величины
ее диэлектрической проницаемости: в =» 6), Например,
аммиак в водном растворе — гораздо более сильное осно-
вание, чем гидроксид свинца (IV); в уксусной кислоте
различие в их силе будет значительно меныпнм.
В среде уксусной кислоты довольно Сильным Основанием
будет вода:
IТИО Н- СI ijCOOl f НаО* Н- CHjOOCT
Воду в этом растворителе можно титровать как основание,
например, хлорной кислотой:
Н^О* +CHiCOO--h 11+ -5-CIO; -+ CHjCOOH + Н^О*+СЮ;
Точку эквивалентности при таком титровании можно уста-
новить различными физико-химическими методами, напри-
мер, по изменению электропроводности раствора.
ЭЕ Точное наследие России"
Таким образом, если для кислот безводная уксусная кис-
лота является дифференцирующим растворителем, то для
оснований это нивелирующий растворитель.
Рассмотрим поведение оснований и кислот в раствори-
телях с еще меньшим сродством к протону, чем уксусная
кислота,-—в безводных Fl^SO.,, HF и НСЮ*.
В этих растворителях вообще ис может существовать
кислот (за исключением хлорной кислоты в H2SO4 if в HF),
Молекулы этих растворителей не могут оторвать протон от
других молекул. Зато в этих растворителях основаниями
будут почти все вещества, содержащие водород. Например,
в жидком фтор водороде основаниями будут CHjCOOH к
HNOa:
Н F+СН^СООН СН/ЗООЩ + F-
HF-f-НЫО,^ HaNOr-HF-
Эти вещества будут реагировать как основания с хлорной
кислотой:
Н, NO, -!- F -|- НF + СЮ; = Н F + f 1ЯЩ|- + СЮ;
В безводном HF сильным основанием будет вода:
Н2О+HF—H7Oh+F-
Основаннями, причем довольно сильными, будут спирты,
например этиловый:
адон -у н и _ о,н*он * 4- р-
Раствор этилового спирта в этой среде хорошо титруется
хлорной кислотой.
Даже углеводороды в безводном HF будут основаниями;
например:
JridJBlE
Их можно титровать хлорной кислотой.
Таковы некоторые примеры, иллюстрирующие протон-
ную теорию кислот и основании. Если теория соль вое истем
может рассматриваться как возрождение на более высоком
уровне кислород ной теории Лавуазье, то протонная теория
есть развитие водородной теории кислот Либиха. Из всех
теорий кислот и оснований протонная теория наиболее раз-
работана с количественной стороны; значительный и клад
в этом н ап р а влей н н внесен советскими учеными, особенно
Н. А. Измайловым. Многие химики считают эту теорию
наиболее совершенной.
239
ЭБ ,:Научнов наследие России"
4. Элоктршпгпя теория
Согласно электронной теории основание— это вещество,
поставляющее электронные пары для образования химиче-
ской связи — донор электронных пар; кислота — вещество,
принимающее электронные пары, — акцептор электронных
пар. Кислотно-основное взаимодействие, согласно электрон-
ной теории, заключается в образовании донорно-акцептор-
ной связи. В результате взаимодействия кислоты с осно-
ванием образуется солеподобный продукт, который назы-
вают аддицнонным соединением, или аддуктам. Обычно
аддукты удается выделить как индивидуальные вещества.
Вот несколько примеров реакций, которые, согласно
электронной теории, представляют кислотно-основное вза-
имодействие;
Л Г Я Т
H:N; -I- H;Q: -> H:N:H I =С|Г
H кисло- L Н J
CCHQ"
ИВ НИС
(CHjhNs + ВС!3 = (СН3ЪК: ВС!Я
осноилни^ кис-
лота
(Н|б:)ЧН+-НЙ:11
кис-
лота
ФСЯОЯВНИС
:О: '
Ю:Й :б:Н = H,J
"iO: Й
кисло-
та
осно-
ва» :ie
Ag+-r2!NH1 = ^:NH9)J]*
кисло- основа-
та мне
Электронная пара, осуществляющая донорно-акцептор-
ную связь, может быть в различной степени смещена к од-
ному из атомов. В предельном случае, когда смещение
является практически полным, образуется ионное соедиЕ1е-
иие (например, NH^Ct). Подобное перетягивание электрон-
ной пары происходит и при образоваЕ1ин соединений гало-
240
ЭБ ,:Научнов наследие России"
генидов алюминия с органическими аминами, например
пиридином (CsHbN),
Таким образом, электронная теория Льюиса рассматри-
вает нейтрализацию в водных растворах, взаимодействие
аммиака с галогенидами бора, комплексообразование, ре-
акции ангидридов с водой как сходные процессы. Действи-
тельно, согласно теории химической связи, во всех этих про-
цессах взаимодействие между частицами имеет одинаковую
природу — образуется донорно-акцепторная связь. Веще-
ства, являющиеся донорами электронЕГЫХ пар, часто назы-
вают основаниями Льюиса, а акцепторы электронных пар —
кислотами Льюиса или L-кцслотйми. Большинство катио-
нов является L-кислатами, а анионов — льюисовскими ос-
нованиями. Соли — типичные кислотно-основные ком-
плексы. Мы видим, что теория Льюиса рассматривает вопрос
о кислотах и основаниях более широко, чем другие теории.
Даже в кристаллической решетке соли MgClj катион метина можно
считан, t-кислотой. Он окружен шестью хлорид-ионам к (основания
Льюиса}. В разбавленном водном растворе ионы Мдг*окружены шестью
молекулами воды (основание), образуя сольватированный ион. В этом
же растворителе СГ-ноны связаны водородной связью с молекулами
воды; гидратированные ноны хлора можно считать аддипленными
соединениями, в которых роль кислоты выполняет вода.
К основаниям Льюиса Относятся вещества, содержащие
амиЕшый азот (аммиак, алифатические и ароматические
змегны, пиридин и т. и.), кислородсодержащие соединения
общей формулы RSCO, где R — органический радикал или
атам галогена, ноны галогенов.
Кислотами Лыоиса считаются галогениды бора, алюми-
ния, кремния, олова, фосфора, мышьяка, сурьмы и многих
других элементов, ноны-комплексообразователи Ag’, Со®4’,
СгЯ4, Р(®+ и др.
Многие аддициэнные соединения находят практическое
применение. Например, широко используемый в промышлен-
ности эфир а г трехфторнстого бора получают из газообраз-
ного BFS (/ки„ —90 X) и диэтилового эфира (^|)П 34,6 X).
Продукт нейтрализации (C2HS)2O : BFS кипит в интервале
126—129 еС.
Кислоты Лъдоса делят гса н Первичными
нааыыхгг те, взаимодс^стбис которых е основан и я ми протекают мгне-
выпю (э пер сил актнягцин здесь очень мала). В этом случае достаточно
первичную кислоту смешать с основа и нем, чтобы произошла кислот-
но-осгсовгтя релкцпя. Напри мер h сразу же взаимодействуют ЙС1 н
21!
ЭБ ,:Научнов наследие России"
NH-t Вторичные кислоты Льюиса реазмруют с Основаниями гальке
и определенных условиях (энергия антнвл ни и этих npoiuxcon имеет
большую величину)
Типичными вторичными £ кислотами явлнютсн, например, альдс
гиды, СОг и CS- Д1,оксид углерода при температуре - АО °C в растворе
этемоного cnbpie мало активен и практически ею реагирует с этилатом
натрия Однако при комнатной температуре СО2 как вторнчЕсая кис^
лота Льюиса Вкгулаег тго азаимодейсгвЕЕе с НО -ношэмк, образуя би-
карбонат нон
+ Л к Л
Cf„ 4-iOH -vC^-QH
4Oj- ‘ Xcii-
Предсказать поведение кислот к оснований Льюиса
можно исходя из знаний кваитовохимнчсских характери-
стик порядков связи, индексов свободных валентностей и
л’Электронных зарядов*' Эти характеристики позволяют
определить реакционный центр (или центры) молекулы,
участвующей в кислотно-основном взаимодействии
Например, для биологически важного гетероцикличе-
ского основания аденина по вычисленным методом Хюккеля
величинам л-электронных зарядов (указаны около атомов
азота) можно определить последовательность участия этих
атомов в реакции нейтрализации
1,810 м-1,
г
1,282 ,|Ж
I
1,267 1,593
Болес основсн атом азота, у которого наибольший л-элек-
троннын заряд Этот атом в первую очередь будет реагиро-
вать с кислотой
Возможность образования аддиционпых продуктов за-
висит от геометрии молекул (или ионов) Так, основания
триэтилямин и хинуклндин одинаково легко реагируют
с протонами- малый размер протонов позволяет им беспре-
пятственно подходить к атомам азота и присоединяться
к ним При замене протона на другую кислоту Льюиса —
три мстил бор (СН5)ЭВ картина взаимодействия меняется.
Плоская молекула три мети л бора легко образует связь с азо-
* См М. X. Карапстьянц. С. И Драхнн. Строение вещества, Зе
hja. М», Высш л я ш почта, 1978, С, 193—203,
ЭЕ H^VS-ice наследие России"
том хинуклидинг, по из-за пространственных затруднений
она не может прочно присоединиться к азоту в трнэтил-
ами не:
I
CHj
гаигуклНл1пг
(я 11 р £ ЬГН да л ь ИМ
окружение ftsjjTiij
трн&1 ил ямин-
[сгпра ыя дв лыем-
окружение взота]
трнметцлбср
(плАекай ип,пс чулй !
В отличие от протонной теории в теории Льюиса поня-
тие о силе кислоты относится не к самой кислоте, а к паре
кислота — основание. Покажем это на примерах. NH3
в водных растворах считается, согласно протонной теории,
значительно более слабым основанием, чем НО-коны. Это
утверждение справедливо в теории Лыоиса, если рассматри-
вать реакцию взаимодействия NHa и НСГ-ионов с ионами
гидроксония. Если же в качестве Л-кислоты взять Ад*-ионы,
то более сильным основанием будет аммиак. Это можно до-
казать, сопоставляя концентр а пни ионов серебра, которые
поставляют в раствор гидроксид серебра AgOH и комплекс-
ный катион [ Ag{ N HjJj 1* — комплекс мен се диссоциирован,
чем AgOH. Эти факты объясняются концепцией жестких
и мягких кислот и оснований.
Представление о жестких и мягких кислотах и основа-
ниях выдвинул Пирсон (1963), Все кислоты и основания
он разделил па два класса — мягкие и жесткие и сформули-
ровал правило: жесткие кислоты предпочитают связы-
ваться (образуют более прочные соединения) с жесткими
основаниями, л мягкие кислоты предпочитают связываться
с мягкими основаниями.
Отличительные свойства мягких и жестких кислот и ос-
нований приведены в табл. 21. Отметим, что одна и та же
кислота не обязательно должна обладать всеми перечислен-
ными свойствами. Это же справедливо и для оснований.
Понятия жесткий и мягкий нельзя отождествлять с терми-
нами сильный и слабый.
С позиции теории жестких и мягких кислот и оснований
рассматривают пять основных типов реакций неорганиче-
ских и органических соединений:
1) перепое электрона, например:
F^+C^r-i-F^+Ce**
S43
ЭБ ,:Научнов наследие России"
Т а б л и eta 21
Важнейшие характеристики и примеры жестких и мягких
кислот и осноэаннй
OtiKMajtice Кислега
Характерное СЛУЙСТЕО Пример™ i X Р[НЧНГ<>рцпс CuoftctB-0 Примеры
Мяг- кие Легка поляризу- ются, низкая алсктроатрмцательг костью легко окис- ляются 1- Н-. hs- сян#ч СО, С*Н41 =S : Большой раз’ | мер часгниы, I малый поло- жительный заряд Ag+, Си+ !Я1 Нй^. Вг-, ВН3, бОЛЕъ- шннство ме- таллов (Ni. PC Pd)- Катионы щелочных, щелочнозе- мельных ме- таллов и М^ь
Лромечуготнмс Вг . sof Fell-, DJI11-, QiH+# CQit, В(СН>)>
Жест- кие Поляризуются с трудомь высокая электроогрица’ ТВЛЬЕЕОСГЬ, ОКИСЛЯ- ЮТСЯ с большим трудом, электро- ны акцептировать НС способны F\ Cl", НО ; О® . NHa, НА CHjCOO’, sor Малый рламер частицы, шлео- кнй положи- тельный заряд, не могут быть донорами электронов Н+, Са*+, BFa, Сг*\ Miilp
2) перенос протона, например:
3) реакция между кислотой и ос пора пнем в общем виде:
А 4- сВ -* А:В
нн с- ос КО’ алдицы-
лйг вавде ciiriiae
та соедине-
на
4) нуклеофильное замещение (в общем виде):
Bf+AB-* В'А-НВ
где В/ — нуклеофил-частица (аннон или молекула), несущая
неподелениую лару вдектропон, за счет которой образуется
241
ЭБ ,:НаучноЕ наследие России"
новая связь; АВ — молекула, испытывающая атаку ну-
клеофила; :В — уходящая группа. Примерами реакций
нуклеофильного замещения могут служить следующие:'
но-+(Ci ььса -* а + (ci ььсон
(СН^Ч + (СН,ЬОС1 -> (СН^ЙСССНЛ+СГ
5) электрофильное замещение (в общем виде):
А' + ВЛ -► A' R + Л
где А — электрофильный агент, имеющий незавершенные
электронные оболочки (NOj, SC^H* и др.} и стремящийся
заполнить их; В А — молекула, испытывающая атаку элек-
трофила. Примером реакции электрофильного замещения
служит реакция нитрования бензола нитроний-ионам и (см.
с. 251):
Жесткие кислоты связывают жесткие основания глав-
ным образом за счет ионных сил. Мягкие же кислоты свя-
зывают мягкие основания главным образом с помощью ко-
валентных связей. Для осуществления прочного ковалент-
ного связывания атомы должны быть близких размеров
и близкой электроотрицательности.
В рядах соединений (/-элементов жесткость кислот изме-
няется в зависимости от степени окисления. Так, Ni° в тет-
ракарбониле никеля (Ni(CO).J является мягкой кислотой,
Ni(IH) — жесткая кислота, a Ni!+ занимает промежуточное
положение.
11роиллюстрируем понятие о жестких и мягких кислотах
и основаниях па нескольких примерах.
Природа отобрала и хранит в земной коре наиболее
устойчивые соединения; так, алюминий встречается в виде
оксида, гидроксида и силикатов, кальций — в виде карбо-
ната (жесткие кислоты связаны с жесткими основаниями),
а медь, ртуть и другие (/-элементы — обычно в виде сульфи-
дов (мягкие кислоты связаны с мягкими основаниями).
245
ЭБ ,:Научнов наследие России"
Иодид-ионы — осиовашгс более мягкое, чем хлорид-
ионы, поэтому легко происходит реакция
п далее
HgCm-2KI-=Hfil,+ 3KCb
Hgll+2Kf = K!(Hgl1|
Фторид- п х л op яд-по л ы — жесткие основания, причем
по жесткости фторнд-ионы превосходят последние, Если
к раствору хлорида железа (III), имеющему желтый цвет,
прибавить фторид-ионы, то окраска раствора исчезает ввиду
образоваЕгия бесцветного стабильного гексафторферрата
<П1):
FeCI, Ч- 6NH« F == 6N HjCl+(N HtM FeFe ]
В качестве катализаторов химических процессов часто
и с пользу еот мелко раздробленные «/-металлы (платину,
палладий, Егикель я др.). Атомы этих металлов—мягкие
кислоты, поэтому они катализируют реакции, в которых
принимают участие умеренно мягкие осЕювания, например
оксид углерода. Другие более мягкие основания — соеди-
нения мышьяка и фосфора — являются каталитическими
ядами, поскольку они образуют более прочные соединения
с этими элементами и блокируют активные центры на ме-
талла х-катализатор а х <
Окись углерода СО, соединяясь с Fe (II) гемоглобина
крови, более прочно связывается с Fe(II), чем кислород
(см, характеристики веществ в табл. 21); этим обусловлена
ядовитость СО,
Мягкие кислоты — катионы тяжелых металлов Hg4*,
Си1*, Pbz+ И др. — сильные яды для организмов, ПОСКОЛЬКУ,
взаимодействуя с —S11 -группами физиологически важных
соединений, блокируют их и исключают участие в жизне-
деятельности организма.
Для аналитического обнаружения и определения ультра-
малых количеств ионов Pb4+, Hgs+ и некоторых других ио-
нов используют дитизон:
С помощью дитизона можно обнаружить в воздухе, морской
воде, биологических объектах (кровь, ткань) до 10 ’—10 * г
наследие России"
ртутя (fl). Активны ft центр молекулы дитизона — атом
серы (мягкое основание). Указанные катионы—мягкие
кислоты, поэтому дитизон дает окрашенные продукты реак-
ция с катионами Pb2*, Hg2* и другими мягкими кислотами.
С дитизоном еге реагируют ионы Са2* и Сг*% которые отно-
сятся к жестким кислотам,
В последние голы опубликовано много работ, где пред-
ложены различные соотношения, количественно выражаю-
щие понятия электронной теории кислот и оснований.
Так, представление о мягких и жестких кислотах и основа-
ниях можно приближенно выразить формулой
lgX=SASD+oArtD, (IV.44)
где К — константа равновесия реакции донора D с акцеп-
тором А:
D-f-А 5± DA
параметры SA и Sp определяют силу (p/f) кислоты и осно-
вания. од н on —их жесткость (или мягкость}.
Весьма эффективным оказалось аналогичное уравнение,
выражающее энтальпию образования Д#до аддукта из
акцептора и донора:
' ^^AD"“^A^D +CACD» (IV 45)
где параметры £ характеризуют ионное взаимодействие;
С — ковалентную связь, При этом считают
ЕиЙр, Cxi'Cf/?,
где р — дипольный момент молекулы; R — ее поляризуе-
мость, а и b — эмпирические константы. Для использования
уравнения (IV. 45) нужно знать четыре входящие в пего
неизвестные йд, йо, Ьд, in. которые Определяют из опыта.
Для этого измеряют четыре значения &Нцо для каких-
либо пар акцепторов н доноров. Уравнение (IV.45) хорошо
выполняется при применении к рядам сходных веществ.
Оно по своему физическому смыслу определяет ДЯ образо-
вания молекулы аддукта AD в газовой фазе из газообраз-
ных А и D. Однако его обычно применяют при изучении
ДЯдв в среде инертного растворителя (СС14, СаН]2), что
проще осуществить, чем определение А НдD в газовой фазе.
С помощью уравнений (IV. 45) было вычислено много (ты-
сячи) значений Atf.w, и они обычно удовлетворительно
(с точвостыо 1—2 к к ал/моль) согласуются с эксперимен-
тальными данными,
247
ЭБ ,:Научнов наследие России"
Возможен н другой путь количественной оценки А//дп<
Он целован на сопоставлении величин ЛЯдр, относящихся
к реакциям двух различных акцепторов с одним и тем же
набором доноров. Этот способ — один ив примеров первого
метода сравнительного расчета* В качестве опорных вели-
чин здесь всегда берут ДН реакций различных допоров со
^стандартным» акцептором SbCl5. При этом ДЯдо измеряют
Рис. 74, кон-
ста шт обраяонання аддукта
с SbCl-, с донорным числом
рис, 7S4 Взаимосвязь зи-
тлльпий реакций, (ккал/моль)
различных веществ с SLBr3 с ах
донорными числами
в среде F3 мертвого в отношении да юр ио-акне игорного взак-
модействия растворителя CsHjCllt СвНй клнСС]<> Тепловой
экзоэффект (в к кал/моль) реакции вещества — донора
с SbCJs называют донорным числом данного вещества и обоз-
начают DN. Величина DN является мерой донорной спо-
собности соединения. Донорные числа некоторых веществ
приведены в табл. 22.
Таблица 22
Донорные чяс-ча некоторых растворителей
ГяетвО^нтсль DN Растворитель DN
Хлористый СуЛъф-урНЛ I ХлорЕССТЫЙ 7ИПНРЛ НктрометдЕЕ Нитро&спзол Оксихлорид селена Мстнлацетзт 0J 0,4 Z7 4Н4 12,2 1G.5 Аеютпге Вода Ли эти лоеый эфир Триметнлф(и?фат Ди метнлсулы|юкс ид ПирНДЮЕ 17,0 18.0 10,2 28,0 293 । 38,1
ЭД
ЭБ ,:Научнов наследие России"
Между донорными числами и константами равновесия
реакций образования аддуктов с SbCJs существует линей-
ная взаимосвязь (рис. 74).
Если вместо SbCI4 взять к а кой-л и би другой акцептор, то
значения А Кат> для реакций доноров С ЭТИМ акцептором
обычно находятся в линейной зависимости от донорных чи-
сел (рис. 75). Зная такую взаимосвязь, по величинам DN
можно предсказывать неизвестные значения ДЯдо- По-
скольку для проведения прямой требуется минимум две
точки, для построения данной Линейной зависимости, так же
как при использовании уравнения (IV.45), требуется че-
тыре опорных значения Д/7до-
5. Значение теории кислот и оснований
У читателя, впервые знакомящегося с различными тео-
риями кислот и оснований, может возникнуть вопрос: За-
чем применять разнообразные подходы к проблеме кислот
и оснований, не являются ли изложенные концепции в ка-
кой-то мере схоластическими упражнениями. Не все ли
равно, причислять ли данную реакцию к кислотно-основ-
ным или нет, — какое это имеет практическое значение?
Необходимость в различных теориях кислот и основании
обусловлена тем, что природа многогранна, каждое даже
самое элементарное явление имеет много сторон и особенно-
стей. В одних задачах важны одни детали химическою вза-
имодействия, в других — другие. Теории кислот и основа-
ний, даже в том кратком и элементарном виде, в котором
они здесь представлены, полезны для химика потому, что
во многих случаях они позволяют предвидеть, какие веще-
ства будут реагировать друг с другом как кислоты и основа-
ния. Кис л отн о-ос н овн ыс р са к ци и i[меют рядспец и фнческих
черт, которые обусловливают важность и широкое приме-
нение этих процессов. Такие реакшш, как правило, идут
быстро даже при низкой температуре, iie сопровождаются
побочными процессами, не требуют для своего проведения ка-
тализаторов и часто протекают полностью, давая 100%-ный
выход продукта. Ясно, что совокупность этих особенно-
стей во многих случаях имеет большое значение. Если ка-
кая-либо теория систематизирует подобные процессы н
позволяет предвидеть новые, то она полезна.
Как вид ио из предшествующего изложения, современная
теория кислот и оснований является более общей, чем тео-
рия Аррениуса, применимость которой по сути дела ограни-
249
ЭБ ,:Научнов наследие России"
чивается процессами, протекающими только в одном раст-
ворителе — воде.
По понятным историческим причинам химики в первую
очередь изучили и взяли на вооружение реакции, происхо-
дящие в видных растворах. Однако далеко не во всех слу-
чаях вода является самым подходящим растворителем;
огромное количество веществ вообще нс может существо-
вать в водной среде, Б последние десятилетия процессы,
протекающие в неводных растворах, получили широчайшее
применение в промышленности и научных исследованиях.
Многие из этих процессов относятся к кислотно-основным.
Одна из важных областей применения реакций в невод-
ных растворах — химический анализ. Здесьшир око исполь-
зуются методы неводного титрования, позволяющие быстро
и точно определять состав таких смесей, которые трудно
анализировать при помощи обычной методики. Как мы уста-
новили в разделе, посвященном протонной теории, в не-
водных растворителях кислотно-основные свойства веществ
могут резко изменяться. Так, в среда жидкого фтор водорода
основаниями будут HNOJ( СН3СООН, спирты и даже угле-
водороды. Наоборот, в жидком аммиаке вода, спирты н
углеводороды будут вести себя как кислоты. Эти изменения
К] [СЛОТ НО-ОС НО В них СВОЙСТВ используются D [{СВОДНОМ тит-
ровании. Подобрав подходящий растворитель, можно тит-
ровать вещества, инертные в вод пой среде, например спирты
или углеводороды.
В неводном титровании широко используется дифферен-
цирующее действие растворителей. Так, например, HCI,
НМО3 и НСЮ4 в водном растворе являются одинаково
сильными кислотами. Если их смесь титровать основанием,
то она будет реагировать как одно вещество, и данным мето-
дом нельзя определить, сколько в смеси содержится НС1,
HNO, и HCtOj. Однако если взять соответствующий
дифференцирующий растворитель (например, безводЕгую
СН3СООН), то в нем можно провести раздельное титрова-
ние указанных веществ и найти содержание каждого ИЗ них.
Современная теория кислот и оснований позволяет пред-
сказать ocoOeFJHQCTH поведен El я веществ в разлцчЕ1ых раст-
ворителях. Она дает пстадковзЕнзе огромного количества
реакций органических веществ. Эти вопросы излагаются
в курсе органической химии. Здесь можно огра|[ичиться
только одним примером — объяснением хорошо известного
химикам-органикам нитрующего действия смеси серной и
азотной кислот.
250
ЭБ ,:Научнов наследие России"
В растворе безводной серной кислоты HNOj ведет себя
как основание (см. с. 239):
HNQ, + Н,5О* Г HSNOJ + HSO;
Серная кислота, будучи сильным водоотш[мающим агентом,
отнимает молекулу I1-0 от иона нитроксония HaNOJ:
н*мо;+HjSo* > no;+h,so4 . h/j
Таким образом, в среде безводной серной кислоты образу-
ется большое количество иолов нитрония NO* (для НаЭОх
е = 101), которые имеют положительный заряд и поэтому
легко могут заменять ноны водорода в органических веще-
ствах, давая иитросоед мнения.
Широко используются многие продукты кислотно-ос-
новиых реакций. Так, например, фторид бора, являющийся
мощным катализатором ряда органических реакций, не-
удобно хранить и транспортировать, так как это газообраз-
ное соединение. Поэтому BFd часто применяют в виде эфи-
рата FSB : О(СаН4)^; как мы знаем, образование данного
вещества представляет типичный случаи кислотно-основ-
ного взаимодействия по Льюису (см. с. 241).
Велико значение теории кислот и оснований в неорга-
ническом синтезе. За последние десятилетия в неводных
средах получено колоссальное количество новых неоргани-
ческих соединении; многие из них весьма своеобразны и не
похожи на вещества, существующие в водных растворах.
Одним из процессов, который получил огромное приме-
нение за последние десятилетня, является фторирование.
Оно широко используется в технологии редких элементов,
в производстве синтетических материалов и биологически
активных пре паратон. В процессе фторирования весьма
трудной задачей является подбор растворителя для про-
ведения этой реакции, поскольку фтор разрушающе дейст-
вует из большинство веществ, Это обстоятельство вызвало
появление значительного числа работ по изучению реакций
кислот и оснований в безводном HF и других фторсодержа-
щнх растворителях, В этих исследованиях было обнаружено
большое количество новых, весьма своеобразных соедине-
ний. В качестве примеров назовем некоторые из этих ве-
ществ.
I IpEJ растворении пептафторида иода IF5 в безводном HF
происходит реакция
25J
ЭБ ,:Научнов наследие России'1
ноч
но-?р
нс/
Эта реакция совершенно аналогична хорошо известному
взаимодействию ангидридов с водой, например:
SOjH-HjO-* HjfSOd
В результате образуется комплексная кислота HHFJ, для
которой могут быть получены многие соли. Данисе вещество
интересно тем, что в его анионе содержатся только атомы
галогенов. Эта кислота является (propистым аналогом йод-
новатой кислоты HJOS.
В среде безводного фторводорода могут быть получены
и другие фторзаметенные кислоты. Например, при раство-
рении ортофосфорной кислоты происходит реакция
=0+11F i JO 7P=0 + HSO
ио/
фтспфосфор-
кая кислота
При дальнейшем действии фторводорода образуется
R
F >Р=О днфторфосфорная кислота
нс/
Можно полностью заменить атомы кислорода в фосфор-
ной кислоте на фтор - в безводном HF растворяют пентахло-
рид фосфора РС14; при этом происходит сольволиз:
PCl(+&HF=PFe+5HCi
Образующийся пентафторид <[юсфора PF5 сразу же при-
соединяет молекулу фтористого водорода:
PFb-|-HF=HPF4
Образуется HPFe — аналог НРО,; для этой кислоты из-
вестно много солей,
В жидком фториде брома BrF3 также могут быть полу-
чены весьма своеобразные комплексные г|гтор]|ДьЕ. Собствен-
ная диссоциация этого вещества выражается уравнением
2BrF,^ BrFf+BrPf
Фторид калия, растворенный в BrF3, будет, согласно тео-
рии сольаоснстем, вести себя как основание:
KF4-BrF3= K[DrF4J
Опять получаем соль, в анионе которой содержатся только
атомы галогенов. Фторид олова в растворе BrF3 ведет себя
ЗБ наследие России"
как кислота:
2i?rFa Ц-SnF,«
При растворении двуокиси азота в ВгЬа происходит реак-
ция окисления— восстановления, аналогичная взаимодей-
ствию металлов с водой;
б1/о4 4- 2 В? Fa = 6(NOt)+ F- + ВгЗ
Сравните эту реакцию с процессом
2bfa -|- 2Н0Н = 2N е1 ОТ 1 - + Н J |
Образующееся вещество NOaF (фторид нитрония), со-
гласно теории солъвосистем, в растворе BrF3 является ос-
нованием; следовательно, оно будет реагировать с кисло-
тами, давая соли, например:
afNCW* F-+(BrF^tSn FJ - (NOt)l ($п Fd»- + 2BrF,
Диалогичным образом можно получить другие соли нитро-
ния: (NOj)BF4, (NOg)AuF4t (NOs)PFfl и т. д.
Как уже указывалось в среде жидкого аммиака также
могут быть получены весьма своеобразные соединения.
Некоторые из них можно получить только в жидком амми-
аке; например, единственным способом получения нитрида
ртути Hg3l\\ является взаимодействие соли ртути с ами-
дами щелочных металлов в жидком аммиаке (см. с. 224).
Аналогично получают нитриды висмута и таллия (T1BN
и BIN). Иными способами приготовить эти соединения не
удается. Вот несколько примеров аммонолиза, приводящих
к образованию весьма своеобразных продуктов:
Si Cl, J-8H;NНг»S 1(NHs), + 4NH,C1
тетр имид
кремлия
NO CI + 3H- NH, » KONH,+NH*Q
ннтрОэил-
аЧнд
so,a,-мн \и4=stMNHa)a+2nh*cl
Сильфа м 'ГД
ИЗ
ЭБ ,:Научнов наследие России"
Таковы лишь некоторые, довольно произвольно выбран-
ные примеры, показывающие, какое многообразие новых
химических соединений и процессов укладывается в схемы
современной теории кислот и оснований.
Глава V]
ФЯЗИ1Ю-ХИМИЧЕСКШ1 АНАЛИЗ
Для изучения свойств соединений часто получают их
н чистом состоянии, применяя для этого кристаллизацию,
вы нар ива пне, сублимацию, фильтрование, перегонку и
другие операции. Это—приемы препаративного метода
исследовании. Использование этого метода ограничено,
С его помощью не всегда удается исследовать растворы,
сплавы, стекла. Часто встречаются и экспериментальные
трудности: например, отделить кристаллы от маточного раст-
вора становится сложным, если он обладает большой вяз-
костью, а соль разлагается под действием растворителей,
служащих для отмывания раствора. Еще труднее отделить
твердое вещество от жидкого при высоких температурах или
разделить сплав на составные части. Для того чтобы выяс-
нить характер взаимодействия веществ, т. е. узнать, дают
ли они между собой механические смеси, растворы или хи-
мические соединения, необходимо либо отделить их друг
от друга, либо применить другой метод, позволяющий уста-
новить прпроду и состав образующихся в системе соедине-
ний, не прибегая к их выделению и анализу, а именно метод
физико-химического анализа. С его помощью устанавливают
зависимость между изучаемым свойством н составом системы
и выряжают результаты'исследован ня в виде диаграммы со-
став—свойство. Это целесообразнее, чем воспроизведение
результатов опытов в виде таблиц (они недостаточно на-
глядны и требуют интерполяции) или формул (их составле-
ние трудоемко и не всегда осуществимо). А главное —
анализ диаграммы состав—свойство позволяет определить
число и химическую природу фаз, границы их существова-
ния, характер взаимодействия ком по центов, наличие соеди-
нений, их состав и относительную устойчивость — словом,
получить обширную и содержательную информацию.
Физико-химический анализ как метод исследования был
предложен М. В. Ломоносовым, широко примененД. И. Мен-
делеевым в его работах по изучению плоти остей растворов
п разработан в самостоятельную научную дисциплину
Н. С, Кур па новым.
254
ЭБ ‘ Научное наследие России"
исследуемой системы
Рис. 76. Крииыс охлаждения:
j — чистя и ж ид копт L-i 2 — растер, dfjin
по поицеитрв-unit or
КйП смёсИк J — раствор, состав которого
СЛ11 падает с составом эд1еындоской евдп
(схемл)
Предметом изучения с его помощью служат более
30 свойств — температура плавления, плотность, вязкость,
электропроводность, давление пара и многие другие. Они
объединены примерно в 10 групп (тепловые, электрические,
оптические, магнитные н др.) *.
1. Термический анализ
Мы ограничимся рассмотрением раздела физико-химиче-
ского анализа, посвященного изучению зависимости темпе-
ратуры кристаллизации
от ее состава (терми-
ческий анализ). Объек-
там и термического
анализа служат самые
разнообразные систе-
мы — разл 11 ч I [ыс л ростыс
вещества (например, ме-
таллы), органические
соединении, растворы,
смеси солей и т. д.
Результатом его про-
ведения является по-
строение диаграммы
плавкости.
Кривые охлаждения.
Пусть происходит ох-
лаждение чистого жид-
кого вещества. Переход из жидкого состояния в
твердое сопровождается выделением теплоты кристалли-
зации и поэтому, пока вся жидкость не отвердеет, темпера-
тура системы остается постоянной. Если охлаждать рас-
плавленное вещество, то зависимость между температурой
и временем может быть выражена в виде кривой охлажде-
ния (рис. 76, /).
Участок ab соответствует практически равномерному
понижению температуры жидкости. Затем начинается про-
цесс кристаллизации (если не происходит переохлаждения),
характеризующийся горизонтальным участком В точке с
отвердевает последняя капелька жидкости. Отвердевшее
вещество вновь охлаждается (участок cd). Можно было бы
осуществить обратный процесс и получить кривую нагре-
вания deba. Мы описали случай, когда изучаемое вещество
• Иллюстрациями истом фнянко хмылчккого анализа могут слу-
жить ряс, 14, 22, 30, 34, 51 и 6].
255
ЭБ ,:Научнов наследие России"
не имеет никаких превращений ниже температуры кристал-
лизации (если при /„Tlt происходят пол л морфные пре-
вращения, то появляются дополнительные температурные
остановки).
Характер кривой охлаждения раствора (расплава) за-
висит от природы системы. Рассмотрим простейший случай,
когда из бинарного раствора кристаллизуются чистые ком-
поненты,
Прн охлаждении такого раствора наблюдается не-
сколько иная картина (рис. 76, 2), Понижение температуры
от а до Ь, как ив случае чистого вещества, происходит
примерно равномерно. Затем из раствора начинают выде-
ляться кристаллы одного из веществ. Так как температура
отвердевания раствора ниже, чем чистого растворителя
(см. с. 161), то это произойдет ниже точки отвердевания
чистого вещества. Прн этом состав жидкости будет изме-
няться, вследствие чего температура се отвердевания непре-
рывно понижается. Но выделяющаяся при кристаллизации
теплота замедляет темп охлаждения; поэтому, начиная сточ-
ки Ь, крутизна лишен уменьшается. Наконец, наступает
момент (точка с), когда раствор делается насыщенным отно-
сительно обоих компонентов. Тогда они начинают кристал-
лизоваться одновременно. Поэтому отвердевание жидкости,
начиная с точки с, происходит при постоянной темпера-
туре; на кривой охлаждения появляется горизонтальный
участок cd. J 1осле отвердевания всей массы (точка d) охлаж-
дение возобновляется (кривая de).
Таким образом, кристаллизация жидкой смеси в отличие
от чистого вещества-растягивается па некоторый темпера-
турный интервал, который тем значительнее, чем ближе со-
став расплава к чистому веществу. Поэтому если взягь
смесь, содержащую второй компонент в несколько большем
количестве, то момент начала выпадения первого компонента
наступает при более низкой температуре.
Последовательно увеличивая в растворе долю второго
вещества, мы дойдем до таких концентраций, когда раствор
в момент начала отвердевания будет насыщен одновременно
обОЕЕми компонентами (кривая 3). При его охлаждении на-
чинает кристаллизоваться сразу смесь веществ при посто-
янной температуре (в точке 6); поэтому кривая охлаждения
будет такая же, как и для чистого вещества. Так как эта
температура ниже температуры начала кристаллизации
любых других смесей, то смесь данного состава будет самой
легкоплавкой. Эта смесь называется жтектической (от греч,
ЭЕ (?^<чсе наследие России"
«хорошо плавящийся»). Она при своем плавлении образует
раствор, насыщен в biff относительно всех ее компонентов.
Если изучать охлажден не предварительно расплавлен-
ных смесей, начиная от второго чистого вещества, то полу-
чится картина, подобная изображенной на рис. 76, при-
чем кривых вида 2 будет столько, сколько взято проб между
чистым компонентом я эвтектической смесью.
На основании кривых охлаждения строят диаграмму
плавкости, перенося с ник точки, отвечающие температур-
ным остановкам или изменению скорости охлаждения, на
Рис, П. Дна г рампа
плавкости вещее in,
образующих 9ВПК1И-
ческую спесь
диаграмму температура—состав. Ниже рассмотрены раз-
личные типы диаграмм плавкости. В каждом случае ня гра-
фике совмещается их схематическое изображение с кон-
кретным примером. Каждый из последних относится к р =
= 1 атм.
Диаграммы плавкости систем, не образующих химиче-
ских соединений. На рис. 77 показана такая диаграмма
для системы Sb—РЬ. Эго случай, когда вещества неограни-
ченно растворимы в жидком состоянии и совершенно нераст-
воримы в твердом; он соответствует рис. 76. Точка а отве-
чает температуре плавления компонента А (сурьмы, 630 °C),
точка h—температуре плавления компонента В (свинца,
327 X).
Допустим, при 650 ’С взята жидкая смесь, состоящая из
80 % Sb и 20 % РЬ (точка /). При ее охлаждении (Кривая 2
на рис. 76) не произойдет никаких изменений до тех пор,
пока не будет достигнута температура, соответствующая
пересечению вертикали охлаждения с кривой аЕ крмстал-
У М. X. К л рл нетьяН ц 257
ЭБ ,:Научнов наследие России"
лизацми Sb (точка g). В этой точке расплав становится па-
сы [ценным сурьмой. Поэтому дальнейшее понижение тем-
пературы вызовет появление кристаллов Sb, и ее концен-
трация в оставшейся жидкости начнет уменьшаться, При
последующем охлаждении состав жидкости изменяется но
кривой кристаллизации Sb (отрезок g£), пока не будет до-
стигнута точка Е (83 % РЬ; 246 °C); В ней начнется совмест-
ная к ристал л и за цяя оставшейся сурьмы н всего взятого
CBiinna с образованием эвтектики — смеси мельчайших
кристаллов. После отвердевания всей системы дальнейший
отвод теплоты будет сопровождаться лишь уменьшением
Температуры.
Если охлаждению подвергают жидкую смесь, состоящую
из 83 % РЬ и 17 % Sb (кривая 3 на рис. 76), то состав обра-
зующемся твердой массы (точка Е) представляет собой чи-
стую эвтектику. При других соотношениях металлов к эв-
тектике будут примешены ранее выпавшие более крупные
кристаллы Sb или РЬ.
Из изложенного следует, что диаграмма плавкости в рас-
смотренном случае делится на пять областей: f — раствор;
Н — смесь раствора, насыщенного веществом А (в данном
случае сурьмой), и кристаллов этого вещества; !// — смесь
раствора, насыщенного В (здесь — свинцом), и кристаллов
В; IV — смесь эвтектики и кристаллов A (Sb); V —смесь
эвтектики и кристаллов В (РЬ). Кривая аЕЬ начала кристал-
лизации (конца плавления) называется линией ликвидуса
(от лат. 1 iquidus — жидкий), линия cEd конца кристаллиза-
ции (начала плавления) — линией солидуса (от лат. soli-
dus— плотный, твердый).
Эвтектика в системе'вода — соль называется криогидра-
том. Эго тонкая смесь кристаллов льда и соли удобна для
поддержания постоянной температуры. Последняя может
быть значительно ниже О °C; гак, эвтектика Н4О ZnCla
(51 % соли) отвечает t = —62 °C.
Итак, намеченная рнс. 61. ДнаграиНа получила свое завершен не
кз рис. 77. Остается лишь Добавить, что да я построения последней
Можно воспользоваться соотношением (11.20). Так как Кривые аЕ (/}
н ЬЕ (2) отиечакл равновесию между данные чистым компонентом и
насыщенным ям раствором, то в согласна с (11.17) k.l ~ Сл к Кл = Сй,
Выразяд концентрации через мольные доли, в доо гиге теки с (II.20J
получаем
-/?Г lnJVA=-iWA-7MA
и
-ЙТ 1п/Уи=йЯв-ГД.$а#
258
ЭБ ,:Научнов наследие России"
Приняв Л// н AS центов, получаем и равными мольегым характеристикам чистых комло- Та - мя.(^"1>А <1VJ6) P^mJa n I . v 1 (Пл)л (дй'4^—• <,v-47' (“ЦПлЪ Dlrt АГ (ГЬ)а R1""“
Эти уравнения выражают взакчосвяэк между температурой и составом
вдоль кривых к ристал л и 1а ин и веществ А и В. Их совместное решение
даст координаты эвтектической точки. Абсциссам точек а (ЛЛ = I)
и b (Л'в — I) в соответствии с этими уравнениями отвечают равенства
и
(^стл ) В = Р пл) пл) В
Следует подчеркнуть, что уравнении (IV. 46) н (IV.47) дают хорошее
совпадение с опытом лишь для сходных веществ (например, смеси
углеводородов}, так как вышеприведенный ВЫВОД основан ио допуще-
нии идеальности растворов.
Диаграммы плавкости систем, образующих химические
соединения. Если при’сплавлении* веществ между ними
образуется соединение, то на диаграмме появляется макси-
мум, отвечающий его составу. Такие диаграммы (рис, 78)
представляют собой как
бы сочетание двух диа-
грамм Езида рис. 77.
Здесь две эвтектики: Е,
и £8; под кривыми E-fi
и сЕг — области сосу-
ществования кристаллов
химического соединения
А,Вл, (в данном случае
TelJ с растворами, на-
сыщенными ими.
Острый максимум
свидетельствует о проч-
ности соединения; оно
плавится без разложе-
ния (конгруэнтно — от
лат. congruens — соот-
ветствующий, совпадаю-
щий), т. е. подобно
Рнс. 78. Диаграмма глаакости веществ,
образующих устойчивое химическое
соединение ДЛВ^
259
9-
ЭБ 'НзуччоЕ наследие России'
чистому веществу. Так, легкоплавкие металлы moi у г дать
тугоплавкий сплав. Примером может служить смесь Mg (fnjl =
— 651 °C) и Sb (#Дл — 630 *С), образующая сплав MggSb2
с /пл = 96! °C. Кристаллизация соединения А.,Вт по обе
стороны прямой СС1 протекает в неодинаковых условиях;
слева от нее молекулы АПВ.Ч находятся в сочетании с моле-
кулами А, справа — с молекулами В. Скачок условии крис-
РН£. 7^г Диаграмму плавкости йе-
шеста, образующих малоустойчивое
л им л четкое соединение А^Влз
таллпзацип на этой границе отражается в том, что с
является точкой пересечения двух кривых (Etc и cEJ,
т. е. в ней происходит излом кривых состав—свойство.
Такне точки называются сингулярными (от. лат, singula-
ris— отдельный, особы и) или дальтоновскими *.
Если же соединение при плавлении испытывает чаетич*
ную диссоциацию, то максимум на кривых становится округ-
лым {рис. 79), причем с ростом степени непрочности соеди-
нения его округлость увеличивается, а высота уменьшается.
Действительно, согласно изложенному на с. 162 и 172 пони-
жение температуры кристаллизации тем значительнее, чем
больше концентрация раствора (в данном случае содержание
свободных А II В в А*ВЛ). Особенно округлен максимум
ДЛЯ органических систем, так как в органических соедине-
ниях частицы связаны, как правило, сравнительно слабыми
молекулярными силами.
* В честь английского физика в хинина Дж- Дальтона соединения
пьстая иного состава называют дальтон «да мн,
дао
ЭБ ,:Научнов наследие России"
Если по оси абсцисс рисунков вида 78—80 концентра’
ция отложена в атомных (мольных) долях (%), то верти-
кали, отвечающие максимумам, непосредственно дают со
став соединений (см., например, рнс. 78).
Если число максимумов на диаграмме плавкости больше
одного, то это означает, что компоненты образуют несколько
соединений; количество нх равно числу максимумов (см.
с. 263), Примером могут служить некоторые кристаллогид-
раты.
Рнс. 80. Диаграмма л ла моста веществ,
образующих неустойчивое химическое
соединение Ал£#,
Наряду со стехиометрическими соотношениями между
компонентами в соединениях, что особенно характерно для
силикатных систем (4PbO-SiOt; 2BaO-SiO4; 3CaO*SiOf;
SCaO-SSiO^ NajO-SiOi и т. д.) и крпсталлосоиьватов (гид-
раты CuSQp HNO3, HiSOj, аммиакаты), существуют соеди-
нения, состав которых Fie отвечает валентности образующих
нх элементов. Таковы, в частности, интерметалличе-
ские соединения. Так, серебро со стронцием образует 4 сое-
динения. Поэтому на диаграмме плавкости системы Ag—Sr
мы обнаружили бы четыре максимума, отвечающих соста-
вам AgiSr, A&Sr, AgSr, Ag,Sra<
Для систем, образующих соединения, плавящиеся с раз-
ложе! шеи, диаграмма имеет вид, изображенный на рис. 80.
Выше температуры 4 соединение разлагается, т. е. его плав-
ление в точке D сопровождается распадом на жидкость d
£С1
ЭБ 'Научное наследие России"
и кристаллы А (точка nJ, т. с. при 1й возникает равновесие:
(к)Х^ W+A (к}
На рис, 80 видно, что CuCI-2КС!(КгСиС19) несколько выше
200 ’С распадается на две фазы: кристаллы КО и насыщен-
ный раствор, содержащий 60 % CuCL
Такое плавление, при котором составы исходной твер-
дой фазы и получающейся жидкости не совпадают, назы-
вается инконгруэнтным. В этом случае кривая кристалли-
зации химического соединения перекрывается кривой кри-
сталлизации компонента, вследствие чего максимум (точка
Л1) Отсутствует,
Рис. 8L. Диаграмма плавкости веществ,
образующих тпердые растворы (изоморф-
ные системы) (схема):
д — <15лаждэехый рдсплав; — состав пер-
вого кристалл ина н «г, — ист** послсЛи^П
цапли жлдкАета: Ьг3 — mpiiLLb я ео^глвов на-
^ыщ£циы1 жидких pdtrfrCipftp: — ирняая
cCKVatWB -рапивндолыг им гие^дыj рвстворои
Во всех рассмотренных случаях диаграммы плавкости
строятся по кривым охлаждения. Их вид для чистых Е;еществ
и химических соединений совпадает с линией 1 на рис. 76 —
варьируется лишь высота изотермического пояса (опреде-
ляемая тугоплавкостью вещества), его протяженность (опре-
деленная природой и количеством вещества), а также наклон
ее кривел инейных участков (производная dt/dt зависит от
перепада температур па границе «вещество — внешняя
среда»), Характер же кривых охлаждения смесей может не-
сколько отличаться от кривой 2 ла рис, 76.
Диаграмма плавкости веществ, образующих твердые
растворы. Вещества, молекулы (атомы) которых обладают
близкими размерами, поляризуемостью, имеют сходную
структуру, кристаллизуются из раствора совместно, обра-
262
ЭБ ,:Научнов наследие России"
зуя непрерывные твердые растворы (изоморфные смеси) —
твердую фазу перемен нога состава. Это объясняется тем,
что компоненты способны замещать друг друга в кристал-
лической решетке, давая смешанные кристаллы. Благодаря
такой замене в каждом кристалле содержатся частицы обоих
компонентов и вся твердая фаза представляет собой одно-
родную систему. Примерами подобного рода систем могут
служить смеси Au—Ад, Mo—W, К В г— KI, KsSO*—K?CrOdT
NaAlSijOg-^CaAijSijOj. Все они дают диаграммы плавкости
типа рис. 81. В данном случае кривая охлаждения для смеси
будет отличаться от линии 2 на рис. 76; после ab получим
наклонный отрезок, близкий к прямолинейному и отвечаю-
щий кристаллизации расплава, а затем—линию охлажде-
ния твердого раствора.
В диаграмме плавкости в случае образования твердых
растворов имеются две кривые. Верхняя (кривая ликвиду-
са) — выражает состав кристаллов, находящихся в равнове-
сии с расплавом. Поэтому область над кривой ликвидуса
отвечает условиям существования жидкой фазы, область
под кривой солидуса — условиям существования твердого
раствора; Область между обеими кривыми соответствует
сосуществованию жидкого сплава и смешанных кристаллов.
Если, например, охлаждать сплав, содержащий 60 % Au
(точка с), то из него начинают выделяться смешанные кри-
сталлы, первая порция которых должна содержать
^75 % Ан (точка 6t). В процессе отвердевания состав насы-
щенного раствора будет меняться вдоль отрезка Ьс\ кривой
Ликвидуса, а состав отвечающей ему твердой фазы— вдоль
отрезка fye кривой солидуса. Последняя капля жидкости
будет иметь составе;, а равновесный ей кристалл — составе.
Дальнейший отвод тепла приведет к охлаждению твердого
раствора (вертикаль cd). Нередко на практике кривая соли-
дуса отвечает неравновесным состояниям (пунктирная кри-
вая на схеме) — сказывается медленность изменения со-
става в твердой фазе, перемешивание расплава в сочетании
с медленным охлаждением * подтягивает» ее к равновесной
кривой.
Наряду с системами, подобными представленной ла
рнс. 81, встречаются изоморфные системы, диаграмма плав-
кости которых схематически изображена на рнс. 82 (Си—Ag,
Cu^AgjS, KI— КВг, NaOH—КОН, KBOj—NaBOj.
CaSiO,,—SrSiOg и др.) и 83 (например, Ni—Мп). Они пред-
ставляют собой как бы две диаграммы типа рис. 81, «срос-
шиеся» по вертикали экстремума.
2€3
ЭБ ,:Научнов наследие России"
Смыкание кривых ликвидуса и солидуса (точка е) озна-
чает тождественность составов сосуществующих фаз.
Поэтому для систем, представленных на рис. 81, и для
любых смесей, представленных на рис. 82 и 83 (кроме смеси е
на последних), охлаждение приводит к кристаллизации
твердого раствора, состав которого отличается от состава
жидкой фазы. Это, в свою очередь, вызывает постепенное
изменение состава расплава и, как следствие, температуры
его отвердевания (см. «елочки» на рис. 82). Для составов же,
Рис. В2. Диаграмма плавкости вешегтв, образую-
щих твердые растиоры (изоморфные системы) с
мз ней чумом точки пламени я (обозначения те же,
что н на рнс. 81)
соответствующих точке е, охлаждение приводит к отверде-
ванию всей массы при неизменности состава. Поэтому для
них кривая охлаждения будет подобна кривой 1 на рис. 76.
Аналогичные рассуждения, применимы и к процессам нагре-
вания (рис. 83).
Для болы ни яства типов диаграмм плавкости начальные
участки кривых кристаллизации ниспадают и ход их прямо-
линеен (см. рнс. 77—80). Так, небольшие добавки РЬ к Sb
или Sb к РЬ приводят к пропории01гальЕ1ым им понижениям
температур плавления (соответственно Sb н РЬ). Этот резуль-
тат непосредствен!]о следует из сказанного на с. 169 (см.
рис. 61). Ис ключей к я ми являются системы, в которых обра-
зуются твердые растворы. Как видно из рис. 81 и 82, участки
линий ликвидуса, примыкающие к осям легкоплавких
компонентов, идут на подъем — температура отвердевания
раствора оказывается даже больше температуры отверде-
264
ЭБ 'Научное наследие России"
взиня чистого растворителя. Проведите мысленно на рис. 60
кривую Ешже кривой DO (давление пара над твердым раст-
вором ниже, чем нал твердым растворителем), и вы при
дсте к тому же результату.
Рассмотренная диаграмма плавкости соответствует про-
стейшему случаю. Чаще встречаются более сложЕзые, соче-
тающие признаки простых, в частности, представленные
на рис. 79 и ЯО (пример — система КаО—ТааО5).
L_________I_________I__________________I________
о го ы) ао loo
Ил мде £ Сй Си
Рис. 83. Диаграмма плавкости веществ^ образу-
ющих твердые растворы (нЗСмСрфЕгые системы)
с Минимумом точки плавления;
а — HarpteacsiMfl твердый раствор. Еч — спстРя
ргервян н*Плн. — состпв. лослодзгего кристалл!;
frci — ьпивья госта ют* <т&ющ.|[к> кпист&ллов, friC —
Ир НАИН составов- pi BJ! O-MCIE Ы Л НМ ЖВДКНЛ р-а створов
Некоторые закономерности. Рассмотрим теперь на срав-
нительно простых примерах связь вида диаграммы плавко-
сти с положением элементов в период!, чес к ой системе.
Химически подобные элементы (соединения) часто дают и
аналогичные диаграммы. В частности, элементы одной под-
группы ИЛИ стоящие рядом в периоде с почти одинаковыми
размерами атомов обычно образуют твердые растворы.
Закономерность в изменении типа диаграмм плавкости на
примере щелочных металлов показана на рис 84: отличие
свойств Li от других элементов подгруппы приводит к тому,
что Li и Rb взаимно нерастворимы ни в твердом, ни в жиД’
ком состоянии; линия ликвидуса представляет собой гори-
зонталь при температуре плавления Rb, линия солидуса —
горизонталь при температуре плавления Li. Сходство Na
с более тяжелыми его аналогами вызывает значительную
Эдб
ЭБ ,:Научнов наследие России"
взаимную растворимость компонентов: диаграмма плавко-
сти для системы Na—Rb имеет вид рнс. 77. Еще больше
сходство К, Rb п Cs, поэтому они образуют изоморфные
смеси, т, е. их диаграммы имеют вид типа рис. 83. Такой же
облик имеет диаграмма для Си и Ag, а для Ag н Аи (сказы-
вается лантаноидное сжатие) она приобретает простейший
облик (рис. 81).
43 —полная ucjjRCTBopHмасть в кристаллическом к жидком состоя-
ниях; б— полипа растворимость s жидком н полная нерастворимость
в криста л.тяясскоч состоя и к и; г — полна н рясгворюгосгь в пристал-
Л11ЧССКОЧ II жидком отсго ннп яХ
Другой пример. Изучение систем ScClff—MeCi(Me=Ltl
Na, К» Rb, Cs) показало» что при переходе от лития к цезию
происходит закономерное изменение типа и характера диа-
граммы» в частности, увеличивается число соединений,
расширяется область концентраций, из которых кристалли-
зуются бинарные соединения» повышается их устойчивость
(что можно связать -с усилением стремления к комплексо-
образованиЕО в связи с изменением поляризующего действия
соответствующих ионов).
2. Вынлд типа диаграмм плавкости
из кривых энергии Гиббса
Теоретический подход к определению облика диаграммы плавкости
возможен нн основании анализа зависимости удельной энергии Гиббса
раствора от его состава (рис. 85}. Для механической смеси изотерма
О — / (Q будет прямой (линия /); дли раствора она криволинейна
с выпуклостью к ося состава (линий 2). так как образование раствора
сопровождается убылью <?1Ютгнциали. Если же, например, прн дан-
ной температуре вещества скЕыинмются ограничение, т. в, имеется
область сосуществования двух тианмко насыщенных растворов (рис. 57,
J < /к)? то на 6 — с = кривой появится участок, обращенный выпуК’
лостыо suepx (линия ? на рис, 85); абсциссы точек cud соответствуют
концентрациям этих растворов.
268
ЭБ ,:Н-аучноЕ наследие России"
Теперь рассмотрим изотерму 6 — / (СУ когда & равновесии с раст-
вором находятся кристаллы данного веществе (белее тугоплавкого,
чем други в) , т. е. раствор насыщен им. В этом случае диаграмма будет
иметь вид, изображенный на рнс 86. Так как при этой температуре
для вещества А более устойчиво тоердсе состоя и на (точка чем жид-
кое (точка д), то Gq Grt. Проведя на точки касательную к кри-
вой раствора, получим точку с. Ее абсцисса отвечает етеыву раствора,.
насыщенного (при данной температуре) компонентом А, а любая точка
между ах и г — механической смеси сосуществующих фаз.
Рис. 85, Зависимость
удельной энергии Гиббса
ог состава (схема);
/ — механическая смеж
2 — htcr pbii я че*| и а я
творммость; J — фгрвямчгН-
ная рэстяоримсниь (erf — об-
ласть расслоения)
Рис, SG- Изотерма удель-
ной ЭЕшргип Гиббса рас.’
твора (точка с) н находя-
щегося л равЕговесин с ним
кристаллического вещест-
ва А (точка ctj) |4 <;
< л1 (с^ма)
Проследим, какой ляд будут иметь изотермы G = ДО, начиная с
такой высокой температуры, когда но всем интервале растаор представ*
ласт собой однородЕЕую жидкость, и кончи я столь пилкой температурой,
когда вся масса будет твердой,
Начнем с fj при условии, что XCir’b- ?таЛ изотерме
отвечает рис. 87, /1, так как точка Л| окажется выше а (и соответственно
выше &). Для более низкотемпературной изотермы /Й1 характеризую-
щейся условием )д= (*Х^оти)в, точки и & приблизятся друг
к другу (но по-прежнему перья л будет ниже второй), а точки Д! н д
совпадут (так как ™ G?; рнс, 87, 5).
Выберем температуру Га так, чтобы (^)л > 4 >
изотерме отвечает рнс* 87, У? (тождествен ееый рис, S6L Оста на ад и на-
емся на еще более низкой температуре /t: пусть она удовлетворяет
требованию (^отн)д > /| = 0отв)в- Тогда ей будет отвечать рис. 87, Г;
она также аеевлогична рис. 86, но чшзг* ог — J стал больше.
При когда {/О„)А > /з < полу43*4 составы растворов,
нйсыщевных как к р еестя л.лам и А (точка с^), тан и кристаллами В (точг
ха d, рнс, 87, Д),
287
ЭБ ,:Научнов наследие России"
При дальнейшем охлаждении движущиеся навстречу друг другу
точки с2 н d сольются
G
Н
©
Ъ
<Ч«1'
£1
й
е; пусть эго произойдет при /* (рис 87„ £).
Обе касательные превратились в общую
Прямую — твсрЛыс компоненты находятся в
равновесии с одним и тем же раствором;
иными словами, при этой температуре раствор
становится насыщенным одновременно обоими
вещегтаамЕк
Последующее понижение температуры
х#тя н позволяет но аналогии г пышен зло-
жеиным провести касательные (мто и сде-
лано era рис. 87н Ж), однако они не будут
отвечать устойчивым равновесиям с жидкое*
тьво;]фп убудет сушеетвонать только
смесь игердш компонентов..
Проектируя теперь с изотерм f(B fJp |Э1
^Вр н t характерные для них точки на
Г =£ongt
Рис. 88. Построение диаграмма плавкости
по зависимости удельной энергии Гиббса
от состава (схема)
л з в
Рис. 87. Построение диаграммы плавкости
по зависимости удел той энергии Гкббса от
состава при разных температурах
график температура — состав, получим диаграмму плавкости. П дан-
ном случае это будет Диаграмма типа представлен пой на рис, 77 —
263
ЭБ 'Научное наследие России"
на ряс плана крисгаллнэуются ко^номонты, не образующие
твердых растворов; иэсггерыа
а точка е — эвтектической точкой.
Аналогичным путем легко пос-
троить и дна граммы ила а кости .любого
другого вида. Так4 для системы с
твердым раствором (диаграмма типа
Ац — Ли} на графике G =f (Q
выгни температуры плавления болм
тугоплавкого компонента будут две
крноые вида ? [см. рнс. 8Б (верк-
ляп для твердого раствора)!, ни-
же точки плавления легкоплавкого
килпюсспита, иаоборогн верхняя изо-
терму будет отвечать жидкой фазе.
Дл ft температуры, лежаихен между
<<„в)а 11 <A>JiP5™ «РЧРВД пересе-
кутся. Проведя к обеим кривым об-
тую касательную и проектируя, как
это Делалось а рассмотренном выше
примере, точки касания (с н d), полу-
чаем диаграмму плаякостн (рис. 88}.
Мы не ставили перед собой
задачу рассмотреть метод физико-
химического анализа во всей его
полноте; даже изложение тер-
мического анализа ограничили
простыми примерами.
11 ренета вл яется весьма пло-
л вляется эвтектической,
О 20 40 СО 60 100
НгО Moai>n.%SOj 50 j
Рис. 89. Сопоставление зэврь
снмости различны* свойств от
состава я системе HjO —SC\;
/ — температура криста л л язл-
цнн; т] — вязкость; р — плот-
ность; г — электрическое соп-
p&TJtfbieHne (а прльим нижнем
углу изображен фрагмент диа-
граммы план кости на участке,
примыкающем к пси H4SO#>
дотворным а ।завизировать кри-
вые к р иста>1 л и за! ш и совмес гн о
с кривымит характеризующими
другие свойства системы. По-
добное сопоста в л е н не, илл юстр и -
руемое на одном примере рис. 89,
позволяет уточнить выводы о
свойствах системы, обнаружить
особенности ее поведения, недостаточно четко выраженные
зависимостью отдельных свойств от концентрации.
ЭБ ''Научное наследие России1
РАЗДЕЛ V
ХИМИЧЕСКОЕ СРОДСТВО И СВОЙСТВА
ЭЛЕМЕНТОВ И ИХ СОЕДИНЕНИИ
ВВЕДЕНИЕ
Выше отмечалась связь различных свойств веществ с по-
ложением элементов в периодической системе Д. И. Мен-
делеева. Здесь мы вкратце рассмотрим этот вопрос на при-
мере — одной из важнейших характеристик вещества —
их химическом сродстве.
Хотя значения AG е отличие от ЛИ известны для немно-
гих соединений, однако рассмотрение различных процессов
в свете периодического закона, т. е. группировка их в род-
ственные реакции (например, в ряды процессов образования
сходных веществ), упрощает оценку реакционной способ-
ности. В таких процессах значения AS близки (см. с, 45),
т. е. значения AG отличаются от ЛИ примерно на одну и ту
же величину (см. с. 64).
Так, расчет AS™ в реакциях
“3fl(SOj)ffl (к) = Э„ОЛ (k)^SO9 (г)
дает для 20 изученных сульфатов ~44э. е, со средним откло-
нением в 3 и с максимальным отклонением в 7 э. е. Если
учесть разнообразие соединений (сульфаты одно-, двух-,
трех- и четырех валентных металлов), а также вероятную
погрешЕюсть значений ЛИ^л и Д(?^Я1 то вполне можно гово-
рить о Том, что ASjjs const. Тем больше оснований
утверждать это, имея в виду группу родственЕ!ых сульфа-
тов, например MgSO4, CaSOj, SrSO41 BaSO.j и RaSO4.
Глалл Г
ПЕРИОДИЧЕСКАЯ СИСТЕМА ЭЛЕМЕНТОВ
И РЕАКЦИОННАЯ СПОСОБНОСТЬ ВЕЩЕСТВ
L Глаьныс подгруппы
Химическая активность. Из учения 0 Строении вещества
известно, что активность металлов с увеличением номера
группы падает, а активность неметаллов возрастает, т, е.
270
ЭБ ,:Научнов наследие России"
наиболее реакционноспособными являются щелочные ме-
таллы, а из неметаллов — галогены. При этом если для
первых химическое сродство в подгруппе растет, то для
последних оно уменьшается.
Эти соображения иллюстрируются следующим прЕ9ме-
рой *:
для металлов
Nal (к) Mgl# (к) АП, (к)
1/2 (-86) 1/3 (—72,671)
К1 (к) Са1( (к) Cal, (к)
—77,66 1/2 (—137^6) 1/3 /—52)
для неметаллов
Ni l, (г) —3,993 HSO (г) —54,638 HF (г) —1x5,203
PH, (г) HjS (г) на (г)
3.2 —8.081 —23.656
AsH, (г) H,Se (г) НВг (г)
16,47 4,71 —12,238
Н2Т& (г) HI (г)
20,353 0,425
Последние величины иллюстрируют уменьшение химической
активности элементов V, VI и VII групп и, как следствие,
рост неустойчивости ЭН,Ъ ЭН5 и Э11 с увеличением поряд-
кового номера Э; в то же время они свидетельствуют об уси-
лении восстановительной активности соединений в этих
вертикальных рядах, Изменение восстанови тельной спо-
собности для ряда НГ можно показать и на примере следую-
щих реакций’
2HF <т>+ HaSO« (ж)=Г! (O+SOi (r)-t-2HjO (ж) {1}
2на (г) + Н^О4 (ж)»Cls (г) + SOfe (г)4-гн^ (ж) (II)
2HI (O + HjSQi (JK)-Ii (K)+SOb (г) + 2НгО (ж) (Ш)
8HI (гЦ-HjSO, (ж) = 4!, (K) + HgS (г)4-4Н,О (ж) (EV)
Дли них находим: (Дб^я)| = Jl 0,231; (ДОзд)ц =
= 25,137; - —21,025 is (AG«S)JV = —73,31 ккал.
Эти результаты, свидетельствующие об усилении восста-
нови тел ьн о й активности в ряду И Г, означают вместе с тем
возможность получения IIF и НС1 взаимодействием фтори-
дов и хлоридов с серной кислотой и невозможность полу-
* Здесь и в дальнейшем под формулами веществ указами я (мне-
ния стандартны» тиснений энергии Гиййсз (ккал/моль) нх
образования. Они, вдх и значения 5^й1, заимствованы из приложе-
ния L
271
ЭБ ,:Научнов наследие России"
чеки я аналогичным путем HI; последнее соединение будет
-< -J +1
восстанавливать S до S и S.
Переход в пределах данной группы элементов от второго
периода к третьему и от третьего к четвертому обычно свя-
зан со значительным изменением свойств; переходы же от
четвертого к пятому, от пятого к шестому и от шестого к седь-
мому сопровождаются сравнительно небольшим и мало
отличающимся изменением свойств соединений. Эта особен-
ность проявляется и на химическом сродстве. Она может
быть показана на примере реакции
'/Ле1° (к)-Н/,Н,0 (ж)~МеОН (к) (V)
Для щелочных металлов имеем: LiOH(—9,632),
NaOH(—17,452}, КОН(—23,749). Для Me = Rb, Cs, Гг
результаты вычислений не могут быть надежными вслед-
ствие недостоверности значений образования соот-
ветствующих оксидов и гидроксидов.
Все возрастающий разрыв в значениях для соеди-
нений второго и третьего периодов достигает своей кульми-
нации для пары F—С1Г что свидетельствует об исключи-
тельно большой реакционной способности фтора и высокой
прочности его соединений, 3>го видно, например, из следую-
щих данных:
CF* (г) CGI, (г) СВг* (г) CI, (г)
-212,33 —14.50 8,6 62,356
Устойчивость CF4 по отношению к углероду и фтору харак-
терна и для других* перфторал каков (ChF^,.^), чем объяс-
няются цепные свойства этих соединений— их инертность,
термическая стабильность и т. д.
Чем же обусловлена большая реакционная способность
фтора — энтальпийным или энтропийным фактором?
Для решения этого вопроса сопоставим фтор с хлором.
Так как
ор9в) г, ” (^мя) г ₽ J 5 — 37.92 ел 10,53 э, С-.
а
Д^лвр=(^*)с1,-(^п)а = йЭДО-39,457= !3.836 Э. е„
т. е. Д54тор отличается от Д5ХЛ0р всего лишь на 3,303 э. е.,
то решающее значение имеет величина Д//, т. е. большая
прочность связи фтора с другими элементами,
ЗБ пЙ1учнов наследие России"
С характером изменения активности элементов по под-
группам связано направление многих процессов, например
реакций вытеснения;
В а (к) -bMgCF* ВаС!г 4- Mg (*) (VI)
Ка I (к) + VjCla (г)=NaQ (Ю+V* к (О (VII}
Из табличных данных следует., что (&G^)vt = —51,671 к
(&G*m)vil = —23,095 ккал, Чем дальше расположены эле-
менты по подгруппе, тем глубже должны протекать про-
цессы вытеснения, подобные (VI) и (VII)
Кислотно-основные свойства. Изменение химической
активности проявляется, в частности, и в изменении к по-
лотно-основных свойств. Для иллюстрации рассмотрим
закономерность изменения значений Ди^ для элементов
подгруппы бериллия в процессах
МеС (к)-|-СОа (r) = MeCOj (к)
И
МеО (к)-!-5Оя (rJ^MeSOi (к)
Для карбонатов стандартные изменения энергии Гиббса
образовании из оксидов равны: ВеССь, (3,791), MgCOa
(—15,776), СаСО5 (—31,231), SrCO;t (—43,941), ВаСО,
(—52,141). Для сульфатов ЭТИ величины таковы: BcSOt
(—36,108), MgSO4 (—49,565), CaSO4 (—82,55), $rS04
(—97,45), BaSO4 (—102,69). Из этих данных вытекает ряд
выводов, в частности рост устойчивости МеСО» к MeSO4
по отношению к оксидам с увеличением порядкового но-
мера металла. На примере карбонатов этот вывод подкреп-
ляется значениями их температур диссоциации (рсо, —
= I атм):
МйСОя СзСОд ЭгСОя BaCOj
230 т ВП'С ИМ °C 1300 т
Единственным соединением, которое легко распадается на
оксиды, является ВеСОа (легкость его разложения — ре-
зультат значительного поляризующего действия Be®*).
Из сопоставления значений разложения карбона-
тов н сульфатов следует, что SOs является более кислот-
ным оксидом, чем СО2. Это проявляется н в том, что суль-
фаты устойчивее к нагреванию, чем карбонаты (они начи-
нают разлагаться при более высоких температурах).
£73
ЭБ ,:Научнов наследие России"
Иллюстрацией закономерного хода свойств по подгруппе
могут служить и следующие данные:
HOG! (р)-НС1 (р}+>/А (О
—19,17 —31,37 О
НОВг (р) = НВг (pH- '/А (г)
—10,7 —24,85 О
НО! С[»)=Н1 (pH1/А (г)
—23.7 —12.33 О
Оеш отвечают росту устойчивости кислот в ряду НОГ
(Г = С1, В г, 1). Легко подтвердить ослабление кислотных
свойств л pit переходе от СО. к SiO< (см. с. 287).
Что' касается усиления основных свойств в ряду оксидов
металлов основных подгрупп, то дли щелочных металлов
это следует из значений Аб2»ч. отвечающих реакциям (V).
Другой иллюстрацией могут служить процессы
GaA (к)+6НС1 (р)=2ваС14 (pJ-f-ЗН.О (ж)
1пА (к)Ч-БНС! (р)«21пС1а (p} + 3HsO (ж)
IIА (к)4-бНС1 (р)-2Т1СЦ (р)-З-ЗН/) (ж)
(VII!)
(IX)
(X)
Хоти использование значений ДСЙЯ для ЭС1Д (к) вместо
ЭС1а (р) (последние неизвестны) делает расчет приближен-
ным, однако из того, что (AijisJvni == (28.69'1) >(ЛО«я)Гх=
= (—39,854) > (Д(?«*)х = (—45,079) ккал, следует, что
растворимость оксидов в кислотах возрастает, т, е. в ряду
Ga2Oj" ln4O3—Tl2Os наиболее основной оксид—по-
следний.
Стремление к проявлению различной степени окисления.
Проанализируем процессы, в которых данный элемент
проявляет различные степени окисления а одной реакции.
Рассмотрим их на примере диспропорционирования, огра-
ничившись оксидами элементов больших периодов, в част-
ности подгруппы германия, т, е. процессам и
Э» (к)+эЬ4 (к)=2Э& (к).
где Э Ge. Sn, РЬ. В данном случае
Ge (k)+GcO, (x)=2GeO (х)
Sn (k)-|-SiiQj (K}a2SnO (к)
Pb (кНРЬО, (K) = 2PbO (к. I)
(Xi)
(XII)
(XIII)
Из табличных данных находим, что значения Абдо для
рассмотренных реакций равны 19,689 (XI), 1,45 (XII) ц
—38,22 ккал (XIII). Эти результаты означают, что пер-
вый процесс должен протекать справа налево, что SnO4
274
ЭБ ,:Научнов наследие России"
лить несколько устойчивее SnO, а последняя реакция
идет слева направо. Обобщая полученный результат, при-
ходим к выводу, что для германия более характерна сте-
пень окисления +4, а для свинца типична степень окнеле-
h в +J
НИЯ +2; иначе говоря, Ge — восстановитель, а РЬ — энер-
гичный окислитель.
Рнс. ЙО. Изменение
стандартной эпе|>1 ин Гиб-
бса Да’ЛЧ об-
разования обидой ме-
M&EI7OB IV ГруПЛЫ
К аналогичным результатам легко прийти и в отноше-
нии других подгрупп, в частности подгруппы мышьяка:
соединения As устойчивее соединении Аз, в то время как
состояние Bi в соединениях висмута нехарактерно*
Рис. 91, Взаимосвязь между нэме-
неиием стандартны к энергий Гиб-
бса (ккал/моль) образования
аналогичных соединений а л Боливия
и бериллия:
/ - AICI (г) к BcCI (г>; 1 S _ A|N u
ВеМ: J — AhS, я Ве$; « — ДПЛ и
Вг|в: fl — Al 1[ ВсВг±; * — Al|CJ-
(г> н (г). 7 — AICIi м ВйСГ.Л -
А1Л+ (pF и ? - ALOj и В^О-
М _ ц<А1 (ОНИ и /7 _
Al (QН)j II ЦС (ОН)« - Alft И
UtFrf /J - Л1л (SQJj BrSO< ь
Остановимся еще на одном примере влияния изменения
степени окисления па свойства химических соединений.
В ряду нею (р) — HClOi (р) — нею, (р) — НС1О4 (р) си-
ла кислот возрастает. Об этом можно судить даже по тем
разрозненным и ориентировочным данным, которыми мы
располагаем, — по ориентировочным значениям для
275
ЭБ 'Научное наследие России1
НСЮ (р) (—19,17) и нею. (р) {—20). Учитывая, что эти
величины войдут в (Лб^)^пИ11[||| с обратным знаком, при-
ходим к выводу, что HClQj — наиболее сильная кислота.
Втор ич на я sтс риоди ч но с т ь- Немец еотон кость измене ни и
свойств элементов и соединений, которая проявляется в так
называемой вторично й иериодаадос/пы, обнаруживаете я и
в характере изменения
значений ДО-™- Проиллюстрируем
эту особенность на приме’
ре оксидов элементов IV
группы; если в ряду СОг
(г) — SiQj (кварц} — Т1О?
(рутил) — ZrO£ (к) — НЮ^
(к) значения ЛО^ меня-
ются монотонно, то в ос-
новной подгруппе монотон-
ность нарушается (рис, 90)-
Нс которые аа иономер-
нести. К рассмотренным
Рис. ЭЙ. Температура начала &заи-
модейетвнл /1ЬЙ (СС) титана с неко-
торыми простыни веществами; и
скобках указаны значения измене-
ний стандартной эдершн Гибйса
(ккад/зиоль) обрззонаиия со-
отйтстеующнх СМДНИСЧИЙ (fl);
взаимосвязь между 1жвн 46^обра-
за ванн я Т1Г( (Г = F. С1, Вг, i) (6}
Рнс. 93* Взаимосвязь между
стандартными зЕпролнямя S;fl*
(э. в.) азота и окиси углерода лрл
равных температурах Г (К)
правильностям можно прибавить и другие. Как известно^
элементы 1 расположенные в периодической системе по диа-
гон ал и т проявляют черты сходства, Так, Al характеризуется
ЭБ <ное наследие России"
диагональным сходством с Be (оба металла имеют близкие
температуры кипения, устойчивы к холодной воде, их кар-
бонаты нестабильны и одинаково взаимодействуют с водой,
оба склонны к комплексообразованию и т. д.)< Об их
сходстве свидетельствует и рис. 91: практически для всех
при веден стык на нем веществ точки тяготеют к прямой,
отвечающей равному химическому сродству
По-видимому, существует енмбатность в температуре
начала взаимодействия данного простого вещества с дру-
гими 11 ХИМ! ЧТИ ОМ сро-
дстве между ними фпс.
92, а); более того, для
сходных веществ эта сим-
бати ость носит количест-
венный характер (рис.
92, б).
Си мбат в ость увеличе-
нии энтропии сходных
веществ с их нагреванием
подчас позволяет восполь-
зоваться приближенным
линейным соотношением
Sm = 4~ fi, в кото-
Рнс. 94, Ваа и посиять между ста-
ндартными энтропиями SJ„ (э.с.)
галогенов и благородных газов
ром при равных температурах сопоставляются значения
энтропии веществ-аналогов Ми N. При большом сходстве
объектов сравнения А в нем близко к единице; это наблю-
дается, например, для азота л окиси углерода; для них
в интервале 50— 10 000 К
(5'т)сой£(^Н+1.5
со средней погрешностью 0,8 э. е. (рис, 93}, т. е. их энтро-
пии в первом приближении одинаковы. Близость свойств
СО и Ns определяется последовательностью расположен ня
С, N и О в периодической системе.
Рис. 94, иллюстрирующий уравнение (Н.12), свидетель-
ствует о том, что водород является аналогом (разумеется,
не полным) галогенов.
2. Побочные подгруппы
Химическая активность. Меньшая реакционная способ-
ность элементов побочных подгрупп по сравнению с эле-
* Ода сгвддает сдойюшемию JL L21, в котором 4^J и
1277
ЭБ ,:Научнов наследие России"
ментями основных подгрупп подтверждается следующим к
примерами:
1. Сродство кадия и меди к галогенам:
KF (к) КС1 (к) КВг (к) KI (к)
— 128,515 —97'0 —90,120 —77,00
Си И (к) CuCL (к) СиВг (к) СиТ (к)
-50 —ЭД,70 — 23^0 —16.65
Ряд, анологкчный нижнему, будет и для СнГв: если
(ДСмн) сир, — 116, 582, то (AG«s)cur, = —3 ккал. Дей-
ствительно, первое соединение начинает разлагаться при
950’С, а второе уже при обычной температуре.
2. Если для реакции
2Rb (kJ+’/A (r) = Rb*O (к)
ДС»»^“-70 ккал, то для реакции
2Ag {к) Ч- VА (г) = AgXl (к)
AG =—2,69 ккал. Первая величина отвечает большому
сродству рубидия к кислороду (он легко окисляется), вто-
рая — непрочности окиси серебра.
3. Если для реакции
SlO (x)-l-SOa (r)mSrSO4 (к)
AGj,u = —97,45 ккал, то для реакции
CdO (к) 4-SA (O-CdSO, (к)
Дб;,8 = —53,39 ккал. Этот результат подкрепляется и
сопоставлением реакции
Sr (K)-f-S (ром6)4-2Оа (r}==SrSOt (к)
для которой AGjm = —323,46 ккал, с реакцией
Cd (K)4-S (ромб)-|-2О, (г)=сазО1 (к)
для которой &GjK — — 196,91 ккал,
В пределак подгруппы устойчивость однотипных соеди-
нений уменьшается, например:
ZnO (к) CdO (к) HgO (к, 1)
—76,64 —54,81 —14,02
Ход значений ДбмВ в этой совокупности веществ соответ-
ствует уменьшению их термической стабильности (ZnO
сублимируется при 1950е С без разложении, a HgO рас-
падается при 430" С), обусловленной ростом поляризации
в ряду Zn—Cd— Hg,
276
ЭБ ,:Научнов наследие России"
Для сульфидов аналогичная картитга:
ZnS (к) CdS {к) HgS (к)
—47,973 —36.51 “12,1
Эти соединения вследствие меньшего сродства металлов
к сере, чем к кислороду* будут разлагаться при более
низких температурах.
Рассмотренное отличие соединений элементов побочных
подгрупп от соединений элементов основных подгрупп еще
на двух примерах иллюстри-
рует рис, 95,
Рассмотрим другой при-
мер:
Сг1я (к)
—55
Мо1а (к)
-13
WI, (К)
Он отвечает тому, что хром
при нагревании соединяется
с любым галогеном, вольфрам
реагирует только с хлором.
Иллюстрируя различия в
свойствах элементов главных
и побочных подгрупп, следу-
ет помнить к о некотором их
сходстве. Так, хотя
(™76,64) отличается от
АСйвМйО (—135,985) чуть ли
не вдвое, однако стремление
к взаимодействию оксидов
цинка и магния с данным со-
реагентом примерно одина-
ково. Об этом можно судить
по близости прямой рнс. 96
к пунктиру, отвечающему
равному химическому срод-
ству (расположение точек
Рис. 95. Зависимость стандарт-
ной Энергии Гиббса
(к нал/моль) образования хлори-
дов (а) и гидроокисей (б) эле-
ментов второй группы от поряд-
кового номера элементов 2
свидетельствует о том, что
MgO несколько более основной оксид, чем ZnO).
Лантаноидное сжатие. На химизме элемеЕгтов VI (VII)
периодов сказывается лантаноидное (актиноидное) сжатие.
Вследствие лантаноидного сжатия многие свойства соеди-
279
ЭБ 'Научное наследие России"
нений V периода, отличаясь от соответствующих свойств
их аналогов (по подгруппе) IV периода, близки к свойствам
соединений элементов VI периода. Например:
TiN (к) VA (и) МнОг (р)
—73,637 —339,4 —105,23
ZrN (к) КЬгО^ (к) ТсО; (р)
—80,47 - 421,6 —150.63
HIN (к) ТаА (к) ReO“ (р)
-8L4 —457,1 —167,1
К этим примерам можно присоединить и многие другие,
рассмотрев, в частности, ход значений в рядах
Рнс. 96. Взаимосвязь между
стандартной Энергией Гиббса
(ккал/моль) взаимодей-
ствия окислив инн на И -магния
с различными веществами:
I — 30 (X) + SQ, (ЖГ 7 - 530 (к) +
4- 510, (Ч :,В»рC1J: 3 — 30 (К) +
7- мео,(fc): 1 - 30 (к) + СО, (Г|:«—
30 (я) н- ЫО, (а-кварц): S — ЭО
<к] + ц,о (Ж): 7 - эо (Н}т-
+ К,0, (г)
Рис. 97, Взаимосвязь между стан-
дартными энергиями Гнббса
(ккад/моль) образования различ-
ных соединений ниобия и тантала
ТЮг—2гО,—НЮ- (с. 276), CrtB—MoL—WI, (с. 279) Близки
и энтропии (S, э. е.) веществ-а налогов:
PbWO. (к) ....... ........40.2
РЬМо04 (к).............. 39,7
На многих примерах лантаноидное сжатие иллюстрирует
рис. 97; для самых различных соединении точки тяготеют
к линии (пунктирная прямая), отвечающей (1.15), в кото-
ром ,4 =» 1'и й = 0.
280
ЭБ ,:НауччоЕ наследие России"
Стремление и проявлению различной степени окисления.
Рост стремления проявить максимальную степень окисле-
ния для элементов побочных подгрупп (в отличие от эле-
ментов основных подгрупп) видна из следующих примеров:
CrO-j (К) Л'оО, (к) WOa (к)
—120,7 —150,7 —1*2,62
С этими цифрами можно связать то, что СгОэ — сильный
окислитель, a WO;1 испаряется без разложения.
Сравнивая
Мп А (к) Тс/Л (к) Re А (к)
— —130 -224,1 —255,0
приходим к выводу о росте устойчивости (падении окисли-
тельной активности) в этом ряду оксидов. Действительно,
первое соединение кипит при 90”, второе—при 260°,
третье — при ^600° С без разложения.
Уменьшение тенденции к низшей степени окисления
вытекает из следующих данных?
VjQi (к) (к)
—315,2 —339,4
NbA (К) Mb А (к)
-354,5 . -421,6
По мере увеличения степени окисления элементов уси-
ливаются кислотные свойства их соединений. Так, в ряду
MnO——Мп О..—Мп4О7 основные свойства ослабевают.
Этот факт подкрепляется следующими данными:
МпО (к) 4- (ж) - Мп(ОН )4 (к), , = —3,047
и
Мп А (к)4-ЗНаО (ж) = SMnfOHj., (к), <10^,-17,559
Другим примером служат реакции
VdCotOHj^+HCI (p)-i/£oCli (P) + H»O (ж)
н
'/AfOHb-f-HCJ (p)-VACU+HiD {ж)
для которых 4G »й равны соответственно —2,62 и
1,15 к кал/моль НгО.
Сходство с элементами основных подгрупп в высших
степенях окисления. Известно, что различие между элемен-
тами основных и побочных подгрупп велико для низших
(в том числе нулевой) степеней окисления и становится
28 [
ЭБ ,:Научнов наследие России"
менее значительным в высших степенна окислении. Это
обстоятельство подтверждается следующим примером:
l/'2Cr3Q, (к) ЗОЙ (г)
—I2G.6 -71,752
Kj&O, (к} К^О4 (к)
—308,26 —314,62
Даже приняв во внимание не тождественные степени окис-
ления н эффективные заряды хрома и серы в их оксидах,
а также различие в агрегатном состоянии последних, можно
по большой разнице ДО«* их образования судить об их
несходстве. В то же время хромат и сульфат сходны. Эго
следует и из близости их стандартных энтропий, они равны
соответственно 47,8 и 41,96 э. е.
Другой пример: Мп ничего не имеет общего с CJ.; соеди-
нения Мп не похожи на соединения С1; но Мп аналог с!
к? +?
(в частности, анионы MnOj и СЮ; близки по свойствам).
Так, (5зм)кмпо< = 41,0, а ($ш)кС1о, = 36,1 э. е. Можно
указать и на такой пример; = 35,7; ($й»)м#,МпО. =
= 38,7 э. е.
3. Периоды
Химическая активность. Химическое сродства элемен-
тов друг к другу усиливается по мере отдаления их по
периоду* Это видно из следующих данных:
y3BN (к) ‘АВеО (к) LiF (к!
i/a г-54,195) Va (— 136,12} —139,746
Ь (к) J/3NaaTe (я) Nal (к)
х/,{----50) Ч» (-S3) —68,193
Значительно различаются свойства соединений как так
и ^-элементов между собой, свойства соединений d-элемен-
тов более близки и сходство соединений /-элементов еще
больше. Это нашло отражение в характеристиках веществ,
представляющих малые и большие периоды. Вот один
пример (см. рис. 25):
КС1 (к) CaC!t (к) ScCh (к)
—97,70 —179,05 —207.8
T1CI, (К) VCh (к) СгС1а (к) МпС14 (к) FeCla (к)
-156,341 -123,45 —118,0 103 —79.S21
СеС1, (к) РгС, (к) NdCl3 (к) PmClj (к)
—239,7 —234,5 -230,1 £34
SmCi3 (к) ЕиС[3 (к) GdCl3 (к) ТЪС13 (к}
—233 —2QO.O —222,5 —224
Ж
ЭБ ,:НаучноЕ наследие России"
Здесь, как и в случае движения по подгруппе, существует
количествен л а я закономерность (рис. 98).
Сходство элементов семейства железа покажем ня при-
мере гидроксидов, хлоридов, карбонатов, оксидов и суль-
фидов элементов семейства железа:
Fe(OH)t (к) — J 14,ЬЙ Co(OH)| Jk) FeCL (к) l’<?COh (к) — 158,96 FcO (к) FeS (к)
—72,28 -21,09
СЮ, (в) —63,884 CoOOj (к) —153,0 СоО (к) —51Л CoS 00 —20,2
Ni(OH)/ (к) NiClf (и} —Gl,67) NiCCs (к) NiO (к) NiS (к)
-100.55 —147,688 —&0.S73 —18,371
Кислот но-основные свойства. Изменение кислотно-ос-
новных свойств рассмотрим на ряде процессов с участием
соединений элементов треть-
его периода:
а) взаимодействие основ-
ного оксида с водой:
NasO (х)+Н.О (K) = 2NaOI4 (к),
Лб;„=—34,904
MgO (кУ+HjO M-MfifOH), (к),
А! А (к) + ЗНаО (ж)=2А1(0Н}а (к),
AG'1]t =-4,721
в расчете на I моль Нй0
получаем соответственно
—34,904; —6,562 и —1,57
ккал. Эти результаты нахо-
дятся в соответствии с тем,
что Na2O — наиболее основ-
ной оксид, MgO обладает
меньщим сродством к воде,
а ALO. практически не взаи-
модействует С ней, это отве-
чает и тому факту, что
NaOH — сильное основа-
ние, Mg(OH)B — основание
средней силы, а Л] (ОН)а
— амфотерное соединение;
Рис. 98. Взаимосвязь между
изменеинем стандартной энергии
Гнббса AG’Bj (ккал/моль) обра-
зледНИЯ кристалл и чески к ди-
хлеридов d-sn&ieKiOB четвертого
периода к их кристаллических
дифторнда к триХлоридов
б) взаимодействие кислотного оксида с водой:
РА» (К) + 6Н.0 {ж}- 4 Н аРО« (и), A G|tB = —84,974
SOi (к) -i- (ж) = 11 jSO4 (ж), AJ = —19,28
2H3
ЭБ 'Научное наследие России1
или в расчете на I мать FLO соответственно —14,16 н
—19,28 ккал, Танни образом, в ряду R*Ojij—SOs растет
реакционная способность (а в ряду НяРО4—H4SO4 воз-
растает сила кислот). Аналогичные результаты получаются
при анализе взаимодействия кислотных оксидов нс с На0,
а С каким-либо основным оксидом.
Стремление к проявлению различной степени окисления.
Эту характерную тенденцию для соединений близких л о
свойствам элементов (т. е. входящих во вставные декады)
покажем на примере семейства железа. Важно не только
то, что устойчивость как Ме(ОН)4, так н Ме(ОН), растет
от никеля к железу, но и вывод, который вытекает из
анализа значений для реакций
Fe(OH)a (K)-f-VA (О + ^НЛ M-FefOH). (к)
Со(ОН)( (к)+>/А (гЦ-^НгО (ж)=Со(О!1), (к)
Ni(CH)a г)Ч-1/*Н/> (*) — Ni(OH)a (к)
На основании табличных данных получаем для них соот-
ветственно Д(г^я = —24,21, —4,897 и +8,839 ккал. Эти
результаты означают, что из шести гидроксидов макси-
мальной окислительной активностью обладает Ni(OH)t, а
миинмалыюй FefOHjj, Обобщая данный вывод на раз,тнч-
1J +1
ные соединения Э и Э (Э = Fe, Со, Ni), приходим к заклю-
чению, что для железа более характерно состояние Fe,
тй 11 И*
а для никеля Ni, поэтому, в частности, Ni будетокислятьре,
Изменение стремления к диспроЕторцисшированию ио
периоду видно иа следующею примера:
2Au (л) + АиС13 (к) =3AvCi (к),
Hg ftKj+HgCl, <к) = НеаС1я (к), ДС;^ = -7Д5
2Т! (H + TlClj (K) = 3T1CL (к). Д0;и=-62a624
Из него можно сделать некоторые выводы, в частности,
&ывод о необходимости располагать активным окислителем
4 | +J
для перевода Т1 в Т1. Мысленно продолжив этот ряд до Bi
включительно, приходим к заключению о предпочтитель-
ности ₽£) ла сравнению с РЬ и о нестабильности соедине-
-I Ь
ний Bi (см. также с. 274).
284
ЭБ ,:Научнов наследие России"
Глапл П
ВЛИЯНИЕ ТЕМПЕРАТУРЫ НА ХИМИЧЕСКОЕ СРОДСТВО
В соответствии с (11.10) влияние температуры ну хими-
ческое сродство определяется знаком и величиной AS, Это
позволяет нагреванием (или охлаждением) увеличить или
уменьшить реакционную способность вещества или сово-
купности веществ, усилить стремление к протеканию нуж-
ного процесса (на совокупности конкурирующих реакций.
Рис. 99. Зависимость измене-
ния стандартной энергии Гиб-
бса AG*,, (ккал/моль)образова-
ния некоторых соединений
кальция от температуры Т'(К):
пр — теылерлура аллотропного
превращенья: п — температура
плавления: р — температура раз-
ложения
Рнс. КМ). Зависимость нзиеве-
мия стандартной энергии Гиб-
бса (ккял/мйл11) образа-
йання ' егс которых соединен и А
црглка о г температуры 7 (К)г
п — температуря плавления, я —
температуру. чпления
т+е. обладающих близкими AG) и т. д. Если AS >0, то
переход на высокотемпературный режим благоприятствует
течению процесса; это особенно важно в тех случаях, когда
при низких температурах, вопреки отрицательному зна-
чению Д0+ реакция в силу инертности реагентов не про-
текает. Так, хотя для реакции
NHa (r}-h CliO (K}=Nf (r)+Cu (к) + Н^ (г)
AG< 0 (AG«H = —19J25), однако на холоду она не идет;
повышение же температуры вызывает течение процесса;
265
ЭБ ,:Научнов наследие России"
500 ЮОО 1&0Q 2000
Г ’
благоприятно сказываясь на его кинетике, оно не умень-
шает сродство NHg к СоО, а, наоборот, увеличивает его,
так как в данном случае AS >0 (AS™ = 43,568
э. е.).
Во всех случаях, когда процесс сопровождается умень-
шением объема, т. е. AS < 0, повышение температуры вы-
зывает возрастание Д<7. В частности, это относится к реак-
циям образования большинства
твердых соединен и II из простых
веществ. На рнс. 99 и 100 это
обстоятельство иллюстрируется
процессами образования ряда со-
единений кальпня и цинка. Отме-
ченные ранее выводы об уменьше-
нии активности в ряду F2—О,—Бгг
— fг (с. 271) и о большой реакцион-
но й с пособнос ти э л еме ej тон 11А - под-
еру ппы по сравЕзению с элемен-
тами 11 Б-подгруппы (с. 278) спра-
ведливы н при высоких темпера-
турах.
В ряду Cls(r)—Вгг (г)— L (г)
повышение температуры в соот-
ветствии с уменьшением теплоты
диссоциации при переходе от С12 к
Вг2 и Ij оказывает ослабевающее
воздействие, так как влияние тем-
пературы па равновесие диссоциа-
ции Г± тем значительнее, чем про-
чнее эта молекула. Эго иллюстри-
руется рис. 101. Сравните,
например, наклоны касательных к
кривым АСР= f (Г) при Т = 1250 К.
Выпуклость ЭТИХ KpEfBbJX К ОСИ
ординат, означающая замедле-
ние роста с Т, вместе с тем свидетельствует о том,
что повышение температуры вызыЕззет уменьшение теп-
лоты диссоциации каждого из этих вещесги. Из рис. 101
следует также, что в ряду Fj—С14—Вгг—ls фтор за в и мает
особое положение; прочность его молекул значительно
меньше прочности молекул хлора (сравните при данной
температуре ординаты и наклоны прямых). С этим связана
н большая убыль энергии Гиббса при образовании фтори-
дов (в частности, CFt( CaFs и ZnFj.
266
ЭБ ,:НлучноЕ наследие России"
Рис. 101. Зависимость
константы равновесия
реакции
Га(г)-2Г(г)
or температуры Г (К)
(Г = F; Cl; fcr; 1)
В тех случаях, когда происходят параллельные реак-
ции, следует учитывать влияние температуры на каждую
из них.
Так, в процессах
NH*NOt (н} |
-Njp (r)4-2HsO (г)
(г)-и'/А (г)+2НаО (с)
(Л)
(Б)
протекающих при нагревании (AS >0), ДО^н равны соот-
ветственно + 14,186 и •—10,698. Во л се и н пи [с । [ внос течсн 11 е
второй реакции должно наблюдаться и при повышенных
температурах, так как Д5в >Д5д. Действительно,
(Д$лв)л = 61,556 э. е . в то время как (А£до)б в 103,78 э. е.
Мы уже отмечали (с. 50), что для реакций, в которых
участвуют вещества-ан а л от и, AS = const, Так, для про-
цессов
Э (K)+VA (г) = 30 (к)
имеем AS;» = — 24,02 (ZnO), —23,77 (CdO) и —25,84 э. е.
(HgO, I). Некоторое отличие AS»R для HgO обусловлено
и тем, что эта величина относится к жидкой ртути. В соот-
нетствни со значениями И;» = -,-83,80 (ZnO), — 61,90 С<10
и —21,72 ккал (HgO) приходим к следующим выводам:
а) падение устойчивости в ряду ZnO—CdO— HgO; б) умень-
шелне стабильности этих оксидов с ростом температуры;
в) наименее устойчивым будет окенд ртути (он должен на-
чать разлагаться при
21 72 10DD
ilw-8I0 К:
— 25,84
точный расчет даст 750 К, опыт -^750 К). Последнее озна-
чает. что окисление ртути возможно лишь при низких
температурах.,
Важно, чтобы во всех ср ай ни вас.мы х процессах агрегат-
ное состояние реагентов было одинаковым, В противном
случае значения &.S не будут совпадать. Так, реакции
и
CaO+COt=CaCQi
CaO+SiC!=CaSlOi
(XIV)
(XV)
аналогичны, однако An^lv=—J, a = 0. Поэтому
с повышением температуры AGxrv становится более поло-
жительной, a AGxv практически не меняется. В резуль-
287
ЭБ ,:Научнов наследие России"
тате если
| > । (^ite)xv [♦
то, например,
|WUxivJ<|№«)xv|.
Другими словами, если при низких температурах СОг яв-
ляется более кислотный оксидом, чем StOs, то при Высоких
температурах наоборот.
Представляет интерес влияние температуры на проч-
ность соединений, полученных При взаимодействии данного
вещества с каким-либо другим, в зависимости от того,
в каком соотношении они входят в состав соединения.
Рассмотрим реакцию вида
Э (к)+"/Л (г>=ЭГл (к)
С увеличением я нагревание будет оказывать все возра-
стающее влияние (так как рост п означает увеличение
абсолютного значения AS). Поэтому В данном случае по-
вышение температуры будет Е±е только понижать устойчи-
вость соединений (AS< 0), по и вызовет уменьшение раз-
личий в их устойчивости. На примере реакции
Pt (k)+v£Ii (r) = PtClff (к)
этот вывод иллюстрирует рнс. 102.
Сродство элементов к кислороду. Остановимся более
подробно на реакциях вида
21
=9 (к)+01 (г)-ЗДОж (XVI)
Температурная зависимость Для некоторых окси-
дов представлена на рис. 103. Из него непосредственно
видно, что разложение оксидов благородных металлов
должна происходить при невысоких температурах, а рас-
пад оксидов активных металлов требует высокотемператур-
ного режима; оксиды элементов ПЛ-нодгруппы прочнее
оксидов элементов 115-подгруппы. Чем ниже линия Дб“ =
= f (7), тем устойчивее 3fOm, тем сильнее восстановитель-
ная способность Э. Так, кремний восстанавливает все выше-
лежащие ОКСИДЫ; А1 восстанавливает $i из SiO^ Са—
наиболее активный восстановитель. Нанеся на этот график
линии для РЬОг, SnOi и GeOj, мы бы убедились, что их
пересечение с абсциссой отвечает растущему значению t.
Действительно, если PbOj легко разлагается, то термиче-
ский распад SnOs происходит при 370* С, а разложение
GeOa — прн t > 70(Г С. Этот вывод соответствует резуль-
татам, рассмотренным на с. 274,
28S
ЭБ ,:Научнов наследие России"
Примерная параллельность низкотемпературных участ-
ков линий Л-G г = / (Г) обусловлена тем, что для любого
элемента в реакциях типа (XV]) величина Длг одна и
та же (Днг =—1), а разность
?s -2's
iG*
Г11-
Рлс. [Q2^ Зависимость
изменен ил стаЕсдартной
энергии Гиббса
(мкал/маль) от темпера-
туры Г (К) для реакции
PtOO+Wh (0=
= ИС1Л(Ю
f - Ft - 1: ? - fr » ft
J — Л FP 3: J - П в *
для различных элементов хотя и не одинакова, однако
гораздо меньше величины 5о,. Исключение составляет
углерод: так как СО и СО2 газо-
образны. то для образования
СО- (Дяс = 0) AS — 0, а для об-
разования 2СО(Дяг = ])AS >-0.
Этим и обусловлена практиче-
ская горизонтальность линии
для СО2 и отрицательный нак-
лон ее для СО, Все это означает,
что с повышением температуры
устойчивость всех оксидов
(кроме СО2 и СО) падает, ста-
бильность COj практически не
меняется, а устойчивость СО
возрастает.
Таким образом, сравнительно
большое абсолютное значение А5
в подавляющем большинстве ре-
акций вида (XVI) означает чув-
ствительность ДО° (и At?) к тем-
пературе, т. е. существенное влия-
ние температуры на химическое
сродство различных веществ к
кислороду.
Примерно одинаковая величина AS для различных реак-
ций типа (XVI) при сравнительно невысоких температурах
приводит к тому, что последовательность расположения
линий f (Г) примерно совпадает с последователь-
ностью значений АН для этих веществ, а в первом прибли-
жении — и с последовательностью значений электроотри-
цательности соответствующих элементов (сравните также
ряс. J2 и 25).
При высоких температурах картина несколько ослож-
няется: в точках фазовых превращений Э или Э(О™ в соот-
ветствии с уравнением Д5фгИ = A//^n/T^n происходит
скачок в величине Л5 (а поэтому и Д5’), что вызывает
10 M. Я, №р^иетьякц
ЭБ ,:Научнов наследие России"
289
ла
изменение наклона прямых,
Так, если до / = Н05°С
(точка кипения магния)
окислению отвечает про-
цесс (XVI), т. е. Длг = —1,
то, начиная с этой темпера-
туры, в реакцию вступает
газообразный магний, в со-
ответствии с чем Длг = —3.
Из рис. 103 можно сде-
лать ряд выводов о направ-
лении различных окисли-
тельно-восстановительных
реакций. При £ = 810° С
(при этой температуре пун-
ктирная линия пересекает
прямую для 2Н2О) A<jp = 0
и в соответствии с уравне-
нием ДО’ = — RT 1пК
(II.20) константа равнове-
сия реакции СОа + Нг —
— СО—НаО равна единице.
Так как здесь Длг = 0, то
равновесная смесь COj, Нг,
СО и [IjO будет состоять
из равного количества мо-
лей исходных и конечных
веществ; при /<810° С
будет преобладать восста-
новительная активность
СО (по отношению к Н4О),
при />810’С—'Восстановительная активность Нг (по
отношению к COj).
Пересечение линий для СОг и 2СО при t = 710° С озна-
чает, что при £<710° С более энергичным песета нови-
тел ем будет углерод, если он превращается в С02 (Дбсо, <
< ЙДбсо), а при / >710’С большему химическому срод-
ству отвечает его окисление в СО. Таким образом, для
восстановления оксидов можно воспользоваться как реак-
цией
О №О НИ) 1500 20D0 25<»
Г
Put, 103, Эависммоеть мзмс-
моиил стандартной энергии
Гиббса (ккал/илль QJ
для реакций
2d 2
-эо+адо- J-X
ХЭАДк) or г ("С)
/ — температура плйлгтгмня
Эс 3 — температура kirir^N4iq
3; Л — текпера турв пллилгций
Э^СЛ: * — ttMncpaTypa KkfW-
ния Э,£|
J ля
С (гр Эфнт)+2ЭО (К) = 23 (К) -L СОа (г) (XVII)
так и реакцией
С ^графыт)-)-ЭО (к)=>Э (KJ4-C0 (г) (XVIII)
290
ЭБ ,:Научнов наследие России1
Промышленное получение ряда металлов {7п, Mg, Ge, Sc
и Др.) и неметаллов (Si. Sb, Р4 и др.) основано именно на
процессах (XVII) и (XVJII).
Течению реакций (XVII) и (XVITI) вправо способствует
повышение температуры, при этом тем более значитель-
ное, чем устойчивее оксид. Так. восстановление ZnO угле-
родом требует сравнительно низких температур, восста-
новление SiO- протекает при более высоких температурах,
a MgO — лишь прн весьма высокой температуре. Из рнс. 103
видно также, что и РЬ м Fe можно получить из их оксидов
высокотемпературным восстановлением оксидом углерода.
Аналогичные рассуждения применимы для обоснования
процессов вида
Э* (к)+Э'О (К)=Э’О (к)-ЬЭ' (к), (XIX)
подобных рассмотренным, и установления их оптимального
температурного режима: это реакция взаимодействия AI
с оксидами Ва, Мп, Сг. Ее и др. (ашметдошл), Mg с SiO4t
К с В2ОЭ, Ва с РЪО и т. д. Так, можно показать, что усло-
вием осуществимости алюмотермии должно быть следую-
щее:
ккал/моль.
Выше линии 2С + Оа — 2СО лежит область Карбынер-
мии (восстановление элемента из его оксидов углеродам),
нижа — область металлотермии (здесь расположены ли-
нии для большинства легких и редких Э). Чем больше
сродство элемента к кислороду, т. е. чем правее пересечс«
иие линии Э/Оот с линией 2СО, тем выше температура вос-
становления оксидом углерода.
Таким образом, с помощью графика вида рнс. 103 можно
решить целый ряд задач, в частности, выбрать на и лучший
восстановитель. Так из него следует, какой из двух воз-
можных восстановителей — кремний или углерод — может
обеспечить лучшее восстановление УгО6 до V при 1000,
1500 и 2000 К- Восстановительная способность Si с увели-
чением температуры падает, При 1000 К углерод нс может
восстанавливать VsOfr, при 1500 К его уже можно при-
менять в качестве восстановителя, а прн 2000 К ОН стано-
вится более эффективным восстановителем, чем кремний.
Подобных примеров очень много. Следует лишь иметь
в виду различные «химические накладки», усложняющие
течение процессов, подобных (XVI), (XVII), (XVIII) и
(XIX), например образование смешанных оксидов (оксиды
1D* 291
ЭБ ‘ Научное наследие России"
металлов в присутствии SiOJ, карбидов (Ti), образование
твердых растворов нт. д.,п, как всегда, проблему скорости
взаимодействия.
На рис. 104 показана зависимость, аналогичная пред-
ставленной на рис. 103 и относящаяся к оксидам азота.
Рис, 104. Зависимость из*
менышя стандартной тргми
Гиббел (шил/ичль) об-
рзЗОВЗ НИ я " раЗЛ!!Ч ник ох ис-
лоа азота от темпера тури
T(KJ
Как известно, последние полу-
чаются косвенным путем. Это
объясняется тем. что для ослаб-
ления весьма прочных К-свя-
зей необходима очень высокая
температура. В то же время для
си!1теза NBO, NOlf N»O*, N;03
И NjO5 Дпг< 0 и поэтому Л5< 0.
Поэтому второй член уравнения
АО = Д/7 — 7AS велик и отри-
цателен (ЛЯ >0), что приводит
к положительному значению Л б,
т, е. к невозможности синтеза
перечисленных оксидов. Прямой
синтез возможен лишь для NO;
это связано с тем, что NO—един-
ственный оксид азота, для ко-
торого AS не отрицательна.
Однако так как AS близка к
нулю (-^3 э, е.), то синтез NO
возможен лишь при очень вы-
соких температурах, столь вы-
соких (-'"•3000 К), что скорость распада образовавшихся
молекул NO будет очень велика; для того чтобы вос-
препятствовать разложению последних, надо равновесную
смесь быстро охлаждать («закаливать»).
ЭБ 'Научное наследие России"
ЛИТЕРАТУРА
1. Айлетт Б., Смит Б, Задач]] и упражнения но неорганической
Хнмйн. — М,: Мир, 1967.
2. Аячстов Н- Q Неорганическая химия. — М.: Высшая школа.
1975. Часть передя. Разделы III и V.
Я. Барнард А, Теоретические основы Eteopгаесической химии.—
М.: Мир, 1968, гл. VI FL IX.
4. Глинка Н. Л. Общая хнИня. — Л .г Химия. 1973.
5. Дей И, K.h Олби и Д. Теоретическая неорганическая химия. —
М-: Химия, 1976, гль 3, 4. 8—10,
6. Зайцев О. <1 Химическая термодинамика к курсу общей хи-
мии. — М*: Изд-во МГУ, J973.
7. Карапетъяиц М. X. Химическое сродство. — М» Знание. 1972#
8. Карапеты) нц М. X. Конспект лекции по неорганической хим нВ
(подгруппы лития, бериллия, бора и углерода). — М,; изд. МХТИ
им. Д. И. Менделеева, 1973.
9. Кяралетькнц М. ХГ1 Драким С. И. Строение вещества. — №.:
Высшая школа. 1978.
10. Кемпбел Дж. А. Почему происходят хи лги четкие реакции? —
м.! Мир, 1967.
II. Коттой Ф.р Уилкинсон Дж. СовремеЕЕнак Etcop га ее и ч-сс кэ я
ккмия. — М.: Мир, 1976.
12. Кудрявцев А. Аь Составление химических уравнений. — М/
Высшая школа, J979,
!3. Мискиджьни С. П, Краткое введение и современную теорию
кислот и оснований и неводное титронаггцс, — Химия, 1966.
14. Михайленко И. Курс о&цей и неорганической химии. —
M.l Высшая школа, [966, рль VI—X.
15. Некрасов Би В. Основы общей хнминч — М.: Химия, т. [, 1963^
т. II. 1967; т. HL 1970.
16, Мсницесиу К- Общая химия. — М.: Мир, 1968.
17. Николаев Л, А. Современная химик. — М.: Просвещение,
1970.
18. Нпвнхов Г, Н, Введение в неорганическую химию, — Минск:
Вышэйшля trriccua. 1973, часть I.
19. Полторак О. М. Химическая термодинамика и кинетика в курсе
общей и неорганической химии, — М.: Изд-во МГУ, !973.
2Q, Полине Л.р Полинг П. Химии. — М.: Мир, 1978.
21. Снейпе Ми. Плейн Рм Хестер Р. Структурная неорганическая
химики — М.: Мир, 1968, гл. IV, V.
2Й. -Спайс Дж. Химическая связь и строение. — М,: Мир, 1966,
гл. 7, 9. J0.
23. Шу^^рсв G А. Неорганическая химип.— М+; Высшая школа,
1970. т. [,
ЭБ ,:Научнов наследие России1
ПРИЛОЖЕНИЕ J
ПЕРЕСЧЕТ» ЫЕ ЗНАЧЕНИЯ ДЛЯ НЕКОТОРЫХ
ЕДИНИЦ ИЗМЕРЕНИЯ
Мсгтрнчесвця ^днинцй Гднянцй СИ
1 л 1 мл 1 г/см’ 1 г/мл (кг/л) 1 CMs/r J мл/г 1 л/молъ л *с 1 кг 1 дни а 1 ат fTexEi) 1 атм (физ) 1 кгм 1 л- атм 1 кал 1 кэлДмояъ < град} 1,0000 -10^ м3 1.0000-10-3 НЭ 1000 кг/м3 1О00 кг/м* 1O'J м^/кг 1.0000- IO"® н»/кг 1,0000. |0“3 мэ/кмаль Г. К-273. 16е 9.80665 И* Ю-s н 980ГЛ5 Н/м* 101325 Н/м* 9.80665 Дж** 101.328 Лис 4,1868 Дж 4.1868 ДжДмаЛь грзд)
* I нъюточ (Н) * (1 кг) * (I и): (L
** L джоуль (Д*1 " (1 Н? [1
ЭБ ,:Научнов наследие России1
ПРИЛОЖЕНИЕ 1L СТАНДАРТНЫЕ ИЗМЕНЕНИЯ ЭНЕРГИИ
ГИББСА ОБРАЗОВАНИЯ НЕКОТОРЫХ НЕОРГАНИЧЕСКИХ
И ОРГАНИЧЕСКИХ ВЕЩЕСТВ И ИХ СТАНДАРТНЫЕ
энтропии
Ниже приводятся значения Ао;и и S^B. Величины Д№« легко
найти из этих данных по уравнению А//;„ = Д<7;„ + 29в.16Д5^я
Приводимые ВЕЛИЧИНЫ El ОСНОВНОМ взяты из справочника «Терми-
ческие константы веществ*/Нод род. В. П. Глушко; вып. 1—VI — Мл
ЛН СССР. ВИНИТИ, 1965—1973. Кроме того, использованы кн.:
В, А- Кнрееа. Методы практических расчетов в термодинамике химиче-
ских реакций. — М.: Химия, 1975; Г, Б. Наумов, Б. ЬГ Рыжснко,
И. П. Ходаковский. Справочник термодинамических величин (для
геологов). — Мл Лтомиздат. 1971: F. (tossing D. VVagniiin^ W, E^an^
S, LevFne> J. Jaffe. Selected values of chemical thermodynamic pro-
perties. Cine. XaL Bur. Stand. 500, Washington, 1952^ Д. (лалл> Э. Be-
стран, Г. Зинке. Химическая термодинамика органических соедине-
ний. — М,: Мир, 1971; М. X, Карапст^внц, М, Л< Клрипстьячц. Основ-
ные термодинамические константы неорганических и органических,
веществ. — Мл Химия, 1968.
Вещества расположены п алфавитном порядке, Водородсодсржя^
щие соединения (в частности, кислоты) помещены по алфавиту элемента
fa конце). Значения Д(/^я и для растворенных веществ (р) отве-
чают гипотетическому раствор у > в котором концентрация растворен-
ного вещества равна 1 М> но его состояние такое же, как при беско-
нечном раэбавлегши. Приводимые в этом случае значения SJt| ab.w
юте я парциальными молмгымн величинами; они часто бывают отри-
цательными. Помимо состояния О), отвечающего бесконечному разфав-
леник?> приводятся н гипотетические состоим и я бссхомсчЕЕо-разбав лен-
ного раствор а с псдиссоцииро1ьаЕ!11ымк частицами: они отмечены звез-
дочкой. Поскольку электролиты при бесконечном разбавленei лол-
яостмю диссоциируют па ноны, термодинамические параметры раство-
ренных электролитов находят суммированием соответствующих вели-
чии для составляющих пл ионов. Следует подчеркнуть, чго точность
прнвсдеЕЕпых величин во многих случаях является н.гиморной. Таб-
лицу составили М. X. Карлпетьинщ С. И, Дрюкни н Г. М- Фролова
на основании своего спр^иочкИК# *Т1зоблриые потеицпалы и энтропии
неорганических веществ». — Мл ИЗД- МлТИ им, Д. И. Менделеева,
1970.
ЭБ ,:Научнов наследие России1
ЭБ ,:НаучноЕ наследие России1
Вещество Эг е,
ККВ.1/ЫОЛВ
АС (Г) 44,93 5
Ас (к) 0 15,00
Ас А 00 —16К 32,0
Ад (г) 58.80 41,32
Ag (к} 0 10,17
AgH (р) 16/13 17,36
AgBr (к) —23,24 25,6
AgCN (к) 37,5 25.62
AgjCNjj (р) 72,1 4ВД2
AgCl (г) IGJ9 58,5
AgCl (к) -2G/25 23,97
AfiClOs (к) 17,1 32,16
AgClOh, (к) 17,0 а!\8
AjjCIO* (л) 21 зв.з
AgF (к) AgF 2НЙО (к) —44,91 -160,1 20.0 40,14
AgF-4H,0 (к) -2733 62,99
AgEI (г) 66,30 48.93
Ag] (к) —1 27.6
AgN3(к) 90,46 2V0
AgfNHjtf (р) -4,20 5Й.86
AfiNO2 Ск) 4,55 30.6
AfiNOi {р J 9,574 50.71
AgNOj (к. II) -8,03 33^68
AgtX (к) —£625 44
Ag/Xb(Ю -101,5 40.0
AgjCrQf (к) —151,78 52,0
Ag.MoQ* (к) —17Л2 5id7
Всщсстю
A1N (К)
Al JKOah ' 6Н,О (К)
а|оЛр}
Л1(0Н)3 (к, Tf)
А1(ОН)Г (Р)
А13Вг€ (г)
AltBre (к)
А1(С1а (г)
A|jCle (к. II)
Л 1,1а [Г)
ЛЫа (к)
AW, (К, а)
ALA-CaO (к)
Л)А -Н/: (к)
AIA' ЗП,0 (к)
А1Д (К)'
АЦЗОЛ {к)
AUfSO^ eHjO (к)
A|jSiObj андалузит (к)
AljSiCX, силлиманит (к)
Л|4Сз (к. гекс.)
A in (к)
Ams+ (р)
Ат" (р)
АтОк (к)
ЛтпвО3 (к)
Аг (г)
Д<?йи*| КМЛЛ/ьгОлЬ Saatt 5- ?-
—6Я691 4,82
—526.658 111,8
15.171 52, Е7
— —10,997*
—276,5 16,75
21,435*
-^232.Ш 122,4
—234,509 8Ъ16
—238,956 106,2
—300,468 52,24
- 135^62 130.7
-145,342 90,6
—378,17 12.17
—527.9 27,3
—435,34 23,15
-552,913 83/5
- 117,7 23
—741J3 57Д7
— иО5.(МЮ H2U
—584,33 22,28
->583,74 22.97
-46.932 21,26
0 17,2
—160Д —38
—110.2 —89
—230 20
—400 38
0 36^982
в
й*
Tf-—а г- со — я мя я с*:—и
ей ЕЕ ia О » о с^сч —«с» — к £ч Я » — «ьО ЩЛ"" оз — и\
Ж И t^rQ*C> К ^r^ry7ih Sir‘Kin пнг «ГГ- я erf — |Q 0?'C?
СфМ" Ю M r- HQ c£> -Ф u5 rt PG 1—1 CM оЗ 1Л ю W Tf Tt Tf c ЬПГ4
я ж — »
я й —S’l’ooSfeS’^^O- !Й ™ 5Й
сч sj о s cn «Г —" *<0 r-f — — irf w cf r< rf . * cf
щп й N ГОФ O-H -и HO I r Z И Ф II- ;?
8 1П77 'j'-ifn---
ГСЩ-SJO • _ _
?J Gi Щ C^T— -t 10 QQ
tt1 qp x“ OCT О Of * ]'
— -T-Г- I CQ^-O —
I 1 I 1 I
'.I.1 h . . I 41.1 _.'
— etc' см о
— O™^C4JCh-3C—'ОЗЙ>«
co"rjf -r ter [' ctf pfl ctf h^o" kO Mfitf -4 PC tc Tf П5 i!*5 ctf’etfef r-j’t^’kri'uS' ^Г"" of
tN ПЭ C*i ЧТ €>э ЧТ Щ P”S US Оф lO r-ea’Tu^C^WrviflUjG?" ч-тг LT о) ТГ
297
ЭБ ,:Научнов наследие России"
ЭБ ,:НаучноЕ наследие России1
tQ В(щестьо 3 ккалумсль Susi э- *
AuCly (р) -56,31* 64,119*
AuF (к) — 14 20,8
AuF, (К) -7Е 1 27
Ли1 (к) -ОД -зад 2<Х552
Au(OH)j (к) 29
AuTcj (к) —2,266 аз,87
ЛиаС8 (к) ]8Ь8 32,1
В (г) 123.71 3B,64S
В* (г) 316U38 33,088
В, (г) 188,468 48,23
В (к) 0 1,40
ВВг (г) 46,924 53,75
ВВгО (г) —63,068 59,5
ВВга (г) 7,016 70,4
ВВ13 (г) —55,349 77,6
ВВг? (ж) —56,775 54,54
В(СЙ,\ (г) —8 ДМ 75,3
ВС1 (г) 37J96 50,94
ВСЮ (г) -76123 56,7
BCIi (г) —17,717 65J
ВС13 (г) 69,2
BCis <*) —02,537 49,3
BF (г) -34Д38 47,89
В КО {г) -I4XIII 54,2
Bh (г) -122Л53 59.3
BF20 {г) —1яада 454,0
BF, (г) —267J40 60,78
вил- (р) —354.2* 42,12»
ЛСям! ЯКЙ-П/«АЛТ> 9в Ci
BafClOth W BaGOj] (к) —128 —31^76 6! 37,2
BaF3 (к) —274,5 23.03
BaH (r} 37/2 52.3
Bal2 (к) Ba(NO3), {к) —145 —190X5 40 51J 56,252
BaO (r) —36,134
BaO (к) -132,0 16,80
ВаОг (к) — [35 16
Ва(ОН), (к) —204 24
В aS (к) —104,5 135
BaSOa («) -226,3 29
BaSQj (к) —Э23Л 31.6
BaSiCb (к) —3tilJ 2Е\2
BaTiO? (к, II) —371,5 Z5<82
BaZrQ, (к) -558,0 29,8
Ba,SiOt (к) —512,3 42, (0
Baa(POd)a (K) —994 85
Вс (r) 69,02 32,з45
Bt (к) 0 2,28
B^Up) ’ —91J 47
ВеВг2 (к) —85 29
BcCOa (к) —226,6 IM.
BeCla (к, a) —106,659 lt\76
BcQa 4Hp (к) —376 58
BtF, (к) —2293 шло
B<?H (г) 69/М 4Ш
ВеИ3 (s) 27,656 41,35
cift Я I «о»
— ГС г- « Л Г'” М СГ’Г- —V та П ГС О Ш — Ct 1 L51rf±S"r^
<4 Ч- ~ сч —?(* — tCHC^»»TiI5 <о I?J L< -
1 U^Ct !"- -tf 4*5 <4 ai Ц5
1 та » —г' ВД таГ-'' ей"
М Ч?Щ ф ТО тТ
"S’
*
_ -м
in щ та о tfj in in о » та
О S С О in 5 " Zi г, Tt х Д -V Г- ^ ?] о тГ г-
—‘ та' id" та'та л' та14 -г «Г—" щГь5 т-" еГсГ** та । с" та oi с^—
t 1TJ Гт Т « Л 1Л МЛ |р -- -т !? ; о ю —
СЭ с-] 4^ 1^ —|£^
ci — о -н ci iq хГ с> ж'ю
ei ’т — та :>] >j кгз
С1
* э п « та <й та —. о
счс — щ та _ 5}^с1татата’*о ч м
г- — — та та^ та ns талщ » т л ® 5&
г- г-_ ^-Г _Г LT. । та сч -7 Г-’ ci та [-" —г -то о ,.q otT та
т м s » 1 та — -г тг т та г- ^1 та та г- се та та
та 1 сч I ”™г'1сч — — сч — та
xicnoQcdjaQ^^ ей®ci'cd’pqд'щеё'сё'(±^±X щщгпя сйсййЬей
2&0
ЭБ ,:Научнов наследие России1
ЭБ ,:НауччоЕ наследие России1
PeuLetnifl ккч.п'моль 5±й»« f-
Bi2Tea (к) —17,008 60
Вг (г) !0,69 4LW
Вг1' (г) 292,523 42,247
Вг- (г) -57,042 39,049
Вг’ (Р> —24,88 19,9
Вг2 (г) 0,749 5В.645
Btj (ж) 0 36^38
ВтС] (г} -0,18 5134
ВгН (г) —13.794 5^70
BrF3 (г) —54,849 (^,9
BrF, {ж} —58,951 42,5?
BrFs (г) -^3,732 76,3
ВгО; (р) 0,44 39т4
НВг (г) -12,238 47J64
HBf (р) -24,85 I9J
НВгО (р) —3 9,7 34
с (Г} 160,056 37,76
С, алчаз, (к) 0,677 0,566
Сн графит, (к) 0 1.372
С, (г) 18&832 47,63
Q (г) 173,599 5^8
G (г) 210,127 74
CBrN (г) ЭОЛ 50,33
СВ г, (г) 1 85,5
CCI (г> 97,442 5ЗД4
CCIN (г) 31,32 56,42
СС14 (г) 50.336 63,6
саао (г> -49,42 «7,82
Вещества Л (7 j п*' кк•л/иоль St84« *- вг
CNI -4GLE8 61,33
CNO- (p) —23.3 2*5
SCN" (p) 21,5 34,906
co (Г) -32,778 47,214
СО(ШЯ), (к) -17,020 25,00
corNHih (pj —48,42 41,975
COS (r) —10,40 55,32
COj (r) —04,'259 51,07
ООГ <P) —I26LI —13395
CS (r) 42,687 50,30
cs, (Г) !5’Zt 58,83
CSg (ж) 15,306 3*10
CjF. (r) -30 i ,489 70,3
CjH, (f) 4027 48,00
(-^1 l-.l'i (rj —74,932 63,38
C^HgFjj (r) —1.50,616 68,60
CJh (Г) 16,334 52,41
QHtF, (r) —103,555 67,48
CsHjCl (r> —13,38 65,99
CjHjF frl -52,583 вад
QHjL (r) 5,199 70,8
CjHs (r) —7,883 51,85
CgH/Э, Этанол (г) —10,17 67,5
CjbUO, этанол (ж) —41,637 38,48
CjH*O. дищгтнловыА эфир (г) —26,956 63,7
C.N* (г) 71,12 57,79
(ООО) Г ГР) —150,366 7.0
СвЩ, бензол (ж) 20,72 41,41
ЭБ ,:НаучноЕ наследие России"
СО, (г) 22.566 71Л
CCU (г) —14.50 74,07
CCI* (ж) —15,445 51,25
CF [г) 52L2I4 50, SO 57,72
CF, (г) —4L655
С1я (г) — 108,796 6:13
CF4 (г) -212.33 62.47
СЯ (г) 131045 43.72
СНВГ4 (г) 7,767 7Й.03
СНВг, (ж) -13 52.8
CHClj (Г) —16^39 70,72
CHF, (г) -158.77 62iO4
СН. (г) 8£\830 43,2
СН.Вг. (г) 0,66 70. [
СНаС12 (г) — 16,50 64,55
СНаС1, (ж) —J6L83 42,7
CH.F. (г) —99.92 58,94
СНг1а (г) 24.403 7107
СНЕО (г) —2^266 52,26
СН, (г} 34,536 46^2
СН,13г (г) СН,СОС- (р) —6,70 -^88.28 58,76 20,93]
СН£! 1г) —15.00 5^02
CTliC! (ж) -12,3 34.64
W (г) —53,29 58.25
СН,[ (г) 3,91 60,67
CHaC4i (г) -.39,04 57,3
СИаОН (Ж) —39,94 30.26
СН4 (Г) —12,146 4153
С!, (Г) 62,356 93,6
CN (г) 95.09 48.41
CN- (р) 4i,0l 23,051
CjHuh циклогексен (ж)
С<Н|Й, циклогексан (ж)
C,HNl АР-гексан (ж)
н-октан (ж}
С^Н|Вк 2’Метилгептан (г)
CgH^, £.3*димстклгекслн (г)
2,2.3ДтстраметиЛ'
бутан (г)
НСК (г)
HCN (ж)
HCNO (р)
НСООН (г)
НСООН (ж)
НСООН (р)
нсо. (р)
Н,СОЯ (р)
Н/\О2, уксусная кислота (г)
Н|С^Ог| уксусная кислота
(*)
HjCaOj. личер муравьиной
кислоты (г)
Са (г)
Са (к, а}
Саз^ (р)
СаВг2 (к)
СаВгв GHtO (к)
CaCj (к, а)
СаОЩ НЙО (к)
СаСО5, кальцит (к)
CaCO3-HgCO3 (к)
СаС13 (к)
СаС1а-6НаО (к)
15,96 51,67
6137 4Ъ,84
—1.04 70,76
W 111,55
3,05 103,81
4/23 106.11
5,26 93.06
29,709 43,21
29.096 27,03
—27,71* 32,024*
—S3JJ8 59,45
—36,57 30Д2
—89.14* 38,909*
—140,19 22,124
—148.93* 45,400*
—89,29 67.5
-93,06 38,2
—171 83
31,31 3^902
0 9,97
—132.10 —13,2
-156.8 32
—506L42 72.83
—16 щв
—360,7 37,28
—269,78 22,2
—514,32 37.09
—179,05 27,2
-529 68J
ЭБ ,:НаучноЕ наследие России1
Вещество Ad jg ti доал/моль 3. e.
СЮгО4 (и) CaFi (к) СаНРО. (к) СаНРО4 2НгО {к) СяНа (к) Cat. (к) Са(ЫОД4 (к) Ca(NO;Ja 4НгО (к) СЮ (к) СЮ-ВгОя (к) СаО-2ВД <к> ЙСаО 1*А (к) ЗСЮ * (к) СД (к} Са(ОНЬ (К) CaS (к) CaSOa (к) CaSQi (к) CaSCVO^HjO (к, а} CaSOv2H,O (к) CjSiOa (к. а) CaSiQg (к, fij CaWO4 (к) O,N,. (к) Са,(РОД (К. «) Са^РО^ (к. 0} Cd (г) Cd (к, а} -305,3 —281,9 -40i,53 —5М,76 —32,6 —J27.56 —177.34 —106,5 — ] 44,3 —459,90 —756,91 —620,59 —779,08 — 143 —214,77 —114,1 -315^6 -349,26 —429,19 —369,05 —370,3 —376,9 ^88, J —929,7 —932 18,46! 0 32 16.46 26,62 45,28 10 31,72 46.2 81 ft50 2M 32,2 31.7 43;9 103 19ДО ! 3r5 24,2 25p air2 46,31 20,9 19,6 30.2 25 57.6 56,6 40.066 12,37
Вещество ЛСЛ*’ ККЗЛ/ЬГОЛЪ S-
CL (г) 0 53.293
C!F (г) ^12.278 52,06
CIF, (г) -29.4 67,28
C]F3 (ж) —15,106 44,95
СЮ (г) 23,326 &4J4
СЮ- (р) —8,75 7,88
СЮа (г} 29,24 6 из
СЮ; (р) 4r 1 2U
СЮ; (р) —0,05 39,3
СЮ, (р) —031 43,9
С1*О (г) 22.52 63/33
на (г) —22,656 44/544
НС! (р) —31,37 135
нею (г) —16,948 5615]
нею (р) -19,17 25.54
НС1О4 (Г) 21,958 70,8
НСЮ4 (ж) 18.69 45,02
Си (г) 01,737 42.879
Со (к) 0 7.18
Со»" (р) —12,82
С<?+ (р) +31,00 —6ft, 122
СоВг* (к) —50 32
СаСО, (к} ^15ад 21,18
CoClj (к) —63381 2G12J
СоРг (к) —148 355 19,59
CoFз (к) “I69 22.3
Col* (к) —24 38
Co(NH,}p (р) -57* —
^ikizijoj эйУвюен аонкАвН:. ЭЕ
сос
2-^-^ <?* .^..5? —
и
...ц. j“ i-W^i Jw? 'uLr' w— ь-F-' J- LHJ J-r‘ “-Л j**- ^-w' i" J--4
ел "-j to CTi t<- ск -A/sk <p тс pj £. SC
Ею >“ I— Gc — — 4< Ca? bJ Л flG —
W — ~jfj K> w
tJJkb DC^
Л —
со;э
щ щ
b*
Q C n n Q Л n n n Q Г) О D r> r> Г:
g^.................................
* "K-Zr
Xh id [< £j — rJ — — —- W M hG M IC — r-J r-J <J? rJ — r-J t<- “ -xj -q
c и ч t<: СЮ -7j ;л -J Я i IG C; I рю □
— o<At"i -j-u co 4* OX' Ъ’о>ыр1лЪ к oo’o ’ct 4 Q — S
hj i5 * ui 5; ?a -J
ЭБ ,:Научнов наследие России1
□с № дф ~ QQ SM йг>
СчЧ’etocjc wo rfc:fC^fjCJ
— iS t1* к Sc C4jrt l>L Cb
tft ’Ф tTW irff^ «Хм ip -7 Q _7
!^i СЧ ™ — I?1 J C& ™ г-1 СЧ CO X-
3 O\ 5c Й S S^CM й ^1 S{ ?5 Si3?^Kc8»E?JC4^^S?5™_ c5 «J-O S Ф
4D_ ОС SO u5 ей Ч' -* ТЯ* dl CO H CM <J> f ui т "Ф Г- 0C ’т |ГО «? Ш H Cl C i <C К
l'J L"; ifi S u: Г- M Г? M —rc: ^]Q - <' t' l; 1^- яэ
i i i--717 i i^77 1^177^1^81^ 77
_x * <4 _ 3Г
«co—1 «я ’т О) — raS
О ц; st go « m ’f -e ci о — m — c — c: —.50 r-i Ci >5
С Г1 Щх C Lffo — (£ ttf c£r ttf — «" o> —r‘» К ji о O hl"? tr^—7 Ц?- cT c£j” чу QZ"|^« L* <J
N CM Li fi W 1^ GM ч " fl ? Щ CN Л Ч<4 Й T — fi чСЧ Ь ’f — 7 4 41
305
11 M_ X. K.D pn [I СТ ЬЯ НЦ
ЭБ ,:Научнов наследие России"
ЭБ ' Научное наследие России1
ЭБ ':НаучноЕ наследие России"
GafOHh (к) —199,88 27.1
GafOH); (р) —241.fi
GjF (к) -22,312 12,3
GaS (к) -4fi.fi —
GaSb (к) -т-9,78? 1BJ8
GaSe (к) -38,579 1^8
GaTe (к) -29,9 2Ir6
Ga«Qj (к) —238.583 20^1
СвД (к> —120,8
Ga^ (к) —93,5 w-и
GasTej (к) —62,7
Ga*S, (М —212.9 ——
Gd (г) 46,416
Gd (к) 16,24
G(F* (р) —160 —47
GcfCl^ (к) —222.5 35
GdCh-eHjO (к) —583,7 93.fi
Gdb (к) — L46 52
Gd/), (к) -413.37 36,0
Ge (г) 80,862 40,103
Gc (к) 0 7,44
Go. (г) 101.48 60,4
GeBr (г) 24.247 61,48
GeBrj (г} ~25£94 i 76.0
СеВг, (г) —77.214 94,7
GeC (г) 137.081 56,2
GeCI (г> 14.298 58,63
GeCI, (г) —15.0X2 70,4
GeCl, (г] —111.379 83,1
GeCj. (ж) — 119 60
GeF (г) — IG.767 55,91
GeF, (г) —! 15.627 64,7
Hf (к) 0 10,41
MFC (к) —40,143 9,58
HfF. (к) —437,16 31,5
HfCl. (к} -215,79 45,6
HfN (к) -81<4 юл
HFQ, (к) —260,1 14,13
Hg (г) 7,624 41±792
hr (ж) 0 18,14
Hr’' (p) 39,36 —6.012
HgJ* (p> 36.71 19.642
HgBr (r) 15,976 64,9
UgBr, (к) -3Z16 42,98
HgCO3 (к) —! 12.0 44,0
HftCl (r) 1438 62,1
HgCl3 (k) —43/23 38.47
HgF2 IK) —89 27,8
HgH (r) 51.724 52,49
Hgi (Г) 2ГД02 67.1
Hgl3 (к, красный) —24,63 41199
HrC (к, красный) —14,022 16.80
HrO (к, желтый, 1) —1336 17.085
Hg(OHh (к) —703 24,6
HgS (к, «) -12,1 19,7
HfiiA (к) -I41J 32.6
НргВг2 (к) —13.34 52,03
Н&СОд (К) -112.0 43Д7
Hg2CJt (к) —50,38 46,8
H&Es (к) —104,2 4!,8
HgjI. (к) —26,57 56,23
FFgsfNjJj (к) (79.3 48,56
Hfeo (к) — 13 3t
H&SO. (к) —! 49.97 47,97
- ? О S3 С* О » Йt « о * S § г£
щu-jг-c-hгг"щ"« ® —о К'г- о сГ-Г’г^л ?f Sa?)(?js sm(с
и
ес । Q £ м tt«
в *
j. W
к * 7
iS
и _А.
22 g г г г'odes “?.Ч>ЧЧ.Ч^ЧЯА.’^^Ч1Ч „ * ъ
tS “ I I S Ф '4? cS ot £{r- M ? S О «з ф® aS 18св — ot S ч Ф
-Two 1 1 a m"» а-Г«fs q;c?c^-4«i’-fla-* О i^f«fфc"inвГс*—-
ЧГ ™ -гр а ф ~ to r <?5 W W ‘T l/;1 т Ф ю й W fi S ft r- *
to t? Cr Г-Г ft Л ~ Г-^СЗ ?7 ft 1Г; ГТ; — if Ch а S i; Sftc S » О « ft
тк К I — ОФ 3C — — ’Ф » Л ГС Г4^ oa 1П Th CftfqoO — C* ™
~’ii i i i i i Г17 j 11- i i 1 17 17" 11
309
ЭБ ,:Научнов наследие России1
ЭБ ’’НауччоЕ наследие России"
Вещество ККЛ л/МЙЛЪ >. c.
LaCl3 (к) —238,41 343
LaClj 7НВО (к) —645.0 i 10,6
Lal3 (к) —16в 51
La(OH);t (к} —307,4 34,6
La^Ci, (к, гсксагоя.) —107,7 30,43
LafS, (к) -302 32
L* (г) 30.602 33,141
Li (М 0 6,753
Li* (г) 135,351 31,767
Li+ (Р) —68.86 2,5
Lia (г) 4O.5J9 47,050
LiA1H4 (к) -11,56 21,0
Li АЮ, (к) -269,429 12,751
Li ВЫ, (к) —29,824 18,12
LiBO, (nj —229,87 12,351
LiBr (г) -42,92 53,78
LiBr (к) -81 16^5
LiBr>2H±O (к) —197 27
LiCl (г) 51,926 50,861
LiCI (к) -91,786 14,173
LiOO4 (к) —60,706 30,0
LiF —139,7441 8.523
LiH (r) 26,654 40,825
LiH (к) —16,368 4,788
LiHF, (к) -208J 16,97
Lil (r) —29,43 55,5
Lil (к) —61 18,3
LiN, (kJ 18,50 17,15
nCLUftflbO КН-В rl/Mrt-П Ь э. е.
MgF (г) —59.28S 52,793
MgF, (к) —256,006 Г 3.683
MgFcjOi (к) —314,9
MgJ-l (г) 33,928 4^141
MfiHt (к) —8.8 7.136
Mgl2 (к) —86 32
MgMoQ. {к) —300,7 28.4
MgfNOjJg (к) —140.63 39.2
Мй(ТО^-ЬН2О (к) -495.3 108.3
MgO (к) —135.985 6,44
Mg(OH)t (К) — 199,25 15J0
Mrs (к) —81,4 Н
MfiSOs (к) —222 22
Meso, (к) —274,26 2L,8t
MgSOj -GHjO (к) —628,9 83.2
MgSiO, (к) —349.33 16,19
МйТА (к) 354,37 1W
MgTijOi (к) —565,65 зол
МйА (к) [7,729 24,00
MgjSiOi (к) -491.72 22J3
М&тю< (к) —489,133 27,52
M&(AsO<)» (к} —679 54
M&Ng (к) -95,82 21,00
М&(РО^ (к) -848,1 45/29
Мп (г) 57,61 41.492
Л1п (к. а) 0 7,65
Л1п=* (р) —54,96 —16.0
МпВга (к) —87,4 33y5
аэ 1П г-* ой Щ in
ft 6S1-^ OJ -Ji Г’; С* 1Г Г' » Г’ « -* Ор Щ u*^tc т 30 О
О Щ>" xTei «5 о ’т otiG ft" с> ч- эс щ —' ^-' ft" ft ?d l? — ft’ сп. гм ?о я
Г'СМТь1р4^’^ — — Т-Т — ?Ч — " М ТО Т “С LQ ft — Г—
“СМ
33
О
tf *
б'Ф’^т' । -'^’'5 -еэ^-
о оо so о и$<я а 4^44%
_____PEcceeceeeeei±w^Ee
~ ^з -р-г -?
- щ ii-QOO
□ □ Q U С- Q
^S 83 8Лй Й5^«8х?.3.-л°S.*§-й 'Г
О- г-г м- —• сэ г-’ ж -J ft с । щ .л — ft"ir: г--' юс: ж сл c-s чо ?: ггГ г--"^ хГ
см — ™ сч —< ej гб in сч сп го см — счж-——-—гчэСкПё^
н-н *** "ь?
И V
X. О а с'и^.
О « "^Х
'sS'S’3
i i g § йз31i 5 7?«|ъМйМ1 WS
—I -J -J r-^-—«£; << ₽t, r<t. -t, ₽t, -t,
311
ЭБ ,:НаучноЕ наследие России1
ЭБ ,:Научнов наследие России"
Вещество Л fi j ЬН ь К К ВЛ/КО.1 Ь SifA* 5- *-
МоС1а (к) —100.4 53,8
МоС1(, (к) -92,88 €3
MoF, (к) -351’0 G2,06l
Мо12 (х) —13 40
No!;, (к) —15 50
Мо10 (к) -20 64
Мо1± (к) -19 78
МаОа (к) — 127,45 1J,G6
МсОа (к) 18,58
МоО; (р) -20^ 14
МлС (х) —|1т2
MoSj (к) —54,03 1Ш
МсиС (к) -и 13.0
N (г) ЗОВ_888 3^613
N+ (г) 443.7S4 3Ba63o
Nt (г) 0 45.769
NJ (г) 358b86J 47,12
Na (г) 118,907 53,2
NT (р) 83^ 25,8
NBr (г) 67,372 5^25
NF (г) ЩЗЮ 51J
NF, (г) 13J66 59,71
NFa (г) -20,178 62,28
МН (г) 7Э,74О 43.30
NH, (г) 43,854 46,52
NHtOH (г) —0.86(3 56^3
NH, (г) -3.993 46,04
КНа (Р) —6,35 2^6
Вещество ккзл/моль S;gii э. e-
NS (г) 40j653 53,06
NtF< (г) 19,005 74,9
NjH. (г) 38,023 57.0
КгН4 (ж) 35.071 29,0
NaH^ Ip) 30,6 33
NsO (r> 24,884 52,55
N4O3 (r) 3M70 73,4
NA (r) 23,522 716
NA (ж) 23,29 50,0
NjQ^ (r) 27,63 85,0
NjQs (к) 27,271 42,6
HNj (r) 78,401 57,08
HNS (ж) 78,202 33,6
HNO± (r) -10,893 60,8
HNOj (p) —13,3 36,5
HNOj (r> —17,889 63,79
UNO, (ж) —19,322 37,20
HN0a-H,0 (ж) —78,531 51,84
НКОЯ . 3H/J (ж) — 193,8)0 82,9
HNaOs (p) 20 34
N»Or (P) 35
Na (r) 18,475 , 30,7141
Na (x) 0 1124
Na+ (r) 137,562 35,337
Na+ (p) —62,65 14,08
Naa (r) 23,807 54.994
NaAlCl, (к) —249.0 45,0
NaAJH4 (x) -11,6 29,6
ЭБ ,:Научнов наследие России"
м
ы
NHj 0,5НйО (ж)
NHa 2НаО (ж)
NHj (г)
NH; (р)
(к)
МН^АЦЯО,)^ ISHjO (к)
NH.Br (и)
NHjSCN (р)
NH^Cl (к)
NH4C10 (р)
NHdClO< (к)
NHtF (к)
NHJ-JCOj {к}
NH4HFB (К)
NH4HS (к)
NH.HjAsO.
NH4H2POl (к)
NHJ (к)
nh4no1 (X)
NH4N3 (х>
NHjOH (ж)
NH4OH (р)
(NJIJ^O. (к)
NO (г)
NO+ (г)
NOR г (г>
NOC! (г)
NOF (г)
NOI (г)
(г)
NOs (р)
NO; (Р>
-31.945 3197
— ! [7,821 55,67
169,474 44.5
—jy.oo 27,32
—487,47 5h7
- 117ЭЛ25 163,9
-41.753 26^97
aja 6Lr6
--18.558 22.9
—27.8 37
-2IJ1 402
—83,28 I7P2O
-159,2 28.9
-155.6 27t61
—12,1 23^
—200.13 41, [Э
—289,10 36,32
—26,9 28
—43,94 36,1
б5л> 26,9
—60,74 39,57
-63J04’ 433*
—215/13 52,6
20,693 50,33
235,204 47h35
19.059 65,38
15,97 62,53
—12,12 59,27
22.09 67,673
I2L32O 57,40
—8,86 33,35
—26^67 35,18
NaA]Oi (к)
NaAISiO4, нефелин (х)
NaAlSiA (к.)
NaAISijOft, альбит (к)
NaBHj {к)
МаВСК (к)
NaBr (к)
Nalk 2На0 (к)
NaBrQj (к)
NaCN (к)
NaCNO (к)
NaSCN (к)
Nad (г)
NaCI (к)
NaClOs (к)
NaClt\ (к)
Na£lO4 (к)
NSF (г)
NaF (к}
NaF HF (к)
NaH (г)
NaHCOa (к)
Nal (к)
МаКОь (к)
МаМОь (к. [I)
NfiOH (к)
NaQi (к)
МаяВД (х)
Na/Д {к}
№ясол ЮНдО (к
(к)
МайМ<Ю4 (к)
-255,561
—468.62
-677,42
—ввадв
—ЗОЛВ
—220^54
—83.49
—]D^t
—60Д8
— 19,227
- J 9,462
-37
—48,117
—Я 1,788
-63.21
-60,785
—74JI
—129,866
—198,0
24,603
—195,0
—6,4.103
-68
—87,45
—30,87
-—52
—736,52
—25030
-820Л5
^294,03
—3233
16,826
29J
31,9
50/2
2-1/232
17,573
21,8
42
3!г2
28,315
29,81
27
54,89?
17,236
27
53Л
34,0
51,983
12,260
21,73
45,00
24,4
24.0
25
27,85
15, =Ю
27,7
15,289
33,17
134,97
43,0
38,1
f'^'C'dd?'? и — С ~ 7-~
Ч^Ч^ЧМЛ-еЛ^—С О
««ye «. -гг-гл
314
ЭБ ,:НауччоЕ наследие Рогсии"
ЭБ ,:Научнов наследие России"
Nd(0Hj3 (к) —304,41 26,5
Nd А (к) —411,1 .46,9
NdjS», (к) —277 32
Nd^SOJa (к) —867
Ne (г) 0 31,91?
Ne+ <г) 496,456 37,801
Ni (г) 90,041 43,519
Ni (к, и) 0 7,14
Nla+ (р) —10,39 —30,126
NiBr, (к) —49 31
NHCOh (г) —135,6 95,4
NiCQ, (к) —147,6ЯК 20,6
NiCl, (к) —61,671 23,44
NiCli-бН/) (к) —410,9 83,7
NiF/ (к) —J 43,873 1Т,£»
NiFejO* (К) -228,713 30,3
NiCjOj (к) -18*6 31 г753
Nil. (к) NifNHJf (р) —2L3 —46,6’ 57’
Ni(NH^< (р) —60,46’ 8з,204
(к) —57 46
NiO (к) —50,573 9,08
Ni(OH), (к) —Ю0Л5 tej
Ni(OH)5 (xj -129,063 23
NiS (к, а) —18.371 12,66
NISO, (к) -182Д15 24,82
NiTiOa (к) —267,8 17,8 25,4
NinC (к) 6
N[> (к) 0 12
NpBra (к) —Е88 49
NpBr, (к) — 175 58
МрС1я (к) —200 Зв
Р+ (г) ЗОЙ,958 39,88
Р (к, белый. Ш) 0 0,82
Р (к, черный. I) — 7,9ЯЙ 5,42
Р (К, красный, три к л,} -2.&41 5^45
Рз (г) 24,705 52,09
Р. (0 5,872
РВг» (г) —37.21 83.2
PCi (г) 29,847 5^6
РС1Э (г) —62.25 7W
PCI, (ж) 65,1 51,9
РСЦ (г) —71.02 87,1
Р& (г) —223,63 65Д5
PF* (г) -362,629 70,0
PH (г) 53JH! 4(^87
PH, fr) 3,2 50.24
Pfl.Br (к) —пл 26,3
PH А (к) — LC.9 ——
PHJ (к) (^02 20,4
PN (г) 1 8,475 50,44
РО (г) — 12.287 53.22
РОВга (г) -92ДЧЗ 86^0
РОС1а (г) —122Л9 77,4
РОГ1 (г) -287,703 6SJ0
PS (г) ] 8>13 56,2*>
РО?* (Р) -218,012 —52,92
P.O}- (р) ~45Я^8 -25
РА {Г} —350,033 i 82,9
РА Г) —532.3 92
РА« (0 ^-635,15 94,3
Р,О1Л (к. Ill) —6-MJ 54,7
P,S3 (к, II) -26,054 4860
Р.^Щ 0s) —7Э.743 91,24
О 1 SS— Ог-SS
I г-г-ъ1 г-^—.-щ — —।- a — —F«J«3 <яйчtfToiofсл
„ ^^ччт—^Й?00?^5с5РОкГЗГЧ — ?з — тГ - ?J ГС U7 LC |£ ?q
316
ЭБ ,:НауччоЕ наследие России"
ЭБ ,:Научнов наследие России"
РЫ (г) 20,483 6^86
РМ4 (F) — 14,109 84,6
РЫ, (к) —41,48 4L9I
РЬМоО, (к) —225/7 за.т
РЬ(МО,)4 (X) —60 51
PbfNj). (х) I2G.3 35,76
РЮ (г) 11,283 57/U
РЮ (к, желтый, 1) —44,98 16,42
РЮ (it, красный, II) —45,20 15.8
PbOj (к) PbfOHh (к) —52,18 -107,85 17,9 21
PbS (к) —23,61 21,8
PbSO4 (к, II) —194,51 ЭД51
PhSc (к) —24,3 24^
PbS)03 {к. II) —253,86 26,2
PbWO4 (к) -250.0 40,2
РЬО РЬСОа (к) -195.6 48,5
PbsSiO4 IK, II) —2Я9.4 44,6
РЬА М —I44?8S 50,5
Pb^POiJj (к) -58L4 8-1,4
PJ (г) 79,765 39,90
Pd (к) 0 9,00
PtlBr. (к) —22 33,5
PJC1-- (р) -102,2- 70,980
PdCI> (р) -10.V- 55,181
РЮ (к) —20/181 9,3
РфОН), (к) —73.4 21
Pd(OHh (к) —110J43 27
Pm (н) 0 17,2
Pm1' (р) —168. -41
РтС1в (к) -234 ззд
Рсь (г) 26^(49 45,127
Ptcis- (pi —i i дозе* 53,4*
Pt(0H)2 (к) —64,283 24
PtS (к) — 1&Л4Д 13J6
PtSs (X) —24.512 17.85
Pu (r) 74,113 42,3166
Pu (к, a.) 0 13,18
PuJ+ (p). -HI
PuClj (к)
PuF, (к) —35^454 27.00
PuHj (K) —24,329 14,30
P«[3 (к) -134 56
PuOa (к) ^-24ода
Ra (Г) 31.2CH 42,15
Rh (x) 0 IZ0
Ra’T (p) -J34£ 13
RaCl. 21'LO (к) —ЗИЛ 50
(1г) —140,3 52
RaO (к) -118 18
RaSOj (к} -32fi 35
fib (rj 1Э.&13 40,63
Rb (x> 0 18.22
Rb+ (p) 67J7 28.8
Rb. (r) [ЯД27 64.59
RbBr (kJ —9115 25.88
RbCl (к) ^97,4Я 23.90
RbClQ, (к) -71.83 36.3
RbClOi (x) —73.57 40
RbHQO, (X> —3Q5 29
Rbl (k> —77 28
RbNQ, (к) —93 34
RbOH (x. I1J —87 19
RbiCO3 (к) —250 41
* S® I* S^3SS iooSS
—" r- CC ft Ю -Л ft p£« В Q Лft'Q ftft ifi^Г4^0 сч pf о сГ-= OS'
, “ — t" т Щ OlSvinSinr’Sc^Tf-^^^^-C’HOL-^rciQ^-^Tf—щиэ
Л ft СЧ “ С4? “ тг in —• О-
1 О Я ® Я ® ft1 О LfJ ft —'
tC£D иГщ И rdro pi “ «fО ttf-ffl C4! c> »«f » *r h* —^"4^0 if? Q
35 ] !7 22Z^^Sj§?iPI I i*£’$! I *g ’•?
a | | 1 I I I J I F 1 II INI
£,
‘Zl'Z4 SSS-S;s5£r.—.“ S
7777* ’ LT;t;„ J„ I '7c a 7s*i ~ r
££4449^4.4° °=o« § S 3 “
Л й / vi uTcn jyv; vj cq l/ь ./г I “ _ц „ X X — сл </] V) c/) cn
“ „„ 7 «*5*
— ~^ГЩ. щ [£ Г-: M <Г^ ’Г с^ L* с^ 5 1.Г} О?
И ift Л И « r^ iT) dt> СП 10 Cj ao M ft <£f тг-1>- C pJ Фф c *: r^ L< s ’*" щ
Cxi СЧ СЧ rt СЧ — ™ vr fJ Гф L*'j f I?; - f. j 1^ ? ЧТ
§
£
7
Ф
c
“ffi □ (^p о y7Q<S “О О о □ о 4^ “ ^ *
j; л .a jj j2i 4i щ q I; Ti < v -_^ ^^-С£._=£:_е^_Е E is s
3ia
ЭБ ,:НаучноЕ наследие России"
ЭБ ,:Научнов наследие России"
RuCl, (к) —35 [0
RuClj (к) -16,9
RuF, (к) —I3S.75 Л8р
RuCk (к) —60,13 14,0
Ruo; (к) —35,993 33,7
Ri*S3 (к) —50,816 Ш5
S (г) 55,516 40,081
S (К, МО1ЮКЛ., II) 0.090 ш
S (к, ромб.) 0 163
S+ (г) 294.643 39М2
S*" (р) 20,41 —3,17
S. (г) ] 8,778 54J50
S« (г) 1 17 . 90
S# (г) 10,915 LOGJ
SCI (г) 23,501 57,2
SCIj (г) -16,7 67.15
SF (г) 11.444 54,5
SF, (г) -31,416 61,4
sf, (г> —173,484 «9,30
SF,C1 (г) —223,44 7^38
SF, (г) -263,67
SO (г) ^6128 53.02
5OCI (г) —27,391 66/2
SOCIt (г) —47.231 73,60
SOF3 (г) -166,359 66,5В
50. (г) -71.752 53,29
SOyClj (г) —75,207 74г4
SOjCl, (ж) —73 51J
SOjFj (г) -194,206 6В,№
5Оа (г) -88,71 6135
5ОЛ (Ж) —89,15 29,1
SL, (к) ^—89 1215
SLBra (к)
S1]Clg (г)
SbCIg (К)
ЭЬаБ (г)
SbCU (ж)
SbF (г)
SbFa (kJ
SMI3 (r)
Sb]3 (r)
Sbl, (к)
SbN (r)
SbO (r)
SbO+ (p)
SbfOHh (к)
oO,2
80,9
43.8
9tZ0
70,5
56,1
25
55,(iB
95/>
51
55.3
56,91
SbTe (r)
Sl.bO-j (кь куби орторомб.)
SliA (к)
Sb2Oa (к)
(К, черный)
Sb/}, (X, I)
Sc (г)
Sc (к)
5cs* (p)
SeB^ 1»
ScClj (к)
ScfOH), (K)
ScA (к)
Se (r)
Se (к, гексзгои., HI)
Se (ст )
Sc1" (r)
Se’" (P)
—57t44l
-71,5»
^77,07
-76,573
—82,^1!
^2X964
—200
35Л86
-14,703
-23
59,862
18,322
—42
—153
36,650
-1.УФ
—1ВД23
-206.6Й
—37,30
—290.W
80.34
0
—I43J
-29Z0
—125.0
44.829
0
0.630
269ЛО0
32,0
25.8
64,1
29,4
30,4
29.9
43,4
67?4
4i.748
8.28
—6:4,2
37
30.7
22,3
18,4
42,212
10,07
J2/<
41763
—6,70
ЭБ ,:Научнов наследие России"
SiCl. (г) ^71ЛЭ5 75f6 SnOa (к) —124,25 I2Jj
SiCLH (г) —1 [8,402 7<UJ& SnS (r) 14,968 58,19
SiCi* (г) —1ВД61 79,1 SnS (K, a) (K) —25,87 18,4
SIQ. (ж) —143,0 седа —34f\8 37,1
SiF, (г) —15D,-19 —375^4 61f3 SrSa (К, H) —[7,708 20,9
SiF. (г) 67,4 SnSc (r) 17,976 61,04
SiFj" (?) —^055 303 SnTc (к) “14,6 24,5
SiH (Г) 79.922 47,31 Sr (Г) 31,3 39,3251
SiH(CHi)? (г) 5iH3(CHa)a (г) —40.793 —29.514 79.01 71,7 Sr (K) Sf” (P) 0 —136(58 12^5 —6,5
SiH£H, (if —ОД77 til.S 86(1 SrBra (к) —169,33 32,4
Si(CHa)4 (г) ^42ьО7Й SrCO, (к) —275,5 23,2
Si(CH3Jj (ж) —4L973 C$26 Srtl, (к) —187,5 30,0
Siltc (г) 13,67 48.89 SrCi. GE-ijO (к) -533 83,73
Sib SiN Cr) 5,719 75.8 SrFa (kJ —282,16 19,63
10B.544 51,79 StH (r) 45,8 50,78
Si0 (r) 12! 50^4 Sr]j (к) —135 38
SiOFj (r) —227,231 64,81 SrMoa (к) —347,6 33,7
SiOa (r) -78,267 54,4 SrfNOjlj (к) — 149 42
SiOa (к. а-к&эри) —204J5 10,00 Sr(NO,)t (к) —188,91 46,50
SiOH (к. сг-криегабаллнт) ^-2О1Д7 !0,2 Sr(NOj)t 4HaO (к) —412 86,9
SiOa (p, а-трклчмнт) —204,42 [0,4 SrO (к) Sr(OA), (K) —137,3 I3,UO
SiOj (ст.) -203,33 11,2 —208 21
SiS (r) 12.384 53,43 SrS (K) —107.0 16,3
Si2He <r> W24 65,6 SrSO. (K) —323,46 26,5
SiaNj (к) —153,6 24,2 SrSeOi (к) —230,84 3036
(NH^SiF, {к, 1П —567,343 66.SS SrSeO. (x) —248,86 27,0
Sm (r) 41,48 43,722 SrSiOs (K> —373,24 23,!
Sin (к) SmS1' (p) 0 J6.6 SrTiQ, (h) —379,0 26,0
—123 StWOj (if) —370,3 32,9
Sm3+ (p) —159 -50 Ta (r) 176(67 44,241
К SmClj (к) — —233 3-1 Ta <K) 0 9,92
Lp m
БгецгСГро I Q AtfJw Ч К 4 л/МО ЛЬ
I § TaC (к) —33.6 10.11
£ ТяС1ь |к1 -179.36 56.4
™ TaF. (к) —12^.01 40.6
о TaN (it) ^53/2 12
S TaA (к) 457J 34,2
I. Tb (r) 83.60 43,5515
Tb (x) 0 (7,52
Tb»’ tft) —165 —13
TbClg (к, (1) -224 35
ThOi (к) -216 19.8
тъА (к) —413,4 71.6
Tc (r) —] 44,40 43^5
Тс (к) 0 8.00
TcO, (к) —91 15
TcO3 (x) —] 10 17,3
TcOr <p) —150.63 —75,0
ToO? (к) —224.1 44
НПО, (к) -141.3 ЗЗЛ
Те (г) 36.336 43J542
Те (к, 11) 0 !1.83
Те+ (г) 244,262 13.19S
Тс. (г) 2H.0I3 64,07
ТеВг, (к) —31) 17
TeCI4 (KJ —ат 50
ТеГ4 (г) —292,078 80.3
ТеО (г) 35,252 57.5
ТеО. <г) -W31 65,3
ТеО3 (к) —63,236 15
ИйЕцеегьо A<J Я п * г кка лумолъ 5*2 va> s- е-
TiN (к) —73,637 7J 93
TiO (к) —118,69 8,33
TiO2 (к, рутил) —21238 13,01
TisOj (к, 11) —342,73 18.83
TIA 00 —553,78! 30,92
TI (г) 34,928 43,225
Т1 (к. я) 0 15,34
ТГ (г) 179,214 41.848
И* (р) -7,75 30,103
TI»*- (р) 51,33 -42,28!)
Т1Е (Г) 60,795 68,5
ТШг (г) —18.518 63.96
Tier (к) —10.01 2ЙЛ0
TICI (г) -32,113 61,20
tics {к) -44.208 26,6»
TICIOj (к) —9.811 40,4
TIO, (к) —70 —
TIE (г) —4&683 58,43
TIF (к) —73,18 22,87
TIF, (к) -[23 1—Г
TIH (г) 43.080 51.41
ТИ (г) - —7,081 6fi,l
TU (к) - 29.95 . 30,52
ТИО, (К) — 19,08 39,672
TINOi (к, III) —36,704 39,3
TIN, (к) 7L8W 30,1
TIQH (г) -25,637 61,0
Т1ОН (к) —46,8 21,0
w w en
gj “ Щ cs T1» щ Ш
re *35 и to w c;- r x и -.^гч еч к *? со- ю i~- U7 ™» гч до чг m
ф г--- м ?е г-7 со' с? сч хгLf?ofcc"'f-:'о оо t> сс*го-ХьscTtf счОг*- ч- с£
— ™ Ж т I СЧ г- 5* СЧ ет *Г СЧ сч тг ,о —• *> СЧ —• —— Г'ОФ£Е
ПЕ । 177 1 । |77Г 7Т|-П|Е?ГП
?8 ОС И 8 1Г? S ?С « СЧ 1>1ш 5^ 5' $ S- S; C ™ -С О S3
И «Г— и*Г^г сч re i>'^q йч »L^feSe оь
— । <ч со 1С — сч to ’Г *о
й со
323
ЭБ ,:Н-аучнов наследие России1
зи
ЭБ ,:НаучноЕ наследие России1
ЭБ ,:Научнов наследие России"
VCI, (к) —95,0 23,2
VCIS (к) — 123,45 31,3
VCL (ж) — 1 AM 56,2
VF, (к) -317,-5 29,3
VN (к) —45,7 8,91
VO (к) —S+R65 9,3
W (р) —1№) -26
VA (к) —187 24
VA (к) —273,35 235
У>0* (к, Ц) ~ 3t^2 24,6
Vft (к) -^30,4 31,3
W (г) 193 41,550
W (к) 0 7,80
WBrs (Н? -17 37
WBrj (к) 27 55
WBfj, (к) 65
WBrD (kJ —7856 72,0
WCl, (к) —131,2 60,70
WF. И) —39tl,0 60.11
Wlj (к) —3 42
WO., (kJ —127,6 12,O3[
WOi (к. a) —182,62 18.144
WO;' (?) —220 15
WSa (it) «48,7 16,3
H,WO, (к) —247,693 28,0
Хе (r) 0 40,529
Xe* (r) 278.393 43,284
Y (r) 91,0 42,868
Y (к. ft) a 10,52
YSr (p) — 160 50
YClj <k) —227,7 33.0
bZi
ti
2 iiO (к) —76/M 10,43
Zn(OH)Ej (к, ромб.] —132^5 ]8
ZnS, сфалерит (к) —47,973 1ЭД0
ZnSO4 (к) —207,96 2ВД2
ZnSOj TH/} (kJ —612,73 92,9
ZnSu (к) -39,294 20,0
ZnSiQa (к) —274,6 21,4
ZnTJ (kJ —28,6 22.06
ZnWO] (к) -270,1 ЗЭ.8
2г (г) 135,278 43,315
Zr (к, л) 0 &Д2
2гВгй (к) —95,768 3lr5
2rBrs (и) —145,213 4IJ22
ZrBr^ (к) -173,14 53,702
ZrC (к) -4^194 Z964
гтС1и (к) —121,386 : 27,0
ZrCh (к) —188,724 31r29D
ZrCh (к) -212,477 4lr358
ZrFj (к) -219,041 21.OQ
ZrFj, (к) —341,425 10r325
ZrF, (К) —432,596 25.024
ZrHa (к) —29,32 B,374
Zrla (к) -61,652 35,9
Zrla (к) —94,389 48 897
ZrJ, (к) —114,882 6lf4]2
ZrN («) —80,47 9,238
Zr(OH). (к) —35S^5I 30,5
ZrO(OH)2 (К) —312 22
Zr(\ (к) -248,502 12,036
Zi'fSOiJj (kJ 24.]
ZrSiO4 (к) —4oG;386 20,033
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Автокатализ 130
Адсорбция лктивировангмп 135
Активаторы (промоторы) 132
А KTJ! BI юсть J 88
— коэффициент 188
Актор 115
Акцептор 115
Алюмотермия 291
АминйК жидкий 224
Аммонолиз 225
Амфолиты 190
Анализ физикохимический 254
и сл<
— химический 250
— термический 255
Бромид сурьмы расплавленный
226
Вероятность состояния 36
Вещества
— а мфп прото иные 236
— протогенные 236
— протофильные 236
Волл
— ионное произведение 193
— электролитическая диссоциа-
ция 177, 192 и слР
Высаливание !56
Гальванический элемент 68
— алемепт Даниэля—Якоби 68
Гидратация 142ч см. также Соль-
ватация
— теплота 23
-----вторичная 178
Гидраты 142
Гидролиз 208 и ел.
— константа 213
— по аниону 209
— ио катиону 209
— По Катиону н аниону 210
— степень 212
----- Зависимость от разбавле-
ния 214
------От температуры 215
ступенчатый 2ГЙ
Граница полной сольватации 179
Давление
— внешнее 7
— насыщенного мара 159
— осмотическое 364
— пара 159
Дальтон ады 260
Диаграмма
— плавкости 255
-----системы, в второй обра^
эуются химические соедине-
ния 2о9
— — — твердые растворы 262
— — — с сингулярными (даль-
тоновенимн) топками 360
— состав — свойство 25*1
— энтальпийная 9, 25—26
Диссоциация
— воды !77, 192 и сл.
— ионных кристаллов 179
— электролитическая Шр 173
— — амфотерных соединений
190
—----кислот 188
-----комплексных ионов [98
----константа 188
----- оснований 194
------ полярных молекул 180
------ солей 188
— — степень 174
— — теория |71
Диэлектрическая проЕ1ицдсмость
растворителя 183
Закои(ы)
— Генрн 153
— Гесса (1 и сл*
Дальтона 7а
— действующих масс 112, 184
— разбавлен и и (Оствальда) 185
— распределения 149
— Рауля, первый 150 и ед,
----- второй 163
Золь 139
ЭБ 'нз^чнав наследие России"
Индикаторы 194
Индуктор 115
Индукция химическая 115
Hriiifh])
— ассоциация 167
— водорода 176
гидратация [76 и сл,
— гидрсксоиня 176
— диссоциация 198 и ел.
— комплсксообразователь 199
— константа нестойкости 199
— координационное число соль-
ватацин J77
— лиата ЙЗЗ
— лиония 233
“ Образование 204
— сольватация 179
Ионное произведение
----- автопротолиза 233
— — виды 233
Калориметр И
— псндоемкость 11
Кярботсрмня 291
Катал [[3, см. таыке Катализа
тор(ы)
— гетерогенная 134)
— гомоаеппый 130
— соединения промежуточные
134 н сл„
— теория 132
Каталин тор (ы) 130 и ел.
— активность 132
— механизм 134
— отравление обратимое 132
— Центры активные- 136
— яды каталитические 132
Клтиоиы фогенй» 230
Кинетика химическая [08
— теория 12L п сл.
Кислоты
— анионные 229
— диссоциация 188
— жесткие и мягкие 243
— катионные 229
— Льюиса 2*11
-----первичные 241
“ — вторичные 24 L
— нейтральные 229
— н Основания сопряжённые 230
— теория 217 и сл.
Комплекс активирлЕзашиай 121а
125 л ел.
Коэффициент трансмиссионный
12В
ЭБ ,:Научнов наследие России"
Константны}, см. также Коэф-
фициенты)
— аоюпроюляза 233 н сл.
— ги Л рол ила 213
— криоскопнческая 162
— нестойкости комплекса 199
— универсальная тазовая 37,
151
— устойчивости комплекса 199
— химического равновесия 75,
П2
— эбуллносконическая 162
— электроянтинескон диссоциа-
ции !89
Коэффициент^
— активности 183
— изотопический 172
— распределении 149
Кривая(ые)
-™ ЛнквпЛуса (лнмня) 253
— нагревания 255
— охлаждения 255
— плавления 167
— растворимости [55
— солидуса (линия) 258
— сублимации 167
Криогидрат 258
Кристаллизация 253
— растворов 255
Макросостоянпе 36
Металлотермия 291
А1стод(ы)
— анализ физико-химический
254
-----термический 255
— колориметрн чё-схн й L95
— неводного титрования 250
— обратною осмоса 169
— препаративный 254
— сравнительного расчета 30
Механизм процесса 108
Мсдалохимпя 108
Мнкросостояиие 36
Молекулы активные 120
Молекулярность реакции 1Е7 н
ел,
Мольная доля 140
Мол ЯЛ ЬЕЕОСТЬ I4E
Молярность 141
Неводное титрование 230
Нормальность 14!
327
Осмос !(i4
Основания
— анионные 229
— катионные 229
— к кислоты сопряженные 230
— Лъюнса 241
Переохлаждение 255
Периодичность
— вторичная 30, £76
— изменения энтропии 49
— стандартных электродных по-
тегсщгалол 70
— стандартных энтропий 43
— термических эффектов 27
Перхлорат гидроксоння 223
Плавление никоигруэнтное 202
Порядок реакции 118
Погеицнал{ы)
— изобарный 52
— электродный 71
— — водорода 71
-----стандартный (нормаль-
ный) 7Е и ел.
Правило
— Еертолле 204
— Вамт-Гоффа ! 19
— Семенченко 15! и сл.
Принцип
— Бертле 59
— Вант-Гофф# 165
— Ле Шателье 7Я н сл*
Проводники
— второго рода 1 70
— первого рода 170
Произведение растворимости 196
— расчет (96
Промоторы I3S
Путь реинини
— — каталитический 133 н сл*
— — бес каталитический 133 и
сл.
Работа 7
Равновесие
— зависимость от концеггтрацни
87
----- давления 84
— — присутствия инертного га-
за 88
— “ температуры 79
— ионное в растворе 184
— и ста иное (устойчивое) 34
— кажущееся (заторможенное)
35
— смешение 79# 84
*— — направление 79
— — степень 79
— химическое 33 и ей.
Раствор(ы) 138 и ей,
— буферные [95
— водные !39
— газа в жидкостях 152
— газообразные 139
— гидратапня 142
— жидкие 139
— идеальные 145
— и тагом и чес к не 172
— ИСТМКНЫО 139
— коллоидные J39
— концентрация 140
— концентрированные 143
— кристаллизаиня £55
— насыщен кый 147
— неводные 220
— ненасыщенный 147
— образование 14t и сл.
пересыщенный 147
— разбавленные 146
-----давлеггие насыщенного па-
ра 159
----- неэлектролитов 158 и сл.
— — осмотическое давление 164
-----температура кипения 161
--------отаердеввиня 161
----- электролитов 176
— сильных электролитов !86
— твердые 139
Растворение 144
— критическая температура 155
Растворенное вещество [40
-----олределсшге молекуляр-
ной массы 166
Растворимость
— влияние посторонних веществ
156
— газов я жидкостях 15£
— жидкостей л жидкостях 154
—-------неограниченная 155
*------ — ограниченная 154
— критическая температура 155
— твердых тел в жидкостях
155
Раствори тел ь(и) 140
— апротонные £30
— дифферент!рующгге 233
— нивелирующие 237
— л рою литические 230
328
ЭБ ' Научное наследие России"
Реакции
— ц среде жидкого HF 225
--------NHa 224
--------NA 22?
-----расплавленного SbBr* 226
— гетерогенные 110
— гомогенные 1!0
— нейтрализации 203
— необратимые 33 н сл.
— обменные 202
— обратимые 33 и ел.
— ОКисл и тельно-восстановитель-
ные 93 и сл.
— — ннутрнмсигсвулярные 98
-----диемутяцнн ЭВ
-----диспропорционирования
£38
-----зависимость от концен-
трации 109
— -----среды Юб
-----межмоле куля рныс 98
----- исправление J04
-----самоокмслеЕЕИя^ само-
восстановления 98
----- составление уравнений 93
— ггара'ллелъЕгые 114
— порядок 117 и слй
“ последовательные 114 -
— простые 111
— сложные 111
— сопряжсинЕде i 14
— термические эффекты 16 н сл,
— цепные 121
-----с неразветвленкой цепцьи
121
-----с разве твлсешоЙ цепью J2J
— экзотермические 8
— эндотерм ноские 8
Рид напряжений 71
Свой-стаа
— амфотерЕЕые 190
— киелотно-осЕЕОвиые 273
— — соеднЕЕеиик элементов ос-
новных подгрупп 270 Ei сл,
—-------— побочных подеру пн
277 и с-1.
-----п зайясимости от уделения
по периоду 282 н ел.
Сжатие лантаноидное 279 и сл.
Снла(ьО
— лритлво^лектродБижущая 68
— электродвижущая fiH
Системами}
— ьодд — трехоксид серы 269
- СНаСООСНя-СНС15 264
— иод — теллур 260
— KQ-CuCl 260—261
— литий — рубидий 266
— марганец — медь 265
— серебро — золото 262 и ел,,
269
— сурьма — свинец 257
цинк — магний 259
Скорость химических реакций
106 LE СЛ*
— BHHKJJHe различных факторов
108
------ концентрации J08 ег сл>
----температуры 119 и сл.
— константа 112
Смесь
— изоморфная 262
— эвтектическая 256
Соединения
— амфнпротонпые 236
— амфотерные 190
— промежуточные 134 и сл.
Сели
— двойные L9U
— кислые 190
— комплексные 190
-г- ЕЕсЯтралыше 190
— ОСНОВЕЕЫС 1ЕЮ
— смешанные 190
Сольват(ы) Е42
Сольвата пкя 142
— граница 178
— коордипацЕЕолЕЕое число 17?
— теплота 144
Солыюлиз 208
Состояние стандартное Е4
СоудареЕгня активЕЕые (результа-
тнвЕЕые) 121 и сл.
— фактор вероятностный (сте-
ричсскин) 122
Сродство
—* к кислороду 288
— К прогону 235
— химическое 33 н ел.
----и свойства аленентов
270
----влияние температуры 285
н слн
------ мера 53! 59
Степень
— гидролиза 212
— полноты резники 89
— электролитической диссоциа-
ции 174
329
ЭБ ,:Научнов наследие России"
Температура
— кипели я 161
------ ПОвЫШНИВ 163
— критическая растйореиия 155
— отвердевания 161
— — понижение 163
Теория
— катализа 132 к сл,
— КИСЛОТ И ОСНСнаИИЙ 217 и сл.
----прогонная 221, 228 и сл.
----электронная 221,240 и сл,
— рзстноров 142—143
— гатыюеистеч 220, 221 я сл.
— электролитической диссоциа-
ции 171 ” ел.
Теплота
— гидратации 23
— — ионов 177
— кристаллизации 255
— нейтрализации 20
— образования 13
----атомарная 23
----и структура 31
— плавления 28
— растворения 2] и сл„ 144
— сгорания 13
— сольватации 144
— стандартная образования 1В,
21
----сгорания 18
----фазного перехода 20
Термохимия 11
Титр 14!
Уравнение
— Борна 183
— Байт-Гоффа 73
— Нернета 78
— составление 93
— термохимические 10
Фак гор
— стернческий 122
— внтальпнйиый 50, 56
— энтропийный 50, 56
Фотохимия 108
Фторирование 251
Фтор водород жидкий 225
Цикл Бор на—Габера 26
Четырехоксид азота жидкий 22?
Число
— А1югадро 37
— координационное сольна та-
йки 177
Зв го к трг на 256
Электролиз |70
Элеппролнт(ы)
— сильна 175
-----растворы 186
— слабые 17S
— — образован Fte ЙОЗ
— — равновесие 184
— средней силы |75
Эле и троп роиодиос! ь 170
— растворов 170 j? сл,
— — удельная 170
— ьквЕгдалетизуг 171
— — зависимость от разбавле-
ния 170, 173
Энергия, см. также Теплота
— aKiивалил 1Й4
-----экспериментальная 121
— внутренняя 7
— Гиббса 52
— ионизации 19
— кристаллической реЕнеткгг 17
— ионной решетки 24
“ ArTCKTpOHFfOrO сродства 19
Энтальпия 8
Энтропия
— зависимость от давления 39
-----от состояния вещества 38
-----от температуры 38
— изменение 46 и сл.
— — В ЭЛВЕЕСИ МОСТИ D1 ОбХ'МЗ
38 и сл,
--------------™ структуры 40
*-------- температуры 38
— — лрл растворении 39
— — при н|мЗовоч переходе 38
и сл.г 47—48
— как мера неупорядоченно
сти 37
— и периодический закон 43
— и природа вещества 41
— стандартная 40
------ некоторых веществ 1|
Энтропийная единица 37
Эффект
— индукционный 231
•* тепловой 12. см< также Теп-
лота
*- — реакции 9, 12
-------влияние давления и
температуры 15
— — стандартный 13
— Тиндали 139
ЭБ гНауччоЕ наследие России1
ОГЛАВЛЕНИЕ
Предисловие............+.....................................3
РАЗДЕЛ I. ЭНЕРГЕТИКА ПРОЦЕССОМ
Введение *............. . .\ . . . ч..............., . . - 7
Глава [. Энергетические эффекты........................ « Й
Глава II. Заком Гесса........................................Н
Глава III. Тепловые эффекты рамочных нррцесспя » » 16
L Химические реакции 16
2. Фазовые превращения . + , . . . .........., . 19
3- Процессы в растворах .......................... 19
Глава IV. Примеры применения закона Гесса ,............ 22
Глава V- Некоторые заноиогч^риости 27
I. Теплоты сходных процессов..................... 27
2. Теплоты образования и структура . > + . > + . . + . 31
РАЗДЕЛ ич -ХИМИЧЕСКОЕ CPU ДСП ВО
Введение.........г + . + < , . х . , + . / L + . . + . . L. 33
Глава I. Необратимые и обратимые процессы ...................... 33
Глава II. Энтрелнй . . . п h ............................... 36
I. Энтропия — мера кеу порядочен ног г и.............. 37
2. Энтропии и природа'вещества............... . . * Л
3, Изменение энтропии . ..................... , . < 16
4. Энтропийный и эггтальггнйпый фа «торы процесса - * оО
Глава Hi. Энергия Гиббса .............................. , . , 5®
I, Критерий направления процесса + .. + 5’2
2. Энтальпийный и энтропийный факторы и направлен
ине процесса............... + . . * ........
3. Стандартные изменения энергии Гиббса . ь + . . + . GI
4. Природа вещестид и итмспеиие элегии Гиббса . . 63
5. Определение изменения энергии Гиббса процесса 67
6. Стандартные электродные потенциалы ....... r 69
7. Константа химического равновесия................... 75
Глава IV. Влияние изменения условии нй химическое ривне?-
весие . > , р ..............................-........... 73
1. ЛрнЕщпп ыЧе Шателъе .,.*.. * . . н < » . . , * ,
2. Влияние температуры . * . *........ « Р « » . Ф . 79
3. Влияние давления . . . . , * ч . *......... . + + ^4
4. Влияние коЕшеитраани .. Я7
5. Влияние инертного газд ..........-............ < йй
6. Оптимальные условия осуществлении процесса . , < 88
Глава V. Й кисли тельное восстанови тельные реакции............. 90
1. Составление уравнений реакций > т * . , . . < , , * 93
2. Типы окислительно-восстаЕЕОвителъкык реакций + » 9S
3. Влияние среды на характер реакций . . < , , t , 100
4. Haup&iitEiUti реакций . ь * ...............в Ю4
331
ЭБ гНаучнов наследие России"
РАЗДЕЛ 1IL СКОРОСТЬ И ХИМИЧЕСКИХ РЕАКЦИИ
ВведеЕше х.............. ь , +.................. 306
Глпа I. Основные понятия.............................« ♦ . 308
Глава 1|, Влиыинс концентрации.............................. ПО
1. Гомогенные и гетеро юееныс реакции ПО
2. Простые и сложные реакции . х , . -..........* П1
3. Молекулярности м порядок реакции ..**.**« 117
Глада III*. Влияние температуры.......................'»» 119
L Теория химической кинетики , , . . .............. 121
Гл а в а IV. Влияние катализатора ... . . ............. 130
1. ОбщгЕе сведеЕгия ........ + ч Р +.............. 330
2. Элементы теории катализа................»..«» 132
РАЗДАЛ IV РАСТВОРЫ
ВведеЕЕие..........• ....................... 138
Глава I. Образование растворов + . . , . , т ................. 141
1. Изменение свойств при обра лопан ни растворов . ... 144
2г Растворимость ..............................♦*.. 346
3. Влияние па растворимость природы komiioeechtoe 149
4. Влияние Eia растворимость внешних условий . . . , 152
5. Влияние на растворимость посторонних веществ 156
6. Некоторые закономерности......................... 156
Г л а в з IL Разбавленные рнстноры неэлектролитов + . - . . - 158
L Давление пара.................................... 159
£Р Температуры кипения я отвердевания .............. 161
3. Осмотическое давление < + ........ 164
4. Определение молекулярной массы растворенегого
вещества , т „ , , 4 , ь .............. . . , < . . * * 166
5. Некоторые выводы 4 ,, + ч ........ ч - - + 167
Глава III. Растворы электролитов . . + ................. - > * 170
|. Электрон роподЕЮСТЬ . , ............ . . * - . • 17D
2. Основы теории электролитической диссоциации , . 171
3. Образование растворов . . ....................... 175
Глава IV. Ионные равновесия я обменные реакции в раство-
рах электролитов......................................... 184
L Равновесие а растворах слабых электролитов , . . , 184
2. Растворы сильных электролитов..................... 186
3. Диссоциация кислот, оснований н солей в воде 188
4, Влияние одноименных ноегов era диссоциации сла-
бых электролитов + + + + 191
5. Диссоциация воды * . ......................... 192
6. Буферные растворы . ♦ .......... . . . , + < , « 195
7+ Произведение растворимости ........... . » . + 196
8. Диссоциация комплексных ионов........... . . . 198
9. Обменегые реакции ......................... - 202
10. Гидролиз...........................* - . - р » * 208
Глава V. Теория кислот я оснований + - . + - k + - - । * 217
1. Краткая история допроса d . + d ь ч . , - . + ► 217
2. Теория сольвоснстем , < . + , , . < . +....* • ^21
3* Протонная теория , , .....................* * - 238
4, Электронная теория . ♦ , . ........ . * . - - . 240
5, Значение теории кислот и оснований * .......... 249
Глава VI. Физикохимический анализ .................... * 254
L Термический анализ ........................* * * 255
ЭБ ATJhhob наследие России"
2. Вывод типа диаграмм плавкости из кривых энергии
Гиббса ................................. , . + + . Ж
РАЗДЕЛ V. ХИМИЧЕСКОЕ С?ОДСТ ПО И СВОЙСТВА ЭЛЕМЕНТОВ
Н ИХ СОЕДИНЕНИЯ
Введение . . . . + . . ...... . ....................♦ . 270
Глам L Периодическая система элементов и реакционная
способность веществ ............ . . . . .... 270
1, Главные подгруппы................. , . > < . ь 270
2. Побочггые подгруппы.........................+ 277
3. Периоды.................................... 282
Глава И. Влияние температуры на химическое средство * . * 2S5
Литература..............♦ ........* , . * , ь * <* 293
Приложение L Переучетные значения доя некоторых единиц
намерения - , .............................. . . + . k . . .- 294
Приложен не Н. Стандартные изменился энергии Гиббса образе^
пани я некоторых неорганических и оргаимче-
скнх веществ к их стандартные энтропии . . » 2£?5
Предметный указатель » я........... .................... 320
ЭБ ,:Научнов наследие России1
Михаил Христофорович Кдрапетьянц
ВВЕДЕНИЕ
В ТЕОРИЮ
ХИМИЧЕСКИХ
ПРОЦЕССОВ
3BS, р+длкцмсft С, Ф. Кяндряшиова
Редактор М. М. ПоидзвсклЯ
Мл. рид л Kt йры С. М. Е|4шнш и Л. С. Макаркина
Художник й- И- П<|иферсв
Худижчх! iLfrimuri редактор Т. М. Ск иорцива
Техничный! ||?лак*Фр Л. К. ЗЗгстероил
Корректор С. К> Задъял^пз
И5 № 2040
Изд. № Хнм-filJ, Сдано н набор ]р. ] 0, ДО. Подо, « печать -
Форыаг 84 к 10в7ц, Бум, ТИП. 2 Глрннт^ра литературная. Печать
лыгоияя. О^ъсм I7.GJ усл. псч. л. 37.74 уел. кр^отт, I8.-3? уч.-нзд. л,
Тираж 27000 эху Зан. Av I&I4. Цена ДО коп.
11 j дй г ел ы? гво «Высшая школ|»|
Muckhj. K-$i. Her лини ан ул„ д. 29U*
OpjltHJj ОктяйрьккиЛ РсвиЛюцнн. йрДела Т^удоиою Красного 3-НамсЯг
Ленинградское в рол з подсть ^-Kilo-Tax mi ч всоде- Объединение * Печатный
Дв&р* имени: Д. М. Горького Спшапалнграфкроыв при ГрО'Дзрствен-
цОм СССР ПО дзлнн издательств, полиграфии к кнвжиой тор’
FPMH. |$7'WP Л«ниг^*д< ПЧЗб, Чвило^СднА пр^ Jm'h
ЭБ ,:Научнов наследие России"
ИЗДАТЕЛЬСТВО
«ВЫСШАЯ ШКОЛ Ди
выл ус тит в. свет
в 1981 году
дпй студентов вузов
следующие учебные пособия:
ГллЭФй В. Mr Основы фнАнЧОСкйй хнмИмГ Учеб
бне. — 30 л,— В пер.: 1 р. 30 я.
Последовательна изложены основные положения химиче-
ской термОдннамнкнг учения о reieporOHHbix р flfl мой СС и яXd хи-
мическом равновесии, термодинамике растворов поверх постны 1
вален и йл и злектра химии, в отличие от имеющихся л-особмй по
физической химии усилены такие разделы курса, кам термо-
динамика гетерогенных равновесий, поверхностные явлений и
электрохимия; при рассмотрении ряда термодинамических ха-
рактеристик г риале каете л периодический закон Д И. Менделе-
ева. Книга рассчитана на повышенный уровень математической
подт о то в к к
Для студентов втузов.
Кнорре Д. Г,„ Крылова /1. Ф.г Музыкантов В. С
Физическая ХИМИЯ: Учебник.— 2S п.р и Л. “В пер.: 1 р. 20 к
Пособие содержит изложений □Гневных понятий, Законов и
методов физической химии, необходимых для углубленного м
ускоренного ус в ой ни я неорганической, органической и биологи-
ческой химии. Книга состоит из 2--Х частой. Первая посвящена
рассмотрению строения и состояния вещества, причем материал
нзЛЛгдйТС я а рамнах единого подход а к веществу кА К к системе
из взанм-одейстеукзщнх электронов к ядер, из которых обра-
эукттсл молекулы, □ затем и макрос кол инее кие системы. Стро-
го и достаточно просто разбирается ряд положений кеаиюзой
механики к статистической физики, на которых базируется изу-
чение строения и состояния вещества о современной химии.
Во второй, части рассмотрены термодинамика и кинетика хм-
мичеккил проц etc о Bl
Для студен г ов химических и биологических факультеты
университетов.
Крестов Г. А. Теоретические основы неорганической №
мин: Учеб, пособий. — 20 л., ил. — В пер,: 90 к.
Пособие содержит обобщение закономерностей химии на
о<ко0с общих методов исследования структурного, гермеди-
ЭБ ,:Научнов наследие России"
Н-ймич&СКОго и кинетического. Дается необходимый теорети-
ческий фундамент для изучения химии элементов и их соеди-
нений, Проводится системное обсуждение современных пред-
ставлений о химической структуре, химической динамике, дина-
мике процессов, в растворах и химических процессах. Особое
вниманий удалено использованию общих методов исследования
для характеристики вещее то и химических процессов.
Для студентов хнми ко-технологи чес к их и соответствующих
факультетов политехнических (технологических) институтов.
Уважаемые читатели!
Издательство «Высшая школ в» выпускает учебмикиг учеб-
ные и методические пособия, плакаты. Подробнее познакомить-
ся с учебной литературой Вам поможет аннотированный план
выпуска литературы на fPSl год (вузы и техникумы), который
имеется в книжных магазинах.
Предварительные заявки на книги Вы можете сделать в ма-
газинах Книготорга млн потребительской кооперации,
ЭБ ,:Научнов наследие России"