/

















































































































































































































































































































































































































































































































































































































































































































Текст
Б. А. ПАВЛОВ и А. П. ТЕРЕНТЬЕВ
КУРС
ОРГАНИЧЕСКОЙ ХИМИИ
попущено Министерством
высшего и среднего специального образования СССР
в качестве учебника для химических техникумов
ИЗДАНИЕ ПЯТОЕ, ПЕРЕРАБОТАННОЕ
ИЗДАТЕЛЬСТВО «ХИМИЯ»
Москва ¦ 1965 • Ленинград
УДК 547@75.3)
П 12
Книга является переработанным и дополненным изданием
известного учебника органической химии Б А. Павлвва и
А. П. Терентьева. В пятое издание включены новые материалы:
о свободных радикалах, хелатных соединениях, органических
перекисях, иенообменных смолах и хроматографии, ферроценах,
нуклеиновых кислотах, стероидных гормонах и др. Обновлены
и переработаны параграфы, касающиеся высокомолекулярных
соединений и пластических масс, органических красителей,
ядохимикатов, номенклатуры органических соединений и др.
Книга предназначена для учащихся химических техникумов,
но может быть использована также студентами нехимических
вузов и втузов. Книга может служить пособием для учащихся
заочных учебных заведений и для самообразования
СОДЕРЖАНИЕ
Предисловие
Введение
1. Органическая химия
1. Предмет и характерные черты органической химии ...
2. Органическая химия и промышленность органического
синтеза
II Работа с органическими веществами
3. Выделение и очистка органических веществ
4. Определение физических констант
5. Состав органических веществ. Качественный анализ ...
6. Количественный анализ
7. Установление простейшей формулы
8. Определение молекулярного веса
III. Теория химического строения
9. Теория радикалов и теория типов
10. Теория химического строения Бутлерова
11. Классификация органических соединений
Часть первая. Соединения с открытой цепью углеродных
атомов
Глава I. Углеводороды
Предельные углеводороды
12. Гомологический ряд предельных углеводородов
13. Углеводородные радикалы (алкилы) ,
14. Строение предельных углеводородов
15. Нахождение в природе и получение предельных
углеводородов
16. Физические свойства предельных углеводородов ....
17. Химические свойства предельных углеводородов ....
18. Метан
19. Общие черты реакций замещения
20. Женевская номенклатура органических соединений ...
21. Электронные схемы строения органических соединений .
22. Дипольные моменты
Нефть и природные газы
23. Состав нефти и нахождение ее в природе ........
24. Происхождение нефти
25. Продукты переработки нефти и их применение
26. Искусственное жидкое топливо
27. Природные газы, Асфальты .
Содержание
Непредельные углеводороды 71
Углеводороды с одной двойной связью. Алкены (олефины) ... 72
28. Двойная связь 72
29. Номенклатура олефинов 74
30. Способы получения и строение олефинов 75
31. Физические и химические свойства олефинов 76
32. Показатель преломления. Молекулярная рефракция . . 84
33. Этилен 87
34. Изобутилен 89
Углеводороды с тройной связью (алкины) 90
35. Строение ацетиленовых углеводородов и их получение . 90
36. Свойства ацетиленовых углеводородов 91
37. Ацетилен 93
Углеводороды с двумя двойными связями. Каучук 95
38. Строение и свойства углеводородов с двумя двойными
связями 95
39. Каучук и его свойства 99
40. Строение и синтез каучука 100
Глава II. Галоидпроизводные углеводородов 106
41. Галоидные алкилы. Изомерия и способы получения . . . 106
42. Свойства галоидных алкилов 108
43. Дигалоидные производные предельных углеводородов . . 111
44. Полигалоидные производные предельных углеводородов 113
45. Фторпроизводные предельных углеводородов ...... 113
46. Галоидпроизводные непредельных углеводородов .... 114
47. Высокомолекулярные соединения. Пластические массы 115
Глава II/. Элементорганические соединения 121
48. Элементорганические соединения и их значение 121
49. Цинкорганические соединения 123
50. Магнийорганические соединения 124
51. Алюминийорганические соединения 125
52. Кремнийорганические соединения 125
53. Фосфор- и мышьякорганические соединения 128
Глава IV. Спирты и их производные 132
Одноатомные предельные спирты 132
54. Строение спиртов 132
55. Гомология и изомерия спиртов 133
56. Способы получения одноатомных спиртов 135
57. Физические свойства спиртов 137
58. Химические свойства спиртов 140
59. Главнейшие представители спиртов Метиловый спирт (ме
та нол) , 144
60. Этиловый спирт (этанол) 148
61. Пропиловые, бутиловые и амиловые спирты 150
62. Оптическая деятельность органических соединений ... 154
63. Стереохимическая теория 157
64. Электронное строение ординарных связей (а-связь) ... 162
Непредельные спирты 165
65. Аллиловый спирт 165
66.' Синтетические пластические материалы на основе
полимеров винилового спирта и его производных . . •. . 167
Содержание
Многоатомные спирты 168
Двухатомные спирты, или гликоли 168
67. Строение, изомерия и номенклатура гликолей 168
68. Физические и химические свойства гликолей 169
Трехатомные спирты, или глицерины 171
69. Глицерин 171
70. Спирты высшей атомности 172
Эфиры 172
Сложные эфиры неорганических кислот 173
71. Получение и свойства сложных эфиров неорганических
кислот 173
72. Нитроглицерин 175
73. Эфиры ортокремневой кислоты 176
Простые эфиры 177
74. Строение и способы получения простых эфиров 177
75. Свойства простых эфиров 178
76. Этиловый эфир 179
77. Органические перекисные соединения 180
Глава V. Меркаптаны, сульфокислоти, тиоэфиры 182
78. Меркаптаны 182
79. Сульфокислоты 183
80. Тиоэфиры 183
Глава VI. Альдегиды и кетоны 185
81. Строение, изомерия и номенклатура 185
82. Получение и некоторые свойства альдегидов 187
83. Получение и некоторые свойства кетонов 189
Реакции альдегидов и кетонов 190
84. Реакции присоединения 190
85. Реакции замещения 194
86. Окисление альдегидов и кетонов 196
87. Реакции с участием водородного атома в а-положении к
карбонильной группе 200
88. Сравнение свойств альдегидов и кетонов 202
Отдельные представители альдегидов и кетонов 204
89. Муравьиный альдегид 204
90. Уксусный альдегид 207
91. Хлораль B,2,2-трихлорэтаналь) 20J
92. Акролеин 210
93. Цитраль 212
94. Ацетон 212
95. Диальдегиды и дикетоны 215
96. Хелатные соединения 217
Глава VII. Карбоновые кислоты и их производные 219
Предельные одноосновные кислоты 219
97. Строение и способы получения карбоновых кислот . . . 219
98. Изомерия и номенклатура предельных одноосновных
кислот 221
99. Физические свойства предельных одноосновных кислот . . 223
100. Химические свойства предельных одноосновных кислот '226
в Содержание
101. Муравьиная кислота . 229
102. Уксусная кислота 230
103. Пропионовая кислота 233
104. Масляная кислота 233
105. Пальмитиновая и стеариновая кислоты 234
106. Мыла н моющие вещества 234
Непредельные одноосновные кислоты 237
107. Гомологический ряд непредельных одноосновных кислот 237
108. Акриловая, метакрнловая и кротоновые кислоты .... 237
109. Цнс-транс-нзомерня 240
ПО. Теория напряжения 243
111. Электронное строение двойных связей (л-связн) .... 243
112. Сопряженные кратные связи. Конъюгены 247
113. Олеиновая, линолевая и лнноленовая кислоты 249
114. Синтетические материалы на основе полимеров
производных акриловой и метакрнловой кислот 251
Сложные эфнры карбоновых кислот 252
115. Строение н способы получения сложных эфиров .... 252
116. Этернфнкацня 252
117. Свойства сложных эфнров 254
118. Отдельные представители сложных эфиров карбоновых
кислот 256
119. Воска 257
120. Жиры 257
121. Расщепление жиров 258
122. Стеариновое производство н мыловарение 259
123. Раст-ительные масла ' . . . . 260
124. Гидрогенизация жиров 262
125. Синтетические жирные кислоты 263
Хлорангндриды кислот 264
126. Строение и получение хлорангндридов кислот 264
127. Свойства хлорангндридов кислот 265
Ангидриды кислот 266
128. Строение и получение ангидридов кислот 266
129. Свойства ангидридов кислот 267
Амиды кислот 267
130. Строение н получение амидов кислот 267
131. Свойства амидов кислот 268
132. Производные кислот н кислотный радикал 269
Предельные двухосновные кислоты 270
133. Строение предельных двухосновных кислот 270
134. Физические и химические свойства двухосновных
предельных кислот 270
135. Щавелевая кислота 272
136. Малоновая кислота 273
137. Синтезы при помощи малонового эфира 275
138. Янтарная кислота 277
Непредельные двухосновные кислоты 277
139. Фумаровая н малеиновая кислоты 277
Глава VIII. Галоидзамещенные кислоты 280
140. Строение и способы получения галоидзамещенных кислот 280
141. Свойства галоидзамещенных кислот 281
Содержание
Глава ЇХ. Оксикислоти 282
142. Способы получения и строение оксикислот 282
143. Окисление многоатомных спиртов 284
144. Свойства оксикислот 285
145. Молочная кислота 286
146. Яблочная кислота 290
147. Винные кислоты 290
148. Расщепление рацемических веществ на оптические
антиподы 295
149. Асимметрический синтез 297
150. Стереоизомерия веществ с двумя или несколькими
асимметрическими атомами углерода в молекуле. Диастерео-
меры 299
151. Лимонная кислота 301
152. Стереоизомерия а-окснкислот 302
Глава Х- Альдегидокислоты и кетонокислоти 305
153. Строение и отдельные представители альдегидо- и
кетонокислот 305
154. Ацетоуксусный эфир 306
155. Таутомерия 310
156. Синтезы при помощи ацетоуксусного эфира 311
Глава XL Углеводы 314
157. Классификация углеводов 314
Моносахариды 315
158. Строение моносахаридов 315
159. Понятие о стереоизомерии альдогексоз 319
160. Нахождение в природе и способы получения
моносахаридов 323
161. Свойства моносахаридов 326
162. Триозы 329
163. Пентозы 330
164. Гексозы 332
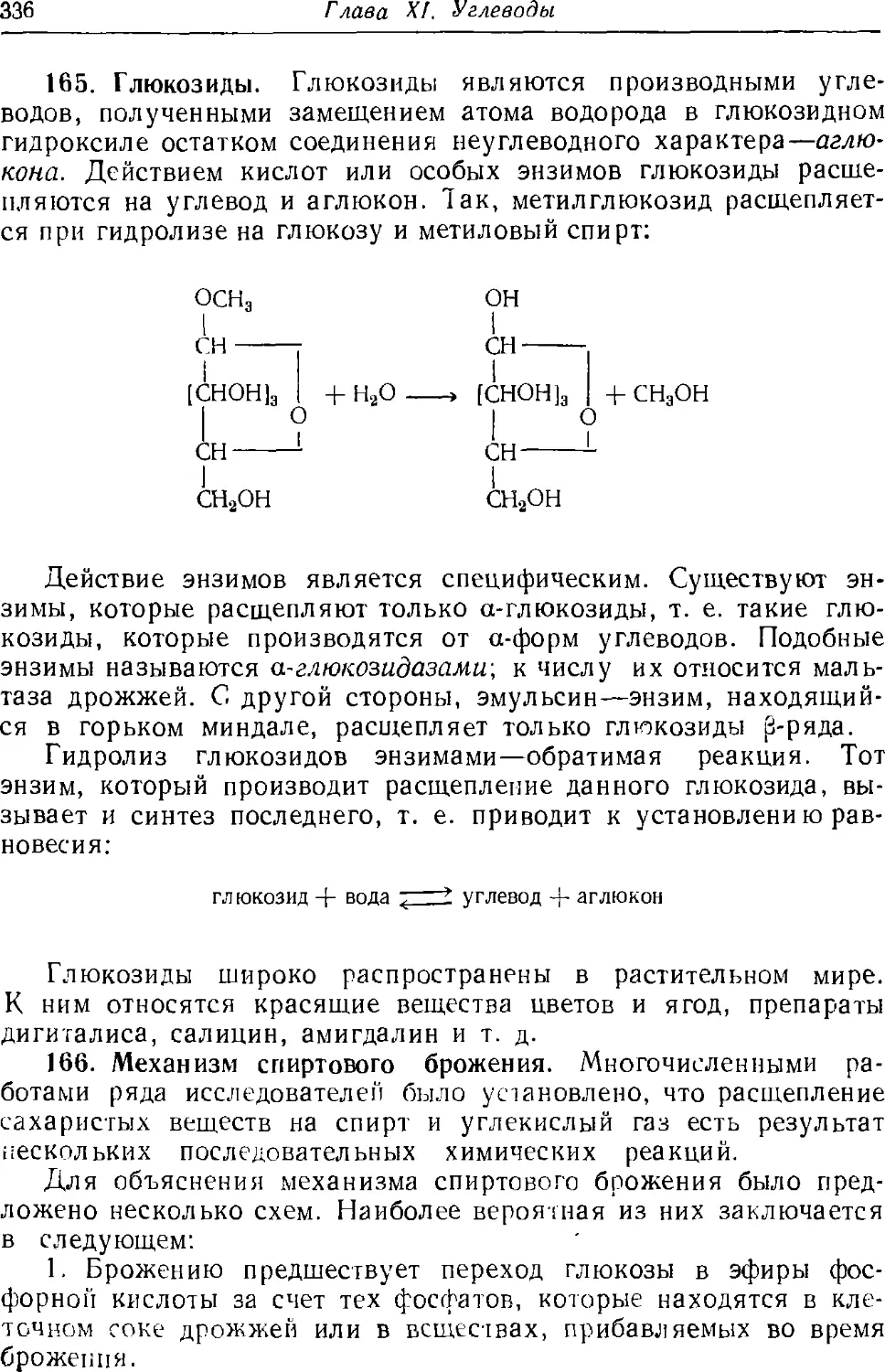
165. Глюкозиды 336
. 166 Механизм спиртового брожения 336
Низкомолекулярные полисахариды (олигосахариды) 338
167. Свойства и CTpoeHHt дисахаридов 338
168. Сахароза 342
169. Мальтоза. Молочный сахар. Целлобиоза 344
Высокомолекулярные полисахариды 344
170. Крахмал 344
171. Декстрины 345
172. Гликоген. Инулин 346
173. Целлюлоза 346
174. Сложные и простые эфиры целлюлозы 346
175. Искусственное волокно 352
176. Гемицеллюлозы и пектиновые вещества 354
Глава XII. Нитросоедииения 356
177. Строение и способы получения нитросоединений .... 356
178. Свойства нитросоедннений 358
Глава ХИІ. Амины 361
179. Строение аминов 361
180. Свойства аминов 361
181. Строение аммонийных солей 364
8 Содержание
182. Получение аминов 365
183. Отдельные представители аминов 367
Глава XIV. Амииоспирты и аминокислоты 370
184. Аминоспирты 370
185. Строение, способы получения и свойства аминокислот . 373
186. Отдельные представители аминокислот 377
187. Обозначение стереоизомерных молекул, содержащих амино-
и оксигруппы 384
Глава XV. Белковые вещества 387
188. Белки в природе 387
189. Свойства белков 388
190. Собственно белки 390
191. Протеиды 391
192. Понятие о химическом строении белков 393
193. О синтезе белков 396
194. Синтетическое волокно 397
195 Работы с веществами, содержащими меченые атомы . . . 398
Глава XVI. Соединения циана 401
196. Цианистые соединения и источники их получения . . . 401
197. Нитрилы. Получение и свойства 402
198. Изонитрилы 404
199. Синильная кислота 405
200. Циан 406
201. Цианамид 406
202. Циановая кислота '. . . . 407
203. Тиоциановая, или роданистоводородная, кислота . . . 408
Глава. XVII. Производные угольной кислоты 411
204. Хлорангидриды угольной кислоты 411
205. Сложные эфиры угольной кислоты 413
206. Производные угольной кислоты, содержащие азот . . . 413
207. Сернистые аналоги производных угольной кислоты . . 418
208. Пластмассы на основе мочевино-формальдегидных (карб-
амидных) смол 420
Часть вторая. Карбоциклические соединения
СОЕДИНЕНИЯ АРОМАТИЧЕСКОГО РЯДА
Глава X VIII. Одноядерные ароматические соединения 422
Бензол и его гомологи 422
209. Свойства бензола 422
210. Строение бензола 425
211. Двузамещенные и трехзамещенные производные бензола.
Изомерия 429
212. Строение н свойства гомологов бензола 430
213. Способы получения гомологов бензола 432
214. Промышленное получение ароматических соединений . 434
Галоидпроизводные бензола и его гомологов 437
215. Получение галоидпроизводных бензольных углеводородов 437
216. Свойства галоидпроизводных бензольных углеводородов 439
217. Некоторые представители галоидпроизводных бензольных
углеводородов 440
Нитросоединения и сульфокислота 440
218. Нитрование 440
219. Направляющее действие заместителей 441
Содержание
220. Свойства нитросоединений 446
221. Отдельные представители нитросоединений 447
222. Сульфирование, сульфокислоты 448
Фенолы и ароматические спирты. Хиноны 448
223. Строение, способы получения и свойства фенолов .... 448
224. Фенол. Крезолы 452
225. Регуляторы роста растений и гербициды 453
226. Феноло формальдегидные смолы 454
227. Пикриновая кислота 457
228. Двухатомные фенолы 458
229. Хиноны 460
230. Грехатомные фенолы 461
231. Ароматические эфиры 463
232. Ароматические спирты 464
Ароматические альдегиды и кетоны 464
233. Альдегиды 464
234. Кетоны 469
Карбоновые кислоты . 470
235. Бензойная кислота 470
236. Сахарин 472
237. Фталевые кислоты 473
238. Полиэфирные смолы 474
239« Фталеины 477
Оксикислоты 479
240. Салициловая кислота 479
241. Галловая кислота 482
242. Дубильные вещества 483
Кислоты с карбоксилом в боковой цепи 484
243. Фенилуксусная кислота 484
244. Миндальная кислота 484
245. ?-Фенилпропионовая кислота 484
246. Коричная кислота 485
Ароматические амины 487
247. Способы получения и свойства первичных ароматических
аминов 487
248. Анилин и его производные 490
249. Вторичные и третичные амины 493
250. Диамины' 495
Аминофенолы и аминобензойные кислоты 497
251. Аминофенолы 497
252. Ионообменные смолы 499
253. Аминобензойные кислоты 501
254. Фенилаланин 503
Диазосоеди нения 503
255. Получение и строение лиазосоединений 503
256. Реакции диазосоединений, проходящие с выделением азота 505
257. Синтезы при помощи диазосоединений 506
258. Восстановление диазосоединений. Гидразины 506
Азосоединенпя. Азокрасители 507
259. Азосоединения 507
260. Аминоазосоединення, оксиазосоединения 509
261. Получение азокрасителей . . 510
262. Красители и крашение . . 512
263. Природные и синтетические красители 515
10 Содержание
Глава XIX. Многоядерные ароматические соединения 518
Соединения, содержащие бензольные ядра, связанные через углерод
или непосредственно 518
264. Днфенил. Бензидин 518
265. Трнфенилметан и его производные 520
266. Красители ряда трифенилметана Строение и свойства . 522
267. Получение красителей ряда трифенилметана 524
268. Свободные радикалы 525
Соединения с конденсированными ядрами 527
269. Нафталин 527
270. Производные нафталина 530
271. Доказательство строения нафталина 534
272. Красители ряда нафталина 535
273. Тетралин. Декалин 536
274. Антрацен 536
275. Антрахинон . , 537
276. Ализарин 539
277. Кубовые полициклические красители (индантрены) . . 541
278. Фенантрен 542
279. Углеводороды со многими конденсированными
бензольными ядрами 543
АЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Глава XX- Циклопарафины 545
280. Строение и способы получения циклопарафинов .... 545
281. Свойства циклопарафинов ... . 548
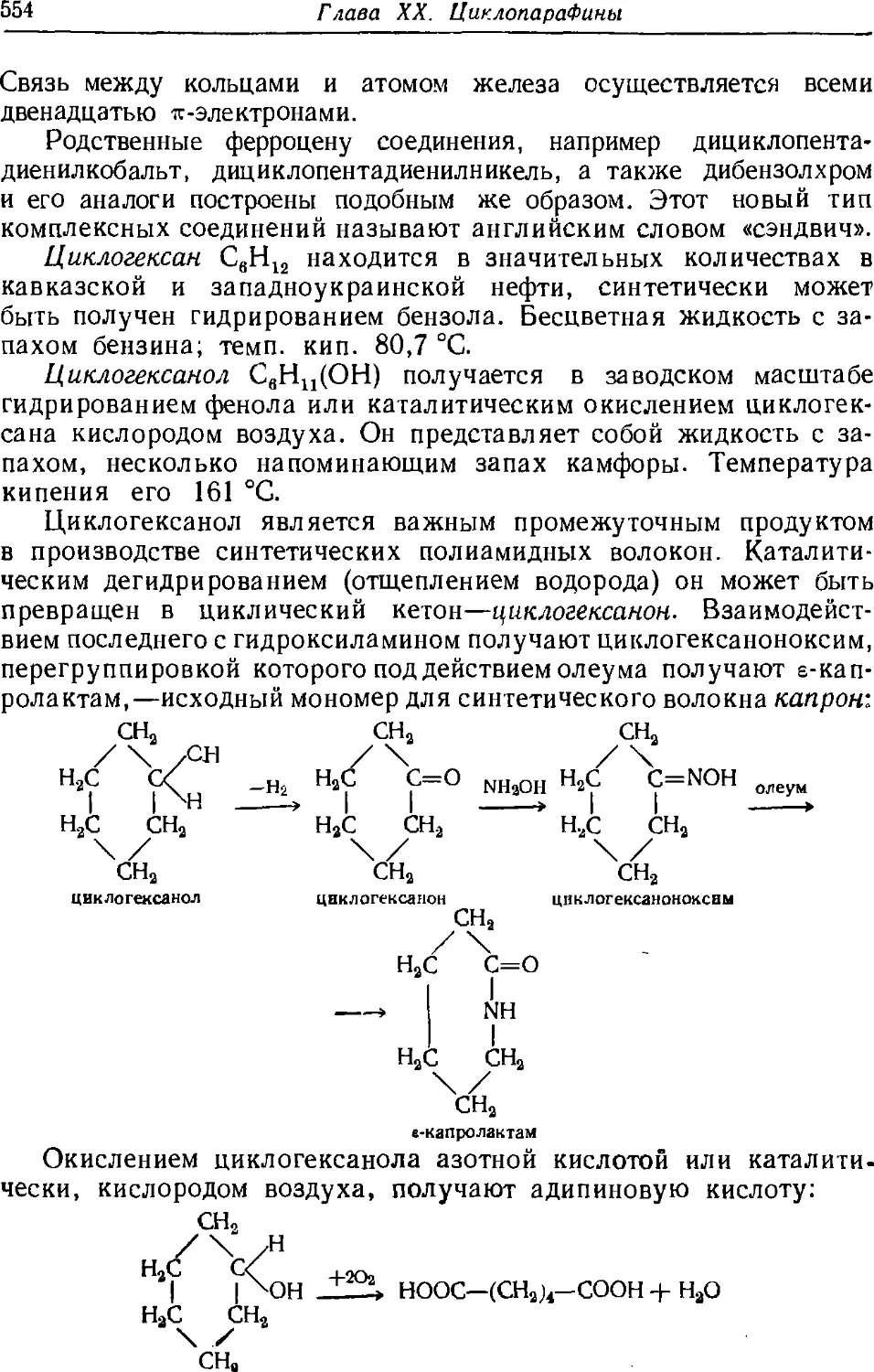
282. Отдельные представители циклопарафинов 549
283. Напряжение в алициклических соединениях 555
284. Стереоизомерия алициклических соединений .... 557
Глава XXI. Терпены 560
285. Терпены в природе 560
286. Классификация терпенон 561
287. Моноциклические терпены 561
288. Бициклические терпены 564
289. Камфора 566
Глава XXII- Каротиноиды 568
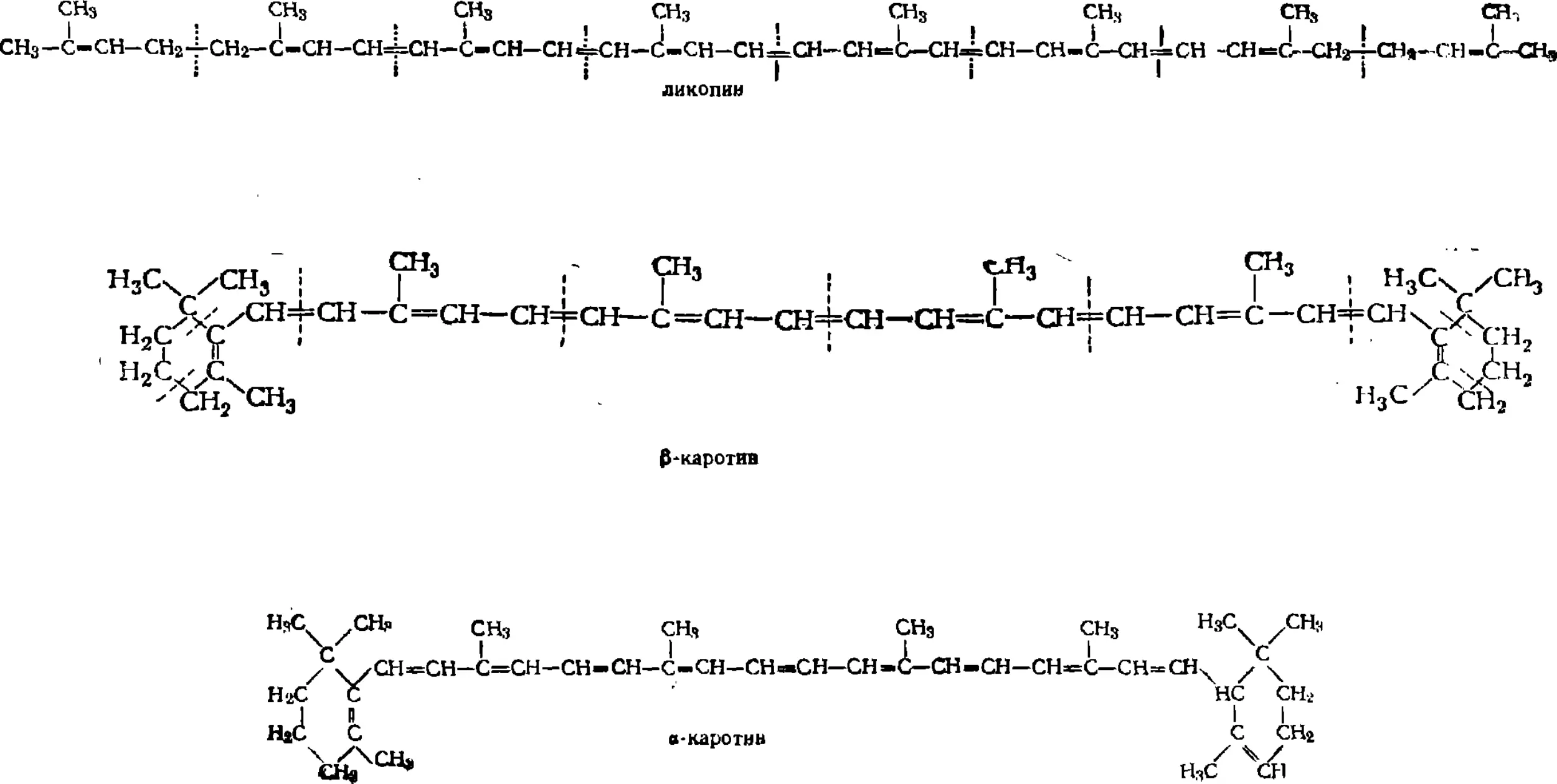
290. Ликопин 568
291. Каротин 568
292. Витамин А .' 570
Глава XXIII. Стерины. Желчные кислоты. Стероидные гормоны .... 571
293. Стерины 571
294. Желчные кислоты 572
295. Стероидные гормоны 574
Часть третья. Гетероциклические соединения
Классификация и общая характеристика 576
296. Классификация и общая характеристика
гетероциклических соединений . 57&
Глава XXIV. Пятичленные гетероциклы 579
297. Общая характеристика пятичленных гетероциклов . . , 579
Содержание
Группа фурана 5SQ
298. Фуран 5^)
299. Фурфурол 5f2
300. Пирослизевая кислота 582
Группа тиофена 583
301. Тиофен 583
Группа пиррола 585
302. Пиррол 5O6
303. Пирролидин 587
304. Гемин и хлорофилл 5j$?
305. Хроматографический анализ 5§0
Группа индола 594
306. Индол 594
307 Триптофан . 596
308. Гетероауксин 5j*6
309 Индиго 597
Группа азолов 60fl
310. Сульфатиазол 900
311. Пенициллин бф
312. Антипирин и пирамидон 602
313. Пилокарпин 603
Глава XXV. Шестичленные гетероциклы 605
Группа пирана . 605
314. у-Пирон и соли пироксония 606
315. Хромон 608
316. Антоцианы 608
Группа пиридина 610
317. Пиридин 610
318. Никотин 613
319. Пиперидин 613
Группа хинолина 614
320. Хинолин 614
321. Хинин и антималярийные препараты 616
Группа пурина ¦ 618
322. Мочевая кислота 618
323. Некоторые другие вещества группы пурина . .... 620
324. Триазин. Цианурхлорид. Селективные гербициды , . . 622
325. Активные красители. Оптически отбеливающие вещества 624
326. Нуклеиновые кислоты, нуклеотиды. РНК и ДНК . . . 686
Заключение 630
Приложение I. Номенклатура органических соединений 633
I «Рациональная» номенклатура 634
2. Женевская номенклатура 635
3 Номенклатура международного союза чистой и прикладной
химии (ЮПАК) 646
4 Принципы систематической номенклатуры 654
Приложение U. Названия часто встречающихся радикалов и
циклических структур 658
Именной указатель 663
Предметный указатель 666
ПРЕДИСЛОВИЕ
Первоначальное изучение курса органической химии, как
правило, труднее курса неорганической химии, основы которого
в какой-то мере закладываются еще в средней школе. И это не
случайно. Неорганическая химия имеет дело с веществами,
различающимися по качественному составу, и с молекулами,
состоящими из немногих атомов. Неорганические вещества в
большинстве случаев растворимы в воде (или гидрофильны), а их реакции
идут с большими скоростями и по ионным механизмам В
органической химии учащийся сразу же сталкивается главным образом
с гомеополярними связями и с химическими процессами,
протекающими по сложным и не всегда ясным механизмам/с малыми
скоростями и в разнообразных условиях. Учащийся встречается
с непривычной классификацией соединений и реакций, с большим
числом новых понятий и громадным числом новых терминов и
названий веществ. Поэтому при изучении начального курса
органической химии решающее значение имеет методически
правильное расположение изучаемого материала, равномерное
внесение нового и систематическое повторение пройденного на
различных примерах.
В настоящем учебнике изучение новых для читателя общих
теорий органической химии (гомология, изомерия и т. д.) и
свойств гомеополярных соединений начинается на
сравнительно простом материале ациклических соединений. Основы
теории химического строения—вот что должно быть усвоено
учащимися прочно и систематически. Этого преподаватель должен
настойчиво добиваться. Главное на первых шагах—понимание и
усвоение структурных формул и их частей (радикалов и
функциональных групп), гомологических рядов и их закономерностей,
а также взаимных переходов соединений различных классов.
Изучение номенклатуры не имеет самостоятельного значения,
а служит в первую очередь основной цели. Именно поэтому в
учебнике в основном излагается женевская номенклатура, тесно
связанная с теорией строения. Вынесение номенклатуры в
приложение не означает, что ее надо проходить в конце курса.
Предисловие 13
Стереохимические представления, значение которых в
последние годы так возросло, вводятся постепенно как развитие теории
химического строения. С целью развития конкретных
пространственных представлений в необходимых случаях введено
изложение с помощью «ро—сигма» системы, обозначения которой
связаны не со схематической проекционной формулой, а с
объемной моделью.
Мы не считаем возможным и полезным перегружать
изложение электронными представлениями. Очень важно, чтобы у
начинающих не создавалось ложного мнения, будто бы электронное
строение органических соединений может само по себе объяснить
все их реакции. Это заблуждение тем более опасно, что уводит
изучающего от изучения конкретных условий реакций и создает
иллюзию того, что все свойства и реакции уже объяснены.
В химии карбоциклических соединений основное внимание
уделено ароматическим соединениям и характерным для них
реакциям злекірофильного замещения. В разделе
гетероциклических соединений охарактеризованы их классы и отдельные
наиболее важные представители. Здесь необходимо обратить
внимание изучающих на то, как меняется химический характер
соединений при переходе от алифатических систем к системам,
полностью сопряженным и ароматизированным.
При изучении практического применения органических
реакций и веществ в технике и быту особенно важно вызывать
полезные ассоциации, привлекая в качестве примеров факты, хорошо
знакомые из повседневной практики. Необходимо, чтобы у
учащихся развилось правильное представление о взаимной
зависимости теоретической науки и проблем производства и
техники.
В основу этого учебника при его первом издании было
положено второе издание книги «Курс органической химии» Б. А.
Павлова, вышедшей в 1951 г. под редакцией и с дополнениями проф.
А. П. Терентьева. При последующем переиздании вносились
различные исправления и значительные дополнения.
Настоящее, пятое издание книги существенно переработано
и дополнено сравнительно с изданием 1961 г. Включены новые
материалы о свободных радикалах, хелатных соединениях,
органических перекисях, ионообменных смолах и хроматографии,
стероидных гормонах, нуклеиновых кислотах и др. Из учебника
исключены устаревшие и маловажные сведения. Существенно
переработаны параграфы, касающиеся высокомолекулярных
соединений, органических красителей, ядохимикатов и др.
При подготовке настоящего, 5-го издания, так же как и при
подготовке двух предыдущих изданий, неоценимую помощь
автору оказала Е. Г. Рухадзе. Ею просмотрен весь текст, переработан
14 Предисловие
и заново написан ряд параграфов, за что приношу ей мою
искреннюю благодарность.
В заключение я должен отметить очень большую работу,
выполненную О. В. Корсунским, обновившим и существенно
дополнившим ряд параграфов, касающихся методов промышленного
производства и применения органических веществ, пластических
масс, каучуков и др. С большой благодарностью я должен
отметить помощь, оказанную им при просмотре всего курса,
позволившую устранить ряд неточных и устаревших положений.
Член-корр. Академии наук СССР,
проф. А. ТЕРЕНТЬЕВ
ВВЕДЕНИЕ
1. ОРГАНИЧЕСКАЯ ХИМИЯ
1. Предмет и характерные черты органической химии. В
начале XIX века название «органическая химия» относилось к
химии веществ, из которых состоят растительные и животные
организмы. Изучение процессов горения (Лавуазье, 1743—1794 г.)
позволило разработать методы элементарного анализа этих
органических веществ. Оказалось, что многочисленные и
разнообразные органические вещества состоят из немногих элементов, из
которых всегда присутствуют углерод и водород и наиболее часто
встречаются кислород и азот.
Из простейших исходных неорганических веществ живые
организмы создают сложнейшие продукты: сахара, жиры, клеї
чатку, крахмал, белковые вещества и др.
Химия юго времени не владела методами получения
органических веществ. Отсюда создалось убеждение, что в живой
природе действуют особые законы, обусловленные нахождением в
живых существах так называемой «жизненной силы»—vis vitalis.
«В живой природе элементы повинуются иным законам, чем
в безжизненной»,—писал в 1827 году знаменитый шведский
химик. Я- Берцелиус A779—1848 г.); поэтому он определил
органическую химию как «химию растительных и животных веществ
или веществ, образующихся под влиянием жизненной силы».
В 1811 году петербургский химик К. С. Кирхгоф A764—
1833 г.) показал, что при нагревании крахмала с небольшим
количеством разбавленной серной кислоты можно осахаривать большие
количества крахмала. Действуя на крахмал малыми
количествами солода, Кирхгоф получил солодовый сахар. Такие процессы,
в которых химические реакции совершаются в присутствии
малых количеств некоторых веществ, не входящих в состав
конечных продуктов реакции, Берцелиус предложил A830) называть
каталитическими.
Жизненная сила, по мнению Берцелиуса, в живой природе
действует при помощи каталитической силы (от гоеческого слова
катализ—развязывание, разрешение).
16 Введение
Таким образом, с точки зрения «виталистического учения»
органическую химию следовало выделить в самостоятельный
раздел химии, в котором рассматриваются процессы,
совершающиеся под действием каких-то особых таинственных
непознаваемых сил. Действуя различными химическими реагентами
на органические вещества природного происхождения, химики
стали получать многочисленные продукты, уже не
встречающиеся в природе.
Эти новые искусственные вещества, однако, по составу и
характеру были подобны природным. Поэтому их также пришлось
называть органическими. Так возникла и развивалась
органическая химия, основанная на синтезе. В 1828 году немецкому
химику Ф. Вёлеру (ученику Берцелиуса) удалось искусственно
получить мочевину. Исходным веществом при этом была
неорганическая соль—цианистый калий, при окислении которой
образуется циановокислый калий:
KCN + О > KOCN
Обменным разложением KOCN с сернокислым аммонием
получается циановокислый аммоний, который при нагревании
превращается в мочевину:
2KOCN + (NH4JSO4 > 2NH4OCN » (NH2JCO
циановокмслый мочевина
аммоний
К концу первой половины прошлого столетия было выделено
и исследовано очень большое число новых веществ растительного
и животного происхождения. Еще больше было получено
искусственным путем новых соединений, не содержащихся в живых
организмах. Эти синтезы и превращения производились обычными
химическими методами.
В результате успехов синтетической органической химии
виталистическое учение теряло почву и было окончательно
оставлено. Тем не менее необходимость выделения органической
химии в особой раздел химии сохранилась, но теперь уже по
совершенно другим причинам.
В начале 60-х годов появилось два учебника органической
химии: «Органическая химия» Д. И. Менделеева A860) и
«Введение к полному изучению органической химии» А. М. Бутлерова
A864—1866). В этих учебниках приводятся почти одни и те же
соображения, заставляющие рассматривать органическую химию
как особый раздел химии.
Сложные соединения углерода, как природного
происхождения, так и синтетические, отличаются рядом специфических
признаков;
/. предмет, и характерные черты органической химии 17
'1.>Для органических соединений характерна меньшая (по
сравнению с неорганическими) устойчивость, легкая изменяемость
при нагревании, горючесть. Однако, пользуясь этими
признаками, невозможно провести резкой границы между соединениями
органическими и неорганическими.
^2. Углерод способен в соединении с водородом, кислородом
и азотом образовывать очень большое число веществ. Для
соединений углерода характерно явление изомерии, что очень
редко встречается среди неорганических соединение
3. В органической химии имеются ряды сходных по составу
и химическому поведению веществ—гомологов.
Таким образом, наличие атомов углерода в соединениях
характерно выделяет их из ряда соединений, не содержащих
углерод. «Совокупность исчисленных свойств и признаков
органических соединений дает некоторую возможность отличить их от
неорганических» (Менделеев). Бутлеров полагал, что для
определения принадлежности вещества к органическим достаточно
наличия в нем углерода: «Все тела, содержащие углерод, должны
тогда войти в область органической химии, или, правильней,
эта последняя должна получить название химии углеродистых
соединений. Без сомнения, описание этих соединений должно было
бы найти свое место в химической системе вообще, но обилие
предлежащего материала, своеобразность химического характера
и самая важность этих веществ дает повод к образованию в науке
особого отдела из описания углеродистых соединений»
Менделеев, определяя предмет органической химии, вводит
некоторое ограничение: «Органическая химия есть отдел химии,
занимающийся изучением свойств и изменений (реакций)
непостоянных углеродистых соединений».
Позднее, после создания Бутлеровым теории химического
строения, К- Шорлеммер определил органическую химию как
«химию углеводородов и их производных». Он хотел этим
определением подчеркнуть: а) способность углерода образоваїь цепи
С—С-атомов; б) присутствие водорода в большинстве
органических молекул; в) способность водорода к замещению со
вступлением на его место разнообразных других атомов (галоиды,
O,S, N, Р...).
Это определение безусловно правильно и хорошо выявляет
особенности громадного числа относительно просто построенных
органических веществ, составлявших к тому времени A880 г.)
главную массу синтезированных органических соединений. На
основе такого определения строится систематика органических
соединений по принципу генезиса: каждое сложное соединение
рассматривается как бы происшедшим из некоторого
углеводорода путем замещения атомов водорода как гетероатомами, так и
2—788
!8 Введение
разнообразными функциональными группами. Однако для
сложных соединений с большим относительным числом неуглеродных
атомов (кремний, азот, кислород...) определение Шорлеммера
становится узким. В этих случаях определения, данные Бутле
ровым и Менделеевым, являются более правильными.
Итак, органическая химия входит как обособленная часть
в химическую науку. Физические и химические свойства
органических веществ и их строение определяются наличием в них
атомов углерода.
В настоящее время мы можем более точно охарактеризовать
особые свойства углерода как элемента и с современной точки
зрения рассмотреть отличия типических углеродистых соедине
ний от соединений других элементов. Эти отличия заключаются
в следующем:
1Л Углерод в периодической системе элементов занимает
серединное положение между типичными металлами и неметаллами
Он имеет одинаковую валентность и по водороду, и по кислороду,
равную 4. Таким образом, он является четырехвалентным эле
ментом почти во всех своих соединениях.
2^ Углерод способен соединяться с большинством других
элементов. Особенно характерно, что углерод способен образовывать
сложные и прочные молекулы, в которых атомы углерода/
соединены между собой в виде непрерывных цепей;
линейных,'разветвленных, сложного кольчатого строения. Связи между этими
атомами могут быть простыми (ординарными), двойными и тройными
3. В типичных химических превращениях, происходящих в
мягких условиях, эти системы углеродных связей обычно не
нарушаются, составляя своеобразный «углеродный скелет»
молекулы. Реакции расщепления или синтеза обычно протекают та
ким образом, что разрыв или образование новых связей
происходит без нарушения остальных связей молекул, участвующих в
реакции—принцип наименьших изменений (Бутлеров, 1861).
4. Способность к образованию сложных молекул из
разнообразно связанных атомов приводит к тому, что молекулы при
одном и том же составе могут иметь различную структуру, а тем
самым и различные свойства: явление изомерии—характернейшая
особенность соединений углерода.
5. Углерод не склонен давать солеобразные соединения ион
ного характера. Растворы органических соединений даже в воде
обычно не являются электролитами (не проводят тока). Как
правило, связи в органических молекулах являются ковалентными.
6. В громадном большинстве органических соединений
содержится водород. Связь С—Н почти никогда не имеет ионного
характера. Атомы водорода, связанные с углеродом, обычно
менее реакционноспособны, чем связанные с кислородом (О—Н),
2. Органическая химия и промышленность органического синтеза 19
азотом (N—Н) и др. Углеводородная часть молекулы
характеризуется большей инертностью, чем группы, содержащие гетеро-
атомы (О, N, S, Р...). Поэтому химический характер сложных
органических соединений определяется наличием
«.функциональных» групп, содержащих гетероатомы.
2. Органическая химия и промышленность органического
синтеза. Органическая химия, возникшая первоначально как наука
о составе и превращениях продуктов живой природы, в
дальнейшем стала развиваться в направлении искусственного получения
углеродистых веществ. На этом пути она достигла больших
успехов и в области теории, и в области практики.
Большинство природных органических веществ—это сложные
соединения-, изучение их было не под силу молодой науке. Первые
успехи были достигнуты при работе с более простыми веществами:
винным спиртом, уксусной кислотой, бензолом и др., из которых
можно было получать множество новых соединений, в живой
природе не встречающихся. Так произошло превращение
органической химии из науки о веществах органического происхождения
в науку о веществах, получаемых путем синтеза.
Каковы же те силы, которые направляют развитие этой науки?
Как, почему и в каком направлении идет развитие отдельных
областей органической химии?
История органической химии тесно связана с историей
человеческого общества, с возникновением новых потребностей, с
созданием новых областей промышленности. Спирты и уксусная
кислота—это продукты пищевой промышленности. Изучение
явлений брожения, использование продуктов этой промышленности
дало в руки химиков доступные материалы, из которых можно
было готовить искусственно множество полезных веществ.
Жировая и мыловаренная промышленность дала толчок развитию
химии органических соединений «жирного»* ряда.
Широкое промышленное применение сухой перегонки
каменного угля для получения светильного газа и кокса привело к
необходимости изучения основных (газы) и побочных продуктов этой
промышленности. Уже в 1825 г. Фарадей выделил из светильного
газа бензол, конденсировавшийся в газопроводных трубах.
Химики вынуждены были заняться проблемой использования
больших количеств каменноугольной смолы, при разгонке которой
были выделены, кроме бензола, многие другие «ароматические»
углеводороды: толуол, нафталин, антрацен, далее ряд фенолов и
много других продуктов. При химической переработке продуктов
выделенных из смолы, были получены нитросоединения, амины и
* Часто соединения нециклического строения называют алифатическими,
от греческого слова «алейфар»—жир.
2*
20 Введение
многие другие соединения, нашедшие применение в качестве
синтетических красителей («анилиновые» красители), взрывчатых
веществ и др. Возникли мощные промышленные предприятия,
использующие продукты переработки угля.
Необходимость быстрейшего решения практических задач,
вызванных развитием промышленности, требовала углубленного
теоретического изучения. «Нет ничего более практического, чем
хорошая теория»,—это известное выражение как нельзя лучше
применимо к органической химии.
Создание и развитие теории химического строения
потребовало громадных усилий нескольких поколений химиков. Эта
теория явилась надежным фундаментом для понимания химических
процессов, для планомерного синтеза, для предсказания свойств
еще не полученных веществ и предвидения их практического
применения.
Теоретическое развитие той или другой области органической
химии чрезвычайно тесно связано с практическими потребностями.
Развитие теоретических исследований в области углеводородов и,
особенно, органического катализа в большой степени связано,
например, с задачами, выдвигаемыми промышленностью
моторного топлива и производством полимерных материалов. Открытие
новых ценных типов красителей и лекарственных веществ в
свою очередь сильно ускорило развитие химии
гетероциклических соединений. Такие отрасли химической промышленности,
как получение искусственного волокна, синтетического каучука,
пластических масс и т. д., возникли и широко развились только
за последние 40—50 лет, и в связи с ними возникла и развивается
химия высокомолекулярных органических соединений.
Так же, как будущему инженеру-строителю или механику,
прежде чем строить, надо изучить свойства строительных
материалов и основы механики, так и химикам—строителям химических
молекул органических веществ—нужно изучить первоначально
на простейших примерах свойства органических соединений и
законы, управляющие их химическими превращениями.
II. РАБОТА С ОРГАНИЧЕСКИМИ ВЕЩЕСТВАМИ
3. Выделение и очистка органических веществ. Для анализа
и исследования органического вещества необходимо получить его
в чистом виде. Главнейшими приемами очистки и выделения
органических соединений из природных веществ или из продуктов
реакций являются: перегонка, извлечение растворителями,
кристаллизация и фильтрование.
Перегонка является наиболее распространенным способом
разделения и очистки органических веществ.
3. Выделение и очистка органических веществ
21
Для перегонки какой-либо жидкости ее наливают в кругло-
донную колбу с боковым отводом (рис. 1); закрепляют термометр
так, чтобы его шарик находился чуть ниже отводной трубки, а
в муфту холодильника по направлению снизу вверх (по принципу
противотока) пускают ток холодной воды. В колбу с жидкостью
помещают узкие стеклянные капиллярные трубочки, запаянные
с верхнего конца. Присутствие воздуха в капиллярах облегчает
образование пара, и кипение происходит равномерно, без
толчков. Вместо капиллярных трубочек можно пользоваться
кусочками необожженной глины (тарелки) или другого пористого
материала.
Рис. 1. Прибор для перегонки.
Смеси веществ, значительно отличающихся по температурам
кипения, можно путем перегонки разделить и получить составные
части смеси в достаточно чистом виде.
При перегонке смеси веществ, точки кипения которых близки,
целесообразно пользоваться дефлегматорами или
ректификационными колонками. Простейшие ректификационные колонки—это
стеклянные трубки, наполненные стеклянными бусами или
короткими металлическими спиралями. В настоящее время имеются
ректификационные колонны, позволяющие разделять смеси
жидкостей, температуры кипения которых различаются между собой
лишь на 1—2 градуса.
Некоторые вещества при перегонке под обычном давлением
разлагаются, поэтому их перегоняют под уменьшенным
давлением. Чем меньше давление, тем ниже температура, при которой
кипит вещество. Так, при понижении давления с 760 до 10—
22
Введение
15 мм рт. ст. (такой вакуум достигается при помощи
водоструйных насосов) температура кипения понижается
приблизительно на 100 °С; при понижении давления до десятых или сотых
долей миллиметра (так называемый глубокий вакуум,
достигаемый применением масляных или ртутных насосов) температура
кипения понижается на 200 °С.
Разновидностью перегонки под уменьшенным давлением
является молекулярная перегонт. Она протекает при давлениях не
выше 0 001 мм рт. ст. Для молекулярной перегонки применяют
приборы (рис. 2), отличающиеся
тем, что молекулы
испарившегося вещества не возвращаются
обратно и не встречают на пути
молекул воздуха, так как
расстояние между нагреваемой и
охлаждаемой поверхностью
очень короткое. Благодаря
этому удается перегнать такие
вещества, как тринитроглицерин
(см. стр. 175) и некоторые
сахара.
Для выделения
малорастворимых в воде веществ, имеющих
при температуре около 100 С
заметную упругость паров,
применяют перегонку с водяным
паром. Водяной пар, необходимый для перегонки, получают
обычно в отдельном кипятильнике-парообразователе и вводят его по
трубке в круглодонную колбу, где находится смесь, содержащая
выделяемое вещество; при перегонке смесь паров конденсируется
в холодильнике и собирается в приемнике. Прибор для
перегонки с водяным паром показан на рис. 3. Перегонкой с водяным
паром удается выделять из смесей и очишать многие вещества.
Перегонка широко применяется во многих отраслях
химической промышленности. Так, например, на спиртовых заводах
при брожении получают смесь спиртов, которую разделяют на
ректификационных колоннах и получают винный, или этиловый,
спирт; оставшуюся смесь «сивушных масел» подвергают
дальнейшей ректификации и выделяют пропиловый, изобутиловыи и изо-
амиловые спирты. При перегонке нефти на перерабатывающих
заводах получают различные бензиновые фракции и фракции
менее летучие: лигроин, керосин, соляровые масла и т. д.
Извлечение веществ (экстракция) из водного раствора
летучими и не смешивающимися с водой растворителями, например
эфиром, петролейным эфиром, хлороформом, сероуглеродом и т. д.,
Рис. 2. Прибор для молекулярной
перегонки, смонтированный из
перегонной колбы.
3. Выделение и очистка органических веществ
23
является широко распространенным приемом выделения веществ.
В лабораторной практике и в производственных условиях
экстракцией выделяют эфирные масла, алкалоиды из растений,
различные антибиотики из грибковых культур и др. В лаборатории
извлечение производят следующим образом. В делительную
воронку наливают водный раствор вещества, прибавляют растворитель,
например эфир, и, закрыв воронку стеклянной пробкой, сильно
взбалтывают. Вещество при этом частично извлекается из воды
эфиром. Содержимому делительной воронки дают отстояться,
Рис. 3. Приборы для перегонки с водяным паром
в результате чего образуются два слоя. После этого открывают
пробку и спускают через кран нижний (водный) слой; верхний
(эфирный) слой сливают через горло воронки. Водный слой
снова обрабатывают эфиром. Если вещество хорошо растворимо в
эфире и плохо—в воде, то, повторяя эту операцию несколько раз,
можно почти полностью извлечь его из раствора. Эфирные
вытяжки соединяют вместе и сушат, добавляя в них поташ, едкое
кали, сплавленный хлористый кальций или обезвоженный
прокаливанием сульфат натрия или магния. Высушенный раствор
фильтруют. Эфир удаляют отгонкой на кипящей водяной бане, а
оставшееся вещество подвергают дальнейшей очистке
перегонкой или кристаллизацией. Экстракцию твердых веществ проводят
в аппаратах Сокслета (рис. 4^.
В лабораторной практике все большее применение находит
фракционная экстракция двумя растворителями, позволяющая
разделять сложные смеси органических веществ, близких по
свойствам, Приборы, применяемые для этой цели, обычно действуют
автоматически.
24
Введение
Кристаллизация и фильтрование. При охлаждении
горячего раствора, содержащего смесь двух или нескольких твердых
веществ, из него выкристаллизовывается то вещество,
относительно которого раствор при данной температуре является
пересыщенным. Выделившиеся
кристаллы отфильтровывают. В
химических лабораториях для
фильтрования часто пользуются
стеклянной или фарфоровой
воронкой, имеющей перегородку с
отверстиями. На эту
перегородку помещают кружок
фильтровальной бумаги. Воронку
вставляют на пробке в горло
склянки для отсасывания (рис. 5).
Склянка соединяется с
водоструйным насосом, который выка-
Рис. 4. Приборы для
непрерывно-периодической экстракции из твердых
тел при температуре кипения
растворителя.
Рис. 5. Соединение
воронки с колбой
для отсасывания.
чивает из нее воздух. Благодаря этому в склянке получается
разрежение, и фильтрование происходит быстро.
В настоящее время для отделения осадка используют
центрифуги и ультрацентрифуги, особенно при работе с
коллоидными растворами и мелкодисперсными осадками.
4. Определение физических констант. Каждое чистое вещество
характеризуется определенными физическими константами. Они
могут служить средством идентификации вещества, т. е.
установления его юждестЕа с известным веществом. В то же время пос-
4. Определение физических констант
25
тоянство значения той или иной физической константы при
повторных очистках вещества во многих случаях может быть
показателем того, что вещество получено в чистом виде.
Из физических констант органических веществ наиболее часто
определяют температуры плавления и кипения, плотность,
показатель преломления.
Температура плавления. Для определения температуры
плавления вещество тщательно измельчают в порошок и насыпают в
запаянную с одного конца капиллярную трубочку диаметром
I—1,5 мм; высота слоя порошка—около 2 мм. Капилляр
прикрепляют (например, каплей серной кислоты) к
термометру так, чтобы вещество находилось в
непосредственной близости с его шариком.
Термометр при помощи пробки, в которой есть вырез
для прохода воздуха, помещают в прибор (рис. 6),
наполненный на 3/4 концентрированной серной
кислотой (темп. кип. около 350 °С). Медленно
нагревают прибор и замечают температуру, при
которой происходит плавление вещества. Если
вещество не содержит примесей, то плавление
происходит в малом интервале температур @,5—1 °С).
Присутствие даже небольшого количества
примесей заметно снижает температуру плавления
вещества; кроме того, в этом случае плавление
происходит не резко, а в значительном
интервале температур. Изменение атмосферного давления
не сказывается в сколько-нибудь заметной
степени на температуре плавления.
Температура кипения. При очистке вещества
перегонкой одновременно определяется и
температура кипения. Если имеется смесь жидкостей,
то температура кипения во время перегонки в
большинстве случаев непрерывно повышается.
Если же перегонке подвергается чистое вещество, то
термометр во время перегонки неизменно
показывает одну и ту же температуру. Изменение
атмосферного давления влияет в заметной степени (иногда на 2—
3 °С) на температуру кипения вещества. Поэтому необходимо
указывать давление, при котором производилась перегонка; если
такого указания нет, это означает, что определение производили
при нормальном атмосферном давлении G60 мм рт. ст.).
Для определения температуры кипения вещества по методу
Сиволобова достаточно одной капли жидкости. Определение
производят следующим образом Жидкость помещают в очень узкую,
запаянную снизу трубку диаметром около 2 лш и опускают в нее
Рис. 6.
Прибор для
определения
температуры
плавления
26
Введение
тончайший капилляр с запаянным верхним концом. Трубку о
жидкостью прикрепляют к термометру прибора для определения
температуры плавления так, как это показано на рис. 7, посуіе
чего начинают медленно нагревать колбу прибора. Непрерывная
цепочка пузырьков, выходящих из капилляра, указывает на то,
что жидкость закипела. Нагревание прекращают и отмечают
температуру, при которой выделение пузырьков внезапно
прерывается. Эта температура и является температурой кипения жидкости.
Плотность. Относительная плотность. Жидкие вещества
удобно характеризовать по плотности определяемой как
отношение массы тела к его объему (р=-гг-, где т—масса в г, а V—
объем в см"; при измерении с точностью,
не превышающей 0,01%, можно объем
выражать в мл).
На практике часто характеризуют
вещества по относительной плотности,
выражающей отношение плотности вещества
к плотности воды. Относительную
плотность определяют с помощью пикнометра
емкостью 1—5 мл (рис. 8). Вначале
определяют массу воды, вмещающейся в
пикнометре. Для этого предварительно
взвешенный пикнометр наполняют
дистиллированной водой и помещают на 20—30
І мин в термостат при температуре 20 °С.
Затем пикнометр обтирают
фильтровальной бумагой и взвешивают. Таким же об-
I разом определяют массу исследуемой жид-
V / кости в пикнометре. Из отношения най-
денной массы жидкости к массе воды в
Рис. 7. Тер- Рис. 8. Пи- том же объеме определяют относитель-
МОМЄТР С КНОМетр. г ~_ О/-Ї /joo\
трубкой и ка- ную плотность при 20 С (d§o)-
пилляром. Обычно принято вычислять
относительную плотность вещества при 20 "G
по отношению к плотности воды при 4 °С, приблизительно
равной 1 г/см3. Так как плотность воды при 20 °С равна
0,9982 г!см\ то значение относительной плотности (df) находят
по следующей формуле:
df =—0,9982
где т—масса вещества в объеме пикнометра при 20 °С;
w—масса воды в том же объеме и при той же температуре.
В лабораторной практике и в технике часто пользуются для
определения плотности жидкости ареометрами и спиртометрами.
5. Состав органических веществ. Качественный анализ 27
5. Состав органических веществ. Качественный анализ. В
состав органических веществ кроме углерода чаще всего входят еще
водород, кислород и азот; эти четыре элемента получили название
органогенов. Наряду с ними в молекулу органического вещества
могут входить и другие элементы.
В составе многих белковых веществ находится сера; казеин
молока содержит фосфор; в гемоглобине крови содержится железо,
в хлорофилле—магний. Для различного рода синтезов большое
значение имеют органические вещества, содержащие галоиды.
Очень интересную и своеобразную группу составляют так
называемые элементорганические соединения, в молекулах
которых содержатся атомы различных элементов, например
металлов, непосредственно связанные с углеродом.
Открытие углерода и водорода. Присутствие углерода во
многих органических соединениях можно обнаружить по
обугливанию вещества при осторожном его прокаливании. Более общим
методом открытия углерода и одновременно с ним водорода
является прокаливание в пробирке органического вещества в смеси
с окислителем, в качестве которого применяют мелкий порошок
окиси меди. При этом происходит окисление углерода
органического вещества в углекислый газ СО2, а водорода—в воду,
например:
С3НаОа + 7СиО = ЗСО, + 4НгО + 7Си
глицерин
Выделяющийся газ пропускают в пробирку со
свежеприготовленной известковой водой. Наличие углекислого газа доказывается
выпадением белого осадка нерастворимого углекислого кальция
СаСО3. Воду в продуктах сжигания можно обнаружить,
пропуская газ в сухую пробирку, на дне которой насыпано немного
порошка безводной сернокислой меди CuSO4 (ее получают в виде
белого порошка путем прокаливания синих кристаллов медного
купороса). Безводная сернокислая медь жадно присоединяет
к себе воду, снова образуя синие кристаллы медного купороса:
CuSO4 + 5Н2О — CuSO4-5H2O
Открытие азота. Для открытия азота небольшое количество
органического вещества сильно прокаливают с металлическим
калием или натрием. При этом калий (или натрий) соединяется с
частью углерода и азота органического вещества, образуя
цианистый калий KCN (или цианистый натрий NaCN). Полученный
плав растворяют в воде и прибавляют к нему раствор соли
двухвалентного железа, например железного купороса FeSO4-7H8O,
и раствор хлорного железа FeCl3, в котором железо трехвалентно.
После подкисления выпадает темно-синий осадок берлинской
лазури.
28 Введение
Образование лазури основано на следующих реакциях:
Сначала происходит обменная реакция между цианистым
калием и железным купоросом, в результате чего образуется
цианистое железо Fe(CNJ и сернокислый калий K2SO4:
2KCN + FeSO4 = Fe(CN), + KaSO4
Образовавшееся цианистое железо реагирует с оставшимся
цианистым калием, образуя железистосинеродистый калий
K4Fe(CN)e. Последний, взаимодействуя с хлорным железом, дает
соединение синего цвета—берлинскую лазурь:
4FeCl3 + 3K4Fe(CN)e = Fe4[Fe(CN)e]3 + 12KCI
берлинская
лазурь
Если вещество содержит много азота, то берлинская лазурь
выпадает в виде осадка, если же азота было немного, то
получается только сине-зеленое окрашивание.
Открытие серы. Для открытия серы органическое вещество
прокаливают, так же как и при определении азота, с
металлическим натрием. При этом натрий соединяется с серой, образуя
сернистый натрий. Плав растворяют в воде и к полученному
раствору прибавляют раствор азотнокислого свинца Pb(NO8)s или
раствор нитропруссида натрия Nas[Fe(CN)sNO]-2H2O.
Азотнокислый свинец с сернистым натрием дает черный
осадок сернистого свинца PbS:
Pb(NO3J + Na2S = 2NaNOa + PbS
Нитропруссид натрия, взаимодействуя с сернистым натрием,
дает красно-фиолетовое окрашивание.
Открытие галоидов. Галоиды проще всего открываются по
Бейльштейну—прокаливанием органического вещества с окисью
меди в пламени горелки. Кислород окиси меди окисляет углерод
и водород органического вещества в углекислый газ и воду, медь
же соединяется с галоидом. Так, например, с хлороформом СНС1:,
эту реакцию можно выразить уравнением:
2СНС13 + 5СиО = CuCl, + 4CuCl + 2СО2 + Н2О
Образовавшаяся галоидная медь, улетучиваясь в пламени
горелки, окрашивает пламя в зеленый цвет. Появление зеленого
окрашивания доказывает присутствие в органическом
соединении галоида.
По Степанову, определение галоида производят нагреванием
спиртового раствора испытуемого вещества с металлическим
натрием. Натрий вытесняет из гидроксильной группы спирта
водород, который в момент выделения отщепляет атом галоида, пе-
6. Количественный анализ 29
реводя его в ионное состояние:
С,Н5ОН + Na = C2H5ONa + Н
СНС13 + 6Н = 3HCI 4- СН4
C,H5ONa + HCI = С2Н5ОН + NaCI и т. д.
По окончании реакции раствор разбавляют водой, подкисляют
азотной кислотой и прибавляют к нему раствор азотнокислого
серебра. При этом выпадает хлопьевидный характерный осадок
галоидной соли серебра:
HCI + AgNO3 = AgCI + HNO3
Установление наличия кислорода. Открытие углерода,
водорода, азота, серы и галоидов не представляет каких-либо
затруднений. Открытие кислорода значительно сложнее, и о его
присутствии чаше всего судят по данным количественного анализа.
Так, если в результате количественного анализа было
установлено, что в состав вещества входят 58,5% углерода, 4,1%
водорода и 11,4% азота и не обнаружено никаких других элементов,
то недостающее до 100% количество относят на долю кислорода.
Так как в нашем примере
58,5 + 4,1 + 11,4 = 74%
то в состав данного вещества входит 100—74=26% кислорода.
Таким образом, для открытия отдельных элементов
органического соединения необходимо .предварительно его разрушить
путем полного сжигания, или окисления, или сплавления с
металлическим натрием для того, чтобы превратить углерод,
водород, азот и другие элементы в простые вещества, удобные для
качественного открытия.
6. Количественный анализ. Определение количественного
содержания отдельных элементов в органических веществах
называется элементарным анализом. Определение главнейших
органогенов G, Н, N, О чаще всего производят сжиганием навески
вещества в трубке из тугоплавкого стекла или кварца. При
макроанализе берут для сжигания навеску в 0,15—2 г, а при
микроанализе—в 2—5 мг. При микроопределениях можно не только
работать со значительно меньшими навесками, но и проводить
анализ гораздо быстрее. Описание аппаратуры для элементарного
анализа, а также необходимые расчеты приводятся в руководствах
к практическим работам по органической химии, поэтому здесь
укажем вкратце только принципы определения тех или иных
элементов.
Определение углерода и водорода. При прокаливании точно
отвешенного количества органического вещества с окисью меди в
струе кислорода образуются углекислый газ и вода. Вода
поглощается в U-образной трубке хлористым кальцием или перхлора-
30 Введение
том магния Mg(C104J («ангидрон»), жадно соединяющимися с
водой, углекислый газ поглощается в кали-аппарате крепким
раствором едкого кали, вступающим в реакцию с углекислым
газом по уравнению
2КОН + СО2 = К2СОа + Н3О
Взвешивая U-образную трубку до и после опыта,
устанавливают количество образовавшейся воды; увеличение в весе кали-
аппарата показывает количество углекислого газа. Из этих
данных можно вычислить процентное содержание углерода и
водорода во взятом для исследования веществе.
Определение азота. Азот в органическом веществе может быть
определен различными методами. По методу Дюма, навеску
вещества сжигают в трубке с окисью меди в струе углекислого газа.
Азот при этом выделяется в элементарном виде (N2) и вместе с
другими продуктами окисления (Н2О и СО2) вытесняется в
аппарат (азотометр), наполненный крепким раствором едкого кали,
поглощающим углекислый газ и конденсирующуюся воду.
Вытесненный азот определяется по объему.
Часто применяется метод Кьельдаля. По этому методу, навеску
вещества кипятят с крепкой серной кислотой, которая обугливает
и далее сжигает вещество, сама частично превращаясь в S02.
Азот превращается в сернокислый аммоний. По окончании
окисления полученный раствор обрабатывают избытком щелочи.
Выделяющийся аммиак поглощают раствором соляной или серной
кислоты известной концентрации.
Определение кислорода. Как указывалось выше, кислород
обычно определяют «по остатку». Однако предложены методы и
«прямого» его определения. По методу, разработанному М. О.
Коршун, навеску вещества прокаливают в кварцевой "трубке в токе
азота. Продукты распада пропускают над сильно накаленным
углем, при этом весь кислород превращается в окись углерода.
Последняя количественно определяется при помощи пятиокиси
иода. Определение основано на восстановлении пятиокиси иода,
нагретой до 150 °С, окисью углерода по уравнению:
JiA + 5СО = 5СО2 + J3
Иод определяют титрованием тиосульфатом.
Определение галоидов и серы. Навеску вещества нагревают в
запаянной толстостенной стеклянной трубке с азотной кислотой
(плотность 1,5 г/см3) в присутствии азотнокислого серебра.
Азотная кислота окисляет углерод и водород органического
вещества в углекислый газ и воду, а галоид выделяется в виде
труднорастворимого осадка (AgCl, AgBr, AgJ). Его
отфильтровывают, промывают, высушивают и взвешивают.
7. Установление простейшей формулы Зі
Определение серы проводят аналогично определению галоидов,
с той лишь разницей, что не прибавляют азотнокислого серебра.
Так как окисление серы облегчается в присутствии брома, то
часто к навеске вещества прибавляют несколько кристалликов чи
стого бромистого калия. Азотная кислота окисляет серу до
серной кислоты, которую осаждают раствором хлористого бария в
виде сульфата бария. Осадок сульфата бария отфильтровывают,
промывают, прокаливают при 500 °С и взвешивают. Этот способ
определения галоидов и серы предложен Кариусом.
7. Установление простейшей формулы. Количественный
анализ показывает состав вещества в процентах. Как же, зная
процентное содержание различных элементов в веществе, выразить
состав вещества не в процентах, а в атомных единицах, т. е.
каким образом установить его химическую формулу?
Допустим, что в результате анализа найдено, что в состав
вещества входит 79,20% углерода, 15,07% кислорода и 5,73%
водорода, т. е. что количества углерода, кислорода и водорода
относятся, как 79,20 : 15,07 : 5,73.
Чтобы установить простейшую формулу вещества, надо знать
отношение чисел, выражающих количества атомов различных
элементов, входящих в молекулу. Это отношение получим, если
числа, выражающие процентное содержание элементов,
разделим на атомную массу, поскольку чем больше атомная масса
элемента, тем меньше атомов соответствует найденному
процентному содержанию. Так как атомная масса углерода 12,
кислорода 16, а водорода 1,01 (округленно), то имеем:
12
0.942
т. є. на 0,942 атома кислорода приходится 5,67 атома водорода
и 6,6 атома углерода. Но в молекуле вещества не может быть
дробного числа атомов. Поэтому узнаем, сколько атомов углерода и
водорода приходится не на 0,942 атома кислорода, а на 1 атом
кислорода. Для этого разделим наши числа на 0,942*:
6,6
q Q.2 = ' (число атомов углерода)
5,67
q q^2 = ° (число атомов водорода)
0,942
0 сц2 = ' (число атомов кислорода)
* Деление надо производить на наименьшее из чисел, показывающих
содержание атомов. В нашем примере таким числом является число,
показывающее количество атомов кислорода.
32 Введение
Следовательно, на 1 атом кислорода в молекуле вещества
приходится 7 атомов углерода и 6 атомов водорода, и его состав можно
выразить формулой QHaO.
8. Определение молекулярного веса. Мы установили, что
в молекуле исследованного вещества на 1 атом кислорода
приходится 7 атомов углерода и 6 атомов водорода, т. е. что состав
его может быть выражен формулой С,НаО. Но в молекулах
веществ С14Н12О2, С21Н18О3, С28Н24О4 и т. п. на 1 атом кислорода
тоже приходится 6 атомов водорода и 7 атомов углерода; эти
вещества имеют одинаковый процентный состав. Следовательно,
формула QHeO не единственно возможная для нашего вещества; она
является только наиболее простой. Для того чтобы установить,
какая из этих формул является истинной, надо определить
молекулярный вес вещества. Так как атомный вес углерода 12,
кислорода 16, водорода 1, то молекулярный вес вещества, формула
которого QHflO, равен 106A2-7+6+16), молекулярный вес
вещества CUH12O7 равен 212 и т. д.
По закону Авогадро, молекулярный вес равен удвоенной
плотности газа (или пара) по водороду: M=2d.
Так, в нашем примере, если плотность пара по водороду будет
равна 53, то молекулярный вес вещества равен 106 и его
формула будет QHgO; если плотность пара по водороду окажется
равной 106, то молекулярный вес равен 212 и формула вещества
будет CUO12O2 и т. д.
В том случае, если вещество не превращается в пар.
молекулярный вес определяют по повышению температуры кипения
или по понижению температуры замерзания раствора вещества в
определенном растворителе
Элементарный анализ и определение молекулярного веса
позволяют, таким образом, установить лишь общую
«молекулярную» формулу. Для большинства типичных неорганических
веществ химический анализ на этом заканчивается. В органической
химии одной молекулярной формуле почти всегда отвечает много
веществ, различных по физическим и химическим свойствам
(«изомеров») Молекулы этих веществ отличаются порядком связи
атомов: они имеют разное «химическое строение». Что такое
химическое строение и каковы методы его определения, мы узнаем
из следующего раздела.
III. ТЕОРИЯ ХИМИЧЕСКОГО СТРОЕНИЯ
9. Теория радикалов и теория типов. В 1861 г. профессор
Казанского университета Александр Михайлович Бутлеров
впервые высказал "гновные идеи теории химического строения
органических соединений. Чтооы \яснить себе значение созданной
9. Теория радикалов и теория типов 33
А. М. Бутлеровым теории химического строения, необходимо
познакомиться с теоретическими воззрениями в органической химии
в период, предшествовавший созданию бутлеровской теории.
Одной из первых теорий органической химии было учение
Берцелиуса об электрохимическом дуализме A820—1840).
Причину химического сродства Берцелиус отождествлял с
притяжением частиц, несущих разноименные электрические заряды.
Молекула сложного вещества состоит из двух частей, заряженных
противоположными зарядами. Такое воззрение возникло из
наблюдений над электролизом солей, кислот, оснований. Самым
электроотрицательным элементом считался кислород, атомы
щелочных металлов—самыми положительными. Сера, соединившись
с кислородом, давала серный ангидрид SO8—молекулу с
преобладанием отрицательных зарядов. В окислах металлов—натрия
и калия—имеется избыток положительных зарядов. Отсюда
следовало, что сернокислый натрий представляет собой молекулу
(+) (-)
(Na2O) • (SOa). Пропуская в раствор этой соли постоянный
электрический ток, мы производим разложение вещества на исходную
едкую щелочь и серную кислоту. Так как частица Na2O
положительно заряжена, она притягивается к отрицательно заряжен-
(-)
ному катоду, a (SOa) направляется к аноду.
Большинство органических соединений не проводит тока в
растворах. Таким образом, объяснение их строения с точки
зрения электрохимического дуализма встречало большие трудности.
Однако Берцелиус упорно утверждал, что и каждое органическое
соединение, подобно неорганическим солям слагается из двух
сложных радикалов, несущих разноименные электрические
заряды. Такие органические сложные радикалы могут существовать
в свободном состоянии подобно окиси кальция СаО, серному
ангидриду SO8 и др.
Отыскание, изучение и выделение таких сложных радикалов
представлялось одной из важнейших научных задач
органической химии.
По мере изучения органических соединений все большее и
большее число наблюдений не находило себе объяснения с точки
зрения этой теории радикалов. Ей на смену пришла так
называемая унитарная теория Жерара*—Лорана A840—1860). Молеку-
* Шарль Жерар (Ch. Gerhardt), французский химик A816—1856).
Родился и работал в Страсбурге. Совместно с А. Лораном A807—1853) является
создателем унитарного учения, теории типов и оригинальной систематики
органических соединений. Из его экспериментальных работ отметим
получение смешанных ангидридов кислот, получение замещенных амидов (форм-
анилид, ацетанилид, бензаннлид и др.) и сульфонампдов.
3—788
34
Введение
ла органического соединения, как утверждало новое учение, не
слагается из определенных противоположно заряженных частей,
а представляет собой единое целое. При реакциях двойного
обмена, наиболее типичных для органических соединений,
молекулы распадаются на остатки, возникающие лишь в процессе
реакции, а не существовавшие заранее в исходных молекулах и не
способные к существооанию в свободном состоянии. По
характерным реакциям все органические соединения могут быть отнесены
к немногим типам.
Так, все амины, получающиеся при действии аммиака на
органические галоидпроизводные, образуют тип сложных
аммиаков:
Н
Н
Н
При реакции углеводорода с хлором процесс совершается по
тому же типу, что и взаимодействие водорода с хлором:
н
н
N
с2н6
сгн:
н
Н
Н
С3Н6
Н
С1
С1
¦+¦ /-, I =
CI
С1
н
С1
н
CI
н
С1
сгн5
С1
Поэтому углеводороды относились к типу водорода, а
галоидпроизводные—к типу хлористого водорода.
Спирты, кислоты, эфиры рассматривались как производные
воды, в которой один или два атома водорода замещены на
органические остатки:
О
сан6
н
о
С2НаО
н
о
вода
сяирт
уксусная
кислота
С2Н5
с2на
эфир
о
Таким образом, в качестве простых основных типов органических
соединений были взяты типы:
Н
N
ні
ні
одорода
ні
а)
хлористого
водорода
9. Теория радикалов и теория типов
35
В 1858 г. А. Кекуле предложил еще один тип—тип метана,
к которому можно было отнести ряд соединений:
н
н
н
н
с
CI
н
н
н
хлористый
м^тил
с
С1
С1
С1
н
хлороформ
с
N0,
С1
С1
С1
хлорпикрин
CI
Сі и др.
О
фосген
Очевидно также, что к типу метана можно было отнести
углеводороды. Интересно отметить, что еще в начале 60-х годов среди
химиков господствовало убеждение, что соединение С2Н„ может
существовать в виде двух изомерных соединений:
с2нБ
водородистый
этил
сн;
СН;
дииетил
Таким образом, первоначально признавали, что органические
соединения аналогичны по своим свойствам реально
существующим неорганическим соединениям. Вскоре, однако, были открыты
органические соединения, не укладывающиеся по свойствам в
эти простые типы. К таким соединениям относится, например,
глицерин, в молекуле которого содержится не один водный
остаток ОН, а три. В молекуле гликоля содержится два водных
остатка. Диамины состоят как бы из двух соединенных молекул
аммиака.
Пришлось допустить существование «.кратных типов»:
Н,
удвоенный
тип воды
удвоенный тип
хлорно того
водорода
о.
утроенный
тип воды
Теперь как бы признавали реальное существование
органических соединений, соответствующих удвоенной воде, удвоенному
хлористому водороду и др., т. е. неорганическим веществам,
которые уже реально не существуют.
Были открыты новые, более сложные органические
соединения, которые следовало бы относить к двум различным типам,
например хлоруксусная кислота, вступающая в реакции то по типу
3*
36 Введение
воды, то по типу хлористого водорода. Так возникло учение о
«смешанных типах»:
сі J с2н2сю | с2нэо
2сю | с2нэо2
„}0 ні»
смешанный тип воды и хлористого водорода
(на примере хлоруксусной кислоты)
При действии аммиака на хлоруксусную кислоту получается ами-
ноуксусная кислота, которую приходится относить к смешанному
типу воды и аммиака:
і
О
н J
н
н
н
1°
N
С2НгН
н
н
н
Неорганические прототипы этих соединений реально не
существуют: смешанный тип воды и хлористого водорода—это не
раствор последнего в воде (т. е. обычная соляная кислота).
Свойства аминоуксусной кислоты также не подобны свойствам водного
раствора аммиака. Но если исходные типы реально не существуют,
необходимо ли, чтобы типические формулы органических
соединений выражали действительное строение молекул этих
соединений?
Жерар и его последователи отвечали на этот вопрос
отрицательно. Типические формулы—это только рациональные формулы,
которые условно указывают лишь на аналогию в химических
превращениях. И так как таких превращений может быть много, то
для одного и того же соединения может быть несколько таких
формул. Изучением химических свойств, по мнению Жерара и его
последователей, невозможно определить действительное строение
молекулы вещества.
Такой вывод, приводящий к философскому агностицизму, тем
более был странен, что теория типов в своем развитии
чрезвычайно много сделала для развития фактического содержания химии.
По существу к 60-м годам были установлены основные
положения для создания правильной теории. Вместо прежних
эквивалентов начали пользоваться истинными атомными весами; было
установлено понятие о валентности элементов. Весьма важными
для создания новой теории органической химии были работы
A858—1860) шотландского ученого Арчибальда Купера и круп-
10 Теория химического строєная Бутлерова 37
нейшего немецкого химика Августа Кекуле. Ими было
установлено, что углерод в своих соединениях имеет постоянную
валентность, был предложен новый тип органическах соединений—тип
метана (Кекуле, 1858) и показана способность углерода к
образованию углеродных цепей. Однако эти отдельные факты и
положения не были сведены в единую стройную теоретическую
систему. Создание этой новой теории принадлежит Александру
Михайловичу Бутлерову.*
10. Теория химического строения Бутлерова. Основные идеи
нового учения были впервые высказаны публично А. М.
Бутлеровым в докладе, произнесенном 19 сентября 1861 г. на съезде
немецких естествоиспытателей и врачей в Шпейере.
«.Химическая натура сложной частицы определяется натурой
элементарных составных частей, количеством их и химическим
строением-»,—таково общее положение Бутлерова. Все атомы,
составляющие молекулу сложного вещества, взаимодействуют друг
с другом; наиболее сильное взаимное влияние наблюдается у
атомов, непосредственно связанных; меньшее взаимное влияние
наблюдается между атомами элементов, связанных лишь через
посредство других атомов. Распределение этого взаимодействия,
определяемое порядком связи атомов в молекуле, Бутлеров
называет химическим строением.
* Александр Михайлович Бутлеров A828—1886) родился в городе
Чистополе Казанской губернии. Химией он начал интересоваться еще в гимназии
и особенно в студенческие годы. Под руководством профессора Казанского
университета Н. Н. Зинина студент А. М. Бутлеров провел свои первые
работы. В 1847 г. Н. Н. Зинин переехал в Петербург, и Бутлеров начал работать
под руководством К. Клауса, который был не только химиком (он открыл
элемент рутений), но п широко образованным естественником. Первую свою
научную работу Бутлеров выполнил на тему, не имеющую отношения к
химии,—«Дневные бабочки волго-донской фауны». Магистерскую диссертацию
Александр Михайлович написал уже на химическую тему: «Об окисляющем
действии осмиевой кислоты на органические вещества» A851).
В 1852 г. Бутлеров стал профессором Казанского университета, заменив
ушедшего Клауса. Свою докторскую диссертацию «Об эфирных маслах»
Александр Михайлович защитил в Московском университете. В 1857—1858 гг.
Бутлеров, командированный за границу, познакомился с рядом химических
лабораторий Германии и Франции. В лаборатории Вюрца он выполнил работу
по получению и химическому исследованию йодистого метилена СЬЫг. Эти
исследования он продолжал уже в казанской лаборатории. А. М. Бутлеров
доказал, что при действии меди на йодистый метилен образуется не свободный
радикал метилен СНг, а углеводород этилен С2Н4. Из этого же йодистого
метилена Бутлеров приготовил полимер муравьиного альдегида («диоксимети-
лен»); действием извести на последний было получено искусственное
сахаристое вещество—«метиленитан» A861).
Бутлеров читал курс органической химии и разрабатывал теоретические
основы этой науки.
В 1868 г. Бутлеров был избран (по представлению Д. И. Менделеева)
профессором Петербургского университета. В 1874 г. ои стал академиком.
38 Введение
Можно ли определить химическое строение? На этот вопрос
Бутлеров отвечает положительно: «Возможность судить о
химическом строении достаточно доказывается настоящим состоянием
наших знаний».
Изомерия соединенний углерода. В органической химии, в
отличие от неорганической, особенно большое значение приобретает
явление изомерии.
У соединений углерода, как было уже ранее указано, одна и
та же молекулярная формула отвечает не одному, а нескольким
соединениям, различающимися по физическим и химическим
свойствам. Это явление было открыто давно. В 1830 г. Бериелиус дал
ему название «изомерия»*. Даже у сравнительно простых
соединений углерода существуют многочисленные изомеры. Так,
например, известно свыше 20 различных веществ состава C4HAO2,
которые резко отличаются по физическим и химическим
свойствам и иногда принадлежат к различным классам; некоторые из
них—кислоты (масляная и изомасляная), другие—нейтральные
вещества (например, диоксан). Составу С6Н12О2 отвечают 80
веществ, описанных в литературе. Подобных примеров можно
привести бесчисленное множество.
Теория химического строения Бутлерова позволила
правильно объяснить причины изомерии.
Рассматривая явления изомерии, Бутлеров указывает, что
при одном и том же молекулярном составе порядок связей атомов
в молекулах изомеров различен: «Единственно только некоторым
различием этого отношения (различием способа связи) и можно
объяснить явления изомерии».
Иными словами, различие в порядке связей в молекуле и
является причиной изомерии.
Основные методы определения химического строения. В
противоположность сторонникам теории типов Бутлеров
утверждает, что для каждого соединения возможна лишь одна формула
строения, которая должна выражать все свойства данного
вещества.
«Заключение о химическом строении веществ, по всей
вероятности, можно лучше всего основывать на изучении способов их
синтетического образования— и преимущественно на тех
синтезах, которые совершаются при температуре мало возвышенной
и—вообще—при условиях, где можно следить за ходом
постепенного усложнения химической частицы». Этот путь постоянно
используется в химии. В указанных условиях, как правило,
наблюдается нарушение одной или нескольких связей; происходит пе-
* От греческого слоиа «иэомерес», что означает «состоящий из равных
частей».
10. Теория химического строения Ьутлерова 39
рераспределение радикалов и остатков без нарушения структуры
последних.
Характерными примерами здесь являются: получение
углеводородов по реакции Вюрца (стр. 51), реакции образования
простых (стр. 172) и сложных эфиров (стр. 252), синтезы с помощью
металлорганических соединений (стр. 121), синтезы с участием
малонового эфира (стр. 275) и др.
«...Аналитические реакции могут также служить для
определения химического строения: так, например, если тело
претерпевает определенное разложение под каким-либо малоэнергичным
влиянием, то необходимо принять, что происходящие из него
простейшие вещества или остатки (радикалы) их находились
готовыми в разложившейся частице».
Первые стадии процессов изучения строения природных
соединений, как правило, начинаются с
разложения—систематической деструкции сложного вещества. Таковы пути изучения
строения жиров, Сахаров, белков, натурального каучука, витаминов,
антибиотиков и многих других.
Прошло уже свыше 90 лет с того времени, когда эти
положения были установлены. История развития органической химии
только подтвердила и дополнила их. Конечно, наряду с реакциями,
подчиняющимися этому основному «принципу наименьишх
изменений», наблюдаются и реакции, сопровождающиеся более или
менее сильными изменениями структуры радикалов. Но и эти
нарушения проходят обычно закономерно.
Бутлеров не мог в то время предложить широко использовать
физические методы, поскольку для этого, не было достаточного
материала и физика того времени еще не владела теми
средствами, которыми она располагает в настоящее время. Однако он
предвидел возможность использования физических свойств
веществ для определения их строения. Именно этим вопросам по-
свящеиа обширная глава в его учебнике «Введение к полному
изучению органической химии».
Изучение оптических и электрических свойств органических
веществ позволяет современной науке часто не только
более'быстро и точно определять относительное расположение атомов в
молекуле, но и устанавливать количественные характеристики
прочности связей между атомами и вычислять расстояния между
атомами в абсолютных единицах.
Пророчески прав был Бутлеров, говоря, что
«...исследование физических свойств имеет, для достижения упомянутой
цели (определения химического строения), огромное значе-
н ие».
В нашем элементарном курсе мы лишь частично касаемся
применения физических методов в о ганической химии^опрсделения
40 Введение
дипольных моментов (стр. 62), молекулярной рефракции (стр. 85),
вращения плоскости поляризации (стр. 156 ел.).
Особенно большое значение для распространения нового
учения имел курс «Введение к полному изучению органической
химии», изданный в Казани A864—1866) и переведенный позднее
A868) на немецкий язык. В нем Бутлеров систематически и
чрезвычайно убедительно доказывал справедливость положений своей
теории, приложимых ко всем классам органических соединений;
он показывал, как в конкретных случаях, пользуясь его теорией,
можно определять химическое строение органических
веществ .
Бутлеров смело предсказывал возможность получения ряда
новых органических веществ и даже целых классов
соединений.
В своих последующих экспериментальных исследованиях
Бутлеров осуществил синтез некоторых предсказанных им
соединений: второго из двух возможных бутанов (изобутан) A867);
первого представителя третичных спиртов (триметилкарбинол)
A864); триметилуксусной кислоты A872).
Трудами его учеников—В. В. Марковникова, А. М. Зайцева,
Е. Е. Вагнера, А. В. Попова и др.—был произведен синтез
многих новых соединений, возможность существования которых
вытекала из бутлеровской теории.
Взаимное влияние атомоё в химических соединениях.
Химический характер того или иного атома и поведение его при реакциях
определяются как природой самого атома, так и положением его
в молекуле.
Так, атомы водорода в метиловом спирте чрезвычайно резко
отличаются по реакционной способности.
Из четырех атомов водорода только один—водород гидроксиль-
ной группы—способен замещаться при действии металлического
натрия:
Н Н
Н—С—ОН + Na » Н—C-ONa + Н
А А
Большое влияние оказывают не только атомы элементов,
непосредственно связанные с водородом, но и соседние с ним
(«посредственно связанные»).
Этиловый спирт и уксусная кислота содержат гидроксильные
группы, однако атомы водорода в этих группах по своему
поведению различны. Это обусловлено тем, что в уксусной кислоте
10. Теория химического строения Бутлерова 41
имеется соседний с гидроксилом атом кислорода:
Н Н Н
Н-С—С—Н Н-С-С=О
Н ОН Н ОН
Чем ближе друг к другу находятся рассматриваемые атомы, тем
в большей степени проявляется их взаимное влияние. Так, эти-
ленхлоргидрин, имеющий строение (I), вполне устойчив, а его
изомер строения (II)—неустойчив и самопроизвольно отщепляет
хлористый водород:
н н н н н н
н-с-с-н Н-С-С-С1 —» н-с-с + на
а он н Ан н ^
і її
Многочисленные примеры взаимного влияния атомов в
молекулах постоянно наблюдаются в химии. Учение о взаимном
влиянии атомов является краеугольным камнем бутлеровской
теории.
Изучению этой проблемы посвящено много работ Бутлерова
и его учеников. Так, диссертация В. В. Марковникова на
соискание ученой степени доктора наук A869) носит название
«Материалы по вопросу о взаимном влиянии атомов в химических
соединениях». Найденные им закономерности присоединения к
двойной связи углеводорода будут изложены в соответствующем месте
учебника.
Итак, говоря о созданной Бутлеровым теории химического
строения, надо ясно представлять себе:
Исходные положения:
1. Атомы и построенные из них молекулы существуют реально.
2. Строение молекул вещества может быть определено
экспериментально.
Основные положения теории:
3. Свойства веществ определяются их качественным и
количественным составом и химическим строением их молекул.
4. Химическое строение—это порядок связи атомов в
молекуле, который может быть выражен химической формулой.
5. Принцип взаимного влияния атомов: атомы, связанные в
одну молекулу, влияют друг на друга. Наибольшее влияние
проявляют атомы, связанные непосредственно, значительно меньшее
взаимное влияние оказывают атомы,, не связанные
непосредственно.
42 Введение
Методы определения химического
строения:
6. Химическое строение может быть определено путем
изучения химических превращений вещества. При химических
процессах, происходящих в мягких условиях, строение «остатков»
не изменяется (принцип наименьших изменений).
7. Распознанию химического строения могут способствовать
учение о постоянстве валентности элементов и учение о
сцеплении однородных атомов (особенно С—С).
Новые возможности, открытые этой тео-
р и е й:
8. Действительное объяснение гомологии и изомерии, а также
предсказание новых органических веществ и их свойств.
9. Создание общей научной классификации органических
соединений.
10. Результатом разработки теории химического строения
явилось создание Бутлеровым учебника «Введение к полному
изучению органической химии» A864—1866), в котором он
изложил все содержание органической химии с позиций нового
учения.
Открытые закономерности и сделанные обобщения все больше
и больше находят себе объяснение в настоящее время в связи с
углублением наших знании о природе химических связей.
Ї1. Классификация органических соединений. «Химическая
классификация будет естественной, если главным основанием
сближения одних тел и разделения других служит аналогия и
различие химической структуры их, а натура определяется натурой
составных частей, их количеством и химическим строением
частицы»,—такие требования к научной классификации
органических веществ выдвинул Бутлеров. Следовательно, к одному
классу должны быть отнесены соединения, сходные по структуре.
Химически наименее активными, обычно мало
изменяющимися в типичных реакциях, являются связи С—С и С—Н. Поэтому
химический характер органических соединений определяется
в первую очередь наличием в молекуле атомов галоида,
кислорода, азота и др. Так, соединения, содержащие группы
О ^
и II
о
являются кислотами. Наличие группы —NH, придает веществу
основной характер.
Подобные группы называют функциональными.
//. Классификация органических соединений 43
Таким образом, наиболее прочной и малоизменяемой частью
сложного органического вещества является его углеводородная
часть и особенно углеродная цепь—углеродный скелет.
Три главных раздела органической химии определяются
структурой этой как бы основной «опоры» соединений—углеродного
скелета.
А. Ациклические (алифатические)
соединен и я—скелет составлен из непосредственно связанных атомов
углерода в виде незамкнутой, прямой или разветвленной, цепи:
Б. Карбоциклические соединения, в
молекулах которых имеются углеродные цепи кольчатого характера:
в.
жащие
других
d—с
Г е т е р о ц і
с с—с—с
V
ІКЛИЧЄСКИ
' С t І
е соединения, содер-
кольчатые системы, включающие, кроме углерода, атомы
элементов
С С
V
с
; с с—с
А і
V
с
/\ N
і і і
w
Образующиеся при этом соединения обладают рядом
характерных особенностей.
Внутри каждого раздела соединения распределяются по
классам в зависимости от имеющихся в них функциональных групп.
Простейшими соединениями являются углеводороды
предельного vi непредельного рядов.
Более сложные соединения можно представить как
образовавшиеся из того или иного углеводорода путем замещения
одного или нескольких водородных атомов соответствующими
атомами или атомными группами. Так возникают классы, оксисоедине-
ний—одноатомных и многоатомных спиртов с одной или
несколькими группами ОН; оксосоединений, содержащих группы С=О
44 Введение
(альдегиды и кетоны); далее—карбоновые кислоты с
карбоксильными группами СООН и др.
В молекуле органического соединения может находиться и
несколько различных функциональных групп.
Таковы оксикислоты:
Н Н о Н Н
и )• ^ / \- г1
н онЧ°н н_/ V-cf
\ / \Н
вксяпрогтионовая Q—Q
(молочная) кислота і ,
н он
оксибензнйная (салициловая)
кислота
Каждая из этих функциональных групп, сохраняя в основном
свой химический характер, оказывает влияние на другие и сама
испытывает воздействие соседних атомов.
Таким образом, классификация органических соединений
основывается на сходстве и различии их строения, отображаемого
структурными формулами; она является генетической, т. е.
показывает развитие (генезис) данного сложного соединения из
простейшего углеводорода.
Отражая именно эту характерную черту, Шорлеммер, как
было указано выше (стр. 17), определил органическую химию как
химию углеводородов и их производных.
ЧАСТЬ ПЕРВАЯ
СОЕДИНЕНИЯ С ОТКРЫТОЙ ЦЕПЬЮ
УГЛЕРОДНЫХ АТОМОВ
ГЛАВА І
УГЛЕВОДОРОДЫ
ПРЕДЕЛЬНЫЕ УГЛЕВОДОРОДЫ
Предельными углеводородами, или парафинами, называются
углеводороды, в молекулах которых атомы углерода связаны
между собой простой (ординарной) связью, причем все единицы
валентности, не затраченные на связь между углеродными атомами,
насыщены атомами водорода.
12. Гомологический ряд предельных углеводородов.
Простейшими предельными углеводородами являются:
СН4 С2На С3Н8 . QH10 C5H12
метан этан пропан бутан пентан
Далее в этом ряду следуют углеводороды: С6Н14(гексан), QHj,
(гептан), С8Н18 (октан), С9Н20 (нонан) и т. д. и еще более сложные,
например Св0Н122 (гексаконтан) и C70HU2 (гептаконтан). Каждый
член этого ряда отличается от предыдущего члена ряда на
группу СН2.
Вещества, которые, будучи сходны по своему строению,
отличаются друг от друга по составу на одну или несколько групп
СН2, называются гомологами. Группа гомологов составляет
гомологический ряд. В данном случае приведен гомологический ряд
предельных углеводородов, или парафинов. Предположим, что
имеется предельный углеводород с неразветвленной цепью из
п атомов углерода:
ИНН Н Н Н
Н-С-С—С--.С-С—С—Н или H(CHS)„H
ААА ААА
Каждый из этих углеродных атомов связан с двумя атомами
водорода; сверх того, каждый из двух крайних атомов углерода
связан еще с одним атомом водорода. Следовательно, число всех
атомов водорода равно 2л+2, и' общий состав предельных
углеводородов может быть выражен формулой С„Н2 2. Как будет
46 Глава І. Углеводороды
ясно из последующего, эта формула применима и к предельным
углеводородам с разветвленной углеродной цепью.
В сравнении с другими углеводородами предельные
углеводороды наиболее богаты водородом; каждый углеродный атом в
молекуле углеводорода соединен с максимально возможным
числом водородных атомов, и, таким образом, достигается высший
предел насыщения углерода водородом. Отсюда и произошло
название предельные, или насыщенные, углеводороды.
Названия углеводородов этого ряда имеют одинаковое
окончание «ан». Первая часть названия углеводорода, начиная от С5
и выше, производится из греческих числительных, указывающих
число атомов углерода, например углеводород С^Н12—пентан,
СвН14—гексан, QH,,.—гептан, С8Н1а—октан, С9Н20—нонан и т. д.
Количественное изменение состава молекулы, вызванное
прибавлением группы СН2 к предыдущему члену ряда, приводит к
образованию качественно отличного вещества. Это является одним
из нагляднейших примеров закона природы о переходе
количества в качество.
Этот закон проходит красной нитью через всю химию. Энгельс
писал: «Химию можно назвать наукой о качественных
изменениях тел, происходящих под влиянием изменения
количественного состава».
Основная заслуга исследования гомологического ряда
парафинов принадлежит Шорлеммеру*. Оценивая значение работ Шор-
леммера по углеводородам парафинового ряда, Энгельс писал:
«Теми знаниями, которыми мы в настоящее время обладаем о
парафинах, мы обязаны главным образом Шорлеммеру. Он
исследовал известные в то время соединения, принадлежащие к группе
парафинов, отделил один вид от другого, многие из них были
впервые получены им в чистом виде. Другие члены ряда, которые
по теории должны были существовать, но в действительности не
были еще найдены, были им "открыты и им же получены. Таким
образом, он стал одним из основателей современной научной
органической химии».
* Карл Шорлеммер A834 — 1892) ролился в Германии В 1858 г. переехал
в Англию. С 1874 г был профессором органической химии в Манчестере.
Участник Первого Интернационала, коммунист, друг Ф. Энгельса и К. Маркса.
Главные работы К. Шорлеммера относятся к изучению и синтезу предельных
углеводородов. Своими исследованиями он опроверг утверждение что этан
существует в двух изомерных формах, доказав, что оба «изомера этана»
тождественны и являются одним и тем же диметилом СНа—СИ:,. Эта работа имела
большое теоретическое значение и сыграла выдающуюся роль в истории
органической химии. Большой известностью пользовались его учебники химии
(составленные частично при участии Роско) Его в нысшей .-тепени ценная
книга «Возникновение и развитие органической химии» переведена в 1937 г.
на русский язык.
ІЗ. Углеводородные радикалы (алкилы) 47
13. Углеводородные радикалы (алкилы). Если от молекулы
предельного углеводорода отнять один атом водорода, то
получится остаток, называемый одновалентным радикалом. В
свободном виде эти радикалы не могут быть выделены*, однако
понятие о радикалах можно использовать при составлении названия
сложных углеводородов и других органических соединений.
Названия радикалов образуются из названий соответствующих
предельных углеводородов заменой окончания ан на ил
Углеводород
СН4 ....... метан
Q,He этан
С3Н8 пропан
С4НЫ бутан
С5Н12 пентан
СвН14 гексан
гептан
—сн, .
-ел
—с3н,
-ел
—C5Hil
-с,н1й
Радикал
метил
ЭТИЛ
пропял
бутил
амил
гексил
гептил
Исключение из правила в случае наименования для радикала С6НИ —
амил произошло потому, что названия простейших радикалов появились
ранее названий самих углеводородов. Они произошли из наименований
алкоголен (метиловый этиловый, амиловый). Именно по этой причине
углеводородные радикалы часто называют общим именем алкилы.
Пользуясь названиями радикалов, можно составлять названия
многих органических соединений:
CH.jJ йодистый метил
С5НпВг бромистый амил
С(СН3L тетраметилметан
И Т. Д.
14. Строение предельных углеводородов. Каждый
предельный углеводород можно произвести от соответствующего
предыдущего члена гомологического ряда путем замещения одного
атома водорода метилом; тем самым состав молекулы усложняется
на группу СНа (метилен). Таким путем от метана производим этан
и от этана—пропан:
Н Н г_[ Н ^ Н
н-с-н н-с-с—н н-с!—с-с—н
* О возможности кратковременного существования в химических
реакциях так называемых «свободных радикалов» будет указано позднее (стр. 525).
48 Глава f. Углеводороды
В молекулах метана и этана все атомы водорода совершенно
равноценны, и безразлично, какой из них заместить метилом. Это
соответствует тому, что первые три члена гомологического ряда
парафинов не имеют изомеров.
Но в молекуле пропана уже не все атомы водорода
равноценны. Атомы водорода, связанные с крайними углеродными
атомами, отличны по своему положению от атомов водорода,
связанных со средним углеродным атомом. Поэтому замещение в
молекуле пропана атома водорода метилом можно произвести двумя
путями: заместить атом водорода у среднего углерода или же
у крайнего атома углерода. При замещении атома водорода у
одного из крайних атомов углерода получается нормальный
бутан:
н Н Н н
\ 1 1 /
Н-С—С—С—С— Н
При замещении атома водорода у среднего атома углерода
образуется изобутан:
Н " Н
Н\ I /Н
Н—С-С—С—Н
н/ і \н
Состав обоих веществ выражается одной и той же
формулой С4Н10: нормальный бутан и изобутан являются
изомерами. Изомерия бутана была открыта А. М. Бутлеровым,
который в 1867 г. синтезировал изобутан.
В молекуле нормального бутана имеются два рода
водородных атомов: одни из них входят в состав групп СН3, симметрично
расположенных на концах цепи, другие—в состав групп СН2,
расположенных в середине цепи. В соответствии с этим от
нормального бутана можно произвести два углеводорода состава
С5Н12—нормальный пенчан и нэопентан:
Н " н
H\ M I /H \ I I /н
Н—С-С—С-С—С—Н Н-С—С-С-С—Н
1 il
нормальный пентод изипентап
14. Строение предельных углеводородов 49
В молекуле изобутана мы вновь встречаемся с двоякого рода
водородными атомами: девять из них входят в состав совершенно
одинаково расположенных групп СН3 и отличаются по своему
положению от десятого водородного атома. Поэтому от
нзобутана также можно произвести два углеводорода:
н " н н н н
н\ I I /Н Н\ ! /Н
Н-С-С-С-С—Н Н-С-С-С—Н
н/ і А \н н/ і \н
111
изопеитан
IV
тетраметнлметан
Из приведенных выше четырех структурных формул две (II
и III) совершенно одинаковы. Таким образом, для углеводорода
состава CSH12 возможны (и на самом деле известны) три изомера:
нормальный пентан, изопентан и тетраметилметан. Последнее
название объясняется тем, что тетраметилметан можно
рассматривать как метан СН4, в молекуле которого все атомы водорода
замещены метильными группами.
По мере перехода к высшим членам ряда число теоретически
возможных изомеров очень быстро возрастает:
Число атомов углерода
в молекуле .... 1234567 8 9 10 11 12 13
Число изомеров ... 1 1 1 2 3 5 9 18 35 75 159 355 803
Приведенные в этом параграфе развернутые формулы
строения наглядно показывают порядок связи всех атомов в молекуле.
В органической химии часто применяется другой способ
изображения формул строения—отмечают черточкой* только связи
между углеродными атомами:
Этан СН3—СНЭ
Пропан CHj—CHj—СН3
Бутан CHg—СНа—СН,—СНа
Пентан СН3—СН,—СНа—СНа—СН3
СНЗЧ
Изобутан >СН—СН3
СН,/
* В старой литературе иногда вместо черточек приводятся точки, например
СК3-СН3.
4-788
50 Глава Л Углеводороды
СН3
Изопентан >
сн/
•г
Гетраметилметан >Х
СН/ \СН3
Соединения с неразветвленной углеродной цепью, как,
например, бутан, пентан, называют соединениями нормального
строения; если же цепь имеет разветвления, то говорят об изо-
строении. Примерами соединений, обладающих изо-строением,
могут служить изобутан, изопентан.
Атом углерода, связанный только с одним другим атомом
углерода, называется первичным, связанный с двумя
другими—вторичным, с тремя—третичным и с четырьмя—четвертичным:
С С С
С—СН3 СН, С—СН С—С—С
і
первичный вторичный третичный :етвертичньй
В приведенных выше примерах первичный, вторичный н т. д.
;ітомьі углерода выделены жирным шрифтом.
В молекулах углеводородов нормального строения бывают
только первичные и вторичные атомы углерода. В молекулах
соединений изо-строения есть третичные или четвертичные атомы
углерода. В молекуле изобутана третичный атом углерода
затрачивает три единицы валентности на связь с тремя другими
атомами углерода и одну единицу валентности—на связь с одним
атомом водорода. Каждый из трех остальных атомов углерода
затрачивает одну единицу валентности на связь с третичным атомом
углерода и три остальные единицы валентности—на связь с тремя
атомами водорода, и поэтому в молекуле изобутана имеются
один третичный и три первичных атома углерода.
15. Нахождение в природе и получение предельных
углеводородов. Предельные углеводороды весьма широко
распространены в природе.
В громадном количестве они содержатся в некоторых видах
ііефти (см. стр. 67). Твердые предельные углеводороды являются
главной составной частью битумов, асфальтов, озокерита
(горного воска). Очень большие количества метана, как это
показывают данные спектрального анализа, находятся в атмосфере
больших планет: Юпитера, Сатурна, Урана и Нептуна.
14 Нахождение в природе и получение предельных углеводородов 51
Из лабораторных способов синтетического получения
предельных углеводородов наибольшее значение имеет открытая в 1854 г.
реакция Вюрца*. Эта реакция заключается в нагревании
галоидных алкилов с металлическим натрием. Приведем два примера.
Этан С2Н„ можно получить действием на йодистый метил
металлического натрия:
Н Н Н Н
H-C-IT+SNa'T П-С-Н » Н—С-С-Н + 2NaJ
А ' ' А А А
Два атома натрия отнимают от двух молекул йодистого
метила по одному атому иода. При этом у атомов углерода
освобождается по единице валентности, которые взаимно насыщаются,
образуя молекулу этана.
Схема реакции не раскрывает ее механизма, который большей
частью значительно сложнее, чем схема. Так, исследования
П. П. Шорыгина и других химиков показали, что при реакции
Вюрца натрий сначала замещает галоид, после чего натриевое
производное реагирует с галоидным алкилом:
2№ » NaJ +- СН3—Na
СН3—Na + J—СН3 » NaJ + СН3—СН3
Пропан С3Н8 можно получить действием металлического
натрия на смесь йодистого метила и йодистого этила**:
Н Н Н Н Н Н
Н—С—j 7+'2Na+'.ІІ—С—С—Н » Н—С—С—С—Н + 2NaJ
А ' ' А А А А А
пропан
Реакцией Вюрца можно воспользоваться и для установления
строения получаемых углеводородов. Как мы знаем, при хнми-
* Адольф Вюрц (А. Wurtz). французский химик A817 —1884). Ьл-о работы
в области органической химии весьма многочисленны. Им была открыта
реакция синтеза предельных углеводородов (действием металлического натрия на
галоидлроизводные), реакции получения и обнаружения жирных аминов.
В 1859 г. Вюрцем был получен первый представитель двухатомных алкого-
лей —этиленгликоль («удвоенный тип воды»),
** Очевидно при реакции образуется также этан (из двух молекул
йодистого метила и бутан (из двуя молекул йодистого этила).
52 Глава І. Углеводороды
ческих реакциях, протекающих в умеренных условиях, строение
радикалов остается неизменным, поэтому строение
углеводорода определяется строением взятых для реакции галоидных ал-
килов.
В качестве примера приведем установление строения бутана.
Как уже было указано, для бутана С4Ні0 известно два
изомера—нормальный бутан и изобутан. Первый из них—газ,
сгущающийся в жидкость при —0,5 °С, другой—также газ, но
сгущающийся в жидкость только при —11,7 °С.
При действии металлического натрия на йодистый этил может
получиться только углеводород, имеющий строение нормального
бутана:
Н Н Н Н Н Н Н Н
Н-С-С—J + 2Na + J-C—С—Н > Н-С—С-С-С-Н + 2NaJ
и а ап
Как показывает опыт, температура кипения его —0,5 °С.
Следовательно, углеводород, сгущающийся в жидкость при —11,7 °С,
должен иметь строение изобутана.
16. Физические свойства предельных углеводородов.
Физические свойства некоторых предельных углеводородов приведены
в табл. 1.
Легко заметить, что первые пять углеводородов и один из
изомерных пентанов—тетраметилметан при обычной температуре
являются газами, следующие—жидкостями, углеводороды же,
начиная с С^Н^,—твердые вещества. С увеличением числа атомов
углерода в молекуле возрастает их плотность, а также повышаются
температуры плавления и кипения углеводородов. Предельные
углеводороды с разветвленной цепью кипят при более низких
температурах, чем изомерные углеводороды с нормальной цепью.
Наоборот, температура плавления тем выше, чем больше
разветвлена углеродная цепь. Так, один из октанов, формула
которого (СН8)8С—С(СНа)8, плавится при +100,6 °С, в то время как
температура плавления нормального октана СН9(СН2NСН8
—56,8 °С, т. е. на 157,4 °С ниже.
В ряду предельных углеводородов с увеличением молекулярного
веса повышаются температуры плавления и кипения.
Это положение, разумеется, справедливо только в том случае,
когда сравниваются углеводороды одинакового строения.
Метан и этан не обладают запахом, жидкие углеводороды
имеют характерный запах, высшие углеводороды—без запаха.
Все углеводороды практически нерастворимы в воде;
плотность их меньше единицы.
Физические свойства предельных углеводородов
Таблаца 1
Название
Этан
Пропан
н-Бутан
Изобутан
«-Пеытан
Изопентан
Тетраметилк.'зтан
н-Гексан
«-Гептан
«-Октан
«-Нонан
«¦Декан
«Пентадекан
н-Гексадекан ....
н-Эйкозан
н-Гексаконтан
н-Гептаконтан
Формул..
сн4
С.Н,
С4Н10
с6н13
C8H1S
С9На0
C1UH23
Физическое
состояние при обычной
температуре
Газ
Жидкость
Гаа
Жидкость
Твердое тело
Температура, °С
плавления
—182,5
—183,3
—187,6
— 138,3
— 159,6
—129,8
— 159,9
—16,6
—95,3
—90,6
—56,8
—53,6
—29,7
+ 10
+ 18
+ 36,4
+99
+ 105,2
кипения
—161,6
—88,6
—42,1
—0,5
— 11,7
+36,07
+ 27,9
+9,5
+68,7
+98,5
+ 125,7
+ 150,8
+ 174,0
+ 270,5
+287,1
+ 309,7
+250
(при 0,00001 мм рт. ст.)
+ 300
.при 0,0001 мм рт. ст.)
Плотность, г/см*
В ЖИДКОМ СОСТОЯНИИ)
0,416 (—161,6 °С)
0,5462 (—88.6 °С)
0.5S24 (—42,1 °С)
0,5788 B0 °С)
0,5592 B0 3С)
0,6264 B0 °С)
0,6199 B0 °С)
0,613 @°Q
0,6594 B0 °С)
0,6838 B0 °С)
0,7028 B0 "С)
0,7179 B0 °С)
0,7298 B0 °С)
0,7658 B0 °С)
0,7749 B0 °С)
0,7777 C7,4 °С)
0,7203 A90,6 °С)
54 Глава І. Углеводороды
17. Химические свойства предельных углеводородов.
Предельные углеводороды отличаются химической инертностью, т. е.
при обычной температуре не окисляются и не реагируют с
концентрированной серной кислотой и рядом других энергичных
реагентов. Этим объясняется их название—парафины (parum
affin is), что в переводе на русский язык означает «мало сродства».
В результате более подробных исследований установлено, что
предельные углеводороды инертны только по отношению к
основаниям, минеральным кислотам средней силы и окислителям в
водном растворе. К реакциям присоединения парафины неспособны,
так как в этих соединениях все связи атома углерода насыщены.
Однако они легко вступают в реакции замещения при
взаимодействии с хлором и бромом, образуя соответстоующие галоидпроиз-
водные. Эти реакции происходят на рассеянном солнечном свету
даже при обыкновенной температуре.
При действии на метан хлора один атом последнего отнимает
от метана один атом водорода, а другой атом хлора становится
на место водорода. В результате реакции образуется хлористый
метил и хлористый водород:
сн JH+cij—а —»снаа + неї
і !
хлористый
метил
Реакции с участием свободных радикалов. Если смешать газообразные хлор
fl водород в темноте при обычной температуре, то взаимодействие совершается
крайне медленно Чтобы ускорить химическую реакцию необходимо нагреть
смесь или воздействовать на нее ярким светом. Как теперь известно в этих
условиях нейтральные молекулы хлора поглотав іепловую или световую
внергию распадаются с образованием свободных атомов или радикалові
:с):сі: —*- :сі- + -а:
Радикал содержит нечетное число электронов в молекуле (имеет неспаренныв
электрон) не несет электрического заряда т. е. не является ионом. Реакции
с участием радикалов называют радикальными. Атомы или радикалы могут ре-
комбинироваться* снова в молекулу Сії с выделением энергии нли могут
воздействовать на молекулу водорода Атом хлора разрывает молекулу водорода
давая молекулу НС1 и свободный атом водорода!
ч:сі- + н:н —>- н.ск + н-
Атомарный водород в свою очередь может атаковать молекулу хлора в
результате чего образуется молекула хлористого водорода и атомарный хлор
Так возникает цепная реакция, которая может привести к вврыву.
Термин рекомбинация—обратное сдваивание (ге—лат. приставка,
означает обратно, com—вместе bine—двойной).
17. Химические свойства предельных углеводородов 55
Аналогично может происходить взаимодействие хлора с предельными
углеводородами, например метаном. Свободный атом хлора отнимает от молекулы
метана атом водорода, образуя НС1 и свободный радикал метил:
Н Н
:су + н:с:н —*- :а:н + -с:н
н " н
При этом так же. как в случае реакции хлора и водорода, происходит
выделение энергии, что приводит к развитию самоускоряющейся реакции и может
привести к взрыву газообразной смеси. Саободный метил или вообще
углеводородные радикалы существуют ничтожные доли секунды. Они могут или
развить реакцию хлорирования встречаясь с хлором или рекомбинироваться.
Цепь радикальных превращений может оборваться при встрече радикалов с
другими радикалами, со стенками сосуда или с посторонними примесями.
Открытие и изучение механизма цепных реакций (Н. Н. Семенов и его
школа) имело громадное значение для понимания ряда фотохимических
процессов, реакции окисления, полимеризации и др.
Наряду с образованием хлористого метила происходит
дальнейшее замещение водорода хлором, в результате чего получается
смесь различных веществ:
С1СН4—hl -fClj—СІ , CH,CIV + НО
С1.СН—і
хлористый
метилен
Н +СІІ—CI » СНС13 + НС1
хлороформ
С|3с—JH+cij—сі —»ссі4 + на
четырех-
XJJOp^Clbfti
углерод
На прямом солнечном свету взаимодействие метана и хлора
происходит со взрывом. При этом образуется хлористый водород
и выделяется углерод:
СН, + 2С1, » 4НС1 + С
Свойство хлора замещать е органических соединениях водород
наблюдалось еще p. 17Q3 р. русским академиком Г. Ё. Ловиием на примере
получения хлоруксусных кислот (см. стр. 281). а в 30-х годах прошлого века —
было вторично открыто французским химиком Дюма.
Серная кислота при обыкновенной температуре не действует
на предельные углеводороды; при слабом нагревании дымящая
серная кислота может действовать на углеводороды, образуя
сульфокислота (см. стр. 183). Реакция сульфирования идет легче с
предельными углеводородами, имеющими изо-строение
(содержащими группу СН).
56 Глава І. Углеводороды
При высокой температуре серная кислота действует как
окислитель, превращая углеводороды в углекислый газ и воду.
В технике для получения сульфопроизводных часто используют реакцию
сульфохлорирования. В керосиновую фракцию нефти, содержащую большое
количество углеводородов нормального строения, пропускают равномолеку-
лярную смесь сернистого газа и хлора. Реакционную смесь освещают
ртутными лампами, дающими большое количество света с короткими волнами
{фиолетового и ультрафиолетового). Из молекул хлора образуются под
воздействием облучения атомы хлора, которые и вызывают реакцию:
R—снэ + so, + сі, —» на + R—сна—S—а
В результате реакции получаются хлорангидриды жирных
сульфокислот, используемые для получения моющих средств (стр. 234).
Азотная кислота при обыкновенной температуре почти не
действует на предельные углеводороды; при нагревании действует
как окислитель. Однако, как показал М. И. Коновалов*, слабая
азотная кислота при нагревании под повышенным давлением
действует «нитрующим» образом. При реакции- нитрования метана
получается почти исключительно нитрометан:
СН4 + НО—N0, » СНЭ—NO, -f H,0
Предельные углеводороды легко нитруются в газовой фазе при
150—175 °С двуокисью азота или парами азотной кислоты; при
этом частично происходит и окисление. В настоящее время
существуют промышленные установки для нитрования предельных
углеводородов в газовой фазе.
Окислители, даже такие, как хромовая смесь и
марганцовокислый калий, при обыкновенной температуре не действуют на
предельные углеводороды с нормальной углеродной цепью. Легче
* Михаил Иванович Коновалов A858—1906) окончил в 1884 г. Москов
ский университет. В 1896—1899 гг.—профессор Московского
сельскохозяйственного института, с 1899 г.—профессор Киевского Политехнического
института. Первые работы М. И. Коновалова были посвящены изучению природы
кавказской нефти. Он разработал методы выделения, очистки и получения
различных производных нафтенов (стр. 545), изучал действие брома и
бромистого алюминия на нафтены. В 1888 г. Коновалов открыл нитрующее действие
разбавленной азотной кислоты при нагревании ее с предельными
углеводородами (стр. 358). Исследования в этой области он обобщил в докторской диссер
гации «Нитрующее действие азотной кислоты на углеводороды предельного ха
рактера» A893). Предложенный им метод позволил получить и исследовать
многочисленные новые нитросоединения. М. И. Коновалов разработал способ
получения из нитросоединений оксимов (стр. 194), спиртов, альдегидов и кето-
нов. Он использовал также реакцию нитрования для определения строения
углеводородов, создал метод разделения нитросоединений и их очистки
18. Метан Ь<
окисляются углеводороды, в молекулах которых имеется
третичный атом углерода (см. стр. 50).
При высокой температуре предельные углеводороды
воспламеняются на воздухе и сгорают с образованием СО2 и Н2О:
СН4 + 20, > СО2 + 2Н,0
При средних температурах эти углеводороды (особенно
твердые) окисляются кислородом воздуха, давая
кислородсодержащие вещества с меньшим числом атомов углерода в молекуле;
следовательно, при этом происходит не только окисление, но и
расщепление молекул углеводородов.
Итак, члены гомологического ряда предельных углеводородов
имеют сходное строение (молекула каждого углеводорода
отличается от молекулы предыдущего углеводорода на группу СН2).
Их состав может быть выражен общей формулой. Члены ряда
обладают сходными свойствами, закономерно изменяющимися с
увеличением числа атомов углерода в молекуле. Это показывает,
какое облегчение для изучения органической химии дает понятие
о гомологических рядах. Достаточно изучить свойства главных
представителей гомологического ряда для того, чтобы иметь
представление об основных свойствах остальных членов ряда.
Введение понятия гомологических рядов в качестве основы
классификации органических веществ является заслугой французскою
ученого Жерара.
18. Метан. Метан СН4 нередко называют болотным, или
рудничным, газом. Образование его на дне болот объясняется
метановым брожением клетчатки, происходящим под влиянием
особых бактерий. Разложение клетчатки в первом желудке жвачных
животных также представляет собой метановое брожение,
поэтому воздух, выдыхаемый животными, употребляющими в пищу
клетчатку, всегда содержит метан. Метан находится в газах
кишечника и крови как животных, так и людей. Газ, содержащийся
в пустотах каменноугольных пластов, состоит на 80—90% из
метана. Метан образуется при сухой перегонке дерева, торфа и
каменного угля и содержится в природном и в светильном газах.
Метан может получаться при высокой температуре
непосредственно из элементов:
С + 2Н2 ^=t CH4
Так, он образуется в вольтовой дуге между угольными
электродами в атмосфере водорода. Если нагревать с водородом мел ко-
раздробленный никель, покрытый слоем сажи, то при 475 °С в
газовой смеси содержится 51% метана; никель является в этом
случае 1*атализатором.
58 Глава І. Углеводороды
При помощи никелевого катализатора в метан могут быть
восстановлены при 250—400 °С окись углерода и углекислый газ:
со + зн, —»сн4 + н,о
СО, + 4Н2 » СН4 + 2Н2О
В лаборатории метан легко получить сплавлением
уксуснокислого натрия с едким натром:
СНЭ— I COONa -f NaO |—Н » СН4 + Na2CO8
Другой удобный лабораторный способ получения метана—
действие воды на карбид алюминия:
А14С3 + 12Н.0 » ЗСН4 + 4А1(ОН)Э
Метан—газ без цвета и запаха, плохо растворимый в воде; горит
бледным пламенем; смесь его с воздухом при воспламенении
сильно взрывает.
Обычно метан получают из природного газа (см. стр. 51 и 65).
Это почти чистое и доступное органическое сырье, которое служит
для получения хлористого метила (СН3С1), хлороформа (СНС13),
четыреххлормстого углерода (СС14) и др. Все эти продукты широко
применяются в химической промышленности (см. стр. 113).
19. Общие черты реакций замещения. При рассмотрении
приведенных реакций углеводородов можно сделать ряд обобщений,
которые помогут в дальнейшем лучше понимать причины
протекания типичных реакций. Это в первую очередь относится к
многочисленным реакциям замещения.
Легко заметить, что в таких реакциях одним из получающихся
продуктов является какое-либо простое и очень прочное
неорганическое вещество: НС1—при хлорировании, NaBr—при реакции
Вюрца, Na2CO3—при получении метана из уксуснокислого
натрия, Н2О—при нитровании. Именно образование этих соединений
d первую очередь и обусловливает выделение энергии (тепла),
а тем самым и направление хода реакции. Таким образом,
например, хлорирование метана вызывается большим сродством хлора
к водороду, ведущим к образованию НС1, а получающийся при
этом СН3С1 с термодинамической точки зрения является лишь
своего рода «побочным продуктом».
Отсюда понятно, что такие реакции, как, например
НС1 » СН4 + СІ,
СН8С| + НС1 > СН2С13 + Н,
не могут протекать по термодинамическим соображениям.
20. Женевская номенклатура органических соединений 59
Чтобы не делать грубых ошибок и лучше понимать
химические процессы в органической химии, очень важно, особенно на
первых порах, писать полностью все продукты реакции.
20. Женевская номенклатура органических соединений.
Ввиду многообразия органических соединений, вопрос об их
номенклатуре приобретает особое значение. Название органического
соединения должно быть составлено так, чтобы оно не только
указывало число атомов, входящих в состав вещества, но и давало
представление о структуре его молекулы, чтобы можно было легко
и точно написать структурную формулу. Так, из названия «те-
траметилметан» следует, что мы имеем дело с веществом, которое
производится от метана замещением каждого из четырех атомов
водорода метилом:
н сн3
Н-І-Н СНд-С-СНз
н in,
метан тетраметнлметан
Названия типа «тетраметилметан» относятся к так
называемой «рациональной» номенклатуре. Правила составления
названий должны быть общими для различных классов соединений.
Наиболее удобной и универсальной является номенклатура,
выработанная в 1892 г. в Женеве Международным совещанием
представителей химических обществ. Она получила название
женевской, или официальной, номенклатуры.
Изложим основные принципы этой номенклатуры.
По женевской номенклатуре предельные углеводороды с
нормальной, т. е. с неразветвленной, цепью имеют названия: метан,
этан, пропан, бутан, пентан, гексан, гептан, октан, нонан, декан
и т. д. (по греческим числительным). Названия всех производных
этих углеводородов образуются от тех же корней. Чтобы
составить название какого-либо углеводорода с разветвленной цепью,
его рассматривают как продукт замещения нормального
углеводорода; за углеродную цепь этого углеводорода принимают
наиболее длинную цепь атомов углерода в молекуле. Так, изогептан
СН3—СН-СН,—СН,—CHj-CHg
СН3
рассматривается как производное гексана (наиболее длинная
цепь), а именно, как продукт замещения в гексане одного из
атомов водорода метилом. Для составления названия этого
углеводорода нумеруют атомы углерода наиболее длинной цепи,
начиная от того конца, к кото ому ближе разветвление, и цифрой ука-
60 Глава І. Углеводороды
зывают, при каком атоме углерода находится метил
СНЭ—СН—CHj-CH,—СНї-Cti,
снэ
Таким образом, изогептан по женевской номенклатуре будет
называться 2-метилгексаном.
Так как тетраметилметан является продуктом замещения в
молекуле пропана двух атомов водорода у среднего атома углерода
двумя группами СНЭ, то его название по женевской номенклатуре
будет 2,2-диметилпропан:
СН3
СНз—С—СНз
сн3
2,2-диметилпропан
Атомы углерода, которые не находятся в наиболее длинной
цепи, составляют боковую цепь. В молекуле 2,2-диметилпропана
имеются две боковые цепи, из которых каждая является метилом.
Те же правила применяются и для обозначения галоидпроиз-
водных. Приведем примеры:
сна—cHsci CHjCi—сн2сі сн3—сна,
хлорэтан 1,2-дихлорэтан 1,1-днхлорэтан
СНа—CHj—CHC1, СНа—СС12—СН3 СН,С1-СН2-СН2С1
1,1-дихлорпропап 2,2-днхлорпропан 1,3-дихлорпропан
СН3-СС1-СНЭ СН3—СН2—СС1—СС1—СНд
СН3 СНд СН3
2-хлор-2-метнлпропан 2,3-дихлор-2,3-днметилпеитан
Если цепь не разветвлена, то счет начинают с того конца цепи,
ближе к которому находится галоид:
СНЭ—CHCl—CHS—СНЭ
2-хлорбутан
Ясно поэтому, что название не изменится, если формулу
переписать в другом порядке. Так, формулы
СНЭ-СН2-СНС12 и СНС12-СН2-СН3
изображают один и тот же 1,1-дихлорпропан.
Общее название предельных углеводородов по женевской
номенклатуре—алканы.
21. Электронные схемы строения органических соединений 61
Более подробно о составлении названий органических
соединений по женевской номенклатуре см. Приложения.
21. Электронные схемы строения органических соединений.
Согласно теории Косселя A916), излагаемой в курсах
неорганической химии, атомы элементов, вступая в химическое соединение,
отдают или получают валентные электроны. В результате этого
они приобретают электрические заряды, т. е. становятся ионами,
формируя вокруг себя устойчивые восьмиэлектронные оболочки.
Обозначая внешние электроны одного атома крестиками, а
другого—точками (значение крестиков и точек совершенно
одинаково, введены они лишь для наглядности), можно изобразить
процесс соединения натрия с хлором следующим образом:
Na« + »сі: —>- Na* + ;сіГ
' • • • *
Атом натрия, отдавая один электрон, становится
положительным ионом с электронной оболочкой, подобной оболочке неона;
атом хлора, принимая один электрон, становится отрицательным
ионом и образует электронную оболочку, подобную оболочке
аргона. Притяжение между разноименными ионами удерживает их
вместе. Натрий проявляет положительную валентность, равную
единице, а хлор—отрицательную валентность, также равную
единице.
Такая связь носит название гетерополярной (электровалентной
или ионной) связи.
Соединения, молекулы которых состоят из ионов, называются
ионными.
Теория Косселя применительно к этим соединениям
подтверждается целым рядом фактов. Различными путями было доказано,
что кристаллическая решетка этих веществ состоит из ионов, а
не из атомов. В водных растворах солеобразных соединений
происходит электролитическая диссоциация, и ионы отделяются
один от другого и перемещаются самостоятельно.
В то же время существует громадное число веществ, в
молекулах которых ионов обнаружить не удалось и которые
неспособны к электролитической диссоциации. Таковы, например,
водород Н2, кислород О2 и подавляющее большинство
органических соединений; в последних никогда не удавалось обнаружить
ионов углерода.
Связь в таких соединениях называется гомеополярной или
ковалентной связью.
Впервые теория ковалентных связей была предложена в 1916 г.
Льюисом. Согл-асно этой теории, устойчивые молекулы из атомов
получаются в том случае, когда осуществляется образование устой-
62 Глава І. Углеводороды
чивой системы электронов. Это достигается не потерей или
получением электронов, а таким строением молекулы, при котором
отдельные электроны являются общими для двух атомов,
причем каждая связь образуется парой общих электронов.
Обозначая по-прежнему электроны одного атома крестиками,
а электроны соседнего атома точками, получаем следующие
схемы:
водород
н
н:с:н
н
метан
:а:
:сі:с:сі:
•а:
• •
четыреххлористыЙ
углерод
н
н;с:о:н
н "
метиловый
спирт
Из приведенных формул видно, что в этих молекулах атомы
углерода, кислорода и хлора окружены восемью электронами,
или, как говорят, октетами электронов, причем каждая связь
осуществляется парой электронов. Так как для образования кова-
лентной связи необходима пара электронов, то атом углерода может
присоединять к себе не более четырех других атомов. В
противном случае число электронов, связывающих этот атом с другими
атомами, превзойдет восемь. Правило октетов хорошо
применимо к углероду, азоту и кислороду. Для соединений других
элементов встречаются исключения, ко многим элементам оно
совсем неприменимо.
Характер органических веществ как неионных соединений
сказывается на их реакционной способности и физических
свойствах.
Реакции между веществами, распадающимися на ионы,
сводятся к реакциям между ионами и протекают практически
мгновенно. Органические вещества не ионизированы, поэтому
время протекания реакций, в которых они принимают участие, часто
измеряется не секундами или минутами, а часами. Во многих
случаях эти реакции идут с заметной скоростью лишь при
повышенных температурах.
Силы притяжения между разноименными ионами, например
между нонами натрия и хлора в кристалле хлористого натрия,
гораздо больше сил притяжения между молекулами в кристаллах
органических веществ, поэтому, например, температуры
плавления большинства органических веществ значительно ниже
температур плавления неорганических.
22 Дипольные моменты. Каждая простая ковалентная связь
осуществляется электронной парой, общей двум атомам. Эта
электронная пара принадлежит обоим атомам в одинаковой мере
лишь в том случае, если связанные между собой атомы совершен-
22. Дипольчые моменты 63
но равноценны по своим электрическим свойствам, как, например,
атомы водорода в молекуле водорода или атомы углерода в
молекуле этана.
В случаях, когда связаны неодинаковые атомы, электронная
пара сдвинута к одному из атомов, и связь между атомами имеет,
как говорят, полярный характер; тот атом, который обладает
способностью сильнее притягивать к себе электроны,
приобретает некоторый избыток отрицательного заряда.
Если условно обозначить полярность связи положением
электронной пары на линии, соединяющей оба атома, то получится
следующая схема:
с ^ ел
сильно
полярная
г *
<- *
полярная
N
неполярная
L. ^. ¦ И g
полярная
С* ^ ... УЛ-
сильно
полярная
Связи в молекуле отличаются, таким образом, своей
полярностью. Это и является одной из причин различной реакционной
способности молекул.
Для количественной характеристики степени полярности
связи введено понятие о дипольном моменте. Если два равных, но
противоположных заряда -\-е и —е находятся на расстоянии /,
то такая система называется диполем и характеризуется диполь-
ным моментом р, которым измеряется произведением величины
заряда е на расстояние между зарядами /:
IX = Є ¦ I
В молекулах водорода Н2 и азота N2 «электрические центры
тяжести» положительных и отрицательных зарядов совпадают,
так как общие пары электронов в
равной мере принадлежат обоим s—\^~\ .-—-^
атомам; такие молекулы не име- (*Х~) vt_~-) С 3
ют дипольного момента, они не- Нврм/1Я Поляр/1ая Н?„иЛйриай
полярны.
В молекулах МНОГИХ ДРУГИХ Рис. 9. Схемы молекул.
веществ отрицательные и
положительные заряды расположены неравномерно, электрические
центры тяжести этих зарядов не совпадают друг с другом; такие
молекулы имеют дипольный момент и называются полярными
(рис. 9). Так как заряд электрона равен 4,77-10~10
электростатических единиц, а длина диполя—величина того же порядка, что
и диаметр молекулы, т. е. порядка 10~8 см, то дипольный момент
всегда имеет величину порядка 1(Г18. Дипольные моменты
вычисляются, как это подробно описывается в курсах физической химии,
64
Глава І. Углеводороды
из определяемой экспериментальным путем зависимости между
диэлектрической постоянной данного вещества и температурой.
Приводим дипольные моменты некоторых веществ:
ц. Ю1
ц-101
Водород
Азот
Двуокись углерода .
Сероуглерод ....
Сероводород ....
Двуокись серы . . .
Вода
Аммиак
0
0
0
0
1,1
1,61
1,85
1,5
Метан ,
Этан
Пропан
Гептан ...
Хлористый метил ....
Хлористый метилен . . .
Хлороформ
Четыреххлористый углерод
0
0
0
0
1,97
1,59
0,95
0
в
Неполярные молекулы, находясь в электрическом поле,
поляризуются, т. е. становятся диполями. Это является результатом
смещения зарядов в молекуле под влиянием
электрических сил внешнего поля. После
прекращения действия электрических сил
молекулы снова становятся неполярными. В отличие
от постоянных диполей такие диполи называют
индуцированными.
Молекулы с постоянными дипольными
моментами также способны к поляризации. Если
такие диполи попадают во внешнее
электрическое поле, то оно оказывает на них
ориентирующее влияние: диполи стремятся
повернуться по направлению силовых линий поля
(рис. 10). Одновременно происходит и
некоторая дополнительная поляризация молекул—к
постоянному дипольному моменту
прибавляется индуцированный.
Зная величину дипольных моментов, можно получить
представление о характере распределения зарядов и тем самым о
симметрии строения мо-
' О ) (С) ( О ) ( S
лекул. В некоторых
случаях это
указывает и на расположение
атомов в молекуле.
Приведем
несколько примеров.
1. Молекулы двуокиси углерода СО2 и сероуглерода CS2 не-
полярны. Следовательно, эти молекулы должны иметь строение
с прямолинейной симметрией (рис. 11).
2. Молекула воды характеризуется значительным дипольным
моментом. Такая молекула не может иметь строения с прямоли-
Рис. 10. Действие
внешнего поля на
полярные
молекулы.
Рис. 11. Схема строения молекул двуокиси
углерода (а) и сероуглерода (б).
25. Состав нефти и нахождение ее в природе 65
нейной симметрией. В молекуле воды атомы водорода связаны с
атомом кислорода так, что их связи образуют между собой
некоторый угол, близкий к 110° (рис. 12).
3. Молекула четыреххлорпстого углерода ССІ4 ^полярна,
несмотря на то, что связь С—С1 является
сильно полярной. Следовательно,
молекула СС14 имеет симметричное строение,
хотя отсутствие дипольного момента не
указывает в данном случае, как
построена молекула—в плоскости или в виде Рис. 12. Схема строения
тетраэдра. молекулы воды.
Значение дипольных моментов для
объяснения свойств веществ и выяснения расположения атомов в
молекулах неоднократно указывается в дальнейшем при
описании различных соединений.
Другим практически важным для химика фактом является
то, что полярные вещества, как правило, растворимы в полярных
растворителях и нерастворимы в неполярных растворителях, а
неполярные вещества растворимы в неполярных растворителях.
НЕФТЬ И ПРИРОДНЫЕ ! АЗЫ
Углеводороды широко распространены в природе. В земной
коре смеси углеводородов находятся в виде нефги, природных
газов, асфальтов, озокеритов.
23. Состав нефти и нахождение ее в природе.
Нефть—маслянистая, темно-коричневая жидкость. По химическому составу
нефть представляет собой смесь углеводородов и,таким образом,
является их главным природным источником. Нефти, в
зависимости от месторождения, отличаются между собой по составу, ио
все они содержат в различных количествах следующиэ кпассы
углеводородов: предельные, эпициклические и ароматические.
В состав нефгей входят твердые, жидкие и газообразные
углеводороды. Последние выделяются из земли, образуя прирогный,
или нефтяной, газ. Различают нефги с парафиновым оснэванием
(содержат главным образом жидкие углеводороды) и нефти с
асфальтовым основанием (содержат большое количество твердых
углеводородов).
Кроме углеводородов, в нефтях содержатся кислородные,
сернистые и азотистые соединения, иногда в значительном
количестве.
24. Происхождение нефти. Для объяснения происхождения
нефти предложен ряд теорий: согласно одним из них, нефть
является продуктом превращения неорганических веществ, другие
5—788
CG Глава I. Углеводороды
приписывают нефти органическое происхождение—из остатков
животных и растений. К теориям первой группы относятся
теория космического происхождения нефти и теория Д. И.
Менделеева.
По теории космического происхождения нефти углеводороды,
составляющие нефть, образовались непосредственно из углерода
и водорода в начальной стадии существования земного шара.
Эта теория объясняет наличие значительных количеств метана
в атмосферах больших планет. По мнению Д. И. Менделеева,
нефть образовалась в результате действия воды на карбиды
металлов (в частности, на углеродистое железо), из которых состоит
ядро земного шара. Действительно, карбиды металлов, реагируя
с водой или разбавленными кислотами, образуют углеводороды,
главным образом метан и ацетилен. Карбид железа и
марганцовистый чугун при взаимодействии с водой дают нефтеподобную
смесь жидких углеводородов. Несмотря на то, что эти факты как
будто подтверждают теорию Менделеева, она в настоящее время
почти совершенно оставлена. Против нее говорит содержание в
нефти азотистых соединений и ее оптическая активность (стр. 154),
что определенным образом указывает на органическое
происхождение нефти.
Теории органического происхождения нефти имеют
наибольшее число сторонников. Одни из исследователей считают, что
нефть образовалась из остатков морских животных, другие—из
остатков морских водорослей; некоторые видят источник
образования нефти в остатках наземных растений. Энглер получил
нефтеподобную смесь жидких углеводородов перегонкой рыбьего
жира под давлением. Н. Д. Зелинский получил подобные же
продукты, разлагая в присутствии хлористого алюминия различные
вещества животного и растительного происхождения:
высокомолекулярные спирты (стерины), жирные кислоты и т. п. Смешанное
растительно-животное происхождение нефти было доказано в
1934 г. Трейбсом, который во всех исследованных им 29 образцах
нефти нашел производные хлорофилла и гемина (последних в
количестве в 20 раз меньшем, чем производных хлорофилла).
Можно предполагать, что нефть образовалась частью из животного,
частью из растительного вещества. Весьма вероятно, что
источником происхождения нефти был морской планктон* и морские
водоросли, громадные количества которых находятся в морях и
океанах.
25. Продукты переработки нефти и их применение. Нефть
является основным источником получения различных видов топ-
* Планктон—мельчайшие морские организмы и водоросли, живущие в
толще воды.
Продукты переработки нефти и их применение 67
лива—ракетного, топлива для дизельмоторов, горючего для
двигателей внутреннего сгорания, сырьем для получения смазочных
масел и высококачественных дорожных покрытий; нефтяные
продукты находят все более широкое применение в качестве
котельного топлива. Кроме того, нефть все в большей и большей
степени используется как сырье для химической переработки: в
первую очередь для получения различных синтетических материалов—
пластических масс, каучуков, волокон, синтетических
растворителей, алкоголей, кислот, моющих веществ и многих других ценных
продуктов. Наиболее распространенным первичным процессом
переработки нефти является перегонка. Продукты перегонки
подвергаются затем химической очистке для удаления сернистых
соединений и смолообразующих веществ.
Первый погон, получаемый при перегонке нефти, называется
сырым, бензином или газолином.. Дальнейшей разгонкой из него
получают различные сорта бензини: авиационный бензин, бензин
1-го сорта, бензин 2-го сорта и т. д. Наиболее легкокипящей
частью этого погона является петро_л?!лныи эфи&. Следующий погон,
кипящий при температуре от 150 до 300 ^С, служит для получения
различных сортов керосина.. Остаток после отгонки керосинового
дистиллята называется мазутом: он служит котельным топливом.
При перегонке мазута получают соляровое масло (которое
частично применяется для изготовления вазелинового масла) и
различные смазочные масла: веретенное, цилиндровое, машинное.
Остаток после отгонки смазочных масел называется гудроном. Его
используют для мощения дорог. При нагревании гудрона с
перегретым водяным паром пли же при нагревании гудрона с
одновременным пропусканием через него воздуха гудрон теряет летучие
примеси и превращается в искусственный асфальт. Из последнего
нефтяного погона получается вазелин, представляющий собой смесь
твердых и жидких углеводородов. Из некоторых сортов нефти
можно выделить смесь твердых предельных углеводородов,
называемую парафина,1^. В СССР парафином богаты грозненская и
западноукраинская нефтн.Ліамі^-иіиі'Ч
Развитие автомобильного и авиационного транспорта
предъявило большой спрос на легкокипяаше фракции нефти—бензины.
Однако путем прямой разгонки нефги из нее редко удается
выделить более 20% бензина. Эго привело к необходимости
разработать специальные технологические процессы, повышающие выход
бензина из продуктов нефтедобычи. Одним из таких процессов
является извлечение бензина, содержащегося в попутных
нефтяных газах, выделяющихся из нефтяных скважин вместе с
добываемой нефтью.
Для получения бензина нефтяной газ сжимают до небольшого
обьема и пропускают через змеевик холодильника, охлаждаемого
.68 Глава І. Углеводороды
холодильными агентами или проточной водой, при этом выделяется
значительная часть бензина. Для извлечения остатков бензина газ
проходит через башни, наполненные коксом и орошаемые сверху
соляровым маслом. Масло поглощает весь бензин, который за-
іем отделяется перегонкой. Газовый бензин отличается малой
плотностью и низкой температурой кипения. Улавливание бензина
из газа позволяет увеличить количество добываемого бензина, но
не повышает его выход из нефти. Последнее достигается в
результате процессов химической переработки нефти, в первую очередь
путем крекинга*, нефти или отдельных нефтяных погонов.
Сущность крекинга заключается в расщеплении тяжелых
молекул высококипящих углеводородов на легкие молекулы низ-
кокипящих углеводородов, образующих бензин. Так, например,
углеводород додекан С,2Н26 можно расщепить на гексан С6НІ4 и
гексен С6Н12:
темп.
кип.
216 °С
темп.
кип.
69 °С
темп
кип.
64 °С
При этом одновременно происходит растепление и другого
типа, в результате чего образуется нефтяной кокс:
Кроме того, при крекинге образуются в значительном количестве
зіилен и другие низкомолекулярные олефины (пропилен, бути-
лены, амилены). Первая промышленная установка для крекинга
нефти была построена В. Г. Шуховым.
В технике различают процессы термического крекинга и
каталитического крекинга. При термическом крекинге расщепление
углеводородных молекул происходит под воздействием высоких
температур D50 °С и выше), причем процесс проводится под
повышенным давлением. В зависимости от исходного сырья и
необходимости получения тех или иных продуктов процесс может
проводиться в различных условиях. Так называемый газофазный
крекинг проводится при низком давлении C—5 am) и температуре
550—600 °С. В этом случае процесс протекает в газовой фазе.
Жидкофазный крекинг ведется при более высоком давлении B0—
70 am) и более низкой температуре D50—500 "С); в этом случае
часть исходных и образующихся углеводородов находится в
жидкой фазе.
В процессах каталихи.ческога крекинга расщепление
углеводородов происходит на катализаторах при более низких
температурах D50—500 °С), чем газофазный крекинг, и при давлении, близ-
* Слово крекинг в переводе с английского означает «расщепление».
26. Искусственное жидкое топливо ' С9
ком к атмосферному. Процесс проводится только в газовой фазе,
в качестве катализаторов чаще всего применяются различные
алюмосиликаты. Установки каталитического крекинга более
сложны и дороги, но позволяют получать бензин более высокого
качества и с большим выходом.
Из других процессов химического облагораживания нефтяных
продуктов особенно широкое распространение нашли процессы
так называемого каталитического риформинга, в результате
которых происходят сложные структурные изменения молекул
взятых углеводородов, но в основном без изменения в них числа
углеродных атомов.
Такие способы переработки нефти позволяют повысить выход
бензина из нефти до 80%, и в настоящее время основная часть
бензинов получается этим путем.
Из нефтяных углеводородов путем последовательной
химической переработки получают целый ряд различных химических
соединений: непредельные углеводороды, спирты, кислоты, эфи-
ры, альдегиды—продукты, играющие огромную роль как для
изготовления предметов бытового потребления, так и для
развития современной техники. Так, содержащийся в газах крекинга
этилен при взаимодействии с хлором образует дихлорэтан,
являющийся исходным сырьем для получения поливинилхлори-
да. Гидратацией этилена под действием катализаторов
получается синтетический этиловый спирт, являющийся важным
исходным сырьем для ряда химических процессов. Эта реакция,
открытая А. М. Бутлеровым и В. Горяиновым, сохранила важное
техническое значение и до настоящего времени.
26. Искусственное жидкое топливо. В связи с ограниченностью
запасов нефти в некоторых странах в 20-х и 30-х годах были
разработаны технические методы получения жидкого топлива из
ископаемых углей. Один из методов заключался в том, что
суспензия каменного или бурого угля в тяжелых маслах нагревалась
с водородом в присутствии железного катализатора примерно
до 400 °С под давлением 200—300 am. При этом происходило
гидрирование угля, т. е. соединение углерода с водородом, и
получались углеводороды, образующие синтетическую нефть.
Другой способ был основан на реакции, открытой в 1908 г.
Е. И. Орловым, который показал, что при пропускании окиси
углерода с водородом над катализаторами (Ni, Pd) образуются
этилен и другие непредельные углеводороды. Основываясь на
реакции Е. И. Орлова, Фишер и Тропш в 1926 г. разработали
технический метод синтеза жидкого топлива. Они показали, что
при пропускании смеси окиси углерода и водорода над
некоторыми катализаторами при 200—300 °С получаются предельные
углеводороды. В качестве катализаторов применялись металлы вось-
70 Глава І. Углеводороды
мой группы (в том числе кобальт, никель и железо) с добавлением
других веществ.
Реакцию образования жидких и твердых углеводородов с
различной молекулярной массой из окиси углерода и водорода можно
выразить уравнениями:
Со. N1
jrCO + 2хНа * (СН2), + *Н2О
Fe
2хСО + хН2 > (СН2)^ + хСО2
Большая часть образующихся непредельных углеводородов
(см. стр. 71 ел.) под действием тех же катализаторов частично
присоединяет водород, превращаясь в предельные углеводороды.
Получаемый по этому методу синтетический бензин—синтин
представляет собой смесь предельных и непредельных
углеводородов преимущественно с нормальной цепью углеродных атомов.
27. Природные газы. Асфальты. В СССР имеются
многочисленные месторождения горючих газов, особенно в
нефтеносных районах. Таковы месторождения близ Саратова. Дашавы,
Ставрополя, Ухты и др. Главной составной частью природных газов
(90—98%) является метан, которому в меньших количествах
сопутствуют этан, пропан, бутаны. Иногда в качестве примеси
присутствует гелий (в некоторых случаях в количестве до
нескольких процентов).
Природный газ представляет собой исключительно ценное
топливо. Он сгорает нацело, не оставляя золы, не образуя окиси
углерода. Его теплотворная способность очень высока (около
11 000—12 00C) ккал/кг), т. е. намного больше, чем у других
видов топлива (дрова 4700—5100, каменный уголь 6000—8000,
керосин 10 00u ккал/кг). Газ не только может быгь использован
в местах добычи, но и транспортируется по трубам газопроводов
на далекие расстояния, например: Саратов—Москва, Дашава —
Киев—Москва, нефтепровод «Дружба» проходит в Польшу, ГДР
и др.
Нагревая метан до 1400—1500 °С, получают водород и углерод
(в виде сажи):
СН4 » С + 2Н3
Получаемая таким образом сажа употребляется в большом
количестве в лакокрасочной промышленности и как добавка к каучуку
в производстве резины.
При взаимодействии метана с водяным паром при температуре
700—800 °С на катализаторе (никель на магнезите) образуется
так называемый синтез-газ:
СН4 + Н2О » СО + ЗН3
2'/. Природные газы. Асфальти 71
являющийся исходным сырьем в производстве синтетического
метилового спирта (см. стр. 144).
Каталитическим частичным окислением природного газа в
присутствии окислов азота получают формальдегид:
сн4 + о2 -* н-с7 +н2о
\н
Из формальдегида можно получить метиловый спирт СН3ОН
(см. стр. 144), пластические массы (см. стр. 420) и др.
Асфальты состоят из углеводородов, молекулы которых
содержат обычно шестнадцать и больше атомов углерода. В СССР
наиболее известные месторождения асфальта находятся на
Сахалине. Иногда асфальт пропитывает рыхлые горные породы. Так,
близ Сызрани и на Кавказе встречается асфальтовый известняк.
Асфальт применяется как изолятор, а также для
приготовления лаков и кровельного толя; большие количества асфальта
расходуются в дорожном строительстве.
Озокерит (горный воск) состоит главным образом из твердых
предельных углеводородов с разветвленной цепью углеродных
атомов. Как было раньше указано, углеводороды с сильно
разветвленными цепями обычно имеют более высокие температуры
плавления, чем соответствующие изомеры нормального строения.
Этим объясняется сравнительно высокая температура плавления
озокерита.
Озокерит почти не окисляется.
Путем очистки из озокерита получают церезин^ применяемый
как заменитель воска при изготовлении мастики для полов,
для вощения бумаги, для изготовления изоляционных материалов.
НЕПРЕДЕЛЬНЫЕ УГЛЕВОДОРОДЫ
Непредельными называются углеводороды, в молекулах
которых число атомов водорода меньше, чем в молекулах
предельных углеводородов с тем же числом углеродных атомов.
Общие формулы углеводородов:
—2Н —2Н
CflH^+j *¦ С„Н2Л — —*- С„Н.2„_2 и т. д.
предельные непредельные
Ряд углеводородов, отдельные члены которого различаются
между собой на BН)„, называется изологическим, например:
QH14l свн12, свн10, свна, свнв...
В таком ряду степень непредельности повышается.
72 Глава I. Углеводороды
Углеводороды с одной двойной связью. Алкены (олефины)
28. Двойная связь. Углеводороды, содержащие в молекуле
на два атома водорода меньше, чем соответствующие парафины,
имеют одну двойную связь. Общая формула таких
углеводородов СпНгп." Простейший член этого ряда — этилен* С2Н4.
Наиболее характерным свойством этилена является его резко
выраженная способность вступать в реакции соединения;
например, молекула этилена способна присоединять два атома галоида:
этилен диоромэтан
или (в присутствии катализаторов) два гтома водорода:
С2Н4 + Н2 -*•¦ С2Н9
этилен этан
Другим характерным свойством этилена является его
способность окисляться. При осторожном окислении этилена
(например, марганцовокислым калием по Е. Е. Вагнеру) образуется
этиленгликоль:
С2Н4 + (Н2О + О) = СН2(ОН)-СН2(ОН)
В молекуле этана СН3—СН3 три единицы валентности
каждого атома углерода насыщены единицами валентности трех атомов
водорода, четвертая единица валентности углерода насыщает
единицу валентности другого атома углерода. Так как в молекуле
этилена на два атома водорода меньше, чем в молекуле этана, то
в молекуле этилена у атомов углерода должны находиться две
свободные единицы валентности. При этом возможны два случая:
1) у каждого атома углерода находится по свободной
единице валентности (формула I);
2) обе свободные единицы валентности находятся у одного
атома углерода (формула II).
Наконец, возможно и предположение, что свободные единицы
валентности атомов углерода в этилене взаимно насыщают друг
друга, т. е. что в молекуле этилена атомы углерода связаны
двойной связью (формула III):
нн нн 1Т т1
и а ' н/
Ш
* Метилен СН2 в свободном виде, не существует. Во всех реакциях,
которые должны были вести к образованию метилена, всегда получался
углеводород с удвоенным составом—этилен C2Hi.
28. Двойная связь 73
Какая же из этих формул выражает строение этилена?
Совершенно ясно, что присоединение к молекуле этилена
других веществ, например хлора или брома, должно происходить
или за счет свободных единиц валентности, или за счет единиц
валентности, освободившихся вследствие разрыва второй связи.
Если строение этилена выражается формулами 1 или III, то
вещество, которое получается при соединении этилена с хлором—
С2Н4С12, должно иметь строение:
Н Н
С1-С—С-С1
U
Если же этилену соответствует формула II, то, соединяясь с
хлором, этилен должен дать вещество иного строения:
Н Н
Н-ІХ
С1
Вещество, получаемое при соединении этилена с хлором,
отличается от вещества, которое образуется при действии на
уксусный альдегид СНН—СНО пятихлористого фосфора и строение
которого может выражаться только формулой СН3—СНС12.
Следовательно, дихлорэтан имеет строение СН2С1—СН2С1, и
поэтому строение этилена не соответствует формуле II.
Далее, молекула этилена присоединяет только два атома
галоида или водорода. Ни разу не удавалось наблюдать, чтобы
присоединился только один атом галоида. Формула I не
объясняет этого явления, и остается совершенно непонятным, почему
свободные единицы валентности углеродных атомов должны
насыщаться одновременно. Поэтому следует признать, что в
молекуле этилена свободные единицы валентности углеродных
атомов насыщают друг друга, т. е. что в молекуле этилена атомы
углерода связаны двойной связью:
нх н
с=с
н/ \н
При действии на этилен водорода (при пропускании смеси
этилена и водорода над нагретым никелем) или галоидов происходит
74 Глава І. Углеводороды
присоединение атомов водорода или галоидов по месту двойной
связи:
Нч /Н Нч уН Нч Лі Нч yti
с=с + н, —» н—с—с—н с=с + вга —»н—с—с—н
н/ н н н н н Вг Вг
этилен этан этилен дибромэтан
Симметричное строение этилена было установлено в 1868 г.
исследованиями А. М. Бутлерова и М. Осокина.
29. Номенклатура олефинов. Подобно тому, как от метана
производится гомологический ряд предельных углеводородов, или
парафинов, так и от этилену производится гомологический ряд
^этиленовых углеводородов, или олесришщ*. Каждый член этого
ряда отличается от предыдущего и последующего члена на группу
СН2. В молекуле этилена все атомы водорода равноценны. Поэтому,
замещая в нем любой атом водорода, можно получить только
углеводород метилэтилен СН2=СН—СН3. В метилэтилене три
ряда водородных атомов, поэтому из него, заменой атома водорода
на группу СНа, можно произвести три разных олефина:
этилэтилен СН2=СН—CHS—СН3, симметричный
диметилэтилен СН3—СН=СН—СН3 и несимметричный диметилэтилен
~/С=СН2. Эти три углеводорода изомерны между собой.
Изомерия этилэтилена и симметричного диметилэтилена
обусловлена различным положением двойной связи между атомами
углерода в нормальной цепи. Третий изомер, в отличие от двух
предыдущих, имеет разветвленную углеродную цепь. Еще
большее число изомеров возможно для амилена (пентена) С5Н10 и
гексена С6Н|2.
Ранее названия этиленовых углеводородов производили от
названий алкоголей с тем же числом атомов углерода в
молекуле: этиловый алкоголь — этилен, амиловый — амилен. Для
обозначения изомеров часто применяются названия, в которых
отображено происхождение этиленовых углеводородов путем
замещения водорода в этилене на алкилы, например:
OV
>СН—СН=СНг Х=СН2
¦з СН/
изсшропилэтилен несимметричный диметилэтилен ,
* Полученному в XVIII в. в Голландии хлористому этилену,
представляющему собой маслянистую жидкость, дали название «масло голландских
химиков». Отсюда и название для этилена—«маслородный газ».
30. Способы получения и строение олефинов 75
По женевской номенклатуре названия этиленовых
углеводородов имеют окончанием«; цифрой в конце названия обозначается
номер первого атома углерода, при котором находится двойная
связь. Счет ведут так, чтобы двойная связь была ближе к началу
нумерации:
этен
пропен
бутен-1
бутен-2
2-метилпропен-1
СН.
СН.
СН,
сн3
СН;
г=сн2
a=CH-CHa
2 3
,-СН=СН-СН
,—с=сн2
сн.
Общее название этиленовых углеводородов—алкены.
Некоторые углеводородные радикалы этиленовых
углеводородов имеют традиционные названия, образованные из названий
спиртов:
СН2=СН— винил
СН2=СН— СН2— ал лил
30. Способы получения и строение олефинов. 1. Олефины
образуются при сухой перегонке многих органических веществ.
Поэтому они содержатся в коксовом газе и газах крекинга нефти.
Газы, образующиеся при жидкофазном крекинге, состоят на 15%
из непредельных углеводородов; выход газа составляет около 8%
от массы сырья.
2. Олефины получают из спиртов отнятием от последних воды
при помощи водуотнимающих средств или разложением паров
соответствующего спирта при высокой температуре в
присутствии катализаторов (А12Оа и др.):
—СН2 * СН,=СН2 -}- Н2О
|
Ін'ОН;
оС—СН2
| | этилен
3. Весьма важным способом получения олефинов является
действие на галоидпроизводные предельных углеводородов
концентрированного раствора едкого кали в этиловом или
метиловом спирте. Если при действии водного раствора щелочи
происходит замещение галоиДа на гидроксил и образование спирта:
с3н,и"+'кЬн —»с3н,-он f kj
76 Глава І. Углеводороды
то при действии спиртового раствора щелочи происходит
отщепление молекулы галоидоводорода и образование олефинов:
н2с-сн-сн3 + кон —> сн2=сн-сн3 + KJ + н2о
| | пропилен
Г1 ні
йодистый пропил
Н2С-СН-СН3 + КОН » СН2=СН-СН3 + KJ + Н2О
| | пропилен
! Н J"";
йодистый изопропил
При этом иод соединяется с калием, а водород от соседнего
атома углерода—с гидроксилом щелочи. Это имеет характер
общего правила: отнятие водорода и галоида происходит от соседних
атомов углерода.
31. Физические и химические свойства олефинов. Этилен,
пропилен, бутилен—газы; следующие члены гомологического ряда—
жидкости; начиная с С18Н36—твердые тела (см. табл. 2).
Плотности олефинов выше, чем у соответствующих предельных
углеводородов. Как и в случае предельных углеводородов, с
увеличением числа атомов углерода в молекуле возрастает плотность,
а также повышаются температура плавления и кипения
олефинов. Олефины с двойной связью на краю цепи имеют более низкую
температуру кипения, например З-метилбутен-1 имеет темп. кип.
+20,1 °С, а 2-метилбутен-2—темп. кип. 38,6 °С; 2,4,4-триметил-
пентен-1—темп. кип. 101,4°, а 2,4,4-триметилпентен-2—темп,
кип. 104, 9 °С.
В ряду олефинов, как и в ряду предельных углеводородов, с
увеличением молекулярного веса повышаются температуры
плавления и кипения. Как и все другие углеводороды, олефины мало
растворимы в воде. Пары олефинов имеют характерный резкий
запах.
Этиленовые углеводороды—исключительно реакционноспо-
собные соединения. В громадном большинстве реакций
принимают участие атомы углерода, связанные двойной связью.
Наиболее характерным свойством олефинов является их резко
выраженная способность вступать в реакции соединения с другими
веществами. При этом разрывается одна из связей между
атомами углерода, связанными кратной связью, и за счет
освобождающихся валентностей к атомам углерода присоединяются другие
атомы или группы.
1. При температуре 150—200 °С в присутствии
мелкораздробленного никеля этиленовые углеводороды присоединяют водород
Физические свойства олефииов
Таблица 2
Название
Этилен, или этен
Пропилен, или пропен
Этилэтилен, или бутен-1
Изобутилен, или 2-метил
пропен-1
Пропил^тилен, или пен-
тен-1
Несимм. метилэтилэтилен.
или 2-метилбутен-1
н-Октадецилен, октаде-
цен-1
Формула
сна=снг
СН3—СН :=СН2
СН3—СНа—СН=СН2
(СН3JС=СН2
СН3СНа—СНаСН=СН г
СН3
1
СгН6-С=СН2
CH3(CHaI5CH=CH2
Физическое
состояние при
обычной тем
пературе
Газ
То же
»
Жидкость
То же
Твердый
Температури, °L
плавления
-169,2
— 185,2
— 185,3
— 140,3
— 165,2
— 137,5
+ 17,6
кипения
-103,7
-47,7
—6,3
—6,9
+30,0
31,2
314,8
Плотность (в жидкой
состоянии) г/смЬ
0,570
(при темп кип.)
1,5139
(при темп, кип.)
0,5951
(прн темп кип.)
0,5942
(при темп кип.)
0,6405
0,6504
0,7888
78 І лава I. Углеводороды
(реакция Сабатье), при этом одна молекула олефина
присоединяет два атома водорода:
СН2=СН2 + Н2 > СНэ-СН;,
этан
В присутствии порошкообразной платины или палладия эта
реакция проходит и при обыкновенной температуре. : Реакция
присоединения водорода к непредельным соединениям
называется гидрированием или гидрогенизацией. ¦
2. Весьма легко олефины присоединяют галоиды, особенно
хлор и бром-
По двойной связи присоединяются два атома галоида:
СН5=СН—СН.з + Вг2 » СНаВг—СНВг—СНЭ
Эту реакцию используют для качественного и количественного
определения двойных связей.
3. Олефины очень легко присоединяют галоидоводород (одну
молекулу галоидоводорода на одну двойную связь молекулы
олефина):
:на=сн—сн~ + ri—j —» сн3—си—сн3
J
аодистый изопропил
При этом атом водорода становится у наиболее гидрогенизи-
рованмого, т. е. у наиболее богатого водородом, атома углерода.
Эта закономерность открыта В. В. Марковниковым* и называется
правилом Марковы икови.
* Владимир Васильевич Марковников A838—1904) родился близ Нижнего
Новгорода (г. Горький). Высшее образонапяе он получил в Казанском
университете A856—1860) по окончании которого работал r лаборатории А М Ijyi
лерова. В. В. Марковников синтезировал ряд новых соединений, предсказанных
структурной теорией и широко развил идеи о взаимном влиянии атомов в
молекулах. Такоиы, например его работы по синтезу иэомасляной оксииас-
лянон, хлормасляной кислот, по изучению реакции присоединения гялоидо-
водородов к двойной связи. Большой научный интерес представляют работы:
«К истории учения о химическом строении» «Об изомерии органических
соединении» (магистерская диссертация, 1865). «Материалы по вопросу о
взаимном влиянии атомов в химических соединениях» (докторская диссертация, 1869).
Глубокие мысли о динамике химических реакций содержатся в его статье
«Принцип химического равновесия» A902). С 1880 г. Марковников перешел к
изучению состава кавказских нефтей. Им было впервые доказано, і что в неф-
гях содержатся в больших количествах циклопарафины, которые были им
названы «нафтенами». '
ЗІ Физические и химические свойства олефинав
79
Правило Марковникова не имеет абсолютного значения. В
некоторых случаях, например в присутствии веществ перекисного
характера, присоединение бромистого водорода происходит вопреки
этому правилу. Так, бромистый винил СНа —СНВг2 в отсутствие
перекисей присоединяет бромистый водород с образованием только
1,1-дибромэтана СН:(—СНВг2, т. е. согласно правилу
Марковникова, в присутствии же перекисей получается главным образом
1,2-дибромэтан СН2Вг—СН.Вг.
А. С концентрированной серной кислотой олефины реагируют
с образованием кислых эфиров серной кислоты:
СН2 н
СН, О—S—
ОН
СН3
СН
—О
ОН
этилсерная кислота
СН8
СН-2 Н
СН
+-О—SO3H
3—С
СН3—СН—OSO3H
изопропилсерная
кислота
При этом водород присоединяется к наиболее гидрогенизирован-
ному атому углерода, т. е. по правилу Марковникова.
Алкилсерные кислоты можно рассматривать как производные
серной кислоты, в молекуле которой один атом водорода замещен
ал килом. При обработке елкилсерных кислот водой образуются
спирты:
НО
—S—O-icHJ—С
+н—ОІ—Н
СНз-CHj-OH + H.2SO4
СН—ОН + H2SO4
СН,
Эту реакцию используют для промышленного получения спиртов из олефи-
нов. В связи с этим любопытно отметить, что еще в 1873 г. А. Л1 Бутлеров и
В. Горяинов показали, что серная кислота при температуре около 160 °С легко
поглощает этилен, и считали это за «факт, обещающий приобрести со временем
практическое значение; если бы удалось открыть дешевый способ
приготовления этилена, то он составил бы материал для добывания спирта».
В настоящее время газы крекинга начали очень широко использоваться
в качестве химического сырья для получения многих ценных
продуктов.
80 Глава І. Углеводороды
5. Олефины присоединяют хлорноватистую кислоту; при этом
к одному углеродному атому (менее гидрогенизированному)
присоединяется гидроксил, а к другому—хлор:
СНа—CH=CHS+HO—СІ > СН3—СНОН—CHjCI
I | пропиленхлоргидрин
6. При действии озона на раствор олефинов в четыреххлори-
стом углероде образуются озониды:
о
R_CH=CH—R' + Os , R—CH CH-R'
l—CH=C<
о
+ 02 » R—CH
¦R"
Как легко видеть из зіих формул, действие озона приводит
к разрыву углеродной цепи. Буквами R, R', R" здесь
обозначены различные углеводородные радикалы. Можно подставить
вместо R, например, СНа—, а вместо R' С„На—, или же вмесю R
СаН7—, а вместо R" QH,,—, но характер реакпии от этою не
изменится. С подобным обобщенным способом написания
химических формул мы будем часто встречаться в дальнейшем.
При действии воды озониды расщепляются с образованием
перекиси водорода и альдегидов или кетонов:
- О .О S)
r-ch^ch—R'+ н2о —*¦ р—с^ + r'-cx + нго2
^Г Н Н
R-CblvC^ + Н2О R-< + О=СЧ + НаО,
AiAV
Если в молекуле, подвергшейся озонированию, углерод при
двойной связи был соединен с водородом, то получаеіся альдегид,
если же углерод не имел при себе водорода, то образуется кегон.
Так как разрыв молекулы происходит в том месте, где была
двойная связь, то, изучая продукты окисления, можно определить
St. Физические и химические свойства олефинов 81
местоположение двойной связи. В качестве примера может
служить окисление трех изомерных бутенов:
О
1) СН,=СН—СН,—СН3 + О8 »Н2С СН—СН8-СН8
о—о
Н,С СН-СНз—СН3 + Н2О » Н—С + СН3—CHj-C7 +
А-А чн \н
муравьиный пропионооый
альдегид альдегид
о
2) СН3—СН=СН—СНЭ + О3 »СН3—НС СН—СН3
О 0
0 о
СН3— НС СН—СН3 + Н,0 » 2СН3—С + Н2О2
уксусный альдегид
о
снзч снзх / \
3) >с=сн2 + о3 —. ус сн,
сн/ СНз А_ А
0 о
СНд. / \ СН8Ч /U
>с сна + н8о —» >со + н—с + н2о2
сн/1__Ъ сн/ \н
ацетон муравьиный альдегид
Как видно из приведенной схемы, при окислении получаются:
в первом случае муравьиный и пропионовый альдегиды, во
втором—уксусный альдегид, а в третьем—ацетон и муравьиный
альдегид.
7. При осторожном окислении олефннов водным раствором
марганцовокислого калия происходит присоединение кислорода
и воды по двойной связи:
СНа=СН2 + (Н2О + О) » СНа(ОН)—СН8(ОН)
зтнленгликоль
В результате получаются гликоли, і. є. спирты с двумя гидрок-
силами в молекуле.
6—788
82 Глава І. Углеводороды
Эта р^кция была открыта Е. Е. Вагнером*.
Более сильное окисление олефинов приводит к расщеплению
молекулы по двойной связи:
СН~ 2О CHj,
>С=СН—СН3 » >СО + СН,СНО
сн/ сн/
олефии кетон альдегид
8. Молекулы одного и того же олефина могут соединяться
друг с другом. Так, бутилен С4Н„ дает углеводороды С8Н1в,
С12Н24 и т. д.
Проиесс соединения нескольких одинаковых молекул в одну,
более сложную, называется полимеризацией, а образующееся
вещество—полимером. При соединении двух одинаковых молекул
получается димер исходного вещества. Так, например, при
соединении двух молекул изобутилена образуется 2, 2, 4-триметилпентен-З.
СНЭ\ і і /СН3 CHf.
>С=СН,+Н-СН=С< » >С-СН
сн/\ і х:нз сн/\
сн3
Присоединение одной молекулы углеводорода к другой
молекуле происходит по двойной связи таким образом, что атом
водорода присоединяется к наиболее гидрогенизированному атому
углерода, в то время как к другому атому углерода
присоединяется остальная часть молекулы.
Эта реакция происходит в грисутствии серной кислоты,
хлористого цинка и других веществ. При действии серной кислоты
в качестве промежуточного продукта образуется бутилсерная
кислота:
СН3ч СНЗЧ
>С=СНо + Н—О—SO,—ОН » >С—СН3
сн/ ' сн/1
O_SO2-OH
которая реагирует со второй молекулой углеводорода:
СНЗ
сн
СНд СН3
Х I ', , /СН3 СНЗЧ [ /СН3
V_;OSOOOH+H!—СН=С< > >С—СН=Г/ +H2SO4
/ і :- ! ч:н3 сн/ Ч^з
* Егор Егорович Вагнер A849—1903)—вышел из казанской школы хи-
микор, ученик А М. Зайцева. Работал в Петербурге и Варшаве. Известен
своими работами в области непредельных соединений и терпенов.
ЗІ. Физические и химические свойства олефинов 83
Изобутилен B-метилпропен) находится в газах крекинга
нефти. Полученный полимеризацией изобутилена димер переводят
действием водорода в присутствии катализаторов в 2,2,4-триме-
тилпентан, называемый в технике изоокгпаном:
3 Щ
/СН3 СНач I /СНа
- сн=с< +н, —» >с—сн2—сн<
\сн3 сн/ ч:н3
СН3
сн:/
изооктан
Промышленное получение изооктана имеет большое значение,
так как изооктан применяют в видр добавки к бензинам для
получения высококачественного, так называемого высокооктанового,
топлива для двигателей внутреннего сгорания.
В цилиндрах двигателей внутреннего сгорания может происходить
детонация, т. е. преждевременное мгновенное разложение молекул топлива еще
в момент, когда происходит сжатие, т. е. до достижения нижней мертвой точки
поршня. Детонация, в отличие от нормального сгорания, вызывается не
электрической искрой от запальной свечи двигателя, а лишь высокой температурой
О1 сильного сжатия газовой смеси, производимого давлением поршня
двигателя. При детонации пары горючего сгорают не полностью, выделяются окись
углерода и водород, образуются клубы дыма, мотор издает характерный стук
и т. д.; это приводит к преждевременному износу, а при сильной детонации —
даже к разрушению двигателя Чем меньше способность моторного топлива к
детонации, тем сильнее можно сжимать горючую смесь под поршнем, тем
большую мощность может развивать двигатель и тем экономнее расходуется
горючее. Аитидетоиационную способность бензина оценивают путем
испытания его иа специальных двигателях с переменной степенью сжатия и выражают
о условных единицах—так называемых октановых числах. Условно принимают,
что октановое число изооктана равно 100, нормального гептана—равно нулю,
октановое число смеси из 80% изооктана и 20% «-гептана равно 80 и т. п. Затем
срапнинают способность к детонации таких смесей и исследуемого бензина;
качество бензина оценивается октановым числом, т. е. процентным содержанием
изооктана в такой смеси его с нормальным гептаном, которая имеет такую же
способность к детонации, как и данный бензин. Если, например, в подобной
смеси содержится 85% изооктана, считают, что октановое число бензина равно
85. Если октановое число горючего больше 100. например 115. это означает,
что для получения из него бензина с октановым числом 100 к нему надо при-
банить и-гептан.
Чем больше октановое число горючего для двигателей внутреннего
сгорания, тем выше его качество. Добавка изооктана к бензину увеличивает
мощность и экономичность двигателей. Другим способом повышения октанового
числа горючего является добавка небольших количеств так называемых
антидетонаторов в частности тетраэтилсвинца (см. стр. 121).
Проблема высокооктанового горючего была особенно актуальной в
авиации до появления реактивных двигателей, но сохранила свое значение и в на-
сгояйіее время для поршневых самолетных и для автомобильных бензиновых
моторов внутреннего сгорания.
Октановое число зависит от строения углеводорода. У парафинов с
нормальной цепью углеродных атомов оно меньше, чем у соответствующих
парафинов с разветвленной углеродной цепью, олефинов и циклических
углеводородов. Так например октановое число и-гексаиа равно 40, в то время как у
84
Глава I. Углеводороды
изомерных с ним 3-метилпентана и 2,2-диметилбутана оно соответственно
равно 80 и 120; октановое число циклогексана 80 бензола—100. Смесь изооктаиа
с бензином и неогексаном B,2- ди мети л бута ном) имеет октановое число 115
(октановое число бензииа прямой гонки равно 40—60).
Двойная связь является наиболее активным местом
молекулы, куда направляется в первую очередь действие реагентов.
Для непредельных углеводородов весьма характерна
следующая качественная реакция: капля брома, добавленная к олефину,
мгновенно обесцвечивается. Так же быстро обесцвечивается и
бромная вода при взбалтывании ее с веществами, в молекуле
которых содержатся двойные связи. Бром—реактив на двойную
углеводородную связь. Другим реактивом на двойную углеродную
связь является разбавленный раствор марганцовокислого калия
и соды. При взбалтывании этого раствора с непредельными
соединениями происходит очень быстрое обесцвечивание раствора
и выделение бурых хлопьев гидрата двуокиси марганца
(реакция Е. Е. Вагнера). Конечно, надо учитывать, что существуют
вещества, не имеющие двойных углеродных связей, но легко
окисляющиеся и обесцвечивающие марганцовокислый калий (как,
например, альдегиды, спирты и др.).
32. Показатель преломления. Молекулярная рефракция. Для
характеристики жидких веществ большое значение имеет
показатель преломления света.
Напомним, что показателем преломления
называется отношение синуса угла падения
луча (а) к синусу угла преломления (?)
(рис. 13):
п =
sin*
sin ?
Рнс. 13.
Преломление луча на
границе двух
прозрачных веществ
Эта величина для света с одной и той
же длиной волны при данной температуре
является постоянной и не зависит от угла,
под которым падает луч. Как правило,
показатель преломления вещества
уменьшается при повышении температуры и при увеличении длины
волны (от фиолетового к красному). Поэтому необходимо всегда
указывать температуру опыта и длину волны света. Последнюю
обозначают или в миллимикронах (ммк), или в ангстремах А
@,1 ммк), или же при помощи заглавных латинских букв,
указывающих длину волны линий солнечного спектра:
На (красная линия спектра водорода)
D (желтая линия спектра натрия) .
A/g (голубая линия спектра водорода)
656 ммк
589 ммк
486 ммк
32. Показатель преломления. Молекулярная рефракция
85
Для определения показателя преломления в химических
лабораториях обычно применяют рефрактометры типа Аббе (рис. 14)
или ИРФ-23. Устройство их таково, что при освещении обычным
дневным светом на шкале прибора можно прочесть показатель
преломления для волны 589 ммк (линия D спектра натрия).
Наиболее часто определение ведут при 20 "С. В таком случае
показатель преломления обозначают ri?
или /г!аэ-
Достаточно 2—3 капель жидкости,
чтобы при помощи рефрактометра
очень точно определить показатель
преломления.
Показатель преломления в большой
степени зависит от состава веществ и
от их химического строения.
В качестве примера приведем
величины ri? для некоторых
соединений:
м-С4Н9С1 . . .
н-С4Н8Вг . . .
H-C4HeJ ....
(С,Н5),0 . . .
и-С4Н,ОН . . .
,4015
,4398
,4998
,3497
,3991
Рис. 14. Внешний вид
рефрактометра.
Таким образом, эта физическая
константа позволяет характеризовать
и определять чистоту органических
веществ и во многих случаях определять
состав смесей двух веществ,
показатели преломления которых значительно различаются по величине.
При химических исследованиях часто оказывается очень
полезной сложная физическая константа, связывающая в общей
формуле показатель преломления п, плотность d и
молекулярный вес М. Это так называемая молекулярная рефракция MR:
л*-1 М
Для каждого вещества молекулярная рефракция является
постоянной величиной, очень мало зависящей от температуры,
так как при изменении температуры п и d изменяются в
противоположных направлениях, что приводит к взаимной
компенсации .
86 Глава І. Углеводороды
Необходимо, чтобы and были определены при одной и той же
температуре. Знак MRd означает, что п определен для линии D
спектра.
Молекулярная рефракция определяется природой вещества,
т. е. его составом и химическим строением. Ее величина может
быть определена для исследуемого вещества практически, если
известны п и d, а кроме того, она может быть вычислена по сумме
так называемых атомных рефракций, которые определяются
путем изучения молекулярных рефракций чистых веществ
известного состава и строения.
Покажем, как могут быть найдены значения атомных
рефракций. В рядах гомологических соединений, например в
гомологическом ряду предельных углеводородов, молекулярная
рефракция при переходе от одного члена к последующему члену ряда
увеличивается приблизительно на одну и ту же величину. Как
среднее из многих определений, эта величина (для линии D
натриевого спектра) равна 4,618. Очевидно, это рефракция
группы СН2.
Для «-гептана С7Н1в молекулярная рефракция равна 34,526.
Вычислим, исходя из этой величины, атомные рефракции водорода
и углерода:
4,618-7 = 32,326 34,526 — 32,326 = 2,200
Следовательно, атомная рефракция водорода равна 1,109, а
атомная рефракция углерода 4,618—2,200=2,418.
Таким же оЗразом могут быть найдены атомные рефракции и
других элементов. При этом оказывается, что атомная
рефракция кислорода зависит от того, находится ли кислород в группе
С=О (карбонильный кислород), С—ОН (кислород гндроксила)
или же в группе С—О—С (эфирный кислород).
Ниже приведены величины атомных рефракций некоторых
элементов (вычисленные как средние из многих определений):
Линия О натрия Линия О нутрия
Углерод 2,418 Хлор 5.967
Водород 1,100 Вром 8.865
Кислород Иод 13,900
карбонильный . 2,211 Инкремент
гидроксильный 1,643 двойной связи С=С 1,733
эфирный . . . 1,525 тройной связи CsC 2,398
Для непредельных соединений молекулярная рефракция,
найденная опытным путем, всегда больше, чем вычисленная как
сумма атомных рефракций. Так, молекулярная рефракция амилена
С5Н10 равна 24,83, в то время как вычисление дает 23,09. Увели-
33. Этилен
чение молекулярной рефракции, вызванное двойной углеродной
связью, называется инкрементом* двойной связи. Еще большее
увеличение молекулярной рефракции вызывает тройная связь.
Таким образом, атомная рефракция углерода зависит от степени
его насыщения.
Определение молекулярной рефракции имеет большое
значение для идентификации вещества. Молекулярная рефракция
является в то же время средством проверки правильности
структурной формулы: если молекулярная рефракция, вычисленная на
основании структурной формулы, т. е. как сумма атомных
рефракций и инкрементов, совпадает с молекулярной рефракцией,
найденной опытным путем (или близко подходит к ней), то это
служит веским подтверждением правильности принятой для
данного вещества структуры.
33. Этилен. Этилен С2Н4 содержится в коксовом газе и в газах
крекинга нефти. В лаборатории его получают отщеплением
элементов воды от этилового спирта при помощи концентрированной
серной кислоты:
сн3—сна—он —»сна=сна + нао
Для этой цели нагревают смесь спирта с концентрированной
серной кислотой. Сначала образуется этилсерная кислота
; _Н.О
Н—СНа—СНа-;ОН -f Н!—О—SOj—ОН — U Н-СНа—СНа—О— ^Оа-ОН
которая' при дальнейшем нагревании распадается на этилен к
серную кислоту:
Н—СНа-СНа—O-SOa-OH » СН2=СНа -f HaSO4
Наряду с этиленом образуются и другие вещества. Это
объясняется тем, что при нагревании серной кислоты со спиртом
протекает одновременно несколько параллельных реакций. Так,
параллельно с отщеплением одной молекулы воды от одной
молекулы спирта (образование этилена) происходит отщепление одной
молекулы воды от двух молекул спирта, в результате чего
образуется простой эфир:
сн3—сна—он + н—о—сна—сн3 —»с2н5—о—сан5 + нао
Наконец, серная кислота, отдавая один атом кислорода, может
окислять спирт, сжигая его в углекислый газ и воду, при этом.
* От латинского incrementum—прирост.
Глава І. Углеводороды
она восстанавливается до сернистой кислоты H2SO3, которая,
будучи неустойчивой, распадается на сернистый газ и воду:
6H2SO4=6H2SOg + 60
6H2SO3=6SO2 + 6Н2О
С2Н5ОН + 6О=2СО2 + ЗН2О
CjHjOH + 6H2SO4=2CO2 + 6SO2 + 9НаО
Таким образом, в результате взаимодействия серной кислоты
со спиртом получаются, кроме этилена, еще эфир, сернистый и
углекислый газы.
Этот пример показывает, что органическое вещество,
взаимодействуя с другим органическим или неорганическим веществом,
может вступать с ним в несколько различных реакций. В
результате этих реакций могут быть получены совершенно различные
продукты Подбирая соответствующим образом условия
проведения реакции (температуру, давление, относительные количества
взятых веществ, катализаторы и т. д.), можно заставить одну из
этих реакций преобладать над другими. Так, например, при 170 °С
этилсерная кислота разлагается с образованием этилена, а при
140 °С она реагирует с этиловым спиртом с образованием эфира
(стр. 178).
Этилен—газ, сгущающийся в жидкость при —103,7 °С; при
—169,2 °С он затвердевает; почти нерастворим в воде; горит более
ярким пламенем, чем метан; с воздухом образует взрывчатую
смесь. Этилен способен полимеризоваться:
лСаН4 » (—CHa—СН2—)п
где п—степень полимеризации (п может иметь значение 1000 и
больше).
Высокомолекулярный полиэтилен—желто-белое роговидное,
пластичное, легкое, морозостойкое вещество.
Концентрированные кислоты, включая фтористоводородную, а также щелочи не
разрушают его.
В последние годы были открыты комплексные металлорганические
катализаторы, позволяющие получать высокомолекулярные твердые полимеры
этилена (полиэтилен) без применения давления. Одним из таких широко
применяемых катализаторов является система, состоящая из триэтилалюминия
А1(С2Н5)з и четыреххлористого титана TiCU. При взаимодействии этих двух
соединений образуется твердое вещество, состоящее из сложного металлорга-
нического комплекса, каталитически воздействующего на полимеризацию
этилена. Полиэтилен, получаемый при помощи этого катализатора, представляет
собой предельный углеводород нормального строения. Он менее эластичен,
чем полиэтилен, получаемый при высоких давлениях, но обладает большей
твердостью и способен выдерживать воздействие более высоких температур.
34. Изобутилен 89
Благодаря сочетанию многих ценных свойств диапазон облагтгй
применения полиэтилена очень широк. Полиэтилен является одним ич лучших
материалов для изоляции кабелей, для применения в радарной технике
радиотехнике, телевидении и др. Из него изготавливают трубы, шланги, сосуды,
пленки различной толщины и многие бытовые предметы.
Кроме того, этилен является исходным сырьем ДЛЯ получения
важнейших полупродуктов, на которых основывается
производство синтетических материалов,—стирола, окиси этилена,
хлористого винила и других соединений.
Из него получают этиловый спирт и ряд ценных
растворителей. Действием этилена на хлористую серу получали боевое
отравляющее вещество—иприт (см. стр. 184).
Было обнаружено, что если снятые с кустов недозрелые помидоры
поместить в атмосферу этилена, то дозревание их значительно ускоряется,
причем для этого достаточно ничтожного содержания этилена в возлухе. В
настоящее время этилен широко используется для ускорения дозренания плодов.
Теломеризация. Интересен недавно открытый и введенный в
технику процесс полимеризации этилена, названный шеломери-
зацией. Если в смесьэтилена иСС14 внести перекись бензоила или
другой инициатор, распадающийся с образованием свободных
радикалов, то происходит следующий процесс:
R.-t-CCI4 » RCI +СС1з-
Радикалы СС13- инициируют цепную полимеризацию этилена:
СС13- + CHa=CHs » СС13—СН3—СН3.
СС13—CH.J—СН3.+СН3=СН3 » ССІз—СН3—СН3—СН3—СН,. и т. д.
При встрече с другой молекулой СС14 рост цепи прекращается:
СО,—(СН.,—СН3)„.+СС14 . СС1„—(СН2—СН2)„—CI+CCI;,-
Радикал СС1, ¦ дает начало новой цепи.
Образующиеся низкомолекулярные продукты полимеризации,
содержащие на концах цепи остатки молекулы растворителя,
называются шеломерами. Получены теломеры со значениями п—2, 3,
4....15. Теломеры путем гидролиза или аммонолиза могут быть
превращены в окси- и аминокислоты:
СООН(СНг—СН2)„ОН СОО1-|(СН3—СН.3)„МН.,
34. Изобутилен B-метилпропен) СН2=С(СНя)г, бесцветный газ
(темп. кип. —Ь,У JC), получается ньгазив нефтеперерабоїки. Впер-
90 Глава І. Углеводороды
вые синтезирован А. М. Бутлеровым в 1868 г. Под влиянием
хлористого цинка, фтористого бора, хлористого алюминия и других-
катализаторов происходит полимеризация изобутилена. В
зависимости от условий полимеризации получаются полимеры с
различным молекулярным весом—от вязких жидкостей до твердых,
эластичных материалов. Техническое применение получил поли-
изобутилгн—высокомолекулярный полимер со средним
молекулярным весом—от 100 000 до 500 000. Он отличается высокой
химической стойкостью и водостойкостью и применяется в
виде обкладочных листов и антикоррозионных защитных
пленок.
Особенно большое техническое значение получил
бутилкаучук—сополимер изобутилена с 2—3% изопрена (см. стр. 96).
Бутилкаучук отличается высокой газонепроницаемостью,
вследствие чего он применяется для изготовления камер
автомобильных шин, внутреннего слоя бескамерных шин и различных
резинотехнических изделий.
Углеводороды с тройной связью (алкины)
35. Строение ацетиленовых углеводородов и их получение.
Простейшим представителем углеводородов с тройной связью
является ацетилен С2Н2 или Н—С=С—Н.
От ацетилена, замещением в его молекуле атомов водорода ал-
килами, выводятся другие члены гомологического ряда
углеводородов с одной тройной связью в молекуле. Их названия по
женевской номенклатуре имеют окончания ин:
Ацетилен—этин СН=СН
Метилацетилен—пропин CHsC—СН3
Этилацетилен—бутин -1 СН=С—CHa—СН3
Диметилацетилен—бутин-2 СН3—С=С—СН3
И т. Д.
Так как молекулы ацетиленовых углеводородов с одной
тройной связью содержат на четыре атома водорода меньше, чем
молекулы соответствующих предельных углеводородов, то состав
их можно выразить общей фо р му л о й С„ Н 2n_g._ J
Ацетиленовые углеводороды образуются* при сухой перегонке
многих органических веществ. Они могут быть получены, как
это показал в 1861 г. Савич, отнятием двух молекул галоидово-
дорода из соединений, содержащих два атома галоида при
соседних атомах углерода:
CHoCI— CHoCl + 2КОН , CH^eCH + 2КС1 + 2HSO
36 Свойства ацетиленовых углеводородов 9*
Так как такие галоидные соединения получаются
присоединением галоидов к олефинам, то приведенная реакция позволяет
перейти от соединений с двойной связью к соединениям с
тройной связью:
СН2=СН—СН3 .. С'а > CHjCI—CHCI—СН3 ~2НС) СН=С—СН3
Ацетиленовые углеводороды изучались многими химиками.
Особенно большое значение в этой области имеют пользующиеся
мировой известностью работы научных школ А. Е. Фаворского* и
Н. Д. Зелинского.
36. Свойства ацетиленовых углеводородов. Ацетилен и ме-
тилацетилен—газы, следующие гомологи—жидкости, высшие—
твердые тела. Ацетиленовые углеводороды имеют более высокие
температуры кипения, чем соответствующие предельные
углеводороды. Так, температура кипения н-пентана 36 °С, пентина-1
39,7 °С.
Ацетиленовые углеводороды являются в еще большей мере
ненасыщенными, чем этиленовые углеводороды. Поэтому
ацетиленовые углеіодородьі легко вступают в реакции присоединения.
Их молекулы способны присоединить четыре атома водорода или
галоида или две молекулы галоидоводорода.
1. Присоединение водорода:
Н Н
H-CSEC-H+2R, > Н-С-С—Н
ацетилен І І
н н
этан
'Алексей Евграфович Фаворский A860—1945) принадлежит к числу
выдающихся учеников А. М. Бутлерова.
Обширны его исследования в области ацетиленовых углеводородов и
продуктов получающихся на основе ацетилена. Фаворский открыл и изучил
явления изомеризации и взаимных переходов ацетиленовых и алленовых
углеводородов; разработал метод получения простых виниловых эфнров действием
спиртов на ацетилен в присутствии порошка едкого кали. Позднее он совместно
со своими учениками развил эту реакцию и разработал производственные
методы получения виниловых эфиров (М Ф. Шостаковский). Широко внедрены-
в практику предложенные им совместно с учениками (И. Н. Назаров) реакции
ацетилена и ацетиленовых углеводородов с кетонами. Этим методом можно-
пэлучить изопрен для синтетического каучука
В 1902 г. А. Е. Фаворский занял кафедру органической химии в
Петербургском университете. В 1929 г. он был избран действительным членом
Академии наук СССР. Его учениками являются многие выдающиеся химики
(С. В. Лебедев, Б. В. Вызов, И. Н. Назаров и др.).
92 Глава I. Углеводороды
2. Присоединение галоидов:
Н—Г=С— Н + 2Вг2
ацетилен
Вг Вг
четырехбром истый
ацетилен
A,1,2,2-тетрабромэтан)
3. Присоединение галоидоводородов происходит согласно
правилу Марковникова:
сн=с—СН3 + 2HCI =. СН3—СС1.2—СН3
меіилацетилен 2,2 дихлорпропа і
4. При пропускании ацетилена в воду, в которой находится
в качестве катализатора соль двухвалентной ртути и серная
кислота, молекула ацетилена присоединяет молекулу воды с
образованием уксусного альдегида. Эта реакция, открытая в 1881 г.
М. Г. Кучеровым*, служит и в настоящее время для технического
получения уксусного альдегида, окислением которого получают
уксусную кислоту. Гомологи ацетилена в этих условиях дают ке-
тоны.
Схема реакции может быть изображена следующим образом:
і ¦ п/П "\ /Н
Н—CsC—H + Н—ОН =. >С=С/ * Н—С—С<С
t :
Нч
уксусный альдегид
Нч уСН3
Н—С=С—СН3 + Н—ОН * >С=С< ;. СН3—С
н/ \он о
ацетон
Вначале образуется непредельный спирт; однако, как показал
А. П. Эльтеков, соединения, содержащие гидроксил при углероде
с двойной связью, изомеризуются в альдегиды или кетоны.
5. Ацетиленовые углеводороды легко окисляются при
действии окислителей. При энергичном окислении углеродная цепь
разрывается по месту тройной связи.
* Михаил Григорьевич Кучеров A850—191!) был профессором
Петербургского земледельческого института. Свои работы по гидратации ацетилена он
начал около 1875 г. Реакцию получения уксусного альдегида открыл в 1881 г.-
В дальнейших работах Кучеровым было показано, что метилацетилен и
фенилацетилен реагируют аналогичным путем, причем получаются уже не
альдегиды, а, как и следовало ожидать, соответствующие кетоны. Им
исследован также процесс взаимодействия олефинов с ртутными солями.
37 Ацетилен 93
6. Если в ацетиленовых углеводородах при углероде с
тройной связью имеется атом водорода, то этот атом способен
замещаться металлом. Так, при пропускании ацетилена через
аммиачный раствор азотнокислого серебра выпадает белый осадок
ацетилен истого серебра Ag—С=С—Ag. Аммиачный раствор
хлористой меди дает красно-бурый осадок ацетиленистой меди.
Ацетиленистая медь и ацетиленистое серебро в сухом виде при
нагревании или при ударе легко взрываются.
При действии кислот на ацетиленистые металлы снова
выделяется ацетилен:
Ag—С=С—Ag + 2HNO3 » H—Q=C—H + 2AgNO3
7. Ацетиленовые углеводороды способны к полимеризации.
Так, ацетилен в присутствии солей закиси меди (катализатор) по-
лимеризуется в весьма интересный продукт—винилацетилен
СН=С—СН=СН2. Его можно рассматривать как продукт,
полученный замещением в ацетилене атома водорода радикалом
винилом —СН=СН2.
Винилацетилен является промежуточным продуктом при
получении одного из видов синтетического каучука (стр. 104).
Температура кипения винилацетилена 5 °С.
37. Ацетилен. Ацетилен образуется при сухой перегонке
многих органических веществ. Синтез ацетилена из элементов
(образование ацетилена в вольтовой дуге между угольными
электродами в атмосфере водорода), проведенный Бертло в 1860 г.,
явился первым синтезом простейшего углеводорода.
До недавнего времени ацетилен получали только действием
воды на карбид кальция:
Ca ;
\i
С
11 +
I
H—
H—
—ОН
» HCseCH + Са(ОН)а
—ОН
Эта реакция открыта в 1852 г. Вёлером, но практическое
значение она приобрела лишь после того, как был разработан способ
получения карбида кальция сплавлением извести и кокса в
электрической печи. В настоящее время большое промышленное
значение приобретают способы получения ацетилена из нефтяного
сырья и природных газов. Метан превращается в ацетилен под
кратковременным (сотые доли секунды) воздействием очень
высоких температур A400 °С и выше):
2СЦ, » С,Н2 + ЗНЭ
94 Глава І. Углеводороды
Реакция сильно эндотермична и технические методы получения
ацетилена различаются по способам подвода тепла, например
посредством вольтовой дуги, путем сжигания части метана
непосредственно в реакционном пространстве и др. Аналогичным
путем, но при несколько более низких температурах, ацетилен
может быть получен из высших углеводородов—пропана, бутана ил if
легких нефтяных погонов. Реакция получения ацетилена из
углеводородов протекает сложно и сопровождается образованием
большого количества побочных продуктов—этилена, углерода
в виде сажи, гомологов ацетилена. Разработанные методы
разделения газовой смеси на отдельные компоненты с последующей
тщательной очисткой позволяют выделить ацетилен в достаточно
чистом виде.
Ацетилен—бесцветный газ, в совершенно чистом виде
имеющий слабый эфирный запах. Неочищенный ацетилен,
полученный из карбида, имеет неприятный запах, обусловленный
примесями фосфористого водорода и сероводорода. Ацетилен
сравнительно хорошо растворим в воде и довольно легко сгущается в
жидкость, кипящую при температуре —83,6 °С.
Так как ацетилен содержит больший процент углерода, чем
этилен и метан, то он горит более ярким и коптящим пламенем,
чем эти два углеводорода. Однако при смешении с большим
количеством воздуха, что достигается специальным устройством
ацетиленовых горелок, ацетилен сгорает ярким пламенем без
копоти.
Так как при сгорании ацетилена, особенно в кислороде,
развивается очень высокая температура (даже более высокая, чем
при сгорании водорода), то ацетилен-кислородным пламенем
широко пользуются для автогенной сварки и резки металлов.
Сжатый и особенно жидкий, ацетилен легко взрывается даже
от ничтожного толчка. Поэтому его хранят и перевозят в
стальных баллонах в виде полученного под давлением раствора в
ацетоне. Взрывчатые свойства ацетилена объясняются тем, что он
представляет собой эндотермическоэ вещество, т. е. при сгорании
ацетилена выделяется больше тепла, чем при сгорании
такого же количества углерода и водорода. При разложении
ацетилена на углерод и водород выделяется 54,8 ккал на
1 моль.
С воздухом ацетилен образует чрезвычайно взрывоопасные
смеси; такая смесь способна взрываться при содержании в ней
от 3 до 82% ацетилена.
Огромное применение находит ацетилен как исходны"! продукт
для получения ряда химических соединений. По ре л кии и Куче-
рова ацетилен переводят в уксусны! альдегид из которого в
свою очередь получаю: уксусную кислоту и другие соединения
38 Строение и свойства углеводородов с двумя двойными связями 95
Получаемые из ацетилена жидкие пгепграхлорэтан СНС12—СНС12
и трихлорэтилен СНС1=СС12 (последний получается кипячением
тетрахлорэтана с известью)
СН=СН + 2С12 > СНС|2-СНС12
1,1,2,2-тетрахлорэтан
2СНСІ2—СНС12 + Са(ОНJ > 2СНС1=СС12 + СаС1а + 2Н./)
1,1,2-трихлорэтилен
являются превосходными растворителями жиров и серы.
В отличие от многих других растворителей, они трудно
воспламеняются и поэтому безопасны в пожарном отношении. Их
применяют для извлечения масел и для обезжиривания металлов.
Весьма значительные количества ацетилена используются
для синтеза каучука (см. стр. 100 ел.) и получения поливинил-
хлоридных пластических масс (см. стр. 117).
При пропускании ацетилена через хлористый мышьяк AsCU в
присутствии хлористого алюминия АІСІз (в качестве катализатора) образуется хлорви-
нилдихлорарсин:
і І АІСІз /СІ
СН^СН + ClAsCI2 * CI-CH=CH-As/
1 '. льюисит Xll
применявшийся в первую мировую воину как отравляющее вещество под
названием льюисит.
Льюисит представляет собой жидкость с запахом герани. Подобно
иприту, он действует на кожу, вызывая образование нарывов; кроме того, он
обладает общеядовитым и раздражающим действием.
Углеводороды с двумя двойными связями. Каучук
38. Строение и свойства углеводородов с двумя двойными
связями. Так как каждая двойная связь уменьшает количество
водорода в молекуле на два атома, а тройная связь—на четыре
атома, то углеводороды с двумя двойными связями имеют тот же
состав, что и соответствующие углеводороды ряда ацетилена,
т. е. состав их выражается формулой С„Н2„_2. По женевской
номенклатуре названия этих углеводородов имеют окончание диен;
цифрами указывают номера углеродных атомов, которые связаны
с последующими атомами углерода двойными связями.
96 Глава І. Углеводороды
Из углеводородов с двумя двойными связями наибольшее
значение имеют:
СНЭ
сна=<:н—сн=сна сна=с—сн=сна
бутадиен-1,3 2-метилбутадиеи-1,3
(изопрен)
сн3 сн3
СН2=С C^
диметилбутадиен-1,3
Бутадиен, или дивинил, СНа=СН—СН=СН2 представляет
собой газ, сжижающийся при —5 °С; 2-метилбутадиен-1,3, или
изопрен,—жидкость, кипящая при 34 °С;
диметилбутадиен—жидкость с температурой кипения 70 °С. Изопрен образуется в
значительных количествах при сухо1" перегонке природного каучука и
обладает способностью полимеризоваться в каучук.
В бутадиене и изопрене двойные связи разделены двумя
атомами углерода, соединенными между собой простой связью:
—С=С-С=С—
Такие системы двойных связей получили название
сопряженных или конъюгированных двойных связей.
Вещества с сопряженными двойными связями обладают
характерной особенностью: они способны вступать в реакции
соединения таким образом, что присоединение происходит к
первому и четвертому атомам углерода, при этом между вторым и
третьим атомами углерода возникает двойная связь:
СН2=СН-СН=СН2 + Вг-Вг » СН2Вг-СН=СН-СЧ,Вг
^ : 1,4-ДИОромоу ieri-i
бута.'іиен 1.3
Теория, объясняющая эту особеннос.ь соединений с
сопряженными двойными связями, бьиа развита в 189Э г. немецким
ученым Тиле*. Согласно этой теории, у кажчого из агомов углерода,
соединенных двойной связью, остается способность к
присоединению, так как при образовании кратной связи сродство атомов
углерода не используется полностью, а сохраняется остаточная,
* Современные представления о строении двойной связи и яьлениях
сопряжения см. стр 243.
38 Строение и свойства углеводородов с двумя двойными связями 97
или парциальная, валентность. Она условно обозначается
пунктиром:
—СНа=СНа—
Атомы брома притягиваются силами парциальных
валентностей, вторая связь между атомами углерода разрывается, и
каждая из освобождающихся единиц валентности насыщается
единицами валентности брома.
В молекуле дивинила имеются парциальные валентности у
каждого атома углерода:
СН3=СН—СН=СН.а
Парциальные валентности гторого и третьего атомов углерода
насыщают друг друга, вследствие чего образуется некоторое
подобие двойной связи. Чтобы отличить эту «двойную» связь от
обыкновенной двойной связи, обозначим ее дугообразной линией:
CH2=QH—СН=СН2
Таким образом, незамкнутые парциальные валентности
остаются у первого и четвертого атомов углерода. К ним и
присоединяются атомы брома. При этом происходит разрыв второй связи
между первым и вторым и между третьим и четвертым атомами
углерода. Освободившиеся валентности второго и третьего
атомов углерода насыщают друг друга, между этими атомами
возникает обычная двойная связь, и у каждого из них остаются
парциальные валентности:
СНгВг-СН=СН-СН3Вг
1,4-дибромбутен-2
Следует отметить, что правило Тиле не имеет общего
характера; в ряде случаев присоединение идет обычным путем, т. е. по
месту двойной связи.
Так, наряду с приведенной выше, идет и другая реакция:
СН3=СН-СН=СН3 + Вг.а » CHjBr—СНВг—СН=СНа
Ч,4-дибромбутен-1
При действии избытка брома происходит присоединение
галоидов по обеим связям:
СН2=СН—СН=СН„ + 2Вг3 > СН3Вг—CHBr-CHBr-CHjBr
1,2,3,4-тетрабромбутан
7—788
98 Глава І. Углеводороды
Особенно характерным свойством углеводородов с
сопряженными двойными связями является их способность к
полимеризации. Проведенная в определенных условиях полимеризация
углеводородов этого типа дает вещества, весьма сходные с каучуком.
Наличие сопряженных двойных связей сильно отражается
на некоторых физических свойствах веществ. Так, оказывается,
что молекулярная рефракция соединений с сопряженными
связями, вычисленная из экспериментальных данных, значительно
больше молекулярной рефракции, полученной из суммы атомных
рефракций и инкрементов.
Покажем это на примере двух изомеров гексадиена (C6HL0,
мол. вес 82,16).
а) Гексадиен-1,5 с двумя изолированными двойными связями
имеет показатель преломления и относительную плотность:
«В'= 1,4044; df = 0,6880
Его молекулярная рефракция:
' 82,16 9q 19
2912
= 1,4044«+й '
б) Гексадиен-1,3 с системой двух сопряженных связей имеет
показатель преломления и относительную плотность:
яЬ°= 1,4384; d? = 0,7108
Его молекулярная рефракция:
[, 43842 — 1 82,16
1,4384'+2 • СШ58
= 30'36
Вычислим молекулярную рефракцию MRd из атомных
рефракций:
Q 2,418x6 = 14,508
Н10 1,100x10= 11,000
1,733x2 = 3,466
28,974
Для изомера с изолированными двойными связями разиость между
экспериментальной величиной молекулярной рефракции и
вычисленной равна 0,15, а для изомера с сопряженными двойными
связями эта разность равна 1,39.
39. Каучук и его свойства 99
Это повышение молекулярной рефракции, называемое
экзальтацией, систематически наблюдается у соединений с
сопряженными двойными связями.
Таким образом, по величине молекулярной рефракции можно
в ряде случаев судить с строении непредельного углеводорода.
39. Каучук и его свойства. Натуральный каучук в очищенном
состоянии является полимером, имеющим общую формулу
(С,Н8)Ж, и получается главным образом из млечного сока
(латекса) некоторых тропических растений, преимущественно
гевеи—громадного дерева, родиной которого является Бразилия.
Туземцы Южной Америки называли этот млечный сок «као
чо»—слезы дерева. Французские ученые, исследовавшие
свойства этого вещества, назвали его каучуком.
В латексе каучук находится в виде водной дисперсии. При
прибавлении небольшого количества уксусной кислоты каучук
свертывается в сплошную массу и отделяется от жидкости. Каучук
вальцуют в виде листов, просушивают и коптят дымом для
предохранения от плесневения и порчи.
Каучук хорошо растворим в бензине, бензоле, сероуглероде.
При низкой температуре он становится хрупким, при
нагревании липким. Для того чтобы улучшить механические и
химические свойства каучука, его превращают в резину, подвергая
вулканизации. Для получения резиновых изделий сначала формуют
изделия из смеси каучука с серой, а также с так называемыми
наполнителями— сажей, мелом, глиной и некоторыми
органическими веществами, которые служат ускорителями вулканизации.
Затем изделия подвергают нагреванию—горячей
вулканизации.
При холодной вулканизации, которая применяется для тонких
и мелких изделий (прорезиненные ткани, тонкие трубки и т. д.),
их непродолжительное время обрабатывают раствором серы в
сероуглероде или хлористой сере. Каучук с большим содержанием
серы (до 32%) представляет собой твердое неэластичное
вещество и называется эбонитом; применяется он как изолятор в
электроприборах.
В результате вулканизации сера химически связывается с
каучуком. Кроме того, в вулканизованном каучуке содержится в
виде мельчайших частиц и свободная сера.
Рост технической вооруженности и моторизации народного
хозяйства делает каучук сырьем первостепенной важности.
Каучук имеет громадное применение: из него производят резиновые
шины для автомобилей, велосипедов и самолетов, резиновые
тормозные рукава поездов, прорезиненные приводные ремни,
изоляцию для электрических проводов, резиновые маски противогазов,
клапаны тракторов и автомобилей, галоши, всевозможные домаш-
7»
100 Глава І. Углеводороды
ниє и медицинские изделия, некоторые предметы лабораторного
оборудования* и т. п.
40. Строение и синтез каучука. По своей химической природе
каучук является высокомолекулярным непредельным
углеводородом и представлет собой смесь сложных полимерных молекул.
Как непредельное соединение, каучук присоединяет бром и гало-
идоводороды, причем на одну группу С3Н8 присоединяются два
атома брома или одна молекула галоидоводорода. Следовательно,
на каждую группу С5Н8 в молекуле каучука приходится одна
двойная связь. При сухой перегонке каучука образуется, наряду
с другими углеводородами, изопрен С5Н8. Первые сведения о
строении каучука были получены в 1905 г., когда Гарриес,
обработав каучук озоном, получил стекловидный озонид состава
СюНібОб. При разложении озонида водой образуется до 90%
левулинового альдегида СН3—СО—СН2—СН2—СНО.
Его образование при разложении озонида указывает, что в
молекуле каучука имеется правильно повторяющаяся группа
СНд
=С—СН2—СНа—СН=
и молекула каучука должна иметь следующий вид:
cH-i СЛі СН-і
І І І
.—сн2—сн=с -сн2—сн2—сн=с -сн2 -сн2—сн=с -сн2
каучук
озонирование Оэ
е Оэ
¦^СНл НС і С—СНг.—СН?—НС ( С—СНг.—СНО—НС і С—СНо —
2 III 2 2 111 2 2 i;i 2
О-ь-О О-гО О-|-О
озонид каучука
расщепление Н2О
і ** О і ^ О
—+ со—сн2—сн2—с^ +¦ со—сн2—сн2—с^ + ¦¦¦
н н
левулиновый альдегид
* В химических лабораториях широко применяются резиновые пробки
и трубки для соединения частей приборов, подвода воды и газов. При работе
с ними надо учитывать свойства резины. Резина мало изменяется от
действия разбавленных щелочей и разбавленной соляной кислоты, но
концентрированная серная и особенно азотная кислоты легко ее разрушают. В
органических растворителях (например, галоидуглеводородах) резина набухает и
растворяется. От паров хлора или брома она делается жесткой и хрупко».
40. Строение и синтез каучука 101
В приведенной выше формуле каучука можно увидеть
повторяющийся скелет изопрена. Можно предположить, что молекулы
изопрена взаимодействуют друг с другом как системы с
сопряженными связями, т. е. присоединение происходит по
концам цепи, а между средними атомами углерода возникают
двойные связи:
СН3 СН3 СН3
СН2=СН—С=СН2 + СН2=СН—С=СН2 + СН2=СН—С=СН2 + • • • »
СН3 СН3 СНз
Состав групп на концах цепи до сих пор не выяснен.
Каучук—типичный пример высокомолекулярного соединения.
Штаудингер на основании определения вязкости растворов
каучука установил, что природный каучук из гевеи имеет
молекулярным вес около 170 000, что соответствует 2500 изопреновым
гр;ппам: следовательно, коэффициент полимеризации п. равен
іГіОО. При выделении каучука из природных веществ
молекулярная цепь каучука разрывается—коэффициент полимеризации
уменьшается, достигая в техническом продукте примерно 400.
Коэффициент полимеризации неодинаков у разных молекул в
одном и том же образце каучука. Молекулы каучука содержат
различное число изопреновых групп. Они являются членами по-
лнмер-гомологического ряда (С,НЧ)П, различаясь между собой
по составу на то или иное число групп С5НЧ. В данном случае
можно говорить только о среднем молекулярном весе.
Как было ранее указано, каучук «вулканизуют», нагревая его
с серой. Механизм вулканизации заключается в том, что
отдельные линейные молекулы каучука связываются между
собой в разных местах атомами серы Образуются
высокомолекулярные соединения, имеющие сетчатую структуру и обладающие
меньшей эластичностью, чем исходный каучук. Чем больше
образуется соединительных мостиков из атомов серы, тем
будет больше твердость и меньше эластичность продукта.
Громадное практическое значение каучука давно привлекло
внимание химиков к проблеме получения этого продукта
синтетическим путем. Однако промышленный синтез самого изопрена
представлял значительные трудности. Поэтому первые попытки
получения каучукоподобных продуктов были сделаны с
аналогами изопрена—углеводородами, содержащими систему
сопряженных связей
Н2С=С—С=СНа
где R и R'—алкилы.
102 Глава I Углеводороды
Так, полимеризацией диметилбутадиена в 1902 г И. Л.
Кондаков впервые получил каучукоподобное вещество. Позднее, в
первую мировую войну 1914—1918 гг., в Германии была сделана
попытка промышленного получения синтетического каучука из
диметилбутадиена; было изготовлено всего около 2000 т «метил-
каучука», который, однако, оказался технически мало ценным
Промышленное получение синтетического каучука было
впервые осуществлено в СССР. Большая заслуга в этом принадлежит
С. В. Лебедеву*. Он предложил и разработал метод
промышленного производства синтетического каучука на основе дивинила.
Исходным сырьем для получения синтетического каучука по
способу Лебедева является этиловый спирт. Пропуская при
400—500 °С пары этилового спирта над катализатором, состав
которого был разработан С. В. Лебедевым, получают смесь
веществ, из которой выделяют дивинил:
2С2Н5-ОН > СН2=СН—СН=СН2 + 2НоО + На
дивинил
Дивинил, отделенный от побочных продуктов, подвергают
полимеризации, например, в присутствии металлического натрия;
при этом получается масса, похожая по свойствам на природный
каучук. Эта масса после вулканизации превращается в резину.
В настоящее время дивинил получают не только из спирта.
Наиболее выгодным оказалось его получение из бутана,
содержащегося в попутных газах нефтедобычи, или из бутиленовой
фракции нефтезаводских газов.
*Сергей Васильевич Лебедев A874—1934) родился в Люблине. Среднее
образование получил в Варшавской гимназии. В 1895 г. С. В. Лебедев поступил
на физико-математический факультет Петербургского университета.
С. В. Лебедев был учеником академика А. Е. Фаворского, под
руководством которого и выполнены его первые работы. Исследования полимеризации
непредельных органических соединений Сергей Васильевич начал в 1906 г.
В декабре 1909 г. на заседании Русского химического общества он сделал
доклад о полимеризации диеновых углеводородов и демонстрировал
каучукоподобное соединение, полученное из дивинила.
В апреле 1913 г. С. В. Лебедев защитил магистерскую диссертацию на тему:
«Исследования в области полимеризации днунепредельных углеводородов».
В 1925 г. Сергей Васильенич организовал в Ленинградском университете
лабораторию по переработке нефти, где были проведены работы по получению
синтетического каучука из спирта (через дивинил).
В дальнейшем под непосредственным руководством С. В. Лебедева был
построен опытный завод синтетического каучука, на котором в яниаре 1931 г.
был выпущен первый блок промышленного продукта.
В 1928 г. Лебедев был избран членом-корреспондентом, а в 1932 г.
действительным членом Академии наук СССР. За особо выдающиеся заслуги по
разрешению проблемы «Получение синтетического каучука» С. В. Лебедев был
награжден орденом Ленина.
40. Строение и синтез каучука 103
При пропускании бутана при температуре 550—600 °С
над катализатором, состоящим из смеси окислов алюминия и
хрома, происходит реакция дегидрирования (отщепления
водорода):
СН3—СН3—СНа—СН3 > СН3—СНа—СН=СН2 + Н2
бутилен
Полученный бутилен вновь подвергают дегидрированию
при более высокой температуре над катализатором другого
состава:
СН;,—СН2—СН=СН2 > СН.2=СН— СН=СНа + Н2
дивинил
Путем подбора соответствующего катализатора и условий
реакции процесс может быть проведен и в одну стадию:
СН3— СНа—СН2—СН3 > СНа=СН—СН=СНа + 2Н2
Впервые путь синтеза дивинила из нефтяных углеводородов
был предложен еще в 20-х годах Б. В. Бызовым. Однако состояние
техники того времени не позволяло еще выделять индивидуальные
углеводороды в достаточно чистом виде. В настоящее время
дегидрирование бутана и бутиленов является наиболее
распространенным способом получения дивинила в СССР и в большинстве
других стран.
Один из видов синтетического каучука получают из ацетилена.
Выше (см. стр. 93) указывалось, что при полимеризации
ацетилена образуется винилацетилен СН=С—СН=СН2. Винилацетилен
присоединяет молекулу хлористого водорода; при этом
получается 2-хлорбутадиен-1,3 {хлоропрен):
СІ
СН==С—СН=СНа + НСІ s. СН2=С—СН=СН2
Хлоропрен—бесцветная жидкость, кипящая при 59 °С. Она
самопроизвольно весьма легко полимеризуется, образуя сначала
пластическую массу, сходную с невулканизованным каучуком, а
в дальнейшем—твердый продукт (вулканизация без серы):
СНа=СН—Ш=СНа + СН3=СН—СС1=СНа + СН,=СН-СС1=СН2+. - ¦ >
— .. .сн2—сн==сс1—сна—сна—снфссі—сна сн2—снфссі—спа- ¦ •
104 Глава І Углеводороды
Такое строение доказывается гем, что при окислении этого
вида синтетического каучука образуется янтарная кислота
НООС—СН2—СН2—СООН. Места разрыва углеродной цепи
показаны пункіиром.
Хлоропреновый каучук благодаря своей негорючести,
термостойкости, светостойкости, а также устойчивости к
воздействию масел находит широкое применение в производстве
резинотехнических изделий.
Техника предъявляет к резиновым изделиям самые
разнообразные требования. В одном случае необходима большая
прочность, в другом—высокая эластичность, в третьем—термическая
устойчивость. Все эти требования невозможно удовлетворить одним
каким-нибудь типом каучука. В связи с этим промышленность
выпускает десятки сортов синтетического каучука, полученных
на основе самых различных химических соединений. Выше
указывались ценные свойства хлоропренових каучуков и бутил
каучука. Каучуки на основе кремнийорганических соединений
отличаются сохранением эластических свойств как при низких, так и
при высоких температурах; каучуки на основе фторорганических
соединений сочетают высокую термостойкость с почти
абсолютной химической устойчивостью; каучуки, полученные сополиме-
ризацией дивинила с акрилонитрилом, хорошо выдерживают
действие бензина и других нефтепродуктов. Наиболее массовым іи-
пом каучука, широко применяемым для изготовления шин,
является каучук, получаемый сополимеризацией дивинила со
стиролом (стр. 486). Эти каучуки отличаются хорошей прочностью и
поэтому изготавливаются в громадных количествах Однако по
эластичности и некоторым другим свойствам они все же уступают
натуральному каучуку, вследствие чего до последнего времени
он являлся незаменимым для целого ряда изделий. Зіи ценные
свойства натурального каучука были связаны со строением
полимерной цепи, которое отличалось строго регулярным
расположением в пространстве отдельных звеньев. Такую структуру долго не
удавалось воспроизвести в синтетических каучуках. Лишь в
50-х годах в СССР и в других странах было найдено, что
проведение полимеризации в присутствии комплексных металлорга-
нических катализаторов приводит к образованию полимеров
регулярной структуры.
Этим методом из бутадиена и из изопрена были получены
каучуки, не только не уступающие натуральному каучуку, но и
превосходящие его по некоторым свойствам.
Открытие метода стереоспецифической полимеризации
послужило новым толчком для развития производства
синтетических каучуков.
40. Строение и синтез каучука 105
Различные виды синтетического каучука обладают
отдельными специфическими свойствами, далеко превосходящими
свойства натурального каучука: масло- и бензостойкостью,
теплостойкостью, газонепроницаемостью, светонепроницаемостью,
химической стойкостью и т. п. Поэтому в настоящее время натуральный
каучук не занимает того монопольного положения, как это
было лет 40—50 тому назад.
ГЛАВА II
ГАЛОИДПРОИЗВОДНЫЕ УГЛЕВОДОРОДОВ
В зависимости от числа атомов водорода, замещенных
галоидом, различают одногалоидные, дву галоидные, трех галоидные и
полигалоидные соединения. В качестве примера можно
привести галоидпроизводные метана:
СН3С1
хлористый
метил
СН2СІ2
хлористый
метилен
СНСіз
хлороформ
СС14
четыреххлорнстый
углеоод
41. Галоидные алкилы. Изомерия и способы получения. Одно-
галоидные производные предельных углеводородов называются
галоидными алкилами; их состав можно выразить общей
формулой СПН2,1+1Х, где X может быть F, С1, Br, J.
В молекуле метана все атомы водорода равноценны, то же
самое справедливо и для атомов водорода в молекуле этана.
Поэтому замещение атома водорода в метане или в этане каким-либо
галоидом, например иодом, дает только по одному галоидпро-
изводному:
Н Н Н
Н—C-J н-С-С—J
A U
йодистый йодистый
метил эгил
Замещением водорода в пропане СН3—СН2—СН3 можно
получить два йодистых (хлористых, бромистых, фтористых)
пропила:
СН3—СН2—СН2—J СН3—CHJ—СН3
первичный йодистый вторичный йодистый
пропил пропил
(Ч-иодпропан) йодистый изопропил
B-иодпропан)
В молекуле первого из этих веществ атом иода находится у
первичного атома углерода, в молекуле второго—у вторичного
41. Галоидные алкилы. Изомерия и способы получения 107
і
атома углерода. Первичный йодистый пропил кипит при 102,5 °С,
температура кипения иодистого изопропила равна 89,5 °С.
Еще большее число изомеров можно произвести от бутанов.
Замещением водорода в нормальном бутане СН3—СН2—СН2—СН3
получают нормальный первичный йодистый бутил и нормальный
вторичный »одистый бутил:
СН3—СН^—СН-2—CH.^J СН3—СН2—CHJ—СН3
норма7ьныЛ первичный нормальный вторичный
йодистый бутил йодистый бутил
A-иодбутам) B-иодбутан)
При замещении атомов водорода иодом в молекуле изобутана
образуются два других изомера—первичный йодистый изобу-
тил и третичный йодистый бутил:
СН3—CH-CHaJ CH3-CJ-CH3
СН3 GH3
первичный третичный
йодистый изобутил йодистый бутил
(і-нод-2-метилпропан) B-иод-2-метилпропан)
В молекуле последнего соединения атом иода находится у
третичного атома углерода.
Таким образом, для вещества состава C4H9J возможно (к в
действительности известно) четыре изомера. Конечно, то же
самое число изомеров получится при замещении атома водорода
в молекуле любым другим галоидом.
Галоидные алкилы можно получить несколькими способами:
1. Действием хлора или брома на предельные углеводороды:
СН4 + С12 » СН3С1 + HCI
Как было ранее указано, реакции хлорирования и бромиро-
вания протекают под действием света.
2. Присоединением галоидоводородов к олефинам.
Присоединение происходит согласно правилу Марковникова:
СН3—СН=СНа— HJ »СН3—CHJ—СН3
3. Замещением в спиртах гидроксила галоидом. Это
замещение можно произвести действием концентрированных галоидо-
водородных кислот на спирты
С,Н5ОН + HBr r=zl С,НдВг + НаО
108 Глава П. Галоидпроизводные углеводородов
или же взаимодействием спиртов с галоидными соединениями
фосфора:
ЗСН3ОН + PJ3 » 3CH3J + Р(ОНK
Реакция между спиртом и галоидоводородом обратима:
СН3ОН + HCI ,—* CH3Cl
Для того чтобы сдвинуть равновесие вправо, берут безволный
спирт. В присутствии крепкой серной кислоты, связывающей
выделяющуюся воду, также увеличивается выход галоидного ал-
кила.
42. Свойства галоидных алкилов. Низшие галоидные алкилы—
газы, следующие—жидкости, высшие—твердые тела. В іабл. З
указаны температура кипения и относительная плотность
некоторых галоидных алкилов; эти данные относятся к нормальным
первичным соединениям.
Рассматривая данные, приведенные в горизонтальных
строках табл. 3, легко заметить, что иодзамещенные соединения имеют
большую относительную плотность и кипят при более высокой
температуре, чем соответствующие бромзамещенные, бромзаме-
щенные имеют большую относительную плотность и кипяі при
более высокой температуре, чем хлорзамещенные соединения.
Таким образом, относительная плотность галоидзамещенных
повышается с увеличением атомного веса галоида, входящего
в состав молекулы.
При сравнении данных вертикальных столбцов таблицы
видно, что относительная плотность галоидпроизводных понижается,
а температуры кипения повышаются с увеличением числа атомов
углерода, входящих в состав молекулы.
Галоидные ал килы почти нерастворимы в воде. Низшие
галоидные алкилы обладают характерным запахом и при
внесении их в пламя на медной проволоке окрашивают его по краям
в зеленый цвет (проба Бейльштейна на галоиды).
Галоидные алкилы принадлежат к числ\ органических ве
ществ с большой реакционной способностью—спи легко
обменивают атом галоида на всевозможные группы.
1. При действии гидроокиси серебра или даже воды
(особенно в присутствии щелочных веществ) галоидные алкилы дают
спирты:
C2H6J + AgOH » QH5—ОН + AgJ A)
CjH,—1-1 + Hi—О—H -^zzl C2H6—OH -+- HJ B)
Таблица 3
Температура кипения и относительная плотность первичных галоидных алкилов
(нормального строения)
Формула
и название
СН3С1
хлористый метил
СгНцСІ
хлорчстый этил
С3Н7С1
хлористый пропил
CiHsCi
хлористый бутил
С5Н,,С)
хлористый амил
Темпе
ратура
кипения
°С
-23,7
+ 12.4
46,6
78,5
108.4
Относи
тельная
плотность
0,952
(при 0°)
0,918
(При 8°)
0.892
0,887
0,878
Формула
к название
СН3Вг
бромистый метил
С,Н5Вг
бромистый зтил
С3Н7Вг
бромне і ы її пропил
С,Н0Вг
бромистый бутил
L5H,,Br
бромистый амил
Температура
кипения
JC
+3,5
38,4
71,0
101,6
127,9
Относительная
плотность
«г
1,7Х-
(iiDa о°;
1,461
1,351
1,276
1,218
Формула
и нааваиие
CH3J
йодистый метил
C2H5J
йодистый этил
C3H,J
иодистыГ) пропил
CjHgJ
йодистый бутил
QH„J
йодистый амил
Температура
кипения
°С
42,5
72,3
102,5
130,4
154,2
Относительная
плотность
, 211
4
2,279
1,936
1,749
1,615
1,510
110 Глава II. Галоидпроизводные углеводородов
Реакция B) обратима. Для того чтобы реакция шла в сторону
образования спирта, необходимо брать большое количество воды
или удалять образующуюся галоидоводородную кислоту
отгонкой или добавлением едкого натра, соды и других щелочей:
СН3—CH2J + NaOH » СН3СН2ОН + NaJ
2. С цианистым калием они образуют цианистые алкилы
(нитрилы):
сн3—м + kl—cn —»сн3—cn +- kj
цианистый
метил
3. При действии на галоидные алкилы металлического натрия
получаются парафины (реакция Вюрца):
СН3—|j + гиг+ji—СНз » СН3—СНЭ
Подобных реакций известно большое число. Поэтому
галоидные алкилы имеют исключительное значение для синтеза других
веществ.
4. При действии цинка на галоидные алкилы образуются ме-
таллорганические соединения:
2CaH5J + 2Zn » (C3H5JZn + ZnJ2
При действии магния на галоидные алкилы в присутствии
безводного диэтилового эфира получаются растворы, содержащие
смешанные магнийорганические соединения (реактив Гриньяра)
CH3J + Mg > CH3MgJ
Далее (стр. 121) мы более подробно познакомимся с металл-
органическими соединениями.
5. Едкие щелочи (спиртовым раствор ) отщепляют от
галоидных алкилов галоидоводород; при этом образуются олефины:
СНЭ—СН2—СН2Вг + КОН » СН3—СН=СН2 + КВг + Н.2О
Присоединяя затем к олефину галоидоводород, можно получить
галоидный алкил другого строения.
Из галоидных алкилов легче всего вступают в реакцию
йодистые алкилы, труднее всех—хлористые алкнлы.
43. Дигалоидные производные предельных углеводородов 111
Хотя атом галоида в галоидных алкилах и обладает большой
подвижностью, галоидные алкилы не являются ионными
соединениями и в растворе или почти не ионизированы, или
ионизированы в крайне незначительной степени. Поэтому при
обыкновенной температуре они весьма медленно реагируют с азотнокислым
серебром.
Хлористый этил С2Н5С1 под небольшим давлением сгущается
в жидкость, кипящую при +12,4 °С. Если налить немного
такой жидкости на кожу, произойдет быстрое испарение хлористого
этила и резкое охлаждение соответствующего участка тела. На
этом основано применение хлористого этила в медицине как
обезболивающего средства при хирургических операциях
(местная анестезия).
В технике хлористый этил расходуется в больших количествах
для получения антидетонатора—тетраэтилсвинца (см. стр. 123)
и в ряде синтезов, где требуется введение этильной группы.
Хранят хлористый этил в стеклянных запаянных или
герметически закрытых ампулах.
43. Дигалоидные производные предельных углеводородов.
Простейшими дигалоидными производными предельных
углеводородов являются производные метана:
СН2С12 CH2Br2 CH2J„
хлористый бромистыП йодистый
метилен метилен метилен
Их можно рассматривать как галоидные соединения
двухвалентного радикала СН2<, называемого метиленом.
Этан С2Н6 может давать два изомерных дигалоидных
производных—хлористый этилен СН2С1—СН2С1 A,2-дихлорэтан) и
хлористый этилиден СН3—СНС12 A,1-дихлорэтан). В молекуле
хлористого этилена атомы хлора находятся у различных атомов
углерода, в молекуле хлористого этилидеиа—у одного атома
углерода.
Двухвалентный радикал —СН2—СН2—называется этиленом,
двухвалентный радикал СН3—СНу называется этилиденом.
Пропану СН3—СН2—СН3 соответствует четыре изомерных
дигалоидных производных:
СН3—СН2—СНС12 СН3—СС12—СН3
1,1-ди хлор пропан 2,2-днхлорпропан
СН3—СНСІ—CHjCI СН2С!-СН2-СН2С!
1,2-дихлорпропан 1,3-дихлорпропан
(хлористый триметилен)
112 Глава II. Галоидпроизводные углеводородов
Дигалоидные производные предельных углеводородов
получают:
1. Присоединением галоида к олефинам:
СН2=СНа + С1а > CHgCl—СНаС1
Таким путем образуются вещества с атомами галоида у
соседних атомов углерода.
2. Действием пятихлористого или пятибромисюго фосфора
на альдегиды и кетоны:
СНз-af + РСІі > СНз-CHClj, + РОС13
уксусный
альдегид
СНз-СО-СНз -
ацегон
f РВг6
»СН,
хлорокиеь
фсс^дра
,_СВг2-СН3 + РОВГз
брюмоі<ись
фосфсша
При этом получаются вещества с двумя атомами галоида при
одном атоме углерода.
Получение дигалоидных производных с атомами галоида не
у соседних углеродных атомов можег быть осуществлено
взаимодействием непредельных галоидных производных с НВг в
присутствии перекисей (см. стр. 180):
СНа=СН—СНаВг + НВг » СН.2Вг—СН2—СН2Вг
В этом случае бромистый водород присоединяется не по
правилу Марковникова.
Дигалоидные производные предельных
углеводородов—тяжелые масла или твердые вещества, нерастворимые в воде.
Йодистый метилен CH2J2—очень тяжелая жидкость;
плотность 3,333 г/см3 при 15 °С; темп. кип. 180 °С. Применяется при
исследовании горных пород для разделения минералов по их
плотности; впервые получен А. М. Бутлеровым в 1858 г.
Дихлорэтан, или хлористый этилен,
СНаО—СНЙО—жидкость; плотность 1,25г/см3 при 20 °С; темп. кип. М SC, темп, плавл.
—35,3 °С. Получается присоединением хлора к этилену, является
хорошим растворителем, но вследствие токсичности применение
его для этих целей ограничено. Используеіся как
промежуточный продукт в одном из методов получения хлористого винила
(см. стр. 115) и в других синтезах.
Хлористый зтилиден СНЯ—CHCt2—жидкость: плотность
1,189 г/см3 при 0 "С, темп. кип'. 58 °С.
44. Полигалоидные производные предельных углеводородов 113
44 Полигалоидные производные предельных углеводородов.
Из галоидпроизводных с несколькими атомами галоида в
молекуле наибольшее значение имеют галоидпроизводные метана—
хлороформ СНС1Я, йодоформ CH.J а и четыреххлористый углерод
СС14.
Хлороформ CHOI., получается из спирта или ацетона действием
щелочного раствора хлора или белильной извести (см стр 209).
Он прі-дічавляет собой жидкость с характерным
приторно-сладким запахом, іемп. кип. 61,2 "С; плотность 1,498 г/см3 при 15 °С.
При действии света хлороформ окисляется кислородом воздуха,
давая Сі,,, НС1, СО2 и фосген СОС12. Вдыхание паров хлороформа
вызывает потерю сознания (наркоз). На этом основано его
применение в медицине при хирургических операциях.
Йодоформ СШ3 получается, подобно хлороформу, из спирта
или ацетона—действием иода и щелочи. Йодоформ—кристаллы
ли мои но-желтого цвета с характерным неприятным запахом;
очень легко возгоняется; плавится при 119 °С; применяется как
¦антисептическое средство при лечении ран.
Четыреххлористый углерод СС14 получают действием хлора
на сероуглерод:
CSj + ЗС1„ » CCi4 4- SjClj
Четыреххлористый углерод—жидкость; темп. кип. 76,5 °С;
ллогность 1,58 г/см3 при 21 °С; его пары совершенно негорючи.
Четыреххлористый углерод применяемся как растворитель смол,
жиров, каучука и других веществ, а также как средство для
тушения пожаров (в специальных огнетушителях).
45. Фторпроизводные предельных углеводородов.
Непосредственное действие свободного фтора на углеводороды проходит
очень бурно, с полным обугливанием углеводородов.
Фторирование углеводородов производят при нагревании, пропуская пары
предельного углеводорода в смеси с азотом через слой трехфтори-
стого кобальта:
К-Н + 2CoFg » R-F + 2CoF2 + HF
200—350 °С
Постепенно заметая в предельных углеводородах все атомы
водорода фтором, можно получить перфторуглеводороды. Фюр
как заместитель водорода занимает совершенно особое место.
Присутствие в молекуле, по меньшей мере, двух атомов фтора
при одном атоме углерода стабилизирует вещество и делает
соединение химически устойчивым. Все перфторуглеводороды —
прочные соединения, при нагревании даже до 400—500 °С они не
расщепляются. В пасюящее время получены: перфторметан
8—788
11 і 1лаво II. Галоидпроизеодные углеводородов
CF4—газ, сгущающийся в тяжелую жидкость с темп. кип.
—128 °С; перфторэтан C2F„—газ, темп. кип. —78,2 °С. Известно
много перфторуглеводородов, вплоть до перфторгексадекана
C„.F34—бесцветные пластинки с темп, плавл. 115 °С.
Из фторпроизводных наибольшее применение нашли легко-
кипящие соединения, в молекуле которых содержатся
одновременно хлор и фтор, например дифторхлорметан. CHF2C1, дифтор-
дихлорметан CF2CI2. Такие вещества в технике получили название
фреонов; они применяются в холодильных машинах в качестве
хладоагентов.
Дифтордихлорметан CF2C12 при обычных
условиях—бесцветный газ без запаха, сгущается в жидкость и кипит при —30 °С.
Получают его из четыреххлористого углерода и трехфтористой
сурьмы в присутствии брома или SbCl6 как инициатора реакции:
ЗСС14 + 2SbF3 * 3CF2C!2 -(- 2SbCl3
Дифтордихлорметан—прочное соединение. Пары его
нетоксичны. В этом соединении хлор и фтор нереакционноспособны.
46. Галоидпроизводные непредельных углеводородов.
Простейшими галоидпроизводными олефинов являются
галоидпроизводные этилена:
СН2=СНС1 CHa=CHBr
хлористый винил бромистый винил
(хлорэген) (бромэтен)
Их можно рассматривать как галоидные соединения
одновалентного радикала винила СН2=СН—
Для одногалоидного производного пропилена возможны гри
изомера:
СНС1=СН—СН3 СН2=СС1—СН3 СН2=СН—СНаС1
а-хлорпропилен ?-хлорпропилен хлористьш аллил
A-хлорпропен-1) B-хлорпропен-1) (З-хлорпропен-1)
По реакционной способности атомов галоида одногалоидные
производные этиленовых углеводородов можно разделить на две
группы:
1) соединения, в которых атом галоида находится при
атоме углерода, связанном с другим атомом углерода двойной
связью;
2) соединения, в которых атом галоида находится при каком-
либо другом углеродном атоме.
К соединениям первой группы относятся хлористый винил, а
также а- и ?-хлорпропилены; примером соединений второй
группы может служить хлористый аллил.
47. Высокомолекулярные соединения. Пластические массы 115
Соединения первого типа получаются:
1. Отщеплением одной молекулы галоидоводорода от дига-
лоидных производных, в молекулах которых атомы галоида
находятся у одного и того же или у двух соседних атомов углерода:
CH.^Cl—СНаС1 + КОН , СН3=СНС1 + КС1 + НаО
2. Присоединением одной молекулы галоидоводорода к
ацетиленовым углеводородам :
НС=СН + НС1 » СН2=СНС1
По своему химическому характеру соединения этого типа
резко отличаются от галоидных алкилов В галоидных алкилах атом
галоида легко может быть замещен гидроксилом и другими
группами. Соединения же, в которых галоид находится при углероде
с двойной связью, почти совершенно не способны к такого рода
реакциям: атом галоида в них мало подвижен. Наоборот,
галоидные соединения типа хлористого аллила вступают в реакции
обмена легче, чем галоидные алкилы.
Хлористый винил СН2 = СНС1—газ; темп. кип. —13 °С;
довольно легко полимеризуется, давая высокомолекулярные
соединения; применяется для изготовления пластических масс.
Хлористый, бромистый и йодистый аллилы СН2=СН—СН2Х
(Х=С1, Вг, J)—жидкости (темп. кип. соответственно 45; 71 и
103 °С, с запахом, напоминающим запах горчицы. Хлористый ал-
лил и бромистый аллил получаются действием пятигалоидных
соединений фосфора или галоидоводородов на аллиловый спирт
СН2=СН—СН2ОН.
В технике хлористый аллил получают непосредственным
хлорированием пропилена при высоких температурах (около
500 °С). В этих условиях происходит не присоединение галоида
к олефину по двойной связи, а замещение водорода у
углеродного атома, не имеющего двойной связи:
сн2=сн—сн3 + сі3 —»сна=ск—сн3—сі + неї
пропилен хлористый й
47. Высокомолекулярные соединения. Пластические массы.
Непредельные углеводороды, их галоидные и другие производные
способны полимеризоваться (см. стр. 82, 116). Продукты
полимеризации—высокомолекулярные соединения с молекулярным весом
от нескольких сотен до миллиона и более, нашли широкое
применение в технике для получения различных синтетических
материалов—пластических масс, синтетических каучуков,
синтетических волокон, клеев, ионообменных смол и др.
116 Глава II. Галоидпроизводные углеводородов
Пластическими массами называют такие высокомолекулярные
соединения, которые под воздействием высокой температуры и
давления способны переходить в пластическое состояние и
принимать любую заданную форму. В отличие от синтетических каучуков
пластические массы при обычной температуре не обладают совсем
или обладают ограниченной эластичностью. Несмотря на то, что
производство пластмасс началось всего 40—50 лет тому назад,
в настоящее время они имеют громадное значение в технике и
в быту. Пластические массы успешно заменяют металлы и дерево,
стекло, драгоценные камни, хрусталь и фарфор. Они стали
теперь незаменимыми материалами в машиностроении, авто-
сіроении, авиастроении, радиотехнике, судостроении,
электротехнике, химической промышленности и во многих других
отраслях народного хозяйства.
Оказалось, что в пластических массах часто сочетается несколько ценных
свойств. Так, примером прочного материала является сталь, легкими и
твердыми веществами являются дерево и алюминий; пример прозрачного
материала—стекло. Однако сталь химически неустойчива, она ржавеет трудно
поддается механический обработке; дерево гниет, непрозрачно, плохой изолятор
электричества; стекло—хрупко, трудно обрабатывается в холодном виде.
Пластмассы же не имеют этих недостатков. Большинству пластмасс присущи
легкость хорошие электроизоляционные свойства, высокая прочность; они
легко поддаются механической обработке. Многие пластмассы прозрачны,
не гниют, стойки к действию сильных кислот и щелочей и др.
Основой в пластмассе является высокомолекулярное вещество.
Высокомолекулярные вещества могут быть природными (пеки,
асфальты) и синтетическими, В настоящее время наиболее
важное значение имеют синтетические полимеры, получаемые
полимеризацией или поликонденсацией.
Полимеризация и поликонденсация—процессы разных типов.
При пилимеризации происходит соединение одинаковых молекул без
выделения простых молекул, и образующийся полимер имеет молекулярный
вес. равный сумме молекулярных весов реагирующих молекул. Если в
образовании полимера участвует два или несколько различных вешеств, то
такая полимеризация называется совместной полимеризацией или сополимери-
зациеіі
При поликонденсации соединение простых молекул и образование
полимера происходит за счет взаимодействия функциональных групп и
сопровождается выделением таких простых веществ, как НгО, НС), NH3 и др., и,
следовательно, молекулярный вес полимера уже не будет равен сумме
молекулярных весов исходных молекул.
Синтетические полимеры получают обычно в присутствии
веществ, которые ускоряют процесс полимеризации.
Так, например, полимер хлористого винила (—СНг—СНСІ—)„—поливи-
нилхлорид получают полимеризацией хлористого винила СНг=СНС1 в водных
41 Высокомолекулярные соединения. Пластические массы 117
эмульсиях в присутствии перекисных соединений. (О полимеризации этилена
и изобутилена см. на стр. 88 и 89)
Высокомолекулярные соединения представляют собой вязкие
жидкости или твердые вещества, часто способные размягчаться
при нагревании. Обычно пластмассы классифицируют и различают
по химическому характеру полимера, например поливинилхло-
ридные, феноло-формальдегидные, меламино-формальдегидные
и т. п.
Некоторые пластические массы, например полиэтилен,
полиамиды, полностью состоят из полимера, в других же содержание
высокомолекулярных соединении не превышает 20—60%, а
остальное составляют так называемые іічполнители (древесная мука,
стеклянное волокно, асбест и др.). Назначение
наполнителей—изменение свойств пластмасс в желаемом направлении—придание
им механической прочности, твердости огнестойкости и проч.
Введение наполнителей широко используется при изготовление
пластических масс из феноло-формальдегидных, мочевино-фор-
мальдегидных, эпоксидных, и некоторых других полимеров.
Для превращения некоторых полимеров, например поливинил-
хлоридных, в пластические массы необходимо применение так
называемых пластификаторов. Пластификаторы—это вещества,
придающие полимеру пластичность, т. е. способствующие
превращению твердых и хрупких смол в тестообразное состояние,
удобное для придания им желаемой формы. Увеличивая
количество пластификаторов, можно получать гибкие, довольно
эластичные материалы, напоминающие резину.
Поливинилхлорид представляет собой мелкий белый порошок.
Смешивая его с пластификаторами и подвергая прессованию или вальцеванию,
получают пластичный материал, который может быть использован для
изготовления различных изделий (обуви, сумок, плащей, клеенки и др.).
Во многих случаях в композицию вводят так называемые
стабилизаторы—вещества, предохраняющие высокомолекулярное
соединение от разложения в процессе переработки и от действия
тепла и света в процессе эксплуатации, а также красители,
придающие изделиям желаемую окраску.
Синтетические материалы на основе виниловых полимеров
Хлористый винил СН2=СНС1 при действии света или при
нагревании образует полимер в виде белого аморфного порошка.
Благодаря дешевизне, доступности, негорючести пластмассы
из поливинилхлорида широко используются в технике и
быту.
118 Глава П. Галоидпроизводные углеводородов
Пластифицированный поливинилхлорид в больших
количествах используется для изоляции кабелей и проводов связи, причем
он одновременно заменяет каучук, свинец и хлопчатобумажную
пряжу. Другие области применения—производство
искусственной кожи, линолеума, плащей, накидок, сумок и других
предметов домашнего обихода. Путем переработки поливинилхлорида
без применения пластификаторов получают винипласт. Это
твердая пластическая масса, которая легко сваривается и поддается
механической обработке. Винипласт применяется для
изготовления вентиляционных труб, насосов и различных частей
аппаратуры. Хлорированием поливинилхлорида получают пер-
хлорвиниловую смолу. В виде лаков и клеев ее применяют для
поверхностных покрытий; из нее готовят волокно (хлорин).
Винишцетат СН2=СНОСОСН3. Получается взаимодействием
ацетилена и уксусной кислоты в газовой фазе:
+ НС=СН * СНзf
СН„
з-Cf
X)—СН=
Полимеризацией винилацетата получают поливинилацетат,
находящий широкое применение для изготовления водяных латекс-
ных красок, не требующих при употреблении растворителей,
Другие области применения поливинилацетата—отделочные
препараты, придающие тканям несминаемость, и клеящие составы для
¦бумаги, кожи, дерева, металлов и проч.
На основе сополимеров хлористого винила и винилацетата
получают высококачественные лакообразующие смолы, а также
смолы для изготовления долгоиграющих граммпластинок.
Сополимеры хлористого винила и винилиденхлорида СН.,=
=СС12 находят техническое применение для изготовления
лаковых смол, труб, арматуры, а также синтетического волокна,
малопригодного для изготовления одежды, но используемого для
получения обивочных, декоративных и некоторых других
материалов.
Фторсодержащие полимеры
Введение в молекулу углеводорода не менее 2-х атомов фтора
резко повышает общую устойчивость соединения. Вследствие
этого фторсодержащие полимеры обладают непревзойденной
стойкостью к химическим и температурным воздействиям.
Непосредственное фторирование углеводородов не удается
осуществить вследствие разрушения молекулы углеводорода.
47. Высокомолекулярные соединения Пластические массы 119
Для получения фтор углеводородов и полностью фторированных
веществ—фторуглеродов исходят из соответствующих хлорпроиз-
водных.
Тетрафторэтилен CF2=CF2. Бесцветный газ с температурой
кипения —76 °С. Для его получения исходят из хлороформа:
СНС13 + 2HF > CHC1F2 + 2НС1
2CHCIF2 * CF2=CF2 + 2НС1
Тетрафторэтилен легко полимеризуется при 60 °С и
умеренном давлении в присутствии перекисных соединений.
Полученный полимер имеет правильную линейную структуру.
F F F F
-LLLL .
FF FF
Пластические массы на основе политетрафторэтилена получили
название фторопласты-4 (в США—тефлон). Они отличаются высокой
термической устойчивостью от —183 °С до +300 °С и высокой
химической стойкостью. На фторопласт-4 не действуют горячая
дымящая азотная кислота, концентрированная серная кислота,
расплавленный едкий натр. Лишь расплавленный металлический
натрий постепенно разрушает его.
Фторопласт-4 является незаменимым материалом для
изготовления ответственных деталей приборов и аппаратуры,
работающих в жестких условиях, в том числе электроизоляционных
материалов, арматуры, применяемой в химическом машиностроении,
специальных пленок и др.
Недостатком фторопласта-4 является сложность его
переработки в изделия, для чего требуются специальные приемы,
поскольку материал нерастворим ни в одном из растворителей, не имеет
определенной температуры плавления и даже в размягченном
состоянии обладает низкой текучестью.
Пластмассы на основе политрифторхлорэтилена
Трифторхлорэтилен CF2=CFC1—бесцветный газ,
конденсируется в жидкость при —27 °С, сравнительно легко
полимеризуется при комнатной температуре в политрифторхлорэтилен:
F F F F
F СІ F СІ
120 Глава II. Галоидпроизводные углеводородов
Пол итрифтор хлор этилен, или фторопласт-3, несколько
уступает фторопласту-4 по устойчивости к термическим и химическим
воздействиям, но обладает способностью легче перерабатываться
в изделия.
Сополимер политрифторхлорэтилена и винилиденфторида
CH2=CF2, содержащий более 50% фтора по весу, обладает
хорошими эластическими свойствами и относится к группе так
называемых фторкаучуков. Применяется в тех случаях, когда изделия
из каучука должны выдерживать высокие температуры или
действие химически агрессивных веществ.
ГЛАВА III
ЭЛЕМЕНТОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
48 Элементорганические соединения и их значение.
Рассматривая состав органических веществ, легко заметить, что, кроме
обязательно присутствующего углерода, они наиболее часто
содержат следующие элементы: водород, кислород, азот, серу,
галоиды. Эти элементы часто называют типичными органогенами.
Однако среди природных и особенно среди искусственно
полученных продуктов насчитывается очень много веществ, в состав
которых входят также и другие элементы (неорганогены):
металлы Na, К, AI, Zn, Mg, Hg, Pb, Sn и неметаллы P, Si, B, As.
Конечно, такое разделение элементор на органогены и неорганогены очень
относительно и условно, но ведь и привычное разделение элементов на металлы
и неметаллы является столь же условным (например, As, Те, J).
Исторически первыми (Франкланд, 1849) среди элементорга-
нических соединений были открыты соединения, содержащие
в своем составе металл. Благодаря своей необычайной
реакционной способности эти соединения приобрели весьма важное
значение и были выделены в особую группу так называемых металл-
органических соединений. С дальнейшим развитием органической
химии были получены соединения, содержащие неметаллы, и
теперь принято говорить о так называемых элементорганических
соединениях.
Элементорганическими соединениями называются только те
из органических соединений, содержащих в своем составе какой-
либо металл (или неметалл), в которых атом этого элемента
непосредственно связан с атомом углерода.
Примерами могут служить
CH3Na (C3H,JZn (C2H6JHg (C2H6LPb
метилнатрий днпропнлциии диэтилртуть тетгаэтилсвинец
Таким образом, к элементорганическим соединениям нельзя
причислять соли карбоновых кислот, алкоголяты, эфиры фосфор-
122 Глава 111. Элементорганические соединения
ной кислоты и пр., где атомы металла, фосфора (или других
неорганогенных элементов) связаны с углеводородным радикалом не
непосредственно, а, например, через атом кислорода:
О
СН3—С—ONa C2H5ONa
Как видно из приведенных примеров элементорганических
соединений, атомы различных элементов соединяются с разным
числом радикалов.
Двухвалентные и многовалентные металлы могут давать два
вида металлорганических соединений—чистые и смешанные. В
соединениях первого типа атом металла связан только с
углеводородными радикалами, в соединениях второго типа, кроме
органического, имеется также и неорганический радикал. Примеры
чистых металлорганических соединений приведены выше.
Примерами смешанных металлорганических соединений могут
служить:
СН3—Zn—J QH,—Mg—J (CH3JSiCI2
цкнкиодіїегил магнийиодпропкл дихлорднмегнлснлан
Чистые металлорганические соединения в большинстве
случаев жидкости, перегоняющиеся без разложения. Натрий- и
магнийорганические соединения при нагревании разлагаются.
Благодаря своей летучести металлорганические соединения были
применены для определения атомного веса и валентности
различных металлов (алюминий, олово и свинец).
Связь атома металла с углеродом малопрочна, в результате
чего металл легко уступает свое место другим атомам или
радикалам.
Большая реакционная способность металлорганических
соединений позволяет попользовать их для получения ряда веществ.
Особенно большое значение для органического синтеза имеют
соединения цинка и магния.
Металлорганические соединения сыграли большую роль в
развитии органической химии, особенно ее синтетических методов.
Трудами А. М. Бутлерова, А. М. Зайцева, Е. Е. Вагнера,
Ф. Ф. Бейльштейна, С. Н. Реформатского и их ученмкои во р.то-
рой половине XIX в. были разработаны методы получения ксто-
нов, третичных и вторичных спиртов и некоторых
металлорганических соединений с помощью цинкорганических
соединений.
49 Цинкорганические соединения 123
Смешанные магнийорганические соединения, впервые
изученные Гриньяром*, имеют для органических синтезов совершенно
исключительное значение. С их помощью могут быть получены
вещества, принадлежащие к самым разнообразным классам
органических соединений (углеводороды, спирты, эфиры, кетоны,
альдегиды, карбоновые кислоты, сложные эфиры, нитрилы),
а также и другие металлорганические соединения.
Совершенно особое место среди металлорганических
соединений принадлежит тетраэтилсвинцу (С2Н5LРЬ. Применение этого-
вещества в качестве весьма эффективного антидетонатора**
привело к созданию в ряде стран специальных производств большой
мощности.
Тетраэтилсвинец (С2Н5LРЬ—бесцветная тяжелая жидкость
(плотность около 1,65 г/см3) с неприятным запахом,
Получают его действием хлористого этила на сплав свинца с
натрием:
4С2Н6С1 + 4NaPb > РЬ(СаН6L + ЗРЬ + 4NaCI
Пары тетраэтилсвинца очень ядовиты.
49. Цинкорганические соединения. Цинкорганические
соединения получаются нагреванием йодистых алкилов с цинком.
Реакция протекает в две фазы. Сначала образуется смешанное
металлорганическое соединение, например кристаллический цинк-
иодэтил
C2H6J + Zn > CjH6—Zn—J
* Виктор Гриньяр (Francois August Victor Grignard)—знаменитый
французский химик-органик A871 —1935). Известны его работы по синтезу и
применению так называемых смешанных магнийорганических соединений. Он
показал, что металлический магний реагирует с йодистыми алкилами,
растворенными в тщательно обезвоженном эфире, давая раствор эфиратов смешанных
магнийорганических соединений.
AlkJ + Mg » Alk—Mg—J
Гриньяр взял за основу известную реакцию Зайцева—взаимодействие
йодистых алкияов с цинком—и заменил цинк магнием, использовав в качестве
растворителя безводный эфир. Этим область применения металлорганических
соединений значительно расширилась, так как магний является более
активным металлом и может реагировать не только с йодистыми (как цинк), но и
с бромистыми и хлористыми алкилами и арилами (см. стр. 124). В 1912 г.
Гриньяру за открытие магнийорганических соединений была присуждена
Нобелевская премия.
** Антидетонаторы—вещества, добавляемые к моторному топливу для
снижения способности последнего к детонации в цилиндрах двигателей
внутреннего сгорания (см. стр. 83).
124 Глава Iff. Элемент органические соединения
который при высокой температуре превращается в жидкий ди-
этилцинк:
СаН6—jZn—j":
'\Zn + ZnJa
C2H6-Zn-j J
С2НЙ
Цпнкорганические соединения на воздухе самовоспламеняются
и горят синим пламенем; вода разлагает их на углеводороды и
гидроокись цинка:
/іСНз + Н:—О—Н
2п< ! ; > 2СН4 + Zn(OH),
х;сн3 + н;-о—н
Ввиду легкой самовоспламеняемости цинкорганических
соединений, всю работу с ними ведут в атмосфере сухого
углекислого газа, водорода или азота.
50. Магнийорганические соединения. Магнийорганические
соединения получают прибавлением галоидного алкила к
стружкам металлического магния, находящимся под слоем тщательно
обезвоженного эфира*. Реакция проходит настолько энергично,
что эфир закипает.
В результате реакции образуется смешанное металлорганиче-
ское соединение R—Mg—X (R—радикал, а X—галоид), например:
C3H,J + Mg » C3H,MgJ
Полученные подобным образом металлорганические соединения
химически связаны с эфиром. Они вступают в те же реакции, что
и индивидуальные., т. е. лишенные эфира, магнийорганические
соединения. Поэтому их обычно не выделяют из раствора, а
непосредственно применяют для дальнейших синтезов.
Индивидуальные магнийорганическиесоединения, как показал
В. В. Челинцев, образуются при прибавлении к раствору
галоидного алкила в бензоле стружек магния и нескольких капель
эфира или диметиланилина, которые в данном случае являются
только катализаторами.
Смешанные магнийорганические соединения не
воспламеняются на воздухе. Вода, спирты и другие вещества, содержащие
гидроксильную группу (а также первичные и вторичные амины),
легко разлагают эти соединения с образованием углеводородов:
2СН3—JMg—J+'н—О|—Н * 2СН4 + Mg(OH)J
СНз-jMg-J + QHg—O]—Н * СН4 + C2H6OMgJ
* Без эфира металлический магний лишь с трудом действует на галоидные
алкилы. Принято считать, что эфир является катализатором этой реакции.
51. Алюминийорганические соединения 12а
На этом основан предложенный в 1920 г. Л. А. Чугаевым и
разработанный Ф. В. Церевитиновым способ количественного
определения гидроксильных групп в органических соединениях:
навеску подлежащего исследованию вещества обрабатывают маг-
нийиодметнлом и измеряют объем получающегося при этом
метана.
51. Алюминийорганические соединения. Обычно
алюминийорганические соединения получают взаимодействием йодистых алки-
лов с алюминием:
2АІ + 3CH3J > CH3AUa + (CH3JAU
2(CH3JAIJ * AI(CH3K + CH3AIJ2
В технике триалкилалюминий получают из олефина, водорода и
мелко раздробленного алюминия при І00—120 °С под даьлением
AI + ЗСН2=СН2 + 1УаН2 „ AlfQH^g
Триметилалюминий А1(СНаK—жидкость с темп. кип. 130 °С,
воспламеняется на воздухе. Триэтилалюминий А1(С,НГ|)а кипит при
194 °С. Для алюминия известны первичные, вторичные и
третичные (полные) производные.
Алюминийорганические соединения применяются в
промышленности, так как с их помощью оказалось возможным
осуществить ряд важных технических синтезов. Так, например, в
присутствии триэтилалюминия получают полиэтилен неразветвлен-
ного строения (см. стр. 88).
52. Кремнийорганические соединения. Кремний, как и
углерод, находится в IV группе периодической системы и по типу
простейших соединений является аналогом последнего. Однако по
химическим свойствам соединения углерода и кремния очень
сильно отличаются друг от друга. Это, в первую очередь,
относится к способности образовывать цепи: в то время как для
углерода характерны соединения, содержащие разнообразные цепи
С—С атомов, кремний не способен давать цепи, состоящие более
чем из шести атомов Si. Кремневодороды—силаны—очень
неустойчивые соединения самовозгорающиеся на воздухе. Первые
члены ряда силанов—газы или легкокипящие жидкости: моноси-
лан SiH4 (темп. кип. —112 °С), дисилан Si2He (темп. кип.
— 44°С), трисилан Si3H8 (темп. кип.+53 °С).
Так же мало устойчивы и очень реакционноспособны хлор-
производные силанов: хлорсилан SiH3Cl, трихлорсилан SiHCla
(силанохлороформ), четыреххлористый кремний SiCl4. Все эти
вещества энергично гидролизуются водой; с цинк- и магнийорга-
126 Глава III. Элементорганические соединения
ническими соединениями они дают кремнийорганические
соединения—алкилсиланы:
Cl|siH3 » QH5-SiH3
fHjgJ 2CH^MgJ
CH3—SiCI3 - (CH8).2—SiCl2 : ' (CH3LSi
метил грих.пор- дихлордиме- гетраметил
силан тилсилаи силан
Практически вместо смешанных магнийорганических соединений
применяют галоидный алкил и магний:
C2H6Br+Mg C2HaBr+g
SiCl4 *¦ С2На—SiCl3 - (C2H5JSiCI2
Более устойчивыми являются те алкилсиланы, в которых нет
атомоп водорода при кремнии,—тетраалкилсиланы.
Практическое применение нашли алкнлхлорсиланы, из
которых гидролизом получаются силанолы (или силнколы):
(CjHsJaSiCIj, + 2Н2О » (C2H5JSi(OHJ + 2НС1
диэтилсиландпол
Диалкилсиландиолы весьма неустойчивы: они легко
уплотняются с выделением воды, давая полисилоксаны,—так
называемые «силиконы»:
_, 7> . I : ; I -
y\r>— Si— ЮН + H!0—Si—ЮН + НЮ—Si—OH >
-' I ; : \ ' ' I
C2H5 СгНа C2H5
» • ¦ ¦O-Si(CaH5)a-O-Si(CaHB)a-O-Si(CaHe)a-
Таким образом, для кремния характерна способность к
образованию силоксановых цепей.
Кремнийорганические полимеры могут быть жидкими,
твердыми и хрупкими, а также высокоэластичными—каучукоподоб-
ными. Наиболее существенным отличием кремнийорганических
полимеров является их более высокая термостойкость по
сравнению с органическими полимерами.
Способность противостоять воздействию как очень низких
(—100 °С), так и очень высоких температур (+300 °С и выше)
сделала кремнийорганические полимеры очень важными
материалами в современной технике, в том числе в электротехнической
промышленности для повышения надежности электродвигателей,
работающих в шахтах, на транспорте и др., а также в
радиотехнике, в авиационной промышленности и в других отраслях.
52. Кремнийорганические соединения 127
Эластичный кремнийорганический полимер, так называемый
силиконовый каучук, получается гидролизом диметилдихлорсилана.
В этом случае силоксановая цепь имеет следующую структуру:
СНд CHj CH9
О—Si—О—Si—О—Si
СН3 СН3 СНз
Силиконовый каучук белого цвета и имеет относительно
невысокую механическую прочность, но в тех случаях, когда от
материала требуется сохранение эластических свойств в широком
интервале температур, он незаменим.
Кремнийорганические полимеры имеют и другие важные для
техники свойства. Широкое применение нашли силиконовые масла,
обладающие высокой термической стойкостью и практически
постоянной вязкостью в широком интервале температур.
Силиконовые смазки используют также в лабораторной практике; они
очень устойчивы и не окисляются даже такими сильными
окислителями, как озон.
Молекулы полимера построены симметрично, и это определяет
отсутствие полярности, поэтому такие вещества являются
изоляторами; углеводородные радикалы сообщают им
водоотталкивающие свойства—гидрофобность. Гидрофобность силиконов в
первую очередь определяет ряд областей технического применения
их. Так, например, в настоящее время получают материалы, не
пропускающие воду, но проницаемые для воздуха (картон,
фарфор, кирпич), подвергая их действию паров кремнийорганиче-
ских соединений. Кремнийорганические соединения
взаимодействуют с водой;
: | : |
—О—IH+ Сі!—Si—ICl +Hi—О—;Н + СІІ—Si— Cl
' j !! :; I
геп
і + і
і..н,
І
[ні
с2н5
H-o-lH'+cil-Si-icT+Hi—о-!н'+сі!-Зі-
! '¦ і ! ' ; '¦ ¦
СіН,
128 Глава III Элементорганические соединения
При этом в порах материала образуются водонепроницаемые
силиконовые пленки:
С2Н5 СзН5
О—Si—О—Si
С2Н5 О CjH,
O-Si-O— Si
СаН5 CjHj
Большое значение в создании и развитии химии полимерных
кремнийорганических соединений имеют работы К. А.
Андрианова.
53. Фосфор- и мышьякорганичгские соединения. Фосфор и
мышьяк, как и азот, находятся в V группе периодической системы
элементов. Они образуют соединения с водородом: РН3, AsH3,
подобно NH,. Фосфористый водород, как и аммиак, может,
соединяясь с кислотами, давать соли фосфония, например йодистый
фосфоний PH4J. По сравнению с солями аммония соли
фосфония менее устойчивы, легко гидролизуются в воде.
Мышьяковистый водород с кислотами солей не образует, так как у AsH.t слабо
выражен основной характер.
Подобно тому как существуют органические производные
аммиака (амины, см. гл. XIII), могут существовать органические
произволные фосфористого водорода и мышьяковистого водорода.
Рассмотрим два типа фосфорорганических соединений:
фосфины и 4осФиновые кислоты.
Фосфины могут быть получены взаимодействием трехгалоид-
ных соединений фосфора с металлорганическими соединениями:
2РС13 + 32п(СНаK » 2Р(СН3K + 3ZnCl
a
Различают первичные, вторичные и третичные фосфины
(аналогично первичным, вторичным и третичным аминам):
/СНз
Р-Н
\н
первичный фосфин
/СН3
Р-СН3
\н
вторичный фосфин
/СНз
Р-СНз
\сн3
третичный фосфин
Введение алкильных групп в молекулу фосфористого водорода
повышает его основность, подобно тому как это имеет место у
производных аммиака. Так, например, третичные фосфины способны
53. Фосфор- и мышьякоргантеские соединения 129
присоединять галоидные алкилы, образуя соли четвертичных фос-
фониевых оснований:
Р(СН3)з + CH3J , [P(CH3L]J
Фосфины—газообразные или жидкие вещества с неприятным
запахом, очень токсичны; в воде, не растворяются. На воздухе
легко окисляются, могут даже самовоспламеняться. Окисляясь,
фосфины дают окиси фосфинов:
СН3. СН3.
СН3—Р + О > СН3-Р=О
сн/ сн/
Водные растворы фосфинов не имеют щелочной реакции (в
отличие от аминов), но с сильными кислотами дают соли:
РНа(СНз) + НС1 »[РН3(СН3)]С1
Третичные фосфины применяются для качественного открытия
сероуглерода, так как образуют ярко-красные малорастворимые
продукты присоединения:
(С2НбKР + CS2 » (QH5KP-CSa
Фосфиновые кислоты получаются при окислении азотной
кислотой первичных и вторичных фосфинов:
/Н уОН
СН3-Р/ + 30 * СН3-Р<
\Н Н Х>Н
первичный метилфосфииовая
фосфин кислота
СН3ч СН^
\p__H + 20 » >Р-ОН
сн/ сн/д
вторичный диметилфосфиновая
фосфин кислота
Алкилфосфиновые кислоты, как это видно из структурных
формул, можно рассматривать как производные ортофосфорной
/ОН
кислоты НО—Р\ , у которой одна или две гидроксильные груп-
II Хон
О
9-788
130 Глава III. Элементорганические соединения
пы замещены на алкилы. Фосфиновые кислоты—бесцветные
кристаллические вещества, легко растворимые в воде.
Из производных фосфиновых кислот следует указать на этиловый эфир
N-диметиламиноцианфосфиновой кислоты, называемый табуном:
СНЧЧ /CN
сн
P
її
Табун—жидкость, мало растворимая в воде при обычной температуре,
обладает слабым фруктовым запахом, сильно ядовита. Пары табуна обладают
токсическим действием на дыхательные пути, проникают через кожу и слизистые
оболочки, вызывают головную боль, сужение зрачков.
При больших концентрациях наступает смерть.
Многие фосфорсодержащие органические соединения
применяются как инсектициды—средства борьбы с насекомыми. Для
уничтожения тлей, растительноядных клещей и других сосущих
вредителей растений широко применяются инсектициды
контактного действия—т иофос и карбофос:
С2Н5О СН3ОЧ
>P-OQH4NO2 >P—S-CH-COOCoH,
С2Н5О/1| СН3О/1| |
тиофос карбофос
В виде водной эмульсии карбофос можно применять в индивидуальных
садах. Карбофос—эффективный препарат, малотоксичный для человека и
домашних животных.
Инсектицидом системного* действия является октаметил:
r(CH3JN и
Много работ по изучению органических соединений фосфора
проведено казанской школой химиков, возглавляемой акад. А. Е.
Арбузовым.
Мышьяк способен давать соединения, аналогичные
фосфорсодержащим органическим соединениям—арсины:
As-H As—CH3
метиларсин диметиларсин
* Инсектициды системного действия проникают в растения и делают их
ядовитыми для насекомых.
53. Фосфор- и мышьякорганические соединения 131
Арсины—малорастворимые в воде вещества с сильным
неприятным запахом, очень токсичны.
Арсины очень легко окисляются кислородом воздуха. Это
нейтральные вещества, не способные образовывать соли.
Некоторые арсины применялись в 1914 г. в качестве
отравляющих веществ.
Арсиновые кислоты можно считать производными мышьяковой
кислоты, в которой гидроксилы замещены на алкилы:
НОЧ СНЗЧ СНз.
>As—ОН >As—ОН \As_OH
НСК || ИСК || СН/ ||
мышьтопая метилмышьяковая диметилмыщьяковая
кислена (метиллрсиновая) (диметиларс-иновая)
кислота кислота
Арсиновые кислоты менее ядовиты, чем арсины. Некоторые
арсиновые кислоты применяются в качестве медицинских
препаратов, например арренал—натриевая соль метиларсиновой кислоты
3N>As—ONa-6H2O
NaO/ у
а также натриевая соль диметиларсиновой (какодиловой)
кислоты—какодиловокислый натрий:
3N>As—ONa-3HaO
сн3/ її
9»
СЛАВА IV
СПИРТЫ И ИХ ПРОИЗВОДНЫЕ
ОДНОАТОМНЫЕ ПРЕДЕЛЬНЫЕ СПИРТЫ
64. Строение спиртов. С представителями класса спиртов, или
алкоголен, мы неоднократно уже встречались. Как было ранее
сказано (см. стр. 34), еще Вюрц и позднее Жерар рассматривали
спирты как производные типа воды, в которой один водород
замещен углеводородным радикалом
°{н «{„"
Подтверждением такого строения спиртов может служить
реакция взаимодействия галоидных алкилов с влажной окисью
серебра, растворенная часть которой реагирует как гидроокись:
CH3J + Ag—ОН > СН3—ОН + AgJ
подпетый метиловый
метил спирт
СН3—CHaJ + Ag—ОН > СН3—СНа—ОН + AgJ
йодистый этиловый
этил спирт
или в общей форме:
R—J + Ag—ОН > R—ОН + AgJ
Даже при низкой температуре атом галоида в галоидных ал-
килах обменивается на гидроксил и получается спирт и
малорастворимое йодистое серебро; благодаря этому реакция образования
спиртов идет до конца (необратима).
Таким образом, алкоголь можно рассматривать как
углеводород, в котором атом водорода замещен водным остатком—гидро-
ксилом. Следует подчеркнуть, что водород гидроксильной
группы, связанный с атомом кислорода, резко отличается по
характеру от Еодородных атомов, связанных с углеродами.
55. Гомология а изомерия спиртов 133
По числу гидроксилов алкоголя бывают одноатомными,
двухатомными, трехатомными и т. д.
55. Гомология и изомерия спиртов. Подобно предельным
углеводородам, одноатомные спирты образуют закономерно
построенный ряд гомологов:
СпНап+1—Н CnH.2n+i—ОН
парафины предельные
одноатомные спирты
Так же как и в других рядах гомологов, каждый член ряда
спиртов отличается по составу от предыдущего и последующего
на гомологическую разность СН2.
Легко видеть, что количественное содержание кислорода
резко уменьшается, а содержание углерода увеличивается при
переходе от низших членов ряда к высшим:
%с %н %о
СН3—ОН 37,48 12,58 49,93
QH5-OH 52,14 13,13 34,73
QH,—ОН 59,96 13,42 26,62
С4Н9-ОН 64,81 13,60 21,59
С6НП-ОН 68,13 13,72 18,15
Понятно поэтому, почему метиловый и этиловый спирты горят
бесцветным пламенем, а высшие—коптящим пламенем.
' В зависимости от того, при каком атоме углерода находится
гидроксил, различают первичные, вторичные и третичные спирты.
В молекулах первичных спиртов находится одновалентная
группа—СН2ОН, связанная с одним радикалом (гидроксил при
первичном атоме углерода). Вторичные спирты характеризуются
двухвалентной группой>СН(ОН), связанной с двумя радикалами
(гидроксил при вторичном атоме углерода). В молекулах
третичных спиртов находится трехвалентная группа -^С—ОН,
связанная с тремя радикалами (гидроксил при третичном атоме
углерода). Обозначая радикал через R, можно написать формулы этих
спиртов в общем виде:
R\ R\
R—CHaOH CH(OH) R'—С—ОН
первичный Р'/ R"/
СПИРТ ^ Л
вторичный третичный
спирт спирт
По числу и характеру изомеров алкоголи общей формулы
СпН2п-и—ОН вполне подобны галоидным алкилам общей
формулы С Н2П+1С1.
134 Глава IV. Спирты и их производные
Для определения числа возможных изомеров
алкоголен для первых членов ряда можно поступить следующим
образом. Возьмем простейший алкоголь—метиловый спирт, или
карбинол, Н3С—ОН; при замене на метильную группу любого из
трех атомов водорода, связанных с углеродом, мы получим один
и тот же алкоголь СНЯ—СН2—ОН метилкарбинол, или этиловый
спирт. Следующий гомолог С3Н7—ОН имеет 2 изомера. Первый
изомер может быть получен заменой водорода этилового спирта
в группе СН3 на метил:
СН3—СН2—СН2ОН
ЭТИ пґм бЧНОЛ,
или н-пропиловый спирт
Второй производится путем замещения водорода в метилено-
вой группе:
СН3СН(ОН)—СН3 или (СН3JСН—ОН
диметилкарбинол, или изопропиловый спирт
Оба эти изомера имеют одинаковое строение углеродного
скелета, отвечающее только углеводороду
СН3—СН2—СН3—пропану, и различаются лишь положением в цепи гидроксила.
Следующий гомолог С4Н9—ОН имеет уже 4 изомера:
I. С углеродным скелетом нормального строения:
СН3—СН2—СН2—СН2ОН СН3—СН2—СН(ОН)—СН3
нормальный бутиловый спирт, вторичный бутиловый спирт,
или пропилкарбинол или метилэтилкарбинол
П. С углеродным скелетом изобутана B-метилпропана):
СН3-СН— СН2(ОН) СН3-С(ОН)-СН3
СНЯ
сн3
изобутиловый спирт первичный, третичный бутиловый спирт,
или изопропнлкарбинол или триметилкарбннол
Число возможных изомеров* очень быстро увеличивается при
переходе к высшим членам гомологического ряда:
Сі—І С5-8
Q-1 С,-17
Сэ-2 Q-39
Cj—4 С?»07
Qo—5622Ї09
* При расчете числа изомеров не учитываются стереоязомеры (см. стр. 161).
56. Способы получения одноатомных спиртов 135
Как было показано на ряде изомеров (например, метилкарби-
нол, этилкарбинол), в простейших случаях названия алкоголей
могут быть построены по старой «карбинольной» номенклатуре:
данный изомер алкоголя рассматривается как полученный из
карбинола СН3—ОН путем замещения его водородов на
соответствующие радикалы. Однако эта система позволяет строить
наименования спиртов только в простых случаях.
Названия по женевской номенклатуре строятся так же, как
для углеводородов и галоидпроизводных. В основе названия
алкоголя лежит наименование углеводорода, наличие же
гидроксильной группы отмечается окончанием ол с цифрой за ним,
указывающей номер атома углерода, при котором стоит гидр-
оксил. При этом надо так расположить нумерацию, чтобы гидрок-
сил имел самый малый из возможных номеров. Очевидно, что
для низших спиртов (метанол СН3ОН, этанол С2Н5ОН) не
требуется ставить цифры.
я і і
СН3—СН3—СН3ОН пропанол-1
СНз—СН(ОН)—СН. прспанол-2
СН3(ОН)—СН—СН3 2-метилпропанол-1
СН3
СН3(ОН)—СНа— СН—СН3 2-метилбу танол- 4
СН3
Как видно из последнего примера, при наличии разветвленной
цепи, счет углеродных атомов начинается с того конца цепи, к
которому ближе разветвление.
56. Способы получения одноатомных спиртов. 1. Одним из
общих способов введения гидроксильной группы в молекулы
органических соединений является гидролиз галоидпроизводных
углеводородов в присутствии водных растворов* щелочей
с„ная+1х + н-он ¦?=* с„н2п+1он + нх
где X—галоид (С1, Br, J).
Например:
СН3ч СН.тч
>СН—Вг + Н—ОН - * >СН(ОН) + НВг
СН/ СН/
* Необходимо вспомнить, что при действии на галоидпроизводные
спиртовых растворов щелочей происходит отшепление галоидоводородов, а это
ведет к образованию непредельной связи (см. стр. 76).
136 Глава IV. Спирты и их производные
Щелочи ускоряют процесс и, нейтрализуя образующуюся
кислоту, делают его необратимым. Легко видеть, что при этом
углеродный скелет остается без изменения. Поскольку галоид
соединен в галоидуглеводородах неионной связью и галоидпроизвод-
ные мало растворимы в воде, эта реакция проходит при
длительном нагревании.
Отметим, что способность к гидролизу зависит от природы
галоида; по легкости отщепления галоиды располагаются в
следующий ряд:
хлориды < бромиды < иодиды
Это значит, что иодиды гидролизуются легче, чем бромиды и
хлориды.
Галоиды, связанные с третичным атомом углерода,
гидролизуются значительно легче, чем вторичные и особенно первичные
галоидпроизводные. >
2. Как было ранее указано (см. стр. 79), спирты можно
получать действием воды на этиленовые углеводороды:
СН2=СН2 + НОН * СН3—СН2ОН
Реакция протекает при нагревании этиленовых углеводородов
с водой в присутствии катализаторов: серной кислоты,
хлористого цинка и др. Из этилена получают первичный спирт.
Реакция образования этилового спирта из этилеиа была подробио изучеиа
А. М. Бутлеровым. Он получил этиловый спирт гидратацией этилена в
присутствии серной кислоты. В настоящее время спирты производят в промышленном
масштабе гидратацией непредельных углеводородов. Так, изопропиловый спирт
получают в больших количествах из пропилена. В связи с необходимостью
замены пищевого сырья этиловый спирт получают из этилеиа; газообразный
этилен, получаемый из газов крекинга, пропускают в серную кислоту при
нагревании до 70 °С под давлением 10 am.
Более совершеиным является метод прямой гидратации этилена в
присутствии твердых катализаторов.
Гомологи этилена образуют вторичные и третичные спирты,
причем присоединение молекулы воды идет по правилу Марковни-
кова: атом водорода становится к наиболее гидрогенизированному
атому углерода (см. стр. 78):
СН3—СН=СН2 + НОН > СН3—СН(ОН)—СН3
пропилеи изопропиловый спирт
В связи с этим рассмотрим процесс получения одного спирта
из другого. Возьмем, например, первичный изобутиловый спирт
B-метилпропанол-1), при дегидратации из него получится изо-
бутилен B-метилпропен). Гидратация (присоединение воды)
57. Физические свойства спиртов 137
2-метилпропена приведет уже к триметилкарбинолу, т. е. к
третичному спирту:
CHjv СНач СНзу
>сн—сна —* >с=сна + н—он —» >с—снэ
сн/ і сн/ сн3/|
он он
2-метнлпропанол-і 2-метилпропен 2-метилпропаиол-2
3. Спирты можно получить восстановлением альдегидов и ке-
тонов. Альдегиды превращаются в первичные, а кетоны—во
вторичные алкоголи. Более подробно об этом см. на стр. 191.
4. Амины с общей формулой R—NH2 (первичные) при
действии азотистой кислоты, выделяя азот, превращаются в спирты
{см. стр. 364). Необходимо иметь в виду, что реакция протекает
сложно и часто сопровождается изомеризацией.
5. Синтез алкоголей при помощи цинкорганических и магний-
органических соединений имеет очень большое значение,
особенно в лабораторной практике. Этим путем получаются спирты с
более сложным строением углеводородного скелета, чем строение
исходных веществ, в качестве которых чаще всего употребляют
альдегиды и кетоны (см. стр. 192), хлорангидриды кислот и
сложные эфиры.
6. В технике большое количество этилового спирта получают из
растительного сырья, содержащего крахмал: картофеля, риса, хлебных злаков и т. д.
Однако в последнее время разработаны различные способы получения спирта
из непищевого сырья. Так, например, этиловый спирт готовят сбраживанием
смеси углеводов (глюкозы), получаемой при кислотном гидролизе клетчатки,
содержащейся в древесных опилках и других отходах лесной
промышленности. Это так называемый «гидролизный спирт» (он содержит также некоторое
количество метилового спирта); его применяют для получения синтетического
каучука. Предложены способы получения спиртов сбраживанием продуктов
гидролиза молодого торфа и сульфитных щелоков—отходов
бумажно-целлюлозного производства.
Значительные количества спирта получаются, как указано выше (см.
стр. 136), гидратацией этилена.
57. Физические свойства спиртов. Сопоставляя некоторые
физические свойства соединений внутри отдельных гомологических
рядов, можно отметить повторяющиеся закономерности. Особенно
ярко проявляется закономерное изменение температур кипения
для соединений нормального строения. По мере увеличения числа
СН2-групп в соединениях, принадлежащих к предельным
углеводородам, хлор-, бром-, иодпроизводным и первичным алкоголям,
их температура кипения закономерно возрастает. Сравнивая
свойства галоидпроизводных, легко видеть, что замена в
молекуле одного и того же углеводорода атома Н на С1, Вг, J ведет
к увеличению молекулярного веса и резкому повышению тем-
138 Глава IV. Спирты и их производные
пературы кипения. Если же заменить атом G1 (экв. вес 35,5)
на более легкую группу ОН (экв. вес 17), то вместо понижения
происходит значительное повышение температуры кипения,
например:
QH5ci
QH6Br
QH5J
QHbOH
Мол ек уляр ный
вес
64,5
109
156
46
Температура
кипения. °С
12,4
38,4
72,3
78,4
В чем же причина этого явления? Вспомним, что галоидово-
дороды (газы) кипят при значительно низшей температуре, чем
вода. При сопоставлении воды и спирта между ними легко
обнаруживается аналогия. Так, например, молекула воды и молекула
спирта содержат гидроксильную группу. Связь между кислородом
и водородом в молекуле спирта, так же как и в молекуле воды,
частично ионная. Напомним (стр. 65), что в молекуле воды связи
между атомами водорода и кислорода направлены так, что
образуют между собой угол, в вершине которого находится кислород.
Вследствие этого распределение электрических зарядов в
молекуле воды несимметрично, и в ней возникает довольно высокий
дипольный момент (fx = 1,85); между кислородом и водородом
устанавливается частично ионная связь; возникающие на
кислороде и водороде частичные отрицательные и положительные
заряды по величине приблизительно равны одной трети заряда
электрона:
(+)Н
Полярный характер молекул воды приводит к тому, что они
взаимно притягиваются; поэтому вода является соединением
ассоциированным (Н2О)Х:
Н
...О—H---O—Н---О— Н--
I I
н н
н
Дипольный момент этилового спирта (ц. = 1,7) близок к диполь-
ному моменту воды. Молекулы спирта также взаимодействуют
между собой и образуют ассоциированные молекулы:
О_н • • -О—Н • • -О-Н • • ¦ О—Н • • •
к і і і
58. Физические свойства спиртов
139
Ненормально высокая температура кипения метилового
спирта и воды в сравнении с температурой кипения йодистого метила
или йодистого водорода как раз и обусловлена явлением
ассоциации.
В ряду спиртов нормального строения, как и в других
гомологических рядах, температуры кипения закономерно
повышаются при переходе к высшим гомологам. Следует подчеркнуть
также, что среди изомеров третичные алкоголи отличаются самой
низкой температурой кипения, а первичные с неразветвленной
структурой—наивысшей.
Сходство спиртов с водой проявляется и в их растворимости.
Метиловый, этиловый и пропиловый спирты смешиваются с
водой во всех отношениях; молекулы воды, так же как и
молекулы спирта, отличаются высокими дипольными моментами, поэтому
между ними также может происходить взаимодействие. Этим
объясняется большая растворимость метилового, этилового и пропи-
лового спиртов в воде. Кроме того, спирт может образовать с
водой гидраты, о чем свидетельствует повышение температуры при
смешивании спирта с водой и то, что объем получаемой смеси
меньше суммы объемов спирта и воды в отдельности.
Бутиловые и амиловые спирты ограниченно растворимы в
воде. Высшие гомологи спиртов не растворяются в воде.
Растворимость третичных спиртов выше растворимости первичных и
вторичных спиртов с тем же числом атомов углерода в молекуле.
Таблица 4
Физические свойства бутиловых спиртов
Спирт
Нормальный первичный
бутиловый ....
Изобутиловый ....
Нормальный вторичный
бутиловый
Третичный бутиловый
Формула
СН3СНаСНаСНаОН
(СН3JСНСНаОН
СН3СН,СН(ОН)СН3
(CH3)SC(OH)
Темпе
рітура
плавления
°С
-89,
— 108
— 114,
+25,
6
7
5
Температура чи
пения, °С
при 76
рт.
117
108
99
82
) ММ
ст.
9
1
5
8
Растворимость
в 100 г воды
при 20 »С
8,3
9,5
13
Смешивается во
всех
отношениях
Плотность спиртов меньше единицы, т. е. все спирты легче
воды. Запах низших спиртов слабый, характерный для алкого-
лей; средние спирты обладают сильным, иногда неприятным
запахом.
140 Глава IV. Спирты и ах производные
58. Химические свойства спиртов. Спирты не обладают четко
выраженными кислотными или основными свойствами; как сами
спирты, так и их растворы в воде не проводят электрического
тока в заметной степени. Связи атомов у них гомеополярны (см.
стр. 61 ел.) и менее поляризованы, чем у воды:
Н н
н:с:с:о:н
н н
Благодаря своей доступности и способности вступать в
многочисленные химические реакции спирты играют громадную роль
в различных синтезах.
Отдельные группы химических превращений спиртов мы
рассмотрим в порядке последовательного участия атомов:
п і
—С—О—Н
III
н
I. Реакции, идущие с участием только водорода гидроксиль-
ной группы.
II. Реакции, обусловленные свойствами самого гидроксила.
III. Реакции окисления, в которых одновременно принимают
участие как гидроксил, так и атомы водорода в радикале, а в
некоторых случаях и атомы углерода (у третичных спиртов).
I. Реакции, идущие с участием водорода
гидроксильной группы. Атом водорода гидроксила
обладает большой подвижностью и способен легко замещаться.
1. Замещение атома водорода в гидроксиле металлом;
вещества, получающиеся в результате такого замещения, называются
алкоголятами:
2R—OH + 2Na > 2R—ONa + На
алкоголят
натрня
Алкоголяты, образуемые метиловым спиртом, называют мети-
латами, а образуемые этиловым спиртом—этилатами.
Алкоголяты—твердые вещества, легко растворимые в спирте.
Алкоголяты натрия—нестойкие соединения, быстро темнеют (ос-
моляются) на воздухе, особенно при нагревании. Наиболее
устойчив метилат натрия. В присутствии следов влаги алкоголят натрия
разлагается, и вновь образуется спирт:
R—ONa + НОН » R—ОН + NaOH
Реакция образования алкоголята показывает аналогию спирта с водой.
Как известно, из воды, при действии металлического натрия, бурно выделяется
58. Химические свойства спиртов 14)
водород. Низшие спирты СНэ—ОН, СгН6—ОН, наиболее близкие к воде,
реагируют с натрием бурио, средние—слабее, а высшие реагируют лишь при
нагревании. Алкоголяты образуютси при действии других металлов, например
магиня. алюминия.
В реакции образования алкоголита спирт проивлиет свойства кислоты,
хоти ионов водорода в спирте не удаетси обнаружить обычными индикаторами.
2. Замещение атома водорода гидроксила радикалами и
образование сложных эфиров. При взаимодействии спирта с
органическими кислотами получаются сложные эфиры:
СН3—О;Н + НОІ—С—СН3 ^п± СНЭ—О—С—СН3+.НаО
1 ' о о
метиловый уксусная уксуснометиловый
спирт кислота эфир
Реакция образования сложных эфиров носит название реак-
ции этерификации (от латинского слова aether—эфир).
Формально реакция этерификации подобна реакции нейтрализации,
причем спирт в этой реакции напоминает щелочь, а эфир—соль.
Однако, хотя спирты и содержат гидроксильную группу ОН, они все же
не являются щелочами. По отношению к лакмусу и фенолфталеину
спирты—нейтральные соединения. Как спирт, так и
эфиры—неионные соединения. Поэтому реакция этерификации лишь формально
похожа на реакцию нейтрализации. Реакция этерификации
является обратимой реакцией: вода разлагает сложные эфиры с об
разованием исходных веществ—кислоты и спирта. Такое
гидролитическое разложение сложных эфиров называется реакцией гидро
лиза.
Реакция этерификации, а также образующиеся в результат*
ее эфиры имеют огромное значение, поэтому в дальнейшем этг>
реакция будет рассмотрена подробнее (см. стр. 252).
II. Реакции, идущие с участием гидро ь-
сила спирта. Гидроксил-ьная группа спирта не
диссоциирует; тем не менее в некоторых реакциях она обладает известной
подвижностью и может замещаться или отщепляться.
1. Замещение гидроксила на галоид и образование галоидпро-
изводных углеводородов. Обычно реакция осуществляется при
действии на спирты галоидных соединений фосфора или галоидо
водородов (см. стр. 108).
ОН + PJ3 ^=± 3QH5-J + НЭРО,
QjH5—;Oh+ НіВг ї=» QjHa—Br + HaO
Получение галоидпроизводных углеводородов взаимодействием спиртої
с галондоводородными кислотами представлиет собой частный случай реакиив
этерификации. Как ранее было упомянуто, соединении, которые получаются
при взаимодействии спирта и кислот, носит название сложных эфнров. Поэто
142 Глава IV. Спирты и их производные
му галоидпроизводные углеводородов можно рассматривать как сложные эфи-
ры. образованные спиртами и галоидоводородными кислотами.
Реакция взаимодействия спирта с галоидоводородными кислотами
является обратимой, так как вода разлагает получающийся галоидный алкил
(бромистый этил) на исходные вещества. Для того чтобы добиться большего
выхода бромистого этила, нужно сдвинуть равновесие слева направо, для чего
необходимо либо увеличить концентрацию исходных веществ, либо удалять
из реакционной смеси воду. Практически поступают так- реакцию ведут в
присутствии водуотнимающих веществ, как, например, концентрированной H2SO(
или же в безводный спирт пропускают газообразный галоидоводород С целью
уменьшить количество присутствующей воды удобнее брать не галоидоводорол-
иую кислоту, а ее соль и выделять из нее сухой галоидоводород действием
концентрированной серной кислоты. Такой способ проведения реакции
является наиболее удобным так как образующийся галоидоводород действует
активнее в момент выделения.
2. Образование олефинов путем отщепления воды. При
нагревании спирта с большим количеством крепкой серной кислоты или
хлористым цинком, а также при пропускании паров спирта при
350—500 °С через фарфоровую трубку с окисью алюминия
происходит реакция дегидратации (отнятие воды) и образуются
этиленовые углеводороды. Так, например, из этилового спирта
получается этилен:
СН3—CHjOH » СНа=СН2 + НаО
Образование молекулы воды происходит за счет гидроксила и
атома водорода у соседнего атома углерода. Легче всего
дегидратируются третичные, затем вторичные и первичные алкоголи:
СН3 СН3
СН3—С—СН—СН3 -~На°> СНз—С=СН—СНЭ
! |
ЇОНЇЇ;
2-метилбутанол-2 2-метилбутеи-2
Реакция образования этиленовых углеводородов из спиртов
дает возможность превращать первичные спирты во вторичные
или третичные. Напомним, что реакция гидратации идет по
правилу Марковникова (см. стр. 78). Так, например, отнимая воду
от первичного пропилового спирта, можно получить пропилен, а
присоединением молекулы воды к пропилену получают изопро-
пиловый спирт:
СН3—СН2—СНа—ОН -» СНЭ—СН=СН2 J—U СНЭ—СН—СН3
пропиловый пропилен
спирт
изопроп иловый
спирт
58. Химические свойства спиртов 143
Отнимая воду у изобутилового спирта и присоединяя молекулу
воды к изобутилену, получают третичный бутиловый спирт:
СН3 СН3 СНэ
СН3-СН-СН.2ОН —ІИ* СН3-С=СНа ^ % СН3-С-СН3
изобутиловый изобутилеи і
он
спирт
третичный
бутиловый
спирт
При нагревании избытка спирта с серной кислотой или при
пропускании паров спирта через порошкообразный безводный
сернокислый алюминий при 20 °С наряду с этиленовыми
углеводородами получаются простые эфиры (см. стр. 177):
QH5O:H + HOj-GA * (СаН5JО + НаО
3. Отщепление водорода (дегидрогенизация, дегидрирование).
При пропускании паров спирта при 200—300 °С над металлами—
железом, цинком, мелко раздробленной медью и др. первичные
спирты распадаются на альдегид и водород, а вторичные спирты—
на кетон и водород:
СН3—СНа—ОН > СН3СНО + На
этиловый спирт уксусный
альдегид
СН3—СН(ОН)—СН3 > СН3—СО—СН3 + На
изопропиловый спирт ацетон
III. Реакция окисления спиртов. Окисление
спиртов обычно производят довольно сильными окислителями,
как, например, K2Cr20, +H2SO4 или KMnO4+H2SO4. При
окислении спиртов действие окислителя направляется на тот
углеродный атом, который связан с гидроксильной группой.
Следовательно, в зависимости от того, какой спирт окисляется—первичный,
вторичный или третичный, получаются различные продукты
окисления.
При окислении третичного спирта, не имеющего водорода у
углеродного атома, связанного с гидроксилом, происходит разрыв
углеродных связей и обычно получается несколько
кислородсодержащих соединений с меньшим, чем в исходном спирте, числом
атомов углерода в молекуле.
При окислении вторичных спиртов происходит образование
кетонов (см. стр. 189). Так, например, при окислении вторичного
144 Глава IV. Спирты и их производные
пропилового спирта (изопропилового) получается ацетон:
СНзч /ОН СНЗЧ
"Хс/ +о —» V=o + нао
сн/ \н ал/
изопропиловын ацетон
спирт
Или в общем виде:
R' R' R'
R—С—ОН + О * R— С-ОІН | » R—С=0 + Н.2О
X N"OH; кетон
Г1 : :
вторичный
спирт
Окисление первичных спиртов происходит аналогичным
образом, но так как у углеродного атома первичных спиртов,
связанного с гидроксилом, имеется на один атом водорода больше, чем
у вторичных, то продуктами окисления в этом случае являются
0
вещества, содержащие группу —С<^ и называемые альдегидами
(см. стр. 187).
Так, например, при окислении этилового спирта образуется
уксусный альдегид:
Н
Н3С—С—ОН + О
н
н
Н3С—С—OJH
ІОН"
н
Н3С—
с=о + нгс
уксусный
альдегид
Или в общем виде:
н н н
' R—С—ОН + О » R—С—ОІН: » R—С=0 + НаО
| ¦ I ! !
н ;он |
первичный- >-альдегид
спирт
59. Главнейшие представители спиртов. Метиловый спирт
(метанол). Строение метилового спирта выражается формулой
Н
Н—С—О—Н или СНа—ОН
н
59 Главнейшие представители спиртов. Метиловый спирт (метанол) 145
До сравнительно недавнего времени метиловый спирт
получали почти исключительно сухой перегонкой дерева. Поэтому его
называли также древесным спиртом.
При сухой перегонке дерева образуется горючий газ,
состоящий главным образом из СО, СО2, СН4 и Н2, древесный-<уголь и
жидкие продукты, разделяющиеся на масляный слой (смола или
деготь) и водный слой—подсмольная вода, в которой растворены
уксусная кислота, метиловый спирт, ацетон и ряд других веществ.
Эту смесь обрабатывают Са(ОНJ, разделяют на фракции
перегонкой в специальных аппаратах и получают метиловый спирт,
содержащий незначительное количество воды и летучих примесей.
В настоящее время получают преимущественно синтетический
метиловый спирт, пропуская смесь водорода и окиси углерода
при высокой температуре и большом давлении над катализатором
(окись цинка в смеси с некоторыми другими веществами):
СО + 2Н2 » СН3ОН
Получение метилового спирта по этому способу коренным
образом отличается от получения метилового спирта путем сухой
перегонки дерева. При сухой перегонке метиловый спирт
образуется как продукт разложения уже готового органического вещества
древесины, а при получении метилового спирта из окиси углерода
и водорода происходит его синтез из более простых веществ.
Промышленное осуществление синтезов такого рода приобретает
в последнее время все большее и большее значение.
Важнейшее место в современном химическом промышленном
синтезе занимают процессы, идущие под влиянием катализаторов.
Гомогенный и гетерогенный катализ. В синтезе многих органических
веществ—как в лаборатории, так н в промышленности—очень большое место
занимают процессы, протекающие под влиянием катализаторов. Термин
катализатор происходит от греческого слова—«развязывать», «разрешать» (Бер-
целиус 1830).
Одним из первых катализаторов была серная кислотаг при нагревании
крахмала с разбавленной серной кислотой К. С. Кирхгоф (Петербург, 1811)
произвел осахаривание крахмала и организовал промышленное производство
глюкозы. Подобным же каталитическим действием обладает серная кислота
(и некоторые другие кислоты) при реакции дегидратации спиртов до простых
эфиров или до олефинов, или при обратной реакции гидратации этих веществ
до соответствующего спирта. Так, при приливаний этилового спирта к нагретой
концентрированной серной кислоте при температуре 140 °С образуется эфир,
при 170 °С—этилен При нагревании эфира или этилена с разбавленной
серной кислотой происходит обратная реакция гидратации до этилового спирта.
Сама же серная кислота прн этом не расходуется, и если бы не наличие
побочных реакций окисления, могла-бы катализировать превращение неограниченно
больших количеств спирта или эфира. Если катализируемое вещество и
катализатор находятся в одном и том же агрегатном состоянии и не имеют
видимых поверхностей раздела (подобно тому, как это было в описанном примере),
катализ называют гомогенным, если же катализатор представляет собой твер-
10-788
!46
Глава IV. Спирты и их производные
дое вещество, а реагирующие соединения—жидкости или газы, катализ
называют гетерогенным. Катализатор способен ускорять не все реакции, а лишь те,
которые являются термодинамически возможными в данных условиях, причем
в случае равновесных реакций катализатор не сдвигает положения равновесия,
г. е. одинаково ускоряет прямую и обратную реакции. Другим характерным
свойством катализаторов является специфичность, избирательность их действия —
из многих возможных реакций превращения катализатор обычно ускоряет
только одну, т. е. способствует получению только одного целеыого продукта.
Важнейшими гомогенными катализаторами, применяемыми в органической
химии, являются кислоты, основания, хлористый алюминий, алюминаты
спиртов, фтористый бор.
ГазаоІЇрпзмйіе
продукты
реакции
Рис 15. Лабораторная установка для каталитических работ:
/—реактор—трубка из каарцепого стекла; 2—трубчатая печь; Л—шприц для
механической подачи исходной жидкости; 4—шкив электромотора; 5—двуххо-
доеый кран (присоединен к реакционной трубке); S—поршень шприца;
7—отверстие, через которое всасывается в шприц исходная жидкость; я
-капилляр, через который стекает исходная жидкость н реактор; 9—кг/ализатор;
10—термопара: //—приемник; 12—холодильник.
Огромное значение, особенно в природных процессах (брожение,
пищеварение, дыхание), имеют разнообразные ферменты (энзимы), представляющие
собой высокоэффективные и избирательно действующие органические
катализаторы. В последние годы все более важное значение приобретают также
искусственно получаемые органические катализаторы.
Из числа важнейших гетерогенных катализаторов, применяемых в
органическом катализе, следует отметить алюмосиликаты, окислы металлов
(алюминия, хрома, меди, цинка и т. п.) и металлы—платину, палладий, никель, медь,
железо и т. п. Схема простейшей лабораторной установки для каталитических
превращений органических веществ приведена на рис. 15.
Катализ играет огромную роль в промышленности органического синтеза.
Процессы получения высокомолекулярных соединений—синтетических кау-
чуков, волокон и пластических масс а также .большинства исходных мономеров
для этих процессов являются каталитическими. Важнейшие химические
соединения имеющие промышленное значение,—кислоты, спирты, альдегиды и
другие—получаются каталитическими методами. Наиболее эффективные
способы переработки нефти также основываются на каталитических процессах.
На рис. 16 изображена схема установки для получения
синтетического метилового спирта.
59. Главнейшие представители спиртов. Метиловый спирт (метанол) 147
Смесь окиси углерода и водорода многоступенчатым
компрессором 1 сжимается до 250 am и поступает в систему фильтров
(на схеме не обозначены), в которых очищается от брызг
машинного масла, попадающего в газ при прохождении его через
компрессор. Затем газовая смесь попадает в аппарат 2, где
смешивается с непрореагировавшим газом, и направляется в контактный
аппарат 3, в котором находится катализатор. Контактный
аппарат представляет собой толстостенный стальной сосуд.
Катализатор помещают в трубе, вставленной внутрь
контактного аппарата таким образом, чтобы между стенками этой
трубы и наружными стенками аппарата оставался кольцевой
проход. Перед началом работы контактный аппарат подогревается
Рис. 16. Схем? установки для получения метилового спирта.
электрическим током; в дальнейшем нагревание прекращают, так
как выделяющегося при реакции тепла достаточно для
поддержания желаемой температуры.
Газ входит в контактный аппарат, поднимается вверх по
кольцевому пространству, подогревается здесь за счет теплоты
реакции, проходящей внутри аппарата, а затем поступает во
внутреннюю трубу. После выхода из аппарата пары метилового спирта
вместе с непрореагировавшими окисью углерода и водородом
направляются в холодильники 4 и 5. Пары спирта сгущаются в
резервуаре второго холодильника и поступают в приемный
резервуар. Непрореагировавшие окись углерода и водород
засасываются циркуляционным компрессором 6 и через фильтр
направляются в смеситель 2 и далее в контактный аппарат.
В зависимости от взятого катализатора температура и
давление процесса могут колебаться в широких пределах. Для часто
применяемых цинк-хромовых катализаторов рабочая
температура лежит в пределах 370—400 °С, а давление 250—300 am.
Синтетический метанол является гораздо более чистым
продуктом, чем метиловый спирт, полученный сухой перегонкой
дерева; последний всегда содержит следы ацетона.
148 Глава IV. Спирты и их производные
Метиловый спирт—легкая (плотность 0,79 г/см3) жидкость,
кипящая при 65 °С, замерзает при —97 °С. Горит синим
пламенем. Принятый внутрь, даже в небольших количествах,
метиловый спирт вызывает тяжелое отравление, сопровождаемое
потерей зрения. При принятии больших количеств его может наступить
смерть. Применяется метиловый спирт при изготовлении лаков и
политур, для производства формальдегида, для денатурирования
винного спирта. Метиловый спирт используют в органическом
синтезе для введения метильной группы в молекулы различных
веществ. Он имеет большое значение в производстве красителей
и химико-фармацевтических препаратов.
60. Этиловый спирт (этанол). В настоящее время этиловый
спирт в громадных количествах синтезируют из этилена (см.
стр. 136), получаемого из газов нефтепереработки и попутных газов.
В промышленности этот метод имеет-наибольшее преимущество
по сравнению со всеми другими методами. Значительные
количества этилового спирта получают из отходов лесной
промышленности (см. стр. 137). Этиловый .спирт называется также винным
спиртом. Такой спирт получается в результате брожения
сахаристых веществ.
Спиртовое брожение сахаристых веществ вызывается
дрожжевыми грибками. Для брожения наиболее благоприятна
температура 25—30 °С.
Сущность спиртового брожения заключается в том, что
виноградный сахар (глюкоза) СвН12Ов, через ряд стадий,
расщепляется на спирт и углекислый газ:
СвН1аО„ * 2СаНбОН + 2СОа
Знаменитый французский ученый Пастер полагал, что брожение
неразрывно связано с присутствием живых дрожжевых клеток. По его мнению,
«брожение есть следствие жизни без воздуха, без свободного кислорода».
Общепризнанное в то время предположение Пастера было опровергнуто русским
врачом М. М. Манассеиной, установившей путем ряда опытов, что и
совершенно убитые дрожжевые клетки способны вызывать брожение. Через 26 лет после
опубликования работы Манассеиной (в 1897 г.) немецкий химик Бухнер
повторил ее опыты и подтвердил, что брожение не требует обязательного
присутствия живой клетки. Для уничтожения клеточной оболочки дрожжей он
растирал дрожжи с песком и инфузорной землей и при отжимании этой массы
под прессом получал сок, совершенно не содержащий живых клеток. Тем не
менее ничтожная добавка полученного таким путем сока вызывала брожение
большого количества сахара. Чтобы опровергнуть утверждение, что брожение
вызывается находящейся в отжатом соке «живой протоплазмой», Бухнер
предварительно убил дрожжи ацетоном и затем показал, что сок, отжатый из
таких дрожжей, ие уступает по действию соку, отжатому из живых клеток.
Таким образом было доказано, что брожение вызывается находящимся
внутри дрожжевых клеток веществом, которое не теряет своей активности и вне
дрожжевой клетки.
60. Этиловый спирт (этанол) 149
Вещество, вызывающее брожение, было названо эимазой.
Зимаза относится к группе веществ, называемых энзимами, или
ферментами. Энзимы—вещества, вырабатываемые живыми
организмами, представляют собой весьма эффективные органические
катализаторы. В отличие от обычных катализаторов энзимы
чаще всего обладают большой специфичностью и способны
воздействовать только на определенные формы органических
веществ.
Приведенное выше уравнение выражает только общий итог
спиртового брожения. На самом деле брожение представляет
собой ряд химических процессов, в результате которых из
виноградного сахара получаются различные вещества (см. стр. 336).
Каждый из этих процессов вызывается, по-видимому, особым
энзимом.
Сырьем для получения пищевого спирта чаще всего служит
картофель и хлебные злаки. При сбраживании белковые вещества,
входящие в состав этих продуктов, также подвергаются
расщеплению и дают ряд примесей. Таким образом, из перебродившей
массы (бражки) отгоняют «сырец», содержащий кроме винного
спирта летучие высшие спирты—сивушные масла. Для отделения от
примесей сырец подвергается дополнительной химической
очистке и повторной перегонке.
Полученный перегонкой бражки спирт называется сырцом.
Его снова подвергают перегонке. В результате получается
ректифицированный спирт, содержащий 95,5% С2Н5ОН. Остатком от
перегонки является сивушное масло—маслянистая жидкость,
состоящая главным образом из пропилового, изобутилового и изо-
амилового спиртов.
Спирт, вырабатываемый для технических целей,
денатурируется путем прибавления веществ, делающих его негодным
для питья. Для этой цели служат формалин, метиловый
спирт, пиридиновые основания, некоторые красящие
вещества.
Этиловый спирт—бесцветная легкоподвижная жидкость,
плотность которой при О °С равна 0,806 г/см3. Кипит при 78 °С и
замерзает при —110,5 °С, поэтому им наполняют термометры,
предназначенные для измерения низких температур (до —50 °С).
Горит слабо светящимся пламенем. Смешивается с водой во всех
отношениях.
Определение процентного содержания спирта в водной смеси
производится при помощи спиртометров. Спиртометр
представляет собой ареометр, н-а шкале которого нанесены значения не
плотности, а содержания спирта в объемных процентах
(градусах). Иногда также применяют спиртометры, показывающие
содержание спирта в весовых процентах.
150 Глава IV. Спирты и их производные
Спирт является весьма важным для народного хозяйства
продуктом. Большое количество его все еще расходуется на
производство синтетического каучука. Он находит применение в
фармацевтической промышленности для изготовления лекарств, в
парфюмерии для изготовления духов и одеколона, в лакокрасочной
промышленности для производства лаков и политур, в
производстве бездымного пороха и т. д.
Этиловый спирт широко применяется при биологических
работах, для консервирования ботанических и биологических
препаратов. Для многих целей требуется хорошо обезвоженный
спирт—«абсолютный» алкоголь. Спирт нельзя полностью
отделить от воды простой перегонкой потому, что он образует с водой
нераздельно кипящую смесь (азеотропная смесь). Ее состав:
95,6% спирта и 4,4% воды, а темп. кип. 78,15 °С (при
760 мм рт. ст.), в то время как абсолютный спирт кипит при
78,37 °С, а вода—при 100 °С. Для удаления воды из такой смеси
нельзя применять высушивание хлористым кальцием, так как он_
образует со спиртом соединение СаС1,-ЗС2Н5ОН, которое
растворяется в спирте. Почти безводный спирт можно получить,
настаивая длительное время ректификат с порошком безводной
сернокислой меди, полученным прокаливанием медного купороса. Эта
соль извлекает почти всю воду из спирта, и сама в спирте не
растворяется. Лучшего обезвоживания можно достичь, если
несколько часов кипятить спирт с большим количеством хорошо
прокаленной извести и затем произвести отгонку, защищая
дистиллят от соприкосновения с влажным воздухом.
61. Проииловые, бутиловые и амиловые спирты. Для про-
пилового спирта известны оба возможные по теории строения
изомера: первичный пропиловый спирт, кипящий при 97 °С, и
вторичный пропиловый (изопропиловый) спирт, температура кипения
которого 82,4 °С:
СН3—СН2—СН2ОН СН3—СН(ОН)—СН8
первичный вторичный
лролиювыП спирт пропиловый спирт
Строение первичного пропилового спирта доказывается
тем, что окислением он может быть переведен в пропионовую
кислоту:
сн3сн2сн2он -u-> сн3сн2соон
пропыоновая кислота
Следовательно, спирт, кипящий при 82,4 °С, является
вторичным.
61. Пропиловые, бутиловые и амиловые спирты 151
Первичный проп иловый спирт в небольшом количестве
содержится в сивушном масле. Вторичный пропиловый спирт
получается из пропилена, содержащегося в газах, образующихся при
крекинге нефти; применяется он как растворитель.
Бутиловые спирты. Как мы-уже ранее говорили, для спиртов
С4Н9ОН на основании теории химического строения возможно
существование четырех изомеров. Все они получены, и строение
их строго доказано.
н-Бутиловый спирт (бутанол-1) кипит выше остальных
изомеров (при 117,9 °С); в 100 г воды растворяется 8,3 части спирта.
Он окисляется с образованием нормальной масляной кислоты,
чем доказывается его строение:
сн3—сн2—сн2—снаон -^-» сн3—сна—сн2—сдан
В технике н-бутиловый спирт может быть получен с помощью
так называемого «ацетонобутилового» брожения сахаристых
веществ, проводимого с помощью специальных бактерий. Более
эффективны чисто синтетические методы, одним из которых
является процесс так называемого «оксосинтеза».
Смесь олефина, окиси углерода и водорода пропускают под
высоким давлением и при повышенной температуре через раствор,
содержащий кобальтовый катализатор. При этом образуется
смесь изомерных альдегидов, имеющих в углеродной цепи на
один атом углерода больше, чем в исходном олефине. Так, из
пропилена получают смесь «-масляного и изо-масляного
альдегидов:
СН3—СН=СН2 + СО + Н2—
СНз CHg CHg '
»масляный альдегид
сн;
" сн3
изо-масляный альдегид
Гидрированием этих альдегидов получают соответствующие
спирты.
Метод оксосинтеза может быть распространен и на получение
высших спиртов. Так, исходя из амиленов, получают смесь гекси-
ловых спиртов, а исходя из гептиленов—смесь изомерных октило-
вых спиртов.
Изобутиловый спирт B-метилпропанол-1) известен давно, так
как он выделяется перегонкой из сивушных масел,
получающихся при спиртовом брожении (см. стр. 149); при окислении .пре-
152 Глава IV. Спирты и их производные
вращается в изомасляную кислоту (т. е. является первичным
спиртом):
СНзч о CH~
>СН—СНаОН > >СН—СООН
OV , СИ/
Изобутиловый спирт сравнительно плохо растворим в воде.
Вторичный бутиловый спирт (бутанол-2) при дегидратации
превращается в бутен-2:
СН3—СН(ОН)СН2—СН3 ^2 СНЭ—СН=СН—
При окислении бутанола-2 образуется соответствующий кетон
(метилэтилкетон СН3—СО—СН2—СН3); этим доказывается
строение бутанола. Применяется в виде эфиров уксусной, масляной
и других кислот для производства искусственных фруктовых
эссенций и для получения некоторых парфюмерных веществ
(искусственный мускус), а также при изготовлении фармацевтических
препаратов.
В больших количествах бутанол-2 применяется для получения
метилэтилкетона, являющегося ценным растворителем.
трет-Бутиловый спирт B-метилпропанол-2, триметилкарби-
нол) был получен в 1864 г. Бутлеровым при действии на ацетон
диметилцин ком:
СНз СН2
I /СН3 I НгО
СНэ—С + Zn< > СНЭ—С—СНз -Ї-*
II ХСНЭ I
О О—Zn—СНЭ
» СНЭ—С—СНЭ + CH3Zn(OH)
ОН
Триметилкарбинол при охлаждении кристаллизуется, темп, плавл.
+25,5 °С; обладает самой низкой температурой кипения (83 °С) из всех
изомерных бутиловых спиртов, смешивается с водой во всех пропорциях; имеет
запах камфары.
Амиловые спирты. Как упоминалось выше (см. стр. 134), для
спирта с пятью атомами углерода известно восемь изомеров (см.
табл. 5).
Эти спирты называются амиловыми спиртами.
В технике амиловые спирты получают из газов крекинга
нефти.
Амиловая фракция сивушных масел содержит
преимущественно «изоамиловый» спирт—2-метилбутанол-4 (IV) с примесью
оптически активного 2-метилбутанола-1 (VII). Технический изо-
61. Пропиловые, бутиловые и амиловые спирты
153
амиловый спирт (часто называемый просто «амиловым») имеет
характерный запах; вдыхание его паров раздражает горло и
вызывает кашель. Амиловый спирт малорастворим в воде B части в
100 частях воды).
Применяется в качестве растворителя и для получения эфиров
органических кислот, которые используются в качестве душистых
веществ, например изоа мил ацетат—грушевая эссенция.
Таблица 5
Изомерия амиловых спиртов
Изомерия
углеродного скелета
5 4 3 2 1
С—С-С—С—С
с
1 [2 3 4
С-С—С—С
с
3 |2 1
С—С—С
і
I
II
III
IV С-
V
VI
VII
VIII
Изомерия положения
гидроксила
5 4 3 2 1
С—С—С—С—С—ОН
5 4 3 2 1
С—С—С—С—С
1
о'н
5 4 3 2 1
С—С—С—С—С
1
он
с
|2 3 4
_С—С—С—ОН
с
1 І2 3 4
С—С—С—С
он
с
І \і 3 4
\_*—y^t—K_t—kj
1
OH
с
I |2 3 4
С—С—С—С
о'н
с
3 І2 1
С—С—С—ОН
і
Формулы изомеров
5 4 3 2 1
СН3-СН2—СН2—СН2—СН2ОН
пеитанол-1
СН3—СНа—СН2—СН(ОН)—СН8
пеитанол-2
5 4 3 2 1
СН3—CHjj—СН(ОН)—СНа—СН3
пентаиол-3
СНз\2 3 4
і /СН—СН2—СН2—СН
сн„/
2-метнлбутанол-4
СНз\2 3 4
і .СИ—СН(ОН)—СН,
сн3/
2-мет нлбутаиол-3
СН*\2 3 4
і )С(ОН)—СН2—СН,
CH/
2-метнлбутанол-2
СНз\2 3 4
і /СН—СНо—GH«
нонас/
2-метилбутанол-1
сн3
3 |2 1
СН3—С—СН2ОН
1
сн3
2,2-диметилпропанол-і
154
Глава IV. Спирты и их производные
62. Оптическая деятельность органических соединений. Для
некоторых спиртов известны случаи замечательной изомерии.
С этим явлением можно познакомиться на примере 2-метилбу-
танола-1 A), который удобнее изобразить в виде формулы И:
HO—CHa—CH—CHa—CH3
сн„
Для этого амилового спирта известны два изомера, молекулы
которых имеют одно и то же строение, выражаемое только что
приведенной формулой. По всем химическим и большинству
физических свойств эти два изомерных спирта ничем не отличаются
друг от друга. Их плотности, температуры кипения, показатели
преломления и т. д. совершенно тождественны между собой. Но
одно свойство отличает их друг от друга: при прохождении через
них поляризованного света один из этих спиртов вращает плоскость
поляризации светового луча вправо, другой—вращает эту
плоскость на равный угол влево.
Некоторые твердые тела, как, например, кварц и двойная
сернокислая соль лития и калия, обладают способностью
вращать плоскость поляризации света, только находясь в
кристаллическом состоянии. Эти вещества образуют кристаллы, которые
являются зеркальными изображениями друг друга и не могут
быть совмещены. Другими словами, они относятся друг к другу
так, как не имеющий плоскости симметрии предмет относится к
своему изображению в зеркале. Точно такое же различие сущест-
вует между формами правой и левой руки или между
одинаковыми спиралями, закрученными в различные стороны. Такого рода
кристаллы называются энантиоморфными кристаллами (рис. 17).
Рис. 17. Энантиоморфные кристаллы.
Один из двух энантиоморфных кристаллов вращает плоскость
поляризации поляризованного луча света вправо, другой—влево
Если разрушить путем растворения или плавления
кристаллическое строение таких іел, они теряют вращательную способность
Таким образом, их вращательная способность зависит от
строения кристалла, а не от строения самих молекул.
62. Оптическая деятельность органических соединений 155
Совершенно иначе обстоит дело с вращательной способностью
амиловых спиртов: они оптически деяіельньї и в растворе, и в
парообразном состоянии. Следовательно, причиной оптической
деятельности их является строение самих молекул.
В молекуле амилового спирта атом углерода (отмеченный в
написанной выше формуле звездочкой) одной связью соединен
с этилом, другой связью—с водородом, третьей связью—с
метилом и четвертой связью—с группой СН2ОН. Таким образом,
в амиловом спирте один из атомов углерода соединен с четырьмя
различными группами атомов. Такой атом углерода называется
асимметрическим атомом углерода.
Вант-Гофф* в 1874 г. нашел, что наличие асимметрического
атома углерода в молекуле является достаточной причиной для
оптической деятельности органических веществ.
В качестве примеров оптически деятельных органических
соединений можно привести следующие:
с—он
с—сн2он
QH5
2-метилбутанол-1
с-он
сн
/\
метилизопропилкарбинол,
или 2-метилбутанол-З
амиловые спирты
вторичный бутиловый спирт,
или метилэтилкарбинол
Все эти соединения содержат асимметрический атом углерода,
отмеченный в структурной формуле звездочкой.
Очевидно, что если заменить в этих спиртах гидроксильную группу на
атом галоида, то получаемые галоидпроизводные будут также содержать
асимметрические атомы углерода.
Надо иметь в аиду, что при синтетическом получении этих алкоголем из
соединений оптически недеятельных возникает равное число молекул «лравых»
* Я. Вант-Гофф (по-голландски произносят Ван-эт-Хоф)—голландский
химик A852—1911). Один из основателей стереохимии и современной
физической химии. До 1895 г. работал в Голландии (Утрехт, Амстердам), а затем в
Германии (Берлин).
Свою знаменитую работу по стереохимии «Расположение атомов в
пространстве» он опубликовал в очень молодом возрасте B2—23 лет). Им были
развиты смелые идеи об асимметрическом атоме углерода, объясняющие
причины оптической деятельности органических соединений. Аналогичные
взгляды одновременно с Вант-Гоффом опубликовал французский химик А. Ле-Бель.
Работами этих двух ученых были заложены основы стереохимии органических
соединений.
В дальнейшем Вант-Гофф перешел к исследованиям в области физической
химии: химическая динамика, учение о химическом равновесии, исследование
растворов солей, изучение законов осмотического давления и многое д угое.
156
Глава IV. Спирты и их производные
и «левых». Таким образом, вращение одних компенсируется обратным
вращением других, и вещество в целом не проявляет оптической деятельности.
Подробнее об этом см. на стр. 158.
Измерение оптической активности. Оптическую активность
органических соединений измеряют при помощи приборов,
называемых поляриметрами.
Рис. 18. Схема устройства полутеиевого поляриметра.
Устройство простейшего, так называемого полутеневого,
поляриметра показано на.рис. 18. Луч света от осветителя / попадает
на неподвижно укрепленную призму Николя 2 (поляризатор)
и выходит из нее в виде поляризованного луча. Затем он попадает
на вторую призму Николя 3 (так называемый анализатор),
которую можно вращать с помощью рукоятки 4, и далее, через лупу 5,
в глаз наблюдателя. Прибор устроен таким образом, что если
между поляризатором и анализатором луч не проходит через
вещество, обладающее оптической активностью, то анализатор
должен стоять на положении О, и при этом наблюдатель видит
через лупу поле, разделенное на две половины, освещенные
одинаково ярко. Если же между поляризатором и анализатором
помещена длинная стеклянная трубка 6, наполненная оптически
активным веществом, то при прохождении через него света плоскость
поляризации этого света изменяется на некоторый угол, и одна
из половин поля зрения становится более яркой. Тогда
поворачивают анализатор 2 таким образом, чтобы обе половины поля
зрения снова стали одинаково яркими. Угол поворота
анализатора (определенный по круговой шкале 7) указывает величину угла
вращения плоскости поляризации света при прохождении через
исследуемое вещество, т. е. величину оптической активности
этого вещества.
63. Стереохимическая теория 157
Величина угла вращения плоскости поляризации света
зависит от температуры, длины волны поляризованного света, от
природы растворителя. Она прямо пропорциональна толщине
слоя и в большинстве случаев прямо пропорциональна
концентрации.
Для того чтобы можно было количественно сравнивать
оптическую деятельность различных веществ, вычисляют так
называемое удельное вращение. Удельным вращением называется угол
вращения, наблюдаемый при толщине слоя жидкости 1 дм и при
концентрации оптически деятельного вещества, равной 1 гімл.
Удельное вращение обозначается [а]. Если при толщине слоя
bL дециметров раствор, в 100 мл которого содержится с граммов
вещества, вращает плоскость поляризации света на угол а, то
удельное вращение его равно:
г . а-100
Га] = —;—
1 ' L-c
Если же оптически деятельная жидкость, плотность которой
равна d, взята в неразбавленном состоянии, то удельное вращение
вычисляют по формуле
г 1 а
1 J L-a
Конечно, при этом надо указать, при какой температуре
производилось измерение, и длину волны света; правое вращение
обозначается знаком плюс, левое—знаком минус. Если, например,
удельное вращение оптически деятельного амилового спирта,
находящегося в сивушном масле, измеренное при 20 °С, для линии D
солнечного спектра (с длиной волны 589 ммк) равно 5,9° влево,
то это записывают так:
Вращение обычно сильно возрастает при переходе к более
коротким длинам волн света. Приборы, позволяющие определять
и регистрировать вращение по всему спектру в видимой и
ультрафиолетовой частях спектра, т. е. от 220 ммк до 780 Ммк,
называются спектрополяриметрами. Получаемые при этом кривые
дисперсии вращения позволяют гораздо глубже характеризовать
оптическую дятельность вещества.
63. Стереохимическая теория. Обозначая строение изомеров
амилового спирта с помощью обычных структурных формул,
изображающих молекулу как бы расположенной в одной плоскости,
нельзя объяснить оптическую изомерию амиловых ^спиртов.
Однако это явление становится легко объяснимым, если предста-
158 Глава IV. Спирты и их производные
вить атомы расположенными в молекуле не в одной плоскости,
а в пространстве.
Еще в 1863 г. творец теории химического строения А. М.
Бутлеров писал: «Если же атомы действительно существуют, то я не
вижу, почему должны быть тщетны все попытки определить
пространственное расположение последних, почему будущее не
должно научить нас производить подобное определение».
В 1874 г. Вант-Гофф и Ле-Бель положили начало
стереохимии, т. е. учению о пространственном расположении атомов в
молекуле. Согласно теории Вант-Гоффа, валентные связи атома
углерода направлены к вершинам правильного тетраэдра, в центре
которого расположен углерод. Эта модель хорошо объясняет
равноценность всех четырех связей атома углерода, так как
наиболее симметричным расположением четырех точек в пространстве
вокруг пятой будет такое, при котором четыре точки расположены
в углах правильного тетраэдра, а пятая в центре его*. Таким
образом, направления валентных связей совпадают с линиями,
соединяющими центр тетраэдра с его вершинами, и составляют
между собой углы, из которых каждый равен 109°28'.
В том случае, если все четыре валентные связи углеродного
атома насыщены четырьмя различными атомными группами, эти
последние, как показано на рис. 19 и 20, можно расположить
двумя различными способами. На рис. 19 изображены тетраэдры,
а на рис. 20—только направления валентных связей.
Как легко видеть, первое расположение является зеркальным
изображением второго и не может быть никаким образом с ним
совмещено.
Различие между ними особенно бросается в глаза, если
мысленно совершить переход от группы СН2ОН через Н к С2Н5.
В одном случае этот переход происходит по часовой стрелке, в
другом—по противоположному направлению.
Таким образом, различие между правовращающим и левовра-
щающим амиловыми спиртами объясняется несимметричностью
их молекул: заключая в себе асимметрический атом углерода, эти
молекулы не имеют плоскости симметрии. Изомеры, подобные
правовращающему и левоврашающему амиловым спиртам,
называются оптическими антиподами. Модель молекулы одного из
оптических антиподов является зеркальным изображением модели
молекулы другого оптического антипода. Один из антиподов
вращает плоскость поляризации света вправо на столько же, на
сколько другой антипод вращает ее влево.
* Центр правильного тетраэдра находится на перпендикуляре,
опушенном из вершины на основание, на расстоянии одной четверти этого
перпендикуляра от основания. Это есть точка пересечения высот тетраэдра.
63. Стереохимическая теория
159
При смешении равных количеств правовращающего и левовра-
щающего амиловых спиртов получается недеятельный амиловый
спирт, не оказывающий влияния на поляризованный свет. Этот
спирт состоит из равного числа молекул правовращающего и ле-
вовращающего изомеров. Такие смеси соединений называются
рацемическими.
СН,ОН
СН2ОН
Рис. 19. Схема пространственного расположения радикалов и
атомов в молекулах правовращающего и левовращающего
амиловых спиртов.
сн„он
СН2ОН
Рис. 20. На'.равления валентных связей радикалов и ато-
моа в молен ,.лах правовращающего и левовращающего
амиловых спиртов.
Поскольку олтические антиподы вращают плоскость
поляризации «вправо» или «влево», их иногда называют «правый изомер»
и «левый изомер». Это в сущности неправильно, так как напраз-
ление и величина вращения плоскости поляризации зависят от
растворителя, кислотности среды и других факторов. Поэтому
для объективного описания зеркальных изомеров нужно
рассматривать схематическую модель молекулы.
Представим себе пару стульев; у стула А отломана передняя
левая ножка, а у стула Б—передняя правая. Понятно, что левой
или правой мы называем ножки с точки зрения наблюдателя,
которого мы мысленно посадили в обычном положении на стул. Сами
160 Глава IV. Спирты и их производные
по себе ножки стула не являются правыми или левыми. Такой
прием описания несимметрических предметов «с точки зрения
наблюдателя», расположенного определенным образом
относительно предмета, применяется часто: берега рек по отношению к
направлению течения, вращение по часовой стрелке и др.
Пространственная модель молекулы одного из антиподов,
подобно сломанному стулу, не является сама по себе правой или
левой, но также может быть описана или условно обозначена
методом «наблюдателя».
Представим себе наблюдателя, идущего вдоль по углеродной
цепи модели одного антипода от СA) к СB) 2-хлор-2-бромбутана:
С1 Вг
н3с сн2сн,
I
Атом СІ находится слева, а атом Вг справа от наблюдателя.
Условимся обозначать положение справа греческой буквой о
(сигма), а положение слева греческой буквой р (ро)*.
Очевидно, что в таком и подобных случаях достаточно, чтобы
описать пространственную структуру, обозначить символами р
или о один из заместителей: 2р-хлор-2-бромбутан или 2-хлор-
2о-бромбутан.
Следуя описанному правилу, легко обозначить
представленные на рис. 20 зеркальные изомеры амилового спирта. Изомер (а)
получит обозначение 2р-метилбутанол-1, а изомер (б) 2о-метил-
бутанол-1.
Теория Вант-Гоффа о тетраэдрическом направлении связей
у атомов углерода в молекулах соединений жирного ряда
получила позднее блестящее экспериментальное подтверждение в
работе Брэгга, исследовавшего прохождение рентгеновских лучей
через кристаллы алмаза. Эти исследования показали, что
кристаллическая решетка алмаза построена таким образом, что
каждый углеродный атом расположен в центре правильного
тетраэдра, в вершинах которого находятся другие углеродные атомы
(рис. 21).
Изомерия, которая зависит от пространственного
расположения атомов в молекуле, называется пространственной изомерией,
или спгереоизомерией.
* Буквы р и о выбраны потому, что они антисимметричны и напоминают
при написании движение против и по часовой стрелке. Кроме того, они никогда
не применяются в номенклатурных названиях для обозначения положения
заместителей, как а, ?, -г, 5.
63. Стереохимическая теория
161
Следует заметить, что если в молекуле вещества имеются два
или несколько асимметрических атомов углерода, то явление
усложняется и число стереоизомеров значительно увеличивается;
кроме того, в этом случае, помимо оптических антиподов,
возможно появление стереоизомеров и другого характера.
Стереохимические представления не находятся в каком-либо
противоречии с теорией химического строения. Н. Д. Зелинский
указывал, что «...стоило только формулам строения придать
стереометрическое значение, как то, что казалось непонятным,
приняло новую и ясную форму, нисколько не подрывая основ теории
строения, но, напротив, все далее ее
развивая и совершенствуя».
Стереохимическая теория
представляет собой дальнейшее развитие идеи,
лежащей в основе теории строения,
идеи о связях между атомами.
Конфигурация углеродной цепи.
Если в молекуле (например, в
молекуле этана) находятся два атома
углерода, связанные между собой, то их
следует изобразить двумя тетраэдрами,
имеющими одну общую вершину,
причем линия, соединяющая их центры, должна проходить
через эту вершину (рис. 22). В трехзвенной углеродной цепи для
н
Рис. 21. Кристаллическая
решетка алмаза.
н
Рис. 22. Схема молекулы этана.
Н
Рис. 23.
Конфигурация цепи из трех
атомов углерода.
атомов углерода возможно также только одно единственное
расположение (рис. 23). Для цепи из четырех или пяти атомов
углерода, если все они лежат в одной плоскости, возможны
два расположения: форма клешни (рис. 24,6) и форма зигзага
(рис. 24,е). Если же предположить что атомы углерода могут
11—788 ¦
162
Глава IV. Спирты и их производные
находиться не в одной плоскости, то число возможностей
неограниченно велико.
ар ?
Рис. 24. Конфигурация цепи из четырех и пяти атомов углерода:
а, б—форма клешни; в—форма зигзага.
Форма зигзага возможна для углеродной цепи любой
длины, форма же клешни становится невозможной у углеродной
цепи, содержащей более пяти атомов углерода, так как уже шесть
атомов углерода начинают препятствовать друг другу в
пространственном отношении (рис. 25). Из всего изложенного следует,
что даже в нормальной углеродной цепи атомы углерода не
расположены на одной прямой.
Следует отметить, что в кристаллическом
состоянии органические соединения (углеводороды,
спирты, кислоты) нормального строения
содержат длинные углеродные цепи, построенные в
форме зигзага. Это в ряде случаев подтверждено
экспериментально. Так, например,
рентгенографическое исследование кристаллов лауриновой
кислоты СН3—(СН2I0—СООН показало, что в ее
молекуле атомы углерода расположены
зигзагообразно, причем угол зигзага очень близок к
109°28', т.е. к углу между направлениями
валентных связей в тетраэдрической модели Вант-Гоф-
фа (рис. 26).
64. Электронное строение ординарных связей (а-связь). Вопрос
о природе химической связи является одним из самых сложных
в органической химии.
Для типичных неорганических соединений на разных этапах
истории химии с большим успехом и сравнительно просто
применялись электростатические теории—теория электрохимического
дуализма Берцелиуса и теория электролитической диссоциации,
в той или иной мере сохраняющаяся в настоящее время.
Рис. 25.
Конфигурация
цепи из
шести атомов
углерода.
64. Электронное строение ординарных связей (а-связь)
163
Однако большие, часто непреодолимые трудности и
противоречия встречались в тех случаях, когда требовалось объяснить
образование связи между однородными атомами. А именно
такого рода связи и существуют главным образом в органических
соединениях.
Теория Льюиса—Лангмюра об электронном строении гомео-
полярных связей представляет собой некоторый исторический
етап в развитии учения о строении вещества.
В основе теории Льюиса—Лангмюра лежит
представление о том, что каждая связь
осуществляется парой электронов,
расположенных между соединяемыми атомами и
принадлежащих им в равной степени (см. стр. 61).
Этими представлениями в органической
химии продолжают пользоваться до
настоящего времени.
Новейшая физика обогатила нас новыми
внаниями о строении ковалентных связей. Два
электрона, участвующие в химической
ординарной связи, движутся с громадными
скоростями.
Быстрое движение светящегося или
освещенного шарика по одному направлению
представляется человеческому глазу светлой
полосой. Если движение такого тела будет
происходить по различным направлениям в
определенном объеме, то глазу наблюдателя этот
объем представится заполненным светящимся
облаком. Это облако имеет неравномерную
плотность: более «непрозрачны» те части
области, в которых электрон пребывает чаще.
Такова грубая зрительная модель
«электронного облака» и «электронной плотности».
Как же в свете нового учения о строении р
вещества представляются химические связи в молекулы лаурино-*
простейших органических молекулах? вой кислоты.
В какой мере современная физика
подтверждает или опровергает развитые ранее представления о
строении вещества?
Что нового и ценного находят химики-органики, используя
выводы современного учения о химической связи?
Единственный электрон атома водорода движется вокруг ядра
(протона), образуя шарообразное электронное облако (рис. 27).
Однако только в простейшем случае электронное облако
может быть шарообразным. У атома углерода имеется 6 электронов,
а=Ю9°ЗО'
164
Глава IV. Спирты и их производные
из которых только 4 внешних электрона участвуют в образовании
ковалентних связей.
В соответствии с представлениями, развитыми классической
органической химией, принимают, что в простейших случаях
(например, у метана и предельных углеводородов) эти четыре
электронных облака одинаковы; они имеют форму
несимметричных фигур (рис. 28) и расположены вокруг ядра углерода
равномерно, образуя между собой углы 109°28/ (рис. 29). Валент-
Рис. 27.
Электронное
облако
атома
водорода.
Рис. 28.
Электронное
облако
атома углерода
(одно из
четырех).
Рис. 29. Схема
строения молекулы
метана.
ные углы углерода могут несколько меняться в зависимости
от взаимного отталкивания или притяжения между атомами:
/Н
С< 109°28'
44
С
111°±30
с
СІ
С
Вг
109o±2a
Вг
¦Вг
112°±2°
Таким образом, углы между валентными связями в
зависимости от природы элементов меняются, но в сравнительно узких
пределах.
Используя физические методы (рентгеноструктурный анализ
и др.), можно определить абсолютные расстояния между атомами
(в ангстремах; 1 А = Ы0~8 см).
В молекуле метана атомы водорода удерживаются атомом
углерода вследствие того, что электроны водорода и углерода,
двигаясь по своим орбитам, притягиваются (хотя и в разной
степени) ядрами обоих элементов. Электронные облака частично
«перекрываются». То же самое относится и к ординарной
связи С—С (рис. 30). Химические связи такого рода называются
«з-связями (сигма-связь).
65. Аллиловый спирт
165
Ординарные связи, изображаемые в структурных формулах
чертой, осуществляются парой электронов, из которых каждый
движется по своей орбите.
При соединении двух различных
атомов электронные облака пары
валентных электронов сдвигаются в
сторону более электроотрицательного
элемента, что можно выразить
условно стрелкой (рис. 31).
С : С
С—С
Рис. 30. Схема
ординарной С—С
сигма-связи.
С :С
С—С
Рис. 31. Сдвиг
электронных
облаков пары
валентных электронов.
Рис. 32. Схема строения
молекулы этана.
Электронное строение молекулы этана может быть
представлено схемой, приведенной на рис. 32. Из рисунка видно, что в
этане имеется шесть а-связей С—Н и одна а-связь С—С.
Новейшие данные о природе химической связи
подтверждают основные представления классической органической химии
о направленности валентных связей, валентных углах, о
круговой симметрии ординарной связи и возможности свободного
вращения по этой связи.
Электронные представления, кроме того, дополняют и
развивают структурное учение, что особенно ярко проявляется,
например, при рассмотрении галоидзамещенных кислот (см.
стр. 281) непредельных (см. стр. 247) и ароматических (см.
стр. 427) соединений.
НЕПРЕДЕЛЬНЫЕ СПИРТЫ
Замещение водорода в углеводородах непредельного ряда
С„Н2П на гидроксильную группу приводит к образованию
непредельных спиртов.
65. Аллиловый спирт. Из непредельных спиртов простейшим
был бы виниловый спирт СН2=СН(ОН), но оказывается, что
спирты с гидроксилом при углероде, связанном с другим атомом
углерода двойной связью, могут существовать только в
исключительных условиях. Такого рода спирты претерпевают изомерное
166 Глава IV. Спирты и их производные
превращение в момент своего образования, и в тех случаях, когда
по ходу реакции можно было бы ожидать группировки атомов
СН2=С(ОН)—, происходит перегруппировка в СН3—СО—
(правило Элыпекова). Так, вместо винилового спирта СН2=СН(ОН)
образуется уксусный альдегид СН3—С<Г .
Простейшим представителем ненасыщенных спиртов является
аллиловый спирт СН2=СН—СН2ОН (по женевской номенклатуре
пропен-1-ол-З).
Доказательством строения аллилового спирта является
получение из него, при окислении, акриловой кислоты:
СНа=СН—СНа—ОН -±2* СН2=СН-СООН
акриловая
кислота
Аллиловый спирт—жидкость (темп. кип. 97 °С), обладающая
резким запахом; хорошо растворим в воде.
Аллиловый спирт находится в подсмольной воде,
получающейся при сухой перегонке дерева. Он может быть получен
нагреванием глицерина с муравьиной или щавелевой кислотой. При
этом промежуточно образуется сложный эфир, который при
нагревании разлагается с выделением углекислого газа:
сн2он ен2он сн2он
сно ;н но;—со .—> сн—о—со —> сн + соа
СН2О;Н НО|—СО СН2-О—СО СН2 + СО3
В технике аллиловый спирт может быть получен частичным
гидрированием акролеина
,0
СН2=СН—C<f + Н2 * СН2=СН—СН2ОН
\н
или омылением хлористого аллила водным раствором щелочи:
СН2=СН-СН2С1 + NaOH > СН2=СН-СН2ОН + NaCl
Химические реакции аллилового спирта обусловливаются как
присутствием гидроксила, так и наличием в его молекуле двойной
связи.
Так, при действии на аллиловый спирт треххлористого или
трехбромистого фосфора получаются соответственно хлористый
аллил СН2=СН—СН2С1 или бромистый аллил СН2=СН—СН2Вг.
Присутствие двойной связи обусловливает, например, реакции
полимеризации.
бе. Пластические материалы на основе полимеров винилового спирта 167
66. Синтетические пластические материалы на основе полимеров
винилового спирта и его производных. Хотя виниловый спирт крайне неустойчив, его
производные сравнительно легко образуют полимеры, которые широко
используются в промышленности.
В технике поливиниловый спирт получают омылением (гидролизом)
сложного эфира поливинилового спирта—поливинилацетата в спиртовом растворе.
Омыление можно вести при помощи кислот (НО, H2SO4) или щелочей:
• •—СНа—СН СНа—СН с2н5он
О—СО—СН3 О—СО—СН3 нсГ~
пол и ви н и л ацетат
¦ • • -СНа-СН—СН2-СН Ь nCHsCOOQHs + mCH3COOH
ОН ОН
поливиниловый спирт
По мере омыления полимер теряет свою растворимость в спирте и осаж-
іается, в зависимости от условий реакции, в виде тонкого бесцветного порош-
<а, нитей или гелеобразной массы. Поливиниловый спирт не пахнет, не ядо-
шт, растворим в воде, в кислотах, в глицерине, гликоле и не растворим в одно-
ітомньїх спиртах, кетонах, эфирах. Он является одним из наиболее водорас«
воримых полимеров.
Поливиниловый спирт благодаря его способности растворяться в воде
іспользован в медицине для изготовления нитей для зашивания ран. После
аживания раны такие нити постепенно рассасываются организмом. Продукты
занмодействия поливинилового спирта с альдегидами обладают высокой
клеящей способностью и нашли широкое применение как непосредственно в виде
:леев, так и в виде электроизоляционных эмалей, отделочных препаратов и др.
1ри взаимодействии поливинилового спирта с масляным альдегидом образует-
я поливинилбуспираль:
—СН—СНа— СН
он он
¦ Чс—сн2—с
снг—сн3 -
ОН ОН иасляный альдегид
СНа—СН—СНа—СН
поливиниловый спирт
СНз-CH-CHj-CH—СНа—СН—СН2—СН
О—СН—О О ОН
> СН3—СН2-СНз СН—СНа-СН3—СН3
он он о он
СН2-СН—СН3—СН-СНз-СН—СН,-СН
поливинилбутираль
168 Глава IV. Спирты и их производные
На основе поливинилбутираля изготовляют универсальные клеи БФ и
прокладочную пленку для безосколочных автомобильных и авиационных
стекол.
МНОГОАТОМНЫЕ СПИРТЫ
Двухатомные спирты или гликоли
Если в предельных углеводородах заместить два атома
водорода на гидроксильные группы, получим двухатомные спирты,
или гликоли.
67. Строение, изомерия и номенклатура гликолей. Первый
представитель этой группы двухатомных спиртов—гликоль—
был получен в 1856 г. А. Вюрцем путем гидролиза дихлорэтана.
Химическое строение этого соединения отвечает следующей
формуле:
Н2С СН2
или СН2(ОН)-СН2(ОН)
ОН ОН
В 1861 г. Бутлеров пытался получить простейший
двухатомный алкоголь гидролизом йодистого метилена:
.ОН
CH2J, + 2Н2О * 2HJ + Н2С<
Х)Н
Однако оказалось, что в свободном состоянии такие гликоли
не существуют. В момент своего образования они теряют воду и
превращаются в альдегиды или кетоны:
СН2(ОН)а » Н2О + НСНО
муравьиный
альдегид
Это свойство является общим для всех органических
соединений: атом углерода может удерживать только один гидроксил,
вещества же с двумя гидроксилами у одного углеродного атома
очень неустойчивы и распадаются с выделением воды.
Введением в молекулу пропана СН3—СН2—СНЭ двух гидрок-
силов получаем два изомера двухатомных спиртов:
СНз—СН—CHq СН2—СН2—СН2
он он он он
пропиленгликоль триметнленгликоль
Приведенные формулы строения подтверждаются получением
этих гликолей из таких галоидпроизводных, в которых атомы га-
68. Физические и химические свойства гликолей 169
лоида находятся у различных атомов углеро/а. Так, при
кипячении бромистого триметилена с водой в присутствии поташа и соды
получается триметиленгликоль:
CI-L—Вг СНа—ОН
СН2 -+- 2Н.,О » СНа + 2НВг
СНа-Вг СН2-ОН
Так как первоначально большинство двухатомных алкоголен
получали гидролизом дигалоидных соединений, образующихся
из олефинов, то этим гликолям были даны названия по
непредельным углеводородам: этиленгликоль, изобутиленгликоль. По
женевской номенклатуре наименования двухатомных спиртов строят
так же, как и для одноатомных спиртов, но в окончании
указывают «диол», а также места гидрокснлов:
СН3
н2с—сн2—сна сн3—с—сна
он он он он
пропандиол-1,3 2-метилпропандиол-1,2
(изобутиленгликоль)
68. Физические и химические свойства гликолей. Гликоли—
большей частью густые, бесцветные, труднолетучие жидкости.
Смешиваются с водой и спиртом. Большинство гликолей обладает
сладким вкусом, некоторые горьковаты.
Гликоли кипят при более высокой температуре и имеют
плотность большую, чем соответствующие им одноатомные спирты.
Так, этиловый спирт кипит при 78 °С и имеет плотность
0,806 г/см3, а температура кипения этиленгликоля 197 °С и
плотность 1,109 г/см3.
Гликоли дают все реакции одноатомных спиртов, с тем лишь
различием, что в реакцию может быть введена либо одна, либо обе
спиртовые группы. Поэтому для гликолей известны два ряда
производных—полные и неполные.
Так, при действии на гликоль металлического натрия
получаются гликоляты:
CHa(OH)—CH2(ONa) и CH2(ONa)—CH2(ONa)
Следует отметить, что в гликолях водород замещается легче, чем
в спиртах.
Точно так же, как в одноатомных спиртах, гидроксил в
гликолях можно заместить галоидом.
170 * ^ Глава IV. Спирты и их производные
При замещении гидроксила в гликоле на атом хлора получают;
СН2(ОН)—СН2С1 и СН2С1—СН2С1
этиленхлоргидрин хлористый этилен
Как было показано А. Е. Фаворским, при нагревании
этиленгликоля с малыми количествами серной кислоты происходит
выделение двух молекул воды из двух молекул гликоля и образуется
циклический эфир—диоксан:
О
/
СН2ОН НОСН2
сн2он носн2
диэтиленовый эфир
(!,4-диоксан)
Диоксан—жидкость, кипящая пр« 101,2 °С. Обладает высокой
растворяющей способностью. Хорошо смешивается с водой и
углеводородами. Практическое применение в качестве
растворителя ограниченно вследствие токсичности.
Этиленгликоль СН2—СН2 широко применяется в технике как
ОН ОН
заменитель глицерина, особенно для приготовления так
называемых «антифризов». Антифризы—вещества, добавляемые к воде
для предупреждения ее замерзания. Их применяют в зимнее
время для охлаждения автомобильных моторов, стволов
пулеметов и др.
Этиленгликоль применяется также в качестве составной части
жидкостей для тормозных гидравлических приспособлений в
артиллерийских орудиях и для получения различных
синтетических материалов на основе полиэфирных смол (см. стр. 474).
Эфиры этиленгликоля широко применяются как растворители
в производстве лаков.
Окись этилена. Действуя спиртовым раствором едкого кали
на этиленхлоргидрин, Вюрц в 1858 г. получил окись этилена:
СН2—О:н ! СН2Ч
і j + КОН! > | >О + KCI + НаО
СН2—|С1 і СН/
В промышленности окись этилена получают окислением
этилена и других низших олефинов воздухом п и 150—350 °С в газо-
69. Глицерин 171
вой фазе над катализатором (металлическое серебро):
2СН.2=СН2 + Оа
2
О
Окись этилена—газ, который сгущается в жидкость, кипящую
при +10,7 °С. При нагревании с водой окись этилена дает этилен-
гликоль. Она является изомером уксусного альдегида.
Окись этилена—чрезвычайно реакционноспособное
соединение, широко используемое для лабораторных и промышленных
синтезов. Так, окись этилена присоединяет при нагревании в
присутствии небольшого количества серной кислоты (катализатор)
спирты, образуя неполные простые эфиры гликоля:
СН2Ч О—С2Н5 СН2-О-С2Н6
I No + | —» [
сн/ н сн2—он
Такие моноэфиры, а также смешанные эфиры гликоля,
называемые целлозольвами, служат в качестве растворителей в
производстве нитратцеллюлозных лаков.
Важную роль играют продукты присоединения нескольких
молекул окиси этилена к алифатическим и ароматическим
спиртам. Эти соединения нашли широкое применение в технике как
поверхностно-активные вещества, на основе которых получаются
так называемые неионогенные моющие средства, не теряющие
моющего действия даже в морской воде.
Трехатомные спирты, или глицерины
Трехатомные спирты содержат три гидроксильные группы.
Они называются также глицеринами. Из них совершенно
исключительное значение имеет первый представитель этой группы, "ha-
зываемый просто глицерином.
69. Глицерин. Строение глицерина С3Н8О3 доказывается тем,
что он образует три ряда металлических производных (глицера-
тов) и в нем можно три гидроксила заместить галоидом.
Следовательно, в его молекуле находятся три гидроксила: С3Н5(ОНK.
Так как каждый атом углерода может удерживать только один
гидроксил, то строение глицерина должно выражаться формулой:
С.ГІ2—О͗ѫò2
он он он
179 Глава IV. Спирты и их производные
Таким образом, глицерин—это производное пропана, в
котором трп атома водорода замещены тремя гидрокснламп. По
женевской номенклатуре его называют пропантриол-] ,2,3.
Глицерин был открыт в ]783 г. Шееле, который нашел, что при
обработке оливкового масла окисью свинца получается
своеобразное сладкое вещество. Он показал, что это же вещество
получается и из других жирных масел, а также из твердых жиров.
Глицерин получают из жиров (на стеариновых, а иногда и
мыловаренных заводах) и брожением сахаристых веществ в
присутствии сернистокислого натрия (стр. 337). В последнее время
его получают также синтетическим путем; исходным сырьем
служит пропилен из газов крекинга нефти.
Глицерин—густая сиропообразная жидкость сладкого вкуса,
плотность 1,26 г/см3. Он кипит при 290 °С, смешивается с водой
во всех отношениях, весьма гигроскопичен.
Для получения кристаллического глицерина его охлаждают
до 0 °С и вносят в него для затравки кристаллик глицерина;
плавится глицерин при +17 °С.
Гигроскопичность глицерина позволяет предохранять
смазанные им предметы от высыхания. Поэтому глицерин применяется
в косметике, в кожевенной промышленности, в текстильной
промышленности при отделке тканей для придания им мягкости и
эластичности. Значительное количество глицерина используется
в пищевой промышленности, а также для приготовления
антифризов.
Глицерин в больших количествах расходуется для получения
нитроглицерина (см. стр. 175) и пленкообразующих лаковых,
так называемых алкидных смол (см. стр. 477), широко
применяемых для окраски станков, машин, вагонов и проч.
70. Спирты высшей атомности. Существуют спирты с еще
большим числом гидроксилов в молекуле. Таков четырехатомный
спирт эритрит С^^ОН)^ известный в виде четырех стереоизо-
меров, различные пентиты С5Н7(ОНM и гекситы СвН8(ОН)в,
образующие большое число стереоизомеров.
Некоторые из них (?)-сорбит, ?)-маннит и дульцит) описаны в
главе об углеводах.
ЭФИРЫ
Замещая в молекуле спирта атом водорода в водном остатке на
радикал R', получаем различные эфиры:
о.
спирты вфиры
71. Получение и свойства сложных яфиров неорганических кислот 173
В зависимости от характера радикала R' различают три типа
эфиров:
1) простые эфиры (R'—углеводородный радикал);
2) сложные эфиры неорганических кислот (R'—остаток
неорганической кислоты: NO2—азотной, N0—азотистой,
SO3H—серной и т. п.);
3) сложные эфиры органических кислот (R'—остаток
органической кислоты; R—во вс:х случаях углеводородный
радикал):
o<
^¦углеводородный
радикал
простой эфир
/
^остаток
неорганической
кислоты
сложный эфир
неорганической кислоты
0/R
достаток
органической
кислоты
сложный эфир
органической кислоты
Сложные эфиры неорганических кислот
'71. Получение и свойства сложных эфиров неорганических
кислот. Спирты способны вступать в реакцию с кислотами;
кислотный водород замещается радикалом спирта:
С2Н5— ОН + Hi—О—NO2 * С,Н.5—О—NO2 + Н2О
Получающиеся при этом вещества называются сложными эфи-
рами. Примерами могут сужить:
О
C2H5-O-N=O
зтилниірит їтилнитрат
СНз-0 /0 CH3-°\s/°
н—о^ ^о сн3~о/
метилсульфат днметилсульфат
(метилсерная кислота)
Сложные эфиры можно получать и другими способами,
например действием солей на галоидные ал килы:
С2Н5—!J + Ag:—ONO > C2H5—ONO + AgJ
Реакция образования эфиров взаимодействием спирта с
кислотой (этерификация) является равновесной и обратимой.
174 Глава IV. Спирты и их производные
В качестве водуотнимающего средства обычно применяют
концентрированную серную кислоту, а получающийся сложный эфир
отгоняют из реакционной смеси.
Если эфир не содержит свободных кислотных или гидроксиль-
ных водородов, то такой эфир называется полным; если часть
гидроксильных групп осталась неэтерифицированной, эфир
называют неполным; при наличии в молекуле эфира свободного
кислотного водорода его называют кислым эфиром.
Например, серная кислота образует два ряда эфиров:
.OR Очч /OR
О^
Азотная кислота кислых эфиров не образует, но с
многоатомными спиртами может давать неполные эфиры.
Фосфорная кислота может образовать три ряда эфиров:
,OR .OR /OR
О=Р—ОН О=Р—OR O=P—OR
Некоторые эфиры фосфорной и тиофосфорной кислот исполь-
вуются как инсектициды (см. стр. 130).
Полные сложные эфиры неорганических кислот для первых
членов ряда спиртов—летучие жидкости с приятным запахом,
не смешивающиеся с водой. При кипячении эфиров с водными
растворами щелочей происходит их расщепление до спирта и
соли кислоты:
ЭСН3 NaOH О. /ОСН3
¦ сн3он
О. УОСН3 NaOH О. ,ONa
>s< * >s< +сн3он
О^ NONa O^ NDNfa
По формулам строения сложные эфиры похожи на соли.
Однако по свойствам они сильно отличаются от последних. Соли,
как правило,—твердые нелетучие вещества, растворимые в воде
и нерастворимые в органических растворителях. Эфиры
же—обычно жидки, летучи, мало растворимы в воде и хорошо растворяются
в органических растворителях. Это различие объясняется тем,
что соли представляют собой соединения ионного типа, в то
время как эфиры построены гомеополярно. Например, водный
72. Нитроглицерин 175
раствор бисульфата натрия полностью подобен смеси
сульфата натрия и серной кислоты:
/ОН ,ONa
O2S< + O2S< = 2SOJ - + 2Na+ + 2H+
X)H NDNa
тогда как в растворах эфиров алкильные радикалы не
образуют ионов. Так, например, кислый эфир серной кислоты
СНдО—SO2OH это вещество, не соответствующее по свойствам
смеси серной кислоты и диметилсульфата:
/
O2S<
ОН /ОСН3 .ОСН.,
O2S< = 2Н+ + SOI - + OaS<
хосн3 хосна
Димепгилсульфапг (CHaOJSO2, или (CHaJSO4—жидкость
характерного запаха, темп. кип. 188 °С; плотность 1,35 г/см3. Часто
применяется при химических синтезах для введения группы СНа.
Работа с диметилсульфатом требует ряда предосторожностей,
так как пары его очень ядовиты.
72. Нитроглицерин. Нитроглицерин CaH5(ONO2K—сложный
эфир глицерина и азотной кислоты—получается действием на
глицерин смеси азотной и серной кислот:
СН2—ОН Н—О—NO2 СНа—О—NOa
I H2so4 I
СН—ОН + Н—О—NO2 > СН—О—NOa + ЗНаО
СН2—ОН Н—О—NOa СНа—О—NO2
нитроглицерин
Серная кислота служит для связывания воды, выделяющейся при
реакции глицерина с азотной кислотой.
Нитроглицерин—густая маслообразная жидкость,
застывающая на холоду. Нитроглицерин—одно из сильнейших
взрывчатых веществ. Он взрывает от удара, сотрясения, взрыва запала
гремучей ртути и в результате саморазложения. Пары его весьма
ядовиты. В чистом виде как взрывчатое вещество нитроглицерин
не применяют. Он служит для изготовления нитроглицериновых
порохов и динамитов.
Динамиты представляют собой порошкообразные,
пластичные или желатинообразные взрывчатые вещества, содержащие
в качестве действующей части нитроглицерин. Получают их
пропиткой нитроглицерином различных поглотителей, т. е. веществ,
впитывающих нитроглицерин и образующих с ним твердую
массу. В качестве поглотителей применяют древесную муку, нитрат
176 Глава IV. Спирты и их производные
целлюлозы, инфузорную землю, селитру и т. д. Динамиты более
устойчивы и более безопасны в обращении, чем нитроглицерин;
они гораздо менее чувствительны к ударам и сотрясениям.
Динамиты находят широкое применение в современной технике.
Причина неустойчивости нитроглицерина объясняется
строением его молекулы. В молекуле нитроглицерина, кроме атомов
углерода и водорода, содержатся атомы кислорода в количестве,
достаточном для того, чтобы сжечь все атомы углерода в
углекислый газ, а атомы водорода—в воду. Препятствием к этому
служат входящие в состав молекулы нитроглицерина атомы азота,
которые как бы барьером отделяют атомы кислорода от атомов
углерода и водорода и сами стремятся соединиться друг с другом
в прочные молекулы. Достаточно, однако, малейшей причины
(например, легкого сотрясения), чтобы произошла перегруппировка
атомов с выделением углекислого газа, воды, кислорода, азота:
4C3H5(ONO2K * 12СО-, + ЮН2О + 6Na + О2
нитроглицерин
При этой реакции выделяется громадное количество тепла.
Эта перегруппировка мгновенно охватывает всю массу
нитроглицерина, и в результате выделения большого количества газов
происходит взрыв колоссальной силы.
За последнее время наряду с нитроглицерином большое
значение в качестве взрывчатых веществ приобрели нитрогликоли,
т. е. сложные эфиры гликолей и азотной кислоты:
СН3
CHa-O-NOa . СН-О—NOa
СН2—О—NOa СНа—О—NO2
нитроэтиленгликоль нитропропилентликоль
73. Эфиры ортокремневой кислоты. При взаимодействии
спирта н хлористого кремния образуются эфиры ортокремневой
кислоты:
4СН3—ОН + SiCl4 > Si(OCHaL + 4НС1
4QH5—ОН + SiCi4 > Si(OCaH6L + 4HC1
Аналогичным образом получают эфиры пропилового,
бутилового и изобутилового спиртов.
Эфиры ортокремневой кислоты нашли широкое практическое
применение.
74. Строение и способы получений простых эфиров УП
Как показал К. А. Андрианов, гидролиз эфиров ортокремне-
вой кислоты сопровождается поликонденсацией, приводящей к
образованию технически ценных полимеров:
Si(OCH3L + 2НОН » (CHaO^SiCOH^ + 2СН3ОН
ОСНЭ
—Si—
НО—Si—О—
осн.
¦ ОСН3 "
—Si—О—
. осн, .
ОСНэ
Si-OH + (п — l)HaO
n—i OCH3
Подобным же образом ведут себя эфиры алкилкремневых кислот, которые
получаются в промышленных условиях из алкилхлоридов и эфиров орто
кремневой кислоты по реакции Гриньяра
СН3С1 + Mg » CH3MgCl
Si(OCaH5L + CH3MgCl » CH3Si(OCaH,)a + QjH5OMgCl
нли непосредственным действием смеси алкилхлорида и эфира ортокремневой
кислоты на металлический магний:
Mg + СН3СІ + Si(OQjH6L * CH3Si(OC2H5K + C2H5OMgCl
Простые эфиры
74. Строение и способы получения простых эфиров. Простые
эфиры, примером которых может служить хорошо всем известный
этиловый (серный) эфир С4Н10О, изомерны со спиртами. Так,
этиловый эфир имеет тот же состав, что и бутиловый спирт;
метиловый эфир С2Н6О является изомером этилового спирта.
Строение этих веществ становится ясным при рассмотрении
способа их получения из алкоголятов и галоидных алкилов
(синтез Вильямсона):
СаН5—О—!_№_+JI—Q)H6 * СаНй—О—QH6 + NaJ
этиловый эфир
Из этого синтеза следует, что в простых эфирах два
углеводородных радикала связаны атомом кислорода.
Названия эфиров составляются по названию радикалов:
СН3—О—СН3 метиловый (диметиловый) эфир
СаН5—О—С2Н6 этиловый (днэтнловый) эфир
СНЭ—О—СаН, метилпропиловый эфир
—О—С3Н7 этилпропиловый эфир
В том случае, когда оба радикала различны, простые эфиры
называются смешанными .эфирами.
Некоторые простые эфиры получаются нагреванием спиртов
с концентрированной серной кислотой. Так получается,
например, имеющий наибольшее техническое значение этиловый эфир.
12—7*88
178 Глава IV. Спирты и их производные
Сначала из спирта и серной кислоты образуется этилсерная
кислота
О О
но—S—о—;н"+" Щ—CjHs —»H0-S-0-QH5 +
II
II II
о о
этилсериая кислота
т. е. продукт замещения одного атома водорода в молекуле
серной кислоты этилом.
В дальнейшем этилсерная кислота реагирует со второй
молекулой спирта:
О
II
НО—S—О—{СІННЕ QH5-O]— Н * СаНб-О—QHb + HaSO4
II
О
Конечный итог реакции выражается уравнением:
2СаН6ОН > QHj-O-QiHs + НаО
Для получения этилового эфира широко используют также и
другой способ—пропускание паров этилового спирта при 240—
260 °С над окисью алюминия, служащей катализатором.
Молекулы даже вполне симметричных простых эфиров имеют
дипольный момент. Это, как и для воды, объясняется тем, что
связи атомов углерода с атомами кислорода образуют между собой
некоторый угол:
(-м-)
(+) О (+)
R/\R
75. Свойства простых эфиров. Низшие представители простых
эфиров кипят при более низкой температуре, чем соответствующие
им спирты:
Температура Температура
Спирты кипении, °С Эфиры кипения, °С
СН3ОН +65 (СН3JО —23,6
QHSOH +78 (QHB)aO +35,6
QHjOH +97 (С3Н,JО +90,7
Плотности эфиров также ниже плотностей
соответствующих спиртов, например: плотность С2НЬОН равна 0,790 г/см*;
плотность (С2Н6JО равна 0,714 г/см3.
Эфиры метиловый и метилэтиловый—при обычной
температуре газообразны, эфиры большего молекулярного
веса—легколетучие жидкости; начиная с (С17.Н38JО—это твердые тела.
76. Этиловый эфир 179
Эфиры—нереакционноспособные вещества. Они не
изменяются при нагревании с водой, щелочам^ и разбавленными
кислотами. В отличие от спиртов, при взаимодействии с металлическим
натрием они не выделяют водорода.
Своеобразно отношение простых эфиров к крепкой соляной
и бромистоводородной кислотам: эфиры растворяются в них с
выделением тепла. Было показано, что при этом образуются
непрочные соединения солеобразного характера—оксониевые
соединения (по аналогии с аммониевыми соединениями). По
электронной теории образование оксониевых соединений можно
представить следующим образом.
У эфирного атома кислорода имеются две пары свободных
электронов; положительно заряженный ион водорода соляной
кислоты, лишенный электрона, может воспользоваться свободной
парой электронов кислорода, образуя непрочный оксониевый
положительно заряженный ион:
СП
з
сн
3
4«?:]'
О '. -г п і; і л: і у : О ІН
сн.
сн.
['-«]¦
При разбавлении водой оксониевых соединений происходит
вытеснение молекулы эфира молекулами воды; это приводит к
разрушению и разложению оксониевых соединений.
При насыщении простых эфиров йодистым водородом, даже
при обыкновенной температуре, происходит расщепление
молекулы эфира по кислородной связи:
СаН6—О—С3Н6 + HJ » QaH6J + С2Н6ОН
При длительном хранении на воздухе и особенно на свету
простые эфиры окисляются, при этом образуются гидроперекиси
эфиров:
СНСНОС2Н6
о—он
этиловый эфир гидроперекись этилового
эфира
Гидроперекись этилового эфира неустойчивая жидкость. При
нагревании разрушается со взрывом.
76. Этиловый эфир. Этиловый эфир (С2Н5JО называется иногда
обыкновенным, или серным*, эфиром. Это подвижная бесцвет-
* Название «серный» сохранилось за этиловым эфиром с тех пор, когда
единственным известным способом получения его было взаимодействие спирта
с серной кислотой.
180 Глава IV. Спирты и их производные
ная жидкость, кипящая при 35,6 °С и замерзающая при —117,6 °С;
плотил :ть 0,714 г/см3. Эфир мало растворяется в воде:
1 л воды растворяет при 16 "С примерно 100 мл эфира; 1 л эфира
растворяет около 30 мл воды. Эфир чрезвычайно летуч и очень
легко воспламеняется. Пары эфира примерно в 2,5 раза тяжелее
воздуха и образуют с ним взрывчатые смеси. При работе с
эфиром в лаборатории надо всегда помнить, что тяжелые пары эфира
могут далеко распространяться по поверхности стола и по полу
и загореться от ближайшей горелки или электроплитки.
Хранят эфир в толстостенных, хорошо закупоренных сосудах, так
как даже незначительное повышение температуры сильно
увеличивает упругость его паров.
Эфир служит для наркоза и как анестезирующее средство.
Значительные его количества используются в производстве
бездымного пороха и коллодия. Являясь превосходным
растворителем, эфир широко применяется в лабораториях для извлечения
и кристаллизации органических веществ. Применение эфира как
растворителя в заводской практике ограничено из-за его крайней
огнеопасности.
Для проведения многих химических реакции (реакция Вюрца, магний-
органические синтезы) в качестве растворителя применяется безводный, так
называемый «абсолютный» эфир. Его готовят из продажного эфира
обработкой сернокислым железом для удаления перекисей и гидроперекисей (см.
стр. 181), отмывая водой от примеси спирта, высушивая плавленым
хлористым кальцием н перегоняя со стружками (или проволокой) металлического
натрия.
77. Органические перекисные соединения. Как было указано (см. стр. 132),
спирты можно рассматривать как производные воды; подобно этому
органические перекисные соединения можно произвести от перекиси водорода:
О—Н О—Alk О—Alk
I 1-І
О—Н О—Н О—Alk
перекись органические органические
водорода гидроперекиси перекиси
Гидроперекись метила получают действием на перекись водорода диметил-
сульфатом:
Н—О—О—Н + СН3—О—SOa—ОСНз » СН3—О—О—Н + СН3—О—SO2—ОН
гидроперекись метила
Гидроперекиси метила н этила—малоустойчивые соединения, сильно
взрывают. Более устойчивы гидроперекиси, содержащие вторичные радикалы,
например гидроперекись изопропила (СН3JСН—О—О—Н и гидроперекись
/npem-бутила (СН3KС—О—ОН, Эти жидкости можно перегонять (оии не
взрывают).
Все гидроперекиси быстро реагируют с йодистым водородом, выделяя иод.
СН3—О—О—Н + HJ > Ja + Н.2О + СН3ОН
На этом основано качественное и количественное нх определение.
П. Органические переписные соединения 181
При нагревании ги дроперекиси образуют свободные радикалы (см.
стр. 54):
СН3 СНа
СН3—С—О-О-Н * СНа-С-О. + »О—Н
I I
СН3 СНз
Эти свободные радикалы вызывают (инициируют) цепные реакции. Так,
например, 60%-ный раствор гидроперекиси трет -бутил а в триметилкарбиноле
является инициатором реакции полимеризации непредельных соединений
(см. стр. 88).
Перекиси алкилов получают действием двух молекул диметилсульфата на
перекись водорода:
Н—О—О—Н + 2СН3О—SOa—ОСН3 * СН3—О—О—СН3 + 2CH3OSOaH
перекись метила
или взаимодействием соответствующей гидроперекиси и диалкнлсульфата:
СН3
СНз—С-О—О-Н + CHaO-SOa-OCHa *
СН3
СНз
I
* СН3—С—О—О—СН3 + НО—SOa-OCH3
Так получают перекиси с различными алкилами. Перекись метила—газ,
конденсируется а жидкость при +13,5 °С Перекись этила кипит при 64 °С.
Перекись mpem-бутила—маслянистая жидкость, темп. кип. 109 °С. Перекиси
труднее реагируют с HJ, чем гидроперекиси. Перекиси легко распадаются.
Конечными продуктами распада перекиси mpem-бутила являются ацетон
и этан:
СНз СНз СНз СНз
СНз-С-О-0—С—СНз * СНз—С—О» + »О—С—СН3 *
II II
СНа СНз СНз СНЭ
СН3
* 2СНа-С=О + СНз—СН3
Перекисные соединения, так же как и ранее указанные гидроперекиси,
находят практическое применение в качестве инициаторов в процессах
полимеризации.
ГЛАВА V
МЕРКАПТАНЫ, СУЛЬФОКИСЛОТЫ, ТИОЭФИРЫ
Спирты и простые эфиры можно рассматривать как
производные воды, в молекуле которой один или оба атома водорода
замещены углеводородными радикалами. Подобно этому
меркаптаны и тиоэфиры можно рассматривать как производные
сероводорода, в молекуле которого один или оба атома водорода
замещены углеводородными радикалами:
Н-0—Н
вода
Н—S—Н
сероводород
R-O-H
спирт
R-S—H
меркаптан
R—O-R
простой
эфир
R-S-R
тноэфир
78. Меркаптаны. Подобно получению спиртов действием
щелочей на галоидные алкилы
C2H5J + КОН » С2Н5ОН -1- KJ
можно получить и меркаптаны, называемые также тиоспиртами.
Для этого на галоидные алкилы действуют гидросульфидом калия:
СН3—1 + К—S—Н » СНз-S—н + KJ
метилмеркаптан
Спирты—производные воды, меркаптаны—производные
сероводорода: вода—нейтральное соединение, сероводород—слабая
кислота. В соответствии с этим спирты не обладают ни
кислотными, ни основными свойствами. Меркаптаны обладают слабыми
кислотными свойствами и образуют металлические производные
(меркаптиды) не только при действии щелочей, но и при действии
окисей тяжелых металлов. Особенно характерны для них ртутные
соединения:
2СаН5—S—Н + HgO » (QH5SJHg -f Н2О
этилмеркаптан
Отсюда и происходит название меркаптанов: corpus mercurio
aptum—тело, забирающее ртуть.
79. Сульфокислоти 18$
Температуры кипения меркаптанов нвдке температур кипения
соответствующих спиртов. Так, метилмеркаптан кипит при 6 °С,
а метиловый спирт—при 65 °С. Меркаптаны обладают
отвратительным, чрезвычайно сильным запахом.
79. Сульфокислоти. При окислении меркаптанов азотной
кислотой они переходят в сульфокислоти:
2СНЭ—SH + ЗО2 і 2СНЭ—SO2OH
метилсульфо-
кислота
Сульфокислоты содержат остаток серной кислоты—сульфо-
группу—SO2OH, атом серы которой связан с углеродом.
Сульфокислоты легко растворимы в воде и являются очень сильными
кислотами.
При действии хлористого фосфора на сульфокислоты
получаются хмрангидриды сульфокислот, или сульфохлориды:
СНэ—SO3—ОН + РС16 * СНЭ—SO2—Сі + РОСІз + HQ
При действии воды сульфохлориды превращаются в
сульфокислоты:
СНЭ—SO„—СІ + НОН »¦ СНЭ—SOa—ОН + HCI
Сульфокислоты при взаимодействии с едким натром или содой
образуют легкорастворимые в воде соли—сульфонаты:
R—СН2—SO2—ОН + NaOH » R—СН2—SO2—ONa + Н2О
Алифатические сульфокислоты находят применение в технике
для получения поверхностно-активных веществ. Так, методом
фотохимического сульфохлорирования (см. стр. 56)
углеводородов с цепью из 10—12 углеродных атомов получают хлорангид-
риды алкилсульфокислот; при нейтрализации этих хлорангид-
ридов щелочами получают растворимые в воде алкилсульфонаты,
которые нашли применение в качестве моющих средств.
80.- Тиоэфиры. Строение тиоэфиров вытекает из способа их
получения взаимодействием галоидных алкилов и сернистого
калия:
СНэ~Ч ± К;—S—|К +.Ji—Щ, * СНэ—S—СНЭ + 2KJ
днметилсульфид
Низшие тиоэфиры—нерастворимые в воде жидкости.
Подобно солям сероводорода их называют сульфидами.
184 Глава V. Меркаптаны, сульфокислоти, тиоэфиры
Из тиоэфиров широкую известность приобрел в войну 1914—
1918 гг. ?,?/-ДlИxлopдиэтилcyльфид:
уСНо—СН2—С1
к
ХСН2-СН2-С1
иприт
получивший название иприта*, или горчичного газа
(неочищенный иприт имеет слабый запах горчицы).
Иприт может быть получен действием этилена на однохлори-
стую серу:
yCHaCHgCl
S2C12 + 2СН2=СН2 > S< + S
NCH2CH2C1
Иприт кипит при 217 °С, чистый иприт плавится при 14 °С,
неочищенный—много ниже. Иприт—отравляющее вещество
нарывного действия. Попадая на кожу, он вызывает образование
трудно заживающих язв. Пары его действуют на слизистые
оболочки, дыхательные пути и кожу человека и животных.
Иприт относится к стойким отравляющим веществам.
Средством индивидуальной защиты от иприта служат
противогаз и защитная одежда, средством дегазации—хлорная известь,
которая одновременно и хлорирует и окисляет иприт, в результате
чего образуются нетоксичные вещества.
* Иприт впервые был получен в 1886 г. Н. Д. Зелинским.
ГЛАВА VI
АЛЬДЕГИДЫ И КЕТОНЫ
81. Строение, изомерия и номенклатура. К классу альдегидов
и кетонов относятся соединения, в молекуле которых имеется
карбонильная (или оксо-) группа Ъс=О
¦Р „О
R-c/ R-c/
ХН NR'
альдегиды кетоны
Соединения этого класса называют также оксосоединениями.
Как было указано ранее (см. стр. 144), альдегиды и кетоны
получаются окислением соответственно первичных и вторичных
алкоголей без изменения углеродной цепи. При этом молекула
спирта теряет два атома водорода. Таким образом, общая
формула гомологического ряда для альдегидов и кетонов одна и та же:
С„Н2П+2О-2Н »С„Н2ПО
алкоголи альдегиды
и кетоны
Число изомерных альдегидов, очевидно, соответствует числу
первичных спиртов, а кетонов—числу вторичных спиртов с тем
же числом углеродных атомов. Третичные спирты не могут
превращаться в карбонильные соединения без расщепления
углеродного скелета.
Приведем некоторые методы расчета числа изомеров на примере альдегидов
в кетонов. Пусть нам известно число всех изомеров спиртов до Q:
с
С3. . .
. 1
1
. . 2
С4. . .
Q .
с6. . .
. . 4
. . 8
. . 1/
Отсюда легко показать, что число альдегидов (или, что то же
самое,—первичных спиртов) для С4 будет равно числу всех спиртов для С3; число альдегидов
186 Глава VI. Альдегиды и кетоны
Сб равно 4 (по числу спиртов С4) и т. д. В самом деле, можно мысленно заменить
.0
/о
группу ОН на—C<f !
NH
СН3—СНа—СНа—ОН СН3—СНа—СН3—СНО
(СН3)аСН—ОН (СН3)аСН—СНО
спирты Сз . альдегиды C^j
Несколько по-иному можно рассчитать число кетонов. Возьмем в качестве
примера расчет для кетонов QH^O.
Возможны следующие типы:
1) CV 1) Сач
>С0 >С0
4) С/ 2) С/
і и
Очевидно, что системе 1 отвечает 4 изомера Ax4), а системе II—2 изомера
AX2). Итого, в сумме 6.
Если п—число нечетное, расчет будет немного сложней. Например, для С,
1) С1Ч 1) С2Ч 2)
>С0 >С0
8)- С/ 4) С/ 2) О
I п Ш
Таким образом, для системы I будет 8 изомеров, для системы II—4 изомера.
Для системы III число изомеров будет ие 4 Bx2), а на один меньше, так как
здесь будет повторение одинаковых изомеров: норм.—норм.; изо—изо; норм.—
изо (и повторно нзо—норм.). Итого сумма всех изомеров кетонов С, равнаї
8+4 + 3= 15
Исторически названия альдегидов были связаны с
наименованиями кислот, в которыеэти альдегиды переходят при
окислении: муравьиный, уксусный, пропионовый, масляный ит. д.
альдегиды. Наименования кетонов обычно составляются из названий
радикалов, связанных карбонильной группой:
QHa—СО—QHs диэтилкетои
СН3—СНа—СН3—СО—СН(СН3J пропилизопропилкетон
По женевской номенклатуре наличие альдегидной группы
отмечается суффиксом аль (реже ал):
/Р
СН3—СН,—C<f пропаналь
ХН
,0
СН3—СН—C<f 2-метилпропаналь
I ХН
СН,
82. Получение и некоторые свойства альдегидов 187
Наличие кетонной группы обозначается суффиксом он:
СН3—CRj—CRj—СО—СН3 пентанон-2
СН3—СНа—СО—СН2—СНЭ пентанон-3
СН3—СО—СН—СН3 2-метилбутанон-З
СН3
В свойствах альдегидов и кетонов много общего. Поэтому
описание этих классов веществ удобно вести параллельно.
82. Получение и некоторые свойства альдегидов. Альдегиды
могут быть получены различными способами; из них укажем три.
1. Окисление первичных спиртов:
.О
R—СН,ОН + О * R—Cf + Н,0
Этот способ получения альдегидов объясняет происхождение
их названия. Слово «альдегид» составлено сокращением
латинских слов a/cohol dehydrogenatus, что в переводе на русский язык
означает спирт, лишенный водорода. Интересно отметить, что
превращение спирта в альдегид можно произвести и в отсутствие
кислорода, взбалтывая спирт с мелкораздробленным палладием,
который способен поглощать водород:
СН3-СН2ОН » СН3-С<^ + На
Отщепление водорода можно проводить, пропуская пары
спирта при повышенной температуре C00 °С) над медной или
серебряной сеткой в тугоплавкой трубке. Реакции отщепления водорода
называются дегидрированием.
2. Сухая перегонка смеси кальциевых и бариевых солей кар-
боновых кислот (см. стр. 227).
3. Нагревание с водой таких дигалоидных производных,
которые содержат оба атома галоида при одном и том же атоме
углерода, находящемся в конце цепи:
'ґ>л ті: ґ\і і
СНз—СНІ + 1 *
\|сі ні-он
/ОН"
СН3—СН
\он
—НгО
* СН
Вначале образуются вещества с двумя гидроксилами при
одном атоме углерода; их следует рассматривать как гидраты аль-
188
Глава VI. Альдегиды и кетоны
дегидов. Выделяя воду, они дают альдегиды. В водных растворах
альдегиды почти всегда частично существуют в виде таких
гидратов.
Простейшим из альдегидов является муравьиный альдегид—
газ с резким неприятным запахом. Средние
гомологи—жидкости—также имеют резкий неприятный запах. Более
высокомолекулярные (С10 и выше) альдегиды в разбавленном состоянии имеют
цветочные запахи; многие из них встречаются в натуральных
эфирных маслах цветов. Так, например, нониловый альдегид
С8Н17СНО содержится в розовом масле. Некоторые из веществ
этого класса применяются в парфюмерии.
Физические свойства простейших альдегидов приведены в
табл. 6.
Таблица 6
Физические свойства простейших альдегидов
Название
Формула
Относительная
плотность
Температура
¦ кипения
"С
Муравьиный альдегид .
Уксусный альдегид . .
Пропиоиовый альдегид
Нормальный масляный
альдегид
Изомасляный альдегид
Валериановый альдегид
Н-СНО
СН3—СНО
СН3СН2—СНО
СН3
На—СНО
ч
>СН—СНО
сн3/
СН3СН2СН2СН2—СНО
0,815
(прн-20 °С)
0,780
0,807
0,817
0,794
0,819
(при II °С)
—19,2
+20,8
+49,1
+75
+64
+ 102
Характерным свойством альдегидов является их способность
давать красное или фиолетовое окрашивание с фуксинсернистой
кислотой*.
При действии щелочей происходит осмоление альдегидов, и
они переходят в красно-желтые вещества—альдегидную смолу.
Муравьиный альдегид щелочами не осмоляется. Точно так же не
осмоляются щелочами и такие альдегиды, в которых соседний
* Фуксиисериистую кислоту получают, пропуская в ярко-красиый
раствор фуксина сернистый газ до полного обесцвечивания жидкости или же
взбалтывая очень разбавленный водный раствор фуксина с несколькими
каплями раствора кислого сернистокислого натрия.
83. Получение и некоторые свойства кетонов
.89
с карбонилом атом углерода не связан с водородом. Примером
таких альдегидов может служить триметилуксусний альдегид
83. Получение и некоторые свойства кетонов. Способы
получения кетонов аналогичны способам получения альдеіадов.
1. Окисление вторичных спиртов:
R-CH(OH)—R' + О
¦ R—СО—R' + Н2О
2. Сухая перегонка кальциевых и бариевых солей карбоновых
кислот (см. стр. 227).
3. Нагревание с водой дигалоидных производных,
содержащих оба атома галоида при одном и том же атоме углерода, не
стоящем в конце цепи:
СН3^ ;C! Hi-OH
с ! + і
сн/ \;сі ні—он
сн/ \он
-н2о
1_П3ч
с=о
сн/
Низшие и средние члены гомологического ряда
кетонов—жидкости. Некоторые из них обладают приятным запахом, другие
имеют неприятный прогорклый запах. Высшие кетоны—твердые
тела. В отличие от альдегидов, кетоны не вызывают окрашивания
фуксннсернистой кислоты.
Таблица 7
Физические свойства простейших кетонов
Название
Диметилкетон (ацетон) .
Метилэтилкетон ....
Диэтилкетоа
Метилпропилкетон . . .
Метилбутилкетон ....
Дипропилкетон
Диизобутилкетон ....
Формула
СН3—СО—СН3
СНз-СО—С2Н5
CoHj-CO-QHj
СНз—СО—С3Н,
СНз-СО—С4Н9
СЭН7-СО-С3Н7
СН3, СН3
СН—СН2—СО—СНа—СН
1 1
сн3 снэ
Относительная
плотность
rfjo
0,7980
0,8058
0,8138
0,8089
0,330
0,818
0,895
Температура
кипения
"С
56,1
79,6
101,8
100,8
127,7
144
166
190 Глава VI. Альдегиды и кетоны
РЕАКЦИИ АЛЬДЕГИДОВ И КЕТОНОВ
Альдегиды и кетоны, в особенности первые,—весьма реак-
ционноспособные соединения. Они способны к реакциям
присоединения, замещения и окисления.
84. Реакции присоединения. Реакции присоединения к
альдегидам и кетонам обусловлены тем, что двойная связь между
атомами кислорода и углерода в карбонильной группе легко
переходит в простую связь; вследствие этого у атомов углерода и
кислорода освобождается по единице валентности, которые и
насыщаются единицами валентности других атомов:
/0— R4 ,0—
R—С— и
\н R
1. Альдегиды и те из кетонов, которые содержат метальную
группу, связанную с карбонилом, т. е. группу СН3—С—, спо-
I!
О
собны присоединять кислый сернистокислый натрий. В
результате получаются так называемые бисульфитные
соединения.
В этих веществах углерод непосредственно связан с серой:
н і ; нх/он
С=0 + NaHSO3 » С
сн^ ; снз4/он
С=0 + NaHSO3 * С
СН,/ \ \ СИ,/ \sOoNa
Реакцию проводят, взбалтывая альдегид или кетон с
возможно более концентрированным раствором NaHSO3.
Бисульфитные соединения—кристаллические вещества,
расщепляющиеся при нагревании с раствором соды или
разбавленными кислотами с образованием альдегидов и кетонов. Они служат
для выделения альдегидов и кетонов из их смесей с другими
веществами и получения их в чистом виде.
84. Реакции присоединения 191
2. Альдегиды и кетоны способны присоединять водород;
альдегиды дают при этом иервичные спирты, кетоны—вторичные:
з-с/ + Н—Н > СНз-СНа-ОН
? н і
СНзХ І І СНзЧ
с=о + н—н —> сн—он
сн8/! і сн/
Эту реакцию можно осуществить, пропуская над
мелкораздробленным никелем смесь водорода с парами альдегида или ке-
тона или действуя водородом в момент выделения.
3. С аммиаком альдегиды образуют кристаллические
соединения—альдегидаммиаки:
СН3—СН— NH2 СН,—СН—NHa
II + I > I
о*- н он
Однако даже свежеприготовленный альдегидаммиак представляет
собой продукт полимеризации и имеет молекулярный вес,
в три раза больший, чем это следует из приведенной формулы,
которая выражает результат только первой фазы реакции.
При действии разбавленных кислот альдегидаммиаки дают
исходные альдегиды и аммонийные соли.
Кетоны с аммиаком дают продукты сложных превращений.
4. Альдегиды и кетоны способны присоединять магнийорга-
нические соединения. Так, при прибавлении уксусного альдегида
к эфирному раствору магнийиодметила выделяется объемистый
осадок продукта присоединения:
СНз—СН СНз СНз—СН—СНд
II + I > I
О MgJ OMgJ
Как видно из уравнения, радикал магнийорганического
соединения присоединяется к атому углерода, а остальная часть
молекулы—к атому кислорода. Реакция ведется в растворе
тщательно обезвоженного («абсолютного») эфира в приборе с
обратным холодильником. Полученный продукт присоединения
представляет собой алкоголят.
192 " Глава VI. Альдегиды и кетони'
При разложении его водным раствором кислоты получается
изопропиловый спирт:
%:н—О—fMgJ Т Н—<3|—Н * ^СН—ОН + MgJ(OH)
снэ/ - сн/
Эта реакция позволяет, исходя из альдегида, получить
вторичный спирт с большим числом атомов углерода в молекуле.
Если взять муравьиный "альдегид, то в результате реакции
образуется первичный спирт:
Н—с/ + СН3—СНа—Mg—Вг » СН3—СНа—СНа—О—MgBr
t ТІ і
СН3-СНа—СНа—О—;Mg—Br ± Н-0|—Н »
* СН3—СНа—СНа-ОН 4- MgBr(OH)
пропиловый спирт
В общем виде реакцию для альдегидов можно написать
следующим образом (R—радикал или, для муравьиного альдегида,
водород):
/Р R\
R—с/ 4- R'—Mg—J * >СН—О—MgJ
R4 R4
>СН—О—MgJ 4-Н—О—Н » >СН-ОН 4-MgJ(OH)
Совершенно аналогично идут реакции с кетонами:
: RN
R4 І і R\
\С=О + R"—Mg—J * R'—С—OMgJ
При разложении продуктов присоединения водой получаются
третичные спирты:
R\ R\
R'—С—О—jMgJ +Н—О:—Н * R'—С— ОН + MgJ(OH)
/ /
84. Реакции присоединения 193
Таким образом, с помощью магнийорганических соединений
можно получить первичные, вторичные и третичные спирты с
большим числом атомов углерода в молекуле, чем у исходных
альдегидов и кетонов:
муравьиный
альдегид
JRMgJ
R-CH2—ОН
первичный
спирт
Другие
альдегиды
R-C<(°
JR'MgJ
R4
>СН-ОН
R'/
вторичные
спирты
кетоны
R\
>с=о
Р' f
JR.MgJ
R\
R'-C-OH
R"/
третичные
спиргьі
Реакция получения спиртов при помощи металлорганических
соединений была открыта А. М. Зайцевым*. Он вместе с
сотрудниками в 1874 г. и в последующие годы синтезировал при помощи
цинкорганичсских соединений значительное число различных
спиртов.
В настоящее время эти синтезы проводят с помощью магний-
органическил соединений.
5. Альдегиды и кетоны присоединяют синильную кислоту с
образованием оксинитрилов:
R-CH CN R-CH-CN
II + I * I
ОН ОН
Оксинитрилы могут быть легко превращены в оксикислоты
см. стр. 282 ел.) и аминокислоты (см. стр. 373 ел.).
* Александр Михайлович Зайцев A841 —1910)—один из ближайших
учеников А. М. Бутлерова—родился, учился и работал в Казани. С 1871 г. до
конца своей жизни Александр Михайлович руководил кафедрой органической
химии Казанского университета.
Зайцев открыл путь превращения хлорангидридов кислот в
соответствующие спирты и получил нормальный бутиловый спирт—четвертый и последний
изомер бутанолов.
Большое значение имеют работы А. М. Зайцева по изучению отщепления
галоидоводородов от галоидных алкилов с образованием олефинов.
Продолжая начатые А. М. Бутлеровым синтезы с применением цинкорга-
нических соединений, А. М. Зайцев нашел простой и доступный метод получения
вторичных и третичных предельных и непредельных спиртов взаимодействием
йодистых алкилов, цинка и альдегидов или кетонов.
13-788
194 Глава VI. Альдегиды и кгтоны
6. Альдегиды весьма легко полимеризуются, т. е. молекулы
одного и того же альдегида соединяются друг с другом с
образованием более крупных молекул. В результате полимеризации
могут получаться: 1) вещества, молекулы которых можно расщепить
на молекулы исходного альдегида, и 2) вещества, не способные к
такому расщеплению (см. стр. 201). Кетоны менее
способны к полимеризации. Полимеризуются они только по второму
типу.
85. Реакции замещения. Из реакций замещения опишем те,
в результате которых происходит замещение атома кислорода
карбонильной группы.
1. С гидроксиламином NH2—ОН альдегиды и кетоны дают
окахм.ы:
Нч
)OH » CH3-CH=NOH + Н2О
;ОНч
i... + :)>N-
Н: НК
оксим
уксусного альдегида
сн.
¦О + !\n—OH > 3\c=NOH + H2O
:' Hi/ CH/
сн3^
оксим ацетона
2. При действии на альдегиды и кетоны гидразина NH2—NH2
получаются гидразоны'.
.10 Н!ч
+ і \N-NH2 > R-CH=N—NHa + H2o
H'L.H.r .
гидразон альдегида
: 'ці р
0+ f>N-NH2 * Nc=N—NH, + HaO
НК R'/
гидразон кстона
Гидразоны под действием твердых едких щелочей или алкого-
лятов разлагаются с выделением свободного азота и образованием
углеводородов (реакция Кижнера):
К R\
\C=N-NH2 » >СН2 +N2
R'/ R'/
85. Реакции замещения 195
Таким образом из альдегидов или кетонов можно получить
углеводороды:
\с=О » 4>C=N—NHa » Nch,
R#/ R/ R'x
альдегид гидразон углеводород
или кетон
Эта реакция была открыта в 1910 г. Н. М. Кижнером, который
производил разложение гидразонов, нагревая их в колбе с
небольшим количеством едкого кали (значительно лучше реакция
протекает в присутствии кусочков платинированной глины).
Полтора года спустя после опубликования первых работ Кижнера
об открытой им реакции немецкий ученый Вольф вновь описал
эту же реакцию. Отличие в методике у Вольфа заключается лишь
в том, что реакция разложения гидразонов ведется в запаянных
трубках.
3. Альдегиды, взаимодействуя с фенилгидразнном*,
образуют фенилгидразоны:
R—С/ + >N—NH—СвН5 . R—CH=N-NH—CgHj + Н,0
фенилгидразон
СН3
сн,
3\С=Ю+ i^N-NH-CgHa > 34>C=N-NH-Q,H6 + НаО
\/ І Ні/ . СИ/
фенилгидразон
ацетона
При действии на фенилгидразоны растворов кислот
происходит обратная реакция, и они, присоединяя воду, расщепляются
на фенилгидразин и альдегид или кетон:
R-C=N-NH-CeHa + Н2О » R—с/ + CeH5—NH—NH»
н
Аналогично гидролизуются и оксимы.
Оксимы и фенилгидразоны большей частью являются
кристаллическими веществами с характерными температурами плавле-
* Фенилгидразин является производным гидразина NH2—NHj, один из
атомов водорода которого замешен на радикал фенил СвН6.. Фенилгидразин
плавится при 20 °С. Он почти нерастворим в воде, но с соляной кислотой
образует растворимые соли.
13*
196 Глава VI. Альдегиды и кетоны
ния. Реакцией их образования пользуются для обнаружения того
или другого альдегида или кетона, или для того, чтобы выделить
альдегиды и кетоны из смеси с веществами других классов.
4. При действии пятихлористого фосфора атом кислорода в
молекуле альдегида или кетона замещается двумя атомами хлора:
/
СНз-Of
/Р
f + РС16 » СН3-СНС12
н
Таким образом получаются галоидпроизводные углеводородов
с двумя атомами галоида при одном атоме углерода. Дигалоид-
ные производные углеводородов, реагируя с водой, способны опять
давать исходные альдегиды или кетоны:
.О
CH3CHC12 -f Н20 > СН3с/ + 2НС1
5. Со спиртами альдегиды образуют ацетали по следующей
схеме:
JO Н|-О-С2Н6 /
СНз-О^-Т"! і »СН3-СН + Н20
н ІЗ2 ^о-с2н5
диэтилацеталь
і ¦¦ уксусного альдегида
Для получения ацеталей нагревают смесь спирта и альдегида
с безводной сернокислой медью или же кипятят альдегид со
спиртовым раствором хлористого водорода.
Ацетали можно рассматривать как простые эфиры гидратов
альдегидов.
Ацетали—бесцветные жидкости с приятным запахом;
водными растворами кислот они быстро расщепляются на альдегид
и спирт. Известны аналогичные соединения кетонов, но они
получаются другим путем.
86. Окисление альдегидов и кетонов. Альдегиды легко
окисляются. Они могут окисляться даже кислородом воздуха
и такими слабыми окислителями, как аммиачный раствор окиси
серебра.
Реакцию альдегидов с аммиачным раствором окиси серебра
называют «реакцией на серебряное зеркало». Ее используют для
обнаружения альдегидов.
Реакцию выполняют обычно так. В тщательно вымытую
пробирку вливают 1—2 мл 5%-ного раствора азотнокислого серебра,
осторожно прикапывают слабый раствор аммиака до растворе-
86. Окисление альдегидов и кетонов 197
ния первоначально образующегося осадка и вносят каплю
альдегида. Перемешав содержимое, опускают пробирку в горячую
F0—70 °С) воду и оставляют на несколько минут.
Восстановленное металлическое серебро выделяется частично в виде черного
осадка, а частично осаждается на стенках пробирки в виде
блестящего зеркала.
Происходящие при этом реакции можно выразить следующими
уравнениями:
NH4OH NH3
AgNO3 >¦ AgOH * (Ag(NH3J]OH
[Ag(NH3)a]OH + R—O( » R—COONH4 + NH4OH + Ag
При окислении альдегиды присоединяют кислород,
получающиеся при этом вещества носят название карбонових кислот:
,0
Н—Of
0
+ О
муравьиный муравьиная
альдегид кислота
о
О/
пропионовый пропиоиовая
альдегид кислота
Как видно из приведенных уравнений, при окислении
альдегидов атом кислорода направляется к углероду, при котором уже
находится кислород.
Карбоновые кислоты характеризуются наличием группы
Ґ или —СООН, которая называется карбоксилом.
Реакция Тищенко. Характерная склонность альдегидов к
реакциям окисления и восстановления очень ярко проявляется в
реакции Тищенко A906). Если к какому-либо альдегиду прибавить
небольшое количество алкоголята алюминия, например этилата
алюминия Al(OCaHaK, то происходит энергичная реакция:
двойная связь углерода с кислородом разрывается и водород
альдегидной группы одной молекулы присоединяется к углероду
другой молекулы, а оставшаяся часть присоединяется к кислороду;
при этом образуется сложный эфир:
0 . О
II II
R—С + О » R—С—О
1 II I
Н СН —R CHaR
198
Глава VI Альдегиды и кетоны
Если гидролизовать сложный эфир, то получается кислота
и спирт. Таким образом, в реакции Тишенко как бы происходит
окисление одной молекулы альдегида за счет восстановления
другой молекулы.
Возьмем для примера уксусный альдегид. В условиях реакции
Тищенко он превращается в этиловый эфир уксусной кислоты:
Р
сн3— се + о=сн—сн3 —»сн3-
ХН ? ЧО-СН5СН3
•і
зтилацетаї
Реакция Тищенко имеет техническое значение для получения
в промышленном масштабе этилацетага. являющегося важным
растворителем.
Расположим в виде таблицы формулы углеводородов,
спиртов, альдегидов и кислот с одинаковым числом атомов углерода
в молекуле:
Н-С<(
муравьиный альдегид
СН3-С^
уксусный альдегид
о
сн4
пропав
бутан
с6н1а
сн3—он
метиловый спирт
QH6—ОН
этиловый спирт
СзН,—ОН
пропиловьіґі спирт
СЛ-ОН
бутиловый спирт
QHU—ОН
амиловый спирт
н-с/°
хон
муравьиная кислота
сн3-с/
хон
уксусная кислота
пропионовый альдегид
ОН
пропионовая кислота
.0
масляный альдегид
У.0
н
валериановый альдегид валериановая ,ислота
4 ОН
масляная квелота
с4н9-с/
хон
В каждом вертикальном столбце находятся члены одного и
того же гомологического ряда. В каждой горизонтальной сіро-
ке—вещества, которые являются производными одного и того же
углеводорода и которые получаются одно из другого при помощи
простых химических реакций. «Если ряд неполный, то мы не
только знаем состав отсутствующих членов ряда и их основные
свойства, но мы можем также найти способы приготовления их,
если бы мы пожелали этот ряд дополнить» (Шорлемме)).
86. Окисление альдегидов и кетонов 199
Окисление кетонов. Кетоны не окисляются ни
кислородом воздуха, ни слабыми окислителями, они не
восстанавливают аммиачного раствора окиси серебра. Кетоны окисляются
под действием более сильных окислителей, например
марганцовокислого калия, причем окисление их происходит иначе, чем
окисление альдегидов. При окислении молекула кетона
расщепляется и из нее получаются две молекулы кислоты или
молекула кислоты и молекула кетона, менее сложного, чем
первоначальный. Разрыв цепи углеродных атомов происходит у
карбонильного углерода:
О Н;
II і і" хО ?>
СН3—СН2—С—!—СН—СН., + 30 » СН-,—СН,— Of + СИ,—Cf
хон хон
диэтилкетон пропионовая кислота уксусная кислота
О Н;
СН3ч II І і ХН3 СНЗЧ /О СН3ч
>СН-С-!-С< +20 » >CH-Of + >С=0
cw/ ¦ х:н3 сн3х хон сн3х
диизопропилкетон изомасляная кнслоіа ацетон
Если в молекуле кетона находятся два различных радикала,
то распад молекулы при окислении может идти по двум
направлениям, например:
Н:
О | |
II
СНз-С-
О
0
он
1
0
—C<f
хон
20
СН2-
20
метилпропилкетон уксусная кислота пропионовая
кислота
О
II /Р jf>
C-CHj—СН,—СН3 » Н -Cf + CH3-CHa-CH.2-C<f
+ N0H NOH
о
метилпропилкетон муравьиная маслянаи кислота
кислота
Таким образом, произведя окисление кетона и узнав, какие
кислоты получились в результате окисления, мы можем
определить строение кетона.
Правила окислительного расщепления кегонов были иыведе-
ны А. Н. Поповым.
200 Глава VI. Альдегиды и кетоны
87. Реакции с участием водородного атома в а-положении
к карбонильной группе. Карбонильная группа в альдегидах и
кетонах оказывает очень большое влияние на водородные атомы,
стоящие у соседнего с карбонильной группой углерода (термин
а-положение подробно объяснен на стр. 280).
Так, эти атомы легко обмениваются при действии хлора или
брома:
,0 .О
СНа— СН2 — СН,—C<f + Вг2 » СН3—СН2—СНВг—с/ + НВг
масляный альдегид а-броммасляный альдегид
CHa-CHjj-CO-CHjj-СНз + С1„ > СН3-СН2-СО-СНС1-СН3 + HCI
пентянон-3 2-хлорпентанон-З
Эти галоиды, находящиеся в ближайшем соседстве с
карбонильной группой, отличаются большой реакционной
способностью. Такие а-галоидзамещенные альдегидов и
кетонов—сильные лакриматоры (по латински lacrima—слеза), так как их пары
вызывают раздражение слизистых оболочек носа и глаз.
Большое значение имеют реакции уплотнения, происходящие
с кетонами и особенно альдегидами под влиянием щелочных
растворов, а также некоторых других реагентов. Так, например,
уксусный альдегид при действии на холоду слабых растворов
щелочей или хлористого цинка превращается в альдоль:
,0 .О
СНа— СН-н- СН,—Of > СН3—СН—СНа—of
'і + | nh і Чі
н он
|
н
альдоль
(бутанол-З-ал-1)
Продукт реакции—жидкость, смешивающаяся с водой,
перегоняется без разложения только при уменьшенном давлении. Как
видно из формулы, это соединение содержит в молекуле как
альдегидную группу, так и спиртовую (альд-оль).
Аналогичные альдоли получаются и из гомологов уксусного
альдегида. Такая реакция называется альдольной конденсацией.
Необходимо при этом заметить, что альдольная конденсация идет
за счет подвижного атома водорода в а-положении к
карбонильной группе. Для пропионового альдегида реакция выразится
следующим уравнением
СН3 СН3
| хО I О
СН3—СН,—СН + СН—Of » СНЭ—СН2—СН—СН—Of
Jl І н і н
он он
2-метилпентанол-З-ал-1
87. Реакции водородного атома в а-положении к карбонильной группе 201
Альдоли представляют собой малоустойчивые соединения;
они легко теряют элементы воды, образующейся за счет
отщепления гидроксильной группы и оставшегося а-водорода:
СНз-СН-СН-с/ ——> CH3-CH=CH-C<f
| | NH -Н2° ХН
он н
альдоль кротоновий альдегид
(бутен-2-ал-І)
Конденсация альдегидов, протекающая с отщеплением воды,
называется кротоновой. Она легко происходит и с гомологами
уксусного альдегида, имеющими свободную метиленовую
группу >СНа в а-положении к карбонилу. Так, с масляным
альдегидом протекает следующая реакция:
СН3-СН2-СНа-СН=О +
і-зниііепссп-г-ші-і
г-ші-і CHO
При действии на альдегиды крепких щелочей процесс
конденсации проходит очень энергично и приводит к образованию
смолообразных продуктов—смеси высокомолекулярных
соединений. Причину такого поведения легко понять. Как при
образовании альдоля, так и кротонового альдегида, сохраняются
альдегидные группы, которые далее могут вступать во взаимодействие
друг с другом и с молекулами исходных альдегидов по типу аль-
дольной и кротоновой конденсации.
Реакциями конденсации принято называть такие реакции
уплотнения, которые приводят к образованию новых
углерод-углеродных связей. При этом молекулы могут конденсироваться без
выделения простых молекул (как при образовании альдолей)
или же с выделением их (как в кротоновой конденсации).
Аналогично альдегидам наиболее активные кетоны способны
к альдольной и кротоновой конденсациям. Так, например,
ацетон при действии гидроокиси бария уплотняется по типу
альдольной конденсации, давая так называемый диацетоналкоголь:
СН3 ¦ СН3
СНз-О- СН8—СО—СН3 * СН3—С—СН„—СО—СН8
II + І І
Н ОН
диацетоналкоголь
B-метштентанол-2-он-4)
202 Глава VI. Альдегиды и кгтоны
Под действием крепкой щелочи реакция протекает по типу
кротоновой конденсации и приводит к образованию окиси мези-
тила:
СН3 СН3ч
^>С=О + HjCH—СО—СН3 * >С=СН—СО—СНо
ал/ -н*° сн3/
окись меэитила
B-МЄТИЛПЄИТЄН-2-ОН-4)
Кетоны, а также альдегиды конденсируются в присутствии
формальдегида со вторичными аминами с образованием аминоке-
тонов:
CHS
СН3—С—CHSH +0 + HNfCHj,), » СН3—С—СН3—СН2— N(CHa)j
Д форм- диметиламнн II
О альдегид О
ацетон (диметиламинометил)-ацетои
D-диметиламннобутанон-2)
Реакция впервые была открыта в 1917 г. и носит название
реакции Манниха Проходит она в водном или спиртовом растворе
при низкой температуре в присутствии каталитических количеств
НО. Вместо вторичных аминов можно применять первичные
амины или даже аммиак.
88. Сравнение свойств альдегидов и кетонов. Как альдегиды,
так и кетоны могут присоединять водород, кислый сернистокис-
лый натрий (из кетонов только те, которые содержат одну ме-
тильную или две метиленовых группы в непосредственном
соседстве с карбонилом), синильную кислоту, магнийорганические
соединения; с гидроксиламином и фенил гидразином альдегиды и
кетоны реагируют с образованием оксимов и фенилгидразонов;
при действии пятихлористого фосфора атом кислорода в
молекулах альдегидов и кетонов замешается двумя атомами хлора
(табл. 8).
Между альдегидами и кетонами имеется также и ряд
существенных различий. Так, в отличие от альдегидов, кетоны не
окрашивают бесцветный раствор фуксинсернистой кислоты, не осмо-
ляются щелочами, не присоединяют аммиака, а реагируют с
ним. выделяя элементы воды и образуя ряд сложных веществ;
не дают со спиртами ацеталей.
Окисляются кетоны труднее, чем альдегиды, причем при их
окислении происходит разрушение молекулы; труднее протекают
и реакции конденсации.
88. Сравнение свойств альдегидов и кетонов
203
Таблица 8
Взаимодействие альдегидов и кетинов с различными реагентами
Реагент
Водород
2Н
Бисульфит
NaHSOj
Синильная кислота
HCN
Магнийгалоидалкил
R"—MgJ
Аммиак
NH3
Гидроксиламии
NHSOH
Гидразин
NH,—NHj,
Фенилгидразин
C„H6NH—NH„
Пятихлористый фосфор
РС1„
Продукты взаимодействия
с альдегидами
Первичные спирты
R—СН,ОН
эисульфитные соединения
юн
R-CH
Циаигидрииы
ЮН
R—СН
\zN
yOMgJ
R—С—R"
\н
Альдегидаммиаки
ЮН
R-C-NH.2
4 1 1
Оксимы
R-CH=NOH
Гидразоны
R—CH=N—NH,
Феиилгидразоны
R-CH=N-NH-C,Ht
Дигалоидны?
производные
R—СНСІ,
о ке гонами
Вторичные спирты
R_CH(OH)—R'
Бисульфитыые
соединения
R\ /он
Циангидрииы
VOH
R'/ ^N
R'/ ^OMgJ
Продукты сложных
превращений
Оксимы
\C=NOH
R'/
Гидразоны
R4
>C=N-NH,
Фенилгидразоны
\c=N—NH— С,Н§
Дигалоидные
производные
R'—CCi,—R"
204
Глава VI. Альдегиды и кетоны
Продолжение
Реагент
Спирт
R'—ОН
Фуксинсернистая
кислота
Аммиачный раствор
окиси серебра
Продукты взаимодействия
с альдегидами
Ацетали
/<Ж
R-CH
Окрашивание
«Серебряное зеркало»
с кетонами
Ацеталн кетонов(кетали)
таким путем не
образуются
ОТДЕЛЬНЫЕ ПРЕДСТАВИТЕЛИ АЛЬДЕГИДОВ И КЕТОНОВ
89. Муравьиный альдегид. Муравьиный альдегид Н—С/
представляет собой газ с резким запахом. Он называется также
формальдегидом (от латинского названия муравьиной кислоты—
acidum formicum); ЗО—40%-ный раствор муравьиного
альдегида известен под названием формалина. Впервые формальдегид
получен А. М. Бутлеровым.
В промышленности формальдегид получают окислением
метилового спирта кислородом воздуха, пропуская смесь паров
спирта и воздуха над медным катализатором при высокой
температуре. В процессе реакции катализатор продолжает
оставаться раскаленным за счет тепла самой реакции.
Прибор для лабораторного получения муравьиного альдегида
каталитическим окислением метилового спирта изображен на
рис. 33.
В колбу, погруженную в стакан с водой, температура которой
поддерживается около 45—48 °С, помещают метиловый спирт.
Сильно накалив медную спираль в трубке, просасывают через
прибор воздух, высушенный в промывной склянке с серной
кислотой. Воздух увлекает пары метилового спирта. Через
некоторое время фуксинсернистая кислота, налитая в правую промывную
склянку, окрашивается в яркий фиолетовый цвет, что указывает
на образование альдегида. Спираль даже после удаления
пламени остается накаленной.
По химическим свойствам муравьиный альдегид сильно
отличается от других альдегидов. В то время как большинство аль-
89. Муравьиный альдегид
205
дегидов жирного ряда осмоляется щелочами, муравьиный
альдегид при действии щелочи дает метиловый спирт и муравьиную
кислоту:
/Р /Р
Н— СЄ + HaO + H-C<f » СН3ОН + НСООН
При этом одна молекула муравьиного альдегида окисляется,
а другая восстанавливается. Такое превращение альдегида в
Ннасасу
Рис. 33. Прибор для каталитического окисления
метилового спирта.
спирт и кислоту называется реакцией Канниццаро по имени
исследователя, открывшего эту реакцию A853). Реакция
Канниццаро, по всей вероятности, играет значительную роль при многих
биологических процессах и протекает в природе при участии
энзимов.
Муравьиный альдегид с аммиаком не образует альдегидам-
ииака, но реагирует с образованием уротропина.
Суммарное уравнение реакции:
6 не/ +4NH3
ЧН
> QHlaN4 + 6HaO
уротропин
Уротропин—белое кристаллическое вещество. Впервые получен А. М. Бут-
яеровым. Применяется в медицине для дезинфекции мочевых путей, как
противогриппозное средство (входит в состав лекарства «кальцекс»). Действием на
уротропин азотной кислотой получают мощное взрычатое вещество
бризантного действия—гексоген.
206 Глава VI. Альдегиды и кетоны
При осторожном выпаривании водного раствора муравьиного
альдегида происходит его полимеризация и образуется твердый
полимер муравьиного альдегида—так называемый параформаль-
дегид- Молекулярный вес его во много раз больше
молекулярного веса муравьиного альдегида. При нагревании параформаль-
легида снова образуется муравьиный альдегид.
Сухой муравьиный альдегид в неионных растворителях
(например, в толуоле или гептане) в присутствии некоторых
катализаторов способен полимеризоваться с образованием устойчивых
высокомолекулярных полимеров, имеющих полиоксиметиленовую
структуру (полиформальдегид):
СН2—О—CHj—О -СН,—О—СН2—О
Полученные на их основе пластические массы отличаются особо
высокой механической прочностью, вследствие чего они
применяются как полноценные заменители цветных и черных металлои.
Конденсацией формальдегида с фенолом, мочевиной или мела
мином получают фенольные или карбамидные смолы; изготовлен
ные на их основе пластические ^ассы исторически явились одним
из первых типов синтетических материалов, получивших широкое
распространение. Вследствие появления поливинилхлорида.
полистирола, полиэтилена и других новых типов пластических масс
они утратили свое универсальное значение, но все еще продолжают
производиться в громадных количествах для изготовления газ-
личных предметов бытового и технического назначения (см.
стр. 454).
В 1870 г. Байер, основываясь на опытах Бутлерова,
получившего первое сахаристое вещество уплотнением формальдегида,
высказал предположение, что муравьиный альдегид является
первым продуктом ассимиляции углерода растениями:
со, + н.,о —> н—с/ + о.г
Эта реакция происходит при участии хлорофилла и связана
с поглощением энергии солнечного света. В лальнейшем
муравьиный альдегид полимеризуется в углеводы:
6H-&f > QH12Oe
Водный раствор муравьиного альдегида—формалин—ядовит
и является сильным дезинфицирующим средством. Он
применяется для дезинфекции помещении и для протравливания зерна
90 Уксусный альдегид 207
(убивает споры головни). В формалине сохраняют
анатомические препараты.
90. Уксусный альдегид. Уксусный альдегид, или ацетальде-
гид, получается в промышленном масштабе различными методами.
В более старом способе исходят из ацетилена (метод основан на
реакции Кучерова, см. стр. 92):
Hg1"
Н-СэС-Н + HjO -—¦
K2SO4
В более поздних методах исходят из этилена. Присоединением
воды к этилену получают этиловый спирт, который затем
подвергают дегидрированию:
+н2о -н2 лО
СНа=СН2 > СН3-СН2ОН * СН3—Of
NH
В последние годы удалось разработать метод прямого
окисления этилена в аиетальдегид кислородом или воздухом в жидкой
фазе в присутствии медно-палладиевого катализатора:
О
H
Уксусный альдегид—я'идкость с резким запахом, кипящая
при 21 °С. В присутствии катализаторов, например солей
марганца, он легко окисляется кислородом воздуха в уксусную
кислоту:
2СН3С/ + О2 » 2СН3СООН
Н
При действии капли концентрированной серной кислоты на
уксусный альдегид он переходит в вещесіво, кипящее при 124 °С,
т. е. при температуре гораздо более высокой, чем уксусный
альдегид.
Это вещество называется паральдегидом.
Относительная плотность пара паральдегида по водороду
равна 66; следовательно, молекулярньи вес его равен 132, в то
время как молекулярный вес уксусного альдегида равен 44. Ясно,
что три молекулы уксусного альдегида дают одну молекулу па-
рал ьдегида.
Характер связи молекул уксусного альдегида в паральдегиде
становится понятным при изучении свойств этого соединения.
208 Глава VI. Альдегиды и кетоны
Паральдегид не показывает характерных альдегидных
реакций, не восстанавливает аммиачного раствора окиси серебра, не
дает окрашивания с фуксинсернистой кислотой, не образует
фенилгидразона и т. д. Это показывает, что в его молекуле нет
характерной для альдегидов группировки —С\
При нагревании со слабой серной кислотой паральдегид
переходит обратно в легко летучий уксусный альдегид.
Следовательно, связь между молекулами уксусного альдегида не
отличается особенной прочностью. Так как связь между атомами
углерода достаточно прочна и не может разорваться при
нагревании вещества со слабой серной кислотой, надо предположить,
что молекулы уксусного альдегида соединяются не через атомы
углерода, а через атомы кислорода.
Образование паральдегида можно представить следующей
схемой:
V V
о
СН- ||
>С С
V
Н,
три молекулы паральдегид
уксусного альдегид
При действии на паральдегид или уксусный альдегид серной
кислоты устанавливается равновесие:
уксусный альдегид zZ. паральдегид
Если осторожно, при сильном охлаждении, прибавить к
уксусному альдегиду немного крепкой серной кислоты, то часть
уксусного альдегида перейдет в паральдегид. Нейтрализовав кислоту
щелочью, легко отогнать уксусный альдегид от паральдегида.
Если к паральдегиду прибавить немного серной кислоты, то часть
его перейдет в уксусный альдегид. При нагревании уксусный
альдегид будет превращаться в пары и улетучиваться из сферы
реакции. Поэтому реакция пойдет справа налево, и час.ь
паральдегида вновь превратится в уксусный альдегид и т. д.
При действии серной кислоты на уксусный альдегид при
низкой температуре образуется другой полимер—кристаллический
метальдегид (С2Н4ОL. Метальдегид, подобно паральдегиду,
может переходить обратно в уксусный альдегид.
91. Хлораль B,2,2-трихлорэтаналь) 209
В технике уксусный альдегид вырабатывают в больших
количествах. Он является исходным продуктом для получения
уксусной кислоты, уксусного ангидрида и этилацетата см.
стр. 198).
91. Хлораль B,2,2-трихлорэтаналь) С18С—С\ . Как было
отмечено выше (стр. 200), водород при а-углеродном атоме
альдегидов и кетонов имеет повышенную активность, что проявляется
и в том, что при действии на них галоидов этот атом водорода
легко замещается галоидом:
+ 3CI2 > CCI3— Of + 3HCI
Хлораль—маслянистая жидкость плотностью 1,5 г/см3; кипит
при 98 °С. Применяется в медицине как снотворное. При
прибавлении к хлоралю воды происходит разогревание и образуется
твердый хАоральгидрат СС13—СН(ОНJ:
-с/ + Й-С
/OH
CCl3-C<f + H-OH » CC13—C—H
Хлоральгидрат—редкий пример устойчивого вещества с
двумя гидроксилами при одном атоме углерода.
Три атома хлора в молекуле хлораля значительно понижают
прочность связи между атомами углерода, поэтом\ хлораль при
действии водных растворов щелочей уже при обыкновенной
температуре расщепляется на хлороформ и соль муравьиной
кислоты:
з—! с/ + К—О:—Н » CHCL, + Н—с/
I ^Н I ' ХI<
Эта реакция позволяет объяснить механизм получения йодоформа и
хлороформа из этилового спирта, .щелочи и галоида.
При действии щелочи на иод образуется йодистый калий и иодноватисто-
кисльш калий KJO:
2КОН + J2 » KJ + KJO + Н8О
14-788
210 Глава VI. Альдегиды и кегоны
Последний окисляет этиловый спирт в уксусный альдегид:
GjH5OH + KJO » СН3—C<f + KJ + НаО
В полученном уксусном альдегиде иод замещает три атома водорода,
результате чего получается иодаль:
СНЭ
э-с<( +3ja—»аэ-с<( +3HJ
Едкое кали нейтрализует йодистый водород:
3HJ + ЗКОН » 3KJ + ЗН,0
Избыток же щелочи расщепляет иодаль на йодоформ и муравьинокислый
калий:
+0
CJ3—C<f + КОН » CHJ3 + HCOOK
Складывая уравнения всех реакции, получаем суммарное уравнение
образования йодоформа:
2КОН + Ja = KJ + KJO + НаО
QH60H + KJO = СН3СНО -f- KJ + HaO
CHgCHO + 3Ja = CI3CHO + 3HJ
3HJ + 3KOH = 3KJ + 3HaO
CJSCHO + KOH = CHJ3 + HCOOK
QH50H + 6KOH + 4Ja = CHJ3 + HCOOK + 5KJ + 5H2O
Реакция является характерной не только для уксусного альдегида, но и
для всех веществ, содержащих группировку СНЭ—С—, например для всех
О
метил кетон ов. Все они в указанных условиях дают йодоформ.
Хлороформ получается аналогичным путем—действием на этиловый спирт
щелочным раствором хлора или хлорной известью.
92. Акролеин. Акролеин (пропеналь) СН2=СН—О^ —
простейший непредельный альдегид.
Акролеин получается отщеплением от глицерина двух
молекул воды при помощи водуотнимающих средств. Так как
водород и гидроксил отнимаются от соседних атомов углерода, то
92. Акролеин 211
в результате реакции должен был бы получиться спирт с гидр-
оксилом при атоме углерода, связанном двойной связью.
ЮН Hj н
Н—С-С-С-ОН * СНа=С=СН-ОН + 2НаО
I I. !
Н ;ОНН!
Но такой спирт неустойчив; в момент своего образования он
изомеризуется в альдегид:
: :
СН2=С=С<^ » СН2=СН—С^
Акролеин—бесцветная жидкость, кипящая при 52 °С; обладает
чрезвычайно острым запахом, отчего и происходит его название
(acre—острое, oleum—масло). Образованием ничтожных
количеств акролеина объясняется резкий запах подгоревших жиров
и масел.
В технике акролеин получают прямым каталитическим
окислением пропилена:
О
СН2=СН-СНа + О2а<^
Он находит применение как промежуточный продукт при
промышленном получении синтетического глицерина, а также в
производстве некоторых синтетических смол.
Акролеин химически очень активен; это связано с наличием
в его молекуле альдегидной группы и двойной связи. Особенно
резко выражена у него способность к реакциям присоединения
как по альдегидной группе, так и по этиленовой связи, а иногда
сразу в обоих направлениях. При действии брома получается,
например, дибромпропионовый альдегид:
СН^СН-СНО + Вг2 » СН2Вг—СНВг—СНО
Хлористый водород присоединяется к акролеину с
образованием 3-хлорпропанала, т. е. процесс идет не по правилу Мар-
ковникова (см. стр. 78).
Акролеин очень легко-полимеризуется в твердую аморфную
массу —дисакрил.
При окислении акролеина окисью серебра получается
акриловая кислота СН2=СН—СООН,
14*
212 Глава VI. Альдегиды и кетоны
Окислением аллилового спирта в специальных условиях
можно получить акролеин, а при окислении акролеина—акриловую
кислоту:
.О
CHa=CH—CHjOH > СН2=СН—С/ =. СН2=СН—СООН
аллиловый спирт акролеин Н акриловая кислота
93. Цитраль. Из других непредельных альдегидов интересен цитраль
С((|Н16О—маслянистая жидкость с приятным запахом лимонов, темп, кип
228 °С. Он встречается во многих эфирных маслах, например в лимонном масле.
Альдегидная природа цитраля доказывается тем, что он может быть получен
окислением первичного спирта гераниола С10Н18О и сам может быть окислен в
гераниевую кислоту С10Н16О2. Строение цитраля выражается формулой:
СН3—С=СН—СНа—CHa—С= СН—О?
I I ЧН
Следует отметить, что молекула цитраля построена как бы из углеродньи
скелетов двух молекул изопентана:
С
С—С—С—С
с
Подобное строение углеродной цепи можно отметить в ряде других
природных соединений—в натуральном каучуке, терпенах, • каротиноидах и др
Цитраль применяется как душистое вещество и служит для синтеза ио
нона—вещества с запахом фиалки.
В последние годы цитраль приобрел важное значение как медицинский
препарат, снижающий кровяное давление (при гипертонии).
94. Ацетон. Из кетонов наибольшее промышленное значение
имеет ацетон СН3—СО—СН3 (пропанон-2).
Ацетон образуется при сухой перегонке дерева, но выделение
его из продуктов перегонки затруднительно. Его можно
получить разложением уксуснокислого кальция, образующегося при
нейтрализации известью уксусной кислоты, которая также
получается при сухой перегонке. При нагревании уксуснокислого
кальция происходит его разложение на ацетон и углекислый
кальций:
СН3— Of: ~|
Хп ' СН3ч
>СО + СаСОэ
СН/
СН3-;С '
94. Ацетн 213
Раньше этот способ получения ацетона был практически
единственным. Получение ацетона может быть осуществлено
непосредственно из уксусной кислоты—пропусканием ее паров над
нагретыми катализаторами (А12О3, ThOa). В настоящее время ацетон
производится главным образом из газов крекинга нефти:
пропиленом алкилируют бензол и из образовавшегося кумола
получают ацетон и фенол (см. стр. 453). Ацетон производят также
дегидрированием изопропилового спирта, получаемого из
пропилена. Для этого пары изопропилового спирта пропускают над
металлической медью или окисью цинка:
СН34 Л\ СН3ч
с —» с=о + н3
сиз--7 \он ел/
изопропиловый ацетон
спирт
Ацетон может быть получен из крахмала в результате ацетоно-
бутилового брожения, вызываемого некоторыми грибками. При
брожении крахмал сначала осахаривается в глюкозу, которая
затем превращается в ацетон, этиловый и бутиловый спирты, причем
происходит выделение больших количеств углекислого газа а
водорода:
QH13Oe > СН3СОСН3 + QHsOH + С4Н9ОН + СО3 + Н3
Дать точное уравнение реакции невозможно ввиду ее
сложности и малой изученности.
Ацетон—легкая жидкость, кипящая при 56 °С; имеет
характерный запах, является превосходным растворителем многих веществ.
С водой смешивается во всех отношениях. Если к водному
раствору ацетона прибавить раствор поташа, то жидкость
разделяется на два слоя: нижний слой—водный раствор поташа с
небольшим количеством ацетона, верхний слой—главным образом
ацетон. Этот процесс называется «высаливанием» ацетона.
Так как ацетон, подобно уксусному альдегиду, содержит в
своей молекуле группу СН3—С—, то он, так же как и уксусный
II
О
альдегид, при совместном действии иода и щелючи дает йодоформ.
Реакции, происходящие при этом, выражаются уравнениями:
СН3СОСН3.+ 3J3 = CJ3COCH3 + 3HJ
CJ3COCH3 + КОН = CHJ3 + СНзСООК
3HJ + ЗКОН = 3KJ + ЗН3О
СН3СОСН3 + 3J„ + 41<ОН = CHJ3 + CH3COOK + 3KJ + ЗН3О
¦2Н Глава VI. Альдегиды и кетоны
Ацетон находит обширное и разнообразное применение, tiro
используют для получения хлороформа, йодоформа, как
растворитель в производстве лаков, негорючих кинопленок, одного из
ендов искусственного волокна (ацетатного). Ацетон применяется
для растворения ацетилена, как желатинпзатор в производстве
¦порохов и как исходный материал для промышленного получения
ряда органических веществ.
Хлор- и бромпроизводные ацетона обладают сильным
слезоточивым действием и применялись в войне 1914—1918 гг. как
¦отравляющие вещества.
Теоретический и практический интерес представляет реакция
конденсации ацетона с углеводородами, имеющими тройную связь
в молекуле. Она была открыта А. Е. Фаворским, который показал,
что ацетиленовые углеводороды способны в присутствии
порошкообразного едкого кали конденсироваться с альдегидами и
кетонами, образуя ацетиленовые спирты. Так, в присутствии едкого
кали ацетилен с ацетоном дают диметилэтинилкарбинол*,
электролитическим гидрированием которого получается диметилви-
«илкарбинол; при отщеплении от последнего элементов воды
образуется изопреи:
І І CHav 2H
CH
>C=O + НС==СН ¦ >C—C=
СН/ Г : CH/ I
І ! O
H
диметилэтннилкарбинол
CH^ _HSO
» >C—CH=CHj » CH,=C— CH=CHj
ch/| I
OH CHa
диметылвиннлкарбинол изопрен
Полимеризацией изопрена получают синтетический каучук
<см. стр. 105 ел.).
Конденсацией винилацетилена с ацетоном в присутствии
едкого кали получается диметилвинилэтинилкарбинол:
СН,ч СН
>С=О + НС=С—СН=СН2 >
сп/ сн
диметилвинилэтиыилкарбниол
И. Н. Назаров разработал способ получения из этого спирта
вини.лацетиленовых смол, которые нашли широкое применение
* Этинилом называется радикал СН sC—.
95 Диальдегиды и дикетоны 215
в качестве склеивающих веществ (так называемый «карбиноль-
ный клей») для разнообразных материалов (стекло, минералы,
пластмассы, металлы) и для производства лаков.
Если пары ацетона пропускать через нагретую до 500—600 °С
трубку с окисью алюминия или над накаленной металлической
проволокой, то происходит распад молекулы ацетона с
образованием метана и кешена:
НСНа-СО , СН4 + СНа=С=О
I кетен
СН3
Кетен—газ с резким запахом, кипит при —41 °С, чрезвычайно ре-
акционноспособен, часто используется в органических синтезах.
Реагирует с водой с образованием уксусной кислоты:
СНг=С=О + Н-ОН > СНЭ-С=О
С хлористым водородом кетен дает хлорангидрид уксусной
кислоты—хлористый ацетил:
сн4=с=о + на —> сн9-с=о
1 1 1
Кетен легко полимеризуется в жидкий димер (темп. кип.
127,4 °С), пиролизом которого можно обратно получить исходный
мономер. В технике кетен применяется для производства
уксусного ангидрида:
О
СНа=С=О + СНа-СООН
II
сн9-сч
\
95. Диальдегиды и дикетоны. Диальдегиды и дикетоны
содержат две карбонильные группы. Примерами этих веществ
могут служить глиоксаль, диацетил и ацетилацетон.
Глава VI. Альдегиды и квтоны
Глиоксаль СНО—СНО является простейшим диальдегидом.
Он получается осторожным окислением этилового спирта
азотной кислотой. Глиоксаль—красивые желтые кристаллы,
которые плавятся при 15 °С, переходя в желтую жидкость, кипящую
при 51 °С. Пары глиоксаля изумрудно-зеленого цвета. Он очень
легко полимеризуется, давая твердые бесцветные полимеры.
Глиоксаль является простейшим представителем окрашенных
веществ, состоящих только из углерода, водорода и кислорода.
Соединения, содержащие две карбонильные группы, связанные
вместе, имеют систему сопряженных двойных связей и окрашены
в желтый цвет.
Диацешил СН3—СО—СО—СН3—простейший дикетон; его мож
но рассматривать как диметильное производное глиоксаля. Диаце-
тил—желтая жидкость с резким запахом; темп. кип. 88 °С. При
действии на диацетил гидроксиламином получают диоксим—du-
непшлглиоксим (темп, плавл. 240 °С).
СНз-С-С-СН,
| + 2 » || ||
О HON NOH
з,
|| || + 2Н2ЫОН » || || + 2Н„О
О О HON NOH
Ацешилацетон СН3—С—СН2—С—СН3, или ?-дикетон, полу-
II II
О О
чается реакцией Клайзена—конденсацией уксусноэтилового эфира
с ацетоном в присутствии алкоголята натрия:
СН3—СООСаНьЧ-СНз—СОСНз с2н6О№ СН,—С—СНа—С—СН,+СаНьОН
——> ' II II
о о
Реакция Клайзена подробнее рассмотрена на стр. 307.
Ацетилацетон—жидкость со своеобразным запахом, темп, кип
137 °С. При кипячении с водой ацетилацетон разлагается нэ
уксусную кислоту и ацетон:
сн3—с—снг—соснэ + н—он —»снэсоон + сн3сосн,
II
о
Как было сказано (см. стр. 200), карбонильная группа
делает реакционноспособными а-водородные атомы. Такое влия-
96. Хелатные соединения 217
ниє карбонильной группы можно проследить почти на всех карбо-
нилсодержащих соединениях:
У3
—сС
карбоновые
кислоты
<ч
С-іН;
/1 ¦-"
сложные эфиры
н\ /
<
/
/
R\^°
\
С—ІН! X-
| / |
і кетоны
альдегиды
R-Сч
.С—:Н!
/ \
^0
Р-днкетоны
с2н6-оч у)
с\
yC-;HJ
/ \
кетоэфиры
-ЇНІ
Rn /0
<
/С-іН!
/ \
/ \
CN N
кетонитрилы
В ?-дикетонах имеются две карбонильные группы, и а-водородные
атомы, находящиеся по соседству с двумя карбонильными
группами, становятся еще более подвижными. В растворе ацетилацетона
существует равновесие:
СН3—С-СН2—С-СН3 ^=t СН3-С=СН-С—СН3
II II I II
0 0 ОН О
кетоннаи форма енольнзи форма
Соединения, содержащие при углероде с двойной связью (ен)
гидроксильную группу (ол), принято называть енолами (или эно-
лами). Такого рода химическая структура обладает рядом
характерных особенностей.
Физико-химическими методами было доказано, что равновесие
в случае ацетилацетона смещено в сторону енольной формы
(в растворе содержится 23,6% дикетона и 76,4% енола).
96. Хелатные соединения. Ацетилацетон образует с медью,
никелем, бериллием, а также алюминием, хромом, железом и др.
соединения следующего строения:
/с=0\
С-0 0=Cv
Х
СН3
Ч
/СН
СН3
В данном случае енольная форма ацетилацетона реагирует с
солями металлов или их гидроокисями так, что металл связывается
218 Глава VI. Альдегиды и кетоны
ковалентно с остатком гидроксильной группы и координационно—
с кислородным атомом карбонила.
Таким образом получаются циклические соединения,
содержащие металл в цикле. Такие соединения называются хелатными
(от греч. слова «хела»—клешня) или внутршсомплексными
соединениями. Эти соединения резко отличаются от типичных солей
с ионной связью между катионом металла и анионом (см. стр. 375,
599).
Хелатные соединения обладают следующими свойствами:
1) Хорошо растворяются в органических растворителях (с
характерной окраской) и не растворяются в воде.
2) Очень слабо диссоциированы, т. е. являются
неэлектролитами.
3) Многие из них способны возгоняться и перегоняться без
разложения.
4) Часто имеют яркую окраску, отличающуюся от окраски
обычных солей соответствующих металлов.
Всеми перечисленными свойствами обладает хелатное
соединение никеля с диметилглиоксимом (см. стр. 216):
О—Н-0
/
™
о-н—о
Наличие четырех 5-членных циклов делает это соединение
устойчивым. Реакция диметилглиоксима с солями никеля была
открыта в 1906 г. Л. А. Чугаевым; она широко применяется для
качественного и количественного определения никеля (в
присутствии кобальта) и ряда других металлов.
Хелатные соединения находят широкое применение в
аналитической химии в качестве реактивов, а также для получения
металлов высокой степени чистоты.
ГЛАВА V!l
КАРБОНОВЫЕ КИСЛОТЫ И ИХ ПРОИЗВОДНЫЕ
ПРЕДЕЛЬНЫЕ ОДНООСНОВНЫЕ КИСЛОТЫ
97. Строение и способы получения карбонових кислот В ма-
°
лекулах карбоновых кислот содержится группа атомов —L<
или —СООН, называемая карбоксилом.
Примерами карбоновых кислот могут служить:
/Р
Н-С<
NOH
СН
/
Р
)Н
/Р
муравьиная
кислота
QH,-c<
масляная
кислота
Р
юн
уксусная
кислота
с4н„-с<°
валериановая
кислота
пропионовая
кислота
Структурная формула и даже само название «карб-оксил»
указывает, что эта группа как бы состоит из карбонильной
группы >С=О и оксигруппы —ОН. Однако кетонный или
альдегидный характер, свойственный группе >С=О, у карбоксила ничем
не проявляется. Атомы кислорода в карбоксиле совершенно
одинаковы. Это подтверждено изучением физических свойств
карбоновых кислот и их рентгеновских спектров.
Поэтому строение карбоксильной группы кислот и их солей
лучше выражается следующими структурами:
R-(
которые показывают, что отрицательный заряд равномерно
распределен между обоими кислородными атомами.
220 Глава VII. Карбоновые кислоты и их производные
Основность кислоты определяется числом карбоксильных
групп в ее молекуле: уксусная кислота СН3—СООН
одноосновная кислота, щавелевая кислота НООС—СООН двухосновная.
Состав одноосновных предельных кислот выражается
формулой С„Н2/1 ,—СООН; они составляют типичный гомологический
ряд.
Для получения карбоновых кислот существует ряд способов.
1. Гидролиз нитрилов. При обработке йодистого
метила цианистым калием образуется цианистый метил (жидкость,
кипящая при 81,5 °С):
СН3—JJ'T'KjC=N » СН3—С=П + KJ
При нагревании с разбавленными неорганическими кислотами
цианистый метил, присоединяя воду, расщепляется на аммиак
и уксусную кислоту:
снз—c=;n + hjo .о
! і » СН3—C<f + NH,
! Ні—ОН NOH
Если же нагревать цианистый метил со щелочами, то выделяются
аммиак и уксуснокислый натрий.
Таким же образом могут быть получены и другие кислоты:
Н—CN + 2Н2О > НСООН + NH3
синильная муравьиная
кислота кислота
С3Н7—CsN + 2НаО > С3Н,—СООН + NHa
цианистый масляная
пропил кислота
Как видно из уравнения, муравьиная кислота может быть
получена из синильной кислоты.
Цианистые ал килы при гидролизе превращаются в кислоты
с тем же числом атомов углерода, поэтому их называют
нитрилами кислот: метилцианид или ацетонитрил, этилцианид или
нитрил пропионовой кислоты. Эта реакция может служить
доказательством строения кислот.
2. Действие углекислого газа на магний-
органические соединения. При действии на ме-
тилмагнийбромид углекислого газа образуется продукт
присоединения
СН3—Mg—Вг + СО2 > СН3—Of
MgBr
98. Изомерия и номенклатура предельных одноосновных кислот 221
который можно рассматривать как смешанную соль уксусной и
бромистоводородной кислот. При обработке этой соли
минеральной кислотой получается уксусная кислота:
уО о
2СНЭ—C<f + 2НС1 » 2СН3— C<f + MgBr2 + MgCls
NOMgBr \Ж
3. Получение кислот при помощи малонового эфира и при
помощи ацетоуксусного эфира описано на стр. 311—313.
4. Окисление органических соединений.
Одноосновные предельные кислоты получаются в результате
окисления многих органических соединений. Часто при этом
происходит расщепление молекул и в молекуле получающихся кислот
содержится меньше атомов углерода, чем в исходном соединении;
например при окислении этилбутилкетона
СНз-CHj-i
І-СО-І
^СН2—СН2—СН3
может получиться:
уксусная кислота СН3—СООН
пропионовая кислота СНЭ—СНа—СООН
«-масляная кислота СН3—CHS—CH2—СООН
«-валериановая кислота СН3—СН2—СН2—СН2—СООН
При окислении первичных спиртов (стр. 143) и альдегидов
(стр. 196) получаются кислоты с тем же числом атомов углерода
в молекуле.
Многие кислоты могут быть также получены из природных
растительных и животных веществ.
98. Изомерия и номенклатура предельных одноосновных
кислот. Одноосновные предельные карбоновые кислоты общей
формулы С„Ная і—СООН нередко называют жирными кислотами,
так как впер+вые представители этого ряда были получены из
жиров.
Муравьиную кислоту Н—СООН можно рассматривать как
соединение водорода с карбоксилом. Уксусная кислота СН3—СООН
является карбоксильным-производным метана СН4; пропионовая
кислота С2Н5—СООН представляет собой карбоксильное
производное этана С2Н„. Так как в молекулах метана и этана все атомы
водорода равноценны, то безразлично, какой из этих атомов
222 Глава VII. Карбоновые кислоты и их производные
заместить карбоксилом; поэтому у уксусной и пропионовой
кислот нет изомеров. Для четвертого члена ряда—масляной кислоты—
возможны два изомера:
СНз.
СН3—СН4—СН.2—СООН >
СН3Х
СН—СООН
нормальная масляная изомасляная
кислота кислота
Для валериановой кислоты возможны и в действительности
известны четыре изомера. Два из них можно рассматривать как
производные нормального бутана, а другие два—как
производные изобутана
f^LJ ґ^ті рм Ґ^ї-І
нормальный бутан
СНЯ—СН2—СНа—СН2—СООН ^СН—СООН
СНз-СН/
нормальним вялериапошя метил9іилуксусная
кислота кислота
сн3—сн—сн3
СНз
изобутан
СН- CHjx
'^>СН—СНа—СООН СНз-С—СООН
сн
и:ювалериановая триметилуксусиая
кислота кислота
Если спирты можно рассматривать как углеводороды, в
которых один атом водорода замещен гидроксилом, то кислоты
будут являться углеводородами, в которых один атом водорода
замещен карбоксилом. Замещая атом водорода карбоксилом, мы
вводим в молекулу один новый атом углерода, поэтому число
изомерных карбоновых кислоте п углеродными атомами в
молекуле равно числу изомерных спиртов с (п—1) атомами углерода в
молекуле.
По женевской номенклатуре названия кислот производятся
от названия углеводородов с тем же числом атомов углерода,
что и в молекуле кислоты, с добавлением слова «кислота».
Муравьиная кислота Н—СООН называется метановой кислотой, уксусная
кислота СНо—СООН этановой кислотой и т. д.
99. Физические свойства предельных одноосновных кислот 223
Таким образом, женевская номенклатура рассматривает
кислоты как производные углеводородов, одно звено которых
превращено в карбоксил:
СН3—СН2—СН3
пропан
сн.
сн3—сн2—соон сн3—сна—сн—соон
пропанооая 2-метилбутановая
кислота кислота
При составлении рациональных названии кислот с
разветвленной цепью атомов углерода их часто рассматривают как
производные уксусной кислоты СНдСООН, в молекуле которой
атомы водорода метильной группы замещены радикалами: метил-
этилуксусная кислота, триметилуксусная кислота.
В большинстве случаев пользуются, однако, традиционными
названиями, которые обыкновенно указывают на материал, из
которого была выделена та или иная кислота, например:
муравьиная кислота, уксусная кислота, масляная кислота (полученная
впервые из коровьего масла), валериановая кислота (из корней
валерианы), лауриновая кислота (из плодов лаврового дерева)
и т. д.
99. Физические свойства предельных одноосновных кислот.
Первые четыре представителя ряда жирных кислота-подвижные
жидкости, смешивающиеся с водой во всех отношениях.
Кислоты, в молекуле которых содержится от пяти до девяти атомов
углерода (а также изомасляная кислота), маслянисты;
растворимость их в воде невелика.
Высшие кислоты—твердые тела, практически нерастворимые
в воде, при перегонке под обыкновенным давлением они
разлагаются.
Муравьиная, уксусная и пропионовая кислоты имеют
острый запах; средние члены ряда обладают неприятным запахом;
высшие кислоты запаха не имеют. Основные физические свойства
предельных одноосновных кислот приведены в табл. 9.
Данные, приведенные в табл. 9, показывают, что с
увеличением молекулярного веса повышается температура кипения и
уменьшается плотность кислот. Если температуры кипения
жирных кислот плавно возрастают по мере увеличения в молекуле
кислоты количества атомов углерода, то для температур
плавления этих кислот отмечено следующее интересное явление:
температура плавления кислоты с четным числом углеродных
атомов в молекуле выше температур плавления двух соседних кислот
224
Глава Vf/. Карбоновые кислоты и их производные
Таблица
Физические свойства предельных одноосновных кислот
Название
Муравьиная кислота
Уксусная ....
Пропионовая . . ,
Масляная ....
Изомасляная . .
Валериановая , .
Капроновая . . .
Энантовая ....
Каприловая . . .
Пеларгоновая . , .
Каприновая . . .
Ундекановая . . .
Лауриновая . . .
Тридекановая . . .
Миристиновая . .
Пентадекановая
Пальмитиновая . .
Маргариновая* ,
Стеариновая . . .
Формула
нсоон
СН3СООН
снасн„соон
СН3СН2СН2СООН
(СН3JСНСООН
СНЭ(СН2KСООН
СН3(СН2LСООН
CHg(CH2MCOOH
СНЭ(СН2NСООН
СНЭ(СН2),СООН
СН3(СН2)8СООН
СН3(СН2)„СООН
СН3(СН2I0СООН
СНЭ(СН2)ПСООН
СН3(СН2I2СООН
СН3(СН2I3СООН
СНЭ(СН2I4СООН
СНЭ(СН.2I6СООН
СН3(СН2IвСООН
Температура
плавления
°С
+8,25
-16,6
-20,7
-3,11
-47
—34,5
-1,5
— 10,5
+ 16,2
+ 12,5
+31,5
+30,5
+44,3
+42,0
+53,7
+52,1
+62,6
+60,8
+69,4
Температура
кипения
°С
100,5
118,5
141,1
163
154,4
186
205,3
223
237,5
253
268,4
280
225
гпри 100 мм
рт. ст.)
236
(при 100 мм
рт. ст.)
250,5
(при 100 мм
рт. ст.)
257
(при 100 мм
рт. ст.)
271,5
(при 100 мм
рт. ст.)
277
(при 100 мм
рт. :т.)
287
(при 100 мм
рт. ст.)
Относительная
ПЛОТРЮСТЬ
420
1,2322
1,049
0,9916
0.У59
0,949
0,9387
0,922
0,9184
0,910
0,9057
0,«858
три 40°С)
0,8907
(при 30 °С)
0,8740
(при 4І °С)
—
0,8533
(при 70 °С
—
0,849
(при /0 °С)
0,848
(при 70 °С)
0,848
(при 70 °С)
* Маргариновая кислота не была найдена в природных продуктах и ие имеет непосредствен,
ного отношения к маргарину. Она была получена синтеті чески Н. Я- Демьяновым и С. М.
Кочерг иным.
99. Физические свойства предельных одноосновных кислот
225
с нечетным числом углеродных атомов. Эта зависимость
наглядно показана на рис. 34.
Определение молекулярного веса кислот показывает, что в
жидком состоянии они имеют удвоенные молекулы. Плотность
75
50
%25
I
V
-25
-50
Рис.
д
А/
V
Л
J
/
3 6 9 t2 16 Ш
Vt/сло атомов углерода S цепи
34. Температура плавления предельных
одноосновных кислот нормального строения.
'//ШЖ///?.
/ /Плоскость спайності///
пара уксусной кислоты при низких температурах больше 30 и
меньше 60, т. е. пары уксусной кислоты представляют собой смесь
простых и удвоенных молекул. С повышением температуры
плотность пара уменьшается, но
только выше 250 °С становится
равной 30. Таким образом,
явление ассоциации молекул
выражено у кислот в еще
большей степени, чем это
наблюдается у алкоголей (см. стр. 138).
Кристаллы стеариновой,
лауриновой и других кислот с
большим молекулярным весом
были исследованы Брэггом при
помощи рентгеновских лучей.
Исследования показали, что в
кристаллах этих кислот молекулы расположены рядами и
несколько наклонно к плоскости спайности, т. е. к той плоскости, по
которой раскалывается кристалл (рис. 35). Эти : яды молекул
образуют двойные слои, причем в каждом слое молекулы двух
рядов обращены друг к другу своими карбоксильными группами
15—788
Рис. 35. Положение углеродных
цепей в кристалле лауриновой кислоты.
226
Главо VII. Карбоновые кислоты и их производные
и прочно связаны за счет сил побочных валентностей.
Молекулы же различных слоев соприкасаются по плоскости спайности
метильными группами, связь между которыми непрочна. Этим
и объясняется скользкость кристаллов жирных кислот и жиров.
100. Химические свойства предельных одноосновных кислот.
І. Карбоновые кислоты обладают кислой реакцией
(обнаруживается индикаторами) и образуют соли со щелочами и щелочными
металлами:
СНаСООН -Ь NaOH = CH^COONa + НаО
2НСООН + Mg = (HCOO)aMg + На
Гидроксильная группа содержится как в молекулах спиртов,
так и в молекулах кислот. Но спирты не обладают кислотными
свойствами, в то время как водород гидроксила в кислотах
обладает всеми свойствами кислотного атома водорода.
Усиление кислотных свойств гидроксила в кислотах объясняет
ся влиянием на нею кислорода карбонильной группы, с
углеродом которой непосредственно связан гидроксил.
Карбоновыр кпслоты в большинстве случаев в водном растворе
диссоциированы лишь в малой степени и являются слабыми
кислотами, значительно уступая таким кислотам, как соляная,
азотная и серная. Так, при растворении 1 грамм-молекулы в
16 л воду степень диссоциации муравьиной кислоты равна 0,06,
уксусной кислоты 0,0167, в то время как соляная кислота при
таком разбавлении диссоциирована почти полностью.
'Аонстанты электролитической диссоциации жирных кислот
приведены в табл. 10.
Таблица 10
Константы электролитической диссоциации жирных кислот
Кислота
Муравьиная
Уксусная
Пропконовая
Констант;.
диссоциации
2Н 10-«
17,6-10-'
13,4-10-«
Кислота
Масляная
Валерманоная
Капроновая
Константа
диссоциации
15,2 Ю-"
16,0 10-е
13,8 10-»
2. При сплавлении солей щелочных металлов жирных кислот
со щелочами происходи! расщепление углеродной цепи, в
результате чего из углеводородного радикала кислоты образуется
углеводород:
СНа—;COONa+'NaOН * СН4 + NaaCO3
100. Химические свойства предельных одноосновных кислот 227
3. Сухая перегонка кальциевых и бариевых солей жирных
кислот (кроме солей муравьиной кислоты) приводит к
образованию кетонов. При этом получаются кетоны с двумя одинаковыми
радикалами.
Так, при перегонке уксуснокислого кальция получается
диметилкетон, при перегонке пропионовокислого кальция—ди-
этилкетон:
СН3—СО—Оч
СНзч
> > >
СН3-СО—(У СП/
Н3—СО—Оч
>Са
Н3-СО—(У
уксуснокислый диметилкетон
кальций (ацетон)
С2НВ-СО-ОЧ СаН5ч
>Са > >СО + СаСО3
QHs-CO-CK ОД/
прогшомовокислый диэгилкетон
кальций
Если взять смесь солей двух различных кислот, то, наряду
с этой реакцией, происходит и реакция между двумя различными
молекулами солей. В результате образуются кетоны с двумя
различными радикалами.
Так, при перегонке смеси уксуснокислого кальция и
пропионовокислого кальция, кроме диметилкетона СН3СОСН3 и диэтил-
кетона CaHgCOCaHg, получается метилэтил кетон:
CHj—СО—О. уО— СО—QH5 CH3v
Са + Са » 2 С=О + 2СаСО3
уксуснокислый пропноповокнслый метилэтилкето«
кальций кальций
Если же взять смесь солей, из которых одна является солью
муравьиной кислоты, то получается альдегид:
R-CO—ІОГ"
Са + Са І » 2R—С + 2СаСО3
R—СО—ОХ ^О—СО!—Н ^Н
Для получения кетонов, вместо сухой перегонки солей, можно
пропускать пары кислот над нагретыми катализаторами,
например: окисью тория, углекислым кальцием и др.; для получения
альдегидов в качестве катализаторов берут закись марганца и
окись титана. Можно представить, что при этом сначала
образуются как промежуточные продукты соли кислот, которые затем
разлагаются, давая снова исходный катализатор. Так, например,
15*
228 Глава VII. Карбоновые кислоты и их производные
при пропускании паров кислот над углекислым кальцием
образуется кальциевая соль карбоновой кислоты, вода и углекислый
газ. Кальциевая соль разлагается с образованием кетона и
углекислого кальция; при взаимодействии последнего с кислотой
снова получается кальциевая соль, вода и углекислый газ и т. д.
Результат реакции выражается уравнением:
2СН3СООН » СН3СОСН3 + Н2О + СО,
4. Водородные атомы углеводородного радикала в кислотах
по реакционной способности подобны атомам водорода у
парафинов. Исключение составляют атомы водорода, расположенные
у атома углерода, непосредственно связанного с
карбоксилом. Это очень отчетливо проявляется при хлорировании и
бромировании жирных кислот. При действии хл"ора или брома
в присутствии переносчиков галоидов (РС13, J2 и др.) на жирные
кислоты или на их хлорангидриды -происходит замещение
а-водородных атомов:
CHsCHjCHjCOOH + Вга » СНзСН^СНВгСООН + НВг
Карбоксил, так же как и карбонильная группа, усиливает
реакционную способность а-водородных атомов (см. стр.-200).
Это впервые было отмечено В. В. Марковниковым.
5. Жирные кислоты, как правило, устойчивы ^действию
окислителей. Легко окисляются лишь муравьиная кислота и кислоты
с третичным атомом углерода в молекуле. При окислении
последних получаются оксикислоты, т. е. вещества, которые являются
одновременно и спиртами и кислотами:
СНз\ а,
а, СНчу ¦
СН-СООН + О » >С—СООН
сн/ сн
изомасляная а-оксиизпмасляная
кислота кисхота
В организмах жирные кислоты способны окисляться, причем
кислород напрарляется в ?-положение, т. е. к атому углерода,
отделенному от карбоксила одним атомом углерода. Так,
например, в организме диабетиков масляная кислота переходит в
?-оксимасляную кислоту:
. • .СН3-СН2-СН2СООН + VaO * СНз-СН(ОН)—СНа—СООН
масляная кислота fJ-оксиммсляная кислота
10/. Муравьиная кислота ?29
Окисление масляной кислоты удалось произвести и в
лабораторных условиях, действуя на нее 3%-ным раствором перекиси
водорода. Масляная кислота при этом переходит в р-оксимасля-
ную кислоту (которая содержит вторичную спиртовую группу),
при дальнейшем окислении из нее получается ацетоуксусная
кислота:
СН3—СН(ОН)—СН2—СООН + О » СНЭ—СО—СН2—СООН + R,0
6. При действии на кислоты галоидных соединений фосфора
получаются галоидангидриды кислот (см. стр. 264):
ЗСНзСООН + РС13 > ЗСНдСО-С) ¦+- Н3РО3
хлористый
ацетил
7. Со спиртами кислоты дают сложные эфиры (стр. 252)
СНзСООН + GjH6OH їгг* СНзСО—О—CjHfi + НаО
уксусноэтиловый (
эфир
8. При нагревании аммонийных солей кислот получаются
амиды (см. стр. 267):
CH3COONH4 » CHjCO—NH2 + НаО
ацетамил
101. Муравьиная кислота. Муравьиная кислота Н—СООН
находится в выделениях желез некоторых видов муравьев,
например красного лесного муравья Formica ruf а. Отсюда ее латинское
название acidum formicum. Соли муравьиной кислоты поэтому
называют формишпами.
В прежнее время муравьиную кислоту добывали из муравьев,
обрабатывая их водяным паром. В настоящее время ее готовят
действием окиси углерода на едкий натр при повышенной
температуре и давлении; при этой реакции получается муравьинокис-
лый натрий:
СО + NaOH ,. Н—COONa
Муравьиная кислота—бесцветная жидкость, с резким
запахом и жгучим вкусом. Муравьиная кислота, как это видно из ее
Ї ° \
Ї // \
структурной формулы I H—С\ I, отличается по своему
строению от других кислот; в ее молекуле находится карбонил> свя-
230 Глава VII. Карбоновые кислоты и их производные
//°
занный с водородом, т. е. альдегидная группа Н—С( . Вследствие
этого она отличается от других кислот и по своим химическим
свойствам.
1. Муравьиная кислота, подобно альдегидам и в отличие от
других карбоновых кислот, легко окисляется окислителями
(КМпО4, СгОа и др.).
В результате окисления получаются углекислый газ и вода:
/°
—О— С » СОа -f- НаО
\
от
Н—С +о
При нагревании раствора сулемы, подкисленного муравьиной
кислотой, происходит восстановление сулемы сначала в каломель,
а затем в металлическую ртуть:
НСООН + 2HgCl.2 » Hg2CI2 + 2HCI + СО,
НСООН + Hg2Cl2 * 2Hg + 2HCI +
При нагревании ртутной соли муравьиной кислоты происходит
восстановление ртути до свободного металла, а половина углерода
окисляется в углекислый газ:
(HCOOJHg » Hg + НСООН + СОа
2. Муравьиная кислота при действии крепкой серной кислоты
разлагается на окись углерода и воду:
НСООН » СО + Н2О
3. Муравьиная кислота является довольно сильной кислотой;
в водном растворе она значительно диссоциирована (см. табл. 10,
стр. 226).
В технике муравьиная кислота находит применение в
текстильной промышленности, а также в некоторых процессах
органического синтеза, в том числе для получения щавелевой кислоты.
102. Уксусная кислота. Уксусная кислота известна с
древнейших времен, так как она образуется при скисании
вина—виноградный уксус. Само слово уксус происходит от греческого
«оксиес»—кислый.
В технике уксусную кислоту готовили из так наз. древесного
уксуса, который получался при сухой перегонке дерева. Уксусная
102. Уксусная кислота 231
кислота СН,—СООН широко распространена в природе; в
свободном виде она содержится в выделениях человеческого
организма—в моче и поте. Эфиры уксусной кислоты находятся во многих
растениях.
До недавнего времени уксусную кислоту получали двумя
способами—сухой перегонкой древесины и бактериальным
окислением спиртовых жидкостей («уксусное брожение»). В первом
случае сырьем является древесная целлюлоза, во втором—винный
спирт.
При сухой перегонке древесины уксусная кислота собирается
в подсмольной воде. Для отделения уксулной кислоты от
древесного спирта и ацетона ее нейтрализуют известью; полученный
уксуснокислый кальций, так называемый «уксусный порошок»,
разлагают соляной кислотой или серной кислотой:
2СН3СООН + Са(ОНJ -> (СН3СООJСа + 2Н2О
(СН3СОО)аСа + HaSO4 > 2СН3СООН + CaSO4
На некоторых заводах уксусную кислоту извлекают из
подсмольной воды растворителями (этиловый эфир, уксусноэтило-
вый зфир и т. д.). После отгонки растворителя им вновь пользуются
для извлечения уксусной кислоты.
Уксусное брожение спиртовых жидкостей состоит в окислении
этилового спирта в уксусную кислоту при помощи особых
бактерий, называемых уксусным грибком. Эги бактерии, размножаясь
на поверхности спирта, окисляют этиловый спирт кислородом
воздуха в уксусную кислоту:
СН3СН2ОН + Оа » СН3СООН + Н.2О
Так как бактериям необходима пища, содержащая азот и
фосфор, то уксуснокислое брожение может про/сходить в
виноградном соке, в вине и пиве, но не может происходить в водных
растворах чистого спирта, не содержащих соединений азота и
фосфора.
Основным техническим методом получения уксусной кислоты
является окисление ацетальдегида кислородом воздуха в
присутствии марганцевых катализаторов:
2СН3--С< +Оа »2СН3-с/
Как указывалось выше (см. стр. 92), ацетальдегнд получают
либо из этилена, либо из ацетилена. Последний способ
постепенно утрачивает свое значение.
232 Глава VII. Карбоновые кислоты и их производные
В последние годы разработаны промышленные методы
получения уксусной кислоты окислением углеводородов, например
бутана, пентана или гексанов (Н. М. Эмануэль и др.).
Уксусная кислота в безводном состоянии—кристаллическое
вещество, плавящееся при 16,5 °С. Кристаллы ее по виду
напоминают лед. Поэтому 100%-ная уксусная кислота называется
ледяной уксусной кислотой. Впервые кристаллическая уксусная
кислота была получена Т. Е. Ловицем.
В продажной уксусной эссенции содержится 70—80%
уксусной кислоты; столовый уксус представляет собой 3—5%-ный
раствор уксусной кислоты.
Уксусная кислота, как и большинство карбоновых кислот,—
слабая кислота. Поэтому многие ее соли легко подвергаются
гидролизу, т. е. легко разлагаются действием воды. В результате
гидролиза происходит образование гидроокиси металла и
основных уксуснокислых солей:
(СН3СОО)зРе + Н,О » (СН3СООJРе(ОН) + СН3СООН
(CH3COOKFe + 2НаО » (CHjCOOjFeCOHfe + 2СН3СООН
(CH3COOKFe + ЗНаО » Fe(OHK + ЗСН,СООН
Как сама уксусная кислота, так и ее соли, называемые
ацетатами, находят обширное применение.
Уксусная кислота применяется как приправа к пище и для
консервирования мясных и рыбных продуктов; из нее получают
уксусный ангидрид (стр. 267), применяемый при изготовлении
искусственного волокна (ацетатного); монохлоруксусная кислота
СН2С1—СООН (получаемая хлорированием уксусной кислоты)
в громадных количествах расходуется в производстве
гербицидов; уксусная кислота служит для синтеза многих душистых
веществ и растворителей; она применяется в кожевенной,
текстильной и других отраслях промышленности. Из солей уксусной
кислоты наибольший интерес представляют соли железа,
алюминия и хрома, применяемые как протрава при крашении тканей.
Соли уксусной кислоты хорошо растворяются в воде; из них чаще
других применяется уксуснокислый свинец (СН3СОО)аРЬ-ЗНаО,
называемый свинцовым сахаром; он применяется в производстве
свинцовых белил, очень ядовит.
Основной уксуснокислый свинец (СН3СОО)аРЬ-РЬ(ОНL, или
свинцовый уксус, применяется в медицине (свинцовая примочка).
Основная уксуснокислая медь под названием ярь-медянки
применяется как зеленая краска.
Соединение уксуснокислой меди с мышья ковистокислой медью,
так называемая парижская (швейнфуртская) зелень,—ядовитое
вещество, используемое как инсектицид (средство, убивающее насеко-
104. Масляная кислота 233
мых) для борьбы с полевыми, лесными и огородными вредителями.
Для этой цели растения опрыскивают взболтанной в воде
парижской зеленью или распыляют ее в сухом состоянии.
Из уксуснокислого кальция получают ацетон (стр. 212).
103 Пропионовая кислота. Пропионовая (пропановая)
кислота может быть получена окислением н-пропилового альдегида
или действием на йодистый этил цианистого калия и последующим
омылением полученного цианистого этила:
CH3CH2J + KCN » CH3CHaCN + K.J
CH3CHaCN + 2HaO » СН3СН2СООН + NH3
Название пропионовой кислоты происходит от греческих слов
«протос»—первый и «пио»—жир; она является первой кислотой
гомологического ряда, дающей жирную на ощупь кальциевую
соль.
104. Масляная кислота. Для масляной кислоты известны оба
возможные по теории строения изомера: нормальная масляная
кислота (бутановая кислота) и изомасляная кислота (метилпро-
пановая кислота). Строение первой из них доказывается ее
получением из йодистого пропила: строение второй—получением из
йодистого изопропила:
KCN НгО
CH3CHaCHaJ * CHaCH^CHjCN » CH3CHaCHaCOOH
йодистый нормальная
пропил масляная кислота
СН3 KCN СНЗЧ Н2О СНгг.
>CHJ > >CHCN > >СНСООН
сн/ сн/ сн/
йодистый изомасляная
изопропил кислота
Кальциевая соль нормальной масляной кислоты, в отличие от
кальциевой соли иэомасляной кислоты, лучше растворима в
холодной, чем в горячей воде.
Изомерия этих кислот была открыта в 1865 г. В. В. Марковниковым,
показавшим, что кислота, которая образуется при окислении бутилового спирта,
кипящего при 108 °С, не тождественна обычной масляной кислоте. Он получил
эту кислоту из йодистого изопропила и тем установил ее строение и строение
того спирта, при окислении которого она образуется.
Нормальная масляная кислота была выделена в 1814 г. Шев-
релем из коровьего масла, где она находится в виде
глицеринового эфира.
В технике масляную кислоту получают окислением н-масля-
ного альдегида или н-бутанола. Она находит применение в
производстве пластических масс и душистых веществ.
234 Г лава VII. Карбоновые кислоты и их производные
105. Пальмитиновая и стеариновая кислоты.
Пальмитиновая кислота С16На1СООН, строение которой выражается
формулой СИ3—(СН2I4—СООН, и стеариновая кислота С17Н36СООН,
структурная формула которой СН3—(СН2Iв—СООН, вместе с
непредельными кислотами (олеиновой и др.) встречаются в
природе в виде глицериновых эфиров, которые являются главными
составными частями растительных и животных жиров. Смесь
стеариновой и пальмитиновой кислот составляет стеарин.
Соли высокомолекулярных органических кислот называются
мылами. Обыкновенные мыла состоят в основном из смеси
натриевых солей пальмитиновой, стеариновой и олеиновой кислот.
Ка іийное мыло имеет вид мази и носит название жидкого мыла.
Калийное мыло, подкрашенное в зеленый цвет (мыло, сваренное
из конопляного масла, имеет натуральный зеленый цвет),
применяется в медицине как антисептическое средство под
названием згггнаго мы га.
Кальциевые соли вышеупомянутых кислот нерастворимы в
воде. Поэтому в жесткой, т. е. богатой солями кальция, воде мыло
образует хлопья нерастворимых кальциевых солей:
SC^HasCOONa + СаС1а > (Ci,Ha5COOJCa + 2NaCI
При действии воды на мыло часть его подвергается гидролизу:
- Н2О » Cl7H35COOH + NaOH
Это доказывается появлением малиново-красного
окрашивания при разбавлении водой спиртового раствора мыла,
содержащего несколько капель фенолфталеина.
106. Мыла и моющие вещества. Обычное мыло получается
«омылением» жиров, т. е. глицеридов высших (С1в—С1а) жирных
кислот. Говяжье сало нагревается с раствором едкого натра. Когда
процесс гидролиза, т. е. образование свободного глицерина и
натриевых солей жирных кислот, заканчивается, к смеси
прибавляют поваренную соль, которая «высаливает» эти соли, при этом
образуется два слоя. Для приготовления «ядрового» мыла слой
натриевых солей отделяют от водного «подмыльного щелока»
(из которого вырабатывают глицерин), охлаждают, превращают
в стружки и подсушивают. После добавления красителей и
отдушек (ароматических веществ) стружки мыла спрессовывают и
штампуют в виде кусков хозяйственного и туалетного мыла.
Если для омыления применять более концентрированные
растворы щелочей и не проводить высаливания, то по охлаждении
вся масса затвердевает. В таком «клеевом» мыле содержится весь
глицерин и другие примеси из жира. В мыла, применяемые лля
106. Мыла и моющие вещества
OOOOOOOOOO-COONa
З6- Распределение молекул мы-
ла на
стирки белья, обычно добавляют соду и силикат натрия, которые
усиливают щелочность мыла.
Моющее действие мыла основано на сложных
физико-химических, точнее—коллоидных, процессах и рассматривается
подробнее в курсах физической химии. Здесь мы ограничимся
объяснением самого общего характера. Мыла являются поверхностно-
активными веществами: прибавление их к воде очень резко
снижает поверхностное натяжение. Это можно продемонстрировать
следующим простым опытом. Нальем в широкий кристаллизатор
чистой воды и покроем всю ее
поверхность порошком талька
или серы, т. е. веществами, не
смачиваемыми водой. Если
теперь в середину чашки капнуть
раствор мыла, то мы увидим,
что мгновенно образуется как
бы разрыв поверхностной
пленки, удерживающей порошок.
Действительно, поверхностные Рис
молекулы воды как бы образу-
ют своеобразную пленку. Каждая молекула мыла состоит из
двух частей: длинной углеводородной цепи, имеющей
гидрофобный характер, т. е. отталкивающейся от воды, и
карбоксильной, солевой, группы гидрофильного характера
(притягивающейся к воде). Молекулы мыла располагаются на
поверхности воды в виде слоя так, что все они плавают на воде, образуя
на поверхности подобие частокола (рис. 36).
Так как между углеводородными частями молекул взаимное
притяжение гораздо меньше, чем между молекулами воды, то
происходит разрыв прочной поверхности пленки воды, что мы
и наблюдаем при помощи порошка талька.
Белье, соприкасаясь с кожей, захватывает частицы жира,
выделяемые сальными железами. Засаленное белье легче
удерживает пыль. Жировые частицы не смачиваются водой и поэтому не
смываются чистой водой. При обработке мылом молекулы мыла
располагаются так, что их углеводородные части обращены
внутрь капелек жира, а к поверхности обращены гидрофильные
карбоксилы. Такие частицы уже смачиваются водой и образуют
прочные капельки эмульсии жира вместе с прилипшими к ним
частицами пыли и в таком виде легко удаляются с белья. Это
описание, конечно, дает лишь примитивное представление о
механизме моющей способности мыла. Однако оно дает нам
возможность понять пути поисков заменителей мыла, т. е. веществ,
которые обладают моющей способностью, но не требуют
использования пищевого сырья (жиры и масла) и не имеют некоторых
236 - Глава VII. Карбоновыв кислоты и их производные
недостатков мыла (понижение моющего действия в соленой или
жесткой воде). Укажем, например, такой путь получения моющих
средств. Если одноатомный спирт С„Н2я ,CHaOH (где п=12г
13... 20) соответствующей обработкой превратить в соль
кислого эфира серной кислоты
RCH2OH + SO3 » RCHoO—S—ОН » RCH,OSO,Na
то образуется молекула, построенная подобно молекуле обычного
мыла из гидрофобного радикала и гидрофильной группы. Такие
мыла имеют то преимущество, что они не дают осадков с солями
кальция и магния и, будучи легче растворимыми, могут
употребляться даже в жесткой воде.
Сульфаты высших жирных спиртов являются одним из
лучших пипов синтетических моющих средств и получили широкое
применение в промышленности и в быту. Исходные жирные
спирты получают из кашалотового жира, кокосового или таллового
масла. Синтетические способы основываются на
непосредственном окислении парафиновых углеводородов, на восстановлении
жирных кислот, полученных окислением парафина, на методе
оксосинтеза (см. стр. 151) или же на получении из низших олефи-
новых углеводородов с помощью алюминийорганических
катализаторов. Последний способ имеет то преимущество, что позволяет
получать спирты с нормальной цепью, что особенно ценно для
эффективности моющего действия.
Другая важная группа синтетических моющих средств
относится к классу алкиларилсульфонатов, содержащих алифатическую
цепь и сульфированное ароматическое ядро (см. стр. 449).
Некоторое значение в производстве моющих средств имеют
уже упоминавшиеся алкилеульфонаты (см. стр. 183).
Следует иметь в виду, что современные высококачественные
моющие средства представляют собой довольно сложные
композиции. Обычно они содержат 20—25% поверхностно-активного
вещества (алкиларилсульфонаты, алкилсульфаты и др.), до 50%
фосфорнокислых солей (например, тринатрийполифосфата),
различные наполнители и 2—3% активизирующих добавок, чаще
всего карбоксиметилцеллюлозу (см. стр. 346). Такие моющие
средства не только полноценно заменяют жировое мыло, но и
превосходят его по моющему действию, нечувствительны к
жесткой воде, не оказывают ослабляющего действия на ткани, меньше
раздражают кожу. В связи с успехами, достигнутыми в
разработке методов синтеза поверхностно-активных веществ и их
применения, в ряде стран производство мыла значительно снизилось,
108. Акриловая, нетакриловая и кретоновые кислоты 237
а производство синтетических моющих средств выросло в
несколько раз.
Необходимо отметить, что мыла применяются в обыденной
жизни и в технике не только для мытья. Мыла обладают большой
дезинфицирующей способностью, особенно при добавлении к ним
таких веществ, как фенолы и др.
При борьбе с вредителями сельскохозяйственных культур,
путем опрыскивания растений ядохимикатами, часто требуется
приготовить стойкие эмульсии, которые бы смачивали
поверхность листьев, имеющих восковой покров; для этого в них
добавляют мыла.
Соли высших жирных кислот образуют с жидкими
углеводородами продукты густой консистенции. Некоторые из них, например
на основе кальциевых мыл, применяются в качестве консистентных
смазок (солидол и др.).
НЕПРЕДЕЛЬНЫЕ ОДНООСНОВНЫЕ КИСЛОТЫ ,
107. Гомологический ряд непредельных одноосновных кислот.
Состав молекулы непредельных одноосновных кислот с одной
двойной связью можно выразить общей формулой QjH^jCOOH.
Как кислоты они образуют соли, как непредельные соединения—
способны присоединять два атома водорода или галоида и
окисляться по месту двойной связи. Энергичное окисление этих кислот
приводит к расщеплению их молекулы. По своей силе а,8-не-
предельные кислоты несколько превосходят соответствующие
жирные кислоты, так как ДЕОйная связь, находящаяся рядом с
карбоксильной группой, усиливает ее кислотные свойства.
1С8. Акриловая, метакриловая и кротоновые кислоты.
Простейшая непредельная одноосновная кислота—акриловая кислота
СН2=СН—СООН может быть получена окислением акролеина
окисью серебра
СН, =СН—C<f + Ag2O > СН^СН—СООН + 2Ag
а также окислением аллилового спирта или омылением нитрила
акриловой кислоты. Один из методов синтеза основывается на
отщеплении воды от ?-оксипропионовой кислоты:
СН2(ОН)СН2СООН > СН2=СНСООН + Н2О
?-оксипропионовая акриловая
кислота кислота
'2M Глава VII. Карбоновые кислоты и их производные
?-Оксипропионовая кислота получается, в свою очередь, из
этилена:
СН2=СН2 + НОСІ » CBjOH-CHaCl
этиленхлоргндрин
СНаОН—СНаС1 + KCN » СНаОН—СНа—C=N
оксинитрил
CHjOH—CH2—C=N + 2HaO » CH2OH—CHa—СООН + NH3
?-оксипропионовая
кислота
В технике акриловую кислоту получают из окиси этилена и
синильной кислоты через этиленциангидрин:
СНа—СНа + HCN » СН2—СН2—CN > СН2=СН—СООН
О ОН
окись этилена этиленциангидрив акриловая кислота
или же прямым синтезом из ацетилена и окиси углерода:
нс^сн ¦+ со + н,р —»сн2=сн—соон
Акриловая кислота (темп, плавл. 13 °С; темп. кип. 140 °С)
легко пол и мер из у етс я, давая высокомолекулярную
полиакриловую кислоту:
пСН=СНа > СН—СН.,—СН—СНа—СН—СНа
і 111
СООН СООН СООН СООН
Полимерные эфиры акриловой кислоты применяются для
получения пленкообразующих веществ, связующих веществ и
пластических масс.
Нитрил акриловой кислоты—жидкость (темп. кип. 78 °С);
получается дегидратацией оксинитрила (см. выше) или
присоединением синильной кислоты к ацетилену в присутствии
катализаторов:
НСзСН + Н—CN » СН2=СН—CN
!
Один из современных промышленных способов получения ак-
рилонитрила основывается на совместном каталитическом
окислении пропилена и аммиака:
2СН4=СН—СНа + 2NH3 -)- 30.2 , 2СН.,=СН—CN + 6НаО
108. Акриловая, метакриловая и кротоновые кислоты 239
Акрилонитрил производится химической промышленностью
в больших количествах, так как он является одним из исходных
мономеров для получения важных высокополимерных
синтетических материалов Путем полимеризации акрилонитрила или со-
полимеризации его с некоторыми другими мономерами получают
ценные синтетические волокна, заменяющие шерсть (типа нитрон
или орлон, акрилан и др.). Сополимеризацией акрилонитрила
с бутадиеном получают бензиностойкие синтетические каучуки,
а тройной сополимер на основе бутадиена, стирола и
акрилонитрила дает особо прочные пластмассы.
Акриловая и другие а.З-непредельные кислоты способны
присоединять галоидоводородные кислоты; при этом получаются
?-галоидзамешенные карбоновые кислоты:
СНа=СН—СООН + НВг , СНаВг—СНа—СООН
Таким образом, присоединение идет не по правилу Марков-
никова (см. стр 79).
Согласно теории химического строения, для кислоты с
открытой цепью углеродных атомов состава С3Н5СООН возможны
три изомера:
СН3
СНа=СН—СН2—СООН СН3—СН=СН—СООН СНа=С—СООН
аинилуксусная критоноаая кислота метаириловая
кислота кислота
Их можно рассматривать как соединения, полученные
отщеплением двух атомов водорода от двух соседних атомов углерода
масляной кислоты.
От нормальной масляной кислоты таким путем производятся
винилуксусная и кротоновая кислоты, от изомасляной кислоты—
метакриловая кислота Из этих кислот практическое значение
имеет метакриловая кислота, а наибольший теоретический
интерес представляет кротоновая кислчта.
Метакриловая кислота (темп. пл. 16 °С; темп. кип. 160—
161 °С) получается отщеплением воды от а-окснизомасляной
кислоты (которая, в свою очередь, получается из ацетона,—см.
стр. 283):
СН3 . СН3
СН3—С—СООН ¦. СН3=С—СООН + Н3О
' он
Высокополимерные сложные эфиры метакриловой кислоты
(например, метиловый эфир) применяются для производства
240
Глава Vii. Карбоновые кислоты и их производные
прозрачных стекловидных пластических масс—органического
стекла (плексиглас).
Полимеризацию можно выразить схемой:
сн3
СН2=С
СООСНз
сн3
СНо=С
снэ
СН2=С +¦
СООСНз
СООСН3
СН-, СНЭ СН3
Г I I
сн.2—с—сн2—с—сн2—с
соосн, соосн, соосн.
СООН
При сухой перегонке полимеров эфиров акриловой и метакрило-
вой кислот происходит обратный процесс «деполимеризации» с
образованием исходных мономеров.
Кротоновая кислота СН3—СН=СН—СООН известна в двух
изомерных формах: кротоновая кислота (темп, плавл. + 72 °С,
темп. кип. +189 °С) и
жидкая изокротоновая kvic-
лота (темп, плавл. +15,5 °С,
темп. кип. +172 °С). При
осторожном окислении они
расщепляются с
образованием щавелевой НООС—СООН
и уксусной кислот;
присоединяя водород, обе кротоно-
вые кислоты переходят в
норРис. 37 Пространственное
расположение атомов в молекуле масляной
кислоты.
мальную масляную кислоту
СН3СН2СН2СООН. Изомерия
этих кислот объясняется раз-'
расположением атомов в их моле-
личным пространственным
куле.
109. Цис-транс-изомерия. Если изобразить при помощи двух
тетраэдров пространственное расположение атомов в молекуле
масляной кислоты, то мы получим модель, представленную на
рис. 37. В этой модели в центре обоих тетраэдров находятся
атомы углерода метиленовых групп СН2. Если вращать один из
тетраэдров вокруг линии, соединяющей их центры, то можно
получить любое число пространственных положений атомов одного
тетраэдра относительно атомов другого тетраэдра. Два из этих
положений и изображены на рис. 37.
Таким образом, казалось бы, для масляной кислоты возможна
изомерия. Однако ни у нормальной масляной кислоты, ни у дру-
109. Uuc-транс-изомерия
24)
гих подобным образом построенных соединений не наблюдается
такого рода «поворотных» изомеров. Это легко объяснить, если
принять предложенный ВантТоффом принцип свободного
вращения атомов по ординарной связи.
Не следует, однако, считать, что
такое вращение действительно
совершенно свободно. Рассмотрим,
например, молекулу дихлорэтана (рис. 38).
Атомы С—С, связанные простой связью,
соединены с атомами водорода и
хлора, которые при повороте вокруг
Рис 38. Молекула
дихлорэтана
соон
Рис 39. Стереоизомерия кротонових
кислот.
линии С—С испытывают в большей или меньшей степени
взаимное притяжение или отталкивание. Это приводит к тому, что
молекула принимает некоторое пространственное положение («кон-
формацию»), которое в этих условиях будет наиболее устойчивым,
например как на данном рисунке, где однородные атомы
располагаются в наиболее удаленных положениях. Существование
такого рода заторможенного вращения было доказано
оптическими и другими физическими методами, особенно при изучении
поведения вещества при низких температурах.
Открыто также много случаев, когда поворот по ординарной
связи вовсе не может произойти из-за помех, обусловленных
большими размерами близко расположенных атомов и групп. В этих
случаях, например у производных дифенила (С6Н5—С6Н6),
поворотные изомеры выделены в устойчивых формах.
Совершенно другая картина получается, если два атома
углерода связаны двойной связью.
Рассмотрим этот случай на примере кротоновой кислоты.
Для получения молекулы кротоновой кислоты надо от
молекулы масляной кислоты отнять два атома водорода;
освобождающиеся при этом валентные связи насыщают друг друга. На тетра-
эдрической модели это изображают складывая тетраэдры по ребру
(рис. 39). Такое расположение атомов делает невозможным сво-
16—788
242 Глава V/I. Карбоновые кислоты и их производные
бодное вращение, так как для этого необходим разрыв связей.
Водородные атомы молекул кротоновых кислот могут находиться
или по одну сторону от плоскости, проходящей через учлеродные
атомы и двойную связь, или же на различных ее сторонах. Для
обозначения первого положения перед названием изомера
ставится приставка цис-, второе расположение обозначается
приставкой транс-.
Цис- и транс-изомеры кротоновых кислот схематично-можно
изобразить следующим образом:
Н-С—СН3 СНЭ—С-Н
Н-С-СООН Н-С-СООН
цис- изомер транс-иломер
изокротоновая кислота кротоновая кислота
(темп, плавл 15,5 °С) (темп, плавл. Т2 °С)
Другим примером цис-транс- изомер и и могут служить
олеиновая и элаидиновая кислоты (см. стр. 250).
Теорию этого вида изомерии разработал в 1887 г. Вислиценус,
исходя из тетраэдрической модели Вант-Гоффа.
Изучение химических свойств обоих изомеров позволяет для
каждого из них выбрать определенную конфигурацию (см.
стр. 279 ел.).
Цис- или транс-конфигурации во многих случаях можно
установить и на основании величины дипольных моментов.
Дис-изомеры 1,2-дигалоидных производных этилена должны
иметь дипольный момент; транс-изомеры (при одинаковых
атомах галоида) не могут иметь дипольного момента вследствие
внутренней компенсации положительных и отрицательных зарядов:
Ver V"
II. II.
с с
н сі а" н
quc-дихлорэтилен тракс-дихлорэтилен
(дипольный момент (дипольный момент=0)
|,85-1(Г18 эл.-стшп. ед.)
Чрезвычайно интересны взаимоотношения между цис- и транс-
изомерами непредельных кислот. Один из этих изомеров
(например, кротоновая или же элаидиновая кислоты) более
устойчив, чем другой (кислоты изокротоновая или же олеиновая).
Неустойчивый изомер (лабильная форма) при нагревании или
воздействии некоторых веществ (H„SO4, КОН, HNOJ может пере-
110. Теория напряжения
243
ходить в устойчивую (стабильную) форму. Это превращение идет
быстро в присутствии следов брома или иода на прямом
солнечном свету. Обратное превращение может происходить при действии
ультрафиолетовых лучей или же может быть осуществлено при
помощи ряда химических реакций.
110. Теория напряжения. Валентные связи атома углерода
направлены от центра к вершинам тетраэдра и образуют между
собой углы, каждый из которых равен 109°28'. Байер
предположил, что при образовании
двойной связи направления
валентных связей
отклоняются до линии, соединяющей
центры атомов. Согласно
этому для нахождения нового
направления валентных
связей, например в молекуле
этилена, мы должны согнуть
чертеж (рис. 40) до пересечения
первоначальных направлений
валентных связей и разделить
пополам угол между ними. Так
как этот угол равен 109°28', то
при образовании двойной связи
между атомами углерода
валентные связи отклоняются на
109°28'
= 54°44'
Рис. 40. Отклонения валентных
углов при образовании двойной связи
между атомами углерода.
Такое положение валентных связей является менее
устойчивым и стремится перейти в прежнее устойчивое тетраэдрическое
расположение. Это и выражается словами напряжение. Другими
словами, сущность теории напряжения заключается в том, что
в молекулах органических соединений наиболее устойчивым является
тетраэдрическое расположение атомов. Всякое другое
расположение менее устойчиво и стремится перейти в тетраэдрическое.
Теория напряжения была предложена Байером в 1885 г. Она
позволила объединить, объяснить и предсказать большое число
весьма важных явлений. Однако ей легко придать неверное,
чисто механическое толкование. Поэтому, пользуясь теорией
напряжения, всегда надо помнить, что химические силы
качественно отличны от механических сил, при помощи которых
объясняют теорию Байера на моделях.
111. Электронное строение двойных связей (it-связи). Как было
ранее сказано, строение обычных гомеополярных связей находит
244 Глава VII. Карбоновые кислоты и их производные
лучшее объяснение с точки зрения электронных представлений.
Ординарная связь—это так называемая а-связь с ее характерным
видом электронных облаков. Отличительными свойствами такой
связи являются: а) относительная химическая инертность; б)
наличие свободного вращения; в) определенные величины
межатомных расстояний—около 1,52—1,54 A; г) определенные величины
междувалентных углов—около 110°.
Классическая органическая химия оперировала с
постоянными механическими моделями связей, не входя в более
глубокое рассмотрение природы химических связей.
Непредельная углерод-углеродная связь представлялась как
система двух атомов, связанных двумя одинаковыми
валентностями. Для того чтобы это представление могло объяснить су-
Рис 41. Схема электронного строения остатка молекулы
этана после отнятия двух атомов водорода
ществование цис-, транс-изомеров, были введены приближенные
модели из тетраэдров. Для объяснения характерной реакционной
способности пришлось допустить особое «расщепление»
валентностей и возникновение «дополнительных», «парциальных»
валентностей. Появление первых электронных представлений, в
соответствии с которыми двойная связь изображалась в виде
четырех общих электронов вместо двух одинаковых черточек,
позволило строить для ее объяснения электронные модели.
Современная физика дает возможность яснее представить
строение кратных связей и объяснить ряд характерных свойств
непредельных соединений.
Представим себе, что в молекуле этана оба метильных
радикала потеряли по одному водородному атому (рис. 41). В таком
случае должны появиться две «свободные» связи. Очевидно,
что характер движения электронов, соответствующих этим
валентностям, должен резко измениться, и произойдут глубокие
изменения пространственного вида и химического характера
молекулы.
Все 6 атомов, образующих молекулу этилена, расположены в
одной плоскости, они по-прежнему связаны а-связями, которые
расположены друг к другу под равными углами C60:3 = 120°).
Движение двух «свободных» электронов совершается в направ-
///. Электронное строение двойных связей (я-связа)
245
лении, перпендикулярном плоскости всей молекулы;
электронные облака их имеют вид симметричных пространственных
«восьмерок», расположенных перпендикулярно к плоскости
молекулы (рис. 42).
Связь, осуществляемая с помощью подобных электронов,
называется «IC-СВЯЗЬЮ».
Наличие в молекуле тс-связи придает ей следующие
особенности:
1. Создается дополнительное притяжение, в результате
которого расстояние между атомами углерода от 1,54 А сокращаете»
до 1,34 А.
2. Появление дополнительных сил, направленных в
плоскости, перпендикулярной к плоскости молекулы, уничтожает или.
затрудняет поворот вдоль линии,
соединяющей атомы углерода.
Возникает возможность существования
геометрических (цис-шранс) изомеров.
3. Связь двух атомов углерода
при помощи системы а- и •х-связей
(«двойная связь» изображается в виде
двух черточек С=С)
термодинамически более устойчива.
При сильном нагревании
предельные углеводороды отщепляют атомы
водорода и превращаются волефины.
4. Воздействие химических реагентов, как правило,
направлено в первую очередь на атомы углерода, связанные двойной
связью. Причина лежит в особом характере электронов,
осуществляющих тс-связь; они обладают значительно большей подвижностью,
их электронные облака легче смещаются под влиянием внешних
воздействий.
Вот как, например, представляется присоединение НВг к
этилену. Изучением этого рода процессов установлено, что реакция
начинается с воздействия более активного протона Н . Его
приближение к одному из атомов углерода вызывает перестройку
облаков подвижных электронов тс-связи так, что этот атом
углерода приобретает более электроотрицательный характер; при
этом уменьшается электроотрицательность соседнего С-атома
(рис. 43,а, б). Это обозначается S— и S+.
Следующий этап состоит в присоединении иона Вг~ к
соседнему С-атому (рис. 43,б),
У этилена или симметричных олефинов оба атома углерода
одинаковы, и электронные облака тс-связей также симметричны.
К несимметричным олефинам присоединение НВг происходит
Рис
42. Схема электронного
строения этилена
246 Гиава VII. Карбоновые кислоты и их производные
по правилу Марковникова так, что водород присоединяется к
наиболее гидрогенизированному атому углерода.
Разберем это на примере изобутилена Он обладает дипольным
моментом 0,49 D. Очевидно, что положительный конец молекули
находится в той стороне, где сосредоточено больше атомов
водорода (рис. 44). Это значит, что
электронные облака, образующие
гс-связь, сдвинуты вправо.
Отсюда понятно, что
последовательное присоединение ионов
Н* и далее Вг приведет к
образованию 2-бром-2-метилпропана
(рис. 45). Аналогично
присоединяются H(SO4H), CI(OH).
При действии окислителей
(например, КМпО4 или озона)
реакция также в первую очередь
идет по месту двойной связи. При
0,4,927
Рис. 43 Схема присоединения Рис. 44. Схема строения молекулы
НВг к этилену изобутилена.
этом образуется уже предельное соединение (например, двухатом-
лый спирт, озонид). Дальнейшее окисление приводит к образо-
н3с
ВГ Н+ Вг Н
Рис. 45. Присоединение НВг к молекуле изобутилена.
оанию кетонной группы с последующим окислением по правил\
А. Н. Попова (см. стр. 199) или по типу расщепления озонидоь
112 Сопряженные кратные связи. Конъюгены
247
(См. стр. 81). В итоге оказывается, что молекула разрывается
по месту двойной связи. Таким образом, мы видим, что по
отношению к химическому воздействию двойная связь является
«слабым» местом молекулы.
5. Наличие двойной связи между атомами углерода
уменьшает подвижность других атомов (Н, CI), соединенных с этими
углеродными атомами (см. стр. 115).
Итак, мы знаем, что двойная связь слагается из в-связи и
л-связи. Химик, продолжая изображать двойную связь двумя
одинаковыми черточками, должен вкладывать в этот символ
иное содержание, более отвечающее современным взглядам.
Понятно также, что рассуждение о причинах «напряжения»
в двойной связи (см. стр. 243) интересно только как исторический
этап, через который шла мысль исследователей при первых
попытках объяснения изучаемых явлений.
112. Сопряженные кратные связи. Конъюгены. Электронные
представления позволяют объяснить ряд особенностей соединений,
содержащих несколько двойных (или, в общем случае,—кратных)
связей, находящихся в сопряженном (конъюгированном)
состоянии
—С=С—С=С—С=Х
ла:
В качестве простейшего примера рассмотрим строение дивини-
12 3 4
НаС=СН—СН=СН»
В молекуле этого соединения имеются четыре смежных атома
углерода с чередующимися о-, о- и it-связями (рис. 46).
Н
;с=с
Н'
н
Рис. 46. Схема строения дивинила.
Взаимное влияние электронных облаков приводит к сильному
изменению связи между вторым и третьим атомами углерода.
Исследование показало, что это уже не обычная о-связь с межатом-
248 Глава VII. Карбоновые кислоты и их производные
ным расстоянием 1,54 А; в дивиниле и его гомологах и аналогах
расстояние между вторым и третьим атомами равняется уже
1,46 А. Кроме того, все атомы дивинила лежат в одной плоскости.
Электронные представления позволяют гораздо лучше
объяснить ряд характерных химических свойств соединений с
сопряженными кратными связями, чем это объясняла гипотеза о
парциальных валентностях (см. стр. 97). Это в первую очередь
относится к реакциям присоединения. Теория Тиле могла
объяснить явление присоединения в положение 1,4, но она была
неспособна дать объяснение реакций присоединения в положение
1,2, что встречается довольно часто.
Рассмотрим, например, реакцию присоединения к дивинилу
молекулы брома.
В отличие от свободнорадикальных цепных реакций замещения
водорода (см. стр. 54), галоидирование в рассматриваемом случае
начинается с распада молекулы галоида на ионы:
—>~ Вг Вг
I
Ион брома с положительным зарядом, приблизившийся к
одному из крайних углеродных атомов, вызовет у этого атома
увеличение электронной плотности и одновременно поляризацию
всей молекулы (круглой стрелкой показан сдвиг электронной
плотности):
н2с=сн-сн=сн, и,с-сн-сн=си2
в <8 І в в в
Вг* Вг
Таким образом, частичный положительный заряд возникает
как у крайнего (четвертого атома), так и у среднего (второго)
атома.
В результате этого атом брома может присоединиться как ко
второму, так и к четвертому атому:
сн2—сн=сн—сн4 сн2—сн—сн=сн2
Вг Вг Вг Вг
1,4дибромбутен-2 1,2-диброу,оутен-3
C,4 ¦ дибромбутеи-1)
Образование продуктов присоединения в положения 1,2 и
3,4 или смеси обоих изомеров зависит, во-первых, от атомов или
113 Олеиновая, липа левая и линоленовая кислоты 24&
групп, которые имеются в такой сопряженной системе,
во-вторых, от присоединяющихся молекул и, в-третьих, от условий
реакции.
Сопряженные системы могут включать не только атомы
углерода, но и атомы иных элементов, например кислорода, азота
и др. Подобными «конъюгенами» из альдегидов являются
акролеин и кротоновый альдегид, из кислот и их
производных—акриловая кислота, ее эфиры и нитрил. У таких систем часто
наблюдается присоединение в положение 1,4. Эти продукты
присоединения иногда неустойчивы и подвергаются последующей
перегруппировке.
Вот как идет, например, восстановление кротонового
альдегида:
Н3С-СН=іСН-СН=О -^ НзС-СН2-СН=СН-ОН п"РегРУ°пяровк»
(ей- ол)
—*- HJZ—СН2-СН2-СН=О
Аналогично можно представить н присоединение молекулы«
аммиака INHJ—[HJ к эфиру акриловой кислоты:
ОС2Н5 OC2HS
н rJ^> XjD, _^ „ г съ-г пн Перегруппировка
г ®,-, ,+ Є ' I I
[nh^ [н] nh2 nh2 ос2н5
В органической химии есть очень много соединений,
содержащих системы из двух, трех и большего числа сопряженных
кратных связей (см., например, стр. 568 ел). Такие конъюгены
встречаются как среди ациклических, так особенно в ряде циклических
соединений. Таковы, например, многочисленные производные
ароматических, а также многих гетероциклических соеди~
нений.
113. Олеиновая, линолевая и лииоленовая кислоты. Олеиновая
кислота С17Н33СООН в виде глицеринового эфира чрезвычайно
распространена в природе. Ее строение выражается формулой:
СНэ-(СН.а^-СН=СН—(CHav- COOH
Олеиновая кислота—бесцветная маслянистая жидкость—легче
воды, на холоду затвердевает в игольчатые кристаллы,
плавящиеся при 14 °С. На воздухе она быстро окисляется, в результате чега
же теет.
250 Глава VII. Карбпновые кислоты и их производные
Молекула олеиновой кислоты способна присоединить два
атома галоида:
СН3—<СНа),-СН=СН-(СН.2),-СООН + Вг9 >
»СН3—(СН.2ь—СНВг—СНВг—(СНа),СООН
дибромстеариновая кислота
В присутствии катализаторов она присоединяет два атома
водорода, переходя в стеариновую кислоту.
При окислении олеиновой кислоты щелочным раствором
марганцовокислого калия происходит присоединение гидроксилов
к углеродным атомам с двойной связью:
СН3—(СН,),—СН=СН—(СН.,),—СООН + Н.2О + О »
»СН3— (СН2O—СН(ОН)—СН(ОН)—(СН.2),—СООН
Более энергичное окисление ведет к расщеплению молекулы:
! Р -по
!
СН3-(СН2O—СН=;=СН-(СН.2O-с
і хон
Р °
>С-(СНа):-С/
он
пеларгоновая кислота азелаиновая кислота
Образование в результате окисления олеиновой кислоты пе-
ларгоновой и азелаиновой кислот, в молекулах которых имеется
нормальная цепь из девяти атомов углерода, доказывает строение
олеиновой кислоты, т. е. наличие в ее молекуле нормальной
углеродной цепи и положение двойной связи между девятым и
десятым атомами углерода.
При действии незначительных количеств азотистой кислоты
олеиновая кислота переходит в твердый изомер—элаидиновую
кислоту.
Изомерия олеиновой и элаидиновой кислот является одним из
случаев цис-пгранс-нзомерцн:
Н—С-(СН,),-СНЭ Н—С-(СНаO—СН,
II II
Н—С—(СН.2O—СООН НООС—(СН2),—С—Н
цис изомер mfjuhu. нзомег:
олеиновоя кислота элаиднновая кислота
Олеиновая кислота получается как побочный продукт на
стеариновых заводах; используют ее для варки мыла, изготовления
пластыря, для замасливания шерсти перед прядением.
114 Митериалы ча основе полимеров акрил, и кетакрил. кислот' 251
Еще более ненасыщенными, чем описанная выше олеиновая
кислота, являются линолевая С,;На)СООН и линоленовая
С17Н29СООН кислоты, которые в виде сложных эфиров с
глицерином (глицеридов) образуют главную составную часть льняного
и конопляного масел. В молекуле линолевой кислоты имеются
две двойные связи. Ее молекула может присоединить четыре
атома водорода или галоида. В молекуле линоленовой кислоты
находятся три двойные связи; она присоединяет поэтому шесть
атомов водорода или галоида. Обе кислоты, присоединяя водород,
переходят в стеариновую кислоту.
114. Синтетические материалы на основе полимеров
производных акриловой и метакриловой кислот. Акриловая, а также мет-
акриловая кислоты в присутствии органических или
неорганических перекисей и кислорода легко полимеризуются при
температурах ниже 100 °С. Полимеры акриловой (так же как
метакриловой) кислоты и ее производных (сложные эфиры, нитрилы,
амиды,—см. стр. 238) называются полиакрилапгами. Это
обширный и разнообразный класс полимеризационных пластических
масс, получивший большое техническое значение. Полимеры
производных акриловой кислоты бесцветны, светостойки и
прозрачны; некоторые из них представляют собой твердые, упругие
стекла; другие—более мягкие, каучукоподобные и даже
воскообразные вещества.
В различных областях техники и быта наибольшее применение
получили полиакрилатные стекла. Ценным техническим свойством
полиакрилатов является способность пропускать
ультрафиолетовые лучи. Так, полиметилметакрилат пропускает свыше99%
солнечного света, и в этом отношении значительно превосходит
силикатные стекла. Преимущество полиакрилатных стекол
становится еще нагляднее, если сравнить их способность пропускать
ультрафиолетовую часть спектра; например, кварцевое стекло
пропускает 100% ультрафиолетовых лучей, полиметилметакри-
латное—73,5%, зеркальное силикатное—3%, обычное
силикатное—0,6%.
Органическое стекло в 10 раз прочнее минерального, легко
поддается механической обработке.
Большое значение приобретают акриловые водные эмульсии
(типа латекса) для пропитки ткани, дерева, бумаги, обработки
кожи и т. д. Они нашли применение в производстве липких
медицинских пластырей, для получения немнущихся тканей и в
качестве композиции при производстве искусственной кожи. Они
используются как покрытия для металла и дерева, а также в
строительной технике для придания водонепроницаемости бетону,
в качестве грунтовки при внутренней окраске стен, для пропитки
пористых строительных материалов и т. д.
25? Глава VII. Карбпновые кислоты и их производные
СЛОЖНЫЕ ЭФИРЫ КАРБОНОВЫХ КИСЛОТ
115. Строение и способы получения сложных эфиров. Из
структурных формул сложных эфиров карбоновых кислот
сн3—с/
ХО—СаН5
уксусноэтиловый
йфнр
С3Н,-С<^
масляноэтиловый
эфир
снэ-с/
ХО-С9Н,
уксу'_нопроинлпвый
^фир
видно, что сложные эфиры карбоновых кислот можно
рассматривать как продукты замещения водорода карбоксила
углеводородным радикалом.
Таким образом, общая формула сложных эфиров:
R-C/
где R и R'—углеводородные радикалы.
Строение эфиров доказывается их получением при действии
галоидных ал кил ов на соли кислот:
.О!
R-c/ I
+ Jj
Р
—R' » К-с/ + AgJ
XOR'
Названия сложных эфиров можно также составлять подобно
названиям солей: соли уксусной кислоты называются ацетатами,
а уксусноэтиловый эфир можно назвать этилацетатом, уксусно-
амиловый эфир—амилацетатом:
CH9COONa CHsCOOQHs CH3COOCBHU
уксуснокнслиіі «атрим уксусноэтиловый эфир уксусноамиловый эфир
(ацетат натрия) (этилацетат) (амилацетат)
Мб. Этерификация. Многие сложные эфиры легко получаются
непосредственным взаимодействием карбоновой кислоты и спирта
в присутствии серной кислоты. Эта реакция носит название
реакции этерификации.
Так, действием серной кислоты на смесь масляной кислоты и
этилового спирта при нагревании получают масляногтиловый
эфир:
О
CgH,—С—iOHT'Hi—О—C.HS » с.Н,—с/ + Н2О
Чэ-СН,
116 Этерификация
253
Из уксусной кислоты и пропилового спирта образуется уксус-
нопропиловый эфир:
О
СНЭ—С—ОН + Н—О—С3Н;
CH.,-
о
о-сэн,
При этой реакции от молекулы спирта отщепляется атом
водорода гидроксильной группы, а от молекулы кислоты—гидр-
оксил.
Такая схема
реакции, которая и раньше
считалась весьма
вероятной, в настоящее время
доказана
непосредственным опытом.
Юрей'A938)
обрабатывал бензойную кислоту
СвН5СООН (в присутствии
небольшого количества
хлористого водорода)
метиловым спиртом,
содержащим некоторое
количество изотопа кислорода
18Q.
Полученная в
результате- реакции вода не
была обогащена тяжелым
изотопом кислорода. Следовательно,
схеме: '
Рис. 47. Прибор для лабораторного
получения уксусноэтилового эфира.
реакция проходила по
QH6—с—О—н + Н-О—СН3
II
О
б-С—О-СНЭ + Н2О
о
т. е. гидроксил отщепляется от молекулы кислоты, а не от
молекулы спирта. ¦
На рис. 47 изображен прибор для лабораторного получения
уксусноэтилового эфира. В колбу, помещенную для
регулирования температуры в масляную баню, наливают смесь 50 мл
этилового спирта и 50 мл концентрированной серной кислоты. Смесь
подогревают до 140 °С и приливают к ней небольшой струей 400 мл
смеси равных объемов спирта и уксусной кислоты. Образующий-
254 Глава VII. Карбоновые кислоты а их производные
ся сложный эфир и воду отгоняют в приемный сосуд. Отогнанный
эфир взбалтывают для удаления примеси кислоты с раствором соды
и для удаления спирта с раствором хлористого кальция. Затем
эфир отдел я ют от воды в делительной воронке (он легче воды и
с водой не смешивается), сушат хлористым кальцием и снова
перегоняют. Этот общепринятый метод получения уксусноэтилового
эфира был предложен в 1873 г. В. В. Марковниковым, им же дано
объяснение механизма протекающей реакции.
Реакция этерификации обратима; она не доходит до конца,
так как наряду с этерификацией идет и обратная реакция
гидролиза (омыление), т. е. расщепление сложного эфира на кислоту и
спирт.
Равновесие, почти не изменяясь от температуры, существенным
образом зависит от природы и количества взятых кислоты и
спирта. Для этилового спирта и уксусной кислоты, в том случае, если
они взяты в равномолекулярных количествах, равновесие
наступает тогда, когда две трети спирта и кислоты обращены и
эфир.
Применяя в этом случае закон действия масс, легко понять,
что прибавление серной кислоты, связывающей воду, значительно
повышает предел этерификации. Точно так же, беря одно из
реагирующих веществ, т. е. кислоту или спирт в избытке, можно
достигнуть почти полной этерификации другого компонента.
Н. А. Меншуткин показал, что скорость образования
сложных эфиров различна для первичных, вторичных и третичных
спиртов. Как правило, первичные спирты, при образовании
сложных эфиров органических кислот, реагируют быстрее вторичных,
и вторичные—быстрее третччных. Если, например, нагревать
различные спирты с эквивалентным количеством уксусной
кислоты в течение одного часа до 155 °С, то первичные спирты
оказываются проэтерифицированными на 46—47%, вторичные—не
более чем на 22%, а третичные—примерно на 2% (данные
Н. А. Меншуткина).
Получение сложных эфиров из хлорангидридов и ангидридов
кислот см. на стр. 266 и 267; получение сложных эфиров из
альдегидов по реакции Тищенко см. на стр. 197.
117. Свойства сложных эмиров. Низшие представители
сложных эфиров—летучие, малорастворимые в воде жидкости.
Многие сложные эфиры имеют очень приятный запах, иногда
напоминающий запах фруктов.
1. Гидролиз (омыление) сложных эфиров. Характерной
особенностью сложных эфиров, определяющей в значительной мере
их свойства, является то, что остатки кислоты и спирта связаны
через атомы кислорода. Если разрыв углеродной цепи происходит
лишь с большим трудом, то связь через кислород в сложном эфи-
117. Свойства сложных эфиров 256
ре обычно легко разрывается даже при действии воды (гидролиз):
О
СН3—С— JO— СДГ+'Н)—°Н > СНзСООН ¦+¦ С2Н5ОН
уксусноэтиловый эфир ук<:уекая этиловый
кислота спирт
о
C3H,-C-;O—С?Н5 -f-Hj-OH > С3Н,СООН + QH5OH
масляноэтиловьш эфир масляная этиловый
кислота спирт
Гидролиз сложного эфира на спирт и кислоту при действии
воды происходит медленно, но при прибавлении небольшого
количества минеральной кислоты реакция значительно ускоряется.
Кислота при этом не подвергается никаким изменениям, она
только ускоряет реакцию, т. е. является катализатором. Точно
так же ускоряют омыление и щелочи, однако в этом случае
реакция несколько усложняется. Щелочь не только ускоряет реакцию
(ионы ОН являются катализатором), но и сама вступает в нее,
реагируя с образовавшейся кислотой, в результате чего
получаются соль и спирт:
CHaCOOGjHu + НоО » CH.COOH + СДШ
СН3СООН + NaOH » СНЯСОО№ + Н.2О
CH3COOCaH5 + NaOH » CH3COONa + QH5OH
Таким образом, присутствие щелочи делает реакцию
гидролиза необратимой.
2. Алкого.шз сложных эфиров. Нагревание сложных эфиров
в растворе спиртов может привести к обмену алкогольных
радикалов (переэтерификация):
NaOH
CH3COOC5HU + СаН5ОН ^z=r CH3COOC2H5 + CjHuOH
Реакцию проводят обычно в присутствии щелочи: можно провести
полный обмен спиртовых радикалов, если брать избыток спирта
и отгонять образующийся (более легкокипящнй) сложный эфир.
Этим путем готовят дфиры жирных кислот из жиров.
3. Аммонолиз сложных эфиров. При действии аммиака, часто
даже на холоду, сложные эфиры подвергаются расщеплению
с образованием амидов кислот:
О О
СН3—С + ІН
І І
СН3—С + QsH6OH
I
NH.
256 Глава VII. Карбоновые кислоты и их производные
4. Действие магнийорганических соединений на сложные эфи-
ры ведет к образованию кетонов и третичных спиртов:
СІ 1:1—О la—С » СН3—CHj—С—CHa
На стадии образования кетона реакцию можно остановить,
ведя процесс при недостатке магнийорганического соединения и
при сильном охлаждении. В противном случае кетон реагирует
далее; при этом образуется третичный алкоголь:
СН3
СН3—СНа—С—СНЭ + CH3MgJ » СН3—СН2-С-СН3
О OMgJ
/СН3 .
,—СН,—С— (
СН3—СН2—С—СН3 + Н2О » СНЭ—Ша—С—СН3 + MgJ(OH)
OMgJ
3 + 2Эа
OM ОН
5. Восстановление сложных эфиров приводит к образованию
первичных спиртов, соответствующих по числу атомов углерода
кислоте, из которой образован сложный эфир:
Ci8H81—COOCjHs » Ci5H31—СН2ОН + СаН5ОН
Реакцию проводят, действуя металлическим натрием на спир-
товый (этиловый или бутиловый) раствор сложного эфира.
В последнее время восстановление проводят газообразным
водородом под давлением в присутствии так называемого скемтного
никелевого катализатора (получаемого из сплава никеля с
алюминием путем обработки его раствором едкого
натра—катализатор Ренея).
118. Отдельные представители сложных эфиров карбонових
кислот. Многие сложные эфиры, как упоминалось выше, обладаюі
приятным фруктовым запахом.
Эфиры муравьиной кислоты. Муравьиноэтиловый эфир
//О
Н—Сс кипит при 55 °С; имеет запах рома.
NJCaH6
Эфиры уксусной кислоты. Уксусноэтиловый эфир, или этил-
ацетат,—жидкость освежающего запаха; темп. кип. 77 "С; мало
119. Воска 257
растворима в воде. Широко применяется как реагент и
растворитель. Как было указано раньше, этилацетат может быть
получен из уксусного альдегида при действии алкоголятов алюминия
(реакция Тищенко).
//°
Уксусноизоамиловый эфир, или изоамилацетат,СНа—(V
ч)с6ни
кипит при 139 "С; обладает запахом груш. В больших
количествах применяется как растворитель для изготовления лаков.
Эфиры масляной кислоты. Этиловый эфир масляной кислоты
СоН,—С<Г кипит при 121 °С, имеет запах абрикосов. Бути-
Х)С2Н5 и -
уР
ловый эфир масляной кислоты С,Н,-~С\ пахнет анана-
' X)CH
сами.
Эфир изовалериановои кислоты и изоамилового спирта пахнет
яблоками.
Перечисленные выше сложные эфиры применяются для
приготовления искусственных фруктовых эссенций, широко
используемых в производстве кондитерских и парфюмерных изделий,
а также фруктовых вод.
119. Воска. Воска состоят главным образом из сложных эфи-
ров высших жирных кислот и высокомолекулярных одноатомных
спиртов. Кроме того, они могут содержать высокомолекулярные
спирты и кислоты в свободном состоянии, а также и высшие
углеводороды. ПчеЛИНЫЙ ВОСК СОСТОИТ Преимущественно ИЗ МИ| ИЦИ-
лового эфира пальмитиновой кислоты С,5Н3|СООС30Н61; кроме
того, он содержит 10—14% церотиновой кислоты Са5Н5,СООН и
12—17% углеводородов.
Спермацет—твердое вещество, добываемое из маслянистой
жидкости, находящейся в черепных полостях кашалота; состоит
главным образом из цетилового эфира пальмитиновой кислоты
НзіС\
120 Жиры. Жиры весьма часто встречаются как в животных
организмах, так и в растениях. Как было указано, они
представляют собой сложные эфиры трехатомного спирта—глицерина и
различных кислот, главным образом стеариновой,
пальмитиновой, олеиновой, линолевой и линоленовой. Это доказано их
расщеплением на глицерин и кислоты и последующим синтезом
жиров из полученных продуктов.
17—788
258 Главо VII. Карбоновые кислоты и их производные
Строение жиров было установлено в начале XIX в. Шеврелем.
Обрабатывая щелочью животные и растительные жиры, он заметил, что масса
продуктов, получаемых при омылении жиров, превышает примерно на 5% массу
исходного жира Масса углерода остается при этом неизменной, масса же кислорода
и водорода увеличиваются в том же самом соотношении, в каком кислород и
водород содержатся в воде Отсюда он сделал вывод, что при омылении жиров
происходит присоединение воды.
Расщепляя жиры, Шеврель получил три кислоты; две
твердые—стеариновую и маргариновую и одну жидкую—олеиновую.
В некоторых жирах (например, в сливочном и касторовом маслах)
Шеврель, кроме указанных трех кислот, нашел и другие кислоты: так, им были
открыты масляная, капронопая и каприновая кислоты. Что же касается
маргариновой кислоты Шевреля, то она оказалась в дальнейшем смесью
стеариновой и открытой в середине XIX в. пальмитиновой кислоты.
Первый синтез жиров был произведен Бертло A854)
нагреванием глицерина с кислотами в запаянных трубках:
СН.2—О— |Н НО:— СО—С„Н35 СНа—О—СОС„Н35
I
СН—О—;Н + НО:
!
СН-О-СОС17НЭ5 + ЗН.,0
сн,-о-:н но-со-с17н35 сн2-о-сос17н35
глицерин стеаринооая кислота жир
Глицерид стеариновой кислоты называется тристеарином,
глицерид пальмитиновой кислоты—трипальмитином. Тристеарин и
трипальмитин—твердые вещества. Триолеин, т. е. глицерид
Олеиновой кислоты,—жидкость, затвердевающая при —4 °С. Гли-
цериды линолевой и линоленовой кислоты—тоже жидкости.
Первые три вещества в различных пропорциях составляют
главную часть сала. Чем больше в сале тристеарина и трнпаль-
митина, тем оно тверже (говяжье и баранье сало). Мягкое сало
(гусиное и свиное) содержит больше триолеина.
Жиры имеют громадное значение как питательные вещества.
По теплотворной способности они занимают в ряду питательных
«еществ первое место,—один грамм жира при сгорании дает
9300 кал. Жиры имеют большое техническое значение—они
служат исходным материалом для получения стеарина, мыла и
глицерина.
121. Расщепление жиров. Техническая переработка жиров
сводится главным образом к их омылению, т. е. к расщеплению
на глицерин и кислоты:
СНа-О-СОС17Нэ5
СН-О-СОС1:Н36 + ЗНаО > C3HO(OHK f ЗС17Н35СООН
сна-о—сос17нм
122. Стеариновое производство и мыловарение 259
Этот процесс ведут обычно в присутствии катализаторов, в
качестве которых применяют окиси некоторых металлов (известь,
магнезия, окись цинка) или специально приготовленные
органические вещества. Из последних широко применяется «контакт
Петрова», который получается при обработке некоторых
нефтяных фракций серной кислотой.
Контакт Петрова представляет собой густую прозрачную
жидкость от темно-желтого до бурого цвета с синеватым отливом.
Он содержит обычно около 40% нафтеновых сульфокислот, 15%
вазелинового масла, немного свободной серной кислоты и воду.
Контакт Петрова эмульгирует жиры, благодаря чему
увеличивается поверхность соприкосновения жира с омыляющей
жидкостью; это ускоряет реакцию.
Расщепление жиров в животном организме происходит под
влиянием особого энзима—липазы. Липаза выделяется
поджелудочной железой и находится в желудке, кишечном соке, крови
и т. д. Под действием липазы жиры расщепляются в кишечнике
на глицерин и кислоты, которые организм снова переводи!
в жиры.
Липаза находится во многих растениях. Особенно богаты
липазой, семена клещевины, которыми пользуются для технического
расщепления жиров.
Расщепление проводят при температуре от 10 до 4Э °С; при
этом жир переводят в эмульсию.
Жир смешивают с небольшим количеством воды, хорошо
перемешивают продуванием воздуха и добавляют к нему эмульсию,
полученную из семян клещевины, и, в качестве активатора для'
липазы, небольшое количество сернокислого марганца. В
течение примерно двух суток расщепление происходит почти
полностью.
Липаза расщепляет не только жиры, но и почти все другие
сложные эфиры.
122. Стеариновое производство и мыловарение. Исходным
материалом для стеаринового производства служат твердые жиры,
из которых при омылении серной кислотой получаются главным
образом стеариновая, пальмитиновая и олеиновая кислоты.
Полученные кислоты, предварительно очищенные перегонкой с
водяным паром, завертывают в салфетки из грубой ткани и
загружают в гидравлический пресс. Под давлением жидкая олеиновая
кислота отжимается, и в салфетках остается смесь стеариновой
и пальмитиновой кислот. Полученная масса называется
стеарином.
Олеиновая кислота перерабатывается на мыло.
В мыловаренном производстве омыление ведется при помощи
щелочей. В этом случае жиры расщепляются на глицерин и
17*
260 Глава VII. Карбоновые кислоты и их производные
мыла, т. е. натриевые или калиевые соли высокомолекулярных
кислот:
СН2-О—СОС17Наг,
СН—О—СОС17Н35 + 3№ОН » С,Н5(ОНK + 3Q,Ha5COONa
I глицерин мыло
СНо—О—COQjHgs
тристеарин
Для варки мыла берут самые разнообразные жиры: сало,
конопляное и хлопковое масла и т. д. Большое значение для
мыловарения имеет олеиновая кислота, получаемая при стеариновом
производстве.
На некоторых заводах варка мыла осуществляется другим
способом. Вначале производят расщепление жира на глицерин и
кислоты, которые затем обрабатывают содой:
2Q7H35COOH + Na2CO, » 2C17H35COONa + Н2О + СО2
Этот способ позволяет получать весьма чистый глицерин и
заменить дорогой едкий натр более дешевой содой.
123. Растительные масла. Растительные жиры называются
маслами, они бывают твердые и жидкие. Твердые масла
(пальмовое, кокосовое) отличаются значительным содержанием тристеари-
на и трипальмцтина, в то время как жидкие масла состоят
главным образом из глицеридов непредельных кислот—олеиновой,
линолевой или линоленовой. По своей способности изменяться на
воздухе жидкие масла разделяются на высыхающие,
полувысыхающие и невысыхаигшие.
Невысыхающие масла (оливковое, миндальное) состоят
преимущественно из триолеина. При хранении, особенно на свету,
они приобретают горький вкус—прогоркают. При прогоркании,
под действием главным образом микроорганизмов, жир
расщепляется на глицерин и кислоты—как предельные, так и
непредельные. Непредельные кислоты, окисляясь, расщепляются на
альдегиды и кислоты с меньшим числом атомов углерода в
молекуле. Предельные кислоты под влиянием микроорганизмов
расщепляются с образованием кетонов. Некоторые из этих кетонов,
например метилгептилкегон, отличаются неприятным
прогорклым запахом. Расщепление кислот происходит по схеме ?-окисле-
ния, т. е. окисляется углерод, находящийся в ?-положении к
карбоксильной группе, например:
СНа—(СН2)„—СН,— СН2—СООН »
! -СОї
» СНЭ-(СН2L—СО—CH2-;COO;H » СН3—(СН2L—СО—СНЭ
метнламилкет он
123. Растительные масла
261
Высыхающие масла (льняное, конопляное) состоят
преимущественно из глицеридов линолевой и линоленовой кислот.
Характерной особенностью этих масел является нх способность на
свету при действии воздуха, особенно в тонком слое, высыхать,
образуя твердую эластичную пленку. При высыхании масла
соединяются с кислородом; при этом температура повышается
настолько, что тряпье, смоченное маслом, может самопроизвольно
загореться. Вес масла при высыхании увеличивается в среднем
на 11—18%. Окисленное и полимеризованное льняное
масло представляет собой линоксин. Это бурая, густая, эластичная
мясса. Применяется в производстве линолеума, линкруста,
клеенки.
Полу высыхающие масла (подсолнечное, хлопковое) отличаются
от высыхающих масел малым содержанием линоленовой кислоты,
а от невысыхающих—сравнительно большим содержанием
линолевой кислоты.
Примерный состав некоторых жиров и масел приведен в
табл. 11.
Таблица 11
Состав некоторых жиров и масел
Название
Говяжье сало . .
Коровье масло (содержит немного гли-
церида масляной кислоты)
Оливковое масло
Хлопковое масло
Подсолнечное масло
Льняное масло
Конопляное масло
Содержание, %
тристеаринй
и трипаль-
митина
75
53
25
25
6,5
8
0
гриолеина
25
39
70
25
33
18
15
трилино
леина
0
0
5
47
58
30
70
других
глицеридов
0
8
0
3
2,5
44
15
Степень непредельности жира или масла определяют по
количеству брома или иода, поглощаемого известным количеством
вещества. На практике предпочитают пользоваться иодом, но так
как прямое присоединение иода по месту двойной связи
наблюдается очень редко, то эту реакцию проводят в специальных
условиях—в спиртовом растворе сулемы. Степень
непредельности выражают так называемым йодным числом, т. е. числом
граммов иода, поглощенных 100 г жира. Очевидно, что это число
гем выше, чем больше непредельных глицеридов содержится в
жире или масле.
262 Глава VII. Карбоновые кислоты и их производные
Приводим йодные числа некоторых жиров и масел:
Говяжье сало .... 35—40 Подсолнечное масло. 129—136
Свиное сало 48—64 Конопляное масло . . 145—162
Оливковое масло . . 77—91 Льняное масло . . . 175—201
Хлопковое масло . . . 104—116
Высыхающие масла в большом количестве применяются для
изготовления олифы—густой жидкости, образующей быстро
сохнущую пченку. Для приготовления олифы льняное масло
нагревают, добавляя катализаторы, называемые сиккативами. В качестве
катализаторов беру і марганцовые, свинцовые или кобальтовые соли
некоторых кислот (льняного масла или канифоли). По одному из
способов для изготовления сиккативов нагревают льняное масло,
канифоль или нефтяные кислоты с определенным количеством
сурика РЬ3О4, двуокиси марганца МпО2 или уксуснокислого
кобальта (СН3СООJСо. Обычно сиккативы используют в виде их
растворов в нефтяных маслах.
Сущность процесса варки олифы состоит в полимеризации и
окислении масла. Иногда до внесения сиккативов в нагретое
масло пропускают воздух, кислород которого производит
окисление глицеридов.
Олифа применяется для изготовления масляных красок и
лаков и в типографском деле. Хорошая олифа высыхает в течение
4—5 ч.
Типографскую олифу варят без применения сиккативов и без
доступа воздуха—в атмосфере углекислого газа.
124. Гидрогенизация жиров. Жидкие растительные масла
состоят главным образом из глицеридов олеиновой, линолевой
и линоленовой кислоты и содержат только незначительное
количество трипальмитина и тристеарина.
Химиков издавна занимал вопрос о превращении жидких
растительных масел в твердые жирої, пригодные для технического
использования и для пищевых целей. Эта задача была решена
в начале XX в., и в настоящее время переработка растительные
масел (и ворвани) в твердые жиры составляет целую отрасль
промышленности.
Сущность этой переработки, называемой гидрогенизацией
жиров, заключается в переводе жидких глииеридов ненасыщенных
кислот в твердые глицериды насыщенных кислот путем
присоединения водорода. Присоединение водорода производится легко
под давлением в присутствии некоторых катализаторов (обычно
мелко раздробленного никеля). Для гидрогенизации жиров
через находящееся в автоклаве нагретое растительное масло, в
котором суспендирован катализатор, пропускают водород (рис. 48).
Полученный продукт для отделения от катализатора фильтруют
125. Синтетические жирные кислоты
263
в теплом состоянии под давлением через полотно и разливают в
бочки, где он и застывает. Этот продукт называется саломасом.
Впервые технологический способ гидрогенизации жиров
разработал С. А. Фокин A865—1917).
Из некоторых сортов растительных масел получают
прекрасные сорта съедобных жиров, которые после смешения с
растительным маслом и молоком идут в пищу (маргарин).Смешение
осуществляется в особых машинах, превращающих загруженную в
них массу в эмульсию.
125. Синтетические
жирные кислоты. С целью
замены пищевых жиров для
мыловарения разработан
способ промышленного
получения карбоновых кислот с
числом углеродов в цепи от
10 до 20 из парафина.
Твердый парафин состои'і из
предельных углеводородов,
имеющих следующий состав:
СН3—(СН2)„— СН3, где п=
от 18 до 38. Окисление
проводят воздухом при 100—
120 DC в присутствии
катализатора, в качестве
которого чаще всего применяют
перманганат калия. В этих
условиях происходит разрыв
углеродной цепи и окисление
концевых групп СН3 до
карбоксила. Теоретически
разрыв цепи может происходить в любом месте, и при
окислении всегда получается вся гамма кислот от муравьиной до
CH3(CH2Ke— COOH. Практически условия окисления подбирают
таким образом, чтобы получить максимальный выход кислот,
пригодных для изготовления мыла (с числом углеродных атомов
оіМО до 20).
Основная происходящая при этом реакция может быть описана
следующим уравнением:
СН3-(СН.2)„-(СН2)т-СН3 + 2VA >
> СН3—(СНз),,-!—СООН + СН3—(СН2)т_,—СООН
Рис. 48. Аппарат для гидрогенизации
жиров.
Н2О
Окисление проводят в вертикальных колоннах, заполненных
расплавленным парафином. Снизу через расплав продувается воздух, в верхнюю часть
колонны непрерывно добавляются небольшие количества перманганате. Во
264 Глава VII. Карбоновые кислоты и их производные
избежание образования побочных продуктов процесс прерывают, когда
окислилось лишь около 40% парафина. После отмывки водой образовавшихся
низших кислот (муравьиной, уксусной и др.) продукты реакции обрабатывают
раствором щелочи; соли жирных кислот переходят в раствор и отделяются от не-
прореагировавшего парафина. После подкисления серной кислотой
полученную смесь жирных кислот разделяют на фракции дистилляцией под
вакуумом.
Синтетические жирные кислоты полноценно заменяют в
мыловарении кислоты, полученные из растительных и животных жиров.
Они отличаются от них только числом углеродных атомов.
Природные кислоты всегда содержат четное число углеродных атомов
в молекуле, тогда как синтетические кислоты представляют
собой смесь примерно равного количества кислот с четным и
нечетным числом атомов в молекуле.
ХЛОРАНГИДРИДЫ КИСЛОТ
126. Строение и получение хлорангидридов кислот.
Замещением в спиртах гидроксильной группы на галоид получают
галоидные алкилы (стр. 141). Гидроксил карбоксильной группы
так же, как и гидроксил в спиртах, может быть замещен
галоидом. В результате такого замещения получаются галоидангид-
риды кислот, из которых наибольшее значение имеют хлоран-
гидриды. Таким образом, хлорангидриды карбонових кислот
можно рассматривать как продукты замещения гидроксила в
кислотах хлором.
Примерами хлорангидридов могут служить: хлорангидрид
уксусной кислоты, или хлористый ацетил, СН3СОС1 и
хлорангидрид пропионовой кислоты, или хлористый пропионил,
С2Н5СОС1. Строение хлорангидридов одноосновных кислот
можно выразить общей формулой
R-c/
.О
С1
Хлорангидриды получают действием на карбоновые кислоты
пятихлористого или треххлористого фосфора:
ЗСН3С-ОН + РС13 » ЗСН3С-С1 + Н3РО3
II II
О О
уксусная хлористый фосфористая
кислота ацетил кислота
127. Свойства хлорангидрадов кислот
265
На рис. 49 изображен прибор для лабораторного получения
хлористого ацетила. В круглодонную колбу с боковым отводом
помещают 50 г безводной уксусной кислоты, а в капельную
воронку, которая закреплена в пробке колбы, наливают 40 г трех-
хлористого фосфора. Колбу погружают в водяную баню и
соединяют с трубкой холодильника, другой конец которой соединен
с приемной склянкой. Приливают в колбу, охлаждаемую водой,
по каплям, треххлористый фосфор. Затем нагревают водяную
баню и отгоняют образовавшийся хлористый ацетил (темп. кип.
55 °С) в приемную склянку.
Рис. 49. Прибор для получения хлористого ацетила,
Для предотвращения взаимодействия хлористого ацетила с
влагой воздуха на боковой отросток приемной склянки надевают
трубку, наполненную хлористым кальцием.
127. Свойства хлорангидридов кислот. Низшие хлорангидриды
кислот представляют собой жидкости тяжелее воды; пары их
обладают острым запахом и раздражают слизистые оболочки
носа и глаз.
Наиболее замечательным свойством хлорангидридов является
чрезвычайная подвижность находящегося в них хлора. Так, хлор-
ангидриды очень быстро разлагаются даже холодной водой,
давая кислоту и хлористый водород
СН3СО-;С1 + Н;-ОН
СН3СООН + НС1
266 Глава VII. Карбоновые кислоты и их производные
Легкокипящие хлорангидриды дымят на воздухе. Это
происходи і потому, что их пары, реагируя с влагой воздуха по
вышеприведенному уравнению, образуют іуман из капелек соляной
и органической кислот.
Хлорангидриды кислот с большим молекулярным весом
реагируют с водой менее энергично.
При действии хлорангидридов на спирты получаются
сложные эфиры:
сНзСО—|сї Тни—о—с2н5 —> сн3со—о—с,н5 + на
уксусноэтилаеып
АНГИДРИДЫ КИСЛОТ
128. Строение и получение ангидридов кислот. Известны
вещества, которые можно представить как результат отнятия
одной молекулы воды от гидроксилов карбоксильных групп двух
молекул карбоновой кислоты. Такие вещества называются
ангидридами кислот:
i_H СН,—С
н2о
,0-Н гн /
/ СН3—С
СН3-С
уксусная ангидрид
кислота уксусной
кислоты
или в сбщем виде:
R-C7
ЮН R—(x.
, >О f И2О
,он R_c/
R-C
ангидрид
Эта реакция может быть осуществлена пропусканием паров
кислот над некоторыми катализаторами.
129. Свойства ангидридов кислот 267
Способом получения ангидридов, который доказывает их
строение, является действие хлорангидридов на натриевую соль
кислоты, например:
СНЧСОЧ
СНЭСО—О—Na + С!—СОСН3 » ' >О + NaCI
СИ-СО '
уксуснокислым
натрнП
хлористый
ацетил
ангидрид
уксуной
кислоти
При помощи этой реакции Жерар в 1852 г. впервые получил
ангидриды одноосновных кислот.
129. Свойства ангидридов кислот. Низшие представители ан-
підридов кислот—жидкости, нерастворимые в воде, имеющие
острый запах.
По химическим свойствам ангидриды подобны хлорангидри-
дам, однако реакции ангидридов, в частности с водой, протекают
менее энергично, чем реакции' хлорангидридов. Так, ангидриды
быстро расщепляются водой только при нагревании:
(СН,СО).2О + Н,0 > 2СНЭСООН
уксусный уксусная
ангидрид кислота
Более быстро реакция идет со щелочами:
(CH3CO)aO + 2NaOH > 2CH3COONa + Н2О
Со спиртами ангидриды образуют сложные эфиры:
(СН;)СОKО + CjHjjOH » СН:,СООС2Н5 + СНзСООН
Уксусный ангидрид—жидкость тяжелее волы; кипит при 140 °С.
В большом количестве применяется для приготовления
искусственного (ацетатного) волокна.
Ангидрид муравьиной кислоты неизвестен; при попытках
получения он в момент образования распадается на СО и HSO.
АМИДЫ КИСЛОТ
130. Строение и получение амидов кислот. Амидами кислот
называются продукты замещения гидроксильной группы кислот
остатком аммиака—аминогруппой NH2.
Н—СО—NH2 амид муравьиной кислоты (формамид)
Н—СО—N(CH3)j диметилформамид
СН3—СО—NHa амид уксусной кислоты (ацетамид)
QHr,—СО—NHa амид пропионовой кислоты
СзН7—СО—NH.j амид масляной кислоты
268 Глава Vif. Карбоновые кислоты и их производные
Общая формула амидов кислот
Р
4NHa
Амиды кислот можно получить действием аммиака на другие
производные кислот, например:
а) Хлорангидриды
О
II
СН3—С—!С1 + Hj-NH2 » CH3CONH2 + HCI
б) Ангидриды
сн3-соч І І
>О + Н—NH2 » CH3CONH2 + СН3СООН
СН3—СО/
t
в) Сложные эфиры
о
II
СН3—С— ;О—С2Н5+ Hi—NHa > CH3CONH2 + QH5OH
Можно получить амиды кислот нагреванием аммонийных солей
карбоновых кислот:
CH3COONH4 > CH3CONH2 + Н2О
а также частичным гидролизом нитрилов кислот:
С2Нб—CseN + Н2О » QHs—CO—NH2
131. Свойства амидов кислот. Амиды кислот, за исключением
жидкого амида муравьиной кислоты,—кристаллические тела.
В отличие от аммиака они почти не обладают основными
свойствами. Только с сильными кислотами амиды кислот дают соли
(например, CH3CONH2-HC1), легко разлагаемые водой.
Водород аминогруппы в амидах кислот способен замещаться металлом;
так, для ацетамида известно соединение состава (CH3CONHJHg.
При кипячении с водой в присутствии кислот и щелочей амиды
кислот образуют аммиак и соответствующую кислоту:
СН3СО—;NH2 + НІ—ОН * СН3СООН + NH,
132. Производные кислот и кислотный радикал 'ib'J
Действие на амиды азотистой кислоты приводит в выделению
азота и образованию карбоновой кислоты:
R—CONH2 + HONO > RCOOH + N2 + Н/)
132. Производные кислот и кислотный радикал. Строение
кислот выражается формулой
R-c/
,0
ОН
Сложные эфиры можно рассматривать как продукты замещения
водорода в гидроксиле кислоты на углеводородный радикал
(формула I).
Хлорангидриды кислот получаются при замещении гидроксн-
ла хлором, их строение можно выразить формулой П.
Ангидриды кислот образуются при отнятии воды от гидрокси-
лов двух молекул кислот и имеют строение, соответствующее
формуле III.
Амиды кислот—это продукты замещения гидроксила кислоты
аминогруппой (формула IV).
Наконец, не требует пояснения, что строение солей можно
выразить формулой V (где Me—металл)
О
,р у.О R-C/
R-Of R-C< >0
ХО—R' NC1 R —C<4
і її in
.0 ,O
R-Gf R— C<f
XNH2 XO—Me
IV V
Таким образом, все эти вещества производятся от кислот
путем изменений только в гидроксильной группе, группа же атомов
R—СО—остается неизменной. Такие вещества, которые
производятся от кислот в результате изменений в гидроксильной
группе, называются производными кислот; неизменяющаяся же часть
молекулы называется кислотным радикалом или ацилом.
Кислотный радикал муравьиной кислоты Н—СО—
называется формилом; радикал уксусной кислоты СН3—СО—
ацетилом; радикал пропионовой кислоты С2Н5—СО— пропионилом;
радикал масляной кислоты С3Н,—СО— бутирилом и т. д.
'270 Глава VII. Карбонояыв кислоты и их производные
ПРЕДЕЛЬНЫЕ ДВУХОСНОВНЫЕ КИСЛОТЫ
133. Строение предельных двухосновных кислот. Примерами
предельных двухосновных кислот могут служить:
НООС—СООН НООС—СН.,—СООН
щавелевая кислота малиновая кислота
НООС—СН2—СН.,—СООН НООС—СН,—CH.j—CH.j—СООН
ангарная к;іслота глугаровая кислота
Гомологический ряд предельных двухосновных кислот может
быть выражен обшей формулой:
(СН2)„(СООНJ
При п=0 получается формула первого члена этого
гомологического ряда Как обычно, первые гомологи имеюі ряд особенностей,
отличаюших их от последующих «типичных» гомологов.
Из кислот этого ряда наибольшее значение имеют те, в
которых функциональные группы находятся на концах нормальной
углеродной цепи, т. е. кислоты, которые имеют строение
НООС-(СН2)„-СООН.
Заметая в молекулах этих кислот атомы водорода метилено-
вых групп алкиламм, можно получить двухосновные кислоты с
разветвленной цепью углеродных аюмов. Таким путем от
малоновой кислоты можно вывести ряд кислот, которые являются ее
непосредственными гомологами:
НО( )С—CHj—СОСН
малонокая кислота
сн3 с,н5 снэ
НООС—СН—СООМ НООС—СН—СООН НООС—С—СООН
метилмалоновая эгилма./юновая I f
кис-лога кислита СН3
ди мет и л ма лоно вая
кислота
Аналогичные ряды можно вывести от янтарной, глутаровон
и других кислот.
134 Физические и химические свойства двухосновных
предельных кислот. Физические свойства некоторых двухосновных
кислот приведены в габл. 12.
Предельные двухосновные кислоты—твердые
кристаллические вещества. Подобно тому, как это отмечалось для
одноосновных кислот (см. стр. 224), предельные двухосновные кислоты
с четным числом атомов углерода плавятся при более высокой
температуре, чем соседние гомологи с нечетным числом атомов
углерода. Растворимость в воде кислот с нечетным числом ато-
134. Физические и химические свойства двухосновных предельных кислот 27\
Таблица 12
Кислота
Щавелевая . .
Ма.вдновая . .
Янтарная . . .
Глутаровая . .
Лднпиновая . .
Пимелиновая .
Пробковая .. .
Азелаииовая . .
Себациновая . .
Физические свойства
Формула
ноос—соон
ноос—сн.,—соон
НООС—(CHa)s—СООН
НООС—(СНа)я-СООН
НООС—(СН2L—СООН
НООС—(СН.,),—СООН
НООС—(СН2)„—СООН
НООС—(CHJ,—СООН
НООС—(СН.2)8—СООН
двухосновных
Температура
плавления
безводного
вещества, °С
189,5
130,3
182,8
97,5
153
105,5
140
107,5
134,5
кислот
Растворимость
в г на 10и г
воды при 20 °С
8,6
73,5
5,8
63,9
1,5
5,0
0,16
0,24
0,10
Первая
константа
электролитической
диссоциации
3,8- Ю-2
1,58-10-а
6,65-10-»
4,75-10-6
3,71-Ю-5
3,49-10-»
2,99 10
2,53-Ю-5
2,38- Ю-5
мов углерода значительно выше растворимости кислот с четным
числом атомов углерода, причем с возрастанием числа атомов
углерода растворимость кислот в воде уменьшается.
Двухосновные кислоты диссоциируют последовательно:
НООС—СООН
ІНООС—СООГ
[НООС—СОО]~
|ООС—СООГ"
-Н'
Они сильнее соответствующих одноосновных кислот.
Особенно сильно диссоциирует на ионы щавелеиая кислота. Степень
диссоциации двухосновных кислот понижается с увеличением их
молекулярного веса.
В молекуле двухосновных кислот содержатся две
карбоксильные группы, и поэтому они дают два ряда производных: средние
и кислые соли, средние и кислые сложные эфиры:
.СООН .COOQH5
СН,< СН./
"чсоос,н6 N:ooc,h5
«и-лый чфнр
маломовои кислоты
среаниГі зфир
малоновом кисло
СО,:
При нагревании щавелевой и малоновой кислот отщепляется
H.OOG'1—СООН —
н;оос1—сн.2—соон
нсоо: і + со.,
-> СН3СООН + СО2
272 Глава VII. Карбоновые кислоты и их производные
Двухосновные кислоты с четырьмя и пятью атомами углерода
в молекуле, т. е. янтарная и глутаровая кислоты, при
нагревании отщепляют элементы воды и дают внутренние циклические
ангидриды:
И—» О
1/0-н; L
янтарная кислота ангидрид
янтарной кислоты
¦P ,p
+ н2о
глугарован кислота ангидрид
глуіаровой кислоты
В молекулах этих ангидридов цепи из четырех или пяти
атомов углерода замкнуты в кольцо атомом кислорода.
Способность янтарной и глутаровой кислот давать
циклические ангидриды можно легко объяснить, пользуясь тетраэдри-
ческой моделью Вант-Гоффа. При расположении атомов углерода
в виде клешни (см. рис. 24,а и б на стр. 162) конечные звенья цепи
из четырех, а особенно из пяти атомов будут весьма близко
подходить друг к другу. Если на концах этой цепи находятся гидрок-
сильные группы (как, например, у янтарной и глутаровой кислот),
то кислород, валентные связи которого направлены под
некоторым углом (см. стр. 65), легко замкнет кольцо.
135. Щавелевая кислота. Щавелевая кислота (этандиовая
кислота) НООС—СООН является простейшей из двухосновных
кислот.
Щавелевая кислота очень распространена в природе: кислая
калиевая соль ее НООС—COOK находится в шавеле и кислице,
щавелевокислый кальций (СООJСа—во многих растениях.
Щавелевая кислота, как первый представитель
гомологического ряда, подобно муравьиной кислоте, отличается по своему
строению и химическому характеру от других кислот. В самом
деле, все карбоновые кислоты—уксусную, пропионовую,
стеариновую и т. д. можно рассматривать как производные
углеводородов, в которых водород замещен карбоксилом. Муравьиная
же и щавелевая кислоты не подходят под это определение. Му-
136. Малоновая кислота 273
равьиная кислота является соединением карбоксила с атомом
водорода; щавелевую кислоту следует рассматривать как два
карбоксила, связанные друг с другом. В свойствах муравьиной
и щавелевой кислот есть много общего.
Щавелевая кислота кристаллизуется с двумя молекулами воды
С2Н2О4 2Н2О; ее можно получить и в виде безводных
гигроскопических кристаллов.
При быстром нагревании щавелевая кислота разлагается:
— СО,, + НСООН
НООС—СООН —
-* COj + СО + НаО
Если нагревание вести в присутствии серной кислоты, то
продуктами реакции являются только углекислый газ, окись
углерода и вода.
Характерным свойством щавелевой кислоты является ее
легкая окисляемость. При окислении щавелевой кислоты
получаются углекислый газ и вода.
Щавелевая кислота образуется при окислении многих
органических веществ. Один из способов ее технического получения
основывается на окислении сахара азотной кислотой в
присутствии ванадиевого катализатора:
С12Н,,ОИ + 12HNO3+ НаО » 6НООС—СООН-2Н2О + 12NO
Щавелевая кислота является довольно сильной кислотой,
константа ее электролитической диссоциации примерно в 2000 раз
больше константы диссоциации уксусной кислоты.
Щавелевая кислота и ее соли—оксалаты—применяются
при ситцепечатании, а также для удаления ржавых пятен с
тканей.
136. Малоновая кислота НООС—СН2—СООН найдена в соке
репы. Для синтетического получения малоновой кислоты в
молекуле уксусной кислоты замещают один из атомов водорода
хлором, получая хлоруксусную кислоту:
сі—ja + н;—сн.2соон —> сіснасоон + на
Взаимодействием хлоруксусной кислоты с цианистым калием
получают циануксусную кислоту.
.СООН .СООН
СНа< > СН,< + КСІ
(9 ±.'.КІ—CN \CsN
18—788
274 Глава VII. Карбоновые кислоты и их производные
которая после омыления превращается в малоновую кислоту:
,СООН УСООН
СН2< + 2Н2О > СЛ/ + NHa
При нагревании малоповой кислоты несколько выше
температуры плавления она отщепляет СО2 и переходит в уксусную
кислоту:
ХООН
С\\/ > СН3СООН + СО,
\соо;н
Таким же образом алкилмалоновые кислоты при нагревании
до температуры выше точки плавления, теряя COS,
превращаются в алкилуксусные кислоты. Так, метилмалоновая кислота дает
метилуксусную, т. е. пропионовую кислоту:
,соон
СНа—СН< > СНаСН2СООН + СО2
Ш9 :hl
Метилэтилмалоновая кислота превращается в метилэтилук-
сусную кислоту, т. е. в один из изомеров валериановой кислоты:
снзч усоон снач
>С< , >СН—СООН + СОа
Cfl/ XiC°.°jH СЛ
В обиіей форме это превращение может быть выражено
уравнениями:
/СООН
R_CH< » R-CHjCOOH+CO,
R4 /СООН R.
>СН—
R4 /
>С<
R'/ \
ХЮН R'
Карбоксильная группа усиливает реакционную способность
атомов водорода у соседнего углерода (см. стр. 273, 228).
В малоновой кислоте и ее эфирах подвижность водородов
группы СН2, связанной с двумя карбоксилами, особенно велика: эти
водороды легко замещаются галоидами:
С /Н 4-Bn HOOQ М
>С< '-* >С<
137. Синтезы при помощи малонового эфира
При действии альдегидов образуются непредельные кислоты:
R—СН = О + Н,С(СООНJ » Hfl + R-CH = C(COOHJ
R-CH=C(COOHJ » СО2 + R-CH=-CH-COOH
137. Синтезы при помоши малонового эфира. Из производных
малоновой кислоіьі наибольшее значение имеет ее диэтиловый
эфир, называемый нередко просто малоновым эфиром:
сн/
чсоос.д,
Он представляет собой жидкость с фруктовым запахом,
кипящую при 199 °С. МалоноЕЫЙ эфир широко применяется в
лабораториях лля различных синіезов.
Замечательное сеойстео этого соединения заключается в том,
что атом водорода в его метиленовой группе способен замещаться
натрием с образованием натриймалонового эфира:
Na—C-H
Для получения натриймалонового эфира пользуются этилатом
натрия.
При действии на натриималоновый эфир галоидных алкилов
образуются эфиры замещенных малоновых кислот, например:
,СООС.,Н5 yCOOQH6
QH6—:J + Na: —СН< ' » QH6—CH< + NaJ
XCOOQHa XCOOC2Ha
этиловый эт)ир
эти лмалоноЕюй
кислоты
При омылении этого эфира едким кали получается калиевая
соль этилмалоновон кислоты
XOOCjH, -COOK
< +2КОН » СгНй—СН< + 2С3Н6ОН
X100QH, ХСООК
калиевая силь
этилма^очоюй
КИСЛОТЫ
276 Глава VII. Карбоновые кислоты и их производные
из которой легко получить свободную кислоту:
.COOK .СООН
С>н5_СН< + 2НС1 » QH6-CH< + 2KCI
NCOOK \ZOOH
атилмалоноьая
л ислота
При нагревании этилмалоновой кислоты происходит
отщепление молекулы СО2 и получается масляная кислота:
/:СОО:Н
QH5-CH< ' , С,Н5СН2СООН + СО3
\соон
масляная
кислота
В молекуле однозамещенного малонового эфира второй атом
водорода может быть также замещен атомом натрия, причем и
последний можно обменять на углеводородный радикал.
В качестве примера приведем синтез одного из изомеров
валериановой кислоты, а именно метилэтилуксусной кислоты:
.COO
Na—CH<
ХЮО
» СН3—С—Na
/ HC / \ /н
¦ СН3—С—GjHs * СН3—С—G}H5 » С
/Cook HC] /іС92Ін с^\ /
3—С—GH* СНСGH С
js3}5
\ХЮК ^СООН
метил этил уксусная
кислота
Таким образом, в общем виде получение при помощи
малонового эфира замещенных уксусной кислоты может быть выражено
следующим уравнением:
,COOQH5 .JCOO'JH
R—CH -¦ R—CH —>~ R—СИ.,—СООН
\с
оон
2 ' /¦• ; R
R—С—R' -¦ R'—С—R -¦ >CH—СООП
\cooh Rv
138. Янтарная кислота 277
138. Янтарная кислота. Янтарная кислота (І) была впервые
получена перегонкой янтаря; она содержится в бурых углях и
во многих растениях.
При перегонке янтарная кислота образует янтарный
ангидрид (II):
сн,—соон сн,— соч
I * I >о
сн.,—соон -Н2° сн2-сск
] II
Янтарный ангидрид—кристаллическое вещество (темп, плавл.
120 °С; темп. кип. 261 °С). Янтарный ангидрид растворяется в
воде, на холоду он лишь медленно присоединяет воду, образуя
янтарную кислоту-
Многие двухосновные карбоновые кислоты приобрели
большое значение в технике. Сложные эфиры бутилового, октилового,
а также других спиртов и адипиновой, себациновой и ортофта-
левой (см. стр. 474) кислот являются пластификаторами для
поливинилхлорида и некоторых других полимеров. Кроме того,
адипиновая кислота вырабатывается в больших количествах в
качестве промежуточного продукта в производстве
синтетических полиамидных волокон.
НЕПРЕДЕЛЬНЫЕ ДВУХОСНОВНЫЕ КИСЛОТЫ
139. Фумаровая и малеиновая кислоты. В сорном растении
дымянке и во многих грибах встречается фумаровая кислота
НООС—СН=СН—СООН, являющаяся простейшим
представителем непредельных двухосновных кислот. Синтетическим путем
может быть получена малеиновая кислота, состав которой
выражается той же формулой.
Обе кислоты образуются при нагревании яблочной кислоты:
яблочная кислота
278 Глава VII. Карбоновые кислоты и их производные
При осторожном нагревании получается главным образом
фумаровая кислота, при более сильном нагревании—малеиновая
кислота*.
Одинаковое строение малеиновой и фумаровой кислот
подтверждается тем, что обе они при восстановлении дают янтарную
кислоту:
НООС—СН = СН—СООН 4- 2Н > НООС—СН,,—Щ,—СООН
Несмотря на одинаковое строение, фумаровая и малеииовая
кислоты резко отличаются одну от другой по своим свойствам.
Фумаровая кислота—кристаллическое вещество, которое,.не
плавясь, возгоняется при 200 °С и плохо растворяется в воде.
Малеиновая кислота плавится при 130 JC, очень легко растворяется
в воде и является более сильной кислотой, чем фумаровая. Она
не была найдена в природе.
При нагревании малеиновая кислота легко отщепляет воду,
образуя малеиновый ангидрид:
не—с/
он
НС—С^
о
малеииовьіГі ангидрид
Малеиновый ангидрид представляет собой игольчатые
кристаллы (темп, пл, П2.6 °С. темп. кип. 202 °С) При действии воды
он медленно переходит в малеиновую кислоту.
Малеиновый ангидрид образуется при неполном окислении
многих оріанических соединений. На этом основываются
технические методы его получения. В промышленности его производят
каталитическим окислением бензола или бутиленов. Он находит
широкое применение в производстве синтетических материалов,
так называемых стеклопластиков, являясь одним из основных
исходных продуктов при получении связующей полиэфирной
смолы для них,
При нагревании фумаровой кислоты до 300 °С также
выделяется вода и образуется малеиновый ангидрид. Фумаровый ангидрид,
т. е. вещество, которое при взаимодействии с водой дает
фумаровую кислоту, неизвестен.
Малеиновая кислота неустойчива и в присутствии
катализаторов (например, следов иода или следов азотистой кислоты) пере-
* Малеиновая кислота получила свое название от латинского
наименования яблочной кислоты—acidum malicuin.
139. Фумаровая и малеичоеая кислоты 279
ходит в фумаровую кислоту. Теплота сгорания малеиновой
кислоты больше теплоты сгорания фумаровой кислоты.
Следовательно, переход малеиновой кислоты в фумаровую связан с
выделением энергии. Эфиры малеиновой кислоты легко переходят
в эфиры фумаровой кислоты в присутствии небольших количеств
иода.
Диметиловый эфир малеиновой кислоты в присутствии
небольшого количества диэтиламина (C2H5JNH превращается в эфир
фумаровой кислоты почти
со скоростью ионной реак- u ^СООН тт -,СООН
ции; при этом выделяется
значительное количество
тепла. Все это указывает
на то, что фумаровая
кислота—вешество, более оед-
ное знеріией, чем малеи-
новая кислота.
Переход фумаровой
кислоты в малеиновую рис. 50. Стереоизомерия фумаровой и ма-
кислоту связан с погло- леиновой кислот:
ЩЄНИЄМ ЭНергИИ. Это Ире- а—малеиновая кпслг.та, б—фумаровая кислота
вращение происходит при
облучении фуыаровой кислоты ультрафиолетовыми лучами.
Отношения между фумаровой и малеиновой кислотами те же, что и
между кислотами кротоновойиизокротоновои или олеиновой и
элаидиновой,—они являются по отношению друг к другу цис-
и транс-изомерами:
н—с—соон н—с—соон
II II
н—с—соон ноос—с.-н
малеи.лшая кислота фумаровая кислота
Выбор формулы для каждой из этих кислот производится на
основании того, что малеиновая кислота, выделяя воду,
образует внутренний ангидрид, в то время как ангидрид фумаровой
кислоты неизвестен. Ясно, что превращение в циклическим
ангидрид может иметь место лишь в том случае, когда оба
карбоксила находятся в г<ис-положении. Следовательно, малеиновая
кислота—^ас-изомер, а фумаровая кислота—транс-изомер.
Правильность такого вывода подтверждают и другие свойства этих
кислот.
ГЛАВА VIII
ГАЛОИДЗАМЕЩЕННЫЕ КИСЛОТЫ
140. Строение и способы получения галоидзамещенных
кислот. Галоидзамещенные кислоты получают замещением
галоидом в карбоновых кислотах одного или нескольких атомов
водорода, связанных с углеродом, например:
CH.fil—COOH монохлоруксусная кислота
CHCI-j—СООН дихлируксусная кислота'
СС1а—СООН трихлоруксусная кислота
Для того чтобы указать, при каком именно атоме углерода
происходит замещение водорода, атомы углерода в молекулах
кислот обозначают первыми буквами греческого алфавита. Атом
углерода, соединенный с карбоксилом, обозначают греческой
буквой а (альфа), следующие атомы—буквами ? (бета), у
(гамма), 8 (дельта) и т. д., например:
СН:)-СН2-СООН СНз-СНСІ-СООН
пропионовая кислота а-хлормропионовая кислота
СНгСІ—СНа—СООН
?-хлорпропиоиовая кислота
У ? «
СН3—СН2—СН2—СООН СН3—СН2—CHCI—СООН
масляная кислота а-хлормаслякая кислота
СН3—CHCI—СН4—СООН
З-хлормасляная кислота
СН,С1—СН2—СН2—СООН
V-хлормасляная кислота
а-Галоидзамещенные кислоты могут быть получены
непосредственным действием галоида на кислоты:
СН3—СООН + С12 » СН2С1—СООН + HCI
Хлорирование и бромирование предельных одноосновных
кислот было изучено в 1868—1870 гг. В. В. Марковниковым. Он
показал, что галоидирование идет в присутствии переносчиков
галоида (иод) и приводит к замещению водородов в а-положении
к карбоксилу.
14t. Свойства галоид замещенных кислот 28 t
Из других способов получения галоидзамещенных кислот
надо указать на присоединение галоидоводородов к непредельным
кислотам. Следует при этом отметить, что если кратная связь
находится у смежного с карбоксилом атома углерода, то
присоединение галоида происходит к атому углерода, находящемуся в
?-положении к карбоксилу:
СНЭ—СН=СН—СООН + НВг » СНЭ—СНВг—СНа—СООН
кротоновая кислота 0-броммасляная кислота
СНа=СН—СООН + HJ > CH2J—СН2—СООН
акриловая кислота ?-иодпропионовая
кислота
Таким образом, в последнем случае присоединение идет не
по правилу Марковникова.
141. Свойства галоидзамещенных кислот. Галоидзамещенные
кислоты вступают во все реакции, характерные для карбоновых
кислот. Введение галоида в молекулу кислоты усиливает
кислотные свойства: монохлоруксусная кислота сильнее уксусной
кислоты, дихлоруксусная кислота сильнее монохлоруксусной
кислоты, трихлоруксусная кислота является весьма сильной
кислотой. Усиление кислотных свойств объясняется тем, что атом
галоида оттягивает к себе электроны не только ближайших, но и более
удаленных атомов. Благодаря этому протон может легче
отделиться от молекулы, т. е. облегчается электролитическая
диссоциация.
Наиболее сильное влияние оказывает галоид в а-поло-
жении; с удалением от карбоксила влияние галоида ослабевает.
Константа
электролитической диссоциации
а-Хлормасляная кислота 140- 1СГ5
?-Хлормасляная кислота 8,9-Ю
Y-Хлормасляная кислота 2,6-10~5
Масляная кислота . . . 1,55-10~*
Наибольшее промышленное значение имеет монохлоруксусная
кислота. Она получается хлорированием уксусной кислоты и
применяется в громадных количествах как полупродукт в
производстве гербицидов (препаратов для химической «прополки»). Она
применяется также в синтезе индиго, при получении некоторых
поверхностно-активных веществ и других соединений.
Монохлоруксусная кислота—бесцветное кристаллическое вещество,
расплывающееся на воздухе (темп, плавл. 61,5 °С; темп. кип.
189 °С).
Монохлоруксусная, а также и трихлоруксусная кислоты были
впервые получены в 1793 г. русским академиком Товием
Егоровичем Ловицем.
ГЛАВА IX
оксикислоты
Оксикислотами, или с пир токис лотами, называются вещества,
которые содержат в молекуле и спиртовий гидроксил, и
карбоксильную группу. Примерами оксикислот могут служить:
Оксиуксусная, или гликолевая, кислота СН2(ОН;—СООН
а-Оксипропионовая, или молочная, кислота СН3—СН(ОН)—СООН
?-Оксипропионовая кислота СН.2(ОН)—СН.2—СООН
Y-Оксимасляная кислота СН.,(ОН)—СН.г—СН.—СООІI
По женевской номенклатуре названия оксикислот образуются
путем введения суффикса «ол», обозначающего наличие оксигруп-
пы в соответствующем месте молекулы. Например, гликолевая
кислота—зтан-ол-овая кислота; а- и ?-оксипропионовые
кислоты—соответственно пропанол-2-овая и пропанол-3-овая кислоты.
Нетрудно перейти от названия к структуре, например: 2-метил-
пропанол-2-овая кислота
СНа—С(ОН)—СООН
СН3
2-метилбутанол-З-овая кислота
СН3—СН—СН(ОН)—СООН
сч
<-'"з
Для оксикислот различают понятия основности и атомности.
Основность оксикислот измеряется числом карбоксилов, а
атомность—числом гидроксилов, включая и гидроксилы
карбоксильных групп. Так, кислоты, формулы которых были приведены
выше, являются одноосновными и двухатомными оксикислотами,
в то время как яблочная кислота НООС—СН2—СН(ОН)—СООН
является двухосновной и трехатомной окснкислотой.
142. Способы получения и строение оксикислот.
Одновременное содержание в оксикислотах гидроксильной и карбоксильной
142. Способы получения и строение оксикислот 283
групп позволяет получать эти соединения как из спиртов, так и
из кислот.
1. Окисление гликолей:
СН3—CHOH-CHaOH Ч- 0.2 » СНз-СНОН-СООН + Н2О
пропиленглико.ль молочная числота
2. Обмен галоида в галоидзамещенных кислотах на гидроксил.
При нагревании галоидзамещенных кислот с водой или со
щелочами протекает реакция:
СН2С1—СООН + Н2О » СН2ОН—СООН + HCI
3. Окисление кислот с третичным атомом углерода,
находящимся в а-положении к карбоксилу, марганцовокислым калием:
СНЗЧ OV
>СН—СООН » >СОН—СООН
сн/ av
изомасляная а-оксиизомасляная
кислота кислота
4. Окисление жирных кислот в ?-положении (стр. 228).
5. Весьма важным является способ получения оксикислот
из альдегидов и кетонов.
Альдегиды и кетоны способны присоединять синильную
кислоту:
он
і і А
,0 + Н -CN » СН3—С—Н
СНЗЧ і : СНзч .ОН
>С=0 + Н—CN > >С<
СН/ f • СН/ XCN
В результате образуются оксинитрилы (циангидрины), при
омылении которых получаются а-оксикислоты:
/ОН ОН
CH3-C-CN + 2Н.,0 > СНз-С-СООН + NH3
молочная кислота
СНЗЧ /ОН СН3ч УОН
>С< +2Н2О „ >С< +NH3
СН3Х NCN СН/ NCO0H
ct-ок' иияомасляная
кислота
284
Глава IX. Оксикислоти
143. Окисление многоатомных спиртов. При окиелении этилен-
гликоля СН2ОН—СН2ОН каждая из групп СН2ОН, характерных
для первичного спирта, может быть окислена в альдегидную
группу СНО или карбоксил СООН. В результате образуются
вещества, формулы и названия которых указаны в приводимой
СН2ОН—СН2ОН
гликоль
\
СН2ОН—СООН
гликолевая
кислота
СН2ОН—СНО
гликолеьый
альдегид
СНО—СНО
СНО—СООН
глиоксилоеая
кислота
НООС—СООН
щавелевая
кислота
Гликолевый альдегид является одновременно и альдегидом
и спиртом, гликолевая кислота—спиртом и кислотой, глиокси-
ловая кислота—альдегидом и кислотой.
В пропиленгликоле СН3—СН(ОН)—СН2ОН находятся
группы СН2ОН и группы СН(ОН); вторая группа, как известно,
характерна для вторичных спиртов, которые дают при окислении
кетоны. Поэтому в результате окисления пропиленгликоля
можно получить ряд веществ, которые, будучи спиртом, альдегидом
или кислотой, являются в то же время кетонами:
СН3—СНЮН)—СН2ОН » СНа—СО—СН2ОН »
пропиленгликоль
» СН3—СО—СНО
метилглиоксаль
(кетоальдегид)
ацетол (кетоспирті
-> СНз-СО—СООН
пиривиноградная
кислота
(кетокислота)
Первыми продуктами окисления глицерина являются
глицериновый альдегид и диоксиацетон:
-•¦ CHsOH—СНОН—СНО
снаон—снон —сн3он—
глицерин
глицериновый альдегид
сн.он—со—снаон
диоксиацетон
144. Свойства оксикислот 285
Таким образом, исходя из многоатомных спиртов, можно
путем окисления получить ряд веществ со смешанными функциями:
альдегидоспирты, кетоспирты, кетоальдегиды, спиртокислоты
(оксикислоти), альдегидокислоты и т. д.
144. Свойства оксикислот. Оксикислота—кристаллические
вещества, легко растворимые в воде. Они плавятся при более
высоких температурах и растворяются в воде легче, чем
соответствующие жирные кислоты. По сравнению с последними окси-
кислоты имеют более высокую константу диссоциации.
Наличие в молекуле оксикислот карбоксильной и гидроксиль-
ной групп приводит к тому, что эти соединения могут реагировать
и как спирты и как кислоты. Например, взаимодействуя с
этиловым спиртом, гликолевая кислота образует сложный этиловый
эфир:
НО—СН2—СООС,Н5
ЭТИЛОВЫЙ 9.|>Ир ГЛИКОЛЄВОН КИСЛОТЫ
В то же время известен сложный эфир, образованный гликолевой
кислотой (как спиртом) с уксусной кислотой:
СН3СО—О—CH.J—СООН
ацетогликолевая кислота
Оксикислоты более или менее легко отщепляют воду, причем,
в зависимости от положения гидроксильной группы, образуются
совершенно различные вещества.
1. а-Оксикислоты при нагревании легко выделяют воду,
образуя циклические сложные эфиры, шестизвенное кольцо которых
состоит из четырех атомов углерода и двух атомов кислорода.
При этом гидроксильные и карбоксильные группы двух молекул
кислоты взаимно этерифицируются. Образующиеся в результате
этерификации эфиры получили название лакшидов (от
латинского названия молочной кислоты—acidum lacticum):
СН3—СН —О—;н" НО:—СО СН3—СН—О—СО
| ' | » I | +2Н2О
СО—IOH " Н|—О—СН —СНа СО—О—СН—СНЭ
молочная кислота молочная кислита пактил
Лактиды—кристаллические вещества. При кипячении с
водой они омыляются с образованием исходной оксикислоты.
2. ?-Оксикислоты, отщепляя при нагревании воду, дают
непредельные кислоты:
СН3— СНОН—СН,—СООН > СН3—СН=СН—СООН ¦+- Н2О
(J-оксимасляиая кислота кротоноьая кислота
286 Глава ІК. Оксикислоти
3. f и й-Оксикислоты в свободном состоянии очень
неустойчивы и в большинстве случаев известны только в виде солей.
При действии на эти соли других кислот должны были бы
образоваться сами окснкислоты, например:
НО—СИ2—СН.,,—СН.2—COONa + HCI » НО—СН2—СН,—СН.2—СООН + NaCl
Y-оксима^лянач кислота
Но они в момент своего образования, вследствие
пространственной близости групп ОН спирта и ОН карбоксила, переходят в
пяти- и шестизвенные циклические лаптопы:
СНо СН2 СН,-СН,
/ \ / ч
СН, СО >СН2 СО
у-оксимас.ляной
кислоты
Лактоны являются циклическими сложными эфирами. Они
обычно представляют собой жидкости или легкоплавкие твердые
вещества, которые перегоняются без разложения.
При действии на лактон едкой щелочи лактонное кольцо
разрывается и получается соль:
СН2—СН.,,—СН.,—СО + КОН » СН2ОН—CHa—СН„—COOK
О уо^-симасляноґі кислоты
лактон
Y-oKc има ляной
ки.лоты
Лактон оксимасляной кислоты был впервые получен в 1873 г.
А. №. Зайцевым.
145. Молочная кислота. Молочная, или оксипропионовая,
кислота СН,—СН(ОН)—СООН была открыта в 1780 г. Шееле
в кислом молоке. Для оксипропионовои кислоты возможны два
изомера:
СН3—СНОН—СООН СН,ОН—СН,—СООН
а-окснпрони'люьйя ? -окснпротюнииая
ки. лота кислота
Синтез молочной кислоты из уксусного альдегида и цианистого
водорода позволил установить ее строение:
/> HCN /°Н Н3О /°Н
СН3-С » CH3-C-CsN > СНз-С-СООН
\н \н ^н
уксусный оксинитрил молочная кислота
альдегид
В уксусном альдегиде кислород связан только с одним атомом
углерода; следовательно, в молочной кислоте гидроксил и карб-
145. Молочная кислота 287
оксил должны находиться у одного атома углерода. Таким
образом, молочная кислота является а-оксипропионовой кислотой.
Изомерная ей ?-оксипропионовая кислота была впервые
получена Вюрцем из этиленхлоргидрина:
KCN Н2О
CH.pH-CH.jCI • CH3OH-CH2-CN »СНаОН-СН.2-СООН
Как видно из структурной формулы молочной кислоты
СН3—СН(ОН)—СООН, в ее молекуле находится
асимметрический атом углерода. Поэтому для нее известны оптически
деятельные формы.
Правовращающая и левовращающая молочные кислоты
плавятся при 25—26 °С и являются оптическими антиподами, т. е.
вращают в разные стороны плоскость поляризации света. Кроме
того, известна и недеятельная молочная кислота—густой сироп,
который при выпаривании под сильно уменьшенным давлением
застывает в кристаллы, плавящиеся при 18 °С. Недеятельная,
рацемическая, молочная кислота может быть получена смешением
равных количеств правовращающей и левовращающей
молочных кислот и, очевидно, является их соединением.
Молочная кислота при нагревании с разбавленной серной
кислотой разлагается на уксусный альдегид и муравьиную
кислоту:
Н
О ,0
CH3-c-;c<f
: >(
он
о—н
СН;
з-С< +Н-С<
хн хон
уксусный мураєьиная
альдегид кислота
молочная кислота
Если же нагревание вести в присутствии концентрированной
серной кислоты, то образующаяся муравьиная кислота разлагается
дальше с выделением окиси углерода.
Способность разлагаться с образованием муравьиной кислоты—
характерное свойство а-оксикислот.
Молочная кислота применяется в кожевенном деле, в
текстильном производстве при крашении тканей и в других отраслях
промышленности.
Недеятельная молочная кислота образуется при
молочнокислом брожении сахаристых веществ, при скисании молока,
при квашении капусты и при силосовании растительных кормов.
Молочная кислота получается также при нагревании глюкозы с.
раствором едкого натра.
Левовращающая молочная кислота получается при брожении,
вызываемом некоторыми бактериями.
288
Глава IX. Оксикислоти
П равовращающая молочная кислота находится в мускулах;
она имеет большое значение как продукт обмена веществ в
мускулах. Она была открыта в 1837 г. Либихом* и известна под
названием мясомолочной кислоты.
В 1869 г. Вислиценус указал, что причину изомерии молочной
и мясомолочной кислот, имеющих тождественное строение,
следует искать в пространственном расположении атомов внутри их
молекул. Статья Вислиценуса (опубликованная в 1873 г.)
натолкнула Вант-Гоффа, как это он сам указывает, на те мысли,
которые нашли выражение в развитой им стереохимической теории.
В последние годы точными физическими методами была
установлена абсолютная пространственная конфигурация оксикислот.
Оказалось, что правовращающая молочная кислота, по принятой
нами системе обозначений (см. стр. 160), это 2о-оксипропноновая
кислота, а левовращающая—2р-оксипропионовая кислота.
При химических синтезах получается недеятельная молочная
кислота, т. е. смесь равных количеств правовращающего и лево-
вращающего антиподов. То же происходит в других случаях,
когда лабораторным путем из оптически недеятельных
соединений создаются вещества с асимметрическим атомом углерода. Это
объясняется тем, что вероятность образования каждого из
антиподов совершенно одинакова.
Рассмотрим три случая образования молекул с
асимметрическим атомом углерода.
1. Реакция присоединения. Нитрил молочной кислоты
получается присоединением цианистого водорода к уксусному
альдегиду:
+ HCN
б-изомер
р-изомер
уксусный альдегид
оптмчрские антиподы нитрила молочной кислоты
* Юстус Либих (J. Liebig)—выдающийся немецкий химик A803—1873).
Был профессором в Гиссенском университете, где создал большую научную
школу химиков.
Либих разработал практическую методику и аппаратуру для определения
углерода и водорода сожжением вещества в тугоплавкой трубке с окисью
меди A831) Им проведено определение состава очень многих органических
веществ. Отметим его работы по производным гремучей кислоты, совместную
работу с Велером над бензальдегидом («масло горьких миндалей») и
производными радикала бенэоила A832). Он изучал действие хлора на спирт, получил и
исследовал уксусный альдегид, хлораль и хлороформ.
145. Молочная кислота 28')
При этом разрывается одна из связей, соединяющих атом
кислорода с атомом углерода: или разрывается связь d, и атом
кислорода остается соединенным с атомом углерода при помощи
связи с , или же разрывается связь с, а связь d остается. В обоих
случаях получаются молекулы с асимметрическим атомом
углерода, но эти молекулы относятся друг к другу, как
несимметричный предмет к своему зеркальному изображению, и являются,
следовательно, молекулами оптических антиподов. Обе связи с
и d полностью тождественны, и вероятность разрыва как той,
так и другой совершенно одинакова; поэтому оба антипода
должны образоваться в одинаковом количестве; следовательно,
полученная молочная кислота будет оптически недеятельна.
2. Реакция замещения. а-Бромпропионовая кислота можеч
быть получена бромированием пропионовой кислоты:
СН3
а
н
ноос-
сн3
а
' хн нс
б-изомер
СН3
а
}QZr Br
р-изомер
пропионовая кислота оптические антиподы а-бромпропиоко вой кислоты
Атом брома может заместить атом водорода или у связи d,
или у связи с. Обе связи тождественны, и вероятность
образования каждого из антиподов совершенно одинакова.
3. Реакция разложения. При отщеплении СОа от метилэтил-
малоновой кислоты образуется метилэтилуксусная кислота; при
этом один из атомов углерода становится асимметрическим:
i5W ЧСООН H5<V "
р-изомер б- изомер
метилэтил малоновая кислота оптические антиподы метилатилуксуслой кислоты
Вероятность отщепления СО2 как у связи d, так и у связи с
совершенно одинакова, поэтому и получаются равные количестса
обоих антиподов.
14-788
290
Глава IX. Оксикислоты
146. Яблочная кислота. Яблочную кислоту, как показывает ее
формула НООС—СН2—СН(ОН)—СООН, можно рассматривать
как производное янтарной кислоты НООС—СНа—СН2—СООН,
в молекуле которой один атом водорода замещен гидроксилом:
она является оксиянтарной кислотой.
Это доказывается образованием янтарной кислоты при
восстановлении яблочной кислоты:
СН,—СООН
НО
'—СН—і
СООН
4-2HJ
СНа—СООН
СНа—СООН
янтарная
кислота
Н2О
Левая її правая яблочные кислоты плавятся при 100 °С; они
различаются только по оптическим свойствам. Недеятельная
яблочная кислота плавится при 130—131 JC; она является
соединением молекул правовращающей и левовращающей кислот.
Последняя встречается в недозрелой рябине, яблоках, виноградном
соке и т. п.
При нагревании выше температуры плавления яблочная
кислота теряет молекулу воды и переходит в фумаровую и малеино-
вую кислоты:
НООС—СН-СН—СООН
іон ¦" н;
ноос—сн=сн—соон + нао
147. Винные кислоты. Винная кислота С4НвОв может быть
получена из фумаровой кислоты следующим образом:
СООН
СН
СН
СООН
фумаровая
к нслота
СООН
<
н
СООН
н
+2AgOH
<
<н
СООН
ОН
н
<н
Х)Н
соон
2AgBr
дибримянгарная
кислота
Следовательно, она является производным янтарной кислоты,
в молекуле которой два атома водорода замещены двумя гидрок-
силами, т. е. диоксиянтарной кислотой.
147. Винные кислоты
291
В молекуле винной кислоты находятся два одинаковых
асимметрических атома углерода:
Н Н
ноос—с—с—соон
он он
Поэтому для нее возможно несколько стереоизомеров.
Пространственное расположение атомов в молекулах винных
кислот представлено на рис. 51.
СООН
СООН
СООН
Н
Л
m
Рис. 5). Пространственное расположение атомон в
молекулах винных кислот:
/—D-винная кислота; //—L-вннная кислота; ///—меэовнн
ная кислота.
1. Правовращающая (+ -винная) кислота, или D-винная
кислота*. Правое вращение обусловлено суммой правых вращений
обоих асимметрических атомов.
2. Левовращающая ( винная) кислота, или /.-винная
кислота*. Левое вращение обусловлено суммой левых вращений
обоих асимметрических атомов.
3. Виноградная кислота. Оптически недеятельна, так как
состоит из равных количеств правовращающей и левовращающей
винных кислот.
4. Мезовинная, или і-винная, кислота. Также оптически
недеятельна. Оптическая недеятельность этой кислоты
обусловлена тем, что в ее молекуле один асимметрический центр
отвечает по конфигурации (и по вращению) D-кислоте, а другой—
L-кислоте. Таким образом, правое вращение одной половины
молекулы компенсируется левым вращением другой половины.
Поэтому мезовинная кислота не может быть расщеплена на
оптически деятельные формы.
Чтобы убедиться в тождественности расположения групп,
связанных с обоими атомами углерода в молекуле оптически
деятельной винной кислоты, надо разрезать схему по горизонтальной
* Сущность обозначений D и L объясняется в § 152 и § 159.
19*
29?
Глава IX. Оксикислоти
плоскости, проходящей через вершины тетраэдров, после чего
перевернуть верхний тетраэдр вершиной вверх и поставить его
на основание рядом с нижним тетраэдром.
Проектируя модели пространственного расположения атомов
в молекулах винных кислот на плоскость, получим следующие
формулы, где точки пересечения линий обозначают
асимметрические атомы углерода:
СООН
СООН
СООН
НІНО—
-он
—н
но-
н-
-н
-он
н-
н—
—он
-он
СООН
1
СООН
II
СООН
III
Правовращающая и лееовращающая винные кислоты, т. е.
D-винная и L-винная кислоты, являются оптическими
антиподами: растворы первой вращают плоскость поляризации
светового луча вправо, растворы второй—на равный угол влево.
Кроме того, они образуют кристаллы, которые являются зеркальными
изображениями один другого и не могут быть друг с другом
совмещены. Обе они плавятся при 170 °С.
Правовращающая винная кислота (I), как видно из модели и
проекционной формулы, это 2р,3а-диоксиянтарная кислота, а
левовращающая (II)—это Зр,2а-диоксиянтарная кислота. Легко
видеть, что обозначения р и о в этом случае не меняются, откуда
бы ни начинался отсчет.
Правовращающая винная кислота, называемая иногда
виннокаменной кислотой, весьма распространена в природе, особенно
много ее в соке винограда. При брожении виноградного сока она
выделяется в виде винного камня, состоящего из кислого
виннокислого калия вместе с небольшим количеством виннокислого
кальция. Для получения винной кислоты винный камень
обрабатывают кислотами; выделенную таким путем кислоту очищают
перекристаллизацией. Правовращающая винная кислота
кристаллизуется в больших прозрачных призмах. Она легко
растворяется в воде и спирте. Соли винных кислот называются тар-
тратами.
Кислый виннокислый калий
НООС—СН(ОН)—СН(ОН)—COOK
применяется в качестве протравы при крашении и печатании
тканей. Эта соль плохо растворима в воде и еще хуже—в спиріе.
147. Винные кислоты 293
Рвотный камень
КООС—СН(ОН)—СН(ОН)—COO(SbO) • VaHaO
находит применение в качестве протравы при крашении и
ситцепечатании, а также в медицине.
Сегнетова соль
NaOOC—СН(ОН)—СН(ОН)—COOK ¦ 4Н2О
применяется в медицине и в лабораторной практике.
При приливаний к щелочным растворам винной кислоты
раствора медных солей, например раствора медного купороса,
следовало бы ожидать выпадения нерастворимого осадка
гидроокиси меди. В действительности же при этом получается темно-
синий прозрачный раствор. Подобные растворы обладают
окислительными свойствами и при действии многих веществ, способных
окисляться, например альдегидов, многих Сахаров, выделяют или
желтый осадок гидроокиси медиA)— СиОН, или же красный
осадок—закись меди Си2О. В лабораториях для определения
восстановителей пользуются так называемой фелинговой
жидкостью, которую готовят следующим образом. В одной колбе
растворяют в 1 л воды 34,6 г медного купороса, в другой колбе
также в 1 л воды растворяют 177 г сегнетовой соли и 60 г едкого
натра. Оба раствора перед употреблением смешивают. Так как
фелингову жидкость нельзя хранить продолжительное время,
то ее готовят в небольших количествах перед каждым опытом.
Левовращающая винная кислота не была найдена в природе
в качестве естественного продукта; впервые она была получена
Пастером* из виноградной кислоты.
Мезовинная кислота, или і-винная кислота, образуется вместе
с виноградной кислотой при кипячении в течение нескольких
часов D-винной кислоты с большим избытком раствора едкого
* Пастер Луи (Pasteur L.)—выдающийся французский ученый A822—
1895). Родился в г. Доль, в семье кожевника. Был профессором Страсбургского,
Лилльского и Парижского университетов. В студенческие годы работал под
руководством Ж- Дюма. Пастер показал, что оптическая активность винной
кислоты и асимметрическое строение ее кристаллов находятся в тесной
зависимости; его работы по асимметрии имеют очень большое значение в
стереохимии. Он провел большие исследования процессов брожения. В 1857 г.
Пастер установил, что молочная кислота образуется при сбраживании сахара в
результате жизнедеятельности молочно-кислых бактерий. Его исследования
в области брожения явились научной основой для использования
микроорганизмов с целью производства пищевых продуктов (уксуса, вина, пива), а также
для разработки метода предохранения их от порчи (пастеризация).
Труды Пастера положили начало развитию микробиологии как
самостоятельной научной дисциплины.
294 Глава IX. Оксикислогы
натра. Ее кислая калиевая соль, в отличие от кислых калиевых
солей других винных кислот, легко растворима в холодной воде.
Мезовинная кислота плавится при 140 °С, т. е. при
значительно низшей температуре, чем другие винные кислоты.
Виноградная кислота может быть получена смешением
растворов право- и левовращающей винных кислот. Если растворы
кислот концентрированные, то при смешении выделяется тепло
и менее растворимая виноградная кислота выпадает в виде
кристаллов.
В отличие от оптически деятельных винных кислот,
кристаллы которых не содержат кристаллизационной воды, виноградная
кислота кристаллизуется из воды в виде гидрата 2С4Н6О6-Н,О;
безводная кислота плавится при 204 °С. Соли ее кристаллизуются
с иным количеством кристаллизационной воды, чем соли право-
и левовращающей кислот, и обладают иной растворимостью.
Так, винограднокислый кальций настолько мало растворим, что
раствор виноградной кислоты мутнеет от прибавления гипсовой
воды, чего не происходит при прибавлении гипсовой воды к
растворам других винных кислот.
Все эти данные показывают, что виноградная кислота является
не смесью оптически деятельных кислот, а их химическим
соединением.
Подобные оптически недеятельные соединения получили
название рацемических веществ, или рацематов, от названия
виноградной кислоты acidum racemicum*, на которой впервые было
замечено явление оптической недеятельности, обусловленной
присутствием равных количеств противоположно вращающих
изомеров. В парах, растворах и расплавах рацематы, по-видимому,
являются смесью обеих оптически деятельных форм. Так, при
определении молекулярного веса виноградной кислоты по
понижению температуры замерзания ее раствора получена величина,
соответствующая формуле С4Н6Ов. Точно так же плотность паров
эфиров виноградной кислоты соответствует простым, а не
удвоенным молекулам. В кристаллической же решетке, как это
показывает рентгенографический анализ, узлы заняты удвоенными
молекулами.
История открытия винных кислот. В 1769 г. Шееле получил из винного
камня «обыкновенную», т. е. правовращающую винную кислоту, а в 1822 г. как
побочный продукт производства этой кислоты была открыта виноградная
кислота.
Виноградная кислота была подробно исследована в 1830 г. Берцелиусом,
который показал, что она является изомером правовращающей винной
кислоты. Было установлено, что виноградная кислота отличается от
правовращающей винной кислоты рядом свойств. Так, в отличие от правовращающей вин-
Racemus по латыни виноград.
148. Расщепление рацемических веществ на оптические антиподы 295
ной кислоты она оптически недеятельна; ее кристаллы не имеют
несимметрично расположенных площадок, характерных для кристаллов правовращаю
щей винной кислоты.
В конце 40-х годов прошлого века Пастер, бывший в то время студентом
Нормальной школы в Париже, обратил внимание на одну из работ немецкого
химика Митчерлиха В этой работе Митчерлих утверждал, что натрийам-
монийная соль правой винной кислоты отличается от найтрийаммонийной
соли виноградной кислоты лишь тем, что раствор первой соли вращает
плоскость поляризации света вправо, а раствор второй соли—оптически недея
телен Все же остальные свойства этих солей, в том числе и их
кристаллическая форма, по утверждению Митчерлиха, одинаковы. По мнению Пастера,
различное отношение этих солей к поляризованному свету можно было бы
объяснить только в том случае, если бы кристаллы соли виноградной кислоты
были в отличие от кристаллов соли винной кислоты лишены несимметрично
расположенных площадок Поэтому Пастер решил повторить работу
Митчерлиха.
Исследования Пастера показали, что кристаллы найтрийаммонийной соли
правой винной кислоты обладают в отличие от такой же соли виноградной
кислоты несимметрично расположенными площадками. Что же касается натрий-
аммонийной соли виноградной кислоты, то при кристаллизации ее из раствора
при температуре ниже 28 °С выпадают кристаллы, тоже обладающие такими
площадками, но одни из этих кристаллов являются зеркальными
изображениями других. Пастер тщательно отделил кристаллы обоих родов и
нашел, что раствор кристаллов одного рода вращает плоскость поляризации
вправо, а раствор кристаллов другого рода—влево. Смешав равные
количества кристаллов обоих родов, он установил, что их раствор оптически
недеятелен.
Из правовращающей соли Пастер получил правую кислоту, а из левопра-
щающей соли впервые выделил левую винную кислоту. Наконец, смешав
концентрированные растворы правой и левой винной кислот, Пастер получил
осадок виноградной кислоты.
В дальнейшем Пастер открыл мезовинную кислоту, а также разработал
способы разделения рацемических соединений на оптически деятельные
изомеры.
148. Расщепление рацемических веществ на оптические
антиподы. Пастер на примере виноградной кислоты разработал три
способа, при помощи которых можно расщеплять рацемат на
оптические антиподы.
1. Самопроизвольное расщепление. При кристаллизации
рацематы иногда распадаются на правовращающий и левовращаю-
щий изомеры, кристаллизующиеся в энантиоморфных
кристаллах, которые можно механически отобрать по их внешнему виду.
Примером самопроизвольного расщепления может служить
кристаллизация двойной натрийаммонийной соли виноградной
кислоты
NaOOC—СН(ОН)—СН(ОН)—COONH4 • 2Н2О
При температурах выше 28 °С из раствора выпадают кристаллы
соли виноградной кислоты; при температурах ниже 28 °С выпадают
отдельно кристаллы солей D- hL-винных кислот. Они отличаются
296
Глава IX. Оксикислоты
несимметрично расположенными гранями (рис. 52) (энаншиомор-
фные кристаллы) и могут быть разделены механическим отбором.
Состав этих кристаллов выражается формулой
NaOOC—СН(ОН)—СН(ОН)—COONH4 ¦ 4Н2О
2. Биохимическое расщепление основано на том, что
микроорганизмы в процессе своей жизнедеятельности способны
потреблять предпочтительно.лишь один из оптических антиподов,
обыкновенно тот, который встречается в виде естественного продукта в
Рис. 52 Кристаллы натрийаммонийных солей винных
(а, 6) и виноірадной (в) кислот.
животном или растительном мире. Так, обычный плесневый
грибок Penicillium glaucum потребляет правую винную кислоту
гораздо быстрее и легче, чем левую винную кислоту. Поэтому
при размножении и прорастании этих грибков в растворах
виноградной кислоты через некоторое время остается только левовра-
щающая винная кислота.
3. Химическое расщепление. Расщепление рацематов по этому
способу основано на том, что соли антиподов оптически активной
кислоты с оптически деятельным основанием обладают различной
растворимостью.
В качестве примера можно указать на расщепление
виноградной кислоты при помощи цинхонина. Цинхонин принадлежит
к группе алкалоидов и находится вместе с хинином в коре
хинного дерева.
Цинхонин—кристаллическое вещество, вращает плоскость
поляризации вправо, обладает сильными основными свинствами и
соединяется с кислотами, образуя соли.
Если обработать виноградную кислоту цинхонином, то
образуются две соли: соли D-винной кислоты с правовращающим
цинхонином и соль L-винной кислоты с правовращающим цнн-
хонином.
Эти соли не являются оптическими антиподами, так как
оптическим антиподом соли правой винной кислоты с правовра-
149. Асимметрический синтез 297
щающим цинхонином будет соль левой винной кислоты с лево-
вращающим цинхонином:
правая кислота леі>ая кислота правая кислота левая кислота
правое основание левое основание правое основание правое основание
оптические антиподы не являются оптическими антиподами
Стереоизомеры обладают одинаковыми свойствами лишь в том
случае, когда различие в их конфигурации сводится только к тому,
что молекулы одного из них являются зеркальным изображением
молекул другого, т.е. если они оптические антиподы. Поскольку
же полученные соли не являются оптическими антиподами, они
должны обладать различными свойствами, в
частности—различной растворимостью. Так, цинхониновая соль I-винной кислоты
менее растворима в воде, чем цинхониновая соль D-винной
кислоты; поэтому эти соли могут быть разделены кристаллизацией.
Обработав в отдельности каждую из солей соляной кислотой,
отделяют цинхонин в виде хлористоводородной соли и получают
из одной соли правую винную кислоту, а из другой—левую
винную кислоту.
149 Асимметрический синтез. Если при химических
синтезах из веществ с симметричными молекулами создаются вещества
с асимметрическим атомом углерода, то они получаются в виде
недеятельных соединений. Однако, пользуясь оптически
деятельными веществами, можно в некоторых случаях из веществ, не
содержащих асимметрического атома углерода, получить
оптически деятельные вещества. Для этого исходное вещество
связывают с оптически деятельным веществом и удаляют последнее
после того, как путем химической реакции получен новый
асимметрический атом. Так можно синтезировать оптически деятельную
молочную кислоту СН3—СН(ОН)—СООН из пировиноградной
кислоты СН3—СО—СООН, в молекуле которой нет
асимметрического атома. Для этого из пировиноградной кислоты и
природного левовращающего спирта борнеола С10Н17ОН (стр. 567)
получают сложный эфир. Восстановлением переводят этот эфир в бор-
неоловый эфир молочной кислоты. При этом в молекуле создается
новы;! асимметрический атом и получаются два
вещества—сложный эфир левовращающего борнеола и левовращающей молочной
кислоты и сложный эфир левовращающего борнеола и
правовращающей молочной кислоты. Эти вещества не являются один по
отношению к другому оптическими антиподами, в связи с чем и
скорости образования их неодинаковы: первое вещество
образуется в несколько большем количестве, чем второе. Поэтому омыление
продукта реакции дает молочную кислоту, обладающую слабым
левым вращением.
298 Глава IX. Оксикислоти
Подобные асимметрические синтезы могут быть произведены
и при помощи энзимов, которые сами оптически деятельны. Так,
при гидролизе рацемического эфира миндальной кислоты
СвН5—СН(ОН)—СООН в присутствии энзима липазы
правовращающая форма гидролизуется быстрее. Если получать нитрил
миндальной кислоты СвН5—СН(ОН)—CN из бензойного
альдегида СвН6СНО и HCN в присутствии эмульсина—энзима горьких
миндалей, то нитрил образуется в оптически деятельной форме.
Итак, в присутствии, энзимов оптически деятельные вещества
могут образоваться как из оптически недеятельных молекул, так и из
рацематов. Вероятно, оба процесса протекают при образовании
оптически активных природных веществ. Таким образом можно
объяснить то, что растение синтезирует из недеятельных веществ
(углекислого газа и воды) правовращающую глюкозу и
бесчисленное множество других оптически деятельных соединений, между
тем как лабораторные синтезы с недеятельными исходными
веществами дают всегда лишь оптически недеятельные вещества.
Однако первоначальное возникновение в природе оптически
деятельных органических веществ остается до сих пор неразрешенной
проблемой.
Ряд исследователей объясняет первичное образование в живой
природе оптически деятельных веществ фотохимическими
процессами. Действительно, если некоторые рацематы подвергать
действию лучей, поляризованных вправо или влево по кругу,
то один из антиподов поглощает больше таких лучей, чем другой
антипод, и разлагается поэтому быстрее. Вещество, оставшееся
после облучения, оказывается оптически деятельным. Применяя
свет, поляризованный по кругу в противоположном направлении,
получают вещество с преобладанием другого антипода. В
последние два десятилетия проведен ряд синтезов под влиянием цирку-
лярно-поляризованных ультрафиолетовых лучей; образующиеся
при этом продукты реакции обладают заметной оптической дея-
тел ьностью.
Асимметрический синтез, проходящий без участия продуктов,
полученных из живой природы, носит название абсолютного
асимметрического синтеза. В отличие от последнего
асимметрический синтез, проходящий только при участии агентов, полу-
чекных из живой природы, называется относительным
асимметрическим синтезом.
В последнее время советскими химиками (А. П. Терентьев, Е. И. Клабу-
невский и В. В. Патрикеев) показано, что абсолютный асимметрический
синтез может быть осуществлен каталитически под влиянием катализирующих
веществ, нанесенных на кристаллы право- и левовращающего кварца, т. е.
вещества, образовавшегося без участия живой природы.
150. Стереоиэом. веществ с несколькими асимметр. атом, углер. 299
!50. Стереоизомерия веществ с двумя или несколькими
асимметрическими атомами углерода в молекуле. Диастереомеры. В
молекуле винной кислоты оба асимметрических атома углерода
равноценны. Для того же, чтобы рассмотреть вопрос о стереоизоме-
рии веществ с несколькими асимметрическими атомами в общем
р.иде, надо, чтобы эти атомы не были одинаковы. Примером
такого вещества может служить хлоряблочная кислота.
ноос—сн(он)—сна—соон
Один из асимметрических атомов углерода в ее молекуле связан
с гидроксилом, другой—с хлором. Поэтому каждый из них
должен вызывать вращение плоскости поляризации света на
неодинаковую величину.
/ п ш iv
Рис. 53. Стереоизомерия хлоряблочных кислот:
I—2о-хлор-3о-оксиянтарная кислота, II—2р-хлор-3р-оксиянтарная кислота;
III—2о-хлор-3р-оксиянтарная кислота; IV—2р-хлор-3о-оксияитарная кислота.
Обозначая один из асимметрических атомов через А, а
другой—через В и подразумевая под знаками плюс и минус
соответственно правое и левое вращение, получим схемы четырех
стереоизомеров хлоряблочной кислоты, которые дадут два
различных рацемата:
12 3 4
-1-А —А +А —А
-fB —В —В -fB
1-й рацемат 2-й рацемат
Один из этих рацематов плавится при 145 °С, другой—при
154 °С. Поскольку в хлоряблочной кислоте асимметрические атомы
равноценны, формы 3 и 4 для нее тождественны и представляют
собой одно и то же вещество, недеятельное вследствие внутренней
компенсации,—мезовинную кислоту.
На рис. 53 представлены модели четырех хлоряблочных
кислот. Формы / и //, так же как и формы /// и IV, являются по-
300 Глава IX. Оксикислоти
парно оптическими антиподами, так как они относятся друг к
другу, как предмет к своему зеркальному изображению. Но
между формами / и /// такого соотношения не существует. Форма /
не является зеркальным изображением формы ///. Эти кислоты
не оказываются одна по отношению к другой оптическими
антиподами и вращают плоскость поляризации света на различные
углы. То же можно сказать и относительно форм // и IV. Такие
стереоизомеры, не являющиеся оптическими антиподами,
называются диаспгереомерами. Примером диастереомерных веществ
могут также служить цинхониновые соли право- и левовращаю-
щей винных кислот или сложные эфиры антиподов оптически
деятельной кислоты с одной из форм оптически деятельного спирта.
Диастереомеры различаются между собой не только
оптическими, но и другими физическими, а также и химическими
свойствами. Это различие возрастает с увеличением числа
асимметрических атомов в мллекуле.
В молекуле хлоряблочной кислоты находятся два
асимметрических атома углерода, и для нее известно 4, т. е. 22, стерео-
изомера. Если в молекуле находится три асимметрических атома,
то число стереоизомеров равно 8, т. є. 23, так как третий
асимметрический атом может комбинироваться с каждой из четырех
предыдущих форм в двух различных конфигурациях:
12 3 4 5 6 7 8
+А —А +А —А +А —А —А +А
+ В —В +В —В —В +В +В —В
+с —с —с +с +с —с +с —с
1-я пара 2-я пара 3-я пара 4-я пара
антиподов антиподов антиподов антиподов
Эти восемь стереоизомеров группируются в четыре пары
оптических антиподов. При наличии четырех асимметрических
атомов ^число стереоизомеров равно 24, т. е. 16. Таким образом, если
в молекуле находится п неравноценных асимметрических
атомов, то число возможных стереоизомеров равно 2й. Кроме того,
2"
возможно -д-, т. е. 2" х рацематов.
В тех случаях, когда в молекулах содержится несколько
асимметрических атомов углерода, для их изображения не
вычерчивают пространственные модели, а пользуются проекционными
формулами.
Для получения этих формул поступают следующим образом.
Вращая атомы пространственной модели молекулы по
ординарным связям, располагают атомы углерода цепи так, чтобы они
находились в одной плоскости и чтобы цепь была согнута дугой
(см. рис. 54, слева). Затем представляют себе, что цепь направле-
151. Лимонная кислота
301
на к зрителю выпуклой стороной дуги и вытянута в указанной
плоскости в прямую линию, а именно так, чтобы заместители,
связанные с углеродной цепью, находились по ту же сторону
плоскости, по которую они находились раньше. Тогда проекция
оказывается в плоскости бумаги (см. рис. 54, справа).
151. Лимонная кислота. Лимонная кислота СвН9О7 в
больших количествах содержится в растениях; она находится в
лимонах, крыжовнике, малине,
в виноградном соке и т. д.
Значительные количества ее
содержатся в листьях
махорки. Получается она из сока
недозрелых лимонов (в
котором находится 6—7%
С,^Н.ЙО7) или путем
брожения глюкозы под влиянием
некоторых бактерий.
Лимонная кислота известна в
форме гидрата (с одной
молекулой воды) и в безводном
состоянии. Гидрат
плавится около 70 °С, безводная
кислота имеет темп, плавл.
153 °С. Лимонная кислота
легко растворима в воде.
Строение лимонной кислоты выражается формулой:
Рис. 54. Модель и проекционная
формула молекулы.
ноос—сн„—с—сн„—соон
но соон
Кроме того, что такое ее строение доказывается синтезом,
наличие в молекуле лимонной кислоты трех карбоксильных групп
подтверждается образованием сложного эфира с тремя
молекулами спирта:
СНа -СО-ЮН f Hj-
со—юн + н.—ос2н6
Х)Н
сна-соос,н6
.COOGjHj
СН2 —СО—ЮН"+"Н{—
он
зн2о
Присутствие в молекуле лимонной кислоты спиртового гидрок-
сила следует из того, что ее сложный эфир, как спирт, вступает
302
Глава IX Оксикис лоты
в реакцию с хлоран гидридом уксусной кислоты, образуя новый
сложный эфир:
сн2—
с
/СООСЛ
<
\о—:н + си—со—сн3
/CQs
¦ С< + НС1
CHj-COOCjHg
CHa—'
O—CO—CH3
a-COOQHs
Из солей лимонной кислоты интересен лимоннокислый
кальций, лучше растворимый в холодной, чем в горячей воде.
Лимонная кислота применяется в кондитерском деле, в
фотографии и для консервирования крови.
152. Стереоизомерия а-оксикислот. В молекулах большинства
оксикислот имеется асимметрический атом углерода, при
котором находится гидроксильная группа. Для установления
единообразия принято обозначать конфигурации этих оксикислот
следующим образом:
СООН
соон
СООН
он н-
D-конфигурация
-он
соон
но-
-н
R
/.-конфигурация
Таким образом, L-a-оксикислоты—это 2с-оксикислоты, а
D-a-оксикислоты—это 2р-оксикислоты.
Обозначения р и а, которыми мы до сих пор пользовались
(стр. 160), обозначения D и L и знаки вращения (~t-) и (—)
независимы и могут употребляться вместе, например D(—) р-мо-
лочная кислота. Разница заключается в том, что обозначения
D и L приписываются в данном случае семейству а-оксикислот,
а обозначения р и а универсальны и введены для наглядного
представления о пространственном строении молекул по их названиям.
Знаки D и L не указывают направления вращения плоскости
поляризации этими кислотами или их производными, а
указывают лишь относительное пространственное расположение
групп у асимметрического атома углерода.
152. Стереоиэомерия а-оксикислот
303
Таким образом, для молочной кислоты:
СООН СООН
ОН
н-
соон
ОН Оконфигурациа
(—(-молочная
кислота
2а-молочная
кислота
ПО
СН3
COOI!
СООН
но-
соон
-н
сп,
L-конфигурацня
(+>молочная
к и лога
2р-молочная
кислота
Необходимо обратить внимание на то основное положение
пространственной модели, исходя из которого принято строить
ее проекцию. Из правила проектирования (стр. 300) следует, что
горизонтальная линия (Н—ОН) при переходе к
пространственной модели изображает ребро тетраэдра впереди
плоскости чертежа
СООН
н —Loh
соон
но !_н
соон
I
он
СН./ЭН
СН4ОН
D(-) Ц+)
глицеринопая кислота
CHjCOOH
Н'
соон
I н
СН2СООН
?•(-)
яблочная кислота
Строение винных кислот на проекционных моделях
изображается гак:
СООН
н—Loh
НО-
соон
но I н
соон
н I он
соон
НО-
I
-Н
Н-
соон
А
-ОН
Н-
-Н
-ОН
НО-
оон
соон
соон
D(+) »инные кислоты И—)
Амезовннная кислота
Очевидно, что конфигурация, а тем самым и обозначение (D
или L), но не знак вращения (-f- или —), останутся неизменными,
304
/ пава IX Оксикислоти
если мы подвергнем исходное вещество химическому
превращению, не затрагивающему связей у асимметрического атома
углерода*.
СООН
Loh
снэ
О( — )-м
кислотя
СООН
но | н
снэ
Ц+)-молочная
кислота
СООС2Н5
н—Lou
снэ
О(+)-этиловыЯ эфир
молочной кислоты
СООН
Н—[—ОН
С.НаВг
D(—)-бром
молочная кислота
Экспериментальным путем было установлено, что молекулы
D-)-молочной кислоты и D-)-глицериновой кислоты имеют одну и
ту же конфигурацию; обе кислоты могут быть получены (с
соблюдением указанного выше условия) из (—)-изосерина, который,
следовательно, имеет ту же L-конфигурацию, что и D-)-глицери-
новая кислота:
СООН
І Н2
но ...н »¦ но.
NOBr
СООН
-Н
но ' н _
CHjNHj
/.(—Іизосерин І р
HNOy
сн.
СООН
J-H
,Вг
СООН
сн3
Ц 4-) -молочная
кислота
HO-
СН2ОН
глицериновая
/.(-Н-кислота
Так как L(—)-изосерин получается из природной (—)-яблоч-
ной кислоты, то очевидно, что и яблочная кислота имеет ту же
L-конфигурацию.
* Как показал П. И. Вальден, при реакциях, затрагивающих связи у
асимметрического центра, часто происходит взаимное превращение
конфигураций (так называемое «вальденовское обращение»).
ГЛАВА X
АЛЬДЕГИДОКИСЛОТЫ И КВТОНОКИСПОТЫ
153 Строение и отдельные представители альдегидо- и
кетонокислот. Альдегидо- и кетонокислотами называются
соединения, содержащие в молекуле карбоксильную группу и
альдегидную или соответственно кетонную группу.
Примерами этих кислот могут служить:
Глиоксиловая кислота (этаноловая кис- Н—СО—СООН
лота)
Пировиноградная кислота (пропаноно СНа—СО—СООН
вая кислота)
Ацетоуксуаіая кислота (бутанон-3-овая СНа—СО—СН.;—СООН
кислота)
Левулиновая кислота (пентанон-4-овая СН3—СО—СНа—СН.,,—СООН
кислота)
Пировиноградная кислота является а-кетонокислотой, ацето-
уксусная кислота—?-кетонокислотой, левулиновая кислота—т-ке-
тонокислотой.
Глиоксиловая кислота Н—СО—СООН-Н2О содержится в
незрелых фруктах; по мере их созревания она постепенно исчезает.
Так как молекула глиоксиловой кислоты прочно связана с
молекулой воды, то эту кислоту рассматривают как гидрат
альдегида. Строение глиоксиловой кислоты доказывается ее получением
при кипячении дихлорук^усной кислоты с водой:
ноін сі
НО;н СИ-
іЧсн—
соон-*нсі
НО.
НО
\сн—соон
|/
¦ нсосоон-н2о + неї
Глиоксиловая кислота при обычных
условиях—сиропообразная жидкость. При кипячении со щелочью она превращается в
соли гликолевой и щавелевой кислоты:
ноос.-с/ + зкон + %с—соон —»
\н ц/
» КО ОС—СНоОП -і- КООС—COOK + ЗН2О
соль гликолевоґі і;оль щавелеюй
кислотм кислоты
^0—788
306 Глава X. Альдегидокислоты и кетонокислоти
При этом происходит восстановление альдегидной группы
одной молекулы кислоты и окисление альдегидной группы другой
молекулы (реакция Канниццаро).
Пировиноградная Кислота СН3—СО—СООН получается при
сухой перегонке виноградной кислоты. Это жидкость (темп,
плавл. 13,6 °С; темп. кип. 165 °С) с запахом, напоминающим запах
уксусной кислоты. Пировиноградная кислота является побочным
продуктом спиртового брожения; энзимом карбоксилазой
(составной частью зимазы дрожжей) пировиноградная кислота
разлагается на уксусный альдегид и углекислый газ:
/Р
СН3—СО—iCOOiri »CH3-C<f + СО2
Альдегид, соответствующий пировиноградной кислоте,
называется метилглиоксалем, так как его можно рассматривать как
метильное производное глиоксаля:
сн/ хн
метилглиоксаль
Метилглиоксаль—маслянистая жидкость с резким запахом,
легко полимеризуется. При действии воды в присутствии
щелочей дает молочную кислоту:
/Р КОН
СНз-СО-с/ +НаО > CHS-CH(OH)-COOH
ХН
При этом кетонная группа восстанавливается, а альдегидная
скисляется. ?
Ацетоуксусная кислота СН3—СО—СН2—СООН. В свободном
виде она представляет собой сиропообразную жидкость. Как и все
?-кетонокислоты, ацетоуксусная кислота неустойчива и легко
разлагается даже при слабом нагревании, отщепляя углекислый
газ:
СН3—СО—СН2—ЇСООіН » СН3—СО—СН3 -f СО2
ацетоуксусная кислота ацетон
154. Ацетоуксусный эфир. Из производных кетонокислот
наибольшее значение имеет этиловый эфир ацетоуксусной кислоты
СН3—СО—СН2—СООС2НА, называемый обычно ацетоуксусным
эфиром. Он находит обширное применение в органической химии для
синтеза и представляет большой теоретический интерес.
154. Ацетоуксусный эфир 307
Ацетоуксусный эфир—жидкость с приятным фруктовым
запахом; кипит, незначительно разлагаясь, при 180 °С, поэтому его
очищают перегонкой под уменьшенным давлением; мало
растворим в воде.
Получение ацетоуксусного эфира. Ацетоуксусный эфир
получается при действии натрия на этиловый эфир уксусной кислоты.
Относительно механизма этой реакции существуют различные
предположения. Согласно одному из них (Клайзен), механизм
реакции состоит в следующем. Алкоголят натрия, образующийся
из этилового спирта, который в ничтожных количествах всегда
бывает примешан даже к весьма чистым препаратам эфира,
присоединяется к молекуле эфира:
+ Na—OCjH» » CH3— C-OC2H5
Продукт присоединения реагирует со второй молекулой эфира,
выделяя две молекулы спирта:
,ONa
СН3 —С—jOCOHs"" Н!\ ХН—COOGjHj
\Lr „ „і >CH-COOQH6 » СНЯ-С/
;OC2HaH:/ NONa
Спирт снова дает алкоголят, который опять присоединяется к
уксусноэтиловому эфиру, и т. д.
В результате реакции получается натриевое производное
этилового эфира оксикротоновой кислоты, который при действии
кислот выделяется в свободном виде
.СН—COOGjHs
СН3—C<f + НС1 > СНз—С(ОН)=СН—COOGjHj + NaCI
xONa
и изомеризуется в ацетоуксусный эфир
CH3-C(OH)=CH-COOC2Ha z=zt СН3-СО-СН2—СООС2Н6
эфир бутен-2-ол-З-овой кислоты эфир бутанон-3-овой кислоты
Такой конденсации могут подвергаться и другие сложные
эфиры кислот, содержащие смежную с карбоксильной группу
20*
308 Глава X. Альдегидокислоты и кетонокислоты
СН2. Именно за счет водородов этой группы и происходит
конденсация с образованием производных ?-кетонокислот:
R ° R
R—CHj—с/ ±!Н|—СН » R—СНа—С-СН—COOR' + R'—ОН
Ni.o?.'. і \coor'
Строение ацетоуксусного эфира. Строение «обыкновенного»
ацетоуксусного эфира было предметом спора химиков в течение
многих десятилетий. Одни рассматривали его как ацетоуксус-
ный эфир (I), другие считали его эфиром оксикротоновой
кислоты (II):
СН3—С 0—СНа—СООС2НЬ СН3—С=СН—СООСгНь
Ан
Согласно первой формуле обыкновенный ацетоуксусный эфир
является и эфиром, и кетоном; согласно второй формуле он,
будучи эфиром, является в то же время и непредельным
спиртом.
В некоторых реакциях обыкновенный ацетоуксусный эфир
реагирует как этиловый эфир ацетоуксусной кислоты. Так,
например, он, подобно другим кетонам, присоединяет кислый сер-
нистокислый натрий. С синильной кислотой он образует циангид-
рин, превращающийся в результате гидролиза и дегидратации в
метилмалеиновую кислоту:
СНз-С— СНг—СООСгН5 GH.,—С—CHjCOOCjH,
|| + HCN , /\
О NC ОН
циангидрин
СНз—С—CHjCOOCjHa гидролиз СН3—С—СН—COOCaHj -н2о
NC ОН НООС ОН
—»сн3—с=сн—соон
соон
метилмалеиновая
кислота
Все эти реакции указывают на кетонное строение ацетоуксусного
эфи ра.
В других реакциях он ведет себя как этиловый эфир
оксикротоновой кислоты. Так, с хлористым ацетилом он в оп еделенных
154. Ацетоуксусный эфир ЗОЯ
условиях образует вещество, в котором кислотный радикал
связан с кислородом:
СН3 СН3
с—о—Ун'Тсї;—со—снэ —> с—о—со—снэ + на
II '• її
СН—СООС2Н6 СН—СООС2Н5
Как непредельное соединение, ацетоуксусный эфир
присоединяет бром, а как спирт, имеющий гидроксил при углероде с дзой-
ной связью, дает с хлорным железом фиолетовое окрашивание
(характерная реакция на енолы). Таким образом, обыкновенный
ацетоуксусный эфир реагирует то как кетон, то как спол.
Многочисленные исследования показали, что обыкновенный
ацетоуксусный эфир представляет собой смесь двух изомеров:
I (кетонная форма) и II (енольная форма). Оба изомера находятся
в равновесии друг с другом и при нарушении равновесия быстро
превращаются один в другой. Если к ацетоуксусному эфиру в
водном растворе прибавить несколько капель хлорного железа,
то изомер в енольной форме дает фиолетовое окрашивание.
Прибавляя затем по каплям бром, можно перевести енольную форму
в бромпроизводное; при этом фиолетовое окрашивание пропадает.
Через некоторое время, однако, фиолетовое окрашивание снова
появляется, так как восстанавливается нарушенное равновесие
и кетонная форма частично переходит в енольную. Прибавлением
брома снова переводят енол в бромпроизводное. Таким образом
можно заставить все взятое количество ацетоуксусного эфира
реагировать в виде енольной формы. С другой стороны, действуя
реагентом, связывающим кетонную форму, например NaHSO3f
можно заставить весь ацетоуксусный эфир реагировать как кетон.
При действии натрия или алкоголята натрия на
ацетоуксусный эфир один атом водорода в его молекуле замещается
натрием.
В результате получается натрийацетоуксусный эфир:
СН3—С=СН-СООС2Н6 + C2H6ONa * СН3—С=СН—СООС2Н6 + С2Н6ОН
ONa
ОН
Если прибавить к натрийацетоуксусному эфиру соляную кислоту,
то выделяется мало растворимая в воде енольная форма
ацетоуксусного эфира:
СН3—С=СН—COOGjHs + HCI > СНз—С=СН—СООСаН6 + NaCI
ONa OH
310 Глава X. Альдегидокислоты и кетонокислоти
Скорость химических реакций, в том числе и скорость
изомерного превращения енола в кетон, сильно уменьшается с
понижением температуры. Поэтому, производя последний опыт при
очень сильном охлаждении, удалось получить почти чистую еноль-
ную форму в виде незастывающего при —78 °С масла. Енольная
форма ацетоуксусного эфира, т.е. афир оксикротоновой кислоты, в
отличие от кетонной формы, моментапьно растворяется в щелочах,
присоединяет бром, дает окрашивание с хлорным железом. С
другой стороны, охлаждая жидким воздухом раствор обыкновенного
ацетоуксусного эфира в петролейном эфире, удалось получить в
кристаллическом виде (темп, плавл. 39 °С) чистый ацетоуксус-
ный эфир, не реагирующий с бромом и не дающий окрашивания
с FeCI3. Обе формы при обычной температуре переходят в
обыкновенный ацетоуксусный эфир, представляющий собой смесь 92, 5%
кетона и 7,5% енола.
Как показали исследования А. Н. Несмеянова, натрийацетоук-
сусный эфир существует только в енольной форме
СН3—С=СН—С—ОС,Н5
I II
ONa О
Двойственная же реакционная способность, т. е. способность нат-
рийацетоуксусного эфира образовывать соединения кетонной и
енольной формы, появляется вследствие наличия в молекуле
сопряженной системы связей и переноса реакционного центра от
атома кислорода (связанного с металлом) к углероду метинной
группы. Это схематически изображают так (см. также стр. 312):
=с-
К
6)
Na
155. Таутомерия. Итак, ацетоуксусный эфир существует в
двух изомерных формах, переходящих одна в другую, и образует
производные обеих форм.
Явление, когда вещество может существовать в виде
нескольких изомерных форм, легко переходящих друг в друга и
находящихся в подвижном равновесии, называют таутомерией:
таутомерия—весьма частое явление в органической химии.
Переходящие друг в друга формы называются таутомерами, а их
взаимный переход—таутомерным превращением.
В том случае, когда таутомерами являются вещества с
карбонильной группой и енолы (например, изомерные формы ацето-
156 Синтезы при помощи ацетоуксусного эфира 311
уксусного эфира—см. стр. 308), таутомерия называется кепго-еноль-
ной.
При таутомерных превращениях атом водорода легко меняет
свое положение, причем происходит изменение характера связи,
например:
—С-С— 7=± -С=С— —С—N- ^=Z —C=N—
II I I II I I
OH OH OH OH
Для некоторых таутомерных веществ (например,
ацетоуксусного эфира, фенилнитрометана) удалось выделить в чистом виде
оба изоцера. Гораздо чаще встречаются такие случаи таутомерии,
когда вещество при химических реакциях дает производные двух
изомерных форм, хотя оно само известно лишь в одной форме.
Так, например, известна только одна синильная кислота, хотя
реагирует она в двух таутомерных формах:
H—C=N -с > Н—N=C
Выделить обе формы синильной кислоты не удается вследствие
чрезвычайно большой скорости таутомерного превращения.
Надо отметить, что еще в 1876 г. А. М. Бутлеров предвидел
возможность явления, названного впоследствии таутомерией.
В докладе Академии Наук, появившемся в печати в 1877 г., он
писал: «Большинство жидких и газообразных веществ находится
в состоянии химического равновесия тогда, когда число молекул
одного рода чрезвычайно велико, а число молекул другого или
нескольких других видов бесконечно мало... Однако, несомненно,
имеются случаи, в которых количество одного из изомеров не
является минимальным и молекулы обоих изомеров находятся
в постоянной «конкуренции»... Ясно, что химические
превращения такого тела должны будут совершаться то в духе одной, то
другой химической группировки, в зависимости от природы
реагирующих веществ и условий опыта».
156. Синтезы при помощи ацетоуксусного эфира. Ацетоуксус-
ный эфир расщепляется при действии на него щелочей. Это
расщепление, в зависимости от условий, происходит различно.
1. При действии на ацетоуксусный эфир разбавленных
щелочей (или кислот) происходит его омыление с последующим
отщеплением СО2 от образовавшейся ацетоуксусной кислоты:
СНаСОСНа —СО—Ю—QHj + НІ—О—Н » СН3СОСН2--СО--ОН + С2НВОН
СНзСОСНа—ІСО—О;—Н » СН3СОСН3 + СО2
Этот вид расщепления ацетоуксусного эфира называется ке-
тонным расщеплением. В результате его образуются углекислый
газ, спирт и ацетон.
i\2 Глава X. Альдегидокислоты и кетонокислоти
2. При нагревании ацетоуксусного эфира с крепкими
щелочами происходит кислотное расщепление:
СНз—С—CHj—С—О—
II і II
О і О
к—о—:н к—
2СН3СООК
о—н
Синтезы кетонов и кислот. Как указывалось ранее (см.
стр. 307), при действии на ацетоуксусный эфир натрия или алко-
голята натрия образуется натрийаиетоуксусный эфир:
СН3—С=СН—СООС,н5
ONa
При действии на него галоидных алкилов металл замещается
алкилом, причем алкил связывается не с кислородом, а с
углеродом, несмотря на го, что в натрийацетоуксусном эфире натрий
был связан с кислородом:
С— О—Ш± J)—СН3 >¦ Ol,—СО—СН—COOCjH8 + NaJ
II
СН—
Это явление и многочисленные аналогичные случаи
вступления радикалов не по месту, ранее занятому металлом, как уже
указывалось на стр. 310, называется «переносом реакционного центра».
В полученном моноалкилированном эфире еще один атом
водорода может быть замещен натрием, который, в свою очередь,
можно обменять на радикал:
СН3 СНа
І І
СН3—СО—СН—СООС2Н5 + CuH5ONa > СН3—С=С—COOQHa + С^ОН
ONa
СН3 СН3
. .. I Н2О
С—О— iNa + Ji-С,Н5 * СН3—СО—С—COOQHa »
QH5
метилэтилацетоухсусный эфир
снэ
—* СНдСО-С—СООН + CjH5OH
С.Н,
Синтезы при помощи ацетоуксусного эфира 313-
В результате получаются двузамешенные производные
ацетоуксусного эфира.
Подобно самому ацетоуксусному эфиру, его одно- и двузаме-
щенные производные способны подвергаться кеюнному и
кислотному расщеплению. Это позволяет синтезировать при помощи
ацетоуксусного эфира кетоны, у которых один из радикалов—
метил, а другой может иметь нормальную или раьвеївлснную
цепь углеродных атомов.
Схема синтезов кетонов (см. стр. 311):
СН3СОСН2СООС2Н6-
СНзСОСНэ
CH3COCHR—COOC2H6 » СНзСОСНа—R
CH3COCRR'—COOCjH6 » CH3COCH<
XR
Из ацетоуксусного эфира и его одно- и двузамещенных
производных можно синтезировать также кислоты нормального и
разветвленного строения.
Схема синтезов кислот (см. стр. 312):
СН3СОСН2СООСаН6—
СНзСООН
CH3COCHR—COOQH6 » RCH.2COOH
CH3COCRR'—COOQHj . >CHCOOH
R'x
ГЛАВА XI
УГЛЕВОДЫ
157. Классификация углеводов. Углеводы в большом
количестве содержатся в растениях и животных. Они играют очень
важную роль в процессах, протекающих в живых организмах.
Примерами углеводов могут служить глюкоза, или виноградный
сахар (С0Н12Ос), тростниковый, или свекловичный сахар
(С12Н22ОП), крахмал и целлюлоза, состав которых выражается
формулой (CcH10Oa)*-
Из приведенных формул видно, что эти вещества состоят из
углерода, водорода и кислорода, причем водород и кислород
находятся в том же отношении, что и в воде, т. е. на два атома
водорода приходится один атом кислорода. Таким образом, их
состав можно выразить общей формулой С„(Н2О)ОТ,—они как бы
состоят из углерода и воды; отсюда и произошло их название
углеводы, впервые предложенное русским химиком К.
Шмидтом A822-1894).
В настоящее время известны как природные, так и
синтетически полученные вещества, у которых не соблюдена указанная
пропорция между водородом и кислородом, но которые,
несомненно, относятся к тому же классу веществ, что глюкоза и
клетчатка. Но все же состав подавляющего большинства углеводов
выражается формулой Cn(H2O)m.
Углеводы делятся на две группы:
1) моносахариды, или монозы, примером которых может
служить глюкоза;
2) полисахариды, или полиозы.
Последние, в свою очередь, разделяются на две группы:
а) олигосахариды (низкомолекулярные полисахариды),
представителем которых может служить свекловичный сахар;
б) высокомолекулярные полисахариды, например, крахмал и
цел толоза.
Молекулы полисахаридов построены из остатков молекул
моносахаридов; полисахариды расщепляются при гидролизе на
более простые углеводы. Моносахариды неспособны к
гидролитическому расщеплению.
158. Строение моносахаридов 315
МОНОСАХАРИДЫ
158. Строение моносахаридов. Со строением моносахаридов
можно познакомиться на примерах глюкозы и фруктозы
(плодовый сахар). Состав как глюкозы, так и фруктозы выражается
формулой С6Н12О6.
Известен сложный эфир, образованный одной молекулой
глюкозы с пятью молекулами уксусной кислоты, получающийся
обработкой глюкозы в соответствующих условиях уксусным
ангидридом. Следовательно, в молекуле глюкозы имеется пять гидрок-
сильных групп. Впервые это было экспериментально доказано
в 1869 г. А. А. Колли*.
Глюкоза вступает и в некоторые реакции, характерные для
альдегидов; например, она дает реакцию на серебряное зеркало;
при осторожном окислении к молекуле глюкозы присоединяется
один атом кислорода и образуется одноосновная кислота.
Следовательно, в молекуле глюкозы находится альдегидная группа.
При энергичном восстановлении глюкозы йодистым
водородом получается 2-иодгексан CH3CH2CH2CH2CHJCH3. Это
свидетельствует о том, что глюкоза имеет нормальную цепь
углеродных атомов.
Таким образом, приняв во внимание, что один атом углерода
может удержать только один гидроксил, можно выразить
строение глюкозы формулой:
СН3(ОН)-СН(ОН)-СН(ОН)-СН(ОН)—СН(ОН)—С<^
Согласно этой формуле, глюкоза одновременно является и
альдегидом, и спиртом; глюкоза—альдегидоспирт.
В молекуле фруктозы, как и в молекуле глюкозы, имеется пять
гидроксильных групп, но, в отличие от глюкозы, фруктоза при
окислении (окисью ртути в присутствии гидроокиси бария)
расщепляется на две кислоты—триоксимасляную кислоту
СН2(ОН)—СН(ОН)—СН(ОН)—СООН и гликолевую кислоту
СН2(ОН)—СООН. Это указывает на наличие в молекуле
фруктозы кетонной группы, находящейся при втором атоме
углерода от начала цепи; следовательно, строение фруктозы
выражается формулой:
СНа(ОН)-СН(ОН)—СН(ОН)-СН(Огі)-СО-СН2ОН
* Александр Андреевич Колли A840—1916) был преподавателем в
Московском университете, а в 1876—1903 гг. занимал кафедру химии в
Московском высшем техническом училище.
Ему принадлежит ряд работ, имеющих принципиальное значение в химии
углеводов. Так, он установил пятиатомность глюкозы, предложил
циклическую форму для моносахаридов A871 г.), первый синтезировал дисахарид и др.
316 Глава Х.І.- Углеводы
Фруктоза одновременно является и кетоном, и многоатомным
спиртом; фруктоза—кетоноспирт.
Окисление фруктозы окисью ртути в присутствии Ва(ОНJ
можно изобразить следующей схемой:
н н н -н н
І І І II I
Н—С—С—С~С—:
I
ОН ОН ОН ОН
—С—С—Н + 20
II I
о он
> СН2(ОН)—СН'ОН)—СН(ОН)—СООН + СН2(ОН)—СООН
гриоксимасляная ;ислота гли:<олевая кислота
Как в молекуле глюкозы, так и в молекуле фруктозы
содержится группа атомов:
Н
-С—С—
I II
НО О
Моносахариды, содержащие альдегидную группу, называются
альдозами; содержащие кетонную группу—кетозами. По числу
атомов кислорода в молекуле различают биозы*, триозы, те/прозы,
пентозы, гексозы, гептозы и т. д. Так:
СН2(ОН)—[СНОН|з—СНО альдопентоза
СН2(ОН)—[СНОН]4-СНО альдогексоза
СН2(ОН)—[СНОН]3—СО—СНаОН кетогексоза
В природе встречаются почти исключительно пентозы и
гексозы. Подавляющее большинство моносахаридов имеет нормальную
цепь углеродных атомов.
В соответствии с выведенной выше формулой
СН2(ОН)—СН(ОН)—СН(ОН)—СН(ОН)—СН(ОН)—СНО
глюкоза является альдегидоспиртом. Известен, однако, ряд
фактов, которые не объясняются достаточно удовлетворительно этой
формулой. Так, глюкоза не дает некоторых реакций альдегидов,
например окрашивания с фуксинсернистой кислотой, не вступает
в отсутствие воды в реакцию с хлором и бромом (А. А. Колли).
При нагревании глюкозы с безводным метиловым спиртом,
содержащим небольшое количество хлористого водорода (в качестве
Биозами иногда называются дисахариды (см. стр. 338 ел.).
158. Строение моносахаридов 317
катализатора), атом водорода одной из гидроксильных групп
глюкозы замещается метилом,—образуется метилглюкозид. Ме-
тилглюкозид не дает никаких альдегидных реакций; он легко
гидролизуется, расщепляясь на метиловый спирт и глюкозу.
Если метилглюкозид обработать йодистым метилом и окисью
серебра, то и атомы водорода четырех остальных гидроксильных
групп замещаются метил ьны ми группами—получается тетра-
метилметилглюкозид. При гидролизе этого соединения сначала
отщепляется одна метильная группа, а образовавшаяся 2,3,4,6-
тетраметилглюкоза вступает во все альдегидные реакции,
характерные для самой глюкозы. Итак, один гидроксил в молекуле
глюкозы имеет особый характер; замещение атома водорода в
этом гидроксиле приводит к потере веществом альдегидных свойств.
Этот особый гидроксил обычно называют глюкозидным гидрок-
силом.
Указанные явления хорошо объясняются формулой, согласно
которой глюкоза имеет циклическое (окисное) строение:
СНа(ОН)_СН—СН(ОН)—СН(ОН)—СН(ОН)—СН(ОН)
-о-
Как легко заметить, циклическая формула образуется из
альдегидной перемещением атома водорода от гидроксила пятого
атома углерода к кислороду, стоящему при первом атоме
углерода. Благодаря этому у первого атома углерода и у кислорода,
связанного с пятым атомом углерода, появляются свободные
валентные связи, которые и насыщают друг друга с образованием
кольца, состоящего из пяти атомов углерода и одного атома
кислорода.
В растворе глюкозы существуют молекулы альдегидной
формы и молекулы окисной формы, т. е. в растворе имеет место тауто-
мерное равновесие:
б 5 4 3 2 1
СН2(ОН)—СН(ОН)—СН(ОН)—СН(ОН)—СН(ОН)—СН=О z=±
I—* СНа(ОН)—СН—СН(ОН)—СН(ОН)—СН(ОН)—СН(ОН)
При образовании метилглюкозида происходит замещение ме-
тильной группой атома водорода гидроксила, связанного с пер-
318 Глава XI. Углеводы
вым атомом углерода, т. е. водорода глюкозидного гидроксила:
—сн—сн(ОН)—сн(он)—сн(он)-сн-о—!н+"но|-сн3—»
I О 1
СН2(ОН)-СН-СН(ОН)-СН(ОН)-СН(ОН)-СН-О-СН3 + НаО
I О 1
метилглкжозид
В молекуле метилглюкозида гидроксильная группа у первого
атома углерода отсутствует; таким образом, в молекуле нет
группировки, могущей переходить в альдегидную группу. Поэтому
метилглюкозид не дает альдегидных реакций. Что же касается
2,3,4,6-тетраметилглюкозы, то для нее возможно таутомерное
превращение в альдегидную форму:
СНа(ОСН3)—СН-СН(ОСНз)—
-о-
ї=^СН2(ОСН3)-СН(ОН)-СН(ОСНз)-СН(ОСНз)-СН(ОСН3)-СНО
Фруктоза также может реагировать в таутомерных формах:
6 5 4 3 2 1
Ш2(он)—сн(ОН)—сн(ОН)—сн(ОН)—со—cHjOh^i^;
^=± СН2-СН(ОН)-СН(ОН)-СН(ОН)-С(ОН )-Ш2(ОН)
II
о
Строение глюкозы и фруктозы в твердом состоянии
выражается окисной формулой, так как из насыщенного раствора
выделяется только окисная форма.
Для того чтобы наглядно показать циклическое строение
глюкозы, ее формулу (I)
CHj.(OH)—СН-СН(ОН)-СН(ОН)-СН(ОН)—СН(ОН)
I
можно изобразить следующим образом (II):
Н ОН
\/
Нч/ ЧуН
но
с
\
он
НОСН/Ч / Х)Н
н
\
н-'с
II
Н-С
ч
1
н
У
Vh
II
с-н
[II
пи рай
/59. Понятие о стереоизомерии альдогексоз 319
Сравнение этой формулы с формулой пирана (III)
показывает, что в глюкозе находится ядро гидрированного пирана, и ее
можно назвать глюкопиранозой.
В устойчивых формах глюкозы 'и фруктозы кольцо образовано
пятью атомами углерода и одним атомом кислорода.
В неустойчивых формах этих углеводов, например в ?-фрук-
тозе, имеется пятичленное кольцо, состоящее из четырех атомов
углерода и одного атома кислорода:
(НО)СНа—СН—СН(ОН)—СН(ОН)—QOH)—СНа(ОН)
I О 1
у-фруктоза
Такое колыю характерно для соединений группы фурана (см.
стр. 580), поэтому фруктозу можно назвать фруктофуранозой:
Н Н
НО-С—С-ОН
Н Н Н—с—С—Н
\ / II 'II
(НО)СН„—С С—СН2(ОН) Н—С С—Н
V V
Y-фруктоза фуран
Как было указано выше, А. А. Колли еще в 1871 г. предложил
циклическую окисную форму строения глюкозы. В то время не
могло быть и речи об определении формы этого цикла. Лишь в
1926 г. Херст и Хэуорс показали, что в устойчивых формах
моносахаридов находится шестичленное кольцо (см. стр. 318).
159. Понятие о стереоизомерии альдогексоз. Глюкоза является
альдогексозой. Формула глюкозы
СНа(ОН)—СН(ОН)—СН(ОН)—СН(ОН)—СН(ОН)—С<^
показывает, что в ее молекуле находится четыре неравноценных*
между собой асимметрических атома углерода. В этом случае
* Т. е. связанных с разными группами. Так, атомы углерода в положении
3 и 4 связаны со следующими группами:
Н Н
) (С,Н6О3)-С-(СзН6О3)
о:
н он
C) D)
320 Глава XI. Углеводы
возможно 24, т. е. шестнадцать стереоизомеров, которые
разбиваются на восемь пар. Члены каждой пары являются по
отношению друг к другу оптическими антиподами, т. е. молекулы одного
из них представляют собой зеркальное изображение молекул
другого: один из них вращает плоскость поляризации света
вправо, другой—на равный угол влево. Каждая пара имеет одно
название, но перед названием одного члена пары ставится буква D,
перед названием другого—буква L, например: D-глюкоза и
L-глюкоза. Что же касается различных пар, то они отличаются
не только оптическими, но и другими физическими и некоторыми
химическими свойствами.
Первоначально строчными латинскими буквами d и / (dexter—
правый, laevus—левый) пользовались для различия
правовращающих и левовращающих форм оптически деятельных
соединений. В настоящее время правое вращение обозначается
знаком плюс (+), левое вращение—знаком минус (—), буквы же
служат для обозначения вида конфигурации, и чтобы не путать
буквенные обозначения с направлением вращения, используют
уже не строчные, а прописные буквы D и L.
Способность вещества к вращению плоскости поляризации
имеет большое значение для его характеристики, однако из
сравнения величин вращения даже двух весьма сходных соединений
нельзя установить, имеют ли они одинаковую или различную
конфигурацию.
Можно привести следующие примеры: мясомолочная кислота
врашает плоскость поляризации вправо, в то время как ее соли
и сложные эфиры обладают левым вращением, хотя из них и
может быть обратно получена правовращающая молочная кислота,
которая, несомненно, имеет ту же самую конфигурацию, что ее
соли и сложные эфиры. Знак вращения изменяется во многих
случаях в зависимости от растворителя и даже от концентрации
раствора.
Э. Фишер, вводя обозначения d и /, относил к d-ряду
соединения, полученные из d-глюкозы.
В настоящее время (по предложению М. А. Розанова в 1906 г.)
в качестве исходного вещества для сравнения конфигураций
оптически деятельных соединений принят глицериновый альдегид,
известный в виде двух антиподов:
СНО СНО
Н—С—ОН НО-С—Н
сн2он снаон
I 1]
159. Понятие о стереоизомерии альдогексоз
321
Из двух возможных конфигураций принимают для
правовращающего глицеринового альдегида конфигурацию (I) и обозначают
его о(+)-глицериновым альдегидом.
Вещества, имеющие конфигурацию, сходную с конфигурацией
D-глицеринового альдегида, относят к D-ряду; вещества L-ряда
имеют конфигурацию, сходную с конфигурацией L-глицерино-
вого альдегида (II).
При окислении О(+)-глицеринового альдегида получается
D(—)-глицериновая кислота. Это указывает на аналогичность
конфигураций обоих соединений:
010 СНО СООН
-ОН
Н-
-он
Н2ОН СН2ОН СН2ОН
Таким же образом устанавливается относительная конфигурация
и других а-оксикислот.
Обозначения семейства у (+)-глицеринового альдегида и
(—)-глицериновой кислоты совпадают, хотя глицериновая
кислота считается D-оксикислотой по конфигурации асимметрического
центра в а-положении от карбоксильной группы, т. е. по «клю-
ЧУ» (П СООН СНО
Н.
-ОН
R CH2OH
і п
а оксиальдегиды относят к семейству D по «ключу» (II) и это
может привести к недоразумениям (см. § 187).
Для углеводов принадлежность к D- или L-ряду определяется
конфигурацией асимметрического атома углерода, наиболее
удаленного от карбонила:
СНО СНО
I I
Н— С— ОН НО—С—Н
СН2ОН
L
НО—С—Н
Н—С—ОН
н—с—он
сн,он
О-глюкоза
НО—С—Н
н-с-он
н—с—он
снаом
О-манноза
о
но—с—н
Н-С-ОН
н—с—он
сн.2он
D-фруктоза
322 Глава XI. Углеводы .
Обозначения D и L для моносахаридов, имеющих по
нескольку асимметрических центров, ничего не говорят о конфигурации
каждого из этих центров. Для такого детального описания
конфигурации удобно воспользоваться системой обозначений «р—о»
(см. стр. 160). В этой системе D-глюкоза будет обозначена,
например, 2р,3а,4р,5р,6-пентаоксигексанал или гексанпента-
ол-B,4,5)р,Зо,6-ал-1. Соответственно фруктоза будет обозначена:
1,Зо,4р,5р,6-пентаоксигексанон-2 или гексанпентаол-D,5)р,3о,
1,6-он-2 и т. д.
а- и ?-Формы моносахаридов. Согласно альдегидной формуле,
возможны шестнадцать стереоизомерных альдогексоз.
Однако их существует больше. Если приготовить раствор
D-глюкозы (обыкновенная природная глюкоза), то наблюдается
замечательное явление: удельное вращение раствора
[a]D = + 113°* постепенно уменьшается, пока не достигнет
постоянной величины [aJD = +52°. Это явление называется мутаро-
тацией.
Следовательно, глюкоза переходит в какое-то другое вещество,
обладающее меньшим удельным вращением. Исследования
показали, что существуют две стереоизомерные D-глюкозы: a-D-глю-
коза и ?-D-глюкоза, причем первая вращает плоскость
поляризации на угол в шесть раз больший, чем вторая. Обыкновенная
D-глюкоза—это a-D-глюкоза.
Обе формы D-глюкозы в растворе или в расплавленном
состоянии легко переходят друг в друга. Так, удельное вращение
свежеприготовленного раствора ?-D-глюкозы [a]D = -fl9c
увеличивается с течением времени. Это увеличение продолжается до
тех пор, пока удельное вращение раствора не достигнет
постоянного значения, т. е. будет равно -t-52°, как и при растворении
a-D-глюкозы. Таким образом, в растворе находятся оба вида
глюкозы, и между ними устанавливается определенное
равновесие. Из раствора, в зависимости от условий кристаллизации,
можно выделить ту или иную форму глюкозы. Так как
a-D-глюкоза менее растворима в воде, чем ?-D-глюкоза, то она и будет
выделяться из водного раствора. Наоборот, из раствора
глюкозы в ледяной уксусной кислоте, если в охлажденный раствор
внести кусочек ?-D-глюкозы, выкристаллизовывается
?-D-глюкоза. Известны производные обеих форм, например а-метил-
глюкозид и ?-метилглюкозид.
Точно так же и для многих других стереоизомеров,
возможных согласно альдегидной формуле, было доказано существование
• [a\D обозначает, что вращение определено для желтой линии D натрия
(длина волиы 589 ммк).
160. Нахождение в природе и способы получения моносахаридов 323
двух форм. Это явление прекрасно объясняется окисной
формулой
СН2(ОН)-СН-СН(ОН)-СН(ОН)-СН'ОН)-СН(ОН)
-О-
Согласно этой формуле, в молекуле альдогексозы имеется не
четыре, а пять асимметрических атомов углерода, и потому сте-
реоизомеров должно быть не шестнадцать, а тридцать два..
Другими словами, для каждого из стереоизомеров, соответствующего
альдегидной формуле, возможно существование двух
модификаций: а- и ?-форм. Сравнение окисной и альдегидной формул
показывает, что согласно окисной формуле, в молекуле
альдогексозы имеется лишь один лишний асимметрический атом углерода,
а именно—первый атом углерода. Таким образом, различие
молекул а- и ?-форм сводится к различному пространственному
расположению групп около этого углеродного атома в плоскости,
составляющей основание тетраэдра для этого атома.
Для того чтобы мысленно перейти от ОН-группы через атом
Н к кислороду, замыкающему кольцо, в молекуле одной формы
надо двигаться по часовой стрелке, в молекуле другой формы—
против часовой стрелки:
Н-С-ОН
Н—С-ОН
но—с-н о
I
Н—С-ОН
Н-І—
СН3ОН
a-D- глюкоза
В молекуле ?-формы гидроксильные группы при перЕом и
втором атомах углерода находятся по разные стороны кольца;
расстояние между ними больше, чем расстояние между этими гидрок-
сильными группами в молекуле а-формы. Этим и объясняется,
что в растворе глюкозы больше ?-формы, обладающей меньшим
запасом энергии.
160. Нахождение в природе и способы получения
моносахаридов. Моносахариды широко распространены в природе как в
свободном состоянии, так и в связанном виде. Они образуются при
21*
324 Глава XI. Углеводы
гидролизе полисахаридов, дубильных веществ типа таннина,
некоторых сложных белков—нуклеопротеидов и глюкопротеидоь.
Так, например, при гидролизе крахмала получается глюкоза:
Первый синтез сахаристого вещества был осуществлен в
1861 г. А. М. Бутлеровым. Нагревая муравьиный альдегид с
известковым молоком, он получил светло-желтый сладкий сироп,
названный им метиленитан, по аналогии с маннитаном (старое
название маннита). Этот сироп обладает обычными реакциями
моносахаридов. Впоследствии из него была выделена а-акроза,
т. е. рацемическая фруктоза.
а-Акроза как индивидуальное вещество была синтезирована
в 1887 г. Э. Фишером* из акролеина СН2=СН—СНО;
присоединением брома к акролеину был получен дибромид; при действии
на последний баритовой воды происходило образование
глицеринового альдегида, который, изомеризуясь частично в диоксиаце-
тон, превращался в а-акрозу:
-2Ha'J +Bra
СН2(ОН)—СН(ОН)—СН,(ОН) » СН2=СН—СНО *
глицерин акролеин
+Ва(ОИ)а
» СН2Вг—СНВг—СНО СН2(ОН)—СН(ОН)—СНО
дибромакролеин глицериновый альдегид
СН2(ОН)—СН(ОН)—СНО > СН2(ОН)—СО—CH.j(OH)
глицериновый альдегид диохспацетон
СН2(ОН)—СН(ОН)—СНО + НСН(ОН)—СО—СН2(ОН)
глицериновый альдегид диоксиацетом
¦ СНа(ОН)-[СН(ОН)]3-СО—СНа(ОН)
а-акроза
• Эмиль Фишер A852—1919)—один из наиболее выдающихся
химиков-органиков. Учился и работал у Байера. В 1892 г. был назначен в качестве
преемника А. В. Гофмана на кафедру химии в Берлинский университет. Работы
Фишера относятся к исследованию класса гидразинов, в частности фенилгидрази-
на. продолжением этих исследований являются работы по углеводам. Своими
работами Фишер доказал, что углеводы предстанляют собой частью альдеги-
доспирты. частью кетоноспнрты. Другой ряд работ Фишера относится к
исследованию розанилина и парарозанплпна. Фишер занимался исследованием
производных пурина и оригинально, со стереохимической точки зрения,
объяснил действие ферментов и процесс брожения. Наибольшее значение из всех
работ Фишера имеют его исследования в области белковых веществ, являющиеся
первым конкретным шагом на пути к синтезу белков.
160. Нахождение в природе и способы получения моносахаридов 325
Акроза Э. Фишера оказалась смесью многих Сахаров; в ней
были обнаружены глюкоза, манноза, фруктоза. Таким образом,
по выражению Фишера, «глицерин оказался теми воротами,
через которые мы дошли до синтеза природных сахаристых
веществ».
Моносахариды с большим числом атомов углерода могут быть
получены при помощи циангидринового синтеза из альдоз с
меньшим числом углеродных атомов. Примером может служить
переход от пентозы к гексозе.
Присоединением синильной кислоты к альдопентозе получают
оксинитрил, который при гидролизе дает одноосновную
оксикислоту:
он
.О 4-HCN / 4-2Н2О
СН2(ОН)— [СНОН]3—С<^ > СН2(ОН)—[СНОН]3—С—CN >
н \н
альдопеитоза оксинитрил
» СН2(ОН)-[СНОН]4—СООН + NH3
оксикислота
Оксикислота переходит в лактон, из которого при
восстановлении амальгамой натрия получают альдогексозу:
~н2о
СН2(ОН)—СН(ОН)—СН(ОН)—СН(ОН)—СН(ОН)—СООН >
оксикислота
+Н2
, СН2(ОН)—СН(ОН)—СН—СН(ОН)—СН(ОН)—СО »
I о 1
СН2(ОН)-СН(ОН)-СН(ОН)-СН(ОН)-СН(ОН)-С^
альдогексоэа
Таким путем были получены гептозы С,Н14О,, октозы СвН16О8,
нонозы С9Н19О9 и даже декозы С10Н20О10.
Получение моносахаридов с меньшим числом атомов углерода
из моносахаридов с большим числом атомов углерода
производится по способу Руффа. Альдозу окислением переводят в
одноосновную оксикислоту и ее кальциевую соль окисляют Н2О2 в
присутствии солей окиси железа.
В качестве промежуточного продукта образуется по всей
вероятности кетонооксикислота, которая отщепляет С02 и пере-
326 Глава XI. Углеводы
ходит в альдозу, содержащую на один атом углерода меньше, чем
исходная альдоза:
О о
СНа(ОН)—[СНОН]3—СН(ОН)—с/ •
альдогексоза
Н2О2
* СН2(ОН)—[СНОН]Э—СН(ОН)—СООН
оксикислота
-СО2 Р
—»сн2(он)—[сноніз—со—:соо:н * сн2(оі;) ^
кетонооксикислота альдопеитоаа
161. Свойства моносахаридов. 1. Моносахариды легко
окисляются; они восстанавливают аммиачный раствор окиси серебра и
фелингову жидкость. При осторожном окислении альдоз
образуются одноосновные оксикислота с тем же числом углеродных
атомов,—так называемые оновые кислоты; глюкоза дает глюко-
новую кислоту, манноза—манноновую кислоту и т. д.
,0 о
CH„OH-[CHOH]4-C<f » СН2ОН-[СНОН]4-СООН
При более сильном окислении (например, концентрированной
HNO3) образуются двухосновные оксикислота; двухосновная
оксикислота, получаемая при окислении глюкозы, называется
сахарной кислотой:
/Р °
CH2OH-[CHOH]4-C<f * НООС-[СНОН]4-СООН + Н2О
2. При восстановлении моносахаридов они переходят в
многоатомные спирты:
СН,(ОН)-[СНОН]4-С/ > СН2ОН-[СНОН]4-СНаОН
н
3. Весьма характерна реакция моносахаридов с фенилгидр-
азином,—она приводит к образованию озазонов, или, точнее,
фени лозазонов.
Эту реакцию можно объяснить на примере глюкозы.
161. Свойства моносахаридов 327
Фенилгидразин, взаимодействуя с альдегидной группой
глюкозы, образует растворимый фенилгидразон:
СН=О + H2N—NH—С„Нб CH=N—NH—QHb
СНОН СНОН
I »I +H2O
[CHOH]3 [CHOH]3
CH2OH CH2OH
фенилгидраэви глюкозы
При нагревании фенилгидразона с избытком фенилгидразнна
происходит перегруппировка, сопровождающаяся отщеплением от
гидразона молекулы анилина. При этом возникает соединение, в
котором на месте карбонильной группы оказывается иминная
группа, а соседняя группа СНОН превращается в карбонильную
группу. Такое соединение можно назвать замешенным иминокетоном.
Избыточный фенилгидразин немедленно реагирует с получившимся
иминокетоном, отщепляя аммиак и воду и образуя озазон:
CH=N—NHQHb CH=NH
СНОН __ А=О +^HsNHNH^
. . . I -NH-,; -H2O
(СНОН)з (СНОН)э
(
СН
:н2он сн2он
фенилгндразон иминпкетон
глюкозы
CH=N—NH—СвН5
C=N—NH—
(СНОН)з
СН3ОН
оаазон глюкозы
Озазон D-глюкозы трудно растворим в воде. Он представляет
собой желтые иглы (темп, плавл. 204—205 °С). D-Глюкоза, D-ман-
ноза и D-фруктоза дают один и тот же озазон. В реакцию с фе-
нилгидразином вступает прежде всего карбонильная группа.
У D-глюкозы и у D-маннозы углеродом карбонильной группы
является первый атом углерода, у D-фруктозы—второй атом
углерода. Озазоны D-глюкозы, D-маннозы и D-фруктозы тождест-
328
Глава XI. Углеводы
I
Н-С-ОН
НО—С—Н
Н—С—ОН
I
Н—С—ОН
сн„он
D- глюкоза
венны. Следовательно: 1) в молекуле озазона остатки фенилгидр-
азина связаны с первым и вторым атомами углерода; 2)
пространственное расположение атомов около третьего, четвертого и
пятого атомов углерода в молекулах D-глюкозы, D-маннозы и
D-фруктозы одинаково:
СН2ОН
U
НО—С—Н
н—с—он
н—с—он
СН2ОН
D-фруктоза
НО—С—Н
но—с—н
Н-С-ОН
Н-С-ОН
СН2ОН
D-мапноза
CH=N-NH-QH6
Н—С—ОН
НО—С—Н
Н—С—ОН
Н-С—ОН
СН2ОН
фенилгидразон
D глюкозы
CH=N—
но-с-н
НО—С—Н
н-с—он
Н—С—ОН
СН2ОН
феннлгидразон
D-маннозы
СН2ОН
C=N—NH— QHS
но—с—н
н-с—он
н—с—он
сн.рн
фенил гндразои
D-фруктозы
CH=N— NH-QHb
*. C=N—NH—QH6 ^
HO—C—H
H-C-OH
H—C-OH
CH2OH
фенилозазон
Конфигурации D-глюкозы и D-маннозы отличаются только
пространственным расположением атомов у второго атома
углерода. Подобные альдозы называются эпимерными.
162. Триозы 329
4. При осторожном нагревании озазонов с крепкой соляной
кислотой происходит отщепление двух молекул фенилгидразина
и образуются озоны:
: СНО
CH=N—NH—СаН5 На|О неї
і
—NH—С6Н5 Н2:О
+ ) * СО -f- 2СвН5ЫНЫНа
[Ш
[СНОН]3
[бЮН],
СН2ОН
Озоны, кроме гидроксильных, содержат также альдегидную и ке-
тонную группы. При обработке озонов цинковой пылью и
уксусной кислотой происходит восстановление альдегидной группы,
в результате чего образуются кетозы:
СН2ОН—[СНОН]3—СО—С<Г -f Н2 > СН2ОН—[СНОН]3—СО—СН2ОН
Через озазон и озон можно от альдозы перейти к кетозе:
C6H5NH-NH2 H2O Н2
альдоза *¦ озазон * озон * кетоза
5. При действии разбавленных щелочей альдозы частично
превращаются в свои эпимеры и соответствующую кетозу. Так,
например, D-глюкоза при этом частично переходит в D-маннозу
и D-фруктозу. При действии же крепких щелочей происходит
разложение моносахаридов; раствор при этом буреет; крепкие
щелочи разлагают гексозы на молочную и другие кислоты.
6. Как и спирты, моносахариды при действии на них
ангидридов кислот образуют сложные эфиры. Со щелочами и с
гидроокисями щелочноземельных металлов они дают соединения
типа алкоголятов. О метилировании моносахаридов было
сказано выше (см. стр. 316).
7. Брожение моносахаридов см. стр. 336.
162. Триозы. Главнейшими триозами являются
глицериновый альдегид и диоксиацешон:
СН2(ОН) _СН(ОН)—с/ СН2(ОН)—СО—СН2(ОН)
глицериновый альдегид диоксиацетон
Смесь обеих триоз получается при осторожном окислении
глицерина, например, перекисью водорода в присутствии солей
закиси железа. Эта смесь представляет собой сиропообразную
жидкость с характерными для моносахаридов реакциями; ее назы-
330 Глава XI. Углеводы
вают глицерозой. При окислении глицерина сорбозными
бактериями получается только диоксиацетон, так как эти бактерии
действуют исключительно на вторичную спиртовую группу.
В молекуле глицеринового альдегида находится
асимметрический атом углерода, для этого соединения известны оптически
деятельные формы. Недеятельный глицериновый альдегид
плавится при 142 °С, обладает сладким вкусом. При кипячении с
безводным пиридином он частично переходит в диоксиацетон:
глицериновый альдегид ( ' диоксиацетон
Диоксиацетон—кристаллическое вещество. Глицериновый
альдегид и диоксиацетон имеют удвоенный молекулярный пес.
В растворах они постепенно переходят в мономолекулярную
форму.
163. Пентозы. Пентозы имеют состав CaH10Oa, т. е. в
молекулах пентоз содержится пять атомов кислорода. Наиболее
распространенные в природе пентозы являются альдопентозами.
Строение их выражается формулами:
СН2(ОН)—СН(ОН)— СН(ОН)—СН(ОН)—О?
хн
или
СН2—СН(ОН)—СН(ОН)—СН(ОН)—СН(ОН)
I 0
Согласно первой формуле, в молекуле альдопентоз находятся
три асимметрических атома углерода, поэтому для альдопентоз
возможны четыре пары стереоизомеров:
сно сно сно сно
Н—С—ОН НО—С—Н НО—С—Н Н—С—ОН
І І І І
Н—С—ОН НО—С—Н Н—С—ОН НО—С—Н
н-с-с
-ОН НО—С—Н Н—С—ОН НО-С—Н
І І І І
СН2ОН СН2ОН СН,ОН СН2ОН
D-рибоза L-рибоза D-арабиноза L арабиноза
сно сно сно сно
I I ! I
НО—С—Н Н—С—ОН Н—С—ОН НО—С—Н
І І І
НО—С—Н Н—С—ОН НО—С—Н Н—С—ОН
Н—С—ОН НО—С—Н Н—С— ОН НО—С—Н
І І І І
СН2ОН СН2ОН СН.2ОН СН2ОН
D-ликсоза Л-ликсоза D-ксилоза /.-ксилоза
163. Пентози 331
Для каждого из этих восьми стереоизомеров возможны а- и
?-формы.
Все альдопептозы могут быть окислены в триоксиглутаровые
кислоты НООС—СН(ОН)—СН(ОН)—СН(ОН)—СООН. В
соответствии с приведенными проекционными формулами, рибоза и
ксилоза дают оптически недеятельные триоксиглутаровые
кислоты:
СООН СООН
н_с-он н-с—он
ПЛ0СК0С1Ь н-с-он но-с-н --•
I I
Н—С—ОН Н—С—ОН
СООН СООН
В молекулах этих кислот два асимметрических атома
углерода, связанные с карбоксильными группами, имеют
противоположные конфигурации и компенсируют друг друга. Молекулы
этих кислот симметричны. Наоборот, триоксиглутаровые кислоты,
получающиеся при окислении арабинозы и ликсозы, имеют
несимметричные молекулы и потому оптически деятельны.
Стереохимия триоксиглутаровых кислот не может быть
описана с помощью символов D и L, но легко описывается
обозначениями о—а (см. § 187).
В природе пентозы встречаются главным образом в виде
полисахаридов пентозанов (C5HSO4)^, а также входят в состав
пектиновых веществ и растительных камедей, например,
гуммиарабика, вишневого клея (см. стр. 354). Пентозаны находятся
в древесине (лиственные деревья содержат 22—27%
пентозанов), в сене A0—15%), соломе, оболочках семян, подсолнечной
лузге и т. д. При гидролизе пентозаны расщепляются на пентозы:
При нагревании с кислотами пентоза переходит в
циклический альдегид—фурфурол. При этом от каждой молекулы
пентозы отщепляются три молекулы воды:
Н н
. . I I .
;но-гс 9—он
:Н ~Т~С
н-с—с-н
г_!_н ; * " " "I" 3HaO
с •¦ н : Н—С С—СНО
Н ЮН Н:О СНО
рентозз фурфурол
332 Глава XI. Углеводы
Фурфурол можно обнаружить при помощи некоторых
цветных реакций. Так, с флороглюцином (см. стр. 462) и соляной
кислотой он дает вишневое окрашивание, с солями
анилина—красное окрашивание.
L(-{-)-Apa6uHO3a получается нагреванием гуммиарабика или
вишневого клея с разбавленной серной кислотой; плавится при
160 °С; обладает сладким вкусом, обнаруживает мутаротацию.
Рацемическая арабиноза иногда встречается в моче (пентозурия)
D(+)-Ксилоза (древесный сахар) получается кипячением с
разбавленными кислотами пшеничной соломы; темп, плавл. 143 °С,
обладает очень сладким вкусом, обнаруживает мутаротацию.
D(—уРибоза (темп, плавл. 95 °С) входит в состав нуклеиновых
кислот (стр. 626).
164. Гексозы. Из гексоз наибольшее значение имеют
D-глюкоза, D-манкоза, D-галактоза и D-фруктоза.
Р(+)-Глюкоза (виноградный сахар) в большом количестве
содержится в растениях и животных. Она находится в соке
винограда, в других сладких плодах, а также в семенах, листьях,
корнях, цветах. В животных организмах она содержится в крови,
лимфе, жидкости спинного мозга. В ничтожных количествах
глюкоза имеется в человеческой моче. Содержание глюкозы в моче
резко увеличивается при так называемой сахарной болезни,
достигая иногда 12%.
В промышленности глюкозу получают из крахмала
кипячением его с разбавленной серной кислотой. После кипячения
кислоту нейтрализуют мелом; раствор глюкозы отфильтровывают
от малорастворимого сернокислого кальция и упаривают под
уменьшенным давлением*. Полученный сироп называется
патокой. Для получения кристаллической глюкозы крахмал осаха-
ривают возможно полнее, и сгущенный сироп разливают в
ящики, где он застывает в белые или желтоватые плитки.
Наличие в молекуле D-глюкозы шестичленного кольца доказано следующим
образом. При осторожном окислении 2,3,4|6-тетраметилглюкозы
СН2(ОСН3)—СН—СН(ОСНз)—СН(ОСНз)—СН(ОСНз)—СН(ОН)
О 1
получается 2,3,4,6-тетраметилглюконовая кислота
СН2(ОСН3) —!СН(ОН)—СН(ОСНз)—СН(ОСНз) —СН(ОСНз)—СООН
* Этот способ осахаривания крахмала был открыт в Петербурге в 1811 г.
адъюнктом Академии наук К. С. Кирхгофом A764—1833). Им же было
показано, что крахмал при действии солода превращается в солодовый сахар
(дисахарид— мальтоза).
164. Гексози 333
При дальнейшем действии окислителей происходит окисление вторичной
спиртовой группы в кетонную группу и затем, как и всегда при окислении ке-
тонов, расщепление молекулы у кетонной группы. При этом получается тримет-
окснглутаровая кислота:
НООС-СН(ОСН3)-СН(ОСН3)-СН(ОСН3)-СООН
Если бы в молекуле глюкозы имелось пятичленное кольцо, то окисление
ее тетраметилпроизводного привело бы к получению 2,3,5,6-тетраметилглюко-
новой кислоты:
СН2(ОСН3)-СН(ОСН3)—СН(ОН)—СН(ОСН3)—СН(ОСН3)—СООН
В этой кислоте гидроксил находится у четвертого атома углерода, при ее
окислении получить триметоксиглутаровую кислоту невозможно.
Глюкоза кристаллизуется с одной молекулой воды, безводная
плавится при 146 °С; хорошо растворима б соде. Глюкоза
примерно в два раза менее сладка, чем сахар.
При окислении глюкоза дает сначала D-глюконовую, а затем
D-сахарную кислоты, при восстановлении переходит в
шестиатомный спирт сорбит:
сн,он сно соон соон
І І І І
Н—С—ОН Н—С—ОН Н—С—ОН Н—С—ОН
3—С—Н .-,,, НО—С—Н
НО—С—Н 2Н НО—С—Н о НО—С—Н 2О НО—С—Н
н—с—он н—с—он н-с—он н—с—он
н—с—он н—с—он н—с—он н—с—он
сн.рн снаон сн2он соон
сорбит D-глюкоза D-глюконовая D-сахарная
кислота кислота
Сорбит находится в ягодах рябины, в соке вишен, слив,
яблок, груш и т. д.; плавится при 110—111 °С; обладает сладким
вкусом.
Глюкоза применяется в кондитерском производстве. В
хлопчатобумажной промышленности она используется в качестве
восстановителя при крашении и печатании, идет для
изготовления протрав и т. д.
D-Манноза встречается в природе главным образом в виде
полисахаридов—маннанов, содержащихся в скорлупе каменного
ореха, плодах некоторых пальм, зернах ячменя и пшеницы,
корнях спаржи, цикория и г. д. Манноза—кристаллическое
вещество сладкого вкуса (темп, плавл. 132 °С), хорошо растворим!
в воде; обнаруживает мутаротацию. При окислении дает D-манно-
334 Глава XI. Углеводы
новую и манносахарную кислоты, при восстановлении переходит
в шестиатомный спирт маннит:
снаон сно соон соон
НО—С—Н НО—С—Н НО—С—Н НО—С—Н
с—н
Н—С—ОН н—С—ОН * Н—С-ОН * н—С—ОН
но—с—н +2Н но—с—н о но—с-н ю но—с—н
н—с— он н—с—он н—с—он н—с—он
снаон сн2он снаон соон
маннит D-маниоза D-манноновая манносахарная
кислота кислота
Маннит находится в больших количествах в так называемой
манне—высушенном соке некоторых южных и тропических
растений. В манне ясеня, произрастающего в Закавказье и в
Сицилии, содержится до 55% маннита. Некоторые водоросли Белого
моря также содержат значительное количество этого вещества.
D-Маннит представляет собой кристаллы, обладающие сладким
вкусом; темп, плавл. 165—166 °С.
D-Галактоза встречается в природе в виде полисахаридов,
например молочного сахара. D-Галактоза—кристаллическое
вещество сладкого вкуса (темп, плавл. 165 °С); сравнительно хорошо
растворима в воде; обнаруживает мутаротацию. При окислении
дает D-галактоновую и слизевую кислоты; при восстановлении
переходит в шестиатомный спирт дульцит:
СН2ОН
Н—С—ОН
О—С—Н +2Н
О-С-Н *
н—с—он
1
снаон
дульцит
сно
Н—С—ОН
НО—С— Н
НО—С— Н
Н—С—ОН
1
СН2ОН
D- галактоза
О
— 1
соон
н—с-он
но-с—н ,п
> 1 —,
НО-С-Н
Н-С-ОН
1
снаон
D-галактоиовая
кислота
соон
Н-С—ОН
НО—С—Н
¦ но—с—н
н—с—он
1
соон
слизевая
кислота
Слизевая кислота—мелкокристаллический порошок, очень
трудно растворимый в воде; оптически недеятельна.
Дульцит встречается в растениях. Он представляет собой
кристаллы сладкого вкуса (темп, плавл. 188 °С); оптически
недеятелен.
164. Гексози 335
D-Фруктоза (плодовый сахар) находится вместе с
D-глюкозой во многих сладких плодах; смесь равных количеств
D-фруктозы и D-глюкозы составляет главную часть (80%) меда; она
входит в состав тростникового сахара и инулина (стр. 346).
D-Фруктоза обычно образует кристаллы состава 2СвН12О6 • Н2О;
она значительно слаще сахара; обнаруживает мутаротацию.
Из других альдогексоз имеют значение, ввиду их близости
к витамину С, эпимерные между собой D-гулоза и D-идоза:
СНО СНО
І І
Н—С-ОН НО—С—Н
Н—С—ОН Н—С—ОН
но-с—н но-с-н
] I
н-с—он н-с-он
сн2он сн2он
D-гулоза D-идоза
ВитаминС» или /.-аскорбиновая кислота, является енольной
формой лактона 2-кето-/.-идоновой B-кето-/.-гулоновой)
кислоты:
СООН СО
І І
СО С(ОН)
I II
НО-С-Н С(ОН)
Н—С—ОН НС
НО—С—Н НО-С—Н
о
сн2с
сн2он сн2он
2-кето-і.-идоновая ^.-аскорбиновая
B-кето-і.-гулоновая) кислота {витамин С)
кислота
Витамин С—кристаллическое вещество (темп, плавл. 190 °С),
оптически деятелен; является сильным восстановителем; весьма
распространен в природе. Он содержится в свежих фруктах,
ягодах и овощах, в меньшем количестве находится в малине и
сливочном масле. Особенно богаты витамином С черная смородина,
лимоны, апельсины, томаты. Недостаток витамина С в пище
вызывает заболевание цингой. В настоящее время аскорбиновую
кислоту (витамин С) получают в промышленных масштабах из
D-глюкозы.
336 Глава XI. Углеводы
165. Глюкозиды. Глюкозиды являются производными
углеводов, полученными замещением атома водорода в глюкозидном
гидроксиле остатком соединения неуглеводного характера—аглю-
кона. Действием кислот или особых энзимов глюкозиды
расщепляются на углевод и аглюкон. Так, метилглюкозид
расщепляется при гидролизе на глюкозу и метиловый спирт:
ОСНЭ ОН
1 I
СН СН
[СНОН];
Н2О
снчон
О I О
н 1 сн L
сн2он сн2он
Действие энзимов является специфическим. Существуют
энзимы, которые расщепляют только а-глюкозиды, т. е. такие
глюкозиды, которые производятся от а-форм углеводов. Подобные
энзимы называются а-глнжозидазами; к числу их относится маль-
таза дрожжей. С другой стороны, эмульсин—энзим,
находящийся в горьком миндале, расщепляет только глюкозиды ?-ряда.
Гидролиз глюкозидов энзимами—обратимая реакция. Тот
энзим, который производит расщепление данного глюкозида,
вызывает и синтез последнего, т. е. приводит к установлению
равновесия.•
глюкозид + вода . у углевод + аглюкон
Глюкозиды широко распространены в растительном мире.
К ним относятся красящие вещества цветов и ягод, препараты
дигиталиса, салицин, амигдалин и т. д.
166. Механизм спиртового брожения. Многочисленными
работами ряда исследователей было установлено, что расщепление
сахаристых веществ на спирт и углекислый газ есть результат
нескольких последовательных химических реакций.
Для объяснения механизма спиртового брожения было
предложено несколько схем. Наиболее вероятная из них заключается
в следующем:
1. Брожению предшествует переход глюкозы в эфиры
фосфорной кислоты за счет тех фосфатов, которые находятся в
клеточном соке дрожжей или в веществах, прибавляемых во время
брожения.
166. Механизм спиртового брожения 337
Затем происходит расщепление фосфорнокислых эфиров на
диоксиацетон и глицериновый альдегид, находящиеся в
динамическом (таутомерком) равновесии*:
.О
С6Н12О6 > 2СН2(ОН)-СО-СН2(ОН) ^=1 2СН2(ОН)-СН(ОН)-С<^
В дальнейшем из глицеринового альдегида образуются по
реакции Канниццаро глицерин и глицериновая кислота:
2СН2ОН—СНОН—СНО + Н2О >
» СН2ОН—СНОН—СН2ОН + СН2ОН—СНОН—СООН
Если к бродильной жидкости прибавить фтористый натрий,
то можно прервать брожение на этой стадии и выделить из
жидкости глицерин и глицериновую кислоту в виде фосфорнокислых
эфиров.
Эта фаза является как бы «затравкой» брожения.
2. Глицериновая кислота переходит в пировиноградную
кислоту по схеме:
СН2ОН—СНОН—СООН » СН3—СО—СООН + Н2О
Под влиянием энзима карбоксилазы пировиноградная
кислота расщепляется на СО2 и уксусный альдегид:
СН3—СО—СООН » СО2 + СН3СНО
3. Уксусный альдегид восстанавливается за счет глюкозы в
этиловый спирт; одновременно происходит образование
глицериновой кислоты:
2СН3СНО + QHj2O6 + 2Н2О » 2С2Н5ОН + 2СН3ОН-СНОН-СООН
Теперь процесс становится стационарным: глицериновая
кислота снова дает (через пировиноградную кислоту) уксусный
альдегид и СО2 и т. д. Таким образом, брожение протекает, минуя
первую фазу. Если же к бродящей жидкости прибавить Na2SOa,
то исключается последняя фаза, и уксусный альдегид переходит
в бисульфитное соединение, на которое энзимы дрожжей не дей-
* Для простоты изложения приведены реакции превращения не фосфор-
нокиоых эфиров, а соответствующих простых сахаристых веществ, кислот
и г. п.
22—788
338 Глава XI. Углеводы
ствуют. В этом случае главными продуктами брожения являются
глицерин, уксусный альдегид и углекислый газ:
Q,HI2O6 > СНзОН-СНОН-СНаОН + СН3СНО + СОа
Дрожжи сбраживают D-глюкозу, D-маннозу, D-фруктозу и
D-галактозу. На другие гексозы, даже на антиподы
упомянутых углеводов, а также на пентозы, дрожжи не действуют. В
молекулах D-глюкозы, D-маннозы и D-фруктозы пространственное
расположение водорода и гидроксила у третьего, четвертого и
пятого атомов углерода одинаково, в то время как в молекуле
D-галактозы у одного из этих углеродных атомов, а именно у
четвертого атома углерода, оно иное, и D-галактоза сбраживается
дрожжами труднее; некоторые же виды дрожжей на нее вовсе
не действуют. По образному сравнению Фишера, энзим должен
подходить к веществу, как ключ к замку.
Спиртовое брожение гексоз вызывается дрожжами. Некоторые
другие микроорганизмы вызывают иные виды брожения:
1. Молочнокислое брожение:
С6Н1аО6 > 2СН3—СНОН-СООН
2. Маслянокислое брожение:
QHvA > QH7-COOH + 2Н2 + 2СОа
3. Лимоннокислое брожение:
QH13O6 + О4 > НООС—СИ,—С(ОН)(СООН)—СН2—СООН + 2НзО
4. Ацетонобутиловое брожение:
2С6Н12О6 > С4Н3ОН + СН3СОСН3 + 4Н2 + 5СО3
Процессы брожения имеют большое значение в
промышленности. Биохимические процессы, происходящие под влиянием
ферментов, в ряде производств используются с практической
целью. В организмах высших животных непрерывно протекают
процессы биохимического расщепления и синтеза моносахаридов.
При мышечном сокращении, в результате расщепления углеводов,
образуется молочная кислота, а также ряд других продуктов.
НИЗКОМОЛЕКУЛЯРНЫЕ ПОЛИСАХАРИДЫ (ОЛИГОСАХАРИДЫ)
167. Свойства и строение дисахаридов. Полисахаридами
называются углеводы, молекулы которых, присоединяя воду,
расщепляются на молекулы моносахаридов или более простых поли-
167. Свойства и строение дисахаридов 339
сахаридов. Они разделяются на две группы: низкомолекулярные
полисахариды (олигосахариды) и высокомолекулярные полисахариды. К
первой группе относятся полисахариды, которые по своим
свойствам приближаются к тростниковому сахару. Они большей частью
хорошо кристаллизуются, растворимы в воде, обладают сладким
вкусом и определенным молекулярным весом. Простейшими из
них являются дисахариды. Дисахаридами называются такие
углеводы, молекула которых, присоединяя молекулу воды,
расщепляется на две молекулы моносахаридов.
Примерами дисахаридов могут служить: I) тростниковый,
или свекловичный, сахар—так называемая сахароза; 2)
солодовый сахар, или мальтоза; 3) молочный сахар, или лактоза;
4) целлобиоза. Состав всех этих дисахаридов выражается одной
и той же формулой С12НиОц.
Сахароза гидролизу ется на D-глюкозу и D-фруктозу:
СцН^Оп "Ь HjO » СбН12Ов +
сахароза D-глюкоза D-фруктоза
Мальтоза и целлобиоза при гидролизе дают только молекулы
D-глюкозы:
мальтоза D-глюкоза
Молочный сахар, гидролизуясь, переходит в D-глюкозу и
D-галактозу:
СиН-дОц + Н2О » СвНіаО(і + CeH12Oe
молочный D-глкмозэ D-галактоза
сахар
Гидролиз дисахаридов происходит под влиянием кислот или
энзимов. Действие последних носит избирательный характер.
Инвертаза—один из энзимов дрожжей—гидролизует сахарозу,
но не действует на молочный сахар; наоборот, эмульсин
расщепляет молочный сахар, но не действует на сахарозу. Гидролиз
дисахаридов является обратимой реакцией:
СіаНаАі + HjO ^=zl .QHiA + CeH12OG
Тот энзим, который в присутствии избытка воды расщепляет
данный дисахарид, в концентрированном растворе вызывает его
образование: мальтоза расщепляется мальтазой дрожжей на
глюкозу; при действии же мальтазы на концентрированный раствор
глюкозы удалось получить мальтозу. Целлобиоза при действии
эмульсина дает глюкозу, но при действии того же эмульсина на
концентрированные растворы глюкозы происходит образование
целлобиозы. Сахарозу синтетически получить до сих по не
удалось.
22*
340
Глава XI. Углеводы
Одни из дисахаридов восстанавливают фелингову жидкость,
образуют фенилгидразоны и озазоны; следовательно, в их
молекулах имеется карбонил или группировка, легко переходящая в
карбонил. В других дисахаридах нет этой группировки, и они не
дают указанных выше реакций. К Дисахаридам первого типа
относятся мальтоза, молочный сахар и целлобиоза. Из
дисахаридов второго типа наибольшее значение имеет сахароза.
Сахароза. Молекула сахарозы расщепляется при гидролизе
на молекулу D-глюкозы и молекулу D-фруктозы. Следовательно,
в молекуле сахарозы остатки молекул D-глюкозы и D-фруктозы
соединены при помощи атома кислорода, а формулу сахарозы
надо выводить из формул глюкозы и фруктозы отнятием
элементов воды от гидроксильных групп. Поскольку сахароза не
обнаруживает ни свойств кетозы, ни свойств альдозы, не
восстанавливает фелингову жидкость, не дает ни фенилгидразона, ни оза-
зона, следует считать, что в ее молекуле нет ни карбонила, ни
группировки, легко переходящей в карбонил. Следовательно,
отщепление элементов воды происходит от двух глюкозидных гидр-
оксилов (образование глюкозид-глюкозидной связи), что
объясняет отсутствие в молекуле сахарозы активных функциональных
групп:
НСО-Н-
\
\
неон
HOCH
СН.2ОН
сш
\
о
HOCH
неон
I
НС
неон
НС.
о
снаон
глюкоза
СН2ОН
фруктоза
НОСЫ | + Н,0
] о
неон
НС-
СНаОН
снаон
сахароза
Мальтоза и молочный сахар. Мальтоза при гидролизе
расщепляется на две молекулы D-глюкозы. Следовательно, в ней два
остатка D-глюкозы соединены при помощи атома кислорода.
Мальтоза окисляется, присоединяя один атом кислорода, в
одноосновную кислоту; с фенил гидразином она образует фенилгидр-
азон. Следовательно, в ее молекуле есть одна альдегидная группа
или группировка, легко переходящая в альдегидную группу.
Поэтому отщепление элементов воды должно произойти в первой
молекуле глюкозы от глюкозидного, а 10 второй молекуле—от
неглюкозидного гидроксила. Доказано, что этим гідроксилом
167. Свойства и строение дисахаридов
является гидроксил при четвертом атоме углерода (возникновение
глюкозидно-глюкозной связи):
HCO;Hj
НСОН
HOCH ,
НСОН
нс —
I
СН2ОН
глюкоза
НСОН
HOCH
I: :O
НСіОН;
I
СН2ОН
глюкоза
НС
о
НСОН
I
HOCH
НСОН
НС—
I
СН2ОН
НСОН
\
о
\
\
НСОН
HOCH
і:
нс-
О + Н2О
СН2ОН
мальтоза
Присутствие ГЛЮКОЗИДНОГО ГИДрОКСИЛЭ ПРИВОДИТ К ТОМу, ЧТО'
молекула мальтозы может реагировать в альдегидной форме:
Н
не-
|\
нсонч
I о
HOCH
I
НСОН
I
нс—
СНцОН
СН2ОН
Строение молочного сахара аналогично строению мальтозы,
отличается лишь тем, что роль молекулы глюкозы в нем играет,
молекула D-галактозы:
НС-
НСОН\
HOCH
I
HOCH
нс—
о
\
\
СНоОН
^О
НСОН
HOCH
\
НС
I
НСОН
СН3ОН
молочный сахар (альдегидная форми)
342 Глава XI. Углеводы
Доказательством того, что альдегидная группа сохранилась
у остатка глюкозы, а не у остатка галактозы, является то, что
при осторожном окислении молочного сахара образуется лакто-
бионовая кислота
НС , СООН
\ о неон
нсон\ \ |
| О \ HOCH
HOCH
HOCH
нс-
нс
неон
СН2ОН
которая при гидролизе расщепляется на молекулу D-галактозы
и молекулу D-глюконовой кислоты
СН2(ОН)—СН(ОН)—СН(ОН)—СН(ОН)—СН(ОН)—СООН
т. е. той кислоты, которая получается при окислении D-глюкозы.
В молекулах молочного сахара и мальтозы тог остаток
молекулы глюкозы, в котором сохранилась группировка,
переходящая в альдегидную группу, может находиться в форме или
остатка a-D-глюкозы, или остатка ?-D-глюкозы. Поэтому молочный
сахар и мальтоза могут существовать в виде а- и ?-форм, легко
переходящих одна в другую, и их растворы обладают
способностью к мутаротации. Что же касается первого остатка
моносахарида, то он входит в состав молекулы дисахарида в одной
определенной форме. Так, в молекуле мальтозы первым остатком
является остаток a-D-глюкозы.
Целлобиоза. Строение целлобиозы отличается от строения
мальтозы тем, что в молекуле целлобиозы первым остатком
является остаток ?-D-глюкозы, в то время как в молекуле мальтозы
первым остатком служит остаток a-D-глюкозы. Это доказывается
следующим. Мальтоза и целлобиоза являются производными
Л-глюкозы, в молекуле которой водород глюкозидного гидроксила
замещен остатком второй молекулы глюкозы; таким образом,
они построены по типу глюкозидов. Глюкозиды a-ряда
расщепляются мальтазой, глюкозиды ?-ряда—эмульсином. На целло-
биозу мальтаза не действует, эмульсин же ее гидролизует. Это
позволяет отнести целлобиозу к ?-ряду.
168. Сахароза. Сахароза, называемая также тростниковым или
свекловичным" сахаром,—это обычный сахар. Она чрезвычайно
167. Свойства и строение дисахаридов
распространена в растительном мире. Она имеет большое
значение в жизни человека, являясь весьма важным питательным
и вкусовым веществом. Сахароза хорошо усваивается
человеческим организмом; при сгорании 1 г сахарозы выделяется
4000 кал.
Сахароза образует бесцветные прозрачные кристаллы (темп,
плавл. 160 °С); расплавленная сахароза застывает в аморфную
прозрачную массу—карамель. Раствор сахара вращает плоскость,
поляризации вправо. При гидролизе сахароза расщепляется
на равно молекулярные количества D-глюкозы и D-фруктозы.
Смесь же равных количеств глюкозы и фруктозы обладает
левым вращением. Таким образом, при реакции гидролитического
расщепления сахарозы происходит перемена направления
вращения плоскости поляризации. Поэтому сам процесс
расщепления сахарозы на глюкозу и фруктозу назвали инверсией
(обращением) сахара.
Сахароза содержится в очень больших количествах в стебляхі
сахарного тростника, а также в сахарной свекле. Впервые сахар*
из сахарного тростника начали получать более чем 21/2 тысячи
лет тому назад. Создание промышленного способа получения
сахара из сахарной свеклы относится к началу прошлого столетия.
Первый сахарный завод в России был построен в 1802 г.„
Для__получения_са24а?а..ха2сарну_ю»свеклу ^ежут машинами на
стружку, которую затем обрабатывают~горячей водой. Вода
извлекает из свеклы почти весь сахар; выщелоченная стружка—
жом—идет на корм скоту. Полученный раствор окрашен в
темный цвет и содержит, кроме сахара, ряд примесей (щавелевая и
фосфорная кислоты, белки, красящие вещества и др.). После
очистки от примесей (обработкой известью) раствор фильтруют,
упаривают в вакуум-аппаратах и отделяют выпавшие из раствора
кристаллы (сахарный песок) на центрифугах.
Жидкость, оставшаяся после выделения сахара, называется
чернод.гіатокой, или мелассой. Она содержит до 50% сахара и
может быть"использована ifй'бо~для выделения последнего, либо для
переработки на спирт. Для выделения из мелассы сахара ее
кипятят с раствором Sr(OHJ; при этом образуется
нерастворимый сахарат Cl2H22Ou¦ 2SrO, который отфильтровывают и
разлагают углекислым газом в водной среде:
Q3H2Ai-2Sr0 + 2СО3 >
Полученный сироп обрабатывают, как это было описано выше.
Белый _сахарный_ песок содержит До_99і9%_??ха?озьі. Для
получения ещ'ё~о'олее чистого сахара—"рафинада—сахарный песок-
344 Глава XI. Углеводы
растворяют в воде, фильтруют через костяной уголь и упаривают
до консистенции густой каши. Последнюю вливают в формы,
промывают после застывания чистым сиропом и высушивают под
уменьшенным давлением.
Из тростника сахар получают так же, как и из свеклы.
169. Мальтоза. Молочный сахар. Целлобиоза. Мальтоза
образуется при действии на крахмал диастаза солода, а также
птиалина слюны. Она является промежуточным продуктом
винокуренной и пивоваренной промышленности.
Мальтоза кристаллизуется с одной молекулой воды; раствор
ее вращает плоскость поляризации вправо; она примерно на
40% менее сладка, чем сахар. Мальтоза сбраживается
дрожжами, так как при действии мальтазы дрожжей она расщепляется
на D-глюкозу.
Молочный сахар (лактоза) содержится в молоке (в коровьем
4—5%, в женском 5—8%). Для получения молочного сахара
пользуются сывороткой, которая остается после удаления из
молока жира и белка.
Молочный сахар кристаллизуется с одной молекулой воды;
величина удельного вращения его +52° (по окончании мутарота-
ции); он примерно на 70% менее сладок, чем сахароза.
Молочный сахар сбраживается молочными дрожжами, которые
содержат лактазу, и расщепляется при этом на D-глюкозу и
D-галактозу. Пивные и винные дрожжи не содержат лактазы, поэтому
молочный сахар не бродит с этими видами дрожжей. Молочный
сахар применяется иногда при диэтическом питании.
Целлобиоза в виде уксуснокислого эфира (октоацетата)
получается при обработке целлюлозы уксусным ангидридом и
концентрированной серной кислотой. Целлобиоза—кристаллический
почти не сладкий порошок, растворимый в воде; она вращает
плоскость поляризации вправо, обладает мутаротацией,
восстанавливает фелингову жидкость.
ВЫСОКОМОЛЕКУЛЯРНЫЕ ПОЛИСАХАРИДЫ
Большинство высокомолекулярных полисахаридов
нерастворимо в воде; некоторые образуют лишь коллоидные растворы.
При нагревании они, не плавясь, разлагаются.
Главнейшими представителями высокомолекулярных
полисахаридов являются крахмал, целлюлоза, гликоген и инулин.
170. Крахмал. Крахмал (С6Н10Оь)х содержится в растениях,
являясь запасным питательным веществом последних. Он
служит одним из главнейших пищевых веществ для человека и
травоядных животных. Крахмал в виде зерен различной величины и
17t. Декстрины 345
формы откладывается преимущественно в клубнях и зернах
растений. Так, клубни картофеля содержат около 20% крахмала и
75% воды; в зернах пшеницы находится до 70% крахмала.
Крахмал—белый порошок, нерастворимый в воде. При
нагревании с водой он образует клейстер. С иодом крахмал дает синее
окрашивание. Это окрашивание исчезает при нагревании и снова
появляется при охлаждении. Крахмал не восстанавливает фелин-
гову жидкость.
Зерно крахмала построено из двух различных веществ:
амилопектина, составляющего оболочку зерна, и
амилозы—внутренней части зерна; количество амилопектина относится к
количеству амилозы примерно как 2:1. Амилопектин дает клейстер;
амилоза образует настоящий коллоидный раствор, не
проявляющий никаких свойств клейстера. Амилоза окрашивается иодом
в чисто синий цвет, амилопектин—в красновато-фиолетовый.
Амилоза—углевод, амилопектин—фосфорнокислый эфир другого
углевода. Молекулы амилозы имеют линейное строение, а
молекулы амилопектина—сильно разветвлены.
Присоединяя воду, крахмал постепенно расщепляется на
другие, более простые углеводы. Вначале он превращается в
растворимый крахмал*, который затем расщепляется на декстрины.
При гидролизе декстринов получается мальтоза. Молекула
мальтозы расщепляется на две молекулы D-глюкозы. Таким образом^
конечным продуктом гидролиза крахмала является D-глюкоза:
(СвН10Об)х + *Н2О » ^СвН12Ов
крахмал глюкоза
Гидролиз крахмала происходит при кипячении его с кислотами
или под действием энзимов. Примерами таких энзимов являются.
диастаз солода и птиалин слюны, которые гидролизуют крахмал
до мальтозы.
Постепенное расщепление крахмала под действием кислот и
энзимов можно наблюдать по реакции с иодом. Первоначальный
раствор окрашивается иодом в фиолетовый цвет; пробы, взятые на
последующих стадиях гидролиза, дают с иодом красно-бурую
окраску. Эти окраски характерны для сравнительно
высокомолекулярных декстринов. Низкомолекулярные декстрины окрашиваются иодом
в желтые цвета. Олигосахариды и моносахариды (низшие
декстрины, мальтоза и глюкоза) иодом не окрашиваются.
171. Декстрины. Декстрины—твердые вещества, растворимые
в воде. Они являются полисахаридами, но менее сложными, чем
крахмал. Декстрины восстанавливают фелингову жидкость. В тех-
• Крахмал, растворяющийся в горячей воде без образования клейстера
346 Глава XI. Углеводы
нике декстрины получают нагреванием крахмала до 180—200 °С.
Их можно также получить, нагревая крахмал до 150 °С, если он
предварительно смочен соляной или азотной кислотой.
Декстрины находят обширное применение в технике в
качестве клея, для загустки красок, в ситцепечатании и т. п.
Блестящая корка хлеба состоит из декстринов. Декстрины содержатся
и во всей массе печеного хлеба. Основное значение процесса
хлебопечения состоит в превращении нерастворимого крахмала
в растворимые и легче усваиваемые организмом декстрины.
172. Гликоген. Инулин. Гликоген (CeH10Os)x играет в
животном организме такую же роль запасного питательного вещества,
как и крахмал в растениях. Он отлагается главным образом в
печени (до 10%), содержится также в мускулах. Гликоген—белый
аморфный порошок, довольно легко растворяется в горячей воде,
давая коллоидный раствор, который свертывается от ничтожного
количества солей. С иодом дает желто-красную окраску. Раствор
гликогена вращает плоскость поляризации вправо. При
гидролизе кислотами и энзимами гликоген превращается в D-глюкозу.
Инулин (CeH10O5)^, подобно крахмалу, служит запасным
питательным веществом некоторых растений, но не имеет такого
широкого распространения, как крахмал. Он встречается в
клубнях георгин A0—12%), в корнях цикория A0%), в артишоках,
в нарциссах и во многих других растениях.
Инулин—белый порошок, легко растворяющийся в горячей
воде, не окрашивается иодом; раствор его вращает плоскость
поляризации света вправо. При гидролизе кислотами и энзимами
инулин дает D-фруктозу.
173. Целлюлоза. Целлюлоза, или клетчатка, (СвН10ОБ)х
является главной составной частью оболочек растительных клеток.
Молекулы целлюлозы, как и молекулы амилозы, входящей в
состав крахмала, имеют линейное строение. Обычно она не
встречается в растениях в чистом виде, а сопровождается так называемыми
инкрустирующими веществами (стр. 354). Гигроскопическая вата,
хлопчатобумажные и льняные ткани, хорошие сорта
фильтровальной бумаги состоят главным образом из целлюлозы,
несколько измененной в процессе выработки. Наиболее чистая
природная целлюлоза—это волокна хлопка, содержащие более 90%
целлюлозы и б—8% воды. В древесине хвойных деревьев целлюлоза
составляет около 50%; в лиственных деревьях ее содержится
значительно меньше.
Целлюлоза не растворяется ни в воде, ни в эфире, ни в спирте;
в обычных условиях довольно устойчива к действию
разбавленных кислот, щелочей и слабых окислителей. Она растворяется в
реактиве Швейцера (раствор комплексного соединения Cu(NH3)rt(OHJ в
173. Целлюлоза 347
аммиаке), в солянокислом растворе хлористого цинка, в
концентрированной серной кислоте (стр. 348).
Выделение целлюлозы в более или менее чистом виде
осуществляется в огромных количествах при производстве бумаги. До^
половины XIX в. для приготовления бумаги употребляли почти
исключительно льняное и хлопчатобумажное тряпье, предстап-
ляющее собой почти чистую целлюлозу. С развитием
книгопечатания и газетного дела тряпичной бумаги оказалось
совершенно недостаточно, поэтому были разработаны способы
получения целлюлозы из древесины. В настоящее время наиболее
простые сорта бумаги получают обработкой еловой древесины.
Такая бумага при хранении, особенно на свету, становится хрупкой.
Более высококачественные сорта изготовляют из бумажной
массы, представляющей собой смесь древесной массы с более или
менее чистой целлюлозой.
Для получения целлюлозы в промышленности пользуются
рядом способов. Наиболее распространенным из них является
сульфитный. По этому способу измельченную древесину (главным
образом еловую) варят при повышенном давлении в громадных
котлах—автоклавах емкостью до 300 м3 и более с раствором кислого,
сернистокислого кальция Ca(HSO3J. Древесина разрушается
и частично переходит в раствор; содержащаяся же в ней
целлюлоза остается в виде волокнистой массы. По окончании варки
содержимое котла передавливают в громадную сцежу—бетонный
резервуаре полом из дырчатых плиток. В сцеже целлюлозу
отделяют от раствора, промывают водой, отжимают на прессах,
высушивают и направляют для дальнейшей переработки на бумажную
фабрику.
Отделенный в сцеже раствор, так называемый сульфитный
щелок, содержит значительное количество сахаристых веществ,
которые могут быть использованы для получения спирта путем
брожения. Это один из вспомогательных источников получения
этилового спирта для технических целей из непищевого сырья
(«гидролизный спирт»).
Кроме производства бумаги, значительное количество
целлюлозы применяется для получения искусственного волокна,
пластических масс, лаков и порохов.
Гидролиз целлюлозы. Подобно крахмалу, целлюлоза при
действии на нее растворов кислот гидролизуется; при этом
образуется D-глюкоза:
(СвН10О5), + *НгО-**С6Н12О6
Это уравнение выражает только конечный итог гидролиза.
На самом деле гидролиз целлюлозы происходит постепенно, в
результате чего пол чаются все более и более простые вещества.
348 Глава XI. Углеводы
При непродолжительном действии крепкой серной кислоты
целлюлоза переходит в амилоид, который синеет от иода. Этой
реакцией часто пользуются для открытия целлюлозы. Иногда
вместо серной кислоты и иода для этой цели берут хлорцинкиод,
т. е. раствор иода и йодистого калия в насыщенном растворе
хлористого цинка.
Образованием амилоида пользуются в технике для
приготовления пергаментной бумаги: непроклеенную бумагу погружают
на несколько секунд в 80%-ную серную кислоту и затем
промывают водой и раствором аммиака; при этом на поверхности
бумаги образуется слой амилоида, делающий бумагу непроницаемой
дл я воды.
При действии минеральных кислот (соляной, серной)
целлюлоза частично гидролиз у ется. Получается хрупкое вещество,
которое легко истирается, восстанавливает фелингову жидкость,
частично растворяется в щелочах. Это—смесь неизмененной
целлюлозы и продуктов ее разрушения и гидролиза.
Если целлюлозу, пропитанную даже разбавленной кислотой,
высушить, особенно в токе горячего воздуха, она легко
рассыпается в порошок. Этим пользуются для выделения шерсти из
старых тканей, сделанных из смешанной пряжи (хлопка и шерсти).
При более энергичном действии кислот происходит
дальнейший гидролиз целлюлозы с образованием все более и более
простых полисахаридов.
Конечным продуктом гидролиза целлюлозы является
глюкоза.
В промышленности для осахаривания целлюлозы используют
древесные отходы деревообделочных заводов, нагревая их под
давлением с 0,1%-ным раствором серной кислоты; полученный
таким путем сироп перерабатывают в винный спирт.
По другому способу осахаривание производят на холоду—
действием на клетчатку соляной кислотой плотностью 1,21 г/см3.
Продукты гидролиза затем нагревают для удаления главной
массы соляной кислоты и нейтрализуют содой. Нейтрализованный
продукт применяют в качестве корма для скота.
Разрушение целлюлозы может происходить и под действием
микроорганизмов. Эти процессы имеют громадное значение в
природе; так происходит разрушение на поверхности земли
растительных остатков. Одним из таких процессов является
разрушение деревянных построек домовым грибком, который при помощи
кислорода воздуха окисляет целлюлозу до СО2 и Н2О. Большое
значение имеет метановое брожение целлюлозы, производимое
некоторыми видами бактерий на дне стоячих вод. Это брожение
происходит без доступа воздуха; при этом получаются метан,
углекислый газ и жирные кислоты.
174. Сложные и простые эфиры целлюлозы 349
Отношение целлюлозы к щелочам. Целлюлоза, сравнительно
легко изменяющаяся под действием кислот, весьма устойчива по
отношению к щелочам.
При действии холодных растворов едких щелочей целлюлоза
сильно набухает и поглощает из раствора щелочь, давая с ней
химические соединения, так называемую щелочную целлюлозу, или
алкалицеллюлозу. При действии 18%-ного раствора едкого натра
образуется молекулярное соединение [(СвНHО5J-NaOH]„, которое
может быть отмыто от раствора едкого натра спиртом и затем
высушено.
Щелочная целлюлоза легко разлагается водой с образованием
гидратцеллюлозы. По химическому составу гидратцеллюлоза не
отличается от исходной целлюлозы, но она более гигроскопична,
труднее теряет гигроскопическую воду, легче окрашивается и
менее устойчива к химическим воздействиям.
Обработка хлопчатобумажных тканей и пряжи щелочью с
последующей промывкой их водой называется мерсеризацией. При
обработке ткани или пряжи щелочью происходит сокращение их
размеров приблизительно на 20%, причем ткань становится
плотнее, а пряжа—толще. Прочность на разрыв значительно
увеличивается. Сокращение размеров и увеличение прочности не
изменяется и после отмывки щелочи водой. Если механически
воспрепятствовать сжатию, т. е. производить мерсеризацию ткани
или пряжи в натянутом состоянии, то волокна приобретают
шелковистый блеск.
Окисление целлюлозы. При действии на целлюлозу различных
окислителей (влажный хлор и его окислы, перекись водорода,
марганцовокислый калий) происходит постепенно окисление
целлюлозы с образованием различных видов оксицеллюлозы. Окси-
целлюлоза представляет собой смесь неизмененной целлюлозы с
продуктами ее окисления. Оксицеллюлоза содержит вещества
с альдегидными и карбоксильными группами; она
восстанавливает фелингову жидкость, окрашивается фуксинсернистой
кислотой в малиновый цвет и часто растворяется в щелочах.
Кислород воздуха при обыкновенных условиях почти не
действует на целлюлозу, нов щелочной среде и при повышенной
температуре действие его усиливается. В процессе сплавления
целлюлозы со щелочами при доступе воздуха происходит ее окисление
в щавелевую кислоту.
174. Сложные и простые эфиры целлюлозы. В молекуле
целлюлозы содержатся гидроксильные группы, причем на каждую
группу С6Н,0О5 приходится три гидрокаип, поэтому простейшую
формулу целлюлозы следует изобразить так: СвН7О2(ОНK.
Известны сложные эфиры целлюлозы, в которых на одну
группу C6HJ0O6 приходится три остатка молекулы одноосновной
350 Глава XI. Углеводы
кислоты. Из сложных эфиров наибольшее значение имеют эфиры
азотной, уксусной и ксантогеновой (стр. 418) кислот.
Азотнокислые эфиры получают обработкой целлюлозы
смесью азотной и серной кислот. Если вести расчет на одну
группу CfiH,0O5, то происходящие при этом реакции можно
изобразить следующими уравнениями:
QHA-o-
\o-
-H + H—OyNOa .O—NO2
-H + H—o4-NOa * C6H,O2—O—NO2 + 3H2O
¦Н + H-O-NO2 \э—N0.2
тринитрат целлюлозы
/O--U + Н—О ]—NO2 .О—N0.2
С0Н,О3—О-рН+Н—О!— NO2 » С0Н,О—О—NO2 + 2Н2О
\э—н \он
динитрат целлюлозы
/w—11 -1-П--О—NO2 /0—N02
/ ~ ¦-¦¦¦¦ ; 4 ГНООН 4-HO
\o—и \эн
мононитрат целлюлозы
Обычно при нитровании* получается смесь азотнокислых
эфиров.
Смесь продуктов нитрования целлюлозы с большим
процентным содержанием азота A3—13,6%) называется пироксилином.
Спрессованный в шашки пироксилин применяется как
взрывчатое вещество для взрывных работ. Для стрельбы из
огнестрельных орудий чистый пироксилин непригоден, так как взрывается
слишком быстро. Для замедления быстроты взрыва пироксилин
обрабатывают спиртом, эфиром и другими веществами, и из
полученной пластичной массы изготовляют ленты и трубки так
называемого бездымного пороха. Бездымный порох был
изобретен в 1886 г.
Нитрат целлюлозы, содержащий 11 —12% азота, называется
коллоксилином. Раствор последнего в смеси спирта и эфира под
названием коллодий применяется в медицине.
* Термин нитрование здесь означает этерификацию азотной кислотой;
обычно нитрованием называют процесс образования ннтросоединений,
имеющих связь нитрогруппы с углеродом: С—NOa.
174. Сложные и простые эфиры целлюлозы 351
Ранее нитрат целлюлозы находил широкое применение для
производства целлулоида, кинопленки и быстро высыхающих
дешевых и прочных нитролаков и эмалей. Исторически
целлулоид явился первой пластмассой, получившей довольно широкое
распространение еще в прошлом столетии. Для его изготовления
коллоксилин смешивают при нагревании с камфорой и
небольшим количеством спирта; затем полученную массу пропускают
через нагретые вальцы, в результате чего образуются тонкие
листы, которые при нагревании спрессовывают гидравлическими
прессами. Целлулоид применялся для изготовления различных
предметов широкого потребления*. При изготовлении кинолент
коллоксилин и камфору растворяют в смеси ацетона и амилового
спирта и выливают полученный раствор через узкую щель на
движущуюся металлическую ленту. Полученную после испарения
большей части растворителей пленку сушат воздухом.
В настоящее время вследствие горючести и связанной с ней
пожароопасностью целлулоид утратил свое значение и
повсеместно вытесняется малогорючими пластмассами. В
производстве кинопленки пожароопасный нитрат целлюлозы также
заменяется негорючим триацетатом целлюлозы.
Нитролаки представляют собой растворы коллоксилина в
органических растворителях со специальными добавками
(разбавители, пластификаторы), служащими для придания пленке
требуемых свойств (прилипаемость к окрашиваемой
поверхности, прочность, эластичность). Внося в состав нитролаков
минеральные или органические пигменты, получают нитрокраски и
эмали.
Нитролаки, краски и эмали широко применяются для окраски
различных предметов. Значительные их количества расходуются
на окраску автомобилей. Сложные эфиры целлюлозы с уксусной
кислотой называются ацетатами целлюлозы:
О—СО—СНЭ /О—СО—СНЭ
ОСОСН СНА
СвН7О2—О—СО—СН3 СвНА—О—СО—СНЭ
\<э—со—сн3 \эн
триацетат целлюлозы диацетат целлюлозы
Из растворов ацетата целлюлозы в ацетоне изготовляют
ацетатное волокно. Пластифицированный ацетат целлюлозы, а также
ацетобутират целлюлозы (смешанный эфир целлюлозы с уксусной
и масляной кислотами) применяются в производстве пластических
* Стекло «триплекс» состоит из двух стеклянных пластинок и
целлулоидной или целлоновой прокладки между ними.
352 Глава XI. Углеводы
масс; на их основе изготавливают штурвалы для автомобилей и
другие изделия методом отливки под давлением.
Кроме сложных эфиров, известны и простые эфиры целлюлозы.
Такова, например, триэтилцеллюлоза С6Н7О2(ОС2Н5K, которая
получается обработкой щелочной целлюлозы хлористым этилом в
присутствии щелочи. Она находит применение в производстве
эфироцеллюлозных пластиков. Водорастворимый эфир
целлюлозы, получаемый при этерификации монохлоруксусной кислотой,
так называемая карбассиметилцеллюлоза, находит широкое
применение в качестве поверхностно-активного вещества—в
нефтяной промышленности при бурении, в производстве моющих
средств для повышения диспергирующей и пенообразующей
способности, в текстильной промышленности при крашении и отделке
тканей.
175. Искусственное волокно. Химическая переработка
целлюлозы получила широкое распространение в связи с
необычайно быстрым развитием промышленности искусственного
волокна.
Для производства искусственного волокна имеется ряд
способов, из которых наибольшее значение приобрел вискозный.
1. Вискозное волокно. При производстве искусственного
волокна по вискозному способу целлюлозу обрабатывают едким натром,
превращая ее в щелочную целлюлозу, которую в больших,
медленно вращающихся барабанах обрабатывают сероуглеродом.
В результате такой обработки образуется масса оранжевого цвета,
представляющая собой эфир целлюлозы и соли ксантогеновой
кислоты (СвН9О4О—CS—SNa)n (cm. стр. 418). Растворяя
ксантогенат в слабом растворе едкого натра, получают вязкий раствор,
называемый вискозой. При действии на вискозу кислот
происходит нейтрализация едкого натра и отщепление от ксантогената
сероуглерода. Образующаяся при этом гидратцеллюлоза
выделяется из раствора.
Приводим схему получения нитей из вискозы.
Вискоза, подаваемая с определенной скоростью через
трубку / {рис. 55), погруженную в осадительную ванну 2, содержащую
раствор сернокислого натрия и серной кислоты, выдавливается
через ряд отверстий диаметром 0,1 мм, имеющихся в специальном
колпачке-фильере 3, закрепленном на конце трубки.
Образующаяся нить, состоящая из 40—60 волокон (по числу отверстий
в фильере), проводится сначала вверх, перекидывается через
вращающийся ролик 4, а затем через стеклянную воронку 5 опускается
внутрь быстро вращающегося E000—6000 об/мин и более)
алюминиевого барабана центрифуги 6. Центробежной силой нить
отбрасывается к стенкам барабана; при этом она укладывается
правильными витками и одновременно скручивается.
175. Искусственное волокно
353
Если вискозу продавливать в осадительный раствор через
узкие прорези, то получаются тонкие прозрачные пленки
целлофана, которые для улучшения механических свойств
пластифицируют глицерином. Целлофан широко применяется для упаковки
различных изделий.
Сырьем для изготовления вискозы служит целлюлоза,
полученная по сульфитному или другому способу из древесины.
Рис. 55. Схема установки для получе- •
ния вискозного волокна:
/—трубка, через которую подается вискоза;
2—осадительиая ваниа; 3—колпачок
фильеры; 4—ролики; 5—стеклянная воронка; 6—цен
трифуга.
2. Ацетатное волокно производят из ацетата целлюлозы, кото*
рый для этой цели растворяется в ацетоне. Полученный раствор
продавливается через отверстия фильер и спускается пучком
нитей навстречу теплому воздуху, уносящему с собой пары
растворителя. Такой прием называется сухим прядением. Сырьем
для ацетатного волокна служит линтер (хлопковые очесы до
5 мм длины).
3. Медноаммиачное волокно готовится из раствора
целлюлозы в аммиачном растворе гидроокиси меди. Раствор целлюлозы
продавливается через отверстия фильер в ванну с теплой водой
и слабой серной кислотой. В этой ванне целлюлоза выделяется из
раствора в виде гидратцеллюлозы, образуя волокно
(медноаммиачное волокно).
Производство искусственного волокна началось в 1884 г.,
когда была построена первая фабрика искусственного волокна
во Франции.
23—788
354 Глава XII. Нитросоединения
В дореволюционной России производства искусственного
волокна практически не существовало. Имелась только одна
фабрика (под Москвой), которая вырабатывала около 100 т
искусственного волокна в год. За последние 30 лет в СССР
создана мощная промышленность искусственного волокна.
176. Гемицеллюлозы и пектиновые вещества. В оболочках
растительных клеток вместе с клетчаткой всегда находятся
углеводы, весьма близкие к клетчатке, которые называются геми-
целлюлозами. Гемицеллюлозы разделяются на гексозаны
(С6Ніо05)ж и пентозаны (С5Н8О4)х. Гексозаны при гидролизе
дают глюкозу и другие гексозы (галактозу, фруктозу);
продуктами гидролиза пентозанов являются ксилоза и арабиноза.
Гемицеллюлозы гидролизуются легче, чем клетчатка; в отличие от
клетчатки, они растворяются в щелочах.
В близком отношении к пентозанам находятся камеди и
пектиновые вещества, или растительные слизи. Камеди
представляют собой отвердевший на воздухе сок растений; к ним
принадлежат вишневый клей и гуммиарабик—застывший сок
тропических акаций.
Пектиновые вещества содержатся в больших количествах в
плодах некоторых растений—крыжовнике, землянике, яблоках.
Благодаря их присутствию сахарные фруктовые сиропы,
нагретые до кипения и затем охлажденные, способны образовывать
желеобразные м^ссы. Этим свойством пектиновых веществ пользуются
в производстве мармелада, желе, пастилы. Пектиновые вещества
являются кальциевыми и магниевыми солями
полигалактуроковой кислоты. D-Галактуроновая кислота—одна из стереоизомер-
ных кислот общей формулы:
НООС—СН—СН(ОН)—СН(ОН)—СН(ОН)—СН(ОН)
-о-
Ее можно представить как продукт окисления в D-галактозе
группы СН2ОН. В поли-О-галактуроновой кислоте остатки D-ra-
лактуроновой кислоты связаны аналогично остаткам глюкозы в
мол е кул е целлюлоз ы.
В пектиновых веществах часть карбоксильных групп этери-
фицирована метальными группами. Природные пектиновые
вещества смешаны с полисахаридами, дающими при гидролизе
D-галактозу (галактаны) и D-арабинозу (арабаны).
Лигнин. Лигнин является одним из главнейших
инкрустирующих* клетчатку веществ. В древесине сосны содержится 34%
примеси
От латинского crusta—кора, скорлупа. Здесь—твердые включения,
:и.
176. Гемицеллюлозы и пектиновые вещества 355
лигнина, в древесине бука—22,5%. Он состоит из углерода,
водорода и кислорода, причем процентное содержание углерода в нем
больше, чем в клетчатке. Состав лигнина точно не определен.
Лигнин менее прочен к химическим воздействиям, чем клетчатка.
Наличие значительного количества метоксильных групп в
молекуле лигнина объясняет, почему при сухой перегонке дерева
образуется метиловый спирт. При сухой перегонке чистой
целлюлозы метиловый спирт не образуется.
При сплавлении лигнина со щелочью *л при окислении
озоном получаются соединения ароматического ряда.
23*
ГЛАВА XII
НИТРОСОЕДИНЕНИЯ
177 Строение и способы получения нитросоединений Нитро-
соединениями называются вещества, содержащие нитрогруппу
NO2, атом азота которой непосредственно связан с атомом
углерода.
Примерами нитросоеди нений являются нитрометан СН3—NO,,
нитроэтан С2Н6—NO2. Они изомерны эфирам азотистой кислоты;
кипят при более высоких температурах, чем последние.
Температу- Температу-
Нитросоединения ра кипения Эфиры азотистой ра кипения
°С кислоты °С
СН3—NO2 101,5 СНа—О— N0 —12
СН3—СН2—NO2 114,0 С.2Н6—О—NO 17
CH3-CH2-CH,-NO2 131,6 С3Н,—О—NO 57
При восстановлении нитросоединения переходят в амины, в
то время как при восстановлении эфиров азотистой кислоты
получаются спирты и аммиак:
C2H5—N02 -
нитроэтан
C2H5-O-NO +
этилнитрит
4-ЗН3
зн2-
> QH5-NH2
этиламин
—* О,н6-он +
+ 2Н2О
¦ NH3 + Н2О
В зависимости от того, с каким атомом углерода связана нит-
рогруппа, различают первичные, вторичные и третичные
нитросоединения:
СН3
CH3CH.2CR2—NO2 СН3—СН—СН3 СН3—С—СН3
перничный нитропропан I I
A-нитропропан) NU2 NO2
пторичный нитропропан третичный ештробутан
B нитронрошн) (?-нитро-2-метилпропан!
Если предположить, что в молекуле нитросоединения атом
азота связан с каждым из атомов кислорода двойной связью, то
177. Строение и способы получения нитросоединений 35?
электронная формула нитросоединения будет находиться в
противоречии с правилом октетов—атом азота будет окружен
десятью электронами:
" /° Я
h-c-n^ h:c:n::o
Поскольку это невозможно, следует принять другое
расположение электронов, что обычно изображают формулой I или II.
н н
It/3 I /°
Н-С—Nf_ H-C-Nf
І Ч> і Ч)
н н
1 U
При этом написании предполагается, что атомы кислорода,
стоящие у азота нитросоединения, как бы неравноценны; один
из них связан с атомом азота двумя парами общих электронов,
другой же—только двумя электронами, причем оба электрона
доставляются лишь одним атомом—атомом азота; эти электроны
дополняют до октета число электронов кислорода. При этом
атом кислорода приобретает отрицательный заряд, атом азота—
положительный заряд, так как пара электронов уже не
принадлежит исключительно ему. Такие связи называются
полуполярными (семиполярными) связями*. Вещества с семиполярными
связями имеют большой дипольный момент; они обыкновенно кипят
при более высокой температуре, чем изомерные им вещества без
семиполярных связей.
Следует отметить, что приведенные выше формулы строения
нитросоединений являются упрощенными; в действительности,
как показали исследования последнего времени, оба атома
кислорода в нитросоединениях совершенно равноценны.
Это принято обычно изображать с помощью изогнутых
стрелок, указывающих направление смещения электронной плотности:
Нитропроизводные предельных углеводородов называются ни-
тропарафинами.
* Латинское semi—полу.
358 Глава XII. Нитросоединения
Нитропарафины были впервые получены действием
азотистокислого серебра на галоидные алкилы:
C2H5J + AgNO2 . QH5—NO2 + AgJ
Одновременно с нитросоединениями при этом образуется и
некоторое количество эфиров азотистой кислоты.
М. И. Коновалов A886) открыл, что нитросоединения
жирного ряда могут быть получены нагреванием предельных
углеводородов с разбавленной HNO3 до 130—140 °С в запаянных
трубках:
СН,
СН3-СН2-СН2-СН2-СН-;Н + HOi—NO2 ->
гексан
сн3
» СНз—СН2—СН2—СН2—СН—NO2 + Н2О
2-ннтрогеисан
Замещение происходит у менее гидрогенизированного атома
углерода.
В последнее время реакция Коновалова используется в
технике для нитрования нефтяных углеводородов.
Нитрование обычно проводят, действуя на пары
углеводородов двуокисью азота.
178. Свойства нитросоединений. Низшие гомологи нитропара-
финов—бесцветные жидкости с приятным запахом, которые
смешиваются со спиртом и с эфиром. Они не проводят электрический
ток, плохо растворимы в воде и являются нейтральными
веществами. Однако первичные и вторичные нитросоединения
медленно растворяются в растворах едких щелочей, причем происходит
нейтрализация щелочи.
В молекулах первичных и вторичных нитросоединений атом
водорода, связанный с тем же атомом углерода, что и нитрогруппа,
способен замещаться металлом. Так, если слить спиртовые
растворы нитроэтана C2H5NO2 и этилата натрия, то выпадает
кристаллический осадок состава C2H4NaNO2. Эти металлические
производные легко растворимы в воде. В водном растворе они,
судя по электропроводности, хорошо диссоциированы и
показывают на лакмус нейтральную реакцию, т. е. являются солями
сильных кислот. Их следует считать солями кислот, таутомерных
нейтральным нитросоединениям:
СН3—СН2—NO3 CH3-CH=N<f CH3-CH=N<
Х)Н XONa
нейтральная кислотная металлическое
форма форма производное
178. Свойства читрасаединений 359
Кислотные формы нитросоединений нередко называют изони-
тросоединениями*. При добавлении кислоты к металлическому
производному образуется кислотная форма, которая затем изоме-
ризуется в нейтральное нитросоединение:
СНа—CH=N<; + HCI
Г *о і
СН,—CH=N< > СНа—СНа—NOa
L xohJ
Совершенно аналогичные явления имеют место и у вторичных
нитросоединений. Они также могут реагировать в двух таутомер-
ных формах:
R\ R\ *°
>сн—NOa и >C=N<
Таким образом, нитрогруппа сообщает подвижность атому
водорода, находящемуся при том же атоме углерода. В третичных ни-
тросоединениях у атома углерода, с которым связана
нитрогруппа, нет атома водорода, и водород в таких нитросоединениях
металлом не замещается.
Кислотные формы нитросоединений жирного ряда изолировать
не удалось, однако это оказалось возможным сделать для
соединений жирно-ароматического ряда.
В 1896 г. М. И. Коновалов и др. показали, что фенилнитроме-
тан может существовать в двух формах.
Фенилнитрометан С6Н5—СН2—N02 можно рассматривать как
нитрометан СН3—N02, в молекуле которого один атом водорода
замещен фенилом. Фенилнитрометан—маслообразная жидкость,
кипящая при 225—227 °С. Если растворить фенилнитрометан в
щелочи и подкислить полученный раствор, то выпадает
кристаллическое вещество, которое плавится при 84 °С и имеет состав
фен илнитрометана:
-1-NaOH „О -I-HCI „О
CeHa-CHa-NO2 * СвН6-СН=№Г » CeH6-CH=N<*
xONa XOH
Через несколько часов оно переходит в жидкий фенилнитрометан:
< > С6Н5-СН3—N02
* Строение изонитросоединений может быть изображено и так:
CHa-CH=N<
хон
360 Глава XII. Нитросоединения
Кислотная форма фенилнитрометана легко растворима в воде;
раствор вначале обладает хорошей электропроводностью,
которая постепенно, по мере изомеризации фенилнитрометана в
нейтральную форму, уменьшается и, наконец, становится
незаметной.
Нейтральные вещества, которые при действии щелочей дают
соли изомерных сильных кислот, называются псевдокислотами*.
По гречески «псевдос»—ложный.
РЛАВА Kill
АМИНЫ
179. Строение аминов. Аминами называются производные
аммиака, в молекуле которого один или несколько атомов водорода
замещены углеводородными радикалами.
По числу атомов водорода, замещенных углеводородными
радикалами, различают первичные, вторичные и третичные амины.
Аммиак
Н\
Н—N
н/
Первичные амины:
CHa-NHa
метиламин
Вторичные амины:
CH3\n
en/
димегиламин
CaH6-NHa
этиламин
'¦>NH
с,н/
диэтиламин
Третичные амины:
СН3—N QHs—N
СН-/ QJ\/
гриметиламиь тричтиламин
Группа —NH2, входящая в состав первичных аминов,
называется аминогруппой. Группа >NH, находящаяся во вторичных
аминах, называется иминогруппой.
180. Свойства аминов. Метиламин, диметиламин и триметил-
амин—газы, средние члены ряда аминов—жидкости, высшие—
твердые тела. С увеличением молекулярного веса увеличивается
плотность, повышается температура кипения и уменьшается
362 Глава л///. Амины
растворимость амина в воде. Высшие амины в воде
нерастворимы. Низшие амины имеют запах, несколько напоминающий запах
аммиака. Высшие амины или не имеют запаха, или имеют очень
слабый запах.
По химическим свойствам амины весьма сходны с аммиаком.
I. Характерным свойством аммиака является, как известно,
его способность, соединяясь с кислотами, давать соли аммония:
Шэ + НС1 » NH4CI
Так же, как и аммиак, амины соединяются с кислотами и
образуют соли, подобные солям аммония:
CH3NH2
метиламин
(CHaJNH -f
ди метила мин
(CH3foN +
гриметиламин
+ на -
на —
на —
* (CH3NH3|CI
хлористый
метиламмониґі
-^ [(CH3),NH.2]a
хлористый
диметиламмоний
-+ [(CH3KNHia
хлористый
грнметиламмониіі
Эти соединения можно рассматривать как производные солей
аммония, в которых один или несколько атомов водорода
замещены углеводородными радикалами.
Обычно формулы солей аминов изображают не так, как
показано выше, а отделяя точкой формулу амина от формулы
кислоты. Так, например, хлористый метиламмоний (CH3NH3)C1
рассматривают как CH3NH2-HC1 и называют хлористоводородным
метиламином, подобно тому как ранее считали, что хлористый
аммоний NH4C1 представляет собой хлористоводородный аммиак
NH.-HC1.
2. Соли аммония, при действии на них щелочей, легко
разлагаются с выделением свободного аммиака:
NH.CI + NaOH * NH, + NaCl + Н2О
Точно так же соли аминов разлагаются щелочами с выделением
свободного амина:
[CH3NHS]CI + NaOH » CH3NH, -f NaCl + H2O
3. Водный раствор аммиака окрашивает лакмус в синий цвет,
т. е. обладает щелочными свойствами; водные растворы аминов
жирного ряда также придают лакмусу синий цвет.
Щелочные свойства водных растворов аминов объясняются
тем, что в этих растворах находятся производные гидроокиси
180. Свойства аминов
363
аммония, в которой один или несколько атомов водорода
замещены углеводородными радикалами:
CH3NH2 + H2O -
метиламин
(CH3JNH + Н2О ¦
диметиламин
(CH3KN + Н20 -
триметиламин
> [CH3NH3]OH
гидроокись
метиламчоння
¦» |(CH3JNH2|OH
гидроокись
диметиламмония
> |(CH3KNH]OH
гидроокись
триметиламмонни
Эти гидроокиси обладают более сильными основными
свойствами, чем гидроокись аммония.
Таким образом, сравнение свойств аммиака и аминов
показывает, что амины, являясь производными аммиака, повторяют
многие его свойства. Другими словами, амины являются
органическими основаниями.
В большинстве случаев органические основания с жирными
радикалами—более сильные основания, чем аммиак, т. е. в
водном растворе они диссоциированы в большей степени и по
основности превышают аммиак. Константы электролитической
диссоциации аммиака и органических оснований приведены в табл. 13.
Таблица 13
Константы электролитической диссоциации аммиака
и органических оснований
Основание
Аммиак
Метиламин
Этиламин
ч-Пропиламин .....
Изапропиламин .....
«-Бутиламин
Изобутиламин
Изоамиламин
Константа
диссоциации
1,79- 10--
4,38-10-«
5,60-10-«
4,70-10"«
5,30-10-«
4,1 10-«
3,1 -10-«
5,0 -10-«
Основание
Диметиламин
Триметиламин
Диэтиламин
Триэтиламин
Дипропиламин
1рипропиламин ....
Диизобутиламин ....
Константа
диссоциации
5,20-10
5,45-10-«
9.6 .10-«
6,4 -Ю-1
1,02-Ю
5.5 -10-*
4,8 .10-'
В отличие от аммиака, амины способны гореть в воздухе.
Горючесть аминов и привела к их открытию A848 г.). Вюрц
первоначально принимал пары этиламина (кипит при 17 °С) за аммиак,
364 Глава XIII. Амины
пока, благодаря случаю, ему не удалось заметить, что газ с
запахом аммиака способен гореть в воздухе.
Действие азотистой кислоты на амины. При действии
азотистой кислоты на первичные амины происходит выделение
свободного азота и образование спиртов:
СН3—;N|/ + O=N-HDH » CH3OH + N2+HaO
і і п
Вторичные амины с азотистой кислотой дают нитрозамины
желтоватые жидкости, мало растворимые в воде:
CH3;2N-;H 4- НО;—N=0 * (CH3)aN—N0 + НаО
^ *
нитрозоди метиламин
При обработке крепкой соляной кислотой нитрозамины снова
дают исходные амины:
(СН3)гН—N0 + НС1 > (CH3JNH + NOCI
Третичные амины устойчивы к действию азотистой кислоты
(образуют соли азотистой кислоты).
Соли четырехзамещенного аммония. При нагревании
третичных аминов с галоидными алкилами образуются соли
четырехзамещенного аммония:
(CH3KN + CH3J »|(CH3LN]J
йодистый іетра
метилам моний
Эти соединения представляют собой кристаллические вещества,
растворимые в воде и хорошо диссоциированные в водном
растворе.
При действии гидроокиси серебра на йодистые соли
четырехзамещенного аммония получаются растворы гидроокисей
четырехзамещенного аммония:
[(CHa^Nl- + .Г + Ag+ + ОН" > AgJ + [(CHaLN,- + 0Н~
Эти гидроокиси—столь же сильные щелочи, как едкий натр
и едкое кали. Гидроокись тетраметиламмония [(CH3LN]OH
может быть получена в виде твердого белого вещества, чрезвычайно
сходного с едкими щелочами.
181. Строение аммонийных солей. Образование хлористого
аммония из аммиака и соляной кислоты можно представить еле-
182. Получение аминов
365
дующим образом. В молекуле аммиака из пяти валентных
электронов азота три идут на образование ковалентних связей с тремя
атомами водорода. Таким образом, у атома азота остается
свободная пара электронов. Ион водорода (протон) кислоты
присоединяется к свободной паре электронов азота, образуя ион
аммония с положительным зарядом:
н
h:n
н
н
h:n:h
н
Очевидно, что после образования иона аммония нельзя уже
различить, который из атомов водорода «воспользовался» неподелек-
ной парой электронов азота. Ясно также, что и ион хлора уже
не относится к какому-либо одному определенному атому
водорода. Аналогично можно представить и строение солей аминов:
сн,
h3c:n:
н
н
н~=
СНз
h3c:n:h
[ко,]'
Присоединение галоидных алкилов к третичным аминам
происходит по той же схеме:
сн,
н3с : N: +
сн,
сн2:
• J «
• •
сні _
ще
сн3
:n:ch2
сн3
:сн3
Соли аминов следует рассматривать как комплексные
соединения. Центральным атомом в них является атом азота,
координационное число которого равно четырем. Атомы водорода и
алкилы связаны с атомом азота и находятся во внутренней
координационной сфере; кислотный остаток или гидроксил находятся
во внешней координационной сфере и при электролитической
диссоциации отделяются в виде ионов.
182. Получение аминов. Амины могут быть получены
различными способами.
1. Действием аммиа ка на галоидные
алкилы (реакция Гофмана). Для проведения этой реакции
нагревают спиртовый раствор аммиака с галоидным алкилом.
Рассмотрим эту реакцию на примере действия аммиака на йодистый метил.
366 Глава XIП. Амины
Аммиак, присоединяя молекулу йодистого водорода, образует
йодистый аммоний:
NH3 + HJ » (NH4]J
Подобно этому, при действии аммиака на йодистый метил
образуется йодистый метиламмоний:
CH3J + NH» » [(CH3NH3)]J
Часть полученного йодистого метиламмония разлагается
избытком аммиака с образованием свободного метиламина
[(CH3NH3)]J + NH3 * NH4.I + CH3NH2
который снова соединяется с йодистым метилом, давая йодистый
диметиламмоний:
СНзЩ, + CH3J > [(CH3JNH,]J
Последний разлагается аммиаком с образованием свободного
диметиламина:
l(CH3JNHa]J + NH3 » NHjJ + (CH3)aNH
В дальнейшем образуется и триметиламин:
(CH3)aNH + CH„J * [(CH3KNH|.l
йодистый
тр и мет и ла м мои и Й
|(CH3KNH|J + NH3 » NH4J +- (CHa),N
гриметиламин
Наконец, молекула триметиламина присоединяет к себе еще одну
молекулу подпетого метила, образуя йодистый тетраметиламмо-
ний
(CH:)),N + CH3J » ((CH3LN]J
который является солью четырехзамещенного аммония. Таким
образом, в результате этих реакций получается смесь
замещенных солей аммония.
2. Нагреванием амидов кислот с
щелочными растворами бромноватистых или
хлорноватистых солей. При этой реакции происходит
перегруппировка, в результате которой аминогруппа
непосредственно связывается с радикалом. Механизм реакции весьма
сложен; конечный результат ее можно выразить уравнением:
R—СО—NHa + 2NaOH + NaCIO » RNH2 + NaXO3 + NaCI + HaO
183. Отдельные представители аминов 367
Эта реакция была открыта в 1881 г. Гофманом*. Она имеет
промышленное значение и применяется, например, для получения
антраниловой кислоты (см. стр. 598), одного из полупродуктов
синтеза индиго.
3. Восстановлением нитросоединени и,
например, при помощи олова и соляной кислоты:
СН3—NOa + 6Н , CH3-NH2 + 2Н2О
нитрометарг метиламин
Получение аминов из нитросоединений жирного ряда имело
до сих пор небольшое значение из-за малой доступности
нитросоединений. Однако успехи в области нитрования парафинов,
достигнутые в результате работ А. В. Топчиева и А. И. Титова,
делают этот способ весьма перспективным.
О получении аминов из нитрилов и эфиров изоциановой
кислоты см. на стр. 403 и 408.
Амины образуются в природе при гниении органических
остатков, содержащих белковые вещества.
183. Отдельные представители аминов. Метиламин CH3NH2
образуется при сухой перегонке костей. В свободном состоянии
метиламин—газ, сгущающийся при —6,3 °С в жидкость;
обладает запахом, похожим на запах аммиака, и одновременно
напоминающим запах рыбы. Весьма хорошо растворим в воде.
Диметиламин (CH3JNH находится в селедочном рассоле и
образуется при гниении рыбы; сгущается в жидкость при
температуре +6,9 °С.
Триметиламин (CH3KN находится в селедочном рассоле и
в некоторых растениях; образуется при сухой перегонке
свекловичной патоки, сгущается в жидкость при +2,8 °С.
Концентрированный триметиламин имеет запах, весьма
сходный с запахом аммиака. В малых концентрациях запах триме-
тиламина отвратителен, он слегка напоминает запах рыбьего
жира и ворвани. Запах триметиламина очень долго удерживается
платьем, кожей и волосами.
* Август-Вильгельм Гофман (A. W. Hoffmann) A818—1892). Родился в Гис-
сене (Германия).
Длительное время работал в Лондоне, а с 1864 г.—в Берлине.
Осуществил синтез первичных, вторичных и третичных жирных аминов
действием аммиака на галоидпроизводные и изучил эти амины, а также
исследовал ароматические амины.
А. В. Гофман также открыл в каменноугольной смоле бензол и превратил
его в нитробензол, исследовал краситель индиго и синтезировал важные
красители—фуксин и розанилин.
368 Глава XIII. Амины
В технике алкиламины получают при взаимодействии
аммиака с соответствующим спиртом при высоких температурах и
давлениях над катализатором, например
СН3ОН + NH3 » CH3NH2 + НаО
2СН3ОН + NH3 * (CHaJNH + 2НаО
ЗСН3ОН + NH3 » (CH3KN + ЗН.р
Монометиламин применяется для получения поверхностно-
активных веществ; диметиламин и диэтиламин являются
исходными продуктами для получения важных ускорителей
вулканизации каучука.
Этилендиамин H2NCH2CH2NH2 A,2-диаминоэтан) является
простейшим примером диаминов, т. е. аминов с двумя
аминогруппами в молекуле. Диамины легко растворимы в воде, обладают
характерным запахом, имеют сильно щелочную реакцию,
взаимодействуют с углекислым газом воздуха. Они образуют
устойчивые соли с двумя эквивалентами кислоты.
Этилендиамин может быть получен действием аммиака на
бромистый этилен:
ВгСН2СН2Вг + 2NH3 » HBr-H2NCH2CH2NH2-HBr
Этилендиамин—жидкость с аммиачным запахом; темп. кип.
116,5 °С; темп, плавл. 8,5 °С.
Тетраметилендиамин A,4-диаминобутан), или путресцин
NH2CH2CH2CH2CH2NH2
и пентаметилендиамин A,5-диаминопентан), или кадаверин
NH2CH2CH2CH2CHaCH2NH2
были открыты в продуктах разложения белковых веществ и
названы птомаинами (от греческого слова «птома»—падаль); их
считали ранее «трупными ядами». В настоящее время выяснено,
что ядовитость гниющих белков не является следствием
наличия в них птомаинов, а вызвана присутствием других веществ
неизвестной химической природы.
Путресцин и кадаверин образуются в результате
жизнедеятельности многих бацилл (например, возбудителей столбняка
и холеры) и грибков, встречаются в сыре, спорынье, мухоморе,
пивных дрожжах.
Путресцин (от слова putreso— гнить) был впервые найден в
гное. Это кристаллическое вещество; темп, плавл. 27—28 °С;
18.3. Отдельные представители аминов 369
темп. кип. 158—160 °С. Кадаверин (от слова cadaver—труп) —
жидкость, кипящая при 178—179 °С; был найден в разлагающихся
трупах. Реакции, выясняющие строение этих диаминов,
приведены на стр. 381. Путресцин и кадаверин—сильные основания.
Спермин NH2[CH2]3NHCH2CH3CH2CH2NH[CH2]3NH2
является тетрамином. Его можно рассматривать как путресцин, в
молекуле которого два атома водорода, по одному в каждой
аминогруппе, замещены двумя аминопропильными группами, причем
NH2-rpynnbi последних находятся у 7-углеродных атомов.
Спермин—это ди-1\1,Ы'-(у-аминопропил)-путресцин. Спермин был
выделен из человеческой спермы. Легче всего его получить из
поджелудочной железы крупного рогатого скота.
Спермин—кристаллическое вещество, обладает сильно щелочной реакцией.
В последнее время некоторые диамины нашли широкое
применение в качестве сырья для получения полиамидных
волокон и пластмасс. Так, из гексаметилендиамина
NH2CH2CH2CH2CH2CH2CH2NH2 можно получить весьма ценное
синтетическое волокно (см. стр. 397).
24-788
ГЛАВА XIV
АМИНОСПИРТЫ И АМИНОКИСЛОТЫ
184. Аминоспирты. Аминоспирты—соединения со
смешанными функциями, в молекуле которых содержатся амино- и окси-
группы. Из аминоспиртов хорошо изучен и представляет большой
интерес этаноламин, или коламин, НО—СН2СН2—NH2.
Строение этаноламина B-аминоэтанол-1) легко представить
из реакций его получения.
1. Действием аммиака на этиленхлоргидрин:
НОСН2СН2С1 + NH3 » HOCH2CH2NH3 + НС1
2. Присоединением аммиака к окиси этилена:
СН2—CHS + NH3 » HOCH2CHSNH2
О
Этаноламин—густое масло (темп. кип. 171 °С); смешивается
с водой во всех отношениях; обладает сильными щелочными
свойствами. В приведенных выше способах получения этаноламина
наряду с моноэтаноламином получаются также диэтаноламин
и триэтаноламин:
HOCHaCHaNH2 + СН2—СН2 » (HOCH2-CH2JNH и (HOCH2CHaKN
О диэтаноламин триэтаноламин
Этаноламины широко применяются в технике в качестве
эмульгаторов и других поверхностно-активных веществ, а также в
различных процессах газоочистки, в том числе для извлечения
углекислого газа из топочных газов при производстве жидкой углекислоты
и сухого льда.
184. Аминоспиріьі
371
Холин представляет собой четырехзамещенное аммониевое
основание. В нем с атомом азота связаны три метильные и одна
оксиэтильная группа:
НОСНа-СН, СНЭ
X
сна сн<.
ОН"
Холин—гидроокись оксиэтилтриметиламмонпя.
Холин входит в состав лецитинов—жироподобных веществ;
весьма распространен в животных и растительных организмах
и может быть выделен из них. Синтетический холин получают
обработкой концентрированного раствора триметиламина окисью
этилена при обыкновенной температуре
н,о
[(CH3KNHpH
"HOCHj—СН, CH,
CH2-CH, +
снэ сн3_
ОН"
Холин представляет собой кристаллическую, весьма
гигроскопическую, легко расплывающуюся на воздухе массу. Он обладает
сильными щелочными свойствами и с кислотами легко образует
соли.
Лецитинами называются сложные эфиры глицерина, в
молекуле которого два гидроксила этерифицированы жирными
кислотами, а один гидроксил этерифицирован ортофосфорной
кислотой, которая в свою очередь образует сложный эфир с холином,
например*:
СН-О-ОС-С17Нз
а—О— Р—О—СН8—СН2—Й(СНэ)э
О"
При нагревании с разбавленными кислотами или щелочами
лецитины гидролизуются с образованием холина, жирных^кислот
* В этой формуле принято, что между фосфорной кислотой и остатком
холина имеется внутримолекулярная связь, аналогичная бетаиновой (стр. 378).
24*
372
Глава XIV. Аминоспирты и аминокислоты
и оптически деятельной глицерин-а-фосфорной кислоты:
СН2—ООС—Ci,HM
СН—ООС—С17Н35
СН2—О—РО—О-СНа—СНа—Й(СН3)а
+4Н2О
О"
гецитин
СН2—ОН
СН—ОН +
СНа—О—РО(ОНJ
глицерин-а-фосфорная
кислота
"HOCH,—СН, СН3
.Н3 СН3
ОН - + 2С17НЗЙСООН
стеариновая
кислота
Менее распространены лецитины, в молекулах которых
фосфорной кислотой этерифицирован ?-гидроксмл глицерина. При
гидролизе таких лецитинов получается оптически недеятельная
глицерин-Р-фосфорная кислота:
СНаОН
СН—О—РО(ОН)а
СНаОН
глицер ии-C -фосфорная
кислота
Лецитины встречаются почти во всех животных и
растительных организмах.
Наиболее богаты лецитинами мозг, нервное вещество, сперма,
яичные желтки (9,4%) (по-гречески «лецитос»—желток); в
растениях лецитины особенно распространены в семенах и ростках.
Многие лецитины получены синтетически.
Лецитины представляют собой плохо кристаллизующиеся
вещества. В воде они обычно набухают и дают коллоидные растворы.
Лецитины обладают очень большой адсорбционной
способностью к самым различным веществам—белкам, сахарам,
алкалоидам и т. д.
При гниении лецитинов или при кипячении холина с
баритовой водой от оксиэтильной группы холина отщепляется вода и
образуется гидроокись винилтриметиламмония, или нейрин:
НОСНаСН2 СН3
\
СН
3_
ОН"
СН2—СН
X
CHg CHgJ
нейрин
ОН" + НаО
/55 Строение, способы получения и свойства аминокислот 37$
Нейрин—сиропообразная масса, обладающая сильными
щелочными свойствами; очень ядовит.
Кефалины, в отличие от лецитинов, вместо остатка холина
содержат остаток коламина (см. стр. 370); они содержатся в
веществе мозга и могут быть из него выделены.
185. Строение, способы получения и свойства аминокислот.
Аминокислотами называются соединения, в молекуле которых
содержатся одновременно аминная и карбоксильная группы. Таким
образом, аминокислоты являются в одно и то же время и
основаниями и кислотами. Примером их может служить аминоуксусная
кислота NHa—СН2—СООН.
В зависимости от положения аминогруппы по отношению к.
карбоксилу различают а-, ?-, 7- и т. д. аминокислоты:
СН3СНСООН
NHa
ос-аминопропионевая
¦< и с лота
СНзСЫ.СНСООН
NHa
а-амнномасляная
кислота
снаснасоон
0-аминопропионовая
кислота
СН2СНаСН2СООН
NHa
V-аминомасляная
кислета
Аминокислоты в настоящее время рассматривают как
продукты взаимной нейтрализации карбоксила и аминогруппы в одной-
и той же молекуле, т. е. как внутренние соли:
A) NHa—СНа—СООН (Ш NH8—CHa—COO
аминауксусная аминоуксусная
кислота кислота как
внутренняя соль
Молекула внутренней соли является ионом с разноименными-
зарядами на противоположных концах молекулы—биполярным
ионом. Молекула внутренней соли ионизирована, но не может
диссоциировать. В растворе часть молекул находится в состоянии,
изображаемом формулой (I), т. е. имеет место равновесие:
NH,—СНа—СООН 7 * NHo—СН2—СОО"
Аминокислоты имеют большое физиологическое значение; они
образуются при гидролизе белковых веществ животных и
растительных о ганизмов.
374 Глава XIV. Аминоспирты и аминокислоты
Подобно тому, как амины образуются при действии аммиака
на галоидпроизводные углеводородов, аминокислоты могут быть
получены действием аммиака на галоидзамещенные кислоты:
С1СНаСООН + NH3 » NHaCH.iCOOH + HCl
а-Аминокислоты получаются действием аммиака на оксини-
трилы; при этом сначала образуются аминонитрилы, которые
при омылении дают соответствующие аминокислоты:
СН,—С—CN + NHa * СН3—С—CN + НаО
\н \н
СН3—С—CN + 2Н2О > СН3—С-СООН + NH3
\н \н
Так как оксинитрилы получаются присоединением синильной
кислоты к альдегидам и кетонам, то эта реакция позволяет
перейти от альдегидов и кетонов к аминокислотам (циангидринный
синтез):
/О /ОН /NH, /NH,
// HCN / NH:: / 2 Н2О / г
СН3—С > СН3—С—CN , СН,—С—CN , СН:,—С—СООН
\н ^н Nh \h
альдегид оксипитрил амиионитри.7! аминокислота
Как показали акад. Н. Д. Зелинский и его ученики,
«-аминокислоты проще всего получаются действием на альдегиды и кето-
ны водных растворов цианистого калия и хлористого аммония.
В результате обмена образуется хлористый калий и цианистый
аммоний:
NH4CI + KCN = KCI + NH4CN
Цианистый аммоний, вступая в реакцию с альдегидом или кетоном,
образует аминонитрил.
Аминокислоты получаются также при каталитическом
восстановлении кетокарбоновых кислот водородом в присутствии
аммиака или аминов:
СН3—СО—СООН + NH3 + \\ » СН3—CH(NH2)—СООН + HaO
пировиноградная а-аминопропиоиовая
кислота кислота
185. Строение, способы получения и свойства аминокислот 375
Следует отметить, что вместо катализатора и водорода можна
применить цистеин (стр. 381), т. е. нормальную составную часть
белков. Возможно, что аналогичным путем аминокислоты
образуются в природе.
Свойства аминокислот. 1. Большинство
аминокислот—бесцветные кристаллические вещества, обычно хорошо растворимые
в воде, часто сладковатые на вкус.
2. В молекулах аминокислот содержатся две группы с прямо-
противоположными свойствами: карбоксильная
группа—кислотная и аминогруппа с основными свойствами. Поэтому они
обладают одновременно и кислотными и основными свойствами. Как
кислоты, аминокислоты образуют со спиртами сложные эфиры,
а с металлами и основаниями—соли:
NHaCHaCOOQjHB [NH.jCH2COO j2Cu
этиловый эфир медная соль
аминщуксусной кислоты аминоуксусной кислоты
Для аминокислот особенно характерно образование медных
солей, обладающих специфической синей окраской. Эти вещества
являются внутренними комплексными солями; в них атом меди
связан не только с атомами кислорода, но и с атомами азота
аминогрупп:
СН2—NH, NH2-CH2
I \ У I
СО—О—Си—О—СО
Связь между атомами меди и азота координационная (за счет
свободной пары электронов азота аминогруппы). Такие
соединения называют хелатными. Напомним, что хе-латными
соединениями является также диметилглиоксимат никеля (см. стр. 218) и
ацетилацетонаты металлов (см. стр. 217).
При действии едких щелочей на медные соли аминокислот не
происходит выпадения гидроокиси меди. Однако при действии
сероводорода происходит разрушение внутрикомплексного
соединения и выпадает труднорастворимая в воде сернистая медь.
Кислотные свойства в моноаминокислотах выражены весьма
слабо—аминокислоты почти не изменяют окраски лакмуса.
Таким образом, кислотные свойства карбоксила в них значительно
ослаблены.
Как амины, аминокислоты образуют соли с кислотами,
например:
HCI-NH2CH2COOH
Но эти соли весьма непрочны и легко разлагаются. Таким
образом, основные свойства аминогруппы в аминокислотах также зна~
чительно ослаблены.
376 Глава XIV. Аминоспирты и аминокислоты
3. При действии азотистой кислоты на аминокислоты образуются
оксикислоты:
NHjCHgCOOH + HNOa > НОСНаСООН + Na + HaO
Эта реакция совершенно аналогична реакции образования
спиртов при действии азотистой кислоты на первичные амины.
4. С галоидангидридами кислот аминокислоты образуют
вещества, которые одновременно являются и аминокислотами и
амидами кислот. Так, при действии хлористого ацетила на амино-
уксусную кислоту образуется ацетиламиноуксусная кислота:
СН3СОС1 + NHaCHaCOOH » CH3CONHCH2COOH + НС1
ацетиламинсуксусная
кислота
Ацетиламиноуксусную кислоту можно рассматривать и как
производное аминоуксусной кислоты, в молекуле которой атом
водорода аминогруппы замещен ацетилом СН3СО—, и как ацет-
амид, в молекуле которого атом водорода аминогруппы замещен
остатком уксусной кислоты —СНаСООН.
5. а-Аминокислоты при нагревании легко отщепляют воду,
причем из двух молекул аминокислоты выделяются две
молекулы воды и образуются дикешопиперазины:
СНа—СО—IOH Hi—NH СИ,—СО—NH
ІОІ— СО-СН, NH—CO—CI
2HaO
NH-ІН НО;-
Дикетопиперазины—циклические соединения, кольцо
которых образовано четырьмя атомами углерода и двумя атомами
азота. Дикетопиперазины—твердые, хорошо кристаллизующиеся
вещества.
?-Аминокислоты при нагревании теряют аммиак, переходя в
непредельные кислоты:
СНа—СН—СООН
I | » СНа=СН—СООН + NH3
І„.Г.:4.....„.І акриловая кислота
(а-аминопропионовая
кислота
¦у-Аминокислоты легко отщепляют воду, образуя лакшамы:
СНа—СН„—СНо—СО
I ... і —
NH jH HO!
186. Отдельные представители аминокислот 377
Лактамы можно рассматривать как внутренние амиды.
6. В молекулах большинства а-аминокислот содержится
асимметрический атом углерода; поэтому аминокислоты существуют
в виде оптических антиподов. Те из антиподов, конфигурация
которых аналогична конфигурации правовращающего
глицеринового альдегида, обозначаются буквой D; буквой L
обозначаются антиподы, конфигурация которых соответствует конфигурации
левовращающего глицеринового альдегида:
СООН СООН
Н—г—NHa NFLj-j—Н
D-конфнгурацня 1-конфнгурация
(природные аминокислоты]
Антиподы с 2р-положением аминогруппы относят к D-ряду,,
антиподы с 2а-положением аминогруппы—к L-ряду. Таким
образом, семейства а-аминокислот родственны по конфигурации
семействам а-окснкислот. Подробнее о стереохимических
обозначениях аминокислот см. § 187.
186. Отдельные представители аминокислот А миноуксусная
кислота. NHaCH2COOH, называемая также гликоколом
(по-гречески «гликос»—сладкий, «колла»—клей») или глицином,
встречается в мускулах низших животных. В большом количестве C6%
от веса исходного материала) образуется при гидролизе
белкового вещества шелка. Получается кипячением животного клея
с разбавленной серной кислотой или баритовой водой, а также
гидролизом гиппуровой кислоты.
Гиппуровая кислота CeH5CONHCHaCOOH находится в
большом количестве в моче лошадей. При кипячении ее с
разбавленными кислотами образуется бензойная кислота и гликокол:
QHeCO-rNHCH4COOH + НаО . С6Н6СООН
Гликокол может быть получен синтетически из аммиака и
хлоруксусной кислоты. Если же в этом синтезе вместо аммиака
взять амин, то получаются производные гликокола, в молекуле
которых атомы водорода при азоте замещены алкилами. Так, при
действии метиламина на хлоруксусную кислоту получается
метилгликокол, или саркозин:
СН3—NH—|н"+С||—CHjCOOH j, СН3—NH—СН2СООН + ПСІ
саркозин
378 Глава XIV. Аминоспирты и аминокислоты
Саркозин образуется при гидролизе креатина (см. стр. 418)—
вещества, находящегося в мускулах.
При действии на хлоруксусную кислоту диметиламина
получается диметиламиноуксусная кислота:
n4>N—iH -j-cii—сн.,соон —» 3\n—сн2соон + на
сн/ - : сн/
диметиламиноуксусная
кислота
Наконец, при действии на хлоруксусную кислоту триметиламнна
образуется соль четырехзамещенного аммониевого основания:
(CH3KN + СІ—СНХООН * KCHjOsN—СН2СООН]СГ
Это вещество называют (не совсем точно) хлористоводородной
•солью бетаина. Соответствующее основание нестойко и, отщепляя
воду, дает бетаин, который следует рассматривать как
внутреннюю соль четырехзамещенного аммониевого основания:
(CH3KN—
(CH3j3N-CH2COCr
бетаин
ІОН"
Бетаин встречается во многих растениях. Так, например, он
содержится в соке сахарной свеклы (по-латински beta—свекла)
и при производстве сахара накапливается в патоке. Бетаин
кристаллизуется с одной молекулой воды. В воде он очень хорошо
растворяется; водный раствор имеет нейтральную реакцию.
Безводный бетаин плавится при 293 °С.
В настоящее время бетаинами называются и другие
внутренние соли четырехзамещенных аммониевых оснований.
#
а-Аланин СН3—CH(NH2)—СООН, а-аминопропионовая
кислота B-аминопропановая кислота). Щ-)-Аланин в большом
количестве получается при гидролизе фиброина шелка;
незначительные количества его образуются при гидролизе многих
белковых веществ.
Лейцин
—СН,—CH(NH2)—СООН
а-аминоизокапроновая кислота D-амино-2-метилпентановая-5-
кислота). Лейцин находится в значительных количествах в про-
186. Отдельные представители аминокислот 379
дуктах гидролиза большинства белковых веществ, например:
гемоглобина крови, казеина, яичного альбумина. Природный
лейцин образует блестящие чешуйчатые кристаллы (по-гречески
«лейкос»—белый блестящий). Раствор его вращает плоскость
поляризации вправо.
Изолейцин
СНаСНаСН—CH(NHa)—COOH
СН3
а-амино-З-метилвалериановая кислота B-амино-З-метилпентано-
вая кислота). Изолейцин в незначительных количествах
содержится в продуктах гидролиза многих белковых веществ и вместе
с лейцином в свекловичной патоке. Его молекула содержит два
асимметрических атома углерода; природный изолейцин вращает
плоскость поляризации вправо.
Лейцин и изолейцин под влиянием некоторых
микроорганизмов, например дрожжей, превращаются в спирты, молекулы
которых содержат на один атом углерода меньше, чем молекулы
взятых аминокислот. При этом от молекулы аминокислоты
отщепляется СО2, а аминогруппа замещается гидроксилом.
Таким путем из лейцина получается оптически недеятельный
первичный изоамиловый спирт, а из изолейцина—оптически
деятельный первичный изоамиловый спирт:
СН3ч /fCOOiH CH34
>снсн.2сн/ : + н,о —> >снснасн3он + nh3 + со,
сн/ \nh3 сн/
оптически недеятельный
изоамиловый спирт
сн
4 /iCOOJH QH54,
>СНСН< ' +Н3О > >СНСНаОН + NHa + СОа
/ \nh3 сн/
оптически деятельный
изоамиловый спирт
Валин (а-аминоизовалериановая кислота) при брожении дает
первичный изобутиловый спирт:
СНЗЧ ІСООІН СН3ч
>СНСН< ' + Н3О > >СНСНаОН + NH3 + СО3
СН3/ \NH3 СН/
СН/
первичный
изобутиловый спирт
Этими реакциями объясняется образование спиртов
сивушного масла при винокурении. Они получаются не из Сахаров, а
380 Глава XfV. Аминоспирты и аминокислоты
-из аминокислот, образующихся в свою очередь из тех белковых
веществ, которые находятся во взятом для брожения материале.
В молекулах аспарагиновой (аминояншарной) кислоты
HOOC-CHaCH(NH2j—СООН
и глутаминовой (а-аминоглушаровой) кислоты
НООС—CHaCHaCH(NH8)—СООН
содержатся два карбоксила и одна аминогруппа; в отличие от
-одноосновных аминокислот они обладают ясно выраженной
кислой реакцией. Неполными амидами этих кислот являются
встречающиеся в растениях аспарагин
NHa-CO—CHaCH(NH.,)-COOH
и глутамин
NHa-CO—CH8CHaCH(NHa)-COOH
Особенно распространен L(—)-аспарагин; он был впервые
найден в ростках спаржи (Asparagus). Значительное количество
L(—)-аспарагина находится и в этиолированных (проросших
в темноте) ростках растений; в ростках вики содержится не
менее 28% Ц—)-аспарагина. В ростках вики находится иО(+)-аспара-
гин. Замечательно различное физиологическое действие обоих
антиподов: L-аспарагин безвкусен, в то время как D-аспарагин
обладает сладким вкусом. Различия в физиэлогическом действии
наблюдаются и среди других оптических антиподов. Так,
например, L(—)-никотин (природный продукт) в 2—3 раза более
ядовит, чем его антипод. Согласно Пастеру, различие в
физиологическом действии оптических антиподов объясняется тем, что они,
реагируя с асимметрическим веществом живой ткани, образуют
соединения, которые являются (один по отношению к другому)
не антиподами, а диастереомерами, и поэтому различаются
между собой по физическим и химическим свойствам.
Из других аминокислот большое значение имеют лизин, или
нормальная а,є-диаминокапроновая кислота B,6-диаминогексано-
вая кислота)
NHa—СНгСНгСНаСНаСН(Шг)—СООН
и орнитин, или нормальная а,8-диампновалериановая кислота
NH,—CH2CH2CHaCH(NH.,)-COOH
186 Отдельные представители аминокислот 381
Лизин и орнитин, в молекулах которых имеются две
аминогруппы и только один карбоксил, обладают сильными основными
свойствами. Под влиянием гнилостных бактерий происходит
отщепление от них молекулы углекислого газа с образованием
птомаинов:
.іСбОІН
NH2-CH2CH2CH„CH2-CH< ¦'¦ : * NH2—CH2CH2CH2CH2CH2—NH2+ COa
XNH,
пентаметилендиамии (кадаверин)
.iCOO'H
NH2—СН2СН2СН2—СН/ ' » NH„—СНгСН8СНаСНа—NH8 + CCv,
ЧМН
орнитив тетраметиленднамин
(путресцин)
L-Лизин и L-орнитин обычно содержатся в продуктах
гидролиза белков.
Орнитин, вероятно, представляет собой вторичный продукт
расщепления белковых веществ. Непосредственным продуктом
гидролиза белков является аргинин (см. стр. 417), при гидролизе
которого получаются мочевина и орнитин.
Циспгвин HS—CH2CH(NH2)—СООН является производным
а-аминопропионовой кислоты, в молекуле которой один атом
водорода замещен группой —SH. При окислении кислородом
воздуха цистеин переходит в цистин:
НООС—CH(NRj)CH2—S— iH !
і +0! »
НООС—CH(NH2)CH2—S—;Н і
» НООС—CH(NH2)CH2—S—S—CH2CH(NHa)—СООН + Н20
цистин
При гидролизе белковых веществ большая часть входящей в
их состав серы выделяется в виде L-цистеина или L-цистина. При
некоторых болезнях цистин образует камни в мочевом1 пузыре и
почках.
Серии НО—CH2CH(NH2)—СООН представляет собой
кислородный аналог цистеина. Он находится в продуктах гидролиза
серицина (шелкового клея) и в незначительных количествах
образуется при гидролизе других белковых веществ.
Таурин NH2—СН2СН2—SO3H является одновременно и
амином и сул ьфо кислотой; он может быть получен из цистеина. При
энергичном окислении цистеина образуется сульфокислота
CHa СН—СООН
SOaH NH.
382
Глава XIV. Аииногпирты и аминокислоты
которая при нагревании в запаянной трубке отщепляет СО2 и
переходит в гаурин. Таурин был впервые открыт в продуктах
гидролиза бычьей желчи (по-гречески «таурос»—бык), главной
составной частью которой является таурохолевая кислота.
В табл. 14 приведены формулы некоторых аминокислот и тех
веществ, производными которых они являются.
Таблица 14
Некоторые г-амииокислотм и соответствующие им карбоновые кислоты
Карбоновая кислота
сна—соон
уксусная кислота
сн3сн,—соон
пропионовая кислота
>СНСНа—СООН
сн/
изовалериановая кислота
>снсн2сн соон
сн/
изокапроновая кислота
CHaCHjCHCHj—СООН
СН3
?-метилвалериановая кислота
НООС—СН2-СН2—СООН
янтарная кислота
HOOC-CHSCH2CH9-COOH
глутаровая кислота
СН3СН2СН2СН2—СООН
валериановая кислота
СНзСНаСН2СН2СНа-СООН
капроновая кислота
Аминокислота
CH.,(NH8)—СООН
глицив
CH3CH(NHa)—СООН
а-аланин
3V>CHCH(NH2)—СООН
сна/
валин
сн.к
XHCHoCHCNH.,)—СООН
сн/
лейцин
СНаСН2СНСН(ЫН2)—СООН
сн3
изолейцин
НООС—CHaCH(NHa)-COOH
аспарагиновая кислоти
NH2—С-СН2СН-СООН
І! І
О NHa
аспарагин
НООС—CH9CH2CH(NH2)— СООН
глутаминовяя кислота
NHa—С—CHjCHjCH—СООН
І] І
II 1
О NHa
глутамин
NH2CH2CHaCH2CH(NH2)-COOH
орнитин
NH2CH2CH2CHsCH3CH(NHa)—СООН
лизин
186 Отдельные представители аминокислот
383
Производными пропионовой кислоты являются также
следующие аминокислоты:
NHaCH2CH2— СООН HOCH2CH(NH2)—СООН HSCH2CH(NH2)—СООН
?-аланин серии цисте иы
НООС—CH(NHa)CH2—S— S—CH2CH(NH2)—СООН
цистин
Физические свойства природных аминокислот указаны
в табл. 15.
Таблица 15
Физические свойства некоторых природных аминокислот жирного ряда
Название
Гликокол
L (+)-Аланин . . .
L (—)Серин . . .
L (—) Цистин . . .
L (-|-)-Валин . . .
L (—)-Лейцин . . .
L (+)-Изолейцин .
L (+)-Аргинин . .
L (+)-Орнитин . .
L (+)-Лизин . . .
L (—)-Аспарагино-
вая кислота . .
L (+)Тлутаминовая
кислота ....
L (—)-Аспарагин .
L D-)-Глутамин . .
Температура
плавления или разложения
°С
289—292
297
228 (разл.)
258—261 (разл.)
315
337
280
228 (разл.)
(сироп)
224 (разл.)
270
248
236
—
Оптическая
активность
Неактивен
+2,7°
—6,8°
—222,4°
(в солянокислом
растворе)
+6,42°
— 10,4°
+9,7°
+26, Г
+ 16,8°
(солянокислая
соль)
+0,3°
(солянокислая
соль)
+6° (при 21,5 °С;
при более
высокой температуре
вращает влево)
+ 12,0°
-5,4°
+6,45°
Растворимость
в воде
Легко растворим
То же
»
Почти нерастворим
на холоду
Очень легко
растворим
1 :45 (при 20 °С)
1 :25,8 (при 15 °С)
Легко растворим
То же
1 : 222 (при 20 °С)
1 : 100 (при 16 °С)
3,1 : 100 (при 28 °С)
1 :27,7 (при 18 °С)
Среди продуктов гидролиза белковых веществ встречаются
более сложные аминокислоты, о которых сказано ниже:
аргинин (см. стр. 417), фенилаланин (см. стр. 503), тирозин (см.
стр. 503), тироксин (см. стр. 503), пролин (см. стр. 587),
триптофан (см. стр. 596) и некоторые другие.
384
Глава XIV. Аминоспирты и аминокислоты
За последнее время большое практическое значение получила
е-аминокапроновая кислота и ее лактам
CHgCHaCHgCHgCHjCO
і і
-NH-
B природе эта кислота не встречается. В значительных
количествах ее синтезируют и используют для получения одного из видов
синтетического волокна—капрона (см. стр. 397).
§ 187. Обозначение стереоизомерных молекул, содержащих амино- и окси-
группы. Для описания зеркальной изомерии первоначально были
использованы понятия «правый» и «левый» изомер Эти понятия условны, так как
предметы или молекулы сами по себе не являются «правыми» или «левыми».
Для многих классов соединений были выбраны конфигурации, которые
условно стали считать правыми и обозначать их буквой D (Dexter—правый),
а их зеркальные отражения—левыми и обозначать их буквой L (Laevo—левый).
Если в молекуле имеется только один асимметрический центр, то условные
обозначения D и L точно характеризуют расположение атомов в молекуле.
Если же в молекуле несколько асимметрических центров, то буквы D и L
описывают конфигурацию только одного асимметрического центра, считающегося
«ключевым».
Для семейств а-окси- и а-аминокислот «ключевым» считают
асимметрический центр а-углеродного атома (т. е. наиболее близкого к карбоксильной
группе). Для семейств оксиальдегилов и оксикетонов «ключевым» считают
асимметрический атом, наиболее удаленный от карбонильной группы
Конфигурацию функциональных производных обозначают тем же знаком, что и
конфигурацию исходных соединений.
«Ключ» семейства аминокислот:
соон
-Н
СООН
Н—p-NRj
Л-ряд
«Ключ» семейства оксикислот!
СООН
HO-J—Н
О-ряд
СООН
н—і-он
L-ряд О-ряд
«Ключ» семейства оксиальдегидов:
СНО СИО
НО-
тн
снаон
L-рял
и~—он
СН2ОН
D-ряд
187. Обозначение стереизомеров, содержащих амино- и оксигруппы 385
Введение асимметрических «ключей» первоначально внесло порядок в
номенклатуру соединений с несколькими асимметрическими центрами, в
частности в номенклатуру альдоз и кетоз. Эти понятия сохранили свою ценность
для многих случаев и до сих пор. Тем не менее очень скоро при переходе от
одного химического класса к другому возникли недоразумения, не изжитые
до сего времени. Так, при окислении D-арабимозы П) получаются
соответствующая D-арабоновая кислота (II), а затем триоксиглутаровая кислота A11)
(по ключу оксиальдегидов они относятся к О-ряду):
СНО
НО—
Н—
Н—
—Н
ОН
—ОН
НО-
н-
соон
-н
-ОН
н—
сн2он
I
-ОН
НО-
н-
н-
СН2ОН
соон
—н
—он
—ОН
соон
ш
Легко видеть, что эти же соединения по ключу а-оксикислот должны быть
отнесены к L-ряду. А триоксиглутаровая кислота (III) может быть отнесена и
по тому и по другому ключу как к D-, так и к Л-ряду.
Дело еще более осложняется, если в соединениях имеется несколько
асимметрических центров, относящихся к различным «ключам». Такова, например,
аминооксиянтарная кислота, которую можно отнести как к ряду О-оксикислот.
(IVa), так и к ряду L-аминокислот AV6):
СООН
СООН
Н—
Н-
-ОН
-NH2
H2N—
НО—
СООН
IV (а)
-н
—н
соон
IV (б)
Не лучше обстоит дело и с широко распространенными в природе амино-
сахарами и оксиаминокислотами, взаимные переходы которых трудно описать
символами D и L. Например, 2-амино-З-оксимасляная кислота—треонин (V)—
может быть отнесена по «ключу» оксиальдегидов к D-ряду, а по ключу
аминокислот—к L-ряду:
СООН
NH2—
Н—
—Н
-ОН
СН3
v
Чтобы устранить путаницу, ввели для таких соединений дополнительные
индексы g и s. Символы Dg и Ig означают, что конфигурация определена в «ключе»
оксиальдегидов (g—глицериновый альдегид, глюкоза), a Ds и Ls—что
конфигурация определена в «ключе» оксиаминокислот (s—серии).
25—788
386 Глава XIV. Аминоспирты и аминокислоты
Используя метод условных «ключей», приходится вводить новые и новые
«ключи» и правила для определения D- и L-рядов для каждой новой группы
соединений. Таким образом получается, что простая система, удобная для
приведения в порядок сравнительно небольшого числа соединений с усложнением
соединений становится громоздкой и противоречивой. Это характерный пример
того, как развитие содержания приводит к необходимости построения новой,
более совершенной формы.
Такой формой, по нашему мнению, является система «ро—сигма» (см
стр. 160 и 322).
Выше при описании изомеров спиртов, оксикислот и оксиальдегидов
было показано, как с помощью обозначений «р—а» сохранить наглядное
представление о пространственном строении молекул. Эта система помогает и тогда,
когда система «ключей» заходит в тупик. Так, написанная выше 2-амино-З-окси-
янтарная кислота (IV) может быть названа 2а-амино-3а-оксиянтарная
кислота, или, по женевской номенклатуре, Зр-аминобутанол-2р-диовая-1,4 кислота.
Большое преимущество системы «ро—сигма» заключается в том, что она
описывает конфигурацию каждого асимметрического центра. Поэтому ею
можно пользоваться для описания всех случаев зеркальной изомерии. В системе
«ро—сигма» обозначения соединений, приведенных в этом параграфе, будут
выглядеть следующим образом:
I—пентантетраол-2а,C,4)р,5-ал-1 или 2а,C,4)р,5-тетраоксипентанал;
II—пентантетраол-2а,C,4)р,5-овая-1 кислота или 2а,C,4)р,5-тетраоксипен-
тановая кислота;
III—пентантриол-2а,C,4)р-диовая-1,5 кислота или 2а,C,4)р-триоксиглута-
ровая кислота. '
Система «ро—сигма» позволяет обозначать конфигурацию, исходя как из
моделей, так и из проекционных формул, и воспроизводить непосредственно
как модель, так и проекционную формулу Нужно только помнить, что при
движении по цепи атомов углерода, как показано на стр. 160, заместитель,
находящийся слева, обозначается буквой р, а заместитель, находящийся справа,
обозначается буквой а. Это означает, что в общепринятой вертикальной
проекции оксикислот, аминокислот и Сахаров обозначение р будет эквивалентно
D-конфигурации, а обозначение а—Л-конфигурации.
Подробное описание системы «ро—сигма» и ее разнообразного применения
можно найти в специальной литературе*.
* См., например, А. П. Т е р е н т ь е в, А. Н. К о с т, А. М. Ц у-
керна», В. М. Потапов, Номенклатура органических соединений,
Изд. АН СССР, 1955.
ГЛАВА XV
БЕЛКОВЫЕ ВЕЩЕСТВА
188. Белки в природе. Белковые вещества, или белки,
находятся во всех растительных и животных организмах. Белки
являются главной составной частью протоплазмы, содержатся в
крови, молоке, мышцах и хрящах животных, составляют
главную часть куриного яйца. Белки входят в состав волос, когтей,
рогов, кожи, перьев, шерсти и шелка. Животный организм более
богат белковыми веществами, чем растительный. В растениях
белки встречаются в протоплазме, ядре, клеточном соке и
семенах. Главную же массу растений составляет клетчатка.
Белковые вещества образуются в растениях, которые
используют для их получения азот, находящийся в солях почвы, или,
как бобовые растения,—азот атмосферы. Животные не способны
к такого рода синтезам. Они получают белковые вещества в
готовом виде, поедая растения или других животных. Белки,
входящие в состав организма, во время его жизни непрерывно
подвергаются процессам разрушения и окисления, а потому они должны
являться необходимой составной частью пищи.
В состав почти всех белков входят пять элементов: углерод,
водород, кислород, азот и сера; некоторые весьма важные
белковые вещества содержат, кроме того, фосфор.
Содержание этих элементов в белках колеблется в следующих
пределах (%):
Углерод . . . 50—55 Кислород . . . 19—24
Водород . . . 6,6—7,3 Сера 0,2—2,4
Азот 15—18
Существуют белки, в которых содержание серы достигает 5%.
С другой стороны, белки, которые находятся в сперматозоидах
рыб, совсем не содержат серы; они получили название протами-
нов.
Один из белков крови—гемоглобин—содержит железо @,3—
0,5%). Наконец, известны белки, содержащие иод и другие
галоиды.
25*
388
Глава XV. Белковые вещества
189. Свойства белков. Белки—высокомолекулярные
нелетучие соединения, не растворимые в обычных растворителях; те
белки, которые растворяются в воде, дают коллоидные растворы.
При сжигании белки обугливаются и при этом наблюдается
характерный запах жженого рога.
Для обнаружения белка обычно используют следующие
цветные реакции.
Ксаншопрошеиновая реакция*. Белок с концентрированной
азотной кислотой дает желтое окрашивание. Желтые пятна,
образующиеся на коже при
неаккуратной работе с
концентрированной
азотной кислотой,— результат
ксантопротеиновой
реакции с белками кожи.
Биурепговая реакция.
Раствор
^ оелна/
4 Мишиллированная веда
Рис. 56. Схема электродиализатора.
При прибавлении к
раствору белка едкого натра
и нескольких капель
разбавленного раствора
сернокислой меди получается
фиолетовое окрашивание. Этой реакцией часто пользуются для
качественного открытия белка.
Белки претерпевают сложные изменения (денатурацию) при
нагревании и воздействии многих реактивов. Такие свойства
белков чрезвычайно затрудняют изучение их химического строения.
Кроме того, в природе белки встречаются всегда в виде
сложных смесей, поэтому выделение и очистка их сопряжены с
большими трудностями. Для подобного рода соединений необходимо
разрабатывать особые методы, с которыми не приходится
встречаться при работе с обычными органическими веществами.
Для разделения и очистки водорастворимых белков обычно
применяется фракционное высаливание при помощи растворов
нейтральных солей: NaCl, Na2SO4, MgSO4, (NH4JSO4 и др.
Отделенные таким образом фракции белков освобождаются от солей
при помощи диализа через полупроницаемые перепонки
(пергамент, пленка коллодия, задубленный желатин и др.), которые
легко пропускают соли и другие низкомолекулярные вещества,
но удерживают высокомолекулярные белки. Часто применяют и
электродиализ.
На рис. 56 изображена схема электродиализатора. В средний
сосуд наливают волный раствор белка, содержащий соли; в бо-
* Слово «ксантопротенн» возникло из двух греческих слов: «ксантос»—
желтый, «протеин»—белок.
189. Свойства белков 389
ковые сосуды, отделенные от среднего пергаментной перепонкой,
медленно пропускают дистиллированную воду. Ионы солей
быстро уносятся через перепонку к соответствующим электродам.
Разделенные и очищенные белки удается даже получить в
кристаллическом состоянии.
Для определения молекулярного веса белков почти не
применимы обычные методы, основанные на измерении упругости
пара, повышения температуры кипения и понижения температуры
замерзания растворов. Чаще всего пользуются специальными
методами, разработанными для исследования
высокомолекулярных веществ: определение скорости диффузии, вязкости
растворов, ультрацентрифугирование и др.
В ряде случаев можно пользоваться и химическими
методами. Покажем это на примере казеина—белка, выделяемого
из молока. Процентный состав его следующий:
Углерод 53,13 Азот 15,78
Водород 7,06 Сера 0,80
Кислород .... 22,37 Фосфор 0,86
Таким образом, в 100 весовых частях казеина находится 0,80
весовых частей серы. Так как известно, что сера в казеине
содержится в виде остатка молекулы цистина I—ОС—CH(NH2)— CH2S12,
то, следовательно, в молекуле белка должно быть ы менее двух
атомов серы, т. е. 64 весовых части. Исходя из этого,
молекулярный вес казеина определяют из пропорции:
0,80—100 юо й4
' х = ^-4 = 8000
64—X 0,80
Если же принять, что в частице казеина содержится 2
остатка цистина, то молекулярный вес будет равен 8000x2=16 000.
Эта величина имеет тот же порядок, который получается при
определении ее физическими методами. Молекулярная формула
казеина в таком случае будет C70sHjj300224N18()S4P6.
Молекулярный вес яичного альбумина («белка»),
определенный методом ультрацентрифугирования, составляет около
34 000; гемоглобина (из крови)—около 68 500. Есть белки,
имеющие еще больший молекулярный вес.
Отсюда ясно, сколь сложно строение этих громадных
молекул и как трудна задача определения химического строения и
тем более проведения систематического синтеза аналогичных
белку соединений.
Белковые вещества можно разделить на две большие группы:
1) собственно белки, или протеины, и 2) протеиды. Молекулы
390 Главо XV. Белковые вещества
последних при осторожном гидролизе распадаются на молекулы
собственно белков и на молекулы веществ, не относящихся к
белкам.
190. Собственно белки. По ряду характерных свойств
собственно белки можно разделить на несколько подгрупп.
1. Альбумины. Они растворимы в воде, свертываются при
нагревании, нейтральны, сравнительно трудно осаждаются
растворами солей. Примерами их могут служить: альбумин белка
куриного яйца, альбумин кровяной сыворотки, альбумин
мускульной ткани, молочный альбумин. Последний находится в
молоке вместе с другим белком—казеином; пенка на кипяченом
молоке состоит нз альбумина.
Техническое значение имеет альбумин кровяной сыворотки,
который применяется для изготовления светочувствительных
бумаг и в ситцепечатании. Он, так жг как и фибриноген,
получается на бойнях из крови животных. При выходе из
кровеносных сосудов фибриноген свертывается в фибрин. Для того чтобы
лучше отделять фибрин, кровь сбивают. При сбивании кровв
фибрин выделяется в виде белых нитей, а в остатке получается
жидкость, красная от плавающих в ней кровяных телец.
Последние удаляют центрифугированием и получают кровяную
сыворотку. При осторожном выпаривании этой сыворотки из нее
выделяются кристаллы альбумина.
2. Глобулины. Они нерастворимы в воде, но растворяются
в очень слабых растворах солей. Более концентрированными
растворами солей они вновь осаждаются; осаждение происходит
при меньшей концентрации, чем та, которая необходима для
осаждения альбуминов. Эти белки являются очень слабыми
кислотами. Примерами глобулинов могут служить: фибриноген,
глобулин кровяной сыворотки, глобулин мускульной ткани,
глобулин Селка куриного яйца. Таким образом, в белке куриного
яйца, і> крови, в мускульной ткани находятся и глобулины и
альбумины. В отличие от этого в молоке почти не содержится
глобулинов. Из глобулинов состоят и многие растительные белки.
3. Гисшоны. Белки основного характера. Находятся в виде
нуклеопротеидов (см. стр. 392) в лейкоцитах и красных
кровяных шариках.
4. Протамины. Не содержат серы, обладают сравнительно
сильными основными свойствами, дают кристаллические соли;
содержатся (в виде нуклеопротеинов) в сперматозоидах рыб.
Некоторые исследователи считают их простейшими из белковых
веществ.
5. Проламины. Находятся в зернах различных хлебных
злаков. Замечательной их особенностью является растворимость
в 80%-ном спирте. Представителем этих белков может служить
191. Протеиды 391
глиадин, составляющий главную часть клейковины. Клейковину
можно легко получить из пшеницы. Для этого пригоршню муки
замешивают в тесто, и, завернув в кисею, отмывают под
краном—остается тягучая, грязно-желтого цвета клейковина.
Клейковина пшеницы—это смесь различных белков, в
которой преобладает уже упомянутый выше глиадин. Другие белки,
входящие в состав клейковины, в спирте не растворяются.
6. Склеропропгеины (прежнее название альбуминоиды).
Нерастворимые белки, которые составляют наружный покров тела
животного и находятся в скелете и в соединительной ткани.
К ним относятся кератин, коллагены, эластин, фиброин.
Кератин является главной составной частью волос, рогов,
копыт, ногтей, перьев и верхнего слоя кожи. Скорлупа куриного
яйца состоит из извести и кератина. Если растворить известь
скорлупы яйца в кислоте, то останется мягкая кожа, состоящая
«з кераіина; из кератина состоит и та кожица, которая следует
за скорлупой яйца. По химическому составу кератин богат
серой.
Коллагены. Чрезвычайно распространены в животных
организмах. Из коллагенов состоит соединительная ткань; они
находятся в хрящах. Кости позвоночных животных состоят из
неорганических веществ (фосфорнокислого и углекислого кальция),
жира и коллагенов
При кипячении с водой или при действии перегретого
водяного пара коллагены образуют клей. Если из костей извлечь жир
и потом, обработав их кислотой, растворить фосфорнокислый
кальций, то останется белковое вещество—оссеин. При
обработке оссеина перегретым водяным паром он переходит в клей.
Чистый костяной клей называется желатином. Особенно чистый
желатин получается из рыбьего пузыря кипячением его с водой.
Эластин входит в состав жил и других эластичных веществ
соединительной ткани.
Нити сырого шелка состоят из белкового
вещества—фиброина, покрытого другим белковым веществом, играющим роль
шелкового клея,—серицином При кипячении с водой шелк
освобождается от клея, который при этом переходит в раствор.
191. Протеиды. 1. Фосфопротеиды содержат в своем
составе фосфор. Они, в противоположность протаминам,
обладающим основными свойствами, имеют определенно выраженный
кислотный характер.
Главнейшим представителем фосфопротеидов является
казеин молока. Он обладает настолько ясно .выраженным
кислотным характером, что разлагает углекислые соли с выделением
углекислого газа. Казеин растворяется в слабых растворах
щелочей, образуя с ними соли. Так, в молоке казеин находится
392 Глава XV. Белковые вещества
в виде своей кальциевой соли. Соли казеина называются казеина-
тами.
При нагревании казеин не свертывается. При действии
кислот на соли казеина он выделяется в свободном виде. Этим
объясняется свертывание молока при прокисании. При стоянии
молока молочный сахар, который входит в его состав, под действием
молочнокислых бактерий превращается в молочную кислоту.
Получившаяся кислота и осаждает казеин из его кальциевой
соли в виде студенистой массы. Нагреванием этой массы до 35—
40 °С получают творог.
При действии на молоко сычужных энзимов казеин
претерпевает значительное изменение и превращается в
нерастворимый параказеин, который выпадает в виде сгустка. При
дальнейшей обработке этого сгустка получается сыр, состоящий
главным образом из параказеина. При изготовлении подобных
сыров, которые в отличие от кисломолочных называются
сычужными, пользуются вытяжкой энзимов из сычуга теленка.
Казеин применяется для изготовления твердой,
напоминающей рог пластмассы—галалита. Для получения галалита
казеин смешивают с водой, красками и наполнителями, прессуют
под давлением, и полученные пластины обрабатывают
формалином. Казеин содержит фосфор в виде сложного эфира
фосфорной кислоты.
Из других фосфопротеидов следует отметить вителлин,
который находится в желтке куриного яйца.
2. Нуклеопротеиды находятся в клеточных ядрах. При
осторожном гидролизе они расщепляются на белок и нуклеиновую
кислоту.
Нуклеиновые кислоты являются весьма сложными
веществами, расщепляющимися при гидролизе на фосфорную кислоту,
углеводы и азотсодержащие органические вещества группы
пиримидина и группы пурина (см. стр. 626 ел.)
3. Хромопротеиды. Под этим названием известны протеиды,
которые представляют собой сочетание белков с окрашенными
веществами. Из хромопротеидов наиболее изучен гемоглобин—
красящее вещество красных кровяных шариков. Гемоглобин,
соединяясь с кислородом, превращается в оксигемоглобин,
который, отдавая свой кислород другим веществам, снова
превращается в гемоглобин. Значение гемоглобина в жизни человека и
животных очень велико. Он играет роль переносчика кислорода
от легких к тканям. Образовавшийся в легких оксигемоглобин
кровью разносится по телу и, отдавая свой кислород,
способствует протеканию в организме окислительных процессов. Кроме
того, гемоглобин вместе с плазмой крови осуществляет
регуляцию величины pH крови и перенос углекислоты в организме.
192. Понятие о химическом строении белков 393
Характерной особенностью гемоглобина является его
способность соединяться с окисью углерода, после чего он теряет
способность соединяться с кислородом. Этим объясняется ядовитое
действие окиси углерода.
Гемоглобин представляет собой соединение белка глобина
с красящим началом гемом. Вне организма гемоглобин,
при действии воздуха, превращается в метгемоглобин, который
отличается от оксигемоглобина прочностью связи с кислородом.
При обработке ледяной уксусной кислотой метгемоглобин
расщепляется с образованием глобина и гематина C34H32O4N4Fe(OH).
Обработкой метгемоглобина тем же реактивом, но в присутствии
NaCI, получается хлористая соль гематина, называемая гемином,
C34H32O4N4FeCi.
Гемин образует характерные красно-коричневые таблички,
которые дают возможность открыть присутствие крови в пятнах
даже через несколько лет.
4. Глюкопротеиды. Некоторые белки этой группы
встречаются в слизистых выделениях животных организмов и
обусловливают свойства этих выделений тянуться в нити даже при
сравнительно большом разбавлении. Эти белки образуются в
подчелюстной железе (подчелюстная железа—одна из слюнных
желез), печени, железах желудка и кишечника. Другие глюко-
протеиды находятся в хрящах, яичном белке, стекловидном
теле глаза и т. д. Исследованные представители глюкопротеи-
дов являются сочетанием белков с олиго- или полисахаридами.
192. Понятие о химическом строении белков. Как мы
неоднократно имели случай убедиться, наиболее устойчивыми к
воздействию химических реактивов в органических молекулах
являются углерод-углеродные связи. Нагревание вещества с водными
растворами кислот или щелочей обычно не нарушает этих
связей; гидролиз, как правило, 'приводит к расщеплению связей
у кислорода или азота. Таковы реакции гидролитического
расщепления сложных эфиров (например, жиров) и амидов.
Белковые вещества при гидролизе распадаются в конечном итоге до
а-аминокислот. Если в состав белка входят только различные
а-аминокислоты, то мы имеем дело с так называемыми
собственно белками, ил и протеинами Но существуют и сложные
белки, или протеиды, в состав которых входят остатки соединений,
принадлежащих к иным классам органических и
неорганических соедннеььр.
Каковы же основные формы связи аминокислот в сложной
молекуле белка?
Еще в 1891 году А. Я Данилевский высказал
предположение, что это—амидные связи, образованные карбок-
394 Глава XV. Белковые вещества
силом одной молекулы аминокислоты и аминогруппой другой:
О
H4N—СНа—C<f -t- HNH—СН3—СООН > H,N—СН,—СО— NH-CHj-COOH
Х)Н
Такого рода связи носят название шептионых связей-». Этим
путем могут быть соединены 2, 3, 4... остатка одинаковых или
разных а-аминокислот в виде дипептида, трипептида. тетрапептида
и т. д.; подобные соединения называют полипептидами.
Это предположение было впоследствии доказано
Гофмейстером и Э. Фишером A902).
Примерами соединений с пептидными связями являются:
HaN—СН—СО—NH— СН—CO-j-NH— СН3—СООН
СН ' СН;,
валилаланнлгпнциі1
H2N—СН2—СО—NH—СН3—СО—NH—СН,—СО—NH—СН—СООН
I
СН,
гоиглицилаланин
И Т. Д.
В полипептидах на одном конце цепи находиіся свободна«
аминогруппа, а на другом—карбоксильная.
В последнее время осуществлен полный си Hied нескольких очень сложны*
природных пол и пептидов, имеющих важное биологическое значение. К ни»
относятся, например, гормоны: инсулин, состоящий из 51 остатка аминокислот
окситоиин и др.
Кроме линейных цепей аминокислот, в некоторых природных
веществах обнаружены цепи, замкнутые в виде циклических
«ангидридов» —циклопептидов. Простейшие циклопептиды—дикетопипе-
NH NH
ОС СН, ОС СН-СН,,
II II
H.JZ СО СНд— НС СО
\/ \/
NH NH
разины, существование которых в молекулах белка предполагали
Н. Д. Зелинский и Э. Абдергальден, как установлено, в белках
не обнаруживаются. В то же время известны природные
циклопептиды, кольца которых составлены из многих остатков различных
192. Понятие о химическом строении белков 395
аминокислот. К ним относятся, например, антибиотик грамицидин
(стр. 602), гормон окситоцин и др.
Очень существенную роль играет дисульфидная связь,
образующаяся путем соединения остатков циеіеина через серу:
НООС—СН—СН2—S—S—СН2—CH—COOH
NH2 NH2
Таким образом, пептидные цепи могут быть «сшиты» дисуль-
фидными мостиками. Расщепление пептидной связи происходит
при гидролизе, дисульфидная же связь разрывается путем
восстановления:
—СО—CH—NH—
СН., -СО—CH-NH—
I ' I
S +2H CH.-SH
S " СН2—SH
СН2 — NH— CH—СО —
—NH—СН—СО—
По всей вероятности, и гидроксильные группы а-амино-Э-окси-
кислот [НО—СН2—CH(NH2)—СООН—серии I также принимают
какое-то участие в образовании цепей по типу сложных эфиров.
Однако это до сих пор в полной мере еще не доказано.
Кроме этих чисто ковалентных связей, между
полипептидными цепями и циклами действуют силы ассоциации*.
Не следует забывать, что среди аминокислот, входящих в
состав природных белков, имеются также моноаминодикарбоновые
кислоты и диаминомонокарбоновые кислоты. Избыток первых
в данном белке увеличивает его кислотный характер. В белках
основного характера содержится некоторый избыток диамино-
кислот.
Ряд характерных реакций белков обусловлен нахождением
феноламинокислот (тирозин—стр. 503) и аминокислот,
включающих гетероциклические системы: триптофан (стр. 586), пролин
(стр. 587) и др.
Все а-аминокислоты (кроме гликокола) имеют
асимметрический атом углерода; при этом существенно отметить, что амино-
* Подобные тем, которые, например, вызывают ассоциацию спиртов.
396 Глава XV. Белковые вещества
кислоты, входящие в состав природных белков, принадлежат к
L-ряду:
СООН .СООН
NH2 —і—Н или NH2'(T^>H
R R
Изучение аминокислотного состава белков является очень
сложной задачей, так как приходится подвергать разделению
смесь близких по составу и строению веществ. Большим
прогрессом в этом вопросе было применение метода хроматографиче-
ской адсорбции, открытого русским ботаником М. С. Цветом.
Изложение этого метода будет дано на стр. 590. Теперь же
отметим, что для изучения продуктов гидролиза белков
наиболее широко применяется метод разделительной хроматографии
на фильтровальной бумаге.
Итак, белковые вещества отличаются большим
разнообразием химических функций: образуют соединения с молекулами
веществ небелкового характера, очень легко в природных
условиях подвергаются распаду и усложнению. Так, если для
гидролиза белка необходимо длительное воздействие растворов
минеральных кислот при повышенной температуре, то подобное
расщепление при помощи органических
катализаторов—ферментов может происходить при 20—40 °С в течение короткого времени.
Примерами каталитического действия органических
катализаторов могут служить процессы пищеварения под действием
пищеварительных ферментов, а также многочисленные процессы
обмена веществ.
193. О синтезе белков. Как мы видели, белковые вещества
слагаются из многочисленных остатков а-аминокислот,
соединенных между собой амидными, дисульфидными и некоторыми
другими системами связей. Современная органическая химия
обладает обширным арсеналом разнообразных синтетических
методов. Предложено большое число остроумных и тонких методов
получения иолипеитидов заданного строения. Это давало и дает
основание химикам пытаться искусственно создать соединения,
подобные по строению белкам.
Несмотря на работы многочисленных исследователей, до сих
пор еще не получены вещества, тождественные природному
белку. Трудность решения этой задачи объясняется не только
физико-химическими свойствами белков, затрудняющими получение
их в чистом виде, но и громадным числом возможных изомеров.
Так, если предположить, что в состав молекул белков каждый
из двадцати продуктов гидролиза входит в количестве только
одной молекулы, то, изменяя порядок сочетания, мы получим
194. Синтетическое волокно 397
количество изомеров, равное 2,3-1010. Если же принять во
внимание, что в молекуле белка могут находиться не одна, а
несколько молекул одной и той же аминокислоты и что белки
могут различаться не только строением, но и пространственным
оасположением атомов, то к этому числу надо приписать справа
еще очень много нулей. Таким образом, может существовать
почти бесчисленное множество различных белков. Однако
изучение продуктов гидролиза дает нам надежное средство,
позволяющее заглянуть в строение белковой молекулы, и делает
трудную задачу синтеза белковых веществ возможной для решения.
О роли нуклеиновых кислот в синтезе белка см. стр. 626.
194. Синтетическое волокно. Полиамидные смолы.
Волокнистые материалы животного происхождения (шелк, шерсть и др.)
являются белковыми веществами. Их молекулы построены из
длинных цепей аминокислот, связанных между собой по типу
амидов. Из растворимых белков можно приготовить
искусственные волокна, пропуская под давлением растворы белков
(например, казеин) через фильеры. Получаемые нити последующей
обработкой формальдегидом переводят в нерастворимое в воде
состояние.
Гораздо большее значение имеют высокопрочные
синтетические волокна из полиамидных смол, в которых компоненты,
образующие полимер, связаны между собой так же, как и
аминокислоты в молекуле белка (шерсти). Из таких полимеров
наибольшее промышленное применение получили: анид (иначе
называют найлон), представляющий собой продукт конденсации
большого числа молекул {поликонденсации) адипиновой
кислоты и гексаметилендиамина:
НООС-(СН2L—СОЮН + HiNH—(CH2N—NH ; Н + •
• • •+ НО і ОС-(СН2L-СОІОН + H;NH-(CH2)e—NH2 »
» HOOC-(CHaL-CO—[NH(CH2NNHOC(CH2LCO]„—NH-(CH2H-NH2
и капрон {перлон), являющийся продуктом поликонденсации
s-аминокапроновой кислоты:
H2N-(CH.,)S—COiOH + HjNH-fCH.Js-COjOH + HjNH—(СН,)—СО і ОНИ
* NH2(CH2K-CO-[NH—(CH2M-CO]„-NH(CH2)—COOH
Пропуская под давлением найлон или капрон в
расплавленном состоянии через фильеры с отверстием диаметром 0,25 мк,
получают волокна, превосходящие по прочности другие
натуральные или искусственные волокна.
398 Глава XV. Белковые вещества
Волокна из полиамидных смол применяются в производстве
авто- и авиапокрышек, для изготовления прочных и негниющих
рыболовных сетей и др. Особенно широко капроновое и найло-
новое волокно используют для изготовления трикотажных
изделий. Хорошо известно, что ткани из этих синтетических смол
нельзя гладить очень горячим утюгом, так как они
расплавляются при нагревании.
Советскими химиками создан новый вид полиамидного волокна —
энант, которое мало отличается по своим свойствам от других
полиамидных волокон. Оно более светостойко и эластично, чем
волокно капрон. Сырьем для получения энанта являются этилен
и четыреххлористый углерод.
195. Работа с веществами, содержащими меченые атомы.
Громадное развитие физики и химии стабильных и
радиоактивных изотопов многих элементов создало необозримые
возможности для изучения многих научных вопросов также в области
органической химии, биохимии, в медицине и др. Пользуясь
точными методами обнаружения и определения изотопных веществ,
можно решать такие вопросы, которые были недоступны для
решения обычными химическими методами. Для проведения
таких работ необходимо во многих случаях иметь органические
вещества, в молекулы которых введены простые или
радиоактивные (рад.) изотопы: дейтерий (D), тритий 3Н (рад.),
тяжелый кислород 18О, сера 34S или 3aS (рад.), 14С (рад.), 31Р (рад.)
и др. Так как соединения с мечеными атомами очень дороги, а
в ряде случаен весьма опасны для здоровья, от химика
требуется большая тщательность в работе с очень малыми количествами
вещества, часто с применением особых мер предосторожности.
Это, однако, не останавливает исследователей, и подобные
работы очень энергично развиваюіся.
Приведем несколько примеров применения метода меченых
а і омов.
Для реакции между уксусным ангидридом и этиловым
спиртом, протекающей с образованием этилацетата и ускусной
кислоты, можно было допустить два рода взаимодействия:
І «|
СНзСО—О—СОСНз + С,Н5—СН-Н » СНзСООН + CHaCOOQHs
і * I
ил и
СНяСО-СН-СОСНа + СгН5-р5—Н » СНзСООН + CH3COOQH5
Применяя меченый атом кислорода A8О), А. И. Бродский и
Н. И. Дедусенко A940) показали, что в действительности
реакция протекает по первой из этих схем.
105. Работа с веществами, содержащими меченые атомы 399
Аналогично был выяснен механизм реакции образования
сложных эфиров из спирта и кислоты.
Как известно, качество шелка во многом зависит от
тончайшего покрытия—олеата натрия, которое наносится на нить при
ее получении. Если нить покрыта олеатом натрия неровным
слоем, то краска ложится неровно и качество вязки резко
снижается. Равномерность покрытия нити олеатом натрия можно
контролировать с помощью радиоактивного натрия, который
вводится в состав соли. Если слой олеата натрия слишком тонок,
радиоактивность будет меньше контрольной величины, если слой
толстый, то и радиоактивность будет больше. Таким образом
контролируется качесіво выпускаемой продукции.
В 1940 г. П. А. Виноградов и Р. В. Тейс, Есспользовавшись
методом меченых атомов, показали, что в процессе
фотосинтеза у растений выделяющийся кислород образуется
из кислорода воды, а не из углекислого газа. А. Л. Кур-
санов нашел, что раеіения могут ассимилировать углекислоту
также через корневую систему.
Весьма интересные биохимические исследования были
проведены с использованием меченой уксусной кислоты состава
CD3—|4СО2Н. Очень много работ было осуществлено в области
изучения обмена веществ с помощью соединений, содержащих
радиоактивный фосфор 31Р. Использованы вешестЕа со
стабильным изотопом азота I5N (мочевина, аминокислоты).
Синтезированы и применяются некоторые содержащие серу
аминокислоты, например СН;,—a4S—CH2—СН„— CH(NH2)—СООН
(меіионин). Но оссбенно широко в области срганическсй
химии и биохимии применяются разнообразные вещестЕа с
радиоактивным изотопом углерода 14С. Исходным веществом для
синтеза в этих случаях часто является 14СО2 (из Ва14СО3). Очень
многие синтезы проводят с использованием реакции Гриньяра:
получение кислот, сложных эфиров, кетонов, алкоголен и др.
Таким образом, приобретают большое значение такие синтезы,
которые казалось бы никогда не было смысла применять в
практике. Так, например, описан путь получения толуола по схеме:
CH3MgJ + "C02 > CH3—14CO2H ' 5 ... CH,-»4COOC2H6
H3C< ' > H2C< ' *
xCH,CHaBr XC HXMaMgBr
Л^лГХл •¦ -Сіп /КА14 - /\-tl-2 v^ri(> Л_іП — 1_.П
/ \/ ¦ / \ / \
H2C liC > HaC /4C—CH3 » MC I4C—CH,
^CHj-CHjNdh ^CH^ -CM ^CH-CM
400 Глава XV Белковые вещества
Получаемые соединения сами могут быть исходными веще"
ствами для синтеза многочисленных соединений, необходимых
для того или иного исследования, например, лекарственных
препаратов. Исследователь может изучать их поведение в организме
животного, так как присутствие радиоактивного элемента A4С, 31Р
и др.) очень точно может быть обнаружено соответствующими
физическими методами.
ГЛАВА XVI
СОЕДИНЕНИЯ ЦИАНА
196. Цианистые соединения и источники их получения.
Цианистыми соединениями называются вещества, содержащие
радикал CN, который может иметь двоякое строение:
—C=N C=N—
циан цзоциан
Простейшими цианистыми соединениями являются дициан (CNJ
и синильная кислота HCN.
Слово «циан» происходит от греческого слова «кюанеос»—
синий. Такое название радикал циан получил потому, что он
входит в состав синей краски—берлинской лазури и турнбулле-
вой сини. Этим же объясняется название «синильная кислота».
Одним из источников получения цианистых соединений
является железистосинеродистый калий K4[Fe(CNN], который
кристаллизуется с тремя молекулами воды K4[Fe(CN)eJ-3H2O.
Эти кристаллы известны под названием желтой кровяной соли.
Железистосинеродистый калий получали раньше главным
образом из животных отбросов (рога, копыта, кровь) путем
сплавления их с поташом в присутствии железных опилок. В настоящее
время большие количества этого вещества получают из
отработанной газоочистительной массы (в состав ее входит гидроокись
железа), образующейся при очистке светильного газа.
При сплавлении железистосинеродистого калия с
металлическим натрием получаются одновременно цианистый калий и
цианистый натрий:
K4[Fe(CNN] + 2Na > 2NaCN + 4KCN + Fe
Цианистый натрий получают также и другими способами,
например прокаливанием соды с углем в атмосфере азота:
Na2CO3 + 4С + N2 > 2NaCN + ЗСО
26—788,'
402 Глава XVI. Соединения циана
Цианистый калий и цианистый натрий—белые
кристаллические чрезвычайно ядовитые вещества. Их водные растворы
обладают способностью в присутствии кислорода воздуха
растворять золото и потому применяются в большом количестве для
извлечения золата из золотоносных руд (цианндный способ):
2Аи + 4KCN + 2HaO + О3 » 2[<[Au(CNJ| + 2КОН + НА
Образующаяся перекись водорода участвует далее в получении
комплексной соли золота:
2Аи + 4KCN + Н2Оа * 2K[Au(CN)a] + 2КОН
Из полученной таким путем комплексной соли золото может
быть выделено различными способами, например при помощи
металлического цинка.
Цианистые металлы находят также применение для
золочения, серебрения, никелирования (гальванопластика). В
органической химии цианистый калий широко применяется для
различных синтезов. Напомним, например, получение карбоновых одно-
и многоосновных кислот, окси- и аминокислот через
промежуточное образование нитрилов и последующий их гидролиз (см.
стр. 220).
При действии кислот на цианистый натрий или цианистый
калий получается синильная кислота HCN:
NaCN + НС1 * NaCl + HCN
197. Нитрилы. Получение и свойства. Нитрилами
называются вещества, в которых органический радикал связан с
углеродом CN-группы:
СН3—CN СаНь—CN
нитрил уксусно.і нитрил пропионовой
кислоты кислоты
(цианистый метил) (цианистый этил1
Нитрилы могут быть получены действием цианистого калия
на галоидные ал килы:
QH5J + KCN * QH5—C=N + KJ
При этом углеродная цепь удлиняется на одно звено.
197. Нитрилы. Получение и свойства 403
Другой способ получения нитрилов состоит в отнятии воды
от амидов кислот при нагревании последних с водуотнимающими
средствами (например, с фосфорным ангидридом):
Ж:
СН3—С—N< і і » СН3—CsN + Н2О
II Nh!
іо
Нитрилы, в молекуле которых содержится до четырнадцати
атомов углерода,—жидкости; высшие нитрилы—твердые тела.
Нитрилы обладают своеобразным запахом, напоминающим
запах горького миндаля. Они ядовиты, но в гораздо меньшей
степени, чем синильная кислота.
Отметим два главнейших свойства нитрилов.
1. При восстановлении нитрилов радикал циан, присоединяя
водород, переходит в аминогруппу:
СН3—CsN + 2На » QH6—NHa
этиламин
Восстановление проводят в спиртовом растворе
металлическим натрием. Выделяющийся из спирта водород разрывает
вторую и третью связи между атомами углерода и азота,
присоединяется (в количестве двух атомов) к каждому из них. Этот очень
важный метод восстановления был предложен впервые в 1880 г.
учеником Бутлерова—А. Н. Вышнеградским.
Реакция восстановления нитрилов служит для
доказательства строения диаминов. Так, строение пентаметилендиамина
NH.2—СН2СНаСН2СН2СН2—NHS
доказывается тем, что он может быть получен из бромистого три-
метилена A,3-дибромпропана) следующим образом:
/СН2—!Вг ""Щ—CN /СН.2— CN
f\-\ ' _l_ Ґ~ЧЛ І Olf Dr
^CHj—!вг к!—cn \:н2—cn
/CH3—CN /CHa—СН2—NHa
CH2 + 8H > CHa
\CH.2—CN \zHj—CHa—NHa
2. При действии растворов кислот и щелочей нитрилы омы-
ляются в кислоты (см. стр. 220).
26*
404 Глава XVI. Соединения циана
198. Изонитрилы. Изонитрилами, или карбиламинами,
называются вещества, в которых органический радикал связан с
азотом CN-группы. Примером изонитрилов может служить ме-
тилкарбиламин СН3—N=C.
Изонитрилы получаются наряду с нитрилами при действии
солей синильной кислоты на галоидные алкилы. Если для
реакции брать цианистый калий, то получаются главным образом
нитрилы, если же проводить ее с цианистым серебром, то
получаются изонитрилы:
CH,J + AgCN » CH3-N=C + AgJ
Другой способ получения изонитрилов заключается в
действии хлороформа и едкой щелочи на первичные амины:
,Н С1Ч УН -2HCI Г № І -неї
C.,H5-N< + >С< - C2H5-N=C< » CH,-N=C
Этой реакцией пользуются для открытия первичных аминов, так
как образование даже небольших количеств изонитрилов легко
обнаруживается по характерному, чрезвычайно неприятному
запаху.
Под действием кислот изонитрилы присоединяют воду и
расщепляются на первичные амины и муравьиную кислоту:
R—N=c/
L H
,0H1 +H20 yOH
R—N=C
J
Эта реакция доказывает, что в изонитрилах углеводородный
радикал связан с азотом.
Изонитрилы способны к реакциям присоединения; это
присоединение происходит только к атому углерода, например:
R—N=C + S » r_N=C=S
эфир изотиоциановой
кислоты
Присоединение исключительно к атому углерода показывает,
что в изонитрилах атом углерода имеет особый характер.
Структуру изонитрилов ранее изображали формулой с
«двухвалентным» атомом углерода. В настоящее время принимают,
что углерод и азот в пзонитрильной группе связаны
своеобразно:! тройной связью: две валентности ее построены из двух
«поделенных» пар электронов; третья же валентная пара целиком
взята от атома азота. Таким образом, атом углерода имеет сво-
199. Синильная кислота 40&
бодную пару электронов и несет отрицательный заряд, а атом
азота несет положительный заряд:
Изонитрилы—жидкости, кипящие ниже изомерных им
нитрилов. Они более ядовиты, чем нитрилы, и имеют отвратительный
запах.
199. Синильная кислота. Синильная кислота
HCN—бесцветная жидкость с запахом горького миндаля (темп. кип. 25 °С).
Очень слабо диссоциирована в водном растворе; является
сильнейшим ядом.
Синильная кислота довольно часто встречается в растениях
как в свободном, так и в связанном виде. Так, в листьях
лавровишни, в горьком миндале, в косточках персиков, абрикосов,
слив, вишен находится глюкозид амигдалин C20H27OuN-3H2Or
молекула которого при гидролизе расщепляется на две молекулы
глюкозы, молекулу бензойного альдегида и молекулу синильной
кислоты:
C()H„NOU + 2Н..0 * 2С6Н12О6 + QHa-CHO + HCN
Синильная кислота является нитрилом муравьиной кислоты;
при длительном стоянии ее водного раствора образуется му-
равьинокислый аммоний:
Г sP Л /
Н-С/ + NH3 * Н—C<f
L ХОН J X
-teN + 2Н„О+ 3 f
L Х J XONH4
Как было уже указано, при действии солей синильной
кислоты на галоидные алкилы получаются одновременно и
нитрилы R—C=N и изонитрилы R—C = N. Поэтому, а также и по
другим причинам синильную кислоту считают легко
превращающейся смесью двух таутомеров:
Н—C=N і—> Н—N=C
Изонитрильная форма находится в синильной кислоте в
небольшом количестве.
В технике синильную- кислоту получают совместным
окислением метана и аммиака воздухом на платиновом катализаторе:
2СН4 + 2NH3 + ЗО, * 2HCN + 6Н2О
406 Глава XV/. Соединения циана
Она применяется как исходный полупродукт в производстве
акрилонитрила и метилметакрилате (для органического стекла).
200. Циан. Циан (CNJ, называемый часто дицианом,
впервые получил в 1815 г. Гей-Люссак, нагревая цианистую ртуть:
Hg<(
CN
» Hg + NsC—CS
CN
Он может быть также получен сливанием растворов
сернокислой меди и цианистого калия:
2CuSO4 -f- 4KCN * 2I<2SO4 -f- 2CuCN -f- (CNJ
Образующаяся вначале Cu(CNJ сейчас же распадается на
CuCN и свободный циан. Небольшие количества циана
образуются при пропускании азота через накаленный уголь.
Спектральным анализом присутствие циана обнаружено в хвостах комет.
Циан—бесцветный, очень ядовитый газ, сгущается в
жидкость при —20,7 °С, замерзает при —34,2 °С.
Циан образуется при нагревании щавелевокислого аммония
с водуотнимающими средствами:
NH4OOC—COONH4 » N=C—GsN + 4H2O
Действием разбавленной соляной кислоты циан может быть
омылен в щавелевую кислоту и аммиак (вместо них, конечно,
получаются хлористый аммоний и щавелевокислый аммоний):
N=C—CssN + 4Н,0 » НООС—СООН + 2NH3
Таким образом, циан является нитрилом щавелевой кислоты.
Циан имеет некоторое сходство с галоидами: при сгорании
калия в атмосфере хлора получается хлористый калий КС1, а в
атмосфере циана—цианистый калий KCN. С едким кали хлор
образует КС1 и КС1О, а при пропускании через раствор КОН
циана получается цианистый калий KCN и циановокислый
калий KCNO. Цианистое серебро AgCN, подобно хлористому
серебру,—белый творожистый осадок, нерастворимый в воде, но
растворяющийся в аммиаке.
201. Цианамид. При действии на синильную кислоту хлора
образуется хлористый циан С1—C=N (темп. кип. 15,5 °С):
CI-jCl + Hi-C=N > Cl—C=N + НС1
202. Циановая кислота 40?
В результате обработки эфирного раствора хлористого циана
аммиаком получается цианамид NHa—(feN:
NH2—;Н + Сії— teH * NHj— C=N + HC1
Цианамид представляет собой бесцветные кристаллы,
расплывающиеся на воздухе, плавящиеся при 41—42 °С. Водород
в цианамиде легко замещается металлом.
Большое значение имеет кальциевая соль цианамида—каль
цийцианамид CaN—C=N, состав которого изображают обычно
формулой CaCN2. Кальцийцианамид получают действием азота
на карбид кальция при температуре 1000—1100'С:
CaQ -4- N2 = CaCN2 + С
При действии• воды кальцийцианамид разлагается по
уравнению:
CaCNa + ЗН-Р > СаСОя 4- 2NHS
Приготовление цианамида кальция является одним из
возможных способов связывания атмосферного азота.
В связи с этим он одно время применялся в качестве азотного
удобрения, но позднее был вытеснен другими, легче
усваиваемыми растениями соединениями (мочевина и аммиачная селитра).
В технике он продолжает изготавливаться в больших
количествах, так как находит применение в качестве дефолианта*
хлопчатника, а также для получения некоторых азотистых соединений*
-NH—CN N N
II II I
NH H2N—С С—NHa
дициандиамид \ #
(циангуанидіш) N
являющихся важными полупродуктами для получения поверхно-
стноактивных веществ и пластических масс.
202, Циановая кислота. При сплавлении цианистого калия
с окисью свинца получается соль циановой кислоты—цианово-
* Дефолианты—химические соединения применяемые для удаления
листьев некоторых растений, что особенно важно при машинной уборке урожай-
таких культур, как, например, хлопчатник.
¦408 Глава XVI. Соединения циана
кислый калий (окись свинца служит в данном случае
окислителем). Циановая кислота—летучая жидкость, чрезвычайно легко
переходящая в твердые полимеры.
Если циановую кислоту рассматривать как продукт окисления
синильной кислоты, то двум таутомерным формам синильной
кислоты должны соответствовать две таутомерные формы циановой
кислоты:
H-C=N —
синильная
^ н—N=C
кислота
НО—C=N
циановая
кислота
-—*Н—N=C=O
нзоцнаноаая
кислота
Обе таутомерные формы этой кислоты не были выделены
однако сложные эфиры их известны:
R-O-C=N R-N=C=O
эфир циановой эфир изоциановой
кислоты иислоты
Эфиры изоциановой кислоты—жидкости с неприятным
запахом. Они получаются при действии галоидных алкилов на соли
циановой кислоты:
G,H5J + AgCNO » QH3—N=C=O + AgJ
Углеводородный радикал в эфирах изоциановой кислоты
связан с азотом. Это доказывается тем, что при действии на эфиры
растворов щелочей образуются первичный амин и углекислый газ:
CaH5—N=;C=O! +Н2Ю
COa
Эфиры циановой кислоты образуются при действии хлористого
циана на алкоголяты:
CjH— О—;Na + СП—C=N > QH5—О—C=N + NaCl
Они сейчас же полимеризуются в вещества с утроенным
молекулярным весом, например (C2H5OCNK. В этих полимерах радикал
связан с кислородом; при гидролизе их получаются спирты. В
мономерном виде эфиры циановой кислоты получены не были.
203. Тиоциановая, или роданистоводородная, кислота. При
кипячении цианистого калия с серой получается роданистый
калий KCNS—соль роданистоводородной, или тиоциановой, кисло-
203. Тиоциановая, или роданистоводородная, кислота 409
ты. Р ода н истов одо роди а я кислота—жидкость; в довольно
значительном количестве оиа находится в свежем соке лука.
Подобно двум таутомериым формам циановой кислоты можно
представить и две таутомериые формы тиоциановой кислоты:
Н—О—C=N Н—S—OsN
циановая кислота тиоциановая
кислота
Н—N=C=O н—N=C=S
нзоциановая изотиоциановая
кислота кислота
Известны сложные эфиры обеих изомерных форм тиоциаио-
вой кислоты:
R—S—C=N R_N=C=S
эфир тиоциановой эфир нзотиоцнановой
кислоты кислоты
Эфиры тиоциановой кислоты—жидкости с запасом лука.
Оии получаются при действии галоидных алкилов на соли рода-
иистоводородиой кислоты:
GjH6J + KCNS . CjH6-S-C=N + KJ
Углеводородный радикал в эфирах тиоциаиовой кислоты
связан с атомом серы, что доказывается образованием меркаптаиов.
и метиламина при восстановлении этих эфиров:
CjH6-S—C=N + ЗН2 > GjH6SH + CH3NH2
Эфиры изотиоциаиовой кислоты называются горчичными
маслами. Оии представляют собой жидкости с чрезвычайно острым
запахом. Горчичные масла получаются нагреванием сложных
эфиров тиоциановой кислоты:
R—S—C=N » R_N=C=S
Радикал в эфирах изотиоциаиовой кислоты связан с азотом. Это
видно из того, что при действии серной кислоты эти эфиры
присоединяют воду и расщепляются на первичные амины и серо-
окись углерода COS:
R_N=;C=S і +Н2Ю і > RNH2 + COS
Горчичные масла встречаются в виде глюкозидов в зернах
и корнях некоторых растений. В этих же растениях обычно
содержатся и энзимы, гидролизующие эти глюкозиды. Если таьие зер-
-410 ' Глава XVI. Соединении циана
¦на растереть с водой и оставить стоять, то через некоторое время
появляются горчичные масла в свободном виде. Так, в черной и
сарептской горчице содержится глюкозид синигрин*, который при
действии энзима расщепляется на глюкозу, алл ил горчичное
масло и кислый сернокислый калий:
Q0HleO9NS2K + Н.2О , СНа=СН—СН2—N=C=S + QH12O6 + KHSO4
синигрин аллилгорчичное масла
Алл ил горчичное масло обладает чрезвычайно резким запахом,
при попадании на кожу вызывает чувство жжения и образование
пузырей
названия черной горчицы Sinapis nigra
ГЛАВА XVII
ПРОИЗВОДНЫЕ УГОЛЬНОЙ КИСЛОТЫ
204. Хлорангидриды угольной кислоты. Угольной кислоте,,
как двухосновной кислоте, соответствуют два хлорангидрида:
полный хлорангидрид—фосген и неполный хлорангидрид—
хлоругольная кислота, называемая также хлормуравьиной
кислотой:
/ОН XI XI
О=С< О=С< О=С<
\он х)н х:і
угольная кислота хлоругольная кислота фосген
(хлормуравьнная
кислота)
Хлоругольная кислота и ее эфиры. Хлоругольную (хлорму-
равьиную) кислоту можно рассматривать как производное
угольной кислоты, в молекуле которой гидроксил замещен атомом
хлора, или как производное муравьиной кислоты Н—СО—ОН, в
молекуле которой подвергся замещению хлором атом водорода,,
связанный с углеродом. Хлоругольная кислота в свободном
состоянии неизвестна. Эфиры ее могут быть получены при
взаимодействии фосгена со спиртами:
XI XI
+ НС1
)СН3
метиловый эфир
хлоругольной
кислоты
XI XI
О=С< + СН3ОН > О=С<
ХС1 Х)
Эфиры хлоругольной кислоты—жидкости с резким запахом,,
напоминающим запах фосгена; их пары вызывают слезотечение.
Хлорированием метилового эфира хлоругольной кислоты
получается монохлорметиловый эфир хлоругольной кислоты (темп.,
кип. 106 °С)
=с<
X)—
о=<
СН..С1
412 Глава XV/1. Производные угольной кислоты
-и трихлорметиловый эфир этой же кислоты—дифосген (.темп,
кип. 127 °С)
С1
о=с<
Ю-СС13
дифосген
Оба эти соединения применялись в войну 1914—1918 гг. как
отравляющие вещества.
При нагревании дифосген разлагается, причем из одной его
молекулы получаются две молекулы фосгена:
/С1
О=С< »2СОС1,
ХОСС13
Фосген СОС12 впервые получил в 1811 г. Дэви соединением
окиси углерода и хлора на солнечном свету (по-гречески «фос»—
свет, «геннао»—рождаю):
СО + С1, » СОС1.,
В промышленности фосген получают пропусканием при
повышенной температуре окиси углерода и хлора над
активированным углем. Фосген—газ, под давлением или при охлаждении
сгущающийся в жидкость, кипящую при 8,2 °С; обладает сильным,
резким запахом.
Фосген является удушающим веществом: действие его на
организм заключается главным образом в поражении тканей легких.
Фосген вступает в реакции, типичные для хлорангидридов
кислот. С водой он образует угольную и соляную кислоты:
ici ні-он он
О=С< 4- ' »О=С< +2НС!
Nici н!-он хон
При обыкновенной температуре эта реакция идет довольно
медленно.
При действии на фосген спиртов образуются эфиры хлоруголь-
ной кислоты, а затем эфиры угольной кислоты:
С1 .С)
» О=С<( + НС!
;СІ+н;-О-с2на ос2н5
jCi'+H!-O-CaH5 OC2H5
О=С< > О=С< +НС1
¦осл-ц хос,н5
206 Производные угольной кислоты, содержащие азот 413
Фосген применяется в технике для получения полиуретано-
вых пластических масс и каучуков, а также в производстве
некоторых красителей. В войну 1914—1918 гг. он был применен
как отравляющее вещество.
205. Сложные эфиры угольной кислоты. Сложные эфиры
угольной кислоты могут быть получены взаимодействием фосгена
со спиртами. Они представляют собой бесцветные жидкости с
приятным запахом, мало растворимые в воде.
Известны сложные эфиры гипотетической ортоугольной
кислоты:
ІНО
ортоуголькая
кислота
он
iH
Н2О
Эфиры этой кислоты образуются при действии алкоголятов
на четырехбромистый углерод:
^ІВг + Nai—OC2H5 QH5O.
C2H6O—jNa + Вг;/ \;Вг + Na:—OCaH5 С^О^ NOC2H5
этиловый эфир
ортвугольноіі
кислоты
Этиловый эфир ортоугольной кислоты—жидкость; водой он
расщепляется на спирт и этиловый эфир угольной кислоты.
206. Производные угольной кислоты, содержащие азот.
.Игольной кислоте соответствуют два амида: полный
амид—мочевина, или карбамид, и неполный амид—карбаминовая кислота:
.он ,он vu
о=с/ о=с/ о=о
угольная карбаминовая м®чезина
кислота кислвта
Карбаминовая кислота в свободном состоянии неизвестна, ее
аммонийная соль—карбаминовокислый аммоний—получается при
взаимодействии углекислого газа и аммиака:
ONH,
O=C=O+2NHS » О=С/
414 Глава XV/1. Производные угольной кислоты
Взаимодействуя с водой, карбаминовокислый аммоний
превращается в углекислый аммоний:
/ONH4 X>NH4
О=С< + Н2О » О=С<
\nh2 Ndnh4
Отнятием от карбаминовокислого аммония воды можно получить
мочевину:
/ONH4 /NH2
О=С< » О=С< + Н,0
xNHa XNH2
Эфиры карбаминовой кислоты называются уретанами. Они
могут быть получены действием аммиака на эфиры хлоругольной
кислоты:
О=С<
+ NH3 . О=С< + НС1
Cl xNHa
Уретаны—кристаллические вещества. Некоторые из них
применяются в медицине как снотворные и болеутоляющие средства.
Мочевина (карбамид) CO(NH2J является главнейшим конечным
продуктом превращения белковых веществ в организме
млекопитающих. Она содержится поэтому в большом количестве в моче
человека и млекопитающих животных (в моче человека около 2%
мочевины). Человек выделяет в сутки около 20—30 г мочевины.
Синтетически мочевина была впервые получена в 1828 г. Ве-
лером* выпариванием водного раствора циановокислого аммония.
Циановокислый аммоний изомеризуется гьри этом в мочевину;
реакция обратима:
NH4OCN ¦; " CO(NH2J
Строение мочевины видно из ее синтеза из фосгена и аммиака:
ІСІ'+Ні—NH, ,NH,
! I < "
ІСІ+Ні—NH, ,NH,
С<! I . О=С< "+2НС1
jCI + Hi-NHa XNH2
О
* Фридрих Вёлер (F. Wohler) —немецкий химик A800—1882), ученик
Я. Берцелиуса. Среди многочисленных работ Вёлера в области органической
химии отметим: синтез мочевины A828), получение и исследование
производных бензоила (совместно с Ю. Либихом, 1832), а также мочевой кислоты.
206. Производные угольной кислоты, содержащие азот ^ 1 о
В промышленном масштабе мочевину, применяемую как
удобрение и как добавка к кормам для скота, получают нагреванием
смеси аммиака и углекислого газа под давлением:
СО, + 2NH3 . CO(NH2J + Н3О
Мочевина—твердое кристаллическое вещество;
кристаллизуется в виде плоских призм, похожих на кристаллы калийной
селитры; плавится при 133 °С; очень хорошо растворима в воде.
Недавно было открыто неожиданное свойство мочевины: в присутствии
небольшого количества метилового спирта кристаллы мочевины способны
адсорбировать парафиновые углеводороды, алкоголи, галоидпроизводные,
содержащие неразветвленные углеводородные цепи. Соединения же с разветвленными
цепями не поглощаются мочевиной. Причина этого явления заключается в
своеобразном строении кристаллов мочевины, образующих «кристаллические
поры», столь узкие, что в них не могут проникнуть углеводороды с
разветвленными цепями. В настоящее время этим путем на практике выделяют
нормальные парафины из нефтяных продуктов после отжимания жидкой фазы,
содержащей изопарафины, кристаллы обрабатывают водой, которая переводит
мочевину в водный раствор, а углеводород отделяется
Будучи амидом, мочевина образует соли с кислотами, но в
реакцию солеобразования вступает только одна аминогруппа:
NH, /NHS
О=с/ + HNO3 » О=С<
XNH2 N
+ 3<
NH2 NNH3-HNO
Азотнокислая соль мочевины трудно растворима в воде.
Как и все амиды, мочевина при нагревании с растворами
щелочей или кислот расщепляется с образованием кислоты и
аммиака:
a + Н:—О—Н Г -ОН
I
o + НІ-О-Н
Г /ОН]
2NH. 4- О=С< » 2NH3 + СО2 + Н2О
L он]
Подобное расщепление мочевины происходит и при действии
уреазы—энзима, находящегося в почках млекопитающих и в
семенах многих растений, например в соевых бобах.
С азотистой кислотой мочевина реагирует подобно другим
амидам, давая азот и кислоту;
/N;H„ O=:N^OH Г ,ОН1
О=С<: і '+ І і , 2Na + 2H„O + р=С<Г »
NN;H2 O=!N:-OH |_ 4OhJ
. 2N, + 3H,0 + СО,
416 Глава XVI!. Производные угольной кислоты
Мочевина может реагировать с кислотами, хлорангидридами,
ангидридами и сложными эфирами. При действии на мочевину
хлористого ацетила атом водорода аминной группы замещается
ацетилом и образуется ацетилмочевина СН3СО—NHCONH2
(темп, плавл. 217 °С). При нагревании с малоновой кислотой
мочевина дает барбитуровую кислоту (кристаллы):
/NH—ІН НОІ—ОСЧ /NH—СО
О=С< І + і >СН2 , О=С >СН, + 2Н2О
NsiH-iH НОІ-ОС/ \KIU /п
При нагревании выше температуры плавления мочевина
выделяет аммиак и переходит в биурет:
NH2—CO—jNH2 + Н !— NH—СО—NH2 . NH2CO—NH—CONH2 + NH3
Биурет плавится при 190 °С. В щелочном растворе биурет дает с
медным купоросом фиолетово-красное окрашивание.
Семикарбазид. При действии на мочевину концентрированной
азотной кислоты в присутствии концентрированной серной
кислоты образуется нитромочевина:
NH2—CO-NH-ІН'Т"НО:—NO2 . NH2CONH-NO2 + Н2О
Нитромочевина—кристаллическое вещество, плавится с
разложением при 150 °С.
При электролитическом восстановлении нитромочевина
переходит в семикарбазид:
NH2CONH—NO2 + ЗН2 ¦ NH2CONH—NH2 + 2Н2О
2CONH—NH2 + 2Н2
Семикарбазид—кристаллическое вещество, плавится при 96 °С.
Он образует соли с одним эквивалентом кислоты.
Реагируя с альдегидами и кетонами, семикарбазид дает се-
микарбазоны:
р : ті- р
р р
\С=ІО+ INn—NH-CONH2 , Nc=N-NHCONH2 + Н2О
Семикарбазоны хорошо кристаллизуются; их применяют для
открытия альдегидов и кетонов, а также для их отделения от
д у г их веществ.
206. Производные угольной кис юты, содержащие азот 417
[уанидин образуется при действии аммиака на эфиры орто-
угольной кислоты по схеме:
Ж QftOL /ЮСаНв Hi-NH, ,NH2
NH<i + :>C<: + і , NH=C< + 4C2H5OH
N;H CM-fi/ NOQHs Hj-NH, \NH2
Его можно рассматривать как производное угольной кислоты,
в молекуле которой два гидроксила замещены аминогруппами,
а карбонильный кислород—группой NH:
/ОН
О
/ОН ,N
=С< HN=C<
NOH ^N
угольная кислота гуаннднн
Гуанидин—бесцветное кристаллическое вещество. Он очень
гигроскопичен, поглощает из воздуха углекислый газ, образует
соли с одним эквивалентом кислоты и обладает сильными
щелочными свойствами.
Из производных гуанидина большое физиологическое
значение имеют аргинин и креатин.
Аргинин
NH2—С— NH—СН3СН2СН2—СН—СООН
II I
NH NH2
можно рассматривать как производное а-аминовалериа новой
кислоты, в молекуле которой атом водорода у a-углеродного
атома замещен остатком гуанидина. Аргинин—это а-амино-&-гуани-
дилвалериановая кислота—один из главнейших продуктов
гидролиза многих белковых веществ; в ростках растений встречается
в свободном виде.
При нагревании со щелочами аргинин расщепляется на
мочевину и орнитин:
NH2—C-j-NH—СН2СН2СН2—СН—СООН + Н-рО—Н »
NH NH2
аргинин
, О=с/ 2+NH2-CH2CH2CH2-CH(NH2)—СООН
XNH2
мочевина . орнитии
Подобное расщепление происходит и при действии аргиназы—
энзима, находящегося в печени.
27—788
418 Глава XVII. Производные угольной кислоты
Креатин
NH2
СН2—СООН
NH2
HN=C<
4N—СН2—
можно рассматривать как производное уксусной кислоты, в
молекуле которой атом водорода замещен остатком
метилированного при азоте гуанидина. Креатин—это N-метилгуанидилуксусная
кислота. Креатин находится в мышечном соке позвоночных; он
плавится при 100 °С и имеет горький вкус. При нагревании с
разбавленными кислотами креатин, отщепляя воду, дает креатинин:
NH—;н
HN=C<
N—сн.2—со—;он
сн.
,NH— СО
HN=C< I +H20
\N CH,
креатинин
Креатинин находится в мышечном соке и моче животных, а также
в растениях.
207. Сернистые аналоги производных угольной кислоты. Если
в молекуле угольной кислоты и ее производных атомы
кислорода заменить атомами серы, то получаются соответствующие
сернистые аналоги. Среди них наиболее важными являются:
/ОН ,NHS
s=c=s s=c< s=c<
NSH ^NHj
сероуглерод днтиоугольная кислота тиомочевина
(ксаитогеновая кислота)
Растворы серы в сероуглероде применяются для так называемой
«холодной» вулканизации каучука.
Ксантогеновая кислота. При взаимодействии сероуглерода
со спиртами в присутствии едкой щелочи или с алкоголятами
происходит образование эфиров солей дитиоугольной кислоты (ксан-
тогенатов, или ксантатов):
S-<- Na SNa
S—C** OC2Hg S=C—OC2H^
Производные ксантогеновой кислоты имеют желтую окраску.
Свободная кислота очень неустойчива и легко разлагается.
207. Сернистые аналоги производных угольной кислоты
419
воздух
— S=C-S-PbS
Как было указано раньше (см. стр. 352), ксантогеновые эфи-
ры целлюлозы применяются в промышленности для получения
искусственного волокна (вискозный способ).
При действии ксантогенатов на соли тяжелых металлов
образуются труднорастворимые осадки.
Этил- и бутилксантогенаты щелочных металлов применяются при
обогащении руд тяжелых металлов (Си, Ni, Pb, Zn и др.)
посредством флотации. Сернистые руды металлов содержат смеси
силикатных пород (силикаты натрия, кальция, магния и др.) и
сернистых соединений тяжелых металлов: PbS (свинцовый блеск),
CuS (медный колчедан), ZnS (цинковая обманка) и др. Для
отделения сернистых соединений
от пустой породы руду
предварительно размалывают и
взмучивают в воде. Если теперь
прибавить к этой «пульпе»
небольшое количество
ксантогенатов, то молекулы их прочно
адсорбируются на поверхности
кристаллов руды (за счет ксанто-
геновой группы). Частицы
руды с адсорбированным на
поверхности ксантогенатом
обладают способностью
накапливаться на границе раздела в о-
д а—в о з д у х (рис. 57).
В флотационной машине к пульпе добавляют вспениватели
(сосновое масло, терпинеол и др.) и подвергают ее сильному
перемешиванию при продувании воздуха. Пузырьки воздуха
покрываются на своей поверхности частицами руды и всплывают на
поверхность воды. Непрерывно вращающиеся деревянные
лопатки флотационной машины отделяют эту пену с рудой от
суспензии пустой породы, поскольку силикатные минералы не имеют
сродства к ксантогенатам и не переходят в пену. Таким образом,
при помощи флотореагентов совершается парадоксальный процесс:
тяжелые минералы «всплывают», а более легкие—силикаты—
остаются внизу.
Применяя такую химическую обработку, можно путем
добавки небольших количеств флотореагентов производить очень
основательное обогащение даже бедных руд. Пользуясь
различными реагентами и химической обработкой, проводят частичное
разделение смесей руд: ¦ медно-железных, свинцово-цинковых
и др.
Тиомочевина NH2CSNH2 по большинству свойств является
полным аналогом мочевины. Впервые она была получена нагре-
Рис. 57. Схема расположения частиц
ксантогената на границе раздела
вода—воздух.
420 Глава XVI! Производные угольной кислоты
ванием роданистого аммония [подобно способу получения
мочевины из циановокислого аммония (см. стр. 414)]:
Ї у™ N
С » С—NH2 »
В настоящее время тиомочевину получают действием
сероводорода на цианамид:
її н //" /NH*
С + I > С—SH , C=S
Тиомочевина—кристаллы (темп, плавл. 180 °С); растворима в
воде. Применяется в фотографии (например, при получении тонов
сепии на отпечатках), для синтеза лекарственных препаратов.
Интересно отметить, что обработка клубней картофеля тиомоче-
виной вызывает прорастание их без предварительного перези-
мования.
208. Пластмассы на основе мочевино-формальдегидных (карб-
амидных) смол. Аминопласты. Мочевина, а также ее
производные—тиомочевина, дициандиамид и др. конденсируются с
формальдегидом и образуют смолообразные продукты—карб-
амидные смолы, из которых приготавливают пластмассы,
называемые аминопластами.
Чтобы иметь некоторое представление о строении сложных
молекул таких аминопластов, рассмотрим примерную схему
образования продуктов конденсации мочевины с формальдегидом.
Одной из первых ступеней этой сложной реакции является
образование диметилолмочевины:
/NHIH + НО!_СН2—ОН ,NH—CH,—ОН
О=С< ' ' , О=С< " + 2Н..0
ЧШ!Н + НО:—СН2—ОН Чш-СВ,—ОН
(гидратированная диметилолмочевина
форма СН-2О)
При нагревании диметилолмочевины с мочевиной в присутствии
кислых добавок (например, щавелевой кислоты) происходит
реакция поликонденсации с выделением воды:
HO-|-CH2NHCONHCH2 ;ОН + HI NH CONHJH + НО: C
. » CHaNHCONHCH2—NHCONH—CHoNHCONHCH, +nH2O
208. Пластмассы на основе мочевино-формальдегидных смол 421
Кроме того, молекулы диметилолмочевины могут вступать между
собой в реакции пол и конденсации по схеме:
HjN—СН2—|ОН + HjN—СН2—
1) •'' I
СО СО
I I
HjN—СН2—ІОН + HjN—СН2—
ОН + H:N—СН2—|ОН + HIN—CH2—ОН -\
¦' I '' * I
со со
I I
ІОН" + HIN—СН2—|0н" + h]n— СН2—ОН + • • •
Присутствующие в смеси молекулы формальдегида могут
«сшивать» между собой линейные молекулы:
• • .СНа—N—СО—NH—СНа—NH—СО—NH—СНа—N—СО—NH—СНа. • •
М !Н!
!о:=сн2 |ОІ=сн2
jHj ;ні
• • .СН„—N—CO—NH—CHa—NH—CO—NH—СНа—N—CO—NH—CH.J- ¦ •
При этом образуется сложная смесь линейных и
пространственных структур.
В зависимости от условий проведения процесса получаются
разнообразного характера смолы, находящие очень широкое
применение в технике.
Карбамидные смолы бесцветны и легко окрашиваются в
любые цвета. Они широко используются для склейки и пропитки
Древесины, для декоративных целей, для изготовления предметов
Широкого потребления (лампы, плафоны, телефонные трубки,
посуда) и в производстве лаков. Из мочевинных смол получают
также легкий пористый материал—мипору, применяемый для
тепло- и звукоизоляции.
Вместо мочевины может применяться тиомочевина, а вместо
формальдегида—другие альдегиды.
ЧАСТЬ ВТОРАЯ
КАРБОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
СОЕДИНЕНИЯ АРОМАТИЧЕСКОГО РЯДА
Соединениями ароматического ряда называют вещества, в
молекулах которых содержится особая группировка из шести
атомов^глерода, называемая бензольным ядром или
бензольным кольцом^ ~"
С
II I
—С С—
ГЛАВА XVШ
ОДНОЯДЕРНЫЕ АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ
БЕНЗОЛ И ЕГО ГОМОЛОГИ
209., Свойства бензола. Главным представителем
ароматических соединений является бензол С6Н6. Бензол—бесцветная
подвижная жидкость (относительная плотность df =0,879); темп,
кип. 80,1 °С; имеет своеобразный запах, не растворяется в воде,
легко воспламеняется и горит ярким коптящим пламенем.
Охлажденный бензол застывает в белую кристаллическую массу,
плавящуюся при 5,5 °С.
Бензол находит обширное применение в технике как сырье
для получения ряда ценных органических веществ и как
растворитель. Большие количества его расходуются в качестве
добавок к жидкому моторному топливу.
Интересна история открытия бензола. В 1812—1815 гг. в Лойдоне впервые
появилось газовое освещение. Светильный газ, добывавшийся из жира
морских животных, доставлялся в железных баллонах. Эти баллоны помещались
обычно в подвале дома; из них газ по трубкам распределялся по всему
помещению. Вскоре было замечено крайне неприятное обстоятельство—в сильные
холода газ терял способность давать при горении яркий свет. Владельцы
газового завода в 1825 г. обратились за советом к Фарадею, который нашел,
209. Свойства бензола 423
что те составные части газа, которые способны гореть ярким пламенем,
собираются на дне баллона в виде прозрачного жидкого слоя. При исследовании
этой жидкости Фарадей* открыл новый углеводород—бензол.
Бензол, его производные и вещества, сходные с ними по
строению, относятся к ряду так называемых ароматических
соединений. Напомним также, что соединения, содержащие открытую
углеродную цепь, относятся к соединениям «жирного ряда». Так
как эти названия могут повести к неправильным представлениям,
необходимо объяснить возникновение этих терминов.
Первые вещества из группы органических кислот, спиртов,
эфиров были тесно связаны с продуктами, получаемыми из
жиров. Значительно поздней было установлено, что эти соединения
содержат незамкнутые цепи углеродных атомов. Поэтому за
соединениями ациклического строения закрепилось название
«соединения жирного ряда».
Производные бензола были впервые выделены из
ароматических смол (бензойная смола, росный ладан и др.). В дальнейшем
выяснилось, что молекулы таких соединений имеют кольчатые
структуры, подобные структуре бензола. Поэтому такие
соединения стали объединять в ряд ароматических соединений.
Особенности ароматических соединений. Бензол является
первым представителем ароматических углеводородов. Необходимо
прежде всего рассмотреть ряд своеобразных свойств бензола,
отличающих его от изученных ранее предельных и непредельных
ациклических (жирных) углеводородов. Мы говорим о так
называемом «ароматическом характере» бензола, который
проявляется в химических свойствах и определяется его химическим
строением,
По составу бензол и его гомологи являются непредельными
углеводородами. Состав бензола выражается формулой С6Н6.
Общая формула гомологов ряда_^ензала_С„Н2„_я^_у
Сравнивая эту формулу с формулой ряда~"прёдельных
углеводородов С„Н2Л+2, легко видеть, что разность между ними равна 8Н.
Следовательно, по химическому составу бензол является сильно
непредельным соединением. Однако непредельный характер
бензола не проявляется в типичных реакциях. Мы вправе были бы
ожидать, что бензол будет вести себя подобно этилену, дивинилу
и другим типичным непредельным углеводородам. Однако ока-
* Михаил Фарадей (М. Faraday), знаменитый английский физик и химик
A791—1867). Родился в Лондоне в семье кузнецз, В юности работал
переплетчиком. Наиболее известны его исследования в области электричества.
Из работ по органической химии укажем: открытие бензола в светильном газе,
открытие бутилена, получение а- н ?-нафталинсульфокислот.
424 Глава XVIП. Одноядерные ароматические соединения
зывается, что он не обесцвечивает бромную воду, т. е. в обычных
условиях не присоединяет бром. Раствор марганцовокислого
калия при взбалтывании с бензолом не обесцвечивается, т. е.
бензол устойчив в этих условиях к окислению. Даже при
длительном кипячении с раствором КМпО4 бензол почти не окисляется.
Для бензола характерны реакции замещения. Бензол
отличается по свойствам от непредельных соединений: он легче вступает
в реакции замещения, чем в реакции соединения.
В присутствии катализаторов (FeCl3, A1C13) хлор и бром не
присоединяются к бензолу, но замещают атомы водорода в его
молекуле:
С6Не + Вг2 _*Шгз> НВг + С6Н5Вг J^> НВг + СвН4Вг2
Эта реакция была открыта в 1877 г. Г. Г. Густавсоном*.
Концентрированная серная кислота не вызывает
полимеризации бензола, а приводит к получению бензолсульфокислоты
С6Н6 + HaSO, * Н2О + CgHsSOaH
Нитрующая смесь (конц. HNO3+H2SO4) производит
нитрование:
С6Н6 + HNO3 , Н2О + C6H5NOo
Далее мы укажем еще на ряд реакций замещения, в которых
бензол и его производные ведут себя как насыщенные соединения.
Вместе с тем в особых условиях бензол полностью проявляет
непредельный характер. Можно указать на различные реакции
присоединения.
Реакции присоединения к бензолу. Гидрирование, т. е.
присоединение водорода, происходит при действии газообразного
водорода в присутствии порошков некоторых металлов (Ni, Pt, Pd).
* Гавриил Гавриилович Густавсон A842—1908)—выдающийся русский
химик. Первые работы Г. Г. Густавсона были проведены под руководством
Д. И. Менделеева и относились к реакциям обмена безводных солей металлов.
Им было открыто каталитическое действие галоидных солей алюминия на
некоторые реакции ароматических соединений. Это открытие было позднее
применено Фриделем и Крафтсом во Франции к известной реакции алкилирования
ароматических соединений. Механизм этой реакции подробно изучил
Густавсон. Он же открыл общую реакцию синтеза циклопропановых углеводородов
отщеплением цинком двух атомов брома от дибромидов:
^СНгВг /сн3
СН2 + Zn * ZnBr2 + СН2|
210. Строение бензола 425
При этом молекула бензола присоединяет 3 молекулы водорода:
100—150 °С
QH6 + ЗН2 — *- С6Н12
Если раствор хлора или брома в бензоле подвергнуть
действию света, богатого фиолетовыми и ультрафиолетовыми лучами
(прямой солнечный свет, кварцевая лампа), то происходит
быстрое присоединение также 3 молекул галоида:
С6Н6 + ЗС12 -—• С6Н6С16
Таким образом, по реакциям присоединения в бензоле
обнаруживается три двойные связи.
Реакции окисления бензола. Если пары бензола пропускать
вместе с кислородом при повышенной температуре D00 °С) над
некоторыми катализаторами (V2O5), то бензол подвергается
окислению с образованием углекислого газа и малеинового ангидрида:
НС—СО
С6Н6 + 4,5О2 , 2СО2 + 2Н2О + ^>О
НС—СО
Молекула бензола присоединяет три молекулы озона.
Следовательно, в этой реакции бензол тоже ведет себя так, как будто
в его молекуле имеется три двойные связи.
Таким образом, ароматический характер бензола выражается
в том, что это соединение по составу является непредельным,
однако в целом ряде химических реакций проявляет себя как
предельное соединение; для него характерна химическая
устойчивость, трудность реакций присоединения. В особых условиях
(катализаторы, облучение) бензол ведет себя так, как будто в
его молекуле имеются три двойные связи. Причина своеобразия
свойств бензола кроется в химическом строении его молекулы.
210. Строение бензола. При замещении в молекуле бензола
одного атома водорода бромом получается только один моно-
бромбензол С6Н5Вг. Точно так же известен только один
монохлорбензол. Это общая закономерность: у однозамещенных
производных бензола нет изомеров. Следовательно, все атомы
водорода в бензоле равноценны, и безразлично, какой из них
замещается.
С другой стороны, продукты замещения в молекуле бензола
двух атомов водорода—двузамещенные производные бензола—
известны в виде трех изомеров (орто, мета, пара, обозначаемые
соответственно о-, м-, п-). Так, существуют три дибромбензола
С6Н4Вг2: о-дибромбензол (темп, плавл. -+-6,7 °С), ж-дибромбензол
(темп, плавл. —7 °С) и n-дибромбензол (темп, плавл. -+-87 °С).
426 Глава XVIII. Одноядерные ароматические соединения
Таким образом, структурная формула бензола должна
объяснять:
1) почему молекула бензола может присоединить только
шесть атомов галоида или водорода;
2) почему не существует изомеров для однозамешенных
производных бензола;
3) почему двузамещенные производные бензола известны в
виде трех изомеров.
Строение бензола чаще всего выражают формулой,
предложенной в 1865 г. Кекуле:*
СН
НС СН
II I
НС СН
СН
Согласно этой формуле, в молекуле бензола шесть атомов
углерода образуют замкнутую углеродную цепь—так называемое
бензольное кольцо, поэтому молекула бензола может
присоединить шесть атомов водорода или галоида. В то же время эта
формула показывает, что в молекуле бензола между атомами
углерода имеются три двойные связи.
В полном согласии с этим находится образование глиоксаля
при разложении водой озонида бензола:
I сГ^ J)
нсГ 1^9 нс
не' \/Ъ не.
-лиоксаль
* Фридрих-Август Кекуле (F. A. Kekule, 1829—1896). Родился в г. Дарм-
штадте Ученик Либиха. Профессор химии Гентского (Бельгия) и Боннского
университетов Основные его работы посвящены теоретической химии В 1854 г
он впервые высказал идею о «двухосновности», т е двухвалентности,
кислорода и серы. Углерод рассматривал как четырехатомный элемент. Разработал
теорию многоатомных радикалов, являющуюся распространением теории
типов Жерара В 1865 г. предложил циклическую формулу строения бензола с
чередующимися простыми и двойными связями
Кекуле получил тиоуксусную кислоту; в 1856 г. разработал метод
получения гликолевой кислоты кипячением хлоруксусной кислоты с водой. Получил
в 1872 г. т ифенилметан и антрахинон
210. Строение бензола 427
На основе формулы бензола, предложенной Кекуле,
объясняли также многие другие опытные данные, однако некоторые
факты объяснить было трудно. Так, например, согласно
формуле Кекуле, должны были бы существовать два ряда изомерных
дву замещенных производных бензола, например:
С—Br CH
НС С—Вг НС С-Вг
II I и (| |
НС СН НС С—Вг
В первой формуле оба атома брома стоят у углеродных
атомов, связанных между собой двойной связью, во второй
формуле—у атомов, связанных простой связью. Поскольку у
производных бензола изомерия подобного рода никогда не
наблюдалась, Кекуле выдвинул гипотезу о том, что в молекуле бензола
простая и двойная связи не закреплены, а могут перемещаться
(осцилл ировать) :
С—Вг С—Вг
НС С—Вг НС С—Вг
її І г—* І і!
НС СН НС СН
\ f \ /
СН СН
Эта гипотеза получила название осцилляционной гипотезы.
Ввиду простоты формулы Кекуле и ее практических удобств
химики продолжают ею
пользоваться до настоящего времени,
хотя ни сам факт чередования
простых и двойных связей в
бензольном кольце, ни гипоте- Lyi^« h,40A
за осцилляции не подтвержда- ^*\ ^*\
ются современной наукой. ^ \-^ \~ °—Н
Современные представления
о строении бензола. С помощью
современных физических мето- Рис 58- Строение молекулы бензола,
дов установлено, что молекула
бензола имеет следующее строение (рис. 58).
1. Атомы углерода образуют правильный плоский
шестиугольник со сторонами 1,40 А. Следовательно, длина С—С-связи в
бензоле меньше простой С—С-связи A,54 А) и больше типичной
двойной С=С-связи A,34 А).
428 Глаза XVIII. Одноядерные ароматические соединения
2. Все шесть атомов углерода и шесть атомов водорода
лежат в одной плоскости.
При рассмотрении электронного строения непредельных
соединений (см. стр. 243) было указано, что двойная связь слагается
из а- и it -связей. Электронные облака ic -связей значительно
более подвижны, чем облака а-связей. Соединения с двумя и
более сопряженными двойными связями (конъюгены) имеют эти
характерные облака и-связей у четырех (и более) стоящих
подряд атомов углерода и обладают рядом особых физических и
химических свойств (см. стр. 247). Бензол по своему строению
является резко выраженным конъюгеном. В дивиниле
наибольшая реакционная способность свойственна крайним
углеродным атомам; то же можно сказать и о гексатриене-1,3,5:
Бензол же можно представить себе как циклическую систему
гексатриена, вследствие чего у него нет особо реакционных
крайних атомов:
или иначе с
\
В действительности же в бензоле все шесть атомов
совершенно одинаковы и связаны между собой а- и ic -связями. Благодаря
полной симметрии все связи полностью выровнены, так что
нельзя сказать, какая пара С—С-атомов связана простой (о)
связью и какая—двойной (<з+іс) связью.
Каждый углеродный атом бензола имеет три а-связи, две из
которых направлены к соседним атомам углерода, а третья—
к атому водорода. Кроме того, каждый углеродный атом имеет
тс-связь, облако которой расположено перпендикулярно
плоскости кольца. Электронные облака ic -связей обладают большой
подвижностью. Такое строение бензола определяет его особые
(см. стр. 421) ароматические свойства.
Для простоты написания формулу бензола часто изображают
в виде шестиугольника, в каждой вершине которого
подразумевается группа СН:
211. Двузамещенные и трехзамещенные производные бензола 429
211. Двузамещенные и трехзамещенные производные бензола.
Изомерия. В молекуле бензола все атомы водорода
совершенно равноценны. Поэтому замещение любого из них,
например атомом брома, даст один и тот же бромбензол:
О-Вг
Если же в полученный бромбензол вводить второй атом
брома, то возможно образование трех различных веществ:
Вг Вг
I I .
V-Вг {\
—Вг
Вг
opmo-дибромбензол дата-дибромбензол пара-двбромбензол
Как видно из приведенных формул, изомерия получающихся
соединений объясняется различным расположением атомов
брома относительно друг друга.
Если в молекуле бензола замещены атомы водорода у
соседних атомов углерода, то такие соединения называются орпго-
изомерами.
Если в бензоле замещены атомы водорода у атомов углерода,
отделенных друг от друга одним атомом углерода, то такие
соединения называются мета-изомерами.
Пара-изомерами называются такие изомеры, в молекулах
которых замещающие группы стоят у атомов углерода,
расположенных в бензольном кольце через два атома углерода.
Для того чтобы указать местоположение заместителя, атомы
углерода в бензольном кольце нумеруют; счет начинают с одного
из тех атомов, при которых произошло замещение.
Формулы изомеров дибромбензола можно также выразить
следующим образом:
.Вг A) Вг A) /Вг A)
С.Н4< Со,Н4< QH4<
ХВг B) ХВг C) \Вг D)
1,2-диброыоензо.л і.^-діюиомбензол 1,4-дибромбензол
430 Глава XVHI. Одноядерные ароматические соединения
При замещении в молекуле бензола трех атомов водорода
одина новыми атомами или группами возможно
образование следующих изомеров:
Вг Вг Вг
^ ,_Br
I ^
Br
1,3.5- или 1.2.4 или 1,2,3- или
симметричный несимметричный смежный
трибромбензол триоромбенэол (рядоввП)
трибромбензол
Для четырехзамещенных производных бензола (с
одинаковыми заместителями) также возможны три изомера, для пяти- и
шестизамещенных производных бензола нет изомеров.
Таким образом, при последовательном замещении в молекуле
бензола от одного до шести атомов водорода хлором возможны
двенадцать хлорзамещенных бензола. Они были получены в
1875—1880 гг. Ф. Ф. Бейльштейном и А. И. Курбатовым.
212. Строение и свойства гомологов бензола. Замещением
в молекуле бензола атомов водорода алкилами можно получить
гомолог бензола, например:
I
А-СНа f\ f
lj U
СН3 СН, СН3
I Г I
ч/ V э
;н3
толуол о-ксилол «-ксилол -и-ксилол этилбензол п метилизопр^іпил-
бензол. или цимол
Бензольные углеводороды—бесцветные жидкости, легче
воды, сильно преломляющие лучи света; они имеют своеобразный
запах и не растворяются в воде (табл. 16).
Бензольные углеводороды по сравнению с предельными
углеводородами имеют большие относительные плотности и
показатели преломления. Так, например:
СНв df = 0,879 п о = 1 ¦ 5017
ССНЫ df = 0,681 4Q=>.3754
Группы атомов, которые можно получить из бензольных
углеводородов отнятием одного атома водорода (из числа атомов, не-
212. Строение и свойства гомологов бензола
431
Таблица 16
Физические свойства гомологов бензола
Название
Бензол
Толуол
о-Ксилол
«-Ксилол
я-Ксилол
Этилбензол . .
Цимол
Формула
С.Н,
С6Н6СН,
С6Н4(СН3)а
С6Н4(СН3J
СН3С6Н4СН(СНз)а
Температура
плавления
°С
+5,5
-95,1
—25,2
—47,9
+ 13,2
—95,0
—67,2
Температура
FMпения
°С
80,1
110,6
144,4
139
138,4
137
177
посредственно связанных с бензольным кольцом), носят
название ароматических радикалов, или арилов. Остаток бензола
СвН5 называется фенилом, остаток толуола СН3О0Н4—толи-
лом, остаток ксилола (СН3JСаН3—ксилилом. Двухвалентный
радикал С„Н4 называется фениленом.
Из рассмотрения формул строения бензольных углеводородов
видно, что в молекулах этих веществ надо различать, с одной
стороны, бензольное ядро (кольцо), а с другой—связанный с ним
углеводородный радикал жирного ряда, который составляет
боковую цепь. Так, в толуоле боковой цепью является метил, в
этилбензоле—этил, в цимоле находятся две боковые цепи:
метил ьная и изопропильная. Такие углеводороды называются жир-
ноароматическими.
В соединениях жирноароматического ряда бензольное ядро
сохраняет особенности, присущие бензолу. Так, например,
оно отличается большой устойчивостью к действию
окислителей.
При действии на жирноароматические углеводороды сильных
окислителей (например, марганцовокислого калия) происходит
окисление боковой цепи, причем ближайший к ядру атом
углерода остается связанным с ядром и образует карбоксильную
группу:
СОа + 2НаО
этилбензол
бензойная кислота
Следует заметить, что в кристалле алмаза, а равно и в
соединениях жи ного ряда, валентные связи атомов углерода направ-
432
Глава XV///. Одноядерные ароматические соединения
лены к вершинам тетраэдра (стр. 158), в графите же (рис. с>9)
атомы углерода располагаются в параллельных плоскостях,
причем в каждой плоскости атомы находятся в вершинах
правильных шестиугольников.
ЬА
Рис 59. Кристаллическая решетка
графита.
213. Способы получения гомологов бензола. 1. Синтез Фат-
тага осуществляется действием натрия на смесь бромбензола и
галоидпроизводного предельного углеводорода:
Hs -f 2NaBr
бромистый
этил
этилбенэол
Для получения этилбензола в колбу с обратным холодильником (рис 60)
помещают 27 г нарезанного тонкими полосками натрия и наливают 200 мл
должным образом просушенного эфира. Через несколько часов, после того,
как натрий окончательно высушит эфир, через трубку холодильника
приливают 60 г бромистого этила и 60 г бромбензола и оставляют смесь
стоять до другого дня Если смесь закипит, колбу охлаждают водой На
следующий день отгоняют сначала эфир, а затем полученный этилбензол Для
получения при помощи этой реакции некоторых углеводородов приходится
нагревать смесь на водяной или масляной бане
Как видно из уравнения, синтез Фиттига вполне сходен с
синтезом предельных углеводородов Вюрца (см. стр. 51). При
помощи этого синтеза было выяснено строение ряда
ароматических соединений—толуола, этилбензола, ксилола и др.
2. Синтез Фриделя—Крафтса. При нагревании смеси
бензола и хлорироизвоДного предельного углеводорода в присутст-
213. Способы получения гомологов бензола
433
вии безводного хлористого алюминия происходит замещение атома
водорода в молекуле бензола углеводородным радикалом и
выделение хлористого водорода:
/
бензол
_H + С1-СН3
HCI
хлористый
метил
твлуол
Эта реакция была открыта в 1877 г. французским химиком
Фриделем и работавшим вместе с ним американским химиком
Крафтсом. Толчком к их исследованиям послужили работы
ученика А. М. Бутлерова—Г. Г. Густавсона.
3. Восстановление ароматических кетонов (стр. 469).
4. Полимеризация непредельных соединений жирного ряда.
Как сам бензол, так и некоторые бензольные углеводороды
могут быть получены из соединений жирного ряда. Так, при
нагревании ацетилена до 550—600 °С последний,
как это показал Бертло в 1866 г.,
превращается в бензол:
СН СН
^ / Ч
НС СН
НС СН
III
НС СН
СН
НС СН
Сущность реакции заключается в том,
что в молекулах ацетилена разрывается по
одной связи в группе С=С, причем
освободившиеся валентные связи одной молекулы
ацетилена насыщаются освободившимися
связями другой молекулы. Таким образом,
из трех молекул ацетилена получается
одна молекула бензола.
Н. Д. Зелинский и Б. А. Казанский
показали, что эта реакция идет очень хорошо
при пропускании ацетилена через нагретый
до 600—650 °С активированный уголь.
5. Синтез из солей ароматических
кислот. При нагревании сухих солей
ароматических кислот с едким натром
происходит разложение солей с образованием углеводородов.
СвН6—COONa + NaOH , QHe + NaaCU:,
Этот способ аналогичен способу получения углеводородов
жирного ряда.
28-788
Рис. 60. Прибор
для лабораторного
получения
гомологов бензола.
434 Глава XVШ. Одноядерные ароматические соединения
214. Промышленное получение ароматических соединений.
Основным источником получения бензола и других
ароматических углеводородов до середины XX века являлись
каменноугольная смола и коксовый газ, получаемые при сухой перегонке
каменного угля. В связи с быстрым ростом химической
промышленности, происходившим одновременно с развитием
нефтеперерабатывающей промышленности, в последние годы
ароматические углеводороды во все возрастающих количествах получаются
также из продуктов переработки нефти.
Сухая перегонка каменного угля осуществляется на
коксохимических заводах с целью получения металлургического кокса.
В этом процессе одновременно образуются: коксовый газ,
каменноугольная смола, аммиачная вода и кокс. Количество
каменноугольной смолы, образующейся при коксовании, составляет
3—6% от массы каменного угля. Она содержит большое число
различных ароматических углеводородов.
Содержание самого бензола в каменноугольной смоле
невелико и составляет всего 0,05—0,1%. Основное количество бензола
извлекается из коксового газа путем абсорбции высококипящими
фракциями каменноугольной смолы (тяжелое масло). Сырой
коксовый газ содержит 25—35 г/м:> смеси ароматических
углеводородов примерно следующего состава: 70—80% бензола, 16—20%
толуола, 5% ксилолов и 2% прочих соединений. Образовавшийся
при сухой перегонке коксовый газ пропускают через ряд
холодильников для отделения каменноугольной смолы, а затем через
орошаемые водой скрубберы для поглощения содержащегося
в нем аммиака. Освобожденный от смолы и аммиака газ подается
на абсорберы для извлечения ароматических углеводородов.
Абсорбированные ароматические углеводороды отделяются от
масла отгонкой, после чего очищаются серной кислотой или
гидрированием под давлением (для освобождения от сернистых и
непредельных соединений). Выделение индивидуальных
углеводородов из полученного сырого бензола производится
дистилляцией.
Каменноугольную смолу подвергают перегонке, разделяя
обычно на следующие фракции:
1) Легкое масло, кипящее при температуре до 170 °С. Содержит
бензол и его гомологи. Количество этой фракции не превышает
3% от массы смолы.
2) Среднее масло, кипящее в интервале 170—2Э0 °С. Содержит
фенол, нафталин, пиридиновые основания.
3) Тяжелое масло, кипящее в интервале 230—270 °С.
Содержит крезолы, ксиленолы, хинолин, многоядерные
углеводороды, а также некоторое количество фенола, нафталина и
антрацена.
2L. Промышленное получение ароматических соединений 435
4) Антраценовое масло, кипящее в интервале 270—340 °С.
Для понижения температуры кипения часто перегоняется под
вакуумом. Содержит антрацен, фенантрен, карбазол и
многоядерные углеводороды.
После перегонки каменноугольной смолы остается густая
масса—пек, применяемый для приготовления лаков, для дорожных
покрытий и как пропиточный материал (например, для
шпал).
Извлечение индивидуальных соединений из полученных
фракций каменноугольной смолы достигается сочетанием методов
ректификации и кристаллизации. Общий выход ароматических
углеводородов, получаемых при сухой перегонке, составляет
примерно 1% на взятый уголь.
Для обеспечения быстро развивающегося производства
синтетических материалов ресурсов коксохимической
промышленности по ароматическим углеводородам оказалось уже
недостаточно. Кроме того, переработка нефтяных углеводородов
позволяет осуществлять более направленные промышленные процессы,
приводящие к получению желаемых соединений, тогда как
сухая перегонка каменного угля предоставляет в этом отношении
значительно меньше возможностей.
Нефти некоторых месторождений (Чусовские Городки на
Урале) содержат около 60% различных ароматических
углеводородов, однако в нефтях главных месторождений СССР
ароматических углеводородов значительно меньше и для их получения
необходимо проведение процессов химической переработки.
Ароматические углеводороды могут быть получены из нефти
или отдельных нефтяных фракций путем пиролиза, т. е.
нагревания до высоких температур G00 °С и выше). Этот способ впервые
был практически осуществлен еще в 70-х годах прошлого
столетия инж. А. А. Летним, который построил в г. Баку завод для
переработки тяжелых нефтяных остатков с целью получения из
них бензина, керосина и ароматических углеводородов. В
настоящее время пиролиз нефтепродуктов осуществляется в
крупных промышленных масштабах для получения этилена и
пропилена; образующиеся в этом процессе побочные жидкие продукты
могут служить вспомогательным источником получения
ароматических углеводородов. Некоторое количество бензола и его
гомологов образуется в процессе каталитического крекинга.
Основным способом получения ароматических углеводородов
из нефтепродуктов являются процессы каталитической переработки,
впервые разработанные Н. Д. Зелинским, Б. А. Казанским и
другими советскими учеными.
В зависимости от исходного нефтяного сырья и желаемых
конечных продуктов процесс может проводиться на различных ката-
28*
436 Глава XVНІ. Одноядерные ароматические соединения
лизаторах и при различных условиях. Наиболее часто применяется
в качестве катализатора платина, нанесенная на окись алюминия,
отчего этот процесс получил название плашформинга. Обычно
он проводится при температуре около 500 °С и давлении 20—30 am.
Содержащиеся в исходном нефтяном сырье нафтеновые и
парафиновые углеводороды подвергаются при этом сложным
химическим превращениям, приводящим к образованию
ароматических углеводородов.
Нафтеновые углеводороды или дегидрируются:
СНа
НаС С
I I —»II +
нас
или дегидрируются с одновременным расширением кольца:
сна
•\
с
1
с
на
1
н
н
сна
1 1
аС СН2
\ /
сн.
Парафиновые углеводороды подвергаются дегидроциклизации
т. е. отщеплению водорода с одновременным замыканием кольца):
СН3—СН2—СНа—CHj—CHj—СНа—СНз * ^Г** — СН3+4Н„
Обычно в продуктах реакции содержится до 50%
ароматических углеводородов. Их отделяют от парафиновых и других
углеводородов экстракцией растворителями и затем очищают
ректификацией.
В промышленном масштабе процессы каталитической
ароматизации нефтепродуктов впервые были осуществлены для
повышения качества бензинов и получили очень широкое
распространение. Значение этого процесса для облагораживания моторного
топлива видно из следующего примера: октановое число толуола
120, т. е. почти в 3 раза выше, чем октановое число 2-метилгек-
сана, содержащего то же число углеродных атомов в молекуле,
октановое число которого 43.
В настоящее время процессы каталитической ароматизации
широко используются также для получения бензола и других
ароматических углеводородов.
215. Получение галоидпроизводных бензольных углеводородов 437
ГАЛОИДПРОИЗВОДНЫЕ БЕНЗОЛА И ЕГО ГОМОЛОГОВ
215. Получение галоидпроизводных бензольных
углеводородов. Хлор- и бромпроизводные бензольных углеводородов
получаются обычно непосредственным действием хлора или брома на
углеводороды. Бензол в присутствии катализаторов легко
реагирует с хлором или бромом. Подбирая условия реакции и
катализаторы, можно последовательно все атомы водорода, связанные
с бензольным кольцом, заместить галоидом.
На рис. 61 изображен прибор для лабораторного получения
бромбензола. Для проведения реакции в колбу помещают
немного крупных железных опилок. Приливают бензол и постепенно
Рис. 61. Прибор для получения бромбензола.
при помешивании добавляют бром. Вначале брома добавляют
немного. После того как начнется выделение бромистого
водорода, количество добавляемого брома увеличивают. Скорость
введения брома должна быть такова, чтобы реакция не шла
слишком бурно. В этой реакции катализатором является
получающееся бромное железо FeBr3.
Прямое введение иода в ароматические соединения
представляет большие трудности, так как образующийся при реакции
йодистый водород частично восстанавливает иодбензол в
исходное вещество:
СЙНЙ -+- J., . HJ -
438
Глава XVIII. Одноядерные ароматические соединения
Для получения иодбензола прямым путем надо нагревать в
запаянной трубке смесь бензола, иода и йодноватой кислоты:
йодноватая кислота окисляет йодистый водород до свободного иода.
Гомологи бензола хлорируются и бромируются легче, чем
бензол. При этом галоид может или вступить в боковую цепь или
заместить водород бензольного кольца. Так, при введении хлора
в боковую цепь толуола получается хлористый бензил СвН6СН2С1,
названный так по радикалу бензилу СвН6СН2—.
Если же хлор замещает водород, связанный с ядром, то
получается хлортолуол (смесь орто- и пара-изомеров):
хлористый
бензил
о-хлортолуол
n-хлортолуол
Как показал еще в 1886 г. Ф. Ф. Бейльштейн, хлорирование
толуола при низкой температуре в присутствии катализаторов
(J2) ведет к образованию хлорзамещенных в ядре; при
повышенной температуре хлор направляется в боковую цепь.
Если действовать на углеводород галоидом на прямом
солнечном свету, галоид замещает атомы водорода исключительно
в боковой цепи.
Определить положение галоида можно путем окисления
исследуемого вещества. Так, хлористый бензил дает при окислении
бензойную кислоту, хлортолуол—хлорбензойную кислоту:
хлористый бензил бензойная кислота
о-хлортолуол
о-хызрбензопная
кислота
216. Свойства галоидпроизводных бензольных углеводородов
439
216. Свойства галоидпроизводных бензольных углеводородов.
Галоидпроизводные бензольных углеводородов—жидкости или
твердые тела (табл. 17).
Таблица 17
Физические свойства некоторых галоидпроизводных бензольных углеводородов
Название
Формула
СвН5С1
С„Н5Вг
CeH5J
СбН5СН2С1
СвН6СН,Вг
СвН5СНС12
С„Н5СС1з
С6Н4(СН3)С1
СХ1в
Температура
плавления
°С
—45
—30,6
—31,4
-39
-3.9
-17.4
-4.8
—36,5
+229
Температура
кипения
°С
132
156,2
188,7
179
198
205.3
220
159,2
322
Хлорбензол
Бромбензол
Иодбензол
Хлористый бензил . . .
Бромистый бензил . . .
Хлористый бензилиден
Бензотрихлорид .. . .
о-Хлортолуол
Гексахлорбензол . . .
Вещества, в которых галоид находится у а-углеродного атома
боковой цепи, имеют резкий, раздражающий запах. Некоторые и$
них, например бромистый бензил С„НьСН.>Вг, применялись в
первую мировую войну 1914—1918 гг. как слезоточивые вещества.
Галоид у а-углеродного атома боковой цепи обладает
большой подвижностью и легко замещается гидроксилом, цианом
и др., т. е. ведет себя так же, как и в галоидпроизводных жирного
ряда. Так, при действии едкого натра на хлористый бензил
получается бензиловый спирт:
<_/—CH2—Cl -t- Na-OH
хлористый бензнл
—СНа—ОН + NaCl
бензнловый спирт
Соединения, в ядре которых содержится галоид, имеют
слабый приятный запах. Галоид в этих соединениях связан с ядром
весьма прочно. Он обладает малой подвижностью и замешается
только в особых условиях. Так, например, в молекуле
хлорбензола хлор замещается гидроксилом лишь при нагревании
хлорбензола с раствором щелочи до 300—350 °С обычно в присутствии
меди. В этом отношении вещества, содержащие галоид,
связанный непосредственно с атомом углерода кольца, подобны галоид-
производным непредельных углеводородов с галоидом при
углероде с двойной связью, галоид которых также малоподвижен.
440 Глава XVII1. Одноядерные ароматические соединения
217. Некоторые представители галоидпроизводных
бензольных углеводородов. Хлорбензол—жидкость, кипяшая при 132 °С;
готовится в больших количествах и применяется как
промежуточный продукт для синтеза многих органических веществ,
например для получения фенола и
динитрохлорбензола—полупродукта, используемого при синтезе красителя—сернистого
черного.
Широкое применение для борьбы с вредными насекомыми
(в качестве инсектицида*) нашел препарат ДДТ, или п,п'-ди-
хлордифенилтрихлорметилметан.
ДДТ получают конденсацией хлорбензола с хлоралем в
присутствии концентрированной, или дымящей, серной кислоты:
и
\ п H2S°l
СН
СС13
^~^-а + Н2О
CCI,
Он представляет собой белые кристаллы.
Сильнейшим инсектицидом является также гексахлорцикг
логексан С6НвС1,, {«гексахлоран» или «666»), который получается
при присоединении хлора к бензолу на солнечном свету или
при освещении ртутными лампами:
QH6 + 3CJ2 QH6C16
НИТРОСОЕДИНЕНИЯ И СУЛЬФОКИСЛОТИ
218. Нитрование. Характерной особенностью ароматических
соединений является их способность легко давать нитросоедине-
ния при действии азотной кислоты.
В то время как предельные углеводороды нитруются лишь
с трудом, ароматические углеводороды легко нитруются
концентрированной азотной кислотой или, чаще, смесью
концентрированных азотной и серной кислот. Последняя служит для
связывания воды, образующейся при реакции.
* Лат.—убивающий насекомых.
219. Направляющее действие заместителей 44!
Реакция между бензолом и азотной кислотой выражается
уравнением:
нитробензол
Как видно из уравнения, от бензольного ядра отщепляется
атом водорода, а от молекулы азотной кислоты—гидроксиль-
ная группа.
В результате реакции получается нитробензол, в котором атом
углерода бензольного ядра соединен с атомом азота. Эта реакция
была открыта в 1834 г. Митчерлихом.
Аналогичным образом нитруются и другие ароматические
соединения: углеводороды, фенолы, кислоты и т. д.
При известных условиях в бензольное ядро можно ввести
не одну, а две и три нитрогруппы.
Реакция идет последовательно: сначала получается
нитробензол, из него—динитробензол:
NO2 NO3
) -
бензол нитробензол jw-динитробензол
219. Направляющее действие заместителей. При нитровании
толуола образуются изомерные мононитросоединения—о- и и-ни-
тротолуолы:
О
толуол
о-нитротолуол
+HNO3
NOa
п-нитротолуол
442 Глава XVIII. Одноядерные ароматические соединения
Что же касается нитробензола, то при его нитровании
образуется почти исключительно jn-динитробензол, а при бромирова-
нии—.м-бромнитробензол:
NO2 NOa NOa NOa
О —6ч 0 —6ч
нитробензол й-динитробензол м-бромннтробензол
Эти примеры показывают, что, в зависимости от природы
первой замещающей группы, вторая замещающая группа вступает
в различные положения относительно первой группы:
находящиеся в ядре группы направляют (ориентируют) второй
заместитель.
Имеются два типа заместителей. Заместители первого рода
ориентируют вторую группу в орто- и пара-положение, причем
обыкновенно образуются оба изсмера. Мета-изомер при этом или
вовсе не образуется, или образуется в небольшом количестве.
Заместители второго рода ориентируют вторую группу
исключительно или почти исключительно в мета-положение.
Заместителями (или ориентантами) первого рода являются
следующие группы, расположенные в порядке убывания
направляющей силы: —ОН, —NH2, СІ, Br, J, —CH3 н другие алкилы.
К заместителям второго рода относятся следующие группы:
,0 .О
-NO2 , —SO3H -с/ —с/ -C=N
Во многих заместителях второго рода имеются двойные или
тройные связи.
Образование различных количеств орто-, мета- и
пара-изомеров объясняется тем, что три конкурирующие реакции, именно
орто-, мета- и пара-замещения, протекают одновременно, но с
разными скоростями.
Правила замещения в бензольном ядре имеют очень большое
значение, так как знание их дает возможность предсказать ход
реакции и выбрать правильный путь синтеза того или другого
нужного вещества. В качестве примера направляющего действия
заместителей можно привести образование различных изомеров
нитрохлорбензола. Для получения нитропроизводного
хлорбензола можно или сначала хлорировать бензол и- затем полученный
хлорбензол нитровать, или же сначала нитровать бензол и затем
полученный нитробензол подвергнуть хлорированию. Хлор—за-
219. Направляющее действие заместителей
443
меститель первого рода, поэтому в результате нитрования
хлорбензола получаются о- и tt-нитрохлорбензолы. Нитрогруппа,
наоборот—заместитель второго рода, поэтому хлорирование
нитробензола дает ж-нитрохлорбензол. Таким образом, изменяя
порядок проведения реакции, можно получить все три изомера
нитрохлорбензола:
а
СІ
Замещающие группы не только направляют (ориентируют)
второй заместитель, но и оказывают влияние на подвижность
атомов водорода, связанных с бензольным кольцом. Заместители
второго рода уменьшают подвижность этих атомов, а
заместители первого рода усиливают ее. Так, гомологи бензола,
например толуол и ксилол, нитруются легче, чем сам бензол.
Правила ориентации с точки зрения электронных
представлений. Как было указано выше (стр. 428), электронные облака
я-связей обладают большой подвижностью; поэтому электронная
плотность одинакова у всех атомов только у незамещенного
бензола СвН6. При вступлении заместителей (например СІ, ОН, NO2)
происходит значительное перераспределение электронной
плотности в зависимости от рода заместителей.
Обозначим условно замещающую группу:
А —атом, непосредственно связанный с бензольным ядром;
Вп—остальные атомы* замещающей группы.
Заместители первого рода А В„(—ОН,—NH2, —СІ, —Вг, —J,
—СН3) характеризуются тем, что в них, как правило, атом А,
связанный с бензольным ядром, более электроотрицателен, чем
атом (или группа атомов) Вл.
* В случае одноатомных заместителей В =0.
444
Главо XVIII Одноядерные ароматические соединения
Свободные (неподеленные) пары электронов этих атомов (А)
увеличивают электронную плотность бензольного ядра
преимущественно в орто- и пара-положениях. Изогнутые стрелки
показывают направление электронного смещения и атомы углерода
бензольного ядра, приобретающие повышенную электронную
плотность (рис. 62).
Рис 62. Схема сдвига
электронных плотностей
в молекуле бензола под
влиянием заместителей
первого рода.
Рис 63. Схема сдвига
электронных плотностей
в молекуле бензола под
влиянием заместителей
второго рода.
Заместители второго рода—А Вп (—СО2Н, —SO3H, —NO2)
характеризуются тем, что в них, как правило, атом А, связанный
с бензольным ядром, более электроположителен, чем атом (или
группа атомов) В„. Заместители второго рода, притягивая к себе
электронные облака, снижают электронную плотность в ядре
бензола, причем особенно сильно в орто- и пара-положениях
(рис. 63). Поэтому электронная плотность у атомов в
мета-положениях относительно больше, чем в орто- и пара-положениях.
Важнейшие реакции замещения в ароматическом ряду, такие,
как галоидирование, нитрование, сульфирование, реакция Фри-
деля—Крафтса и др., относятся к так называемым реакциям
электрофильного* замещения, т. е. вступающие группы**
[С1(С1), NO2(OH); SO3H(OH); CH3CO(C1)) имеют
электроположительный характер и направляются в первую очередь к атомам,
имеющим повышенную электронную плотность. В бензольном
ядре, как мы видели из рис. 62, повышенная электронная
плотность при наличии заместителей первого рода создается в орто-
и пара-положениях, а при наличии заместителей второго рода
(рис. 63)—в мета-положениях. Отсюда вытекают правила
ориентации для электрофильных заместителей.
* По гречески «филео»—любить.
** В круглую скобку взята часть молекулы, отщепляющаяся при реакции
замещения.
919. Направляющее действие заместителей
445
1. Место вхождения вступающих заместителей определяется
природой ориентанта (атома или группы, находящейся в ядре)
и не зависит от природы входящей группы (так как, будучи
электрофильными заместителями, все они несут положительный
заряд).
2. Ориентанты первой группы, увеличивая электронную плот
ность бензольного ядра, усиливают его реакционную способность
и направляют заместители в орто- и пара-положения.
Действительно, толуол, фенол, анилин легче сульфируются, нитруются,
галоидируются, чем сам бензол. При этих реакциях получаются
по преимуществу орто- и пара-изомеры.
3. Ориентанты второй группы, оттягивая на себя
электронные облака бензола, уменьшают реакционную способность
бензольного ядра; они допускают вступление групп преимущественно
в мета-положение. Такие производные бензола, как нитробензол,
бензолсульфокислота, бензойная кислота, более устойчивы к
химическим воздействиям, чем сам бензол (и особенно фенол,
анилин и др.); при реакциях с электрофильными заместителями
получаются преимущественно мета-изомеры.
Необходимо отметить, что эти правила не имеют абсолютного
значения. В большинстве реакций, наряду с преобладающим
количеством основного продукта (или основных продуктов)
реакции (согласно правилу), образуются и другие изомеры, но в
значительно меньших количествах. Ориентирующее влияние
заместителей видно из табл. 18,
Таблица 18
Ориентирующее влияние заместителей
Имеющийся
заместитель
в положении і
он
NH2
сн3
SO3H
соон
Положение входящего заместителя
CI
4, 2
4, B)
4, 2
3
3, 2, D)
Вг
4,2
4,2
3 -
3
3
SO3H
2, 4, C)
2, 4
4, 2, C)
3, D)
3, B), D)
3, D)
NO-,
2, 4
4, C), B)
2, 4, C)
3, B), D)
3, B), D)
3, 2, D)
* Цифрами, взятыми в скобки, обозначены изомеры, образующиеся лишь
в очень небольших количествах.
Относительные количества получающихся изомеров в
значительной мере зависят также и от условий реакции (температура,
концентрация реагентов в реакционной смеси и др.).
446
Глава XVIII. Одноядерные ароматические соединения
220. Свойства нитросоединении. Нитросоединения
ароматического ряда в большинстве случаев—кристаллические вещества
желтого цвета. Некоторые их константы приведены в табл. 19.
Таблица 19
Физические свойства
Название
Нитробензол
.и-Динитробензо.л
о-Нитротолуол
«-Нитротолуол
.«-Нитротолуол
2,4-Динитротолуол
2,4|6-Тринитротолуол
ароматических нитросоединении
Формула
C0H6NO2
C«H4(NO2K
CHaCsH4NOo
CH8CeH4NO2
CHsQHjNOj
CH,C,Ha(NOa)a
CH8CeH2(NO2)8
Температуру'
плавления
'С
+- 5,7
+89,6
-3,2
-t-51,6
+ 15,5
+69.6
+80,1
Температура
кипения
210,9
.102
222,3
238
232,6
300
Укажем следующие основные химические свойства
ароматических нитросоединении:
1. Нитросоединения восстанавливаются до первичных аминов:
C«H6NO2 + ЗН2
2Н.р
2. В молекулах нитросоединении замещение атома водорода
происходит в мета-положении по отношению к нитрогруппе.
Так, например, при действии на нитробензол дымящей серной
кислоты получается лі-нитробензолсульфокислота:
NO,
NO,
О
1 +so3
^
-SOaH
Но эти реакции с нитросоединениями идут гораздо труднее, чем
с углеводородами,— нигпрогруппа уменьшает посважность
водородных атомов бензольного ядра. Так, нитробензол C6H5NO2
образуется при взбалтывании бензола со смесью азотной и
серной кислот. Для получения диниіробензола C6H4(NO2J
требуется нагревание и применяется дымящая азотная кислота.
Введение же в молекулу бензола третьей ншрогруппы
осуществляется с очень большим трудом.
221. Отдельные представители нитросоединений
447
'Л. Галоид, связанный с атомом углерода бензольного ядра,
отличается малой подвижностью, и замещение его происходит
с большим трудом. Если же, как это было впервые отмечено
А. Н. Энгельгардтом и П. А. Лачиновым A870 г.), ввести нитро-
группу в орто- или пара-положение по отношению к галоиду,
то последний приобретает значительную подвижность. Так, у
о- и n-нитрохлорбензолов хлор обменивается на гидроксил при
нагревании до 100 °С с раствором едкого натра:
/NO2
о- хлорннтробензол
-ОН
NaCl
9-НИТрофЄНОЛ
4. Нитрогруппа, введенная в молекулу фенола или карбоно-
вой кислоты, усиливает кислотные свойства вещества. Так,
константа диссоциации о-нитробензойной
кислоты в тысячу раз больше константы
диссоциации бензойной кислоты.
Таким образом, введение нитрогруппы
оказывает сильное влияние на
реакционную способность вещества; оно в
значительной мере изменяет свойства атомов
водорода бензольного ядра и их
заместителей. Это становится понятным,
если вспомнить, что нитрогруппа,
являясь заместителем второго рода,
оттягивает на себя электроны и понижает
электроннув) плотность бензольного
ядра.
221. Отдельные представители
нитросоединений. Нитробензол CeH3NO2—
желтоватая жидкость, плотность 1,203
гГсм* (при 20 °С); имеет запах,
напоминающий запах горького миндаля; Рис 64 Нитратор (раз
ядовит, особенно в виде паров. Нитро- рез).
бензол готовится в очень больших
количествах и перерабатывается главным образом на анилин (см.
стр. 487).
Нитрование производится в больших, обыкновенно чугунных
котлах, называемых нитраторами (рис. 64). Нитратор снабжен
мешалкой. Для охлаждения реагирующей смеси служит
внутренний холодильник. Охлаждение может быть достигнуто и при
помощи воды, протекающей между двойными стенками нитра-
тора.
448 Глава XVIIІ. Одноядерные ароматические соединения
Тринитротолуол, тротил, тол, или ТНТ, CH3CeH2(NO2K
получается нитрованием толуола. Сначала образуются о- и гс-нит-
ротолуолы, затем—динитротолуолы и наконец—тротил:
СН3 СН3
NO2 02N ' ,N02
СН„
^У Ч
сн3 сн3 Ч.
N0a
,N0, NC
Ч/1 Ч>"
S\ r^V " '5°а
N0
2
Тротил—желтые иглы (темп, плавл. 80, 1 °С). Он обладает
сильными взрывчатыми свойствами и применяется для снаряжения
боеприпасов. Тротил стали использовать как взрывчатое
вещество с 1905 г., хотя он был открыт еще в 1863 г.
222. Сульфирование, сульфокислоти. Столь же характерной
для ароматических соединений реакцией, как и нитрование,
является действие концентрированной серной кислоты.
Реакция между бензолом и серной кислотой выражается
уравнением:
—ІН ± Щ—s°i—он ^=^ \У~S°2°H
бензолсульфокислота
Как видно из уравнения, от бензольного ядра отщепляется
один атом водорода, а от молекулы серной кислоты—водный
остаток. Освободившиеся валентности атомов углерода и серы
взаимно насыщают друг друга. В результате реакции получается
бензолсульфокислота, в которой атом углерода бензольного
кольца соединен с атомом серы.
Введение в молекулу сульфогруппы действием серной кислоты
называется сульфированием. Реакция сульфирования обратима,
поэтому для того, чтобы по возможности весь бензол вошел в
реакцию, следует брать большой избыток концентрированной
серной кислоты. Для получения дисульфокислот применяют
дымящую серную кислоту (олеум).
В технике процесс непрерывного сульфирования бензола
проводят, пропуская его пары через 100%-ную серную кислоту,
помещенную в специальный сульфуратор; образующаяся вода
уносится избытком парообразного бензола.
222. Сульфирование, сульфокислоти 449
Сульфокислота—бесцветные кристаллические вещества, легко
растворимые в воде. В отличие от карбонових кислот,
сульфокислоти являются сильными кислотами, так как в них кислотные
свойства обусловлены присутствием остатков сильной (серной)
кислоты. Натриевые соли сульфокислот обычно хорошо1
растворимы в воде, но малорастворимы в насыщенном растворе
поваренной соли.
В реакцию сульфирования вступают и другие ароматические
соединения—углеводороды, кислоты и т. д. Так, при
сульфировании толуола получаются о- и /г-толуолсульфокислоты.
При сульфировании в более жестких условиях (повышение
температуры, применение серной кислоты в виде олеума
различных концентраций) в бензольное ядро можно ввести не одну, а
две или три сульфогруппы. Таким путем, например, получается
л«-бензолдисульфокислота, служащая исходным продуктом для
получения резорцина (см. стр. 458).
СН3
6
о
ІС
ЗО2ОН SO2OH
о-толуолсульфокислота п-толуолсульфокислота м-бензолдисульфокислота
По окончании сульфирования в реакционной массе обычно
находится значительное количество избыточной серной кислоты.
В технике процесс отделения серной кислоты от продуктов
сульфирования называется известкованием. Реакционную смесь
разбавляют водой, нейтрализуют известью и фильтруют. На
фильтре остается сульфат кальция, а бензолсульфокислый кальций
(CeHbSO3JCa как легко растворимая соль переходит в фильтрат.
К полученному фильтрату прибавляют соду и снова раствор
отфильтровывают от выпавшего углекислого кальция:
(C6H5SO8).2Ca + Na2CO3 . гс^БО^а + СаСО3
бензолсульфо-
кнслый натрий
Фильтрат, содержащий растворимую натриевую соль бензол-
сульфокислоты, выпаривают досуха, и в кристаллическом виде
получают бензолсульфокислый натрий.
Характерным свойством сульфокислот является способность
замещать сульфогруппу на гидроксил, водород, нитрильную
29—788
450 Глава XVIII. Одноядерные ароматические соединения
группу. Так, например, при сплавлении натриевой соли
сульфокислота со щелочами получается фенол:
.................. ЧЛП °С
С,Н6—;SO3Na + Na jOH + NaOH > Q,H5—ONa + Na^SO,
QH6—ONa + SOa +Нз°> QH6-OH + NaHSO,
фенол
Эта реакция служит для синтеза многих оксисоединений
ароматического ряда.
При действии на сульфокислоти перегретого водяного пара
происходит отщепление сульфогруппы и образование
ароматического углеводорода:
НО!—Н » QH, +
Сульфокислота ароматических углеводородов имеют большое
значение для получения ряда важных соединений (фенолов,
ароматических кислот и др.). Кроме того, сульфогруппа часто
вводится в молекулу для того, чтобы сделать соединение
растворимым в воде. Бензолсульфокислота и толуолсульфокислота иногда
употребляются как катализаторы (например, при этерификации)
вместо серной кислоты, вызывающей нежелательные побочные
процессы.
ФЕНОЛЫ И АРОМАТИЧЕСКИЕ СПИРТЫ. ХИНОНЫ
223. Строение, способы получения и свойства фенолов. Мы
выяснили различие между галоидпроизводными, содержащими
галоид, связанный с бензольным ядром, и соединениями с
галоидом в боковой цепи. Точно так же существует значительное
различие между веществами, в которых гидроксил связан с ядром,
и веществами с гидроксилом, находящимся в боковой цепи.
Соединения, содержащие гидроксил, который связан с
атомом углерода бензольного ряда, называются оксисоединениями или
фенолами. Простейший из фенолов—гидроксильное производное
бензола—называется фенолом, или карболовой кислотой:
Синтетически фенолы получают:
1. Сплавлением солей сульфокислот со щелочами.
2. Разложением диазониевых солей в определенных условиях
(см. стр. 505).
223. Строение, способы получения и свойства фенолов 45!
3. Обработкой галоидпроизводных бензольных углеводородов
растворами щелочей при высокой температуре.
Фенолы обладают слабокислотными свойствами. Если спирты
дают алкоголяты только при действии на них свободного металла,
то водород гидроксильной группы фенолов замещается не только
при действии металла, но и при действии на фенолы едких
щелочей. Получающиеся при этом вещества называются фенолятами:
СН5ОН + КОН z=l CeH5OK ¦+¦ НаО
фенолят
калия
Так как феноляты растворимы в воде, то фенолы, будучи
сами в воде нерастворимы, растворяются при действии на них
щелочей, в то время как нерастворимые в воде спирты не
растворяются и в щелочах. Однако фенолы обладают очень слабыми
кислотными свойствами, и феноляты разлагаются
разбавленными кислотами с выделением свободного фенола:
СвН5ОК + НС) » СвН5ОН + KCI
Даже угольная кислота разлагает феноляты с выделением
свободного фенола:
CeH6ONa + СОа + НаО » NaHCO3 + С„Н6ОН
Радикал фенил и гидроксильная группа оказывают друг на
друга известное влияние: под влиянием фенила гидроксильная
группа приобретает кислотные свойства. Кислотные свойства
фенольного гидроксила особенно усиливаются под влиянием
отрицательных групп, например —NO2. Нитрофенолы являются уже
более сильными кислотами (см. стр. 457), чем фенол и его
гомологи. С другой стороны, под влиянием гидроксильной группы
атомы водорода в бензольном ядре приобретают большую
подвижность и большую способность к реакциям замещения. Так,
для замещения водорода в бензоле бромом приходится
применять катализаторы, водород же бензольного ядра в феноле
замещается бромом весьма легко Даже при действии на феноль
бромной воды:
ОН ОН
А ._ Вг\А/Вг
о
| + ЗВга , IJ + ЗНВг
Y
Вг
трибромфенол
29*
452 Глава XVIII. Одноядерные ароматические соединения
Отметим при этом, что атомы галоида вступают в пара- и орто-
положения по отношению к ОН-группе, являющейся сильным
ориентантом первого рода.
Фенол нитруется и сульфируется легче, чем бензол. Та« же,
как и в предыдущей реакции, нитро- и сульфогруппы входят в
пара- и орто-положение по отношению к ОН-группе. Так, при
действии на фенол разбавленной азотной кислотой получается
смесь /г-нитрофенола и о-нитрофенола. о-Нитрофенол легко
может быть выделен из смеси перегонкой с водяным паром (см.
стр. 23). Пара-изомер с водяным паром не перегоняется.
Известны сложные эфиры фенолов, например С6Н5О—СОСН3.
Они могут быть получены действием на фенолы хлорангидридов
или ангидридов кислот:
CgH5—о—|Н+сії—ос—сна —» CdH5—о—со—снэ + на
Простые эфиры фенолов получаются действием галоидных
алкилов на феноляты щелочных металлов:
CH3-<
п-крезолят натрия бромистый аллвл
* СН3—^=^—О—СНа—СН=СН2 + NaBr
аллиловый эфир п-крезола
Для получения метиловых и этиловых эфиров обычно
применяют диметил- и диэтилсульфат:
CdH5ONa + (CH3O)aSOa > С6Н5—О—СНЯ + NaO—SOa—OCH3
анизол соль метилсерной
кислоты
Гидроксил фенолов, в отличие от гидроксила спиртов, с
трудом замещается галоидом.
С хлорным железом растворы большинства фенолов дают
характерные окрашивания.
224. Фенол. Крезолы. Фенол С6Н6ОН—кристаллическое
бесцветное вещество, которое плавится при 42,3 °С и кипит при 182 °С;
на воздухе, вследствие окисления, он принимает сначала
розовую, а затем бурую окраску. Фенол обладает характерным и
очень сильным запахом; он плохо растворим в воде. С водой
образует кристаллогидрат С6Н5ОН-Н3О, плавящийся при 16 °С.
При приливаний к фенолу воды образуются два слоя: нижний
слой—раствор воды в феноле, верхний—раствор фенола в воде.
При повышении температуры увеличивается растворимость как
СНз
</~Х_сн -^
сн3
изопропилбензол
сн3
^~^>—С—О—С
1
гидроперекись
225 Регуляторы роста растений и гербициды 453
воды в феноле, так и фенола в воде, и при 68 °С они растворяются
друг в друге в любых пропорциях. С хлорным железом фенол
дает фиолетовое окрашивание. Фенол находится в
каменноугольной, древесной и торфяной смоле. Количество фенола,
добываемого из смолы, только частично удовлетворяет потребность в нем,
поэтому большая часть фенола получается в настоящее время
синтетическим путем. Из синтетических способов важнейшие—
сплавление натриевой соли бензолсульфокислоты с едким натром
и нагревание до 350 °С хлорбензола с водой и известью. По
новому способу фенол получается путем окисления изопропил-
бензола (кумола) с последующим разложением образующейся
гидроперекиси; при этом образуется также ацетон:
H2SO4 Л N
> ^ ^_ОН + СН3—СО—СН3
фенол ацетон
Фенол является важнейшим техническим полупродуктом. Он
идет для приготовления пластических масс, пикриновой кислоты,
салициловых препаратов, красителей и др. Эфиры
фенола—анизол СвН5ОСН3 (темп. кип. 153,8 °С) и фенетол СвН5ОС2Н6 (темп,
кип. 172 °С)—приятно пахнущие жидкости, нерастворимые в воде
и водных щелочах. Фенол—продукт нормального обмена веществ
в организме, и в виде солей кислых эфиров серной кислоты,
например C6H5OSO2ONa, находится в моче.
Ближайшими гомологами фенола являются крезолы
СН3С(.Н4ОН—гидроксильные производные толуола. Их получают
из каменноугольной смолы, п-Крезол является одним из
продуктов гниения белков.
Все фенолы обладают сильным бактерицидным действием
даже в очень слабых растворах. Поэтому фенол и крезолы
широко применяются в медицине и ветеринарии для
обеззараживания. В чистом состоянии и в концентрированных растворах они
вызывают труднозаживающие ожоги кожи. Очень часто
применяется для дезинфекции лизол,—мыльный раствор смеси о-, м-
и /г-крезолов, выделяемой из каменноугольной смолы и из
продуктов сухой перегонки дерева и торфа.
225. Регуляторы роста растений и гербициды. Многие
вещества даже в очень малых количествах оказывают сильное
воздействие на растения. Формы этого воздействия весьма
разнообразны. Важное значение имеют вещества, которые
значительно ускоряют прорастание семян и рост корней. Такие вещества
«азываются обычно регуляторами роста растений. Из природных
454 Глава XVIII Одноядерные ароматические соединения
веществ этого типа укажем индол илу ксусную кислоту,
гетероауксин (см. стр. 596). Очень большое практическое применение
получили производные феноксиуксусной кислоты. Они могут
быть получены общим способом: действием соответствующих
фенолятов натрия на натриевую соль хлоруксусной кислоты:
QH6ONa + СІ—СН2—COONa » С6Н6О—СН2—СООН
феноксиуксусная
кислота
ОСН2СООН ОСНаСООН ОСНаСООН
1 С1 J
Практическое значение имеют:
ОСНоСООН ОСН
(Xа аХУ" <Г
сі сі а
2,4дихлорфеноксн- 2.4,5-трнхлорфенокси- 2-метил-4-хлорфеноксв-
уксусная кислота уксусная кислота уксусная кислота
B.4-Д) B,4,5-Т) BМ4Х)
При действии очень слабых растворов @,001%) этих веществ
на корневую систему, а также и на листья, наблюдается
значительное ускорение роста и более энергичное развитие всего
растения.
Отметим, что эти же вещества B,4-Д и др.) с успехом
применяются для укоренения черенков древесных растений. При
опрыскивании ими завязей цветов плодовых и огородных растений
получают больший выход плодов, причем созревание, например
помидоров, ускоряется и получаются плоды без косточек.
Если же применять эти вещества в больших концентрациях,
то вместо усиления развития растений наблюдается его
угнетение вплоть до полной гибели. Наиболее чувствительны к
повышению концентрации двудольные растения (многие из сорняков),
тогда как однодольные растения (злаки) устойчивы по отношению
к этим веществам. Таким образом, можно использовать эти
препараты в качестве гербицидов, т. е. производить «химическую
прополку» пшеницы, ржи и других злаков. Для этой цели посевы
подвергают опрыскиванию (обычно с самолета.). Так как стоимость
самих веществ невысока, а обработка не требует большой затраты
рабочей силы, то химическая-прополка оказывается очень
мощным практическим средством борьбы с сорняками.
226. Феноло-формальдегидные смолы. В 70-х годах
прошлого века установили, что фенол может вступать в реакцию
конденсации с муравьиным альдегидом, образуя
смолоподобные вещества. В 1902 г., т. е. через тридцать лет, появились
226. Феноло-формальдегидные смолы
455
первые искусственные смолы, получившие практическое
применение.
Эти смолы были получены реакцией поликонденсации
фенола с формалином путем нагревания в присутствии кислот:
H(CeH3(OH)CHaj„CeH4OH
(где я=4, 5...8). Они оказались прекрасно растворимыми в
спирте и под названием «новолаки» быстро вошли в употребление.
В настоящее время нагреванием фенолов с альдегидами
получают весьма разнообразные смолы. Одни из них растворимы в
спирте, другие—в маслах.
Структурная формула новолака на основе фенола и
формальдегида:
ОН
ОН
Формула новолака на основе я-крезола и ацетальдегида:
ОН
сн3
он
сн3
Таким образом, новолаки представляют собой продукты
поликонденсации и имеют линейное строение.
В 1907 г. были открыты отверждающиеся смолы, т. е. такие,
которые при нагревании превращаются в твердые, неплавкие
и практически ни в каких растворителях не растворяющиеся
пластические массы. Примером последних является бакелит. Его
готовят, нагревая фенол с формалином в присутствии аммиака.
Через некоторое время выпадает густая масса, которая затем
превращается в смолу (резол), размягчающуюся при 50—60 °С.
При дальнейшем, более сильном, нагревании она переходит в
твердое неплавкое и нерастворимое вещество (резит). Общая
схема образования резола и резита может быть представлена
следующим образом.
456
Глава XVIII. Одноядерные ароматические соединения
Вначале фенол и формальдегид реагируют в присутствии
щелочного катализатора с образованием смеси фенолоспиртов:
он
I
ОН
+ сн2о
сн2он
он
I
ч
2СН2О
он
I сь
СН2ОН
он
I
он
НОСНоч і-
ЗСН2О
СН2ОН
ч
СН2ОН
Далее молекулы фенолоспиртов конденсируются с
выделением воды, образуя линейные молекулы резола:
ОН
ОН
ОН
он
СН.ОН
сн,он
СН2ОН
7
СНоОН
и т. д.
Затем идет конденсация резольных молекул с образованием
твердого полимера—резита:
ОН
он
I
сн,
он
он
он
сн.
Чх
сн,
01!
он
227. Пикриновая кислота 457
Резит имеет сетчатую, пространственную структуру.
Новолачные смолы могут быть превращены в резольные
смолы и резит при добавлении к ним, при нагревании,
формальдегида или гексаметилентетрамина (уротропина).
Феноло-формальдегидные смолы и пластические массы на их
основе, так называемые фенопласты, явились первым типом
пластических масс, получившим широкое распространение. Из
них изготавливают многочисленные электротехнические и
радиотехнические детали, телефонные аппараты и трубки, детали машин,
шестерни, предметы домашнего обихода. Важное применение
имеют эти смолы в качестве связующего вещества при
изготовлении древесно-волокнистых плит и фанеры. Несмотря на
появление новых видов пластических масс, феноло-формальдегидные
смолы благодаря доступности, дешевизне и универсальности
свойств не утратили и в настоящее время своего значения и
продолжают изготавливаться в больших количествах.
227. Пикриновая кислота. Пикриновая кислота B,4,6-тринит-
рофенол)
ОН
' NO,
NO,
получается нитрованием фенола. Она представляет собой твердое
кристаллическое вещество; плавится при 122 °С, имеет горький
вкус; в прежнее время служила для окрашивания шерсти и
шелка в желтый цвет.
Пикриновая кислота обладает сильными кислотными
свойствами. Водород гидроксильной группы в этой кислоте может быть
замещен металлом, в результате чего получаются соли
пикриновой кислоты, называемые пикр~атами:
QH2(NO2KONa {QH2(NO2KO]2Ca
пикрат натрия пикрат кальция
Пикриновая кислота является взрывчатым веществом и под
названиями мелинита, лиддита и шимозы применялась для
снаряжения артиллерийских снарядов.
Отрицательным свойством пикриновой кислоты как
взрывчатого вещества является то, что ее соли, особенно пикрат
свинца, взрываются от совершенно незначительных причин, что
делает производство пикриновой кислоты и снаряжение ею
боеприпасов весьма опасным.
458 Глава XV111. Одноядерные ароматические соединения
Пикриновая кислота является сырьем для получения
хлорпикрина, который образуется при действии хлора на щелочной
раствор пикриновой кислоты:
QH2(OH)(NO2K+ ПС12 + 5Н2О ¦ 3CC13NO2 + I3HC! + ЗСО,
Хлорпикрин — жидкость (темп. кип. 112 °С); пары ее сильно
слезоточивы и ядовиты.
228. Двухатомные фенолы. Простейшими из двухатомных
фенолов являются двугидроксильные производные бензола,
которые называются диоксибензолами СбН4(ОНJ.
Известны все три возможные по теории строения изомера:
n-диоксибензол (гидрохинон), .и-диоксибензол (резорцин) и
о-диоксибензол (пирокатехин):
ОН ОН
1\ I Ч,ОН
О СХ0Н СХ0Н
он
гидрохинон резорцин пирокатехин
Гидрохинон получается восстановлением хинона (см. стр: 461);
применяется в фотографии как проявитель.
Резорцин получается сплавлением .и-бензолдисульфокислоты
с едким натром:
.SOjONa A) /ОНA)
QH<< + 2NaOH » QH/ + 2Na2SO8
\SOsONa C) XOH C)
Он применяется для получения некоторых видов пластиче«
ских масс, синтетических красителей и лекарственных
препаратов.
Пирокатехин был впервые получен сухой перегонкой
различных видов тропического растения катеху; синтетически он
получается нагреванием до 196 °С смеси о-хлорфенола с
разбавленным раствором едкого натра и небольшим количеством медного
купороса.
228. Двухатомные фенолы 459
Монометиловый эфир пирокатехина называется гваяколом; он
содержится в большом количестве в буковом дегте. Гваякол
и калиевая соль его сульфокислот (тиокол) применяются в
медицине при заболеваниях дыхательных путей:
он
rV
гваякол
ОСНз
ОН
1
f\
і І
К)
sos
/ОСНз
он
1
^Х/ОСНз
и I
А/1
SO3K
Двухатомные фенолы—кристаллические вещества. Они дают
с хлорным железом яркое окрашивание: пирокатехин—зеленое,
резорцин—фиолетовое, гидрохинон—грязно-зеленое,
переходящее, вследствие окисления при дальнейшем прибавлении
реактива, в желтое. Двухатомные фенолы легко окисляются в
щелочном растворе кислородом воздуха. При окислении их
азотнокислым серебром выделяется металлическое серебро. Труднее
Других окисляется резорцин.
Антиоксиданти. Многие алкилфенолы нашли широкое
применение в качестве отрицательных катализаторов для реакций
окисления. Такие вещества получили название антиоксидантов или
ингибиторов окисления. Практическое применение нашли
следующие соединения:
СНз 0Н °Н
СНз L .ОН
сн
3 L. I
НзС—С—СНз
СНз
2,4-диметил-б-трет-бу п-трет- бутилпи-
тилфенол. Ингибитор рокатехин. Инги-
окисления бензинов битор
полимеризации дивинила
(вызываемой
окислением при хранении)
460
Глава XVIII. Одноядерные ароматические соединения
229. Хиноны. При окислении гидрохинона образуются
золотисто-желтые кристаллы хинона С6Н4О2:
НС
НС
Ч\
0-Н
1
1
', ч
\
СН
II
СН
чс
+ 0
vJ -П
гидрохинон
о
с
/\
НС СН
І! 1!
* НС СН
\/
с
II
II
0
хиной
н2о
Как видно из приведенной формулы, хинон является дикетоном;
кроме того, в его кольце нет характерного для бензола
чередования двойных связей.
Такое строение хинона подтверждается тем, что к одной его
молекуле присоединяются четыре атома брома или хлора, а
также и тем, что с гидроксиламином хинон образует диоксим:
О
Н
1С СН
НС СН
\/
с
о
+ 2NH3—ОН
N-OH
II
С
НС СН
ІІ II
НС СН
у
N—ОН
хннондиоиснм
2Н3О
Система двойных связей, которая имеется в молекуле хинона,
называется хиноидной группировкой:
хнноидная группировка
Наличие хиноидной группировки характерно для многих
красителей.
Хинон был открыт в 1838 г. А. А. Воскресенским. Он имеет
резкий запах, плавится при 116 °С и легко возгоняется. Его
получают окислением анилина.
230. Трехатомние фенолы 461
При восстановлении хинон легко переходит в гидрохинон:
О ОН
с
/\
НС СН
II II + 2Н <
НС СН
\/
с
II
0
кинон
/
НС
1!
НС
\
с
' \
СН
СН
с
он
гидрохинон
Хинон обладает способностью образовывать с фенолами ярко
окрашенные соединения. Соединение молекулы хинона с
молекулой гидрохинона носит название хингидрона. Хингидрон имеет
вид красивых темно-зеленых кристаллов.
Другие хиноны. Описанный выше хинон правильнее назвать
л-бензохиноном. Существует ряд других хинонов, в которых
атомы кислорода находятся в пара- или орто-положении. Так, при
осторожном окислении пирокатехина получается о-бензохинон:
СН СН
НС С—О—ІЇЇ 1 НС С=О
II | І + О ! < | | + HSO
НС С—О—|Н ! НС С=О
\ / ; : \ /
СН СН
пирокатехин о-?ензохинон
Наибольшее значение имеют л-хиноны. Они окрашены в
золотисто-желтый цвет, обладают своеобразным резким запахом и
перегоняются с водяным паром. ж-Хиноны не существуют, что
вполне согласуется со строением бензола.
230. Трехатомные фенолы. Известны все три возможных по
теории строения триоксибензола: смежный пирогаллол,
симметричный—флороглюцин и несимметричный—оксигидрохинон:
ОН
гУ0Н
V-Чон
,2,3-триоксибензел
^пирогаллол!
он
Л
онААон
1,3,5-триоксябензол
(флороглюцин)
он
rV0H
и
он
(,2,4-тривксибензол
(оксигидрохинон)
Все они—кристаллические вещества.
462
Глава XVIII. Одноядерные ароматические соединения
Рис. 65. Прибор для
демонстрации поглощения
кислорода воздуха
пирогаллолом
Пирогаллол получается обычно нагреванием галловой
кислоты (см. стр. 482), от которой при этом отщепляется СО2.
Пирогаллол весьма быстро окисляется в щелочном растворе
кислородом воздуха. Для того чтобы показать легкую окисляемость
пирогаллола, в колбу насыпают небольшое количество его, приливают
раствор едкого натра и быстро закрывают отверстие колбы
пробкой, соединенной с трубкой, другой ко-
нец которой опущен в стаканчик с
подкрашенной водой (рис. 65). При
взбалтывании пирогаллол начинает быстро
буреть, а вода по трубочке поднимается
вверх, занимая место кислорода,
пошедшего на окисление пирогаллола.
Пирогаллол применяется в
фотографии как проявитель, а также при
анализе газов для определения
содержания кислорода в газовых смесях.
Флороглюцин получается
сплавлением многих природных смол с едким
кали. Он кристаллизуется с двумя
молекулами воды, обладает сладким
вкусом, с хлорным железом дает
темно-фиолетовое окрашивание.
Флороглюцин может реагировать в двух таутомерных формах: как
трехатомный фенол или как циклический трикетон (трикетоге-
ксаметилен):
С—ОН ; С=О
/ \ \ /\
; -НС С- ¦ Н,С СН,
І II І 7=ї І І
но—с с—он о=с с=о
\/ \ \/
сн- ; сн2
Как трикетон флороглюцин с гидроксиламином дает триоксим,
как трехатомный фенол он реагирует с хлористым ацетилом с
образованием сложного эфира СвН3(ОСОСН3)а. Таким образом,
флороглюцин реагирует то как фенол, то как кетон.
Таутомерные формы флороглюцина не были разделены,
и известен только один флороглюцин. Его спектр поглощения
сходен со спектрами фенолов, а не кетояов. Поэтому можно думать,
что большая часть его находится в фенольной форме.
Флороглюцин применяется как реактив на лигнин.
Смоченный соляной кислотой лигнин от капли 1%-ного спиртового
раствора флороглюцина окрашивается в вишнево-красный цвет.
При помощи этой реакции можно обнаружить примесь лигнина
в дешевых сортах бумаги.
231. Ароматические эфиры
463
Физические свойства фенолов приведены в табл. 20.
Таблица 20
Физические свойства фенолов
Название
Формула
Температура
плавления
"С
42,3
31
11,9
35
105
ПО
170,5
133
219
140,5
Температура
4ИПЄННЯ
"С
182
191
202,2
202
245
277
286
309
—
Растворимость в г
на 100 г воды
Фенол .
о-Крезол
jw-Крезол
п-Крезол
Пирокатехин A,2-диоксибен-
зол)
Резорцин A,3-диоксибензол)
Гидрохинон A,4-диоксибен-
зол)
Пирогаллол A,2,3-триокси-
бензол)
Флороглюцин A,3,5-триокси-
бензол)
Оксигидрохинон A,2,4-три-
оксибензол)
СвН6ОН
СНэСвН4ОН
СНэСвН4ОН
СНэСвН4ОН
СвН4(ОНJ
С„Н4(ОН),
С,Н4(ОНJ
с,нэ(ОН)8
СвНэ(ОНK
СвН,(ОН)8
8 (при 25 °С);
смешивается
при 68 "С
3 (при 35 °С)
2,4 (при 25 °С)
2.4 (при 40 °С)
24 (при 20 *С)
147 (при 20 °С)
6.5 (при 20 °С)
6,0 (при 20 °С)
Очень хорошо
растворим
231. Ароматические эфиры. Различают чисто ароматические
эфиры, как, например, дифениловый эфир, и
жирноароматические, в которых один радикал принадлежит к жирному ряду, а
другой—к ароматическому:
СвН6-О-С,Н6
дифениловый эфир
с,н6-о-снэ
метилфениловый эфир
(анизол)
Жирноароматические эфиры получают взаимодействием га-
лоидпроизводных углеводородов с фенолятами:
СвН6—О— JNa + Л—СН3
СвН6-О-СН3 + NaJ
Более дешевым и удобным метилирующим средством является
диметилсульфат, реагирующий с фенолами в щелочной среде:
CeH6—ONa + CHSO—SO8—OCHS > С„Н6ОСН8 + NaOSOa—OCH,
464 Глава XV'111. Одноядерные ароматические соединения
Чисто ароматические эфиры можно получать, действуя на
феноляты ароматическими галоидпроизводными в присутствии
порошкообразной меди. Так, дифениловый эфир получают в
присутствии этого катализатора, нагревая до 210 °С смесь бромбен-
зола с фенолятом калия.
Другим путем их можно получить, нагревая фенолы с воду-
отнимающими средствами, например с безводным хлористым
алюминием или хлористым цинком:
С,Н6—О—ІН+Н—OI—С6Н5 ^2 Q,H5—О-С6Н5
232. Ароматические спирты. Соединения, представляющие
собой продукт замещения гидроксилом водорода в боковой цепи
ароматического углеводорода, называются ароматическими
спиртами.
Простейшим ароматическим спиртом является бензиловий
спирт-
~\СНаОН или СвН5СН2ОН
Бензиловый спирт—жидкость со слабым приятным запахом
(темп. кип. 205,5 °С), нерастворим ни в воде, ни в щелочах.
Бензиловый спирт и некоторые сложные эфиры его
применяются в парфюмерии.
Ароматические спирты обладают типичными свойствами
предельных спиртов. Так, бензиловый спирт образует простые и
сложные эфиры; при действии хлористого водорода он переходит
в хлористый бензил:
С„Н5СНг—ОН + НСІ » СвН5СН2—СІ + Н2О
Бензиловый спирт способен окисляться, давая в результате
реакции бензойный альдегид:
СЛСНаОН + О » С6Н6-СНО + HaO
Ароматические спирты, в противоположность фенолам, не
дают окрашивания с хлорным железом.
АРОМАТИЧЕСКИЕ АЛЬДЕГИДЫ И КЕТОНЫ
233. Альдегиды. Простейшим ароматическим альдегидом
является бензойный альдегид (бензальдегид) СвН5—СНО. Он
встречается в природе в виде глюкозида—амигдалина Ca0Ha7NOu
который находится в горьком миндале и листьях лавровишни.'
233. Альдегиды 465
Амигдалин под влиянием содержащегося вместе с ним энзима
эмульсина подвергается гидролизу с образованием глюкозы,
бензойного альдегида и синильной кислоты.
Эту реакцию можно провести, растирая с водой горький
миндаль или листья лавровишни.
В промышленности бензойный альдегид получают из толуола.
Для этого толуол хлорируют. Первоначальным продуктом
хлорирования является хлористый бензил С„Н5СН2С1, при дальнейшем
хлорировании которого получается хлористый бензилиден
С6Н5СНС12. При нагревании последнего с водой и небольшим
количеством гидроокиси кальция или серной кислоты
образуется бензойный альдегид:
id Hj-0-н _2НС, /)!Н!
С.Н.СН/! + і , C,HSCH<...! і »CeH6CHO+H2O
Na ні—о—н Noh;
Другой путь получения бензойного
альдегида—непосредственное окисление толуола путем пропускания при повышенной
температуре паров толуола и воздуха через трубку с
катализаторами (окислы железа):
3 + , ^^ НаО
толуол
Гомологи бензойного альдегида могут быть получены
реакцией Гаттермана—действием на ароматические углеводороды
окиси углерода и сухого хлористого водорода в присутствии
хлористого алюминия и хлористой меди; окись углерода и хлористый
водород реагируют при этом как хлорангидрид муравьиной
кислоты НСОС1:
-H + сі -с/ —» сн3-^~^—сно + на
\н \/
і -с/
\н
талуол п-толуиловый
альдегид
Альдегидная группа вступает при этой реакции в
пара-положение к заместителю в бензольном кольце.
Бензойный альдегид—жидкость, кипящая при 178,1 °С;
плотность 1,05 г/см3; обладает сильным запахом горького миндаля.
Отметим следующие свойства этого вещества.
30— 788
466 Глава XVIII. Одноядерные ароматические соединения
1. Бензойный альдегид обладает типичными свойствами
альдегидов жирного ряда. Так, с бисульфитом натрия он дает
кристаллическое соединение. С гидроксиламином образует оксим:
QH,
Н
;_с=|о + |\noh —» QHj—c=
: ynwi і — т ч^ді 15—v^—NOH -
І оксим бензойного
альдегида
С фенилгидразином получается фенилгидразон:
Н
СаН6-С=
Ні
О + { >N—NHC„H6 » CeH5—C=N—NHCeH6 + H2O
„6e5e6
Нг фенилгидразон бензойного
альдегида
(темп. пл. 157 °С)
При взаимодействии бензойного альдегида с семикарбазидом
получается семикарбазон:
Н
1 н\
Q,H6—С=О + ^N—NHCONHj » CeH5—CH =N— NHCONHa H • Н2О
Н' семнкаобаэон бензойного
альдегида
(темп. пл. 222 °С)
2. Кислородом воздуха бензойный альдегид довольно быстро
окисляется, застывая при этом в сплошную массу кристаллов
бензойной кислоты С„Н5СООН.
Механизм реакции заключается в следующем. Бензойный
альдегид поглощает кислород, образуя промежуточное
соединение, по всей вероятности гидроперекись бензоила:
¦Р ,0
¦н '* * ' 6 \о—он
Гидроперекись бензоила окисляет затем другую часть
бензойного альдегида с образованием бензойной кислоты:
/О ,0
CeH5-C<f +QH6-C/ >2CeH6-COOH
\Н N3—ОН
Первоначальное образование перекисей характерно для
реакций самоокисления, т. е. окисления различных веществ
газообразным кислородом, протекающего даже при комнатной темпе-
233. Альдегиды 467
ратуре. Если наряду с веществом, способным к самоокислению,
ввести какое-нибудь легко окисляющееся вещество, то оно будет
окислено образующейся перекисью—происходит активирование
кислорода воздуха. Этим объясняется окисление в присутствии
бензойного альдегида некоторых веществ (например, сульфо-
кислоты индиго), на которые кислород при обыкновенных
условиях не действует. При этом на окисление введенного
вещества расходуется столько кислорода, сколько затрачивается его
на окисление бензойного альдегида в бензойную кислоту, что
находится в согласии с приведенными выше уравнениями.
3. Бензойный альдегид, подобно другим альдегидам, не
содержащим а-водородного атома, не осмоляется щелочами. Под
влиянием растворов щелочей он образует бензиловий спирт и
бензойную кислоту:
C,H6-Gf + КОН + С„Н6-С/ * САСН2-ОН + QH.-COOK
бенэиловый соль
спирт бензойной
кислоты
Эта реакция была открыта в 1853 г. Канниццаро.
Если взять для этой реакции смесь ароматического альдегида
и формальдегида, то получаются муравьиная кислота и
соответствующий ароматический спирт (В. М. Родионов, 1936):
;О X)
+ КОН + HC<f » С6Н5СН2—ОН + НСООК
4. Бензойный альдегид легко вступает в реакции конденсации
с другими веществами. Так, с альдегидами, кетонами и кислотами
жирного ряда, в молекуле которых содержатся два атома
водорода у а-углеродного атома, он реагирует с образованием
вещества с ненасыщенной боковой цепью:
Н
СН5—С=ІО+ hQCH— СНО ¦ QH5CH=CH—СНО + Н2О
/ксусный коричный альдегид
альдегид
н
I
С„Н5—С=|О+ Н2;СН— СО--СН3 . С6Н5СН=СН—СО—СН3 + НаО
ацетон оензальацеток
30*
468 Глава XVI!І. Одноядерные ароматические соединения
Конденсация бензальдегида с ацетоном происходит в
присутствии едкого натра. В зависимости от соотношения
реагирующих веществ образуются или бензальацетон, или дибензаль-
ацетон:
С„Н6СНО Н2СНЧ
+ J>CO * СбН.,СН=СН—СО—СН=СНС„НВ + 2Н2О
СбН6СНО Н^СЬи яибензальацетов
Бензальацетон—светло-желтые кристаллы, обладающие
приятным запахом; применяется в парфюмерии. Дибензальацетон—
желтое кристаллическое вещество, которое легко выделить и
идентифицировать по температуре плавления A12 °С). Поэтому
реакция получения дибензальацетона может быть использована
в качестве метода определения небольших количеств ацетона.
5. Бензойный альдегид при действии хлора превращается в
хлористый бензоил:
0
CeH5-C-:H
+С1
:—СІ
0
—» с,н5—с—сі + на
хлористый бензоин
6. В присутствии цианистого калия как катализатора из
бензойного альдегида получается бензоин СвН5СОСН(ОН)СвН5:
ЧН (X KCN
+ V-CgHg * CeH.-CO-CH(OH)C6Hj
О Н бензоин
При этой реакции атом водорода альдегидной группы одной
молекулы бензальдегида переходит к атому кислорода
альдегидной группы другой молекулы бензойного альдегида.
Освобождающиеся при этом валентности атомов углерода обеих
альдегидных групп взаимно насыщают друг друга.
Бензоин—бесцветное кристаллическое вещество, плавящееся
при 137 °С. Он был впервые получен в 1839 г. Н. Н. Зининым. При
окислении бензоина (например, при помощи концентрированной
азотной кислоты) получается дикетон—дибензоил, называемый
бензилом. СвН5—СО—СО—СвН5. Бензил—желтое
кристаллическое вещество с темп, плавл. 95 °С.
Бензойный альдегид применяется для изготовления душистых
веществ и красителей.
Может показаться, что ароматические альдегиды, в силу своей
способности вступать в реакцию Канниццаро и реакцию бен-
234. Кетоны 46&
зоиновой конденсации, принципиально отличаются от
альдегидов жирного ряда. Однако при более внимательном
рассмотрении можно заметить, что причина различного отношения к
щелочам у ароматических и жирных альдегидов состоит не в свойствах
самой альдегидной группы, а в наличии активного атома
водорода в а-положении у альдегидов жирного ряда. Мы видели,
что при действии едких щелочей на формальдегид (не имеющий
а-водорода) он превращается в муравьиную кислоту и метанол;
подобное же превращение уксусного альдегида в кислоту и спирт
совершается в условиях реакции брожения (стр. 336). В ряде
других биологических процессов из указанного альдегида
образуется ацетоин СН3СОСН(ОН)СН3.
234. Кетоны. В ароматических кетонах с карбонильной
группой могут быть связаны или два ароматических радикала, или
же ароматический радикал и алкил, т. е. радикал жирного ряда.
Примером кетонов второй группы является метилфенилкетон
(ацешофенон) С6Н;,СОСН3; представителем первой группы
служит дифен ил кетон (бензофенон) С6Н5СОС6Н5.
Ароматические кетоны могут быть синтезированы по методу
Фриделя—Крафтса действием хлорангидридов кислот на
ароматические углеводороды в присутствии безводного хлористого
алюминия:
СНз-СО-С! 4- СвН„ А1С13.> СН3—CO-QHs + НС1
ацетофенон
Ароматические кетоны, подобно кетонам жирного ряда, с
гидроксиламином образуют оксимы, а с фенилгидразином дают
фенилгидразоны.
При действии на жирноароматические кетоны
амальгамированного цинка и концентрированной соляной кислоты они
восстанавливаются в гомологи бензола:
С6Н5-СО-СН3 + 2Н2 » СбН6-СН4СН3 + Н2О
Производное ацетофенона—хлорацетофенон С6Н5СОСН2С1
является одним из сильнейших слезоточивых отравляющих
веществ. Он ¦ получается действием хлора на ацетофенон:
QH5COCH3 + С1а > С6Н6СОСН2С1 + НС1
Хлорацетофенон—кристаллическое вещество (темп, плавл.
52 °С) с запахом фиалки.
470 Глава XVIII. Одноядерные ароматические соединения
КАРБОНОВЫЕ КИСЛОТЫ
235. Бензойная кислота. Бензойная кислота С6Н5—СООН
содержится в некоторых смолах; ее производное—гиппуровая
кислота C6H5CONHCH2COOH—обнаружено в лошадиной моче.
В промышленности бензойную кислоту получают из толуола.
При полном хлорировании боковой цепи толуола образуется бен-
зотрихлорнд (фенилтрихлорметан) СвН5СС13.
При нагревании фенилтрихлорметана с водой получается
бензойная кислота:
QHs-CCIa + 2НаО * QHs-C^f + 3HCI
Х)Н
Кроме того, бензойная кислота получается при окислении
ароматических углеводородов с одной боковой цепью:
QH6CH3 + ЗО » С,Н6СООН + НаО
QH6-CHaCH3 + 6О * QH6COOH + 2НаО + СОа
Бензойная кислота—кристаллическое вещество, плавящееся
при температуре 122,4 °С. Она легко возгоняется с образованием
белых пластинок. Бензойная кислота плохо растворяется в
холодной воде; с повышением температуры растворимость ее
значительно увеличивается. Как кислота, бензойная кислота
несколько сильнее уксусной (соответствующие значения константы
диссоциации равны 6,8- Ю~5 и 1,8-10~5).
Бензойная кислота по своим химическим свойствам подобна
карбоновым кислотам жирного ряда. Так, с основаниями она
образует соли. Ее соли с щелочными металлами хорошо
растворимы в воде, поэтому бензойная кислота растворима в щелочах:
QH6COOH ¦+- NaOH . C^COONa + НаО
При сплавлении бензойнокислого натрия с едким натром
образуется бензол:
СбН6—jCOONa + NaOjH » QH,, + Na2CO,
Эта реакция аналогична реакции получения метана из
уксуснокислого натрия и едкого натра.
Со спиртами бензойная кислота образует сложные эфиры:
qHb-CO-ОіН + HOj-СНз » СбН6СООСНз + НаО
бензойно мет иловый
эфир
235. Бензойная кислота 471>
При действии на бензойную кислоту хлористых соединений
фосфора она переходит в хлорангидрид СвН6СОС1, называемый
хлористым бензоилом; из него и бензойнокислого натрия можно
получить бензойный ангидрид (С„Н6СОJО:
СНв-с/.
Хі^ і С„Н6
>—:Na
О
NaCl
Хлористый бензоил—жидкость с резким запахом,
кипящая при 198 °С. Легко вступает в реакции, типичные для хлоран-
гидридов, но реагирует при этом менее бурно, чем хлористый1
ацетил. Хлористым бензоилом часто пользуются для введения
бензоильной группы в молекулы спиртов, фенолов, аминов. Так,
например, при взбалтывании хлористого бензоила со щелочным*
раствором фенола легко образуется твердый бензойнофениловый
эфир (фенилбензоат):
СвН5СОС1 + СвН6ОН + NaOH * CeHsCOOQHe + NaCl + HSO
Бензойная кислота и ее соли находят применение в медицине;,
она используется как полупродукт в некоторых промышленных
синтезах, применяется также для консервирования пищевых
продуктов.
Бензоат натрия применяется как ингибитор коррозии.
Рял производных бензойной кислоты был получен в 1832 г. Вёлером н
Лнбихом в их работе по изучению горькоминдального масла (бензойного
альдегида).
При обработке бензойного альдегида [С,Н5О]Н хлором или бромом оии
получили хлористый беизоил [C^HjOlCl и бромистый бензоил (C^HjOJBr,
которые при действии йодистого калия или пиаиистого калия образуют йодистый
бензоил [C^HaOlJ или, соответственно, нитрил [C^HsOjCN.C аммиаком
хлористый беизоил и бромистый бензоил образуют беизамид |С,Н6О)ЫН2, а со
спиртом дают этиловый эфир бензойной кислоты '-»та работа имела в свое время
большое значение. Она показала, что во всех полученных Либихом и
Вёлером веществах находится радикал C^HsO, назнаниый этими исследователями
бензоилом, который может, не изменяясь, переходить из одного соединения-
в другое.
Амид бензойной кислоты C„H6CONH2—кристаллическое
вещество, темп. пл. 130 °С. Получается теми же методами, что и амиды,
жирных кислот (см. стр. 267).
Некоторые амиды бензойной кислоты и ее гомологов находят
применение как епелленты (см. стр. 474). Эти соединения отли-
472 Глава XVIII Одноядерные ароматические соединения
чаются более длительным отпугивающим действием на
кровососущих насекомых и клещей. Так, диэтилтолуамид (ДЭТА)
CON(C2H6J
СНЭ
действует в течение 5—8 ч.
236. Сахарин. Из соединений, содержащих одновременно
карбоксил и сульфогруппу, укажем на о-сульфобензойную кислоту.
Производным этой кислоты является сахарин:
„ ,SO,OH
1 II
о-сульфобензойная
кислота
so2
II NH
CO
сахарун
Такого рода циклические амиды двухосновных кислот, со-
держащие>ЫН группу, называют имидами кислот.
Для получения сахарина нельзя исходить из бензойной
кислоты, так как карбоксильная группа является заместителем
второго рода, поэтому при сульфировании бензойной кислоты
получается почти исключительно л?-сульфобензойная кислота.
Чтобы получить сахарин, толуол обрабатывают хлорсульфо-
новой кислотой C1SO2OH. При этом образуется смесь хлорангид-
ридов о- и n-толуолсульфокислот. При сильном охлаждении
более высокоплавкий пара-изомер выпадает в виде кристаллов и
может быть отделен от жидкого орто-изомера. Полученный таким
путем хлорангидрид о-толуолсульфокислоты действием аммиака
переводят в о-толуолсульфамид, а затем окисляют
марганцовокислым калием метильную группу в карбоксил. Образующийся
в результате окисления о-сульфамид бензойной кислоты при
нагревании отщепляет элементы воды и переходит в сахарин:
уСН3 A) ,СН3 A)
СеНз—СН3 » СцН^ » С6Н4\ »
толуол NoU2U \l) NoU2lNn3 (і.)
хлорангидрид
о-толуолсульфо
кислоты
/СООН
бН4\
XSO3NH2
о-сульфамид
бензойной
кислоты
A)
B)
0 -толуолсульфамид
СО
. СвН4<^ ^NH
so2
имид о-сульфобен-
ІОЙНОЙ КИСЛОТЫ
(сахарин)
237. Фталевые кислоты
В продажных таблетках сахарина содержится его натриевое
производное:
so2
О
известное под названием криспгаллозы; она в несколько сот раз
слаще сахара. Сахарин не усваивается человеческим организмом
и является только вкусовым средством.
237. Фталевые кислоты. При окислении ароматических
углеводородов, в молекулах которых находятся две боковые цепи,
получаются кислоты с двумя карбоксильными группами,
называемые фталевыми кислотами:
сн3
I
иэофталевая
кислота
терефталевая
кислота
Фталевые кислоты—твердые кристаллические вещества.
При нагревании выше температуры плавления о-фталева»
кислота легко теряет воду и переходит во фталевий ангидрид:
СО-О-М ,с?
QH4< v-: ! » С,Н4< >0 + Н20
\CO-rO-H; \^
фталевый
ангидрид
Две другие изомерные фталевые кислоты не дают ангидридов.
Это различие между фталевыми кислотами объясняется легко:
в молекуле о-фталевой кислоты карбоксилы расположены
наиболее близко один от другого, поэтому может произойти
отщепление воды и образоваться циклический ангидрид, содержащий
пятичленное кольцо.
¦474 Глава XVIfL Одноядерные ароматические соединения
Фталевый ангидрид кристаллизуется в виде блестящих игл
шли призм, плавится при 130,8 °С, кипит при 285 °С, легко
возгоняется.
При пропускании через нагретый фталевый ангидрид сухого
аммиака образуется имид фталевой кислоты, известный под
названием фпгалимида:
QH4^
со со
\О + NH3 » QH4<^ ^>
со со
Фталимид—белые листочки (темп, плавл. 238 °С). При
действии на фталимид спиртового раствора едкого кали водород имид-
ной группы замещается калием—образуется фталимидкалий:
СО
qh
СО
Фталевые кислоты имеют важное промышленное значение.
о-Фталевая кислота (в виде фталевого ангидрида) в технике
получается окислением нафталина или о-ксилола кислородом
воздуха на катализаторе из пятиокиси ванадия. Диэфиры о-фта-
левой кислоты (диметилфталат, дибутилфталат, диоктилфталат
и другие) находят широкое применение.
Диметилфталат—жидкость (темп. кип. 282 °С) со слабым
запахом; она может служить для отпугивания комаров. Такие
вещества на зы ва юте я репеллен тами.
Дибутилфталат (жидкость, темп. кип. 340 °С) и
диоктилфталат (жидкость, темп. кип. выше 350 °С) применяются как
пластификаторы для пластических масс. Конденсацией
фталевого ангидрида с многоатомными спиртами получают важные
синтетические смолы (см. стр. 477). Фталевый ангидрид является
полупродуктом в производстве некоторых синтетических
красителей.
Терефталевая кислота производится во все возрастающих
количествах, так как она является основным исходным продуктом для
получения полиэфирных волокон.
Изофталевая кислота широкого технического значения не
имеет. Ее эфиры применяются как пластификаторы.
238. Полиэфирные смолы. Поликонденсацией двухосновных
кислот (адипиновой, малеиновой, фталевой, терефталевой и др.) с
многоатомными спиртами (этиленгликоль, глицерин и др.)
получаются сложные полиэфиры, многие из которых приобрели весьма
238 Полиэфирные смолы 47&
важное значение в технике для получения синтетических
волокон, пластических масс, защитных покрытий и других
синтетических материалов.
Смолы на основе терефталевой кислоты и этиленгликоля.
Поликонденсацией терефталевой кислоты и этиленгликоля
получают волокнообразующую смолу полиэтилентерефталат,
нашедшую широкое применение для изготовления ценных
полиэфирных волокон.
В технике, ввиду трудности получения терефталевой
кислоты требуемой высокой степени чистоты, обычно исходят
не из свободной кислоты, а из ее диметилового эфира—диметил-
терефталата, проводя переэтерификацию этого эфира этиленгли-
колем;
СН3О—С— /~\—С—OCHS + 2СН,ОНСН2ОН »
4
днметилтерефталат
• НОСН2СН,О—С—f ^—С—ОСН2СН/)Н + 2CHSOH
A W A
днэтнлолтерефталат
о
-С—СН»СН2ОН
w 4
—* нГ-ОСН,,СН2—О—С— /~Ч—С— 1 ОСН2СН2ОН + (п—1)Н0СНгСН20В
II N=/ II
L о о Jn
полиэтнлентерефталат
Полиэфирное волокно на основе полиэтилентерефталата в СССР
получило название лавсан*, в Англии терилен, в США дакрон.
Оно отличается высокой разрывной прочностью и
водостойкостью.
Наибольшее распространение получили ткани из смеси
природных волокон (шерсти, хлопка) и лавсана, так как
прибавление 30—50% лавсана, не ухудшая потребительских свойств
материала, придает тканям такие ценные свойства, как высокая
прочность и несминаемость.
Ненасыщенные полиэфиры. При этерификации смеси
насыщенных и ненасыщенных двухосновных кислот гликолями образу-
* Сокращенное название: ла (лаборатория), в (высокомолекулярных),
с (соединений), а (академии), н (наук).
476 Глава XVIII Одноядерные ароматические соединения
ются низкомолекулярные жидкие полиэфиры линейного
строения (молекулярный вес 1000—3000):
сн—соч
НООС—(СНаL—СООН + 2НОСН2СН2ОН + || , >О »
СН—СОЛ
С—(СН2ь—С—О—СН,—СН2—О—С-СН=СН-С-О-СН2-СН2-О
ЛИ 4 4
С(СН2ьСОСН,СН2ОС
ЛИ 4 4
При повышенной температуре и в присутствии инициаторов
полимеризации эти жидкие полиэфирные смолы способны сополи-
меризоваться с другими ненасыщенными соединениями, например
со стиролом, с образованием полимеров сетчатой, трехмерной
структуры:
С—(СН2L—С—О—СН2—СН2—О—С—СН—СН—С—О—СН2—СН2—О
4 A А I &
гсн2
\cn-f\
L| \=/Jn
С—(СН2L—С—О—СН2—СН2—О—С—СН—СН—С—О—СН,—СН,—О—
Я її її і її
її
0
її і її
0:0
Ненасыщенные полиэфирные смолы приобрели большое
значение для получения особо прочных синтетических материалов,
так называемых стеклопластиков. Для их получения стеклянное
волокно пропитывают смесью жидкого полиэфира, стирола (или
другого винильного мономера) и инициатора полимеризации,
помещают в форму, соответствующую конфигурации изделия,
и нагревают до температуры около 100 °С. При нагревании
происходит сополимеризация ненасыщенного полиэфира и стирола,
так называемое отверждение полимера. Полученные материалы не
уступают по прочности стали и значительно превосходят ее по
легкости.
В качестве исходных кислот для полиэфиров применяют ади-
пиновую, себациновую, азелаиновую и другие двухосновные
кислоты, в качестве гликолей—этиленгликоль, диэтиленгликоль,
глицерин и другие многоатомные спирты. Вместо ненасыщенной
кислоты чаще всего берут малеиновый ангидрид.
Смолы на основе фталевого ангидрида и глицерина. При
поликонденсации глицерина и фталевого ангидрида получаются
смолы, известные под названием глифталевых или алкидных смол.
939. Фталеины 477
Реакция гюликонденсации идет постепенно. В первой стадии
образуются кислые эфиры:
О + НО—СНа—СИ—СНаОН
со он
Во второй стадии кислые эфиры реагируют друг с другом, и
в результате получаются полимеры по схеме:
—СН(ОН)СН2ОІН + НО;СО СООСН2—СН(ОН)СН2ОН
::| I
\
НОСОСООСН2—СН(ОН)СН2—О—COCOOCHj—СН(ОН)СН2ОН + Н,0
1—1 1-4
-о о
Сначала образуется низкомолекулярная плавкая смола, которая
содержит реакционные группы ОН и СООН; постепенно, при
длительном нагревании A50—180 °С), смола переходит в неплавкое
твердое состояние.
Чистые глицерино-фталевые смолы не применяются, так как
они образуют очень хрупкую и нестойкую к воде пленку. Обычно
к ним добавляют смоляные и жирные кислоты, тогда получаются
эластичные маслорастворимые смолы, обладающие хорошей
водостойкостью.
Глифталевые смолы широко применяют как
электроизоляционный материал в виде антикоррозионных атмосферостойких лаков.
239. Фталеины. Фталевый ангидрид под действием водуот-
нимающих веществ способен вступать в реакцию конденсации
с фенолами; в результате образуются вещества, которые
называют фталеинами. Простейшим из них является фенолфталеин.
Фенолфталеин получается нагреванием с серной кислотой
смеси фталевого ангидрида и фенола:
о
/
фенолфталеин
478
Глава XVIIІ. Одноядерные ароматические соединения
Фенолфталеин—бесцветное твердое вещество, растворяется
в щелочах с образованием солей, окрашенных в
малиново-красный цвет.
Применяется как индикатор в анализе и под названием
«пурген» в медицине как слабительное.
Изменение окраски при применении фенолфталеина как
индикатора обусловлено образованием хиноиднои группировки
связей при взаимодействии фенолфталеина со щелочью:
—ОН
=/=и +нао
COOK
Примером фталеинов является также флуоресцеин.
Получается он сплавлением с хлористым цинком смеси фталевого
ангидрида и резорцина:
О
;н + н
>со
Флуоресцеин растворяется в щелочах, придавая жидкости
желтую окраску и желто-зеленую флуоресценцию. Эта
флуоресценция еще ясно обнаруживается при содержании одной части
флуоресцеина в 40 000 000 частях воды.
Флуоресцеин был применен для изучения путей протекания
подземных вод.
Флуоресцеин окрашивает волокна животного происхождения
в ярко-желтый цвет.
Производные флуоресцеина, в которых атомы водорода
замещены галоидами и другими группами, обладают ценными крася-
щиии свойствами. Простейшим из таких производных флуорес-
240. Салициловая кислота 479
ценна является тетрабромфлуоресцеин, или эозин (по-гречески
«эос»—утренняя заря):
С
Эозин окрашивает шелк в красивый желто-розовый цвет,
отличающийся прекрасной флуоресценцией.
Различные препараты эозина применяются при
микроскопических исследованиях в медицине и биологии.
оксикислоты
240. Салициловая кислота. Салициловая кислота
одновременно является и кислотой и фенолом. Гидроксил и карбоксил
находятся в ее молекуле в орто-положении друг к другу—она
является о-оксибензойной кислотой:
•СООН
Производные салициловой кислоты содержатся в некоторых
растениях, например в листьях и коре ивы. От латинского названия
ивы Salix и происходит ее наименование.
В технике салициловую кислоту получают, нагревая фенолят
натрия с углекислым газом при 130 °С в автоклавах под
давлением. При этом получается салициловокислыи натрий:
/Н /ОН
QH4< + СО2 » QH4<
^ 4\О-№ \COONa
Полученную соль разлагают соляной кислотой:
.ОН /ОН
QH4< + НС1 » QH4< + NaCl
\OON \COOH
480 Глава XVIII. Одноядерные ароматические соединения
Салициловая кислота—кристаллическое вещество,
труднорастворимое в холодной воде; плавится при 159 °С. Подобно
фенолу, она дает с хлорным железом фиолетовое окрашивание.
При быстром нагревании салициловой кислоты от нее
отщепляется СО2 и она превращается в фенол:
+с0,
При действии на раствор салициловой кислоты бромной воды
происходит вытеснение карбоксила бромом и выпадение белого
труднорастворимого осадка 2,4,6-трибромфенола:
ОН
BVvBr
—* Ї J + СОа + ЗНВг
Вг
Салициловая кислота образует сложные эфиры как со
спиртами и фенолами, так и с кислотами.
Сложный эфир салициловой кислоты (по фенольной группе)
с уксусной кислотой известен под названием аспирина. Он
может быть получен действием хлористого ацетила или уксусного
ангидрида на салициловую кислоту:
/СООН
/ , /СООН
X)—:Н+ С1І—СО—СН3 » С6Н4< + НС1
; ' X)—СО— СН3
3
аспирин
Аспирин (темп, плавл. 135 °С)—один из широко применяемых
медикаментов (жаропонижающее).
Сложный эфир салициловой кислоты (по карбоксильной
группе) и фенола называется салолом:
QH.
і /CO-O-QHs
/CO-jOH + Hj-O-QHs > QH4<^ + HaO
нЗН
Для получения салола нагревают смесь салицилата натрия с
треххлористым фосфором и фенолом; таким образом,
промежуточным иродуктом является хлорангидрид салициловой кислоты.
240. Салициловая кислота 481
Салол (темп, плавл. 43 °С) применяется как дезинфицирующее
средство при кишечных заболеваниях. Он проходит без измене^
ния через желудок (в котором реакция кислая), но подвергается
гидролизу в щелочной среде кишечника.
Салициловая кислота препятствует брожению и гниению.
Поэтому ее используют для консервирования пищевых
продуктов (икра) и напитков (пиво). Она является полупродуктом
при изготовлении многих красителей. Салициловой кислоте
соответствуют салициловый альдегид и салициловый спирт:
СООН /^ ^СНО
¦юн ^
салициловая кислота салициловый альдегид салицилевый спирт
Салициловый альдегид—жидкость (темп. кип. 197 °С),
обладающая приятным запахом. Он может быть получен реакцией
Реймера—Тимана, которая заключается в нагревании фенолов с
хлороформом и едким кали. Реакция идет в две стадии:
СНС12
r Y + неї
оС
чАон
+2KOH »[ ft " 4- 2КС1 + HSO
ОН
Альдегидная группа при этом становится в орто- и
пара-положения к гидроксилу, причем преимущественно получается орто-
изомер.
Салициловый спирт, или салигенин,—кристаллическое
вещество. Получается при гидролизе салицина С^Н^О,—глюкозида,
содержащегося в коре ивы:
НаО
.сн
QH/
Х)Н
Салигенин образуется-также при действии формальдегида кя
фенол как промежуточный продукт при производстве феноло-
формальдегидных смол (см. стр. 456).
31—788
482 Глава XVIII. Одноядерные ароматические соединения
241. Галловая кислота. Из оксикислот большое значение имеет
также галловая кислота
СООН
В свободном состоянии она содержится в чернильных орешках
дуба, листьях чая, дубовой коре; в форме эфиров и глюкозидов
галловая кислоіа в значительных количествах находится в
дубильных веществах типа таннина. Галловая кислота
кристаллизуется с одной молекулой воды, образуя шелковистые иглы.
Отщепляя при нагревании СО2, она переходит в пирогаллол
(стр.462).
С раствором солей окиси железа галловая кислота дает
жидкость черного иьега применявшуюся ранее как чернила. С
животным клеем образует нерастворимое в воде соединение; этим
обусловлено действие дубильных веществ, применяемых при
выделке кож. Раствор галловой кислоты имеет кисловато-вяжущий
вкус.
Молекулы оксикислот могут вступать в реакцию между собой
с образованием сложных эфиров.
Примером такого эфира является дигалловая кислота
С6Н2(ОН)Э—CO-O-QH2( OHJCOOH
образование которой можно выразить следующей реакцией:
НОЧ о НОЧ ,ОН
но/ чсоон
н<\ о ноч ,он
н<\ о
\ II
-/)-С-
w w +H'°
но/ хсоон
дигалловая кислота
Эта кислота называется метадигалловой кислотой, так как в ней
этерифицирован гидроксил, находящийся в мета-положении к
карбоксилу.
242. Дубильные вещества 483
Сложные эфиры ароматических оксикислот, подобные дигал-
ловой кислоте, были названы депсидами (от греческого слова
«депсис»—дубление); они имеют ближайшее отношение к
некоторым дубильным веществам.
242. Дубильные вещества. Дубильные вещества представляют
собой соединения, которые имеют вяжущий вкус, образуют с
хлорным железом темно-синие и темно-зеленые осадки; они
осаждают белки и превращают шкуру животных в кожу. Легко
растворимы в воде.
Дубильные вещества находятся во многих растениях,
например в коре ивы и дуба; особенно богаты дубильными веществами
некоторые тропические растения—катеху, турецкий чернильный
орешек диви-диви, акация, квебрахо-колорадо.
Типичным дубильным веществом является таннин, который
легче всего получить из чернильных орешков. Чернильные
орешки—это вздутия на листьях и ветвях дуба, которые образуются
от ранения растения насекомым орехотворкой. Таннин находится
в чае и придает ему горький вкус. Таннин—белый порошок,
легко растворимый в воде. При кипячении с соляной кислотой он
целиком превращается в галловую кислоту и глюкозу.
Китайский таннин является сложным эфиром глюкозы (окисная
форма) и дигалловой кислоты. Его формула
OR OR OR OR
RO-CH,—CH—CH-CH-CH—CH
О 1
где R—остаток дигалловой кислоты
QH2(OHKCOO-CeH2(OH)aCO-
Таннины, полученные из различных растений, имеют разный
состав. В молекуле таннинов некоторых растений остатки
дигалловой кислоты частично заменены остатками галловой кислоты-
Таннин служит протравой при крашении; в медицине
применяется как вяжущее средство.
По своему строению дубильные вещества находятся в тесном
родстве с ароматическими оксикислотами. Они делятся на два
класса. Дубильные вещества одного класса легко подвергаются
гидролизу, расщепляясь на более простые соединения, имеют
характер эфиров. Дубильные вещества другого класса при
сплавлении со щелочами дают различные ароматические оксикислоты
и фенолы, главным образом пирокатехин и флороглюцин.
31*
484 Глава XV111. Одноядерные ароматические соединения
КИСЛОТЫ С КАРБОКСИЛОМ В БОКОВОЙ ЦЕПИ
243. Фенилуксусная кислота. В описанных выше кислотах
карбоксил непосредственно связан с бензольным кольцом
Простейшей кислотой с карбоксилом в боковой цепи является
фенилуксусная кислота:
>—CHa—СООН
Она легко может быть получена из хлористого бензила;
QH5CHa-Ci +KCN> CeH6CHa-CN +2Ш ¦, СПН5СН,-СООН
Фенилуксусная кислота плавится при 76,9 °С, кипит при 266,5eG.
Ее этиловый эфир применяется в парфюмерии
244. Миндальная кислота. Миндальная кислота является
эксифенилуксусной кислотой
- СН(ОН)—СООН
В ее молекуле имеется асимметрический атом углерода; в
соответствии с этим она сущесчвует в виде оптических антиподов.
Миндальная кислота может быть получена из бензойного
альдегида и синильной кислоты:
.о /он
tu^ QH5—С— CN itfiU С.Н.5—СН(ОН)—СООН
\н
Температура плавления миндальной кислоты 118 *G.
245. ?-Фенилпропионовая кислота. ?-Фенилпропионовая, или
гидрокоричная, кислота
>—СН2—СНа—СООН
может быть получена восстановлением коричной кислоты,
например, при помощи амальгамы натрия; плавится при 48,6 *G;
кипит при 280 eG.
?-Фенилпропионовая кислота и ее пара-оксипроизводное
имеют прямое отношение к некоторым продуктам гидролиза
белковых веществ.
246. Коричная кислота 485
246. Коричная кислота: Коричная кислота
>—СН=СН-СООН
может служить примером ароматических кислот с двойной связью
в боковой цепи. Коричная кислота получается нагреванием
бензойного альдегида с уксусным ангидридом в присутствии
уксуснокислого натрия:
-н2о
СвН6—СН=О + Н2СН—СО—О—СО—СНЭ »
>• СаН6—СН=СН—СО—О—СО—СНЭ »
» СаН5—СН=СН—СООН -ЬС
Нагревание ароматических альдегидов с натриевыми солями
жирных кислот в присутствии ангидридов этих кислот является
общим способом получения ароматических кислот с
карбоксилом в ненасыщенной боковой цепи. Эта реакция известна под
названием реакции Перкина. Обозначая ароматический радикал
через Аг, а алифатический—через R, можно написать общую
схему этой реакции:
/°
Ar—Cf + H2CR—COONa » Ar—CH=CR—COONa + H,0
\h
Для коричной кислоты имеет место цис-пгранс-изомерия:
СаНв-С—Н СаНв-С—Н
НООС—С—Н Н—С—СООН
¦ллокоричная кислота обычная коричная кислота
(темп плавл. 68 °С) (темп планл 134 °С)
Обычная коричная кислота входит в состав бальзамов
(перуанского, толуанского), получаемых из некоторых растений, в
большинстве случаев произрастающих в тропической зоне. В
этих бальзамах, кроме смолистых веществ, содержатся главным
образом эфиры коричной и бензойной кислот с бензиловым и
коричным спиртами.
Эфиры коричной кислоты применяются в парфюмерии.
Коричная кислота обладает всеми свойствами непредельных
соединений: легко присоединяет борм, бромистый водород,
окисляется марганцовокислым калием и т. д.
486 Глава XVIII. Одноядерные ароматические соединения
При медленной перегонке коричной кислоты происходив
отщепление СО2 и получается углеводород стирол:
—СН=СН—JCOOjH <^
стирол
Стирол—приятно пахнущая жидкость, кипит при 145 ГС;
легко полимеризуется, образуя высокомолекулярные полимеры
состава (СвНв)„, известные под названием полистиролов.
Полимеризация медленно протекает даже при обыкновенной температуре;
при нагревании же она значительно ускоряется. Полимеризация
стирола, протекающая по схеме
СН=СН3 + СН=СНа + СН=СНа Н »
QH6 C6Ha QHs
» СН-СНа-СН—СНа-СН-СНа —...
QH5 С6Н6
приводит к образованию стекловидной массы, находящей
применение в качестве изолятора в электротехнике, для изготовления
химически стойкой посуды и аппаратуры, типографского шрифта
и др. При нагревании полистирола до 300 °С происходит его
деполимеризация, в результате чего снова образуется стирол.
Так как полистирольные пластические массы оказались очень
ценным материалом, то они готовятся в громадных количествах.
Стирол получают из бензола и этилена (газы крекинга) при
действии безводного хлористого алюминия. Образующийся при этом
этилбензол подвергается дегидрированию над катализатором
(Сг2О3) при 400 °С:
АІСІз Сг2О3
QHaH + СН2=СН2 * QH5-CHa-CH3 —^ СвНб-СН=СНа
бензол этилен этилбензол стирол
При совместной полимеризации стирола и бутадиена
получают ценные сорта синтетического каучука.
Коричной кислоте соответствует коричный алкоголь и
коричный альдегид:
О-СН=СН-СН2
20Н
корнчньїіі алкоголь коричный альдегид
247. Способы получения и свойства первичных аром, аминов 487
Коричный алкоголь—твердое вещество (темп, плавл. 33 °С;
темп. кип. 257 °С) с запахом гиацинтов; коричный
альдегид—жидкость; кипит при 251 °С, частично разлагаясь; он является
душистым веществом корицы.
АРОМАТИЧЕСКИЕ АМИНЫ
247. Способы получения и свойства первичных ароматических
аминов. Простейшим ароматическим амином является фениламин,
или аминобензол, CeHs—NH2, называемый анилином:
О
—NHa
Ближайший гомолог анилина—аминопроизводное толуола—
известен в виде четырех изомеров. Три из них содержат
аминогруппу, связанную с бензольным кольцом; они называются то-
луидинами:
СНо
NHa
а-толуидин л-толуидин
Четвертый изомер, содержащий аминогруппу в боковой цепи,
называется бензиламином:
(
бенэиламин
Амины, в боковой цепи которых содержится аминогруппа,
сходны по химическим свойствам с аминами жирного ряда.
Поэтому в дальнейшем описываются лишь амины, содержащие
аминогруппу, связанную с бензольным кольцом.
Основным методом получения первичных ароматических
аминов является реакция, открытая в 1842 г. знаменитым русским
488 Глава XVIII. Одноядерные ароматические соединения
химиком Н. Н. Зининым*, впервые получившим синтетический
анилин (и, аналогичным путем, а-нафтиламин и метафениленди-
амин) восстановлением нитросоединений:
—NO2 + ЗН3 * vIy~NH2 + 2На°
нитробензол анилин
В качестве восстановителя Н. Н. Зинин применял раствор
сернистого аммония. В настоящее время восстановление
нитросоединений производят обычно металлами (Fe, Sn, Zn) в кислой
среде или пропуская пары нитробензола с водородом над
катализатором (Си) при 300—400 °С.
Если амины жирного ряда получаются действием аммиака на
галоидные алкилы, то ароматические амины получать таким
путем затруднительно—галоид прочно связан с бензольным
ядром и с трудом вступает в реакцию с аммиаком. Однако при
действии аммиака на хлорбензол при нагревании под высоким
давлением получается анилин:
> /^_NH3 + HC1
Свойства первичных ароматических аминов:
1. Первичные ароматические амины—высококипящие
жидкости или твердые тела со своеобразным запахом; они
малорастворимы в воде.
2. Амины жирного ряда обладают сильными основными
свойствами: они образуют с кислотами соли; водный раствор их имеет
сильнощелочную реакцию. Ароматические амины, в отличие от
* Николай Николаевич Зинин A812—1880) родился в г. Шуше, в
Закавказье. В 1830 г. Н. Н. Зинин поступил в Казанский университет на физико-
математический факультет. В 1836 г. защитил диссертацию на тему «О
явлениях химического сродства и о превосходстве теории Берцелиуса о
постоянных химических пропорциях перед химической статикою Вертолета». В 1841 г.
в Петербургском университете Николай Николаевич защитил докторскую
диссертацию «О соединении беизоила и об открытых новых телах, относящихся
к беизоиловому ряду».
Возвратись в Казань и получив кафедру химической технологии, Н. Н.
Зинин начал исследования в новой области органических соединений, и уже в
октябре 1842 г. в «Известиях Академии Наук» была напечатана его работа о
превращении нитробензола н нитронафталина в «бензидам» (анилин) и «наф-
тилидам» (нафтиламин) действием сероводорода в аммиачной среде.
Из казанской лаборатории Н. Н. Зннииа вышел ряд блестящих работ по
синтезу продуктов восстановления моно- и динитросоедииений ароматического
ряда. Были получены азоксибензол, гидразобензол, бензидин, лі-нитроани-
лин. Значение этих работ прекрасно характеризуют слова А. В. Гофманг на
заседании немецкого химического общества. «Если бы Зинин не сделал ничего
более, кроме превращения нитробензола в анилин, то и тогда его имя осталось
бы записанным гигантскими буквами в истории химии».
247. Способы получения и свойства первичных аром, аминов 489
аминов жирного ряда, представляют собой слабые основания:
водный раствор ароматических аминов не окрашивает лакмуса
в синий цвет, хотя с кислотами эти амины образуют соли. Так,
при взаимодействии анилина с соляной кислотой получается
хлористый фениламмоний:
QH5NH2 + HCI > [QH5NH3]C1
Обыкновенно эту соль называют солянокислым анилином и
состав ее изображают формулой C6H6NH2-HCJ.
Таким образом, основные свойства аминогруппы в
ароматических аминах ослаблены. В этом сказывается влияние
бензольного остатка—фенила, усиливающего кислотные свойства
вещества.
3. При действии на первичные амины жирного ряда азотистой
кислоты получаются спирты:
C2H5-NH2 + HNO2 * QH5-OH + N2 + Н2О
Первичные же амины ароматического ряда при действии на их
соли азотистой кислоты переходят в гидроксильные производные
не сразу, а образуют вначале имеющие очень большое
техническое значение диазосоединения (см. также стр. 503 ел.):
C6H5NH2-HC1 + HNO2 > C6H5N2C1 + 2Н2О
диазосоединение
4. При действии на первичные ароматические амины как
самих кислот (при длительном нагревании), так и их ангидридов
(на холоду) получаются анилиды. Так, например, при действии
на анилин уксусной кислоты получается ацетанилид:
QH5-NH-:H + НО!—СОСНз » QH5-NH—СОСН3 + Н2О
Как видно из формулы, ацетанилид—производное анилина,
в молекуле которого один атом водорода замещен на ацильную
группу. Его также можно рассматривать как производное
уксусной кислоты, в которой гидроксил замещен остатком анилина.
Анилиды при кипячении с кислотами или щелочами
присоединяют воду и расщепляются обратно на амин и кислоту:
QH5—NH—:СОСН3+ НО:-Н > С6Н5—NH2 + СН3СООН
5. Бензольное ядро в ароматических аминах в общем
сохраняет свои свойства: водород бензольного ядра ароматических
аминов можно заместить галоидом; ароматические амины могут
сульфироваться и нитроваться.
490 Глава XVIII. Одноядерные ароматические соединения
Подобно фенолам, в аминах замещение водорода бензольного
ядра галоидом происходит более легко, чем в ароматических
углеводородах. Напомним, что группа—NH2 является сильным
ориентантом I рода (см. стр. 442 ел.). Так, при действии на
анилин бромной воды происходит образование триброманилина, в
котором атомы брома находятся в орто- и пара-положениях по
отношению к аминогруппе:
NH,
Вг
2.4,6-триброманилин
6. Ароматические амины легко окисляются.
248. Анилин и его производные. Анилин—бесцветная
маслообразная жидкость, тяжелее воды, кипит при 184,4 °С; в воде
анилин малорастворим (в 100 г воды растворяются 3,2 г анилина).
Анилин довольно легко окисляется, при этом наблюдается
характерное изменение окраски. Так, при действии на анилин
белильной извести получается фиолетовое окрашивание. Если
анилин растворить в крепкой серной кислоте и прибавить дву-
хромовокислый калий, получается зеленое окрашивание, которое
переходит в синее и, наконец, в черное.
Краска, получающаяся при окислении анилина двухромово-
кислым калием, называется черным анилином. При крашении
ткани в черный цвет анилином на ткань наносится солянокислый
анилин (анилиновая соль), затем ткань обрабатывают раствором,
содержащим двухромовокислый калий, серную кислоту и
другие вещества, и, наконец, подвергают специальной обработке,
называемой вызреванием.
Соли анилина, например хлористоводородный анилин или
сернокислый анилин, окрашивают древесину в желтый цвет. Чем
хуже бумага, т. е. чем больше в ней древесины и меньше чистой
целлюлозы, тем сильнее окрашивание.
Анилин является одним из основных продуктов химической
промышленности. Он широко применяется как исходное сырье
для получения красителей, фармацевтических препаратов,
ускорителей вулканизации каучука и для синтеза многих органических
веществ.
Сульфаниловая кислота. Сульфирование анилина не
представляет каких-либо затруднений. Оно осуществляется
нагреванием анилина с концентрированной серной кислотой до 180—
248. Анилин и его производные 491
190 °С, в результате чего получается я-аминобензолсульфокислота,
названная сульфаниловой кислотой. Вначале происходит
присоединение серной кислоты к анилину с образованием соли,
затем отщепляется вода и образуется анилид; наконец,
происходит обмен сульфогруппы с водородом в пара-положении:
+H2SO4 /гь 2 ,*».
NHHs°* У~NHS02°H
HOOjS— <f\—NH2
Суммарно реакция сульфирования анилина выражается
уравнением:
/NHa
СН5—NH2 + H2SO4 » CeH4< + Н2О
\SOOH
сульфаниловая
кислота
Сульфаниловая кислота кристаллизуется в блестящих
пластинчатых кристаллах .состава C6H4(NH2)SO2OH-2H2O, трудно
растворимых в холодной воде; эти кристаллы выветриваются на
воздухе
Сульфаниловая кислота обладает ясно выраженными
кислотными свойствами и при действии щелочей или соды образует
растворимые соли. С кислотами сульфаниловая кислота солей не
образует.
Сульфаниловая кислота представляет собой своеобразную
«внутреннюю» соль; в ней солеобразование осуществляется через
взаимодействие основной аминной группы с кислотной сульфо-
группой внутри одной молекулы:
сульфаниловая кислота
как внутренняя соль
Образованием внутренней соли объясняется малая
растворимость сульфаниловой кислоты в воде (сульфокислота
ароматических углеводородов, как правило, хорошо растворимы).
Сульфаниловая кислота широко применяется для синтеза
многих красителей.
Очень большое значение в современной медицине имеют
производные амида сульфаниловой кислоты—сульфамидные
препараты (см. стр. 512).
492 Глава XVIII. Одноядерные ароматические соединения
Нишроанилины. содержат и нитрогруппу и аминогруппу:
о-нитроанилин "' м-китроанилин л-нитроанилив
Нитроанилины—кристаллические вещества. Их пара- и мета-
изомеры желтого цвета; о-нитроанилин оранжево-желтый.
Получение нитроанилинов нитрованием анилина встречает
некоторые затруднения, так как азотная кислота не только
нитрует анилин, но частично и окисляет его. Кроме того, в сильно
кислой среде, вследствие образования четвертичной
аммонийной соли, меняется направляющее действие заместителя. Если
аминогруппа, как было указано, ориентирует преимущественно
в орто- и пара-положения, то аммонийная соль направляет
заместители главным образом в мета-положение. Поэтому для
получения л-нитроанилина анилин сначала переводят в ацетанилид
С6Н5—NH—СОСН3 (анилид уксусной кислоты), который труднее
окисляется, чем анилин, этим достигается как бы «защита»
аминогруппы от окисления и сохраняется преимущественная
ориентация в пара-положение.
Полученный ацетанилид подвергают нитрованию, в
результате которого нитрогруппа вступает в пара-положение по
отношению к аминогруппе:
CH3CONH-<f \-;'н"+ HO:-NO2 » CHaCONH-f ?—N0„ + Н20
ацетанилид л-нитроацетанилид
Омыляя /г-нитроацетанилид щелочью, получают л-нитроани-
лин, являющийся одним из важнейших продуктов в
производстве красителей. Процесс омыления протекает по схеме:
>-NH2+CH3COONa
n-нитроацетанилид n-иитроанилин
При нитровании, наряду с я-нитроанилином, образуется в
небольшом количестве и о-нитроанилин.
лі-Нитроанилин получается частичным восстановлением* лег-
* Восстановление только одной нитрогруппы производится нагреванием
днннтросоединения с сернистыми нлн многосерннстыми соединениями
щелочных металлов нлн аммония:
N02 NH2
+ Na2S3 + НаО * CeH4<^
249. Вторичные и третичные амины
493
кодоступного ж-динитробензола:
NO,
3H» (~
я-динитробенам к-нитроанилив
249. Вторичные и третичные амины. Анилин содержит
аминогруппу NH2, яляясь, таким образом, первичным амином.
Существуют вторичные и третичные ароматические амины, примерами
которых могут служить кристаллические дифениламин
(C6H5hNH и трифениламин (C6HsKN:
\
дифеннламнв трнфениламин
Дифениламин получается нагреванием в автоклаве до 200—
220 °С хлористоводородной соли анилина со свободным анилином:
СвНв-ІЙН^ЇНСІТнІ-^Н-СвНб » CeH6-NH-CeH6 + NH4CI
Трифениламин можно получить кипячением дифениламина
с иодбензолом, углекислым калием и медным порошком:
Си
(CeHJjNH + CeH5J > (CeH5KN + HJ
Дифениламин обладает еще более слабыми основными
свойствами, чем анилин, его соли легко разлагаются водой.
Трифениламин основными свойствами почти не обладает. Таким
образом, накопление фенильных групп вызывает ослабление
основных свойств.
Раствор дифениламина в концентрированной серной кислоте—
чувствительный реактив на азотную кислоту. При прибавлении
к этому раствору даже следов азотной кислоты получается яркое
синее окрашивание. Эта реакция может служить для открытия
азотной кислоты только при отсутствии других окислителей, так
как дифениламин дает синее окрашивание и с бромной водой,
марганцевокислым калием, перекисью водорода и т. д. Дифенил-
аімин служит промежуточным продуктом для синтеза некоторых
красителей; кроме того, его применяют для получения
стабилизаторов бездымных порохов. Нитрат целлюлозы, составляющий
494 Глава XVШ. Одноядерные ароматические соединения
главную часть бездымного пороха, склонен к саморазложению.
Это саморазложение, постепенно усиливаясь, может привести
к взрыву всей массы пороха. Стабилизаторы, введенные в состав
бездымных порохов в количестве 0,5—1,5%, предохраняют нитрат
целлюлозы от саморазложения.
Примером смешанного жирноароматического амина является
димешиланилин:
О
-N(CH3)j
Он получается нагреванием в автоклаве смеси сернокислой соли
анилина с метиловым спиртом:
CeH5NHs.H2SO4 + 2СН3ОН » CeH5N(CH3)a-HsSO4 + 2Н2О
или пропусканием смеси паров анилина и метанола над
катализатором.
Диметиланилин—маслянистая бесцветная жидкость со
своеобразным запахом. Подобно другим жирноароматическим аминам,
он обладает более сильными основными свойствами, чем чисто
ароматические вторичные и третичные амины. Водородный атом
в пара-положении к группе —N(CHSJ необычайно подвижен и при
обработке диметиланилина разбавленным раствором азотистой
кислоты замещается нитрозогруппой; при этом образуется
я-нитрозодиметиланилин:
+ HONO » (CH3)aN-<^^-NO + HaO
n-нитро зо диметиланилин
л-Нитрозодиметиланилин—зеленые кристаллы (темп, плавл.
85 °С). При кипячении со щелочами он расщепляется на /г-нитро-
зофенол (белые иглы) и диметиламин:
(CH3)aN—<f]3~NQ + На° * НО—
Эта реакция служит для получения диметиламина без
примеси монометил- и триметиламинов.
Повышенная подвижность водородного атома в
пара-положении проявляется и в других реакциях диметиланилина. В
качестве примера можно указать реакцию диметиланилина с
фосгеном, в результате которой получается кетон Михлера:
2HCI
250. Диамины 495
Кетон Михлера можно рассматривать как бензофенон, в
молекуле которого водородные атомы обоих бензольных ядер,
находящиеся в пара-положенин к карбонильной группе, замещены
группами —N(CH3J. Его можно назвать ди-(п-диметиламино)-
бензофеноном.
Кетон Михлера представляет собой бесцветные кристаллы;
применяется для синтеза некоторых красителей.
При нитровании диметиланилина азотной кислотой три нитро-
группы замещают водородные атомы кольца, а четвертая
становится на место одной из метильных групп. В результате
получается тринитрофенилметилнитрамин, или тетрил:
Тетрил—бледно-желтые кристаллы (темп, плавл. 130 °С);
является одним из сильнейших взрывчатых веществ.
250. Диамины. Простейшими ароматическими диаминами
являются диаминопроизводные бензола; они называются фенилен-
диаминами:
HaN-<Q-NH3
о-фенилендиамин и-фенилендиамин п-фенилендиамин
о-Фенилендиамин и гс-фенилендиамин получаются
восстановлением соответствующих нитроанилинов, лг-фенилендиамин—
восстановлением лг-динитробензола.
Фенилендиамины—кристаллические вещества, очень хорошо
растворяющиеся в "горячей воде. Они легко окисляются и при
стоянии на воздухе окрашиваются в темный цвет.
Фенилендиамины являются слабыми основаниями; они образуют соли с двумя
эквивалентами кислоты. лг-Фенилендиамин и «-фенилендиамин
применяются для синтеза красителей.
Принадлежность двузамещенных производных бензола к
орто-, мета- или пара-ряду можно определить по числу
изомерных веществ, получающихся из этого соединения при замещении
третьего атома водорода бензольного ядра на какой-либо иной
атом или радикал. Известно шесть диаминобензойных кислот
состава C6H3(NH2JCOOH. Эти кислоты, отщепляя СО2, дают
фенилендиамины.
496
Глава XVIII Одноядерные ароматические соединения
лі-Фенилендиамин с темп. кип. 284 °С получаеіси из трех ди-
аминобензойных кислот:
NH,
NHa
NH2
j >. л-фениленднамнн
о-Фенилендиамин с темп. кип. 257 °С получается из двух диами-
нобензойных кислот:
HaN !HB
NHa_Q>
HaN
NHa
о-фенилеьдиаыин
{COOiH
п-Фенилендиамин с темп. кип. 267 °С получается из одной ди-
аминобензойной кислоты:
JCOOjH
n-фенилендиамнн
Следовательно, для двузамещенных с двумя одинаковыми
заместителями три трех замещенных изомера соответствуют
мета-соединению, два трехзамещенных—орто-соединению и один трех-
замещенный изомер—пара-соединению.
Физические свойства ароматических аминов приведены в
табл. 21.
241. Аминофенолы
497
Таблица 21
Физические свойства ароматических аминов
Название
Формула
Темпера
тура
плавления
°С
—6
—24,4
—31,2
+44
+2.4
+53,5
+ 127
+ 102
+62.8
+ 139,7
Температура
квпення
•С
184,4
200.3
203,4
200,5
194
302
365
257
284
267
Растворимость в г
на 100 г воды
Анилин
о-Толуидин . . .
л-Толуидин . . .
п-Толуидин . . .
Диметилааилин .
Дифениламин . .
Трифениламин . .
о-Фенилендиамин
.м-Фенилен диамин
п-Фенилендиамин
C„H6NHS
CH3CeH4NH2
CH3CeH4NHa
(C.HJ.NH
(CeH6KN
CeH4(NH2)a
C„H4(NH2J
3,5 при 25 *C
1,5 при 25 *C
Мало растворим
0,730 при 21 °С
Очень мало
растворим
Нерастворим
Очень хорошо
растворимы
в горячей воде
АМИНОФЕНОЛЫ И АМИНОБЕНЗОЙНЫЕ КИСЛОТЫ
251. Аминофенолы. Аминофенолами называются вещества,
которые являются одновременно и ароматическими аминами и
фенолами.
Аминофенолы—твердые кристаллические вещества. о-Амино-
фенол плавится при 172 °С, л-аминофенол—при 186 °С,
температура плавления .и-аминофенола 122 °С. Являясь одновременно и
аминами и фенолами, аминофенолы образуют соли и с кислотами и
со щелочами:
NaO-CeH4-NH2
+НС1
+NaOH
+HCI
,—NH2 г=± НО
+NaOH
—СН4->Ш2 ¦ НС1
Эти соли более растворимы в воде, чем сами аминофенолы.
Водные, особенно щелочные, растворы аминофенолов легко
окисляются кислородом воздуха. Благодаря сильным
восстановительным свойствам аминофенолы, наряду с гидрохиноном и
пирогаллолом, применяются в фотографии как проявители. При
проявлении главным процессом является восстановление галоидных
соединений серебра до металлического серебра. Ввиду
сложности процесса дать здесь уравнение реакции невозможно.
Можно лишь указать, что в бензольном ядре органического прояви-
32-788
498
Глава XVIII. Одноядерные ароматические соединения
теля должно содержаться не менее двух групп ОН или группа ОН
и группа NH2 в орто- или пара-положении. В первой стадии
окисления происходит отщепление двух атомов водорода от
этих двух групп.
Формулы и названия некоторых проявителей приведены в
табл. 22.
Таблица 22
Фотографические проявители
Химическое наименование
Формула
Название
проявителя
я-Аминофенол солянокислый . ,
я-Метиламинофенол сернокислый
2,4-Диаминофеиол солянокислый
Гидрохинон ¦
Пирогаллол
НО—QH4—NHg.HCI
НО—С„Н4—NH(CHa) • H2SO4
HO-CeHs(NH2J.HCI
Парааминофенол
Метол
Амидол
Гидрохинон
Пирогаллол
Парааминофенол получается главным образом
электролитическим восстановлением нитробензола. Для этой цели
подвергают электролизу раствор серной кислоты, в котором в виде
суспензии находится нитробензол. Выделяющийся на катоде
водород восстанавливает нитробензол в фенилгидроксиламин:
4Н
О
_NH—ОН -f H2O
фенилгидроксиламин
Фенилгидроксиламин С6Н5—NH—ОН можно рассматривать
как производное гидроксиламина NH2—ОН, в молекуле которого
атом водорода при азоте замещен на фенил.
Фенилгидроксиламин в кислом растворе претерпевает перегруппировку и
переходит в ге-аминофенол:
фенилгидроксиламин
rt-аминофенол
При этой реакции происходит обмен местами между гидроксилом
и атомом водорода в пара-положении.
252. Ионообменные смолы
499
я-Аминофенол можно получить и другим путем. Так,
например, он образуется при восстановлении /г-нитрофенола:
H0~V—/~
+ 6Н
НО—
2Н2О
252. Ионообменные смолы. Ароматические диамины, амино-
фенолы и амины, подобно фенолу, легко вступают в реакцию
поликонденсации с формальдегидом. При определенном
соотношении компонентов, а также pH среды получаются
высокомолекулярные соединения с сетчатой пространственной структурой.
Обычно это черные или темно-бурые стекловидные вещества,
нерастворимые в воде и других растворителях. В воде и водных
растворах солей и кислот они набухают.
Благодаря тому что эти вещества содержат полярные группы
основного характера: —NH—, —NH2, при набухании в воде онн
образуют нерастворимый макромолекул ярный катион и ион ОН".
Таким образом, их можно рассматривать как
высокомолекулярные слабые основания.
Отличительной особенностью этих полимеров является то, что
они вступают в реакцию обмена с растворами электролитов с
образованием солей по следующей схеме:
ОН
ОН
он
I
СНз
I U
СНа—N—СН3 ОН"
СН3
ОН
неї
І-СН.,-,
I ¦ 1+
CH3-N-CH3 C|-
СН3
нон
Вследствие набухания полимерного основания в водных
растворах солей повышается скорость диффузии ионов в полимере,
а это ускоряет реакцию ионного обмена и вовлекает в реакцию
все имеющиеся основные группы полимера, несмотря на его
нерастворимость. При обработке полученной соли водой можно
обратно выделить исходное соединение.
500 Глава XVIII. Одноядерные ароматические соединения
Свойство полимеров вступать в обратимую реакцию с
электролитами дает возможность использовать эти полимеры в качестве
ионообменных фильтров для извлечения анионов из растворов.
Высокомолекулярные основания получили название анионооб-
менных смол или анионипгов.
Если проводить конденсацию гс-фенолсульфокислоты с
формальдегидом, получаются сульфополифенолы:
. ОН ОН
SO,H
SO3H
Это нерастворимые, но набухающие в воде высокомолекулярные
кислоты. Полимеры легко поглощают катионы из водных
растворов солей:
лМеХ"
он он
, h nHX
so*
|яМе SO,Me
При обработке полимерных солей кислотами можно выделить
соли металлов в чистом виде. Высокомолекулярные соединения,
содержащие в своем составе ионные группы кислого характера
—ОН, —SO3H, —СООН, называются кашионообменными
смолами или катионитами.
Ионообменные смолы нашли большое техническое применение.
С введением в практику ионитов стало возможным проводить
разделение близких в химическом отношении соединений, например
разделять смеси аминокислот, алкалоидов и др. Кроме того;
легко осуществляется задача избирательного извлечения кислот или
оснований из растворов их смесей.
253. Аминобензойные кислоты 50)
Промышленность выпускает большое число разнообразных
синтетических ионитов (например, КУ-1, КУ-2—катионит
универсальный; АН-1, АН-2—анионит низкоосновной).
Аниониты и катиониты обычно характеризуются обменной
емкостью (большей частью в мг-экв/мл). Последняя тем больше,
чем больше ионит содержит ионообменных групп
(соответственно амино-, гидроксильных, сульфо- или карбоксильных
групп).
Частицы ионита имеют пористую структуру. Для
рационального выбора ионита надо знать его плотность, набухаемость,
статистическую и полную обменную емкость.
Замечено, что катионы лучше всего вытесняются ионами
водорода, а анионы—ионами гидроксила. Поэтому для получения
водородной формы катионита его обрабатывают разбавленной
НС1. Аниониты применяют в форме оснований, содержащих ОН-
группы, которые получаются путем обработки смолы
разбавленным раствором едкого натра.
253. Аминобензойные кислоты. Из трех возможных (по
теории строения) аминобензойных кислот наибольшее значение
имеет о-аминобензойная, или анпграниловая, кислота:
соон
Антраниловая кислота плавится при 145 °С, имеет сладкий
вкус, легко растворяется в воде и спирте. Она применяется для
изготовления некоторых красителей (индиго) и фармацевтических
препаратов; ее метиловый эфир находит применение в
парфюмерии.
Промышленный способ получения антраниловой кислоты
заключается в действии щелочи и хлорноватистых солей на фтал-
имид. Вначале образуется соль фталаминовои кислоты, которая
затем по реакции Гофмана (см. стр. 365) дает соль антраниловой
кислоты:
СО CONH,
COONa
4<^ \МН + NaOH , QH4/
CO .
+ NaaCO3 + NaCI + Н„О
lOONa
502 Глава XVIIІ. Одноядерные ароматические соединения
п-Аминобензойная кислота
NFi,
СООН
бесцветные кристаллы (темп. пл. 187—188 °С). Получают
rt-аминобензойную кислоту из толуола по следующей схеме:
СНа СН3 СООН СООН
6Н
толуол п нитро п-нитробенэом і ачинобензей
ная кислота ная кислита
я-Аминобензойная кислота является исходным веществом для
синтеза ряда местнодействующих анестезирующих
(обезболивающих) препаратов: анестезина (этиловый эфир гс-аминобензой-
ной кислоты)
NH2
и новокаина (сложный эфир гс-аминобензойной кислоты и ди-
этиламиноэтилового спирта):
NH„
Производное «-аминобепзойной кислоты—п-аминосалицило-
вая кислота
применяется под названием «ПАСК» как препарат для лечения
туберкулеза.
Диазосоединения 503
254. Фенилаланин. Фенилаланин
>—СН„—CH(NH2)—COOH
можно рассматривать как производное аланина (см. стр. 378),
в молекуле которого атом водорода при ?-углеродном атоме
замещен фенилом.
Левовращающие фенилаланин (темп. пл. 280 °С) и л-оксифе-
нилаланин, или тирозин, НО—С6Н4—СН2—CH(NH2)—COOH
(темп, плавл. 316—320 °С) образуются при гидролизе почти всех
белковых веществ. Из производных тирозина очень большое
значение имеет тироксин:
НО—<(%—О—\/— СН2—CH(NHa)—COOH
Тироксин (темп, плавл. 321 °С) был выделен в 1919 г. из
щитовидной железы. Он является гормоном, регулирующим обмен
веществ. Его строение было установлено в 1926 г. как изучением
продуктов расщепления, так и синтезом.
ДИАЗОСОЕДИНЕНИЯ
255. Получение и строение диазосоединений.
Диазосоединения получаются действием азотистой кислоты на соли первичных
ароматических аминов*. Так, из хлористоводородной соли
анилина получается вещество состава C6H5N2C1, называемое
хлористым фенилдиазонием, из азотнокислой соли этого
амина—азотнокислый фенилдиазоний CeHsN2NO3:
CH.-N—:н 4
сі Nh
"
QHs-N-IH 4
О—NO3
HOk
<yf
•••••••••••¦
но!ч
(У?
і
N * CeHa-N=N +
СІ
хлористый
феннлдиазоний
б—NO2
азотнокислый
фенилдиааоний
2НаО
2Н2О
* Эту реакцию проводят, прибавляя к соли амина минеральную кислоту
и эквивалентное количество азотистокислого натрия. При действии кислоты
на азотистокислий натрий образуется азотистая кислота, которая затем и
реагирует с солью амина.
504 Глава XVIII. Одноядерные ароматические соединения
Диазосоединения, как это видно из приведенных уравнений,
можно рассматривать как производные солей аммония, в которых
один атом водорода замещен ароматическим радикалом, а три
других—атомом азота. Такое производное аммония называется
диазонием.
Диазосоединения, подобно солям аммония, представляют
собой солеобразные вещества, в водном растворе почти полностью
+ —
диссоциированные на ионы CeH6N2 и СІ и имеющие, несмотря
на то что один из ионов—кислотный остаток, совершенно
нейтральную реакцию.
Если раствор хлористого фенилдиазония обработать влажной
окисью серебра, происходит выпадение творожистого осадка
хлористого серебра; сам раствор приобретает при этом
сильнощелочную реакцию. Образовавшаяся очень неустойчивая
гидроокись фенилдиазония представляет собой основание, по силе
равное едким щелочам (NaOH, КОН):
QHj-N СГ + AgOH » QHj—N ОН + AgCI
Я В
гидроокись
диазония
Раствор быстро теряет щелочные свойства. Это объясняется
переходом гидроокиси диазония в изомерный ей диазогидрапг.
Последний обладает свойствами слабой кислоты:
QH5N + ОН ?=i [QH6N ОН] ^ZZi QHj—N=N—ОН ї=* QH6N=N—O +H
II
N
При действии щелочей это превращение совершается очень
быстро, в результате чего получаются соли диазогидратов—ди-
азотаты:
QH5— N=N—ОН + КОН » QHs—N=N-OK + Н2О
При прибавлении избытка минеральной кислоты диазотаты
снова превращаются в соли диазония.
Соли диазония весьма неустойчивы, многие из них в сухом
виде обладают способностью взрывать*; они легко разлагаются
и способны к многочисленным реакциям. При помощи солей
диазония можно приготовить большое число самых разнообразных
* Поэтому обычно работают с водными растворами солей диазония.
256. Реакции диазсоединенип, проходящие с выделением азота 505
веществ. Некоторые из реакций- диазосоединений проходят с
выделением свободного азота; при других реакциях азот не
выделяется.
256. Реакции диазосоединений, проходящие с выделением
азота. 1. Нагревание водных растворов солей диазония приводит
+
к отщеплению кислотного остатка; группа —N=N замещается
гидроксилом и образуются фенолы:
QH6!Nai ІСІ +НІОН * QH6OH + N2 + НС1
:: :
¦
2. При нагревании диазосоединений со спиртами протекают
одновременно две реакции, причем в отдельных случаях идет
преимущественно одна из них. По одной из этих реакций
молекула спирта отдает два атома водорода, которые переводят ди-
азосоединение в ароматический углеводород, при этом спирт
окисляется в альдегид:
QH6N2C1 + СН3СН2ОН * QHe + N2 + СНЭСНО + НС1
В результате второй реакции получаются простые эфиры:
QH5N2CI + QH6OH » QH6OQH6 + N, + HC1
3. При нагревании растворов диазосоединений с йодистым
калием получаются иодзамещенные в ядре:
QH6N2C1 + KJ » QH6J + N2 + KCI
4. При нагревании растворов хлористых или бромистых солей
диазония в присутствии соответствующих галоидных солей
одновалентной меди (или медного порошка) происходит выделение
азота и галоид становится на место диазогруппы:
Сіі2СІ2
QH6N,C1 »• QH6C! + N2
5. При действии цианистого калия на соли диазония в
определенных условиях получаются нитрилы. Так, при действии
цианистого калия на хлористый фенилдиазоний получается бензо-
нитрил:
QH5NaC! + KCN * QH6CN + N, + КС!
6. Диазосоединения образуют с хлорной ртутью двойные
соли, которые, как показал А. Н. Несмеянов, в присутствии порош-
506 Глава XVIII. Одноядерные ароматические соединения
ка меди разлагаются с образованием ртутьорганических
соединений:
CeHBNaCl HgCla + Си » QHsHgCI + СиС1а + Na
Аналогично могут быть получены ароматические производные
сурьмы и олова (А. Н. Несмеянов, К. А. Кочешков, О. А. Реутов).
257. Синтезы при помощи диазосоедииений. Пользуясь
легкодоступными ароматическими аминами, можно синтезировать
через диазосоединения многочисленные производные бензола,
которые не получаются прямой реакцией. Ниже даны примеры
таких синтезов.
Синтез п-иодтолуола. В качестве исходного вещества для
приготовления л-иодтолуола берут я-толуидин:
/СИ, П) д„рова^ .СН:, ^ ,СН3 О) ш
XNH2 D) \N„CJ \j D)
л-иодтолуол
Синтез м-ншпрофенола. Прямым нитрованием фенола нельзя
получить jn-нитрофенол, однако его можно получить через диазо-
соединение из легко доступного jn-нитроанилина:
A) диазотироваиие /NaOSO3H HaO
C) ЧМОа кипячение
)Н (і,
+ Na + HaSO4
NOa C)
Синтез о-толуиловой кислоты. Исходным веществом является
о-толуидин, получаемый обычно из онитротолуола. Диазотиро-
ванием о-толуидина получают хлористый о-толилдиазоний. При
действии на него цианистой медью Cua(CN)a в растворе
цианистого калия происходит разложение диазосоединения и
образование нитрила. Последний может быть гидролизован
нагреванием с соляной или серной кислотой:
C?M4<T »• СйН4ч >• СбН4<ґ ~г l»a
XNHa B) \NaCI XCN B)
1 +HaO(Hc^ q,h /СНз
" ХЮОН
258. Восстановление диазосоединений. Гидразины. При
восстановлении диазосоединения переходят в производные гидр-
255 Азосоединения 507
азина NH2—NH2. Так, при восстановлении хлористого фенил-
диазония получается солянокислый фенилгидразин:
QH5N2CI + 4Н » QHg—NH—NHj-HO
Фенилгидразин образует пластинчатые кристаллы (темп.
плавл. 23 °С; темп. кип. 241 °С), легко окисляется, буреет на
воздухе, обладает основными свойствами. Фенилгидразин
применяется при исследовании альдегидов, нетонов и Сахаров (образование
фенилгидразонов и озазонов). Им пользуются для синтеза
многих веществ, имеющих широкое применение, например
антипирина, пирамидона, некоторых красителей.
Некоторые другие реакции диазосоединений, происходящие
без выделения азота, приведены в разделе об азокрасителях.
АЗОСОЕДИНЕНИЯ. АЗОКРАСИТЕЛИ
259. Азосоединения. Азосоединениями называются
соединения, содержащие в молекуле группу «азо» —N=N—, свободные
валентности которой связаны с углеводородными радикалами.
Примерами этих веществ могут служить азобензол и
бензолазотолуол
O~N=N~<O
азобензол . бензолазотолуол
Азосоединения получаются восстановлением нитросоединений.
Так, азобензол может быть получен восстановлением
нитробензола хлористым оловом в присутствии избытка едкого кали.
На состав и характер продуктов восстановления
ароматических соединений решающее влияние оказывают характер среды,
взятый восстановитель и другие условия реакции.
В кислой среде восстановление протекает по следующей схеме:
QH5NOa -^-* QH5NO -^-» QH5bIHOH —?-* C6H5NHa
—Hau —nW
нитробензол ннтрозобензол фенилгидр анилин
окси мамин
В нейтральной среде реакция восстановления обычно доходит
лишь до стадии образования фенилгидроксиламина. В щелочной
среде, в результате конденсирующего воздействия щелочи, про-
508 Глава XVIIІ. Одноядерные ароматические соединения
исходит образование двухядерных соединений с отщеплением
воды и установлением связи между двумя атомами азота:
2C»HeNOs -^-» Q,HBNO + QANHOH —^U
нитроза фен илгидр-
бекзол океиламин
.—N = N—Q,H5 —U СаНв—N=N—QH6 -—¦ C,H6NH—NHCeH5
азобензол гндразебенза."
азоксибензол
Конечным продуктом восстановления нитросоединений в
щелочной среде является гидразобензол, в кислой среде—первичный
амин.
Фенилгидроксилашн CeH6—NHOH может быть получен восстановлением
нитробензола цинковой пылью в растворе хлористого аммония Феннлгидр-
оксиламин—бесцветные иглы (темп, плавл 81 °С). О перегруппировке его в
парааминофенол см стр 498
Ншпрозобензол С„НВ— N0 получается окислением фенилгидроксилаыина
хромовой смесью Нитрозобензол—бесцветные иглы (темп плавл. 68 °С);
в расплавленном состоянии а в растворах окрашен в сине-зеленый овет
В твердом состоянии ннтрозобензол бимолекулярен. При плавлении н
растворении происходит диссоциация с образованием окрашенной
мономолекулярной формы
+
Аэоксибензол QH,-,— N = N—С„Н- обыкновенно получают кипячением ни-
О"
тробензола с метилатом натрия, который при этом окисляется в муравьино
кислый натрий;
4QH6NO2 + 3CH3ONa > 2С„Н,—N=N—С«НВ + 3HC00Na + 3H,0
і
і"
Азоксибензол—бледно-желтые иглы (темп, плавл. 36 °С), При нагревании с
железными опилками он восстанавливается в азобензол.
Гидразобензол CgHj—NH—NH—СвН5 получается восстановлением
нитробензола или азобензола цинковой пылью и едким кали
Гидразобензол—бесцветные кристаллы (темп плавл. 131 °С). При действии сильных
восстановителей гидразобензол переходит в анилин; легко окиелнется кислородом
воздуха, снова превращаясь в азобензол О бензидиновой перегруппировке см.
на стр. 518 ел.
Простейшие незамещенные азосоединения—вещества красного
или оранжевого цвета. Так, азобензол представляет собой
оранжево-красные кристаллы, плавящиеся при 68 "С. Азосоедине-
ния хотя и обладают окраской, но не являются красителями, так
как они не удерживаются тканями.
250. Аминоазосоедияения, оксиазосоединения 509
260. Аминоазосоединения, оксиазосоединения. Аминопроиз-
водные азосоединений называются аминоазосоединениями.
Примерами их могут служить:
—N=N—C.H.,—NHg QiH6—N = N—CH/
аминоазойснзол диаминоазооензо^
Гидроксильные производные азосоединений называются окси-
азосоединениями. Примерами соединений такого рода являются:
/ОН
Q,HO—N=N—Q,H,— ОН CeH6—N=N—CeH./
оксиа^оЛензол диоксназобензол
Как видно из приведенных формул, аминоазосоединения
содержат аминогруппу, поэтому они способны образовывать соли
с кислотами; оксиазосоединения содержат в ядре гидроксил,
т. е. являются фенолами, и потому способны образовывать соли
со щелочами. Эти соли растворимы в воде. В виде этих солей
аминосоединения и оксиазосоединения находят широкое
применение в качестве красителей. Красители такого типа
называются азокрасителями. Примерами простейших азокрасителей
могут служить: солянокислый n-аминоазобензол, называемый
также анилиновым желтым
~N = N-<^~)>-NHa - HCI
и солянокислый 2,4-диаминоазобензол, или хризоидип
4~У—n=n—/==^—nh, -на
Более сложными азокрасителями являются соединения, в
которых атомы водорода аминогруппы замешены углеводородными
радикалами, например п-диметиламиноазобензол
»—N=N
солянокислая соль которого является желтым красителем, или
510 Гл. XVIII. Одноядерные ароматические соединения
соединения, содержащие сульфогруппу, например диметилами-
ноазобензо лсул ьфокис лота
которая известна под названием гелиантина, или метилового
оранжевого* ¦
261. Получение азокрасителей. Аминоазосоединения
получаются из диазосоединений и ароматических аминов. При действии
на третичный амин хлористых солей диазония хлор соединяется
с водородом бензольного ядра амина, остатки диазония и амина
образуют аминоазосоединение:
диметнламиноазобенаол
Оксиазосоединения получаются действием солей диазония на
фенолы:
-|аТн|-/~\-он —, <G}>-n=n-/~\-oh + на
хлористый фенол оксиазобензол
фенил диазоний
Взаимодействие между солями диазония и аминами или
фенолами может происходить в щелочной или слабокислой среде.
Поэтому зту реакцию проводят в присутствии едкого натра, соды
или уксуснокислого натрия. Едкий натр и сода нейтрализуют
образующуюся соляную кислоту. При взаимодействии
уксуснокислого натрия с соляной кислотой получается хлористый натрий
и уксусная кислота. Таким образом, в результате прибавления
уксуснокислого натрия в растворе образуется вместо сильной
соляной кислоты слабая уксусная кислота.
Реакцию получения азосоединений взаимодействием пары
компонентов—диазосоединения и амина (или
фенола)—называют азосочетанием.
Как видно из структурных формул диметиламиноазобензола
и оксиазобензола, аминогруппа и гидроксил в их молекулах
находятся в пара-положении к азогруппе. Это является общим
правилом; при реакции солей диазония с фенолами и аминами
* Применяемый в аналитической химии индикатор метиловый оранжевый
является натриевой солью этой сульфокислоти.
261. Получение аяокрасителей 511
в большинстве случаев замещается водород, находящийся в
пара-положении относительно аминогруппы или гидроксила. Если
пара-положение занято какой-либо группой, то замещается
водород в орто-положении.
Строение аэокрасителей устанавливается путем
идентификации аминов, образующихся при восстановлении красителей. При
восстановлении разрываются связи между атомами азота, и из
каждого из них образуется аминогруппа:
оксиазобеизал анилин л-аминофенол
Азокрасители применяют главным образом для крашения
текстильных волокон, некоторые из них находят применение для
окраски кожи, а также в полиграфической и лакокрасочной
промышленности Азокраситель метиловый оранжевый широко
применяется в аналитической химии как индикатор.
Метиловый оранжевый, или диметиламиноазобензолсульфокис-
лота
готовится диазотированием сульфаниловой кислоты и сочетанием
полученного диазосоединения с диметиланилином:
NaOH »
_. + H,0
Красный стрептоцид (пронтозил красный)
HaN—f~\—N=N—<^~>)-SOa—NHa
получается сочетанием диазотированного амида сульфаниловой
кислоты с лі-фенилендиамином. Красный стрептоцид был первым
эффективным средством борьбы против заболеваний, вызываемых
болезнетворными кокками. Его активность при стрептококковой
инфекции на опытах с белыми мышами открыл в 1935 г.
бактериолог Домагк.
512 Гл. XVIII. Одноядерные ароматические соединения
Амид сульфаниловой кислоты
известен под названием белого стрептоцида (пронтозил белый).
Домагк в течение ряда лет изучал действие на стрептококки самых
разнообразных соединений как ранее описанных, так и впервые синтезированных
работавшими с ним химиками Несмотря на то, что некоторые из этих
соединений оказались весьма активными, они по разным причинам не могли быть
использованы для лечения стрептококковых заболеваний. Одни из них,
например ряд соединений золота, вызывали явления общего отравления
организма; другие (некоторые производные хинина), будучи активны при местном
применении, не оказывали действия при общей стрептококковой инфекции;
третьи проявляли активность только in vitro, т. е нне живого организма, и не
оказывали действия на стрептококки in vivo, т. е. в живом организме
Последней особенностью обладают, как это было известно еще с 1913 г., многие азо-
красители. Домагк добился успеха, только перейдя к азокрасителям,
содержащим сульфамидную группу SO^NHa; одним из них и был красный стрептоцид.
В отличие от других азокрасителей, эти вещества активны только in vivo, a
не in vitro Носителем противострептококковой активности в азокрасителях
является сульфамидная группа атомов. Удаление этой группы приводит к азо
красителям, не активным в организме. Если же сульфамидная группа сохра
няется, то активность остается и при изменении остальной части молекулы.
Вскоре удалось показать, что активным средством против стрептококков
является один из продуктов расщепления красного стрептоцида в организме,
представляющий собой амид сульфаниловой кислоты, синтезированный еще
в 1908 г.
Амид сульфаниловой кислоты (белый стрептоцид), подобно красному
стрептоциду, применяется при лечении рожи, острой аигины и ее осложнений,
стрептококкового менингита и других стрептококковых заболеваний.
Стрептоцид с большим успехом применяется не только при общей стрептококковой
инфекции, по и как местное средство при лечении гнойных ран и ожогов
Открытие противострептококковой активности
сульфаниламида послужило толчком для синтеза новых соединений,
содержащих сульфамидную группу атомов; за пять лет было
синтезировано около 3000 этих веществ. К ним относятся сульфидин
(см. стр. 612) и сульфатиазол (см. стр. 600), которые находятся
в самом близком отношении к белому стрепюциду.
262. Красители и крашение. Аминоазосоединения и оксиазо-
соединения, как уже было указано, являются красителями, Под
красителями разумеются вещества, способные так окрашивать
ткани, что краску нельзя удалить трением или мытьем. Следует
различать окрашенное вещество от красителя; так, хотя
азобензол окрашен в оранжево-красный цвет, он не являеіся
красителем, т. е. не способен окрашивать волокно.
Естественно, возникает вопрос, от каких причин зависит
появление окраски вещества и каким условиям должно
удовлетворять окрашенное вещество, чтобы оно являлось красителем.
262. Красители и крашение 513
Некоторые вещества поглощают совершенно равномерно лучи всех цветов.
Если через такое вещество пропустить пучок белых лучей, то последние,
пройдя через него, лишь ослабеют в своей яркости, но останутся белыми. Такие
вещества—бесцветны. Окрашенные же вещества поглощают преимущественно
лучи определенных цветов, т. е. определенной длины волны; они, как говорят,
обладают избирательным поглощением. Направим на такое вещество (или
его раствор) пучок белых лучей и предположим, что у нас ие будет происходить
никаких других явлений, кроме поглощения света. Тогда лучи, которые
пройдут через вещество, уже не будут белыми лучами, а приобретут ту окраску,
которая получается при смешении всех цветов солнечного спектра, кроме
поглощенных. Например, если вещество поглотит сине-зеленые лучи, то
прошедшие через вещество лучи будут окрашены в красный цвет, так как красный
цвет может быть получен смешением всех цветов солнечного спектра, кроме
сине-зеленых. Некоторые вещества обладают избирательным поглощением
только в области инфракрасных и ультрафиолетовых лучей. Так, например,
бензол обладает избирательным поглощением в ультрафиолетовой части
спектра. Такие практически бесцветные вещества, строго говоря, тоже «окрашены».
С обычной же точки зрения окрашенными считаются лишь те вещества,
которые обладают избирательным поглощением в видимой части спектра.
Согласно хромофорной теории Витта, для того чтобы
органическое соединение являлось красителем, необходимо
наличие в его молекуле особых групп: хромофорных и ауксохром-
вых.
Хромофоры («носители цветности»)—атомные группировки,
наличие которых придает соединению окраску. Характерной
структурной особенностью хромофоров является наличие
ненасыщенных атомов и двойных связей. Сильными хромофорами
являются, например, нитрогруппа —NO2, нитрозогруппа —N=0,
азогруппа —N=N—. Относительно более слабый1 хромофор—
карбонильная группа >С=0. Еще более слабым, но весьма
важным хромофором является двойная связь >С=С<.
Дальнейшим развитием теории цветности органических
соединений явилось представление о сложных хромофорах.
Наиболее важным видом сложных хромофорных систем являются
конъюгированные системы двойных связей ациклического или
циклического строения. По мере удлинения сопряженной цепи
спектр поглощения переходит из ультрафиолетовой области
(короткие волны) все дальше и дальше в сторону красной части
спектра (более длинные волны). Примером окрашенных
углеводородов являются: каротин—оранжевого цвета,
ликопин—оранжево-красного цвета (см. стр. 568), дифенилгексадекаоктаен
G6Hя—(СН=СН)8--С6Н5—медно-красного цвета.
Бензольное ядро и ароматические углеводороды с
конденсированными ядрами являются сложными хромофорными
системами, имеющими большое значение в химии красителей. Сами по
себе бензол и нафталин бесцветны, но при соединении с другими
хромофорами получаются окрашенные вещества.
514 Г л XVIII Одноядерные ароматические соединения
Важным видом сложных хромофорных систем являются па-
рахиноидная (I) и ортохиноидная (II) группировки. Они
содержатся, например, в я-бензохиноне (III) и о-бензохиноне (IV):
О О
II II II II г,
О (/ О (Г
II II
о
п и о хиноидньге п-бензохинон о-бензохинон
хромофорные системы (желтый) (красный)
1 11 III IV
Для превращения окрашенного вещества в краситель
необходимо ввести в его молекулу, кроме хромофоров, добавочные
группы—«ауксохромы» («усилители цветности»), способствующие
«углублению« цвета (сдвигу области поглощения от
ультрафиолетовой области в сторону красной части спектра). При этом
наблюдается появление сродства к волокну. Необходимо,
однако, строго различать влияние групп на цветность и на сродство
к волокну (способность окрашивать волокно), которое зависит
от разных причин. Типичными ауксохромами являются группы
—NH2, —ОН, —Ы(СНЯJ- Кислотные группы — SO3H, —СООН
в настоящее время не включают в число ауксохромных. Они
оказывают лишь незначительное влияние на цветность, но имеют
большое значение для крашения, сообщая красителям
растворимость в воде и сродство к шерстяному и шелковому волокну.
Некоторые из красителей окрашивают хлопчатобумажную
ткань непосредственно из водного раствора. Такие красители
называются субстантивными* или прямыми.
Для крашения тканей некоторыми красителями (так
называемыми протравными) ткань необходимо предварительно
пропитать веществом, которое образует с краской нерастворимое
соединение, что способствует закреплению ее на ткани. Вещества,
служащие для закрепления красителя на волокне, называются
протравами. В качестве протрав применяют уксуснокислый
алюминий, соли окиси железа и окиси меди, соединения олова, а
также таннин. Для крашения ткань погружают в раствор
протравы и затем подвергают действию пара при высокой темпера-
* Первый субстантивный краситель был получен и применен в 1883 г.
О. К. Миллером и С. И. Прохоровым на Трехгорной мануфактуре в Москве.
Краситель получался окислением роданистого калия бертолетовой солью.
Он был назван «канарином», так как окрашивал ткани в канареечно-желтый
цвет.
263. Природные и синтетические красители 515
туре. При этом происходит гидролиз примененного в качестве
протравы вещества. Образовавшаяся при гидролизе гидроокись
металла в мелкораздробленном виде закрепляется на волокне;
соединяясь с ней, краситель образует нерастворимый лак.
Применяя различные протравы, можно одним и тем же красителем
окрасить ткань в различные цвета (см. также стр. 535, 542).
Протравное крашение было известно очень давно. Живший в I веке до
нашей эры римский писатель Плиний писал:
«В Египте замечательно раскрашивают и одежды, для чего белую материю
предварительно пропитывают не красками, но поглощающими краску
составами. При этом на материи ничего не выявляется, но вскоре после погружения
в котел с кипящей краской она оттуда вынимается раскрашенной.
Удивительно, что хотя в котле краска одна, одежды приобретают тот или иной цвет в
зависимости от свойств состава. После этого краску смыть нельзя. В
котле краски не смешиваются; там, где запечатлеется рисунок, он при
кипячении выйдет одного цвета. От кипячения одежды становятся более
прочными».
263. Природные и синтетические красители. В прежнее
время органические красители добывались исключительно из
растений и животных. Из корней растения марены добывали
ализарин, который с незапамятных времен применялся для
кумачового окрашения; из индигоносных растений получали синее
индиго; из высушенных насекомых, живущих на некоторых
породах кактуса,— красный кармин. Знаменитый в древности
«античный пурпур», применявшийся для окраски царских мантий
и драгоценных одежд, добывался из одного вида моллюска,
живущего по берегам Средиземного моря; моллюск выделяет
бесцветное вещество, которое на свету окисляется кислородом
воздуха и превращается в краску. Строение этого красителя было
установлено в 1909—1911 гг. Фридлендером (см. стр. 600).
Первый искусственный краситель (мовеин) был получен в
Англии в 1856 г. В. Перкиным, который случайно получил это
вещество при окислении нечистого анилина. Вскоре стали
готовить заводским способом и ряд других красителей, в том числе
ализарин и индиго.
Были получены также и многие сотни красящих веществ, не
встречающихся в природе. В настоящее время синтетические
красители почти совершенно вытеснили природные.
Синтезы первых красителей имели случайный характер;
только после создания А. М. Бутлеровым теории строения,
установления структурной фомулы бензола и выяснения строения и
закономерности образования красящих веществ разработка
методов получения искусственных красителей получила широкую
научную базу.
В настоящее время синтезированы многие тысячи
индивидуальных красителей, принадлежащих к различным классам со-
33*
516 Гл. XVIII. Одноядерные ароматические соединения
единений. Классификация их и рациональное наименование были
связаны с затруднениями как вследствие многообразия
химического состава, методов получения и областей применения, так
и в результате того, что зарубежные фирмы, выпускавшие на
рынок тот или иной краситель, часто присваивали ему случайное
наименование, ничего общего не имеющее с его составом или
строением.
В настоящее время приняты 2 системы классификации
красителей: химическая и техническая.
Химическая классификация красителей основана на общности
хромофорных групп, строения и химических свойств. В
соответствии с ней красители подразделяются на такие группы, как
нитрокрасители (хромофор—нитрогруппа), нитрозокрасители
(хромофор—нитрозогруппа), азокрасители, арилметановые
красители, индиго и индигоидные красители и другие.
Химическая классификация наиболее удобна для изучения
химии красителей, разработки рациональных методов их
получения, изыскания новых марок красителей.
Техническая классификация подразделяет красители в
зависимости от областей и методов их применения Так, в одну группу
кислотных красителей объединяются все красители, красящие
шерсть из кислой ванны, хотя химически это могут быть самые
различные по химическому составу соединения: простые или
сложные азокрасители, трифенилметановые красители, производные
антрахинона и др. Техническая классификация дает возможность
судить о назначении красителя и об основных методах его
применения.
Техническая классификация предусматривает следующие
важнейшие группы красителей: прямые
(субстантивные)—окрашивающие целлюлозные волокна из нейтральных растворов;
сернистые—окрашивающие целлюлозные волокна из водных растворов
сернистого натрия; кислотные—окрашивающие шерсть и шелк из
кислой ванны; протравные для шерсти—окрашивающие шерсть
подобно кислотным красителям, но с последующим закреплением
окраски обработкой, например, хромовыми солями; кубовые
красители—окрашивающие целлюлозные волокна в форме
бесцветных продуктов восстановления (так называемых лейкосоедине-
ний) из слабощелочного раствора (куба) с последующим
образованием красителя при окислении кислородом воздуха
непосредственно на ткани; пигменты и лаки—нерастворимые в воде
красители, часто употребляемые в видесолей (они широко применяются
в лакокрасочной и полиграфической промышленности).
В соответствии с технической классификацией в СССР
разработана рациональная номенклатура красителей. Первое слово
в наименовании красителя казывает на группу, второе слово—
263. Природные и синтетические красители . 517
на цвет. Буквенное обозначение указывает на оттенок цвета
красителя или на особые условия его применения. Например,
название красителя «Прямой красный ЖХ» означает, что он
предназначен для крашения протравленных солями хрома хлопка
или вискозы в красный цвет с желтоватым оттенком.
Лишь для отдельных марок красителей сохранены
исторически сложившиеся индивидуальные наименования в тех
случаях, когда они стали международными, например индиго, тио-
индиго, фуксин, хризоидин, конго красный и некоторые другие.
ГЛАВА XIX
МНОГОЯДЕРНЫЕ АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ
Многоядерными ароматическими соединениями называются
такие вещества, в молекулах которых находятся два или несколько
бензольных ядер, связанных между собой.
Различают два вида многоядерных бензольных соединений.
В одном из них бензольные кольца соединены друг с другом
или непосредственно или при помощи атомов углерода. К
другому виду соединений относятся вещества, в которых бензольные
кольца связаны друг с другом так, что некоторые атомы
углерода являются общими.
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ БЕНЗОЛЬНЫЕ ЯДРА,
СВЯЗАННЫЕ ЧЕРЕЗ УГЛЕРОД ИЛИ НЕПОСРЕДСТВЕННО
264. Дифенил. Бензидин. Простейшим представителем
соединений, в которых бензольные ядра непосредственно связаны друг
с другом, является дифенил СвН5—С6Н6 (темп, плавл. 70 °С).
Его молекула состоит из двух связанных друг с другом
бензольных колец.
Строение дифенила доказывается способом его получения—
действием металлического натрия на бромбензол:
+ 2NaBr
броыбензол бромбензол дифенил
Оба бензольных кольца в дифениле полностью сохраняют свои
свойства.
Из производных дифенила наибольшее значение имеет
бензидин
полученный впервые в 1845 г. Н. Н. Зининым.
Как видно из формулы, бензидин является диамином
дифенила; аминогруппы в нем находятся в пара-положении по
отношению к связи, соединяющей бензольные ядра.
264. Дифенил. Бензидин 519
Бензидин получается действием сильных минеральных кислот
на гидразобензол. При этом атомы водорода, находящиеся в
пара-положении к группам NH, переходят к азоту. Каждая из групп
NH образует группу NH2, связь между атомами азота
разрывается, бензольные же кольца связываются между собой:
— NH— NH—¦¦
гидразобензол бензидин
Это превращение называется бензидиновой перегруппировкой.
Как всякий первичный ароматический амин, бензидин при
действии на него азотистой кислоты диазотируется и переходит
в диазосоединение:
У^=Ч / —А.
-NjCl
Сочетанием этого диазосоединения с фенолами и аминами
получаются вещества, в составе которых содержатся две азогруппы.
Эти вещества являются важными красителями, окрашивающими
непосредственно (без протравы) хлопчатобумажные ткани.
Примером красителей такого рода может служить конго красный-
Он получается сочетанием диазотированного бензидина с наф-
тионовой кислотой (см. стр. 532):
NH,
k
,ОН SOjOH
Натриевая соль этого соединения и является красителем.
Как ясно из названия самого красителя, он имеет красный
цвет. При действии минеральных кислот на конго красный
образуется кислота красителя, окрашенная в синий цвет.
Для изменения цвета конго красного требуется большая
концентрация водородных ионов, чем это необходимо для изменения
цвета метилового оранжевого. Поэтому конго пользуются как инди-
520
Глава XIX. Многоядерные ароматические соединения
катором на сильные (минеральные) кислоты. Концентрация ионов
водорода даже в очень разбавленных растворах этих кислот
достаточна для того, чтобы вызвать изменение красного цвета
конго в фиолетовый цвет, в то время как органические кислоты
(уксусная) изменяют его окраску в синий только в том случае,
если их концентрация велика.
265. Трифенилметан и его производные. Простейшими
примерами соединений, содержащих бензольные ядра, связанные при
помощи атомов углерода, могут служить дифенилметан,
трифенилметан и тетрафенилметан.
Эти углеводороды можно рассматривать как производные
метана, в молекуле которого два или несколько атомов водорода
замещены фенильными группами.
Физические свойства фенильных производных метана,
включая и толуол, приведены в табл. 23.
Таблица 23
Физические свойства фенильных производных метана
Название
Толуол
Дифенилметан
Трифенилметан
Тетрафенилметан ....
Формула
(СвН6)СН3
(СвН6JСН2
(СвН5KСН
(СаН5LС
Температура
плавления
°С
-95
26
92,5
285
Температура
кипения
°С
110,6
262
359
431
Из этих соединений наибольшее значение имеет
трифенилметан и его производные:
QH6
или QH6—С—Н
трнфеннлметам
Трифенилметан можно получить, действуя бензолом на
хлороформ в присутствии безводного хлористого алюминия:
СаН6-;Н С1;^
С6Н5—|н +С1І—С—Н
j—JH O/
АІСІз
C„H5—С—H + JHC1
265. Трифенилметан и его производные 521
Если вместо хлороформа взять четыреххлористый углерод,
то получается трифенилхлорметан:
QH5—:Н Сік С6Н6ч
qh—;н +сіі—с—сі -і^> с6н5—с—сі + знсі
с5н6—ін а/ с6н5/
Трифенилхлорметан—бесцветные кристаллы (темп, плавл.
113 °С). Он легко разлагается водой с образованием
трифенилкарбинола:
(QHsJaCCl + Н2О » (С6Н5KСОН + НСІ
Трифенилкарбинол может быть также получен действием маг-
нийбромфенила на бензофенон:
>
б5\ убй -1-НоП
СО + QHsMgBr » ус/ _L21» (QHJaCOH + Mg(OH)Br
С6Н/ \QM
Трифенилкарбинол—бесцветные кристаллы (темп, плавл.
162,5 °С). Гидроксильная группа в его молекуле очень легко
замещается галоидом. Так, при пропускании сухого хлористого
водорода через раствор трифенилкарбинола в хлороформе
образуется трифенилхлорметан:
(С6Н5KС-ОН + НС1 ¦ (С6Н5KСС1 + Н2О
Трифенилкарбинол и его производные обладают основными
свойствами; они растворяются в концентрированных минеральных
кислотах с образованием интенсивно окрашенных солей. Так,
при растворении трифенилкарбинола в концентрированной
серной кислоте образуется раствор, окрашенный в желтый цвет:
(СбН5Kс-он + h2so4 —> [(С6н5Kс]f + hsoj + н2о
Однако эта окрашенная соль легко разлагается водой с
выделением осадка бесцветного трифенилкарбинола
Здесь, как и в других случаях, появление окраски связано
с изменением строения молекулы.
522 Глава XIX. Многоядерные ароматические соединения
266. Красители ряда трифенилметана. Строение и свойства.
Трифенилметан является родоначальником ряда красителей, к
числу которых относятся фталеины (см. стр. 477).
Особое значение из красителей ряда трифенилметана
имеют те, которые являются аминопроизводными как самого
трифенилметана, так и его гомологов. Выясним характерные
особенности веществ этой группы на примере некоторых
производных триаминотрифенилметана и триаминодифенилтолил-
метана.
Первое из этих веществ можно рассматривать как
производное трифенилметана, в каждом бензольном ядре которого атом
водорода в пара-положении замещен аминогруппой:
триаминотрифенилметан
Второе вещество является ближайшим гомологом
предыдущего, причем метил находится в орто-положении по отношению
к аминогруппе:
\Q>-NHa
триаыинодифенилтолилметан
Следует отметить, что в красители могут переходить только
те амины, в которых находятся по меньшей мере две
аминогруппы, причем каждая из этих групп стоит в пара-положении
по отношению к метановому атому углерода.
Амины ряда трифенилметана—бесцветные вещества; они
являются так называемыми лейкооснованиями красителей. При
окислении они легко переходят в гидроксильные производные.
Этим путем из триаминотрифенилметана получается триами-
нотрифенилкарбинол, называемый парарозанилином. При окис-
266. Красители ряда трифенилметана. Строение и свойства 523
лении триаминодифенилтолилметана образуется розанилин:
П)
СН3 B)
Н3<
НО-С—CeH4—NH3 НО—С-СбН4—NH2
\Vii—NHj, \СбН4—NH2
парароэанилин розанилин
Эти гидроксильные производные называют псевдооснованиями
красителей.
При действии на них кислот происходит следующий процесс:
NHa,
СН,
-NH.,
-C6H/'
NHa-C6H/ NOh+ Hj-Cl
/ 3xc/==
=NH2
сі-+нао
фуксан
т. е. выделяется вода и одно из бензольных колец принимает
хиноидное строение*. В результате получаются солеобразные
вещества, имеющие свойства красителей.
При этом из розанилина образуется фуксин, а из парароз-
анилина—парафуксин
Lnhs-qh/
парафуйся н
Эти красители нельзя рассматривать как соли тех веществ,
из которых они получаются при действии кислот. Поэтому-то
гидроксильные производные и являются псевдооснованиями, а
не истинными основаниями красителей.
При действии на красители щелочей сначала получаются
окрашенные истинные основания, например
6 4^C=(==N=NHa ОН-
NHo—СЛ\/ л—'
истинное основание
которые очень быстро переходят в бесцветные псевдооснования.
При восстановлении трифенилметановые красители переходят
в бесцветные лейкооснования. Превращения трифенилметановых
* Вопрос о правильном написании структурных формул этих красителей
нельзя считать окончательно решенным.
524
Глава XIX. Многоядерные ароматические соединения
красителей могут
NfIl2CeH44
«ейкоосноиа
быть
NH,Ce.
NH,C6
- \
выражены
./ ^^
краситель
1
>-NH2 u
о
нма
и.
NU
NH
следу
ющей
С1
'J
ч
2^6 4\ /
чСпН)С<
осноиание
схемой;
=4. ¦
] _
он
NH2C6H4 ^
nh2c6h/ ^h
псевдооснование
267. Получение красителей ряда трифенилметана. Красители
ряда трифенилметана получаются двумя способами: 1)
окислением одноядерных ароматических производных и 2)
конденсацией с участием производных дифенилметана. По первому
способу получают фуксин, парафуксин и малахитовый зеленый, по
второму способу—кристаллический фиолетовый.
Фуксин может быть получен окислением смеси анилина,
о-толуидина и п-толуидина, причем метановый атом углерода
образуется из СН3-группы молекулы /г-толуидина:
Q>
СН;
HjN-<T~\n
с-<
HsN-\_/Xoh
сн,
268. Свободные радикалы 525
В результате получается розанилин, который при действии
соляной кислоты дает фуксин.
Парафуксин получается окислением смеси n-толуидина и
анилина:
CH3-<C>-NHa 4- V,O,
2HaO
Парарозанилин с соляной кислотой образует парафуксин.
Фуксин образует кристаллы, отливающие с поверхности
зеленоватым цветом; с водой он дает интенсивно красный раствор.
При добавлении к этому раствору избытка концентрированной
соляной кислоты он принимает желто-оранжевый цвет, при
разбавлении водой снова появляется красное окрашивание.
При введении в молекулы фуксина и парафуксина метиль-
ных групп получаются фиолетовые красители, при введении фе-
нильных групп образуются синие красители.
268. Свободные радикалы. В 1900 г. Гомберг, действуя на
трифенилметилбромид серебряным порошком или цинковой
пылью при полном отсутствии воздуха, получил гексафенилэтан:
QHe\ с6Нвч c6h5v ^с6н5
C6H5-C-iBrT2Ag+'Bri C-QH6 » QH6— С—С—QH5 4- 2AgBr
од/ qh6/ qh/ \:6h6
Гексафенилэтан—бесцветные кристаллы, дающие при
растворении, например в бензоле, желтый раствор. Этот раствор
необыкновенно легко окисляется и мгновенно реагирует с бромом
и иодом; при этом образуются (С6Н6)аСВг и (C„H5KCJ. Ни одна
нз этих реакций не является типичной для обычных
углеводородов. Дальнейшие исследования показали, что в растворе
находится свободный радикал трифенилметил
или СНй—С*
626 Глава XIX. Многоядерные ароматические соединения
в котором центральный углеродный атом (отмеченный жирной
точкой) трехвалентен.
В растворе имеет место равновесие:
2С.Н.-С«
Это означает, что гексафенилэтан диссоциирует на два
свободных радикала, что было доказано путем определения
молекулярного веса гексафенилэтана в расплавленном нафталине при
80 °С. Молекулярный вес гексафенилэтана 486, трифенилмети-
ла 243. Найденное на опыте значение молекулярных весов
соответствовало содержанию в растворе 30% гексафенилэтана в
диссоциированном состоянии. Повышение температуры
приводит к увеличению процентного содержания трифенилметила
в растворе.
К насосу
Рис. 66. Прибор для получения свободных алкильных радикалов:
/—пробирка: 2—кварцевая трубка; 3—ловушка; 4—свинцовое зеркало; 5—нагреваемый
участок трубки.
Были получены и другие вещества, распадающиеся иа
радикалы с трехвалентным атомом углерода. Некоторые из них уже
при обыкновенной температуре полностью распадаются на
радикалы. В этих веществах трехвалентный атом углерода связан
с группами, обладающими большим молекулярным весом.
Наконец, в 1929 г. Панет и Гофедиц путем разложения
тетраметилсвинца при нагревании доказали весьма кратковременное
(тысячные доли секунды) существование свободного радикала
метила:
РЬ(СН3L ^=t РЬ + 4СН3.
Для обнаружения легких свободных радикалов Панет и Гофедип
разработали метод зеркал, заключающийся в следующем: пропускают быстрый ток
чистого водорода (или азота) при давлении в 1—2 мм и насыщают его парами
тетраметилсвинца путем пропускания через пробирку / (рис. 66), содержащую
РЬ(СНзЬ, охлажденный твердой углекислотой. Затем газ пропускают через
длинную кварцевую трубку 2 и откачивают через ловушку 3, погруженную
в жидкий воздух. Вначале местным нагреванием на участке 4 получают евин-
269. Нафталин 527
цовое зеркало; эатем трубку нагревают во второй точке 5, ближе к входу газов,
а область доставляют холодной. При этом оказалось, что на участке 5
откладывается свежее зеркало, а первое зеркало одновременно исчезает (если только
расстояние между 4 и 5 не превышает 30 см). Отсюда следует, что одним из
продуктов термического разложения РЬ(СНзL на участке 5 должен быть газ,
способный реагировать с холодным металлическим свинцом на участке 4.
Таковым может быть только свободный метил СНз, так как экспериментальным
путем было установлено, что все другие газообразные продукты разложения—
водород, метан, этилен—не оказывают никакого влияния на свинцовое зеркало.
В 1931 г. Панет и Лаутц этим же методом получили из тетраэтилсвинца
свободный радикал этил С2Н5.
Используя электронные представления, можно выразить
строение свободного метильного радикала следующим образом:
н
н:с-
н
Таким образом, свободные радикалы имеют неспаренный
электрон, не несут электрического заряда (не являются ионами)
и способны соединяться между собой (рекомбинация
радикалов):
Н Н Н Н
н:с- + -с:н —5- н:с:с:н
н н н i-i
Многочисленными исследованиями советских и иностранных
ученых (Н. Н. Семенов с сотрудниками, А. Н. Теренин, В. Н.
Кондратьев, Райе, Нориш, Уотерс и мн. др.) установлено, что очень
многие химические реакции, как, например, окисление,
полимеризация, термическое разложение углеводородов, синтезы на
основе водяного газа и т. п., протекают через промежуточное
образование нестойких свободных радикалов.'
Представление о существовании свободных радикалов лежит
в основе так называемой цепной теории химических процессов.
В ряде случаев существование короткоживущих свободных
радикалов удалось непосредственно подтвердить на основе
спектроскопических данных.
СОЕДИНЕНИЯ С КОНДЕНСИРОВАННЫМИ ЯДРАМИ
269. Нафталин. Простейшим представителем ароматических
соединений со срощенными (конденсированными) ядрами
является нафталин СМН8.
628 Глава XIX. Многоядерные ароматические соединения
Нафталин кристаллизуется в блестящих листочках, плавится
при 80 °С, кипит при 218 °С; он практически нерастворим в воде,
имеет характерный запах.
Несмотря на высокую температуру кипения, нафталин очень
летуч и легко возгоняется.
Нафталин получается из той части каменноугольной смолы,
которая перегоняется при температуре от 170 до 230 °С и которая
называется средним маслом. После обработки этого масла
раствором едкого натра (для удаления из него фенолов) масло
перегоняют. Получающийся при этом дистиллят, состоящий
преимущественно из нафталина, почти нацело застывает.
Застывшую массу отжимают под прессом, промывают кислотой и
раствором едкого натра; окончательно нафталин очищают
перегонкой с водяным паром или возгонкой.
В последние годы все большее значение начало приобретать
получение нафталина из нефтяного сырья. В кубовых остатках,
получаемых при переработке продуктов каталитического
крекинга и каталитической ароматизации нефтепродуктов, содержатся
вначительные (до 30% и выше) количества метилнафталинов.
Поскольку сами метилнафталины технического применения не
имеют, они используются для получения из них нафталина; для
этого содержащие метилнафталины фракции подвергаются
каталитическому дезалкилированию в среде водорода:
Катализаторами для этого процесса служат различные металлы
на носителях. Из нефтяного сырья легче получить нафталин
высокой степени чистоты, чем при извлечении его из
каменноугольной смолы.
При сравнении состава нафталина (С10Н8) с составом
предельного углеводорода с десятью атомами углерода в
молекуле (СюН22) видно, что в молекуле нафталина на четырнадцать
атомов водорода меньше, чем требуется для полного насыщения.
Несмотря на это, нафталин легко вступает в реакции замещения,
что характерно для ароматических соединений.
Элементарный состав нафталина установил А. А.
Воскресенский. Впоследствии Эрленмейер предложил для него
следующую структурную формулу:
СН СН
НС С Сп
і її і
НС С СН
СН
269. Нафталин
529
Согласно этой формуле, в молекуле нафталина находятся два
бензольных ядра, соединенных друг с другом так, что два
соседних атома углерода являются общими для обоих бензольных
ядер.
Весьма часто строение нафталина выражают упрощенной
формулой, нумеруя атомы углерода, связанные с атомами
водорода, следующим образом:
При замещении одного атома водорода в молекуле нафталина
каким-либо другим атомом, например атомом галоида, могут
получаться два ряда производных. Во-первых, может заместиться
атом водорода у атома углерода, который связан
непосредственно с одним из двух общих углеродных атомов, т. е.
замещение может произойти у 1-го, 4-го, 5-го или 8-го углеродного
атома. Эти производные нафталина обозначаются буквой а. Во-
вторых, замещение может произойти у 2-го, 3-го, 6-го или 7-го
углеродного атома. Такие производные нафталина обозначаются
буквой ?:
Вг
а-бремнафталан
?-бромнафталин
Таким образом, для однэзамещенных производных нафталина
возможны два изомера.
Сильными окислителями нафталин окисляется во фталевую
кислоту СвН4(СООНJ. В течение многих лет это окисление
производилось в промышленности нагреванием нафталина с
дымящей серной кислотой в присутствии сернокислой ртути как
катализатора:
+ 9SO3
Н
2СО2 + Н2О + 9SOa
34—788
530 Глава XIX. Многоядерные ароматические соединения
Серный ангидрид восстанавливается при этом в сернистый
газ, а фталевая кислота превращается далее во фталевый
ангидрид.
Значение сернокислой ртути для этой реакции было открыто случайно.
При сульфировании нафталина в одном из опытов разбился термометр и ртуть
попала в сульфомассу. К удивлению работавшего химика, из аппарата стал
выделяться в виде дыма фталевый ангидрид.
Более совершенный метод окисления нафталина во фталевый
ангидрид был разработан в 1918 г. При работе по этому методу
пары нафталина в смеси с воздухом пропускают при 450 °С над
катализатором, в качестве которого служит пятиокись ванадия
v,o6.
Нафталин гидрируется водородом в присутствии
катализаторов легче, чем бензол (см. стр. 424).
270. Производные нафталина. Для нафталина известны
многочисленные производные.
При действии галоидов нафталин бромируется и хлорируется,
причем в первую очередь получается а-производное:
Н
При действии на нафталин крепкой серной кислоты
происходит сульфирование, т. е. водород нафталина замещается суль-
фогруппой —SOjOH.
В результате реакции образуются нафталансульфокислоты
состава С,0Н,—SO2OH:
С10Н,-|Н +¦ НО;—SO2OH * Q0H,-SO2OH -f- H2O
В зависимости от условий реакции получаются ос- или
?-сульфокислоты. Если реакция происходит при низкой
температуре F0 °С), то получается главным образом а-нафталинсуль-
фокислота. При высокой температуре (порядка 160 °С) получается
главным образом ?-нафталинсульфокислота, так как при этой
температуре ос-сульфокислота превращается в ?-сульфокис-
лоту.
При сплавлении солей сульфокислот с едкими щелочами
происходит замещение сульфогруппы гидроксилом и получаются
270. Производные нафталина 531
гидроксильные производные нафталина—нафтолы С10Н7—ОН:
Q0H7—|SO2ONa + Nai—ОН » С10Н,—ОН + Na2SO„
ОН
.ОН
а-нафтол
Нафтолы по своим свойствам сходны с фенолами. Как и
фенолы, они растворяются в едких щелочах и дают с хлорным
железом характерные окрашивания. Водный раствор а-нафтола
с хлорным железом образует фиолетовый осадок; ?-нафтол
окрашивается в присутствии хлорного железа в зеленый цвет.
Для производства красителей имеют большое значение суль-
фокислоты нафтолов. Примерами их могут служить 2-нафтол-
3,6-дисульфокислота и 2-нафтол-6,8-дисульфокислота. Первая
кислота называется Р-кислотой, вторая—Г-кислотой:
SOaOH
A
Р-кислота Г-кнслота
Входя в качестве компонента в молекулу красителя,
Р-кислота придает последнему красный, а Г-кислота—желтый
оттенки*.
При действии на нафталин концентрированной азотной
кислоты происходит замещение водорода нитрогруппой NO2. В
результате реакции получается а-нитронафталин С10Н7—NOa:
а-НБтронафталин
?-Нитронафталин трудно доступен и получается другим
путем.
* Р и Г первые буквы немецких слов rot и gelb.
34*
532 Глава XIX. Многоядерные ароматические соединения
Аминопроизводные нафталина С10Н,—NH2 называются ни-
фшиламинами. а-Нафтиламин получается восстановлением я-ннт-
ронафталина:
NO,
-6Н »[ |j | + 2НэО
а-нитронафталин а-нафтиламин
Он получен впервые Н. Н. Зининым (стр. 487).
Для получения ?-нафтиламина ?-нафтол нагревают под
давлением с аммиаком и с сернистым аммонием:
?-нафтилачин
Технический а-нафтиламин имеет неприятный запах
экскрементов; ?-нафтиламин запаха не имеет.
Ввиду сильного канцерогенного (вызывающего рак) действия
?-нафтиламина производство его в СССР в настоящее время
прекращено; необходимые для получения ценных марок красителей
производные ?-нафтиламина вырабатываются обходными
путями.
Физические свойства некоторых производных нафталина
приведены в табл. 24.
По своему химическому характеру нафтиламины сходны с
анилином: они обладают слабыми основными свойствами,
способны нитроваться, сульфироваться и т. д. Так, сульфированием
а-нафтиламина может быть получена нафшионовая кислота:
NH,
so:
Нафтионовая кислота практически нерастворима в воде, но
легко растворяется в ней в присутствии соды вследствие
образования хорошо растворимой натриевой соли.
270 Производные нафталина
533
Таблица 24
Физические свойства производных нафталина
Название
а-Хлорнафталин
?-Бромнафталин
?-Нитронафталин
а-Нафталинсульфокислота . . .
?-Нафталинсульфокислота . . .
о-Нафтол
?-Нафтол
о-Нафтиламин
Формула
С10Н,С1
С10Н,С1
QoHjBr
СиН^О,
C1(,H,NO2
Q^jSOaOH
C10H7SQjOH
СщН,ОН
C10H,OH
C10H,NH2
Q0H,NH2
Температура
плавлення
°С
— 1?
56,7
6,2
59
61,5
79
88
83
96
122
50
113
Температура
кипения
°С
259
266
281
281
304
165
(при 15 мм
pm ст.)
288
294
301
306,1
Из других производных нафталина, содержащих
аминогруппу, следует указать на так называемую Аш-кислоту—1-амино-
8-нафтол-3,6-дисульфокислоту:
HOOaS
,ОаОН
Она является одним из главнейших полупродуктов в
производстве азокрасителей.
Хиноны нафталина называются нафто канонами Известно
три нафтохинона:
О
оо аґ ?o
zO
II
о
а-нафтохинон
В-нафтохинон
S34
Глава XIX. Многоядерные ароматические соединения
Нафтохиноны могут быть получены окислением
соответствующих аминонафтолов. Нафтохиноны—кристаллические
окрашенные вещества: а-нафтохинон—желтый, два других нафтохинона—
красные; а-нафтохинон легколетуч и обладает острым
запахом.
271. Доказательство строения нафталина. Строение
нафталина доказывается окислением его производных. При окислении
нафталина получается фталевая кислота:
:'_•»
н
н н
Точно так же при окислении а-нитронафталина получается
яитрофталевая кислота:
н с соон
NO,
Следовательно, в молекуле нафталина имеется бензольное ядро А.
При восстановлении а-нитронафталина получается нафтил-
амин. Совершенно ясно, что этот нафтиламин является а-наф-
тиламином, и группа NHg находится в том же ядре, в котором
находилась группа ЫО2.Если подвергнуть окислению а-нафтил-
амин, то получается фталевая кислота:
ноос
НООС
і-нафтиламив фталевая кпслота
N0
а-нитронафталнн
При этом происходит разрушение ядра А. Следовательно, в
молекуле нафталина, кроме бензольного ядра А, имеется и бен-
ТЇ2. Красители ряда нафталана 535
зольное ядро Б, т. е. строение нафталина выражается
следующей формулой:
СН СН
не с сн
! II I ™ (
нс с сн Ч
Попутно отметим, что наличие заместителей первого рода (NH2.
и др.) усиливает, а второго рода (NO2 и др.)—ослабляет
реакционную способность данного бензольного ядра.
272. Красители ряда нафталина. Подобно другим
ароматическим аминам, нафтиламины при действии азотистой кислоты
диазотируются и переходят в диазосоединения. Так, в
результате диазотирования а-нафтиламина получается хлористый а-на-
фтилдиазоний C1OH7N2C1:
NH, HCI NaCI
+ HONO.
¦00
Диазосоединения ряда нафталина имеют тот же химически»
характер, что и диазосоединения бензольного ряда. Подобно
последним, диазосоединения ряда нафталина с нафтолами и
аминами образуют амино- и оксиазосоединения, многие-из которых
являются весьма важными красителями. Некоторые красители-
получаются сочетанием диазосоединений бензольного ряда с
нафтолами и нафтиламинами.
Примером красителей ряда нафталина может служить пара-
красный, получаемый сочетанием диазотированного га-нитро-
анилина с ?-нафтолом:
Крашение тканей этим красителем производится следующим
образом. Ткань пропитывают щелочным раствором ?-нафтола,.
подсушивают, а затем опускают в охлаждаемый льдом раствор
диазотированного паранитроанилина; нерастворимый краситель
образуется на самой ткани. Такое крашение называется ледя-
536 Глава XIX. Многоядерные ароматические соединения
ным, или холодным крашением, так как во избежание
разложения диазосоединений оно ведется при охлаждении.
273. Тетралин. Декалин- При нагревании нафталина с
водородом в присутствии никелевого катализатора под давлением
получаются продукты присоединения водорода к нафталину—
тетралин С10Н12 и декалин С10Н18:
СН СН2 СИ2 СН2
не7 с сн4 н2с сн снг
I I! I III
НС С СН2 Н2С СН СН2
\/\/ \/\/
ГТ-І ГТ-J ГТ-J /"""И
\_i\i ^П2 vjtly V-jfl.T
тетралин декалиг:
Тетралин и декалин—жидкости легче воды. Первый из них
кипит при 207,6 °С, второй—около 190 °С; они применяются как
растворители для лаков.
274. Антрацен. Антрацен С14Н10 получается из фракции
каменноугольной смолы, кипящей выше 270 °С, называемой
антраценовым маслом.
Антрацен кристаллизуется в белых листочках с голубой
флуоресценцией; плавится при 217 °С; кипит при 354 °С; в воде
нерастворим.
Антрацен имеет следующую структурную формулу:
СН СН СН
НС С С СН
\/\/\s
сн сн сн
Обычно строение антрацена выражают более простой
формулой, нумеруя в ней атомы углерода, связанные с водородом,
следующим образом:
Атомы углерода, связанные с водородом, разделяются по
своему положению на три группы: положения 1, 4, 5 и 8
обозначаются буквой а; положения 2, 3, 6 и 7—буквой ?;
положения 9 и 10—буквой у- Таким образом, для однозамещенных
производных антрацена возможны три изомера.
275. Антрахинон
537
275. Антрахинон. При действии хромовой смеси на антрацен
он окисляется в антрахинон С14Н8О2.
Антрахинон образует желтые кристаллы, плавится при 286 °С,
кипит при 382 °С, легко возгоняется, нерастворим в воде и лишь с
трудом окисляется.
Строение его выражается формулой:
О
II
сн с
сн
НС С
I II II
НС С С
сн с "сн
II
о
сн
сн
или
Такое строение антрахинона подтверждается его синтезом
из фталевого ангидрида и бензола. Реакцию проводят в две фазы.
Сначала из фталевого ангидрида и бензола образуется в
присутствии хлористого алюминия о-бензоилбензойная кислота,
затем последняя при действии концентрированной серной
кислоты дает антрахинон:
О
'Ч
С
^
AICIs
О
фталевый
ангндрнв
С
бензол
HaSO4
о-бенэоялбеняойная
кислота
Эта реакция, как и окисление антрацена, служит для
технического получения антрахинона. Антрахинон получается
окислением антрацена кислородом воздуха при 400—500 °С в
присутствии катализаторов (V2O5 и др.).
Антрахинон можно бромировать, нитровать и сульфировать.
М. А. Ильинский открыл, что при сульфировании
антрахинона в присутствии даже небольшого количества ртути сульфо-
группа вступает почти исключительно в а-положение. Если же
сульфировать антрахинон в отсутствие ртути, то получается
?-сульфокислота. Это позволяет получать а-сульфокислоту почти
без примесей ?-изомера.
538 Глава XIX. Многоядерные ароматические соединения
Цинковой пылью или газообразным водородом в присутствии
никеля в щелочной среде антрахинон быстро восстанавливается
в антрагидрохинон:
О
II ОН
0СО-Ч)р0
он
о
антрахинон антрагвдрохвнон
Антрагидрохинон растворяется в щелочах, давая
темно-красный раствор. На воздухе этот раствор обесцвечивается, при этом
выпадает осадок антрахинона.
Реакция окисления антрагидрохинона обратима:
О
Окисление антрагидрохинона воздухом сопровождается
образованием перекиси водорода. На этой реакции основаны
методы получения перекиси водорода в технике. В качестве
исходного соединения используют 2-этилантрахинон или какое-либо
другое алкильное производное антрахинона. Реакция протекает
по следующей схеме:
О
2-Этилантрахинон растворяют в органическом растворителе и гидрируют
водородом в присутствии катализатора (палладий на носителе). Отфильтрован-
276 Ализарин 53$
ный от суспензии катализатора раствор охлаждают и окисляют продуванием
воздуха. Образовавшуюся перекись водорода экстрагируют водой и
направляют на концентрирование, а раствор этилантрахинона возвращают на
гидрирование.
276. Ализарин. Главнейшим производным антрахинона
является ализарин QjHgCv Ализарин образует красные иглы,
плавится при 290 °С, легко возгоняется. Он почти нерастворим вводе.
Строение ализарина выражается формулой:
Ализарин—типичный протравно;'} краситель. По
алюминиевой протраве он окрашивает хлопок в ярко-красный цвет, с
гидроокисью железа он дает темно-фиолетовый, а с гидроокисью
хрома—зеленый лак.
До 1869 г. ализарин добывался из корней растения,
известного под названием марены, разводившегося на юге
Франции и на Кавказе. В 1868 г. Гребе и Либерман, производя
восстановление ализарина перегонкой с цинковой пылью, перевел»
его в антрацен. Это дало необходимые сведения для выяснения
строения ализарина. Еще раньше было установлено, что
ализарин образует сложные эфиры, в молекулах которых находятся
два остатка одноосновной кислоты, и растворяется в щелочах,
т. е. что ализарин является двухатомным фенолом. Состав
антрацена выражается формулой С14Н10, состав антрахинона—
С14Н8О2, а состав ализарина—С14Н6О4. Исходя из этих данных^
Гребе и Либерман решили, что ализарин является двугидр-
оксильным производным антрахинона. Свое предположение они
подтвердили синтезом ализарина. Это был первый случай
получения синтетическим путем красителя, встречающегося в
растительном мире.
Синтетический ализарин стал впервые готовиться в
заводском масштабе в 1869 г., т. е. сейчас же после работ Гребе и Ли-
бермана. В 1873 г. был разработан промышленный способ
получения ализарина, который с некоторыми изменениями
применяется и в настоящее время. Сущность этого способа
заключается в следующем.
540
Глава XIX. Многоядерный ароматические соединения
Сульфированием антрахинона получают антрахинонсульфо-
кислоту:
НО|—SOa—ОН
О„ОН
+ Н„О
антрахинансульфокислвта
Сплавляя натриевую соль этой кислоты с едким натром в
присутствии бертолетовой соли или селитры, действующих как
окислители, получают ализарин:
о
II
с
О
он
'SOoONa
+ Nai—ОН + О
С
II
О
Ч.
NaaSO3
Получаемый этим способом синтетический ализарин
совершенно вытеснил природный продукт. Если в 70-х годах прошлого
столетия ежегодный сбор марены составлял около 500 тыс. т,
то теперь это растение можно видеть только в коллекциях и
музеях.
В своей книге «Возникновение и развитие органической химии» Шорлем-
мер писал: «Открытие искусственного ализарина отразилось не только иа
земледелии, но еще большее влияние оно оказало на производство
каменноугольного дегтя, каустической соды и хлор нов атокислого калия; что же
касается трехокиси серы, применяемой для получения сульфокислоты то ее
производство открыло совершенно новую отрасль химической промышленности»
Установление строения и синтез ализарина были первым
применением теории строения для решения практических
вопросов. Эта теория была путеводной звездой в исследованиях Гребе,
Либермана и других ученых, и мы видим, какое блестящее
подтверждение она нашла в практике. Синтез ализарина произвел
очень большое впечатление на современников. Он дал мощный
толчок исследованию красителей, что привело к ряду открытий,
имевших первостепенное значение как для науки, так и для
промышленности. В результате этих исследований синтезировано
277. Кубовые полициклические красители (индантрены)
541
и выпускается промышленностью очень много производных антра-
хинона, не встречающихся в растительном мире и являющихся
превосходными красителями.
Сам ализарин в настоящее время утратил свое прежнее
значение как краситель для хлопка и производится лишь в
ограниченном масштабе для получения некоторых лаков.
Получение прочных кубовых и азокрасителей способствовало его
вытеснению. Однако многие производные антрахинона имеют
первостепенное значение в химии красителей. Сульфокислота амино-
н оксипроизводных антрахинона образуют группу так
называемых кислотных антрахиноновых красителей, отличающихся
высокой прочностью и яркостью окрасок. Примерами могут служить
красители кислотный красный ализариновый и кислотный синий
антрахиноновый:
О
SO3Na
кислотный красный
ализариновый
О.Н
ОН О NH2
кислотный синий
знтрахиненовыР
277. Кубовые полициклические красители (индантрены). При
сплавлении ?-аминоантрахинона с едким кали и селитрой в
качестве окислителя при 220 °С получается краситель кубовый синий О
(индантрон):
0
11
II
с
и
с
II
0
Y
н
4NH2
+-2О
-2Н2О
0
її
II
С
II
0
H,Nv
Н
О
ьубовый синий О
с
II
о
542 Гл. XIX. Многоядерные ароматические соединения
Более сложным красителем, получающимся из антрахинона,
является кубовый ярко-зеленый С:
СНЭО
СН3О
Известно много других кубовых полициклических красителей.
Сырьем для их получения служат в большинстве случаев антра-
хинон и его производные.
Эти красители имеют большой молекулярный вес, содержат
большой процент углерода и малый процент водорода; они
нерастворимы в воде, нелетучи и отличаются высокими
температурами плавления. В отношении прочности они почти не имеют
себе равных.
Антрахинон восстановлением можно перевести в растворимый
в щелочах антрагидрохинон, который кислородом воздуха снова
окисляется в нерастворимый антрахинон. Подобно этому, при
восстановлении кубовых красителей происходит присоединение
водорода к карбонильному кислороду. В результате образуются
гидроксильные производные, растворимые в щелочах. Эти
щелочные растворы на воздухе снова образуют красители.
Крашение индантренами производят следующим образом.
Красители восстанавливают гидросульфитом натрия в
щелочном растворе. В полученный таким путем «куб» опускают ткань,
выдерживают ее там некоторое время, вынимают и подвергают
действию воздуха. Кислород воздуха производит на ткани
окисление щелочного-раствора, и краситель окрашивает ткань,
причем цвет окрашенной ткани резко отличается от цвета «куба».
Такое крашение, когда краситель образуется на самой ткани
путем окисления щелочного раствора кислородом воздуха,
называется кубовым крашением*, а красители, служащие для этой
цели,—кубовыми красителями. Древнейшим кубовым
красителем является индиго (см. стр. 597).
278. Фенантрен. Фенантрен С14Н10 является изомером антра-
иена и, подобно антрацену, находится в каменноугольной смоле.
* Это название произошло оттого, что крашение проводилось раньше в
больших чанах четырехугольной формы—«кубах».
279. Углеводороды со многими конденсирован бензольными ядрами 543
Фенантрен—бесцветные кристаллы; темп, плавл. 101 °С;
темп. кип. 340 °С.
Строение фенантрена выражается формулой, которую можно
написать двумя различными способами:
ила
Обе формулы тождественны. Каждая из них показывает,
во-первых, что в молекуле фенантрена имеются два бензольных кольца
(А и Б), непосредственно связанных между собой, и, во-вторых,
что каждое из этих колец связано в орто-положении с группой
—СН=СН—, образующей вместе с частью углеродных атомов
первых двух колец третье шестичленное кольцо.
Фенантрен не имеет технического применения, но
представляет большой интерес, так как в близком к нему отношении
находятся некоторые природные вещества, имеющие громадное
физиологическое значение. Таковы, например, различные стероиды,
витамин D, половые гормоны.
279. Углеводороды со многими конденсированными
бензольными ядрами. В каменноугольной смоле содержатся
углеводороды хриэен С18Н12 (темп, плавл. 255 °С) и парен С^Ню (темп,
плавл. 156 °С):
и
хризен пирен
Как легко видеть, оба углеводорода содержат кольцевую систему
фенантрена.
В каменноугольной смоле в незначительном количестве
содержится также и 1,2-бензпирен С20Н12:
544 Гл. XIX. Многоядерные ароматические соединения
1,2-Бензпирен обладает канцерогенными свойствами, т. е.
действует как возбудитель рака*.
Кроме 1,2-бензпирена, известен ряд других канцерогенных
углеводородов, которые в большинстве случаев содержат
кольцевую систему 1,2-бензан/працена
'Y4
1,2-бензантрацен
Нанесенные на кожу, они вызывают явление накожного рака,
подкожное впрыскивание этих углеводородов ведет к
образованию рака соединительной ткани.
* В последние годы американскими химиками были проведены
тщательные исследования дегтеобразного вещества, полученного из табачного дыма.
Оказалось, что в нем содержатся, помимо никотина, ароматические
углеводороды (типа бензпирена),- обладающие сильными канцерогенными свойствами.
Табачный деготь при нанесении на кожу или в легкие мыши вызывал
образование раковых опухолей.
АЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Алициклическими углеводородами называются углеводороды
кольчатого строения, не бензольного типа. Большинство из них
по химическому характеру лишь в незначительной степени
отличается от соответствующих соединений алифатического ряда.
Этим и объясняется их название—алициклические, т. е.
алифатические циклические углеводороды.
ГЛАВА XX
ЦИКЛОПАРАФИНЫ
280. Строение и способы получения циклопарафинов.
Циклопа рафины иначе называются полиметиленовыми углеводородами.
Их состав выражается формулой С„Н2я; они имеют такой же
состав, как и соответствующие углеводороды ряда этилена.
Молекулы основных углеводородов циклопарафинового ряда
состоят из метиленовых групп СН8, углеродные атомы которых
связаны между собой:
^2
/ \
н2с сн2
триметилен
(циклопропан)
сн2—сн2
тетраметилен
(циклобутан)
|
сн3
пентаметилен
(циклопентан)
/CHg СН2
н2с ^
—сн2
гексаметилен
(циклогексан)
СН2 CHg CHfl
І ^
сн2-сн2—сн,
гептаметнлен
(циклогептан)
Производные циклопентана и циклогексана содержатся в
нефти. Поэтому углеводороды циклопарафинового ряда нередко
называют нафтенами. Многие из циклопарафинов были выделены
35-788
54G Глава XX Цифлопарафины
из бакинской нефти и получены искусственно В. В. Марковни-
ковым и Н. Д. Зелинским*.
В исследовании циклопарафинов, кроме В. В. Марковникова
и Н. Д. Зелинского, видное участие принимали многие русские
химики: Г. Г. Густавсон, Н. Я. Демьянов, М. И. Коновалов,
Н. М. Кижнер, С. С. Наметкин, Б. А. Казанский и др.
Циклическое строение этих углеводородов доказывается каь
способами их получения, так и их свойствами. Так, циклопропан
может быть получен действием цинка на 1,3-дибромпропан:
<?.
¦CH,f Br
nch.
+ I
Br
/СН2
ZnBr
,
Эта реакция была открыта Г. Г. Густавсоном.
Циклическое строение гексаметилена доказывается тем, что
при его окислении получается двухосновная адипиновая кислота:
/СНа—CHa СН2—CHa—СООН
Н2С ЧсН2 -t- 50 * I + НаО
N:h2—ch2 CHa—ch,—cooH
* Николай Дмитриевич Зелинский A861 — 1953) родился 6 февраля 1861 р.
в г. Тирасполе. В 1884 г. он окончил естественное отделение Новороссийского
университета в Одессе. В 1888 г. начал преподавательскую деятельность
в качестве приват-доцента в Новороссийском университете. В 1889 г. он
защитил магистерскую диссертацию «К вопросу об изомерии в тиофеновом ряду»,
а в 1891 г.—докторскую диссертацию «Исследование явлений стереоизомерии
в рядах предельных углеродистых соединений».
В 1893 г. Николай Дмитриевич получил кафедру органической и
аналитической химии в Московском университете, в котором он проработал до самой
смерти C1 июля 1953 г.).
В 1926 г. Николай Дмитриевич был избран членом-корреспондентом и в
1929 г.—действительным членом Академии наук.
Н. Д. Зелинский создал самую крупную научную школу химиков. Его
учениками являются выдающиеся химики: Л. А. Чугаев, Н. А. Шилов,
С С. Наметкин, А. Н. Несмеянов, Б. А. Казанский, А. А. Баландин, В. С.
Садиков и многие другие. Научная деятельность Зелинского очень разнообразна;
и» совместно с учениками опубликовано свыше 500 научных трудов.
Наибольшее число работ Николая Дмитриевича посвящено химии углево-
дородон н химии нефти, в частности синтезу и изучению свойстр
разнообразных представителей предельных и непредельных циклических углеводородов
(изучение каталитических процессов гидрирования и дегидрирования,
ароматизации нефтяных углеводородов и их превращении при действии хлористого
алюминия). Широко известно изобретение Зелинским первого угольного
противогаза,- спасшего жизнь многим тысячам людей. Очень важны работы
Зелинского по синтезу жидкого топлива на основе окиси углерода.
280 Строение и способы получения циклопарафинов 547
Если бы гексаметилен имел не циклическое строение, а был
етиленовим углеводородом
СН3-СН=СН-СН„-СНа-СН1,
то при его окислении не могла бы получиться двухосновная
кислота с тем же числом атомов углерода, а произошел бы разрыв
цепи и получились кислоты, в молекулах которых содержалось
бы меньшее число углеродных атомов:
СН3—СН=СН—СНа—СНа—СН„+ 40 »
» СН3—СООН + НООС—СНа—СНа—СНЭ
Получение в результате окисления циклогексана
двухосновной кислоты с тем же числом углеродных атомов в молекуле
является несомненным доказательством циклического строения
этого вещества.
Синтетически циклопарафины могут быть получены
различными способами.
1. Отщеплением галоида от таких двугалоидных
производных, в которых атомы галоида находятся не у соседних атомов
углерода, например по реакции Густавсона.
2. Сухой перегонкой кальциевых солей двухосновных кислот.
При этом сначала получается кетон, который при восстановлении
переходит в углеводород:
СНа—СНа4-С0— О
СН,—СНа —
:са
сна—сна
сна—сна
СаСОа
адипиновокислый кальций циклепентанон
СНа—СН^ СНа—CHg
| \>СО + 4Н , | ^>СН, + НаО
сна—Сна сн2—сн2
циклопентанон циклопентан
Пользуясь ториевыми солями, Л. Ружичка таким путем
получил кетоны, в кольце которых содержалось до тридцати атомов
углерода.
35*
548 ГЛава XX. Цик лопарафины
3. Циклогексан и его производные могут быть получены
гидрированием соответствующих соединений ароматического ряда:
СН СНа
НС ХСН НаС СНа
|| | +ЗНа -, | |
НС СН Н„С СНа
\// \/
СН СНа
Для этой цели смесь водорода и паров гидрируемого
вещества пропускают над металлическими катализаторами—никелем,
платиной, палладием.
Большое число непредельных алициклических соединений с
шестичленными кольцами было получено в последнее время
конденсацией конъюгированных диеновых углеводородов с
различными непредельными соединениями (кислотами, их ангидридами,
альдегидами или кетонами), в молекулах которых двойная связь
расположена непосредственно между двумя карбонильными или
карбоксильными группами. В качестве примера можно привести
конденсацию бутадиена с малеиновым ангидридом:
сна
НС
1 -1
НС
сна
сн-со
41 >
СН—СО
сна
/\
НС СН-СО
НС (
:н—со
сна
ангидрид
тетрагидрофталевой кислоты
При этих реакциях концевые атомы углерода сопряженной
системы связываются с атомами углерода другого непредельного
соединения, а между средними атомами углерода сопряженной
системы возникает двойная связь. Синтез веществ на основе этой
реакции называется диеновым синтезом.
Реакции диенового синтеза были открыты A928 г.) и
исследованы немецкими химиками О. Дильсом и К. А ль дером.
281. Свойства циклопарафинов. Циклопропан и циклобутан—
газы, средние циклопарафины—жидкости, высшие—твердые тела.
Физические свойства некоторых циклопарафинов приведены
в табл. 25.
Циклопарафины, несмотря на то что они на два атома
водорода беднее предельных углеводородов, по своим свойствам
очень сходны с последними. Они весьма устойчивы к действию
окислителей. В большинстве случаев они лишь очень медленно
282. Отдельные представители щиклопарафинов
649
Таблица 25
Физические свойства циклопарафинов
Название
Циклопропан
Циклобутан
Циклогексан
Циклооктан
Формул.»
с4н,
О.н1а
с,н14
О,н1в
Температура
плавления
°С
— 127,6
-90
—93,2
+6,5
-8,1
+ 14,8
1 Температура
кипения
°С
—32,8
+ 12,6
+49,5
+80,7
+ 118,5
+ 150,6
(при 709 мм
рт. ст.)
Плотность
при 20 °С
0,720
(при —79 °С)
0,7038
0,7454
0,7786
0,811
0,8362
присоединяют бром или вообще его не присоединяют. Это
объясняется устойчивостью циклопарафиновых колец. Особенно
устойчивы кольца, состоящие из пяти или шести атомов
углерода. Однако циклопропановое кольцо легко разрывается
действием йодистого водорода или брома:
Нґ* '
N
йодистый
пропил
циклопропан
н2с
вг2
BrCHjCHaCHgBr
1.3-днбромпропан
циклопропан
Циклопентановое и циклогексановое кольцо можно разорвать
только с большим трудом. Так, циклопентан в присутствии
платины присоединяет водород, переходя в нормальный пентан,
только при 305—310 °С, тогда как кольцо циклопропана в
присутствии того же катализатора расщепляется уже на холоду.
Объяснение различной устойчивости циклических соединений см.
на стр. 243 и 555.
282. Отдельные представители циклопарафинов. Циклопентан
С5Н,0 входит в состав некоторых нефтей. Это бесцветная
подвижная жидкость с температурой кипения 49,5 °С.
550 Глава XX. Циюлопарафины
На свету циклопентан реагирует с бромом с образованием
бромциклопентана:
ХНа—СН2 уСН2—СНВг
[2С -f- Вг2 > НаС
-сна \сна—сна
бромциклопентан
НВг
Карбоксильными производными циклопентана являются
некоторые нафтеновые кислоты, которые в значительных
количествах содержатся в кавказской нефти. Их состав выражается
общей формулой СпНая_,СЮОН. Нафтеновые кислоты являются
соединениями, углеводородный радикал которых содержит цик-
лопарафиновое кольцо. В качестве примера можно указать ме-
тилциклопентанкарбоновую кислоту, выделенную из нефти
В. В. Марковниковым. Натриевые соли нафтеновых кислот
получаются при очистке керосина и некоторых других нефтяных по-
гонов щелочью. Эти смолы могут давать пену и обладают моющей
способностью. Технический продукт известен под названием
мылонафта.
Циклопентану СБН,0 соответствуют непредельные
углеводороды циклопентен СБН8 и циклопентадиен СБНв:
yCHg—СНа >СН=СН >СН=СН
нас н2с нас |
\сн.,-сна \сна-сна \сн=сн
циклопентан циклопентен циклопентадиен
Циклопентен может быть получен обработкой
бромциклопентана спиртовым раствором едкого кали. Циклопентен—жидкость,
темп. кип. 44 °С; он обладает всеми свойствами олефина.
Из производных циклопентена наиболее важны ауксины—стимуляторы
роста растений:
СНд СНо СНЧ
І /\ І
СНзСНаСН—НС СН—СНСН3СН3
НС=С—СН(ОН)СНаСН(ОН)СН(ОН)СООН
ауксин А
СН3 СНа СН»
І /\ І
СНаСНаСН—НС СН-СНСНаСН,
НС=С—СН(ОН)СНаСОСНаСООН
ауксин В
282. Отдельные представители циклопарафинов 551
Они находятся во всех растениях. Интересно отметить, что действие,
аналогичное действию ауксинов, производят и некоторые другие вещества
совершенно иного состава и строения (см. стр. 454).
Гиднокарповая и хаульмугровая кислоты
СН=СН СН=СН
| ^>СН—(СНаI0—СООН I \СН-(СН2I2-СООН
СНа—СН, СНа—СН2
гиднокарповая кислота хаульмугровая кислота
содержатся в виде глицеридов в хаульмугровом масле, находящемся в
семенах растений, произрастающих в Восточной Азии, Африке и Южной Америке.
Оба вещества обладают сильным бактерицидным действием. Их соли и
этиловые эфиры применяются для лечения больных проказой.
Циклопентадиен образуется при сухой перегонке каменного
угля и при пиролизе некоторых нефтепродуктов. Синтетически
он был получен Н. Д. Зелинским совместное сотрудниками;
присоединением брома к циклопентену ими был получен 1,2-дибром-
циклопентен, при нагревании которого до 182 °С со сплавленным
уксуснокислым натрием и уксусной кислотой образовался
циклопентадиен:
н/ II +Вгг н/ НВг иг і
2—сн ^Hj—снвг \:н=сн
циклэпентен 1.2-дибромцикло циклопентадиен
пеитан
Циклопентадиен—бесцветная жидкость; темп. кип. 42,5 "G.
В его молекуле находится система сопряженных двойных связей,
которые придают ему высокую реакционную способность.
Как и другие диеновые соединения, циклопентадиен легко по-
лимеризуется. Его молекулы присоединяются к молекулам
многих непредельных соединений по схеме диенового синтеза. Так,
с акролеином он образует аналог тетрагидробензоиного альдегида
и/Г
сн
НС/1 ^СНа
II І І
| а + 2
НС I II Л
L
Водороды метиленовой группы легко вступают в реакции за-
мещения. Например, в кипящем ксилоле циклопентадиен реагирует
552 Глава XX Цик-лопарафины
с порошкообразным натрием и образуется циклопентадиенил-
натрий:
НС СН Na НС СН
II II * II II
НС СН НС СН
\/ \
чснг сн
Na
циклопентадиенилнатрий
С альдегидами и кетонами циклопентадиен конденсируется
с образованием окрашенных углеводородов, называемых фуль-
'венами. С муравьиным альдегидом получается фульвен:
С ацетоном циклопентадиен реагирует с образованием диметил-
фульвена:
НС СН О НС СН
НІ СН + СНз-с'-СНз^А НІ СН
\і -Н2° V
It
CHs-C-CH,
дииетилфульвен
Сам фульвен весьма неустойчив, но его гомологи достаточно
устойчивы и хорошо изучены. Фульвены окрашены, и их цвет
меняется от светло-желтого у фульвена до интенсивно-красного у ди-
фенилфульвена. Фульвены очень легко присоединяют кислород.
В 1951 г. Кили и Посон получили своеобразное производное
циклопентадиена—дициклопентадиенилжелезо, называемое чаще
ферроценом. Его можно получить из магнийорганического
соединения циклопентадиена и хлористого железа:
2CBH4MgBr + FeCla * (CBH6)Fe + MgBr3 + MgCl3
ферроцен
282. Отдельные представители циклопарафинов 553
Другой более простой метод получения ферроцена заключается
в том, что газообразный циклопентадиен пропускают над
восстановленным железом при 300 °С:
2С6Н6 Jl» (CsHsJFe + H.2
Ферроцен представляет собой желто-оранжевые кристаллы
с темп, плавл. 174 °С. Он чрезвычайно устойчив, его можно
нагревать до 470 °С и кипятить с водным раствором НС1 и NaOH без
разложения. Легко возгоняется, не растворяется в воде,
растворяется в органических растворителях. Фундаментальные
исследования в области химических превращений ферроцена были
выполнены А. Н. Несмеяновым.
В реакциях замещения ферроцен ведет себя подобно бензолу и
фенолу. Он вступает в реакцию Фриделя—Крафтса,
сульфируется, вступает в реакцию Манниха (конденсируется с
формальдегидом) и в реакцию азосочетания: в него можно ввести и
альдегидную группу. Замещению подвергаются атомы водорода одного или
обоих колец. Таким образом, ферроцен проявляет свойства,
совокупность которых химики называют ароматичностью.
Ферроцен был первым из небензоидных ароматических
соединений. Вслед за ним были получены аналогичные соединения
циклопентадиен ила с другими металлами: Со, Mo, Ni, Mn, О, Ті,
V, Ru и др. Позднее было выяснено, что ароматические
углеводороды (бензол и др.) также образуют с металлами
устойчивые кристаллические вещества, по характеру близкие к ферроцену.
Рентгеноструктурный анализ доказал, что ферроцен имеет
симметричное относительно центра двухплоскостное строение. Атом
железа расположен в центре между двумя параллельными цик-
лопентадиенильными кольцами на равном расстоянии от всех 10
атомов углерода.
Расстояния между атомами углерода в каждом цикле
одинаковы и равны 1,41 А, т. е. близки к расстояниям между
атомами углерода в бензоле A,39 A). Атом железа представляет
собой двухзарядный катион, а в кольцах сосредоточены
отрицательные заряды, так что молекула в целом электрически нейтральна.
554 Глава XX. ЦиклопараФины
Связь между кольцами и атомом железа осуществляется всеми
двенадцатью ^-электронами.
Родственные ферроцену соединения, например дициклопента-
диенилкобальт, дициклопентадиенилникель, а также дибензолхром
и его аналоги построены подобным же образом. Этот новый тип
комплексных соединений называют английским словом «сэндвич».
Циклогексан СвН12 находится в значительных количествах в
кавказской и западноукраинской нефти, синтетически может
быть получен гидрированием бензола. Бесцветная жидкость с
запахом бензина; темп. кип. 80,7 °С.
Циклогексанол СвНи(ОН) получается в заводском масштабе
гидрированием фенола или каталитическим окислением циклогек-
сана кислородом воздуха. Он представляет собой жидкость с
запахом, несколько напоминающим запах камфоры. Температура
кипения его 161 °С.
Циклогексанол является важным промежуточным продуктом
в производстве синтетических полиамидных волокон.
Каталитическим дегидрированием (отщеплением водорода) он может быть
превращен в циклический кетон—циклогексанон.
Взаимодействием последнего с гидроксиламином получают циклогексаноноксим,
перегруппировкой которого под действием олеума получают є-кап-
ролактам,—исходный мономер для синтетического волокна капрон:
сн, сна сн„
\ /СН /\ /\
НС Г —NOH
Н2С СНа HjC CH.j H..C СНа
Хсн \н Х н
циклогексанол цвклогексаион цпклогексаноноксвы
СНа
/\
нас с=о
NH
сна
в-капролактам
Окислением циклогексанола азотной кислотой или
каталитически, кислородом воздуха, получают адипиновую кислоту:
нас с/ +2О2
I | ХОН -Т ¦> НООС—(СН^-СООН + НаО
Ґ*1 1
Ч_іПо
\ У
сн„
283. Напряжение в алициклических соединениях 555
Адипиновая кислота применяется для получения
полиэфирных смол и некоторых пластификаторов и в больших
количествах—для получения полиамидных волокон типа найлон.
Циклогексанол находит некоторое самостоятельное
применение в качестве растворителя.
283. Напряжение в алициклических соединениях. Для
объяснения большой устойчивости пентаметиленового и гекеамети-
ленового колец и малой устойчивости циклов из четырех и
особенно из трех атомов углерода, а также этиленовых и
ацетиленовых углеводородов А. Байер* предложил так называемую
теорию напряжения (см. стр. 243).
На рис. 23 и 24 (см. стр. 161, 162) представлены схемы цепей из
трех, четырех и пяти атомов углерода. Из рисунков видно, что
для замыкания цикла из трех атомов углерода валентные связи
атомов углерода должны быть выведены из своего
первоначального направления. Что касается пятичленного цикла, то
замыкание его происходит без всякого отклонения валентных связей.
Вычислим угол отклонения этих связей в различных циклах.
Так как в трехчленном цикле направления связей между
атомами углерода образуют равносторонний треугольник, то угол
между ними равняется 60° (рис. 67), а так как угол между
направлениями валентных связей в тетраэдре равен 109°28', то угол
отклонения каждой валентной связи равен
ЮУ28'-60° 24о44,
Направления валентных связей в четырехчленном цикле
образуют квадрат. Следовательно, угол отклонения равен
Ю9°28'-90° = дО44,
Каждый из внутренних углов пятиугольника равен** 108°.
Отсюда угол отклонения равен 0°44' (см. рис. 68). Произведя
* Адольф Байер (А. Bayer)—выдающийся немецкий химик A835—1917).
Работал в Берлине и в Страсбурге.
Из многочисленных работ Байера в различных областях органической
химии назовем: установление строения пиррола и индола; установление
строения и первый синтез индиго A880), исследования в области ацетиленовых,
ароматических и алициклических углеводородов, приведшие его к созданию
теории напряжения A885).
В 1870 г. Байер высказал предположение о том, что ассимиляция углерода
зелеными частями растений протекает через промежуточное образование форм
альдегида, который далее полимеризуется в сахаристые вещества.
2d(n-2)
** Каждый внутренний угол правильного многоугольника равен ,
где я—число сторон многоугольника.
556
' Глава XX ЦиклопараФины
чакие же вычисления для шестичленного и восьмичленного
циклов и принимая, что все атомы углерода лежат в одной
плоскости, получим соответственно 5°36' и 12°5Г (рис. 69).
Тетраэдр и чес кое расположение связей, при котором углы
между ними равны 109°28', наиболее устойчиво. Угол
отклонения от этого направления является, по теории Байера, мерой
«напряжения», г. е. мерой неустойчивости цикла.
Рис 67. Отклонения валентных
связей в трехчленном цикле
Рис. 68. Отклонения валентных
связей в пятичленном цикле.
Согласно теории Байера, шестичленный цикл должен быть
менее устойчив, чем пятичленный; высшие циклы должны быть
так же неустойчивы, как и триметиленовое кольцо. Однако это
Рис. 69, Отклонения
валентных связей в плоском вось-
мичленном цикле
Рис. 70. Схема
шестичленного кольца без напряжения
не соответствует действительности: шестичленный цикл устойчив
не менее пятичленного цикла; известны также весьма устойчивые
циклы с очень большим числом атомов углерода. Это
противоречие теперь устранено, так как доказано, что, начиная
с шестичленного кольца, атомы углерода в цикле нахо-
284. Стереоизомерия алициклических соединений
557
дятся не в одной плоскости; тогда можно построить циклы из
многих атомов углерода без отклонения направления валентных
связей, т. е. без напряжения (рис. 70). Подобные циклы не
имеют «байеровского» напряжения.
284. Стереоизомерия алициклических соединений. Атом
углерода кольца является асимметрическим в том случае, если он
связан с двумя различными атомами или радикалами и с двумя
различными остатками кольца. Это можно пояснить на примере
циклопропана:
VH
ІХХн
циклопропан
ч ХХЮН
\
С
СООН
\:оон
циклопропанкарбоновая
кислота
циклопропан диларбоновая-і.ї-
кислота
і 11 111
Если в молекуле циклопропана (I) заместить один атом
водорода карбоксилом, то атом углерода, при котором произошло
замещение, не становится асимметрическим, так как две его
единицы валентности насыщены совершенно одинаковыми
остатками кольца (II). Если замещение атома водорода карбоксилом
произвести у двух углеродных атомов, то оба атома углерода
становятся асимметрическими, так как остатки кольца, с
которыми связан каждый из них, теперь неодинаковы (III); в то же
время легко видеть, что оба асимметрических атома равноценны
между собой. Как и у винных кислот, в данном случае также
возможны четыре изомера—два оптических антипода,
недеятельная вследствие внутренней компенсации форма и рацемическое
соединение.
Вывод о конфигурациях стереоизомеров можно сделать
следующим образом. Так как в молекуле циклопропана атомы
углерода лежат в одной плоскости, то из шести атомов водорода
три должны находиться по одну сторону плоскости кольца и
три—по другую сторону этой плоскости. Если у двух атомов
углерода молекулы циклопропана заместить по одному атому
водорода карбоксилом, то последние могут находиться или по
одну сторону плоскости кольца (цис-расположение), или же по
разные стороны (транс-расположение):
НОО
ООН
ноос
658
Глава XX. ЦиклопаоаФины
Цис-форма циклопропандикарбоновой-1,2 кислоты плавится
при 139 °С; выделяя воду, она дает ангидрид. Температура
плавления рацемической транс-формы 175 °С; ангидрид ее неизвестен.
Молекула цис-циклопропандикарбоновой-1,2 кислоты
симметрична, поэтому эта кислота оптически недеятельна. Молекула
транс-циклопропандикарбоновой-1,2 кислоты не имеет плоскости
симметрии, она не совпадает со своим зеркальным отражением,
поэтому для нее возможны два оптических антипода, а также и
рацемическая форма.
Оптические антиподы /яранс-циклопропандикарбоновой-1,2
кислоты Aа1В=±84,8°)
ноос
н
н \ соан ноос /н\н
н
соон
были получены расщеплением рацемической формы при помощи
оптически деятельного основания (бруцина).
Большой интерес представляет изомерия инозита (гексаокси-
циклогексана) СвНв(ОНN. Несмотря на то что в молекуле
инозита нет асимметрического атома углерода, инозит известен в
оптически деятельных формах. Молекулы оптически деятельных
веществ несимметричны—они не имеют плоскости симметрии и
поэтому несовместимы со своими зеркальными изображениями.
При наличии в молекуле хотя бы одного асимметрического атома
углерода молекула несимметрична. Однако, как это показано на
примере инозита, молекула может быть несимметричной, и не
имея асимметрического атома углерода. Для инозита возможно
восемь пространственных изомеров; из них семь обладают
симметрично построенными молекулами и оптически недеятельны.
Один из стереоизомеров, в молекуле которого 1,2,4-гидроксилы
находятся по одну сторону кольца, а 3,5,6-гидроксилы—по
другую сторону кольца, известен в виде двух оптических антиподов:
н
н
н
284. Стереоизомерия алициклических соединений 559
Молекулы этих антиподов не имеют плоскости симметрии;
являясь зеркальными изображениями друг друга, они несовместимы
одна с другой.
Левовращающий и правовращающий инозиты в виде
метиловых зфиров находятся в некоторых смолах. Один из
недеятельных, симметрично построенных инозитов,
называемый мезоинозитом, находится как в животных организмах
(например, в мышечном соке сердца, в печени, в мозговом
веществе—в виде сложных соединений), так и в растениях. В
растениях он находится в виде инозитфосфорной кислоты, т. е.
кислого эфира фосфорной кислоты. Его выделяют из растений,
например из конопляных жмыхов, в виде кальциевой соли,
известной под названием фитина. Мезоинозит—кристаллическое
вещество сладкого вг.уса.
ГЛАВА XXI
ТЕРПЕНЫ
285. Терпены в природе. Терпенами называются природные
углеводороды состава С10Н1в. Как сами терпены, так и близкие к
ним кислородсодержащие вещества весьма распространены в
растительном мире и находятся в так называемых эфирных маслах.
Эфирные масла содержатся в различных частях растений—
корне, стволе, листьях, цветах и плодах. Они обыкновенно
имеют сильный и приятный запах и потому находят обширное
применение.
Некоторые эфирные масла, например лимонное, апельсинное
и скипидар, или терпентинное масло, представляют собой почти
исключительно смесь терпенов.
По своим физическим свойствам терпены—бесцветные
жидкости легче воды, кипящие при температуре от 140 до 190 °С. Они
сильно преломляют лучи света и нерастворимы в воде.
Для установления строения терпенов большое значение имели
работы Е. Е. Вагнера и его учеников. Для многих терпенов
Е. Е. Вагнер предложил структурные формулы, которые в
настоящее время считаются общепринятыми.
В области терпенов много и плодотворно работал С. С.
Наметкин*.
* Сергей Семенович Намегкии A876—1950) родился в Казани. Окончил
Москоиский университет. С 1905 г. началась педагогическая деятельность
С. С. Наметкина.
Магистерская и докторская диссертации С. С. Наметкина посвящены
изучению реакции нитрования углеводородов и исследованию с помощью этой
реакции бициклическмх углеводородов.
В 1918 г. Наметкин начал проводить исследования в области терпенов и
продолжал их до конца своей жизни. Им синтезированы и изучены многие
производные камфена.
Большая часть работ С. С. Наметкина посвящена химии и технологии
нефти. Он разработал ряд проблем химии нефти (каталитическая
ароматизация нефтяных фракций, синтез хлорпроизводных и спиртов на основе нефтяных
углеводородов, окисление парафинов в спирты и альдегиды, получение
моющих средств и др.), составил руководство по химии нефти.
Ряд работ С. С. Наметкина посвящен вопросам стереохимии алициклнче-
ских соединений, а также синтезу душистых веществ, гербицидов и
стимуляторов роста растений.
В 1932 г. С. С. Наметкин был избран членом-корреспондентом Академии
наук СССР, а в 1939 г.—действительным ее членом.
286. Классификация терпенов 561
286. Классификация терпенов. В молекулах терпенов
содержится на шесть атомоз водорода меньше, чем в молекуле
соответствующего предельного углеводорода С10Н2а.
Исходя из юго, что каждая двойная связь, а равно и
замыкание кольца уменьшают количество водорода на два атома,
терпены можно разделить на четыре группы:
1. Терпены, в молекулах которых имеется открытая цепь ато-
тов углерода и три двойные связи.
2. Моноциклические терпены, в молекулах которых имеется
одно кольцо из атомов углерода и две двойные связи.
3. Бициклические терпены, в молекулах которых содержится
два кольца из атомов углерода и одна двойная связь.
4. Трициклические терпены,—в молекулах нет двойных
связей, а атомы углерода образуют три кольца.
Наибольшее значение имеют моноциклические и
бициклические терпены. Большая часть этих терпенов имеет близкое
отношение к n-метилизопропилбензолу, или цимолу.
В эфирных маслах, кроме терпенов—С]0Н16, часто
встречаются и более сложные углеводороды того же состава, но более
высокого молекулярного веса. Состав их можно выразить общей
формулой (С5НЯ)Я. Для терпенов п—2, для политерпенов п
больше двух. Политерпены подразделяют на сесквитерпены—С15Н24,
дитерпены—С2оН32 и т. д. К производным политерпенов
относятся абиетиновая кислота, содержащаяся в канифоли,
смоляные кислоты и другие природные вещества.
287. Моноциклические терпены. Обзор моноциклических
терпенов и родственных им веществ удобно начать с углеводорода мен-
шана С10Н20, который можно получить из цимола, присоединив
к его молекуле шесть атомов водорода. Это осуществляют,
пропуская пары цимола вместе с водородом через стеклянную трубку
с мелко раздробленным никелем:
Ч_.Пз *-Г1з
СН
нс хсн н2с с
|| | + ЗНа » | | -
НС СН Н2С СНа
V
Л
Н3С СНз Н3С СН3
цимол меитан
Ментан—жидкость, кипящая при температуре около 170 °С.
Сам ментан в природе не встречается, но его спирт, который на-
36—788
562 Глава XXI. Терпены
зывается ментолом, составляет главную часть мятного масла.
При восстановлении ментол переходит в ментан,
следовательно, в нем атомы углерода связаны так же, как и в ментане:
VjO? *-«»g ^-*Пд
СН СН СН
нас сн2 , н нас сна j^ н2с сна
I I *-^ I I
IX СН, Н,С СН
н2с сн2 н2с сн н2с с=о
сн сн он сн
сн сн сн
н3с сн3 н3с снэ н3с сн„
ментан менто/і ментон
Свойства ментола как вторичного спирта сказываются в том,
что при окислении его получается кетон—ментон, который
также содержится в мятном масле. Наконец, положение гидроксила
в его молекуле определяется тем, что путем ряда реакций-ментол
можно перевести в л-крезол.
Ментол представляет собой призматические кристаллы,
плавящиеся при 45 °С; он кипит при 212 °С; обладает сильным
(мятным) запахом; применяется ментол в парфюмерии (входит в
состав зубных порошков и паст) и как обезболивающее и
дезинфицирующее средство при мигрени, насморке и т. п.
Двухатомный спирт, производный от мента на,— терпин
С10Н1а(ОНI имеет строение:
СН3
С—ОН
Н2С СН,
І і
І
Н2С
Y
с
—он
н3с сн3
терпин
Терпин—жидкость. Он может быть получен обработкой
скипидара 25%-ной серной кислотой*.
При действии на терпин разбавленной серной кислоты
происходит отщепление молекулы воды и образуются одноатомные
* При этом получается кристаллический терпингидрат СюН18(ОНJ-НїО,
который легко теряет молекулу кристаллизационной воды.
287. Моноциклические терпены
563
спирты с одной двойной связью в молекуле, например a-niepnu-
неол С1вН17(ОН):
СН:, СН3
с—он
н.2с
сн
HuSO4
нас
:—он
V
терпвн
/ ч
н2с сн
I I
Н2С СН-2
*сн
с—он
н3с сн8
а-терпинеол
а-Терпинеол—кристаллическое вещество (темп, плавл. 38—
409 С; темп. кип. 219 °С) с запахом сирени. Он известен в
оптически деятельных формах. В молекуле а-терпинеола находится один
асимметрический атом (отмечен звездочкой): он связан с атомом
водорода, с группой—С(ОН)(СН3J и двумя неодинаковыми
половинками кольца. Наличие в а-терпинеоле асимметрического
атома углерода доказывает правильность его изображенной выше
формулы, так как при отщеплении от молекулы терпина воды
во всех других возможных направлениях не может образоваться
асимметрический атом углерода.
При отщеплении от а-терпинеола одной молекулы воды
образуются терпены общей формулы СгоН16—терпинолен (темп,
кип. 185—187 °С) и лимонен (темп. кип. 175 3С):
Н,С
н3с
СНа
С
сн.
СН
—н2о
Ч
НаС uij
D и L-лиманен
СН.
\
а-терпннео-п
сн3
терпинолен
В одном случае получается вещество, в молекуле которого
имеется асимметрический атом углерода; в другом
случае—вещество, в молекуле которого нет такого атома. Из двух
приведенных формул первую принимают для лимонена, а вторую—
для терпинолена.
36*
564
Глава XXI. Терпены
Правовращающий лимонен почти нацело составляет эфирное
масло померанцевой корки и является главной составной
частью масел: лимонного, бергамотного, тминного и укропного.
Лево вращающий лимонен находится в масле сосновой хвои.
Недеятельный лимонен получил название дипентена. Он
находится в эфирном масле цитварного семени и может быть
получен сухой перегонкой натурального каучука.
Так как в молекуле лимонена находятся две двойные связи,
то он может присоединять четыре атома галоида или две
молекулы галоидоводорода.
288. Бициклические терпены. Большинство бициклических
терпенов С]0Н1в можно произвести от ментана С]0Н20, если
замкнуть в нем второе кольцо, и, отняв два атома водорода,
образовать двойную связь. Состав получающихся при замыкании
кольца углеводородов выражается формулой С10Н18.
Замыкание кольца можно произвести различным образом.
При этом в зависимости от местоположения атомов углерода,
являющихся общими для обоих колец, получаются углеводороды
каран, пинан и камфан:
СН3
СН
НаС СНа
I |
НаС СН4
СН
СН
сн3
нас
сна
с
V
н3с-с—сн
н2с
СН,
СН»
каран
258. Бициклические терпены
565
Модель основного скелета молекулы бициклического терпена—
камфана изображена на рис. 71.
Удалением из описанных выше биииклических углеводородов
двух атомов водорода с образованием двойной связи
получаются терпены общей формулы С,0Н1(,.
Примером терпенов такого типа может служить получаемый
из пинана а-пинсн:
Н
а-Пинен составляет главную массу скипидара; он представляет
собой жидкость, кипящую при 156 °С. а-Пинен легко окисляется.
Так, если смешать дымящую азотную
кислоту с концентрированной серной
кислотой и прибавлять по каплям
скипидар, то в результате быстрого
окисления происходит вспышка. Он
окисляется и кислородом воздуха.
При этом к его молекуле
присоединяйся молекула кислорода и
образуется вещество состава С10Н1вО„.
Это вещество имеет характер
перекиси и легко окисляет другие
вещества, отщепляя от своей молекулы
один атом кислорода. Поэтому в
присутствии скипидара кислород
веществ: раствор индиго обесцвечивается, мышьяковистая
кислота окисляется в мышьяковую кислоту; ускоряется
«высыхание» лаков, содержащих непредельные кислоты.
При пропускании в охлажденный скипидар сухого хлористого
водорода выделяется соединение пинена с хлористым водородом
Рис. 71. Основной скелет
молекулы камфана.
воздуха окисляет ряд
566 Глава XXI. Терпены
С10Н17СІ, весьма напоминающее по внешнему виду и по запаху
камфору и поэтому иногда неправильно называемое
«искусственной камфорой». Это вещество отнюдь не тождественно камфоре,
полученной синтетическим путем.
Скипидар. Скипидар получают из хвойных деревьев
следующим образом. Счищают часть коры с хвойного дерева и делают
на стволе надрез; из надреза вытекает смолистый сок, который
застывает в вязкую массу, называемую терпентином или
живицей. При перегонке терпентина с водяным паром в приемном
сосуде собирается скипидар, или терпентинное масло, а в
перегонном аппарате остается твердая масса, которая называется
канифолью или гарпиусом.
Скипидар получается также при сухой перегонке сосновых
пней. Смола, содержащая скипидар, извлекается из осмоленной
древесины различными растворителями, главным образом высо-
кокипящим бензином.
Скипидар применяется в медицине; в технике он служит для
изготовления лаков и красок.
Канифоль* представляет собой смесь трициклических кислот
состава С20Нзо021 из которых самой важной является
абиетиновая кислота. Щелочные соли этих кислот обладают поверхностно-
активным действием, поэтому канифоль часто применяют в
качестве добавки при изготовлении некоторых сортов мыла с
большой пенообразующей способностью, а также в качестве
эмульгатора в производстве синтетических каучуков. Она
применяется также в производстве лаков и вместе с квасцами—для
проклейки бумаги.
289. Камфора. Из веществ, близких к терпенам, наибольшее
значение имеет обыкновенная японская, или лавровая, камфора
С10Н,6О. Она получается при перегонке с водяным паром
древесины и листьев камфарного дерева.
Камфора кристаллизуется в блестящих призмах; обладает
сильным характерным запахом. Температура плавления
камфоры 175 °С, температура кипения 209 °С. Несмотря на высокую
температуру плавления, камфора возгоняется уже при обыкновенной
температуре.
Обычная природная камфора оптически деятельна; вращает
плоскость поляризации вправо: [а]Ь0=+44°.
Многочисленными исследованиями доказано, что строение
камфоры соответствует приведенной ниже формуле. Согласно
этой формуле, камфора представляет собой кетон.
• Название происходит от греческого города Колофан (колофонийская
смола).
289. Камфора
567
Камфора может быть получена окислением природного
вторичного алкоголя—борнеола (темп, плавл. 202—203 °С; темп,
кип. 212 °С):
СНа
/
Н,С
Н3С~С—СН3
С<н
Н-С
*сн
камфора
борнеол
В хвое сибирской пихты, растущей в большом количестве &
Сибири и на востоке Европейской части СССР, содержится
уксуснокислый эфир борнеола:
О—СО—СНа
Омылением этого эфира получают борнеол, который затем может
быть окислен в камфору.
В очень большом количестве камфора получается из
скипидара. Для этого а-пинен при помощи ряда реакций переводят
в борнеол, который затем окисляют в камфору. В СССР способ
получения камфоры из скипидара разработан и внедрен в
промышленность В. Е. Тищенко.
Механизм перегруппировки соединений ряда пинана и камфа-
на (синтез камфоры из а-пинена) установлен С. С. Наметкиным.
Камфора применяется в медицине и в значительных
количествах в производстве целлулоида и бездымного пороха.
ГЛАВА XXI)
КАРОТИНОИДЫ
Каротиноидами называются природные окрашенные вещества,
сходные по своему строению с каротином—желто-красным
веществом, которое обусловливает цвет моркови (Daucus carota).
В молекулах каротиноидов находится большое число
сопряженных двойных связей; они являются полиеновыми красящими
веществами. Каротиноиды растворимы в растительных и
животных жирах, обладают характерными спектрами поглощения,
с крепкой серной кислотой дают индигово-синее окрашивание.
Большинство их легко окисляется кислородом воздуха.
290. Ликопин. Окраска томатов и плодов шиповника зависиі
главным образом от присутствия в них ликопина—углеводорода
состава С40Н56. Ликопин—карминово-красные призмы (темп,
плавл. 168—169 °С). В молекуле ликопина находится тринадцать
двойных связей. При действии водорода в присутствии
катализаторов ликопин присоединяет на одну молекулу 26 атомов
водорода и переходит в предельный углеводород С40Н82. Изучение
реакций расщепления ликопина позволяет принять для него
формулу (см. стр. 569), согласно которой в молекуле ликопина
восемь раз повторяется чередование атомов углерода,
характерное для углеродного скелета изопрена; при этом чередующиеся
группы атомов (в приведенной формуле они отделены
пунктиром) не все одинаково связаны между собой; в середине
молекулы происходит их перестановка, вследствие чего молекула
ликопина состоит как бы из двух равных, симметрично
расположенных частей.
291. Каротин. Каротин С40Н66 изомерен ликопину. Он
находится в моркови, во многих цветах, плодах, молоке, кровяной
сыворотке. Вместе со своим двугидроксильным
производным—желтым ксантофиллом C40HS4(OHJ он содержится в зеленых
листьях и обусловливает осеннюю окраску листвы. Для каротина
известны три изомера: ?-каротин (темп, плавл. 183 °С), а-каротин
(темп, плавл. 187 °С) и r-каротин (темп, плавл. 178 °С).
Изучение продуктов расщепления ?-каротина позволяет
изобразить его строение формулой, приведенной на стр. 569.
Формулу а-каротина можно вывести из формулы ликопина,
замыкая в последней оба конца цепи атомов углерода в кольцо;
замыкание кольца происходит у десятых атомов углерода, считая
от середины молекулы.
СНз _ CHg к СНз СН3 СН3 СНЧ СН-, СН-,
СНз-С-СН-СН2-І-СН2-С-СН-СНІСН-С-СН-Сн4=СН-с!-СН-СнІ=СН~СН=С—СН=СН-СН-С-СН=СН -СН^СІ-СНг-І-
і і і І і І і
ликопин
н3сч /сн3 і f f , , 34/
—сн==сн-сн=с-сн4=снччк..
Ч^з
?-каротив
СН, СН9 СН3 «зС СНа
ї її X '
/Яч/СН«=СН-С=СН-СН-СН-С-СН-СН—СН-СН-С-СН-СН-СН«=С-СН=СНХ
Н,С С№ СНз СН, СН9 СН3 «зС
>* її її X '
/Яч/СН«=СН-С=СН-СН-СН-С-СН-СН—СН-СН-С-СН-СН-СН«=С-СН=СНХ / \
НаС С ¦ НС СН
IP II
HC С о-кл doth в С СН
н/
370 Глава XXII. Каротиноиды
Строение а-каротина (см. стр. 569) отличается от строения
?-каротина лишь иным расположением двойной связи в одном из
колец.
1-Каротин по своему строению занимает промежуточное
положение между ?-каротином и ликопином: одна половина его
молекулы аналогична половине молекулы ?-каротина,
другая—половине молекулы ликопина (т. е. в молекуле у-каротина
содержится только одно кольцо).
292. Витамин А. Витамин А очень близок по строению к
?-каротину; представляет собой светло-желтое масло состава
СМН2§ОН. Он растворим в жирах, перегоняется в глубоком
вакууме. Его строение изображают формулой:
НС СНз СН3 ._ СН3
2c-Ch4=CH—С=СН— СН=«=СН— С=СН—СН2ОН'
н2сх>с-сн3'
'СН2
Из сопоставления формулы витамина А с формулой
?-каротина видно, что молекула витамина А соответствует половине
молекулы ?-каротина, причем к месту разрыва присоединены
гидроксильная группа и атом водорода.
Витамин А содержится в коровьем молоке (особенно .летом,
когда коровы питаются свежей травой), в масле, в яичном
желтке, в рыбьем жире, в большинстве овощей и фруктов. Он
является фактором роста, Недостаток его в пище человека вызывает
убыль в весе, высыхание роговицы глаза и понижение
сопротивляемости организма к инфекциям.
В пищевом рационе витамин А можно заменить каротином.
Каротин является провитамином А: в животном организме он
переходит в витамин А, который часто в значительных количествах
накапливается в печени.
ГЛАВА ХХШ
СТЕРИНЫ ЖЕЛЧНЫЕ КИСЛОТЫ. СТЕРОИДНЫЕ ГОРМОНЫ
293. Стерины. Стеринами называется группа одноатомных
полициклических спиртов, весьма распространенных как в
животном, так и в растительном мире. В молекулах этих спиртов
находятся четыре углеродных кольца, из которых три являются ше-
стичленными, но не имеют ароматического характера.
Наиболее давно известным стерином является холестерин
(по-гречески «холе»—желчь, «стереос»—твердый) С27Н45ОН. Он
находится, частично в виде эфиров, почти во всех органах
человека, но особенно в больших количествах в мозгу и в
веществе нервов. Холестерин был впервые выделен из
желчных камней, которые содержат его в качестве главной составной
части. Холестерин—кристаллическое вещество (темп, плавл.
149 °С); его строение было выяснено в 1932 г. главным образом
работами Виланда и Виндауса. Как видно из формулы:
I -
сн-сн3
холестерин является вторичным спиртом; в его молекуле
находится система гидрированного фенантрена, сконденсированная с
кольцом циклопентана. Подобно холестерину, и другие стерины
характеризуются наличием в их молекулах гидрированного
углеродного скелета циклопентанофенантрена:
НаС СН
\
с
сна
I
I I I
CHa CH CH-CHa
h/^Yh
HaC CH CHa
CH2 CH2
гидрированный циклопентанофепантсеа
572 Глава XXIII. Стерины. Желчные кислоты. Стероидные гормоны
Примером стеринов может служить эргостерин
(темп, плавл. 163 °С):
Н3С СН—СН=СН—СН—СН—СН,
/\
II
н
¦ч/
эргостерин
Эргостерин можно выделить из дрожжей и некоторых других
продуктов. При облучении эргостерина ультрафиолетовыми
лучами в его молекуле происходит размыкание одного из шести-
членных колец; при этом образуется вещество, аналогичное по
действию антирахитическому витамину, находящемуся в рыбьем
жире. Это вещество было названо витамином Da и выпущено
в продажу под названием кальциферола (темп, плавл. 115—117 °С);
его строение выражается формулой:
СН8 СН3 СН3
I I I
Н3С СН—СН=СН—СН—СН—СН,
нас
но.
нХ/
V
Известно очень много природных веществ, в молекулах
которых, подобно, стеринам, содержится скелет циклопентанофе-
нантрена. Таковы желчные кислоты, половые гормоны, сердечные
яды из листьев, семян и корней дигиталиса (наперстянки) и
различных видов строфанта и ряд других веществ. Вся группа этих
веществ получила название стероидов.
294. Желчные кислоты. Желчные кислоты находятся в желчи
человека и многих животных. Так, желчь человека содержит:
холевую кислоту
дезоксихолевую кислоту
антроподезоксихолевую кислоту
литохолевую кислоту
СазН37(ОНJСООН
С,3Нз8(ОН)СООН
294 Желчные кислоты
573
Все эти кислоты являются гидроксильными производными
колановой кислоты СгзНзвСООН, в которую и могут быть
переведены путем восстановления
-СНо—СООН
холановая кислота
Холевая и дезоксихолевая кислоты находятся в желчи в виде
«парных желчных кислот» в сочетании с гликоколом и таурином.
Такова, например, гликохолевая кислота, которая расщепляется
на холевую кислоту и гликокол:
СазНзв(ОНKСОЫНСН2СООН + Н8О
гликохолевая кислота
холеваая квелета
NHaCH„COOH
гликокол
Желчные кислоты обладают способностью эмульгировать
жиры. Холевая и дезоксихолевая кислоты могут соединяться с
самыми различными веществами (высшие жирные кислоты,
углеводороды, высшие кетоны), образуя так называемые холеиновые
кислоты, щелочные соли которых растворимы в воде.
Из дезоксихолевои кислоты был получен углеводород—ме-
тилхолантрен (твердое вещество желтого цвета):
НООС СНа
СН3 СН—і
сна
н3с
Н3С
/\
дезоксихолевая кислота
574 Глава ХХШ. Сгерины. Желчные кислоты Стероидные гормоны
Метилхолантрен обладает очень сильно выраженными
канцерогенными свойствами.
Некоторые исследователи полагают, что многие виды раковых
заболеваний развиваются вследствие образования метилхолан-
трена и близких к нему веществ из желчных кислот или из сте-
ринов и половых гормонов организма.
295. Стероидные гормоны. Стероидные гормоны выделяются
половыми железами и корой надпочечников. В организме они
играют исключительно важную роль, регулируя процессы обмена
веществ, роста, размножения и старения.
В зависимости от биологического действия стероидные
гормоны разделяют на половые и кортикоидные гормоны.
Половые гормоны. Половые гормоны выделяются половыми
железами, от них зависит развитие специфических мужских
и женских половых признаков.
Примеры половых гормонов;
Н3С
со-сн,
эстрон прогестерон
женские палоеые гормоны
няс
о
н,с
/V
но
J
н3с
А
нэс
он
андростерон
гестостеров
шужскне половые гормоны
Если в молекуле женского полового гормона—прогестерона
группу СОСН3 заместить гидроксилом, то получим структурную
формулу мужского полового гормона—тестостерона.
295 Стероидные гормоны
575
Кортикоидные гормоны. Важнейшими кортикоидными
гормонами являются кортизон и гидрокортизон:
сн2он
Н3с с=о
о /х|/1чон
кортизон
гидрокортизон
В медицине кортикоидные гормоны применяются прежде всего
при заболеваниях надпочечников. Кроме того, они нашли
широкое применение для лечения ревматических заболеваний,
воспалительных процессов, бронхиальной астмы и целого ряда других
болезней.
В последнее время было найдено, что введение двойной связи
в положение 1—2 молекулы кортизона и гидрокортизона
значительно увеличивает их активность.
Таким путем были получены новые препараты преднизон и
преднизалон:
Н
иредназои
преднизалон
ЧАСТЬ ТРЕТЬЯ
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
КЛАССИФИКАЦИЯ И ОБЩАЯ
ХАРАКТЕРИСТИКА
296 Классификация и общая характеристика
гетероциклических соединений. В предыдущих разделах книги были
рассмотрены соединения с открытой цепью углеродных атомов, так
называемые ациклические соединения и карбоциклические
соединения, в молекулах которых имеются циклы из углеродных атомов.
Настоящий раздел охватывает гетероциклические соединения
(или гетероциклы). В молекулах гетероциклических соединений
имеются циклические группировки атомов, включающие, кроме
атомов углерода, и другие (гетеро) атомы. Так же, как и среди
карбоциклических соединений, среди гетероциклов наиболее
распространены пяти- и шестичленные циклы. Причина большей
устойчивости и легкости образования таких циклов—отсутствие
напряжения (см. стр. 555). Гетероциклические соединения могут
образовывать системы с двумя и большим числом циклов,
сочленяясь, например, с ароматическими кольцами.
Рассмотрение гетероциклических соединений в последнем
разделе курса объясняется тем, что строение их более сложно, чем
строение соединений жирного и карбоциклического рядов.
Отсюда более сложными и разнообразными являются и их
химические свойства и превращения.
Значение гетероциклических соединений в природе и технике
весьма велико. К этому классу относятся такие важные
природные вещества, как хлорофилл растений, гемин крови,
гетероауксин, индиго, пенициллин и др. Чрезвычайно большую группу
среди гетероциклических соединений составляют растительные яды—
алкалоиды: хинин, морфин, никотин и др.
Среди гетероциклов мы встречаем также важнейшие группы
так называемых кубовых и сернистых красителей и очень
большое число разнообразных лекарственных веществ: сульфидин,
сульфазол, синтетические антималярийные препараты (акрихин,
плазмохин) и мн. др.
Физические и особенно химические свойства
гетероциклических соединений в большой степени зависят от характера связей
в цикле. Вещества этого класса, в цикле которых нет двойных
296. Классификация и общая характеристика 577
связей, как правило, напоминают по своим реакциям
ациклические и алициклические соединения.
Так, например, по формальным признакам янтарный и фта-
левый ангидриды
Н2С СНа J\с=0
А
янтарным ангидрид фталевыП ангидрид
должны быть отнесены к гетероциклическим соединениям.
Между тем по химическим свойствам они существенно* не отличаются
от обычных ангидридов, разве только легкостью образования.
Точно так же такое соединение, как тетрагидрофуран, обладает
всеми свойствами обычных простых эфиров, а
пиперидин—свойствами обычных вторичных жирных аминов.
аи
Н,С CH.a Н,С СН2
II II
НаС СН2 НаС CHa
О NH
тетрагидрофуран пиперидин
Ряд специфических свойств гетероциклических соединений
проявляется в том случае, если в цикле имеется сопряженная
система двойных связей, например в циклах фурана и пиридина:
НС СН
II II
НС СН
V
фураь
СН
/\
НС СН
II 1
НС СН
V
пиридин
Такие соединения имеют «ароматический» характер. Для
гетероциклических соединений, в цикле которых имеются сопряженные
двойные связи, при действии галоидов, азотной и серной кислот
(и других подобных реагентов) характерны не реакции
присоединения, а реакции замещения.
Номенклатура гетероциклических соединений подобна
номенклатуре соединений ароматического ряда. Корнем слова служит
37-788
578 Часть третья. Гетероциклические соединения
название гетероцикла; заместители называются в том же
порядке, что и в ароматических соединениях. Так, группа ОН в цикле
называется «окси», а соединения, содержащие группу СООН,—
карбоновыми кислотами. Место атома или группы в цикле
отмечается цифрой, причем счет начинается, как правило, с
гетероатома. Если в соединении имеется несколько гетероатомов, счет
начинается с атома кислорода, затем нумеруют атомы серы и
далее азота.
ГЛАВА XXIV
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
297. Общая характеристика пятичленных гетероциклов.
Основными группами гетероциклических соединений с пятичленным
циклом являются группы фурана, тиофена и пиррола.
Родоначальники этих групп—фуран, тиофен и пиррол
НС СН
І! II
НС СН
V
фуран
НС СН
II II
НС СН
V
тиофен
НС СН
II II
НС СН
пиррол
близки по строению и имеют ряд сходных признаков.
Если обозначить гетероатом буквой X, то общую формулу
этих соединений можно написать следующим образом:
?' НС CH?
II II
а' НС СНа
\/
X
Близкое родство фурана, тиофена и пиррола наглядно
проявляется в возможности их взаимного перехода. Как показано
Ю. К. Юрьевым, при пропускании паров фурана в смеси с
сероводородом или аммиаком над А12О3 при температуре 400—500 СС
происходит взаимное превращение:
Фуран, тиофен и пиррол имеют систему сопряженных двойных
связей и по своему поведению в ряде реакций близко стоят к непре-
37*
580 Глава XXIV. Пятичленные гетероциклы
дельным соединениям типа дивинила (осмоление при действии
кислот, полимеризация и т. п.). С другой стороны, они
напоминают ароматические соединения (способность к замещению атомов
водорода при галоидировании, нитровании, сульфировании и
ацилировании). Такие типичные реакции, как гидрирование,
идут с ними так же, как и с дивинилом (водород становится
первоначально в положение 1,4):
| X 1 | Х- 1
сн=сн—сн=сн + н2 —> сна—сн=сн—сна
Реакции, характерные для ароматических соединений
(сульфирование, нитрование, ацилирование), протекают с замещением
атомов водорода, стоящих в а-положении. В качестве примера может
служить реакция сульфирования. Как показал А. П. Терентьев,
эта реакция может быть осуществлена с фураном, тиофеном,
пирролом и рядом их гомологов путем нагревания сульфируемого
соединения с продуктом присоединения серного ангидрида к
пиридину (пиридинсульфотриоксид):
НС СН
НС С—SO3H- W \
V
ГРУППА ФУРАНА
298. Фуран. Фуран—жидкость, малорастворимая в воде (темп,
кип. 32 °С)
НС4 itCH
II II
нс5 асн
фуран
Пары фурана окрашивают древесину, смоченную соляной
кислотой, в зеленый цвет. Фуран вместе со своими ближайшими
гомологами а-метилфураном (сильван) и а,а'-диметилфураном
находится в легколетучих фракциях продуктов сухой перегонки
дерева. Чистый фуран приготовляют обычно из пирослизевой
кислоты (см. стр. 582), нагревая ее с дифениламином.
По структуре и свойствам фуран несколько сходен с эфирами
винилового спирта. Так же как и они, фуран и его гомологи
устойчивы к действию щелочей, но гидролизуются при нагревании
разбавленной соляной кислотой. Из фурана образуется при этом
298. Фуран 581
очень легко изменяющийся янтарный диальдегид, а из 2,5-диметил-
фурана—аиетонилаиетон (гександион-2,5):
НС СН
Н II
СН?-С С-СН3 -±^-»
Хс/
2,5-диметилфуран
сн-сн
II II
он он
СНа СН,
» СНз—С С—СН
II II
О О
ацетони лацетон
Диеновый характер фурана проявляется в том, что он осмо
лнется под действием серной кислоты. Окислители (азотная
кислота) превращают его в малеиновую кислоту. При
гидрировании фурана получается сначала 2,5-дигидрофуран, а затем те-
трагидрофуран—фуранидин, имеющий характер типичного эфира.
Подобно дивинилу, фуран легко присоединяется к малеино-
вому ангидриду (диеновый синтез):
СН
НС | С
|| |
НС=СН СН—СО НС | СН—СО
о
/ ¦ II / II
НС=СН СН—СО НС СН—СО
Ароматический характер фурана сказывается в его
поведении при галоидировании, нитровании и сульфировании.
При хлорировании фурана хлористым сульфурилом
образуется 2-хлорфуран—жидкость с темп. кип. 77 °С; при действии
на фуран азотной кислоты в растворе уксусного ангидрида
образуется 2-нитрофуран с плохим выходом; при сульфировании
фурана получается фурансульфоновая-2 кислота; ацетил и рование
уксусным ангидридом в присутствии хлористого цинка
(Я. Л. Гольдфарб и Л. М. Сморгонский) дает 2-ацетилфуран и
2,5-диацетилфуран.
Заместители первого рода (ОН, NH2) увеличивают
реакционную способность и тем самым неустойчивость бензольного
ядра. Такое же влияние они оказывают на ядро фурана, и
неудивительно, что окси- и аминофураны малодоступны
(вследствие своей неустойчивости). Заместители второго рода, как и в
ароматическом ряду, дезактивируют ядро фурана. Поэтому
альдегиды, кетоны и кислоты фурана гораздо более устойчивы, чем
сам фуран и его гомологи.
Из производных фурана наиболее известны фурфурол и пи-
росл изевая кислота.
582 Глава XXIV. Пятичленпые гетероциклы
299. Фурфурол. Фурфурол—бесцветная жидкость с темп.
кип. 162 °С; легко темнеет и осмоляется при хранении под
действием воздуха. Фурфурол является альдегидом:
НС СН
II II
НС С—С
V
фурфурол
Запах фурфурола несколько напоминает запах свежевыпечен-
ного хлеба.
Фурфурол получается при нагревании с разбавленными
кислотами оболочек семян, содержащих пентозы (см. стр. 330).
Сырьем для его получения могут служить также отруби
(А. Н. Энгельгардт, 1870), лузга подсолнечника и др.
В фурфуроле реакционная способность атомов водорода,
находящихся в цикле, ослаблена альдегидной группой. Поэтому по
химическим свойствам фурфурол очень похож на бензойный
альдегид.
В технике фурфурол находит применение в качестве
селективного растворителя, например при разделении сложных
углеводородных смесей, а также как полупродукт для получения феноло-
альдегидных смол, в которых исходным альдегидом является не
формальдегид, а фурфурол.
Окислители (KMnOJ превращают фурфурол в фуранкарбо-
новую (пирослизевую) кислоту. Под действием крепких
щелочей фурфурол превращается по реакции Канниццаро—Тищенко
в эквимолекулярную смесь пирослизевой кислоты и фурилового
спирта:
2ч )~C<C +KOH »' )- COOK +
\ / \Q \ / N / Yh ПН
фурфурол калиевая соль фуриловыи
пирослизевой спирт
кислоты
Можно обнаружить в смеси очень малые количества
фурфурола, действуя на нее ацетоном, а затем крепкой серной
кислотой,—получается интенсивное малиновое окрашивание (В. В. Че-
линцев); реакция пригодна и для открытия ацетона. Столь же
чувствительной реакцией является нагревание фурфурола с
уксуснокислым анилином (красное окрашивание).
300. Пирослизевая кислота. Пирослизевая кислота
получается обычно из фурфурола, но может быть получена нагре-
301. Тиофен 583
ванием слизевой или сахарной кислот. Она представляет собой
бесцветные кристаллы (темп, плавл. 130 °С) и является кислотой,
более сильной, чем бензойная.
При нагревании пирослизевая кислота теряет СО2,
превращаясь в фуран.
Наличие в фурановом кольце пирослизевой кислоты
карбоксильной группы придает кольцу значительную устойчивость.
В безводной среде пирослизевая кислота хлорируется, бро-
мируется, нитруется и сульфируется преимущественно в а-поло-
жение.
Нитропроизводные фурана применяются а медицине.
Наиболее известен препарат фурацилин, являющийся семикарбазоном
нитрофурфурола:
2o = N—NH—СО—NHj
Его успешно используют для лечения дизентерии, брюшного
тифа, дифтерии.
Тешрагидрофуран, или фуранидин, в технике может быть
получен гидрированием фурана или дегидратацией 1,4-бутандиола:
HqC СН2
СН.ОН-СНа—СН3—СНаОН »НаО+ | |
Н2С CH.j
о
Тетрагидрофуран—жидкость, темп. кип. 65 °С; является
прекрасным растворителем, находит применение в качестве полупродукта
для получения полиамидных волокон.
ГРУППА ТИОФЕНА
301. Тиофен. Тиофен—бесцветная жидкость с темп. кип. 84 °С
НС4—зСН
II II
НС5 2СН
тиофен
имеет характерный запах, напоминающий запах неочищенного
керосина.
584 Глава XXIV. Пятичлечные гетвроциклы
История открытия тиофена весьма любопытна. В 1883 г. немецкий химик
Виктор Мейер, показывая на лекции получение бензола перегонкой бензойно-
кислого натрия с натронной известью, проделал качественную реакцию «нз
бензол». Такой реакцией считалось появление синего окрашивания при
действии на бензол раствора изатина в концентрированной серной кислоте. Однако
опыт не удался. При проверке реактива с обычными сортами бензола все
проходило нормально. Заинтересовавшись этим явлением и проведя ряд опытов,
Мейер вскоре выяснил, что синее окрашивание дает не сам бензол, а
трудноотделимая от него примесь, содержащаяся в небольшом количестве в
каменноугольном бензоле. Исследование этой примеси показало, что она представляет
собой сернистое соединение C4H4S, очень близкое по свойствам к бензолу. По*
лученному веществу Мейер дал название тнофен, что означает сернистый
аналог бензола.
Тиофеновые соединения содержатся в каменноугольной смоле.
Сернистые соединения, присутствующие в нефти, также частично
состоят из тиофеновых гомологов.
Тиофен с хорошими выходами получается по реакции
Ю. К. Юрьева—пропусканием паров фурана с сероводородом
над окисью алюминия при 450 °С.
Образование тиофенов происходит также при пропускании
дивинила или изопрена в пары кипящей серы:
НС С—СН3
І! ІІ + 2S => !| |! ~ + H2S
Н2С СН2
Тиофен устойчив к нагреванию. По многим свойствам он очень
напоминает бензол, но отличается от последнего сравнительно
легкой окисляемостью при реакции с хлорноватистой кислотой,
азотной кислотой и др. В этом отношении тиофен похож на фу-
рановые соединения. Ароматический характер тиофена
проявляется в различных реакциях замещения. Соблюдая некоторые
предосторожности (разбавление инертными растворителями,
охлаждение), действием хлора и брома можно получить а-галоид-
производные тиофена. При нитровании и сульфировании в
мягких условиях образуются соответственно а-нитротиофен и а-суль-
фотиофен.
Тиофен более реакционноспособен, чем бензол; этим
пользуются для удаления из бензола примеси тиофена.
Для очистки сырого бензола его взбалтывают с небольшим количеством
85%-ной серной кислоты, которая извлекает тиофен в виде сульфотиофена.
При восстановлении а-нитротиофена получается а-аминотио-
фенг напоминающий по свойствам анилин. В отличие от анилина,
аминотиофен, взаимодействуя с азотистой кислотой, не образует
диазосоединений, а подвергается глубокому окислению.
302. Пиррол 585
ГРУППА ПИРРОЛА
302. Пиррол. Пиррол—жидкость (темп. кип. 131 °С),
малорастворимая в воде
НС4—зСН
II I!
НС6 оСН
f
пиррол
Впервые пиррол был найден в каменноугольной смоле и в
масле, получающемся при сухой перегонке костей. Свое
название пиррол (по-гречески «пир»—огонь) получил вследствие того,
что его пары окрашивают лучину, смоченную соляной кислотой,
в красный цвет.
Пиррол может быть выделен из костяного масла. Его можно
также получить по методу Юрьева (см. стр. 579) действием
аммиака на фуран. Если вместо аммиака взять метиламин или
анилин, то образуются соответственно 1-метил- или 1-фенилпиррол.
При нагревании сахарнокислого аммония в глицерине (Е. С. Хо-
тинский) также происходит образование пиррола:
ОН ОН
I I
СН—СН—COONH4 СН=СН
» NNH + 2СО2 + NH3 + 4НаО
СН—СН—COONH4 СН=СН
ОН ОН
Наличие группы NH характеризует пиррол как вторичный
амин. Пиррол реагирует с металлическим калием, образуя
пиррол-калий. При действии йодистого метила на пиррол-калий он
превращается в 1-метилпиррол, а при действии уксусного
ангидрида—в 1-ацетилпиррол. Основные свойства NH-группы
проявляются очень слабо.
Пиррол в еще большей степени, чем фуран, имеет характер
диенового соединения. Он исключительно легко, даже в
присутствии небольшого количества минеральной кислоты,
превращается в темно-красную смолу—смесь полимеров. При
хранении, особенно на свету и в присутствии воздуха, он быстро
осмоляется. При действии окислителей (азотная кислота,
хромовая кислота и т. п.)'гТиррол легко превращается в малеинимид:
СН=СН л СН-СО
-4-ЗО її v
11 Ч"Ч + Н2О
СН=СН СН—СО
586 Глава XXIV. Пятичленные гетероциклы
По отношению к щелочам пиррол вполне устойчив.
Так же как и фуран, пиррол проявляет «ароматический»
характер, напоминая часто по легкости превращений фенол. Так,
при действии иода в щелочном водном растворе на пиррол легко
образуется тетраиодпиррол—иодол. Хлор и бром в тех же
условиях окисляют пиррол.
Обычное нитрование азотной кислотой приводит к полному
разрушению пиррольного кольца. Концентрированная серная
кислота вызывает сильное осмоление пиррола. Действием на
пиррол продукта присоединения серного ангидрида к пиридину с
количественными выходами получается а-пирролсульфоноеая
кислота. Соли ее являются вполне устойчивыми соединениями.
Введение в пиррольное кольцо карбоксильной группы
осуществляется действием на пиррол-калий углекислого газа; при этом
сначала получается соль N-пирролкарбоновой кислоты, а при
200 °С—соль а-пирролкарбоновой кислоты,—реакция, подобная
реакции получения салициловой кислоты из фенолята натрия
(см. стр. 479).
Аналогично идет реакция для смешанных магнийорганиче-
ских производных пиррола, получаемых, например, при действии
магнийбромэтила. Из продукта реакции уже без значительного
повышения температуры образуется а-пирролкарбо новая кислота.
При действий на магнийбромпиррол хлорангидридов и
ангидридов кислот или сложных эфиров образуются кетоны. Так,
например, при взаимодействии магнийбромпиррола с хлористым
бензоилом получается а-бензоияпиррол (темп, плавл. 79 °С):
НС СН НС СН
|| || + С1СОС0Н5 » || || + MgClBr
НС СН НС С—COQjH5
\/ \ /
N—MgBr NH
магнийбромпиррол а-бензоилпиррол
Взаимодействием магнийбромпиррола с эфиром муравьиной
кислоты получается а-пирролальдегид (В. В. Челинцев и
А. П. Терентьев), твердое вещество с темп, плавл. 50 °С:
:н ,н не—сн
> II II .
О НС С-С<
tf—MgBr NH
магнийбромпиррол а-пирролальдегид
Альдегиды и кетоны легко конденсируются с пирролом,
причем в конденсации участвуют атомы водорода в ос-положении.
303, Пирролидин 587
Так, при взаимодействии пиррола с формальдегидом в
присутствии щелочи получается двухатомный спирт (В. В. Челинцев,
Б. В. Максоров):
НС
II
НС
\/
NH
СН
II + 2Н—С<
СН
НС СН
—* II II
Н0СН2-С С-СНа0Н
NH
Весьма подробно изучено действие на пиррол жирных и
ароматических кетонов, причем показано, что в первую очередь
образуются продукты конденсации:
Таким образом, у незамещенного пиррола наиболее реакцион-
носпособными являются атомы водорода в а-положении.
Производные пиррола по устойчивости разделяются на две
группы. Малоустойчивы производные с заместителями первого
рода, т. е. гомологи, а также окси- и аминопроизводные, в которых
группы —ОН и —NH2 стоят у углеродов пиррольного ядра.
Более устойчивы производные с ориентантами второго рода
—СООН, —SO3H, —СНО, —СОСН3 и др. Такое же положение
мы наблюдали у ароматических соединений и производных фу-
рана.
Соединения, содержащие пиррольное кольцо, широко
распространены в животном и растительном мире.
303. Пирролидин. При восстановлении молекула пиррола
присоединяет два атома водорода (в положение 2,5) и образует
дигидропиррол—пирролин. Дальнейшее гидрирование дает те-
трагидропиррол—пирролидин. Пирролидин по своим свойствам
подобен жирным вторичным аминам. Это—жидкость (темп. кип.
88,5 °С) с характерным аммиачным запахом, сильно дымит на
воздухе. Из производных пирролидина отметим а-пирролидин-
карбоновую кислоту—пролин:
СН2
I I
Н2С СН-СООН
\1Н
\i
Эта аминокислота получается, наряду с другими аминокислотами,
при гидролизе белковых веществ.
588 Глава XXIV. Пятичленные гетероциклы
Важные производные пирролидина могут быть получены при
действии аммиака или алкиламинов на т-бутиролактон:
H2C СНд ^а^'
CH3NH2 Н2С СО +NH3 Н2С
"~ V
СН3
N-метил- у-бутиролакт«н а-пврролидон
пирролидон
N-Метилпирролидон является высокоэффективным селективным
растворителем. Он находит применение в технике для четкого
разделения сложных углеводородных смесей, в том числе для
выделения ацетилена из смеси с другими углеводородами.
При действии на а-пирролидон ацетилена под давлением
образуется N-винил-а-пирролидон:
НаС СНа Н3С СНа
| | -і-HCsCH | |
Н2С СО НоС СО
\/ \/
NH N—СН=СНа
х-пврролпдои N-ввнвл-а-пврролндов
Введение винильной группы в молекулу пирролидона придает
ей способность к полимеризации. Низковязкие полимеры винил-
пирролидона (молекулярный вес около 40 000) растворимы в
воде; их водные растворы нашли применение в качестве
заменителей кровяной плазмы:
СН,—СН
Н2С СН,
Перед консервированной кровью поли-М-винилпирролидон
имеет такие важные преимущества, как независимость от
группы крови н возможность хранения без охлаждения.
304. Гемин и хлорофилл. Особенно важными производными
пиррола являются пигменты крови и зеленых растений.
Красящее вещество крови, гемоглобин, играющий роль передатчика
кислорода, является сложным белком. При гидролизе он
распадается на протеин—глобин и небелковое вещество—гемин. В мо-
304. Гемин и хлорофилл
589
лекуле гемина содержится четыре ядра пиррола, связанные ме-
тинньши (СН=) группами. Существенной частью молекулы
гемина является координационно связанный атом железа (см.
стр. 217, 375):
CHj
СН2-СН2-СООН
ci-
СН2—СН2-СООН
Хлорофилл—зеленый пигмент листьев—состоит из двух
близких по строению веществ: хлорофилла а—сине-зеленого и
хлорофилла b—желто-зеленого цвета. Роль хлорофилла была в
значительной степени выяснена работами К. А. Тимирязева.
Разделение обоих весьма близких по свойствам пигментов удалось
впервые осуществить русскому ботанику М. С. Цвету, который
применил для этого изобретенный им метод (см. стр. 590).
Хлорофиллы по структурной формуле весьма близки к геми-
ну. Точно так же, как и в гемине, в их молекулах имеется по
четыре пиррольных ядра и атом металла (железа—в молекуле
гемина и магния—в хлорофиллах). Приводим формулу
хлорофилла а:
НзвС20ООС-гН2С-СН2 НС С=О
COOCHj
590 Глава XXIV. Пятичленные гетероциклы
Большое структурное сходство с этими природными
пигментами имеют синтетические красящие вещества фталоцианины,
получаемые из фталевого ангидрида:
фталевыи ангидрид
фталацианин меди
На основе фталоцианина меди синтезированы весьма ценные
пигменты и красители синего, голубого и зеленого цветов. Они
отличаются высокой химической устойчивостью и красивыми,
яркими оттенками.
305. Хроматографический анализ. При химическом
исследовании хлорофилла большие трудности встречаются при
разделении и очистке близких по свойствам растительных пигментов.
Они не перегоняются и не разделяются перекристаллизацией.
Кроме того, они весьма легко изменяются при химической обработке.
Поэтому очень большое значение приобрел особый метод
разделения этих пигментов, изобретенный в 1906 г. русским ботаником
М. С. Цветом A872—1919) и названный им хроматографическим
адсорбционным анализом.
Метод заключается в следующем. Раствор (чаще всего
бензиновый или бензольный) разделяемых веществ пропускается через
хроматографическую колонку—стеклянную трубку,
наполненную порошком белой глины, окиси алюминия, мела и т. п. (рис. 72).
При этом растворенные вещества адсорбируются в верхних
частях слоя сорбента. Затем таким же образом пропускают через
колонку чистый растворитель, который постепенно смывает ме-
305. Хроматографический анализ
591
нее прочно удерживаемые пигменты и переноси? их на более низко
лежащие слои адсорбента. После такого «проявления» пигменты
оказываются расположенными в виде отдельных цветных зон,
хорошо видных на белой колонке сорбента. Затем осторожно
выталкивают деревянным пестиком весь столбик адсорбента и
разрезают его на отдельные, различно
окрашенные части.
Обрабатывая растворителем (бензином или
бензолом с прибавлением спирта) каждую часть
колонки адсорбента, можно полностью извлечь
сорбированные пигменты (отдельно
друготдруга) и подвергнуть их дальнейшему
исследованию.
Метод М. С. Цвета широко используется
для разделения смесей веществ, которые
невозможно отделить другим путем. При
помощи этого метода выделены и изучены
разнообразные вещества, встречающиеся в весьма
малых количествах в растениях и организмах
животных: пигменты плодов и цветов,
витамины, пигменты крыльев бабочек (птерины),
алкалоиды и многие другие вещества.
Хроматографические методы анализа,
основанные на различной адсорбционной
способности индивидуальных химических веществ,
получили широкое распространение в
лабораторной и промышленной практике для быстрого
и точного определения состава
многокомпонентных газообразных и жидких смесей
соединений, близких по физическим и химическим
свойствам. Специально сконструированные для
этой цели приборы называются
хроматографами.
В настоящее время хроматографические
методы применяются для выделения и очистки
различных продуктов в промышленности.
В лабораторной практике широко применяется восходящая
хроматография на бумаге.
Из специальной хроматографической бумаги вырезают
полоску длиной 40—60 см. Каплю анализируемой смеси (концентрация
0,5—1 мг/мл) из капиллярной пипетки наносят на нижнюю часть
полоски бумаги. После высушивания ее подвешивают в
специальную камеру (рис. 73) так, чтобы конец ее погружался в налитый
на дно растворитель (например, для аминокислот применяют смесь
Рис. 72. Прибор
для проведения
хроматографа чес-
кого анализа:
/—хроматографи ческая
колонка с
окрашенными зонами сорбента;
2—сетка и ела Я ваты;
3—колба для
отсасывания.
59?
Глава XXIV. Пятичленные гетероциклы
бутилового спирта, воды и уксусной кислоты). Когда
растворитель поднимется почти до верха бумажной полоски, ее вынимают
и карандашом отмечают верхнюю
границу (фронт) растворителя. После
высушивания хроматограммы ее
проявляют, обрызгивая бумагу из
пульверизатора специально подобранным
индикатором. Появляются окрашенные
пятна, характерные для каждого
компонента анализируемой смеси (рис. 74).
Если анализируется смесь окрашенных
компонентов, то на хроматограмме сразу
появляются характерные цветные пятна
и проявлять ее не требуется.
Для идентификации компонентов
по хроматограмме используется то
обстоятельство, что индивидуальные
вещества имеют в соответствующих
системах растворителей определенную
величину коэффициента распределения
Rf. Для вычисления Rf измеряют: расстояние х от линии
нанесения вещества (старт) до центра пятна и расстояние у от той же
Рис. 73. Камера для хро-
матографирования на
бумаге.
а і г з ь 5 в 7 в а з ю п іг сліз та
Рис. 74. Хроматограмма Сахаров на бумаге.
линии старта до линии фронта растворителя (рис. 75). Значение
Rf вычисляется по формуле:
По таблицам Rf индивидуальных веществ находят, какому
веществу соответствует найденная величина Rf.
j. Хрояатографическай аяалиа
593
Так как величина Rt зависит от многих факторов
(растворителя, температуры и др.), часто проводят хроматографирование
в присутствии «свидетелей». Для этого на расстоянии 3—4 см
от капли исследуемого вещества, на той же линии старта,
наносят каплю или несколько капель раствороп чистых ве-
I
I
Рис, 75. Определение Ry при хрома- Рис. 76. Прибор для хроматографи-
тографировании на бумаге. рования в «тонком слое»:
/—пластинка для равномерного распределения
сорбента; 2—держатель; 3— хроматограмма,
помещенная для проявлення в закрытый сосуд.
шеств, предположительно входящих в состав анализируемой
смеси, и сравнивают расположение пятен на проявленной
хроматограмме.
Обычно хроматографирование на бумаге занимает 12—18 ч.
Более удобным и быстрым методом оказалось хромато-
графнрование в «тонком елее». В этом случае
порошкообразный сорбент, например специальным образом
приготовленную окись алюминия, равномерно распределяют на
пластинке (рис. 76) и наносят на линию старта каплю раствора
исследуемой смеси.
После распределения в слое сорбента компонентов смеси и
их разделения хроматограмму проявляют подходящим реактивом,
образующим с исследуемым веществом окрашенное соединение
(чаще всего используют для этой цели пары иода). Этим методом
удается провести хроматографирование за 15—20 мин.
38—788
594 Глава XXIV. Пятичленные гетероциклы
ГРУППА ИНДОЛА
306. Индол. Индол—твердое бесцветное вещество, темп,
плавл. 52,5 °С; темп. кип. 254 °С
СН
не7 с—сн р
I I! 1!
НС С СН а
ИНДОЛ
Индол входит в состав эфирных масел некоторых цветов
(жасмин); он образуется в заметных количествах при гниении
белковых веществ, а также при сухой перегонке каменного угля;
кроме того, он может быть выделен из соответствующих
фракций каменноугольной смолы. Очищенный индол применяется в
парфюмерии, так как сообщает смеси душистых веществ
характерный приятный запах, хотя сам он в чистом состоянии
имеет сильный фекальный запах.
В структурной формуле индола ядро пиррола
сконденсировано с бензольным, подобно тому, как в нафталине
сконденсированы два ядра бензола. Наличие бензольного ядра в
молекулах индольных соединений доказывается методами их
получения. Так, индол может быть получен (А. Е. Чичибабин) путем
пропускания паров анилина с ацетиленом через нагретую до
600—700 °С трубку:
H сн
Выход увеличивается, если реакционную смесь разбавить
углекислым газом. Гомологи индола обычно получают из фенилгид-
разонов альдегидов или кетонов при нагревании их с хлористым
цинком (Э. Фишер) или хлористой медью (А. Е. Арбузов). При
работе по этому методу из фенилгидразона ацетона образуется
а-метилиндол (метилкетол), а из фенилгидразона пропионового
альдегида—?-метилиндол (скатол):
СНа—СНЭ
I —
?-метилиндол
(скатол)
306. Индол 595
По-видимому, процесс идет через таутомерно образующийся
промежуточный нестойкий продукт, имеющий строение и свойства,
аналогичные гидразобензолу. Тогда реакция (для фенилгидразона
ацетона), как и в случае образования бензидина, идет по
следующим стадиям:
Х-СН3
tfH V\NHa H
Таким же образом можно представить схему реакции
превращения фенилгидразонов ацетофенона (образование а-фенилиндо-
ла) и пропионового альдегида (образование ?-метилиндола).
Реакционная способность гетероциклической половины индола
более ярко выражена, чем у бензольной части молекулы.
По химическим свойствам индол во многом напоминает
пиррол: он быстро темнеет на воздухе, под действием минеральных
кислот уплотняется, окрашивает древесину, смоченную соляной
кислотой, в красный цвет. Наличие бензольного ядра, как
обычно, повышает кислотные свойства—индол почти не обнаруживает
основных свойств. Наоборот, водород NH-группы замещается на
металл, например при действии металлического калия или даже
при сплавлении с едкими щелочами. Сходство индола с пирролом
обнаруживается также по его реакции с пиридинсульфотриокси-
дом. В этом случае при 100 °С образуется индолсульфоновая-2
кислота, тогда как при других реакциях обычно замещается атом
водорода, находящийся в ?-положении и имеющий свойства,
аналогичные свойствам а-водородного атома нафталина. Так,
например, в слабощелочном растворе иод реагирует с индолом,
давая 3-иодиндол. Магнийорганические производные индола
образуют с ангидридами кетоны, а с этилформиатом—альдегид. Ке-
тонная и альдегидная группы становятся в ?-положение.
Производные индола имеют большое биологическое значение;
некоторые из его производных используются в технике.
В 1952 г. из корня растения Rauvolfia serpentina был выделен
алкалоид резерпин.
В настоящее время установлено строение и осуществлен
полный синтез резерпина (Вудворд); полученный резерпин по
свойствам соответствует природному алкалоиду.
38*
596 Глава XXIV. Пятичленные гетероциклы
Резерпин—сложное соединение, скелет его молекулы состоит из
5 циклов (А, В, С, D, Е), содержащих 19 атомов углерода и два
атома азота (из которых один принадлежит индольному ядру):
Резерпин приобрел большое значение как гипоіенсивкое и се-
дативное (успокаивающее) лекарственное вещество, действующее
ка центральную нервную систему, им пользуются для лечения
гипертонии и некоторых психических заболеваний.
307. Триптофан. Среди аминокислот, получаемых при
гидролизе белковых тел, важное значение имеет а-амино-Р-(индолил-З)-
пропионовая кислота—триптофан (темп, плавл. 289 СС):
Так, в казеине молока содержится около 1,85%
L(—^триптофана. Животный организм не может сам синтезировать
триптофан, поэтому в составе пищи животных необходимы белки,
содержащие это вещество.
308. Гетероауксин. Гетероауксин, или а-индолилуксусиая
кислота (темп, плавл. 165 °С)
()пгСНа-С00Н
N
принадлежит к группе природных веществ, влияющих на скорость
роста растения и обусловливающих явление фототропизма
(стремление к свету). Эти вещества называются ауксинами; они играют
роль гормонов растений (фитогормоны).
Первые опыты, которые привели к открытию этих веществ,
были проделаны еще Дарвином; однако химической природы их
тогда не знали. Ауксины (см. также стр. 550) были обнаружены в
верхушках первых трубчатых листьев злаковых растений,
появляющихся при прорастании семян (колеоптилях). Сам гетероауксин
был сначала выделен Кеглем из мочи человека и лишь потом
найден в растениях.
309. Индиго 597
Ауксины содержатся в растениях в весьма незначительном
количестве. Так, для выделения 0,25 г гетероауксина из проростков
кукурузы надо было бы переработать примерно 2 миллиарда этих
проростков.
Для выделения гетероауксина в чистом виде был использован,
хроматографический метод (стр. 590).
Надо отметить, что название «ростовые вещества»,
применяющееся иногда для этих соединений, не особенно удачно и
может дать повод думать, что в этих веществах как бы заложено
свойство роста растений. Примерно такую «теорию» в прошлом^
веке развивал немецкий физиолог Ю. Сакс. Он полагал, что
причиной образования органов растений—стебля, корня,
цветов—являются особые «стебле-, корне- и т. п. образовательные
вещества». Ошибочность и вредность такой теории вскрыл К. А.
Тимирязев. Описанные выше фитогормоны являются лишь
веществами, которые могут оказывать тормозящее или
стимулирующее (возбуждающее) действие на процессы, естественно
протекающие в растительном организме. Более углубленное изучение
механизма действия фитогормонов—еще далеко не разрешенная
вадача, однако использование этих веществ в сельском
хозяйстве дает ^же теперь большие результаты.
309. Индиго. Индиго было известно еще в древней Греции.
Оно ввозилось в Грецию из Индии, где его добывали из
растений рода Indigofera. В Европе, и в частности в России, индиго
получали из растения вайда—Isatis Unctoria. Яркий и прочный
цвет тканей, окрашенных индиго, придавал этому красителю
большую ценность.
В изучении индиго принимали участие многие химики. В
сороковых годах прошлого столетия русский академик Ю. В. Фрии-
ше A808—1871) очистил индиго и получил его впервые в
кристаллическом состоянии. Сплавлением индиго со щелочью он
получил из него масло, названное им от арабского названия
индиго «alnil» анилином A840). Когда в 1842 г. Н. Н. Зинин
восстановлением нитробензола получил свой «бензидам», Фрицше
указал на идентичность последнего с открытым им ранее
анилином. Фрицше также была получена из индиго антраниловая
(о-аминобензойная) кислота. Позднее изучением индиго
занимался М. В. Ненцкий A847—1901), определивший плотность
пара индиго и отметивший, что индол при окислении частично
превращается в индиго.
Решающими для изучения индиго явились работы А. Байера,
завершившиеся в 1880 г. определением строения и синтезом этого
красителя.
S98 Глава XXIV. Пятчленные гетероциклы
Промышленные методы получения индиго были разработаны
несколько лет спустя К. Гейманом. Один из предложенных им
способов основывался на следующей схеме: нафталин (из
каменноугольной смолы) окислялся до фталевой кислоты, которая
переводилась во фталимид и далее по реакции Гофмана (см. стр. 367)
в антраниловую кислоту:
Обработкой хлоруксусной кислотой антраниловая кислота
превращалась в о-фенилглицинкарбоновую кислоту:
соон /ч/соон
Г I +С1-СН2СООН_Г Т сн2_сООН+НС1
антраниловая о-фенилгляцин-
кислота каобчнивая кислота
При сплавлении полученной кислоты со щелочью происходит
выделение воды и замыкание цикла:
Na0H
sj. /ч ХН L " уСН— COONa
NH XCOOH NH
2Н2О
соль индоксиловаЯ
кислоты
Образовавшаяся соль индоксиловой кислоты при пропускании
в ее щелочные растворы воздуха отщепляет COa с образованием
индоксила; последний окисляется в индиго, выпадающее в виде
синих хлопьев:
JS то А- „
со2
н-соон
синее индиго (транс-форыа)
309. Индиго 599
Связывающая две индоксильные группировки двойная связь
обусловливает возможность существования двух геометрических
изомеров индиго (цис- и транс-формы). Однако во всех реакциях
образуется только транс-форма:
СО NH
Синее индиго, или индиготин, представляет собой синий
порошок. Он плавится при температуре 390 °С и возгоняется, частично
разлагаясь. Индиго не растворяется в обычных растворителях,
но может растворяться при нагревании в анилине. При
окислении азотной кислотой индиго переходит в изатин—оранжево-
красные кристаллы (темп, плавл. 201 °С)
со
NH
При перегонке индиго с цинковой пылью Байер получил
индол, строение которого было им позднее доказано синтезом.
Так как индиго не растворяется в воде, то для нанесения на
ткань его обычно переводят в так называемое белое или лейкоин-
диго. Это достигается осторожным восстановлением индиго в
щелочной среде (при этом восстанавливаются карбонильные
группы, входящие в систему сопряженных связей):
С—ONa NH
MX)
С—ONa
синее индиго лейкоинднго
Для крашения ткань пропитывают бесцветным щелочным
раствором лейкоиндиго и затем, вынув из красильной ванны,
подвергают действию воздуха. Лейкоиндиго очень легко
окисляется кислородвм воздуха и вновь превращается в
нерастворимый синий пигмент—«кубовое» крашение (стр. 542).
Бромируя или хлорируя индиго, получают более прочные к.
трению и свету красители. При этом галоиды становятся в 5,5'
и 7,7'-положения.
Глава XXIV. Пятичленные гетероциклы
В 1909—1911 гг. Фридлендер исследовал и установил
строение античного пурпура. Этот краситель получали в древности
из средиземноморских моллюсков; приготовление его было очень
дорогим и сложным. Все исследование было проведено Фридлен-
дером с 1,4 г пурпура, который был извлечен из 12 000
моллюсков. Было доказано, что античный пурпур—это диброминдиго,
в котором атомы галоидов стоят в 6- и б'-положениях:
JH
античный пурпур
В сравнении с имеющимися в настоящее время красителями
окраски, достигаемые с помощью античного пурпура, являются
тусклыми и недостаточно прочными.
ГРУППА АЗОЛОВ
Азолами называются пятичленные гетероциклы, содержащие
два гетероатома, из которых по крайней мере один—азот.
Главнейшими из азолов являются:
НС4—-3N НС,—;N HCj—;N HC4—jCH
II II II II 11 il II' II
НС5 2CH HCS ^H НС6 2СН ¦ НС5 2N
6 s NH кн
оксазол тиазол нмилазол пиразол
Производные циклов этой группы многочисленны и
разнообразны. Мы рассмотрим только некоторые из них, имеющие значение
в медицине.
310. Сульфатиазол. Сульфатиазол (темп, плавл. 202
°С)—лечебный препарат, близкий по строению к белому стрептоциду
<стр. 512):
НС N
II II
НС С—NH—SO2—<f~~^— NH,
Он получается из ацилированного хлорангидоида сульфанило-
вой кислоты и 2-аминотиазола с последующим омылением
полученного соединения.
Sit. Пенициллин 60t
311. Пенициллин. В 1928 г. английский микробиолог
Флеминг наблюдал задержку роста культуры стафилококка
колонией плесени, случайно попавшей в лабораторный препарат.
Флеминг приписал это действие образуемому плесенью веществу,
которое было названо пенициллином. Пенициллин является одним
из наиболее важных антибиотиков, т. е. веществ,
вырабатываемых различными организмами и в малых дозах угнетающих
жизнедеятельность микробов*. Пенициллин вырабатывается
некоторыми видами плесени Penicilliutn. Во время второй мировой
войны группой английских биологов и химиков были разработаны
промышленные способы выделения пенициллина из продуктов
жизнедеятельности плесени.
Оказалось, что пенициллин способен угнетать деятельность
гнилостных бактерий даже при разведении одной его
части в 50 000 006 частях воды. В то же время даже более высокие
концентрации пенициллина не являются ядовитыми для человека.
Химический анализ показал, что пенициллин представляет
собой смесь близких по строению веществ. Общая его формула
включает гетероциклическую систему тиазолидина, т. е.
гидрированного тиазола:
НООС—СН—N —СО
СН3.
СН—СН—NH—COR
СН/ \ /
В разных пенициллинах радикал R различен:
Пенициллин F содержит радикал —СН2СН=СН—СН2СН3 пентенил
»
»
»
»
G
X
К
V
»
»
>
»
» —CH2C6Ha
» -СН2С6Н4ОН
» —CH2(CH2)aCH3
» —СН2ОС6Н5
бензил
оксибензил
к-гептил
феноксиметил
В 1957 г. в США Шиханом с сотрудниками был осуществлен
полный синтез феноксиметилпенициллина.
Как показывает структурная формула пенициллина, это
соединение должно быть химически мало устойчивым—в нем
имеется четырехчленный лактамньїй цикл. При действии кислот
пенициллин легко гидролизуется.
* Впервые антибиотическое действие плесени было установлено в 1870—
1871 гг. русскими врачами В. А. Манассеиным и Н. Г. Полотебневым, которые
в ряде работ показали возможность применения плесени для лечения гнойных
язв, фурункулеза и т. д.
Глава XXIV. Пятичленные гетероциклы
Одним из продуктов гидролиза пенициллина является а-ами-
HO-?-тиокислота (диметилцистеин), названная пеницилламином:
>с
3х |
—сн—соон
| |
SH NHj
пеницилламин
Как оказалось, эта кислота принадлежит к ряду
D-аминокислот, а не к ряду L-аминокислот, к которому принадлежат все
аминокислоты обычных природных белков:
Н-
СООН СООН
NH.j H
(CH3JCSH (CH3)aCSH
D-димегилцистеип L-димети лцистеин
и_1 пенициллина
В СССР проводятся большие работы по приготовлению пенициллина и
других антибиотиков. Много работ по изучению пенициллина и его
производству в СССР выполнено проф. 3. В. Ермольевой.
Весьма важным и широко применяемым для лечения ряда болезней
является антибиотик, выделяемый одним почвенным микробом. Молекула этого
антибиотика, открытого в 1942 г. Г. Ф. Гаузе и М. Г. Бражниковой и
названного грамицидином С (советский), представляет собой сложный циклический
декапептид, в состав которого входит по два остатка следующих пяти
аминокислот: пролий, валин, орнитии, лейцин, фенилалаикн. Четыре первые
аминокислоты имеют ^.-конфигурацию, а фенилаланин принадлежит к D-ряду. Полный
синтез этого антибиотика был осуществлен в 1956 р. в Швейцарии (Р. Швицер
и П Зибер).
312. Антипирин и пирамидон. Ряд лекарственных веществ
принадлежит к группе замещенных пиразолона:
н2с—
о=с
-СН
II
II
N
пиразол он-5
НС СН
* НО—С N
NH
Нагреванием фенил гидразина с ацетоуксусным эфиром
получается 1-фенил-З-метилпиразолон:
СН2—С—СН3
II
'H,;N НаС С—СН3
?:| „ I II
H;N-CaH6 °=C N
І9МІ N-CH,
1-фенил- 3- метили иразолон
313. Пилокарпин 603
Из его производных надо отметить антипирин, применяемый как
жаропонижающее средство. Антипирин получается при обработке
фенилметилпиразолона йодистым метилом или диметилсульфа-
том. При этом фенилметилпиразолон реагирует в таутомерпой
форме:
Н2С С—СН0
I II 7=
О=С N
N-
C6H6
Диметиламиноантипирин носит название пирамидона и
применяется как жаропонижающее и антиневралгическое средство.
Получают его следующим образом. Антипирин обрабатывают
азотистой кислотой и вводят в положение 4 нитрозогруппу, а
затем восстанавливают ее до аминогруппы. На полученный
продукт действуют йодистым метилом:
НС=С-СНЭ +нш, ON-C=C-CH
О=С N-СНз * О=С N-СНз
N-QH6 t
H.N-C—С-СН, +2СН.г,
I I *
0=C N—CH3 0=C N—CH3
пирамидон
С N
N-
Интересна история открытия лечебных свойств антипирина и его
производных. В 80-х годах прошлого столетия Л. Кнорр, изучая один из продуктов-
конденсаций ацетоуксусного эфира с фенилгидразином, ошибочно принял его
за производное гидрированного хинолина. Полагая, что это вещество имеет
свойства хинина, он решил испытать его в качестве лечебного препарата, а для
улучшения растворимости обработал его йодистым метилом. Прн испытании
было обнаружено жаропонижающее действие препарата, после чего ои был
введен в медицинскую практику под названием «антипирин». Это открытие
вызвало большой интерес к производным пиразолона как к медицинским
препаратам.
313. Пилокарпин. К группе имидазола принадлежит весьма
ценный алкалоид пилокарпин (темп, плавл. 34 °С), выделяемый.
604 Глава XXIV. Пятичленные гетероциклы
из некоторых видов африканских растений
СН—N
II II
Q,H6—СН—СН— СН2—С СН
О=С СН2 N—СН3
о
пилокарпин
Пилокарпин возбуждает деятельность слюнных, потовых и
пищеварительных желез. Особенно большое значение он имеет для
лечения глазных болезней, вызывая сужение зрачка и понижая
внутриглазное давление. Синтез пилокарпина был осуществлен
в 1936 г. Н. А. Преображенским.
ГЛАВА XXV
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
ГРУППА ПИРАНА
Из шестичленних гетероииклов с одним атомом кислорода
наиболее важными являются производные т-пирана A,4-пира-
на). Сам упирай до сих пор не получен. Его тетрагидропро-
изводное представляет собой внутренний эфир пентандиола-1,5,
из которого он может быть получен нагреванием с 60%-ной
серной кислотой
Y
сн2 сн2
/4\ /\
НС5 3СН ? Н2С СНа
II II II
НСР КИ а Н8С СНа
V V
Y-пиран тетрагндропирав
(неизвестен)
Напомним, что гексозы—как альдозы, так и кетозы (стр. 318)—
представляют собой циклические системы, чаще всего шести-
членные гетероциклы—«пиранозы»:
сн—он сн—он
НО—НС СН—ОН НО—НС СН—ОН
II І І /ОН
НО-НС ' СН-СНаОН Н2С С<
\ / \ / \:наон
о о
вльдогехсозы кетогексозы
Широко распространенными в природе являются производные
окисленного пирана—-рпиРона' Перед их описанием необходимо
остановиться на особых свойствах простейших производных
7-пирона.
606 Глава XXV. Шестичленные гетероциклы
3L. у-Пирон и соли пироксония. -рПирон (темп, плавл.
32,0 °С; темп. кип. 217 °С) был впервые получен декарбоксилиро-
ванием хелидоновой кислоты:
С=О С=О
/\ /\
НС СН -^со2 НС СН
I! II * II II
НЮОСі—С С— ІСООІН НС СН
v — v
хелидоновая у-пирон
кислота
Хелидоновая кислота (-рпирон-2,6-дикарбоновая кислота)
получила свое название потому, что была впервые выделена из
растения чистотел—Chelidonium majus. Она была синтезирована из
этилового эфира щавелевой кислоты и ацетона по следующей
схеме:
COOQjHg COOCjHg COOQ,H5
I : : I I
CO;OQjH6 H:CHa CO—CH2 C=CH
: 4 I -H2o / \
+ C=O » C=O > О CO
coiOQjHj "н;сна со—сна с=сн
la v-w—v_na
COOQH5 COOQjH5 COOCaHe
Первая стадия реакции проводится по типу конденсации Клай-
зена при действии этилата натрия; замыкание кольца
совершается при кипячении полученного продукта с соляной кислотой.
7-Пирон хорошо растворим в воде; раствор имеет
нейтральную реакцию. Свойства 7-пиРона и его производных довольно
своеобразны. Так, карбонильная группа не может быть
обнаружена при помощи обычных реактивов—гидроксиламина, фенил-
гидразина. Восстановление идет с трудом. Гидрирование 7-пи-
рона водородом в присутствии палладия приводит к тетрагидро-
пирону. 7'Пирон обладает некоторыми свойствами
ароматического характера: при обработке его бромом в присутствии
хлорного железа образуется желтый продукт присоединения—пер-
бромид, который при перегонке с паром превращается в 3-бром-
и 3,5-дибромпирон; кислородный мостик при этом не размыкается.
К щелочам кольцо 7-пиР°на более чувствительно и легко
расщепляется по кислородной связи. Так, при обработке
314. Пирон и соли пиооксония
607
2,6-диметилпирона щелочью образуется трикетон—гептантрион:
О
Н
С
С
СН
Н2О
сн3—с с—снэ
О
НС
СН
II II
снэ—с с—сна
он
он
О
II
С
СНз—С С—
о
о
Наиболее своеобразной является способность f-пиронов об-
разовывать с сильными кислотами соли по типу оксониевых (так
называемые соли пироксония). В этом случае соли оксония
получаются путем присоединения иона водорода к различным
кислородным соединениям (за счет свободной пары электронов
атома кислорода); реакция идет подобно образованию солей аммония.
Образование солей пироксония происходит следующим
образом:
С=О
/ \ +
НС СН Н
II II +_
нс сн а
V
С—ОН'
/ \
НС СН
I II
НС СН
\/
о^
В результате присоединения протона атом кислорода получает
положительный заряд, а структура пирона становится подобной
структуре бензола, чем, видимо, и может быть объяснена
устойчивость Y-ішрона к действию кислот.
Доказательством того, что превращение идет именно таким
образом, является присоединение к пирону и его производным
йодистых алкилов. Так, 2,6-диметилпирон с йодистым метилом
образует йодистый 4-метокси-2,6-диметилпироксоний, который
при действии аммиака превращается в хорошо известный 4-мето-
кси-2,6-диметил пиридин:
с=о
/\
НС СН
II II
+CH3J
с—осн3 -
Н(/ СН
I II
сн3—с с—снэ
о+
+2NHj
-NH^J; -H2O
С-ОСНз
НС
I II
э—С С—СНа
N
608
Глава XXV. Шестичленные гетероциклы
315. Хромон. Известно много природных
пигментов—производных бензопирона, или хромона (темп, плавл. 59 СС):
ал с=о
НС« С ^
I
НС
W
"СН
W
сн
хромон
Многочисленные желтые и коричневые красящие вещества
из коры и древесины многих растений, а также из цветов,
являются оксипроизводными 2-фенилхромона, или флавона (по-ла-
тыни flavus—желтый). Флавон содержится в пыльце,
покрывающей поверхность некоторых цветов и листьев. Хризин (диокси-
флавон)—желтый пигмент из почек тополей. Лутеолин (тетра-
оксифлавон) выделен из лепестков желтой резеды и некоторых
других цветов. Кверцетин (пентаоксифлавон)—пигмент
американского красильного дуба (Quercus unctoria). Кора этого дуба
служит для крашения шелка и шерсти. Кверцетин найден во
многих растениях, например, таких, как, хмель, чай, желтый левкой,
цветы мать-и-мачеха и др.
ОН О ОН О
I II ОН
хризин лутеолии
он о
он
кверцетин
Большинство флавонов—желтые твердые вещества с высокой
температурой плавления, растворимые в спирте, водных
растворах щелочей (фенольные свойства) и кислот (оксониевый
характер). Хлорное железо образует с ними характерные
окрашивания—темно-зеленое, красно-коричневое.
316. Антоцианы. Яркая окраска многих цветов и плодов
обусловлена наличием в них многочисленных пигментов, часто
весьма близких по строению и химическому характеру. Разделение
этих пигментов было осуществлено применением хроматографи-
ческого метода (стр. 590).
316 Антоцианы
609
Вильштеттер*, положивший начало исследованиям этих
веществ, назвал их антоцианами**. Интересно, что пестрая
окраска цветов создается довольно однотипно построенными
соединениями.
Антоцианы представляют собой глюкозиды, т. е. вещества, в
составе которых содержатся углеводы и соединения
неуглеводного строения—антоцианидины. Желтые пигменты, как
указывалось выше, являются производными группы 2-фенилхромона
(флавона). Пигменты красных и синих цветов относятся к группе
оксониевых солей 2-фенилхромена (не содержащего карбонильной
группы). Из них чаше всего встречаются в виде солей пеларго-
нидин, цианидин и дельфинидин:
—ОН
СІ"
ОН
НО'
он
он
с
о
-О-°н
СІ"
хлористый пеларгонидпн
хлористый цианидин
>Н
сі-
хлористый дельфииидин
Сходство и различие между этими антоцианидинами выявляются
при сплавлении их со щелочами. Пирановое кольцо при этом
разрушается. Левая часть молекулы, сходная у всех соединений,
превращается во флороглюцин A,3,5-триоксибензол). Правые
части молекул дают соответственно: пеларгонидин—гс-оксибензой-
ную, цианидин—протокатеховую, дельфинидин—галловую C,4,5-
триоксибензойную) кислоты. Строение антоцианов было
подтверждено их синтезом.
Антоцианы—окрашенные вещества. Их цвет зависит от ряда
факторов. Так, например, увеличение числа гидроксильных група
в молекуле изменяет окраску оксониевых солей от
оранжево-красной до фиолетово-красной (изменение окраски оснований менее
характерно). Соединение солей металлов с основаниями антоциа-
* Рихард Вильштеттер A872—1942 гг.)—выдающийся немецкий химик-
органик. Основные работы его связаны с исследованием строения хлорофилла,
изучением химии ферментов и процессов ассимиляции углекислоты
растениями. За научные заслуги Вильштеттер был избран иностранным членом
Академии наук СССР.
** «Антос»—греч. цветок.
39—788
610 Глава XXV. Шестичленные гетероциклы
нов дает синие оттенки. Большое значение имеет также
кислотность среды. Резкое изменение цвета в зависимости от pH среды
связано, по-видимому, с изменением структуры цианидина:
ОС6НпОб
сг
оксониевая соль цианидина основание цианидина
При хранении консервированных плодов в жестяной таре
часто происходит изменение окраски вследствие образования
соединений пигмента с солями олова.
Содержание пигмента в лепестках колеблется в очень
широких пределах. Так, содержание пигмента у василька, считая на
сухой вес, всего лишь 0,75%, у красного георгина—до 20%, у
темно-синих анютиных глазок—до 33%.
ГРУППА ПИРИДИНА
317. Пиридин. Пиридин, его гомологи и другие пиридиновые
основания были впервые открыты в масле, получающемся при
сухой перегонке костей. В настоящее время источником
получения пиридина является каменноугольная смола.
Пиридин—бесцветная жидкость с характерным неприятным
запахом (темп, плавл.—42 °С; темп. кип. 115 °С; df=0,982)
у
сн
НО ЭСН S
ні ін.
V
пиридин
Пиридин смешивается с водой во всех отношениях. Он является
очень хорошим растворителем многих органических соединений.
В нем хорошо растворяются также многие неорганические соли:
CuCl2, Cu2Cl2, ZnCl2, HgCl2, AgNO3. Раствор марганцовокислого
калия в пиридине часто применяют для окисления органических
веществ.
По некоторым свойствам пиридин напоминает бензол.
Подобно бензолу, он очень устойчив к действию кислот и
окислителей. Пиридин является третичным амином и обладает замет-
317. Пиридин 611
ными, но не очень сильными основными свойствами: он
образует соли с кислотами; дает многочисленные продукты
присоединения с ангидридами кислот и с различными солями и гало-
генидами.
Присоединение осуществляется за счет пары свободных
электронов у азота:
N
н|а сн, Г :o:s:o:
солянокислый нодмегилат пиридин комплекс
пиридин пиридина сульфо- пиридина с
триоксид фтористым
бором
Водородом в момент выделения (например, при действии
металлическим натрием в спиртовой среде), а также водородом в
присутствии платины пиридин восстанавливается до гексагидро-
пиридина—пиперидина:
СН
не хсн
11 1 +ЗНа
НС СН
V
пиридин
(
нас
*нас
сна
сна
<н
пиперидин
При действии на пиридин хлора или брома сначала
образуются непрочные продукты присоединения галоида к азоту.
Вступление галоидов в ядро, а именно в ?-положения, происходит
только при нагревании до 300—400 °С. Аналогично, только при
нагревании пиридина с концентрированной серной кислотой выше
300°С можно получить пиридинсульфоновую-3 кислоту. З-Нитро-
пиридин получается нитрованием пиридина смесью селитры и
серной кислоты при 300 °С.
Гораздо легче идут реакции с щелочными реагентами. При
нагревании пиридина с амидом натрия легко образуются а- и
1-аминопиридины (А. Е. Чичибабин):
( 1 + NaNHa -^l ( l_NHNa +№ ul_NH
N NN
а-амннопиридин
39«
612 Глава XXV. Шестичленные гетероциклы
а-Аминопириоин (темп, плавл. 57,5 °С) применяется для
получения многих производных пиридина. В частности, он применялся
для приготовления сыгравшего большую роль сульфамидного
препарата—сульфидина*:
j?\ -неї
CH:iCO—NH—!^~4-SO2C] + NH3—I ) *
N=/ N
хлорангидрид
ацетилсульфоииловой кислоты
^=5^ f\ +NaOH
» CH3CONH—( ^—SO3—NH-I ) *
¦ CH3COONa + NHa—<f ^—SOa—NH— I
"N
сульфидин
Кдкие щелочи при нагревании действуют на пиридин
аналогично амиду натрия. При этом происходит выделение водорода
и образование а- и 7-оксипиридинов. Эти же соединения могут
быть получены при действии на соответствующие аминопиридины
азотистой кислоты. Оксипиридины подобны фенолам: они дают
цветную реакцию с хлорным железом, растворяются в водных
щелочах, а- и 7-Оксипиридины—твердые вещества (темп, плавл.
а-оксипиридина 107 °С; темп, плавл. f-оксипиридина 148 °С);
они таутомерно превращаются в кетосоединения—пиридоны:
ОН О
„0-0=0.0 0 0
NN NN
і і
а-оксипиридин и-пиридон V-оксипириДі і у-пиридон
Карбоновые кислоты пиридина могут быть получены при
действии сильных окислителей на гомологи и другие
производные пиридина. Так, из 2-метилпиридина (а-пиколина)
образуется пиколиповая кислота. Отметим никотиновую (пиридин-
карбоновую-3) кислоту (темп, плавл. 234 °С), которая может
быть получена окислением никотина азотной кислотой или при
* В настоящее время вместо сульфидина используются менее токсичные
для человека сульфамидные препараты—производные аминотиазола, амннопири-
мидина, аминопиразола и др.
318. Никотин 613
нагревании хинолиновой кислоты (см. стр. 615). Никотиновая
кислота и ее амид являются одними из важнейших составных
частей смеси веществ, входящих в состав витаминов группы В.
Никотиновую кислоту и никотинамид обычно называют
витамином PP. Недостаток этого витамина вызывает тяжелое
заболевание—пеллагру.
318. Никотин. К производным пиридина относится очень
важный алкалоид никотин C10Hl4N2:
,1 I
—сн сна
сн3
Никотин (темп. кип. 247 °С) в виде лимоннокислой соли
содержится в количестве до 2—3% в листьях табака (Nicotiana tabacum) и
махорки. Впервые в Европу из Америки табак был привезен
португальскими моряками в 1550 г. и распространен французским
послом в Португалии Нико; от этой фамилии и произошло
название растения и алкалоида. Природный алкалоид вращает
плоскость поляризации влево. Первый синтез никотина был
осуществлен Пикте в 1904 г.
Никотин—один из сильнейших ядов, по силе
приближающийся к синильной кислоте. Интересно отметить, что левовращаю-
щий (природный) никотин в два с лишним раза более ядовит,
чем правовращающий изомер. Дешевым источником никотина
является махорка и табачная пыль, получающаяся как отход
на табачных фабриках. Водные экстракты, содержащие
никотин, применяются как сильнейшее инсектицидное средство.
319. Пиперидин. При восстановлении пиридина натрием в
спиртовом растворе (реакция Вышнеградского) получается гек-
сагидропиридйн, или пиперидин (темп, плавл. —9 °С; темп. кип.
106 °С). Он представляет собой сильное вторичное основание
СН.2
НаС СНа
Н2С CrLj
NH
пиперидин
Ядро пиперидина входит в состав многих алкалоидов (А. Н. Вьг-
шнеградский). Одним из простейших по составу является
алкалоид ядовитого растения болиголова Conium maculatum—ко-
614 Глава XXV. Шестичленные гетероциклы
ниин (темп. кип. 166 °С). Этот алкалоид по строению представляет
собой а-пропилпиперидин. В формуле кониина имеется один
асимметрический атом углерода. Природный алкалоид вращает
плоскость поляризации вправо.
В 1930 г. А. П. Орехов выделил и исследовал алкалоид,
содержащийся в широко распространенном в Средней Азии
сорном растении Anabasis aphytta, названный им анабазином
сна сна
II. /<*,!!
Н2С С—CHjCHjCH,, 11 1—НС СНа
Vх» у \t
кониин анабазин
Анабазин—жидкость, кипящая при 145 °С при давлении 15 мм
ptn. cm. По химическому составу он представляет собой a-(?-
пиридил)-пиперидин. Анабазин является изомером никотина:
вместо метилпирролидина, содержащегося в никотине, в его
молекулу входит пиперидин. Анабазин оказался очень действенным
средством для борьбы с насекомыми—вредителями в сельском
хозяйстве. Его применяют в виде свободного основания или,
чаще, в виде сернокислой соли—анабазинсульфата.
ГРУППА ХИНОЛИНА
320. Хинолин. Хинолин C„H,N является бензопиридином.
По строению и ряду свойств он подобен нафталину
СН СН
НС« С ЭСН
I II I
НС1 С 2СН
СН N
хинолин
Хинолин был получен Жераром в 1842 г. при перегонке с
едким натром смеси алкалоидов хинной коры. Строение хинолина
устанавливается как рядом синтезов, так и его превращениями.
Хинолин обычно получается по методу Скраупа—нагреванием
смеси анилина, глицерина и нитробензола в серной кислоте.
Процесс, no-видимом , идет по следующим стадиям. Под действием
320 Хинолин
615
серной кислоты глицерин теряет воду, превращаясь в акролеин:
СНдССН
Гон'н: іон"ніон
:: :і
сна=с=с/ —» сна=сн-с/
I NOHJ акролеин X)
глицерин
Акролеин с анилином дает продукт присоединения, который под
действием серной кислоты конденсируется в дигидрохинолин.
Последний окисляется нитробензолом в хинолин:
о=сн
СН,
- енолизация
ІНОІ—СН
анрвлеин
-н2о
дигидрохинвлвн гииолив
Хинолин и его гомологи содержатся в каменноугольной смоле.
Хинолин—высококипящая жидкость (темп, плавл. —19,5 °С;
темп. кип. 237,7 °С), мало растворимая в воде, имеет характерный
запах.
Химические свойства хинолина очень хорошо объясняются,
если рассматривать его как бензопиридин. Подобно пиридину,
хинолин является слабым третичным основанием: образует соли
с минеральными кислотами; йодистые алкилы дают с ним соли
четвертичных аммониевых оснований. При окислении, например,
марганцовокислым калием пиридиновая половина, как более
устойчивая, не изменяется, и образуется пиридиндикарбоно-
вая-2,3 кислота—хинолиновая кислота:
-Юі
соон
;оон
2СОа + НаО
хинолиновая
ьнслота
616 Глава XXV. Шестичленные гетероциклы
Как и пиридин, хинолин при действии амида натрия дает а- и
7-аминохинолины (реакция Чичибабина). При сплавлении
хинолина с едким кали образуется а-оксихинолин, который, подобно
оксипиридину, способен превращаться в кетоформу—карбо-
апирил.
Кислородные реагенты с большим трудом атакуют
пиридиновое кольцо, поэтому обычные реакции замещения направляются
в хинолине к бензольному ядру. При этом наблюдаются
определенные закономерности, подобные тем, которые известны для
нафталина: аналогично а-положениям у нафталина, наиболее ре-
акционноспособными положениями у хинолина являются 5 и 8.
Так, при нитровании хинолина в первую очередь получаются мо-
нонитрохинолины в виде смеси примерно равных количеств 5-
и 8-нитрохинолинов. Сульфирование хинолина впервые
осуществлено Н. Н. Любавиным A870). На холоду дымящая серная
кислота образует 8-сульфохинолин, который при нагревании
перегруппировывается в 6-сульфохинолин,—переход, подобный
превращению а-нафталинсульфоновой кислоты в -f-изомер.
Восстановление хинолина впервые было осуществлено
А. Н. Вышнеградским. При гидрировании в первую очередь
происходит вступление атома водорода в пиридиновую
половину и образуются дигидро- и тетрагидрохинолины.
Из производных хинолина надо отметить 8-оксихинолин
который может быть получен действием едкой щелочи на
соответствующее сульфопроизводное. 8-Оксихинолин называется
также оксином. Он оказался очень ценным аналитическим реакти-
Еом. При помощи оксина можно открывать и определять
количественно медь, цинк, кадмий и другие металлы в виде
малорастворимых осадков (см. хелатные соединения, стр. 217).
221. Хинин и антималярийные препараты. Хинин—очень
важный медицинский препарат, служащий для лечения малярии;
является представителем группы алкалоидов. Опираясь на свои
исследования хинина и других растительных оснований, А. Н. Вы-
шнеградский в 1878—1880 гг. высказал впервые мысль, что
многие алкалоиды растений принадлежат к производным
пиридина и хинолина. Это и было подтверждено впоследствии, когда
были изучены многочисленные алкалоиды.
321. Хинин и антималярийные препараты 617
Хинин, наряду со многими другими близкими ему по
структуре алкалоидами, находится в коре хинного дерева, лечебные
свойства которой стали известны в Европе еще в XVII веке.
Исследования строения и свойств хинина, проведенные
рядом ученых, имели большое практическое значение. Из смеси
алкалоидов, содержащихся в хинной коре, удалось выделить
хинин и установить его химическое строение.
В настоящее время формула хинина вполне выяснена и
подтверждена полным синтезом (ВуДворд):
СН
Н2С Лм СН—СН=СНа
I
СН С—СН(ОН)—НС 9На
сн3о—с с сн \1/
I II I N
НС С СН
СНа
/
Н
хшіпн (темп, плавл. 177 °С)
Как видно из формулы, в молекуле хинина содержатся два
третичных атома азота; хинин может образовывать соли с
минеральными кислотами. В его молекуле четыре асимметрических
атома углерода. Спиртовый раствор природного хинина способен
сильно вращать плоскость поляризации влево [а]о=—144 °.
Несмотря на то, что синтез хинина в лабораторных условиях
осуществлен, приготовление синтетического хинина в
промышленном масштабе представляется еще мало реальным.
При изучении многочисленных синтетических соединений,
близких по химическому строению к хинину, было открыто
большое количество препаратов, по лечебным свойствам подобных
хинину. Среди них следует отметить ряд производных хинолина,
например, плазмоцид, полученный почти одновременно в СССР,
в Германии и во Франции:
YV
NHCH2CH3CH3 N(Cs НбK
плазмоцид
618 Глава XXV Шестичленные гетероциклы
Для лечения малярии иіироко применяется акрихин (агебрин}:
ЧСН„)а—N
акрихин
Акрихин является производным не хинолина (бензопиридина),
а акридина (дибензопиридина); в его строении сохраняются
некоторые черты сходства с хинином и препаратами типа
плазмоцида. Нельзя, однако, установить какой-либо простой
зависимости между химическим строением и лечебными свойствами. В
качестве примера соединения, не имеющего какого-либо сходства по
строению с хинином, но применяемого в качестве
антималярийного препарата, может служить бигумаль (палудрин):
Cl—<f >—NH—С—NH—С—NH—CH-CHj
N=/ II II I
NH NH CH3
бигумаль
ГРУППА ПУРИНА
Основным веществом этой группы является пурин (темп,
плавл. 216 °С). В его молекуле кольцо пиримидина конденсировано
с кольцом имидазола:
СН СН
СН СН—NH N1 0С—NH
I II II >сн | || Г
НС СН СН—W НС2 <C-N^
N N
пиримидин нмндазол пурин
Пурин не был найден в качестве природного продукта, но
многие вещества, в молекуле которых содержится ядро пурина,
широко распространены в органическом мире и имеют большое
физиологическое значение.
322. Мочевая кислота. Формулу мочевой кислоты C5H4N4O8
можно вывести из формулы пурина путем замещения трех
атомов водорода гидроксильными группами. Мочевая кислота яв-
322. Мочевая кислота 619
ляется 2,6,8-триоксипурином, но для нее возможна и таутомер-
ная форма:
С—ОН С=О
Nf7 С—NH HN С—NH
I 1 >С-ОН^ 1 I >С=О
НО—С С— W О=С С—NH
\/ \/
N NH
мочевая кислота
При таутомерном превращении происходит перегруппировка:
—N=C—ОН (—* NH—С=О
I I
В молекуле мочевой кислоты два остатка мочевины связаны с
цепочкой из трех атомов углерода.
Мочевая кислота вместе с мочевиной является главным
продуктом азотистого обмена веществ в животном организме. Она
содержится в небольших количествах в моче человека. У птиц
и пресмыкающихся мочевая кислота является основной
составной частью экскрементов; в экскрементах удава ее количество
достигает 90%. При подагре мочевая кислота отлагается в
суставах; мочевые камни состоят главным образом из мочевой
кислоты. В технике мочевая кислота обычно добывается из гуано—
скопления экскрементов птиц на островах Южной Америки.
Мочевая кислота—бесцветный кристаллический порошок,*очень
мало растворимый в воде. Она обладает слабыми кислотным
свойствами; в ее молекуле два атома водорода способны замещаться
металлом. Если к мочевой кислоте прибавить азотную кислоту и
смесь упаривать, то получается желтовато-коричневый остаток,
который после прибавления небольшого количества аммиака
дает красивое пурпурное окрашивание. Эта «мурексидная»
реакция служит для качественного определения мочевой кислоты. Она
объясняется образованием мурексида C„HsNeOe—аммонийной соли
пурпуровой кислоты CSH6N6O6.
При окислении мочевой кислоты азотной кислотой получается
аллоксан и мочевина:
С=О С=О
HN С—І—NH HN C=O ^
j ^>С=О + Н2О 4- О * + С=О
О=С С—I—NH О=С С=О L.
\ / I \ / NH*
NH NH
мочевая кислота аллоксан
620
Глава XXV. Шестичленные гетероциклы
Строение аллоксана доказывается тем, что при обработке
щелочами он расщепляется на мочевину и мезоксалевую кислоту*:
NH—
і
o=c
1
NH—
-C=0
|
C=OH
1
—c=o
ноно—
—H
—H
NH3
1
, C=O
1
COOH
1
+ c=o
1
COOH
Образование аллоксана при окислении мочевой кислоты
указывает на то, что в молекуле мочевой кислоты находится кольцо
пиримидина.
При окислении мочевой кислоты марганцовокислым калием
образуется алланшоин (темп, плавл. 238 °С):
С=О
/ \
HN С— NH
О=С С
\/
NH
! >
,—NH
,С=О + Н3О + О
NH., О
I " И
О=С С—NH
Н—CH—NH
аллантоин
Строение аллантоина доказывается его получением из глиокси-
ловой кислоты и мочевины:
О
>=i
С ІОН + Н;—NH
NH—|н НО;-СН-;ОН + H1-NH
NH2 О
I II
O=C С—NH
I I ><
NH—CH—NH
ЗНаО
гидрат глиокси-
ловоЛ кислоты
Образование аллантоина при окислении мочевой кислоты
указывает на то, что в молекуле мочевой кислоты находится кольцо
имидазола.
323. Некоторые другие вещества группы пурина Гипоксан-
тин C5H4N4O и ксантин C5H4N4O2, подобно мочевой кислоте,
* Мезоксалевую кислоту можно получить, обрабатывая диброммалоновую
кислоту НООС—СВгг—СООН щелочью.
323. Некоторые другие вещества группы пурина 621
можно рассматривать как оксипроизводные пурина. Гипоксантин
является 6-оксипурином; ксантин представляет собой 2,6-ди-
оксипурин; для обоих соединений возможны и таутомерные
формы:
с—он с=о
l/ C-NH HN С—NH
I II >СН = I I
НС С—N^ НС С—N
V V
гипоксантин
с—он с=о
N С—NH HN С—NH
I II >СН -?=* | || >СН
НО—G С— N* О=С С—N«^
N NH
іяантаь
Ксантин содержится во многих животных тканях, в крови,
моче, печени, мочевых камнях. Ксантин—кристаллическое вещество,
плохо растворимое в воде; образует соли с кислотами и
основаниями. Гипоксантин обладает амфотерними свойствами; обычно
совместно с гипоксантином в животном организме содержится
ксантин.
Кофеин C„H10N4O2 (или теин) и теобромин CjH9N^Os
являются метильными производными ксантина:
С=О С=О
/ \ /СН3 / \ /СН8
СН3—N С—N< HN С—N<
І II >СН І II J>CH
o=c с—w o=c c—w
N—CH3 N—CH3
кофеин ІгеинІ гевбромин
Кофеин A,3,7-триметилксантин) находится в листьях и
бобах кофейного кустарника, в листьях чая. Он кристаллизуется
с одной молекулой воды, образуя гонкие иглы (темп, плавл.
622 Глава XXV. Шестичленные гетероциклы
237 °С). Кофеин обладает горьковатым вкусом, легко
растворяется в горячей воде; действует возбуждающе на нервную систему.
Теобромин C,7-диметилксантин) содержится в бобах какао
A heobroma cacao). Это кристаллическое вещество (темп, плавл.
351 °С), плохо растворимое в воде; применяется как мочегонное
средство.
Аденин C5HbN5, или 6-аминопурин, и гуанин C5H5ON5, т. е.
2-амино-6-оксипурин, принадлежат к числу наиболее
распространенных веществ ряда пурина:
С—NH, C=NH
-NH HN C—NH
N
v_i ш к;
/v;
>СН tzzi \ || \СН
у ИГ Г.—МГ
НС С—N^ НС
V V
аденин
с—он с=о
N С—NH HN С—NH
| | > ^ | ||
HaN—С С—N^ H3N—С С—IS
Л/ ^/
N N
гуанин
Аденин вместе с ксантином, гипоксантином и гуанином
образуется при гидролизе нуклеиновых кислот (см. стр. 626). Пу-
риновые основания этой группы в значительных количествах
содержатся во многих растениях—чае, свекловице, хмеле и т. д.,
в большом количестве находятся в дрожжах, встречаются
в тканях животных организмов, а также в моче и
гуано. Значительные количества гуанина находятся в рыбьей
чешуе и коже рыб, пресмыкающихся и амфибий, своеобразный
металлический блеск чешуи которых вызывается этим
веществом. Аденин—кристаллическое вещество; темп, плавл. около
360 °С; обладает довольно сильными основными свойствами.
Гуанин нерастворим в воде, с кислотами образует соли, которые
легко гидролизуются.
324. Триазин. Цианурхлорид. Селективные гербициды. Из гете-
роциклов с тремя атомами азота в кольце наибольшее значение
324 Триазин. Цианурхлорид. Селективные гербициды 623
имеет шестичленний гетероцикл—симметричный іриазин:
Н
С
N N
II I
н—с с—н
Важнейшее его производное—цианурхлорид—получается
полимеризацией хлорциана:
С1
С
3C1CN » || |
С1-С С—С1
\//
N
Процесс проводится в газовой фазе при температуре 350—
400 °С над катализатором—активированным углем, пропитанным
хлоридами металлов.
Чистый цианурхлорид—кристаллическое вещество белого
цвета с темп, плавл. 146—147 °С. ? воде растворим мало, хорошо
растворяется в большинстве органических растворителей. Атомы
хлора в молекуле цианурхлорида обладают большой
подвижностью и относительно легко могут быть замещены другими груп
пами. Замещаться может один, два и три атома хлора. Под
действием воды, особенно легко при кипячении, атомы хлора
заменяются на гидроксильные группы и образуется циануровая
кислота:
С1 ОН
N І
N \l NN
II І + ЗН2О » II І + ЗНСІ
Cl-C C-Cl HO—С С—ОН
\/ \/
N N
Аналогично протекает реакция со спиртами или фенолами,
причем образуются соответствующие эфиры.
624 Глава XXV. Шесгичленные гетероциклы
Многие производные цианурхлорида нашли важное
техническое применение в различных областях органической
химии.
При действии на цианурхлорид этиламина в щелочной среде
при небольшом нагревании происходит замещение двух атомов
хлора и образуется 2-хлор-4,6-бис-(этиламино)-симметричный
триазин:
Сі
/\
+ 2C,H5NH, + 2NaOH
Сі—С С—СІ
V
СІ
І II + 2NaCl + 2НаО
—NH-C С—NH—СН
І II
СаН5—NH-C С—NH—С2Н5
N
Это соединение под техническим названием симазин нашло
широкое применение в сельском хозяйстве в качестве селективного
гербицида для посевов кукурузы. Оно эффективно уничтожает
сорняки, не оказывая вредного действия на основную культуру.
325. Активные красители. Оптически отбеливающие вещества.
За последние 20 лет синтезированы особо прочные, так
называемые активные красители, образующие химическую (ковалентную)
связь с волокнами растительного происхождения (хлопок,
вискоза).
Для их получения сначала проводится реакция
цианурхлорида с каким-либо соединением, уже имеющим в своем составе
хромофорную, ауксохромную и кислотную (для придания
растворимости) группы. Обычно для этой цели берут красители, обладающие
красивой, яркой окраской, но не находящие самостоятельного
применения вследствие малой прочности. После взаимодействия
с цианурхлоридом образуется краситель, имеющий в своем
составе триазиновое кольцо с одним или двумя подвижными атомами
•32.5 Активные красители. Оптически отбеливающие вещества 625
хлора. Примером может служить активный краситель желтого
цвета:
ОН HH—cf ^N
\n=c<
=<с,
NaO3S SOsNa
Такой краситель может химически реагировать с гидр-
оксильными группами целлюлозы. В общем виде схему
образования окраски на целлюлозном волокне с помощью активных
красителей можно представить следующим образом:
I Краситель) [Кра
N м"^м
ІІ I + 2НС1
сААа
О N О
J 1_
9Н 9Н Целлюлоза
Целлюлоза
В дальнейшем было найдено, что кроме триазинового кольца
в качестве связующего звена между молекулой красителя и
целлюлозой могут быть применены и некоторые другие группировки
(например, оксиэтилсульфоновые и пиримидиновые).
Большим преимуществом активных красителей является их
высокая прочность, а также возможность получения очень ярких,
светлых окрасок.
Кроме того, цианурхлорид применяется для получения так
называемых оптически отбеливающих средств, иначе называемых
«белыми красителями-». Действие их основано на явлении
флуоресценции—поглощении части невидимых лучей солнечного
спектра (ультрафиолетовых) и трансформации их в видимые лучи с
большей длиной волны (синие или фиолетовые). Поскольку в
большинстве случаев обычные примеси в тканях, бумаге,
синтетических моющих веществах придают этим материалам
нежелательную желтоватую или желтовато-грязную окраску, добавление
флуоресцирующего синим цветом вещества выравнивает цвет
материала до чистого белого оттенка. В настоящее время известно
40—788
626 Глава XXV. Шестичленные гетероциклы
много оптических отбеливателей, принадлежащих к различным
классам органических соединений. Одним из наиболее
распространенных является производное цианурхлорида, имеющее
следующее строение:
N N
, „ ! Ц
NN \он <^н NN
С С
I I
ОН ОН
326. Нуклеиновые кислоты, нуклеотиды. РНК и ДНК. Как
было сказано выше (см. стр. 392), во всех живых клетках и
вирусах содержатся нуклеопротеиды—соединения, построенные из
белков и нуклеиновых кислот (НК)- Нуклеиновые кислоты, или
поли нуклеотиды, являются главными составными частями
клеточных ядер и представляют собой очень сложные
высокомолекулярные соединения. Как показали исследования химиков, биологов
и физиков, полинуклеотиды состоят из большого числа мононук-
леотидных остатков: молекулярный вес НК достигает нескольких
десятков миллионов.
В настоящее время известно, что нуклеиновые кислоты
принимают участие в осуществлении ряда важнейших
жизненных процессов.
Различают два типа НК: рибонуклеиновые кислоты (РНК)
и дезоксирибонуклеиновые кислоты (ДНК). Обе эти нуклеиновые
кислоты близки по химической природе и содержат углевод—
пентозу (см. стр. 330). В молекуле РНК содержится рибоза,
а в ДНК—дезоксирибоза, отличающаяся от рибозы тем, что
имеет меньшее число гидроксильных групп:
ОН ОН ОН
J kj?»
H0H2C
рибоза дезоксирибоза
ДНК составляет основу хромосом и непосредственно связана с
передачей наследственных признаков; РНК принимает участие в
синтезе белка. В клетках ДНК и РНК связаны с белками
непрочными связями, по всей вероятности солеобразного характера.
326 Нуклеиновые кислоты, нуклеотиды, РНК и ДНК
627
В результате гидролиза РНК и ДНК кислотами или
ферментами было установлено, что в них содержатся четыре
гетероциклических основания.
В РНК входят пиримидиновые основания—урацил (У) л
цитозин (Ц) и пуриновые основания—аденин (А) и гуанин (Г):
О NU
V)
N
I
урацил (У)
пиримидиновые
°ч NH
1 II
і
Y
NHj
ЦИТОЗИН (Ц)
основания
N
/ N—NH
N
YV
NH2
аденин (А)
пуриновые
rlulN N
\^ \_
N
ч/\
он
гуанин (Г)
основания
NH
/У
В ДНК урацил (У) отсутствует, а вместо него имеется гомолог
урацила—тимин (Т).
N II
Wh
ОН
тимин (Т)
пиримндиновое основание
В полимерной молекуле нуклеиновой кислоты мононуклеотиды
связаны между собой остатками фосфорной кислоты.
. Мономерное звено полимерной цепи РНК можно представить
так:
Основание
(У, Ц, А, Г)
Рибоза—О—
-р/
о-
)Н
Схема мономерного звена ДНК:
Основание
(Т, Ц, А, Г)
Дезоксирибоза—О—
-У
о-
р
он
40*
628
Глава XXV. Шестичленные гетероциклы
Строение отрезка цепи ДНКі
PO2H
5U- О^ 4N-^N^^H3
Рис. 77. Модель
молекулы ДНК.
Следует иметь в виду, что хотя цепи ДНК и РНК содержат
всего четыре основания, огромный молекулярный вес,
достигающий нескольких десятков миллионов, обусловливает
чрезвычайно большое число всевозможных комбинаций различных
индивидуальных нуклеиновых кислот, отличающихся набором и
чередованием оснований.
Рентгеноструктурными исследованиями установлено строение
РНК и ДНК, способ связи мономерных звеньев, их
последовательность, характер концевых групп, конфигурация и конформация
этих макромолекул. Так, оказалось, что ДНК представляет
собой сдвоенные полимерные цепи, расположенные параллельно
друг другу по виткам спирали (рис. 77).
326. Нуклеиновые кислоты, нуклеоіидьі. РНК и ДНК 629
Гетероциклические основания этих двух параллельных
витков ДНК направлены внутрь спирали и соединены попарно
межмолекулярными водородными связями. Установлено, что эти
водородные связи могут возникнуть только между парами вполне
определенных оснований, например А и Т, Г и Ц.
Конформация РНК резко отличается от конформации ДНК.
РНК представляет собой единичную, несдвоенную нить, причем
в зависимости от внешних условий она может иметь вид
перепутанного клубка, палочки или вытянутой нити.
В настоящее время создана правдоподобная гипотеза об
участии ДНК и РНК в синтезе белка и механизме передачи
наследственности.
Таким образом, нуклеиновые кислоты вместе с белками
являются носителями важнейших биологических функций и они
определяют все те признаки, которые отличают живую материю
от неживой, а именно: обмен веществ, способность реагировать на
изменения окружающей среды и, наконец, воспроизводимость.
ЗАКЛЮЧЕНИЕ
Заканчивая изложение курса, следует еше раз остановиться
на значении теории химического строения для изучения
органической химии.
Возникшая, как и всякая другая наука, из практических
потребностей производства, органическая химия свыше 150 лет
назад выделилась в самостоятельную отрасль химии—отрасль,
изучавшую вначале процессы превращения и свойства веществ,
вырабатываемых в живых организмах.
Первые удачные попытки научно подойти к химии природных
соединений углерода были сделаны на примере наиболее простых
по составу и легко доступных соединений. Объектами
исследования являлись жирные кислоты, спирты, углеводороды. Изучение
характерных особенностей этих веществ привело к синтезу
соединений, не встречающихся в природе. Среди них надо назвать
хлорангидриды кислот, галоидпроизводные углеводородов, ди-
азосоединения и многие другие вещества. Развитие
промышленности в первой половине XIX столетия и расширение области
применения всевозможных органических веществ природного
происхождения (красители, дубильные вещества и т. п.)
значительно способствовало усилению интереса к органической
химии и стимулировало проведение специальных исследований.
Накопление экспериментального материала в свою очередь
вызывало настоятельную необходимость в теоретических
обобщениях, позволяющих объяснить многообразие органических
веществ и различные явления, наблюдаемые при их
превращениях.
Возникновение во второй половине XIX столетия глубоко
материалистической и диалектической по своему существу
теории химического строения А. М. Бутлерова подняло
органическую химию на новую, высшую ступень. Эта теория не только
объяснила весь накопленный до ее возникновения опытный
материал, но и дала возможность предсказывать новые факты, новые
соединения, а также сознательно выбирать пути синтеза этих
соединений. Основные положения теории химического строения
надолго определили направление развития органической химии
и являются ее незыблемым фундаментом.
Заключение 63 t
Обогащенная в своем развитии новыми фактами и понятиями,
почерпнутыми из стереохимии, химической кинетики и других
разделов химической науки, теория химического строения
является основным методом познания структуры и свойств
сложных органических молекул и позволяет строго научно
подходить к сложнейшим проблемам органической химии.
Для химика-органика большое значение имеет знакомство с
методами, позволяющими индивидуализировать и определять
органические соединения. Еще более важным является для него
глубокое понимание структурной формулы соединения; он должен
уметь по структурной формуле составить себе представление о
физических и химических свойствах изображенного формулой
соединения. Так, например, наличие в молекуле карбоксильной или
аминогруппы свидетельствует о том, что вещество обладает
кислым или, соответственно, основным характером; большой вес
углеводородной части молекулы указывает на малую
растворимость вещества в воде и значительную растворимость его в
органических растворителях. Обратное заключение можно сделать
при большом числе гидроксильных или сульфо-групп. Из
рассмотрения структурной формулы часто становятся ясными такие
свойства соединения, как легкая окисляемость, способность
подвергаться гидролитическому расщеплению; наличие
характерных «хромофорных» групп (азогруппы, хиноидные системы и-
др.) показывает, что соединение обладает окраской.
Нельзя, однако, забывать, что, отражая действительное
строение молекулы, структурная формула, в обычном ее написании,
является только схематическим изображением молекулы и не
учитывает влияния ряда факторов, сказывающихся на свойствах
вещества. К факторам такого рода .следует отнести в первую
очередь пространственное расположение отдельных атомов в
молекуле, а также взаимовлияние отдельных частей молекулы.
Поэтому правильное «чтение» структурных формул должно
основываться не на заучивании отдельных общих положений курса, а
на глубоком знании фактического материала современной
органической химии. Такое знание дается не сразу и приобретается
не только запоминанием многих частностей, но в большей мере
пониманием общего строя, системы и методов органической химии.
Далеко не всегда ясное понимание и усвоение материала
приходит с первого чтения.
В последних строках своего знаменитого «Введения к полному
изучению органической химии» А. М. Бутлеров, обращаясь к
читателю, писал: «Учащийся будущий химик пусть позволит
заключить это сочинение советом: окончив чтение, приняться за
него еще раз сначала. То, что успел извлечь он, внимательно
прочитав эту книгу до конца, будет достаточно для того, чтобы*
€32 Заключение
многое при повторном чтении представилось ему с большей
отчетливостью и ясностью, чтобы ко многому он мог отнестись
теперь с более самостоятельной, критической точки зрения и,
не принимая на веру прочитанного, оценил, с одной стороны,
взаимные отношения различных факторов и соображений, с
другой—достоинства и недостатки этих последних».
ПРИЛОЖЕНИЕ I
НОМЕНКЛАТУРА ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ*
Для удобства пользования справочниками и указателями, для
свободного восприятия химической литературы необходимо хорошо понимать
названия химических соединений, т. е. уверенно владеть химической
номенклатурой. Изучение номенклатуры полезно также и тем, что помогает понять
и усвоить систему классификации химических соединений и структурные
взаимоотношения соединений разных классов.
Чтобы действительно служить общим языком химиков, номенклатур*
должна описывать химическое строение соединений и обеспечивать
однозначное соответствие названия со строением молекулы; такую
номенклатуру называют структурной. Современная структурная номенклатура — это,
в сущности, номенклатура не веществ, а формул строения.
Первой структурной номенклатурой была применяемая и поныне так.
называемая «старая рациональная номенклатура». Однако развитие
органического синтеза уже в последних десятилетиях прошлого века показало,
что, несмотря ка логичность и простоту праиил, старая рациональная
номенклатура непригодна для называния сложных органических соединений.
В J892 г. в Женеве специальный международный конгресс химиков
утвердил основные принципы и правила новой официальной международной
номенклатуры, известной под названием жецевскощ Строго логичная и
тесно связанная с научной систематикой химических соединений, женевская-
номенклатура была развита для веществ, в которых все атомы углерода
соединены непосредственно между собой, а не через гетероатом, и
преимущественно для соединений с открытой цепью атомов углерода. Женевской
номенклатурой не были предусмотрены названия некоторых типов сложных
полифункциональных, элементоорганических и разнообразных циклических
соединений. Поэтому крупнейшие справочники и реферативные журналы
вносили свои дополнения к официальной номенклатуре, иногда произвольные.
Ближе всего к женевской номенклатуре стоит номенклатура,
применяемая' в 4-м издании справочника «Beilstein's Handbuch der Organischen
Chemie», основанного русским академиком Ф. Ф, Бейльштейном,
Работа по унификации номенклатуры, предпринятая Международным
союзом чистой и прикладной химии**, привела _§.. 1.951О г. к созданию нового
свода правил, известных под названием льежскихі Льёжская номенклатура,
варианты которой нашли широкое применение в современной химической
литературе, в отличие от женевской допускала для каждого соединения
не одно, я несколько названий. Не удовлетворяясь льежской номенклатурой.
* Раздел, посвященный номенклатуре, написан совместно с А. М. Цукер-
маном.
** International Union of Pure and Applied Chemistry, сокращенно IUPAC,.
по-русски произносится ЮПАК.
634 Номенклатура органических соединений
крупнейшие справочники и периодические издания придерживались своих
вариантов номенклатуры.
Создавшееся положение потребовало нового пересмотра номенклатуры,
который был начат в 50-х годах и продолжается по сей день. В 1957 г. на
конференции в Париже приняты правила номенклатуры углеводородов и
циклических структур, известные под названием «Правила ЮПАК 1957 г.»
В 1962 г. ЮПАК опубликовал для обсуждения проект правил
номенклатуры соединений с неуглеродными заместителями, содержащими галогены, О,
N, S, Se, Те.
Широкое развитие специальных отделов химии (химии стероидов,
углеводов, аминокислот и пептидов, нуклеиновых кислот и др.) вызвало
появление «областных» номенклатур, приспособленных к определенным узким
областям химии. Это облегчает взаимопонимание специалистам, но
затрудняет понимание непосвященным.
Химия нераздельна, и у нее должен быть помимо областных наречий
также и обший официальный язык. Химики нуждаются в такой структурной
номенклатуре, которая была бы проста, однозначна, последовательна и
свободна от внутренних противоречий. Ни одна из действующих
номенклатур не отвечает полностью этим требованиям. Им должна отвечать подлинно
систематическая номенклатура.
В настоящее время в мировой химической литературе нет единства
номенклатуры. Применяют как старую рациональную, так и женевскую,
и льежскую, и новую официальную номенклатуру ЮПАК, которая еще
окончательно не доработана.
Ниже дается характеристика основных видов современных номенклатур.
Основы наиболее логичной — женевской номенклатуры излагаются
подробно, потому что все последующие системы используют те же
выразительные средства.
1. «РАЦИОНАЛЬНАЯ» НОМЕНКЛАТУРА
В основе «рационального» названия лежит название какого-либо
простейшего «ядра» молекулы:
СН4 СН2=СН, СН=СН
метан этилен ацетилен
СН3ОН СН3СНО —С— СН3СООН NHa
карбинол уксусный Д уксусная амин
альдегид О кислота
кетон
Названия образуют, прибавляя к названию «ядра» названия радикалов.
Примеры:
СНЯ-СН3 (СН3JСНВ (СНЭKСН
метнлметан диметилметан триметнлметан
сн3 снэ
I I сн^
СН9-СН-СН-СН3 >С=СН5 СН3-СН=СН-СН3
сн/
диметилизопропил- несимметричный симметричный
метай диметилэтилен циметилэтилен
Женевская номенклатура635
СН
СН3.
\сн-с=сн сн3сн2—сн2он с
^"з этилкарбинол
изопропилацетилен
СНз-C-CjHj
II
0
метилэтилкетон
C3H,-NH2
пропиламин
триметилуксусная
кислота
CH3-NH-C2HB
метилэтила мин
2. ЖЕНЕВСКАЯ НОМЕНКЛАТУРА
Ниже излагаются основы женевской номенклатуры так, как она
принята в справочнике Бейлыптейна. Достоинством ее является логичность и
тесная связь с научной систематикой органических соединений. Тем самым
в значительной мере уменьшается возможность употребления произвольных
названий.
Наиболее основательно эта номенклатура разработана для «цельных»
(Бутлеров) соединений, в которых все атомы углерода соединены
непосредственно между собой. В таких соединениях имеются только чистые
(незамещенные) функциональные группы (ОН, NH2...).
Построение названия для таких соединений может быть представлен»
единой простой схемой (см. стр. 645). Наименование органического
соединения построено как сложное слово, слагающееся из корня (главная цепь)
и префиксов и суффиксов (функциональные и другие группы). Каждый
элемент названия имеет определенное место в сложном слове. Схема
показывает, как название соединения зависит от научной систематики
органических соединений Нам неоднократно приходилось убеждаться на практике
в том, что, поняв эту схему и принцип ее использования, учащиеся легко
запоминают ее и с интересом выполняют упражнения по номенклатуре.
Общие принципы
Название соединения по женевской номенклатуре определяется:
1. Строением основного скелета соединения.
2. Характером и числом замещающих групп с учетом их «старшинства».
3. Принципом наибольшей простоты при прочих равных условиях.
По строению основного скелета органические соединения делятся
следующим образом:
а) ациклические системы;
б) карбо-, или изоциклические системы;
в) гетероциклические системы.
Название веществ по женевской номенклатуре составляется по принципу
обычного грамматического построения слов. Его «корнем» является
название самой длинной непрерывной цепи углеродных атомов, соответствующей
какому-либо углеводороду. Формально путем замены атомов водорода в
таком углеводороде на функциональные группы и другие заместители в
строго определенном порядке можно прийти к полному названию соединения.
Для каждого функционального и нефункционального заместителя имеются
твердо фиксированные названия и места — или перед названием главной
цепи, или после него.
€36 Номенклатура органических соединений
Названия основных соединений (углеводородов, спиртов, первичных
аминов и др.) состоят обычно из одного слова; производные основных
соединений (эфиры, хлорангидриды, амиды и т. п.) обозначают несколькими
словами. Степень сложности названия определяется сложностью структуры
называемого соединения.
Для построения названия соединения по женевской номенклатуре
прежде всего следует установить, какая цепь углеродных атомов является в
соединении самой длинной.
Углеводородные радикалы являются старшими при определении
нумерации главной цепи. При равном положении двух радикалов старшим
является радикал с меньшим числом углеродных атомов. Название радикала
строится из названия углеводорода с соответствующим числом атомов
углерода, но с заменой суффикса ан на ил.
Если радикал содержит кратные связи, т. е соответствует
непредельному углеводороду, название которого оканчивается на ен или на ин, то в
этих случаях суффикс не отбрасывают, а добавляют второй суффикс: ил
{енил, инил и т. п.).
Названия радикалов ставят перед названием главной цепи. Перед
названием радикала ставится отделенная дефисом цифра, обозначающая, к
какому атому углерода главной цепи присоединен этот радикал.
Кратные связи обозначаются суффиксами ен (для двойной связи) или
ин (для тройной связи), которые ставятся вместо суффикса ан,
соответствующего насыщенному углеводороду.
Кратные связи занимают второе место по старшинству, т. е. определяют
начало нумерации в том случае, если его не определяют разветвления цепи.
Тройная связь старше двойной.
Функциональные группы*, содержащие кислород или серу, являются
следующими, третьими по старшинству. Обозначения, соответствующие этим
группам, ставятся в конце слова.
Если в соединении можно выбрать две или более длинные цепи с
равным числом атомов углерода, то выбирают самую сложную, т. е. цепь,
включающую наибольшее число разветвлений или заместителей.
Выбрав главную цепь, определяют начало нумерации, руководствуясь
наличием и старшинством заместителей главной цепи.
Заместители делятся на углеводородные радикалы, функциональные и
нефункциональные группы. Кроме того, на правах заместителей
учитываются и обозначаются в названии кратные (двойные, тройные)
углерод-углеродные связи.
Азотсодержащие функциональные группы занимают следующее (более
низкое) место по старшинству. Обозначения этих групп ставятся в
названии "соединения перед названием углеводородных радикалов, т. е. в форме
префиксов (приставок).
Нефункциональные группы занимают последнее место по старшинству,
т. е. определяют начало нумерации только при отсутствии или при
симметричном расположении всех предыдущих заместителей. Обозначение
нефункциональных групп ставится в самом начале названия соединения.
С порядком применения основных принципов женевской номенклатуры
мы лучше всего познакомимся на практике и прежде всего изучая
номенклатуру простейших органических веществ — углеводородов с открытой цепью
* Функциональными группами называются такие группы атомов,
которые содержат группировку [С]—А—Н, где А — атом или группа атомов
кислорода, серы, азота (но не углерода). К функциональным относят и
карбонильную группу.
Женевская номенклатура
637
Функциональные группы, содержащие кислород
(по возрастающему старшинству)
Формула
—ОН
-SH
^>[С]=О
^>[C]=S
-|С|<°
Название
ОЛ-
тиол-
он-
тион-
ал- (аль)
тиал- (тиаль)
Формула
нс<°
NOH
-(С]/
NOH
-[с/
-SO2H
-SO3H
или серу
Название
-овая
кислота
-тионовая
кислота
-тиолтионовая
кислота
-сульфиновая
кислота
-сульфоновая
кислота
Азотсодержащие функциональные группы
(по возрастающему старшинству)
Формула
-NH2
—NHOH
-NH-NH2
и т. д.
Название
-амино
-гидроксиламино
-гидразино
Нефункциональные группы
(по возрастающему старшинству)
Формула
-F
-CI
-Вг
—J
Название
-фтор
-хлор
-бром
-иод
Формула
—NO
-NO2
-N,
Название
-нитрозо
-нитро
-азидо
638
Номенклатура органических соединений
углеродных атомов. Далее мы изложим схему и способ составления
названий ациклических соединений с различными заместителями.
В номенклатуре карбоциклически.ч и гетероциклических соединений
имеется ряд дополнительных правил, но в основном она следует принципам
и правилам, установленным для ациклических систем
Общая последовательность расположения элементов названий сложного
соединения дана на схеме, представленной на стр. 645.
Ациклические соединения
Предельные углеводороды нормального строения (алканы, парафины)
Первый раздел схемы
1. Название предельных углеводородов характеризуется суффиксом а«.
Первые четыре углеводорода носят исторические названия; начиная с
пятого, в основу названия углеводорода берется греческое числительное,
соответствующее числу углеродных атомов.
2. Углеводороды с нормальной цепью углеродных атомов имеют
следующие названия:
Формула
сн4
сн3сн,
СНЭСН2СН3
CH,(CHSJCH8
СН,(СН2)ЭСНЭ
СН^СН^СНэ
CHd(CH2NCH3
CH3(CHs)eCH3
СЩСН^Нэ
СН,(СН2)вСН3
СНЭ(СН2),,СН3
СНЭ(СН2IAСН3
сьусн^сн,,
Название
Метан
Этан
Пропан
Бутан
Пен тан
Гексан
Гептан
Октан
Нонан
Декан
Ундекан
Додекан
Тридекан
Формула
СНЭ(СН2I2СНЭ
СНЭ(СН2IЭСН3
СНэ(СНаI4СН3
СНз(СН2I6СН,
СНэ(СН2IвСН3
СЩСНаЬСН,,
СН3(СН2IвСН3
СНэ(СНаI9СН3
СН3(СН2J(,СН8
СНэ(СН2JвСНэ
СНэ(СН2)э8СНа
СНЭ(СН2L8СНЭ
СН3(СН2MвСН3
Название
Тетрадекан
Пентадекан
Гексадекан
Гептадекан
Октадекан
Нона декан
Эйкосан (эйкозан)
Генэйкосан (генэй-
коаан)
Докосан (докозан)
Триаконтан
Тетраконтан
Пентаконтан
Гексаконтан и т. д.
Предельные углеводороды с разветвленными цепями (изоалканы)
Второй раздел схемы
Названия изоалканов строятся следующим образом:
1. Указывают название главной цепи.
Женевская номенклатура
639
11 Перед названием главной цепи пишут название алкила.
III. В конце названия ставят суффикс ан.
алкилы
главная цепь
ш
суффикс ан
3. За основу названия данного соединения (корень слова) принимают
название углеводорода, соответствующее числу углеродных атомов главной
цепи Главной цепью углеродных атомов считают:
а) самую длинную;
б) самую сложную.
Если в углеводороде можно выделить две или несколько одинаково
длинных цепей, то за главную выбирают ту из них, которая имеет
наибольшее число разветвлений.
4. После того как установлена главная цепь, необходимо
перенумеровать углеродные атомы. Нумерация начинается с того края цепи, к
которому ближе примыкает любой из алкнлов. Если разные алкилы находятся
в одинаковом положении по отношению к краям цепи, то нумерацию
начинают от того края цепи, к которому ближе радикал с меньшим числом
углеродных атомов (метил, этил, пропил и т. д).
Примеры:
I 2 3 4 о б 7 8
() СН3—СНа—СН—СН2—СН—СН2—СН2—СН3
[ [
СН2СН3 СН3
2) СН3—СН,.—СН—СНа—СНа—СН—СНа—СН3
| [
|
СН3-СН2
[
СН3
начало нумерации определяет
ближайший к краю радикал—
этил
начало нумерации определяет
младший радикал—метил
Если же одинаковые радикалы, определяющие начало нумерации,
находятся на равном расстоянии от обоих концов цепи, но с одной стороны их
имеется большее количество, чем с другой, то нумерация начинается с того
края, где число разветвлений больше.
Примеры:
СН,
і г' з 4б
3) СН3-С-СН2-СН-СН3
j СН3
4) СН3—СН-СНг-СНг-СН-СН-СНз
СНЯ СН3 СН3
5. После того как определена главная цепь и углеродные атомы ее
перенумерованы, строится и само название. Первыми указывают наиболее
простые заместители: метил или в отсутствие его этил и т. д. Перед названием
радикала ставят цифру, соответствующую номеру углеродного атома
главной цепи, при котором находится данный радикал. Цифра от названия ра«
640
Номенклатура органических соединений
дикала отделяется дефисом. Вслед за метилом называют этил и т. д. —
в порядке увеличения числа углеродных атомов в радикале, после чего
называют углеводород, соответствующий главной цепи углеродных атомов.
Если углеводород содержит несколько одинаковых радикалов, то их
число пишут прописью перед названием радикалов; место присоединения
радикалов указывают цифрами. Цифры разделяют запятыми, располагают
по возрастающей величине и ставят перед названием данных радикалов.
Так, приведенные в этом разделе углеводороды носят следующие
названия:
1) 5-метил-З-зтилоктан;
2) З-метил-6-этилоктан;
3) 2,2,4-триметилпентан;
4) 2,3,6-триметилгептан.
Непредельные углеводороды (алкены, алкины)
Третий раздел схемы
6. Названия непредельных углеводородов строятся по следующей схеме
ал килы
кратные связи
7. Нумерация определяется алкилами главной цепи, как указано для
предельных углеводородов. При отсутствии алкилоЕ счет определяется той
из кратных связей, которая ближе к краю цепи. При одинаковых же
положениях от краев цепи двойной и тройной связей старшей считается тройная
связь, и поэтому она определяет начало нумерации.
8. Названия непредельных углеводородов образуются из названий
предельных углеводородов с тем же числом углеродных атомов путем замены
суффикса ан (у алканов) суффиксами ен (для алкенов) и ин (для алкинов).
Место расположения каждой из кратных связей в главной цепи
обозначается цифрами, которые ставят после суффикса и отделяют от него
дефисом. Цифра соответствует номеру одного из углеродных атомов,
связанного с другим атомом углерода данной кратной связью, причем обозначают
тот атом углерода при кратной связи, который имеет наименьший номер.
9. Название углеводорода, содержащего несколько кратных связей,
имеет сложное окончание, в котором прописью указывают число кратных
связей и их характер; последовательность слогов названия — сначала ен,
потом ин — сохраняется независимо от нумерации.
Примеры:
1) СН2=СН—СН—СН=СН2 начало нумерации
| безразлично
СН3
З-метилпентадиен-1,4
» Ь і 3 2 1
2) СН=С—СН2—СН2—СН—СН3 начало нумерации определяет
1 боковая цепь
^•метилгексин-6
сн3
Женевская номенклатура Є41
СНЬ двойная и тройная связи на
і і |з 4 а равных расстояниях от краев
3) СН=С—С—СН=СН2 цепи;
| начало нумерации определяет
СНа тройная связь
З.З-диметилпентен-4-ин-і
Соединения, содержащие кислородные функциональные группы
(спирты, альдегиды, кетоны, кислоты) и их сернистые аналоги
Четвертый раздел схемы
10. Выбор главной цепи определяется, как было указано ранее Начало
нумерации определяется прежде всего алкилами, затем кратными связями
При отсутствии их или при их симметричном расположении начало
нумерации определяют кислородные функции, при этом преимущество имеет группа:
а) расположенная ближе к одному из краев цепи;
б) старшая в данном разделе.
Старшинство групп определяется возрастающей степенью окисления, при
чем функциональная группа, содержащая серу, старше, чем соответствующая
кислородсодержащая группа.
11. Названия этих групп обозначают окончаниями, которые ставят за
названием главной цепи после обозначения кратных связей Углерод
функциональной группы учитывается в главной цепи. Его номер ставится после
названия соответствующей функции. В случае сернистых аналогов ставится
номер углерода, непосредственно связанного с атомом серы.
Обозначения функциональных групп, содержащих кислород или серу,
и их расположение по старшинству даны на стр. 637.
Примеры:
1) СН,—СН2-с/ 2) СНз-СН,—С-СНз-СН-,
ХН ||
О
пропаналь пентанои-Э
3) СН3—CH-CHj-COOH
сн3
2-меі'Илбутановая-І
12. Если соединение содержит несколько функциональных групп, то их
обозначения располагаются в порядке возрастающего старшинства.
Примеры:
1) СН3—СН—СН=СН—С^ начало нумерации от ближай-
Ч)Н шего к краю радикала—
меіила
CHS
2-метилпентен-і-овіЯ-5
кислота
41—788
642 Номенклатура органических соединений
2) СН3—СН—СН2—С—СН— С</ начало нумерации определяет-
I || | ХОСН3 ся группой СООСНа
СН3 О СН3
метиловый эфир
2.5-днметилгексанон-З-овой-І
кислоты
5 4 a si
3) CHj,—СН— СНа—С—CHj начало нумерации определяется
|| старшинством кетонной группы
ЭН О
пентанол-4-он-2
4j*
А
13 Если кислородные функции входят а боковые цепи (алкилы), то их
названия входят в названия этих же алкильных групп.
Примеры:
/°
—СН2ОН метилол —Of метиловая кислота
—СН2—CHjOH этилол —СНг—С^ этилал (этилаль)
14. При равном положении алкилов, определяющих нумерацию,
преимущество имеют алкилы, включающие кислородные функции (усложненные
алкилы).
Примеры:
НООС—СН—СН2—CHj—СН—СООН начало нумерации определяется
| | усложненной метильной
СН8 СН2ОН группой
б-метил-2-метилолгексаидиовая-1,6
кислота
Азотсодержащие функциональные группы
Пятый раздел схемы
15. Главнейшие представители этого раздела в порядке возрастающего
старшинства при определении нумерации указаны в таблице на стр. 637.
Начало нумерации главной цепи эти заместители определяют при отсутствии
представителен второго, третьего и четвертого разделов схемы.
Названия этих чаместителей помещают перед названиями радикалоз.
Первой называют аминогруппу, затем гидроксиламиио и, наконец, гидра-
зиногруппу. Впереди помещают отделенную дефисом цифру, обозначающую
номер углеродного атома главной цепи, при котором находится этот
заместитель.
Примеры:
NH2
1) СН3—СН—CHj—СН—СН3 2) СН3—С—СООН
NH2 NH-NH, СН3
4-амиио-2-гидразинопентан 2-амнно-2-метнл-
аропаиовая-1 кислота
Женевская номенклатура 643
3) NK,—СН,—CHa—СН.,—SO3H
1-эминопропа ісульфоновая-І
КИСЛОТУ
16. Если аминогруппа находится в боковой цени, то название ее
помещают перед названиями радикалов, с обозначением соответствующего
номера углеродного атома боковой цепи, прн котором стоит аминогруппа.
Примеры:
СН2ОН
і г а її 5 j скелет несимметричный,
1) СНа—СН—СН—СН—СН2—СООН начало нумерации определяет
| | метильная группа
СНЬ СН2—NHa
3'-амино-^,3-диметил-4-метилол-
гексавовая-6 кислота
2) CHS—СН—СН—СНг-СН,
I Jv
NH2 CHa—NH,
2,3'-дитмино-3-мети лпеятан
Нефункциональные заместители
Шестой раздел схемы
17. В таблице на стр. 637 указано семь нефункциональных
заместителей, они ке содержат водородных атомов и распределяются по
возрастающем) старшинству.
По влиянию на начало нумерации нефункциональные заместители іани'
маки последнее место, т е. они определяют начало нумерации только в том
случае, если оно еше не определено последовательно заместителями второго,
третьего, четвертого и т. д разделов.
18. Если все же начало нумерации определяется нефункциональными
заместителями, то преимущество имеет тот из них, который стоит ближе
к краю главной цепи. При равных положениях преимущество имеет
старший заместитель.
Примеры:
I) CH2=CH—CHjBr начало нумерации определяет
t-бромпропен-1 кратная связи
2) CH-jCl—CHa—CHJ—СН, начало нумерации определяет заме-
і-хл®р-з-иодбуіан ститель, стоящий ближе к краю
3) CH2J— CHj—СН—СН,— СНаО начало нумерации определяет
і„ старший заместитель
41*
641
Номенклатура органических соединений
19. Названия заместителей шестого раздела помещают перед
названиями всех других заместителем. Цифра, отделенная дефисом, указывает на
номер атома углерода главной или боковой цепи, при котором находится
данный заместитель. Порядок расположения названий заместителей шестого
раздела схемы — обратный по отношению к их старшинству при определении
нумерации.
Выше мы рассмотрели основные правила наименования органических
ациклических соединений. Выбор главной цепи определяется количеством
атомов углерода и сложностью цепи, а порядок нумерации главной цепи —
наличием заместителей.
Влияние заместителей на определение порядка нумерации зависит от
старшинства разделов в порядке ТТ—III—IV—V—VI и от старшинства групп
в разделах. На стр. 645 приведена схема построения названия сложного
органического соединения ациклического ряда.
Карбоциклические соединения
Основные положения и правила официальной номенклатуры,
изложенные в разделе об ациклических соединениях., сохраняются и в раздела
о карбоциклических соединениях. Отметим лишь ряд особенностей;
20. Название моноцнклического предельного углеводорода дается по
названию соответствующего (по числу углеродных атомов) нормального
углеводорода с присоединением приставки цикло.
21. Карбоксильная группа, связанная с циклом, обозначается
окончанием «карбоновая кислота». Если боковая цепь длинная и сложная, то
вещество называют как ациклическое, обозначая наличие цикла, как и любого
другого заместителя, соответствующими префиксами (например, «циклолен-
тил», «циклогексил» и т. п.).
Пример ы:
СН—СО СН2—СН2
СН—СООН 2) Н2с/ \СН-СН-СН2-СООН
Ґ*\А Ґ*\Л /~Ч-Ї ҐЧ-J I
ьп—lxi2 ы-12—^п2 СООН
цик лопентен-З-он-2- 2-циклогекснлбутандиовая-1,4
карбоновая-1 кислота кислота
22. Ароматические соединения принято называть как производные
соответствующих углеводородов.
Примеры:
< -бром-12-ннтро-1 -эте-
ннлбеизол
SO3H
4-амннонафталинсуль-
фоновая-I кислота
23. Если гидроксильная группа связана непосредственно с
ароматическим ядром, то ее обозначают приставкой окси и ставят эту приставку
перед названием радикалов (т. е. переносят в V раздел схемы).
Схема построения названия сложного органического соединения ациклического ряда
СН3
метил
NH2
амино
CI
F
фтор хлор бром
Вг
J
иод
NO
нитрозо
NO2
нитро
VI
NHOH
гидро-
ксил-
амино
NHNH2
гидра-
зино
V
с2н5
этил
II
1 I I
-с-с-с-
I I I
c=cc=
-ОН
ол
III
=О
он
О
он
овая
кислота
(СҐ
SO3H
IV
Несруннциональные заместители
Азотсодержащие
функциональна /е
группы
Углеводо-
родные
радикалы
Главная
цепь
Нратные
связи
Функциональные группы, содержащие
кислород, и их сернистые аналоги
Префиксы
Называются перед главной цепью. Цифра, обозначающая
место, ставится перед обозначением заместителя.
Примечание. Стрелки укавывают направление возрастания старшинства.
Корень Суффиксы
Обозначаются с соответствующими
окончаниями после главной цепи. Цифра, показывающая
место, ставится после данного окончания.
Номенклатура органических соединении
Примеры:
4-амино-і-окскбензол 4-о сиЛензолкарбоновая-і
шслота
?4. Если боковая цепь жирноароматического соединения длинная и
разветвленная, то за основу названия берут боковую цепь, рассматривая
ароматическое ядро как заместитель,
Примеры:
I) /"^-СН-СН-СН,
СН ОН NH2
4-амнио- '-Фенилбутанол-і І,а-дифенилпропанон-і
3. НОМЕНКЛАТУРА МЕЖДУНАРОДНОГО СОЮЗА ЧИСТОЙ
И ПРИКЛАДНОЙ ХИМИИ (ЮПАК)*
В правилах ЮПАК I93U—1949 гг. подробно разработана номенклатура
элементоорганнчески.ч и смешанных цикло-алнфатических соединений; в
номенклатуре 1957 г. — номенклатура всех видов циклических соединений.
Номенклатура ЮПАК использует те же выразительные средства и
приемы построения названий, «то и женевская номенклатура; различие іа-
ключаегся лишь в способе их применения
1. Льежские правила 1930 г, допускаю-і для каждого соединения не
одно, а несколько названий, среди которых рекомендуется выбрать
«наиболее простое» или «удобное» название. Так как критерии «простоты» и
«удобства» субъективны, то это порождает множественность названий
В правилах 1957 г. углеводороды получают единственное ^аяван.иг.
2, Нумерация главной цепи в женевской номенклатуре определяется
в первую очередь углеводородными радикалами, т е. радикалы считаются
старше связей и функций. В номенклатуре ЮПАК 1930, 1957 и 1961 гг.
последовательно проведен принцип нарастающего старшинства связей и
функций, которые считаются старше радикалов. Это изменяет подход к
каждому новому классу соединении и может заметно изменить название при
незначительных химических превращениях.
Углеводороды
Предельные углеводороды называют в соответствии с жененскими коа-
внлами (см. стр. 635). Подробно разработаны правила старшинства
радикалов
Главную цепь непредельных углеводородов выбираю] так, чтобы н нес
попа 1о не наибольшее число атомов углерода, а наибольшее число кратных
гвяіеі'і В остальном действуют правила для предельных углеводородов.
* Русский перевод правил льежской номенклатуры можно найти в книге
А П. Терентьева и др. «Номенклатура органических соединений»,
Під АН СССР. 1955, стр 278—289.
Русский перевод правил ЮПАК '.957 г, опубликован в «Справочнике
димнка», т. 2, Мзд «Химия», 1964.
Номенклатура ЮПАК 647
Пример:
СН,=СН—C=Cri,
0 "її
2-Децилбутадиеи-1.3
Углеводородные радикалы, образованные отнятием одного атомя
водорода, обозначают суффиксом ил, отнятием двух атомов водорода от одного
атома углерода — суффиксом илиден, трех атомов водорода от одного
атома углерода — суффиксом илидин. Счет атомов главной цепи радикала
начинают от атома с ненасыщенной валентностью. Радикалы, образованные
отнятием нескольких атомов водорода от нескольких атомов углерода
исходной молекулы, обозначают суффиксами диил, триал, диилиден. и т. п.
с соответствующими цифрами. a.w-Бирадикалы предельных углеводородов
нормального строения называют полиметиленами; подобные радикалы
непредельных углеводородов обозначают суффиксами енилен., инилен, диенилен
и т п Допускаются к применению традиционные названия ряда радикалов
Примеры-
СН3—СН2—СН2— пропил
СН3—СНг—Ct\/ пропилиден
СН3—СН2—С— пропилидин
\
СН3— СН—СНа 1-метилэтил
—СН2—СН—СН2— пропантринл -1,2,3
j—СН2—СН2— триметилен
—СН=СН2—СН2— пропенилен
Алициклические углеводороды называют по системе Байера.
Циклическое строение углеводорода обозначают префиксом цикло и указывают число
атомов в цикле (циклопентан, циклогексан и т. п.). Для бицнклических и
трициклических соединений этого класса применяется следующий метод:
І В структуре выбирают самый большой цикл и нумеруют его атомы,
начиная с узловых, сначала самый длинный отрезок (мостик).
2. В названии указывают: а)—число циклов; б)—длину мостиков
(число атомов), соединяющих узловые атомы; в) — число атомов в цикле.
Заместители и кратные связи обозначают по общим правилам.
648
Номенклатура органических соединений
Примеры:
6 5
бицикле |t з.1\ уидекаи
1.7.7-триметилбицикло 2:1.\\ гептан
бицихло {4.4.01 пекан
2,6.6-триметилбиииило 13.1.11 гептен -2-ОН-4
3) В названиях грициклических углеводородов при четвертом (самом
коротком) мостике указывают верхним индексом номера атомов, которые
он соединяет.
Пример:
"Ъ 4
в
трицикло D.2.l.O^i 5) нонав
Номенклатура терпенов и стероидов регламентируется специальными
правилами.
Для ароматических углеводородов отобрано определенное число
основных структур (см. стр. 660), из названий которых конструируют названия
сложных структур, пользуясь системой Паітерсона.
По системе Паітерсона для нумерации структуры ее ориентируют в
плоскости чертежа так, чтобы: а)—самый длинный ряд колеи располагался
горизонтально; б) — большая часть колец располагалась вверху и справа,
а меньшая часть — слева и внизу.
Пример;
неправильно
Номенклатура ЮПАК
649
Затем нумеруют неузловые атомы структуры, начиная с верхнего
«свободного угла и двигаясь по часовой стрелке. Узловые атомы обозначают
номером предшествующего неузлового с добавлением латинских букв в
порядке алфавита.
Чтобы назвать структуру по системе Паттерсона:
1) выделяют фрагменты, из которых будут строить название структуры,
например:
нафталин
антрацен
2) нумеруют каждый фрагмент по обшим правилам, причем грани
большего фрагмента, считающегося основным, обозначают буквами
латинского алфавита, грань 1—2 буквой а, грань 2—3 буквой Ь и т. д.:
3) определяют «координаты» места срощения фрагментов, следя за тем,
чюбы атомы обоих фрагментов в месте срощения перечислялись в одном
направлении:
3,2,1-нафталин
b de -антрацен
4) образуют название, беря младший фрагмент в сокращении;
«координаты» места срощения указывают в скобках между названиями
фрагментов
нафт 13,2,1—de\ антрацен
Даже небольшая разница в шифре «координат срощения»
свидетельствует о различии структуры. Например, название нафт[1,2,3—гіфнтрацен.
?50
Номенклатура органических соединений
отличающееся от предыдущего только порядком цифр в шифре, соответствует
совершенно иной структуре:
Если структура не полностью «ароматизирована», т. е. если в ней
вместо каких-либо групп —СН= имеются группы —СН2— или вместо группы
=С< имеется группа —СН<, то положение такого «лишнего» водорода
обозначают перед названием цифрами и прописной курсивной буквой Н.
Примеры:
/Н-бенз \ef] нафталин
9ЬН-беяз \ef] нафталин
Простые спирановые структуры называют по Байеру, обозначая в скоб«
¦кал цифрами длину мостиков. Например:
9 Ю
отаро Ч.5І декан
и ю
диспиро E.1.7.2] гептадекан
Сложные спирановые структуры называют по образующим их
фрагментам, указывая в скобках номер атома срощения в каждом из фрагментов:
флуорен-спнро [9,1'| циклопропан
Для основных гетероциклических структур номенклатура ЮПАК. ис*
пользует традиционные названия (см. табл. на стр. 661),
Номенклатура ЮПАК 651
В качестве универсальной номенклатуры гетероциклических структур
используется так называемая «ав-система.
По этой системе название любого гетероцикла можно образовать из
названия соответствующего по форме карбоцикла и приставки,
обозначающей сокращенно гетероатом:
О—окса
S—тиа
N—аза
Р—фосфа
Si—сила
В—бора
Перед гласными буква «а» в прнставкал опускается.
Примеры:
о
эксациклогептан і,7,9-триазяфенантоеи
Для моноциклических гетероциклов широко применяется
комбинированная номенклатура, в которой гетероатомы обозначают префиксами по
«а»системе, а размер цикла и степень насыщенности—суффиксами. Пяти-
членные циклы обозначают суффиксом ол, шестичленные — ин
Насыщенность циклов, содержащих азот, обозначается суффиксами ин и идин.
Насыщенность прочих циклов обозначается суффиксом ан. Например:
N Р
II II
| II 1,3-оксазол || || оксафосфол
О О
, NH I РН
| I 1,3-оксазол и дин I I оксафосфолан
14 О
f J 1,4-фосфазин ( J
Р О
NH S
Г J пергидрофосфазии Г ]
оксатиин
оксатнан
РН О
Насыщенные гетероциклические системы часто называют, присоединяя
к названию ненасыщенного соединения префиксы дигидро, тетрагиоро
и т. Д. до пергидро, что означает полное гидрирование.
Названия сложных ерошенных гетероциклических структур образуют по
тем же правилам, что н названия ерошенных карбоциклнческих структур.
Названия различных гетероциклических соединений строятся подобно
названиям ароматических соединений. Начало нумерации в моноцикличе-
652
Номенклатура органических соединений
ских системах идет от гетероатома, в конденсированных системах — от
атома, ближайшего к одному нз узловых атомов, так чтобы гетероатом получил
самый низший номер.
П
Примеры:
1) JC CJ
J I
2)
H2C CH2
і«
-соон
3,4-дииодпирролкарбв-
новая-2 кислота
3)
4)
2,2,5, Б-тетраметил гетра
гидрофуран
хниолинкарбоьовая-і кислота
5)
NH,
5-ннтро-2-аминопиридин
8-ОКСИ-4-МЄТИЛХИНОЛИН
Для соединений с двумя гетероатомами нумерацию начиняют со
старшего гетероатома. По старшинству гетероатомы располагаются в следующем
порядке: О, S, N и дальше элементы в порядке латинского алфавита.
Нумерацию ведут по кратчайшему пути от старшего гетероатома к младшему.
Пример:
О С—СИ,
2 411
Н3С-С СН
2,5-диметнлоксаяол
Правила номенклатуры соединений, содержащих характеристические
группы*, еще не узаконены, но намечена общая тенденция развития этой
области номенклатуры:
1) устанавливается строгий порядок старшинства характеристических
групп;
2) главную цепь выбирают так, чтобы в нее попала старшая и
наибольшее число следующих по старшинству характеристических групп;
3) суффиксами обозначают только кратные связи и старшую
характеристическую группу; все остальные группы обозначают префиксами. В
соответствии с этим каждая группа имеет два обозначения', для употребления
в качестве суффиксов и в качестве префиксов. Важнейшие примеры
приведены в таблице на стр. 653.
* Этим термином охватываются все неуглеродиые заместители —
нефункциональные и функциональные.
Номенклатура ЮПАК
653
Обозначения некоторых
Функция, группа
ICI-OH
ici=o
1С<н
—сно
хзн
—соон
RO-
-SH
|C]=S
RS-
—SO3H
—COOR
-CN
=N
—NH„
ФУНКЦИЙ И групп 8
„РефИ.
гидрокси-(окси-)
оксо-(кето-)
жсо-(альдо-)
формил-
—
карбокси-
алкокси-
тиоло-, меркапто-
тиоксо-
тиоксо-
алкилтио-
сульфо-
алкилоксикарбонил-
циано-
нитрило-
амино-
иоменклатуре ЮГТАК
Суффикс
ол
-он
•ал
-карбальдегид
овая кислота
•карбоновая кислота
—
¦тиол
-тиал
-тмон
—
•сульфоновая кислота
-алкилкарбоксилат
-карбонитрил
-нитрил
¦амин
Цифры, обозначающие положение в молекуле связи или группы, в
разных странах помешают по-разному. Так, в США принято все цифры ставить
перед обозначением групп, а положение кратной связи указывать перед
корнем слова названия; во Франции принято ставить цифры после
префиксов и перед суффиксами; в немецкой литературе цифры помещают перед
префиксами и после суффиксов.
Пример:
СН8-СН=С—СНаОН
СНа-СН3
американская литература ...... 2-этил-2-бутен-1-ол
французская < этил-2-бут-2-ен-1-ол
немецкая - 2-этилбутен-2-ол-1
женевс кая номенклатура З-метилолпентен-2
654 Номенклатура органических соединений
4. ПРИНЦИПЫ СИСТГ МАТИ ЧЕСКОЙ НОМЕНКЛАТУРЫ
Общим официальным языком химии может быть только
последовательно систематическая номенклатура.
Такая номенклатура должна удовлетворять по крайней мере четырем
требованиям:
1. Должны существовать простые правила, при помощи которых можно
было бы построить систематическое название каждого органического
соединения, а по названию — формулу. Такое название должно находиться в
однозначном соответствии с формулой строения, т. е. каждой формуле
должно соответствовать одно систематическое название, а названию — одна
формула
2. Систематические названия органических соединений должны
отражать научную классификацию соединений, иначе говоря классификацию,
основанную на теории химического строения.
3 Номенклатура должна базироваться на некоторых общих
положениях, и ни одно частное правило не может противоречить предыдущим,
ранее установленным правилам.
4. Названия следует строить в соответствии с особенностями языка.
Систематическая номенклатура должна опираться на общепринятую
классификацию органических соединений, подробно разрабоїэнную в
справочнике Бейлышейна В основе этой классификации лежит строение
углеродного скелета или гетероциклического ядра молекулы.
Гетероциклические и гетероцгпные соединения, содержащие большое
число неугл?родны.\ атомов, например кремния, бора или других
элементов, в систематической номенклатуре следует рассматривать как производные
этих элементов.
Требованиям систематической номенклатуры ближе всего отвечаеі
женевская номенклатура Ниже излагаются основы более современной и
последовательной системы*
Настоящая система не узаконена Она приводится в учебнике для того,
чтобы показать возможность создания и употребления последовательно
систематической номенклатуры и ее преимущества, однозначность названий,
логику, краткость и немногочисленность правил.
Углеводороды
Предельные углеводороды называют в основном по женевским правилам.
Существенное отличие состоит в выборе начала нумерации и названия*
радикалов
Нумерацию атомов углерода в главной цепи начинают и тою конца, к
которому ближе старший заместитель.
СНа—СН—СН3—СН—СН,—СН, начало нумерации
| | определяет этил
СН;. СНа—СН„
*А П Терентьев, А Н Кост, A S\ Уукермяи, В М
Потапов, Номенклатура оріанических соединений, Изд. АН СССР, 1955.
Принципы систематической номенклатуры 655-
Связанный с главной цепью радикал рассматривают и нумеруют, как
независимый углеводород, в котором старший заместитель — главная цепь
основного углеводорода «Автономные» номера атомов радикала помещают
в виде верхнего индекса к «федеральному» номеру — номеру атома главно»
цепи основного углеводорода, к которому присоединен радикал.
Пример:
CHS—CHj—СНа—СН2—СН—СНа—СН2—СНа—СН„
в1 І52 б" 5*
СНЭ—СН—СН—CHS
СНЯ
5 (З-метилбутил-2) ноная
Непредельные углеводороды называют подобно предельным. Положение
кратных связей обозначается суффиксами в конце слова-названия с
указанием числа связей и номеров атомов.
Пример:
СН2=СН
СН„—С=СН—С—СНа—СН—СН,—СН,—СН„
I II
СНЭ СН—СНЭ
2-метил-4.6-диэтилнонатриея-2,4°.б1
Названия ациклических соединений, содержащих характеристические
заместители (функциональные и нефункциональные группы), строят по
общим правилам с добавлением суффиксов и префиксов. Никакие изменения-
функционального характера соединения не могут изменить способ выбора
главной цепи и порядка нумерации, определенных углеродным скелетом
Для обозначения карбоксильной функции используется суффикс ацид.
Примеры:
СН=СН, ОН
СНЭ—С—СН2—СН—СН2—СН—СНа НООС—CHj—С—СНа—СООН
II I I
О СН„ СООН
з1
'2-иетил-4-9тилгеатен-41вОИ-6 З-ыегилпентанод-З-триацид-l.b, З1
Циклические структуры
В названиях циклических соединений указывают!
1. Число атомов в циклической структуре
2 Число циклов (в структуре столько циклов, сколько связей мпжн»
разорвать, не расчленяя полиостью структуру, т. є. не ілделяя часіей от
молекулы).
656
Номенклатура органических соединений
3. Порядок соединения атомов (от высшего номера к низшему) и
характер их связей (ароматические связи обозначают суффиксом арен, сопря-
жечные — коен).
4. Гетероатомы указывают по «а»-системе.
Основное в системе — правила нумерации атомов циклической структуры.
Все атомы циклической структуры делят на 4 класса:
Класс а — неузловые атомы.
Класс б — узловые атомы, принадлежащие двум смежным кольцам.
Класс в — узловые атомы, принадлежащие трем смежным кольцам.
Класс г — спирановые атомы и узловые атомы, принадлежащие 4
смежным кольцам.
Старшинство классов: а>б>а>г.
Пример:
Важной характеристикой циклической структуры является главный
контур— самая длинная замкнутая цепь атомов, содержащая наибольшее число
атомов старшего класса.
Нумерацию атомов начинают с главного контура с атома старшего
класса (обычно а). Начало и направление нумерации выбирают так, чтобы
подряд нумеровалось наибольшее число атомов старшего класса
(«алфавитный принцип»). В случае нескольких возможностей нумерацию ведут так,
чтобы атомы в порядке нумерации образовывали «старшее по алфавиту
слово».
Пример:
В данном примере возможны на первый взгляд 4 направления
нумерации, где в каждом случае подряд идут 4 атома класса а. Однако одно
из этих направлений выгоднее:
Атомы
Направление
I
11
III
IV
1
а
а
а
а
2
а
а
а
а
3
а
а
а
а
4
а
а
а
а
5
6
6
б
б
' б
б
а
а
а
7
б
б
а
8
¦Ї .
* •
Принципы систематической номенклатуры
657
Направление II выгоднее направления I F-й атом о); направление И(
вьігоднрє направления П (8-й атом и), направление IV выголнрр всех
остальных (атомы № 6 и № 7— два а подряд). Таким образом, структура должна
быть пронумерована следующим образом:
В структуре 4 иикла, образуемые 18 го атомами. Все атомы входяг
в главный контур Вне- последовательности нумерации связаны между
собой атомы 18—5; 17—8; 15—10.
Систематическое название структуры будет:
тетрацикло [IS—5: 17—8; 15—10] октадекан
Приведем еще несколько примеров нумерации и названий циклически»
структур;
В главный контур входит 12 атомов;
13(8,4) означает, что атом № 13
связан с атомами № 8 и № 4
трицикло [12. 13 (8. 4>; тридекан
Знак «х» перед атомом № 10
означает, что этот атом не
связан с атомом № 9
трицикло("9.x 10 (S. й); 9—4| декан
788
Названия радикалов и циклич. структур
ПРИЛОЖЕНИЕ II
НАЗВАНИЯ ЧАСТО ВСТРЕЧАЮЩИХСЯ РАДИКАЛОВ
И ЦИКЛИЧЕСКИХ СТРУКТУР
Одновалентные радикалы насыщенных углеводородов
СНз-
>— 1Ї3СГІ2—
GH3CH2CH2—
СНзч
>сн-
гн /
^п3
СН3СН2СН.,СН2-
%:н-сн2-
сн3/
^СНд
метил
этил
пропил
нзопропил
бутил
изобутял
o/nop-бутил
СНз
СНз-С-
1
сн>
СНз(СН2KСН2-
^СН—СН2—СН
сн3/
СН3
1
сн3сн2-с-
СНз
СН3(СН2),СН2-
mpem-бутил
амил
2— изоамил
трет-шип
гексил
метилен
сн3сн
Двухвалентные радикалы насыщенных углеводородов
СНа,
сна
—CHjCHaCHa— триметилен
/
этил идеи
пропилиден
изопропилиден
СН,=СН—
СН=С—
Радикалы непредельных углеводородов
винил (этенил)
ацетиленил
(эти нил)
СНа—СН=СН— пропенил
СН,=СН—СН2— аллил
СН2=С—
| изопропенил
СН3
СН
,=<
винилпден
CHsC—СН2— пропаргил
СН3—СН2—СН=СН— бутенил
СН3—СН=СН—СН2— кротил
СНЭ
>с=сн—
сн/
—СН=СН— винилен
изокротил
Названия радикалов и циклич. структур
659'
QHfi-
С«Н4<(
CfiHa—CH2—
С, „- <^
CH,O-
QH.O-
Радикалы
CH2—СН2ч
алициклических углеводородов
СИ— циклопентил
СН\сн~с
Радикалы
фенил
фенилеп
(о-, и-, п-
бензнл
оенчилиден
(бензаль)
метокси
этокси
СН— ииклогекси,-
2
ароматических углеводородов
СН3-СЙН4-
СН3-СAН3<(
с1Он,—
с6н5-сн=сн-
Радикалы спиртов
С3Н,О-
с4н,о-
Радикалы кислот
толил
(о-, м-, п-)
тол у ил ен
нафтил
(а- и ?-)
стирил
пропочси
бутокси
О
о
СН3СН2-
СН3
СН3-
Р
формнл
ацетил
пропионил
бутирил
;О
сн
СН3
рил
валерил
изова-
лерил
^>С-СН2-
-СН2-СН3-С^
оксалил
малонил
О _ ...
0
бензонл
/СО—
х:о—
толуил
(о-, м-, п-)
фталил
42*
вбО
Названия радикаюв и циклич. структур
Радикалы аминокислот
О
NH-—СНа—C<f глицил
сн3.
,0
алапил
NH,
СН,—СН—C<f серил
І І Ч
ОН NHa
Важнейшие конденсированные карбоциклические структуры
СН
СН
СН
СН
О
3\СН—СН—с/ валил
NH»
—СНа—СН—С<^ лейцил
О
NH,
СНЯ—CHj-CH-CH-c/
СН3 NH»
•О
изолеицил
Г-А;
6 і
и і
пентален
инден
нафталин
азулен
аценафтилен
флуорен
6 І'І 4
Ю 1
фенантрен*
антра[іен*
пирен
,/V4Aya
In II И 1
'654
хризен
нафтаиен
* Нумерация аюмов традициинная — исключение иэ пранил.
Названия радикалов и циклич структио
Важнейшие гетероциклические структуры
S
о
о
2сн,
о
=сна
ГЇН
О:
NH
NH
\2
4
тиофеи
фуран
2Я-пиран
2/У-хромен
пиррол
имидазол
пиразол
пыридни
пиразин
N
в/ 42
N3
7
бЧ/\ >
1 NH
NH
пиримидин
индол
пурин*
N
К Ii ПОЛИН
NH
:(P0
8 9 ]
карбазол
акридин
in
9 1N 1
N
б
феназин
* Нумерация атомов традиционная — исключение из правил.
662 Названия радикалов и циклич. структур
Радикалы гетероциклических соединений
(«I
О О
С4Н3О- фурил fjT ( і)
О
СвНйО— фурфурил | | '
(N) 'а) @)
N— NU NH
C4H4N_ пиррші О j_Jj~ (
s <ai s (W
C4H3S- тиенил |fjl~ II И
NN N
C6H4N- пиридил
CjH.N— индолил
ч/—- \/—= v
(N) («I 0
N- NH NH
ИМЕННОЙ УКАЗАТЕЛЬ
Абдергальден Э. 394
Авогадро А. 32
Альдер К. 548
Андрианов К. А 128, 177
Арбузов А. Е 130, 594
Байер А. 206, 243, 555, 556, 597, 599.
647, 650
Баландин А. А. 546
Бейльштейн Ф. Ф. 28, 108, 122, 430,
438, 633
Бертло М. 93, 258, 433
Берцелиус И. Я. 15 33, 38, 294, 414
Бражникова М. Г. 602
Бродский А И. 398
Брэгг 160, 225
Бутлеров А. М. 16, 32, 37 ел., 48, 69,
74, 78, 79, 90, 112, 122, 136. I6S,
193, 204, 205, 206, 311, 324, 4U3,
515, 630, 631
Бухиер Э 148
Вызов Б. Б. 91, 103
Вагнер Е. Е. 40, 72, 82. 84, 122, 560
Вальден П. И. 304
Вант-Гофф Я. 155, 158, 160, 162, 241,
272, 288
Велер Ф. 16, 93, 288, 414, 471
Виланд Г. 571
Вильштеттер Р. 609
Вильямсон А. 177
Виндаус 571
Виноградов А. П. 399
Вислиценус И. 242, 288
Витт О И. 513
Вольф 195
Воскресенский А А. 460, 528
Вудворд Р. 595, 617
Вышнеградский А. Н. 403, 613, 616
Вюрц А. 51, 58, ПО, 132, 168, 170,
180, 363, 432
Гарриес Ц. 100
Гаттерман Л 465
Гаузе Г. Ф. 602
Гей-Люссак Л. Ж 406
Гейман К. 598
Гольдфарб Я Л. 581
Гомберг М. 525
Горяинон В. 69, 79
Гофедии 526
Гофман А. В. 324, 365, 367, 488 501
598
Гофмейстер К. К 394
Гребе К. 539, 540
Гриньяр В ПО, 123
Густавсон Г. Г. 424, 433, 546
Домагк 511, 512
Данилевский А. Я. 393
Дарвин Ч. 596
Дедусенко Н. И. 398
Демьянов Н. Я. 224, 546
Дильс О. 548
Дэви 412
Дюма Ж. Б. А. 30, 55, 293
Ермольева 3. В. 602
Жерар Ш. 33, 36, 57, 132, 267. 426
614
Зайцев А. М. 40, 82, 122, 123, 193. 286
Зелинский Н. Д. 66, 91, 161, 184, 374,
394, 433, 435, 546, 551
Знбер П 602
Зииин Н. Н. 37, 463, 488, 518,
532, 597
Ильинский М. А. 537
Казанский Б. А. 433, 435, 546
Канниццаро С. 205, 306, 467, 582
Кариус 31
Кегль 596
Кекуле А. 35, 37, 426
Кижнер Н. М. 194, 546
Кили 552
Кирхгоф К. С 15, 145. 332
Клабуновскиїі Е. И. 298
Клайзеи Л. 307
Клаус К 37
Кнорр Л 603
Колли А. А. 315, 319
664
Именной указатель
Кондаков И Л. 102
Кондратьев В. Н. 527
Коновалов М И. 56, 358, 359, 546
Коршун М. О. 30
Кохгсель В. 61
Кост А. Н. 386
Кочергин С. М. 224
Кочешков К. А 506
Крафтс Дж М. 424, 433, 469, 553
Купер А 37
Курбатов А. И. 430
Курс а нов А Л 399
Кучеров М Г. 92
Кьельдаль И. 3U
Ляв\азье А. Л. 15
Лангмюр И. 163
Л а ути 527
Лачипов П. А. 447
Лебедев С. В 91, 102
Ле-Бель А. 155, 158
Летний А. А 435
Либермаи 539, 540
Либи.х Ю. 288, 414, 426, 471
Ловии Т. Е. 55, 232, 281
Лоран А. 33
Лькжс 61, 163
Любіївин Н. Н. 616
Максоров Б В. 587
Манассеин В А. 601
Манассеина М. М. 148
Манних 202, 553
Марковникои В В. 40, 41, 78, 79. 112,
136, 142, 228, 233, 254, 280, 546,
550
Маркс К. 46
Маііер В. 584
Менделеев Д. И. 16, Ь6, 424
Меншуткин Н. А. 254
Миллер О К 514
Митчерлих Э 295, 441
Михлер В. 495
Наметкин С. С 546, 560, 567
Назаров И Н 91. 214
Неникий М В. 597
Несмеянов А. Н. 310, 505, 506, 546,
553
Нико 613
Нориш 527
Орехов А П 614
Орлов Е И. 69
Осокин М. 74
Панет Ф. ,526. 527
Пастер Л. 148, 293, 295, 380
Патрикеев В В. 298
Пагтерсон 648
Перкин В. 485, 515
Петров Г. С. 259
Пикте 613
Плиний 515
Полотебнев Н. Г. 601
Попов А В. 40
Попов А Н. 199, 246
Посон 552
Потапов В. М. 386
Преображенский Н. А. 604
Прохоров С И. 514
Раис Ф О 527
. Pf-ймер К. 481
Реней 256
Реутов О. А 506
Реформатский С. Н. .122
Родионов В. М. 467
Розанов М. А. 320
Роско Г. Е. 46
Ружнчка Л. 547
Руфф 325
Сабатье П. 78
Савич В. 90
Садиков В. С. 546
Сакс Ю 597
Семенов Н. Н. 55, 527
Сивплобов 25
Скрауп 614
Сморгонский Л. М. 581
Сокслет 23
Степанов А. В. 28
Тейс Р. В. 399
Теренин А. Н. 527
Терентьев А П 298, 386 580, 586
Тиле 96, 97, 248
Тиман Ф 481
Тимирязев К А. 589, 597
Титов А. И 367
Тищенко В Є 197, 254, э67, 582
Топчиев А. В. 367
Трейбс В. 66
Тропш X. 69
Уотерс 527
Фаворский А. Е 91, 102, 170, 214
Фарадей М. 19, 422. 423
Фишер Фр 69
Фишер Э 320. 324, 325, 338, 394, 594
Флеминг 601
Фмкнн С А 263
Франкланд 121
Именной указатель
665
Фридель Ц. 424, 433, 469. 553
Фридлендер 515, 600
Фрицше Ю. Ф. 597
Херст 319
Хоти некий Е. С. 585
Хэуорс 319
Цвет М. С. 396, 589, 590, 591, 597, 608
Церевитинов Ф. В 125
Цуккерман А. М. 386
Челиниев В. В. 124 582, 586, 5S7
Чичибабпн А. Е. 594, 611. 616
Чугаев Л. А. 125, 218, 346
Швейиер 346
Ulnimep Р 602
Шеврель 233, 258
Шееле К. В. 172, 286, 294
Шилов Н. А. 546
Шихан 601
Шмидт К. 314
Шорлеммер К. 17, 44, 46, 198, 540
Шорыгин П П 51
Шостаковокмй М Ф. 91
Штаудмшер Г. 101
Шухов В. Г. 68
Эльтеков А. П. 92, 166
Эммануэль Н. М. 232
Эшельгардт А. Н. 447, 582
Энгельс Ф. 46
Энглер 66
Эрленмейер 528
Юрей 253
Юрьев Ю. К. 579, 584, 585
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абиетиновая кислота 566
Абсол вотирование
спирта 150
эфира 180
Аглюкон 336
Аденин F-Аминопурин) 622. 627
Адипиновая киспота 271, 554, 555
Азелаиновая кислота 250, 271
Азобензол 507
Азокрасители 509 ел.
Азоксибензол 508
Азолы 600 ел.
Азосоединения 507 ел.
Азосочеіание 510
Азот, определение 27, 30
Азулен 660
Акридин (Дибензопиридин) 618,661
Акриловая кислота 166, 211, 237 ел.,
249. 281
Акрилонитрил 238
Акрихин (Агебрин) 618
я-Акроза (Фруктоза рацемическая)
324
Акролеин (Пропеналь) 210, 237
Аланил 660
а-Аланил (а-Амииопропионовая
кислота) 373, 378, 382
Ализарин 539 ел.
Алифатические соединения 19, 45 ел.
Алиииклические соединения 545 ел.
Алкалицеллюлоза (Щелочная
целлюлоза) 349
Алкалоиды 576, 603, 613
Алканы 60
Алгсены 75
Алкндпые смолы 172, 476
Алкиларилсульфонаты 236
Ллкилкремневые эфиры 177
Алкилсерные кислоты 79
Алкилсулі.фатьі 236
Алкилсульфонаты 183, 236
Алкилхлорсиланы 12Ь
Алкилы 47
галоидные 106 ел., 132 ел., 173
радикалы 526
Алкоголиз 255
Алкоголяты 140
Аллантоин 620
Алл ил 75, 658
бромистый 115
йодистый 115
хлористый 115, 166
Аллиловын спирт 165, 212, 237
Аллокоричная кислота 485
Аллоксан 619
Альбумины 390
Альдегидаммиаки 191, 203
Альдегидокислоты 305 ел.
Альдегидоспирты 315
Альдегиды 144, 185 ел.
Альдогексозы 316, 319, 605
Альдозы 316
эпимерные 328
Альдоли и альдольная конденсация
200
Альлопентозы 330 ел.
Алюминийорганические соединения
125
Амигдалин 464, 465
Амидол 498
Амиды кислот 229, 267 ел., 413
А мил 658
бромистый 109
йодистый 109
Амилацетат 252
Амиловые спирты 152 ел.
Амилоза 345
Амилоид 348
Амилопектин 345
Аминоазобензол 509
Аминоазосоединения 509 ел.
Аминобензойные кис/юты 501 ел.
п-Аминобензолсульфсжислота (Су ль-
фаниловая кислота) 491
Аминогруппа 361
а-Аминоизовалериановая кислота
(Валин) 379, Г-82
a-AMHHO-(? индолил-3)-пропионова
кислота (Триптофан) 596
а-Аминокапроновая кислота 384
Аминокислоты 370 ел.
алифатические 370 ел.
ароматические 501
Аминомасляная кислота 373
Предметный указатель
667
1-Амино-8-нафтол-3,6-дисульфокис-
лота (Аш-кислота) 533
2-Амино-6-оксипурин (Гуанин) 622,
627
а-Аминопиридии 611
Аминопласты 420
а-Аминопропионовая кислота
(а-Аланин) 373, 378, 382
6-Амичопурин (Аденин) 622, 627
'ьАминосалициловая кислота
(ПАСК) 502
Аминоспирты 370 ел.
и-Амино^-тиоизовалериановая
кислота (Пеницилламин, Диме-
тплцистеин) 602
Аминоуксусная кислота (Глицин,
Гликокол) 373, 377, 382
я-Аминофенол солянокислый (Пара-
аминофенол) 498
Аминофенолы 497 ел.
Амины
алифатические 361 ел.
ароматические 487 ел.
Аммониевые основания 363
Аммонолиз 255
а-ифи-Нафтохинои 533
Анабазин 614
Ангидриды кислот 266 ел.
Ангидрон 30
Андростерон 574
Анестезин 502
Аннд (Найлон) 397, 555
Анизол 463
Анилиды 489
Анилин 487, 490 ел., 497
Анилиновый желтый о09
Аииониты (Анионообменные смолы)
500
Антибиотики 602
Антидетонаторы 83, 123
Антиоксиданти 459
Антипирин 603
Антиподы оптические, см. Оптические
антиподы
Антифризы 170, 172
Антоцианидины 609
Аитоцианы 608
Антрагидрохинон 538
Аитраниловая (о-Аминобензойная)
кислота 501
Антрахинон 537
Антрахиноновые красители 541
Антрацен 536
Антраценовое масло 435, 480
Лнтроподезоксихолевая кислота 572
Ц )-Арабиноза 332 ел., 354
Арабаны 354
Аргиназа 417
Аргинин 383, 417
Арил 431
Ароматизация нефти 435
Ароматические соединения 19, 422 ел
многоядерные 518 ел., 527 ел.
небензоидные 553
Арренал 131
Арсиновые кислоты 131
Арсины 130
Асимметрический атом углерода 155
158, 288
Асимметрический синтез 297 ел.
L-Аскорбииовая кислота (Витамин
С) 335
Аспарагин 380, 382
Аспарагиновая кислота 380, 382
Аспирин 480
Асфальты 70, 71
Атебрин (Акрихин) 618
Атомная рефракция 86
Ауксины 550, 596, 597
Ауксохромы 514
Аценафтилеч 660
Ацетали 196
Ацетальдегид (Уксусный альдегид)
92, 188, 207, 231
Ацетамид 229, 267
Ацетаиилид 489, 492
Ацетаты 232
целлюлозы 351
Ацетил 269, 659
хлористый 229, 264
Ацетилацетон 2І6
Ацетилен 93 ел.
Ацетиленил 658
Ацетиленистые медь и серебро 93
Ацетиленовые углеводороды 90 ел.
Ацет:ілмочевина 416
Ацетобутират целлюлозы 351
Ацетогликолевая кислота 285
Ацетоии 469
Ацетол 284
Ацетон (Пропанон-2) 144, 189, 212 ел.
Ацетонилацетон (Гександион-2,5) 581
Ацетонитрил 220
Ацетоио-бутиловое брожение 213, 338
Ацетоуксусиая кислота 229, 306
Ацетоуксусный эфир 306 ел.
синтезы 311 ел.
Ацетофенон (Метилфенилкетон) 469
Ациклические соединения 43
номенклатура 638
Ли ил 269
668
Предметный указатель
Лш-кислота (І -Амино-8-нафтол-З,'
днсульфокислота) 533
Бальзамы 485
карбитуровая кислота 416
Ьездымный порол 351
Белки 387 ел.
Бензальацетон 467 ел.
1,2-Беіпаитрацсн М4
Бензидин 518 ел.
Бензидиновая перегруппировка 519
Бензил (Дибснзоил) 468
бромистый 439
радикал 65?1
хлористий 439
Ьеизилиден 659
хлористый 439
Бензиловий спирт 439, 464
Бензины 67 ел,
Бензоил 471. 6Ї9
гидроперекись 466
хлористый 471
о-Брнзоилбензойная кислота 537
«Брнзоил;іиррол 586
Бензоин 468
Бензойная кислота 470 ел.
Бензойнометиловый эфир 470
Бензойный альдегид 464, 467 ел.
Бензойный ангидрид 471
Бензол 422 с.п„ 430 ел.
галоидпроизводные 437 ел.
строение 425 ел., 427
Бензолазотнлуол 507
Бензолсульфокислоты 449
Бензольные углеводороды 430
Бсизопирон (Хромон) 608
Бензотрихлорид 439
Бензофенон (Дифенилкетон) 469
с-Бензолипон 461
1,2 Бензпирен 543
Берлинская лазурь 28
Бетаин 378
Бигумаль (Палудрин) 618
Биозы 316
Бисульфитные соединения 190, 203
Битумы 65 ел., 71
Бнурет 416
Биуретовая реакция 388
Борнеол 567
Брожение 148, 231, 335, 338
Бромбензол 437, 439
Бромистые алкилы 109, 115
? Броммасляная кислота 281
а-Бромнафталин 529, 533
я-Бромнитробс нзол 442
Броминклонентан 550
Бутадиен (Дивинил) 96, 102, 103, 248
Бутан 45, 49, 53
Бутановая (Масляная) кислота 224.
233
Бутен (Бутилен) 75, 76, 77, 103
Б>тенил 658
Бутил 658
бромистый 109
йодистый 109
х-Бутиламин 363
Бутилкаучук 90
Бутиловые спирты 139, 143, 151
п-трет Бутилпирокатехин 459
Бутилсерная кислота 82
Бутирил 269, 659
у-Бутиролактон (Оксимасляная
кислота, ^лактон) 282, 286
Бутоксы 659
Вазелин 67
Вазелиноаое масло 67
Валентные углы 164, 243
Валериановая кислота 224, 382
Валерианоный альдегид 188
Валерил 659
Балил 660
Валин @,-Лминоизовалериановая
кислота) 379, 382
Вальдсновское обращение 304
Взаимное влияние атомов 40, 41
Нинил ,5, 114, 658
бромистый 79
хлористый 117
йинилацетат 118
Биниланетилен 93, 103
Пинилсн 658
Винилиден 658
хлористій 118
фтористый 120
Винилуксусная кислота 239
Винипласт 118
Виннокаменная кислота 292
Винные кислоты 290 ел
Винограиная кислота 291, 294
Виноградный сахар 332
Вискозное волокно 352
Витамины
А 570
С (L-Аскорбиновая кислота) 335^
Do (Кальциферол) 572
РР 613
Вителлин 392
Витти теория 513
Водород
определение 29
открытие 27
электронное облако 164
Предметный указатель
669
Воска 257
Воск горный (Озокерит) 71
Восстанонлечия реакция 256, 326,
367, 403, 506 ел.
Вращение удельное 157
Вторичные спирты 133
Вулканизация 99
механизм 102
холодная 99, 4 [8
Высокомолекулярные соединения
115 ел.
Газолин 67
Галакганы .354
Ь Галактоза 334, 354
D-Галактоновая кислота 334
Галловая кислота 482
Галоидангидриды кислот 229,
264 ел
Гэлоидчамешенные кислоты 280 ел.
Галоидироьание 54, 74, 78, 91, 228
424, 437
Галоидные ал килы 106—111
Галои.чмронзводные предельные
углеводородов 106 ел.
изомерия 106
свойства 108
способы получения 107
Гваякол 459
Гексадекан 53, 638
Гексадиены 98
Г'ексаконтан 45, 53, 638
Гексамечилен (Цикчогексан) 545,
548, 549, 554
Гексаметилендиамин 369
Гексан 45, 53
Гексан дион-2,5 (Ацетон и л ацетон)
581
Гексафснилэтан 525
Гексахлорбензол 439
Гексахлорциклогексан (Гексахлоран)
440
Гексил 658
Гексит 172
Гексоген 205
Гексозы 3IG, 332 гл.
Гелнантин (Метилоранж) 510 сл.
Гем 393
Гематин 393
Гемиїї 588. 393
Гемииел.иолозы 354
Гемоглобин W, 393, 588
Гемохромоген 393
Генэнкосап (Генчйкозан) 638
Гепталєкаи 638
Гептаконтан 45, 53
Гептаметилен (Циклогептан) 545, 549
Гептан 45, 53, 638
Гєптатриом 607
[ептозы 316
Гераниевая кислота 212
Гераниол 212
Гербициды 454
Гетероа>ксин (а-Инлолилуксусная
кислота) 596
Гетероциклические соединения 43,
576 ел.
Гидчокарповая кислота 551
Гилразнни 506 ел
Гидразобензол 508
I идразоны 194, 203
Гидратация олефииов 79, 136
Гндратцеллюлоза 349, 352, 353
Гидрирование (Гидрогенизация) 78,
92, 262, 424, 436
жиров 262 ел.
угля 69
Гндрокоричная (?-феиилпропионо-
вая) кислота 484
Гидрокортиюн 575
Гидролиз 135. 141, 202, 254
олигосахаридой 339
полисахаридов 345. 347
Гидролизный спирт 137
Гидроперекиси 180 ел.
метила 180
этилового ->фира 179
Гидрохинон И-Диоксибензол) 458,
463, 498
Гипоксантин 620
Гипп\ровая кислота 377
Гистоны 390
Г-кислота B-Нафгол-6,8-лисул1.фо-
кнелота) 531
Гликоген 346
Гликокол (Аминоуксусняя кислота,
Глицин) 373, 377, 382
Гликолевач кислота 284
Г.'іиколевьій альдегид 284
Гликочи 81, 168 сл„ 284
Гликоляты 169
Глі^кох-олевая кислота 573
Глноксяль 216, 284
Глнокспловая кислота 284, 305. 620
Глифталевые смолы 476
Г.'шиериды 258
Глниерин 171. 284
Глицериновый альдегид 284, JA), JZ»
Глицеринфосфорная кислота 372
Глицерсна 330
Глицил 660
Глицин (Аминоуксусная кислота,
Гликокол) 373, 377, 382
Глобин 393, 588
570
Предметный указатель
Глобулины 390
Глутамин 380, 382
Глутаминовая кислота 380, 382
Глутаровая кислота 271, 382
Глюкоза 314 ел.
D-форма 332
Глюкозидный гидроксил 317
Глюкозиды 336
D-Глюкоиовая кислота 333
Глюкопротеиды 393
Гомология 45, 133
Гормоны 503, 574 ел.
Горный воск (Озокерит) 71
Горчичное масло 409
Горчичный газ, см. Дихлордпэтил-
сульфид
Грамицидин С 602
Гуанидин 417
Гуанин B-Амино-б-оксипурин) 622,
627
Гудрон 67
D-Гулоза 335
Гуммиарабик 354
Дакрон (Лавсан, Тернлеи) 475
Двойные связи 72, 84
инкремент 87
сопряженные (коньюгироваиные)
96, 247 ел.
экзальтация 99
электронное строение 243 ел.
Двойственная реакционная
способность 310, 312
ДДТ (п, я-Дихлордифеиилтрихлор-
метилметан) 440
Дегидратация 142
Дегидрирование (Дегидрогенизация)
143, 187
каталитическое 435, 436
Дегидроциклизация 436
Дезоксирибоза 626
Дезоксирибонуклеиновая кислоту
(ДНК) 626, 628
Дезоксихолевая кислота 572, 573
Декалин 536
Декаи 53
Декстрины 345 ел.
Дельфинидин 609
Депсиды 483
Детонация 83, 123
Дефолианты 407
Диазогндрат 504
Дмазоний 504
Диазосоединения 503 ел.
Диазотаты 504
Диальдегиды 215
Диаминоазобензол 509
а, е-Диаминокапроноеая кислота
(Лизин) 380, 382
2,2-Диамннофенол (Амидол) 498
Диамины 495 ел.
Диастаз 345
Диастереомеры 300, 380
Диацетил 216
Диацетоналкоголь 201
Дибензальаиетон 468
Дибензоил (Бензил) 468
Дибензолхром 554
Дибензопиридин (Акридин) 618, 661
Дибромбензол 429
Дибромбутены 97
1,2-Дибромпропан 78
Дибромстеарииовая кислота 250
Дибромэтан 72
Дибутилфталат 474
Дивинил см. Бутадиен
Дигалловая кислота 482
эфир 483
Дигидрохинолин 615
Диеновый синтез 548, 551, 581
Диизобутиламин 363
Диизобутилкетон 189
Днизопропилкетон 199
Дикетоны 215 ел.
Дикетопиперазины 376, 394
Димер 82
Диметиламин 361, 363, 367
Диметмламиноазобензол 510
Д и мети лами ноазобензол
сульфокислота 501 ел.
Диметиламиноуксусная кислота 378
Диметиланилин 494, 497
Диметиларсин 130
Диметилбутадиен-1,3 96
2,4-Диметил-6-тр<?т-бутилфенол 459
Диметилвинилкарбннол 214
Диметилвинилэтинилкарбинол 214
Диметилглиоксим 216, 218
Диметилкетон 144, 189, 212 ел.
3,7-Диметилксантин (Теобромин) 621
Диметнлмышьяковая кислота 131
Дпметилолмочевина 420
2.2-Диметилпропанол-1 153
Диметилсульфат 173. 175, 463
Диметилсульфид 183
Диметилтерефталат 475
Диметилфталат 474, 475
Диметилфульвен 552
Диметилцистенн (а-Амино-Р-тиоизо-
валериановая кислота, Пепи-
цилламин) 602
Диметилэтилен 74
Предметный указатель
671
Диметилэтинилкарбинол 214
Динамиты 175
Динитробензол 441, 442, 446
2,4-Динитротолуол 446
Дпоксан 170
Диоксиазобензол 509
Диоксиацетон 284, 329
ж-Диоксибензол (Резорцин) 458, 463
о-Диоксибензол (Пирокатехин 458, 461
«-Дноксибензол (Гидрохинон) 458,
463, 498
Диоксифлавон (Хризин) 608
Дпоктилфталат 474
Дипентен 564
Дипольные моменты 62 ел.
Дипропиламин 363
Дипропилкетон 189
Дисакрил 211
Дисахариды 339 ел.
Дисилан 125
Дитиоугольная (ксантогеновая)
кислота 418
Дифенил 241, 518
Дифениламин 493, 497
Дифенилгексадекаоктан 513
Дифенилкетон (Бензофенон) 469
Дифенилметан 520
Дифениловый эфир 463 ел.
Дифосген 412
Дифтордихлорметан 114
Дифторхлорметан 114
Дихлордиметилсилан 122
п,л'-Дихлордифенилтрихлор метил -
метан (ДДТ) 440
Дихлордиэтилсульфид (Горчичный
газ, Иприт) 184
Дихлорпропан 92, 111
2,4-Дихлорфеноксиуксусная кислота
454
Дихлорэтан 112, 241, 242
Дихлорэтилены 242
Дициан 406
Дициандиамид 407
Дициклопентадиени л железо
(Ферроцен) 552 ел.
Диэтиламин 361. 363
Диэтилкетон 189
Днэтилолтерефталат 475
Диэтилтолуамид (ДЭТА) 472
Диэтилиинк 124
ДНК (Дезоксирибонуклеиновая
кислота) 626. 628
Додекан 638
Докосан (Докозан) 638
Древесный спирт, см. Метиловый
спирт
Дубильные вещества 483
Дульцит 334
Душистые вещества 212, 256
Енолы 217, 309
Желатин 391
Желчные кислоты 572
Женевская номенклатура 59 ел.,
635 ел.
Живица (Терпентин) 566
Жирноароматические углеводороды
431
Жирные кислоты 219 ел.
синтетические 263
Жиры 257 ел., 262
Зеленое мыло 234
Зимаза 149
U-Идоза 335
Изатин 599
Изоамил 658
Изоамиламин 363
Изоамилацетат (Уксуснонзоам иловый
эфир) 257
Изобутан 50, 53
Изобутил 658
Изобутиламин 363
Изобутилен B-Метилпропен) 75. 77,
89, 136, 137, 246
Изобутиленгликоль B-Метилпропан-
диол-1,2) 169
Изобутиловый спирт B-Метилпро-
панол-1) 136, 137, 151
Изобутирил 659
Изовалериановая кислота 382
Изовалерил 659
Изокапроновая кислота 382
Изокротил 658
Изокротоновая кислота 240, 242
Изолейцмл 660
Изолейцин 379, 382
Изолопіческий ряд 71
Изомасляиая кислота 224
Изомасляный альдегид 188
Изомерия 18, 37
поворотная 241
цис-транс- 240
Изонитрилы 404
Изонитросоедпнения 359
Изооктан B, 2,4-Триметилпентан) 83
Изопентан 49, 50, 53
Изопрен 96, 100. 214
Изопропемил 658
672
Преометчый указатель
Изопрогшл 65S
йодистый 78, 106
Изопропиламин 363
Изопропилбензол (Кумол) 213, 453
ИзопропплоБый спирт 136, 142
Изопропллсерная кислота 79
И.юпропилэтилен 74
Ичотиишіа мешая кислота 409
Изофталеиая кислота 473
Изоцианоиая кислота 408
Имидазол 600, 6J8, 061
Имиды кислот 4/2
Иминогрунна 361
Иминокетон 327
Инверсия сахара 343
Индатрены 541 ел.
Ипдеп 660
Индиго 597 сл„ 599
Индоксил 598
Индол 594, fifil
u-Индилллуксусная кислота
(Гетероауксин) 596
Инициаторы полимеризации 181
Инкрустирующие вещества 354
Инозиты 55Я. 5г,9
Инсектициды 130, 174, 232, 440
Инсулин 394
Инулин 346
Иодаль 210
Иодбензол 439
Йодистые алкилы 106, 109
Полное число 261
Подол (Тетранодииррол) 586
Йодоформ 1 13. 209 ел.
[і Иодіфопнонопая кислота 281
м-Иодтилуол 506
Иониты (Ионообменные смолы)
49Я ел.
Ионные соединения 61
Ионон 212
Иприт (Горчичный газ, Дихлор-
диътилсульфид) 184
Кадаоернн (Пентаметилендиамин)
368. 381
Казеин 391
Какодиловочислый натрий 131
Ка.пьимйцианамид 407
Кальциферол (Витамин D2) 572
Камеди 354
Каменноугольная смола [9, 434, 528
Каменный уголь, сухая перегонка 434
Камфаи 564
Камфора 566
«искусственная» 566
Каин оль 566
Канцерогенные углеводороды 544
Каприловая кислота 224
Каприновая кислота 224
е-Капролактам 554
Капрон 397, 554
Капроновая кислота 224, 382
Каран 564
Карбазол 661
Карбаминовая кислота 413
Карбинол см. Метиловый спирт
Карбинольный клей 215
Карбоксил 197, 219, 228
Карбоксилаза 306
К а рбокси метил целлюлоза 352
Карболовая кислота см. Фенол
Карґюнизанмя 384
Карбонильная группа 185
Карбоичвые кислоты 197, 219
ароматические 470 ел.
двухосновные 270 ел., 277 ел.
непредельные 237 ел., 277 ел.
однооснокные 219 ел. 237 ел.
предельные 219 ел., 270 ел.
Карбофос 130
Карбсшпклическне соединения 43,
422 ел.
номенклатура 644
Каротин 568 ел.
Каритинонды 568 ел.
Катализ 15, 145 ел., 435
Катмопмты (катионообменные смолы)
5П0
Каучук 100 ел.
натуральный 99
синтетический 102 ел., 214,239,486
Качественный анализ 27 ел.
Квлрнетин (Пентаоксифлавон) 608
Кератин 391
Керосин 67
Кстали 204
Кетен 215
Кетогексозы 605
Кето-енольная таутомерия 311
Кетозы 316
Кетон Мнхлера 494 ел.
Кетонное расщепление 311
Кетонокислоты 305 ел.
Кетоны 143, 185 ел.
ароматические 469
Кетоспирты 316
Кефалины 373
Кислотное расщепление 312
Кислотный красный ализариновый
541
Кислотный синий антрахнноиовый
541
Предметный указатель
673
Классификация органических
соединений 42 ел.
Клетчатка см Целлюлоза
«Ключи» стереохимпческие 384
Ковалентная связь 61, 163
Коксопый газ 434
Коламин (Этаниламин) 370
Количественный анализ 2У ел.
Коллагены 391
Коллодий 350
Колликсилин 350
Конго красный 519
Коннин 613, 614
Контакт Петрова 259
Конфигурация цепи 161
Конфирмационные представления 241
Коньюгены 96, 247 ел.
Коричная кислота 484, 485
Коричный алкоголь 486, 487
Коричный альдегид 467, 486, 487
Кортизон 575
Кортикоидмые гормоны 575
Кофеин (Теин, 1, 3, 7-Триметилксан-
тии) 621
Красители 512 ел., 515, см. 519,
522 ел.
активные 624
антрахинопивые 54J
«белые» 625
бен.іиднішвьіе 519
индиго 5У7
классификация 516 ел.
кубовые 542, 599
ледяные 535
нафталнноные 535 ел.
номенклатура 516 ел.
полипиклические 541
природные 515
протравные 514 ел., 539
синтетические 515
субстантивные 514
трифенилметановые 522 ел.
Крахмал 15, 344 ел.
Крашение
кубовое 542, 599
ледяное (холодное) 535
Креатин и креаіинин 418
Крезолы 453. 463
Крекинг нефти 68
Кремневодороды (Снланы) 125
Кремнийорганическне соединения
125 ел.
Кристаллизация 24
Кристаллический фиолетовый 524
Кристаллоза 473
Кротил 6о8
Т88
Кротоновая кислота 239, 240, 241
Кротоновая конденсация 201
Кротоновий альдегид 201, 249
Ксантии 620 ел.
Ксантогеновая (дитиугольная)
кислота 418
Ксантогенаты (Ксантаты) 352, 418 ел.
Кгантопротеиновая реакции 388
Ксантофил 568
Ксилил 431
?>( + ) Ксилоза 330, 332
Ксилолы 430 ел.
Кубовый синий О 541
Кубовый ярко-зеленый С 542
Кумол (Изонропилбензол) 213, 453
Лавсан (Дакрон, Терилен) 475
Лактамы 376
Л.НК1ИДЫ 285
Лактоза (Молочный сахар) 341,
344 ел.
Лактоны 286
Латекс 99
Ла^риноная кислота 162, 163, 224, 225
Левулиновая кислота W5
Левулиновый альдегид 100
Лепкоиндию 599
Ленкооснования красителей 522
Лейцил 660
Лепнин 37S. 382
Лс-иитины 371
Лигнин 354, 462
Лизин (а, е-Днаминокапроновая
кислота) 380—382
Личол 45.)
Лпкиїїин 568
Лимонен 563
Лимонная кислота 301
Лимонлокислое бро/к.'Нне 338
Линолевая и линоленовая кислоты
251
Липаза 259
Лпгохолсвая кислота 572
Л^теолин 608
Лыоисит (Хлорвинилдихлорарсии) 95
Маіниііиодгіропил 122
Уінгнийорганические соединения 124
Мазут 67
Малахитовый зеленый 524
Малеипимид 585
Малеиновая кислота 277 ел.
Ma.ir-нновый ангидрид 278
Мялонил 659
Малоновая кислота 271, 273 ел.
Малоновый зфир 275
674
Предметный указатель
Мальтоза 340, 344
D-Маннит 334
Маннитан 324
D-Манноза 333
D-Манноновая кислота 330, 334
D-Манносахарная кислота 334
Маргарин 263
Л'іаргариновая кислота 224
Масла
антраценовое 435, 536
голландских химиков 74
горчичное 409
легкое 434
растительные 260
сивушные 22, 149
силиконовые 127
смазочные 67
соляровое 67
среднее (карболовое) 434
тяжелое (креозотовое) 434
Масляная (бутановая) кислота 224, 233
Маслянокислое брожение 338
Масляноэтиловый эфир 252, 257
Масляный альдегид 188
Медноаммиачное волокно 353
Мезовинная кислота 291, 293
А'іезоинозит 559
Мезоксалевая кислота 620
Меламин 407
Меласса 343
Ментан 561, 564
Ментол 562
Ментон 562
Меркаптаны (Тиоспирты) 182
Меркаптиды 182
Мерсеризация 349
Метадигалловая кислота 482
лега-Изомеры 429
Метакриловая кислота 239
Металлоорганические соединения
ПО, 121 ел.
Метальдегид 208
Метан 45, 47, 50, 53, 57
электронное строение 164
Метанол (Метиловый спирт) 144 ел.,
147
Метансульфокислота 183
Метгемоглобин 393
Метил 658
бромистый 109
йодистый 109
радикал 526
Метиламин 361, 363, 367
п-Метиламинофенол сернокислый
(Метол) 498
Метиларсин 130
2-Метилбутадиен-1,3 (Изопрен) 96
Метилбутанолы 153
2-Метилбутен-1 77
Метилбутилкетон 189
?-Метилвалериановая кислота 382
Метилглиоксаль 284, 306
Метилглюкозид 317
Метилен 72, 658
бромистый 111
йодистый 111, 112
Метиленнтан 37, 324
л-Метилизопропилбензол (Цимол)
430, 561
а-Метилнндол (Метилкетол) 594
fi-Мєтилиндол (Скатол) 594
Метилкаучук 102
Метнлмалоновая кислота 270
Метилмеркаптан 182
Метнлмышьяковая кислота 131
Метилнафталины 528
Метиловый спирт (Метанол) 144 ел.,
147
Метиловый оранжевый 510 ел.
2-Метилпиридин (а-Пиколин) 612
2-Метилпропандиол-1,2 (Изобутилен-
гликоль) 169
2 Метилпропачол-1 (Изобутиловый
спирт) 136, 137, 151
2-Метилпропен (Изобутилен) 75, 77,
89, 136, 137, 246
Метилпропилкетон 189
Метилсульфат 173
Метилуксусная (пропионовая)
кислота 224, 233, 382
Мет илфен ил кетон (Ацетофенон) 469
Метилфениловый эфир 463
а-Метилфуран 580
2-Метил-4-хлорфеноксиуксусная
кислота BМ-4Х) 454
Метилхолантрен 573
Метилэтилен 74
Метилатилкетон 189
Метилэтилмалоновая кислота 274
Метилэтилуксусная кислота 274, 276
Метокси 659
4-Метокси-2,6-диметилпиридин 607
Метол (rt-Метиламинофенол
сернокислый) 498
Миндальная кислота 484
Мипора 421
Миристиновая кислота 224
Михлера кетон 494 ел.
Молекулярная перегонка 22 '
Молекулярный вес, определение 32
Молочная (оксипропионовая)
кислота 238, 282, 286
Предметный указатель
675
Молочнокислое брожение 287, 338
Молочный сахар (Лактоза) 341,
344 ел.
Моносахариды 315 ел.
Мочевая кислота 619
Мочевина 16, 413 ел., 619
Моющие вещества 234 ел., 236
неионогенные 171
Муравьиная кислота 224, 229
Муравьиноэтиловый эфир 256
Муравьиный альдегид
(Формальдегид) 188, 204 ел.
Мурексид 619
Мутаротация 322, 332
.Мыла 234 ел.
Мыловарение 259
Мылонафт 550
Мышьякорганические соединения 128
Найлон (Анид) 397, 555
Наполнители 117
Нафталин 527 ел.
строение 534
Нафталинсульфокислоты 530, 533
Нафтацен 660
Нафтеновые кислоты 550
Нафтены, см. Циклопарафины
Нафтил 659
Нафтиламины 532
Нафтионовая кислота 532
Нафтолы 531, 533
Нафтолдисульфокислоты 531
Нафтохиноны 533
Небензоидные ароматические
соединения 553
Нейрин 372
Нефть 65
ароматизация 435
Нефункциональные заместители 637,
643
Никотин 613
Никотиновая (пирндинкарбоновая-3)
кислота 612, 613
амид 613
Нитратор 447
Нитраты целлюлозы 350 ел.
Нитрилы 402
Нитроанилины 492
¦•2-Нитроацетанилид 492
Нитробензол 442, 446, 447, 50?
Нитрование 350, 440
Ннтрогликоли 176
Нитроглицерин 175
Нитрозамины 364
Нитрозобензол 508
я-Нитрозоднметиланилчн 494
Нитролаки 351
Нитромочевина 416
Нитрон 239
а-Нитронафталкн 531
Иитропарафины 357
Нитросоединения 356, 440
ароматического ряда 440 ел.
жирного ряда 356 ел.
Нитротолуолы 441, 446
Нитрофенолы 447, 506
Нитроцрллюлоза см. Нитраты
целлюлозы
Новокаин 502
Новолаки 455
Номенклатура 59 ел , 633 ел.
«а»-система 651
альдегидов 186
ароматических соединений 645
Байера 647. 650
галоидпроиэводных 60
гетероииклов 577, 650 ел.
гликолей 168
диенов 95
женевская 59, 75, 635
карбоновых кислот 221, 280
кетонов 186
красителей 516 ел.
л ье жск а я 633
оксикислот 282
олефинов 74
Паттерсона 648
рациональная 59, 634
систематическая 654 ел.,
спиртов 135
стереоизомеров 160, 320, 381
углеводов 316
углеводородов 46. 59, 636
— с двойной связью 74
— с тройной связью 90
ЮПАК 646
зфиров 177, 252
Нонадекан 638
Нонан 45, 53, 638
Ноннловый альдегид 188
Нуклеиновые кислоты 626 ел.
Нуклеопротеиды 392, 626
Озазомы 326 ел.
Озокерит (Горный воск) 71
Озонилы 80. 100, 246
Озонирование 80
Озопы 329
Окись этилена 170, 238
Окса.юл 000, 651
Оксазолидин 651
676
Предметный указатель
Оксалил 659
Оксаіиин и оксатиан 651
Океафосфюл и оксафосфолан 651
Оксиалобснзол 509
Оксиа н>соединения 509 ел.
Оіииіемоїлобин 392
Оксигидро.хннон (І, 2, 4-Триоксибен-
зол) 461
«Оксиизомасляная кислота 283
Оксикислоти 228
аромаїические 479 ел.
жирные 282 ел,, 285 ел,, 302 ел.
Оксимасляпая кислота (у Бутиролак-
тон, у-Лакюн) 282, 286
Оксимы 194, 203
Оксинитрилы (Циангидрины) 193,
238, 281
Оксипиридины 612
Оксипромионовая (молочная)
кислот а 238, 282, 286
Окситоиин 344
Оксиуксуснан кислота 282
8 Оксилинолин (Оксин) 016
Океице/ілюлоза 349
Оксониевые соединения 179, 607
Оксоеингез 15|
Океосоединения 185
Октадскан 638
Октадсцилен (Октадецен-1) 77
Окіамеї ил I3U
Октан 45. 53
Октаноьое число 83, 436
Олтекю правило 62
Олеиновая кислота 242, 249, 250
О.К'фииы 72 ел.
О.іиі оса.хариды 338 ел.
Олифа 262
Омыление
жиров 258
сложных іфиров 174, 254
Ононые кислоты 326
Оптическая активность 154 ел.
Оптические антиподы 158, 2Я8 ел.,
2%, 300, 377
Оптические отбеливатели 625 ел.
Органическая химия 15 ел 17
Органические соединения 15, 17
выделение и очистка 20
Органическое стекло 240
Органогены 27
Ординарные связи 1U2, 244
Орлон 239
Орииткн )80 ел.
Орто-и«)меры 429
Ортсжремиевые эфиры 176
Оссеин 391
Осцилляциокная гипотеза 427
Палудрин (Бигумаль) 618
Пальмитиновая кислої а 224, 234
Паральдегид 207 ел.
Парааминофенол (л Аминофенол
солянокислый) 498
Пара-изомеры 429
Ппракрасный 535
Парарозанилин (Триамипотрифенил-
карбинол) 522, 525
Парафин 67
Парафины 45 ел.
Параформальдегід 206
Псірафуксин 523, 525
Парциальные валентности 97
ПАСК (я-Аминосалицпловая
кислота) 502
Пектиновые вещества 354
Пеларгониаин 6С9
Пеларгоновая кислота 224, 250
Пеницилламин (u-Амино ? тиоичова-
лериановая кислота, диметил-
цистеин) 602
Пенициллин Ь01
Пеніадекан 53
Пентадекановая кислота 224
Пиніаксштан 638
Пснгалеи ouO
Пентаметилем (Циклопентан) 545,
547. 549
Пентамеї млендиамин (Кадаверин)
368. 381
Пентан 45, 50, 53
Пентаиолы 153
Гіентаоксифлавоп (Кверцетин) 6U8
Пентен-1 (Пропилэтилен) 77
П^нтит 172
Пентозани 331
Пентозы 316. 330 ел,
Пептидные связи 394
Перегонка 20 ел.
Перекиси органические 180 ел.
Перенос реакционного центра 310,
312
Переэтерификапия 255
Перлон 39?
Перфторгексадекан 114
Перфторметан 113
Перфторэтап 114
Петролейний эфир 67
а Пиколин B-Метилгіііридин) 612
Пнколиновая кислота 612
Пикраты 457
Предметный указатель
677
Пикриновая кислота B,4,6-1 риинтро-
фенол) 457
Пилокарпин OU3
Пиыелинивая кислота 271
Пинан 5O4
а-Пішен 565
Пиперидин fill, 613 ел.
Пиразин 661
Пиразол oou
Пиразолон Ь02
Пирамидон 6U2 ел.
Пирам 605
Пнрамозы 605
Пирен 543
Пиридпл 662
Пиридин (I0 ел.
сульфотриоксид 580, 595, 611
Пиридиикгірбоновая-3 (никотиновая)
кимита 612, 613
Пирндинсульфоновая-3 кислота 611
Пиридоны 612
Пиримидин 618, 661
Пировнноградиая кислота 284, 305,
3U6
Пирогаллол A,2,3-Триоксибензол)
¦ 461. 462, 498
Пирокатехин (о-Диоксибензол) 4э8,
461
Пироксилин 350
Пироксония соли 607
Пиролиз 435
у-Пирон 605
Пирослизекая кислота 580, 582
Пиррил 662
Пиррол 579. 585. 661
а-Пирролальдегид 586
Пнрролі-дни (Тетрагидропиррол) 587
Пиррол и дин 588
Пирршшн 58?
а-Пнрролкарбоновая кислота 586
о-Пирролсульфоновая кислота 586
Плазмоцид 617
Пластификаторы 117. 474
Пластические массы 115 ел., 251, 420,
486
Платформинг 436
Плексиглас 240
Плодовый сахар (Фруктоза) 325, 335
Поворотные изомеры 241
Показатель преломления 84
Полиакриловая кислота 238
Полиакрилаты 238, 251
Поливпниланетат I 18. 167
Полпвииилбутираль 167
Поливиниловый спирт 167
Поливинилпирролидон 588
Поливинилхлорид 116, 117
Полигалактуроновая кислота 354
Полпизобутилен 90
Поликонденсация 116, 397
Полимер 82
Полимеризация 82, 88, 98, 116 194
433
Полиметиленовые углеводороды, см.
Циклопарафпмы
Полиметилметакрилат 251
Полпнуклеотиды 626
Полнокспметилсн 206
Полипептиды 394
Полисачариды 338 ел.
Полисилоксаны (Силиконы) 126
Полистирол 486
Политетрафторэтилен 119
Полиформальдегид 206
Полиэтилен 88
Полиэтилентерефталат 475
Полиэфирные смолы 476
Поляриметр 156
Порох бездымный 350
Правила
Марковникова 78
октетов 62
Попова 199
Тиле 97
Эльтекова 166
Предельные углеводороды 45 ел., 636
Преднизолон и преднизон 575
Пробкооая кислота 271
Прогестерон 574
Проекционные формулы 301
Проламнны 390
Пролин S87
Промтозил (Стрептоцид) 511, 512
Пропан 45, 47, 49. 51 ел.
Пропандиол-1.3 169
Пропановая кислота 223
Проланои-2, см. Ацетон
Пропяргил 658
Пропен 75, 77
Пропеналь (Акролеин) 210, 237
Пропеиил 658
Пропил 658
бромистый 109
йодистый 109
н-Пропиламии 363
Пропилен 76. 77
Пропнленгликоль 168, 283
Пропиленхлоргидрпн 80
Пропилиден 658
Проппловые спирты 150
Промилчтилеи (Пентен-1) 77
Пропионпл 269, 659
678
Предметный указатель
Пропионовая (метилуксусная)
кислота 224, 233. 382
Пропионовый альдегид 188
Промокси 659
Протамины 387, 390
Протеиды 389, 391
Протеины 389
Протравы 514 ел.
Проявители 497, 498
Псевдооснования 523
Псевдокислоты 360
Птиалин 345
Птомаины 368, 381
Пурин 618. 661
Пурпур античный 600
Пурпуровая кислота 619
Путресцин (Тетраметилендиамнн)
368, 381
Радикалы 47, 525, 647 ел.
свободные 525 ел., 527
Радикальные реакции 54, 527
Рацемические смеси 159, 294 ел.
Рвотный камень 293
Реактив Гриньяра 110
Реакции
азосочетания 510
биуретовая 388
бромирования 228
Вагнера 82, 84
Вильямсона 177
восстановления 256, 326, 367, 403,
506, 507
Вышнеградского 613
Вюрца 51, НО
і'алоиднрования 54, 74, 78, 91,
228, 324, 437
Гаттермана 465
гидрирования 78, 91, 92, 424
гидролиза 126 ел., 135, 141, 202,
254, 339, 347
Гофмана 365, 367, 501
Густавсона 546
Реакции
дегидрирования 142, 187, 436
Зайцева 193
замещения 58
изомеризации 106
Кяпниццаро 467
Канниццаро—Тищенко 582
Кнжнера 194
Коновалова 358
ксантопротеииовая 388
Кучерова 92
мурексидная 619
Реакции
нитрования 56, 440
озонирования 80
окисления 56, 81, 92, 143, 187,
196 ел., 221, 229, 283, 326
омыления 174, 254, 258, 311
Орлова 69
Перкина 485
поликонденсации 116
полимеризации 82, 93, 98, 116,
194, 433
радикальные 54, 527
Реймара—Тимана 481
Сабатье 76
самоокисления 466
сульфирования 55, 79, 448
сульфохлорирования 56, 183
Тищенко 197
хлорирования 228
Чичибабина 611, 616
Чугаева 218
этерификации 141, 173, 252
Юрьева 579, 584
Регуляторы роста растений 453 ел.
Резерпин 595
Резина 99
Резиты и резолы 455
Резорцин (лі-Диоксибензол) 458, 463
Рекомбинация 54
Репелленты 471, 474
Рефракция 84, 86, 87
О(—) Рибоза 330, 332, 626
Рибонуклеиновая кислота (РНК)
626 ел.
Р-кислота B-Нафтол-3,6-дисульфо-
кислота) 531
Риформинг 69
РНК (Рибонуклеиновая кислота)
626 ел.
Роданистоводородная (тиоциановая)
кислота 408 ел.
Розанилин 523
«Po-сигма» система 160, 322, 386
Ростовые вещества 597
Ртутьорганическне соединения 506
Салигенин (Салициловый спирт) 481
Салициловая кислота 479 ел.
Салициловый альдегид 481
Салициловый спирт (Салпгенин) 481
Салицин 481
Салол 480 ел.
Саломас 263
Саркозин 377
Сахараты 343
Предметный указатель
679
Сахарин 472
D-Сахарная кислота 333
Сахароза 340, 342 ел. с^Хр
Свинцовый сахар и свинцовый уксус
232
Свободные радикалы 47, 54, 525 ел.,
527
Связи
гетерополярная и гомеополярная
61
двойная 72
ионные 61
ковалентная 61
координационная 218, 375, 589
пептидные 394
семиполярные 357
сопряженные (коньюгированные)
96, 247 ел.
Себациновая кислота 271
Сегнетова соль 293
Семикарбазид 416
Семикарбазоны 416, 583
Сера, определение 28, 30
Серил 660
Серии 381, 383
Серный (этиловый эфир) 179
Сероуглерод 418
Сивушные масла 22, 149
Сиккативы 262
Силанолы 126
Силанохлороформ 125
Силаны (Кремпеводороды) |25
Силиконы (Полисилоксаны) 126
Сильван (<і Метилфуран) 580
Симазин 624
Синигрин 410
Синильная кислота 405
Синтез
асимметрический 297
Вильямсона 177
Вюрца 51, ПО
диеновый 548
при помищи . малонового зфира
275 ел.
Скраупа 614
Фиттига 432
Фриделя—Крафтса 432, 469
Синтез-газ 70
Синтетический каучук 102 ел., 214,
239. 486
Синтетическое волокно 397 ел.
Синтин (Синтетический бензин} 70
Скатол (?-Метилиндол) 594
Скелетный никелевым катализатор
256
Скипидар 566
Склеропротеины 391
Слизевая кислота 334
Смазочные масла 67
Солидол 237
Соляровое масло 67
Сопряженные двойные связи 96, 243
D-Сорбит 333
Спермацет 257
Спермин 369
Спираны 650
Спиртовое брожение 148, 336
Спирты 132 ел., 168 ел., 284
высшие 172, 284
двухатомные 168 ел.
непредельные 165 ел.
одноатомные 132 ел., |65 ел.
трехатомные 171 ел.
Стабилизаторы 117
Стеарин 234
Стеариновая кислота 224, 234, 259
Стеклопластики 476
Стереоизомерия 160, 240, ел., 279 ел.,
302 сл„ 319 ел., 384 ел., 557 ел.
Стерины 571
Стероидные гормоны 574
Стероиды 572
Стирил 659
Стирол 486
Стрептоцид
белый 512
красный 511 ел.
Сукцинил 659
Сульфамидные препараты 491, 512
Сульфаниловая кислота (я-Амнпо-
бензолсульфокислота) 490
Сульфатиазол 600
Сульфидин 612
Сульонды (Тиоэфиры) 183
Сульоирование 55, 79, 448
Сульфитный щелок 347
о-Сульфобензоиная кислота 472
Сульфокислоты 183
ароматические 448 ел.
Сульфонаты 183, 236
Сульфохлориды (Хлорангидрнды
сульфокислот) 183
Сульфохлорированне 56, 183
Сульфуратор 448
Сухая перегонка каменного угля 434
«Сэндвичевые» соединения 554
Табун 130
Таннин 483
Тартраты 292
Таурин 381, 382
680
Предметный указатель
Таурохолевая кислота 382
Таутомерия 310, 408
Теин, см. Кофеин
Теломеризацпя 89
Теобромин C,7-Диметилксантин) 621
Теория
Бутлерова 16, 37 ел.
Косселя 61
Льюиса—Лаигмюра 163
напряжения 243, 555 ел.
обраюнания нефти 66
радикалоч 32
стереохимическая 157 ел.
Тнле 96
тииив ,і2 сл.
унитарная 33
химического строения 37 сл.
хромофорная 513
Терефталенач кислота 473
Терпентин (Живица) 566
Терпены 560 сл
Терилен (Дакрон, Лавсан) 475
Термин 562
а-Терпинеол 563
Терпинолен 563
Тестостерон 574
1,2,3,4-Тетрабромбутан 97
1,1,2,2-Тетраоромэган (Четырехбро
мистый ацетилен) 92
Тетрагидропиран 605
Тетрагидропиррол (Пирролидин) 587
Тетрадекач 638
Тетраиодпиррол (Иодол) 586
Тетраконтан 638
Тетралин 536
Тетраметиламмоний йодистый 366
Тетраметнлен (Циклобуїан) 545, 549
Тетраметиленлиамин (Путресцин)
368, 381
Тетраметилметан 49, 50, 53
Тетраметилсвинеи 526
Тетрафенилметан 520
Тетрафторэтилен 119
Теїрахлорэтан 95
Тетраэтилгнинец 123, 527
Тетрил 495
Тетрозы 316
Тефлон I19
Тиазол 600
Тиенил 662
Тимин 627
Тиокол 459
Тиомоченина 413, 419 сл.
Тиоспирты (Меркаптаны) 182
Тноугольная кислота 418
Тиофен 579, 583
Тиофос 130
Тиоциановая (роданистоводородная)
кислота 408
Тиоэфиры (Сульфиды) 183
Тирозин 503
Тироксин 503
Тол, см Тринитротолуол
Толил 431, 659
Толуидины 487, 497
Толуил 659
Толуи.пен 659
о-Толуиловая кислота 506
Толуол 399, 430. 431, 520
Толуолсульфокислоты 449
Триазин 623
Триакоитан 638
Триаминотрифенилкарбинол f Пара-
розанилин) 522. 525
2,4,6-Триброманитин 490
Три^ромбензол 430
Трибромфенол 431
Тридекан 638
Тридекановая кислота 224
Трнметиламин 361, 363, 367
Т[шметилалюминий 125
Триметилен (Циклопропан) 545, 549
Триметиленгликоль 168
Триметилкарбинол 134
1,3,7-Тримети'іксантин (Кофеин,
Теин) 621
2,2,4-Триметилпентан (Июоктан) 83
2,2.4Триметитпентен-3 82
Триметилуксусный альдегид 189
2,4,6-Тринитротолуол (Тол, Тротил)
446. 448
2,4,6-Тринитрофенол (Пикриновая
кислота) 45/
Триозы 316, 329
1 2 3-Триоксибензэл (Пирогаллол)
461, 462, 498
1,2,4-Триоксибензол (Оксигидрохи-
нои) 461
І 3 5-Триоксиоензол (Флороглюцин)
332. 461, 462
Триолеин 258, 260
Триплекс 351
Трипропиламин 353
Триптофан [а-Амичо- fP-индолип-З)-
пропионовая кислота] о9Ь
Трисилан 125
Тристеарнн 258, 2Р0
Трифениламин 493, '97
Трифенилкарбинол 521
Трифенилметан 520
Трифенипметил 525
Трифенилхлпрметч» 521
Трифтор\лораіилен 119
Трихлорсилан 125
Предметный указатель
681
Трихлоруксусная кислота 281
2,4,5-Трихлорфенокси уксусная
кислота B,4.5-1 ) 454
2,2,2 Трихлорэганол (Хлораль) 209
Тричтиламин 361, 363
Трихлорчтилен 95
Триэтилалюминий 125
Триэгилпеллюлоза 352
Тростниковый сахар см. Сахароза
Тротил см. Тринитротолуол
Углеводороды
ароматические 19, 422 ел.
ацетиленовые 90 ел.
непредельные 71 ел.
предельные 45 ел.
циклические 545 ел.
Углеводы 314 ел.
Уголь каменный 434
Угольная кислота, амид 413
Уксусная кислота 219, 221, 230 ел.,
382
Уксусноизоамиловый эфир (Изоамил-
апегат) 257
Уксусноэл иловый эфир см. Этилаце-
тат
Уксусным альдегид см. Ацетальде-
гид
Уксусный ангидрид 267
Ундекан 638
Ундекановая кислота 224
Урацил 627
Уреаза 415
Уретаны 414
Уротропин 205, 457
Фелингова жидкость 293
Феназин 661
Фенантрен 542 ел., 660
Фенил 431, 659
Феиилаланин 503
Фенилбензоат 471
Фенмлінлразин 195, 203. 507
Фенилгидразоны 195, 203
Фенилгмдроксиламин 498, 507 ел.
Фенилдмэзопий 503
Фенмлен 431, 659
Фенилендиамины 495, 497
|. Фенил -Зметилпиразолон 602
Фени питрометан 359
Фенмлочазоны 326
Ч Фени іпропиоионая (гидрокорич
ная) кислота 484
Фенилум-Усная кислота 484
2 Фени.іхромон (Флавон) 608
Фенокси\кс\г'ная кислота 454
Фенол (Кчроолпвая кислота) 450,
452, 453, 463
Фенолфталеин 477 ел.
Фенолы 450 ел.
двухатомные 458
трехатомные 461 ел.
Феноляты 451
Фенопласты 457
Ферменты, см. Энзимы
Ферроцен (Дипиклопентадиепил-
железо) 552 ел,
Фибрин 390
Фиброин 391
Фильтрование 24
Фитин 559
Флаион B-Фенилхромон) 60fi
Флороглюцин A,3,5-ірииксибензол)
332, 461, 462
Флотация 419
Флуорен 660
Флуореспин 478
Формалин 206
Формальдегид (Муравьиный
альдегид) 188, 204 ел.
Формамид 267
Формиаты 229
Фор мил 269, 659
Фосген 412
Фосфиновые кислоты 128, 129
Фпсфины 128, 129
Фосфопротеиаы 391
Фосфорные эфмры 174
Фосфорорганнческме соединения 128
Фотопроявнтели 498
Фреоны 114
Фруктоза (Плодовый сахар) 324, 335
Фіаламинопая кислота 501
Фталевые кислоты 473 ел., 529
Фталеыый ангидрид 473, 474, 530
Фталеины 477
Фталил 659
Фталимид 474
Фторопласлы 119, 120
Фторуглероды 114
Фуксин 523, 524 ел.
Фу.мьвены 552
Фумаровая кислола 277
Функциональные группы 19, 42, 634,
635
Фуран 579. 580, 661
Фуранидин 581, 58?
Фурапилин 583
Фу рил 66^
фуриловый спирт 582
Фурфурил 662
фурфурол 331, 582
Хаульмугровая кислота 551
Хелатные соединения 217 ел.
682
Предметный указатель
Хелидоновая кислота 606
Химическое строение 41
Хингидрон 461
Хинин 616 ел.
Хиноидная группировка 460
Хинолин 614 ел.
Хинолиновая кислота 615
Хинондиоксим 460
Хиноны 460 ел., 533
Хлораль B,2,2-Трихлорзтаналь) 209
Хлоралыидрат 209
Хлорангидриды
карбоновых кислот 264, 268 269,
411
сульфокислот (Сульфохлориды)
183
Хлорацетофенон 469
Хлорбензол 439, 440
2-Хлорбутадиен-1,3 (хлоропрен) 103
Хлорвинилдихлорарсин (Льюисит) 95
Хлорин 118
Хлористые алкилы 109—111
Хлористый
аллил 114, 115
ацетил 229, 264
бензил 438
бензилиден 439
бензоил 471
винил 114, 115, 117
випилиден 118
метилен 55, 111
этилиден 112
Хлормасляная кислота 280
Хлормуравьимая (хлоругольная)
кислота 411
Хлорнафталины 533
Хлоропрен B Хлорбута.аиен-1,3) 103
Хлорофилл 588, 589
Хлороформ 55, 113, 209 ел.
Хлорпикрин 458
Хлорпропилен 114
Хлорпропионовые кислоты 280
Хлорсилан 125
Хлортолуолы 438
Хлоругольная (хлормуравьиная)
кислота 411
трихлорметиловый эфир
(Дифосген) 412
Хлоруксусные кислоты 280, 281
2-Хлорфуран 581
Хлоряблочная кислота 299
Холановая кислота 573
Холевая кислота 572
Холеиновые кислоты 573
Холестерин 571
Холин 371
Хризен 543
Хризин (Диоксифлавон) 608
Хризоидин B,4-Диаминоазобензол)
509
Хроматография 590 ел., 608
Хромен 661
Хромон (Бензопирон) 608
Хромопротеиды 392
Хромофоры 513
Целлобиоза 342, 344
Целлозольвы 171
Целлофан 353
Целлулоид 351
Целлюлоза (Клетчатка) 346 ел.
Церезин 71
Циан 406
Цианамид 406 ел.
Циангидрины см. Оксинитрилы
Цианидин 609
Цианистые соединения 401 ел.
Циановая кислота 407 ел.
Циановокислый аммоний 16
Циануксусная кислота 273
Циаиуровая кислота 623
Цианурхлорид 623
Циклизация каталитическая 436
Циклобутан (Тетраметилен) 545, 549
Циклогексан (Гекса метилен) 545,
54o, 549, 554
Циклогексанол 554
Циклогекснл 659
Циклогептан (Гептаметилен)
549
545,
Циклооктан 549
Циклопарафины (Нафтены) 545
стереоизомерия 557 ел.
Цнклопентадиен 551
Циклопентадиенилы металлов 552 ел.
Циклопентан (Пентаметилен) 545,
547, 549
Циклопентапон 547
Циклопентанофенантрен 571, 572
Циклопентен 550
Циклопентил 659
Циклопентиды 394
Циклопропан (Триметилен) 545, 549
Циклопропанкарбоновые кислоты 557
Цимол (Метилизопропилбензол) 430,
431, 561
Цинкорганические соединения 122,
123 ел.
Предметный указатель
683
Цистеин її цистин 383
//ис-тринс-изомерия 240 ел., 242, 279,
557 ел.
Цитозин 627
Цитраль 212
Черный анилин 490
Четыреххлористый
кремний 125
углерод 55, 113
Щавелевая (этандиовая) кислота
271, 272, 284
Щелок сульфитный 347
Щелочная целлюлоза (Алкалицеллю-
лоза) 349
Эбонит 99
Эйкоэан (Эйкосан) 53, 638
Экзальтация 99
Экстракция 22
Элаидиновая кислота 242, 250
Эластин 391
Электронное строение
бензола 428
двойных связей (я-связей) 243 ел.
коньюгированных связен 247, 248
органических соединений 61
ординарных связей (а-связей) 162
Электрофилыюе замещение 444
Элементарный анализ 29 ел.
Элементорганнческие соединения 27,
121 ел.
Энант 398
Энантноморфные кристаллы 154, 296
Энантовая кислота 224
Энзимы (Ферменты) 146, 149
аргиназа 417
диастаза 345
зимаза 149
карбоксилаза 306
липаза 259
мальтаза 336, 339, 342
птиалин 345
уреаза 415
<н^льсин 336, 339, 342
Энпл см Енол
Эозин 479
Эпимеры 328
Эргостерин 572
Эритрит 172
Эстрон 574
Этан 45, 47, 49, 53
электронное строение 165
Этандиовая (щавелевая) кислота
271, 272, 284
Этанол см. Этиловый спирт
Этаноламин (Коламин) 370
Этен 75
Этернфикация 141, 173, 252
Этил 658
бромистый 109
йодистый 109
радикал 527
Этнламнн 361, 363
Этилацетат (Уксусноэтиловый эфир!
229, 252, 256
Этилбензол 431, 486
Этилен 72, 77, 87, 245
окись 170
Этиленгликоль 72, 81, 170
Этилендиамин 368
Этиленхлоргидрин 170, 238
Этиленииангидрнн 238
Эгилнден III, 658
Этилмалоновая кислота 270, 276
Этилмеркаптан 182
Этилнитрат и этнлнитрнт 173
Этиловый спирт 148, 149
Этиловый эфир 179 ел.
Этилсерная кислота 79, 178
Этилэтилен (Бутен-1) 74, 77
Этинил 214, 658
Этокси 659
Эфиры
ортокремневой кислоты 176 ел,
простые 177 ел.
сложные 173 ел.
смешанные 177
угольной кислоты 413
целлюлозы 349
Яблочная кислота 290
Янтарная кислота 271, 277, 382
Янтарный ангидрид 277
Ярь-медянка 232
Рекомендуемые книги
по общим и теоретическим вопросам органической химии,
методам органического синтеза и лабораторной технике
Неницеску К. Д., Органическая химия, перевод с
румынского, Издатинлит, том I, 1962; том II, 1963.
Кар pep П., К>рс органической химии, перевод с немецкого,
Госхимиздаг, 1962.
Физер Л., Физер М., Органическая химия, перевод с
английского, Издатинлит, 1949.
Крам Д., Хэммонд Дж., Органическая химия, перевод о
английского, Издатинлит, 1963.
Сер рей А., Справочник по органическим реакциям. Именные
реакции в органической химии, перевод с английского,
Госхимиздат, 1962.
Терентьев А. П., К ост А. Н., Потапов В. М, Ц у-
керман А. И., Номенклатура органических соединений,
Изд. АН СССР, 1955.
Ар бузо в А. Е., Краткий очерк развития органической химии
в России, Изд. АН СССР, 1948.
Быков Г. В., История классической теории химического
строения, Изд. АН СССР, 1960.
Жданов Ю. А., Очерки методологии органической химии,
«Высшая школа», I960.
Тодд А. (ред.), Перспективы развития органической химии,
перевод с английского, Издатинлит, 1959.
Органические реакции (Organic reactions), перевод с англий«
ского, Издатинлит, сб. 1, 1948; сб. 2, 1950; сб. 3, 1951; сб. 4,
•1951; сб. 5, 1951; сб. 6, 1953; сб. 7, 1956; сб. 8. 1956; сб. 9,
1959; сб. 1С, 1963.
Реакции и методы исследования органических соединений
(РИМИОС), Госхимиздат, кн. 1, 1951; кн. 2, 1952; кн. 3,
1954; кн. 4, 1956; кн. 5, 1957; кн. 6, 1957; кн. 7, 1958: кн 8,
1959; кн. 9, 1959; кн. 10, 1961; к-н. 11, 1962; кн, 12,
1963; кн. 13, 1964; кн. 14, 1964.
Рекомендуемые книги 6?5
Кочетков Н. К-, Торгов И. В., Ботвини к М. М.,
Химия природных соединений, Изд. АН СССР, 1961.
Вацулик П., Химия мономеров, том I, Издатинлит, I960.
Катрицкий А., Лаговская Дж., Химия
гетероциклических соединений, перевод с английского, Издатинлит, 1963.
Рох о в Ю., X е р д Д., Льюис Р., Химия металлоорі анпческих
соединений, перевод с английского, Издатинлит, 1963.
Гинсбург Д. (ред.), Небензоидные ароматические
соединения, перевод с английского, Издатинлит, 1963.
Реутов О. А., Теоретические основы органической химии, Изд.
ЛІГУ, 1964.
Тем ни ков а Т. И., Курс теоретических основ органической
химии, 2-е изд., Госхимиздат, 19G2.
X юкке ль В., Теоретические основы органической химии,
перевод с немецкого, Издатинлит, том I, 1955; том II, 19)8.
Мюллер Е., Новые воззрения в органической химии,
перевод с немецкого, Издатинлит, I960.
Теоретическая органическая химия, Доклады, прочитанные на
симпозиуме по теории органической химии памяти Кекуле
в Лондоне в 1958 г., Издатинлит, 1963.
И н гольд К. К., Механизм реакций и строение органических
соединений, перевод с английского, Издатинлит, 195У.
Матье Ж., Алле А., Принципы органического синтеза.
Введение в изучение механизма органических реакций,
перевод с французского, Издатинлит, 1962.
Чубар Б., Механизмы органических реакций, перевод с
французского, Издатинлит, 1963.
Ньюмен М. С. (ред.), Пространственные эффекты в
органической химии, перевод с английского, Издатинлит, I960.
Терентьев А. П., Потапов В. М., Основы стереохимии,
изд-во «Химия», 1964.
Клайн (ред.), Успехи стереохимии, перевод с английского,
Госхимиздат. 1961.
Стереохимия производных циклогексана, Сборник статей,
перевод с английского и французского, Издагинлнт, 1958.
Препаративная органическая химия, перевод с польского,
изл во «Химия», 1964.
Шенберг А., Препаративная органическая фотохимия,
перевод с немецкого, Издатинлит, 1962
Вейганд К., Методы эксперимента в органической химии,
перевод с английского, Издатинлит, ч. 1, 1952, ч. II 1952-
ч. III. 1951.
Линстед Р., Элвидж Дж., Вол л и И. М., Вилки н-
сон Дж., Современные методы исследования з
органической химии, перевод с английского, Издагинлит. 1959.
686 Рекомендуемые книги
Современные методы эксперимента в органической химии,
перевод с английского, Госхимиздат, 1960.
Берлин А. Я., Техника лабораторной работы в органической
химии, Госхимиздат, 1963.
Вайсбергер А. (ред.), Физические методы в органической
химии, Издатинлит, том I, 1950; том П, 1952; том 111,
1954; том IV, 1955, том V, 1957.
Черонис Н., Микро- и полумикрометоды органической
химии, перевод с английского, Издатинлит, 1960.
Губен-Вейль, Методы органической химии, том П, Методы
анализа, перевод с немецкого, Госхимиздат, 1963.
Коршун М. О., Гельман Н. Э., Новые методы
элементарного микроанализа, Госхимиздат, 1949.
Файгль Ф., Капельный анализ органических веществ,
перевод с английского, Госхимиздат, 1962.
Роберте Дж., Ядерный магнитный резонанс. Применение в
органической химии, перевод с английского, Издатинлит,
1961.
Роберте Дж., Введение в анализ спектров ЯМР высокого
разрешения (спин-спиновое взаимодействие), перевод с
английского, Издатинлит, 1963.
Роберте Дж., Расчеты по методам молекулярных орбит,
перевод с английского, Издатинлит, 1963.
Хай с И. М., Маце к К. (ред.), Хроматография на бумаге,
перевод с чешского, Издатинлит, 1962.
Томпсон Г. У. (ред.), Успехи спектроскопии, перевод с
английского, Издатинлит, 1963.
Уолдрон Дж. (ред.), Успехи масс-спектрометрии, перевод с
английского, Издатинлит, 1963.
Беллами Л., Инфракрасные спектры сложных молекул,
перевод с английского, Издатинлит, 1963.
Джерасси К., Дисперсия оптического вращения.
Применение в органической химии, перевод с английского,
Издатинлит, 19C2.
Борис Алексеевич Павлов,
Александр Петрович Терентиев
Курс органической химии
А\., Издательство «Химия», 1965 р
G86 с. УДК 547@75.3)
Редактор А. И. Зицер
Техн редактор В. В Коган
Т 01505 Подписано к печати 2/VII 1965 г.
Гпм.га 60x901/18=21,5 бум. л,—43 печ. л. Уч.-изд. л. 40,14
Заказ 788 Цепа [ р. 11 к. Тираж 105 000 экз.
(Третий эппод 700О1 —І05 0О0 экз.)
Московская типография № 2! «Гла'.полиграфпрома»
Государстпе^'ного комтітетя Сонета Министров .-ССР
по псптл. Москва, R8, Угисшская ул., [2.
ИЗДАТЕЛЬСТВО «ХИМИЯ;
ГОТОВЯТСЯ К ПЕЧАТИ
ЖИРЯКОВ В. Г. Органическая химия. Изд. 2-е, пер. и доп.
Изд-во «Химия», 25 л., 50000 экз., цена 90 к.
Книга представляет собой второе издание краткого руководства по
оргзнической химии В ней на современном уровне изложены в
сжатой и доходчивой ферме основные теоретические положения и
фактический материал курса органической химии В книгу включены
специальные разделы, посияшеиные промышленности основного
органического синтеза, высокомолекулярным соединениям, пластмассам,
средствам защиты растений, синтетическим волокнам и каучукам,
химическим основам процессов жизнедеятельности и др.
Книга рассчитана на широкий круг лиц. желающих самостоятельно
ознакомиться с основами органической химии. Она может быть
использована также учащимися техникумов и студентами-заочниками
нехимических высших учебных заведений.
ФИЗЕР Л.. ФИЗЕР М. Органическая химия. Углубленный
курс (США, 1961). Перевод с английского мод ред. докт.
хим. наук Вульфсона И. С. Изд-во «Химия», ПО л.,
15 000 * кз.
Том 1, 55 л., цена 3 р. 50 к. в пер.
Том К, 55 л., цена 3 р. 50 к. в пер.
Ноиая книга хорошо известных советскому читателю американских
химиков, супругов Л и М. Фщрр. представляет собой систематиче
ское ученное и справочное руководство по органической химии. Е ней
оспешены последние достижения теоретической и прикладной оогани-
ческой <ичии причем особенно большое внимание уделено
современным промышленным методам и рракниям органического синтеза Бла
гояаря зтому книга можрг служить »счерпывающим справочником для
иняи-нерно-технических рабіцчикпв самих различных отраслей хим»
ческой промышленности и ценным руководством для органиков-иссле
дова гелей всех направлений.