



























































































































Автор: Орехова В.А. Лашковская Т.А. Шейбак М.П.
Теги: общая генетика общая цитогенетика иммуногенетика эволюционное учение видообразование филогенез общая патология медицина учебное пособие наследственные болезни наследования нормальных и патологических признаков генные болезни клиническая картина причины возникновения методы диагностики врожденные пороки развития
ISBN: 985-06-0228-7
Год: 1997




В.А.Орехова Т.А.Лашковская
М.П.Шейбак
Медицинская генетика
В.А.Орехова Т.А.Лашковская
М.П.Шейбак
Медицинская генетика
Допущено Министерством образования и науки Республики Беларусь в качестве учебного пособия для учащихся медицинских училищ
Библиотека — УЧИДИЩа
Минск "Вышэйшая школа" 1997
УДК 575 (075.32)
ББК 52.5я723
0-65
Рецензенты:
канд. мед. наук, зав. кафедрой медицинской биологии и генетики МГМИ Р. Г. Заяц; канд.
биол. наук, преподаватель Минского медицинского училища № 2 В. М. Ростовцева; директор Минского медицинского училища №» 1 Н. Н. Солодухо
Орехова В. А. и др.
0-65 Медицинская генетика: Учеб, пособие/ В. А. Орехова, Т. А. Лашков-ская, М. П. Шейбак. — Мн.: Выш. шк., 1997. — 123 с.: ил.
ISBN 985-06-0228-7.
Освещаются основные законы, понятия и закономерности наследования нормальных и патологических признаков. Подробно изложены генные болезни с описанием клинической картины и объяснениями причин их возникновения; методы диагностики врожденных пороков развития и наследственных болезней, организация и этапы медикогенетического. консультирования.
Для учащихся медицинских училищ, преподавателей медицинской генетики, а также выпускников школ и абитуриентов. Книга может быть использована студентами биологических специальностей вузов.
ББК 52-.5я723
© В. А. Орехова, Т. А. Лашковская,
М. П. Шейбак, 1997
ISBN 985-06-0228-7
© Издательство «Вышэйшая школа», 1997
Введение
Генетика — это наука о наследственности и изменчивости организмов. Она раскрывает сущность того, каким образом каждая живая форма воспроизводит себя в следующем поколении и как в этих условиях возникают наследственные изменения, которые передаются потомкам. Известно, что из оплодотворенной яйцеклетки развивается только особь данного вида. Это показывает, что наследственность на клеточном уровне представлена генетической программой, которая детерминирует развитие особи.
Медицинская генетика изучает закономерности наследственности и изменчивости с точки зрения патологии. Она выявляет причины возникновения наследственных болезней, разрабатывает меры по профилактике действия мутагенных факторов на организм человека. При их действии на половые клетки здоровых людей происходят нарушения структуры генотипа (мутации), проявляющиеся у потомства наследственными болезнями.
Задачами медицинской генетики являются изучение характера наследственных болезней на молекулярном, клеточном уровнях и уровне целостного организма, а также дальнейшая разработка и усовершенствование методов генной инженерии с целью получения лекарственных веществ (инсулин, интерферон и др.) и генотерапии (замещение патологических генов их нормальными аллелями), интенсивное развитие методов пренатальной (дородовой) диагностики, позволяющих предотвратить рождение ребенка с тяжелой наследственной патологией.
Особый раздел медицинской генетики — клиническая генетика, исследующая вопросы патогенеза, клиники, диагностики, профилактики и лечения наследственных болезней.
з
В последние годы на фоне общего снижения заболеваемости и смертности увеличился удельный вес врожденных и наследственных болезней. В связи с этим роль генетики в практической медицине значительно возросла. Без знания медицинской генетики нельзя эффективно проводить диагностику, лечение и профилактику наследственных и врожденных заболеваний.
Глав al
КРАТКАЯ ИСТОРИЯ МЕДИЦИНСКОЙ ГЕНЕТИКИ
Первые представления о передаче патологических наследственных признаков отражены в Талмуде (собрание догматических, религиозно-этических I правовых положений иудаизма, сложившихся в IV в. до н. э. — V в. н. э.), в котором указана опасность обрезания крайней плоти у новорожденных маль-шков, старшие братья которых или дяди по материнской линии страдают сровотечениями.
В XVIII в. описано наследование доминантного (полидактилия — много-1альность) и рецессивного (альбинизм у негров) признаков. В начале XIX в. несколько авторов одновременно описали наследование гемофилии.
Особого внимания заслуживает книга лондонского врача Адамса, вышедшая в 1814 г. под названием «Трактат о предполагаемых наследственных свойствах болезней, основанных на клиническом наблюдении». Через год она была переиздана под названием «Философский трактат о наследственных свойствах человеческой расы». Это был первый справочник для генетического консультирования. В ней сформулированы несколько принципов медицинской генетики: «Браки между родственниками повышают частоту семейных (то есть рецессивных) болезней» или «Не все врожденные болезни являются наследственными, часть из них связана с внутриутробным поражением плода (например, за счет сифилиса)». Мотульски А. Г. (1959) справедливо назвал Адамса «забытым основателем медицинской генетики».
В середине XIX в. в России над проблемами наследственных болезней работал В. М. Флоринский. Он изложил свои взгляды по усовершенствованию человеческого рода. Однако ряд положений был противоречив и неверен. В то же время В. М. Флоринский поднял и осветил некоторые вопросы медицинской генетики. В своих трудах он правильно оценил значение среды для формирования наследственных признаков, подчеркнул вред родственных браков, показал наследственный характер многих патологических признаков (глухонемота, альбинизм, заячья губа, пороки развития нервной трубки). Однако книга не нашла отклика среди медиков и биологов того времени, так как ученые еще не были подготовлены к восприятию этих идей.
5
В последней четверти XIX в. наибольший вклад в становление генетики человека внес английский биолог Ф. Гальтон (двоюродный брат Ч. Дарвина). Он первым поставил вопрос о наследственности человека как предмете для изучения, обосновал применение геналогического, близнецового и статистического методов для ее изучения и заложил основы для будущего развития генетики человека. Принципиальная ошибка Ф. Гальтона заключается в том, что во всех евгенических мероприятиях он рекомендовал не столько избавиться от патологических генов человека, сколько повысить количество «хороших» генов в человеческих популяциях путем предоставления преимущественных условий для размножения более одаренных, гениальных людей.
Существенный вклад в изучение генетики человека внес выдающийся английский клиницист А. Гаррод, хорошо знавший биологию и химию. Он первым обнаружил взаимосвязь между генами и ферментами и, применив эти знания к изучению патологических признаков, открыл врожденные нарушения обмена веществ.
Работы Адамса и других исследователей того времени не привлекли внимания широкого круга специалистов потому, что наследственность тогда в основном изучалась на растениях. Наблюдения над человеком не учитывались. Между тем, если бы результаты исследований по генетике человека были известны Г. Менделю и другим ученым, изучавшим наследование на ботаническом материале, то открытие законов генетики и их признание могли бы произойти гораздо раньше.
В 1865 г. чешский ученый Г. Мендель глубоко и последовательно с . математическим описанием в опытах на горохе сформулировал законы доминирования для первого поколения гибридов, расщепления и комбинирования наследственных признаков в потомстве гибридов. Этот важнейший вывод доказал существование наследственных факторов, детерминирующих развитие определенных признаков. Работа Г. Менделя оставалась непонятой 35 лет.
В 1900 г. три ботаника независимо друг от друга, не зная работы Г. Менделя, на разных объектах повторили его открытие: Де Фриз из Голландии — в опытах с энотерой, маком и дурманом, Корренс из Германии — с кукурузой, Чермак из Австрии — с горохом. Поэтому 1900 г. считается годом рождения генетики. С него начался период изучения наследственности, отличительной чертой которого стал предложенный ранее Г. Менделем гибридологический метод, анализ наследования отдельных признаков родителей в потомстве.
В 1905 г. В. Бэтсон предложил термин «генетика», а в 1909 г. В. Иогансен предложил термин «ген» (от греческого genes — рождающий, рожденный) для обозначения наследственных факторов. Совокупность всех генов у одной особи ученый назвал генотипом, совокупность признаков организма — фенотипом.
В 1908 г. Г. Харди и В. Вайнберг показали, что менделевские законы
6
объясняют процессы распределения генов в популяциях (от лат. populus — население, народ). Ученые сформулировали закон, который описывает условия генетической стабильности популяции.
В России в 1919 г. Ю. А. Филипченко организовал первую кафедру генетики в Ленинградском университете. В это время работал молодой Н. И. Вавилов, сформулировавший один из генетических законов — закон гомологических рядов наследственной изменчивости.
Н. К. Кольцов, Ю. А. Филипченко и некоторые другие ученые в рамках евгенической программы проводили работы по генетике одаренности, изучая родословные выдающихся личностей. В этих исследованиях были допущены некоторые методические ошибки. Однако по сравнению с генетическими исследованиями в других странах в период расцвета евгеники подходы наших ученых были во многом верными. Так, Н. К. Кольцов и Ю. А. Филипченко правильно поставили вопрос о значении социальной среды в реализации индивидуальных способностей. Они полностью отвергли насильственный путь улучшения природы человека. В период проведения евгенических исследований в СССР были собраны интересные родословные выдающихся личностей (А. С. Пушкина, Л. Н. Толстого, А. М. Горького, Ф. И. Шаляпина и др.).
Конец 20 — начало 30-х годов характеризуются довольно большими успехами в развитии генетики. К этому времени стала общепризнанной хромосомная теория наследственности. Т. Морган и его ученики экспериментально доказали, что гены расположены в хромосомах в линейном порядке и образуют группы сцепления.
Теоретическая и экспериментальная работы С. С. Четверикова (1926, 1929) положили начало современной генетике популяций. Большой вклад в изучение этого раздела внесли труды Р. Фишера (1931), С. Райта (1932), Н. П. Дубинина и Д. Д. Ромашова (1932), Дж. Е. Холдейна (1935) и др.
В ряде стран начала развиваться медицинская генетика. В нашей стране особого упоминания заслуживает Медико-генетический институт, который функционировал с 1932 по 1937 г. При нем был организован центр близнецовых исследований, в котором широко изучались количественные признаки у человека и болезни с наследственным предрасположением (диабет, гипертоническая болезнь, язвенная болезнь и др.). Правильное применение разных методов исследования (клинико-генеалогического, близнецового, цитогенетического, популяционно-статистического) позволило коллективу занять передовые рубежи генетики.
В 20—30-х годах работал талантливый клиницист и генетик С. Н. Да-виденков (1880—1961), который внес свой вклад в изучение наследственных нервных болезней, а также первым в нашей стране начал проводить медико-генетическое консультирование и разрабатывать методику этого вида медицинской помощи.
К концу 30 — началу 50-х годов интерес к генетике человека снизился.
7
В это время в СС С Р медико-генетические исследования были практически прекращены. В течение этого периода вышла только книга С. Н. Давиденкова «,)вол1оционно-гс11С1'и»1ескис проблемы в невропатологии» (1947). Возобновились исследования лишь в начале 60-х годов.
(' I 959 но 1962 г. количество публикаций, конференций, симпозиумов по гене। икс человека быстро возросло. Стало ясно, что наследственные болезни но своей природе гетерогенны, различны не только с клинической, но и с 1спсгичсской точки зрения. Один и тот же фенотип болезни может быть обусловлен мутационным изменением различных белков (генокопия).
После того как было установлено, что ДНК является носителем наследственной информации, ученые направили усилия на изучение молекулярной природы и генетической значимости ее отдельных компонентов.
Исследование ДНК проводилось многими учеными. Весь накопленный комплекс биологических и физико-химических знаний привел к тому, что в 1953 г. Д. Уотсон и Ф. Крик открыли двухценочечную спиральную (пространственную) структуру молекул ДНК. Затем бурно начала развиваться молекулярная и биохимическая генетика человека, а также иммуногенетика.
Развитие цитогенетики человека является ярким примером значения фундаментальных исследований для практического здравоохранения. Так, в 1956 г. А. Леван и Дж. Тио установили, что у человека хромосомный набор состоит из 46 хромосом, а через три года были открыты хромосомные болезни. Очередным переломным моментом в цитогенетике человека была разработка методов дифференциальной окраски хромосом.
Следующим шагом в развитии современной генетики явилось картирование (определение места положения) генов в хромосомах человека. Успехи цитогенетики, генетики соматических клеток обеспечили прогресс в изучении групп сцепления (групп генов, наследующихся совместно). В настоящее время у человека известно 24 группы сцепления. Работы по изучению сцепления генов дают новые практические возможности в диагностике наследственных болезней и медико-генетическом консультировании.
В Республике Беларусь основы медицинской генетики заложил широко известный патологоанатом, член-корреспондент АМН СССР Ю. Г. Гулькевич. Дальнейшее развитие медицинской генетики в нашей республике продолжили Г. И. Лазюк, В. П. Кулаженко, И. Н. Усов. Благодаря Г. И. Лазюку созданы Институт врожденных и наследственных болезней, Республиканский медикогенетический центр и областные медико-генетические консультации.
Таким образом, в истории медицинской генетики можно выделить несколько основных этапов:
1) открытие законов Г. Менделя и изучение наследственности на уровне целостного организма;
2) изучение генетики на хромосомном уровне и открытие сцепленного наследования Т. Морганом и его учениками;
8
3) начало развитию современной генетики популяции дали теоретические экспериментальные работы С. С. Четверикова;
4) развитие молекулярной генетики началось с построения пространствен-1Й структуры молекул ДНК Д. Уотсоном и Ф. Криком.
В настоящее время наследственность изучается на всех уровнях: молекурном, клеточном, организменном и популяционном.
Гл а в all
КЛЕТКА — ОСНОВНАЯ ГЕНЕТИЧЕСКАЯ И СТРУКТУРНО-ФУНКЦИОНАЛЬНАЯ БИОЛОГИЧЕСКАЯ ЕДИНИЦА
Чтобы понять основы наследственности и изменчивости, необходимо знать, хотя бы кратко, строение и функцию клетки.
Клетка была открыта в 1665 г. Робертом Гуком. Более подробно она была изучена во второй половине XX в. благодаря тому, что наряду со световым микроскопом стали широко использоваться другие методы исследования: гистохимический, электронной микроскопии, рентгеноструктурного анализа, авторадиографии (введение меченых радиоактивных атомов в клетки), люминесцентной и ультрафиолетовой микроскопии, замедленной киносъемки. В настоящее время известно, что клетка является основной структурной и функциональной единицей всех живых организмов: многоклеточных (растений, животных, человека) и одноклеточных (микробов, простейших).
Однако в природе существуют и неклеточные формы живого — вирусы.
Вирусы. Вирусы (от лат. virus — яд) — это неклеточные формы жизни с простым строением. Они состоят из нуклеиновой кислоты (ДНК или РНК) и белковой оболочки — капсида. Они самостоятельно жить не могут и являются облигатными (обязательными) паразитами. Вирусы паразитируют в клетках человека (вирусы гриппа, полиомиелита, бешенства, оспы, герпеса, вирус иммунодефицита человека — ВИЧ и другие), животных, растений и бактерий. Вирусы, которые паразитируют в клетках бактерий, называют бактериофагами. Форма вирусов весьма разнообразна: сферическая (шаровидная), палочковидная (нитевидная).
Более сложное строение имеют прокариоты.
Прокариоты. Прокариоты (от лат. pro — перед, раньше и греч. karyon — ядро, буквально «предьядерные») — это бактерии и синезеленые водоросли (цианобактерии). Основные признаки прокариот: 1) отсутствие ядерной оболочки; 2) одна молекула ДНК замкнута в кольцо; 3) нет белков гистонов, которые упаковывают ДНК; 4) ДНК деспирализована (раскручена); 5) не мозаична, то есть информативные гены расположены непрерывно (нет спейсеров — неинформативных участков, которые у эукариот находятся между информативными генами); 6) трансляция быстро
ю
следует за транскрипцией; 7) и-РНК хранится недолго; 8) отсутствие органоидов, имеющих мембранное строение, клеточного центра; 9) наличие мезосом (впячивании клеточной мембраны), выполняющих функции органоидов.
Еще более сложно организованы эукариоты.
Эукариоты. Эукариоты (от греч. эу — полностью и карион — ядро) — это организмы, клетки которых имеют: 1) оформленное ядро (есть ядерная оболочка), 2) цитоплазму с органоидами (рис. 1). К эукариотам относятся все животные, растения и грибы. У эукариот: 1) ДНК может спирализоваться и упаковываться белками-гистонами, а при делении клеток образуются хромосомы (их несколько), 2) ДНК мозаична, то есть между информативными генами располагаются спейсеры (неинформативные участки) и внутри информативных генов — интроны (неинформативные участки), 3) у эукариот в ядре и при выходе из него происходит
Рис. 1. CxcMh строения клетки по данным электронной микроскопии
И
дозревание информационной РНК — процессинг (неинформативные участки вырезаются с помощью ферментов, а концы информативных сшиваются), 4) и-РНК может сохраняться относительно долго.
ОСНОВНЫЕ КОМПОНЕНТЫ ЭУКАРИОТИЧЕСКОЙ КЛЕТКИ
Эукариотическая клетка состоит из оболочки, цитоплазмы и ядра.
Плазматическая мембрана отделяет внутреннее содержимое клетки — цитоплазму от окружающей среды (рис. 2). Она состоит из двух слоев липидов, молекулы которых расположены таким образом, что их головы (гидрофильные участки) обращены к внутренней и внешней водной среде, а их хвосты (гидрофобные участки) находятся в глубине мембраны и обращены друг к другу. В отдельных местах оба липидных слоя пронизываются белковыми молекулами насквозь, образуя в них поры, через которые проходят водорастворимые вещества. Другие белковые молекулы погружаются в липидный слой, а некоторые находятся на внешней или внутренней поверхности
Рис. 2. Схема строения плазматической мембраны:
1 — два слоя липидов; 2 — гидрофильная часть липидных молекул; 3 —гидрофобная часть липидных молекул; 4 — мембранный белок; 5 —периферический мембранный белок; 6 — гликокаликс (углеводный компонент)
мембраны. На наружной поверхности мембраны имеется углеводный компонент (гликокаликс), который присоединяется чаще к белковым, чем к липидным молекулам.
Аналогичные мембраны образуют большинство органоидов клетки. Они
12
являются не просто физическими границами. На мембранах осуществляются многочисленные биохимические процессы: активное поглощение органических и неорганических веществ, синтез ряда соединений и др.
Цитоплазма — полужидкая слизистая бесцветная масса сложного строения. Это коллоид, который может быть в состоянии геля (вязкое состояние) и в состоянии золя (разжиженное состояние). В цитоплазме расположены ядро, органоиды и включения. Основное вещество цитоплазмы называется матриксом или гиалоплазмой (цитозоль). Цитоплазма связывает все органоиды, обеспечивает обмен веществ между ними, в ней протекает также ряд биохимических реакций (синтез нуклеотидов, некоторых аминокислот, жирных кислот, расщепление веществ).
Органоиды (клеточные органы) — это компоненты, которые постоянно присутствуют в клетке, выполняя определенную роль. В зависимости от функции они имеют соответствующее строение. К органоидам относятся: рибосомы, аппарат Гольджи, митохондрии, эндоплазматическая сеть, лизосомы.
Одним из основных компонентов клетки является ядро (рис. 3).
Рис. 3. Схема строения ядра:
/ — хроматин; 2 — ядрышко; 3 — ядсрный сок; 4 — ядсрная оболочка
Ядро состоит из: 1) хроматина; 2) ядрышка; 3) ядерного сока; 4) ядерной оболочки.
Хроматин (от греч. chroma — цвет, краска) — интенсивно окрашенные глыбки, гранулы и сетевидные структуры ядра. Этот видимый в световой микроскоп элемент ядра представляет собой спирализованную ДНК и белок. В делящихся клетках ДНК сильно спирализуется, уплотняется, упаковывается и с помощью белков приобретает определенную форму хромосом. Спирализованные (скрученные) участки ДНК не активны (на них не происходит синтез и-РНК). Передачу генетической информации осуществляют деспирализованные (раскрученные) участки ДНК, которые не видны в световой микроскоп.
Ядрышко формируется определенными участками хромосом (13—15, 21, 22) с генами, кодирующими синтез р-РНК; в нем образуются субчастицы
13
рибосом. Ядрышко обнаруживается только в недедящихся клетках, а во время деления оно исчезает.
Я д е р н ы й сок — кариоплазма (от греч. karyon — ядро) — это бесструктурная масса, в которой содержатся белки, различные РНК, свободные нуклеотиды, аминокислоты, промежуточные продукты обмена веществ, субчастицы рибосом.
Я д е р н а я оболочка состоит из двух мембран, пронизанных многочисленными порами. Каждая из мембран имеет строение, аналогичное плазматической мембране (см. рис. 2).
Эндоплазматическая сеть — это система соединенных между собой канальцев и цистерн (полостей), вакуолей (пузырьков) различной формы и величины, их стенки представляют собой мембраны. Эндоплазматическая сеть контактирует со всеми органоидами клетки, ядром и наружной ядерной мембраной. Она осуществляет транспорт и обмен веществ внутри клетки. На наружной стороне мембран шероховатой эндоплазматической сети расположены рибосомы, в которых синтезируются белки. Гладкая эндоплазматическая сеть не содержит рибосом, в ней находятся ферменты синтеза и расщепления углеводов и липидов.
Рибосома состоит из двух субчастиц: большой и малой, в состав которых входят рибосомальная РНК и белки. Через малую субчастицу проходит и-РНК, в большой происходит образование пептидных связей между аминокислотами в процессе синтеза белка. У эукариот рибосомы образуются в ядрышке, но полностью сформированных рибосом в ядре нет. В эукариотических клетках часть рибосом связана с мембраной эндоплазматической сети (эти рибосомы синтезируют белки, которые поступают в комплекс Гольджи и секретируются клеткой), а часть расположена в гиалоплазме (в них синтезирует белки для собственных нужд клетки).
Аппарат Гольджи — это стопка уплощенных мембранных цистерн (мешочков) и связанных с ними систем пузырьков. В аппарате Гольджи происходят: 1) химические модификации (превращения) поступающих в него клеточных продуктов, 2) транспорт и секреция (выделение) белков, гликопротеинов, углеводов, липидов и 3) формирование лизосом.
Лизосомы — это простые мембранные мешочки, наполненные различными ферментами, которые осуществляют внутриклеточное переваривание. Иногда ферменты выделяются из клетки наружу.
Митохондрии (от греч. mitos — нить и chondrion — зернышко, крупинка) — это энергетическая станция клетки. Она образует и накапливает энергию в виде АТФ (аденозинтрифосфорная кислота). Митохондрия имеет две мембраны: наружную гладкую и внутреннюю, образующую кристы (гребни), что увеличивает площадь внутренней поверхности митохондрии. Внутренняя мембрана окружает матрикс, в котором содержатся кольцевые молекулы ДНК, специфические и-РНК, т-РНК и рибосомы, отличные от цитоплазматических.
14
Клеточный центр — представляет собой пару центриолей, оторые расположены под прямым углом друг к другу. Каждая центриоль — щлиндр, по окружности которого располагается девять триплетов микро-рубочек. Клеточный центр растягивает хроматиды (хромосомы) во время юления клетки, тем самым обеспечивая равноценное распределение генети-юского материала между дочерними клетками.
Кроме органоидов, в цитоплазме встречаются включения — непостоянные омпоненты клетки. Их можно разделить на несколько групп: 1) трофические питательные): жиры, углеводы; 2) секреторные (нужные организму): горюны, ферменты; 3) экскреторные (ненужные и подлежащие выделению из ►рганизма): мочевая кислота и др.; 4) пигментные: меланин (коричневый 1игмент).
Химический состав клетки. В состав клетки входит большинство эле-юнтов периодической системы Д. И. Менделеева. Они находятся в виде 1сорганических (вода, минеральные соли) и органических (белки, нуклеи-овые кислоты, АТФ, углеводы, жиры) соединений.
Для понимания генетики особенно хорошо надо знать структуру молекул слков и нуклеиновых кислот и их роль в клетке.
Белки — это полимеры (от греч. polis — многочисленный), состоящие г» мономеров. Роль мономеров выполняют аминокислоты. В состав боль-1инства белков входит до 20 различных аминокислот. Соединения из не-кольких аминокислот называют пептидами. В зависимости от их количества ывают ди-, три-, тетра-, пента- или полипептиды (содержат от 6—10 до сскольких десятков аминокислот). В состав многих белков входит 300—500 минокислотных остатков, есть и более крупные белки. Молекулярная масса слков колеблется примерно от 5000 до многих миллионов. Различия белков иределяются не только составом и числом аминокислот, но и последова-сльностью чередования их в полипептидной цепи.
Уровни организации белковых молекул:
1) первичная структура — это полипептидная цепь (т. е. нить аминокислот, вязанных ковалентными пептидными связями);
2) вторичная структура — белковая нить, закрученная в виде спирали;
3) третичная структура — спираль, которая далее свертывается, образуя побуду (клубок) или фибриллу (пучок нитей), специфичную для каждого елка;
4) четвертичная структура — состоит из нескольких глобул (например, гмоглобин состоит из 4 глобул).
Функции белков весьма разнообразны:
1) каталитическая: белки-ферменты являются ускорителями биохимичес-их реакций;
2) строительная: белки участвуют в образовании всех клеточных мембран органоидов;
3) двигательная: белки обеспечивают сокращение мышц, мерцание рес-ичск и др.;
15
4) защитная: антитела (гамма-глобулины) распознают чужеродные для организма вещества и способствуют их уничтожению;
5) транспортная: белки переносят .различные соединения (перенос кислорода гемоглобином, гормонов и лекарств белками плазмы и др.);
6) регуляторная: белки участвуют в регуляции обмена веществ (например, инсулин, гормон роста и др.);
7) энергетическая: при распаде 1 г белка до конечных продуктов выделяется 17,6 кДж.
БИОХИМИЧЕСКИЕ ОСНОВЫ НАСЛЕДСТВЕННОСТИ. ДОКАЗАТЕЛЬСТВА ГЕНЕТИЧЕСКОЙ РОЛИ НУКЛЕИНОВЫХ КИСЛОТ
В 1869 г. швейцарский биохимик Ф. Мишер впервые описал вещество содержащееся в ядрах клеток, и назвал его нуклеином, а позже оно был( переименовано в нуклеиновые кислоты (от лат. nucleus — ядро). К ниь относятся дезоксирибонуклеиновая кислота — ДНК (в ее состав входи сахар дезоксирибоза) и рибонуклеиновая кислота — РНК (входит саха] рибоза).
В 1928 г. бактериолог Ф. Гриффит изучал бескапсульные невирулентньи пневмококки (не вызывающие заболевания) и вирулентные в полисахаридное капсуле (вызывающие воспаление легких) для получения вакцины проти! пневмококка. Он показал, что при инъекции мышам живых бескапсульньг пневмококков мыши выживали, а при введении живых капсульных — по гибали. При введении смеси убитых при нагревании капсульных и живы
- бескапсульные живые пневмококки;
- капсульные живые пневмококки;
- капсульные убитые нагреванием пневмококки
Рис. 4. Схема опыта, демонстрирующего явление трансформации
16
бескапсульных пневмококков мыши погибали, из них удалось выделить живых капсульных пневмококков. Таким образом, способность образовывать капсулу перешла от убитого капсульного пневмококка к живому бескап-
сульному (рис. 4).
В 1944 г. О. Эвери с сотрудниками выяснили природу этого загадочного явления. Фактором, превращающим непатогенные (бескапсульные) в патогенные (капсульные) пневмококки, является ДНК, а само явление назвали трансформацией (от лат. transformatio — преобразование, превращение). Следовательно, трансформация — это преобразование признака у одного штамма бактерии в результате проникновения в нее ДНК другого штамма. Явление трансформации стало одним из основных доказательств того, что ДНК является носителем генетической (наследственной) информации.
Позже, в 1952 г. Дж. Ледербергом и Н. Циндером была выявлена передача генетического материала от одного штамма бактерий другому с помощью бактериофага, это было названо трансдукцией (от лат. transductio — Перемещение, передача) (рис. 5). U-об-разная трубка в нижней части разделена бактериальным фильтром. В одну Половину были помещены штаммы сальмонеллы (S. typhi murium),
Рис. 5. Схема опыта, демонстрирующего явление трансдукции:
/ — бактериальные клетки; 2 — ген T+;
3 — бактериофаг; 4 — фильтр
не синтезирующие аминокислоту триптофан (Т-), а в другую — сальмонеллы, синтезирующие триптофан (Т+) и бактериофаги. После инкубации среди сальмонелл, не синтезирующих триптофан, были выделены бактерии Т+. Это объясняется тем, что бактериофаги проходили через бактериальный фильтр и переносили части ДНК от бактерии Т+ к бактериям Т-.
Строение молекул ДНК и РНК
Исследование структуры молекулы ДНК проводилось многими учеными. И только в 1953 г., используя все накопленные биологические и физико-химические знания, Д. Уотсон и Ф. Крик открыли двухцепочеч-
2. Зак. 5285
17
(Библиотека м<АНЦи:<смсго училища
ную спиральную (пространственную) структуру молекулы ДНК.
Каждая цепь — это полимер, мономерами которого являются нуклеотиды. Каждый нуклеотид состоит из сахара дезоксирибозы, остатка фосфорной кислоты и одного из четырех азотистых оснований (аденин, гуанин, тимин, цитозин).
Две цепи ДНК соединяются слабыми водородными связями между азотистыми основаниями по принципу комплементарности: аденин дополняется тимином, гуанин — цитозином (рис. 6).
Рис. 6. Схема строения ДНК
Перед делением клетки ДНК способна удваиваться (реплицироваться). Сначала с помощью фермента ДНК-полимеразы разрываются слабые водородные связи между двумя цепями ДНК, а затем к каждой уже отдельной цепочке достраиваются по принципу комплементарности нуклеотиды (А—Т, Г—Ц), образуются уже две двухцепочечные молекулы ДНК. Репликация ДНК обеспечивает высочайшую точность воспроизведения генетической информации в поколениях клеток и организмов в целом.
Кроме ДНК, в клетке имеются РНК.
Молекула РНК — полимер, ее мономерами являются нуклеотиды.
В отличие от ДНК рибонуклеиновая кислота — это:
1) одноцепочечная молекула;
2) вместо сахара дезоксирибозы в РНК входит сахар рибоза;
3) в состав нуклеотидов входит азотистое основание не тимин, а урацил;
4) состоит из меньшего количества нуклеотидов, чем ДНК.
В зависимости от выполняемых функций выделяют несколько видов РНК: и-РНК (информационная), или м-РНК (матричная), — переносит информацию о структуре белка от ДНК к рибосомам. На долю и-РНК приходится примерно 0,5—1,0 % от общего содержания РНК клетки;
18
т-РНК (транспортная) — переносит аминокислоты в рибосомы. Из общего количества РНК клетки на долю т-РНК приходится около 10 %;
р-РНК (рибосомальная) составляет существенную часть структуры рибосомы. На долю р-РНК приходится около 90 % ог общего количества РНК клетки.
ДНК выполняет разнообразные функции:
1) хранит генетическую (наследственную) информацию, записанную в Ьиде последовательности нуклеотидов;
! 2) передает наследственную информацию из ядра в цитоплазму. Для этого
t ДИК снимает копию и-РНК и переноси!' информацию к рибосомам — месту синтеза белка;
3) передает наследственную информацию от материнской клетки к дочерним клеткам, для чего перед делением клетки ДНК реплицируется.
Далее рассмотрим подробнее каждое из трех указанных положений.
ДНК — носитель генетической информации. Впервые понятие ген было сформулировано в 1941 г. Д. Бидлом и Э. Татумом: ген это участок молекулы ДНК, несущий информацию об одном белке-ферменте. В настоящее рремя геном называют участок молекулы ДНК, кодирующий первичную Структуру полипептида, и понятие о гене расширилось. Известны гены, кодирующие:
J а) белки-ферменты;
' б) структурные белки;
в) т-РНК (много копий);
г) р-РНК (много копий);
д) регуляторные (или функциональные) — включают и выключают другие гены;
е) гены-модуляторы — усиливают или подавляют проявление других Генов.
ДНК непосредственного участия в синтезе белков не принимает. В клетках человека (животных и растений) молекулы ДНК находятся в ядре и отделены ядерной мембраной от цитоплазмы, где происходит синтез белка. Информацию несет посредник — и-РНК, которая по принципу комплементарности считывает (копирует) с ДНК информацию при участии фермента РНК-поли-меразы. Переписывание информации происходит с одной нити ДНК и называется транскрипцией (лат. transcriptio — переписывание). Если в переписываемой нити ДНК стоит нуклеотид гуанин (Г), то фермент РНК-полимераза включает в РНК цитозин (Ц); если тимин (Т) — РНК-полимераза включает аденин (А); если стоит аденин (А), фермент включает урацил (У). По длине каждая из молекул и-РНК в сотни раз короче ДНК. Информационная РНК является копией не всей молекулы ДНК, а только ее части — одного гена, несущего информацию о структуре белка. Готовая и-РНК отходит от ДНК и направляется к месту синтеза белка.
Благодаря процессу транскрипции в клетке осуществляется передача ин
19
формации от ДНК к белку по цепочке: ДНК —> и-РНК —> белок.
Перевод информации с «языка» нуклеотидов на «язык» аминокислот осуществляется с помощью генетического кода.
Генетический код — это система записи информации о последовательности расположения аминокислот в белках с помощью последовательности расположения нуклеотидов в ДНК и и-РНК. Участок молекулы ДНК, состоящий из трех нуклеотидов, называется триплетом или кодоном.
Свойства генетического кода:
1. Код триплетен — каждая из 20 аминокислот зашифрована последовательно расположенными тремя нуклеотидами. Из 4 нуклеотидов (так как существует 4 варианта азотистых оснований) можно создать 64 различные комбинации по 3 нуклеотида в каждом (4x4x4 = 64).
2. Код вырожден — каждая аминокислота шифруется более чем одним кодоном (от двух до шести), исключение составляют аминокислоты: метионин, который кодируется только триплетом АУГ, и триптофан УГГ.
3. Код специфичен — каждый кодон шифрует только одну аминокислоту.
4. Код универсален — один триплет кодирует одну и ту же аминокислоту у всех живых организмов.
5. Код неперекрываем — каждый нуклеотид входит лишь в какой-либо один триплет и переписывание информации происходит строго потриплетно.
6. Триплеты УАА, УАГ, У ГА обозначают прекращение синтеза одной полипептидной цепи, так как к ним нет аминокислот. Они находятся в конце каждого гена.
ПРОГРАММИРОВАНИЕ СИНТЕЗА БЕЛКА В КЛЕТКЕ
В ДНК запрограммирована вся наследственная информация, и-РНК переписывает информацию с участка ДНК (гена) и переносит ее в цитоплазму на рибосому. У эукариот и-РНК еще незрелая, поэтому в ядре и при выходе из него происходит ее процессинг — дозревание (вырезание неинформативных участков и другие процессы), в результате чего РНК укорачивается.
Дозревшая и-РНК переносит информацию о синтезе белка в рибосому (рис. 7). Информация закодирована в виде триплетов. Один триплет (кодон) кодирует йесто одной аминокислоты в белковой молекуле, а последовательность триплетов кодирует последовательность аминокислот в белковой молекуле. Перевод информации с и-РНК на последовательность аминокислот называется трансляцией (от лат. translatio — передача). В и-РНК существуют триплеты: инициирующий АУГ (определяет начало синтеза белка) и терминирующие УАГ, УАА, УГА (заканчивают синтез белка).
Одномоментно в рибосоме помещается 2 триплета: один — в пептидиль-ном, другой — в аминоацильном (аминокислотном) участке рибосомы. К аминоацильному участку во время синтеза белка подтягиваются аминокислоты, а в пептидильном находится пептид (полипептид).
В цитоплазме клетки всегда имеется не менее 20 различных аминокислот
20
Рис. 7. Схема биосинтеза белка
и соответствующих им т-РНК. С помощью специфических ферментов аминокислоты узнаются, активируются и присоединяются к т-РНК, которая переносит их к месту синтеза белка в рибосому. В рибосоме в и-РНК находится кодон, а у т-РНК есть антикодон.
Если в рибосоме на и-РНК будет триплет АУГ, то к нему подойдет т-РНК с комплементарным антикодоном УАЦ; если ГГГ — то т-РНК с антикодоном ЦЦЦ. После этого между аминокислотой, находящейся в пеп-тидильном участке, и аминокислотой, находящейся в аминоацильном участке, происходит образование пептидной связи. Данная реакция осуществляется на большой субъединице рибосомы. Затем т-РНК, находящаяся в пептидиль-ном участке, вытесняется из него и «уходит» в цитоплазму за другой аминокислотой, а рибосома передвигается на следующий триплет (ААА), который будет в аминоацилкном участке рибосомы, триплет же ГГГ окажется в пептидильном участке. Так происходит считывание информации. Когда рибосома окажется на терминирующем триплете, синтез белка заканчивается.
Наращивание аминокислот в белковой молекуле в процессе синтеза белка называется элонгацией (удлинением). Синтез одной молекулы белка длится всего 3—4 с. Каждый этап синтеза катализируется соответствующими ферментами и снабжается энергией за счет расщепления АТФ.
После окончания синтеза белка и образования первичной структуры формируется вторичная, третичная, а иногда и четвертичная структура белка и он становится способным выполнять свои функции.
Сходство и различие организмов определяется набором белков. Каждый
21
вид имеет только ему присущий набор белков, то есть они являются основой видовой специфичности, а также обусловливают индивидуальную специфичность организмов. На Земле нет двух людей, у которых все белки были бы одинаковыми (за исключением монозиготных близнецов). ДНК каждой клетки несет в себе информацию не только о структурных белках, определяющих форму клетки, но и всех белках-ферментах, белках-гормонах и др. Практически все признаки клеток и организма в целом определяются белками. Таким образом, в ДНК заключена вся информация о структуре и деятельности клеток, о всех признаках каждой клетки и организма в целом. Эта информация называется наследственной.
Гл а в а III
ЦИТОЛОГИЧЕСКИЕ ОСНОВЫ НАСЛЕДСТВЕННОСТИ
СТРОЕНИЕ И ТИПЫ МЕТАФАЗНЫХ ХРОМОСОМ ЧЕЛОВЕКА. ПОНЯТИЕ О КАРИОТИПЕ
Хроматин (от греч. chroma — цвет, краска) — это ДНК, связанная с белками гистонами (основными белками) и интенсивно окрашенная. Кроме гистонов, в состав хроматина входят и негистоновые белки (нейтральные или кислые), а также ферменты: для репликации ДНК, для транскрипции и репарации (восстановления поврежденных участков ДНК).
Хромосома — это интенсивно окрашенное тельце. Общая длина молекулы ДНК в хромосоме человека (средней по размерам) достигает примерно 4 см, а суммарная длина этих молекул в клетке с диплоидным (двойным) набором — около 180 см. Благодаря спирализации ДНК и упаковке белками длинная молекула ДНК укорачивается примерно в 5000 раз.
Хромосомы формируются в начале деления клеток. Однако удобнее их изучать в метафазе митоза, когда хромосомы располагаются в плоскости экватора и хорошо ридны в световой микроскоп, так как в этот момент ДНК достигает максимальной спирализации. Метафазные хромосомы состоят из двух сестринских хроматид (удвоенных молекул ДНК), соединенных друг с другом в области первичной перетяжки — центромеры. Центромера делит хромосому на два плеча. В зависимости от расположения центромеры хромосомы бывают:
1) метацентрические — центромера расположена по середине и плечи примерно равной длины;
2) субметацентрические — центромера смещена от середины хромосомы и одно плечо несколько короче другого;
3) акроцентрические — центромера расположена близко к концу хромосомы и одно плечо значительно короче другого.
В некоторых хромосомах есть вторичные перетяжки, отделяющие от плеча хромосомы участок, называемый спутником.
Правила хромосом. 1. Правило постоянства числа хромосом — соматические клетки организма каждого вида имеют строго определенное число
23
хромосом (у человека — 46, у кошки - 38, у мушки дрозофилы — 8, у лошадиной аскариды - - 2, у собаки — 78, у курицы — 78).
2. Правило парности хромосом — каждая хромосома в соматических клетках с диплоидным набором имеет такую же гомологичную (одинаковую) хромосому, идентичную по размерам, форме, но не одинаковую по происхождению: одну — от отца, другую — от матери.
3. Правило индивидуальности хромосом — каждая пара хромосом отличается от другой пары размерами, формой, которая зависит от расположения центромеры, чередованием светлых и темных полос, которые выявляются при дифференциальной окраске.
4. Правило непрерывности — перед делением клетки ДНК удваиваются: к каждой из двух исходных нитей достраиваю гея по принципу комплемен-тарности новые нити ДПК, в результате образуются две молекулы ДНК, из которых получаются две сестринские хроматиды. После деления в дочерние клетки попадает по одной хроматиде, таким образом, хромосомы непрерывны: хромосома от хромосомы.
П Л ял и и и
1 2 3 4 5 6
1 2 3 4 5 б
м н м а м н
7 8 9 10 11 12
04 АО АО XX XX лл
13 14 15 16 17 18
дм х< АЛ ЛК т
19 20 21 22 Jt л
Я М як к М W
1 2 3 4 5 6
7 XX в 9 10 XX 11 ы 12
ла Ай АО JX м
13 14 15 16 17 18
IX XX М АА Ха
19 20 21 22
Рис. 8. Кариотип человека: слева — женщины, справа — мужчины; вверху — хромосомные комплексы, внизу — идиограммы
24
Нее хромосомы подразделяют на аутосомы и половые хромосомы.
На новые — это хромосомы, определяющие формирование мужского и фгнгкого полов.
Аутосомы — все хромосомы в клетках, за исключением половых хромосом
В соматических клетках присутствует диплоидный (двойной) набор хромосом, в половых — гаплоидный (одинарный).
('овокупность хромосом клетки, характеризующаяся их числом, размером и формой, называется кариотипом (рис. 8).
Дня того чтобы легче разобраться в сложном комплексе хромосом, сос-। П1Н1Я1О1ЦИХ кариотип, их располагают в виде идиограммы.
Идеограмма (от греч. idios — своеобразный, gramme — запись) — это * нс юматизированный кариотип. По Денверской классификации (Денвер, । IIIA, 1960 г.) хромосомы располагаются попарно по мере убывания их нспичипы, с учетом положения центромеры, наличия вторичных перегяжек и спутников. Исключением являются половые хромосомы, которые выделя-н и си особо (см. рис. 8). В основу Парижской классификации (1971 г.) поло-•hciiii дифференциальная окраска хромосом, при которой в каждой паре хромосом выявляется характерный только для нее уникальный порядок чередо-IUIIIия темных и светлых полос гетеро- и эухроматиновых районов (рис. 9).
СОВРЕМЕННЫЕ МЕТОДЫ ЦИТОЛОГИЧЕСКОГО АНАЛИЗА ХРОМОСОМ. ПОНЯТИЕ О ГЕТЕРО- И ЭУХРОМАТИНЕ, ПОЛОВОМ ХРОМАТИНЕ
Наиболее подходящей фазой для исследования хромосом является метафиза митоза. Для изучения хромосом чаще используют препараты кратковременной культуры крови, полученные через 48—72 ч после взятия крови, но могут быть использованы клетки костного мозга и культуры фибробластов.
При приготовлении препаратов хромосом к культуре клеток добавляют колхицин, который разрушает веретено деления и останавливает деление и истки в метафазе. Затем клетки обрабатывают гипотоническим раствором, после чего их фиксируют и окрашивают.
Для окраски хромосом чаще используют краситель Романовского—Гимзы, * % ацеткармин или 2 % ацетарсеин. Они окрашивают хромосомы целиком, рппномерно (рутинный метод) и могут быть использованы для выявления численных аномалий хромосом человека (45, 47 и т. д ).
Для получения детальной картины структуры хромосом, идентификации (определения) отдельных хромосом или их сегментов используют различные • пособы дифференциального окрашивания. Один из них — G-метод: по чиипе хромосомы выявляется ряд окрашенных и неокрашенных полос. Чередование этих полос и их размеры строго индивидуальны и постоянны для мокдой пары гомологичных хромосом, поэтому при дифференциальной ок-риске можно легко определить, к какой паре относится хромосома, если
25
даже пары сходны между собой по размерам и форме. Например, хромосомы I 13, 14, 15-й пар трудно отличить при равномерной окраске, а при диффе- i ренциальной — рисунок исчерченности (чередование и размер темных и i светлых полос) неодинаков (см. рис. 9). |\
Хроматин клеточного ядра подразделяется па два основных типа: на эу- и гетерохроматин. Это наследственный материал различной степени и спирализации и упаковки белками различной степени конденсации. и
Эухроматин (от греч. си — полностью и chroma --- цвет) в метафазных }\ хромосомах виден в виде светлых полос. В интерфазс (между делениемн клетки, когда ядро оформленное) находится в деспирализовапном состоянии, qj то есть образует невидимые в световой микроскоп фибриллы (от новолат. ц(
Х /
Рис. ,9. Схематические карты хромосом человека при их дифференциальной окраске '
26
ibrilla волоконце, ниточка). Как правило, в эухромагине находятся струк-урные активные уникальные гены, которые контролируют развитие приз-iukob организма. Эухроматин менее плотно упакован и доступен для фер-1СНТОВ РНК-полимераз, обеспечивающих синтез и-РНК, а затем синтез белков.
Гетерохроматин определяется в метафазных хромосомах при дифференциальном окрашивании в виде темных полос различных размеров, состоящих । конденсированной (спирализованной) плотно упакованной молекулы ДНК. 1,аже в интерфазном ядре гетерохроматин в виде глыбок хорошо виден в ютовой микроскоп. Чаще всего он расположен вокруг ядрышка и около (ерной оболочки. Переписывания информации и-РНК с данных участков : происходит. Эти гены неактивны.
Различают также структурный и факультативный гетерохроматин. Струк-рный гетерохроматин в интерфазном ядре спирализован, плотно упакован в метафазных хромосомах постоянно обнаруживается вокруг центромеры всех 46 хромосомах (составляет около 13 % от генома). Расположение иных полос для каждой пары хромосом строго индивидуально. Функция )уктурного гетерохроматина в целом пока неясна.
Факультативный гетерохрома тин появляется в интерфазном ядре не всегда, представляет собой спирализованный эухроматин. В метафазных хромо-iax факультативный гетерохроматин не обнаруживают. Например, в ядрах ток женщин в диплоидном наборе имеется две Х-хромосомы, одна из орых полностью инактивирована (спирализована, плотно упакована) уже ранних этапах эмбрионального развития и видна в виде глыбки гетеро-матина, прикрепленного к оболочке ядра. Благодаря этому женские и муж-г организмы уравновешиваются по количеству функционирующих генов, тленных с полом, так как у мужчин одна Х-хромосома и одна доза генов эомосомы. Инактивированная Х-хромосома называется половым хрома-тм или тельцем Барра. Половой хроматин обычно определяют путем ана-эпителиальных клеток в соскобс слизистой оболочки щеки. Отсутствие ца Барра у женщин свидетельствует о хромосомном заболевании — син-ie Шерешевского—Тернера (кариотип 45, ХО). Присутствие у мужчин тель-арра свидетельствует о наследственном заболевании синдроме Клайн-тера (кариотип 47, XXY).
ченыс считают, что в клетках по мере специализации все большее число j инактивируется (выключается), эухроматин переходит в гетерохрома-
ОСНОВНЫЕ ТИПЫ ДЕЛЕНИЯ ЭУКАРИОТИЧЕСКИХ КЛЕТОК. КЛЕТОЧНЫЙ ЦИКЛ И ЕГО ПЕРИОДЫ
гществуют различные типы деления клеток: амитоз, митоз, мейоз.
пнь клетки от момента ее возникновения в результате деления мате-ой клетки до ее собственного деления или смертиНазывается жизненным
27
циклом. В течение веей жизни клетки растут, дифференцируются, выполняв^ специфические функции.
Амитоз (от греч. а — отрицание, mitos — нить) -- прямое делен и клетки. При этом сохраняется интерфазное состояние ядра (видны ядерна оболочка и ядрышко, хромосомы не выявляются, веретено деления не об разуется). Ядро делится путем перетяжки, может возникнуть и двухъядерна клетка, иногда перешнуровывается и цитоплазма. Описано амитотическо деление в клетках кожного эпителия, скелетной мускулатуре, в стареющи и патологически измененных клетках. После амитоза клетки не способш приступить к митотическому делению.
Для того чтобы в ряде клеточных поколений сохранялось и строго no/i держивалось определенное количество ДНК, делению обязательно предшсс твует удвоение хромосом. (
Если количество хромосом в гаплоидном (одиночном) наборе обозначь через п, а содержание ДНК — с, то в диплоидном (двойном) наборе д редупликации будет — 2п2с, а после редупликации — 2п4с. 1
Митотический цикл включает подготовку клетки к делению, или интс[ фазу (состоящую из пресинтетического, синтетического и постсинтетическог периодов), и само деление. В эмбриональный период, когда клетки быстр делятся, жизненный цикл совпадает с митотическим. После дифференцировк клеток, когда каждая из них выполняет специфическую функцию, жизненны цикл продолжительнее митотического.
Рассмотрим подробнее три периода подготовки клетки к делению. (
Пресинтетический (Gj от англ, gap — интерва] следует непосредственно за делением. В этот период синтезируются PHI* ( различные белки, увеличивается количество рибосом и митохондрий. Вс , это приводит к тому, что клетка интенсивно растет и может выполнять cboi основную функцию (набор генетического материала будет 2п2с). ,
В синтетический период (S) происходит репликаци (удвоение) количества ДНК, а также синтез РНК и белков (2п4с).
В постсинтетический период (G2) клетка запасаете , энергией, синтезируются белки ахроматинового веретена, идет подготовь । к митозу (2п4с). ।
гМ ито з (от греч. mitos — нить) — непрямое деление клеток, сопр( । /вождающееся спирализацией хромосом. Митозом делятся соматические кла । ки, в результате чего дочерние клетки получают точно такой набор хромосом ।
। какой имела материнская клетка. В митозе выделяют несколько фаз: профаз' шрометафазу, метафазу, анафазу, телофазу. i
Профаза (от греч. pro — до, перед и греч. phasis — появление) ( начальная фаза митоза. Происходит спирализация и укорочение хромосо! которые из тонких длинных невидимых нитей к концу профазы становяп , короткими толстыми видимыми и расположены в виде клубка. Ядрышко ядерная оболочка исчезают, центриоли расходятся к полюсам клетки, i ,
28
diKa тубулина формируются микротрубочки — нити веретена (2п4с).
Впрометафазе (от греч. pro — до, перед и греч. meta — южду, после) хромосомы оказываются в цитоплазме, к центромерам прицепляются нити веретена с обоих полюсов и хромосомы движутся к плос-ости экватора (2п4с).
В метафазу (от греч. meta — между, после) все хромосомы •псполагаются в плоскости экватора; хорошо видно, что они состоят из двух роматид. В этой фазе уже можно сосчитать число хромосом в клетке (2п4с).
В анафазу (от греч. ana — вверх) сестринские (возникшие при цвоении хромосом) хроматиды расходятся и сосредоточиваются у полюсов истки (2п2с).
Телофаза (от греч. telos — конец) обратна профазе: хроматиды хромосомы) из коротких толстых видимых становятся тонкими длинными свидимыми в световой микроскоп; формируются ядерная оболочка и яд-ышко. Заканчивается телофаза разделением цитоплазмы и образованием нух дочерних клеток (2п2с).
Биологическое значение митоза. 1. В результате митоза дочерние клетки олучают точно такой же набор хромосом, который был у материнской истки, поэтому во всех клетках тела (соматических) поддерживается пос-оянное число хромосом.
2. Митозом делятся все клетки, кроме созревающих половых клеток: ) за счет митоза происходит рост организма в эмбриональном и постэм-рионалыюм периодах; б) все функционально устаревшие клетки организма и меняются новыми, делясь митотически (эпителиальные клетки кожи, фор-1спные элементы крови и др.); в) процессы регенерации (восстановление траченных тканей) происходят при делении клеток митозом.
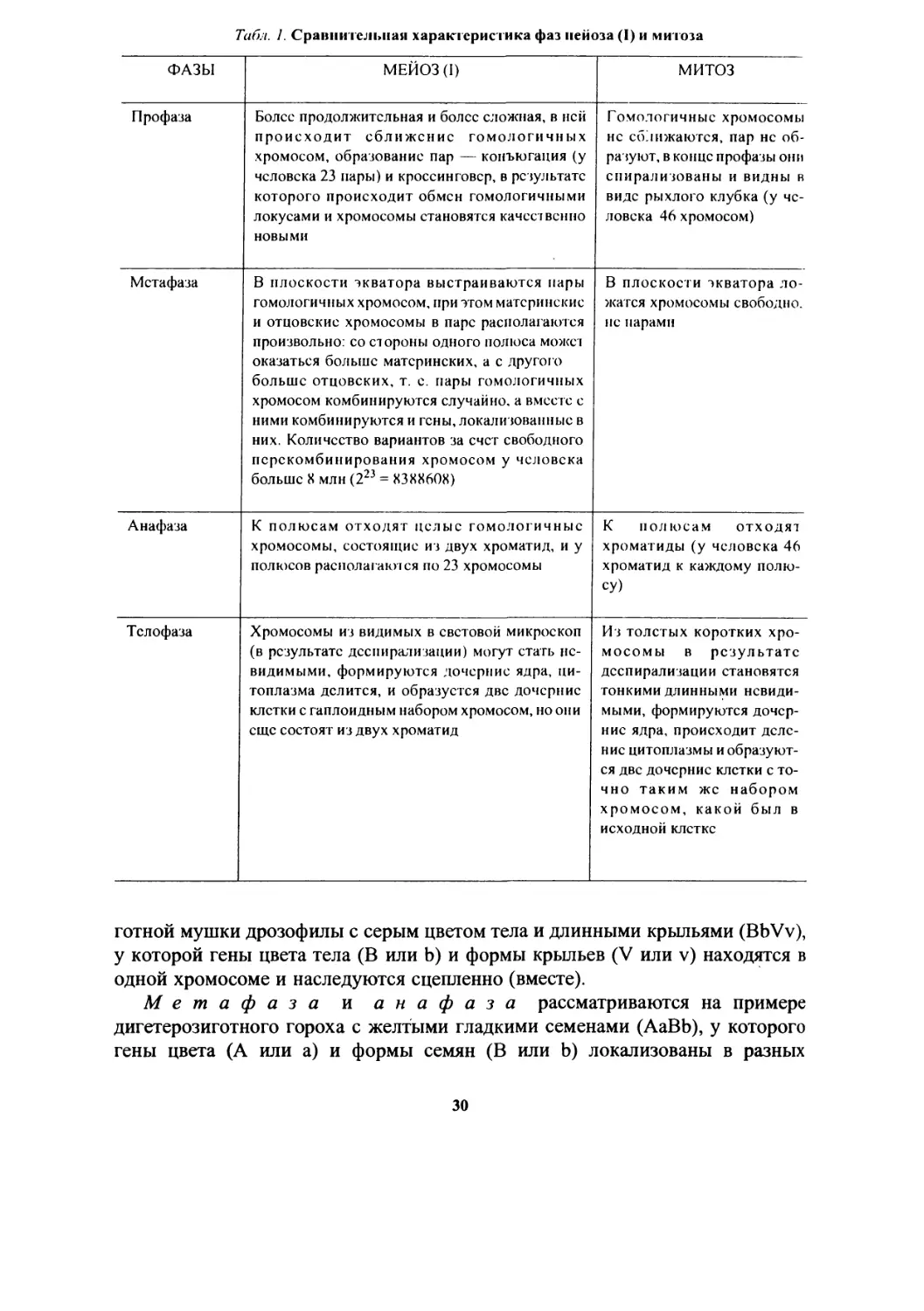
М е й о з (от греч. meiosis — уменьшение) — деление, приводящее к мсныпению числа хромосом вдвое. С помощью мейоза происходит обра-эвание и созревание половых клеток (яйцеклеток и сперматозоидов) из собых соматических клеток яичников и семенников. В результате мейоза пело хромосом уменьшается вдвое (из диплоидных клеток образуются галоидные). Мейоз состоит из двух последовательных делений: первого и горого, причем удвоение ДНК происходит только перед первым делением. I мейоз так же, как и в митоз, вступают клетки с хромосомами, состоящими । двух сестринских хроматид. После первого деления быстро наступает горое деление, без подготовки и без синтеза ДНК. Второе мейотическое сление протекает по типу митоза, с той лишь разницей, что во всех фазах удет половинное число хромосом. В мейозе и митозе фазы называются динаково: профаза, метафаза, анафаза, телофаза (табл. 1).
Чтобы легче понять поведение хромосом при мейозе и митозе, целесо-Празно проанализировать упрощенную схему мейоза (I) и митоза (рис. 10).
Для более доступного восприятия цитологических основ законов наслед-гвснности профаза мейоза рассматривается на примере дигетерози-
29
Табл. 1. Сравни тельная характеристика фаз нейоза (I) и митоза
ФАЗЫ МЕЙ03(1) митоз
Профаза Более продолжительная и более сложная, в ней происходит сближение гомологичных хромосом, образование пар — конъюгация (у человека 23 пары) и кроссин говор, в результате которого происходит обмен гомологичными локусами и хромосомы становятся качественно новыми Гомологичные хромосомы нс сближаются, пар нс образуют, в конце профазы они спирализованы и видны в виде рыхлого клубка (у человека 46 хромосом)
Метафаза В плоскости экватора выстраиваются пары гомологичных хромосом, при этом материнские и отцовские хромосомы в парс располагаются произвольно: со стороны одного полюса можст оказаться больше материнских, а с другого больше отцовских, т. с. пары гомологичных хромосом комбинируются случайно, а вместе с ними комбинируются и гены, локализованные в них. Количество вариантов за счет свободного псрскомбинирования хромосом у человека больше X млн (223 = ХЗХХ60Х) В плоскости экватора ложатся хромосомы свободно, не парами
Анафаза К полюсам отходят целые гомологичные хромосомы, состоящие из двух хроматид, и у полюсов располагаю'!ся по 23 хромосомы К полюсам отходят хроматиды (у человека 46 хроматид к каждому полюсу)
Телофаза Хромосомы из видимых в световой микроскоп (в результате дсспирализации) могут стать невидимыми, формируются дочерние ядра, цитоплазма делится, и образуется две дочерние клетки с гаплоидным набором хромосом, но они еще состоят из двух хроматид Из толстых коротких хромосомы в результате дсспирализации становятся гонкими длинными невидимыми, формируются дочерние ядра, происходит деление цитоплазмы и образуются две дочерние клетки с то-чно таким же набором хромосом, какой был в исходной клетке
готной мушки дрозофилы с серым цветом тела и длинными крыльями (BbVv), у которой гены цвета тела (В или Ь) и формы крыльев (V или v) находятся в одной хромосоме и наследуются сцепленно (вместе).
Метафаза и анафаза рассматриваются на примере дигетерозиготного гороха с желтыми гладкими семенами (АаВЬ), у которого гены цвета (А или а) и формы семян (В или Ь) локализованы в разных
30
Митоз
Рис. 10. Упрощенная схема мейоза (I) и митоза.
Профаза: В — доминантный ген серого цвета мушки дрозофилы; b — рецессивный ген черного цвста;У — доминантный ген длинных крыльев мушки дрозофилы; v— рецессивный ген коротких крыльев. Метафаза: А —доминантный ген желтого цвета семян гороха; а — рецессивный ген зеленого цвета; В — доминантный ген гладкой формы семян гороха; b — рецессивный ген морщинистой формы семян гороха
парах гомологичных хромосом и комбинируются независимо друг от друга (произвольно).
Биологическое значение мейоза. 1. Мейоз приводит к уменьшению числа хромосом вдвое, что обусловливает постоянство видов на Земле. Если ьы число хромосом не уменьшалось, то в каждом последующем поколении происходило бы увеличение хромосом вдвое (у родителей — 46, у детей — *>2. у внуков — 184, у правнуков — 368 и т. д.).
2. Мейоз обеспечивает разнородность гамет по генному составу (в профа-10 — кроссинговер, в метафазе — свободное перекомбинирование хромосом).
3. Случайная встреча гамет (сперматозоида и яйцеклетки) с различным набором генов обусловливает комбинативную изменчивость (гены родителей
31
комбинируются, вследствие чего у детей могут появиться признаки, которых не было у родителей). Комбинативная изменчивость обеспечивает большое разнообразие человечества и дает возможность приспосабливаться к изменяющимся условиям окружающей среды, тем самым способствуя выживанию вида.
Итак, в результате мейоза у человека образуются половые клетки с гаплоидным набором хромосом.
ПОЛОВЫЕ КЛЕТКИ.
РАЗВИТИЕ СПЕРМАТОЗОИДОВ И ЯЙЦЕКЛЕТОК
Г о и а д ы (от греч. gone — пораждающее) — половые органы, образующие половые клетки. Мужские гонады семенники, женские — яичники.
Г а м е т ы (от греч. gamete — жена, gametes муж) — половые клетки, обеспечивающие передачу наследственной информации от родителей потомкам. Мужские гаметы — сперматозоиды, женские яйцеклетки.
Процесс развития (образования) гамет называется гамегогенезом, развития сперматозоидов — сперматогенезом, развития яйцеклеток — овогенезом.
Сперматогенез. Семенник состоит из многочисленных канальцев. Развитие сперматозоидов цроисходит в стенке извитых канальцев семенников. На поперечном разрезе канальца (рис. 11) видно, что в нем имеется несколько слоев клеток (зон).
1. Зона размножения — в наружном слое семенного канальца происходит деление клеток митозом (это диплоидные клетки — 2п2с). Они размножаются на протяжении всего периода половой зрелости мужской особи и называются сперматогонйями. Некоторые из них перемещаются ближе к просвету канальца в зону роста.
2. Зона роста — сперматогонии растут и образуется сперматоцит первого порядка (2п4с).
3. Зона созревания — сначала происходит 1-е мейотическое деление и образуется сперматоцит второго порядка (п2с); затем — 2-е мейотическое деление и образуются сперматиды (пс).
4. Зона формирования — хорошо выражена, из сперматиды (у которой еще много цитоплазмы) формируются головка, шейка и хвостик, образуется сперматозоид (см. рис. 11).
При половом акте мужчина выделяет около 200 млн сперматозоидов. За время половой жизни он продуцирует не менее 500 млрд сперматозоидов.
Овогенез. Он происходит в яичниках. В эмбриональный период клетки яичника делятся митозом и образуются овогонии (2п2с), которые к моменту рождения превращаются в овоцит первого порядка и задерживают свое развитие, сохраняясь без изменений до полового созревания.
С наступлением половой зрелости (обычно ежемесячно) отдельный овоцит переходит к росту: удваивается ДНК (2п4с), увеличивается его размер, на-
32
Рис. 11. Участок семенного канальца семенника млекопитающего и строение сперматозоида (схема):
А — участок семенного канальца: / — спсрматогонии; 2 — сперматоциты первого порядка; 3 — сперматоциты второго порядка; 4 —спсрматидь!; 5 — сперматозоиды; Б — строение сперматозоида (по данным электронного микроскопа): / — акросома; 2 — ядро; 3 — центросома; 4 — митохондрии; а — головка; б — шейка; в — хвост
птливаются белки, жиры, углеводы, пигменты. Каждый овоцит окружается глкими фолликулярными клетками, обеспечивающими его питание. Сначала >разуется первичный, затем вторичный и зрелый фолликулы. Зрелый фол-1кул (1раафов пузырек) заполнен жидкостью, а внутри его находится яй-жлетка (рис. 12).
Далее происходит овуляция (стенка зрелого фолликула лопается, яйце-[етка попадает в воронку маточной трубы) и наступает созревание яйцеклет-[ — 1-е мейотическое деление. Из овоцита первого порядка образуется юцит второго порядка и направительное тельце (которое при 2-м мейоти-jckom делении может разделиться еще раз). В направительное тельце уходит лько избыток хромосомного материала (п2с), а запас питательных веществ тается в овоците второго порядка.
2-е мейотическое деление заканчивается при проникновении сперматозо-(а в овоцит второго порядка. Образуется овотида, которую называют зрелой
Зак. 5285
33
Рис. 12. Яичник млекопитающего и яйцеклетка (схема):
А — яичник млекопитающего: / — первичный фолликул; 2, 3 рост и развитие первичного фолликула; 4 — созревший фолликул (граафов пузырек); 5 — кровеносные сосуды; Б — яйцеклетка человека, окруженная клетками лучистого венца и сперматозоидами
яйцеклеткой (см. рис. 12), и направительное тельце с половиной генетического материала (пс).
К началу полового созревания в яичниках обнаруживается примерно 100 000 овоцитов, однако за весь репродуктивный период в яичниках женщины образуется 300—400 овоцитов.
Отличие сперматогенеза от овогенеза. 1. При сперматогенезе (рис. 13) из одной исходной клетки образуется 4 сперматозоида, а при овогенезе из одной исходной клетки образуется одна яйцеклетка и три направительных тельца (в которые уходит только избыток хромосомного материала, и набор хромосом в яйцеклетке становится гаплоидным).
2. При сперматогенезе зона роста относительно короткая, а при овогенезе — длинная (накапливается запас питательных веществ для будущего зародыша).
3. При сперматогенезе зона формирования хорошо выражена, при овогенезе — не выражена.
Отличие половых клеток от соматических. 1. В половых клетках гаплоидный набор хромосом, в соматических — диплоидный.
2. В половых клетках ядерно-цитоплазматическое отношение не такое, как в соматических (в сперматозоиде оно высокое; в яйцеклетке — низкое).
3. Форма и размеры половых клеток отличаются от соматических: у сперматозоида имеется головка, шейка и хвостик, яйцеклетка — крупная, с большим запасом питательных веществ (в 85 000 раз больше сперматозоида).
При соприкосновении с яйцеклеткой (чаще в верхней трети яйцевода) сперматозоид выделяет ферменты гиалуронидазу и муциназу, которые разрушают оболочки яйцеклетки. Сперматозоид проникает в яйцеклетку, про-
34
Зоны
Сперматогенез
Овогенез
I Размножения
Сперматогонии
Сперматоцит I порядка
II Роста
Овогонии
III Созревания
IV
Формирования
Сперматоцит
Овоцит II порядка
Яйцеклетка
Рис. 13. Гаметогенез (схема). Прослежены изменения двух пар хромосом
н ходит оплодотворение, образуется зигота с диплоидным набором хромосом: • цин набор — от отца, другой — от матери (2п2с). Таким образом, во всех нотках тела имеется диплоидный набор хромосом, а в половых — гаплоидный нс).
Вся наследственная информация передается от родителей к детям через юловые клетки.
Глава IV
ЗАКОНОМЕРНОСТИ НАСЛЕДОВАНИЯ ПРИЗНАКОВ
ОСНОВНЫЕ ПОНЯТИЯ И ТЕРМИНЫ В СОВРЕМЕННОЙ ГЕНЕТИКЕ
Для изучения медицинской генетики необходимо знать основные термин! и понятия, которые используются и в общей генетике.
Наследственность — это свойство живых организмо! сохранять генетическую информацию и признаки предков и передавать и) в ряду поколений.
Наследование — это процесс воспроизведения признаков предко! в последовательных поколениях.
Гомологичные хромосомы — одинаковые по размерам по форме, по составу генов, но разные по происхождению: одна — от отца другая — от матери.
Ген — это участок молекулы ДНК, кодирующий первичную структур; полипептида.
Аллельные гены — гены, которые локализованы в гомологичны) хромосомах в одинаковых локусах и кодируют один и тот же признак ил1 его вариации.
Гомозигота — организм, в котором данная пара аллельных гено! одинакова: АА или аа.
Гетерозигота — организм, в котором пара аллелей неодинакова Аа.
Г емизигота (от греч. hemi — полу- и зигота)— когда в диплоидно! организме присутствует один ген из пары аллелей и он всегда проявляется Например, в Х-хромосоме у мужчин в локусе, которого нет в Y-хромосоме находится один ген гемофилии, а в Y-хромосоме такой ген отсутствует.
Доминантный ген (от лат. dominans — господствующий) преобладающий, который подавляет проявление других аллелей; обозначаю прописной буквой латинского алфавита.
Рецессивный ген (лат. recessus — отступление) — он проявляете) только в гомозиготном состоянии; обозначают строчной буквой латинскоп алфавита.
Закон чистоты гамет: в процессе образования гамет i
36
i< иисдую из них попадает только один ген из аллельной пары. Цитологически • ю объясняется мейозом: в анафазе мейоза гомологичные хромосомы расходятся и вместе с ними расходятся аллельные гены.
Генотип — совокупность генов данного организма. Однако часто под генотипом понимают одну или две пары аллелей (гомозиготы и гетерозиготы).
Фенотип — совокупность признаков данного организма (внешних и внутренних). Он развивается в результате взаимодействия генотипа с внешней средой. В фенотипе реализуются не все генотипические возможности, к лишь часть их, для которых были конкретные оптимальные условия. Фенотип — это частный случай реализации генотипа в конкретных условиях.
Медицинская генетика (и генетика человека) опирается на общие принципы, полученные первоначально в исследованиях на растениях и животных. Глзобраться в простых случаях наследования у человека было невозможно ne t учета опытов Г. Менделя над горохом.
ЗАКОНОМЕРНОСТИ НАСЛЕДОВАНИЯ ПРИЗНАКОВ, УСТАНОВЛЕННЫЕ Г. МЕНДЕЛЕМ
В своих исследованиях Г. Мендель применил гибридологический метод 11 ибрид — это помесь):
1) скрещивал особей, отличающихся по одной паре изучаемых аллелей (моногибридное скрещивание), по двум парам (дигибридное скрещивание), но трем и более (полигибридное скрещивание);
2) производил тщательный количественный подсчет полученных потомков (сколько желтых, сколько зеленых);
3) проводил тщательный анализ полученных результатов на протяжении нескольких поколений и делал выводы.
Скрещивание у растений и животных обозначают знаком умножения — х. У человека тем же знаком обозначают бракосочетание.
При написании схемы скрещивания (у человека — бракосочетания) принято на первом месте ставить женский пол, который обозначают знаком j ( юркало Венеры), затем мужской — cf (щит и копье Марса).
Для того чтобы записать скрещивание, необходимо знать символы, которыми обозначаются родители, гаметы и потомки:
Р — родители (от латинского parents — родители),
G — гаметы (от латинского gametos — гаметы),
Fj — дети (от латинского fillii — дети),
F2, F3 и так далее — последующие поколения.
Моногибридное скрещивание. Мендель скрещивал гомозиготные горохи, • сличающиеся по одной паре признаков: растения с желтыми и зелеными • сменами гомозиготные, и получил F| — 100 % желтые.
37
желт. зел.
Р—^)ААх о* аа
G—@ (af
F j — Аа — 100 % желтые
Согласно закону чистоты гамет, в каждую гамету попадает один ген и пары аллелей.
Ген желтой окраски семян полностью подавляет зеленый, то есть доми нирует, поэтому все первое поколение одинаковое.
Первый закон Менделя: при скрещивании гомозиготны) особей, отличающихся по одной паре альтернативных (взаимоисключающих признаков, наблюдается единообразие гибридов первого поколения как in фенотипу, так и по генотипу.
При скрещивании гибридов первого поколения между собой было полу чено расщепление:
F1 (jAa х d1 А а желт. зел.
G — по фенотипу 3 : 1
F2 АА, Аа, Аа, аа по генотипу 1 :2 : 1
Второй закон Менделя (расщепления г и бридов второго поколения): гибриды второго поколение дают расщепление по фенотипу 3:1 (3 части желтых и 1 часть зеленых) а по генотипу 1:2:1.
Затем Г. Мендель провел дигибридное скрещивание: скрещивал растения отличающиеся по двум парам альтернативных признаков.
Дигибридное скрещивание. Скрещивались растения гороха с желтым» гладкими семенами и растения с зелеными морщинистыми семенами. И Fi получено единообразие гибридов, так как желтый цвет доминировал на; зеленым, а гладкая форма — над морщинистой.
Р — ААВВ х (/a abb А — ген желтого цвета, а — зеленого'
G— ^В) В — ген гладкой формы, b — морщинистой
F1 —АаВЬ—100% желтые гладкие
> При скрещивании гибридов первого поколения между собой (гетерозиготь по форме и цвету) Г. Мендель получил различные семена гороха (желтые гладкие, желтые морщинистые, зеленые гладкие, зеленые морщинистые) по тому, что гены цвета и формы семян гороха и детерминируемые (опреде ляемые) ими признаки комбинировались свободно — цвет не зависел oi формы и наоборот.
С помощью решетки Пеннета (рис. 14) удобнее записать и установит! все возможные сочетания мужских и женских гамет при скрещивании гиб ридов первого поколения между собой, а также определить фенотип и генотип особей Fp
38
9 А В9 9а ьа
SB SB
- - - -
SB ев
О a b О а а в9
9
Гаметы (ав) (Ab) (ав) (ab)
(ав) ААВВ ААВЬ АаВВ АаВЬ
(аь) ААВЬ ААЬЬ АаВЬ АаЬЬ
(ав) АаВВ АаВЬ ааВВ ааВЬ
(ab) АаВЬ АаЬЬ ааВЬ aabb
Рис. 14. Схема построения решетки Пениста при дигибридном скрещивании
Третий закон Менделя (независимого комбинирования генов): гены различных аллельных пар и ^терминируемые ими признаки комбинируются независимо друг от друга, ПК как они локализованы в различных парах гомологичных хромосом. Ци-ологически это объясняется мейозом: в метафазе мейоза (I) пары гомоло-ичных хромосом располагаются в плоскости экватора, свободно комбини->уясь, а вместе с ними комбинируются и гены.
Как в метафазе (I) пары гомологичных хромосом расположатся (то к >дному полюсу больше материнских, то больше отцовских), так в анафазе I) они и разойдутся в разные гаметы.
В первом случае в гаметы попадут гены АВ и ab, во втором — АЬ и iB, то есть гены цвета и формы горошин комбинируются свободно, независимо ipyr от друга.
Согласно закону чистоты гамет, всегда в Гамету попадает один из пани аллелей, сколько бы пар аллелей при скрещивании ни учитывал ис-
39
следователь (если три признака: желтый цвет, гладкая форма семян, длинный стебель — ААВВСС, то в гамету попадут — АВС).
Чтобы проводить скрещивание, Менделю нужно было знать, гомо- или гетерозиготное растение гороха по цвету или по форме семян. Для этого он проводил анализйрующее скрещивание.
Анализирующее скрещивание. Генотип организма, имеющего рецессивный признак, определяется по его фенотипу (он всегда гомозиготный).
Особи с доминантным признаком (гомозигота или гетерозигота) по фенотипу не отличимы. Для установления генотипа особи с доминантным признаком применяется анализирующее скрещивание.
Анализирующее скрещивание заключается в том, что особь с неизвестным генотипом скрещивается с рецессивной особью и по потомству узнают генотип анализируемой особи.
Если при таком скрещивании все потомство окажется единообразным, то анализируемая особь гомозиготна (1), если произойдет расщепление, то она гетерозиготна (2).
1. желт.
Р — £ АА G- @ Fj - Аа
зел. 2. желт. зел.
— 100 % желтые Ft - Аа — 50%, аа — 50%
Анализ генотипов важен не только при селекционной работе, но и для медицинской генетики. Так как на людях эксперименты запрещены, то антропогенетики и врачи прибегают к анализу родословных и ищут браки, которые позволяют проанализировать неизвестный генотип. Например: 1) у обоих родителей полидактилия, их ребенок имеет нормальное строение кистей. Следовательно, родители гетерозиготны по этому признаку; 2) мать имеет резус-отрицательную группу крови (генотип — dd), а отец — резус-положительную группу крови с неизвестным генотипом (DD или Dd). У них родился ребенок, имеющий резус-отрицательную группу, значит, отец гетерозиготен.
МЕНДЕЛИРУЮЩИЕ ПРИЗНАКИ ЧЕЛОВЕКА
Менделирующими признаками называются те, наследование которых происходит по закономерностям, установленным Г. Менделем. Менделирующие признаки определяются одним геном — моногенно (от греч. monos — один), то есть когда проявление признака определяется взаимодействием аллельных генов, один из которых доминирует (подавляет) другой. Менделевские законы справедливы для аутосомных генов с полной пенетрантностью (от лат. penetrans — проникающий, достигающий) и постоянной экспрессивностью (степенью выраженности признака) (см. гл. V).
Если гены локализованы в половых хромосомах (за исключением го-
40
mi логичного участка в X- и Y-хромосомах), или в одной хромосоме сцеплено, пни в ДНК органоидов, то результаты скрещивания не будут следовать законам Менделя.
()бщие законы наследственности одинаковы для всех эукариот. У человека иноке имеются менделирующие признаки, и для него характерны все типы и к наследования: аутосомно-доминантный, аутосомно-рецессивный, сцепленный с половыми хромосомами (с гомологичным участком X- и Y-хромосом).
ТИПЫ НАСЛЕДОВАНИЯ МЕНДЕЛИРУЮЩИХ ПРИЗНАКОВ
I. Аутосомно-доминантный тип наследования. По аутосомно-доминан-нюму типу наследуются некоторые нормальные и патологические признаки:
I) белый локон над лбом;
2) волосы жесткие, прямые (ежик);
1) шерстистые волосы — короткие, легко секущиеся, курчавые, пышные;
4) кожа толстая;
5) способность свертывать язык в трубочку;
6) габсбургская губа — нижняя челюсть узкая, выступающая вперед, нижним губа отвислая и полуоткрытый рот;
7) полидактилия (от греч. polys — многочисленный, daktylos — палец) — многопалость, когда имеется от шести до девяти пальцев на руке или ноге;
8) синдактилия (от греч. syn — вместе) — сращение мягких или костных । кннсй фаланг двух и более пальиев;
9) брахидактилия (короткопалость) — недоразвитие дистальных фаланг пальцев;
10) арахнодактилия (от греч. arahna — паук) — сильно удлиненные «паучьи» пальцы.
11. Аутосомно-рецессивные менделирующие признаки у человека. Если рецессивные гены локализованы в аутосомах, то проявиться они могут при Ираке двух гетерозигот или гомозигот по рецессивному аллелю.
По аутосомно-рецессивному типу наследуются следующие признаки: во-иосы мягкие, прямые, кожа тонкая, 0 (I) группа крови, группа крови Rh-, исощущение горечи вкуса фенилкарбамида, неумение складывать язык в трубочку. По этому типу наследуются также многие болезни обмена веществ: фенилкетонурия, галактоземия, гистидинимия и др. (см. гл. IX).
III. Менделирующие признаки, сцепленные с полом (неполно). X- и Y-хромосомы имеют общие гомологичные участки (рис. 15). В них локали-юваны гены, детерминирующие признаки, наследующиеся одинаково как у мужчин, так и у женщин (подобно признакам, сцепленным с аутосомами).
Гены, локализованные в гомологичных участках X- и Y-хромосом, обусловливают развитие некоторых болезней.
Пигментная ксеродерма — заболевание, при котором под влиянием уль-
41
S Красно-зеленая слепота, гемофилия, атрофия
►зрительного нерва и другие сцепленные с полом признаки
Jchthyosis hystrix gravior и другие голандрические признаки
Общая цветовая слепота
14 '17
18
20
Пигментная ксеродерма
t___________Болезнь Огучи_________________
- — _________Спастическая параплегия----------
-----.— Рецидивирующий буллезный эпидермолиз-
28
Пигментный ретинит
- Геморрагический диатез------
Судорожные расстройства-------
34
Рис. 15. Наиболее вероятное расположение в гомологичном участке X- и Y-хромосом генов, нс полностью сцепленных с полом (по Ниль и Шеллу)
трафиолетовых лучей на открытых частях тела появляются пигментированные пятна. Вначале они в виде веснушек, затем в виде более крупных папиллом различной величины и, наконец, опухолей. Для большинства больных пигментная ксеродерма заканчивается летально.
Болезнь Огучи — в слое палочек и колбочек, пигментном эпителии наблюдаются дегенеративные изменения (болезнь чаще встречается в Японии).
Спастическая параплегия — спастика и слабость нижних конечностей, возникающая в результате дегенерации пирамидных путей в области грудного и поясничных отделов спинного мозга, изредка в стволе головного мозга.
Эпидермолиз буллезный — образование пузырей после механических травм кожи.
Полная (общая) цветовая слепота — полное отсутствие цветового зрения.
Наследование групп крови. Большое значение для медицинской практики имеет изучение групп крови, которые зависят от антигенов, расположенных на поверхности эритроцитов.
Антигены — это высокомолекулярные вещества, в ответ на введение которых в организме вырабатываются антитела (гамма-глобулины — одна из фракций белков крови, которая синтезируется лимфоцитами). Следует отметить, что на собственные антигены организм с нормальной иммунной системой антител не вырабатывает.
42
В настоящее время хорошо изучены группы крови систем: ABO, Rh, MN, Р( Даффи, Льюис, Лютеран, Келл, Кидд и др. В систему входят группы крови, iuнорме детерминируются (определяются) аллелями одного гена.
Множественные аллели — количество аллелей в природе больше двух. | )дним из примеров множественных аллелей у человека являются группы крови системы АВО.
В зависимости от антигенов, которые находятся на поверхности эритроцитов, все люди земного шара делятся на четыре группы. У одних людей пи поверхности эритроцитов нет антигенов А и В — это О (I) группа, у других есть антиген А — А(П) группа, у третьих есть антиген В — В (III) । руина, а у четвертых есть антигены А и В — АВ (IV) группа.
В процессе длительной эволюции живые организмы приспособились к < охранению постоянства своего антигенного состава и не допускают вмешательства других антигенов. Поэтому у людей 0(1) группы крови, не имеющей на поверхности антигенов А и В, есть антитела аир против антигенов А и В; у людей А (II) группы крови есть антитела Р против антигена В; у людей В(Ш) группы есть антитела а йротив антигена А; у АВ (IV) группы пег антител против антигенов А и В.
Четыре группы крови (системы АВО) определяются аллельными генами, которые располагаются в девятой паре хромосом человека. Обозначаются иллсльные гены разными буквами алфавита (1А, 1в, 1°), как исключение из правил генетики.
0(1), А(П) и В(Ш) группы наследуются как менделирующие признаки. Гены 1А и 1в по отношению к гену 1° ведут себя доминантно.
Аллельные гены 1А и 1в у лиц IV группы ведут себя независимо друг от друга: ген 1А детерминирует антиген А, а ген 1в — антиген В. Такое взаимодействие аллельных генов называется кодоминированием (каждый аллель детерминирует свой признак). Наследование AB(IV) группы крови не следует закономерностям, установленным Менделем.
Группы крови А(П) и В(Ш) системы АВО наследуются по аутосомно-доминантному типу, а 0(1) группа — по аутосомно-рецессивному типу.
Рассмотрим, как наследуются группы крови системы АВО.
1) . Если гомозиготная женщина А(П) группы крови выйдет замуж за мужчину с 0(1) группой, то все дети будут А(П) группы крови.
р —5 1А1А G- ©
х 1° 1° 0*
($
Е] — 1А 1° — 100 % А(П) группы
2) . Женщина А(П) группы крови гетерозиготная вышла замуж за мужчину с 0(J) группой крови. Вероятность рождения детей будет: 50 % с 0(1) группой и 50 % с А(П) группой крови.
43
p —J IAI° g— ©(? F]— IAI°
50%A(II)
I°I°d* (?) l“l“
50 % 0(1)
3) . Если женщина B(III) группы крови гомозиготная, а мужчина 0(1) группы, то все дети будут В(Ш) группы гетерозиготные.
Р —§ IBIB X 1°1°с/
G- ® ®
F । — 1В 1° — 100 % В(Ш) группа
4) . Женщина В(Ш) группы крови гетерозиготная, а мужчина 0(1) группы крови. Вероятность рождения детей от этого брака составит: 50 % В(Ш) группы крови гетерозиготных на 50 % 0(1) группы крови.
р — 2 1В1°
G — О©
F, — 1В 1° 50%В(Ш)
х 1° 1° /
1°1°
50 % 0(1)
5). Если женщина А(П) группы крови вышла замуж за мужчину В(Ш) группы крови (оба гомозиготные), то от этого брака все дети будут AB(IV) группы крови.
Р-?
G —
F(-
1А 1А ©
1А1В
х IB IB d*
(3
100 % AB(IV) группа крови
6). Женщина А(П) группы крови вышла замуж за мужчину В(Ш) группы (оба гетерозиготные). От этого брака равновероятно рождение детей 0(1), А(П), В(Ш), AB(IV) группы крови, так как происходит случайная встреча гамет родителей и свободная комбинация генов.
Р —о 1А 1°
G — ©©
F| — 1А 1В,
х 1В 1° (У*
О®
jB j0 jA j0 j0 j0
AB(IV) B(III) A(II) 0(1)
44
Кроме антигенов А, В, 0 на поверхности эритроцитов у людей расположи’ пы антигены групп системы резус. Если на эритроцитах ниходится антиген Rh, то такие люди относятся к группе Rh+ (их около 85 %), й если отсутствует данный антиген, то они относятся к группе Rh- (их око-ин 15 %).
1 руппы крови Rh+ и Rh- системы резус детерминируются генами, которые покализованы в первой паре хромосом человека. Группа крови Rh+ может Oi.rn» гомозиготная (DD) и гетерозиготная (Dd), группа Rh- — только гомо-шгогная (dd).
I руппы крови резус-системы наследуются как менделирующие признаки. Проследим, какие могут быть последствия для детей, если мать имеет ре-iy^ отрицательную группу крови.
Женщина с группой крови Rh- вышла замуж за мужчину, у которого । руина крови Rh+ гомозиготная.
Р dd х DDcT1
(i - (d) (of
I-,— Dd — 100%
()т этого брака все дети будут резус-положительные гетерозиготные, так как ген D полностью доминирует над геном d и F] — единообразно. Во прсмя беременности Rh+ эритроциты плода могут попасть в кровь матери, и материнский организм начнет выработку антител против этих эритроцитов. (* каждой последующей беременностью увеличивается риск иммунизации и возрастает вероятность гемолитической болезни новорожденных и ее тяжести.
Если женщина с группой Rh- вступает в брак с мужчиной гетерозиготным (что чаще встречается), то вероятность рождения детей от этого брака будет равна 50 % Rh+ гетерозиготных и 50 % Rh-.
Р — £ dd х Dd(^
G— (3) (D^
Fl— Dd, dd
ХРОМОСОМНАЯ ТЕОРИЯ НАСЛЕДСТВЕННОСТИ T. МОРГАНА
Установленные Г. Менделем принципы независимого наследования и комбинирования признаков проявляются только тогда, когда гены, определяющие пи признаки, находятся в разных хромосомах (относятся к разным группам сцепления).
Т. Морганом и его группой (1910—1916 гг.) была сформулирована хромосомная теория наследственности. Они подтвердили, что гены находятся в хромосомах. Гены, локализованные в одной хромосоме, образуют группу
45
сцепления и передаются потомству совместно. Групп сцепления столько, сколько пар хромосом, или число групп сцепления равно числу хромосом в гаплоидном наборе. У женщин 23 группы сцепления, так как у них 23 пары гомологичных хромосом, а у мужчин 24 группы сцепления, так как у них 22 пары аутосом, Х-, У-хромосомы — две отдельные группы сцепления.
Следует подчеркнуть, что гены, локализованные в одной хромосоме, сцеплены не абсолютно. В профазе мейоза гомологичные хромосомы конъюгирую! (сближаются), происходит кроссинговер, в результате которого несестринские хромотиды обмениваются гомологичными (одинаковыми) участками. Кроссинговер может происходить в разных участках хромосомы. Чем дальше друг от друга расположены локусы в одной хромосоме, гем выше вероятность кроссинговера между ними.
Например: пара гомологичных хромосом имеет расположение генов А, В, С — в одной хромосоме и а, Ь, с — в другой. Вероятность кроссинговера выше между А и С, чем между А и В.
В своем экспериментальном исследовании, доказывающем явление полного и неполного сцепления, Т. Морган использовал мух дрозофил. Сначала он скрестил гомозиготную самку с серым телом и длинными крыльями с гомозиготным самцом — черным короткокрылым.
Р —jBBVV X BBVVd1
G— (gf
Fj — BbVv— 100%
все единообразные (серые длиннокрылые), так как гены серого цвета тела и длинных крыль-
В — серый цвет тела b — черный цвет тела V — длинные крылья v — укороченные (редуцированные)
ев доминантные
Далее было проведено анализирующее скрещивание, то есть скрещивание Fj с мухами, гомозиготными по рецессивным генам. При анализирующем скрещивании фенотип потомства отражает типы гамет, которые образуют гетерозиготные родители.
I. Анализирующее скрещивание. При скрещивании самки, имеющей черное тело и укороченные крылья, с гетерозиготным самцом из F j образуется 50 % мух с серым телом и длинными крыльями (BbVv) и 50 % — с черным телом и укороченными крыльями (bbw).
46
У самцов дрозофилы кроссинговер практически не происходит, поэтому ины, расположенные в одной хромосоме, обнаруживают полное • ц с п л е н и е, то есть наследуются совместно.
II. Анализирующее скрещивание. При скрещивании дигетерозиготной • «мхи с гомозиготным рецессивным самцом образуется четыре типа потомков: • грыс с длинными крыльями — 41,5 %, черные с укороченными крыльями — 11,5 %, а также с новыми кобинациями признаков (возникшие из кроссоверных । имсг, отличные от родителей) — серые с укороченными крыльями — 8,5 % и черные с длинными крыльями — 8,5 %. В сумме потомков, образовавшихся in кроссоверных гамет, оказалось 17 %. Процент кроссинговера определяют по потомкам: число особей, возникших из кроссоверных гамет, делят на оГицее число особей, возникших из кроссоверных и некроссоверных гамет, и умножают на 100.
Г
И
||
[в V ! । . !
Л----------41,5%
® ! -*--------•“ I 41, 5%
BbVv, bbvv, Bbvv, bbVv । i
lb vi
41,5% 41,5% 8,5% 8,5%'
без кроссинговера (некроссоверные)
! в V |
Z---------------- J 8,5%
l'-1--------------*“[ 8,5%
I I
| b V |
с кроссингов ером (кроссоверные)
Таким образом, у самок дрозофилы в мейозе происходит кроссинговер и поэтому наблюдается неполное сцепление.
В результате анализа опытов Т. Морган сформулировал закон: гены в хромосоме расположены линейно и наследуются сцеплено. Сила сцепления Н1ИИСИТ от расстояния между генами. Расстояние между генами измеряется и процентах кроссинговера: 1 % кроссинговера соответствует 1 морганиде инпвана в честь Моргана). Процент кроссинговера для разных пар генов иппсблется от долей единицы до пятидесяти. Уже при расстоянии в 50 морганид признаки наследуются практически независимо, несмотря на то •по гены локализуются в одной хромосоме.
47
Частота кроссинговера может сильно увеличиваться под влиянием как внутренних (видовая принадлежность, возраст, гормональный фон и др ), так и внешних факторов (температура, радиация, химические мутагены, некоторые лекарственные препараты и др.).
ЛИНЕЙНОЕ РАСПОЛОЖЕНИЕ ГЕНОВ.
КАРТЫ ХРОМОСОМ ЧЕЛОВЕКА
Существование кроссинговера позволило школе Г. Моргана разработан, в 1911—1914 гг. принцип построения генетических карт хромосом. В основу построения карт положен тот факт, что гены расположены по длине хромосомы в линейном порядке. За единицу расстояния между генами принимается 1 % кроссинговера, или 1 морганида.
Генетическая карта хромосомы - это отрезок прямой, на котором обозначен порядок расположения генов и указано расстояние между ними в процентах кроссинговера. Она строится на основе результатон анализирующего скрещивания.
Например: гены А и В локализованы в одной хромосоме, между ними происходит кроссинговер с частотой 10 %. Чтобы узнать расстояние и место в хромосоме третьего гена С, необходимо выяснить, какой процент кроссинговера он дает с обоими из двух уже известных генов. Если с А — 4 % кроссинговера, то можно предположить, что ген С находится либо справа от А (1), либо слева от него (2).
а ю% в
1. АВ=10% < \
АС=4% А 4% с 6% В
ВС=6% Г V Л
С 4% А 10% В
2. ВС=14% Г л----4Z
Для уточнения места расположения гена С необходимо знать процент кроссинговера между В и С. Если между ними 6 % кроссинговера, то ген С расположен справа от А, а если 14 %, то ген С расположен слева от А. У человека анализ сцепления генов классическими методами, разработанными на дрозофиле, невозможен, поскольку невозможны экспериментальные бракосочетания.
- Для изучения групп сцепления и составления карт хромосом у человека используют более 15 методов.
48
Анализ родословных. С этого метода началось изучение сцепления генов
*п поиска. Если точно установлены генотипы родителей и набрано доста-111'11101’ количество информативных семей (точнее детей), то определяют он но у кроссоверных генотипов.
I сим ический анализ гибридных соматических клеток (см. гл. VI).
I нпридпая клетка («мышь человек») при последующих делениях теряет хро-мо| ими человека. Если при потере какой-либо хромосомы признак исчезает, in icii, дсгерминирующий этот признак, расположен в данной хромосоме.
( гйчас для построения карт хромосом широко используют методы анализа рпмосомных перестроек, гибридизации ДНК и др.
II настоящее время у человека изучены все 24 группы сцепления. Что । «н пен я 1 хромосомы, то генетическое расстояние, оцениваемое в результате ।ни низа родословных, хорошо (но не абсолютно) соответствует расстоянию, ннцучасмому на основе картирования с помощью клеточных гибридов «че-nilirh мышь».
Нокусы, сцепленные с Х-хромосомой (а их больше 200), отнесены к этой • римосоме на основе анализа родословных (многие из них подтверждены н» । одами гибридизации соматических клеток). Лишь небольшая часть генов и типизована с помощью методов гибридизации соматических клеток.
Построение карт хромосом имеет большое практическое значение, так I'Hii способствует выявлению наследственных заболеваний у плода, ранней iiini постике этих болезней, установлению носителей генетических нарушении, находящихся еще в бессимптомной фазе.
ОСНОВНЫЕ ПОЛОЖЕНИЯ ХРОМОСОМНОЙ ТЕОРИИ НАСЛЕДСТВЕННОСТИ
I. Гены располагаются в хромосомах, различные хромосомы содержат неодинаковое число генов, набор генов каждой из негомологичных хромосом .ппкален.
2. Гены в хромосоме расположены линейно, каждый ген занимает в хромосоме определенный локус (место).
1 Гены, расположенные в одной хромосоме, образуют группу сцепления и имеете (сцеплено) передаются потомкам, число групп сцепления равно । ппиоидному набору хромосом.
4. Сцепление не абсолютно, так как в профазе мейоза может происходить мрпссинговер и гены, находящиеся в одной хромосоме, разобщатся. Сила • копления зависит от расстояния между генами в хромосоме: чем больше рнсстояние, тем меньше сила сцепления, и наоборот. Расстояние между генами 1нморяется в процентах кроссинговера. 1 % кроссинговера соответствует нцпой морганиде.
I Зик. 5285
49
ГЕНЕТИКА ПОЛА
У женщин 22 пары аутосом и две одинаковые половые хромосомы XX; у мужчин 22 пары аутосом и половые хромосомы X и Y (неодинаковые). В процессе мейоза каждая из пары гомологичных хромосом уходит в разные гаметы. Так как у женщин 23 пары гомологичных хромосом, то во все гаметы попадает 22 аутосомы и одна Х-хромосома (гаметы одинаковы), поэтому женский пол гомогаметный. У мужчин образуется два типа гамет: 22+Х и 22+Y, поэтому мужской пол гетерогаметный. Вероятность рождения девочек так же, как и мальчиков, составляет 50 %.
Р — $ 22х2+ХХ
G— @2+Х)
Fj— 22х2+ХХ
X 22 X2+XY <Г (22+Х%22+У 22x2+XY
50% девочки 50% мальчики
Пол будущего ребенка определяется сочетанием половых хромосом в момент оплодотворения. Если яйцеклетку оплодотворяет сперматозоид с X-хромосомой, то рождается девочка, а если яйцеклетку оплодотворяет сперматозоид с Y-хромосомой, то рождается мальчик.
Признаки организма, связанные с полом, подразделяют на три категории (см. рис. 15):
1) признаки, сцепленные с половыми хромосомами. Это признаки, развитие которых обусловлено генами, расположенными в одной из половых хромосом. Х-хромосома по размерам значительно больше, чем Y-хромосома.
В Х-хромосоме есть большой участок, которого нет в Y-хромосоме, в нем расположены рецессивные гены (гемофилии, дальтонизма, миопатии Дюшена, атрофии зрительного нерва) и доминантные (рахита, не поддающиеся лечению витамином D, темной эмали зубов). Гены этого участка полностью сцеплены с полом, передаются исключительно через Х-хромосому. Доминантные гены из этого участка одинаково проявляются у обоих полов; рецессивные — у женщин только в гомозиготном состоянии, а у мужчин — в гемизиготном состоянии.
Рассмотрим наследование, сцепленное с полом, на примере гемофилии. У мужчин ген гемофилии находится в гемизиготном состоянии (один из пары), и поэтому гемофилия у музкчин всегда проявляется. У женщин, если и есть, этот ген в гетерозиготном состоянии, он не проявляется, так как доминантный ген нормальной свертываемости его подавляет. В брак вступают здоровая женщина-носительница и здоровый мужчина:
Р — <jj XHXh x XHY rf H — ген нормальной
G— XHXh свертываемости крови
Fj— XHXH, XHXh, XHY, XhY h — ген гемофилии
девочка девочка- мальчик мальчик здоровая носитель- здоровый больной
ница
В непарном участке Y-хромосомы, кроме генов, детерминирующих пол, расположено небольшое число генов, которые могут встречаться только у ниц мужского пола и передаются от отца всем сыновьям (голандрические гены): волосатость ушей, перепонки между пальцами ног, ихтиоз (кожа в ниде рыбьей чешуи).
В парном сегменте (гомологичном для X- и Y-хромосом) локализованы гены, детерминирующие пигментную ксеродерму, болезнь Огучи, спастическую параплегию, эпидермолиз буллезный, общую цветовую слепоту и /фугие. Их называют неполно или частично сцепленными с полом. Они могут передаваться как с Х-, так и с Y-хромосомой;
2) признаки, контролируемые полом. Развитие этих признаков обусловлено генами, расположенными в аутосомах обоих полов, но степень проявления их (экспрессивность) разная у мужчин и женщин (особенно у гетерозигот, гик как происходит сдвиг доминантности). Изменение доминирования гена обусловлено половыми гормонами. Например: а) у мужчин ген облысения цоминантный, а у женщин — рецессивный и проявляется только в гомози-ютном состоянии, поэтому лысых мужчин значительно больше, чем женщин; 0) пенетрантность гена подагры у мужчин 80 %, а у женщин 12 %, значит, подагрой чаще болеют мужчины;
3) признаки, ограниченные полом. Развитие этих признаков обусловлено юнами, расположенными в аутосомах обоих полов, но проявляются они юлько у одного пола (гены удойности и жирности молока имеются у коров н быков, но проявляются только у коров).
Гл а в а V
ВЗАИМОДЕЙСТВИЕ ГЕНОВ
Признаки появляются в результате взаимодействия генотипа с окружающей средой. Генотип — это совокупность генов организма, но не сумма, а система генов, находящихся во взаимодействии. Гены взаимодействуют продуктами, которые они кодируют (определяют). Упрощенная схема: ген -» и-РНК белок-фермент биохимические реакции —> признак.
Различают взаимодействие аллельных и неаллельных генов.
ВЗАИМОДЕЙСТВИЕ АЛЛЕЛЬНЫХ ГЕНОВ
Известны следующие взаимодействия между аллельными генами:
1) полное доминирование;
2) неполное доминирование;
3) ко доминирование;
4) сверхдоминирование.
Полное доминирование — когда один ген полностью подавляет действие другого. Рассмотрим полное доминирование на примере цвета глаз у человека.
карие голубые
Р— £ АА х (faa А — карие глаза
G —4 а — голубые глаза
F j — Аа — 100 % карие глаза
Неполное доминирование — когда один ген не полностью, подавляет другой и появляется промежуточный признак. Неполное доминирование рассмотрим на примере формы волос у человека.
курчав. прямые А А — курчав, волосы
р— 9 АА х cf аа Аа — волнистые
G — & аа — прямые
Fi- Аа — 100 % волнистые
52
Кодоминирование — когда в гетерозиготном состоянии каждый аллель детерминирует свой признак. Рассмотрим данное взаимодействие на примере АВ (IV) группы крови человека, где ген 1А определяет антиген А, ген 1в — антиген В.
Сверхдоминирование — когда доминантный ген в гетерозиготном состоянии проявляется сильнее, чем в гомозиготном. Это взаимодействие рассмотрим на примере продолжительности жизни у мушки дрозофилы. АА — нормальная продолжительность жизни; Аа — увеличенная продолжительность жизни; аа — летальный исход.
ВЗАИМОДЕЙСТВИЕ НЕАЛЛЕЛЬНЫХ ГЕНОВ
Во многих случаях на проявление признака могут влиять две (или более) пары неаллельных генов. Это приводит к отклонению от законов Менделя. Основные формы взаимодействия неаллельных генов: 1) комплементарность; 2) эпистаз; 3) полимерия; 4) эффект положейия.
Большинство признаков человека, возникающих при взаимодействии неаллельных генов, недостаточно изучено, поэтому взаимодействие неаллельных генов изучалось на растениях и животных.
Комплементарность — взаимодействие неаллельных генов, при котором один доминантный ген дополняет действие другого доминантного гена и появляется новый признак, отсутствовавший у родителей. Рассмотрим комплемен гарность на двух примерах.
1. Наследование окраски цветков душистого горошка.
При скрещивании двух сортов душистого горошка с белыми лепестками цветов, у одного из которых были доминантные гены АА и рецессивные bb, у другого — рецессивные аа и доминантные — ВВ, а в F] все гибриды оказались с красно-фиолетовыми цветками.
бел. бел.
Р — <j> ААЬЬ х rf ааВВ
G— АЬ (аВ^
А — белый
В — белый
F j — АаВЬ — 100 % красно- фиолетовый А + В —
красно-фиолетовый
При скрещивании Fj между собой:
кр.-ф. кр.-ф.
F1 — <j> АаВЬ х J* АаВЬ
53
G:
@ @
(ав}~ ААВВ ААВЬ АаВВ АаВЬ
©г ААВЬ ААЬЬ АаВЬ Aabb
АаВВ АаВЬ ааВВ ааВЬ
АаВЬ Aabb ааВЬ aabb
Получили расщепление: 9 растений с красно-фиолетовыми цветками и 7 — с белыми (9:7).
2. Синтез интерферона у человека.
Для защиты от вирусов в иммунокомпетентных клетках человека вырабатывается специфический белок интерферон. Его образование в организме связано с комплементарным взаимодействием двух неаллельных генов, локализованных в разных хромосомах.
Э п и с т а з (от греч. epistasis — остановка, препятствие) — взаимодействие неаллельных генов, при котором ген одной аллельной пары подавляет действие гена другой пары. Подавляющий ген называется эписта-тическим (супрессором), а подавляемый — гипостатическим. В случаях, когда ген-подавитель доминантный, имеет место эпистаз.
Примером доминантного эпистаза служит наследование окраски оперения у кур. Куры, имеющие в генотипе доминантный ген окраски, в присутствии эпистатического гена оказываются белыми.
Р — <j) IICC х ccii О*
G— © ©f
F j — liCc — 100 % белые
I — эпистатический ген (ген-подавитель) i — рецессивный ген С — гипостатический ген (ген пигментации) с — рецессивный ген
При скрещивании Fj между собой происходит расщепление. Fi —g ПСс х liCc d*
54
G:
X (ю) © © (к?)
(jcf ИСС ИСс нсс IiCc
©Г НСс Цсс ИСс lice
нсс НСс НСС iiCc
НСс lice iiCc iicc
Получили: 13 белых кур и 3 черных (13:3)
В случаях, когда ген-подавитель рецессивный, имеет место криптомерия (от греч. kryptos — тайный, скрытый).
У человека примером криптомерии может служить «бомбейский феномен». В этом случае редкий рецессивный аллель «х» в гомозиготном состоянии (хх) подавляет активность гена 1в (который детерминирует В(Ш) группу крови системы АВО), поэтому женщина, получившая от матери аллель 1В, фенотипически имели первую группу крови — 0(1).
Полимерия (от греч. polymeria — многосложность) — когда . несколько доминантных генов определяют один и тот же признак примерно в одинаковых количествах. С помощью полимерных генов наследуются количественные признаки. Чем больше число доминантных полимерных генов, тем сильнее выражен признак. В связи с тем, что полимерные гены влияют на проявление одного признака, они обозначаются одной буквой алфавита с индексом. Примером может служить окраска зерен пшеницы:
красная белая
Р— $ А|А|А2А2 х арр^сб
G — ^Ар^^ ^р^У
F j — A pi А2а2 — интенсивность окраски в два раза меньше, чем при генотипе AjAiA2A2
Fj —5 ApiA2a2 х ApiA2a2 с/
У человека полимерными генами определяется пигментация кожи. У коренных жителей Африки (негроидной расы) преобладают доминантные аллели, у представителей европеоидной — рецессивные. Мулаты имеют промежуточную пигментацию. От брака двух мулатов могут родиться как более, так и менее пигментированные дети.
55
Чем большее число неаллельных генов влияет на количественный признак, тем более плавные переходы этих признаков.
Плейотропия (от греч. pieion — более многочисленный и tropos — направление) — множественное действие гена (способность одного гена воздействовать на проявление нескольких признаков). Это обусловлено тем, что генотип представляет собой систему генов, взаимодействующих на уровне продуктов реакций, контролируемых ими. У людей с тонкими длинными «паучьими» пальцами наблюдаются аномалии хрусталика и аневризма аорты (синдром Морфана). Ген, определяющий рыжую окраску волос, одновременно обусловливает более светлую окраску кожи и появление веснушек.
Для большинства генов с той или иной степенью плейотропии характерно сильное влияние на один признак и более слабое — на другие.
В результате анализа различных форм взаимодействия генов становится очевидно, что генотип является сбалансированной системой взаимодействующих генов, на которую существенно влияют факторы внешней среды.
ВЛИЯНИЕ ГЕНОТИПИЧЕСКОЙ СРЕДЫ
И ФАКТОРОВ ВНЕШНЕЙ СРЕДЫ НА ПРОЯВЛЕНИЕ ПРИЗНАКОВ
Признаки проявляются под влиянием генотипической среды (сочетание с другими генами в организме) и условий внешней среды. При наличии гена (или генов), детерминирующего признак, степень выраженности его может иногда изменяться в сторону усиления или ослабления. Это называется экспрессивностью.
Экспрессивность (от лат. expressio — выражение) — степень выраженности признака. Так, совокупность признаков заболевания может проявляться от легких (едва уловимых) до тяжелых: различные формы шизофрении, гипертонии, сахарный диабет и др.
Пенетрантность (от лат. penetrans — проникающий, достигающий) — это частота фенотипического проявления гена среди носителей этого гена (организм может быть гетерозиготным или гомозиготным по доминантным и рецессивным генам). Если среди ста человек-носителей гена признак проявился у всех, то это полная 100 % пенетрантность. Пример полной пенетрантности: шизофрения у гомозигот, гипертрихоз (повышенное оволосение).
Если среди ста человек-носителей гена признак проявляется только у некоторых, то это неполная пенетрантность (частичная); пример: шизофрения гетерозигот — 20 %, сахарный диабет — 20 %, врожденный вывих бедра — 25 %, ретинобластома (злокачественная опухоль сетчатки) — 60 %. Пенетрантность сахарного диабета зависит от условий среды и продолжительности жизни (чем выше продолжительность жизни, тем выше пенетрантность).
Полидактилия у гетерозигот варьирует как в смысле пенетрантности, так в смысле экспрессивности с образованием различной формы конечностей.
56
Пенетрантность может отсутствовать, то есть признак не проявляется: степень выраженности признака может быть различная, количество пальцев на руках и ногах соответственно: 5,5—6,6; 5,6—5,7 и 6,6 -6,6.
Знание пенетрантности имеет .значение в медико-генетическом консультировании для определения возможного генотипа «здоровых» людей, родственники которых имели наследственные заболевания.
По мере развития медицины возможности выявления и раннего лечения наследственных заболеваний увеличиваются. В настоящее время нет фатальной неизбежности некоторых наследственных болезней у человека. Меры, принятые при раннем выявлении наследственных болезней, могут предотвратить их развитие. Например, диетическая коррекция позволяет предупредить развитие тяжелых клинических проявлений при галактоземии, фенилкетонурии и других наследственных болезнях обмена веществ.
Глава VI
ИЗМЕНЧИВОСТЬ
ИЗМЕНЧИВОСТЬ — это общее свойство живых организмов приобретать новые признаки-различия между особями одного вида. Например: у людей различается цвет глаз, волос, форма носа, ушей, темперамент, интеллект, восприимчивость к различным болезням и т. д.
Изменчивость бывает:
1) фенотипическая (модификационная, ненаследственная);
2) генотипическая (наследственная): а) комбинативная; б) мутационная.
МОДИФИКАЦИОННАЯ ИЗМЕНЧИВОСТЬ
Модификационная изменчивость происходит под воздействием факторов внешней среды без изменения структуры генотипа. Если два монозиготных близнеца (с одинаковыми генотипами) будут находиться в разных условиях: один в сельской местности (повышенная физическая нагрузка, продолжительность сна 4—5 ч в сутки, увеличенная продолжительность ультрафиолетового облучения), другой — в городской местности (меньше физическая нагрузка, сон 7—8 ч в сутки, меньшее воздействие ультрафиолетовых лучей), то они будут отличаться друг от друга. У близнеца, проживающего в сельской местности, может быть ниже масса тела, чем у городского, интенсивнее пигментация кожи, более раннее появление морщин.
Но какие бы благоприятные условия ни создавали городскому близнецу, он увеличит массу тела только до определенного уровня, так как существует норма реакции на воздействие факторов внешней среды.
Норма реакции — это норма реагирования на изменяющиеся условия внешней среды (предел модификационной изменчивости), она контролируется генотипом. Норма реакции может быть широкая и узкая.
Широкая норма реакции имеет место в том случае, когда под влиянием факторов внешней среды признак (например, масса тела) изменяется в широких пределах.
Узкая норма реакции имеет место в том случае, когда под влиянием факторов внешней среды признак изменяется в узких пределах (например, цвет волос).
58
Благодаря модификационной изменчивости организмы адаптируются (приспосабливаются) к изменяющимся условиям внешней среды.
Фенокопия имеет место в тех случаях, когда признак под влиянием факторов внешней среды изменяется и копирует признак организма с другим генотипом. Например: у европеоида (с белой кожей) при продолжительном воздействии ультрафиолетовых лучей кожа становится пигментированной и копирует цвет кожи монголоида, у которого другой генотип. У ребенка, мать которого во время беременности перенесла коревую краснуху (вирусное заболевание), может возникнуть помутнение хрусталика, которое будет фенокопией генетически детерминированного помутнения хрусталика (рецессивная мутация).
КОМБИНАТИВНАЯ ИЗМЕНЧИВОСТЬ
Комбинативная изменчивость — это изменчивость, при которой комбинирование генов родителей приводит к появлению новых признаков у детей. Она обеспечивается кроссинговером в профазе мейоза, свободным комбинированием хромосом и генов в метафазе мейоза, случайной встречей гамет с различным набором генов при оплодотворении, а также интенсивной миграцией людей. Чаще стали заключаться браки между супругами, родившимися на значительном расстоянии друг от друга, а чем больше расстояние, тем больше вероятность, что их гаметы будут отличаться по набору генов. Увеличивается также число межнациональных браков, которые ведут к встрече гамет с различающимися генами.
Комбинативная изменчивость дает возможность приспосабливаться к изменяющимся условиям окружающей среды, тем самым способствуя выживанию вида.
Примером комбинативной изменчивости может служить формирование фенотипов групп крови системы АВО: если мать гетерозиготная А(П) группы крови и отец гетерозиготный В(И) группы, то у детей могут быть 0(1) и AB(IV) группы крови, которых не было у родителей.
МУТАЦИОННАЯ ИЗМЕНЧИВОСТЬ
Мутационная изменчивость (мутации) — это изменение генетического материала. Мутации происходят под влиянием факторов внешней или внутренней среды. Процесс образования мутации называется мутагенезом, а факторы, вызывающие мутации, —мутагенными. Мутагены могут быть экзомутагенами (факторы внешней среды) и эндомутагенами (продукты метаболизма в организме). Экзомутагены можно разделить на следующие: 1) физические (ионизирующее излучение, ультрафиолетовые лучи, температура и др.); 2) химические (формалин, горчичный газ, колхицин, многие смолы, соли тяжелых металлов, некоторые лекарственные
59
вещества и др.); 3) биологические (вирусы, токсины бактерий и паразитов животного происхождения).
КЛАССИФИКАЦИЯ МУТАЦИИ
I. По причинам: спонтанные и индуцированные.
1. Спонтанные — это мутации, которые происходят в природе без видимых причин. Например: ген гемофилии может возникнуть спонтанно (когда при анализе родословной не выявлено больных гемофилией).
2. Индуцированные — это мутации, происходящие при направленном воздействии мутагенных факторов.
II. По мутировавшим клеткам: генеративные и соматические.
7. Генеративные — это мутации, возникающие в половых клетках и передающиеся потомкам при половом размножении.
2. Соматические — это мутации, происходящие в соматических клетках и проявляющиеся только у самой особи (например, разный цвет глаз).
III. По изменению генетического материала мутации подразделяют на следующие: генные, хромосомные перестройки, геномные.
7. Генные мутации — это изменения в пределах одного гена: а) вставка или выпадение нуклеотида; б) замена одного нуклеотида на другой.
В результате генных мутаций возникает большинство болезней обмена веществ (фенилкетонурия, галактоземия, алькаптонурия, гистидинимия) и другие наследственные болезни (см. гл. IX). Они чаще выявляются биохимическими методами.
2. Хромосомные перестройки (аберрации) — это мутации, обусловленные изменением структуры хромосом. Они могут быть внутрихромосомными (делеции, дупликации и инверсии) и межхромосомными (транслокации).
Внутрихромосомные перестройки происходят внутри хромосом
Если расположение генов в А В С D Е К тогда:
исходной хромосоме представить 1—-1-1- 1J
а) делеция (нехватка) — выпадение кусочка хромосомы в средней части АВЕК 1—1—1—1 или
концевого участка хромосомы ABCDE I_________________________________I__I . L. J
(примером нехватки концевого участка является синдром кошачьего крика — делеция короткого плеча 5-й хромосомы у человека);
б) дупликация — удвоение участка хромосомы ABCDCDEK ।_________________________________।__।_1_।_।_।_I
в) инверсия — разрыв участка хромосомы, поворот его на 180 градусов и прикрепление к месту отрыва
60
ЛВС’ К El)
i 1 । । i i
Межхромосомные перестройки происходят между негомологичными хромосомами:
транслокации — отрыв участка одной хромосомы и присоединение сю к другой негомологичной
ABCDEKLM
1 I 1_I—I_L—I_1
При инверсии и транслокации затрудняется конъюгация (сближение) гомологичных хромосом. Хромосомные аберрации обнаруживаются цитогенетическими методами. У человека изучена транслоцированная форма болезни Дауна (см. гл. VIII), когда 21-я хромосома присоединяется к 15-й и вместе с ней передается.
3. Геномные мутации — это мутации, обусловленные изменением числа хромосом: полиплоидии и гетероплоидии (анэуплоидии).
Полиплоидия — это кратное гаплоидному увеличение числа хромосом: п — гаплоидное число, 2п — диплоид (норма), Зп — триплоид, 4п — тетраплоид, 5п — нентаплоид и т. д. Полиплоидия у человека представляет собой летальную мутацию. У растений полиплоиды жизнеспособны и обладают повышенной урожайностью (более крупные листья, стебли, корнеплоды, плоды, цветки).
Гетероплоидия — это не кратное гаплоидному увеличение или уменьшение числа хромосом: если одна в паре лишняя — это трисомия, если одной хромосомы из пары не хватает — моносомия, а может быть и две-три лишние или в паре нет двух хромосом — нулесомия (летальная мутация). У человека хорошо изучено несколько заболеваний, причиной которых является изменение числа хромосом (см. гл. VIII).
Трисомия по аутосомам:
по 21-й хромосоме — синдром Дауна;
по 18-й хромосоме — синдром Эдвардса;
по 13-й хромосоме — синдром Патау.
Трисомия по половым хромосомам:
XXY — синдром Клайнфельтера (но может быть XXXY, XXYY и другие изменения числа хромосом).
Моносомия по половым хромосомам:
ХО — синдром Шерешевского—Тернера.
Геномные мутации обнаруживаются цитогенетическими методами. Фенотипически проявляются всегда.
IV. По изменению фенотипа:
1. Аморфные — мутация произошла и признак исчез (отсутствует). Например: альбинизм, безволосость, анофтальмия.
2. Гипоморфные — уменьшение выраженности признака (карликовость, микрофтальмия, микроцефалия).
61
3. Гиперморфные — усиление выраженности признака (гигантизм, полидактилия, гипертрихоз).
4. Неоморфные — в процессе эволюции появляется новый признак, которого ранее не было (гемоглобин, хорда, позвоночник, головной мозг и др)-
5. Антиморфные — вместо одного признака появился другой (в процессе эволюции вместо потовых у млекопитающих появились молочные железы).
Могут также изменяться физиологические признаки (работа систем органов), биохимические (нарушение синтеза ферментов).
V. По исходу для организма:
1. Летальные — смертельные. Например: гетероплоидии по крупным хромосомам (1, 2, 3-й и др.), полиплоидии. Смерть наступает на ранних этапах индивидуального развития, в эмбриональный период.
2. Полулетальные — снижающие жизнеспособность организма. Человек, как правило, не доживает до репродуктивного возраста. Например: синдром Дауна, гемофилия у девочек.
3. Нейтральные — не влияющие на процессы жизнедеятельности, продолжительность жизни. Например: цвет радужной оболочки, пигментация кожи (если ген не обладает плейотропным действием).
4. Положительные — повышающие жизнеспособность. Возникают редко, но имеют большое значение для прогрессивной эволюции.
Мутации в естественных условиях возникают с частотой 10~5—10~7, но при воздействии мутагенных факторов частота их может сильно возрастать.
Несмотря на то что в природе постоянно идет мутационный процесс, существуют механизмы, обеспечивающие устойчивость генетического материала:
а) диплоидный набор хромосом;
б) двойная спираль ДНК;
в) вырожденный генетический код;
г) повторы некоторых генов;
д) репарация молекулы ДНК.
РЕПАРАЦИЯ МОЛЕКУЛЫ ДНК
Репарация (от лат. reparatio — восстановление) — это восстановление поврежденной структуры молекулы ДНК. Она осуществляется специфическими ферментами клетки и имеет несколько разновидностей.
Фоторепарация. Под действием ультрафиолетового облучения между двумя пиримидиновыми основаниями одной нити ДНК (чаще Т—Т) образуются химические связи (возникают димеры), препятствующие считыванию информации (рис. 16). Эти дополнительные связи расщепляет фермент (дезоксипиримидинфотолиаза), активируемый видимым светом. Этот процесс называется фоторепарацией.
Темновая, или эксцизионная (вырезающая) репарация. Она происходит
62
о
Рис. 16. Образование тиминового димера в результате возникновения ковалентных связей между смежными основаниями
СНз
последовательно: а) фермент (эндонуклеаза) «узнает» поврежденный участок нити ДНК; б) фермент (экзонуклеаза) «вырезает» поврежденный участок; в) с помощью фермента (ДНК-полимеразы) синтезируется фрагмент ДНК по принципу комплементарности; г) фермент (лигаза) «сшивает» концы вновь синтезированного участка с основной нитью ДНК.
По времени осуществления репарации различают дорепликативную, пострепликативную и репликативную (рис. 17).
Дорепликативная репарация. Представляет собой восстановление поврежденной нити ДНК до ее удвоения. В простейших случаях разрывы могут быть воссоединены лигазой. В других случаях используется полная ферментативная система репарации (приведена выше).
Пострепликативная репарация. Ее механизм точно не изучен, предполагают различные варианты синтеза ДНК на поврежденной матрице. При пострепликативной репарации происходит лишь вырезание поврежденного участка и сшивание концов, изменяя таким образом ген. При этом клетка может сохранять жизнеспособность и передавать дефектную ДНК дочерним клеткам.
Репликативная репарация. Представляет собой восстановление ДНК в процессе репликации. Этот тип репарации осуществляется удалением поврежденного участка в ходе репликации в зоне роста цепи либо элонгацией цепи в обход повреждения. Как и при пострепликативной репарации, последовательность нуклеотидов в данном участке изменяется.
Существуют мутации, которые нарушают восстановление поврежденных участков молекулы ДНК (нарушают репарацию). Примерами таких мутаций являются пигментная ксеродерма, анемия Фанкони, атаксия-телеангиэктазия. При пигментной ксеродерме в клетках больных отсутствует фермент дезоксипиримидинфотолиаза, необходимый для репарации ДНК, 'поврежденной ультрафиолетовыми лучами. Под действием солнечного света появляются веснушки, расширение капилляров, ороговение эпидермиса, поражение глаз,
63
Путь исправления любого повреждения
qffqqffqqffqqffq
Основание i
повреждено I
qffqqffqqffqqffq
Разрезание;
эндонуклеаза делает
разрез с 5-й стороны от поврежденного основания *
<id<£<d<£<£d<^d<£
-qffqqff qqff qqff q
Эксцизия; экзонуклеаза I удаляет отрезок ДНК ▼
qffqqffqqffqqffq
Полимераза синтезирует новую ДНК ▼
q^qq^qq^qq^q
Лигаза зашивает I
разрез ▼
<6 d<i'6 d<6<6
qffqqffqqffqqffq
Puc. 17. Схема эксцизионной репарации
развитие раковых опухолей кожи, которые приводят к преждевременной смерти.
Если мутация произошла в половой клетке и не была устранена в результате репарации, то она будет передана потомкам.
Мутации, которые определяют появление менее приспособленных особей, но сохраняются в популяции, называются генетическим грузом. Источниками генетического груза служат мутационные и сегрегационные (от позднелат. segregatio — отделение, вы-щепление) процессы. Сегрегационный груз возникает в результате выщепле-ния гетерозиготными родителями менее приспособленных гомозиготных потомков.
Изучение генетического груза человека (наследственные заболевания) важно для решения практических вопросов медицинской генетики. Ряд авторов выделяют общий генетический груз, обусловленный вредными мутациями, присутствующими в геноме человека, и выявляемый генетический груз (ту часть мутаций, которую удается обнаружить).
При отсутствии точных знаний природы большинства генетических систем, лежащих в основе распространенных заболеваний, невозможно предсказать эффект повышения частоты мутаций. Необходимы обстоятельные исследования, чтобы выяснить конкретный вклад генетических факторов в эти заболевания.
К таким факторам, повышающим
частоту мутаций, можно отнести промышленные отходы, выбросы и выхлопы
транспорта, электромагнитные излучения, ионизирующие излучения, лекарственные препараты, токсические вещества, накапливающиеся в пищевых продуктах при их неправильном хранении и обработке, и др. В целях снижения мутагенной нагрузки на человека необходимо внедрять технологии, которые
64
не дают мутагенного загрязнения окружающей среды. Для снижения действия мутагенов рекомендуется диета, богатая естественными антимутагенами: витаминами (Е, С, А, В15, К) и провитаминами. Некоторые пищевые продукты содержат эти вещества в значительном количестве (экстракт капусты, яблок, мятного листа, зеленого перца, ананаса, баклажана и др.). Сбалансированность пищи по незаменимым аминокислотам также снижает спонтанный уровень мутагенеза и придает организму устойчивость к мутагенам.
В связи с возрастающим загрязнением окружающей среды потенциальными мутагенами перед органами здравоохранения поставлена задача предотвращения рождения детей с наследственной патологией. Особое внимание должно уделяться пренатальной диагностике и методам, на которых она основана.
5. Зак. 5285
Гл ав а VII
МЕТОДЫ ИЗУЧЕНИЯ НАСЛЕДСТВЕННОСТИ ЧЕЛОВЕКА
ОСОБЕННОСТИ ИЗУЧЕНИЯ НАСЛЕДСТВЕННОСТИ ЧЕЛОВЕКА
Генетика человека изучает закономерности наследования нормальных и патологических признаков в зависимости от генотипа и факторов внешней среды. Задачей медицинской генетики является выявление, лечение, профилактика наследственных болезней, а также прогнозирование потомства с наследственной патологией.
Генетика человека имеет ряд особенностей: а) на людях запрещены экспериментальные браки; б) рождасгся малое количество потомков; в) наблюдается позднее половое созревание и большая продолжительность смены поколений (25—30 лет); г) у человека сложный кариотип (много хромосом и групп сцеплений); д) невозможность создания одинаковых условий жизни исследуемых.
Несмотря на перечисленные трудности, генетика человека изучена сегодня лучше, чем генетика многих других организмов.
ОСНОВНЫЕ МЕТОДЫ ИЗУЧЕНИЯ НАСЛЕДСТВЕННОСТИ ЧЕЛОВЕКА
Клинико-генеалогический метод. Он был введен в конце XIX века Ф. Гальтоном и основан на составлении и анализе родословных (см. гл. X).
Цитогенетический метод. Он включает: а) методы экспресс-диагностики пола — определение X- и Y-хроматина; б) кариотипирование —определение количества и качества хромосом с целью диагностики хромосомных болезней (геномных мутаций и хромосомных аберраций).
Основными показаниями для цитогенетического исследования являются:
1) пренатальная диагностика пола плода в семьях, отягощенных. заболеваниями, сцепленными с Х-хромосомой;
2) недифференцированная олигофрения (слабоумие);
3) привычные выкидыши и мертворождения;
4) множественные врожденные пороки развития у ребенка;
5) бесплодие у мужчин;
66
6) нарушение менструального цикла (первичная аменорея);
7) пренатальная диагностика при возрасте матери старше 35 лет.
Материалом для цитогенетического исследования могут быть: клетки периферической крови (лимфоциты); фибробласты кожи; клетки, полученные при амниоцентезе или биопсии хориона; клетки абортусов, мертворожденных и др.
Экспресс-диагностика — это исследование полового Х-хроматина (тельца Барра) в ядрах клеток слизистой оболочки ротовой полости, вагинального эпителия или клетках волосяной луковицы. Для выявления Х-полового хроматина мазки окрашивают ацетарсеином и препараты просматривают с помощью обычного светового микроскопа. В норме у женщин обнаруживается одна глыбка Х-хроматина, а у мужчин ее нет.
Для выявления мужского Y-полового хроматина (F-тельце) мазки окрашивают акрихином и просматривают с помощью люминесцентного микроскопа. Y-хроматин это сильно светящаяся точка, по величине и интенсивности свечения отличающаяся от остальных хромоцентров. Он обнаруживается в ядрах клеток мужского организма. Число Y-телец соответствует числу Y-хромосом в кариотипе.
Окончательный диагноз хромосомной болезни выставляется только после исследования кариотипа (см. гл. III).
Генетика соматических клеток. Она изучает наследственность и изменчивость соматических клеток. Благодаря тому, что эти клетки содержат весь объем генетической информации, на них можно также изучать генетические особенности целостного организма. Генетика соматических клеток позволила включить человека в группу экспериментальных объектов.
Соматические клетки человека для генетических исследований получают из материала биопсий (прижизненное иссечение тканей или органов) и аутопсий (кусочки тканей или органов от трупов). Чаще всего используют клеточные культуры фибробластов и лимфоидных клеток.
В настоящее время применяют следующие методы генетики соматических клеток человека:
1) простое культивирование;
2) гибридизация;
3) клонирование;
z) селекция.
Простое культивирование — размножение клеток на питательных средах с целью получения их в достаточном количестве для цитогенетического, биохимического, иммунологического и других методов исследования.
Гибридизация соматических клеток — это слияние клеток двух разных типов. Гибридизацию могут проводить между клетками, полученными от разных людей, а также клетками человека с клетками мыши, крысы, китайского хомячка, морской свинки, обезьяны,
67
курицы. Спонтанное (произвольное) слияние происходит редко, поэтому в смешанную культуру добавляют полиэтиленгликоль или чаще вирус Сендай. При слиянии клеток образуется гетерокарион (гибридная клетка с двумя ядрами разных типов клеток). Затем ядра этой клетки могут слиться с образованием синкариона (от греч. syn — вместе).
Особый интерес представляют гибридные клетки «человек—мышь», так как при последующих делениях они имеют тенденцию к утрате многих хромосом человека. Примерно через 30 поколений можно найти клетки, содержащие только одну—две пары человеческих хромосом. Если в гибридной клетке отсутствует какая-либо хромосома и не происходит синтез каких-то белков, то можно предположить, что гены, детерминирующие синтез этих белков, локализованы в данной хромосоме. Этот метод позволяет установить группы сцепления, а используя хромосомные перестройки (нехватки и транслокации), выяснять последовательность расположения генов и строить генетические карты хромосом человека.
Клонирование — получение потомков одной клетки (клона), взятой из общей клеточной массы. Все клетки будут с одинаковым генотипом. Одним из примеров метода клонирования является получение гибридом (аг лат. hibrida — помесь и греч. ота — опухоль). Гибридома — это клеточный гибрид, получаемый слиянием нормального лимфоцита и опухолевой клетки.
Селекция (от лат. selectio — отбор, выбор) — отбор клеток с заранее заданными свойствами при культивировании их на селективных питательных средах. Например, если использовать питательную среду без лактозы (но с добавлением других сахаров), то из большого числа клеток, помещенных в нее, может оказаться несколько, способных существовать без лактозы. В дальнейшем можно будет получить клон этих клеток.
Метод дерматоглифики. Представляет собой изучение папиллярных узоров пальцев, ладоней и стоп. На этих участках кожи имеются крупные дермальные сосочки, а покрывающий их эпидермис образует гребни и борозды. Дерматоглифические узоры обладают высокой степенью индивидуальности и остаются неизменными в течение всей жизни. Поэтому дерматоглиф ический анализ используется для определения зиготности близнецов, диагностику некоторых геномных и хромосомных мутаций (например, болезни Дауна, Патау и других); для идентификации личности в криминалистике, установления отцовства в судебной медицине.
Близнецовый метод. Он позволяет оценить относительную роль (удельный вес) генетических и средовых факторов в развитии конкретного признака или заболевания. Близнецы бывают монозиготные (однояйцевые) и дизигот-ные (разнояйцевые).
Монозиготные близнецы развиваются из одной оплодотворенной яйцеклетки (зиготы) в результате ее разделения надвое с образованием двух эмбрионов. Монозиготные близнецы имеют одинаковые генотипы, различие их признаков зависит только от факторов внешней среды.
68
Дизиготные близнецы рождаются, когда образуется одновременно две яйцеклетки, оплодотворяемые двумя сперматозоидами. Дизиготные близнецы имеют различные генотипы. Они сходны между собой не более, чем братья и сестры, рожденные порознь. Но благодаря одновременному рождению и совместному воспитанию у них будут общие средовые факторы. Различие их признаков в основном обусловлено различными генотипами.
Для доказательства роли наследственности в развитии признака необходимо сравнить долю (%) конкордантных пар (одинаковых по конкретному признаку) в группах моно- и дизиготных близнецов. Рассмотрим это на примере сахарного диабета. Если один из монозиготных близнецов болен сахарным диабетом, то второй заболевает в 65 % случаях (в 65 % — они кон-кордантны). Если один из дизиготных близнецов заболел, то второй заболевает в 18 % случаев. Большая конкордантность в группе монозиготных пар доказывает, что в этиологии диабета наследственное предрасположение играет существенную роль.
Биохимические методы. Они позволяют выявить изменения в обмене веществ для уточнения диагноза заболевания, установления гетерозиготного носительства. Например, гетерозиготные носители рецессивного аллеля фенилкетонурии реагируют на введение фенилаланина более сильным повышением концентрации аминокислоты в плазме, чем нормальные гомозиготы. Этот метод используют в медико-генетическом консультировании для определения вероятности рождения ребенка с наследственным заболеванием.
Заболевания, в основе которых лежит нарушение обмена веществ, составляют значительную часть наследственной патологии (фенилкетонурия, галактоземия, алкаптонурия и др.).
Предположить наличие у больного наследственного дефекта обмена можно по следующим признакам:
1) умственная отсталость, изолированная или в сочетании с патологией других органов;
2) нарушение психического статуса;
3) нарушение физического развития;
4) судороги, мышечная гипо- или гипертония, нарушение походки и координации движений, желтуха, гипо- или гиперпигментация;
5) непереносимость отдельных пищевых продуктов и лекарственных препаратов, нарушение пищеварения и др.
Иммуногенетический метод. Позволяет поставить или уточнить диагноз: 1) при врожденных иммунодефицитных состояниях; 2) при подозрении на антигенную несовместимость матери и плода по тем или иным системам групп крови.
Популяционно-статистический метод. Он дает возможность рассчитать в популяции частоту нормальных и патологических генов и генотипов: гетерозигот, гомозигот доминантных и рецессивных, а также частоту нормальных и патологических фенотипов.
69
Частота генотипов и фенотипов рассчитывается по формуле Харди— Вайнберга: р2 + 2pq + q2 = (р + q)2 = 1 , где р — частота доминантного гена; q — частота рецессивного гена; q2 — частота гомозигот по рецессивному гену.
Очень важно, что по этой формуле можно рассчитать частоту гетерозигот по патологическим рецессивным генам, которые находятся в скрытом состоянии. Например, частота фенилкетонурии в популяции — 1:10000, значит, q2 = 0,0001, q = 0,01; р 4- q = 1, тогда р = 1 - q = 1 - 0,01 = 0,99, a 2pq= = 2 х 0,99 х 0,01 = 0,0198 ~ 0,02, или 2 %. Значит, частота гетерозигот по гену фенилкетонурии в изучаемой популяции составляет 2 %.
Следует помнить, что наследственные заболевания распределены по различным регионам земного шара, среди разных рас и народностей неравномерно. Например, частота фенилкетонурии в Республике Беларусь равна 1:6500, в Австрии — 1:12000, в Финляндии — 1:43000, а у евреев-ашкенази — 1:180000. Знания частоты заболеваний в данном регионе способствуют правильной организации профилактических мероприятий.
Методы пренатальной диагностики. Представляет собой совокупность исследований, позволяющих обнаружить заболевание до рождения ребенка. К основным методам пренатальной диагностики относятся: ультразвуковое исследование, амниоцентез, биопсия хориона, определение ос-фетопротеина и др. (см. гл. X).
Метод моделирования. Изучает болезни человека на животных, которые могут болеть этими заболеваниями. В основе лежит закон Вавилова о гомологичных рядах наследственной изменчивости. Например, гемофилию, сцепленную с полом, можно изучать на собаках; эпилепсию — на кроликах; сахарный диабет, мышечную дистрофию, ахандроплазию — на крысах; неза-ращение губы и неба — на мышах.
Глава УШ
НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ И ИХ КЛАССИФИКАЦИЯ
Все наследственные болезни принято разделять на три большие группы: моногенные, болезни с наследственным предрасположением (мультифакто-риальные) и хромосомные.
Причиной развития моногенных болезней является поражение генетического материала на уровне молекулы ДНК, в результате чего повреждается только один ген. К моногенным болезням относится большинство наследственных болезней обмена (таких, как фенилкетонурия, галактоземия, муковисцидоз, адреногенитальный синдром, гликогенозы, мукополисахаридозы и многие другие). Моногенные болезни наследуются в соответствии с законами Менделя и по типу наследования могут быть разделены на аутосомно-доминантные, аутосомно-рецессивные и сцепленные с Х-хро-мосомой (табл. 2).
Болезни с наследственным предрасположением (мультифакториальные) являются полигенными, и для их проявления требуется влияние определенных факторов внешней среды. Общими признаками мультифакториальных заболеваний являются: 1) высокая частота среди населения; 2) выраженный клинический полиморфизм; 3) сходство клинических проявлений у пробанда и ближайших родственников; 4) возрастные отличия; 5) половые отличия; 6) различная терапевтическая эффективность; 7) несоответствие закономерностей наследования простым менделевским моделям.
Хромосомные болезни могут быть обусловлены количественными аномалиями хромосом (геномные мутации), а также структурными аномалиями хромосом (хромосомные аберрации).
Наследственные болезни нельзя отождествлять с врожденными заболеваниями, с которыми ребенок рождается и которые могут проявляться сразу же после рождения или в более поздний период жизни. Врожденная патология может возникнуть в критические периоды эмбриогенеза под воздействием внешнесредовых тератогенных факторов (физических, химических, биологических и др.) и не передается по наследству. Например, врожденные пороки сердца обусловлены тем, что в период его закладки мать в первом
71
Табл. 2. Классификация наследственных заболеваний
Хромосомные Монотонные Мультифакториальныс (полигонные)
А. Аномалии количества.Половых хромосом:синдром Шсрсшсвского—Тернера, Клайнфсльтсра, синдром трисомии X и др. Аутосом: Болезнь Дауна, синдром Эдвардса, Патау и др. А. Аутосомно-доминантные: синдром Марфана, ахондроплазия, анемия Минковского—Шоф-фара, полидактилия и др. Б. Аутосомно-рсцсссивныс: фенилкетонурия, галактоземия, целиакия, муковисцидоз, адрено-генитальный синдром и др. А. ЦНС:эпилспсия, шизофрения и др. Б. Сердечно-сосудистые: ревматизм, атеросклероз и ДР
Б. Структурные аномалии хромосом:синдром «кошачьего крика» и др. В. Х-сцсплснныс рецессивные: гемофилия, миопатия Дюшенна, ихтиоз и др. В. Кожные: атопической дерматит, псориаз и др.
Г. Х-сцсплснныс доминантные: витамин Д-рсзистснтный рахит, коричневая окраска эмали зубов и др. Г. Дыхательной системы: бронхиальная астма, аллергический альвеолит и др.
Д. Выделительной системы: нефриты, мочекаменная болезнь, энурез и др.
Е. Пищеварительной системы: язвенная болезнь, цирроз печени, нсспсцифичсский язвенный колит и др.
триместре беременности переносит какое-то заболевание (чаще всего вирусную инфекцию, тропную к тканям закладывающегося сердца). Однако не у всех беременных женщин, страдавших такой же вирусной инфекцией в ранние сроки беременности, рождаются дети с врожденными пороками. Это можно наблюдать и в отношении почек, пищеварительного тракта, легких, мозга, желез внутренней секреции. Вредными факторами для беременной женщины являются вирусная и бактериальная инфекции, неблагоприятные производственные и бытовые условия, употребление лекарственных препаратов, алкоголя и многие другие.
Медикам хорошо известен факт появления уродств у детей после употребления беременными женщинами лекарственного препарата талидомида. Этот препарат выпущен в 1958 г. в ФРГ в качестве успокаивающего, про-тиворвотного и снотворного средства. Было обнаружено, что применение его в ранние сроки беременности приводит к разнообразным аномалиям
72
развития: отсутствию конечностей (или конечности в виде ластов), аномалиям ушных раковин и глаз, порокам сердца, почек, пищеварительного тракта и другим уродствам. Применение талидомида после закладки органов не приводило к появлению уродств.
В настоящее время известен алкогольный синдром у плода, когда беременные женщины употребляли алкогольные напитки.
Разграничение врожденных и наследственных заболеваний имеет важное значение для прогнозирования потомства в данной семье.
Наследственные заболевания не могут быть названы «семейными», так как последние могут появляться под влиянием различных факторов среды обитания, характера питания, условий производства и др.
Таким образом, при изучении наследственных заболеваний следует различать хромосомные, моногенные болезни и мультифакторильные, или болезни с наследственнкм предрасположением, а также патологические реакции на действие вредных факторов внешней среды.
В общем виде классификация наследственных заболеваний представлена в табл. 2.
ХРОМОСОМНЫЕ БОЛЕЗНИ
Хромосомный набор у человека принято разделять на семь групп: А (1, 2, 3 пары хромосом), В (4, 5 пар), С (6, 7, 8, 9, 10, 11, 12 пар, а хромосома X по размеру сходна с хромосомами 6 и 7), D (13, 14, 15), Е (16, 17, 18), F (19, 20), G (21, 22 пары хромосом и Y-хромосома).
Хромосомные болезни — частая форма патологии. Было показано, что примерно у 40 % спонтанных абортов имеются хромосомные изменения, а 6 % всех мертворожденных также имеют нарушения в количестве и структуре хромосом. В то же время около 6 из 1000 новорожденных имеют хромосомные нарушения, а удельный вес хромосомных болезней в группе детей с врожденными аномалиями составляет около 50 %. Среди недоношенных детей хромосомные нарушения встречаются в четыре раза чаще, чем среди доношенных.
Клинически почти все хромосомные болезни проявляются: а) нарушением интеллектуального развития; 2) множественными врожденными пороками.
Эти нарушения обычно являются тяжелыми и касаются физического, психического, полового развития. Это может быть умственное и физическое недоразвитие, пороки развития скелета, деформация черепа, микроцефалия, эпикант, характерные черты лица (например, при болезни Дауна лицо однотипное у различных человеческих рас), косолапость, крипторхизм, аномалии грудной клетки, наличие пороков сердца и т. д. В настоящее время внутриутробная диагностика хромосомных заболеваний возможна при исследовании околоплодных вод. Это позволяет предупредить рождение детей с хромосомными заболеваниями.
73
Какие именно причины приводят к хромосомным мутациям — каждый раз установить практически невозможно. Это могут быть физические, химические, биологические факторы. Кроме того, мутации в яйцеклетках у женщины могут иметь место даже во внутриутробном периоде развития. Однако риск развития хромосомных заболеваний больше среди пожилых женщин. Так, риск иметь ребенка с трисомией 13-й, 18-й или 21-й хромосомы для женщин 45 лет и старше в 60 раз превышает эту патологию у женщин 19—24 лет. Очень молодые женщины также имеют повышенный риск рождения ребенка с хромосомным заболеванием.
Числовые аномалии хромосом происходят в мейозе или митозе во время первых делений зиготы. В этих случаях может наблюдаться мозаицизм, так как имеется клон с нормальным и измененным кариотипом. Клиническая картина у таких больных может быть менее выраженной.
Рассмотрим наиболее часто встречающиеся хромосомные синдромы.
СИНДРОМЫ С ЧИСЛОВЫМИ АНОМАЛИЯМИ ПОЛОВЫХ ХРОМОСОМ
Синдром Шерешевского—Тернера. Основой развития этого заболевания
Рис. 18. Внешний вид больной с синдромом Шершевского—Тернера (выраженный шейный птеригиум; широкая грудная клетка; соски молочных желез гипопластичны, расположены кнаружи от средней ключичной линии)
служит нарушение расхождения половых хромосом. Типичные клинические проявления связывают с кариотипом 45X0. При этом синдроме возможны различные варианты мозаицизма (45X0/46XY; 45Х0/47ХХХ и др.). Частота рождения больных составляет 1 из 3000. Масса тела детей при рождении снижена, отмечается лимфатический отек стоп, голеней, кистей рук, низкий рост волос на шее. Отек стоп и голеней может держаться до двух—трех лет. Характерной также является крыловидная складка на боковой поверхности шеи — птеригиум шейный (рис. 18). В течение первого года жизни ребенок плохо набирает длину тела. Отставание в росте особенно заметно в возрасте 9—10 лет, а взрослые женщины имеют длину тела от 114 до 145 см. Аномалии развития скелета становятся более заметными с возрастом ребенка
74
(короткая шея, широкая и короткая грудная клетка, грудина часто деформирована, избыточное разгибание локтевых и коленных суставов, укорочение 4—5-го пальцев на руках, широко расставленные соски). На коже имеется множество пигментных пятен, витилиго. Выявляется своеобразная структура лица: микрогнатия, птоз, эпикант, низкорасположенные и деформированные ушные раковины, высокое твердое небо. У таких больных можно выявить косоглазие, катаракту, миопию, нарушение цветовых восприятий. Отмечается склонность к заболеваниям носоглотки, отиту.
В период полового созревания не появляются вторичные половые признаки, наружные половые органы инфантильные, недоразвитые влагалище и матка. Менструаций нет, больные бесплодны. Анатомически на месте яичников обнаруживают соединительнотканные тяжи. Лабораторные исследования показывают, что выделение эстрогенов снижено в 10 и более раз, а продукция гонадотропинов резко повышена (в 10—15 раз). В буккальном эпителии не находят полового хроматина. Психическое развитие страдает в меньшей степени. Средняя продолжительность жизни таких больных близка к норме.
Лечебные мероприятия проводят обычно эндокринологи с целью стимуляции роста до периода полового созревания и уменьшения полового инфантилизма (длительные курсы введения половых гормонов). Появление менструаций, вторичных половых признаков оказывает большое положительное психологическое воздействие, хотя такие женщины остаются бесплодными.
Синдром Клайнфельтера. Синдром описан Клайнфельтером в 1942 г., а в 1959 г. была установлена его хромосомная природа (47, XXY). В буккальном соскобе обнаруживаются тельца Барра (Х-половой хроматин). Частота встречаемости составляет 2 из 1000 новорожденных мальчиков. Однако клинически аномалия проявляется после пубертатного периода. Обязательный признак заболевания — гипоплазия гонад. Для мужчин с синдромом Клайнфельтера характерен высокий рост, пропорции тела —евнухоидные. Скелет развит по женскому типу (широкий таз, узкие плечи), отмечается гинекомастия и ожирение, слабый рост волос на лице (ичи он отсутствует); на лобке отмечается рост волос по женскому типу. Половой член уменьшен или нормальных размеров, нарушен сперматогенез и в результате этого мужчины бесплодны (рис. 19). Умственное развитие отстает, и до наступления периода полового развития этот признак является ведущим. Однако встречаются лица и с нормальным интеллектом. Помимо хромосомного набора 47, XXY могут быть лица с синдромом Клайнфельтера и с набором хромосом 48, XXXY, 49, XXXXY и др. Замечено: чем больше число Х-хромосом, тем больше умственная отсталость, вплоть до итиотии. Изменение кариотипа может быть и в виде 47, XYY, 48, XXYY. У этих лиц помимо клинических черт синдрома Клайнфельтера в поведении могут наблюдаться агрессивность, немотивированные поступки.
Синдром трисомии X. Впервые синдром трисомии по Х-хромосоме был
75
Рис. 19. Внешний вид брльного с синдромом Клайнфельтера (высокий рост, непропорционально длинные конечности)
описан в 1959 г., когда в ядрах эпителия слизистой оболочки щеки больной было обнаружено два тельца полового хроматина.
Клинически картина этого заболевания чрезвычайно разнообразна. Такие женщины, как правило, имеют недоразвитые яичники, гипоплазию матки, нерегулярный менструальный цикл, бесплодие, у них рано наступает вторичная аменорея. Однако около 30 % таких больных сохраняют генеративную функцию и могут иметь детей.
У женщин с трисомией X довольно часто имеется незначительное снижение интеллекта. Отмечена повышенная вероятность развития у таких больных психозов: наиболее часто обнаруживается шизофрения с неблагоприятным типом течения.
Примерно у одной трети женщин с кариотипом 47, XXX описаны умеренно выраженные соматические аномалии, высокий рост, кифосколиоз, укорочение и искривление мизинцев на руках, черепно-лицевые. диспропорции.
При тетра- и пентасомии-Х характерны тяжелые нарушения интеллекта, выраженные соматические аномалии, недоразвитие гениталий.
Предварительный диагноз синдрома трисомии X основан на исследовании полового хроматина, а окончательный устанавливается только после определения кариотипа. Лечение в основном симптоматическое и в первую очередь направлено на устранение нарушений функции яичников.
СИНДРОМЫ С ЧИСЛОВЫМИ АНОМАЛИЯМИ АУТОСОМ
Синдром Дауна. Данная аномалия является самой частой формой хромосомной патологии человека и проявляется трисомией по 21-й паре хромосом. Заболевание встречается с частотой 1 из 700—800 новорожденных. Простая трисомия составляет около 95 % от общего числа больных с синдромом Дауна, а 4 % приходится на транслокационный вариант и 1 % на мозаицизм.
В основе болезни Дауна лежит нерасхождение по 21-й паре хромосом
76
либо в яйцеклетке во время мейоза, либо на ранних стадиях дробления зиготы. Кариотип больного при трисомии содержит 47 хромосом, при этом лишней является 21-я хромосома. При транслокационном варианте в кариотипе содержится 46 хромосом, а лишняя 21-я хромосома оказывается транспонированной чаще всего на хромосому группы D или G. Иногда подобная транслокация в сбалансированном состоянии обнаруживается у одного из родителей (чаще всего у матери). Для такой семьи имеется повышенный риск повторного рождения ребенка с болезнью Дауна, так как в мейозе у таких родителей наряду с нормальными гаметами будут возникать гаметы с несбалансированным кариотипом.
Как правило, клиническая картина трисомного и транслокационного вариантов неразличима. При мозаичном варианте (норма—трисомия) выраженность клинических симптомов болезни Дауна зависит от соотношения нормального и патологического клонов: чем меньше процент нормальных клеток с 46 хромосомами, тем более выражена клиническая картина.
Рис. 20. Внешний вид больных с синдромом трисомии 21
77
Рис. 21. Внешний вид больного с синдромом трисомии 13: а — аномалии лица; б — двусторонняя полисиндактилия стоп
Мальчики и девочки с болезнью Дауна рождаются с одинаковой частотой. Масса и длина тела при рождении обычно соответствуют доношенному ребенку. Голова меньших размеров со скошенным затылком. Лицо плоское, с косым монголоидным разрезом глаз, широкой переносицей, маленьким носом, большим языком, часто не вмещающимся во рту (рис. 20). У больных рот полуоткрытый, на губах часто трещины, могут быть аномалии зубов, ушных раковин. Суставы имеют большую подвижность, пальцы короткие, на ладони пролегает глубокая борозда («обезьянья борозда»). Мышцы гипотоничные, живот увеличен, часто имеются врожденные пороки сердца. Грудная клетка деформирована. Умственное развитие больных отстает, возможно развитие тяжелой идиотии. Синдром Дауна сопровождается расстройствами эндокринных желез и нарушением обмена веществ.
Продолжительность жизни больных с синдромом Дауна ограничена. Однако при нормализации эндокринных функций и коррекции пороков развития продолжительность жизни может быть удлинена.
Синдром Патау. Этот синдром был описан К. Патау с соавт. (1960) как синдром множественных врожденных пороков развития, сопровождающийся трисомией по 13-й хромосоме (рис. 21). При рождении эти дети имеют малую массу тела, хотя рождаются
78
в срок, у беременных ими женщин отмечается многоводие. Характерен внешний вид больного: окружность черепа уменьшена, низкий лоб, узкие глазные щели, запавшая переносица, типична расщелина губы и неба. Характерна микрофтальмия, помутнение роговицы. Из аномалий костно-мышечной системы наиболее постоянны полидактилия и флексорное положение кистей. Интеллект нарушен, 95 % таких больных умирают в возрасте до года, этому способствуют врожденные пороки сердца (дефекты межпредсердной и межжелудочковой перегородки), органов пищеварения, поликистоз почек. При синдроме Патау всегда поражены гениталии: у мальчиков обычно отмечается крипторхизм, а у девочек дупликация матки и влагалища.
Синдром «кошачьего крика». Этот синдром связан с делецией короткого плеча 5-й хромосомы (описан в 1963 г.). Плач новорожденных похож на крик кошки, что связано с аномалиями развития гортани и голосовых связок. Дети плохо растут, отстают в психическом развитии. Внешний вид больных имеет особенности: микроцефалия, лицо круглое с гипертелоризмом, микрогнатия, эпикант, уши неправильные и низко расположенные, короткая шея. Врожденные пороки внутренних органов встречаются сравнительно редко, наиболее часто порочно развитым оказывается сердце. Большинство детей умирает в раннем возрасте, однако описаны больные старших возрастов, в частности 55-летняя женщина.
Таким образом, изложенная клиническая картина заболеваний при различных хромосомных нарушениях сопровождается в первую очередь отставанием в умственном развитии и множеством пороков развития. Предположительная диагностика возможна на основании клинической картины, а окончательный диагноз устанавливается только после исследования хромосомного набора. Всем этим больным необходима консультация врача-генетика.
Глава IX
ГЕННЫЕ БОЛЕЗНИ
Генные болезни встречаются чаще, чем хромосомные. Диагностика этих заболеваний обычно начинается с анализа клинических и биохимических данных, родословной пробанда, типа наследования. Моногенные болезни могут иметь аутосомно-доминантный, аутосомно-рецессивный и Х-сцеплен-ный типы наследования.
При аутосомн о-д оминантном типе наследования патологический признак встречается в каждом поколении родословной и распределение между больными и здоровыми часто составляет 50:50, но вероятность может быть 100, 75 %. Однако пенетрантность патологических проявлений почти всегда ниже 100 %. Клинические проявления заболеваний и их выраженность могут быть различными не только между семьями, но и внутри одной семьи. Кроме того, клинические признаки могут появиться не сразу после рождения, а спустя много лет.
При аутосомн о-p ецессивном типе наследования правильному анализу способствуют указания на родственный брак (двоюродный брат и сестра), данные биохимических исследований дефектов обмена веществ (выявление энзимопатий). Научной группой ВОЗ разработана и рекомендована к практическому применению следующая классификация наследственных заболеваний обмена веществ:
1) наследственные нарушения обмена аминокислот (фенилкетонурия и др);
2) наследственные нарушения обмена углеводов (гликогеновая болезнь, галактоземия и др.);
3) наследственные нарушения обмена липидов (болезни Ниманна—Пика, болезнь Гоше и др.);
4) наследственные нарушения обмена стероидов (адреногенитальный синдром и др.);
5) наследственные нарушения обмена пуринов и пиримидинов (синдром Леша—Найяна и др.);
6) наследственные нарушения обмена соединительной ткани (мукополисахаридозы, синдром Марфана и др.);
80
7) наследственные нарушения гема- и порфирина (гемоглобинопатии и др);
8) наследственные нарушения обмена в эритроцитах (анемия Минковского—Шоффара и др.);
9) наследственные аномалии обмена металлов (болезнь Коновалова—Вильсона и др.);
10) наследственные нарушения обмена билирубина (синдром Криглера— Найяра и др.);
11) наследственные нарушения всасывания в пищеварительном тракте (муковисцидоз, целиакия, непереносимость лактозы и др.).
Рассмотрим наиболее часто встречающиеся ферментопатии, возникающие в результате генных мутаций.
ЗАБОЛЕВАНИЯ, СВЯЗАННЫЕ С РАССТРОЙСТВОМ АМИНОКИСЛОТНОГО ОБМЕНА
Фенилкетонурия (ФКУ, фенилпировиноградная олигофрения, болезнь Феллинга). Это заболевание обусловлено биохимическим дефектом превращения аминокислоты фенилаланина. Тип наследования аутосомно-рецессивный. Больные являются гомозиготами по гену фенилкетонурии, а родители гетерозиготами. Биохимический дефект состоит в ферментном (энзимном) блоке нормального превращения фенилаланина в аминокислоту тирозин из-за недостаточности фермента фенилаланингидроксилазы. Количество фенилаланина в организме накапливается и концентрация его в крови увеличивается в 10—100 раз. Далее он превращается в фенилпировиноградную кислоту, являющуюся нейротропным ядом.
Накопление фенилаланина в организме идет постепенно и клиническая картина развивается медленно. В первом полугодии жизни у ребенка бывают срыгивания, могут развиваться дерматиты и судорожные припадки. Судо-р /жный синдром развивается по типу малой эпилепсии. В последующем соматическое развитие ребенка мало страдает, но психическое развитие, моторика все больше отстают или деградируют. Только 0,5 % больных сохраняют нормальный интеллект. В характере выявляется импульсивность, резкая возбудимость, склонность к агрессии.
Почти все дети блондины с голубыми глазами. С мочой и потом выделяются продукты обмена фенилаланина (фенилуксусная кислота) и от ребенка исходит неприятный запах («мышиный», «волчий», «затхлый»). Частота этого заболевания составляет 1 на 5600 новорожденных.
Исключение из питания фенилаланина с первых месяцев жизни способствует нормальному развитию ребенка. В настоящее время все новорожденные обследуются на уровень фенилаланина в крови: для этого несколько капель крови на фильтровальной бумаге посылают в лабораторию, где с помощью хроматографического метода определяют содержание данной аминокислоты.
6. Зак. 5285
81
Реже используется проба Феллинга: к 2 -5 мл свежей мочи ребенка добавляют 10 капель 10 % раствора треххлористого железа. Появление сине-зеленого окрашивания свидетельствует о наличии заболевания и ребенок должен быть обследован количественными методами для установления окончательного диагноза.
ЗАБОЛЕВАНИЯ, СВЯЗАННЫЕ С НАРУШЕНИЕМ ОБМЕНА УГЛЕВОДОВ
Одной из, основных частей пищи человека являются углеводы, которые выполняю! энергетическую и структурную функции и являются составными частями некоторых биологически активных веществ (ферментов, гормонов, иммунных тел), мукополисахаридов. Основным углеводом пищи для детей грудного возраста является лактоза, а в последующем — крахмал.
Представителем в нарушении межуточного обмена углеводов является гликогеновая болезнь.
Гликогеновая болезнь. Характерной особенностью этого заболевания является нарушение энзимного звена в сложной цепи процессов, связанных с синтезом и разложением гликогена (животного крахмала). В норме образующийся из глюкозы гликоген при голодании должен снова превратиться в глюкозу и только в этом виде организм человека может использовать углеводы для других целей. Различают несколько типов гликогенозов.
Первый тип гликогеноза — гепаторенальный, или болезнь Г и р к е. При этом заболевании накопившийся гликоген в печени и почках не может обратно превратиться в глюкозу и в организме развивается гипогликемия, так как в печени отсутствует фермент глюкозо-6-фосфатаза, которая играет важную роль в регуляции уровня глюкозы в крови. Наследуется заболевание по аутосомно-рецессивному типу.
В различные возрастные периоды клиническая картина болезни Гирке неодинакова.
В период новорожденности основными симптомами являются гипогликемические судороги и гепатомегалия. Задержка роста начинает отмечаться с 1-го года жизни. Характерен внешний вид больных: большая голова, «кукольное» лцдо, короткая шея, выступающий живот (рис. 22). Уровень глюкозы в крови почти всегда ниже нормы, а липидов и молочной кислоты увеличен. На почве гипогликемии отмечаются обморочные состояния или судороги. Умственное развитие почти не изменяется, половое развитие задерживается. В крови повышается уровень мочевой кислоты и после пубертатного периода нередко наблюдаются признаки подагры.
При нагрузке глюкозой отмечается либо нормальная гликемическая кривая, либо диабетическая, когда уровень сахара в крови к 90—120 мин не приходит к исходному уровню и имеется высокий подъем. При введении адреналина уровень глюкозы в крови увеличивается незначительно. Диагноз подтверждают после исследования биопсийного материала печени.
82
Рис. 22. Внешний вид ребенка, страдающего гликогенезом первого типа (большая голова, короткая шея, “кукольное лицо”, выступающий живот)
В лечебном плане большое значение уделяют диетотерапии. Частоту прие-юв пищи необходимо увеличить для предупреждения выраженной гипогли-:емии. В этих же целях повышают количество углеводов в диете. Жир граничивают, белки назначают в соответствии с возрастной нормой. Лечение люкагоном несколько улучшает прогноз заболевания.
Второй тип гликогеноза, или болезнь Помпе, самый небла-оприятный. Его называют генерализованным, так как накопление гликогена [роисходит не только в печени, но и в скелетных мышцах, миокарде, легких, елезенке, надпочечниках, стенках сосудов, нейронах.
У новорожденных детей размеры сердца нормальные. Спустя 1—2 месяца ребенка появляется мышечная слабость, теряются уже имеющиеся двига-ельные навыки. При кормлении и плаче отмечается цианоз. В это время ыявляются кардиомегалия и макроглоссия. Сухожильные рефлексы исчезают, о плотность мышц остается нормальной. Накопившийся секрет в дыхатель-ых путях приводит к затяжному течению пневмонии, а затем и к гибели ольного.
Диагностика заболевания возможна еще до рождения ребенка. Для этого пределяют активно лъ специфических ферментов в амниотической жидкости ее клетках (определение кислой а-1,4-глюкозидазы).
Третий, четвертый, пятый, шестой и другие типы гликогеноза напо-мнают клиническую картину первого типа и без биохимических исследо-аний дифференциальная диагностика затруднительна.
Галактоземия. Эго заболевание характеризуется накоплением в крови алактозы и проявляется отставанием в физическом и умственном развитии, ижелым поражением печени, нервной системы, глаз и других органов. Набедуется галактоземия по аутосомно-рецессивному типу, частота патологии оставляет 1 из 16000.
Галактоза является составной частью молочного сахара лактозы, при гид-
83
релизе которой в пищеварительном тракте образуются глюкоза и галактоза. Галактоза тормозит всасывание глюкозы и этим создает углеводную среду в кишечнике. Она необходима для миелинизации нервных волокон. Однако избыточные ее количества для организма нецелесообразны, и поэтому она превращается в глюкозу с помощью фермента галактоза- 1-фосфат-уридил-трансферазы. При низкой активности этого фермента происходит накопление галактоза-1-фосфата, который оказывает токсическое действие на функцию печени, мозга, хрусталик глаза.
Начало заболевания может проявляться с первых дней жизни расстройствами пищеварения, интоксикацией (понос, рвота, обезвоживание), развитием гипотрофии. Печень увеличивается, при пальпации она плотная, появляется желтуха, нарастают признаки печеночной недостаточности. Обнаруживается помутнение хрусталика глаза (катаракта). При тяжелом течении и без лечения дети погибают на первом году жизни, а при вскрытии обнаруживают цирроз печени. У выживших отмечается резкое отставание психомоторного развития, гепатомегалия, катаракта. Наиболее точным методом диагностики галактоземии является исследование в эритроцитах ферментов галактоза-1-фосфата и галактоза-1-фосфатуридилтрансферазы, галактозы в крови и моче, где уровни ее увеличены.
Исключение из пищи молока (источника галактозы) дает возможность нормально развиваться больному ребенку.
НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ, СВЯЗАННЫЕ С НАРУШЕНИЕМ ЛИПИДНОГО ОБМЕНА
В группу липидов входят триглицериды, холестерол (холестерин), эфиры холестерола, фосфолипиды, сфинголипиды, свободные или неэстерифициро-ванные жирные кислоты (НЭЖК) и др.
Нарушение обмена липидов может происходить на уровне расщепления, всасывания, транспорта, а также при межуточном обмене. Исследования показали, что можно выделить два основных типа наследственных нарушений обмена липидов: 1) липидозы или сфинголипидозы — болезни, приводящие к накоплению сфинголипидов в клетках разных тканей (внутриклеточные липидозы); 2) болезни с нарушением липопротеидов, содержащихся в крови.
Среди наследственных заболеваний, приводящих к накоплению липидов внутри клеток, наиболее изучены болезнь Ниманна—Пика, Гоше и амавротическая идиотия.
Болезнь Ниманна—Пика. Это заболевание связано с накоплением сфингомиелина в мозге, печени, ретикуло-эндотелиальной системе. Сущность нарушения обмена сфингомиелинов состоит в утрате ферментативной активности сфингомиелиназы. Тип наследования аутосомно-рецесСивный. В патогенетическом плане заболевание связано с включением в состав сфингомиелина жирных кислот, что не свойственно нормальному сфингомиелину. Атипичный сфингомиелин накапливается в клетках. Заболевание развивается
84
в раннем детском возрасте с отказа от еды, появления рвоты, увеличения живота, печени, селезенки (рис. 23). У таких больных отмечаются мышечная слабость, судороги, снижается слух и зрение. На глазном дне у 20—30 % детей отмечается пятно вишневого цвета. В анализах крови — анемия, тромбоцитопения, вакуолизированные лимфоциты, увеличение холестерина, лецитина, сфингомиелина. Прогноз заболевания неблагоприятный. Дети погибают в раннем возрасте.
Болезнь Гоше. При этом заболевании в клетках нервной, ретикулоэндотелиальной системы накапливаются цереброзиды. В основе болезни Гоше лежит утрата активности фермента глюкоцеребро-зидазы, приводящая к накоплению в клетках ретикулоэндотелиальной системы глюкоцереброзида. Различают детскую и ювенильную формы. В пунктате костного мозга,
Рис. 23. Внешний вид ребенка с болезнью Ниманна—Пика.
селезенки находят крупные клетки
Гоше. Накопление цереброзидов в клетках нервной системы приводит к их
разрушению.
Детский тип проявляется в первые месяцы жизни задержкой умственного и физического развития, увеличением живота, печени и селезенки, развитием дыхательной недостаточности (инфильтрация легких клетками Гоше), гипертонией мышц, наблюдаются судороги и ребенок погибает на первом году жизни.
Ювенильная форма поражает детей различных возрастов и заболевание носит хронический характер. В клинической картине отмечаются анемия, спленомегалия, пигментация кожи (коричневые пятна), остеопороз, переломы и деформация костей. Развитие иммунодефицитных состояний приводит к смерти.
Амавротическая идиотия (болезнь Тей—Сакса). Это заболевание связано с резким увеличением в клетках мозга, а также печени и селезенки ганглиозидов из-за дефицита гексозаминидазы А в организме.
При рождении и в первые 3—4 месяца жизни дети не отличаются от здоровых сверстников. Заболевание развивается медленно, ребенок становится
85
менее активным, теряет приобретенные навыки. Рано появляются расстройства зрения, слуха. Психические изменения прогрессируют вплоть до идиотии. Развивается гипотония мышц, возникает паралич конечностей. Часто бывают тонические судороги.
Диагноз основывается на определении активности гексозаминидаз, типичных изменениях глазного дна (атрофия сосков зрительных нервов, вишнево-красное пятно в макулярной области). Несмотря на лечебные мероприятия, прогноз неблагоприятный.
НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ ОБМЕНА СТЕРОИДОВ
Образование кортикостероидов имеет сложный ферментативный путь. Из коры надпочечников выделено более 40 соединений, однако в кровь поступает только несколько гормонов: гидрокортизон, альдостерон, кортикостерон и небольшое количество андрогенов и эстрогенов. На промежуточном пути образования кортикостероидов могут возникать различные ферментативные блоки, что приводит к развитию заболеваний.
Адреногенитальный синдром (АГС). Развивается в результате наследственного дефекта фермента 21-гидроксилазы, приводящего к нарушению биосинтеза гормонов коры надпочечников. Распространенность мутантного гена в популяции составляет 1:20—50. Тип наследования данного заболевания аутосомно-рецессивный. В клинической картине выделяют три формы: соль-теряющую, вирильную и форму с гипертензией.
Сольтеряющая форма развивается в первые недели жизни. Проявляется отказом ребенка от груди, рвотой, а иногда и учащением стула, что способствует значительному обезвоживанию организма. При этом у ребенка могут развиться коллаптоидное состояние, судороги, нарушение сердечной деятельности, что приводит к летальному исходу. Лабораторное исследование сыворотки крови выявляет гипонатриемию, гипохлоремию и гиперкалиемию. Дифференциально-диагностическое значение имеет гиперкалиемия, так как при пилоростенозе, кишечном токсикозе с эксикозом всегда отмечается низкий уровень калия в сыворотке крови. У новорожденных* девочек отмечается различная степень маскулинизации от умеренной гипертрофии клитора до полного срастания губно-мошоночных складок с формированием мошонки. Внутренние половые органы таких больных сформированы правильно, кариотип — 46ХХ. Отмечается гиперпигментация околососковой и генитальной областей. У мальчиков основным клиническим симптомом является преждевременное половое развитие, закрытие зон роста эпифизов, в связи с чем больные имеют низкий рост. Во всех случаях отмечается повышение в моче уровня 17-кетостероидов. Если таким больным не оказывается правильная и своевременная терапевтическая помощь (введение гидрокортизона, физиологического раствора), то заболевание заканчивается смертью. На вскрытии, помимо общего истощения, обезвоживания, специфичным является обнаружение увеличенных надпочечников.
86
Вирильная форма АГС развивается вследствие избыточного образования андрогенов при недостатке гидрокортизона. Значительное увеличение клитора, полового члена, оволосение по мужскому типу характеризуют эту форму АГС, которая может наблюдаться у детей младшего и дошкольного возраста. Истинное половое развитие задерживается, так как андрогенные гормоны тормозят секрецию гонадотропина гипофизом и половые железы не развиваются.
Гипертоническая форма имеет такую же клиническую картину, как и вирильная, но сопровождается увеличением артериального давления, гипертрофией левого желудочка, что ведет в последующем к изменению в сосудах, поражению почек. Лабораторные исследования выявляют повышение экскреции с мочой 17-оксикортикостероидов и 17-гидроксикортикостероидов, снижение в крови кортизола и альдостерона.
НАСЛЕДСТВЕННЫЕ НАРУШЕНИЯ БИОСИНТЕЗА ТИРЕОИДНЫХ ГОРМОНОВ
Гипотиреоз. Это заболевание, обусловленное понижением функции щитовидной железы. Оно может развиться вследствие поражения щитовидной железы — первичный гипотиреоз, поражения гипоталамо-гипофизарной области, регулирующей тиреоидную функцию, — вторичный гипотиреоз. Гипотиреоз может быть как врожденный, так и приобретенный.
Первичный врожденный гипотиреоз чаще возникает в результате внутриутробного порока развития щитовидной железы (аплазия, гипоплазия и др.) или из-за генетического дефекта в синтезе тиреоидных гормонов.
Больные дети рождаются с большой массой тела, что связано с отеком тканей. После рождения у ребенка длительно держится желтуха. Они плохо растут и имеют малую прибавку массы тела, отстают в нервно-психическом развитии, отмечаются запоры. Кожа при гипотиреозе сухая, бледная, шелушащаяся, холодная на ощупь. Частота сердечных сокращений ниже нормы, границы сердца расширены. У многих больных имеется систолический шум. Отмечаются грубый голос и скудный рост волос на голове. Рентгенологическое исследование костей запястья выявляет отставание костного возраста.
Для диагностики используют исследование в крови гормонов щитовидной железы (тироксина и трийодтиронина), а также тиреотропного гормона гипофиза. Вспомогательное значение имеет повышение уровня холестерина в крови (более 6,8 ммоль/л).
Гипотиреоз может быть и у взрослых людей, клиническими проявлениями которого служат заторможенность, гиподинамия, бледность, сухость и отечность кожи, брадикардия, снижение температуры тела, артериального давления. После инфекций, охлаждения, приема снотворных препаратов и проведения оперативных вмешательств может развиться гипотиреоидная (мик-сематозная) кома.
Наследование гипотиреоза аутосомно-рецессивное.
87
Лечение гипотиреоза проводят с помощью заместительной терапии препаратами щитовидной железы, начиная с малых доз. Можно использовать и синтетические препараты — тироксин и трийодтиронин. При недостатке тиреотропного гормона используют его синтетический аналог. Гипотиреоидная кома требует неотложных мероприятий, в том числе и введения гидрокортизона.
При своевременной диагностике и адекватной терапии прогноз для больных с гипотиреозом благоприятный.
НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ СОЕДИНИТЕЛЬНОЙ ТКАНИ
Соединительная ткань выполняет в организме опорную, трофическую и защитную функции, обеспечивает постоянство внутренней среды живого организма. Выделяют три компонента соединительной- ткани: а) клеточные элементы, б) коллагеновые, эластические и ретикулярные волокна, в) аморфное основное вещество.
Сложная структура соединительной ткани имеет генетическую природу и дефекты ее биосинтеза и распада приводят к развитию различных наследственных заболеваний.
Болезнь Марфана. Это наследственное заболевание, характеризующееся системным поражением соединительной ткани. Тип наследования — аутосомно-доминантный. В развитии этого заболевания имеет значение поражение эластина и коллагена, выражающееся в нарушении внутри- и межмолекулярных связей в этих структурах. Для больных типичны высокий рост, длинные (паукообразные) пальцы, воронкообразная или килевидная грудная клетка, плоскостопие (рис. 24). Нередко встречаются бедренные и паховые грыжи, гипоплазия мышц и подкожной клетчатки, мышечная гипотония. При обследовании выявляются врожденные пороки сердца, а с возрастом развивается расслаивающаяся аневризма аорты. Зрение у таких больных снижено, при осмотре выявляется миопия, отслойка сетчатки, подвывих хрусталика, катаракта, косоглазие. В моче определяется повышенное количество мукополисахаридов и их составных частей, которые играют важную роль в формировании коллагена и эластических волокон.
Лечение проводят только симптоматическое. Значительные деформации грудной клетки требуют оперативного лечения, что способствует лучшему функционированию сердца, а также улучшению общего самочувствия, показателей электрокардиограммы.
Мукополисахаридозы. Гаргоилизм. Для заболеваний этой группы общим является нарушение метаболизма кислых гликозаминогликанов. Это приводит к тому, что патологические продукты обмена откладываются в соединительной ткани, а также в печени, селезенке, роговице, клетках центральной нервной системы.
Название «гаргоилизм» произошло от французского слова «gargoille», которым называли уродливые фигуры, украшающие собор Нотр-Дам в Па-
88
Рис. 24. Внешний вид больной с синдромом Марфана в вофастс 9 лет: а -- диспропорциональное телосложение, длинные тонкие конечности, узкое лицо; б воронкообразная грудная клетка, плоскостопие; в - арахнодактилия
риже.
В клинической картине заболевания уже в первые месяцы жизни отмечаются типичные изменения конфигурации черепа («башенный» череп), грубые черты лица с крупными губами и языком, короткая шея, запавшая переносица, укороченное туловище. В дальнейшем развивается кифоз груд
7. Зак. 5285
89
ного или поясничного отделов позвоночника (рис. 25). Эпифизы костей утолщены. Границы сердца расширены, могут выслушиваться шумы. Окружность живота увеличена, отмечается увеличение печени и селезенки, наличие пупочной и паховой грыжи. Нервно-психическое развитие отстает. У таких больных снижены зрение и слух. В процессе жизни развивается помутнение роговицы.
В настоящее время выделяют 8 основных типов мукополисахаридозов, характеризующихся утратой активности разных ферментов и определенными особенностями клинической картины. Ниже приводится описание некоторых из них.
Первый тип (синдром Гурлера). Для данного синдрома характерны деформации скелета, отставание в росте, контрактуры крупных суставов, ге-патоспленомегалия, кардиомегалия, помутнение роговицы, прогрессирующая умственная отсталость (см. рис. 25). Больные погибают в возрасте до 10 лет от бронхолегочных инфекций и сердечной декомпенсации. Наследование данного синдрома аутосомно-рецессивное.
Второй тип (синдром Хантера). Сопровождается глухотой, костно-суставными изменениями, менее выражено отставание нервно-психического развития. Продолжительность жизни около 30 uci Насчечованис синчрома
Рис. 25 Внешний вид больных с мукополисахаридозом первого типа: а грубые черты лица; б - кифоз, макроцефалия
90
рецессивное, сцепленное с Х-хромосомой, поэтому болеют только мальчики.
Третий тип (синдром Санфилиппо). Характеризуется умственной отсталостью, незначительными гепатосплсномегалисй и катарактой, менее выраженными костными изменениями.
Естественно, расшифровка типа мукополисахаридоза возможна только при помощи биохимических исследований кислых гликозаминогликанов в крови и моче.
НАСЛЕДСТВЕННЫЕ НАРУШЕНИЯ ОБМЕНА В ЭРИТРОЦИТАХ
Наследственный микросфероцитоз (семейная гемолитическая анемия Минковского—Шоффара). Заболевание обусловлено генетическим дефектом эритроцитов, в частности врожденной недостаточностью липидов оболочки, что приводит к проникновению в клетку ионов натрия и потере АТФ. Измененные эритроциты разрушаются в селезенке, в результате чего происходит образование токсического непрямого билирубина. Заболевание характеризуется триадой синдромов: анемия, желтуха и спленомегалия. В клинической картине следует выделять хроническое течение с триадой синдромов и острые формы, связанные с усиленным гемолизом. Особые трудности для диагностики представляет болезнь в периоде новорожденное™. Изучение родословной в таких случаях оказывает существенную помощь. Для диагностики используют исследование крови: в анализах отмечаются микросфероцитоз, ретикулоцитоз, снижается осмотическая стойкость эритроцитов, изменяется структура кислотной эритрограммы.
Тип наследования — аутосомно-доминантный. При наличии заболевания у одного из родителей риск рождения ребенка с гемолитической анемией Минковского—Шоффара составляет 50 %.
В лечебном плане проводят симптоматические мероприятия, а при гемолитических кризах радикальным методом лечения является спленэктомия.
НАСЛЕДСТВЕННЫЕ СИНДРОМЫ НАРУШЕННОГО ВСАСЫЙАНИЯ
К данной группе энзимопатий относятся непереносимость лактозы, целиакия, муковисцидоз и др.
Непереносимость лактозы. Это наследственное заболевание, связанное с отсутствием или снижением активности фермента лактазы, расщепляющей молочный сахар до глюкозы и галактозы. Негидролизованная лактоза почти не всасывается и ускоряет перистальтику кишечника. При лактазной недостаточности тонкой кишки с первых дней жизни у ребенка наблюдается упорное послабление стула, беспокойство (колики), срыгивание, обезвоживание, отсутствие прибавки в массе тела. В крови отмечаются значительные электролитные нарушения (снижение уровней калия, натрия, кальция). Нагрузка лактозой выявляет низкий (менее 1,1 ммоль/л) подъем уровня гликемии
8. Зак. 5285
91
(при сравнении с исходной концентрацией), pH кала сдвигается в кислую сторону.
Непереносимость лактозы встречается у 15 % здоровых детей, а при различных хронических заболеваниях пищеварительного тракта у 40—78 %. Исследования на близнецах и их родителях показали, что наследуется данный ферментный дефект по аутосомно-доминантному типу.
Лечебные мероприятия связаны с симптоматической терапией (ликвидация обезвоживания), а также переводом ребенка на безмолочное вскармливание (детские смеси, приготовленные на основе сои).
Целиакия (болезнь Ги—Гертера—Гейбнера, глютеновая энтеропатия). Это наследственное заболевание, обусловленное дефицитом ферментов, расщепляющих глиадин злаков до аминокислот, и накоплением в организме токсигенных продуктов его неполного гидролиза.
Ведущим клиническим признаком данного заболевания является тяжелая дистрофия, а также увеличение живота, учащение стула, анорексия, отставание в росте. Первые признаки заболевания появляются после включения в питание ребенка блюд, приготовленных из круп, муки злаковых (рожь, пшеница, овес, ячмень).
Наследование заболевания — аутосомно-рецессивное. Частота встречаемости этой патологии составляет от 1 : 800 до 1 : 3000.
В родословной семьи, имеющей детей с целиакией, можно выявить хронические заболевания пищеварительного тракта, бесплодие, онкологические заболевания пищеварительного тракта и женских половых органов.
В лечебном плане необходимо исключить из питания блюда из злаковых продуктов и проводить симптоматическую терапию. Так как при целиакии всегда имеется дефицит цинка, то его назначение дает положительный эффект. Аглиадиновую диету необходимо практически соблюдать всю жизнь.
Муковисцидоз (кистофиброз поджелудочной железы). Это наследственное заболевание, причиной которого является нарушение секреторной функции всех эндокринных желез, выражающееся в повышении вязкости секрета. Тип наследования муковисцидоза — аутосомно-рецессивный. Частота заболеваемости по данным различных авторов от 1 из 2000 до 1 из 2500.
Выделяют следующие основные клинические формы заболевания:
1) мекониальный илеус (у новорожденных);
2) бронхолегочная форма с развитием хронического воспалительного процесса и потерей массы тела;
3) желудочно-кишечная форма с развитием дистрофии;
4) смешанная форма.
Среди всех случаев муковисцидоза мекониальный илеус составляет 5—10 %. В просвет кишечника мало поступает трипсина и меконий не подвергается ферментативному воздействию, в результате чего он становится очень вязким, часто скапливается в илеоцекальной области, прилипает к стенкам кишечника. У ребенка после рождения появляется рвота с желчью,
92
вздутие живота, отсутствие выделения первородного кала, может произойти заворот или перфорация кишечника.
При лечении этой формы муковисцидоза применяют панкреатические ферменты, назначают очистительные клизмы, а при перфорации или завороте кишечника — оперативное лечение.
Бронхолегочная форма муковисцидоза проявляется повторными заболеваниями легких, трудно поддающимися лечению. При этой форме нередко у ребенка развивается дистрофия. Наряду с поражением легких имеется нарушение переваривающей способности поджелудочной железы. В кале обнаруживается нейтральный жир, стул обильный с неприятным запахом.
При желудочн о-к ишечной форме нарушается переваривающая способность ферментов поджелудочной железы и кишечника. Потребление большого количества пищи с достаточным количеством белков, липидов, углеводов и витаминов не ликвидирует дистрофию. У больных муковисцидозом могут быть и непостоянные симптомы: серо-коричневая окраска зубов, неприятный запах изо рта, длительные боли в верхней половине живота, выпадение прямой кишки, отеки, поражение печени, синуситы.
Для диагностики заболевания в периоде новорожденное™ можно исследовать меконий на концентрацию в нем белка — альбумина (в норме содержание альбумина в меконии до 20 мг/г сухой массы). У более старших детей исследуют потовую жидкость на концентрацию натрия и хлора. Потовую жидкость получают с помощью электрофореза с пилокарпином. Для муковисцидоза характерно увеличение в 2—5 раз концентрации электролитов в поту (норма 35 ммоль/л).
Лечение симптоматическое. Назначают диету, обогащенную белком и натрием, с ограничением липидов. С заместительной целью назначают ферментные препараты поджелудочной железы (панкреатин и его аналоги), пищу подсаливают.
Смешанная форма муковисцидоза сочетает в себе симптомы бронхолегочной и желудочно-кишечной формы.
Изложенные наследственные заболевания обмена веществ, естественно, не исчерпывают все встречающйеся нозологические формы. Дальнейшее развитие клинической генетики и биохимии позволит расшифровать патогенез многих известных наследственных заболеваний, а также описать новые их формы, разработать рациональные методы лечения.
ОСНОВНЫЕ ПРИНЦИПЫ УХОДА ЗА БОЛЬНЫМИ С НАСЛЕДСТВЕННЫМИ ЗАБОЛЕВАНИЯМИ
Дети с наследственными заболеваниями требуют большего ухода, чем здоровые. Чем раньше начата реабилитация таких больных, тем большая вероятность положительного результата лечения.
Наследственные болезни сопровождаются различными синдромами. При
93
отставании в умственном развитии необходимо более частое общение с детьми. Это будет способствовать развитию всех органов чувств ребенка. Его нельзя оставлять одного на длительное время, так как в этих случаях легко развивается госпитализм. Смена обстановки, игрушек, беседа и обучение простейшим навыкам способствуют интеллектуальному развитию детей.
Вторым важным элементом ухода за такими детьми является правильное кормление. Нельзя забывать, что пища должна быть полноценной и соответствовать возрасту ребенка. Иногда дети получают только жидкую пищу и мучнистые блюда. У них резко нарушается обмен веществ, происходит отставание в массе и длине тела. Немного позже присоединяются различные инфекции пищеварительной и дыхательной системы из-за сниженной имунной защиты, которые могут привести к летальному исходу. У дегей, которые не могут глотать, кормление проводится через зонд, однако состав пищи должен быть сбалансированным. Например, нельзя ребенка в возрасте одного года кормить только молоком или кашами. Ребенок этого возраста даже при кормлении через зонд должен получать протертое мясо, мясные бульоны, протертые фрукты и овощи. Только при полноценном питании можно надеяться на положительный успех всех реабилитационных мероприятий. При некоторых наследственных заболеваниях (фенилкетонурия, галактоземия, лактазная недостаточность) для детей необходимы специальные диеты.
Терморегуляция у детей с наследственными заболеваниями может быть нарушена. Они склонны к перегреванию или переохлаждению. Создание оптимального режима температуры окружающей среды, ее влажности (при центральном отоплении в помещении очень низкая влажность), подвижности воздуха (зоны комфорта) - важный элемент ухода. При всяких отклонениях в состоянии ребенка необходимо измерять температуру тела. При повышении температуры тела необходимо тщательное врачебное обследование. Если температура тела поднимается до 38—-38,5 °C, то срочное назначение жаропонижающих лекарственных препаратов не обязательно. Однако при склонности ребенка к судорожным реакциям необходимо внимательно наблюдать за его состоянием, снижая повышенную температуру тела.
При наследственных заболеваниях необходимо следить и за естественными отправлениями. Иногда у этих детей по несколько дней нет стула, хотя пищи ребенок получает достаточно. В таких случаях необходимо обратить внимание на меню ребенка и увеличить количество клетчатки, овощей, фруктов, соков. И все же при задержке стула и до консультации врача можно сделать ребенку очистительную клизму.
Задержка мочеиспусканий можег быть связана с недостаточным поступлением жидкости в организм, нарушением деятельности сердца, почек, а также с атонией мочевого пузыря. Врачебное обследование ребенка поможет выявить причину задержки мочеиспускания или мочеобразования.
У детей, которые все время находятся в постели в лежачем положении, могут развиться пролежни. Частая смена нательного и постельного белья, отсутствие на нем складок, уход за кожей — все это может предотвратить
94
появление пролежней или способствовать их ликвидации. После дефекации и мочеотделения необходимо подмыть ребенка, кожу высушить (не протирать) и обработать пудрой или масляными составами, которые рекомендует врач. Нельзя забывать и о том, что у таких детей кожа и складки могут поражаться грибками, поэтому назначение противогрибковых препаратов окажется своевременным.
Нередко при наследственных заболеваниях может быть судорожный синдром. При его появлении существует опасность западения языка и остановки дыхания. Ребенка необходимо положить на бок, руками вывести челюсть вперед, ввести противосудорожные препараты, которые назначил врач. Во время судорог ребенок может падать, поэтому таких дсгей не следует оставлять одних на диване, а в кроватке должно быть боковое ограждение (сетка, доска).
Наличие врожденного порока сердца с развитием недостаточности кровообращения вызывает необходимость возвышенного положения верхней части тела ребенка, притока свежего воздуха, а иногда и кислородотерапии.
Наследственные заболевания, сопровождающиеся кровоточивостью, всегда требуют не только местных мероприятий для остановки кровотечения, но и консультации врача, так как быстрая потеря крови может приводить к шоку и гибели ребенка.
Таким образом, правильное питание больных с наследственной патологией, гигиеническое содержание, воспитательные мероприятия и специальные элементы ухода будут способствовать возможно быстрейшей реабилитации детей.
Гл аваХ
КЛИНИКО-ГЕНЕАЛОГИЧЕСКИЙ МЕТОД ИЗУЧЕНИЯ НАСЛЕДСТВЕННОСТИ ЧЕЛОВЕКА
Клинико-генеалогический метод, или метод сбора и анализа родословных, является одним из основных в медицинской генетике. Задачами данного метода являются:
а) установление наследственного характера заболевания;
б) определение типа наследования болезни и пенетрантности гена;
в) выявление в родословной лиц, являющихся гетерозиготными носителями мутантного гена;
г) определение прогноза потомства в семьях, где есть или предполагается рождение ребенка с наследственной патологией.
В генеалогическом методе можно выделить два этапа: составление родословной и генеалогический анализ.
ПОРЯДОК СБОРА ГЕНЕАЛОГИЧЕСКОЙ ИНФОРМАЦИИ И МЕТОДИКА СОСТАВЛЕНИЯ РОДОСЛОВНОЙ
Начинать сбор сведений о родословной следует от пробанда, то есть человека (больного, здорового), обратившегося за консультацией. Сбор генеалогической информации о наличии среди его родственников различных заболеваний может проводиться разными методами: опросом, анкетированием, личным обследованием членов семьи.
В генетическую карту подробно записываются все сведения о пробанде (анамнез настоящего заболевания, его начало, последующее течение, возраст, в котором появились первые признаки заболевания). В дальнейшем собирают данные о его сибсах (братьях и сестрах) и родителях. Далее опрос продолжается о родственнйках по линии матери. Запись сведений удобнее вести в следующем порядке: родители матери, их дети по порядку рождения с указанием потомков (внуков). Очень важно выяснить у женщин, как протекала беременность, на каком фоне она наступила, подробности о всех случаях выкидышей, мертворрждений, о наличии бесплодных браков и ранней детской смертности. В такой же последовательности собираются сведения о род-
96
ственниках отца пробанда. Естественно, чем больше родственников пробанда будет непосредственно опрошено или обследовано, тем выше шансы на получение более достоверных и полезных сведений.
При сборе данных о родословной необходимо отмечать девичьи фамилии женщин и место жительства семьи. Это помогает выявить кровно-родственные браки, которые влияют на появление наследственных болезней и их частоту. Если родители пробанда проживали в одном небольшом по числу жителей районе (особенно изолированном географически), можно предположить, что они имеют общих предков, а следовательно, и большее количество общих патологических генов.
При составлении родословной очень важно учесть наличие и характер профессиональных вредностей (особенно для родителей, имеющих детей с врожденными пороками развития), время их действия (до беременности или во время нее). Следует также выявить факторы, которые могли повлиять на возникновение эмбриопатии (прием лекарственных препаратов в первые недели беременности, заболевания матери в данный период, рентгеновское облучение женщины и др.).
ГРАФИЧЕСКОЕ ИЗОБРАЖЕНИЕ РОДОСЛОВНОЙ
После сбора генеалогической информации переходят к графическому изображению родословной. Наиболее распространенные символы, используемые при ее составлении, представлены на рис. 26.
При графическом изображении родословной необходимо соблюдать определенные правила.
1. Составление родословной начинают с пробанда. Братья и сестры располагаются в родословной в порядке рождения слева направо, начиная со старшего.
2. Все члены родословной должны располагаться строго по поколениям в один ряд.
3. Поколения обозначаются римскими цифрами слева от родословной сверху вниз.
4. Арабскими цифрами нумеруется потомство одного поколения (весь ряд) слева направо последовательно. Таким образом, каждый член родословной имеет свой шифр (например: I—3, II—2 и т. д.).
5. Необходимо указывать возраст членов семьи, так как некоторые наследственные заболевания проявляются в разные периоды жизни.
6. Супруги родственников пробанда могут не изображаться в родословной, если они здоровы.
7. Важно отметить лично обследованных членов родословной знаком (!).
Составляя родословную, желательно получить сведения о максимальном количестве родственников 3—4 поколений. Если рассматриваемых признаков несколько, то можно прибегать к буквенным или штриховым различиям внутри символов (рис. 27).
97
- мужчина
-пробанд
- поп неизвес ген
выкидыш
медицинский аборт
- брак
здоровые । —г-----1 связь
О □ О сибсы
мертворожденный
- кровнородственный брак
потомство
- больные
- умершие
Рис. 26. Символы, испо пмусмыс при составлении родословной
- дизиготные близнецы
- гетерозиготные
- носители рецессивного гена
- лично обследованные
- бесплодный брак
- монозиготные близнецы
Рис. 27. Родословная семьи с сахарным диабетом (обозначено штриховкой) и с синдромом Марфана (обозначено черным цветом)
Все полученные данные о состоянии здоровья родственников, причинах и возрасте смерти записываются внизу под родословной (легенда) с указанием даты ее составления.
98
Тщательно собрав все данные, можно приступи и» к анализу родословной. Рассмотрим основные типы наследования моногснных болезней
АУТОСОМНО-ДОМИНАНТНЫЙ ТИП НАСЛЕДОВАНИЯ
При данном типе наследования наиболее часто встречаются браки между больными (Аа) и здоровыми членами семьи (аа), где А доминантный ген, определяющий развитие наследственного заболевания, а рецессивный ген.
Для аутосомно-доминантного типа наследования характерны следующие признаки:
1) передача заболевания из поколения в поколение (наследование по вертикали);
2) передача заболевания от больных родителей детям;
3) здоровые члены семьи обычно имеют здоровое потомство;
4) оба пола поражаются одинаково часто (рис. 28)
Рис 28 Родословная < мьи с брахидактилией
Пробанд (Ш-4) унаследовала дефект от своей матери (II 3) и деда (I и передала его своим детям (IV—2, 3).
По аутосомно-доминантному типу наследуются синдром Марфана, поли дактилия, брахидактилия, синдактилия, ахондроплазия, анемия Минковского Шоффара и многие другие заболевания.
В случае доминантного типа наследования, если один из родителей болен (Аа), прогноз потомства может быть определен еще до рождения ребенка Он равен 50 % при условии гетерозиготности больного и полной пенегран тности гена.
Если ген встречается в популяции часто, то при аутосомно-доминантном типе наследования возможны браки типа: АахАа, в которых мотут появиться дети гомозиготы по данному заболеванию. В таких семьях вероятность рождения больного ребенка равна 75 %. Однако многие аутосомно-доминантные заболевания в гомозиготном состоянии протекают значительно тяжелее, чем у гетерозигот, и дают, как правило, летальный исход.
99
Иногда при анализе родословной возникает следующая ситуация: пробанд страдает доминантной болезнью (например, отосклерозом), его мать здорова, а бабушка также страдала той же болезнью. В этом случае мать пробанда — носитель патологического гена, так как она передала его своему ребенку, но у нее он не проявился, то есть не пенетрировал. В результате вид родословной изменяется и появляются пропуски поколений.
Пенетрантность — это частота проявления гена среди носителей данного гена. Она представляет собой отношение особей, имеющих данный признак, к особям, имеющим данный ген, выраженное в процентах. Так, пенетрантность отосклероза — 40 %, синдрома Марфана — 30 %, ретинобластомы — 80 %.
Собирая анамнез и анализируя родословную, нельзя забывать и о другом свойстве гена — его различной экспрессивности. Экспрессивность — степень выраженности гена. Понятие экспрессивности аналогично понятию тяжести заболевания. При высокой экспрессивности гена развивается тяжелая форма заболевания, часто с летальным исходом, при низкой — создается впечатление, что человек здоров.
Рассмотрим понятие экспрессивности гена на примере наследственной патологии соединительной ткани — синдроме Марфана. При этом в одной семье можно встретить как очень тяжелые формы с классическим поражением костной системы (в виде сколиоза или кифосколиоза, деформации грудины, арахнодактилии); нарушением зрения (двусторонний подвывих хрусталика) и сердечно-сосудистой системы (расширение аорты), так и стертые формы заболевания, протекающие легко (астеническое телосложение, арахнодакти-лия, сколиоз I степени, небольшая миопия). Таких людей без дополнительного обследования можно отнести к здоровым.
Слабовыраженные клинические формы болезни могут легко просматриваться, тогда родословная также теряет свой «классический» вид, появляются пропуски поколении. Поэтому очень важно личное обследование всех членов семьи.
АУТОСОМНО-РЕЦЕССИВНЫЙ ТИП НАСЛЕДОВАНИЯ
Для данного типа наследования характерны следующие признаки.
а) больные дети рождаются от фенотипически здоровых родителей, являющихся гетерозиготными носителями патологического гена.
Генотипы родителей
Мать Аа Отец Аа
Генотипы детей
АА Аа, Аа аа
(здоровый) (здоровые, но носи- (больной) тели аномального гена)
100
Как видно из схемы, болеют только гомозиготы по рецессивному гену;
б) больные чаще встречаются в одном поколении: среди родных или двоюродных сибсов (наследование «по горизонтали») или среди дядей и племянников (наследование <<по ходу шахматного коня»);
в) в родословной отмечается более высокий процент кровно-родственных браков;
г) одинаково часто болеют мужчины и женщины (рис. 29).
К заболеваниям с аутосомно-рецессивным типом наследования относятся фенилкетонурия, галактоземия, альбинизм, муковисцидоз, целиакия, большинство мукополисахаридозов и др.
Рассмотрим родословную семьи с альбинизмом (см. рис. 29).
Рис. 29. Родословная семьи с альбинизмом
Альбинизм — редкая аутосомно-рецессивная патология аминокислотного обмена, при которой страдает синтез пигмента меланина. Альбиносы имеют белую кожу, белые волосы, обесцвеченную радужную оболочку глаз (красные глаза). Кожа и глаза очень чувствительны к солнечному свету. Как видно из рис. 29, больные чаще встречаются в одном (IV) поколении среди родных и двоюродных сибсов; в III поколении родословной имеются родственные браки. Больные в данной семье появились только в III и IV поколениях. Это—особенность рецессивного гена существовать во многих поколениях в гетерозиготном состоянии, никак не проявляясь фенотипически. Поэтому первый больной рецессивной болезнью может появиться через многие поколения после возникновения мутации, так как рождение больного ребенка возможно только в том случае, если оба родителя являются гетерозиготами (Аа).
Риск рождения больного ребенка в такой семье равен 25 %.
Однако при аутосомно-рецессивном типе наследования редко, но возможны и другие варианты браков:
аа х аа — у таких родителей все дети будут больными (аа).
101
Аа х аа — 50 % детей будут больными (генотип аа), 50 % фенотипически даровыми (генотип Аа), но будут являться носителями мутантного гена.
Следует отметить, что частота рецессивных наследственных болезней особенно повышается в изолятах и среди населения с высоким процентом кровнородственных браков. Об отрицательном влиянии родственных браков на потомство свидетельствует и тот факт, что умственная отсталость среди детей от этих браков в 4 раза выше, чем в семьях с неродственными браками.
Х-СЦЕПЛЕННЫЙ ТИП НАСЛЕДОВАНИЯ
В Х-хромосоме могут локализоваться как доминантные, так и рецессивные гены. У человека известно более 200 генов, в основном патологических, сцепленных с Х-хромосомой.
У женщины аномальный ген может находиться в одной (гетерозигота) или обеих (гомозигота) Х-хромосомах; у мужчин — только в одной Х-хромосоме, поэтому не только доминантный, но и рецессивный ген в его X-хромосоме всегда будет проявляться. Такое состояние гена называется ге-мизиготным.
Если в Х-хромосоме локализуется рецессивный ген, то такой тип наследования называется Х-сцепленным рецессивным. Для этого типа наследования характерны следующие признаки:
а) болеют преимущественно лица мужского пола;
б) больные дети рождаются от фенотипически здоровых родителей, но мать больного является гетерозиготной носительницей патологического гена («кондуктор»);
в) больные мужчины не передают заболевания своим сыновьям, но все их дочери становятся «кондукторами».
, г) редкие случаи заболевания женщин возможны, если их отец болен, а мать — носительница (рис. 30).
Если в брак вступает здоровый мужчина и гетерозиготная женщина, то вероятность рождения больного мальчика в данной семье составляет 50 % от всех мальчиков (или 25 % от всех детей), а все девочки этих родителей будут здоровы, но половина из них станут носительницами патологического гена.
В случае, если в брак вступают больной мужчина и здоровая женщина, все дети будут здоровыми, но дочери получат от отца мутантный аллель и станут «кондукторами».
Третий вариант браков, который встречается редко при рецессивном сцепленном с Х-хромосомой наследовании, — это брак между больным мужчиной и женщиной гетерозиготой. В этом случае ожидается рождение половины больных мальчиков; половина девочек также будет больна, а половина — станет носительницами патологического гена.
Классическим примером рецессивного сцепленного с Х-хромосомой наследования является гемофилия.
102
Клиника заболевания связана с тем, что из-за недостатка необходимых для свертывания крови факторов больные страдают кровоточивостью. Даже после небольших травм возникают значительные кровопотери, развиваются анемия, гемартрозы. На рис. 30 изображена родословная семьи с гемофилией Л.
Рис. 30. Родословная семьи с гемофилией А
Пробанд (III—1), его двоюродный брат (III—8) и их дядя (II—3) больны гемофилией. Бабка пробанда (I—1), его мать (II—2) и его тетка (II— 5) — носители рецессивного сцепленного с Х-хромосомой гена гемофилии А.
Кроме гемофилии, рецессивный сцепленный с Х-хромосомой тип наследования характерен для псевдогипертрофической миопатии Дюшенна, агаммаглобулинемии Брутона, некоторых форм дальтонизма и др.
Если в хромосоме X локализуется доминантный ген, такой тип наследования называется Х-сцепленным доминантным. Для него характерны следующие признаки:
а) заболевание прослеживается в каждом поколении;
б) если болен отец, то все его дочери будут больными, а все сыновья здоровыми;
в) если больна мать, то вероятность рождения больного ребенка равна 50 % независимо от пола;
г) болеют как мужчины, так и женщины, но в целом больных женщин в семье в 2 раза больше, чем больных мужчин;
д) у здоровых родителей все дети будут здоровыми.
Анализ родословной, изображенной на рис. 31, показывает, что дети с коричневыми зубами рождаются от браков, в которых один родитель болен, а другой здоров. Все здоровые члены родословной имеют здоровое потомство. Обращает на себя внимание третье поколение, где все мужчины (III—2, III—3) унаследовали нормальную окраску эмали зубов, а все женщины (III—4, III—6, III—7) унаследовали дефект окраски эмали. Их отец (II—2) имел коричневые зубы, а мать (II—1) — белые. Следовательно, ген коричневой окраски эмали зубов находится в Х-хромосоме. Только в этом случае он не мог оказаться у сыновей, но обязательно должен был попасть к дочерям. При этом он проявился у всех дочерей.
103
Рис. 31. Родословная семьи с доминантным сцепленным с Х-хромосомой типом наследования (коричневой окраской эмали зубов)
Сцепленное с Y-хромосомой наследование характеризуй гея тем, что гены, локализованные в Y-хромосоме, передаются только сыновьям поражение, о отца, а его дочери остаются здоровыми, так как они никогда не получают Y-хромосомы от отца (голандрическое наследование). По такому типу у человека наследуются «мохнатые уши» — наличие волос но краю ушных раковин.
МУЛЬТИФАКТОРИАЛЬНОЕ НАСЛЕДОВАНИЕ
Мультифакториальные болезни, или болезни с наел еде гвенным предрасположением, составляют в настоящее время 92 % от общей патологии человека. Они вызывается изменением чаще нескольких генов и для своего проявления требуют влияния факторов внешней среды.
К наиболее часто встречающимся мультифакториальным заболеваниям относятся: ревматизм, ишемическая болезнь сердца, гипертоническая болезнь, эпилепсия, мигрень, язвенная болезнь, цирроз печени, неснецифический язвенный колит, сахарный диабет, бронхиальная астма, псориаз, шизофрения и др. При перечисленных болезнях из поколения в поколение передается предрасположенность к определенному заболеванию.
Мультифакториальные болезни при всем их разнообразии имеют некоторые общие черты:
1) высокая частота в популяции (например, сахарным диабетом страдает около 5 % населения промышленно развитых стран, аллергическими заболеваниями — более 10 %, шизофренией — 1 % населения, гипертонией около 30 %);
2) несоответствие наследования законам Менделя;
3) существование клинических форм от скрытых субклинических до резко выраженных проявлений;
104
4) более раннее начало заболевания и некоторое усиление клинических проявлений в нисходящих поколениях.
Генетический прогноз при мультифакториальном типе наследования зависит от следующих факторов:
а) частоты встречаемости заболевания в популяции (чем она ниже, тем выше риск возможности заболевания для родственников пробанда);
б) степени выраженности болезни у пробанда (чем она выше, тем больше риск развития болезни у родственников, так как тяжесть заболевания определяется суммарным эффектом более чем одного гена). Например: человек, получивший два гена, от которых зависит артериальная гипертензия, может иметь более тяжелую степень заболевания и в два раза большую вероятность передачи патологического гена потомству;
в) степени родства с пораженным членом семьи, так как это определяет число общих генов у данного человека с больным.
Наибольшее число общих генов (100 %) имеется у монозиготных близнецов. Различия в фенотипе у них могут быть вызваны только факторами внешней среды, поэтому близнецовый метод является ведущим в определении роли наследственных и средовых факторов в развитии мультифакториальных заболеваний.
Каждый родитель передает своему ребенку половину своего хромосомного набора, то есть половину генов, затем с каждым новым поколением у потомков число генов, одинаковых с каким-то общим предком, уменьшается вдвое. Чем больше общих генов, тем больше возможность однотипного влияния двух или более патологических генов, если они были у общего предка. Поэтому важно оценить степень родства с больным, то есть число общих генов (табл. 3).
Табл. 3. Общность генов у родственников разных степеней родства
Степень родства Показатель общности генов
Монозиготные близнецы 100 % (1,0)
I степень родства (родители — дети, родные братья — сестры) 50 % (1/2)
II степень (дядя, тетя — племянники, дедушка, бабушка — внуки) 25 % (1/4)
III степень (двоюродные братья — сестры) 12,5 % (1/8)
IV степень (троюродные братья — сестры) 3,125 % (1/32)
105
При анализе родословных риск проявления заболевания велик при болезни родственников I и II степеней родства. При наличии болезни среди родственников III степени родства он может рассматриваться как умеренный. Единичные, спорадические случаи болезни среди родственников IV и более дальних степеней родства указывают на малую степень риска;
г) число больных родственников также определяет прогноз для пробанда.
Чем больше в родословной больных, тем выше риск проявления болезни. Так, при сахарном диабете риск для сибсов пробанда в зависимости от числа больных родственников будет следующим: 1) если родители здоровы, вероятность заболевания равна 5—10 %; 2) если болен один из родителей, риск увеличивается до 10—20 %; 3) если больны оба родителя, риск заболеть сахарным диабетом для сибсов пробанда будет равен 20- 40 %;
д) в случаях разницы в частоте заболевания по полу риск для родственников будет выше, если пробанд относится к менее поражаемому полу.
Так, в двух семьях, отягощенных язвенной болезнью желудка (рис. 32) и имеющих по двое детей разного пола, в каждой семье вероятность передачи гена одинакова для обоих детей (1/2), но вероятность заболеть больше для лиц мужского пола, так как язвенная болезнь относится к заболеваниям с преимущественным поражением лиц мужского пола. Если же сравнить такую вероятность между сыновьями двух семей, то она выше в первой семье (а), так как язвенной болезнью здесь страдает женщина, то есть в данном случае больной родитель относится к менее поражаемому полу.
Рис. 32. Фрагменты родословных семьи (а, б) с язвенной болезнью желудка
Основу оценки риска при мультифакториальной патологии составляют эмпирические данные о популяционной и семейной частоте каждого заболевания или порока развития (табл. 4).
Такие таблицы составлены для большинства мультифакториальных заболеваний и пороков.
Проводя анализ родословных, можно выявить лиц, генетически предрасположенных к тому или иному заболеванию. Это позволит эффективно проводить лечебно-профилактические мероприятия, направленные на предупреждение развития у них патологии. Так, при наличии гипертонической болезни у одного из родителей необходимо с раннего возраста контролировать уровень артериального давления у детей, в дальнейшем рекомендовать им щадящий режим (избегать нервных и физических перегрузок, употребления
106
алкоголя, курения и др.). В таких семьях необходимо правильное соблюдение режима дня, увеличение физической активности за счет умеренных физических нагрузок, аутотренинг, ограничение приема поваренной соли, регулирование массы тела и др.
Выполнение этих рекомендаций с раннего возраста окажет благотворный эффект на профилактику развития гипертонии.
Табл. 4. Эмпирический риск при некоторых мультифакториальных заболеваниях и пороках
N п/п Заболевания, пороки Риск для сибсов (и в отдельных случаях для потомства)
1. Анэнцефалия 2—5 %
2. Врожденные пороки сердца 2—4 % (в зависимости от формы)
3. Косолапость 2%
4. Опухоль Вильмса 5%
5. Рак молочной железы 6—7%
6. Эпилепсия 3—12%
7 Шизофрения:
если болен один из родителей; 10%
если больны оба родителя; 40%
для сибсов в спорадических случаях 12,5—20%
8. Неосложненная миопия высокой степени 10 15 % (для детей и сибсов)
9. Ревматоидный артрит 5%
10. Псориаз 16 % (для сибсов) 20 % (для детей пробанда)
11. Язвенная болезнь желудка 7,5 %
12. Атопический дерматит 16%
13. Бронхиальная астма 8—9 %
Таким образом, анализ семейных данных с целью выявления групп лиц, склонных к мульфакториальным заболеваниям, — важное и перспективное направление в профилактической медицине.
107
Гл ава XI МЕДИКО-ГЕНЕТИЧЕСКОЕ КОНСУЛЬТИРОВАНИЕ
ЗАДАЧИ И ОРГАНИЗАЦИЯ МЕДИКО-ГЕНЕТИЧЕСКОГО КОНСУЛЬТИРОВАНИЯ
Наиболее распространенным и эффективным методом профилактики наследственных болезней является медико-генетическое консультирование, которое представляет собой один из видов специализированной медицинской помощи населению, направленной на предупреждение появления в семье больного ребенка.
Медико-генетическая служба Республики Беларусь включает пять областных (Гомель, Могилев, Брест, Гродно, Витебск) и Республиканский медикогенетический центры (МГЦ). МГЦ созданы на основании приказов областных органов здравоохранения как самостоятельные учреждения или отделения, входящие в состав крупных лечебно-профилактических учреждений. Республиканский медико-генетический центр входит в структуру 7-й клинической больницы Минска и функционирует в масштабе республики с целью оказания населению всех современных видов медико-генетической помощи. Научноконсультативное руководство медико-генетической службой осуществляет НИИ наследственных и врожденных заболеваний Минздрава Республики Беларусь.
Главная цель генетического консультирования — предупреждение рождения больных детей. Это в первую очередь касается тяжелых и плохо поддающихся лечению пороков развития и наследственных болезней, приводящих к физической или психической неполноценности.
Задачами медико-генетического консультирования являются:
1) ретро- и проспективное консультирование семей и больных с наследственной или врожденной патологией;
2) пренатальная диагностика врожденных и наследственных заболеваний ультразвуковыми, цитогенетическими, биохимическими и молекулярно-генетическими методами;
3) помощь врачам различных специальностей в постановке диагноза наследственного или врожденного заболевания, если для этого требуются специальные генетические методы исследования;
108
4) объяснение пациенту и его семье в доступной форме о величине риска иметь больное потомство и оказание им помощи в принятии решения;
5) ведение территориального регистра семей и больных с врожденной и наследственной патологией и их диспансерное наблюдение;
6) пропаганда медико-генетических знаний среди населения.
Чаще всего за консультацией в МГЦ обращаются семьи, в которых уже есть один или несколько больных детей с наследственным или врожденным заболеванием и родителей беспокоит вопрос дальнейшего деторождения. Другая группа включает семьи, где болен один из супругов, и родителей интересует прогноз здоровья будущих детей. К третьей группе относятся семьи практически здоровых детей, у которых по линии одного или обоих родителей имеются родственники с наследственной патологией. В четвертую группу входят родители, желающие узнать, какова судьба здоровых братьев и сестер больного ребенка (не возникнет ли аналогичное заболевание у них в дальнейшем, а также у их детей).
Кроме перечисленных случаев, необходимо заподозрить наследственную патологию и направить семью в медико-генетический центр при следующих показаниях:
— наличие аналогичных заболеваний или симптомов у нескольких членов семьи;
— первичное бесплодие супругов;
— первичное невынашивание беременности;
— отставание в умственном и физическом развитии;
— рождение ребенка с врожденными пороками развития;
— первичная аменорея, особенно в сочетании с недоразвитием вторичных половых признаков;
— наличие кровного родства между супругами и др.
Особенностью работы МГЦ является то, что исследуется не только человек, обратившийся за консультацией (пробанд), но и члены его семьи. Для генетической консультации требуются подробные сведения о родственниках пробанда, часто возникает необходимость в углубленном их обследовании. Это приводит к гораздо большим затратам времени, чем на прием больного любым другим специалистом. На первичный прием семьи с составлением родословной требуется 1 ч 20 мин, на повторный прием — 30 мин.
ОСНОВНЫЕ ПРИНЦИПЫ КОНСУЛЬТИРОВАНИЯ
Консультации по прогнозу состояния здоровья потомства можно разделить на две группы: 1) проспективное консультирование; 2) ретроспективное консультирование. Наиболее эффективный вид профилактики наследственных болезней — это проспективное консультирование, то есть консультирование супружеских пар до рождения ребенка или молодых людей до вступления в брак. Такие консультации проводятся при наличии кровного родства между супругами, при неблагоприятном семейном анамнезе, при воздействии вред
9. Зак. 5285
109
ных средовых факторов на кого-либо из супругов (профессиональные вредности, лечебное облучение, тяжелые инфекции и др.). Ретроспективное консультирование — это консультирование семей, в которых уже имеется больной ребенок, относительно здоровья будущих детей.
Чтобы правильно рассчитать генетический риск, нужно поставить точный диагноз и определить тип наследования заболевания в данной семье. Для установления или уточнения диагноза обратившемуся за консультацией или его родственникам бывает необходимо назначить ряд инструментальных и лабораторных исследований. В связи с этим в штат медицинского персонала областных МГЦ кроме врача-генетика (педиатра) введены должности врача акушера-гинеколога, врача ультразвуковой диагностики, биохимика-генетика, врача лаборанта-цитогенетика и др.
Средний медицинский персонал (медицинские сестры, фельдшера-акушерки, фельдшера-лаборанты) обеспечивают работу врачей, участвуют во врачебном приеме, проводят забор и подготовку материала для цитогенетического и биохимического исследования, ведут медицинскую документацию.
Минимальный перечень лабораторных исследований, выполняемый в областных МГЦ, включает: определение полового хроматина, кариотипирование с использованием культуры лимфоцитов периферической крови, амниоцитов, клеток хориона и плаценты, иммуноферментное определение в сыворотке крови альфа-фетопротеина и хориогонина, лотовый тест .для диагностики муковисцидоза и др.
ЭТАПЫ КОНСУЛЬТИРОВАНИЯ
Медико-генетическое консультирование можно разделить на три этапа.
Первый этап — это уточнение диагноза заболевания. В ряде случаев точный диагноз заболевания может быть установлен уже перед направлением в МГЦ. Это бывает возможно в случае хорошо изученной или часто встречающейся патологии (гемофилия, сахарный диабет и др ). Однако чаще больных направляют в консультацию для установления диагноза. Так, рождение ребенка с множественными пороками развития может явиться результатом эмбрио- или фетопатии, следствием нарушения кариотипа или результатом мультифакториальной патологии. В этом случае правильный диагноз может быть поставлен только после тщательного анализа родословной, обследования пробанда и его родственников, применения цитогенетического и других специальных методов исследования.
Второй этап — определение генетического прогноза для потомства. Установив диагноз наследственного заболевания, закономерности его передачи в'семье и определив, является ли данная патология следствием новой мутации или возникла как результат скрытого носительства цатологической мутации на генном или хромосомном уровне, производится расчет повторного риска рождения больного ребенка в семье. Это входит в функции врача-генетика.
по
Генетический риск выражает вероятность появления определенной аномалии у обратившегося за консультацией или его потомков и определяется двумя способами: либо путем теоретических расчетов, основанных на генетических закономерностях, либо с помощью эмпирических данных. Некоторые принципы расчета генетического риска при моногенных и мульти-факториальных заболеваниях представлены в главе X.
При хромосомных болезнях, вызванных числовыми аномалиями хромосом, вероятность повторного рождения больного ребенка в семье крайне мала (не превышает 1 %), если известно, что ни у одного из родителей нет хромосомной аномалии, а также отсутствуют другие факторы риска (например, возраст матери). Прогноз для потомства в семье, в которой родился ребенок с транслокационной формой болезни Дауна, неблагоприятен. В таком случае необходимо определить кариотип у обоих родителей, установить, кто является носителем сбалансированной транслокации, и только после этого определить повторный риск рождения ребенка с болезнью Дауна. Так, при транслокации 14/21 риск для потомства равен 10 %, если носитель транслокации мать, и 2,5 %, если носитель отец. При транслокации 21 -й хромосомы на ее гомолог риск повторного рождения больного ребенка составляет 100 % независимо от того, отец или мать несут эту транслокацию.
Прогнозирование потомства неразрывно связано с диагностикой в семьях гетерозиготных носителей патологического гена. Выявление гетерозиготных носителей особенно важно в случаях патологии с аутосомно-рецессивным типом наследования, в семьях, отягощенных заболеваниями, сцепленными с Х-хромосомой; при кровнородственных браках.
В некоторых случаях носительство можно установить уже при анализе родословной: например, если у отца женщины имеется рецессивное заболевание, сцепленное с Х-хромосомой (гемофилия, миопатия Дюшенна и др.), то такая женщина со 100 % вероятностью гетерозиготна по этому гену и риск заболевания для ее сыновей равен 50 %. При заболеваниях с аутосомно-рецессивным типом наследования родители больного и его будущие дети также являются гетерозиготными носителями данного гена. При аутосомнодоминантном наследовании с неполной пенетрантностью носителями патологического гена являются все лица, имеющие больных детей и больных родителей одновременно.
Если гетерозиготность является вероятностной на основании генеалогического анализа, можно воспользоваться клиническими и биохимическими методами. Так, повышенный уровень креатинфосфокиназы в сыворотке крови матери или сестер больного псевдогипертрофической формой миопатии Дюшенна указывает на гетерозиготность по гену миопатии. Снижение в сыворотке крови у сестер или матери больного гемофилией уровня антигемо-фильного глобулина может явиться вполне убедительным доказательством наличия гетерозиготного носительства гена гемофилии. В некоторых случаях при выявлении гетерозиготных носителей патологического гена необходимо обращать внимание на «малые» клинические признаки основного заболевания.
111
Например, женщины-гетерозиготы по гену миопатии Дюшенна часто жалуются на небольшую слабость в ногах и быструю утомляемость; при носительстве гена гемофилии можно выявить в анамнезе легкую или умеренную тенденцию к кровоточивости.
В настоящее время для ряда наследственных заболеваний возможна ДНК-диагностика гетерозиготных носителей патологического гена.
Если гетерозиготный носитель вступает в брак, то следует определить вероятность гетерозиготности будущего супруга (супруги) и информировать семью о риске рождения у них больного ребенка. Гетерозиготным носителям также рекомендуется избегать родственных браков, поскольку в таких случаях увеличивается риск рождения больного ребенка.
На третьем, заключительном этапе консультирования обратившихся за консультацией в МГЦ знакомят с генетическим прогнозом для потомства, то есть с величиной риска рождения больного ребенка, и дают им соответствующие рекомендации.
Генетический риск до 5 % расценивается как низкий и нс является противопоказанием к продолжению деторождения. Риск от 6 до 20 % принято считать средним. В этом случае рекомендации относительно планирования дальнейших беременностей зависят от тяжести наследственного или врожденного заболевания и возможности его пренатальной диагностики. Генетический риск свыше 20 % относится к категории высокого риска, и при отсутствии методов пренатальной диагностики соответствующей патологии дальнейшее деторождение в данной семье не рекомендуется.
Таким образом, давая заключение, необходимо учитывать нс только величину риска рождения больного ребенка, но и тяжесть заболевания, эффективность его лечения, возможности пренатальной диагностики. Учитывая комбинацию этих признаков, в каждом конкретном случае дастся оптимальный совет. Необходимо объяснить родителям случайность распределения генов и отсутствие их вины за рождение больного ребенка, а также сообщить им, что вероятность появления аномалии во время каждой беременности составляет 4—6 % (общепопуляционный риск). Давая свои рекомендации семье, врач не имеет права настаивать на воздержании от деторождения, прерывании беременности или необходимости супругам расстаться, если они являются гетерозиготными носителями одного и того же патологического гена. Все решения по дальнейшему планированию семьи принимаются только супругами.
СОВРЕМЕННЫЕ МЕТОДЫ
ПРЕНАТАЛЬНОЙ ДИАГНОСТИКИ ВРОЖДЕННЫХ ПОРОКОВ РАЗВИТИЯ И НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ
Прогноз потомства, осуществляемый в медико-генетической консультации, является вероятностным и не позволяет ответить однозначно, завершится ли данная беременность рождением здорового или больного ребенка. Эффек
112
тивность медико-генетического консультирования значительно повышается с применением современных методов дородовой диагностики, которые не только позволяют определить заболевание задолго до рождения ребенка, но и прервать беременность при поражении плода в I или II триместре. Своевременное прерывание беременности необходимо при таких наследственных болезнях и врожденных пороках, лечение которых на современном этапе не дает должных результатов или при которых изменения в организме, возникшие в период внутриутробного развития, необратимы (болезни накопления, ахондроплазия, анэнцефалия и др ).
Основными показаниями для проведения пренатальной диагностики являются:
1) наличие в семье точно установленного наследственного заболевания;
2) возраст будущей матери старше 35 лет, отца — старше 40 лет;
3) наличие у матери Х-сцеплснного рецессивного патологического гена;
4) беременные, имеющие в анамнезе спонтанные аборты, мертворождения неясного генеза, детей с множественными врожденными пороками развития и с хромосомной патологией;
5) наличие структурных перестроек хромосом (особенно транслокаций и инверсий) у одного из родителей;
6) гетерозиготность обоих родителей по одной паре аллелей при аутосомно-рецессивных заболеваниях;
7) беременные из зоны повышенного радиационного фона, с тератогенным воздействием и др.
К основным методам пренатальной диагностики относятся: ультразвуковое исследование (УЗИ), амниоцентез, биопсия хориона, фетоскопия, определение альфа-фетопротеина.
Из всех методов пренатальной диагностики наиболее распространено ультразвуковое исследование плода (эхография). Метод основан на способности ультразвуковой волны отражаться от поверхности раздела двух сред, отличающихся различной плотностью, что позволяет получить их изображение на экране электронно-лучевой трубки. Это исследование проводится всем беременным женщинам трехкратно на 14—16-й, 20—21-й и 26—27-й неделях беременности. Женщин из группы риска по рождению детей с пороками развития осматривают в более ранние сроки.
С помощью ультразвукового исследования можно диагностировать грубые пороки мозга (анэнцефалию, гидроцефалию, черепно- и спинномозговые грыжи, микроцефалии); пороки конечностей (отсутствие конечности или ее части, системные скелетные дисплазии); пороки почек (агенезию или гипоплазию почек, гидронефроз, поликистоз), атрезии желудочно-кишечного тракта, пуповинные и диафрагмальные грыжи, некоторые врожденные пороки сердца.
Следует отметить, что разные формы пороков диагностируют в разные
из
сроки беременности. Если анэнцефалия может быть распознана уже на 14— 16-й неделе, то для диагностики гидронефроза необходимо исследование в значительно более поздние сроки беременности. Одни пороки развития (черепно-мозговые грыжи) могут быть выявлены уже при однократном исследовании, другие (микроцефалия, некоторые формы скелетных дисплазий) диагностируются лишь при динамическом наблюдении.
Если у плода при ультразвуковом исследовании выявляется несовместимый с жизнью порок развития, то беременность прерывают. Этот метод пренатальной диагностики практически безопасен как для матери, так и для плода.
Большое распространение получил метод определения альф а-ф е -топротеина (АФП) в сыворотке крови беременной или в амниотической жидкости. АФП-кислый гликопротеид продуцируется печенью плода. Оптимальным сроком определения АФП в сыворотке крови матери является 15—16-я неделя беременности. При выявлении изменений в уровне данного гликопротеида повторное исследование проводят через 2—3 недели.
Уровень АФП повышается при ряде аномалий развития плода, таких как спинномозговые грыжи, анэнцефалия, гидроцефалия, врожденный нефроз, спонтанная внутриутробная гибель плода, при угрозах выкидыша, атрезиях пищевода и двенадцатиперстной кишки и др. Снижение концентрации АФП отмечается при хромосомной патологии (синдром Дауна, Эдвардса и др.). Необходимо помнить, что уровень АФП в крови беременной может быть повышен и при заболеваниях матери: опухолях печени, гератокарциноме яичников, хроническом гепатите, циррозе и др.
Амниоцентез с цитогенетическим или биохимическим исследованием амниотической жидкости имеет важное значение в диагностике наследственных заболеваний обмена веществ, хромосомной патологии и определении пола плода при гетерозиготном носительстве матери Х-сцеплепного рецессивного гена. Амниоцентез проводят под контролем ультразвукового исследования, с помощью которого точно определяют локализацию плаценты. Пункцию амниона производят трансабдоминально, реже трансвагинально и извлекают шприцем около 10—15 мл жидкости. Оптимальный срок для проведения амниоцентеза — 14—16-я неделя беременности, когда накапливается достаточное количество амниотической жидкости и в ней увеличивается количество клеток. Необходимо помнить, что при проведении амниоцентеза возможны осложнения со стороны плода или матери. Наиболее серьезное — гибель плода вследствие разрыва плодного пузыря с последующим спонтанным абортом. Очень редкое — инфицирование полости матки, приводящее к амниониту и выкидышу. Поскольку амниоцентез представляет хотя и небольшую, но все-таки операцию, для его проведения необходимо получить согласие женщины.
Фетоскопия (а м н и о с ко п и я) — это метод визуального наблюдения плода в полости матки через эластический зонд, оснащенный оптической системой. Оптимальный срок исследования — 18—22-я неделя
114
беременности. Этот метод применяется для диагностики видимых врожденных пороков конечностей, лица (расщелины), ахондроплазии, а также для выявления ихтиоза, буллезного эпидермолиза путем биопсии кожи плода.
Перспективным методом пренатальной диагностики, позволяющим начинать исследование плода в более ранние сроки, является биопсия х о р и о н а. Ее проводят обычно на 7—9-й неделе беременности. Ворсинки берут биопсийными щипцами с помощью специального гибкого катетера, вводимого через шейку матки. В дальнейшем их исследуют с помощью цитогенетических и биохимических методов, как при амниоцентезе. При выявлении наследственного заболевания у плода беременность прерывают в первом триместре. Недостатком данного метода является сравнительно более высокая частота выкидышей после взятия биопсии ворсин хориона.
Методы дородовой диагностики с каждым годом совершенствуются. В настоящее время применяется «ранний» амниоцентез, проводимый на 11— 13-й неделе беременности (здесь для анализа берут лишь 1—2 мл околоплодной жидкости), плацентоцентез (получение ворсин плаценты во II триместре беременности); хордоцентез, позволяющий получить пуповинную кровь плода.
Современные методы пренатальной диагностики основаны на молекулярно-генетических подходах. Используя пробы, комплементарные к тем или иным участкам ДНК, удается установить, не является ли данный плод гетерозиготой или гомозиготой по патологическому гену. При этом используются два подхода: непосредственное определение наличия или отсутствия патологического гена (что возможно, когда известно строение гена) или выявление каких-либо других полиморфных участков ДНК (на которые есть зонды), тесно сцепленных с искомым геном неизвестного строения.
В настоящее время пренатальная диагностика при медико-генетическом консультировании семей нашла широкое применение, постоянно увеличивается число распознаваемых наследственных болезней и врожденных пороков развития. Таким образом, выполняется одна из главных задач современной медицинской генетики — предупреждение рождения детей с тяжелой наследственной или врожденной патологией.
Гл а в а XII
ЗНАЧЕНИЕ ГЕНЕТИКИ ДЛЯ ТЕОРИИ И ПРАКТИКИ МЕДИЦИНЫ
ГЕННАЯ И КЛЕТОЧНАЯ ИНЖЕНЕРИЯ. БИОТЕХНОЛОГИЯ
Для лечения многих болезней необходимы различные биологически активные вещества. При выделении их из тканей человека возникаем опасность загрязнения полученного материала различными вирусами (гепатита В, иммунодефицита человека и др.). Кроме того, эти вещества производятся в небольших количествах и являются дорогостоящими. Биологически активные вещества животного происхождения низкоэффективны из-за несовместимости с иммунной системой больного человека. Только развитие новой отрасли — генной инженерии помогло обеспечить получение чистых биологически активных веществ в больших количествах по более низкой цепе.
Генная инженерия — это создание гибридных, рекомбинантных молекул ДНК, а стало быть, и организмов с новыми признаками. Для этого необходимо выделить ген из какого-либо организма или искусственно синтезировать его, клонировать (размножить) и перенести в другой организм.
Инструментами генной инженерии являются ферменты: рестриктазы (разрезающие молекулу ДНК) и лигазы (сшивающие ее). В качестве векторов-переносчиков используются вирусы.
С помощью генной инженерии созданы штаммы кишечных палочек, в которые встроены гены человеческого инсулина (необходимого для лечения сахарного диабета), интерферона (противовирусного препарата), соматотропина (гормона роста).
С помощью генной инженерии созданы дрожжевые клетки, продуцирующие человеческий инсулин. Биосинтетический метод производства человеческого инсулина с помощью дрожжевых клеток широко используется в фармацевтическом производстве (в Дании, Югославии, США, Германии и других странах).
В настоящее время ученые разных стран работают над получением с помощью генной инженерии ряда других необходимых биологически активных веществ, вакцины против гепатита В, активатора профибринолизина (противосвертывающий препарат), интерлейкина-2 (иммуномодулятор) и др.
116
В клетки животных чужеродные гены вводят в виде отдельных молекул ДНК или в составе векторов-вирусов, способных вносить в геном клетки чужую ДНК. Обычно применяют два метода: 1) ДНК добавляют в среду инкубации клеток; 2) производят микроинъекции ДНК непосредственно в ядро (что более эффективно).
Первоочередными задачами генной инженерии у человека являются создание банков генов человека для их изучения и поиск путей генотерапии, то есть замены мутантных генов нормальными аллелями.
Клеточная инженерия — это метод конструирования клеток нового типа на основе их культивирования, гибридизации или реконструкции. При гибридизации искусственно объединяются целые клетки (иногда далеких видов) с образованием гибридной клетки. Клеточная реконструкция — это создание жизнеспособной клетки из отдельных фрагментов разных клеток (ядра, цитоплазмы, хромосом и др.).
Изучение гибридных клеток позволяет решать многие проблемы биологии и медицины. Так, например, биотехнология использует гибридомы. Гибри-дома — это клеточный гибрид, получаемый слиянием нормального лимфоцита и опухолевой клетки. Она обладает способностью к синтезу моноклональных (однородных) антител желаемой специфичности (свойство лимфоцита) и к неограниченному росту в искусственной среде (свойство опухолевой клетки).
Биотехнология — это производство продуктов и материалов, необходимых для человека, с помощью биологических объектов.
Термин «биотехнология» получил распространение в середине 70-х годов XX в., хотя отдельные отрасли биотехнологии известны давно и основаны на применении различных микроорганизмов: хлебопечение, виноделие, пивоварение, сыроварение. Достижения генетики создали большие дополнительные возможности для развития биотехнологии.
В середине XX в. и во второй его половине, используя индуцированный мутагенез, были получены антибиотики (пенициллин, стрептомицин, эритромицин и др.) — с помощью микробов; фермент амилаза — с помощью сенной палочки, аминокислоты — с помощью кишечной палочки; молочная кислота — с помощью молочно-кислых бактерий; лимонная кислота — с помощью аспергилловой плесени; витамины группы В — с помощью дрожжей.
В последние десятилетия в результате успехов, достигнутых генной инженерией, происходит скачок в биотехнологии — развивается микробиологическая промышленность (микробиологическая индустрия), которая позволяет в промышленных условиях с помощью кишечной палочки или дрожжей получать человеческий инсулин, интерферон, соматотропин и другие вещества.
ДОСТИЖЕНИЯ ГЕНЕТИКИ В ДИАГНОСТИКЕ И ПРОФИЛАКТИКЕ ЗАБОЛЕВАНИЙ
В настоящее время в Республике Беларусь проводится массовый скрининг новорожденных в роддомах для выявления фенилкетонурии и врожденного
117
гипотиреоза. Данные исследования позволяют поставить диагноз в ранние сроки и своевременно назначить эффективное лечение.
Больших успехов в последнее десятилетие достигла пренатальная диагностика наследственных заболеваний и врожденных пороков развития. Широкое распространение в медицинской практике получили следующие методы: ультразвуковое исследование, амниоцентез, биопсия хориона, хордоцентез, определение альфа-фетопротеина и хориогонина, ДНК-диагностика. При выявлении методами пренатальной диагностики тяжелых наследственных заболеваний или врожденных пороков развития врач рекомендует прерывание беременности, тем самым вносит большой вклад в профилактическую медицину.
Огромный вклад в диагностику хромосомных болезней внесли генетики, внедрив в практику медицины метод дифференциальной окраски хромосом. С помощью этого метода можно определить количественные и структурные перестройки хромосом, которые невозможно выявить при руги иной окраске, и точно поставить диагноз.
Большое теоретическое и практическое значение имеет изучение групп сцепления у человека и построение карт хромосом. В настоящее время у человека относительно изучены все 24 группы сцепления и установлен ле новых локусов генов продолжается.
В последнее время в практическую медицину внедрены достижения клеточной инженерии. С помощью гибридом получают моноклональные антитела, которые широко используются в диагностике заболеваний.
Наиболее распространенным и эффективным методом профилактики наследственных болезней и врожденных пороков развития является медико-генетическое консультирование, направленное на предупреждение появления в семье больных детей. Врач-генетик рассчитывает риск рождения ребенка с тяжелой наследственной патологией и при высоком риске, при отсутствии методов пренатальной диагностики дальнейшее деторождение в данной семье не рекомендуется.
С целью предупреждения рождения детей с наследственно детерминированными болезнями необходимо объяснять вред близкородственных браков молодым людям, планирующим создание семьи.
Беременным женщинам в возрасте старше 35 лет необходимо обследование у врача-генетика для исключения у плода генетической патологии.
Таким образом, применение достижений генетики в практической медицине способствует предупреждению рождения детей с наследственными заболеваниями и врожденными пороками развития, ранней диагностике и лечению больных.
ТЕРМИНОЛОГИЧЕСКИЙ СЛОВАРЬ
Агенезия — дефект развития или отсутствие части тела.
Акроцефалия — высокий «башенный» череп.
Алопеция — полное или частичное выпадение волос.
Аллели — различные формы одного и того же гена.
Аллельные гены — гены, локализованные в гомологичных хромосомах в одинаковых локусах и кодирующие один и тот же признак или его вариации.
Анкилоз — неподвижность суставов.
Ангиоматоз — патологическое состояние сосудов с формированием множественных ангиом.
Анэнцефалия — полное или почти полное отсутствие головного мозга.
Анофтальмия — отсутствие глазных яблок.
Атрезия — отсутствие или смыкание естественного канала или отверстия.
Аплазия — врожденное отсутствие органа.
Арахнодактилия — длинные и тонкие пальцы кистей и стоп (паукообразные).
Аутосомы — все хромосомы клеток за исключением половых.
Брахидактилия — короткопалость.
Витилиго — очаговая депигментация кожи.
Гамета — половая клетка.
Гаплоидный набор хромосом — одинарный набор хромосом, содержащийся в зрелых половых клетках.
Ген — участок молекулы ДНК, кодирующий первичную структуру полипептида.
Генотип — совокупность генов данного организма.
Гетерозигота — организм, несущий два разных аллеля одного гена.
Гирсутизм — аномальное оволосение.
Гипоплазия — врожденное недоразвитие органа, проявляющееся в дефиците массы или размеров органа.
Гипотрихоз — уменьшение количества волос.
Гипертрихоз — избыточный рост волос.
Гипергидроз — избыточное потоотделение.
Гипертелоризм — аномальное расстояние между двумя органами или частями тела (глазной, сосковый).
Гомозигота — организм, в котором пара аллельных генов одинакова.
Долихоцефалия — удлиненная форма черепа.
119
Зигота — оплодотворенная яйцеклетка, образовавшаяся в результате слияния мужской и женской гамет и содержащая диплоидный набор хромосом.
Инбридинг — близкородственный брак.
Клинодактилия — искривление одного или более пальцев кисти или стопы.
Косолапость — деформация стопы, при которой она вывихнута из нормального положения.
Крипторхизм — задержка яичка на его естественном пути при спускании в мошонку.
Кифосколиоз — искривление позвоночника назад и вбок.
Краниостеноз — уменьшение объема черепной коробки, обусловленное преждевременным зарастанием швов.
Локус — место расположения гена в хромосоме.
Макроглоссия — чрезмерное увеличение языка с выраженной складчатостью слизистой оболочки.
Макросомия — увеличение размеров тела.
Микрофтальмия — уменьшение всех размеров глазного яблока.
Микрогнатия — недоразвитие нижней челюсти.
Микроцефалия — аномально уменьшенная голова.
Монголоидный разрез глаз — опущены внутренние углы глазных щелей.
Олигодактилия — отсутствие нескольких пальцев кистей или стоп.
Плейотропия — способность гена оказывать влияние на развитие нескольких признаков.
Полидактилия — увеличение количества пальцев на кистях и стопах.
Пробанд — человек (больной, здоровый), обратившийся за консультацией.
Прогнатия — выступание верхней челюсти вперед по сравнению с нижней вследствие ее чрезмерного развития.
Птеригиум шейный — толстая складка кожи на боковой поверхности шеи, распространяющаяся от сосцевидного отростка к акромиальному.
Сибсы — братья и сестры пробанда.
Синдактилия — сращение двух или более пальцев кисти или стопы.
Синофриз — увеличение и сращение бровей.
Страбизм — косоглазие.
Телеангиэктазии — расширение капиллярных сосудов и мельчайших артерий, формирующих разнообразные ангиомы.
Тетраплегия — паралич верхних и нижних конечностей.
Фенотип — совокупность признаков данного организма (внешних и внутренних).
Фокомелия — прямое прикрепление кистей и стоп к туловищу. Термин применяется также при частичном отсутствии некоторых проксимальных частей конечностей.
Эпикант — полулунная вертикальная складка у внутреннего угла глаза.
Эписпадия — верхняя расщелина уретры.
Энофтальм — аномальное западение глазных яблок.
120
ЛИТЕРАТУРА
Айала Ф., Кайер Дж. Современная генетика: В 3 т. Пер. с англ. М., 1987. — Т. 1.
Бадалян Л. О., Таболин В. А., Вельтищев Ю. Е. Наследственные болезни у детей. М., 1971.
Барашнев Ю. И., Вельтищев Ю. Е. Наследственные болезни обмена веществ у детей. Л., 1987.
Бочков А. П, Захаров А. Ф., Иванов В. П. Медицинская генетика. М., 1984.
Бочков А. П. Генетика человека. М., 1978.
Врожденные и приобретенные энзимопатии / Под ред. Т. Ташева. М., 1980.
Гершензон С. М. Основы современной генетики. Киев, 1983.
Грин Н., Стаут У., Тейлор Д. Биология. М., 1990.
Дубинин Н. П. Генетика. Кишинев, 1985.
Евграфов О. В., Макаров В. Б. ДНК-диагностика наследственных заболеваний // Итоги науки и техники. ВИНИТИ. Сер.: Генетика человека. 1991. — Т. 9.
Клиническая педиатрия / Под ред. Б. Братанова. София. 1987. — Т. 1.
Козлова С. И., Семенова Е., Демикова Н. С., Блинникова О. Е. Наследственные синдромы и медико-генетическое консультирование. Л., 1987.
Ленц В. Медицинская генетика. М., 1984.
Лильин Е. Т, Богомазов Е. А., Гофман-Кадошников П. Б. Генетика для врачей. М., 1990.
Лобашов М. Е. Генетика. Л., 1969.
Льюин Б. Гены. М., 1987.’
Никитин Ю. П, Лисиченко О. В., Коробкова Е. Н. Клинико-генеалогический метод в медицинской генетике. Новосибирск, 1983.
Перспективы медицинской генетики / Под ред. Н. П. Бочкова. М., 1982.
Попеску О. Синдромы в педиатрии. Бухарест, 1977.
Рыбчик В. Н. Основы генетической инженерии. Мн., 1986.
Тератология человека. Руководство для врачей / Под ред. Г. И. Лазюка. М., 1991.
Фогель Ф., Мотульский А. Генетика человека (История. Хромосомы человека. Формальная генетика): В 3 т. Пер. с англ. М., 1989. — Т. 1; 1990. — Т. 2, Т. 3.
Цветкова И. В. Пренатальная диагностика наследственных дефектов обмена // Итоги науки и техники. ВИНИТИ. Сер.: Генетика человека, 1991. — Т. 9.
Штерн К Основы генетики человека. М., 1965.
ОГЛАВЛЕНИЕ
Введение.......................................................................3
Глава I. КРАТКАЯ ИСТОРИЯ МЕДИЦИНСКОЙ ГЕНЕТИКИ. В. А. Орехова 5
Глава II. КЛЕТКА — ОСНОВНАЯ ГЕНЕТИЧЕСКАЯ И СТРУКТУРНО-
ФУНКЦИОНАЛЬНАЯ БИОЛОГИЧЕСКАЯ ЕДИНИЦА. Я. А. Орехова...................... 10
Основные компоненты эукариотической клетки................................ 12
Биохимические основы наследственности. Доказательство генетической роли нуклеиновых кислот.................................................... 16
Строение молекул ДНК и РНК................................................ 17
Программирование синтеза белка в клетке....................................20
Глава III. ЦИТОЛОГИЧЕСКИЕ ОСНОВЫ НАСЛЕДСТВЕННОСТИ. В .А. Орехова.................23
Строение и типы метафазных хромосом человека. Понятие о кариотипе..........23
Современные методы цитологического анализа хромосом. Понятие о гетеро- и эухроматинс, половом хроматине......................................................25
Основные типы деления эукариотических клеток. Клеточный цикл и его периоды.27
Половые клетки. Развитие сперматозоидов и яйцеклеток.......................32
Глава IV. ЗАКОНОМЕРНОСТИ НАСЛЕДОВАНИЯ ПРИЗНАКОВ. В. А. Орехова...................36
Основные понятия и термины в современной генетике. ........................36
Закономерности наследования признаков, установленные Г. Менделем...........37
Менделирующие признаки человека............................................40
Типы наследования менделирующих признаков..................................41
Хромосомная теория наследственности Т. Моргана.............................45
Линейное расположение генов. Карты хромосом человека.......................48
Основные положения хромосомной теории наследственности.....................49
Генетика пола..............................................................50
Глава V. ВЗАИМОДЕЙСТВИЕ ГЕНОВ. В. А. Орехова...............................52
Взаимодействие аллельных генов.............................................52
Взаимодействие иеаллельных генов...........................................53
Влияние генотипической среды и факторов внешней среды на проявление признаков.56
Глава VI. ИЗМЕНЧИВОСТЬ. В. А. Орехова.........................................58
Модификационная изменчивость...............................................58
Комбинативиая изменчивость.................................................59
Мутационная изменчивость...................................................59
122
Классификация мутации.......................................................60
Репарация молекулы ДНК......................................................62
Глава VII. МЕТОДЫ ИЗУЧЕНИЯ НАСЛЕДСТВЕННОСТИ ЧЕЛОВЕКА. В. А. Орехова...........................................................66
Особенности изучения наследственности человека.ж............................66
Основные методы изучения наследственности человека..........................66
Глава VIII. НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ И ИХ КЛАССИФИКАЦИЯ. М. П. Шейбак............................................................71
Хромосомные болезни.........................................................73
Синдромы с числовыми аномалиями половых хромосом............................74
Синдромы с числовыми аномалиями аутосом.....................................76
Глава IX. ГЕННЫЕ БОЛЕЗНИ. М. П. Шейбак.........................................80
Заболевания, связанные с расстройством аминокислотного обмена...............81
Заболевания, связанные с нарушением обмена углеводов........................82
Наследственные заболевания, связанные с нарушением липидного обмена.........84
Наследственные заболевания обмена стероидов.................................86
Наследственные нарушения биосинтеза тиреоидных гормонов.....................87
Наследственные болезни соединительной ткани.................................88
Наследственные нарушения обмена в эритроцитах...............................91
Наследственные синдромы нарушенного всасывания..............................91
Основные принципы ухода за больными с наследственными заболеваниями.........93
Главах. КЛИНИКО-ГЕНЕАЛОГИЧЕСКИЙ МЕТОД ИЗУЧЕНИЯ НАСЛЕДСТВЕННОСТИ ЧЕЛОВЕКА. Т. А. Лашковская.............................96
Порядок сбора генеалогической информации и методика составления родословной.96
Графическос изображение родословной.........................................97
Аутосомно-доминантный тип наследования.................................... 99
Аутосомно-рецессивный тип наследования.................................... 100
Х-сцепленный тип наследования............................................. 102
Мультифакториальное наследование.......................................... 104
ГлаваХ!. МЕДИКО-ГЕНЕТИЧЕСКОЕ КОНСУЛЬТИРОВАНИЕ. Т. А. Лашковская............108
Задачи и организация медико-генетического консультирования................ 108
Основные принципы консультирования........................................ 109
Этапы консультирования.................................................. 110
Современные методы пренатальной диагностики врожденных пороков развития и наследственных болезней.................................................. 112
Глава XII. ЗНАЧЕНИЕ ГЕНЕТИКИ ДЛЯ ТЕОРИИ И ПРАКТИКИ МЕДИЦИНЫ. В. А. Орехова......................................................... 116
Генная и клеточная инженерия. Биотехнология............................... 116
Достижения генетики в диагностике и профилактике заболеваний.......*.......117
Терминологический словарь.....................................................119
Литература.................................................................. 121
123
Учебное издание
Орехова Вера Александровна Лашковская Татьяна Алексеевна Шейбак Михаил Петрович
МЕДИЦИНСКАЯ ГЕНЕТИКА
Редактор В. В. Такушевич
Художник обложки и художественный редактор В. А. Ярошевич
Технический редактор И. П. Тихонова Компьютерная верстка Л. И. Счисленок Корректоры Т К. Хваль, Н. И. Бондаренко
Подписано в печать с оригинала-макета издательства “Вышэйшая школа” 16.06.97 г. Формат 60x90/16. Бумага газетная. Гарнитура Тип Таймс. Офсетная печать.
Усл.печ. л. 7,75. Усл. кр.-отт. 8,25. Уч.-изд.л. 8,17. Тираж 5000. Заказ 5285.
Государственное предприятие издательство “Вышэйшая школа”, Государственного комитета Республики Беларусь по печати. Лицензия ЛВ № 5. 220048, г. Минск, проспект Машерова, 11.
Типография “Победа”. 222310, г. Молодечно, ул. Тавлая,!.