/






























































































































Текст
54 г
0-5 i
Ч. ДЖ. ОВЕРБЕРРЕР, Ж-П. АНСЕЛМ, ДЖ. Г. ЛОМБАРДИНО
ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
СО СВЯЗЯМИ АЗОТ —АЗОТ
ИЗДАТЕЛЬСТВО «ХИМИЯ»
1970
MODERN С О N С Е PIS IN CH Е М 1 S Г R Y
ORGANIC COMPOUNDS WITH
NITROGEN —NITROGEN BONDS
C. G. OVERBERGER
Polytechnic Institute of Brooklyn
.IP. ANSELME
University of Massachusetts, Boston
.1. G. LOMBARDINO
Chas. Pfizer & Co. Inc.
THE ROLAND PRESS COMPANY
NEW YORK
1966
Ч. ДЖ. 0ВЕРБЕР1 ЕР, ж-ll. АНСЕЛМ, ДЖ. Г. ЛОМБАРДИНО
ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
СО СВЯЗЯМИ АЗОТ —АЗОТ
Перевод с английского
канд. хим. наук Ю. П. АРЦЫБАШЕВОЙ
и канд. хим. наук К. Н. ЗЕЛЕНИНА
Под редакцией и с дополнениями
проф. Б. В. ИОФФЕ
Днепр. еХН
ИНч-.ИТУТА
ИЗДАТЕЛЬ „ХИМИЯ"
ЛЕНИНГРАДСКОЕ ОТДЕЛЕНИЕ
1970
УДК 547.2
Ч. Дж. Овербергер, Ж-П. Анселм, Дж. Г. Лом-
бардино. Органические соединения со связями
азот — азот. Изд-во «Химия», 1970, Л., стр. 128.
Книга содержит лаконичное изложение неко-
торых современных аспектов химии гидразинов,
гидразонов, азо- и диазосоедииений, гидразидов,
нитрозаминов и азидов. Приводится много лите-
ратурных ссылок на обзоры и первоисточники.
Монография может служить дополнением к
учебникам по органической химии и представляет
интерес не только для лиц, работающих в обла-
сти производных гидразийа и родственных ему
соединений, но и для широких кругов химиков-
органиков, преподавателей и аспирантов.
2-5-3
24-70
СОДЕРЖАНИЕ
Г лава I. ВВЕДЕНИЕ....................................... 10
Энергия диссоциации связей азот — азот ................ 10
Реакционная способность насыщенных связей азот — азот ... 10
Реакционная способность двойных связей азот — азот......10
Литература .............................................14
Глава II. ГИДРАЗИНЫ ..................................... 15
Монозамещенные гидразины................................17
Дизамещеиные гидразины..................................18
1,2-Дизамещенные гидразины.......................... 18
1,1-Дизамещеиные гидразины............................20
Три- и тетразамещенные гидразины........................24
Гидразиниевые соли......................................24
Восстановительная способность гидразина ............... 26
Литература .............................................27
Глава III. ГИДРАЗОНЫ И АЗИНЫ........................ 30
Получение ........................................ 30
Строение. Изомерные и таутомерные превращения......32
Реакции ...........................................34
Литература .......................................~38
Глава IV. АЗОСОЕДИНЕНИЯ..................................41
Ароматические азосоединеиия...........................41
Получение...........................................41
Реакции........................................... 42
Алифатические азосоединения...........................43
Получение.......................................... 43
Реакции ............................................45
Литература ........................................,..52
Глава V. ДИАЗОСОЕДИНЕНИЯ .................................55
Соли диазония..........................................57
Строение ............................................57
Получение............................................59
Реакции .............................................60
Диазоалканы ...........................................63
Получение ............................................63
Реакции диазоалканов как нуклеофильных реагентов......66
Реакции диазоалканов как электрофильных реагентов .... 73
Диазоалканы в качестве промежуточных или конечных продук-
тов реакций...........................................76
Изомерное превращение диазоалканы — диазирины...79
Диазирины ..............................................81
Литература .............................................81
5
Глава VI. ГИДРАЗИДЫ 86
Получение 86
Реакции моцоацилгидразинов ........................ 88
Реакции 1,2-диацилгидразинов...........................91
Перегруппировки . 91
Литература 90
Глава VII. N-НИТРОЗАМИНЫ И НИТРАМИНЫ ....... 98
N-Нитрозамнны ........................................98
Получение......................................... 98
Реакции ............................................99
Нитрамины ............................................109
Получение...........................................109
Реакции . ......................................Ш
Литература . . ..................111
Глава VIII. АЗИДЫ И РОДСТВЕННЫЕ ИМ СОЕДИНЕНИЯ . .114
Получение .........................................114
Реакции.............................................114
Ацилазиды ............... ................ 118
Тетразены и триазены.................................120
Литература 120
Указатель ...........................................122
ОТ РЕДАКТОРА
Последние десятилетия ознаменовались резким повыше-
нием интереса к неароматическим гидразинам, гидразонам,
гидразидам, азо- и диазосоединениям, прежде всего в связи
с новыми областями их применения — в качестве ракетного
топлива, инициаторов полимеризации, гербицидов, противо-
туберкулезных и противораковых средств. Алифатические
производные гидразина и родственные им вещества стали
объектами специальных систематических исследований, ко-
торые привели к открытию новых путей синтеза, неожидан-
ных перегруппировок и новых классов соединений.
Небольшая книга известных американских авторов имеет
целью в какой-то мере восполнить существующий в литера-
туре пробел и отразить быстрое развитие некоторых аспектов
химии гидразо-, азо- и диазосоединений. Весьма ограничен-
ный объем книги, естественно, не позволяет претендовать на
достаточную полноту, и основное внимание уделяется новым
и необычным реакциям. Выбор тем для более подробного из-
ложения носит довольно субъективный характер, и, как от-
мечают сами авторы, многие важные вопросы не затраги-
ваются или лишь бегло упоминаются. Однако поскольку соб-
ственные работы и интересы авторов относятся к таким
актуальным проблемам, как окисление гидразинов, гомоли-
тический распад азосоединений и роль нитренов в механиз-
мах реакций азотистых соединений, субъективизм изложения
не становится существенным недостатком книги. Перевод до-
полнен изложением наиболее важных и интересных работ
1966—1968 гг. и многими ссылками на новейшую литературу.
Дополнения редактора в тексте набраны петитом, а в
списках литературы отмечены звездочкой; краткие примеча-
ния заключены в квадратные скобки. Английский текст носил
следы поспешности подготовки монографии, поэтому при ре-
дактировании перевода оказалось целесообразны^ заменить
названия некоторых разделов на более точные и сделать не-
большие перестановки. Одновременно были произведены не-
которые сокращения за счет изъятия элементарных сведений,
не снабженных ссылками па оригинальную литературу и со-
держащихся в учебниках (например, раздел об озазонах).
7
Б. А. Порай-Кошицем | сделав-
Глава о диазосоединениях была просмотрена профессо-
рами И. К. Коробицыной и
шими ряд критических замечаний и дополнений. Два кратких
параграфа этой главы, посвященные строению и получению
ароматических солей диазония, содержали неудачно изло-
женные отрывочные данные и были заменены новым текстом,
написанным проф. Б. А. Порай-Кошицем . При составлении
дополнений к главам V и VIII были учтены также предло-
жения кандидатов химических наук И. В. Целинского,
Л. Л. Родиной и О. П. Студзинского.
Краткие дополнения и примечания, конечно, не устраняют
полностью недостатков книги, тем не менее можно надеяться,
что она послужит полезным дополнением к имеющимся на
русском языке учебникам и монографиям и будет использо-
вана в качестве введения к изучению современной литера-
туры по химии органических производных гидразина и род-
ственных им соединений со связями азот — азот.
Профессор Ленинградского университета
Б. В. ИОФФЕ
ИЗ ПРЕДИСЛОВИЯ АВТОРОВ
...Нами написана краткая монография, охватывающая ин-
тересующие нас вопросы, включая области, в которых мы
работали сами и по которым хотели бы выразить наше мне-
ние. Была сделана попытка как можно полнее охватить но-
вую литературу. Однако мы лишь бегло упомянули или опу-
стили работы ряда превосходных исследователей по вопро-
сам, весьма интересным, но н*е имеющим отношения к
изложению наших взглядов.
Представляло интерес отразить быстрое развитие неко-
торых аспектов химии азо- и диазосоединений. Новые успехи
в области циклических азосоединений относятся преимуще-
ственно к соединениям неароматического характера, хотя
значительный интерес проявляется и к ароматическим соеди-
нениям со структурами иными, чем обычные пяти- или шести-
членные кольца. Открытие трехчленных колец, содержащих
двойную связь азот — азот, стимулировало интерес к стерео-
химии этой связи.
Концепция азотного секстета в классических перегруппи-
ровках Гофмана и Курциуса уже много лет привлекала вни-
мание, но очень быстро химия азотных секстетов разрослась
именно за последнее десятилетие, что нами особо и подчер-
кивается. Разновидности образующихся из гидразинов «ди-
азоподобных» промежуточных соединений, содержащих такой
азотный секстет, занимают важное место в наших исследо-
вательских работах.
Ч. Дж. Овербергер,
Ж-П. Анселм.,
Дж. Г. Ломбардино
Глава Г ВВЕДЕНИЕ
ЭНЕРГИЯ ДИССОЦИАЦИИ СВЯЗЕЙ АЗОТ - АЗОТ
Обсуждение стабильности и реакционной способности
связи азот — азот обязательно должно включать сравнение
со стабильностью и реакционной способностью связей ато-
мов других элементов. Экспериментальные значения энергий
диссоциации связей1 приведены в таблице.
Энергия днссоциацин связей
Связь Энергия, ккал/моль । Связь Энергия, ккал/моль Связь Энергия, ккал/моль
N— N 37 К= N 612 N = N 2252
С-С 80 С=С 145 CsC 198
C-N 66 C = N 1123 C = N 209
S—S 64 0 = 0 96
Обращает на себя внимание то, что простая связь азот —
азот имеет минимальную энергию диссоциации среди других
простых связей, и, наоборот, связь наибольшей кратности
между атомами азота (N=N) обладает максимальной энер-
гией диссоциации. Эта аномально высокая устойчивость си-
стемы N=N, благодаря которой элементарный азот образует
самые стабильные из известных двухатомных молекул, дает
возможность объяснить многие из реакций связи азот — азот,
обсуждаемых в следующих главах.
РЕАКЦИОННАЯ СПОСОБНОСТЬ
НАСЫЩЕННЫХ СВЯЗЕЙ АЗОТ — АЗОТ
Реакции насыщенной связи азот — азот с окислителями и
восстановителями очень часто проходят одним из двух путей:
либо непрочная связь азот — азот подвергается восстанови-
тельному расщеплению с образование;! соответствующих
10
аминов, либо вещество окисляется до некоторого промежу-
точного соединения, которое, обладая достаточной энергией,
разлагается с выделением элементарного азота. Примеры
восстановительного разложения:
R-C-NH—NH2 + Н2 R—С—NH2 + NHS (1-1)
II II
О О
NO
Ar—N—Аг - Na2S4?-6-» Ar-NH—Ar (1-2)
Как видно из таблицы, энергия разрыва связи азот — азот
значительно меньше энергии диссоциации углерод-углеродной
связи. Вследствие этого реакции (1-1) и (1-2) проходят при
температурах до 80° С. т. е. много ниже температур, обычно
необходимых для разрыва углерод-углеродной связи в угле-
водородах. Как и для всех типов связей, прочность данной
связи азот — азот изменяется в зависимости от заместителей
у атомов при связи. Так, для связей азот — азот в группиров-
ках O2N—NO и O2N — NO2 приводились такие низкие ве-
личины, как 10 и 13 ккал/моль4 соответственно. В этих слу-
чаях стерические и связанные с электроотрицательностью
воздействия атомов кислорода способствуют дальнейшему
ослаблению связи азот-—азот.
Продуктами окисления насыщенной связи азот — азот ча-
сто являются элементарный азот или такое промежуточное
соединение, которое при передаче ему достаточной энергии
может выделить азот. Это видно на следующих примерах:
R-NH—NH—R — -> R—N = N—R R—R + N2 + RH (1-3)
R2N—NH2 ——R2N—N = N—NR2 —* > N2 + азотсодержащие coe-
- 'динения (1-4)
Ar—C—C—Ar
|| || — -> Ar-C^C-Ar + 2N2 " (1-5)
.NN,
H2Nz xNH2
Ar—CH2—N—CH2—Ar —Ar—CH2CH2—Ar 4yN2 (1-6)
NH2
Промежуточные соединения реакций (1-3) и (1-4) иногда
можно изолировать, а в реакциях (1-5) и (1-6) сразу обра-
зуются элементарный азот и указанные продукты. Промежу-
точные соединения превращений (1-3) и (1-4), называемые
соответственно азосоединениями и тетразенами, будут рас-
смотрены в последующих главах.
РЕАКЦИОННАЯ СПОСОБНОСТЬ
ДВОЙНЫХ СВЯЗЕЙ АЗОТ-АЗОТ
Ненасыщенная связь в цепи углеродных атомов обычно
является реакционным центром молекулы. Почти все реакции
олефиновых и ацетиленовых производных проходят по крат-
ной связи, причем наиболее сильные реагенты разрывают ее:
RCH = CHR + KMnO4 —2RCOOH (1-7)
Однако в соединениях с ненасыщенной системой азот —
азот (N = N) связь между атомами азота никогда не разры-
вается во время реакции, скорее проявляется тенденция к
выделению фрагмента —N = N— в виде азота. Это явление,
лучше всего наблюдаемое у азидов (1-8), тетразенов (1-4),
азо- и диазосоединений (1-9), будет особо рассмотрено в
дальнейшем:
Ае+
RCON3 + Н2О ——> N2 + RNH2 + СО2 . (1-8)
RCHN2 + HX —>N2 + RCH2X (1-9)
Как выше отмечалось, эта тенденция к образованию очень
устойчивого элементарного азота весьма сильна !. Превраще-
ние насыщенной связи азот — азот (например, в гидразине)
в элементарный азот
а б в
является выгодным с точки зрения энергетических изменений.
Питцер1 отмечал, что между р-орбиталями насыщенной
азотной системы (а) существует отталкивание, которое де-
лает связь N—N более длинной (1,40А), чем сумма радиусов
двух атомов азота (0,53 А). Однако в элементарном азоте (в)
р-орбитали взаимно притягиваются, что дополнительно ста-
билизирует связь между атомами. Вследствие этого химиче-
ские реакции гидразина (а), приводящие к азоту (а), проте-
кают легко. Высокая устойчивость элементарного азота ока-
зывает большое влияние на ход реакций, затрагивающих
связь азот — азот. Поскольку азот — вещество со столь низ-
кой энергией, его образование в реакции почти всегда при-
водит к уменьшению свободной энергии реакционной системы.
Важно также отметить, что газообразный азот уже в момент
образования покидает сферу реакции. Таким образом, рав-
новесие в реакции с образованием элементарного азота не-
возможно, и вследствие этого исходное соединение можно
полностью превратить в конечные продукты.
Сопоставление реакций ненасыщенных связей С = С, C = N
и N = N свидетельствует о большей электроотрицательности
атома азота по сравнению с углеродным атомом.
12
В двойной связи углерод — азот (C=N) поляризация свя-
зи в сторону азота допускает легкое присоединение нуклео-
фильных реагентов к атому углерода:
\ п + I _ ।
XC = N— +НВ: —> НВ—С—N— —► В—С—NH— (1-10)
Так, металлоорганические реагенты присоединяются и к
алифатическим и к ароматическим двойным связям угле-
род— азот (например, в пиридине). Сравнение двойной связи
C = N с изолированной углерод-углеродной двойной связью
указывает на повышенную реакционную способность первой
по отношению к различным нуклеофилам и восстановителям.
Разнообразные гидриды металлов, не восстанавливающие
С = С-связь, успешно используются для восстановления C = N-
связей, что также иллюстрирует ее более полярную природу.
Присоединение по двойной связи азот — азот хорошо из-
вестно. Особенно хорошо изученным является присоединение
нуклеофилов к эфирам азодикарбоновых кислот, происходя-
щее экзотермически при комнатной температуре.
Тройные связи углерода и азота (С=С, C = N, N^N)
очень сильно различаются по реакционной способности. Ацети-
леновая система (С = С) обнаруживает высокую реакционную
способность в реакциях окисления (часто со взрывом), вос-
становления и некоторых реакциях присоединения. Например,
гидратация ацетилена позволяет осуществить промышленное
производство ацетальдегида, а реакция с галогенводородны-
ми кислотами является удобным источником получения ви-
ниловых мономеров:
НС = СН + НХ —► СН2=СНХ
(Ml)
Нитрильные группы (—С = Х') проявляют многие из ра-
нее отмеченных свойств функции ^C = N—. Эти свойства
также обусловлены поляризацией связи в направлении атома
азота. Гидролиз нитрилов до амидов, восстановление в амины
и превращение в иминоэфиры являются лишь небколькими
примерами общих реакций, обусловленных полярностью ни-
трильной группы.
Ацетиленовые связи, как и следовало ожидать, менее ус-
тойчивы и более реакционноспособны, чем простые и двойные
углеродные связи. Напротив, элементарный азот уникален
своим сочетанием ненасыщенности с высокой устойчивостью.
Он инертен ко всем обычным реагентам и значительно менее
реакционноспособен, чем его насыщенные (гидразины) или
обладающие двойной связью (азосоединения) предшествен-
ники. Действительно, газообразный азот во многих реакциях
используется как инертная среда.
13
Обычные окислители и восстановители в лабораторных
условиях не действуют на элементарный азот. Однако в про-
мышленности легкая доступность азота, получаемого фрак-
ционированием воздуха, делает рентабельными некоторые
процессы, проводимые в условиях высоких температур и дав-
лений. Например, в процессе восстановления азота водоро-
дом по Габеру с образованием аммиака необходимо давле-
ние порядка 1000 атм и температура 600° С. Азот окисляется
кислородом при 2000° С, для чего требуется электрическая
дуга. Последний процесс, протекающий также и в атмосфере
при грозовых разрядах, дает различные окислы азота, уно-
симые затем дождем в почву и порождающие важные ком-
поненты растительных и животных организмов. Другая реак-
ция элементарного азота, также осуществляемая при очень
высоких температурах, — это соединение с такими метал-
лами, как литий, магний, кальций и бор, образующими соот-
ветствующие нитриды.
Итак, высокая реакционная способность простой и двой-
ной связи азот—азот является следствием, во-первых, не-
прочности связи N—N и, во-вторых, высокой стабильности
элементарного азота — продукта реакций связей азот — азот.
Литература
Общая
N. S i d g w i c k. The Organic Chemist-y of Nitrogen, London, 1966.
Цитируемая
1. К. S. P i t z e r, J. Am. Chem. Soc., 70, 2140 (1948).
2, N. Sidgwick, The Organic Chemistry of Nitrogen, London, 1966.
3. Л. Физер, M. Физер, Органическая химия, Изд. «Мир», т. 1, 1966,
стр. 646; т. 2, стр. 735.
4. Т. Коттрелл, Прочность химических связей, ИЛ, 1956.
Глава II. ГИДРАЗИНЫ
Так как очень многие органические соединения содержат
два связанных между собой атома азота, лучше всего начать
с обсуждения таких соединений, у которых атомы азота на-
ходятся в наименее окисленном состоянии. Первый в этой
категории веществ --- их родоначальник, гидразин H2N—NH2.
Возросший интерес 1а-6 к гидразину и его производным
обусловлен отчасти использованием некоторых гидразинов в
военной технике [и космических исследованиях] в качестве
ракетных топлив, а также разнообразным применением про-
изводных гидразина в медицине 1в и сельском хозяйстве 1г.
Гидразин—весьма реакционноспособное соединение: он
окисляется на воздухе, окисление протекает через промежу-
точное образование диимида, давая азот. Как уже отмеча-
лось, превращение гидразина в элементарный азот сопровож-
дается выделением большого количества энергии. Поэтому,
а также в результате легкости его получения по методу Ра-
шита
2NH3 + NaOCl —> H2N—NH2 + NaCl + Н2О (II-l)
гидразин нашел широкое применение в. качестве {Макетного
топлива. Если использовать его в сочетании с азотной кисло-
той как окисляющим агентом, то газообразные продукты
окисления гидразина (азот, окислы азота) развивают очень
эффективную тягу. Некоторыми недостатками гидразина как
топлива являются [высокая температура плавления], малая
стабильность на воздухе и коррозионная активность, затруд-
няющие хранение и работу с ним.
Из трехфтористого азота при повышенной температуре
был получен тетрафторгидразин, но, как и ожидалось, нали-
чие сильно электроотрицательных атомов фтора делает это
соединение еще менее стабильным, чем гидразин. Метилги-
дразин, превосходя гидразин по некоторым физическим по-
казателям, по-видимому, вытеснит последний как жидкое ра-
кетное топливо.
15
Производные гидразина можно
три- и тетразамегценные:
разделить на монс-, ди-,
RNH—NH2 RNH—NHR
I Па
R\
XN—NH2
R^
Пб
R\
XN—NHR
RZ
ш
R\ ,R
'N—N
RZ 4R
IV
Дизамещенные гидразины II следует подразделить па два
класса и рассматривать их отдельно, так как первичная
аминная функция в 1,1-дигамещенных гидразинах Пб обусло-
вливает свойства, которыми не обладают 1,2-дизамещенные
гидразины Па. Методы синтеза различных замещенных ги-
дразинов описаны в ряде книг2-4 и статей5 и здесь рассмо-
трены не будут.
Из новых лабораторных методик синтеза гидразинов следует отметить
получение мопозамещенных гидразинов реакцией галогеналкилов с боль-
шим избытком гидразингидрата 6 7, которая может быть распространена и
на непредельные галогенпроизводные8, а также гидролизом 1-алкилдиази-
ридинов, получаемых в свою очередь из шиффовых оснований и хлорамина
или О-гидроксиламин-сульфокислоты 9'10. Описан также переход к моно-
замещенным гидразинам через сидноны и.
1,1-Диалкилгидразины с не очень сложными радикалами целесообраз-
но получать восстановлением нитрозаминов амальгамированным цинком
в кислой среде |2. Они могут быть также получены из алифатических шиф-
фовых оснований и N-хлоралкиламинов через Н,Н'-диазиридины
Удобным способом синтеза 1.2-диалкилгидразинов может служить вос-
становление ставших теперь доступными алифатических азосоединений гид-
разингидратом в присутствии никеля Ренея *4.
Триалкилгидразины получаются с удовлетворительными выходами из
реактивов Гриньяра и диалкилгидразонов альдегидов в диизоамнловом
эфире при 100° С 15.
Был описан 16 удобный метод аминирования вторичных и
третичных аминов до гидразинов и гидразиниевых солей 0-
гидроксиламинсульфокислотой:
NH2OSO3H + R2NH —> R2N—NH2 + H2SO4 (П-2)
Этот реагент является удобным источником частиц NH2
и может найти в будущем более широкое применение.
Сильно нуклеофильный характер гидразина и алкилгид-
разинов проявляется в различных реакциях. Так, ряд акти-
вированных ароматических галогенпроизводных можно вве-
сти в реакцию с гидразином, в результате образуются арил-
гидразины:
O2N—\ Cl + H2N—NHj —> O2N—NH—NH2 (П-3)
"'•NO, \NO2
16
Аналогично гидразин атакует олефины, обедненные элек-
тронами, например а, p-ненасыщенные сложные эфиры, с по-
следующей циклизацией в пиразолидоны:
ArCH=CHCOOR + H2N—NHS —> ArCHCH2COOR —>
nh-nh2
Ar—j г
—> HN I (II-4)
NH u
Интересный вариант приведенной выше реакции был най-
ден при взаимодействии 1, 1-диалкилгидразинов и акролеина.
Здесь начальная атака более нуклеофильного трехзамещен-
пого атома азота с последующей циклизацией приводит к
четвертичной пиразолиниевой соли. Мягкое разложение этой
соли щелочью разрывает связь N—N, давая р-аминонитрил ,7:
R2N-NH2 + CH2=CHCHO —N —R2NCH2CH2CN (11-5)
R/ ^R
Мнение авторов, что реакция начинается с нуклеофильной атаки атома
азота по двойной связи, не доказано. Вероятно, первой стадией этой реакции
является образование диметилгидразона, циклизующегося в кислой среде.
Последняя стадия представляет собой один из частных случаев «амино-
цитрильной перегруппировки» четвертичных альдогидразониевых структур
под действием щелочей, подробнее описываемой в гл. III.
Реакции гидразида натрия (NaNH—NH2) с различными
соединениями были исследованы Кауфманбгл и сотр.18. Мно-
госторонность этого реагента предполагает его бодце широ-
кое применение в будущем.
МОНОЗАМЕЩЕННЫЕ ГИДРАЗИНЫ
Монозамещенные гидразины I по химическим свойствам
подобны незамещенному родоначальнику и также легко оки-
сляются многими окислителями, включая воздух. Другие
окислители легко реагируют с монозамещенными гидрази-
нами; так, бром окисляет фенилгидразин до бромбензола и
азота. Алкилирование монозамещенных гидразинов дает 1,1-
диалкил- и более замещенные гидразины. Фенилгидразин ал-
килируется по первому [замещенному] атому азота (П-6),
хотя многие утверждали, что он метилируется иодистым ме-
тилом по второму атому азота, образуя 1-фенил-2-метилгид-
разин.
Следует отметить, что из возможных переходных состояний
при алкилировании замещенного гидразина возникающий
положительный^ за^яд- будет больше стабилизирован при
। ' 17
» Днепр. Xi!.11. к<' :ехн.
| ИНСТИТУТА )
С^МШМИЗи'*- 't
замещенном атоме азота; промежуточное соединение уравне-
ния (П-6) стабилизировано индуктивным эффектом арома-
тического кольца, что невозможно в альтернативном 1,2-ди-
замещенном промежуточном соединении:
c8h5nh—nh2 + сид —>
~с6н5, +
XNH— NH2I~
CH3Z
N—NI12 + HI
(11-6)
CH3NH-NH2 - CH3I —> (CHJ2N-NH2 + HI (II-7)
Монозамещенные гидразины реагируют с различными
карбонильными соединениями. В реакции с альдегидами или
кетонами продуктами будут гидразоны (см. гл. III) и вода.
С карбоновыми кислотами, хлорангидридами или сложными
эфирами образуются 1-замещенные гидразиды (гл. VI).
Из многих гидразинов типа I фенилгидразин нашел при-
менение в химии углеводов (например, образование озазо-
нов), а 2,4-динитрофенилгидразин широко используется при
идентификации альдегидов и кетонов в виде твердых дини-
трофенилгидразонов.
ДИЗАМЕЩЕИНЫЕ ГИДРАЗИНЫ
1,2-Дизамещенные гидразины
Отщепление азота при окислении 1,2-дизамещенных ги-
дразинов Па включает две стадии, и промежуточное соеди-
нение часто можно выделить, особенно когда R или R' (или
же оба) — ароматические группы:
RNH— NHR' —RN = NR' —> R—R' + N2 (П-8)
В этой реакции были использованы различные окисли-
тели, включая окись ртути, хлорное железо и перманганат
калия. Некоторые 1,2-дизамещенные гидразины, особенно те,
в которых гидразинный фрагмент заключен в циклическую
структуру, окисляются до соответствующих азосоединений
при стоянии на воздухе. Многие из промежуточных азосоеди-
нений были выделены и затем разложены при нагревании
или на свету до азота и углеводородов. Природа заместите-
лей R и R' в этих азосоединениях определяет их устойчивость.
Если R и R/— простые алкильные группы, для выделения
азота требуется повышенная температура; некоторые цикли-
ческие и бензилзамещенные азосоединения разлагаются при
комнатной температуре; ароматические же азосоединения
вполне устойчивы (см. гл. IV),
18
1,2-Диалкилгидразины реагируют с алифатическими аль-
дегидами, давая 1,3,4-оксадиазолидины, которые можно пре-
вратить в 1,2,4-триазолидипы реакцией с первичным ами-
ном ,9:
CHR CHR
H3C-N/ \ 1 О H3C-N\ / CHR р,к,п„ НчС—NZ \ н+ > 1 N + R' (П-9) н Н3С-N\ / CHR
Соединения с двум;! атомами азота, связанными через ме-
тиленовую группу, можно рассматривать как 1,2-дизамещен-
ные гидразины. Недавние литературные данные20 демонстри-
руют большой йрогресс, достигнутый в области трехчленных
циклических гидразинов (диазиридинов) *.
• Было показано, что трехчленные циклические гидразины
можно легко приготовить общим методом из хлорамина и
азометинов:
ZNH
RCH=NR' + NH2C1 —> RCH
^NR'
(IMO)
Диазиридины растворяются в органических растворите-
лях, медленно реагируют с кислотой, устойчивы к щелочи и
нагреванию (до 100° С). Альтернативным синтетическим ме-
тодом может служить присоединение реактивов Гриньяра к
диазиринам (трехчленным циклическим азосоединениям) с
образованием N-алкилдиазиридинов, последние могут дальше
гидролизоваться в алкилгидразины 21:
C8H„MgBr ?
/XN~C6H,r
CeHnNH—NH2
(П-11)
В аналогичной реакции 2-метилдиазирина с этилмагний-
бромидом получается 1-этил-2-метилдиазиридин, который мо-
жет также быть приготовлен из хлорамина и этилиденэтил-
1мнна:
и,С, N
'с !1
CgHsMgBr
Н3С< ,NH
XI
XN—С2Н5
, NHaCl
CH3CH=NC2H5
(П-12)
* [См. также Э. Шмитц, Трехчленные циклы с двумя гетероатомами,
I13J л-Мир», 1970.
2’ 19
Известны многие другие примеры циклических гидрази-
нов, в которых оба атома азота включены в цикл. Обсужде-
ние шестичленных циклических гидразинов (пиперидазинов)
выходит за пределы данной книги. О синтезе четырехчленных
циклических гидразинов в литературе сведений мало, изве-
стна, например, реакция активированных олефинов с диэтил-
азокарбоксилатом (см. также стр. 92):
Н—С—R \Т-СООС2П5 RCH—N—СООС2Н5
11+11 —* I I
Н-- С—Н N—СООС2Н5 НСН—N—СООС2Н5
(П-13)
Под влиянием кислот ароматические 1,2-дизамещенные
гидразины подвергаются перегруппировкам типа бензидино-
вой (П-14). Механизм этих реакций был предметом интен-
сивных исследований22, и, по-видимому, он включает образо-
вание «протонного сандвича»23:
----” (11-14)
[Аналогичные превращения, осложненные образованием
азосоединений и аминов, происходят также при нагревании
ароматических гидразосоединений в отсутствие кислот24.]
Изучалось 25 поведение 1,2-диалкилгидразинов в условиях
реакции Манниха. Реакция гидрохлоридов 1,2-дизамещенных
гидразинов с формальдегидом и ацетофеноном приводит к
З-фенил-1,2-диалкил-Д3-пиразолинам:
С6НБСОСН3 + (СН2О)Л + RHN—NHR • НС1 —>
—> [C6H5COCH2CH2NR-NHR] —> I | СбН5 (П-15)
N\
\N/ R
I
R
[Алициклические кетоны — циклогексанон, циклогептанон
и циклооктанон (но не циклопентанон)—также конденси-
руются с 1,2-диметилгидразином и формальдегидом, образуя
с хорошими выходами- соответствующие Д3-пиразолины26.]
1,1-Дизамещенные гидразины
Благодаря первичной аминной функции 1, 1-дизамещен-
ные гидразины Пб способны к некоторым реакциям, невоз-
можным у изомерных 1,2-дизамещенных гидразинов. Окисле-
ние 1,1-дизамещенных гидразинов может привести к двум
20
продуктам и, вероятно, протекает через промежуточный
N-нитрен (см. гл. VII):
R\ zR
—> ^N-N=N—
R' vi 1Г
—R-R' + N2
^N—NH2 — -> K—N
Rz/ L R'Z
Сочетание двух частиц нитрена или, что более вероятно,
реакция нитрена с непрореагировавшим гидразином (по типу
а) дает тетразены VI. Наблюдалось также разложение ни-
трена с образованием азота и углеводорода (по типу б). Про-
дукты окисления зависят от природы заместителей R и Rz,
однако обычно окисление дает тетразен VI (П-16). В некото-
рых случаях окисление 1,1-дизамещенных гидразинов при-
водит непосредственно к выделению азота и образованию
углеводородов (П-16, б). Этот последний путь, называемый
«аномальным окислением»27, требует, чтобы замещающие
группы могли стабилизировать промежуточные фрагменты,
образующие новую углерод-углеродную связь. Этими свойст-
вами обладают такие группы, как бензильная (П-17) и циа-
нометиленовая:
C6H5CH2NCH2C6H5 + HgO —► C6H5CH2CH2C6H5 + N2 + Hg+H2O (1Ы7)
NH2
Такие аномальные реакции окисления легко происходят
в гетерогенных окисляющих системах (например, спирт —
окись ртути) с большой поверхностью окислителя, увеличи-
вающей скорость выделения азота. Аналогичные результаты
были получены с гетерогенными системами и в других реак-
циях с выделением азота28. Гомогенная среда часто благо-
приятствует образованию тетразена. Например, в методе
получения тетрабензилтетразена из дибензилгидразина ис-
пользуется спиртовой раствор ацетата ртути.
Твердая поверхность, по-видимому, способствует сниже-
нию, энергии активации выделения азота и, вероятно, помо-
гает изолировать промежуточные частицы N-нитрена друг от
друга, препятствуя образованию тетразена. Если в 1, 1-диза-
мещенном гидразине присутствуют метильные, этильные,
циклогексильные или подобные алкильные группы, то неспо-
собность этих групп стабилизировать промежуточные углерод-
ные фрагменты (ионы или свободные радикалы — еще не
выяснено) подавляет разрыв связей углерод—азот. В этих
случаях даже в гетерогенной * среде образование азота не-
выгодно энергетически, и вместо этого получается тетразен.
* [В английском тексте — гомогенной.]
21
Такие соединения, как 1-бензил-1-бутилгидразин, окисляются
окисью ртути в хлористом метилене, давая с низким выходом
амилбензол; однако в этанольном растворе та же реакция
приводит к соответствующему тетразену. Другими примерами
аномального окисления могут служить:
окисление N-аминоизоиндолина до бензоциклобутена и
1,2-диметилен-3,5-циклогексадиена
z^\/\ (О) \—
I || N__Nh2 I | I +
\/\/
1
I 1
^Z^CH2 .
окисление N-амино-!, 3-дифенилизоиндолина в 1,2-дифе-
нилбензоциклобутен 29
(П-19)
С6Н6
(транс)
(транс 81%)
окисление цис и тра«с-1Ч-амино-2,6-дифенилпиперидинов
до цис- и транс-1,2-дифенилциклопентанов27 и соответствую-
щего олефина
nh2
(ф?с)
н' Н
(цис)
+ С6Н5СН = СН(СЙ2)3С6Н3 (П-20)
(11-18)
Предварительное сообщение30 об окислении 1, 1-диалкил-
гидразинов подтверждает и расширяет представление о
протекании реакции через диалкилнитрен (диалкилдиазен),
который может или димеризоваться в тетразен, или подвер-
гаться катализируемой щелочами перегруппировке в алкил-
гидразоны, или же'алкилировать другие основания, присут-
ствующие в смеси (П-21):
r2chx
N—NH2
r2chz
I-2H
У
R2CHx
XN-N
. r2chz
r2chx zchr2
—-> N— N = N —N
r2chx zchr2
(П-21)
— > r2c=n-nh-chr2
—-> r2chs + r2choh + r2ch-n=n-chr2 +n2
22
При окислении 1,1-дипропилгидразина главными продук-
тами являются пропилгидразон пропионового альдегида, ди-
пропилгидразоп'пропионового альдегида и тетрапропилтетра-
зсн. Второстепенные продукты этой реакции — пропанол,
азопропан, пропан и азот. Здесь следует отметить, что
гетразены, будучи нагреты до достаточно высокой темпера-
туры, выделяют азот; другие продукты такого распада неопре-
деленны и, ио-видимому, состоят из тетразамещенного гидра-
зина и азометинов.
Реакцией, аналогичной аномальному окислению гидрази-
нов Пб, является щелочное разложение сульфонилгидрази-
нов31'32. Эти реакции, по-видимому, протекают через нитрен
тождественный рассмотренному выше (П-21), и продукты ча-
сто идентичны продуктам окисления соответствующих гидра-
зинов (П-22):
xn-nh2
R'Z
R\ в-
XN— NHSO2Ar -->
R'Z
N—N
+ ArSO2
(11-22)
vn
Здесь, как и в реакциях аномального окисления, для обра-
зования азота и углеводорода желательны стабилизирующие
заместители (например, бензил). Аналогично, обработка не-
которых нитрозаминов гидросульфитом натрия в щелочной
среде дает азот и углеводороды/ Так как продукты реакции
идентичны получаемым при окислении соответствующего ги-
дразина окисью ртути, то промежуточное образование нит-
рена Предполагалось27 и для этой реакции:
Na3s,o4, он
lT
NO
(11-23)
Наличие первичной аминной функции в 1,1-дизамешен-
пых гидразинах обусловливает и ряд других реакций, невое-
ложных с 1,2-диза.мещенными, Например, 1,1-дизамещенные
гидразины дают с карбонильными соединениями гидразоны,
а азотистая кислота дезаминирует 1,1-дизамещенные гидра-
нты, образуя азот и вторичные амины.
ТРИ- И ТЕТРАЗАМЕЩЕННЫЕ ГИДРАЗИНЫ
Характерной особенностью алифатических трехзамещенных гидрази-
нов III является гладкое окисление (при стоянии на воздухе) в д:т = амещен-
ные гидразоны 8-16:
R2N—NHCHR'—R2N-N=CR' (П-24)
Тетразамещенным гидразинам IV уделяется по сравнению
с другими классами меньше внимания. Тетрафенилгидразин,
однако, интересен проявлением особых свойств из-за пере-
крывания объемистых фенильных групп, окружающих ма-
ленькие атомы азота. Даже при температуре жидкого воз-
духа тетрафенилгидразин разлагается в присутствии кислоты
с образованием ионов и, возможно, радикалов. Очевидно, в
этом соединении связь азот — азот ослаблена вследствие сте-
рического перекрывания, так как другие тетразамещенные
гидразины совершенно устойчивы по отношению к кислотам:
С6Н5х /С6Н5 + с6н5Х _ С6Н5- С6Н6, .
2 N( +н —► )N+ )NH + 2 ;N (П-25)
С5Н5 С3Н5 С6Н5 CeH5 CjHg
Для сочетания двух атомов азота с образованием 1,1-бис-
азиридина VIII был использован циклический хлорамин, по-
лученный из этиленимина, что иллюстрирует возможный путь
синтеза других тетразамещенных гидразинов:
|/NLi+ ,
|\NH --- \ (П-26)
VIII
Гидразин VIII — слабое основание и разлагается при на-
гревании в присутствии кислорода со взрывом.
ГИДРАЗИНИЕВЫЕ СОЛИ33
Моноалкилгидразины атакуются простейшими галоген-
алкилами по замещенному атому азота, если не возникает
пространственных препятствий. Когда гидразин реагирует с
избытком простого галогеналкила (например, йодистого ме-
тила), получается 1,1, 1-тризамещенный гидразин — гидрази-
ниевая соль. Большие алкильные группы, такие как изопро-
пил, не могут размещаться по три у одного атома азота и
обычно образуют смеси моно-, 1,1- и 1, 2-дизамещенных ги-
дразинов. Лучшим способом получения гидразиниевых солей,
допускающим большее разнообразие заместителей, является
обработка третичного амина хлорамином 34:
R3N + NH2C1 —> R3N+—NHjCr (11-27)
24
Обычные электронодонорные свойства алкильных групп
привели некоторых исследователей к предсказанию более вы-
сокой основности (и, следовательно, более низкой кислот-
ности протонированной формы) замещенных гидразинов по
сравнению с самим гидразином.
Однако, вопреки ожиданию, метилирование гидразина не
повышает основности; гидрохлорид гидразина — более сла-
бая кислота (р/Са = 7,95; 8,1), чем гидрохлорид метилгидра-
зина (р/Са = 7,87), который в свою очередь слабее гидрохло-
рида диметилгидразина как кислоты (рАа = 7,21) 35. Такую,
кажущуюся аномальной, кислотность гидрохлоридов гидрази-
нов можно объяснить следующим образом: в протонирован-
ной форме 1,1-диметилгидразина первый атом азота имеет
тетраэдрическую конфигурацию, которая вызывает стериче-
ское отталкивание между метильными группами и вторым
атомом азота. Потеря протона и, следовательно, потеря тет-
раэдрической конфигурации способствует уменьшению напря-
жения в молекуле. Так как напряжение тетраэдрической кон-
фигурации в диметилгидразине больше, чем в метилгидра-
зине, и еще больше, чем в самом гидразине, потеря протона
в случае гидрохлорида диметилгидразина энергетически бо-
лее выгодна, чем в случае гидрохлоридов метилгидразина
или незамещенного гидразина. Равновесие (П-28) смещается
поэтому вправо, и это увеличивает кислотность гидрохлорида
диметилгидразина:
(CH3)2NH-NH2 (CH3)2N-NH2 + Н+ (П-28)
Так как экспериментальные факты показывают, что даль-
нейшее алкилирование алкилгидразинов идет по замещен-
ному атому, можно сделать заключение, что замещенный
атом азота менее основен, но более нуклеофилен, чем неза-
мещенный.
Кондон36 иначе объясняет аномальную основность алкилгидразинов,
связывая ее с гидратацией их в водных растворах по обоим атомам азота и
потерей воды при протонировании
'О—Н
Hz
R\ i /R"
)n—
R'Z = \r"'
H
I
,OX
z H....
(11-29)
W
Замещение водорода у любого атома азота на алкильную группу
с этой точки зрения должно способствовать гидратации, как реакции, кон-
курирующей с протонированием, и приводить к снижению рКа (т. е.
25
\ меныиепл.о основности;, анп;,лнру‘. прямое влияние и-щуктиввого эф-
фекта зачестителей на ос;:овном,-,, В пледполлженьи, что несимметричные
Е1.:килзамегценньге гидразины протонируютгя по азоту, имеющему большее
1исло алкильных групп, удается коррелировать рЛч с константами Тафта
заместителей. Однако изучение основности недавно синтезированных фтор-
замешенных моноалкилгидразинов и других гидразинов с электроотрица-
тельными заместителями показало37, что лучшая корреляция получается
в предположении протсннровання незамещенного атома азота. Проблема
основности гидразинов остается, таким образом, достаточно запутанной.
ВОССТАНОВИТЕЛЬНАЯ СПОСОБНОСТЬ ГИДРАЗИНА
Восстановительную способность пробовали использовать в
различных системах [см. указанную в списке литературы мо-
нографию А. П. Грекова]. Имеются подробные обзоры по вос-
становлению гидразином олефиновых соединений в присут-
ствии кислорода34-г8-зэ. В этой системе, которая особенно
удобна для восстановления ненасыщенной части олефиновых
кислот, необходимым компонентом является кислород, вводи-
мый энергичным перемешиванием на воздухе. Оптимальные
выходы достигаются при 50е С и в узком интервале основно-
сти; з атмосфере чистого азота восстановление не идет.
Предполагаемое образование в системе гидразин—кисло-
род промежуточного диимида нашло серьезное подтвержде-
ние в работах других исследователей с аналогичными систе-
мами 40’41. При использовании систем гидразин — окислитель
было установлено, что непредельные соединения могут вос-
станавливаться с гщг-присоединением водородных атомов, ве-
роятно через циклический промежуточный комплекс (П-30):
Н
(11-30)
Реакция (П-31) иллюстрирует стереоспецифичность этого
метода:
С5Н5С^ССООН —> С6Н5СН = СНСООН (в основном ^uc-форма) (11-31)
Было показано, что следы ионов меди оказывают уско-
ряющее действие на эти процессы. Селективность действия
на двойные связи диимида, получаемого разными методами,
иллюстрируется его способностью восстанавливать диаллил-
дисульфид в дипропилдисульфид.
Доказательство образования диимида из гидразина было
получено при масс-спектрографическом изучении электриче-
ского разряда в гидразине42. Способность восстанавливать
олефиновые связи в мягких условиях, а также в щелочных
26
растворах указывает, что при -псбых реакциях с гидразином
н присутствии кислорода следует принимать в расчет воз-
можное восстановление олефина в результате образования
нестойкого диимида. Это затруднение можно обойти, проводя
восстановление в атмосфере азота.
В некоторых 1,2-дизамещенных гидразинах наблюдалось
восстановительное расщепление связи азот — азот сильными
восстановителями (например, гидразобензол расщепляется
до анилина). \
Основательно было изучено действие никеля Ренея на
гидразин43. В таких мягких условиях, как кипячение в мета-
юле, эта смесь вызывает разрыв N—N-связн в различных
гидразинах и М.М'-диацилгидразйна.х (до амидов).
[Расщепление связи N—N наблюдалось в таких условиях
лишь у ароматических гидразосоединений с аминогруппой в
пара-положении4,1'45 или в сильно разбавленных спиртовых
растворах гидразингидрата 46.
Азобензол и другие ароматические азосоединения гладко
восстанавливаются гидразингидратом в присутствии никеля
Ренея до гидразосоединений 48’47. Недавно было показано,
что алифатические азосоединения также гладко гидрируются
до 1,2-диалкилгидразинов 14.]
Эта же восстановительная система была использована
для превращения нитросоединений в амины 48’49. Сообща-
лось 50 об удобном способе получения первичных аминов из
нитрилов также при применении гидразингидрата и никеля
Ренея в спирте:
RCs=N + NH2—NH2 "|<"-|!'.?|,|.'я> rch2NH2 (11-32)
Л итература
Общая -
Л. Одрит, Б. О г г. Химия гидразина, ИЛ, 1954.
А. Н. Кост, Р. С. С а г и т у л л и н, Моноалкилгидразины, Усп. хим., 33,
361 (1964).
*А. П. Греков, Органическая химия гидразина. Изд. «Техника», Киев,
1966.
Ж. М. Федорова, Применение гидразина в различных областях на-
родного хозяйства, Научно-техническая информация ГИПХ, Л., 1957.
Н. W i е 1 а п d, Die Hydrazine, Stuttgart, 1913.
H. H. Sisi er, G. M, Omie tanski, B. Rudner, The Chemistry of Qua-
terr.ized Hydrazine Compounds, Chem. Rev., 57, 1021 (1957).
¥P. A. S. Smith, The Chemistry of Open-Chain Organic Nitrogen Com-
pounds, vol. 11, ch. 9, 1966. p. 119—148.
*E, Muller, Aliphatische Hydrazine und Hydrazoverbindungen, (Houben-
Weil) Methoden der organischen Chemie, 4 Aufl., X, 2, I—71 (1967).
’E. Enders, Arylhydrazine und Arylhydrazone, (Houben-Weil) Methoden
der organischen Chemie, 4 Aufl., X, 2, 169—692 (1967).
*E. Enders. Aromatische Hydrazoverbindungen, Tri, und Tetraarylhydra-
zine, (Houben-Weil) Methoden der organischen Chemie, 4 Aufl., X, 2,
693—756 (1967).
27
Цитируемая
1. а) А. Н. Кост, Р. С. Сагитуллин, Усг. хим., 33, 361 (1964).
б) A. Furst. R. С. Berio, S. Н о о t о n, Chem. Rev., 65 . 51 (1965).
в) Е. J u с к.е г, Angew. Chem., 71, 321 (1959).
*г) Н. М. Федорова, Применение гидрааина з различных областях
пародисте хозяйства. Научно-техническая информация ГИПХ, Л.,
1957.
2. Л. Од р и т, Б. О г г, Химия гидразина, ИЛ, 1954
3. L. F. Deleting (ed.), An Outline of Organic Nitrogen Compounds,
Michigan. 1950, p. 376.
4. R. Wagner, H. Zook, Synthetic Organic Chemistry, New York, 1956.
5. a) R. F. Evans, Rev. Pure Appl. Chem, 12, 146 (1962).
6) A. Ebn о (her, E. J tick er, A. L i n d e n m a n n, E. R i s s i,
R. Steiner, R. Suess, A. Vogel, Helv. Chim. Acta, 42, 533
(1959).
* 6. A. H. К о ст, P. С. Сагитуллин, ЖОХ, 33, 867 (1963).
* 7. Б. В. Иоффе, В. С. Сто некий, 3. И. Сергеева, ЖОрХ, 4, 986
(1968).
* 8. Б. В. Иоффе, 3. И. Сергеева, А. П. Кочетов, ЖОрХ, 3, 983
(1967).
* 9. Е. Schmitz, D. Н a b i s ch, Вег., 95, 680 (1962).
* 10. R. Ohme, E. Schmitz, L. S t e r k, J. prakt. chem., [4], 37, 257 (1968).
“11. В. Г. Я ш У н с к и й, В. Ф. Васильева, ЖОХ, 30, 2764 (1960).
“12. Б. В. И о ф □ е, ЖОХ, 28, 1296 (1958),
* 13. Е. Schmitz, К. Schinkowski, Вег., 97, 49 (1964).
* 14. Б, В. Иоффе, 3. И. Сергеева, Ю. Я. Д у м и и с, ЖОрХ, 5, 1735.
(1969).
* 15. Б. В. Иоффе, Л. Е. Порошин, ЖОХ, 29, 3154 (1959).
16. R. G о s 1, A. Mewsen, Chem. Вег.. 92, 2521 (1959).
17. Б. В. Иоффе, К- Н. Зеленин, ДАН СССР, 134, 1094 (1960); Tetra-
hedron Lett., 1962, 481.
18. Т. К a u f I m а п и, Angew. Chem., 76, 206 (1964).
19. J. Str a ting et al., Rec. Trav. Chim.. 84, 408 (1965); 83, 387, 877
(1964).
20. E. Schmitz, R. Ohme, Chem. Ber., 94, 2166 (1961); E. Schmitz,
ibid., 95, 676 (1962).
21. E. Schmitz, R. Ohme, Angew. Chem., 73, 220 (1961); E. Schmitz,
ibid., 76, 197 (1964).
22. M. Vecera, Chem. Listy, 52, 1373 (1958); M. J. S. Dewar, in
P. de Mayo, Molecular Rearrangements, New York, 1963, p. 323.
23. L. L. Frestanding, Tetrahedron Lett., 1963, 1235.
*24. В. О. Лукашевич, Усп. хим., 36, 2053 (1967).
25. R. L. Hinman, R. D. E 11 e f s о n, R. D. Campbell, J. Am. Chem.
Soc., 82, 3988 (1960).
*26. Набил Махмуд Омар, А. В. Ельцов, ЖОрХ, 4, 1294 (1968).
27. С. G. О ver ber ger, J. G. Lombardi no, R. G. Hiskey, J. Am.
Chem. Soc., 79, 1510, 6430 (1957); 80, 3009 (1958); см. также
D. M. King, A. J. Bard, ibid., 87, 419 (1965); C. D. Campbell,
C. W. R e e s, Proc. Chem. Soc., 1964, 296.
28. M. S. Newman, E. C a f 1 i s h, J. Am. Chem. Soc., 80, 862 (1958).
29. L. H. Carpino, J. Am. Chem. Soc., 82, 2728 (1960); 84, 2196 (1962).
30. W. H. Urry, A. L. Olsen, E. M. Bens, H. W. Kruse, C. Ikoku,
AD 622785, Avail. CFSTI, 43 pp. (1965); C. A., 64, 14078e.
31. L. H. Carpino, J. Am. Chem. Soc., 79, 98, 4427 (1957).
32. R. L. Hinman, K. Hamm, J. Am. Chem. See., 81, 3294 (1959).
33. H. H. S i s 1 er, G. M. О m i e t a n s k i, B. R nd n er, Chem. Rev., 57,
1021 (1957).
34 G. M. Ornie tanski, H. H. S i s 1 e r, J. Am. Chem. Soc., 78, 1211
(1956).
35. R. L. H i nm a n, J. Org. Chem., 23, 1587 (1958).
28
).
IX
I.,
S,
3.
i,
3
3
i
<36 F. E. Condo n, J. Am. ChemJ Soc., 87, 4491 (1965):
i;37. R Pol let, H. van der. Eynde, Bull. Soc. ch.m. belg.. 77, 341
(1968).
38, S. Aylward. M. Sawjstowska, Chem. a. Ind. (London), 1962,
484.
39. S. Hilnig, H. R. Muller. W. Th (er, Angew. Crtem., 77, 368 (1965).
40. E. J. Corey, W L. Mock, D. J, Paste, J. Am Chem. Soc., 83,
2957 (1961),
41. S, Hilnig, H. R. Muller, W. Tliisr, Tetrahedron Lett., 1961, 353.
12. S. N. Foner. R. L. Htdson, J. Chem, Phys, 28, 719 (1958).
43. D. Robinson, R. K. Brown, Can. J, Chem, 39, 1171 (1961).
* 44. S. Kubota, A. Akita, T. J о ко shim a, J. Pharm. Soc. Japan, 78,
1194 (1958).
* 45. W. H. Stafford, M. Loss, N. T. Thomson, Chem. a. Ind., 1956,
1277.
* 46. A. Frust. R. E. Moore, J. Am. Chem. Soc., 79, 5492 (1957).
* 47. S. Hornsby, W. L. P e а с о о k, Chem. a. Ind., 1958, 858.
48. L. P. Kuhn, J. Am. Chem. Soc., 73, 1510 (1951).
49. B. Balcom, E, Furs:, J. Дт. Chem. Soc., 75, 4334 (1953).
50. А. П. Терентьев, M, H. Преображенская, Хим. наука и
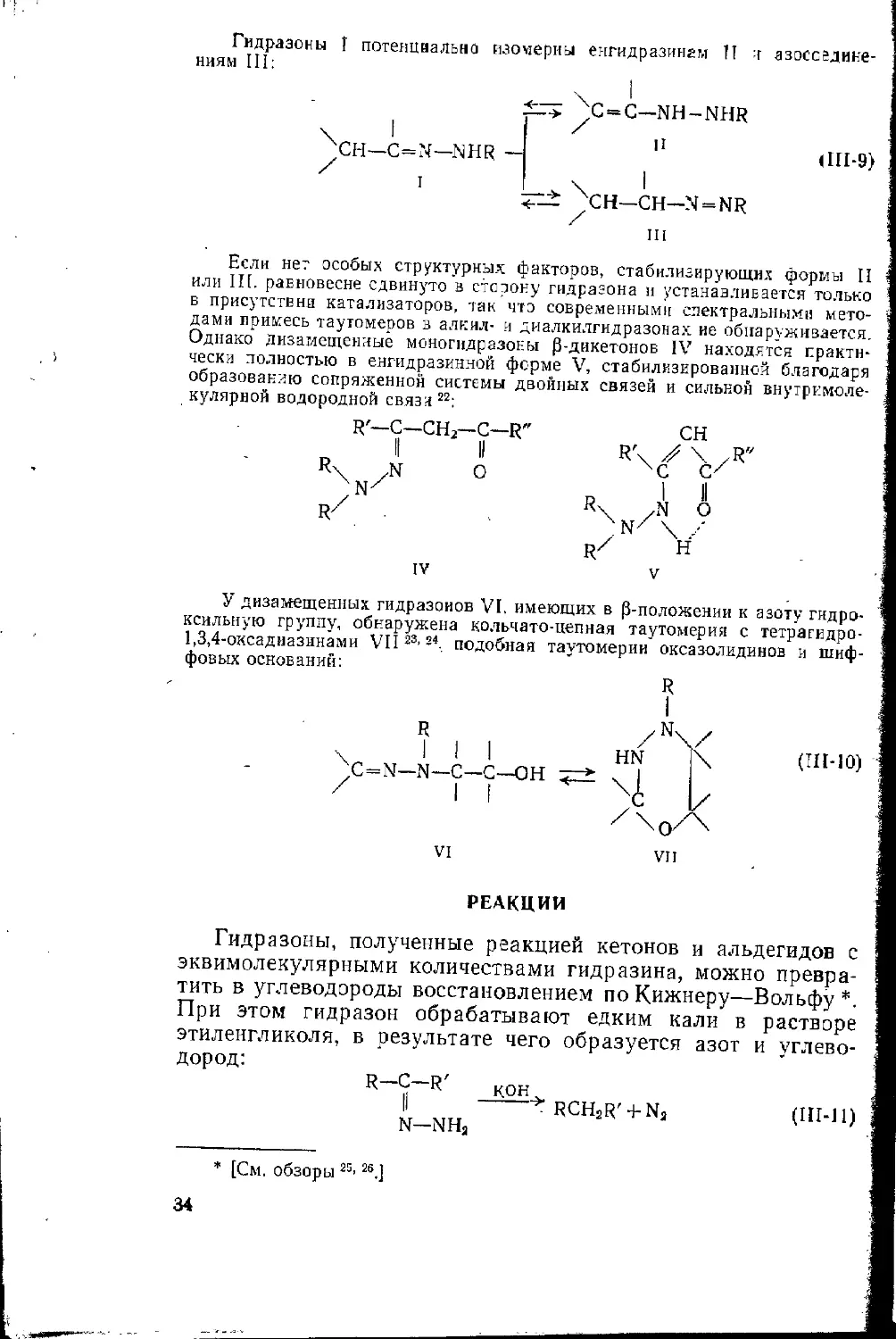
пром.. 4, 281 (1959).
Глава 111. ГИДРАЗОНЫ И АЗИНЫ
ПОЛУЧЕНИЕ
Гидразин и его производные, содержащие хотя бы одну
первичную аминную функцию, присоединяются к карбониль-
ным группам альдегидов и кетонов. Свободная электронная
пара концевого атсма азота начинает атаку на поляризован-
ный карбонил с последующим отщеплением воды:
С—Р + R'NH—NH2 — -> II О . ’ R. .OH 1 X+ L RX xNH2-NHR'j I (111-1)
R\ —н~о Y=n—nhr' <—5— RZ Rx .OH* C\ . RZ -NH—NHR' _
Кислые катализаторы ускоряют первую стадию (II1-1) в
результате образования с кетонами сопряженных кислот, что
еще сильнее поляризует карбонильную группу. Эта реакция
оказалась чрезвычайно полезной для превращения многих
жидких альдегидов и кетонов в твердые гидразоны; темпера-
туры плавления твердых 2,4-динитрофенилгидразонов сведены
в таблицы
Азины (дигидразоны) легко получаются реакцией гидра-
зина с двумя молями альдегида или кетона:
R\ ,R
2R—C-R + NH2-NH2—► XC=N-N=C/ +2H2O (Ш-2)
И RZ \R
О
Альдегиды легко реагируют с гидразином уже при обыч-
ных условиях, образуя сначала гидразон альдегида, который
быстро конденсируется со второй молекулой альдегида в азин.
Гидразоны альдегидов можно выделить только при осторож-
ной работе в мягких условиях с использованием избытка
гидразина.
30
При конденсации гидразина с формальдегидом получается
полимерный продукт. Однако нагревание полимера в инерт-
ной атмосфере дает мономерный азин формальдегида —очень
реакционноспособное вещество, устойчивое лишь при низких
температурах2.
Простейший из грилгидразонсв— фенил гидразон формальдегида уда-
,’ioCj получить лишь недавно3. Принимавшиеся ранее за сенилгидразов
формальдегида кристаллические вещества состава CjHsN2 являлись болеа
или менее загрязненными препаратами его димера, имеющего структура
1.4-дифенилгексагидро-с1ьил-тегразииаУ
сбн5.
/N—NH^
2C6H5N’H—N = CHj CH2 CH2
\\H-~ bjf
4cGH5
(HI-3)
Склонность к димеризации является общим свойством монозамегцен-
1!Ых гидразонов и самих гидразонов простейших альдегидов 5~8. При этом,
по видимому, сначала образуются нециклические, сохраняющие одну гидра-
:<г:пную группировку димеры, и лишь затем замыкается гексагидротетрази-
;i >ное кольцоs:
R
I
СИ
./ \
HN N'
I I
HN N
R/Z НС^
"R
СН
/ \ XR'
HN N'
I I
,N NH
R' \ /
СН
R
I
R
R (Ш-4)
(R — Н пли алкил; R'—Н, алкил или арил)
С а,р-иепредельными карбонильными соединениями гидразин и моно-
алкилгидразины образуют Д2-пиразолины. Тенденция к замыканию пиразо-
липового кольца столь велика, что даже с 1,1-диалкилгидразинами непре-
дельные гидразоны получаются лишь при неконцевых или расположенных
в месте разветвления связях С = С, или же в случае тяжелых и разветв-
ленных алкильных радикалов гидразина9-11. 1,1-Диметилгидразин с про-
стейшими непредельными карбонильными соединениями образует в слабо-
кислой среде четвертичные пиразолиниевые соли °, легко подвергающиеся
в щелочной среде аминонитрильной перегруппировке (см. стр. 35).
В 1967 г. было описано несколько изопропилгидразонов непредельных
альдегидов R Незамещенные непредельные гидразоны и моноалкилзаме-
щенные гидразоны аф-непредельных кетонов продолжают оставаться не-
известными. У непредельных кетонов более отчетливо проявляется склон-
ность к присоединению гидразинов по связи С=С с образованием гидра-
шнгидразонов 13.
Дикетоны реагируют с гидразином, образуя циклические
азины или изомерные им соединения; 1,3- и 1,4-дикетоны
31
были применены для приготовления пиразолов (Ш-5), дигид-
ропиридазинов и пирролов (Ш-6):
I TR
NHj—NHS4-RCOCH2COR —> R-t (II1-5)
VlH
NHs-NH2 + RCOCHjCHsCOR —► R—J J-R + R-<f ^-R
\nz 'N—
I
nh2
(III-6)
Еще большие циклические системы можно получить кон-
денсацией а, <в-диароилалканов с гидразином |4:
Аг—С—(СН2)ге—С—Ar + NHj—NH2 —>
О О
/(CH.)nx
Аг—С С—Аг
"Ч,-—
(Ш-7)
(п = 3; 4)
18-, 20-, 24- и 28-членные циклические бцсазипы были по-
лучены 14 в условиях сильного разбавления.
СТРОЕНИЕ. ИЗОМЕРНЫЕ И ТАУТОМЕРНЫЕ ПРЕВРАЩЕНИЯ
Фиксированное положение заместителей у двойной связи
C=N делает возможной геометрическую (син-анти) изоме-
рию гидразонов:
R\
C=N
R'Z
-NHR" R\
или C=N
R,/Z X-NHR"
Значительные успехи в изучении стереоизомерии гидразонов были до-
стигнуты благодаря применению ПМР в систематических исследованиях
Карабатсоса и сотрудниковIS. Оказалось, что дизамещеиные гидразоны
альдегидов представляют собой снн-изомеры *, неспособные к превраще-
нию в анти-формы даже при действии кислот. Производные кетонов и моно-
замещенные производные альдегидов существуют в обеих (сип и анти-)
формах, равновесие между которыми быстро устанавливается в растворах
в присутствии следов кислот. Равновесные отношения си«-форма : анти-
форма почти не зависят от природы гидразинного фрагмента и опреде-
ляются строением исходного карбонильного соединения, причем обычно
преобладает син-форма (60—96%).
Стереоизомерия производных непредельных карбонильных соединений
гораздо сложнее. Кроме син-анти-форм здесь возможны s-цис- и s-транс-
конформации вследствие заторможенности вращения по центральной связи
* [саи-Изомером считается имеющий в щщ-положении к группе NR"RW
атом водорода или меньший из радикалов фрагмента RR'C=N].
32
С—С и геометрическая Изомерия по этиленовой связи. В изученных пока
2,4-динитрофенилгидразэнах а,0-неп радельных альдегидов и кетонов В * * * * * * 15 на-
блюдались только з-традс-фармы.
Известны случаи успешного разделения двух возможных
геометрических изомеров: например, реакция трет-бутилфе-
нилглиоксиловой кислоты с л-толидгидразином дает два про-
дукта 17, имеющих различные точки плавления, УФ-спектры
и константы кислотности:
Ат, ХСООН
fl
II
о
+ H2N—NH—СИ;
Аг = Жгрег-ЩНзСзН^
анти (А)
т. пл. 135-137 °C
pKa = 6,S
син (Б)
т. пл. 118—120 °C
рКа = 5,1
(III-8)
В этом случае пространственное расположение соседних
групп влияет на кислотность карбоксила, возможно из-за об-
разования водородной связи в син-форме, что способствует
ионизации карбоксильной функции.
Азины имеют дипольный момент 1,2—1.9 D—промежуточный между
ожидаемым для s-цис- и s-трйкс-конформацнй, так что фрагменты их мо-
лекул повернуты вокруг N—N-связн приблизительно на 70° 18.
В гидразонах имеется возможность р— л-сопряжеиия, т. е. перекрыва-
ния р-орбнталей неподеленной электронной пары азота и орбиталей
.т-электронов связи группы N1—№ = С3.
р — л-Сопряжеиие проявляется в существовании спин-спинового взаимо-
действия протонов через гидразонную группировку 19 * а также в высокой
интенсивности, большей длине волны полос л л*-переходов в электронных
спектрах16-21, экзальтации молекулярной рефракции и снижении частот ва-
лентных колебаний связи С = \.
Однако, как показали исследования спектров ПМР и электронных спек-
тров 19~21, р — л-сопряжение не осуществляется в диалкилгидразоиах кето-
нов, по-видимому, нз-за пространственных затруднений, препятствующих
надлежащей ориентации р- и л-орбиталей и вызываемых заместителями
у атомов N1 и С3.
3 Зак. 334
33
Гидразоны I потенциально изомерны енгидразингм 11 т азосседиие-
ниям Ш:
;C=C—NH-NHR
;СН—C=N—NHR -
(Ш-9)
CH—CH—N = NR
Ш
Если нет особых структурных факторов, стабилизирующих формы II
или III. равновесие сдвинуто а сторону гидразона и устанавливается только
в присутствии катализаторов, так что современными спектральными мето-
дами примесь таутомеров в алкил- и диалкилгидразонах ие обнаруживается.
Однако Дизамещеиные моногидразоны 0-дикетонов IV находятся практи-
чески полностью в енгидразинной ферме V, стабилизированной благодаря
образованию сопряженной системы двойных связей и сильной внутримоле-
кулярной водородной связи 22:
R'—С—СН2—С—R"
II II
R\ ,N о
zN/
RZ
У дизамещенных гидразонов VI, имеющих в р-положезии к азоту гидро-
ксильную группу, обнаружена кольчато-цепная таутомерия с тетрагидро-
1,3,4-оксадиазинами VII23,-4 подобная таутомерии оксазолидинов и шиф-
фовых оснований:
R
VII
РЕАКЦИИ
Гидразоны, полученные реакцией кетонов и альдегидов с
эквимолекулярными количествами гидразина, можно превра-
тить в углеводороды восстановлением поКижнеру—Вольфу*.
При этом гидразон обрабатывают едким кали в растворе
этиленгликоля, в результате чего образуется азот и углево-
дород:
R-C-R' кон
И --------^RCHsR' + Nj (III-11)
N—NHa
* [См. обзоры 25' 2S.J
34
Изучение кинетики2’ разложения гидразонов диарилкето-
нов показало, что реакция Кижкера—Вольфа имеет первый
порядок по гидразону и основанию; для суммарной реакции
предложен следующий механизм:
быстро
R2C=N— NH2 + В~ ....R2C=N—NH" + ВН
медленно
R2C = N-NH‘ ± RjCH—N-=N" (Ш-12)
RsCH—N=N” -быст?% R2CH’+N2
быстро
RjCH' + ВН Z==± R2CH2 + B~
В этом ряду второй стадией, определяющей скорость реак-
ции, является таутомерный сдвиг, дающий нестойкий анион
замещенного диимида. Интересное восстановление этого типа
в очень мягких условиях28 происходит при обработке 3-индо-
лилоксалата гидразингидратом в отсутствие щелочи. Образо-
вание в этой реакции индол-3-ацетгидразида указывает на
необычное восстановление а-карбонильной группы, вероятно
через такое же промежуточное соединение, как и в реакции
Кижнера—Вольфа; а-галогенкетоны при обработке гидрази-
ном дают соответствующие олефины29.
Ряд кетогидразонов был превращен в азоацетаты дейст-
вием, тетраацетата свинца30 в очень мягких условиях:
ZN=NR"
RR'C=N-—NHR" + Pb(CH3COO)4 —> RR'C^ (111-13)
-ОСОСНз
Для реакции был постулирован механизм, включающий
как медленную стадию, определяющую скорость, отрыв отно-
сительно реакционноспособного водородного атома от азота.
Перегруппировка в азосоединение с последующим отщепле-
нием ацетоксигруппы углеродным радикалом дает азоаце-
таты30.
Был описан31 интересный метод превращения альдегидов
в нитрилы через гидразониевые соли:
RCHO + (CH3)2N— NHa —> RCH=N—N(CH3)2 —?I->
—> RCH=N—N(CH3)3 QCH0BaHK% RC^N + N(CH3)3 (III-14)
Метод использует малую прочность связи N—N в кватер-
низованном гидразоне; эта связь легко разрывается с отры-
вом протона (при атоме углерода) под действием основания.
Хорошие выходы, легкость приготовления солей, а также при-
менимость к ароматическим и алифатическим альдегидам
Делает это превращение удобным методом приготовления
нитрилов.
3* 35
Способ получения нитрилов (Ш-14), предложенный почти одновремен-
но несколькими исследователями 31-33, является одним из частных случаев
аминонитрильной перегруппировки 34, впервые наблюдавшейся у незамещен-
ных в положении 3 четвертичных №-пиразолиниевых солей, гладко превра-
щающихся под действием щелочей в замещенные [3-диалкиламинопропиони-
трилы. Способность к аминонитрильной перегруппировке по схеме
—CH=N—N— Х~ + В~
N— +— C^N + HB + X" (Ш-15)
характерна для всех типов соединений, содержащих альдогидразониевый
фрагмент —CH=N—N—, вне зависимости от того, находится ли он в от-
крытой цепи (Ш-14), пиразолиниевом (П-5) или тетрагидропиридазиние-
вом35 кольцах. Щелочное расщепление четвертичных альдогидразониевых
солей и их аналогов может применяться не только для синтеза нитрилов
и амиионитрилов, но и для определения строения пиразолинов и тетрагидро-
пиридазинов (см. обзор 36 и указанную там литературу).
Гидразоны альдегидов также могут быть расщеплены на нитрилы и
амины при действии кислот или солей металлов 37, но в отличие от амино-
нитрильиой перегруппировки при этом требуется сильное нагревание, наблю-
даются побочные превращения и выходы получаются гораздо меньше.
Арилгидразоны ароматических альдегидов при нагревании с амидами
щелочных металлов или реактивами Гриньяра в кипящем ксилоле пере-
группировываются в амидины38. Доказан радикальный механизм этой
перегруппировки 39’40:
ArCH=N—NHAr' —> [ArCH=N •] + [• HNAr']
[ArCH=N •] —> [ArC=NH]
[ArC = NH] + [• HNAr'] —> АгС< (Ш-16)
XNHA/
Четвертичные кетогидразониевые соли [rRzC = N—NR"]x~ к амино-
нитрильному расщеплению не способны, и при действии на них оснований
в зависимости от природы радикалов R и R', щелочного агента и условий
имеют место различные реакции. Так, производные ацетофенона, фенилаце-
тона и дибензилкетона при нагревании с этилатом натрия в спиртовых
растворах превращаются в а-аминокетоны (Смит и Мост41):
C8H5-C=N-N(CH3)3r С6Н5—C==N +N(CH3)3 (Ш-17)
СН3 сн2
|н2о
CeH5COCH2NHa
Необходимым условием перегруппировки Смита и Моста является на-
личие в исходном ароматическом соедииеиии а-метиленовой группы, уча-
ствующей в образовании азиринового кольца.
При действии изопропилата натрия на иодметилат диметилгидразона
изобутирофеноиа удалось выделить 2-фенил-3,3-диметилазирин 4S,
36
Из производного пропиофенона в зависимости от количества изопро-
пилата натрия получается 2,5-дифенил-3,6-диметилпиразин VIII или (с из-
бытком изопропилата) 2-фенил-2-изопропокси-3-метилазиридин IX43;
CSH6CCH2CH3
II +
N—N(CH3)3I”
ызо-СдНуСЖа
изо-СзНуОН
csh5X/n. /сн.)
II I
СН3/'\n^\c6H5
избыток uso-CsHyONa
U3O-C3H7OH |
ф
(Ш-18)
с6н,х____уН
«зо-СзНуО-^^-^^СНз
«зо-С3Н70\
с6н/\/\сн3
IX
Новые усовершенствования в методике получения арил-
диазоалканов разложением n-толуолсульфонилгидразонов ще-
лочью по Бэмфорду—Стивенсу расширяют сферу распростра-
нения и применения этой реакции (см. гл. V):
IX
ArCH=N— NHSO2-^_^>-CH3 -£-> ArCHN2 + СН3— \ J/~S02 + ВН
~ (Ш-19)
Доступность исходных гидразонов44 и достаточно высокие
выходы арилдиазоалканов могут сделать этот метод полез-
ным. Прямое окисление гидразонов в диазоалканы будет рас-
смотрено в гл. V.
1,4-Присоединением галогена (например, хлора) к соответ-
ствующим азинам можно получить промежуточное [дихлор]-
азосоединение45:
Аг-. /Аг Аг-. /Аг
V-N-N=(/ + С!2 —> ^CC1N = NC1C/
RZ XR R/ ^R
(III-20)
Atx ,Ar
XCC1-CICZ + Nj
RZ XR
Термическое разложение этих азосоединений дает дихло-
рид, а в присутствии хлористого цинка азосоединение обра-
зует инден.
При окислении гидразоны, как сообщалось, дают элемен-
тарный азот и альдегиды, хотя выходы часто незначительны:
СН3
X ,CH=N-NH2
сн3
(Ш-21)
С активной двуокисью марганца в мягких условиях (бензольные рас-
творы. при комнатной температуре) превращение азинов и гидразонов в
карбонильные соединения идет гладко и почти количественно 46.
ФеннлгидразоНы в растворах очень легко окисляются кислородом воз-
духа с образованием азогидроперекисей 47’48 (раньше появление в спектрах
полос азогруппы в этих условиях ошибочно считалось следствием таутомер-
ного равновесия с азосоединениями).
Общая для феннлгидразонов типа C6H5NH—N = CRCHR2 реакция ката-
литического превращения в индолы (реакция Фишера) подробно освещена
в обзорах 4S’50 и в данной книге ке рассматривается.
Дигидразоны а-дикетонов окисляются до ацетиленовых
производных, вероятно через бисдиазосоединение:
R\ /R
\с— с/
II И
,N N,
H2NZ xnh2
n2 n2
—> RCsCR + 2N2 (III-22)
[Окислительное разложение дигидразонов а-дикетонов
было использовано для введения тройной связи в 8-, 9- и 10-
членные кольца Б|~53.
При окислении .«оногидразонов бензилкетонов трифтор-
ацетатом ртути также получаются ацетиленовые углеводо-
роды 54].
Реакцией диазониевой соли с азометином, получаемым из
альдегида и гидразина, были синтезированы формазаны: *
ArCH=N— NH—S—NO2 + ArN2Cl —>
>N—NH — <f~S—NO2
Ar-CX '=/
(III-23)
Эти соединения представляют особый интерес из-за воз-
можности их легкого превращения (при окислении) в тетра-
золиевые соли:
NH— NO2 yN\ ч
Аг— СГ Ar— С N-<^_^>-NO2 + ОН’
\n = N- Аг * I I ~ (Ш-24)
N==N— Ar
Образующиеся тетразолиевые соли имеют яркую окраску,
и поэтому равновесие (Ш-24) можно использовать для бы-
строго колориметрического определения окислителей или вос-
становителей.
Литература
Общая
Н. Ниньо, Химия и аналитические свойства гидразонов, Фармация (Со-
фия), 12, № 2, 21 (1962).
[О методах получения н реакциях формазанов см.м.]
88
A H. Кост, И. И. Грандберг, Альдазины и кетазины, Усп. хим., 28,
921 (1959).
*Ю. П; Китаев, Строение и свойства гидразонов. Доклад по работам,
представленным на соискание ученой степени доктора хим. наук, Ро-
стовский н/Д государственный университет, 1967.
‘ В. С, С т о [I с к и й, Моноалкилгидразоны, их свойства и изомерные пре-
вращения. Автореф. канд. дисс., ЛГУ, 1967.
*D. К о 1 b а с h, D. Koruncev, Azine, (Houben-Weii) Methoden der orga-
nischen Chemie, 4 Aufl,, X, 2, 85—122 (1967).
Цитируемая
1. P. Шрайнер, P. Фьюсон, Систематический качественный анализ
органических соединений, ИЛ, 1950.
2. N. Р. N е и г е i t е г, J, Am. Chem. Soc., 81, 2910 (1959).
*3 Б. В. Иоффе. В. С. Стопе кий, ДАН СССР, 175, 1064 (1967).
’4. Е. Schmitz, R. Ohme, Ann. Chem., 635, 82 (I960).
* 5. W. Skorianetz, E. Kovats. Tetrahedron Lett., 1966, 1960.
6. T. Kaufmann, G. Ruckelhauss, J. Schulz, Angew. Chem., 75,
1204 (1963).
* 7. Г. С. Гольдин, T. А. Балабина, С. Г. Федоров, ЖОрХ, 1,
1723 (1965).
* 8. Б. В. Иоффе, В. С. Стопский, 3. И. Сергеева, ЖОрХ, 4, 986
(1968).
* 9. Б. В. Иоффе, К. Н. Зеленин, ЖОХ, 33, 3589 (1963).
* 10. Б. В. Иоффе, Е. И. Сабинина, ЖОХ, 33, 2188 (1963).
* 11. Н. Л. Зеленина, Б. В. Иоффе, Тезисы И Всесоюзной конферен-
ции по химии пятичленных азотистых гетероциклов, Ростов-на-Допу,
1966.
* 12. Б. В. Иоффе, 3. И. Сергеева, В. В. Цибульский,
Н. И. Клюева, ЖОрХ, 3, 988 (1967).
* 13. Б. В. И оф фе, К. Н. 3 е ленин, ЖОрХ, 5, 183 (1969).
14. С. G. Overberger, I. Т a s h 1 i с k, J. Am. Chem. Soc., 81, 217
(1959); C. G. Overberger, M. Lapkin, ibid., 77, 4651 (1955).
*15. G. J. Karabatsos, R, A. Taller, Tetrahedron, 24, 3923 (1968) и
предыдущие статьи этой серин.
*16. M.-L. Filleux-Blanchard, G. J. Martin, Bull. Soc. chim.
France, 1968, 2618.
17. C. Vogel, M.. Matter, Helv. Chim. Acta, 42, 527 (1959).
* 18. Ю. П. Китаев, Л. E. H и в о p о ж к и н, С. А. Ф л е г о н т о в,
О. А. Раевский, 3. С. Титова, ДАН СССР, 178, 1328 (1968).
* 19 G, J. Karabatsos, R. A Taller, F. М. Vane, Tetrahedron Lett.,
1964, 1081.
* 20. Б. В. Иоффе, О. В. Свердлова, Л. М. Коржиков а, Теор. и
эксп. хим., 1967, 119.
* 21. .1. А. В а г 11 г о р, М. С о n 1 о n g. J. Chem. Soc., (В), 1967, 1081.
“22. И. А. Домнин, С. И. Я к и м о в и ч, ЖОрХ. 1, 658, 1024 (1965).
* 23. А. А. Потехин, Б. В. Иоффе, Tetrahedron Lett., 1967, 3505; ДАН
СССР, 179, 1120 (1968).
* 24. L. С. D о г m а п, J. Org. Chem., 32, 255 (1967).
25. В. М. Родионов, Н. Г. Я р ц е в а, в кн.: Реакции и методы иссле-
дования органических соединений, т. 1, Госхимиздат, 1951, стр. 7.
*26 D. Todd, The Wolf-Kishner Reduction. Org. Reactions, 4, 378 (1948).
27. H. S z m a n t, H. Harnsberger, T. Butler, W. В a r i e, J. Am.
Chem. Soc., 74, 2724 (1952); [H. H. Szmant, Angew. Chem., 80, 141.
(1968)].
28. J. S e m u s z k о v i c z, J. Med. Pharm. Chem., 4, 274 (1961).
29. P, S. Wharton, S.-Dunny, L. S. Krebs, J. Org. Chem., 29, 958
(1964); см. также N. J. Leonard. S. Gelfand. J. Am. Chem. Soc.,
77, 3269 (1965).
30. D. S. Iff land, L. Salisbury, W. R. Schafer, J. Am. Chem.
Soc., 82, 747 (1961).
39
*49.
31. R. F. Smith, L. E. Walker, J. Org. Chem, 27, 4372 (1962).
* 32. Б. В. Иоффе, 3. И. Сергеева, К- М. Д е р в и н с к а й т е, ЖОХ,
33, 2794 (1963).
* 33. И. И. Гр андберг, ЖОХ, 34, 569 (1964).
* 34. Б. В. Иоффе, К. Н. Зеленин, ДАН СССР, 144, 1303 (1962); ЖОХ,
33, 3231 (1963).
* 35. К. Й. Зеленин, В. Г. Камердинеров, ЖОрХ, 1, 1899 (1965);
ХГС, 1968, 530.
* 36. К. Н. Зеленин, Б. В. Иоффе, Вести. ЛГУ, № 16, 159 (1968).
* 37. Ю. А. Наумов, И. И. Г р а н д б е р г, Усп. хим., 35, 21 (1966).
* 38. Ст. Робев, ДАН СССР, 101, 277 (1955); Докл. Болг. АН, 7, 37
(1954); 8, 39 (1955); 12, 137, 141 (1959); 14, 353 (1961); Изв. Хим.
инет. Болг. АН, 3, 495 (1955); Вег., 91, 244 (1958).
* 39. Ст. Робев, Д. Д. Цитович, Докл. Болг. АН, 17, 738 (1964).
* 40. И. И. Грандберг, Ю. А. Наумов, А. Н. Кост, ЖОрХ, 1, 805
(1965).
* 41. Р. S m i t h, E. M о s t, J. Org. Chem., 22, 358 (1957).
* 42. R. F. Par cell, Chem. a. Ind., 1963, 1396.
* 43. Sh. Sato, Bull. Japan Chem. Soo., 41, 1440 (1968).
44. D. Q. F a г n u m, J. Ogr. Chem., 28, 870 (1963); G. L. Closs,
R. A. Moss, J. Am. Chem. Soc., 86, 4042 (1964); H. Shechter et al.,
ibid., 87, 935 (1965).
45. S. Goldschmidt, B. Ackstqiner, Chem. Ber., 91, 502 (1958).
* 46. G. Maier, U. Hepp, Angew. Chem., 77, 967 (1965).
* 47. A. J. Bellamy, P. X. Guthrol, Chem. a. Ind., 1964, 1575; J. Chem.
Soc, 1965, 2788, 3528; 1966 (C), 1989.
* 48. А. В. Чернова, P. P. Шаги дул л ин, Ю. П. Китаев, ЖОрХ, 3,
916 (1967).
Н. Н. Суворов, В. П. Мамаев, В. М. Родионов, в кн.: Реак-
ции и методы исследования органических соединений, т. 9, Госхимиз-
дат, 1959, стр. 1.
* 50. Ю. П. Китаев, Усп. хим., 28, 336 (1959).
* 51. А. Т. Blomquist, R. Е. В и г d е, Jr., А. С. S и с s у, J. Am. Chem.
Soc., 74, 3636 (1952).
* 52. А. Т. Blomquist, L. Н. Liu, Y. С. Bohrer, J. Am. Chem. Soc.,
74, 3643 (1952).
* 53. A. T. Blomquist, L. H. Liu, J. Am. Chem. Soc, 75, 2135 (1953).
* 54. R. J. Theis, R. E. D e s s y, J. Org. Chem, 31, 624 (1966).
* 55. R. P ii 11 e r, Formazane, (Houben-Weil) Methoden der organischen Che-
mie, 4 Aufl, X, 3, 627 (1965).
Глава IV. АЗОСОЕДИНЕНИЯ
Для азосоединений R—N = N—R' характерна ненасыщен-
ная группа азот—азот.
Ароматические азосоединения (где R и R' — ароматиче-*
ские заместители)—ярко окрашенные вещества, чем объяс-
няется их широкое использование в производстве красителей.
Напротив, большинство алифатических азосоединений (R, R'
или оба — алифатические группы) бесцветны. Еще большие
различия наблюдаются как в свойствах, так и в методах по-
лучения алифатических и ароматических азосоединений. При
обсуждении эти два типа будут рассмотрены отдельно, хотя
очевидно, что некоторые свойства могут быть общими.
По азосоединениям имеется превосходная книга Цоллин-
гера 1 и другие менее полные обзоры2-4.
АРОМАТИЧЕСКИЕ АЗОСОЕДИНЕНИЯ
В этом разделе будут рассмотрены только соединения,
имеющие оба ароматических заместителя; все прочие будут
обсуждены вместе с алифатическими.
Получение
Более века назад Грисс открыл, что ароматические соли
диазония (гл. V) сочетаются с активированными ароматиче-
скими соединениями, приводя к ярко окрашенным веществам,
которые стали известны как «азокрасители»:
Эти синтетические красители нашли широкое применение,
и значение их все возрастает. Хотя применение реакции азо-
сочетцния ограничивается активированными ароматическими
соединениями (аминами, фенолами), она является наиболее
широко используемым методом синтеза ароматических
41
азосоединений. Существуют хорошие обзоры этой реакции '•4-5.
Достаточно упомянуть, что атака имеет место почти исключи-
тельно в пара-положение к активирующей группе и в орто-,
если пара-положение блокировано. Сочетание проходит мгно-
венно. Активные метиленсвые группы также могут реагиро-
вать с солями диазония (см. гл. V).
Несмотря на популярность и практичность реакции азосо-
четания, были найдены многочисленные способы синтеза аро-
матических азосоединений, дополняющие этот метод и заме-
няющие его, когда он неприменим. Например, восстановление
нитро-, N-нитрозо- и азоксисоединений приводит к азосоеди-
иениям:
2Ar—NO2
восстановление
------------> Аг—N —N—Аг
(IV-2)
Окисление ароматических аминов или их конденсация с
ароматическими нитрозосоединениями также дает азосоеди-
нения:
„. окисление , конденсация .
2Аг—ЫН2 --------> Ar—N = N—Аг <----------- Аг—N = O + Ar—NH2
(IV-3)
Азосоединения могут быть получены окислением арома-
тических гидразинов, конденсацией их же с хинонами, раз-
ложением солей диазония или перегруппировкой триазенов
(перегруппировка типа Фишера—Геппа). Азоксисоединения
претерпевают перегруппировку Валлаха с образованием окси-
азосоединений. Интересным, хотя практически и невыгодным
является диспропорционирование гидразосоединений. Было
показано, что гидразобензол пиролизуется на анилин и азо-
бензол без разрыва связи N—N в получающемся азобензоле,
т. е. гидразобензол окисляется в азосоединение.
Реакции
Под влиянием света тр««с-азосоединения (устойчивая фор-
ма) могут изомеризоваться в цис-изомеры. Азо-связь очень
устойчива к нагреванию и не подвергается воздействию мно-
гих реагентов. Систематических исследований реакций азо-
соединений не проводилось. Как и следовало ожидать,
азогруппа является орто-, пара-ориентантом в реакциях
электрофильного замещения. Азосоединения можно восста-
навливать в соответствующие гидразосоединения, а при даль-
нейшем восстановлении связь N—N разрывается с образова-
нием аминов.
При окислении азосоединений образуются азоксисоеди-
нения.
Метилзамещенные ароматические соединения могут при
высоких температурах расщеплять азосоединения на шиф-
42
фово основание и амин, вероятно с промежуточным образова-
нием продукта присоединения:
С»Н5—N=N—С6Н5 + С6Н5СН3 —[С6Н6—N—NH—С6Н5] —>
СН2С6Н5
—> C6H5-N = СНС6Н5 + С6Н5—NH2 (IV-4)
Продукт присоединения арилсульфиновых кислот по свя-
зи —N = N— может быть выделен. Щелочные металлы дают
производные гидразосоединений. цмс-Азобензол присоединяет
дифенилкетен (IV-5):
СбН5 С6Н5Х ,СеН5
N—С6Н5 I „ XN-NZ
|| + С=С = О —>С6Н5, | | (IV-5)
N— С6Н, I С—С=О
с6н5 с6н/
Следует упомянуть об образовании бензо-[с]-циннолина I
при получении цис-азобензола:
/N=N\ /n=n-x
\ \ °2 (?) > \ у-\ у (IV-6)
I
АЛИФАТИЧЕСКИЕ АЗОСОЕДИНЕНИЯ
В этом же разделе будут рассмотрены все неароматиче-
ские (по определению на стр. 41) азосоединения, включая
чисто алифатические (оба заместителя — алкилы) и «смешан-
ные» (один заместитель—алкил, другой — арил). Эфиры и
амиды знилазосоединений рассматриваются в гл. VI.
Получение
Важнейший метод синтеза алифатических азосоединений
состоит в окислении соответствующих гидразинов:
R-NH-NH-R' окисление> R_N==N_R' (IV.7)
Для этой цели применялось много окислителей, лучшим
оказалась окись ртути. Однако есть и другие пути введения
азогруппы. Например, реакция солей диазония с цианид- или
бензолсульфинат-ионами дает азосоединение:
C6H5-N = NC1~ + X~ —► C6H5-N = N—X (IV-8)
Х= —CN; —SO2C6H5 (см. гл. V)
Иногда возможно окислить N-галогенамины в соответ-
ствующие азосоединения. Неясно, проходит ли реакция че-
рез «нитрен» или по механизму замещения—отщепления.
«Смешанные» .азосоединения можно получить взаимодей-
ствием солей диазония с цинкалкилами. Эта реакция
43
приводит к тому же результату, что и реакция азосочетания
с карбанионами (см, гл. V), хотя, возможно, и по другому
механизму:
R—ZnCl + С6Н5—N+ = NCF —> R—N = N—CsH5 + ZnCl2 (IV-9)
Бенциг6 расширил и изучил хлорирование кетазинов, впер-
вые описанное Гольдшмидтом и приводящее к а, а'-дихлор-
азосоединениям. Из этих дихлорпроизводных можно полу-
чить многочисленные а, а'-дизамещенные азосоединения:
Ry /Я Ry ZR
\:=N—N = C/ + Cl2 —► VC1N=NC1C/ (IV-10)
R/Z \R, R,Z \RZ
В некоторой степени сходным является окисление гидразо-
нов тетраацетатом свинца до а-ацетоксиазосоединений (азо-
ацетатов) :
Ry Ry ОСОСНз
J)C=N— NH—R" + Pb(CH3COO)4 —> (IV-11)
R'Z RZ \n=N—R"
Эта реакция была впервые описана Ифлендом, Солсбери
и Шефером7 и распространена на Д2-пиразолины Фриманом 8:
_______________________________/
R'\| N + Pb(CH3COO)4 —> R'\| nXOCOCH3 (IV-12)
r"/xnh r'"/Xi/z
Фторированием нитрилов9 были синтезированы перфтор-
азоалканы, которые при термическом и фотохимическом раз-
ложении дают перфтор алкильные радикалы9’10.
Было показано, что реакция диазоалканов с олефинами
(см. гл. V) приводит к пятичленным циклическим азосоедине-
ниям (А’-пиразолинам) даже в случае, когда структура бла-
гоприятствует образованию более устойчивых [циклических]
гидразонов 12:
+ i ГСбНй
С6Н5СН = СН2 + С6Н5СН—N = N —> r „ | N (IV-13)
СбН5—\ zz
Недавно 13 сообщалось
в азосоединение III*
о прямом восстановлении
азина II
__РИМ, _
CHjCOOH
in
(IV-14)
* [Образование аэосоединеаий при каталитическом гидрировании ази-
нов наблюдалось еще Тайпале 144
44
Удобным и во многих случаях наиболее целесообразным способом полу-
чения алифатических и жирпоароматических азосоединений может служить
изомеризация монозамещенных гидразонов при нагревании со щелочами
с последующей постепенной отгонкой более летучего азосоединения на ко-
лонке Аналогичный способ может быть использован и для получения
некоторых циклических азосоединений (5,5-диметил-Д’-пиразолина из Д2-нзо-
мера 18).
аф-Ненасыщенные алифатические азосоединения были получены толь-
ко недавно при реакции моноалкилгидразинов с а-хлорзамещенными карбо-
нильными соединениями 19'2<|:
RCHC1COR' + 2H2N—NHR" —>
—► H2N—NHR" • НС1 + H2O + RCH = CR'—N=N—R" (IV-15)
IV
Реакция фтористого серебра с дицианом дает перфторсо-
единение, для которого была предложена четырехчленная
циклическая структура, включающая азо-связь2’. Оно терми-
чески разлагается до тетрафторэтилена:
F F
AgF + (CN)2 —> F—1---LF l50°c-> F2O=CF2 + N2 (IV-16)
N = N
О получении и реакциях диазиринов (трехчленных цикли-
ческих азосоединений) см. гл. V..
Реакции
В противоположность ароматическим аналогам большин-
ство алифатических азосоединений относительно непрочно и
претерпевает гомолитический распад под влиянием тепла и
света. Лабильность алифатических азосоединений будет, оче-
видно, зависеть от природы заместителей; «смешанные» арил-
алкилазосоединения, как и следовало ожидать, являются
более стойкими. Газ азометан (СН3—N = N—СН3) заметно
не разлагается до 200° С. Однако, если ввести а-заместители,
способные стабилизировать получающиеся свободные ради-
калы, гомолитический распад происходит легче. Простран-
ственные факторы также могут играть решающую роль, как
будет показано на примере циклических и стерически затруд-
ненных азосоединений.
Кроме склонности алифатических азосоединений к гомо-
литическому распаду досадным фактором, с которым следует
считаться при наличии ос-водорода, часто является изомери-
зация в гидразоны (особенно, если в результате такой изоме-
ризации образуется сопряженная система). Этот вопрос был
изучен О’Коннором22.
Положение равновесия азосоединение гидразон в алифатическом
ряду было установлено методом газо-жидкостной хроматографии, причем
оказалось, что содержание азосоединений в равновесной смеси при 100° С
43
достигает нескольких процентов23. Оценка содержания азо-формы в рав- j
новесии с фенилгидразонам бензальдегида дает величину в миллион раз
меньшую24, так как в этом случае гидразонная структура дополнительно
стабилизирована длинной цепью л — л — р — л-сопряжения.
Реакции Дильса—Альдера для азодиацилов с диенами и
другими соединениями будут рассмотрены в гл. VI.
[Непредельные азосоединения могут вступать в реакцию
Дильса—Альдера в качестве диенового компонента, образуя
д2-тетрагидропиридазины25.]
Азоалканы дают устойчивые продукты присоединения с со-
лями одновалентной меди26.
Гомолитическое разложение. Важнейшей по зна-
чению реакцией алифатических азосоединений является их
термическое разложение и фотолиз до азота и свободных ра-
дикалов. Интерес к механизму этого разложения — как прак-
тический, так и теоретический — вновь возник благодаря из-
вестным исследованиям Льюиса27 и Овербергера28, посвя- '
щенным кинетике разложения азобис(изобутиронитрила) и
его применению в качестве инициатора полимеризации. Хотя
проблема одновременного разрыва двух связей углерод—азот
была трудноразрешимой, теперь установлено, что обе С—N-
связи вносят вклад в стадию, определяющую скорость. Это
находится в соответствии с предположением Рамспергера29,
который изучал парофазное разложение серии азоалканов.
Коэн и Вэнг31 сравнили скорость разложения азометана,
азобис(изопропана), 1-азобис(1-фенилэтана) и азобис(дифе-
нилметана) и установили, что замещение а-водорода метиль-
ными или фенильными заместителями оказывает приблизи-
тельно аддитивное влияние на уменьшение энергии активации.
Это наблюдение в сочетании с дополнительным сравнением
бензолазодифенилметана с бензолазотрифенилметаном и азо-
бис (дифенилметаном) привело их к заключению, что такой
эффект вызван в основном резонансной стабилизацией ради-
калов, образующихся при разложении. Дополнительное под-
тверждение вывода, что оба заместителя азо-связи участвуют
в лимитирующей стадии, можно найти в данных табл. IV-1.
При сравнении соединений V, IX и XIII видно, что замена
метильной группы в XIII фенилом V уменьшает Еа прибли-
зительно на 4 ккал/моль, последующее замещение двух ме-
тилов в XIII двумя фенилами IX вызывает снижение на
8 ккал/моль, т. е. эффект, как отмечалось выше, аддитивен.
Увеличение реакционной способности предсказать труднее,
так как она зависит как от Еа, так и от S, но вклад замести-
телей в обоих случаях будет значительно больше статистиче-
ского фактора 2, ожидаемого, если имеет место ступенчатое
разложение. Как аддитивный эффект, так и увеличение ско-
рости согласуются с представлением, что оба радикала вно-
сят вклад в стадию, определяющую скорость. Эта интерпре-
46
Таблица IV-1
Разложение арилалкилазосоединеиий30
I I
В—С—N=N—С—F
I I
D G
Соедине- ние Заместители Скорость‘104 при 120° С, сек~1 Энергия активации Еа- ккал/молъ Энтропия активации S, а. е.
А в D Е F G
V СНз С6Н5 н СНз . Н СНз 0,132 36,5 9,3
VI СНз С6н5 СНз СНз н СНз 1,06 36,7 14
VII с2н5 С6н5 с2н5 С2Н5 н С2Н5 0,78 39,0 19
VIII СНз С6Н6 мзо-С4Н9 СНз н мзо-С4Н9 8,51 35,2 15
IX СНз С6н5 н СНз С6Н5 н 4,85 32,6 7
X с2н5 С6Н5 н С2Н5 С6н5 н 2,06 32,3 —
XI мзо-С4Н9 С6Н5 н «зо-С4Н9 С6н5 н 7,11 33,3 9
XII Н С6Н5 н Н С6н5 н 0,045 35,0 5
XIII СНз н сн3 СНз н СНз 0,00001 40,9 1
тация в дальнейшем была подтверждена величиной вторич-
ного изотопного эффекта а-дейтерия, найденной для азобис-
а-фенилэтана-а, a'-Df. Наблюдаемый эффект Ah/^d = 1,27,
т. е. приблизительно вдвое больше, чем у других «мономоле-
кулярных» реакций; это показывает, что обе С—N-связи
рвутся одновременно в переходном состоянии лимитирующей
стадии. В итоге, механизм разложения может быть изобра-
жен следующим образок:
СН3 СНз
Н—С—N = N—С—Н
I I
С6н5 С6Н5
СНз ~
N.С-Н
I
с6н5_
—> 2С6Н5—С • + М2 —> 2,3-дифенилбутан
Н
(IV-17)
Как предсказывали Коэн и Вэнг, главным фактором, опре-
деляющим устойчивость алифатических азосоединений, яв-
ляется резонансная стабилизация фрагментов — радикалов,
образующихся при разложении. Таким образом, по относи-
тельной скорости приведенные ниже группы заместителей
можно расположить в ряд:
СН3 < изо-С3Н7 < С6Н5СН2 < С6Н5СНСН3 < (CH3)2CCN «
« (СНз)2ССООСНз < (С6Н5)2СН < (С6Н5)зС
Влияние стерического фактора на скорость разложения
изучено Овербергером с сотрудниками33 в ряду азонитрилов
формулы
CN
XIV
(R и R' — алкильные заместители)
Когда R и R'—метил, этил, изопропил или трет-бутил, на-
блюдаемые скорости одинаковы, что означает отсутствие ка-
ких-либо изменений в переходном состоянии. Однако при раз-
ветвлении у p-углеродного атома (от азо-связи) наблюдалось
резкое увеличение скорости. Эти результаты в сочетании с
аналогичными данными, полученными Хьюсоном с сотруд-
никами34 для карбамилазонитрилов [RR'(CN)C—N = N—
—CONH2], у которых стерическое взаимодействие между
двумя половинами молекулы невозможно, указывает, что
пространственный эффект обусловлен, вероятно, «В-напря-
жением», т. е. снятием напряжения на а-углеродном атоме
48
при переходе от тетраэдрической к плоскостной конфигура-
ции в переходном состоянии. Этот эффект ясен из табл. VI-1,
где представлено последовательное увеличение р-замещения
в соединениях VI и VII.
Поскольку разложение сравнительно нечувствительно к
влиянию растворителя35, была предложена изящная мето-
дика для демонстрации «/-напряжения» (циклическое напря-
жение) в свободнорадикальных реакциях36. В табл. IV-2 сум-
Таблица IV-2
Скорости разложения азонитрилов
CH2 CN
(сн2)„с СНз
СН2 N С (СН2)„
CN СН2
XV
Соединение Скорость-104 при 80° С, сек~1 Энергия активации Еа, [ккал!моль] Энтропия активации S, [э. е.|
XV, п -1 0,00173 32,1 1,3
XV, п = 2 0,726 33,8 18,9
XV, п = 3 0,063 35,4 17,8
XV, п - 4 12,12 27,5 6,1
XV, п = 5 83,5 25,9 5,9
XV, п = 7 18,12 28,0 8,5
мированы результаты, полученные для ряда циклоалкилазо-
нитрилов. Повышенная скорость разложения соединения XV,
п = 2, по отношению к XV, п = 3, определяется тенденцией к
снятию циклического напряжения в переходном состоянии
для циклопентильного производного. Скорости разложения
соединений с большими циклами также согласуются с пред-
положением «/-напряжения» по Брауну.
Такое же исследование было проведено в ряду метилци-
клоалкилазонитрилов37’38, и никакого заметного изменения
в скорости для циклов различных размеров, исключая цикло-
пропановое кольцо, не наблюдалось. При разложении 2,2'-
азобис(циклопропилпропионитрила) наблюдается двадцати-
кратное увеличение скорости, что было интерпретировано как
пример стабилизации радикала вследствие гиперконъюгации
в циклопропильном кольце.
В общем, продукты этих разложений — вещества, которые
получаются при сочетании двух образующихся радикалов или
в результате отрыва водорода от молекулы растворителя.
Предположение о промежуточном образовании диметил-М-(2-
циано-2-пропил) кетенимина XVI было недавно подтвержде-
но его выделением39. Как показано ниже, кетенимины
4 Зак. 334
49
образуются после лимитирующей стадии, включающей выде-
ление азота, и, следовательно, не влияют на ранее обсужден-
ные константы скоростей первого порядка:
СН3
CN |
I - ,N4X Х-СНз
сн3-с/ %Z|
I CN
СНз
СН3.
—> 2 'C-C^N —>
медленно /
СНз
СН3 СНз
СН3-С С—СН3
CN CN
(IV-18)
СНзх I
C = C = N—С—СНз
сн/ 1
CN
Н3С-С‘
I
CN „
СНзч
‘C = C«=N-
CH3Z
XVI
Роль такого промежуточного соединения была продемон-
стрирована сравнением способности инициировать полимери-
зацию стирола исходным азосоединением и соответствующим
ему кетенимином. Кетенимин оказался менее эффективным,
и, следовательно, пониженную активность азобис(изобутиро-
нитрила) можно гораздо лучше объяснить этим фактором,
чем ранее предложенным нерадикальным циклическим меха-
низмом (IV-19):
снзч
/С,) ,,-С—СНЭ
сн3 C-N CN
СНз СН,
N2 + ^C==C = N —С—СН3
CH3 CN
Циклический механизм несостоятелен, так как известны
только транс-конфигурации линейных азосоединений, а обра-
зование шестичленного цикла, содержащего транс-азо-связь,
трудно представить.
Влияние стерических факторов было продемонстрировано
эффектом включения азо-связи в циклическую структуру
XVII, где она оказывается в zfuc-конфигурации (табл. IV-3):
XVIU
Можно отметить и некоторые другие особенности. Исклю-
чительно низкая энергия активации для пятичленного цикли-
ческого соединения XVII, п=1, подтверждает квазипланар-
ную конфигурацию этого цикла и связанное с ней напряже-
но
Таблица IV-3
Данные по разложению циклических азосоединений XVII и XVIII
в сравнении с нециклическими
Скорость Относи- Энергия активации Лите-
Соединение тельная Е„, ратура
сек 1 скорость ккал1моль
С6Н5Х /CSH5
Vh-n=n—ch 1,9- 10“’ 1 36,7 41
С2Н5/ XCeH5
XVII, и = 1 1,2-10“3 631 17,9 12
XVII, и = 2 6,0- 10“4 375 — 44
XVII, н = 3 4,3- 10“4 268 29,7 45
XVII, и=4 3,5- 10“ь 0,02 36,7 41
ние12’40. Аномально малая скорость разложения восьмичлен-
ного цикла объясняется затруднениями системы С—N = N—С
стать копланарной с a-заместителями41'42. В соединениях
типа XVIII можно полагать транс-конфигурацию азо-связи и
поэтому [энергия активации]* такая же, как и у азобис(1-фе-
нилциклопропана) .
Термический распад циклических азосоединений типа
XVII дает интересные результаты. Семи- и восьмичленные
циклические гомологи (п = 3, 4; предположительно один изо-
мер в каждом случае) приводят к смеси цис и транс-1,2-
дифенилциклоалканов и соответствующих олефинов, в то
время как шестичленный цикл (и = 2) дает стирол. Напротив,
транс-пятичленный цикл (и = 1) дает транс- 1,2-дифенилцик-
лопропан в качестве основного продукта разложения. При
этом предполагается, что промежуточные бирадикалы, обра-
зованные из семи- и восьмичленных колец, являются свобод-
ными и достаточно устойчивыми, чтобы «рацемизоваться» до
их сочетания; в случае же соединения с пятичленным цик-
лом образование связи происходит очень быстро40. Недавно
было показано12, что цис-3,5-дианизил-А!-пиразолин дает
смесь соответствующих цис- и транс-циклопропанов.
Большая лабильность 9-фенилфлуоренилазобензола по
сравнению с соответствующими трифенилметилазобензолами
приписывалась большей стабилизации получающихся свобод-
ных радикалов43.
Эффект напряжения, очевидный из табл. IV-3 (скорость
падает для XVII при изменении и от 1 до 4), был дополни-
тельно иллюстрирован сравнением скоростей разложения
XIX и XX. Оказалось46, что XIX разлагается в 380 ра.^
* [В оригинале — скорость.]
4*
51
быстрее, чем XX, и его энергия активации ниже на
7 ккал/моль', кинетических данных для интересного квадрици-
клического азосоединения XXI47 нет-
Берсон и сотрудники 4« недавно показали, что получаются
одинаковые продукты из соединений XXII и XXIII:
В зависимости от природы и реакционной способности об-
разующиеся свободные радикалы будут реагировать с рас-
творителем (такие активные радикалы, как СН3-), димеризо-
ваться или атаковать реакционноспособный субстрат. Невоз-
можность уловить некоторые активные свободные радикалы
с помощью чувствительных к ним веществ привела к пред-
положению о действии «клеточного эффекта» 49; окруженные
молекулами растворителя свободные радикалы не могут
диффундировать через так называемую «клетку» растворителя.
Это было подробно показано Хэммондом и другими50’51.
Литература
Общая
Н. Zollinger, Diazo and Azo Chemistry: Aliphatic and Aromatic Com-
pounds, New York, 1961.
C. G. Overberger, New Reactions of 1,1- and 1,2-disubstituted Hydrazi-
nes, Record Chem. Progr., 21, 21 (1960).
*E. M u 11 e r, Aliphatische Azo- und Azoxyverbindungen, (Houben-Weil)
Methoden der organischen Chemie, 4 Aufl., X, 2, 757—802 (1967).
*E. Muller, Azoalylverbindungen, (Houben-Weil) Methoden der organi-
schen Chemie, 4 Aufl., X, 2, 803—822 (1967).
Цитируемая
1. H. Zollinger, Diazo and Azo Chemistry: Aliphatic and Aromatic
Compounds, New York, 1961.
2. C. G. Overberger, Record Chem. Progr., 21, 21 (1960).
3. A. Guillemonat, A. Gislon, in V. Grignard (ed.), Traite de
Chimie Organique, vol. 15, Paris, 1948, r. 257.
4. E. H. Rod d, Chemistry of Carbon Compounds, vol. 3-A, New York
1954, p. 319.
5. К. H. Saunders, The Aromatic Diazo Compounds, London, 1949.
6. E. Benzig, Chemia, 13, 89 (1959); Angew. Chem., 72, 571 (I960).
62
7. О. С. I f f 1 a n d, L. Salisbury, W. R. Schafer, J. Am. Chem.
Soc., 83, 747 (1961).
8. J. P. Freeman, J. Org. Chem. ,28, 885 (1963).
9. J. A. Young, W. S. Du г re 11, R. D. Dresdner, J. Am. Chem.
Soc., 82, 4553 (1960); W. J. C h a m b e r s, C. W. T u 11 о c k, D. D. С о 1 f-
man, ibid., 84, 2337 (1962); J. H. Attaway, R. H. Groth,
L. A. Bigelow, ibid., 81, 3599 (1959).
10. J. A. Young, R. D. Dresdner, J. Org. Chem., 28, 833 (1963).
11. C G. Overberger, J.-P. Aaselme, J. Am. Chem. Soc., 84, 869
(1962); 86, 658 (1964).
12. C. G Overberger, N. Weinshenker, J.-P. A n s e 1 m e, J. Am.
Chem. Soc., 86, 5364 (1964); 87, 4119 (1965).
13. J. Kossanyi, Compt. rend., 257, 929 (1963); Bull. Soc. chim. France,
1965, 722.
* 14. К- А. Тайпале, ЖРХО, 56, 81 (1924).
* 15. Б. В. Иоффе, 3. И. Сергеева, В. С. Стопский, авт. свид. 174188,
1964; Бюлл. изобр., № 17 (1965); ДАН СССР, 167, 831 (1966).
* 16. Б. В. Иоффе, В. С. Стопский, ДАН СССР, 175, 1064 (1967).
* 17. Б. В. Иоффе, Л. М. Герштейн, ЖОрХ, 5, 268 (1969).
”18. Б. В. Иоффе, Д. Д. Ц и т о в и ч, ХГС, 1965, 559.
* 19. В. Т. G i 11 i s, J. D. H a g a r t y, J. Am. Chem. Soc., 87, 4576 (1965).
* 20. 3. M. Матвеева, А. П. Кочетов, 3. И. Сергеева, К. Н. Зе-
ленин, Б. В. Иоффе, ЖОрХ, 5, 424 (1969).
21. Н. J. Emeleus, G. L. Hurst, J. Chem. Soc., 1962, 3276.
22. R. O’C о n n о r, J. Org. Chem, 26, 4375 (1961); R. O’C о n n о r, W. R o-
senbrook, J. Am. Chem. Soc., 26, 5208 (1961).
* 23. Б. В. Иоффе, В. С. Стопский, ЖОрХ. 4, 1504 (1968).
* 24. Н. Simon, W. Moldenhauer, Ber., 100, 1949 (1967).
* 25. К. Н. Зеленин, 3. М. Матвеева, ЖОрХ, 4, 532 (1968); ДАН
СССР, 184, 1105 (1969).
26. О. D е i 1 s, W. К о 11, Ann, 443, 262 (1925).
27. F. М. Lewis, М. S. Matheson, J. Am. Chem. Soc, 71, 747 (1949).
28. C. G. Overberger, M. T. O’S haughnessy, H. Shalt, J. Am,
Chem. Soc, 71, 2661 (1949).
29. H. C. R a m s p e r g e r, J. Am. Chem. Soc, 51, 2141 (1929).
30. C. G. Overberger, A. V. D i G i u 1 i o, J. Am. Chem. Soc, 81, 2154
(1959).
31. S. G. Cohen, С. H. Wang, J. Am. Chem. Soc., 77, 2457 (1955); 75,
5504 (1953).
32. S. Seltzer, J. Am. Chem. Soc, 85, 14 (1963).
33. C. G. Overberger et al, J. Am. Chem. Soc, 73, 2618, 4880, 4883
(1951); 76, 6185 (1954).
34. A. M. Hyson et al. Abstracts of the 129th Meeting of the American
Chemical Society, Dallas, Tex, April 8 to 13, 1956, p. 6R.
35. J. E. Leffler, R. A. Hubbard, J. Org. Chem, 19, 1089 (1954);
J. G. Adler, J. E. Leffler, J. Am. Chem. Soc, 76, 1425 (1954);
R. C. Peterson, J. H. M a r k g r a f, S. D. Ross, ibid, 83, 3819
(1961).
36. C. G. О v e r b e r g e r et al, J. Am. Chem. Soc, 75, 2078 (1953).
37. C. G. Overberger, A. L e b о v i t s, J. Am. Chem. Soc, 76, 2722
(1954).
38. C. G. Overberger, M. T о r k e s, A. Zweig, J. Org. Chem, 28,
620 (1963).
39. M. Talat-Erben, S. В у w a t e r, J. Am. Chem. Soc, 77, 3710, 3712
(1955); G. S. Hammond et al, ibid, 81, 4878 (1959); 82, 5386, 5394
(1960).
40. C. G. Overberger, J. P. Ansel me, J. Am. Chem. Soc, 86, 658
(1964).
41. C. G. Overberger, I. Tash lick, J. Am. Chem. Soc, 81, 217
(1959).
53
42. C. Q. Overberger, J. P. Ansel me, J. R. Hall, J. Am. Chem.
Soc, 85, 2752 (1963).
43. S. Q. Cohen, F. Cohen, С. H. Wang, J. Org. Chem, 28, 1479
(1963).
44. S. G. Cohen, S. Hsiao, E. S а к I a d, С. H. Wang, J. Am. Chem.
Soc., 79, 4400 (1957).
45. C. G. Overberger, J. C. Lombardin o, J. Am. Chem. Soc, 80,
2317 (1958).
46. S. G. Cohen, R. Zand, J. Am. Chem. Soc, 84, 586 (1962).
47. R. M. Mo r la r ty, J. Org. Chem, 28, 2385 (1963).
48. J. A. В e r s о n, C. J. Olson, J. S. Walia, J. Am. Chem. Soc. 84,
3337 (1962).
49. J. Frank, E. Rabinovitch, Trans. Faraday Soc, 30, 120 (1934).
50. G. S. H a m mo n d, R. C. N eu m a n, J. Am. Chem. Soc, 85, 1501
(1963).
51. L. Herk, M. Feld, M. Szwarc, J. Am. Chem. Soc, 83, 2998 (1961);
R. K. Lyon, D. H. Levy, ibid, 83, 4290 (1961); S. Ko da ma, Bull.
Chem. Soc. Japan, 35, 827 (1962).
Глава V. ДИАЗОСОЕДИНЕНИЯ
Алифатические и ароматические диазосоединения содер-
жат группу из двух атомов азота, связанную с одним угле-
родным атомом. В диазоалканах группа — N2+ связана с пер-
вичным или вторичным С-атомом, который, теряя протон,
дает нейтральную стабилизированную резонансом молекулу:
R\ + _ н+ R\- + R\-
/СН—NsN /С—NsN -<-> V—N = N+ <->
R'Z +н R'/ R'/
I Па Пб
R\ + Rx +
V = N=N~ С—N = N"
R'z R'Z
Пв Hr
Свойства, а также синтез диазоалканов можно понять,
лишь учитывая возможность потери а-протона, так как без
этой важной стадии соединения типа I должны немедленно
выделять азот. Третичные алифатические диазосоединения
+
R3CN = NX~ не способные к такой резонансной стабилиза-
ции, неизвестны, хотя им и придается большое значение как
активным промежуточным соединениям в реакциях алифа-
тических аминов с азотистой кислотой. Резонансное взаимо-
действие диазогруппы с ароматическим ядролСв солях диазо-
ния III до некоторой степени подавляет тенденцию к потере
азота.
Nsn]x" \=N = N-jx" ^~\ = N = N"]x"
Ilia Шб Шв
Тем не менее большинство солей диазония могут суще-
ствовать только при низких температурах и уже при слабом
нагревании разлагаются со взрывом, выделяя азот *.
* Недавно было сообщено о получении устойчивых неароматических
солей диазония *>2 [см. также 3~5].
55
В некоторых особых случаях протон отщепляется от ато-
ма, значительно удаленного от диазогруппы (при образова-
нии диазооксидов IV и диазоиминов V):
NHTos
IVa IV6
N=N—Tos
— TosH
N—Tos
Va V6
При добавлении даже очень слабокислых агентов к рас-
творам диазоалканов образуются формы I, которые немед-
ленно теряют азот (см. раздел «Реакции»),
Стабильность диазоалканов повышается, когда радикалы
R и R' в II способны к резонансному взаимодействию с
а-углеродом. При замещении водорода и алкильных групп
на ароматические или карбонилсодержащиие радикалы VI
алифатические диазосоединения становятся более устойчи-
выми:
ZN = N
С
II
С
R'Z \О~
Бис (трифторметил) диазометан устойчив к действию кис-
лот. Особенно интересен случай чрезвычайной устойчивости
диазоциклопентадиена, так как здесь отрицательный заряд
принимает участие в образовании бл-электронной системы
цикла
f®4)»—N«N
VII
56
Волее стабильны, чем соединение VII, производные циклопентадиена,
имеющие электроноакцепторные заместителя в цикле4’6'7. Устойчивым
является также диазоциклонойатрибн, кольцо которого образует 10л-элек-
троннуй) систему.
Хотя многие диазокарбонильные соединения устойчивы
при комнатной температуре и могут храниться без особых
предосторожностей, было отмечено, что ароматические диазо-
оксиды — исключительно взрывчатые вещества, которые
обычно не удается выделить. Простейшие алифатические
диазосоединения трудно и опасно выделять вследствие их
токсичности и взрывчатости. Обычно их готовят при низких
температурах в инертных растворителях и немедленно ис-
пользуют, чтобы избежать проблемы хранения.
Долгое время диазоалканам приписывали циклическую
структуру VIII:
R\ /N
С ||
R,/Z ' N
VIII
Спор двух школ продолжался до тех пор, пока физиче-
скими методами не показали8-11, что диазоалканы имеют ли-
нейное расположение атомов азота и лучше всего могут быть
описаны резонансными гибридами9-11 нескольких структур
II (а, б, в, г). Главный вклад в основное состояние алифати-
ческих диазосоединений вносит карбанионная структура
Па12-13.
Однако химическое доказательство их линейной струк-
туры получили лишь в 1960 г., когда Паульсен 14 и Шмитц 15>16
независимо друг от друга синтезировали первые диазирины
VIII. До синтеза диазиринов, которые являются бесцветными
веществами в противоположность сильноокрашенным диазо-
алканам, предполагалось, что такие циклические структуры
могут вносить некоторый, хотя и малый, вклад в резонанс-
ный гибрид диазоалканов. Однако было убедительно дока-
зано17, что они реально существуют самостоятельно. Следо-
вательно, циклическая структура, VIII не вносит вклада в
резонансный гибрид диазоалканов.
По химии диазосоединений имеются хорошие обзоры 8’ 18~20,
поэтому общие реакции и методы получения их будут только
упомянуты при дальнейшем изложении, а интересные осо-
бенности — рассмотрены подробнее.
СОЛИ ДИАЗОНИЯ
Строение
Значение солей диазония в получении азокра'сителей и их
разнообразные реакции сильно способствовали развитию ис-
следований в этой области. Превращения солей диазония,
57
особенно в водных растворах, зависящие иногда от тонких
изменений внешней среды (значения pH растворов, темпера-
туры и т. д.), очень разнообразны: включают таутометрию,
стереохимические превращения, кислотно основные равнове-
сия.
Подробно описана химия солей диазония в монографиях
Саундерса18 и позднее Цоллингера8. Однако химическое
строение солей диазония настолько интересно и сложно, что
не коснуться этого вопроса, хотя бы в нескольких словах,
представляется невозможным.
Положительный заряд в катионе солей диазония располо-
жен на обоих атомах азота: на связанном с ароматическим
ядром в силу его sp-состояния и на крайнем атоме азота
из-за сильного индукционного эффекта соседнего атома. Ди-
азониевая группа — настолько сильный акцептор, что арома-
тическое ядро со всеми имеющимися в нем заместителями
является донором электронов. Это положение сохраняется
даже при наличии в ядре такого сильного акцептора, как
нитрогруппа. Конечно, характер заместителей в ароматиче-
ском ядре так или иначе изменяет величину положительного
заряда на крайнем атоме азота, так как заместители в пара-
и орто-положениях сопряженье с ним из-за того, что одна из
трех пар электронов, связывающих атомы азота, расположена
в плоскости л-электронного облака кольца. Другие же связи
способны передавать лишь индукционный эффект от одного
атома азота к другому. Все сказанное иллюстрируется сле-
дующей схемой:
В результате сопряжения крайнего атома азота с арома-
тическим ядром в ароматических солях диазония склонность
к выделению элементарного азота, по сравнению с алифати-
ческими диазосоединениями, значительно понижена. Поэтому
атомы азота в катионе диазония не являются равноценными.
Однако недавно было предположено21, что при гидролизе
некоторых солей диазония образуется промежуточный ком-
плекс строения , в котором оба атома азота
одинаковы.
При обработке солей диазония щелочами образуется ам-
фотерный диазогидрат Аг—N = NOH, мгновенно диспропор-
пионирующий на катион диазония и анион диазотата. Таким
образом, обработка эквивалентным количеством щелочи при-
водит не к диазогидрату, а к смеси диазокатиона и диазо-
аниона, как-будто бы половинное количество катиона диазо-
ния не реагирует со щелочью вовсе, а вторая половина
58
нейтрализуется, потребляя двухэквивалентное количество ще-
лочи.
Быстрое прибавление двухэквивалентного количества ще-
лочи к катиону диазония приводит к полному его превраще-
нию в анион диазотата, имеющий цис(син)-конфигурацию.
При нагревании qtzc-диазотата, а иногда и без нагревания
происходит перегруппировка в транс(или анти)-цназотат.
Обратное превращение осуществляется лишь в исключитель-
ных случаях, при действии ультрафиолетового света. Анти-
Диазотат, в отличие от смн-изомера, устойчив, образованные
им щелочные соли применяются в технологии крашения.
Чрезвычайно интересно, что при подкислении щелочного рас-
твора анти-диазотата обратное превращение его в катион ди-
азония происходит медленно и не тем путем, по которому он
был получен. Таким образом, равновесие между катионом
диазония и смн-диазотатом устанавливается мгновенно, а
равновесие между тем же катионом и а«т«-диазотатом —
медленно. Это происходит потому, что первым продуктом ре-
акции анти-диазотата с кислотой является нитрозамин IX,
который сравнительно медленно таутомерно перегруппировы-
вается в анта-диазогидрат, под действием кислоты превра-
щающийся в катион диазония. Все сказанное выше можно
иллюстрировать следующей несколько упрощенной схемой
взаимных превращений различных форм ароматических ди-
азосоединений:
—Ar— N=N—ОН—
ZN=NX Ar.
Аг ЧГ \N = N
hv
± Ar—NH—NO <-— —у
(V-1)
При обработке солей диазония другими, кроме гидро-
ксильного иона, нуклеофилами образуются устойчивые
соединения, в которых оба атома азота находятся уже в со-
стоянии хр2-гибридизации и которые способны, подобно ди-
азотату, к ^ыс-транс-изомерии. К ним принадлежат извест-
ные диазоцианиды Аг—N = N—CN, диазосульфинаты
Аг—N=N—SO2—Аг, диазоэфиры Ar—N=N—OAlk и самые
важные в этом ряду вещества — азосоединения Аг—N =
= N—Аг. \
Получение
Соли диазония можно получить многими методами, но
только два из них нашли широкое применение (см. моногра-
фии Цоллингера, Саундерса и Родда). Самый удобный
69
метод, применимый по отношению к сравнительно сильнооснов-
ным ароматическим аминам, заключается во взаимодействии
ароматических аминов с нитритами щелочных металлов в
растворе минеральной кислоты. На первой стадии катион
нитрозония, образующийся в результате взаимодействия азо-
тистой кислоты с минеральной кислотой, реагирует с ами-
ном (содержащимся в равновесных системах даже в сильно-
кислой среде) с образованием нитрозамина, который после
таутомерной перегруппировки в диазогидрат при действии
протонов превращается в катион диазония:
Ar—NH2 + NO —> Аг—NH—NO + H+ (V-2)
н+
Ar—NH—NO Aj—N=N—OH Ar— N* + H,0 (V-3)
OH~
Избыток минеральной кислоты необходим для растворе-
ния амина и образования энергичной нитрозирующей части-
цы— иона нитрозония. Недостаток или избыток нитрита ще-
лочного металла нежелателен, так как это приводит к
побочным реакциям. Температура реакционной массы при ди-
азотировании обычно поддерживается, во избежание разло-
жения, в пределах 0ч-5°С, однако имеются случаи, когда ее
можно значительно повысить (хлориды n-нитрофенил- и на-
фтилдиазония). По второму способу в качестве растворителя
для реакции используют серную кислоту, нитрозируя амин
заранее приготовленной нитрозилсерной кислотой. Этот спо-
соб применяют по отношению к очень слабоосновным ами-
нам и он приводит к образованию бисульфатов диазония 22’23.
Прямое введение диазониевой группы, описанное Теддером,
ограничивается фенолами и диалкиланилинами и не имеет,
по-видимому, технического значения
+ 2H0N0 + нз° —► n2—он
(V-4)
Реакции
Так как эти соединения несут положительный заряд, ну-
клеофильная атака различными веществами является глав-
ным типом реакций солей диазония. Они могут быть разде-
лены на два общих класса: а) с нуклеофильной атакой по
концевому атому азота диазогруппы без потери азота и б)
с нуклеофильной атакой по а-углероду (т. е. связанному с
—NiT-группой) и выделением азота
Ar—N*C1" + ВН —> Ar—N = N—В + НС1 (V-5)
Ar-N+СГ + ВН —> Аг—В + N, + НС1 (V-6)
60
Реакции без выделения азота. Различные ну-
клеофилы атакуют соли диазония по концевому атому азота,
образуя арилазосоединения. Этим способом были получены
арилазоцианиды (диазоцианиды), арилазосульфонаты (ди-
азосульфонаты) и ряд побочных веществ.
Одним из интересных примеров является реакция солей
диазония с азид-ионом23. Промежуточный «арилазоазид»
выделить невозможно, он циклизуется в соединение с реак-
ционным пентазольным кольцом, которое, в свою очередь,
теряет азот, превращаясь в соответствующий арилазид:
Г Ar—N = N-
•N=N
ArN+Cl’+N"—> _ +/N —> Ar—NZ | ——> ArN3
N=N-
\n=n
(V-7)
Важность реакции сочетания солей диазония с активи-
рованными ароматическими соединениями, в результате
которой образуются яркие красители, не нуждается в обсу-
ждении* (см. обзоры8’18-20). Может также иметь место со-
четание с активированными метиленовыми группами [алифа-
тических соединений]21 (V-8) и [активированными метано-
выми группами] — реакция Яппа — Клингемана (V-9) 25:
- /R ZR
Ar—N*Cr + CH; —> Ar—N=N-CH^ + СГ —>
(V-8)
Ar-N+Cl’ + ;c
Ar—N = N—CH:
(V-9)
(R' и R" — электроноакцепторные группы)
При реакции Яппа — Клингемаиа одна из электроноакцепторных групп
отщепляется при сольволизе первичного продукта азосочетаний:
-zR I X
ArN* + C—R' —> Ar—N=N—C—R' H2°-» R"OH + соединение X (V-10)
В некоторых случаях реакции Яппа — Клингемана удает-
ся выделить промежуточное азосоединение26. Реакция
* [Азокрасители — самый распространенный класс органических кра-
сящих веществ].
61
азосочетания оказалась полезной и для практического полу-
чения альдегидов и кетонов 27:
ОН
R'
О
II /-Х /СНз
—> R—С—R' + HO3S-^ Ъ—N = N— " У~N( (V-11)
\=/ \СНз
Восстановление солей диазония до сих пор является наи-
более широко применяемым методом синтеза арилгидрази-
нов.
Реакции с выделением азота. Широкое примене-
ние реакций этого типа говорит об их многосторонности. Ди-
азотированием аминов и последующими реакциями с соот-
ветствующими реагентами можно ввести в ароматическое
кольцо ряд групп. Иногда это — единственный метод получе-
ния нужного продукта:
ArNj + HB —> Ar—B + N2 + H+ (V-12)
Предполагают, что некоторые реакции этого класса про-
ходят через гомолитическое расщепление связи арил — азот,
однако при других условиях только ионный механизм можно,
по-видимому, согласовать с экспериментальными данными.
Таблица V-1
Продукты реакций солей диазония с выделением азота
Получаемые соединения Реагенты [или катализаторы] Реакции
[Фенолы] Ароматические углеводороды Ароматические галогенпроиз- водные Ароматические галогенпроиз- водные и нитрилы Ароматические фториды Ароматические мышьякови- стые и мышьяковые кислоты Биарилы [Ароматические металлоорга- нические соединении (ртути, сурьмы, мышьяка, висмута, олова, свинца, таллия)] Н2О Н3РО2 [и другие восстановители] НХ 1 СиХ 1 Си, НХ ArNjf, BF4~ Производные мышь- яка Основания Галогениды и окси- галогениды ме- таллов н металли- ческие порошки Дезаминирова- ния28 Грисса Зандмайера29 Гаттермана30 Шимана31 Барта32 Г омберга — Бах- мана33 Диазометод Не- смеянова34
62
Почти все такие реакции катализируются солями металлов,
особенно солями меди. Эти реакции, описываемые в большин-
стве учебников по органической химии, перечислены в
табл. V-1.
Элементы АгХ могут присоединяться по активированным
двойным связям, в этом состоит реакция арилирования по
Меервейну с помощью солей диазония35:
. \ / I 'l
ArN^X’ + С = С —> Аг—С—С—X + N2 (V-13)
/ Х II
Представлены доказательства в пользу как ионного, так
и радикального механизмов реакции Меервейна. Ни один из
этих механизмов не может объяснить всех эксперименталь-
ных данных, что привело к предположению об образовании
промежуточного комплекса соли диазония, олефина и хло-
рида меди. Такой комплекс может разлагаться путем внут-
реннего переноса одного электрона36.
ДИАЗОАЛКАНЫ
Получение
Существует несколько способов синтеза диазоалканов.
Гутше37 обсудил их относительные преимущества и дал об-
зор литературы до 1954 г. Эти методы можно разделить на
три общие группы.
1. Действие щелочей на нитрозамиды. Дей-
ствие водных щелочей или алкоголятов щелочных металлов
на нитрозамиды дает диазоалканы согласно уравнению
(V-14):
\h-N-R" -°свов-анне> \-N = N+ (V-14)
Rz/ I R'Z
NO
(R" = —CONH2, -COOC2H5, —COR"',
NH
—SO2R'", —C—NH—NO2)
Некоторые неудобства представляют нестойкость нитро-
зометилмочевины, а также нарывное действие нитрозометил-
уретанов. Однако N-нитрозометил-п-толуолсульфамид, пред-
ложенный Боером и Беккером38, весьма устойчив и прост в
приготовлении:
RCH2—N—SO2—/ S-СНз -2£225^>RCHN2++ SO3-/ %-СН3
NO
(V-15)
63
2. Окисление гидразонов. Гидразоны альдегидов
и кетонов можно окислить в соответствующие диазоалканы.
Этот метод особенно пригоден для синтеза диарилазоал-
канов:
R\ _ R\
C=N—NHj + HgO C--N«r (V-16)
R'Z R'Z
[Он дает также хорошие результаты при синтезе а-диазо-
КеТОНОВ 39-41 J
3. Действие азотистой кислоты на аминосо-
единения. Этот метод применим только к соединениям с
аминогруппой по соседству с электроноакцепторной функ-
цией:
NH2CH2COOC2H5 N+=N—СНСООС2Н5 (V-17)
Диазоалканы можно получать и другими способами.
Представляют интерес реакции, дающие диазометан: дей-
ствие хлороформа на гидразин в присутствии едкого кали и
реакция N, N-дихлорметиламина с гидроксиламином. Ката-
лизируемое щелочами разложение диазобензила дает фенил-
диазометан (V-18):
CeH5CCOC6H5 C6H5CHN2 + С5Н5СОО~ (V-18)
N = N+
[Разложение других диазокетонов щелочами происходит
значительно сложнее (см., например, 42’43).]
Заслуживает упоминания уникальный метод синтеза ди-
азоциклопентадиена VII и родственных ему соединений:
NjSO2CgHs »-
^x,N = N“N-SO2C6H5
Ч/ш
>=-N=N-^HSO2C6Hs
N = N*
VII
(v-19)
[В последние годы метод синтеза диазосоединений с по-
мощью тозилазида нашел широкое применение44.]
Катализируемое щелочами разложение п-толуолсульфо-
нилгидразинов (см. гл. IV и VI), о котором сообщили Бэм-
форд и Стивенс45, было использовано для превращения ди-
азоалканов in situ, но, как было недавно показано46, этот
метод не является общим препаративным; он не дает прием-
лемого выхода /г-метоксифенилдиазометана 47:
R\ R\ -
\:=N-NH-SOsR" V-N=N+ + SO2R" (V-20)
R'Z R'Z
64
Некоторое применение, по-видимому, находит реакция
Форстера (V-21) (а-оксиминокетоны дают а-диазокетоны);
недавно было показано42, что ее можно использовать как
общий метод синтеза диазоалканов:
R\
XC = N—ОН + Х—NH2
R'Z
-
?сн°в_а.н.и^ xc-n=n+
Rz/
(V-21)
(X=—Cl, —OSO3H)
Перспективны два новых метода синтеза диазометана —
разложение N, N'-нитрозодиметилоксамида49 и устойчивой
калиевой соли N-нитрозометиламина50:
СН3—N—СОСО—N-СНз |-снова|"|0-> 2CH2N2 (V-22)
I I
NO NO
CH3NH2 + NOCI + KOC2H5 —> CH3—N = N—OK —> CH2N2 (V-23)
1,3-Биполярное присоединение51 закиси азота к трифенил-
фосфинметилену приводит к диазометану и окиси трифенил-
фосфина:
(свн5)3р-снг <- n2o —
(с.н,),р-гСнх
—* (СвН5)3Р-О~ +
CH2N2
(V-24)
Окисление гидразонов (метод 2) является самым общим
способом, но неудобным из-за неустойчивости гидразонов,
которые могут окисляться в процессе их получения. Однако
для большинства диазоалканов, кроме диазомегана, этот ме-
тод, вероятно, практически наиболее пригоден. Препаратив-
ное нитрозирование аминов Имеет структурные ограничения,
упомянутые ранее.
Несмотря на существующие различия, между методами
синтеза диазоалканов есть одна общая черта — все они
включают отщепление HOR" от исходного вещества:
R\ R\
СН—N=N— OP" —XC-N = N+
R' Z R'Z
Rx ZOR"
«— C=N—N (V-25)
R'Z "'H
(R"=-H, -HgOH, -COR, -SO2R и t. Д.)
5 Зак. 334
65
Приведенные примеры еще раз подчеркивают важность
отщепления а-протона и тем самым глубокое различие ме-
жду диазоалканами и солями диазония.
Подробное обсуждение многочисленных и разнообразных
реакций диайоалканов выходит за пределы этой книги. В слу-
чае необходимости детального рассмотрения и использования
реакций таких соединений читатель может обратиться к об-
зорам б-52. Будут рассмотрены более новые и интересные ре-
акции. Следует обратить внимание на нуклеофильный ха-
рактер диазоалканов и на некоторые другие особенности.
Понимание этих свойств поможет объяснить большинство
реакций.
Реакции диазоалканов как нуклеофильных реагентов
Типичными для диазоалканов являются реакции, в кото-
рых они выступают в качестве нуклеофилов, причем нуклео-
фильным центром чаще всего является а-углерод. В целом,
легкость таких реакций падает с уменьшением основности
а-углерода, что видно из большой разницы реакционных спо-
собностей диазометана и диазоуксусных эфиров (это сказы-
вается и на их получении, см. «Получение»), Как и следо-
вало ожидать, если а-углерод имеет «объемистые заместите-
ли», большую роль играют пространственные эффекты.
Реакции с кислотами. Минеральные кислоты про-
тонируют а-углерод, и получающийся ион диазония XI может
подвергаться атаке различными нуклеофилами. р-Элимини-
рование протона дает олефин, но может произойти и пере-
группировка. Природа растворителя часто является решаю-
щим фактором, определяющим направление реакции53:
R\_
/С—N = N+ + H+ —>
Rz/
Г
'CH—N
. Rz/
/R
HONO + HSNCH
^R'
-Na, —H+
CH—X
олефин
(V-26)
Диазоалканы, особенно диазометан, часто используют как
алкилирующие агенты, преимущество которых заключается
в мягких условиях алкилирования и высоких выходах про-
дуктов. Если органические кислоты алкилируются без ката-
лизаторов, то при алкилировании таких слабокислых соеди-
66
нений как спирты, амины и енолы, требуется присутствие
кислот Льюиса *:
RCOOH + CH2N2 —► RCOOCH3 + N2 (V-27)
ROH + CH2N2 ROCH3 + N2 (V-28)
Реакции с ненасыщенными системами. Сле-
дующая схема отражает нуклеофильные реакции диазоалка*
нов с карбонильными соединениями. Образующийся в пер-
вой стадии биполярный ион может претерпевать в дальней-
шем ряд превращений:
R\
ХС = О
R'Z
+
ch2n2
' R\ z0’
. R'Z XCH2-N+
—> RCOCH2R'
---> RCH2COR'
Rx
—-> C----CH2
R'Z \OZ
(V-29)
(V-30)
(V-31)
RCOCH2N+ R.COCH-N+ RCOCH2R'
(V-31a)
Реакции (V-29) — (V-31) происходят с альдегидами и ке-
тонами. Расширение кольца в циклических кетонах 37 служит
удобной альтернативой реакции Тиффено — Демьянова56.
В случае а-дикетонов или а-оксиминокетонов часто полу-
чаются циклические продукты:
(V-32)
[Образование 1,3-диоксолов типично лишь для реакции
диазоалканов с о-хинонами, в случае а-дикарбонильных со-
единений иной структуры взаимодействие протекает в соот-
ветствии со схемами (V-29) — (V-31) 57-59.]
Кетен с диазометаном дает циклопропанон, который даль-
ше реагирует с диазометаном, образуя циклобутанон. Эту
реакцию следует сравнить с реакцией дифенилкетёна с ди-
фенилдиазометаном, идущей иначе и приводящей к окса-
диазолину60. В этом случае важны стерические факторы,
упомянутые ранее. Интересно отметить, что с циклопропено-
* [Енолы, обладающие достаточно кислым харашером, алкилируются
без катализатора — см., например,54’55.]
5’ 67
ном диазометан взаимодействует не по карбонильной группе,
а по связи ZC=C4 :
снг,с=о + сн2й2 —~ сн2=</ —- УСНз
'CH2~N2 q
Н2С-СН-
.----- I I 2 ГЦ.ЯЯ
,0,
с6н5. СвН5. + С6Н5. / \ хсвнв
^С = С = О+ N = N —> ХС С=(Г
свн/ с6н/ c6h5z \n=nz XCeH5
(V-34)
Реакции с хлорангидридами представлены превращением
(V-36):
.О /О ,0
—> R< RC< R(7
xch2n+ \ch-n+ \ch2ci
(V-36)
Получающиеся диазокетоны до тех пор реагируют с вы-
деляющимся галогенводородом с образованием а-галогенке-
тона, пока присутствующее основание не нейтрализует гало-
генводород. Обычно для этого добавляют избыток диазомег
тана, но был использован и триэтиламин 62.
Диазокетоны могут участвовать во многих реакциях63,
одной из которых является перегруппировка Вольфа — про-
межуточная стадия в синтезах Арндта — Эйстерта64. [См.
также обзор65.] Эфиры, амиды и подобные соединения, у ко-
торых положительный заряд карбонильного углерода пони-
жен, не взаимодействуют с диазоалканами. Галогенангидри-
ды сульфокислот также не реагируют с диазоалканами.
Фар 66 недавно сообщил об образовании «внутренних солей»
с кислотами Льюиса. >
68
Типичные примеры реакций диазометана с нитрилами,
азометинами и нитрозосоединениями приведены ниже:
R—C==N
RC = N + CH2N2—> | I
H2C N
CH2N2
R-C=N
HC N—CH3
'W
(V-37)
R-CH—N—R'
RCH = N—R' + C H2N2 —> I I
H2C N
(V-38)67
R—N = O + CH2N2 —> FR—N CH21
I 4OZ J
R—N = CH2
O"
(V-39)
Сообщалось о некоторых интересных аномальных реак-
циях. Диазометан не присоединяется по связи —C = N в со-
единении XII, а имеет место N-метилирование68:
CH3SO2X CH3SO2x_ .
\h-C^N —> V=C=NH -- 2
CH3SO/ CH3SO/
XII
CH3SCK
V=C=N—СНз
CH3SO/
(V-40)
С эфиром оксима XIII происходит присоединение по связи
C = N- 69:
CH3SO2.
'C = N—OCH3 + CH2N2 —>
CH3S0/
XIII
CH3SO2x /SO2CH3
CH2 N—OCH3
\n = N/
CH3SO2. zso2ch3
Nr
Hac—-N-OCHa
(V-41)
69
Важные работы Леонарда с сотрудниками по
диазометана с солями иминов
:c = n
привели
тию расширения кольца в циклических иммониевых
реакции
к откры-
солях70:
Было показано71, что реакция диазоалканов с азодикар-
бонильными соединениями дает скорее оксадиазолины, а не
диазациклопропаны, недавно советские исследователи 72 уста-
новили, что гексафторазометан в реакции с диазометаном
образует два продукта присоединения:
СГ3—N = N—CF3 + CH2N2 —>
CF3—N---N—CF3
H2C N
CF3—N—N—CF3
\hs
(V-43)
Реакция диазокетонов с эфирами азодикарбоновых кис-
лот включает ряд перегруппировок73. В упрощенном виде
реакция представлена схемой (V-44):
RCOCH— N = N + C2H5COON=NCOOC2H5 —>
,СООС2Н5
—> RCOCH=N—N^- (V-44)
\соос2н5
Диазоалканы реагируют с диарилкарбимидами, изоциа-
натами, изотиоцианатами и тиокарбонильными соединениями:
Присоединение диазоалканов к ненасыщенным связям
углерод — углерод известно давно. В большинстве реакций
70
выделяют продукт первичного присоединения (без выделения
азота) или его изомер. Такие же аддукты образуются при
многих реакциях диазоалканов и ненасыщенных систем с ге-
тероатомами. Присоединение этого типа составляет часть ис-
ключительно важных реакций 1,3-биполярного присоедине-
ния9 (V-48). Вопрос в целом был обобщен Хьюзгеном74,
который определил и расширил сферу этой очень многосто-
ронней реакции:
R\ _
XC~N=N+ + -a = b
R'Z
(V-48)
В общем, активированная двойная связь С = С реагирует
с диазоалканами с образованием гетероциклических соеди-
нений. Легкость протекания и направление присоединения
обусловливаются как электронными, так и стерическими
факторами*. Алкины в реакциях с диазоалканами образуют
пиразолы (V-49), олефины — Д’-пиразолины, которые обычно
изомеризуются в Д-2-изомеры XV (при наличии а-водорода),
что приводит к конъюгированной системе (V-50):
RC^CR' + CH2N2
R—С=С—R' R—С=С—R'
” H2<i N ” НС NH (V-49)
\N /
Однако имеются случаи получения стабильных Д’-пиразо-
линов, даже когда изомеризация энергетически выгодна78.
Присоединение диазоалканов к олефинам требует согласо-
ванного присоединения77 и рассматривается как цис-присо-
единение79.
Последние данные80 по стереоспецифическому присоеди-
нению диазометана к изомерным метиловым эфирам
* [Наряду с «нормальным» присоединением углеродного атома диазо-
алкана в ^-положение к активирующей группе возможно «аномальное»
присоединение в это положение непредельного соединения концевого атома
азога диазогруппы. Тот или иной вид присоединения определяется специ-
фическими электронными и стерическими факторами диазоалкаиа и оле-
фина 75~7Т]
71
ангеликовой и тиглиновой кислот показали, что из каждого эфи- 1
ра получается только один соответствующий изомер, что, по- |
видимому, и должно подтверждать предложенный механизм. |
Однако этот механизм неприменим ко всем реакциям 1,3-би- 1
полярного присоединения диазоалканов: так, в реакции и-мет- 1
оксистирола с п-(метоксифенил) диазометаном получается 1
смесь соответствующих цис- и трянс-Д'-пиразолинов 47166: j
сн3о—ch=ch2 + n = n—СН—ОСН3 —►
/СНг-^
—> СН3О-^ ^-СН СН—/ V-OCHj (V-5I)
Сообщалось81 о первых примерах метилирования виниль- j
ного водорода в бензальмалононитриле при трехстадийной i
реакции с диазометаном: |
C6H5CHO + CH2(CN)2—> J
СН3 •
—-> C6H6CH = C(CN)2 C6H5C = C(CN.)2 —> СвН5-СО-СН3 з
(V-52) |
Диазоалканы взаимодействуют по углероду со многими
электрофильными реагентами52. Особенно интересным яв-
ляется образование хлордиазометана из диазометана с трет-
бутилгипохлоритом при — 100°С82. Дигалокарбену реагируют
с диазоалканами, образуя галогенпроизводные83. Катализи-
руемая трехфтористым бором полимеризация диазоалканов
предусматривает два варианта нуклеофильной атаки: ката-
лизатором (BF3) и исходным карбониевым ионом84. Инте-
ресные результаты были получены при реакции трифенилме-
тильного катиона с диазоалканами85. Озонолиз дифенилди-
азометана до бензофенона 86 описывается схемой:
- + - (С.НЛ.С-.Ч,
(СвН5)2С—Х = \' + 0. —> 0-0-0—C(CSH5)2 ----
N+
zN = N+
—> (С8Н5)2СО + N2 + (СвН5)2с/ —►
О— О'
,N=N
—* (СеН6)2С | + N2O + (CeII5)2CO (V-53)
О— О
72
При действии S02 на диазоалканы сначала образуются
промежуточные «сульфены»'87:
- + (С.Н ),C-N*
(С6Н5)2С—N = N+SO2 —> [(C6H5)2C = SO2] - — -
—* (С6Н5)2С---С(С6Н5)2 (V-54)
SO2
Другие примеры электрофильной атаки — реакции с йоди-
стым цинком88 и хлоридом n-нитрофенилдиазония. В этих
немногих случаях наряду с продуктами электрофильной ре-
акции по концевому атому азота образуются азины, которые
получаются при разложении диазоалканов89.
Бис (трифторметил) диазометан реагирует с циклогексаном,
по-видимому, по радикальной схеме с образованием азосо-
единения80:
_ /ч ZN=N—CH(CF3)2
| | +(.CF3)2C—N = N+ — + I 'l (V-55)
Реакции диазоалканов как электрофильных реагентов
Поведение диазоалканов как электрофилов менее ясно, и
таких примеров немного. Диазоацетофенон реагирует с ци-
анидом, причем образуется цианогидразон фенилглиоксаля.
При реакции диазоалканов с третичными фосфинами по-
лучаются фосфазины:
R\ ’ + R< _ +
XC = N=N" + Р(С6НБ)3 —> )C=N—N-P(C6H5)3 (V-56)
R'Z . R'Z
[Эта реакция может применяться для идентификации ди-
азоалканов91.] В присутствии медн получаются илиды, хотя
возможно образование и азинов^2. Интересной является ре-
акция дифенилдиазометана с триизопропилфосфитом93:
с-свн.
(c6H,)2CN=N+ (uw-C3H; (’ж-С3Н7о)эРО D Э g В Э । N (цзо-С3Н7о)2 p+-nhn- с (c6h5)2 t/SNK % + ,o)2P H ch3ch-ch2 £cH2 (V-”) СИ CH’ (c,H,)2 CrN-NrC^Hs), 73 «1
Амины и гидразины94 также могут атаковать диазоалка-
ны. Сообщалось95, что диазодимедон подвергался «сочета-
нию» с активированными ароматическими соединениями.
Реакция диазокетонов с соединениями, содержащими метиле-
новые группы, обычно приводит к пиразолам. Фарнум и
Уэтс96 сообщили о реакции диазосоединений с переносом
центра:
C6H.COCH-N-N
_ZCOOCH,
нс( 3
XC6H5
С6н5со-Сн2
о
CBH5C О
k=N-Cc 6 5
хсоосн,
с н \*$.
C,H.COCH,N-N-C( 6 5
6 5 2 XCOOCH, \
(v-5S)
QgHgCO—<j:---C-OH
N C
NH P»HS
Реактивы Гриньяра атакуют диазоалканы с образованием
замещенных гидразонов:
R\l-
;с—N =N+ + R"MgX —>
Rz/
R\
\: = N—NHR"
R,Z
(V-59)
Такие сильные основания, как метиллитий, превращают
диазометан в диазометильный анион, который при гидролизе
в кислой среде дает изодиазометан XVI97~99:
CH2 = N = N“+ CH3Li —>-HC = N = N~
—> HCsN-N-H ---С0°— -> RCONH-NHCHO
XVI
(V-60)
Реакции * диазоалканов со свободными радикалами не-
многочисленны. В реакции трифенилметильного радикала с
диазометаном был выделен гексафенилпропан '°4. Включает
ли реакция диазоалканов с тетракарбонилом никеля свобод-
* [Для диазоалканов возможно разложение с образованием карбенов.
Обычно такое превращение проводится фотолитически или термически в
присутствии катализаторов и дает соответствующие карбены, которые под-
вергаются реакциям, рассмотренным в обзорах 10°_ 103 и выходящим за рам-
ки данной книги.]
74
ные радикалы — неясно 105. Действие окиси азота приводит к
ряду интересных реакций106:
Сообщалось и о других подобных реакциях 107. Аналогич-
ные результаты получены при действии металлического на-
трия на диазофлуорен ,08:
U\- +
I V-N^N+Na
А7
\А
н2о
—> Дифлуоренилэтан (V-62)
При восстановлении диазоалканов получаются различные
продукты в зависимости от метода восстановления и природы
заместителей 109:
H2N-N = CHCOOC2H, <-
СН3СООС2Н5
N = N—СН—СООС2Н5
(V-63)
NH3 + h2nch2cooc2h5 ^Н007спСНз 1 Zn; он-> H2N—NHCH2COOC2H5
n2 + csh5ch2c6h5 (C6H5)2C-N=N+
c6H5x
АИДА CH—nh2
csh/
C2H5O2Cy _
'C—N = N+ —>
C2H50aC
XC=N—NH2
С2Н3О2С/7<
(V-65)
75
Диазоалканы в качестве промежуточных или конечных
продуктов реакций
В одних случаях они были выделены и охарактеризованы;
в других их образование в процессе реакции строго доказы-
валась косвенными методами. Свободнорадикальное окисле-
ние гидразонов в азины не включает промежуточного образо-
вания диазоалканов 10, но механизмы могут быть различ-
ны111: окисление гидразона бензофенона иодом привело к
азину, в то время как в присутствии триэтиламина удалось
выделить дифенилдиазометан:
С6Н5,
C=N— NH2 + I2
свн/
(CaHshN
~С6Н5Х
C = N— NHI
_с6н/
(C2H5)3N >
с8н5х _
С—N = N+
С6н/
(V-66)
Продукты реакции гидразонов с галогенированными о-хи-
нонами предполагают промежуточное образование диазоал-
канов112:
X
Х\А^° /сбнз-
| | +nh2n = cz
х/'улэ ЧС6Н5
X
X
Х\А/ОН
I [| + С—N = N+ —>
Х^^ОН СвИб7"
х\А/°\ /с«н'
I II С
X-ZYX°/ ХС*Н5
X
(V-67)
Циммерман и Сомасекара 113 показали, что при пиролизе
азинов до соответствующих олефинов диазоалканы высту-
> пают в роли переносчиков цепи:
ArHC=N_N-CHAr + ArCH-N, ------- ArCOyN-N-CHAr _____~
ArCH-Olj
(V-68)
---- ArCH-Nj + ArCH-CHAr + N2
76
Бридсон-Джонс и сотрудники114 нашли, что при окисле-
нии олефинов и ацетиленов окисью азота при высоких тем-
пературах и давлении получаются соединения, образование
которых можно объяснить только промежуточным возникно-
вением диазоалканов (V-69):
R4 zR
R'z ?XR'
°
R l 4R
O'
k’R'C—CR'R”' (v-70)
о
Разнообразие получающихся соединений зависит в зна-
чительной мере от природы олефина. Первичный аддукт
имеет такое же строение, как и при присоединении диазоал-
канов к альдегидам и кетонам. Установлено115, что диазо-
алканы возникают в результате окисления гидразонов над-
уксусной кислотой и разложения моноокисей азинов XVII
(V-71):
Недавно сообщалось об изомеризации дифенилдиазирина
XVIII в дифенилдиазометан Н6:
(C6H6)2C = NH + H2NOSO3H
NH3
-70 °C
ГОД5\ /NH1
1<ОД/ XNHJ
ОДвх _
С—N = N+
ОД5Х
(V-72)
XVIII
77
Вибо и Бон 117 постулировали промежуточное образование
диметилдиазометана во время озонолиза азина ацетона:
(V-7,)
CH3 -—- )C-№N + CHj o5 + ©co o3 CH3 ©co + Oz + N;
CH3Z CH3Z СН/
Перегруппировка азотриазолов XIX в тетразолы, по мне-
нию Педерсона ”8, также включает промежуточный диазо-
алкан:
Ar-N=N-C=C-OH
N (N-R
4Ny
Ar-№N-C-CO-NHR
XIX
Ar—N C-CONHR
XN=N/
(V-74)
Косвенное доказательство образования диазоалканов было
получено в реакции азотистой кислоты с а-аминокетонами "9:
он
RCOCH-NH, R—СО—СН-₽' R- С-Озн-Р'
I <11
HO N (on
RCH—Nj + RCOOH (v-zs)
Деникер и Дрюй120 недавно сообщили об образовании
N-ацетилдиазоацетатамида при каталитическом восстановле-
нии сиднонимина XX:
W^N—СН
N0C = N-COCH3 СвН,СНз
vo/
XX
йг- CHCONHCOCHj ——
HN—-— СН
N © C=N-COCHj
4oz
n-^Lch
I Nil
N^C—NHCOCH3
4OZ
(v-7«)
78
Пространственно затрудненные N-нитрозамины восстанав-
ливаются в соответствующие диазоалканы алюмогидридом
лития (см. гл. VII).
Ненасыщенные диазоалканы могут получаться при раз-
ложений натриевых солей тозилгидразонов «^-ненасыщен-
ных кетонов 121:
СНз.
Хс=снх СН3
CH3Z с
II
N
Tos—N7" Na+
' CH3v
C = CI-r
СНз/ С—СНз
+ I
, N = N
(V-77)
Сообщалось 116 об отщеплении диазоалканов некоторыми
соединениями, образующимися через 1,3-биполярное присо-
единение:
>N7
C-CH1
+ n=N-N-Tos
(V-7S)
C2H5O-C^c-H + N-N-N—Tos -
сгн5о-с=^рн
Tos-n) n
--» Tos-N=C
ZOC2H5
4ch-n2
(v-79)
ИЗОМЕРНОЕ ПРЕВРАЩЕНИЕ ДИАЗОАЛКАНЫ - ДИАЗИРИНЫ
Ранее упоминалось (см. раздел «строение»), что недавно
осуществленный синтез диазиринов VIII исключил возмож-
ность даже малого вклада этой циклической структуры в ре-
зонансный гибрид диазоалканов XXI:
R\-
С—N = N+
XXI
VIII
Однако здесь наблюдается особый случай валентной
Изомерии *, включающий только электроны; XXI можно
* [В оригинале — таутомерии.]
79
рассматривать как диазирин, если допустить, что между поло-
жительным и отрицательным концами молекулы существует
«ионная» связь. Априори диазоалканы и диазирины должны
быть взаимнопревращаемы, так как химия нитронов служит
прецедентом такого поведения. Действительно, Сплиттер и
Кельвин 123 превратили нитроны XXII в оксазираны XXIII
при УФ-облучеЕИи:
О
)c=N-R" — > ;С—N-R"
R’- + R'Z
XXII
XXIII
Позднее Боннер, Кларк и Тодд сообщили о подобной изо-
меризации 124. Известно и обратное термическое превращение
оксазиранов в нитроны. Некоторые другие реакции соедине-
ний, имеющих структурный фрагмент = —О”, можно
объяснить только предварительной изомеризацией в цикли-
ческую структуру 106,197 ’109,115,125.
Выделение дифенилдиазометана 116 при реакции имина
бензофенона и О-гидроксиламинсульфокислоты приписывали
такому же раскрытию кольца соответствующего диазирина
[см. (V-72)]. Аналогичное превращение наблюдали, когда
флуоренонимин реагировал с дифторамином126, причем ди-
азофлуорен был единственным выделенным продуктом. При
УФ-облучении диазирина VIII (R = R' = H) получен и диазо-
метан 127> 128:
/N .
н /|| Лукаргон> СН2—N = N+ (V-80)
Эксперименты с мечеными атомами (V-81) подтверждают
промежуточное образование диазиринов, хотя не следует ис-
ключать и возможность обмена с последующей рекомбина-
цией (V-82) 128:
hv, аргон - + (
СН2—,5N = N+------* СН2—N = N15 (V-81)
СН2—15N=N —СН2—N = N + №N15 (V-82)
Вероятно, диазирины обладают большим запасом энергии,
и изомеризация диазоалканов в диазирины состоит в накоп-
лении электромагнитной энергии в виде энергии химической
связи, как это предполагалось Сплиттером и Кельвином для
превращения нитрон — оксазиран. Термины электроизомери-
зация и электроизомеры, по-видимому, лучше всего соответ-
ствуют этим особым случаям валентной изомерии 129.
80
ДИАЗИРИНЫ Vю- 131
Шмитц с сотрудниками провели подробное исследование
диазиринов, которые они получили окислением соответствую-
щих циклических гидразинов (диазиридинов). В противопо-
ложность соответствующим диазоалканам диазирины — бес-
цветные вещества, значительно менее склонные к термиче-
скому разложению, приводящему к единственному продукту —
олефину. Это следует противопоставить поведению ди-
азоалканов, которые часто дают соответствующие азины8Э.
Химическую инертность диазиринов можно объяснить склон-
ностью к образованию азинов. Диазирины.чрезвычайно устой-
чивы к действию сильных кислот и щелочей. Однако их
можно восстановить в соответствующие гидразины или про-
дукты их расщепления. Реакции диазиринов с реактивами
Гниньяра подтверждают их циклическую структуру [ср. с
(V-59)]:
с6нимйВг> /-^/N-CeHH
Z\NH
CgHji N —
(V-83)
УФ- и ПК-спектры соответствуют спектрам гщс-азосоеди-
нений (гл. IV) с отсутствием поглощения в видимой области.
При сильном нагревании диазирины взрывают. Фрей и Сти-
венс 132 изучали их фотолитическое разложение.
Литература
Общая
К. Saunders, The Aromatic Diazo Compounds, -London, 1949.
H. Zollinger, Diazo and Azo Compounds: Aliphatic and Aromatic Com-
pounds, New York, 1961.
R. Huisgen, Aliphatic Diazo Compounds, Angew. Chem., 67, 439 (1955).
E. Schmitz, in A. R. К a t r i t z k y, Advances in Heterocyclic Chemistry,
vol. 2, New York, 1964, p. 83.
*B. E i s t e r t, M. Regitz, G. Heck, H. S c h w a 11, Aliphatische Diazo-
verbindungen (Houben-Weil) Methoden der organischen Chemie, 4
Aufl., X, 473—894 (1968).
*И. А, Дьяконов, Алифатические диазосоединения. Изд. ЛГУ, 1958.
"R. Putter, Methoden zur Herstellung und Umwandlung aromatisch^r
Diazoniumsalze, (Houben-Weil) Methoden der organischen Chemie, 4
Aufl., 10, III, 1—212 (1965).
Цитируемая '
1. H. R e i m 1 i n g e r, Angew. Chem., 75, 788 (1963).
2. K. Bott, Angew. Chem., 76, 992 (1964).
3. K. Bott, Tetrahedron, 22, 1251 (1966).
4. F. К 1 a ges, K. Bott, Chem. Ber,. 97, 735 (1964).
5. E. Fahr, W. D. Hermann, Ann., 682, 48 (1965).
6. D. G. Cram, R. D. P a r t о s, J. Am. Chem. Soc., 85, 1273 (1963).
7. 0. W. Webster, J. Am. Chem. Soc., 88, 4055 (1966).
8. H. Zollinger, Diazo and Azo Compounds:1 Aliphatic and Aroma-
tic Compounds, New York, 1961.
9. L. I. Smith, Chem. Rev., 23, 193 (1938).
6 Зак. 334
§1
10. В. Eisterl, in Newer Methods of Preparative Organic Chemistry,
New York, 1948, p. 513.
11. A. L e d w i t h, E. C. Friedrich, J. Chem. See., 1964, 504.
*12. A. Led with, E. C. Friedrich, J. Chem. Soc., 1964, 505.
*13. P. Schuster, О. E. Polansky, Monatsh., 96, 396 (1965).
14. S. R. Paulsen, Angew. Chem., 72, 781 (1960).
15. E. Schmitz, R. Ohme, Chem.-Ber, 94, 2166 (1961).
*16. E. Шмитц, ЖВХО, 7, 343 (1962).
17. E. Schmitz, R. Ohme, Tetrahedron Lett., 1961, 612; Chem. Ber.,
95, 795 (1962); E. Schmitz, R. Ohme, R. D. Schmidt, ibid.,
95, 2714 (1962); L. Pierce, V. Dob у ns, J. Am. Chem. Soc., 84,
2657 (1962).
18. K. Saunders, The Aromatic Diazo Compounds, London, 1949.
19. Б. И. Белов, В. В. Козлов, Усп. хим., 32, 121 (1963).
20. Е. Н. R о d d, Chemistry of Carbon Compounds, vol. III-A, New York,
1954, p. 256.
21. J. M. Insole, E. S. Lewis, J. Am. Chem. Soc., 85, 122 (1963);
E. S. L e w i s, J. M. I n s о 1 e, ibid., 86, 34, 107 (1964); см. также
R, A. Abramovich et al., Tetrahedron Lett., 1963, 1507; A. K. Bo-
se, I. Kugajevsky, J. Am. Chem. Soc., 88, 2325 (1966).
22. M. R. Piercey, E. R. Ward, J. Chem. Soc., 1962, 3841.
23. I. Ugi in A. R, К a t r i t z k y, Advances in Heterocyclic Chemistry,
vol. 3, New York, 1965, p. 373.
24. С. M. П a p м e т e p, в кн.: Органические реакции, сб. 10, ИЛ, 1963,
стр. 7.
25. Р. Филлип с, там же, стр. 148.
26. Н. С. Ya о, Р. Resnick, J. Am. Chem. Soc., 84, 3514 (1962).
27. M. Stiles, A. S i s t i, J. Org. Chem., 25, 1691 (1960); A. S i s t i,
J. В u r g m a s t e r, M. F u d i m, ibid., 27, 279 (1962); A. S i s t i,
J. Sawinski, R. Stout, J. Chem. Eng. Data. 9, 108 (1964).
28. H. Корнблюм, в кн.: Органические реакции, сб. 2, ИЛ, 1950,
. стр. 285.
*29. Н. Н. Hodgson, Chem. Rev., 40, 251 (1947); W. С о w d г а у,
P. D avies, Quart. Rev., 6, 358 (1952).
*30. Л. Бигелоу, в кн.: Органические реакции, сб. 1, ИЛ, 1949, стр. 136..
31. А. Роэ, там же, сб. 5, 1951, стр. 155.
32. К. Гамильтон, Д. Морган, там же, сб. 2, 1950, стр. 448.
33. В. Е. Б а х м а н н, Р. А. Г о ф м а н н, там же, сб. 2, 1950, стр. 244,
см. также J. I. G. С a d о g г a m, J. Chem. Soc., 1962, 4257.
34. Л. Г. Макарова, в кн.: Реакции и методы исследования органи-
ческих соединений, т. 3, Госхимиздат, 1954, стр. 73.
35. X. С. Р о н д е с т в е д т, в кнл Органические реакции, сб. 11, ИЛ,
1965, стр. 1’99.
36. R. Koelsch, V. Boekelheide, J. Am. Chem. Soc., 66, 412 (1944)';
C. S. Rondestvedt, H. Vogl, J. Am. Chem. Soc., 77, 2313 (1955).
37. К. Д. Гутше, в кн.: Органические реакции, сб. 8, ИЛ, 1956, стр. 469.
38. Т. J. De Boer, Н. J. Backer, Rec. Trav. Chim., 73, 229 (1954);
C. G. Overberger, J.-P. A n s e 1 m e, J. Org. Chem., 28, 592 (1963).
* 39. B. Ei s tert, W. Sch a de, Ber, 91, 1411 (1958).
* 40. H. M о r r i s о n, J. Org. Chem, 26, 2617 (1961).
* 41. И. К- Ко p о б иц ы н а, Л. Л. Родина, ЖОХ, 34, 2851 (1964).
* 42. P. dates, B. Shapiro, J. Am. Chem. Soc, 81, 212 (1959).
* 43. Л. Л. Родина, И. К- Коробицына, ЖОрХ, 4, 2212 (1968).
* 44. М. Regitz, Angew. Chem, 79, 786 (1967).
45. W. R. Bamford, T. S. Stevens, J. Chem. Soc, 1952, 4735.
46. D. G. F a mum, J. Org. Chem, 28, 870 (1963).
47. C. G. Overberger, N. We in sh en к er, J.-P. A n s e 1 m e, J Am.
Chem. Soc, 87, 4119 (1965).
48. J. M e i n w a 1 d, P. G. G a s s m a n, E. G. Miller, J. Am. Chem.
Soc, 81, 4757 (1959); W. Rundel, Angew, Chem, 74, 592 (1963).
82
49. H. R е i m 1 i n g е г, Chem. Ber., 94, 2547 (19S1).
50. E. M u 1 1 e r, H. H a i s s, W. Riindel, Chem. Ber., 93, 1541 (1950).
51. W. R u n d e 1, Angew. Chem. 76, 603 (1964).
52. R. H u i s g e n, Angew. Chem., 67, 439 (19o5).
53. D. Bethell, J. D. C a 1 1 i s t e r, J. Chem. Soc., 1963, 3801, 3808.
*54. R. M. A c h e s о n, J. Chem. Soc., 1956, 4232.
*55. W. W i s 1 i c e n u s, F. M e 1 m s, Ann., 436, 101 (1924).
56. П. А. С. Смит, в кн.: Органические реакции, сб. 11, ИЛ, 1£6б,
стр. 165; для а, P-ненасыщенных кетонов см. W. S. Johnson et al.,
J. Am. Chem. Soc., 84, 989 (1962).
* 57. И. К. Коробицына, К. К. Пивницкий, ДАН СССР, 132, 127
(1960).
* 58. И. К. Коробицына, О. П. Студзинский, ЖОрХ, 4, 524 (1968).
* 59. A. Schonberg, К. Junghaus, Вег., 99, 531 (1966).
60. W. К i г m s е, Chem. Вег., 93, 2357 (1960).
61. R. Т. I z z о, A. S. Ke n d е. Chem. Ind. (London), 1964, 839; R. В res-
low et al., J. Am. Chem. Soc., 87, 1320 (1965); H. L. Logothetis,
J. Org. Chem., 29, 3049 (1964).
62. M. S. Newman, P. Beal, J. Am. Chem. Soc., 71, 1506 (1949).
63. F. Weygand, H. J. В e s t m a n, Angew. Chem., 72, 532 (I960).
64. В. Бахман н, В. Струве, в кн.: Органические реакции, сб. 1, ИЛ,
1948, стр. 53.
*65. Л. Л. Родина, И. К- Коробицына, Усп. хим., 36, 611 (1967).
66. Е. F a h г, Angew. Chem., 73, 766 (1961). .
67. A. Mustafa, J. Chem. Soc., 1949, 234; G. D. Buckley, ibid., 1954,
1850; P. К. К a d a b a, J. O. Edwards, J. Org. Chem., 26, 233
(1961).
68. R. Dijkstra, H. J. Backer, Proc. Konikl. Ned. Akad. Wetenschap,
55 B, 382 (1952).
69. H. J. Backer, B. Bos, Rec. Trav. Chim., 69, 1233 (1950).
70. N. J. Leonard, K. J a n n, J. V. P a u k s t e 1 i s, С. K. S t e in-
ha r d t, J. Org. Chem., 28, 1499 (1963).
71. E. Fahr, Ann., 638, 1 (1960); Angew. Chem., 73, 536 (1961).
72. E. Гинзбург, А. Я- Якубович, А. С. Филатов, Г. E. Зе-
ленин, С. П. Макаров, В. А. Шпанский, Г. П. Котельни-
кова, Л. Ф. Сергиенко, Л. Л. Мартынова, ДАН СССР, 142,
354 (1962).
73. Е. Fahr, F. S с h е с k е n b a ch, Ann., 655, 86 (1962); Е. Fahr,
К. Doppert, F. Scherkenbach, Angew, Chem., 75, 670 (1963).
74. R, Hu is gen, Proc. Chem. Soc., 1961, 357; Chem. Weekblad, 59, 83
(1963); Angew. Chem., 75, 604, 741 (1963); P. X ь ю з г e н, P. Г p э-
ш и, Дж. Сойер, в кн.: Химия алкенов, под ред. С. Патая, гл. X,
Изд. «Химия», 1969.
* 75. W. Е.’ Р а г h a m, С. S е г г е s, Р. R. О’С о n п о г, J. Am. Chem. Soc.,
80, 588 (1958).
* 76. W. Е. Parham, С. S е г г е s, Н. G. Braxton, Р. R. O’Connor,
J. Org. Chem., 26, 1805 (1961).
* 77. R. Hilt tel, L. L i e d 1, H. Martin, Ber., 93, 1427 (1960).
78. C. G. О ver ber ger, J.-P. A n s e 1 m e, J. Am. Chem. Soc., 84, 869
(1962); 86, 658 (1964).
79. R. Huis gen, H. Stangl, H. J. Strum, H. Wagenhofer, An-
gew. Chem., 73, 170 (1961).
80. T. V. Van A u k e n, K. L. Reinehar t,'J. Am. Chem. Soc., 84, 3736
(1962); C. G. Overberger, J.-P. A n s e 1 m ё, J, R. Hall, ibid., 85,
2752 (1963); C. G. Overberger, N. Weinshenker, J.-P. An-
sel m e, ibid, 86, 5364 (1964).
81. M. Alguero et al. Tetrahedron, 18, 1381 (1962). J. В a s t u s, Tetra-
hedron Lett, 1963, 955; C. Belil, J. Pascual, F. Serratos a,
Anales Real Soc. Espan. Fis. Quim, Ser. B, 59, 507 (1963).
82 G. L. Class, J. J. Coyle, J. Am. Chem. Soc, 84, 4350 (1962).
6* g8
83. H. Reimlinger, Angew. Chem., 74, 153 (1962); H. Reimlinger
F. Bi Ilian, M. R e i p e n, Chem. Ber., 97, 339 (1964); см. такж<
A. Schonberg, E. Frese, Angew. Chem., 76, 343 (1964).
84. С. E. H. В awn, Proc. Chem. Soc., 165, 174 (1962).
85. H. M. Whitlock, .1. Am. Chem. Soc., 84, 2807 (1962).
86. A. M. Reader, P. S. Bailey, Chem. Ind. (London), 1961, 1620
A. M. Reader, P. S. Bailey, H. M. White, J. Org. Chem., 30
784 (1965).
87. H. Standing er, F. P f e n n i n g e r, Ber., 49, 1941 (1916);
L. V. Vargha, E. К о r a e s, ibid., 75,, 794 (1942); G. Hesse,
E. M a j u m d a r, Chem. Ber., 93, 1129 (1960); см. также G. Opitz
’ К- T i s c h e r, Angew. Chem., 74, 41 (1965).
88. D. E. Applequist, H. В a b a d, J, Org. Chem., 27, 288 (1962).
89. C. G. Overberger, J.-P. An sei me, J. Org. Chem., 29, 1188
(1964).
90. D. M. Gale, W. J. M i d d 1 e t о n, C. G. Rrespan, J. Am. Chem.
Soc., 87, 657 (1965).
*91. H Schwa 11, M. Regitz, Ber., 101, 2633 (1968).
92. G, Wittig, M. Schlosser, Tetrahedron, 18, 1023 (1962).
93. A. C. Poshkus, J. E. Herweh, J. Org. Chem., 27, 2700 (1962).
94. W. R. Bamford, T. S. Stevens, J. Chem. Soc., 1952, 4675.
95. T. S e v e r i n, Angew. Chem., 70, 745 (1958); Chem. Ber., 92,1517 (1959).
96. D. G. Farnum, P. Yates, Proc. Chem. Soc., 1960, 244.
97. E. Muller, W. Rflndel, Chem. Ber., 90, 2673 (1957): E. M ii 11 e r,
P. Kastner, W. R 0 n d e 1, ibid” 98, 711 (1965); J.-P. A n s e 1 m e,
J. Chem. Educ, 43, 596 (1966).
* 98. E. Muller, D. L u d s t e d, Ber, ,87, 1887 (1954).
* 99. E. Muller, W. Run del, Ber, 88, 917 (1953).
* 100. P. Migniniac, Bull. Soc. chim. France, 2111 (1962); В. Кирмсе,
Химия карбенов, Изд. «Мир», 1966; W. Kirmse, Angew. Chem, 71,
537 (1959); 73, 161 (1961); E. Chinoporos, Chem Rev, 63, 235
(1963); И. JI. Кнунянц, H. П. Гамбарян, E, M. Pox л н и, Усп.
хим, 27, 1361 (1958); J. Hine, Divalent Carbon, New York, 1964.
* 101. И. А. Дьяконов, Журн. ВХО, 7, 436 (1962).
* 102. Г. Г. Розанцев, А. А. ,Ф а й н з и л ь б е р, С. С. Новиков, Усп.
хим, 34, 177 (1965).
* 103. Е. Фогель, Усп. хим, 30, 95. (1961).
104. W. Schlenk, С. Bornhardt. Ann, 394, 183 (1912); см. также
Е. М й 1 1 е г, А. М о о s m а у е г, A. R i е с h е г, Z. Naturforsch, 18b,
982 (1963).
105. G. Ruchardt, G N. Schranzer, Chem. Ber, 93, 1840 (1960).
106. L. Horner, L. H о c k e n b e r g e r, W. Kirmse, Chem. Ber. 94,
290 (1961).
107. E. F a h r, H. L i n d, Angew. Chem, 75, 1122 (1963).
108. T. Kauffmann, S. M. H a g e, Angew. Chem, 75, 248 (1963).
109. A. G и i 11 e m о n a t, in V. Grignard (ed.), Traite de Chimie
Organique, vol. 15, Paris, 1948, p. 25.
110. M. Z. Barakat, M. F. A. E 1 - W a h a b, M. M. E 1 - S a d r, J. Am.
Chem. Soc, 77, 1670 (1965).
111. D. H. R. Barton, R. E. O’Brien, S. S t e r n h e 11, J. Chem. Soc,
1962, 470.
112. N. Latif, J. Fathy, J. Org. Chem, 24, 1883 (1959).
113. H. E. Zimmerman, S. Somasekhara, J. Am. Chem. Soc. 82,
5865 (1960).
114. F. S. В r i dso n - Jo ne s et al, J. Chem. Soc, 1951, 2999, 3009, 3016.
115. L. Horner, W. Kirmse, H. Fernekess, Chem. Ber, 94, 279
(1961); L. Horner, H. Fernekess, ibid, 94, 712 (1961).
116. C. G. Overberger, J.-P. A n s e 1 m e, Tetrahedron Lett, 1963, 1405;
см. также E. Schmitz, A. Stark, С. H о n i g, Chem. Ber, 98,
2509 (1965).
84
117. J. P. Wibaut, J. W. P. Boon, Helv. Chim. Acta, 44, 1171 (1961);
R. E. Mil ler, J., Org. Chem., 26, 2327 (1961).
118. C. Pedersen, Acta Chim. Scand., 12, 1236 (I960).
119. H. E. Baumgarten, С. H. Anderson, J. Am. Chem. Soc.,' 83,
399 (1961).
120. H. U. Daeniker, J. Qruey, Helv. Chim. Acta, 46, 805 (1962).
121. G. L. С 1 о s s, W. Boll, Angew. Chem., 75, 640 (1963); G. L. С 1 о s s,
L. E. Cl oss, W. Boll, J. Am. Chem. Soc.. 85, 3796 (1963);
G. L. Closs, W. Boll, ibid., 85, 1904 (1963); G. Ege, Tetrahedron
Lett., 1963, 1665, 1667.
122. R. Fusco, G. Biancetti, D. F о c a r, R. U g o, Chem. Ber., 96,
802 (1963); P. Griinanger, P. V. F i n z i, Tetrahedron Lett., 1963,
1053.
123. J. S. Splitter, M. Calvin, J. Org. Chem., 23, 651 (1958).
124. R. Bonnett, V. M. Clark, A. Todd, J. Chem. Soc., 1959, 2012.
125. C. J. P e d e r s e n, J. Am. Chem. Soc., 79, 5014 (1957).
126. W. H. G r a h a m, J. Am. Chem. Soc., 84, 1063 (1962).
127. M. J. Amrich, J. A. Bell, J. Am. Chem. Soc., 86, 292 (1954).
128. G. C. Pimentel, С. B. Moore, неопубликованные результаты.
129. G. W. Wheland, Advanced Organic Chemistry, New York, 1960,
p. 728.
130. E. Schmitz, Angew. Chem., 76, 197 (1964).
'131. Э. Шмитц, Трехчленные циклы с двумя - гетероатомами, Изд.
«Мир», 1970.
132. Н. М. Frey, I. D. R. Stevens, Proc. Chem. Soc., 1962, 79; J. Am.
Chem. Soc., 84, 2547 (1962); J. Chem. Soc., 1962, 3865; 1963, 3514;
1964, 4700; 1965, 1700.
Глава VI. ГИДРАЗИДЫ
Гидразиды — производные гидразинов, имеющие ацильные
заместители при атомах азота. Под общим названием «ацил»
имеются в виду такие группировки, как RCO—.RSCb—,
ROCO— и т. д. Для удобства изложения гидразиды разде-
лены на: моноацилгидразины * Acyl-NRNR'R" и диацил-
гидразины * — Acyl-NRNR'-Acyl.
Все обсуждаемые соединения будут рассматриваться как
производные этих двух классов.
Получение
Моноацилгидразины — простейшие соединения этого клас-
са — обычно получают реакцией соответствующих эфиров с-
избытком гидразина:
rcooc2h5 + h2n—nh2 —► RCONH—NH2 + C2H5OH (VI-1)
[Гидразинолиз сложных эфиров N-замещенных аминокис-
лот широко используется при синтезе пептидов для получе-
ния исходных гидразидов в азидном методе (см.1 и указан-
ную там литературу).] Этот* способ становится менее при-
годным с увеличением радикала R в эфире. В таких случаях
необходимо применять более активные галогенангидриды,
хотя такой путь обычно приводит к 1,2-диацилпроизводным
(VI-4). Моносульфонилгидразины получают реакцией суль-
фонилгалогенида с гидразином. Для получения моноацил-
гидразинов также-удобно взаимодействие амидов с гидрази-
ном. При соединении гидразина с кислотой сначала по-
лучается соль, которая образует моноацилгидразин при
пиролизе.
1,1-Дизамещенные ацилгидразины можно получать из га-
логенангидридов кислот:
R'R"N—NH2 + RCOC1 —► RCONH-NR'R" (VI-2)
* [В английском тексте употребляются неточные термины «моноацил-
гидразиды» и «диацилгидразиды», которые нами всюду заменены на ацил-
гидразины.]
86
Аналогичные реакции с производными а,р-непредельных карбоновых
кислот протекают различно в зависимости от реагентов и условий и ослож-
няются возможностью образования гетероциклов:
RCH — CR1
(CH3)2NwCO
МН
(ch3)2n-nh2
a)R=R'“H
б)р„Н.Р.'-СН3 (CHj)2N-NHCH-CHCOOCH3
6)R=CH3;R = H R r'
При ацилировании несимметричного диметилгидразина ангидридами
акриловой и кротоновой кислот наряду с диметилгидразидами I образуются
изомерные им 1,1-диметилпиразолиний-З-оксиды II2'3, а с хлорангидри-
дами тех же кислот образуются хлоргидраты соединений 11. Реакция ди-
метилгидразина с эфирами кислот зависит от природы среды: в полярном
растворителе (вода, спирт) с метилакрилатом получается Па, практически
с количественным выходом, а в неполярном (бензол) — продукт присоеди-
нения по двойной связи С = С III4. Между I и II установлено явление свое-
образного таутомерного превращения 5.
Хлорангидриды и ангидриды акриловой, метакриловой и кротоноцой
кислот, реагируя с Х-бензпл-М-фенилгидразином, дают соответствующие
гидразиды. Кольчатые соединения в этом случае совсем не образуются6.
1,1 -Диметнлги др азиды акриловой и метакриловой кислот и все синте-
зированные из них производные полимеризуются с инициаторами свободно-
радикального типа, образуя высокомолекулярные полиэлектролиты7.
Как упоминалось выше, реакция галогенангидридов с ги-
дразином дает очень хорошие выходы 1,2-диацилгидразинов,
которые могут также быть получены окислением соответ-
ствующих моноацилгидразинов:
2RCOC1 + H2N—NH2—► RCONH—NHCOR ' (VI-4)
2RCONH—NH2 + I2 —> RCONH—NHCOR (VI-5)
«Смешанные» 1,2-диацилгидразины синтезируют реакцией
моноацилгидразинов с соответствующими галогенангидри-
дами:
RCONH—NH2 + ArSO2Cl —> RCONH—NHSO2Ar (VI-6)
[Кинетика реакции ацилирования гидразидов карбоновых
кислот галогенангидридами, ангидридами и сложными эфи-
рами была систематически исследована в работах Грекова
и сотрудников8’9.]
Методы введения карбо-трет-бутоксигруппы были развиты
главным образом в работах Карпино10. Недавно было най-
дено, что карбо-трет-бутоксигруппу можно легко удалить
87
пиролизом н. Это превращение протекает, вероятно, Через ше-
стичленное переходное состояние и имеет перспективы при-
менения в качестве альтернативного метода удаления карбо-
трет-бутоксигруппы.
сн2 /Сен5
о-с( СН н—сн2
N—N:—/С(СН3),
о
СН2 ZC6H5
О = С\ ^сн
NH-NH
+ сО2Ч- (СН3)2С — CHj;
(VI-7)
РЕАКЦИИ МОНОАЦИЛГИДРАЗИНОВ
Моноацилгидразины являются источником получения ацил- •
азидов. В качестве нитрозирующего агента чаще всего при-
меняют нитрит натрия, хотя употребляют и алкилнитриты:
RCONH—NH2 + HNO2 —► RCON3 + 2H2O (VI-8)
[Реакция идет настолько гладко, что может быть исполь- -
зована и для количественного определения гидразидов12-13.] ,
Семикарбазид — хорошо известный реагент на карбониль- j
ную группу, но также реагирует и большинство гидразидов. I
Каталитическое восстановление гидразидов в присутствии 1
никеля Ренея приводит к разрыву связи азот—азот 14. '
Ранее было упомянуто (VI-5), что моноацилгидразины :
могут окисляться до 1,2-диацилгидразинов. Было описано ис-
пользование для такого окисления надуксусной кислоты15. !
Алкилирование моноацилгидразинов проходит по неаци-
лированному атому азота. Вторая алкильная группа направ-
ляется к тому же самому атому и даже из 1,1-диалкил-2-ацил- .
гидразинов при алкилировании получаются гидразидиниевые '
соли: i
RCONH—NH2 + 2R'X —> RCONH—NR' — > 1
—> [RCONHNR']X’ --,оиапие> RCON-NR3 (VI-9) 1
iv ,
Эти гидразидиниевые соли в присутствии щелочей дают 4
соединения типа «илидов» IV1б.
Гидразиды можно восстановить до соответствующих ги- '
дразинов с помощью комплексных гидридов. 1
Реакции с выделением азота. Здесь объединены :
реакции, в результате которых замещается «гидразинный ;
остаток» в моноацилгидразине.
Окисление до альдегида по Кальб—Гроссу 17: ;
RCONH-NH2 —rcho (VI-10) J
88
Превращение в галогенангидриды по Карпино 18’ 19:
RCONH—NH2 - -Н-С1; 2С1г> RC0C1 + N2 + ЗНС1 (VI-11)
ArSO2NH-NH2 ArSO2Br + N2 + 3HBr (VI-12)
[Ароматические сульфонилгидразины с бромом реагируют
быстро и количественно в кислых растворах (реакция идет 1
как в неводных, так и в водных растворах). Выходы алифа-
тических хлоридов в неводных растворах составляли 48—1
79%.]
Аналогичную'реакцию применили Вольман и Геллоп для
синтеза пептидов20 [когда на смесь гидразида N-защищенной
аминокислоты или пептида с соответствующим аминокомпо-
нентом в растворителе действуют на холоду окислителем —
N-бромсукцинимидом, иодом, перекисью водорода или окисью
серебра].
Дезаминирование по Никону — Зипуу2'-.
RNH2+R'SO2C1 —> RNHSO2R' NH2°s°3H>
—> RN—SO2R' —> RH + R'SO2H + N2 (Vi-13)
NH2
Арилгидразиды расщепляются щелочами до соответствую-
щих углеводородов:
ArNH— NHSO2R ArH + N2 + RSO3H (VI44)
Этот прямой метод восстановительного дезаминирования (RNH2~>RH)
дает для алифатических и жирноароматических первичных аминов колеб-
лющиеся выходы, которые, однако, в пересчете на прореагировавший сульф-
амид обычно очень высоки (>90%). Ароматические амины реагирует
хуже. Вероятным промежуточным продуктом является монозамещенный
диимид, разлагающийся с выделением азота.
Разложение сульфонилгидразинов; диимид:
ArSO2NH—NH2 —> ArSO2H + [HN = NH] (VI-15)
Разложение обычно идет при 100—150° С, но гидразиды нитробензол-
сульфокислот разлагаются уже при 25—75° С.
Алкилазиды из [2-алкилсемикарбазидов]22:
R—N—CONH2 H0N°> RN3 + СО2 + N2 (VI-16)
NH2
Гексагидробензилазпд был получен этим методом с выходом 49%. но
выход изобутилазида мал.
89
a, ft-Ненасыщенные кислоты из пиразолинонов23:
Cl. С-С—С6Н5
I II
0=С N
СсН5С^ССООН
Г12О и °
(VI-17)
С1
I
R—С С-СвН5
I II
0=С N
\н
он-
-Що* с6н5сн=с;
хсоон
(VI-18)
цис- и транс-
[Выходы фенилпропиоловой и тетроловой кислот дости-
гали 75—76%, а а-метилкоричной, a-фенилкоричной и а-фе-
нилкротоновой — 40—60%.]
Было показано, что эти реакции протекают через проме-
жуточный диазадиклопентадиенон 24.
Олефины из дигидрооксадиазинонов25:
R'
I
R—С—ОН + NH2—NHCOOC2H5 —>
R"—С = О
200° С
-------->
—-> RR'C = CHR" + CO2 + N2
(VI-19)
[Метод дает возможность перехода от a-оксикетонов и
a-дикетонов к олефинам в невосстановительных условиях.]
Превращение тозилгидразонов в диазоалканы по Бэмфор-
ду — Стивенсу уже упоминалось в гл. III и V, а описанное
Карпино24 катализируемое щелочами разложение 1,1-диза-
мещенных тозилгидразинов — в гл. II и VII. Восстановление
тозилгидразонов алюмогидридом лития до насыщенных угле-
водородов26 является полезным дополнением к методу Киж-
нера — Вольфа:
RR'C = N—NH—Tos RR'Ch2 (VI-20)
Недавно сообщалось27 об интересном случае образования
пиразоленинов V из солей тозилгидразонов а,р-ненасыщен-
ных карбонильных соединений:
R"
R\ I _
ХС = С—C = N—N—Tos —>
R,/Z I
R'" Na+
R'/^N^
(VI-21)
V
90
РЕАКЦИИ 1,2-ДИАЦИЛ ГИДРАЗИНОВ
Алкилирование 1,2-диацилгидразинов— удобный метод
получения 1,2-диалкилгидразинов. Синтез альдегидов по Мак-
фадиену—Стивенсу28 был распространен на алифатические
альдегиды29 с использованием диметилсульфоксида в каче-
стве растворителя. Для этой реакции предложен30 модифи-
цированный механизм (VI-22), подобный механизму реакции
Гофмана:
RCO-NH-NH-SO2Ar RCONH—N-^sbgAr
--- RCONH-N
RCO —N=N—H (vt-гг)
Из 1,2-диацилгидразинов
гетероциклы, например из
нил-1,3,4-оксадиазол VI:
легко получаются пятичленные
дибензоилгидразина — 2,5-дифе-
С6НЬС—NH-NH—СС6Н5
N---N
PoOs II И
—С6Н5-с С—С6Н5
VI
(VI-28)
Циклодегидратация 1,2-диацилгидразииов происходит под действием
водуотнимающих реагентов (Р2О5, РС15, ZnCl2 и др.) или просто при на-
гревании в вакууме и является важнейшим методом получения производ-
ных 1,3,4-оксадиазо ла (см. обзор 31).
Полигидразиды, получаемые конденсацией гидразина с двухоснов-
ными карбоновыми кислотами, также способны к циклодегидратации, пре-
вращаясь при этом в поли (1,3,4-оксадиазолы)32:
“О О ~
II II
R---С—NH—NH—С--
— — п
(VI-24)
Подобным же образом могут быть получены тиадиазолы
и триазолы. 1,2-Диацилгидразины можно превратить в а,а'-
бисхлоразины действием пятихлористого фосфора:
О о С1 С1
1! 11 2РС16 । 1
RC-NH-NH—CR ——-> RC = N— N-CR +2НС1 + 2РОС13 (VI-25)
91
Диацилгидразины легко окисляются до соответствующий
азодиацилов33 — чрезвычайно активных диенофилов34, даю-
щих производные шестичленных тетрагидропиридазинов *:
RCO—NH—NH—COR — > RCON = NCOR .бутадие,1>
/\ /COR
Ж
\/N\cor
(VI-261
Пример образования четырехчленного кольца присоедине-
нием олефина — реакция с тетрафторэтиленомЗб:
CF2=CFS + СН3ООС—N=N— СООСНз —► CF2—CF2
II
СНзООС—N N—COOCHj
(VI-27)
Однако с подвижным водородом могут иметь место реак-
ции замещения. Ниже даются некоторые интересные при-
меры:
-
R”OOC XCOOR"
yN-NHR'
C)n-nhcoor"
R"OOC
R. .N-N-R’
Hz yN—NHCOOR
R'OOC
NHCOOR”
(vt-28)36
+ c2h5o2c—n=n-co2c2h5
—c2h5o2c-n-nhco2c2h5
dH
(VI-Z3)
О О
II II
. CeH6—C—N=N—С—CeH6 + ROH
—► C6H5COOR +
о
II
С6Н5—С—N = NH
—>C6H5COOR + [HN = NH]
—> C6H5CONH—NHCOCeH5
алкен v
----N2 + алкан
(VI
* [Подробнее о реакциях диенового синтеза
монографии39.]
с
диацилдиимидом
92
[Аналогичным образом реагирует и фенилбензоилдиимид,
образуя бензоат и разлагающийся далее фенилдиимид.]
ТуОвг-Бутилмагвийиодид присоединяется по связи N=N
г^ег-бутилазокарбэксилата40. Недавно Гофман41 сообщил о
методе, который может оказаться полезным для получения
арилкарбанионов:
О
11 'О
R4/\/N=N-c-R-" R0- R'\Z\ И
| II f II . + ROCR"' + N2 (VI-31)
R„/'4/\Br R'//\/\Br
Реакция азодизфиров с диазоалканами упомянута в
гл. V [см. (V-44)]; механизм ее достаточно сложен:
rcoch-n2
c2h5o2c-n-n-co2c2h5
^C-OCjH,
C2H6O2C—N—N
ch-n2+
COR
) oc2H,
C2H 5O2C-N--N- С H— COR
c2rtsO2c
N— N
// \ „
CjHjOq ^chn2
Х О COR
»нв
С2Н5ОС СН-СОВ
хо7
о
С2Н5О2СЧ
)n-n=chcor
С солями диазония Г,2-диацилгидразин образует гекса-
аз адиены42:
COR -
+ I
ArN2Cr + RCONH—NHCOR —» Ar—N=N—N—N—N = N—Ar (VI-33)
COR
N, М'-Диалкокси-1,2-диацилгидразины, приготовленные
окислением М-карбалкокси-О-алкилгидраксиДЭминов, окисью
серебра, приобретают необычные свойства под действием
кислот и оснований43.
93
ПЕРЕГРУППИРОВКИ
Из многих известных перегруппировок гидразидов упо-
мянем несколько наиболее интересных.
При перегонке М-бензил-N, N-диметилацетимидов VII про-
исходит миграция бензильных групп 44:
СН3 О
1+ - И
СН3—N—N— С—СН,
I "
X—С6Н4СН2
о
II
сн3>> хссн3
перегонка в вакууме
------------------> N—N
СН3/ \сн2свн4—X
VII
(VI-34)
Недавно было показано45, что реакциям 1-метил-, 1,1-ди-
метил- и 1,1-дибензил-2-тозилгидразонов в кислой среде
предшествует диссоциация на диазений-ион VIII и сульфи-
нат-ион:
СНзх
' N—NH—SO2—^>-СНз
СНз^ Х=
СНз-
СН3'
vnr
(VI-35)
Продукты реакции получаются из этих двух частиц. Во-
прос существования иона диазения будет обсуждаться в
гл. Vlk
Катализируемая щелочами перегруппировка 2-аллил-2-
бензолсульфонилбензоилгидразина в бензоилгидразон 3-бен-
золсульфонилпропаналя происходит через отщепление — при-
соединение 46:
,СН2СН=СН2
С6Н6СО—NH-N^
SO2CeH5
t. основание
—► С6Н5СО—N=N—CH2CH = CH2 + CeH6SO2H °сн°вани---
—► С6Н5СО—NH—N = CHCH=CH2 -C-^5-foH-
—> CeHsCO—NH-N = CH(CH2)2SO2C6H5
(VI-36)
Основное вещество, полученное при гидролизе в кислой
среде аддукта дифенилкетена с 2-карбэтоксиазобензолом47, —i
04
продукт перегруппировки IX:
с6н8Х
С = С=О
свн/
* +
с2н3о2с—n=n-c6h5
на|у
С6Н&\___
—> CsH5XN- N— С6Н5
I
СО2С2Н3
,NHCO2CaH5
(VI-37)
Перегруппировка a-N-ацетил-А-тирозингидразинэ. возмож-
но явится новым методом идентификации некоторых а-амико-
кислот48:
R\
/СН—cf
NH
ХС--I
\ ^NH,
OJ сн3 2
RCHCO NH-N НС О СН 3
I (vi-зв)
ЫНг (R~n-HOC6H4CH2-)
К интересным результатам привело распространение на
гидразиды метода окисления тетраацетатом свинца 49:
^c-n-nh-с<СНз
с6н5 сн3
с=*о
I ^сн,
3
сн3
II /СН3
СН. OCNC
6 \с/ СН3
сбН5Х хс=о
сн3
о
СбН\- + С ! СН.
/С-О^
СсНе I ^СН
6 5 СН£-О
(vi-зз)
lJ5
Катализируемая основаниями перегруппировка 2-бензил
2-фенилтозилгидразина включает промежуточное образова
ние «нитрена»50 (см. гл. VII):
/С6Н5
Tos—NH—N
•сн2с6н5
Tos—N—N
csH5
—Tos
5
С6н5
N:
—-2-> [С6Н5- + С6Н5СН2-]
1с6н5сн2
продукты
стабилизации
радикалов
(VI-40]
Литература
Общая
Н. Wieland, Die Hydrazine, Stuttgart, 1913, S. 180.
С. С. С 1 a r k, Hydrazine, Baltimore, 1953, p. 43.
* E. МОИ er, Di- Tri- und Tetraacylhydrazine, (Houben-Weil) Methoden
der organischen Chemie, 4 Aufl., X, 2, 123—168 (1967).
* A. П. Греков, Органическая химия гидразина, Изд. «Техника», 1966
Цитируемая
* 1. Э. Шрёдер, К. Любке, Пептиды, т. 1, ИЛ, 1967, стр. 121.
* 2. Т. А. Соколова, Н. П. Запев а лов а; Усгг. хим., 38, 2239 (1969)
* 3. Т. ‘ ~ .........
Соколова, Н. И. Запевалова, А. И. Кольцов
Л. А. Овсянникова, Изв. АН СССР, сер. хим., 1964, 1727
Н. П. Запевалова, Т. А. Соколова, Изв. АН СССР, сер. хим.
1965, 1442; 1966, 865.
* 4. Н. П. Запевалова, Л. А. Овсянникова, Т. А. Соколова,
Изв. АН СССР, сер. хим., 1966, 2200; авт. свид. 165732; бюлл. изобр.,
№ 6,137 (1965).
* 5. Т. А. С о к о л о в а, И. Н. Осипова, Л. А. Овсянникова, ЖОрХ,
3, 2252 (1967).
* 6. Н. И. Запевалова, Т. А.
1966, 2197.
* 7. Т. А. Соколова, Л. А. О
Высокомол. соед., А, 10, 956
№ 21, 93 (1968).
* 8. А. П. Греков и др., Укр.
ЖОХ, 32, 542 (1962); 33, 1469, 1474, 1552 (1963);' ЖОрХ, 3, 1251', 1287,
1844 (1967); 4, 286, 1077, 1270 (1968).
* 9. А. И. Греков, М. С. М а р а х о в а, Труды конференции по пробле-
мам применения корреляционных уравнений в органической химии
Тарту, 1962, стр. 177.
10. L. А. С а г р i п о, J. Am. Chem. Soc., 82, 2725 (1960); J. Org. Chem.,
28, 1909 (1963); 29, 2820 (1964).
11. C. G. Overberger, N. Weinshenker, J.-P. A n s e 1 т с, не-
опубликованные результаты.
Л. M. Литвиненко, Д. Г. А р л о з о р о в, В. И. Королева, Укр
х-им. ж.. 22, 527 11956).
А. П. Греков н др., ЖАХ. 16. 643 (1961); 19, 260 (1964); 21, 1276
(1966).
С. Ainsworth, J. Am. Chem. Soc., 76, 5774 (1954); R. L. Hin-
man, ibid., 78, 2463 (1956); J. Org. Chem., 22, 148 (1957); T. Ueda.
T. T s u j u, Chem. Pharm. Bull. (Tokyo), 9, 71 (1961).
L. Horner, H. Fernekess, Chem. Ber.. 94, 712 (1961).
H. H. S i s 1 e r, G. M. О m i e t a n s k i, B. R u d tier, Chem. Rev., 57,
1021 (1957); R. L. Hinman, M. C. Flores, J. Org. Chem, 24, 660
(1959).
С о к о л о в а,'Изв. АН СССР, сер. хим.
вс я ннико в а, Н. П. Запева лова
(1968); авт. свид. 221285; бюлл. изобр.
»
хим. ж., 27, 384 (1961); 28, 98 (1962);
14.
15.
16.
96
17, Э. Мозетти, в кн.: Органические реакции, сб. 8, ИЛ, 1956, стр. 288.
18. L. А. С а г р i п о, J. Am. Chem. Soc., 79, 98 (1957).
19. А. С. Р о sh k u s, J. E. H e r weh, F. A. Magnotta, J. Org. Chem.,
28, 2766 (1963).
20. Y Wolman, P. M Gallop, H. Patch or nick, A. Berger, J.
Am. Chem. Soc, 83, 1263 (1961); 84, 1889 (1962); Y. Wolman,
P. M. Gallop, J. Org. Chem, 27, 1902 (1962); D. M. Brown,
N. K. Kramer, Proc. Chem. Soc, 1960, 212.
21. A. Nick on A. S i n z, J. Am. Chem. Soc, 82, 753 (1960); A. N i-
cko n, A. S. H i 11, ibid, 86, 1152 (1964).
22. P. A. S. Smith,. J. M. Glegg, S. L a k r i t z, J. Org. Chem, 23, 1595
(1958).
23. L. A. C a r p i n o, J. Am. Chem. Soc, 80, 599, 601 (1958).
24. L. A. C a r p i n o, J. Am. Chem. Soc, 79, 4427 (1957).
25. M. Rosenblum et al, Chem. Ind. (London), 1956, 1480; J. Am.
Chem. Soc, 85, 3874 (1963).
26. L. G a g 1 i о 11 i, M. M a g i, Tetrahedron, 19, 1127 (1963).
27. G. L. G 1 о s s, W. В о 11, Angew. Chem, 75, 640 (1963).
28. Cm. [17, стр. 289].
29. I, Friedmann, American Chemical Society Abstracts, Fall 1962
Meeting, p. 96Q, no. 174; M. S p r e c h e r, M. F e 1 d k i n e 1, M. W i 1-
chek, J. Org. Chem, 26, 3664 (1961).
30. U. M. Brown, P. H. Carter, M. Tomlinson, J. Chem. Soc, 1958,
1843.
*31. E. П. He сынов, А. П. Греков, Усп. хим, 33, 1184 (1964).
*32. А. П. Греков, С. А. М а л ютенко, К. А. Корнев, ХГС, 1966,
352.
33. Е. F a h г, Н. L i n d, Angew. Chem, 78, 376 (1966).
34. S. В. N e e d 1 e m a n, M. C. Chang Kuo, Chem. Rev, 62, 405 (1962).
35. J. С. К a u e r, H. F. Schneider, J. Am. Chem. Soc, 82, 852 (I960);
R. W. Hoffmann, H. Hauser, Angew. Chem, 76, 346 (1964).
36. В. T. G i 11 i s, F. A. D a n i s h e r, J. Org. Chem, 27, 4001 (1962).
37. J. M. Cinnamon, K. Weiss, ibid, 26, 2644 (1961).
38. J. F. N e u m e r, J. T. G e r i g, American Chemical Society Abstracts,
Fall 1962 Meeting, p. 23Q, no. 40; J. E. Leffler, W. В. B o n d, J. Am.
Chem. Soc, 78, 335 (1956); D. Mackay, U. F. Mark, W. A. Wa-
ters, J. Chem. Soc, 1964, 4793; S. G. Cohen, I. Nicholson, J.
Org. Chem, 30, 1162 (1965).
*39. В. T. Gillis, in J. Hamer, 1,4-Cycloaddition Reactions, New York,
1967, Chapter 6, p. 143.
40. L. A. Carpino, P. H. Terry, P. J. С г о w 1 y, J. Org. Chem, 26,
4336 (1961).
41. R. W. Hoffmann, Angew. Chem, 75, 168 (1963); Chem. Ber, 97,
2763, 2772 (1964).
42. J. p. Horwitz, V. A. G r a k a u s a s, J. Am. Chem. Soc, 79, 1249
(1957).
43. R. J. Crawford, R. R a ap, J. Org. Chem, 28, 2419 (1957).
44. S. W a w z о n e k, E. Y e a k e y, J. Am. Chem. Soc, 82, 5718 (1960).
45. S. W a w z о n e k, W. M с К i 11 i p, J. Org. Chem, 27, 3946 (1962).
46. M. S. Newman, I. Ungar, J. Org. Chem, 27, 1238 (1962).
47. C. W. В i r d, J. Chem. Soc, 1963, 674; 1964, 5284.
48. A. N. Kurtz, C. Niemann, J. Am. Chem. Soc, 83, 3309 (1961).
49. D. C. If flan d, T. M. Davis, ibid, 85, 2182 (1963).
50. Р. C a r t e r, T. S. Stevens, J. Chem. Soc, 1961, 1743; D. Lem al,
F. M e n g e r, E. Coats, J. Am. Chem. Soc, 86, 2395 (1964).
7 Зак. 334
Глава VII. N-НИТРОЗАМИНЫ И НИТРАМИНЫ
N-НИТРОЗАМИНЫ
N-Нитрозамины I, как показывает само название, имеют ,
нитрозо-функцию, непосредственно связанную с аминным
атомом азота:
R\
'N—N=O—► R—N = N—OR' (R'-H)
R'Z
I II
В том случае, когда один из заместителей амина — водо- J
род, предпочтительна таутомерная форма II, как и оксим- ;
ная форма для С-нитрозосоединений. Диазогидраты неустой-
чивы и образуются в первой стадии нитрозирования первич-
ных аминов (см. гл. V, VII, VIII). Родоначальное соединение,
где R = R' = H, неизвестно. Поэтому изложенная здесь химия
N-нитрозаминов ограничена такими представителями, в ко-
торых ни R, ни R' не являются атомами водорода.
Получение
Самый общий и распространенный на практике метод по-
лучения N-нитрозаминов заключается в действии нитрита
натрия на слабокислый раствор вторичного амина. Величина
pH должна быть подобрана так, чтобы ион нитрозония по-
лучался в присутствии достаточного количества свободного
амина, необходимого для протекания реакции.
Если этот метод неудобен из-за структурных особенностей
аминов, в качестве нитрозирующего агента с успехом приме-
няют окислы азота (еще лучше — хлористый нитрозил). В не-
которых случаях представляет интерес использование тетра-
нитрометана:
R\ R4
'N—CH2R" + C(NO2)4 —► N-N = O + R''CHO + CH(NO2)3 (VII-l)
R'X R'Z
Тетраарилгидразины реагируют с окисью азота до соот-
ветствующих N-нитрозаминов, с солями оксимов окись азота
98
образует N-нитрозосоли
RCH
/NO
N\0Na
Удобный син-
тез азоксисоединений основан на реакции реагентов Гриньяра
с О-тозилпроизводными N-нитрозогидроксиламинов2. N-Ни-
трозариламиды можно также готовить реакцией галогенан-
гидридов с арилдиазотатами.
Реакции
Применяется несколько методов восстановления N-нитроз-
аминов до соответствующих 1,1-дизамещенных гидразинов.
При этом иногда в небольших количествах образуются те-
тразены.
При действии едкого кали на арилалкил-Ы-нитрозамины
получается арилдиазотат и спирт, соответствующий алкиль-
ной группе.
R\
N—N=₽O + KOH —> Ar— N==N— O~K+ + ROH (VH-2)
ArZ
Пиролиз соединений III аналогичен реакциям расщепле-
ния ацетатов и ксантогенатов:
О
II
СН3-С\
сн2
СНз—С N—N = O
СНз R
Ш
О
II
CH3CI
RN = N—<
Действие дегидратирующих
N-нитрозоглицины приводит к С1
/СН3
i=c;
\СН3 (VH-3)
I
агентов на N-замещенные
нонам IV*:
(СНзСО)гО ч
R—N---С-Н
I ± I
N С=О
(VII-4)
IV
Хлоргидраты сиднониминов были также приготовлены из
нитрозонитрилов в присутствии сухого хлористого водорода.
* [О сиднонах см. обзор 3.]
7*
99
Эфиры Ы-нитрозо-М-тозилглицина расщепляются под влия-
нием щелочей4 на тозиламид, щавелевую кислоту и гидро-
ксиламин:
Tos-N-CH2COOCH3—- Tos —N—CHCOOCHj-
NO
Tos—N—СНСООСН,
t\/ 3
N
о~
---- TOS -N-CHCOOCH,-^— TOS — NH-CCOOCH,
I 3 II 3
NO N-OH
---- Tos — NHj + HOOC-COOH + NHjOH
f vn - s )
N-Нитрозоациланилиды ацилируют фенилгидразин до
арилдиазогидрата, который реагирует дальше с фенилгидра-
зином, образуя бензол и азот:
С6Н6—N—COR + С6Н6—NH—NH2 —> C6H5NH—NHCOR + CaH5N = NOH
(VII-6)
NO
Реактивы Гриньяра обычным образом присоединяются к
связи N = O нитрозамина с образованием соответствующего
гидразона (VII-7):
С6Н5. С6Н5 /OMgl
^N-N=o + c2HsMgi —► n; —>
С6н/ свн/ ХС2Н5
СаН5у
—> ;n—n=chch3 (vii-7)
С6н/
Если гриньяровский реагент лишен а-водорода, образует-
ся диазиридин V, который присоединяет еще одну молекулу
магнийорганического реагента с раскрытием цикла (VII-8):
CsHsy
N = O + C6H5MgBr —> С2Н5—N---N—С6Н5 —>
С2Н/ I I
С2Н5 OMgBr
—► C2H5-N N-C6HB
СН
СНз (VII-8)
|caH5MgBr
у
C2H5—N—NH—C6HS
I
CSH6CHCH3
100
Расщепление N-нитрозаминов до соответствующих ами-
нов (денитрозирование) можно проводить несколькими спо-
собами. Обычно пользуются серной или соляной кислотами,
иногда в комбинации с хлористым оловом, спиртом, цинко-
вой пылью или хлоридом меди.
Реакция нитрозаминов с серной кислотой в присутствии
фенола представляет собой пробу Либермана на нитрозо-
группу.
При пиролизе N-нитрозамины могут разлагаться по двум
возможным направлениям (VII-9; а, б) в зависимости от при-
роды заместителей5. Наблюдаемые продукты получаются в
результате взаимодействия с растворителем или комбинации
фрагментов обеих реакций:
R\
Чг—N=O
R"7
R\
Чр + -N = O
R'Z
(VII-9)
—-> [R-N=N-OR'J —* R- + N2 + -OR'
Когда R = CH3, a R'— трифенилметил, расщепление идет
только по типу б, а если R' — бензгидрил, происходит реак-
ция а. Если Р = СбН5, a R' — трифенилметил, реакция идет
сразу по обоим направлениям.
При фотолизе алкилнитрозаминов в присутствии кислот
образуются амидоксимы. Разрыв молекулы осуществляется
в комплексе, содержащем водородную связь6:
R-N-CH2R'+HX—> R—N—CH2R' hv R—NH RNH
I ' I > I I
N = O N=O---H—X CHR' C=NOH
O=N—H R'
(VII-10)
Под действием кислот арилнитрозамины подвергаются пе-
регруппировке Фишера — Геппа. Нитрозогруппы мигрируют
от аминного азота в пара-положение ароматического ядра
(если это положение свободно):
/ S—N—R
\=/ ।
NO
—O = N-<^J>—NHR
(VII-I1)
[Перегруппировка происходит под действием кислот с
межмолекулярной миграцией нитрозил-катиона 7.]
101
Дифенилнитрозамин с фенилгидразином дает дифенил-
амин, анилин и закись азота:
(c6H8)2N-N=O + CgHjNH—NHa ----- (ceH5)2NH +
н
К
С8Н5— N—N
= ' II.---- CeH,NH2 + NjO
CjHjNH- NH— NO
( Vil-it)
Недавно сообщалось о подобной реакции с активными ме-
тиленовыми соединениями8:
R R
R\J\ R\J\
I СН2 + (C8H6)2N-N = О —> I С = N—ОН + (C8H5)2NH
r/'Y r/\Z (VII-13)
R R
(R = C8H5, Cl)
N-Нитрозамины окисляются до соответствующих N-нитр-
аминов9.
Реакции N-ацил-М-нитрозаминов зависят от природы за-
местителей при атоме азота. Арилзамещенные соединения
обычно разлагаются с образованием арильных радикалов.
Таутомерное превращение в азосоединение предшествует раз-
ложению 10:
О О 0
I I I
Al—Nj-Cf? —* Ar—N —N—OCR —- Аг» + Nj + • OC-R (vii-k)
N-O
В реакции N-нитрозалкиламидов со щелочами, как уже
описано в гл. V, получаются диазоалканы. Уайт11 изучил
термическое разложение этих веществ. Было показано, что
диазоалкан образуется, если молекула нитрозамида содер-
жит первичный алкильный радикал. Рекомбинация диазоал-
кана и получившейся кислоты приводит к эфиру (VII-15).
Этот метод был предложен как замена реакции дезаминиро-
вания азотистой кислотой:
г о
RCH2-N — N — OCR’
О
RCHN; + R'COOH —RCH.OCR’ CVJI-15)
102
В присутствии вторичных или третичных алкильных групп,
вероятно, образуется карбониевый ион; обсуждался п-12 ме-
ханизм реакции в различных условиях:
о
R2CHN—N=O —►
O=CR'
О
II
R2CHN=NOCR'
—> LRaCHOCR'J —>
О
II
—► RjCHOCR'
(VII-16)
Можно сделать заключение, что по своим свойствам N-ни-
трозамины совершенно непохожи на С-нитрозосоединения
VI, для которых, как и для альдегидов, характерны многие
реакции конденсации. Эта разница между С- и N-нитрозо-
соединениями, несомненно, обусловлена значительным вкла-
дом резонансной структуры VII в нитрозаминах, что доказано
спектральными исследованиями:
J>N-N-0
R3C-N = O
VII
[Конфигурации и конформации N-нитрозаминов опреде-
ляли методом ЯМР-спектроскопии13.] Последние результа-
ты14 указывают на тенденцию N-нитрозаминов реагировать
по кислородному атому. Продукты таких реакций являются
алкилирующими агентами. С натрийциклопентадиенилом по-
лучается соответствующий гидразон 15:
RCH2. rch2, +
ЧТ—N=0 + RjOX- —> ^N^N^O-R'
RCH/ RCH/
(VII-17)
Вопреки прежним утверждениям, триалкиламины реаги-
руют с азотистой кислотой (при pH = 4-*-5). Продукты рас-
щепления содержат альдегид, нитрозамины и закись азота
(VII-18). Некоторые из предложенных промежуточных
103
соединений можно выделить, если нитрозировать хлористым
нитрозилом в безводной среде6-.
(RCH2)3N + HON = O -СНбо^°ЛсН50> I(RCH2)3N - N=O] + Н2О
—-(rch2)2n=chr
|нон
(rch2),nh+rcho
| HOMO
(rch^n-n^o
+ [noh]
I 2 X
г ’ т (vn-ia)
[hon=noh]
h2o+n2o
Реакция Овербергера — Ломбардии о; N - н и-
трены16-17. При восстановлении активированных вторичных
N-нитрозаминов в щелочной среде происходит выделение
азота и сочетание получающихся фрагментов 15:
С6Н5СН2—N—СН2СвН5 — > N2 + С6Н6СН2-СН2С6Н5 (VII-19)
NO
Сходство $тих результатов 16-18 с данными по окислению
соответствующих гидразинов (VI1-20) свидетельствует о воз-
можности образования одинакового промежуточного соеди-
нения. • Чаще всего в качестве реагента применяется гидро-
сульфит натрия в присутствии оснований, и было высказано
предположение, что реакция проходит следующие стадии:
(C6H5CH2)2N-N=O
(C6H5CH2)2N-NH2
2g4-> (C6H5CH2)2N-NHOH ———
—> (C6H5CH2)2N—NHHgOH
-CeH6CH24
/N-N
.CgHsCHj/ “
—> C6H5CH2CH2C8H5 + N2 (VII-20)
viii
Природа промежуточной частицы VIII и механизм ее раз--
ложения в настоящее время не выяснены. Несколько имею-
щихся исследований не позволяют решить окончательно во-
прос механизма. Состав продуктов при введении заместителей
в ароматический остаток изменяется столь незначительно,
что этому не следует придавать значения 19. Условия (гете-
рогенная среда), в которых проводятся реакции восстановле-
ния и окисления, не благоприятствуют кинетическим определе-
ниям. Если принять VIII или частицу, стремящуюся к
образованию VIII как промежуточное соединение, тогда
трехцентровый механизм лучше соответствует полученным
104
результатам, чем радикальный или ионный. Это заключение
основано на том факте, что М-нитрозо-2,6-дифенилпиперидины
и соответствующие гидразины обнаруживают большую сте-
реоспецифичность, чем нециклические соединения в тех же
условиях 16~18’ 20' 21.
Дальнейшим подтверждением трехцентрового механизма,
по крайней мере в случае циклов, является получение 1,2-ди-
фенилциклобутана при окислении 2,5-дифенил-М-аминопирро-
лидина21. Между тем, если допустить образование 1,4-бута-
диильного радикала, что, по-видимому, имеет место при тер-
мическом разложении 3,6-дифенил-1,2-диазациклогексена-1
в стирол, то нельзя ожидать образования каких-либо цикли-
ческих углеводородов:
СН2-СН2
I I
с6н5-сн сн-с8н8
nh2
сн2—сн2
СбН5—CH СН—С8Н6
I
СН2—CHj
I I
CH—CH+
I I
c6H5 C8H8
HgQ ?
+ олефин
(VII-21)
ZCH2-CH2^
C8H8 = CH CH—C8H5
^N==N/Z
CH2—CH2
I I
CSH5-CH CH-C8H5
• •
—> C8H5CH=CH2
Большая стереоспецифичность в циклических производных
по сравнению с нециклическими указывает на эффект, вы-
званный присутствием, кольца:
CbHs
с6н5 n^,n
с6н5
(vn-22)
Напротив, термическое разложение 3,7-дифенил-1,2-диаза-
циклогептена-1 совершенно нестереоспецифично.
Трехцентровый механизм разложения, представленный
здесь, является лишь усовершенствованным вариантом, кото-
рый в отдельных случаях лучше объясняет образование про-
дуктов реакции. Он не исключает реакций и по другим меха-
низмам, идущих одновременно и дающих разные продукты.
Во многих реакциях N-нитрены (R2N—N) получаются в
качестве гипотетических промежуточных частиц, хотя никаких
конкретных доказательств их существования пока еще нет.
105
Единственным экспериментальным подтверждением служит
работа Макбрайда и Крузе22, которые представили убеди-
тельные данные в пользу существования соответствующих им
кислот — ионов диазения (R2N = NH).
[Образование N, N-диалкилдиазениевых ионов при окисле-
нии 1,1-диалкилгидразинов в сильнокислой среде доказано
спектральными данными, возможностью введения их в реак-
цию Дильса—Альдера 22’23 и образованием «смешанных»
тетразенов из смеси продуктов окисления двух разных гидра-
зинов.] Существование С-нитренов (R—N) установлено24, а
теоретически N-нитрен, если учитывать его сходство с ди-
азоалканами и азидами, должен быть более, стабилен:
диазоалканы
R\_
^С—N = N+
Rz/
R\ .. R\ +
?C = N—N C = N==N"
Rz/ " R'Z
азиды
R—N—N=N+ R—N = N—N R—n = N = N-
N-нитрены
R\ .. Rx +
^N—N XN = N~
Rz/ ” Rz/
ABB
Формально диазоалканы и азиды также можно рассмат-
ривать как N-нитрены (см. формулу Б).
Другое косвенное доказательство получено при аналогич-
ном восстановлении N-нитрозимина в соответствующий N-ни-
трен 25; N-нитрен в этом случае можно выделить, так как он
представляет собой соответствующий диазоалкан:
\ = N-N = O —!--ЬН4->
Аг
'А1ч
,C = N—N—ОН
Аг/ I
Аг\
ArZ
C = N—Ns
основание
Н2О
(VI1-23)
Как уже ранее упоминалось, промежуточное образование
N-нитренов постулировалось и для некоторых других реак-
ций. При окислении бромом и трет-бутилгипохлоритом акти-
вированных 1,1-дизамещенных гидразинов26 преимущественно
106
образуются N-галогенгидразины, которые теряют элементы
галогенводорода и дают N-нитрены:
СбН5СН2, . CgHgCHav
Si—NH2 4- Br2 —> /N~NHBr
CeH5CH2/ CeH5CH/
~C6H5CH2X 1 (C6H5CH2)2N-N=N-N(CH2CeH5)2
—> N—N —> + (VII-24)
_CelisCH27 " C6H5CH2CH2C6H5
Описанное Карпино27 катализируемое щелочами разложе-
ние тозилгидразинов аналогично реакциям элиминирования
N-нитрозаминов и гидразинов:
(свн5снДм-NH-Sо2 -ОсНз-^2^ (c^ch^n-HAoj-O-ch,1^
c.h»ch^n_^
с^сн/
(Vi,-г»)
Сообщалось28 об интересном применении этой реакции
для синтеза ацетиленов:
RVN4
R'ANZ
' I
.•n:
*-•- RC=Cs' (vu-2e)
Другие реакции, проходящие по этому же пути, — дезами-
нирование дифторамином (VII-27 ) 29, окисление тетраацета-
том свинца30 лактама Небера в 3-циннолинол (VII-28),
озонолиз азометинов (VII-29) 31 и превращение диаллиламина
в 1-аллил-Д2-пиразолин (VII-30) 32:
107
о
о
СН3
I /СНз + I /СНз СНз—с = о
CH3COC = N—N СНз—с—К—N —> | +
^Аг I Аг СН3—С = О
СНз—С
II
О
—-+ СНз—N—Аг + оа
I
NO
(VII-29)
СН2 = СНСН2/
\ih + hnf2
СН2=»СНСН/
'CH2 = CHCH2
XN— N
СН2СН = СН2-
СН2 = СНСН2/
N—N
сн2 \н
'Ч'СН2/
(VII-30)
Последняя реакция до сих пор является самым суще-
ственным доказательством промежуточного образования
N-нитрена.
В противоположность N-нитрозоазетидину33 N-нитрозо-
азиридин очень нестабилен30'31. При попытке выделения он
разлагается на закись азота и этилен (VII-31a), Однако при
низких температурах был снят его УФ-спектр в растворе и
вещество подвергнуто интересным превращениям с реакти-
вом Гриньяра34:
JnH + NOC1 — ^-N = O CHi СНг
csHsx
(C,H,N-O)+ N-N-CH-CjH, +
с4нэ
n2o
(vn-31a)
(VII-310 )
Моноалкил- и 1,2-диалкилгидразины также образуют
N-нитрозопроизводные. Динитрозосоединения могут терять
108
две молекулы азотистой кислоты, превращаясь в соответ-
ствующие азосоединения35:
СН3—N—NH2
(CH3Q)2SO4
СН3—N—NHCH3 <-НО-Х° СНз—NH—NH—СН8
О
|hono
ch3nh—nh2
N
Ihono
2HONO
(VII-32)
СН3—N—N—СН3 —СНз—N=N-CH3
NO NO
НИТРАМИНЫ
N-Нитрамины IX родственны по структуре N-нитрозами-
нам, только нитрозогруппа замещена нитрогруппой:
R\ +Z°
\N—N
\0-
+ /OR'
R—N = N
СТ
IX IX a
(R'=H)
Химия нитраминов изучена мало и обзор Ламбертона36
обобщает литературу до 1951 г. Следует указать некоторые
особенности, отличающие их от N-нитрозаминов. Они не
проявляют основных свойств, а первичные нитрамины (1Ха,
R = Н) являются кислотами. В IX как R, так и R' могут
быть атомами водорода, что является существенным отли-
чием от нитрозаминов. Хотя группе —NO2 и необяза-
тельно свойственна нестойкость, для многих N-нитросоедине-
ний характерны взрывчатые свойства (циклонит, нитрогуани-
дин и др.)
Структура и таутомерия нитраминов рассмотрены Лам-
бертоном 36.
Получение
Из-за неустойчивости первичных нитраминов в кислой
среде для их получения используют косвенный способ. Са-
мым общим является метод нитрования ацилпроизводных с
109
последующей щелочной обработкой получающегося при этом
продукта:
0 0 О
„ n ln, HONOs n 1 1) основание
R—NH2 4- Cl—CR —> R—NH—CR > R—N—C—R кнслота *•
no2
—> R—NH—N02 • (VII-33)
Другой метод синтеза первичных нитраминов — присоеди-
нение диазоалканов к нитроуретану с дальнейшим разложе-
нием:
N02
r2c-n2 + no2—nhco2c2h5 —> r2chn—со2с2н5 -4)-ки^оатГ'^
—> R2CHNH—N02 (VII-34)
Еще один способ состоит в нитровании N, N-дихлорами-
нов в присутствии уксусного ангидрида с последующим вос-
становлением полученных N-хлорнитраминов:
RNC12 + HONO2 - (СНзС0.)2°-> r-N-CI -^aa-S2-?-3» RNH—N02 (VII-35)
NOj
Два более новых метода, по-видимому, в равной степени
пригодны для получения как первичных, так и вторичных
нитраминов 37’38:
R2NH2-NO- -JCHsCOhOijnCH^ R2N—N02 + СНэСООН (VII-36)
CH3\ zono2
R2NH+ —> R2N—N02 + CH3COCH3 + HCN (VI1-37)
CH3Z XC=N
Нитролизом диалкиламидов получают вторичные нитр-
амины, хотя иногда могут образоваться нитрамиды:
г-2-* RaN—N02 + R'COOH
R'CONR2 + HON02 - 6 (VII-38)
i-2* R'CONR + ROH
I
no2
Ароматические первичные нитрамины можно синтезиро-
вать окислением диазотатов. Недавно было осуществлено
прямое N-нитрование ароматических аминов с использова-
нием фениллития в качестве основания и амилнитрата как
нитрующего агента 35.
N-Нитроазетидин получается при окислении N-нитрозо-
азетидина 33.
110
Реакции
Действие кислот. В присутствии кислот первичные
алифатические нитрамины разлагаются на соответствующие
спирты и закись азота; в то же время ароматические нитр-
амины подвергаются перегруппировкам типа Фишера — Геп-
па. Вторичные алифатические нитрамины с трудом поддаются
действию кислот даже при 100°С. Механизм перегруппировки
ароматических нитраминов изучался много лет39, и недавно
было показано, что действуют как межмолекулярный, так и
внутримолекулярный механизмы40. Вода превращает диал-
килнитрамины в соответствующие гидроксиламины и закись
азота. Первичные алифатические нитрамины конденсируются
с формальдегидом в присутствии кислот41.
Действие щелочей. Отщепление азотистой кислоты
от вторичных алифатических нитраминов, имеющих а-водо-
родные атомы, ускоряется щелочами; образующиеся шиф-
фовы основания расщепляются на амины и альдегид:
R—N—CH2R'
N+ -'^анм* RN = CHR' + HONO (VII-39)
Большинство первичных алифатических нитраминов со
щелочами дают соли; они могут присоединяться к активиро-
ванной двойной связи по типу реакции Михаэля 42.
Прочие реакции. Восстановление приводит к разрыву
молекулы или к соответствующим гидразинам. О-Замещенные
первичные алифатические нитрамины (которые можно полу-
чить алкилированием соответствующих калиевых или сереб-
ряных солей) разлагаются кислотами43:
+/°~ н+
r—n=n; —
ROH + N2O + R'OH
(VII-40)
Был исследован пиролиз N-нитраминов, который проте-
кает аналогично пиролизу N-нитрозамидов44:
О О" О о
II |_ II II
R—N—С — R'—> LR—N-N-OCR'J —► ROCR' + N2O (VII-41)
NO2
Литература
Общая
И. Губен, Методы органической химии, т. IV, вып. 1, Госхимиздат, 1949,
стр. 97.
А. Н. Lamberton, Chemistry of Nitramines, Quart. Rev., 5, 75 (1951).
Ill
Цитируемая
1. М. J. Danzig, R. F. Martel, S. R. Rice ti ell о, J. Org. Chem., 26,
3327 (1961).
2. T. E. S t e v e n s, J. Org. Chem., 29, 311 (1964).
*3. F. H. C. S t e w a r t, Chem. Rev., 65, 129 (1964).
4. W. К i г m s e, L. Horner, Chem. Ber., 89, 1674 (1956); см. также
H. U. D a e n i k e r, Helv. Chin. Acta, 47, 33 (1964).
5. W. Rickatson, T. S. Stevens, J. Chem. Soc., 1963, 3960.
6. E. M. Burges, J. M. Lavanish, Tetrahedron Lett., 1964, 1221;
Y.-L. Chow, ibid., 1964, 2333.
*7. H. H e n e c k a, Methoden cer organischen Chemie, (Houben-Weil),
4 Aufl., IV, 2, 57 (1955); F. Moller, там же, 11, I, 834 (1957).
8. С. H. Schmidt, Angew. Chem., 75, 169 (1963).
9. C. L. Bumgardner, K- S. McCallum, J. P. Freeman, J. Am.
Chem. Soc., 83, 4417 (1961).
10. R. Huisge n, Ann., 574. 184 (1951); R. Huisgen, H. Reim lin-
ger, Ann., 599, 161 (1956).
11. E. H. White, С. H. Au f derma rsh, J. Am. Chem. Soc., 83, 1179
(1961).
12. E. H. White, J. E. Stuber. J. Am. Chem. Soc., 85, 2168 (1963).
13. G. Karabatsos, R. A. Taller, ibid., 86, 4373 (1964) [P. Rade-
macher, R. Stolevik, W. Liittke, Angew. Chem., Intern. Ed., 7,
806 (1968)].
14. S. Hilnig, L. Geld er n, E. Lucke, Angew. Chem., 75, 476 (1963);
Rev. Chim. Acad. Rep. Populaire Roumaine (Romania), 7, 935 (1962);
A. Schmidpeter, Tetrahedron Lett., 1963, 1421; Chem. Ber., 96,
3275 (1963); D. Klaman, W Kos er, Angew. Chem., 75, 1104 (1963).
15. K. Hafner, K- Wagner, Angew. Chem., 75, 1104 (1963); R. N. Lo-
epp ky, Diss. Abstr., 24. 2273 (1963).
16. R. A. Abramovitch, B. A. Devis, Chem. Rev., 64, 149 (1964).
17. C. G. Overberger, J. G. Lombardino, R. G. Hiskey, J. Am.
Chem. Soc., 80, 3009 (1958).
18. C. G. Overberger, J. G. Lombardino, R. G. Hiskey, J. Am.
Chem. Soc., 79, 6430 (1957).
19. C. G. Overberger, L. P. Her in, J. Org. Chem., 27, 417, 2423
(1962).
20. C. G. Overberger, N. P. M a r u 11 o, J. Am. Chem. Soc., 83, 1378
(1961).
21. C. G. Overberger, M. Valentine, неопубликованные результаты.
22. W. R. McBride, H. W. Kruse, J. Am. Chem. Soc., 79, 572 (1957);
W. H. Urry, H. W. Kruse, \V. R. McBride, ibid., 79, 6568 (1957);
W. R. McBride, E. M. Bens, ibid., 81, 5546 (1959); W. H. Urry,
P. Szecsi, C. Ik oku, D. W. Moore, ibid., 86, 2224 (1964).
*23. К. H. Зеленин, И. П. Бежав, ХГС, № 2 (1970).
24. G. S m о 1 i n s k у, E. Wasserman, W. A. Yager, J. Am. Chem.
Soc., 84, 3220 (1962).
25. H. E. Zimmerman, D. H. Paskovich, J. Am. Chem. Soc., 86,
2149 (1964).
26. C. G. Overberger, B. S. Marks, J. Am. Chemi Soc., 77, 4104
(1955).
27. L. A. Carpino, J. Am. Chem. Soc., 79, 4427 (1957); D. M. Lem al,
T. W. Rave, S. D. McGregor, ibid, 85, 1944 (1963); D. M. L e-
mal et al., ibid., 86, 2395 (1964); 87, 393 (1965).
28. F. G. Wiley. Angew. Chem., 76, 144 (1964); см. также C. D. Cam-
pell, C. W. Rees, Chem. Comm., 1965, 192; C. W. R e e s, R. C. S t о r r,
ibid., 1965, 193; см. также ссылку [27] гл. II.
29. C. L. Bumgardner, К. J. Martin, J. P. Freeman, J. Am. Chem.
Soc., 85, 97 (1963).
30. H. E. Baumgarten, P. L. C r e g e r, R. L. Z e y, J. Am. Chem. Soc.,
82,3977 (1960),
ll?
31. J. P. Wibaut, J. W. P. Boon, Helv. Chim. Acta, 44, 1171 (1961).
32. C. L. Bumgardner, J. P. Freeman, J. Am. Chem. Soc., 86, 2233
(1964).
33. W. N. White, E. F. W о 1 f a r t h, J. R. Klink, J. К i n d i g, C. Hat-
haway, D. L a у d i n s, J. Org. Chem., 26, 4124 (1926).
34. W. R fl n d el, E M ii 11 er, Chem. Ber., 96, 2528 (1963).
35. J. Thiele, Ann., 376, 239 (1910); C. G. Overberger, G. Kesslin,
J. Org. Chem., 27, 3898 (1962).
36. A. H. Lamberton, Quart. Rev., 5, 75 (1951).
37. G. F. Wri gh t et al., Can. J. Res., 26B, 89, 114 (1948).
38. W. D. Emmons, J. P. Freeman, J. Am. Chem. Soc., 77, 4387 (1955).
39. S. Brown stein, C. A. Bunton, E. D. Hughes, J. Chem. Soc.,
1958, 4354; Б. А. Геллер, Л. H. Дуброва, ЖОХ, 30, 2627 (I960)-
W. N. White, J. R. Klink, D. L a у d i n s, C. Hathaway^
J. T. Golden, H. S. White, J. Am. Chem. Soc., 83, 2024 (1961);
W. N. Wh ite, J. T. Golden, Chem. Ind. (London), 1962, 138.
40. A. H. Lamberton, J. Chem. Soc., 1961, 1797.
41. L. Goodman, J. Am.. Chem. Soc., 75, 3019 (1953).
42. L. W. Kissinger, M. Schwary, J. Org. Chem., 23, 1342 (1958).
43. P. Bruck, A, H. Lamberton, J. Chem. Soc., 1955, 3997; P. Bruck,
J. N. Denton, A. H. Lamberton, ibid., 1956, 921.
44. E. H. White, D. W. G r i s 1 e y, J. Am. Chem. Soc., 83, 1911 (1961).
§ Зак. 334
Глава VIII. АЗИДЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ 1
АЗИДЫ |
Азиды R—N3 характеризуются линейным расположением !
трех атомов азота, соединенных друг с другом. Ковалентную 1
структуру для азидной группы написать невозможно; ее луч- |
ше представить резонансным гибридом нескольких структур: 1
R—N = N—N «*-> R—N—N=N R—N = N = N~ 1
la 16 I в ]
Группа R может быть алкилом, арилом или ацилом I
(R'CO—, RSO2— и т. д.). Имеется ряд подробных обзоров по |
синтезу и реакциям азидов 1-е. Мы ограничимся наиболее су-
щественными особенностями и новейшими открытиями. Син- |
тезы и реакции ацилазидов обсуждены отдельно. а
ПОЛУЧЕНИЕ
Самый распространенный метод синтеза азидов состоит в |
замещении галогена или сульфогруппы действием азотисто- I
водородной кислоты или ее солей. Найдено, что замещению |
подвергаются также многие другие группы: 1
^X + NaNs—> R-N3 + NaX (VIII-1) 1
Присоединение HN3 по активированной двойной связи— I
другой часто используемый путь синтеза алкилазидов. Ино-- 1
гда ввести азидогруппу такими способами оказывается не- |
возможным (арилазиды) и требуется постадийное построение j
азидной функции. Для этого имеется несколько методов1-2. 1
[Бром- и иодазиды присоединяются и к неактивированной |
двойной связи; этим методом возможно получение непред ель- 1
ных азидов7. Удобным методом синтеза азидоспиртов может 1
служить расщепление а-окисных циклов азотистоводородной |
кислотой или азид-ионами8-10.] |
Реакции |
Гидролиз. Азидогруппа, как псевдогалоген, может ги- |
дролизоваться в кислых условиях, давая азотистоводородную 1
кислоту. Однако щелочи не разлагают большинства азидов I
114
даже при высоких температурах (но азотистоводородную ки-
слоту можно отщепить с образованием олефина). Сопряже-
ние азидогруппы с активирующими заместителями способ-
ствует щелочному гидролизу а-азидокарбонильных соедине-
ний.
Разложение. В отличие от кислот, основания не ката-
лизируют разложения азидов за исключением отдельных слу-
чаев. При катализируемом кислотами разложении можно
получить разнообразные продукты. Реакции эти интенсивно
изучаются1’3. Катализируемое щелочами разложение азидо-
карбонильных соединений обычно состоит в отщеплении «-во-
дорода и последующем образовании имина:
( VI1I-2)
Интенсивно изучалось термическое и фотолитическое раз-
ложение азидов. Чаще всего потеря- азота приводит к обра-
зованию электронодефицитных частиц II, изоэлектронных
карбенам и называемых нитренами (или азенами и именами):
R—N~—N = N+ -~v R—N —► R—N:
(VIII-3)
Ila 116
Реакции нитренов, основное состояние которых, как пока-
зано, — триплетное [II б], рассмотрены в обстоятельных обзо-
рах Хорнером и Христианом н, а также Абрамовичем и Де-
висом 12 и будут упомянуты здесь в самых общих чертах
(схема VIII-4)
r'/Vn/
I
rnh2 н
А
I §н_____R - R'(CH2)4-1
\ О
СОС2Н5 *• R _ с2НбОС— (К N]
7 II
о
R - СвН5(СН2)з--> । J. ।
'NZ
Н
8*
115
К-(СНз)зС- I r°c6h5ch2-
I !
CHS—NH C6H5— N = CH2
\c/ +
/' \ c6h5ch = nh
CH3 CH3
(V11I-4)
Первый устойчивый нитрен был выделен недавно Смитом
и сотрудниками 13:
N=N
I I
Ar—N С—Аг
\с^
:N:
В некоторых случаях может иметь место отщепление азо- ;
тистоводородной кислоты с образованием карбена: 1
СНз—N3 —> СН2: + HN3 (VIII-5) 1
Реакции присоединения. Азиды склонны к много- I
численным реакциям присоединения. Присоединение водо- 1
рода протекает очень легко с образованием соответствующих |
аминов и азота. Используются каталитические и. некаталити- 1
ческие методы гидрирования. Иногда такой метод выбирают |
для синтеза аминов. Среди соединений, которые можно при- I
соединять к азидам, следует упомянуть гриньяровские ре- |
агенты. Продукт присоединения метилмагнийиодида к фенил- Я
азиду — З-метил-1-фенилтриазен является очень хорошим ме- 1
тилирующим агентом и к тому же устойчив 14: |
С6Н5-N3 + CH3Mgl —> CeHs—N=N—N—СН3 |
Mgl 1
—> С6Н6—N = N—NHCH3 (VIII-6) J
Образование фосфиниминов из азидов и фосфиновых про- I
изводных, впервые отмеченное Штаудингером и Мейером15, I
вероятно, идет через первичный продукт присоединения: 1
CeH5-N3 + (C6HS)3P —> [(CeH5)3P—N—N = NC6H5] |
—> (CeH6)3P-NCeHs (VIII-7) 1
С ненасыщенными системами азиды вступают в 1,3-бипо- |
лярное присоединение. Такие реакции идут с олефинами, аце- 1
тиленами и нитрилами3'18. , |
Н6 1
Реакции органических азидов с комплексами Иоцича и ацетиленидами
лития требуют более мягких условий, чем с другими ацетиленовыми соеди-
нениями, и производные 1,2,3-триазола получаются в некоторых случаях
с большими выходами |7.
О присоединении к олефинам см. также новейшие работы 18~22. Пере-
группировка аллилазидов, протекающая через циклическое переходное со-
стояние23, также может рассматриваться как пример внутримолекулярного
присоединения азидогруппы по двойной связи:
I
| | + | |
ХС=С-C=N = N- q=± /\ I \ N = N = N—С—С = С^ (VIII-8)
/ | N N | \
Недавно опубликованная реакция фенилазида с бензаль-
дегидом24, по-видимому, включает присоединение фенилни-
трена (С6Нб—N) к карбонильной группе с образованием ок-
сгзирана, который окисляет бензальдегид до бензойной кис-
лоты:
/°\
С6Н5—N3 + С6Н6СНО —► C6H6N-----СН-С6Н5 СбЩСН0->
—► C6H5COOH + C6H5N = CHC6H5 (VII1-9)
Натрий образует с фенилазидом ион-радикал, который
при гидролизе превращается в гидразобензол. При дальней-
шем действии натрия на этот ион-радикал получается динат-
риевое производное анилина 25:
С6Н5—N3 + Na —+ CeHj-N-7 'Чг
’ Na+
c6h5nh—nhc6h6
—> C6H5-N: Na+ - Na (VIII-10)
L£L~> C6H5— N— Na2
Реакции фенилазида с фосфоранами и литиевыми солями
бензилбензолсульфината или дифенилбензилфосфиноксидом
приводят к интересным результатам 26:
(С6Н6)3Р—CHR + 2С6Н5—N3 —> (CeHs)3P-NCeH5 + C6H-,N = CHR + N2
(VIII-11)
C6H6SO2CHCeHsLi+ + CeH5—N3 —► C6H6-N = CHCeHs + CJUSO2Li + N2
(VIII-12)
(C6H5)2P—CHCeH5Li++ CSH5—N3 —► (C8H5)2P—CH— N = N—NLi+
O’ O’ C6H5 C6H5
(VIII-13)
117
. Бойер и Морган47 нашли, что алкилазиды в присутствш
кислот образуют с бензальдегидом замещенные амиды:
ОН Л',+ г ОН п
АгСН0 н+ I С1 । +
+ ----> Ar—СН—NCH2R —> [Ar—CH—N—CH2R_
rCH2N3 I (VIII-14
k k
ArCNHCH2R CHO
О Ar—N—CH2R
Подобный азониевый ион был постулирован в реакции бу-
тилазида с карбониевыми ионами для объяснения продуктов
реакции (VIII-15)28:
c4h9n, + ch+ —► С4Н9—N—СН3 —> C3H7-CH=NCH3 и c4h9n=ch2
(VIII-15)
Открытая недавно реакция карбенов с азидами29 приводит к образо-
ванию производных изоциановой кислоты;
+ :СС1, --------- R-N^T,N RN=CC12 (viu-ie)
v Cl,
АЦИЛАЗ ИДЫ
Чаще всего ацилазиды готовят взаимодействием соответ-
ствующих галогенангидридов с азидом натрия (метод 1). Ре-
акция протекает легко и с высокими выходами:
RCOCl + NaNj —>RCON3 + NaCl (VIII-17)
Нитрозирование ацилгидразинов (метод 2) применяют, ко-
гда эфир кислоты легче доступен, чем сама кислота, или если
последняя слишком чувствительна к условиям, необходимым
для получения галогенангидридов:
RCO2R'+H2N—NH2 —► RCONH—NH2 - ——ч- RCON3 + H2O (VIII-18)
Более новый метод30 включает использование «смешан-
ных ангидридов», что избавляет от необходимости синтезиро-
вать хлорангидрид:
О О
RCOOH + C1COOC2H6 —► RC—О—С—ОС2Н5 RCON3 (VIII-19)
Этот метод дает высокий выход амина (по VIII-20) без
рацемизации. Более того, кислоту можно непосредственно
превратить в амин без выделения промежуточного азида. Не-
давнее усовершенствование метода 2 заключается в исполь-
зовании хлористого нитрозила или алкилнитритов в качестве
118
питрозирующих агентов31. Основная реакция азидов карбо-
новых кислот — разложение до изоцианатов — известна каг
реакция Курциуса. Последовательность реакций (VIII-20),
рассмотренная в обзоре Смита н, приводит к превращениям
кислоты в амины с меньшим на единицу числом углеродных
атомов:
RCOOH —> RCOC1 —► RCON3 —>
R—С—N
—► R—N = C=O + RNH2
(VIII-20)
Наличие промежуточного ацилнитрена было доказано об-
разованием аддуктов 1,3-биполярного присоединения с бен-
зонитрилом и фенилацетиленом32. Изоцианат можно выде-
лить или же превратить в амин или его производные (кар-
бамат или мочевину, если изоцианат разлагать в присутствии
спирта или амина). Сульфазиды не претерпевают в обычных
условиях превращения, подобного реакции Курниуса33.
С фосфинами ацилазиды дают фосфинимиды. Некоторые
из первичных аддуктов III были недавно выделены34:
RS.O2N3 + (C8H5)3P —> RSO2-N=N-N-P(CeH6)3 — ->
in
—> RSO2N-P(C6H6)3 (VIII-21)
[Замещение азидогруппы на аминогруппу лежит в основе
весьма важного «азидного метода» синтеза пептидов, при ко-
тором не наблюдается рацемизации35.]
Ацилазиды — очень хорошие ацилирующие агенты. Арил-
галогениды можно с высокими выходами превратить в со-
ответствующие азиды36 реакцией Гриньяра с сульфазидами:
Аг— Вг —> ArMgBr—R——ArN3 + RSO2MgBr (VIII-22)
Бензолсульфазид реагирует с антраценом, образуя с 55%
выходом 1-бензолсульфамидоантрацен и только по 5—15%
каждого из 2- и 9-изомеров, и таким образом ведет себя ско-
рее как реагент на кратную связь, чем как свободнорадикаль-
ный реагент37. Азиридин был предположен здесь в качестве
промежуточного соединения. Участвует ли в реакции ацил-
нитрен (C6H5SO2—N) не установлено:
(vnt-гз)
119
ТЕТРАЗЕНЫ И ТРИАЗЕНЫ38
Тетразены имеют цепь из четырех атомов азота с одной
двойной связью. Лучше других известны Д2-изомеры IV, ко-
торые могут быть получены осторожным окислением соответ-
ствующих гидразинов и в некоторых случаях контролируе-
мым [неполным] восстановлением N-нитрозаминов:
Ч (О) R\ /R (Н) /R
\j-NH2 —XN-N = N-NZ O=N-N (V1II-24)
RZ RZ XR ^R
IV
Хотя известно некоторое число Д2-тетразенов, их химия не
изучалась, и сведения о реакциях этих соединений очень огра-
ниченны. Они разлагаются при нагревании, давая соответ-
ствующие свободные азотные радикалы39. Основное превра-
щение этих свободных радикалов — димеризация в гидразин,
хотя могут образоваться и продукты перегруппировки:
R\ ZR'
xn-n=n-nz
R'Z XR
R\ ZR
xn—nz
Rz/ \R'
(VIII-25)
При реакции Гриньяра с Д2-тетразенами образуются ами-
ны 40.
Триазены V получаются из аминов и солей диазония (гл.
V) или из азидов и гриньяровских реагентов (VIII-26):
ArN2X" + RNH2 —> Ar—N = N—NH—R -<— ArN3+RMgX
(VIII-26)
Ar—NH—N = N—R
Эти соединения существуют в таутомерных формах и мо-
гут расщепляться кислотами 14:
ArN2Cl" + ArNH2.HCl ArN = N—NH—R --°c^lk>
—► RC1 + N2 + ArNH2.HCl (VIII-27)
Восстановление цинком в кислом растворе приводит к со-
ответствующим гидразину и амину. Триазены можно алкили-
ровать; при быстром нагревании они взрывают.
Литература
Общая
П. А. С. Смит, в кн.: Органические реакции, сб. 3, ИЛ, гл. 9, 1951,
стр. 322.
*В. Я. Починок, Триазены, Изд. Киевского государственного универси-
тета, 1968.
J. Н. Boyer, F. С. Canter, Alkyl and Aryl Azides, Chem. Rev., 54, 1
(1954).
E. Lieber, R. L. Minnis, C. N. Rao, Carbamoyl Azides, Chem. Rev.,
65, 377 (1965).
*C. Grundmann, Organische Azidoverbindungen, (Houben-Weil) Metho-
den der organischen Chemie, 4 Aufl., X, 3, 777 (1955),
120
*E, Mil 11 er, Aliphatische Triazene und Tetrazene, (Ilouber-Weil) Methoden
der organischen Chemie, 4 Aufl,, X, 2, 823—832 (19671.
Цитируемая
1. J. H. Boyer, F. C. Canter, Chem. Rev., 54, 1 (1954)
2. N. P. В u u - H о i, P. P. C a g n i a n t, in V. Grignard (ed.), Traite
de Chimie Organique, vol. 15, Paris, 1948, p. 715.
3. L. 1. Smith, Chem. Rev., 23, 193 (1938).
4. П. А. С. С м и т, в кн.: Органические реакции, сб. 3, ИЛ, 1951, стр. 322.
5. Р. A. S. Smith, Р. de Mayo, Molecular Rearrangements, vol. 1,
ch. 8, New York, 1963, p. 457.
* 6. E. Lieber J. S. Curtice, C. N. Rao, Chem. a. Ind., 1966, 589.
* 7. A. Hassner и др., J. Am. Chem. Soc., 87, 4203 (1965); 89, 2077
(1967); 90, 216 (1968); J. Org. Chem., 33, 2686 (1968).
* 8. J. D. I n g г a m, J. Org. Chem., 21, 1373 (1956).
* 9. C. A. Van der W e f, R. H u s 1 e r, W. M с E v e n, J. Am. Chem. Soc.,
76, 1231 (1954).
*10. G. Swift, D. S w e r n, J. Org. Chem., 32, 511 (1967).
11. L. Horner, A. Christman n, Angew. Chem., 75, 707 (1963).
12. R. A. Abramovitch, B. A. Davis, Chem. Rev.. 64, 149 (1964).
13. P. A. S. Smith, O. L. К r b e c h e k, W. Res eman, J. Am. Chem.
Soc, 86, 2025 (1964).
14. E. H. W h i t e, H. S h e u e r, Tetrahedron Lett, 1960, 758.
15. H. Staudinger, J. Meyer, Helv. Chim. Acta, 2, 635 (1919);
H. Staudinger, E. Hauser, ibid, 4, 861 (1921).
16. R. Huis gen, Angew. Chem, 75, 604, 741 (1963).
* 17. Г. С. Акимова, В. H. Ч и с т о к л e т о в, А. А. Петров, ЖОрХ,
3, 986, 2241 (1967); 4, 389 (1968).
* 18. S. К- Khetan, М. V. George, Can. J. Chem, 45, 1993 (1967). -
*19. A. S. В е i 1 е у, J: J. Wedgwood, J. Chem. Soc, (Cl, 1968, 682.
* 20. К. R. H e n e г у - L о g a n, R. A. Clark, Tetrahedron Lett, 1968, 801.
* 21. R. F. Bleiholder, H. Shechter, J. Am. Chem. Soc, 90, 2131
(1968).
* 22. J. S. Meek, J. S. Fowler, J. Org. Chem, 33, 985 (1S68).
* 23. C. A. Van der W e r f, V. S. H e a s 1 e y, J. Org. Chem, 31, 3534
(1966).
* 24. JI. А. Нейман, В. И. M а й м и н д, М. М. Шемякин, Изв. АН
СССР, ОХН, 1962, 1498.
25. Т. К a u f f m а п п, S. М. Н a g е, Angew. Chem, 75, 248 (1963),
26. Н. Н о f f m а п п, Chem. Вег, 95, 2563 (1962).
27. J. Н. Boyer, L. R. Morgan, J. Org. Chem, 24, 561 (1959).
28. W. Pritzkow, G. Pohl, J. prakt. Chem, 20, 122 (1963); N. Wi-
berg, К. H. Schmid, Angew. Chem, 76, 381 (1964).
*29. J. E. Baldwin, I. E. Patrick, Chem. Commun, 1968, 968.
30. J. Weinstock, J Org. Chem, 26, 3571 (1961).
31. J. Honzl, J. Rudinger, Coll. Czech. Chem. Ccmmun, 26, 2333
(1961).
32. R. Huis gen, J.-P. A n s e 1 m e, Chem. Ber, 98, 2998 (1965); см.
также W. L w о w s k i, G. T. T i s u e, J. Am. Chem. Soc, 87, 4022
(1965).
33. W. L w о w s k i, E. S c h e i f f e 1 e, J. Am. Chem. Soc.. 87, 4359 (1965).
34. J. E. Franz, С. О s u c h, Tetrahedron Lett, 1963, 84; H. Bock,
W. W e i g r a b e, Angew. Chem, 75, 779 (1963).
*35. Э. Шрёдер, К- Любке, Пептиды, т. I, ИЛ, 1967, стр. 120.
36. L. В. Bruner, Diss. Abstr, 19, 438 (1958).
37. J. F. Т i 1 п е у - В a s s е 11, J. Chem. Soc, 1962, 2517.
*38. C. S ii s 1 i n g. Aromatische Triazene und hohere Azagomologe, (Houben-
Weil) Methoden der organischen Chemie, 4 Aufl, X, 3, 695 (1965).
39. В G. G о w e n 1 о c k, P. P. Jones, D. R. Snelling, Can. J. Chem,
41, 1911 (1963).
40. R. M. Briggs, Diss. Abstr, 21, 752 (1960).
121
УКАЗАТЕЛЬ
Азиды
алкил и арил 114
получение 61, 89, 114
реакции 114
ацил 118
Азины
гидрирование 44
озонирование 78
N окиси 77
пиролиз 76
реакция с галогенами 37, 44
Азоксисоединения 42
Азосоединения
алифатические 43
изомеризация 45
а, р-ненасыщенные 45, 46
получение 43, 73
разложение 45—52
реакции 16, 46, 76
цис-транс 42, 50
ароматические
получение 41, 61
расщепление 42
реакции 27, 42
днацилзамещенные 72, 90
реакции 90, 92, 93
Азосочетание 61
Алкилирование
гидразинов 16, 17, 18
диазоалканами 66, 69, 72
триазенами 116
Альдогидразониевые соли 17, 35,
36
Аминонитрильная перегруппировка
17, 35, 36
Амины
из гидразинов 27
из нитрилов 27
из нитросоедииений 27
окисление в азосоединения 42
Биполярное присоединение, 1,3- 65,
71, 79, 119
Валентная таутомерия 79
122
Гекса азадиены 93
Гексагидротетразины 31
Гидразндиниевые соли 88
Гидразиды — см. Гидразины, моно-
и диацил
Гидразин(ы)
алкилирование 16—18
ацилирование 86, 87
восстановление гидразином 26,
27
диацил 91
перегруппировки 94
1,1- дизамещеиные 20
1,2- дизамещенные 18-
моноацил
алкилирование 88
окисление в альдегиды по
Кальбу—Гроссу 88
расщепление до углеводоро-
дов 89
реакций 88—90
монозамещенные 17
окисление 18, 43, 87, 120
основность 25, 26
реакция с карбонильной груп-
пой 10, 30—32
тетразамещенные 24, 98, 120
трехзамещенные 24
Гидразиниевые соли 24—26
Гидразониевые соли 17, 35, 36
аминонитрильная перегруппи-
ровка 17, 35, 36
щелочное разложение кетогид-
разониевых солей 36
Гидразоны
из 1,1-дизамещенных гидрази-
нов 23
из монозамещенных гидразинов
31
окисление 21, 23, 37, 38, 64, 76
реакция с тетраацетатом свин-
ца 35, 44
Г пдроксиламино-О-сульфокислота,
реакция с
амииамн 16
Гидроксиламино - О - сульфокисло-
та, реакция с
кетиминами 77, 80
Глицины, N-нитрозо 99
Дезаминирование диазониевых со-
лей 62
Диазения ион 94, 106
Диазнридин 16, 19, 100
Диазирины
получение 94, 106
свойства 19, 57, 77, 81
таутомерия 80
Диазоалканы
восстановление 75
полимеризация 72
получение из
аминосоединений 64
гидразина и хлороформа
64
гидразонов 64
нитрозоамидов 63
оксимов 65
тозилазида 64
тозилгидразоиов 64
получение, различные способы
76—79
промежуточные соединения или
продукты из
азинов 76
азотриазолов 78
аминокетонов 78
гидразонов и хинонов 76
диазиринов 77, 80
N-окисей азииов 77
олефинов и окиси азота 77
сиднониминов 78
реакции с
аминами 74
гидразинами 74
кислотами 66
ненасыщенными соединения-
ми 67—73
радикалами 74, 75
соединениями трехвалентного
' фосфора 73
стабильность 55, 56
таутомерия 55
третичные 55
Диазоимины 55
Диазония соли
получение 59, 60
реакции без выделения азота
38, 61
восстановление в гидразины
с активными метиленовыми
соединениями 61
с нуклеофилами 59, 61
сочетание 61
Диазония соли
реакции с выделением азота 62
галогеном, циано- и другими
группами 59, 61
получение диарилов 62
присоединение к олефинам 63
стабильность 55
строение 57
Диазооксиды 56
Диазотаты 59
Диимид 26, 89
Енамины 70
Енгидразины 34
Закись азота, присоединение к оле-
финам 77
трифеиилфосфинметилену 65 '
Изодиазометан 57
Изомеризация
азосоединений 34, 35
гидразонов 34
Карбены из
азидов 116
диазоалканов 74
Кетеи
дифенил, реакции 43, 68, 95
реакция с диазометаном 68
Кетеиимии 49
Метиллитий, реакция с диазомета-
ном 74
Нитрены
ацил 119
С- 115, 117
N- 22—23, 96, 104—108
N-Нитроамины
получение 109, 110
реакции 111
N-Нитрозоамины
ацил, разложение 63
получение 98
реакции
Овербергера — Ломбардиио
104, 105
пиролиз 99, 101
расщепление 101-
фотолиз 101
строение 103
Нитрозогндразины 109
С-Нитрозосоединения 103
Нитросоедииения 27, 42
Озазоны 18
Оксадиазиноны, реакция с этплкар-
базатом 90
123
Оксадиазолидины 1,3,4- из 1,2-диал-
килгидразинов 19
Оксадиазолы 1,3,4- из 1,2-диацил-
гидразинов 91
Основность гидразинов 25
Перегруппировка
аминонитрильная 17, 35, 36
бензидиновая 20
Валлаха 42
Вольфа 68
гидразидов 94
Демьянова — Тиффено 67
Курциуса 119
Фишера — Геппа 42, 101, 111
Пиразоленины 79, 90
Пиразолидоны 17
Пиразолиний, 1,1-диалкил-, соли 17,
31, 36
Пиразолин-З-оксиды 87
Пиразолиноны 90
Пиразолины
Д'- 44, 71
Д2- 31, 71, 72, 107
А3- 20, 44
Пиразолы 32, 71
Радикалы
из азосоединений 46—52
реакции с диазоалканами 74,
75
Ракетное топливо 15
Рашига синтез 15
Реакции
арилирования по Меервейну 63
Арндта — Эйстерта 68
Барта 62
Бэмфорда — Стивенса 37, 64
Гаттермана 62
Гомберга — Бахмана 62
Гриньяра с
азидамн 116, 119
азосоединениями 93
гидразонами 16
диазиринами 19, 81
диазоалканами 74
дезаминирования по Никону —
Зинцу 89
Дильса — Альдера с
1,2-диацилазосоединениями 92
Реакции
а, р-ненасыщенными азосое-
динениями 46
Зандмайера 62
Карпино 89, 107
Кижнера — Вольфа 90
Макфадиена— Стивенса 91
Манниха с 1,2-диалкилгидрази-
нами 20
Несмеянова 62
Овербергера — Ломбардино 104
Форстера 65
Шимана 62
Яппа — Клингемана 61
Сиднонимины 78, 99
Сидноны 99
Сопряжение, р, л- 33
Сульфены 73
Таутомерия
азосоединений и гидразонов 34,
45
валентная 79, 80
1,2,4-гексагидродиазинов и
р-оксиэтилгидразонов 34
диазоалканов и диазиринов 80 '
енгидразинов и гидразонов 34 '
нитронов и оксазиранов 80
Тетразены 21, 107, 120
Тетразолия соли 38
1,2,4-Тиадиазолы 91 "
Триазены ;
алкилирование 120 4
из азидов 116 j
из аминов и солей диазония 120 ,
Триазолидин, 1,2,4- 19
Тропилий бромид 92
4
Фенилгидразон формальдегида 31 j
Формазаны 38 i
Фосфора производные, реакции 65, 1
73, 116, 117
Хлорамин, реакция с I
третичными аминами 24
шиффовыми основаниями 19
Хлордиазометан 72 j
Электроизомерия 80 1
Ч. Дж. ОВЕРБЕРГЕР, Ж-П. Анселм, Дж. Г. Ломбардино
с. 128
ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
СО СВЯЗЯМИ АЗОТ —АЗОТ
Издательство „Химия", Ленинградское отделение
Невский пр„ 28
Редактор Л. Б. Мясникова
Технический редактор Ф. Т. Черкасская
Корректор Л. А. Любовнч
Сдано в набор 1/х 1969 г. Подписано к печати 15/1 1970 г. Пей. л. 8,0.
Уч.-изд. л. 8,09 Формат 60 X 901/,«. Бумага тил. № 2 Тираж 6000 экз.
Цена 56 коп. Зак. № 334.
Ордена Трудового Красного Знамени
Ленинградская типография № 2 им. Евгении Соколовой
Главполиграфпрома Комитета по печати при Совете Министров СССР
Измайловский пр., 29.