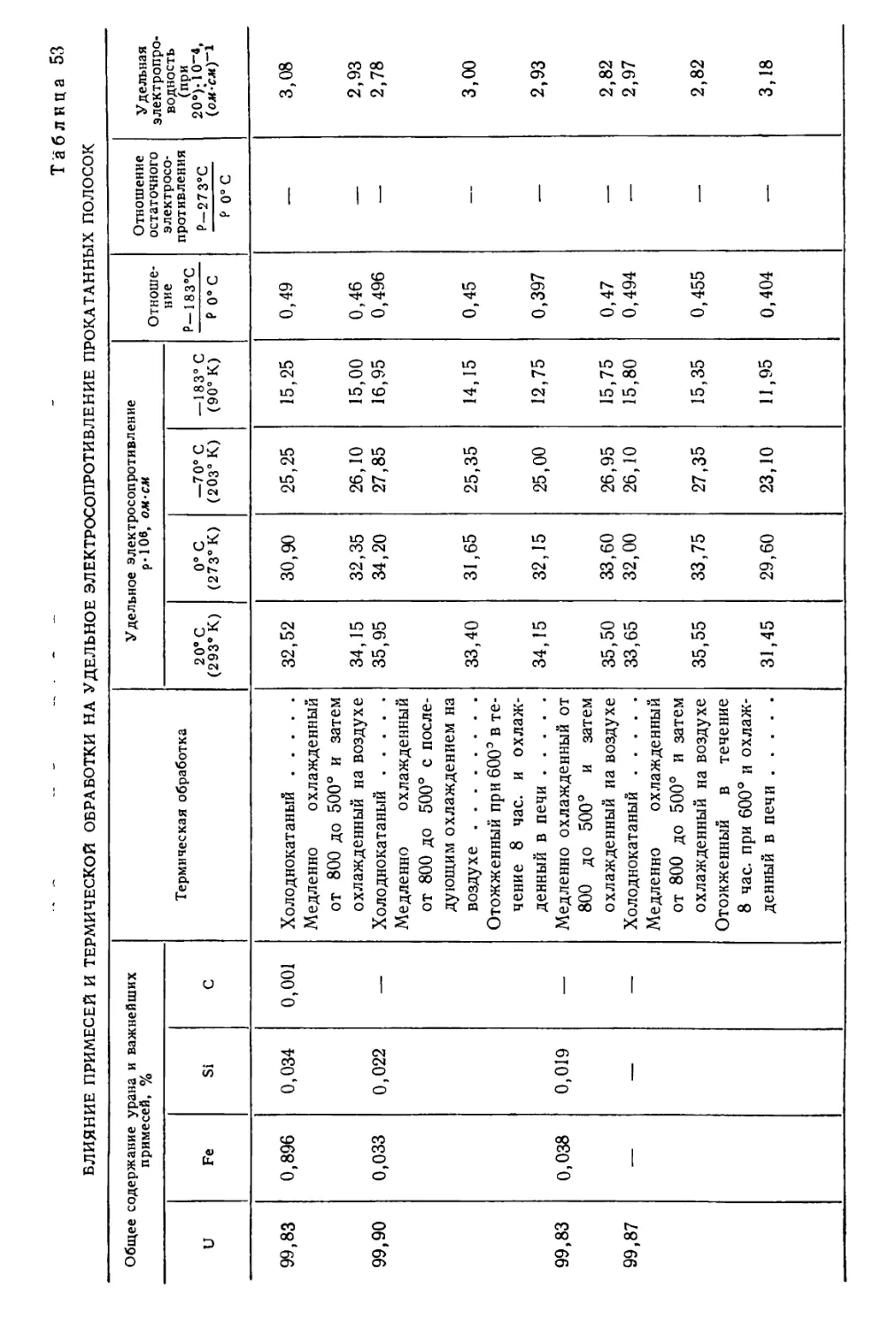
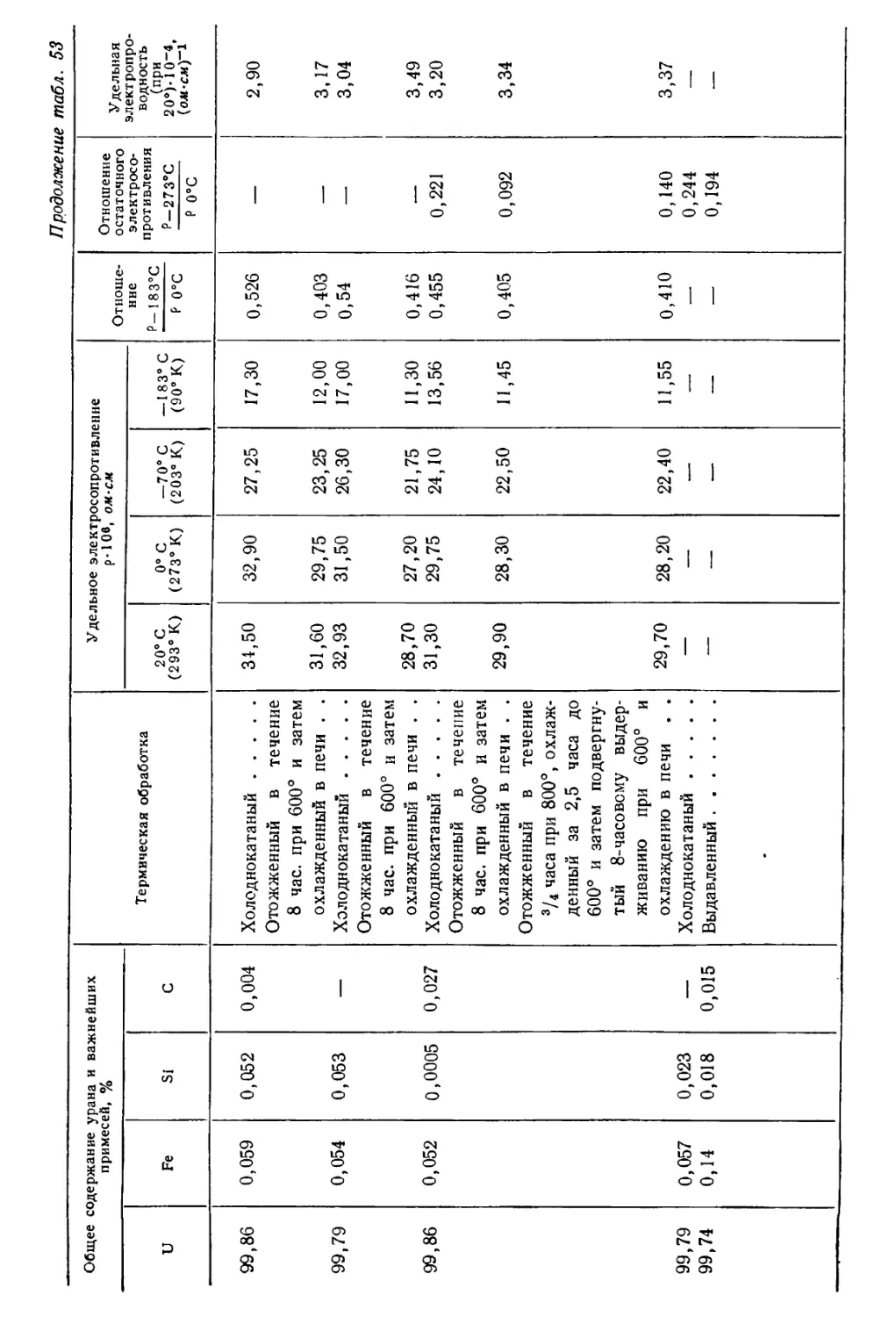
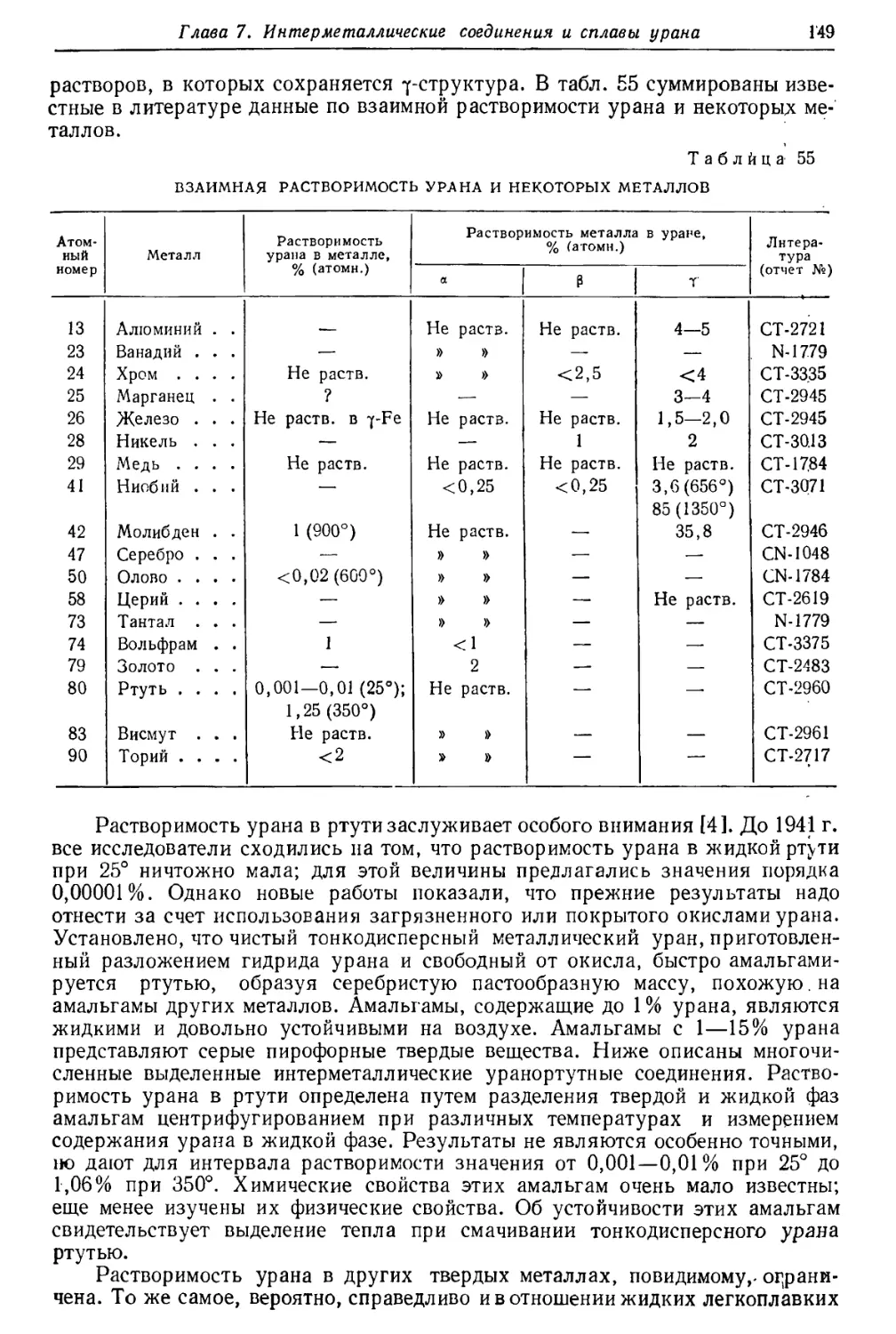
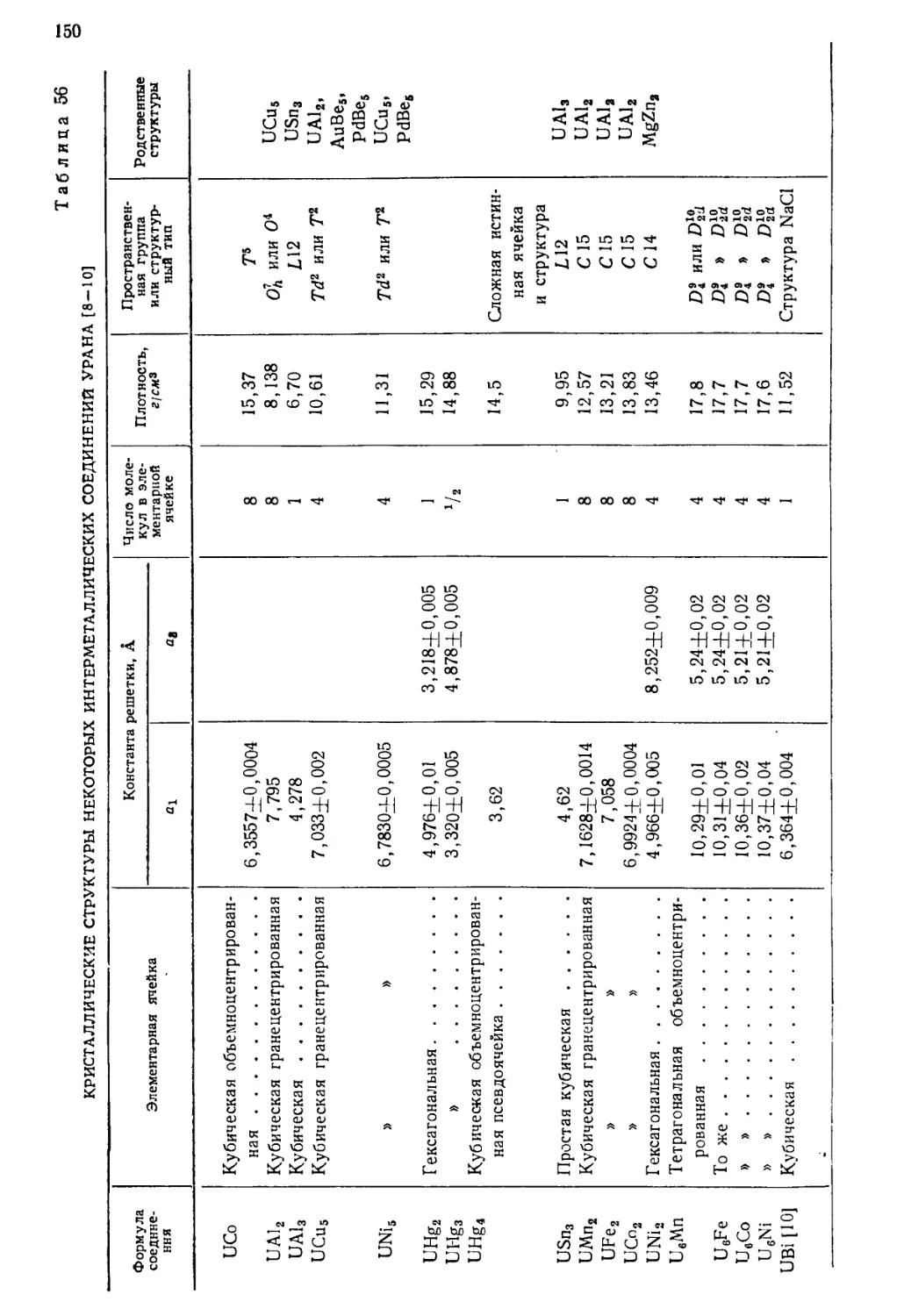
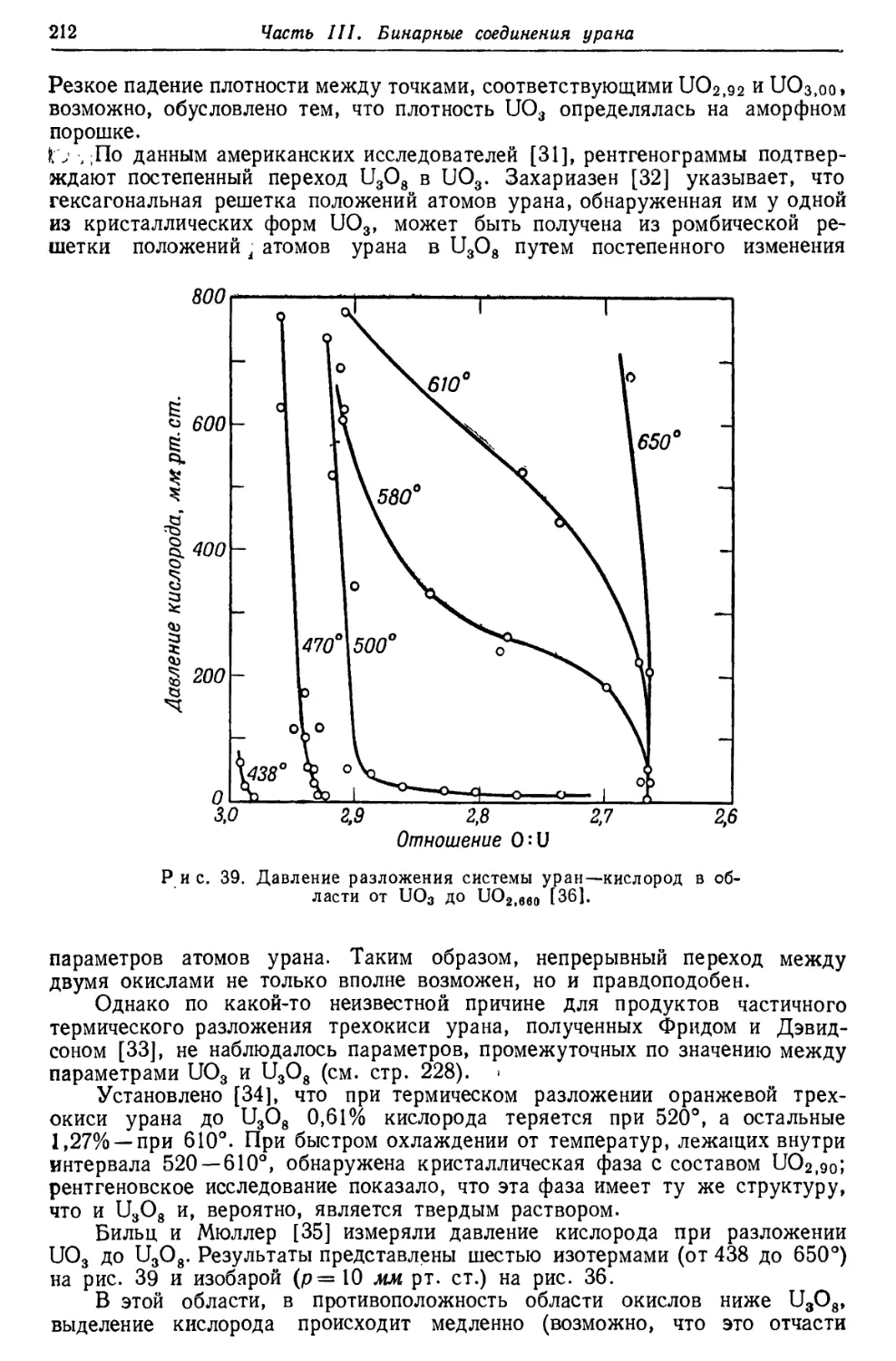
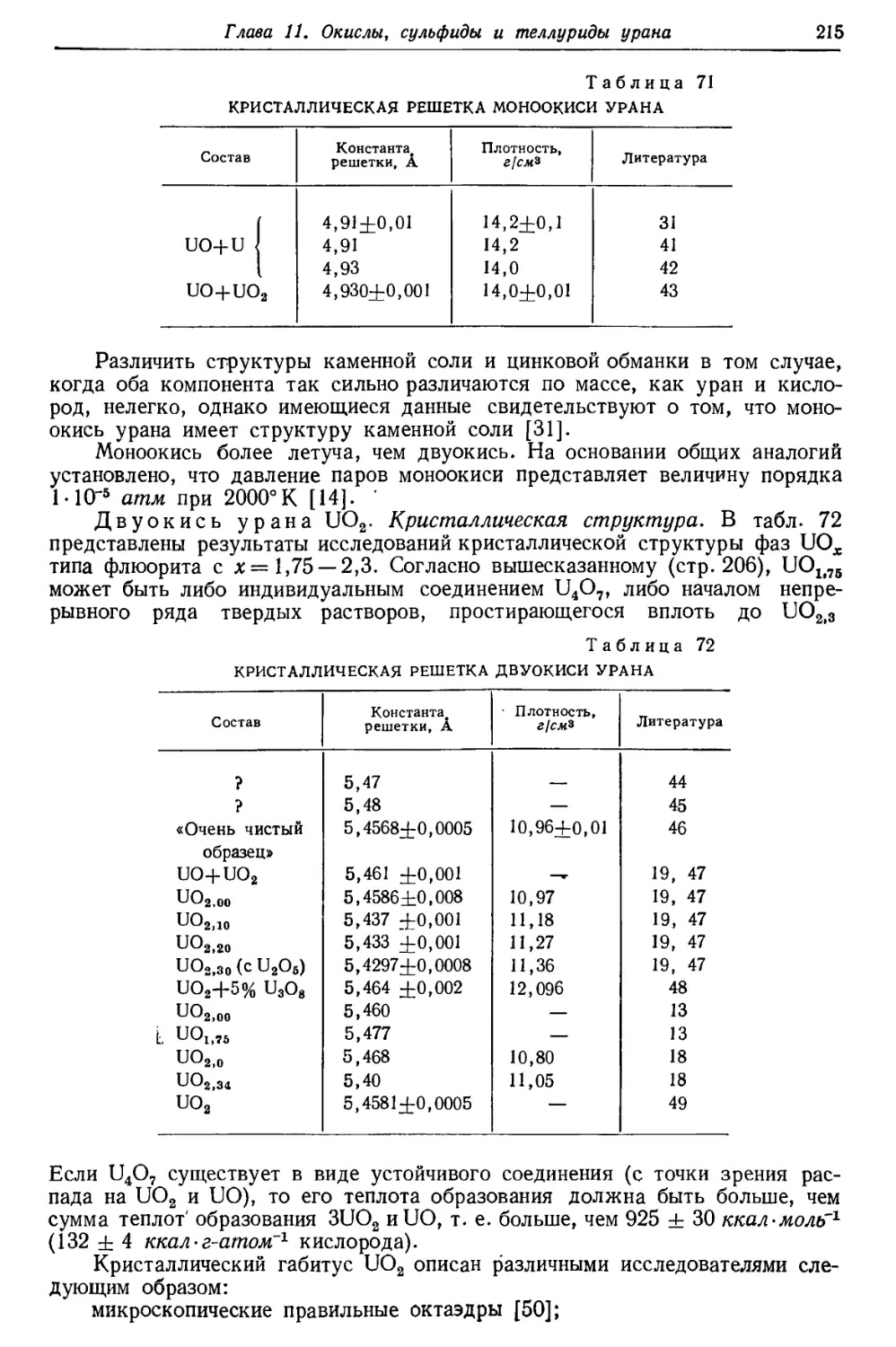
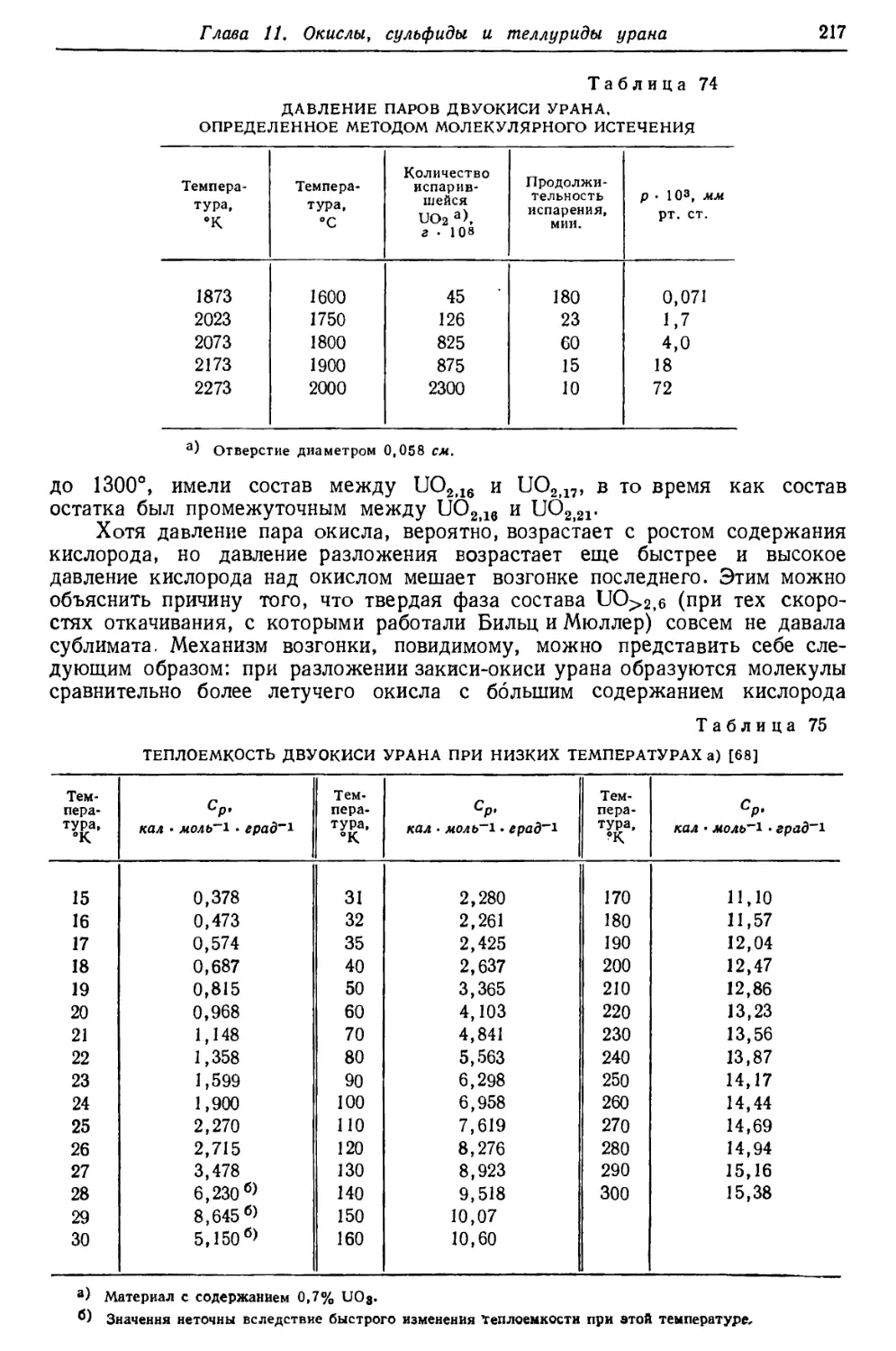
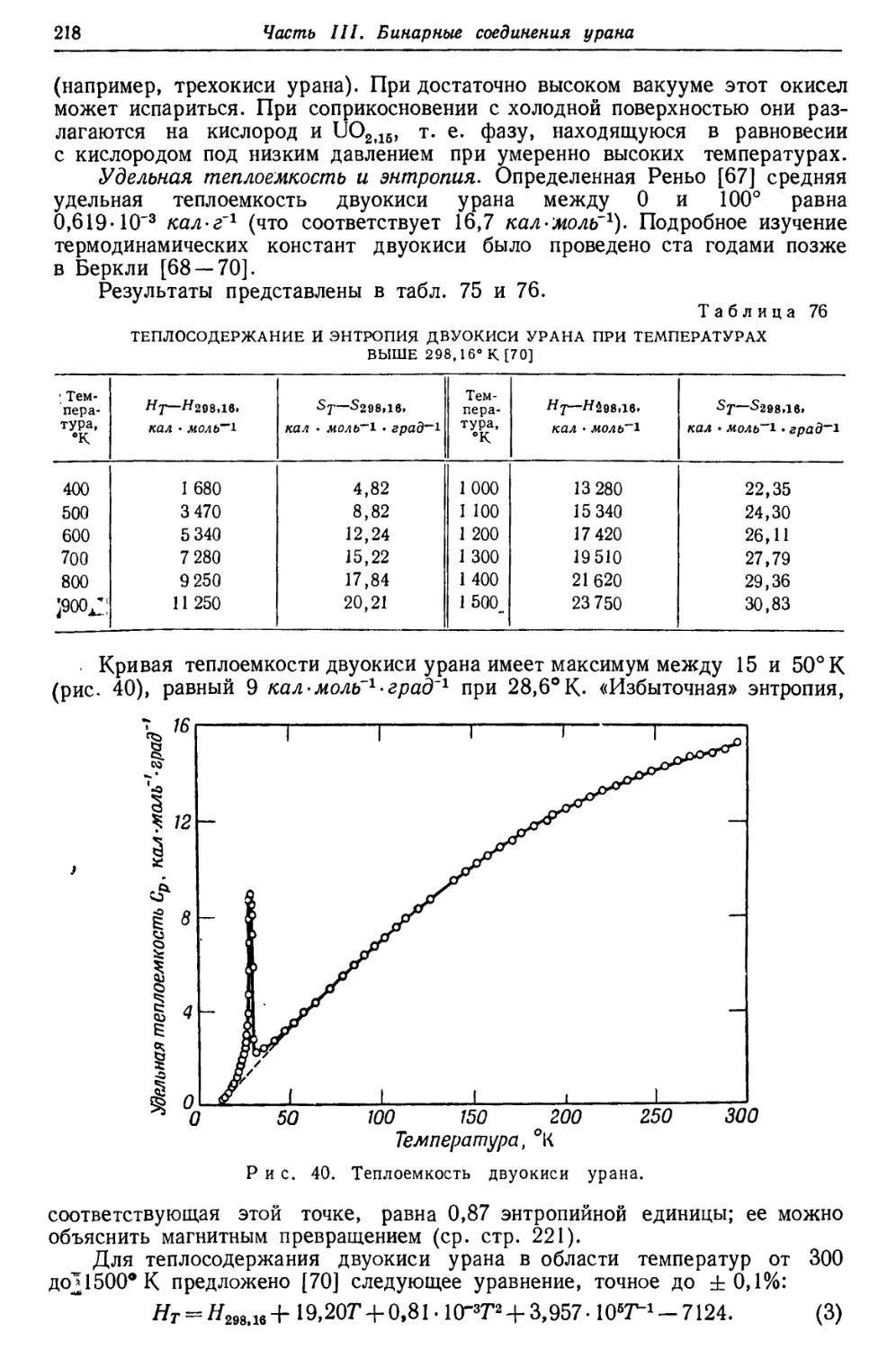
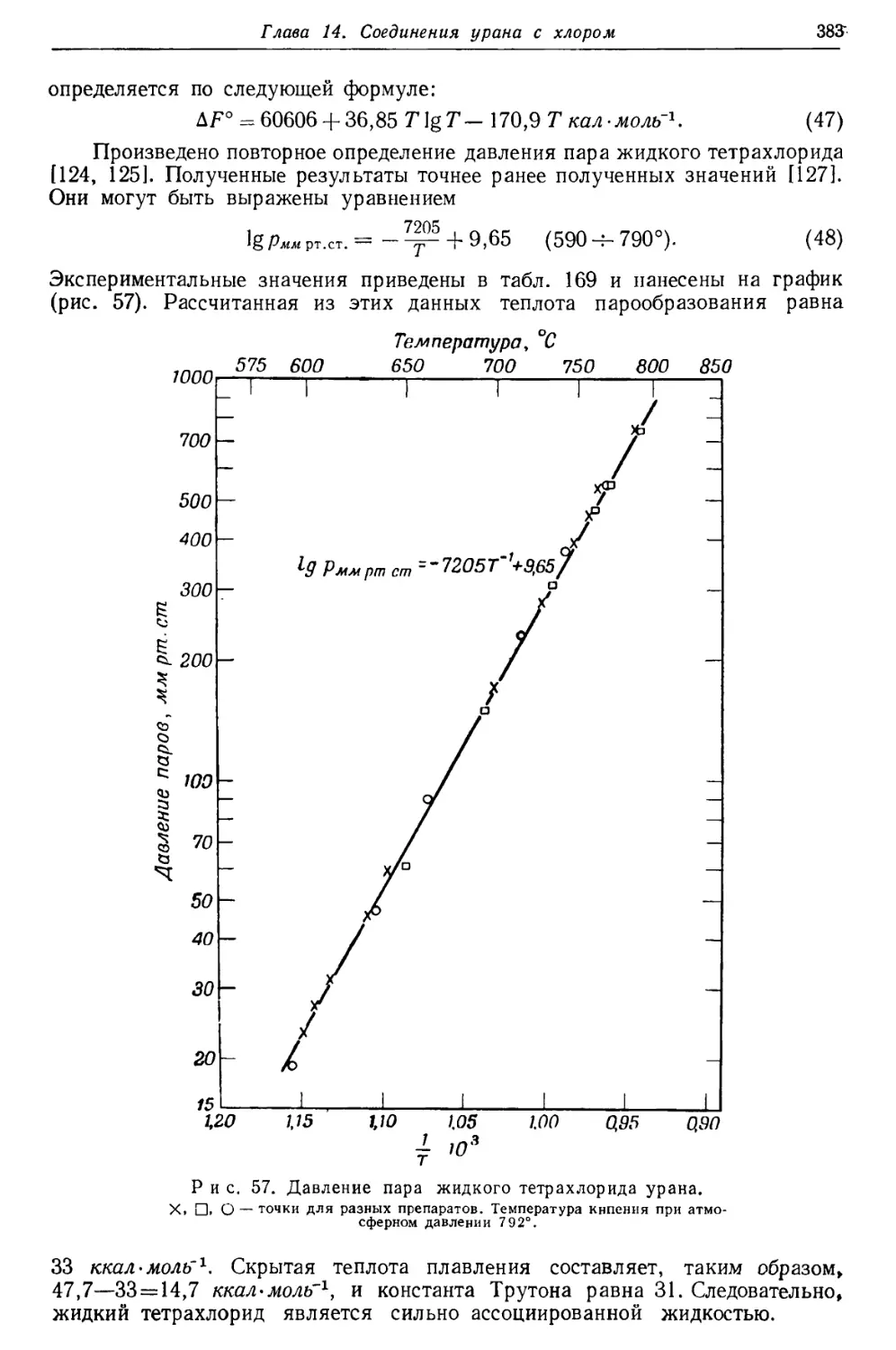
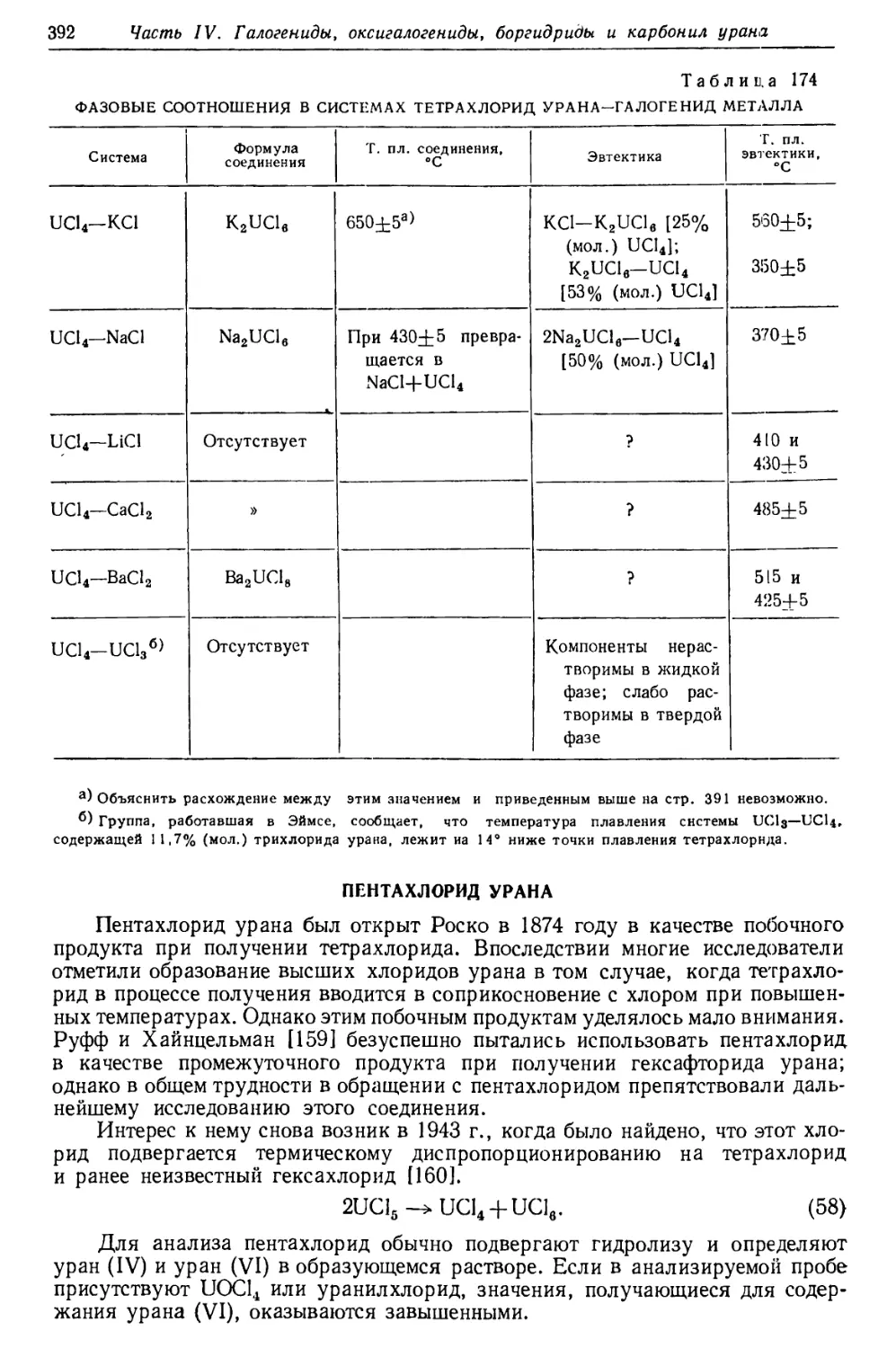
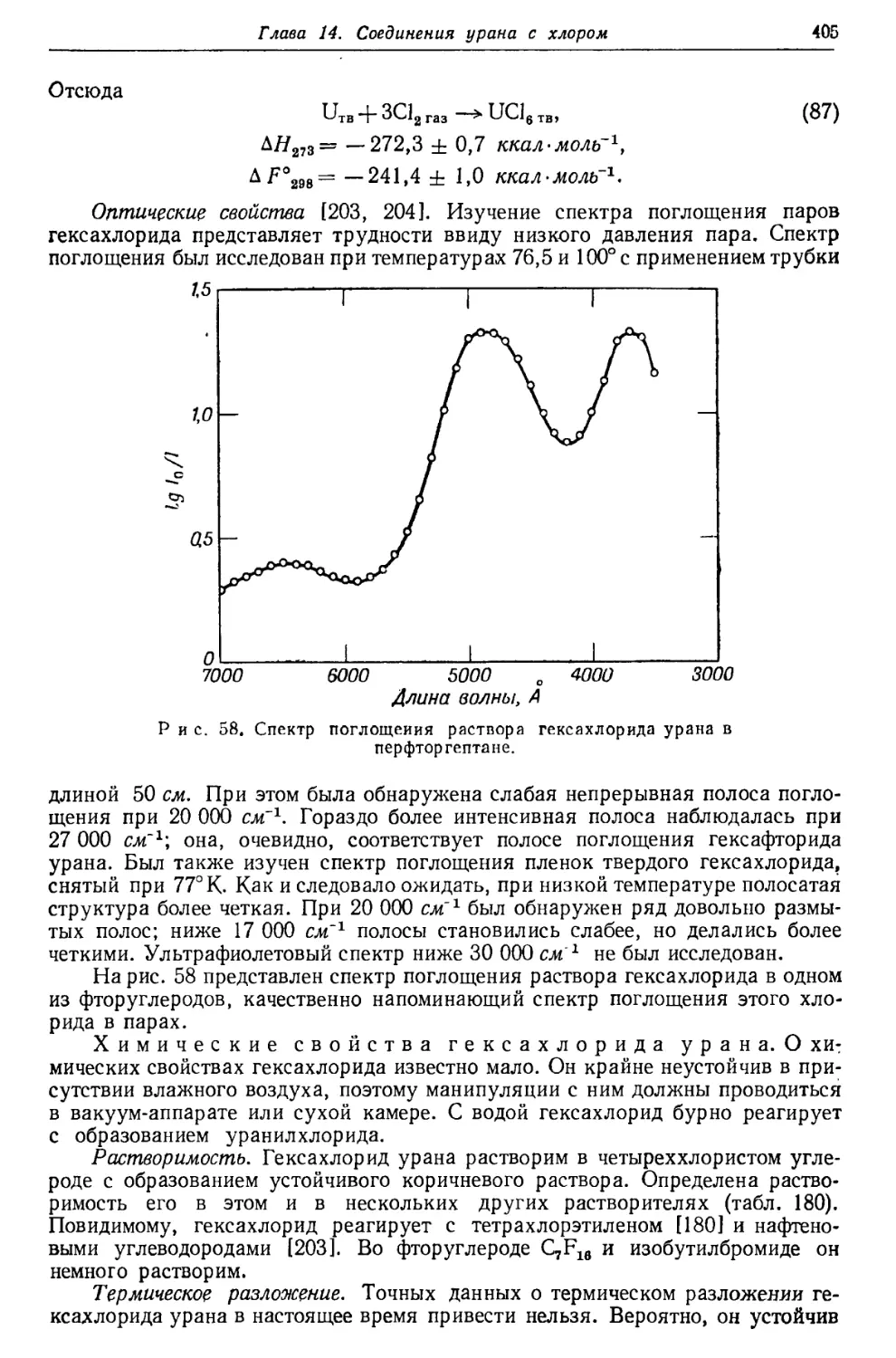
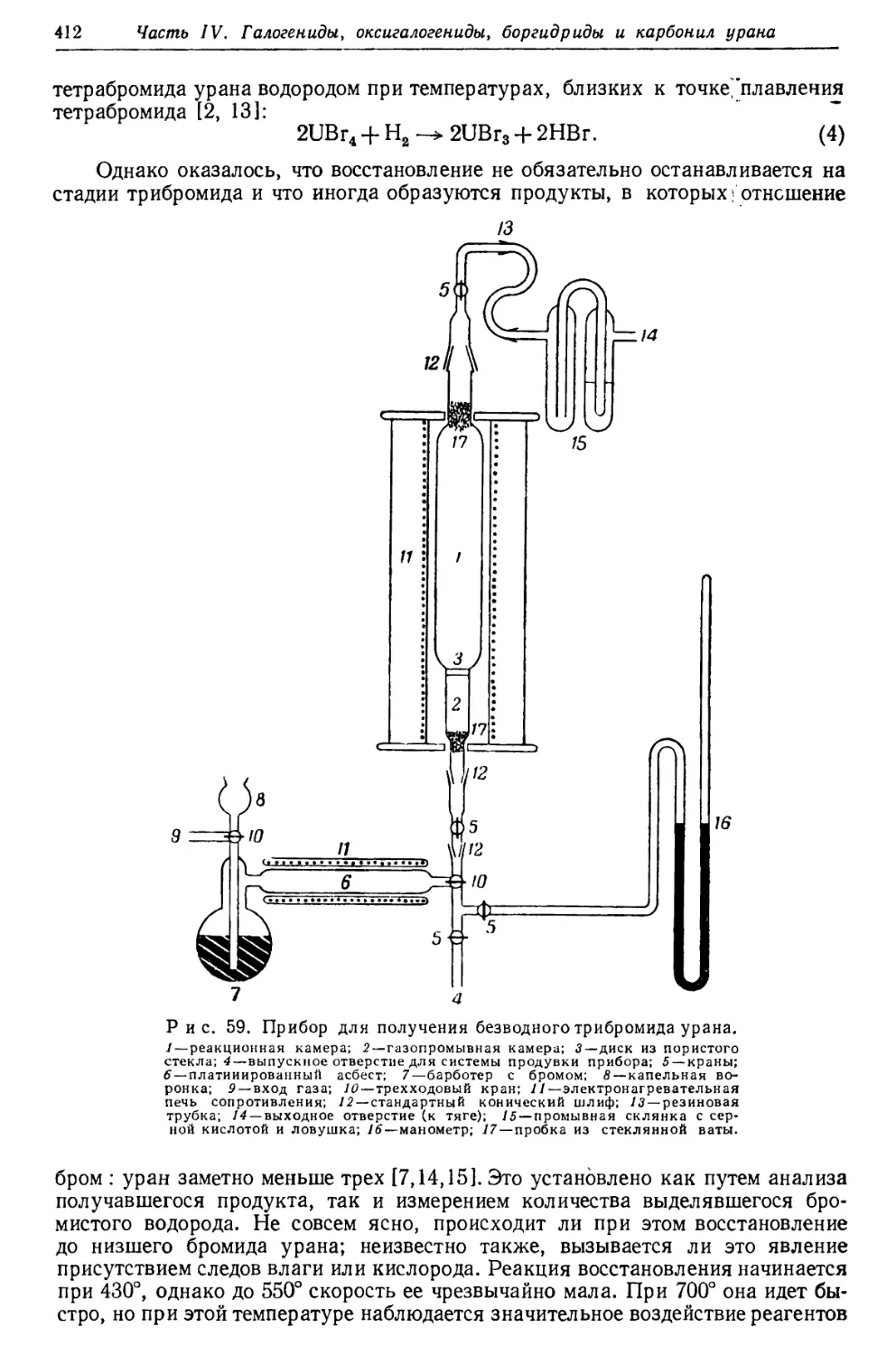
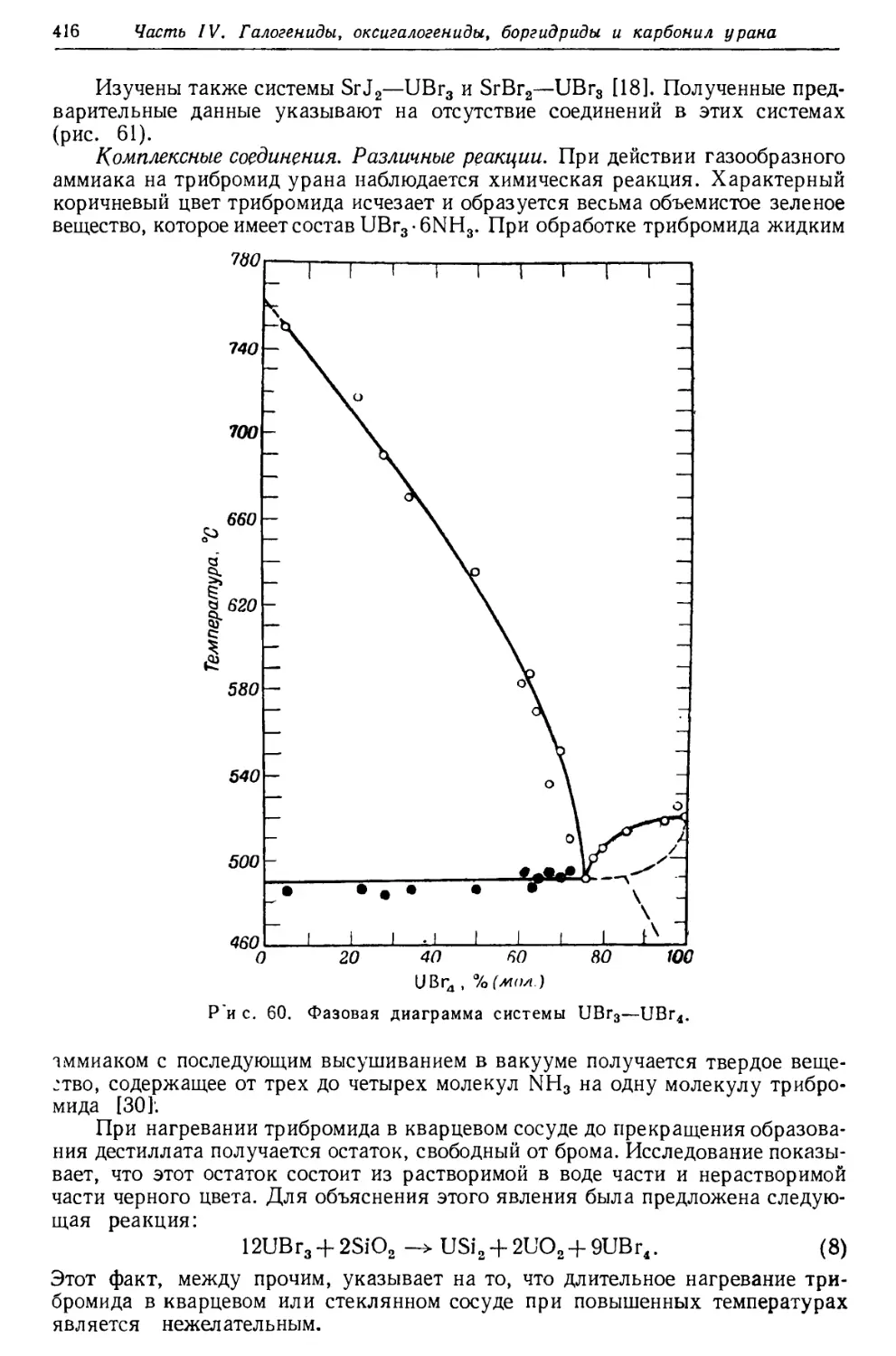
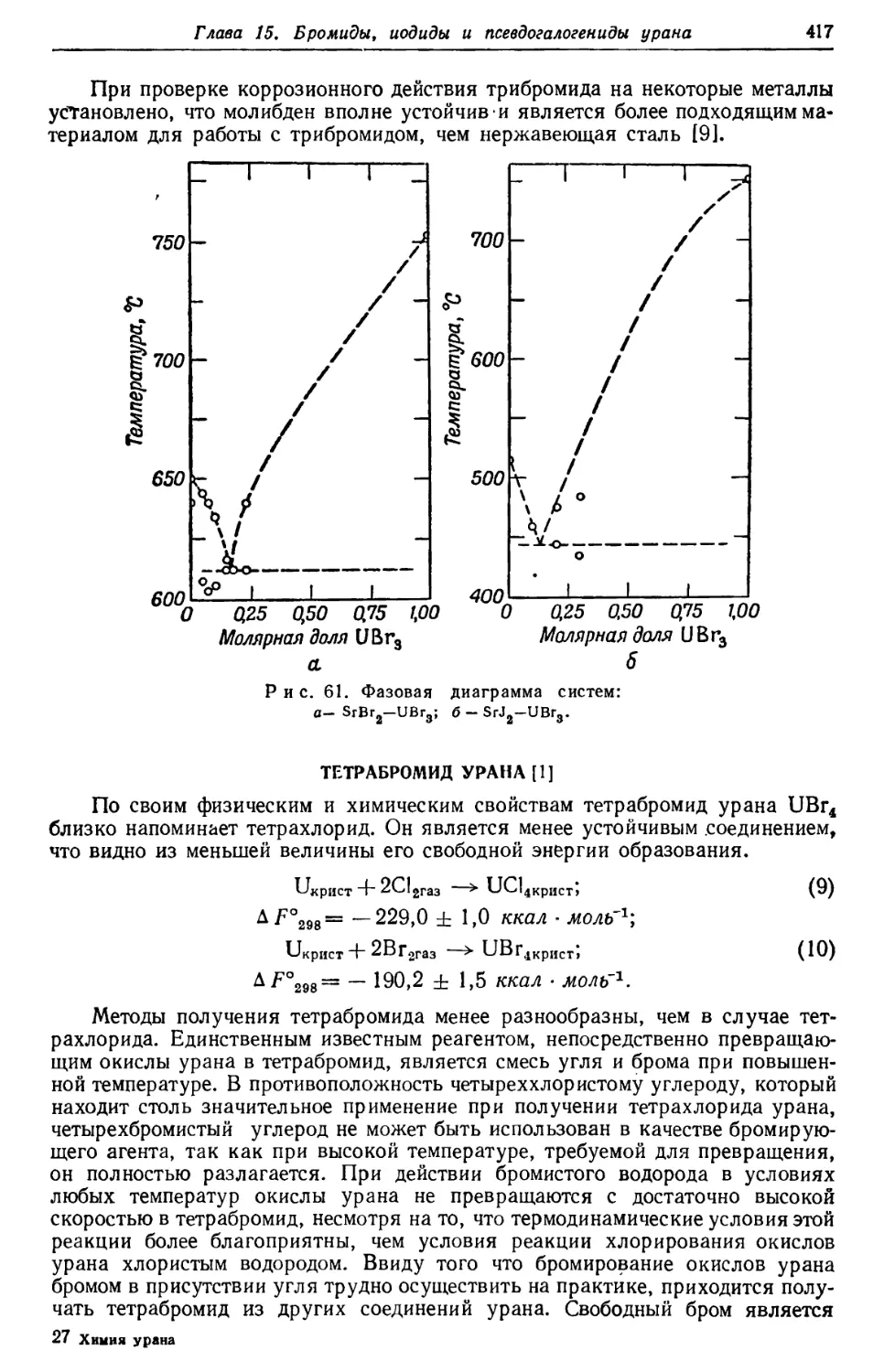
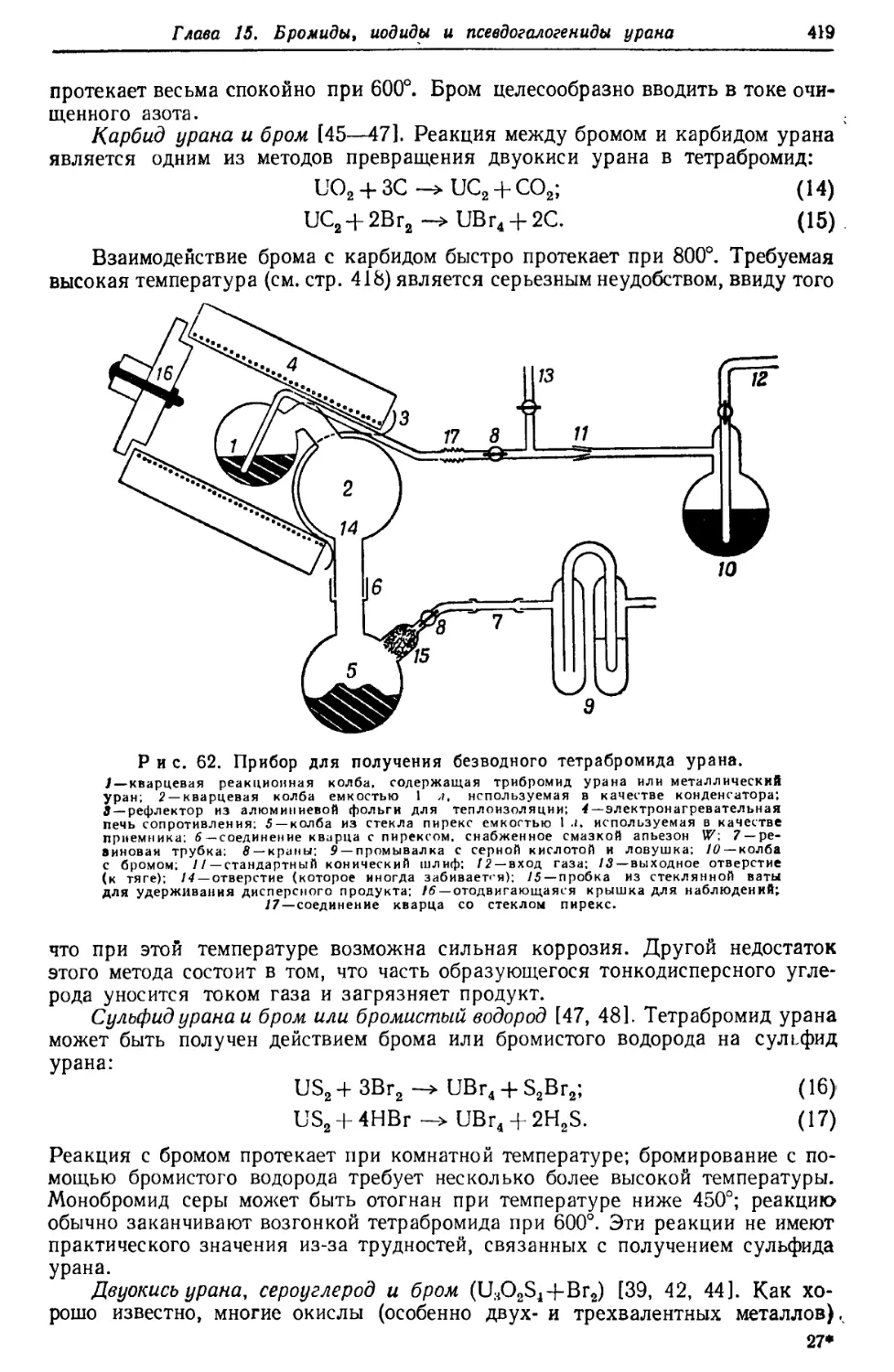
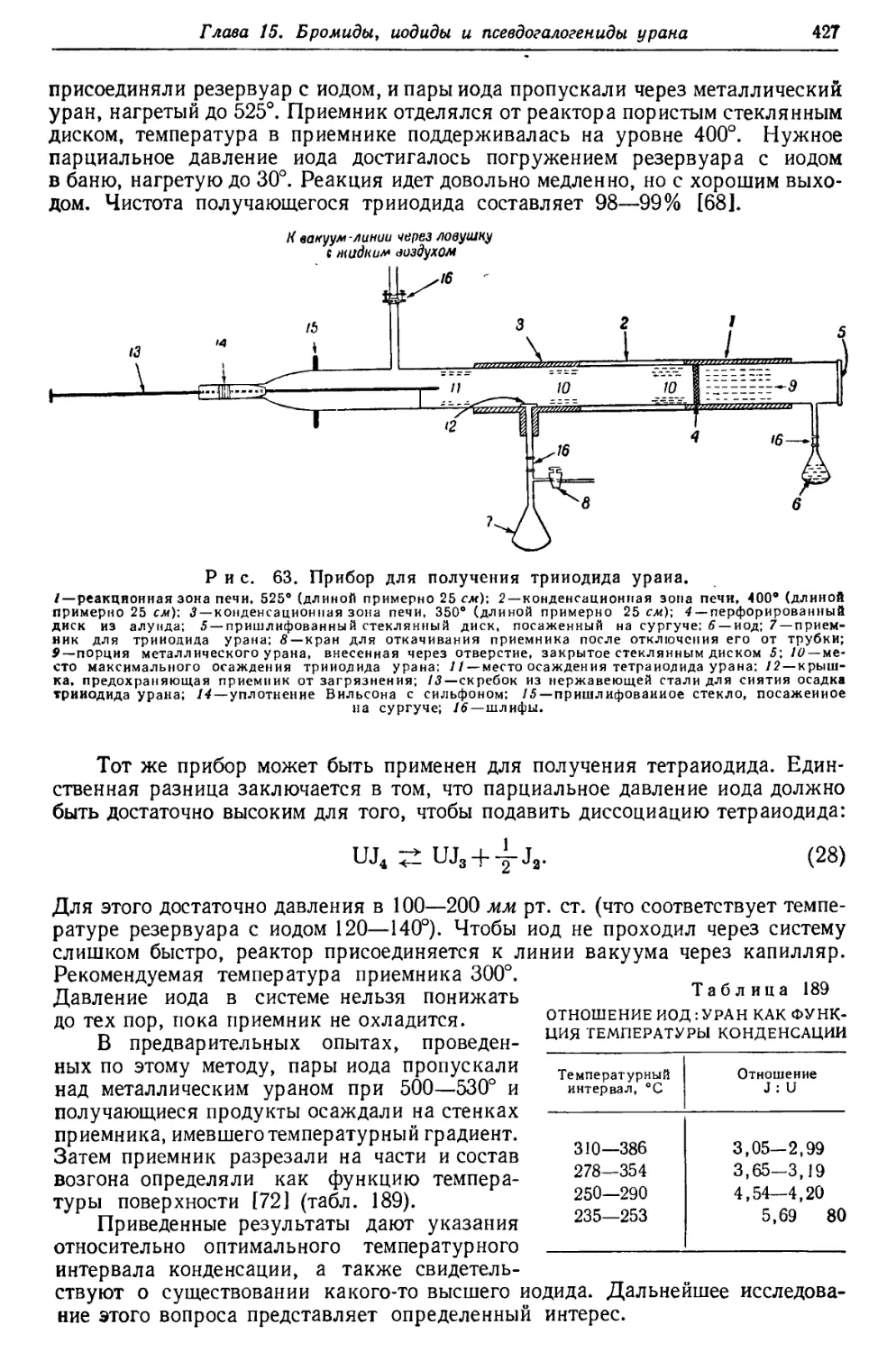
/
































































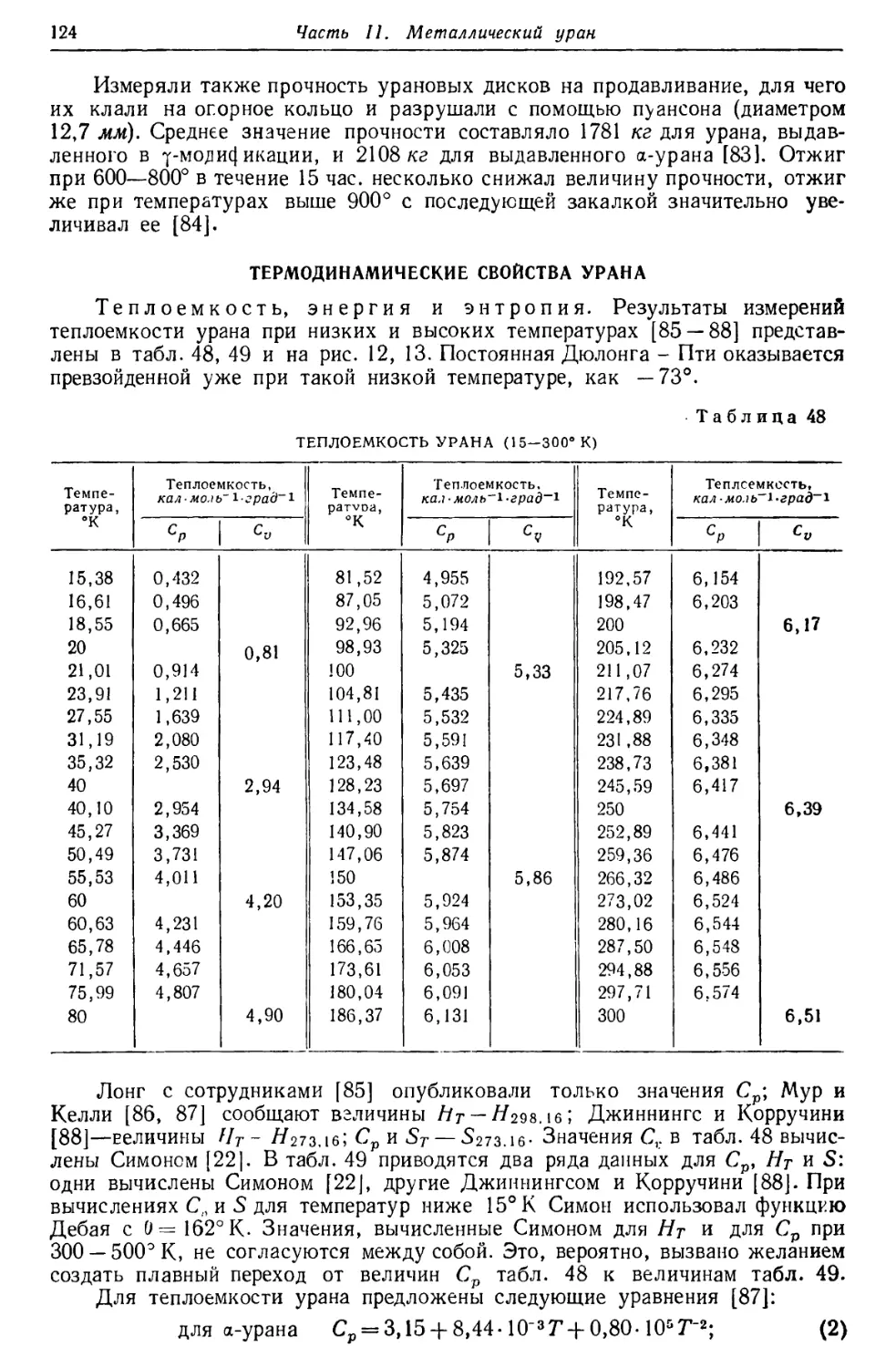
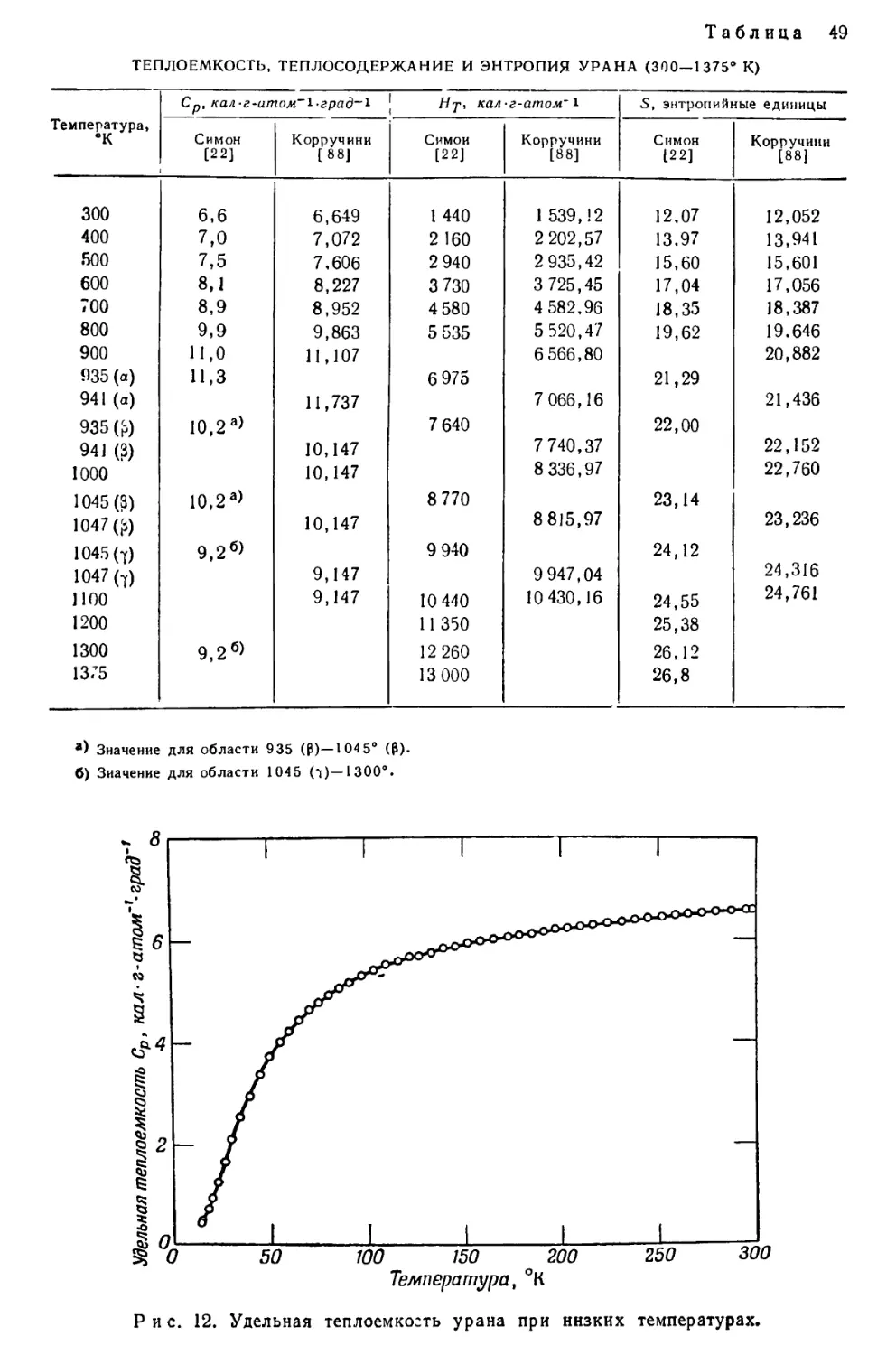
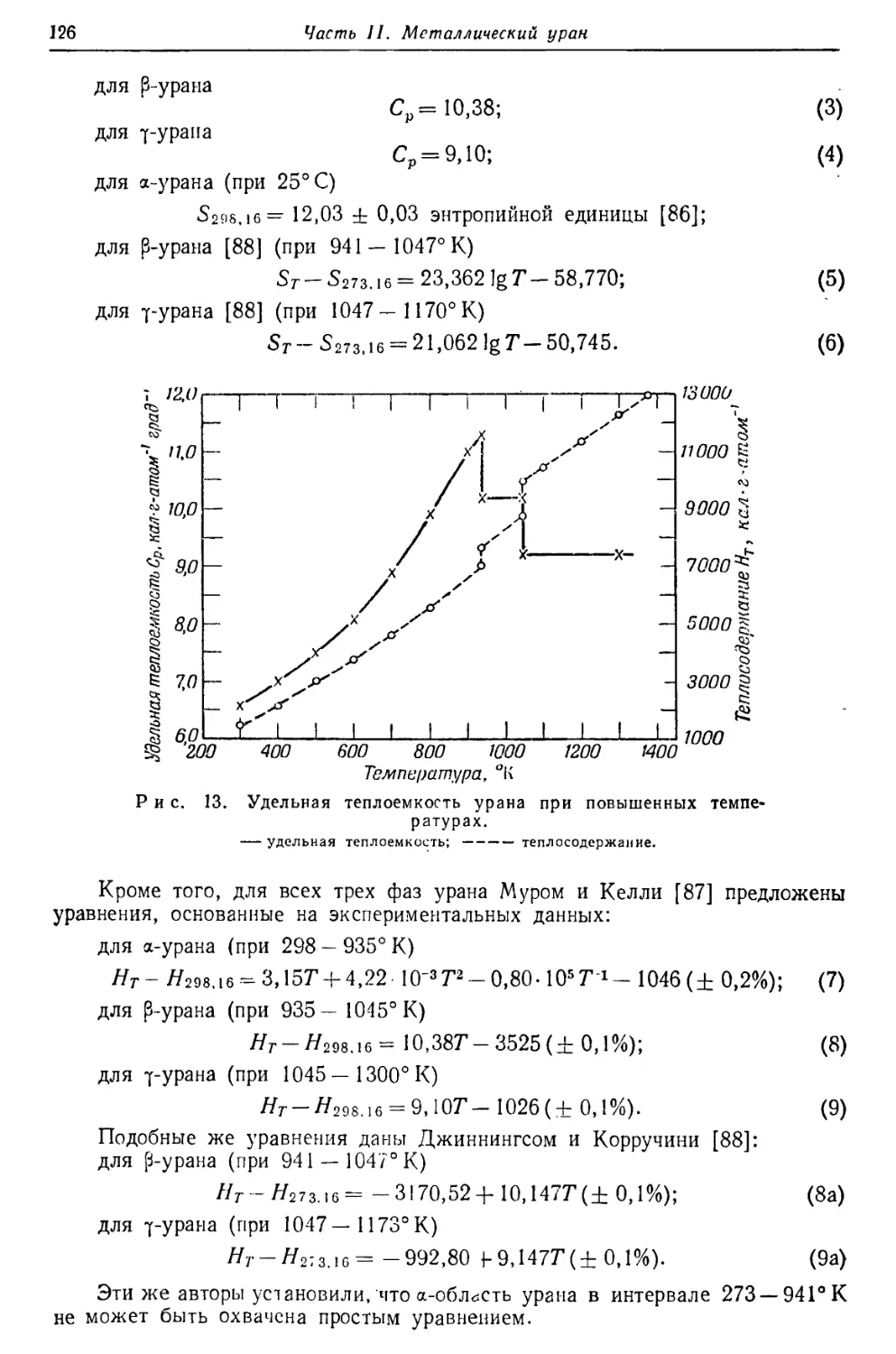
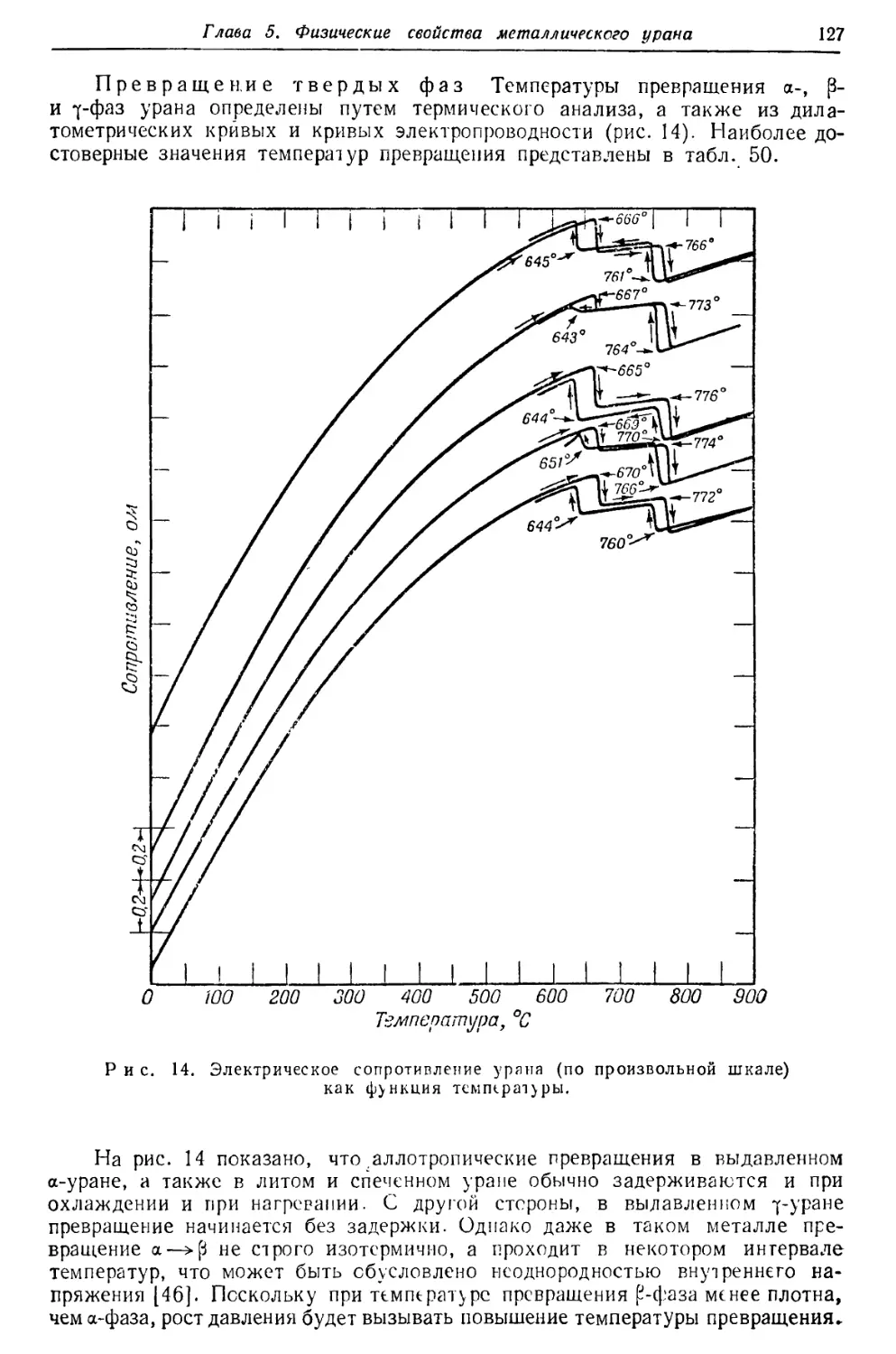



















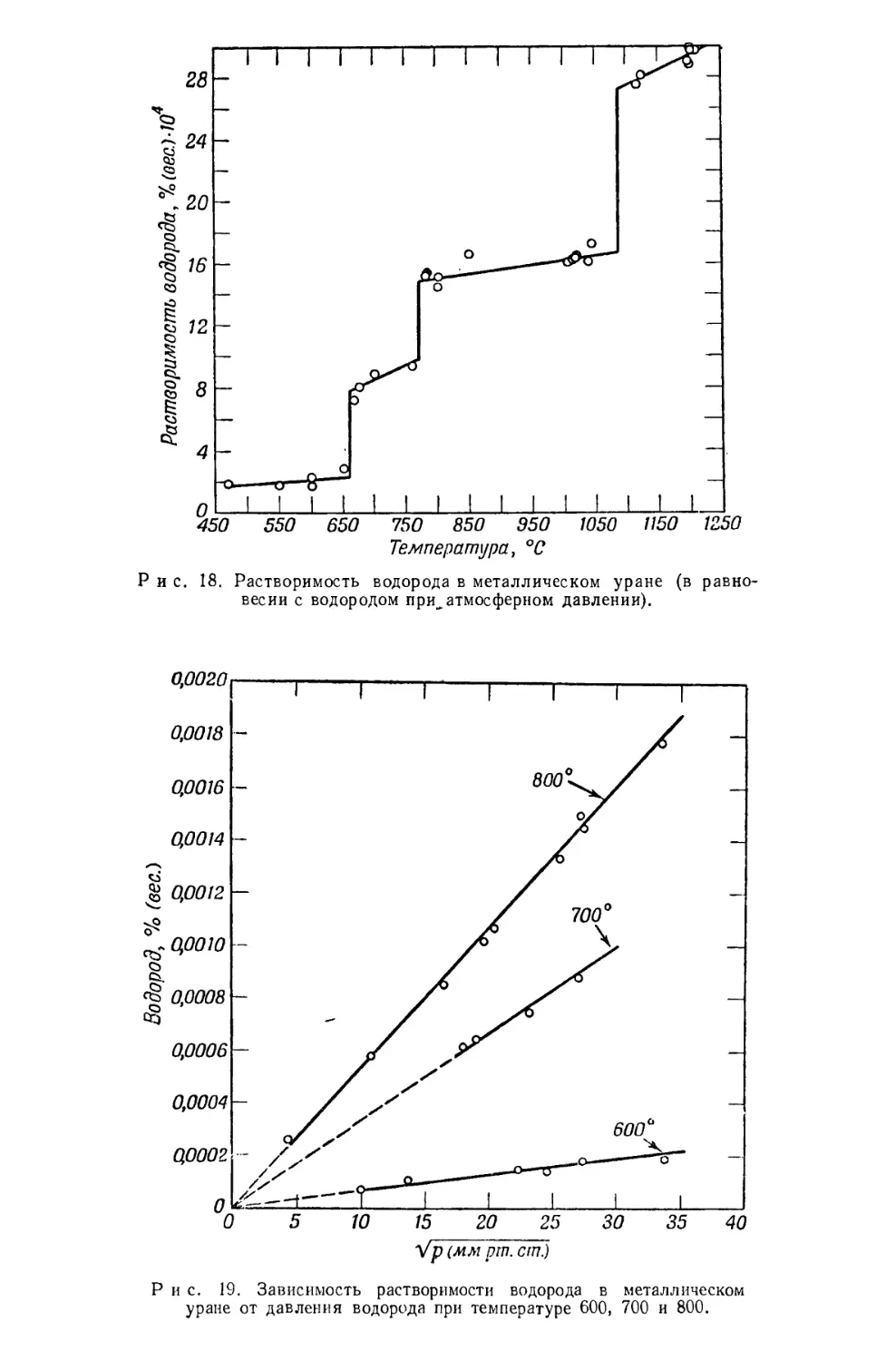

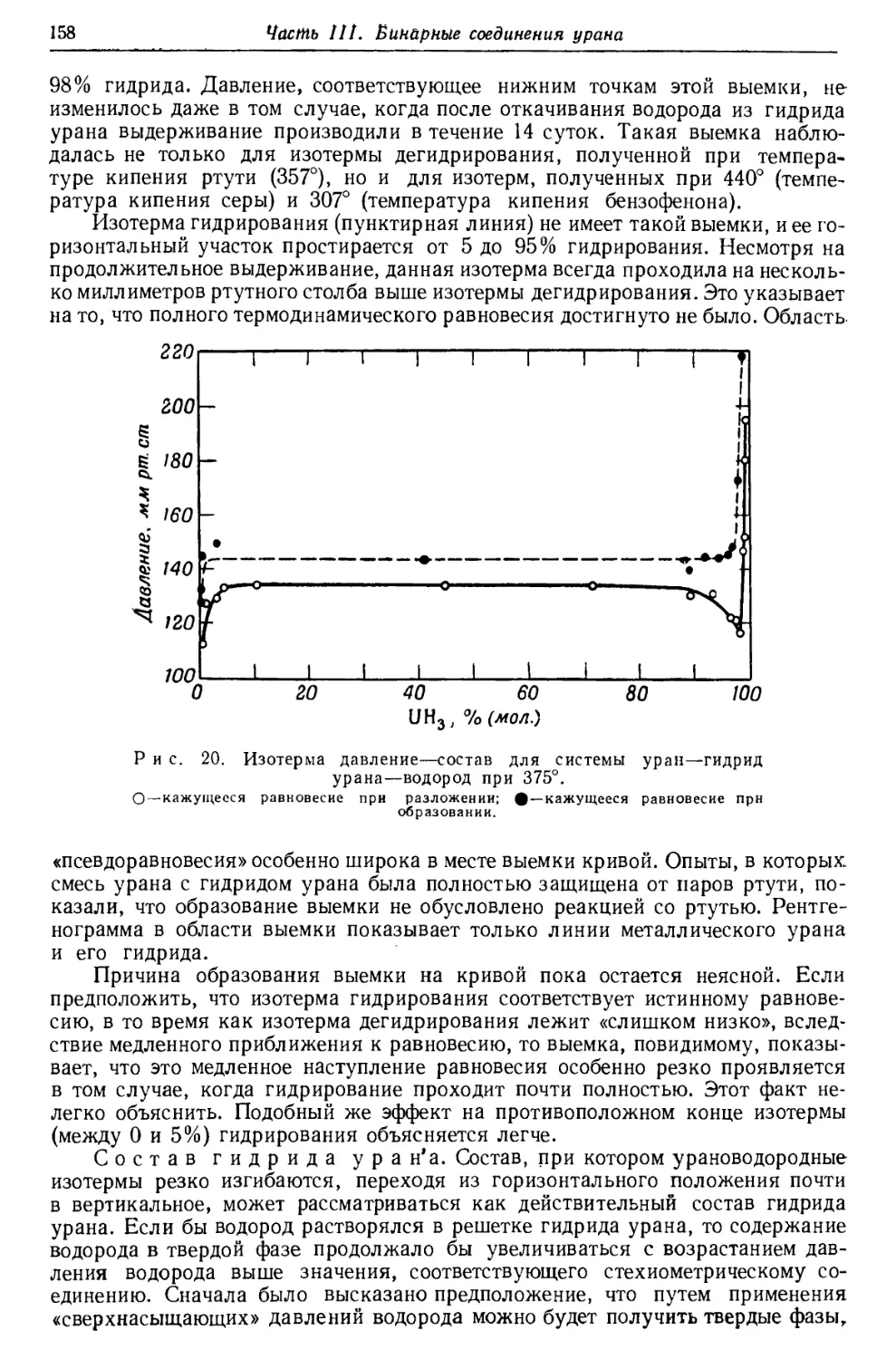

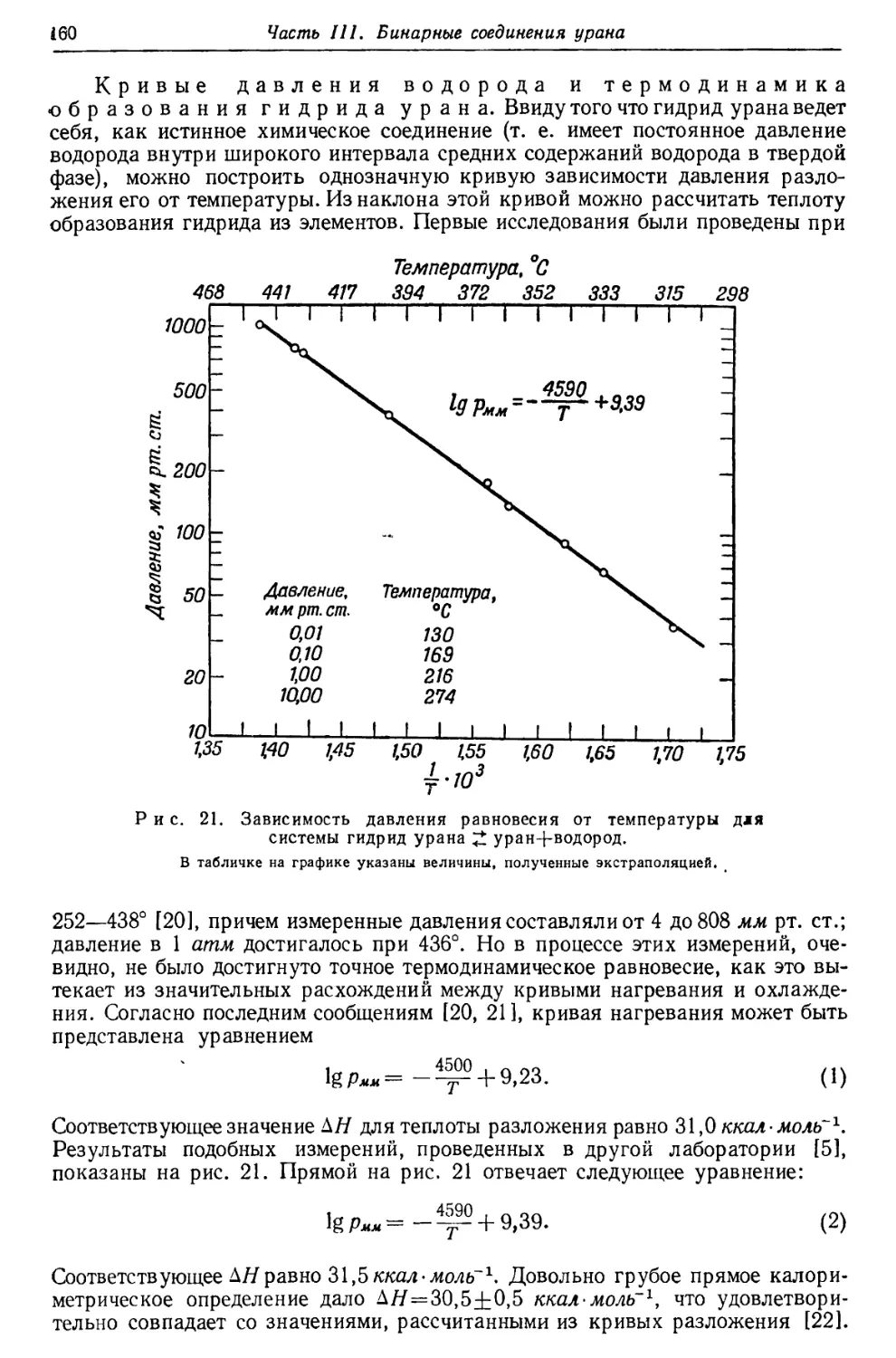

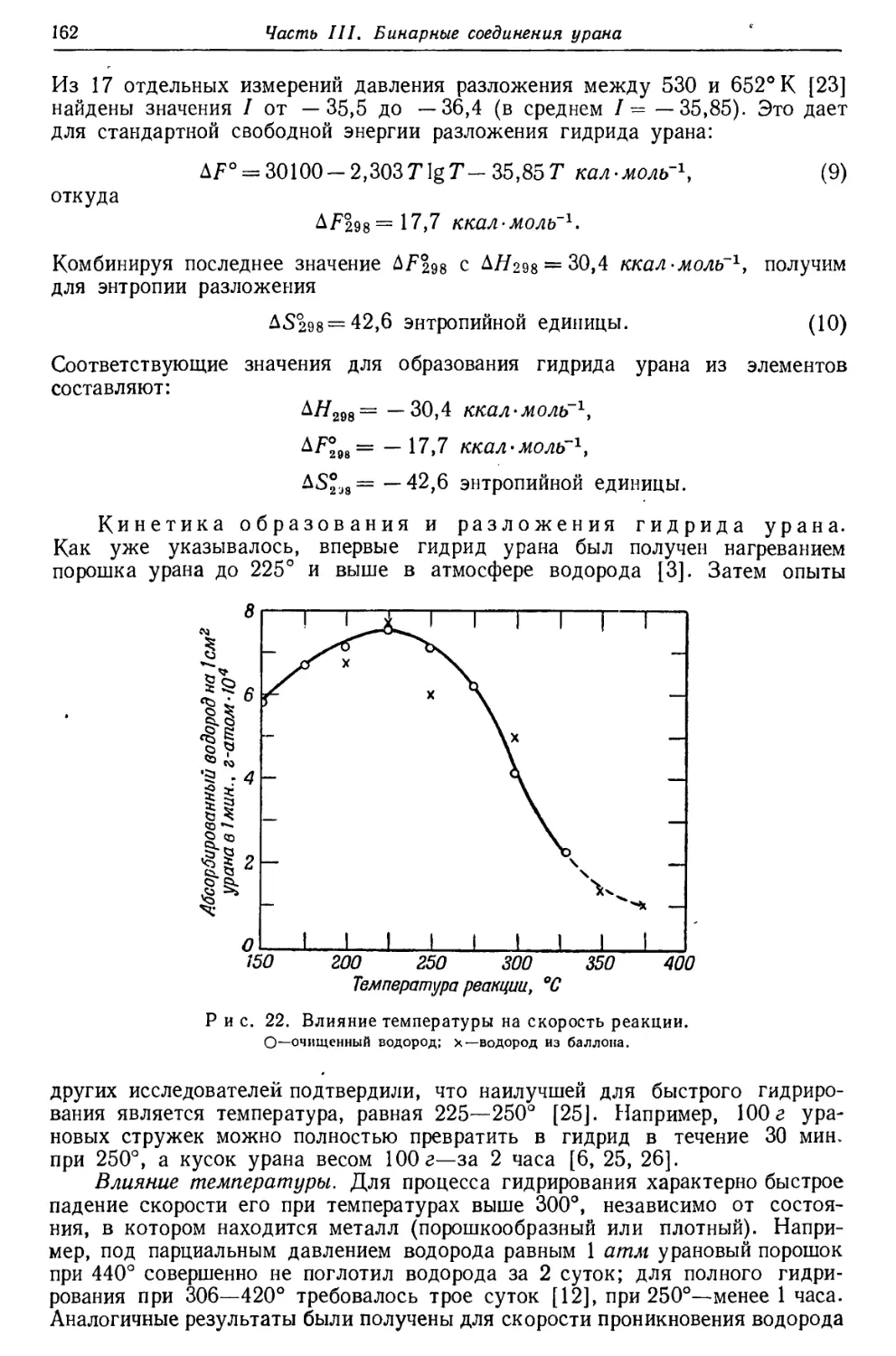































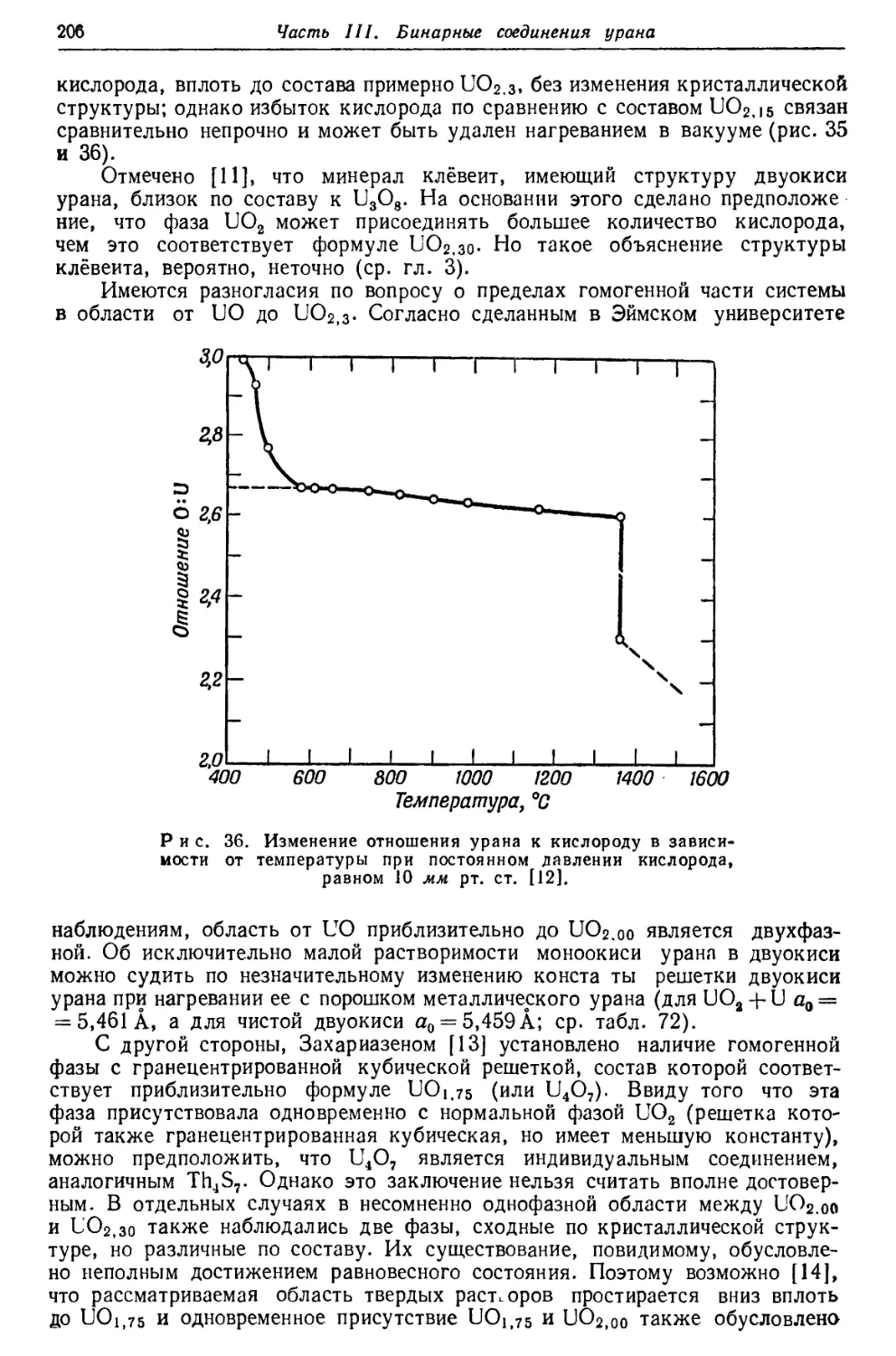

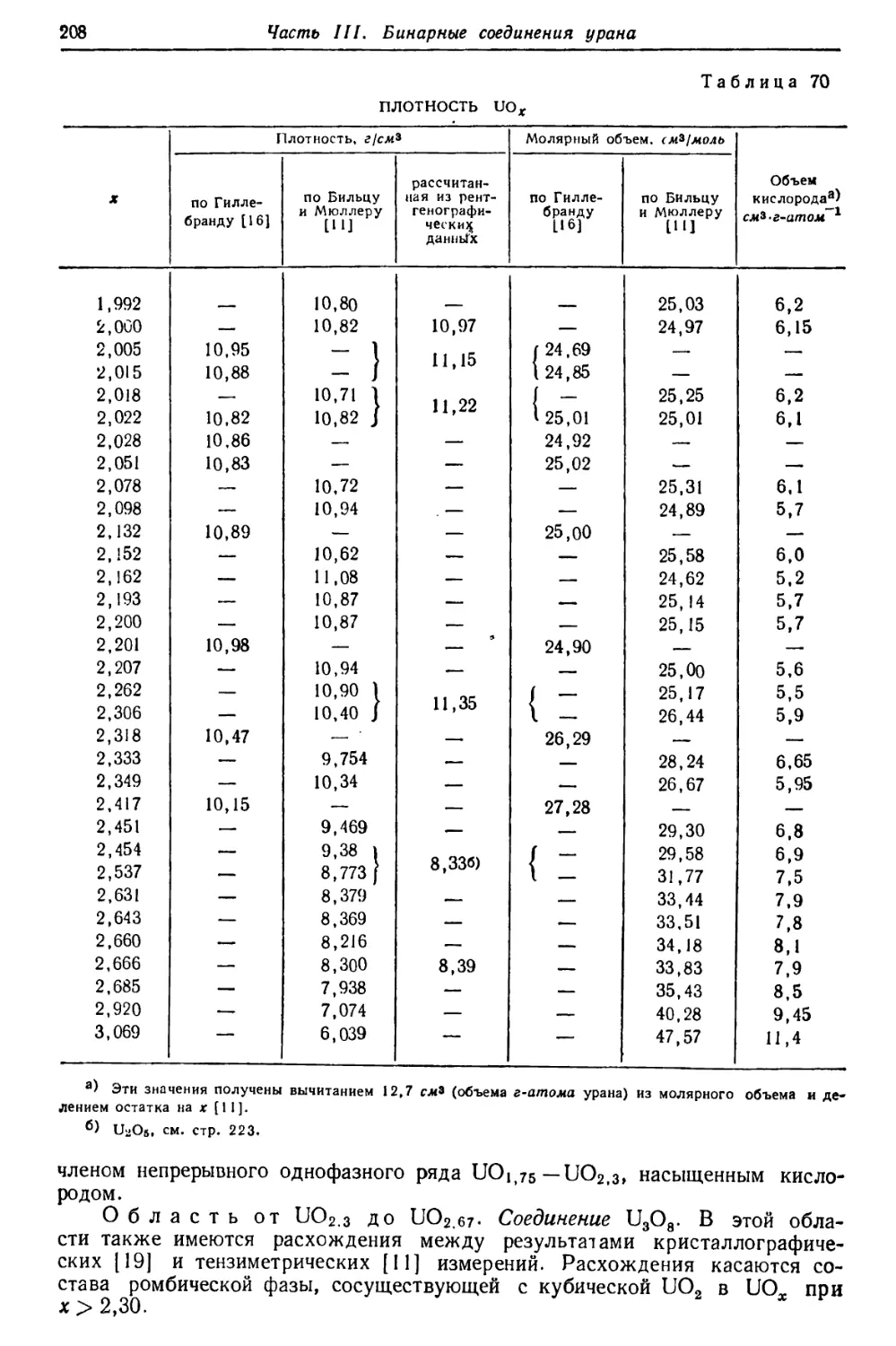

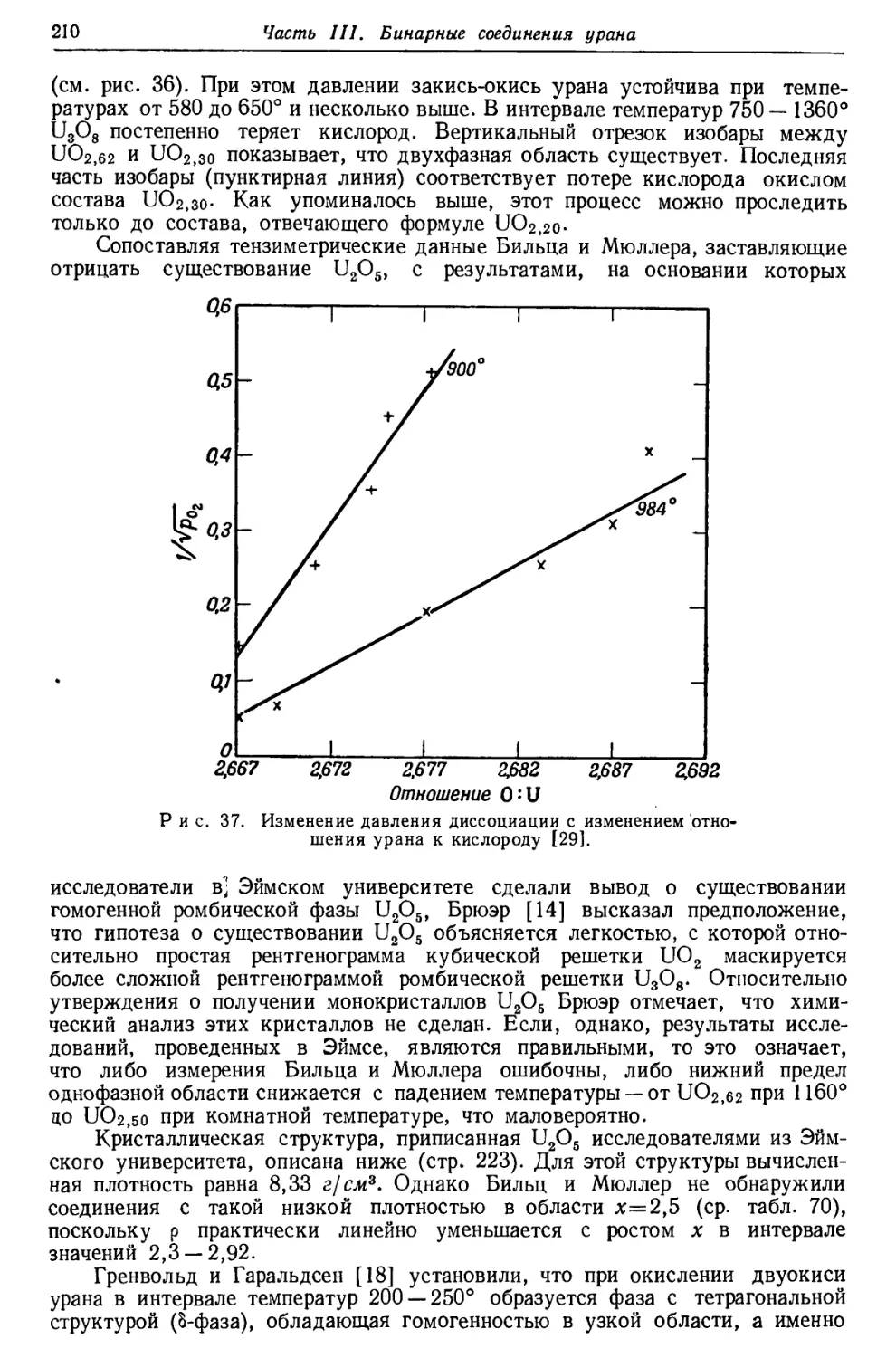
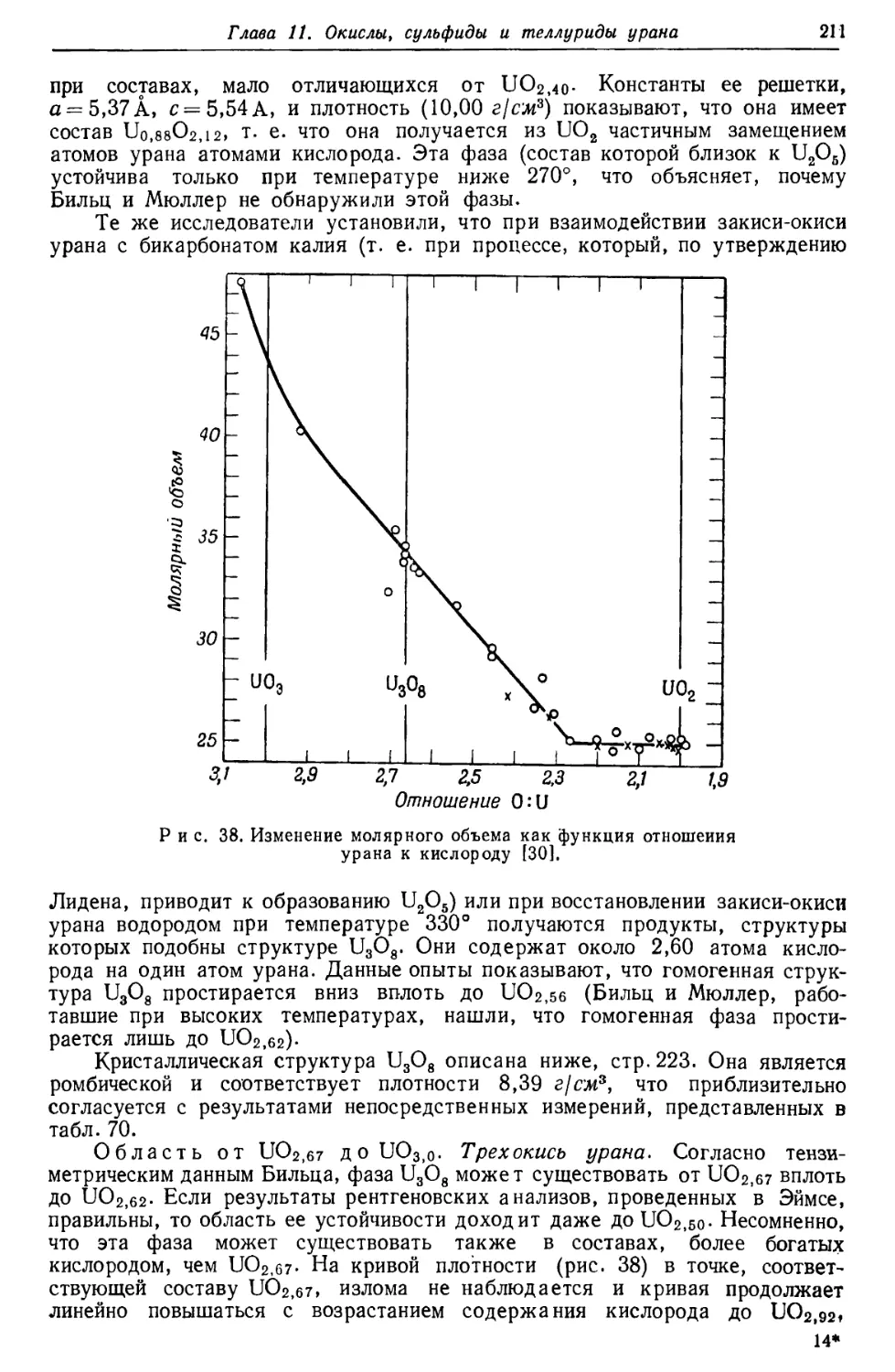




















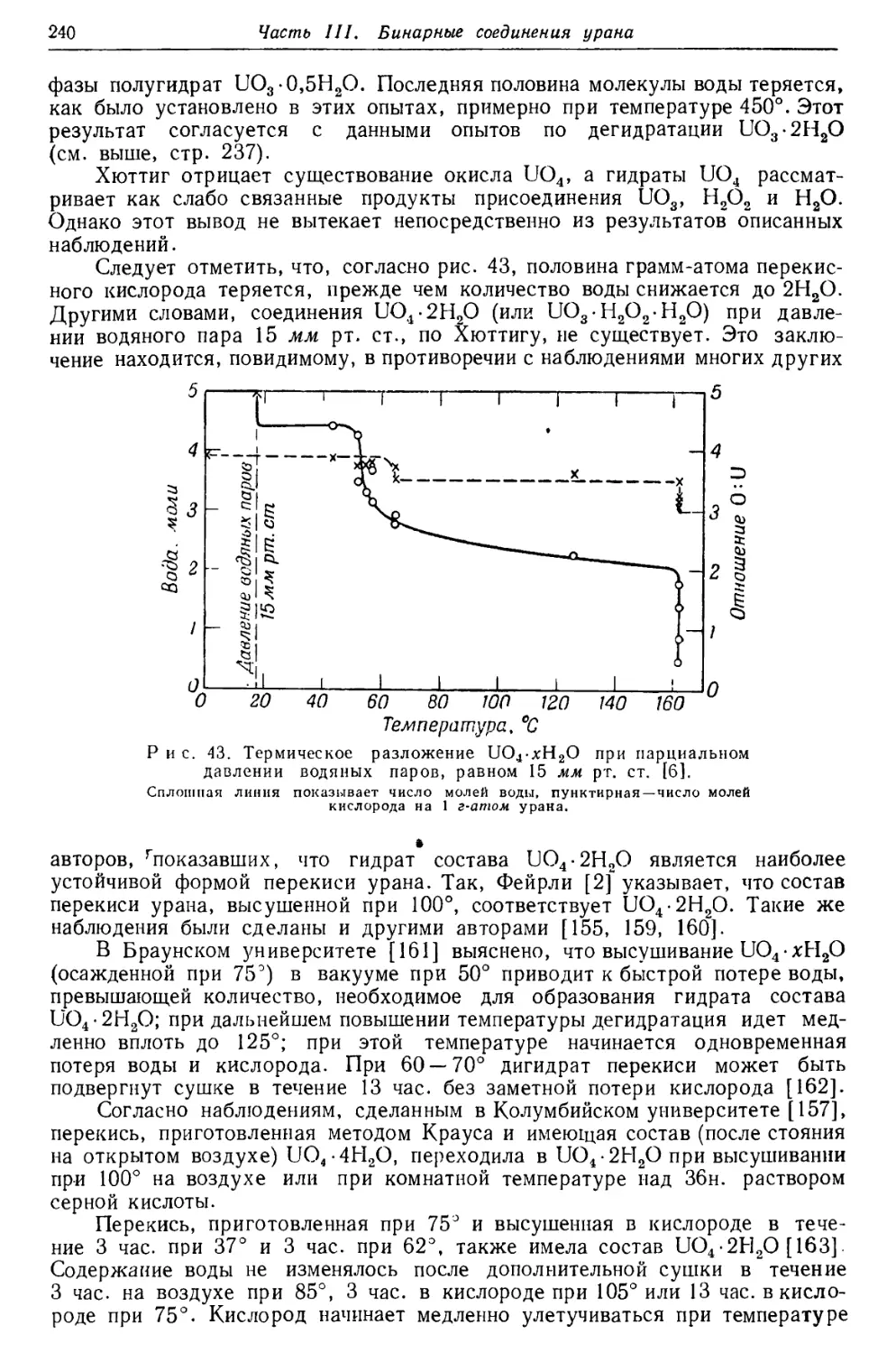




























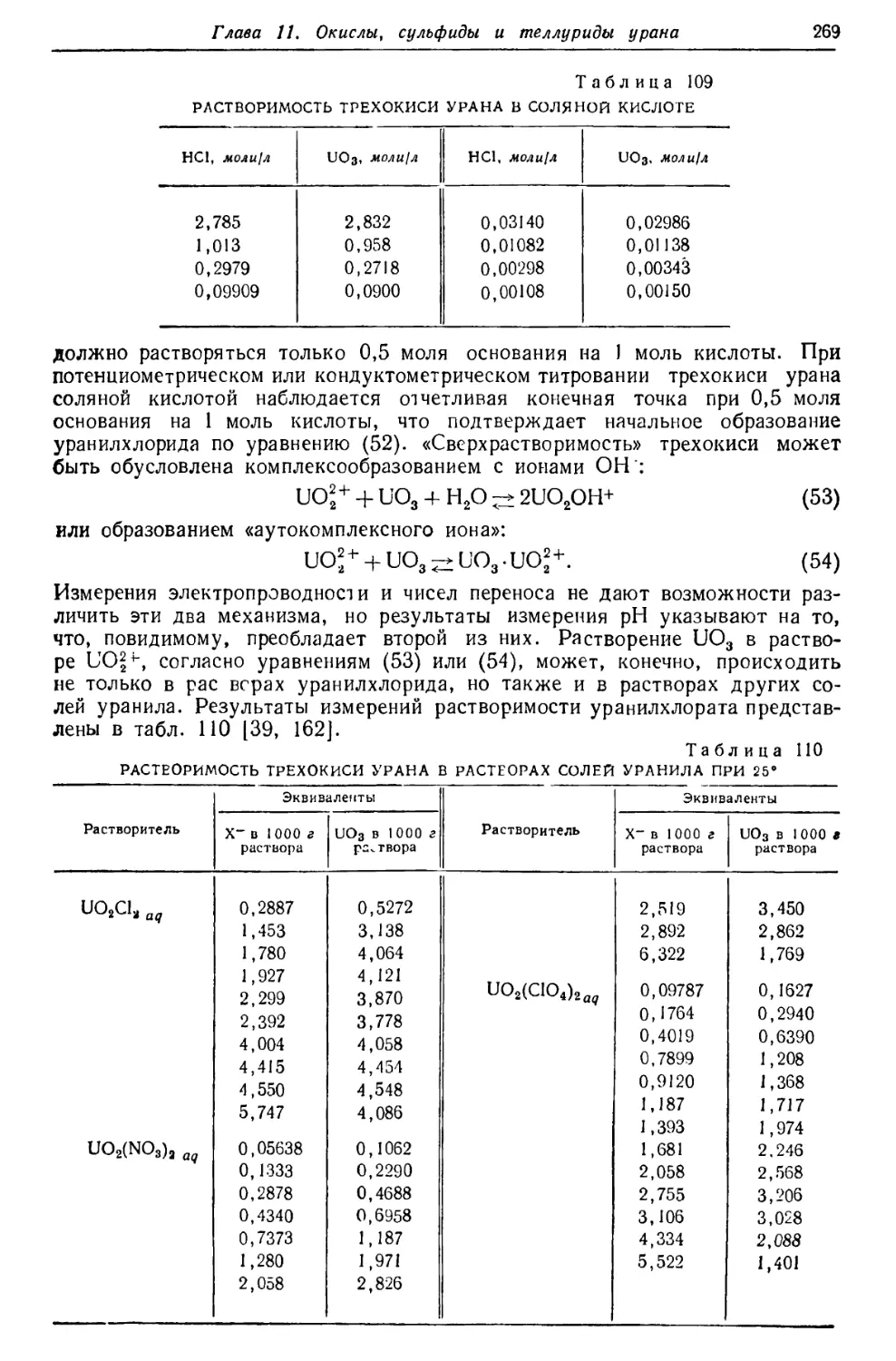
















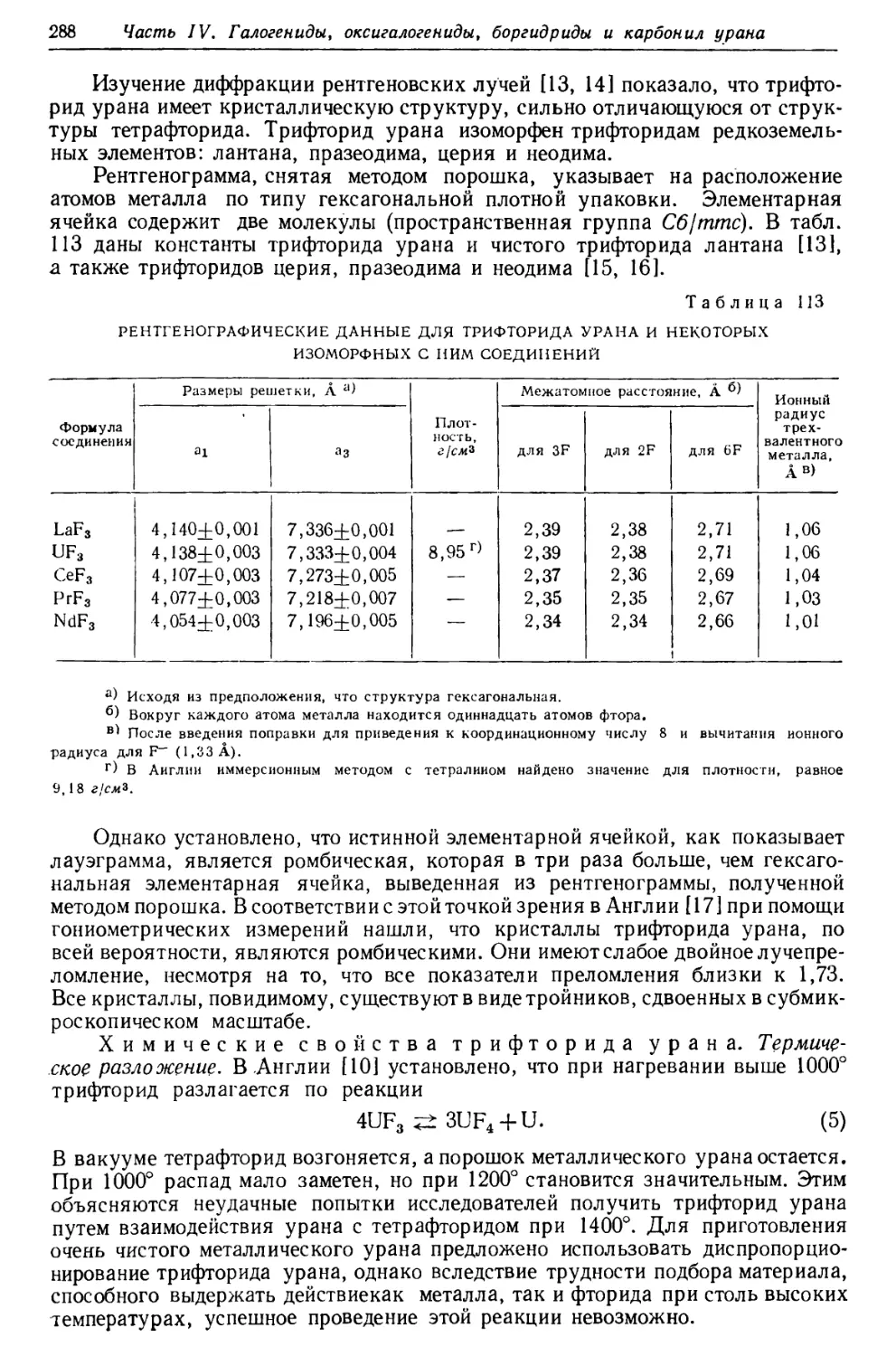







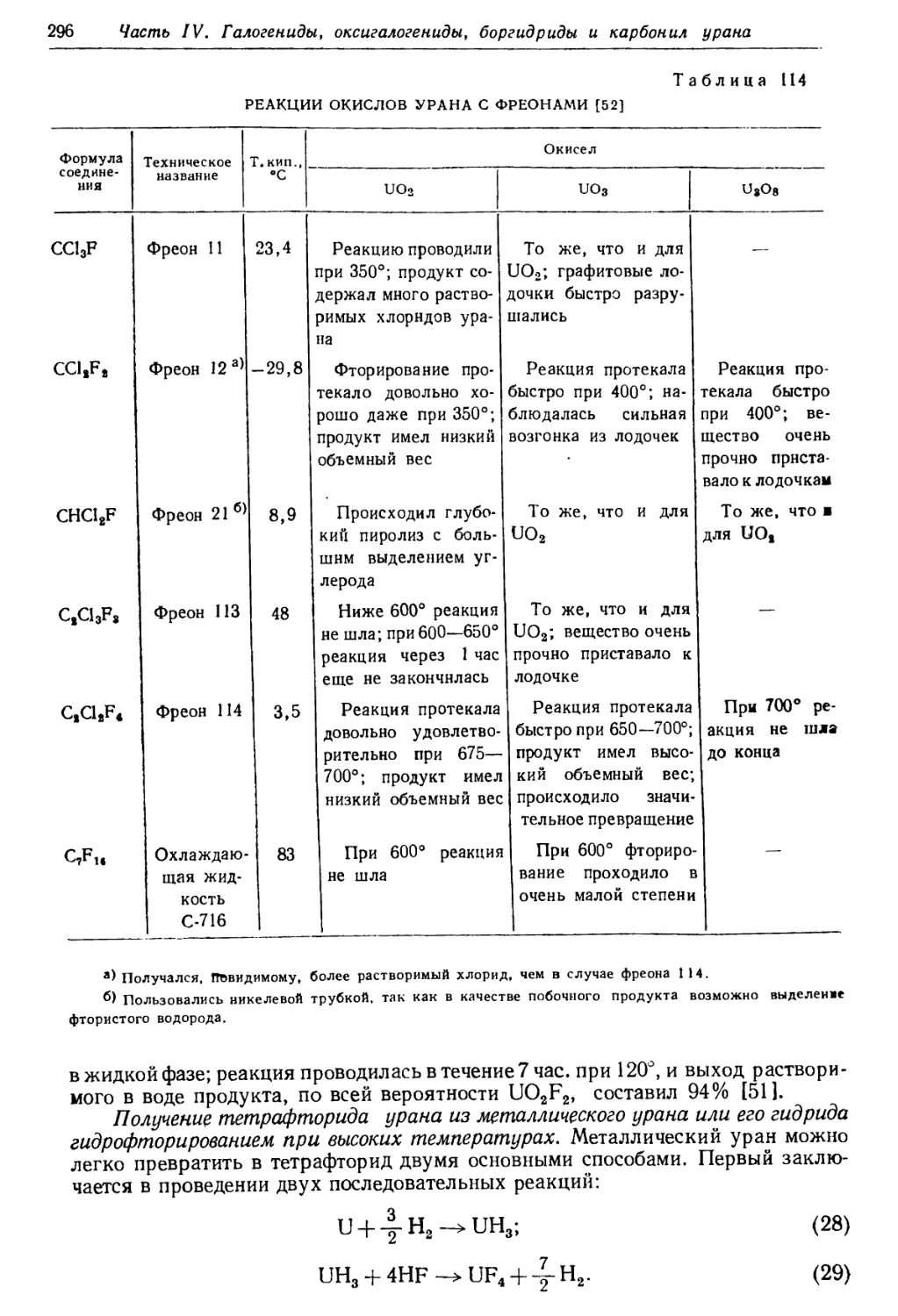


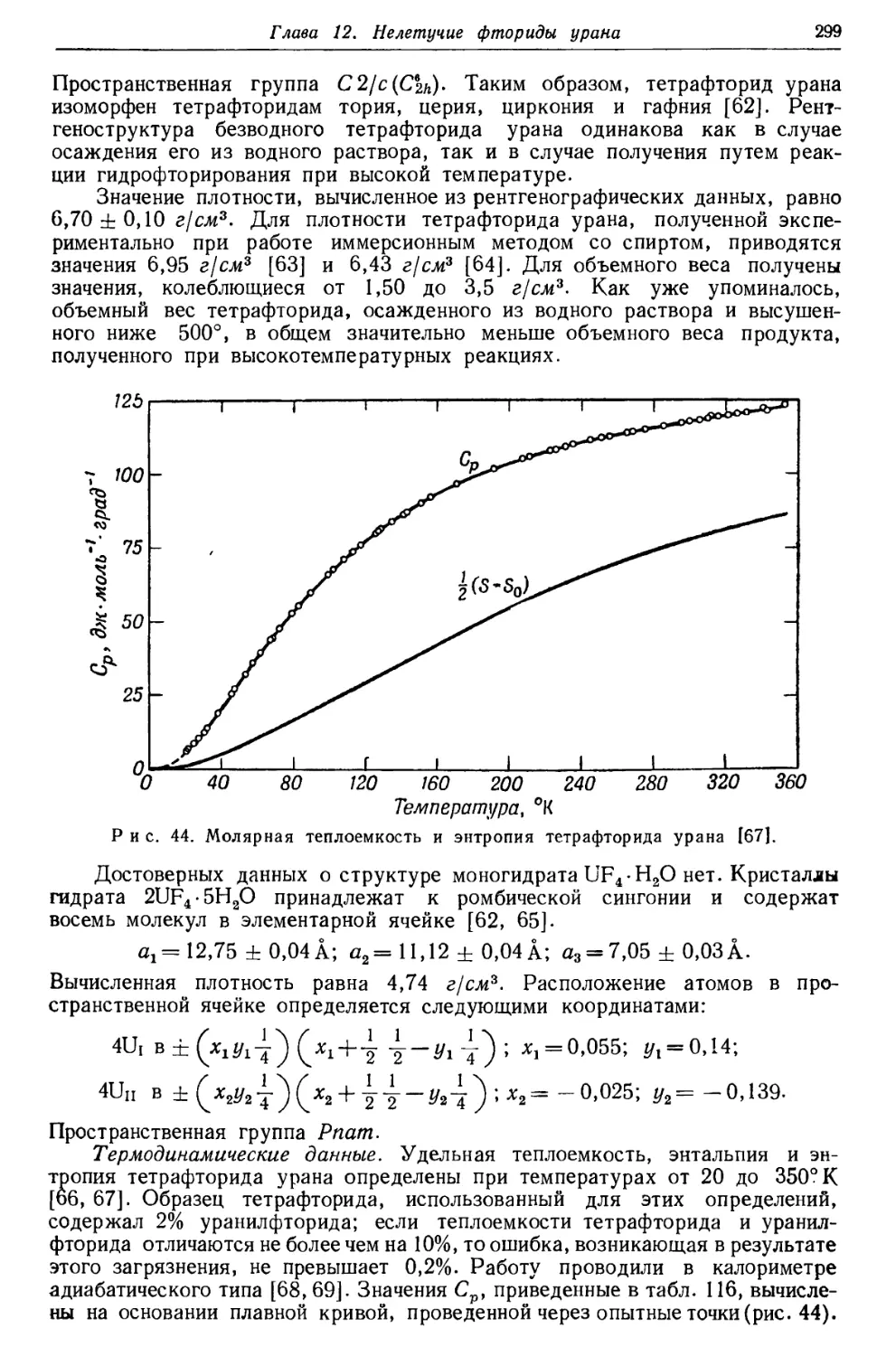
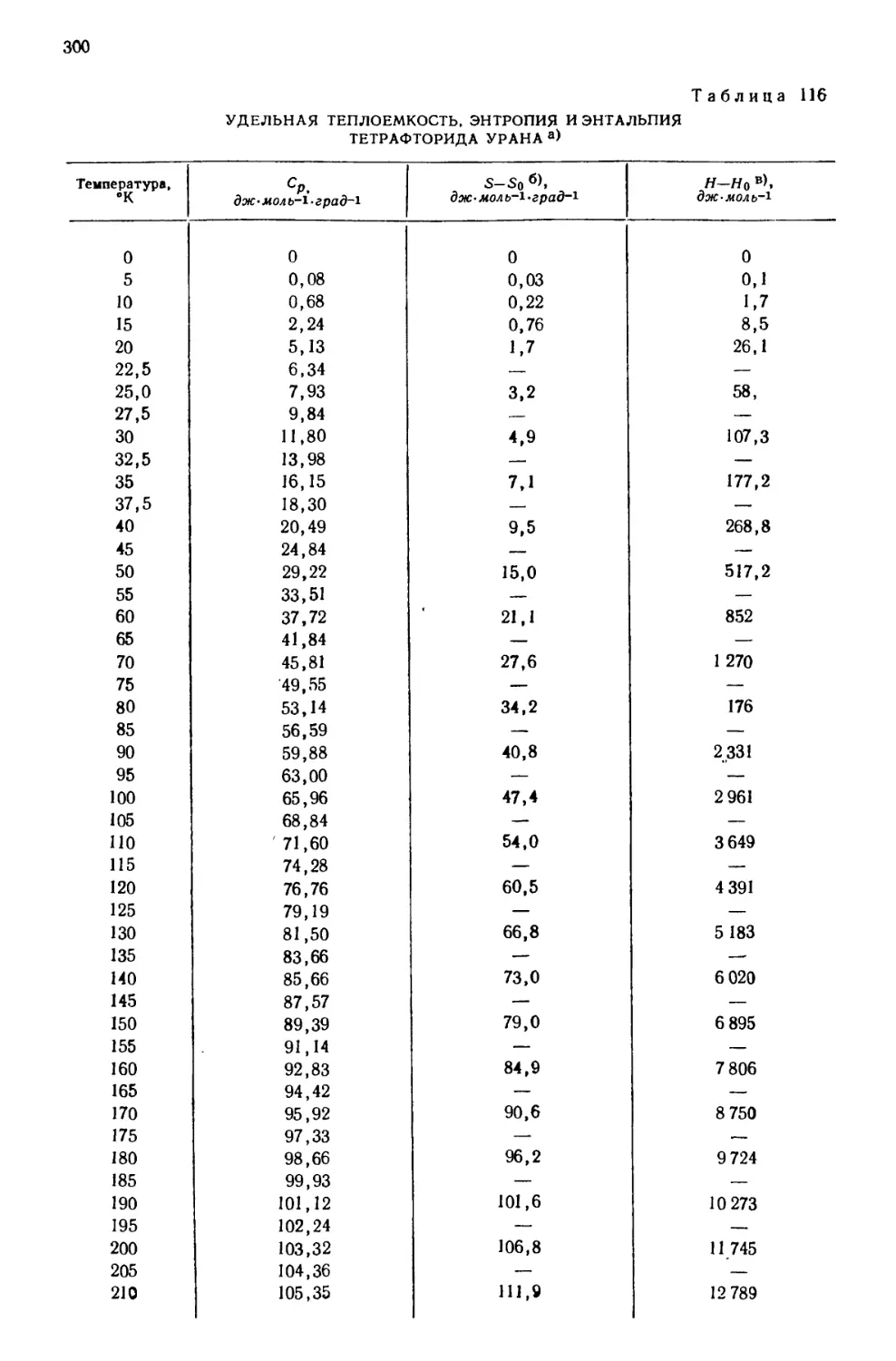
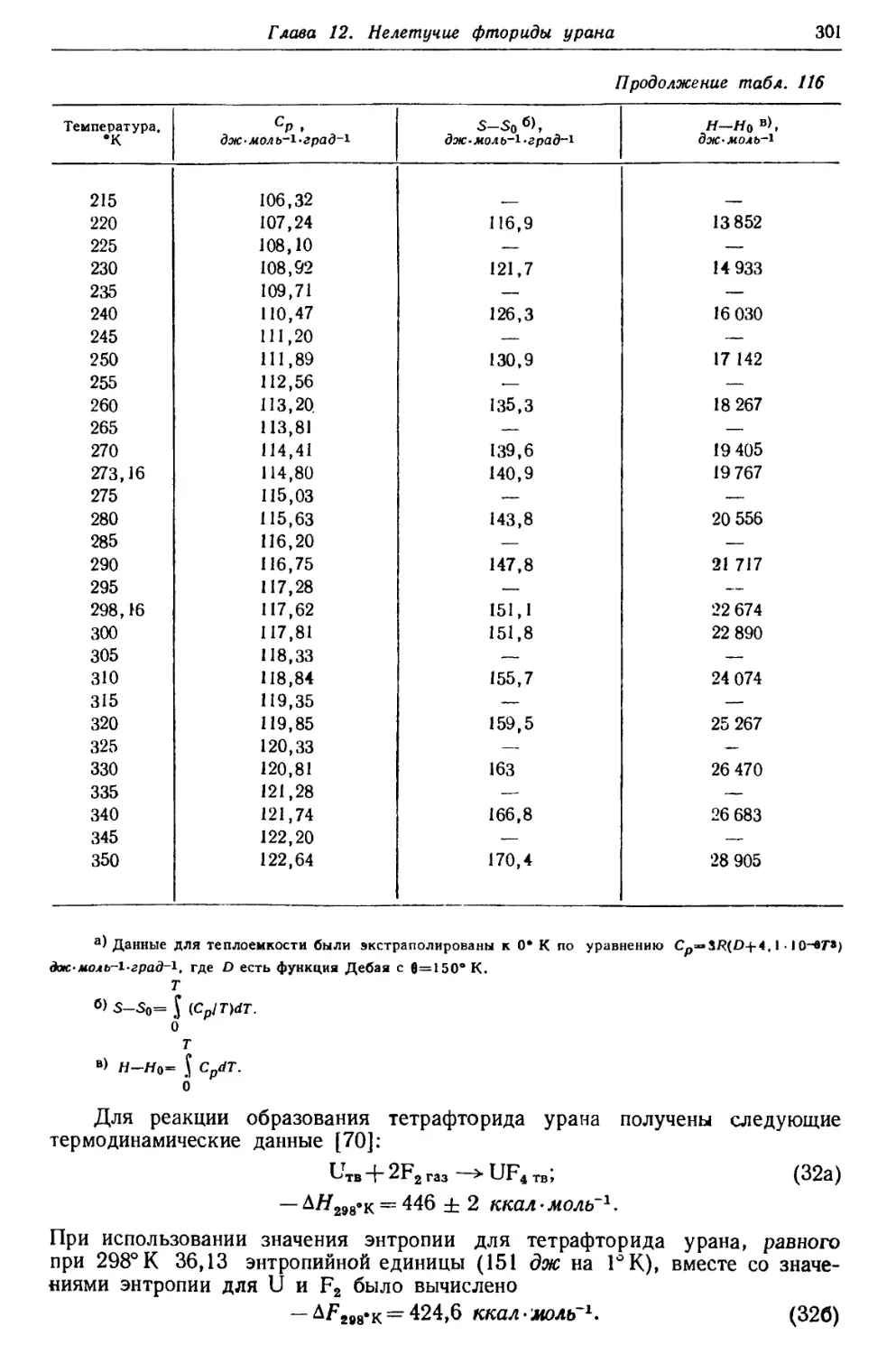







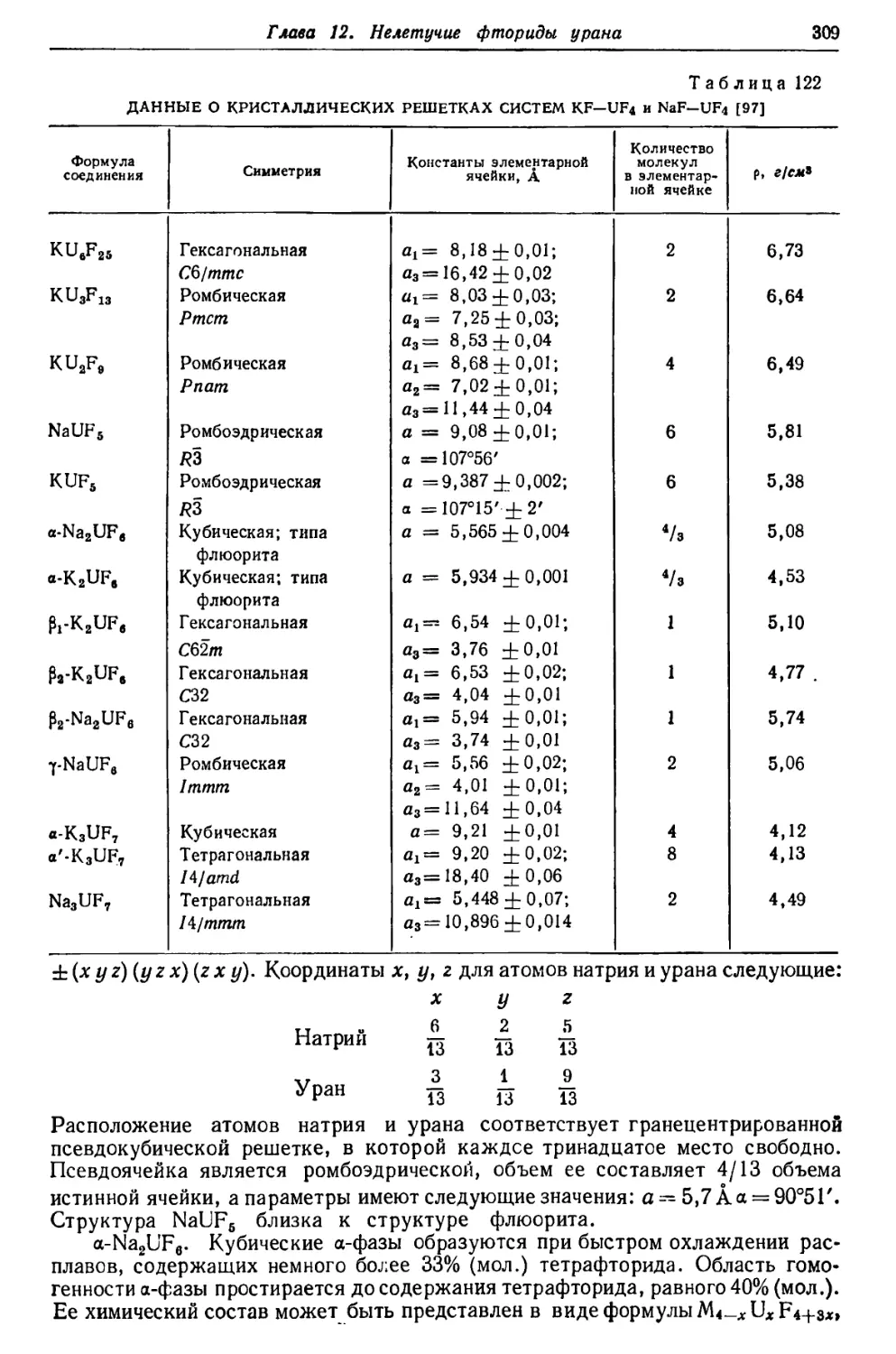

















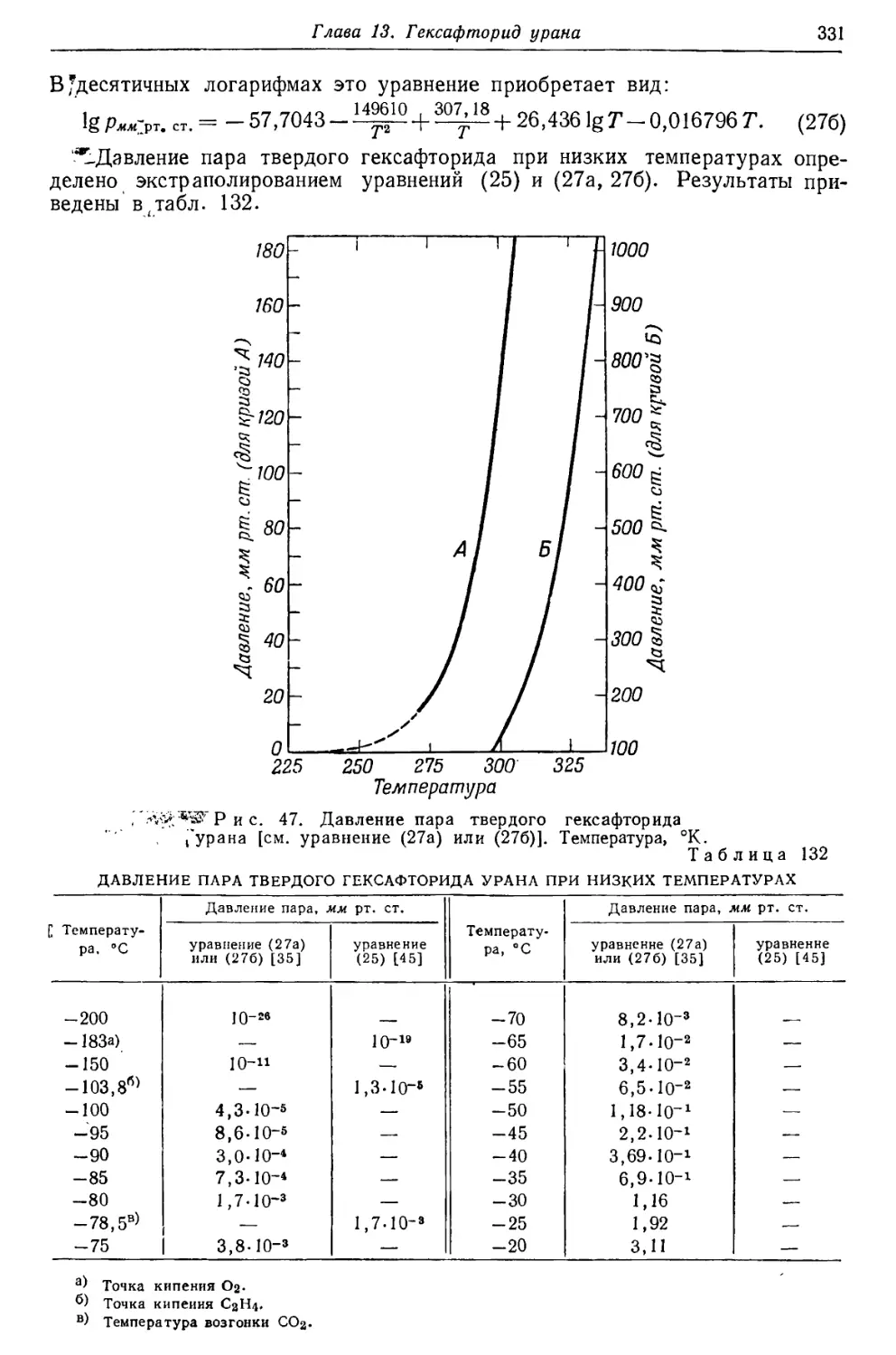

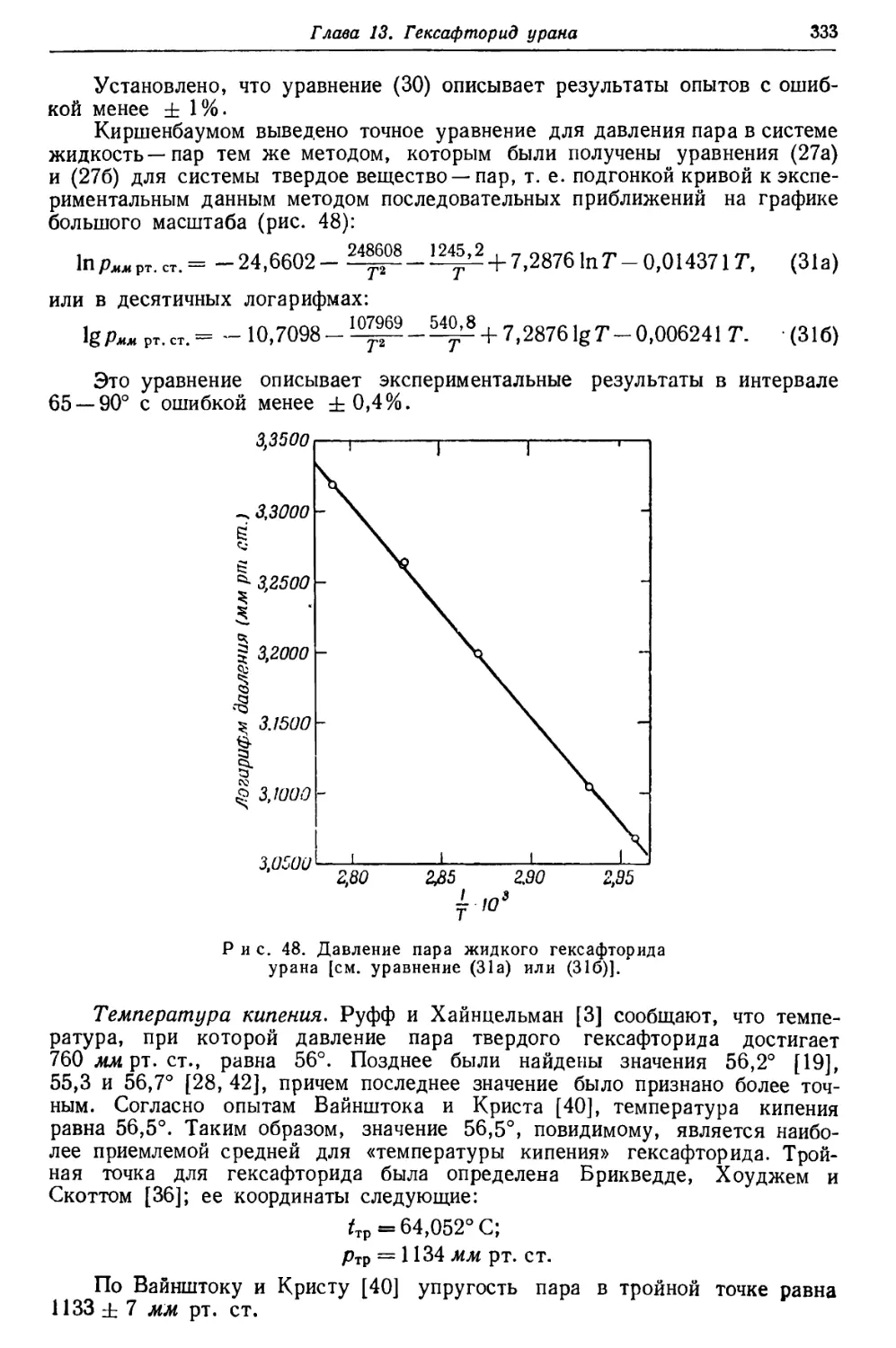


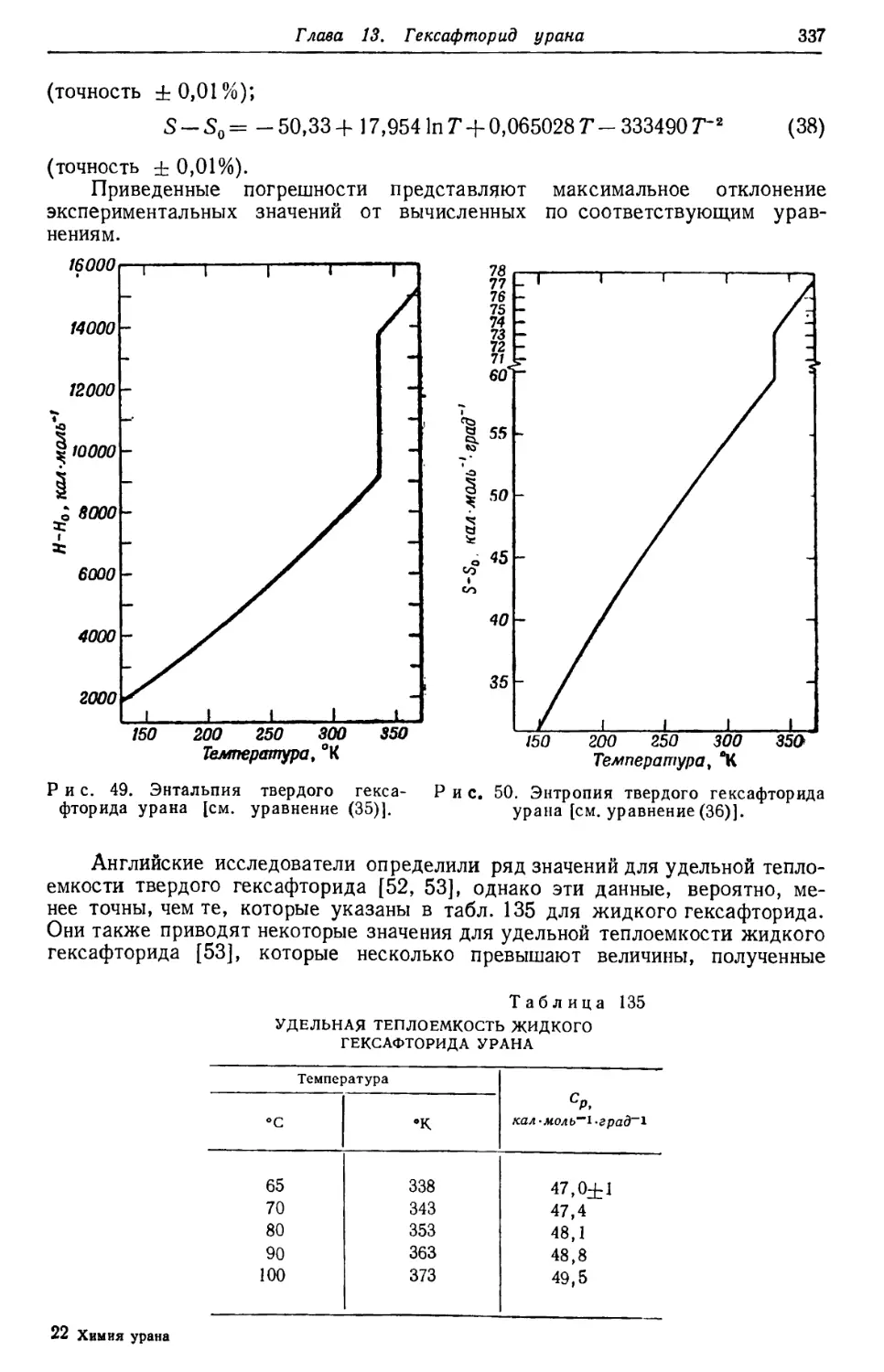





































































































































Текст
Дж. КАЦ и Е. РАБИНОВИЧ
ХИМИЯ УРАНА
УРАН КАК ЭЛЕМЕНТ,
ЕГО БИНАРНЫЕ СОЕДИНЕНИЯ,
ГИДРАТЫ ОКИСЛОВ
И ОКСИГАЛОГЕНИДЫ
Перевод с английского
И * Л
ИЗДАТЕЛЬСТВО
ИНОСТРАННОЙ ЛИТЕРАТУРЫ
Москва, 1954
THE CHEMISTRY OF URANIUM
Part I
THE ELEMENT, ITS BINARY
AND RELATED COMPOUNDS
by
J. J. KATZ and E. RABINOWITCH
First Edition
New York—Toronto—London
McGraw-Hill Book Company, Inc.
1951
ОТ РЕДАКЦИИ
Книга Дж. Каца и Е. Рабиновича «Химия урана», выпущенная в США
в 1951 г., является первой из двух книг, намеченных авторами к изданию по
этой теме, и представляет литературный обзор работ, выполненных в основном
зарубежными исследователями за период с конца XVIII века по 1950 г.
Книга состоит из четырех частей.
В первой части сообщаются краткие сведения об естественных и
искусственных изотопах урана, о результатах рентгеновских исследований по
определению длин волн, соответствующих главным границам поглощения урана для
различных валентных состояний, об эмиссионном характеристическом
рентгеновском спектре и фотоэлектрическом эффекте этого элемента. Приводятся также
данные по изучению оптического спектра, результаты измерений наиболее
важных длин волн в дуговом и искровом спектрах урана, данные по ядерным
эффектам, по влиянию электрического и магнитного полей на спектр, а также
анализ термов (даются таблицы их символов и значений). Кроме того, в этой
части приводятся сведения по географии и геологии месторождений
урановых руд.
Во второй части книги описываются работы по получению и исследованию
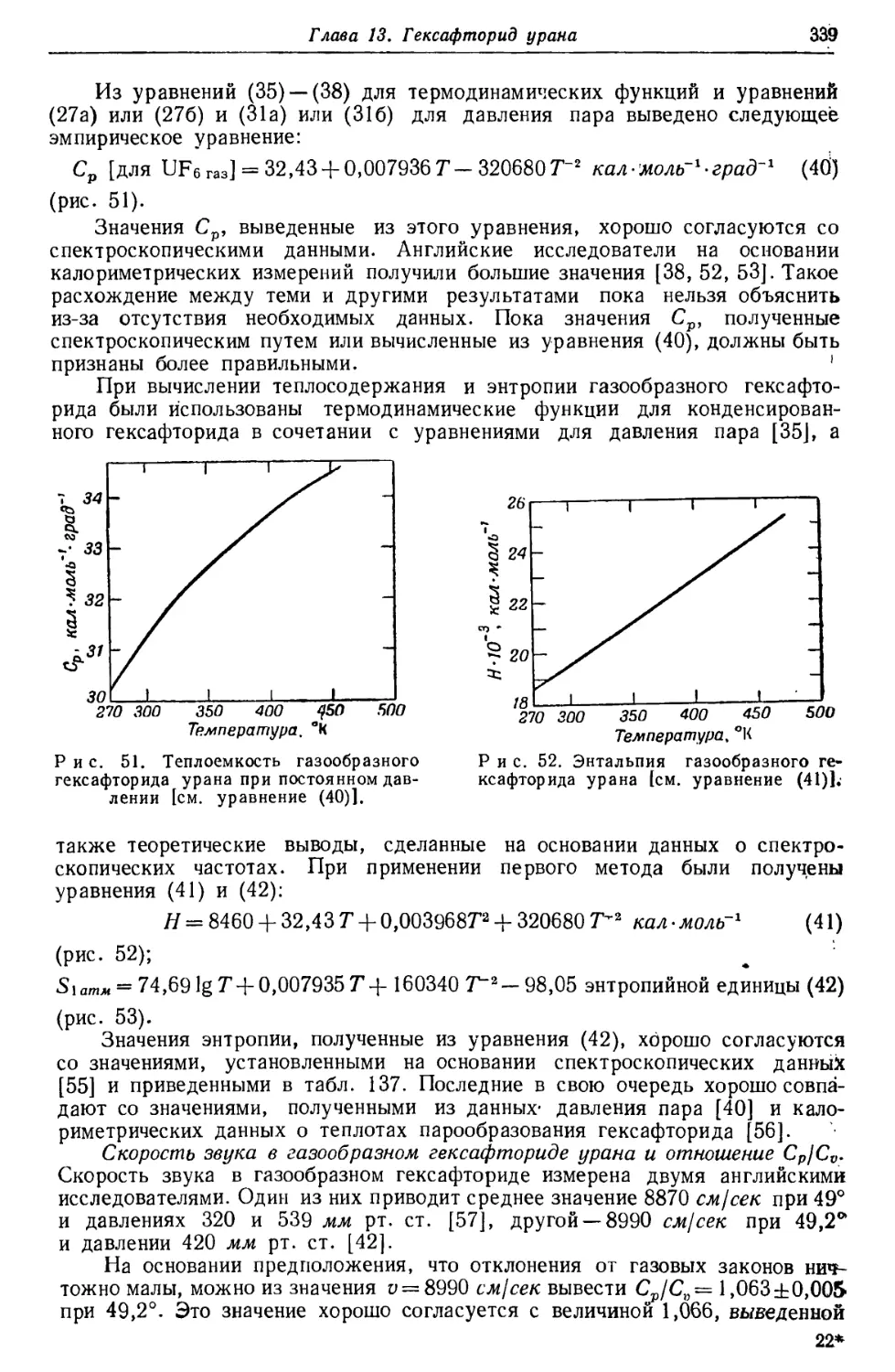
металлического урана —методы переработки урановых руд, получение
металлического урана, — рассматривается кристаллическая структура, размеры атома,
плотность, термическое расширение, твердость, упругая и неупругая
деформации, результаты механических испытаний, теплоемкость,
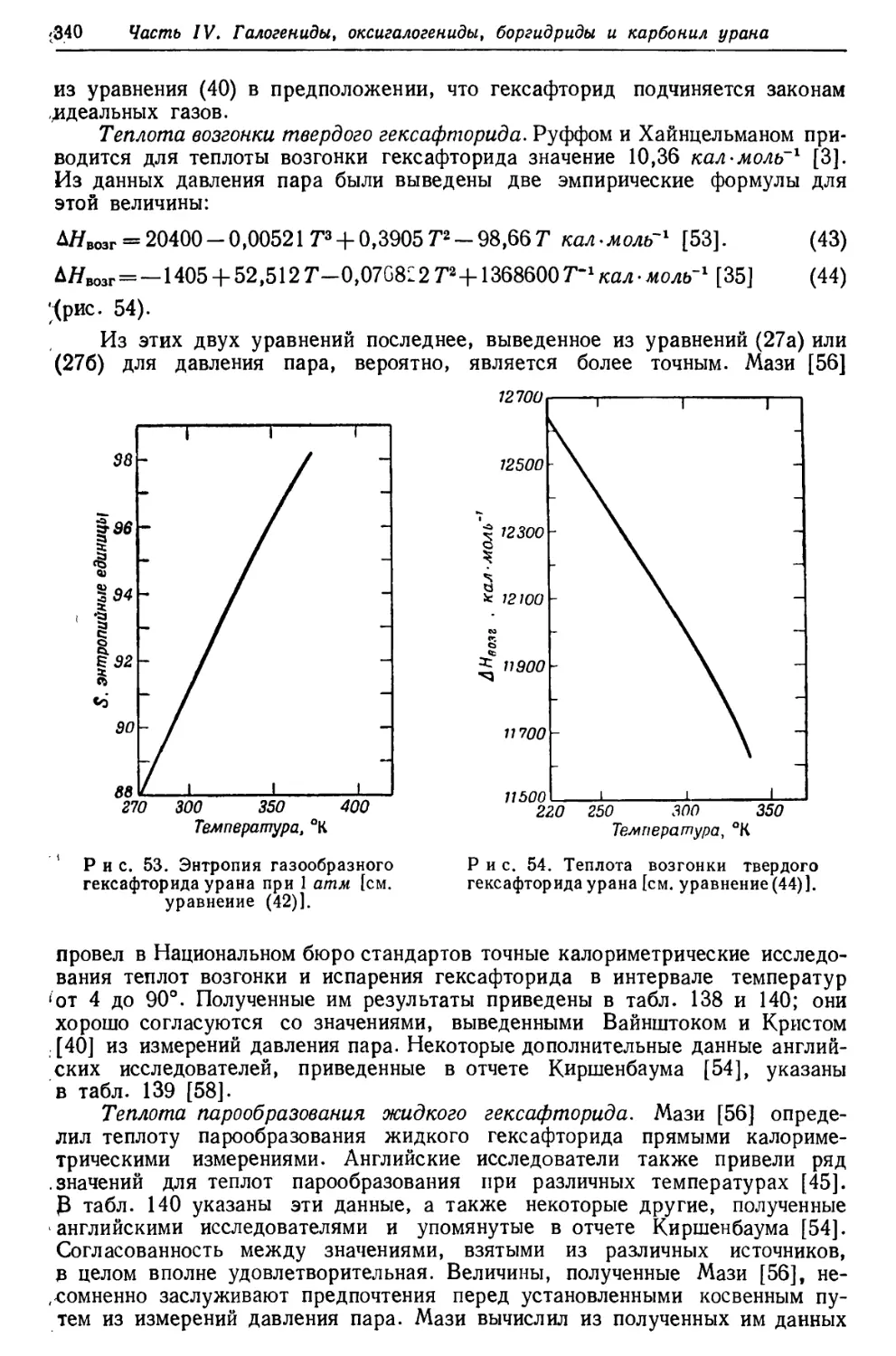
термоэлектродвижущая сила, магнитная восприимчивость, оптическая излучательная
способность и другие физические свойства. Приводятся также химические
свойства металлического урана — отношение его к неметаллам,
неорганическим и органическим кислотам, водным растворам щелочей и некоторых
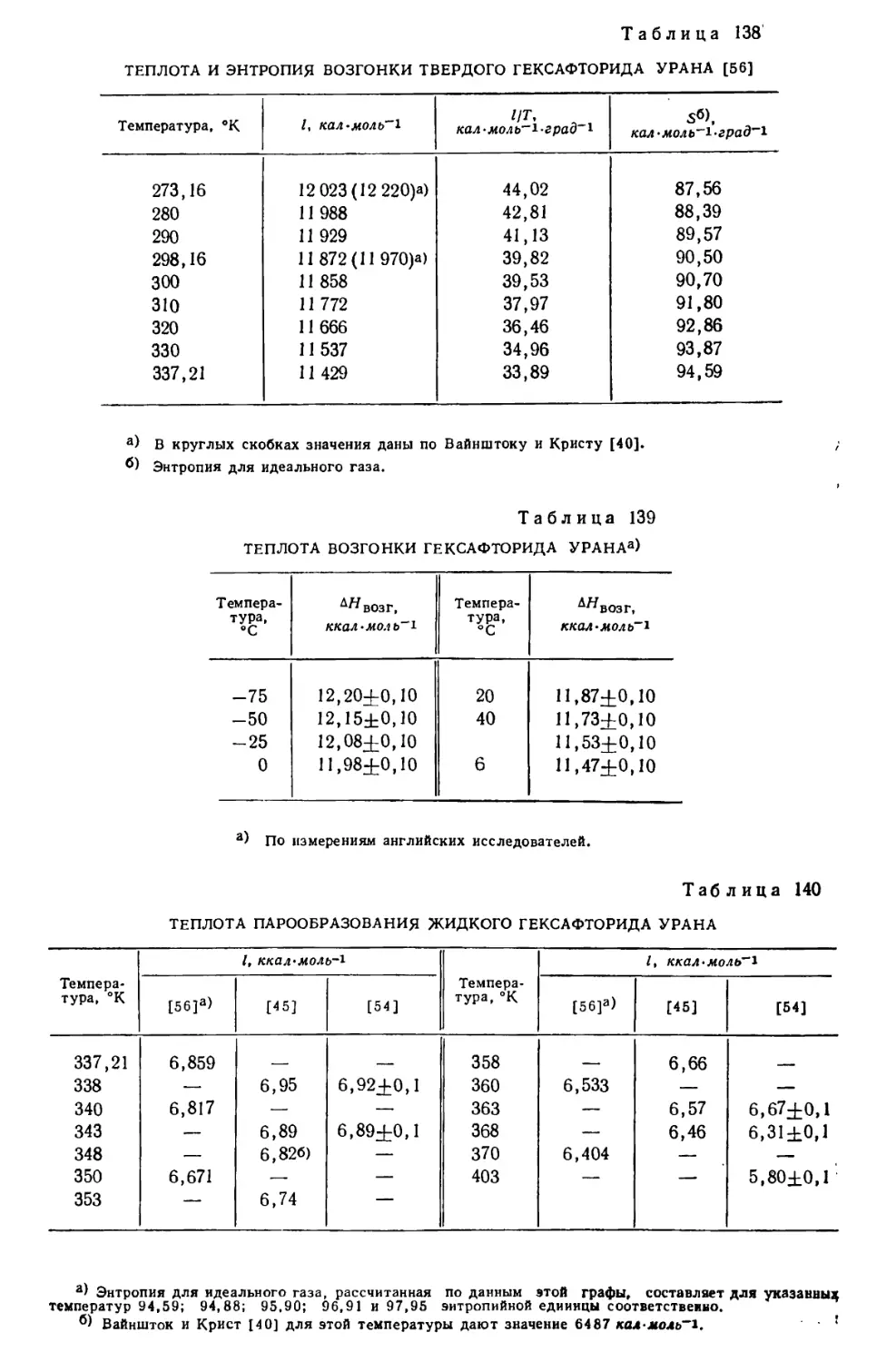
солей.
В третьей части книги обсуждаются методы получения и свойства
гидридов и дейтеридов, боридов, карбидов, силицидов, нитридов, фосфидов, ар-
сенидов и антимонидов урана, а также его окислов, сульфидов, селенидов
и теллуридов.
В четвертой части рассматриваются галогениды и оксигалогениды, бор-
гидриды и карбонилы урана.
В каждом разделе книги даются результаты исследования физических
свойств соединений урана (кристаллическая структура, термодинамические
данные и т. п.).
4
От редакции
Ценность книги заключается в том, что в ней собран большой фактический
материал, причем приводятся данные не только из опубликованной литературы,
но также частично сообщается и о результатах исследований, проведенных
по программе, известной в США под названием Манхэттенского проекта.
Книга представляет несомненный интерес для химиков и физиков,
работающих в области химии урана.
Перевод книги издается с небольшими сокращениями текста,
содержащего материал, не представляющий интереса для советского читателя.
Часть I
УРАН КАК ЭЛЕМЕНТ
Глава 1
ИЗОТОПНЫЙ СОСТАВ И АТОМНЫЙ
ПРИРОДНОГО УРАНА
ВЕС
В этой главе даются краткие сведения об изотопном составе природного
урана. Подробное рассмотрение ядерных свойств всех естественных и
искусственных изотопов выходит за пределы тематики данного труда (по этому
вопросу см. [1]). Здесь обсуждаются лишь некоторые основные данные об
изотопном составе, атомном весе и взаимосвязи между тремя
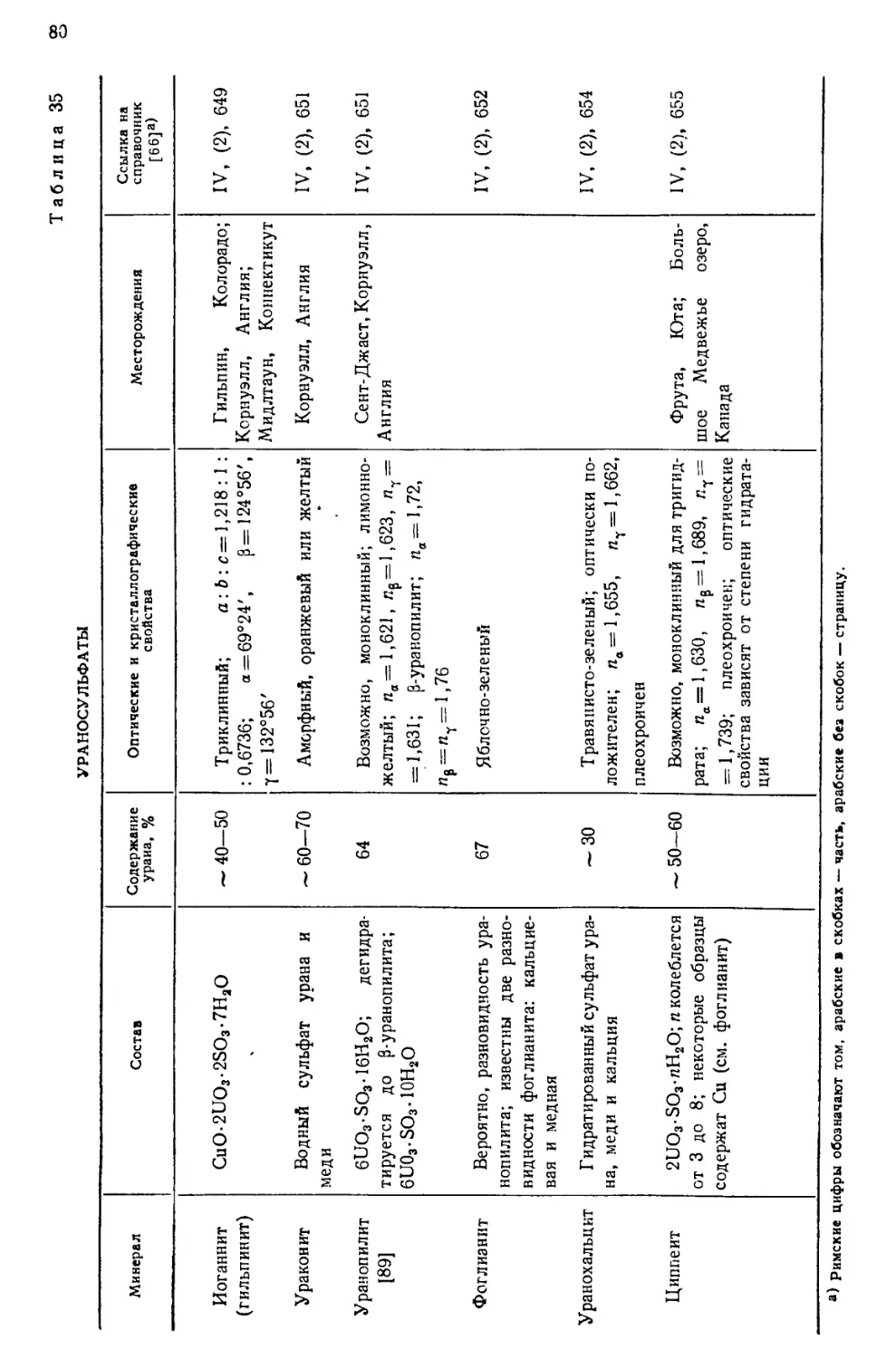
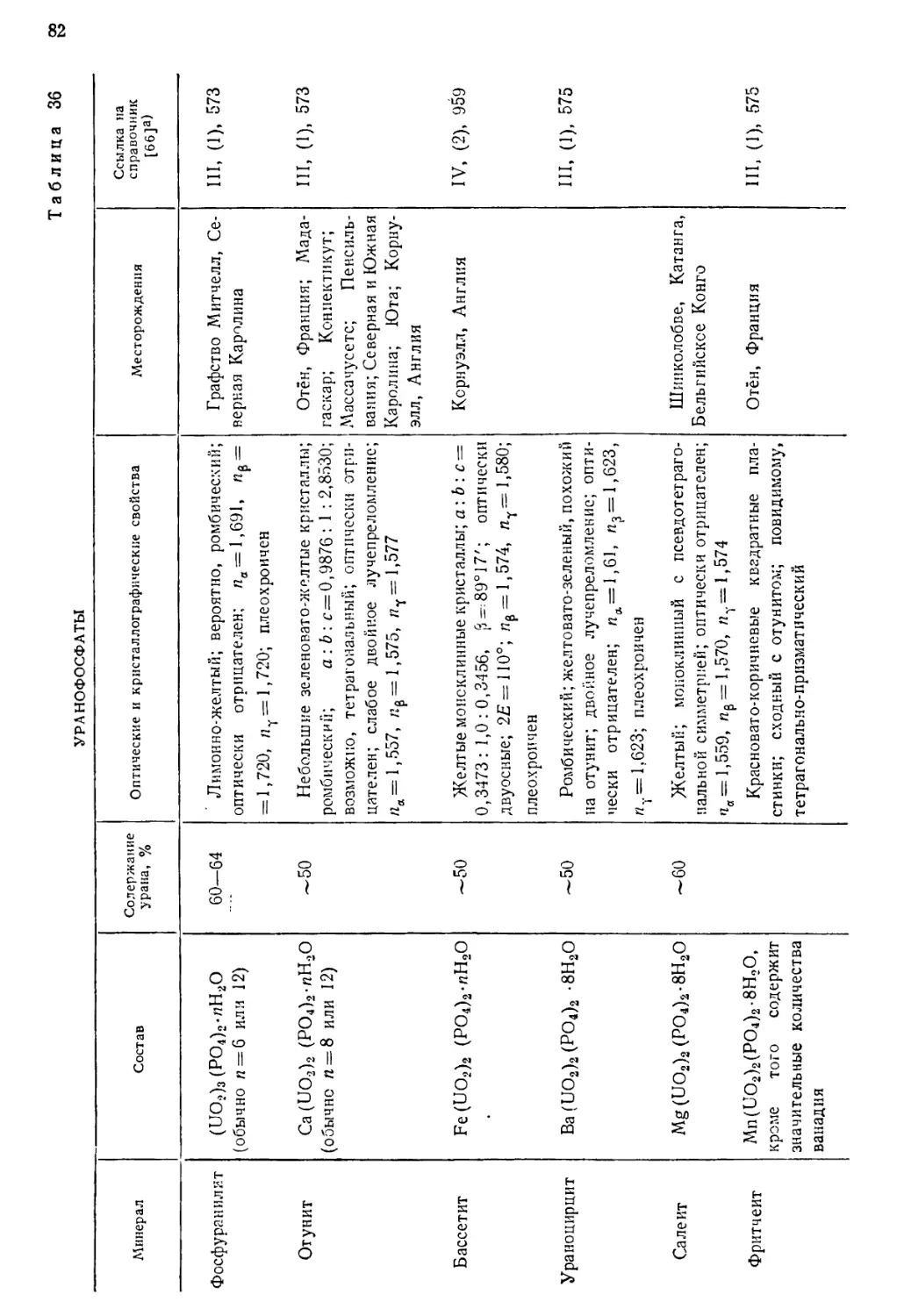
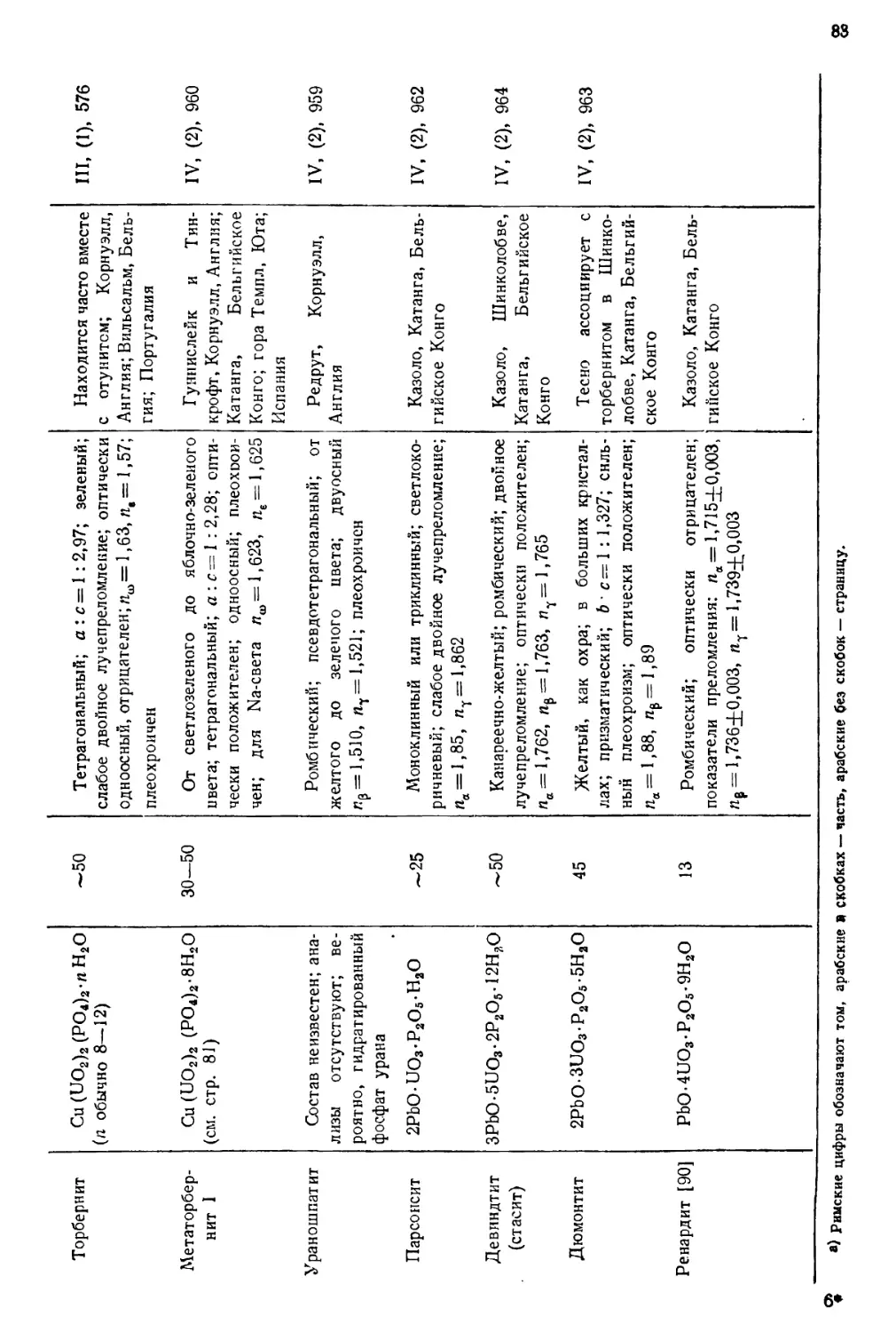
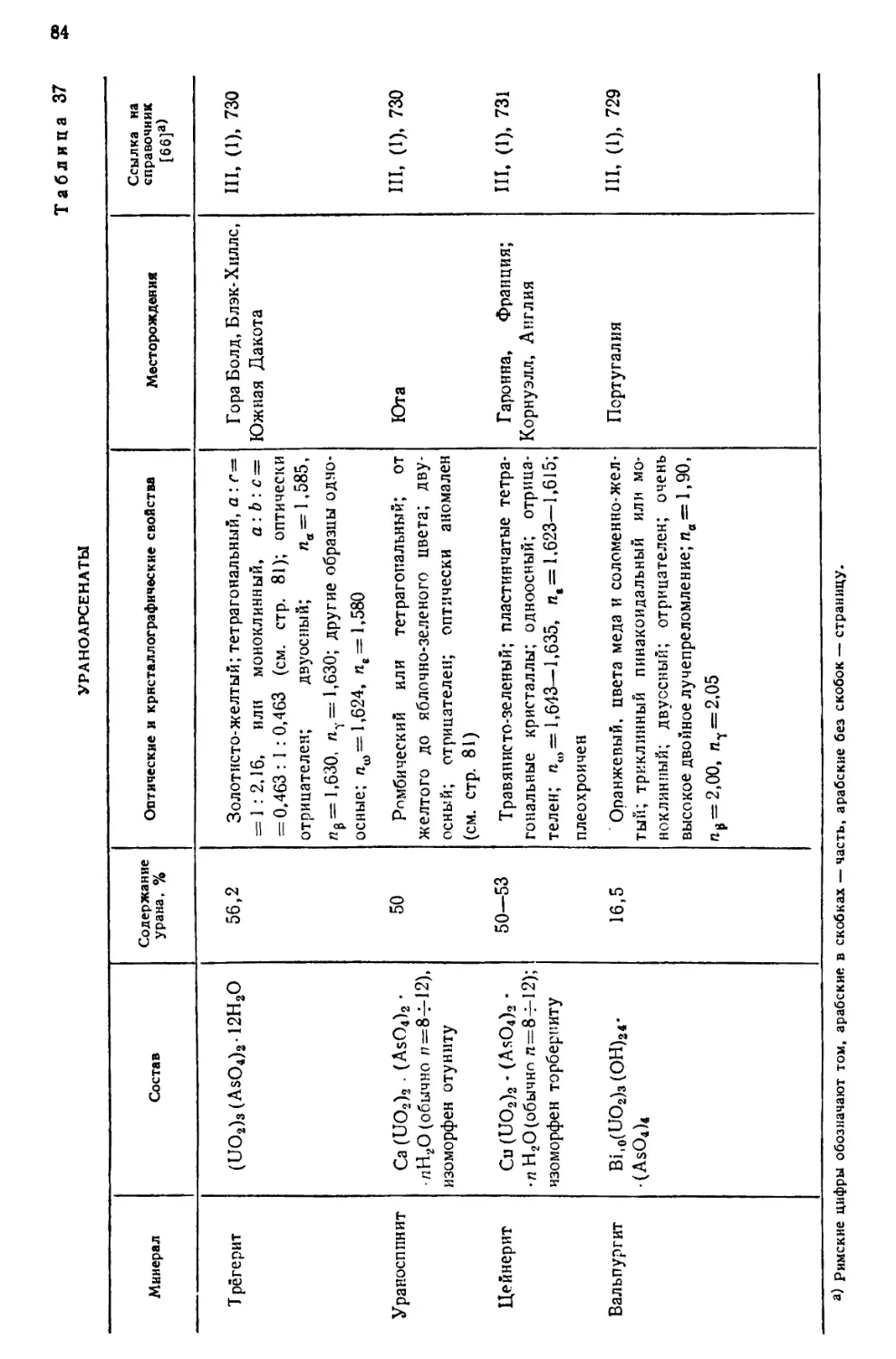
встречающимися в природе изотопами урана.
ИЗОТОПНЫЙ СОСТАВ
Природный уран содержит три изотопа U238(UI), U234(UII) и U235 (акти-
ноуран AcU). Несколько сравнительно короткоживущих изотопов получено
искусственно—путем различных ядерных реакций, в которых исходными
веществами служили актиний, торий, протактиний или долгоживущие
изотопы урана. Радиоактивные постоянные естественных и искусственно
полученных изотопов урана даны в табл. 1. Изотопы U238(UI) и U234(UII)
Таблица 1
РАДИОАКТИВНЫЕ ПОСТОЯННЫЕ ЕСТЕСТВЕННЫХ
И ИСКУССТВЕННЫХ ИЗОТОПОВ УРАНА L2J
Массовое
число
Излучение
характер
энергия,
Мэв
период
полураспада
Содержание
в природном
уране, %
234 (UH)
235 (AcU)
238 (UI)
228
229
230
[231
232
233
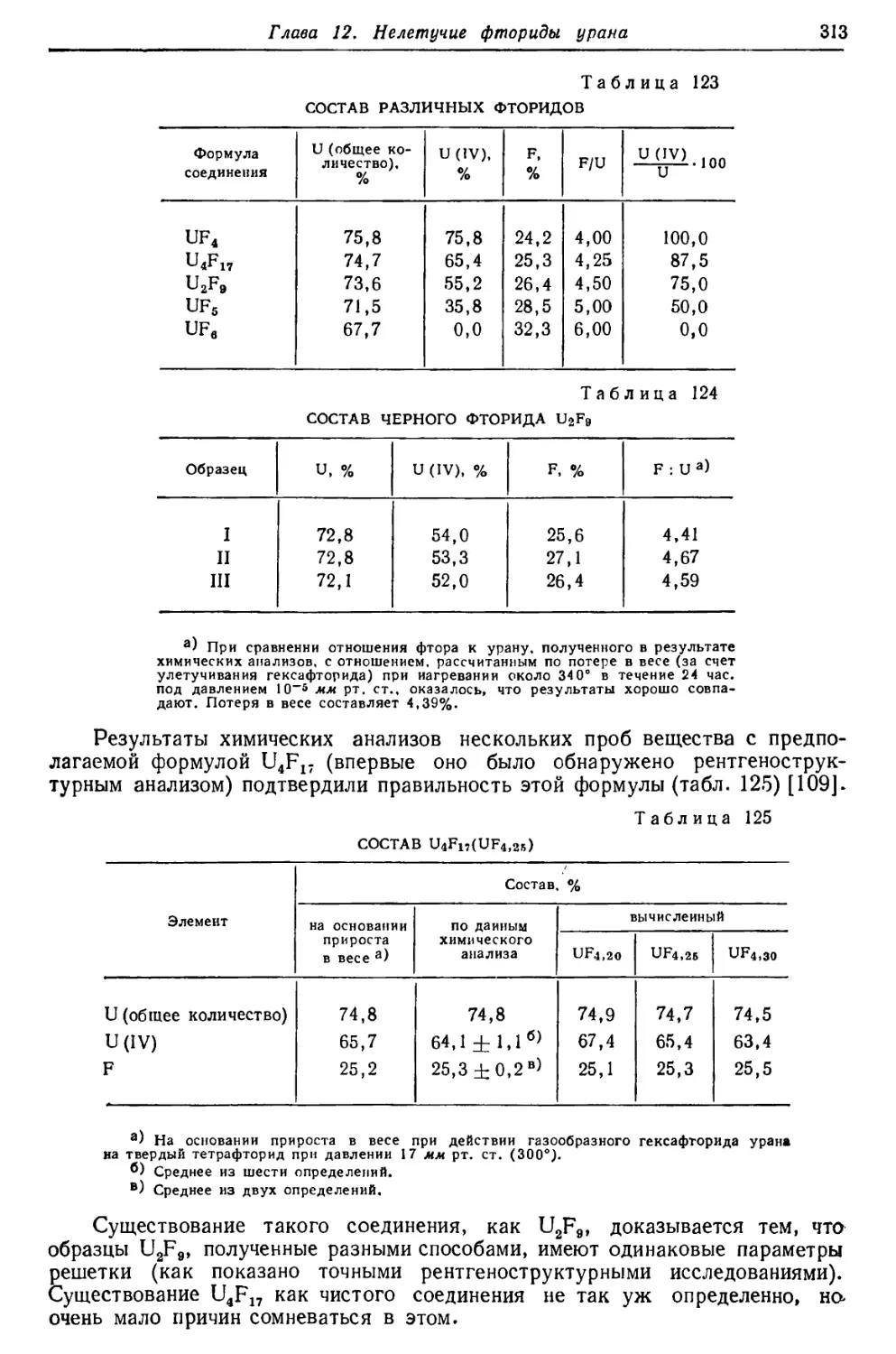
'237
239
1 4»?6
4,52
4,21 |
Естественные изотопы
2,35-106 лет
8,91-Ю8 »
7,07-Ю8 »
8,52.108 »
4,5Ы09 »
Искусственные изотопы
9,3 мин.
58 »
20,8 суток
4,2 »
70 лет
1,6- 105 »
6,8 суток
23,5 мин.
а (80о/0)
'К (20% Г
а (-20%)
/С (-80%)
а
к
а
а, •(, е-
£"» Vе'
Р~. Ъ ег
6,72 1
6,42
5,85
5,3
4,8
0,005
0,71
99,28
8
Часть 1, Уран как элемент
являются членами одного и того же радиоактивного ряда, так называемого ряда
(4/г+2):
UI.^->UX1JL^UX2J_»UII_JL-».
Поэтому во всех рудах урана, в которых установилось радиоактивное
равновесие, эти изотопы должны находиться в постоянном отношении, равном
отношению их периодов полураспада.
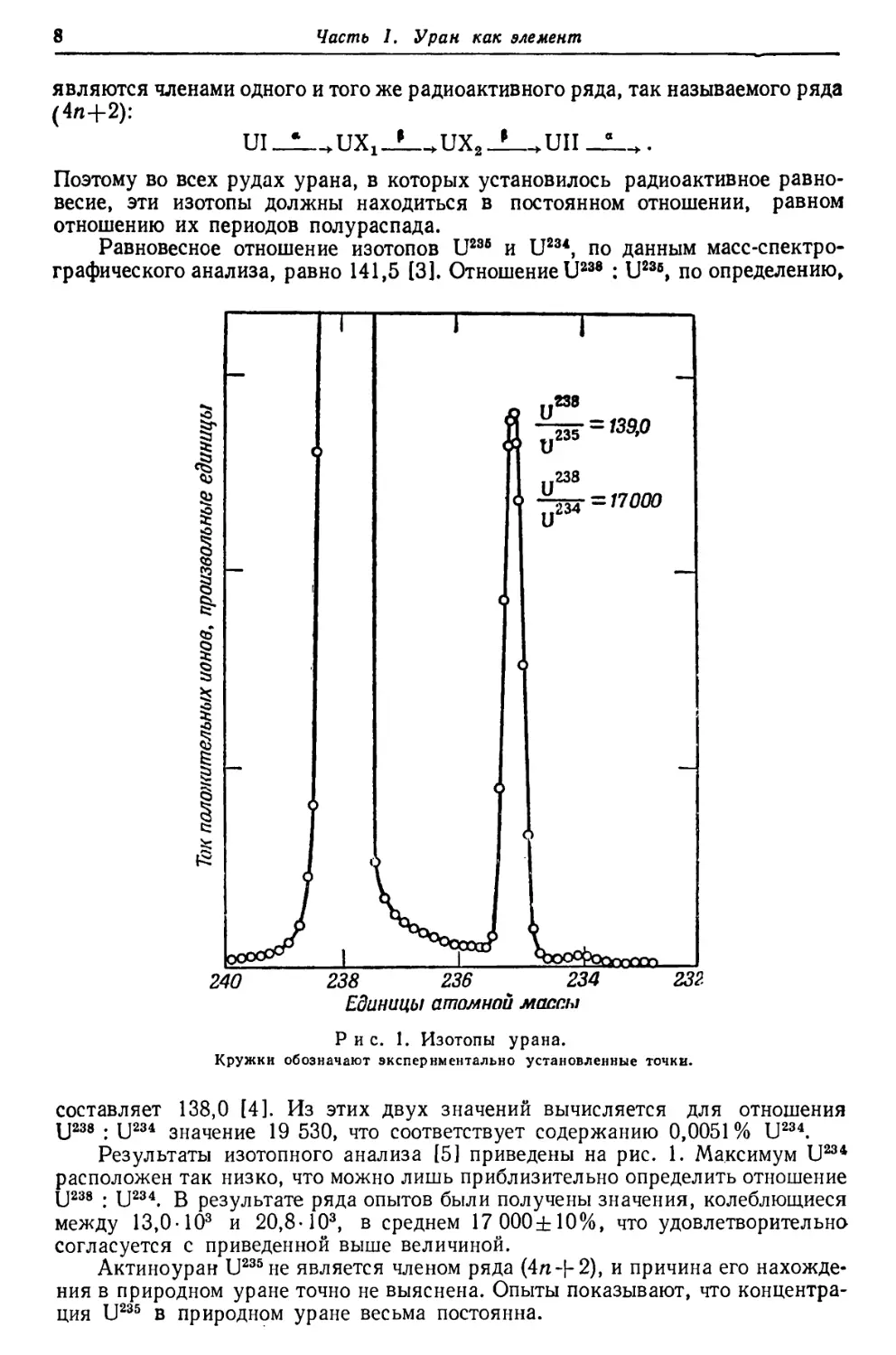
Равновесное отношение изотопов U236 и U234, по данным масс-спектро-
графического анализа, равно 141,5 13]. Отношение U238 : U236, по определению,
If.
1
V
£
1:
$
§
сэ
£■'
с: |
сь 1
§
1;
51
5
§
ё
6
§
§
§
t:
f§
<
в
nr~
\
?
1
J I 1
6
a
' I
T
T
J,4i
i u238 1
1 гш=т>
T "
II238
k 7Ш = 17000
I
1
о
^boOOtbrywy»
240 238 236 234
Единицы атомной массы
232
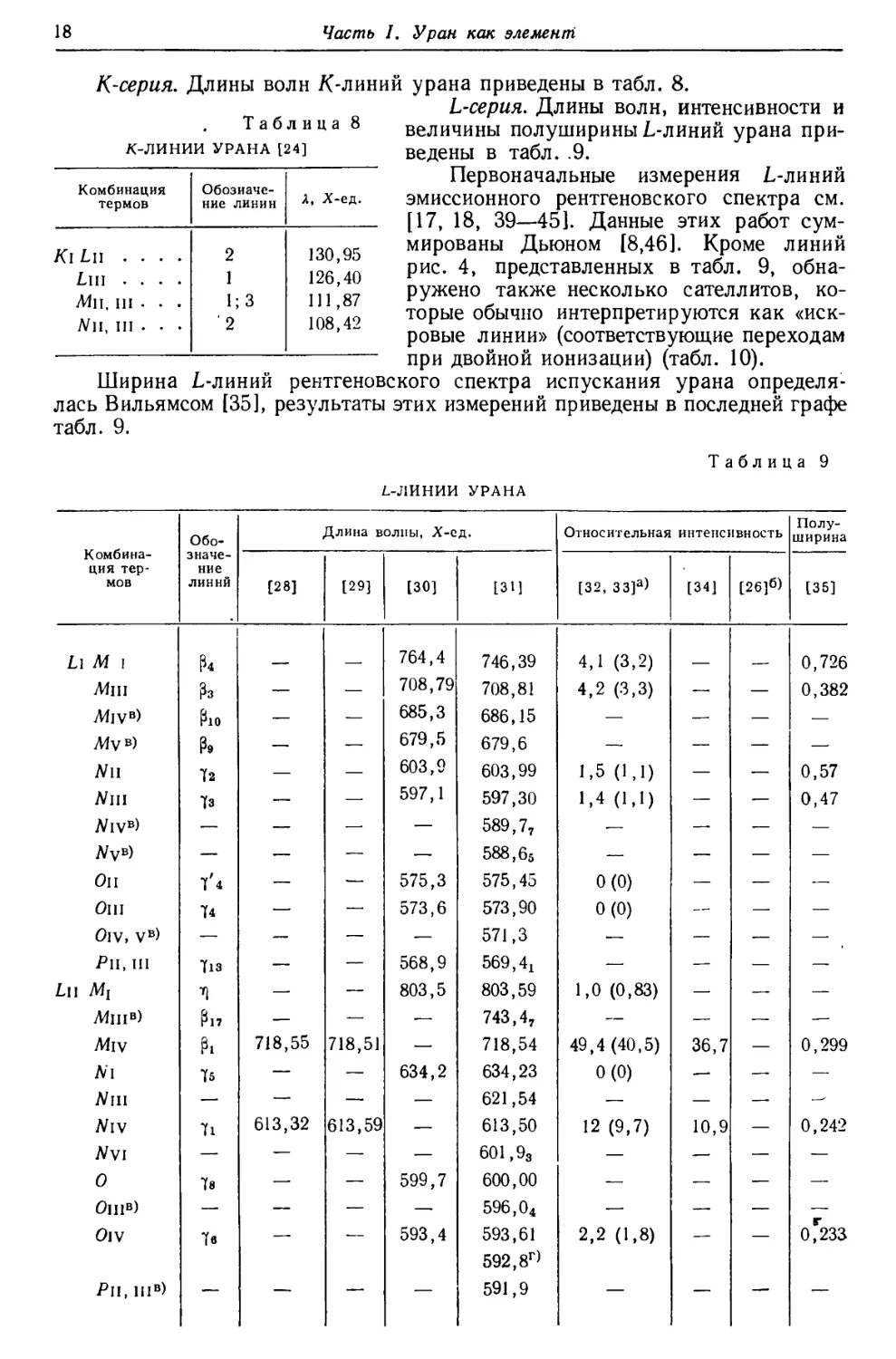
Рис. 1. Изотопы урана.
Кружки обозначают экспериментально установленные точки.
составляет 138,0 [4]. Из этих двух значений вычисляется для отношения
U238 : U234 значение 19 530, что соответствует содержанию 0,0051% U234.
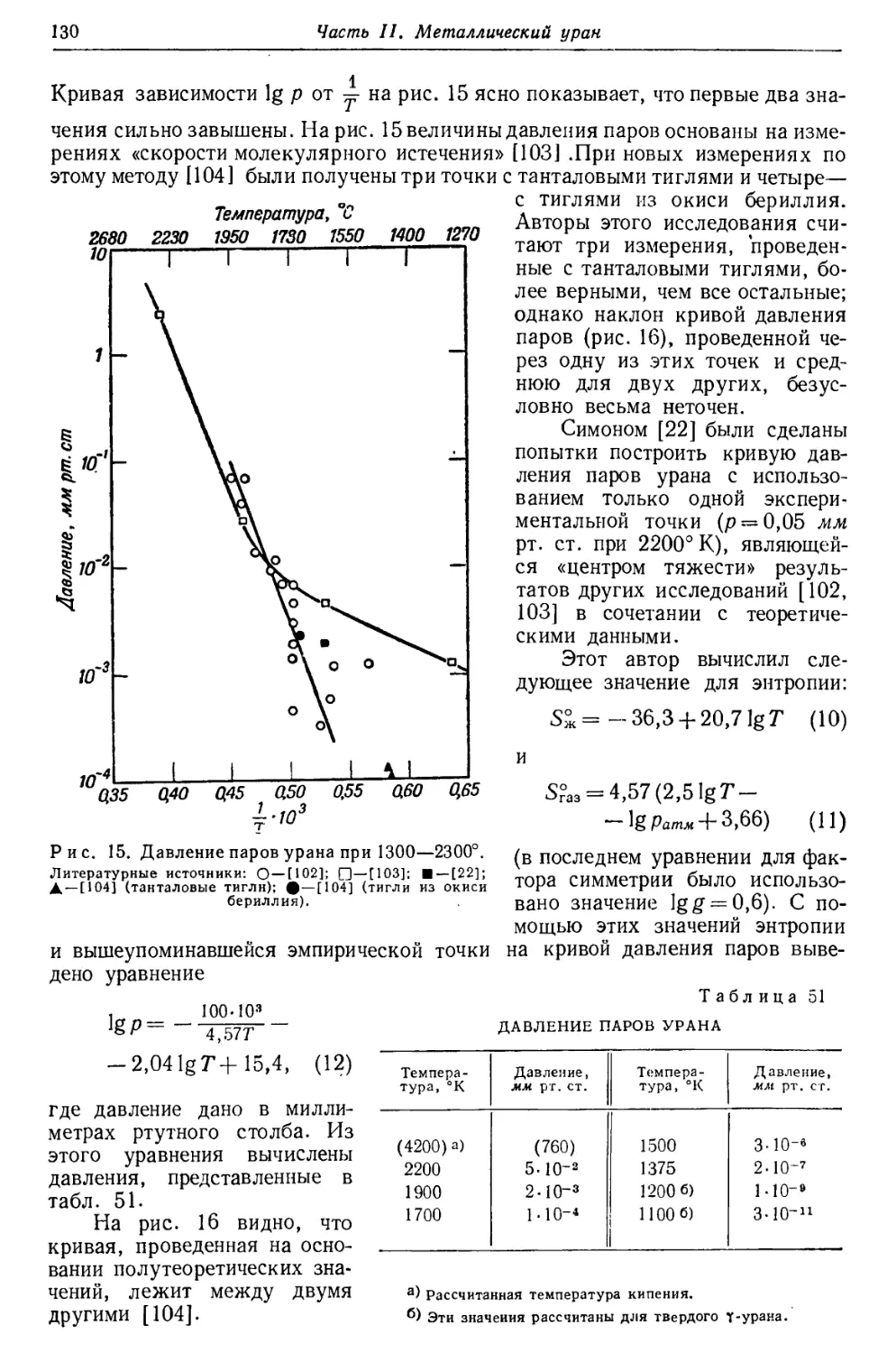
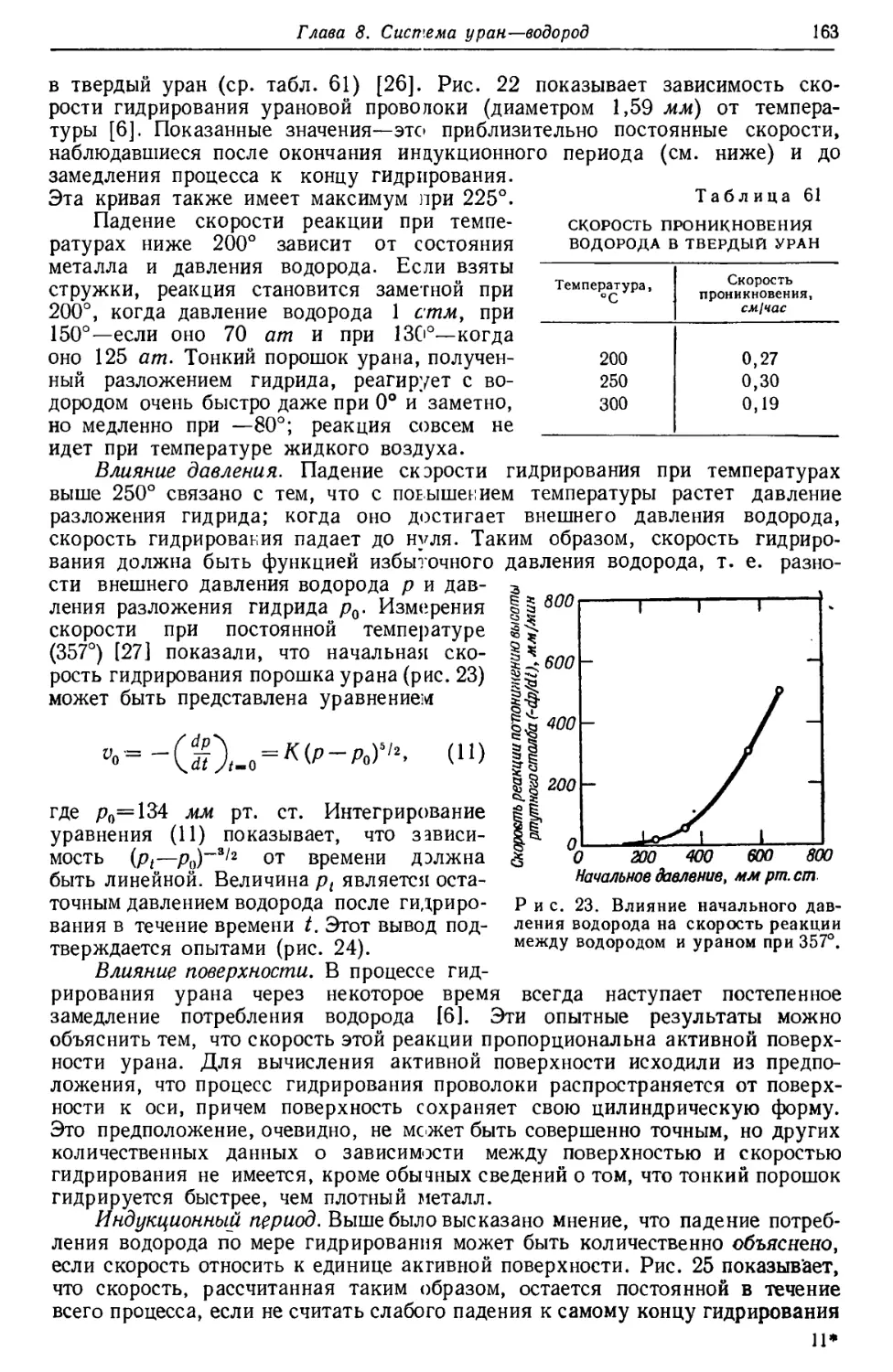
Результаты изотопного анализа [5] приведены на рис. 1. Максимум U234
расположен так низко, что можно лишь приблизительно определить отношение
U238 : U234. В результате ряда опытов были получены значения, колеблющиеся
между 13,0-103 и 20,8-103, в среднем 17 000±10%, что удовлетворительно
согласуется с приведенной выше величиной.
Актиноуран и235не является членом ряда (4л+ 2), и причина его
нахождения в природном уране точно не выяснена. Опыты показывают, что
концентрация U235 в природном уране весьма постоянна.
Глава 1. Изотопный состав и атомный вес природного урана 9
Впервые присутствие U236 в природном уране было установлено
масс-спектрографическим анализом [6]. При этом было показано, что концентрация U235
меньше 1%. Позднее было произведено точное определение [51, результаты
которого для различных урановых минералов представлены в табл. 2. В
среднем эти значения равны 139+1- Из данных табл. 2 видно, что возраст минерала
не оказывает заметного влияния на отношение U238 : U235.
Таблица 2
ОТНОШЕНИЕ ИЗОТОПОВ U238 : U235
Минерал
Уранинит (Онтарио) ....
Возраст,
годы
4. 108
1,0- 109
1 - 103
Отношение U288 : U235 а) \
пределы значений
137,5—140,7
135,3—140,8
136,6—141,3
среднее |
значение!
139,0
138,9
138,8
а) В качестве источника ионов пользовались тетрахлоридом и тетра-
бромидом урана.
Это отношение было вторично определено сотрудниками Колумбийского
университета [4]. Они использовали ионы UF* и UF* из UFe (стандартный
источник) и ионыи+ (источник с ионизацией с поверхности). Результаты
представлены в табл. 3. При сравнении двух африканских руд, одной канадской
и колорадского карнотита разница между значениями отношения U238 : U235
не превышала 0,03%.
Таблица 3
ОТНОШЕНИЕ U288 : U285, ОПРЕДЕЛЕННОЕ С ДВУМЯ РАЗЛИЧНЫМИ
ИСТОЧНИКАМИ
Источник
Отношение U238 : U235
предельные
значения
135,0-138,6
137,9-139,0
среднее значение
137,0±0,7
138,0±0,3
Значения, полученные с этими двумя источниками ионов, совпадают
в пределах статистической ошибки; в данной работе принято значение
138,0±0,3, полученное в Колумбийском университете [4]. Прежнее
значение (139±1) [5] также совпадает с данным в пределах статистики. Возможно,
что разница в значениях, полученных с различными источниками,
объясняется обогащением смеси одним изотопом из-за различной летучести
изотопов.
Постоянство содержания U235 в природных урановых минералах
различного возраста может быть объяснено двояко: по одной гипотезе предполагается,
что во время образования земли содержание U235 в естественной смеси
изотопов было значительно больше, чем в настоящее время, и что теперь его
концентрация является остаточной от первоначальной. Согласно второй гипотезе,
10
Часть I. Уран как элемент
Таблица 4
ОТНОШЕНИЕ U238 : U235 В РАЗНЫЕ
ГЕОЛОГИЧЕСКИЕ ПЕРИОДЫ [7]
Возраст
минерала.
годы • 1СГ8
0
4
8
12
16
20
ц238 . у235
139,0
100,2
72,2
52,1
37,5
26,9
Отношение
активностей рядов
актиния и урана
0,046
0,065
0,089
0,123
0,171
0,238
принимают, что U235 образовался (и, возможно, образуется и сейчас) из U238,
например за счет ядерных реакций
U238 + п > U239 2fi v Pu239 ' , U235.
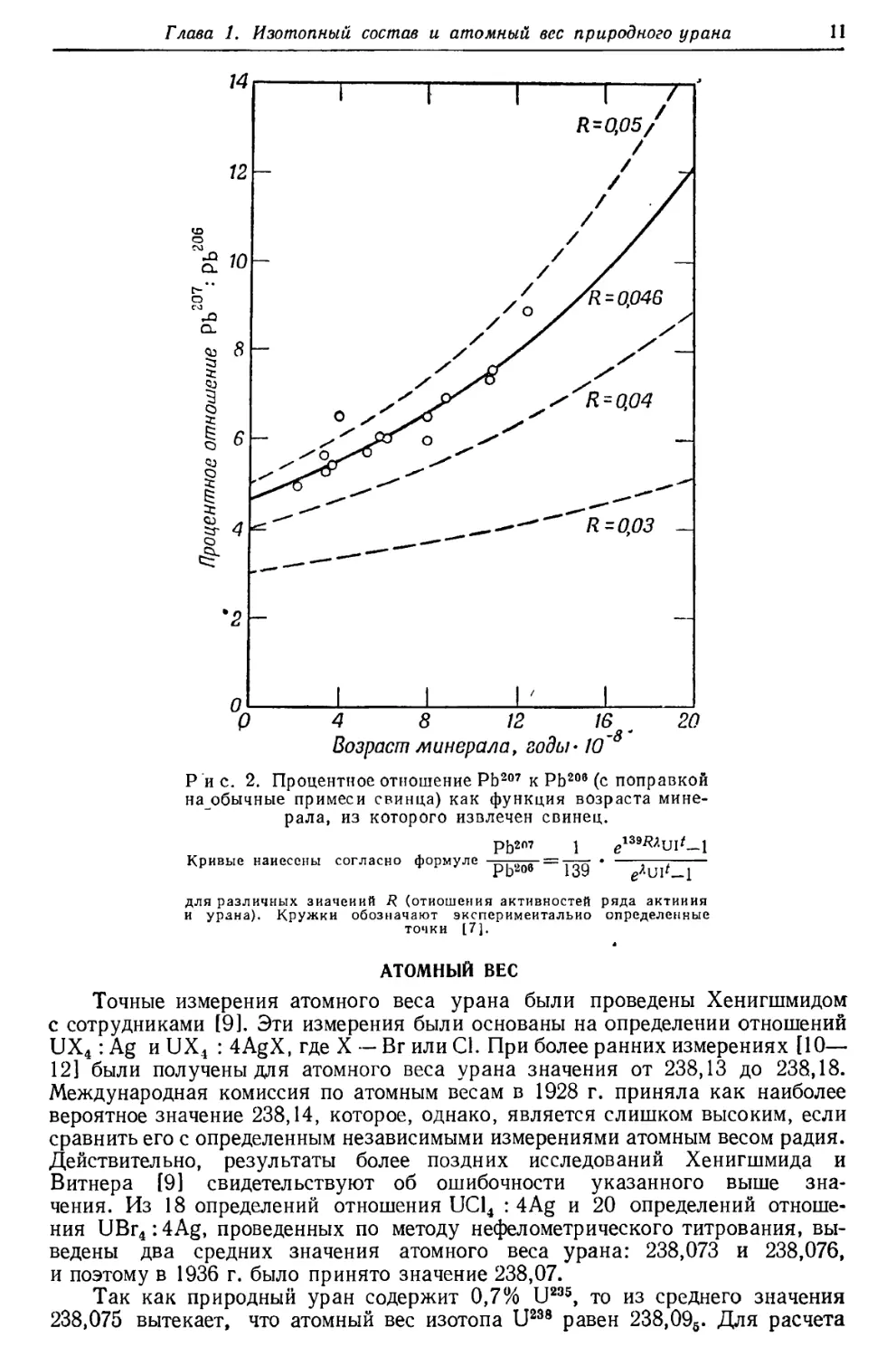
Первая гипотеза может быть проверена определением отношения урано-
свинца Pb206 (RaG) к актиносвинцу Pb207 (AcD) в урановых минералах
различного возраста. Это отношение
значительно меньше имеющегося в настоящее
время отношения U238 : U235 (5—10%
Pb207 по сравнению с 0,7% U235) и
увеличивается с возрастом минерала.
Результаты, полученные для минералов с возрастом
от 2-Ю8 до 13-108 лет [7],
соответствуют теоретической кривой,
основанной на современном отношении
активностей ряда Ас и ряда U, равном 0,046,
и на отношении концентраций U238 и U235,
равном 139. Из этих констант были
вычислены значения отношения U238 : U235
в прошедшие геологические периоды
(табл. 4).
Значения в последней графе табл. 4 меньше значений, полученных при
некоторых прежних подсчетах, в которых исходили из большего отношения
U238 : U235 и меньшей относительной активности ряда актиния.
Следовательно, роль ряда актиния в тепловой истории земли не так велика, как эю
предполагалось прежде.
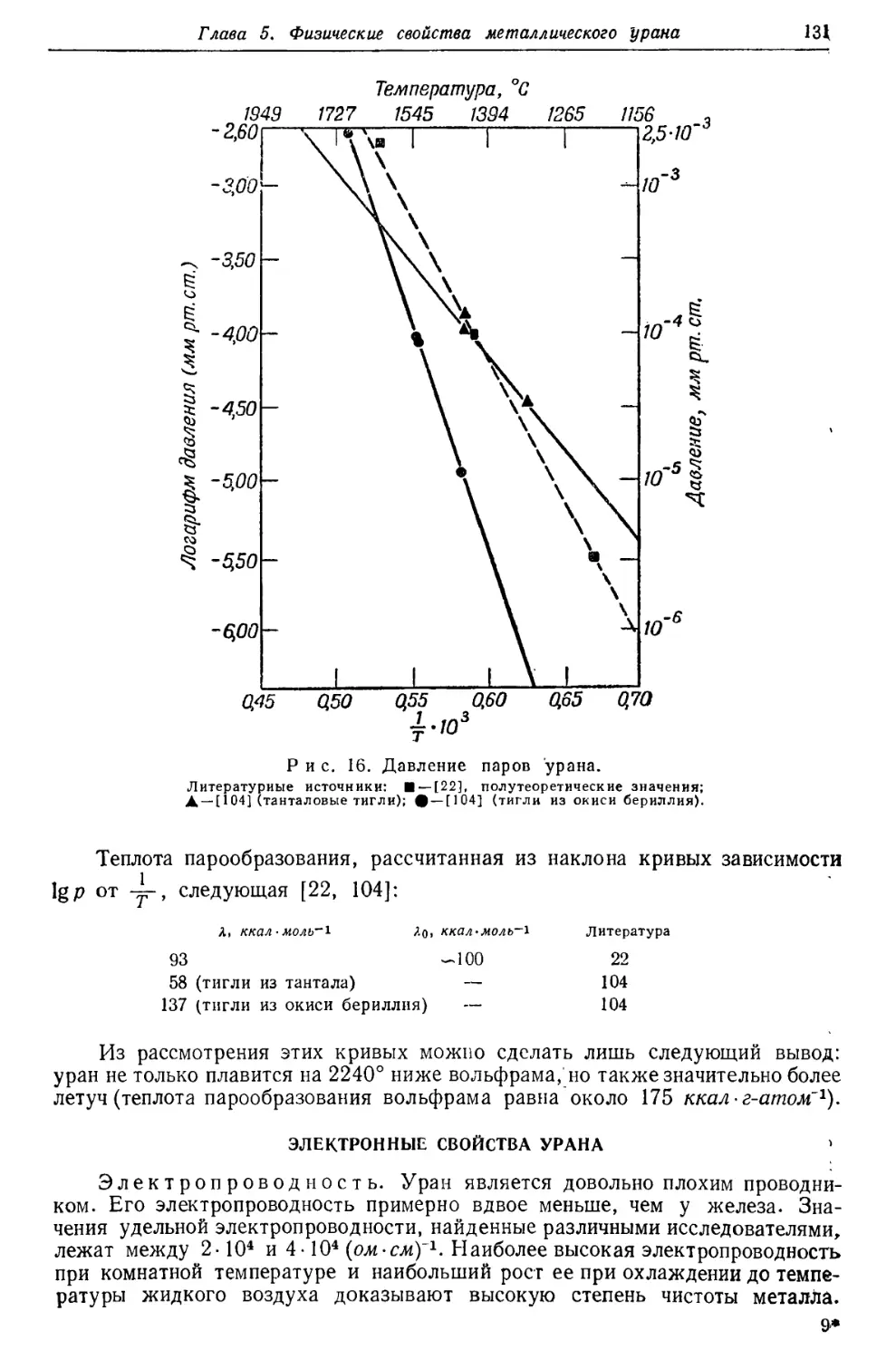
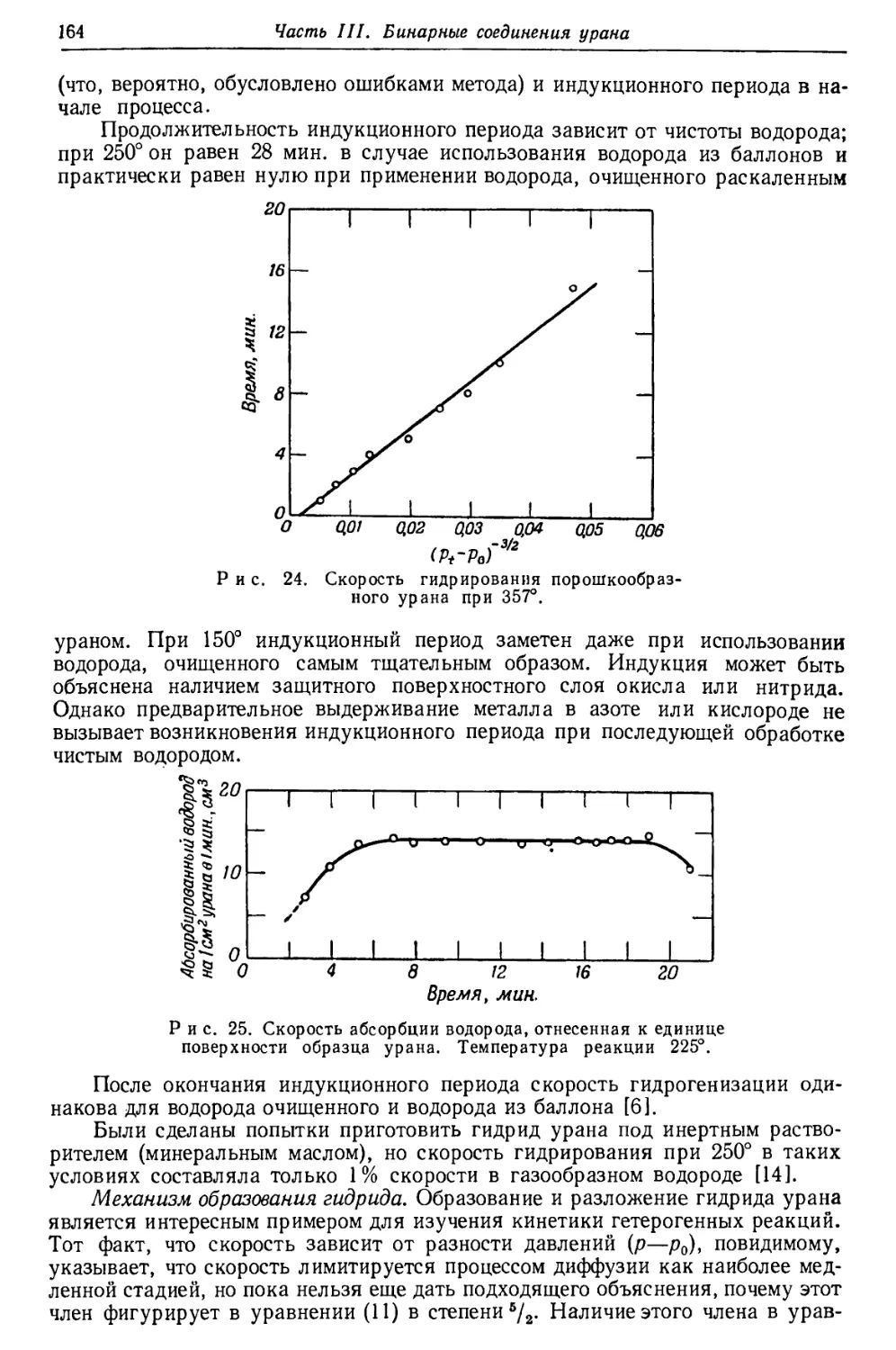
Для того чтобы полученные цифры были возможно ближе к отношениям
Pb207 : Pb206, найденным в минералах различного возраста, для современного
отношения активностей ряда актиния и ряда урана была выбрана
упомянутая выше величина 0,046 (рис. 2). На основании этой величины отношения
концентраций U235: U238, равного 1 : 139, и значения 4,5Ы О9 лет для периода
полураспада U238 выводится, что период полураспада U235 равен 7-Ю8 лет.
С другой стороны, прямые измерения активностей упомянутых двух рядов
приводят к отношению в 0,040, откуда вычисленный период полураспада U235
составляет 8, ЫО8 лет. Как видно на рис. 2, наблюдавшиеся отношения
Pb207: РЬ20в примерно на 20% выше вычисленных из отношения
активностей 0,040. Причина этого расхождения, которое может быть обусловлено
или избытком Pb2'J7, или недостатком Pb206, пока еще не ясна. Высказано
предположение, что экспериментальное значение отношения активностей
занижено [7]. Однако в более поздних работах [8] указывается, что
период полураспада U235, вероятно, равен 8,9-108 лет, откуда следует, что
отношение активностей должно быть даже ниже, чем 0,040.
В общем, результаты масс-спектрографических анализов урана и свинца
согласуются с гипотезой о первоначальном содержании U235b земле; вопрос
же о размерах дополнительного образования U235 в течение времени
существования земли остается открытым. Атомы U235 образуются всякий раз, когда
атомы U238 подвергаются действию нейтронов, которые всегда имеются в земле,
возникая при поглощении космических лучей, спонтанном делении ядер и по
реакциям (а, п). Очевидно, отсутствуют какие бы то ни было причины для того,
чтобы количество U235, образующегося в результате бомбардировки U238
нейтронами, было одинаковым для различных месторождений урана. Поэтому
доказательство образования U235 из U238 после затвердевания земной коры
может быть найдено именно в небольших отклонениях содержания U235
в различных минералах от постоянства, а не в наличии приблизительно
одинакового соотношения U235 : U238 во всех рудах (~0,7% U235).
Глава 1. Изотопный состав и атомный вес природного урана 11
'2
R=0№ -J
1
5
12
16
Возраст минерала, годы- Ю
20
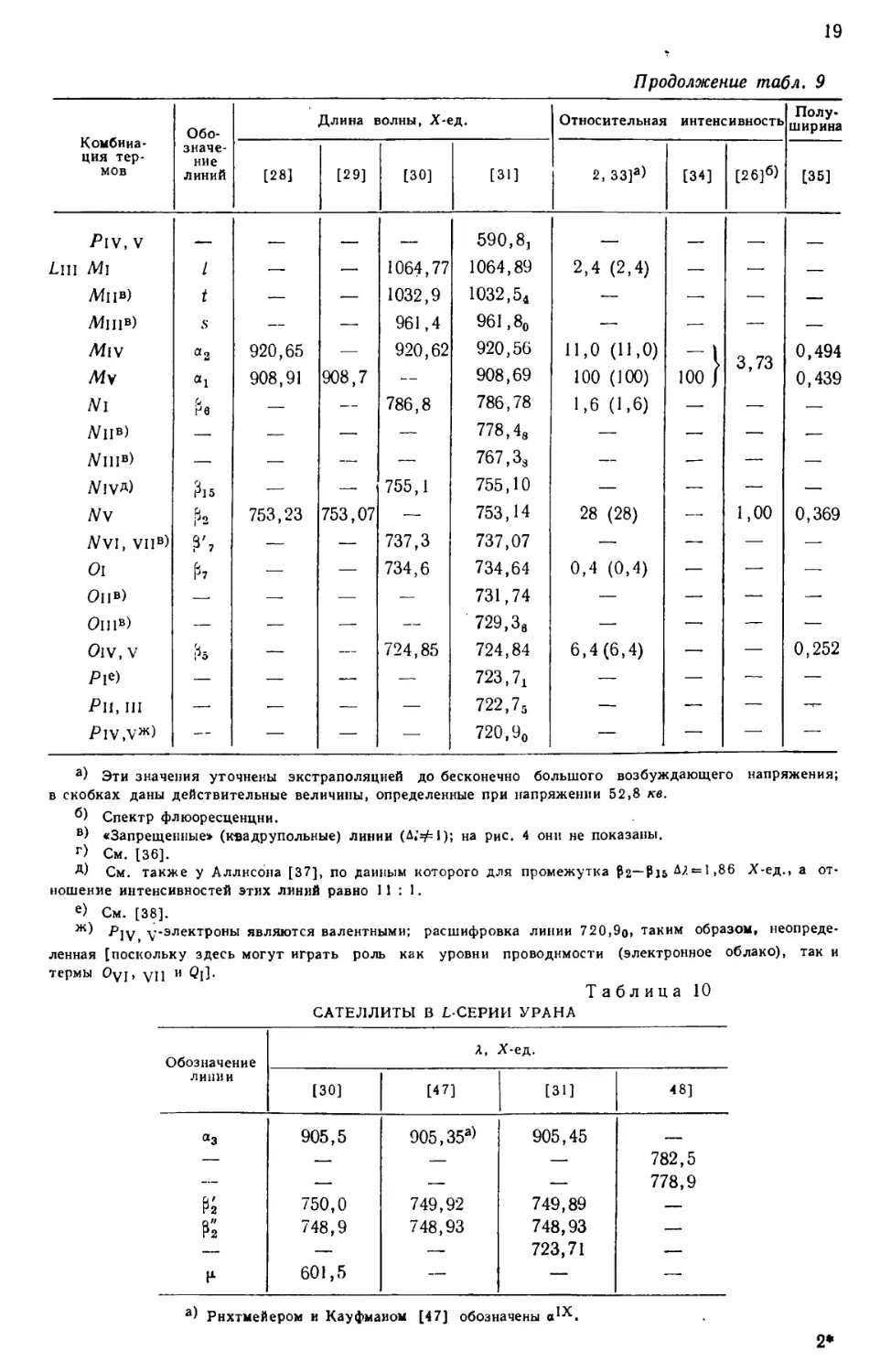
Р и с. 2. Процентное отношение РЬ207 к РЬ206 (с поправкой
на обычные примеси свинца) как функция возраста
минерала, из которого извлечен свинец.
РЬ207 1 e139i?Aui'_l
Кривые нанесены согласно формуле
РЬ2°б 139
eAui'_i
для различных значений R (отношения активностей ряда актиния
и урана). Кружки обозначают экспериментально определенные
точки [7].
АТОМНЫЙ ВЕС
Точные измерения атомного веса урана были проведены Хенигшмидом
с сотрудниками [9]. Эти измерения были основаны на определении отношений
UX4: Ag и UX4 : 4AgX, где X — Вг или С1. При более ранних измерениях [10—
12] были получены для атомного веса урана значения от 238,13 до 238,18.
Международная комиссия по атомным весам в 1928 г. приняла как наиболее
вероятное значение 238,14, которое, однако, является слишком высоким, если
сравнить его с определенным независимыми измерениями атомным весом радия.
Действительно, результаты более поздних исследований Хенигшмида и
Витнера [9] свидетельствуют об ошибочности указанного выше
значения. Из 18 определений отношения UC14 : 4Ag и 20 определений
отношения UBr4:4Ag, проведенных по методу нефелометр и чес ко го титрования,
выведены два средних значения атомного веса урана: 238,073 и 238,076,
и поэтому в 1936 г. было принято значение 238,07.
Так как природный уран содержит 0,7% U235, то из среднего значения
238,075 вытекает, что атомный вес изотопа U238 равен 238,09б. Для расчета
12
Часть /. Уран как элемент
упаковочного коэффициента этот «химический» атомный вес (основанный на
атомном весе 16,0000 для природного кислорода) должен быть умножен на
1,000272 для перевода в «физический» атомный вес (основанный на 16,0000
для О16). Это дает ифИ8з=238,16 и приводит к значению упаковочного
коэффициента
Pv = ^ = o,00067.
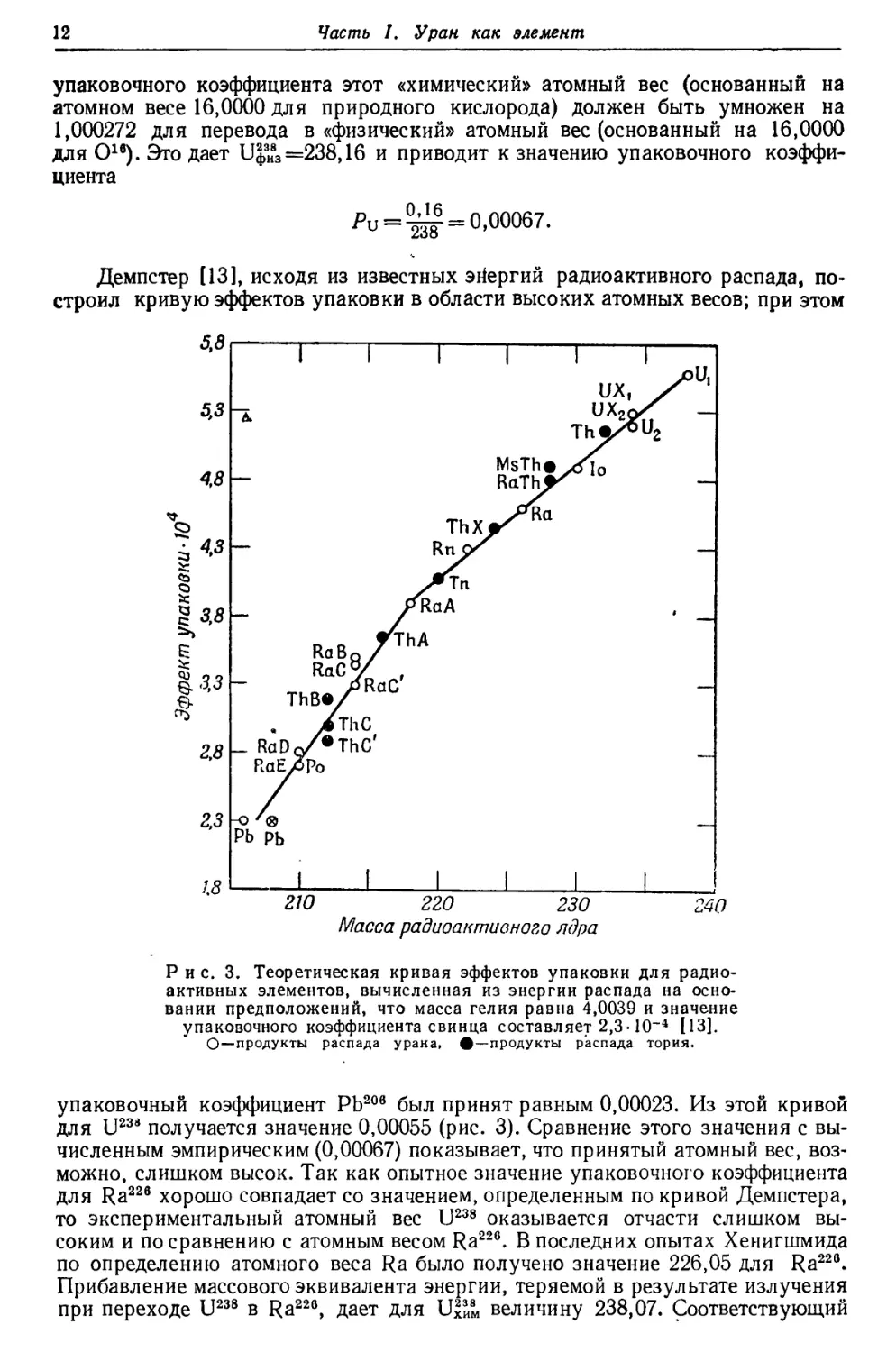
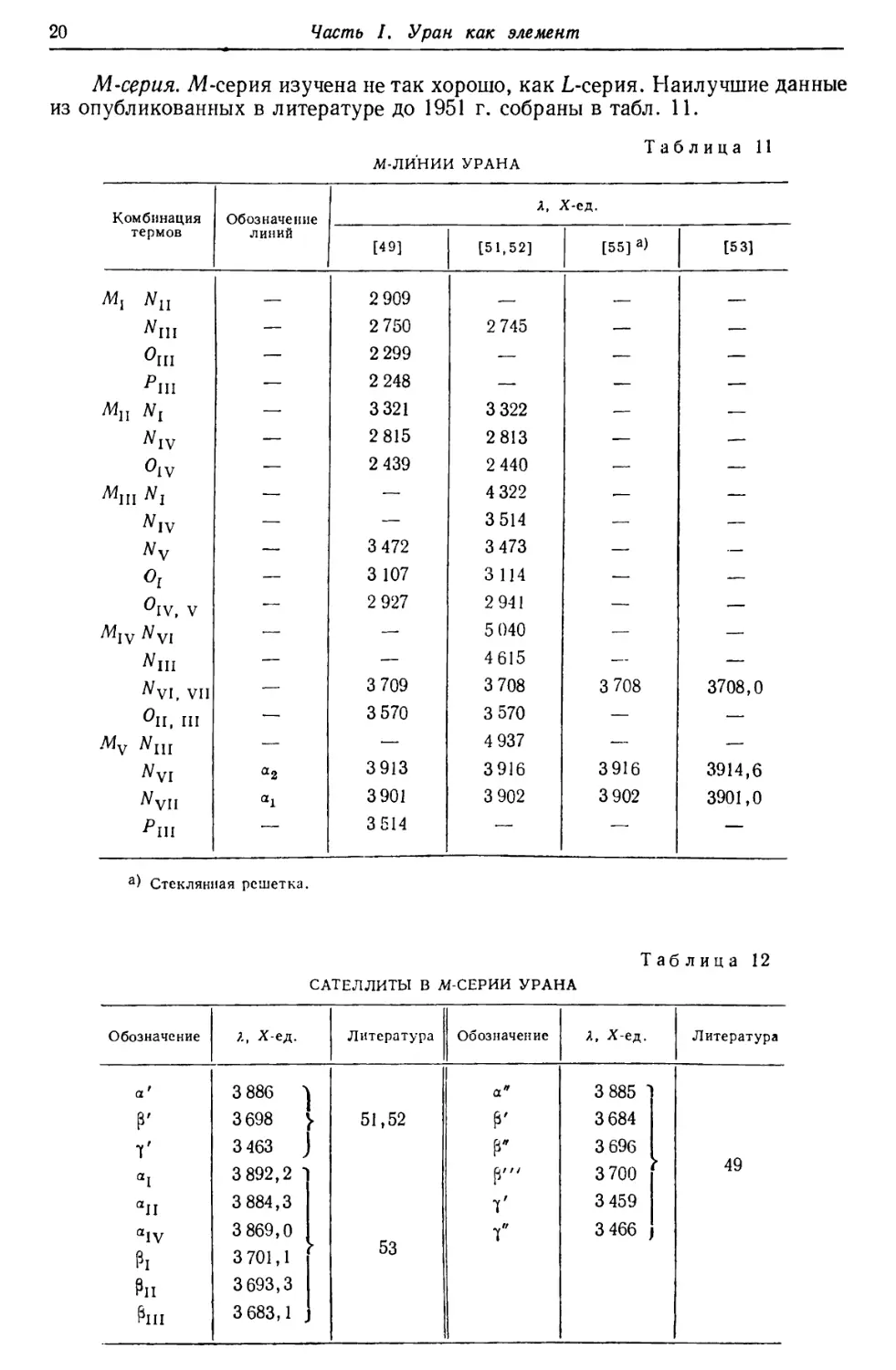
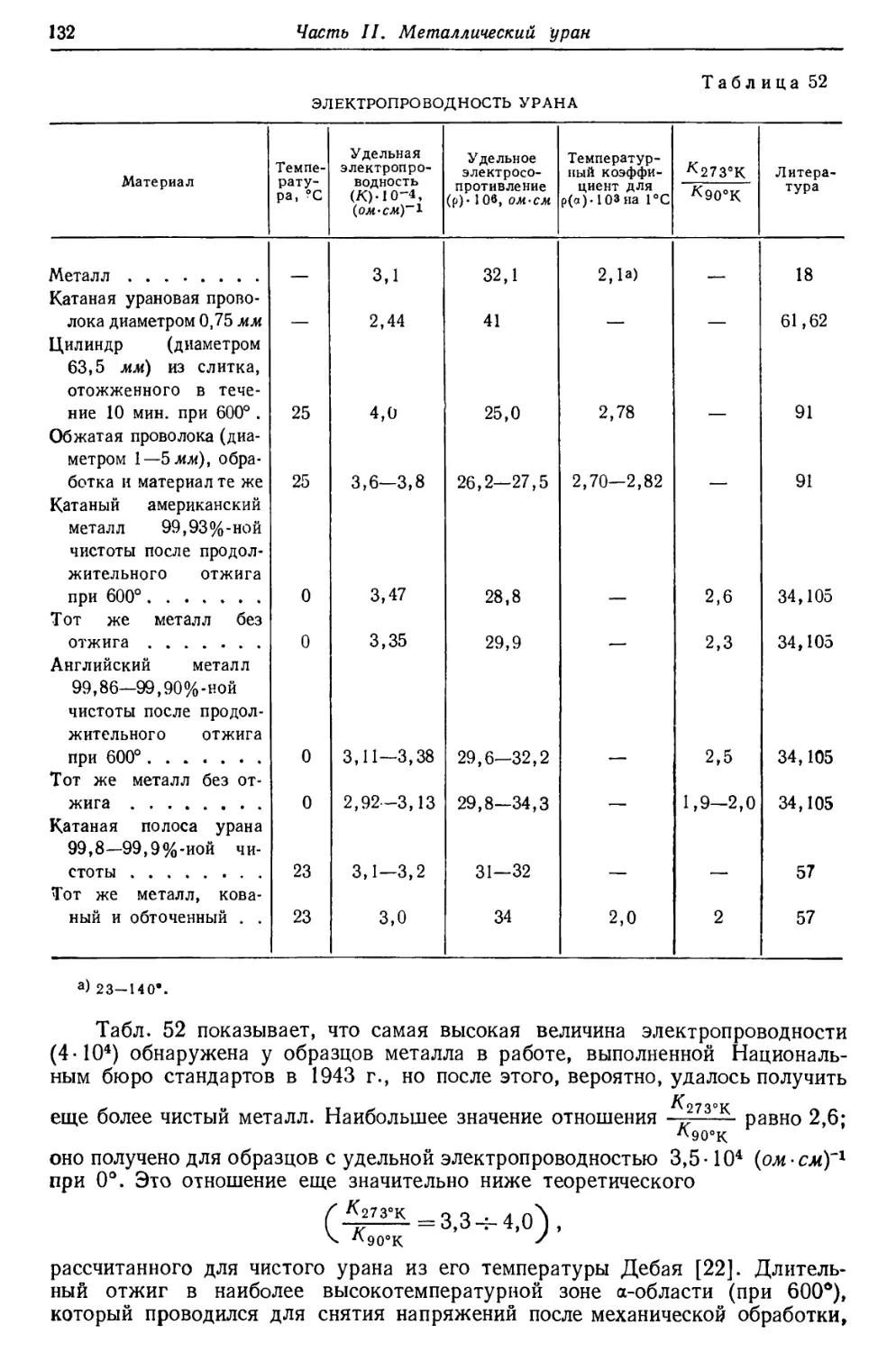
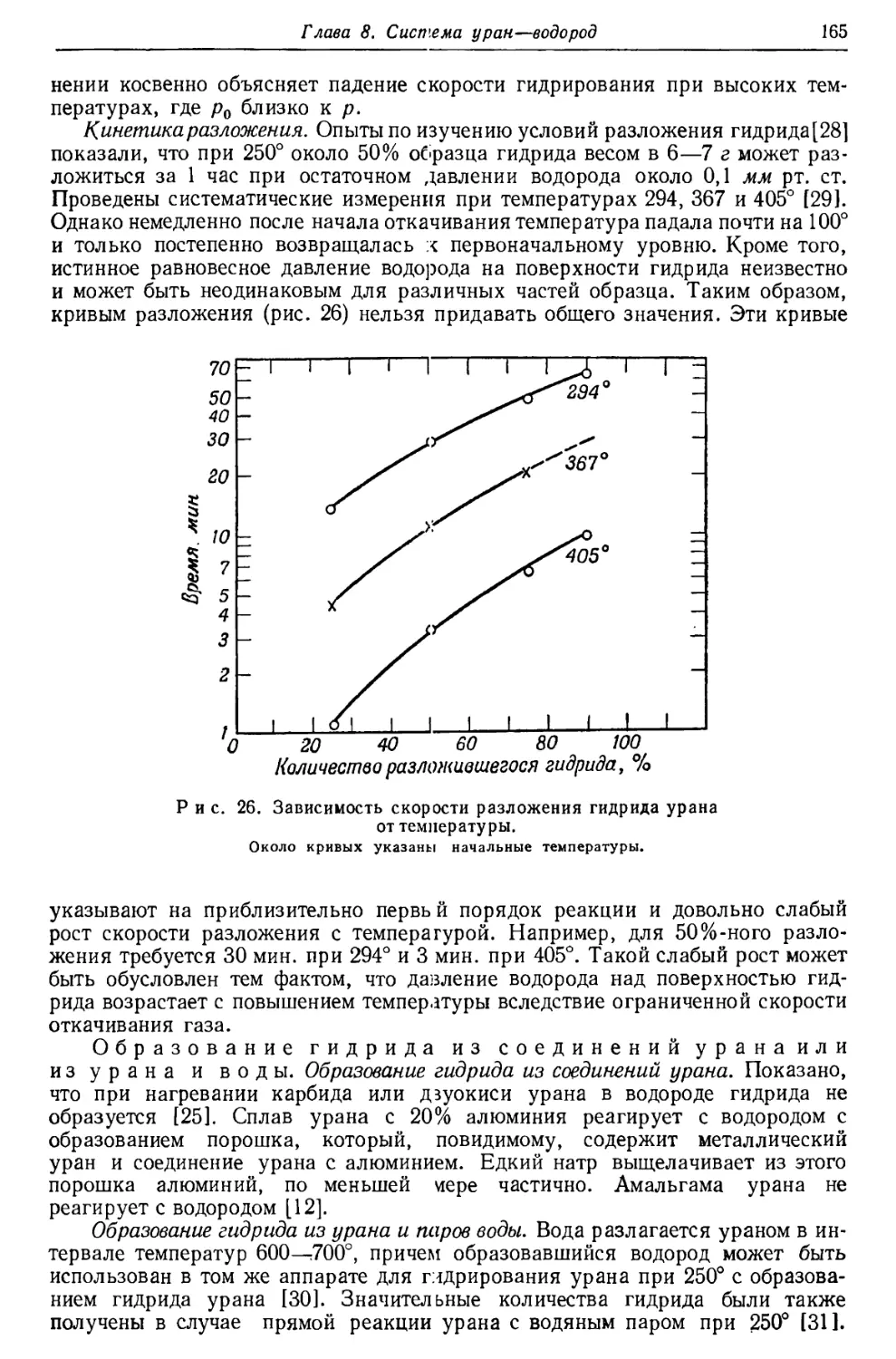
Демпстер [13], исходя из известных энергий радиоактивного распада,
построил кривую эффектов упаковки в области высоких атомных весов; при этом
210
220 230
Масса радиоактивного ядра
240
Рис. 3. Теоретическая кривая эффектов упаковки для
радиоактивных элементов, вычисленная из энергии распада на
основании предположений, что масса гелия равна 4,0039 и значение
упаковочного коэффициента свинца составляет 2,3-10~4 [13].
О—продукты распада урана, #—продукты распада тория.
упаковочный коэффициент РЬ20в был принят равным 0,00023. Из этой кривой
для U23d получается значение 0,00055 (рис. 3). Сравнение этого значения с
вычисленным эмпирическим (0,00067) показывает, что принятый атомный вес,
возможно, слишком высок. Так как опытное значение упаковочного коэффициента
для Ra22e хорошо совпадает со значением, определенным по кривой Демпстера,
то экспериментальный атомный вес U238 оказывается отчасти слишком
высоким и по сравнению с атомным весом Ra226. В последних опытах Хенигшмида
по определению атомного веса Ra было получено значение 226,05 для Ra225.
Прибавление массового эквивалента энергии, теряемой в результате излучения
при переходе U238 в Ra22e, дает для 1Л3И8М величину 238,07. Соответствующий
Глава U Изотопный состав и атомный вес природного урана 13
упаковочный коэффициент равен
п 238,07-1,00027—238 0,135 л ЛППС7
Ри = 238 = "238" = 0>00057-
ЛИТЕРАТУРА
1. National Nuclear Energy Series, Div. IV, vol. 17A.
2. Seaborg G. Т., P e r I m a n I., Revs. Modern Phys,, 20, 585*-667 (1948).
3. С h a m b e r 1 a i n O., W i 1 1 i a m s D., Y u s t e г Р., Phys. Rev., 70, 580
(194G).
4. F о x M.f Rustad В., Report. A-3828, Jan. 16, 1946 (SAM Columbia 1).
5. N i er A. O., Phys. Rev., 55, 150—153 (1939).
6. Dempster A. J., Nature, 136, 180 (1935).
7. N i er A. О , Phys. Rev., 55, 153 — 163 (1939).
8. С1 a r k F. L., Spencer-Palmer H. J., Woodward R. N., Report
BR 521, Oct., 1944 (British I).
9. H 6 n i g s с h m i d O. et al., Z. anorg. u. allgem. Chem., 226, 289 (1936).
10. H 6 n i g sc h m i d O. et al., Monatsh., 36. 51 (1915).
11. Honigschmid O. et al., Monatsh., 37, 185 (1916).
12. H 6 n i g s с h m i d O. et al., Z. anorg. u. aligem. Chem., 170, 145 (1928).
13. Dempster A. J., Phys. Rev., 53, 869—874 (1938).
Глава 2
СВОЙСТВА АТОМА УРАНА
РЕНТГЕНОВСКИЙ СПЕКТР
Являясь одним из наиболее тяжелых элементов, уран отличается очень
сложным рентгеновским спектром. Нейтральный атом урана в своем наиболее
низком энергетическом состоянии имеет целиком законченные электронные
оболочки К (2 электрона), L (8 электронов), М (18 электронов), N (32 электрона)
и частично заполненные оболочки О (21 электрон), Р (9 электронов) и Q (2
электрона). Распределение шести наружных электронов по группам 5/ (Ovi—vnj,
6d(P\V-v) и 7s(Qi) соответствует, вероятно, конфигурации f3ds2 (см. стр. 49).
Эти шесть электронов являются валентными электронами урана; в результате
их возбуждения получается оптический спектр. Остальные 86 электронов
представляют собой «внутренние электроны», и их возбуждение дает рентгеновский
спектр, который, таким образом, должен состоять из серий К, L, M, N, О и Р.
Однако линий, принадлежащих последним двум «ультрамягким» сериям, до
сих пор еще не обнаружено.
Рентгеновский спектр поглощения. В табл. 5
представлены некоторые результаты проведенных за последнее время опытов по
определению длин волн, соответствующих главным границам поглощения урана.
Измерения длин волн производились частично с металлическим ураном,
частично с двуокисью урана в качестве поглощающего вещества. Влияние
образования химического соединения и его природы на положение главных
/.-границ поглощения иллюстрируется данными табл. 6 [1]. Кроме определений,
в результате которых получены эти данные, измерения длин волн,
соответствующих главным границам поглощения, проводились также и другими
исследователями [2—11].
Таблица 5
ГРАНИЦЫ ПОГЛОЩЕНИЯ УРАНА
Обозначение
Кг
in {
ini {
Mi
Ми
Положение
границы
Х-ед.
106,58
568,0
568,2
591,3
590,7
720,8
720,7
2228
2385
4R
8550
1604,3
1603,7
1541,0
1539,5
1264,2
1264,3
408,9
382,1
Ширина,
Х-ед.
4,5
4,3
4,2
Литература
15
16
13
16
13
16
13
] 17 18,19
Обозначение
Mm {
Miv j
My j
Положение
границы
Х-ед.
2873
2877
3326
3327
3326
3491
3491
3491
\IR
317,18
316,7
273,99
273,9
273,99
261,03
261,03
261,03
Ширина,
Х-ед.
да)
13б>
Литература
20
21
20
21
14
20
21
14
а) От 3322 до 3331 Х-ед.
б) От 3:87 до 3500 Х-ед.
Глава 2. Свойства атома урана
1БГ-
Т абл ица 6
ТОНКАЯ СТРУКТУРА L-ГРАНИЦ ПОГЛОЩЕНИЯ УРАНА [1]
Граница
Минимум
и
U (металл) . . .
ио2
ио3
U (металл) ♦ . .
ио3
U (металл) . . .
UOo
ио3
568,28
568,10
568,04
590,71
590,62
590,50
1 720,76
720,64
720,54
—
—
590,17
590,12
590,00
1 720,17
720,1G
720,0.,
—
—
Lu
589,7
589,7
589,4
Liii
719,50
719,50
719,30
—
—
589,о
588,7
588,8
718,7
718,e
718,5
—
—
588,5
588,3
588,х
1 717,8
717,0
717,7
587,з
586,9
586,7
716,з
716,2
716,,
586,2.
585, в
586,!
714,7-
714,Б
714,9
Тонкая структура границ поглощения зависит от химического состояния,
урана, и результаты ее изучения могут служить для анализа связей,
валентных электронов в кристалле. Было отмечено [12] существование
«белой линии» у Lin-границы. Впоследствии подобную линию удалось
обнаружить как у Lur, так и у Ln-границ поглощения металлического урана (с
длиной волны соответственно 720,4 и 590,4 Х-ед.); у Li-границы белой линии не
было замечено [13]. Для М\\- и AIv-грапиц поглощения в случае двуокиси
урана тонкой структуры не наблюдалось [14].
Первые подробные исследования тонкой структуры Lu- и Lm-границ.
поглощения урана были выполнены Манеску [1 ]. Из табл. 6 следует, что у этих
границ поглощения со стороны коротких волн имеется сначала минимум (белая
линия) и затем несколько максимумов (черные линии) а, р и ?, разделенные
минимумами А и В. В пределах первого минимума должно лежать
несколько линий, которые соответствуют электронным переходам из группы 2р в
частично заполненные наружные s- и d-группы; и действительно, в спектре
металлического урана, полученном с большей дисперсией, была обнаружена
тонкая структура белой линии, соответствующая этим переходам.
Длины волн, указанные в табл. 6 для двуокиси урана, являются
типичными для тех соединений урана (IV), которые исследовались Манеску; сюда
относятся, помимо двуокиси, кристаллические фосфат и сульфат урана (IV).
Точно так же приведенные в таблице данные о спектре трехокиси урана
типичны для исследованных соединений урана (VI); к ним относятся нитрат,
ацетат и сульфат уранила.
Кроме смещения всех L-границ поглощения в сторону коротких волн
в ряду U (металлический)—>U(IV)—>U(VI) (что предсказывается теоретически),
было также установлено, что положение и относительная интенсивность линий
а, р и | определенным образом изменяются с изменением валентности. Так,
например, в Lin-границе поглощения для соединений урана (IV) а-линия
является резкой и интенсивной, р-линия — сравнительно слабой и -(-линия—
хотя и заметной, но диффузной. В случае трехокиси урана и солей уранила
а-линия тоньше и слабее, чем в первом случае (уран (IV)), (3-линия
интенсивнее и 7-линия сдвинута ближе к главной границе.
16
Часть I. Уран как элемент
Манеску изучил также рентгеновские спектры поглощения водных растворов
солей урана и установил, что они совершенно подобны спектрам
соответствующих твердых гидратированных солей. В спектрах, создаваемых ионами урана,
находящимися в растворе, можно отметить наличие тонкой структуры,
занимающей полосу энергии шириной примерно 200 эв\ это является свидетельством
упорядоченного (псевдокристаллического) расположения молекул воды вокруг
иона.
Коэффициенты поглощения ураном монохроматических рентгеновских
лучей различных длин волн в области L-границ поглощения определены Стоу-
нером и Мартином [10], в областях /С- и TW-границ поглощения—Алленом [22];
результаты этих определений представлены в табл. 7. Коэффициент
поглощения падает в 2,9 раза у /(-границы поглощения [23] и в целом в 3,52 раза у трех
L-границ поглощения [10,25]. По данным Кюстнера [25], у границ Lu Lu и
Lm поглощение уменьшается соответственно в 1,11, 1,31 и 2,17 раза. Скачок
поглощения у Lm-границы составляет2,27 [26], у Му-границы—2,4раза [14],
Таблица 7
МАССОВЫЕ КОЭФФИЦИЕНТЫ ПОГЛОЩЕНИЯ (|х/р) РЕНТГЕНОВСКИХ
ЛУЧЕЙ МЕТАЛЛИЧЕСКИМ УРАНОМ
А, Х-ед.
64
72
98
107,5
107,5
130
175
200
400
450
500
550 !
600 1
631
708
7о0
800
Ю80
ц/р. см*1г [23]
1,80
2,25
3,90
4,65 К-граница
1,62
2,10
3,95
5,40
Относительные единицы [10]
0,34
0,45
0,59
0J7 Li, н-граиицы
0,64
0,73
/,00 Lin-граница
0,48
0,56
И/Р. см21г [22]
™
Я, Х-ед
]1
1240
1450
154„
1800
1930
1970
206о
2180
' 2240
235»
249„
2600
2970
3040
318„ |
3220
337„
3440
*-660 1
3740
3930
ц/р, смЩ [22]
190
285
360
442
470
485
495
516 Mi -граница
525
540 Ми-граница
560
570 Мш-граннца
600
600
600
600 Miv-граница
580
564 Mv-граница
410
424
490
По данным Аллена [22], коэффициент поглощения урана пропорционален
X2»92 в ^-области и X2'6 в Af-области.
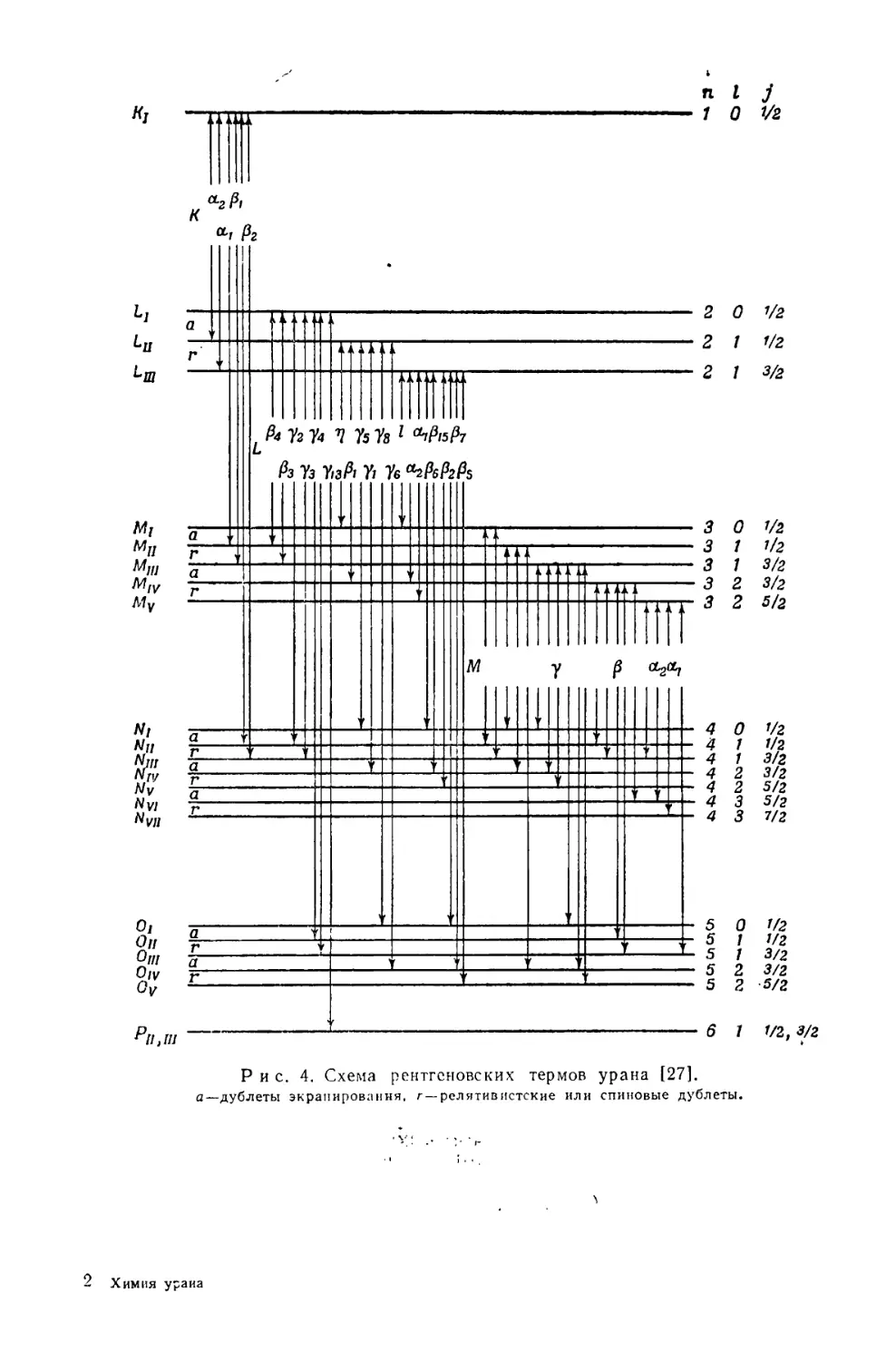
Характеристический рентгеновский спектр.
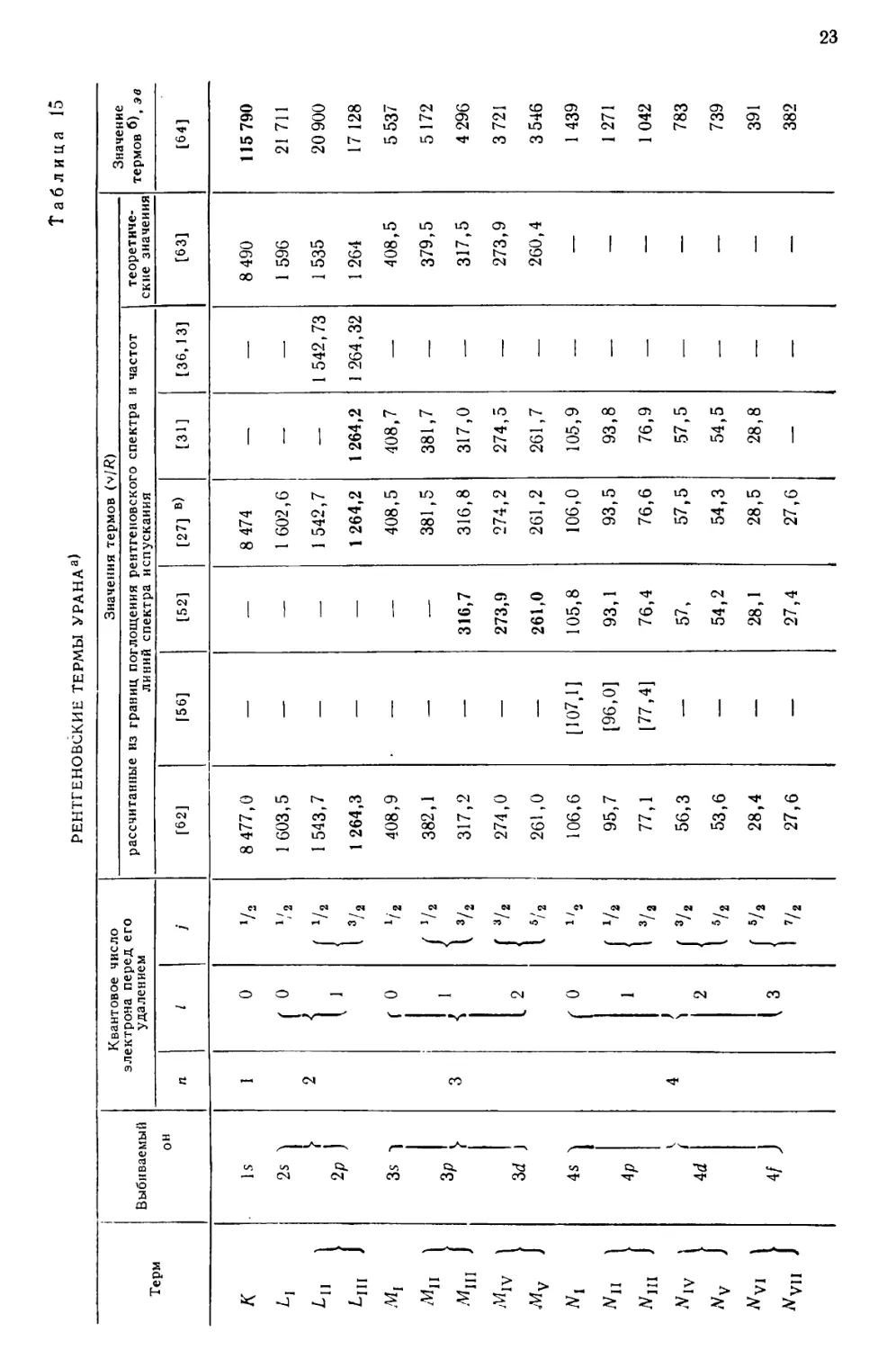
В рентгеновском спектре урана измерены линии, принадлежащие четырем
сериям—К, L, М и Л/. На рис. 4 дается объяснение возникновения наиболее
важных линий первых трех серий [27]. В табл. 8—13 приведены значения для
длин волн всех измеренных линий.
ч
Mi
МИ
My
N,
N11
ft"
w
Nvi
Oi
On
От
Oiv
Ov
n I j
1 0 1/2
*2Pt
11,111
CLj
я 11 1
г M
a \
r
a
r
a
r
ft
l| |i 1 1 11 1
1 АГгГ4 V 7s'
ЛГзУ/зАГ»
IT
I] T 1 1 1 11 i Ml
Y 1 1 11 1
)| Mill т I I
II Mill Y1
Y Y 1 11 1 1
\ III 1 1
a in у
г
a
г
a y j
Г Y
a
r
1
•
11
1 1 ill 111111
Ys l ЪМг
Ув «2ftAft
11 yI1111111
|| 11111 f|j ||
II llllllll 11 т t f
II tIIIIIII 1 1 1 1 1 ftt 1 f
11 il 1 1111 1 1 1 1 1 1 1 1 1 1
Г У
11 III Y Y 1 | |
II II II II ill 1
II III1' Y II 1 II 1
{ T M
T T 1 T
Y 71 Y T
Y
i
11 411
1111
/3 <4h
1 T || 1 1 || 1
T 1 1 1 T 1 1 1
It t
II r |
Y
T Y
\
2 0 1/2
2 1 1/2
2 / 3/2
3
3
3
3
3
0
7
1
2
2
1/2
1/2
3/2
3/2
5/2
4
4
4
4
4
4
4
0
1
1
2
2
3
3
1/2
1/2
3/2
3/2
5/2
5/2
7/2
5
5
5
5
5
0
/
/
2
г
1/2
1/2
3/2
3/2
5/2
6 1 1/2, 3/2
Рис. 4. Схема рентгеновских термов урана [27].
а—дублеты экранирования, г—релятивистские или спиновые дублеты.
Химия ураиа
18
Часть I, Уран как элемент
Таблица 8
К-ЛИНИИ УРАНА [24]
К-серия. Длины волн /(-линий урана приведены в табл. 8.
L-серия. Длины волн, интенсивности и
величины полуширины L-линий урана
приведены в табл. .9.
Первоначальные измерения L-линий
эмиссионного рентгеновского спектра см.
[17, 18, 39—45]. Данные этих работ
суммированы Дьюном [8,46]. Кроме линий
рис. 4, представленных в табл. 9,
обнаружено также несколько сателлитов,
которые обычно интерпретируются как
«искровые линии» (соответствующие переходам
при двойной ионизации) (табл. 10).
Ширина L-линий рентгеновского спектра испускания урана
определялась Вильямсом [35], результаты этих измерений приведены в последней графе
табл. 9.
Комбинация
термов
KiLu . . . .
Lin ....
Ми, ш . . .
Nu, ш . . .
Обозначение линии
2
1
i;3
2
А, Х-ед.
130,95
126,40
111,87
108,42
Таблица 9
Комбинация
термов
и м i
Мш
MivB)
MvB)
Nu
Nm
NivB)
NvB>
On
Oin
Oiv, vb>
Pu, in
Ln M{
Miiib)
Miv
A'i
Nm
Niv
Nvi
0
Ощв)
Oiv
Pu,m*)
значение
линий
h
h
PlO
p*
72
7з
—
—
7 4
74
—
7i3
-ц
Pit
Pi
7s
—
7i
—
78
—
7e
—
L-ЛИНИИ
УРАНА
Длина волны, Х-сд.
[28]
—
—
—
—
—
—
—
~
—
—
—
—
—
718,55
—
—
613,32
—
—
—
—
—
[29}
—
—
—
—
—
—
—
—
—
—
—
—
—
718,51
—
—
613,59
—
—
—
—
—
[30]
764,4
708,79
685,3
679,5
603,9
597,1
—
—
575,3
573,6
—
568,9
803,5
—
—
634,2
—
—
—
599,7
—
593,4
—
134
746,39
708,81
686,15
679,6
603,99
597,30
589,77
588,65
575,45
573,90
571,3
569,4Х
803,59
743,47
718,54
634,23
621,54
613,50
601,9а
600,00
596,04
593,61
592,8Г>
591,9
Относительная
[32, 33]а)
4,1 (3,2)
4,2 (3,3)
—
—
1,5 (1,1)
1,4(1,1)
—
—
0(0)
0(0)
—
—
1,0 (0,83)
—
49,4(40,5)
0(0)
—
12 (9,7)
_
—
—
2,2 (1,8)
—
интенсивность
[34]
~г
—
—
—
—
—
—
—
—
—
—
—
—
—
36,7
—
—
10,9
—
—
—
—
—
[26]6)
~
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
Полуширина
[35]
0,726
0,382
—
—
0,57
0,47
—
—
—
—
—
—
—
—
0,299
—
-
0,242
—
—
—
оГгзз
—
19
Продолжение табл. 9
Комбинация
термов
Piv, v
Lm Ml
Ml IB)
Мщв)
Miv
Mv
JVi
yViiB)
iVlHB)
iVlVA)
Nv
Nvi, vnB)
Oi
Оцв)
0111»)
Oiv, v
Pie)
Pli.III
PlV.V^
значение
линий
—
/
t
s
«2
«1
i^6
—
—
?15
(Bo
¥l
fc
—
—
f*5
—
—
—
Длина волны, Х-ед.
[28]
—
—
—
920,65
908,91
—
—
—
—
753,23
—
—
—
—
—
—
—
—
[29]
—
—
_
908,7
—
—
—
_ ,
753,07
—
—
—
—
___
—
—
— ,
[30]
—
1064,77
1032,9
961,4
920,62
786,8
—
—
755,1
—
737,3
734,6
—
—
724,85
—
—
—
[31]
590,8j
1064,89
1032,54
961 Л
920,56
908,69
786,78
778,48
767,33
755,10
753,14
737,07
734,64
731,74
729,3e
724,84
723,7X
722,75
720,90
Относительная
t 2, 33]a)
—
2,4 (2,4)
—
—
11,0 (11,0)
100 (100)
1,6 (1,6)
—
—
—
28 (28)
—
0,4 (0,4)
—
—
6,4(6,4)
—
—
— ]
интенсивность
[34]
—
—
—
—
100 /
—
—
—
—
—
—
—
—
—
—
—
—
[26]б>
—
—
—
—
3,73
—
—
—
—
1,00
—
—
—
—
—
—
—
- 1 - 1
!
Полуширина
[35]
—
—
—
0,494
0,439
—
—
—
—
0,369
—
—
—
—
0,252
—
—
—
a) Эти значения уточнены экстраполяцией до бесконечно большого возбуждающего напряжения;
в скобках даны действительные величины, определенные при напряжении 52,8 кв.
") Спектр флюоресценции.
в) «Запрещенные» (квадрупольные) линии (Мф\); на рис. 4 они не показаны.
г) См. [36].
д) См. также у Аллнсона [37], по данным которого для промежутка р2—Pis Д-Я = t ,86 Х-ед., а
отношение интенсивностей этих линий равно 11:1.
е) См. [38].
ж) Я]у у-электроны являются валентными; расшифровка линии 720,9о, таким образом,
неопределенная [поскольку здесь могут играть роль как уровни проводимости (электронное облако), так и
термы Oyj, уп и Q|].
Таблица 10
САТЕЛЛИТЫ В L-СЕРИИ УРАНА
Обозначение
линии
аз
—
—
Pi
К
—
Р
[30]
905,5
—
_
750,0
748,9
—
601,5
я,
[47]
905,35а)
—
—
749,92
748,93
—
—
Х-ед.
[31]
905,45
—
—
749,89
748,93
723,71
—
48]
—
782,5
778,9
—
—
—
—
1) Рнхтмейером и Кауфманом [47] обозначены о
2*
20
Часть /. Уран как элемент
М-серия. М-серия изучена не так хорошо, как L-серия. Наилучшие данные
из опубликованных в литературе до 1951 г. собраны в табл. 11.
Л1-ЛИНИИ УРАНА
Таблица И
Комбинация
термов
Mi Nn
*ш
От
Рщ
Ми Л/,
N1V
01V
Мш Nt
NiV
Nv
°l
°IV, V
Mlv Nvl
Nm
Nyl, VII
°II, HI
Щ Nm
Nvi
NW
Piu
Обозначение
линий
—
—
! —
—
—
—
—
—
__
—
—
—
—
—
—
—
a2
ai
—
[4 9]
2 909
2 750
2 299
2 248
3 321
2815
2 439
—
—
3 472
3 107
2 927
—
—
3 709
3 570 1
— !
3913
3 901
3514
Я, Х-ед.
[51,52]
2 745
—
—
3 322
2813
2 440
4 322
3 514
3 473
3114
2 941
5 040
4615
3 708
3 570
4 937
3916
3 902
—
[55] a)
—
—
—
—
—
—
—
—
—
—
—
—
—
3 708
—
—
3916
3 902
—
[5 3]
—
—
—
—
—
—
—
—
—
—
—
—
—
3708,0
—
—
3914,6
3901,0
—
a) Стеклянная решетка.
Таблица 12
САТЕЛЛИТЫ В Af-СЕРИИ УРАНА
Обозначение
а'
Р'
ап
Pi
Рп
Рш
Я, Х-ед.
3 886 ^
3698 }
3 463
3 892,2 "
3 884,3
3 869,0
3 701,1
3 693,3
3 683,1
1
"
Литература
51,52
53
Обозначение
а"
Р'
Р"
Р'"
Г
ч"
Я, Х-ед.
3 885 ]
3 684
3 696
3 700
Г
3 459
3 466 J
Литература
49
Глава 2, Свойства атома урана
21
Результаты первоначальных определений длин волн [49, 50] суммированы
Дьюном [8, 46]. В TW-серии также были измерены сателлиты (искровые
линии) [51—53] (табл. 12). Шесть линий /И-серии урана были использованы
в измерениях полуширины линий на рентгенограмме кристалла
кальцита [54].
N-серия. Единственные имевшиеся к моменту написания этой книги
данные [49, 56] являются ненадежными, тем более что интерпретации
двух исследователей в некоторых случаях расходятся о(табл. 13). Несколько
«ультрамягких» линий с длиной волны большей, чем 30А, измерены Зигбаном
и Магнуссоном [57].
Таблица 13
W-ЛИНИИ УРАНА
Комбинация термов [56] а)
*10ц WiAv)
Ощ Wi Ош)
^11 (*1 Рщ)
Рщ
NuOl
01V
Ov WmOv)
NmOv
Pi (NmPu)
NlvNvl
NVNVl,VU
^VI°IV
Nvifiv
wVIov
Длина волны, Х-ед.
[4 9]
10 385
9619
8 691
—
—
—
12 874
—
12 250
—
—
—
—
—
[56]
—
9 873
8 704
8 605
12850
1 008o
—
1 270o
—
—
—
—
—
—
[57]
—
—
—
—
—
—
—
—
—
3 1780
348)0
4 208o
—■
4 3280
a) В скобках дана расшифровка Хальмара [4 9].
Флюоресценция. Флюоресцентное испускание L- и М-линий в
рентгеновском спектре урана было изучено Стефенсоном [26], а также Хевеши и Лэем
[58, 59]. Стефенсон установил, что при поглощении в 1ш-уровне
выход флюоресценции составляет 0,67. Отношение интенсивности линий
Lau2 и L$2 в спектре флюоресценции составляет 3,73, что согласуется
с величиной, найденной для характеристического эмиссионного
рентгеновского спектра. По Хевеши и Лэю [58, 59], выход флюоресценции ниже (0,45
в Lin-уровне и 0,06 в Af-уровнях).
Фотоэлектрический эффект. Испускание ураном
электронов при облучении его рентгеновскими лучами наблюдалось де-Бройлем [60]
и Робинсоном [61].
Изучая поведение в магнитном поле электронов, испускаемых двуокисью
урана под действием CuKa-изл учения, Робинсон нашел среди различных
электронных групп урана 10 таких, появление которых может быть отнесено
за счет фотоэлектрического эффекта (табл. 14).
22
Часть. I. Уран как элемент
Т а б лица
ФОТОЭЛЕКТРОННАЯ ЭМИССИЯ ОКИСИ УРАНА ПРИ ОБЛУЧЕНИИ
CuKq-РЕНТГЕНОВСКИМИ ЛУЧАМИ а)
Группа
1
2
3
4
5
6
7
8
9
10
Л
Интенсив*
ность б)
3
4
5
2
3
5
6
2-3
. 3
о
2—
Энергия
электронов
276,9
318,8
332,4
486,8
500,3 |
517,3
540,4
555,8
566,8
577,1
589,
Потеря
энергии
(592,8—v/R)
315,9
274,0
260,4
106,0
92,5
75,5
52,4
37,0
26,0
15,7
2,9
Соответствующий рентгеновский
терм
обозначение
Miv
Miv
Mv
"г
"и
А'ш
^IV, V
Кислород К (?)
^VI, VlA
°и, ш
°iv, vp
v/K
317
274
261
106
93
76
57; 54
27
18; 15
7; 2
а) Энергия vo/K=592,8.
б) Произвольная шкала от 1 до 6.
Система термов. Ввиду того что границы поглощения в
рентгеновском спектре не резки, обычно для расчета рентгеновских термов выбирают
одну или несколько наиболее точно известных границ поглощения и сочетают
их с гораздо более резко обозначенными частотами испускаемых линий.
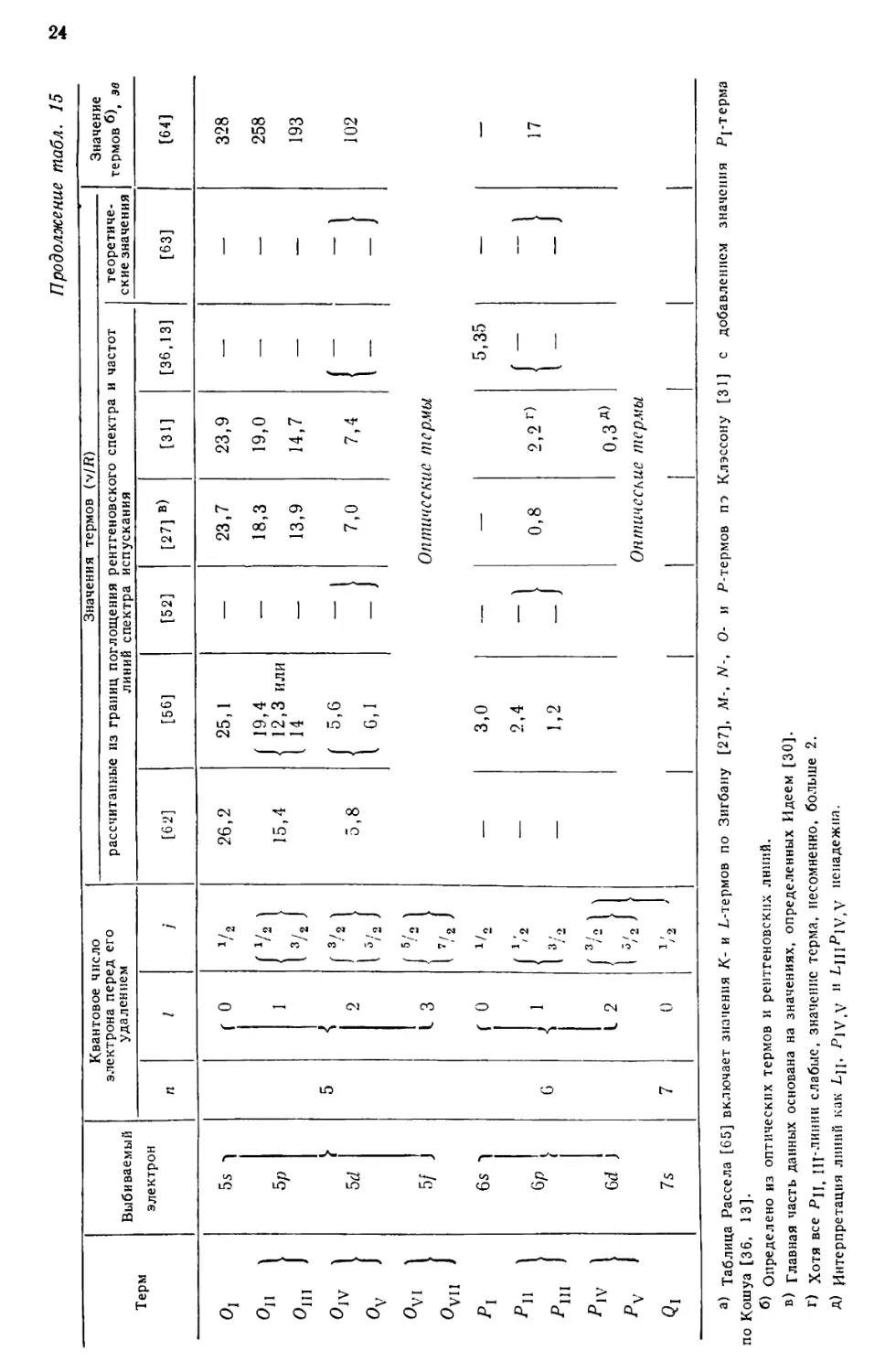
Полученные таким образом данные [13, 27, 31, 36, 52, 56, 62] приведены в табл. 15.
Основные значения, полученные из измерений спектров поглощения, даны
жирным шрифтом. Величины первоначальных измерений, взятые за основу при
последующих расчетах, заключены в скобки. Предпоследняя графа содержит
значения термов, рассчитанные [63] из общих уравнений термов с
использованием констант экранирования, значения которых приведены в табл. 16;
в последней графе даны величины термов (в электрон-вольтах), полученные [6,4]
путем сочетания частот рентгеновских линий со значениями оптических термов
(какие оптические значения были использованы в случае урана, не ясно).
Величина /(-терма, представленная в последней графе, рассчитана по границе
поглощения, так как в этом случае расхождение между результатом
непосредственных измерений поглощения (v//?==8550, см. табл. 5) и значением,
полученным из L-границы и /(-линий спектра испускания (v//?=8477), может
рассматриваться как результат ошибок в измерениях /(-линий.
Рассел [65] попытался с помощью значений термов, приведенных в табл. 15,
установить, какая из валентных электронных групп, P\v,v или Qvi.vii,
имеет более низкую энергию, иными словами, заполняются ли сначала Ы-
или 5/-подуровни в атоме урана. Он приводит два довода в пользу
предположения, что у элементов, стоящих за торием, 5/-электроны связаны сильнее,
чем 6й(-электроны. Прежде всего разность термов 0\—Оц (5s—Ър) для урана
лишь немногим больше, чем для тория [A(v/i?) равна 4,9 для Th и 5,0 для U].
В предшествующем периоде периодической системы увеличение разностей
Ni—Nu (т. е. 4s—4р) в ряду Hf—Pt (где достраивается 5я(-подуровень)
несколько больше и сохраняет приблизительно постоянное значение (около2^//?-еди-
ниц на элемент). В ряду редкоземельных элементов, у которых происходит
Таблица 15
РЕНТГЕНОВСКИЕ ТЕРМЫ УРАНА3)
Терм
к
h
L" 1
Чи J
Mt
Л«11 J
^JII 1
^1V 1
•My i
"l
<VIV 1
*v J
i
Выбиваемый
он
Is
2s \
2p)
3s ^
3pl
3d J
4s ^
4p
Ad
4/ .
Квантовое 1
электрона пер
удаление
n
1
2
3
4
/
0
(°
t,
\ j
f о
<
>
1
1
. 2
f 0
1
2
ч 3
1НСЛО
ед его
м
/
Vs
Vi
| V,
u
V.
Г'1
U
{V.
1 */.
!'s
IV.
1 3u
1 5/2
1 72
Значения термов (v/Я)
рассчитанные из границ поглощения рентгеновского спектра
линий спектра испускания
[62]
8 477,0
1603,5
1 543,7
1 264,3
408,9
382,1
317,2
274,0
261,0
106,6
95,7
77,1
56,3
53,6
28,4
27,6
[56]
—
—
—
—
—
—
—
:
[107,1]
[96,0]
[77,4]
:
—
[52]
—
—
—
—
—
—
316,7
273,9
261,0
105,8
93,1
76,4
57,
54,2
28,1
27,4
[27] в>
8 474
1 602,6
1 542,7
1 264,2
408,5
381,5
316,8
274,2
261,2
106,0
93,5
76,6
57,5
54,3
28,5
27,6
[31]
—
—
—
1 264,2
408,7
381,7
317,0
274,5
261,7
105,9
93,8
76,9
57,5
54,5
28,8
л частот
[36,13]
—
—
1542,73
1 264,32
—
—
—
—
—
—-
—
—
теоретические значения
[63]
8 490
1 596
1535
1264
408,5
379,5
317,5
273,9
260,4
—
—
—
^ияиАии^
ипаЧСНИС
термов б), эв
[64]
115 790
21 711
20 900
17 128
5 537
5 172
4 296
3 721
3 546
1 439
1271
1042
783
739
391
382
Продолжение табл. 15
Терм
01
От )
°IV )
ov J
°vn 1
Л
P» 1
Ли
Qi
Выбиваемый
электрон
5s !
5/,
5d
5/ .
6s -
6p
Gti .
r
7s
ЭЛС
n
5
G
7
Квантовое число
;ктрона перед его
удалением
/
г о
^
1
2
L 3
г о
J
1
1
. 2
0
/
V2
г;:)
!.;:)
{::;)
!/0
i::
Ы
М
1 'о '
Значения термов (v/Я)
рассчитанные из границ поглощения рентгеновского спектра
линий спектра испускания
[62]
26,2
15,4
5,8
—
|
[56]
25,1
[ 19'4
\ 12,3 или
1 и
( 5,6
1 G,l
[52]
—
—
.
[27] в)
23,7
18,3
13,9
7,0
[31]
23,9
19,0
14,7
7,4
Оптические термы
3,0 j
2,4
1,2
—
:}
—
0,8
2,2 г)
0,3 д)
Оптические термы
1 1 1
и частот
[36,13]
—
—
I:
5,35
{:
1.
теоретические значения
[63]
—
—
:)
—
:}
Значение
термов ^\ эв
[64]
328
258
193
102
—
17
а) Таблица Рассела [65] включает значения К- и L-термов по Зигбану [27], Af-, N-, О- и Р-термов по Клэссону [31] с добавлением значения Pj-термз
по Кошуа [36, 13].
б) Определено из оптических термов и рентгеновских линий.
в) Главная часть данных основана на значениях, определенных Идеем [30].
г) Хотя все Рц iij-линии слабые, значение терма, несомненно, больше 2.
А) Интерпретация линий как Ljj, Pjy.V H ^111^*1 V,V ненадежна.
Глава 2. Свойства атома урана
25
Таблица J6
КОНСТАНТЫ ЭКРАНИРОВАНИЯ УРАНА [63]
Терм
°2
31
h
2,0
21,4
L1L, III
3,49
22,4
мх
6,8
38,9
MU, III
8,5
40,7
-MIV, V
13,0
44,2
заполнение 4/-уровня, это увеличение ничтожно мало и изменяется
неупорядоченно. Из сказанного может быть сделан вывод, что элементы рядаТЬ—U
напоминают ряд редкоземельных элементов больше, чем они напоминают
последовательность элементов Ш—Pt. Однако приведенные факты сами по себе
еще недостаточны, чтобы иметь полную уверенность в правильности этого
вывода.
Второй довод более убедителен. Табл. 15 показывает, что значения термов
для границ поглощения М\\ и My (273,9 и 261,0) примерно на 0,5 v/^-единиц
меньше, чем значения М\у- и Л1у-термов (274,5 и 261,6), вычисленных из Ьщ-
границы поглощения и L-линий спектра испускания. Объяснение этой разницы,
предложенное Расселом, состоит в том, что поглощение и в Lin- и в Afiv.v ■
уровнях приводит к переходу электрона в незаполненные валентные
подуровни, а не к полной ионизации. Согласно правилу /-отбора, Д/ = ±1, Ьщ (2р)-
электроны (/ — 1) могут перескакивать в Piv.v-валентный подуровень (6б(-электро-
ны, /^2), но Л^у.уэлектроны (Зс(-электроны, 1 = 2) могут переходить только
в валентный подуровень Ovi.vii (группа 5/, / = 3). Если электроны последней
группы связаны более прочно (приблизительно на 5 эв), чем в 6d-rpynne, то
для поглощения в Miv.v-группе требуется меньшее количество энергии, чем
вычисляемое из разницы между энергией поглощения в Lm-подуровне и
энергией /.щЛ^у.улинии спектра испускания. Таким образом может быть
объяснена указанная выше разница между рассчитанным и наблюдаемым значениями
термов Afiv.v
Однако если даже величина этой разницы, повидимому, и превышает
ошибку опыта, нет полной уверенности в правильности этого объяснения.
Анализ электронных уровней кристалла, особенно металлического кристалла, на
основании данных об электронных состояниях свободных атомов не является
достаточно надежным по причине хорошо известного превращения этих
уровней в уровни проводимости («электронное облако»), которое, вероятнее всего,
принадлежит кристаллу в целом, а не отдельным атомам.
Следует отметить, что Af-границы поглощения (см. табл. 5) имеют такую
ширину, что различие между рассчитанными и найденными значениями М\\>-
и Му-термов практически исчезает, если значение каждого терма определяется
из длины волны, соответствующей не началу или середине границы поглощения,
а максимуму поглощения (см. [14]). v"
Брейт [66] допускает возможность измеримого влияния ядерного^спина
на рентгеновские термы для наиболее тяжелых элементов. Он рассчитал, что
если уран имеет ядерный спин, равный <J/2 единицы, то его /(-уровень должен
расщепляться на две составляющие с разностью энергий в 22,5 эв, a Ln-ypo-
вень—на две составляющие с разностью в 4 эв. Вильямсом [35] было отмечено,
что естественная ширина Li-линии урана слишком велика для того, чтобы
можно было обнаружить расщепление такого порядка.
Атом U238, атомный номер и атомный вес которого являются величинами
четными, повидимому, не имеет ядерного магнитного момента; ядро же U23*
с нечетным атомным весом обладает ядерным моментом, результатом чего
26
Часть I. Уран как элемент
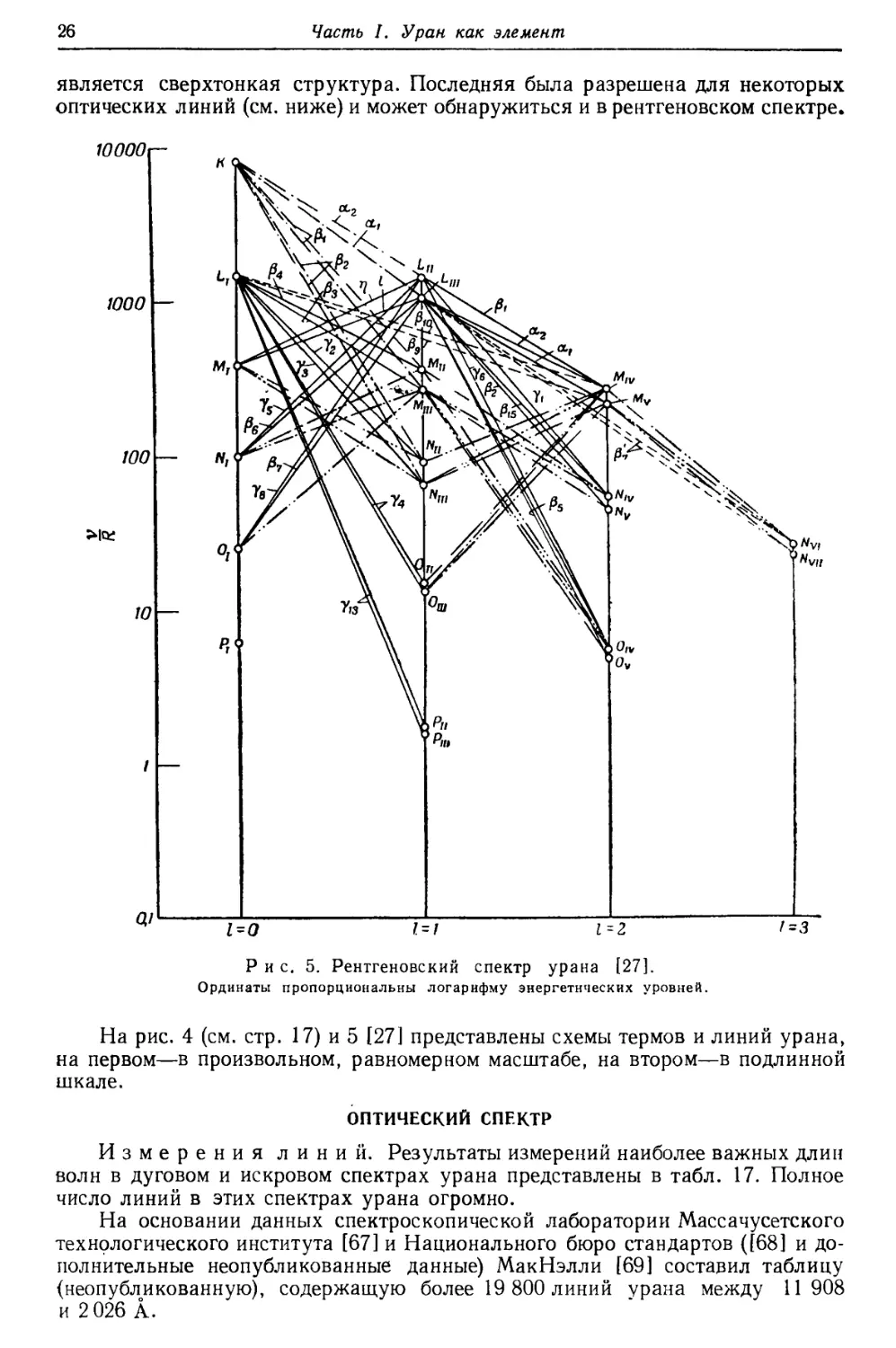
является сверхтонкая структура. Последняя была разрешена для некоторых
оптических линий (см. ниже) и может обнаружиться и в рентгеновском спектре.
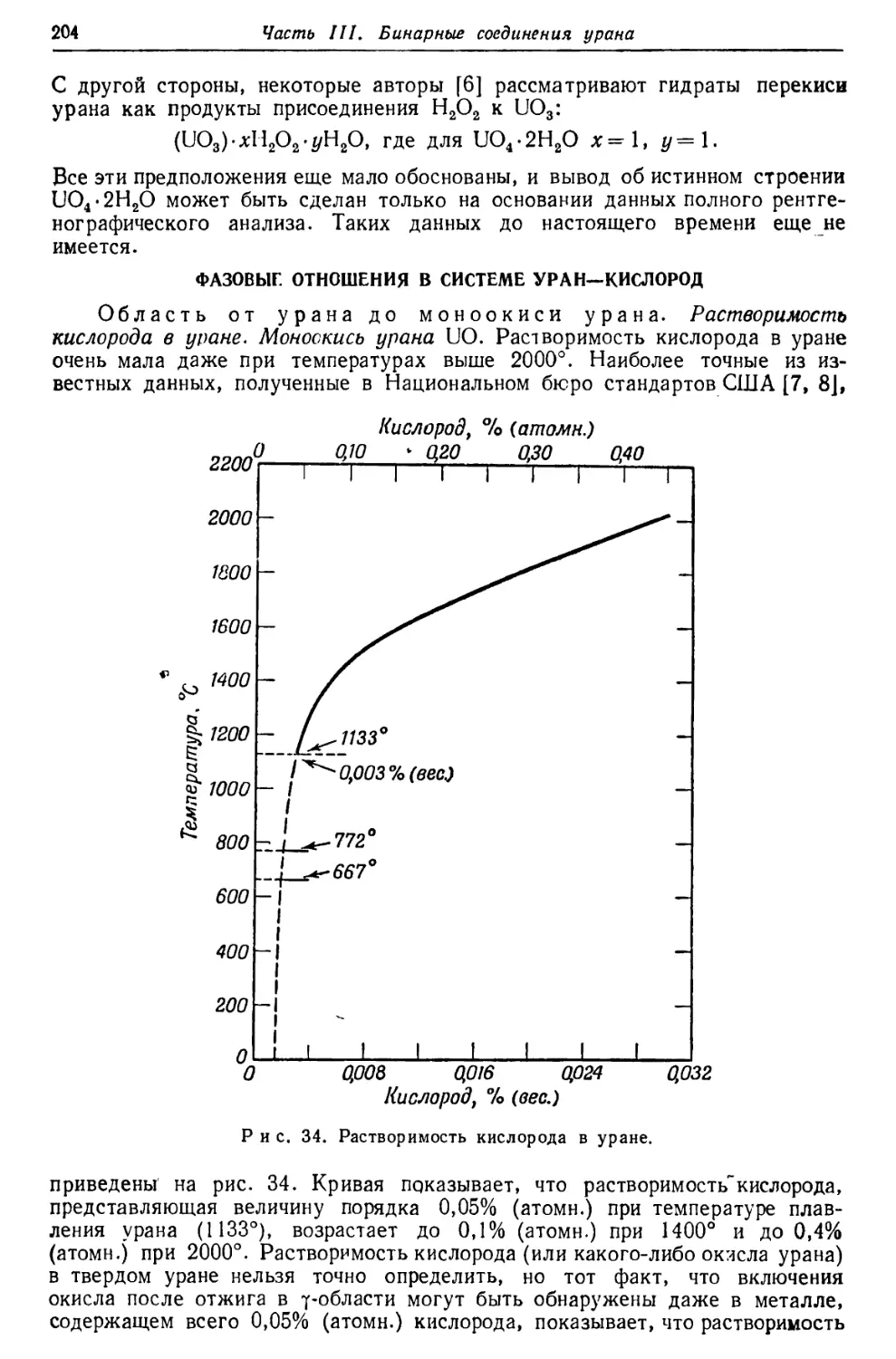
ЮОООг-
1000
>|с*
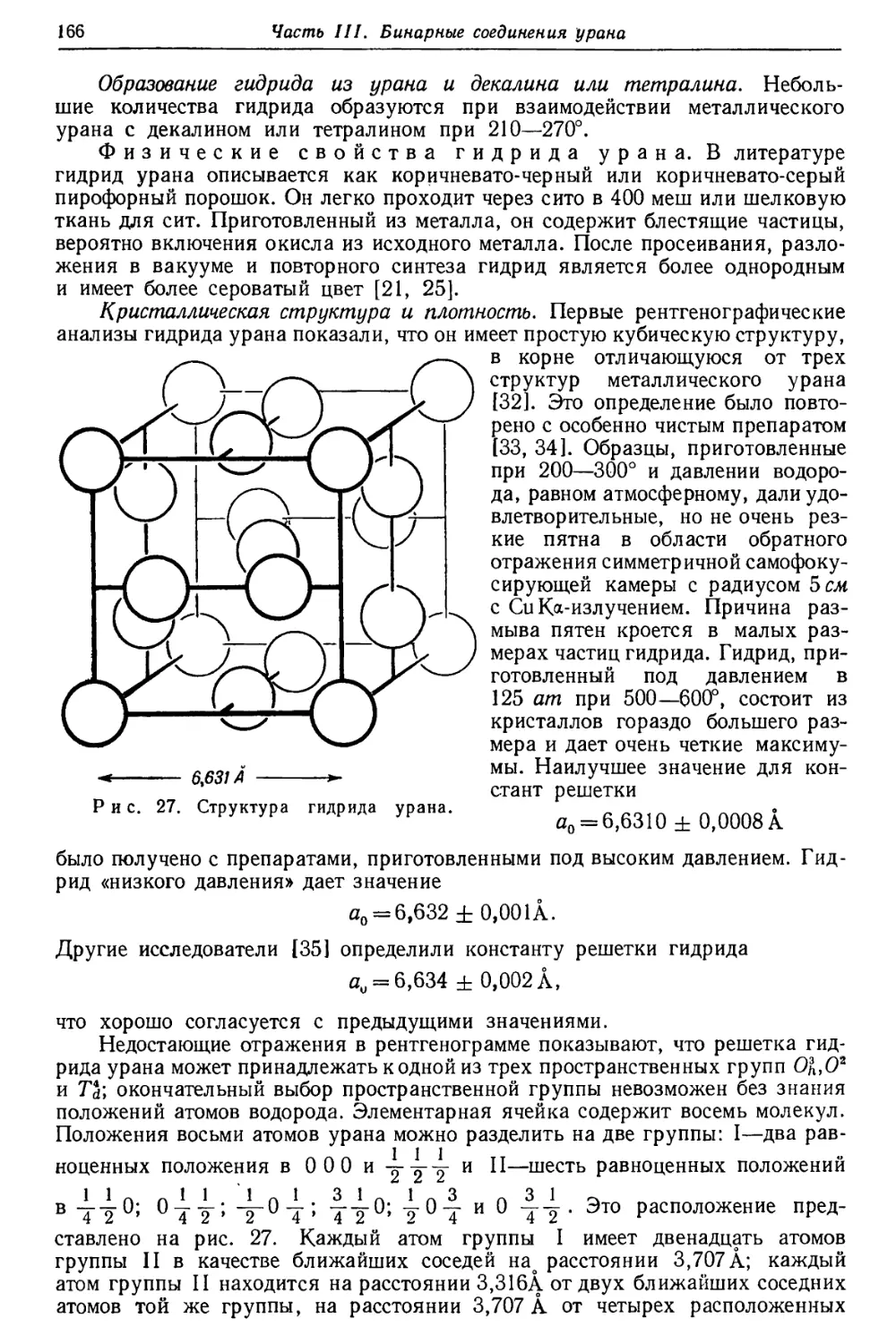
Рис. 5. Рентгеновский спектр урана [27].
Ординаты пропорциональны логарифму энергетических уровней.
На рис. 4 (см. стр. 17) и 5 [27] представлены схемы термов и линий урана,
на первом—в произвольном, равномерном масштабе, на втором—в подлинной
шкале.
ОПТИЧЕСКИЙ СПЕКТР
Измерения линий. Результаты измерений наиболее важных длин
волн в дуговом и искровом спектрах урана представлены в табл. 17. Полное
число линий в этих спектрах урана огромно.
На основании данных спектроскопической лаборатории Массачусетского
технологического института [67] и Национального бюро стандартов ([68] и
дополнительные неопубликованные данные) МакНэлли [69] составил таблицу
(неопубликованную), содержащую более 19 800 линий урана между 11908
и 2 026 А.
Глава 2. Свойства атома урана
27
Таблица 17
ДАННЫЕ ПО ИЗМЕРЕНИЮ ДЛИН ВОЛН СПЕКТРА УРАНА
Интервал
длин „волн,
А
Число
измеренных
линий
Дуговой спектр
4 642— 7 130
3 583- 5 871
2 264— 6 827
8 223— 8 758
5 500— 9 530
2 000—10 000
2 900—11 000
-600
2 200
4 960
17
674
5 238а)
9 000б)
Литература
70
71
72
73
74
67
68
Интервал
длни „волн,
А
Число
измеренных
линий
Искровой спектр
2 195— 6 449
374— 2 000
2 000—10 000
2 865— 5 600
5 632
93
5 238а)
~300в)
Литература
72
75
67
76
а) Общее число линий, полученных в дуговом и искровом спектрах.
б) Согласно Ма^кНэлли [69], общее число измеренных лнний [G8] превысило 14 000.
в) Только идентифицированные линии (ср. с табл. 23 на стр. 4 8).
Из примерно 20 тысяч известных линий урана к моменту написания
данной книги было идентифицировано несколько тысяч линий в отношении
принадлежности к переходам между определенными термами U+ -иона и
нейтрального атома урана (спектры UII и UI соответственно).
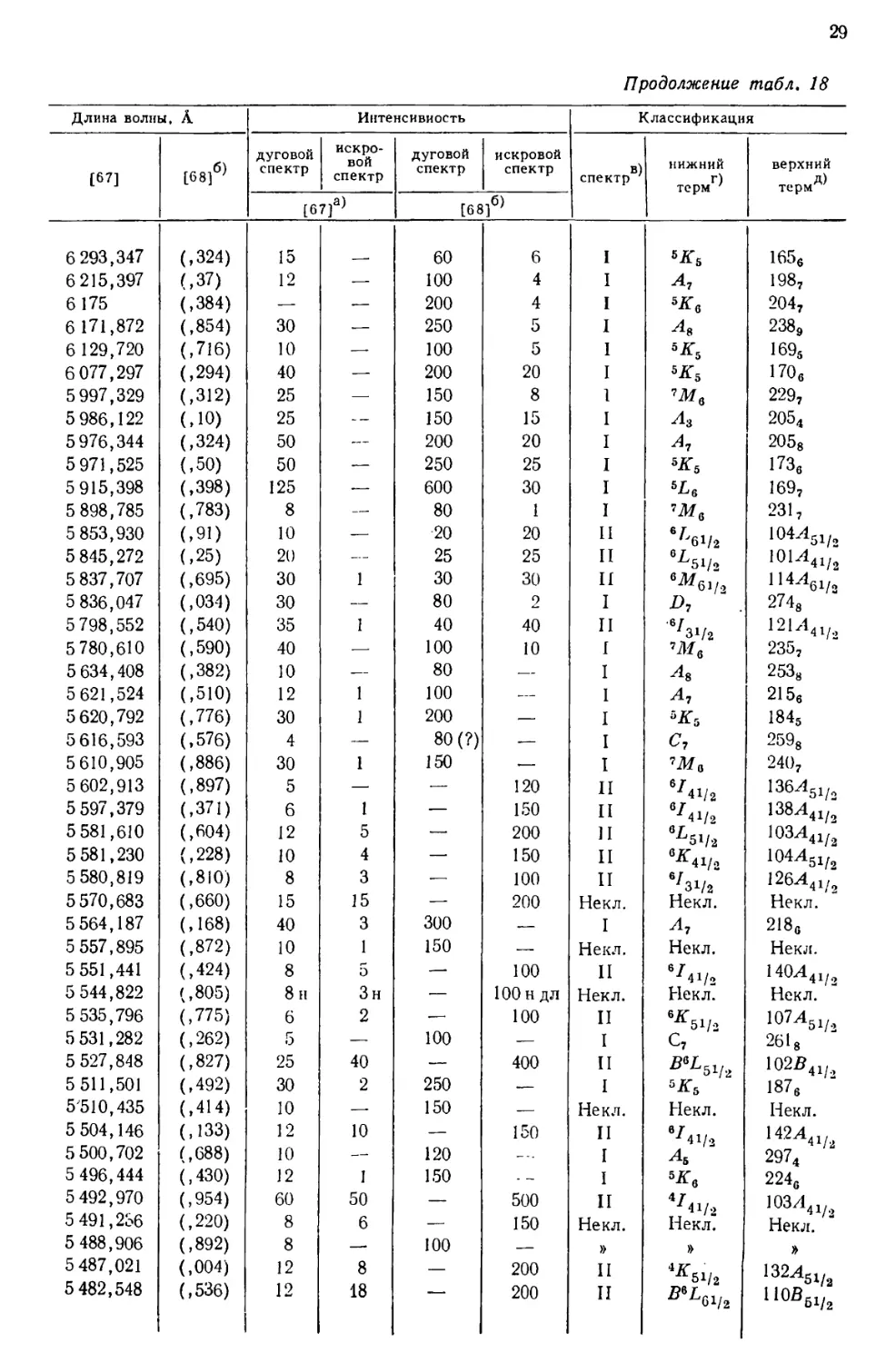
Перечень 430 наиболее интенсивных линий урана, расположенных в
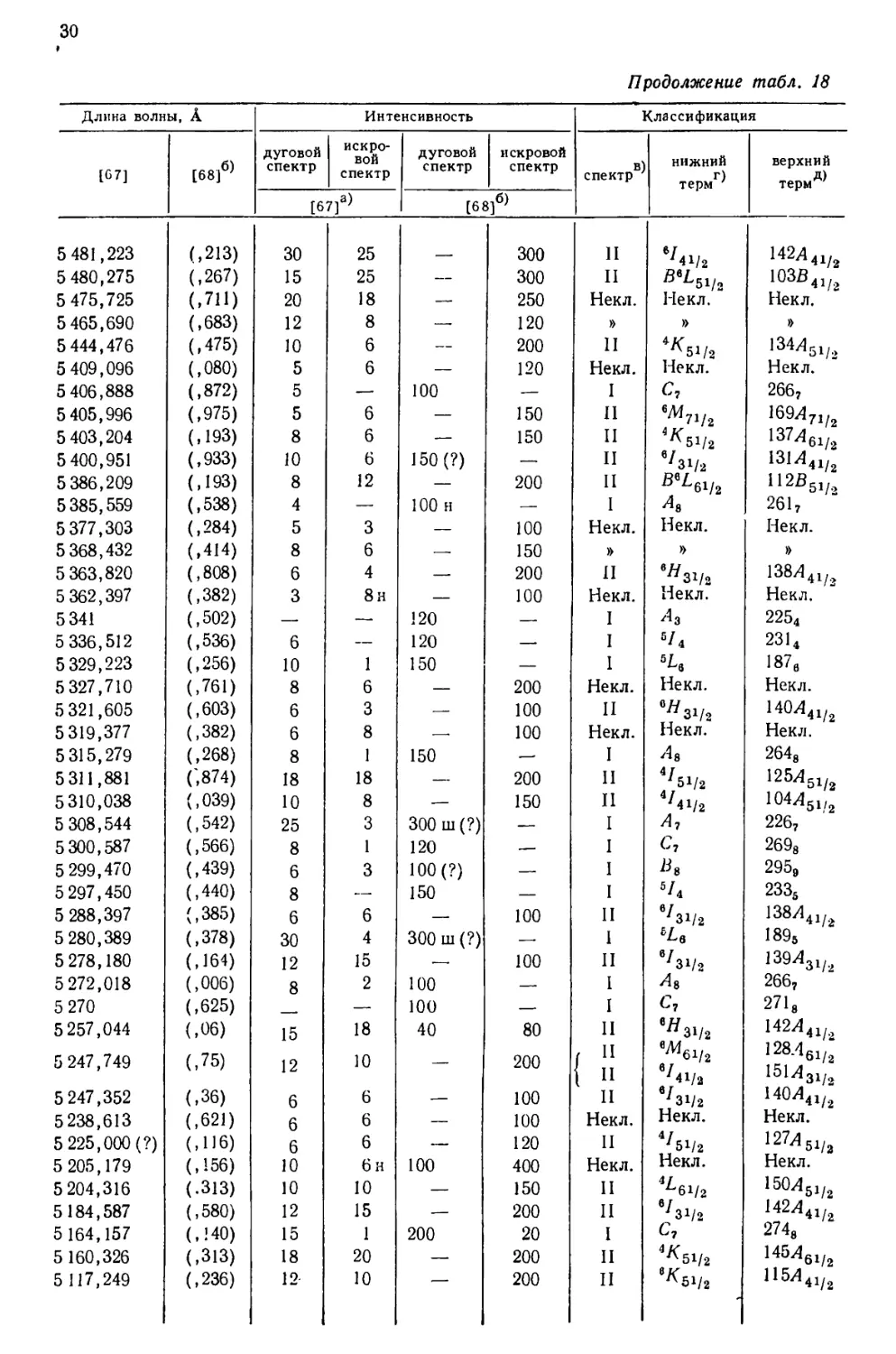
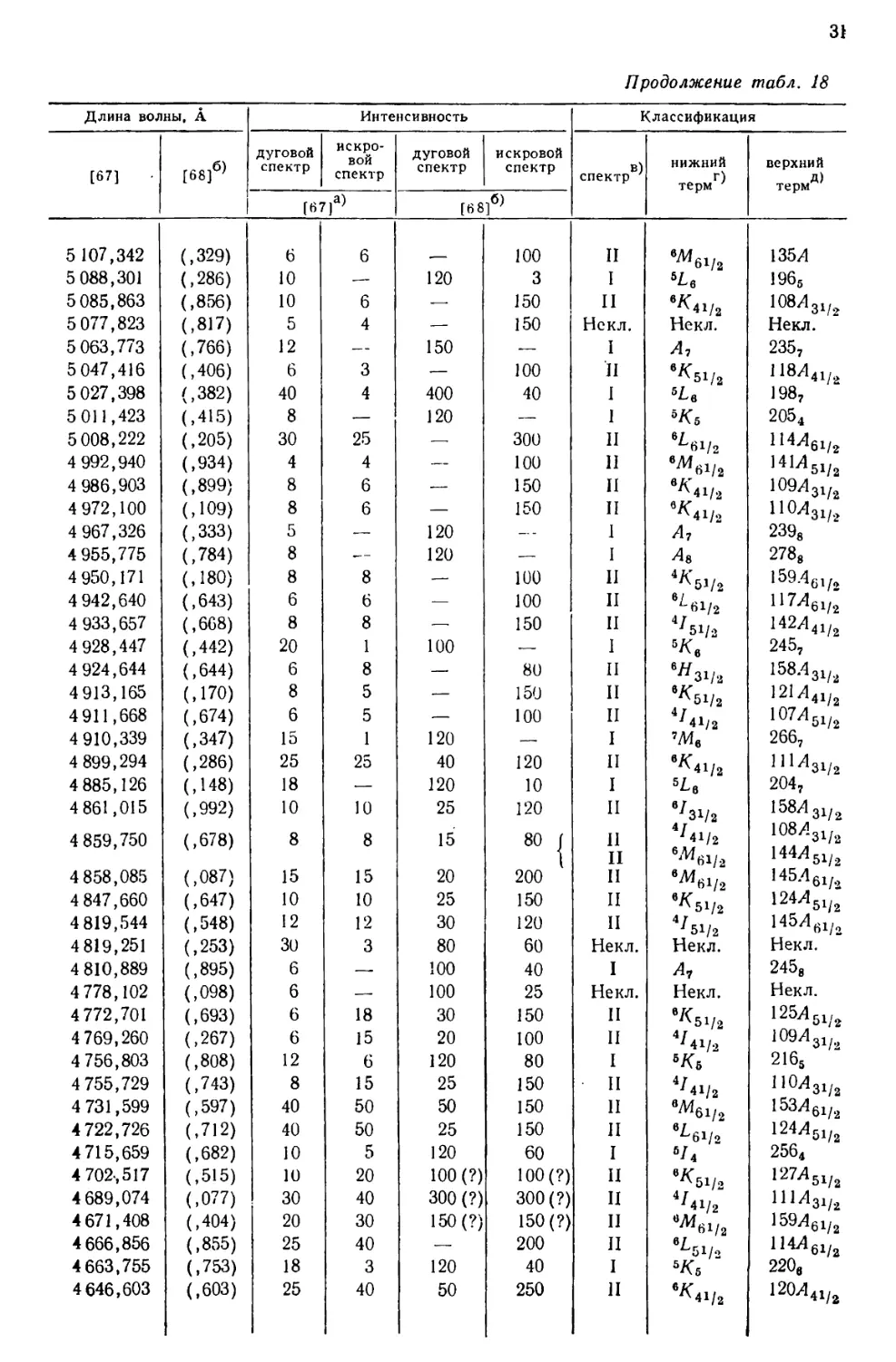
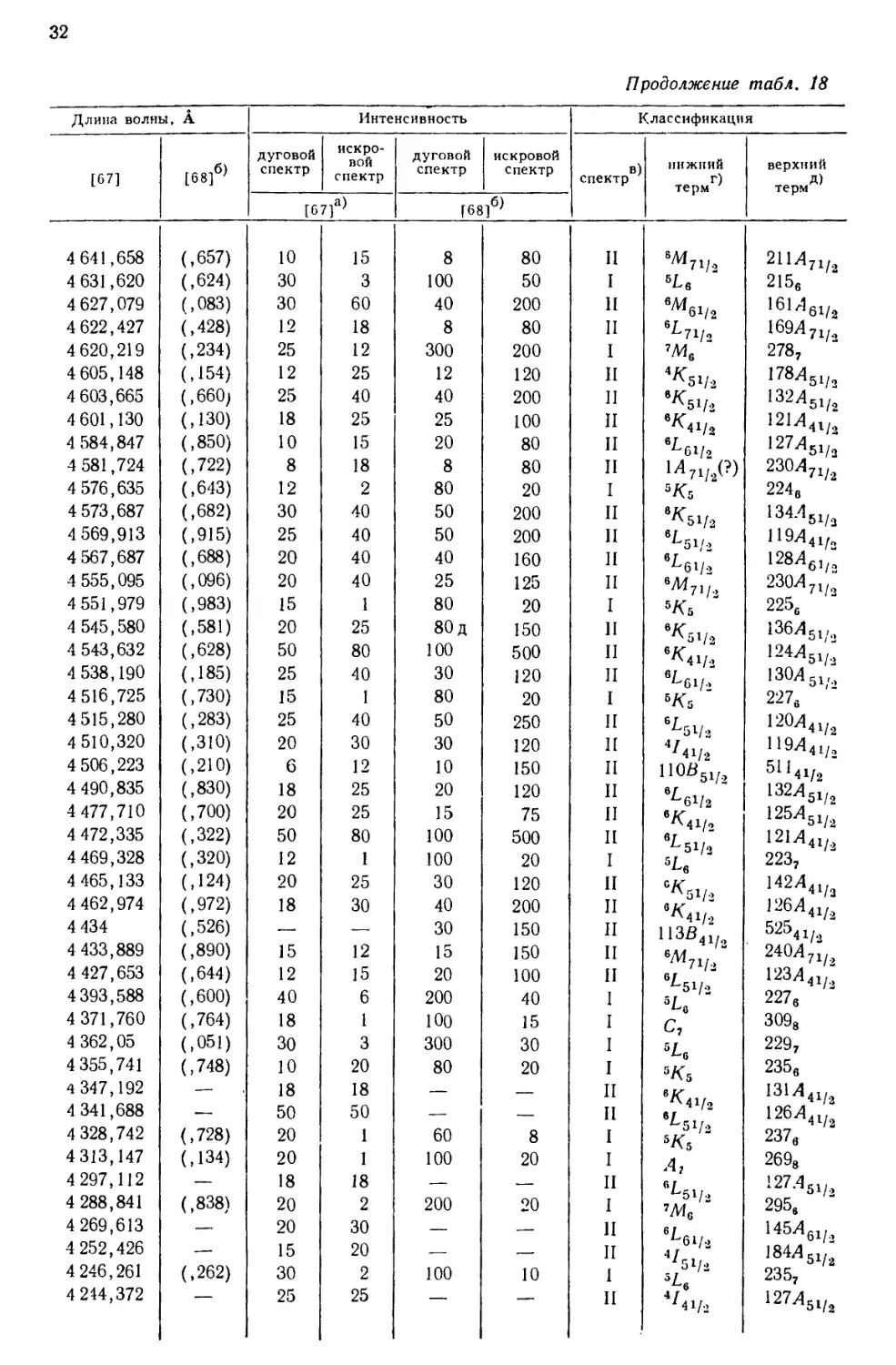
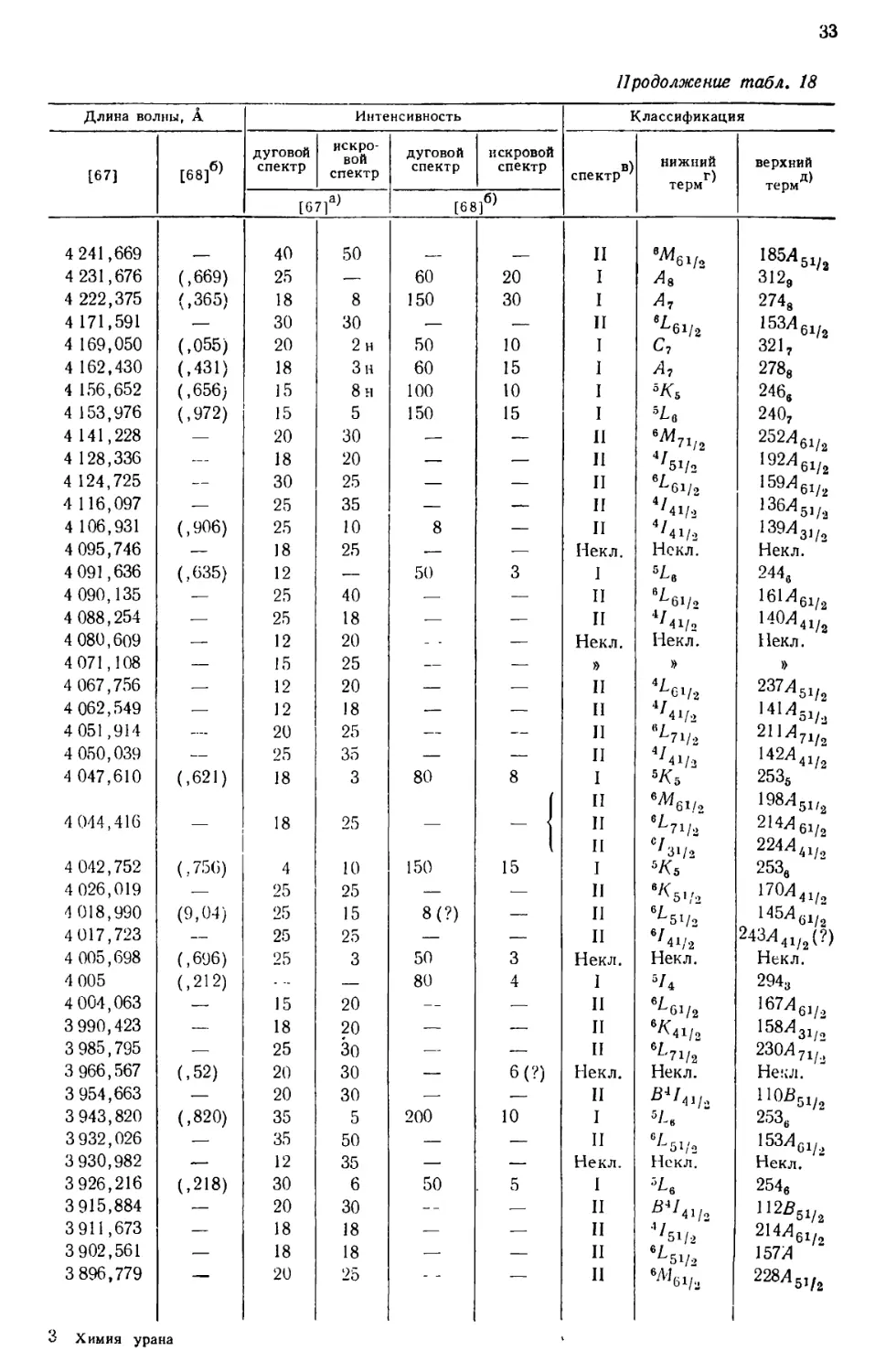
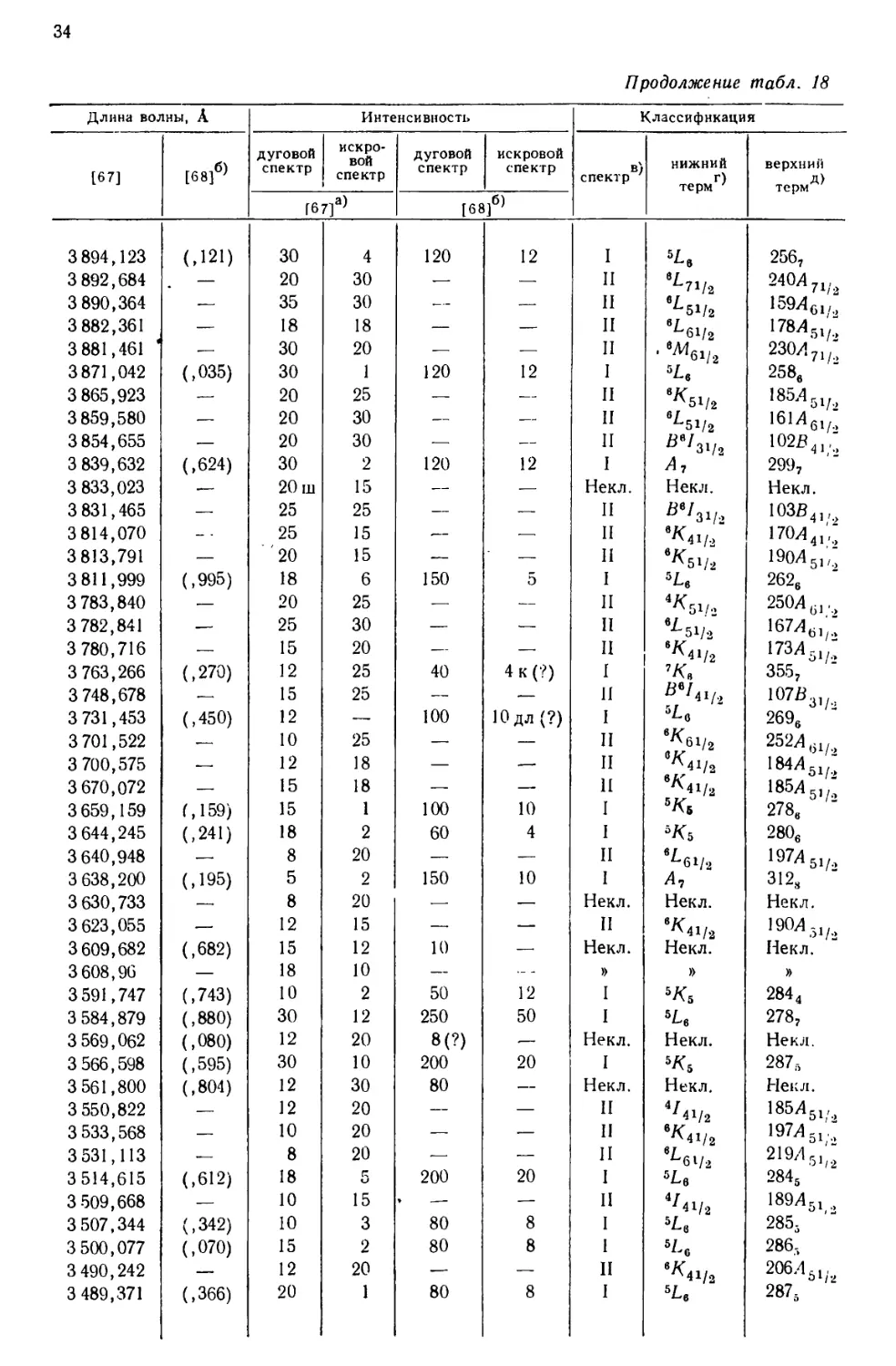
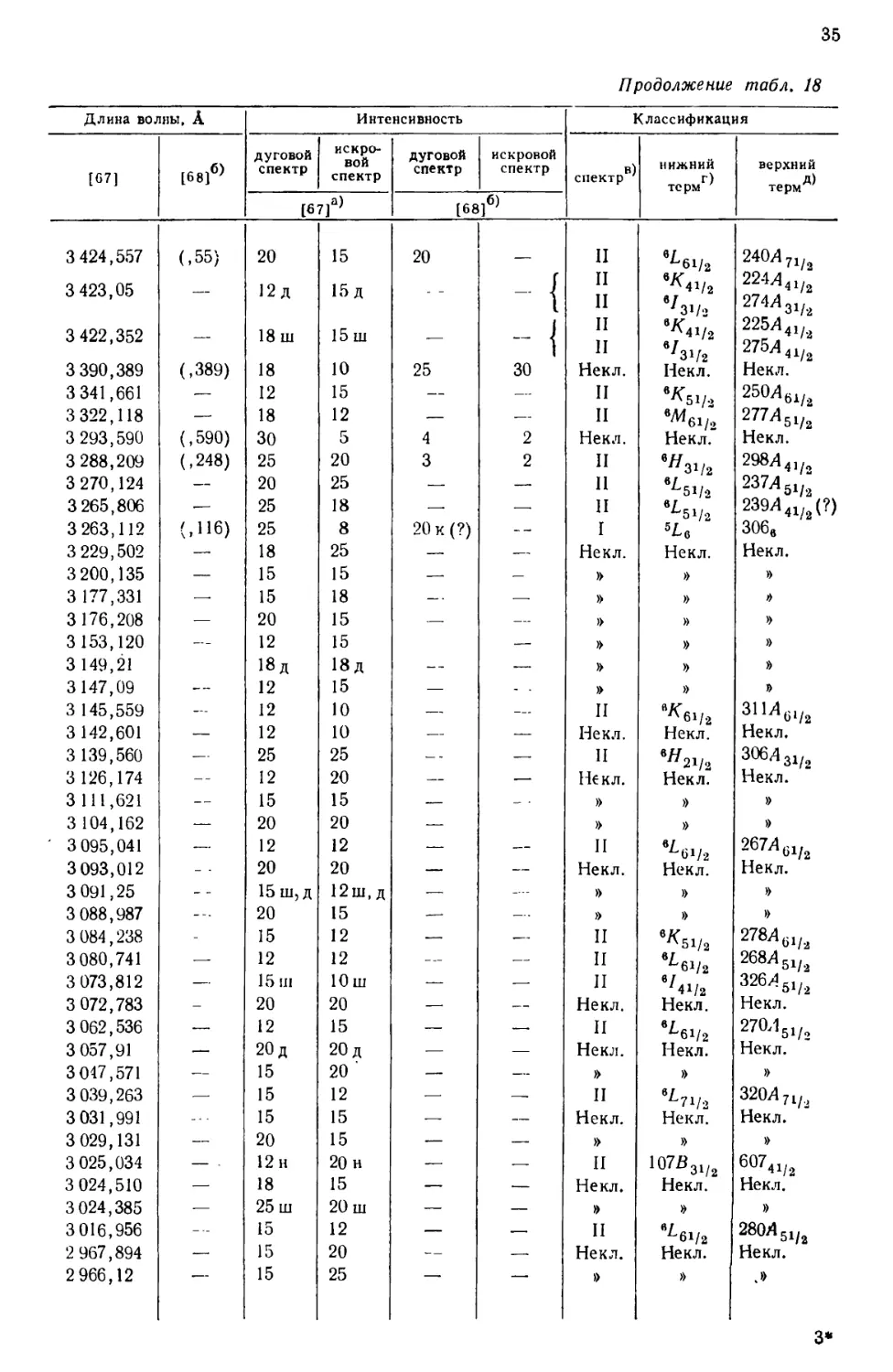
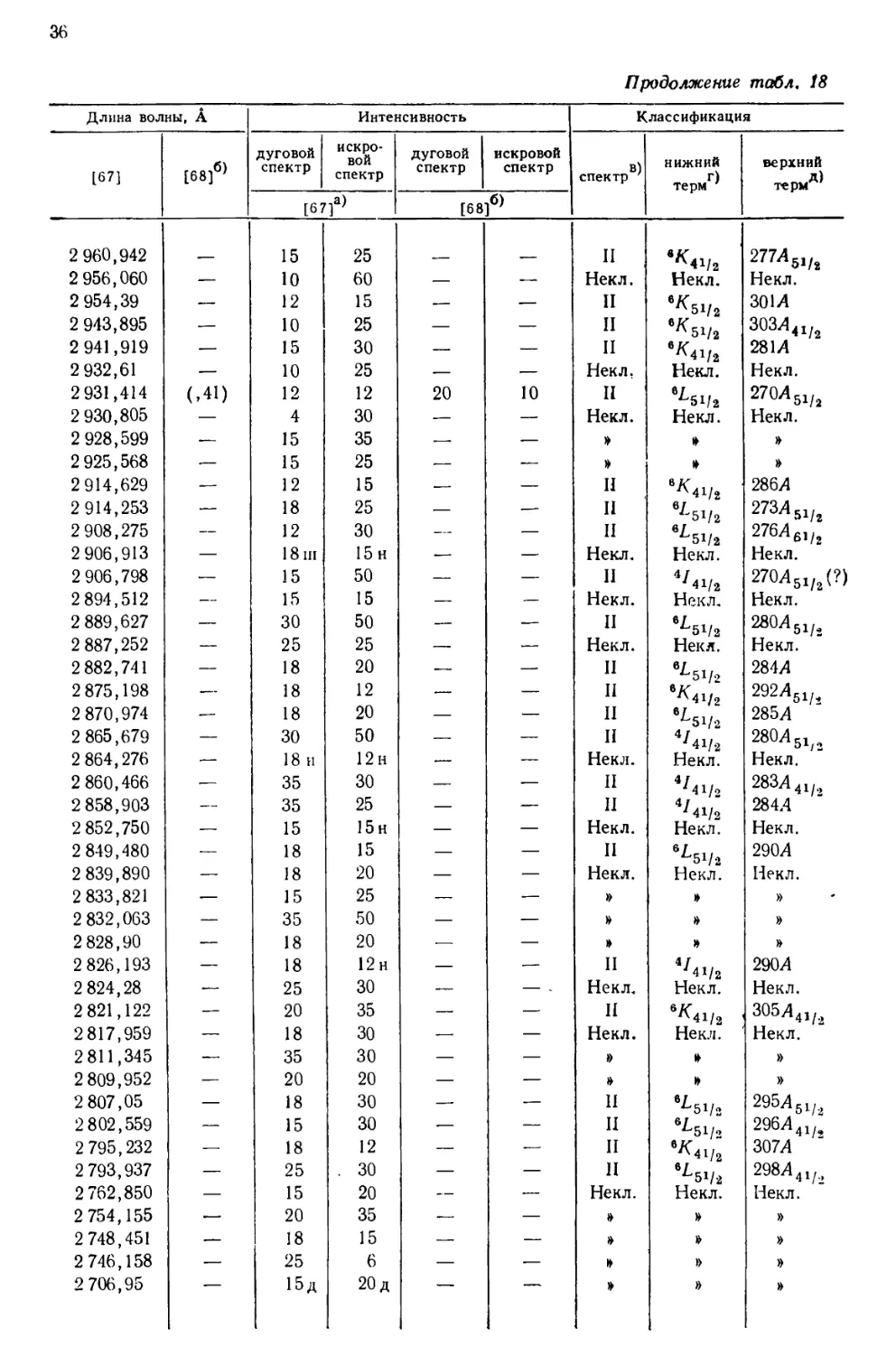
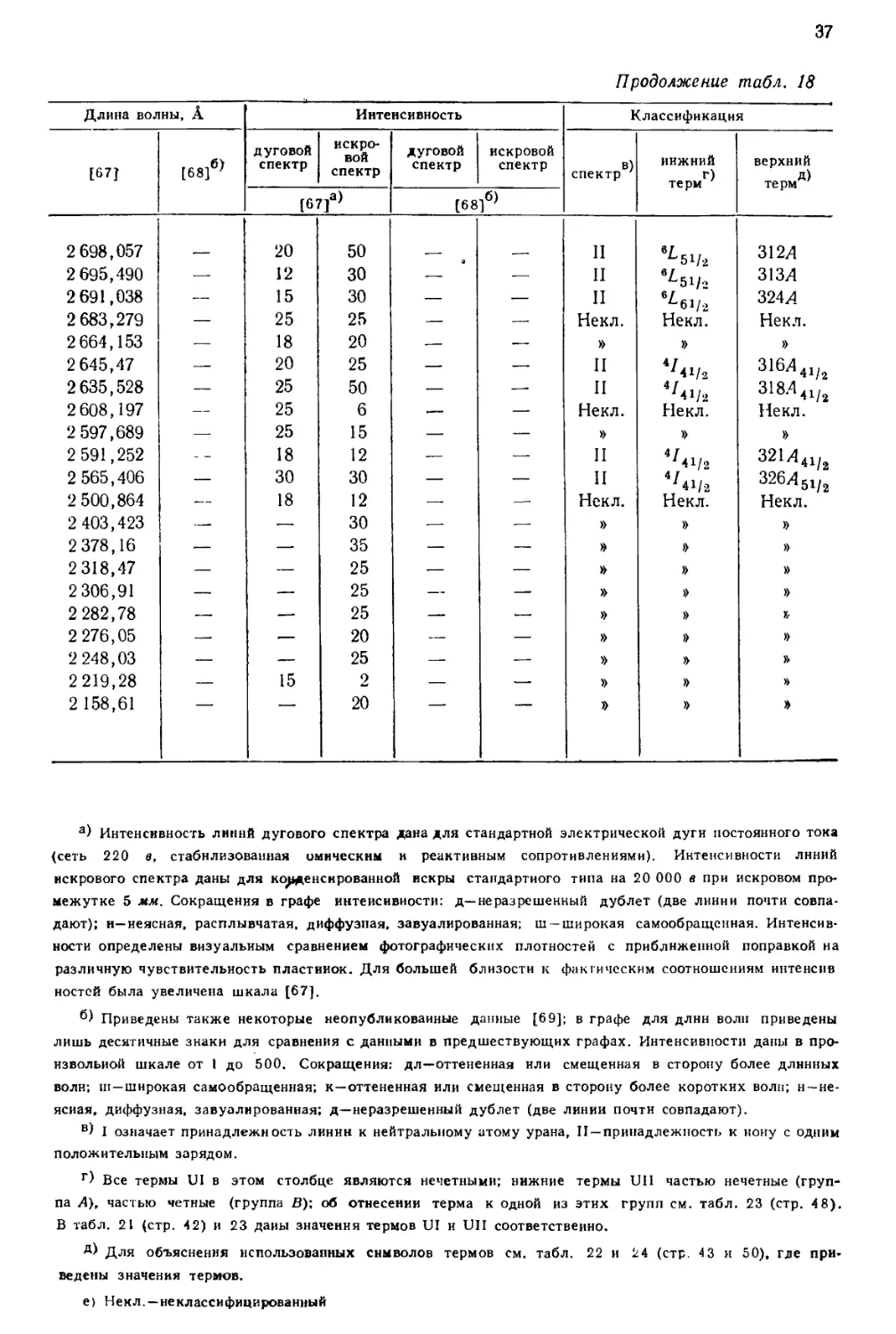
интервале от 10555 до 2158 А, приведен в табл. 18, составленной [69] по
данным измерений, выполненных в Массачусетском технологическом
институте и Национальном бюро стандартов. В других источниках можно найти
более подробные перечни: 5238 линий UI и UII [67], 1240
классифицированных линий UI [68] и 743 классифицированных линий UII [77].
Более трех четвертей (78%) всех интенсивных линий урана, приведенных
в табл. 18, классифицированы как вызванные переходом между известными
термами U и U+. Из остающихся 84 линий 3%, повидимому, принадлежат U2+,
15%—U+ и 4%—нейтральному атому урана [69].
Вопрос об использовании спектра урана для определения этого
элемента, а также примесей в уране здесь не обсуждается (см. сборники по
аналитической химии из американской серии работ по ядерной энергии).
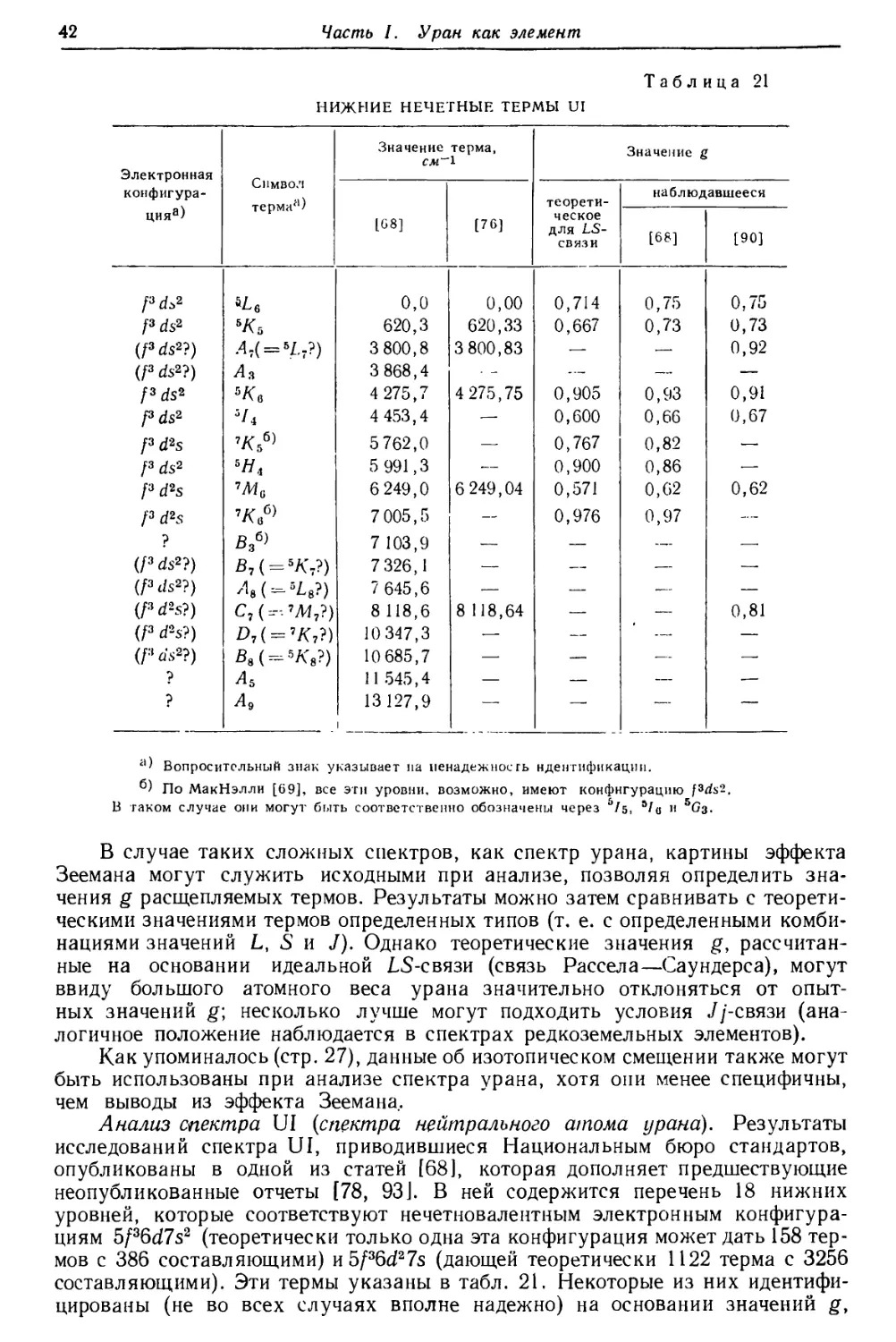
Наиболее «устойчивые» (постоянно встречающиеся) линии урана (они не
обязательно являются наиболее интенсивными линиями) приведены в табл. 19.
Первоначальные наблюдения проводились с загрязненным металлом [80, 81]
и некоторые линии, отмеченные при этом, повидимому, совсем не принадлежат
урану. Фотоснимки дугового и искрового спектров урана, приведенные в
атласе Эдера и Валенты [82], также, очевидно, содержат много линий,
принадлежащих примесям.
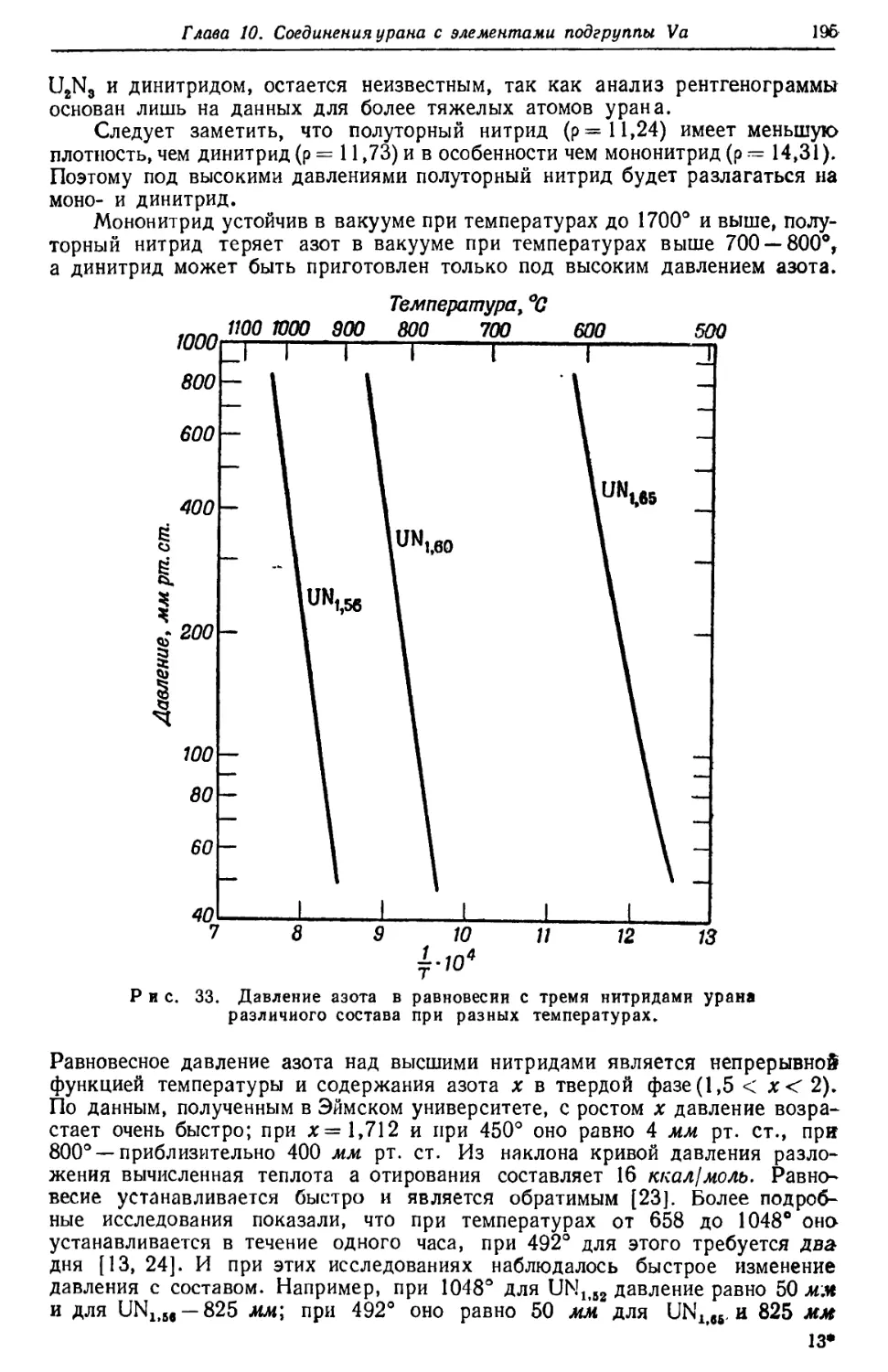
Ядерные эффекты в спектре урана. В атомных
спектрах наблюдаются два типа ядерных эффектов: 1) смещение линий и 2) их
расщепление (так называемая «сверхтонкая структура»), вызванное
взаимодействием между ядерным спиновым моментом и вращательными моментами
валентных электронов.
Смещение линий. Сообщается о смещении линии 5027,40 А в спектре UI
для U235 (по сравнению с ее положением для U238) на 0,426 см'1 [83]. Было
сравнено поведение нескольких линий спектра UII и показано, что смещения
особенно велики для тех линий, испускание которых вызывается переходом
электрона на одну из проникающих орбит 7s [84]. Измерение смещений,
обусловленных изотопическим эффектом, может поэтому оказаться полезным при анализе
термов спектра.
сп сп сп
W GO GO
СЛ "-4 00
CD Ю СО
СО 4* ОО
О СП О
СЛ СО 4*.
Ю 4* -si
ОО СО -4
и*- 4^ 4*.
СО СП —
О О 00
1 1 1
*— to
О О 00
О О О
— to
О О 00
(л Ц, СЛ
— — ю
СЛ CD —
-ч ^ сп
V се «л
СГ>
СО
CD
ГО
-si
00
-si
ф-
СЛ
to
о
1
00
о
1
СП
СО
CD
СЛ
4^
4*
СП
4^
to
to
о
о
1
to
о
о
to
о
СП СП
4^ 4*
4J" СП
CD СЛ
— О
СП О
-si СЛ
СЛ CD
00 -si
О N?
О СЛ
1 1
СО —
О СЛ
О О
СО —
О СЛ
о» о сп
^
СП
сп
а
^
СЛ
СП
«Л
— to
СП Ю
— -si
tfb Q0
СП
СЛ
00
СО
4^
СЛ
СО
СО
-s!
^—
СП
1
00
о
00
а
Ю
СП
а
СП -si -41 -si "41
00 О — *■ СЛ
Ю -si tO tO СО
СП 4^ 00 СЛ 00
CD O0 CD СЛ CD
СО — — О —
со ч оо ел со
— 00 СО О —
to to to >— to
СЛ Ol О 00 О
! 1 1 1 1
4*. tO •— Ю
О 00 О СЛ СЛ
О О О О О
л. to — to
О 00 О СЛ СЛ
^1
ГП
О
СО
^_
СП
СП
4*
1
^
О
о
.—
0 дл (?)
ел ел с;» t-i к. (>vj
Ь t> >! Ь - -
а ст. сп °»
, t. t , ti r , . ¥_^
4J* 00 4х 00 -vj
СП 4^ СП 4^ О
С» СЛ О С О
to
о
4*
•о
—) —J —5 —-1
СП СП СП -si
— СО СО 00
СО n— СО 4*
СО -si СЛ —
СЛ •— 4* 00
СО -si Ol •—
4^ Ю tO CO
to
O0 00 4* О
Mil
со to to ел
О СЛ О О
о о о о
— to
О 00 СП СЛ
Ч Jv С" ь<
с. «"
to — ►— ►—
О СП -J СО
•v] CD W А
«ч! si с> С"
S S ОО
00 СО *—
00 -*J -J
— О 4^
СО 4^ СО
4* СП О
СО 4*> СО
—* СП О
•—
СЛ 00 4».
1 1 1
4* »— tO
О О СЛ
О О О
to —
О О СЛ
^j rv ел
н~ Ю —
00 О СП
CO СП СЛ
с» о» »
00 OO
to to
to en
CO tO
о
СЛ
о о
CD СЛ
1
1 4^
1 1
to ►—
О СП
о о
СО 00
•— ю
00 О
4* Ю
СЛ О
00
СО
00
СО
4*
СО
4*
to
1
*—
О
О
СЛ
ГО
О)
и
X
а>
fa
000000000000000000
СОС04^4*.4^4хСлСЛСЛ
4*004*4*СЛС0О4хСЛ
СП — •— СП О СП 4^ О --)
vl ОО Ю 00 О S Ю 00
^ О) СЛ СЛ СО 004*
v
SOOlOOJOOCJi^CO
слспо-sitocooocoto
1
t04^t04^4^l 4^4^ЬО
1 1 1 1 1 11 1 1
— 4* 4i» •— >—
OOtOOOOOOOOOCO
ооооооооо
ЮСП4>О0О0О04*Ю
1
•""'
(ь а> ■£ ел
lw
О
СО
S 00 CD СЛ СЛ СО *^ -si tO
СО СО 4* СП СП ОО'Г 4^ СО
«Cii?0»M«lv,*4
—о
-W
00 ОО
СЛ СЛ
СП -si
-vl О
-si СЛ
СО —
-si СЛ
— to
Ю 4*
1 1
, .
4^ tO
О О
1 1
i— .—
СП СО
— -s|
*ь О»
00
СЛ
^)
4*
СП
СЛ
со
to
1
со
о
1
■sj
6;
^
^1
со
00
СП
о
-s]
СО
4-v
со
СП
00
1
СП
о
о
СП
СЛ
ь
о»
h—
^~-
СП
Ol
00
СП
со
to
00
to
со
ф>
1
,_.,
СЛ
о
1
X
5s
X
(V
и
X
•X
и
00
^1
о
-si
->1
-si
СП
4^
1
4*.
о
1
Ol
1^
^-
•s.1
4^
00
^1
СЛ
со
СП
со
СП
00
to
1
to
СЛ
1
00
-si
СЛ
-si
-si
СЛ
-si
-si
4»
I
_
СЛ
о
1
t-J-l
a»
fa
ta x
si а»
h*)
00
fa
X
ж
fa
00
со
СЛ
со
-si
со
4»
Ю
1
K*)
о
1
CO
о
CO
CO
СП
00
1
1
1
to
СЛ
1
vl Ci
СЛ СЛ
^-
СП
со
►—
►—
СП
Ol
со
to
-si
СП
to
4ь
4»
CO
Ю
i
to
о
1
l-J-l
54
fa
X
n
fa
X
(V
fa
о
СЛ
СЛ
СЛ
—
о
to
1
1
1
1
to
о
1
•si
^-
СЛ
СП
с»
-si
'а>
00
3
о
"^w
'о»
о\
ГОВОЙ
ектр
искровой
спектр
«5
"1
31
^g,
SS
скровой
спектр
о
3
н
•о
00
X
•о ^
2 =
% =
н а>
а> хз
*3 х
> s
X
W
волны, А
Интенси
td
X
о
о
н
ег
г.
Вз
о
о
я
S
fa
S
СЛ СЛ СЛ СП СП СП СЛ
•^ Д ^ 4^ ^ ^ СП
ОО 00 ОО Ю УЗ Ю О
to -g oo — to аз о
СЛ О CD ГО CD 4* -v)
^ Ю О ОГ' Ч 4^ О
оо •— аз аз о 4* to
СЛ О 00 to CD 4*. CD
СО О CD Ю СЛ СО ОО
О) ^ Ю О ^ О 00
to to оо оо о to о
»-*■ 1 ел 1
ОО ОО 1 Оз О — I
I 1 о 1 1 сл to
1 ' О ' ' О О
Ю Ю | ^- СЛ , .
о о ел о , !
о о ' о о '
X
— ■— ^ *-*
ел
СЛ
о
4*
_1
4^
аз
h_
со
CO
to
о
1
►—
СЛ
о
1Г^сл я £ » •£
N
*-* ГТ ^ *— to to
;- со ZC о to CD
p to <i> со 4* -o
CD ^ * g 4 e» *.
en en г «t»
_
4^
(О
>
ел
СЛ
о
4*
СО
ел
4^
4*
о
1
1
ел
о
1
X
*л
X
Он
CD
СЛ
СЛ
*—
СЛ
о
4^
<о
to
8
to
to
СЛ
о
.
1
ел
ел
ко
~о
00
4ь>
00
оо
К")
■<!
го
СП
4*
о
1
4*
о
о
СП J-4
ел
tv
00
-ч
OS
_
о
to
to
l-i
СЛ
Сл
со
ю
00
to
to
аз
ю
От
1
1
о
о
.
1
СЛ
СЛ
со
СЛ
^1
CD
аз
■^1
-О
СЛ
СТ:
to
1
►—
о
о
ел
го
Оз
00
о
-vj
ел
li.
СП
СЛ
4ь
4*
ОО
Ю
to
ОО
о
сл
оо
Со
1
о
о
X
X
Sa
ел
ел
СЛ
4^
4^
4^
ю
4^
00
СЛ
1
1—•
о
о
X £
St
X
о
_
4^
о
СЛ
ел
СЛ
■-J
00
CD
СП
00
-^
(О
о
Сл
о
,
1
X
и
ел
ел
аз
►U
^-
00
^
^~
аз
00
4^
о
со
со
о
о
ел
СЛ
--4
о
»
со
аз
аз
о
СП
СЛ
1
ю
г>
о
з:
ia
сл ел ел ел сл
сл ел сл ел аз
00 ОО 00 CD О
о — — -*j to
оо ю аз со cd
— СО — ^1 —
CD О О CD СО
оо to аз со оо
- ЮО М0
О 00 4* — --J
оо о ю аз сл
1
СО 4^ СП *- 1
1 1 1 1 I
^— ^— to ^— •—*
О СЛ О СЛ (О
о о о о о
HthKwhOOCSCSO»
• • М ю- ^* ^ м
X
О)
5
to
оо
о
X
1>
to о о со со
аз 4^ со оо аз
{Ь. |Ь 4^ »Ь. ^
ни сл *■ Ji. сл
ч5 tO U> 1чЭ 10
сл ел
аз аз
о аз
CD Сл
О CD
СЛ СО
00 СЛ
00 --О
аз аз
со
О 4*
1
gg
,—^ч
-о
а
to ю
4^ СЛ
О CD
-si 00
СЛ СЛ
аз аз
to to
о >-
-vl СЛ
CD Ю
tO 4*
~<l СЛ
~<J ^-
а> о
Co ~
о to
tO ta-
о о
о о
ел сл сл
^ ^ *^
СО 00 CD
4* О ОО
4^ аз сл
о — сл
оо о to
со сл сл
ОО CD 4^
to о о
— 4^ СО
о о ел
1 1 1—
00 О 4^
о о о
1 — 4*
1 О О
ц^ ^ ^ к^, s
to
•— to
00 •—
4^ СЛ
С" О»
to to ►—
ел со to
СО СЛ —
ос о s^
t-
СЛ
00
со
аз
о
4*.
^
о
со
со
о
!
1
00
о
to
сл сл ел сл сл ел ел
00 ОО 00 ОО CD CD CD
СО 4ь СЛ CD — -vJ -J
vi сл со оо сл — аз
-vj to cd -vi со ел со
О ^1 СО ОО CD tO 4^
-Ч tO О СЛ 00 СЛ 4*
аз to cd ч w сл со
CD СП — ОО CD О tO
СЛ^ ^ СО 00 'w 4i.
со to *- to сл сл
о с: О оо сл о О
— 'I'll!
м i 1 1 1 1 !
аз to to
со to to оо о сл о
О СЛ О О О О О
со to to со to to
О СП О ►— О СЛ О
ел сл
CD CD
00 CD
аз -<j
•— со
to to
tO CD
~ CO
о —
w tO
to to
СЛ СЛ
' 1
1 1
СЛ СЛ
о о
сл оо
CJ »— )_> С» О
ro
-<J
►>>
00
— — — to — »— to
— о о со аз ч о
4*. ■— 4^ — Ю СО СЛ
^ ^ ^ ^ ч » »
О *. Сл
t-Э U> t<9
to to
о со
СЛ CD
#» -^
аз
о
-v|
-ч
to
^>.
-ч
to
CD
4^
4^
о
1
1
to
о
о
to
о
СЛ
•-J
о
с»
аз
i—'
to
CD
-vl
to
о
-ч
CJ>
1
1
о
о
Сл
СП
<т>
fO
СП
аз аз аз аз
•-* >— to to
-v] <J — cD
•— сл сл со
00 СО СО
-<1 (О *>
tO ~vj -Ч
00 СО СО СО
сл оо ч to
4i. 4*. ^-^ 4^.
СО I — ~
о 1 to сл
1 1 1 1
1111
to to —
сл о о аз
о о о о
ел 4^ 4^ аз
С» СП
to ю — ь-
со о со аз
00 4^ 00 СЛ
<е ч -л о»
«71
-4
1—'
'о
оо
СЗ
"~DJ
1—1
СЗ
оо
3
дуговой
спектр
искровой
спектр
дуговой
спектр
я
° 2
я я
Я о
н и
тз о
»
Г)
a
ю
нижний
г)
терм '
, ев
а ^
•О х
S я
*3 X
X
о
£
н
о
я
ев
я
о
ег
ссификаци
я
слслслслслслслслсл
_*-.— *— tO tO Ю tO tO
-ojoiooootocoji.
wwi-mw-ocnw
^.ЮСЛСЮ'-ЧО-СЛ
CDCTi-vj^JCTiCOOCOND
•о
tOCO'-СЛСО»— *— CDCO
CO— 4хО0~СЛ — tOCD
5 W О О W 05 Oi -^
tOOOOitOOOCDCDCD
00»-СЛОО:СЛ(Х05
as
N3 —
о о
1 1 ° 1 1 ^ I ! 1
II II III
Ю Ю tO *— 4* — •— •—
ооюослоюоо
ooooooooo
X X
5a 5a
(л ел ^^«.-«м
-^ -^ **" to" . **" . ►*
to ы N
1—
— ►— Ю>— »— НИ — If- ►—
ел ел ^ to о 2 /-4 2 о
♦^ O) *■ СЛ • СЛ • rffc.
(в К t* Kl tO tO
СЛ СЛ
to to
4* СЛ
^Д -4
-vl О
4*. 4^
CD 4*
^1 О
СЛ CD
to сл
О ОО
4*
1 °
1
ю
О 00
о о
XXX
а а о»
и О О:
to -^ Ci!
to to
СЛ tO 4*
— 00 tO
►h» »£» J4*
WO)*-
to to to
СЛ
to
-si
О
CD
Ю
СЛ
t
1
I
I
, .
О
о
1
•—
/■Ч
to
СП
to
~vl
to
о
00
о
о
со
00
to
, .
о
о
1
—
оо
to
*»0 CD
00
ст
■"■
СЛ СЛ СЛ
ю to to
-j оо оо
оо о оо
»— со со
00 00 CD
О CD -4
СЛ -^ 00
.^ 00 СЛ
to о cd
СЛ 4^ CD
со
о
1 ° I
•■о
о о
о ' о
X — X
б» Сп О»
*-ч £•», '-ч
Со а Со
to to
со оо со
CD CD ОС
Гь4* ^
СО *>
ю to
СЛ
ю
CD
-vl
4*
СЛ
о
4^
4^
о
00
1
. .
СЛ
о
1
~
СЛ
to
со
W
СЛ
сл
to
CD
CD
4*
-•4
о
4*
со
CD
CD
со
. .
о
о
•-о
1
—'
00^
ю
CD
сг
*о
СЛ
К
О
8
--4
СЛ
CD
CD
00
•—
t .
ю
о
1
•—
СЛ
СР
00
СЛ
4^
4*
СЛ
4^
to
to
СЛ
со
со
о
о
в
■■о
\
•—
ю
CD
СС
оо
to
to
CD
«л
СЛ СЛ
со со
о —
О 00
£2
о оо
СО -4
CD 4*
О 00
00 00
1 1
1 !
•— to
СЛ О
о о
X X
to to
о to
4* СП
2ь ^
сл сл
to to
СЛ
со
СЛ
ю
-J
CD
to
CD
ОО
00
—
. .
сл
о
1
~
QD
to
CD
4х
QD
сл сл
со со
— to
CD —
СО CD
■^j о
-^1 СЛ
СО CD
00 О
to со
CD CD
00 СО
1 1
1 1
о о
о о
X
Sa
« со
* to
• *.
to
СЛ
СО
to
-vi
^J
о
^J
CD
00
CD
1
1
Ю
о
о
X
S3
X
к
Sa
X
О
£а
СЛ
со
to
CD
IO
го
со
to
Cl
CD
О
♦—
^
СЛ
о
1
—
сл
Г-
_
00
-s
сь
СЛ
со
со
CD
СЛ
to
сл
8?
CD
1
Ю
О
1
~
сл
--ч
to
СО
4ь
Сл
со
4*
СЛ
о
to
1
I
1
1
,,_
to
о
1
~
со
to
to
сл
IU
СЛ СЛ
88
to со
СО 00
CD Ю
^1 О
со оо
00 О
to оо
СО CD
00 4^
и
1 1
1 1
— to
о о
о о
X
CD i-^
ь
^fM О)
« со
• to
Зн го
is
• *>
to
СЛ
со
г
4*
со
to
4*
4х
00
CD
1
1
Сл
о
*
сл
со
•^1
•^1
со
о
со
ю
оо
4^
СЛ
со
1
1
о
о
X
Sa
*
и
X
п>
сл сл сл сл сл
СО СО 4х 4* 4*.
00 00 О О О
СЛ CD О СО СЛ
СЛ tO CD Ю CD
СЛ О СЛ О CD
CD CD — 4* CD
СЛ — CD — CD
CO CD CO CD -4
00 CO CO CO СЛ
4* 00 О 00 СЛ
1 tO CD CD CD
t , ■
о сл
О | О . |
я -i М
. to . — —
о сл сл
1 о ' о о
~ X X X X
Г" Ь- СЛ -VJ
°* uT Jt -It
-^ to to
to
N0 •— n- — —
CD — CO CO CD
— tO •— -4 CD
-* to ^ ^ ^
ел * en -g
to to to to
СЛ
4x
О
CD
00
00
00
00
^1
to
СЛ
1
1
t .
о
о
1
■—
to
CD
CD
СЛ СЛ
4^ 4^
St
О 4*
CD ~^1
CD CD
О 4^
ОО -4
О СЛ
сл о
CD CD
1 ]
1 1
•— to
to о
о о
X
5з
X *
« СЛ
1=1 -£
to
3=»
• СЛ
Ю
СЛ
*
сл
CD
CD
О
CD
00
СО
to
00
1
1
to
о
*
n?
СЛ СЛ СЛ |
4^ Ф- U^
ч оо оо
СЛ О —
**J Ю tO
to *-J to
сл сл со
ч ю ю
м- CD —
— ^J СО 1
to — со
о сл о
— ю to
оо сл сл
1 1 1
1 I I
to со со
СЛ О О
о о о
X
CD 1—1 1—
£а
^ ^2 ^
К С^ м
to
{? О 4*
S со to
g to ^
* *. *.
to to
о
с»
Ov
'о»
t»
"о»
сю
3
s»й
«gc5
■3 ?
rtS
я т
•3 о
о о
Я ?!
п тэ
?! О
'1
о
го
t3
OB
н s
5 *
M
c»
«
Ч П)
л t3
42 *
=
-2
S
X
го
3
о
о
X
о
«1
01
О (
я ;
я
*
00
я
о
3
3i
Продолжение табл. 18
Длина волны, А
[67] •
5 107,342
5 088,301
5 085,863
5 077,823
5 063,773
5 047,416
5 027,398
5 011,423
5 008,222 ;
4 992,940 !
4 986,903
4 972,100
4 967,326
4 955,775
4 950,171
4 942,640
4 933,657
4 928,447
4 924,644
4913,165
4 911,668
4 910,339
4 899,294
4 885,126
4861,015
4 859,750
4 858,085
4 847,660
4 819,544
4819,251
4 810,889
4 778,102
4 772,701
4 769,260
4 756,803
4 755,729
4 731,599
4 722,726
4 715,659
4 702,517
4 689,074
4 671,408
4 666,856
4 663,755
4 646,603
[68]б)
(,329)
(,286)
(,856)
(,817)
(,766)
(,406)
(,382)
(,415)
(,205)
(,934)
(,899)
(,109)
(,333)
(,784)
(,180)
(,643)
(,668)
(,442)
(,644)
(,170)
(,674)
(,347)
(,286)
(,148)
(,992)
(,678)
(,087)
(,647)
(,548)
(,253)
1 (.895)
(,098)
(,693)
(,267)
(,808)
(.743)
(,597)
(,712)
(,682)
(,515)
(,077)
(,404)
(,855)
(,753)
(,603)
дуговой
спектр
[6?
6
10
10
5
12
6
40
8
30
4
8
8
5
8
8
6
8
20
6
8
6
15
25
18
10
8
15
10
12
30
6
6
6
6
12
8
40
40
10
10
! зо
1 20
25
18
25
Интенсивность
искровой
спектр
iZLZJ
6
~~
6
4
—
3
4
—
25
4
6
6
—
~~
8 !
6
8
1
8
5
5
1
25
—
10
8
15
10
12
3
—
—
18
15
6
15
50
50
5
20
40
30
40
3
40
дуговой
спектр
искровой
спектр
ГВ8]б>
120
—
—
150
—
400
120
—
—
—
—
120
120
—
—
—
100
—
—
—
120
40
120
25
15
20
25
30
80
100
100
30
20
120
25
50
1 25
120
100 (?)
300 (?)
150 (?)
—
120
50
100
3 |
150
150 |
__
100
40
—
300
100
150
150
--
__
100
100
150
„
80
150
100
—
120
10
120
80 Г
200
150
120
60
40
25
150
100
80
150
150
150
60
100 (?)
300 (?)
150 (?)
200
40
250
Классификация
в)
спектр
II
I
II
Нскл.
I
II
I
I
II 1
II
II ,
II
I
I
II
II
II
I
II
II
II
I
II
I
II
II
II
II
II
II
Некл.
I
Некл.
II
II
I
" И
II
II
I
II
II
II
II
I
11
нижний
г)
терм '
w6./2
6U
вК4./2
Некл.
А,
в*51/г
5£в
bKs
6LBi/2
eA,6i/2
в*41/2
**41/,
А,
АЙ
4K5i/2
'^l/,
4/51/2
5*.
e"3i/a
в*51/2
4/41/2
'М,
в*41,2
ьи
в/31/2
У41/2
в^61/2
6/^51/2
4/
У51/2
Некл.
л7
Некл.
*Кьщ
4^41/2
>Кь
4/41/2
•Мв1/,
^61/2
bh
•*61/,
4/41/2
Ш«1/2
'L51/2
bKs
•*41/,
верхний
Д)
терм
135 Л
19б5
108Л31/2
Некл.
235,
П8Л41/2
198,
2054
Н4Л61/2
14^5./2
109Л31/г
пол31/2
2398
278,
159Л61/,
1'7Л61/2
142^41/2
245,
158Л31/2
12М41/1
1°™51/2
266,
"^31/,
204,
158Л31/2
108/.31/2
Н4Л51/2
Н5Л61/.,
»2^51/,*
Н5Л61/2
Некл.
2458
Некл.
125Л51/2
109Л31/,
2165
1Ю^31/2
«5^61/,
124Л51/2
1 2564
1 127Л51/2
И^31/2
1 159^6i/2
П4Л61/а
220,
120Л41/а
32
Продолжение табл. 18
Длина волны, А
[67]
4 641,658
4 631,620
4 627,079
4 622,427
4620,219
4 605,148
4 603,665
4 601,130
4 584,847
4 581,724
4 576,635
4 573,687
4 569,913
4 567,687
4 555,095
4 551,979
4 545,580
4 543,632
4 538,190
4 516,725
4 515,280
4 510,320
4 506,223
4 490,835
4 477,710
4 472,335
4 469,328
4 465,133
4 462,974
4 434
4 433,889
4 427,653
4 393,588
4 371,760
4 362,05
4 355,741
4 347,192
4 341,688
4 328,742
4313,147
4 297,112
4 288,841
4 269,613
4 252,426
4 246,261
4 244,372
[б8]б)
(,657)
(,624)
(,083)
(,428)
(,234)
(,154)
(,660)
(,130)
(,850)
(,722)
(,643)
(,682)
(.915)
(,688)
(,096)
(,983)
(,581)
(,628)
(,185)
(,730)
(,283)
(,310)
(,210)
(,830)
(,700) ;
(,322)
(,320)
(,124)
(,972)
(,526)
(,890)
(.644)
(,600) !
(,764)
(,051)
(,748)
(,728)
(,134)
(,838)
(,262)
дуговой
спектр
Интенсивность
искро-
спектр
[67f>
10
30
30
12
25
12
25
18
10
8
12
30
25
20
20
15
20
50
25
15
25
20
6
18
20
50
12
20
18
—
15
12
40
18 :
30 1
10
18
50
20 1
20
18
20
20
15
30
25
15
3
60
18
12
25
40
25
15
18
2
40
40
40
40
1
25
80
40
1
40
30
12
25
25
80
1
25
30
—
12
15
6
1
3 1
20
18
50
1
1
18
2 1
30
20
2
25
дуговой
спектр
искровой
спектр
Г68]б)
8
100
40
8
300
12
40
25
20
8
80
50
50
40
25
80
80 д
100
30
80
50
30
10
20
15
100
100
30
40
30 i
15 1
20 1
200
юо
300
80
60
100
200
100
—
80
50
200
80
200
120
200
100
80
80
20
200
200
160
125
20
150
500
120
20
250
120
150
120
75
500
20
120
200 '
150
150
100
40
15
30
20
8
20
20
10
—
Классификации
в)
спектр
II
I
11
II
I
II
II
II
II
II
I
II
II
II
II
I
II
II
II
I
II
II
II
II
II |
II
I
II
II
II
II
II 1
I
I
I
I
II
II
I
I
II
I
II
II
I
II
нижний
г)
терм
*М7Щ
5Le
ввМ61/2
в^-71/»
'/Ив
4*51/2
•*5V.
1К*1<*
6^61/2
W7i/t(?)
5АГ5
;K5i/,
51/2
6^61/2
6^71/,
5#5
вК51/.,
6*41/."
* А/2
4-61/.
4/41/2
L6l/2
6^41/2
в^51/2
5^в
*КгЛ1ъ
У1/2
Ml/a
113Д41/о
71/2
6Г
51/2
c7
5Lr
*^6 1
5к5
or
b5i/2
5K5
A?
л*1
6L61/, i
4W
5LR
41/2
я
верхний
термД)
21M71/2
215e
16M61/2
169Л71/о
i 278,
1 178Л51/2
I3^5»/2
12M4i/,
12™61/S
23M71/
224e
'34ЛБ1/3
Л9Л41/„
128Л6]/:
2зол71/:
2256
136^sl/o
124ЛМ/:
130i451/4
227e
120^41/,
П9Л41/,
51141/2"
132Л51/2
125Л51/2
12М41/,
223,
142Л41
126Л41/2
52541/3
' 240Л71/,
123Л41/"
2276
3098
229,
235в
131441/,
126Л41/3
237в
2698
127.45i/,
295,
145^*61/.
235,
12™5.,2
33
Продолжение табл. 18
Длина волны, А
[67]
4 241,669
4 231,676
4 222,375
4 171,591
4 169,050
4 162,430
4 156,652
4 153,976
4 141,228
4 128,336
4 124,725
4 116,097
4 106,931
4 095,746
4 091,636
4 090,135
4 088,254
4 080,609
4 071,108
4 067,756
4 062,549
4051,914
4 050,039
4 047,610
4 044,416
4 042,752
4 026,019
4 018,990
4017,723
4 005,698
4 005
4 004,063
3 990,423
3 985,795
3 966,567
3 954,663
3 943,820
3 932,026
3 930,982
3926,216
3915,884
3 911,673
3 902,561
3 896,779
[б8]б>
(,669)
(,365)
—
(,055)
(,431)
(,656;
(,972)
—
—
—
—
(,906)
—
(,635)
—
—
—
—
—
—
—
—
(,621)
—
(,75(>)
(9,04)
—
(,696)
(,212)
—
.._.
—
(,52)
—
(,820)
—
—
(,218)
—
—
—
дуговой
спектр
Интенсивность
искровой
спектр
[67]а>
40
25
18
30
20
18
15
15
20
18
30
25
25
18
12
25
25
12
15
12
12
20
25
18
18
4
25
25
25
25
15
18
25
20
20
35
35
12
30
20
18
18
20
50
—
8
30
2н
Зн
8 н
5
30
20
25
35
10
25
—
40
18
20
25
20
18
25
35
3
25
10
25
15
25
3
—
20
20
30
30
30
5
50
35
6
30
18
18
25
дуговой
спектр
искровой
спектр
[68]б)
60
150
—
50
60
100
150
—
—
—
—
8
—
50
—
—
- -
—
—
—
—
—
80
—
150
—
8(?)
—
50
80
--
—
—
—
—
200
—
—
50
--
—
—
20
30
—
10
15
10
15
—
—
—
—
—
—
3
—
—
—
—
—
—
—
—
8
-1
15
—
—
—
3
4
—
—
—
6(?)
—
10
—
—
5
—
—
—
Классификаци
в)
спектр '
II
I
I
II
I
1
I
I
II
II
II
II
II
Некл.
I
II
II
Некл.
»
II
II
II
II
I
II
II
II
I
11
II
II
Некл.
I
II
II
II
Некл.
II
I
II
Некл.
I
II
II
II
II
нижний
г)
терм
W6i/.
Aa
А7
e^6i/2
с,
А,
5К5
ъц
6Л171/2
4/
6 /2
6^6l/2
4/
1 41/2
4/
7 4 1/2
Некл.
Б/
6V2
4/Г41/2
Некл.
»
4^С1/2
4'41/,
'W.
4/
741/а
5К5
с/з./2
"Кь
I*5'''8
^51/2
'41/2
Некл.
5/4
в/-6./2
$^41/о
8L71/2"
Некл.
Я4'41/,
*L«
J i/ 2
Некл.
:'Ц
*4'41/.
751/2
6/
51/-> 1
•Л,6.,;
я
верхний
терм
3129
2748
153i46i/2
3217
2788
246,
2407
252^6i/2
'92Л61/!,
159i461/2
13^51/,
139Л3)/2
Некл.
244,
161^61/в
140^4./3
Некл.
»
237Л61/2
'4^51/..
2im71/:
142<V/
2535
198Л51/2
214^!61/2
224Л41/.
253,
170^41,.
145^01/2
2«л41/2(?)
Некл.
2943
167Л61/„
'58^31/,
230Л71/^
Не:;л.
П0Я51/2
253,
'53^01/г
Некл.
254,
)12^51/2
214<V/2
157.4
2^5./2
3 Химия урана
со со
4*> 4^
00 СО
СО О
со to
-vj 4^
н- Ю
СО 1
cd 1
>S
to —
о to
to
— о
00 1
о 1
1
00 1
Ю Ю
2S8
01 ^
сл
м
СО
сл
8
О
-vj
-nJ
о
-vj
^о
сл
to
00
о
оо
Сл
с»
ю
оо
CD
СО
сл
о
->J
со
4^
4^
СО
4^
to
о
со
00
о
00
СО
сл
о
СО
cd
cd
оо
I
1
о
сл
1
1
1
1
Сл ,U
се »ь.
to"
Ю
ОО
сл
о<
00
со
ь.
СЛ
i
СО
сл
4^
cd
СЛ
CD
lO
00
СЛ
to
о
о
to
о
d
Г-
to
00
4^
сл
СО
ОТ
СО
, .
►—
со
1
1
оо
to
о
1
1
1
1
о
to
CD
!b»
т
л
СО
СЛ
СО
СО
СЛ
CD
оо
1
1
о
to
о
1
1
1
1
в»
ах
CD
>1
а>
сл
'"
со
СП
СЛ
о
оо
to
to
1
1
to
to
о
1
1
1
00
сл
4*
СЛ
*
СО
сл
cd
~
00
о
о
оо
о
►£*
to
со
о
оо
о
1
ПС
го
^
£з
X
ГС
fa
2
2Г
се
Р
СО
СЛ
a
сл
CD
00
СЛ
CD
СЛ
со
о
о
to
о
о
ю
о
Сл
>:
сл
to
00
-vj
w-l
СО
СЛ
CD
CD
Ы
to
8
з
to
to
о
оо
•>D
1
1
X
Т>
т:
Sa
X
rt>
fa
г™
со
сл
00
4*.
оо
■^J
CD
8
з
СО
О
to
to
сл
о
сл
о
Сл
г-
OS
to
-V)
00
СО
сл
CD
-vl
4^
-vl
-vl
4^
CO
О
to
сл
о
to
Сл
ел
to
оо
4^
«к
СО
CD
О
00
CD
О
1
1
00
о
1
*
«
со
CD
О
СО
$
to
CD
00
to
СЛ
to
о
1
1
X
п>
я
fa
CO
CD
to
CO
о
сл
сл
1
I
to
сл
1
1
1
1
X
fa
CD
О
Ь.
o>
1
CO
a
о
-nJ
со
со
1
1
00
to
о
1
1
1
1
ГС
а>
«
fa
X
fa
X
fa
со со
CD CD
СО 4^
00 О
Ю СО
3 оо
»—i |
СО 1
СЛ
СЛ 00
to
to о
СЛ 1
о 1
о 1
to
со —
— СО
tO -vj
« ^
СЛ
li
со
CD
4>-
4^
to
4^
сл
to
4^
•—•
00
to
CD
О
4^
со со со со
CD CD ->J -vj
СЛ ->4 О О
CD О О >—
— о сл сл
СЛ -v) -v) tO
СО tO СЛ tO
— 1 ! !
s
сл ел to о
— — to
^- 00 00 СЛ
8 111
■—111
о 1 1 1
со со со со
-vj -vj -<1 -Ч
СО 4^ CD 00
— оо со о
4^ CD tO -vj
СЛ -vj CD ►—
CO 00 CD CD
^ 1 Ю 1
сл 1 s I
3 3
to сл to сл
1 to to to
1 o^ сл о
О 1 4^ 1
о i о !
ё 1 « 1
£=я 1 . . 1
f СЛ О» в OS с Ч, 41 О»
СЛ в1 rfk. О». О в "N* £ь
К- l_ »- ►£». >-
to to ьэ -С ю
to
s
А
to — — го
Ч 00 Q0 СП
00 СЛ 4^ tO
* 4* ^ Ь.
Сл Сл с
li lv К,
ч>
ю — со —
CD О СЛ "^J
СО *Ч СЛ СО
« Сс"41 ^
Со См
со
-v}
оо
to
оо
4>ь
1
1
to
сл
со
о
1
1
1
1
СЛ
~
CD
-vj
^
ст.
СО
^1
оо
со
оо
4^
о
1
1
to
о
to
сл
1
1
1
1
>;
сл
to
сл
о
4^
с
* tv
со
00
*-*
со
СО
СО
со
СО
сл
00
CD
сл
о
сл
со со
оо оо
СО 4^
■^J О
со -^J
— о
1 1
1
to to
о сл
сл сл
1 1
1 1
1 1
1 1
Ст СЬ в>
* СЛ ^
К. 1С
ю
CD
to
ся
1
CD -*J
о о
:ь ^
СЛ *U
V 1*
со
9°
СО
•-'
4^
CD
сл
1
1
to
сл
to
сл
1
1
1
1
CO
со
оо оо
со
со
о
to
со
1
1
to
о
в
Сл
1
1
1
1
X
гт>
?;
fa
Сс X
• CD
4
о
со
to
Ох
X
<о
CD
со
to
CD
to
4^
со
о
to
to
to
«J
CO
00
СЛ
4*.
CD
СЛ
СЛ
1
1
to
о
CO
о
1
1
1
CO CO
00 00
СЛ CD
CO СЛ
СЛ CO
oo to
о со
1 1
1 [
to to
о о
CO to
о сл
1 1
i 1
1 1
tC k2 ^
^2 cP
to
to
CD
CO
(
о
to
tX3
J^
CD 00
м- СП
!ь ^
О СЛ
* lv
со
оо
■^
"^
о
4^
to
^^
р
Со
СЛ
со
о
to
о
to
со со со со
оо оо оо оо
оо оо со со
— to о ю
4^ СО CO CD
CD CD CD 00
— .— 4^ 4^
4-
1111
1 1 [ 1
со — со to
о оо сл о
to — со со
О 00 О О
1 1 1 !
Mil
1111
мм
_
сл о» с* а» о»
г- > г- г- г-
^- t«" w" w"
to
СЛ
оо
сь
1
to »— — to
СО S СЯ Д
О 00 СО О
^ ^ ^ :ь
М СЛ С) S
i lv 1С tv
со
оо
CD
4=»
|—>
to
со
1—1
to
^—
СО
о
4^
to
о
to
Сл
г-
а»
to
сл
CD
^»
'аз
"о>
оо
'"З1
"ci
1—w
00
"Тэ
дуговой
спектр
искровой
1 спектр
8$
н »
•a °
S
о о
X О
ч го
п
a
тэ
11
3»
а
о тэ
2 =
>з 2.
5=3
ь
X
Си
ш
>•
S
3
о
н
Ь 1
п
■е-
х
JS
S
»
^3
о
о
3
35
Продолжение табл. 18
Длина волны, А
[G7]
3 424,557
3 423,05
3 422,352
3 390,389
3 341,661
3 322,118
3 293,590
3 288,209
3 270,124
3 265,806
3 263,112
3 229,502
3 200,135
3 177,331
3 176,208
3 153,120
3 149,21
3 147,09
3 145,559
3 142,601
3 139,560
3 126,174
3 111,621
3 104,162
' 3 095,041
3 093,012
3 091,25
3 088,987
3 084,238
3 080,741
3 073,812
3 072,783
3 062,536
3 057,91
3 047,571
3 039,263
3 031,991
3 029,131
3 025,034
3 024,510
3 024,385
3 016,956
2 967,894
2 966,12
[б8]б>
(,55)
—
—
(,389)
—
—
(,590)
(,248)
—
—
(.Н6)
—
—
—
—
—
—
—
—
—
--
--
—
—
--
---
-
—
—
-
—
—
—
—
_..
__
— -
—
—
--
—
дуговой
спектр
Интенсивность
искровой
спектр
[*7]а>
20
12д
18ш
18
12
18
30
25
20
25
25
18
15
15
20
12
18Д
12
12
12
25
12
15
20
12
20
15ш,д
20
15
12
15 ш
20
12
20 д
15
15
15
20
12 н
18
25 ш
15
15
15
15
15 д
15ш
10
15
12
5
20
25
18
8
25
15
18
15
15
18д
15
10
10
25
20
15
20
12
20
12ш,д
15
12
12
10ш
20
15
20 д
20'
12
15
15
20 н
15
20 ш
12
20
25
дуговой
спектр
искровой
спектр
[68]б>
20
—
25
—
—
4
3
—
—
20к(?)
—
—•
_.
—
--
—
—
—
--
__
—
—
_
_
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
:{
-{
30
_..
—
2
2
—
—
--
—
— •
—
_..
—
__
- -
—
—
—
—
—
—
_...
_..
—
—
—
___
_
—
_..
—
— i
—
—
—
—
—
—
"
Классификация
в)
спектр '
II
II
II
II
II
Некл.
II
II
Некл.
II
11
II
I
Некл.
»
»
»
»
»
»
и
Некл.
II
Некл.
»
»
II
Некл.
»
»
II
II
II
Некл.
II
Некл.
»
II
Некл.
»
II
Некл.
*
II
Некл.
»
нижний
г)
терм
'L61/2
в*4Х/2
в/
в#41/2
в/
1 31/2
Некл.
в*51/->
«ме1;2
Некл.
в.Яз1/2
в^51/2
в^51/2
5и
Некл.
»
»
»
»
»
»
•*«„
Некл.
в//21/3
Некл.
»
»
в^61/2
Некл.
»
»
в^51/2
в^61/2
41/2
Некл.
в^61/2
Некл.
»
в£71/2
Некл.
»
'07В3./2
Некл.
»
"L6i/2 I
Некл.
»
верхний
Д)
терм
240Л71/2
224Л41/2
|274Л31/2
225Л41у2
275Л41/2
Некл.
250Л61/2
277Л51/2
Некл.
298Л41/о
,237Л51/;
239Л41/2(?)
306в
Некл.
»
»
1 »
| »
»
г>
31М6./,
Некл.
30бЛ31/2
Некл.
»
»
267Л61/2
Некл.
»
»
278ЛС1/з
268Л51/2
326/<61/1
Некл.
27W51/S
Некл.
»
320Л71/а
Некл.
»
60741/2
Некл.
)>
28ол51/а
Некл.
.»
3*
36
Продолжение табл. 18
Длина волны, А 1
167]
2 960,942
2 956,060
2 954,39
2 943,895
2 941,919
2 932,61
2931,414
2 930,805
2 928,599
2 925,568
2 914,629
2 914,253
2 908,275
2 906,913
2 906,798
2894,512
2 889,627
2 887,252
2 882,741
2 875,198
2 870,974
2 865,679
2 864,276
2 860,466
2 858,903
2 852,750
2 849,480
2 839,890
2 833,821
2 832,063
2 828,90
2 826,193
2 824,28
2 821,122
2817,959
2811,345
2 809,952
2 807,05
2 802,559
2 795,232
2 793,937
2 762,850
2 754,155
2 748,451
2 746,158
2 706,95
[68]б)
—
—
—
—
—
(.41)
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
•—
1 __
—
—
—
—
—
—
—
—
—
~
дуговой
спектр
Интенсивность
искровой
спектр
[67]3)
15
10
12
10
15
10
12
4
15
15
12
18
12
18 ш
15
15
30
25
18
18
18
30
18 и
35
35
15
18
18
15
35
18
18
25
20
18
| 35
20
18
15
18
25
15
20
18
25
15д
25
60
15
25
30
25
12
30
35
25
15
25
30
15 н
50
15
50
25
20
12
20
50
12н
30
25
15н
15
20
25
50
20
12н
30
35
30
30
20
30
30
12
. 30
20
35
15
6
20д
дуговой
спектр !
[68
_
—
—
—
—
—
20
—
—
—
—
—
—
—
' —
—
__
i —
__
—
—
—
—
—
—
—
~
—
~
—
—
—
__
—
—
—
_
—
—
—
—
—
—
__
—
~
искровой
спектр
7s '
_
—
—
—
—
—
10
—
—
__
—
—
, —
—
—
—
—
—
—
—
—
_
_
—
—
—
"
—
—
—
—
—
— -
—
—
—
—
—
—
—
—
—
—
—
—
| "
Классификация
в)
спектр
II
Некл.
II
II
II
Некл,
II
Некл.
»
»
II
II
II
Некл.
II
Некл.
II
Некл.
II
II
II
II
Некл.
II
II
Некл.
II
Некл.
»
»
»
II
Некл.
II
Некл.
»
»
II
II
И
II
Некл.
*
*
*
*
нижний
г)
терм
•*41/,
Некл.
в*51/2
•*51/,
в^41/2
Некл.
в^5Ч2
Некл.
w
w
'в*41'2
6^51/2
в^51/2
Некл.
4^41/2
Некл.
6^51/2
Некл.
^51/2
>'2
^51/9
^41/а
Некл.
4^41/2
4/41/2
Некл.
6/
51/2
Некл.
»
»
»
4/
41/2
Некл.
Д41/2
Некл.
w
W
?51/3
6^51/2
А41/2
1 в^51/2
Некл.
»
»
»
»
верхний
термА'
277Л51/!
Некл.
301Л
зозл41/2
281Л
Некл.
270Л51/2
Некл.
»
»
286Л
273Л51/2
276Л61/2
Некл.
270Л51/2(?)
Некл.
280Л51/з
Некл.
284Л
292Л51/,
285Л
28^51/2
Некл.
283Л41/1>
284Л
Некл.
290Л
Некл.
»
»
»
290Л
Некл.
305Л41/2
Некл.
»
»
295Л51/2
296Л41/,
307Л
298Л41/2
, Некл.
»
»
»
»
37
Продолжение табл, 18
Длина волны, А
[67]
2 698,057
2 695,490 !
2 691,038 |
2 683,279
2 664,153
2 645,47
2 635,528
2 608,197
2 597,689
2 591,252
2 565,406
2 500,864
2 403,423
2 378,16
2318,47
2 306,91
2 282,78
2 276,05
2 248,03
2219,28
2 158,61
[68]6>
—
™
—
—
—
—
—
--
—
_-
—
—
—
—
—
__
—
—
Интенсивность
дуговой
спектр
искровой
спектр
[67]а) 1
20
12
15
25
18
20
25
25
25
18
30
18
—
—
—
—
—
—
—
15
50
30
30
25
20
25
50
6
15
12 !
30 :
12
30
35
25
25
25
20
25
2
20
дуговой
спектр
искровой
спектр
[68]б> ~
_ ;
—
—
—
"
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
__
,
—
—
—
—
—
—
—
—
—
—
—
—
—
Классификация
в)
спектр '
II
II
II
Некл.
»
II
II
Некл.
»
II
II
Некл.
»
ъ
»
»
»
»
»
»
»
нижний
г)
терм
6^51/2
;*5i/2
6^61/2
Некл.
»
4/4i/2
4/41/2
Некл.
»
4/41,2
4/
М1/-2
Некл.
»
»
»
»
»
»
»
»
»
верхний
д)
терм
312Л
313Л
324,4
Некл.
»
316Л41/2
318Л щ%
Некл.
»
32Ы41/2
326/l5i/2
Некл.
»
»
»
»
У/
»
»
»
ъ
а) Интенсивность линий дугового спектра дана для стандартной электрической дуги постоянного тока
(сеть 220 в, стабилизованная омическим и реактивным сопротивлениями). Интенсивности линий
искрового спектра даны для конденсированной искры стандартного типа на 20 000 в при искровом
промежутке 5 мм. Сокращения в графе интенсивности: д— неразрешенный дублет (две линии почти
совпадают); н—неясная, расплывчатая, диффузная, завуалированная; ш — широкая самообращенная.
Интенсивности определены визуальным сравнением фотографических плотностей с приближенной поправкой на
различную чувствительность пластинок. Для большей близости к фактическим соотношениям интенсив
ностей была увеличена шкала [67].
б) Приведены также некоторые неопубликованные данные [69]; в графе для длин волн приведены
лишь десятичные знаки для сравнения с данными в предшествующих графах. Интенсивности даны в
произвольной шкале от I до 500. Сокращения: дл—оттененная или смещенная в сторону более длинных
волн; ш —широкая самообращенная; к—оттененная или смещенная в сторону более коротких волн; н —
неясная, диффузная, завуалированная; д—неразрешенный дублет (две линии почти совпадают).
в) I означает принадлежность линии к нейтральному атому урана, II —принадлежность к иону с одним
положительным зарядом.
г) Все термы LJI в этом столбце являются нечетными; нижние термы UII частью нечетные
(группа А), частью четные (группа В); об отнесении терма к одной из этих групп см. табл. 23 (стр. 4 8).
В табл. 21 (стр. 42) и 23 даиы значения термов UI и UII соответственно.
д) Для объяснения использованных символов термов см. табл. 22 и 24 (стр. 4 3 и 50), где
приведены значения термов.
е) Некл.—неклассифицированный
3S
Часть I. Уран как элемент
Таблица 19
УСТОЙЧИВЫЕ (ПОСТОЯННО ВСТРЕЧАЮЩИЕСЯ) ЛИНИИ УРАНА
ПО ДАННЫМ РАЗЛИЧНЫХ АВТОРОВ
Длина
волны, А
3019
3090
3102
3552,172
3672,579
3859,58
3932,02
3966,57
4090,14
4241,669
4341,69
4620,22
5027,38
5915,40
5919,61
6395,42
6449,10
Интенсивность [67]а)
дуговой
спектр
8
8
40
искровой
спектр
12
15
50
Интенсивность6) [78]
600
400
500
200
300
Порядок появления6)
[78]
(5)
(4)
0)
(3)
(2)
[79J
(3)
0)
(4)
(2)
(3)
Таблица 20
СРЕДНИЕ СМЕЩЕНИЯ ЛИНИЙ
(U236-U238) в UII-спектре [92]
а) Определено фотографически.
б) Определено визуально.
В табл. 20 приведены средние значения смещений для линий спектра UII
различными нижними термами. Изучены изотопические смещения для
U235 и U233 [85]. В дуговом спектре в области от 2500 до 4800 А
заметные изотопические смещения
отчетливо наблюдаются более чем для 900 линий,
а в области с длиной волны большей, чем
4800 А, смещение заметно только для двух
линий (X равна 6101,74 и 6465,0).
Смещение наблюдается для одних и тех же
линий и в случае U235 и в случае U233.
В ряду U238 -> U235 -> U233 все смещения
наблюдаются в сторону более коротких
волн. (Как видно по табл. 20, МакНэлли
наблюдал также смещения в
противоположном направлении.) Никаких
количественных зависимостей между разностью
масс и величиной смещения не обнаружено.
Одиннадцать приведенных в таблице
типичных смещений занимают интервал 0,14—
1,37 см1 для пары U239—U235 и 0,07—1,06 см'1 для пары U236—U233; никакой
пропорциональности или хотя бы строгого параллелизма между обоими
рядами смещений не имеется.
Как видно из сравнения с табл. 18, все эти 11 линий [85] принадлежат
спектру UII; в согласии с табл. 20, наиболее сильные смещения (от 0,93 до
1,37 еж"1) проявляются у линий с нижними термами, имеющими конфигурацию
/3 s2. Показано, что величина изотопического смещения может быть исполь-
Электронная
конфигурация
нижнего терма
/3S2
Pds
f3d2
/*s
f<d
Смещение линии
и235—и238
см~1
0,9
0,2
-0,2
огз
-0,4
Глава 2. Свойства атома урана
39
зевана для спектрографического определения соотношения изотопов урана
в данном образце, и приведены примеры, показывающие применимость этого
метода [85].
На рис. 6 показана линия 4244,4 А в спектрах трех чистых изотопов и трех
двойных смесей. Смещения /,v равны соответственно 1,37 см'1, 0,70 см'1
и 1,37+0,70, или 2,07 см'1. Соседняя линия 4241,7 А претерпевает лишь едва
заметное смещение.
Сверхтонкая структура. Изотопы урана с нечетным массовым числом
U235 и U233 должны иметь ядерный спин, отличный от нуля, и их линии должны
поэтому обладать сверхтонкой структурой. По первоначальным наблюдениям,
сделанным в Англии [86], ядерный спин IF5 равен 9/2, но по более современным
данным [83] значение спина
5/
/2
или, что также возмож-
равно /2
по, 7/3. В интервале от 4000 до 8500 А при помощи метода,
основанного на применении эталона Фабри—Перо, были
обнаружены только две линии (5915 и 6926 А), тонкую
структуру которых оказалось возможным разрешить и измерить.
Первая из этих линий, с величиной интервала тонкой
структуры 0,21 см'1, была использована для оценки ядерного спина.
Влияние электрического и
магнитного полей н а спектр. Был измерен эффект
Штарка для некоторых линий дугового спектра урана [87], а
также смещение из-за остаточного давления, являющееся по
существу результатом влияния электрических полей (эффект
Штарка) соседних атомов [881.
Эффект Зеемана для линий спектра урана изучался очень
подробно, так как результаты такого исследования дают
возможность наилучшим образом идентифицировать
спектральные термы сложных спектров. Первые измерения были
сделаны Россом [89]. Совсем недавно точные измерения были
выполнены с магнитом Вейсса (42 000 эрстед) в лаборатории
Зеемана (Амстердам), с электромагнитом Биттера (85 000
эрстед) в Массачусетс ком технологическом институте и с
магнитом Вейсса с водяным охлаждением (28 000—35 000 эрстед)
в Национальном бюро стандартов.
Подробно опубликованы лишь результаты исследования
голландскими исследователями [76, 90]. Наиболее современный обзор данных
по изучению эффекта Зеемана (имевшийся к моменту написания настоящей
главы") можно найти в статье ван-ден-Боша и ван-ден-Берга [90]. В ней
приводятся данные о полностью или частично разрешенных структурах
эффекта Зеемана для 115 линий U1 в интервале 5511—2971 А и 158 линий UII
в интервале 5504—3232 А, а также даются значения квантового числа J и
величины g термов. В статье, кроме того, имеются фотографии и
фотометрические кривые эффектов Зеемана для следующих линий UII:
Рис. 6.
Изотопические
смещения линий
4241,7 (Л) и
4244,4 (В). в
спектре урана.
Приведенные
спектры (сверху вниз)
снимались со
следующих образцов:
,_и238. 2_и238 +
Т233.
+ и2ЯЗ; 3-
4_и23Г,+иа
5_и233;6—U23
+ и233.
проведенного
4646,61 [76,90]
4543,64 [76,90]
4515,29 [76,901
4472,34 [76,90]
4393,60 [90]
4341,69 [76,90]
4050,05 [90]
3932,03 [76,90]
3890,37 [76]
3859,57 [76]
3831,47 [76]
3670,08 [76,90]
3270,13 [90]
Результаты изучения эффекта Зеемана в Массачусетсом технологическом
институте не опубликованы, за исключением данных о 19 линиях, имеющихся
п
п
U\\X=4341,69 \}\\ Х-4472,34 \}\\ Х-4515,29 U1I Х-4543,64
233g/-fJdsL
ч/г
226g/2-f dsL„/2
224g/2-fadsL11/2
2291l/2-fdsK9/2
U I! A -4846,61
224g/2-f3dsKg/2
\5\\X-3270,13 UllX-3670,08 Ш1 X-3932,03 UU X=4050,05 ШХ-4393,60
308
-*з«24,
?3. 2$.
11/2-rdsLu/2 281,1/2-fJdsK9/2 257w/2-fJdsLn/2 247g/2~fJs2 Ig/2 227g-f3ds2%
Рис. 7. Эффект Зеемана для нескольких линий в спектре Uf[90].
Глава 2. Свойства атома урана
41
в раннем сообщении [91]. В более позднем сообщении [92] приведены рисунки
эффекта Зеемана для линий UII 4244,372; 4341,688 и 4543,632 А.
Эффект Зеемана определен также для нескольких тысяч линий спектра UI
[68]. Большинство линий давало или триплеты, или неразрешенные картины,
но те немногие, хорошо разрешенные картины расщепления, которые
удалось получить, оказали существенную помощь при анализе спектра Ш,
позволив определить для термов значения g и квантовые числа«/. В указанной выше
статье [68] в качестве примеров приведены картины эффекта Зеемана только
для семи линий.
На рис. 7 показаны 10 примеров магнитного расщепленияслиний урана [90].
На рис. 8 показаны^- и о-составляющие линии UII4341,69A; снимки сделаны
в Массачусетском технологическом институте.
t i й t t t ;i- г г -1
I t ш | 1 § I i i i § § § I
Рис. 8. Картина эффекта Зеемана в спектре UII для
длины волны 4341,688 А.
Анализ термов спектра урана. Причина
чрезвычайной сложности спектра урана та же, что и в случае редкоземельных
элементов. Эта сложность обусловлена тем, что в наиболее часто встречающихся
состояниях атом урана и его ион с одним положительным зарядом
[обладают несколькими (обычно тремя, а иногда четырьмя) /-электронамиЛ'Орби-
тальный момент этих электронов велик (/ = 3), и разрешенные изменения в их
взаимной ориентации приводят к большому разнообразию общих
орбитальных моментов L. В табл. 21 показаны, например, термы Ш
конфигурации f3d2s со значениями L вплоть до 9 (М-термы). При параллельной
ориентации орбитальных моментов всех шести валентных электронов значение L может
дойти до 13, но до сих пор термов такого типа в урановом спектре не было
обнаружено. Параллельная ориентация спинов всех шести валентных
электронов (s=1/2) приводит к общему спиновому моменту S, равному 3. Таким
образом, проявлением высшей мультиплетности могут быть септеты (7=2-3-f-l).
Максимальное значение общего спинового момента S иона U+ равно 2х/2> и
поэтому максимальная мультиплетность равна 6(2-21/2-|-1). Установлено
наличие септет- и квинтет-термов в спектре UI и секстет- и квартет-термов в
спектре UIL Термы меньшей мультиплетности пока обнаружить не удалось.
42
Часть I. Уран как элемент
Таблица 21
НИЖНИЕ НЕЧЕТНЫЕ ТЕРМЫ UI
Электронная
конфигурация3)
Pdb*
Pds*
(р ds2?)
(Р ds2?)
f*ds*
Pds*
pd*s
Pds2
Pd4
P d2s
?
(P ds2?)
(pds2?)
(Pd2s>)
(Pd4?)
(Pas2})
?
?
Символ
терма'1)
*L6
6/c5
.4,( = 5Z.-?)
A*
5/Co
J/4
'/C56'
bHt
7MU
**.e>
5,6)
B7(=5AV)
He(-5V)
С,(,-,'УИ7?)
Z>7( = '/C,?)
Я. ( = «*.?)
A,
A,
Значение
с Af-
iGS]
0,0
620,3
3 800,8
3 868,4
4 275,7
4 453,4
5762,0
5 991,3
6 249,0
7 005,5
7 103,9
7 326,1
7 645,6
8118,6
10347,3
10 685,7
11545,4
13 127,9
терма,
l
[76]
0,00
620,33
3 800,83
4 275,75
—
—
—
16249,04
—
—
—
—
8 118,64
—
—
—
—'
теоретическое
для LS-
связи
0,714
0,667
—
_-
0,905
0,600
0,767
0,900
0,571
0,976
—
—
—
__
—
—
—
—
Значение Q
г
наблюдавшееся
[68]
0,75
0,73
—
_..
0,93
0,66
0,82
0,86
0,62
0,97
—
—
—
—
._.
—
—
—
[90]
0,75
0,73
0,92
—
0,91
0,67
—
—
0,62
_.._
—.
—
—
0,81
—
—
—
—
л) Вопросительный знак указывает па ненадежность идентификации.
б) По МакНэлли [69], все эти уровни, возможно, имеют конфигурацию f3ds2.
В таком случае они могут быть соответственно обозначены через ь1ь, а/« и 5бз.
В случае таких сложных спектров, как спектр урана, картины эффекта
Зеемана могут служить исходными при анализе, позволяя определить
значения g расщепляемых термов. Результаты можно затем сравнивать с
теоретическими значениями термов определенных типов (т. е. с определенными
комбинациями значений L, S и У). Однако теоретические значения g,
рассчитанные на основании идеальной 15-связи (связь Рассела—Саундерса), могут
ввиду большого атомного веса урана значительно отклоняться от
опытных значений g\ несколько лучше могут подходить условия Jy-связи
(аналогичное положение наблюдается в спектрах редкоземельных элементов).
Как упоминалось (стр. 27), данные об изотопическом смещении также могут
быть использованы при анализе спектра урана, хотя они менее специфичны,
чем выводы из эффекта Зеемана..
Анализ спектра UI (спектра нейтрального атома урана). Результаты
исследований спектра UI, приводившиеся Национальным бюро стандартов,
опубликованы в одной из статей [68], которая дополняет предшествующие
неопубликованные отчеты [78, 93J. В ней содержится перечень 18 нижних
уровней, которые соответствуют нечетновалентным электронным
конфигурациям 5/36d7s2 (теоретически только одна эта конфигурация может дать 158
термов с 386 составляющими) H5/36d27s (дающей теоретически 1122 терма с 3256
составляющими). Эти термы указаны в табл. 21. Некоторые из них
идентифицированы (не во всех случаях вполне надежно) на основании значений g,
43
ВЕРХНИЕ ЧЕТНЫЕ ТЕРМЫ UI
Таблица 22
Обозначение
терма и
значение Уа)
П6,
1225
1345
14бб
15б7
15б6
1575
1614
1656
1697
Н395
170б
1736
1744
1785>0
1784
1795
1793
1814
182в
1845
J854
185.,
187в
1895
1914
1924
1945
1948
1954
1965
1963
1974
1976
1977
1987
2015
2014
202в
2035
2033
2046
2047
2058
2054
20б5
206с
2077
Уровень6)- см~1
11614,0
12 227,8
13 463,4 (,42)
14643,9(,87)
15 631,9 (,84)
15638,4(,36)
15 720,7
16121,9
16505,8(,80)
16900,4(,42)
16 929,8
17070,5(,47)
17361,9(, 84)
17 468,2
17 848,12г)
17893,8
17 908,2
17968,7
18 186,0
18 253,9
18406,5 (,51)
18526,9
18 530,8
18 759,2 (,20)
18932,8 (,72)
19127,2
19 192,4
19 471,9
19 489,0
19552,5
19 647,5
19668,4
19740,7
19 783,4
19826,7
19885,4(,46)
20114,3
20 148,0
20218,8(,83)
20 311,5
20 391,5
20 420,5
20 464,5
20 528,9
20 569,2
20 621,2
20 661,5
20 766,5
Значение gB) ,
(0,66)
(0,63)
(0,90)
(0,87)
v0,90)
(0,93)
(0,88)
(0,86)
(0,80)
0,91(0,90)
(0,64)
0,98
0,96
0,88(0,92)
(0,80)
-
Обозначение
терма и
значение Уа)
2085
2096
2104
210.5
212G
2135
2154
215б
216*
1 2176
2186
2193
2204
2206
2234
2237
2235
2238
2244
2246
2256
2254
2267
2276
2273
2278
228в
2297
2307
2318
2314
2317
2335
2344
2345
2343
2353
2357
2354
2356
237G
2377
2384
2389
2387
2398
2396
Уровень °}' см 1
20 851,6
20 943,4
21 062,3
21 078,7
21 265,1 (,04)
21330,0
21 545,1
21 584,7(,71)
21637,0
21 766,5
21 768,0
21 940,6
22 038,0
22 056,3
22 365,0
22 368,4(,44)
22 377,7
22 383,1
22 383,4
22 464,3
22 582,7 (,67)
22 584,5
22 633,2
22 754,1 (,08)
22 774,1
22 789,8
22 8b2,4(,45)
22 918,6(,51)
23 057,7
23 110,8
23 186,9
23 197,0
23 325,2
23 430,1
23 432,8(,79)
23 464,1
23 486,7
23 543,5(,50)
23 560,6
23 572,0
23 715,3
23 779,2
23 825,4
23 843,7
23 848,6
23 926,7
23 932,8
Значение gB)
0,90(0,88)
1,04
0,98
0,97
0,83
0,95(0,91)
0,96(0,93)
0,97
1,02(1,00)
0,92
to
CD
CD
IX
Ю
CD
СП
СО
-vl
о
to
00
00
О»
N3
00
00
CD
О
СО
N3 Ю
CD О)
ел ел
to го
CD CD
ел ел
CD СП
05 О
СО 4^
ГО N3
00 00
*-g *-j
«J Cn
КЗ N3
00 00
-vj 0>
со ел
00 О
CO 00
NO
CO
*«—'
3
"*
00
to
tO
CD
ел
*
N3
о>
ел
0>
00
N3
00
ел
-j
N3
00
ел
о>
0>
4^
ю
05
ел
00
N3
0>
4^
СО
N3
"
N0
00
о>
Сл
N3
00
ел
CD
N3
-J
N0
OS
£ь
Ю
о>
4*.
Сп
4^
N3
00
ел
*»
N3
00
ел
4*.
оо
4>
Ю
CD
ОО
~д
tO
CD
оо
СО
*—
СО
N3
00
ел
N3
00
СЛ
О
оо
ел
ю
CD
оо
00
Ю
CD
оо
оо
оо
N3
00
4*.
ез
N3
00
4*
-<1
О
N3
N3
а
сл
N3
CD
оо
о
СП
о
ю
00
4*
I*
N3
00
4ь
ел
4*.
О
N0
CD
N3
«а
N3
о>
ю
->1
4*
00
N3
00
4ь
-J
Ю
00
4^
ел
^
^—
N3
05
N3
<л
tO
CD
ГО
to
ел
сл
ъ
оо
N3
00
4ь
сл
ГО
00
4*.
4^
4*.
СЛ
Ю
0>
•л
N3
0>
ГО
о
00
00
N3
00
оо
о
to
00
4ь
оо
О
СП
N3
05
ее
tO
CD
>—
СО
N3
4*.
to
00
оо
«J
N3
00
N3
00
ел
00
ю
О)
№
N3
CD
i—.
О
оо
-vl
to
00
СЛ
to
00
»—»
00
00
оо
tO N3
ел сл
СО СО
а о»
N3 N3
СЛ СЛ
СО СО
00 00
КЗ —
tO N3
00 00
ю •—
■vl -3
to to
00 00
1 , .
сл •—
N3 00
-J 00
00
CD
to го
ел ел
00 00
О) Сл
N3 tO
ел ел
00 00
N3 О
сл ел
CD О0
О
СО
tO КЗ
00 00
— О
сэ а»
Ю Ю
00 00
О О
СО СЛ
00 О0
СО —
~
ъ
оо
N3
СП
-J
<э»
N3
СП
"*4
СО
*-*
Oi
to
00
О
■а
tO
00
о
4^
00
КЗ
ю ю
ел си
--а о5
оо <а
N3 N3
ел ел
-J CD
оо -g
СО N3
О СП
о
СО
CD
to ю
-vl -.!
СО СО
OS «О
to ю
оо -д
О СО
N3 CD
N3 СО
СО --4
Ю
a
*»
to
ел
CD
СП
оо
со
to
"-J
СО
N3
-vl
СО
CD
СП
СО
го
ел
CD
«D
to
ел
СП
tO
CD
CD
Ю
"^1
00
а
tO
-J
СО
4^
—
to
to
сл
4».
СП
N3
СП
4ь
CD
N3
0>
N3
-J
00
-а
ю
-J
00
00
-J
о
0>
'со
00
О
Ъо
CD
о
Ъо
о
to
ел
со
со
N3
СП
СО
00
00
СО
N3
--J
00
00
to
-0
00
00
СП
J-
ъ
СЛ
N0
СП
со
А
to
СП
со
4^
СО
О
'оо
Vl
-vl
0,96(0,93) |
N3
--J
00
СЛ
to
--J
-vl
СО
to
СЛ
оо
to
сл
со
со
to
to
--J
--а
сп
Ю
--а
--а
-<i
00
О
О
to
СП
to
Сп
to
СП
to
сл
СЛ
4^
to
*-0
"-J
о»
to
-vl
-vl
4^
4^
О
to to
сл ел
to to
a i*
to to
Сл СП
to to
CO КЗ
СП 4^
-g _
О
CO
00
to to
■^ ^
CD CD
сл а
to to
-vl -vl
CD CD
00 —
to сл
to oo
to
ел
сл
to
ел
•—>
-j
00
~
to
-J
СП
№
Ю
-*^
OJ
о
Сп
-J
to
ел
u
to
ел
i—>
OJ
о
->J
to
-J
4^
a
to
-J
4^
CO
CO
4*.
to
СП
о
CJi
to
СП
о
-vl
to
-<l
4^.
oo
to
-<l
4^>
-vl
-<l
CD
J-
ъ
to
4».
CO
«k.
to
4».
CO
4».
О
СЛ
to
-J
oo
сл
to
->J
oo
00
1—
-vl
to
4».
CO
Сл
to
4*
CO
О
CD
00
to
-vl
oo
•4
to
-vl
oo
to
4*
СЛ
to
4^.
->4
Ck.
to
4^.
-J
СЛ
-J
to
to
-J
*b
to
-J
1—
00
4*.
"
to
4^.
CD
o>
to
4^.
CD
-J
►—
4^.
О
"cD
СЛ
to
-J
to
00
to
-J
•—
СЛ
О
СЛ
to
4ь
СП
00
to
4*.
СП
00
►—
to
to
-J
со
to
-J
о
00
CD
4^
to
4*.
СЛ
to
4^.
СП
CD
О
4*.
to
~>J
О
C2
to
"^J
о
"^J
to
4^.
to
4^.
СЛ
СЛ
to
4».
СЛ
oo
Cn
CO
to
-vl
О
00
to
CD
CO
00
CO
to
4*.
СЛ
CD
to
4*.
СП
-vl
oo
to
CD
CO
*
to
CD
CO
-vl
CO
oo
to
4ь
4*.
ас
to
4*.
4*.
СП
•—
-1
to
CD
CO
Сл
to
CD
CO
to
о
--0
to
4*.
4ь
СЛ
to
4*.
4^.
4^.
00
О
to
CD
00
u
to
CD
00
Cn
СЛ
4*.
to
4^
4^.
cn
to
4^
4^.
CO
oo
to
о
4>
to
CD
CO
a
to
CD
-J
CO
~"
OJ
to
4^.
oo
-j
to
4*.
oo
oo
oo
00
to
o>
00
o>
to
OJ
-J
СП
00
00
to to
4ь 4^
— О
■^ ^i
to to
4^. 4k>
— О
00 Oi
C51 05
00 05
СЛ
4^.
>—'
1,02(0,98) 1
to to
05 СП
-J CD
OJ C3
to to
CD CD
-J CD
.— oo
СЛ —
СП 4s»
to
4k>
О
a>
to
4k>
О
to
CD
to
to
CD
CD
«J
to
CD
CD
О
00
СЛ
5 w =
ig5
4
-Z)
О
Ш |
V
3
1
w
X
Обозн
терма
| чение
; s»
ш ы 2
—'X J
¥ n
a
fD
X
V
1
n
Ш |
"a
eii
45
Продолжение табл. 22
Обозначение 1
терма и
значение
289в
2887
2898
290в
2908
2917
291 в
292в
2925
2924
2939
294в
2948
2943
2953
2959
295в
2968
2963
2974
2977
2987
2997
298б
3007
л 2990
2996
2995
3008
3015
3023
п 302в
3027
300б
3034
303»
301 в
и 303в
п 3046
302G
303в
3044
304в
п 305 б
305«
30б5
30б0
30б7
Уровень б), см~1
28 874,9
28 895,6
28 993,7
29 033,6
29 037,2
29 107,1
29 109,8
29 126,1
29 232,6
29 250,5
29 339,3
29 400,9
29 413,2
29413,7
29 430,3
29 550,3
29 558,8
29612,7
29 644,6
29719,9
29 790,7
29 797,2
29 837,6 (,60)
29 865,5
29 958,1
29 978,060
29 986,4
29 999,580
30 027,2
30 143,1
30 222,4
30 245,620
1 30 279,1
30 294,3
30 303,7
30 306,100
30 335,0
30 353,300
30 435,920
30 451,4
30 490,3
30 499,1
30 500,1
30 538,680
30 586,7
30 619,880
30 636,7
30 642,8
Значение gB)
0,92
1,04(1,03)
Обозначение
терма и
значение
3076
п 306 6
3085
3078
307б
308в
3095
3094
3098
зю5
3107
3115
зив
3117
3124
3134
3129
3127
312;
3137
313б
3147
3146
3148
3145
315г
315G
3157
3166
3164
3175
3198
319,
3187
3197
3207
3217
3219
3214
3235
3248
3246
325в
3267
3277
3290
3340
3357
Уровень б), смг!
30 681,690
30 687,700
30707,230
30 737,1
30 875,6
30 894,5
30 936,6
30 979,7
30 986,3
30 993,0
31 024,8
31 129,5
31 135,0
31 166,2(,26)
31221,3
31243,5
31270,3
31276,0
31279,1
31301,1
31 339,8
31 358,6
31 408,480
31 445,3
31 467,6
31488,2
31551,5
31 568,400
31 603,570
31633,9
31744,2
31 923,1
31946,0
31968,1
31974,4
32016,7
1 32 098,2
32 108,4
32 141,2
32 317,8
32 477,8
32 490,6
32 495,7
32 641,8
32 774,2
32 955,080
33412,2
33 570,6
Значение gB)
1,09
46
Часть I. Уран как элемент
Продолжение табл. 22
Обозначение
терма и
значение
336в
3377
3388
3407
Уровень б)» см~1
33 639,6
33 770,23г)
33 850,78г)
34 059,9
Значение gB)
0,83
Обозначение
терма и
значение
3403
3418
3426
Уровень б), см х
34 065,4
34 105,4
34 285,190
Значение gB)
а) Термы, как правило, обозначаются первыми тремя цифрами их волновых чисел, значение ./ дастся
в индексе.
б) Первое (шестизначное) число—волновое число по Киесу [68]; в круглых скобках помещены два
десятичных знака волнового числа по Шуурмансу [76].
в) Значения g без скобок даны по ван-ден-Бошу и ван-ден-Бергу [90], в круглых скобках—по
Шуурмансу и др. [76].
г) Частное сообщение Кнеса, переданное МакНэлли (июль 194 9 г.); значком «п» в первой графе
отмечен терм (по Киесу), данный в обозначении, которое повторяет уже встречавшееся.
выведенных с помощью эффекта Зеемана. В табл. 21 приведены также значения
термов, независимо найденные Шуурмансом [76], и значения g no другим
источникам [68, 90]. Изменения в обозначении некоторых приведенных в таблице
термов, отмеченные в'сноске, были предложены МакНэлли [69] ввиду ожидаемого
для конфигурации f6ds спектра UII расщепления s-типа, а также и потому,
что по схеме Киеса предполагается, что какой-то другой уровень конфигурации
f3d2s более устойчив, чем наиболее низкий из возможных составляющих
терма 7М6.
В табл. 22 указаны идентифицированные верхние термы спектра UI. Можно
предполагать, что эти термы соответствуют переходам электронов Ы и 7s
в бр-состояние и принадлежат четновалентным электронным конфигурациям
f*dps, f3ps2 \\f*d2p. Опубликовано 275 таких термов [68]; позднее найдено
дополнительно 18 термов (ср. [69]). В табл. 22 указано еще 30 термов, определенных
Шуурмансом [76]; для 42 термов определены значения g [76, 90],
Терм 6L—самый низкий из установленных термов UI—может
принадлежать конфигурациям fsds2 или f3d2s. Были приведены различные доводы в пользу
первого предположения [68]. Но, во всяком случае, наличие этого терма
указывает на то, что в наиболее низком из известных состояний свободный
атом урана имеет три /-электрона. Самым низким термом конфигурации d4s2
(которая имеется у вольфрама и первоначально предполагалась также для
урана) должен быть терм 6D.
Ван-ден-Бош и ван-ден-Берг [90] сомневаются в том, что самый низкий
из установленных термов UI действительно является основным термом
нейтрального атома урана. Они отметили, что пока еще не известны термы,
относящиеся к четной конфигурации /4s2 (которые комбинировались бы с нечетными
верхними термами и были бы, таким образом, причиной появления второй
системы линий в спектре UI). Проведение аналогии с неодимом, для которого
было установлено, что наиболее низкий терм принадлежит конфигурации
4/46s2 (по Киесу [68] наиболее устойчивой конфигурацией неодима является
4/35d6s2), указывает на возможность наличия у нейтрального атома урана в его
основном состоянии четырех 5/-электронов вместо трех. По Киесу, практически
все интенсивные линии UI могут считаться принадлежащими к одной известной
системе, но ван-ден-Бош и ван-ден-Берг указывают, что около тридцати из
наиболее интенсивных линий остаются неклассифицированными (ср. табл. 18).
Глава 2. Свойства атома урана
47
Они считают также, что поскольку, как теперь известно, в дуговом спектре
урана наблюдается большое число искровых линий, дальнейшее
усовершенствование метода возбуждения может привести к получению более чистого
дугового спектра и к обнаружению еще ряда новых линий.
Абсолютное значение наиболее низкого из известных термов UI (т. е.
ионизационный потенциал атома урана в состоянии f3ds2 °L6) неизвестно. Ни одного
терма серии Бальмера, который дал бы возможность экстраполировать предел
сходимости, пока еще не обнаружено. Однако на основании того факта, что ниже
2900 А Для нейтрального атома урана не было найдено никаких линий, сделан
вывод, что ионизационный потенциал урана должен быть порядка 4 эв [68].
Это заключение подтверждается легкостью ионизации атома урана в
электрической дуге.
Анализ спектра UII (спектр Псиона). Изучение спектра и+-иона
проводилось независимо в Массачусетском технологическом институте [84, 92, 94] и в
лаборатории Зеемана в Амстердаме [76, 90, 95—97]. Результаты первого
исследования были представлены в отчете [91], в котором описаны 14 нижних
термов и около 150 верхних термов с указанием для всех термов значений У,
а для некоторых из них и значений g. Было замечено, что нижние термы
образуют две отдельные системы из 12 и 2 термов соответственно; из верхних термов
к первой группе принадлежат 140, ко второй—10. Нижние термы первой
системы были первоначально отнесены к четной конфигурации f2d2s. Самым низким
из наблюдавшихся термов был терм hj29 за которым сразу же (с интервалом
289 см~1) следовал терм Ln/2. Гаррисон сначала предположил, что последний
может являться наиболее низкой составляющей действительного основного
терма и+-иона (самым низким термом конфигурации f2d2s теоретически является
терм6!), а наинизший из наблюдавшихся уровней /9/2 может быть случайно
пониженной составляющей терма 6/ той же конфигурации, центр тяжести
которого выше, чем у терма 6L.
Позже эта предварительная интерпретация была изменена [69, 92].
Указанная выше группа из 12 нижних термов (группа А) отнесена к нечетным
конфигурациям /V, f3ds и f3d2 (как это показано в табл. 23). Два остальных
нижних терма, идентифицированных в Массачусетском технологическом
институте (группа В), должны быть в таком случае четными термами. Их положение
относительно термов системы А нельзя было определить, так как
«интеркомбинационных» линий не наблюдалось (линий, обусловленных взаимными
комбинациями термов этих систем). В табл. 23 перечень нижних термов
группы А пополнен восемью термами, идентифицированными позднее в
Массачусетском технологическом институте и в Амстердаме; к нижним термам группы В
добавлено 7 новых термов, 5 из которых найдены амстердамской группой
исследователей.
Опубликована таблица [76], в которой имеются данные о 16 нижних
нечетных и 57 верхних четных термах (система Л), и таблица с данными о 7 нижних
четных и 5 верхних нечетных термах (система В)\ около 350 линий
идентифицированы как возникающие в результате комбинаций этих термов (системы А и В
в голландских работах обозначены в обратном порядке — В и А).
Ван-ден-Бош [77] в дополнение к данным о спектре UII сообщил о наличии
еще трех верхних нечетных и 17 «очень высоких» четных термов системы В.
Правильность этих сведений была, однако, подвергнута сомнению со стороны
МакНэлли [98]. Ван-ден-Бош [77] установил также, что в системе А имеется
еще 63 верхних четных терма. Несмотря на прибавление группы «очень
высоких» термов, взаимных комбинаций термов систем А и В все-таки обнаружить
не удалось.
Ван-ден-Бош, как и МакНэлли, считает, что нижние термы системы А
относятся к нечетным конфигурациям /V, f3ds и /3d2; верхние термы этой группы
в таком случае могут быть отнесены к четным конфигурациям f2d2s> f2ds2,.
48
Часть I. Уран как элемент
Таблица 23
НИЖНИЕ ТЕРМЫ U+-HOHA (1Л1)[69, 77, 90, 92]
Система
А
(нечетная)
В
(четная)
Конфигу-
1 рация
/3S2
f4s
1 /3d2
(?)
(?)*
(?)
/4s
fxd
Символ
терма
4/4l/2
У51/2
$Lb4z
*L*4t
*Ln,t
*Lulh
•*4i/,
'Кы,*
°K&ih
'«ЫП
У31/2
;41/3
в'5./4
•tf2l/4
6//31/2
'M61/4
™7l„
1/47l/2
^7l/s
3^81/2
7 31/2
6/41/2
6/"б1/2
4/
41/2
L5l/2
G^oi/2
6/<41/2
1B0l/2
2B5l/2
Значение терма, смг*
[91]
0,00
4 420,88
289,05
1 749,14
5 259,67
6 283,47
914,77
2 294,72
5 526,76
5 790,66
5 401,55
6 445,03
8510,91
4 706,31
5 667,34
4 585,48
8 394,36
8 521,99(?)
9 291,77(?)
12 645,67 (?)
0,00
1 052,65
3 683,82
3 759,55
7 850,08
10 728,59
11 547,90
6853,37 (?)
7 695,85 (?)
[77]
0,00
4 420,85
289,04
1 749,11
5 259,64
6 283,44
914,76
2 294,69
5 526,73
5 790,62
5 401,52
6 445,04
—
4 706,28
5 067,33
4 585,43
8 394,40
—
—
—
0,00
1052,64
3 683,88
3 759,59
7 850,15
10 728,67
11 547,93
—
—
Значение g
опытное
[91]
: 0,755
0,973
0,651
0,863
1,01
0,785
0,605
0,870
1,03
0,849
0,694
0,84
#—
0,473
0,737
0,780
0,975
1,05 (?)
—
—
0,495
0,826
—
__
—
—
—
—
—
[90]
0,75
0,93
0,66
0,84
0,99
0,79
0,60
0,86
1,02
0,84
0,67
0,85
—
0,45
0,72
0,79
0,95
—
—
—
0,48
0,83
1,03
0,81
0,67
0,88
0,66 [76]
—
—
теоретическое
для
LS-связи
0,727
| 0,965
, 0,615
0,851
1,004
0,800
0,546
: 0,839
1,015
0,769
0,444
0,828
1,035
0,286
0,825
0,667
0,879
—
—
—
0,444
0,755
1,035
0,727
0,615
0,851
0,546
—
—
Pdp и f3sp. В системе В нижние термы, повидимому, обусловлены четными
конфигурациями /4s и /4d, а верхние термы—нечетной конфигурацией /4р; очень
высокие термы (если они действительно существуют) могут, повидимому,
быть отнесены к четной группировке /47d.
Поскольку пока не обнаружены взаимные комбинации между термами
систем А и В (такие «интеркомбинации» известны для атома тория в том же
электронном состоянии), вопрос о действительно основном терме, а
следовательно, и о наиболее устойчивом состоянии изолированного и+-иона остается
открытым. Возможна либо конфигурация /3s2 4/э/2 (система Л), либо /4s
9 /т/о (система В). МакНэлли [98] предположил, что первый из названных термов
Глава 2. Свойства атома урана
49
может лежать на 4000 смг1 ниже второго; голландская группа исследователей
склоняется к тому, чтобы рассматривать второй терм как основной терм U+,
терм же /V 4/и/2 в таком случае расположен выше на 20 000 смг1.
В табл. 23 собраны данные как МакНэлли, так и голландских
исследователей относительно уровней и+-иона. Термы системы А приписаны
конфигурациям /3s2, fdds и /3d2 с тремя 5/-электронами; термы системы
В—конфигурациям /4s и /4d с четырьмя электронами группы 5/. В табл. 24,
составленной МакНэлли [69], собраны таким же образом данные, относящиеся к
средним и очень высоким термам обеих известных систем спектра UII.
Попыток определения значений L, S, J или электронных конфигураций для
верхних термов UII не делалось, хотя, согласно МакНэлли [69], возникновение
некоторых из них может быть выведено из данных интенсивностей,
эффекта Зеемана и изотопических смещений. В качестве примеров можно
указать на уровень 103Л, который, вероятно, допустимо рассматривать как
f2ds2 4/41/2-уровень, и уровень 161 Л, который, вероятно, представляет собой
f3dp *Мб1/2-УРОвень.
Электронная конфигурация. Химические свойства
элементов ряда Th—U более сходны со свойствами ряда Zr, Nb, Mo, чем ряда La,
Се, Рг. Другими словами, постепенное заполнение электронами d-оболочки у
этих элементов происходит, повидимому, прежде чем достраивается f-оболочка,
заполнение которой характерно для семейства редкоземельных элементов.
Первые теоретические расчеты прочности электронных оболочек [99], повидимому,
подтверждают высказанное предположение, так как они указывают на более
прочную связь 6^-электронов по сравнению со связью для 5/-электронов.
Однако последующие расчеты [100] с использованием приближенного метода
Венцеля—Крамера—Бриллюэна показали, что при Z, равном 92 (уран), 5/-элек-
троны могут быть почти столь же прочно связаны, как и 6^-электроны, и что
при Z, равном 93, в основном состоянии, вероятно, находится по крайней мере
один 5/-электрон. Кроме того, с помощью статистического метода
приближения Томаса—Ферми [101 ] установлено, что заполнение 5/-оболочки электронами
должно начинаться при Z, равном 91 или 92.
Отмечено [102, 103] сходство между спектрами поглощения соединений
редкоземельных элементов с их резкими линиями и спектрами соединений урана:
спектры некоторых соединений урана также имеют узкие отчетливые полосы
поглощения. Можно считать, что этот факт указывает на наличие таких
состояний, в которых роль оптических электронов играют хорошо экранированные
f-электроны. Влияние анионов на спектр катиона также сходно для солей
редкоземельных элементов и солей урана. Однако спектры, о которых идет речь,
являются спектрами ионов U(III), U(IV), а многовалентные ионы имеют
большую тенденцию к распределению электронов без образования незаполненных
внутренних оболочек, чем нейтральные атомы. Поэтому предположение о том,
что у нейтрального атома урана имеется два /-электрона и что от тория
начинается ряд «торидов», у элементов которого достраивается 5/ч)болочка, не было
обоснованным. Надежные сведения по этому вопросу получены в последнее
время путем вышеописанного анализа спектра. Этот анализ показал, что
свободный ион U+ и свободный нейтральный атом урана имеют следующие
основные термы:
U+ : 5/3 7s2 Ч (или, возможно, 5/4 7s e/);
U:5/36d7s25L.
(Как упоминалось выше, ван-ден-Бош и ван-ден-Берг считают возможным, что
основным термом атома урана является неизвестный до сих пор терм
конфигурации 5f47s2.)
Атом урана и ион U+ имеют в наиболее устойчивом состоянии по крайней
мере три 5/-электрона.
4 Химия урана
Таблица 24
Ш-РХПИ1- ТЕРМЫ В СПЕКТРЕ UII
Значение g
UN III С
[91]
[90]
[76]
А. Верхние четные термы (система А)в
100 Л
101 Л
102Л
103 А
104 Л
105 Л
106Л
107 Л
108 Л
109Л
ИОЛ
111 Л
112Л
113Л
114Л
П5Л
116Л
117Л
118Л
П9Л
120Л
121 Л
122Л
123Л
124Л
125Л
126Л
127 Л
128Л
129Л
130Л
131 Л
132 Л
133Л
134 Л
135 Л
136 Л
137Л
138Л
139Л
3%, 4J4
4У2
5J4
4У2
5й
Щ
ЗУ2
5Й
3%
зу2
ЗУ2
зу2
4J4
ЗУ2, 4%,
5у2
6У2
4У2
зу2
еу2
4У2
454
4*/2
4У2
5У2
4У2
5%
5У2
4У2
5У2
еу2
зу2, щ
Ьуг
4У2
5У2
5У2
5%
5У2, бу2
5й
еу2
4У2
зу2
I 15 679,52 (?)
17 392,22
17 434,37
18 200,08
18 827,01
19 097,56 (?)
19517,73
20 354,00
20 571,73
20 961,72
1 21021,40
21320,22
21555,28
21 679,17 (?)
21 710,76
21 831,05
21 860,06
21 975,57
22 101,37
22 165,18
22 429,84
22 642,49
22 764,93
22 868,06
22 917,46
23 241,36
23 315,09
23 554,00
23 635,94
23 741,32
23778,14
23 911,66
24 010,48
24019,29
24 152,82
24 159,73
24 288,00
24 293,13
24 305,62
24 342,21
—
0,783
или 0,405
0,810
0,774
0,94
—
—
1,00
—
0,855
0,885
0,827
1,023
—
0,907
0,890
—
1,029
или 0,697
—
0,885
или 0,621
0,929
0,872
0,980
или 0,760
0,98
0,849
0,957
0,874
1,033
0,912
—
0,865
1,056
0,969
—
0,906
0,959
1,012
1,019
0,974
0,763
или 0,625
1 ~~
--
—
0,78
—
~
—
_
0,97
0,92
0,86
0,85
—
| —•
0,89
—
—
0,86
0,92
0,94
0,86
—
0,<;8
0,85
0,95
0,89
1,03
0,94
~
0,89
—
0,98
—
0,92
—
1,01
—
—
—
] —
—
—
0,78
—
—
—
—
0,92
0,86
0,88
0,80
—
—
0,91
—
—
—
0,87
0,90
0,93
0,8/
—
0,98
0,86
0,96
0,88
1,04
0,91
—
0,86
—
0,97
—
0,91
—
1,01
—
0,97
0,75
5Ь
Значение
J
4У2
5J/2
щ
6У2
Ьуг
6%
ЬУг
5%
Щ
4%
5й
зу2
5>/2
6У2
2%
3'/2
5И
■Щ, ЬУг
3%
еу2
•г,у2
6У2
5У2
5'/2
6'/2
6У2
5й
6%
7у2
4У2
5Й, 6%
б)
выше
Продолжение табл. 24
Значение g
3J4
5У2
4%
5%
4%
51/2
5у2
зу2
4%
4У2, 5у2
5%
5у2
5У2
24 453,45
24 608,16
24 684,16
24 923,59 (?)
25 156,89
25 163,96
25 200,82
25 357,02
25418,56
25 437,59
25 492,92
25 495,47
25 543,99
25714,06
25 737,60
25 764,70 (?)
25 770,59
25 906,06
25 967,70
25 986,35
26 084,82
26 191,34
26 285
26 330
26 397
26415
26 595
26 716,74
26 879,25
26 887,29
27 126,11
27 143,73
27 290,25
27 357,30
27 361,05б)
27 390,51
27 446,35
27 453,53
27 499,38
27 583,35 (?)
27 698,03
27 708,38 (?)
27 725,02
27912,14
27 930,28
28 154,46
18
17
89(?)
18
51
[91]
1,100
0,904
0,918
1,088
0,962
1,030
•—■
0,94
0,993
0,958
—
1,010
0,923
1,05
0,855
0,979
0,900
_
1,035
—
.—
0,990
1,012
0,93
или 0,54
1,036
или 0,662
0,730
1,012
—
0,997
0,885
[90]
_..
—
_._
—
—
—
—
—
--
—
--
....
—■
—
—
—
—
1,03
—
1,00
1,05
1,06
0,90
1,05
—
_..
---
1,01
0,89
[7G]
1,10
0,89
0,93
---
_-
1,05
,
-*-
-J-
—
-л-
1,01
—
'
0,84
0,99
0,90
—
--
1,01
—
0,99
1,08
1,01
0,97
—
1,06
—
^
1 1,01
! 0,89
Продолжение табл. 124
Обозначение
терма а)
186Л
187 Л
188 Л
189Л
190 Л
191 Л
192 Л
193 Л
194 Л
195 Л
196Л
197 Л
198 Л
199Л
200 Л
201 Л
202 Л
203 Л
204 Л
205 Л
206 Л
207 Л
208 Л
209 Л
210Л
211 Л
212Л
213 Л
214Л
215Л
216Л
217Л
218Л
219Л
220 Л
221 Л
222 Л
223 Л
224 Л
225 Л
226 Л
227 Л
228 Л
229 Л
230 Л
231 Л
232 Л
233 Л
Значение
J
щ
3%
5Н
554
5 К
5%
6Н
554
4%
5J4
454, 5 54
554
5 К
454, 554
6%
454
ЗУ2, 454
5*4
5»/2
6%
554
4%
зи
454
5%
7Н
354
5%
6У2
6%
5у2, 654
5%
4V4
5'/2
5'/2
ay.
5Й. 654
4 К
4%
4%
6J4
6>/2
БН
454
7»/2
5*4
5У2
5У2
Уровень б)
выше
4/41/2' сл1_1
28 159,72
28 347,28
28 444,51
28 484,51
28 507,96
28 587,30
28 636,89
28 758,11
28 763,026>
29 162,14(?)
29 205,39 (?)
29 206,71
29 303,92
29 349,22
29 352,89 (?)
29 388,68
29 396,82
29 402,00
29 403,74
29 476,76
29 557,96
29 707,21б>
29 824,93б)
29 827,43
29 858,49
29 932,41
29 933,94б>
29 936,47
29 978,19
30 000,16
30015,84
30 037,11
30 042,93
30 060,72
30 061,77(?)
30 066,59
30 085,80
30 086,78 (?)
30 120,14
30126,00
30 129,88
30139,07
30 240,45
30 263,98
30 341,70
30 374,11
1 30 438,55
J 30 468,75
[91]
1,02
—
_
1,020
—
1,064
—
__
0,977
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
1,06
—
—
—
—
—
—
„
—
1,017
—
—
—
—
—
—
__
—
—
1,019
—
—
—
Значение g
[901
0,98
._
—
1,03
—
—
—
—
—
—
1,03
—
—
—
—
—
—
—
1,09
—
—
—
1,04
—
1,04
—
—
—
—
—
—
—
1,07
—
—
—
—
—
—
—
—
1,06
—
1,01
—
—
—
[76]
1,02
—
—
1,04
1,07
—
—
—
—
_
1,05
—
—
—
—
—
—
—
1,10
—
—
—
1,08
—
1,06
—
—
—
—
—
—
—
1,15
—
—
—
—
—
—
—
; —
1,06
—
1,02
—
—
—
53
Продолжение табл. 24
Обозначение
11
терма а»
234 Л
235 Л
236 Л
237.4
238 Л
239 Л
240 Л
241 Л
242 Л
243 Л
244 Л
245 Л
246 Л
247 Л
248 Л
249 Л
250 Л
251 Л
252 Л
253 Л
254 Л
255 Л
256 Л
257 Л
258 Л
259 Л
260 Л
261 Л
262 Л
263 Л
264 Л
265 Л
266 Л
267 Л
268 Л
269 Л
270 Л
271 Л
272 Л
273 Л
274 Л
275 Л
276 Л
277 Л
278 Л
279 Л
280 Л
Значение
J
6й
5й |
6% !
5У2
бу2
Щ
7Уг
5%
5%
4/2
6У2, 7й
4J4
3%
6У2
щ
5%
бй
5У2
6%
4>/2
6И
5%
6%
зу2
7%
4У2
6%
6%
4%
5%
зу2
5У2
зу2
i 6У2
I 5J4
6%
5У2
5у2, 6й
5й
5%
зу2
4J4
бу2
5У2
бу2
7%
5У2
Уровень б)
выше
4/41/2' см-1
30 550,39
30 561,516>
30716,23
30 860,13
30 863,44б>
30 900,65 (?)
30 941,64
31083,68
31219,22
31 327,76 (?)
31 346,11 (?)
31670,33(?)
31 784,81 (?)
31 936,69
32 028,36 (?)
32 187,68
32 211,36
32 416,69
32 535,07
32 717,24б)
32 838,74
33018,86
33 104,46
33 184,00б)
33 224,14(?)
33 359,62б>
33 431,61
33 475,06
33 516,22б>
33 577,83
33 648,16
33 748,79
33 795,02
34 049,54
34 199,47
| 34 268,16б)
34 392,34
34 402,25
34 439,33
34 593,06
34 607,01
34 612,86
34 663,67
34 678,05
34 708,25
34 791,40
34 885,51
[91]
—
—
0,977
—
—
1,03
—
1,040
0,905
—
—
0,934
—
1,023
_.
—
—
0,997
—
__
—
—
__
1,068(?)
—
—
—
—
—
—
—
0,977
—
—
—
—
—
—
—
—
0,958
или 0,562
—
—
_
—
1,004
Значение g
[90]
—
—
0,98
—
—
1,04
—
1,00
—
—
—
.__
—
—
—
1,03
—
0,98
—
—
—
—
—
—
—
—
—
—
—
—
—
—
! —
—
—
! —
—
1,05
—
—
—
—
—
1,12
—
0,84
[76]
т—
—
0,99
—
__
1,03
—
1,04
—
—
—
—
—
—
—
1,02
—
1,00
—
—
—
_
—
—
—
—
„
—
—
—
—
—
1,08
—
—
—
—
1,03
—
—
—
~~
—
1 1,04
—-
—
54
Продолжение табл. 24
Обозначение
о\
;герма а*
281 Л
282 Л
283 Л
284 Л
285 Л
286 Л
287 Л
288 Л
289 Л
290 Л
291 Л
292 Л
293 Л
294 Л
295 Л
296 Л
297 Л
298 Л
299 Л
300 Л
301 Л
302 Л
303 Л
304 Л
305 Л
306 Л
307 Л
308 Л
309 Л
310 А
ЗИЛ
312Л
313Л
314Л
315Л
316Л
317Л
318Л
319Л
320 Л
321 Л
322 Л
323 Л
324 Л
325 Л
326 Л
327 Л
328 Л
Значение
J
4Й, 5у.
5J4, 6%
4 И
4%, 5 И
4%, 5%
4%, 5%
Щ, 5%
4V4, 5%
3%
4У2, 5» г
6'/2
5й
5Н, 6У,
5Н, 6Й
5й
4й
4'/2> 5'/2
4Н
4й, 5fc
4У2, 5Н
4Й, 5у2
щ
4 И
4Н, 5J4
4i/2
3'/2
4'/2, 5й
6%
4V4
4>/2, 5Н
6Й
4У2, 5Ji
4й, б И
4'/2) 5'/»
5%
4%
44. 5%
4J4
4Й, 5%
7%
4%
Зй
5У2
5'/2, 6«/2
3%
5%
5У2
6J4, 7й
Jy ровень
б)
БЫШС
4141/2* СМ~Х
34 896,18
34 923,62
34 949,12
34 968,14
35 110,22
35214,41
35 234,08
35 324,38
,35 326,16 б)
35 372,90
35 446,89б)
35 684,83
35 790,18
35 899,45
35 903,18
35 960,19
36 029,20
36 070,31
36 089,94
36 105,39
36 132,82
36150,57
36 253,41
36 273,86
36 351,21
36 548,656)
36 679,45
36 707,40
36 782,70
37 149,71
37 308,35б)
37 341,79
37 377,10(?)
37 659,53
37 684,33
37 789,18
37 869,85
37 931,77
38 128,90
38 152,90
38 579,87
38 681,86
38 788,39
38 898,43
38 903,29
38 968,45
39 108,95
39 508,25б)
Значение g *
l01]
—
—
—
—
—
—
—
: —
—
—
—
—.
--
—
„_
—
—
—
—
—
—
—
__
—
_._
—
—
—
—
—
- -
—
--
[00]
„
—
—
—
—
—
—
—
—
—
.—.
■—
—
—.
—
—
—
—
—
—
—
—
—
__
—
—
—
—
—
—
—
—
—
—
._-
—
._
--
—
.__
.__
—
_
—
—
[70]
—
—
—
—
—
—
—
—
.—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
_
—
—
—
—
—
—
—
55
Продолжение табл. 24
Обозначение
терма а)
329 Л
330 Л
331 Л
332 Л
333 Л
334 Л
335 Л
Значение
J
4У2
4]/2
зу2
ъуг
ЗУ2, 4%
6У2
6У2
Уровень
б) !
выше
414ij2, см-1
40 356,31
40 391,36
40 731,32
41317,04
41 542,60
44 174,01
45 533,53
[01]
—
-._
1,059
—
—
—
Значение #
[9 0]
—
■—
—
—
—
—
[76]
—
—
—
—
—
—
Б. Верхние нечетные термы (система В)г^
1 Щ
4й
щ
1 4й
4V4
3%, 4у2
зу2
зу2
4у2, 5у2
5У2
5J4
4У2, 5у2
| 5}4
Щ
1 4У2, 5у2
j 354
5J4
5У2
! ЗУ2, 4У2
4у2
4у2, 5Н
4J4, 5У2
3J4, 4й
5У2
5У2
5J4
1 5}4
В. Очень высок
1 4У2
4У2
4%
4%, 5У2
4У2
4У2
4У2
4У2
454
25 805,44 (?)
25 869,43
25 935,34
26 092,29
26 332,33
27 065,42 (?)
27 297,27
27 721,19
28 693,86
28 777,79б)
28 963,30
29 058,92б>
29 289,40
29 970,82б)
32 586,02
32 840,28
33 056,48
33 070,85
33 593,76
33 663,38
33 960,47
35 325,03
36 353,41
37 503,66
37 785,87
43 911,61
47 210,23
ие четные терм
51 148,616)
52514,616)
53 873,536)
54 082,256)
54 082,566)
54 691,556)
54 923,776)
55 237,966)
55 692,516)
—
—
0,905
0,973
—
—
0,576
0,955
—
—
1,01
—
1,08
—
—
—
—
—
—
—
—
—
—
—
—
—
—
ыА^ (сист
—
—
—
—
—
—
—
—
\ ■
0,91
0,99
0,93
1,01
1,08
0,92
0,99
0,96
1,00
1,07
56
Часть I. Уран как элемент
Продолжение табл. 24 %
Обозначение
терма а)
559
560
563
575
578
589
590
607
Значение
J
Щ
4Н
4У2
3%, 4J4
4J4
4%
4%
4*4
Уровень 6)
выше
4/4i/2f сл-1
55 935,176)
55 972,256)
56 306,806)
57 589,806)
57 840,156)
58 901,526)
59 055,976)
60 769,096)
[91]
—
—
—
—
—
—
—
Значение g
[90]
—
—
—
—
—
._
—
[76]
—
_
—
—
—
—
—
а) Ввиду неопределенности действительного основного терма возбужденные термы в частях
таблицы А и Б обозначены последовательно возрастающими числами от 100 А н 100 В соответственно, а не
первыми тремя цифрами волнового числа, как в табл. 23; в части же В [7 7] обозначение такое же, как
в табл. 23.
б) Значения термов взяты из данных, полученных в Массачусетском технологическом институте [91],
за исключением тех, которые отмечены значкомб); последние значения приведены в статье вап-ден-
Боша [77].
в) Система В голландских исследователей [76, 7 7, 90].
г) Система А голландских исследователей [7 6, 77, 90].
Д) Существование всех этих термов МакНэлли ставит под сомнение [98].
Результаты химических экспериментов, полученные за последнее время,
подтверждают мнение о том, что в последнем периоде периодической системы
существует ряд элементов, подобный ряду лантанидов, хотя последовательное
увеличение главной валентности в «семействе лантанидов» наблюдается
только для первых двух элементов—La (III) и Ce(IV), а в «семействе актинидов»—
для большого числа элементов Ac (III), Th(IV), Pa(V), U(VI).
Однако в результате заполнения /-оболочки не обязательно должны
получаться ряды элементов, химически подобные редкоземельным. Близкое
сходство элементов лантанидов обусловлено узостью интервала изменения их
ионизационных потенциалов и ионных радиусов. Качественная аналогия
актинидов и лантанидов в отношении распределения электронов еще не говорит
об обязательном количественном сходстве свойств у элементов первой из
названных групп. Аналогия между элементами ряда актинидов и
редкоземельными элементами становится более заметной для трансурановых элементов,
у которых происходит понижение главной валентности: U(VI), Np(V),'Pu(IV),
Am(III). Физические и химические свойства элементов обоих семейств
рассматриваются в работах Сиборга [104] и Захариазена [105].
ЛИТЕРАТУРА
1. Manescu I., Compl. rend., 225, 537 (1947).
2. de В г о g 1 i e M., Compt. rend., 162, 597; 163, 354 (1916).
3. de В г о g I i e M., J. phys., 6, 161 (1916).
4. de Broglie M., Compt. rend., 168, 854; 169, 964 (1919).
5. Siegbahn M„ Jonsson E., Physik. Z., 20, 255 (1919).
6. D u a n e W., Patterson R. A., Proc. Natl. Acad. Sci. U.S., 6, 512 (1920).
7. D u a n e W., F r i с k H., Stenstrom W., Proc. Natl. Acad. Sci. U S., 6,
611 (1920).
8. D u a n e W., Bull. Natl. Research Counsil, 1 (6), 389, 396 (1920).
9. D u a n e W., Proc. Natl. Acad. Sci. U. S., 7, 270 (1921).
Глава 2. Свойства атома урана
57
10. S t о п е г Е.С., Martin С. Н., Proc. Roy. Soe. L., A107, 312 (1925).
11. D a u v i 1 1 i e r A., Rev. sci., 65, 707 (1927).
12. D a u v i 1 1 i e r A.. Compt. rend., 173, 37 (1921).
13. С a uc hois Y., Manescu I., Compt. rend., 210, 172 (1940).
14. P о 1 а с z e к W., Sitzber. Akad. Wiss. Wicn, Math, naturw. Klasse, Abt. Ha, 148,
81 (1939).
15. Mack J.E., Cook J. M., Phys. Rev., 30, 741 (1927).
16. S a n d s t г 6 m A., Z. Physik, 65, 632 (1930).
17. Coster D., Compt. rend., 172, 1176 (1921).
18. Coster D., Z. Physik, 5, 143 (1922).
19. С о s t e г D., Phys. Rev., 19, 20 (1922).
20. Stenstrora W., Dissertation Lund (1919); см. также [17—19,39].
21. Li n db erg E., Z. Physik, 54, 632 (1929).
22. A 1 1 e n S. J. M., Phys. Rev., 27, 266; 28, 907 (1926).
23. С о m p t о n A. H., Allison S. K., X-rays in Theory and Experiment,
D. Van Nostrand Company, Inc., New York, 1935, p. 801, 803.
24. Rechou G., Compt. rend., 180, 1107(1925).
25. Kustner H., Physik. Z., 33, 46 (1932).
26. Stephenson R. J., Phys. Rev., 43, 527—533 (1933).
27. Siegbahn M., Spektroskopie der Rontgenstrahlen, Verlag Julius Springer,
Berlin, 1931, 2d ed., p. 332, 335, 346.
28. S с h r 6 г., Ann. Physik, 80, 302 (1926).
29. F r i m a n E., Z. Physik, 39, 825 (1926).
30. I d e i S., Science Repts. Tohoku Imp. Univ., First Ser., 19, 572 (1930).
31. Claesson H., Z. Physik, 101, 499 (1936).
32. A 1 1 i s о n S. K., Phys. Rev., 32, 1 (1928).
33. С о m p t о n A. H., Allison S. K., X-rays in Theory and Experiment; D.
Van Nostrand Company, Inc., New York, 1935, p. 645.
34. A 1 1 i s о n S.K., Andrews V. J., Phys. Rev., 38, 452 (1931).
35. W i 1 1 i a m s J. H., Phys. Rev., 37, 232, 1431 (1931).
36. С a u с h о i s Y., A 1 1 a i s M. L., J. Phys. Radium, 1, 44 (1940).
37. A 1 1 i s о n S. K., Phys. Rev., 34, 176 (1929).
38. H u 1 u b e i H., J. Phys. Radium, 8, 260 (1937).
39. Coster D., Z. Physik, 4, 184 (192П.
40. D a u v i 1 1 i e r A., Compt. rend., 172, 915 (1921).
41. D a u v i 1 1 i e r A., Compt. rend., 173, 647 (192П.
42. D a u v i 1 1 i e r A., A u g e r P., Compt. rend., 176, 1298 (1923).
43. Siegbahn M., Friman E., Phil. Mag., 31, 405 (1916).
44. S i e g b a h n M., Friman E., Phil. Mag., 32, 40 (1916>.
45. S i e g b a h n M., Friman E., Physik. Z., 17, 17, 6'l (1916).
46 D u a n e W., P a t t e r s о n R. A., Proc. Natl. Acad. Sci. U. S., 8, 88 (1922).
47. R i с h t m у е г F. К., Kaufman S., Phys. Rev., 44, 606 (1933).
48. С a u с h о i s Y., Compt. rend., 222, 1484 (1946).
49. Hjalmar E., Z. Physik, 15, 79, 65 (1923).
50. Stenstrom Wr., Ann. Physik, 57, 347 (1918).
51. Lin db erg E., Z. Physik, 50, 83 (1928).
52. L i n d b e r g E., Nova Acta Regiae бос. Sci. Upsaliensis, 7 (7), 21 (1931).
53. H i r s с h F. R., Jr., Phys. Rev., 38, 919, 923 (1931).
54. P a r r a t t L. G., Phys. Rev., 41, 561 (1932).
55. P u r d о m E.G., Cork Т. М., Phys. Rev., 44, 328, 975 (1933).
56. Dolejsek V., Z. Physik, 21, 111 (1924).
57. Siegbahn M., M a g n u s s о n Т., Z. Physik, 88, 559 (1934).
58. H eves у G. V., L а у Н., Nature, 134, 98 (1934).
59. Lay H., Z. Physik, 91, 533 (1934).
60. de Broglie M., Compt. rend., 173, 1157 (1922).
61. R о b i n s о n H. R., Phil. Mag., 50, 241 (1925).
62. Bohr N., Coster D., Z. Physik, 12, 342 (1923).
63. M о n k А. Т., Allison S. K., Reports CP-2120, Sept. 9, 1944 (MP Chicago 1).
64. R u a r k A. E., Maxfield F. A., Phys. Rev., 47, 107 (1935).
65. Russell H., Jr., Report LA-145, Sept. 22, 1944 (Los Alamos 1).
66. Breit G., Phys. Rev., 35, 1447 (1930).
67. H a r r i s о n G. R. et al., Massachusetts Institute of Technology, Wavelength
Tables, John Wiley a. Sons, Inc. New York, 1939.
68. К i e s s С. С, Humphreys С. J., L a u n D. D., J. Research Natl. Bur.
Standards, 37, 57 (1946).
69. M с N a 1 1 у J. R., Jr., personal communication, Jan. 1950 (Carbide and Carbons 3).
70. E d e r J. M., V a 1 e n t a E., Sitzber. Akad. Wiss. Wien. Math, naturw. Klasse,
Abt. IIA 119 39 (1910).
71. Has'selber'g В., Klg. Svenska Vetenskapsakd. Handl., 45, (1) (5), 39 (1910).
58
Часть I. Уран как элемент
72. Е х п е г F., H a s с h е к Е., Die Spectrcn der Elemente bei normalen Druck,
F. Deutickc, Leipzig und Wien, 1911, vol. 2, p. 282; vol. 3, p. 256.
73. Meissner K. W., Ann. Physik, 50, 727 (1916).
74. К i e s s C. C, Meggers W. F., Natl. Bur. Standards U. S., Sci. Papers,
16 (372) (1921).
75. Lang R. J., Trans. Roy. Soc. L., A224, 419 (1924).
76. S с h u u r m a n s P., van-den-В osch J. C, D i j к w e 1 N., Physica, 13,
117 (1947).
77. van-den-B osch J. C, Physica, 15, 503 (1949).
78. К i e s s C. C, Humphreys C. J., L a u n D. D., Report A-1747, Feb. 7,
1944 (Natl. Bur. Standards 1).
79. F r e d M., personal communication, Febr. 1945 (MP Chicago 2).
80. H a r t 1 e у W. N„ Moss H. W., Proc. Roy. Soc. London, A87, 46 (1912).
81. Meyer G., Physik. Z., 22, 583 (1921).
82. E d e r J. M., V a I e n t a E., Atlas typischer Spektren, A. Holder, Vienna,
1924, 2d ed., p. 61, 118 plates 15, 21, 24, 29, 38, 50, 52.
83. A n d e r s о n O.E., White H. E., Phys. Rev., 71, 911 (1947).
84. Mc N a 1 1 у J. R., Jr., Report Y-232, Sept. 9, 1948 (Carbide and Carbon 1).
85. В u г к h a r t L. E., St ukenbroeker G., A d a m s S., Phys. Rev., 75,
83 (1949).
86. Personal communication through S. K. Allison, Jan. 1945 (British 1).
87. N a g а о к a H., S u g i u r a Y., Japan J. Phys., 3, 71 (1924).
88. H u m p h г е у s W. G., Astrophys. J., 6, 169 (1897).
89. Ross A. D., Proc. Roy. Soc. Edinburgh., 30, 448 (1910).
90. van-den-B osch J. C, van-den-Berg G. J., Physica, 15, 329—350 (1949).
91. H a r r i s о n G. R., unpublished data reported in a personal communication, Jan.
1945 (MIT 2).
92. M с N a 1 1 у J. R., Jr., Harrison G. R., Report Y-340, Feb. 11, 1949
(Carbide and Carbon 2).
93. Spectroscopy Section, Report [A] M-2187, undated. Received May 14, 1945 (Natl. Bur.
Standards'2).
94. H a r r i s о n G. R., personal communication, Jan. 1945 (MIT 1).
95. Sc h u u r m a n s P., Physika, 11, 419 (1946).
96. S с h u u r m a n s P., Physika, 11, 475 (1946).
97. S с h u u r m a n s P., Physika, 12, 589 (1946).
98. M с N a 1 1 у J. R., Jr., Phys. Rev., 77, 417 (1950).
99. S u g i u r a Y„ Urey H. C, Kgl. Danske Videnskab. Selskab. Math. fys. Medd.,
7, 3 (1926).
100. Ta-You Wu, Goudsmit S., Phys. Rev., 43, 496 (1933).
101. G о e p p e r t-M а у е г М., Phvs. Rev., 60, 184 (1941).
102. Ephraim F.,Mezener X, Helv. Chim. Acta, 16, 1257 (1933).
103. Ephraim F., J. Indian Chem. Soc, Rav-Memorial Vol., p. 243 (1933).
104. S e a b о r g G. Т., Paper 21.1 oi «The Transuranium Elements», National Nuclear
Energy Series, Division IV, Vol. 14B, McGraw-Hill Book Co. Inc., New York, 1949;
Nucleonics, 5, (5) 16 (1949).
105. Z а с h a r i a s e n W. H., Science, 111, 460 (1950).
Глава 3
УРАН В ПРИРОДЕ
ОБЩИЙ ОБЗОР
Изверженные породы. В настоящее время уран считают
«вездесущим» элементом. Тот факт, что о широкой распространенности урана нам
известно больше, чем о распространенности других элементов, объясняется его
радиоактивными свойствами, а также радиоактивностью продуктов его
распада, например радия, всегда сопутствующего урану в природе. Эта
радиоактивность позволяет легко открывать и определять даже минимальные
количества этого элемента. Обычным является косвенный метод определения малых
количеств урана в минералах. Он состоит в измерении при помощи
электроскопа количества эманации радия, выделяемой известным весовым
количеством минерала. По данным этого измерения определяют содержание радия,
количество же урана можно рассчитать, если допустить наличие
радиоактивного равновесия между ураном и продуктом его распада—радием [1 ]. В
результате многих независимых друг от друга экспериментальных работ было
доказано постоянство отношения урана к радию в невыветренных породах,
а именно 2,84 • 106 : 1, что согласуется с теоретическими расчетами*.
Содержание урана в различных минералах и рудах определяется также
непосредственно, методом флюоресцентного анализа, который является чрезвычайно
чувствительным аналитическим методом [5—7] (см. стр. 86). Установлено, что
изверженные породы, богатые кремнекислотой, часто называемые кислыми,
фельзическими, содержат значительно больше урана, чем породы с меньшим
содержанием кремнезема (основные, мафические породы). Это показано в
табл. 25, где минералы размещены приблизительно в порядке убывающего
содержания Si02. Помимо урана и тория в таблице дано еще содержание
калия, имеющего довольно большое геофизическое значение в качестве третьей
радиоактивной составляющей земли [8].
Наиболее вероятное значение среднего содержания урана в поверхности
земной коры равно 4-10~6г на 1 г породы. Концентрация в обычных
породах лежит в пределах от 0,2-10~8 до 25-10~8 г урана на 1 г породы. Эти
значения, данные Хевеши [9, 10], были признаны Гольдшмидтом [11] наиболее
вероятными** (см. также [12]).
Табл. 26, в которой приведено содержание некоторых элементов в
изверженных породах, показывает, что такие металлы, как кадмий, висмут, ртуть
и серебро, не считающиеся особенно редкими, содержатся в земной коре в
значительно меньших количествах, чем уран. Однако простой зависимости между
средней концентрацией элемента в земной коре и вероятностью нахождения
экономически важных залежей этого элемента не имеется.
* В некоторых случаях отношение Ra : U отличается от рассчитанного по закону
радиоактивного равновесия. Это объясняется выветриванием и избирательным
выщелачиванием [2—4].
** Необходимо указать не неправильность некоторых данных в часто цитируемой
известной работе Кларка и Вашингтона [13], так как вследствие математической ошибки
и использования устаревшего значения для отношения урана к радию эти авторы дали
для распространенности урана в земной коре величину в 20 раз большую, чем истинная.
А. Е. Ферсман [14] предлагает величину 9-10~вг на 1 г, а Андерсон [15]—2«10~7г
на 1 г породы.
60
Таблица 25
РАДИОАКТИВНЫЕ СОСТАВЛЯЮЩИЕ ИЗВЕРЖЕННЫХ ПОРОД
Тип породы
Базальты центрального
типа:
континентальные . .
океанические ....
Плато-базальт
Дунит
Приблизительное
содержание
Si02> о/0
70
66
60
50
50
43
40
Количество на 1 т
уран, г
9,0
7,7
4,0
3,5
3,6
2,2
2,4
1,0
1,5
1,4
торий, г
20,0
18,0
6,0
9,1
7,1 •
5,0
5,1
1,8
3,3
3,4
калий, кг
34
25
17
19
18
8
7
4
8
0,3
Таблица 26
СОДЕРЖАНИЕ НЕКОТОРЫХ ЭЛЕМЕНТОВ В ИЗВЕРЖЕННЫХ ПОРОДАХ [16].
Элемент
Li
Be
Na
Al
Si
Cr
Cu
Zr
As
Ag
Cd
Содержание
на 1 m
породы, г
65
ба)
28 300
81300
277 200
200
100
40
5
о,ю
0,5
на 100 атомов Si,|
атомы
0,091
0,0067
12,4
30,5
100
0,039
0,016
0,0062
0,00067
0,000009
0,000045
Элемент
In
Gd
W
Pt
Au
Hg
Pb6>
Bi
Th
U
Содержание
на 1 m ,
породы, г
0,1
6,36а>
69
0,005
0,005
0,5а>
16
0,2
11,5а'в>
4
на 100 атомов Si,
атомы
0,000007
0,000394
0,0038
0,00000027
0,00000026
0,000025
0,00080
0,000009
0,00050
0,00016
Это значение основано на результатах анализов осадочных пород.
е) По вопросу о вычислении возраста солнца на основании отношения Pb : U на земле
см. [17].
в) Это значение основано главным образом на анализах осадочных пород. Отношение
Th : U изучено многими исследователями. На основании ста определений для обычных
изверженных пород можно для этого отношения принять значение 3,2, если не считать
типичных ториевых и урановых минералов [18, 19]. В более поздней работе [20] для
отношения Th : U в гранитах найдены значения от 2,4 0 до 3,98, среднее—3,39; для
промежуточных пород среднее 3,98. Геохимическое распределение тория, повидимому,
немного отличается от распределения урана [2 1, 22]. Установлено [17], что среднее
отношение Th : U для всей земли равно 6,5—7, в то время как для верхней части земной коры
оно составляет 3—4; следовательно, относительная концентрация тория в центре земли
должна быть гораздо выше (см. также [23]).
Глава 3. Уран в природе
61
Возникает вопрос, соответствует ли содержание урана вблизи поверхности
земной коры концентрации урана во всей земной коре. Ответ заключается в том,
что верхняя часть коры, вероятно, обогащена ураном. Как указывалось выше,
уран встречается преимущественно в кислых изверженных породах. Ввиду
того что важнейшие компоненты глубоких слоев земной коры сравнительно
бедны кремнеземом, можно полагать, что распространенность урана там ниже,
чем вблизи поверхности. Другой аргумент в защиту этого предположения может
быть получен путем анализа теплового баланса земли. При радиоактивном
распаде выделяется тепло. Если предположить, что радиоактивные элементы уран
и торий находятся в земной коре до глубины в 16 км в тех же концентрациях,
что и вблизи поверхности земли, и что калий довольно равномерно
распространен по всей литосфере, то тепло, выделяющееся при радиоактивном распаде,
должно возмещать все потери, происходящие из-за излучения землей тепла
в пространство. Если же значительные количества урана и тория существуют
еще и в более глубоких слоях, то становится совершенно непонятной
приблизительная устойчивость теплового состояния земли [24, 25].
Не следует, однако, думать, что более глубокие слои земной коры
совершенно лишены радиоактивных элементов. Наоборот, еще в 1909 г. было
высказано предположение [26], что геологические революции (периоды
вулканической деятельности и горообразования) в истории земли связаны с
существованием урана в этих слоях. Можно принять, что, в то время как основная масса
урана содержится в пределах 16—20 км от поверхности земли, некоторая часть
его может находиться ниже—до глубины от 40 до 48 км, которая считается
толщиной литосферы.
Вес земной коры толщиной в 20 км равен приблизительно 3,25-1019 т.
Если предположить, что среднее содержание урана равно 4-10~6 г на 1 г
породы, то вес всего урана, находящегося в земной коре, должен составлять
около 1,3-1014 т. Основное количество урана содержится в богатых
кремнеземом изверженных породах, покрывающих материки, и только относительно
небольшие количества, вероятно, находятся в породах (главным образом
основных), образующих дно океанов.
Осадочные породы. Изверженные и метаморфические породы
составляют 95% веса верхнего шестнадцатикилометрового слоя земной коры,
на долю осадочных пород (сланцев и т. д.) приходятся остальные 5%.
За исключением карнотитовых пород Колорадо и Юта, осадочные породы
содержат намного меньше урана, чем изверженные (в среднем, пожалуй,
раза в два).
Океаны, реки и горячие источники. Уран содержится
в измеримых концентрациях в воде океанов. Старые значения концентрации
урана в морской воде [27] были основаны на определениях радия. Однако при
этом предполагалось существование вокеанах радиоактивного равновесия между
ураном и радием, что нельзя считать доказанным. Наоборот, имеются
веские аргументы в пользу того, что концентрация радия в морской воде равна,
вероятно, только 0,1 равновесной концентрации. Это понятно, так как
большая часть радия, образующегося в результате распада растворенного урана,
вероятно, осаждается из морской воды в виде сульфата и карбоната. Поэтому
прежние данные для концентрации урана в морской воде лишены значения,
так как они рассчитаны по содержанию радия. Применение прямого
флюоресцентного анализа дало для содержания урана в морской воде величины от
0,36- 10"в до 2,3«10_вг/л [28]. Содержание урана пропорционально общей
солености. Вода океанов, содержащая 3,5% солей, имеет около 2-10-6 г урана
в 1 л (2-10~9 г/г) [29], что составляет около 0,05% содержания урана в равном
по весу количестве породы. Если считать объем океанов равным 2-109 кмв, то
окажется, что полное содержание урана в океанах составляет 4-Ю9 т, или
0,003% (вес.) количества его в земной коре (1,3-1014 т).
62
Часть I. Уран как элемент
Известно, что многие горячие источники радиоактивны. Но их активность
обусловлена, повидимому, главным образом присутствием радия;
относительно содержания урана в этих водах известно очень мало. На основании
некоторых отрывочных данных предполагается, что содержание урана в реках того
же порядка, что и в морской воде.
Много исследований проведено по определению содержания урана в
океанических отложениях [30,31]. В последних отсутствует радиоактивное
равновесие; верхние слои, в частности, содержат избыточные количества радия.
Причина этого явления, как указывалось выше, заключается в осаждении из
морской воды нерастворимых сульфата и карбоната радия. Вследствие этого
количество урана, находящегося в осадках, не может быть точно рассчитано из
определений количеств радия. Содержание урана в океанических осадках
считают равным приблизительно 10б т (крайне незначительное количество по
сравнению с содержанием в материковых породах).
Живая материя. .Уран является составной частью живой материи
(«биосферы» по терминологии В. И. Вернадского); биологическое значение
повсеместного распределения урана в растениях и животных рассматривается
в биологических томах американской серии работ по ядерной энергии. Уран,
вероятно, является нормальным компонентом протоплазмы [32]. Установлено,
что уран встречается в протоплазме в концентрациях от Ь10~4 до Ы0"9%
(вес.) [33—37]. Связывание урана морскими водорослями, возможно, имело
определенное значение для образования некоторых залежей урана [38] (см.
стр. 89).
Нахождение урана в космическом пространстве.
Многочисленные анализы метеоритов показали, что они содержат уран [39—44].
Однако в соответствии с правилом, согласно которому основные, бедные
кремнеземом, породы на земле в общем отличаются низким содержанием урана,
установлено, что каменные метеориты (так называемые «аэролиты»), которые
имеют еще меньше Si02, чем даже сильноосновные земные породы, содержат
только около 3,6-10"7 г урана на 1 г (это среднее значение, основанное на
анализах 20 аэролитов). Содержание урана в железных метеоритах (сидеритах)
еще меньше. Показано, что такие метеориты содержат в среднем 9-Ю"8 г
урана на 1 г, т. е. только около 0,5% среднего содержания урана в
изверженных породах. Это согласуется также с данными по геохимическому
распределению урана, так как железное ядро земли, вероятно, почти не содержит
урана. Относительно большое содержание гелия в малых метеоритах частично
объясняется разрушением ядер урана и тория космическими лучами [45].
Путем спектроскопических определении еще не удалось установить с
достаточной точностью присутствие урана на солнце и других звездах.
Мы не будем здесь останавливаться на возможности использования
радиоактивности урана для определения возраста минералов. Этому вопросу
посвящено много работ [1, 46—49]. Открытие явления деления ядер урана дало
новое толкование роли радиоактивных элементов в геохимии и геофизике.
Для знакомства с этими вопросами мы отсылаем к обзорной статье Гудмэна
[50] «Приложение ядерной физики к геологии», которая содержит подробную
библиографию (см. также [51, 52]).
РАСПРОСТРАНЕНИЕ И СОСТАВ МИНЕРАЛОВ, СОДЕРЖАЩИХ УРАН [53]
В настоящее время трудно дать полную последовательную картину
происхождения огромной массы урановых минералов, найденных в природе. Эти
трудности объясняются многими причинами. Во-первых, большинство этих
минералов настолько сложно и отличается таким непостоянством составных
частей, что даже химический состав известен достаточно точно только для
немногих из них. Кроме того, данные анализов кристаллической структуры
Глава 3. Уран в природе
63
большинства урановых минералов до сих пор носят отрывочный характер, если
считать, что они вообще существуют. Наконец, некоторые стороны химии
урана только теперь становятся ясными. Это особенно верно для системы
уран—кислород, имеющей большое значение для понимания природы
минералов уранинита и урановой смоляной руды.
Большинство урановых минералов найдено в пегматитах, в частности
в гранитных пегматитах. Главными исключениями являются урановая
смоляная руда (которая встречается в жилах и пластах), карнотит и некоторые
урановые залежи, ассоциированные с органическими веществами, как
например, кольм (разновидность битуминозного сланца, найденная в Швеции).
Присутствие урана в гранитных пегматитах может быть объяснено на основании
геохимических принципов Гольдшмидта [54]. Согласно его взглядам, после
того как однородно расплавленный земной шар начал остывать, вещество его
распределилось между одной газовой и тремя концентрически сгустившимися
фазами. Распределение элементов по различным фазам, согласно Гольдшмидту,
показано в табл. 27.
Таблица 27
РАСПРЕДЕЛЕНИЕ ЭЛЕМЕНТОВ ПО РАЗЛИЧНЫМ ЗОНАМ ЗЕМЛИ а)
Элемент ы
сидеросферы
(железная фаза)
Fe, Co, Ni
Р, С
Mo, (W?)
Pt, Ir, Os?, (Pd)
Ru, Rh
халькосферы
(сульфидная фаза)
[0], S, Se, Те,
Fe, (Ni), (Co), Mn?
Cu, Zn, Cd, Pb
(Sn?), Ge, (Mo?)
As, Sb, Bi
литосферы
(силикатная фаза)
0, (S), (P), (Я)
Si, Ti, Zr, Hf, Th
F, CI, Br, J
B, A!, Sc, Y, La, Ce,
Pr, Nd, Sm, Eu, Gd,
Tb, Dy, Ho, Er, Tu,
Yb, Lu
Li, Na, K, Rb, Cs
Be, Mg, Ca, Sr, Ba
(Fe), V, Cr, Mo, [Ni],
[Co]
Nb, Та, W, U, Sn
(QB)
атмосферы
(газовая фаза)
H, N6), (0), (CI?)
He, Ne, Ar,
Кг, Хе
а) Круглые скобки обозначают малые, а квадратные—очень малые количества.
б) Вероятно, также в бидс нитридов в сидеросфере при высокой температуре и большом давлении.
в) В виде карбонатов.
Сидеросфера составляет земное ядро; халькосфера образует промежуточный
слой; литосфера является внешней земной корой. Элементы,
концентрирующиеся в литосфере,—это те, которые образуют термохимически наиболее
устойчивые окислы, в том числе урановые. Во время остывания жидкой-силикатной
магмы первыми начали кристаллизоваться высокоплавкие твердые фазы,
содержащие главные компоненты смеси. В случае возможности образования
твердых растворов между кристаллизующимися веществами и
второстепенными компонентами последние выпадали совместно на ранней стадии. Если
кристаллохимические константы элемента затрудняли внедрение его в
решетку образовавшихся ранее минералов, то этот элемент постепенно все
более концентрировался в остаточной жидкой фазе. В третьей графе табл. 27
64
Часть I. Уран как элемент
указаны некоторые элементы, кристаллохимические свойства которых
препятствовали их вхождению в кристаллы пород, образованных из массы
силикатной магмы; это—редкоземельные элементы, цирконий, торий, гафний, ниобий,
тантал, вольфрам, олово, литий, бериллий, бор и уран. Эти редкие элементы
должны были концентрироваться в последней части жидкой магмы. Породы,
образовавшиеся из остаточной силикатной магмы, являются хорошо
известными геологическими видами, богатыми полевым шпатом и носящими название
пегматитов. Таким образом, можно дать рациональное объяснение присутствию
в пегматитах урана вместе с торием, танталом, ниобием, цирконием, гафнием
и редкоземельными элементами.
После первоначального осаждения урановых минералов из магмы они могли
подвергнуться многочисленным реакциям. Всегда существовала вероятность
окисления. Могло иметь место избирательное выщелачивание пегматита, или
последний мог быть залит горячими растворами различных веществ. Таким
путем многие первичные минералы претерпевали значительные изменения.
Следовательно, геологическая история тех или иных отдельных урановых
минералов часто является очень сложной, и только для некоторых из них мы имеем
более или менее ясную картину. Однако с возобновлением интереса к этому
вопросу положение может улучшиться.
Кроме урановых минералов, найденных в пегматитах, имеются два других
типа, требующие особого внимания ввиду их экономической важности. Один
из них—тЭто карнотит, он подробно рассматривается ниже. Другой—урановая
смоляная руда, жилы которой найдены в Канаде, Бельгийском Конго и в
других местах. Предполагают, что жилы урановой смоляной руды образовались
из той же остаточной магмы, что и пегматиты. Эти магмы могли содержать
заметные количества воды, следовательно, в дополнение к минералам, которые
выкристаллизовались из магмы как пегматиты, могли также образоваться
и водные растворы. При высоких температурах и давлении большинство
элементов (перечисленных во второй графе табл. 27), которые находились
в остаточной магме, должно было концентрироваться в водных растворах, Это
произошло и с ураном, многие соединения которого хорошо растворима
в воде. При соприкосновении этих горячих растворов с изверженными породами
происходили химические реакции, в результате которых возникли
месторождения так называемых «гидротермальных» рудных жил. Таким образом, уран
в жилах смоляной руды ассоциируется с халькофильными элементами—медью,
висмутом, серебром, оловом и золотом. Большинство этих элементов выпадало
в виде сульфидов, уран же, согласно своим химическим свойствам, почти всегда
встречается в виде кислородных соединений.
Классификация урановых минералов. Скудность
хороших рентгеновских кристаллографических данных приводит к тому, что
классификация урановых минералов до некоторой степени является
произвольной.
Прежде минералоги применяли термины «уранинит» и «урановая
смоляная руда» без разбора для обозначения почти всех уран-кислородных
минералов. Это неправильно, так как между этими двумя минералами существуют
определенные химические различия. Использование обоих терминов как
синонимов создало значительную путаницу. Уранинит находится только в
пегматитах, и он всегда содержит значительные количества тория и редкоземельных
элементов. Урановая смоляная руда обнаружена только в гидротермальных
жилах, она обычно не содержит тория и имеет лишь следы редких земель.
Уранинит первоначально, вероятно, представлял собой чистую двуокись урана
U02; весь шестивалентный уран, находящийся в уранините, вероятно,
образовался в результате последующего окисления. Наоборот, урановая смоляная
руда скорее всего вначале имела состав, близкий к закиси-окиси урана Ua0R.
Уранинит — кристаллический минерал, урановая смоляная руда обычн'
Глава 3. Уран в природе
65
аморфна. В дальнейшем термин «уранинит» мы будем применять только
для обозначения кристаллической двуокиси урана, найденной в
пегматитах* [55].
Рассматриваемые здесь минералы помещены в табл. 39. Для них
принята следующая классификация (ср. [57]):
A. Пегматитовые урановые минералы:
1) уранинит,
2) урансодержащие ниобаты и танталаты.
Б. Непегматитовые урановые минералы:
1) урановая смоляная руда,
2) окислы урана, возникшие в результате окисления урановой смоляной
руды (часто одинаковые по составу с минералами, образовавшимися
из уранинита).
B. Вторичные урановые минералы:
1) уранаты, силикаты, карбонаты, сульфаты,
2) урановые «слюдки» типа М(Н) (U02)2(X04)2-ftH20, где М—Са, Си,
Fe, Pb, Mn или UO;2, a X—Р, V или As,
3) органические вещества, содержащие уран.
Вторичные минералы, образовавшиеся в результате окисления или
других химических реакций, могут встречаться как в пегматитах, так и в жилах
урановой смоляной руды. Некоторые наиболее важные представители этих
групп будут более подробно рассмотрены в следующих разделах.
Для того чтобы легче было разобраться в составе урановых минералов,
в табл. 28 даны ионные радиусы наиболее часто встречающихся в них
элементов. Существует общее правило, что ионы, радиусы которых различаются не
более чем на 15%, могут замещать друг друга в кристаллической решетке при
соблюдении некоторых условий, например при сохранении общей суммы
зарядов; применение и ограничения этого правила см. у Гольдшмидта [11, 16].
При сопоставлении ионных р'адиусов (см. табл. 28) видно, что практически
Таблица 28
ИОННЫЕ РАДИУСЫ а) НЕКОТОРЫХ ЭЛЕМЕНТОВ, ОБЫЧНО ВСТРЕЧАЮЩИХСЯ
В УРАНОВЫХ МИНЕРАЛАХ б)
Ион
U (IV)
Th (IV)
Er (III)
Yb(III)
Y (HI)
Са (II)
Радиус, А
1,05
1,10
1,02
1,00 ]
1,06 1
1,06
Ион
Nb(V)
Ta(V)
Ti (IV)
Mo (IV)
Fe(III)
Радиус, А
0,69
0,68
0,64
0,68
0,67
Ион
P(V)
V(+V)
As (V)
Cr(VI)
S(VI)
Радиус, А
0,3—0,4
-0,4
0,4
0,3-0,4
-0,34
а) Для координационного числа 6.
б) Значения взяты у Эванса [58].
все минералы, содержащие заметные количества редкоземельных элементов,
вероятно, должны содержать немного и урана.
Уранинит (табл. 29). Уранинит [56] можно рассматривать как
первичный урановый минерал. Он встречается в гранитных и сиенитовых
пегматитах совместно с цирконом, турмалином, монацитом, полевым шпатом,
слюдой и т. д. Часто он также тесно ассоциирует с минералами, содержащими
* Иногда урановая смоляная руда рассматривается как разновидность уранинита
[56]; это принято в современной минералогии. Однако различия в химических свойствах
этих двух минералов являются основанием для данной здесь трактовки.
5 Химия урана
Таблица 29
УРАНИНИТОВЫЕ МИНЕРАЛЫ
Минерал
Уранинит
Брёггерит
(ториевый
уранинит)
Клевеит, ни-
венит (ит-
трово-це-
риевые
ураниниты)
То же
Торианит
Структура
Главным образом
U02 с U03,
образующимся в
результате окисления;
структура флюорита,
изоморфен Th02 i; Ce02;
плотность 8,0—
—10,6 г/см*
(U, Th)02;
содержит до 14% Th02,
замещающего уран;
этот минерал
отличается тем, что Th >
>Y, Се и т. д.;
содержание редких земель
0-8 %
[U(IV), Z(III)]02,
где Z—редкие земли
5,17%; присутствуют
Y, Ег, Се, La;
отношение U02 : U03
меняется от 0,5 до 0,7
[U(IV), Z(1II)]02,
где Z—редкие земли
11,2—11,8%;
присутствуют Y, Ег, Се,
La; отношение U02:
:U03—2,5; плотность
8,29 г/см3
[Th(IV), U(IV)]02;
отношение Th()2 :
: U3Os от 6 : 1 до 2 : 1;
плотность 8—9,7 г/см3
Содержание
урана, %
65,2—74,5
48,7—74,9
53,3—66,4
57
9,5-28,2
Оптические и
кристаллографические
свойства
Кристаллический непрозрачный;
октаэдрические или
кубические
изотропные
кристаллы; цвет
переменный; иногда имеет
полуметаллический
блеск; наименее
измененные
образцы железио-серого
цвета
Изотропные
кристаллы
Кристаллический
»
Кубические
кристаллы; цвет от
желтовато-коричневого до черного;
показатель
преломления 1,8
Типичные
месторождения
Масаки, Япония;
Гайя, Индия; Граф-
тон Сентер, Нью-Гэмп-
шир; Портланд,
Коннектикут; Бедфорд,
Нью-Йорк; графство
Митчелл, Северная
Каролина; графство
Пенингтон, Южная
Дакота; Квебек,
Канада; Капская
провинция, Южная
Африка
Полуостров Анне-
род, Норвегия
Норвегия
Графство Льяно,
Техас; пегматиты
Ясака на о. Хондо,
Япония
Цейлон и
Япония (?); Мадагаскар;
Истоп, Пенсильвания
Глава 3. Уран в природе
67
редкие земли и ниобий или тантал. Гольдшмидт и Томассен [59], которые
изучали структуру кристаллов природного уранинита из Южной Норвегии при
помощи рентгеновских лучей, первые показали, что уранинит является
практически двуокисью урана U02. Согласно этим авторам, уранинит изоморфен
двуокиси церия и двуокиси тория; он имеет кубическую структуру типа
флюорита. Константа его решетки 5,460 А. Подобные же результаты были получены
и Скупом [60], установившим, что природный уранинит дает ту же
рентгенограмму, что и синтетическая двуокись урана. Ураниниты из различных
месторождений кристаллографически идентичны. Первая графа табл.28 показывает, какие
замещающие ионы обычно встречаются в уранините. Близость ионных
радиусов объясняет постоянное присутствие редкоземельных элементов в кристаллах
уранинита.
Исходя из химических свойств двуокиси урана, можно предполагать, что
природный уранинит всегда должен быть более или менее окислен, так что его
действительный состав лежит между U02 и U02>67(U308) обычно с
преобладанием четырехвалентного урана. Это окисление может быть результатом
выветривания, однако оно может быть и следствием действия кислорода,
выделяющегося при радиоактивном распаде урана до свинца:
UOa-^PbO + VaOa ( + 8Не).
Экспериментально установлено, что количество кислорода, освобождающегося
при радиоактивном распаде урана, находящегося в кристаллах, близко
соответствует количеству образовавшейся РЬО (окиси RaG) [61]. Таким образом,
радиоактивность играет важную роль в изменении состава минералов,
содержащих двуокись урана. Это также доказывается их общим метамиктным
характером.
Было изучено влияние выветривания на состав монокристаллов уранинита
путем определения отношения U(IV) : U(VI) в нескольких слоях кристаллов
(клевеита) как функции расстояния от поверхности. Это отношение возрастает
в направлении центра [62—64]. Аналитическое исследование указывает, что
уранинит может подвергаться избирательному выщелачиванию, которое
особенно действует на радий и свинец, содержащиеся в кристаллах. Минерал,
подвергшийся вторичным изменениям, чернеет; менее изменившиеся образцы
имеют железно-серый цвет. Более резко выраженное изменение ведет к
появлению продуктов разложения алого, оранжевого, желтого, зеленого, серого или
коричневого цвета. Были сделаны попытки воспроизвести процесс
выветривания уранинита [65]. Образцы клевеита и брёггерита нагревались с водой в
автоклаве до температуры 200°. При этом клевеит (с высоким отношением U03: U02)
подвергался очень небольшому изменению, тогда как брёггерит (с низким
отношением U03 : U02) довольно сильно изменился.
Ряд типичных анализов минералов уранинита дан в табл. 30 (см. также [66]).
Рассмотрение этих данных подтверждает мнение, что все эти минералы
образовались в результате изменений и замещения ионов в обычной чистой двуокиси
урана (названной Киршем «ульрихитом», этот термин иногда применяется в
литературе и для обозначения наименее измененных разностей уранинита).
Другие урановые минералы пегматитовых
месторождений. Ниобаты, тантал аты и титанаты. Вероятно,
наиболее часто встречающиеся урановые минералы в пегматитах являются уран-
содержащими ниобатами, танталатами и титанатами. Ниобий, тантал и титан
находятся среди элементов, которые кристаллизуются на последних стадиях
затвердевания магмы (см. выше). Величины ионных радиусов (см. табл. 28)
указывают на возможные взаимные замещения в соединениях, содержащих
эти элементы. Структура и состав образующихся минералов здесь не
рассматриваются детально, однако соответствующие данные могут быть взяты из
литературы [56]. Достаточно указать, что эти минералы являются окислами
5*
68
Часть I. Уран как элемент
Таблица 30
СОСТАВ НЕКОТОРЫХ УРАНИНИТОВЫХ МИНЕРАЛОВ, %
Компоненты
СаО
ЛпО
РЬО
MgO
<Y, Er)303
(Се, La)203 ....
СеОэ
иоа
и3о8
U03
Th02
ZrOo
Si02
А1203
Fe203
С02
Н20
Нерастворимые . . .
Плотность, г/см3 . .
Брёгге-
рит а)
0,37
—
9,04
Следы
1,11
—
0,27
0,18
46,13
—
30,63
6,00
i 0,06
0,22
—
0,25
—
0,74
4,42
0,19
99,61
8,893
Нивенит")
, 0,32
—
10,08
—
9,46
—
2,36
0,34
44,17
—
20,89
6,69
0,34
0,46
—
0,14
—
1,48
1,47
0,08
98,28
8,29
Клевеит в)
0,86
—
10,92
0,14
9,99
2,25
—
—
23,07
—
40,60
4,60
—
—
—
1,02
—
4,96
2,34
—
100,75
7,49
Ураиннитг)
1,01
0,03
10,95
0,08
2,14
1,88
—
—
39,10
—
32,40
10,60
—
0,19
0,09
0,43
—
0,70
0,15
0,31
100,06
9,062
УранииитД)
0,46
0,001
16,42
0,01
1,01
—
0,80
0,265
48,87
—
28,582
2,15
0,22
0,055
—
0,30
—
0,44
0,15
| 0,39
100,123
9,182
а) Из рудника Густав, Аннерод, Норвегия [67].
<*) Из Бариигер-Хилл, графство Льяио, Техас [68].
в) Из Арендала, Норвегия [69]; состав пересчитан [67].
г) Из Вильберфорса, графство Халибуртон, Онтарио [7 0]»
Д) Из рудника Ингерсолл, графство Пеннингтои, Южная Дакота [71].
изодесмического типа. Ввиду того что для большей части этих урансодержащих
ниобатов, танталатов и титанатов кристаллографические данные отсутствуют,
в настоящее время их можно классифицировать только по химическим
признакам. Химический состав большинства из них может быть выражен
формулой AmBn02(m+n), где отношение т:п лежит между 1 и 0,5,
А—редкоземельные элементы, U, Ca, Th, Fe(II), Na, Mn или Zr; В—Nb, Та, Ti, Sn, W(?),
Zr(?) или Fe (III). Эти минералы чрезвычайно сложны, весьма непостоянны
по составу и с трудом поддаются анализу; поэтому предлагаемые формулы
должны во многих случаях рассматриваться как весьма приближенные.
В табл. 31 даны наиболее важные урансодержащие минералы этого типа,
а в табл. 29 были рассмотрены некоторые наиболее существенные урановые
минералы, встречающиеся в пегматитах. Несмотря на широкое
распространение всех этих минералов, они не имеют практического значения. Вернее, они
не приобрели значения вплоть до настоящего времени, т. е. до момента
написания этой главы. Для получения урана добываются только два из этих минералов
и в очень ограниченных размерах: бетафит на Мадагаскаре и эвксенит-поликраз
в Западной Австралии и Бразилии. Ввиду возросшего значения урана это
положение может измениться. Например, пегматиты Ясака на о. Хондо (Япония)
содержат около 100 г урана на 1 т породы. Они мало интересны как источник
Таблица 31
ПЕГМАТИТОВЫЕ НИОБИЕВЫЕ, ТАНТАЛОВЫЕ И ТИТАНОВЫЕ МИНЕРАЛЫ, СОДЕРЖАЩИЕ УРАН
Минерал
Состав и строение
Содержание
урана, %
Оптические и кристаллографические
свойства
Месторождения
Ссылка на
справочник а)
[66]
[56]
Ряд пкрохлора, микролита [А2В206(0, ОН, F)]
A—Na, Са, К, Mg, Fe (II), Мп (II), Pb (?), Се, La, Dy, Ег, Y, Th, Zr, U
B — Nb, Та, Ti, Sn (?), Fe (III) (?), W (?)
Пирохлор
Микролит
Гатчеттолит
Эльсвортит
В основном
NaCaNb2OeF
В основном
(Na, Ca)aTa2Oe (О, ОН, F)
Урановый пирохлор
Кальциево-железный
пирохлор с высоким
содержанием урана; сильно
измененный; родственен
гатчеттолиту
2,5—8,1
0—1,3
10—15
15-17
Блеск от стеклянного до смоляного;
от бурого до почти черного цвета
Небольшие октаэдры; от золотисто-
желтого до бурого цвета
Прозрачные изотропные октаэдры;
блеск смоляной; п= 1,98 ^
Массивный; от янтарно-желтого до
шоколадно-коричневого цвета;
изотропный; п= 1,89^
В нефелиновых
сиенитовых пегматитах и
гранитных пегматитах
Швеция; Виргиния;
Гренландия; Западная
Австралия; [ Массачусетс
Хкбла, Онтарио;
графство Митчелл,
Северная Каролина;
Мадагаскар
Хибла, Онтарио; Ха-
либуртон, Онтарио
ш. (о.
95
Ш. (1).
250
Ш. (1).
250
IV, (2),
955
Продолжение табл. 31
Минерал
Состав и строение
Содержание
урана, %
Оптические и кристаллографические
свойства
Месторождения
Ссылка на
справочник а)
[66]
Фергусоиит
Ретзерфорднт
Иттротанталит
Ишикавапт
Браннерит
Ряд фергусонита, форманита (АВ04)
A —Y, Ег, Се, La, Dy, U (IV), Zr, Th, Ca, Fe (II)
B —Nb, Та, Ti, Sn, W
В основном ниобат-
танталат Y, Ег
Измененный
фергусоиит
В основном железо-
иттро-урано-ниобиевын
танталат
Главным образом уран
(1\')-железо-редкоземель-
ный ниобат-танталат,
близок к самарскиту
(U, Ca, Fe, Y, Th)3
Ti5O10
0,2-8,16
1,4-3,96
20
40
Тетрагональный; с =1,4643;
пирамидальные кристаллы; черно-бурый
Ромбический; а : Ъ : с : -— 0,5566 :
: 1,0 : 0,5173; призматический; от
желтовато-бурого до черного;
изотропный; /2 ---2,15 ±0,02
Пластинчатые ромбические
кристаллы; а : 6 : с = 0,9451 : 1 : 1,1472;
черный; с восковым блеском; уд. вес
6,2—6,4
Изотропный; nD = 2,30 ± 0,02;
черный, просвечивает лишь в тонких
обломках
Гренландия; Швеция;
Норвегия; Массачусетс;
Льяно, Техас; Цейлон;
графство Митчелл,
Северная Каролина
Графство Ретзерфорд,
Северная Каролина
Норвегия; Швеция,
Алабама
Провинция Иваки,
Япония
Графство Кастер,
Айдахо
III (Г
252
Ш, (1),-
256
Продолжение табл, 31
Минерал
Состав и строение
Содержание
урана, %
Оптические и кристаллографические
свойства
Месторождения
Ссылка на
справочник а)
[06]
Ряд эвксенит-
поликраза, куда
входит п
разновидность лин до-
кит; близки кт-
трокразит, эшве-
гит, но плохо
идентифицируются
Ряд эшиннта-
прнорита-блом-
страндина
Самарскит б)
(вникит и хьел-
мит являются
плохо
идентифицируемыми
минералами, возможно,
родственными
самарски ту)
(Y, Са, Се, U, Th)
(Nb, Та, Ti)2Oc; крайним
представителем ряда,
богатым титаном, является
полнкраз
[Y, Ег, Са, Fe (II),
Th, U (IV)] (Ti, Nb)206
(Y, Er, Се, U, Са,
Fe, Pb, Th) (Nb, Та, Ti,
Sn)2O0
Ряд АВ2Ос
A —Y, Ce, Ca, U, Th, Er, La, Pb
B—Ti, Nb, Та, Fe (III), Sn, W, Zr (?)
2,3-14,5
1—5
3,5—14,0
Ромбические; черно-бурые
Подобны эвксениту
Ромбические; а: Ь: с = 0,5456 : 1,0 :
: 0,5178, от желтовато-бурого до
черного цвета; изотропные; /1 = 2,20^
4-0,05
Вудсток, Западная
Австралия; Ниписсинг,
Онтарио; Норвегия;
Свазиленд (Трансвааль);
Гренландия; Северная и
Южная Каролина;
Бразилия; Мадагаскар
Норвегия; Свазиленд
Бертье, Квебек;
Балтимора, Мидленд;
графство Митчелл, Северная
Каролина; Колорадо;
Мадагаскар
Ш, (1),
102
Ш, (1),
106
256
Продолжение табл. 31
Минерал
Состав и строение
Содержание
урана, %
Оптические и кристаллографические
свойства
Месторождения
Ссылка на спра
вочник а)
[661
[56]
Ряд AmBnXx
т:п= 1,3
A —U, Ca, Th, Pb, Се, Y, Er
В —Ti, Nb, Та, Fe (III) Al (?)
Группа бетафи-
та, в нее входят
родственные
минералы бетафит,
самирезит, блом-
страндит,
менделеевит, джалмаит
Ампангабеит
Делореицит
Цирке лит
(U.Ca)
(Nb, Та, Ti)309/iH20
Окисел ниобия с
редкими землями и ураном;
формула неизвестна;
вероятно, аналогичен бета-
фиту
Окисел Ti, Y, U, Fe
и Sn, возможно, АВ308
(Ca, Fe, Th, U)2
(Ti, Zr)2Os (?)
| Обычно
20—26,
но может
упасть
до 10
10-17
-10
1
1 Зеленовато-бурые; изотропные;
п= 1,91-1,97
Ромбический; изотропный;
п = 2,13 ± 0,03; все оттенки
коричневого
Ромбический; черный; a: b : с =
= 0,3375: 1,0:0,03412
Черный; п = 2,19 ±0,01
Мадагаскар
Ампангабе,
Мадагаскар
Краведжия, Италия
Сан-Пауло, Бразилия
Ш, (1),
97
IV, (2),
957
Ш, (1),
52
I, 803
I, 806
I, 808
I, 740
а) Римские цифры обозначают том, арабские в скобках—часть, арабские без скобок—страницу,
б) Анализ и определение возраста образца австралийского самарскита см. [72]; анализ ряда образцов самарскита округа Неллоре, Индия, см. [73].
Глава 3. Уран в природе
73
радия, но могут эксплуатироваться для извлечения урана [74]. В общем,
однако, количество урана, которое может быть извлечено из пегматитов,
невелико.
Урановая смоляная руда и продукты ее
изменения. Урановая смоляная руда является наиболее важным урановым
минералом с точки зрения богатства месторождений. Ее состав различен, но при
отсутствии сильного выветривания ее состав приближается к закиси-окиси
урана U308. Урановая смоляная руда встречается в рудных жилах вместе
с сульфидами и арсенидами железа, меди, свинца, кобальта, никеля, мышьяка
и висмута. Она найдена в гидротермальных (так называемых
«гипертермальных») оловянных жилах в виде коллоформной корки совместно с
касситеритом, пиритом, галенитом и минералами кобальта никеля, висмута
и мышьяка, особенно в Корнуэлле (Англия). Другие и более важные типы
месторождений находятся в «мезотермальных» жилах, содержащих кобальт,
никель, висмут, серебро и мышьяк (гидротермальные жилы, образовавшиеся
при умеренных температурах). В этом случае урановая смоляная руда залегает
совместно с пиритом, халькопиритом, баритом, флюоритом, самородными
висмутом и серебром, соединениями кобальта, никеля и мышьяка. Месторождения
около Большого Медвежьего озера в Канаде относятся к этому типу. Урановая
смоляная руда также найдена в виде коллоформной корки с пиритом,
сфалеритами и т. п. в гидротермальных сульфидных жилах, образовавшихся при
умеренных температурах и не содержащих кобальто-никелевых минералов. Залежи
в графстве Гильпин (Колорадо) служат примером месторождений последнего
типа. Месторождения урановой смоляной руды в районе Большого Медвежьего
озера и в Бельгийском Конго более детально будут рассматриваться в
следующем разделе.
Как установлено раньше, образование месторождений урановой смоляной
руды связано с минерализацией жил халькофильными и сидерофильными
элементами, приведенными в табл. 27. Пока еще не известно, какие химические
реакции происходили при переносе урана горячими водными растворами и при
его осаждении в виде U3Os, т. е. в процессах, неизбежно происходивших на
последних стадиях кристаллизации магмы. Как уже отмечено, урановая
смоляная руда ассоциирует с сульфидами, но сам уран в этом минерале содержится
в форме окисла.
Урановая смоляная руда всегда встречается в виде плотного
тонкозернистого характерного черного вещества, лишенного признаков
макроскопической кристалличности. Содержание кислорода переменно, хотя во многих
случаях оно приближается к требуемому формулой U308. Различия в составе
становятся понятными, если учитывать данные кристаллографического
изучения системы U — О. Эти исследования (см. гл. 11) показали, что отношение
уран : кислород может колебаться приблизительно между U02>5 и U03 без
фазовых превращений. В урановой смоляной руде обычно находятся
небольшие количества железа, марганца, алюминия, кальция, магния, кремния
и т. д. вместе с гелием и свинцом, образующимися в результате
радиоактивного распада. Очень существенным различием между урановой смоляной рудой
и уранинитом является то, что первая практически лишена тория и содержит
менее 1 % редких земель. Содержание урана в смоляной руде колеблется от 40
до 76% [66, 75].
Химические свойства урановой смоляной руды практически те же, что
и закиси-окиси урана (см. гл. 11). При нагревании минерала выделяется гелий
[76]. В случае урановой смоляной руды из Бельгийского Конго образуется
также сублимат селена [77]. Все разновидности урановой смоляной руды
растворимы в серной или соляной кислоте.
Известно значительное число продуктов окисления урановой смоляной
руды. Они встречаются или в сложных смесях минералов, или в виде отдельных
74
Часть I. Уран как элемент
зон вторичных изменений в урановой смоляной руде или уранините.
В большинстве случаев анализы этих продуктов бывают неудовлетворительны,
а в случае янтинита они почти всегда заведомо неправильны. Это обстоятельство
затрудняет точное определение строения этих минералов. В табл. 32
представлены гидратированные окислы, которые могли образоваться в результате
окисления урановой смоляной руды. Ввиду того что эти минералы
подвергались сильному выщелачиванию, они обычно содержат небольшие количества
других элементов. Все эти элементы, кроме свинца, могут рассматриваться
лишь как примеси, а не как существенные компоненты минерала.
Месторождения урановой смоляной руды в Бельгийском Конго особенно
богаты продуктами изменения. Минералы этих месторождений, представленные
в табл. 32, содержатся в виде сложных смесей. Помимо минералов,
перечисленных в этой таблице, описаны два новых минерала [78] (названные параскупит
и эпиянтинит) неизвестного состава, происшедшие от скупита и янтинита или
родственные им. Хотя точные отношения неизвестны, эти минералы
представляют различные степени окисления. Термин «гуммит» применяется к минералам,
которые являются практически гидратизированными окислами урана
неизвестного состава и, вероятно, представляют собой конечные продукты
окисления и гидратации урановой смоляной руды. Обычно присутствуют небольшие
количества щелочных металлов, щелочных земель, редких земель, кремнезема,
и т. д., но они, повидимому, являются посторонними примесями. Физические
и химические свойства гуммита меняются в широких пределах.
Вторичные урановые минералы [79]. Образование ура-
натов (а также силикатов, карбонатов, фосфатов, ванадатов, арсенатов и
сульфатов урана), вероятно, происходило путем полного растворения уранинита
или урановой смоляной руды и последующего отложения вторичных
соединений. Некоторые, но не все, из этих вторичных минералов могли
образоваться в водных растворах при температурах, близких к критической
температуре воды, но в настоящее время мало известно о химических реакциях,
которые могут идти при таких условиях.
Силикаты и уранаты. Для удобства и в соответствии с принятой
классификацией [47] эти два класса минералов рассматриваются вместе. Их состав
часто устанавливается неточно. Ряд характерных минералов этого типа
приводится в табл. 33. В литературе описаны [80] биллиетит (гидратированный
уранат бария), вандеыдрисшеит, мезуиит и рихетит (гидратированные уранаты
свинца, найденные в месторождениях Бельгийского Конго).
Рентгенографически определена структура уранотила CaO-2U03-2SiO.,-
•6Н20. Он образует ромбические кристаллы, изоморфные склодовскиту [81].
Карбонаты. Большей частью эти минералы плохо распознаются. Наиболее
типичным представителем этого класса, повидимому, является ураноталлит.
О происхождении этих минералов известно мало, но, вероятно, они
образовались не в процессе простого выветривания на месте залегания, а путем
растворения и вторичного осаждения из водных растворов. Ряд уранокарбонатных
минералов дан в табл. 34. Химические формулы этих минералов недостаточно
точно установлены. То же самое относится и к гидратированным карбонатам
урана—студтиту и дидерихиту [80].
Сульфаты. Минералы этого типа (иногда они называются «урановые охры»)
довольно распространены, но обычно залегают только в очень малых
количествах. Многие из них растворимы в воде и поэтому быстро подвергаются
изменению, что затрудняет установление точных химических формул. Ввиду этого
химическое строение минералов, помещенных в табл. 35, сомнительно.
Главным представителем этих минералов является уранопилит.
Фосфаты, арсенаты и ванадаты. Большое число урановых минералов
вторичного происхождения имеет состав, который может быть выражен общей
формулой M(II)(U02)a(X04)2, где M-Cu, Fe, Mn. Ш3> Са или РЬ, a X-P,As
Таблица 32
продукты окисления урлновоп смоляной руды
Минерал
Беккерелит
Скупит
Янтинит
Гуммит
Состав
2U03-3H20?; формула
известна неточно; воду
теряет при 500°
4U03«9H20?;
полного анализа нет; воду
теряет при 325°
2U02«7HoO; формула
определенно
неправильна; легко изменяется
в беккерелит и скупит
и03-/гН20 (см. стр.
74)
Содержание
урана, %
74
65—70
70
40—70
Оптические и
кристаллографические свойства
Ромбические пластинки;
от канареечно-желтого до
оранжевого цвета
Небольшие желтые
кристаллы; ромбический;
а0 = 14,40, ^о=1б,89,
с0= 14,75; а0: Ь0 : с0=
=0,852 : 1 : 0,873;
элементарная ячейка содержит
и820„в-72Н20?
Ромбический;
фиолетово-черный;
полуметаллический блеск
Желтый, оранжевый,
красный,
красновато-коричневый до черного
Месторождения
Казоло, Катанга,
Бельгийское Конго;
Вельзендорф, Бавария
Казоло, Катанга,
Бельгийское Конго
Шинколобве, Казоло,
Катанга, Бельгийское
Конго
Найден во многих
месторождениях
уранинита и урановой
смоляной руды в
Бельгийском Конго,
Северной Каролине,
Коннектикуте, Квебеке ив
других местах
Ссылка на справочник а)
[66]
IV, (2),
937
IV, (2),
939
IV, (2),
941
IV, (2),
950
[50]
I, 625
I, 627
I, 633
I, 622
Продолжение табл. 32
Минерал
Кларкеит
Фурмарьерит
Кюрит
Состав
По существу гуммит;
гндратнрованная
двуокись U с РЬ,
щелочными и щелочио-зе-
мельными элементами
Возможно,
PbO-4U(V5H20
Гидратированный
окисел РЬ и U,
вероятно, 2PbO-5U03-4H20;
полностью теряет воду
при 450°
Содержание
урана, %
-80
60—70
60—70
Оптические и
кристаллографические свойства
Красновато-кор и чневы й
Ромбический; от
красного до коричневого цвета
Оранжево-красный;
ромбический; а0=12,52,
60=12,98, с0=8,35;
элементарная ячейка
содержит Pb0U15O5i-12H2O
Месторождения
Графство Митчелл,
Северная Каролина
Казоло, Катанга,
Бельгийское Конго
Казоло, Катанга,
Бельгийское Конго
Ссылка на справочник а)
[66]
IV, (2),
944
IV, (2),
942
[56]
I, 625
I, 628
I, 629
L) *нмские цифры обозначают том, арабские в скобках — часть, арабские без скобок — страницу.
Таблица 33
УРАНАТЫ И УРАНОСИЛИКАТЫ а)
Минерал
Ванденбрандит (ураио-
лепидит)
Ураносферит
Соддиит
Уранотил [82] (ураио-
фан, ламбертит);
существует еще
диморфный (3-уранотил
Состав
2CuO-2U03.5H20
(BiO)2Ua07-3H20
12U03-5Si02.14H20 или
5U03-2SiOa-6H20
CaO-2UO,.2SiOa.6H20
Содержание урана,
%
-60
-50
-70
-50
Оптические и
кристаллографические свойства
Триклииные кристаллы; от
темнозеленого до черного
цвета: п% = 1,77, лт<1,80
Желтые ромбические
кристаллы; оптически
положителен; па = 1,955, Па —1,985,
я?=2,05
Желтые ромбические
кристаллы; а:Ь : с : =0,7959 : 1 :
: 1,6685; некоторые
призматические кристаллы обладают
сильным плеохроизмом,
бесцветным вдоль оси а, темно-
фиолетовым вдоль оси 3 и
фиолетовым вдоль оси i
Зеленый или желтый;
ромбический; а: Ъ : с=0,3705 :1:1;
оптически отрицателен, плео-
хроичен
Месторождения
Калонгве, Катанга,
Бельгийское Конго (вместе с ка-
золитом, склодовскитом и др.)
Вместе с вальпургитом,
трёгеритом, цейнеритом,
ураноспинитом
Казоло, Катанга,
Бельгийское Конго (вместе с кюри-
том)
Арен даль, Норвегия;
Вилльнев, Канада; графство
Митчелл, Северная
Каролина; Катанга, Бельгийское
Конго; Мадрид, Испания;
Пенсильвания
Ссылка на
справочник
[6 б]6)
IV, (2), 946
И, (2), 164
Продолжение табл. 33
Минерал
Склодовскит
(шинколобвит)
Казолит [83—85]
Пкль барит
Состав
MgO-2U03-2SiOa.6H20
PbO.U03-Si02-H2O
PbO-U03*Th02.2Si02.4H20
Содержание урана,
%
-50—60
-40
-27U
~30Th
Оптические и
кристаллографические свойства
Ромбические желтые
кристаллы; п* = 1,613, Яд=1,635,
rtf=1,657; плеохроичен;
а : 6 : с=0,3114 : 1 : 1,0554
Моноклинный; желтый;
а:6:с=1,8566: 1 : 1,0811;
Э=103°40'; па = 1,89, лр = 1,90,
пт=1,967
Месторождения
Казоло, Катанга,
Бельгийское Конго
Казоло, Катанга,
Бельгийское Конго
Золотые прииски Пильбара,
Западная Австралия
Ссылка на
справочник
[6 6]б>
IV, (2), 947
IV, (2), 949
П, (1), 804
а) Не вполне достоверные виды минералов макинтошит, метланднт и николаит аналогичны минералам, рассмотренным в этой таблице.
б) Римские цифры обозначают том, арабские в скобках — часть, арабские без скобок — страницу.
У РАНО КАРБОНАТЫ
Таблица 34
Минерал
Резерфордин
Ураноталлит
Либигит
Шрёкингерит (дакеит)
Фоглит
Шарпит [86—88]
Состав
и02С03 (установлено
только для одного образца)
2СаО-ио3'ЗСОг10Н2О
Гидратированные
карбонаты Са и U; точный состав
неизвестен
3CaO.Na2O.U08-3C02-
• SO3-10H2O;
натрий-кальций-урановый
карбонат-сульфат
Возможно, Cu(U02) (С03)2-
• 10Н2О
. 6U03-5C02.8H20
Содержание
урана, %
~75
-30
-30
25
-30
1 67
Оптические н кристаллографические
свойства
Ромбические (?) желтые
кристаллы
Яблочно-зеленые ромбические
кристаллы; а : b : с=0,954 : 1 : 0,783;
сильное двойное лучепреломление;
л« = 1,50, лр= 1,503, лт=1,537
Псевдогексагональиые пластинки;
зеленовато-желтый; л« = 1,49,
/7^=1,54; отрицателен, 2V 0—25°
От яблочно-зеленого до темно-
зеленого цвета, вероятно,
ромбический; оптически положителен; плео-
хроичен
Ромбический (?);
желтовато-зеленый; погасание прямое;
удлинение положительное; а= 1,633,
у—1,72; очень сильное двойное
лучепреломление; слабый
плеохроизм; х—коричневатого цвета; z—от
светложелтого до слабозеленова-
1 того цвета
Месторождения
Морогоро, Восточная
Африка
Адрианополь, Турция
Вайоминг
Бельгийское Конго
Ссылка на
справочник
[663а>
I, 547
1, 545
I, 546
I, 546
I, 546
Римские цифры обозначают том, арабские—страницу.
Таблица 35
УРАНОСУЛЬФАТЫ
Минерал
Иоганнит
(гильпинит)
Ураконит
У ранопилит
[89]
Фоглианит
Уранохальцкт
Циппеит
Состав
CuO-2U03-2S03-7HaO
*
Водный сульфат урана и
меди
6U03-SCV16H20;
дегидратируется до р-уранопилита;
6U03.SO3-10H2O
Вероятно, разновидность ура-
нопилита; известны две
разновидности фоглианита:
кальциевая и медная
Гидратированный сульфат
урана, меди и кальция
2U03-S03-/iH20; n колеблется
от 3 до 8; некоторые образцы
содержат Си (см. фоглианит)
Содержание
ураиа, %
— 40—50
~ 60—70
64
67
-30
- 50—60
Оптические и кристаллографические
свойства
Триклинный; а : Ъ: с =1,218 : 1 :
: 0,6736; а = 69°24', р=124°56',
7=132°56'
Аморфный, оранжевый или желтый
Возможно, моноклинный; лимонно-
желтый; па— 1,621, Па = 1,623, п^ =
= 1,631; {3-уранопилит; /га=1,72,
ria = n^ = l ,76
Яблочно-зеленый
Травянисто-зеленый; оптически
положителен; яв= 1,655, ят = 1,662,
плеохроичен
Возможно, моноклинный для тригид-
рата; па= 1,630, Яо = 1,689, п^ =
— 1,739; плеохроичен; оптические
свойства зависят от степени
гидратации
Месторождения
Гильпин, Колорадо;
Корнуэлл, Англия;
Мидлтаун, Коннектикут
Корнуэлл, Англия
Сент-Джаст, Корнуэлл,
Англия
Фрута, Юта;
Большое Медвежье озеро,
Канада
Ссылка на
справочник
[66]*)
IV, (2), 649
IV, (2), 651
IV, (2), 651
IV, (2), 652
IV, (2), 654
IV, (2), 655
а) Римские цифры обозначают том, арабские в скобках — часть, арабские без скобок — страницу.
Глава 3, Уран в природе
81
или V (см. табл. 28). Логично было бы поместить и сульфаты в этот же класс,
но аналитические данные, имеющиеся в настоящее время, не позволяют этого
сделать. Для удобства (а также вследствие их значения) фосфаты, арсенаты
и ванадаты помещены отдельно в табл. 36—38.
Оптические и морфолого-кристаллографические свойства многих фосфатов
и арсенатов представляют своеобразную проблему. Торбернит, цейнерит
и искусственный ураноспинит оптически одноосны. Отунит, ураноцирцит
и естественный ураноспинит оптически двуосны с ромбической симметрией.
Трёгерит по своим оптическим свойствам должен быть моноклинным, а по своей
морфологии и кристаллографической структуре—тетрагональным.
Предполагают, что все эти минералы тетрагональны, хотя это приводит к ряду аномалий
[91]. Эго мнение подтверждается тем, что в природе иногда встречаются
одноосные образцы трёгерита и ураноспинита. Аномальные оптические свойства
трёгерита Гольдшмидт объясняет искажением кристаллической решетки этого
минерала цейнеритом и деформацией ребер и граней кристалла из-за смещения
слоев, возникающего в результате скольжения по плоскостям спайности.
Для решения этого вопроса желательна была бы постановка дополнительных
исследований. Минералы этого класса химически исследованы мало. Для
большинства из них определена растворимость, проведены также некоторые
работы по дегидратации этих гидратных минералов. Изучены также
изменения отунита и цейнерита [92].
Особого упоминания заслуживают отношения, встречающиеся в
торберните [93].
60-65° 100°
Торбернит ;r^zr Метаторбернит I Т~^± Метаторбернит II
Cu(U02)2(P04)a • 12H20 Cu(U02),(P04)2 ■ 8H20 Cu(L'0?)2(P04)2 • 4НХ>
Тетрагональный, одноос- Тетрагональный, оптически Ромбический. лвусаый,
ный, оптически отрица- положительный оптически отрицательный
тельный
Эти превращения, вероятно, являются также и функцией давления [941.
Минералы, содержащие кальций вместо меди (отуниты), ведут себя подобным
же образом.
Органические урановые минералы. Известно много органических веществ»
богатых ураном. Так, кольм, являющийся углеподобным веществом (с
высоким содержанием золы), залегает в виде дискообразных линз в глинистых
[95, 96] кембрийских сланцах Вестерготландской равнины в Швеции. После
его сжигания остается 27% золы, которая содержит до 2,87% урана (в среднем
1,87% U308). Эга зола используется для промышленного получения радия.
Известно также много битумов, которые содержат небольшие количества
урана [97—99J. Тухолит является веществом с неизвестным химическим
составом, которое содержит углерод, водород, кислород, торий, уран, церий,
иттрий, эрбий, ванадий, фосфор, кальций, кремний и много других обычных
элементов, а также воду. Он найден в Канаде и Швеции [I00J.
Несмотря на широкое распространение многочисленных урановых
минералов, описанных в этом разделе, только карнотит уже давно имеет большое
экономическое значение. Этот минерал рассматривается на стр. 89.
Прочие минералы, содержащие уран. Минералами, содержащими некоторое
количество урана, являются: алланит, гадолинит, иттриалит, корвузит, ксено-
тим, монацит, наегит, роуландит, циркон, циртолит, чевкинит. Многие другие
редкоземельные минералы также, без сомнения, содержат небольшие
количества урана. Поскольку монацит используется как источник тория, может
приобрести значение получение урана в качестве побочного продукта из этого
минерала, а также из ксенотима. Список минералов со ссылкой иа таблицы,
в которых они встречаются, дан в алфавитном порядке в табл. 39.
6 Химия урана
Таблица 36
УРАНОФОСФАТЫ
Минерал
Фосфуранилит
Отунит
Бассетит
Ураноцирцит
Салект
Фритчеит
Состав
(иОг)3(Р04)2.лН20
(обычно л = 6 или 12)
Ca(U02)2 (Р04)2-/гН30
(обычно /г = 8 или 12)
Fe(UO.,)2 (Р04)2.«Н20
Ba(U02)2(P04)2 -8H20
Mg(U02)2(P04)2-8H20
Mn(U02)2(P04)2-8HoOf
кроме того содержит
значительные количества
ванадия
Содержание
урана, %
60—64
-50
-50
-50
-60
Оптические и кристаллографические свойства
Лимонно-желтый; вероятно, ромбический;
оптически отрицателен; па = 1,691, rta =
= 1,720, лY= 1,720; плеохроичен
Небольшие зеленовато-желтые кристаллы;
ромбический; а : b : с= 0,9876 : 1 : 2,8530;
возможно, тетрагональный; оптически
отрицателен; слабое двойное лучепреломление;
па= 1,557, Па — 1,575, /7^=1,577
Желтые моноклинные кристаллы; а: Ъ : с =
0,3473: 1,0:0,3456, р==89°17'; оптически
двуосные; 2£ = 110°; лр = 1,574, п^= 1,580;
плеохроичен
Ромбический; желтовато-зеленый, похожий
на отунит; двойное лучепреломление;
оптически отрицателен; ял=1,61, По = 1,623,
«..= 1,623; плеохроичен
Желтый; моноклинный с
псевдотетрагональной симметрией; оптически отрицателен;
ая = 1,559, яр =1,570, л у= 1,574
Красновато-коричневые квадратные
пластинки; сходный с отунитом; повидимому,
тетрагонально-призматический
Месторождения
Графство Митчелл,
Северная Каролина
Отён, Франция;
Мадагаскар; Коннектикут;
Массачусетс;
Пенсильвания; Северная и Южная
Каролина; Юта;
Корнуэлл, Англия
Корнуэлл, Англия
Шипколобве, Катанга,
Бельгийское Конго
Отён, Франция
Ссылка на
справочник
[66]а)
III, (1), 573
III, (1), 573
IV, (2), 959
III, (1), 575
III, (1), 575
Торбернит
Метаторбер-
нит 1
Ураношпатит
Парсонсит
Девиндтит
(стасит)
Дюмонтит
Ренардит [90]
Cu(U02)2(P04)2-nH20
(п обычно 8—12)
Cu(U02)2 (P04)2.8H20
(см. стр. 8J)
Состав неизвестен;
анализы отсутствуют;
вероятно, гидратированный
фосфат урана
2PbO-U03-P2<VH20
3Pb0.5U03-2P205.12H?0
2РЬО-ЗиОз'Р205-5Н20
Pb0.4U03-P205-9H20
-50
30-50
-25
^50
45
13
Тетрагональный; а : с= 1 : 2,97; зеленый;
слабое двойное лучепреломление; оптически
одноосный, отрицателен; ^=1,63, яе= 1,57;
плеохроичен
От светлозеленого до яблочно-зелеиого
цвета; тетрагональный; а: £=1:2,28;
оптически положителен; одноосный; плеохоои-
чен; для Na-света лш = 1,623, пе —1,625
Ромбический; псевдотетрагональный; от
желтого до зеленого цвета; двуосный
/jp = 1,510, пт= 1,521; плеохроичен
Моноклинный или триклинный; светлоко-
ричневый; слабое двойное лучепреломление;
па = 1,85, лт = 1,862
Канареечно-желтый; ромбический; двойное
лучепреломление; оптически положителен;
яв = 1,762, лр = 1,763, пY = 1,765
Желтый, как охра; в больших
кристаллах; призматический; Ь • с = 1 : 1,327;
сильный плеохроизм; оптически положителен;
/га= 1,88, лр = 1,89
Ромбический; оптически отрицателен;
показатели преломления: па = 1,715^0,003,
Яр = 1,736±0,003, лт = 1,739±0,003
Находится часто вместе
с отунитсм; Корнуэлл,
Англия; Вильсальм,
Бельгия; Португалия
Гуниислейк и Тин-
крофт, Корнуэлл, Англия;
Катанга, Бельгийское
Конго; гора Темпл, Юта;
Испания
Редрут, Корнуэлл,
Англия
Казоло, Катанга,
Бельгийское Конго
Казоло, Шинколобве,
Катанга, Бельгийское
Конго
Тесно ассоциирует с
торбернитом в
Шинколобве, Катанга,
Бельгийское Конго
Казоло, Катанга,
Бельгийское Конго
III, (1), 576
IV, (2), 960
IV, (2), 959
IV, (2), 962
IV, (2), 964
IV, (2), 963
а) Римские цифры обозначают том, арабские я скобках — часть, арабские 0еэ скобок — страницу.
Таблица 37
УРАНОАРСЕНАТЫ
Минерал
Т рёгерит
Ураиосппнит
Цейнерит
Вальпургит
Состав
(U02)3(As04)212H20
Са (U02)2 . (As04)2 •
■лН20 (обычно /7=84-12),
изоморфен отуниту
Си (U02)2 • (As04)2 •
•п Н20(обычнп л=8^12);
изоморфен торберниту
Bil0(UO2)3(OH)24.
(As04)4
Содержание
урана, %
56,2
50
50—53
16,5
Оптические и кристаллографические свойства
I Золотисто-желтый; тетрагональный, а : г=
= 1:2,16, или моноклинный, а:Ь:с =
= 0.463:1:0,463 (см. стр. 81); оптически
отрицателен; двуосный; па = 1,585,
i лр = 1,630, п^.— 1,630; другие образцы
одноосные; п^ = 1,624, яв= 1,580
Ромбический или тетрагональный; от
желтого до яблочно-зеленого цвета; дву-
;осный; отрицателен; оптически аномален
(см. стр. 81)
Травянисто-зеленый; пластинчатые
тетрагональные кристаллы; одноосный;
отрицателен; лю= 1,64а-1,635, яв= 1.623—1,615;
плеохроичен
Оранжевый, цвета меда и
соломенно-желтый; триклинный пинакоидальный или
моноклинный; двуссньтй; отрицателен; очень
высокое двойное лучепреломление; ла= 1,90,
rip = 2,00, лт = 2,05
Месторождения
Гора Болд, Блэк-Хиллс,
Южная Дакота
К>га
Гаронна, Франция;
Корнуэлл, Англия
Португалия
Ссылка на
справочник
[66]а>
III, (1), 730
III, (1), 730
Ш. (I), 731
III, (1), 729
1) Римские цифры обозначают том, арабские в скобках — часть, арабские без скобок — страницу.
УРАНОВАНАДАТЫ
Таблица 38
Минерал
Тюямунит
(рандит)
Раувит
Карнотит
Уванит
Состав
Ca0.2U03-V2CVrtH20
(обычно л = 9—10, но
может упасть и до 4)
CaO-2U03-6V205-
♦20НаО
K20.2U03-V205-rtH20
(обычно /z = 1—3)
2U03-3Va05-I5HaO
Содержание
урана, %
50—60
17
-50
33
Оптические и кристаллографические свойства
Ромбический; а : 6 — 0,77: 1; от канарееч-
ио-желтсго до зеленоватого; отрицателен;
очемь высокий показатель преломления
и двойное лучепреломление; яа=1,67,
^ =1,87, мт= 1,895; плеохроичец
Красновато-пурпурный; анизотропный;
показатель преломления —1,88
Ромбический; а : 6 = 0,81 : 1; желтый;
двойное лучепреломление; оптически
отрицателен; яа = 2,06, А1а = 2,06, /гт = 2,06—
2,08; плеохроичен
Коричневато-желтый; ромбический, дву-
осный; ла = 1,817, «р = 1,879, nY = 2,057;
плеохроичец
Месторождения
Долина Парадокс,
Колорадо; горы Генри, Юта
Река Грин, Юта
Колорадо; Юта;
Западная Австралия (землисто-
порошкообразные
вкрапления в песчанике);
Пенсильвания; Катанга,
Бельгийское Конго
Гора Темпл, Юта
Ссылка на
, справочник
[66]а)
III, (1), 848
IV, (2), 968
III. (1), 844
IV, (2), 967
Римские цифры обозначают том, арабские в скобках — часть, арабские без скобок — страницу.
Часть I. Уран как элемент
Таблица 39
АЛФАВИТНЫЙ СПИСОК МИНЕРАЛОВ, ПРИВЕДЕННЫХ В ТАБЛИЦАХ
Минерал
Минерал
Минерал
Ампангабеит .
Бассетит . .
Беккерелит .
Бетафит . .
Бломстрандин
Бломстрандит
Браннерит . .
Брёггерит . .
Вальпургит .
Ванденбрандит
Виикит ♦ . .
Гатчеттолит .
Гильпинит
Гуммит . . .
Дакеит . . .
Девиндтит . .
Делоренцит .
Джалмаит . .
Дюмонтит . .
Иоганнит . .
Иттрокразит .
Иттротанталит
Ишикаваит .
Казолит . . .
Карнотит . .
Кларкеит . .
Клевеит . . .
Кельм . . .
Кюрит . . .
Ламбертит .
31
36
32
31
31
31
31
29,
30
37
33
31
31
35
32
34
36
3]
31
36
35
31
31
31
33
38
32
29,30]
Стр
81
32
33
Либигит . . .
Линдокит . . .
Макинтошит .
Менделеевит .
Метаторбериит
Метландит . .
Микролит . .
Нивенит . . .
Николаит . . .
Отупит . . . .
Парсомсит . .
Пильбарит . .
Пирохлор . . .
Приорит . . .
Рандит . . . .
Раувит . . . .
Ренардит . . .
Резерфордин .
Ретзерфордит .
Салеит . . . .
Самарскит . .
Самирезнт . .
Склодовскит .
Скупит . . . .
Соддннт . . .
Стасит . . . .
Торбернит . .
Торианит . .
Трёгерит . . .
Тюямунит . .
Уванит . . . .
Ураконит .- . .
34
31
33
31
36
33
31
29,
30
33
36
36
33
31
31
38
38
36
34
31
36
31
31
33
32
33
36
36
29
37
38
38
35
Уранинит
Урановая смоляная руда
Уранолепидит . . . .
Уранопилит
Ураноспинит
Ураносферит
Ураноталлнт
Уранотил
Уранофан
Уранохальцит
Ураноцирцит
Ураношпатит
Фергусонит
Фоглианит
Фоглит
Фосфурапилит . . . .
Фрнтчеит
Фурмарьерит . . . • .
Хьельмит
Цейнерит
Циппеит
Циркелит
Шарпит
Шинколобвит
Шрёкингерит
Эвксенит-поликраз . .
Эльсвортит
Эшвегит
Эшинит
Янтинит
Флюоресценция урановых минералов [101—104].
Давно известно, что многие урановые минералы флюоресцируют. За редким
исключением, интенсивная люминесценция наблюдается только у вторичных
минералов, и лишь очень немногие урановые минералы пегматитового
происхождения также люминесцируют. Сильно люминесцирующими урановыми
минералами являются фосфаты, сульфаты и арсенаты, которые под
ультрафиолетовыми лучами светятся интенсивным характерным желто-зеленым цветом.
К этим минералам относятся отунит, ураноспинит, ураноцирцит и уранопилит.
Карбонаты урана и ураноталлит обладают интенсивной зеленой
флюоресценцией, шрёкингерит—яркой желто-зеленой. Слабое желтоватое свечение
наблюдается у карнотита, девиндтита, соддиита, беккерелита и некоторых образцов
циппеита, уранотила и гуммита. Силикаты урана и урановые слюдки, содержа-
Глава 3. Уран в природе
87
щие медь, свинец, висмут, марганец и железо, нелюминесцируют;так же ведут
себя и цейнерит, фритчеит, бассетит, трёгерит, торбернит, фосфуранилит, тюя-
мунит, вальпургнт, купросклодовскит, яптинит, казолит, кюрит, фурмарьерит,
иоганнит (гнльпинит), урапосферит, бетафит, гатчеттолит и эльсвортит. До сих
пор еще не удалось обнаружить определенной взаимозависимости между
флюоресценцией и химической структурой*. Установлено, что при нанесении кислот,
например серной, азотной, соляной, уксусной или фосфорной, на несветящиеся
соединения урана последние начинают светиться при действии
ультрафиолетовых лучей. Из этих кислот фосфорная действует слабо, уксусная же
вызывает наиболее интенсивную флюоресценцию [106].
Эги наблюдения могут иметь практическое применение при поисках и
разведках на уран. Помимо флюоресценции, все возрастающее значение для
разведки урановых месторождений имеют методы, основанные на открытии гамма-
излучения [107—114].
ПРОМЫШЛЕННЫЕ МЕСТОРОЖДЕНИЯ УРАНОВЫХ МИНЕРАЛОВ
До 1942 г. урановые руды никогда не разрабатывались исключительно
с целью добычи урана; в первую очередь при этом стремились к извлечению
радия. При производстве ванадия из карнотита получались значительные
количества урана в виде побочного продукта. Соединения урана не имели широкого
спроса, поэтому экономика добычи некоторых руд определялась только
стоимостью получаемого радия и ванадия. С открытием процесса деления ядра и его
технического применения уран приобрел огромное значение. Экономические
критерии, которые раньше определяли выгодность эксплуатации урановых
руд, потеряли свое значение, и месторождения урана, которые раньше не
эксплуатировались, стали интенсивно разрабатываться. Авторы не имеют
возможности описать современный процесс добычи, сообщить количества
добываемой руды, оценить запасы сырья или дать результаты изысканий,
которые проводились начиная с 1940 г.
Хотя уран и не является особенно редким элементом земной коры, в
прошлом найдено относительно мало урановых месторождений, пригодных для
разработки. Богатые месторождения находятся в Канаде, в районе
Большого Медвежьего озера, и в Бельгийском Конго, в Катанге. Значительные
месторождения имеются также в США.
Месторождения урановой смоляной руды у
Большого Медвежьего озера (Север о-З ападная
территория, Канада) [115—119]. Бэлыное Медвежье озеро находится на самом
полярном круге, восточнее реки Маккепзи, на расстоянии 2200 км водой или
1300 км по воздуху от конечного пункта железной дороги в Уоэтервейсе,
Альберта. Месторождение было открыто в 1930 г. на восточной стороне озера.
Уран встречается в виде урановой смоляной руды в богатых минералами
зонах. Руда находится в метасоматических жилах и штокверках вдоль зон
разлома и сдвигов, которые пересекают осадочные докембрийские и измененные
вулканические породы, прорезанные гранодиоритом. Известно несколько зон,
которые имеют ширину от 10 до 250 м. Процесс минералообразования, повиди-
мому, был очень сложным; в месторождениях найдено около 40 минералов.
Основными из них являются урановая смоляная руда, самородное серебро,
пирит и халькопирит. Найдены также соединения железа, кобальта, никеля, меди,
свинца, цинка, серебра, молибдена, висмута и марганца. Считают, что это
месторождение возникло из гидротермальных растворов, образовавшихся при
затвердевании гранитной магмы [120]. Минералообразование шло в несколько
стадий. Урановая смоляная руда, повидимому, осаждалась на самых ранних
ф Обзор по флюоресценции урановых минералов, сделанный Де-Меитом [105],
расходится в некоторых деталях с данными Мейкснера, цитированными выше.
88
Часть /. Уран как элемент
стадиях гидротермального процесса, затем отлагались кобальто-никелевые,
свинцово-цинково-медные и, наконец, медно-серебряные минералы. Урановая
смоляная руда образует коллоидные отложения в трещинах. Предполагается
[121], что месторождения у озера Контакт в районе Большого Медвежьего
озера образовались в результате указанного выше процесса. Сбоггщенная
урановая смоляная руда, отгружаемая из рудника Эльдорадо на Большом
Медвежьем озере, содержит от 30 до 62% закиси-окиси урана [122].
В Канаде, кроме месторождения у Большого Медвежьего озера, усиленно
разрабатывались еще только месторождения Вильберфорс Кардифского
района (графство Халибуртон, Онтарио). Добывается там уранинит, который
в пегматитах дает руду со средним содержанием урана около 0,1% [47].
Описано много небольших пегматитовых месторождений.
Месторождения в Бельгийском Конго [123—125].
Месторождения были открыты в 1915 г. Систематическая разработка началась
в 1921 г. Месторождения представлены изолированными окремненными
брекчиями в 25 км южнее Камбове, на водоразделе между бассейнами рек Мура
и Панда. Горные породы образуют хребты высотой до 12—15 му длиной 75 м.
Урановая руда в некоторых местах находится очень близко от поверхности;,
сплошные массы урановой смоляной руды покрыты слоем песчано-глинистой
почвы толщиной всего в несколько сантиметров. Вероятно, тонкий слой
почвы, покрывающей невыветренный жильный материал, имеет наносный
характер.
Ввиду того что весь данный район богат минералами, можно не сомневаться,
что источники растворов, из которых образовались месторождения, являются
магматическими. Окружающие породы, в которых залегает смоляная руда,
имеют сходство с многочисленными породами в месторождениях меди в Катанге.
Они состоят из измененных карбонатных пород свиты Серия де Мин,
которые окружены сравнительно мало разрушенными глинистыми и тальковыми
сланцами (формации Кунделунгу). В Шинколобве, помимо урана, найдены
медь, кобальт, никель, ванадий, железо и драгоценные металлы. Встречается
также и молибден (в виде молибденита и вульфенита). Ни один из этих
элементов не найден в формации Кунделунгу в отдельности.
Свита Серия де Мин, в которой найден уран, имеет мощность около 200 м
и состоит главным образом из доломитизированного известняка. Жилы,
содержащие урановую смоляную руду, тянутся параллельно главным сбросам,
которые прорезают доломит, но не находятся в сбросах. Жилы непостоянны
по своей мощности; их ширина на коротких расстояниях изменяется от
нескольких сантиметров до 1 м. Они дают массы сплошной смоляной руды
весом в несколько тонн. Урановые минералы не содержат видимой пустой
породы. Для снижения потерь извлекают всю рудную массу, которую затем
сортируют вручную. Неравномерность жил затрудняет оценку запасов руды.
Кроме жил, уран встречается также в рассеянном состоянии в соседних
с жилами боковых породах. В этом случае он находится в различных минералах,
образовавшихся в результате полного вторичного изменения смоляной руды.
Другие вкрапления, содержащие преимущественно торбернит, не имеют, по-
видимому, отношения к жилам. Более половины окисленного урана находится
в форме торбернита, но вместе с ним присутствуют и многочисленные другие
продукты окисления урановой смоляной руды [126]. Казолит и склодовскит
находятся в рассеянном состоянии; склодовскит находится в трещинах,.
подобно торберниту. Встречается также и уранинит [127]. Кроме того,
в урановых жилах найдено немного сульфидов меди, кобальта и никеля,
которые содержатся в значительных количествах в других месторождениях
свиты Серия де Мин.
Последовательность минералообразования сложна. Считают [123], что
источником возникновения руд являлись растворы, содержавшие кремнезем..
Глава 3. Уран в природе
89
Эти растворы проникали в карбонатные породы и дали при этом ряд
силикатных минералов; затем отложилась урановая смоляная руда и после
этого—сульфидные минералы. В результате последующих процессов происходило
изменение руде образованием вторичных минералов. Таким образом, предполагается,
что урановая смоляная руда возникла на ранней высокотемпературной
стадии образования медных минералов. Такая последовательность
образования минералов является общей для всего района. Урановая смоляная
руда или продукты ее изменения найдены в семи месторождениях от Рауши
на юго-востоке до крайней северо-западной части медной зоны;
промежуточными пунктами являются Луршиа и Камбове [128].
Месторождения в Колорадо и Юте [129—131 ].
Месторождения карнотита в Колорадо и Юте являются единственными
месторождениями в США, из которых до настоящего времени получали уран в
промышленных масштабах. Уранинит встречается в месторождениях Северной
Каролины, Коннектикута, Нью-Гэмпшира [1321, Блэк-Хиллса (Южная Дакота),
Барингер-Хилла (Техас) и в различных других местах, но вследствие
незначительных размеров этих месторождений возможность развития промышленной
добычи урана из них раньше считалась исключенной.
Месторождения карнотита протягиваются от Коул-Крик, вблизи Микера,
(Колорадо) до горы Каризо, на границе между Нью-Мексико и Аризоной,
и от графства Уэрфано (Колорадо) по направлению на запад к Сильвер-Риф
и юго-западной части Юты, но основные месторождения распределены по
площади длиной 200 км и шириной 80 км в юго-западном Колорадо и
юго-восточной Юте. Месторождения распределены неравномерно, пятнами и содержат
в отдельных агрегатах большие количества урановых и ванадиевых минералов.
Ванадия в них содержится гораздо больше, чем урана. Руда встречается в
пластах и гнездах, часто в виде корок и вкраплений в песчанике. По общему
мнению, крупнейшие месторождения находятся в области между долиной Парадокс
и рекой Сан-Мигель. Здесь главная часть рудоносного песчаника образует
линзы около 30 м толщиной, составляющие только часть гораздо более
толстой формации (Моррисон) сланца и песчаника.
Урановые минералы, найденные в этой главной области, преимущественно
состоят из карнотита и тюямунита; кроме того, там обнаружены и другие
сложные ванадиево-урановые минералы (уванит, раувит, цейнерит).
Месторождения относятся к залежам осадочного происхождения [1331. Значительная
часть руды ассоциирована с окаменелыми деревьями, остатками растений и
костями древних ящеров или асфальтом. Согласно Гессу, процесс образования
залежей на Колорадском плато можно представить следующим образом: пови-
димому, уран и ванадий в этом районе прежде находились в жилах, которые
содержали также^и пирит. В результате окисления пирита образовались серная
кислота и сульфат железа (111), которые растворили ванадий и уран с
образованием их сульфатов. Природа моррисонских песчаников, в которых
находится большая часть месторождений, указывает на сеязь их происхождения
с мелкими прибрежными водами в результате перемещения островов, отмелей
и берегов. Растворы сульфатов урана и ванадия проникали в эти воды, где
изобильная растительность типа водорослей погибала под ударами прибоя.
Реки переносили в эти прибрежные воды стволы деревьев. Вокруг них
скапливался песок и остатки омертвелых водорослей. Под действием органических
веществ происходила концентрация урана и ванадия из очень разбавленных
растворов путем восстановления или ионного замещения. Затем, когда
началось окаменение органических остатков, могло происходить еще замещение
органических веществ кальцитом и урано-ванадиевыми минералами. На роль
растительного материала в сбразовании карнотитов указывает чрезвычайно
высокая концентрация урана и ванадия в некоторых окаменевших деревьях,
найденных вблизи реки Сан-Мигель. Например, один из окаменевших стволов
90
Часть I. Уран как элемент
около 30 м длиной и 1,2 м диаметром содержал приблизительно 105 т
карнотита. Предполагают [133], что и микроорганизмы также играли роль в
концентрации урана и ванадия из разбавленных растворов (см. также [134]).
В наиболее богатых рудах, селективно разрабатывающихся в этом районе
для извлечения закиси-окиси урана, содержится 1,25—l,5%Ua08 и около 3,5%
V205. Состав типичной карнотитовой руды приведен в табл. 40. Несомненно,
что в этом районе находятся значительные количества руды, которые прежде
считались слишком низкокачественными, чтобы оправдать добычу.
Таблица 40
СОСТАВ КАРНОТИТОВЫХ РУД
Компонент
v2o5
и3о8
SiO,
Fe203-bAl203
СаСОз + CaS04
Содержание, %
2,25—10,5
0,25- 3,0
70—80
5—12
От следов до 3
Компонент
BaC03 + BaS04
С11СО3 1-CuS
Pb
Na, K,Mg, Ra и т. д.
Н20
Содержание, %
От следов до 2
» » » 2
~1
Небольшие
количества
5 — 25
Месторождения карнотита представляют геологическую редкость. С другой
стороны, самые известные в мире месторождения ванадия (в Мииасрагра,
Перу) не содержат урана, несмотря на присутствие органического вещества
(в виде асфальта).
Месторождения урана меньшего значения. Кроме
трех главнейших месторождений, описанных выше, имеется много других,
которые в различное время разрабатывались с целью получения урана. Они
кратко перечислены ниже.
США. До разработок карнотитовых месторождений небольшие количества
урана добывались в графстве Гильпин (Колорадо), где урановая смоляная руда
ассоциирует с пиритом и сульфидами цинка и свинца [135, 136].
Португалия [137—139]. Урансодержащая зона залегает в области
сплошных гранитов, которые расположены почти во всей северной части
Португалии, между Галисией и Кастелло Бранко, и простирается в провинции Миньо,
Траз-уж-Монтеш и Бейра. Богатейшая часть месторождений расположена между
городами Гуарда и Сабугал и в области Виллар-Фермоза. Жилы около Гуардо
особенно богаты вольфрамитом.
Урановые минералы встречаются в узких пегматитовых жилах мощностью
от 0,5 до 1 м. В районе Розманейра, помимо урановых, имеются
промышленные руды вольфрама и олова. Отунит Ca(U02),-(P04)2-12Н20 является там,
вероятно, старейшим урановым минералом, которому сопутствуют
многочисленные продукты его изменения [140]. Отунит образует небольшие скопления
квадратных пластинок интенсивно желтого цвета или образует во впадинах
горных пород желтый слой, состоящий из небольших пятнистых наслоений;
еще чаще отунит встречается в виде светложелтых вкраплений, рассеянных по
всей тускложелтоватой породе. В глинистых частях жил урановый минерал
часто незаметен и может быть обнаружен только по радиоактивности. Имеются
желтые вкрапления и на поверхности гранита, придающие ему интенсивно
желтый цвет, хотя действительное содержание урана в граните очень мало.
Содержание урана в жилах колеблется в очень широких пределах. Руда,
содержащая 2% закиси-окиси урана, считается самой лучшей, а содержащая 1% —
довольно хорошей [141, 142].
Глава 3. Уран в природе
91
Англия [143, 144]. Минеральные месторождения в Корнуэлле и в Южном
Девоне разрабатывались с древних времен для добычи олова и меди. Главные
месторождения урана находятся в Уил-Тренвите, Сент-Айве и в руднике
Саут-Террас (Сан-Стефан). Последнее месторождение—это единственное
известное в Корнуэлле, где основные минералы представляют собой соединения
урана. Жилы этого месторождения содержат достаточное количество урана для
промышленной добычи.
Жила Южной террасы лежит в сланце, интрудированном гранитом.
Соседние жилы содержат главным образом олово и медь и другие минералы, в состав
которых входят кобальт, никель, свинец, уран, железо, изредка мышьяк
и халькопирит. Вероятно, жилы с высоким содержанием урана, кобальта
и никеля образовались не одновременно с жилами, в которые входит железо.
Норвегия и Швеция. Давно известны и хорошо изучены пегматитовые
месторождения в Норвегии севернее Эвьо и Хрпстианзунда.
В Швеции [145] государственный нефтеслапцевый завод в Кванторпе
(провинция Нарке) перерабатывает сланцы, содержащие кольм; получаемый
остаток содержит уран и другие металлы [146].
Мадагаскар [147]. Мадагаскар является одним из немногих мест, где
в прошлом разрабатывались пегматитовые урановые минералы. Большинство
из них встречается в древних горных породах в средней части острова. Там
найдены следующие урановые минералы: эвксепит-поликраз, бетафит, ампанга-
беит, фергусонит, самарскит и бломстрандит. Минералы, имеющие
экономическое значение, залегают главным образом в богатых калием и только изредка
в богатых натрием пегматитах. Отунит и урапоцирцит найдены на дне
высохшего озера Антсираб, куда стекали воды через разрушенные пегматиты,
содержащие бетафит и другие сложные минералы урана, ниобия и тантала.
Африка (за исключением Бельгийского Конго) [148]. Урановая руда
найдена в районах Улугура, округ Морогоро (Восточная Африка), в Лолдайга,
округ Нангаки (Британская Восточная Асррика). Имеются также
месторождения вблизи Мессины (Северный Трансвааль) и в районе Гордонии (Капская
провинция).
Австралия [149—153]. Описаны месторождения урана в Австралии. Одно
из них у Радиум-Хилл (гора Пайнтер) вблизи Олари в Южной Австралии,
другое в золотых приисках Пильбара в Западной Австралии. У Радиум-Хилла
(30 км юго-восточнее Олари, 440 км северо-восточнее Аделаиды) найдены
торбернит, отунит, уранофан, гуммит и фергусонит. Они, повидимому, произошли
от урано-титано-тантало-ииобиевых минералов близлежащих пегматитов.
Пильбарит, который является сложным гидратированным силикатом
свинца, урана и тория, содержащим небольшие количества церия и
иттрия [154], добывается в Пильбара в Западной Австралии.
Мексика [155, 156]. Месторождения большей частью находятся в 200 км
на северо-восток от Гвадалупы в штате Чиуауа. Вместе с ураном находится
золото. Жилы имеют ширину от нескольких сантиметров до 5 м [157].
Бразилия. Недавно был найден уран в Пройио (Минас Жераэс). Руда,
содержащая после обогащения 13—18% закиси-окиси урана, добывается из
пегматитов, разрабатывавшихся ради добычи слюды, турмалина и берилла. Руда
из Уба (Минас Жераэс) после обогащения содержит до 75% самарскита,
15% монацита и до 10% колумбита [158]. Эвксепит найден в Помбе (Минас
Жераэс) еще в 1911 г., но до сих пор он не разрабатывался [159]. Недавно
был сделан подсчет запасов бразильских месторождений урана [160]. Основные
залежи урана в Бразилии находятся в штате Минас Жераэс [161, 162].
Франция. В литературе имеется обзор месторождений во Франции [163].
Статистика. В настоящее время имеется мало статистических данных
о производстве урана и ценах на него. Весьма приближенные сведения об
экономической истории урана можно получить из последовательных томов
92
Часть I. Уран как элемент
Minerals Yearbook. В качестве иллюстрации в табл. 41 представлены данные
из последних изданий этого труда.
Таблица 41
РУДЫ И СОЕДИНЕНИЯ УРАНА, ИМПОРТИРОВАННЫЕ
ДЛЯ ПОТРЕБЛЕНИЯ В США С 1939 ПО 194 3 Г.
Год
1939
1940
1941
1912
1943
Урановые руды
вес, т
0,002
1090
—
246
—
цена.
доллары
10
2 110 927
—
806 919
—
Окислы
вес, т
654
109
176
171
96
п соли урана
цена.
доллары
1 197 786
388 355
501 370
851 098
431410
ЛИТЕРАТУРА
1. Kirsch G., Geologie und Radioaktivitat, p. 35—42. Verlag Julius Springer,
Leipzig, 1928.
2. С т а р и к И. Е., Академику В. И. Вернадскому к пятидесятилетию научной и
педагогической деятельности, 1, 455—462, М., 1936.
3. С т а р и к И. Е., Труды Гос. ралиевого института СССР, 3, 211—217 (1937).
4. С е г е л ь Н. М., Труды Гог. рал.иевото института СССР, 4, 350—383 (1938).
5. U г г у W. D., Am. J. Sci., 239, 191—203 (19-11).
6. Умовская В., .ПАН СССР, 29, 380—383 (1940).
7. National Nuclear Energy Scries, Division VIII, vol 4.
8. Hess V.F., Roll J. D., Phys. Rev., 73, 916—918 (1948).
9. H с v e s v G., Alexander E., Wiirst 1 in K., Z. anorg. u. allgem.
Chem., '194, 316 (1930)
10. H e v e s у G., Chemical Analysis by X-rays and its Applications, Ch. XVI, McGraw-
Hill Book Company, Inc., New York, 1932.
11. Gol dsc h mi dt V. M., Geochemische Verteilungsgesetze der Elemente, vol. 9,
J. Dybwad, Oslo, 1938, p. 60.
12. E v a n s R.D., Goodman C, Bull. Geol. Soc. Am., 52, 459 (1941).
13. С 1 а г k e F. W.f Washington H. S., U. S. Geol. Survey, Profess. Paper
127 (1924).
И. Ферсман А. Е., Геохимия, Л., 1933.
15. A n d e r s о n J. S., J. Proc. Roy. Soc. N. S. Walles, 76, 329—345 (1942).
16. G о 1 d s с h m i d t V. M., Geochemische Verteilungsgesetze der Elemente, vol. 9,
J. Dybwad, Oslo, 1938, p. 99.
17. Meyer S., Naturwissenschaften, 25, 764—765 (1937).
18. Keevil N. В., Econ. Geol., 33, 685—696 (1938).
19. К e e v i 1 N. В., Am. J. Sci, 242, 309—320 (1944).
20. S e n f t 1 e F. E., Keevil N. В., Trans. Am. Geophys. Union, 28, 732—738
(1947).
21. Mever S., Sitzber. Akad. Wiss. Wien, Math, naturw. Klasse, Abt. Ha, 146,
175—197 (1937).
22. К i r sc h G., Hecht F., Z, anorg. u. allgem. Chem., 236, 157—164 (1938).
23. M u 1 d e г С J., Atoom, 1, 133 — 139; 161—163 (1947).
24. H о 1 m e s A., Phil. Mag., Dec, 1225 (1926).
25. H о 1 m e s A., Geol. Mag., July (1926).
26. J ol у J., Radioactivity and Geology, Ch. VII, Constable and Co., Ltd., London,
1909.
27. G m e 1 i n L., Handbuch der anorganischen Chemie, Svstem No. 55, Verlag Chemie,
Berlin, 1936, p. 11.
28. H e r n e g g e r F., К а г 1 i k В., Sitzber. Akad. Wiss., Wien, Math, naturw.
Klasse, Abt. Ha, 144, 217—226 (1935).
29. F о у n E., Karlik В., Pettersson H., Ron a E., Nature, 143,
275—276 (1939).
30. P i g g о t C.S., Urry \V. D., Am. J Sci., 240, 1 — 12; 93—103 (1942).
31. Hoffmann J., Chem. Erde, 14, 239—252 (1942). .
32. Д р о б к о в А. А., ДАН СССР, 17, 229—232 (1937).
Глава 3 Уран в природе
93
33. Hoffmann J., Wien klin. Wochschr., 54, 1055 — 1059 (1941).
34. Hoffmann J., Chem Ztg., 66, 181— 183 (1942).
35. Hoffmann J., Z. physiol Chem., 276, 275—279 (1942).
36. Hoffmann J., Biochem. Z., 313, 277— 287 (1943).
37. Hoffmann J., Bodenkunde u. Pflanzenernahr., 32, 295—336 (1943).
38. Hoffmann J., Naturwissenschaften, 29, 403—404 (1941).
39. P a n e t h F., Z. Elektrochem., 34, 645 (1928).
40. P a n e t h F , Z. Elektrochem., 36, 727 (1930).
41. Pa net h F., Naturwissenschaften, 19, 164 (1931).
42. Q u i г к е Т. T , Finkelstein L.f Am. J. Sci., [4] 44, 237 (1917).
43. N о d d а с к I., N о d d а с к W., Naturwissenschaften, 18, 757 (1930).
44. Hoffmann J., Zentr. Mineral. Geo!., 1941 A, 31—37.
45. В a u e г С. A., Astron. J., 53, 1 10 (1948).
46. L a n e A C, Report of the Committee on the Measurement of Geologic Time, Natl.
Research Council, Washington, D С , 1924.
47. E 1 1 s w о г t h H. V., Rare Element Minerals of Canade, Can. Dept. Mines
Resources Geol Survey, Econ. Geol. Ser. No 11, Ch. X, 1932, p. 136—257.
48. H о I m e s А., К о v a r i к A., The Age of the Earth, IV, Bull. Natl. Research
Council, No. 80, 1931.
49. Holmes A., The Age of the Earth, Thomas Nelson and Sons, New York, 1937.
50. Goodman C, J. Applied Phys., 12, 276—289 (1942).
51. Siegl \V., Berg.-u. huttenmann. Monatsch. montan. Hochschule Leoben, 92,
112 — 114 (1947)
52. X л о n и н В Г., Г е р л и н г Е. К., Барановская Н. В., Изв. АН
СССР, ОХН, 599—604 (1947).
53. Tomkeief f S. I., Science Progress, 34, 695—712 (1946).
54. G о 1 d s с h m i d t V. M., Geojhemische Verteilungsgesetze der Elemente, No. 3,
J Dybwad, Oslo, 1923.
55 van A u b e I R., Compt. rend., 185, 586 (1927).
56. P a 1 а с h e С, В е г m a n H., F г о n d e 1 C, Dana's System of Mineralogy,
vol. I, John Wiley and Sons, Inc., New York, 1944, p. П1 1—620.
57. Tyler P.f U. S. Bur. AAines Inform. Ore. 6312, 16 (1930)..
58 Э в а н с Р., Введение в кристаллохимию, Госхимизлат, М.—Л., 1948.
59. G о 1 d s с h m i d t V M., Thomassen L., Videnskapsselskapets-Skrifter I.
Mat.-naturv. Klasse Kristiana. 2, 12 (1923).
60. Schopp А., В i 1 1 i e t V., Ann. So-, geol. Belg. Bull.. 58, B198—206 (1935).
61. Bakken R., Glcdi tsc hE, Nord. Kemi Kermode Forh., 5, 200—201 (1939);
from Chem. Zentr., 1942, II, 2572.
62. Bakken R., Gleditsch E., Am. J. Sci., |5] 36, 95—106 (1938).
-63. A 1 t e r CM, К i p p E. M , Am. J. Sci., [5] 32, 120—128 (1936)
64. В a k k e n R, Gleditsch E., Pappas A. C., Bull. soc. chim. France,
1948, 515-517.
65. F 6 у n E., Norsk Geol. Tids., 17, 197—292 (1939); from Chem. Abstracts, 32, 6583.
66 D о с 1 t e г С, Leil meier H., Hnndbuch der Mineralchemie, Bd. 4, Tl. 2,
909, Th. Steinkopff., Dresden—Leipzig 1929.
67. H i I 1 e b r a n d F. W , U. S. Geol. Survey Bull., 78, 43 (1891).
68. H i 1 I e b г a n d F. W., Am. J. Sci , 42, 390 (1891).
69. H i d d e n W. E., Mackintosh J. В., Am. J. Sci., 38, 474 (1889).
70. Ellsworth H. V., Natl. Research Council Can. Am. Repts., App. H. Exhibit A,
1929—1930.
71. Davis C. W., Am. J. Sci., 11, 201 (1926)
72. К 1 e e m a n A. W., Trans. Roy. Soc. S Australia, 70, 175 — 177 (1946).
73. К а г u n a k а г a n C, N e e 1 a k a n t a m K-, Proc. Indian Acad. Sci., 25A,
404—407 (1947).
74. I i m о г i Т., Am. J. Sci., 239, 819—821 (1941).
75. Hint z e C, Handbuch der Mineralo^ie, Bd. I, Abt. 3, 2 Halfte, p. 4152; Abt. 4,
1 Halfte, p. 971, Berlin und Leipzig, 1930.
76. H i 1 1 e b г a n d W. F., Am. J. Sci , |3] 38, 329 (1889).
77. S t e i n k u h 1 e г W., Bull. soc. chim. Belg., 32, 233 (1923).
78. Schoep A., S t г a d i о t S.. Am. Mineral., 32, 344— 350 (1947).
79. G m e 1 i n L., Handbuch der anorganischen Chemie, System No. 55, Verlag Chemie.
Berlin, 1936, p. 17—37.
80. V a e s J. F., Ann. soc. geol. Belg. Bull., 70, B212—225 (1946).
81. В i 1 1 i e t V., Natuurw. Tijdschr., 18, 79 (1936): from Chem. Abstracts, 31. 4236.
82. В i 1 1 i e t V., Mineralou. Abstracts, 7, 108 (1938).
83. Mineralog. Abstracts, в, 429 (1937).
84. AAineraloe Abstracts, 8. 15 (1941).
85 Chem Abstracts, 35, 7888 (1941).
86. Mineralog. Abstracts, 7, 225 (1939).
94
Часть. I. Уран как элемент
87. Am Mineral., 24, 658 (1939).
88. J. Melon. Bull. Inst. roy. colonial belg., 9, 333 (1938).
89. Novacek R., Mineralog. Abstracts, 9, 211 (1946).
90. Bull. soc. franc, mineral., 51, 247 (!928).
91. G о 1 d s с h m i d t V. M., Z. Kryst. Mineral., 31, 468 (1899).
92. С т а р и к И. E.f С а м а о ц е в а А. Г., Я щ е н к о М. Л., ДАН СССР, 31,
909—910 (1941).
93. Rinne F., Centr. Mineral. Geol., 1901, 618 (1901).
94. H a 1 1 i m о n d A. F.f Mineralog., Mag., 17, 326 (1916).
95. W i с k m a n F. E., Geol. Foren. Forh., 64, 466 (1942).
96. \V e 1 1 s R.C., Stevens R, E., J. Wash. Acad. Sci., 21, 409 (1931).
97. Longobardi E., Florentine N.. Mercader A., Anales asoc.
quim. argenlina, 35, 131 — 136 (1947).
98. L e x о w S. G., Maneschi E. P. P., Univ. nacl. Cuyo, Facultad ing. у cienc.
exact., fis. у nat., Inst, petrol. (Mendoza), Pub. I, p. 5—11 (1948).
99. Fobs F. J., Bull. Am. Assoc. Petroleum Geol., 32, 317—350 (1918).
100. Grip E., О d m a n О. Н., Svcriges Geol. Undersokn. Arsbok, 38, No. 6 (1944).
101. Meixner H., Naturwissenschaften, 27, 454 (1939),
102. Meixner H., Chem. Erde, 12, 433—450 (1940).
103. Meixner H., Z. Krist. Mineral. Pctrog., Abt. A, 52, 275—277 (1940).
104. H a b er 1 a n d t H., Karlik В., Przibram K., Sitzber. Akad. Wiss.
Wien, Math, naturw. Kiasse, Abt. Ila, 144, 135 — 140 (1935).
105. De M e n t J., Fluorochemistry, Chemical Publishing Company, Inc., Brooklyn, N. Y.f
1945, p. 476—182.
106. M e л ь к о в В. Г., Свердлов 3. М., ДАН СССР, 31, 361—362 (1941).
107. Dreblow W. D., Z. Instrumentenk., 62, 60—66; 85—93 (1942).
108. Sill С W., Peterson H. E., U. S. Bur. Mines Inform. Circ, 7337 (1945).
109. Sill С W„ Peterson H. E., Anal. Chem., 19, No. 9, 646—651 (1947).
110. N or t h u p M. A., Ind. Eng. Chem. Anal. Ed., 17, 664—670 (1945).
111. De Ment J., J. Chem. Education, 23, 213—219 (1946).
112. Haberlandt H., Hernegger F., Anz. Akad. Wiss. Wien, Math, naturw.
Kiasse, No. 13, 116 (1946).
113. P r z i b г a m K., Bull, classe sci., Acad. roy. Belg., 32, 363—369 (1946).
114. G i b b T. R.f Jr.. Evans H. Т., Jr., Science, 105, 72—73 (1947).
115. К i d d D. F., Can. Dept. Mines Resources Geol. Survey, Econ. Geol. Ser. No. 11
(1932).
116. К i d d D. F., Can. Dept., Mines Resources Geol. Survey Mem. 187 (1936).
117. Krusch P., Die metallischen Rohstoffe, vol. 1; F. Enke, Stuttgart, 1937,
p. 97—103.
118. L e m a i r e E., Genie civil, 123, 201—204: 213—215 (1946).
119. Deri b ere M., La Nature, 1947, 41—43.
120. Ridd D.F., Haycock M. H., Bull. Geol. Soc. Am., 46, 879—960 (1935).
121. F u r n i v a 1 G. M., Econ. Geol., 34, 739—776 (1939).
122. P a r m e 1 e e H. C, Eng. Mining J., 139, 31—35 (1938).
123. Hess F. L., U. S. Bur. Mines, Minerals Yearbook, p. 449 (1934); from J. Thoreau
and R. du Trieu de Tcrdonck, L'Institut colonial Beige, section sci, nat. et med.,
T. 1, fasc. 8, coll. 4.
124. L e g г а у e M., Ann. soc. geol. Belg. Bull., 68, B157—174 (1944).
125. С h a r r i n V., Genie civil, 124, 386—389 (1947).
126. Buttgenbach, Chimie and industne, 35, 79 (1935).
127. HitchenS.C, van Aubel R., Compt. rend., 199, 1133 — 1135 (1934).
128. Vandendriessche A., Natuurw. Tijdschr., 17, 197—203 (1935).
129. Hess F. L., Ore Deposits of the Western States, Am. Inst. Mining Engrs., 1933,
p. 455—480.
130. Krusch P., Die metallischen Rohstoffe, vol. 1, F. Enke, Stuttgart (1937),
p. 95-97 *
131. Stokes W. L., Bull. Geol. Soc. Am., 55, 951—992 (1944).
132. Shaub В. М., Am. Mineral., 22, 207 (1937); 23, 334—341 (1938).
133. Fischer R. P., Econ. Geol., 32, 197 — 198; 906—951 (1937).
134. Frederick son A. F.f Science, 108, 184—185(1948).
135. В a s t i n E. S., Econ. Geol., 10, 262 (1915).
136. A 1 s d о r f P. C, Econ. Geol., 11, 266 (1916).
137. Moore R. В., Kittril K. L., U S. Bur. Mines Bull., 70, 49 (1913).
138. Krusch P., Die metallischen Rohstoffe, vol. 1, p. 113—116; F. Enke,
Stuttgart, 1937.
139. Ferreira A. Bernardo, Neiva J. M. Cotelo, Portugal, Direc.
gcral. minas e serv. geol., 1, 177 — 189 (1945).
140. L e p i e г г е С , Bull. soc. chim. France, f4| 53, 72 (1933).
141. Dorpinhaus W. Т., Met. Engr., 11, 302 (1914).
Глава 3. Уран в природе
95
142. Е b 1 е г Е., Bender W., Z. angew. Chem., 28, 30 (1915).
143. Dines H. G., Mining Mag., 42, 213—217 (1930).
144. Ellsworth H. V., Rare Element Minerals of Canada, Can. Dept. Mines
Resources Geo!. Survey, Econ. Geol. Scr. No 11, 1932, Ch. X, p. 132.
145. U. S. Bur. Mines, Minerals Yearbook, 1943.
146. Krusch P., Die Metallischen Rohstoffe, vol. 1, Enke F., Stuttgart, 1937,
p. 119—120.
147. Krusch P., Die Metallischen Rohstoffe, vol. I, p. 94—95.
148. Krusch P., Die Metallischen Rohstoffe, vol. I, p. 93.
149. U. S. Depart. Commerce, Bur. Mines, Mineral Resources of the U. S., U. S.
Government Printing Office, Washington, D. C, 1925, p. 617; 1926, p. 267—268; 1927,
p. 441—442; 1928, p. 136; 1929, p. 108—109; 1931, p. 188.
150. U. S. Bur. Mines, Minerals Yearbook, 1932—1933, p. 188, 333; 1934, p. 506; 1935, p. 558.
151. Krusch P., Die metallischen Rohstoffe, vol. 1, F. Enke, Stuttgart, 1937,
p. 121.
152. M a w s о n D.f Trans. Roy. Soc. S. Australia, 68, 334—357 (1944).
153. Carroll D., Row ledge H. P., Western Australia, Dept. Mines, Mineral
Resources W. Australia Bull., 3, 71 — 150 (1945).
154. Simpson E. S., Chem. News, 102, 283 (1910).
155. Krieger P., Econ. Geol., 27, 051 (1932).
156. Krusch P., Die metallischen Rohstoffe, vol. 1, Enke F., Stuttgart, 1937,
p. 103.
157. Gonzalez Reyna J., Comite direct, invest, recursos mineral. Мех., Bol. 5
(1946).
158. Nabuco de А г a u j о С. Е., Jr., Chem. Eng. News, 23, 1900 (1945).
159. Krusch P., Die metallischen Rohstoffe, vol. 1, Enke F., Stuttgart, 1934,
p. 104.
160. Leonardos О. Н., Ministry Agr. (Rio de Janeiro), Servico fomento produccao
mineral, Bull. U (1930); from Chem. Abstracts, 30, 8093.
161. Hess F. L., Henderson E. P., J. Franklin Inst., 200, 235 (1925).
162. Da S i 1 v a Pinto M., Mineralog. Abstracts, 10, 115 (1947).
163. L e m a i г е Е., С h a r r i n V., Genie civil, 123, 86—87 (1946).
Часть II
МЕТАЛЛИЧЕСКИЙ УРАН
7 Химия урана
Глава 4
ИЗВЛЕЧЕНИЕ УРАНА ИЗ РУД И ПОЛУЧЕНИЕ
МЕТАЛЛИЧЕСКОГО УРАНА
ИЗВЛЕЧЕНИЕ УРАНА ИЗ РУД
Еще совсем недавно главной целью переработки урановых руд было
извлечение из них радия. Это положение теперь изменилось, и радий, вероятно,
станет побочным продуктом при выделении урана. В старой литературе
описывались почти исключительно способы эффективного извлечения радия и
мало уделялось внимания наиболее рациональным методам выделения
урана.
Интенсивно разрабатывались только два типа урановых руд: урановая
смоляная руда и карнотит. Разнообразие применяемых методов переработки
зависело от природы руды и характера присутствующих в ней элементов. Для
всех руд общими являются следующие важнейшие операции: 1)
выщелачивание руды серной, азотной или соляной кислотой для растворения урана (иногда
для разложения руды применяется обработка щелочными растворами или
сплавление со щелочами); 2) перевод урана в растворимый комплексный
карбонат с целью отделения железа, алюминия и марганца; 3) осаждение из
уранового раствора сульфидов свинца и меди; 4) выделение урана в виде Na2U207
или (NH4)2U207. В случае карнотита для отделения ванадия и фосфора от
урана применяли особые операции. С деталями переработки можно
ознакомиться, рассмотрев ряд специальных процессов. Описан [1] процесс извлечения
урана из бетафита (стр. 68), путем выщелачивания урана концентрированной
серной кислотой, за которым следовала обычная операция отделения урана
от ниобия, тантала и титана.
Извлечение урана из канадской урановой
смоляной руды [2—5]. Процесс извлечения усложняется присутствием
в руде значительных количеств золота и серебра. Чтобы предупредить
возможное вспенивание, карбонаты и сульфиды, также содержащиеся в руде,
должны быть разрушены до кислотной обработки. Кроме того, должны быть
удалены значительные количества мышьяка и меди.
Руда, добываемая в районе Радиум-Сити на Большом Медвежьем озере,
подвергается обогащению механическими методами и флотацией с выходом
концентрата 1 : 50. В добываемой руде находится около 1 % закиси-окиси
урана к U308. Концентрат содержит около 50% закиси-окиси урана и
1—7% серебра в зависимости от разрабатываемого участка вруднике. Этот
концентрат направляется в порт Хоп (Онтарио) для дальнейшей переработки.
Этот процесс переработки концентрата показан на схеме 1 [6].
Концентрат сначала измельчают в шаровой мельнице, затем он поступает
в магнитный сепаратор для отделения магнетита. Более легкие компоненты
руды удаляются в флотационных камерах. Продукт флотации прокаливают
в печи при температуре 600°, причем сульфиды и карбонаты
разлагаются, а мышьяк и сурьма частично улетучиваются. Затем добавляют хлорид
натрия и температуру повышают до 800°; в этих условиях серебро
превращается в хлорид серебра. После охлаждения продукт обжига выщелачивают
серной кислотой, в результате чего извлекаются уран, марганец, медь и железо.
7*
Концентрат обогащения
I
Шаровая мельница
I
Магнитный сепаратор
• Магнетит
Флотационные камеры
-Легкая пустая порода
NaCl
Сушильная печь (600°)
I > С02, S02, As, Sb (в пылеуловитель)
Печь Веджа(800°)
Стержневая мельница
i i
Грохот <40 меш
Выщелачивание серной кислотой
I
Вакуум-фильтр
Нерастворимый остаток
Ra, Ва, Pb, Ag, Cu, SiOo
Фильтрат после кислотного ^_ | Са(ОН)2 до рН=2,8
выщелачивания ^~\ FeCl3
На извлечение радия
CaS04, As
Раствор U02S04 <— Избыток Na2C03
Со(ОН)2, Fe(OH)3, Al(OH), Раствор
Na4[U02(C03)3]
■NaOH
HC1-
H2S-
-^ Технический Na2Uo07 <-
Фильтрат (в отвал)
-> Раствор U02C12
CuS, AsoS5 Очищенный раствор UOoCl2 «— NH3
I
Трехкратная обработка НС1
и переосаждение
аммиаком
-► (NH4)2U2Q7 Фильтрат ^- NaOH
В отвал Na2U207
UoOa
Схема 1. Технологическая схема производства U308 из канадской урановой смоляной
руды на заводе в порту Хоп (Онтарио).
Глава 4. Извлечение урана из руд и получение металлического урана 101
В стадии выщелачивания обычно вводят хлорид бария в качестве носителя для
радия, с тем чтобы последний остался в нерастворенной части руды.
Добавлением гидроокиси кальция рН кислотной вытяжки доводят до 2,8; затем вводят
хлорное железо для удаления мышьяка в виде нерастворимого арсената
трехвалентного железа. К фильтрату добавляют избыток карбоната натрия,
достаточный для сохранения урана в растворе и осаждения гидроокисей железа,
алюминия, марганца и т. п. После декантации к раствору трикарбоната урана
добавляют едкий натр для осаждения Na2U207. Диуранат натрия очищают
растворением в соляной кислоте и насыщением раствора сероводородом,
осаждающим сульфиды меди и мышьяка. Избыток сероводорода удаляют
кипячением и добавляют аммиак, в результате чего уран осаждается в виде
(NH4)2U207. Последний прокаливанием при 1000° переводят в закись-окись
урана U308. Одно время применяли трехкратную обработку закиси-окиси
горячей соляной кислотой для удаления растворимых в кислоте примесей, но
этот способ оставлен из-за больших потерь урана. Полученный таким путем
окисел содержал от 97 до 99% U308.
Описанная выше обработка хлоридом натрия проводится для отделения
содержащегося в руде серебра. Вместо прокаливания с хлоридом натрия
измельченную руду после магнитного обогащения можно подвергать цианидной
обработке; в этом случае удаляется не только серебро, но и золото, если оно
присутствует. Этот способ предпочитают, когда в руде содержатся заметные
количества золота.
Обработка серной кислотой производится в керамиковой аппаратуре. Как
было указано выше, хлорид бария добавляется в качестве носителя для радия.
На 375 частей (вес.) обожженой руды требуется 350 частей серной кислоты
(уд. вес 1,8), 300 частей воды и 20 частей нитрата натрия. Последний
добавляют для того, чтобы обеспечить полное окисление урана до
шестивалентного состояния.
Извлечение урана из африканской смоляной
руды. Химические проблемы, связанные с извлечением урана из африканской
смоляной руды, в целом менее сложны, чем при выделении его из канадских
руд. Вследствие отсутствия золота и серебра стадия обжига с хлоридом
натрия (или заменяющая его цианидная обработка) может быть опущена.
Кроме того, при переработке африканской руды некоторые операции
канадского процесса, в частности стадия извлечения, также несколько
видоизменяются. Например, вместо нитрата натрия в качестве окислителя часто
пользуются двуокисью марганца. После стадии выщелачивания все
процессы, применяемые для различных видов урановой смоляной руды,
идентичны. Типичная последовательность переработки африканской руды показана
на схеме 2.
Извлечение урана из других смоляных руд.
Для извлечения урана и радия из других смоляных руд целым рядом
поколений химиков были разработаны многочисленные процессы. Эти руды
в настоящее время не имеют большого значения, а поэтому детального
описания способов их переработки можно не давать. Переработка является
очень сложной, так как руда содержит около 30 металлов. Обогащение
руды обычно состоит из ручной разборки и флотации. Концентрат
измельчают и обжигают со смесью соды и сульфата натрия при 800° в течение 10 час.
При этом удаляется сера в виде сернистого газа и образуются уранаты, антимо-
наты, вольфраматы, молибдаты и ванадаты натрия. Обожженную руду опять
размалывают и выщелачивают сначала серной, а затем азотной кислотой.
Большая часть урана переходит в растворимый в воде сульфат уранила.
Дальнейшая операция очистки в каждом отдельном случае зависит от природы
остальных присутствующих элементов [ 7, 8]. Применяют также непосредственное
извлечение серной и азотной кислотами [9, 10].
Рудный концентрат
Шаровая мельница < Количество воды, достаточное для образования
пульпы, которую можно перекачивать насосом
[Н2504(уд. вес 1,8)
< ВаС12
1Мп02
Выщелачивание кислотой (3 часа при 90°)
Na2C03 до рН^Ю; обработка при 60°
Ra, Ba, Al, Fe и т. д.
На извлечение радия
Разбавленная ЫС1 —> Na4[U02(C03)3
—->С02
NaOH —> Раствор U02C12
Na2U207
В отвал
или
Концентрирование
кипячением до 26°Вё
Фильтрат <r- NaOH
Na*U.,07
В отвал
Раствор Na4[U02(C03)3] <- Na2S
PbS
Очищенный раствор ^ МпЛи
Na4[U02(C03)3]F^NaOH
H2S04 \ кт т т п
(NH4)2S04 ]->т*иМ
(NH4)2U2Q7
В отвал
и3о8
Схема 2. Технологическая схема производства U308 из африканской смоляной руды.
Глава 4. Извлечение урана из руд и получение металлического урана 103
Извлечение урана из карнотита*. Извлечение урана-из
карнотитовых руд представляет значительно более трудную проблему, чем
выделение его из урановой смоляной руды, так как в данном случае приходится
перерабатывать относительно большие количества сырой руды.
Количественное отделение ванадия и фосфора от урана также представляет
значительные трудности. ,
Прежде на получение богатых ураном концентратов из низкосортного
карнотита обращалось мало внимания, но в последнее время в этом направлении
проведено много работ. Карнотит встречается в виде небольших агрегатов
светлых рыхлых частиц как мелкозернистый компонент глинистой маточной породы
(лимонита), скрепляющей отдельные зерна кварца, а также в виде тонкой
прочной оболочки на зернах кварца. Измельчая породу и пропуская ее через сито
в 200 меш (при этом получают частицы приблизительно размера отдельных
кварцевых зерен), можно извлечь большое количество карнотита [12]. Чем
тоньше измельчение, тем выше степень извлечения. Можно достигнуть
65—78%-ного извлечения урана, причем при просеве достигается обогащение
в 2—4 раза по сравнению с исходной рудой [13, 14]. Мокрое измельчение дает
лучшие результаты, чем сухое. Остатки карнотита, прилипшие к частицам
кварца, предложено отделять перемешиванием размолотой руды
воздушной струей, в процессе которого отдельные частицы будут подвергаться
истиранию; мелочь затем можно отделить на циклонном сепараторе [15, 16].
В результате предварительных исследований различных рудообогатитель-
ных операций предложена схема обогащения, основанная] на дефлокуля-
ции и флотации (схема 3) [17, 19]**.
Указывают [21], что предварительный обжиг карнотитовых руд имеет
много преимуществ, так как эти руды часто содержат значительные количества
углеродистых веществ (асфальт и т. п.), которые затрудняют измельчение и
мешают последующей кислотной обработке. При обжиге органические вещества
разрушаются, а железо одновременно превращается в нерастворимую в
кислотах окись. Утверждают, что при использовании обжига можно достигнуть
необычайно высокого выделения ценных компонентов минералов [21].
Для извлечения урана, ванадия и радия из карнотита пользуются
различными методами. Большинство широко применяемых операций включает
обработку руды разбавленной или концентрированной серной кислотой при
обыкновенной [22] или повышенной температуре (100—300°) [23—27]. Если процесс
ведут при обыкновенной температуре, то порошкообразную руду смачивают
5—10% (вес.) серной кислоты (60° Вё) и оставляют на 20—90 суток.
При повышенных температурах процесс извлечения происходит гораздо
быстрее. После кислотной обработки производят выщелачивание
разбавленной серной кислотой. Уран и ванадий получаются в виде водных растворов
сульфатов.
Горное бюро США одно время предлагало применять для выщелачивания
соляную или азотную кислоту вместо более дешевой серной кислоты [28].
Оказалось, что для большинства руд извлечение урана соляной кислотой может
быть успешно осуществлено только после предварительной обработки содой
или едким натром. Запатентовано много методов с применением соляной
кислоты, но они используются редко [29, 30]. Предлагается также использовать смесь
соляной и щавелевой кислот [31]. Обычно в тех случаях, когда желательно
извлечь радий, применение соляной кислоты не рекомендуется.
Обработка азотной кислотой не эффективна для руд, содержащих
углеродистые вещества [29, 32—34]. Выход ванадия из таких руд очень мал, и
* См. статью Дорнера [11], где имеется библиография патентов, относящихся к
получению радия.
** Обогащение руд с менее распространенными металлическими минералами,
включая урановые, описано Митчеллом [20].
Измельченная руда (200 меш)
• Шаровая или галечная мельница
Дражный классификатор
Пески
Слив (<200 меш)
Чаше вый классификатор
Пески
Мешалка
Слив
Чашевый классификатор
i i
Пески Слив
—> Сгуститель -
Вода Разгрузка
Машина для
первичной флотации
Концентрат
Машина для очистной флотации
Хвосты Концентрат
12—15% твердого
0,6—1% NaOH на \т*
для дефлокуляции
' Собиратель: олеиновая кислота
0,3—0.5 кг/т
Пенообразователь: технический
крезол 0,1 кг/т
Хвосты
Сгуститель
Вода
Разгрузка
I
Фильтр
Осадок
Вода
т/лиа11, о „ ,_ Г Уменьшение массы в 5 раз 1
Конечный продукт j Коэффициент обогащения U308 3,5 J
Схема 3. Схема обогащения карнотитовых руд (предложенная).
* Здесь, повидимому, в иностранном оригинале опечатка: вероятно, должно быть
0,6—1 фунта на 1 длинную тонну или 0,3—0,5 кг/т.—Прим. ред.
Глава 4. Извлечение урана из руд и получение металлического урана 105
значительные количества урана и ванадия теряются вследствие осаждения при
последующих операциях фильтрования, протекающих весьма медленно и с
большим трудом. Метод выщелачивания азотной кислотой считается
неудовлетворительным, особенно для пылевидных концентратов [21, 35].
Предварительный обжиг руды значительно облегчает обработку азотной кислотой (схема 4).
Добавление к кислоте, применяемой для обработки, небольшого
количества плавиковой кислоты помогает извлечению всех ценных составных
частей руды. В общем серная кислота остается наиболее употребительным
выщелачивающим агентом.
Выщелачивание можно также проводить растворами щелочных
карбонатов или щелочами [36—38]. Например, топкоизмельченную руду нагревают
до 90° с раствором карбоната натрия или калия, при этом около 80% урана
и 60—65% ванадия могут перейти в раствор. Концентрацию раствора карбоната
меняют в зависимости от содержания в руде урана и ванадия и также от их
соотношения. Щелочную обработку можно проводить и под давлением [39].
Если пользуются смесью карбонатов и гидроокисей щелочных металлов,
то ванадий извлекается в виде растворимого ванадата, а уран остается в виде
нерастворимого ураната. После удаления большей части ванадия уран можно
извлечь выщелачиванием кислотой [40, 41].
Предложена также обработка руды раствором едкого натра при 200—300°
[42—44]. Полное отделение урана от ванадия этим путем вряд ли возможно,
однако этот метод может быть использован, если ванадий присутствует в
значительно больших количествах, чем уран. Введение перекисных соединений
(например, перекиси водорода) в раствор едкого натра и карбоната натрия
является видоизменением щелочного метода выделения. При этих условиях
уран и ванадий переходят в раствор [45].
Кроме вышеописанных способов извлечения урана и ванадия, когда
процессы протекают в водных растворах при умеренных температурах,
предложено много методов разложения руды сплавлением при высоких температурах.
Измельченную руду (или концентрат) прокаливают со смесью хлорида натрия
и едкого натра. Ванадат натрия выщелачивают водой, а уран отделяют от
остатка серной кислотой [46, 47]. Рекомендуется также сплавление с
трехкратным избытком бисульфата натрия [48, 49] или с твердым карбонатом натрия
[50]. Недавно предложено .пользоваться сульфатом аммония при 380—430 .
Этот метод имеет то преимущество, что обжиг и разложение руды
объединяются в одну операцию. При указанных температурах сульфат аммония
эквивалентен серному ангидриду, который также может быть применен
непосредственно [51]. При обработке продукта реакции водой как уран, так и
ванадий переходят в раствор. Имеется сравнительно мало данных об
эффективности этих, повидимому, редко используемых методов. Они могут быть пригодны
в случае богатых руд или концентратов, но применение их при переработке
больших количеств низкосортных руд явно затруднительно. Большие
количества кремнезема, вероятно, также могут быть переведены в растворимое
состояние щелочным плавлением, что может вызвать затруднения при
последующих операциях.
Уран и ванадий предложено разделять нагреванием смеси сульфатов ура-
нила и ванадила, получаемых выпариванием сернокислотной вытяжки
карнотита. При 500—650° сульфат ванадила разлагается и, реагируя с
присутствующим железом, образует нерастворимый ванадат железа. Уранилсульфат вновь
извлекается выщелачиванием [52]. Разделение урана и ванадия может быть
также достигнуто доведением кислотности раствора сульфатов уранила и
ванадила (из сернокислотной вытяжки) до рН= 1ч-2 и окислением ванадия хлоратом
натрия или двуокисью марганца. При этих условиях большая часть ванадия
осаждается в виде ванадиевой кислоты HV03 [53, 54].
Способ переработки отунита описан Глазером [55].
Руда, обожженная прн 700°
Размол в галечной мельнице с горячим
раствором соды, фильтрование
и промывка
Остаток
Обработка азотной
кислотой, фильтрование
и^промывка
Щелочной раствор,
содержащий некоторые
количества U и V
Остаток
Отход
Раствор нитратов
неметаллов <-
ЫаОНдо достижения 0,25тн.
кислотности, ВаС12, Na2S04
Ra, BaSQ4
На извлечение радия
Нитраты урана < NaOH до нейтрализации, "
н ванадия нагревание, добавление FeS04
Na2S04 —
сплавление,
выщелачивание
водой
-»U+V
Регенерация
нитратов
H2S04-
' Остаток
Раствор U02S04
Очистка через
Na4[U02(C03)3]
Раствор ванадилсульфата
Нагревание
HV03
(NH4)2U207
U03
Схема 4. Схема переработки карнотитовых руд азотнокислотным методом, разработанным
Горным бюро США (метод, видоизмененный Дорнером [11, 21]).
Глава 4. Извлечение урана из руд и получение металлического урана Ю7
Указанные методы являются большей частью экспериментальными или
взяты из патентной литературы. Метод, имеющий техническое применение,,
состоит в обжиге сырого карнотита с хлоридом натрия и в последующем
выщелачивании водой для удаления образующегося во время обжига ванадата
натрия. Хвосты затем выщелачивают разбавленной серной кислотой, причем в
раствор переходит уран и нерастворившийся в воде ванадий. Уран потом
отделяется методами, описанными выше.
Специальные методы извлечения урана из
низкосортных руд [56—59]. Для обработки бедных урановых руд
предложен ряд интересных методов. До настоящего времени эти способы имели мало
значения, тем не менее ниже дается их краткое описание, так как они могут
быть полезны при разработке новых методов в будущем.
Хлорирование. Этим методом пользовались при переработке мадагаскарского
бетафита. Тонкоизмельченный минерал смешивают с древесным углем и
обрабатывают хлором при температуре красного каления. Образовавшиеся
летучие пентахлориды урана, ниобия и тантала отгоняются из реактора [60].
Аналогичный метод применяли для американской урановой смоляной руды,
содержащей пирит. При обработке хлором в условиях повышенной
температуры хлориды урана и железа отгоняются из реакционной смеси [61].
Хлорирующим агентом является, повидимому, SC12 (см. гл. 14). Фосген
реагирует с отунитом следующим образом [62]:
800°
Са(1Ю2)2(Р04)2 + 10СОС12 > 2UC14 + 2РОС13 + 10СО2 + СаС12 + 2С12.
Другие хлорирующие агенты, например тионилхлорид или четыреххлористый
углерод, также могут представить интерес при разработке эффективных
методов для обработки низкокачественных руд.
Извлечение при помощи S02. Сообщается, что при обработке превращенного
в порошок карнотита насыщенным водным раствором сернистого газа при
нормальных температуре и давлении уран и ванадий переходят в раствор [58, 63].
Для этой цели пытались также использовать жидкий сернистый ангидрид [64].
Извлечение растворителями. Выпущено много патентов с описанием
процессов извлечения урана (и других металлов) из руд методом экстрагирования
растворителями [65]. Этот метод в общем заключается в том, что ценные
компоненты минералов выщелачиванием переводятся в водный раствор, а затем
извлекаются из него противоточным экстрагированием растворителем, не
смешивающимся с водой. Для повышения эффективности может быть
осуществлена рециркуляция. На последнюю возлагаются большие надежды, и она,
несомненно, в дальнейшем будет предметом интенсивных исследований.
Исследован [66] процесс экстрагирования растворителями урановых руд. Оказалось,
что уран можно легко очистить от кремния, титана, ванадия, ниобия, тантала,
молибдена и вольфрама экстрагированием эфиром из раствора нитратов. Бор
и фосфор отделяются неполностью.
Подобный метод применяют в течение некоторого времени для очистки
технической закиси-окиси урана. Для некоторых целей необходимо удалять из
урана все следы редких земель. Удаление достигается методом экстрагирования
растворителями, основанным на растворимости уранилнитрата в этиловом эфире,
в котором нитраты редкоземельных элементов нерастворимы [67]. Этим
способом может быть получен очень чистый окисел урана.
ПОЛУЧЕНИЕ МЕТАЛЛА
Впервые металлический уран получен более ста лет назад, но производство
чистого металла в больших масштабах стало возможным только с 1940 г.
До разработки методов, описанных в этой главе, металлический уран получали
108
Часть II. Металлический уран
в небольших количествах главным образом для научных целей. Процессы
получения были непроизводительны, кроме того, выделенный металл не являлся
достаточно чистым. Современная металлургия урана детально описана в
других томах этого издания. В этой главе дается только краткий обзор методов
получения металлического урана.
Исторический обзор. В 1789 г. Клапрот восстановил трех
окись урана углем в условиях высокой температуры. Продукт восстановления
имел металлический вид и поэтому был принят за свободный металл. Такое
мнение было общепринятым до 1840 г., когда Пелиго доказал, что выделенный
Клапротом продукт является низшим окислом урана (U02). Металлический уран
вначале получали восстановлением тетрахлорида урана металлическим калием,
а затем в лабораторном масштабе применяли следующие методы получения:
а) восстановление окислов урана углем; б) восстановление окислов урана
алюминием, кальцием или магнием; в) восстановление галогенидов урана
щелочными или щелочноземельными металлами; г) электролитическое
восстановление галогенидов урана. За небольшим исключением, металл при этом
получался в виде порошка.
Восстановление окислов урана углем. Восстановление окислов урана до
металла углем возможно только при очень высоких температурах
электрической дуги. Такое восстановление было проведено Муассаном [68—74]
в электродуговой печи. Смесь закиси-окиси урана и угля (полученного
из сахара) подвергали сильному прессованию в графитовом тигле.
Восстановление проходило в течение нескольких минут (450 а, напряжение 60 в).
Муассан предполагал, что этим путем можно получить свободный от углерода
металл, но это предположение оказалось неверным, так как уран легко
реагирует при повышенных температурах с углеродом с образованием карбидов
урана. Необходимо тщательное удаление из реакционной смеси как
кислорода, так и азота. Если применить меньшие количества углерода
(40 г угля на 500 г закиси-окиси урана), чтобы избежать образования
карбидов, то полученный металл будет содержать окисел урана [75].
Продолжительное нагревание смеси в графитовых тиглях при повышенной температуре
неизбежно приводит к образованию карбидов урана. При повторении этих
опытов [76] установлено, что восстановление урана возможно и в более
мягких, чем у Муассана, условиях. Недавно опыты Муассана были
проведены с двуокисью и закисью-окисью урана, причем в качестве
восстановителя применяли сахарный уголь, ацетиленовую сажу, графит и другие
формы углерода. Во всех случаях в заметных количествах образовывался
карбид урана. Изменение соотношения между окислом урана и углеродом
в широких пределах не устраняет образования карбида [75].
Восстановление окислов урана алюминием, кальцием или магнием. Согласно
термодинамическим расчетам, двуокись урана восстанавливается кальцием,
магнием или алюминием при довольно высоких температурах. Подобные
реакции обычно проводят в эвакуированной стальной аппаратуре. При
восстановлении двуокиси урана кальцием при температуре 950—1250° в герметически
закрытых железных тиглях образуется тонкий порошкообразный металл высокой
степени чистоты [77].
По аналогичному методу [78] закись-окись урана и кальций нагревали
в стальной трубке, вставленной в эвакуированную кварцевую трубку. При
этом, так же как и в опытах других исследователей [79, 80], ранее применявших
подобный метод, металл получался в виде очень тонкого порошка.
Предположение о том, что при восстановлении окиси урана кальцием получается металл
высокой степени чистоты, не нашло подтверждения в дальнейшем; например,
при нагревании закиси-окиси урана с теоретическим количеством кальция при
1000° в течение 45 мин. в эвакуированной стальной бомбе [81 ] главным
продуктом оказался коричневый порошок; в образовавшихся шариках металлического
Глава 4. Извлечение урана из руд и получение металлического урана 109
урана содержалось довольно много железа и кислорода. Эти наблюдения были
подтверждены и последующими работами [82].
В дальнейшем восстановление металлическим кальцием проводилось в
присутствии щелочных или щелочноземельных галогенидов. Эти методы описаны
в многочисленных патентах [83—85]. Добавка хлорида кальция или хлорида
магния в качестве флюса способствует реакции, причем металл получается
в форме крупных зерен. Для восстановления двуокиси урана может быть
также использован металлический магний, но вследствие высокого давления
паров он может испариться из реакционной массы, а поэтому процесс
надо вести под давлением [86]. Начавшись, реакция идет очень бурно и с
трудом поддается регулированию. Имеется указание на то, что применение
сплава, состав которого приблизительно выражается формулой Ca3Mg4 [84],
представляет некоторые преимущества.
Двуокись урана можно также восстанавливать гидридом кальция
[87—89]:
U02 + 2CaH2 -» U + 2CaO + 2H2.
Смесь двуокиси урана и гидрида кальция помещают в нихромовый
цилиндрический сосуд, который затем вставляют в обогреваемую газом реторту. Реакция
начинается при 960°. Загрузку охлаждают под вакуумом для удаления
водорода. Продукт реакции выгружают, подвергают дроблению и размолу, а затем
выщелачивают известь разбавленной уксусной кислотой. Порошок урана
промывают водой, сушат, прессуют в плотную массу и подвергают спеканию.
Процесс восстановления окислов урана металлическим алюминием
изучали многие исследователи [71,76, 90—93]. Закись-окись урана
восстанавливают порошкообразным алюминием до низшего окисла при 600°, но для
получения чистого металла, очевидно, необходима более высокая температура.
Сделана попытка достигнуть полного восстановления с применением
жидкого воздуха в качестве окислителя*. Сообщалось, что металл удалось
получить в виде королька, но если принять во внимание тенденцию
металлического урана реагировать с кислородом и азотом, то не приходится удивляться,
что указанное сообщение не подтвердилось. Описано [94] алюминотермическое
восстановление окислов металлов, в том числе и окислов урана в
центрифужном аппарате. Отмечаются различные преимущества этого метода. Хотя
алюминий и может быть выделен из сплава урана с алюминием сплавлением
в вакууме, все же эта способность урана давать подобные сплавы при алю-
минотермическом восстановлении его окислов усложняет процесс.
Восстановление галогенидов урана щелочными или щелочноземельными
металлами. Как указывалось выше, металлический уран был получен
Пелиго [67] восстановлением тетрахлорида урана металлическим калием. С тех
пор часто применяли восстановление галогенидов урана щелочными или
щелочноземельными металлами. Полученный Пелиго металл содержал примесь
платины, так как восстановление проводилось в платиновом тигле. Замена
калия натрием почти не улучшила процесса [95]. По методу Пелиго в качестве
флюса применялся хлорид калия, а реакционная смесь в платиновом тигле
защищалась от воздуха слоем древесного угля. Циммерман [96] изменил метод
Пелиго. Он использовал железную бомбу [97]. Для предохранения от
разъедания ее защищали слоем расплавленного хлорида натрия. Тетрахлорид урана,
хлорид натрия и металлический натрий загружали в бомбу, которую затем
завинчивали, а реакционную смесь нагревали до белого каления. Циммерман
утверждал, что этим методом он получил чистый, плотный металл. Муассан [72]
повторил опыты Циммермана и установил, что металлический уран, полученный
* Жидкий воздух применяется здесь в качестве окислителя алюминия с целью созда?
ння высокой температуры, необходимой для получения спекшегося металла.—Прим. ред.
100
Часть //. Металлический уран
таким способом, содержит 2% железа. Вместо тетрахлорида урана он
предпочитал пользоваться менее гигроскопичной двойной солью Na2UCle.
Восстановление металлическим натрием Муассан проводил также в железном цилиндре
с завинчивающейся пробкой. Продукт реакции получался в виде тонкого
порошка довольно сомнительной чистоты. Ряд других исследователей применял
такие же методы с бомбами, но результаты по существу не улучшались.
Микстер [98], точно воспроизводя метод Муассана и применяя Na2UCl6,
получил порошкообразный уран 97—99%-ной чистоты.
Родербург [99] провел обширные исследования по проверке методов
выделения чистого урана, в частности, метода восстановления галогенидов урана
щелочными металлами. С целью повышения чистоты металла применяли бомбы
из различных легированных сталей, но во всех случаях металлический уран
получался со значительным содержанием железа. Родербург пытался также
восстановить тетрафторид урана натрием и калием, но реакция проходила лишь
частично. Фишер [100] и Райдил [101] также исследовали различные методы
получения металлического урана. Наилучшим из значительного количества
изученных методов они считали способ восстановления тетрахлорида урана
магнием или натрием в присутствии хлорида кальция. Однако партии металла,
полученные этим путем, имели различную степень чистоты, о чем можно судить
по колебаниям электропроводности. Для того чтобы свести загрязнения
кислородом и азотом к минимуму, восстановление тетрахлорида урана металлическим
натрием проводили в эвакуированной металлической бомбе [102]. Продукт
получился в виде тонкого порошка и попрежнему содержал значительные
количества кислорода. Имеется детальное описание подобного процесса
восстановления в вакууме [103].
Исследованы методы восстановления галогенидов урана кальцием,
магнием, сплавами кальция с магнием, а также алюминием; они описаны в ряде
патентов [104]. Согласно этим методам, тетрафторид урана KUF5 и некоторые
другие нелетучие галогениды или двойные соли урана восстанавливали одним
из ранее упоминавшихся металлических восстановителей, обычно в
присутствии хлорида кальция в качестве флюса. После выщелачивания и промывки
реакционной массы уран получался в виде тонкого порошка.
Ряду исследователей [81], повидимому, удалось получить плотный
металлический уран высокой степени чистоты. Реакция
UCl4 + 2Ca -> 2CaCl2 + U
проводилась в эвакуированной стальной бомбе. При использовании
повторно перегнанного кальция можно получить плотный металл без примеси железа.
Успех этих исследователей частично может быть объяснен тем, что опыты
проводились в довольно большом масштабе, что способствовало меньшему
загрязнению и минимальной потере тепла системой. Эта работа, вероятно, наиболее
успешная из всех старых работ, опубликованных в литературе. В последнее
время изучен процесс восстановления галогенидов урана металлическим
калием, полученным взаимодействием карбида кальция с хлоридом
калия [105]. Установлено, что окислы урана могут быть восстановлены
ферросилицием при высокой температуре.
Электролитическое восстановление галогенидов урана. Последним методом,
использованным в старых работах в этой области, является электролитическое
восстановление галогенидов урана в расплавленных солевых ваннах [106].
Не считая первых опытов Муассана [74], описанная ниже работа по
электрохимическому получению металлического урана [107—112] является наиболее
значительной*. Электролит состоял из двойной соли KUF5 или тетрафторида
• Методы получения металлических тория и урана описаны в журнале Steel»
120. 93—4, 124, 126, 130, 133 (1947).
Глава 4. Извлечение урана из руд и получение металлического урана 111
урана, растворенных в расплавленной смеси из 80% хлорида кальция и 20%
хлорида натрия. Электролитической ванной служил графитовый тигель,
являвшийся одновременно анодом; катод был сделан из молибдена. Ванна
работала при 900° с плотностью тока около 150 а/дм2. Уран получался
зернистым и пирофорным. Для удаления окклюдированных солей требовалась
сложная система операций промывки и сушки.
Многие исследователи пытались выделить металлический уран из водного
раствора, но никаких доказательств осуществимости этого процесса не имеется.
Во всех случаях на катоде получалась сложная смесь гидратированных
окислов. Неудачная попытка исследователей электролитически осадить уран из
водного раствора не вызывает удивления, так как самым
электроположительным металлом, который еще можно выделить из водного раствора, является
марганец, в то время как уран почти несомненно стоит в ряду напряжений
выше марганца. Имеются некоторые указания на то, что при электролизе
водных растворов с ртутным катодом при высоких плотностях тока могут
образоваться разбавленные амальгамы урана [113].
Сделаны также многократные попытки электролитического осаждения
урана из растворов в неводных растворителях [114, 115]. Электролизу
подвергали насыщенные растворы тетрабромида урана в бензоле, бромистом этиле,
диэтиловом эфире, ацетоне, диоксане, ацетоне, насыщенном S02, нитробензоле,
формамиде и формамиде, содержащем бромистый водород [116]. Заметный
осадок на катоде получен только из раствора в формамиде, но не установлено,
являлся ли он металлическим ураном. Поэтому полученные результаты не
внесли ясности в это положение, и можно сделать вывод, что в настоящее время
электролиз водных или неводных растворов солей урана, повидимому, не может
служить практическим методом получения металлического урана. Однако
в данной области проведено сравнительно мало исследований, а поэтому
дальнейшие работы могут изменить это заключение.
Выводы. В табл. 42 приводится большое количество возможных реакций
получения металлического урана. По сравнению с восстановлением окислов
урана восстановление его галогенидов щелочными или щелочноземельными
металлами дает лучшие результаты. В этом случае получаются шлаки, которые
плавятся большей частью при относительно низких температурах. Некоторые
из этих реакций достаточно экзотермичны для того, чтобы реакционная смесь
полностью расплавилась. Значения теплот реакций показывают, что тетрафто-
рид, тетрахлорид и тетрабромид можно восстанавливать натрием, кальцием или
магнием, восстановление же алюминием самопроизвольно не происходит.
Наиболее экзотермичны реакции восстановления хлоридов или бромидов
урана металлическим натрием и реакция восстановления фторида урана
кальцием. Реакции восстановления магнием по сравнению с восстановлением
кальцием во всех случаях менее экзотермичны, хотя значения теплот этих
реакций могут быть все же близкими (см. табл. 42).
Получение металлического урана термическим
разложением галогенидов урана [117—121 ]. Многие
металлы получены термическим разложением паров галогенидов
соответствующих металлов на раскаленной нити. Этот метод, в частности, применяется для
получения циркония, титана, вольфрама и тория [122]. Таким путем можно
получить очень чистый металл.
В случае применения этого метода для получения чистого металлического
урана возникает много трудностей. По сравнению с металлами, которые были
успешно получены этим методом, уран имеет низкую точку плавления (1133°).
Вначале процесс проводили таким образом, чтобы расплавленный металл
стекал каплями с проволоки [123]. Однако этот метод считается
неудовлетворительным; лучше применять такие низкие температуры разложения, при
которых уран остается твердым [124].
112
Часть II. Металлический уран
Таблица 42
ВОЗМОЖНЫЕ РЕАКЦИИ ПОЛУЧЕНИЯ МЕТАЛЛИЧЕСКОГО УРАНА
Реакция
U02 + 2Ca->U + 2CaO
U02 + 2Mg -* U + 2MgO
U02+-iA1-^U+4A12°3
U308 + 8Са -* 3U + 8СаО
U308 -f 8Mg -> 3U + 8MgO
из08 + ^А1->Зи + |-А12Оз
UF4 + 4Na->U + 4NaF
UF4 + 2Ca->U + 2CaF2
UF4 + 2Mg-*U + 2MgF2
UF4 + iAl->U + yAlF3
UCl4 + 4Na->U + 4NaCI
UCl4+5Ca->U + 2CaCl2
UCl4 + 2Mg->U+2MgCI2
UCl4+-iAI->U + ^-AlCl3
UBr4 + 4Na-»U + 4NaBr
UBr4 + 2Ca -* U + 2CaBr2
UBr4 + 2Mg -> U + 2MgBr2
UBr4 + |-Al->U + -|-AlBr3
UCl3 + 3Li-^U + 3LiCl
ДН на 1 моль
урана при
298°, кал
- 47
- 35
- 11,4
-123,1
-108,1
- 76,4
- 98
-134
- 82
6
-141
-131
- 55
28
-144
-124
- 44
31
—
Т. пл.а) шлака,
°С
2572
2500—2800
2050
2572
2500—2800
2050
980—997 (1040)
1330
1225
1040
801
772
708
190 (при 2,5 am)
755
765
695
97,5
613
Т. кип.а) шлака
при 760 мм,
°С
2850
2250
2850
2250
1700
2500
2260
—
1413 (1490)
1925
1420
182,7
177,8 (возг.1
1390
1200 (?)
(806—812)
1125
263,3
1353
а) В скобках даны другие приводимые в литературе значения. Они указываются во всех тех
случаях, когда нет основания для выбора определенной величины.
Наиболее подходящим галогенидом для этого процесса является тетра-
иодид урана. Для разложения других галогенидов урана либо требуется
слишком высокая температура нити, чтобы можно было применить на практике,
либо галогениды недостаточно летучи.
Большое значение для этой проблемы имеет равновесие (см. гл. 15):
Щ4 ^ UJ3 + |J2. (1)
Для предотвращения диссоциации тетраиодида урана в паровой фазе требуется
определенное парциальное давление паров иода (тетраиодид почти нелетуч).
На нити накала идет следующая реакция:
Щ4 ^± U + 2J2. (2)
Высокое парциальное давление паров иода, с одной стороны,
благоприятствует протеканию прямой реакции, а следовательно, и осаждению металла,
так как приводит к повышению концентрации тетраиодида в паровой фазе по
уравнению (1), с другой стороны, тормозит ее, так как способствует смещению
равновесия реакции (2) в сторону образования тетраиодида урана, вследствие
Глава 4. Извлечение урана из руд и получение металлического урана ИЗ
чего осаждение металла замедляется. В результате скорость осаждения металла
достигает максимума при определенном оптимальном давлении иода. Это
давление является функцией температуры нити накала и может быть рассчитано
из констант равновесия обеих реакций.
Получение металлического урана разложением тетраиодида на
раскаленной проволоке успешно осуществляли при температуре нити накала более
низкой, чем температура плавления урана. Восстановление проводили в
стеклянном шарообразном сосуде, помещенном в воздушную отражательную печь;
вводили твердый трииодид урана и систему эвакуировали. Область, где
находится трииодид, окружали медным охлаждающим змеевиком для поддержания
любой заданной температуры. Сосуд, содержащий иод, соединяли с
шарообразным сосудом длинной вертикальной трубкой, что позволяло поддерживать
также и иод при любой нужной температуре. Таким образом регулировали
парциальное давление иода в системе. Условия проведения реакции были
следующими: температура нити накала 1030—1100°, температура в
шарообразном сосуде 520—560°, температура трииодида урана 500—540°, Pj2 = 7 • Ю'гмм
рт. ст. В этих условиях на нити накала осаждался твердый уран.
Довольно подробно разобран процесс термической диссоциации
тетраиодида урана. Указывается [1181, что если температура в колбе поддерживается
равной 800° К, металл осаждается при давлении иода в сообщающемся сосуде
меньшем критического значения, получаемого по эмпирическому уравнению
lg Pj, = 7,860 — 1,234 4^-.
Произведен теоретический анализ процесса термической диссоциации
тетраиодида урана, а также детально описываются возможные механизмы
процесса [125, 1261. В старых работах высказывалось предположение, что весь
иод, испаряющийся с нити накала, находится в форме молекул иода; в
действительности же при температуре нити накала идет полная диссоциация до атомов
иода. Кроме того, при соприкосновении с нитью тетраиодид урана превращается
в трииодид, а последний, если не разлагается до металла, то снова превращается
в тетраиодид при температуре, которая поддерживается внутри шарообразного
сосуда. Приведенная выше зависимость для критических условий отложения
металла, повидимому, согласуется с принятыми в настоящее время
измеренными и установленными для условий равновесия величинами.
ЛИТЕРАТУРА
1. В а с h е 1 е t M., Bull. soc. chim. France, 628—632 (1947).
2. McCoy H. N., Anderson H. C, Report CS-70, без даты (MP Chicago 1).
3. P о с h о n M.f Chem. Met. Eng., 44, No 7, 362—365 (1937).
4. S afford W. H., Kucbel A., J. Chem. Education, 20, 88—91 (1943).
5. Kuebe! A., J. Chem. Education, 21, 148—149(1944).
6. Mactaggert E. F.f Chem. Met. Eng., 50, No. 7, 178 — 181 (1943).
7. H e n г i с h F., Chemie radioaktiver Stoffe, Verlag Julius Springer, Berlin, 1918,
p. 298 ff.
8. U I г i с h C, Z. angew. Chem., 36, 41, 50 (1923).
9. M a re k w a 1 d W., Ber., 41, 1529 (1908).
10. Marckwald W., Russel A. S., Ber., 44, 772 (1911).
11. D о е г n e r H. A., U. S. Bur. Mines Repts. Invest., No. 3057 (1930).
12. D e V a n e у F. E., Eng. Mining J., 139, 43—45 (Nov., 1938)
13. N у e R.D., Demorest D J., пат США 2173523, 1939.
14. Engel A. L., Shelton S. M.t U. S. Bur. Mines Repts. Invest., No. 3628 (1942).
15. Dunn H.E, Rees С P., S p г о u 1 А. А., пат. США 2175484, 1939.
16. Dunn H. E., пат. США 2175457. 1940.
17. D a v i s et all., U. S. Bur. Mines Repts. Invest., No. 3370, 92 (1938).
18. Shelton S.M., Engel A. L., U. S. Bur. Mines Repts. Invest., No. 3636(1942).
19. McCov H. N.f пат. США 1195698, 1916.
20. M i t с h e 1 1 F. В., Mine a. Quarry Eng., 13, 305—330, 334—342 (1947).
о Химия урана
114
Часть II. Металлический уран
21. Doer пег Н. A., U.S. Bur. Mines Repts. Invest., No. 2873 (1928).
22. F 1 e с к Н., Н а 1 d i n e W. G>( White E. L., пат. США 890584, 1907.
23. В г с d t О Р. С, пат. США 1154231, 1914.
24. McCoy H. N.. пат. США 1098282, 1914.
25. Daniorth С. W., Samuels W., Martersteck \V\, пат. США 1126182,
1915.
26. Sc h 1 u n d t H., J. Phvs. Chem., 20, -185 (1916).
27. Schlundt H., пат. США 1181 Ш, 1194569, 1916.
28. P а г s о n s С L, Moore R. В., L i n d S. C, Schaefer О. С, U.S.
Bur. Mines Bull , No. 101, p. 17, 27, 30 (1915).
29. Moore R. В., пят. США 1 If.569:3, 1916.
30. Bell W. A. J., пат. США 1522010, 1922.
31. В е 1 1 W. A. J., пат. США 15269-13, 1921.
32. P 1 u m H. M., J. Am. Chem. So.-., 37, 1797 (1915).
33. P а г s о n s С L., Ind. Eng. Chem., 8, 469 (1916).
34. Viol С. Н., Ind. Eng. Chem., 8. 284, 660 (1916).
35. Doerner H. A., Ind. Eng. Chem., 22, 185—188 (1930).
36. Hayness J. H„ Engle W. D., пат. США 808839, 1905.
37. В 1 e ее к e r W. F., пат. США 1065581, 1913.
38. Thews К. В., Heinle F. J., Ind. Eng. Chem., 15, 1159 (1923).
39. В 1 e ее к e r W. F.f пат. США 1399216, 1919.
40. В 1 e ее к e r W. F., пат. США 1068730, 1913.
41. М о о г е R. В., пат. США I li 5692, 1913.
42. Fischer S., пат. США 1054102, 1912.
43. Ebler E., Bender \V.f Z. япаеш. Chem., 28T, 34 (1915).
44. S с h 1 e s i n g e г W. A.f пат. С ША 1435180, 1918.
45. G i b b s H. L.f пат. США 199980/, 1935.
46. В 1 e e с к e r \V. F., пат. ("ША 1015469, 1912.
47. Vogt L., пат. США 1129029, 1915.
48. R a d с 1 i f f e S., пат. США 1019145, 1911.
49. R a d с 1 i f f с «6., J. Soc. Chem. Ind., 33, 229 (1914).
50. P а г s о n s C.L., Moore R. В., Lind S. C, Schaefer O. C, U. S.
Bur. Mines Bull., No. 104, 24 (1915).
51. M с С о г m а с к Н., пат. С ША 2176609, 1939.
52. Potter J. S., пат. США 2180F92, 1939.
53. S t a m b е г g С. J., пат. США 2176610, 1939.
54. Fleck H., пат. США 2199696, 1940.
55. G 1 a s е г F., Chem. Ztg., 36, 1167 (1912).
56. U 1 z е г F., Sommer R., герм. пат. 254241, 1908.
57. Ebler E., герм. пат. 296132, 1913.
58. Loo m is A. G., Schlundt H., Ind Eng. Chem., 8, 990 (1916).
59. U 1 г i с h С, Z. angevv. Chem., 36, 51 (1923).
60. Curie M., Le radium et les radioelements, J. B. Bailliere e. fils, Paris, 1925,
p. 205.
61. Cable R., Schlundt H., Chem. Met. Eng., 18, 1 (1918).
62. В а г 1 о t J., С h a u v e п с t E., Compl. rend., 157, 1153 (1913).
63. В u г I e i n d J. H., пат. США 1095377, 1914
64. Hedstrom И.О., Chem. Zentr., II, 936 (1922).
65. Hixon A. W., Miller R., пат. США 2227833, 1941; 2202525, 1940; 2211119,
1940.
66. В а с h e 1 e t M., Cheylan E., LeBris J., J. chirn. phys., 44, 302—305
(1947).
67. P ё 1 i g о t E., Ann. chirn. et phvs. (3) 5 7, 42 (1842); Ann., 43, 257, 283 (1842).
68. Moissan H., Compt. rend., П6, 347 (1883).
69. Moissan H., Bull. soc. chirn. Paris, (3) 11, I I (1894).
70. Moissan H., Compt. rend., 122, 1088 (1895).
71. Moissan H., Compt. rend., 122, 1302 (1896).
72. Moissan H., Ann. chirn. et phvs., (7) 9, 264 (1895).
73. Moissan H., Bull. soc chim. Paris, (3) 25, 344 (1901).
74. Moissan H., The Electric Furnace, E. Arnold and Co., London, 1904, p. 162—167.
75. Sped ding F. H., Reports CN-127, June 13, 1942; CC-177, July 8, 1942
(MP Ames 1).
76. A i о у J , Bull. soc. chim. Paris (3), 25, 344 (1901).
77. J a n d e r \V\, Z. anorg. u. allgern. Chem , 138, 321 (1924).
78. В о i о 1 f s e n E., Bull so:, chim. France, 45, 626 (1929).
79. W e d e к i n d E., Z. a"gew. Chem., 24, 1Г/9 (1911).
80. W e d e к i n d E , пат. США 1С88909, 1914.
81. James С , G о g g i n s J. F., С г о n i n J. J., F о g g H.C., Ind. Eng. Chem.r
18, 114 (1926).
Глава 4. Извлечение урана из руд и получение металлического урана 115
82. W i 1 h е 1 m Н. A., Keller W. H., Report CC-238, Aug. 15, 1942 (MP. Ames 2).
83. М а г d e n J. W., пат. США 1659209, 1923.
84. Rich M. N.. пат. США 1738669, 1927 >,
85. С а с h e m a i 1 1 е А., англ. пат. 238663, 1924.
£6. М а г d e n J. W., пат. США 1602542, 1926.
87. Alexander P. P., Metals a. Alloys, 8, 263—264 (1937); 9, 45—48 (1938).
88. D a v i s Т. W., Р е п п е m a n R., Report CC-276, Sept. 15, 1942 (MP
Chicago 2).
89. F о о t e F., Reports CA-278, Sept. 20, 1942: CT-422, Jan. 15, 1943 (MP Chicago 3).
90. S t a v e n h a g с n A., Bit., 32, 3065 (1899).
91. S t a v e n h a g e n A., Schuchard E., Ber., 35, 909 (1902).
92. G i о 1 i t t i F., T a v a n t i G., Gaz/. chim. ital., 38 (II), 239 (1908).
' 93. M a r d e n J. W., пат. США 1648954, 1927.
94. Merle J. M., пат. США 2395286, 1946.
95. P ё 1 i g о t E., Ann. chim. et phys. (4), 17, 368 (1869).
96. Z i m m e r m a n n C.t Ann., 216, 14 (1883).
97. N i 1 s о n L. F., P e t t e г s s о n O., Ber., 11, 381 (1878).
98. M i x t e г W. G., Z. anorg. Chem., 78, 231 (1912).
99. R о d e г b u r g A., Z. anorg. Chcm., 81, 122 (1913).
100. Fischer A., Z. anorg. Chcm., 81, 189 П913).
101. R i d e a 1 E., J. Soc. Chem. Ind., 33, 673 П914).
102. Lely D., Jr., Hamburger L., Z. anorg. Chem., 87, 220 (1914).
103. Moore R. W., Trans. Am. Electrochem. Soc., 43,319(1923).
104. M a r d с n J. W., пат. США 1437984, 1922; 1646734, 1927; 1814721, 1931.
305. L a u t i e R., Bull. soc. chim. France, 974—977 (1947).
106. D г i g g s F H., L i 1-i e n d a h 1 W. C, Ind. Eng. Chem., 22, 516 (1930).
107. M a r d e n J. W., Report A-605, Mar. 23, 1942 (Westinghouse 1).
108. L i 1 i e n d a h 1 W. C, Meistcr G., N a g у R., W г о u g h t о n D.,
В е е s e N. С, М а г d e n J. \V., Papers Relating to the Production of Uranium
by the Eleotrolvtic Method, June 21, 1946 (Westinghouse 2).
109. D г i g g s F. H., et all., пат. США 1821 176, 1928; 1842254, 1928; 1861625, 1932.
110. D r i g g s F. H., Eng. Mining J., 130, 119 (1930).
111. L i 1 1 i e n d a h 1 W. C, H i g h г i t e r H. W., Am. Inst. Mining Met. Engrs.
Tech. Pubs., 630 (1935).
112. M а г d e n J. W., Trans. Electrochem. Soc., 66, 8 (1934).
113. Ferec M. J., Bull. soc. chim. Paris (3), 25,.622 0901).
114. Pier 1ё С Kahlenberg L., J. Phys. Chem., 23, 517 (1919).
115. A u d г i e t h L. F.t Nelson H, W., Chem. Revs., 8, 338 (1931).
11G. Eastman E. D., F о n t a n a B. J., Electrolysis of Non-aqueous Solutions ot
Uranium Bromides, and Reaction of Alkali Metals with Uranium Bromides in Liquid
Ammonia, in National Nuclear Energv Series, Div. VIII, vol. 6 (MP Berkeley 1).
117. P r esc о t t С. Н., Jr., Reynolds F. L., Report RL-4.G.206, Sept. 29, 1943
(UCRL1).
118. P г e s с о t t С H., Jr., H о 1 m e s J. A., Report RL-4.6.260, Apr. 27, 1944 (UCRL 2).
119. Reynolds F. L., Holmes J. A., Report RL-4.6.2G5, June 5, 1944 (UCRL 3).
120. M a g e 1 Т. Т., Reports CK-1240, Jan. 19, 1944; CK-1130, Dec. 10, 1943; СК-Ю40,
Nov. 6, 1943 (MP Chicago 4).
121. M a g e 1 Т. Т., F о s t e г L. S.t Reports CK-963, Nov. 11, 1943; CK-897, July 28,
1943 (MP Chicago 5).
122. van А г к e 1 A. E., Reine Metalle, Verlag Julius Springer, Berlin, 1939, p. 183, 193,
269; Edwards Brothers, Inc., Ann Arbor., Mich., 1943.
123. M a g e 1 Т. Т., Report CK-li:;0, Dec' 10, 1943 (MP Chicago 6).
124. Foote F.f Reports CT-26 16, Dec, 1944: CT-2668, Jan., 1945'(MP Chicago 7).
125. Prescott C. H., Jr., Report RL-4.6.273, July 27, 1944 (UCRL 4).
126. Prescott C. H., Jr., F. L. R e у n о 1 d s, H о 1 m e s J. A., The Preparation of
Uranium A\etal by Thermal Dissociation of the Iodide, in National Nuclear Energy
Series, Division V11I, vol. 6 (UCRL 5).
Глава 5
ФИЗИЧЕСКИЕ СВОЙСТВА
МЕТАЛЛИЧЕСКОГО УРАНА
СТРУКТУРА И МЕХАНИЧЕСКИ СВОЙСТВА ТВЕРДОГО УРАНА
Уран представляет собой очень плотный металл; он на 80% тяжелее свинца.
Свежеотполированная поверхность металлического урана имеет блестящий
серебристый цвет, но на воздухе через несколько часов она тускнеет.
Низкотемпературная форма урана (а-уран) обладает некоторой ковкостью; ее упругость
настолько мала, что она описана как полупластическая. Среднетемпературная
форма (£-уран) хрупка, а высокотемпературная (;-уран)—пластична.
Металлический уран при высоких температурах можно ковать, волочить или
выдавливать, его можно также подвергать холодной обработке.
Кристаллическая структура
металлического урана. Первые анализы рентгенограмм а-урана, на основании которых
этому металлу была приписана сначала объемноцентрированная кубическая [1 ],
а позже моноклинная решетка [2], оказались ошибочными. Теперь [3]
твердо установлено, что а-уран имеет ромбическую кристаллическую решетку
(пространственная группа V17). Эту структуру лучше всего рассматривать как
искаженную гексагональную плотнейшую упаковку. Элементарная ячейка
содержит четыре атома в положениях (OOOjyyOWfOy-^-; Oy-^jc
параметром */- 0,105 ± 0,005. В табл. 43 приведены значения констант решетки,
найденные различными исследователями. Наиболее точные определения
были проведены на двух образцах урана максимальной чистоты. После
закалки от температур 750—800° структура этих образцов определялась
в симметричной фокусирующей камере с обратным отражением.
Таблица 43
КОНСТАНТЫ КРИСТАЛЛИЧЕСКОЙ РЕШЕТКИ а-УРАНА ПРИ
КОМНАТНОЙ ТЕМПЕРАТУРЕ
ао
2,852
2Я*>2
2,8482а)
Константа, V
Ьо
5,865
5,859
5,8565а)
с*
4,945
4,944
4,9476а)
Плотность,
г/см*
18,97
19,00
19,050
Литература
3
4
5
а> ±0,01%.
Точная структура р-урана неизвестна. Она не может быть зафиксирована
яакалкой в чистом уране, но, согласно проведенным исследованиям, этого
можно достигнуть в урано-хромовых сплавах [6]. Изучение этих сплавов
показывает, что р-фаза является, возможно, кубической и отличается гигантской
элементарной ячейкой (а0 = 12,88 А), содержащей 58 атомов. Подобные же
опыты по закалке проводились с урано-молибденовыми сплавами [7]. При
Глава 5. Физические свойства металлического урана
117
выдерживании сплава, содержащего 5% молибдена, при 650° в течение 90 мин.
с последующим охлаждением в печи была получена кубическая гранецен-
трированная фаза. Сначала предполагалось^ что она именно и представляет
р-фазу урана, но констамл решетки (4,93 А) показьвает, что образуется,
вероятно, моноокись (см. табл. 71 на стр. 215). Впослсд:твии оказалось,
что сплав, содержащий 0,6% молибдена и закаленный ст 690°, содержит
новую, вероятно ромбическую фазу, которую предположительно приняли
за Р-фазу. Аллен [8] подтвердил, что при закалке -j- и (3-фаз структуры,
характерные для этих областей, не сохраняются. Некоторые закаленные
от 600° образцы металла показали наличие дополнительной фазы, но ее
присутствие приписывают примесям в металле, вероятно железу. Попытки снять
рентгенограмму чистого р-урана при помощи высокотемпературной камеры
оказались безуспешными вследствие малой интенсивности линий [9]. В
снимках, сделанных при высокой температуре, наблюдаются обычно только
линии гранецентрированной кубической фазы, вероятно моноокиси урана [10],
Недавно появилось сообщение [111 о том, что р-уран, стабилизованный
присутствием 2% (атомн.) хрома, имеет ромбическую элементарную ячейку (а0=
= 7,51 А, &0= 15,00 А, с0 = 5,59 А), содержащую 30 атомов. Несмотря на
существующую сейчас неопределенность в этом вопросе, можно считать вероятным,
что р-уран по структуре родственен а-урану, но имеет гораздо большую
элементарную ячейку и заметно более низкую симметрию.
Структура -[-фазы урана определена в высокотемпературной камере при
785 [9] и 680° [121; она является кубической объемноцентрированной. Подобно
Р-фазе, и эта фаза не может быть сохранена в чистом уране путем
закалки [13]. По определению, при температуре 785° константа решетки для
чистого урана равна 3,48 А [14], после охлаждения до комнатной температуры
а0 = 3,43 А.
Константа решетки 7~УРана ПРИ комнатной температуре изучена как
функция содержания молибдена [15], поскольку оказалось, что некоторые
металлы, в том числе хром и молибден, стабилизуют структуру ^-урана при
комнатной температуре. Графической экстраполяцией ок чистому урану определена
константа решетки, которая равна 3,467i 0,005 А, а плотность урана при
комнатной температуре оказалась равной 18,89i0,05 г/см*. Результаты,
полученные ранее Мак-Леннаном и Мак-Кеем, показывают, что эти авторы также
получили -[-форму, вероятно, стабилизованную случайными примесями [1].
Размеры атома. Большинство металлических элементов, за
исключением таких нетипичных металлов, как висмут и сурьма, имеют
кристаллические структуры, лишь слабо отклоняющиеся от кубической или
гексагональной плотнейшей упаковки твердых шаров. Уран является исключением.
Вероятно, вследствие сильной атомной связи в решетке между атомами урана и его
четырьмя ближайшими соседями атомы урана в а-фазе ведут себя как несфери-
ческие тела. Следовательно, атомный радиус а-урана нельзя точно определить.
В этой фазе каждый атом^ урана окружен двумя ближайшими соседними
атомами на расстоянии 2,75 А (при комнатной температуре), двумя другими—на
расстоянии 2,85 А, четырьмя—на расстоянии 3,25 А и еще четырьмя—на
расстоянии 3,34 А. Таким образом, можно приближенно считать, что атомы урана
имеют эллипсоидную форму с малой полуосью 1,4 А и большой—1,65 А.
Паулипг [16] предположил, что а-уран содержит прямые цепи прочно
связанных атомов, так что его структура подобна предложенной этим же автором
для ^-вольфрама. С другой стороны, существует мнение [11], что а-уран
имеет структуру, состоящую из гофрированных слоев. Связи внутри слоев
сильнее, чем между слоями, как это наблюдается и для структур мышьяка,
сурьмы и висмута. Кроме того, связи внутри слоев заметно зависят от направления.
Атомный радиус урана в кубической ?-фазе, вычисленный из константы
решетки, экстраполированной к комнатной температуре, равен 1,485 А-
118
Часть II. Металлический уран
Плотность. В табл. 43 приведены значения плотности, рассчитанные
на основании структуры решетки а-урана. Наиболее вероятное значение равно
19,050 г/см3. Если бы -[-фаза существовала при комнатной температуре, то ее
плотность должна была бы составлять 18,7 г/см*. Экспериментально
определенная плотность урана часто немного ниже значения, вычисленного из
рентгенографических данных для а-фазы, что, вероятно, обусловлено присутствием
примесей, особенно углерода. Показано, что плотность металлического
,___ УРана пропорциональна содержа-
"] | i j 1 1 | ^ | | нию углерода 117]. Эта
зависимость может быть выражена
уравнением
р= 19,05 —2,14[С], (1)
где [С]—содержание углерода, %
(вес). Наибольшее из измеренных
значений составляет 19,02 г/см3
(0,01 %С), наименьшее—18,58 г/см3
(0,23% С). Путем экстраполяции
к [С] = 0 вычислено, что плотность
чистого урана равна 19,05 г/см*. Это
хорошо совпадает со значением,
рассчитанным из
рентгенографических данных. Другие измерения
Таблица 44
КОЭФФИЦИЕНТ ЛИНЕЙНОГО
РАСШИРЕНИЯ с-УРАНА В ТРЕХ
ГЛАВНЫХ КРИСТАЛЛОГРАФИЧЕСКИХ
НАПРАВЛЕНИЯХ
200 400 600
Температура. °С
800
Направление,
параллельное
оси
а (100)
Ь (010)
с (001)
Средней коэффгцгснт линей*
нот расширения,
1 0ьсм-см~1'град~"1
25-300°
23 ±3
-3,542
17 ±2
25—650е
28 ±2
-1,4±1
22 ±1
Р и~с. 9. Зависимость констант решетки а-урана
от температуры.
дали значения для плотности урана от 18,7 до 19,08 г/см9 [18 — 23]; более
низкие цифры относятся к образцам, содержащим 0,05— 0,06% углерода [24].
Средняя плотность двадцати двух катаных прутков технического а-урана
равнялась 18,882 г/см3 [20, 21].
Плотность урана, полученного спеканием порошка металлического урана,
составляет всего 15 г/см3, после прессования она возрастает до 17 г/см3 [25].
Насыпной вес порошка урана после прессования под давлением 2,8 т/см2
равен 9,4 г/см3 [26], под давлением 11,1 т/см2—12,3 г/см3, под давлением
19,6 т/см2—15,0 г/см3.
Термическое расширение. Линейный коэффициент
термического расширения урана в анизотропной а-форме сильно зависит от
кристаллографического направления [27—29]. Табл. 44 и рис. 9 иллюстрируют
эту зависимость, определенную рентгенографическим методом при высоких
температурах [29, 30]. Объемный коэффициент расширения, вычисленный из
Глава 5. Физические свойства металлического урана
119
Таблица 45
ОТЖИГ НАКЛЕПАННОГО
УРАНА [37Jn)
определенных дилатометрическим методом линейных коэффициентов, равен
44 10е см3-см'6-град 1 в области 25—300°. Из рентгенографических данных
табл. 44 вычисляется объемный коэффициент, составляющий 37-Ю"6; это
более или менее удовлетворительно согласуется с результатами
дилатометрических измерений.
МЕХАНИЧЕСКИЕ СВОЙСТВА УРАНА
Механические свойства урана подробно рассмотрены в другом томе
данной серии [31J. Здесь же будут приведены только наиболее важные данные.
Твердость. Уран, отлитый при 1200°, обычно имеет поверхностную
твердость около 100 по Роквеллу (шкала В), но уже на глубине в 1,25 мм она
уменьшается до 85 RB [32]. Металлический уран с максимальной твердостью
(70—71 /?л, что соответствует 115 Rn) может быть получен нагреванием урана
при 900° в течение 5 час. или более с
последующей закалкой в холодной воде [33]. Такое
повышение твердости, возможно, обусловлено
присутствием в металле карбида [34]. Найдено,
что средняя твердость литого рафинированного
металла равна 200—220 по Бринелю [34].
Уран подвержен наклепу при штамповке
или прокатке; при этом его поверхностная
твердость может возрасти, например, от 90 до \\5Rb
(т. е. от 57 до 71 Ra) [35, 36]. Были проведены
опыты, в которых удалось уменьшить толщину
металла высокой чистоты на 80% без
промежуточного отжига или снятия напряжений [34].
Основная часть наклепа происходит при нервом
обжатии на 20%. Наклепанный металл начинает
размягчаться при 150°, как видно из табл. 45;
полный отжиг наклепанного металла достигается
при 650—700° [35, 37—39]. Ряд наблюдений показывает, что для полного
уничтожения наклепа требуется отжиг по меньшей мере при 600° [40].
Проведены измерения микротвердости урана методами Кнопа и Эбер-
баха[41—45J. В табл.46 представлены некоторые результаты этих измерений
141, 44].
Темперах
°С
310
490
600
720
770
Ура,
Твердость по
Роквеллу
(шкала С)
36,6
24
22
9
0
а) Образцы отжигали в течение
1 часа при эгих температурах.
Таблица 46
ТВЁРДОСТЬ КАТАНОГО УРАНА
Шкала
Роквслл В
Рокнслл G
Виккерс, с алмазной пирамидон (на-
Виккерс, с алмазной шфамндой (на-
Бринель, вычислено из значения Rb
» » » » RG
» » 500 г
» » 1000 г
Сечен не обра ща
поперечное
97
80,5
220
240
250
260
250
240
270
260
продольное
98
83
240
250
260
280
320
300
320
300
290
120 Часть II. Металлический уран
Твердость урана уменьшается с возрастанием температуры [46—48];
до 200° она изменяется очень мало, но в интервале 200—650° твердость по Бри-
нелю падает от 252 до 13. Затем она опять возрастает до 30—40 по этой же
шкале при переходе урана в (^-состояние (660—700°). Твердость пластичного
Tf-урана (выше 770°) настолько мала, что ее нельзя измерить по Бринелю,
Упругая и неупругая деформация. Уран в а-состоянии
был выше назван полупластичным; действительно, его кривая нагрузка—
деформация часто имеет кривизну даже при нагрузках ниже 7 кг/мм2, [49,50].
С другой стороны, получены некоторые кривые нагрузка — деформация,
обладавшие прямолинейностью до 35 кг/мм2 [51]. Однако модуль
упругости, рассчитанный из наклона этих линейных отрезков, был так мал
40
35
с 30
К 25
«
его
I
<0
5
"0 200 400 600 800
Температура, °С
Рис. 10. Зависимость предела пропорциональности урана от
температуры.
(приблизительно 3500 кг/мм1), что линейность, вероятно, была только
кажущейся, т. е. она была вызвана недостаточной точностью измерений при низких
напряжениях. Бо.г.ее надежными можно считать определения [24, £3, 52—59],
которые показали линейность кривых нагрузка—деформация в пределах
7—10,5 кг/мм2 и обладали начальным наклоном, на основании которого
вычисленный модуль упругости составлял 10 500—17 500 кг/мм2. При
использовании динамического метода модуль продольной упругости оказался равным
20 900 кг/мм2 с возможной ошибкой 1% [60]. Для а-фазы вследствие ее
анизотропности модуль упругости, вероятно, зависит от кристаллографического
направления. Имеющиеся данные по измерению модуля упругости а-уд_ана
недостаточны для установления точной зависимости, но немного более низкое
значение модуля упругости (10 500—14 000 кг/мм2), полученное, повидимому,
для продольных образцов катаного металла, может быть следствием
ориентации кристаллов, обусловленной наклепом [37, 61, 62]. Упругость урана, как
и твердость, быстро уменьшается с температурой в а-фазе, но вновь возрастает
при переходе в {з-фазу [51, 63] (рис. 10). В литературе приводится значение
6300 кг/мм2 для модуля упругости урана при 20° [64], но, к сожалению,
неизвестно, с каким металлом проводилась работа, а поэтому ценность этого
измерения сомнительна.
Для модуля сдвига Снайдером и Каммом найдены значения 4620 кг/мм*
для холоднотянутой и 4760кг/мм1 для отожженной урановой проволоки [61,651.
Глава 5. Физические свойства металлического урана
121
Лякер дает величину для модуля сдвига, равную 8470 кг/мм2, с точностью
0,5% [60], что почти вдвое больше предыдущих значений.
Модуль сдвига для образцов с модулем упругости 13 300 кг/мм2,
определенный Волланом и Стефенсоном [66], равен 5950 кг/мм2.
Измерения Снайдера и Камма дают следующие значения объемного
модуля упругости: для наклепанного металла 6300кг/лш2, для отожженной
проволоки 10500 кг/мм2. Эти значения соответствуют коэффициенту
сжимаемости 17,1-Ю-7 и 9,1-Ю-7 см2/кг. По Бриджмэну [67], средний коэффициент
сжимаемости урана, вероятно, не очень чистого, при 12 000 am и 30°
равен 9,7-10"7 см2/кг. В более поздней работе этого же автора [68]
показано, что объем металлического урана уменьшается на 5,9% при давлении
в 100 000 кг/см2, в этих же условиях объем тория уменьшается на 12,4%,
а объем калия на 50%.
Коэффициент Пуассона (отношение относительного поперечного сжатия
к относительному продольному удлинению) является высоким в соответствии
с полупластичной природой урана. Для технического урана, выдавленного
в ^-модификации, этот коэффициент составляет 0,32—0,43 [55,56]. Для
катаного а-урана, гыдержанного свыше 12 час. при температурах 565, 750 и 800°,
он равен соответственно 0,42, 0,49 и 0,27 [61]. Коэффициент Пуассона,
вычисленный на основании измерений Снайдера и Камма, равен 0,20 для
наклепанного металла и 0,31 для отожженной проволоки. Лякер [60] приводит
для коэффициента Пуассона значение 0,23, что хорошо согласуется с
предыдущими. Модули упругости и сдвига, найденные Волланом и Стефгнсонсм [66],
соответствуют неправдоподобно низкому значению коэффициента Пуассона,
а именно ~ 0,1.
Вычислено, что скорость звука в а-уране равна 1500 м/сек [22]. Она,
вероятно, зависит от кристаллографического направления.
Будучи пол^пластичным, а-уран не имеет определенной точки начала
текучести. В связи с этим предел текучести [24, 34, 52—54, 56, 57, 63, 69—711
определяется как напряжение, при котором длина растягиваемого образца
превышает на известную величину (на величину остаточной деформации,
составляющую обычно 0,02, 0,1 или 0,2%) ту длину, которую имел бы образец,
если бы деформация была упругой (т. е. если бы кривая
нагрузка—деформация была линейной). Вследствие упоминавшейся выше некоторой
неопределенности начального наклона кривой нагрузка—деформация (т. е. значения
модуля упругости) определение предела текучести по этому методу очень
неточно. Различия в значениях предела текучести, найденные американскими
и английскими исследователями, очень велики. Большинство значений,
приведенных для предела текучести урана при комнатной температуре, находится
в пределах от 17,5 до 24,5 кг/мм2 для остаточной деформации, равной 0,2%
[24, 52, 53], однако отдельные значения колеблются в пределах от 5,6—9,1
до 53 кг/мм2 [56].
Наибольший предел текучести наблюдается для обработанного а-урана[53].
Медленное охлаждение после отжига уменьшает предел текучести [71], а
закалка, особенно от температур ^-фазы, увеличивает его. Например, предел
текучести при испытании на растяжение (при остаточной деформации, равной
0,2%) у ряда катаных образцов а-урана составлял 17,5 кг/мм2 для
материала, охлажденного в печи, 22,4 кг/мм2 для образцов, закаленных от 570°,
и до 29,4 кг/мм2 для образцов, закаленных от 800° [52]. В другом примере
предел текучести (для остаточной деформации 0,1%) возрастал от 11,9 кг/мм2 для
необработанного катаного образца до 39,9 кг/мм2 для подобного же образца,
закаленного от 1000° [70]. Предел текучести а-урана быстро уменьшается с
возрастанием температуры [63, 72, 73], например от 30,1 кг/мм2 при комнатной
температуре до 6,2 кг/мм2 при 600°. На рис. 11 видно, чтор-уран имеет
определенный предел текучести: 12,6 кг/мм2 при 700° и 9,1 кг/мм2 при 750° [51].
122
Часть //. Металлический уран
Подобно кривым нагрузки, кривые разгрузки урана также нелинейны [55],
так что остаточная деформация после разгрузки меньше остаточной деформации,
измеренной под напряжением. Остаточная реформация после сжатия
урановых цилиндрических образцов была измерена в трех лабораториях [33, 51,74].
Таким образцам сжатием придавали бочкообразную форму без повреждения их,
причем высота при этом уменьшалась на 35%. Давление, которое требуется
Деформация, мм/мм
Рис. 11. Сжатие урана при 700 и 750°.
для сообщения остаточного сжатия в 10%, уменьшается с температурой
[51, 72] от 78 кг/мм1 при 25° до 4,9 кг/мм2, при 650°, но в области р-фазы
возрастает вновь до 11,9 кг/мм" при 700° и до 8,4 кг/мм2 при 750° [51 ].
Деформация под напряжением и остаточная деформация после снятия
напряжения зависят от продолжительности нагрузки; этот факт указывает
на явления ползучести. В табл. 47 приведены результаты измерений
ползучести урана [75].
Предел прочности урана при испытании на
растяжение. Предел прочности урана при испытании на растяжение
колеблется от 35 до 140 кг/мм2 в зависимости от наклепа и термообработки образца
[21, 37, 38, 41, 49, 53, 57, 74]. Наклепанный металл (катаный или кованый
Глава 5. Физические свойства металлического урана 123
Таблица 47
ПОЛЗУЧЕСТЬ УРАНА
Температура,
°С
350
356
356
355
355
353
Нагрузка,
K3JMM-
6,33
2,46
3,71
2,48
3,70
6,20
Продолжительность
ИСПЫ ГаННЯ,
часы
260
263
934
-1000
1000
1000
Среднее
удлинение за 100 час, %
1,3
0,19
0,07 (300 час.)
0,037
0,089
0,22
а-уран) обладает наибольшим пределом прочности, а именно: 120—140 кг/мм?
[37, 38, 41, 491. Отжиг уменьшает предел прочности", например от 120 кг!мм*
после ковки до 95 кг/мм2 после отжига при 625° и до 45,7 кг/мм2 после отжига
при 830° [41, 57]. Литые или выдавленные в ? модификации образцы урана
имеют почти такой же низкий предел прочности, как и отожженные в 7~мо-
дификаиии и медленно охлажденные образцы того же металла, а именно
приблизительно от 45,7 до 63,3 кг/мм2 [34, 46, 54, 57, 76, 77]. Предел прочности
немного возрастает при закалке, в особенности от высоких температур в а- или
^-области; для катаных, кованых или штампованных образцов, закаленных от
600 до 1000°, были получены значения приблизительно от 63,3 до 91,4 кг/мм2
[21, 49, 53, 59].
Предел прочности быстро уменьшается при повышенных температурах:
от 37,3 кг/мм1 при комнатной температуре до 19,0 кг/мм2 при 150° и
8,4 кг/мм2 при 600° [78].
Зейтц [79] исследовал вопрос о причине низкого отношения между
пределом прочности при испытании на растяжение и модулем упругости урана,
а именно 140 :14 000=0,01, тогда как теоретическая величина равна
приблизительно 0,3. Он объясняет указанное расхождение наличием в металле
мельчайших трещин, значительно снижающих площадь приложения нагрузки.
У наклепанного металлического урана относительное удлинение и сужение
при испытании на растяжение малы, а именно 0—3% [34, 57] и 0—8% [53]
соответственно. Они несколько выше для урана, выдавленного в
-[-модификации, а именно 5—10% и 3—15% [54, 77]. Вязкость значительно выше
у катаного а-урана, подвергнутого отжигу и закалке от высоких температур
а-области (350—600°); в этом случае удлинение при разрыве и сужение шейки
доходят до 20%. Закалка от температур (3- или 7*Фазы (700—800°) вызывает
падение этих величин до 10% или меньше. Испытания на копре показывают,
что вязкость в низкотемпературной области ^-фазы (660—670°) очень мала;
при 705° она несколько выше [80].
Ударная вязкость по Шарпи трех урановых образцов литого металла
равнялась 1,9—2,3 кгм [81]. Для литого металла приводится также значение
0,7 кгм [57], т. е. такая же величина, как для выдавленного металла, или
немного ниже [34].
Измерена усталостная прочность выдавленных образцов а-урана на кон-
сольно-балочной машине Краузе на 10 000 об/мин [82]. Предел усталости
лежит в области 17,6 кг/мм2; при 24,5 кг/мм2 образцы разрушались через
1,2- 10б—2,5-105 циклов. Предварительная выдержка под нагрузкой 17,6 кг/мм2
увеличивала предел усталости. Отношение предела усталости к пределу
прочности на растяжение составляет около 0,2, что для металла является низкой
величиной.
124
Часть II, Металлический уран
Измеряли также прочность урановых дисков на продавливание, для чего
их клали на опорное кольцо и разрушали с помощью п>ансона (диаметром
12,7 мм). Среднее значение прочности составляло 1781 кг для урана,
выдавленного в 7-м°/ш4икации, и 2108 /сг для выдавленного а-урана [831. Отжиг
при 600—800° в течение 15 час. несколько снижал величину прочности, отжиг
же при температурах выше 900° с последующей закалкой значительно
увеличивал ее [84].
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА УРАНА
Теплоемкость, энергия и энтропия. Результаты измерений
теплоемкости урана при низких и высоких температурах [85 — 88]
представлены в табл. 48, 49 и на рис. 12, 13. Постоянная Дюлонга - Пти оказывается
превзойденной уже при такой низкой температуре, как —73°.
Таблица 48
ТЕПЛОЕМКОСТЬ УРАНА (15-300° К)
Температура,
°К
15,38
16,61
18,55
20
21,01
23,91
27,55
31,19
35,32
40
40,10
45,27
50,49
55,53
60
60,63
65,78
71,57
75,99
80
Теплоемкость,
кал -MO.ib'l- град" 1
°р
0,432
0,496
0,665
0,914
1,211
1,639
2,080
2,530
2,954
3,369
3,731
4,011
4,231
4,446
4,657
4,807
Cv 1
0,81
2,94
4,20
4,90
Темпе-
paTvoa,
°К
81,52
87,05
92,96
98,93
100
104,81
111,00
117,40
123,48
128,23
134,58
140,90
147,06
!50
153,35
159,76
166,65
173,61
180,04
186,37
Теплоемкость,
кал • моль-l -град-1
С \ С \
4,955
5,072
5,194
5,325
5,435
5,532
5,591
5,639
5,697
5,754
5,823
5,874
5,924
5,964
6,008
6,053
6,091
6,131
5,33
5,86
Температура,
°К
192,57
198,47
200
205,12
211,07
217,76
224,89
231,88
238,73
245,59
250
252,89
, 259,36
266,32
273,02
280,16
287,50
294,88
297,71
300
Теплоемкость,
кал - моль~1 >град~1
СР
6,154
6,203
6,232
6,274
6,295
6,335
6,348
6,381
6,417
6,441
6,476
6,486
6,524
6,544
6,548
6,556
6?574
Cv
6,17
6,39
6,51
Лонг с сотрудниками [85] опубликовали только значения Ср; Мур и
Келли [86, 87] сообщают величины hj — #298.16; Джиннингс и Корручини
[88]—Ееличины ИТ - #273,16; Ср и ST — S273.i6- Значения Сг в табл. 48
вычислены Симоном [221 - В табл. 49 приводятся два ряда данных для Ср, Нт и S:
одни вычислены Симоном [22J, другие Джиннингсом и Корручини [88]. При
вычислениях С, и 5 для температур ниже 15° К Симон использовал функцию
Дебая с 0= 162°К. Значения, вычисленные Симоном для Нт и для Ср при
300 —500° К, не согласуются между собой. Это, вероятно, вызвано желанием
создать плавный переход от величин Ср табл. 48 к величинам табл. 49.
Для теплоемкости урана предложены следующие уравнения [87]:
для а-урана Ср = 3,15 + 8,44.10"3r+0,80.105Г2; (2)
Таблица 49
ТЕПЛОЕМКОСТЬ, ТЕПЛОСОДЕРЖАНИЕ И ЭНТРОПИЯ УРАНА (300-1375* К)
Температура,
°К
300
400
500
600
700
800
900
935 (а)
941 (а)
935 (й
94 J (3)
1000
1045(3)
1047(»
1045(7)
1047(Т)
1100
1200
1300
1375
Ср, кал-г-атом~1'град~1
Симон
[22]
6,6
7,0
7,5
8,1
8,9
9,9
11,0
11,3
10,2а)
10,2а>
9,2б)
9,2б>
Корручини
[88]
6,649
7,072
7,606
8,227
8,952
9,863
11,107
11,737
10,147 ,
10,147
10,147
9,147
9,147
Hfi кал
Симой
[22]
1440
2 160
2 940
3 730
4 580
5 535
6 975
7 640
8 770
9 940
10 440
11 350
12 260
13 000
•г-атом' i
Корручини
[88]
1539,12
2 202,57
2 935,42
3 725,45
4 582,96
5 520,47
6 566,80
7 066,16
7 740,37
8 336,97
8815,97
9 947,04
10 430,16
S, энтропийные единицы
Симон
[22]
12,07
13.97
15,60
17,04
18,35
19,62
21,29
22,00
23,14
24,12
24,55
25,38
26,12
26,8
Корручини
[88]
12,052
13,941
15,601
17,056
18,387
19,646
20,882
21,436
22,152
22,760
23,236
24,316
24,761
а) Значение для области 935 О)—104 5° (р).
б) Значение для области 1045 (i)—1300°.
100 150 200
Температура, °К
300
Рис. 12. Удельная теплоемкость урана при низких температурах.
126
Часть 11. Металлический уран
для [3-урана
для т-урапа
Ср= 10,38;
СР = 9,Ю;
для а-урана (при 25° С)
5296,16= 12,03 ± 0,03 энтропийной единицы [86];
для р-урана [88] (при 941-1047° К)
Sr-5273.i6 = 23,362 IgT-58,770;
для т-урана [88] (при 1047-1170° К)
Sr-S273.i6 = 21,062 ]gT-50,745.
(3)
(4)
(5)
(б)
7 12X1
110
? 10,0 У—
I ад
I
а
§ 6,0
Е§ 200
—1—г—i—1—1—I—i—
Г А
Е /1-
1 х
Е / ''
Е //
Е />
1 f_J l... I I I I
1—Г
i
J L
1 1
1
muoo
11000 Ц
9000^
7000^
acts
5000%
§
С:
4ДО
/200
/400
/000
£00 <500 /000
Температура, °К
Рис. 13. Удельная теплоемкость урана при повышенных
температурах.
— удельная теплоемкость; теплосодержание.
Кроме того, для всех трех фаз урана Муром и Келли [87] предложены
уравнения, основанные на экспериментальных данных:
для а-урана (при 298 - 935° К)
Яг-Я298лб-3,157, + 4,22 10-3Г2-0,80*105Г-1-1046(±0,2%);
для бурана (при 935- 1045° К)
Яг-Я298,1б= 10,38Г-3525(±0,1%);
для т-урана (при 1045 -1300° К)
Яг-Я298.1б = 9,10Г-1026(±0,1°/о).
Подобные же уравнения даны Джиннингсом и Корручини [88]:
для р-урана (при 941 — 1047°К)
//г-//273.16= -3170,52+10,147Г(± 0,1%);
для т-урана (при 1047— II73°К)
//г-//27з.1б= -992,80 f 9,147Г(±0,1%). (9а)
Эти же авторы установили, что а-обл^сть урана в интервале 273 —941° К
не может быть охвачена простым уравнением.
(7)
(8)
(9)
(8а)
Глава 5. Физические свойства металлического урана 127
Превращение твердых фаз Температуры превращения а-, |3-
и f-фаз урана определены путем термического анализа, а также из
дилатометрических кривых и кривых электропроводности (рис. 14). Наиболее
достоверные значения температур превращения представлены в табл. 50.
0 100 200 300 400 500 600 700 800 900
Температура, °С
Рис. 14. Электрическое сопротивление урана (по произвольной шкале)
как функция TCMntpai>pbi.
На рис. 14 показано, что .аллотропические превращения в выдавленном
а-уране, а также в литом и спеченном уране обычно задерживаются и при
охлаждении и при нагрерапии. С другой стороны, в выдавленном *[-УРаие
превращение начинается без задержки. Однако даже в таком металле
превращение а—>р не строго изотермично, а проходит в некотором интервале
температур, что может быть обусловлено неоднородностью внутреннего
напряжения [46]. Поскольку при температуре превращения р-фаза менее плотна,
чема-фаза, рост давления будет вызывать повышение температуры превращения.
128
Часть П. Металлический уран
Таблица 50
ТЕМПЕРАТУРЫ ПРЕВРАЩЕНИЯ УРАНА
Материал
г
Кованые круглые
прутки литого
металла диаметром
5 мм
Не указан
Уран, содержащий
0,03% С
Уран, содержащий
0,39о/0 С _ . ,
Уран, содержащий
1,5% С
Не указан
Выдавленный пруток
(продольный
образец, обработанный в
-(-модификации) . .
Выдавленный пруток
Выдавленный пруток
(99,9%-ный уран,
обработанный в
^-модификации) ....
Экстраполировано
для чистого урчна
из значений, полу
ченкых для 98,6—
99,9%-но го ме-
Металлический уран
(99,9%-ный) . . .
Температура превращения,
нагревание
а.->Р
*
,„
673
6G2±3
—
•
665
660,5±1,5
665—670
668—676
669-671,5
644±4
667
Р-т
775
772±3
—
__
—
776
—
766-776
775-783
779-785
765±10
772
°с
охлаждение
WP
770
—
775
785
766
770
—
760—770
767—760
765
752+3
764
р-*«
~~—
—
645
652
633
644
—
643-651
644—639
642-643
624±3
645
Метод определения
Термические
измерения
Измерение
теплосодержания
Термические
измерения
То же
»
Метод
электросопротивления
Термические
измерения
Метод
электропроводности
Дилатометрический мегод
То же
Термические
измерения
Метод
электросопротивления
ратура
59
86
89
90
55
91
28
8
34
92
Влияние содержания углерода на температуры превращения, показанное
в табл. 50, незначительно [91J, но другие присадки могут влиять на эти
температуры гораздо сильнее. Выше упоминалось, что ^-фаза может быть
сохранена в этих сплавах при комнатной температуре путем быстрого
охлаждения. Излучательная способность урана резко изменяется при
температуре 1048 — 1050° |93J; возможно, что это явление обусловлено третьим
Глава 5. Физические свойства металлического урана 129
аллотропическим превращением. Действительно, имеются сообщения о
наличии еще и третьего аллотропического превращения, но при температурах
значительно ниже 1040° [48].
При превращении а—>р поглощается 665 кал• г-атомг1, при
превращении р—>? 1170 кал-г-атомг1 (см. табл. 49). Мур и Келли [87] дают
для второго превращения значение, близкое к указанному, для первого,
однако, они предлагают значение 680 кал-г-атом'1, заметно отличающееся
от вышеприведенного. Кроме того, по их данным, температура превращения
а—>(3 равна 662°, тогда как по данным большинства других исследователей
она выше. Энтропия первого превращения равна 0,71, а второго 0,98
энтропийной единицы [86].
Температура и теплота плавления урана. Долгое
время считали, что металлический уран имеет высокую температуру
плавления. Это мнение, повидимому, находилось в согласии с положением урана
в периодической системе, а именно в самой нижней части подгруппы, занятой
чрезвычайно тугоплавкими металлами—хромом, молибденом и вольфрамом.
Экспериментальные определения дали для температуры плавления урана
величины 1690°* [18] и 1700±25° [94]. Однако вскоре в связи с развитием работ
по производству чистого металла было установлено, что эти значения
завышены [95]. По определению английских исследователей, температура
плавления урана равна 1150° [96]. Более поздние определения дали значения 1105—
1116° для урана 99,8 и 99,9%-ной чистоты [34, 57]. В Америке вначале для этой
температуры было найдено значение 1300° [97, 98], но последующие, более
точные измерения [99] привели к еще более низким значениям, чем
английские, а именно 1080±20° для 99,1%-ного урана и1125±25°для 99,7%-ного
металла. Другие величины лежат в интервале от 1123° (1,5% С) до 1134°
(0,03%С) [89]. Из диаграммы состояния системы уран—алюминий
экстраполяцией найдено значение 1125° [100].
Повидимому, наиболее точной можно считать работу, проведенную в
Национальном бюро стандартов [101], в которой было установлено, что уран,
содержащий 0,03—0,05% углерода, вначале плавится при 1125°, но если металл
выдержать в течение 15 час. при температуре, очень мало превышающей его
точку плавления, то температура затвердения металла постепенно повышается
до ИЗО—1132°. За это время содержание углерода уменьшается до 1%
первоначальной величины (от 0,05% до 0,0005%), а остающийся углерод
сегрегируется в виде карбида в образующейся на металле корке. Таким образом,
можно отнести рост точки плавления за счет самоочистки металла.
Следовательно, наиболее вероятным значением для температуры плавления чистого
урана будет
^пл=1133±2°С;
ГПЛ=1406±2°К.
Вычислено, что теплота плавления урана Д#пл составляет от 2,5 до
3,0 ккал-г-атом"1 [22]. Отсюда значение энтропии плавления AS=2
энтропийным единицам.
Давление паров и теплота парообразования.
Все известные измерения давления паров урана носят предварительный
характер, и найденные значения сильно колеблются. Измерения, проведенные по
методу определения «скорости испарения» [102], дали значения 0,0013 мм рт.
ст. при 1300°, 0,0044 мм при 1620°, 0,026 мм при 1900° и 2,4 мм при 2300°.
* Впоследствии неправильная температура плавления, найденная при этом
определении, была приписана не недостаточной чистоте металла, а случайному выбору
неудачного метода определения (пирометрического).
9 Химия урана
130
Часть II. Металлический уран
Кривая зависимости lg р от — на рис. 15 ясно показывает, что первые два
значения сильно завышены. На рис. 15 величины давления паров основаны на
измерениях «скорости молекулярного истечения» [103] .При новых измерениях по
этому методу [104] были получены три точки с танталовыми тиглями и четыре—
-. «£ с тиглями из окиси бериллия.
2680 2230 Ю50 1730 *1550 1400 1270 ^°РЫ ЭТ0Г0 исследования счи-
/2р ==j 1 j 1 1 1 тают три измерения,
проведенные с танталовыми тиглями,
более верными, чем все остальные;
однако наклон кривой давления
паров (рис. 16), проведенной
через одну из этих точек и
среднюю для двух других,
безусловно весьма неточен.
Симоном [22] были сделаны
попытки построить кривую
давления паров урана с
использованием только одной
экспериментальной точки (р = 0,05 мм
рт. ст. при 2200° К),
являющейся «центром тяжести»
результатов других исследований [102,
103] в сочетании с
теоретическими данными.
Этот автор вычислил
следующее значение для энтропии:
SS<=-36,3 + 20,71gr (Ю)
Рис. 15. Давление паров урана при 1300—2300°.
Литературные источники: О — [102]; П—[103]: ■ —[22];
А — [Ю4] (танталовые тиглн); #—[104] (тигли из окиси
бериллия).
и вышеупоминавшеися эмпирической точки
дено уравнение
IgP
100-103
4,57Г
2,04 \ёТ+ 15,4,
Sr°as = 4,57 (2,5 lgT-
-lgPam* + 3,66) (11)
(в последнем уравнении для
фактора симметрии было
использовано значение lgg = 0,6). С
помощью этих значений энтропии
на кривой давления паров выве-
Таблица 51
ДАВЛЕНИЕ ПАРОВ УРАНА
(12)
где давление дано в
миллиметрах ртутного столба. Из
этого уравнения вычислены
давления, представленные в
табл. 51.
На рис. 16 видно, что
кривая, проведенная на
основании полутеоретических
значений, лежит между двумя
другими [104].
Температура, °к
(4200)а)
2200
1900
1700
Давление,
мм рт. ст.
(760)
5.10'2
2-Ю-3
МО"4
Температура, °К
1500
1375
1200 6)
1100 6)
Давление,
мм рт. ст.
3-10-*
2. Ю-7
ью-»
3-Ю-11
а) Рассчитанная температура кипения.
б) Эти значения рассчитаны для твердого Y-урана.
Глава 5. Физические свойства металлического урана
131
1949
'2,60
Температура, °С
1727 1545 1394 1265
1156
Рис. 16. Давление паров урана.
Литературные источники: ■ —[22], полутеоретические значения;
А —[Ю4] (танталовые тигли); # — [104] (тигли из окиси бериллия).
Теплота парообразования, рассчитанная из наклона кривых зависимости
Ыр от f~y следующая [22, 104]:
А, ккал -моль-1
;.0i ккал'Моль-i
93 -100
58 (тигли из тантала) —
137 (тигли из окиси бериллия) —
Литература
22
104
104
Из рассмотрения этих кривых можно сделать лишь следующий вывод:
уран не только плавится на 2240° ниже вольфрама, но также значительно более
летуч (теплота парообразования вольфрама равна "около 175 ккал-г-атом'1).
ЭЛЕКТРОННЫЕ СВОЙСТВА УРАНА >
Электропроводность. Уран является довольно плохим
проводником. Его электропроводность примерно вдвое меньше, чем у железа.
Значения удельной электропроводности, найденные различными исследователями,
лежат между 2-104 и 4-104 (ом-см)'1. Наиболее высокая электропроводность
при комнатной температуре и наибольший рост ее при охлаждении до
температуры жидкого воздуха доказывают высокую степень чистоты металла.
9*
132
Часть II. Металлический уран
Таблица 52
ЭЛЕКТРОПРОВОДНОСТЬ УРАНА
Материал
Катаная урановая
проволока диаметром 0,75 мм
Цилиндр (диаметром
63,5 мм) из слитка,
отожженного в
течение 10 мин. при 600° .
Обжатая проволока
(диаметром 1— Ьмм),
обработка и материал те же
Катаный американский
металл 99,93%-ной
чистоты после
продолжительного отжига
Тот же металл без
Английский металл
99,86—99,90%-кой
чистоты после
продолжительного отжига
Тот же металл без от-
Катаная полоса урана
99,8— 99,9%-иой чи-
Тот же металл,
кованый и обточенный . .
ратура, ?С
—
25
25
0
0
0
0
23
23
Удельная
электропроводность
(К) -10-4,
3,1
2,44
4,0
3,6—3,8
3,47
3,35
3,11—3,38
2,92—3,13
3,1—3,2
3,0
Удельное
электросопротивление
(р)-106, ом-см
32,1
41
25,0
26,2—27,5
28,8
29,9
29,6—32,2
29,8—34,3
31—32
34
Температурный
коэффициент для
Р(а).103на 1°С
2,1а)
—
2,78
2,70—2,82
—
—
—
—
—
2,0
*273°К
^90°К
—
—
—
2,6
2,3
2,5
1,9—2,0
—
2
Литература
18
61,62
91
91
34,105
34,105
34,105
34,105
57
57
а) 23—140*.
Табл. 52 показывает, что самая высокая величина электропроводности
(4-104) обнаружена у образцов металла в работе, выполненной
Национальным бюро стандартов в 1943 г., но после этого, вероятно, удалось получить
еще более чистый металл. Наибольшее значение отношения ^273°к равно 2,6;
К
90°К
оно получено для образцов с удельной электропроводностью 3,5-104 (ом-см)"1
при 0°. Это отношение еще значительно ниже теоретического
( #273°К _о
i,3-s-4,0),
^ ^90°К
рассчитанного для чистого урана из его температуры Дебая [22].
Длительный отжиг в наиболее высокотемпературной зоне а-области (при 600°),
который проводился для снятия напряжений после механической обработки,
Глава 5. Физические свойства металлического урана
133
несколько улучшил электропроводность при комнатной температуре и, в част-
IS
ности, вызвал рост отношения „273 к , но все же не в такой мере, чтобы
^90°К
достигнуть теоретического значения.
Установлено, что электропроводность уменьшается при отжиге в (3- или
а-области (см. также табл. 53, последняя графа [74]). Для одного образца
урана 99,9?/о-ной чистоты Эбертом и Шульце [106] найдена удельная
электропроводность 3,45» 104 (ом-см)"1. Это значение, так же как и температурный
коэффициент электросопротивления 2,18-10~3, в общем совпадает с данными
других литературных источников. Это же верно и для значения,
определенного Бальцем [107]. Проведено измерение электропроводности образцов
урана, содержащих примеси других элементов: 0,03% Са, 0,02% Hg, 0,002%
Sn, 0,14% Si, 0,19% Си, 0,60% Fe, 0,04% А1 и 0,30% С при 13,9—373° К [108].
Подробные данные не опубликованы, но указывается, что результаты
могут быть с точностью до 0,5% представлены следующим уравнением:
Rt = Ra + (1~Ra) 273^0(8/273,2)' ^
где RT=—-—, т. е. отношению сопротивления при температуре 7ю К
Р273,2
к сопротивлению при 273,2° К, a G является так называемой функцией
Грюнэйзена, значения которой можно взять из таблиц. Для 0 было
предположено значение 175° К, которое удовлетворительно совпадает с
характеристической температурой 162° К, вычисленной из измеренных величин
теплоемкости (см. стр. 124). Величина Ra может быть выражена уравнением
. %А = 0,194 + 0,0005117\ (14)
Остаточное электросопротивление 0,194 Р27з,2 гораздо больше, чем можно
было бы ожидать, исходя из наличия 1% примесей, упоминавшихся выше.
Следовательно, взятый металл либо содержал заметные количества водорода
или кислорода (для которых нет данных), либо имел большое число
физических нарушений кристаллической решетки.
Величина остаточного удельного электросопротивления была впоследствии
вновь определена для образца урана 99,86%-ной чистоты [77]. Металл
отжигали 4 часа при 595е, охлаждали с печью и подвергали холодной
прокатке с обжатием на 75%. В отсутствие отжига л р0 =0,221; после отжига
Р273,2
в течение 3/4 часа при 800° и охлаждения до 600° за 2,5 часа, последующего
отжига в течение 8 час. при 600° и охлаждения с печью это отношение
упало до 0,140.
Данные табл. 53 взяты из одного из последних английских отчетов [64\.
Изменение электросопротивления при повышенных температурах
иллюстрируется рис. 14 [90]. Резкие скачки в температурных интервалах
640 — 670° и 760 — 780°* соответствуют превращениям а—>р и Р—>?• Все
кривые отличаются гистерезисом, т. е. перегревом на восходящей ветви
кривой и переохлаждением на нисходящей; возможно, что причиной
гистерезиса является только перегрев или только переохлаждение, но не исключено,
что имеют место и оба эти явления. Температурный коэффициент
электросопротивления падает в a-фазе примерно от 3-Ю"3 при комнатной
температуре до 0,5-Ю"3 при 650° и снижается дальше до 0,14- 1(Г3 в р-фазе [90, 91]-
В более поздних работах Национального бюро стандартов с пятью
* Более низкое значение в каждом температурном интервале соответствует
условиям охлаждения, т. е, превращениям ?->« и ч-*ф, более высокое—превращениям при
нагревании.
"" ~ " " ----- - Таблица 53
ВЛИЯНИЕ ПРИМЕСЕЙ И ТЕРМИЧЕСКОЙ ОБРАБОТКИ НА УДЕЛЬНОЕ ЭЛЕКТРОСОПРОТИВЛЕНИЕ ПРОКАТАННЫХ ПОЛОСОК
Общее содержание урана и важнейших
примесей, %
Fe
Si
Термическая обработка
Удельное электросопротивление
р* 10вг ом-см
20е С
(293°К)
0°С
(273°К)
—70° С
(203° К)
— 183°С
(90° К)
Отношение
Р-183°С
Р 0°С
Отношение
остаточного
электросопротивления
Р-273°С
Р 0°С
Удельная
электропроводность
(при
20°):10-4,
99,83
99,90
99,83
99,87
0,896
0,033
0,034
0,022
0,001
0,038
0,019
Холоднокатаный
Медленно охлажденный
от 800 до 500° и затем
охлажденный на воздухе
Холоднокатаный
Медленно охлажденный
от 800 до 500° с
последующим охлаждением на
воздухе
Отожженный при 600° в
течение 8 час. и
охлажденный в печи
Медленно охлажденный от
800 до 500° и затем
охлажденный иа воздухе
Холоднокатаный
Медленно охлажденный
от 800 до 500° и затем
охлажденный на воздухе
Отожженный в течение
8 час. при 600° и
охлажденный в печи
32,52
34,15
35,95
33,40
34,15
35,50
33,65
35,55
31,45
30,90
32,35
34,20
31,65
32,15
33,60
32,00
33,75
29,60
25,25
26,10
27,85
25,35
25,00
26,95
26,10
27,35
23,10
15,25
15,00
16,95
14,15
12,75
15,75
15,80
15,35
11,95
0,49
0,46
0,496
0,45
0,397
0,47
0,494
0,455
0,404
2,93
2,78
3,00
2,93
2,82
2,97
2,82
3,18
Продолжение табл. 53
Общее содержание урана и важнейших
примесей, %
Fe
Si
Термическая обработка
Удельное электросопротивление
р-10в, ом-cm
2 0° С
(293°К)
0° С
(273° К)
—70° С
(203° К)
— 183°С
(90° К)
Отношение
Р-183°С
Р 0°С
Отношение
остаточного
электросопротивления
Р-273°С
Р 0°С
0,059
0,054
0,052
0,052
0,053
0,0005
0,057
0,14
0,023
0,018
0,004
0,027
0,015
Холоднокатаный
Отожженный в течение
8 час. при 600° и затем
охлажденный в печи . .
Холоднокатаный
Отожженный в течение
8 час. при 600° и затем
охлажденный в печи . .
Холоднокатаный
Отожженный в течение
8 час. при 600° и затем
охлажденный в печи . .
Отожженный в течение
3/4 часа при 800°,
охлажденный за 2,5 часа до
600° и затем
подвергнутый 8-часовсму
выдерживанию при 600° и
охлаждению в печи . .
Холоднокатаный
Выдавленный
34,50
31,60
32,93
28,70
31,30
29,90
29,70
32,90
29,75
31,50
27,20
29,75
28,30
28,20
27,25
23,25
26,30
21,75
24,10
22,50
22,40
17,30
12,00
17,00
11,30
13,56
11,45
11,55
0,526
0,403
0,54
0,416
0,455
0,405
0,410
0,221
0,092
0,140
0,244
0,194
136
Часть II. Металлический уран
образцами очень чистого урана температурный коэффициент
электросопротивления оказался равным 2,76• 10~3 в интервале 0—100° для металла,
отожженного при 600°; для металла, отожженного при 910°, средний
температурный коэффициент в этой же области температур равнялся 2,61 • 10~3
[92]. Это, пожалуй, наиболее точное значение температурного коэффициента.
При высоком давлении (до 12 000 am) сопротивление урана уменьшается
в среднем на 4,36• 10~4% на 1 am [109], однако для этой работы брали
не особенно чистый образец урана, о чем можно судить по его высокому
удельному электросопротивлению при 0° (76-Ю-6 ом-см).
Ниже 1,3° К уран становится сверхпроводником [ПО]. Е. Алексеевский
и Л. Мигунов [111] подтвердили это наблюдение для некоторых образцов
металла, однако они сообщают, что сверхпроводимость отсутствует в случае
менее чистых образцов. Сначала сверхпроводимость урана, отмеченная для
В
о
1*3
и,иос
0,078
0,074
Q070
0,066
0,062
ппья
—
—
1 г~ 1 1 i
о
1 _.!.._ i. . I L ...
1 1
<р
» °
1 1
Т~
О
J_
1 1
о
-J
J
-J
—
1
О 50 ЮО
150 200 250 300 350 400 450
Температура, °С
Рис. 17. Теплопроводность урана.
некоторых его образцов, приписывалась наличию в нем сверхпроводящих
примесей [112], но в более поздней работе [113] было установлено, что
уран действительно является сверхпроводником, однако точки перехода
в сверхпроводящее состояние зависят от степени чистоты металла. Для
проявления сверхпроводимости иногда могут требоваться столь низкие темпе-г
ратуры, как 0,75° К-
Теплопроводность. Первые измерения теплопроводности
урана, проведанные над спеченным металлом, дали значения порядка
50- КГ8 кал -см'1 -сек'1 -град'1 или еще меньше [25, 61, 66, 114]. Другие
измерения [57, 66, 115, 116] приводят к значениям приблизительно от 60-10 3 до
65-10~3 кал-см'1-сек'1-град'1 для комнатной температуры и немного более
высоких (для 30 — 60°). Средний температурный коэффициент теплопроводности,
согласно рис. 17, равен приблизительно 1,5-10~3 на 1° в интервале 100 — 225°
и 0,4-10~3 на 1° в интервале 225 — 450°. Довольно резкий перегиб на
кривой при 225° еще не нашел объяснения. Для таких же образцов
(выдавленные в ^-модификации прутки урана) отмечен линейный рост теплопроводности
между 125 и 300°; температурный коэффициент составляет около 1,3-10~3
на 1° [116].
Теплопроводность a-урана, вероятно, зависит от кристаллографического
направления; поэтому теплопроводность поликристаллического вещества
с преобладающей ориентацией кристаллов (катаный или выдавленный металл)
может быть неодинаковой для продольного и поперечного образцов. Однако
Глава 5. Физические свойства металлического урана
137
измерения радиальной теплопроводности (проводимость в перпендикулярном
направлении к выдавливающим напряжениям для урана, выдавленного в -j-mo-
дификации) дали значения, заметно не отличающиеся от полученных для других
образцов при 84-241°, а именно 62-10'3 — №-№'* кал-см'1-сек1-град^ [115].
Если принять, что электропроводность чистого урана при комнатной
температуре равна 4-Ю4 (ом-см)'1 и пользоваться для отношения Видемана—
Франца значением 5,8-10~9, то окажется, что теплопроводность урана должна
составлять 70-10~3 кал-см'1-сек'1-град'1. Это немного выше наибольшего
экспериментального значения (66-10~3).
Термоэлектродвижущая сила. В табл. 54 даны значения,
найденные для термоэлектродвижущей силы кованой урановой проволоки
(металл 99,9%-ной чистоты), отожженной в течение 10 мин. при 900°[81].
Оказалось, что уже относительно небольшое изменение чистоты урана довольно
сильно влияет на термоэлектродвижущую силу [92]; разница в составе двух
образцов на 0,07% Fe и 0,02% О обусловила разницу на 1,92 мв при 800°.
Значение dE/dt относительно меди составляло около 5 мкв-грасГ1 [106]. Эта
величина показывает, что в термоэлектрическом ряду уран занимает то же
положение, что и молибден.
Кривая зависимости ЭДС от температуры показывает только очень
слабые прерывности в точках превращения.
Электронная эмиссия. Все имеющиеся определения работы
выхода электронов для урана были проведены до 1940 г. с металлом
сомнительной чистоты. Из эмиссии вольфрамовой проволоки, содержащей уран,
вычислен верхний предел для работы выхода электронов урана, равный 3,28 в
[117]. Измерения электронной эмиссии полосы и прутка урана при 680—1030°
дали для работы выхода электронов значения между 3,60 и 3,15 в, причем
эта работа, как правило, падала с повышением степени обезгаживания. Для
чистого металла наиболее вероятной величиной считается вычисленное
значение 3,27 ±0,05 в [94].
На основе порога фотоэлектрического эффекта было вычислено значение
3,63 в для фотоэлектрической работы выхода урана [118]. Разница между
этим значением и величиной, найденной для работы выхода электронов (3,28в),
превышает экспериментальную ошибку [117].
Таблица 54
ТЕРМОЭЛЕКТРОДВИЖУЩАЯ СИЛА УРАНА ОТНОСИТЕЛЬНО
ПЛАТИНЫ
Температура,
°С
0
100
200
300
400
Термоэлектродвижущая сила,
мв
0,00
1,19
2,87
5,03
7,59
Температура,
°С
500
600
700
800
900
Термоэлектродвижущая сила,
мв
10,54
13,90
17,51
21,35
25,50
Магнитная восприимчивость. Уран слабо парамагнитен.
Основные работы в этой области проводились в 1910—1912 гг. [119—121],
и взятый для опытов металл был, вероятно, сильно загрязнен, особенно
железом. Этим, возможно, объясняется отмеченное небольшое падение удельной
восприимчивости (х = 2,6-10~6 при 18°) с температурой [121].
Оптическая излучательная способность. Средний
коэффициент излучательной способности равен 0,51 при 670 гац [94]. Путем
138
Часть II. Металлический уран
сравнения кажущейся температуры поверхности урана с истинной,
определенной по излучению из полости в металле, которая служила абсолютно черным
телом, вычислено, что коэффициент излучательнои способности урана равен
0,453 при 97—1050° и 0,415 при 1052—1097° [93]. Резкий скачок при 1050^
1052°, повидимому, указывает на аллотропическое превращение. Поскольку
оба эти коэффициента излучательнои способности малы, вряд ли возможно
приписать один из них какому-либо окислу урана.
ЛИТЕРАТУРА
l.McLennan J.C., McKay R. W., Trans. Roy. Soc. Can., (3) 24, HI, 1 (1930).
2. Wilson T. A., Physics, 4, 148; Phys. Rev., 43, 781 (1933).
3. J а с о b С. W., Warren В. Е., J. Am. Chem. Soc, 59, 2588 (1937).
4. Battelle Memorial Institute, Report CT-2144, Sept. 1, 1944, p. 218 (Battelle 1).
5. G о r d о n P., Report CT-2780, Mar. 3, 1945 (MIT 1).
6. Metallurgical Project Information Meeting, Report CS-2745, Feb. 21, 1945, p. 6
(MP Ames 1).
'/. Battelle Memorial Institute, Reports CT-2374, Nov. 1944, p. 249, and CT-2700, Feb.,
1945, p. 33 (Battelle 2).
8. Allen N.. Reports BR-579, Febr. 26, 1945 and BR-592, Apr. 11, 1945 (British 7).
9. Battelle Memorial Institute, Reports CT-1795, June 1, 1944, p. 143, andCT-2483, Dec. 1,
1944, p. 282 (Battelle 3).
10. Wilson A. S., Reports CN-1495, Apr. 10, 1944, p. 24, and CT-1501, May 10, 1944,-
p. 14 (MP Ames 2).
11. Tucker C. W., Jr., Report AECD-2716 (1949).
12. Wilson A. S., Report CT-1985, Nov. 10, 1944, p. 20 (MP Ames 3).
13. Wilson A. S., Report CT-1775, June 10, 1944, p. 19 (MP Ames 4).
14. Battelle Memorial Institute, Report CT-2374, Nov. 1, 1944, p. 252 (Battelle 4).
15. Wilson A. S., R u n d 1 e R. E., Acta Crystal lographica, 2, 126 (1949).
16. P a u 1 i n g L., J. Am. Chem. Soc, 69, 542—553 (1947).
17. К a u f m a n n A. R., Report CT-685, May 29, 1943, p. 15 (MIT 2).
18. Driggs F. H., Lilliendahl W. C, Ind. Eng. Chem., 22, 516 (1930).
19. Peterson D. Report CC-682, May 15, 1943, Part V, p. 7 (MP Ames 5).
20. L a u 1 e t t a P, A., Hamilton N. E., Report N-1611a, July 3, 1944 (MP
Chicago I).
21. V a n Echo A., Report N-I61 lc, Julv 8, 1944 (MP Chicago 2).
22. S i m о n F. E., Report BR-280, Aug. '16, 1943 (British 1).
23. Thompson J. G., Report CT-539, Mar. 27, 1943, Part F. p. 1 (Natl. Bur. Stan-
dards 1).
24. S e у b о 1 t A. U., Stark L. В., Arnold W. R, Schnettler F. J.,
Report LA-68, Feb. 15, 1944, p. 6 (Los Alamos 1).
25. Plot t R. F., Raeth C. H., Report CP-228, Aug. 14, 1942 (MP Chicago 3).
26. M а г d e n J. W., Report A-605, Mar. 23, 1943 (SAM Columbia 1).
27. Allen N., Report BR-679, 1946 (British 11).
28. Allen N.. Report BR-703, 1946 (British 12).
29. Allen N., Report BR-717, 1946 (British 13).
30. Battelle Memorial Institute, Reports CT-2002, Aug. 1, 1944, p. 194; CT-2144, Sept.
1944, p. 216 (Battelle 5).
31. National Nuclear Energy Series, Division IV, vol. 12A.
32. С r e u t z E. C, Simmons J., Report CP-322, Oct. 29, 1942, p. 4 (MP Chi-
cago 4).
33. S e у b о 1 t A. U., Stark L. В., Arnold W. F., Report LA-55, Jan. 14,
1944, p. 7 (Los Alamos 2).
34. Imperial Chemical Industries, Report BR-658, Sept. 21, 1945 (British 3).
35. Battelle Memorial Institute, Report CT-393, Dec. 15, 1942, p. 2 (Battelle 8).
36. Battelle Memorial Institute, Report CT-428, Jan. 1, 1943, p. 10 (Battelle 9).
37. К a u f m a n n A. R., Report CT-422, Jan. 15, 1943, Part D (MIT 3).
38. К a u f m a n n A. R., Report CT-539, Mar. 27, 1943, Part G, p. 7 (MIT 4).
39. С 1 e a v e s H. E., Report CT-2375, Oct., 1944, p. 1 (Natl. Bur. Standards 2).
40. Alexander W. O., Forrest K., Report BR-727, 1946 (British 14).
41. Thompson J. G., Report CT-2478, Nov. 1944, p. 2 (Natl. Bur. Standards 3).
42. Thompson J. G., Report CT-2252, Sept. 1944, p. 32 (Natl. Bur. Standards 4).
43. Thompson J. G., Report MUC-FF-135B (N-1404), July 10, 1944 (Natl. Bur.
Standards 5).
44. T h о m p s о n J. G., Report CT-2692, Jan. 1945 (Natl. Bur. Standards 6).
45. G о r d о n P., Private communication to F. Foote, Dec. 7, 1944 (MIT 5).
Глава 5. Физические свойства металлического урана
139
46. Battelle Memorial Institute, Report CT-1697, Mav 1, 1944, p. 91, 102 (Battelle 10).
47. Battelle Memorial Institute, Report CT-688, Mav'10, 1943 (Battelle 11).
48. Sykes C, Report BR-203, Apr. 19, 1943 (British 4).
49. Thompson J. G., Report CT-750, June 26, 1943, p. 22 (Natl. Bur. Standards 7).
50. Or ow an E., Report BR-685, 1946 (British 15V
51. V a n E с h о A., Report CT-26G8, Jan., 1945, p.' 10—14 (MP Chicago 5).
52. Hamilton N. E.,Lauletta P. A., Van Echo A., Report N-1611c,
July 8, 1944 (MP Chicago 6).
53. V a n Echo A., Report CT-2743, Febr., 1945, p. 5, 6 (MP Chicago 7).
54. S ey b ol t A. U., Report LA-I80, Dec. 6, 1944, p. 25 (Los Alamos 3).
55. Battelle Memorial Institute, Report CT-1571, Apr. 1, 1944 (Battelle 6).
56. Battelle Memorial Institute, Report CT-1697, May 1, 1944, p. 113 (Battelle 12).
57. Directorate of Tube Alloys, Report BR-403, Mar. I, 1944 (British 2).
58. G r e e n w о о d H., Report BR-78, Dec. 18, 1942 (British 5).
59. К a u f m a n n A. R., Report CE-345, Nov. 15, 1942, Part B, p. 5 (MIT 6).
60. Laquer H., Report AECD-2606 (1949).
61. Snyder T. M., Kamm R. L., Report CP-124, June 13, 1942, p. 3 (MP Chicago 8).
62. S n у d e r T. M., Kamm R. L., Report CT-192, p. 38 (MP Chicago 9).
63. Battelle Memorial Institute, Report CT-468, Feb. 1, 1943, p. 56, 57 (Battelle 13).
64. К б s t e r W., Z. Metallkunde, 39, 1—9 (1948).
65. S n у d e г Т. M., Kamm R. L., Report CT-96, Part E, p. 5 (MP Chicago 10).
66. W о 1 I a n E.O., Stephenson R. L., Report CP-76, May 16, 1942 (MP
Chicago 11).
67. В r i d g m a n P. W., Proc. Natl. Acad. Sci. U. S., 8, 361 (1922).
68. В r i d g m a n P. W., Proc. Am. Acad. Arts Sci., 76, 55—70 (1948).
69. Battelle Memorial Institute, Report CT-1937, July 1, 1944, p. 150, 165 (Battelle 7).
70. Battelle Memorial Institute, Reports CT-893, Aug. 10, 1943, p. 279; CT-956, Sept. 10,
1943, p. 321 (Battelle 14).
71. С о 1 b e с к E. W., G а г n e г R. P., Report BR-707, April, 1946 (British 16).
72. M о r r i s о n, Parker, Report BR-735, 1946 (British 17).
73. Alien N., Report BR-718, 1946 (British 18).
74. Cleaves H. E., Report CT-1179, Jan. 1, 1944, p. 7, 8 (Natl. Bur.
Standards 8).
75. P a 1 m er R. N.. Report CT-2606, Apr. 7, 1945, p. 12; CT-3060, May, 1945, p. 13;
CT-3122, Aug. 9, 1945, p. 9; CT-3194, July 10, 1945, p. 8; CT-3459, Mar. 14, 1946, p. 8
(MIT 7).
76. Battelle Memorial Institute, Report CT-893, Aug. 10, 1943 (Battelle 15).
77. Directorate of Tube Alloys, Report (B) LRG-42, Mar., 1945 (British 6).
78. Battelle Memorial Institute, Report CT-753, June 10, 1943, p. 197 (Battelle 16).
79. Seitz F., Report CP-1598, Apr. 21, 1944, p. 4 (MP Chicago 12).
80. Battelle Memorial Institute, Report CT-611, Apr. 30, 1943, p. 122 (Battelle 17).
81. Thompson J. G., Report CT-890, Aug. 28, 1943, p. 16, 18 (Natl. Bur.
Standards 10).
82. Battelle Memorial Institute, Report CT-I795, June 1, 1944, p. 160 (Battelle 18).
83. С r e u t z E. C, Report CP-1507, Mar. 27, 1944, p. 34 (MP Chicago 13).
84. Creutz E. C, Report CP-1576, Apr. 24, 1944 (MP Chicago 14).
85. Long E. A., Jones W. M., Gordon J., Report A-329, Oct., 1942 (U. S. Bur.
Mines 2).
86. Moore G. E., Long E. A., Report A-502, Dec. 31, 1942; L о n g E. А., К е 1-
1 e у К. K-, M о о г е G. E., Report СТ-385, Dec. 24, 1942 (U. S. Bur. Mines 1).
87. Moore G.E., К е 1 1 e у К. К-, J. Am. Chem. Soc., 69, 2105—2107 (1947).
88. С о г г и с с i n i R. J., G i n n i n g s D. C, Report A-3947, undated, received
July 25, 1946 (Natl. Bur. Standards 14).
89. С a r t e r J. H., Report CT-G09, Apr. 24, 1943, Part. A, p. 5 (MP Ames 6).
90. Thompson J. G., Report CT-685, May 29, 1943, p. 25 (Natl. Bur.
Standards 9).
91. Thompson J. G., Report CT-815, July 24, 1943 (Natl. Bur. Standards 11).
92. D a h 1 A. I., V a n Dusen M.S., J. Research Natl. Bur. Standards, 39 [1],
53—58 (1947).
93. W a h 1 i n H. В., Report CT-2149, Sept. 12, 1944 (Wisconsin П.
94. H ol e W. L., Wright R. W., Phys. Rev., 56, 785 (1939).
95. A 1 e x a n d e г P. P., Metals and Alloys, 9, 270—274 (1938).
96. Ferguson J., Report B-37, Jan., 1942 (British 8).
97. S z i 1 а г d L., Report A-24, Aug. 16, 1941 (MP Chicago 15).
98. R о d d e n C. J., Report A-36, p. 4 (Natl. Bur. Standards 12).
99. Wensel H. Т., Roeser W. F., Report A-67 (Natl. Bur. Standards 13).
100. К a u f m a n n A. R., Gordon P., Palmer R. N., Report CT-953, Sept. 25,
1943, p. 18 (MIT 8).
101. D a h 1 A. I., С 1 e a v e s H. E., J. Research Natl. Bur. Standards, 43, 513 (1949).
140
Часть II. Металлический уран
102. Creutz E. С, Report A-95 (MP Chicago 16).
103. Creutz E. С, Report CP-255, Sept. 15, 1942 (MP Chicago 17).
104. Derge G., Cefola M., Report CT-2277, Oct. 26, 1944^(MP Chicago 18).
105. Directorate of Tube Alloys, Report (B) LRG-41, Febr., 1945 (Britisch 9).
106. E b e г t H., S с h u 1 z e A., Metallforschung, 2, 46—49 (1947).
107. Balz G., Metallforschung, 2, 144—146 (1947).
108. Denton W. H., Report (B) LRG-39, Dec, 1944 (British 10).
109. Bridgman P. W., Proc. Am. Acad. Arts Sci., 58, 158 (1923).
110. A sc h e г m a n n G., Justi E., Physik. Z., 43, 207 (1942).
111. А л e kc e e вс к и й Е., Мигунов Л., J. Phys. USSR, 11, 95 (1947).
112. S h о е n b е г g D., Nature, 159, 303 (1947).
113. G о о d m a n В. В., S h о e n b e г g D., Nature, 165, 441 (1950).
114. P 1 о t t R. F., R a e t h С. Н., Report CE-236, Aug. 15, 1942 (MP Chicago 19).
115. Kratz H. R., Raeth C. H., Report CT-539, Mar. 27, 1943, Part B, p. 8;
Kratz H. R., Report CT-861, July 20, 1943, p. 11; Creutz E. C, Report
CT-890, Aug. 28, 1943, p. 7; Kratz H. R., Report CT-953, Oct. 2, 1943;
Raeth C.H., King E., Report CP-1087, Nov. 27, 1943, p. 18; К г a t z H. R.>
Report CP-1728, May 25, 1944, p. 30; Raeth С H., Reports CP-2332, Dec. 7,
1944; MUC-RS-4 (N-1880), Jan. 20, 1945, and MUC-RS-9 (N-1568), Feb. 21, 1945;
Kratz H. R., Raeth С H., Report CP-2315, Jan. 27, 1945; R a e t h C.H,
Report MUC-RJM-2 (N-1936), Mar. 9, 1945 (MP Chicago 20).
116. Battelle Memorial Institute, Report CT-2700, Febr. 1, 1945, p. 31 (Battelle 19).
117. Dushman S., Phys. Rev., 21, 623 (1923).
118. R e n t s с h 1 er H.C., Henry D.E., Smith К. О., Rev. Sci. Instruments,
3, 794 (1932).
119. H о n d a K., Ann. Physik, (4) 32, 1047 (1910).
120. Honda K-, Science Repts. Tohoku Imp. Univ., 1, (I), 21 (1912).
121. Owen M., Proc. Acad. Sci. Amsterdam, 14, 637 (1912); Ann. Physik, (4) 37, 667,
668 (1912).
Глава 6
ХИМИЧЕСКИЕ СВОЙСТВА
МЕТАЛЛИЧЕСКОГО УРАНА
В этой главе дан обзор химических реакций металлического урана.
Приводится почти один лишь перечень различных реакций, поскольку практически
отсутствуют какие-либо количественные данные по кинетике этих реакций
или механизму их. Во избежание повторений даются ссылки на другие
главы этого тома, где можно найти более подробные описания отдельных
реакций.
Металлический уран является весьма реакционноспособным элементом,
он легко реагирует со всеми неметаллами и, кроме того, образует
интерметаллические соединения с ртутью, оловом, медью, свинцом, алюминием,
висмутом, железом, никелем, марганцем, кобальтом, цинком и бериллием (эти
соединения рассмотрены в гл. 7). Основным химическим свойством урана является
его сильная восстановительная способность, которая проявляется особенно
в водных растворах. Место урана в ряду напряжений точно не известно, но,
повидимому, он стоит близко к бериллию.
Приводимые ниже значения скорости реакций были получены в опытах
с плотными отливками металлического урана (99,9%U), очищенными от окиси
разбавленной азотной кислотой. Тонкодисперсный металл (например,
полученный разложением гидрида урана) часто реагирует гораздо энергичнее
плотного. Многие реакции, приписываемые гидриду урана (см. гл. 8), особенно
протекающие при температуре выше 400—500°, на самом деле являются реакциями
тонко дисперсного металлического урана.
РЕАКЦИИ С НЕМЕТАЛЛАМИ
Элементы рассматриваются ниже в порядке их расположения в
периодической таблице Д. И. Менделеева.
Водород и дейтерий (см. гл. 8, где эта реакция рассмотрена
более подробно). Стружки или куски урана превращаются в гидрид урана
действием газообразного водорода при 250° или выше.
U + |h2->UH3. (1)
Бор (см. гл. 9). Тонкодисперсный уран реагирует с аморфным бором
при температуре электрической печи с образованием борида урана [1 ].
Углерод (см. гл. 9). Нагреванием тесной смеси порошкообразного
урана с соответствующим количеством порошкообразного угля до 800—1200°
можно получить тот или другой из двух известных карбидов урана, UC и UC2.
Когда уран плавят в графитовом тигле, то тигель защищается пленкой
карбида урана, образующейся на его внутренней поверхности. Поэтому
разрушение тигля делается заметным только тогда, когда температура достигает
1500—1650°.
142
Часть II. Металлический уран
Кремний (см» гл. 9). При совместном плавлении порошкообразных
урана и кремния образуются сплавы. Фазовая диаграмма, являющаяся очень
сложной, указывает на существование не менее пяти урано-кремниевых
соединений.
Азот (см. гл. 10). При атмосферном давлении урановые стружки
медленно реагируют с азотом при 450°. При повышении температуры до 700°
реакция ускоряется, причем образуется соединение UNi,75- Реакция с
порошкообразным ураном протекает быстро даже при 520°. При более высоком
давлении азота можно получить нитрид UN2. Если температура реакции поднимается
выше 1300°, то образуется мононитрид UN, так как в этой температурной
зоне все высшие нитриды неустойчивы по сравнению с мононитридом.
Фосфор (см. гл. 10). Если тонкодисперсный уран нагревают с
порошком фосфора до 600—1000°, то образуется фосфид U3P4 12].
Мышьяк (см.. гл. 10). В качестве продукта реакции мышьяка с
ураном были идентифицированы два соединения, U2As и UAs.
Кислород (см. гл. 11)*. Уран в виде стружек или небольших кусков
ярко горит в кислороде при температуре 700—1000°, испуская белый свет.
3U + 402^U308. (2)
При очень низком парциальном давлении кислорода (менее 1-Ю"4 атм) на
металлическом уране образуется пленка моноокиси UO.
На воздухе при комнатной температуре плотный металлический уран
медленно окисляется [4,5]. Вначале он приобретает золотисто-желтую окраску»
затем пленка окиси темнеет и через 3—4 суток металл кажется уже черным.
Пленка окиси, которая образуется на уране на воздухе, не предохраняет его
от дальнейшего действия кислорода воздуха.
Урановые стружки окисляются с умеренной скоростью при 125°. Если их
прокаливать на воздухе, то они сгорают без пламени. Плотный металл медленна
окисляется при 500-700°, но при 700-1000° достигается полное окисление
в течение 1 часа.
Порошкообразный металлический уран является обычно пирофорным
и горит ярким пламенем. При обработке металлического урана напильником
или на точильном камне происходит эффектное образование искр.
По наблюдениям английских исследователей [6], при окислении урана на
воздухе при температуре ниже 100° образуется только двуокись урана, в
интервале температур между 100 и 200° получается и двуокись и закись-окись.
Сера и селен [7—9] (см. гл. 11). Уран медленно реагирует с
расплавленной серой при 250—300°, а при 500° он горит в парах серы. В зависимости
от условий получается дисульфид US2, полуторный сульфид урана U2S3> или
смесь обоих соединений. Селен реагирует аналогичным образом.
Фтор (см. гл. 12 и 13). Фтор энергично взаимодействует с
металлическим ураном при комнатной температуре с образованием гексафторида урана.
Тонкодисперсный металл в атмосфере фтора легко может раскалиться добела.
Хлор (см. гл. 14). Хлор реагирует с компактным металлическим ураном
с умеренной скоростью при 500—600°. Тонкодисперсный металл горит в
хлоре при 150—180°. Продукты сгорания содержат тетра-, пента- и гекса-
хлорид урана. Хлориды возгоняются и собираются на более холодных частях
аппаратуры.
Бром (см. гл. 15, стр. 417 и ел.). При 650° бром спокойно реагирует
с урановыми стружками с образованием тетрабромида урана, который
отгоняется из сферы реакции:
U+2Br2->UBr4. (3>
* См. [3], где приводятся результаты прежних наблюдений, относящихся к
загрязненному металлу.
Глава 6. Химические свойства металлического урана
143
При меньшем количестве брома получается трибромид (см. гл. 15, стр. 411).
Иод (см. гл. 15). Металлический уран реагирует с парами иода
при 350°. При этом получается трииодид или тетраиодид в зависимости от
условий опыта.
Инертные газы (см. гл. 8). Вследствие способности металлического
урана реагировать с примесями, обычно присутствующими в этих газах,
он может быть использован для получения очень чистых гелия, аргона и
Других инертных газов [10].
РЕАКЦИИ С СОЕДИНЕНИЯМИ НЕМЕТАЛЛОВ
Вода. Поведение металлического урана при соприкосновении с водой
и паром изучено очень тщательно [11]. Однако результаты всех
многочисленных опытов здесь не могут быть приведены. Кипящая вода медленно
действует на плотный уран.
U+2H20~>U02 + 2H2 (4)
Водород ускоряет коррозию урана вследствие образования гидрида. В
аэрированной дестиллированной воде скорость реакции вначале меньше, чем
в воде, насыщенной водородом. Это явление, вероятно, зависит от
образования защитной окисной пленки. Но затем скорость реакции возрастает и
приближается к значению в насыщенной водородом воде [12—14].
Водяной пар реагирует с ураном при 150—250°. Существуют
разногласия относительно природы получающихся продуктов. Американские ученые
предполагают, что при этом протекает следующая реакция:
7U + 6Н2Огаз -^1- 3U02 + 4UH3. (5)
При 600—700° образуется чистая двуокись урана и водород. Образования
закиси-окиси урана не наблюдалось даже при 1000° [15,16]. Однако
английские ученые утверждают, что основным продуктом реакции между водяным
паром и ураном при температурах выше 300° является закись-окись урана [6].
Причина указанных противоречивых результатов пока неизвестна. Все
исследователи согласны с тем, что пар действует гораздо более энергично, чем
кислород [17, 18].
Фтористый водород (см. гл. 12). Порошкообразный
металлический уран реагирует с безводным фтористым водородом при повышенных
температурах с образованием тетрафторида урана:
U + 4HF-»UF4 + 2H2 (6)
(при взаимодействии гидрида урана с фтористым водородом тетрафторид
образуется в температурном интервале 200—400°). Путем использования смеси
водорода и фтористого водорода можно компактный металл ввести в реакцию
при 250°. Ввиду того что водород образуется в самой реакции, внешний
источник водорода может быть удален сразу же после начала реакции, которая
затем будет продолжаться до тех пор, пока не израсходуется весь металл.
Хлористый водород (см. гл. 14). При взаимодействии
тонкодисперсного урана с хлористым водородом образуется трихлорид урана:
U+ ЗНС1 _» UCla + -| Н2. (7)
Эта реакция идет неполностью с компактным металлом.
Бромистый и йодистый водород (см. гл. 8). Реакции
этих соединений с металлическим ураном подробно не изучены. При
обработке гидрида урана бромистым водородом образуется трибромид урана»
144
Часть II. Металлический уран
Йодистый водород с гидридом урана, повидимому, дает смесь три- и тетра-
иодида.
Окись углерода (см. гл. 9). При температуре ниже 400° реакция
между порошкообразным металлическим ураном и окисью углерода мало
заметна, но она, несомненно, идет со стружками урана при 750°, в результате ее
получается смесь двуокиси и карбида урана [19, 20].
Двуокись углерода (см. гл. 9). При 750° уран и двуокись
углерода быстро реагируют с образованием окислов и карбидов урана.
В некоторых случаях наблюдалось самопроизвольное воспламенение
тонкого порошка металлического урана в двуокиси углерода [20].
Аммиак (см. гл. 10). Порошкообразный уран быстро реагирует с
аммиаком при 400° (уран в виде стружек реагирует при 700°) с образованием
нитрида UNi,75-
Окись азота. Уран в виде стружек сгорает в окиси азота при
400—500° с образованием закиси-окиси урана и азота [21].
Метан. Тонкодисперсный уран реагирует с метаном при 900°, причем
получается монокарбид UC [22].
РЕАКЦИИ С ВОДНЫМИ РАСТВОРАМИ КИСЛОТ
Плавиковая кислота. На компактный металлический уран
концентрированная плавиковая кислота действует медленно, даже при
температуре 80—90°, вероятно вследствие образования на поверхности урана
нерастворимого тетрафторида. Окислители, например перекись водорода,
повидимому, заметно не ускоряют реакцию.
Соляная кислота. Металлический уран подвергается разрушению
концентрированной соляной кислотой с очень большой скоростью. Скорость
реакции в 1 н. растворе кислоты намного меньше, чем в 6 н. растворе. Эта
реакция, повидимому, является сложной, так как различные количества металла
превращаются в нерастворимое черное вещество. Было высказано
предположение, что этот продукт представляет собой гидратированную двуокись урана
[23]. В случае применения большого избытка кислоты образуются только
небольшие количества черного вещества, при меньшем же количестве кислоты
до 20% металла превращается в этот продукт [24]. Изменяется также и
отношение уран (III) : уран (IV) в конечном растворе. В 12 н. растворе кислоты
практически весь металл окисляется до четырехвалентного состояния, но
в 6 н. растворе валентность окисленного продукта в среднем составляет
3,2—3,4. Степень окисления зависит от концентрации кислоты, отношения
количеств кислоты и металла, температуры, времени и, вероятно, от других
еще неизвестных факторов [25].
Для полного растворения металлического урана можно использовать
смесь соляной кислоты и окислителей. Можно брать для этого перекись
водорода, азотную кислоту, персульфат аммония, хлорат калия или хлорную
кислоту. В присутствии 0,05 М раствора кремнефтористоводородной кислоты
металлический уран полностью растворяется в концентрированной соляной
кислоте без образования черного осадка [26]. Такой раствор свободен от
трехвалентного урана. Метиловый спирт, насыщенный хлористым водородом,
растворяет уран с умеренной скоростью, но около 28% металла превращается
в черный остаток. Образуется фиолетовый раствор, устойчивый в течение
некоторого времени [23].
Бромистоводородная и иодистоводородная
кислоты. Бромистоводородная кислота по своему действию на уран сходна
с соляной, но реагирует она медленнее. В этом случае также образуется черный
остаток. Иодистоводородная кислота реагирует еще медленнее.
Глава 6. Химические свойства металлического урана
145
Азотная кислота. Плотный металл растворяется в
разбавленной или концентрированной азотной кислоте с умеренной скоростью, при этом
образуется уранилнитрат. Ввиду того что пары азотной кислоты или двуокиси
азота могут реагировать с ураном со взрывом, необходимо во избежание
несчастных случаев тонкоизмельченный уран вводить в азотную кислоту
небольшими порциями [27—29].
Серная кислота. Разбавленный (6 н.) раствор серной кислоты не
реагирует с ураном, при температуре кипения действие кислоты аналогично
действию одной кипящей воды. Горячая концентрированная серная кислота
медленно реагирует с ураном с образованием бисульфата урана (IV), двуокиси
серы, элементарной серы, сероводорода и других продуктов. В присутствии
окислителей, например перекиси водорода или чазотной кислоты,
разбавленная серная кислота растворяет уран. Растворение урана происходит также
при электролитическом окислении его в растворе серной кислоты [30].
Фосфорная кислота. Холодная 85%-ная фосфорная кислота
действует на уран медленно. При нагревании скорость реакции вначале
возрастает лишь немного. При дальнейшем нагревании вследствие
испарения воды концентрация фосфорной кислоты повышается настолько, что
начинается быстрая экзотермическая реакция, в результате которой
образуется зеленый раствор кислого фосфата урана (IV). Продолжительное
нагревание раствора фосфата урана (IV) в фосфорной кислоте может окончиться
образованием стекол, чрезвычайно устойчивых к последующим химическим
воздействиям.
Хлорная кислота. Разбавленный раствор хлорной кислоты
довольно инертен по отношению к урану. Когда концентрация хлорной
кислоты достигает 90%, начинается энергичная реакция вследствие
выкипания воды. Окисление идет очень бурно, поэтому рекомендуется не
пользоваться этим реагентом для растворения больших количеств металла.
Разбавленная хлорная кислота спокойно растворяет уран в присутствии
окислителей.
Органические кислоты. Хотя муравьиная, уксусная, про-
пионовая и масляная кислоты (разбавленные или безводные) и не реагируют
с металлическим ураном, но в присутствии хлористого водорода или соляной
кислоты начинаются быстрые экзотермические реакции, заканчивающиеся
образованием соответствующих солей четырехвалентного урана [31]. Ацетат
урана может также быть получен действием уксусного ангидрида или ацетил-
хлорида на металл. Уран реагирует с бензойной кислотой в эфирном растворе
с образованием бензоата урана (IV).
РЕАКЦИИ С ВОДНЫМИ РАСТВОРАМИ ЩЕЛОЧЕЙ
Растворы гидроокисей щелочных металлов слабо действуют на
металлический уран [5, 9, 29]. Растворы едкого натра, содержащие перекись водорода
(или смеси перекиси натрия с водой), растворяют уран. Образуются
растворимые перуранаты натрия [23]. О подобной реакции с двуокисью урана см. гл. 11.
РЕАКЦИИ С РАСТВОРАМИ СОЛЕЙ ТЯЖЕЛЫХ МЕТАЛЛОВ
Металлический уран является достаточно сильным восстановителем для
того, чтобы вытеснять многие металлы из растворов их солей. При действии
металлического урана на растворы нитратов ртути (II) или серебра, сульфата
меди, хлоридов олова (II), платины (IV) и золота (III) образуются осадки
соответствующих металлов [9]. Эта реакция изучена при разработке способов
определения урана в металлах. Предложен метод, основанный на растворении
урана в соляной кислоте и измерении количества выделяющегося водорода, но
10 Химия урана
146
Часть II. Металлический уран
сложность реакции (см. стр. 144) делает этот метод непригодным. Поэтому были
сделаны попытки определять уран измерением количеств других металлов,
вытесненных из растворов; до настоящего времени эти исследования еще не
привели к удачным результатам.
Соли серебра [23, 32]. Растворы сульфата серебра медленно
реагируют с ураном. Поверхность урана, повидимому, покрывается серебром,
которое практически препятствует дальнейшей реакции. Растворы
перхлората серебра реагируют энергичнее. Реакция сложна, так как получается не
только серебро, но и хлорид серебра, вследствие одновременного
восстановления перхлората до хлорида:
U + 6AgC104 + 2H20 --> 6Ag + U02 (С104)2 + 4НС104; (8)
U + 4AgC104 -> U (С104)4 + 4Ag; (9)
2U + 5AgC104 —> 4Ag + AgCl + 2U02 (C104)2. (10)
Около 20% урана реагирует по уравнению (10) и лишь менее 0,1%—по
уравнению (9). Одновременное образование серебра и хлорида серебра в различных
отношениях делает невозможным использование этой реакции для определения
урана. В толуоле (в котором перхлорат серебра хорошо растворим) реакция
происходит тем же путем с образованием как серебра, так и его хлорида.
Соли меди [33—37]. Хотя уран лишь слабо реагирует с сульфатом
меди (II) (ср., однако, [9]), в растворахтетрамминкупрохлорида он растворяется
быстро. Осажденная вначале медь растворяется при встряхивании с избытком
хлорида аммония, присутствующего в растворе. Ввиду того, что карбиды и
окислы урана, так же как и уран, заметно растворимы в этом реактиве,
данный метод не может иметь аналитического применения.
ДРУГИЕ РЕАКЦИИ
Тетрафторид урана (см. гл. 12). Порошкообразный уран
восстанавливает тетрафторид до трифторида при температуре 1100°:
3UF4 + U->4UF3. (11)
Двуокись урана (см. гл. 11). При 2400° металлический уран
восстанавливает двуокись урана до моноокиси:
U02 + U->2UO. (12)
Вещества, содержащие кремнезем. Стекло, фарфор и
кварц вступают в реакцию с тонкодисперсным ураном при 700—800° с
образованием смеси двуокиси и силицида урана.
Борная кислота [38]. Металлический уран практически инертен
при сплавлении с борной кислотой.
Растворы персульфатов калия и аммония [39].
Если металлический уран обрабатывать водным раствором персульфата калия,
начинается энергичная реакция. С персульфатом аммония реакция идет
медленнее.
Метиловый спирт [40]. Сообщается, что порошкообразный уран
не реагирует с безводным метиловым спиртом. Это наблюдение нуждается
в подтверждении.
Хлорированные углеводороды. При температуре 150—200°
металлический уран медленно реагирует с парами четыреххлористого углерода,
хлороформа и трихлорэтилена. Реакция с четыреххлористым углеродом
быстро идет при температуре выше 1000°. При этом необходимо соблюдать
Глава 6. Химические свойства металлического урана
147
осторожность, так как тонкодисперсный уран бурно реагирует с жидким
галогенированными углеводородами, особенно если металл приготовлен
разложением гидрида и поэтому еще содержит небольшое его количество.
ЛИТЕРАТУРА
1. Wedekind E.( Jochem О., Вег., 46, 1204 (1913).
2. D г i g g s F. H., L i 1 1 i e n d a h 1 \V. С, пат. США 1893296, 1929.
3. «Gmelins Handbuch der anorganischen Chemie», System No. 55, p. 65, Verlag Chemie,
Berlin (1936).
4. Moore R. W., Trans. Am. Electrochem. Soc, 43, 223 (1923).
5. L e 1 у D., Hamburger L., Z. anorg. Chem., 87, 220 (1914).
6. W a t h er Т., Report BR 223, May 13, 1943 (British 1).
7. P ё 1 i g о t E., Ann. chim. et phys., (3) 5, 5 (1842).
8. M о i s s a n H., Compt. rend., 122, 1092 (1896).
9. ZimmermannC, Ber., 15, 849 (1882).
10. Newton A. S., The Use of Uranium and Uranium Compounds in Purifying Gases,
in N. N. E. S. Division VIII, vol. 6 (MP Ames 1).
11. National Nuclear Energy Series, Division IV, vol. 6A.
12. M о 1 1 i s о n W. A., English G. C, Nelson F., Report CT-3055, Aug. 8,
1945 (MP Chicago 1).
13. В e n s e n N., S t r a e t z R. P., D r a 1 e у J. E., Report CT-3043, June 4, 1945
(хЧР Chicago 2).
14. Hopkins J. M., N e 1 s о n F., Binger W. W., Report CT-3031, May 31, 1945
(MP Chicago 3).
15. Feibeg J. G., Warf J. C, Report CT-1524, Mar. 10, 1944 (MP Ames 2).
16. Newton A. S., Report CC-695, May 26, 1943 (MP Ames 3).
17. Report [BJ LRG-14, Nov., 1942 (British 2).
18. R os n er G., Report CT-2548, Jan. 15, 1945 (MP Chicago 4).
19. Warf J. C, Report CC-580, Apr. 15, 1943, CC-587, Apr. 19, 1943 (MP Ames 4).
20. W i 1 h e 1 m H. A., H о x e n g R., Report CC-238, Aug. 15, 1942 (MP Ames 5).
21. E m i с h F., Monatsh., 15, 375 (1894).
22. L i t z L. M., Garrett А. В., С г о x t о n F. C, J. Am. Chem. Soc., 70, 1718
(1948).
23. Fischer R. W., Powell J., Warf J. C, Report CC-1194, Dec. 9, 1943
(MP Ames 6).
24. Peterson D. W., Report CC-1061, Oct. 8, 1943 (MP Ames 7).
25. Fulmer R., Report CC-1194, Dec. 9, 1943 (MP Ames 8).
26. В a n k s С V., Noyce W. K., Patterson J. H., Warf J, C, Report
CC-2942, July 18, 1945 (MP Ames 9).
27. S u t t о n J. В., Report CN-566, Apr. 12, 1943 (MP Chicago 5).
28. С u n n i n g h a m T. R., Report N-42, undated (MP Chicago 6).
29. H у d e A. C, Report CN-1751, May 15, 1944 (MP Chicago 7). «
30. S a f r a n s k i L, Straetz R. P., S p e nc e R., Report CC-934, Sept. 11,
1943 (MP Chicago 8).
31. F e i b i g J. G., Report CC-1504, Jun. 10, 1944 (MP Ames 10).
32. Fi sh er R. W., Report CC-1057, Nov. 6, 1943 (MP Ames 11).
33. R i о t t J. P., Ind. Eng. Chem., Anal. Ed. 13, 546 (1941).
34. W i 1 1 e m s F. W., Z. anorg. u. allgem. Chem., 246, 46 (1941).
35. S a f a r a s k i L., Report CC-1047, Nov. 6, 1943 (MP Chicago 9).
36. Safraski L., Potratz H.A., Report СК-Ю64, Nov. 6, 1943 (MP Chicago 10).
37. Fry x ell R. E., Report CC-1448, Mar. 14, 1944 (MP Chicago 11).
38. T e v e b a u g h R., Report CC-1194, Dec. 9, 1943 (MP Ames 12).
39. Levi M. G., Miglorini E., Ergolini G., Gazz. chim. ital., 38, (1),
599 (1908).
40. Brown H. D., Report CC-1524, Mar. 10, 1944 (MP Ames 13).
10*
Глава 7
ИНТЕРМЕТАЛЛИЧЕСКИЕ СОЕДИНЕНИЯ
И СПЛАВЫ УРАНА
Подробное описание различных сплавов урана можно найти в другом томе
этой серии [1], здесь же рассматриваются лишь некоторые вопросы,
представляющие особый интерес в химическом отношении. Будет приведен лишь
краткий обзор взаимной растворимости урана и различных металлов, а также
описание некоторых интерметаллических соединений, обнаруженных в
процессе изучения фазовых соотношений в интерметаллических системах.
ПОЛУЧЕНИЕ СПЛАВОВ И ИНТЕРМЕТАЛЛИЧЕСКИХ СОЕДИНЕНИЙ
Сплавление металлических компонентов почти всегда необходимо проводить
в вакууме или инертной атмосфере аргона или гелия. В настоящее время часто
применяются тугоплавкие тигли из окислов бериллия, циркония или тория;
в отдельных случаях пользуются и тиглями из окиси алюминия. Для
предотвращения окисления требуется создание очень хорошего вакуума. ЕсЛи один
из. металлов весьма летуч, то. для сведения к минимуму потерь из-за дестил-
ляции можно применять атмосферу из хорошо очищенного аргона. Лучше всего
.пользовлться индукционным нагревом; это особенно желательно при
сплавлении металлов, сильно различающихся по удельному весу, так как при этом
происходит их более полное перемешивание. В случае легкоплавких металлов,
.например свинца или висмута, применяются электролитические процессы.
Так, тетрахлорид урана растворяли в расплавленной смеси хлоридов натрия
и кальция (т. пл. 750°), затем смесь подвергали электролизу в ванне со
стальным катодом, покрытым слоем жидкого свинца или висмута [2]. Для
получения ртутных амальгам необходимо применять очень чистый металлический
уран, приготовленный разложением гидрида. Некоторые сплавы были случайно
получены при одновременном восстановлении тетрафторида урана и фторидов
других металлов. Но этот метод не рекомендуется для систематического
изучения, так как при нем затруднительно заранее определить конечный состав
и структуру сплавов.
ВЗАИМНАЯ РАСТВОРИМОСТЬ УРАНА И РАЗЛИЧНЫХ МЕТАЛЛОВ
Имеются многочисленные данные по растворимости урана в различных
металлах и растворимости различных металлов в уране. В большинстве случаев
эти данные основаны на применении рентгеновской и металлографической
техники. Как было указано в гл. 5, а-уран обладает необычайно сложной
кристаллической структурой. Эго затрудняет применение обычных
кристаллографических и химических критериев для оценки его способности образовывать
твердые растворы [3]. Насколько известно, ни один металл не обладает хорошей
растворимостью в а-уране. Однако многие металлы хорошо растворимы в
7-уране, который устойчив при температуре выше 770°. В случае молибдена
резкое охлаждение до комнатной температуры ведет к образованию твердых
Глава 7. Интерметаллические соединения и сплавы урана 149
растворов, в которых сохраняется ^-структура. В табл. 55 суммированы
известные в литературе данные по взаимной растворимости урана и некоторых
металлов.
Таблица 55
ВЗАИМНАЯ РАСТВОРИМОСТЬ УРАНА И НЕКОТОРЫХ МЕТАЛЛОВ
Атомный
номер
13
23
24
25
26
28
29
41
42
47
50
58
73
74
79
80
83
90
Металл
Алюминий . .
Ванадий . ♦ .
Хром ....
Марганец . .
Железо . . .
Никель . . .
Медь ....
Ниобий . . .
Молибден . .
Серебро . . .
Олово ....
Церий ....
Тантал . . .
Вольфрам • .
Золото . . .
Ртуть ....
Висмут . . .
Торий ....
Растворимость
урана в металле,
% (атомн.)
.
—
Не раств.
?
Не раств. в -y-Fe
—
Не раств.
—
1 (900°)
—
<0,G2(6G0°)
—
—
1
—
0,001—0,01(25°);
1,25(350°)
Не раств.
1 <2
Растворимость металла
°L (атомнЛ
а
Не раств.
» »
» »
—
Не раств.
— ■
Не раств.
<0,25
Не раств.
» »
» »
» »
» »
<1
2
. Не раств.
» »
» »
со.
Не раств.
—
<2,5
—
Не раств.
1
Не раств.
<0,25 !
—
—
—
—
—
—
—
—
—
в уране,
Г
4—5
—
<4
3-4
1,5—2,0
2
Не раств.
3,6(656°)
85(1350°)
35,8
—
—
Не раств.
—
—
—
—•
—
1 —
Литература
(отчет ■№)
СТ-2721
N-1779
СТ-3335
СТ-2945
СТ-2945
СТ-30.13
СТ-1784
СТ-3071
СТ-2946
CN-I048
CN-1784
СТ-2619
N-1779
СТ-3375
СТ-2483
! СТ-2960
| СТ-2961
СТ-2717
Растворимость урана в ртути заслуживает особого внимания [4]. До 1941 г.
все исследователи сходились на том, что растворимость урана в жидкой ртути
при 25° ничтожно мала; для этой величины предлагались значения порядка
0,00001%. Однако новые работы показали, что прежние результаты надо
отнести за счет использования загрязненного или покрытого окислами урана.
Установлено, что чистый тонкодисперсный металлический уран,
приготовленный разложением гидрида урана и свободный от окисла, быстро
амальгамируется ртутью, образуя серебристую пастообразную массу, похожую, на
амальгамы других металлов. Амальгамы, содержащие до 1 % урана, являются
жидкими и довольно устойчивыми на воздухе. Амальгамы с 1—15% урана
представляют серые пирофорные твердые вещества. Ниже описаны
многочисленные выделенные интерметаллические уранортутные соединения.
Растворимость урана в ртути определена путем разделения твердой и жидкой фаз
амальгам центрифугированием при различных температурах и измерением
содержания урана в жидкой фазе. Результаты не являются особенно точными,
но дают для интервала растворимости значения от 0,001—0,01% при 25° до
1,06% при 350°. Химические свойства этих амальгам очень мало известны;
еще менее изучены их физические свойства. Об устойчивости этих амальгам
свидетельствует выделение тепла при смачивании тонкодисперсного урана
ртутью.
Растворимость урана в других твердых металлах, повидимому,-
ограничена. То же самое, вероятно, справедливо и в отношении жидких легкоплавких
КРИСТАЛЛИЧЕСКИЕ СТРУКТУРЫ НЕКОТОРЫХ ИНТЕРМЕТАЛЛИЧЕСКИХ СОЕДИНЕНИЙ УРАНА [8-10]
Таблица 56
Элементарная ячейка
Константа
*1
1 6,3557+0,0004
7,795
4,278
7,033+0,002
6,7830+0,0005
4,976+0,01
3,320+0,005
3,62
4,62
7,1628+0,0014
7Д)58
6,9924+0,0004
4,966+0,005
10,29+0,01
10,31+0,04
10,36+0,02
10,37+0,04
6,364+0,004
решетки, А
*а
3,218+0,005
4,878+0,005
8,252+0,009
5,24+0,02
5,24+0,02
5,21 + 0,02
5,21+0,02
Число
молекул в
элементарной
ячейке
8
8
1
4
4
1
V.
1
8
8
8
4
4
4
4
4
I
Плотность,
г/см*
15,37
8,138
6,70
10,61
11,31
15,29
14,88
14,5
9,95
12,57
13,21
13,83
13,46
17,8
17,7
17,7
17,6
11,52
Пространственная группа
или
структурный тип
Т5
0\ или О4
112
Td2 или Г2
Td2 или Г2
Сложная
истинная ячейка
и структура
L12
С 15
С 15
С 15
С 14
Dl или D\°d
Dl » Dfd
Dl % D\%
Dl » 0Й
Структура NaCl
Кубическая объемноцентрирован-
ная
Кубическая гранецентрированная
Кубическая
Кубическая гранецентрированная
Гексагональная
»
Кубическая объемноцентрирован-
ная псевдоячейка
Простая кубическая
Кубическая гранецентрированная
» »
» »
Гексагональная
Тетрагональная объемноцентри-
рованная
То же
» »
» »
Кубическая
Глава 7. Интерметаллические соединения и сплавы урана 151
металлов. Так, например, плотный уран заметно не изменяется даже при
многодневном действии жидкого натрия при 500° [5]. Уран также, повидимому,
нерастворим в жидких сплавах калия и натрия. Сплавы или
интерметаллические соединения урана с щелочными или щелочноземельными металлами
неизвестны [6]. Бериллий, вероятно, образует с ураном и нтер металл и чес кое
соединение, содержащее 93% (атомн.) Be(UBei2-i3) [7]. Была доказана [4,6]
чрезвычайная трудность растворения магния в уране в количестве,
превышающем несколько десятитысячных долей процента.
ИНТЕРМЕТАЛЛИЧЕСКИЕ СОЕДИНЕНИЯ
В настоящее время получено и идентифицировано (главным образом
рентгенографическими методами) значительное число интерметаллических
соединений урана. В табл. 56 помещены все интерметаллические соединения,
для которых известны рентгенографические структуры. Кроме того, методом
химического анализа или микроскопическим исследованием доказано
существование следующих соединений: UA15, UNi, UFeNi, UBi2, U6Sn4 и U3Sn5,
но они не были точно идентифицированы в виде чистых фаз. В табл. 57
Т а б л и а 57
ТЕМПЕРАТУРЫ ПЛАВЛЕНИЯ ИЛИ РАЗЛОЖЕНИЯ
ИНТЕРМЕТАЛЛИЧЕСКИХ СОЕДИНЕНИЙ УРАНА
Формула
соединения
UA12
UA13
UA1$
UCu5
UNi2
UNi6
UeNi
UFe2
Т. пл. или т.
разл., °С
1590
I350a>
730a>
1052a>
810a>
1295
754a>
1235
1-
Формула
соединения
UeFe
1 UMn2
U„Mn
UHg4
UHg,
UHg2
U5Sn4
USn3
Т. пл. или т.
разл., °С
815Q>
1120
726a>
360a>
390a>
450a)
1500
1350a>
l) Разлагается по перитектической реакции.
содержатся известные в настоящее время данные о температурах плавления
или разложения и нтер металлических соединений.
Интерметаллические соединения весьма отличаются друг от друга по
своим физическим свойствам и химической активности. Соединения типа U6M
являются чрезвычайно хрупкими. Они легко разбиваются при ударе молотком,
но недостаточно тверды, чтобы царапать стекло. Соединения этого типа легко
выделить путем использования их инертности к азотной кислоте. При
обработке сплавов урана с железом, кобальтом или марганцем приблизительного
состава U6M разбавленной или концентрированной азотной кислотой в раствор
переходит избыток урана. Системы уран—ртуть, уран—олово, уран—свинец
и уран—висмут отличаются химической активностью. Амальгамы, содержащие
до 15% урана, легко окисляются на воздухе с образованием черного порошка,
в состав которого входят уран и ртуть; сплавы, содержащие более 15% урана,
самопроизвольно воспламеняются на воздухе. Пирофорные сплавы открыты
также в системе уран—олово; 50%-ный сплав очень легко воспламеняется
на воздухе. Это же относится и к системе уран—свинец. В системе с
висмутом высокой реакционной способностью обладают и UBi и UBi2. Они за
152
Часть II. Металлический уран
несколько минут разогреваются на воздухе и реагируют с водой, спиртами,
минеральным маслом, керосином, бензолом и четыреххлористым углеродом.
Систематические исследования реакций этих интерметаллических соединений
не проводились, и, как следует из вышеприведенного материала, имеющиеся
сведения по ним весьма отрывочны.
Исследовано коррозийное действие воды и воздуха на многочисленные
сплавы урана. Более или менее подробно изучены системы из урана со
следующими элементами: натрий,, калий, медь, серебро, золото, бериллий, магний,
цинк, кадмий, ртуть, алюминий, галлий, индий, церий, лантан, неодим, титан,
германий, цирконий, олово, торий, ванадий, ниобий, тантал, висмут, хром,
молибден, вольфрам, марганец, рений, железо, кобальт, никель, рутений,
родий, палладий, осмий, иридий и платина. В большинстве случаев полная
фазовая диаграмма еще не разработана. Недавно опубликованы описания
систем уран—алюминий и уран—железо [И], уран—вольфрам и
уран—тантал [12], уран—марганец и уран—медь [13]. g . Jj
ЛИТЕРАТУРА
1. National Nuclear Energy Series, Division IV, vol. 12.
2. J. Ferguson, Report B-38, Febr. 1942 (British 1).
3. H u m e-R о t h e г у W., MabbotG. W., Evans К. М. С, Trans. Roy.
Soc., London, 233, 1—98 (1933).
4. A h m a n n D. H.f Baldwin R. R., Wilson A. S., Report CT-2960, Dec. 21,
1945 (MP Ames 1).
5. F о о t e F., Report CT-2857, Mar. 24-31, 1945 (MP Chicago 1).
6. A h m a n n D. H., Report CT-2959, Dec. 5, 1945 (MP Ames 2).
7. Cunningham R. L., Report MX-180. Oct. 16, 1945, p. 2 (Montreal 1).
8. R u n d 1 e R. E., Wilson A. S., Acta Crystallographica, 2 148 (1949).
9. Baenziger N. C, R u n d 1 e R. E., Snow A. I., Wilson A. S., Acta
Crystallographica 3, 34 (1950).
10. В r e w e r L., Edwards R. K., Templeton D. H., Report UCRL-433
(revised), Nov. 15, 1949.
11. Gordon P., К a u f m a n n A. R., Report AECD-2683, 1949.
12. SchrammCH., Gordon P., К a u f m a n n A. R., Report AECD-2686, 1949.
13. W i 1 h e 1 m H. A., Carlson O. N., Report AECD-2717, 1949.
Часть III
БИНАРНЫЕ СОЕДИНЕНИЯ
УРАНА
(за исключением галогенидов)
Глав а 8
СИСТЕМА УРАН—ВОДОРОД
РАСТВОРИМОСТЬ ВОДОРОДА В ТВЕРДОМ И ЖИДКОМ
МЕТАЛЛИЧЕСКОМ УРАНЕ
Вследствие образования гидрида урана (см. стр. 157 и ел.) нельзя изучать
растворимость водорода в уране при низких температурах и высоком
давлении. Например, при парциальном давлении водорода в 1 атм растворимость
может быть измерена только при температуре выше 435°, т. е. выше
температуры разложения гидрида при атмосферном давлении. Растворимость водорода
в уране гораздо меньше, чем в таких металлах, как железо, однако она заметно
возрастаете температурой, особенно для жидкого урана. Следовательно, в
отливках урана могут остаться окклюдированными значительные количества
водорода, растворенные в расплавленном металле. Растворимость водорода
в уране, поглощение его и выделение урана изучены довольно подробно [1].
В ранних исследованиях содержание водорода в образцах урана
определялось путем нагревания металла в вакууме до 750° и собирания откачанного
газа. Однако позднее было установлено, что при этой технике часть водорода
образуется за счет восстановления влаги, присутствующей в системе.
Поэтому в более поздних работах количество водорода, окклюдированного ураном,
устанавливали путем измерения давления в процессе нагревания металла
в закрытых сосудах. Сравнимые результаты получаются путем сожжения
металла в кислороде и взвешивания образовавшейся воды после поглощения
ее каким-либо осушителем. Необходимо вводить поправку на растворимость
водорода при температуре обезгаживания, что определяется первым из
указанных выше методов. Аналитические методы более подробно освещены в
другом томе данной серии [2].
На рис. 18 воспроизведена изобара растворимости при давлении водорода
в 1 атм. Она показывает, что равновесное содержание водорода в а-фазе урана
составляет около 2-10~4% и мало изменяется
с температурой. Это соответствует
приблизительно 0,4 см? водорода на 1 см3 металла,
или 1 атому водорода на 2000 атомов урана.
Превращение а-урана в р-уран при 660°
вызывает рост растворимости от 2-Ю"4,
примерно, до 8-10"4% (вес), переход (3—>т ПРИ
770°—дальнейшее повышение до 15 • 10"4 %.
Резкое увеличение растворимости с 17-10~4
до 28-10"4% соответствует температуре
плавления металла (1133°). Содержание водорода в
жидком металле быстро возрастает с
температурой, достигая 30- Ю-4 % при 1250°.
В табл. 58 даны значения коэффициента
диффузии водорода в а-уране, выведенные
из опытов по скорости обезгаживания стержней различного размера.
Как указано выше, растворимость водорода в уране пропорциональна
квадратному корню из значения парциального давления водорода.
Таблица 58
ЗАВИСИМОСТЬ КОЭФФИЦИЕНТА
ДИФФУЗИИ ОТ ТЕМПЕРАТУРЫ
Температура,
°С
566
593
640
Коэффициент
диффузии, смЦчас
0,0258
0,0413
0,0806а)
а) Экстраполированное значение.
28\-
t
24
eg
с*.
<§ W\
CD
i
12\-
4X-
III
I о -riftn—
V-
I I i I
^ 0
IMM
Э
Г 1
-j
о
i i i i i i i i l_j.._lJ
450 550 650
750 850 950
Температура, °С
1050 1150 1250
Рис. 18. Растворимость водорода в металлическом уране (в
равновесии с водородом при^атмосферном давлении).
0,0020
0,0018
Q0016
0,0014
| 0,0012
IsQOow
g.
<§ 0,0008 \~
с§
0,0006
0,0004
Q0002
О
л/р (мм рт. ст.)
Рис. 19. Зависимость растворимости водорода в металлическом
уране от давления водорода при температуре 600, 700 и 800.
Глава 8. Система уран—водород
157
Это иллюстрируется рис. 19, который показывает, что изотермы растворимости
как функции квадратного корня давления являются прямыми линиями.
Такая пропорциональность растворимости квадратному корню из значения
давления показывает, что водород растворяется в уране в форме свободных
атомов. Это правило имеет силу для всех трех фаз твердого урана (a, [J и ^)>
так как для каждой из них изотерма имеет на рис. 19 вид прямой линии.
ГИДРИД УРАНА
Гидрид урана впервые получен нагреванием порошка урана в атмосфере
водорода до 225° [3]. При этом сделано наблюдение, что дальнейшее нагревание
гидрида до 350—400° при парциальном давлении водорода 1 атм ведет к его
разложению. Подробное изучение гидрида урана ведется с 1943 г. [4—11].
Простота стехиометрического состава и постоянство давления разложения
при каждой данной температуре указывают на то, что гидрид урана является
истинным химическим соединением. При выдерживании двух сообщавшихся
сосудов, содержавших соответственно порошок урана и гидрид урана, в
эвакуированной системе в течение 10 час. обмена водородом между ураном и
гидридом при 269, 288, 300 или 325° не происходило [12]. Такое поведение типично
для истинных соединений, так как в случае даже небольшой зависимости
давления* равновесия от концентрации водорода последний диффундировал бы
из гидрида в металл. С другой стороны, по внешнему виду и физическим
свойствам гидрид урана напоминает металлоподобные гидриды церия, лантана
и других редкоземельных металлов. Для всех этих гидридов в ряде опытов
отмечено более или менее постепенное изменение давления разложения с изменением
содержания водорода в металле. Это непостоянство давления разложения,
согласно правилу фаз, указывает на то, что металл и водород образуют одну
твердую фазу переменного состава; в связи с этим гидриды редкоземельных
металлов можно считать «твердыми растворами» [13]. Однако изотермы разложения
этих гидридов, обладающие более или менее пространным, хотя и не
строго горизонтальным участком кривой, не типичны для истинных
растворов. Возможно, что гидриды редкоземельных металлов, подобно гидриду
урана, тоже являются истинными соединениями с точным стехиометриче-
ским составом. Тот факт, что они не дают горизонтальных изотерм, может
быть объяснен применением недостаточно чистых и гомогенных металлов:
использованные металлы могли содержать две или большее количество твердых
фаз, например аллотропные формы самого металла, сплавы, окислы или
карбиды.
Изотермы разложения гидрида урана. Как
упоминалось выше, интерпретация гидрида урана как химического соединения основана
на форме его многократно снятых изотерм разложения [14]. Первые
измерения показали, что равновесие устанавливается чрезвычайно медленно. Изо-
' терма для 357°, полученная в результате откачивания водорода из гидрида
и выдерживания в течение 30 мин., имела горизонтальный участок на уровне
давления 120 мм рт. ст.; однако подобная же изотерма, полученная путем
насыщения металла водородом и выдерживания в течение такого же времени,
имела горизонтальный участок примерно при 170 мм рт. ст. Позднее эта изотерма
■была повторно определена, причем для каждой точки кривой выдерживание
производили до тех пор, пока давление не оставалось постоянным в течение
24 час; обычно для достижения этой степени постоянства требовалось два дня.
Результаты показаны на рис. 20. Если равновесие достигалось «снизу», т. е.
при разложении гидрида, то на кривой имелся горизонтальный участок при
134 мм рт. ст., простирающийся в пределах 5—90% разложения. Наиболее
заметной особенностью этой кривой является небольшое, но воспроизводимое
падение давления в области 90—98% гидрирования урана с минимумом около
158
Часть lit. Бинарные соединения урана
98% гидрида. Давление, соответствующее нижним точкам этой выемки, не
изменилось даже в том случае, когда после откачивания водорода из гидрида
урана выдерживание производили в течение 14 суток. Такая выемка
наблюдалась не только для изотермы дегидрирования, полученной при
температуре кипения ртути (357°), но и для изотерм, полученных при 440°
(температура кипения серы) и 307° (температура кипения бензофенона).
Изотерма гидрирования (пунктирная линия) не имеет такой выемки, и ее
горизонтальный участок простирается от 5 до 95% гидрирования. Несмотря на
продолжительное выдерживание, данная изотерма всегда проходила на
несколько миллиметров ртутного столба выше изотермы дегидрирования. Это указывает
на то, что полного термодинамического равновесия достигнуто не было. Область
5
CJ
5'
С».
*
*
<Ъ
5
*
$
<s*
<аэ
«3
^
220
200
180
WO
140
по
100
I—I—I—1—1—
\—
•
F "* "*"
r
1 l_ 1 1. J_
1 1 1 1 i fl
I
44
4
in
41
J
Ф jr\
i i i.. i .... i
0 20 40 GO 80 100
UH3, %(мол.)
Рис. 20. Изотерма давление—состав для системы уран—гидрид
урана—водород при 375°.
О —кажущееся равновесие при разложении; ф —кажущееся равновесие прн
образовании.
«псевдоравновесия» особенно широка в месте выемки кривой. Опыты, в которых,
смесь урана с гидридом урана была полностью защищена от паров ртути,
показали, что образование выемки не обусловлено реакцией со ртутью.
Рентгенограмма в области выемки показывает только линии металлического урана
и его гидрида.
Причина образования выемки на кривой пока остается неясной. Если
предположить, что изотерма гидрирования соответствует истинному
равновесию, в то время как изотерма дегидрирования лежит «слишком низко»,
вследствие медленного приближения к равновесию, то выемка, повидимому,
показывает, что это медленное наступление равновесия особенно резко проявляется
в том случае, когда гидрирование проходит почти полностью. Этот факт
нелегко объяснить. Подобный же эффект на противоположном конце изотермы
(между 0 и 5%) гидрирования объясняется легче.
Состав гидрида уран* а. Состав, при котором урановодородные
изотермы резко изгибаются, переходя из горизонтального положения почти
в вертикальное, может рассматриваться как действительный состав гидрида
урана. Если бы водород растворялся в решетке гидрида урана, то содержание
водорода в твердой фазе продолжало бы увеличиваться с возрастанием
давления водорода выше значения, соответствующего стехиометрическому
соединению. Сначала было высказано предположение, что путем применения
«сверхнасыщающих» давлений водорода можно будет получить твердые фазы,
Глава 8. Система уран—водород
159
содержащие значительный избыток этого газа [15]. Однако позднее доказано,
что содержание водорода заметно не возрастает даже при таком высоком
давлении водорода, как 136 am [16J. Это показывает, что за точкой насыщения
изотермы практически вертикальны. Единственным видимым результатом
действия высокого давления на структуру гидрида является рост
кристаллов, который можно наблюдать при температурах выше 600°. Рентгеновские
линии в этом случае оказываются более резкими, и в порошке гидрида,
полученного под высоким давлением водорода, можно заметить волокнистые
кристаллы.
Ввиду того что гидрид урана является истинным соединением и почти не
растворяет водорода, анализы дают для него один и тот же состав независимо
от температуры и давления водорода, при котором он был приготовлен. При
первых определениях был установлен состав между UH3,85 (для препарата,
охлажденного в вакууме) и UH/bi5 (для препарата, охлажденного в водороде);
в связи с этим состав гидрида был принят за UH4 [17]. Позже, однако,
найдено [5, 18], что эти результаты являются неточными и действительный состав
близок к UH3. Такой вывод сделан на основании ряда измерений,
проведенных при температуре 300° и давлении водорода от 253 до 1265 мм рт. ст.
Полученное значение соответствовало UH2,9t[5]. В других опытах [18]
состав колебался от UH2,9i до UH3,io (в среднем UH2,9o)« В'этих опытах
измеряли привес урановых стружек при нагревании их в атмосфере водорода до
250°, привес порошкообразного урана (полученного от разложения гидри-.
да урана) при комнатной температуре, а также объем газа,
абсорбированного в процессе гидрирования или выделившегося при разложении гидрида,.
На основании результатов шести опытов определения количества воды,,
образовавшейся при сжигании гидрида, приготовленного при 250°, установлен
состав между UH2i94 и UH2>96 [19]. Продукт, приготовленный при 420°, имед
тот же состав. Отклонения от формулы UH3, отмеченные в этой работе, хотя,
и небольшие, выходят за пределы ошибки опыта, однако их можно объяснить,
присутствием окислов, карбидов или других различных примесей в исходном
металлическом уране. Например, при наличии в порошке металла 0,14%
углерода в виде монокарбида урана отношение водород: уран должно
уменьшиться до 2,91 (при условии, что монокарбид совершенно не восстанавливается
водородом) [5]. В одном из исследований [20] показано, что количество
примесей в данном известном образце было таким, что при гидрировании его
должен был получиться гидриде отношением Н : U, очень близким к
экспериментальной величине 2,97. В другой работе было показано, что при реакции
известного количества урана с водородом после введения поправки на
содержание окисла в металле (которое достигало 11,7%) должен получиться гидрид
состава UH3.0e 18]. В табл. 59 дано отношение водорода к урану, найденное
при различных температурах [6]. Таким образом, формула UH3 может
считаться точно установленной.
Таблица 59
ОТНОШЕНИЕ ВОДОРОДА К УРАНУ В ГИДРИДЕ УРАНА
Температура,
°С
150
175
200
225
Н : U
3,03
2,99]
2,96
3,04]
Температура,
°С
250
275
300
350
Н : U
3,03
3,03
2,99
2,92
160
Часть 111. Бинарные соединения урана
Кривые давления водорода и термодинамика
образования гидрида урана. Ввиду того что гидрид урана ведет
себя, как истинное химическое соединение (т. е. имеет постоянное давление
водорода внутри широкого интервала средних содержаний водорода в твердой
фазе), можно построить однозначную кривую зависимости давления
разложения его от температуры. Из наклона этой кривой можно рассчитать теплоту
образования гидрида из элементов. Первые исследования были проведены при
468 441
1000Y-
500
к 200
tf 100
I 50
20
1 I Г
Температура, С
417 394 372 352 333 315 298
I I I I I I I I I I
IffP^-^+ZSS
Давление,
мм pm. от.
0,01
0,10
100
10t00
Температура,
°С
130
169
216
274
I I I I I
701
135 140 1/15 150 155
±Ч03
I I I I
160 1,65 1,70 1,75
Рис. 21. Зависимость давления равновесия от температуры для
системы гидрид урана £ уран+водород.
В табличке на графике указаны величины, полученные экстраполяцией. #
252—438° [20], причем измеренные давления составляли от 4 до 808 мм рт. ст.;
давление в 1 атм достигалось при 436°. Но в процессе этих измерений,
очевидно, не было достигнуто точное термодинамическое равновесие, как это
вытекает из значительных расхождений между кривыми нагревания и
охлаждения. Согласно последним сообщениям [20, 21], кривая нагревания может быть
представлена уравнением
Ыр*
-^ + 9,23.
(О
Соответствующее значение А# для теплоты разложения равно 31,0 ккал-моль*1.
Результаты подобных измерений, проведенных в другой лаборатории [5],
показаны на рис. 21. Прямой на рис. 21 отвечает следующее уравнение:
lgp**=-^ + 9,39.
(2)
Соответствующее А#равно 31,5ккал-моль"1. Довольно грубое прямое
калориметрическое определение дало А#=30,5±0,5 ккал'моль'1, что
удовлетворительно совпадает со значениями, рассчитанными из кривых разложения [22].
Глава 8. Система уран—водород
161
Однако величины давления разложения, использованные в
вышеприведенных расчетах, вероятно, являются все же заниженными вследствие того,
что полного равновесия достигнуто не было [21]. Три точки на кривой
разложения, полученные в результате болез точных опытов с выдерживанием в
течение нескольких суток при постоянной температуре, лежали заметно выше
точек, полученных в более коротких но времени опытах. Однако при этом не
наблюдалось заметного изменения наклона кривой давление—температура
и, таким образом, значение Д# почти не изменилось. Прямой, проходящей
через эти три точки, соответствует следующее уравнение:
Ырмм= -^ + 9,28. (3)
Попытки приблизиться к равновесию со стороны более высоких
температур, т. е. путем гидрирования ypata, а не дегидрирования его гидрида,
дали менее удовлетворительные результаты. Конечные значения не были
достигнуты даже после выдерживания в течение недели.
В табл. 60 представлены давления, полученные в Калифорнийской
радиационной лаборатории при приближении к состоянию равновесия с обеих
сторон [8, 23J. Эти результаты могут быть выражены уравнением
1 4480 . л ол
Ырмм= --у-+ 9,20,
(4)
соответствующим теплоте разложения 30,7 ккал • моль'1.
Из этих давлений разложения были вычислены Д#0, А/7 и AS для
образования гидрида урана из элементов [23, 24]. Этот расчет основан на
приближенном значении ДСр=М кал-моль'1 для реакции
иня
>и + £нг
(5)
Указанное значение АСр найдено с помощью правила Коппа и дает для
теплоты разложения следующие значения:
Д# = Д#0 +Ы0"3Г ккал• моль'1,
Д#о = 30,1 ккал-моль"1, (6)
Д#2е8 = 30,4 ккал-моль'1-
Из уравнения
ДЯо
#1пК = #1пр8/2==ДСр1пГ-^-/
выводится для ДСр = 1
ад-1пр = 1г.Г-^—/.
2 >„н Т
Таблица 60
ДАВЛЕНИЕ РАЗЛОЖЕНИЯ ГИДРИДА УРАНА [23]
(7)
(8)
Температура,
°С
200
250
300
Давление Нг.
мм рт. ст.
0,6 1
4,5
24,8
Температура,
I ?С
1
350
400
| 436
Давление H2t
мм рт. ст.
103,0
345
760а)
11 Химия урана
а) Экстраполированное значение.
162
Часть III. Бинарные соединения урана
Из 17 отдельных измерений давления разложения между 530 и 652° К [23]
найдены значения / от —35,5 до —36,4 (в среднем / = —35,85). Это дает
для стандартной свободной энергии разложения гидрида урана:
откуда
AF° = 30100-2,303TlgT-35,85 Г кал-моль'1,
ДЛ>98=17,7 ккал-моль'1.
(9)
Комбинируя последнее значение 4F°>98 с Д#298 = 30,4 ккал-моль \ получим
для энтропии разложения
Д5298 = 42,6 энтропийной единицы. (10)
Соответствующие значения для образования гидрида урана из элементов
составляют:
Д#298= —30,4 ккал-моль'1,
А^гэв— —17,7 ккал-моль"1,
Д5° =
■42,6 энтропийной единицы.
Кинетика образования и разложения гидрида урана.
Как уже указывалось, впервые гидрид урана был получен нагреванием
порошка урана до 225° и выше в атмосфере водорода [3]. Затем опыты
150
J I L
J L_L
200 250 300 350
Температура реакции, °С
400
Рис. 22. Влияние температуры на скорость реакции.
О—очищенный водород; х—водород из баллона.
других исследователей подтвердили, что наилучшей для быстрого
гидрирования является температура, равная 225—250° [25]. Например, 100 г
урановых стружек можно полностью превратить в гидрид в течение 30 мин*
при 250°, а кусок урана весом 100 г—за 2 часа [6, 25, 26].
Влияние температуры. Для процесса гидрирования характерно быстрое
падение скорости его при температурах выше 300°, независимо от
состояния, в котором находится металл (порошкообразный или плотный).
Например, под парциальным давлением водорода равным 1 атм урановый порошок
при 440° совершенно не поглотил водорода за 2 суток; для полного
гидрирования при 306—420° требовалось трое суток [12], при 250°—менее 1 часа.
Аналогичные результаты были получены для скорости проникновения водорода
Глава 8. Система уран—водород
163
Таблица 61
СКОРОСТЬ ПРОНИКНОВЕНИЯ
ВОДОРОДА В ТВЕРДЫЙ УРАН
в твердый уран (ср. табл. 61) [26]. Рис. 22 показывает зависимость
скорости гидрирования урановой проволоки (диаметром 1,59 мм) от
температуры [6]. Показанные значения—это приблизительно постоянные скорости,
наблюдавшиеся после окончания индукционного периода (см. ниже) и до
замедления процесса к концу гидрирования.
Эта кривая также имеет максимум при 225°.
Падение скорости реакции при
температурах ниже 200° зависит от состояния
металла и давления водорода. Если взяты
стружки, реакция становится заметной при
200°, когда давление водорода 1 стм, при
150°—если оно 70 am и при 130°—когда
оно 125 am. Тонкий порошок урана,
полученный разложением гидрида, реагирует с
водородом очень быстро даже при 0° и заметно,
но медленно при —80°; реакция совсем не
идет при температуре жидкого воздуха.
Влияние давления. Падение скэрости гидрирования при температурах
выше 250° связано с тем, что с повышением температуры растет давление
разложения гидрида; когда оно достигает внешнего давления водорода,
скорость гидрирования падает до нуля. Таким образом, скорость
гидрирования должна быть функцией избыточного давления водорода, т. е.
разности внешнего давления водорода р и дав- 3
ления разложения гидрида р0. Измерения || 800\
скорости при постоянной температуре
(357°) [27] показали, что начальная
скорость гидрирования порошка урана (рис. 23)
может быть представлена уравнением
Температура,
°С
200
250
300
Скорость
проникновения,
cMJnac
0,27
0,30
0,19
"'--(МХо-х^-роУ*' <п>
800
Начальное давление, мм рт. ст.
Рис. 23. Влияние начального
давления водорода на скорость реакции
между водородом и ураном при 357°.
где р0=134 мм рт. ст. Интегрирование
уравнения (11) показывает, что
зависимость (pt—Ро)~3/2 от времени должна
быть линейной. Величина pt являетс51
остаточным давлением водорода после
гидрирования в течение времени /. Этот вывод
подтверждается опытами (рис. 24).
Влияние поверхности. В процессе
гидрирования урана через некоторое время всегда наступает постепенное
замедление потребления водорода [6]. Эти опытные результаты можно
объяснить тем, что скорость этой реакции пропорциональна активной
поверхности урана. Для вычисления активной поверхности исходили из
предположения, что процесс гидрирования проволоки распространяется от
поверхности к оси, причем поверхность сохраняет свою цилиндрическую форму.
Это предположение, очевидно, не может быть совершенно точным, но других
количественных данных о зависимости между поверхностью и скоростью
гидрирования не имеется, кроме обычных сведений о том, что тонкий порошок
гидрируется быстрее, чем плотный металл.
Индукционный период. Выше было высказано мнение, что падение
потребления водорода по мере гидрирования может быть количественно объяснено,
если скорость относить к единице активной поверхности. Рис. 25 показывает,
что скорость, рассчитанная таким образом, остается постоянной в течение
всего процесса, если не считать слабого падения к самому концу гидрирования
11*
164
Часть III. Бинарные соединения урана
(что, вероятно, обусловлено ошибками метода) и индукционного периода в
начале процесса.
Продолжительность индукционного периода зависит от чистоты водорода;
при 250° он равен 28 мин. в случае использования водорода из баллонов и
практически равен нулю при применении водорода, очищенного раскаленным
20 г
0.01 0.02
Q03 Q04
0.05 Q06
Рис.
24. Скорость гидрирования
порошкообразного урана при 357°.
ураном. При 150° индукционный период заметен даже при использовании
водорода, очищенного самым тщательным образом. Индукция может быть
объяснена наличием защитного поверхностного слоя окисла или нитрида.
Однако предварительное выдерживание металла в азоте или кислороде не
вызывает возникновения индукционного периода при последующей обработке
чистым водородом.
•а *
Г"
V-
1 1
/
/
•
J L
1 1
шП—п—
£>^"" *■ U
1 1
Г
1
1
1 и
1
1 1
! 1
1 ! |
I
1 1
в 12
Время, мин.
16
20
Рис. 25. Скорость абсорбции водорода, отнесенная к единице
поверхности образца урана. Температура реакции 225°.
После окончания индукционного периода скорость гидрогенизации
одинакова для водорода очищенного и водорода из баллона [6].
Были сделаны попытки приготовить гидрид урана под инертным
растворителем (минеральным маслом), но скорость гидрирования при 250° в таких
условиях составляла только 1% скорости в газообразном водороде [14].
Механизм образования гидрида. Образование и разложение гидрида урана
является интересным примером для изучения кинетики гетерогенных реакций.
Тот факт, что скорость зависит от разности давлений (р—р0), повидимому,
указывает, что скорость лимитируется процессом диффузии как наиболее
медленной стадией, но пока нельзя еще дать подходящего объяснения, почему этот
член фигурирует в уравнении (11) в степени 5/2- Наличие этого члена в урав-
Глава 8. Система уран—водород 165
нении косвенно объясняет падение скорости гидрирования при высоких
температурах, где р0 близко к р.
Кинетика разложения. Опыты по изучению условий разложения гидрида[28]
показали, что при 250° около 50% образца гидрида весом в 6—7 г может
разложиться за 1 час при остаточном давлении водорода около 0,1 мм рт. ст.
Проведены систематические измерения при температурах 294, 367 и 405° [29].
Однако немедленно после начала откачивания температура падала почти на 100°
и только постепенно возвращалась :< первоначальному уровню. Кроме того,
истинное равновесное давление водорода на поверхности гидрида неизвестно
и может быть неодинаковым для различных частей образца. Таким образом,
кривым разложения (рис. 26) нельзя придавать общего значения. Эти кривые
70
50
40
30
20
4
3
2
О 20 40 60 80 100
Количество разломившегося гидрида, %
Рис. 26. Зависимость скорости разложения гидрида урана
от температуры.
Около кривых указаны начальные температуры.
указывают на приблизительно первьй порядок реакции и довольно слабый
рост скорости разложения с температурой. Например, для 50%-ного
разложения требуется 30 мин. при 294° и 3 мин. при 405°. Такой слабый рост может
быть обусловлен тем фактом, что давление водорода над поверхностью
гидрида возрастает с повышением температуры вследствие ограниченной скорости
откачивания газа.
Образование гидрида из соединений урана или
из урана и воды. Образование гидрида из соединений урана. Показано,
что при нагревании карбида или двуокиси урана в водороде гидрида не
образуется [25]. Сплав урана с 20% алюминия реагирует с водородом с
образованием порошка, который, повидимому, содержит металлический
уран и соединение урана с алюминием. Едкий натр выщелачивает из этого
порошка алюминий, по меньшей viepe частично. Амальгама урана не
реагирует с водородом [12].
Образование гидрида из урана и паров воды. Вода разлагается ураном в
интервале температур 600^700°, причем образовавшийся водород может быть
использован в том же аппарате для гидрирования урана при 250° с
образованием гидрида урана [30]. Значительные количества гидрида были также
получены в случае прямой реакции урана с водяным паром при 250° [31].
166
Часть IIL Бинарные соединения урана
Образование гидрида из урана и декалина или тетралина.
Небольшие количества гидрида образуются при взаимодействии металлического
урана с декалином или тетралином при 210—270°.
Физические свойства гидрида урана. В литературе
гидрид урана описывается как коричневато-черный или коричневато-серый
пирофорный порошок. Он легко проходит через сито в 400 меш или шелковую
ткань для сит. Приготовленный из металла, он содержит блестящие частицы,
вероятно включения окисла из исходного металла. После просеивания,
разложения в вакууме и повторного синтеза гидрид является более однородным
и имеет более сероватый цвет [21, 25].
Кристаллическая структура и плотность. Первые рентгенографические
анализы гидрида урана показали, что он имеет простую кубическую структуру,
в корне отличающуюся от трех
структур металлического урана
[32]. Это определение было
повторено с особенно чистым препаратом
[33, 34]. Образцы, приготовленные
при 200—300° и давлении
водорода, равном атмосферному, дали
удовлетворительные, но не очень
резкие пятна в области обратного
отражения симметричной
самофокусирующей камеры с радиусом 5 см
с Си Ка-излучением. Причина
размыва пятен кроется в малых
размерах частиц гидрида. Гидрид,
приготовленный под давлением в
125 am при 500—600°, состоит из
кристаллов гораздо большего
размера и дает очень четкие
максимумы. Наилучшее значение для
констант решетки
а0 = 6,6310 ± 0,0008 А
было получено с препаратами, приготовленными под высоким давлением.
Гидрид «низкого давления» дает значение
а0 = 6,632±0,001А.
Другие исследователи [35] определили константу решетки гидрида
а0 = 6,634 ±0,002 А,
что хорошо согласуется с предыдущими значениями.
Недостающие отражения в рентгенограмме показывают, что решетка
гидрида урана может принадлежать к одной из трех пространственных групп 0^уОъ
и Td\ окончательный выбор пространственной группы невозможен без знания
положений атомов водорода. Элементарная ячейка содержит восемь молекул.
Положения восьми атомов урана можно разделить на две группы: I—два
равноценных положения вОООиуууИ II—шесть равноценных положений
в ТТ0; °ТУ; 4~°Т; ТТ0; Т°Т и ° ТТ- Это Расположение
представлено на рис. 27. Каждый атом группы I имеет двенадцать атомов
группы II в качестве ближайших соседей нао расстоянии 3,707 А; каждый
атом группы II находится на расстоянии 3,316А от двух ближайших соседних
атомов той же группы, на расстоянии 3,707 А от четырех расположенных
6,631 А
Рис. 27. Структура гидрида урана.
Глава 8. Система уран—водород
167
по четырехугольнику соседних атомов группы I и на расстоянии 4,06 А
от восьми атомов группы II.
Структура гидрида урана недавно вновь сделалась предметом
исследования [36, 371. Была выдвинута теория электронно-недостаточных структур,
которая, повидимому, позволяет правильно отобразить структуру гидрида
и качественно объяснить физические свойства этого соединения.
Кристаллическая структура гидрида в корне отличается от структур различных форм
металлического урана. Судя по расстояниям между атомами металла
в гидриде, можно вывести, что в этом соединении практически отсутствуют
металлические связи. Тем не менее гидрид обладает явно металлическими
свойствами, например высокой электропроводностью и металлическим
блеском. Высокая температура плавления, хрупкость и твердость также
несовместимы с очень слабыми металлическими связями и, наоборот, говорят о
структуре непрерывного типа, созданной ковалентными связями. Поэтому была
предложена [36] структура, согласно которой каждый атом урана типа I
связан с двенадцатью атомами урана типа II водородными мостиками U—Н—U,
каждый же атом урана типа II непосредственно связан с двумя другими
атомами этого типа и при помощи водородных мостиков с четырьмя атомами урана
типа I [37, 381.
Плотность гидрида, рассчитанная на основании рентгенографических
данных, равна 10,92г/сж3. Этот результат прекрасно совпадает с экспериментальным
определением плотности методом гидростатического взвешивания в гелии [21,39],
давшим 10,95 г/см3. Прежние, менее точные определения плотности (под гек-
саном) дали значение 11,4 г/см3. Насыпной вес сухого порошка гидрида равен
3,4 г/см3', смоченный гексаном порошок уплотняется до 3,5 г/см3.
Центрифугирование увеличивает плотность до 4,0, давление в 3940 кг/см2 повышает ее до
7,3 и давление в 22 500 кг_/см2—до 8,4 г/см3 [21 ]. Порошок гидрида урана,
приготовленный при 700—800° под давлением водорода 125 am, может быть
спрессован до насыпного веса, превышающего 9,0 г/см3 [16].
Низкий насыпной вес гидрида урана является причиной большого
увеличения объема металла при гидрировании. Это разбухание металла необходимо
учитывать в конструкции гидрогенизационных аппаратов.
В частично разложенных или неполностью прогидрированных образцах
гидрида плотность с точностью до ± 1 % является линейной функцией
содержания водорода [35]. Константа решетки гидрида не изменяется при
нагревании смеси гидрида с металлическим ураном; так, после нагревания смеси,
состоявшей изо1 части гидрида и 1 части урана, константа а0 равнялась
6,630±0,002 А. Эти результаты типичны для механической смеси двух твердых
фаз, не обладающих взаимной растворимостью. Как указывалось выше,
образец гидрида, приготовленный под давлением водорода в 125 am, заметно не
отличался по максимумам на рентгенограмме от гидрида, приготовленного под
низким давлением водорода. Другими словами, отсутствуют какие-либо
признаки искажения решетки растворенным или окклюдированным водородом.
Гидрид, выделенный из амальгамы гидрида урана путем аэрации (см. стр. 171
и ел.), имеет ту же константу решетки, что и обычный гидрид.
Электропроводность. Порошок гидрида, впрессованный между
электродами, обладает проводимостью металла. Удельное сопротивление гидрида
урана, равное 0,47 ом-см, не очень сильно отличается от удельного
сопротивления порошка металлического урана, полученного разложением гидрида
(0,68 ом-см) [17]. Спрессованный порошок проводит ток лучше рыхлого [21].
Химические свойства гидрида урана. Гидрид урана
является весьма реакционноспособным веществом и может быть использован
для получения различных соединений урана. Во многих случаях в ходе
реакции в качестве промежуточного продукта образуется, вероятно, свободный
металлический уран, получающийся в результате разложения гидрида в
Таблица 62
РЕАКЦИЯ ГИДРИДА УРАНА С ГАЗАМИ
Температура,
°С
Продукт
Примечания
Элементарные газы
225
250
900
Воспламенение
200
200—300
UN,,.
UN(?)
u3o8
UCI4
UBr4
400
Ш<4)(?)
Медленная реакция, неполная
Быстрообразуется пирофорный нитрид [45]
Найдено 3,6% N (вычислено для UN
5,5%) [43]
При недостатке воздуха может
образоваться двуокись урана
Продукт плавится во время реакции
Коричневый, частично сплавленный
порошок
Продукт неустойчив, состав точно
неизвестен [46]
350
500
400—500
100
250
400
20-400
250—300
200
300—400
ио2
ио2
US2
UN(?)
Гидриды
Черная двуокись, состав, вычисленный
по привесу U02li4 [17] (избыток
против—U02,o обусловлен, вероятно,
просачиванием воздуха)
Реакционная масса раскаливается
Черный порошок; в качестве
промежуточного продукта может быть выделен»
U2S3; реакция медленная
«Низкотемпературный» нитрид;
медленная реакция
UN1>6—UN2 Пирофорный (?); реакция более быстрая/
U2P3 Черный порошок
UF4 | Зеленый сухой порошок; типичная
«зеленая соль»
UC13 I Сухой порошок; оливково-зеленый при
25°; красновато-бурый при более
высоких температурах
иВг3' | Красновато-бурый сухой порошок;
наилучшие результаты получены
пропусканием НВг через UH3 снизу при
250—275° [47]
Ш4(?) I Продукт неустойчив; анализы дают
Ш8-4
СО
С02
До 400
200
U
ио2
Окислы
Летучих карбонилов не образуется
Загрязненный продукт (карбидом);
после начала реакции гидрид все
время гсрит
а) Для лучшего понимания реакции UH3 с азотом и аммиаком следует прочитать
описание нитридов урана в гл. 10.
Глава 8. Система уран—водород
169
Продолжение табл. 62
Газ
Температура,
°С
Продукт
Примечания
Органические газы
С2Н4
HCN
С0С12
CH3J
CCI4
500
400
250
275—300
250
UC(?)
Карбид,
нитрид
исц
Ш(?)
UC14+C(?)
Неизвестный продукт, содержащий
1,5% С; реакция, вероятно,
неполная
Черный порошок; пирофорный; не
содержит CN
Желто-зеленый сухой порошок; слабо
загрязненный углеродом
Состав точно неизвестен; реакция
протекает с умеренной скоростью
Вероятные побочные продукты—метан
и этан [48, 49]
тонкодисперсном и химически активном состоянии. Реакции гидрида изучены
рядом исследователей [17, 21, 40—43].
Реакции с газами. В табл. 62 даны реакции гидрида урана с различными
газами [21, 44]. Гидрид часто является пирофорным [25] и требует
осторожного обращения. Образцы, не обладающие пирофорными свойствами, вероятно,
защищены поверхностным слоем двуокиси; они могут быть получены путем
предварительной экспозиции гидрида на воздухе при температуре сухого
льда. Двуокись углерода и азот являются инертными средами для гидрида
вплоть до 200—225°; при более высоких температурах эти газы тоже реагируют
с гидридом. Раз начавшись, реакция с двуокисью углерода или азотом не
может быть остановлена, однако взрыва при этом можно не опасаться, в отличие
от поведения гидрида в кислороде. На открытом воздухе гидрид спокойна
сгорает до воды и закиси-окиси урана U308; при недостатке воздуха может
образоваться двуокись.
Можно измерить скорость окисления гидрида урана на воздухе, если
только предотвратить возможность воспламенения путем предварительной
осторожной экспозиции на воздухе [50]. Начальная скорость увеличения в весе
устойчивого образца гидрида урана за счет реакции с кислородом составляла
0,25 мг/г в 1 сутки; через 45 суток эта скорость все еще составляла около
половины начальной величины. Так называемый квази-гидрид (см. стр. 171 и ел.)
увеличивался сначала в весе на 0,83 мг/г в 1 сутки, но через 45 суток скорость
падала до одной пятой этой величины.
Из табл. 62 видно, что при взаимодействии гидрида урана с хлористым
водородом, бромистым водородом, фосфином и аммиаком образуются
соединения трехвалентного урана; реакция с хлором, бромом, водой, фтористым
водородом, фосгеном и, вероятно, йодистым водородом ведет к образованию
соединений четырехвалентного урана.
Реакция гидрида с фтористым водородом подробно изучена [42]. При 270°
образуется поверхностный слой тетрафторида урана, который препятствует
дальнейшему прониканию фтористого водорода в гидрид. При 500°
образования плотного поверхностного слоя не происходит и весь гидрид превращается
в тетрафторид. В случае взаимодействия урана со смесью равных объемов
водорода и фтористого водорода при температуре 250° имеет место спокойное
Л 70
Часть III. Бинарные соединения урана
образование тетрафторида; несомненно, что в качестве промежуточного
соединения в этой реакции получается гидрид урана [51].
Реакция с водой и водными растворами кислот и оснований. При
смешивании с водой небольшого количества порошка гидрида урана реакция не идет;
иногда все же при этом гидрид воспламеняется, но если порошок уже смочен
водой, этого можно не опасаться [52]. Большие количества гидрида всегда
воспламеняются, если не приливать к ним воду очень медленно [53]. При
200—300° гидрид урана медленно реагирует с парами воды; образующийся
окисел, вероятно, является двуокисью урана [45].
Гидрид урана растворяется в 6н. или 12н. растворе соляной кислоты
гораздо медленнее металлического урана, причем образуется светло-
-зеленый раствор [52, 54]. Гидрид легко растворим в хлорноватой кислоте
(KC103+H2S04) [55]. Разбавленная хлорная кислота не растворяет гидрида
урана или растворяет его очень медленно, концентрированная реагирует при
нагревании с образованием уранилперхлората U02(C104)2. Эта реакция протекает
спокойно в небольшом масштабе, но при количествах, превышающих 1—2 г,
становится опасной [52]. Разбавленная фосфорная кислота не реагирует с
гидридом, концентрированная растворяет его с образованием фосфата
урана (IV) [52]. Гидрид урана не растворяется в холодном 8 н. растворе уксусной
кислоты, при кипячении образуется коричневая суспензия, которая является
пептизированным гидридом или продуктом его химического превращения.
Ледяная уксусная кислота не действует на гидрид [52]. Гидрид легко
восстанавливает азотную кислоту (6 н. или концентрированную) до двуокиси азота,
причем уран превращается через зеленое промежуточное соединение
в желтый уранилнитрат [52]. Разбавленная серная кислота реагирует с
гидридом очень медленно, горячая концентрированная быстро
восстанавливается до серы и сероводорода; уран в этих условиях образует сульфат
урана (IV) U(S04)2[52].
Горячие или холодные растворы гидроокисей калия, натрия или аммония,
а также цианида натрия не реагируют с гидридом. Он не растворяется также
и в жидком аммиаке [52].
Реакции с органическими растворителями. Органические растворители,
не содержащие галогенов (например, бензол, толуол, гексан, диоксан, спирт,
ацетон, этилацетат), не растворяют гидрида и не реагируют с ним [52]. С
другой стороны, галогенированные растворители представляют опасность в случае
соприкосновения с гидридом. Например, при внесении гидрида урана в че-
тыреххлористый углерод может произойти сильный взрыв, в результате
которого образуются тетрахлорид урана, хлористый водород, водород и углерод.
Пары четыреххлористого углерода реагируют с гидридом только при
температуре выше 200° с образованием три- и тетрахлорида урана и углерода [49].
Окисление гидрида урана слабыми неорганическими окислителями. Многие
слабые неорганические окислители окисляют гидрид до солей четырех- и
шестивалентного урана с выделением водорода. Особенно эффективны соли
серебра [56], например в следующих реакциях:
2UH3 + 8AgF-^2UF4 + 8Ag + 3H2. (12)
Эта реакция протекает энергично.
2UH3 + 12AgN03 + 4H20 -* 12Ag + 2U02 (Ш3)2 + 8HN03 + 3H2. (13)
Эта реакция идет очень быстро; раствор разогревается до кипения.
Окрашивание получающегося раствора указывает на образование комплексов.
2UH3 + 6Ag2S04 + 4H20 -* 2U02S04 + 12Ag + 4H2SO, + 3H2. (14)
.Эта реакция идет медленнее двух предыдущих.
Глава 8. Система уран—водород
171
2UH3 + 12Ag (СН3СОО) + 4Н20 -> 2U02(CH3COO)2 +
+ 12Ag + 8CH3COOH + ЗН2. (15)
Это медленная реакция.
2UH3 + 6Ag2(OOCCHOHCHOHCOO) + 4H20->2U02(OOCCHOHCHOHCOO) +
+ 12Ag + 4(СНОН)2(СООН)2 + ЗН2. (16)
Эта реакция протекает еще медленнее.
С перхлоратом серебра идут три параллельные реакции:
2UH3 + 12AgC104 + 4H20 ->12Ag + 2U02(C104)2 + 8HCI04 + 3H2 (80%); (17)
2UH3 + 8AgC104 -> 8Ag + 2U(C104)4 + 3H2 « 0,1%); (18)
2UH3 + 5AgC104 -» 4Ag + AgCl + 2U02(C104)2 + 3H2 (20%). (19)
При взаимодействии гидрида с перхлоратом серебра в толуоле
образуются серебро и хлорид серебра, возможно, согласно следующему
уравнению [57]:
2UH3 + 6AgC104 -> 2Ag + 2AgCl + Ag20 + 2U02(C104)2 + 3H20. (20)
Подобные же реакции идут и с солями ртути. Например, сулема
энергично реагирует с гидридом с образованием тетрахлорида урана (15%)
и уранилхлорида (35%); остаток урана находится в осадке серого цвета,
состоящем из смеси металлической ртути с каломелью. Быстро реагирует
и нитрат ртути (II) с образованием уранилнитрата и ртути.
Трихлорид сурьмы в растворе 3 H.HC1 окисляет гидрид урана до
тетрахлорида, причем выделяется водород:
8SbC]3 + 6UH3->8Sb + 6UCl4 + 9H2. (21)
Сульфат меди (II) не реагирует с гидридом при комнатной температуре,
но при 80—90° медленно восстанавливается с образованием металлической
меди и сульфата урана. При взаимодействии гидрида урана со смесью
хлоридов меди (II) и аммония происходит энергичное выделение водорода и
образование зеленого раствора [50].
Сульфат железа (III) окисляет гидрид при нагревании до U02+[56].
С солями висмута и свинца реакции не идут [56].
Окисление гидрида урана сильными окислителями. Насыщенный раствор
гипохлорита натрия (NaOCl) слабо действует на гидрид [55].
Тридцатипроцентный раствор перекиси водорода энергично реагирует с гидридом
со вспышкой и образованием черной двуокиси [58]. При реакции перекиси
водорода с суспензией гидрида урана в соляной кислоте получается ура-
нилхлорид. Эта реакция идет медленнее в серной кислоте. В присутствии
органических кислот действие перекиси водорода приводит к образованию
соответствующих солей уранила; с избытком перекиси может получиться
надурановая кислота. Соли церия (IV) окисляют уран в гидриде до
шестивалентного состояния, причем выделяется водород. Бихромат реагирует только
при нагревании с образованием солей уранила. Неподкисленный перманганат
не взаимодействует с гидридом на холоду, в присутствии кислоты
холодный перманганат дает соли уранила. Это же верно и в отношении бро-
мата [170].
Реакции гидрида с хлорной, азотной и серной кислотами рассмотрены
выше (стр. 170).
Амальгама гидрида урана, подобные ей системы
и к в а з и-г и д р и д. Гидрид урана может быть диспергирован в ртути,
причем заметного разогревания или выделения водорода при этом не происхо-
172
Часть III. Бинарные соединения урана
дит [21, 59]. Получающаяся «амальгама» имеет различную консистенцию в
зависимости от содержания гидрида. При 10% гидрида или меньше она жидкая,
при 30%—пастообразная, при 40%—полутвердая. Если содержание гидрида.
превышает 60%, то гидрид впитывает ртуть, как древесные опилки масло.
Амальгама, содержащая 90/6 гидрида, представляет собой серый пирофорный
порошок.
Фильтрованием через алунд или пористое стекло амальгаму можно
разделить на практически чистую ртуть и остаток, который по виду похож на более
концентрированную амальгаму. Можно также выжать чистую ртуть из 35%
амальгамы путем приложения давления в 5600 кг/см2 [60]. Эти результаты
говорят о том, что амальгама является коллоидной системой, а не истинным
молекулярным раствором или химическим соединением.
Поведение амальгамы на воздухе подтверждает предположение о том, что
она является капиллярной системой. Гидрид урана, подвергнутый действию
воздуха, больше не смачивается ртутью. При аэрировании амальгамы,
приготовленной без доступа воздуха, имеет место выпадение гидрида. В процессе этого
осаждения происходит частичное окисление, в связи с чем осадок несколько
отличается от исходного гидрида. Это оправдывает его название квази-гид-
рид [61 ]. Его рентгенограмма аналогична рентгенограмме настоящего гидрида,
но он имеет коричневый цвет и, как показывает типичный анализ, содержит
97,4% урана и 1,15% водорода; остальное, вероятно, приходится на долю
кислорода. Это соответствует составу UH2>82O0i2; следовательно, квази-гидрид
является, повидимому, тесной смесью UH3 и двуокиси урана. Квази-гидрид
может быть устойчивым или пирофорным.
Вследствие частичного окисления воздухом в процессе осаждения
квазигидрида из ртути образуется вода [62]; поглощение кислорода и образование
воды сопровождаются выделением тепла. Если амальгама подвергается
действию неподвижного или медленно циркулирующего воздуха в открытом
сосуде, то поглощение кислорода и образование воды протекают быстро в
первый день и продолжаются с убывающей скоростью в течение месяцев
или даже лет [60].
Свежеосажденный квази-гидрид содержит большое количество тонкодис-
пергированной ртути, от которой он может быть освобожден путем взвешивания
в гексане или петролейном эфире и фильтрования через пористое стекло [63].
Амальгамирование гидрида урана ртутью не нарушается при наличии в ртути
магния [64].
Гидрид диспергируется не только в ртути, но также и в легкоплавких
металлах и сплавах. Так, можно «растворить» 2 г гидрида в 20 мл жидкого нат-
риевокалиевого сплава [65]. И в этом случае тепловой эффект ничтожно мал-
Обработка гидрида урана жидким сплавом Вуда или чистым оловом ведет
к образованию губчатой массы. При этой реакции выделяется водород и
образуются интерметаллические соединения [60]. Были проведены некоторые опыты
с гидридом, полученным из ураножелезного сплава [64]. Он, вероятно,
содержал UH3 вместе с каким-то неизмененным ураножелезным- соединением.
Применение гидрида урана. Первое и наиболее очевидное
использование гидрида урана состоит в приготовлении из него чистого,
тонкодисперсного металлического урана [25]. Получающийся таким образом
металл является реакционноспособным и может быть использован для
приготовления сплавов и многих других соединений, например карбида урана [66].
Синтезом и последующим разложением гидрида урана можно также получить
чистый водород или дейтерий: этот метод находит практическое применение [211-
Интерметаллические и аналогичные соединения урана не реагируют с
водородом, поэтому металлический уран может быть отделен от включений
гидрированием и просеиванием. Этот метод был приложен к ураноалюминиевым
сплавам [67]. Сплавы урана можно протравливать путем образования гидрида [68L
Глава 8. Система уран—водород
173
Предложены методы приготовления различных соединений трех- и
четырехвалентного урана из гидрида урана [69]. Многие из этих методов,
основанные на данных, представленных в табл. 62, были действительно использованы.
Так, был получен тетрафторид урана действием фтористого водорода на
гидрид [70] или одновременной обработкой урана водородом и фтористым
водородом [51, 71]. Можно также получить некоторые соли уранила с
использованием гидрида урана в качестве исходного вещества [21]. Например,
фторосиликат и лактат уранила могут быть получены по уравнениям:
2 Ag2SiF6 + 2UH3 + 4 Ag20 -> 2U02SiF6 + 12 Ag + 3H2; (22)
4(CH3CHOHCOO) Ag-t- 2UH, + 4Ag20 ->
-^ 2(CH3CHOHCOO)2U02+ 12Ag + 3H2. (23)
Попытки использовать уран в качестве катализатора гидрирования
(с нафталином и малеиновой кислотой в качестве акцепторов водорода)
были безуспешны [72].
Предложен аналитический метод определения урана, основанный на
образовании и разложении гидрида и измерении объема полученного из
гидрида водорода [73]. Подобный же процесс был использован для анализа
тяжелой воды путем превращения обыкновенной и тяжелой воды в водород и
дейтерий [21].
ДЕЙТЕРИД УРАНА
Дейтерид урана UD3 впервые был приготовлен и изучен специально
с целью выяснения возможности его использования для отделения дейтерия
от водорода.
Дейтерид урана может быть приготовлен разложением тяжелой воды
ураном при 600—700° с введением полученного дейтерия в реакцию с
ураном при 250°:
U + 2D20^=Ii°;U02 + 2D2; (24)
U-t-fD2 UD3. (25)
Оба эти процесса могут проводиться в одной и той же аппаратуре [30].
Измерено давление разложения смеси гидрида и дейтерида урана
<с 20—80% каждого из них) при 357° [74]. Результаты (рис. 28)
показывают, что эта система ведет себя как идеальный раствор, т. е. давление
является линейной функцией состава. Давление разложения чистого
дейтерида при 357* равно 185 мм рт. ст., т. е. оно значительно выше, чем для
гидрида (134 мм рт. ст.). Отношение /\jd3 : Ллн3 является постоянным
в области температур от 250 до 430° и составляет около 1,4 [75]. Для
давления разложения может быть предложено следующее уравнение:
lgp^=-^ + 9,43 (26)
Jcp. уравнение (3)].
Теплота образования дейтерида урана равна 31 ккал/моль, т. е. та же,
что и гидрида.
Скорость образования дейтерида из урана и дейтерия при 357° гораздо
меньше, чем для гидрида (рис. 29) [27]. Разница может быть объяснена
большей энергией молекулы водорода при 0° К по сравнению с энергией
молекулы дейтерия. Однако вопреки ожиданию эта разница не возрастает, а
уменьшается в том случае, когда синтез проводится при низких температурах.
Измерения при —76° дали весьма колеблющиеся результаты [76], но
174
Часть III. Бинарные соединения урана
скорости потребления водорода и дейтерия были приблизительно одинаковы
и равны 7 см3 газа в 1 час на 1 г урана (начальная скорость при 357°
составляла около 30 см3 водорода в 1 мин).
!89
Рис. 28. Давление при равновесии водорода и дейтерия
над смесями гидрида и дейтерида урана при 357°.
Более медленное образование дейтерида урана и более высокое
давление его разложения открывают, как будто, возможность разделения водорода
и дейтерия, однако опыты разложения смеси гидрида и дейтерида урана [771
900 r
6
J 2 18
Время, мин.
Р н с. 29. Скорости реакций водорода и дейтерия с
порошкообразным ураном при 357°.
привели только к слабому обогащению газовой фазы. Например, если
исходная твердая фаза содержала 19,8% (мол.) дейтерия в расчете на весь
водород, то отношение (D2 : H2) к (UD3 : UH3) было равно 1,2. Первая газовая1
фракция (из десяти) содержала 23,2% D2, десятая (последняя)—16,8% D2.
Разница в скорости образования гидрида и дейтерида урана не может быть
использована для разделения, так как обмен изотопами между твердым
Глава 8. Система уран—водород
175
гидридом и газом является очень быстрым и полностью завершается за 5 мин.,
при 300° [77J.
Описано несколько опытов по разделению амальгам гидрида и дейте-
рида. Установлено, что в этом случае обмен изотопами между газовой
и жидкой фазами идет медленно. Например, при нагревании водорода
с амальгамой, содержащей 25% дейтерида и 75% гидрида урана,
концентрация дейтерия в газе составляла всего 2,7% через 15 мин. и 10,3% через
2 часа. Обратный обмен, т. е. замещение водорода в гидриде дейтерием
из газовой фазы, протекал еще медленнее. Этот медленный обмен
препятствует использованию колонки с амальгамой для эффективного разделения
дейтерия и водорода.
Константа решетки дейтерида урана [61] а0 = 6,625А, т. е. она на
0,006 А меньше такой же константы для гидрида. Рассчитанная плотность,
равна 11,16 г\смъ вместо 10,91 г/см3 для гидрида.
ЛИТЕРАТУРА
1. Battelle Memorial Institute, Reports CT-611, Apr. 10, 1943, p. 131 — 134; CT-688, May
10, 1943, p. 144—154; CT-753, June 10, 1943, p. 176—186; CT-8I8, July 10, 1943,
' p. 222—229; CT-893, Aug. 10, 1943, p. 239—261; CT-956, Sept. 10, Г943, p. 314—319;
CT-1009, Oct. 10, 1943, p. 345—350; CT-1388, Feb. 10, 1944, p. 3—6; CT-2374, Nov.
1, 1944, p. 252—255; CT-2483, Dec. 1, 1944, p. 283—284 (Battelle I).
2. National Nuclear Energy Series, Division VIII, vol. 2.
3. Driggs F. H., пат. США 1816830, 1929; 1835024, 1929; канад. пат. 325501, 1930,.
transferred to Canadian Westinghouse Co., Ltd. (1929).
4. Warf J. C, Newton A. S., Butler T. A., Ay res J. A., Report CC-580,
Apr 15, 1943; Spedding F. H., Johns I. B., Report CC-587, Apr. 19, 1943;
Newton A. S., Warf J. C, Johnson O., N о t t о г f R. W., Report
CC-I201, Jan. 1, 1944; Warf J. C, Iowa State College, Report 1SC-50, Sept. 29,
1949. In National Nuclear Energy Series,Division VIII, vol. 6: Spedding F. H.,
Newton A. S., Johns I. В., Johnson O., D a a n e A. H., Not-
torf R. W., Warf J. C, Preparation and Physical Properties of Uranium
Hydride; Spedding F. H., Warf J. C, Newton A. S., Johnson O.,
Johns I. В., А у r e s J. A., Butler T. A., F i s h e г R. W., Not-
t or f R. W., The Chemical Properties of Uranium Hydride; R u n d 1 e R. E.,
Wilson A. S., McDonald R. A., The X-ray Investigation of the Uranium-
Hydrogen System; The Structure of UH3. The substance of the above papers has.
appeared recently in the open literature: Spedding F. H., Newton A. S.,
Warf J.C, Johnson O., Not torf R. W., Johns I. B., Daa-
n e A. H., Nucleonics, 4 (1), 4—15(1949); Newton A. S., Warf J.C,
Spedding F. H., Johnson O., Johns I. B., Not torf R. W.r
Ay res J. A., Fisher R. W., Kant A., Nucleonics, 4 (2); 17—25 (1949);
Warf J.C, Newton A. S., Butler T. A.', Spedding F. H.,
Nucleonics, 4 (3); 43—47 (1949) (MP Ames I).
5. Battelle Memorial Institute, Report CT-818, July 10, 1943 (Battelle 2).
6. Burke J. E., Smith С S., Report LA-37, Nov. 13, 1943 (Los Alamos I).
7. Perlman, Report CN-1025, Nov. 8, 1943 (MP Clinton I).
8. M а с W о о d G. E., Report RL-4.6.234, Dec. 22, 1943 (UCRL I).
9. Burke J.E., Smith С S., J. Am. Chem. Soc., 69, 2500—2502 (1947).
10. Gueron J., Yaffe L., Nature, 160, 575 (1947).
11. Newt on A. S., пат. США 2446780, 1948.
12. J oh n son O., Report CC-1059, Oct. 9, 1943 (MP Ames 2).
13. S i e v e r t s A., numerous articles in Z. anorg. u. allgem. Chem., from 1925 to 1935;.
Emeleus H. J., A nderson J.S., Modern Aspects of Inorganic Chemistry,
Ch. 6, D. Van Nostrand Company, Inc., New York, 1942.
14. Spedding F. H., Johns I. B.f Report CC-803, July 15, 1943; Newt onA.S.,,
Daane A.H., Johnson O., Nottorf R. W., Report CC-1059, Oct. 19,
1943; Newton A. S., Warf J.C, Johnson O., Nottorf R. W.,
Report CC-1201, Feb. 8, 1944; Nottorf R. W., Report CC-1212, Dec. 12, 1943;.
Spedding F. H. et. al., Preparation and Physical Properties of Uranium
Hydride, in National Nuclear Energy Series, Division VIII, vol. 6 (MP Ames 3).
15. W a r f J. C, Report CC-862, Aug. 8, 1943 (MP Ames 4).
16. Tucker W, Figard P., Reports CC-1781, Oct. 18, 1944; CC-1.975, Oct. 24s
1944 (MP Ames 5).
176
Часть III. Бинарные соединения урана
17. W а г ! J.C., Newton A. S., Butler Т. A., Ayres J. A., Johns I. В.,
Reports CC-580, Apr. 15, 1943; СС-587, Apr. 19, 1943 (MP Ames 6).
18. N e w t о n A. S., N о t t о г f R. W., D a a n e A. H., Johnson O., Report
CC-858, Aug. 7, 1943 (MP Ames 7).
19. Warf J. C, Johnson O., Reports CC-1059, Oct. 1943; CC-1061, Oct. 1943
(MP Ames 8).
20. S p e d d i n g F. H. et al., Preparation and Physical Properties of Uranium
Hydride, in National Nuclear Energy Series, Division VIII, vol. 6 (MP Ames 9).
21. N e w t о n A. S., Warf J. C, Johnson O., N о t t о г f R. W., Report
CC-1201, Feb. 8, 1944 (MP Ames 10).
22. Newton A. S., Report CC-1212, Dec. 10, 1943 (MP Ames II).
23. M а с W о о d G. E., A 1 t m a n D., Report RL-4.7.600, Oct. 24, 1944 (UCRL 2).
24. M а с W о о d G. E., Private communication, May 22, 1945 (UCRL 3).
25. Warf J. C, Newton A. S., Butler T. A., Ayres J. A., Johns I. B.,
Report CC-580, Apr. 15, 1943 (MP Ames 12).
26. Hubble H. H., McCul lough F. C, Report CN-1025, Nov. 8, 1943
(MP Clinton 2).
27. J о h n s о n O., Newton A. S., Report CC-1063, Nov. 6, 1943 (MP Ames 14).
28. Warf J. C, Report CC-1061, Oct. 8, 1943 (MP Ames 15).
29. Newton A. S., Johnson O., Daane A. H., Nottorf R. W.,
Warf J. C, Report CC-803, July 15, 1943 (MP Ames 13).
30. N e w t о n A. S., Report CC-695, May 27, 1943 (MP Ames 52).
.31. Warf J. C, Some Reactions of Uranium Metal, in National Nuclear Energy Series,
Division VIII, vol. 6 (MP Ames 57).
32. R u n d I e R. E., Report CT-609, Apr. 24, 1943 (MP Ames 16).
33. R u n d 1 e R. E., Wilson A. S., McDonald R. A., Report CC-1131, Dec.
18, 1943; Rundle R. E., Baenziger N. C, Wilson A. S„
McDonald R. A., Report CC-2397, Apr. 2, 1945, p. 6—10 (MP Ames 17).
34. R u n d 1 e R. E., Wilson A. S., McDonald R. A., The X-ray
Investigation of the Uranium-Hydrogen System; The Structure of UH3, in National Nuclear
Energy Series, Division VIII, vol. 6 (MP Ames 18).
35. Z а с h а г i a s e n W. H., M о о n e у R. С. L., Report CK-1096, Nov. 27, 1943
(MP Chicago 1).
36. R u n d 1 e R. E, J. Am. Chem. Soc, 69, 1719 (1947).
37. Pauling L., E w i n g F. J., J. Am. Chem. Soc, 70, 1660 (1948).
38. Pauling L., J. Am. Chem. Soc, 69, 542 (1947).
39. Nottorf R. W., Reports CC-1063, Nov. 6, 1943; CC-1212, Dec. 10, 1943 (MP
Ames 19).
40. В u t 1 e г Т. A., Report CC-664, May 14, 1943; Newton A.S., Johnson O.,
Kant A., Nottorf R. W., Reports CC-705, June 7, 1943; CC-725, June 15,
1943; Johnson O., Kant A., Nottorf R. W., Warf J. C,
Report CC-803, July 15, 1943; Daane A. H., Johnson O., Warf J. C,
N о 11 о r f R. W., ReportsCN-852, Aug. 8,1943; CC-1059,|Oct. 9, 1943; F i s h eг R. W.,
Warf J. C, Report CC-1091, Jan. 7, 1944; Warf J. C, G о 1 d b 1 a t t M.,
D e v e b a u g h R. D., Powell J., Report CC-1194, Dec 9, 1943; Ayres J. A.,
Report CN-1943, Jan. 8, 1944; L у о n W., I 1 i f f J., L i p к i n d H., Report
CK-1494, Apr. 29, 1944 (MP Ames 20).
41. S p e d d i n g F. H. et al., The Chemical Properties of Uranium Hydride, in National
Nuclear Energy Series, Division VIII, vol. 6 (MP Ames 21).
42. Lindner M., Report CN-1025, Nov. 8, 1943 (MP Clinton 3).
43. Foster L. S., Report CT-2106, Sept. 14, 1944, p. 29 (MIT 1).
44. JohnsonO., Kant A., Nottorf R. W., Report CC-705, June 7, 1943
(MP Ames 22).
45. N о t t о г f R. W., Report CC-803, July 15, 1943 (MP Ames 24).
46. Kant A., Report CC-803, July 15, 1943 (MP Ames 25).
47. Lyon W., Iiiff J., Lip kind H., Report CK-1494, Apr. 29, 1944 (MP
Ames 26).
48. А у г e s J. A., Report CN-1243, Jan. 8, 1944 (MP Ames 27).
49. Daane A. H., Johnson O., Nottorf R. W., Report CC-1059, Oct. 9,
1943 (MP Ames 28).
50. F i s h er T. W., Report CC-1091, Feb. 14, 1944 (MP Ames 23).
51. Johns I. В., Tevebaugh R. D., Report CC-1059, Oct. 9, 1943 (MP
Ames 29).
52. Warf J.C., Newton A. S., Butler T. A., Ayres J. A., Report
CC-580, Apr. 15, 1943 (MP Ames 30).
53. T e v e b a u g h R. D., Report CC-1194, Dec. 9, 1943 (MP Arnes 31).
54. V о i g t A.F., Walter F. J., Ayres J. A., HeinR.E., Report CN-578,
Apr. 15, 1943 (MP Ames 32).
55. G о 1 d о 1 a t t M., Report CC-1194, Dec. 9, 1943 (MP Ames 33).
Глава 8. Система уран—водород
177
56. А у г е s J. A., Reports CN-853, Aug. 8, 1943; CN-858, Aug. 7, 1943; CN-925, Sept.
8, 1943 (MP Ames 34).
57. Warf J. C, Report CC-1194, Dec. 9, 1943 (MP Ames 35).
58. Warf J. C, Newton A. S.f Butler Т. А., А у г e s J. A., Johns I. B.,
Report CC-580, Apr. 15, 1943; G о 1 d b 1 a t t M., Report CC-682, May 15, 1943
(MP Ames 36).
59. В u t 1 e r T. A., V о i g t A. F., Walter F. J., А у г e s J. A., Report
CN-925, Sept. 8, 1943 (MP Ames 37).
60. W a r f J. G., Report CC-1059, Oct. 9, 1943 (MP Ames 38).
61. Rundle R. D., Wilson A. S., McDonald R. A., Report CC-1131, Dec. 18,
1943 (MP Ames 39).
62. W a r f J. C, Report CC-I091, Oct. 9, 1943 (MP Ames 40).
63. F i s h e r R. W., Report CC-1059, Oct. 9, 1943 (MP Ames 41).
64. Powel J., Report CC-1194, Dec. 9, 1943 (MP Ames 42).
65. D a a n e A. H., Report CC-1059, Oct. 9, 1943 (MP Ames 43).
66. D a a n e A. H., Report CT-686, May 22, 1943 (MP Ames 44).
67. С a r t e г J. H., Report CC-664, May 15, 1943 (MP Ames 45).
68. Butler T. A., Report CC-725, June 15, 1943 (MP Ames 46).
69. Newton A.S., Johnson O., Kant A., N о t t о г f R. W., Report
CN-725, June 15, 1943 (MP Ames 47).
70. Newton A. S., Johnson O., S p e d d i n g F. H., Report CN-717, June 17,
1943 (MP Ames 48).
71. Tevebaugh R. D., Walsh K. A., II iff J., Keller W. H.,
Johns I. В., Report CC-1063, Nov. 6, 1943 (MP Ames 49).
72. Warf J.C, Nottorf R. W., Report CC-803, July 15, 1943 (MP Ames 50).
73. W a r f J. C, Report CC-1782, Aug. 10, 1944 (MP Ames 51).
74. Johnson O., Report CC-1063, Nov. 6, 1943 (MP Ames 53).
75. N e w t о n A. S., Johnson O., D a a n e A. H., Nottorf R. W., Report
CC-803, July 15, 1943 (MP Ames 54).
76. JohnsonO., Report CC-1212, Dec. 10, 1943 (MP Ames 55).
77. N e w t о n A. S., DanneA.H., Johnson O., Nottorf R. W., Report
CC-1059, Oct. 9, 1943 (MP Ames 56).
12 Химия урана
Глава 9
БОРИДЫ, КАРБИДЫ И СИЛИЦИДЫ УРАНА
СИСТЕМА УРАН—БОР
В литературе имеется описание трех боридов урана. Диборид урана UB2
получен путем плавления в электрической дуге электродов из смеси порошка
урана и аморфного бора, спрессованной под давлением 200 am при температуре
1000° [1]. Образовавшийся серебристо-серый продукт промывали
разбавленной кислотой. Диборид устойчив против водных растворов щелочей и кислот,
за исключением азотной и плавиковой. В расплавленной щелочи он
растворяется с выделением водорода.
Проведены опыты по электролизу при 1000° смеси расплава закиси-окиси
урана U308 и окиси бора с окислами и фторидами щелочноземельных металлов
в качестве плавней; при этом получены кристаллы с металлическим блеском,
по составу отвечающие UB4 [2, 3]. Полученный таким путем тетраборид урана
растворяется в холодной плавиковой или соляной кислоте, восстанавливает
концентрированную серную кислоту и легко растворяется в азотной кислоте
и концентрированной перекиси водорода. При сплавлении с гидроокисями
или карбонатами он быстро разлагается, с перекисями бурно реагирует.
Два борида урана UB4 и UB12 изучены методами рентгенографического
анализа [4]. Найдено, что UB4 имеет тетрагональную решетку с константами
а1=7,066 А и а3 = 3,97 А. Элементарная ячейка содержит четыре молекулы.
Для этой структуры предложена пространственная группа P4Imbm. На
основании интенсивностеи отражений принимается, что атомы урана находятся
в положении 4 (g) с х=0,1875; положения атомов бора еще не определены.
Описание борида UB12 появилось недавно [5]; он имеет кубическую
решетку с а = 7,473 А. Элементарная ячейка содержит четыре молекулы;
рассчитанная плотность равна 5,825 г/см3, что, повидимому, подтверждается
непосредственными измерениями плотности. Вероятная пространственная группа
OlrFm&m с атомами урана в положении 4 (а), атомами бора в 48 (i) и х = г1ь.
Каждый атом бора окружен пятью другими атомами бора на расстоянии
В—В = 1,76 А и двумя атомами урана на расстоянии U— JB = 2,79 А. Каждый
атом урана окружен двадцатью четырьмя атомами бора, расположенными
в вершинах правильного кубооктаэдра. Радиус атома урана, 1,91 А, необычно
велик; это приписывают высокой координации. Подобное же увеличение
атомного радиуса наблюдается у тория в соединении ThBe.
Термическим разложением боргидрида урана (IV) U(BH4)4 получен
борид, состав которого еще точно не определен (см. гл. 15).
СИСТЕМА УРАН—УГЛЕРОД
Впервые карбид урана получен нагреванием закиси-окиси урана с углем
в электрической печи [6]. Для этого продукта вначале была предложена
формула U2C3[6,7], позднее показано, что карбид имеет формулу UC2[8,9].
Последующие исследования доказали существование монокарбида UC и
подтвердили формулу дикарбида UC2. Монокарбид термодинамически устойчив
Глава 9. Бор иды, карбиды и силициды урана
179
при комнатной температуре, а дикарбид, вероятно,—только при высоких
температурах. Высказано предположение, что при еще более высоких
температурах (выше 2000°)' существует полуторный карбид U2C3, но это нельзя
считать доказанным [10].
Фазовые отношения в системе ура н—у г л е р о д.
Растворимость чистого углерода (графита) в жидком уране была определена
путем нагревания металла в графитовых тиглях с длительным выдерживанием
при постоянной температуре для того, чтобы обеспечить равновесие между
расплавом и графитом [11,12]. Полученные результаты представлены в табл. 63.
Таблица 63
РАСТВОРИМОСТЬ УГЛЕРОДА (ИЛИ UC) В ЖИДКОМ УРАНЕ
Температура,
°С
1350—1400
1450—1500
1550—1600
1650—1700
1750—1800
1850—1900
1950—2000
2080—2130
Максимальная 1375
» 1400
» 1500
» 1600
» 1800
» 1900
» 2000
Продолжительность
нагрева, мин.
40
20
60
20
30
30
30
Углерод,
% (вес)
о,ю ■)
0,17 }
0,36
0 42 1
0,Ы МП]
1,60
1,74
2,92
0,137 Л.
0,168
0,385
0,626 i [12l
0,775 [
1,20
1,50 j
Атомное
отношение,
С: U
0,02
0,034
0,072
0,084
0,102
0,325-
0,354
0,592
Изучена также фазовая диаграмма системы уран—углерод между UC
(4.8%С) и UC2 (9,16%С) (рис. 30) [13]. Доказательства в пользу существования
высокотемпературного соединения U2C3 нельзя считать серьезными. Видман-
штеттовы фигуры для сплавов с составом между U2C3 и UC2 сейчас
рассматриваются как доказательство существования твердых растворов Ш2С3 и eUC2.
Карбидов с числом атомов углерода более двух на один атом урана, повиди-
мому, не существует. Однако некоторые рентгенографические данные
доказывают растворимость углерода в решетке дикарбида, особенно при
температурах, близких к точке плавления (2375°).
Физические свойства монокарбида урана. Темпе-
ратура плавления. Доказано, что монокарбид урана плавится при 2250°
[13, 14].
Кристаллическая структура монокарбида урана. Монокарбид имеет
гранецентрированную кубическую решетку, а=4,951 ±0,001А [15] или
4,955 А [16]. Экспериментально установленные константы решетки приведены
в табл. 64. Элементарная ячейка содержит четыре молекулы, плотность
монокарбида, рассчитанная на основании рентгенографических данных, равна
13,63 г/см3 [15]. Монокарбид урана изоморфен мононитриду и моноокиси
урана, и углерод в этом карбиде (на основании данных Эймского университета .
может быть замещен кислородом или азотом, что сопровождается небольшим
уменьшением постоянной решетки (до4,936 А при наличии еще двуокиси урана):
12*
ISO
Часть III. Бинарные соединения урана
Таблица 64
КОНСТАНТА РЕШЕТКИ И РЕНТГЕНОГРАФИЧЕСКАЯ
ПЛОТНОСТЬ МОНОКАРБИДА УРАНА
fll, A
4,98
4,966^-0,005
4,948+0,001
от 4,948 до 4,951±0,001а>
4,94
4,995
а) Подробности см. [15].
б) Для ai = 4,951 A.
Плотность,
г/см*
13,56
13,66
13,63б>
—
—
Литература
17
18
19
14
20
16
Вероятно, монокарбид обладает способностью образовывать твердые растворы
при любых соотношениях компонентов с мононитридом и моноокисью. Ввиду
того что замещение углерода кислородом или азотом уменьшает константу
решетки, можно предполагать, что наивысшее значение константы 4,955 А
&OUUI
2400\
<j 2300
а
§;
I 2200
пер
2100
2000
[- г 1, — г' 1
ис2
и2р3 1
\/ / 11!
А
/\
/
L
\-
V-
f—f 1 I1
\ 1
*
\ в / 1
\ / 1
\ ' 1
*+р \ 1 1
\JL-J
т—|
Н
—
—
-\
J 1
4 5 6 7 8 9 Ю
Углерод, %
Рис. 30. Система уран—углерод (4,8— 9,16%С)..
и является наиболее правильным. Константа решетки монокарбида мало
зависит от присутствия избытка урана или углерода. Это доказывает низкую
растворимость обоих компонентов в карбиде при комнатной температуре.
Вследствие того, что рассеяние рентгеновских лучей углеродом очень
мало по сравнению с рассеянием их ураном, трудно решить, к какой из двух
Глава 9. Бориды, карбиды и силициды урана
181
возможных структур, к каменной соли или цинковой обманке, относится
решетка монокарбида. Однако на основании измерений интенсивности
отражений вероятно, что монокарбид имеет решетку типа каменной соли {On-Fm 3m)
со следующими положениями атомов:
(000; 0-^у; lo|; -i40) +
U 4 (а) 0 0 0;
С4 (6)111.
Уран и карбид урана. Сплавы урана с углеродом, содержащие менее 4,8%
второго, являются смесями урана (содержащего менее 0,01 % углерода) с
монокарбидом. Вследствие негомогенности этих сплавов определение
физических констант дает различные результаты. В табл. 65 приводятся значения
твердости [11, 21].
Таблица 65
ТВЕРДОСТЬ СПЛАВОВ УРАНА С УГЛЕРОДОМ
Содержание
углерода, %
0,06
0,08
0,168
0,25
0,30
0,7
1,99
Твердость
по Роквеллу
А
__
—
—
—
—
—
по Роквеллу
В
88
88
91
95
95
93
111
Содержание
углерода, %
1 0,17
0,36
0,42
0,51
1,60
1,74
2,92
Твердость
по Роквеллу
А
52,8а>
54,8
56,0
58,7
63,1
63,8
63,6
по Роквеллу
В
—
—
—
—
—
а) Среднее из нескольких измерений в различных местах поверхности исследованного
образца.
Один образец карбида, по составу отвечающий U3C2, обладал плотностью
10,52 г/см? и теплопроводностью 0,082 кал-см'1 • сек'1 • град'1 при 56° [22, 23].
Полуторный карбид урана. Рентгенографические исследования
образцов карбида урана с отношением U : С=2 : 3 показали, что они всегда
состоят из моно-и дикарбида [24]. В пользу существования при высоких
температурах соединения U2C3 говорит факт образования как будто кубических
монокристаллов при закалке сплава, соответствующего по составу U2C3, хотя
рентгенограммы показывают, что эти кристаллы двухфазные. При
микроскопическом исследовании этих монокристаллов была обнаружена видманштеттова
структура. Отсюда можно заключить, что U2C3, вероятно, является
кубической фазой, устойчивой при некоторой температуре выше 2400°; однако
закалка от этой температуры до сих пор была безуспешной. Вследствие
хрупкости карбида состава U2C3 его твердость была определена только
приблизительно. Она составляет 396 по Роквеллу [25]. Температура плавления образца
карбида с 6,95% углерода (U2C3 должен содержать 7,04% углерода) та же,
что и температура плавления UC2, т. е. 2350—2400° [14].
Физические свойства дикарбида урана.
Температуры плавления и кипения. Образец сплава урана с углеродом,
содержащего 9,18%С (UCo содержит 9,16% С), показал признаки плавления пр
182
Часть III. Бинарные соединения урана
2350—2400° [13, 14]. Вычисленная температура кипения составляет 4370°
при 760 мм рт, ст. [26].
Температура плавления равна 2425° [27, 28], 2200° в электрической
вакуумной печи и 2260° в катодной [29].
Кристаллическая структура [15]*. Дикарбид UC2 был впервые [31]
описан как плотное тонкокристаллическое металлоподобное вещество
плотностью 11,28 г/см3. Изучение кристаллической структуры дикарбида показало,
что он имеет гранецентрированную тетрагональную решетку [32, 33].
Структура подобна структуре дикарбида лантана, с дв^мя молекулами в
элементарной ячейке. Константы решетки^=3,517^=0,00^ и а3 = 5,987±0,001JL Раствоо-
рение углерода в дикарбиде, повидимому, уменьшает константы до ^=3,505 А
и а3=5,951 А. Указывается [33], что это снижение свидетельствует о том, что
дикарбид не является твердым раствором внедрения углерода в уране.
Дикарбид в соответствии с этим предположением построен из небольших
положительных ионов урана и больших отрицательных ионов С2, причем уран
находится в свободных узлах анионной решетки, в сплавах же с составом
UC>2 часть ионов урана отсутствует. Как будет показано ниже, в гл. 11 (при
разборе аналогичной интерпретации уменьшения констант решетки при
переходе от UO2.0 к иОг.з), указанное толкование требует заметного снижения
плотности с ростом содержания углерода. Повидимому, такого рода
уменьшения (при росте содержания кислорода) в случае окислов не обнаружено;
систематических измерений плотности карбидов не проводилось.
Рассчитанная из рентгенографических данных плотность равна 11,68 г/см3.
Экспериментально измеренная плотность равна всего 9,97 г/см3±2% [22, 23].
Теплоемкость. Для вычисления удельнойi теплоемкости дикарбида
предложено следующее уравнение [34]:
Ср = 8,92 +3,95-10"3 Г яал-шшг1. (1)
Значение Ср также вычислено экстраполяцией из известных значений
теплоемкости для других карбидов, в частности для монокарбида тантала [35].
Эта величина равна 75% величины Дюлонга—Пти при 21,3°. Предполагая,
что для дикарбида урана соответствующая доля составляет 80%, получили
Cp(UC2)=15 кал-моль"1 при 1940°К.
Теплопроводность. Теплопроводность образца дикарбида с плотностью
10 г/см3 (см. выше) равна 0,082 кал-см'1 • сек"1 • град"1 [23].
Получение карбидов урана. Термодинамика образования
монокарбида урана. Вычислено, что теплота образования монокарбида из
элементов равна —30 кал-моль"1. Вычисление основано на наблюдении, что
дикарбид при низких температурах разлагается на уран и монокарбид [36].
Термодинамика образования дикарбида урана. Вычислена [35] энергия
и свободная энергия реакции
U02 + 4C (графит) ^ UC2 + 2CO. (2)
Необходимые экспериментальные данные взяты из ранее проведенных
измерений равновесия реакции (2) [37]. При этих измерениях определяли
равновесное давление окиси углерода над смесью двуокиси урана с углеродом
в интервале температур 1480—1801°. Из рис. 31 видно, что давление при
этом менялось от 18 мм рт. ст. до 1 атм и что T\gp изменяется в
зависимости от Т приблизительно линейно. Уравнение
lgp= -^+12,09 (3)
* В литературе [30] опубликована несколько другая интерпретация систеыы уран—
углерод, но в общем она менее удовлетворительна.
Глава 9. Бориды, карбиды и силициды урана
183
отражает опытные данные лишь приблизительно; Д#, повидимому, заметно
изменяется с температурой. Из уравнения (3) выводится, что среднее
значение АН в изученной области равно 174,6 ккал на 1 г-атом урана. Для
реакции (2) вычислено также и ДСр [35]:
ACp = 0,8-8,1.10-3r + 4,68<105r-2. (4)
Из [этого уравнения в сочетании с результатами измерения равновесных
давлений окиси углерода для реакции (2) были выведены следующие
800
бОООг
1400 1600 1800
Температура, °С
1750 1870 1990 2110
Температура, °К
Рис. 31. Давление при равновесии окиси углерода над смесью
двуокиси урана с углеродом [42].
термодинамические уравнения:
ДЯ=188540 + 0,87,-4,05-10-3Г2-4,68.10бГ-1 кал • г-атом'1 U (5)
AF°= 188540- 1,8427 lgr+4,05- 10"3Г2-
- 2,34-lO5^1-93,47* кал-г-атом-1 U. (6)
Для изменения энтропии в той же реакции выводится
AS§98 = 93,8 энтропийной единицы.
Подобные вычисления были сделаны [34] с использованием тех же
данных по равновесию, но с применением величины Ср для дикарбида,
вычисленной из уравнения (1).
AF°-202500+16,1677 lgF+2,775-1(Г3Т2-
-3,495- 1057^x— 156,74 Т кал-г-атом-1 V;
Д#298 = 199,000 ккал • г-атом~1\}\
AF*29s= 166,790 ккал-г-атом1' U;
Д5298 = 108,05 энтропийной единицы.
(7)
(8)
Комбинированием термодинамических данных для реакции (2) с
известными термодинамическими константами для двуокиси урана, углерода и
окиси углерода можно рассчитать теплоту, свободную энергию и энтропию
образования дикарбида урана из элементов. Таким путем получены
значения, представленные в табл. 66.
J 84
Часть III. Бинарные соединения урана
Таблица 66
ТЕРМОДИНАМИЧЕСКИЕ КОНСТАНТЫ ОБРАЗОВАНИЯ UC2: U + 2C—► UC2
—Af°, кал -г-атомг^- U
3925+6,3 Tig T~~
—3,48-10-372+7,545- 104Г"1 +
+ 19,85 Т
ккал .г-
атом U-1
—
8,8
9,83
—Д#°298»
ккал-г-
атом-1 U
—
—
3,92
AS°298,
энтропийные
единицы
23,6
15,1
19,8
Литература
35
38
34
Дикарбид неустойчив при комнатной температуре (по отношению к моно-
карбиду и углероду); вероятно, медленный распад на монокарбид и углерод
происходит уже при температуре ниже 2400°. При закалке дикарбида от
температур выше 2400° получается чистый дикарбид, при закалке от 1000° или
медленном охлаждении получается дикарбид с примесью монокарбида и углерода
независимо от того, какой компонент находился в избытке в расплаве.
Получение карбида урана из металлического урана и углерода. Найдено,
что при нагревании металлического урана в графитовых тиглях до 1800—1900°
стенки тиглей разрушаются и образуется твердый хрупкий карбид [25, 39].
Образование карбида происходит, повидимому, путем диффузии урана в
графит, а не графита в уран. Поэтому реакция протекает в присутствии избытка
углерода и в результате ее получается богатый углеродом дикарбид [40]. Для
получения такого продукта можно также графитовый порошок растворить
в расплавленном уране или спрессовать с урановыми стружками перед
плавлением [21].
Для получения ураноуглеродистых сплавов с высоким содержанием
углерода рекомендуется спекание смеси порошкообразного металлического урана
с избытком углерода [17]. Этим методом (прессованием под давлением 45 ту
спеканием при 1800—2000° в вакууме) удалось получить сплавы с содержанием
углерода 0,174—8,54% (вес.) [24]. Карбид урана был приготовлен также
нагреванием стружек урана с чистым углеродом до 1200—1600° [20].
Все сплавы урана с углеродом, полученные этим путем, по составу
негомогенны, они содержат дендритные включения карбида в основной массе
насыщенного раствора углерода в уране. Карбидные включения можно отделить
путем обработки сплава 3 н. соляной кислотой, содержащей немного перекиси.
Нерастворенный остаток представляет практически чистый монокарбид UC,
что подтверждается рентгенографическим и химическим анализами (4,70%
углерода). Монокарбид урана можно также получить нагреванием урана и
дикарбида UC2 выше 1800°. Сообщается, что при взаимодействии
металлического урана с углеродом в зависимости от температуры может образоваться
монокарбид или дикарбид [16]:
л 2100е
U + C ->UC; (9)
2400°
U + 2C >UC2. (10)
Предполагается, но точно не установлено, что продукты являются
гомогенными.
Получение монокарбида урана из металлического урана и метана.
Монокарбид может быть получен взаимодействием метана с металлическим
ураном [16]:
U + CH4^=^:UC+2H2. (11)
Этот метод является, вероятно, наиболее удобным для получения чистого
Глава 9. Боридыу карбиды и силициды урана
185
монокарбида; металлический уран сначала должен быть превращен через
гидрид в тонкодисперсную форму.
Получение карбида урана из металлического урана и окиси углерода. Были
поставлены опыты по обработке обезгаженного куска металлического урана
окисью углерода с одновременным нагреванием до 1200°. Вначале скорость
реакции была велика, затем она падала. Через 45 мин., когда расплавленный
уран еще медленно поглощал окись углерода, его охладили и исследовали.
На поверхности находилась черная чешуйчатая корка; рентгенографическое
исследование показало, что она состоит из двуокиси и монокарбида урана [41].
Получение карбида урана из окислов урана. Муассан [31 ] для этой цели
смешивал 50 г закиси-окиси урана и 50 г сахарного угля в графитовом тигле и
нагревал смесь 8—10 мин. в электрической печи (45 000 дж). Позднее установлено,
что продукт, полученный этим путем, всегда содержит графит 18]. Руфф и Гейн-
цельман [27] постепенно нагревали в вакуумной печи до 2450° смесь двуокиси
урана с сахарным углем. Реакция идет медленнее, чем с закисью-окисью урана,
но получаемый продукт почти не содержит графита. Описана также реакция
двуокиси урана с избытком углерода в вакуумной печи при 1800° [42];
получаемое соединение не плавилось. Выделявшаяся окись углерода откачивалась.
Проведен ряд опытов, в которых смесь 1 части графита Ачесона и 5—7 частей
закиси-окиси урана нагревали в графитовых тиглях в атмосфере водорода до
образования карбида [43]. Позже установлено [25, 44], что при весовом
отношении UOg : С, находящемся между 5 и 6,4, получался дикарбид урана (9,15%С,
а вычислено для UC2 9,16% С), при U02 : С от 7 до 8 продукт имел состав,
близкий к U2C3 (7,21% С, вычислено для U2C3—7,04%).
Из двуокиси урана и углерода не может быть получен чистый монокарбид.
Попытки получить карбид урана восстановлением смесей тетрафторида урана
и углерода кальцием оказались безуспешными. При нагревании двуокиси
урана с дикарбидом образуется монокарбид UC, но не совсем чистый.
Установлено, что при нагревании закиси-окиси урана со стехиометриче-
ским количеством графита до 1800° образуется монокарбид, при более
высоких температурах—дикарбид [16]:
U308 + 11С —- 3UC -f 8CO; (12)
LL08 + 14С —I 3UC2 + 8CO. (13)
Химические свойства карбидов урана. Данные по
химическому поведению карбида урана относятся к продуктам, состав которых
указан как UC2 или U2C3. Выше многократно упоминалось, что существование
гомогенной фазы полуторного карбида является весьма сомнительным, за
возможным исключением области высоких температур.
Отношение дыкарбида к кислороду. Карбид урана является пирофорным
веществом. При сбрасывании его на твердую поверхность или при ударе по нему
молотком образуются снопы искр [31, 45]. При растирании в агатовой ступке
карбид может воспламениться [31]. В литературе указывается, что нэ воздухе
карбид воспламеняется при 400° [7], в кислороде воспламенение имеет место
при 370° и карбид сгорает до закиси-окиси урана и двуокиси углерода [31].
На воздухе в течение недели дикарбид полностью разлагается, вероятно,
за счет первичной реакции с водяным паром [39].
В Эймском университете было сделано наблюдение, что дикарбид не
окисляется при 300°, но полностью окисляется в токе воздуха при температуре
400—500° в течение 4 час. Образец состава U2C3 после такой же обработки только
покрылся черной пленкой. После нагревания в течение 5х/2 час. до 600° этот
образец состава полуторного карбида распался на гранулы, еще содержавшие
металлоподобные включения.
186
Часть III. Бинарные соединения урана
Отношение дикарбида к азоту. Азот реагирует с карбидом урана при
температуре 1100° [31]. Реакция идет с большой скоростью при 1180° [42]. При
этом карбид поглощает азот до достижения насыщения. Через 12 час. весь
карбид превращается в нитрид.
Отношение дикарбида к фтору. При комнатной температуре реакция
не идет, но уже слабого нагревания достаточно для того, чтобы вызвать
взрыв [31].
Отношение дикарбида к хлору. Карбид воспламеняется в хлоре при
температуре 350°, при этом получается летучий хлорид [31 ]. Позже найдено, что
смеси двуокиси и дикарбида урана реагируют с хлором при 600° с образованием
тетрахлорида, но при этом получается большой остаток [46]. При 800—1 000°
образуются хлориды, отвечающие более высокой степени окисления урана.
Отношение дикарбида к брому. Карбид урана воспламеняется в парах
брома при температуре 390° [31]. Позже показано, что дикарбид реагирует с
бромом при температурах выше 300°; при 900° образуется тетрабромид урана
[47]. В дальнейшем эта реакция
UC2 + 2Br2—>UBr4 + 2C (14)
наблюдалась уже при 800° [48], Образующийся углерод получается в
тонкодисперсном состоянии, и его трудно отделить. При этой реакции разрушается
большинство аппаратостроительных материалов.
Отношение дикарбида к иоду. Карбид урана реагирует с иодом при
температуре ниже красного каления без воспламенения [31]. Карбид с составом
U2C3 реагирует с иодом при 600° с образованием иодида урана [47]. При
пропускании паров иода с парциальным давлением 100 мм рт. ст. над дикарбидом
при 500° образуется тетраиодид [34]. Высказано предположение, что карбид
может служить подходящим исходным веществом для приготовления три- и тет-
раиодида урана.
Отношение дикарбида к сере и селену. Карбид урана горит в парах серы,
образуя сульфид и сероуглерод. Подобная же реакция идет и с селеном [31].
Отношение дикарбида к воде. Карбид урана медленно разлагает воду при
комнатной температуре и быстро при нагревании [31]. В отсутствие воздуха
образуется зеленая гидроокись, на воздухе—серовато-черный продукт.
Углерод карбида превращается при этом в углеводороды, частично (одна треть)
в газообразные, частично (две трети) в жидкие и твердые (состав этой смеси
см. [49—51].) Механизм образования различных углеводородов помимо
ацетилена (образования которого можно было бы ожидать в качестве главного
продукта, так как кристаллические структуры дикарбида урана и карбида
кальция аналогичны) рассматривается Шмидтом [52]. Разнообразие
продуктов взаимодействия карбида урана с водой этот автор объясняет
изменением валентности урана во время разложения.
С водяным паром карбид реагирует при температуре тёмнокрасного
каления, образуя черную двуокись [31 ]. Во влажном воздухе карбид урана
полностью разлагается в течение недели [39].
Отношение дикарбида к аммиаку. При температуре красного каления
аммиак частично разлагает дикарбид [31].
Отношение дикарбида к сероводороду. В сероводороде при
температуре 600° дикарбид воспламеняется, при этом образуется какой-то
сульфид [31].
Отношение дикарбида к хлористому водороду. При температуре 600°
дикарбид воспламеняется в хлористом водороде с образованием какого-то
разлагаемого водой хлорида [31].
Отношение дикарбида к кислотам. Разбавленные растворы соляной,
азотной и серной кислот разлагают дикарбид подобно воде, при этом
получаются желтые растворы солей уранила. Концентрированные кислоты (кроме
Глава 9. Бориды, карбиды и силициды урана
187
азотной) при комнатной температуре реагируют медленно, но при
нагревании—очень энергично [31]. При обработке дикарбида и полуторного
карбида 85%-ной фосфорной кислотой реакция шла медленно в условиях
комнатной температуры и энергично при нагревании, в результате получена
смесь газообразных, жидких и твердых углеводородов [24].
Отношение дикарбида к щелочам. Карбид легко разлагается
щелочами [7].
Отношение дикарбида к солям. При сплавлении дикарбида с нитратом
или перхлоратом калия происходит воспламенение и образуется уранат
калия. Нагреванием смеси поваренной соли, дикарбида и двуокиси урана
до 1000° получено комплексное соединение Na2U02Cl4 [46].
СИСТЕМА УРАН—КРЕМНИЙ
Система уран—кремний исследована термическими, микроскопическими
и рентгенографическими методами. Она оказалась чрезвычайно
сложной [53].
Фазовая диаграмма. На рис. 32 изображена фазовая диаграмма
системы уран—кремний. Она показывает наличие пяти соединений, которым
первоначально приписывали состав U5Si3, USi, U2Si3, USi2 и USi3. Кроме
того, обнаружено шестое соединение, получающееся в результате перитек-
тоидной реакции между свободным от углерода ^-ураном и U-Si3 при 940е;
это соединение способно к существованию при содержании 4 —28% (атомн.)
кремния, и оно идентифицировано как U10Si3 [54]. Таким образом, в системе
уран—кремний считалось всего лишь шесть соединений; три эвтектоидных
и три перитектоидных. Недавно, однако, проведено рентгенографическое
исследование системы уран—кремний [55] и установлено, что некоторые из
первоначальных формул являются ошибочными.
Так, U5Si3 в действительности отвечает составу U3Si2, соединение U2Si3
является кристаллической модификацией USi2, a U10Si3 имеет состав U3Si.
Таким образом, установлено наличие следующих соединений: U3Si2, USi,
P-USi2, a-USi2, USi3 и U3Si.
Методами микроструктурного анализа установлено наличие
эвтектической точки между U3Si2 и USi при 1570°, соответствующее 47% (атомн.)
кремния. Между ^-ураном и U3Si2 имеется эвтектика при 985° с 9% (атомн.)
кремния. Максимальная растворимость кремния в ^-уране составляет около
1,75% (атомн.) при 980°; растворимость в р-уране менее 1% (атомн.).
Присутствие кремния повышает температуру перехода (3-урана в ?-уран
с 770 до~795°; температура перехода а—>(3 остается без изменения (660°).
Дисилицид урана USi2 впервые был приготовлен методом
алюминотермии [56]. Он представляет собой светлосерый металлический
микрокристаллический (кубический) порошок; нерастворим ни в холодных, ни в горячих
кислотах —соляной, азотной или серной,—ни в царской водке, но
растворим в концентрированной плавиковой кислоте. Под действием
расплавленных щелочей или щелочных карбонатов при температуре красного каления
дисилицид превращается в силикат и уранат, расплавленный бисульфат калия
медленно разлагает его. Дисилицид горит в кислороде при 800° и
реагирует с хлором при 500°.
Соединение USi3, трисилшщд урана, идентифицировано путем
микроскопического и рентгенографического анализов; оно перитектически
разлагается при температуре около 1515°. Существует эвтектика между триси-
лицидом и кремнием (содержащая очень мало растворенного урана) с 86%
(атомн.) кремния и температурой плавления 1315°.
Высокотемпературная область на рис. 32 между 35 и 75% (атомн.)
кремния не вполне изучена. Область между ураном и U3Si2, показанная
188
Часть III. Бинарные соединения урана
на рис. 32, соответствует метастабильному равновесию, поскольку в ней
отсутствует упоминавшаяся выше перитектоидная реакция между 4 и 28%
(атомн.) кремния при 940°.
Сплавы, содержащие более 30% (атомн.) кремния, хрупки и трудно
шлифуются.
1700Y-
1500
1300
l
с:
1 1 1
Жидкая фаза
-1700°
Шдкл\5ъЪ\г
■ п—*о п Оо и и и
V + ^St^ л
1665°
•MudK.+ urlJSL9
985"
930°
795°
665°
со
Г)
+
см
СО
3
VS
USU
+3
8
40
Кремний,
60
% (атомн.)
80
100
Рис. 32. Фазовая диаграмма системы уран—кремний.
О—температурные задержки, полученные при нагревании; X—двухфазный сплав
(по данным микроскопического исследования); ф —однофазный сплав (по
данным микроскопического исследования); А—е-перитектоидная температура.
Кристаллические структуры силицидов урана. Захариа-
зен [55] опубликовал подробные анализы кристаллических структур
силицидов урана. Кристаллографические данные представлены в табл. 67 и 68.
Из этих данных делается вывод о наличии ковалентных связей между
атомами кремния во всех структурах, кроме U3Si. Наблюдаемые расстояния
Si —Si равны приблизительно 2,3 А, что указывает на то, что они являются
фактически простыми связями. Связь между атомами кремния в U3Si2
способствует образованию групп Si2, в USi — зигзагообразных цепей кремния,
Глава 9. Бориды, карбиды и силициды урана
189
в p-USi2 — графитоподобыых слоев кремния ив oc-USi2 —трехмерной решетки
атомов кремния. Что касается связей между атомами урана, то они,
несомненно, имеются в U3Si и U3Si2 и, вероятно, отсутствуют в USi.
Таблица 67
КРИСТАЛЛОГРАФИЧЕСКИЕ ДАННЫЕ ДЛЯ СИЛИЦИДОВ УРАНА [55]
Формула
соединения
USi
a -USi2
;j-usi2
USi3
U3Si
U3Si2
Симметрия
Ромбическая
Pbnm
Объемноцентрированная
тетрагональная JAjamd
Гексагональная С6/ттт
Кубическая
Объемноцентрированная
тетрагональная JAjtrcm
Тетрагональная P4/mbm
Константы элементарной
ячейки, А
a2=7,65±0,01 I
я3 =3,90+0,01 j
fll= 3,97+0,03 1
a3=13,7J±0,08 J
fll==3f85±0,0i 1
a3^4,06±0,01 /
a = 4,03
fl",-6,017±-0f002 \
a3=8,679±0,003 J
a,-7,3151±0,0004 1
a3=3, 8925 ±0,0005 ]
Число
молекул в
элементарной
ячейке
4
4
1
—
4
2
Плотность,
г/см*
10,40
8,98
9,25
—
15,58
12,20
Таблица 68
МЕЖАТОМНЫЕ РАССТОЯНИЯ В СИЛИЦИДАХ УРАНА [55]
Формула
соединения
a-USi2
p-USi2
USi
U3Si2
U3Si
Межатомные расстояния, А
U —12Si=3,03
U —12Si=3,01
U — 7Si=2,98
U -U =3,62
Ui —4Si =2,96
Ui —8U =3,32
Uii—6Si =2,92
Uii—4U =3,32
Ui —4Si =3,01
Ui —8U =3,04
Uii—2Si =2,92
Uii—2Si =3,17
Uii—4U =3,02
Un—4U =3,04
Si—6U=3,03
Si—3Si=2,29
Si-6U=3,01
Si-3Si=2,22
Si—7U=2,98
Si—2Si=2,36
Si—2U=2,96
Si—6U=2,92
Si—lSi=2,30
Si—4U=2,92
Si—4U=3,01
Si—4U=3,17
190
Часть III. Бинарные соединения урана
Захариазен рассмотрел структуру этих соединений в свете идей Паулин-
га [57] о природе металлических связей. Наблюдаемые межатомные
расстояния в этих структурах, повидимому, удовлетворительно объясняются
для a-USi2, (3-USi2, USi и U3Si2 допущением существования нормального
числа четырех валентных электронов для кремния и 2,3 валентных
электронов для урана. Атомный радиус урана с 2,3 валентными а электронами
и с координационным числом 12 равен 1,636 А, вместо 1,516 А, найденного
для металлического урана. Этот большой радиус обнаружен во многих
соединениях урана металлического характера. Результаты, полученные при
такой интерпретации структур силицидов урана, указывают, что в этих
силицидах, как и в гидриде урана, присутствует низковалентная форма
урана (см. стр. 166). С другой стороны, структура U3Si может быть
объяснена наличием в ней урана с валентностью 5,78.
ЛИТЕРАТУРА
1. Wedekind E., J о с h e m О., Вег., 46, 1204 (1913).
2. A n d r i e u х J. L., Ann. Chim., 12, 423 (1929).
3. A n d r i e u x J. L., Rev. met., 45, 49—59 (1948); J. four elec, 57, (3); 54 (1948).
4. Bertaut F., Blum P., Compt. rend., 229, 666 (1949).
5. A n d г i e u x J. L., Blum P., Compt. rend., 229, 210 (1949).
6. Moissan H., Compt. rend., 122, 274 (1896).
7. R i d e a I E. K., Dissertation, University of Bonn, 1913.
8. L e b e a u P., Compt. rend., 152, 955, 956 (1911).
9. H e u s 1 e r 0., Dissertation, University of Frankfurt am Main, 1925.
10. National Nuclear Energy Series, Division IV, vol. 12A.
11. Snow 'A. I., Report CT-954, Oct. 2, 1943 (MP Ames 1).
12. Carter J. H., Report CT-609, Apr. 24, 1943 (MP Ames 2).
13. S n о w A. I., Report CT-1102, Nov. 28, 1943 (MP Ames 4).
14. S n о w A. I., Report CT-816, July 24, 1943 (MP Ames 9).
15. R u n d 1 e R. E., В a e n z i g e r N. C, Wilson A. S., McDonald R. A.,
J. Am. Chem. Soc., 70, 99—105 (1948).
16. L i t z L., Garrett А. В., С г о x t о n F. C, J. Am. Chem. Soc., 70, 1718
(1948).
17. R u n d 1 e R. E., Report CT-686, May 22, 1943 (MP Ames 6).
18. R u n d 1 e R. E., Wilson A. S., McDonald R. A., Report CC-1131,
Dec. 18, 1943 (MP Ames 7).
19. R u n d 1 e R. E., Report CT-1270, Mar. 9, 1944 (MP Ames 8).
20. P e i s e r H. S., A 1 с о с k Т. С, Report BR-589, Mar. 3, 1945 (British 1).
21. С a r t e r J. H., Report CT-490, Feb. 20, 1943 (MP Ames 5).
22. P 1 о t t R. E., R a e t h С H., Report CP-228, Aug. 14, 1942 (MP Chicago 1).
23. P 1 о t t R. E., R a e t h C.H., Report CE-236, Aug. 15, 1942 (MP Chicago 2).
24. Daane A.H., Snow A. I., Report CT-751A, June 2, 1943 (MP Ames 10).
25. Baker R. P., Carter J. H., Report CT-393, Dec. 15, 1942 (MP Ames 11).
26. M о t t W. R., Trans. Electrochem. Soc., 34, 279 (1918).
27. R u f f О., Н e i n z e 1 m a n n A., Z. anorg. Chem., 72, 72 (1911).
28. Ruff O., Goecke O., Z. angew. Chem., 24, 1461 (1919).
29. T i e d e E., В i r n b r a u e r E., Z. anorg. Chem., 87, 165 (1914).
30. Esch, Schneider, Z. anorg. Chem., 527, 254—256 (1948).
31. M о i s s a n H., Bull. Soc. chim. Paris, 17, 14 (1897).
32. H agg N. G., Z. physik. Chem., 12, 42 (1931).
33. В a e n z i g e r N. C, Report CT-1515, Apr. 1944 (MP Ames 12).
34. M а с W о о d G. E., A I t m a n D., Report RL 4.7.600, Oct. 24, 1944 (UCRL 1).
35. Davis Т., Burton M., Report CC-231, Aug. 15, 1942 (MP Chicago 3).
36. В r e w e r L., Bromley L. A., G i 1 1 e s P. W., L о f g r e n N. L., Report
CC-3234, Oct. 8, 1945 (MP Berkeley 1).
37. H e u s 1 e r O., Z. anorg. u. allgem. Chem., 154, 353 (1926).
38. Brewer L., Report CC-672, May 15, 1943 (MP Berkeley 2).
39. S p e d d i n g F. H., Report CP-42, Apr. 25, 1942 (MP Ames 13).
40. Carter J. H., Report CT-542, Mar. 27, 1943 (MP Ames 3).
41. Battelle Memorial Institute, Report CT-1388, Feb. 10, 1944 (Battelle 1).
42. H e u s 1 e r O., Z. anorg. u allgem. Chem., 154, 366 (1926).
43. W i 1 h e 1 m H. A., Daane'A. H., Report CC-205, July 16, 1942.
44. D a a n e A. H., Report CT-422, Jan. 15, 1942 (MP Ames 15).
45. T i e d e E., Birnbrauer E., Z. anorg. Chem., 87, 166 (1914).
Глава 9, Бориды, карбиды и силициды урана 191
46. A h m a n n D. H., Report СТ-393, Sec. 1. Part ВЗ, Dec. 15, 1943 (MP Ames 16).
47. S p e d d i n g F. H., Report CC-298, Oct. 16, 1942 (MP Ames 17).
48. Powell Т., Report CC-1778, Aug. 18, 1944 (MP Ames 18).
49. L e b e a u P., Da mi ens A., Compt. rend., 156, 1987 (1913).
50. L e b e a u P., Da mi ens A., Bull. soc. chim. France, 15, 367 (1914).
51. Lebeau P., Da mi ens A., Ann. Chim., 8, 221 (1917).
52. Schmidt, Elektrochem., 40, 171 (1934).
53. Gordon P., Cullity В., Report CT-1101, Dec. 4, 1943; Cohen M.r
Report CT-1384, Mar. 1, 1944; Cullity В., Bitsianes G., К a u f-
m a n n A. R., Reports CT-1696, May 1, 1944; CT-1819, June 1, 1944; CT-1938,
July 1, 1944; CT-2106, July 1, 1944; CT-2145, Aug. 1, 1944 (MIT 1).
54. В i t s i a n e s G., Cullity В., В о s t i a n R. В., К a u f m a n n A. R.r
Report CT-2699, Feb. 8, 1945 (MIT 2).
55. Z ac h а г i a s e n W. H., Acta Crystallographica, 2, 94—99 (1949).
56. Def acqz, Comt. rend., 147, 1050 (1908).
57. Pauling L., J. Am. Chem. Soc., 69, 542 (1947).
Глава 10
СОЕДИНЕНИЯ УРАНА С ЭЛЕМЕНТАМИ
ПОДГРУППЫ Va
НИТРИДЫ УРАНА
Реакция металлического урана со свободным азотом была открыта Муасса-
ном [1], который установил, что эти два элемента образуют при Ш00° желтый
нитрид. Но еще значительно раньше было показано, что при нагревании тетра-
хлорида урана в атмосфере аммиака до красного каления получается
соединение урана с азотом бурого цвета [2]. Позже установлено, что состав этого
соединения выражается формулой U3N4 [3]. Этот же состав был приписан нитриду
Муассана, т. е. соединению, полученному прямым взаимодействием урана
с азотом [4]. На основании этих данных до недавнего времени считалось,
что U3N4 является основным нитридом урана с рациональной химической
формулой.
В 1901—1927 гг. в литературе появились описания новых методов
получения, а также физических и химических свойств этого соединения [5—10].
Согласно последним данным [10], нитрид представляет собой черно-бурый или
черный порошок плотностью 10,09 г/ан3у разлагающийся при температуре
выше 1400°.
Хейзлер [4] сообщает, что в результате разложения U3N4 в вакууме при
1650° получается низший нитрид состава U5N4, повышение температуры до
1900° ведет к образованию нитрида U5N2, еще более бедного азотом. Но другие
авторы отвергли существование этого соединения [И]. Они наблюдали
постепенное, без скачков, изменение давления азота над нитридом (при 1280 и 1480°)
при изменении отношения U : N в интервале от UNo,i до UNi>0. Отсюда сделан
вывод, что продукты с содержанием менее одного атома азота на один атом
урана являются твердыми растворами нитрида урана в металлическом уране.
Рентгенографические исследования нитридов урана, проведенные в
последнее время, показали, что соединения U3N4 не существует [12, 13]. Фаза нитрида
урана, содержащая наименьшее количество азота, представляет собой
мононитрид UN. Между ураном и мононитридом отсутствуют промежуточные сте-
хиометрические соединения или же твердые растворы с большим интервалом
смешиваемости. Мононитрид не имеет заметного давления разложения при
температурах 1280 и 1480°, использованных в вышецитированном
сообщении [11] по исследованию давления азота над нитридом в зависимости от
отношения U : N; поэтому давления азота, измеренные в этой работе, не являются
равновесными давлениями разложения систем UNX с х < 1.
Между UN и следующим нитридом с более высоким содержанием азота,
полуторным нитридом U2N3, система уран—азот является гетерогенной,
состоящей из двух отдельных фаз—UN и U2N3. От полуторного нитрида U2N3 вплоть
до динитрида UN2, третьего «стехиометрического» нитрида урана, система
гомогенна; это значит, что кристаллическая структура U2N3 при увеличении
содержания азота постепенно переходит в структуру UN2.
Кристаллическая структура и фазовые
отношения. Область от U до UN. Растворимость азота в металлическом уране
детально не изучена, имеются лишь указания, что она мала. Вследствие устойчивости
Глава 10. Соединения урана с элементами подгруппы Va 193
мононитрида урана UN система уран—растворенный азот метастабильна, за
исключением области очень высоких температур. Измерения растворимости
азота в уране осложняются тем, что скорость образования нитрида из
плотного урана является значительной уже при температуре 600—700°, а из
тонкого порошка урана, полученного разложением гидрида,—даже при
300—350°. Растворимость урана в мононитриде также не может быть
большой, так как константа пространственной решетки кристаллов
мононитрида заметно не меняется с изменением отношения U : N (в препаратах,
полученных сплавлением урана с мононитридом при высокой температуре).
Небольшие наблюдаемые колебания (например, а = 4,899 А вместо а=4,880А)
обусловлены, вероятно, частичным замещением азота кислородом или углеродом, а не
содержанием урана в решетке мононитрида. Так же, вероятно, мала и
растворимость мононитрида урана в уране, по крайней мере в твердом
состоянии. Наличие нитрида (или окисла) вызывает рост вязкости сплавов
урана [14].
Мононитрид представляет порошок светлосерого цвета [15], имеет гранецен-
трированную кубическую решетку с константой а=4,880±0,001 А [12, 13,
16, 17]. Элементарная ячейка содержит четыре атома урана. Вещество имеет
структуру типа каменной соли, а не цинковой обманки; это подтверждается
тем, что на порошкограммах отражение (4 2 0) заметно интенсивнее, чем
отражение (3 31) [16]. Плотность мононитрида, рассчитанная из
рентгенографических данных, равна 14, 32 г/см3. Мононитрид полностью изоморфен
монокарбиду UC (см. гл. 9).
Если нитрид урана получается путем введения азота в реакцию с ураном
при температурах до 1300°, то после образования мононитрида реакция не
останавливается и не замедляется, а идет до образования фазы U2N3+UN2,
находящейся в условиях опыта в равновесии с азотом. Чистый мононитрид может
быть получен разложением высших нитридов при температуре выше 1300°.
Если нагревание проводится медленно, выделение азота происходит спокойно.
При этом надо проводить реакцию в отсутствие даже следов кислорода, иначе
могут образоваться моно- и двуокись урана. Мононитрид, остающийся после
разложения полуторного нитрида, является очень устойчивым соединением.
Он заметно не разлагается при температуре 1700° [18]. Давление
разложения его при 1200° меньше 110~влш рт. ст. [13]. Мононитрид спекается при
температуре 2300° и плавится в пламени атомарного водорода при
2630±50^ [19].
Область UN—U2N3 (полуторный нитрид урана). Системы, содержащие
на один атом урана от одного до полутора атомов азота, являются
двухфазными, что подтверждается рентгенографическим анализом. Вторая фаза U2N3
тоже кубическая, но объемноцентрированная, с константой решетки 10,678 ±
±0,005 А [13, 17, 20, 21]. Если в рентгенограмме учитывать только
интенсивные отражения, то решетка полуторного нитрида окажется гранеиентри-
рованной с константой, равной приблизительно 5,3 А. При учете и слабых
отражений получается объемноцентрированная решетка с вдвое большей
константой. Эта структура (D53) известна для окиси марганца (III) и
некоторых окислов редкоземельных элементов и близка к структуре флюорита [22].
Она допускает постепенный переход от решетки полуторного нитрида к решетке
динитрида типа флюорита [12].
Атомы урана в элементарной ячейке полуторного нитрида, содержащей
16 молекул, занимают следующие положения:
(000; |44) +
8UI B(6):(Iii;-I|4).>
13 Химия урана
194
Часть III. Бинарные соединения урана
24UII в (d):±(* О 1; * у т))-; *=-0,018;
48N в (е): ± (*#z; y-*yz; Яу — уг; ^^-г^.
Параметры атома азота х, у, г не могут быть выведены из рентгенограммы
вследствие сравнительно слабой рассеивающей способности атомов азота,
но они, повидимому, подобны параметрам кислорода в окиси марганца (III).
Для последней параметры кислорода соответствуют х^ 0,385, # = 0,145
и z — 0,380. Плотность полуторного нитрида, рассчитанная из
рентгенографических данных, равна 11,24 г/см3\ межплоскостные расстояния в полуторном
нитриде и мононитриде урана остаются постоянными при изменении
отношения U:N, что свидетельствует об отсутствии взаимной растворимости этих
двух фаз.
Область U2N3 — UN2. Постепенное превращение полуторного нитрида в ди-
нитрид без скачкообразного изменения фаз возможно потому, что структура
полуторного нитрида, подобная структуре окиси марганца (III), может
рассматриваться как искаженная структура флюорита, в которой кристаллизуется
динитрид. Атомы металла в последней находятся в положениях 0 00; 0 у
1- 1 0
2 ' 2 U
и
1°:
атомы азота —в положениях
4
2
3 3 3 113
и т. д.; у -£ -£-; -у -£ ~ ; и т. д. Если считать ячейку объемноцентрирован-
ной (в восемь раз большей), то атомы металла будут находиться в положениях
OOOjyOO; у-2-^;0Т~2;~2°~2; Т"20' ПРИ пеРех°Де к структуре
полуторного нитрида эти атомы урана остаются фиксированными; другие
24 атома металла слегка смещаются от осей четвертого порядка, что требует
большей по размеру элементарной ячейки. Постепенное изменение размеров
ячейки иллюстрируется табл. 69, в которой приводятся данные для меньшей
псевдогранецентрированной ячейки, которая может быть выведена на
основании одних интенсивных отражений.
Таблица 69
КОНСТАНТЫ РЕШЕТОК U2N3 И UN2
Состав
UN1>435
UNli52
UNIf7e
UN2a)
Константа решетки, А
объемноцентри-
рованная ячейка
10.678±0,005
10,658^-0,005
10,580±0,005
псевдограие-
центрированная
ячейка
5,339±0,003
5,3294-0,003
5,290±0,003
5,31 ±0,01
Плотность,
11,24
11,73
а) Эта фаза является, вероятно, истинной гранецентрированной
кубической с малой элементарной ячейкой.
По мере возрастания содержания азота слабые рентгеновские линии,
указывающие на искажение гранецентрированной ячейки, исчезают.
Распределение атомов азота в продуктах, промежуточных по составу между
Глава 10. Соединения урана с элементами подгруппы У а
196
U2N3 и динитридом, остается неизвестным, так как анализ рентгенограммы
основан лишь на данных для более тяжелых атомов урана.
Следует заметить, что полуторный нитрид (р = 11,24) имеет меньшую
плотность, чем динитрид (р = 11,73) и в особенности чем мононитрид (р = 14,31).
Поэтому под высокими давлениями полуторный нитрид будет разлагаться на
моно- и динитрид.
Мононитрид устойчив в вакууме при температурах до 1700° и выше,
полуторный нитрид теряет азот в вакууме при температурах выше 700 — 800%
а динитрид может быть приготовлен только под высоким давлением азота.
1100 Ю00 900
Температура, °С
800 700
Т
600
500
Рис. 33. Давление азота в равновесии с тремя нитридами урана
различного состава при разных температурах.
Равновесное давление азота над высшими нитридами является непрерывной
функцией температуры и содержания азота х в твердой фазе (1,5 < х< 2).
По данным, полученным в Эймском университете, с ростом х давление возра-
стает очень быстро; при х = 1,712 и при 450° оно равно 4 мм рт. ст., при
800° —приблизительно 400 мм рт. ст. Из наклона кривой давления
разложения вычисленная теплота а отирования составляет 16 ккал/моль.
Равновесие устанавливается быстро и является обратимым [23]. Более
подробные исследования показали, что при температурах от 658 до 1048° она
устанавливается в течение одного часа, при 492° для этого требуется два
дня [13, 24]. И при этих исследованиях наблюдалось быстрое изменение
давления с составом. Например, при 1048° для UNltB2 давление равно 50 мм
и для UN1>M — 825 мм; при 492° оно равно 50 мм для UNltW, и 825 мм
13*
196
Часть III. Бинарные соединения урана
для UNliW. На рис. 33 показаны кривые равновесия lgp = /Y —J для трех
продуктов с различным содержанием азота.
Вышеупомянутое значение (400 мм рт. ст. при 800° для UNb71)
расходится с рис. 33, эта разница объясняется [13, 24J ошибками в определении
азота.
Разложение UNX идет быстро при к > 1,5, но после достижения
состава U2N3 оно заметно замедляется.
Получение нитридов урана. Выше упоминались следующие
два метода синтеза нитрида урана:
1. Прямое взаимодействие урана с азотом при температуре 1000° или
ииже [1].
2. Взаимодействие UC14 с аммиаком при температуре красного
каления [2].
Кроме того, в литературе описаны еще следующие способы:
3. Взаимодействие закиси-окиси урана с магнием в токе азота [5].
4. Взаимодействие 2NaCMJCl4 с аммиаком при температуре красного
каления [7J,
5. Взаимодействие порошкообразного урана с аммиаком при
температуре 1000° или ниже [8].
6. Реакция между металлическим ураном или его сплавами и нитридом
магния в вакууме [9J.
7. Реакция между дикарбидом урана и азотом при 1180° [4].
8. Реакция между гидридом урана и аммиаком при 200° [12].
9. Реакция между гидридом и азотом при 350* [12j.
Согласно последним данным [ 12], чистый нитрид, не загрязненный
примесями других трудно удаляемых твердых веществ, получается только в результате
реакций между ураном, его гидридом или тетрахлоридом и азотом или
аммиаком. Во избежание загрязнения окислами реагенты предварительно надо
тщательно очистить от кислорода.
Согласно фазовым отношениям, рассмотренным в предыдущем разделе
(см. рис. 33), синтез нитрида под атмосферным давлением азота должен давать
смесь кристаллов полуторного нитрида и динитрида со средним составом
между UNli5 и UN1<8, в зависимости от температуры реакции. При
использовании аммиака вместо азота получаются те же результаты. Более низкие
значения для азота (U3N4, т. е. UNb33), полученные при прежних анализах,
возможно, объясняются присутствием окислов или ошибками при определении
азота по Дюма.
Взаимодействие металлического урана с азотом недавно было изучено
более подробно. При этом пользовались как ураном в куске, так и порошком
металлического урана, полученным разложением гидрида. Стружки урана,
очищенные 8 н. азотной кислотой, были превращены в UNlt712 (порошок серо-
стального цвета, р = 11,3 г/см3) путем нагревания при 450° под давлением
азота в 1 атм [23]. Из порошка металлического урана был получен нитрид
срстава UN1)76 путем нагревания его в течение трех дней до 520° в токе
чистого азота [251. Было сделано наблюдение, что в результате частичного
окисления толстых урановых стружек при прокаливании их на воздухе
блестящий металл покрывается слоем нитрида [26]. Установлено, что
скорость азотирования урана возрастает с температурой, особенно быстро
около 800°, вероятно в связи с переходом металла из р- в *j- модификацию [271.
Согласно другим наблюдениям, скорость реакции между плотным
металлическим ураном и азотом резко возрастает при температуре выше 450° [28].
В работах, проведенных Национальным бюро стандартов, показано, что
реакция между ураном и азотом мало заметна при 400°, ускоряется при 500—
€00° и быстро протекает выше 800° 1291. При температуре 750° и ниже образцы
Глава 10. Соединения урана с элементами подгруппы Va 197
<- , .
покрываются черной, плотно пристающей коркой с металлическим блеском.
Небольшие кусочки этой корки отслаиваются при охлаждении. После удаления
всей корки соскабливанием оказывается, что в блестящей сердцевине
содержится 0,012% азота вместо 0,003% до реакции. При 800° или выше вместо
корки образуется темносерый или черный неметаллический порошок; путем
анализа установлено, что и корка и порошок имеют состав UN1|6. Окисная
пленка не защищает уран от действия азота при 700° [29].
Взаимодействие между ураном и аммиаком начинается при температуре
около 400° [12]. Для быстрого превращения стружек урана в нитрид требуется
температура выше 700°. Нагреванием урана в атмосфере аммиака при 800° в
течение 24 час. получается продукт состава UN1>747.
Реакция между гидридом урана и азотом идет даже при 200° [30],
причем в результате ее также получается продукт с приблизительным
составом UN1|75. Реакция требует 10—12 час. при 250° и 1—2 час. при 350°.
Реакция между гидридом урана и аммиаком идет, хотя и медленно, уже при
100° (в течение 1 часа 5 г урана присоединили 1,5 мг азота) [12]. При
температуре выше 200° скорость реакции делается значительной (за 1 час 5 г урана
присоединяет 316 мг азота). Это, повидимому, наиболее легкий метод получения
нитрида. Продукт реакции представляет чрезвычайно тонкий порошок
(насыпной вес 3,4 г/см3, истинная плотность 11,3 г/смг). Вопреки прежним
предположениям [31], низкотемпературные реакции между гидридом и азотом или
аммиаком не останавливаются и не замедляются по достижении состава,
отвечающего мононитриду UN, а продолжаются до образования полуторного
нитрида и выше. Пока неизвестно, образуется ли мононитрид в качестве
промежуточного продукта при низкотемпературном азотировании [32]. Как
указывалось прежде, способ получения мононитрида состоит в разложении
высших нитридов в вакууме при температуре выше 1300°. Так, например,
мононитрид был получен нагреванием UN1(67 в вакууме до 1650° в графитовом
тигле [15].
Динитрид UN2 до сих пор еще не получен в чистом состоянии. Если уран
нагревать в атмосфере азота под давлением в 125 ати при 600°, то получается
смесь моно- и динитрида, что доказано рентгенографическим методом [33).
Хотя средний состав этого продукта (UN7|75) и одинаков с составом нитрида,
полученного при атмосферном давлении азота, однако рентгенографическим
методом показано, что он не содержит полуторного нитрида. Выше (стр. 194)
уже упоминалось, что полуторный нитрид под высоким давлением диспропор-
ционирует на моно- и динитрид вследствие большей плотности динитрида и
особенно мононитрида по сравнению с полуторным нитридом.
Физические и химические свойства нитридов
урана. Нитриды по внешнему виду описаны как черно-бурые,
серо-стальные, темносерые или черные порошки. Кристаллическая структура нитридов
рассмотрена выше (стр. 193). Плотность, вычисленная из
рентгенографических данных, равна 14,31 г\смъ для мононитрида, 11,24 г\смг для полуторного
нитрида и 11,73 г\смъ для динитрида. Прямым определением плотности для
UN1>8, приготовленного из гидрида урана й аммиака, получено значение
11,3* г/ом8. Насыпной вес этого продукта составляет 3,4 г/cmz, электрическое
сопротивление непрессованного продукта—около 200 ом-см [34].
Теплоты образования нитридов известны недостаточно точно. Найдено,
что при образовании нитрида, содержащего 3,5% азота, выделяется 68,5 ккал
на \г-атом поглощенного азота [35]. Указанное количество азота гораздо
меньше содержания его даже в мононитриде UN (5,55% N). Следовательно,
совершенно неизвестно, какая фаза образовалась в действительности, но можно
в качестве гипотезы выдвинуть предположение, что образовавшийся нитрид
представляет собой U2N3, а низкое содержание азота обусловлено присутствием
в продукте непрореагировавшего металла. Исходя из этого предположения*
198
Часть III. Бинарные соединения урана
можно вычислить теплоту образования полуторного нитрида по реакции
2U+|~N2~^U2N3; (1)
она составляет 256 ккал* моль'1.
Из наклона трех прямых (рис. 33) вычислены следующие значения для
суммарных теплот разложения UNX на UN и l(x—1)/2] N2:
UN 1,65 АЯ= 15,69 ккал-г-атом'1^};
UN1>e0 АЯ-= 18,79 ккал-г-атом^и;
UNi ,56 АЯ = 19,55 ккал • г-атом"1 U.
Ввиду того что зависимость АН от х не является линейной, эти
значения нельзя точно экстраполировать к х= 1,5. Тем не менее такое
экстраполирование было проведено; этим путем было выведено значение А# = 21,9,
откуда получается следующее термохимическое уравнение [24]:
2UN + yNa-^U2N3 + 43,8 ккал. (2)
Если зависимость АЯ от х, показанная на рис. 33, правильна, то
дальнейшее азотирование полуторного нитрида будет эндотермическим процессом.
Поэтому можно ожидать, что динитрид будет устойчив при более высоких
температурах.
На основе вычисленных выше значений (256 ккал для полуторного нитрида
и 44 ккал для разницы U2N3 — 2UN) теплоты образования мононитрида
получается значение 106 ккал-моль1, что объясняет устойчивость этого
соединения. Таким образом, мы имеем следующие предварительные значения:
U + -g.Na->UN+106 ккал\ (3)
2UN+lNa->U2N3 + 44 ккал\
U^Ng+^Na—>2UN2 — х ккал (эндотермическая реакция). (4)
Вычисления AF и AS для образования нитрида [36, 37] сделаны в
предположении, что состав образующегося нитрида соответствует U3N4; поэтому
они лишены значения. Вряд ли имеет смысл вводить поправки в эти
значения для расчета А/7 и AS для мононитрида и полуторного нитрида ввиду
отсутствия точных термохимических данных.
Химические реакции нитридов урана. Термическое
разложение высших нитридов урана, полуторного нитрида и динитрида, рассмотрено
выше (стр. 197). Эти нитриды можно восстановить водородом [25]. В случае
взаимодействия UNi,75 с водородом уже при температуре 100° наблюдается
образование аммиака. При 500° в течение 4 час. теряется 10% азота. При
йагревании в атмосфере водорода в течение 1 часа при 800° UNi,75
восстанавливается до UN 1,52; дальнейшее нагревание в течение 18 час. ведет
к образованию продукта с составом UNii43- Однако при этой температуре
(800°) помимо восстановления может иметь место и значительное
термическое разложение.
Нитрид легко окисляется. Он в обычных условиях не пирофорный, хотя
в литературе и имеется указание, что порошкообразный нитрид, полученный
из гидрида урана, обладает этим свойством [15J. На воздухе при
температуре 150-200° он воспламеняется и сгорает с образованием двуокиси
или закиси-окиси урана и азота (но не окислов азота) [34]. При этом было
Глава 10. Соединения урана с элементами подгруппы Va 199
отмечено образование еще аммиака, возможно, в результате взаимодействия
нитрида со следами воды [38]. При нагревании с водяным паром нитрид
разлагается [5]. Такие соединения, как окись меди, хромат свинца, окись
железа, двуокись урана (см, [39]), хлорат и нитрат калия окисляют
нитрид [5].
Кроме того, нитрид медленно окисляется концентрированной азотной
кислотой. На него не действует горячая или холодная соляная или серная
кислота или раствор едкого натра [5, 12], однако он реагирует с
расплавленными щелочами с выделением аммиака [5] и с газообразным хлористым
водородом [40]. Если последняя реакция проводится при 400 — 500°, то
образуется хлорид аммония, a UNi,75 превращается в темную гигроскопическую
спекшуюся массу зеленого и коричневого цвета. Рентгеновские анализы
показывают, что эта масса содержит тетрахлорид, двуокись и закись-окись
урана и неизвестную фазу, возможно, двойную соль (NH4)2UC16; кроме того,
в ней содержится еще 2% аммиака. При подобной же обработке моионитрида
получается продукт, содержащий менее 1% аммиака. Нитрид легко
реагирует с горячей 85%-ной фосфорной кислотой с образованием фосфата
урана (IV) и с концентрированной хлорной кислотой, окисляющей его в ура-
нилперхлорат [12].
Химические свойства мононитрида подробно не изучены; сообщается
лишь, что «они подобны свойствам высших нитридов». Мононитрид
реагирует с углеродом (тонкодисперсным графитом) при 2250° и с двуокисью
урана при температуре около 2300° [39], но продукты реакции не
исследованы.
ФОСФИДЫ УРАНА
В 1872 г. было высказано предположение, что при разложении
1Ю2(Н2Р02)2Н20 образуется монофосфид урана [41]; позднее монофосфид-
ная фаза была идентифицирована рентгенографическим методом. Она имеет
структуру каменной соли, а0 = 5,589А [42).
Путем взаимодействия фосфина с 2NaCI-UCl4 получен фосфид состава
изР4 с небольшим выходом [7]. Эту реакцию можно было осуществить
пропусканием тока сухого водорода над расплавом, содержащим фосфид
алюминия и избыток 2NaCI-UCl4. Продукт выщелачивали водой и соляной
кислотой, затем промывали последовательно водой, спиртом и эфиром.
Остаток представляет собой тонкий черный кристаллический порошок,
содержащий немного алюминия. Этот фосфид медленно окисляется на воздухе
до желтого уранилфосфата. Он горит на воздухе и медленно разлагается
водой, особенно при доступе воздуха. На него не действует разбавленная
соляная кислота, но кипящая концентрированная азотная кислота, царская
водка или расплавленны i едкий натр мгновенно разлагают фосфид.
В качестве промышленного метода приготовления фосфида урана было
предложено нагревание смеси 3 частей порошкообразного металлического
урана и 1 части порошка фосфора до 600 - 1000° [43j. Непрореагировавший
фосфор удаляется при помощи спирта и эфира или отгоняется.
АРСЕНИДЫ И АНТИМОНИДЫ УРАНА
Арсенид и антимонид урана получены по методам, аналогичным тем,
которыми пользовались для синтеза фосфида этого металла. Арсенид урана
приготовлен путем пропускания сухого водорода, насыщенного парами
мышьяка, над двойной солью 2NaCl-UCl4 [44j. В результате образовалось
небольшое количество квадратных или шестиугольных черных блестящих
пластинок. При сплавлении арсенида натрия с избытком мышьяка и 2NaCI-UCl4
в токе сухого водорода образовался микрокристаллический арсенид серо-
200
Часть III. Бинарные соединения урана
стального цвета. Для этой же цели можно прокаливать смесь
порошкообразного мышьяка, трехокиси мышьяка, трехокиси урана и алюминия, хотя
применение алюминотермического метода для приготовления фосфида урана
оказалось неудачным. Арсенид урана состава U3As4 [44J почти не разлагается
на сухом воздухе, несколько быстрее он разлагается при контакте с влажным
воздухом. Он легко растворим в азотной кислоте и горит в пламени бунзе-
новской горелки.
Система уран —мышьяк изучалась в Эймском университете методом
рентгенографического анализа [42]. Идентифицированы два соединения:
моноарсенид UAs (кубическая структура каменной соли, яп = 5,767 А,
р= 10,77 г/см3) и арсенид состава U2As [45], рентгенограмма которого
отличается от рентгенограмм урана, двуокиси урана и арсенида UAs.
Антимонид получен Колани [7] прокаливанием сухого 2NaCl-UCl4
с эквивалентной смесью сурьмы и алюминия в токе водорода. Анализ
продукта (свободного от алюминия) показал, что он содержит 58% Sb и 42% U,
что приблизительно соответствует формуле U3Sb8. Нагревание этого
препарата в токе водорода ведет к постепенной потере сурьмы, но при этом
не удается получить продукт состава U3Sb4, которого Колани ожидал на
основании аналогии с арсенидом и фосфидом. Антимонид разлагался
концентрированной соляной кислотой и растворялся в концентрированной
азотной с образованием какого-то окисла сурьмы.
ЛИТЕРАТУРА
1 Moissan. H., Compt. rend., 122, 1092 (1896).
2. R a m m el s b erg C, Pogg. Ann., 55, 323 (1842).
3. U h r I a u b G. E., Dissertation, University of Gottingen, 1859, p. 27.
4. H e u s 1 e r O., Z. anorg. u. allgem. Chem., 154, 353, 366 (1926).
5. Kohlschutter V., Ann., 317, 166 (1901).
6. Colani A., Compt. rend., 133, 383 (1903).
7. С о 1 a n i A., Ann. chim. et phys., 12, 88 (1907).
8. H а г d t u n g H., Dissertation, Hannover Techn. Hochschule, 1912.
9. M i n e r C. G., пат. США 1631544, transferred to the Anglo California Trust Co.
(1922).
10. H e r z e r H., Dissertation, Hannover Techn. Hochschule, 1927.
11. Lore nz R., Wool cock J., Z. anorg. u. allgem. Chem., 176 (1928).
12. R u n d 1 e R. E., В a e n z i g e г N. С, Newton A. S., D a a n e A. H.,
Butler T. A., Johns I. B., Tucker W., Figard P., The System
Uranium—Nitrogen in National Niclear Energy Series, Division VIII, vol.
6 (MP Ames 1).
13. Battelle Memorial Institute, Report CC-2700, Feb. 1, 1945 (Battelle 1).
14. Mallinckrodt Chemical Works, Report A-1072, Feb. 15, 1945 (Mallinckrodt 1).
15. D a a n e A. H., С h i о t t i P., Report CT-I775, Aug. 1, 1944 (MP Ames 2).
16. R u n d 1 e R. E., Baenziger N. C, Wilson A. S., McDonaldR. A.,
Report CC-2397, Feb. 17, 1945, p. 22 (MP Ames 3).
17. RundleR. E„ Baenziger N.C., WilsonA.S., McDonaldR. A.,
J. Am. Chem. Soc, 70, 99—105 (1948).
18. Daane A. H., Carlson N., Report CC-1496, Apr. 10, 1944, p. 14 (MP Ames 4).
19. DaaneA. H., Car lson N.. Report CC-1500, May 10, 1944, p. 10 (MP Ames 5).
20. Reports CT-686, May 22, 1943; CN-1495, Mar. 10, 1944; CC-1514, Mar. 10, 1944
(MP Ames 6).
21. Battelle Memorial Institute, Report CT-1009, Oct. 10, 1943 (Battelle 2).
22. Strukturbericht, 2, 38 (1937).
23. Newton A. S., Report CC-178I, Oct. 18, 1944 (MP Ames 7).
24. Battelle Memorial Institute, Report CT-I697, May 1, 1944 (Battelle 3).
25. Newton A. S., Nottorf R. W., DaaneA. H., Report CC-1524, Apr. 14,
1944 (MP Ames 8).
26. Peterson D. W., Wimmer E. J., Report CC-1779, July 10,1944
(MP Ames 9).
27. Foot e F., Howe J. P., Report CT-1269, Jan. 29, 1944 (MP Chicago 1).
28. Battelle Memorial Institute, Reports CT-688, May 10, 1943; CT-956, Sept. 10, 1943.
(Battelle 4).
Глава 10. Соединения урана с элементами подгруппы Va
201
29. L i п d 1 i ef W. E., Report CT-1I01, Dec. 4, 1943. Report CT-1179, Jan. 1, 1944,
Holm V. С F., L i n d 1 i e f W. E., Report CT-2733, Mar. 19, 1945 (Natl.
Bui\ Standards 1).
30. T u с к е г W., F i g а г d P., Report CC-1500, May 10, 1944 (MP Ames 10).
31. В u t 1 e г Т. A., Fischer R., Newton A. S., Report CC-664, May 14,
1943 (MP Ames 11).
32. Figard P., Tucker W., Report CC-H96, May 11, 1944; Tucker W.,
Figard P., Report CC-1500, May. 10, 1944 (MP Ames 13).
33. В a e n z i g e r N. C, Report CC-1984, Dec. 19, 1944 (MP Ames 14).
34. J о h n s I. В., Report CC-587, Apr. 19, 1943 (MP Ames 12).
35. Neumann В., Kroger C, Haebler H., Z. anorg. allgem. Chem., 207,
146 (1932).
36. Davis J. W., В u г t о n M., Report CC-231, Aug. 15. 1944, p. 17 (MP Chicago 2).
37. Brewer L., Report CC-672, May 15, 1943 (MP Berkeley 1).
38. Sea b org G. Т., Report CK-591, Apr. 15, 1943 (MP Chicago 3).
39. R u n d 1 e R. E., Baenziger N. C, Report CC-1524, Mar. 10, 1944 (MP
Ames 15).
40. Warf J. C, Ericson R. P., Report CC-1504, Aug. 10, 1944, p. 24 (MP Ames 16).
41. Rammelsberg C, Sitzber, kgl. preuss. Akad. Wiss., 1872, p. 449.
42. Rundle R. E., Baenziger N. C, Report CC-1778, Aug. 18, 1944 (MP
Ames 17).
43. L i 1 1 i e n d a h 1 W. C, Driggs F, H., пат. США 1893296, assigned to Wes-
tinghouse Electric Corp., Lamp Division, 1929.
44. Cola ni A., Ann. chim. et. phys., 12, 93, 95 (1907).
45. Baenziger N. C, Report CC-1781, Oct. 18, 1944 (MP Ames 18).
Глава 11
ОКИСЛЫ, СУЛЬФИДЫ, СЕЛЕНИДЫ
И ТЕЛЛУРИДЫ УРАНА
ОКИСЛЫ И ГИДРООКИСИ УРАНА
Три окисла урана —двуокись U02, закись-окись U308 и трехокись U03—
известны более 100 лет, но систематическое изучение системы U — О началось
лишь в 1920 - 1928 гг. Работы английских исследователей, а также
американские работы, связанные с манхэттэнским проектом, дали много нового; так,
например, была открыта моноокись UO *.
Растворимость кислорода в уране незначительна, так же невелика
растворимость урана в моноокиси; таким образом, в процессе окисления
от урана до моноокиси система по существу является двухфазной. Раньше
предполагалось, что система уран — кислород является двухфазной и в
интервале от моноокиси до двуокиси, однако в 1946 г. была обнаружена
гомогенная фаза состава UOi,75- Это могло быть или новое соединение U407,
или начало ряда твердых растворов, существование которых было уже раньше
доказано для интервала от U02too до LT02,3o- За U02t3o система становится
двухфазной, но верхний предел этой двухфазной области очно не установлен.
В некоторых ранних работах утверждалось существование окисла U02,5
(или U205). Впоследствии в результате исследований при помощи
рентгеновского анализа было подтверждено наличие гомогенной фазы с таким
составом. Согласно последним наблюдениям, проведенным в Эймсе, U02,5
является нижним пределом монофазной области, простирающейся до U02,67
(или U308) и выше. Однако тензиметрические исследования, проведенные
Бильцем и Мюллером, не дали никаких указаний на существование
соединения U205. Из результатов этих анализов следует, что нижний предел
существования третьей у рано-кислородной гомогенной фазы (помимо
моноокиси и двуокиси) соответствует U02i62. Этот состав очень близок к U02)67
(или U308). Хотя таким образом точно и не установлено, начинается ли
монофазная область с U02t5o или с U02,62, но несомненно, что верхний
предел этой гомогенной области простирается далеко за 1Ю2,67> по всей
вероятности, вплоть до U03.
Фазы UO и U02 (более точно от UOi,75 До U02,3o) имеют кубическую
гранецентрированную решетку. Моноокись UO имеет структуру каменной
соли, U02 — структуру флюорита. При изменении состава в пределах от
ЬО2>50 (илии02,б2) дои03роо происходит непрерывный переход от ромбической
решетки к гексагональной. Помимо гексагональной формы U03 (I),
трехокись урана существует также в аморфном состоянии и по крайней мере
еше в двух, а возможно, и в четырех (пока еще не идентифицированных)
кристаллических формах.
Сродство к воде возрастает при переходе от двуокиси урана к трех-
окиси. Известны гидраты 1Ю2-л:Н20 и U308-xH20 (причем, вероятно, je = 2)>
* Систематическое изучение системы U—О должно было проводиться в
лаборатории Гледича в О^ло [I], но результаты этих исследований, если последние и были
осуществлены, не были известны во время составления данного обзора.
Глава 11. Окислы, сульфиды и теллуриды урана
203
но они недостаточно изучены. С другой стороны, твердо установлено как
кристаллографическим, так и аналитическим методами существование
нескольких гидратов трехокиси урана. К ним относятся полугидрат 2U03-H20
(или H2U207), четыре аллотропические формы моногидрата 1Ю3-Н20 (или
H2U04) и две аллотропические формы дигидрата U03*2H20 (или H4U05).
Двуокись урана обладает сильноосновными свойствами. Трехокись урана
амфотерна, т. е. она взаимодействует как с кислотами с образованием
солей уранила (например, U02S04), так и с щелочами с образованием ура-
натов (например, Na2U04). Окисел U308 часто рассматривается как уранат
урана (IV) U02-2U03 или U4+(UOJ~)o. В соответствии с з?тим U308 дает
с кислотами смесь солей четырехвалентного урана и уранила. Однако
формулу U02-2U03 нельзя истолковать в том смысле, что в U308 существуют
два типа атомов. Рентгеновские анализы показывают, что все атомы урана
в U308 структурно эквивалентны; таким образом, они, вероятно, несут также
и равный средний заряд (573+)-
Магнитные исследования показали (ср. ниже стр. 226), что U308 содержит
ионы U5+, а не U4+, поэтому для данного соединения вместо формулы
U02-2U03 была предложена формула U205-U03.
Природа окислов, содержащих более трех атомов кислорода на один
атом урана, еще полностью не изучена. Известны безводные окислы с
отношением кислорода к урану, равным 3,2 — 3,5; они получаются
дегидратацией U04-2H20 или разложением диураната аммония в токе кислорода
(см. стр. 263), а по некоторым данным также- электролизом (ср. стр. 233).
При действии воды эти окислы разлагаются на трехокись урана и кислород,
при растворении их в кислотах образуются соли уранила и также
выделяется кислород. Исследователи Браунского университета полагают, что
U03f5 представляет собой эквимолекулярную смесь трехокиси и безводной
четырехокиси урана, однако неясно, почему нельзя иОз.в рассматривать
как отдельный окисел U207, особенно если принять во внимание, что все
попытки приготовить чистую безводную четырехокись U04 до сих пор не
увенчались успехом (ср. стр. 239).
Гидратированная четырехокись урана может содержать в воздушно-сухом
состоянии до 4,5 молекулы воды. Однако, как установлено большинством
исследователей, определенный состав имеет только дигидрат 1Ю4-2Н20.
В противоположность безводному U03,5, гидрат U04-2H20 не разлагается
ни водой, ни кислотами; в 4 н. растворе серной кислоты он образует
слабый раствор, способный восстанавливать пермангаиат калия. В связи
с этим возможно, что U04-2H20 следует рассматривать как свободную
«надурановую кислоту» H4UOc, или UO(OH)3OOH. Эта кислота при
равновесии с водой образует небольшое количество перекиси водорода:
UO(OH)3OOH + H20 ^ Н202 + Ш(ОН)4. (1)
Надурановая кислота Урановая кислота
Предложение рассматривать U04-2H20 как свободную надурановую кислоту
было высказано уже много лет назад [2, 3], но впоследствии другие
авторы [4] выступили против этого мнения, указывая, что U04-2H20 нельзя
нейтрализовать щелочами с образованием перуранатов. Эти авторы
определили «перекисный» кислород в U04-2H20 титрованием перманганатом
калия или иодом по методу Ризенфельда и May [5j, причем было
установлено, что один атом кислорода является перекисным. На основании
этого была предложена следующая структура U04-2H20:
•2Н20.
204
Часть III. Бинарные соединения урана
С другой стороны, некоторые авторы [6] рассматривают гидраты перекиси
урана как продукты присоединения Н202 к U03:
(U08)-*H202-yH80, где для U04-2H20 x=\, г/=1.
Все эти предположения еще мало обоснованы, и вывод об истинном строении
1Ю4'2Н20 может быть сделан только на основании данных полного
рентгенографического анализа. Таких данных до настоящего времени еще не
имеется.
ФАЗОВЫЕ ОТНОШЕНИЯ В СИСТЕМЕ УРАН—КИСЛОРОД
Область от урана до моноокиси урана. Растворимость
кислорода в у vane. Моноокись урана UO. Растворимость кислорода в уране
очень мала даже при температурах выше 2000°. Наиболее точные из
известных данных, полученные в Национальном бюро стандартов США [7, 8],
2200
Кислород, % (атомн.)
Q10 * 020 Q30 0,40
О Q003 0,016 Qp24 0,032
Кислород, % (вес.)
Рис. 34. Растворимость кислорода в уране.
приведены на рис. 34. Кривая показывает, что растворимость"кислорода,
представляющая величину порядка 0,05% (атомн.) при температуре
плавления урана (1133°), возрастает до 0,1% (атомн.) при 1400° и до 0,4%
(атомн.) при 2000°. Растворимость кислорода (или какого-либо окисла урана)
в твердом уране нельзя точно определить, но тот факт, что включения
окисла после отжига в -^области могут быть обнаружены даже в металле,
содержащем всего 0,05% (атомн.) кислорода, показывает, что растворимость
Глава 11. Окислы, сульфиды и теллуриды урана
205
кислорода в твердой фазе еще меньше, чем в жидкой. Это отмечено
пунктирной линией на рис. 34.
Согласно данным Национального бюро стандартов США [9], из
растворов кислорода в расплавленном уране при охлаждении выделяются окислы
урана; выделение происходит
в Р- или -[-областях, в
зависимости от первоначальной
концентрации кислорода-
Были приготовлены [10]
пробы окислов урана UOx
различного состава
нагреванием в вакууме прессованных
смесей порошка
металлического урана и двуокиси урана.
В области составов с х — 0 ^-1
рептгечофазовым анализом
было установлено наличие
одно-о только урана и
моноокиси урана. Константы
пространственной решетки а-ура-
на заметно не изменяются в
присутствии кислорода;
подобно этому избыток урана
не влияет на константу
пространственной решетки
моноокиси урана. Первый факт
находится в согласии с
упомянутой выше низкой
растворимостью кислорода в а-уране,
второй указывает на то, что
растворимость урана в
моноокиси тоже очень мала.
Хотя чистая моноокись
урана известна только в виде
тонкого поверхностного слоя
на металлическом уране
(кроме одного случайного
наблюдения, см. стр. 248), ее
существование с
несомненностью установле о
рентгенографически. Она имеет
кубическую гранецентрированную
решетку (типа каменной со-
2JS2 2.60 2,40
Отношение 0-U
Рис. 35. Изотермы разложения окислов урана от
UO2>0(j5 ДО U2,2J0 [11].
ли), константа которой аь
равна 4,91 А (ср. табл. 71 на
стр.215) Эта структура
отличается от структуры металлического урана в любой из трех его
аллотропических форм, но очень сходна со структурой мононитрида UN и
монокарбида UC Эти три соединения — UO, UN и UC — легко образуют
смешанные кристаллы, т. е. их неметаллические компоненты, О, N и С,
способны неупорядоченно замещать друг друга.
Область^ от UO до U02.3- Двуокись урана. Область
уран-кислородных соединений, отдающих кислород только при очень высоких
температурах, простирается значительно дальше U02,oo, вероятно, приблизительно
до иС>2,15. Двуокись может присоединить даже большее количество
206
Часть III. Бинарные соединения урана
кислорода, вплоть до состава примерно U02,3, без изменения кристаллической
структуры; однако избыток кислорода по сравнению с составом U02j5 связан
сравнительно непрочно и может быть удален нагреванием в вакууме (рис. 35
и 36).
Отмечено [11], что минерал клевеит, имеющий структуру двуокиси
урана, близок по составу к U308. На основании этого сделано предположе
ние, что фаза U02 может присоединять большее количество кислорода,
чем это соответствует формуле иОг.зо- Но такое объяснение структуры
клевеита, вероятно, неточно (ср. гл. 3).
Имеются разногласия по вопросу о пределах гомогенной части системы
в области от UO до U02,3- Согласно сделанным в Эймском университете
3>°Г*Г\ I I I I I I I 1 1 Г
*Д
э
6 2,6
<ь
S
*
<ъ
а
§2/7
6
О
2.2
- \
-^О-О-О—О—<w__
ч
J
«^
5 J
—J
н
- К -1
■~
__
\
\ J
^_|
2JD\ 1 I I I I I I L_l I I I
400 600 800 Ю00 1200 1400 1600
Температура, °С
Рис. 36. Изменение отношения урана к кислороду в
зависимости от температуры при постоянном давлении кислорода,
равном 10 мм рт. ст. [12].
наблюдениям, область от UO приблизительно до U02,oo является
двухфазной. Об исключительно малой растворимости моноокиси урана в двуокиси
можно судить по незначительному изменению конста ты решетки двуокиси
урана при нагревании ее с порошком металлического урана (для UOa + U а0 =
= 5,461 А, а для чистой двуокиси а0 = 5,459А; ср. табл. 72).
С другой стороны, Захариазеном [13] установлено наличие гомогенной
фазы с гранецентрированной кубической решеткой, состав которой
соответствует приблизительно формуле UOi,75 (или U407). Ввиду того что эта
фаза присутствовала одновременно с нормальной фазой U02 (решетка
которой также гранецентрированная кубическая, но имеет меньшую константу),
можно предположить, что U407 является индивидуальным соединением,
аналогичным Th4S7. Однако это заключение нельзя считать вполне
достоверным. В отдельных случаях в несомненно однофазной области между U02.oo
и Ь02,зо также наблюдались две фазы, сходные по кристаллической
структуре, но различные по составу. Их существование, повидимому,
обусловлено неполным достижением равновесного состояния. Поэтому возможно [14],
что рассматриваемая область твердых растворов простирается вниз вплоть
до U0i,75 и одновременное присутствие UOi,7s и U02,oo также обусловлена
Глава 11. Окислы, сульфиды и теллур иды урана
207
недостижением равновесия. Это предположение подтверждается данными
рентгенографического анализа. Все окислы с составом между L'0li75 и U02,3
имеют гранецентрированную кубическую структуру типао флюорита. Согласна
табл. 72, константа решетки L'Oi,75 оравна 5,477А, стехиометрически
чистой ЬО2-5,460А и U02,3o — 5,430А. Такое равномерное уменьшение
константы решетки показывает правильность предположения о
существовании непрерывного ряда твердых растворов. Это закономерное падение
константы решетки с увеличением содержания кислорода позволило
работникам Эймского университета сделать вывод, что увеличение отношения
кислорода к урану обусловлено недостатком урана, а не присутствием
лишнего кислорода. Если это предположение справедливо, то плотность
окислов должна значительно снизиться в интервале от L02,oo (или UOi,75) До
и02,3о- С другой стороны, было высказано предположение [15], что
атомы кислорода, избыточные против формулы LT02, могут свободно
перемещаться в кристаллической решетке двуокиси или что они находятся
в пустотах решетки 1Ю2 [11]. Согласно этим взглядам, плотность Ь02,зо
должна быть выше, чем плотность Ь02, так как элементарная ячейка Ь'Ог.зо»
несмотря на ее немного меньшие размеры, содержит то же самое число
атомов урана, что и элементарная ячейка U02,oo, и на 15% больше атомов
кислорода.
Экспериментальные данные по плотности окислов урана с составом
между U02,oo и U03,oo представлены в табл. 70. Во-первых, они
показывают, что эмпирическая плотность LT02,oo хорошо согласуется с плотностью,
рассчитанной из рентгенографических данных при допущении, что каждая
элементарная ячейка содержит полный комплект из четырех атомов урана.
Во-вторых, из таблицы видно, что экспериментально найденные значения
плотности для окислов немного увеличиваются при переходе от U02,oo
к UO2,30- Это противоречит предположению о том, что как в U02,oo, так и
в высших окислах урана вплоть до и02,зо недостает заметной доли урана;
на основании измерений электропроводности, вероятно, небольшой
недостаток урана все-таки имеет место, но слишком малый для того, чтобы
сказываться на значении плотности (см. стр. 219).
Наблюдаемое возрастание плотности окислов от U02.oo к U02t3o
несколько менее резко выражено, чем этого можно было ожидать на
основании рентгенографических данных (ср. четвертую графу табл. 70), если
отвергнуть предположение о недостатке как атомов урана, так и кислорода*
Молярные объемы, рассчитанные Бильцем и Мюллером (шестая графа табл. 70),
практически постоянны для окислов в интервале от U02 0 до 1Ю2.26, в то /
время как из значений констант решетки вытекает требование, чтобы эти
объемы снизились в указанном интервале приблизительно на 1,7%. Однако
не исключено, что измерения плотности не настолько точны, чтобы по ним
можно было обнаружить ожидаемое уменьшение на такую величину.
Независимо от предыдущих исследований Жолибуа [17] установил, что
при низкотемпературном окислении двуокиси урана вначале образуется
синевато-черный окисел с х = 2,33, который он рассматривал как новый
окисел урана U307 (или U03-2и02). Однако этот автор заметил, что
кристаллическая структура этого нового окисла почти идентична структуре U02.
Подобные же наблюдения были сделаны и другими исследователями [18],
которые установили, что в атмосфере кислорода при 150° U02 окисляется
до U02,34 со сжатием решетки {а = 5,40 А вместо 5,468 А) и частичным
расширением диффракционных линий рентгенограммы, что указывает на
некоторое снижение симметрии. Это уменьшение размеров решетки
больше того, которое было найдено при переходе от U02,oo к U02t3o; тем не
менее нет оснований полагать, что окисел 1Ю2.зз [17, 18] представляет собой
новую фазу. Согласно ранее приведенным данным, он является, вероятно,.
208
Часть III. Бинарные соединения урана
Таблица 70
плотность vox
X
1,992
2,000
2,005
2,015
2,018
2,022
2,028
2,051
2,078
2,098
2,132
2,152
2,162
2,193
2,200
2,201
2,207
2,262
2,306
2,318
2,333
2,349
2,417
2,451
2,454
2,537
2,631
2,643
2,660
2,666
2,685
2,920
3,069
Плотность, г\см.ъ
по Гилле-
бранду [16]
—
10,95
10,88
—
10,82
10,86
10,83
—
—
10,89
—
—
—
—
10,98
—
—
—
10,47
_
—
10,15
—
—
—
—
—
—
—
—
—
—
по Бильцу
и Мюллеру
[И]
10,80
10,82
= }
10,71 1
10,82 J
—
—
10,72
10,94
—
10,62
11,08
10,87
10,87
—
10,94
i 10,90 1
10,40 J
— '
9,754
10,34
—
9,469
9,38 1
8,773/
8,379
8,369
! 8,216
i 8,300
i 7,938
7,074
6,039
рассчитанная из рент-
I!)
10,97
11,15
11,22
—
—
—
. —
—
—
—
—
—
*
—
11,35
—
—
—
—
—
8,336)
—
—
—
8,39
—
—
—
Молярный объем, см^/моль
по Гилле-
бранду
L16J
—
( 24,69
124,85
125,01
24,92
25,02
—
—
25,00
—
—
—
—
24,90
—
Я
26,29
—
—
27,28
—
{Z
—
—
—
—
—
—
—
по Бильцу
и Мюллеру
111]
25,03
24,97
—
—
25,25
25,01
, —
—
25,31
24,89
—
1 25,58
24,62
25,14
25,15
—
25,00
25,17
26,44
—
28,24
26,67
—
1 29,30
29,58
31,77
33,44
33,51
34,18
33,83
35,43
40,28
47,57
Объем
кислорода3)
смЗ-г-атом х
6,2
6,15
—
—
6,2
1 6,1
—
—
6.1
5,7
—
6,0
5,2
5,7
5,7
—
5,6
5,5
5,9
—
6,65
5,95
—
6.8
6,9
1 7,5
7,9
7,8
8,1
7,9
8,5
9,45
11,4
а) Эти значения получены вычитанием 12,7 см* (объема г-атома урана) из молярного объема и
делением остатка на х [11].
б> Uii05, см. стр. 223.
членом непрерывного однофазного ряда UOi>75 — Ш2,3, насыщенным
кислородом.
Область от U02.3 до Ш2.67- Соединение U308. В этой
области также имеются расхождения между результатами
кристаллографических [19] и тензиметрических [И] измерений. Расхождения касаются
состава ромбической фазы, сосуществующей с кубической U02 в 1ЮЯ при
Глава 11. Окислы, сульфиды и теллур иды урана
209
Согласно данным рентгеновского анализа [19], эта фаза имеет состав
UO2,50- Чистая гомогенная фаза такого состава была получена нагреванием
смеси равных количеств двуокиси и закиси-окиси урана U308.
Кристаллическая структура новой фазы несколько отличалась от структуры закиси-
окиси, хотя обе они ромбические. Поэтому ее можно считать отдельным
соединением состава U205. Игольчатые монокристаллы, которые давали
рентгенограмму U205, были случайно получены (вместе с октаэдрическими
кристаллами U02,3o) при разложении уранилхлорида при 900°. Несмотря
на различие в кристаллической структуре, окисел U205 может быть
превращен в закись-окись урана постепенным насыщением его кислородом без
образования новой фазы.
Некоторые другие авторы [20 — 25] также подтверждают
существование гомогенной фазы U205; иные же отрицают его [26 — 28].
Бильц и Мюллер измеряли давления кислорода над окислами урана
различного состава. В интервале между U02,67 (или U308) и UO2,20
давления разложения устанавливались быстро и обратимо; повидимому, они
являются истинными равновесными давлениями. Вследствие быстрого
изменения давления (р) кислорода с изменением состава х надежные данные
о зависимости р от х в этой области можно получить только в том
случае, если состав твердого вещества точно установлен- Бильц и Мюллер
определяли этот состав непосредственно анализом или вычисляли его по
количеству кислорода в газовой фазе. Результаты прежних исследований
давления разложения окислов урана, в которых необходимость точного
определения состава не учитывалась, лишены существенного значения,
особенно те из них, в которых использовались относительно малые
количества твердого вещества в большом объеме газа [24].
На рис. 35 представлены 5 изотерм разложения (от 752 до 1160°)^
которые показывают очень быстрое изменение давления диссоциации с
изменением состава, в особенности в пределах от U02,67 (или U308) до U02)62.
Вагнер и Шотки [29] попытались дать объяснение такому падению
давления на основании своей теории «упорядоченных смешанных фаз». Они
приписывают изменение давления разложения с уменьшением содержания
кислорода увеличению числа «дырок» в кислородной части ячейки U3Os. Эта
„ о I
теория приводит к линейной зависимости между и отклонением от
V ро2
стехиометрического состава; такое отношение действительно обнаружено
для изотерм 900 и 984°, но не для изотермы 1160° (рис. 37), в области
между U02,67 и U02?69-
За бесспорно однофазной областью в пределах от U02>67 до U02t62
следует, согласно Бильцу и Мюллеру, область постоянного давления
(от U02>6i до 1Ю2,зо)> которая по правилу фаз должна соответствовать
двухфазной системе. Согласно этим измерениям, отсутствует гомогенная
фаза состава 1Ю2,5о- Давление диссоциации начинает вновь падать с
уменьшением х только в хорошо известной однофазной области ниже и02>3о»
Это падение можно было проследить при 1160° вплоть до UO2)20, где
давление было равно 0,022 мм рт. ст. — наименьшее давление, измеренное
Бильцем и Мюллером. Дальнейшее разложение, ведущее к превращению
UO2,20 в U02too, происходит при температурах, значительно
превышающих 1160°.
Бильц и Мюллер показали, что давления разложения окислов U02>26,
U02,46 и U02i6i одинаковы не только при 1160°, но даже при некоторых
более высоких температурах, вплоть до 1240°. Пользуясь опытными и
экстраполированными значениями давлений, эти авторы построили урано-кислород-
ную изобару, соответствующую давлению кислорода в 10 мм рт. ст.
14 Химия урана
210
Часть III. Бинарные соединения урана
(см. рис. 36). При этом давлении закись-окись урана устойчива при
температурах от 580 до 650° и несколько выше. В интервале температур 750 — 1360°
U308 постепенно теряет кислород. Вертикальный отрезок изобары между
UC>2,62 и и02,зо показывает, что двухфазная область существует. Последняя
часть изобары (пунктирная линия) соответствует потере кислорода окислом
состава 1Х)2,зо- Как упоминалось выше, этот процесс можно проследить
только до состава, отвечающего формуле UO2.20-
Сопоставляя тензиметрические данные Бильца и Мюллера, заставляющие
отрицать существование U205, с результатами, на основании которых
2,667 2,672 2,677 2,682 2,687 2,692
Отношение 0-U
Рис. 37. Изменение давления диссоциации с изменением
отношения урана к кислороду [29].
исследователи в'; Эймском университете сделали вывод о существовании
гомогенной ромбической фазы U205, Брюэр [14] высказал предположение,
что гипотеза о существовании U205 объясняется легкостью, с которой
относительно простая рентгенограмма кубической решетки U02 маскируется
более сложной рентгенограммой ромбической решетки U308. Относительно
утверждения о получении монокристаллов U205 Брюэр отмечает, что
химический анализ этих кристаллов не сделан. Если, однако, результаты
исследований, проведенных в Эймсе, являются правильными, то это означает,
что либо измерения Бильца и Мюллера ошибочны, либо нижний предел
однофазной области снижается с падением температуры — от U02,62 при 1160°
до UO2,50 при комнатной температуре, что маловероятно.
Кристаллическая структура, приписанная U205 исследователями из Эйм-
ского университета, описана ниже (стр. 223). Для этой структуры
вычисленная плотность равна 8,33 г/см3. Однако Бильц и Мюллер не обнаружили
соединения с такой низкой плотностью в области х=2,5 (ср. табл. 70),
поскольку р практически линейно уменьшается с ростом х в интервале
значений 2,3 — 2,92.
Гренвольд и Гаральдсен [18] установили, что при окислении двуокиси
урана в интервале температур 200 — 250° образуется фаза с тетрагональной
структурой (5-фаза), обладающая гомогенностью в узкой области, а именно
Глава 11. Окислы, сульфиды и теллуриды урана
211
при составах, мало отличающихся от U02>4o- Константы ее решетки,
а = 5,37А, с=5,54А, и плотность (10,00 г/см3) показывают, что она имеет
состав U0,88O2,i2, т. е. что она получается из Ш2 частичным замещением
атомов урана атомами кислорода. Эта фаза (состав которой близок к U205)
устойчива только при температуре ниже 270°, что объясняет, почему
Бильц и Мюллер не обнаружили этой фазы.
Те же исследователи установили, что при взаимодействии закиси-окиси
урана с бикарбонатом калия (т. е. при процессе, который, по утверждению
3
о
I
45
40
35
30
25
1 l i I I
1 1 1 1 1 1
1
- иоэ
1
1 1
О
1 1
цо8
_1 L
_J L
■ гтн—1—
и
А
-j
—
-А
Л^ щЦ
, Vt*v4n
V 2,9 2,1 ZJ5 2,3 2,1
Отношение 0:{]
19
Ри
с. 38. Изменение молярного объема как функция отношения
урана к кислороду [30].
Лидена, приводит к образованию U205) или при восстановлении закиси-окиси
урана водородом при температуре 330° получаются продукты, структуры
которых подобны структуре U308. Они содержат около 2,60 атома
кислорода на один атом урана. Данные опыты показывают, что гомогенная
структура U308 простирается вниз вплоть до 1Ю2,56 (Бильц и Мюллер,
работавшие при высоких температурах, нашли, что гомогенная фаза
простирается ЛИШЬ ДО U02,62)-
Кристаллическая структура U308 описана ниже, стр.223. Она является
ромбической и соответствует плотности 8,39 г/см3, что приблизительно
согласуется с результатами непосредственных измерений, представленных в
табл. 70.
Область от U02,67 до U03,o- Трехокись урана. Согласно тензи-
метрическим данным Бильца, фаза U308 может существовать от U02>67 вплоть
до U02,62- Если результаты рентгеновских анализов, проведенных в Эймсе,
правильны, то область ее устойчивости доходит даже дои02,бо- Несомненно,
что эта фаза может существовать также в составах, более богатых
кислородом, чем U02i67- На кривой плотности (рис. 38) в точке,
соответствующей составу U02,67, излома не наблюдается и кривая продолжает
линейно повышаться с возрастанием содержания кислорода до U02,92t
и*
212 Часть III. Бинарные соединения урана
Резкое падение плотности между точками, соответствующими U02,92 и U03,oo»
возможно, обусловлено тем, что плотность U03 определялась на аморфном
порошке.
I; -л ;По данным американских исследователей [31], рентгенограммы
подтверждают постепенный переход U308 в U03. Захариазен [32] указывает, что
гексагональная решетка положений атомов урана, обнаруженная им у одной
из кристаллических форм U03, может быть получена из ромбической
решетки положений i атомов урана в U308 путем постепенного изменения
800
S 600
Б
§. 400
а:
1 200
О
3,0 2,9 2,8 2,7 2,6
Отношение 0-U
Рис. 39. Давление разложения системы уран—кислород в
области от U03 до U02,eeo [361.
параметров атомов урана. Таким образом, непрерывный переход между
двумя окислами не только вполне возможен, но и правдоподобен.
Однако по какой-то неизвестной причине для продуктов частичного
термического разложения трехокиси урана, полученных Фридом и
Дэвидсоном [33], не наблюдалось параметров, промежуточных по значению между
параметрами U03 и U308 (см. стр. 228). «
Установлено [34], что при термическом разложении оранжевой
трехокиси урана до U308 0,61% кислорода теряется при 520°, а остальные
1,27% — при 610°. При быстром охлаждении от температур, лежащих внутри
интервала 520 — 610°, обнаружена кристаллическая фаза с составом U02,9o;
рентгеновское исследование показало, что эта фаза имеет ту же структуру,
что и U308 и, вероятно, является твердым раствором.
Бильц и Мюллер [35] измеряли давление кислорода при разложении
U03 до U308. Результаты представлены шестью изотермами (от 438 до 650°)
на рис. 39 и изобарой (р= 10 мм рт. ст.) на рис. 36.
В этой области, в противоположность области окислов ниже U308,
выделение кислорода происходит медленно (возможно, что это отчасти
Глава 11. Окислы, сульфиды и теллуриды урана
213
объясняется проведением реакции при сравнительно низких температурах)
и полнее равновесие часто не достигается даже после многодневного
нагревания. В некоторых опытах кислород выделялся только в том случае,
когда процесс начинали при давлении, которое было намного ниже
равновесного; если же его начинали при давлении, сравнительно близком к
равновесному, хотя все же более низком, выделения газа вообще не
происходило. В других случаях началу выделения газа предшествовали
индукционные периоды продолжительностью 1 сутки или больше. Подобный
замедленный процесс частично наблюдался и в области от U03,oo до U02,92- В этом
случае он, вероятно, связан с медленным переходом аморфной трехокиси
в кристаллические низшие окислы. Этим явлением «индукции» можно
объяснить некоторые противоречивые данные о термической устойчивости 1Ю3>
приведенные в литературе. Однако более вероятно, что эти противоречия
обусловлены главным образом неодинаковой устойчивостью различных
кристаллических форм U03 (ср. стр. 243 и ел.).
Андерсон [37] изучал условия равновесия нестехиометрических
химических соединений. Он исследовал систему U03 —U308 как систему, где
наблюдаются большие отклонения от стехиометрического состава. Хотя
модель Андерсона не дает возможности количественно воспроизвести изотермы
разложения, измеренные Бильцем и Мюллером, он все же высказывает
предположение, что соединения со стехиометрическим составом, точно
соответствующим формуле U03, вообще не существует. Андерсон объясняет
данные измерений Бильца и Мюллера как следствие перехода от
неполной смешиваемости двух соединений к полной смешиваемости при
возрастании температуры; изотерма для 580° имеет форму, близко
соответствующую ожидаемой для критической температуры смешиваемости. Таким
образом, область U03 — U02,67 может быть гомогенной или гетерогенной
в зависимости от «термической истории» вещества.
Вследствие медленности разложения высших окислов урана большая
часть значений давления, использованных для построения изотерм на рис. 39,
была получена экстраполяцией к бесконечному времени. Возникает вопрос,
насколько близки эти давления к действительно равновесным. Результаты
нельзя было проверить путем подхода к равновесию со стороны окислов,
менее богатых кислородом, так как, например, U02,7o совсем не
присоединяла кислород в течение 24 час. при 400° под давлением 72 мм рт. ст.
(обычный метод определения урана в виде U308 основан на предположении,
что этот окисел не присоединяет кислород при прокаливании и охлаждении
на воздухе). Однако процесс превращения U03 в U308 термодинамически
не является необратимым. Действительно, Лебо [26] показал, что тонко-
диспергированный порошок U308, полученный прокаливанием уранилоксалата,
присоединяет кислород при температуре 350°, причем за 12 час. при этой
температуре цвет порошка изменяется от темносерого до
оранжево-коричневого. Это же подтверждают Бильц и Мюллер [28], установившие, что продукт
реакции действительно имел состав, близкий к U03. С другой стороны,
препараты U308, прокаленные при 800°, были обожжены «намертво» и не
присоединяли больше кислорода. Найдено [33], что прокаленную закись-окись
урана можно превратить в трехокись нагреванием до 500° под давлением
кислорода в 28 am в течение одних или нескольких суток. Цвет в этом
случае изменяется от черного до тёмнокрасного.
Булле и Домине-Берже [38] подтвердили наблюдения Лебо о
сравнительно быстром окислении закиси-окиси урана, приготовленной
прокаливанием уранилоксалата при низких температурах (или разложением трехокиси
в вакууме), а также о возможности превращения этой легко окисляющейся
модификации в практически неокисляемую микрокристаллическую
модификацию, которая дает несколько иную рентгенограмму.
214
Часть III. Бинарные соединения урана
Бильц и Мюллер указывают, что обратимость реакции
ЗиОз^ВД + т (2)
подтверждается также существованием в природе желтых минералов трех-
окиси урана, например гуммита или ретзерфордита, которые, по их
предположению, являются продуктами окисления минералов U308 (ср. стр. 256).
Даже если обратимость реакции (2) при использованных температурах
считать установленной, все же остается открытым вопрос о том,
действительно ли давления кислорода, измеренные Бильцем и Мюллером,
соответствуют равновесным давлениям разложения. Эти авторы указывают, однако,
что эти давления были по крайней мере близки к равновесным, так как
теплота разложения, рассчитанная на основании изотерм давления, хорошо
совпадала с теплотой, измеренной калориметрически (ср. стр. 243).
При температурах выше 580°, как видно на рис. 39, отсутствует
область постоянного давления между U02,67 и U03, что ясно указывает на
однофазность системы. Брюэр [14] полагает, что результаты, полученные
Бильцем, указывают на существование при температурах ниже 580°
двухфазной области между U02,67 и U02,92 и однофазной с быстро
изменяющимся давлением между U02,92 и U03,oo- По его мнению, при низких
температурах трехокись, использованная Бильцем и Мюллером, была
аморфной, и ее состав мог изменяться непрерывно только до U02,92, где
начинается образование кристаллического низшего окисла. При более высоких
температурах (500° и выше) возможно образование кристаллической
модификации U03, что делает возможным непрерывный переход на всем пути
от U03 до U308.
Область выше U03. Табл. 70 показывает, что трехокись урана
может присоединить некоторое избыточное количество кислорода по
сравнению с составом U03,oo> но это приводит к быстрому «разбуханию»
кристаллической структуры и резкому снижению прочности ее связей.
Область иОл, где х > 3,00, не очень хорошо изучена. Попытки получить
безводную перекись урана U04 окончились неудачей. Однако
сотрудники Браунского университета нашли, что при нагревании U04-2H20
до 130° в течение 24 час. (пока не разложится приблизительно половина
общего количества U04 до U03) повышение температуры до 300° приводило
к потере всей воды без дальнейшей потери кислорода; безводный остаток
имел средний состав, близкий к U03,5 [39]. Это соединение является
перекисным соединением, но по свойствам оно отличается от гидрата перекиси
U04-2H20; при действии воды или кислоты оно разлагается с выделением
кислорода и образованием гидратированной трехокиси урана. Безводное
перекисное соединение урана было получено в Браунском университете [40]
также прокаливанием диураната аммония в быстром токе кислорода. В
интервале температур 250 — 350° отгонялись NH3 и Н20. При повышении
температуры до 550° получался красный порошок, устойчивый, повидцмому,
при высоких температурах. Он имел состав от U03ji4 до U03(38 и терял
кислород при действии воды.
ФИЗИЧЕСКИЕ СВОЙСТВА БЕЗВОДНЫХ ОКИСЛОВ УРАНА
Моноокись урана UO. Свойства моноокиси урана недостаточно
хорошо изучены. Согласно литературным данным [14], она обладает
хрупкостью и имеет серый цвет с металлическим блеском. В табл. 71
приведены константы решетки, найденные различными исследователями.
Повидимому, последний десятичный знак для константы решетки
моноокиси урана не является достоверным, так как взятый для испытания
образец, возможно, содержал небольшое количество карбида или нитрида.
Глава 11. Окислы, сульфиды и теллуриды урана
215
Таблица 71
КРИСТАЛЛИЧЕСКАЯ РЕШЕТКА МОНООКИСИ УРАНА
Состав
UO+ U
ио+ио2
Константа
решетки, А
4,91±0,01
4,91
4,93
4,930±0,001
Плотность,
14,2±0,1
14,2
14,0
14,0±0,01
Литература
31
41
42
43
Различить структуры каменной соли и цинковой обманки в том случае,
когда оба компонента так сильно различаются по массе, как уран и
кислород, нелегко, однако имеющиеся данные свидетельствуют о том, что
моноокись урана имеет структуру каменной соли [31].
Моноокись более летуча, чем двуокись. На основании общих аналогий
установлено, что давление паров моноокиси представляет величину порядка
МО'5 атм при 2000° К [14]. '
Двуокись урана U02. Кристаллическая структура. В табл. 72
представлены результаты исследований кристаллической структуры фаз UOx
типа флюорита с я =1,75 — 2,3. Согласно вышесказанному (стр.206), U0lt76
может быть либо индивидуальным соединением U407, либо началом
непрерывного ряда твердых растворов, простирающегося вплоть до U02|3
Таблица 72
КРИСТАЛЛИЧЕСКАЯ РЕШЕТКА ДВУОКИСИ УРАНА
Состав
?
?
«Очень чистый
образец»
ио+ио2
ио2,00
ио2)10
ио2,20
UO2f30 (с U206)
U02+5% U308
ио2,00
L иоьи
ио2(0
U02,34
ио2
Константа
решетки, А
5,47
5,48
5,4568±0,0005
5,461 ±0,001
5,4586±0,008
5,437 -Ь0,001
5,433 ±0,001
5,4297±0,0008
5,464 ±0,002
5,460
5,477
5,468
5,40
5,4581 ±0,0005
* Плотность,
г}см*
—
Ю,96±0,01
—г
10,97
11,18
11,27
11,36
12,096
__
—
10,80
11,05
—
Литература
44
45
46
19, 47
19, 47
19, 47
19, 47
19, 47
48
13
13
18
18
49
Если U407 существует в виде устойчивого соединения (с точки зрения
распада на U02 и UO), то его теплота образования должна быть больше, чем
сумма теплот' образования 3U02 и1Ю, т. е. больше, чем 925 ± 30 ккал-моль~г
(132 ± 4 ккал-г-атомг1 кислорода).
Кристаллический габитус U02 описан различными исследователями
следующим образом:
микроскопические правильные октаэдры [50];
216
Часть III. Бинарные соединения урана
блестящие металлические чешуйки [51];
мелкие сильно отражающие октаэдры [52];
ромбические пластинки, обычно закругленные по четырем вершинам [53];
блестящие кубики [54].
Кристаллы двуокиси урана изоморфны кристаллам двуокиси тория.
Оба окисла образуют смешанные кристаллы [52].
Плотность. В дополнение к систематическим данным Гиллебранда,
а также Бильца и Мюллера по плотностям окислов VOx (см. табл. 70)
в табл. 73 приводятся отдельно разные значения плотности двуокиси урана.
Таблица 73
ПЛОТНОСТЬ ДВУОКИСИ УРАНА
Исходное вещество
UOa из оксалата (ср. табл. 70) . . .
Аморфный продукт из оксалата+Н2 .
Окисел с составом, средним между
Ш2,00 и U02,oe (ср. табл. 70) . .
Порошок Малинкродта не уплотненный
Порошок Малинкродта уплотненный .
Расплавленное спеченное при 2200°
Спеченное при 1400—2000° вещество .
Продукт Малинкродта 98,5% U02:
44,5 [х, не обезгаженный ....
Плотность,
г/см*
Г 10,J5
1 10,95—11,0
8,2
10,8
—
Г ~
{ 10,28
1 10,9—11,1
10
8,11
10,37
9,30
10,47
Насыпной
вес,
г(смЪ
—
—
—
3,76
4,51
4,96
—
—
—
—
—
Литература
55
16
56
12
57
57
58
59
59
60
61
61
61
Различие в плотности между обезгаженным и необезгаженным порошком
объясняется способностью двуокиси урана поглощать газы [62]. При
продолжительном нагревании до 130° плотность возрастает, например, от 10,4
до 10,6 г/см* [12].
Твердость. Твердость двуокиси урана равна 3,5 по шкале Мооса. Она
зависит от условий получения окисла [63].
Температура плавления. В литературе приводятся различные данные
о температуре плавления двуокиси урана, а именно: 2176° (в атмосфере
азота) [64] и 2500-2600° [65].
Давление паров. Давление паров двуокиси урана было измерено в
Чикаго [66] методом молекулярного истечения с использованием счета
ос-частиц для определения количества урана. Результаты сведены в табл. 74.
Из данных табл. 74 следует, что теплота испарения двуокиси урана
должна быть равна приблизительно 137 ккал-моль"1. Брюэр в своей сводке
термодинамических данных [14] приводит расчетное значение для давления
паров двуокиси урана (роя=Ь10~8 атм при 2000° К), которое намного
ниже эмпирических значений табл. 74.
Окислы со средним составом UO>2t2o имеют измеримую скорость возгонки
при гораздо более низких температурах, чем использованные в опытах,
представленных в табл. 74. Согласно Бильцу и Мюллеру [11], сублиматы
коричневатого цвета, которые были получены нагреванием UO2t30, ЬтОа>4в и U02 в7
Глава 11. Окислы, сульфиды и теллуриды урана
217
Таблица 74
ДАВЛЕНИЕ ПАРОВ ДВУОКИСИ УРАНА,
ОПРЕДЕЛЕННОЕ МЕТОДОМ МОЛЕКУЛЯРНОГО ИСТЕЧЕНИЯ
Температура,
1873
2023
2073
2173
2273
Температура,
°С
1600
1750
1800
1900
2000
Количество
испарившейся
ио2 а>,
г • 108
45
126
825
875
2300
Продолжительность
испарения,
мии.
180
23
60
15
10
р • 103, мм
рт. ст.
0,071
1,7
4,0
18
72
а) Отверстие диаметром 0,058 см.
до 1300°, имели состав между U02il6 и U02a7, в то время как состав
остатка был промежуточным между U02iie и U02t21.
Хотя давление пара окисла, вероятно, возрастает с ростом содержания
кислорода, но давление разложения возрастает еще быстрее и высокое
давление кислорода над окислом мешает возгонке последнего. Этим можно
объяснить причину того, что твердая фаза состава UO>2,6 (при тех
скоростях откачивания, с которыми работали Бильц и Мюллер) совсем не давала
сублимата. Механизм возгонки, повидимому, можно представить себе
следующим образом: при разложении закиси-окиси урана образуются молекулы
сравнительно более летучего окисла с большим содержанием кислорода
Таблица 75
ТЕПЛОЕМКОСТЬ ДВУОКИСИ УРАНА ПРИ НИЗКИХ ТЕМПЕРАТУРАХ а) [68]
пература,
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
СР'
кал • моль~1 » ерад~1 ,
0,378
0,473
0,574
0,687
0,815
0,968
1,148
1,358
1,599
1,900
2,270
2,715
3,478
6,230 б)
8,645 б>
5,150б>
пература,
31
32
35
1 40
50
60
70
80
90
100
по
120
130
140
150
160
ср,
кал • моль~1 • ерад~1
2,280
2,261
2,425
2,637
3,365
4,103
4,841
5,563
6,298
6,958
7,619 ,
8,276
8,923
9,518
10,07
10,60
пература,
?К
170
180
190
200
210
220
230
240
250
1 260
270
280
290
300
Ср.
кал • моль~1 • град""1
11,10
11,57
12,04
12,47
12,86
13,23
13,56
13,87
14,17
14,44
14,69
14,94
15,16
15,38
а) Материал с содержанием 0,7% UOs-
б) Значения неточны вследствие быстрого изменения Теплоемкости при этой температуре.
218
Часть III. Бинарные соединения урана
(например, трехокиси урана). При достаточно высоком вакууме этот окисел
может испариться. При соприкосновении с холодной поверхностью они
разлагаются на кислород и U02il6, т. е. фазу, находящуюся в равновесии
с кислородом под низким давлением при умеренно высоких температурах.
Удельная теплоемкость и энтропия. Определенная Реньо [67] средняя
удельная теплоемкость двуокиси урана между 0 и 100° равна
0,619-10"3 кал-г'1 (что соответствует 16,7 кал-моль'1). Подробное изучение
термодинамических констант двуокиси было проведено ста годами позже
в Беркли [68 — 70].
Результаты представлены в табл. 75 и 76.
Таблица 76
1
пература,
400
500
600
700
800
зад;
ТЕПЛОСОДЕРЖАНИЕ И ЭНТРОПИЯ ДВУОКИСИ УРАНА ПРИ ТЕМПЕРАТУРАХ
Hj— #208,16.
кал • моль~1
1680
3 470
5 340
7 280
9 250
11250
ВЫШЕ 298,16° К [70]
ST—S298,ie,
кал ■ моль~1 . град~1
4,82
8,82
12,24
15,22
17,84
20,21
пература,
°К
1000
1 100
1200
j 1300
1 1400
1 500^
#7W~^298»16.
кал • моль"!
13 280
15 340
17 420
19510
21620
23 750
Sy—5-2 ft 8 lie»
кал ♦ моль~~1 • град~1
22,35
24,30
26,11
27,79
29,36
30,83
. Кривая теплоемкости двуокиси урана имеет максимум между 15 и 50° К
(рис. 40), равный 9 кал-моль'1-град'1 при 28,6° К- «Избыточная» энтропия,
58
50 100 150 200
Температура, °К
Рис. 40. Теплоемкость двуокиси урана
250
300
соответствующая этой точке, равна 0,87 энтропийной единицы; ее можно
объяснить магнитным превращением (ср. стр. 221).
Для теплосодержания двуокиси урана в области температур от 300
до21500°К предложено [70] следующее уравнение, точное до ±0,1%:
Яг = Я298,1в+19,20Г + 0,8Ы(Г3Г2 + 3,957.106Г-1-7124.
(3)
Глава 11. Окислы, сульфиды и теллуриды урана
219
При дифференцировании получается следующее уравнение для теплоемкости:
Ср= 19,20+ 1,62- КГ3Т- 3,957- 105ГЛ (4)
Другое уравнение [71], удовлетворяющее данным Мура и Лонга [69],
имеет вид:
Ср=18,45 + 2,43Ы0-3Г-2,272.105Г-2. (5)
Энтропия при 298,1° К была вычислена [68] путем графического
интегрирования (ниже 15° К была использована функция Дебая с б =160° К):
5298,1 = 18,63 ± 0,1 энтропийной единицы.
В табл. 77 приведены некоторые дополнительные значения энтропии [14].
Таблица 77
ЭНТРОПИЯ ДВУОКИСИ УРАНА [14]
Fj—tf298
Температура, - .
°к Т
иоль~1 • град~1
500
1000
1500
20,48
27,67
33,6
S-p—5298.
кал • моль~1 • град~1
8,82
22,35
30,83
Теплопроводность, Значения, указанные в табл. 78, были найдены для
порошка двуокиси урана [72 — 74].
Таблица 78
ТЕПЛОПРОВОДНОСТЬ ВУОКИСИ УРАНА
^Температура,
°С
50
100
20—225
270—610
18—160
Неизвестна
Теплопроводность
10- кал • слг"1 • се/с~1 .
3,5
3,4
3,4
1,9
3,3 а)
50+5 б)
град'*
Литература
72
72
73
73
73
74
а) Порошок спрессован до р=6.
б)
Спеченный порошок.
Электрические свойства. Подобно всем темноокрашенным окислам,
двуокись урана является полупроводником. Ее электропроводность сильно
зависит от точного состава, чистоты и степени агломерации (кажущейся
плотности). Наименьшая электропроводность наблюдалась Фридрихом и Зит-
тигом [65]. Эти авторы применяли пруток из бурой двуокиси урана,
приготовленный путем спекания в водороде при 1100°, и нашли для удельной
электропроводности значение, равное всего лишь 4-Ю"8 (ом-см)'1 при
комнатной температуре; для двуокиси урана темносинего цвета (ср. стр. 223),
спеченной в азоте также при 1100°, удельная электропроводность
равнялась приблизительно 3-Ю"6 (ом-см)'1. Эти результаты показывают, что
электропроводность падает по мере приближения к составу UO200. Возникает
220 Часть III. Бинарные соединения урана
вопрос о причине обнаружения некоторыми другими исследователями
значительно более высокой проводимости двуокиси урана [60,75 — 77]: было
ли это вызвано присутствием «избыточного» кислорода или какими-то
другими условиями (например, более компактной формой окисла или лучшим
электрическим контактом). Данные этих авторов представлены в табл. 79.
Полученные ими значения находятся в пределах от 2,4-10~4 до 0,1 (ом-см)~г
при комнатной температуре. Имеются данные [77], указывающие на то, что
присоединение кислорода сверх стехиометрического количества,
соответствующего UO200 (достигнутое нагреванием образца двуокиси урана,
предварительно прокаленного в высоком вакууме, приблизительно до 500° при
1 — 100 мм 02), увеличивает электропроводность от 3-10~4 или 3-10~3 (ом-см)'1
перед обработкой до 1,2-10~2 (ом-см)"1 'после обработки. Электрическое
удельное сопротивление двуокиси урана [77] может быть выражено
уравнением
# = 0,1804е1867/г. (6)
Таблица 79
УДЕЛЬНАЯ ЭЛЕКТРОПРОВОДНОСТЬ ДВУОКИСИ УРАНА
Температура, °с
-60
-46
-25
-15
12
20
20
50
100
150
200
250
300
350
400
450
500
Электропроводность,
l-10-з (ом-см)-1
2,09 л
3,16
5,25 1
7,08 f
11,7
12,7 J
0,24 )
0,4
1,0
5,0
10
15 >
25
35
40
55
70
Литература
76
75
Температура, °с
28
28
28
28
ПО
157
295
478
793
992
22
40
60
80
1 96
131
155
182
223
270
327
Электропроводность,
1.10-3 (ом-см)~1
46,7 а> ч
61 б>
95 в>
114г)
80,6
118
173
247
403
>
1282 J
9,3 1
14,0
20,5
29,5
37,2
56,8 }
73,0
94
126
171
235
Литература
60
77
а) Двуокись спрессована и спечена при 1900—2000°, кажущаяся плотность р=8,11 г/сле*.
б) Кажущаяся плотность р=7,78 г\см*.
в) Кажущаяся плотность р =8,11 г/лиа.
Г) Кажущаяся плотность р=9.17 г}см*.
Обработка кислородом не влияет на значение предэкспоненциального
множителя, но «энергия активации» при этом уменьшается на 20 — 30% [77].
Возрастание электропроводности при увеличении количества кислорода
сверх стехиометрического позволяет отнести двуокись урана к классу
«дырочных» проводников, в которых электропроводность обусловлена отсутствием
Глава 11. Окислы, сульфиды и теллур иды урана
221
в решетке некоторых ионов металла, причем заряд последних переходит
к другим ионам металла, валентность которых временно увеличивается,
достигая, например, Ue+ [76]. Эта гипотеза подтверждена измерениями
эффекта Холла. Наличие этого эффекта позволяет различать «дырочные»
и избыточно-электронные проводники.
Предположение, что в окислах UO>2,oo (вплоть до 1Ю2,зо) существует
недостаток урана, а не избыток кислорода, рассматривалось выше (стр. 207)
с точки зрения рентгенографических наблюдений и было указано, что
значения плотности не позволяют представлять UO2)30 в виде U0j85O2. Однако
возможно, что небольшой недостаток урана все'же существует (независимо
от присутствия избыточного кислорода) и этот недостаток и является
основной причиной повышенной электропроводности. Исходя из эффекта Холла,
вычислено, что «окисленные» препараты двуокиси содержали около 3-Ю18
«центров нарушения» [76]. Это значит, что на каждые 105 атомов урана
нехватает приблизительно одного; такого небольшого недостатка нельзя
установить путем измерения плотности.
Изучены [78] некоторые окислы, сопротивление которых уменьшается
с температурой таким образом, что когда плотность тока превышает
известный предел, потенциал падает с увеличением силы тока («отрицательная
характеристика»). Такие результаты были получены для спеченной двуокиси
урана (плотность 9,2 г/см3), спрессованной в вакууме в небольшие прутки.
Проведено [79] довольно подробное изучение электрических
характеристик двуокиси урана с точки зрения ее применения в качестве термистора.
Термисторы используются как детекторы инфракрасного излучения и волн
ультравысокой частоты; они являются окисными полупроводниками с
большим положительным температурным коэффициентом сопротивления. Первые
термисторы изготовляли из двуокиси урана. Изучено действие давления,
растяжения и температуры на сопротивление кристаллической и аморфной
двуокиси урана, приготовленной различными способами. Измерения трудно
воспроизводимы, и поэтому был сделан вывод в согласии с данными
прежних исследований, что окислы урана являются неудовлетворительными
материалами для изготовления термисторов. Однако в этой работе не
учитывалась большая изменчивость состава окислов урана, даже в том случае,
когда они приготовлены в условиях, кажущихся идентичными.
Исследована ионная эмиссия вольфрамовой проволоки, покрытой
двуокисью урана [80]. Испускаемые ионы представляли собой U+ и,
вероятно, UO+. Определялся также фотоэлектрический эффект U02 [81].
Магнитные свойства. Данные о магнитной восприимчивости двуокиси
урана приведены в табл. 80. На рис. 41 представлены результаты
исследований Гаральдсена и Баккена [85]. Кривая зависимости х от температуры
может быть выражена уравнением Кюри — Вейсса
_ С
где 9 = —310° К по Суксмиту [84] и — 180° К по Гаральдсену и Баккену [85].
Значение константы С, найденное Суксмитом, соответствует магнитному
моменту /гв = 4,4 магнетона Бора (вычислено Гаральдсеном и Баккеном),
в то время как значение, данное Гаральдсеном и Баккеном, приводит
к моменту яв = 2,92 магнетона Бора. Последняя величина очень близка
к теоретическому значению для магнетизма, обусловленного одним эффектом
спина иона U4+ (пв = 2,83). Следует отметить, что при расчетах пв для,и4+
Суксмит использовал только значения, полученные с применением и(504)2
и UC14 при низких температурах, которые дают пв, соответственно равное
3,2 и 2,8.
222
Часть III. Бинарные соединения урана
Таблица 80
МАГНИТНАЯ ВОСПРИИМЧИВОСТЬ ДВУОКИСИ УРАНА
Температура, °С
17
16
—183
—75
17
97
157
226
275
Удельная
магнитная
восприимчивость г (УД.).
CGS-106
7,51
_
—
—
—
—
—
—
'
Объемная
магнитная
восприимчивость х
(объемн.),
CGS-10в
2,46
—
—
—
—
—
—
"
Молярная
магнитная
восприимчивость а)
Х(мол.),
CGS-103
_
—
5,96
4,70
3,96
3,51
3,21
2,75
2,43
Литература
82
83
84
а) С поправкой на диамагнетизм.
Цвет. Цвет двуокиси урана меняется от бурого до черного. Согласно
Лебо [86], чистая кристаллическая двуокись урана представляет бурый
порошок независимо от метода ее приготовления. Бильц и Мюллер [87]
r^WOOi
§
?
W
^800\
с?
Г>
g
53
§ 600
1
§
§•
ё 4оо
CD
5
1
б
5»
5
§ 200
§
! „
* -2L
—
10 С
1 г—
/j
j/
1 1
1 200 400
Температура, "С
1
\)0г/
SWb
i
600
-|
80
5000 ^
§
i
^
4000 о00
i
8
QO
3000 §
1
g-
с
2000 §
6
5
*
£5
/000 |
9
3:
I |
ю *
Рис. 41. Магнитная восприимчивость двуокиси и закиси-окиси
урана [85].
получили двуокись бурого цвета при восстановлении водородом зеленой
закиси-окиси урана и черно-бурую двуокись со слабым фиолетовым оттенком
при таком же восстановлении черной закиси-окиси. Твердые растворы типа
иО2Д0 — UO8f80, полученные разложением закиси-окиси урана в вакууме,
Глава 11. Окислы, сульфиды и теллур иды урана
223
имеют черный цвет. Жолибуа [17] описал окисел U307 (или 1Ю2>33) синевато-
черного цвета. де-Конинк [88,89] сообщает о кирпично-красной модификации
двуокиси урана, полученной разложением уранилбромида (стр. 259), однако
точно неизвестно, не представляла ли эта модификация высший окисел
урана (U03?). Фридрих и Ситтиг [65] получили U02 голубого цвета
нагреванием закиси-окиси до 1100° в токе азота. На основании известных данных
о давлении разложения окислов урана (см. стр. 243) можно сделать вывод
о том, что этот окисел, наверно, представлял собой не UO2t0, а, повиди-
мому, U0215; возможно также, что он содержал азот.
Соединение U205 [49]. Выше (стр. 209) сообщалось о том, что данные
рентгеновского анализа [19] указывают на существование окисла U206,
но результаты тензиметрических исследований при 1160° [28] не
подтверждают этого. Если гомогенная фаза U206 действительно существует при
комнатной температуре, то она может представлять собой нижний предел
области твердых растворов, простирающейся до U308 и далее.
Рентгенограмма U205 (снятая для продукта, полученного совместным нагреванием
равных количеств двуокиси и закиси-окиси урана) подобна рентгенограмме
U308, но не идентична ей [90]. Такая рентгенограмма может быть
приписана ромбической ячейке с константами а = 4,135А, ft = 3,956 А и с = 6,72А^
Рентгеновское исследование монокристаллов U205 (игл, случайно
полученных вместе с октаэдрическими кристаллами U(\30 в результате
термического разложения уранилхлорида при 900°) показало, что вышеуказанные
размеры относятся к псевдоячейке. Для полной элементарной ячейки
константа а должна быть удвоена, а константа b увеличена в восемь раз.
Поэтому действительные размеры элементарной ячейки U206 оказываются
следующими:
я = 8,27 ± 0,02 А; Ь = 31,65 ± 0,1 А; с = 6,72 ± 0,02 А.
В ячейке содержится шестнадцать молекул U205. Плотность U206,
вычисленная на основании рентгенографических данных, равна 8,35 г/см3. Выше
указывалось, что такого низкого значения плотности среди измеренных
плотностей UOx (представленных в табл. 70) не встречается.
Гренвольдом и Гаральдсеном [18] также наблюдалась гомогенная фаза
с составом, близким U206, но не идентичным ему. Она была получена
окислением двуокиси урана кислородом при 200—-250°. Эта фаза (названная
ими 8-фазой) является тетрагональной с а —5,37 А и с = 5,54А (с:а = 1,03);
плотность ее (установленная экспериментально) равна 10,0 г/см3. Эта
величина указывает на состав U088O2)12, что соответствует частичному замещению
атомов урана кислородными атомами, приводящему к деформации кубической
решетки U02. Гомогенная ромбическая фаза U3Og, полученная при низких
температурах, может иметь состав, доходящий до U02>56 (ее получают
восстановлением закиси-окиси урана водородом при 330°).
Таким образом, оказывается, что при низких температурах между
кубической фазой U02 (с составом, доходящим до U02,34) и ромбической
фазой U308 (с составом, могущим дойти вниз до U02j56) существует еще
гомогенная тетрагональная фаза U0i88O2j12.
Закись-окись урана U308.' Кристаллическая структура.
Кристаллическая решетка закиси-окиси урана по Захариазену [91] содержите атома
урана и 5Х/3 атома кислорода в ромбической ячейке с размерами
а = 6,70 ± 0,01 А; 6 = 3,98 ± 0,01 А; с = 4,14 ± 0,01 А.
Эти значения очень близки к размерам, указанным выше для псевдоячейки
U205. Согласно данным американских исследователей [19, 31], константу Ь
для U308 следует утроить, чтобы получить точные размеры ячейки.
224
Часть III. Бинарные соединения урана
Следовательно, действительная элементарная ячейка закиси-окиси урана
содержит 6 атомов урана и 16 атомов кислорода и имеет размеры
а = 6,70 ±0,01 А; 6= 11,94 ±0,03А; с = 4,14 ± 0,01 А.
Таблица 81
КРИСТАЛЛИЧЕСКАЯ СТРУКТУРА ЗАКИСИ-ОКИСИ УРАНА
Препарат
Продукт прокаливания
U04-2H20 при 700°
в течение 40 час. . .
UFe -f Н20 —> осадок,
высушенный при 700°
в течение 40 час. . .
UFe + HN08-fl<7 ->
осадок, высушенный при
700° в течение 40 час.
до» А
6,7023 ±0,0005
6,7030 ±0,0006
6,7036 ±0,0007
Ьо,Аа)
3,9803 ±0,0004
3,9812 ±0,0004
3,9815± 0,0004
со. А
4,1385±0,б007
4,1407 ±0,0006
4,1406 ±0,0005
р, г(см*
8,39
8,39
8,39
0 Одна треть истинного значения.
Табл. 81 показывает результаты точных измерений, выполненных
в Колумбийском университете [48]. Результаты всех трех измерений
совпадают с точностью до ± 0,05%. Шесть атомов урана в элементарной ячейке
U308 занимают положения (0 0 0) f I -i о) (0 0 0) (о -J- (Л ( 0 -у-0) •
Атомы кислорода, вероятно, распределяются^следующим образом:
4 0, в(Ооо)(11-о)(о|-4);
120,,B(ooo)(IIo)(io,)(il,)(I|,)-.
где х = 0,17.
Кристаллическая структура закиси-окиси урана была также
исследована Гренвольдом [92]; ему не удалось приготовить монокристаллов U308,
но он смог получить для одного препарата закиси-окиси рентгенограмму,
индицированную как ромбическая фаза с размерами
а = 6,703 А; Ь = 3,969 А; с-4,136а.
Отмечено, что плотность, рассчитанная на основании этих размеров
в предположении, что ячейка содержит одну молекулу U308, не совпадала
с плотностью, установленной экспериментально (8,34 г/см3; ср. табл. 82).
Совпадения не наблюдалось также и в том случае, если при расчете
принимали, что ячейка содержит две молекулы трехокиси с дополнительным
количеством урана в пустотах решетки (Ulfl203), две молекулы U03 с
кислородом, частично замещенным на уран (Ub(j902t91), или две таким же образом
модифицированные молекулы двуокиси урана (U082O2>18). Для получения
истинного значения плотности при расчетах оставалось предположить, что
в каждой ячейке содержится две молекулы U03 с частичным недостатком
кислорода (U0267) или две молекулы 1Ю2 с избыточным кислородом в
пустотах решетки.
* В оригинале, повидимому, допущена ошибка.—Прим» ред.
Глава 11. Окислы, сульфиды и теллур иды урана
225
Таблица 82
ПЛОТНОСТЬ ЗАКИСИ-ОКИСИ УРАНА
Препарат
Литература
Не указан
» »
Из U04-2H20
С частицами, размером 44 — 74 |х,
обезгажен (продукт Малинкродта)
То же, но не обезгажен
С частицами, размером г/2 р-,
обезгажен
Не указан
Согласно другим данным [38J, рентгенограмма сравнительно легко
окисляемых препаратов U308, полученных при низких температурах (ниже 350°)
из оксалата или трехокиси урана, до некоторой степени отличается от
рентгенограммы практически неокисляемого микрокристаллического продукта,
полученного при более высоких температурах.
Плотность. Плотность, рассчитанная из рентгенографических данных,
составляет 8,39 г/см3. Значения, полученные экспериментальным путем,
несколько меньше (ср. табл. 82).
Твердость. Согласно данным Колумбийского университета [63], закись-
окись урана имеет по шкале Мооса твердость, равную 3,5, которая отчасти
зависит от способа приготовления образца.
Термодинамические свойства. Значения, определенные для теплоемкости,
приведены в табл. 83. В кратком сообщении радиационной лаборатории
Таблица 83
ТЕПЛОЕМКОСТЬ ЗАКИСИ-ОКИСИ УРАНА
Температура, °К
273—373
82—195
196—250
276—314
25—100
Ср.кал.
на 1 г
0,0798
0,0429
0,0616
0,0710
0,0750
градг\
на 1 моль
67,2
36,2
52,0
59,8
63,3
Л итература
93
94
94
94
95
Калифорнийского университета [71] указывается, что теплоемкость закиси-
окиси урана известна для интервала температур 213—333° К, но данных
или ссылок не приводится. В этом сообщении дается также следующее
уравнение:
Ср = 62,6 + 6,6.10'3Г-2,5.1С5Г-2 кал-моль'1-град'1. (7)
Указывается, что оно выведено на основании экспериментальных данных для
U308, относящихся к низким температурам, и интерполированных данных
из значений для U02 и U03 при более высоких температурах.
15 Химия урана
226
Часть III. Бинарные соединения урана
В литературе для энтропии U308 приводятся следующие значения: 72,7
[96] и 66 [97] энтропийных единиц при 298° К; были вычислены также
значения ST — S298 и НТ — Нт для 500, 800, 1100 и 1300°К [97]. Брюэр [14]
путем интерполяции получил значения для 1000 и 1500° К, а также вычислил
величину функции Т т . Эти данные представлены в табл. 84.
Таблица 84
ВЫЧИСЛЕННЫЕ ЗНАЧЕНИЯ ТЕРМОДИНАМИЧЕСКИХ ФУНКЦИЙ
ДЛЯ ЗАКИСИ-ОКИСИ УРАНА
Температура, °К
298
500
1000
1500
IJW298,
кка.7 ■ моль-i
0
12
42
81
ка.1 -мо.1ь~1'град~1
0
28
72
105
FT—H2ds
кал • моль~1-град~1
66J>
1 70
96
117
а) Равна S'208-
Термоэлектродвижущая сила. Измерена [98] ЭДС термопары,
составленной из закиси-окиси урана и платины при температурах холодного спая
от 165 до 1155° и горячего спая от 320 до 1265°. ЭДС была
положительной, т. е. ток шел от платины к окислу через горячий спай вплоть до 700°
(температура горячего спая), выше этой температуры направление тока
изменялось на обратное.
Электрические свойства. Диэлектрическая постоянная. Для порошка
закиси-окиси урана, предварительно нагретого до 200° в течение 1 часа
и спрессованного между двумя пластинками (10000 aw), C= 41,77 [99].
Электропроводность. Подобно двуокиси, закись-окись урана является
полупроводником. Однако последняя отличается от двуокиси тем, что при
пропускании через нее тока возникает явление поляризации, которое
указывает, что в данном случае электропроводность по своей природе является
не чисто электронной [75]. Эта особенность, а также возможность того, что
препараты закиси-окиси, использованные разными наблюдателями, вероятно,
значительно различались по содержанию кислорода, объясняют тот факт,
что величины электропроводности, найденные разными исследователями,
отличаются иногда в 1-108 раз. Как и в случае двуокиси урана, наименьшее
значение электропроводности Фридрихом и Зиттигом установлено [65] для
прутка из закиси-окиси, спеченного в кислороде при 1000°; пруток имел
удельное сопротивление в 40-108 ом-см при комнатной температуре. При 300°
сопротивление его было в десять раз меньше [65]. Гораздо более высокую
электропроводность показывал образец закиси-окиси, полученный окислением
двуокиси на воздухе при 500° [75]. Удельное сопротивление гидратирован-
ной закиси-окиси урана, полученной электролизом, а затем спрессованной,
было еще ниже, составляя только около 2700 ом-см при комнатной
температуре [100]; наконец, еще более высокая электропроводность наблюдалась
Вигандом [101] у прутка, полученного спеканием при 1200°. Некоторые
результаты приведены в табл. 85 [75, 101].
Магнитные свойства. Закись-окись урана парамагнитна. Результаты
измерений магнитной восприимчивости приведены в табл. 86 (без поправки
на диамагнетизм). Данные, представленные на рис. 41, показывают [85]„
Глава 11. Окислы, сульфиды и теллур иды урана
227
Таблица 85
ЭЛЕКТРОПРОВОДНОСТЬ ЗАКИСИ-ОКИСИ УРАНА
Темпера-
20
50
100
150
200
250
300
350
Электропроводность,
104 . (
[75]
0,001
0,0025
0,0090
0,030
0,15
0,50
1,3
3
эм-см)~1
[101] |
500 1
750
—
2 500
—
5 000
■
Темпсра-
400
450
500
600
700
800
900
950
Электропроводность,
(ом-см)-1
[75]
0,0007
0,0014
0,0024
—
—
—
—
—■
[101]
1,05
—
1,88
2,95
4,08
5,60
7,86
9,35
Таблица 86
ПАРАМАГНЕТИЗМ ЗАКИСИ-ОКИСИ УРАНА
Температура,
°с
15
16
Магнитная восприимчивость,
CGS
удельная
0,95-10-6
объемная
0,35Ы0-в
Литература
82
83
что парамагнитная восприимчивость (с поправкой на диамагнетизм) следует
Q
закону Кюри—Вейсса % = (Т_{)) » гДе характеристическая температура 0 =
= — 170° К. Молярный магнитный момент пв, вычисленный из постоянной С,
равен 1,39 магнетона Бора. Это значение очень близко к теоретическому
значению пв для U03U205, равному 1,42; при этом предполагается, что
магнитный спин основного состояния 2F иона U45 является единственным
источником парамагнетизма. Другими словами, закись-окись урана ведет
себя в магнитном поле так, как будто она содержит только шести- и
пятивалентный уран, а не четырехвалентный (обсуждение этого вывода см. в
начале данной главы.
Оптические свойства. Цвет. Окраска закиси-окиси урана может быть
от оливково-зеленой до черно-зеленой или черной, однако даже черные
препараты дают зеленую черту на фарфоре. Цвет зависит не столько
от точного состава, сколько от условий приготовления [102]. Более
темные образцы получаются при более высокой температуре
прокаливания. Например, закись-окись урана, приготовленная из U04-2H20 при
температуре ниже 800°, была мшисто-зеленого цвета, окисел же,
полученный при 900—1000°, —почти черного [11]. Спектр испускания U308 (?)
в кислородно-водородном пламени представлял собой обычный сплошной
спектр раскаленного твердого тела (см. ниже) [103]. Инфракрасный спектр
испускания обладал слабыми максимумами при 2,8 и 3,4р. [104].
Исследована излучательная способность U308 [105, 106] (вследствие
неустойчивости закиси-окиси урана при использованных температурах
результаты этих опытов относятся к окислам неизвестного состава, в которых,
несомненно, на один атом урана приходится меньше 2,67 атома кислорода).
Коэффициент излучательной способности при 650 тц составлял 0,30 для
15*
228
Часть III. Бинарные соединения урана
твердого окисла при 1650° и 0,31 для жидкого (?) при 1700°.ПоВиганду [101],
полная излучательная способность U308 составляла 77—79% излучательной
способности черного тела. Это отношение практически не изменяется в
интервале 900—1400°. Закись-окись урана была использована в качестве
стандарта при изучении селективной излучательной способности других окислов,
причем предполагалось, что спектральное распределение излучения закиси-
окиси урана мало отличается от распределения для черного тела [107].
Однако некоторые данные [108] указывают, что излучательная способность
закиси-окиси урана аналогично излучательной способности окиси церия
и окислов других элементов в синей части спектра (467 тр) выше, чем для
черного тела при той же самой яркости в красной части спектра.
Трехокись урана. Кристаллизация. Трехокись урана, обычно
получаемая прокаливанием U04-2H20, ранее считалась аморфной [109].
Микрокристаллические препараты трехокиси впервые были получены
прокаливанием уранилнитрата. Английские исследователи [ПО] нашли, что
трехокись кристаллизуется при стоянии; ими были идентифицированы две
кристаллические формы. Фрид и Дэвидсон [33, 111] установили, что
трехокись может существовать по крайней мере в трех кристаллических формах:
оранжевой гексагональной U03 (I), красной U03 (II) и желтой U03 (III).
Последняя форма (окись Малинкродта) является, вероятно, наиболее
устойчивой.
Вследствие того что аморфная трехокись урана теряет кислород на
воздухе при температуре выше 450°, Фрид и Дэвидсон проводили опыты по ее
кристаллизации при высоком давлении кислорода (30—150 am). Аморфную
трехокись (или закись-окись) нагревали до 500—750° в запаянных трубках
из стекла пирекс или кварца.
Таблица 87
КРИСТАЛЛИЗАЦИЯ ТРЕХОКИСИ УРАНА
Обработанное
вещество
ио3
и3о8
и3о8
ио3
и3о8
Температура,
• С
450—500
] 500—560
530-560
530—560
700—750
am
28
28
30
30
70—150
жительность,
часы
12
36
112
112
1,5—2
Полученная
фаза
I
1 + И
II + III
I + H
III
Формула продукта UO^
значение х по
количеству
присоединенного
кислорода
2,97
2,997
2,993
значение х по
изменению в весе
при
прокаливании до U308
3,048
2,99
3,01
В табл. 87 указан состав окислов, полученных при различных
условиях. Фаза I проанализирована Захариазеном (см. ниже). Фаза III содержится
в окиси Малинкродта; она наиболее устойчива из всех трех фаз. Причину
первоначального получения фазы I можно объяснить ее
кристаллографическим сходством с закисью-окисью урана, что способствует образованию
этой фазы из закиси-окиси путем постепенного присоединения кислорода.
Кристаллическая структура. Кристаллическая трехокись урана (I),
полученная нагреванием аморфной безводной трехокиси до 500° в течение
8 час. при давлении кислорода в 20 am, имеет, согласно Захариазену [32, 112],
гексагональную решетку с константами
а = 3,963 ± 0,004 А; с = 4,160 ± 0,008 L
Глава 11. Окислы, сульфиды и теллур иды урана
229
Она характеризуется пространственной группой СЗ т и следующим
положением атомов:
1 U в 1 (а);
1 О, в 1 (Ь);
20ц в 2(d) с z = 0,17.
Ближайшими соседями каждого атома урана являются два атома Оь
расположенные от него на расстоянии 2,08 А, и шесть атомов Оц —на
расстоянии 2,39 А. Атомы Oi можно рассматривать как кислород уранила,
хотя в структуре и нет групп 1Ю2. Вместо них по оси с расположены
бесконечные цепи уранила — О — U — О — U — О —U....
Фрид и Дэвидсон [111] сообщают о двух рентгенографических
исследованиях частично раскисленной трехокиси урана, давших довольно
неожиданные результаты (табл. 88). Значения констант решетки при ху
равном 2,82 и 2,96, не оказались промежуточными между значениями констант
при х, равном 2,67 и 3,00.
Таблица 88
КОНСТАНТЫ а) РЕШЕТКИ UOT
X
2,67
2,82
2,96
3,00
a\t А
6,70±0,01
6,90±0,02
6,90±0,02
6,864-f0,004
й2, А
3,98±0,01
3,91±0,02
3,91.+0,02
3,963±0,004
«з, А
4,14±0,01
4,15±0,02
4,15±0,02
4,160+0,008
Литература
32,112
111
111
32,112
') Все значения относятся к ортогексагональным осям.
Рентгенограммы U03 (II) и U03 (III) еще не проанализированы, но они
отличаются от рентгенограммы гексагональной 1Ю3 (I). Окислением закиси-
окиси урана азотной кислотой (в парах) при 350—400° были приготовлены
[33, 111, 113] три образца U03, которые дали две различные
рентгенограммы, отличавшиеся от рентгенограмм вышеуказанных фаз I, II и III.
Таким образом, возможно, что U03 существует в пяти кристаллических
аллотропических формах.
Плотность. Вычисленная плотность U03 (I) равна 8,34 г/см3.
Экспериментальные значения плотности гораздо меньше: большинство из них
относится к аморфной трехокиси, некоторые, вероятно, — к гидратам (например,
U03-H20) (см. стр. 238). В результате опытов были получены следующие
значения плотности: 5,92 г/см3 (16°) [82]; 7,29 г/см3 (15°) [114]; 7,54 г/см3
(25°) [58]; 7,37 г/см3 (25°) [115]; 6,04 г/см3 для UO3f07 и 7,07 г/см3 для
U02j92 [12]; насыпной вес неутрясенного порошка трехокиси урана
составляет 3,63 г/см3, слабо утрясенного —4,26 г/см3 [57].
Давление пара. Выше (см. стр. 209) было указано, что известная
летучесть окисла урана в двухфазной области между U02t62 и U0225,
обнаруженная Бильцем и Мюллером при температуре выше 1160°, может быть
объяснена диспропорционированием U308 по схеме
U308-*U02 + 2UOe
(8)
230
Часть III. Бинарные соединения урана
и испарением U03. При содержании кислорода большем, чем в U02i62, его
парциальное давление гораздо выше, чем давление пара U03, так что
испарение соответствующего окисла становится слишком медленным, чтобы его
можно было заметить. На основании данных Бильца и Мюллера
вычислено [14], что при 1600° К рио3 составляет 1 • 10"5 атм в интервале от1Ю2г25
до U02)62 и 110~4атж в случае чистой 1Ю3.
Термодинамические свойства трехокиси урана. Теплоемкость трехокиси
урана, вероятно в форме U03 (III) [33, 69], измерена в интервале
температур от 15 до 300° К (табл. 89). Энтропия при 298, Г К вычислена путем
Таблица 89
ТЕПЛОЕМКОСТЬ ТРЕХОКИСИ УРАНА
Температура,
°к
15
20
25
30
40
50
60
70
80
90
100
ПО
120
130
140
150
Ср,
кал • моль-1 • град~1
0,608
1,040
1,553
2,103
3,231
4,370
5,505
6,602
7,675
8,704
9,693
10,63
11,49
12,29
13,00
13,70
Температура,
°к
160
170
180
190
200
210
220
230
240
J 250
260
270
! 280
: 290
300
Ср,
-кал • моль-1 . град~1
14,33
14,93
15,49
16,01
16,49
16,94*
17,38
17,82
18,24
18,66
19,05
19,42
19,73
20,02
20,30
графического интегрирования с использованием для температур ниже 15° К
функции Дебая с 6 = 140° К:
J Cpd (In T) для 15 -г- 300° К 23,378
Функция Дебая для 0ч-15°К 0,196
23,57 ± 0,06 энтропийной
единицы
Для интервала температур 400—900° К определено теплосодержание
образца, дегидратированного при 600° в течение 79 час. (диссоциация могла
обусловить ошибку около 1%) [70]. Точки, соответствующие полученным
значениям, лежат на плавной кривой (табл. 90).
Ят-Я298Л6 = 22,09 Г+1,27-10'3Г2 +2,973-105 Г"1-
- 7696 (± 0,1%) кал-моль'1. (9)
Путем дифференцирования можно получить следующее значение для Ср:
Ср= 22,09 + 2,54-10-3Г-2,973.105Г-2. (10)
Глава 11. Окислы, сульфиды и теллуриды урана
231
Таблица 90
ТЕПЛОСОДЕРЖАНИЕ И ЭНТРОПИЯ ТРЕХОКИСИ УРАНА
ПРИ ТЕМПЕРАТУРАХ ВЫШЕ 298,16° К [70]
Температура,
0 К
400
500
600
700
800
900
Нт—#298, 16*
кал • моль-1
2 090
4 260
6510
8 820
11 160
13 540
Sr-5298,I6.
кал • моль~1 • град~1
6,01
10,86
14,96
18,51
21,64
24,44
На основании полученных результатов были вычислены значения функции
р jj
I _ т 298 ^ представленные в табл. 91 [14].
Таблица 91 Таблица 92
функция /= Fy~~^2fl8 для теплопроводность трехокисп
ТРЕХОКИСИ УРАНА УРАНА
Температура,
25—150
160-340
310-600
К, кал • слг-1 • се/с~1 • град~1
0,00067
0,00063
0,00061
Температура,
°К
298
500
1000
f, кал • моль~± • град !
23,57 а>
25,7
34,4
а) Равна S°298
Теплопроводность. Данные по теплопроводности порошка трехокиси
урана, плотно спрессованного с помощью винтового пресса во время
нагревания, представлены в табл. 92 [72, 73].
Электрические свойства. Электропроводность трехокиси урана вплоть
до 300° является неизмеримо малой. Проводимость, которая иногда
наблюдается при комнатной температуре и исчезает при 100—150°, может быть
приписана наличию влаги [75]. Электропроводность сильно, увеличивается
при 350—490°, особенно после продолжительного нагревания. Это, возможно,
обусловливается потерей кислорода и превращением в низшие окислы,
являющиеся полупроводниками. После нагревания в течение 30 час. при 400°
продукт с первоначальным составом и02.9ч-з,о имел удельную
электропроводность 1,4 • 10~5 (ом-сму1. Подобные значения были получены и при
другом исследовании [116].
Диэлектрическая постоянная трехокиси урана определялась на осадке
окиси, отмытом от электролита [117]. Были получены следующие значения:
1,86 (U03 оранжевого цвета, высушенная при 150°, р = 2,0 г/см3, 79,19%
урана), 4,36 (U03 — продукт Малинкродта кирпично-красного цвета) и
9,41 — 11,4 (два образца бурой U03).
Магнитные свойства. По Саксмиту [84], трехокись урана диамагнитна,
другие же авторы считают ее слабо парамагнитной, причем парамагнетизм
не зависит от температуры. Для удельной магнитной восприимчивости ее
232
Часть III. Бинарные соединения урана
предложена величина +1,08-10"4 при 16° [82], для объемной — величина
4-0,146- Ю~в при той же температуре [83].
По Тилку и Клемму [118], х(УД-)= +0,26- 10"в и х(мол.)= +74-КГ6;
с поправкой на диамагнетизм х (мол.) = -h 128- 10~в (среднее из значений,
полученных для образцов трехокиси урана из нитрата, карбоната и перекиси).
Значения диамагнитного эффекта, использованные для поправок, были
приняты в 20-Ю"6 для Ueb и 11,25- 10"в для 02~. Гаральдсен и Баккен [85]
дают для х(мол.) значение +157-10~6 (с поправкой на диамагнетизм).
Цвет. Трехокись урана, образующаяся в виде аморфного порошка при
прокаливании U04-2H20, серно-желтого цвета [11], по другим данным она
яркооранжевого цвета [33]. Гексагональная кристаллическая -U03 (I),
приготовленная [33] из аморфного продукта, была более светлого цвета. Фаза
U03 (II), полученная из аморфной трехокиси или закиси-окиси урана
нагреванием до 500° при давлении кислорода 28 am, была тёмнокрасной.
Кирпично-красный продукт, полученный (ср. стр 254) продолжительным
нагреванием оранжевой трехокиси урана до 600° на воздухе, возможно,
имел ту же кристаллическую форму. Фаза 1Ю3 (III) [33J, образовавшаяся
при весьма длительном нагревании при еще более высоком давлении
кислорода, а также присутствующая в микрокристаллическом веществе (продукте
прокаливания уранилнитрата), является яркожелтой. Две новые
кристаллические формы U03, полученные окислением закиси-окиси урана четырех-
окисью азота N204 (см. стр. 256), были соответственно кирпичпо-красного
(или оранжевого) и желтого цвета.
При частичном раскислении U03 образуются продукты рыжевато-красного
(U02>96), рыжевато-зеленого (U0282) и зеленовато-черного (U02(7()) цвета.
Таким образом, наблюдается постепенный переход к фазе U308 черного цвета.
Соединение С04. Этот окисел неизвестен в безводном состоянии.
Физические свойства его гидратов описаны на стр.241. Порошкообразный
продукт U03>5, полученный Краусом с сотрудниками (ср. стр. 214) и
рассматриваемый ими как U03 + U04, имеет красный цвет.
ГИДРАТЫ ОКИСЛОВ УРАНА
Гидраты двуокиси урана. При действии аммиака на зеленые
растворы солей урана (IV) Пелиго [51] наблюдал образование хлопьевидного
объемистого осадка красновато-коричневого цвета. Согласно
Циммерману [119], при действии щелочей или аммиака на растворы солей урана (IV)
вначале получаются осадки светлозеленого цвета, но на воздухе они быстро
превращаются в бурые гидраты закиси-окиси. Сообщается [120], что
наилучшим методом получения неокисленного свободного от щелочи гидрата
двуокиси урана является гидролиз разбавленного, лишенного воздуха
раствора хлорида или ацетата урана (IV) путем нагревания до
обесцвечивания. Выпадающий черный осадок является основной солью, но его можно
отмыть от анионов кипящей водой. После высушивания над серной кислотой
гидрат имеет состав U02.2H20. Соли урана (IV) в растворе можно
разложить также путем облучения [121]. При фотохимическом разложении
эфирного раствора уранилнитрата получается слизистый черный или зеленовато-
черный гидрат U02 [122]. Черный кристаллический гидрат двуокиси урана
был также получен [123] действием щелочи на кристаллический сульфат
урана (IV).
Влажный аморфный U02-2H20 окисляется на воздухе до UOo-H20;
окисление происходит медленно на холоду и быстро при нагревание
Наоборот, кристаллический гидрат двуокиси устойчив на воздухе при комнатной
температуре в течение нескольких дней. При нагревании он превращается
в зеленую закись-окись урана [123].
Глава 11. Окислы, сульфиды и теллур иды урана
233
1Ю2-#Н20 дегидратируется в воде при температуре выше 285° с обра-
зованием бурой безводной двуокиси [124]. На воздухе гидрат за полчаса
теряет всю воду при 200°, одновременно окисляясь до U308 [125].
Свежеприготовленная гидроокись урана (IV) легко растворима в
кислотах, но при стоянии ее растворимость уменьшается [51, 126—128].
Растворы содержат коллоидные агрегаты или полимерные ионы, а не простые
соли урана (IV).
Гидраты закиси-окиси ура на. Существование гидрата U308, о чем
впервые упоминал Арфведсон [129,а], было установлено Эбельменом [129,6],
получившим его путем фотохимического разложения уранилоксалата; при
этом выпадал хлопьевидный коричневато-фиолетовый осадок гидрата, легко
окислявшийся на воздухе. Чтобы предотвратить разложение, препарат
необходимо было высушить в вакууме. Было предложено [53] вместо
малорастворимого оксалата использовать в качестве исходного вещества
уранилацетат. При действии света на водный раствор уранилацетата,
содержащий эфир или спирт, образуется фиолетовый осадок, труднее окисляемый,
чем гидрат закиси-окиси урана, полученный Эбельменом [129] р»з оксалата.
Подобный же фиолетовый осадок образуется пз растворов любых солей
уранила с концентрацией 1 — 5% при действии света в присутствии
органических веществ, функционирующих как восстановители (ацетальдегид,
эфир, спирт, глюкоза) [130]. Непосредственно после осаждения гидроокись
содержит анионы кислоты, но их можно отмыть кипящей водой. При
применении ураниловых солей органических кислот добавки других
органических восстановителей не требуется.
Показано [63], что закись-окись урана не реагирует с водой даже при
нагревании в течение 11 суток при 185°, однако, если она получена
прокаливанием при температуре красного каления, в ней возможно содержание
трехокиси; вследствие этого препарат поглощает влагу из воздуха, образуя
U03-2H20 [26, 131].
Состав фиолетового гидрата, образующегося при вышеописанных
условиях, выражают обычно формулой U308-xH20. Согласно Смиту [132, 133]
и де-Конинку и Камо [134], при электролизе растворов солей уранила
вначале получается желтый моногидрат U308H20, а затем черный диги-
драт. Однако, по Пирле [135], черный осадок имеет состав U033-2H20,
а не U02>G7'2H20 (стр. 262). Высокий потенциал электрода, покрытого этой
черной гидроокисью (стр. 247), Пирле считает дополнительным
доказательством того, что этот осадок не является гидратом закиси-окиси урана.
Процесс образования осадка гидрата закиси-окиси на металлическом уране при
электролизе раствора уранилнитрата исследован им и другими авторами
[136, 137].
Сообщается [53], что аморфный фиолетовый гидрат из08-л:Н20
не кристаллизуется при охлаждении или нагревании под давлением. При
сушке в вакууме он превращается в твердую черную массу, при растирании
дающую черный порошок. При нагревании в токе азота эта масса теряет
воду, внешне не изменяясь, но при измельчении после сушки она
превращается не в черный порошок, а в зеленый.
Гидрат U308- *Н20 на воздухе легко окисляется до гидрата трехокиси.
При растворении его в кислотах образуется смесь солей урана (IV) и урана
(VI) [53].
Гидраты трехокиси урана. Способность к образованию гидратов
возрастает с валентностью катиона. Гидраты трехокиси гораздо более
устойчивы, чем гидраты двуокиси и закиси-окиси, и поэтому лучше изучены.
Фазовые отношения в системе U03 —Н20. О системе 1Ю3 —Н20
в растворе известно очень мало. Установлено [138, 139], что гидрат UOs-2Н20
слабо растворим в воде (0,16 г/л при 27°). Как показали исследования,
234 Часть III. Бинарные соединения урана
проведенные в Колумбийском университете, взвесь U03H20 в воде при
комнатной температуре имеет рН = 4,8 — 5,2; при этом твердая фаза может
иметь ромбическую или триклинную сингонию.
Известен ряд гидратов трехокиси урана в твердом состоянии.
Соединения U03-H20 (или H2U04) и U03-2H20 (или Н4Ш5) хорошо изучены
химически. На основании исследования изобар гидратации было сделано
заключение о существовании полугидрата 2U03H20 (или H2U20-) и о
возможности существования полуторного гидрата 2U03-3H20 (или H6U209) [6].
3
"О 40 80 120 160 200 240 280 320
Температура, °С
Рис. 42. Термическое разложение 1Ю3-л:Н20 при
парциальном давлении водяных паров, равном 15 мм рт. ст. [6].
Путем нагревания гидрата U04-2H20 до 500° (для разложения всего
нитрата, который мог остаться в перекиси, приготовленной из уранилнитрата)
была получена безводная трехокись урана [6]. Трехокись полностью
дегидратировалась в вакууме при 450° (в этих условиях кислород не терялся,
см. стр. 245 и ел.) и давала равновесную систему с парами воды при комнатной
температуре. При гидратации происходило заметное выделение тепла,
сопровождавшееся небольшим увеличением объема; в качестве конечного
продукта при 30° получался дигидрат U03-2H20. Это соединение
дегидратировалось при нагревании под постоянным давлением паров воды, равном
15 мм рт- ст. Результаты представлены на рис. 42. Первая половина моля
воды выделяется в . интервале 30—100°, вторая сразу теряется при 100°,
третья вновь выделяется постепенно в интервале 100 - 300е, последняя
половина моля воды улетучивается сразу при 300°. Если рассматривать
изобару в направлении справа налево, можно отметить существование двух
стёхиометрически точных соединений U03 и UO30,5H2O, не образующих
между собой смешанных кристаллов. Затем начинается область смешанных
кристаллов UO3-0,5H2O + UO3-H2O. Кривая не дает ясного представления,
существует ли в интервале между 1Н20 и 2Н20 другая сплошная область
смешанных кристаллов моно- и дигидрата трехокиси урана или же в
интервале между U03H20 и U03l,5H30 смешанных кристаллов не образуется.
Если правильно последнее предположение, то U03-1,5H20 является еще
одним чистым гидратом, область же смешанных кристаллов простирается
от этого гидрата до U03-2H20.
Глава 11. Окислы, сульфиды и теллур иды урана
235
Термодинамические свойства гидратов трехокиси урана. Тензиметри-
ческие данные (см. рис. 42) были использованы для расчета теплот
гидратации при помощи приближенной формулы Нернста [140]. В табл. 93
Таблица 93
ТЕПЛОТА ГИДРАТАЦИИ ТРЕХОКИСИ УРАНА
Реакция
ио3+1 н2огаз -> ио3 • ~ н2о
ио3+н2огаз-*ио3-н2о
U03 + l4H=0ra3-*UCVl4H20
и03 + 2Н2Огаз-*и03-2Н20
—&Н, ккал\моль
[140]
13,3
23,4
31,7
39,2
[138]
.
15,5
—
28,5
Уравнение
№
(И)
(12)
(13)
(14)
«сопоставлены результаты этих вычислений со значениями, выведенными
[138, 139J из калориметрических измерений теплот нейтрализации трехокиси
урана, ее моногидрата и дигидрата разбавленной азотной кислотой
(см. стр. 268). Теплоты нейтрализации гидратов трехокиси щелочами или
кислотами с образованием ураиатов или солей уранила рассматриваются
ниже (стр. 265 и ел.).
Результаты опытов по измерению потенциала платинового электрода,
покрытого пастой из желатины, содержащей моногидрат трехокиси урана,
см. на стр. 245 и ел. [135].
Физические свойства гидратов трехокиси урана. Полугидрат U03 • 0,5Н2О.
Этот гидрат кристаллизуется в виде моноклинных оранжевых игл
толщиной 5—15 |л и длиной 2Q — 80 (i, дающих отчетливую рентгенограмму.
Кристаллы обладают характерной спайностью по базису [48, 141].
U03'H20. Цвет моногидрата желтый или оранжево-желтый. Выделена
одна аморфная и четыре аллотропические кристаллические модификации;
все они устойчивы при комнатной температуре [48, 141]. Эти четыре
модификации имеют следующую характеристику:
Модификация а — большие шестигранные ромбические пластинки с
развитым основанием.
Модификация р — микроскопические ромбические призматические
пластинки (до 50—100 fi) с элементарной ячейкой, несколько большей, чем
ячейка а-формы.
Для ромбической формы моногидрата известны более точные данные [91]:
а -6,86 ± 0,03 А; 6-4,27 ± 0,03 А; с= 10,19 ± 0,06 А.
Элементарная ячейка содержит четыре молекулы U03-H20. Атомы урана
находятся в положениях (000) (ууОГуОу; С^Т"0' вычисленная
плотность равна 6,73 г/см3.
Модификация 7 —гексагональные столбики, также микроскопических
размеров; рентгенограмма их аналогична рентгенограмме а- и р-форм,
однако возможно, что кристаллы относятся к другой системе. По данным
анализа этой модификации на один моль U03 приходится меньше 0,9 моля
воды.
Модификация о - триклинная со сложной рентгенограммой. Возможно,
что эта сложность обусловлена наличием примесей. Условия образования
этих четырех форм указаны ниже (стр. 237 и ел.).
236
Часть III. Бинарные соединения урана
По Алюа [53, 142] и Лебо [143], моногидрат трехокиси урана
встречается в аморфной и ромбической формах; Рибан [144, 145] получил этот
гидрат в форме гексагональных призм путем гидролиза уранилацетата;
Цехентер [146] по тому же методу получил его в виде блестящих
гексагональных пластинок. Наконец, в литературе описан еще метод получения
гидрата путем гидролиза уранилнитрата под высоким давлением водорода
в виде прозрачных призм с развитыми гранями [147]. Лебо превратил
аморфный моногидрат в ромбические пластинки путем растворения его в растворе
уранилнитрата (стр. 237) с последующим выпариванием его при 100° досуха
и извлечением эфиром [143].
U03-2H20. Этот желтый или зеленовато-желтый гидрат встречается
в двух модификациях [48, 141]. а-Форма, полученная только в виде
субмикроскопических кристаллов, дающих простую рентгенограмму, имеет,
возможно, гранецентрированную тетрагональную решетку. Р-Модификация,
также известная пока только в форме субмикроскопических кристаллов,
вероятно, является ромбической. Следует отметить, что рентгенограмма
(3-формы дигидрата идентична рентгенограмме (З-формы моногидрата:
размеры элементарных ячеек их различаются менее чем на 0,5%. Из этого
можно сделать вывод, что вторая молекула воды является «цеолитной».
Таблица 94
ПЛОТНОСТЬ U03-*H20 [61]
Шз-яИзО
VOr~ Н20
U03-H20 аморфный
иОз-НоО ромбический (fi)6)
U03-H30 ромбический (а)
U03-H20 триклинный (о)
и03-Н20 триклинный (о)
и03-2Н20
X
0,47
1,04
0,82
1,00
1,00
2,12
Размер
частиц,
И-
> 105
44—74
0—10
—
< 105
1—2
~
р (обезгажен-
ного гидрата),
г\см*
6,47
6,32 а)
6,07
6,73 в)
5,74
5,85
4,87
а) В литературе приводится также значение 5,93 г\смЬ [148].
б) 1% U04.
в) Плотность рассчитана из рентгенографических данных [91].
Механические свойства. В табл. 94 приведены значения плотности для
гидратов трехокиси [61].
Твердость гидратов трехокиси вплоть до дигидрата по шкале Мооса
равна 2,5 [63].
Оптические свойства. Цвет. Полугидрат трехокиси урана имеет
оранжевый цвет, моногидрат — оранжево-желтый или желтый, дигидрат —чисто
желтый или зеленовато-желтый.
Флюоресценция. Безводная трехокись урана не флюоресцирует даже
при—196°; это верно также и для двуокиси, закиси-окиси и гидрата
перекиси урана. С другой стороны, все гидраты трехокиси флюоресцируют при
комнатной температуре и ниже. По структуре спектры флюоресценции
тригидрата в общем похожи на спектры уранил-иона, но полосы
сдвинуты в направлении более длинных волн и занимают более узкую область
спектра (500 — 600 тц). Цвет флюоресценции в отличие от спектра солей
Глава 11. Окислы, сульфиды и теллуриды урана
237
уранила не зеленовато-желтый, а зеленый. Спектры флюоресценции
и03-2Н20(а и р), U03-H20(a, р и S) и UO3-0,5H2O сходны, по их легко
различить [149, 150]. Каждый спектр содержит три или четыре полосы
на расстоянии 810 см'1. Первые две полосы— яркие, третья— слабее,
четвертая—еще слабее. Флюоресценция гидратов трехокиси будет более
подробно рассмотрена во второй книге этого труда, в главах, посвященных
флюоресценции и фотохимии соединений уранила.
Гидратация и дегидратация трехокиси урана. Изобара гидратации,
представленная на рис. 42, показывает, при каких условиях можно ожидать
образования и разложения различных гидратов трехокиси при парциальном
давлении водяного пара 15 мм рт. ст. В литературе по манхэттэнскому
проекту имеется много и других данных по условиям равновесия и
скоростям образования и дегидратации этих гидратов.
Дигидрат U03-2H20. Дигидрат трехокиси урана устойчив при
комнатной температуре. Он образуется из красновато-оранжевой «активной»
трехокиси урана при действии насыщенного водяного пара в области
5 — 75° [63]. В других опытах [141] а-форма получена гидратацией
аморфной трехокиси урана, приготовленной разложенцем гидрата перекиси,
а (В-форма — гидратацией продукта Малинкродта, который является
микрокристаллической трехокисью урана U03 (III) (см. стр. 228).
Показано [151], что оранжевая или кирпично-красная трехокись урана,
приготовленная термическим разложением диураната аммония, при
действии воздуха за несколько дней превращается в желто-зеленый гидрат,
повидимому —дигидрат. При нагревании этот гидрат быстро переходит
в кирпично-красную безводную окись.
Опытами установлено [138, 139], что непосредственная гидратация
трехокиси урана легко приводит к образованию продуктов, содержащих
до 2,5Н20. При дегидратации потеря воды легко может превысить
расчетную для образования дигидрата величину. Поэтому стехиометрический
состав U03-2H20 может быть получен только при прекращении
дегидратации в соответствующий момент.
Вместо гидратации водяным паром дигидрат трехокиси урана можно
получить также действием воды на безводную трехокись или ее
моногидрат. Так, дигидрат был получен растворением моногидрата в
концентрированном водном растворе уранилнитрата (стр. 236), выпариванием воды
над серной кислотой при комнатной температуре и экстракцией ^уранил-
нитрата эфиром [143]. Остаток представлял собой светложелтый
порошок состава U03-2H20. Наивысшая температура, при которой дигидрат
устойчив в соприкосновении с водой, значительно ниже 100°, а именно
по ранее опубликованным данным Колумбийского университета она
составляет 75° [63], по более поздним данным 60° [141]. Прямыми измерениями
при 61 и 77° установлено, что давление водяного пара над дигидратом
выше, чем над водой при температурах, превышающих приблизительно 60°.
Таблица 95
СКОРОСТЬ ДЕГИДРАТАЦИИ U03-2H20
Препарат
U03-2H20 из U04
U03-2H20 из трехокиси
Малинкродта
Время, требующееся для потери одной молекулы
воды, мин.
в вакууме
при '28°
< 123
в атмосфере водяного пара при
61°
> 518
> 100
77° | 87°
<95
< 122; >48
> 12
< 2
100° | 118°
< 2
< 1
238
Часть III. Бинарные соединения урана
Данные табл. 95 характеризуют скорость дегидратации дигидрата
трехокиси урана при различных температурах.
Еще ранее [138] было установлено, что дигидрат трехокиси урана
медленно теряет воду над серной кислотой при комнатной температуре,
но быстро дегидратируется до моногидрата в токе сухого воздуха при 80°.
При температуре около 135° вода выкипает. По Лебо [143], дигидрат
превращается в моногидрат при кипячении в воде, однако Дренкман [152]
считает, что превращение в моногидрат происходит лишь при 160°.
Моногидрат U03-H20. Нагревание дигидрата трехокиси урана до
температур выше 60° должно сначала привести, согласно рис. 42, к
образованию смешанных кристаллов дигидрата и моногидрата (или дигидрата
и полуторного гидрата). Для превращения в чистый моногидрат 1Ю3Н20
необходимо нагревание до 100° при давлении водяного пара, равном
15 мм рт. ст. Если дигидрат находится под водой или в
соприкосновении с насыщенным водяным паром, то необходима еще более высокая
температура для превращения его в моногидрат. Образование смешанных
кристаллов не учтено в первых работах, проведенных в Колумбийском
университете [63], поэтому для моногидрата вначале были даны следующие
пределы устойчивости в контакте с водой: 85 —300° для ромбической формы
и 300 — 310° для триклинной [141]. В этом же сообщении [141] указывается,
что активная красно-оранжевая трехокись урана превращается в
ромбический моногидрат действием насыщенного водяного пара при 85 — 300°
и в триклинный моногидрат при температурах выше 300°. Неактивная
оранжево-желтая трехокись гидратируется при 150 — 300° с образованием
триклинных пластинок моногидрата.
В результате последующих исследований в Колумбийском
университете [141] удалось получить ромбическую а-форму моногидрата трехокиси
непосредственно из моногидрата перекиси нагреванием в воде до 158°
в течение 117 час, а также нагреванием (3-формы дигидрата трехокиси
в воде до 100° в течение 100 — 600 час.
Оранжевая [3-форма моногидрата была получена из а- или ^-дигидрата
путем нагревания до 100° в течение 90 час; 7"Ф°Рма моногидрата
получена нагреванием U04>2H20 или U03-2H20 в воде до 185 — 310°; наконец,
о-форма моногидрата приготовлена путем гидратации a-U03-2H20 в воде
при 185—310°.
Согласно тем же исследованиям, моногидрат трехокиси урана не
изменяется при нагревании с водой в запаянных трубках до 60 — 325°.
Кристаллы моногидрата не гидратировались при стоянии в воде в продолжение
месяца.при 28°, несмотря на то, что дигидрат устойчив в воде при
температурах до 60°. Все четыре кристаллографические формы моногидрата
весьма устойчивы при низких температурах. Качественные наблюдения
давления пара показывают, что гексагональная форма наиболее устойчива при
температурах до 125°, ромбическая — между 125 и 300° и триклинная — при
300 — 310°. Моногидрат трехокиси теряет 85% своей воды за 3 часа при 400°
и бо/iee 95% за 1 час при 450°.
Можно отметить еще следующие, более ранние наблюдения. Моногидрат
трехокиси урана не теряет воду в вакууме или в сухом воздухе при 100°
[53, 142]; он отдает 5% воды за 30 мин. при 300°, 50% при 400° и 94% при
500° [143]. Моногидрат кипит при температуре около 220° [138] и
превращается в безводную окись при 300° и в закись-окись при температуре
красного каления [153].
Лебо получил моногидрат гидратацией трехокиси урана на воздухе
при 25° в течение 24 час. и при 100° за 1 час; очевидно, это соединение
образовалось в виде промежуточного продукта превращения безводной
трехокиси в дигидрат [143]. Отмечен рост гексагональных пластинчатых
Глава 11. Окислы у сульфиды и теллур иды урана
239
кристаллов (очевидно U03*H20?) после 30-минутного выдерживания водной
суспензии трехокиси урана при 100° [154].
Полугидрат UO3-0,5H2O. Согласно ранним наблюдениям [6] (ср. рис. 42)
полугидрат можно получить при давлении водяного пара 15 мм рт. ст. в
интервале температур 160 — 300°. Позже полугидрат был приготовлен нагреванием
U04-2H20 в воде до 310 —350° [141], а также гидратированием неактивной
оранжево-желтой трехокиси урана при 350 — 380° [63]. Полугидрат устойчив
при нагревании в запаянной трубке под водой выше 325° и медленно гидра-
тируется до моногидрата ниже 300°; гидратация заканчивается при 100° за
21 час. Содержание воды падает от 2,8 до 0,5% общего веса при нагревании
на воздухе при 450° в открытой чашке в течение 4 час. Полностью вода
может быть удалена при 500 — 550° [125]. Полугидрат превращается в
красную трехокись урана, содержащую небольшое количество закиси-окиси,
нагреванием на воздухе до 450 — 600°, при 700° он переходит в чистую
закись-окись урана [63].
Гидраты перекиси у paua. Дегидратация U04-*H20. Перекись
урана U04 в безводном состоянии не получена. При дегидратации ее
гидратов они теряют кислород одновременно с водой и образуют трехокись или
низший окисел урана в зависимости от температуры и продолжительности
процесса [6, 155, 156].
Изучение системы U04 —Н20 осложняется тем, что гидраты перекиси
одновременно теряют воду и кислород. Систематические определения
равновесия дегидратации следует проводить при достаточно высоком давлении
кислорода для предотвращения возможности раскисления.
Свежеосажденный воздушно-сухой гидрат перекиси урана может
содержать до четырех и более молекул воды на один атом урана.
Согласно данным различных исследователей, воздушно-сухой гидрат
перекиси урана имеет следующий состав: U04-4H20 [2], U04-3H20 [156]
и U04-4,5H20 [6]. Препарат последнего состава был получен при
высушивании на воздухе при 35°. В металлургической лаборатории Колумбийского
университета [157] найдено, что воздушно-сухая перекись урана,
осажденная из уранилнитрата 3%-ным раствором перекиси водорода при 75°,
содержит четыре молекулы воды (см., однако, ниже данные рентгеновских
анализов, стр. 241). С другой стороны, в университете в Беркли были
получены [158] для препаратов воздушно-сухой перекиси следующие
аналитические данные:
Препарат, промытый водой, спиртом и эфиром,— 1Ю3|98-2,11 Н20;
Препарат, промытый только водой,— U03i96-2,18H20.
Хюттиг и Шредер изучали дегидратацию UO,-л:Н20 под постоянным
давлением водяного пара (около 15 мм рт. ст.) по тому же способу, по
которому была исследована дегидратация U03-;eH20 (см. выше, стр. 237) [6].
На рис. 43 приведены изобары процесса дегидратации и раскисления.
Исследования проводились с воздушно-сухим препаратом, содержавшим около 4,5
молекулы воды на атом урана; было сделано предположение, что эта фаза
является продуктом присоединения перекиси водорода и воды к трехокиси
урана (UO3-H2O2.3,5H20). Кривые показывают, что половина перекисного
кислорода теряется при температурах около 60°, когда соединение еще
содержит 3,5 молекулы воды. Для образующейся фазы состава 1Ю35-ЗН20
предложена формула 1Ю30,5Н2О2-2,5Н2О. При температурах от 54 до 163°
происходит постепенная потеря второй молекулы воды и получается
соединение состава U03--2H20 (по Хюттигу UO30,5H2O2- 1,5H20). Этот состав
может соответствовать индивидуальному соединению или же смешанным
кристаллам U03)5-3H20 и U03-1,5H20. Остаточный перекисный кислород
теряется вместе с 1,5Н20 при 163°; при этом получается в виде твердой
240 Часть III. Бинарные соединения урана
фазы полугидрат UO3-0,5H2O. Последняя половина молекулы воды теряется,
как было установлено в этих опытах, примерно при температуре 450°. Этот
результат согласуется с данными опытов по дегидратации U03-2H20
(см. выше, стр. 237).
Хюттиг отрицает существование окисла U04, а гидраты U04
рассматривает как слабо связанные продукты присоединения U03, Н202 и Н20.
Однако этот вывод не вытекает непосредственно из результатов описанных
наблюдений.
Следует отметить, что, согласно рис. 43, половина грамм-атома перекис-
ного кислорода теряется, прежде чем количество воды снижается до 2Н20.
Другими словами, соединения Ш4-2Н20 (или U03-H202-H20) при
давлении водяного пара 15 мм рт. ст., по Хюттигу, не существует. Это
заключение находится, повидимому, в противоречии с наблюдениями многих других
i-i5
•44
CD
л21
14/
О 20 40 60 80 100 120 140 WO
Температура, °С
Рис. 43. Термическое разложение U04-xH20 при парциальном
давлении водяных паров, равном 15 мм рт. ст. [б].
Сплошная линия показывает число молей воды, пунктирная —число молей
кислорода на 1 г-атом урана.
*
авторов, доказавших, что гидрат состава 1Ю4-2Н20 является наиболее
устойчивой формой перекиси урана. Так, Фейрли [2] указывает, что состав
перекиси урана, высушенной при 100°, соответствует 1Ю4-2Н20. Такие же
наблюдения были сделаны и другими авторами [155, 159, 160].
В Браунском университете [161] выяснено, что высушивание U04• хН20
(осажденной при 75°) в вакууме при 50° приводит к быстрой потере воды,
превышающей количество, необходимое для образования гидрата состава
U04-2H20; при дальнейшем повышении температуры дегидратация идет
медленно вплоть до 125°; при этой температуре начинается одновременная
потеря воды и кислорода. При 60 — 70° дигидрат перекиси может быть
подвергнут сушке в течение 13 час. без заметной потери кислорода [162].
Согласно наблюдениям, сделанным в Колумбийском университете [157],
перекись, приготовленная методом Крауса и имеющая состав (после стояния
на открытом воздухе) 1Ю4-4И20, переходила в 1Ю4-2И20 при высушивании
при 100° на воздухе или при комнатной температуре над 36н. раствором
серной кислоты.
Перекись, приготовленная при 75° и высушенная в кислороде в
течение 3 час. при 37° и 3 час. при 62°, также имела состав U04-2H20 [163]
Содержание воды не изменялось после дополнительной сушки в течение
3 час. на воздухе при 85°, 3 час. в кислороде при 105° или 13 час. в
кислороде при 75°. Кислород начинает медленно улетучиваться при температуре
53
со
ЗУ-
71 1
1 °">:
к"4 -;
^1
U tie
Г sis
4> .
^£
L °&l CL
col ^
SM'o
5; l*<
\— ^1
Г ^
Ъ|
J3
L3 l_
-^—|
H
_i i
~i i—i—
•
X
J 1 1
Глава 11. Окислы, сульфиды и теллур иды урана
241
немногим ниже 100°. Подробности превращения перекиси урана в низшие
окислы см. ниже, стр. 258.
Теплота образования дигидрата перекиси урана. Писаржевский [164]
измерял теплоту реакции дигидрата перекиси урана, высушенного при 100°,
и моногидрата трехокиси (метод приготовления не указан) с 2 н. раствором
серной кислоты. Предполагалось, что первая реакция дает уранилсульфат
и перекись водорода, вторая— уранилсульфат и воду. Писаржевский
вычислил теплоту образования U04-2H20 из и03-Ы20, кислорода и воды,
используя известное значение теплоты окисления Н20 в Н202:
U04.2H20 -+ U03.H20 + ~02 + H20 + 6,15 ккал. (15)
Физические свойства гидратов U04. Плотность обезгажеп-
ного дигидрата перекиси урана, содержащего 79% U04, 11% Н20 и 10% U03,
составляет 4,31 г)см3 [61]. Насыпной вес высушенного дигидрата перекиси
составляет 0,72 г J см3, вес после утряски—1,00 г/см* [57].
Кристаллическая структура. Дигидрат перекиси урана обычно
считается аморфным. Имеются указания [156] о возможности получения
кристаллической перекиси путем приливания 30%-ного раствора перекиси водорода
к равному объему 14%-ного аммонийуранилоксалата при комнатной
температуре и нагреванием полученного студенистого осадка с двойным объемом
воды почти до кипения. При этом получается темножелтый раствор, из
которого при медленном охлаждении выпадают тонкие игольчатые кристаллы.
Последние имеют состав тригидрата перекиси урана; они превращаются
в дигидрат при высушивании на воздухе при 100° или в вакууме над пяти-
окисью фосфора при комнатной температуре. Однако повторные попытки
приготовить эту кристаллическую перекись в Беркли [158] привели к
образованию соединения, содержавшего ионы аммония и оксалата, повидимому,
U04.0,5(NH4)2C2O4 .(1,5 -:- 2)Н20.
Возможно, что продукт, полученный в более ранних исследованиях [156],
имел такой же состав.
Согласно данным Колумбийского университета, U04-xH20, осажденная
при 75° путем приливания 1 л 3%-ного раствора перекиси водорода к
водному раствору уранилнитра а (210 г/л воды), был микрокристаллическим и
давал размытую рентгенограмму [141, 165]. Был также рентгенографическим
методом исследован образец воздушно-сухой 1Ю4-л:Н20, который должен был
содержать 4Н20 (см. выше, стр. 239). Линии рентгенограммы были размыты,
а это указывает на то, что кристаллиты имели размер 100 — 200А [32, 91].
Показано, что элементарная ячейка гидрата перекиси урана является
ромбической гранецентрированной со следующими константами:
а1 = 8,74 ±0,05А; а2 = 6,50 ± 0,03А; а3 = 4,21 ±0,2А.
Два атома урана в элементарной ячейке находятся в положениях (0 00)
и С -к -д- y) • При такой структуре, однако, недостаточно места для четырех
молекул воды. Значения плотности, вычисленные для различного возможного
содержания воды, приведены в табл. 96.
Твердость. Твердость по шкале Мооса равна 2,5; в случае влажного
или рыхлого гидрата она составляет 2 [63].
Парамагнетизм. Исследования парамагнитных свойств 1Ю4-*Н20
показали, что удельная магнитная восприимчивость, рассчитанная для безводного
окисла, равна 0,17- 10е; х (мол.) = 51 • 10~6; х (мол.) с поправкой на
диамагнетизм равна ПЬЮ"6 [118]. Значения, использованные для Ue+ и О2"
при введении поправки на диамагнетизм, составляли соответственно
16 Химия урана
242
Часть III. Бинарные соединения урана
Состав
U04-4H20
ио4.зн2о
U04-2H20
р, г\см*
5,15
4,90
4,66
Таблица 96 — 11,25- 10~в и — 20- 10_в. Парамагнитныесвой-
плотность гидратов ства не зависят от температуры.
перекиси урана Оптические свойства. 1Ю4л;Н20 имеет
светложелтую окраску. В противоположность
U03*H20, гидрат перекиси не флюоресцирует
ни при комнатной температуре, ни при
температуре жидкого азота [48, 150].
Гидрозоли окис л ов у р ана.
Гидрозоль двуокиси урана. Электролитическим
восстановлением раствора, содержащего 50 г уранил-
хлорида в 100 мл 2н. раствора соляной кислоты,
был получен черный осадок [166]; этот осадок
легко переходил в неустойчивый гидрозоль, коагулировавший приблизительно
через 24 часа. Его устойчивость повышалась при добавке тетрахлорида урана.
Частицы золя видимы в ультрамикроскоп. Они отличаются интенсивным
броуновским движением и несут положительный заряд, что подтверждается их
поведением в электрическом поле и характером коагуляции анионами.
Гидрозоль содержит до 0,2 моля U02 на 1 л. Подобный же гидрозоль можно
приготовить восстановлением уранилхлорида цинком или медью в
разбавленной соляной кислоте.
Гидрозоль трехокиси урана. Этот гидрозоль впервые был получен
осаждением раствора уранилнитрата или уранилхлорида едким кали в
присутствии сахара с последующим диализом [167]. Оранжево-желтый
гидрозоль, совершенно свободный от кислоты или щелочи, очень устойчив. Он
может коагулировать при действии солей, но вновь пептизируется водой.
Коллоидная трехокись урана была получена также из осадка,
образовавшегося при гидролизе уранилхлорида в присутствии окиси серебра [168J.
Желтый слабокислый коллоидный раствор содержит немного соляной кислоты,
которая не может быть удалена диализом; он устойчив при 0°, но
разлагается при комнатной температуре с выделением дигидрата трехокиси урана.
Проведены опыты по разложению раствора уранилацетата в водно-
эфирной смеси действием света. Выпадавший фиолетовый осадок из08-л;Н20
окислялся на воздухе до желтого гидрата трехокиси [169]. Из последнего
приготовляли водную суспензию, которую постепенно приливали к горячему
разбавленному раствору уранилнитрата или нитрата тория. При этом получался
очень устойчивый гидрозоль —оранжево-желтый в случае уранилнитрата и
зеленовато-желтый в случае нитрата тория.
Изучено действие различных Сахаров на пептизацию гидрата трехокиси
урана [170]. Осаждение последнего щелочью может быть предотвращено
добавлением сахарозы, лактозы, глюкозы или фруктозы.
Гидрозоль трехокиси урана можно легко получить осаждением диура-
ната аммония из раствора уранилнитрата аммиаком при перемешивании
с последующей пептизацией в большом объеме воды [171].
С целью получения гидрозоля трехокиси урана производились также
опыты осаждения щелочных уранатов из раствора уранилхлорида 0,1 н
растворами едких щелочей [172, 173]. Осадок промывали горячей водой до
тех пор, пока он не начинал проходить через фильтр, и затем пептизировали
его в большом объеме воды. Щелочь удаляли диализом. Частицы золя
заряжены отрицательно. Они имели размер приблизительно 5-Ю"5 см и
плотность 7,45 г/см2. Золь коагулировал при действии хлористого калия или
хлористого бария; приливанием раствора хлористого алюминия можно было
изменить знак заряда на обратный. Вязкость 15%-ного золя при 15° была
в 1,0393 раза выше вязкости воды. Гидрозоль был устойчив при кипячении,
но коагулировал при замораживании. Поверхностное натяжение 1%-нога
золя проходило через максимум при 30 — 40°.
Глава П. Окислы у сульфиды и теллур иды урана
243
ОКИСЛЕНИЕ И ВОССТАНОВЛЕНИЕ УРАНА И ЕГО ОКИСЛОВ
Термодинамика образования и взаимных превращений
окислов урана. Теплоты образования и взаимного превращения.
Проведены прямые калориметрические измерения [174] теплоты сгорания
урана и двуокиси урана в закись-окись и образования ураната натрия
Na2U04 из урана, двуокиси, закиси-окиси или трехокиси его и перекиси
натрия (см. также стр. 266). Таким образом, можно было вычислить
теплоты образования двуокиси и закиси-окиси урана либо на основании
данных сжигания с кислородом, либо на основании реакции с перекисью
натрия, теплоту же образования трехокиси можно было установить только по
реакции с перекисью. Независимое вычисление теплоты образования
трехокиси урана было проведено следующим образом [175]. Сравнивали теплоту
образования растворов уранилхлорида из трихлорида или тетрахлорида урана
с теплотой образования тех же растворов из трехокиси урана, а также
непосредственно измеряли теплоту образования три- и тетрахлорида из
элементов. Результаты были критически рассмотрены и исправлены в
радиационной лаборатории Калифорнийского университета [71]. Данные,
полученные при двух разных калориметрических исследованиях, представлены
в табл. 97.
Таблица 97
ТЕПЛОТА ОБРАЗОВАНИЯ ОКИСЛОВ УРАНА
Реакция
и+о2 - ио2
u+-io2 - 1и3о8
U + y02-U03
ио2+-^о2 -* -^и3ов
uo2+-J-o2 - иоз
3-u3o8+i-o2->uo3
—Д#, ккал-г-атом 1 урана
Микстер [174]
по
методу
сжигания
256,6
281,7
—
25,1
—
—
по
реакции
с ЫагОг
269,7б>
298,5
303,9
28,8
34,2
5,4
Бильц и Фендиус 1175]
через
UC13
—
—
294
—
(27) г>
(12) г)
через UCli
—
—
292 (288) в)
—
(25) г>
(10) г>
Брю-
эра)
[14]
(257)
285
291
28
34
6
Уравнение
(16)
(1.7)
(18)
(19)
(20)
(21)
а) Рассчитано из тензиметрических данных Бильца и Мюллера [11] (о подробностях расчета см.
в тексте).
б) Это значение было впоследствии пересчитано с использованием лучших данных для окиси и
перекиси натрия, что дало ДЯ-^—270,4 ккал\м.оль L176].
в) Исправлено [71].
г) Использованы значения для теплоты сгорания, указанные в работе Микстера [174].
Теплоты реакции по уравнениям (19) и (21) (табл. 97) вычислены также
из значений давления разложения трехокиси и закиси-окиси урана [11J.
Позже Брюэром [14] были проведены более детальные расчеты на
основании тех же экспериментальных данных. Тензиметрические данные Бильца
и Мюллера [11] недостаточны для точного расчета из-за образования твердых
16*
244
Часть III. Бинарные соединения урана
растворов. Для того чтобы получить точные результаты, необходимо
приложить уравнение Вант-Гоффа не только к изохорам разложения закиси-
окиси и трехокиси, но и к изохорам разложения всех окислов
промежуточного состава; полная теплота окисления получается в этом случае
интегрированием дифференциальных теплот, вычисленных при помощи этого
уравнения в пределах соответственно для # = 2,0^-2,67 и х = 2,67-:- 3,0.
Такие точные расчеты, однако, не были проведены. В области между
закисью-окисью и трехокисью урана Бильц и Мюллер [11] использовали
уравнение Вант-Гоффа с применением средних значений давления, полученных
совершенно произвольным усреднением данных изотерм при 500, 580 и 610°
(ср. рис. 39). Этим путем найдено, что средняя теплота разложения равна
17 ккал-г-атом'1 кислорода или 5,67 ккал-г-атом'1 урана. Это значение
мало отличается от приведенной в табл. 97 величины Микстера [174],
составляющей 5,4 ккал-г-атом"1 урана. Из тех же данных Брюэром [14]
было вычислено значение 17,5 ккал-г-атом'1 кислорода.
Бильц и Мюллер применили уравнение Вант-Гоффа в пределах между
двуокисью и закисью-окисью урана в двухфазной области U02,3o — U02i62
(температурный интервал 1160—1240°) и получили значения — А//= 39,5
ккал-г-атом'1 кислорода для теплоты превращения 1Ю2,зо в U02,62- Эта
величина близка к указанному в табл. 97 значению Микстера для
превращения двуокиси урана в закись-окись (25 — 29 ккал-г-атом'1 урана или
37,5 — 43,5 ккал• г-атом'1 кислорода). Брюэр [14] рассчитал из данных
Бильца, что — А//= 40,5 ккал-г-атом'1 кислорода.
Начальная дифференциальная теплота окисления двуокиси, конечно,
гораздо больше, чем 40 ккал • г-атом'1 кислорода. На основании давления разложения
UO2,20, найденного Бильцем, Брюэр рассчитал, что средняя теплота
окисления между U02too и U02)25 равна приблизительно 48 ккал-г-атом'1
кислорода. Для средней теплоты окисления в области U0262 —U02t67 Брюэр
вычислил значение 30 ккал-г-атом'1 кислорода. Конечная
дифференциальная теплота окисления двуокиси в закись-окись, определенная Бильцем и
Мюллером, равна всего 15 ккал• г-атомг1 кислорода.
На основании трех приведенных выше значений (48 ккал-г-атом'1
кислорода для интервала х = 2,00 ~~ 2,25, 40,5 ккал для х = 2,25-^-2,62 и
30 ккал для # = 2,62-^-2,67) и значения Микстера (257 ккал-г-атом'1 урана)
для теплоты образования двуокиси Брюэр рассчитал, что теплота
образования закиси-окиси урана равна 285 ккал-г-атом'1 урана. Если прибавить
еще 17,5 ккал-г-атом'1 кислорода для окисления закиси-окиси урана в
трехокись, то общая теплота образования трехокиси урана составит
291 ккал-г-атом'1 урана. Эти значения помещены в предпоследней графе
табл. 97.
Экстраполяцией из теплот образования окислов других элементов
периодической системы получено значение 270 ккал - моль'1 для теплоты
образования трехокиси урана [177].
Теплота образования трехокиси должна зависеть и от ее
аллотропической формы. По Брюэру, значения, полученные выше, вероятно,
действительны для гексагональной формы U03 (I); аморфная форма немного менее
устойчива, в то время как кристаллические формы 1Ю3 (II) и U03 (III),
возможно, на 1—2 ккал-г-атом'1 урана устойчивее, чем кристаллическая
форма U03 (I).
Возможность образования моноокиси UO из урана и двуокиси урана
указывает на то, что — Д#ио больше, чем 129 ккал-моль'1, т. е половины
теплоты образования двуокиси. С другой стороны, ввиду того что магний
восстанавливает высшие окислы урана до металла, а не до UO, — АН должна
быть меньше 142 ккал-моль'1. Поэтому наиболее вероятным значением
— АЯио будет 135 ± 5 ккал.моль"1.
Глава 1L Окислы, сульфиды и теллур иды урана
245
Свободные энергии и энтропии образования и взаимных превращений
окислов урана. Для энтропии моноокиси Брюэр дает величину —20
энтропийных единиц при 1000° К.
Двуокись урана. Из величины Д# по Микстеру [174] и подсчета
энтропии урана и двуокиси урана, сделанного Латимером, — ASuo2 = 42,l
энтропийной единицы, Брюэр [178] вычислил свободную энергию образования
двуокиси урана:
-Д/Чю2 (298,15° К) -244 ккал-моль'1.
Позже Брюэр [14] дал следующие значения для ASuo2:
Температура,0 К 298 500 1000 1500
ASuo2 42>5 41>9 40,5 40,1
Вышеупомянутое значение свободной энергии образования двуокиси
урана, а именно —244 ккал ■ моль"1, показывает, что двуокись необходимо
нагреть до 4200° К для того, чтобы равновесное давление кислорода над
ней достигло ЫО"2 мм рт. ст.
Вычисленная энтропия образования U02,25 (по предположению Брюэра,
этот окисел является верхним пределом области твердых растворов
кислорода в двуокиси урана) равна 45,5 энтропийной единицы при 1000° К-
Закись-окись урана. Для вычисления свободной энергии образования
этого окисла из двуокиси и кислорода [см. реакцию (19)] могут быть
использованы тензиметрические данные Бильца и Мюллера [11]. Маквуд и
Альтман [71] предположили, что постоянное давление разложения,
экспериментально установленное для области от 1Ю2,зо До U02,62> действительно
для более широкого интервала —от U02,o до U03,o, й отсюда вывели
уравнение
- AF° = 74432 -2,35 Tig T -4,75- 1(Г4 Г2 +
+ 3,096-105 Г"1-28,61 Ткал-моль'1 U308> (22)
откуда следует, что —\F°298 = 65,07 ккал -моль1 U3Og.
При выводе уравнения (22) были использованы уравнения (5) и (7)
соответственно для теплоемкостеи двуокиси и закиси-окиси урана, что
дает (для образования 1 моля U308 из 3 молей U02 и 1 моля 02)
ДСр=1,02 + 9,5Ы0-47,-6,193-105Т"2 (23)
и
-ДЯ-74432+1,02Г + 4,75-10-4Г24-6,193.105Г1. (24)
Отсюда
— Д//298 = 76,75 ккал • моль'1 U308.
При применении приведенного выше значения — А/7°298 (65,07 ккал -моль'1)
было найдено, что — AS298 равно 39,2 энтропийной единицы.
Использованное значение — А#(76,8 ккал моль"1 U308 или 25,6
ккал • г-атом~х урана) удовлетворительно совпадает со значениями,
указанными в табл. 97.
Для этой же реакции было предложено еще следующее уравнение [165]:
_ДГ> = +76890-63,1 Г + 2,58-10"3 Г2 — 0,32- Ю-6Г3 —
-0,62-105 Г"1+ 3,22 Г In Г кал-моль1 U308. (2оЬ
Значения AFf, вычисленные по этому уравнению, представлены
в табл. 98. Уравнение (25) основано на данных Дэвиса и Бартона [179]
для реакции
U308 + 2Н2 -> 3U02 + 2Н2Огаз. . (26)
246
Часть III. Бинарные соединения урана
Таблица 98
СВОБОДНАЯ ЭНЕРГИЯ
ОКИСЛЕНИЯ ДВУОКИСИ УРАНА
ДО ЗАКИСИ-ОКИСИ
Таблица 99
ТЕРМОДИНАМИКА ОБРАЗОВАНИЯ ЗАКИСИ-ОКИСИ
Температура,
•к
300
400
500
600
1000
-">•
ккал-моль~1 U3O8
63,45
59,6
55,8
52,1
38,2
Термодинамические функции
-А^298> ккал-моль~х U308
-AF°29s> ккал-моль~~1 U308
-AS, энтропийные единицы
Брюэр
[14]
855 б>
805
166
а) По Микстеру [174].
б) Вывод см. выше, стр. 24 3.
Для теплоты образования закиси-окиси из элементов Маквуд и Альтман
рассчитали на основании значения Микстера для АЯ298 и вышеупомянутых
уравнений для теплоемкости
-ДЯ = 850000- 19,01 7 + 9,65-10"3 Г2-3,984- Ю5 ТК (27)
При использовании приближенного уравнения Нернста они получили
- AF° = 850000 + 43,79 Т lg T- 9,65-10"3 Т2 + 1,992-105 Г"1 -303,1 7\ (28)
откуда
— Д/?й98 = 791,7 ккал-моль'1 U308
AS298= 184,2 энтропийной единицы.
В табл. 99 сопоставлены результаты Маквуда и Альтмана с цифрами,
данными Брюэром.
В табл. 100 помещены значения функции
AFT — АН2М
для образования U308 из элементов (по Брюэру).
Таблица 100
ЭНТРОПИЯ ОБРАЗОВАНИЯ
ЗАКИСИ-ОКИСИ УРАНА ПРИ
РАЗЛИЧНЫХ ТЕМПЕРАТУРАХ
Таблица 101
Температура,
0 К
298
500
1000
1500
Д/^—Я-208
для U308,
кал >моль~~1.- град"1
166 а)
167
163
161
СВОБОДНАЯ ЭНЕРГИЯ ОКИСЛЕНИЯ ДВУОКИСИ
И ЗАКИСИ-ОКИСИ УРАНА ДО ТРЕХОКИСИ УРАНА
Температура,
UK
300
400
500
600
1000
(29)]
[для реакции
ккал-моль~1
ио2
29,1
27,2
25,2
23,3
15,6
—bF° [для реакции
(30)], ккал-моль~1
и3о8
22,6
21,0
19,1
17,2
8,0
а) Равно Д$298.
Трехокись урана. Для стандартной свободной энергии образования
трехокиси урана из двуокиси или закиси-окиси и кислорода
ио2 + |о2
>ио.
Глава 11. Окислы, сульфиды и теллуриды урана
247
предложены следующие уравнения [48]:
- AF0 = 33830 - 9,32 7 + 0,401 • 10"3 Г2 + !, 16 • 105 Г"1 - 1,37 Г In Г;
и3о8+4о2
3UO
з>
-LF° = 24600 + 35,1 Г-1,38-10-3Г2 + 0,32-10-вГ3 +
+ 4,10-105 Г1 -7,33 Т In T,
(29)
(30)
откуда получаются значения, представленные в табл. 101.
Маквуд и Альтман [71] использовали значение — Д# = 288 ккал-моль"1
для образования трехокиси урана из элементов; эту величину они получали
из данных Бильца и Фендиуса (см. табл. 97), но с исправлением значения
для теплоты образования тетрахлорида урана. Далее, они считали S298 = 22
энтропийным единицам (см. стр. 231). Пользуясь уравнениями теплоемкости
для урана и трехокиси урана, они вывели следующие уравнения для
теплоты и свободной энергии образования U03:
Д# = -289400 + 6,39 Т- 5,04 -КГ3 Г2- 1,29-104 Г"1 кал-моль'1] (31)
Аро^ ~289400-!4,72Г^Г + 5,04-10-3Г2-б,5-103Г1 + 68,2Г. (32)
Из этих уравнений следует, что
Д#298= —288 ккал-моль"1;
4^298= —269,1 ккал-моль"1;
AS298= —63,6 энтропийной единицы.
В табл. 102 приведены значения функции I т " J для образования
трехокиси урана из элементов. Были приготовлены пасты из желатины и
тонкоизмельченных окислов урана (или гидроокисей) и измерен потенциал
платиновых электродов, покрытых этими пастами и погруженных в раствор
уранилнитрата [14,3 г/л U02 (N03)2], по отношению к нормальному
каломельному электроду [135]. Полученные значения, смысл которых неясен, приведены
в табл. 103.
Таблица 102
ЭНТРОПИЯ ОБРАЗОВАНИЯ ТРЕХОКИСИ
ПРИ РАЗЛИЧНЫХ ТЕМПЕРАТУРАХ
Таблица 103
ПОТЕНЦИАЛ ПЛАТИНОВОГО ЭЛЕКТРОДА,
ПОКРЫТОГО ОКИСЛАМИ УРАНА
Температура,
вК
298
500
1000
AFr— АН 2
для UO3,
кал-моАЬ~1 • град-1
62,0 а)
61,7
60,6
Окисел урана
U02 ИЛИ U308
U03-H2Oa>
U3O10.2H2O6>
Потенциал, в
От —0,773 до —0,787
— 0,860
-0,687
а) Равно S298-
а) Желтая гидроокись, полученная электролизом
уранилацетата.
б) Черная гидроокись, полученная электролизом
(см. стр. 262).
Значения свободных энергий восстановления трехокиси и закиси-окиси
урана водородом приведены ниже, стр. 250.
Восстановление окислов урана до металла*
Восстановление окислов урана до металла рассмотрено в гл. 4. Вследствие
248
Часть 111 Бинарные соединения урана
высокой устойчивости двуокиси урана (см. табл. 97) восстановления до
металла можно достигнуть только применением очень сильных восстановителей,
например щелочноземельных металлов.
Образование моноокиси урана окислением
металлического урана или восстановлением двуокиси.
Металлический уран под действием воздуха при комнатной температуре
покрывается коричневато-черной двуокисью урана; при нагревании металла
(см. гл. 6) образуется зеленовато-черная закись-окись урана. Однако если
давление кислорода очень мало (МО9 атм) и температура выше 800°, то
образуется серая пленка, содержащая некоторое непостоянное количество
моноокиси урана, которую можно узнать по ее характерной
рентгенограмме [180]. Отношение моноокиси к двуокиси возрастает при
продолжительном нагревании. После 5-часового отжига в вакууме при 800° на
рентгенограмме окисной пленки почти исчезают линии, характерные для двуокиси
[41, 181]. При взаимодействии двуокиси и порошкообразного
металлического урана также образуется моноокись урана:
U02 + U -> 2UO. (33)
Но эта реакция идет настолько медленно, что даже при продолжительном
нагревании до 2000° равновесие не достигается [182]. В проведенном до настоящего
времени наиболее удачном опыте [43, 182] нагревания смеси порошкообразных
U02 и U до 1900° удалось получить продукт, содержавший 90,6% урана
и 0,24% углерода. Если предположить, что остаток являлся кислородом, то
этот препарат состоял на одну треть из UO и на две трети из U02.
При изучении ураномолибденовыхо сплавов [42] была обнаружена
кубическая фаза с константой решетки 4,93 А; предполагалось, что эта фаза
представляет собой моноокись урана, образованную окислением металлического
порошка урана остаточным газом, находящимся во время термической
обработки в эвакуированных кварцевых трубках.
Интересно отметить, что фаза UO выражена гораздо сильнее в том случае,
когда вакуум создается посредством ртутного диффузионного насоса, а не с
помощью механического насоса.
Непосредственное термическое разложение двуокиси на моноокись и
кислород, которое идет только при очень высоких температурах, еще подробно не
изучено. Сообщается [183], что двуокись урана начинает терять кислород,
в вакууме приблизительно при 1600°.
Образование двуокиси урана из урана и высших
окислов урана. Окисление металлического урана до двуокиси.
Окисление металлического урана воздухом при комнатной температуре приводит
к образованию бурой двуокиси; при продолжительном окислении кислородом
воздуха и нагревании образуется зелено-черная закись-окись. В случае куска
металла первичный окисный слой защищает остальной металл от окисления
[59] (более подробно о поведении урана на воздухе см. в гл. 6).
Ввиду того что свободная энергия образования двуокиси (—122 ккал-г-
атом'1 кислорода) гораздо более отрицательна, чем эта энергия для воды
(—57 ккал • г-атомг1 кислорода), уран термодинамически способен вытеснять
водород из воды, причем образуется двуокись урана. Плотный металл
взаимодействует с холодной водой лишь очень медленно [184, 185]. Порошок урана
медленно реагирует с водой при комнатной температуре и более быстро при 100а
[186, 187]. Весьма тонкодисперсный уран (полученный разложением гидрида
UH3) быстро разлагает даже холодную воду.
Согласно данным Колумбийского университета [63], двуокись,
полученная обработкой металлического урана водой, является пирофорной, т. е. она
самопроизвольно окисляется на воздухе до закиси-окиси.
Глава 11. Окислы, сульфиды и теллур иды урана
249
Термическое разложение закиси-окиси или трехокиси урана до двуокиси
и кислорода. Как указывалось выше (стр. 245), стандартная свободная энергия
разложения закиси-окиси при комнатной температуре на двуокись и кислород
равна 21,5 ккал • г-атом'1 урана (21,7 ккал по Маквуду и Альтману [711 и
21,2 ккал по Киршенбауму [48]). Свободная энергия разложения трехокиси на
двуокись и кислород равна 29,1 ккал-г-атом'1 урана. Можно было поэтому
предполагать, что закись-окись и трехокись легко превращаются в двуокись
при нагревании в вакууме. Вышеприведенные значения представляют, однако,
средние или интегральные свободные энергии разложения. Действительная или
дифференциальная свободная энергия разложения намного выше среднего
значения в области твердых растворов с меньшим содержанием кислорода, чем
в 1Ю2,зо (см- изотермы разложения на рис. 39). Как показывают результаты
опытов Бильца и Мюллера [11], описанные выше (стр. 209), разложение
закиси-окиси урана по этой причине обычно прекращается (в условиях
нормального вакуума и при температурах до 1300°) после достижения состава
приблизительно ио2Л5.
На воздухе разложение закиси-окиси урана начинается при 900°; при
этой температуре давление разложения чистой UO2,e07 достигает парциального
давления кислорода в воздухе [188, 11 ]. Ввиду того что для окислов с составом,
промежуточным между UO2i0G7 и U02 G1, давление разложения быстро падает
с уменьшением содержания кислорода, разложение закиси-окиси урана
прекращается после потери очень небольшого количества кислорода, если только
давление кислорода над окисью не уменьшить весьма значительно по
сравнению с парциальным давлением его в воздухе или если не поднять
температуру значительно выше 1000°. Согласно рис. 35, в интервале постоянного
давления, U02t62—U02,no, давление разложения кислорода равно только
0,3 мм рт. ст. при 1160°. При достижении состава UO2(20 это давление
падает до 0,02 мм рт. ст.
Изотермы, представленные на рис. 35, показывают как далеко может
согласно Бильцу и Мюллеру пойти разложение при различных давлениях
кислорода и температурах вплоть до 1160°.
В других опытах по термическому разложению закиси-окиси урана [1891.
с использованием в качестве исходных веществ UO3t0, U02,64 и U02f6l
разложение в эвакуированных запаянных трубках при 700° останавливалось на
стадии образования U02>Gl. Рост давления кислорода составлял 34 мм рт. ст.
при U03 и 2,4 мм в случае U02>64. При использовании U02((U выделения
кислорода не наблюдалось. После нагревания U03 при 750—830° в высоком вакууме
в течение 12 час. остаток имел состав от U02t55 до U02>62. Эти результаты
подтверждают отмеченный выше вывод о том, что термическое разложение U308
при температурах ниже 1000° прекращается при содержании кислорода,
значительно большем, чем для состава U02,5 даже в высоком вакууме.
Согласно Жолибуа и Босье [27], полное разложение закиси-окиси до
двуокиси и кислорода может быть достигнуто при нагревании до 2000° в вакууме.
Имеются указания, что закись-окись можно полностью раскислить не
только в условиях высокого вакуума, но и в токе инертного газа, свободного от
кислорода. Например, сообщается о получении чистой бурой двуокиси урана
прокаливанием закиси-окиси на газовом пламени в платиновом тигле в
атмосфере азота или двуокиси углерода [190], а в другой работе [65] описано
образование бурой двуокиси при нагревании трехокиси в токе водорода до 1000°
и темносиней двуокиси (см. стр. 223) при нагревании трехокиси в токе азота
при 1100°. Утверждение, что последний продукт имел состав UO2,00, является
сомнительным, если принять во внимание трудность полного превращения
закиси-окиси в двуокись, как это сейчас стало известно. Раскисление трехокиси
урана будет подробно описано ниже (см. стр. 254), при рассмотрении
превращения этого окисла в закись-окись. Некоторые наблюдения, однако, указывают
250
Часть III. Бинарные соединения урана
на то, что трехокись может быть превращена в двуокись без промежуточного
образования закиси-окиси урана (см. ниже).
Восстановление трехокиси и закиси-окиси
урана до двуокиси. Восстановление трехокиси или закиси-окиси урана
водородом до двуокиси. Металлический уран способен вытеснять водород из воды
с образованием двуокиси, в свою очередь водород восстанавливает высшие
окислы урана UO:? и U308 до двуокиси. Свободная энергия восстановления
(с образованием Н20), согласно вычислениям [165], выражается следующими
уравнениями:
ио3 + н2->ио2 + н2оГа3;
-AF° = 22890+ 16,42 Г-0,79-10"3Г2+ 0,22-Ю^Г3-
-0,69.105Г-1-М7Г1пГ/сал.жол&-1; (34)
U308 + 2Н2 — 3U02 + 2Н2Огаз;
- AF° = 36545 + 77,3 Т- 3,36- 10"3Г2 + 0,762- 10"в Г3 +
+ 1,56 ■ 105 Т~1 + 11,32 Г In Г кал • моль'1. (35)
Значения А/7, вычисленные из этих уравнений, представлены в табл. 104.
Таблица 104
СВОБОДНАЯ ЭНЕРГИЯ ВОССТАНОВЛЕНИЯ ТРЕХОКИСИ
И ЗАКИСИ-ОКИСИ УРАНА ДО ДВУОКИСИ ВОДОРОДОМ
Восстанавливаемый
окисел
ио3
и3о8
300° К
25,5
45,8
—AF, ккаЛ'МОль—!
400° К
26,4
47,5
500°К | 600° К
27,2
49,0
27,9
50,3
1000° К
30,6
54,0
Уравнение
№
(34)
(35)
Отнесенная к 1 г-атому урана свободная энергия восстановления высших
окислов урана водородом до двуокиси при комнатной температуре равна
25,5 ккал для трехокиси урана и 15,3 ккал для закиси-окиси.
Вышеприведенные значения основаны на вычисленной свободной
энергии для реакции (35) [179]; они были использованы затем для вычисления
свободной энергии образования закиси-окиси урана [уравнение (25)]. Этот расчет,
конечно, может быть проведен и в обратном порядке: свободную энергию
образования закиси-окиси по Маквуду и Альтману [см. уравнение (28)]-можно
использовать для вычисления свободной энергии восстановления закиси-окиси
водородом.
Получение двуокиси восстановлением высших окислов водородом было
описано рядом исследователей [26, 27, 50, 53, 191—197]. Имеются некоторые
расхождения во мнениях в отношении температур, требуемых для полного
восстановления до UO 2<00) особенно в случае закиси-окиси как исходного материала.
По Лебо [26], закись-окись быстро и полностью восстанавливается водородом
до бурой двуокиси при 900—1000° без образования промежуточных окислов.
Бильц и Мюллер [11] тоже приготовили точно стехиометрическую двуокись
восстановлением закиси-окиси водородом при 900—1000°. Этим способом получена
бурая двуокись из зеленой закиси-окиси и более темная,
пурпурно-коричневая, двуокись из черной, сильно прокаленной закиси-окиси (как показали
результаты анализа, оба продукта имели состав UO2>0). Жолибуа и Босье
127J утверждают, что при использовании фосфорного ангидрида для
связывания паров воды достаточна температура 625—650° для полного
восстановления закиси-окиси водородом.
Глава И. Окислы, сульфиды и теллур иды урана
251
С другой стороны [198], утверждают, что восстановление-закиси-окиси
урана водородом при 650—680° оканчивается образованием 1Ю2Л4 (или
иОд-биСу, т. е. продукта с таким же составом, как и получаемый
разложением закиси-окиси в вакууме при температурах до 1300°. Сделано
предположение, что восстановление закиси-окиси водородом состоит из двух стадий,
а именно: термического разложения окисла и восстановления выделившегося
кислорода водородом. Поэтому реакция прекращается, если давление
разложения окисла становится ничтожно малым, что происходит тогда (см. стр. 249),
когда содержание кислорода снижается приблизительно до U02rl5.
Однако маловероятно, чтобы водород не мог непосредственно реагировать
с твердым окислом. Если даже действительно верно, что при 680°
восстановление закиси-окиси водородом прекращается при достижении состава U02il5,
то это можно скорее объяснить высокой энергией активации прямой реакции
1Ю2Л5 с водородом; термическое разложение закиси-окиси прекращается при
том же составе из-за высокой энергии активации реакции даже при таких
высоких температурах, как^ЗОО0. Утверждение английских ученых о том, что,
•согласно Ньюбери и Прингу [199], для полного восстановления U02,15 до
UO2.00 требуется температура 2000° и давление водорода 150 am,
противоречит результатам вышеприведенных опытов [11, 26], в которых
восстановлением закиси-окиси водородом при 900° и давлении этого газа 1 атм была
получена чистая UO2<0.
Восстановление закиси-окиси водородом при низкой температуре (330°)
приводит к образованию только UO ^ 2>5б, т. е. наинизшего окисла, в котором
еще сохраняется ромбическая структура закиси-окиси (см. стр. 211) [18].
При восстановлении закиси-окиси или трехокиси водородом реакция,
повидимому, не может идти далее образования U02,o- Как указывалось
выше (стр. 243), двуокись урана термодинамически устойчива в водороде,
поскольку это относится к восстановлению до урана и, вероятно, также до моио-
окиси. Что касается восстановления доиОя(1<л;< 2), то нельзя считатьдоказан-
ным, что оно термодинамически невозможно (см. стр. 248), однако до сих пор
на опыте такое восстановление водородом не удавалось осуществить даже при
таких жестких условиях, как давление 25 am и температура плавления
вольфрама [200], 150 am и 2500° [201] или 100 am и 1100° [202].
Восстановления двуокиси водородом не наблюдалось также в условиях дуги высокого
напряжения. Водная суспензия двуокиси устойчива в атмосфере водорода
до 250° [63].
Восстановление закиси-окиси или трехокиси урана металлами и окислами
металлов. Закись-окись урана восстанавливается до двуокиси металлическим
калием при температуре 150° с небольшим выделением тепла [204].
Восстановление можно производить посредством магния [205]. Магний восстанавливает
оранжевую трехокись до бурой двуокиси при 500°; восстановления двуокиси
магнием в этих условиях не наблюдается. Отмечено [206] восстановление
трехокиси закисью железа при повышенных температурах.
Восстановление трехокиси урана до двуокиси окисью углерода. Окись
углерода быстро восстанавливает трехокись урана (но не закись-окись) до двуокиси
при 350° [207].
Восстановление закиси-окиси или трехокиси урана углеродом. Двуокись
урана была впервые приготовлена Клапротом [208] восстановлением закиси-
окиси урана углем. Из-за черного цвета и металлического блеска порошка
двуокиси она была принята за металл. Ошибка была обнаружена Пелиго [184,
209]только через 50 лет. Согласно Муассану [210], восстановление закиси-
окиси урана до урана углеродом возможно только в электрической печи при
3000°. Если температура в электрической печи недостаточно высока для
образования карбида урана, то из закиси-окиси урана (или трехокиси) в
присутствии углерода образуется двуокись [120].
252
Часть III. Бинарные соединения урана
Восстановление закиси-окиси и трехокиси урана метаном. Превращение
светлокраснои трехокиси урана в двуокись, которое медленно происходит
при 500° на воздухе, в азоте или кислороде, ускоряется и быстро приводит
к образованию черной двуокиси в атмосфере метана [151, 176]. Реакция
начинается при 400°. При 450° продукт является смесью двуокиси и закиси-
окиси.
Восстановление закиси-окиси или трехокиси урана серой, сероводородом
и сероуглеродом. Двуокись урана была получена Германом [127]
прокаливанием закиси-окиси (или ураната натрия) с серой и хлоридом аммония и
промывкой продукта реакции водой, а также нагреванием трехокиси или
закиси-окиси в сухом углекислом газе, насыщенном парами сероуглерода. Однако теперь
известно, что эта реакция является сложной и вместо двуокиси урана могут
образоваться оксисульфиды. Трехокись урана восстанавливается до двуокиси
сероводородом в щелочном, нейтральном или даже слабокислом растворе [211 ].
Восстановление закиси-окиси урана аммиаком или хлоридом аммония.
Были проведены опыты по нагреванию закиси-окиси урана до белого каления
в течение 6 час. с большим избытком хлорида аммония в фарфоровом тигле,
помещенном в большей неглазурованный тигель и окруженном древесным
углем [212]. При этом была получена красновато-коричневая двуокись урана,
не содержащая азота или хлора.
Восстановление трехокиси или закиси-окиси аммиаком представляет собой
удобный метод для превращения соединений урана (VI) в соединения урана
(IV) [213]. Трехокись урана почти не восстанавливается аммиаком при
температуре ниже 250° (если не считать случайного образования зеленого
поверхностного слоя). При 250—400° восстановление происходит только до закиси-
окиси. Этот процесс представляет собой более энергичную реакцию
восстановления, чем происходящая при прямом термическом разложении в тех же
температурных условиях. Выше 450° начинается восстановление до двуокиси.
Оно завершается (более полно, чем с водородом) при 550°.
Восстановление закиси-окиси урана щавелевой кислотой. Нагреванием
закиси-окиси со щавелевой кислотой без доступа воздуха получена легко
окисляющаяся двуокись урана черного цвета [214]. Закись-окись урана
восстанавливается до двуокиси оксалатом кальция при температуре
красного каления [215].
Восстановление парами этилового спирта. Закись-окись урана можно
восстановить до двуокиси парами этилового спирта при температуре 200° или
выше [189].
Превращение двуокиси и трехокиси урана в
закись-окись. Окисление двуокиси до закиси-окиси. Окисление двуокиси
кислородом. В термодинамическом отношении закись-окись урана устойчива
только в довольно узком интервале температур, например от 580 до 750° при
Ро2==10 лшрт. ст. (см. рис. 37 на стр. 210) или от 650 до 900° на воздухе (Ро2—
150 мм рт. ст.) [11, 188]. Однако практически закись-окись получается в более
или менее чистом состоянии прокаливанием обоих окислов урана (высшего
и низшего) на воздухе (на этом факте основан обычный весовой метод
определения урана в виде закиси-окиси). Такая устойчивость закиси-окиси обусловлена
разницей между скоростью окисления низших окислов урана до закиси-окиси
и закиси-окиси до трехокиси. Первый процесс легко идет даже при довольно
низких температурах, а второй, наоборот, очень медленно. Согласно сказанному
выше (стр. 212), закись-окись урана не окисляется до трехокиси на воздухе при
низких температурах, за исключением, может быть, геологических периодов
времени. При повышенных температурах (порядка 350°) закись-окись может
быть окислена в лабораторных условиях, если она находится в особенно
активной, тонкодисперсной форме. Для того чтобы быстро окислить обычную закись-
окись в трехокись, необходимо применять давление кислорода порядка 7 am.
Глава П. Окислы, сульфиды и теллур иды урана
253
Вследствие этого различия в скорости окисления соединений с составом
ниже и выше U02,67 прокаливание низших окислов урана на воздухе приводит
к образованию практически чистой закиси-окиси, даже если при данной
температуре трехокись урана термодинамически устойчива (ниже 650°). Если
в процессе прокаливания температура превышает температуру разложения
закиси-окиси (около 900° на воздухе), то кислород, которого нехватает до
формулы tT308, как правило, вновь присоединяется во время охлаждения (если
только охлаждение происходит не очень быстро [102]); однако кислорода
присоединяется не больше, чем это соответствует формуле U02,67.
Температура, до которой необходимо нагреть двуокись урана на воздухе
для окисления ее в закись-окись, зависит от активности образца, т. е., вероятно,
от степени его дисперсности. Согласно Жолибуа и Босье [27] быстрое
окисление обычной двуокиси начинается при температуре около 185°. Однако
Жолибуа [17] нашел, что при низких температурах образуются продукты
с составом не выше U02>33 (приблизительно U307). На кривых нагревания
имеется разрыв между 220° (где достигается состав U3Os) и 300° (где
начинается превращение кубической решетки двуокиси урана в ромбическую
решетку U205 или U308).
Гренвольд и Гаральдсен [18] показали, что препараты двуокиси урана
реагируют с кислородом при столь низких температурах, как, например, 120°.
Продукты, полученные окислением в чистом кислороде при температурах от
100 до 265°, исследованы этими авторами рентгенографически. При 150°
присоединение кислорода приводило к образованию продукта с составом по
меньшей мере U02,34; препарат дал рентгеновский снимок со смещенными и
частично расширенными линиями, что указывает на понижение симметрии. Для
образца с составом y02,3i (который сохранил еще, повидимому, кубическую
симметрию) а = 5,40 А, в то время как для U02 a™5,468 А (данные табл. 72,
стр. 215, показывают, что и в Эймском университете обнаружено изменение
в том же направлении, но разность по величине была меньше—от 5,459 до
5,430 А). Гренвольд и Гаральдсен не решили, является ли изменение решетки
при переходе от UO2i00 к U02t34 непрерывным (как это было указано выше,
стр. 206) или же оно происходит с образованием новой фазы.
Окисление при 200—250° приводит, по Гренвольду и Гаральдсену [18],
к продуктам с я > 2,34. При этом наблюдается образование новой отдельной
фазы, названной 8-фазой (в отличие от кубической р-фазы); она является
тетрагональной и имеет узкие пределы гомогенности при составе, близком
к UO2,40.
Константы ее решетки следующие: а = 5,37А и с=5,54А (с:а=1,03);
наблюдаемая плотность равна 10,00 г/см*. Это значение нельзя объяснить
присоединением кислорода к U02 или отнятием урана от U02, а лишь
замещением урана на кислороде образованием продукта U0j88O2tl2, для которого
теоретическое значение плотности 10,04 г/см3. Данный продукт представляет
собой, вероятно, соединение U205, предложенное Лиденом [25] и другими, более
ранними исследователями, но не обнаруженное Бильцем и Мюллером [11] при
тензиметрических измерениях; это понятно в данном случае, так как
тетрагональная фаза, вероятно, существует только при температурах ниже 270°.
Пирофорная двуокись урана, которая раскаливается, окисляясь на воздухе
при комнатной температуре, приготовлена разложением оксалата [51]. Такая
же неустойчивая двуокись урана получается в результате реакции
металлического урана с кипящей водой [63]. Двуокись урана, полученная
восстановлением трехокиси урана природным газом, тоже является пирофорной [216].
Таким же свойством обладает и бурая двуокись урана, полученная
восстановлением трехокиси водородом при 650° [198].
Окисление двуокиси урана до закиси-окиси другими окислителями, помимо
кислорода. Свободная энергия образования закиси-окиси из двуокиси и
254
Часть III, Бинарные соединения урана
кислорода (AF°=— 64 ккал-моль'1 U308 при 300°К и —38 ккал-моль'1 при
1000°К, см. стр. 245) значительно менее отрицательна, чем свободная энергия
образования эквивалентного количества воды (Д/70^—109 ккал для 2 молей воды
при 300°К и —92 ккал при 1000°К); поэтому нельзя предполагать, что двуокись
урана будет восстанавливать воду. По Реньо [67], водяной пар окисляет
двуокись до закиси-окиси, но это наблюдение не было подтверждено Шодроном
[217]. В Колумбийском университете [63] также показали, что водяной пар
не действует на двуокись при 310°. При изучении [194, 218, 219] окисления
двуокиси урана окислами азота получены следующие результаты: закись азота
не окисляет бурую двуокись урана, приготовленную восстановлением
трехокиси водородом при температуре красного каления, но медленно реагирует
с черной двуокисью, приготовленной при более низких температурах; окись
азота реагирует с бурой двуокисью урана при температуре ниже красного
каления с выделением тепла и образованием черного продукта, который, повиди-
мому, представляет собой U205. Двуокись азота реагирует и с бурой и с черной
двуокисью урана на холоду с большим выделением тепла.
Разложение трехокиси до закиси-окиси. Термическое разложение. Как
описано выше (стр. 252), закись-окись урана можно получить термическим
разложением трехокиси. Изотермы (см. рис. 35 на стр. 205) указывают, что при
давлении кислорода 1 атм разложение трехокиси начинается немного ниже
470° и состав U308 достигается при температуре несколько выше 650°. При
свободном доступе воздуха (ро^ЛЪО мм рт. ст.) полное разложение до закиси-
окиси происходит при температуре около 650°. В вакууме разложение доходит
до конца ниже 580°. Выше упоминалось (стр. 212), что на практике разложение
трехокиси может задержаться на неопределенно долгое время или начаться
только после одно- или многодневного индукционного периода. Иногда оно-
происходит только тогда, когда температура значительно превышает
температуру разложения или когда давление гораздо ниже равновесного давления
разложения.
Эти явления обусловлены существованием по крайней мере четырех (а
возможно, и шести) аллотропических модификаций трехокиси, имеющих
различную устойчивость. Согласно Фриду и Дэвидсону [33], данные Бильца и
Мюллера получены при низких температурах с применением аморфной трехокиси,
приготовленной термическим разложением гидрата перекиси; при более
высоких температурах они, вероятно, имели дело с гексагональной формой U03 (I).
де-Конинк [20, 220] отметил, что кирпично-красная форма трехокиси,.
полученная продолжительным нагреванием аморфной трехокиси на воздухе,
является гораздо более устойчивой, чем исходный аморфный оранжево-красный
окисел, который, вероятно, представлял собой U03 (II) Фрида и Дэвидсона.
Согласно этим двум авторам, желтая форма U03 (III), которая может быть
получена продолжительным нагреванием трехокиси или закиси-окиси при
высоком давлении кислорода и образуется также при прокаливании уранилнит-
рата (окисьМалинкродта), является еще более устойчивой. При давлении
кислорода, равном 1 атму U03 (III) не изменяется даже после часового
прокаливания при 700°. Это можно объяснить или замедленным разложением, или, что
более вероятно, истинной термодинамической устойчивостью. Фрид и Дэвидсон
нашли, что аморфная трехокись разлагается до UC^jo ПРИ 650° и давлении
кислорода, равном 1 атм. Это примерно соответствует результатам Бильца;
и Мюллера [35]. Красная трехокись, полученная [100] из растворов солей
уранила электролизом и дегидратацией, ведет себя подобным же образом. Она
начинает разлагаться на воздухе при 470° и более или менее полно
восстанавливается до закиси-окиси при 600°. Хюттиг и Шредер [6] отметили начальные
признаки выделения кислорода из трехокиси в вакууме при 400—500°. Жоли-
буа и Босье [27] обнаружили быстрое разложение трехокиси в вакууме при
502°. Гольдшмидт и Томассен [221] наблюдали линии U308 на рентгенограмме
Глава 11. Окислы, сульфиды и теллуриды урана
255*
трехокиси после нагревания до 270° на воздухе. Этот результат трудно
объяснить, так как, согласно Захариазену, U03 (I) превращается в закись-окись.
без образования новой фазы.
Повидимому, Лебо [26], отметивший, что трехокись урана совсем не
изменяется на воздухе при температуре ниже 600°, имел дело с одной из
устойчивых модификаций трехокиси. При температуре выше 600° эта трехокись
превращалась в оливково-зеленые или зелено-черные промежуточные окислы
и только при 800° она переходила в серо-черную закись-окись урана.
Аналогично, Тамман и Розенталь [206] также отметили, что трехокись начинает
разлагаться на воздухе только при 670° с образованием промежуточных зеленых
окислов и полностью превращается в черную закись-окись урана при 865°.
Некоторые авторы наблюдали своеобразное явление: образец трехокиси,
который оставался почти неизмененным после 12 суток прокаливания на
воздухе при 600°, восстановился до U02,68 за 12 час, когда над ним пропустили
ток кислорода [11]. Отметим также наблюдение Фрида и Дэвидсона [33], что
кирпично-красная трехокись (полученная окислением закиси-окиси урана
двуокисью азота) при получасовом нагревании до 600—650° под давлением
кислорода, равном 1 атм, перешла на 87% в желтую U03 (III), остальное
представляло зеленую закись-окись.
Эти же авторы обнаружили также образование окислов, промежуточных
по составу между трехокисью и закисью-окисью при 600—650°; действительно,
они получили U02f96 и 1Ю2,82 при нагревании аморфной трехокиси в течение
0,5—1 часа до 620 и 650° соответственно [33]. По Тейлору [222], Ш3,о4
перешла при 580° за 15 мин. в U02,9i, но для достижения состава U02t84
потребовалось 123 часа.
Отмечено также [63], что аморфная безводная трехокись урана при
нагревании до 600° не превращалась в закись-окись; техническая гидратированная
желтая трехокись урана (7% Н20, 14% 1Ю4) теряла воду и кислород
при 600° и превращалась в смесь трехокиси и закиси-окиси.
Полугидрат трехокиси U03 • 0,5 Н20 терял воду и кислород при температуре
выше 430° и постепенно превращался в смесь трехокиси и закиси-окиси.
Измерена начальная скорость этой потери
кислорода трехокисью урана (неопреде- Таблица 105
ленного происхождения), результаты даны начальная скорость разложения
в табл. 105 [189]. трехокиси урана
Оранжевая трехокись,
приготовленная дегидратацией желтого гидрата
(полученного электролизом уранилнитрата) на
воздухе при 380—390° (см. стр. 261),
превращается при 520° в промежуточное
соединение приблизительного состава U02,9o,
а при 610°—в закись-окись урана [34].
Образование промежуточного соединения
(вероятно, твердого раствора с той же
кристаллической структурой, что и у закиси-
окиси) может быть установлено путем непрерывного исследования потери
кислорода с помощью термовесов или же по записи кривых нагревания.
Суммируя вышеприведенные данные, можно сделать вывод, что
неустойчивые формы трехокиси, например аморфная трехокись, начинают разлагаться
на воздухе при 400—500° и могут быть полностью превращены в закись-окись
при 650°, в то время как устойчивые формы, например U03 (HI), начинают
разлагаться только при 600° и требуют нагрева до 800° для полного превращения
в закись-окись. Остается исследовать, до какой степени формы U03 (II) и
U03 (III) должны быть раскислены, прежде чем перейти в ромбическую фазу
закиси-окиси.
Температура, °С
500
590
640
800
Количество
кислорода на 1 моль урана
в 1 мин., молн
0,007
0,031
0,086
0,192
256
Часть III. Бинарные соединения урана
Образование трехокиси и ее гидратов из других
окислов урана. Получение трехокиси окислением двуокиси или закиси-
окиси урана. Трехокись является термодинамически устойчивым на воздухе
окислом урана при температурах от комнатной до 450—600° (в зависимости
от аллотропической формы). Между этими температурами и 650° устойчивая
фаза имеет состав, промежуточный между 1Ю3 и U02>67, но выше 650°
единственным устойчивым на воздухе окислом становится закись-окись урана. Тем
не менее окисление двуокиси кислородом обычно приводит к образованию
только закиси-окиси урана.
Выше (стр. 212 и 252) упоминалось о медленности окисления закиси-окиси
урана до трехокиси воздухом или кислородом. В литературе сообщается только о
нескольких случаях прямого окисления закиси-окиси в трехокись. Так, Лебо
[86] и Бильц и Мюллер [11] установили, что тонкодисперсная серо-черная
закись-окись урана, полученная разложением уранилоксалата при низкой
температуре (350°), становится оранжево-бурой на воздухе после нагревания
в течение 12 час. при 350°. Путем химического анализа продукта реакции
установлено, что он имеет состав, близкий к U03.
В дальнейшем было проведено более тщательное исследование [38, 207]
процесса окисления тонкодисперсной закиси-окиси, полученной разложением
уранилоксалата при 350°, разложением UOa в вакууме при 450° или
восстановлением UOg окисью углерода при 350°. Обратное превращение в трехокись на
воздухе или в кислороде при температурах 300 и 500° можно изучать с помощью
самопишущих термовесов, а также путем рентгеновского анализа образцов,
взятых на различных стадиях окисления. Термовесы, например, показывают,
что привес, соответствующий полному превращению закиси-окиси
(приготовленной из оксалата) в трехокись (1,87%), достигается в случае нагревания
в атмосфере кислорода при 350° за 12 час. Рентгенограммы показывают
постепенное исчезновение максимумов, характерных для закиси-окиси, по мере того
как совершается превращение кристаллического вещества с ромбической
решеткой в аморфную трехокись.
Если действовать кислородом на предварительно нагретую закись-окись
урана, то изменения решетки начинаются при 450° и протекают особенно
интенсивно при температурах выше 575°; при этой температуре ширина диффрак-
ционных максимумов увеличивается вдвое. Эта перекристаллизация с
образованием мелкокристаллического продукта увеличивает трудность окисления.
Твердый раствор состава U02,9o (стр. 2J2) дает рентгенограмму,
идентичную рентгенограмме сравнительно легко окисляющейся формы закиси-
окиси; при 610° он также превращается в практически неокисляющуюся
микрокристаллическую форму закиси-окиси.
Изучалось окисление взвешенной в воде закиси-окиси под высоким
давлением кислорода [223]. Процесс проводился в бомбе из нержавеющей стали,
выложенной стеклом пирекс. В этих условиях окисление идет сравнительно
быстро: при 100—250° и давлении кислорода, равном 3,2—11,6 am, 75%
закиси-окиси окислилось в течение 0,5—2 час. В табл. 106 приведены некоторые
типичные результаты.
Закись-окись урана в сухом состоянии также была превращена в
трехокись при действии кислорода под высоким давлением (30—150 am) [33, 111].
При этом использовались температуры до 500—750° и в зависимости от условий
опыта получалась 1Ю3 (I), Ш3 (II) или U03 (III) (см. табл. 87 на стр. 228).
Наиболее устойчивая форма U03 (III) была получена при самых высоких
температурах (700—750°) и давлениях кислорода (70—150 am).
Закись-окись урана удалось превратить в трехокись действием паров
азотной кислоты при 350—400° или, что еще лучше, двуокиси азота (которую
получали пропусканием воздуха или кислорода через азотную кислоту при
65—80°).
Глава И. Окислы, сульфиды и теллур иды урана
257
Таблица 106
ОКИСЛЕНИЕ ЗАКИСИ-ОКИСИ УРАНА КИСЛОРОДОМ
Начальное давление.
am
Температура, °С
Количество U (IV),
окисленного в U (VI)
за 2 часа. %
Влияние давления кислорода при 150?
4,2
6,3
8,4
П,б
23
26
31
38
Влияние температуры при 8,4 am
100
150
200
250
7,5
30
56
76
Окисление двуокиси до трехокиси перекисью водорода изучено
сравнительно подробно. Уравнение свободной энергии процесса
U02 + Н202 ж -> U03 + Н2Ож;
имеет следующий вид:
-Af = 58360-10,15 Г 4 0,465-10"3 Г2+ 0,69-105Г"1+
+ 1,10 Г In Г кал -моль'1. (36)
Значения А/7, вычисленные из этого уравнения, представлены в табл. 107.
Таблица 107
СВОБОДНАЯ ЭНЕРГИЯ ОКИСЛЕНИЯ ДВУОКИСИ УРАНА
ПЕРЕКИСЬЮ ВОДОРОДА ДО ТРЕХОКИСИ
Реакция
ио3 + н2о2ж -> ио3 + н2ож
ио3 + н2о2а, -> 1Юз + н.2ож
и3о8 ^ н.,о2ж -*зио3 + н,ож
и3о8 + н2о2ш7=зио3+н2ож
—Д/\ ккал-моль' 1
300° К
57,5
54,3
51,0
! 47,8
400° К
57,2
51,0
Уравнение
№
(28)
(29)
(30)
(31)
Образование гидратов трехокиси урана окислением двуокиси и закиси-окиси.
В свое время Эбельмен [153J утверждал, что гидрат закиси-окиси
(полученный фотохимическим разложением уранилоксалата) при высушивании на
воздухе превращается в дигидрат трехокиси U03-2H20, однако позже
было показано [53, 120], что этот процесс ведет к образованию
моногидрата U03-H20.
Дигидрат трехокиси урана был получен действием теплой перекиси
водорода на двуокись. При комнатной температуре та же реакция приводила к
образованию моногидрата трехокиси с небольшой примесью дигидрата [195].
17 Химия урана
258
Часть III. Бинарные соединения урана
Дигидрат трехокиси урана получали также нагреванием смеси закиси-
окиси и бертолетовой соли, после чего продукт промывали кипящей водой
[152].
Алюа [53, 142] рекомендует получать моногидрат U03H20 окислением
гидратов закиси-окиси урана на воздухе. В этом случае при комнатной
температуре образуется аморфный моногидрат, а при 100°—кристаллическая форма
(ромбические пластинки и призмы). Можно также пользоваться и чистым дигид-
ратом двуокиси, но он окисляется гораздо медленнее, чем гидрат закиси-окиси.
Получение трехокиси урана разложением дигидрата перекиси урана.
Для приготовления трехокиси и других низших окислов урана в качестве
исходного материала часто пользуются перекисью урана. Гидрат перекиси урана
U04xH20 легко получается осаждением при действии па растворы солей
урана (IV) или уранила перекиси водорода.
Температура раскисления гидрата перекиси зависит не только от давления
кислорода, но также и от давления водяного пара (так как свободная энергия
этого процесса изменяется с изменением содержания воды). Данных по
скорости и температуре разложения много, но они не систематизированы и
результаты различных исследователей полностью не совпадают.
Ряд авторов [159, 224, 225] утверждает, что на воздухе при 100°потери
кислорода не наблюдается даже после многочасового нагревания, однако,
по Хюттигу и Шредеру [6], при давлении водяного пара в 15 мм рт. ст.
половина грамм-атома кислорода теряется при 54°, а вторая половина—при 163°
(см. рис. 42 на стр. 234). По Алибегову [155], разложение начинается в токе
кислорода при 115° и при 140° делается интенсивным.
Брунк [226] не обнаружил потери кислорода при нагревании в токе
углекислоты при температурах до 150°. По Розепгейму [156], нагревание до
187° ведет к полному разложению дигидрата перекиси до оранжевой трехокиси.
Бильц и Мюллер [11 ] приготовили чистую трехокись урана разложением
небольших количеств дигидрата перекиси урана в токе кислорода в течение
0,5—1 часа при 350—400°. При нагревании той же перекиси до 600° в токе
азота или до 800' на воздухе они получили закись-окись урана.
Дигидрат перекиси урана превращается в дигидрат трехокиси при
температуре выше 110' и в красно-оранжевую трехокись при 250°, притом как в
кислороде, так и па воздухе [63]. Твердый дигидрат перекиси [141, 163] начинает
терять кислород в случае парциального давления кислорода, равного 1 атм,
при температуре немного ниже 100°. Разложение заканчивается через 3 часа
при 200°, но составляет только 75% через 13 час. при 143°. На основании
зависимости скорости разложения от температуры вычисленная теплота активации
составляет 28 ккал-моль1.
Образование U03,5 из дигидрата перекиси.
Описывается [39, 161, 227] получение безводной «перекиси» урана, близкой по
составу к 1Ю3,5> нагреванием дигидрата перекиси вначале при 125—130° в течение
24 час. (до превращения около половины U04 в UO.,), с последующим
повышением температуры до 325°. При соприкосновении продукта разложения с водой
или кислотами выделялся кислород. Подобные же перекиси были получены
из ураната аммония (см. стр. 214).
Образование гидрата перекиси урана U04 • *Н20
окислением низших окислов. Описанные выше исследования
Колумбийского университета по разложению и04-л:Н20 в запаянных трубках
[163] указывают па то, что окисление трехокиси кислородом до 1_К},-л;Н20
вряд ли возможно даже при очень высоком давлении кислорода. В некоторых
опытах конечное давление кислорода в трубке при 133° достигало 36 am, но
и это не препятствовало полному разложению дигидрата перекиси.
При действии перекиси водорода на двуокись или закись-окись урана может
образоваться соединение с большим содержанием кислорода, чем в трехокиси.
Глава 11. Окислы, сульфиды и теллуриды урана
259
Опыты по окислению трехокиси перекисью водорода до U04xH20
проводились следующим образом. Твердую трехокись оставляли в нейтральном
растворе с 50 мл 30%-ной перекиси водорода на 12 час. при комнатной температуре;
слой жидкости над осадком сливали и твердый осадок сначала промывали
небольшим количеством воды, а затем спиртом и эфиром. В результате была по-
лучена светложелтая перекись урана [225].
ПЕРЕХОД ДРУГИХ СОЕДИНЕНИЙ УРАНА В ОКИСЛЫ
Превращение соединений трехвалентного
урана в окислы. В рентгенограмме продукта окисления трифторида урана
отмечены линии, характерные для моноокиси [41 ]. Это соединение образовалось
вследствие того, что аппаратура, в которой чистый трифторид нагревали до
1200°, дала течь [41 ]. Та же самая рентгенограмма была ранее отмечена у
продукта, полученного нагреванием трифторида урана до 1000° в течение 7 суток.
Как показал анализ, в этом продукте содержалось 92,8% урана и 0,1% фтора.
Основными компонентами его, повидимому, были двуокись и моноокись.
Гидрид урана сгорает в кислороде или на воздухе с образованием двуокиси
и воды [10]. Он реагирует с жидкой водой, иногда со взрывом, а также с парами
воды, и при этом получается двуокись урана и водород.
Превращение соединений четырехвалентного
урана в двуокись и закись-окись. Переход тетрафторида
урана в свободную от фтора закись-окись описан ниже (см. гл. 12) [40,
228—230].
Если хлориды урана (IV) или такие двойные соли, какЫа2иС16, нагревать
в парах воды, то они подвергаются гидролизу с образованием двуокиси урана.
Для предотвращения окисления последней до закиси-окиси процесс следует
проводить без доступа воздуха. Эги реакции будут рассмотрены в гл. 14,
где описываются свойства хлоридов урана.
Описывается получение двуокиси урана нагреванием сульфата урана
(IV) U(S04)2 в токе водорода, а также нагреванием этого же соединения до 200°
с K2S5 с последующей промывкой продукта реакции водой [55, 127].
Переход соединений шест и валентного урана
в окислы. Переход солей у ранила в двуокись урана. Двуокись урана может
образоваться из солей уранила путем непосредственного термического
разложения их, например
U02Br2 ->U02 + Br2, X37)
путем восстановления, например
U02C12 + Н2 -» U02 + 2HC1, (38)
или путем предварительного разложения до трехокиси с последующим
термическим разложением последней, например
U02C12 + СаО -» U03 + СаС12; (39)
иОя-»иО, + 1оя (40)
(см., однако, выше, на стр. 251, относительно трудности полного
восстановления трехокиси урана до двуокиси).
де-Конинк [88] получил при термическом разложении уранилбромида
на воздухе двуокись урана кирпично-красного цвета. В противоположность
продукту, полученному из уранилхлорида, это вещество не окислялось вновь
на воздухе до закиси-окиси. Нагреванием в водороде кирпично-красный
17*
260
Часть Iff. Бинарные соединения урана
продукт превращался в двуокись черного цвета. Это описание наводит на мысль,
что кирпично-красный окисел был в действительности трехокисью урана, а не
двуокисью.
Двуокись урана получена также восстановлением водородом уранилхло-
рида [88] и уранилсульфата [195], а также нагреванием уранилхлорида с
порошком магния или алюминия [231]. Восстановление магнием описано и
другими исследователями [205]. При использовании в качестве восстановителей
калия и натрия вместе с двуокисью, вероятно, образуются и щелочные ура-
наты [231]. Нагревание уранилхлорида с окисью кальция или гидроокисью
бария без доступа воздуха приводит к образованию двуокиси урана черного
или буровато-красного цвета [231].
Двуокись была получена также прокаливанием 2KC1U02C12 в токе
сухого водорода с последующим выщелачиванием хлорида калия водой [50,
232].
При продолжительном прокаливании уранилсульфида на воздухе
образовались закись-окись урана и сернистый газ [234].
Уранилнитрат в растворе восстанавливался до двуокиси урана уже при
360° путем обработки в течение 5 суток водородом под давлением 50—80 am
[147].
При прокаливании органических солей уранила восстановителем может
быть сам анион органической кислоты. Так, например, сухой перегонкой
уранилформиата U02(COOH)2 получена черная пирофорная двуокись урана
[195, 235]. Этот окисел образовывался и тогда, когда суспензию
уранилформиата в метиловом спирте выдерживали на рассеянном свету в течение трех
месяцев [23], а также в случае прокаливания уранилоксалата без доступа
воздуха [236]. Пелиго [51] и Эбельмен [55] рекомендуют прокаливать ура-
нилоксалат в токе'сухого водорода. Продукт Пелиго вначале был черным,
а затем становился бурым, пирофорным. Продукт Эбельмена был медно-
красным,, блестящим, он имел кристаллическую структуру и тем большую
устойчивость, чем выше была температура его приготовления. Указывается, что
двуокись урана, полученная из оксалата, всегда содержит углерод [237].
Описано [207] получение двуокиси урана нагреванием уранилоксалата в
вакууме или в атмосфере окиси углерода до 275—300°.
Превращение уранатов в двуокись урана. Раствор диураната аммония
в соляной кислоте обрабатывали избытком хлоридов аммония и натрия,
выпаривали досуха и остаток расплавляли [238]. В результате обработки плава водой
получали в остатке черные кристаллы двуокиси урана (о содержании азота в
таких продуктах см. [16, 239]).
Чистая двуокись урана в виде хорошо образованных черных блестящих
кубиков может быть получена сплавлением одной части ураната натрия с
четырьмя частями безводного хлорида магния [54]. Описан технический метод
превращения диураната натрия в двуокись плавлением с хлористым натрием
и древесным углем [240]. При этом методе можно использовать U.?05,
содержащуюся в уранате. Данная реакция находит некоторое применение при
извлечении урана из руд.
Превращение солей уранила в закисъ-окись урана. Все растворимые соли
уранила можно превратить в закись-окись путем термического разложения
диураната аммония; последний в свою очередь получается прямым осаждением
аммиаком или обработкой аммиаком полиураната щелочного металла. Можно
также осадить из растворов гидрат перекиси урана перекисью водорода и
разложить этот осадок при повышенных температурах.
Соли уранила летучих или неустойчивых кислот можно превратить
в закись-окись урана непосредственным нагреванием. Так, например, этот
окисел был получен прокаливанием уранилнитрата в фарфоровом тигле
11871. Ниже описаны различные способы получения закиси-окиси урана.
Глава 11. Окислы, сульфиды и теллуриды урана 261
Чистый уранилнитрат превращали в оранжевую трехокись нагреванием
ниже температуры красного каления и затем в закись-окись повышением
температуры до 700° [241]. Чистый уранилнитрат или оксалат (а также
гидроокись или перекись) превращали в чистую закись-окись разложением,
восстановлением продукта реакции водородом до двуокиси урана и последующим
окислением до закиси-окиси путем нагревания в чистом кислороде [102]. Как
показали опыты [147], нагревание в течение двух суток 10%-ного раствора
уранилнитрата при 360° и начальном давлении водорода, равном 50—80 am,
приводит к образованию черной закиси-окиси (при более продолжительном
нагревании получается двуокись, см. стр. 260).
Описано получение чистой закиси-окиси урана из технического
уранилнитрата экстракцией сухой соли эфиром, содержавшим 5% (объемн.) воды,
и переводом ее в водную фазу последующей многократной промывкой водой.
Этим способом можно получить чистую закись-окись из урановой смоляной
руды, карнотита и смесей, содержащих редкоземельные элементы [242].
По Пелиго [51 ] и де-Конинку [20, 21], при термическом разложении солей ура-
нила образуется вместе с зеленой закисью-окисью урана черный окисел,
принятый ими за U205; это оспаривается [102], и существование U205 является
вообще сомнительным (ср. стр. 210).
Проведен электролиз 10%-ного раствора уранилнитрата [34]. В анодном
пространстве электролизера находился раствор нитрата, в катодном—чистая
вода. После прохождения в течение нескольких часов через этот электролизер
тока 5 мка (800 в) в катодном пространстве образовался желтый, очень чистый
гидрат трехокиси. Нагревание этого продукта на воздухе до 380—390° дало
оранжевую безводную, но очень гигроскопичную трехокись; при нагревании
до более высокой температуры получалась оливковая или черная закись-
окись.
Превращение уранатов в закись-окись урана. Обычно применяемый метод
приготовления или очистки закиси-окиси урана состоит в термическом
разложении диураната аммония (NH4)2U207 (полученного осаждением аммиаком
из раствора уранилнитрата)
9(NH4)2U207 -> 2Nf + 14NH, + 6U&Ot+ 15H20. (41)
Как хороший метод приготовления закиси-окиси рекомендуется процесс
прокаливания диураната аммония на воздухе [135]. Получаемую закись-
окись можно очищать растворением в азотной кислоте, повторным осаждением
аммиаком и разложением чистого диураната аммония [243]. В Браунском
университете [244] диуранат аммония промывали, сушили при 130° на воздухе
и прокаливали в течение 4 час. при 350° для получения закиси-окиси.
Английские исследователи [245,а] осаждали диуранат аммония из раствора
уранилнитрата (предварительно очищенного от железа), промывали горячей водой,
растворяли в соляной кислоте, очищали осаждением примесей сероводородом,
вновь превращали в диуранат аммония, а затем в закись-окись прокаливанием
при 700° в электрической печи. По другому методу [245,6] гексафторид урана
превращали в диуранат аммония следующим путем: вначале добавлением
едкого натра осаждали диуранат натрия, получаемый осадок растворяли
в азотной кислоте и переосаждали аммиаком, после чего прокаливанием
осадка диураната аммония при 600° на воздухе получали закись-окись с общим
выходом 99%.
Превращение солей уранила в трехокись урана U03. Приготовление
трехокиси термическим разложением уранилнитрата наталкивается на трудности.
Действительно, с одной стороны, необходимо поддерживать высокую
температуру, чтобы обеспечить достаточно полное разложение уранилнитрата до
трехокиси, а с другой—температура должна быть низкой для предотвращения
разложения трехокиси до закиси-окиси. Арфведсон [50, 233, 246] утверждал,
262
Часть III. Бинарные соединения урана
что создать такие условия невозможно и что нитрат нельзя полностью
разложить без того, чтобы часть трехокиси не превратилась в закись-окись урана.
Наоборот, Пелиго [247] предполагал, что уранилнитрат можно превратить
в свободную от азота трехокись прокаливанием при 250°. Предполагалась также
температура 400° [248J. Ряд авторов [27, 143, 249] считал, что температура
350° является недостаточной, а 500°—удовлетворительной. Кроме того,
рекомендовалось нагревание соли уранила в токе кислорода для того, чтобы
избежать образования закиси-окиси [143].
Де-Форкран [250] считал целесообразным проводить разложение при
низкой температуре (290—300°), так как, по его мнению, тёмнокрасные окислы,
получаемые при 500—600°, являются «продуктами полимеризации»
трехокиси с неизвестной степенью полимеризации; под этим можно понимать
аллотропические модификации трехокиси (см. выше, стр. 228).
В Колумбийском университете [58] чистую трехокись получали
прокаливанием гидрата уранилнитрата при 300°. Лонг, Джонс и Гордон [68]
нагревали уранилнитрат в течение 8 час. при 300° в закрытой печи для обеспечения
окислительной атмосферы (N02). Продукт был свободен от примесей,
способных восстанавливать перманганат калия, в нем нельзя*было обнаружить
нитрат испытанием с серной кислотой и сульфатом железа (II).
Приготовление желтого гидрата трехокиси из уранил нитрата и его
дегидратация до оранжевой трехокиси урана электролизом с чистой водой в
катодном пространстве по Булле и Домино-Берже [34] описано выше (стр. 261).
Сообщалось [251] о превращении двойного аммонийуранилкарбоната
в трехокись путем продолжительного нагревания при 300°. Трехокись урана,
полученную прокаливанием уранилнитрата при 300° и еще содержащую немного
азота, можно очистить путем превращения ее в аммонийуранилкарбонат и
разложения путем нагревания до 300° на воздухе [226].
Трехокись урана может быть получена нагреванием трехводного ура-
нилсульфата до красного каления [22, 195] или уранилоксалата до 330° на
воздухе [207].
Превращение солей уранила в гидраты трехокиси гидролизом или
электролизом. Гидраты U03-H20 и U03-2H20 (или промежуточные твердые растворы)
можно получить гидролизом или электролизом растворов солей уранила.
Согласно данным, приведенным выше (стр. 234), можно ожидать, что ниже 60°
будет получаться дигидрат 1Ю3-2Н20, а выше 100°—моногидрат;
промежуточные твердые растворы (или полуторный гидрат, если он существует) должны
получаться в промежуточных областях температур. Экспериментальные
результаты являются не очень четкими.
Эбельмен [153] и Дренкман [152] сообщили, что ими был получен дигидрат
трехокиси продолжительным кипячением раствора аммонийуранилкарбоната.
Рибан [144, 145] получил кристаллический моногидрат UO(S-H20
(гексагональные призмы) нагреванием 2%-пого раствора уранилацетата до 175° в откачанной
и запаянной трубке в течение ЮОчас.ПоЦехентеру [146], достаточно нагревания
до 140—150° в течение 8 час. По данным этого автора, при кипячении с
обратным холодильником раствора натрийуранилацетата также образуется
моногидрат U03-H20, немного загрязненный ионами натрия. Продукт состоит из
блестящих серно-желтых гексагональных пластинок.
В связи с этими результатами следует учесть, что гидролиз ураниловых
солей органических кислот может сопровождаться восстановлением и, таким
образом, образованием гидратов двуокиси или закиси-окиси урана, а не
трехокиси (см. стр. 260).
В случае гидролиза ураниловых солей минеральных кислот всегда
образуются гидраты трехокиси. Образование U03H20 впервые наблюдалось при
выпаривании раствора уранилнитрата и нагревании остатка еще в 1843 г. [148].
Позже был получен кристаллический моногидрат в форме прозрачных призм
Глава 11. Окислы, сульфиды и теллуриды урана
263
гидрированием под высоким давлением при 300° раствора уранилнитрата
в азотной кислоте [147].
Моногидрат можно получать и гидролизом твердого гидратированного
уранилнитрата при нагревании на песчаной бане [252]. Осторожным
прокаливанием UOo(NO:,)o-6H20 с последующим промыванием теплой водой и
высушиванием удалось получить чистый оранжевый моногидрат UO.rH20
без следов желтого гидрата UO;»-2H20 [253]. Гидролиз является причиной
того, что при взаимодействии солей уранила с синильной кислотой также
образуется моногидрат трехокиси урана [254].
Как упоминалось выше (стр. 233), Смит показал, что электролитическое
разложение солей уранила в растворе сопровождается образованием гидрата
закиси-окиси урана [132, 133]. Он считал, что вначале получается желтый
моногидрат трехокиси, который затем превращается в черный гидрат окиси
{U.j08-2HX>?). Электролизом раствора уранилнитрата получен моногидрат
и6;,-Н20 вместе с дигидратом U03-2H26 [134]. Пирле получал моногидрат
электролизом раствора уранилацетата при комнатной температуре и
плотности тока 0,1 а\дм1. Черный окисел, полученный при продолжительном
электролизе и принятый Смитом за U308-2HX>, имеет, согласно Пирле
1135], состав изО10'2НаО.
Рядом авторов изучалось электролитическое отложение гидратов закиси-
окиси на урановых электродах [136, 137].
Превращение уранатов в UO:, и UO3.5. Для того чтобы превратить диуранат
аммония в трехокись урана без дальнейшего разложения до закиси-окиси,
необходимо использовать низкие температуры. Э5ельмен 1251] получил
трехокись длительным нагреванием до 300J, однако по Брунку [226] получаемая
при этом трехокись еще содержала азот. Для очистки рекомендуется превратить
ее в аммонийуранилкарбонат и последний разложить (см. выше).
Описывается также [196] нагревание диураната аммония до 230° в течение 16 час,
а затем до 260—370° в течение 20 час. Продукт реакции содержал еще некоторое
количество воды.
Исследования в Браунском университете [162] подтвердили тот факт, что
трехокись урана, приготовленная низкотемпературным разложением
диураната аммония, содержит немного аммиака, который не может быть удален
простым нагреванием без одновременного превращения препарата в закись-окись
урана; поэтому рекомендуется лучше исходить из дигидрата перекиси урана.
Подобные же наблюдения сделаны в Клинтонской лаборатории [255]. Позже
исследователи Браунского университета [244] описали приготовление
трехокиси из диураната аммония высушиванием на воздухе при 130°,
прокаливанием при 350° в течение 4 час. и последующей промывкой водой. Гидратиро-
ванная трехокись, полученная таким образом, затем подвергалась повторному
прокаливанию в течение 4 час. при 350° для получения безводной трехокиси
урана. В радиационной лаборатории Калифорнийского университета [216]
в результате прокаливания диураната аммония получена трехокись урана
светлокрасного цвета. По другому сообщению прокаливание производилось
при 500° [256].
Как указывалось выше (стр. 214), исследователи Браунского университета
[40] установили, что если диуранат аммония нагревать в быстром токе кислорода,
постепенно повышая температуру с 250 до 550°, то после удаления аммиака
и воды (которое происходит при 250—350°) наблюдается образование красного
порошка, устойчивого при высоких температурах. По данным анализов он
имеет состав, промежуточный между 1Юз,м и 1Ю3,з8. Этот продукт является
перекисиым соединением; при действии на него кислоты или воды происходит
выделение «избыточного» кислорода. Возможно, что подобно безводной
перекиси, полученной в Браунском университете из гидрата перекиси, он
представляет смесь U03 и и63>5 (см. стр. 203).
264
Часть III, Бинарные соединения урана
Превращение солей уранила в 1Ю4хН20. Все растворимые соли уранила
превращаются в гидрат перекиси U04 -хН20 взаимодействием с перекисью
водорода. Это же справедливо и для солей четырехвалентного урана (IV), которые,
повидимому, окисляются сначала в соли урана (VI), а затем до перекиси урана.
Эта важная реакция будет рассмотрена во второй книге данного тома.
ПРЕВРАЩЕНИЕ ОКИСЛОВ В ДРУГИЕ СОЕДИНЕНИЯ УРАНА
Превращение окислов урана в галогениды и окси-
г а л о г е н и д ы (ср. гл. 12, 14—16). Фторирование. Двуокись урана.
Двуокись урана может перейти в тетрафторид при действии газообразного
фтористого водорода, фторированных углеводородов (фреонов) и бифторида
аммония. Эти реакции подробно описаны в гл. 12.
Двуокись урана не реагирует с расплавленным фторидом натрия или
бериллия, но взаимодействует с другими фтористыми металлами [257] (см. гл. 12).
Закись-окись урана. Закись-окись урана реагирует с водной плавиковой
кислотой с образованием растворимого уранилфторида и нерастворимого
тетрафторида урана [258],
Трехокись урана. Трехокись урана растворяется в чистом безводном
жидком фтористом водороде, вероятно с образованием уранилфторида [259].
Хлорирование. Двуокись урана. Двуокись урана нерастворима в
разбавленной и концентрированной холодной или горячей соляной кислоте [51, 191,
129,а]. Горячая дымящая соляная кислота медленно переводит двуокись в
зеленый тетрахлорид [54]. Газообразный хлористый водород не действует на
двуокись даже при высоких температурах [51].
Двуокись урана можно хлорировать многими неорганическими или
органическими хлорирующими агентами, например монохлоридом серы [260],
фосгеном, четыреххлористым углеродом и лерхлорпропиленом (более подробное
описание и список литературы см. в гл. 14).
Превращение двуокиси урана в уранилхлорид можно осуществить
посредством хлорирования сухим хлором при температуре красного каления
[51]. Бурая двуокись урана превращается в уранилхлорид также в случае
использования ее в качестве анода при электролизе соляной кислоты [162].
При растворении двуокиси в избытке расплавленного тетрахлорида
урана при 600° в атмосфере сухого азота образуется безводный
уранилхлорид [40].
Закись-окись урана. Этот окисел даже при повышенных температурах
очень медленно реагирует с разбавленной соляной кислотой, особенно после
обжига [129,а, 233]. Взаимодействие с концентрированной соляной кислотой идет
быстрее; нагревание с соляной кислотой (уд. вес 1,135) в атмосфере углекислого
газа в запаянной трубке при 180—200° приводит к быстрому превращению
в зеленую жидкость [102].
В присутствии окислителей, например азотной кислоты, закись-окись
урана довольно быстро растворяется в соляной кислоте с образованием уранил-
хлорида [261].
Черная продажная закись-окись урана реагирует с хлором при 900° с
образованием желтых кристаллов уранилхлорида [262].
Закись-окись урана превращается в тетрахлорид нагреванием до 230—
250° с монохлоридом серы [263]. Описано также получение тетрахлорида
урана и уранилхлорида из закиси-окиси и фосгена при 500° [160]. Нагревание
закиси-окиси с четыреххлористым углеродом в запаянной трубке дает темно-
красные кристаллы пентахлорида урана, которые при дальнейшем
нагревании легко диссоциируют на тетрахлорид и хлор [264]. Для
хлорирования закиси-окиси урана четыреххлористым углеродом предложена
температура 360° [265].
Глава И. Окислы, сульфиды и теллуриды урана 265
Процесс хлорирования закиси-окиси урана рассматривается в гл. 14.
Трехокись урана. Этот окисел приобретает светложелтую окраску в
газообразном хлористом водороде |266]. Он не растворяется в соляной кислоте,
но остаток от обработки растворяется в воде. Трехокись урана не переходит
в тетрахлорид при обработке безводным хлористым водородом вплоть до 400е
[267]. При действии спиртового раствора хлористого водорода получается,
повидимому, уранилхлорид, но не тетрахлорид урана.
По исследованиям, проведенным в Браунском университете [39, 162],
газообразный хлористый водород реагирует с гидратированной трехокисью урана
(или с безводной в присутствии влаги) с образованием гидратированного ура-
нилхлорида.
Реакция трехокиси урана с водной соляной кислотой рассматривается
ниже (см. стр. 268).
Процесс хлорирования монохлоридом серы изучен в Браунском универ-
ситетеи [268]. Реакция может быть выражена уравнением
2U08 f 4S2C12 -* 2UC14 + 5S + 3S02.
Трехокись урана (полученную разложением гидрата перекиси) удалось
[267] превратить в тетрахлорид урана путем обработки тионилхлоридом при
350° за 1 час.
При 8-часовом нагревании трехокиси урана до 370—380° в запаянной
трубке с тетрахлоридом кремния образуются тетрахлорид урана,
уранилхлорид, двуокись кремния и хлор [269].
Квантеиом [270] проведено хлорирование трехокиси урана смесью хлора с
окисью углерода; он применил также для хлорирования трехокиси четырех-
хлористый углерод, для чего нагревал этот окисел до красного каления в
парах СС14. Позже [271] показано, что трехокись урана реагирует с парами
четыреххлористого углерода при 360° с образованием тетра- и пентахлорида
урана; другие авторы [264] отметили образование неустойчивых тёмнокрасных
кристаллов пентахлорида при нагревании в запаянной трубке трехокиси урана
с парами четыреххлористого углерода (в присутствии хлора или без него)..
Хлорирование трехокиси урана четыреххлористым углеродом в жидкой
фазе описывается ниже (см. гл. 14).
Бронирование. Двуокись урана. Согласно имеющимся данным, двуокись
урана, приготовленная восстановлением закиси-окиси водородом при 300 или
900°, не может быть превращена в тетрабромид действием бромистого водорода
(даже при 800°) или метилбромида (при 550°) [272]. Тетрабромид урана получен
при действии паров брома на смесь двуокиси и угля [127].
Закись-окись урана. При нагревании закиси-окиси в парах брома или
в сухом бромистом водороде образуется смесь уранилбромида и низших
окислов [273]. Дальнейшие подробности превращения окислов урана в бромиды
указаны в гл. 15.
Другие реакции. Превращение окислов урана в сульфиды и окси-
сулъфиды. Нагревание двуокиси или закиси-окиси в парах сероуглерода
приводит к образованию оксисульфида U02-2US2[127]. Смесь двуокиси углерода
и сероуглерода не реагирует с двуокисью урана, но восстанавливает закись-
окись и трехокись при температуре красного каления до двуокиси (стр. 252).
В случае взаимодействия двуокиси с сероуглеродом при 900° образуется
оксисульфид U02-2US2 [2741. Результаты исследований условий образования
дисульфида и оксисульфида урана при взаимодействии окислов урана с
сероводородом и последующего разложения дисульфида до полуторного
сульфида см. на стр. 272 [275].
Превращение окислов урана в карбиды. Двуокись урана реагирует с
углеродом в вакууме, притом, повидимому, даже при столь низких температурах^
как 1600°, с образованием карбида урана [276]. По Гринвуду [183], реакция
266
Часть III. Бинарные соединения урана
двуокиси с углеродом начинается в электрической вакуумной печи при
температуре около 1490° (об образовании при этой температуре окиси углерода можно
судить по возрастанию давления).
Условия равновесия системы
UOa + 4C -TUC2 + 2CO (42)
между 1480 и 1800° изучены количественно [277]. Равновеоюе давление окиси
углерода равно 18 мм рт. ст. при 1480' и достигает 1 атм при 1800°. Уравнение
lgPco=-^+12,09 (43)
(гдер—давление, лшрт. ст.) приблизительно верно представляет опытные данные,
но отклонения указывают, что Mi должно заметно изменяться в исследованном
температурном интервале. Среднее значение Д# равно 87,3 ккал-моль1.
Подробности этих реакций см. в гл. 9.
Превращение окислов урана в уранаты и псруранаты при взаимодействии
с окислами, перекисями и солями металлов. В противоположность
двуокиси урана, имеющей основной характер, трехокись является амфэтерным
окислом и даст два ряда солей: уранила U02X2 и уранатов и полиурапатов Х21Ю4,
X2U207 и т. д.; LLOH (или U02-2UO.;) может быть частично превращена в
уранаты, например путем реакции с окислами металлов.
U808 + 2ВаО —> 2BaU04 + UOa. (44)
Окись бария энергично реагирует с закисыо-окисыо урана при
температуре 330° или выше |278); в присутствии окислителей превращение
может дойти до к'жца, например
ЦА + 6КСЮ4 -» 3KaU04 4- ЗС12 + 10 03. (45)
Закись-окись урана реагирует с твердым перхлоратом калия при 390°
с выделением кислорода и хлора [279 — 281J. Трехокись урана дает с
перхлоратом калия уранат калия и свободный хлор [226].
Другая реакция этого же типа состоит в превращении двуокиси, закиси-
окиси и трехокиси урана в уранат натрия действием твердой перекиси
натрия. Об этой реакции упоминалось выше (стр. 213) в связи с
определениями теплот образования различных окислов урана. Согласно Микстеру [1741
при нагревании окислов урана с перекисью натрия в калориметрической
бомбе образуется Na2UOr Измеренные тепловые эффекты составляют*:
Ш2 + Na202 —> NaaU04 +110,9 ккал; (46)
yU308 + Na2Oa-> Na2U04 + lo2 + 82,l ккал; (47)
U03 + Na202 —> Na2U04 +102 + 76,7 ккал. (48)
Результаты опытов показали, что в бомбе получается не перуранат,
а только нормальный уранат. Однако при извлечении продукта реакции
водой он реагирует с избытком перекиси натрия, причем образуется раствор
перураната. При растворении двуокиси или закиси-окиси в водном растворе
перекиси натрия также образуются перуранаты [282].
Изучена растворимость двуокиси урана в водных растворах перекисей
щелочных элементов [40J. Двуокись урана быстро растворяется в них при
* «Экспериментальные» значения Микстера для нейтрализации закиси-окиси и
трехокиси урана окисью и перекисью натрия в соответствующих отношениях вычислены из
предположения, что Na202->Na20 + -() 02—19,4 ккал.
Глава 11. Окислы, сульфиды и теллуриды урана
267
25 — 45°; наиболее эффективно действует смесь теплых 10%-ных растворов
перекиси водорода и карбоната калия. Результаты, полученные при
встряхивании раствора, содержащего 2 г окисла в 100 мл растворителя, в
течение 2 мин., представлены в табл. 108. Теплый (45°) 5%-ный раствор перекиси
натрия является наилучшим растворителем закиси-окиси урана.
Таблица 108
РАСТВОРИМОСТЬ ДВУОКИСИ УРАНА В ПЕРЕКИСЯХ
ЩЕЛОЧНЫХ МЕТАЛЛОВ
Реагент
10% H,O2 + 10%(NH4)2C2O4
10% HaOa + I0%(NH4)2COs
10% Н2Оа + 10% К2С03
10% Н2О2 + 10% К2СОз
5% Na202
Температура,
°С
25
25
25
35
45
Количество
растворенной
ио2. %
35
69
46
83
17
Реакции взаимодействия трехокиси урана с основными окислами
изучены в 1882 г. [283J. Впоследствии исследованы реакции трехокиси урана
с 25 окислами металлов [206J. Эквивалентные количества трехокиси урана
и порошков окислов металлов смешивали и нагревали до 600° дважды по
10 мин. Окислы бериллия, лантана, церия (IV) и молибдена (IV) совсем
не реагировали- С карбонатом лития (или с окисью лития и двуокисью
углерода), окислами серебра, кальция, бария, стронция, магния, цинка,
кадмия, ртути (II), меди (II), свинца (II), кобальта (II), марганца (II),
никеля (II), алюминия, хрома (III), железа (III) и ванадия (III) про.екала
экзотермическая реакция, которая иногда начиналась уже при столь низкой
температуре, как 125° (например, с окисью стронция), а иногда только при
450° (например, с окисью алюминия). Реакции проходили на 10 — 90%.
Ввиду того что при этом расходовались почти точно эквивалентные
количества U03 и Х20, можно было заключить, что при этом образуются только
нормальные уранаты X2U04. Реакцию можно было обнаружить по
изменению цвета. Желтый продукт реакции окиси кальция с трехокисыо урана
выше 505° принимает зеленую окраску и теряет кислород. Окислы железа (II)
и олова (II) при нагревании восстанавливают трехокись урана (см. стр. 251).
Изучена также реакция взаимодействия трехокиси урана с закисью
кобальта при 1100—1300°; при некоторых опытах получен желтый
кристаллический уранат [284].
При растворении трехокиси урана в растворе хлорида калия частично
образуется полиуранат калия вследствие малой растворимости этого
соединения (см. стр. 270).
Дигидрат U03-2H20 реагирует с хлоратом калия с выделением хлора
и образованием диураната калия [280, 281]. Вычисленная Л. В. Писаржев-
ским [285] теплота нейтрализации полугидрата трехокиси урана едким натром
2UOa ■ Н20 + 2NaOH -> Na2U207 + 2H20 : (49)
равна 17,8 ккал. Это значение получено вычитанием измеренной теплоты
разложения диураната натрия серной кислотой на сульфаты натрия
и уранила (44,5 ккал) из суммы теплот нейтрализации 2С03-Н20 и 2NaOH
3 молями серной кислоты \2-15,35 ккал, согласно измерениям Л. В. Писаржев-
ского (см. стр. 268), и 31,4 ккал соответственно].
268
Часть III. Бинарные соединения урана
Превращение окислов урана в если урана (IV) и уранила. Превращение
окислов урана в уранилхлорид и уранилбромид описано выше (см. стр. 264).
Двуокись урана растворяется в азотной кислоте, причем образуется
лимонно-желтый раствор уранилнитрата [50, 51, 191]. Сильно
разбавленная азотная кислота не действует на двуокись, способствуя лишь ее
гидратации [195]. Двуокись урана легко растворяется в концентрированной
азотной кислоте или царской водке [56, 128].
Ее можно также окислить до уранилнитрат! посредством нитрата
серебра [286,287]. Двуокись урана восстанавливает нитрат серебра в
аммиачном растворе до металлического серебра. В нейтральном растворе вначале
осаждается окись серебра, которая затем превращается в металлическое
серебро.
По Сабатье [219], двуокись урана превращается в основной нитрат
при действии двуокиси азота ка холоду (реакция сопровождается
выделением тепла). Однако Кац и Грюн [288] сомневаются в том, действительно
ли образуется основной уранилнитрат.
Закись-окись урана растворяется в азотной кислоте с образованием
уранилнитрата и с выделением окислов азота. Превращение закиси-окиси
урана в уранилнитрат происходит согласно следующему уравнению [198]:
2U308 + 14HNO, -* 6U02(N03)2 + 7H20 + NO + N02. (50)
Но в этом уравнении не учитывается образование комплексных ионов,
в результате чего увеличивается количество окисла, переведенного в раствор
данным количеством азотной кислоты (см. ниже). Прокаленная двуокись
урана только слабо растворима в разбавленной серной кислоте, но легко
растворяется в концентрированных кислотах и полностью — в кипящей
концентрированной серной кислоте [50, 129,а]. Закись-окись урана нацело
превращается в уранилсульфат и сульфат урана (IV) при продолжительном
нагревании с концентрированной серной кислотой [102]. Закись-окись урана
восстанавливает нитрат серебра до металлического серебра, но очень
медленно [287]. Трехокись урана растворяется во всех минеральных
кислотах [ИЗ] с образованием соответствующих солей уранила.
Теплота нейтрализации трехокиси урана разбавленной азотной
кислотой (без учета образования комплексов) по уравнению
U03 + 2H^->UO*^ + H20, (51)
определенная экспериментально, равна 19,80 ккал-г-атолС1 урана (при 18°)
[138, 139].
Найдено, что теплоты нейтрализации U03-H20 и 1Ю3-2Н20 равны
соответственно 14,85 и 12,38 ккол-г-атсм х урана [138, 139]. Эти
значения использованы (см. стр. 235) для расчета теплот образования обоих
гидратов из трехокиси и воды (см. там же табл. 93). Установленная
теплота нейтрализации 1Ю3-Н20 2н. раствором серной кислоты составляет
15,35 ккал-г-атсмГ1 урана [164]. Теплоты нейтрализации моногидрата
0,5н. растворами различных кислот составляют: НС1 — 16,8 ккал\
НВг - 17,6 /c/om;HN08- 16,8 ккал\ H2S04 - 19,0 ккал\ СН3СООН - 15,0 ккал
на 1 г-атом урана [289]. Трехокись урана реагирует с расплавленным
нитратом аммония с образованием уранилнитрата, аммиака и воды [290].
Определение количества трехокиси урана, растворяющегося в известном
количестве соляной кислоты, показало «сверхрастворимость», которую
можно объяснить комплексообразованием (см. табл. 109) [291].
Согласно этим данным, растворимость составляет приблизительно 1 моль
U03 на 1 моль НС1, в то время как по реакции
U03 + 2HC1->U02C12 + Н20
(52)
Глава И. Окислы, сульфиды и теллур иды урана
269
Таблица 109
РАСТВОРИМОСТЬ ТРЕХОКИСИ УРАНА В СОЛЯНОЙ КИСЛОТЕ
НС1, MOAUJA
2,785
1,013
0,2979
0,09909
UO31 moauJa
2,832
0,958
0,2718
0,0900
НС1, моли!а
! 0,03140
0,01082
0,00298
0,00108
1Юз, MOAUJA
0,02986
0,01138
0,00343
0,00150
должно растворяться только 0,5 моля основания на I моль кислоты. При
потенциометрическом или кондуктометрическом титровании трехокиси урана
соляной кислотой наблюдается отчетливая конечная точка при 0,5 моля
основания на 1 моль кислоты, что подтверждает начальное образование
уранилхлорида по уравнению (52). «Сверхрастворимость» трехокиси может
быть обусловлена комплексообразованием с ионами ОН*:
U022+ + U03 + Н20 ^ 21Ю2ОН+ (53)
или образованием «аутокомплексного иона»:
ио;+ + и085-и08-ио;+. (54)
Измерения электропроводности и чисел переноса не дают возможности
различить эти два механизма, но результаты измерения рН указывают на то,
что, повидимому, преобладает второй из них. Растворение U03 в
растворе UO^4-, согласно уравнениям (53) или (54), может, конечно, происходить
не только в рас верах уранилхлорида, но также и в растворах других
солей уранила. Результаты измерений растворимости уранилхлората
представлены в табл. ПО [39, 162].
Табл ица ПО
РАСТЕОРИМОСТЬ ТРЕХОКИСИ УРАНА В PACTEOPAX СОЛЕЙ УРАНИЛА ПРИ 25°
Растворитель
ио2с1а ая
UOa(N03), aq
Эквиваленты
Х- в 1000 г
раствора
0,2887
1,453
1,780
1,927
2,299
2,392
4,004
4,415
4,550
5,747
0,05638
0,1333
0,2878
0,4340
0,7373
1,280
2,058
U03 в 1000 г
рс^твора
0,5272
3,138
4,064
4,121
3,870
3,778
4,058
4,454
4,548
4,086
0,1062
0,2290
0,4688
0,6958
1,187
1,971
2,826
Растворитель
U02(C104)2a?
Эквиваленты
Х-в 1000 г
раствора
2,519
2,892
6,322
0,09787
0,1764
0,4019
0,7899
0,9120
1,187
1,393
1,681
2,058
2,755
3,106
4,334
5,522
U03 в 1000 *
раствора
3,450
2,862
1,769
0,1627
0,2940
0,6390
1,208
1,368
1,717
1,974
2,246
2,568
3,206
3,028
2У088
1,401
270
Часть III. Бинарные соединения урана
Трехокись урана растворима также в растворах солей щелочных
металлов. Механизм этой реакции, возможно, выражается уравнением:
2UO:) + 2КС1 + Н20 -» К.А07 (или Другие полиуранаты) + 2НС1; (55)
2НС1 + U03 -> UOoCla + Н20. (56)
Полиураиат калия выпадает в осадок, в то время как часть трехокиси
урана переходит в раствор в виде урапилхлорида [162]. В табл. 111
представлены данные по растворимости трехокиси урана.
Таблица Ш
РАСТВОРИМОСТЬ ТРЕХОКИСИ УРАНА В РАСТВОРЕ ХЛОРИДА КАЛИЯ
KCI.
моли /л
0,01
0,025
1,0
2,0
3,0
Температура,
°С
Комнатная
»
»
»
»
Растворенная UO:j,
эка{л
0,024
0,021
0,054
0,047
0,020
КС1.
моли 1л
0,362
0,375
0,468
| 0,571
Температура,
°С
80
80
80
80
Растворенная UO3,
же} л
0,036
0,025
0,024
0,028
Одно из измерений дало 0,016 мол. на 1 л 1,0 н. раствора KN03 при
комнатной температуре (после встряхивания в течение 24 час).
СУЛЬФИДЫ УРАНА
СИСТЕМА УРАН—СЕРА
Фазовые отношения в системе уран—сера. Лучше всего
изучен в системе уран- сера дисульфид US2, открытый еще в 1842 г. [51).
Позже был описан метод приготовления U2S3 путем введения трибромида
урана в реакцию с сероводородом, а также метод получения моиосульфида
US восстановлением U2S3 водородом при температуре красного каления, но
правильность второго утверждения оспаривается (см. стр. 272) [155).
Согласно исследованиям, проведенным в последнее время, моиосульфид может
быть приготовлен, например, восстановлением оксисульфида урана UOS
углем при температурах порядка 1900°. Проведено рентгенографическое
исследование образцов сульфида с отношением уран : сера от 1,2 до 1,6 [292].
Образец с отношением S:U^1,5 содержал одну фазу, что подтверждает
существование U2S,r Образец с отношением S : U -= 1,22 дал линии фазы U2S3
и кубической фазы US, что указывает на отсутствие других соединений
между US и U2S3. Пока не выяснено, переходит ли непрерывно
ромбическая фаза U2S3 в ромбическую фазу US2 (II) или же эти два сульфида
разделяются двухфазной областью.
Сульфиды урана (VI) (аналогичные закиси-окиси и трехокиси
урана) неизвестны, но в литературе описан оксисульфид U02S (см. стр. 275).
Физические свойства сульфидов урана. Кристаллическая
структура и плотность. Моносульфид урана. По Алибегову [293J, он
представляет собой черный аморфный порошок, по другим данным -
сероватое, металлоподобпое, твердое вещество. Изучение кристаллической
структуры моносульфида урана показало, что она является кубической гране-
центрированной (типа каменной соли) с четырьмя атомами урана в
элементарной ячейке и изоморфна CeS и ThS (и другим сульфидам) [32, 292,
294]. Константа решетки а = 5,473 ± 0,002 А. Рассчитанная плотность
составляет 10,87 г/см*.
Глава 11. Окислы, сульфиды и тсяяуриды урана
271
^ = 0,125;
z2 = 0,432;
z, = 0,820.
Рентгеновские данные доказывают отсутствие промежуточных фаз
между мопосульфидом и полуторным сульфидом.
Полуторный сульфид урана представляет ссбсй серсвато-чергые
игольчатые кристаллы [ 155] или же чернее кристаллическое вещество [275J.
Исследование кристаллической структуры этого сульфида показало, что фаза V2S3
является ромбической (изоморфной Th2S3 и близкой к Sb2S3 и Bi?S3) [13].
Элементарная ячейка содержит восемь атомов урана и имеет следующие
размеры:
а2= 10,63 + 0,02 А; а, = 10,39 ± 0,02 А; а3 = 3,88 ± 0,01 А.
Дисульфид урана. Он обычно образует черные или серо-стальные
тетрагональные кристаллы с металлическим блеском [188, 295]. Имеются
указания, что помимо тетрагональной формы L'S2 (1) существует вторая,
ромбическая форма US2 (II).
Элементарная ячейка тетрагональной формы US2 (I) имеет следующие
константы [296]:
ах= 10,25 ± 0,05А; а3 -6,30 ± 0,03А.
Если предположить, что элементарная ячейка содержит 10 атомов
урана, рассчитанная плотность будет составлять 7,54 г/см'3.
Ромбическая форма USo (II) изоморфна ThS2. Пространственная
группа Pmnb. Элементарная ячейка содержит четыре атома урана. Атомы
занимают следующие положения:
4U в 4 (с) ^ = 0,250,
4S, в 4 (с) #2 = 0,375,
4Sn в 4 (с) Уъ = 0,465,
Константы решетки:
0,-4,22 +0,02 А; я2 = 7,08 ± 0,04А; а3 = 8,45 ± 0,04А; р = 7,90 г/см3.
Каждый атом >раиа имеет в качестве ближайших соседей девять
атомов серы, среднее расстояние между этими атомами серы и ураном
равно 2,91 А.
Высказано предположение [275], что одна из двух форм дисульфида
(а именно тетрагональная), возможно, является тем промежуточным
соединением между US2 и L'Si.5, которое упоминается ниже (см. стр. 275).
Вследствие аналогии между US и ThS, U2S3 и Th2S3, а также между
ромбической US2 и ThS2 можно предположить, что промежуточный сульфид урана
имеет состав U4S7 аналогично ThaS7. Однако последнее соединение имеет
гексагональную решетку, а «промежуточный»
сульфид урана- тетрагональную.
Температура плавления. Мопосульфид
плавится значительно выше 2000°;
полуторный сульфид плавится при температуре 1850
±100°, а дисульфид - при 1850 ± 200°
[275].
Магнитные свойства. Результаты
определения магнитной восприимчивости
препаратов сульфида урана представлены
в табл. 112 [297]. Эти данные указывают,
что ион U2+ в моносульфиде имеет
конфигурацию 5/2.
Получение сульфидов урана
[155] путем восстановления сульфида
, Таблица 112
МАГНИТНАЯ ВОСПРИИМЧИВОСТЬ
СУЛЬФИДА УРАНА
Формула
соединения
US
U0S3
us2
Молярная магнитная
воспршммпвость на 1 г-атом
урана, CGS
4180
2630
3050
ю-6
J0-8
Мснссульфид урана. Алибегов
водородом при температуре
красного каления получил вещество, которое он принял за моносульфид US.
U2S3
272
Часть III. Бинарные соединения урана
Но позднее в опытах восстановления дисульфида водородом было
установлено, что процесс заканчивается на стадии образования полуторного сульфида,
поэтому выводы Алибегова признаны ошибочными [298j. Попытки
восстановления дисульфида атомарным водородом в кварцевых трубках при
температуре 200 и 700° были безуспешны [299].
Моиосульфид получен восстановлением оксисульфида урана углем при
1900° в вакууме [300J.
UOSf C->US + CO. (57)
Можно предполагать, что равновесие реакции
UO + CS^US + CO (58)
смещено в сторону образования сульфида урана.
Для получения моносульфида урана предложены два следующих
удобных метода [275, 301J.
Первый метод состоит в обработке тонкодисперсного порошка
металлического урана (полученного разложением гидрида урана) рассчитанным
количеством сероводорода. Гидрид разлагают при температуре 300°, а остаток
обрабатывают сероводородом при 400 - 500°. Циркуляцию сероводорода
осуществляют до тех пор, пока не прореагирует все взятое количество
урана. Вначале продукт не является вполне однородным, так как содержит
высшие сульфиды и непрореагировавший металл, но его можно нагреть
до высокой температуры для гомогенизации (моносульфид не разлагается
при нагревании выше 1800°).
Вторым методом является обработка дисульфида урана рассчитанным
количеством гидрида. Процесс аналогичен вышеописанному, т. е. он состоит
в разложении гидрида при 300°, нагревании до 400 — 600° для
восстановления дисульфида и последующем нагревании до высоких температур для
гомогенизации продукта.
Моносульфид может быть также получен разложением полуторного
сульфида в высоком вакууме при 1800° [293].
U2S3. Алибегов [293] приготовил U2S3 в виде серовато-черных игл
путем 8 - 10-часового нагревания трибромида урана в токе сероводорода в
условиях полного отсутствия воздуха. В результате восстановления
дисульфида водородом, свободным от кислорода, при температуре красного
каления образуется серовато-черный пирофорный порошок, анализ которого
(83, 2% U) показывает, что его состав ближе к U2S3 (82, 6% U), чем к
моносульфиду (88, 4% U) [298].
Для приготовления полуторного сульфида разработаны три метода [275,
302]; два из них идентичны методам, описанным выше для моносульфида.
1. Введение в реакцию урана (из гидрида) с рассчитанным
количеством сероводорода.
2. Введение в реакцию урана (из гидрида) с рассчитанным количеством
дисульфида. Вследствие неустойчивости полуторного сульфида при высоких
температурах конечная гомогенизация продукта должна проводиться путем
продолжительного нагревания при температурах гораздо ниже 1800°.
3. Термическое разложение дисульфида (который может быть получен
восстановлением окислов урана углем в присутствии сероводорода; ср.
ниже). В одном из опытов дисульфид полностью разложился до U2S3
за 20 мин. при температуре 1600° под давлением около 1 • 10~4 мм рт. ст.
Однако, поскольку последний сам сравнительно легко разлагается в
вакууме (он может быть превращен в высоком вакууме при 1800° в моносульфид),
этот метод меньше подходит для приготовления продукта с составом, точно
отвечающим формуле и.Д3, чем первые два. С другой стороны, однако, продукт,
полученный по высокотемпературному методу, состоит из больших кристаллов
Глава 11. Окислы, сульфиды и теллуриды урана
273
и значительно более устойчив на воздухе, чем продукты обоих
низкотемпературных методов—первого и второго. Низкотемпературные препараты
необходимо получать и хранить в атмосфере инертного газа.
USii5<x<2- Сульфиды с составом, промежуточным между USi,5 (или U2S3)
и US2, получены тремя методами, описанными выше. Из них наиболее легким
является метод термического разложения дисульфида, пригодный, правда,
лишь для получения продуктов, которые не должны обладать особенно точным
составом [275, 302].
US2. Уже давно известно, что уран горит в кипящей сере, образуя
дисульфид [303]. Однако этот метод нельзя считать удобным для приготовления
дисульфида. Другой метод основан на реакции тетрахлорида урана с
сероводородом при температуре красного каления [127]:
UCI4 + 2H2S]-^ US2 + 4HC1. (59)
Предложено также [188, 295] использовать вместо тетрахлорида менее летучее
соединение 2NaCl • UC14, а вместо сероводорода—водород, смешанный с парами
серы (ввиду того что сероводород трудно осушить). Реакция должна
проводиться без доступа воздуха. Она начинается уже при температуре 500°. Продукт
промывают водой, не содержащей воздуха, спиртом и эфиром и высушивают
в вакууме. Дисульфид может быть также получен путем взаимодействия
2NaCl-UCl4 с сульфидами натрия, магния, алюминия, сурьмы и, что еще
лучше, с сульфидом олова при высоких температурах. Но все эти методы нельзя
рекомендовать, так как конечный продукт бывает загрязнен следами воды.
Кроме того, известно еще несколько методов приготовления дисульфида
урана. Так, он был получен действием сероводорода на гидрид урана (или,
точнее, на тонкодисперсный уран, полученный разложением гидрида [304]).
Дисульфид приготовляли еще из закиси-окиси урана, которую
предварительно превращали в тетрахлорид. После возгонки последний превращали в той же
реакционной трубке в дисульфид действием чистого сероводорода. Продукт
реакции представлял собой черную массу [298].
В университете в Беркли [275, 301] пользовались двумя методами
получения дисульфида: вышеупомянутым, т. е. из гидрида урана, и методом
восстановления двуокиси или закиси-окиси урана углем в присутствии
сероводорода. Последняя реакция протекает довольно быстро при 1200—1300°.
Вначале получается оксисульфид (ср. стр. 274), но когда температура достигает
максимума, оксисульфид под действием сероуглерода (образовавшегося из
сероводорода и углерода) восстанавливается до дисульфида урана.
При температурах, используемых в конечной стадии процесса, может
произойти частичное термическое разложение дисульфида урана. Поэтому для
предотвращения загрязнения дисульфида низшими сульфидами температура
реакции должна быть ниже 1000°, в этих условиях низшие сульфиды
присоединяют серу из циркулирующего сероводорода.
Если имеется чистый металлический уран, то гидридпый метод синтеза
дисульфида превосходит все другие вследствие его простоты. Однако и здесь,
как и в случае полуторного сульфида, продукт, полученный при
высокотемпературном процессе, устойчивее полученного из гидрида в условиях более низких
температур.
Химические свойства сульфидов урана.
Моносульфид урана. Алибегов [293] сообщает, что полученный им моносульфид почти
не реагировал с концентрированной соляной или разбавленной азотной
кислотой и слабо растворялся в растворе соляной кислоты, содержащей бром.
Дымящая азотная кислота окисляет моносульфид, который при этом
воспламеняется; царская водка реагирует менее энергично. Но, как уже упоминалось
выше, весьма вероятно, что Алибегов получил не моносульфид, а полуторный
■18 Химия урана.
274
Часть III. Бинарные соединения урана
сульфид (ср. стр. 271). Согласно данным университета в Беркли, моносульфид
очень медленно растворяется в разбавленных кислотах.
Полуторный сульфид. Этот сульфид часто получается пирофорным, на
воздухе он разлагается [293, 298]. В присутствии влаги выделяется сероводород.
Другие свойства полуторного сульфида, по Алибегову, подобны описанным выше
для моносульфида [293]. По сообщению этого же автора, U2S3
восстанавливается водородом при температуре красного каления до моносульфида. Позже
показано, что давление разложения полуторного сульфида при температуре
выше 1800° достаточно высоко для возможности превращения последнего в
моносульфид путем нагревания в вакууме [275].
Дисульфид урана. Во влажном воздухе дисульфид очень медленно
окисляется, при этом выделяется сероводород и образуется основной сульфат урана
желтоватого цвета [127]. Дисульфид урана очень медленно вступает во
взаимодействие с холодной водой, но быстро разлагается парами воды [127, 188, 295].
Разбавленная соляная кислота действует на него очень медленно,
концентрированная—быстрее, особенно при нагревании; азотная кислота реагирует очень
быстро. В вакууме при температуре 1300° дисульфид диссоциирует на серу
и низший сульфид [305].
Дисульфид, приготовленный из урановых стружек через гидрид,
реагирует с бромом при 300° с образованием монобромида серы. После нагревания
до 600° был получен расплавленный тетрабромид урана [304].
Оксисульфиды урана. В старой литературе описаны два окси-
сульфида урана: U302S4 (или U02-2US2) и U02S (или 2U03-US3). В
университете в Беркли, однако, высказано сомнение в существовании первого из этих
оксисульфидов. В этом же университете открыто третье соединение UOS (или
U02 • US2).
Оксисульфид урана (IV) UOS. Впервые это соединение получено
нагреванием двуокиси урана в атмосфере сероводорода и водорода при 1000 —
1200° [301, 306]. Во время этого процесса из сероводорода и двуокиси
урана образуется небольшое количество воды, которого, однако,
достаточно для предотвращения образования дисульфида урана. Позже показано
[275], что удобным способом получения является нагревание двуокиси или
закиси-окиси урана с углеродом в токе сероводорода. Для
предотвращения образования дисульфида необходимо ограничить температуру и
продолжительность восстановления.
Оксисульфид UOS имеет сине-черную окраску. Он обладает
тетрагональной структурой, содержит две молекулы в элементарной ячейке
(типа PbFCl), изоморфен ThOS, пространственная группа PJnmm [32, 307]:
2U в (OO^y(yy^i); *i = 0,200
20 в (<>у 0)(у00)
2S в (00х2) (}ух21; х2 = 0,65.
Константы решетки составляют:
аг = 3,835 ± 0,002 А; а3 = 6,682 ± 0,002 А.
Расстояние уран — кислород равно 2,34А, расстояние уран—сера
составляет 2,93А. Рассчитанная плотность 9,60 г/см3.
Оксисульфид UOS растворим в концентрированной серной кислоте; он
реагирует с бромной водой с образованием ураната и сульфата [275, 306].
Оксисульфид U02-2US2 открыт Розе [308], впервые анализирован
Германом [127]. Он описан как вещество цвета от темного свинцово-серого
до черного [127]. Оксисульфид получен путем нагревания закиси-окиси или
Глава 11. Окислы, сульфиды и теллур иды урана
275
двуокиси урана или ураната аммония в парах сероуглерода. Установлено
[309], что рентгенограмма порошка продукта, полученного из двуокиси
урана и сероуглерода, отличается от рентгенограмм двуокиси и дисульфида.
Обнаруженная фаза (U02-2US2?) сложна, и ее структура еще не определена.
Это соединение загорается в хлоре, разлагается дымящей азотной
кислотой и медленно окисляется хлорной водой; оно легко разлагается
концентрированной соляной кислотой [127].
Ураиилсульфиди02Б(или 2U03-US3). Это соединение впервые получено
осаждением солей UOg+ сульфидом аммония [127, 310]. Осадок на воздухе
постепенно превращается в сложные, интенсивно окрашенные продукты,
известные под названием урановых красных (ср. гл. об уранатах во
второй книге этого тома). Чистый кристаллический уранилсульфид получен
путем взаимодействия роданида калия (12 частей) с двуокисью урана
(3 части) и серой (5 частей) в виде черных тетрагональных игольчатых
кристаллов [234]. При прокаливании на воздухе он превращается в закись-
окись урана с выделением сернистого газа. Царская водка или азотная
кислота быстро его окисляют. Соляная кислота не действует на
уранилсульфид при комнатной температуре, но разлагает его при нагревании.
СЕЛЕНИДЫ И ТЕЛЛУРИДЫ УРАНА
С е л е и и д ы. Селенид урана (IV) USe2 получен нагреванием двойной
соли 2NaCl-UCl4 в водороде, насыщенном парами селена при умеренном
красном калении [311, 312]. Он также образуется при взаимодействии SnSe
с 2NaCbUCl4 в токе сухого водорода. Селенид урана представляет
небольшие, хрупкие, сильно преломляющие черные кристаллы и часто бывает
пирофорным. Химические свойства диселенида урана подобны свойствам
дисульфида, но он легче окисляется; при обработке азотной кислотой
воспламеняется. Если во время процесса приготовления селенида берется слишком
мало селена, а температура достигает 1000°, то образуется U2Se3.
Селенид уранила получен методом, аналогичным использованному для
приготовления уранилсульфида, а именно: прокаливанием 1 части закиси-
окиси урана с 5 частями цианида калия и 7 частями селена в двойном
тигле при температуре яркокрасного каления [313]. Продукт реакции после
промывания водой, спиртом и последующего высушивания при 98°
представляет небольшие, черные, шестигранные призмы с красноватым оттенком и
металлическим блеском. Он медленно разлагается водой с образованием
красного раствора. Не реагирует с разбавленными щелочами, легко растворим в
холодной НС1 (с образованием уранилхлорида) и бурно реагирует с азотной
кислотой, причем вначале получается селен, который затем окисляется.
Т е л л у р и д ы. Теллурид урана приблизительного состава UTe. был
получен нагреванием 2NaC\'\JC\l до 1000° в токе сухого водорода,
насыщенного парами теллура [311, 314].
Второй теллурид урана U2Te:J получен нагреванием до 1000° 2NaCbUCl4
с теллуридом натрия, содержащим избыток теллура. Он образует черные
пластинки с металлическим блеском. Предполагаемое соединение UTe9
получить не удалось.
В литературе имеется описание теллурида урана с приблизительным
составом UTe.2,o [315], полученного сплавлением смеси металлического
теллура, трехокиси урана и цианида калия при температуре, красного каления.
Это вещество нерастворимо в обычных растворителях, реагирует с азотной
кислотой с образованием уранилнитрата, при прокаливании на воздухе
превращается в трехокись. Он реагирует с бромом с образованием бромидов; при
сплавлении его с карбонатом калия получаются теллурат и уранат калия.
Системы уран—селен и уран—теллур требуют дальнейшего изучения.
18*
576
Часть III. Бинарные соединения урана
ЛИТЕРАТУРА
1. Gleditsch E., Nature, 28, 127 (1940).
2. Fair ley Т., J. Chem. Soc., 31, 133 (1877).
3. Писаржевский Л., Z. physik. Chem., 43, 161 (1903); ЖРФХО, 35, 44 (1903).
4. Rosenheim A., D a e h r H., Z. anorg. u. allgem. Chem., 208, 81 (1932).
5. Riesenfeld E. H., Mau W., Ber., 44, 3589 (1911).
6. H u t t i g G. F., Schroeder E., Z. anorg. u. allgem. Chem., 121, 243
(1922).
7. Cleaves H. E., Holm, Kimble, Report CT-1696, Apr. 1944 (Natl. Bur.
Standards 1).
8. Cleaves H. E., Cron M. M., Sterling J. Т., Report CT-2618, Feb.
1945 (Natl. Bur. Standards 2).
9. С 1 e a v e s H. E., Report CT-1819, July 6, 1944 (Natl. Bur. Standards 3).
10. R u n d 1 e R. E., В a e n z i g e r N. C, Wilson A. S., X-ray study of the
Uranium-Oxygen System, in National Nuclear Energy Series, Division VIII, vol. 6;
J. Am. Chem. Soc, 70, 99—105 (1948) (MP Ames 1).
11. В i 1 t z W., Miiller H., Z. anorg. u. allgem. Chem., 163, 279 (1927).
12. В i 1 t z W., Miiller H., Z. anorg. u. allgem. Chem., 163, 295 (1927).
13. Zachariasen W. H., Report N-1973, Apr. 24, 1945 (MP Chicago 1).
14. Brewer L., Bromley L. A., Gilles P. W., Lofgren N. L., The
Thermodynamic Properties and Equilibria at High Temperatures of Uranium Hali-
des, Oxides, Nitrides, and Carbides, in National Nuclear Energy Series,
Division VIII, vol. 6 (MP Berkeley 1).
15. Hiittig G. F., Fortschr. chem. Physik u. physik Chem., 18 (1), 12, (1924).
16. H i 1 1 e b r a n d W. F., Z. anorg. Chem., 3, 243 (1893).
17. J о 1 i b о i s P., Compt. rend., 224, 1395 (1947).
18. G r 0 n v о 1 d F., H a r a 1 d s e п Н., Nature, 162, 69 (1948).
19. R u n d 1 e R. E., В a e n z i g e r N. C, Report CC-1980, Nov. 23, 1944 (MP
Ames 2).
20. de Coninck W., Bull, classe sci. Acad. roy. Belg., 226 (1901).
21. de Coninck W., Ann. chim. et phys., [7] 28, 6 (1903).
22. de Coninck W., С a m о М., Bull, classe sci. Acad. roy. Belg., 321 (1901).
23. de Coninck \V., Raynaud A., Bull. soc. chim. France, 11, 1037 (1912).
24. Schwa rz R., Helv. Chim. Acta, 3, 345 (1920); Abbegg R., Handbuch der
anorg. Chemie, vol. IV, 1, 2d part, p. 918, Hirzel S., Leipzig, 1921 (1920).
25. Lyden R., Finska Kemiststamf. Medd., 48, 124 (1939).
26. Lebeau P., Compt. rend., 174, 388 (1922).
27. J о 1 i b о i s P., В о s s u e t R., Compt. rend., 174, 386 (1922).
28. В i 1 t z W., Miiller H., Z. anorg. u. allgem. Chem., 163, 263 (1927).
29. Wagner C, S с h о t t k у W., Z. physik. Chem., (B) 11, 163 (1931).
30. В i 1 t z W., Miiller H., Z. anorg. u. allgem. Chem., 163, 293 (1927).
31. R u n d 1 e R. E., В a e n z i g e r N. C, Wilson A. S., McDonald R. A.,
Report CC-2397, Feb. 17, 1945, p. 35 (MP Ames 3).
32. Zachariasen W. H., Reports CK-2667, Jan. 15, 1945; CC-2768, Mar. 12,
1945 (MP Chicago 2).
33. F r i e d S., Davidson N. R., The Ignition of U308 in Oxygen at High
Pressures and the Crystallization of U03, in National Nuclear Energy Series,
Division VIII, vol. 6 (MP Chicago 3).
34. В о u 1 1 ё A., Dornine-Berges M., Compt. rend., 227, 1365 (1948).
35. В i 1 t z W., Miiller H., Z. anorg. u. allgem. Chem., 163, 291 (1927).
36. В i 1 t z W., M u 1 1 e r H., Z. anorg. u. allgem. Chem., 163, 266 (1927).
37. A n d er s о n J. S., Proc. Roy. Soc. London, A185, 69 (1946).
38. Boull e A., D о m i n e - В e r g ё s M., Compt. rend., 228, 72 (1949).
39. Kraus С A., Report A-281, Sept. 7, 1942 (Brown 1).
40. Kraus С A., Report A-1096, July 29, 1944 (Brown 2).
41. Bunn С W.f Sp епсег-Pal mer H. J., Report BR-502, Sept. 7, 1944
(British 1).
42. Battelle Memorial Institute, Report CT-2632, Jan. 1, 1945 (Battelle 1).
43. R u n d 1 e R. E., В a e n z i g e r N. С, С h i о t t i P., Report CC-1984, Nov. 10,
1944 (MP Ames 10).
44. G о 1 d s с h m i d t V. M., ThomassenL., Videnskapsselskapets-Skrifter, (I) 2, 12
(1923).
45. van А г k e 1 A. E., Physica, 4, 297 (1924).
46. R о s e n b a u m В., Ped erza ni G., Report CP-1168, May 25, 1943 (MP Chicago 4).
•47. В a e n z i g e r N. C, Report CC-1781, Aug. 10, 1944 (MP Ames 4).
48. К i r s с h e n b a u m J., Summary Report SAM Laboratory. Work by G r e n a 1 1 A.,
Cohen B.,Ostrovsky В., Palter R. (SAM Columbia 1).
Глава П. Окислы, сульфиды и теллур иды урана
277
49. R ifn d I e R. Е., В а е n z i g е г N. С, W i 1 s о n A. S., М с D о n a i d R. A.f
J. Am. Chem. Soc., 70, 99—105 (1948).
50. А г f v e d s о n J. A., Pogg. Ann., 1, 250 (1824).
51. P ё 1 i g о t E., Ann. chim. et phys., 5, 24; Ann., 43, 268 (1842).
52. H i 1 1 e b r a n d W. F., Z. anorg. Chem., 3, 249, (1893); U. S. Geol. Survey Bull..
113, 41 (1893).
53. A 1 о у J., Bull. soc. chim. Paris, [3] 23, 368 (1900).
54. H о f m a n n К. А., Н б s с h e 1 e K., Ber., 48, 21 (1815).
55. E b e 1 m e n J. J., Ann. chim. et phys., 5, 195 (1842); Ann., 43, 290 (1842).
56. Raynaud A., Bull. soc. chim. France, 11, 802 (1912).
57. J e n к i n s F. A., Report RL-4.6.47, Mar. 9, 1943 (UCRL 1).
58. Priest H. F., P г i e s t G. L., Report A-257, Aug. 10, 1942 (SAM Columbia 2).
59. S p e d d i n g F. H., Report CP-42, Apr. 25, 1942 (MP Ames 5).
60. С h i о t t i P., Report CT-1985, Jan. 4, 1945 (MP Ames 6).
61. Kirshenbaum I., Summary Report SAM Laboratory, Sec. 8, Work by H i s-
k e у С. F., E i d i n о f f M. L.Mallan J., N a d e 1 h a f t J., Report A-777,
Aug. 14, 1943 (SAM Columbia 3).
62. S с h m i d t O., Z. physik. Chem., 133, 263, 289 (1928).
63. Urey H. С and coworkers, Report A-743, June 30, 1943 (SAM Columbia 4).
64. Ruff O., G oec к e O., Z. angew. Chem., 24, 1459—1461 (1911).
65. Friederich E., Sittig L., Z. anorg. u. allgem. Chem., 145, 127, 138 (1925).
66. R e h n J., С e f о 1 a R., Report CK-1240, Jan. 19, 1944 (MP Chicago 5).
67. R e g n a u 1 t V., Ann. chim. et Phys., 73, 50 (1840); Pogg. Ann., 51, 225 (1840).
68. L о n g E. A., J о n e s W. M., G о г d о n J., Report A-329, Oct. 28, 1942 (U. S.
Bur. Mines 1).
69. Moore G. E., L о n g E. A., Report A-502, Dec. 31, 1942 (U. S. Bur. Mines 2).
70. Moore G. E., Kelley K. K., J. Am. Chem. Soc, 69, 2105—2107 (1947).
71. M а с W о о d G. E., A 1 t m a n D., Report RL-4.7.600 (UCRL 2).
72. Snyder Т. М., К a m m R. L., Reports CP-76, May 16, 1942; CP-92, May 23, 1942
(Princeton 1).
73. S n у d e г Т. M., Ka m m R. L., Report CT-96 (Princeton 2).
74. S n у d e r T. M., К a m m R. L., Report CP-124, June 13, 1942 (Princeton 3).
75. LeBIanc M.,Sachse H., Ber. Verhandl. sachs, Akad. Wiss. Leipzig, 82, 155
(1930).
76. H а г t m a n n W., Z. Physik, 102, 709 (1936).
77. Meyer W., Z. tech. Physik, 14, 126 (1933); Z. Physik, 85, 287(1933).
78. A m г e i n W., Schweiz. Arch, angew. Wiss. u. Tech. , 8, 85, 109 (1942).
79. P г i g e n t J., J. phys., radium, 10, 58—64 (1949).
80. W a h 1 i n H. В., Phys. Rev., 39, 183 (1932).
81. P oc h e t t i n о A., Atti accad. nazl. Lincei, 15, 505 (1932).
82. Wed ek i n d E., H ors t C, Ber., 48, 111 (1915).
83. Meyer St., Wied. Ann., 69, 245 (1899).
84. S uc k sm i t h W., Phil. Mag., 14, 1122 (1932).
85. Нага Id sen H., Bakken R., Naturwissenschaften, 28, 127 (1940).
86. Le'beau P., Compt. rend., 174, 390 (1922).
87. В i 1 t z W., M u 1 1 e г Н., Z. anorg. u. allgem. Chem., 163, 261 (1927).
88. de С о n i n с k W. O., Compt. rend., 135, 900 (1902); Bull, classe sci. Acad. roy.
Belg., 1025 (1902).
89. de Co n i n с k W. O., Bull, classe sci. Acad. roy. Belg., 448 (1904).
90. Run die R. E.,Baenziger N. C, Report CC-1524, Mar. 10, 1944 (MP Ames 7).
91. Z а с h а г i a s e n W. H., Reports CP-1249, Jan. 22, 1944; CK-1367, Feb. 25, 1944
(MP Chicago 6).
92. G г 0 n v о 1 d F., Nature, 162, 70 (1948).
93. D о n a t h J., Ber., 12, 743 (1879).
94. R u s s e 1 1 A. S., Physik. Z., 13, 61 (1912).
95. E g g 1 e s t о n, reported by Snyder Т. М., Report CP-36, Apr. 17, 1942 (Natl.
Bur. Standards 4).
96. К e 1 1 e у К. К., U. S. Bur. Mines Bull., 350, 47 (1932).
97. Davidson N. R., Report N-1617, Sept. 18, 1944 (MP Chicago 7).
98. Bid well С. С, Phys. Rev., 3, 204 (1914).
99. Keller F., Lehmann W. R., Z. Physik, 88, 682 (1934).
100. Fischer A., Z. anorg. Chem., 81, 201 (1913).
101. W i e g a n d E., Z. Physik, 30, 40 (1924).
102. Zimmermann C, Alibegoff G., Kruss G., Ann., 232, 283 (1886).
103. H u g g i n s W., Proc. Roy. Soc. London, 18, 548 (1870); Phil. Mag., 40, 302 (1870).
104. С о b 1 e n t z W. W., Bull. Bur. Stand., 5, 173 (1908).
105. В u r g e s s G. K., W a I t e n b u г g R. G., J. Wash. Acad. Sci., 4, 567 (1914).
106. Burgess G. K., Waltenburg R. G., Bull. Bull. Stand., 11, 591 (1915).
107. Nichols E. L.Howes H. L., Phys. Rev., 19, 300 (1922).
278
Часть III. Бинарные соединения урана
108. Р h i l i p p s M. L., Phys. Rev., 32, 832 (1928).
109. GoldschmidtV. M., Thomassen L., Videnskapsselskapets-Skrifter, (I)
2, 16 (1923).
110. Report [B] LRG-42, March 1945 (British 2).
111. Fried S., Davidson N. R., Reports CN-2689, Mar. 3, 1945; CN-3053, June 28,
1945 (MP Chicago 8).
112. Zachariasen W. H., Acta Crystallographies, 1, 265 (1948).
113. Gates J. W.,Andrews L. J., P i t t R. В., Report CD-495, Jan. 16, 1945
(CEW-TEC 1).
114. Beck G., Z. anorg. u. allgem. Chem., 174, 40 (1928).
115. Schroeder E., Dissertation, University of Gottingen (1922).
116. Guillery P., Ann. phys., 14, 218 (1932).
117. van Wazer J. R., Report CD-462, Aug. 28, 1944 (CEW-TEC 2).
118. T i 1 к W., К 1 e m m W., Z. anorg. u. allgem. Chem., 240, 355 (1939).
119. Z i m m e г m a n n C, Ann., 216, 12 (1882).
120. Al oy J., Recherches sur Turanium et ses composes, Theses Toulouse, No. 21, 23(1901)
121. A 1 о у J., R о d i e г Е., Bull. soc. chim. France, 31, 249 (1922).
122. R о w e 1 1 S. W., R u s s e 1 1 A. S., J. Chem. Soc., 127, 2901 (1925).
123. A 1 oy J., Bull. soc. chim. Paris, [3] 21, 613 (1899).
124. H i s к е у С. F., Report A-123, Aug. 20, 1943 (SAM Columbia 5).
125. Christ R. H., Report A-146, Sept. 4, 1943 (SAM Columbia 6).
126. Rammelsberg C, Pogg. Ann., 59, 12 (1843).
127. Hermann H., Dissertati6n, University of Gottingen, p. 31 (1861).
128. Raynaud A., Compt. rend., 153, 1480 (1911).
129. a) Arfvedson J. A., Kgl. Svenska Vetenskapsakad. Handl., p. 413 (1822);
6) Eb el men J. J., Ann., 43, 295 (1842).
130. A 1 о у J., R о d i e г Е., Bull. soc. chim. France, 26, 101 (1920).
131. S t a e h 1 i n g H., Compt. rend., 173, 1470 (1921).
132. Smith E. F., Am. Chem. J., 1, 329 (1879).
133. Smith E. F., Ber., 13, 751 (1880).
134. de С о n i n с к W„Camo M., Bull, classe sci. Acad: roy. Belg., 332 (1901).
135. Pierle C. A., J. Phys. Chem., 23, 549 (1919).
136. Francis M., Tscheng-Da-Tschang, Compt. rend., 200, 1024 (1935).
137. Francis M., Compt. rend., 201, 473 (1935).
138. de F о г с г а п d R., Compt. rend., 156, 1956 (1913).
139. de F о г с г a n d R., Ann. chim., 3, 36 (1915).
140. H u t t i g G. F., Z. angew. Chem., 35, 391 (1922).
141. V i e г D, Т., Report A-1277, May 26, 1944 (SAM Columbia 7).
142. Al oy J., Ann. chim. et phys., [7] 24, 418 (1901).
143. Lebeau P., Compt. rend., 154, 1808 (1912); Bull. soc. chim. France, 11, 800 (1912).
144. Riban J., Compt. rend., 93, 1141 (1881).
145. Riban J., Bull. soc. chim. Paris, 38, 157 (1882).
146. Zehenter J., Monatsh., 21, 241 (1900).
147. Муромцев Б., Ber., 63, 164 (1930).
148. Malaguti F. I., Ann. chim. et phys., 9, 463 (1843).
149. V i e г D. TM Schultz M. L., В i g e 1 e i s e n J., J. Optical Soc. Am., 38,
811—814 (1948).
150. V i e г D. Т., S с h u 1 t z M. L, В i gel ei se n J., Report A-2176, Dec. 21, 1944
(SAM Columbia 8).
151. Polissar M. J., Kilner S. В., Report RL-4.6.42, Nov. 7, 1942 (UCRL4).
152. Drenckmann В., Z. ges. Naturw., 17, 131 (1861).
153. E b e 1 m e n J.. J., Ann., 43, 294 (1842); Ann. chim. et phys., 5, 198 (1842).
154. E i d i n of f M. L., Report CC-1536, Apr. 6, 1944 (SAM Columbia 9).
155. A 1 i b e g о f f G., Ann., 233, 123 (1886).
156. Rosenheim A., D a e h r H., Z. anorg. u. allgem. Chem., 181, 181 (1929).
157. H i s к е у С. F., Report CC-1198, Jan. 5, 1944; H i s к е у С. F., E i d i n о f f
M. L., Report CC-1383, Feb. 28, 1944 (SAM Columbia 10).
158. Hamaker J. W., К о с h С W., Note on the Composition ot Uranium Peroxides,
in National Nuclear Energy Series, Division VIII, vol. 6 (MP Berkeley 2).
159. Rosenheim A., D a e h г Н., Z. anorg. u. allgem. Chem, 181, 178 (1929)
160. R о s e n h e i m А., К e 1 m у M., Z. anorg. u. allgem. Chem., 206, 31 (1932).
161. К r a u s С A., Report A-328, Oct. 15, 1942 (Brown 3).
162. Kr a u s С A., Report A-360, Oct. 26, 1942 (Brown 4).
163. Kirshenbaum I., Summary Report SAM Laboratory, part 11 (SAM Columbia 11).
164. Писаржевский Л., Z. anorg. Chem., 24, 108 (1900).
165. Kirshenbaum I., Summary Report SAM Laboratory, part 10 (SAM Columbia 12),
166. Самсонов A.,Z. Chem. u. Ind. Kolloide, 8, 96 (1911).
167. Graham Т., J. Ghem. Soc, 15, 254 (1862); Ann., 121, 52 (1862); Ann. chim. et phys.,
65, 183 (1862).
Глава 11. Окислы, сульфиды и теллуриды урана
279
168. My litis F., Dietz R., Ber., 34, 2777 (1901).
169. S z i 1 a r d В., J. chim. phys., 5, 493, 641 (1907).
170. S e n K. C, Z. anorg. u. allgem. Chem., 174, 61, 63, 69 (1928).
171. Каргин В. A., Z. anorg. u. allgem. Chem., 198, 80 (1931).
172. Дьяковский С, Укр. хем. ж., 2, 340 (1926).
173. Дьяковский С, Kolloid. Z., 54, 280 (1931).
174. Mixter W. G., Am. J. Sci., 34, 155 (1912); Z. anorg. Chem., 78, 237 (1912).
175. В i 1 t z W.( Fendius C, Z. anorg. u. allgem. Chem., 176, 49 (1928).
176. M а с W о о d G. E., Report RL-4.7.602, Dec. 18, 1945 (UCRL 3).
177. Августин ик А. И., Ж. прикл. хим., 20, 327 (1947).
178. Brewer L., Report CC-672, May 15, 1943 (MP Berkeley 3).
179. Davis T. W., В u r t о n M., Report CC-231, Aug. 15, 1942 (MP Chicago 9).
180. Wilson A. S.,Rundle R. E., Report CN-1495, Apr. 10, 1944 (MP Ames 8).
181. Report [B] LRG-33, June 1944 (British 3).
182. R a e u с h 1 e R., W a r f J. C, Report CC-1496, Apr. 10, 1944 (MP Ames 9).
183. Greenwood H. C, J. Chem. Soc., 93,. 1492 (1908).
184. P e 1 i g о t E., Ann. chim. et phys., 5, 5 (1842).
185. L e 1 у D., Jr., H a m b u r g e r L., Z. anorg. Chem., 87, 220 (1914).
186. Moissan H., Compt. rend., 116, 347 (1893).
187. Moissan H., Compt. rend., 122, 1088 (1896).
188. С о 1 a n i A., Ann. chim. et phys., 18] 12, 79 (1907).
189. Та n f or d C, T i с h e n о r R. L., Report CD-3.385.1, July 16, 1945 (CEW-TEC3).
190. Zimmermann C, Alibegoff G., Kruss G., Ann., 232, 290 (1886).
191. A r f v e d s о n J. A., Kgl. Svenska Vetenskapsakad. Handl., 408 (1822).
192. L e с a n u L. R., J. pharm. chim., 11, 279 (1825).
193. Laugier, Boudet, J. pharm. chim., 11, 286 (1825).
194. Sabatier P., Senderens J. В., Compt. rend., 120, 618 (1895); Bull. soc.
chim. Paris, 13, 870 (1895).
195. de С о n i n с k W. O., Bull, classe sci. Acad. roy. Beige., 992 (1908).
196. Goldschmidt V. M., Thomassen L., Videnskapsselskapets-Skrifter, (1) 2,
8 (1923).
197. В i 1 t z W., Miiller H., Z. anorg. u. allgem. Chem., 163, 161 (1927).
198. Report BR-250, July 23, 1943 (British 4).
199. N e w b e г у E., P r i n g J. N.. Proc. Roy. Soc. London, (A) 92, 276 (1916).
200. Wartenberg H., Broy J., Reinicke R., Z. Elektrochem., 29, 215
(1923).
201. N e w b e г у E., Pring J. N.. Proc. Roy. Soc. London, (A)92, 282 (1916).
202. R i d e a 1 E. K., J. Soc. Chem. Ind. London, 33, 674 (1914).
203. F a e h r P., Dissertation, Munchen Techn. Hochschule, p. 46 (1908).
204. Gay-Lussac, Thenar d, Recherches nhysico-chimiques, Paris, vol. 1, p. 262,
1811.
205. L а с h e r J. R., Report A-1034, Feb. 19, 1944 (Mallinckrodt 1).
206. Tammann G., Rosenthal W., Z. anorg. u. allgem. Chem., 156, 20
(1926).
207. Boulle A., J a r у R., D о m i n e-B e r g ё s M., Compt. rend., 250, 300
(1950).
208. К 1 a p г о t h M. H., Crell Ann., 11, 396 (1789).
209. P e 1 i g о t E., Ann. chim. et phys., 5, 5 (1842); Ann., 43, 255 (1842).
210. Moissan H., Compt. rend., 115, 1033 (1892).
211. Kohl sch utter V., Ann., 314, 333 (1901).
212. Smith E. R.Matthews J. M., J. Am. Chem. Soc., 17, 637 (1895).
213. Pitt B. M., W a g n e r E. L., Mil 1 e г A. J., Report CD-2.355.2, Jan. 4, 1946
(CEW-TEC 4).
214. Werthei m J., J. prakt. Chemie, 29, 211 (1843).
215. de С о n i n с k W. O., Raynaud A., Bull. soc. chim. France, 9, 304 (1911).
216. R о s e n f e 1 d S., Report RL-4.6.52, Mar. 22, 1943 (UCRL 5).
217. Chaudron G., Ann. chim., 16, 251 (1921).
218. Sabatier P., S e n d e r e n s J. В., Compt. rend., 114, 1431 (1892).
219. Sabatier P., Senderens J. В., Ann. chim. et phys., 7, 356,384, 396
(1896).
220. de С о n i n с k W. O., Bull, classe sci. Acad. roy. Belg., 363 (1904).
221. G о 1 d s с h m i d t V. M., Thomassen L., Videnskapsselskapets-Skrifter, (1)
2, 18 (1923).
222. Taylor N. W., J. Am. Chem. Soc., 53, 4459 (1931).
223. Gillies D. M., Report A-1262, Apr. 7, 1944 (SAM Columbia 13).
224. Fairley Т., J. Chem. Soc, 31, 127 (1877).
225. S i e v e r t s A., M u 1 1 e r E. L., Z. anorg. u. allgem. Chem., 173, 297 (1928).
226. В r u n с k O., Z. anorg. Chem., 10, 246 (1895).
227. К r a u s С A., Report [A] M-7, Apr. 19, 1944 (Brown 5).
280
Часть III. Бинарные соединения урана
228. Tevebaugh A. D., T e v e b a u g h R. D., С 1 i n e W. D., Warf J. C.,
TheConversion of UF4 to U308 in National Nuclear Energy Series, Division VIII, vol.
6 (MP Ames 11).
229. Те v eb a ug h R. D.,Cline W. D., Warf J. C, Report CC-1981, Oct. 10,
1944; ClineW. D., Report CC-1983, Nov. 10, 1944; Warf J. C.,Cline W. D.,
Report CC-2723, June 30, 1945 (MP Ames 12).
230. Gates J. W.,Clewett G. H.,Andrews L. J., Y о и n g H. A., Report
CD-457, Aug. 4, 1944 (CEW-TEC 5).
231. de С о n i n с к W. О., Bull, classe sci. Acad. roy. Belg., 744 (1909).
232. Arfvedson J. A., Kgl. Svenska Vetenskapsakad. Handl., 410 (1822).
233. Arfvedson J. A., Pogg Ann., 1, 256 (1824).
234. M i 1 b а и e r J., 2. anorg. Chem., 42, 448 (1904).
235. Miiller A., Z. anorg. u. allgem. Chem., 93, 269 (1915).
236. В е г z e 1 i и s J. J., Pogg. Ann., 1, 362 (1824).
237. Rammelsberg C, Pogg. Ann., 59, 9 (1843).
238. Wohler F., Ann., 41, 345 (1842).
239. U h r 1 а и b G. E., Dissertation, University of Gottingen (1859).
240. P а г s о n s C. L., J. Ind. Eng. Chem., 9, 466 (1917).
241. McCoy H. N., Ashman G. C, Am. J. Sci., 26, 522 (1908).
242. Hoffman J. I., J. Wash. Acad. Sci., 38, 233 (1948).
243. G i о 1 i t t i F., T a v a n t i G., Gazz. chim. ital., (II) 38, 239 (1908).
244. К г а и s С A., Report A-2300, Oct. 5, 1944 (Brown 6).
245. a) Haworth W. N., Chackett, Report B-26, Jan. 5, 1945 (British 5), 6) Haworth
W. N., A m p h lett С. В., T h о m a s L. F., Report B-27, Oct. 10, 1941 (British 6).
246. Arfvedson J. A., Ann. chim. et phys., 29, 160 (1825).
247. P e 1 i g о t E., Ann. chim. et phys., 12, 562 (1844).
248. M i x t e г W. G., 2. anorg. Chem., 78, 234 (1912).
249. H и t t i g G. F., S с h г о e d e г Е., Z. anorg. u. allgem. Chem., 121, 250 (1922).
250. de F о г с г a n d R., Ann. chim., 3, 34 (1915).
251. E b e 1 m e n J. J., Ann. Chim. et phys., 5, 207 (1842); Ann., 43, 302 (1842).
252. Berzelius J.J., Jahresbericht uber d. Fortschritte der physischen Wissenschaften
d. Chemie u. Mineralogie (Berzelius), 24, 118 (1845).
253. de С о n i n с к W. О., Compt. rend., 152, 1179 (1911).
254. F i sc h e 1 V., Dissertation, University of Bern., p. 10 (1889).
255. G о 1 d г i n g L. S., Report N-2294, May 23, 1946 (MP Clinton 1).
256. Wood W. C, Bradley H., Carroll H. S., Report RL-4.6.287, Aug. 29,
1944 (UCRL 6).
257. К о 1 о d о n у M., Report LA-35, Nov. 1, 1943 (Los Alamos 1).
258. Bolton H. C, Dissertation, University of Gottingen, p. 20 (1866); Z. Chem., 2,
353 (1866); Bull. soc. chim. Paris, 6, 450 (1866); Monatsber. d. Kgl. Preuss. Akad. d.
Wissenschaften, 299 (1866).
259. Gore G., J. Chem, Soc., 22, 393 (1869).
260. Brewer L., Report AECD-2307, July 31, 1948 (MP Berkeley 4).
261. M о г г i s G. O., Report B-50, Mar. 20, 1942 (British 7).
262. К a n g г о W., J a h n R., Z. anorg. u. allgem. Chem., 210, 335 (1933).
263. В о и r i о n F., Ann. chim. et phys., 21, 58 (1910).
264. Michael A., M и r p h у A., Jr., Am. Chem. J., 44, 384 (1910).
265. V e n a b 1 e F. P., J а с к s о n D. H., J. Elisha Mitchell Sci. Soc., 35, 88 (1919—1920)
266. Gore G., Phil. Mag., 29, 546 (1865).
267. M с В е е Е. Т., Beport [A] M-2102, Mar. 21, 1945 (Purdue 1).
268. К г а и s C. A., Reports [A] M-1060, Nov. 2, 1942; CC-365, Nov. 17, 1942 (Brown 7).
269. R а и t e r G., Ann., 270, 254 (1892).
270. Q и a n t i n H., Compt. rend., 106, 1075 (1888).
271. Camboulives P., Compt. rend., 150, 177 (1910).
272. К a t z J. J., DavidsonN. R., Report CK-1221, Jan. 15, 1944 (MP Chicago 10).
273. Richards T. W., Merigold B. S., Z. anorg. Chem., 31, 235 (1902).
274. Powell J., Report CC-1781, Aug. 10, 1944 (MP Ames 13).
275. Brewer L.Bromley L. A., E a s t m a n E. D., G i 1 I e s P. W., L о f -
g r e n N. L.f Preparation and Properties of Sulfides and Oxysulfides of Uranium, in
National Nuclear Energy Series, Division VIII, vol. 6 (MP Berkeley 5).
276. T i e d e E., В i г n b г а и е г Е., Z. anorg. Chem., 87, 165 (1914).
277. Ней si er O., Z. anorg. u. allgem. Chem., 154, 363 (1926).
278. В a 1 а г e v D., Z. anorg. u. allgem. Chem., 136, 217 (1924).
279. Fowler G. J., G r a n t J., J. Chem. Soc., 57, 275 (1890).
280. H о d g к i n s о n W. R., L о w n d e s F. K., Chem. News., 58, 309 (1888).
281. Hodgkinson W. R., Lown desF. K., Chem. News., 59, 64 (1889).
282. Warf L., Report CC-1194, Dec. 9, 1943 (MP Ames 14).
283. Zimmermann C., Ann., 213, 290 (1882).
284. Hedvall J. A., Z. anorg. Chem., 93, 319 (1915).
Глава 11. Окислы сульфиды и теллуриды урана
281
285. Писаржевский Л., Z. anorg. Chem., 24, 111 (1900).
286. Smith E. F., S h i n n O. L., Z. anorg. Chem., 7, 49 (1894); J. Am. Chem. Soc.,
16, 571 (1894).
287. Isambert M., Compt. rend., 30, 1087 (1875).
288. К a t z J. J., G г u e n D. M., J. Am. Chem. Soc. , 71, 2106 (1949).
289. Aloy J., Compt. rend., 122, 1542 (1896).
290. A u d г i e t h L. F.,Schmidt M. Т., Proc. Natl. Acad. Sci. U. S., 20, 223 (1934).
291. Best R. J., Taub D., Longsworth L. G., Report A-380, Nov. 24, 1942
(Rockefeller I).
292. Zachariasen W. H., Report CF-2926, Apr. 15, 1945 (MP Chicago 11).
293. A 1 i b e g о f f G., Ann., 233, 135 (1886).
294. Zachariasen W. H., Report CP-2160, Sept. 23, 1944; Crystal Structure Studies
of Sulfides and Oxysulfides of Uranium, Thorium, and Cerium, in National Nuclear
Energy Series, Division VIII, vol. 6 (MP Chicago 12).
295. Co 1 a n i A., Compt. rend., 137, 382 (1903).
296. M о о n e у R. C, Report CP-1507, Mar. 27, 1944 (MP Chicago 13).
297. Calvin M., Report CK-2411, Oct. 1, 1944 (MP Berkeley 6).
298. F 1 a t t R., H e s s W., Helv. Chim. Acta, 21, 525 (1938).
299. К a r 1 e J., Report CL-1512, Apr. 6, 1944 (MP Chicago 14).
300. S e a b о r g G. T. et al., Report CS-2793, Apr. 10, 1943 (MP Chicago 15).
301. Bromley L. A., L о f g r e n N. L., Report CT-1344, Feb. 14, 1944 (MP
Berkeley 7).
302. Brewer L., В г о m 1 e у L. A., L о f g г e n N. L., Reports CT-1714, Apr. 14,
1944; CT-2139, Aug. 15, 1944 (MP Berkeley 8).
303. P ё 1 i g о t E., Ann. chim. et phys., 5, 20 (1842).
304. Powell J., Report CC-1778, Aug. 18, 1944 (MP Ames 16).
305. Picon M., Compt. rend., 189, 96 (1929).
306. Lofgren N. L., Bromley L. A., Report CK-941, Sept. 1943 (MP Berkeley9).
307. Zachariasen W. H., Reports CN-2615, Jan. 9, 1945; CC-2768, Mar. 12, 1945
(MP Chicago 16).
308. Rose H., Gilb. Ann., 73, 139 (1823).
309. R u n d 1 e R. E., M с D о n a 1 d R., Report CC-1504, Aug. 10, 1944 (MP Ames 15).
310. В e г z e 1 i u s J. J., Pogg. Ann., 1, 373 (1824).
311. Col a ni A., Compt. rend., 137, 383 (1903).
312. Col a ni A., Ann. chim. et phys., [8] 12, 85 (1907).
313. M i 1 b a u e r J., Z. anorg. chem., 42, 450 (1904).
314. С о 1 a n i A., Ann. chim. et phys., [8] 12, 87 (1907).
315. M о n t i g n i e E., Bull. soc. chim. France, 748—749 (1947).
Часть IV
ГАЛОГЕНИДЫ, ОКСИГАЛОГЕНИДЫ,
БОРГИДРИДЫ И КАРБОНИЛ УРАНА
Глава 12
НЕЛЕТУЧИЕ ФТОРИДЫ УРАНА
ТРИФТОРИД УРАНА
Трифторид урана, в противоположность трихлориду и трибромиду,
которые довольно легко получаются и известны уже давно, является соединением,
получение которого связано с некоторыми трудностями. Синтез его осуществлен
лишь недавно. Вначале трифторид пытались получить методом, прежде
успешно применявшимся для приготовления трибромида и трихлорида, а именно
восстановлением тетрафторида урана водородом при повышенных температурах.
Тетрафторид, применявшийся при первых опытах [1, 2], получали из
водного раствора, и поэтому он, вероятно, содержал небольшие количества воды.
При действии на него сухим водородом в условиях температуры красного каления
выделялся фтористый водород и получалось вещество красновато-бурого цвета.
Последнее не растворялось в воде, и на него почти не действовали никакие
кислоты, за исключением концентрированной азотной кислоты. Анализа
полученного вещества не производили, но предполагали, что это—фторид урана
низшей валентности. Однако результаты недавней работы показали, что в
присутствии следов кислорода или воды реакция между тетрафторидом урана и
водородом приводит к образованию двуокиси урана, а не трифторида. Так,
при обработке тетрафторида урана водородом в интервале температур 450—550°
получена [3] в качестве продукта реакции только загрязненная двуокись урана,
что, вероятно, вызвано присутствием кислорода в применявшемся для
восстановления водороде. В Англии [4] также пытались получить трифторид действием
водорода на тетрафторид урана при 600°. При этом наблюдалось выделение
фтористого водорода, однако установить, что полученный продукт представляет
трифторид, не удалось.
В последующих работах проведено повторное исследование реакции между
водородом и тетрафторидом. Водород тщательно очищали пропусканием над
нагретым металлическим ураном (стр. 141). При проведении реакции в
кварцевой трубке при температуре выше 600° выделялись фторсодержащие газы.
Продукт реакции, однако, в значительной степени состоял из двуокиси урана.
Полученные данные объясняли следующим образом: в присутствии следов влаги
небольшое количество тетрафторида подвергается гидролизу, причем образуются
двуокись урана и фтористый водород; последний действует на кварц с
образованием тетрафторида кремния и некоторого количества воды. Цикл
повторяется до полного превращения тетрафторида урана в двуокись. Такой механизм
реакции наиболее вероятен в том случае, когда водород медленно пропускается
через систему. При проведении реакции между тетрафторидом урана и
водородом в трубке из монель-металла выделения фтористого водорода не
наблюдается совсем. Тетрафторид может быть выделен практически неизмененным даже
после 48-часовой обработки чистым водородом при 980°. Эти результаты
непонятны, поскольку, как указывается ниже, в Англии добились успеха в
получении трифторида по существу тем же самым методом.
Несколько безуспешных попыток получить трифторид реакцией
"обменного разложения фтористого водорода с гидридом, трихлоридом или
286 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
трибромидом урана были сделаны в различных лабораториях. Оказалось, что
в то время как трибромид и трихлорид легко образуются при действии
бромистого или хлористого водорода на гидрид, реакция фтористого водорода с
последним приводит только к образованию тетрафторида урана [5J:
UH3+4HF _1^> UF4 + ^H2. (1)
Обработкой трихлорида урана сухим фтористым водородом при 450°
также получен тетрафторид урана [3]:
UC13 + 4HF—>UF4 + 3HCl + iH2. (2)
При обработке трихлорида урана жидким безводным фтористым водородом
при 25° [6] наблюдалось выделение хлористого водорода. Подробности
эксперимента не приведены, но указывается, что получившийся продукт быстро
окисляется сухим воздухом и является, повидимому, трифторидом. Однако
легкая окисляемость продукта совсем не согласуется с известной устойчивостью
трифторида, и поэтому реакция требует дальнейшего изучения.
На основании прежних наблюдений [7] указывалось, что красноватый,
быстро окисляющийся осадок образуется также при добавлении плавиковой
кислоты к водному раствору трихлорида урана. Это наблюдение подтвердилось
при дальнейших исследованиях [8]. Однако хотя осадок и окрашивается в
быстро исчезающий красновато-бурый цвет, из реакционной смеси удалось
выделить лишь тетрафторид урана. Возможной причиной неудачи могла быть быстро-
протекающая реакция трифторида урана с водой, поэтому опыты были
повторены с неводным растворителем [9]. Раствор трибромида урана в диметилформ-
амиде обрабатывали безводным фтористым водородом (предыдущие опыты
показали, что фтористый водород не действует на диметилформамид). Однако
вместо ожидаемого красного осадка трифторида урана в результате
взаимодействия с растворителем" образовалась лишь зеленая смолообразная масса.
Таким образом, трифторид урана не удалось получить ни при одной из этих
галогенообменных реакций.
Получение трифторида урана. Из разработанных до
настоящего времени методов получения трифторида урана лишь два являются
успешными. Первый из них, предложенный в Англии, состоит в восстановлении
тетрафторида водородом, причем особое внимание обращается на влияние
кислорода и влаги, а также на чистоту тетрафторида урана [10]. Второй метод
заключается в восстановлении тетрафторида металлическим ураном.
Восстановление тетрафторида урана водородом [10]. Успех этой операции
зависит от полного отсутствия следов влаги и кислорода. Водород тщательно
очищается. После предварительной осушки хлористым кальцием он
пропускается над палладированным асбестом при 350° для удаления кислорода и затем
окончательно осушается пропусканием последовательно через хлорид кальция,
фосфорный ангидрид и ловушку, охлаждаемую жидким азотом. Следы
взвешенных твердых загрязнений удаляются пропусканием газа через пористый
стеклянный фильтр.
Реакционная трубка состоит из внешней кварцевой трубки, выложенной
нержавеющей сталью (18 : 8 : 1 : 1) во избежание взаимодействия
образующегося фтористого водорода с кварцем. При получении трифторида урана в
больших масштабах достаточно применения одной трубки из нержавеющей стали.
Тетрафторид может быть помещен в молибденовую лодочку. Вся открытая
поверхность кварца в более холодных частях прибора защищается церезиновым
покрытием. Аппаратура перед работой откачивается, тщательно высушивается
и обезгаживается.
Глава 12. Нелетучие фториды урана
287
Для получения чистого трифторида урана необходимо брать тетрафторид,
свободный от кислородсодержащих примесей. При применении тетрафторида,
полученного из водного раствора и содержащего в качестве главных примесей
сульфаты олова и меди, а также воду, образуется смесь трифторида урана
с двуокисью, потому что в условиях высоких температур и сульфаты и вода
реагируют с тетрафторидом. Последний, даже полученный из двуокиси
действием фтористого водорода при высоких температурах и обычно
считающийся безводным, содержит достаточно влаги, чтобы продукт реакции
оказался загрязненным двуокисью урана. Оказалось, что тетрафторид достаточной
степени чистоты можно получить возгонкой обычного тетрафторида в очень
высоком вакууме при 1000° в аппаратуре из молибдена. По этому методу
можно получить очень чистый кристаллический продукт, пригодный для
превращения в трифторид урана.
Восстановление тетрафторида водородом протекает довольно быстро при
1000° (т. е. выше температуры его плавления) согласно уравнению
2UF4 -f H2 ^± 2UF3 + 2HF. (3)
Ниже 900° реакция практически не идет. Дальнейшее восстановление
трифторида при 1000° идет очень медленно. Продолжительное нагревание при
температуре 1000° не рекомендуется, так как в этом случае появляется опасность
термического диспропорционирования трифторида (см. ниже). Продукт, полученный
восстановлением тетрафторида водородом, является кристаллическим и имеет
густой фиолетово-красный цвет. Состав продукта реакции устанавливали
путем химического анализа и рентгенографическим методом.
Трифторид урана получали также, хотя и в очень небольших количествах,
действием атомарного водорода на тетрафторид в кварцевом сосуде [11].
Рентгенограмма полученного продукта указывала на присутствие в нем 60%
трифторида и 40% двуокиси урана. Кислород, вероятно, образовывался из воды,
получающейся в результате реакции между атомарным водородом и двуокисью
кремния.
Восстановление тетрафторида урана металлическим ураном [12].
Тетрафторид урана восстанавливается тонкоизмельченным металлическим ураном
при 1050°:
3UF4 + U^± 4UF3. (4)
При проведении этой реакции большое значение имеет
регулирование температуры, так как уравнение [4] обратимо и выше 1050° равновесие
заметно смещается влево. Реакция может быть проведена в никелевом сосуде,
в который загружается смесь тетрафторида со стехиометрическим количеством
урана в виде стружек. Смесь нагревают до 250° и затем вводят водород для
превращения металла в гидрид. После этого гидрид разлагают при 400° и получают
таким образом тесную смесь тетрафторида урана с тонкодисперсным
металлическим ураном. Эту смесь сплавляют при 1050° в течение 2 час. в атмосфере
аргона. При этом получается черное плотное коксоподобное вещество, которое,
как показывает анализ, представляет трифторид с примесью 1 % двуокиси урана
и уранилфторида.
Физические свойства трифторида урана.
Трифторид, полученный восстановлением тетрафторида водородом, представляет
сплавленную кристаллическую массу, имеющую в крупных кусках почти
черный цвет. Под микроскопом заметна фиолетово-красная окраска кристаллов.
Точку плавления трифторида урана определить не удалось, так как выше 1000°
он подвергается реакции диспропорционирования с образованием урана и
тетрафторида урана, но наблюдения показывают, что точка плавления трифторида
должна лежать выше 1140°. Трифторид выше 1000°, повидимому, должен
обладать весьма слабой летучестью [10, 12].
288 Часть IV. Галоген иды, оксигалогениды, бор гидр иды и карбон ил урана
Изучение диффракции рентгеновских лучей [13, 14] показало, что
трифторид урана имеет кристаллическую структуру, сильно отличающуюся от
структуры тетрафторида. Трифторид урана изоморфен трифторидам
редкоземельных элементов: лантана, празеодима, церия и неодима.
Рентгенограмма, снятая методом порошка, указывает на расположение
атомов металла по типу гексагональной плотной упаковки. Элементарная
ячейка содержит две молекулы (пространственная группа Сб/ттс). В табл.
113 даны константы трифторида урана и чистого трифторида лантана [131,
а также трифторидов церия, празеодима и неодима [15, 16].
Таблица 113
РЕНТГЕНОГРАФИЧЕСКИЕ ДАННЫЕ ДЛЯ ТРИФТОРИДА УРАНА И НЕКОТОРЫХ
ИЗОМОРФНЫХ С НИМ СОЕДИНЕНИЙ
Формула
соединения
LaF3
UF3
CeF3
PrF3
NdF3
Размеры решетки, А а)
ai
4,140+0,001
4,138±0,003
4,107+0,003
4,077+0,003
4,054+0,003
аз
7,336+0,001
7,333+0,004
7,273+0,005
7,218+0,007
7,196+0,005
Плот-
г[см*
_
8,95 г>
—
—
Межатомное расстояние, А °/
для 3F
2,39
2,39
2,37
2,35
2,34
для 2F
2,38
2,38
2,36
2,35
2,34
для 6F
2,71
2,71
2,69
2,67
2,66
Ионный
радиус
трехвалентного
металла,
Ав>
1,06
1,06
1,04
1,03
1,01
а) Исходя из предположения, что структура гексагональная.
б) Вокруг каждого атома металла находится одиннадцать атомов фтора.
в^ После введения поправки для приведения к координационному числу 8 и вычитания ионного
радиуса для F"~ (1,33 А).
г) В Англии иммерсионным методом с тетралииом найдено значение для плотности, равное
9,18 г}смъ.
Однако установлено, что истинной элементарной ячейкой, как показывает
лауэграмма, является ромбическая, которая в три раза больше, чем
гексагональная элементарная ячейка, выведенная из рентгенограммы, полученной
методом порошка. В соответствии с этой точкой зрения в Англии [17] при помощи
гониометрических измерений нашли, что кристаллы трифторида урана, по
всей вероятности, являются ромбическими. Они имеют слабое двойное
лучепреломление, несмотря на то, что все показатели преломления близки к 1,73.
Все кристаллы, повидимому, существуют в виде тройников, сдвоенных в
субмикроскопическом масштабе.
Химические свойства трифторида урана.
Термическое разложение. В Англии [10] установлено, что при нагревании выше 1000°
трифторид разлагается по реакции
4UF3^3UF4 + U. (5)
В вакууме тетрафторид возгоняется, а порошок металлического урана остается.
При 1000° распад мало заметен, но при 1200° становится значительным. Этим
объясняются неудачные попытки исследователей получить трифторид урана
путем взаимодействия урана с тетрафторидом при 1400°. Для приготовления
очень чистого металлического урана предложено использовать диспропорцио-
нирование трифторида урана, однако вследствие трудности подбора материала,
способного выдержать действиекак металла, так и фторида при столь высоких
температурах, успешное проведение этой реакции невозможно.
Глава 12. Нелетучие фториды урана
289
Действие воздуха. При комнатной температуре трифторид урана довольно
медленно подвергается воздействию влажного воздуха [10]. Он мало
гигроскопичен и напоминает этим трихлорид. При нагревании трифторида урана на
воздухе происходит его окисление, по всей вероятности до оксифторида.
При 900° последний количественно превращается в закись-окись урана.
Действие воды. В воде трифторид урана нерастворим. В холодной воде
он медленно окисляется с образованием зеленого желатинообразного продукта.
При 100° вода действует гораздо энергичнее, в результате чего раствор над
осадком приобретает желтый цвет, свидетельствующий об окислении урана
до уранилфторида [ 10].
Действие кислот [10, 12]. Подобно фторидам редкоземельных элементов
трифторид урана довольно инертен по отношению к кислотам. Как и фториды
редкоземельных элементов (и в противоположность тетрафториду), он
нерастворим в оксалате аммония. Кислоты—окислители переводят его в соли урани-
ла и таким образом растворяют его. Разбавленные соляная, серная и азотная
кислоты на холоду медленно действуют на трифторид урана. Горячая азотная
кислота растворяет его довольно быстро, причем выделяются окислы азота.
Горячая разбавленная серная кислота также растворяет трифторид, но
медленнее, чем азотная. Под действием горячей хлорной кислоты образуется
прозрачный раствор перхлората уранила 1Ю2(СЮ4)2. Полагают, что реакция между
трифторидом и соляной кислотой является окислительно-восстановительной:
4UF3 + 4НС1 -> 3UF41 + UC14 + 2H2. (6)
Однако установлено, что при взаимодействии трифторида урана как с
разбавленной, так и с концентрированной соляной кислотой получаются растворы
густого красного цвета, что свидетельствует о присутствии ионов
трехвалентного урана. Последнее наблюдение находится в соответствии с известным
стабилизирующим действием соляной кислоты на растворы соединений
трехвалентного урана. Добавление к соляной и другим кислотам борной кислоты приводит
к образованию фтороборатных комплексов, что заметно облегчает растворение.
Действие окислителей [10, 12]. Смесь хлорида меди (И) и хлорида
аммония, быстро растворяющая металлический уран, на трифторид урана
действует лишь в очень слабой степени. Нитрат железа (III) растворяет трифторид.
Перхлорат серебра в растворе быстро восстанавливается им с образованием
серебряного зеркала. Хлор реагирует с трифторидом с образованием UF3C1
(ср. гл. 15) [18]. Действие фтора не изучено, однако несомненно, что
конечным продуктом будет гексафторид урана, хотя может иметь место и образование
представляющих интерес промежуточных продуктов.
Действие восстановителей [19]. Металлический кальций энергично
восстанавливает трифторид до металлического урана. При восстановлении вместе
с 30%-ным избытком кальция добавляли в качестве вспомогательного
реагента иод (0,5 моля иода на 1 моль трифторида). Этим путем получался хорошо
образованный и плотный металл. Выход составлял 98%.
ТЕТРАФТОРИД УРАНА
Имеется два типа методов получения тетрафторида урана. Первый основан
на осаждении практически нерастворимого в воде тетрафторида из водных
растворов других соединений четырехвалентного урана:
U^f + 4F^ ->UF4J. (7)
Второй основан на взаимодействии различных соединений урана, в
частности двуокиси урана, с газообразными фторирующими агентами при
повышенных температурах, например
U02 + 4HF —^> UF4 + 2НгО. (8)
19 Химия урана
290 Часть IV, Галогениды, оксигалогениды, боргидриды и карбонил урана
Этот метод отличается простотой операций и сравнительной легкостью
получения чистого безводного продукта.
В следующем разделе будет дано описание различных методов
приготовления тетрафторида, причем большее внимание будет уделено химизму, а не
технике эксперимента.
Первые работы. Сначала тетрафторид урана получали действием
плавиковой кислоты на закись-окись урана U308 [20]. Реакция идет очень
энергично; при этом получается желтый раствор и нерастворимый зеленый осадок.
Болтон [1, 2] установил, что в осадке находится тетрафторид:
U308 + 8HF -» 2U02F2 + UF4 \ + 4Ы20. (9)
Полученный этим путем тетрафторид весьма трудно отфильтровать и промыть.
Легче фильтруемый продукт Болтон получил восстановлением в кипящем
растворе уранилфторида U02F2 хлоридом олова и периодическим добавлением
плавиковой кислоты. Быстро отмывающийся осадок тетрафторида образуется
в результате такого же метода восстановления ураната аммония или аммоний-
уранилкарбоната, растворенных в плавиковой кислоте. Болтон нашел, что
добавление плавиковой кислоты к раствору тетрахлорида вызывает появление
объемистого осадка тетрафторида урана. Прокаленная двуокись урана очень
медленно реагирует с плавиковой кислотой; свежеосажденная 1Ю2 хН20
реагирует гораздо быстрее. Это наблюдение подтверждено и другими работами
[21]. Установлено, что тетрафторид урана не образуется даже после
длительного взаимодействия безводной двуокиси урана с горячим, постоянно
кипящим раствором плавиковой кислоты (43,2% HF). Превращение двуокиси
урана в тетрафторид, повидимому, можно ускорить разбавлением плавиковой
кислоты. Изучалось также действие плавиковой кислоты на закись-окись
урана [22], причем результаты этих исследований расходились с данными
ранее проведенных работ. Однако они не имеют ценности ввиду несовершенства
аналитической методики. Повторение этого эксперимента [23] подтвердило
первоначальные выводы.
Получение тетрафторида из водных растворов.
Метод получения тетрафторида из водных растворов разработан в Англии.
Результаты, полученные Болтоном, послужили основанием для различных
видоизменений этого метода. Фторид, хлорид или сульфат уранила
восстанавливают до четырехвалентного состояния и осаждают тетрафторид урана
добавлением плавиковой кислоты. В качестве исходных реагентов используют
различные соединения урана и различные восстановители.
Процессы с применением хлорида олова (II) и фтористого водорода.
Гроссе [24] получал тетрафторид методом, во многом сходным с тем, которым
пользовался Болтон. Метод заключался в растворении ураната натрия в
избытке плавиковой кислоты и восстановлении его хлоридом олова (II) при
температуре кипения раствора. Выход составлял 99%.
Na2U04 + 4HF -* U02F2 + 2NaF + 2H20; (10)
U02F2 + SnCl2 + 4HF ->UF4j + SnCl2F2 + 2H20. (11)
Опыты Гроссе были повторены другими исследователями [25, 26], однако
полученный ими тетрафторид урана оказался загрязненным фторидом натрия
(4—5% после высушивания при 100°). Количество фторида натрия не
соответствовало никакому определенному соединению фторида натрия стетрафторидом
урана. Повидимому, осадок был смесью тетрафторида урана и двойной соли
тетрафторида урана с фторидом натрия, причем точное соотношение обоих
компонентов зависело от относительных растворимостей двойной соли и
тетрафторида в плавиковой кислоте. Фторид натрия является нежелательной примесью,
а поэтому были сделаны попытки приготовить тетрафторид урана, свободный от
натрия. Оказалось, что этого можно достигнуть, применяя уранат натрия (или
Глава 12. Нелетучие ф.ториды урана
291
более доступный диуранат Na2U207) в соляно- или сернокислом растворе
(а не в плавиковой кислоте). По английским данным [26—28], например, тетра-
фторид может быть получен с содержанием натрия меньше 0,1%, если
предварительно растворить диуранат натрия в избытке соляной кислоты:
Na2U207 + 6HCl -» 2U02Cl2 + 2NaCl + 3H20; (12)
U02C12 + SnCl2 +- 4HC1 -> UC14 + SnCl4 + 2H20; (13)
UC14 + 4HF -» UF4 J + 4HC1. (14)
Чтобы получить легко фильтрующийся осадок тетрафторида, требуется очень
сильное перемешивание и энергичное кипячение. Плавиковую кислоту следует
прибавлять медленно и в большом избытке (100%).
Была изучена [25] кинетика восстановления уранил-ионов ионами олова(П);
результаты показали, что эта ионная реакция протекает медленно. Ее скорость
быстро увеличивается с повышением температуры в интервале 17—Д10°,
поэтому для быстрого восстановления необходимо применять как можно более
горячий раствор. Наличие избытка соляной кислоты также приводит к
увеличению скорости восстановления. Этим способом можно достигнуть достаточно
хороших выходов (92—97%) тетрафторида; однако приходится преодолевать
трудности, которые связаны с коррозией, возникающей в результате
действия на аппаратуру горячих растворов, содержащих соляную и плавиковую
кислоты. Применение эбонита может частично разрешить затруднения, так
как он выдерживает действие раствора, содержащего 10% НС1 и 5% HF [29],
но сосуды из эбонита нельзя нагревать до достаточно высокой температуры.
Поэтому процесс был изменен—вместо соляной кислоты стали применять
серную [30, 31], что позволило работать в сосудах, выложенных внутри
свинцом:
Na2U207 + 3H2S04 -* 2U02S04 + 3H20 + Na2S04; (15)
2U02S04 + 2SnCl2 + 4H2S04 -> 2U(S04)2 + 2SnCl2S04 + 4H20. (16)
Однако при этом возникло осложнение, заключающееся в том, что умеренно
растворимый U(S04)2-4H20 может выпасть в осадок и загрязнить тетрафто-
рид (см. стр. 292).
Диуранат натрия не является удобным исходным материалом для
получения тетрафторида, поэтому метод восстановления хлоридом олова (II) был
изменен таким образом, чтобы вместо диураната можно было использовать
более доступную закись-окись урана. Предварительное исследование
процесса растворения закиси-окиси урана в различных кислотах показало, что в
присутствии окислителя, например азотной кислоты, в результате
реакции между закисью-окисью урана и соответствующей кислотой может быть легко
получен уранилхлорид или уранилсульфат [32, 33]:
U308 + 6HC1 + 2HN03 -» 3U02C12 + 4H20 + 2N02, (17)
Применяя азотную кислоту в количестве, несколько меньшем стехиометриче-
ского (85%), можно получать растворы, свободные от избытка этой кислоты.
Соляная кислота, как упоминалось выше, затрудцяет борьбу с коррозией
реакционного сосуда. Ввиду этого соляную кислоту, несмотря на некоторые ее
преимущества, заменяют серной кислотой [34].
В смеси серной и азотной кислот закись-окись урана растворяется очень
легко, особенно при умеренном нагревании. При стехиометрических
количествах используемых кислот согласно уравнению
2U308 + 6H2S04 + 2HN03 -> 6U02S04 + N02 + NO + 7H20 (18)
происходит быстрое превращение закиси-окиси урана в уранилсульфат [35].
19*
292 Часть IV. Галогениды, оксигалогениды> боргидриды и карбонил урана
Полученный уранилсульфат может быть восстановлен указанным выше
способом [36]. Из полученного при этом раствора можно осадить тетрафторид по
методу, подробно описанному ниже.
Получение тетрафторида урана электролитическим восстановлением
соединений уранила. Растворенный уранилсульфат можно также восстановить
до U(S04)2 электролитическим способом, а затем добавлением плавиковой
кислоты осадить тетрафторид урана. При этой операции обходятся без солей
олова (II) и получают более чистый продукт, не содержащий загрязнений,
вносимых химическими восстановителями.
Механизм осаждения тетрафторида урана из растворов U(S04)2.
Выше 70° твердая фаза U(S04)2-4H20 находится в равновесии с раствором.
Эта соль так мало растворима, что выпадает из раствора, содержащего
более 5 г соли на 100 мл, при нагревании выше 70°. Установлено, что
U(S04)2-4H20 растворяется в плавиковой кислоте. Тонкая суспензия
U (S04)2-4H20 также растворяется при добавлении суспензии фторида натрия.
Растворение обусловлено образованием сульфатно-фторидного комплекса, что
подтверждается необходимостью при осаждении тетрафторида добавлять на
1 моль U(S04)2 более 2 молей HF до начала образования осадка [37].
При добавлении к раствору U(S04)2 менее 2 молей HF меняется только
цвет раствора [38].
Все эти наблюдения свидетельствуют о том, что осаждению
тетрафторида предшествует образование растворимого комплексного соединения.
U (S04)2 + 2HF —* Растворимый комплекс; (19)
Растворимый комплекс + 2HF —> UF4 J. (20)
Как видно из уравнения (19), комплексное соединение, повидимому,
содержит U (S04)2 и HF в отношении 1 :2. Общая формула поэтому должна
быть U(S04)2'2HF. По аналогии с другими соединениями
четырехвалентного урана, в которых координационное число урана равно 6, строение
данного комплекса может быть представлено формулой Н2 [U(S04)2 F2].
Согласно этой структуре, данное комплексное соединение должно быть
кислотой, следовательно, имеется возможность получения ее солей.
Действительно, из U(S04)2 и KF получено соединение К2 [U(S04)2F2]-2H20.
Устойчивость комплекса Н2 [U(S04)2F2] детально изучена. Растворы
этого соединения дают качественную реакцию на четырехвалентный уран —
выпадает осадок U[Fe(CN)e]. Следовательно, комплекс должен быть заметно
диссоциирован на UF4 и U(S04)2. В растворе, содержащем два моля HF
на 1 моль U(S04)2, должны существовать следующие равновесия,
включающие диссоциацию комплекса фтористого водорода и U(S04)2:
U (S04)2 + 2HF Zl H2 [U (S04)2 FJ (21)
it It
U4+ + 2SOJ- + 2H+ + 2F" Z- 2H+ + [U (S04)2 F2]2~. (22)
Если такому раствору дать постоять (при любой температуре в интервале
между 20 и 100°), то происходит медленное осаждение тетрафторида урана.
Тщательное изучение показало, что это обусловлено не частичным
окислением U (IV) в U (VI) кислородом воздуха, а, вероятно, медленным
разложением комплекса
2Н2 [U (S04)2 F2] -* UF4 + U (S04)2 + 2Н2 S04. (23)
После стояния раствора в течение 48 час. при 20° осаждается только
тетрафторид урана, возможно в виде 2UF4-5H20. Сульфат урана, образующийся
согласно уравнению (23), остается в растворе, так как его октагидрат
Глава 12. Нелетучие фториды урана
293
U(S04)2-8H20, являющийся устойчивой твердой фазой при температурах
до 70 — 80°, хорошо растворим. При температуре 100° в осадок выпадает
моногидрат тетрафторида урана UF4-H20. Когда достигается предел
растворимости U(S04)2-4H20, в виде смешанных кристаллов осаждаются UF4-H20
hU(S04)2-4H20.
Замечено [39], что суспензии тетрафторида урана, подвергавшиеся в
течение короткого времени действию дымящей серной кислоты, переходят
в раствор при разбавлении кислоты водой. Это может служить
дополнительным доказательством образования комплекса H2[U(S04)2F2].
Осаждение и дегидратация 2UF4-5H20 [40]. Осажденный при
комнатной температуре тетрафторид урана представляет собой аморфную зеленую
массу, которую чрезвычайно трудно отфильтровать и промыть. Для
получения кристаллического, легко фильтрующегося и промывающегося осадка
была предложена следующая операция: аморфный осадок взбалтывается
с разбавленной плавиковой кислотой (0,1 — 3%) в течение промежутка
времени от нескольких часов до нескольких дней. В результате этого он
превращается в объемистую, иногда студенистую массу длинных тонких
шелковистых игл, окрашенных в бирюзовый цвет. Когда превращение
заканчивается, кристаллы можно легко отфильтровать и промыть. Воздушно-
сухие кристаллы имеют постоянный состав и не теряют веса в вакууме при
комнатной температуре. Анализ показывает, что состав соединения
выражается следующей формулой: 2UF4-5H20. Соединение очень устойчиво на
воздухе и практически не окисляется до шестивалентного урана даже при
стоянии на воздухе в течение нескольких недель.
Получение безводного тетрафторида урана из его гидратов. При
высушивании влажного тетрафторида на воздухе при 100° образуется
соединение, приближающееся по составу к моногидрату UF4-H20.
При нагревании на воздухе до 400° вода удаляется полностью, но
при этом в значительной степени происходит гидролиз и окисление [41].
Была сделана попытка высушить тетрафторид при 400° в токе инертного
газа (азота). Но этот способ был оставлен из-за трудности очистки газа
от кислорода, следов которого достаточно, чтобы вызвать окисление.
Дегидратация моногидрата при температуре 400° и давлении ниже 50 мм
рт. ст. дала удовлетворительные результаты. Если нужно свести к
минимуму окисление с поверхности, необходимо, чтобы продолжительность
нагревания и поверхность высушиваемого моногидрата были как можно
меньше. Таким путем можно получить продукт с 0,20% воды и
количеством кислорода, соответствующим содержанию 3% U02F2.
Оказалось также, что удовлетворительная дегидратация UF4H20
может быть достигнута нагреванием его в токе фтористого водорода при
атмосферном давлении. Еще успешнее проходит дегидратация под
пониженным давлением (50 мм рт. ст). при температуре 400° в присутствии
небольшого количества фтористого водорода (5 мм рт. ст.) При этих
условиях оксисоединения практически не получаются, так как присутствие
даже небольших количеств фтористого водорода в весьма заметной степени
задерживает их образование [42].
Как сообщают, гидрат тетрафторида урана UF4-2,5H20 легко теряет
большую часть своей воды при медленном нагревании в вакууме
(давление 2 мм рт. ст.) при 200° в течение 24 час; при этом побочной реакции
не происходит. Последние следы воды удаляются, однако, лишь при
500 — 550° [40]. Иногда в отходящих при дегидратации газах
обнаруживаются следы фтористого водорода. Рекомендуется применять как можно
более глубокий вакуум и постепенно повышать температуру. Согласно
более поздним исследованиям, UF4-2,5H20 дегидратируется нагреванием
в атмосфере сухого фтористого водорода при 450° [43]. Безводный продукт
294 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
сохраняет объемистый внешний вид гидрата и поэтому имеет малый
объемный вес (1,5 г/см3). Гидратирование такого продукта происходит гораздо
легче, чем гидратирование тетрафторида, полученного гидрофторированием
при высокой температуре. Большая удельная поверхность
дегидратированного тетрафторида (от 6 до 8 м2/г) делает его особенно пригодным для
использования в гетерогенных реакциях (т. е. для получения UF6, UF6, U2Fg
и т. п.)
Ниже будет показано (см. стр. 304), что вследствие сравнительно
легкой гидролизуемости тетрафторида желательно осушку его производить
в атмосфере, содержащей фтористый водород.
Получение тетрафторида урана при реакциях в
газовой фазе в условиях повышенных температур. Будучи
приготовлен из водного раствора, тетрафторид почти всегда содержит
небольшие количества воды, а часто и значительные примеси оксисоединений,
которые образуются в процессе дегидратации в результате либо окисления,
либо гидролиза. Могут также присутствовать такие примеси, как
соединения, содержащие олово, цинк или серу (например, сульфаты). Теперь
следует рассмотреть методы, которыми пользуются для получения
свободного от кислорода безводного тетрафторида. Они состоят в обработке
урановых соединений (обычно окислов) газообразными фторирующими агентами
(чаще всего сухим фтористым водородом).
Получение тетрафторида урана гидрофторированием двуокиси урана.
Предпочитают сначала производить операцию восстановления трехокиси
урана водородом, а затем обрабатывать получившуюся двуокись урана
сухим фтористым водородом при атмосферном давлении:
Ш3 + Н2-»и02 + Н20; (24)
U02 + 4HF -> UF4 + 2H20. (25)
Восстановление трехокиси урана проводится при 600 — 700°.
Экспериментальное значение теплоты реакции (25) АН в интервале 600 — 800°
составляет —42,08 ккал [44].
Гидрофторирование является относительно медленной реакцией, и ее
скорость выше 350° мало зависит от температуры [45]. Рекомендуется
применение большого избытка фтористого водорода.
Согласно уравнениям (24) и (25), в качестве исходного материала для
получения тетрафторида пользуются либо трехокисью, либо двуокисью
урана. Возможность применения легко доступного промежуточного окисла,
а именно закиси-окиси урана, зависит от полноты реакции восстановления:
U308 + 2Н2 -> 3U02 + 2Н20. (26)
Утверждают [46], что в обычно применяемых условиях (температура 650—
680° и атмосферное давление) закись-окись урана не может быть полностью
восстановлена водородом до UC^o- Продукт восстановления, полученный
при таких условиях, имеет состав U02,u (или U03-6U02); после обработки
фтористым водородом образуется смесь тетрафторида урана и уранилфторида:
U03 - 6U02 + 26HF -> U02F2 + 6UF4 + 13H20. (27)
Для некоторых целей уранилфторид является весьма нежелательной
примесью.
Получение тетрафторида урана взаимодействием трехокиси урана с
газообразными аммиаком и фтористым водородом. Для получения тетрафторида
в одну стадию смесь аммиака и фтористого водорода пропускают над
трехокисью урана при температуре 500—750° [47]. Реакция идет очень быстро, и
Глава 12. Нелетучие фториды урана
295
в результате ее образуется тетрафторид высокой степени чистоты. С точки
зрения выхода продукта, этот аммиачно-фтористоводородный метод лучше, чем
одностадийный водород-фтористоводородный способ, защищающийся рядом
исследователей.
Получение тетрафторида урана взаимодействием окислов урана с
фторированными углеводородами (фреонами) при высоких температурах. Многие
окислы металлов могут быть превращены во фториды путем взаимодействия
с фторированными углеводородами [48, 49]. Поведение трехокиси урана
изучали в присутствии некоторых фторированных углеводородов [50]. Результаты
изучения находятся в полном соответствии с данными последующих, более
широких исследований, приведших к успешной разработке методов получения
тетрафторида путем взаимодействия фторированных углеводородов с
различными окислами урана [51, 52]. Чистота полученного продукта пока еще не
проверена, но этот метод, повидимому, имеет несомненные преимущества.
Необходимая аппаратура очень проста; она может быть выполнена из стекла,
хотя, возможно, следует предпочесть аппаратуру из графита. Реакция,
повидимому, применима ко всем окислам урана. Образующийся продукт по своим
физическим свойствам отличается от получаемого методом гидрофторирования
и для некоторых целей может оказаться более желательным.
Результаты, полученные при опытах с рядом фторированных
углеводородов и различными окислами урана, представлены в табл. 114. Эти опыты
проводили в трубках из стекла пирекс или вайкор. Для изготовления лодочек можно
применять фторид кальция и полированный графит. В большинстве случаев
пользовались имевшимся в распоряжении графитом. Газообразные фреоны
тщательно осушали, пропуская их над фосфорным ангидридом, так как следы
влаги являются причиной разъедания стеклянных стенок аппаратуры в
начальной стадии реакции. Окислы урана осушали нагреванием при 200—300° либо
в вакууме, либо в токе сухого азота. Во всех случаях применяли большой
избыток фреонов. При использовании жидких фреонов в прибор для
фторирования вводили их пары в токе азота.
Наиболее успешно протекала реакция получения тетрафторида из
трехокиси урана и дихлортетрафторэтана C2C12F4 (фреон 114) при 700°. Этот фреон
при высоких температурах разъедает кварц в меньшей степени, чем какие бы то
ни было другие из исследованных веществ. Тетрафторид хорошего качества
можно получить реакцией между дихлорфторметаном (фреон 12) и двуокисью
урана, но образующийся продукт имеет более низкий объемный вес, чем тот,
который получается в случае фреона 114, и, кроме того, он часто прочно
пристает к лодочкам. Тетрафторид, получаемый при реакциях с фреонами, почти
свободен от хлора, а потери урана в виде летучих хлористых соединений
урана очень незначительны.
Как уже упоминалось, полированный графит является наиболее
подходящим материалом для изготовления лодочек, в которых проводится
фторирование с помощью фреонов. Для изготовления лодочек подходящим материалом
является также фторид кальция, хотя при высоких температурах он,
повидимому, адсорбирует тетрафторид [53]. Металлы заметно разрушаются; никель,
медь, монель-металл, платина и нержавеющая Cr-Ni и Cr-Ni-Mo сталь
подвергаются коррозии и, кроме того, способствуют пиролизу с выделением
углерода. Фреоны в некоторой степени разрушают кварцевую посуду, причем
меньше других в этом отношении действуют фреоны 114 и 12. Чтобы облегчить
проведение реакции, был сконструирован реактор по типу вращающейся
обжиговой печи, в которой реакционная масса находится все время в движении,
благодаря чему все новые и новые поверхности ее непрерывно подвергаются
действию фторирующего агента. Для предохранения кварца от разрушения
графитовую трубку вращают в кварцевой, выложенной внутри графитом [54].
Испытано также действие на трехокись урана трихлорфторметана CC13F (фреон И)
296 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Таблица 114
РЕАКЦИИ ОКИСЛОВ УРАНА С ФРЕОНАМИ [52]
Формула
соединения
CC13F
CC1.F,
CHC18F
С4С1зР8
C.C1.F.
C7F„
Техническое
название
Фреон 11
Фреон 12 а)
Фреон 21 б)
Фреон ИЗ
Фреон 114
Охлаждающая
жидкость
1 С-716
Т.кип.,
°С
23,4
-29,8
8,9
48
3,5
83
Окисел
ио2
Реакцию проводили
при 350°; продукт
содержал много
растворимых хлоридов
урана
Фторирование
протекало довольно
хорошо даже при 350°;
продукт имел низкий
объемный вес
Происходил
глубокий пиролиз с
большим выделением
углерода
Ниже 600° реакция
не шла; при 600—650°
реакция через 1 час
еще не закончилась
Реакция протекала
довольно
удовлетворительно при 675—
700°; продукт имел
низкий объемный вес
При 600° реакция
не шла
ио3
То же, что и для
U02; графитовые
лодочки быстро
разрушались
Реакция протекала
быстро при 400°;
наблюдалась сильная
возгонка из лодочек
То же, что и для
ио2
То же, что и для
U02; вещество очень
прочно приставало к
лодочке
Реакция протекала
быстро при 650—700°;
продукт имел
высокий объемный вес;
происходило
значительное превращение
При 600°
фторирование проходило в
очень малой степени
и8о8
—
Реакция
протекала быстро
при 400°;
вещество очень
прочно
приставало к лодочкам
То же, что ■
для UOt
При 700°
реакция не шла
до конца
—
а) Получался, Невидимому, более растворимый хлорид, чем в случае фреона 114.
*) Пользовались никелевой трубкой, так как в качестве побочного продукта возможно выделение
фтористого водорода.
в жидкой фазе; реакция проводилась в течение 7 час. при 120°, и выход
растворимого в воде продукта, по всей вероятности U02F2, составил 94% [51].
Получение тетрафторида урана из металлического урана или его гидрида
гидрофторированием при высоких температурах. Металлический уран можно
легко превратить в тетрафторид двумя основными способами. Первый
заключается в проведении двух последовательных реакций:
U + 4h2->UH3; (28)
UH3 + 4HF^UF4 + 4h2. (29)
Глава 12. Нелетучие фториды урана
297
Плотный металлический уран превращается сначала в гидрид при 250°. Чтобы
достигнуть полного превращения гидрида в тетрафторид, необходимо
тщательное перемешивание. Для второй стадии, повидимому, достаточна температура
200° [5]. Однако если реакционную смесь не перемешивать, то реакция между
гидридом урана и фтористым водородом при 270° идет неполно вследствие
спекания. Установлено, что если гидрид разложить при 500° и на полученный
тонкораздробленный металл действовать фтористым водородом при той же
самой температуре, то происходит превращение в тетрафторид с хорошим
выходом [55].
По второму способу обе стадии объединены в одну. Произведено
исследование одновременно протекающих реакций водорода и фтористого водорода
с металлическим ураном [56]. Небольшие количества (50 г) металлического
урана при 250° легко превращаются в тетрафторид при действии смеси водорода
и фтористого водорода, взятых примерно в равных молярных концентрациях.
Большие количества урана не могут быть обработаны таким простым путем.
На поверхности плотного куска металла происходит реакция между гидридом
и фтористым водородом, при этом за счет выделяющегося тепла температура
настолько поднимается, что скорость образования гидрида становится
незначительной (эта скорость быстро уменьшается при приближении к 430°, как указано
на стр. 162). Отсюда следует, что для проведения реакции необходимо
охлаждение до 350°. Когда имеют дело с большими количествами урана, следует
применять механическое перемешивание для удаления корки тетрафторида по
мере его образования [57]. С учетом всех этих предосторожностей, можно
считать, что применение одновременно протекающих реакций водорода и
фтористого водорода с ураном целесообразнее, чем применение ступенчатого процесса.
Другие способы получения тетрафторида
урана. В специальных случаях могут быть использованы следующие реакции,
при которых получается тетрафторид.
Реакция между трихлоридом урана и фтористым водородом [3]. При 450°
трихлорид реагирует с сухим фтористым водородом следующим образом:
UC13 + 4HF -* UF4 + 3HC1 + -1Н2 (30)
(см. реакцию между UH3 и HF, стр. 169). Интересно отметить, что при этом
образуется тетрафторид урана, а не трифторид.
Реакция между 1Ю2НР04Н20 и фтористым водородом [58]. При
температурах 350 — 500° единственным продуктом реакции между
1Ю2НР04Н20 и фтористым водородом является уранилфторид U02F2.
Выше 500° происходит довольно неожиданное восстановление; при 800°
в качестве главного продукта реакции получается тетрафторид урана.
Процесс протекает очень быстро.
Реакция между окислами урана и фторидом аммония [21,47,59].
Тетрафторид урана можно легко получить путем реакции между фторидом
или бифторидом аммония и трехокисью урана:
3U03 + 6NH4HF2-^ 3UF4 + 9H20 + N2+4NH3. (31)
Реакция лучше всего идет при температуре 700°. Получающийся продукт
свободен от аммиака. При' более низких температурах (450°) получаются
соединения типа NH4UF5. Для проведения этой реакции над окислом
необходимо пропускать пары фторида аммония, простого сплавления
недостаточно. Большое сходство между этим процессом и описанным выше (с
фтористым водородом и аммиаком) очевидно.
Реакция между тетрахлоридом урана и жидким фтористым
водородом [6]. Реакция между тетрахлоридом урана и жидким безводным
фтористым водородом количественно протекает при комнатной температуре
298 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
с образованием темнозеленого кристаллического продукта. Оказалось, что
полученный тетрафторид урана удерживает значительные количества
фтористого водорода, который не может быть удален даже продолжительным
откачиванием при комнатной температуре. Анализ одной пробы показал,
что состав соединения приблизительно соответствует формуле UF4-HF.
Если это вещество нагревать в течение небольшого промежутка времени
в вакууме (Ь10~3 мм рт. ст.) при 625° (в никелевой трубке, выложенной
платиной), можно получить тетрафторид, совершенно свободный от
фтористого водорода.
Получение тетрафторид а урана из гексафторида. О получении
тетрафторида из гексафторида урана см. на стр. 322.
Физические свойства тетрафторида урана. Температура
плавления. Из горизонтального участка кривой нагревания тетрафторида,
построенной по показаниям оптического пирометра, получено значение
температуры плавления, равное 960 ± 5° [60].
Летучесть. Надежных данных об упругости пара тетрафторида урана
не имеется. Единственное значение 1,9-10"4 мм рт. ст. при 760°,
указанное в литературе [61J, повидимому, согласуется с общими свойствами
тетрафторида. Это соединение не является в заметной степени летучим
даже при нагревании до 800 — 880° в сухом азоте. Наблюдаемая слабая
летучесть может быть объяснена присутствием уранилфторида [52]. В
Англии для очистки тетрафторида применяли возгонку в высоком вакууме
(1-10-6 мм рт. ст.) при температуре 1000° [10]. При опыте пользовались
кварцевой трубкой, выложенной молибденом.
Кристаллографические данные, рентгеноструктура и плотность.
Тетрафторид урана, полученный возгонкой в вакууме при 1000°,
представляет зеленые иглообразные кристаллы длиной в несколько миллиметров
и диаметром около 0,5 'мм. Большинство игл оказывается сдвоенными
параллельно длине. Будучи помещены между скрещенными николями, они
часто создают неполное гашение, которое изменяется от одного участка
кристалла к другому и, вероятно, вызывается искажениями. Кристаллы три-
клинные (см. табл. 115), но все они сдвоены таким образом, что имеют
вид моноклинных.
Таблица 115
ПОКАЗАТЕЛИ ПРЕЛОМЛЕНИЯ И ПЛЕОХРОИЗМ КРИСТАЛЛОВ
ТЕТРАФТОРИДА УРАНА fl7]
Кристаллографическое
направление
а
о
т
п
1,500
1,585
1,598
Плсохроическая окраска
Светлозеленая, почти бесцветная
Изумрудно-зеленая
Изумрудно-зеленая
В результате гониометрического исследования псевдомоноклинных
кристаллов, имеющих грани (310) и (111), были получены следующие
значения для отношений отрезков осей и для угла:
a:b:c=l,282: 1: 1,760(± 0,001); р = 98°49' ± 2'.
Рентгенограммы небольших монокристаллов тетрафторида урана
показывают моноклинную симметрию с двенадцатью молекулами в одной
элементарной ячейке» Приблизительные константы решетки следующие:
ах = 12,79 ± 0,06А; а2= 10,72 ± 0,05 А;
аз = 8,39 ± 0,05 А; а2= 126° 10' ± 40'.
Глава 12. Нелетучие фториды урана
299
Пространственная группа С2/с(С2л). Таким образом, тетрафторид урана
изоморфен тетрафторидам тория, церия, циркония и гафния [62]. Рент-
геноструктура безводного тетрафторида урана одинакова как в случае
осаждения его из водного раствора, так и в случае получения путем
реакции гидрофторирования при высокой температуре.
Значение плотности, вычисленное из рентгенографических данных, равно
6,70 ±0,10 г/см3. Для плотности тетрафторида урана, полученной
экспериментально при работе иммерсионным методом со спиртом, приводятся
значения 6,95 г/см3 [63] и 6,43 г/см3 [64]. Для объемного веса получены
значения, колеблющиеся от 1,50 до 3,5 г/см3. Как уже упоминалось,
объемный вес тетрафторида, осажденного из водного раствора и
высушенного ниже 500°, в общем значительно меньше объемного веса продукта,
полученного при высокотемпературных реакциях.
О
80
280 320
120 160 200 240
Температура, °К
Рис. 44. Молярная теплоемкость и энтропия тетрафторида урана [67].
Достоверных данных о структуре моногидрата UF4-H20 нет. Кристаллы
гидрата 2UF4-5H20 принадлежат к ромбической сингонии и содержат
восемь молекул в элементарной ячейке [62, 65].
ах= 12,75 ±0,04 А; а2= 11,12 ± 0,04 А; а3 = 7,05 ± 0,03А.
Вычисленная плотность равна 4,74 г/см3. Расположение атомов в
пространственной ячейке определяется следующими координатами:
4Ui в±(х1у1±^(^х1 + ± 1-^1^; *,== 0,055; ^ = 0,14;
4Un в ±^x2y2j^x2 + ~^y2-\y>X2^-0t025-t y2= —0,139-
Пространственная группа Рпат.
Термодинамические данные. Удельная теплоемкость, энтальпия и
энтропия тетрафторида урана определены при температурах от 20 до 350° К
[об, 67]. Образец тетрафторида, использованный для этих определений,
содержал 2% уранилфторида; если теплоемкости тетрафторида и уранил-
фторида отличаются не более чем на 10%, то ошибка, возникающая в результате
этого загрязнения, не превышает 0,2%. Работу проводили в калориметре
адиабатического типа [68, 69]. Значения Ср, приведенные в табл. 116,
вычислены на основании плавной кривой, проведенной через опытные точки (рис. 44).
ю to
О СЛ
слослослослослосло
со со
СЛ О
N3 N3 *- •—
СЛ О СЛ О
а
CD CD 00 00 -J ^J
СЛ О СЛ О СЛ О
8
ослосло^сльоо^сл^ослосл
OOOOOcDCDcDcDcDCDcD0000000000«>j->j
СЛ -£* СО Ю ►— (D 00 *«g СЛ J*. Ю •-. CD -4 СЛ СО »- CD СЛ
>J40)050)CnCnCn*.4k4bWWlOtON3b-'- — —
rf^t— OOCnCOCDO^COCDCn >-^W(D^00005W-CD^OiOlM
CO CO CO to •— CDC7>COCD4*.OOb-COCnC7^C7>
CnOt04*.tOCOC>COtOtOCO А. Ю NJ Ф O)
СЛ •— -g to О 00
О CD 0> 00 О 4*.
8
оосл^-слоооо-дслюоо^со'— cdoooocdco •— to o> о
00(0*CЛ►-'4kЮ-^э4k^DOCлOoo*Wl^W*Oooo
о
^. .*. CO
*»4 l О I *>
to to ~
-«4 I — I СЛ
JO I ««4
"ел 1-
co
"to
»— о о о о
£
с»
S&
1
Со
1
со
До
"О
0\
J""*
to
789
*-
745
о
273
CD
724
00
750
->i
806
а>
895
СЛ
020
СЛ
183
4ь
391
со
649
to
961
со I
со 1
_ 1
*»4 1
о>
to
"^ 1
о
00
СЛ 1
to
СЛ
-J 1
to
СП 1
00
Км
•^ 1
-J I
*_
о
->|
83
ю
СП 00 — О О
- СЛ М >-
"i
0 а:
JS.O
1 3-
m
ъ
сг
>
Н
ГО
н а
га «я
а °
> >
§§
£--
> о?
>я
Я
Ci)
Я
н
>
tr
я
я
Я
Глава 12. Нелетучие фториды урана
301
Продолжение табл. 116
Температура,
•к
215
220
225
230
235
240
245
250
255
260
265
270
273,16
275
280
285
290
295
298,16
300
305
310
315
320
325
330
335
340
345
350
СР.
дЖ'М0ль-1'град-1
106,32
107,24
108,10
108,92
109,71
110,47
111,20
111,89
112,56
113,20.
113,81
114,41
114,80
115,03
115,63
116,20
116,75
117,28
117,62
117,81
118,33
118,84
119,35
119,85
120,33
120,81
121,28
121,74
122,20
122,64
5-5об>,
дЖ'Мол ь-1 -град-*
116,9
—
121,7
—
126,3
—
130,9
—
135,3
—
139,6
140,9
—
143,8
—
147,8
—
151,1
151,8
_
155,7
_
159,5
—
163
—
166,8
—
170,4
//-Яов>,
дж^моль-1
13 852
—
14 933
—
16 030
—
17 142
—
18 267
—
19 405
19 767
—
20 556
—
21 717
—
22 674
22 890
—
24 074
—
25 267
—
26 470
—
26 683
—
28 905
а) Данные для теплоемкости были экстраполированы к 0* К по уравнению Ср =-SJR(D+ 4,1.10-er»)
дас*молъ-1'град-1, где D есть функция Дебая с 6=150е К.
Т
6)S-50= I (CpIT)dT.
0
Т
в> Н-Н0= I CpdT.
6
Для реакции образования тетрафторида урана получены следующие
термодинамические данные [70]:
UTB + 2F2ra3->UF4TB; (32а)
— АЯ298*К = 446 ± 2 ккал-моль'1.
При использовании значения энтропии для тетрафторида урана, равного
при 298° К 36,13 энтропийной единицы (151 дж на Г К), вместе со значе-
«иями энтропии для U и F2 было вычислено
— Д^мвчс = 424,6 ккал-моль'1. (326)
302 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Вышеуказанное значение АН получается измерением теплоты
растворения U(S04)2, теплоты образования UF4 • Н20 в водном растворе, теплоты
гидратации UF4 и использованием значений теплот образования U(S04)2,
SOJ- и F", взятых из литературы (табл. 117).
Таблица 117
ТЕРМОДИНАМИЧЕСКИЕ ФУНКЦИИ
Реакция
U(S04)2TB -^U(IV)a, + 2SOra,
U + 2S + 402-> U(S04)2
U (IV)aq + 4Faq + H20 -* UF4.H20TB
uf4.h2otb-^uf4tb+h2o
2S0J-^ 2STB+402
2F2 -> 4Fa9
i/f298°K для UF^'
ккал 'Моль-i
- 24,1±0,2
-534
- 10,3±0,2
+ 3,3+0,3
+ 431,6
-312,8
Уравнение
№
33
34
35
36
37
38
Ввиду большой важности значений термодинамических функций
значения последних в уравнениях (32а) и (326) были проверены [71] другим
методом. UC14TB был превращен в гидратированный UF*TB обработкой
раствором фторида калия (взятым в избытке) и была измерена теплота
гидратации тетрафторида урана (удельная поверхность 6 м2/г) в растворе
фторида калия той же концентрации. Для теплоты образования тетрафторида
было получено значение — 444^2,5 ккал-моль'1. Наиболее вероятным
можно считать среднее из двух определенных значений Д#, а именно
— 443,5 ккал-моль'1. Это значение очень хорошо согласуется с величиной,
полученной при изучении высокотемпературного гидролиза тетрафторида [44].
Теплопроводность твердого тетрафторида урана. Теплопроводность
сплавленного тетрафторида урана при 60° оказалась равной
0,0047±4,3 ккал-см'1-сек-1-град-1 [72].
Магнитная восприимчивость тетрафторида урана и некоторых двои-
ных фторидов урана. Значения магнитной восприимчивости для UF4, KUF6,
K8UF6 и Na3UF7 в интервале от 74 до 300° К были определены [73] по
методу Гая. Результаты опытов приведены в табл. 118.
Таблица 118
МАГНИТНАЯ ВОСПРИИМЧИВОСТЬ ТЕТРАФТОРИДА
УРАНА И НЕКОТОРЫХ ДВОЙНЫХ СОЛЕЙ
Формула
соединения
UF4
KUF5
K2UF6
CaUFe
Na3UF7
Константы Кюри—Вейссаа)
С
1,36
1,30
1,47
1,31
1,45
е, °с
147
122
108
101
290
ц., магнетоны
Бора
3,30
3,30
3,45
3,25
3,40
а> *=с/очеь
Глава 12, Нелетучие фториды урана
303.
Рассчитанный магнитный момент для свободного иона урана с двумя
5/-электронами (состояние 3Н4) равен 3,58 магнетона Бора. Моменты,
полученные опытным путем, довольно близки к этому значению. Хотя все
вышеупомянутые соединения, за исключением K2UF6, во всем интервале
температур подчиняются закону Кюри —Вейсса, исключительно большие значения,
найденные для температуры Вейсса, делают затруднительной интерпретацию
константы Кюри.
Химические свойства тетрафторида урана. Тетрафторид
урана представляет твердое кристаллическое вещество зеленого цвета.
Полученный при высокой температуре, он обычно бывает более темным и
плотным, а также гораздо менее гигроскопичным, чем тетрафторид, полученный
по методу Гроссе [40]. В химическом отношении тетрафторид урана
является устойчивым, довольно неактивным соединением. По физическим свойствам
он напоминает фториды других четырехвалентных элементов, особенно
изоморфные с ним тетрафториды циркония, гафния и тория. Химическое
различие между ними проявляется главным образом в том, что уран может
существовать в целом ряде валентных состояний, в то время как цирконий,
гафний и торий в соединениях с фтором исключительно четырехвалентны.
Действие воды. Растворимость тетрафторида в воде при 25°
приблизительно равна 0,10 г, т. е. 0,32 миллимоля на 1 л [40J. Таким образом, его
молярная растворимость такого же порядка, как и растворимость фторида
кальция. Значение растворимости по английским данным равно 600 мг/л [74].
Наиболее надежными являются следующие значения растворимости
тетрафторида в воде: при 25° —0,004% (вес.) или 1-10~4 моля/л, при 0° — 3-10~5
моля/л и при 60° — 4-10~4 моля/л [75]. При добавлении к воде плавиковой
кислоты до 30%-ной (вес.) концентрации растворимость увеличивается
примерно до значения 0,03 г тетрафторида на 100 г раствора. Вопрос о том,
реагирует ли тетрафторид с водой (кроме гидратации), пока еще остается
сомнительным. По английским данным [76] при кипячении с водой в
течение длительного времени (72 часа) тетрафторид не теряет фтора и не
подвергается гидролизу. По другим данным [77] в течение 24 час. при
комнатной температуре тетрафторид разлагается водой на 85%, переходя при
этом в раствор. При температуре кипения реакция проходит в течение
1 часа. Анализ показывает, что уран в таких растворах уже не являетея
четырехвалентным. Одной из причин расхождения в результатах
упомянутых работ, возможно, является то, что в первой из них тетрафторид
брали в количестве 5 г на 100 мл, а во второй — только 0,3 г на 100 мл,
и в последнем случае количества кислорода, растворенного в воде, могло
быть достаточно для окисления значительной части пробы. Известно, что
небольшие количества избытка фтористого водорода препятствуют
гидролизу. Это подтверждается успешным осаждением тетрафторида из
кипящего водного раствора. Основные фториды урана неизвестны.
Гигроскопичность. Безводный тетрафторид урана на воздухе при
комнатной температуре не обладает заметной гигроскопичностью. В одном случае
через 564 часа наблюдалось увеличение в весе на 0,05% [78]. В связи с этим
важно напомнить, что тетрафторид, полученный из водных растворов, в
частности при дегидратации UF4-2,5H20, гораздо более гигроскопичен, чем тот,
который получается высокотемпературным гидрофторированием.
Действие водяного пара. Непосредственное определение константы
равновесия реакции
U02 + 4HF Z1UF4 + 2H20 (39)
производилось в интервале температур 600—800° [44]. Результаты этих
определений сведены в табл. 119. Теплота реакции АЯ, которая довольно
постоянна в этом температурном интервале, согласно вычислению равна —42,08 ккал.
304 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Это равновесие изучали также в интервале от 200 до 500° [79], для чего
пропускали с различными скоростями водяной пар надтетрафторидом, исследовали состав
газа при каждой скорости и экстраполировали до нулевой скорости. Из
полученных таким образом значений константы равновесия было вычислено среднее
значение А// для этого температурного интервала, которое оказалось равным
—30 кшл. Это, повидимому, достаточно надежная величина, так как теплота
образования тетрафторида, выведенная из этих данных, довольно хорошо
согласуется с общепринятым значением.
Таблица 119
КОНСТАНТЫ РАВНОВЕСИЯ РЕАКЦИИ
ГИДРОФТОРИРОВАНИЯ ДВУОКИСИ УРАНА
Температура,
°С
600
705
800
юооа)
Т
1,145
1,021
0,932
К6)
14,0
1,47
0,155
\пК*
2,639
0,385
-1,865
а) Г—температура, *К.
б) «""Hao/PHF-
в) 1пК=(42080/ЯГ)-21,5.
Очевидно, если тетрафторид урана обработать водяным паром при таких
условиях, чтобы получающийся фтористый водород отводился из системы, то
весь тетрафторид будет превращен в двуокись урана. Константы равновесия
показывают, что гидролиз заметно увеличивается при повышении температуры.
Определены теплоты реакций гидролиза фторидов некоторых двух- и
трехвалентных катионов [80]. Имеющихся в распоряжении авторов
термодинамических данных недостаточно для того, чтобы провести детальное сравнение
свободных энергий реакций гидролиза всех фторидов металлов. Однако по
степени гидролиза все фториды металлов можно разбить на две категории:
1) трудно гидролизуемые фториды одно- и двухвалентных катионов, такие как
фториды натрия, калия, дифториды меди и бериллия; 2) легко Гидролизуемые
фториды трех- и четырехвалентных катионов, такие как трифториды лантана,
алюминия и тетрафториды тория и урана. Экстраполяция свободной энергии
реакции гидролиза тетрафторида урана до 500° приводит к значению
Af500o=-f-2,8 ккал. Столь небольшое значение указывает на то, что гидрати-
рованный тетрафторид следует высушивать при 500° в атмосфере фтористого
водорода, и дает основания полагать, что дегидратацию лучше проводить при
более низких температурах.
Обезвоживание гидратированного тетрафторида, кроме того, осложняется
исключительной его чувствительностью к присутствию следов кислорода. Если
процесс дегидратации продолжается долго и имеются хотя бы следы кислорода,
то тетрафторид окисляется до у ранил фторида. Коль скоро присоединение
кислорода произошло, его не удается удалить и дальнейшей обработкой
фтористым водородом [42, 77].
UF4 + НОН Zl UF3 (ОН) + HF; (40)
UF3(OH) + HOH^UF2(OH)2 + HF; (41)
2UF2 (ОН), + 02 -» 2U02F2 + 2H20. (42)
Гидролитическое превращение тетрафторида урана в окислы урана
использовано как аналитический метод определения фтора в тетрафториде [811. Чтобы
Глава 12. Нелетучие фториды урана
305
достигнуть полноты реакции, гидролиз производят в платиновой аппаратуре
действием перегретого пара при 900°. Пар предварительно перегревается в
кварцевом нагревателе. Плавиковая кислота собирается в платиновом
конденсаторе и определяется титрованием.
Последующие исследования [82, 83] показали, что те фториды (и хлориды),
которые обычно трудно гидролизуются, могут быть легко разложены паром при
1000°, если их смешать с закисью-окисью урана. Равновесие смещается
настолько сильно, что даже такие вещества, как фторид калия или фторид
кальция, при этих условиях очень быстро гидролизуются.
Действие окислителей. В старых работах указывалось, что при
прокаливании на воздухе тетрафторид урана превращается в закись-окись урана, но
предотвращение свободного доступа воздуха (нагревание в закрытом тигле)
приводит к образованию уранилфторида [1, 2, 23]. Совсем недавно появились
указания на то, что тетрафторид урана сравнительно устойчив при нагревании
на воздухе до 200° в течение короткого промежутка времени. При более
высоких температурах он заметно разлагается [41, 77]. Изучено поведение
тетрафторида в чистом сухом кислороде [52, 84], причем получились очень
интересные результаты. При 800° наблюдалась следующая реакция:
2UF4 + 02 -* UFe + U02F2, (43)
При работе с металлической аппаратурой может быть собран гексафторид
урана (более подробно эта реакция будет разбираться на стр. 325). Эти
наблюдения приводят к выводу, что прокаливание тетрафторида в сухом воздухе
рекомендовать нельзя, так как из-за летучести гексафторида можно потерять
заметные количества урана.
Хлор при 500—675° действует на тетрафторид очень слабо [52, 85],
наблюдавшуюся незначительную реакцию можно объяснить присутствием в хлоре
кислорода. Раствор хлора в четыреххлор истом углероде при 125—130° (под
давлением в течение 9 час.) на тетрафторид не действует. Добавление пента-
хлорида урана на их взаимодействие не влияет [86].
Фтор выше 250° реагирует с тетрафторидом, образуя гексафторид [45]
(см. стр. 323):
UF4 + F2 -» UFe. (44)
Фторид кобальта CoF3 переводит тетрафторид урана в гексафторид;
пентафторид сурьмы SbF5 [87] образует с тетрафторидом урана (в эквимо-
лярных количествах) комплекс, термически устойчивый в вакууме до 200°.
При его нагревании до 650° летучих урановых соединений в заметных
количествах не образуется; в остатке же получается 15 — 20% растворимых
в воде соединений урана. В воде комплекс разрушается с образованием
нерастворимого тетрафторида урана и растворимых солей сурьмы.
Кислоты — окислители быстро растворяют тетрафторид урана, и в
растворе появляются ионы уранила. Одним из лучших реагентов для
растворения тетрафторида является дымящая хлорная кислота. Азотная кислота
действует на него довольно медленно; добавление борной кислоты усиливает
растворяющее действие азотной кислоты (так же как и других кислот)
вследствие удаления ионов фтора по мере их образования [77] по
следующей реакции:
2UF4 + 2Н3В03 + 6HNO3 -> 2U02(N03)2 + 2HBF4 + N203 + 5H20. (45)
Другие сильные окислители также реагируют с тетрафторидом с
образованием растворимых солей уранила. Так, тетрафторид растворяется в
растворе сульфата церия. Перекиси щелочных металлов (например, перекись
натрия) и смеси раствора аммиака с перекисью водорода энергично
реагируют с ним с образованием растворимых пероксиуранатов. Реакция с аммиаком
20 Химия урана
306 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонад урана
и перекисью водорода идет очень энергично и должна тщательно
регулироваться. Перекись водорода лучше добавлять к суспензии тетрафторида
урана в 7 н. растворе аммиака с такси скоростью, чтсбы едва
поддерживалось кипение раствора [39]'.
Хлорид и фторид железа (III) реагируют с тетрафтсридом урана с
образованием растворов солей уранила; связывание фтора железом в комплекс
ускоряет реакцию. Низкое значение рН, повидимсму, способствует
окислению и растворению тетрафторида урана фторидом железа (III) [74]. Тетра-
фторид урана быстро растворяется также в кипящем растворе нитрата или
хлорида алюминия, повидимому благодаря образованию устойчивых
комплексных фторалюминат-ионов, например A1F;J~. Одновременно
происходит окисление урана до шестивалентного состояния [88].
Изучено превращение тетрафторида урана в закись-окись при
сплавлении с солями [89]. При сплавлении тетрафторида с KJ03, KBr03, КСЮ3
и со смесью КСЮ3 и KHS04 образуются легко растворимые продукты,
которые, однако, еще содержат фтор. Наиболее удовлетворительно это
превращение проходит в случае нитрата калия. Такие соли, как NH4N03,
(NH4)2C204-H20, KN03, (NH4)2S208 и NH4Br, дают при сплавлении с тет-
рафторидом урана легко растворимые продукты, не содержащие фтора.
Ус ановлено, что оксалат аммония является наиболее эффективным
реагентом из этой группы. Для превращения тетрафторида урана в закись-окись
урана часто применяется также сплавление с борной кислотой. Тетрафюрид
и гексафторид урана могут реагировать друг с другом (см. стр. 314, где
рассматривается получение U2F9, UFC и U4F17).
Реакции с тетрафторидом урана, происходящие без изменения
валентности урана. Холодные концентрированные соляная, серная, фосфорная и
хлорная кислоты лишь едва заметно действуют на тетрафторид. Растворимость
последнего в растворах соляной кислоты резко возрастает с увеличением ее
концентрации. Так, в 1 н. растворе соляной кислоты растворяется 0,18%,
в 6 н. растворе—1,1% и в 12 н. растворе—3,6% тетрафторида. Как уже
отмечалось, добавление борной кислоты сильно увеличивает растворяющее действие
упомянутых кислот, что приводит к образованию растворов соответствующих
солей урана (IV). При выпаривании с серной кислотой до выделения паров
S03 и последующем разбавлении водой тетрафторид растворяется [39].
Тетрафторид довольно легко растворяется в смеси разбавленной серной
кислоты и кремнезема; при этом получаются U(S04)2 и H2SiF6.
Горячая фосфорная кислота растворяет тетрафторид с образованием фосфата
урана (IV). Тетрафторид урана также умеренно растворим в 0,4%-ном
растворе оксалата аммония. При нагревании с раствором едкого натра он
превращается в гидратированную двуокись и02-л:Н20 [1, 2].
С безводным хлоридом алюминия тетрафторид реагирует при умеренно
повышенных температурах с образованием тетрахлорида [90]:
3UF4 + 4А1С13 '50-500U 3UC14 + 4A1F3. (46)
Для этой же цели можно также использовать двойную соль NaAlCl4 [91 ]. Этот
метод с успехом может быть применен для лабораторных условий. Хлориды
металлов, например калия, марганца (II) и цинка, а также кремния,
как можно ожидать на основании термохимических данных, не способны
вступать в подобную реакцию.
Действие восстановителей. Чистый водород при 1000° восстанавливает
тетрафторид урана до трифторида (см. стр. 28Ь). В результате появления
следов влаги при действии атомарного водорода на кварц, из которого сделан
реакционный сосуд, водород, взаимодействуя с тетрафторидом при 700°,
дает смесь трифторида и двуокиси урана [92].
Глава 12. Нелетучие фториды урана
307
Металлический уран восстанавливает тетрафторид до трифторида (см.
стр. 287). Щелочные металлы, так же как и щелочноземельные, особенно
кальций и магний, восстанавливают тетрафторид до металлического урана
(см. стр. 112).
Комплексные соединения тетрафторида урана. Тетрафторид образует
с фторидами металлов ряд двойных солей. Впервые соединения этого типа
были обнаружены еще на ранней стадии развития химии урана. Так, в 1866 г.
Болтон наблюдал образование зеленого нерастворимого в воде соединения
в результате действия на раствор уранилфторида, содержащий фторид калия
и муравьиную кислоту, сильного солнечного света [1, 2]. Фотохимическое
восстановление ионов уранила в растворе, содержащем фторид натрия или
фторид калия, может быть также достигнуто (на солнечном свету) с помощью
спирта, этилового эфира или глюкозы [93]. Во всех случаях получается зеленое
вещество, по внешнему виду напоминающее тетрафторид урана. Это соединение
плавится на воздухе с выделением фтористого водорода; остаток,образующийся
после длительного нагревания, состоит из ураната калия или натрия. С сухим
водородом эти комплексные соли реагируют лишь медленно, в воде или в
разбавленных кислотах практически не растворяются; они разлагаются горячей
концентрированной серной кислотой с выделением фтористого
водорода. Концентрированная соляная кислота растворяет эти соли медленно.
В старых работах для этих соединений были предложены формулы NaUF5
и KUF6.
Совсем недавно система NaF — UF4 была подвергнута более подробному
изучению [94]. При помощи термического анализа установлено, что
существует только одно соединение NaUF6 (т. пл. 714 ±10°). Найдены две
эвтектические смеси: NaF — NaUF6 (т. пл. 600±10°) с 26% (мол.) тетрафторида
и NaUF6 — UF4 (т. пл. 650±10°) с 67% (мол.) тетрафторида.
Системы NaF — UF4 и KF — UF4 были еще раз исследованы рентгено-
структурным методом, в результате чего было установлено, что как
данные, так и другие подобные системы являются гораздо более сложными,
Таблица 120
ФАЗОВЫЙ СОСТАВ СИСТЕМЫ NaF—UF4 [97]
UF4,
% (мол.)
67
50
40
36
33 а)
33 б>
33 в)
31
29
27
25
■20
Присутствующие фазы
•'
основная
NaUF5 + UF4
NaUF5
a-Na2UFe
<x-Na2UFe
T-Na2UF6
rNa2UFe
P2-Na2UFe + a-Na2UFe + 7-Na2UFe
7-Na2UF6
Na3UF7
Na3UF7
Na3UF7
, Na3UF7
второстепенная
p2-Na2UFe + a-Na2UF6
NaF
а) Наименьшая скорость охлаждения.
б) Средняя скорость охлаждения.
в) Высшая скорость охлаждения.
20*
308 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
чем это ранее предполагалось [95 — 97]. Фазы, которые были
идентифицированы в этих системах, указаны в табл. 120 и 121. Эти фазы получали
быстрым плавлением компонентов в платиновом сосуде и затем давали им
охладиться.
Таблица 121
ФАЗОВЫЙ СОСТАВ СИСТЕМЫ KF—UF4 [97]
UF4,
% (мол.)
89
86
83
80
75
67
60
50
45
40
36
33а>
33б)
33 в)
33г>
29
25а>
25б>
22
20
17
14
Присутствующие фазы
основная
KU,FM+UF4
KUbF25
KU6F25
KU2F9 + KUeF25
KU2F9 + KU6F25
KU2F9
KU2F9+UF5
KUF5
1 KUF5
KUFs + pi-KjUFe
Pi-K,UF,
Pi-K,UF,
P,-K2UF,
pJ.K2UFe«>
Pt-K.UF,
fii-KaUF. + a-KsUFT
<x'-KsUF7
a-K3UF7
a-K3UF7
a-K3UF7
a-K3UF7
a-K3UF7
второстепенная
KU2F, + UF4
KU2Fe
PrK2UFe
a-K2UFe
a-K2UF,
KF
KF
в виде следов
UF4
UF4 + KU3F,S
KU3F„ + UF4
KU,F„+KU,F1S+KUF»
KF
а) Наименьшая скорость охлаждения.
б) Большая скорость охлаждения.
в) Высшая скорость охлаждения.
г) Наивысшая скорость охлаждения.
Д) p[A2XF(j является неупорядоченной структурной формой fiAfcXFe, образование
которой вызывается изоморфным взаимным замещением атомов А и X.
В некоторых случаях применяли быстрое охлаждение. Фазы были
идентифицированы рентгеноструктурным методом, причем оказалось возможным
на основании рентгеноструктурных данных установить химические формулы.
Данные о кристаллической решетке отдельных соединений приведены
в табл. 122.
Ниже кратко отмечены некоторые особенности кристаллических
решеток части этих соединений [98].
NaUF6. Эта фаза является ромбоэдрической. В элементарной ячейке
содержится одна группа из шести атомов натрия, одна группа из шести
атомов урана и пять групп из атомов фтора с расположением атомов
Глава 12. Нелетучие фториды урана
309
Таблица 122
ДАННЫЕ О КРИСТАЛЛИЧЕСКИХ РЕШЕТКАХ СИСТЕМ KF—UF4 и NaF-UF4 [97]
Формула
соединения
KUeF26
KU3F13
KU2F9
NaUF6
KUF6
«-Na2UFe
a-K2UFe
PrK2UFe
Pa-K2UFe
P2-Na2UFe
T-NaUFe
a-K3UF7
a'-K3UF7
Na3UF7
Симметрия
Гексагональная
СЪ/ттс
Ромбическая
Pmctn
Ромбическая
Рпат
Ромбоэдрическая
Ромбоэдрическая
R2>
Кубическая; типа
флюорита
Кубическая; типа
флюорита
Гексагональная
С62т
Гексагональная
С32
Гексагональная
С32
Ромбическая
Immtn
Кубическая
Тетрагональная
lAjamd
Тетрагональная
П/ттт
Константы элементарной
ячейки, А
fli= 8,18±0,01;
а3 = 16,42±0,02
д1== 8,03 ±0,03;
аа= 7,25 ±0,03;
а3= 8,53 ±0,04
аг= 8,68±0,01;
а2= 7,02±0,01;
а3=11,44±0,04
а = 9,08 ±-0,01;
а =107°56/
а =9,387 ±0,002;
а = 107°15'±2'
а = 5,565 ±0,004
а = 5,934 ±0,001
ai== 6,54 ±0,01;
а3= 3,76 ±0,01
fli= 6,53 ±0,02;
а3= 4,04 ±0,01
fli= 5,94 ±0,01;
а3= 3,74 ±0,01
а1= 5,56 ±0,02;
а2= 4,01 ±0,01;
а3=11,64 ±0,04
а= 9,21 ±0,01
fll=: 9,20 ±0,02;
а3=18,40 ±0,06
fll= 5,448 ±0,07;
а3 = Ю,896±0,014
Количество
молекул '
в
элементарной ячейке
2
2
4
6
6
4/з
4/з
1
1
1
2
4
8
2
Р, г/см*
6,73
6,64
6,49
5,81
5,38
5,08
4,53
5,10
4,77 .
5,74
5,06
4,12
4,13
4,49
±(xyz)(yz х) {zx у). Координаты х, у, г для атомов натрия и урана следующие:
X
6
13
3
13
У
2
13
1
13
Z
5
13
9
13
Расположение атомов натрия и урана соответствует гранецентрированной
псевдокубической решетке, в которой каждое тринадцатое место свободно.
Псевдоячейка является ромбоэдрической, объем ее составляет 4/13 объема
истинной ячейки, а параметры имеют следующие значения: а — 5,7Аа = 90°5Г.
Структура NaUF6 близка к структуре флюорита.
a-Na2UFe. Кубические а-фазы образуются при быстром охлаждении
расплавов, содержащих немного более 33% (мол.) тетрафторида. Область
гомогенности а-фазы простирается до содержания тетрафторида, равного 40% (мол.).
Ее химический состав может быть представлен в виде формулы М4-.* Ц* F-и-з*»
310 Часть IV. Галогениды оксигалогениды, боргидриды и карбонил урана
где х может иметь значения между 1,33 (что соответствует Na2UFe)
и 1,60. Фаза является гранецентрированной кубической. В случае
соединения Na2UFe константы решетки изменяются вместе со значением х
следующим образом:
х а, А р, г/см9
1,33 5,565 ±0,005 5,08
1,45 5,578 ±0,005 5,35
1,60 5,601 ±0,001 5,66
Такая структура является неупорядоченной структурой типа флюорита.
В каждой элементарной ячейке имеется четыре атома натрия и один атом
урана, статистически распределенных в положениях (OOOKyyOjf^O-o-y
С 0 у у )• Там же содержится от 8,0 до 8,8 атома фтора. При # = 1,60
элементарная ячейка должна содержать больше восьми атомов фтора, но не
меньше четырех атомов металла. Это заключение является прямым
следствием увеличения периода решетки по мере повышения содержания
фтора. Из всех атомов фтора в ячейке восемь атомов находятся в положе-
/111\/133\/313\/331\
ниях ± (^ТТТДТТТ) (дтт)(,ТТ4\)' т' е" занимают такие
же места, как и анионы в структуре флюорита. Остальные атомы
фтора статистически распределены в положениях (о 0 2; (^"2" О
( ~2~ ^ ® )\ ~2~~2~~2~ )' ^сли более 20% этих мест занято, система становится
нестабильной относительно NaUF5.
• Соединение a-K2UF6, изоморфное соединению a-Na2UF6, получается
осаждением из водного раствора, содержащего большой избыток ионов калия.
P-K2UF6. (3-фазы K2UF6 образуются в виде чистых фаз из расплавов,
содержащих 33% (мол.) тетрафторида урана. При медленном охлаждении
получаются ргфазы. При быстром охлаждении образуется неупорядоченная
Р^-фаза K2LrFG, при мгновенном же охлаждении образуется р2-фаза. Быстро или
мгновенно охлажденные расплавы Na2UF6 дают смесь а-, р2- и ?-фаз Na2UFe.
prK2UF6 имеет гексагональную симметрию и следующие
пространственные координаты атомов:
1U в (000)
2Kb ±(44т)
3F, в (*о|) (0*^(^*1)
3Fn в (х20 0)(0*20)(х2х20),
где ^ = 0,220 и *2 = 0,640.
В полностью неупорядоченной р^-структуре атомы урана и калия
распределены в положениях (0 0 0) и ± Г "т "з" Т У статистически- Были
получены образцы с различными степенями неупорядоченности.
Структура р2-фазы также гексагональная, но принадлежит к другому
(хотя и близкому) структурному типу с более низкой симметрией.
1U в (0 0 0)
2 К или 2Na в ± (^ ^и)
3F, B(x10|)(0x1|)(i1i1i)
3Fii в (x20 0)(0x20)(*2x20).
Глава 12. Нелетучие фториды урана
311
Для P2-K2UFe « = 0,450, jq = 0,190 и *8= 0,640; для p2~Na2UFe « = 0,385,
хх = 0,250 и х2 0,600.
Соответственно ионному радиусу координационное число натрия по
отношению к ионам фтора не может быть равно 9, как у калия в (VK2UFe.
Поэтому при замене калия натрием структура изменяется от (Зх к р2.
7-Na2UFe. Химический состав этой фазы вполне определенный. Это
соединение получается в чистом виде при медленном (без принудительного
быстрого охлаждения) остывании расплава, имеющего состав Na2UFe. Фаза
имеет ромбическую симметрию. Пространственные координаты атомов
следующие:
(ooo)(ttt) + 2Ub (00°)*
4Na в ±(0 0«), где tt = -g*
4F в ±Qj—0jt где а = —
8F в ± (х 0 z)(x 0 г), где я = -^- и z = -g-.
Каждый атом урана и натрия связан с восемью атомами фтора, причем
с четырьмя на расстоянии Na — F = U — F = 2,38 А и с четырьмя другими на
расстоянии Na~F = U- F = 2,44 A.
Na3UF7. Эта фаза является единственной, получающейся из расплавов,
содержащих от 25 до 29% (мол.) тетрафторида урана. Тонкие зеленые
кристаллы этого вещества оптически одноосны. Химическая формула для этой
фазы может быть представлена следующим образом: (Nae.x + Ux) UaF14+3xt
где 0 < х < 0,30. Значение х^0 соответствует формуле Na3UF7; когда х
превышает 0,3, фаза Na3UF7 становится неустойчивой относительно фазы
Na2UF6. Фаза Na3UF7 является тетрагональной, период решетки изменяется
с изменением х, константы решетки для крайних в приведенном интервале
значений х следующие:
х alt А а3» А р, г/смг
0 5,448 ±0,007 10,896 ± 0,014 4,49
0,29 5,503 ±0,005 11,006 ±0,100 4,85
Каждый атом металла связан в среднем с 7,0 — 7,4 атомами фтора при
расстоянии между атомами Na — F = U — F = 2,36-;- 2,38 А. Элементарная
ячейка и структура Na3UF7 соответствуют двум элементарным ячейкам
флюорита; атомы урана и натрия занимают соответственно четверть и три
четверти всех мест, занимаемых атомами металла. Два из шестнад iara
атомов фтора удаляются.
Осаждением из водных растворов был получен ряд соединений типа
MUFe(M = Ba, Pb, Sr). Все эти соединения имеют такую же структуру,
как трифторид лантана и следующие константы решетки [99]:
flit A a3, A
BaUFe 4,265 ± 0,005 7,456 ± 0,015
PbUF6 4,175 ± 0,005 7,337 ± 0,015
SrUFe 4,103 ± 0,005 7,290 ± 0,015
Ввиду низких температур плавления (по сравнению с тетрафторидом
урана) системы NaF-UF4 и KF — UF4 приобрели большое значение как
возможные электролиты, применимые для получения металлического урана
высокотемпературными электрохимическими методами (см. стр. НО).
* Здесь, повидимому, авторами допущена ошибка.—Прим. ред.
312 Часть. IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
ПРНТАФТОРИД УРАНА И ПРОМЕЖУТОЧНЫЕ ФТОРИДЫ U2Fe и U4F17
Впервые пентафторид урана описан Руффом и Хайнцельманом [100],
получившими его по реакции между безводным фтористым водородом
и пентахлоридом. Долгое время пентафторид урана был единственным
известным соединением с составом, промежуточным между тетрафторидом
и гексафторидом урана.
Однако существуют по крайней мере два промежуточных соединения,
для которых предложены формулы U2F9 и U4F17. Открытие этих
соединений явилось результатом изучения реакции между тетрафторидом и
гексафторидом, оказавшейся неожиданно сложной.
Исторический обзор. В 1941 г. Гроссе [101, 102] предложил
для получения пентафторида следующую реакцию:
UF4 + UF6 125° , 2UF5. (47)
При попытке синтезировать пентафторид по этому методу [103] был получен
продукт черного цвета (вместо ожидаемого белого или серого).
Химический анализ показал, что это черное вещество соответствует приблизительно
формуле UF4, однако на основании рентгеноструктурных исследований [11]
было установлено, что соединение имеет кубическую симметрию (а0 = 8,455 А.),
в то время как тетрафториду соответствует моноклинная, а пентафто-
риду —тетрагональная симметрия. Объемные соображения, основанные на
величине ионного радиуса F" в других урановых соединениях, показали,
что отношение числа атомов фтора к числу атомов урана в этом черном
веществе равно 4 ± 0,3. Поэтому было сделано предположение, что это
новое вещество является кристаллической модификацией тетрафторида
урана. Оно получило название «черный тетрафторид урана». Некоторым
подтверждением этого взгляда явилась возможность превращения черного
тетрафторида при стоянии или при умеренном нагревании с добавлением
в качестве затравки кристаллов моноклинного тетрафторида в обычный
тетрафторид [104].
Вскоре выяснилось, что вещество типа черного фторида образуется
при самых разнообразных условиях. Так, при пропускании смеси гексафто-
рида урана с водородом через нагретую трубку (при 400 — 600°)
получалось черное, как смоль, твердое вещество с составом, приближающимся
к тетрафториду. В этом случае также был сделан вывод о том, что
образуется новая кристаллическая модификация тетрафторида [105].
Подобное же вещество было получено в Англии при опытах по
изучению коррозии. На образцах мягкой стали, подвергнутых действию паров
гексафторида урана, образовывалась черная пленка, имевшая состав FeF2- 2UF6
(или FeF3'U2F9) [106]. Исследованию подвергалось также комплексное
соединение UF4~UFG. Это соединение было получено обработкой
тетрафторида жидким гексафторидом при 150 — 200°; комплекс представлял
коричневый порошок, имевший состав UF4.4 [107].
Изучение реакции между твердым тетрафторидом и газообразным
гексафторидом показало, что при подходящих условиях в результате этой
реакции может образоваться а- или р-форма UF5, U2F9 или U(JF]7. Состав
соединений U2F9 и U4F17 был установлен химическим анализом. Для справки
в табл. 123 приведены вычисленные значения общего количества урана,
урана (IV) и фтора, содержащихся в различных фторидах урана.
Общее содержание урана и фтора сравнительно мало зависит от
изменений в составе соединений от тетрафторида до пентафторида. Поэтому
гораздо более удобным показателем состава является содержание урана (IV)
(табл. 123). В табл. 124 приведены результаты нескольких анализов
черного фторида [108].
Глава 12. Нелетучие фториды урана
313
Таблица 123
СОСТАВ РАЗЛИЧНЫХ ФТОРИДОВ
Формула
соединения
UF4
U«F„
U2F„
UF5
UFe
U (общее
количество),
%
75,8
74,7
73,6
71,5
67,7
и (IV),
%
75,8
65,4
55,2
35,8
0,0
F,
%
24,2
25,3
26,4
28,5
32,3
F/U
4,00
4,25
4,50
5,00
6,00
-H-OXLioo
100,0
87,5
75,0
50,0
0,0
Таблица 124
СОСТАВ ЧЕРНОГО ФТОРИДА U2F9
Образец
I
II
III
и, %
72,8
72,8
72,1
U (IV), %
54,0
53,3
52,0
F, %
25,6
27,1
26,4
F : Ua)
4,41
4,67
4,59
а) При сравнении отношения фтора к урану, полученного в результате
химических анализов, с отношением, рассчитанным по потере в весе (за счет
улетучивания гексафторида) при нагревании около 34 0° в течение 24 час.
под давлением 10~5 мм рт. ст., оказалось, что результаты хорошо
совпадают. Потеря в весе составляет 4,39%.
Результаты химических анализов нескольких проб вещества с
предполагаемой формулой U4F17 (впервые оно было обнаружено рентгенострук-
турным анализом) подтвердили правильность этой формулы (табл. 125) [109].
Таблица 125
СОСТАВ U4Fi7(UF4,25)
Элемент
U (общее количество)
U (IV)
F
на основании
прироста
в весе а)
74,8
65,7
25,2
Состав
по данным
химического
анализа
74,8
б4,1±1,1б>
25,3±0,2В>
%
вычисленный
UF4,2o
74,9
67,4
25,1
UF4,26
74,7
65,4
25,3
UF4,3o
74,5
63,4
25,5
а) На основании прироста в весе при действии газообразного гексафторида урана
на твердый тетрафторид при давлении 17 мм рт. ст. (300°).
б) Среднее из шести определений.
в) Среднее из двух определений.
Существование такого соединения, как U2F9, доказывается тем, что
образцы U2F9, полученные разными способами, имеют одинаковые параметры
решетки (как показано точными рентгеноструктурными исследованиями).
Существование U4F17 как чистого соединения не так уж определенно, но-
очень мало причин сомневаться в этом.
314 Часть IV. Гпюгвниды, оксигалогениды, боргидриды и карбонил урана
Получение пентафторида урана. Реакция между
тетрафторидом и гексафторидом.
Реакция между тетрафторидом ургна и газообразным гексафторидом
протекает в несколько стадий, и в зависимости от температуры и
парциального давления гексафторида образуются следующие соединения: U4F17,
U2F9, a-UF5 или P-UF6. Условия равновесия для образования
различных продуктов приведены в табл. 126 и на рис 45.
Таблица 126
УСЛОВИЯ ПОЛУЧЕНИЯ ПЕНТАФТОРИДА УРАНА И ПРОМЕЖУТОЧНЫХ
ФТОРИДОВ ПО РЕАКЦИИ МЕЖДУ ГАЗООБРАЗНЫМ ГЕКСАФТОРИДОМ
И ТВЕРДЫМ ТЕТРАФТОРИДОМ
РиРб. мм рт. ст. а)
17,7
120—140
100°
200°
U2F9
a-UF5
320°
U4F17
U2F9
а) Два указанных давления равны давлению паров твердого
гексафторида при 0е и комнатной температуре соответственно.
б) Вероятно, при этих условиях должно происходить образование
Скорость, с которой достигается равновесие (и, следовательно, чистота
продукта при данных условиях), зависит главным образом от природы
тетрафторида урана.
Последний, приготовленный из водного раствора и затем
дегидратированный, имеет удельную поверхность 6-^-8 ж2/г (в то время как
поверхность тетрафторида, приготовленного высокотемпературным
гидрофторированием, равна 0,25 м2)г). Для быстрого достижения равновесия
предпочтительно применять тетрафторид с большой удельной поверхностью. На
скорость реакции сильно влияет также спекание, которое наблюдается при
температурах выше 200°, когда устойчивыми фазами являются UF5 или
U2F9-
Реакция между тетрафторидом и жидким гексафторидом подробно не
изучена. При 100—125° получается P-UF5, при условии использования
тетрафторида с большой удельной поверхностью В случае тетрафторида,
имеющего малую удельную поверхность, или при проведении реакции в течение
времени, недостаточного для достижения равновесия, получается U2F9. Эта
разница в реакционноспособности тетрафторида и послужила, повидимому,
причиной первоначального открытия черного фторида. Хорошим методом
получения P~UF6 является реакция между тетрафторидом, имеющим большую
поверхность, и жидким гексафторидом, проводимая при 125° в течение
недели. В общем, однако, можно считать реакции с газообразным
гексафторидом более предпочтительными, по крайней мере до получения более
подробных сообщений о реакциях в жидкой фазе.
Первоначальный метод Руффа и Хайнцельмана [100] для получения
пентафторида урана был повторно подвергнут исследованию [52, 86, ПО].
Пентахлорид урана обрабатывали избытком жидкого фтористого водорода
в течение 12 час. в никелевом или платиновом сосуде при комнатной
температуре, после чего избыток фтористого водорода и хлористого водорода
удалялся в токе азота. Пентафторид урана получался в р-форме. Для
проведения подобной реакции может быть также применен гексахлорид урана.
Глава 12. Нелетучие фториды урана
315
В этом случае реакция протекает следующим образом:
2UCle+10HF -> 2UF6 + 10HC1 + C12.
{48)
Чтобы получить пентафторид урана, свободный от тетрафторида, гексафто-
рид должен быть очень тщательно очищен от примеси органических
веществ (возгонкой), фтористый водород должен быть освобожден от следов
.влаги (предварительной обработкой пентахлоридом урана). Другим удобным
10*
F-1—I—Г
1—I Г
1 Г
| В
Тройная точка
Рис. 45. Давления диспропорционирования U4F17, U2F9,
a-UF5 и 3-UF5.
методом получения пентафторида урана является действие газообразного
фтористого водорода на пентахлорид при 300°. При этом получается
а-форма.
Действием^ фтора на тетрафторид урана также может быть получен
пентафторид:
UF4 + 4-F2
150—250°
UF6.
(49)
При более высоких температурах (приблизительно при 370°) главным
продуктом реакции является гексафторид, но при температуре 150—250° может
быть получен a-UF5 высокой степени чистоты, особенно если применять
тетрафторид с большой удельной поверхностью. Применением фтора под
316 Часть /V. Галогениды, оксигалогениды, боргидриды и карбонил урана
давлением 7 am [102] превращение тетрафторида в пентафторид может
быть достигнуто при температуре ниже 100°. Эта реакция тщательно не
изучена; иногда в результате ее образуются темноокрашенные продукты.
Реакцию можно затем закончить в более мягких условиях в токе фтора.
Диспропорционирование пентафторида урана и
промежуточных фторидов [111]. Измерены давления гексафторида урана
над а- и fMJF6, U2Fg и U4F17 при их диспропорционировании. Результаты
представлены в табл. 127 и на рис. 45.
Таблица 127
РАВНОВЕСНЫЕ ДАВЛЕНИЯ И ТЕРМОДИНАМИЧЕСКИЕ КОНСТАНТЫ ПРОЦЕССОВ
ДИСПРОПОРЦИОНИРОВАНИЯ
Реакция
3a-UFt^U2Fe+UFera3
3p-UF8^±U2F9+UFera3
ViUjF.Ji'/iUJ'M+UF.ra,
2U4F17^7UF4+UFera3
Равновесное
давление
паров а)
А
2942
4166
7315
7143
В
7,634
10,71
13,68
12,75
Температурный
интервал.
100—200
100—152
225—320
270-350
Термодинамические константы
АН208.
ккал >моль-1
UFe Газ
13.46 ± 0,2
19,06 + 0,5
33.47 ± 1
32,68 ±0,4
AS2 8.
энтропийные
единицы
21,7±0,3
35,8±0,7
49,4± 1,4
45,2±0,6
AF2D8.
ккал - моль-\
UF6 газ
6,99±0,4
8,40±0,8
18,8 ± 1,7
19,2 ±0,7
а) ^Рммрт. ст.=—+£.
Термохимия промежуточных фторидов [112]. Из данных
табл. 127 и 128 могут быть вычислены термодинамические функции для
промежуточных фторидов (табл. 129).
Таблица 128
ТЕРМОХИМИЧЕСКИЕ КОНСТАНТЫ УРАНА И НЕКОТОРЫХ ФТОРИДОВ
Формула соединения
UF6 газ
UF4
и
F2 газ а>
АН298»
ккал -моль-1
— 504 ±3
-443,5 ±2
S298*
энтропийные
единицы
90,3 +1
36,1 ±0,1
12,0 ±0,03
48,58 ±0,1
AF298,
ккал -моль-1
483,9 ±3
421,7 ±2
а) По Келли [113].
Таблица 129
ТЕРМОХИМИЧЕСКИЕ КОНСТАНТЫ ПРОМЕЖУТОЧНЫХ ФТОРИДОВ
Формула
соединения
a-UFs
p-UF5
U2F,
U4F17
ДН|В8|
ккал-моль-i
— 483,7± 1,3
— 485,2 ±1,4
— 933,8 ±3
— 1820,5 ±4
S298.
энтропийные
единицы
48,0 + 0,4
43,3±0,5
75,3±0,6
149,0±0,6
AF298,
ккал -мол6-1
— 458,2 + 1,4
— 458,7 ±1,5
— 884,0 ±3
— 1727,5 ±4
Глава 12. Нелетучие фториды урана
317
Измерена теплоемкость ot-UF6[112]. Исследованный образец
содержал только 82,82% пентафторида урана, и, следовательно, результаты не
являются очень точными. Значение 5298 (45 ± 3 энтропийные единицы),
полученное из измерений теплоемкости, является промежуточным между
значениями, вычисленными выше для а- и P-tJF6. Результаты измерений
теплоемкости представлены в табл. 130.
Таблица 130
ТЕПЛОЕМКОСТЬ И ЭНТРОПИЯ ДЛЯ «-UF6 a)
_%.
Температура,
°к
0
25
50
75
100
125
150
175
200
225
250
275
/300
310
сР,
кал -моАЬ-1-град-1
0
4,23
10,24
15,43
19,11
21,79
23,97
25,77
27,30
28,61
29,71
30,74
31,69
32,04
Я-Яо,
кал-моль-*
0
39
219
544
978
1490
2 063
2 685
3 349
4 049
4 778
5 534
6314
6 633
S-Sq.
кал-моль-1-град-1
0
2,23
6,99
12,18
17,16
21,72
25,89
29,72
33,27
36,56
39,63
42,52
45,23
46,28
(tf-tfo)-r(S-So).
кал»моль-1
0
- 17
-130
-370
-737
-1 224
-1821
-2516
-3 305
-4 178
-5131
-6 158
-7 255
-7713
1) Кристаллографическая модификация этого образца неизвестна.
Физические свойства и кристаллические структуры
промежуточных фторидов. Чистый как а-, так и [3-UF6, вероятно,
бесцветен, так как полученные образцы были почти белыми. Однако
получены вещества (данные анализа которых свидетельствовали о том, что это
пентафториды), имевшие зеленую (UF4 + UFr,ra3)» светлосерую (UF4-fF2)
или даже коричневую (HF+UC15) окраску. Все эти различно окрашенные
образцы дают рентгенограмму, характерную для пентафторида урана.
U2F9 и U4F17 имеют черную окраску.
Структура ct-UF5 [114, 115J. ot-UF5 имеет тетрагональную симметрию,
размеры элементарной ячейки его следующие:
ах = 6,512 ± 0,001 А; а3 = 4,463 ± 0,001 А.
В каждой элементарной ячейке находятся две молекулы; вычисленная плот-
кость равна 5,81 г/см3. На основании геометрических соображений
сделано заключение, что структура ct-UF5 относится к пространственной группе
/4//и (CJh) со следующими координатами атомов:
2U в (000) (44 т)
2F, в (0<4ХТТ0)
8F„ в ±(и»0)Й0)(и + уо4т)(о + ТТ-а т)»
где ы = 0,315 и 0 = 0,113.
318 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Каждый атом урана окружен октаэдрической группировкой из шести
атомов фторао с междуатомными расстояниями U — 2Fi = 2,23A и
U —4Fh = 2,18A. Таким образом, октаэдр вытянут вдоль оси а3; каждый
атом Fi входит в два последовательных октаэдра, и при этом образуются
длинные нити октаэдров вдоль оси а3. Из расстояний между атомами урана
и фтора при допущении в основном ионной связи вычислен радиус иона
иБ+, который оказался равным 0,87 А; он немного меньше радиуса иона
U4+, равного 0,89 А.
Все атомы урана в этой структуре эквивалентны, а поэтому соединение
нельзя считать двойным фторидом четырех- и шестивалентного урана, т. е.
UF4UF6. Этот вывод подтверждается тем, что пентафторид урана
бесцветен.
Структура p-UF5 [115]. P-UF5 также имеет тетрагональную симметрию;
размеры элементарной ячейки следующие:
аа= 11,450 ±0,002 А; а3 =-5,198 ± 0,001 L
В каждой элементарной ячейке находится восемь молекул; вычисленная
плотность равна 6,45 г/см3, т. е. она приблизительно на 10% больше, чем
у а-формы. В этом случае также не было проведено достаточно полных
измерений, чтобы можн<з было указать точную структуру. Однако на
основании данных, включающих ионные объемы, сделано предположение,
что структуры p-UFc относятся к пространственной группе /42d (D").
В предполагаемой структуре каждый атом урана связан с семью атомами
фтора, причем расстояния между ними следующие:
U-lFi = 2,18A; U-2Fn- 2,23 A; U-2Fn = 2,29A; U-2Fm = 2,18 A.
Среднее значение для расстояния U —7F равно 2,23А. Из семи углов
многогранника, образованного атомами фтора вокруг каждого атома урана,
четыре принадлежат одновременно смежным многогранникам.
Как и в случае a-UF5, P~UF6 нельзя рассматривать как UF4-UF6,
так как здесь все атомы урана также эквивалентны друг другу.
Взаимное превращение <ь и p-UF5. Температура перехода p-UF5
в a-UF5 (125° при давлении гексафторида урана 1,76 мм рт. ст.)
вычислена из уравнений равновесных давлений гексафторида над а-и P-UF5,
причем a-UF6 устойчив выше 125°. Чтобы произвести полное превращение (З-формы
в a-форму, может потребоваться 12-часовое нагревание при 185°, так как
при более низких температурах скорость перехода очень мала. Обратное
превращение a-формы в Р-форму пока не было достигнуто.
Структура U2F9 [116, 117]. Кристаллы U2F9 имеют кубическую
симметрию с a0 = 8,45384 ± 0,0019 А. Элементарная ячейка содержит восемь
молекул; вычисленная плотность равна 7,06 г/см3. Сделано предположение,,
что структура имеет симметрию /43 т и следующие пространственные
координаты атомов по системе обозначений, принятой в Международных
таблицах для определения кристаллической структуры [118]:
8U в положениях 8(c) *^ 0,187 ± 0,004;
12Fi» » 12(0 хс^ 0,225;
24Fn» » 12(g) х^0,20, г^0,46.
В этой структуре каждый атом урана связан с девятью атомами фтора,
а каждый атом фтора связан с двумя атомами урана. Около каждого атома
урана на расстоянии U — Fi 2,26 А находятся три ближайших атома-
фтора; на расстоянии U —Fn —2,31А находятся три из шести атомов Fiu
Глава 12. Нелетучие фториды урана
31»
остальные атомы фтора находятся на расстоянии 2,34 А. Незначительным,
изменением параметров атомов фтора все три расстояния могут быть
сделаны одинаковыми. В LJ2F9 каждый атом фтора связан с двумя атомами
урана, а поэтому он должен быть значительно менее летуч, чем гекса- или
пентафторид, в которых все или некоторые атомы фтора связаны
непосредственно только с одним атомом урана. Летучесть U2F9 сравнима с
летучестью тетрафторида.
Все атомы урана в U2F9 структурно эквивалентны друг другу.
Поэтому формулы UF4-UF5 или 3UF4-UFe, в которые входят атомы урана двух
видов, неприемлемы. U2F9 можно рассматривать как смешанные кристаллы
с рациональными отношениями:
(Uft + UgftF*,» или (U^ + U6o^5)F4,5.
Структура U4F17 [109]. Кристаллическая структура этого соединения
пока еще не установлена. Диф фракционная картина его напоминает
в общем картину для тетрафторида, но отличается от нее в деталях
настолько, что U4F17 нельзя считать просто искаженной структурой тетрафторида,
а следует рассматривать его как вполне определенное соединение.
Плотность U4F17 (определенная гидростатическим взвешиванием) равна 6,94 г/см3.
Температура плавления и летучесть пентафторида урана. Хотя
температура плавления пентафторида пока еще точно не определена, установлено,
что она лежит ниже 400°. Имеются некоторые доказательства испарения
пентафторида без химического изменения, но данные эти отрывочны и
определенных значений давления пара пентафторида не может быть дано.
Химические свойства. Кроме уже разобранного выше термического
диспропорционирования, о химических реакциях промежуточных фторидов
мало что можно еще сказать. Все эти соединения подвергаются гидролизу.
По чувствительности к следам влаги пентафторид урана напоминает гек-
сафторид. Во влажном воздухе a-UF6 более чувствителен к гидролизу, чем
P-UF6; свойство это независимо от размера частиц. При взаимодействии
пентафторида с водой образуется уранилфторид и тетрафторид урана:
2UFe + 2H80->UF4 J+UOJ++ 2F" + 4HF. (50)
U2F9, повидимому, гораздо более устойчив к гидролизу, чем пентафторид.
В воде черный цвет этого соединения может сохраняться в течение 1
часа и более; на воздухе проба может оставаться окрашенной в черный
цвет в течение двух недель.
U4F17 реагирует с водой, но данных для сравнения скорости
взаимодействия этого соединения со скоростями взаимодействия других
промежуточных фторидов не имеется. Вероятно, U4F17 менее реакционноспосо-
бен, чем U2F9. Реакция с водой протекает следующим образом:
2U4F17+ 2H20 -►UO;*+ 7UF4+ 4HF+2F". (51)
ЛИТЕРАТУРА
l.Bolton Н. С, Z. Спет., (2) 2, 353 (186С).
2. Bol ton Н. С, Bull. soc. Chim. Paris, (2) 6, 450 (1866).
3. Andrews L. J., G a t e s J. W., Report CD-477, Nov. 9, 1944 (CEW-TEC 1).
4. Ferguson J., Report B-55, Mar. , 1942, p. 5 (British 1).
5. Newton A. S.,Warf J. C.Johnson O., N о t t о г f R., Report CC-1201,
Jan. 1, 1944 (MP Ames 1).
6. К г a u s C. A., Report CC-1772, Apr. 28, 1944, p. 4 (Brown 1).
7. К r a u s С A., Report CC-342, Nov. 15, 1942, Part B, p. 8 (Brown 2).
8. В г о w n H. D., Report CC-1525, Mar. 14, 1944 (MP Ames 2).
9. L i p k i n d H., Report CC-1525, Mar. 14, 1944 (MP Ames 3).
lO.Spencer.Palmer H. J., Report BR-422, date received, June 10, 1944 British 2).
320 Часть /V. Галогениды, оксигалогениды, боргидриды и карбонил урана
П. К а г 1 е J., Report СК-1367, Feb. 25, 1944 (MP Chicago 1).
12. С h i о t t i P., R a e u с h 1 e R., Report CC-1525, Mar. 14, 1944 (MP Ames 4).
13. Z а с h a r i a s e n W. H., Report CN-2526, Jan. 2, 1945 (MP Chicago 2).
14. В a e n z i g e г N. С, R u n d 1 e R. E., Report CC-1525, Mar. 14, 1944 (MP
Ames 5).
15. О f t e d a 1 I., Z. physik. Chem., 5, 272 (1929).
16. Of ted a 1 I., Z. physik. Chem., 13, 190 (1931).
17. Report [B] LRG-33, June, 1944 (British 3).
18. В a e n z i g e r N. C, R u n d 1 e R. E., Report CN-1495, Mar. 10, 1944,
p. 26—27 (MP Ames 6).
19. Keller W., L у о n W., Report CC-1525, Mar. 14, 1944 (MP Ames 7).
20. H er m a n n H., Dissertation, University of Gottingen, S. 32(Gmelin, p. 120) (1861).
21. В r a d d о с k Т. R., С о p e n h a f e г D., Report N-17, Jan. 30, 1943, p. 6 (Maliin-
ckrodt 1).
22. D i t t e A., Compt. rend., 91, 116 (1880).
23. S m i t h e 1 1 s A., J. Chem. Soc, 43, 125—135 (1883).
24. Gross e A. V., S. F. 314, June 1941 (from B. 54, p. 1) (SAM Columbia 1).
25. Harvey B. G., Report B-52, Mar. 12, 1942, p. 3 (British 4).
26. M о г г i s G. 0., Report B-66, Feb. 17, 1942, p. 2 (British 5).
27. McLeishN., Report B-54, Mar. 12, 1942 (British 6).
28. Roberts A. M., Report B-60, Feb. 6, 1942 (British 7).
29. Roberts A. M., Report B-65, Feb. 6, 1942 (British 8).
30. H а г г i s С G., Roberts A. M., J e f f e г i e s E. J., Report B-59, Apr. 8,
1942 (British 9).
31. Roberts A. M., Report B-61, Feb. 6, 1942 (British 10).
32. M о г r i s G. O., Report B-50, Mar. 20, 1942 (British 11).
33. Morris G. O., Report B-51, Feb. 25, 1942 (British 12).
34. R о b e г t s A. M., H a r v e у В. G., Report B-62, Mar. 4, 1942 (British 13).
35. M о г г i s G. О., Н а г v e у В. G., Report B-69, May 4, 1942 (British 14).
36. R о b e г t s A. M., Morris G. O., Report B-63, Mar. 20, 1942 (British 15).
37. Report BR-446, Apr. 1944 (British 16).
38. Report BR-454, date received, July 6, 1944 (British 17).
39. Gates J. W., Andrews L. J.,Schaap W. В., Report CD-4005, Feb. 22,
1945 (CEW-TEC 2).
40. Gross e A. V., Report A-99, Mar. 1941 (SAM Columbia 2).
41. Roberts A. M., Report B-67, Feb. 16, 1942 (British 18).
42. W a t h e n Т., Report BR-483, Aug. 22, 1944 (British 19).
43. S e a b о r g G. Т., Report CK-1514, Mar. 6, 1944 (SAM Columbia 3).
44. J.o h n s J. В., W a 1 s h K. A., Report LA-381, Aug.. 30, 1945 (Los Alamos 1).
45. Brown H. S., Report CC-408, Jan. 8, 1943, p. 5 (MP Chicago 3).
46. Morris G. O., Report BR-250, July 23, 1943 (British 20).
47. P i 11 B. M., W a g n er E. L., M i 11 e г A. J., Report CD-2. 355. 3, Jan. 3, 1946 (CEW-TEC3).
48. Ruff O., Keim R., Z. anorg. u. aligem. Chem., 201, 245 (1931).
49. Henne A. L., пат. США 2136741 (1938).
50. Booth H. S., Krasny-Ergen W„ Heath R., J. Am. Chem. Soc,
68. 1969—1970 (1946).
51. Kraus С A., Report A-1090, Dec. 1, 1943, Part II (Brown 3).
52. К г au s C. A., Report CC-1717, July 29, 1944 (Brown 4).
53. К га us С. A., Report [A] M-ll, June 15, 1944 (Brown 5).
54. К r a u s C. A., Report A-2309, Mar. 24, 1945 (Brown 6).
55. Lindner M., Report CN-1025, Nov. 8, 1943, p. 15 (MP Clinton 1).
56. Report CC-1059, Sept. 9, 1943, p. 23 (MP Ames 8).
57. T e ve baug h A. D., W a 1 s h K., I 1 1 i f J., J о h n s I. B., Reports CN-1199,
Dec. 10, 1943, p. 7, and CC-1063, Nov. 16, 1943, p. 8 (MP Ames 9).
58. G a t e s J. W., A n d r e w s L. J., P i t t В. М., Report CD-4001, Jan. 30, 1945
(CEW-TEC 4).
59. В г a d d о с k Т. R., С о p e n h a f e г D., Report N-24. Jan . 23, 1943, p. 9 (Mai-
linckrodt 2).
60. С r eu t z E., Report CE-301, Oct. 15, 1942 (MP Chicago 4).
61. Davidson N. R., Report CK-932, Sept. 11, 1943, p. 5 (MP Chicago 5).
62. Z а с h а г i a s e n W. H., Report CC-2768, Mar. 12, 1945 (MP Chicago 6).
63. Roberts A. M., Harvey B. G., Report B-58, Mar. 30, 1940 (British 21).
64. P r i e s t H. F., P г i e s t G. R., Report A-257, Aug. 10, 1942 (SAM Columbia 4).
65. Z а с h а г i a s e n W. H., Report CP-1954, July 29, 1944, p. 22 (MP Chicago 7).
66. S с о t t R. В., Н о g e H. J., В г i с k w e d d e F. G., Report A-201, July 14,
1942 (Natl. Bur. Standards 1).
67. Br i с k wed de F. G., H о g e H. J., Scott R. В., J. Chem. Phys., 16,
429 (1948).
68. S о u t h a r d J. С, В r i с k w e d d e F. G., J. Am. Chem. Soc, 55. 4378 (1933).
Глава 12. Нелетучие фториды урана
321
69. В с к к е d a h 1 N.. Scott R. В., J. Research Natl. But. Standards, 29, 87 (1942).
70. L i b b у W. F., Report A-1228x, Aug. 29, 1944; U г е у H. C, Report A-330, Oct.
20, 1942 (SAM Columbia 5).
71. Z e 1 d e s H., reported by F. T. Miles in Division D, Monthly Report M-2589 (DE),
Jan. 5, 1946 (SAM Carbide and Carbon 1).
72. P 1 о t t R. F., R a e t h C. H., Report CP-228, Aug. 14, 1942 (MP Chicago 8).
73. E 1 1 i о t t N.. Phys. Rev., 76, 431 (1949).
74. Report (B)LRG-27, Dec. 1943 (British 22).
75. Kunin R., to be published (SAM Carbide and Carbon 2).
76. Roberts A. M., Report B-64, Feb. 6, 1942 (British 23).
77. D о b г a t z. J. W., Downing R. C, Richardson C. L.,Sp i egler L.,
Report N-34, Apr. 9, 1943 (Du Pont 1).
78. Roberts A. M., Report B-68, Feb. 16, 1942 (British 24).
79. Domange L, Wohlhuter M., Compt. rend., 228, 1591—1592 (1949).
80. Do mange L., Ann., 7, 225—297 (1937).
81. С 1 i n e W. D.Jevebaugh R.,Warf J. C, Report CC-1981, Oct. 10, 1944
(MP Ames 10).
82. W а г f J. С, С 1 i n e W. D., Report CC-2723, June 30, 1945 (MP Ames 11).
83. Feibig J. G., Warf J. C, Report CC-2939, June 29, 1945 (MP Ames 12).
84. Fried S., Davidson N. R., Report N-1722, Nov. 14, 1944 (MP Chicago 9).
85. D a a n e A., Report CC-298, Oct. 16, 1942 (MP Ames 13).
86. Kraus C. A., Report CC-1519 , Feb. 22, 1944 (Brown 7).
87. D о w n i n g F. В., В e n n i n g A. F„ Finch A. L., Report A-728, June 17,
1943 (Du Pont 2).
88. Lord E. J., Andrews L. J., G a t e s J. W., Report CD-1.365.4, Aug. 22,
1945 (CEW-TEC 5).
89. Tevebaugh A. D., Report CC-682, May 15, 1943 (MP Ames 14).
90. Fried S., Report N-2128, Oct. 12, 1945 (MP Chicago 10).
91. Calkins V . P., Reports CD-0.385.2, Oct. 6, 1945, and CD-0.350.4, Sept. 25, 1945
(CEW-TEC 6).
92. К а г 1 e J., Report CK-1512, Apr. 6, 1944 (MP Chicago 11).
93. Aloy J.,Rodier E., Bull. soc. chim. France [4], 31, 247 (1922).
94. Kraus С A., Report [A] M-251, July 13, 1943 (Brown 8).
95. Z а с h а г i a s e n W. H., Report CC-3401, Jan. 10, 1946, in National Nuclear Energy
Series, Division VIII, vol. 6 (MP Chicago 12).
96. Z а с h а г i a s e n W. H., Report CC-3426, Feb. 9, 1946, in National Nuclear Energy
Series, Division VIII, vol. 6 (MP Chicago 13).
97. Z а с h а г i a s e n W. H., J. Am. Chem. Soc., 70, 2147 (1948).
98. Z а с h а г i a s e n W. H., Reports AECD-1798, AECD-2089, AECD-2162 and AECD-
2163 (1948).
99. Z а с h а г i a s e n W. H., Report CF-2796, Mar. 15, 1945, p. 12 (MP Chicago 14).
100. Ruff O., H e i n z e 1 m a n n H., Z. anorg. Chem., 72, 64, 71 (1911).
101. Gross e A. V., Report A-83, June 25, 1941, p. 11 (SAM Columbia 6).
102. Priest H. F., Report A-719, June 3, 1943 (SAM Columbia 7).
103. Livingston R., В u r n s W., Report CN-982, Oct. 9, 1943 (MP Chicago 15).
104. Livingston R., Report N-781, Mar. 10, 1944 (MP Chicago 16).
105. Tevebaugh A. D., Walsh K.,Johns I. B., Report CC-1245, Jan. 8, 1945
(MP Ames 15).
106. Parks J., Report BR-480, Aug. 8, 1944 (British 25).
107. Report [B]LRG-34, July, 1944 (British 26).
108. Weller S., Grenall А., К u n i n R., Report A-3226, Mar. 19, 1945 (SAM
Carbide and Carbon 3).
109. Grenall A., Agron P., Kunin R., Weller S., Report A-3271, June
27, 1945 (SAM Carbide and Carbon 4).
110. Kraus C. A., Report A-1090, Dec. 1, 1943, Part 1 (Brown 9).
111. A g г о n P., Thermodynamics of Intermediate Uranium Fluorides from Measurements
of the Disproportionate Pressures, in National Nuclear Energy Series, Division
VIII, vol. 6 (SAM Carbide and Carbon 5).
112. В r i с к w e d d e F. G., personal communication to W. F. Libby; these data appear
in SAM Carbide and Carbon document DA R-53, Sec. VI, 1. 23. 9 (no date). (Natl.
Bur. Standards 2.)
113. К e 1 1 e у К. К., U. S. Bur. Mines Bull., 434 (1941).
114. Zachariasen W. H., in National Nuclear Energy Series, Division VIII, vol. 6
(MP Chicago 17).
115. Zachariasen W. H., Acta Crystallographica, 2, 296 (1949).
116. Z а с h а г i a s e n W. H., Report CC-2753, Feb. 28, 1945 (MP Chicago 18).
117. Z а с h а г i a s e n W. H., J. Chem. Phys., 16, 425 (1948).
118. International Tables for the Determination of Crystal Structure, Chemical Catalog
Company, New York, 1935.
21 Химия урана
Глава 13
ГЕКСАФТОРИД УРАНА
Гексафторид урана открыт Руффом и Хайнцельманом в 1909 г. [1], но
еще в 1900 г. Муассан [2] заметил, что фтор энергично реагирует с ураном
с образованием какого-то летучего соединения. Однако количество полученного
вещества было слишком мало для того, чтобы его можно было исследовать.
Руфф, ранее изучавший летучие фториды молибдена MoF6 и вольфрама WF6 [1 ],
предположил существование аналогичного соединения и для урана.
Действительно, его предположение оправдалось, и ему удалось получить
гексафторид этого металла путем реакции между фтором и металлическим ураном,
карбидом урана или его 'пентафторидом. Им были определены некоторые
физические и химические свойства гексафторида [3], однако современные
работы показали, что часть результатов его первых работ требует пересмотра.
В течение тридцати лет после исследований Руффа гексафторид урана
привлекал мало внимания, и в немногих препаративных работах по получению
гексафторида, проведенных в этот период времени, применяли метод Руффа.
Гроссе [4] приготовил небольшое количество гексафторида для Астона [5],
который изучал на этом препарате изотопы урана с помощью
масс-спектрографа; были также получены небольшие его количества для различных
магнитных и электронографических исследований. Открытие в 1939 г. явления
распада ядра урана, естественно, привлекло особое внимание к гексафториду
урана как к единственному известному устойчивому газообразному соединению
урана. После этого достигнуты большие успехи в разработке новых
лабораторных и промышленных методов получения гексафторида и приемов
обращения с ним, а также в определении его физических свойств. Химические
свойства гексафторида изучены слабее, и они требуют дальнейшего исследования.
ПОЛУЧЕНИЕ ГЕКСАФТОРИДА УРАНА
Существуют два основных метода получения гексафторида. Один из них
основан на прямом или косвенном применении элементарного фтора, другой—
на реакциях диспропорционирования. Первый метод основан на наблюдении,
что при обработке в соответствующих условиях любого соединения урана
фтором образуется гексафторид. Среди различных вариантов первого метода
наиболее употребительными как в лабораторном, так и в промышленном
масштабе являются те, которые требуют наименьшего количества фтора. Способы,
основанные на применении второго процесса, пока мало изучены, но они
весьма многообещающие.
Фторирование соединений урана. Руфф и Хайнцель-
ман [3] впервые получили гексафторид урана действием фтора на
металлический уран или его карбид:
U + 3F2 -> UFe АН = - 505 ± 3 ккал - моль1 (1)
(см. стр. 334)
UC2 + 7F2-»UF6+2CF4. (2)
Глава 13. Гексафторид урана
323
Реакция (1) протекает очень энергично с выделением большого количества
тепла. Для того чтобы реакция началась (если уран предварительно тщательно
не измельчен), необходимо нагревание, но после того как реакция уже
возникла, скорость ее зависит только от скорости подачи фтора в систему. Руфф
утверждает, что в качестве катализатора этой реакции необходим
газообразный хлор, а поэтому он вводил в реактор хлор или хлорид кальция. Это же
делали и последующие исследователи [4, 6]. В более поздних работах было,'
однако, показано, что хлор или другие катализаторы не требуются 17, 8]
и что присутствие хлора приводит лишь к образованию вместо белого гекса-
фторида загрязненного продукта желтого цвета. Реакция (2) протекает менее
энергично, чем первая, и при этом требуется гораздо больше фтора.
Подходящей температурой для ее проведения является 350°.
Руфф и Хайнцельман получали гексафторид также путем введения в
реакцию фтора с пентахлоридом урана при —40°. Они предполагали, что реакция
протекает следующим образом:
2UC15 + 5F2->UF4 + UF6 + 5C12. (3)
Однако точный состав образующихся продуктов не был установлен. При
проведении этой реакции существует опасность образования взрывчатых фторидов
хлора, а поэтому требуется интенсивное охлаждение. После окончания реакции
гексафторид отделяется возгоцкой. Подобным же путем могут быть гТревращены
в гексафторид и другие галогениды урана. Однако следует отметить, что Руффу
и Хайнцельману действием свободного фтора не удалось превратить пентахло-
рид урана, взвешенный в жидком хлоре, в гексафторид. Они сделали вывода
что или при температурах жидкого хлора ( — 33,7°) скорость реакции очень
мала или же фтор практически нерастворим в жидком хлоре.
Методы получения гексафторида фторированием были затем значительно
усовершенствованы [9]. Вместо металлического урана был использован
тетрафторид:
UF4 + F2->UF6. (4)
Для проведения этой реакции Абельсон рекомендовал температуру 275° и
применение плавленого хлорида натрия в качестве катализатора. В Англии,
однако, установили, что для успешного фторирования присутствие хлорида натрия
или другого какого-либо катализатора не только не является необходимым, но
оно даже вредно [8, 10]. Следует отметить, что применяемый в реакции (4)
тетрафторид урана должен быть как можно чище.
Тетрафторид легко фторируется при относительно низких температурах.
В условиях комнатной температуры реакция идет медленно, но при 250°
протекает очень быстро. При температуре ниже 250° реакция, повидимому, идет
в две стадии:
2UF4 + F2-^2UF5; (5)
2UF5 + F2~>2UF6. (6)
Реакция (5) проходит гораздо быстрее, чем реакция (6), а поэтому к тому
моменту, когда весь тетрафторид превратится в пентафторид, гексафторида либо
совсем не образуется, либо получается очень мало. В отношении использований
фтора реакция (5) выгодно отличается от реакции (6), в процессе которой
большие количества фтора проходят через систему, не реагируя. Повышение
температуры не имеет существенного практического значения, так как скорость
реакции (6), повидимому, очень мало зависит от изменений температуры в
интервале 250—650°; причина такого своеобразного поведения неизвестна к
Одним из методов увеличения использования фтора является пропускание газг{
из реактора, в котором идет процесс по уравнению (6), в реактор, где имеет
место реакция (5). В последнем будет поглощаться практически весь непроре^
21*
324 Часть IV. Галогениды, оксигалог гниды % боргидриды и карбонил урана
агировавший фтор. Направление тока фтора может быть затем изменено на
обратное. Однако высказываются сомнения [8] относительно осуществимости
этого приема, так как опыт показывает, что реакция (5) во втором реакторе
заканчивается раньше, чем реакция (6) в первом.
Повидимому, все соединения урана при нагревании с фтором до достаточно
Высокой температуры дают гексафторид [11]. Например, уранилфосфат легко
превращается при 400° в гексафторид урана и трифторид или пентафторид
фосфора. Окислы урана, по имеющимся данным, требуется нагревать с фтором до
температуры 400—500°, и в общем они довольно трудно фторируются. Однако
закись-окись урана U308 может быть превращена в гексафторид при 300°,
если ее предварительно смешать с углем [7, 12]:
U308 + 4C.+ 9F2 -> 3UF6 + 4C02. (7)
Согласно некоторым отчетам, чистая закись-окись реагирует с фтором при 360—
370° [131:
U308 + 17F2 -> 3UF6 + 80F2. (8)
Другой метод превращения закиси-окиси урана в гексафторид заключается
В предварительном гидрофторировании [7]:
U308 + 8HF
J0HL+ 2U02F2 + UF4 + 4H20;
UF4 + 2U02F2 + 5F2 -» 3UF6 + 202.
(9)
(10)
На рис. 46 указаны условия, при которых происходит образование гексафтори-
да из различных соединений урана.
2U02F2+UF4
U02HP04
Рис. 46. Превращение соединений урана в UFe (показано число
молей фтора, требующееся для образования 1 моля гексафторида
урана).
Реакции фторирования можно проводить в сосудах из монель-металла;
можно использовать также медь,алюминий и магний. Платина для этой цели
не годится, так как при температуре выше 450° она быстро разъедается фтором.
Весьма пригодны реакционные трубки и лодочки из фторида кальция, но они
трудно доступны. Стандартные раструбные соединения, применяемые в
холодильной промышленности, очень удобны для монтажа медной аппаратуры.
При низких температурах фтор можно держать в течение значительного
промежутка времени в тщательно высушенных и обезгаженных сосудах из
стекла пирекс или кварца без опасения заметного их разъедания. Мягкое
стекло обладает большей устойчивостью по отношению к фтору, чем пирекс.
Остальные вопросы о том, как следует обращаться с гексафторидом, будут
освещены несколько ниже.
Глава 13. Гексафторид урана
325
Фторирование соединений урана до гексафторида может быть успешно
достигнуто при помощи не только элементарного фтора, но и других фторирующих
агентов [14—16]. Однако для получения этих силыгофторирующих агентов
требуется свободный фтор, без которого, следовательно, и в этом случае нельзя
полностью обойтись. Иногда использование этих агентов для синтеза
гексафторида дает еще и другие преимущества, помимо возможности обходиться без
свободного фтора. Так, тетрафторид урана может быть превращен в
гексафторид реакцией с трифторидом кобальта:
UF4 + 2CoF3 (250~2751> UF6 + 2CoF2. (11)
Реакционная смесь готовится растиранием тетрафторида урана с пятикратным
избытком трифторида кобальта в сухой камере. Реакция может быть проведена
в цельностеклянной аппаратуре, причем реакционная смесь помещается
в платиновую лодочку. Следует избегать повышения температуры более чем
до 275°, так как трифторид кобальта термически неустойчив. Последний может
быть приготовлен различными способами [17]:
СоС12 . 6Н20 400° ^ СоС12 + 6Н20; (12)
СоС12 + 2HF 250° ,» CoF2 + 2HC1; (13)
2CoF2 + F2 250° + 2CoF3; (14)
2СоС12 + 5F2 400° „ 2CoF3 + 4C1F. (15)
Вероятно, с таким же успехом, как трифторид кобальта, можно применять
и дифторид серебра или трифторид палладия. С другой стороны, пентахлорид
сурьмы, который получается без применения элементарного фтора, не
превращает тетрахлорид урана в гексафторид [18].
Реакции без применения элементарного фтора.
Найдены две реакции получения гексафторида, не требующие применения
свободного фтора. Первая заключается в диспропорционировании UF6:
3UF5 175S ->U2F9 + UF6; (16)
2UaF9 >20QO(?U 3UF4 + UF6. (17)
Необходимый для реакции (16) пентафторид может быть приготовлен из легко
доступного пентахлорида:
UC16+5HF -> UF6 + 5HC1. (18)
Руфф и Хайнцельман [3], которые впервые применили этот метод, встретились
со значительными трудностями при отделении гексафторида от фтористого
водорода, образовавшегося из использованного ими соединения UF5-HF.
Последующие исследования показали, что гексафторид может быть отделен от
фтористого водорода фракционированной перегонкой или даже более простым
способом—декантацией, так как при низких температурах гексафторид лишь
слабо растворим в жидком безводном фтористом водороде [19, 20]. Эта реакция
невыгодна тем, что в этом случае используется лишь одна треть общего
количества урана, а остальная часть должна быть заново переработана.
Второй метод основан на следующей интересной реакции (21). При
действии сухого кислорода на тетрафторид урана при повышенных температурах
(приблизительно 800°) происходит следующая реакция*:
2UF4 + 02 -> UFe + U02F2. (19)
* Подобный опыт до этого проведен в трубке из плавленого кварца. Наблюдалось
появление тяжелого коричневого вещества и отмечена потеря в весе остатка. Высказана
предположение, что при этом процессе происходит образование фтора или гексафторид»
урана, но дальнейших исследований в то время не проводилось (22].
326 Часть IV. Галогениды, оксигалоген оды, боргидриды и карбонил урана
Реакция (19) проводилась в никелевой трубке. Возможно, что фторид кальция
в качестве материала для трубки даже лучше никеля, так как он более инертен.
При 800° выход гексафторида урана составлял 20—40% теоретического.
Сравнительно низкий выход, возможно, обусловлен реакцией между гексафторидом
урана и никелем. Реакция (19) представляет удобный метод лабораторного
Получения гексафторида при отсутствии свободного фтора.
Были сделаны также другие попытки получить гексафторид по реакциям,
не требующим применения свободного фтора [23]. В качестве фторирующего
агента пользовались фторидом ртути (II). Исходными соединениями служили
{Гётрафторид и пентахлорид урана, которые вводились в реакцию сдифторидом
ртути или монофторидом и хлором; в обоих случаях гексафторид получался
в очень небольших количествах. Фторид ртути (II) был приготовлен 124] по
методу Руффа и Балау [25] действием хлора на фторид ртути (I). Пентахлорид
урана реагирует с фторидом ртути (II) при «умеренных» температурах с
образованием белого возгона, по всей вероятности гексафторида урана. При
действии сухим хлором на тесную смесь тетрафторида урана и монофторида ртути
впри 400° в стеклянной аппаратуре также получали гексафторид урана в
небольших количествах. Низкий выход объяснялся побочными реакциями со
стеклом. Сделана также попытка получить гексафторид урана пропусканием смеси
хлора и дихлордифторметана над смесью трехокиси урана и монофторида
ртути; однако трехокись заметно в реакцию не вступала.
Из вышеизложенного ясно, что получение гексафторида урана без
применения свободного фтора вполне возможно.
Очистка гексафторида урана и правила
обращения с ним. Ввиду того что гексафторид является весьма реакционноспособ-
ным веществом, обращение с ним как в лабораторном, так и в промышленном
масштабе требует соблюдения известных правил. Для работы с ним
применяется металлическая аппаратура, но если принимать известные меры
предосторожности, можно с успехом пользоваться и стеклом. Гексафторид быстро разъедает
стекло лишь в присутствии следов воды или фтористого водорода. Для
осушения гексафторида урана можно применять фосфорный ангидрид, но это
довольно сложная операция [4]. Взаимодействие гексафторида со стеклом происходит,
повидимому, следующим образом:
UFe + 2Н20 -» U02F2 + 4HF; (20)
Si02 + 4HF -* SiF4 + 2H20. (21)
Вода, потребляемая в первой реакции, выделяется во второй, и, таким
образом, следов влаги достаточно для разложения больших количеств гексафторида.
Для обрыва этой цепи реакций Гроссе ввел применение «геттеров» [19]. Фториды
натрия или калия соединяются со всеми компонентами, участвующими в
реакциях (20) и (21), с образованием KF-HF, KF-2H20, K2SiF6 и т. д., и поэтому
их можно применять для обрыва упомянутой последовательности реакций.
.Чистый гексафторид урана не дает устойчивого продукта присоединения
с фторидом калия, но при наличии еще фтористого водорода образуются
стабильные тройные соединения. Однако количество гексафторида, теряющееся этим
путем, соответствует лишь стехиометрическому количеству присутствующего
фтористого водорода, которое в большинстве случаев невелико. Поэтому для
возможности хранения гексафторида урана в стеклянном сосуде единственно
необходимым условием является наличие сухого порошкообразного фторида
калия или натрия в количестве, составляющем примерно 5% (вес.)
гексафторида. Ввиду сильной гигроскопичности фторида калия перед употреблением
его следует сплавить; фторид натрия не расплывается во влажном воздухе,
и поэтому он может оказаться более целесообразной добавкой, чем калиевая
соль [26]. Сообщается, что чистый гексафторид даже в отсутствие «геттера» не
Глава 13. Гексафторид урана
327
разлагается в течение неопределенно долгого времени в стеклянном сосуде,
предварительно высушенном нагреванием в вакууме. Гексафторид должен
быть сначала очищен многократной перегонкой через ряд U-образных трубок,
содержащих фторид калия [27]. Довольно подробно описана [28] лабораторная
методика очистки гексафторида и правила обращения с ним.
Процесс конденсации гексафторида в стеклянной аппаратуре должен
проводиться особенно осторожно. Ловушки, охлаждаемые жидким воздухом, не
могут быть рекомендованы, так как они часто растрескиваются при повышении
температуры. Для устранения такой опасности ловушки сначала должны быть
охлаждены сухим льдом; при этом гексафторид оседает в виде пушистой
снегообразной массы. Если намечено использование жидкого воздуха,
рекомендуется применять ловушку с внутренним приспособлением для
охлаждения [29].
Чтобы сделать чистое стекло устойчивым к действию гексафторида, не
содержащего примесей, необходима следующая 'последовательность операций
[30]: 1) промывание ксилолом или трихлорэтиленом, 2) обжиг, 3)
предварительная обработка парами гексафторида урана в течение нескольких часов при
комнатной температуре. Наиболее важной из этих операций является эта
предварительная обработка, которая, повидимому, гораздо более эффективна, чем
обжиг. Ввиду того что для полного высушивания путем нагревания в вакууме
аппаратура должна быть цельностеклянной, краны и соединения необходимо
заменить магнитными затворами (называемыми еще затворами Бриско).
Если пользуются стеклянными кранами и шлифами, то можно применять апье-
зоновую смазку, или апьезоновый воск, или, что еще лучше, фторированные
углеводороды, но с гексафторидом должно соприкасаться только небольшое
количество этой смазки.
Удовлетворительными материалами для изготовления металлической
аппаратуры являются медь, никель и алюминий. Для медных трубок используются
обычные раструбные соединения, применяемые в холодильной
промышленности; тщательно сделанные, они хорошо держат вакуум. Для измерения
давления должны применяться манометры Бурдона с трубками из металла,
устойчивого к гексафториду.
Оказалось, что в гексафториде в качестве примеси обычно присутствует
фтористый водород [31]. Для связывания последнего рекомендуется применять
гораздо большее количество фторида калия, чем указано выше, а именно: на
2% (мол.) фтористого водорода необходимо 50% этого фторида от веса
гексафторида урана. Установлено также, что существует постоянно кипящая смесь из
гексафторида и фтористого водорода (с давлением пара 17,91 мм рт. ст. при 0°)
и что содержание последнего в загрязненном гексафториде можно довести до
2% (мол.) откачиванием при —80°. Таким образом, не следует непосредственно
очищать фторидом калия смеси, содержащие более 2% (мол.) фтористого
водорода, а необходимо их сначала откачать при —80°.
Присутствие фтористого водорода в гексафториде урана и его количество
могут быть определены измерениями давления пара или точки замерзания
смеси. Ввиду малой растворимости фтористого водорода в гексафториде величины
давления пара являются аддитивными. Наличие 1 % (мол.) фтористого
водорода при 0° увеличивает давление пара на 12 мм рт. ст. [32]. Для определения
очень малых количеств фтористого водорода следует применять криоскопиче-
ский метод как более точный. Экспериментально установленное значение
криоскопической константы для гексафторида равно 0,065° на 0,01 % (вес.)
фтористого водорода. Теоретическое значение, рассчитанное по закону Рауля,
равно 0,0839° [33].
В Англии была произведена проверка различных методов, предложенных
для определения содержания фтористого водорода в гексафториде. Сделано
заключение, что наиболее удовлетворительным методом является следующий:
328 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
смесь обоих веществ кипятят с обратным холодильником при таких условиях,
что фтористый водород остается в газовой фазе. Его объем при равновесии
может быть измерен с точностью 0,01% (мол.) [34].
ФИЗИЧЕСКИЕ СВОЙСТВА ГЕКСАФТОРИДА УРАНА
При комнатной температуре гексафторид представляет собой бесцветное
летучее твердое вещество, образующее прозрачные кристаллы с высоким
показателем преломления. При атмосферном давлении он возгоняется, не плавясь.
При более высоких давлениях он плавится с образованием прозрачной
бесцветной подвижной жидкости, обладающей большой плотностью. Несмотря на
высокий молекулярный вес гексафторида, свойства его пара приближаются
к свойствам идеального газа.
Физические свойства гексафторида в его различных состояниях будут
рассмотрены в следующих гГодразделах:
Фазовые соотношения: температура плавления, давление пара в твердом
и жидком состояниях, температура кипения, тройная точка и критические
постоянные.
Термические свойства: удельная теплоемкость, энтропия и энтальпия,
скорость звука, теплоты возгонки, испарения и плавления, теплопроводность.
Механические свойства: плотность, кинетические свойства пара, вязкость
и поверхностное натяжение.
Оптические свойства: показатель преломления, спектр поглощения и спектр
комбинационного рассеяния.
Электрические и магнитные свойства: диэлектрическая постоянная,
магнитная восприимчивость и ионизационный потенциал.
Строение молекулы: определенное методами электронографии,
рентгеновской кристаллографии и измерений дипольного момента.
Диаграммы, приведенные в этой главе, взяты из краткого отчета
Киршенбаума [35], которым авторы настоящей книги широко пользовались.
Фазовые соотношения. Температура плавления. Показано, что
наиболее раннее определение температуры плавления (69,2 — 69,5°),
произведенное Руффом и Хайнцельманом [3] и цитированное Гроссе [19], весьма
неточно. Брикведде, Хоудж и Скотт [36, 37] определили эту* температуру
в 64,052° С (337, 212° К) для препарата гексафторида урана, содержавшего
0,02% (мол.) примесей. Английские исследователи установили для двух
препаратов гексафторида, содержавших меньше чем 0,015% (мол.) примесей,
значения 64,5±0,3° и 64,8±0,4° [38]. Зная удельные объемы жидкого и
твердого гексафторида в тройной точке, Уж и VTB (см. стр. 343 и ел.),
можно вычислить зависимость температуры плавления от давления:
dT^ T(Vж-Утв) =0?0о505 град/am. (22)
"Р л^пл
Давление пара твердого гексафторида урана. Значения давления пара
гексафторида, принадлежащие Руффу и Хайнцельману [3], были
ошибочными, вероятно, благодаря загрязнению гексафторида фтористым водородом
или тетрафторидом кремния.
Рядом исследователей произведено повторное измерение давления пара
гексафторида. В целом, если принимать во внимание трудности измерений,
можно сказать, что результаты различных авторов удовлетворительно
согласуются между собой. Трудно получить гексафторид урана, совершенно
свободный от фтористого водорода, а примесь последнего приводит к
завышенным значениям давления пара. Затруднительно также обеспечить тепловое
равновесие между термостатом и рыхлой массой твердого гексафторида.
Наконец, затруднения вызывает и коррозия материала, из которого
Таблица 131
ДАВЛЕНИЕ ПАРА ТВЕРДОГО ГЕКСАФТОРИДА УРАНА
Температура,
°С
-14,2
-12,4
-1,5
0,0
0,1
1,7
3,6
5,5
6,8
8,5
11,3
12,5
14,3
14,5
14,7
15,0
17,6
20,4
21,0
21,6
22,7
24,9
25,0
25,1
26,4
28,2
29,0
29,5
30,4
31,2
32,7
34,9
35,3
35,5
36,8
38,3
39,7
40,0
40,5
41,7
44,7
45,0
45,2
47,2
49,6
60,0
[41]
6,85
7,80
17,80
—
20,10
23,30
26,60
30,40
32,90
37,00
44,00
Д*
[42, 28]
45,3
52,0
52,5
53,6
54,4
66,1
80,8
—
—
—
—
—
113,5
—
—
—
149,5
216,8
—
—
—
—
—
—
—
—
—
—
_
—
изление пара, мм р
[43]
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
83
—
93
108
—
—
123
138
__
—
158
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
_
т. ст.
[44]
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—-
—
—
—
—
119,5
—
—
—
153,3
178,4
199,0
224,3
—
—
259,5
282,8
310,5
—
316,5
348,9
—
—
418,8
484,1
549,1
[39, 4 0]
—
—
16,9
—
—
—
—
—
—
—
—
—
—
—
—
—
__
—
88,3
—
—
—
—
—
—
—
—
—
—
—
—
216,8
—
—
—
295,4
—
—
—
395,8
—
—
—
522,1
330 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Продолжение табл. 131
Температура,
°С
50,2
51,6
51,8
53,9
55,0
55,7
60,0
63,1
[41]
[42, 28]
521,0
—
—
—
—
—
—
—•
Давление пара, мм рт. ст.
[43]
—
—
—
—
—
—
—
[44]
559,2
605,0
612,5
685,5
„
751,2
—
• [39. 40]
—
—
—
697,2
—
910,0
1,072
изготовлен прибор. Американские исследователи обычно пользовались медной
аппаратурой, а английские, повидимому, предпочитали стеклянную.
Для измерений давления нулевым методом в Колумбийском
университете пользовались латунными сильфонами [39, 40]. Давления выше
атмосферного отсчитывались по многоколенчатому ртутному манометру, в
котором в качестве промежуточной жидкости служил дибутилфталат. Ошибка
при отсчете давления составляла около 2 мм рт. ст. в верхнем интервале
давлений и менее 0,5 мм в нижнем.
Табл. 131 содержит сводку всех значений давления пара твердого
гексафторида, полученных прямым измерением.
Для построения графика давление пара — температура (для
ограниченного температурного интервала) были предложены следующие линейные
уравнения, связывающие lgp и 1/Т:
ЫРммрт. ст.= 10,74-^(12,5-50,2°) [28, 42]; (23)
IgP.
мм рт. ст.
= 21,87103-
3123,479
2714
3,77962 lgT(0-63,Г) [40]; (24)
(25)
ЫРммрт. ст. = 11,19 у- (Для низких температур) [45].
Для точного отображения кривой давления пара в широком интервале
температур было выведено два более сложных уравнения. Уравнение
irt — 2751
lg Рмм рт. ст. —
-75,0е-256°/г_ 1,01 \gT-\- 13,797
(26)
охватывает все значения упругости пара, полученные английскими
исследователями для интервала 0 — 65° с точностью 0,5% [46]. Значения,
полученные в Колумбийском университете, согласуются с этим уравнением
с точностью 1,5%, но все они лежат немного выше кривой, отображаемой
этим уравнением.
В Колумбийском университете [35] выведено другое уравнение,
основанное на всех имеющихся данных. Все опытные точки нанесены на
график в большом масштабе. По ним путем ряда аналитических и
графических приближений проведена кривая; при этом для высших приближений
были использованы уравнения для теплоемкости и энтальпии твердого
гексафторида. Как сообщается, выведенное уравнение отвечает
экспериментальным результатам с точностью ±0,5%, а возможно, даже большей
(рис. 47).
1пр^рт.ст.= -13218690-?^ + 1^ + 26,4361пГ-0,038674Г. (27а)
Глава 13. Гексафторид урана
331
В'десятичных логарифмах это уравнение приобретает вид:
lgp^:pT.cT.= -57J043~i^ + ?^+26,4361gr--0,016796r. (276)
^Давление пара твердого гексафторида при низких температурах
определено экстраполированием уравнений (25) и (27а, 276). Результаты
приведены вLтабл. 132.
180
160
^140
§Г120
^100
J 60
са
§ 40
20
О
225 250 275 300 325
Температура
^.^ Рис. 47. Давление пара твердого гексафторида
1'урана [см. уравнение (27а) или (276)]. Температура, °К.
Таблица 132
ДАВЛЕНИЕ ПАРА ТВЕРДОГО ГЕКСАФТОРИДА УРАНА ПРИ НИЗКИХ ТЕМПЕРАТУРАХ
С
Температура, °С
-200
-183а)
-150
-103,8б)
-100
-95
-90
-85
-80
-78,5В)
-75
Давление пара, мм рт. ст.
уравнение (27а)
или (276) [35]
Ю-2в
—
10-11
—
4,3-10-5
8,6-10-5
3,0-10-*
7,3-10"*
1,7-Ю-3
1 —
| 3,8-10-з
уравнение
(25) [45]
10~19
—
1,3-Ю"5
_
—
—
_
— 1
1,7.Ю-8
—
Температура, °С
-70
-65
-60
-55
-50
-45
-40
-35
-30
-25
1 -20
Давление пара, мм рт. ст.
уравнение (27а)
или (276) [35]
8,2-Ю-3
1,7.10-2
3,4-10-2
6,5.Ю-2
1,18-Ю-1
2.2.10-1
3,69. Ю-1
6,9-10-1
1,16
1,92
3,11
уравнение
(25) [45]
—
—
—
—
—
—
—
—
—
—
а) Точка кипения Ог-
б) Точка кипения С2Н4.
в) Температура возгонки COg.
332 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Помимо значения 16,9 мм рт. ст. для упругости пара твердого
гексафторида при 0°, указанного в табл. 131, в литературе приводятся и
некоторые другие значения. Исследователи, работавшие в Колумбийском
университете, в результате своих опытов определили это давление
в 18,0 'мм рт. ст. [32], однако английские авторы в результате очень
тщательных измерений нашли значение 17,54 ± 0,02 мм рт. ст. [31].
Последняя величина, вероятно, более надежна, так как английскими
исследователями найдено, что существует постоянно кипящая смесь гексафторида
урана и фтористого водорода, которая при 0° имеет давление пара
17,91 ± 0,02 мм рт. ст.
Давление пара жидкого гексафторида урана. Значения давления пара
гексафторида для системы жидкость —пар являются более точными, чем
для системы твердое вещество—пар. Возможно, это объясняется тем, что
в системе жидкость — пар легче достигается тепловое равновесие.
Приведенные в табл. 133 значения давления пара гексафторида получены в
Колумбийском университете с использованием той же аппаратуры, которая
применялась для системы твердое вещество —пар [39]. В эту таблицу
включены также результаты, полученные английскими исследователями
[31, 46].
Таблица 133
ДАВЛЕНИЕ ПАРА ЖИДКОГО ГЕКСАФТОРИДА УРАНА
Температура,
°С
65,0
67,9
70,0
70,1
70,2
75,0
75,2
75,3
80,0
80,3
85,0
85,4
90,0
95,0
Давление пара, мм рт.
опытные
значения
[31, 46]
1175
—
1385
—
—
1620
—
—
1870
—
2165
_
2455
2765
опытные
значения
[39, 40]
1169
1273
—
1360
1366, 1370,
1376
—
1582
1568
_
1830, 1838
_
2087
—
—
ст.
значения,
рассчитанные по
уравнению (30)
1167
1274
—
1361
1365
—
1577
1582
—
1820
—
2089
—
—
Разность
между рассчитанным
и опытным
значениями.
мм рт. ст.
-2
+ 1
—
+ 1
-1, -5, —11
—
-5
+ 14
—
-10, -18
—
+ 2
—
—■
Уравнения (28) и (29), выражающие зависимость между давлением
пара жидкого гексафторида и температурой, были предложены
английскими исследователями, а уравнение (30) принадлежит Вайнштоку и
Кристу [40].
lgft«PT. ст. = ^ + 56,0 lgr-0,0394 7'- 131,64; (28)
lg Рлшрт.ст. =7,73 y~ » (29)
lg Рлшрт.ст. = 18,60033-^^-3,72662 lg7\ (30)
Глава 13. Гексафторид урана
333
Установлено, что уравнение (30) описывает результаты опытов с
ошибкой менее ±1%.
Киршенбаумом выведено точное уравнение для давления пара в системе
жидкость—пар тем же методом, которым были получены уравнения (27а)
и (276) для системы твердое вещество —пар, т. е. подгонкой кривой к
экспериментальным данным методом последовательных приближений на графике
большого масштаба (рис. 48):
In р.
мм рт. ст,
= -24,6602-
248608 1245,2
у-2
- 7,28761пГ- 0,014371Т, (31а)
или в десятичных логарифмах:
lgPju.PT. ст. = -10,7098- 107969
540,8
65-
Т2 т , 7,2876 lgT —0,006241 Т. (316)
Это уравнение описывает экспериментальные результаты в интервале
• 90° с ошибкой менее ±0,4%.
3,3500
$ 3.1500
I
Щ 3,1000
3,0500^—-
Рис. 48. Давление пара жидкого гексафторида
урана [см. уравнение (31а) или (316)].
Температура кипения. Руфф и Хайнцельман [3] сообщают, что
температура, при которой давление пара твердого гексафторида достигает
760 мм рт. ст., равна 56°. Позднее были найдены значения 56,2° [19],
55,3 и 56,7° [28, 42], причем последнее значение было признано более
точным. Согласно опытам Вайнштока и Криста [40], температура кипения
равна 56,5°. Таким образом, значение 56,5°, повидимому, является
наиболее приемлемой средней для «температуры кипения» гексафторида.
Тройная точка для гексафторида была определена Брикведде, Хоуджем и
Скоттом [36]; ее координаты следующие:
7тр = 64,052° С;
ртр = 1134 мм рт. ст.
По Вайнштоку и Кристу [40] упругость пара в тройной точке равна
1133 ± 7 мм рт. ст.
334 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Критические температура, давление и плотность. Для критической
температуры гексафторида английские исследователи приводят значение
245 ± 5° [47]. В Колумбийском университете эта температура определена
в 232° [35], а в Национальном бюро стандартов США [48] на основании
кривой зависимости плотности от температуры установлено, что
критическая температура должна быть равна приблизительно 217°. Другое
приближенное значение критической температуры (243°) вычислено из
поверхностного натяжения при помощи уравнения
„_ 2,12(^-6-/) ^ (32)
где 7=15,6 дин/см при 80°, d = 3,57 г/см3 при 80°. Наконец, из значения
температуры кипения и отношения
-^- = 0,63
* Крит
найдено, что Гкрит = 249°.
Критическое давление, определенное английскими исследователями,
равно 63 am [49], определенное группой исследователей при
Колумбийском университете ~ 50 am [50], а найденное Национальным бюро
стандартов [48] —44 am. Критическая плотность определена в 1,9 г/см3 [49] и
1,39 г/см3 [48]. Последнее значение, вероятно, более надежное. Очевидно,
что необходимы дальнейшие работы по определению критических констант
гексафторида.
Термические свойства. Теплота и свободная энергия образования
гексафторида урана. Теплота реакции гидролиза гексафторида в воде
определена калориметрически в —50,5 ккал/моль при 298,16° К [51]:
UF6kphct + 2Н2Ож -> UO|+ + 2Р + 4HFag; (33)
АН= —50,5 ± 1,7 ккаЛ'Моль'1.
Путем использования значения
Д#возг = — П,2 ± 1 ккал-моль~1
для теплоты возгонки гексафторида [39] и сочетания этих величин со
следующими, известными из литературы:
U022+ + 3H2^ 2H+ + 2H20 + U АЯ= 106,3 ккал\
2H+ + 2F- -> 2HFag АЯ= 4,0 ккал\
6HFa, -> 3H2ra3 + 3Fora3 Atf--= 456 ккал,
установлена теплота образования гексафторида урана, приводимая ниже:
UTB + 3F2ra3 -» UF6ra3 Д# = — 505 ± 3 ккал• моль-1. (34)
Используя значения энтропии (S298), равные 90,3 ± 1 энтропийной
единицы для гексафторида и 12,8 энтропийной единицы для металлического
урана, можно вычислить свободную энергию образования гексафторида:
AF298^= —485 ккал-моль'1.
Теплоемкость, энтропия и энтальпия гексафторида урана.
Теплоемкости твердого и жидкого гексафторида в равновесии с паром определены
очень точно [36, 37]. Рассчитанные из этих данных значения энтропии и
энтальпии приведены в табл. 134; точность их, повидимому, равна ± 1%-
Результаты, приведенные в табл. 134, описываются следующими
эмпирическими уравнениями [35]:
Таблица 134
ТЕПЛОЕМКОСТЬ, ЭНТРОПИЯ И ЭНТАЛЬПИЯ ГЕКСАФТОРИДА УРАНА
Температура,
°К
каЛ'М0ль~1-град~1
Н-Н0,
кал'моль"!
S-S0,
кал-моль-1-град~1
Т (S-S0),
каЛ'Моль~1
О
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
105
ПО
115
120
125
130
135
140
145
150
155
160
165
170
175
180
185
190
195
200
205
210
215
220
225
230
235
Твердый гексафторид урана
0
4,06
5,53
6,92
8,27
9,64
11,00
12,34
13,66
14,90
16,07
17,16
18,22
19,20
20,14
20,99
21,76
22,49
23,17
23,82
24,46
25,08
25,66
26,23
26,77
27,30
27,80
28,30
28,78
29,24
29,71
30,15
30,60
31,03
31,42
31,87
32,27
32,65
33,05
33,46
33,78
34,22
34,60
34,96
35,34
0
28,0
52,0
83,2
121
165
217
275
340
412
489,8
572,8
661,
754,
853,
956,
1063
1 174
1288
1405
1 526
1650
1 777
1907
2 039
2 174
2312
2 452
2 595
2 740
2 887
3 037
3 189
3 343
3 499
3 657
3818
3 980
4 144
4 310
4 479
4 648
4 820
4 994
5 170
0
1,96
3,04
4,16
5,33
6,53
7,72
8,96
10,21
11,45
12,69
13,91
15,13
16,35
17,55
18,72
19,86
20,99
22,11
23,21
24,29
25,34
26,37
27,39
28,40
29,38
30,33
31,29
32,22
33,16
34,04
34,95
35,83
36,69
37,55
38,39
39,23
40,04
40,85
41,67
42,45
43,24
44,01
44,77
45,51
О
39
76
125
187
261
347
448
562
687
825
974
1 135
1308
1492
1685
1 887
2 099
2 322
2 553
2 793
3 041
3 296
3561
3 834
4113
4 398
4 694
4 994
5 306
5617
5 942
6 270
6 604
6 947
7 294
7 650
8 008
8 374
8 751
9 127
9513
9 902
10 296
336 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Продолжение табл. 134
Температура,
°К
240
245
250
255
260
265
270
273,16
275
280
285
290
295
298,16
300
305
310
315
320
325
330
335
337,212
СР.
1 кал-моль~ 1-град~1
35,70
36,07
36,43
36,78
37,14
37,50
37,86
38,08
38,22
38,56
38,90
39,24
39,61
39,86
40,00
40,42
40,87
41,33
41,80
42,27
42,77
43,27
43,49
Я-Я<ь
кал'МОль~ 1
5 348
5 527
5 708
5 892
6 067
6 263
6 451
6 571
6 640
6 833
7 027
7 223
7 420
7 545
7619
7 820
8 023
8 229
8 437
8 648
8 860
9 076
9 172
S-S0,
кал • мол б"*1 -граЪ~^
46,28
47,02
47,74
48,48
49,20
49,89
50,61
51,05
51,30
51,99
52,69
53,36
54,02
54,45
54,69
55,36
56,03
56,68
57,35
57,99
58,64
59,28
59,57
Г (S-So).
кал-мол ь"^
U 107
II 520
11935
12 362
12 792
13 221
13 665
13 942
14 108
14 557
15017
15 474
15 936
16 235
16 407
16 885
17 369
17 854
18 352
18 847
19 351
19 859
20 088
Жидкий гексафторид урана
337,212
340
345
350
355
360
365
370
45,59
45,68
45,84
46,02
46,20
46,33
46,48
46,62
13 760
13 888
14118
14 348
14 579
14811
15 044
15 278
73,17
73,55
74,22
74,89
75,54
76,18
76,83
77,45
24 674
25 007
25 606
26212
26 817
27 425
28 043
28 657
Для твердого гексафторида
Н -Я„ = 9865,0-20,082 Г+0,080790 Г2- 1047920 Г"1 (35)
(рис. 49) (точность ±0,01% в интервале от тройной точки до 265* К и
± 1% от 265 до 225° К);
5-S0= 126,59 - 20,082 In Г+0,16162 Г-523960 Г"2 (36)
(рис. 50) (точность ±0,01% в интервале от тройной точки до 273,16°К).
Для жидкого гексафторида
Н - Н0 = 5986,6 Н-17,954 Т+ 0,032514 Г2 - 666990 Г"1 (37)
Глава 13. Гексафторид урана
337
(точность ±0,01%);
S-S0= -50,33+ 17,954 In T + 0,065028 Т - 333490 Г*2
(38)
(точность ± 0,01%).
Приведенные погрешности представляют максимальное отклонение
экспериментальных значений от вычисленных по соответствующим
уравнениям.
16000\
f4000h
12000\-
| ЮООО
о 8000h
а:
6000 Ь
4000 V
2000
150
200 250 300
Температура, °К
350
150 200 250 300
Температура, ЛК
350
Рис. 49. Энтальпия твердого гекса- Рис. 50. Энтропия твердого гексафторида
фторида урана [см. уравнение (35)]. урана [см. уравнение(36)].
Английские исследователи определили ряд значений для удельной
теплоемкости твердого гексафторида [52, 53], однако эти данные, вероятно,
менее точны, чем те, которые указаны в табл. 135 для жидкого гексафторида.
Они также приводят некоторые значения для удельной теплоемкости жидкого
гексафторида [53], которые несколько превышают величины, полученные
Таблица 135
УДЕЛЬНАЯ ТЕПЛОЕМКОСТЬ ЖИДКОГО
ГЕКСАФТОРИДА УРАНА
Температура
°С
65
70
80
90
100
•К
338
343
353
363
373
СР,
кал-моль~1 -ерад~1
47,0±1
47,4
48,1
48,8
49,5
22 Химия урана
338 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
американскими исследователями (например, Ср = 50 кал-моль"1-град'1 при
353° К), и могут быть представлены следующей формулой:
Ср-47,3+ 0,347 (*-70) кал-моль~1-град'1 у (39)
где ^ —температура, °С. Дополнительные данные английских
исследователей для жидкого гексафторида [54] представлены в табл. 135.
Удельная теплоемкость и энтропия газообразного гексафторида
рассчитаны из инфракрасного спектра (см. стр. 349), причем предполагалось,
что уран находится в центре правильного октаэдра с расстояниями U — F,
равными 2,0 А [55]. Вычисленные таким образом величины удельных тепло-
емкостей представлены в табл. 136, а энтропии —в табл. 137.
Таблица 136
УДЕЛЬНАЯ ТЕПЛОЕМКОСТЬ ГАЗООБРАЗНОГО ГЕКСАФТОРИДА
УРАНА (ПО СПЕКТРОСКОПИЧЕСКИМ ДАННЫМ)
Температура,
°К
100
150
200
273
298
323
с
р 1
кал-моль ь
•град~1
18,93
23,45
26,74
30,13
31,00
31,75
Температура,
°К
348
373
400
500
750
1000
с
кал'моль 1*
•град"*
32,38
32,93
33,46
34,80
36,35
36,94
Таблица 137
ЭНТРОПИЯ ГАЗООБРАЗНОГО ГЕКСАФТОРИДА УРАНА3)
Температура,
°К
100
150
200
273
298
323
348
373
400
500
750
1000
[55]
62,99
71,58
78,81
87,65
90,34
92,89
95,22
97,47
99,86
107,45
122,37
132,58
S°, кал*моль~1*град~
определено по
уравнению (40)
—
—
82,27
—
93,59
—
98,46
100,46
_
—
~
1
рассчитано из
значений давления
пара по уравнению
(40)
—
—
88,21
90,76
92,96
94,97
—
—
—
—
о о
го—Е0
Т
кал -моль~~1- град"1
[40]
50,91
56,41
61,14
67,08
68,92
70,69
72,33
73,93
75,65
81,25
92,99
101,37
а) Ср. со значениями энтропии газообразного гексафторида урана (см. табл. 138 и 139),
полученными нз теплоты сублимации. Найденные таким путем [56] значения очень хорошо согласуются с
полученными на основании спектроскопических данных значениями, приведенными во второй графе этой
таблицы [55].
Глава 13. Гексафторид урана
339
Из уравнений (35) —(38) для термодинамических функций и уравнений
(27а) или (276) и (31а) или (316) для давления пара выведено следующее
эмпирическое уравнение:
Ср [для UF6ra3] = 32,43 + 0,007936T-320680r-2 кал-моль-1-град-1 (40)
(рис. 51).
Значения Ср, выведенные из этого уравнения, хорошо согласуются со
спектроскопическими данными. Английские исследователи на основании
калориметрических измерений получили большие значения [38, 52, 53]. Такое
расхождение между теми и другими результатами пока нельзя объяснить
из-за отсутствия необходимых данных. Пока значения Ср, полученные
спектроскопическим путем или вычисленные из уравнения (40), должны быть
признаны более правильными. '
При вычислении теплосодержания и энтропии газообразного гексафто-
рида были использованы термодинамические функции для
конденсированного гексафторида в сочетании с уравнениями для давления пара [35J, а
7 34
^
а
со
-.- 33
•о
§
? 32
5
*
к 31
&
30
I I
— X
— X
L /
Г i .. 1
1 U 1
—1
А
—
л ) 1
270 300 350 400 $50
Температура. °Н
500
270 300
Рис. 51, Теплоемкость газообразного
гексафторида урана при постоянном
давлении [см. уравнение (40)].
350 400
Температура,
Энтальпия газообразного ге-
450
К
500
Рис. 52
ксафторида урана [см. уравнение (41)1-
также теоретические выводы, сделанные на основании данных о
спектроскопических частотах. При применении первого метода были получены
уравнения (41) и (42):
# = 8460+ 32,43 Г+ 0,003968Г2 +320680 7^2 кал>моль~х (41)
(рис. 52);
S\amM = 74,69 lg T + 0,007935 Т + 160340 Г"2~ 98,05 энтропийной единицы (42)
(рис. 53).
Значения энтропии, полученные из уравнения (42), хорошо согласуются
со значениями, установленными на основании спектроскопических данных
[55] и приведенными в табл. 137. Последние в свою очередь хорошо
совпадают со значениями, полученными из данных* давления пара [40] и
калориметрических данных о теплотах парообразования гексафторида [56].
Скорость звука в газообразном гексафториде урана и отношение CP)CV.
Скорость звука в газообразном гексафториде измерена двумя английскими
исследователями. Один из них приводит среднее значение 8870 см/сек при 49°
и давлениях 320 и 539 мм рт. ст. [57], другой —8990 см/сек при 49,2*
и давлении 420 мм рт. ст. [42].
На основании предположения, что отклонения от газовых законов
ничтожно малы, можно из значения v = 8990 см/сек вывести Ср/Сп= 1,063 ±0,005-
при 49,2°. Это значение хорошо согласуется с величиной 1,066, выведенной
22*
(340 Часть IV» Галогениды, оксигалогениды, боргидриды и карбонил урана
из уравнения (40) в предположении, что гексафторид подчиняется законам
^идеальных газов.
Теплота возгонки твердого гексафторида. Руффом и Хайнцельманом
приводится для теплоты возгонки гексафторида значение 10,36 кал-моль'1 [3].
Из данных давления пара были выведены две эмпирические формулы для
этой величины:
(43)
(44)
ДЯВ03г = 20400-0,00521 Т3 + 0,3905Т2-98,66Т кал-моль-1 [53].
Д//ВОзг = -1405 +52,512 7-0,070812 7»+1368600 Т'1 кал-моль'1 [35]
'{рис. 54).
Из этих двух уравнений последнее, выведенное из уравнений (27а) или
(276) для давления пара, вероятно, является более точным. Мази [56]
$8
§96
а
а:
Cg
Q)
494
*
♦:з
э
с:
о
с*.
s яг
*
с*
«о
90
88
■ 1
— /
- /
/ А
1 ' 1 1
ч
J
_J
—1
-J
1 ... II
270 300 350 400
Температура, °К
Рис. 53. Энтропия газообразного
гексафторида урана при 1 атм [см.
уравнение (42)].
i£. /UU,
12500
'^ 12300
1
к 12 ЮО
«о
f»;
О
*j 11900
11700
11500
\ \
\ 1
\ т
\ -1
\ 1
\ \
Г \ 1
1 L 1
220 250 300 350
Температура, °К
Рис. 54. Теплота возгонки твердого
гексафторида урана [см. уравнение (44)].
провел в Национальном бюро стандартов точные калориметрические
исследования теплот возгонки и испарения гексафторида в интервале температур
<от 4 до 90°. Полученные им результаты приведены в табл. 138 и 140; они
хорошо согласуются со значениями, выведенными Вайнштоком и Кристом
[40] из измерений давления пара. Некоторые дополнительные данные
английских исследователей, приведенные в отчете Киршенбаума [54], указаны
в табл. 139 [58].
Теплота парообразования жидкого гексафторида. Мази [56]
определил теплоту парообразования жидкого гексафторида прямыми
калориметрическими измерениями. Английские исследователи также привели ряд
.значений для теплот парообразования при различных температурах [45].
Э табл. 140 указаны эти данные, а также некоторые другие, полученные
английскими исследователями и упомянутые в отчете Киршенбаума [54].
Согласованность между значениями, взятыми из различных источников,
3 целом вполне удовлетворительная. Величины, полученные Мази [56],
несомненно заслуживают предпочтения перед установленными косвенным
путем из измерений давления пара. Мази вычислил из полученных им данных
Таблица 138
ТЕПЛОТА И ЭНТРОПИЯ ВОЗГОНКИ ТВЕРДОГО ГЕКСАФТОРИДА УРАНА [56]
Температура, °К
273,16
280
290
298,16
300
310
320
330
337,21
/, кал-моль"!
12 023(12 220)а)
11988
11929
11872(11970)а)
11858
11772
11666
11537
11429
//г,
кал*моль~1-град 1
44,02
42,81
41,13
39,82
39,53
37,97
36,46
34,96
33,89
5б),
кал 'Моль-1-град~1
87,56
88,39
89,57
90,50
90,70
91,80
92,86
93,87
94,59
а) В круглых скобках значения даны по Вайнштоку и Кристу [40].
б) Энтропия для идеального газа.
Таблица 139
ТЕПЛОТА ВОЗГОНКИ ГЕКСАФТОРИДА УРАНАа)
Температура,
°С
-75
-50
-25
0
д^возг,
ккал>моль~1
i
12,20±0,10
12,15±0,10
12,08±0,10
11,98±0,10
Температура,
°С
20
40
6
АЯВОЗГ,
ккаЛ'МОль~1
П,87±0,10
11,73±0,10
11,53±0,10
11,47^0,10
а) По измерениям английских исследователей.
Таблица 140
ТЕПЛОТА ПАРООБРАЗОВАНИЯ ЖИДКОГО ГЕКСАФТОРИДА УРАНА
Температура, °К
337,21
338
340
343
348
350
353
[56]а)
6,859
—
6,817
—
—
6,671
—
/, ккал'Моль-г
[45]
6,95
—
6,89
6,826)
—
6,74
[54]
6,92±0,1
—
6,89±0,1
—
—
—
Температура, °к
358
360
363
1 368
370
403
[56]а)
6,533
—
—
6,404
—
/, ккаЛ'Моль~*
[45]
6,66
—
6,57
6,46
—
—
[54]
__-.
—
6,67±0,1
6,31±0,1
—
5,80±0,1
а) Энтропия для идеального газа, рассчитанная по данным этой графы» составляет для указанных
температур 94,59; 94,88; 95.90; 96,91 и 97,95 энтропийной единицы соответственно.
б) Вайншток и Крист [40] для этой температуры дают значение 6487 кал-моль'*. г
342 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Таблица 141
ТЕПЛОТА ПЛАВЛЕНИЯ ГЕКСАФТОРИДА УРАНА
дяпл
калориметрическое
определение
—
—
4,58
4,588
ккал-моль~
д"пл=
5,11
4,60
4,55
=дя
(=
(=
(=
1
возг-д"испар
=12,00-
= 11,63-
=11,47-
—
■
-6
-7
-6
89)
03)
,92)
Литерач
45,
39
35
53
52,
37
гура
59
(табл. 138 и 140) энтропию гексафторида урана как идеального газа, и
найденные им значения хорошо совпали с вычисленными по
спектроскопическим данным (см. табл. 137). Из уравнения давления пара (31а) или (316)
Киршенбаумом [35] выведено следующее уравнение для теплоты
парообразования жидкого гексафторида:
ДЯпар = 2473,4+ 14,476 Т-0,028546 Г2+ 987670Г"1 кал-моль'1
(45)
(рис. 55).
Теплота плавления гексафторида урана. Теплота плавления может
быть определена прямыми калориметрическими измерениями или вычислена
как разность между теплотой возгонки
твердого гексафторида и теплотой
парообразования жидкого. В табл. 141
представлены результаты, полученные
этими двумя способами.
Теплопроводность газообразного и
жидкого гексафторида урана. Прямое
измерение теплопроводности
проводилось методом «нагретой проволоки» [60].
Измерения делались с аргоном и
азотом как эталонными газами; в
соответствующей работе, проведенной в Англии,
эталонный газ не указан. Этим,
возможно, объясняется расхождение
между результатами работ английских и
американских исследователей. В табл.
142 приведены значения
теплопроводности газообразного гексафторида при
нескольких температурах. Для
сравнения там же приведены значения,
вычисленные из данных о вязкости по
формуле Эйкена
335
375
345 355 365
Температура, °\1
Рис. 55. Теплота парообразования
жидкого гексафторида урана [см.
уравнение (45)1.
К=\ (9т -5)^, (46)
где 4 = Cp/Cv, т] — вязкость; остальные символы имеют свои обычные значения.
Как видно из табл. 142, значения, полученные американскими
исследователями в результате непосредственных измерений и вычисленные по вязкости,
прекрасно совпадают. Данные по теплопроводности, полученные в резуль-
Глава 13. Гексафторид урана
343
Таблица 142
ТЕПЛОПРОВОДНОСТЬ ГАЗООБРАЗНОГО ГЕКСАФТОРИДА
УРАНА
Температура,
°С
0
50
100
125
Теплопроводность* 1 0-
[60]
1,450
1,787
2,125
2,294
[61, 62]
1,54
1,84
2,13
2,26
&, каА»см-1-сек-
[54]
1,39
1,65
1,91
2,04
-1-граЭ-1
из данных по
вязкости
[63]
1,454
1,79
2,12
2,28
тате четырех исследований, можно аналитически выразить следующим
образом:
Данные американских работ:
/С = 1,450 + 0,00675/(10"5 Кал ■ см'1 • сек'1 • град-1). (47)
Данные английских работ (старые):
К = 1,542+ 0,0059/(10"5 кал ■ см'1 • сек'1 • град'1). (48)
Данные английских работ (новые):
К= 1,39 + 0,0052ф0~5 кал • слг1 • сек'1 • град'1). (49)
Данные, вычисленные по вязкости:
К= 1,454 +0,0067/(10"6 кал ■ слс1 • сек~х • град'1). (50)
На основании средних из приведенных в этих уравнениях значений
было выведено уравнение (51), которое, вероятно, лучше всего отражает
имеющиеся данные по теплопроводности газообразного гексафторида:
К= 1,459 + 0,00614/(10-5 кал • смг1 • сек'1 . град'1). (51)
Для теплопроводности жидкого гексафторида имеется только одно
значение [64] в 3,83 (± 3%) • 10"4 кал • см'1 • сект1 • град-1 при 72°. Данные
по теплопроводности твердого гексафторида отсутствуют.
Механические свойства.. Плотность твердого гексафторида
урана. Руффом и Хайнцельманом [3] определена плотность твердого
гексафторида, равная 4,68 г/см3 при 20,7°. Из рентгенографических данных было
сначала вычислено значение 4,95 г/см?, а позже более точное: 5,060 ±
± 0,005 г/см3 [65]. Английские исследователи приводят для плотности
величину 5,09 + 0,06 г/см3 при 20,7° [46], а Хоудж и Векслер
[66]-величину 4,87 г/см3 при 62,5°.
Плотность жидкого гексафторида урана. Наиболее ранние измерения
плотности жидкого гексафторида дали значение 3,667 ± 0,05 г/см3 при
65,1° [67]. В Национальном бюро стандартов [68] отношение удельных
объемов жидкого гексафторида при 65,1° и твердого при 63,5° определено
в 1,343 + 0,002; там же выведено уравнение для отношения объема при
температуре / к объему при температуре тройной точки /тр (64,052°):
V/Утр =1 + 1,727 . 10-»(f-*Tp)+3f59 • 10-в(/-гтр)2. (52)
344 Часть /V. Галогениды, оксигалогениды, боргидриды и карбонил урана
В табл. 143 приведены измеренные и вычисленные величины для этого
отношения в интервале температур от 62,45 до 92,4°.
Таблица 143
ОТНОСИТЕЛЬНЫЙ УДЕЛЬНЫЙ ОБЪЕМ ЖИДКОГО ГЕКСАФТОРИДА
УРАНА [РАССЧИТАНО ПО УРАВНЕНИЮ (52)]
t *а)
*—*™Л
тр
-1,60
-0,02
0,92
7,26
9,59
9,59
11,93
18,95
21,29
25,98
28,33
Относительный удельный объем,
V
~
найдено
0,997
1,000
1,001
1,013
1,076
1,017
1,022
1,033; 1,034
1,038
1,048
1,053; 1,051
^тр
разность между 3
найденным и
вычисленным значениями
0,000
0,000
-0,001
0,000
-0,001; 0,000
0,000
0,001
-0,001; 0,000
0,000
0,001
0,001; 0,001
''1 тр=64,052°.
В табл. 144 содержатся измеренные и вычисленные значения
абсолютной плотности жидкого гексафторида. В графе 1 указаны значения,
вычисленные на основании данных табл. 143 для VIVTP при приравнивании
плотности гексафторида в тройной точке (64,052°) к 3,630 г/см3. Данные в графе 2
вычислены тем же одособом, но при этом использовано значение плотности
3,667 г/см3 при 65,1°. Данные в графе 3 получены в результате прямых
измерений [48, 66j, а также путем интерполяции экспериментальных
значений посредством эмпирического уравнения
9 = 3,630-5,805- Ю-3(*-*Tp)-1,36- 105(^-^тр)2, (53)
где tTp = 64,052°. Это уравнение выражает плотность насыщенного жидкого
гексафторида в пределах ошибки опыта в интервале температур 68,66 —
162,59°. Значения, полученные опытным путем, заключены в круглые скобки.
В графе 4 приведены величины, взятые из различных обзорных отчетов,
составленных английскими и американскими группами, ввиду отсутствия
оригинальных работ [35].
Плотность газообразного гексафторида. Газообразный гексафторид
ведет себя подобно идеальному газу. Ассоциации молекул не наблюдается.
Руфф и Хайнцельман [3] определили из двух значений плотности пара
при 448° средний молекулярный вес гексафторида 338. Амфлетт, Маллингер
и Томас [28, 42, 69, 70] вновь определяли плотность пара гексафторида при
различных температурах и давлениях. Полученные ими результаты
представлены в табл. 145 и 146.
Таблица 144
ПЛОТНОСТЬ ЖИДКОГО ГЕКСАФТОРИДА УРАНА
Температура,
°С
65
68,66
70
72,91
75
77
80
85
90
92,65
95
100
112,67
120
131,12
140
156
160
162,59
203
215
225
230
1
3,624
—
3,593
—
3,561
—
3,530
3,498
3,464
—
—
—
—
—
—
—
—
—
—
—
—
—
Плотность, г\смъ
2
3,667
—
3,636
—
3,604
—
3,572
3,540
3,508
_.
—
—
—
—
—
—
—
—
—
—
—
—
"
за>
3,624
(3,604)
3,595
(3,576)
3,565
—
3,532
3,502
3,470
(3,452)
3,437
3,404
(3,316)
3,263
(3,183)
3,111
—
2,948
(2,920)
—
—
—
~
4
—
—
—
—
3,63+0,18
—
—
—
—
—
—
—
—
—
—
3,П±0,16
—
—
2,62±0,13
2,32±0,12
2,09±0,10
1,63±0,08
а) Значения в круглых скобках получены экспериментально.
Подробности см. [66].
Таблица 145
ПЛОТНОСТЬ ПАРА ГЕКСАФТОРИДА
УРАНА [28]
Темпера-
тура,°С
14,1
14,8
17,4
29,7
50,0
50,0
50,0
Давление, мм
рт. ст.
52,0
54,4
66,1
157,5
521,0
419,0
392,1
Плотность
(Н = 1)
176
178
174
174
176
175
174
Молекулярный
вес
(0=16)
355
357
352
352
355
353
352
Таблица 146
ПЛОТНОСТЬ ПАРА ГЕКСАФТОРИДА УРАНА
В ЗАВИСИМОСТИ ОТ ДАВЛЕНИЯ ПРИ
ТЕМПЕРАТУРЕ 49,2° [28]
Давление,
мм рт. ст.
523,0
470,0
424,0
378,0
316,0
259,3
213,3
186,4
Плотность пара-
• 10-», г/ли з
9,016
5,295
6,482
7,240
8,119
4,278
3,627
3,130
Давление-105,
duHbtJCM^
7,027
4,246
5,072
5,717
6,315
3,479
2,862
2,534
346 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Повидимому, закономерная зависимость молекулярного веса от
температуры отсутствует. Арифметическая средняя экспериментальных значений
молекулярного веса гексафторида при комнатной температуре равна 355 ± 3,
а при 50° —353,3 ± 1. Вычисленный молекулярный вес равен 352,8. Из
данных о зависимости плотности пара от давления было выведено
(-р-*) =1,32 ±0,01 . Ю-8.
\др У* = 49,2°
Величина, вычисленная на основании кинетической теории для идеального
газа с молекулярным весом гексафторида, равна 1,314 - 10~8. Поэтому можно
считать, что при обычных температурах и давлениях парообразный гекса-
фторид состоит из одиночных неассоциированных молекул.
Ввиду того что гексафторид ведет себя как идеальный газ, его
плотность при различных температурах и давлениях может быть вычислена по
уравнению (54) [50]:
Р=13,28.^.^, (54)
где р — плотность, г/л, р — давление, см рт. ст., Г —температура, °К.
Коэффициент самодиффузии газообразного гексафторида.
Произведение pD, где р —плотность и D — коэффициент самодиффузии, было
определено в 234 ± 9 микропуаз при 30° [71, 72]. Согласно уравнению (54),
плотность при 30° равна
откуда
Зная эти величины, можно вычислить значение е в уравнении pD = eTj,
где т] — коэффициент вязкости. Согласно английским данным, т\ (30°) =
= 180 микропуаз. По данным Колумбийского университета, г\ (30°) = 206
микропуаз (см. стр. 347). Отсюда значение е для гексафторида равно 1,30
или 1,14. Большая разница между этими двумя цифрами указывает на
необходимость дальнейшего исследования. Согласно кинетической теории газов,
е должно быть равно 1,200, если считать молекулы твердыми упругими
шариками, и 1,504, если принять, что между молекулами существует
взаимодействие, величина которого обратно пропорциональна пятой степени
расстояния. Была выведена теоретическая формула для D(UFe), причем
значение е было принято равным 1,50 [50]:
В = 0,0606- 20,° см [1+6,29(* - 10-8) + 9,6(*. 10-3)2] см2/сек. (57)
Рем рт. ст.
Поскольку, однако, значение е точно неизвестно, применимость этого
уравнения ограничена.
Средний свободный пробег. Для среднего свободного пробега L
молекул гексафторида в газовой фазе дается следующая формула:
L= 20,0c* • 6,32 [1+4,45 (^ 10"3) + 3,1(*. Ю"3)2] • 10~в см (58)
Рем рт. ст.
для t в пределах от 0 до 200°.
Значения для среднего свободного пробега, вычисленные по формуле
1
L =
Y2nNo
где а —диаметр молекулы, выводимый из измерений вязкости,
удовлетворительно совпадают с полученными из уравнения (58).
Глава 13. Гексафторид урана
347
Вязкость гексафторид а урана. Вязкость жидкого гексафторида при
температурах 67,2 — 75,0° была измерена Колумбийской группой;
найденные значения приведены в табл. 147 [73]. Абсолютная вязкость
гексафторида почти в два раза больше вязкости воды (0,422 сантипуаз при 67,2°);
кинематическая вязкость составляет около половины вязкости воды
(т)н2о при 67,2° равно 0,431 сантистокс).
Приведенные в табл. 147 (в круглых скобках) результаты английских
-исследователей не согласуются с данными, полученными в Колумбийском
университете [54].
Таблица 147 Таблица 148
ВЯЗКОСТЬ ЖИДКОГО ГЕКСАФТОРИДА ВЯЗКОСТЬ ГАЗООБРАЗНОГО
УРАНА ГЕКСАФТОРИДА УРАНА
Температура,
°С
0
20
40
50
60
80
100
НО
140
150
170
200
Вязкость, микропуазы
[61, 62, 74]
167
175
—
189
—
—
211
—
—
233
—
255
[76]
—
178,7
—
189,0
199,9
—
216,1
231,9
—
248,0
261,1
Температура,
°С
67,2
70
72,5
72,9
73,4
74,7
75,0
80
90
100
Абсолютная
вязкость,
сантипуазы
1 0,731
(0,91)
0,692
0,685
0,679
0,669
0,663
(0,85)
(0,80)
(0,75)
Кинематическая
вязкость,
сантисток-
сы
0,200
—
0,191
0,189
0,188
0,185
0,184
—
—
Английскими исследователями измерена также вязкость газообразного
гексафторида [61, 62, 74]. Значения для интервала температур 0 — 200°
(приведены в табл. 148) могут быть выражены с точностью до ± 2%
следующим уравнением:
т] . 104 (пуазы) =1,67 — 0,0044 U (59)
где< — температура, °С. Однако результаты опытов по определению вязкости
газообразного гексафторида, проведенных в Колумбийском университете [75],
отличаются на 20% от вышеуказанных. Проведенная проверка [76] показала,
что английские данные в основном правильны. Из этих данных вычислена
константа Сузерленда, которая оказалась приблизительно равной 101 [46].
Поверхностное натяжение жидкого гексафторида урана. Измерено
поверхностное натяжение жидкого гексафторида в равновесии с паром
методом капиллярного поднятия в интервале температур от 70,0 до 100° [47].
Результаты этих измерений представлены в табл. 149.
Оптические свойства. Показатель преломления [77]. Показатель,
преломления жидкого гексафторида определен путем измерений угла
полного внутреннего отражения в интервале температур 70—100° с точностью
примерно 0,4% для абсолютных значений. Относительные значения, вероятно,
еще точнее. Результаты определений приведены в табл. 150. Молекулярная
3 48 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Таблица 149
Таблица 150
ПОВЕРХНОСТНОЕ НАТЯЖЕНИЕ
ЖИДКОГО ГЕКСАФТОРИДА
УРАНА
ПОКАЗАТЕЛЬ ПРЕЛОМЛЕНИЯ ЖИДКОГО
ГЕКСАФТОРИДА УРАНА
Температура,
°с
70
80
90
100
Поверхностное
натяжение,
дины 1см
16,8±0,3
15,6+0,3
14,3±0,3
13,1±0,3
Температура,
°с
70
80
90
100
Показатель преломления
4360А
1,383
1,374
1,365
1,355
5890А
1,367
1,358
1,350
1,342
рефракция R была рассчитана по хорошо известной формуле
^ М п2-\
(60)
где М — молекулярный вес, р — плотность, а п — показатель преломления
при длине волны падающего света. Были получены следующие значения:
#4збоА = 22,59±0,08; /=85°;
/?Б89оА = 21,83±0,08; / = 85°.
Если предположить, что молекулярная рефракция для газообразного гекса-
фторида такая же, как и для жидкого, то для показателя преломления
газа при длине волны света 5100 А получается следующее выражение [46J:
п- 1 = 5,2- 10~4 р/Т,
(61)
где р — давление, мм рт. ст., а Г—температура, °К.
Ультрафиолетовый спектр поглощения гексафторида урана.
Ультрафиолетовый спектр поглощения паров гексафторида впервые снят [78] при
помощи большого прибора Хилджера (разрешающая способность 5 А на 1 мм).
Применялся однометровый слой пара в равновесии с твердым гексафтори-
дом при комнатной температуре. Сплошной спектр поглощения начинался
примерно около 3300о А; ему предшествовал полосатый спектр поглощения
между 3400 и 3800 А. Анализа спектра не сделано.
Мартин и Амфлетт [79] также изучали поглощение ультрафиолетового
излучения парами гексафторида при комнатной температурео и обнаружили
сильное избирательное поглощение с краем полосы при 3701 А и минимумом
около 3417 А. Совсем недавно Амфлетт, Маллингер и Томас [28]
опубликовали некоторые предварительные данные о поглощении излучений парами
гексафторида в интервале между 2000 и 4000 А. В противоположность
результатам, полученным в Колумбийском университете, они наблюдали
сплошное поглощение без структуры и полное поглощение ниже примерно
3100 А. Так как результаты исследований, проведенных в Колумбийском
университете, основаны на гораздо большем экспериментальном материале,
чем данные Амфлетта, то они заслуживают большего доверия.
Спектр поглощения на участке 3500—4000 А повторно изучен в Колум-
•бийском университете [80]. Газообразный гексафторид находился под
давлением 36 мм рт. ст., толщина поглощающего слоя равнялась 102 см. Пять
наиболее четких полос отстояли друг от друга примерно на 590 см'1.
Полосы поглощения в спектре газообразного гексафторида урана
характеризуются следующими частотами (см~г): 24 364, 24 460, 24 504, 24 794,
Глава 13. Гексафторид урана
349
24 860, 24 972, 25 108, 25 211 (несколько резче), 25 344, 25 478, 25 538,
25 720, 26 031, 26 175, 26 227, 26 379, 26 573, 26 644, 26 755, 26 935
(сомнительное значение), 27 015 (умеренно сильное поглощение у фиолетового
края), 27 135, 27 184, 27 234 (наиболее сильное поглощение у фиолетового
края), 27 342, 27 420, 27 562, 27 655, 27 820 (сильное поглощение у
фиолетового края), 27 907, 28 080, 28 415 (середина).
Вокруг каждой главной полосы находились группы тесных полос,
отличающихся по частоте примерно на 100 см'1. Кроме того, повидимому,
имелось еще четыре полосы, отстоящие от главных полос примерно на 200 см"1.
Сообщается также, что толстый слой (12 см) прозрачного кристаллического
гексафторида полностью поглощал излучения с длиной волны ниже 4271 А
{81]. При частоте 42 000 еж-1 поглощение примерно в тысячу раз больше,
чем на участке рассматривавшегося полосатого спектра. Область
сплошного поглощения с постепенно убывающей интенсивностью простирается
до частоты 31000 еж1. Было найдено, что магнитное поле с индукцией
19 000 гаусс не влияет на спектр.
Спектр флюоресценции гексафторида урана [82]. Твердый гексафторид,
возбужденный излучением с длиной волны 3660 А, светится при 77° К ярко-
фиолетовым светом; при комнатной температуре флюоресценция исчезает-
Пары гексафторида не флюоресцировали даже после длительного облучения-
В спектре флюоресценции наблюдаются размытые полосы, расстояние между
которыми составляет примерно 220 см'1*
Полосы в спектре флюоресценции твердого гексафторида урана при 77° К
характеризуются следующими частотами (см'1): 22 230, 22 450, 22 662,
22 883, 23 098, 23 319, 23 537, 23 739, 23 994, 24 166. Максимум
интенсивности приблизительно соответствует частоте 23700 еж"1; исследованный
интервал составлял 22 000—24 700 еж-1. Магнитное поле с индукцией 25 000
гаусс не оказывает влияния на спектр флюоресценции.
Инфракрасный спектр поглощения и спектр комбинационного рассеяния.
Данные по инфракрасному спектру газообразного гексафторида урана [35, 55]
представлены в табл. 151.
Таблица 151
ПОЛОСЫ ПОГЛОЩЕНИЯ В ИНФРАКРАСНОЙ ЧАСТИ СПЕКТРА
ГАЗООБРАЗНОГО ГЕКСАФТОРИДА УРАНА
Частота,
см-1
623
675
713—719
825
Длина
волны, [L
16,05
14,82
14,03—13,91
12,12
Интенсивность
100
10
0,5
2
Частота,
см-1
850
1 163
1 295
2 053
Длина
волны, {X
11,68
8,60
7,72
4,87
Интенсивность
0,25
4
3
0,5
Исследован также спектр комбинационного рассеяния. Сначала было
сообщено, что жидкий гексафторид при 70,9° показывает спектр с 2 сателлитами,
отстоящими на 603 и 228 см"1 от основной линии (при возбуждении ртутной
линией 4358,34 А путем экспозиции в течение 3 час.) [83]. Наиболее
интенсивной была линия 603 см'1. Сделано предположение о существовании линии
572 еж"1, но она не обнаружена, возможно ввиду маскировки ее линией 603 см'1.
Позже проведено более подробное исследование комбинационного
рассеяния гексафторида [55, 84]. Несколько опытов было выполнено с применением
возбуждающего излучения с длиной волны 4358 А. Несмотря на попытки умень?
шить степень фотохимического разложения жидкого гексафторида, экспозицию
350 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
пришлось прекратить через 15 мин. из-за рассеяния света образовавшимися
мелкими пушистыми частицами твердого вещества. Определить наличие линий
комбинационного рассеяния с частотой ниже 300 см'1 не представляется
возможным. За этим участком найдены две линии. Средние значения, полученные
при экспозициях жидкого гексафторида, составляли 656±3 и 511 ±3 см'1.
Линия с большей частотой была четкой и интенсивной, напротив, другая
линия была слабой и несколько размытой. Сделаны попытки получить
дополнительные данные путем постановки опытов с растворами гексафторида во фтор-
углеродах, например перфторгептане. При длине волны возбуждающего
излучения 4358 А фоторазложение еще имело место, так же как и в случае
чистого гексафторида. При длине волны 5460 А фоторазложения не наблюдалось
и получен удовлетворительный спектр комбинационного рассеяния при 20°.
Кроме первоначально найденной линии 656 см'1 обнаружена еще интенсивная
линия 202 см'1. Линия 511 см'1 в спектре перфторгептанового раствора не
видна, вероятно ввиду низкой интенсивности, а линия 656 см'1 смещена
к 666 см'1.
Данные по спектрам инфракрасному и комбинационного рассеяния были
проанализированы двумя группами ученых [84, 55]. Инфракрасный спектр
говорит о возможности некоторого вырождения. Сделано предположение, чта
основная частота равна примерно 640 см'1 и что примерно на ту же область
падает двойной обертон, так что линии 623 см'1 и 675 см'1 образуются как
результат взаимодействия двух уровней примерно при 640 см1. Расшифровка
разных частот, данная в табл. 152, была сделана на основе этого
предположения. В табл. 153 сопоставлены вычисленные и экспериментальные значения
инфракрасных активных двойных обертонов. На основании числа активных
линий в спектрах инфракрасном и комбинационного рассеяния, а также
поглощения можно сделать вывод о том, что гексафторид имеет полностью
симметричную структуру.
Таблица 152
КЛАССИФИКАЦИЯ ЧАСТОТ В СПЕКТРЕ ГАЗООБРАЗНОГО ГЕКСАФТОРИДА
УРАНА, ОСНОВАННАЯ НА ДАННЫХ СПЕКТРОВ КОМБИНАЦИОННОГО
РАССЕЯНИЯ СВЕТА [55]
Обозначение
^1
^2
^3
V4
V5
ve
Частота,
см-i
656
511
200
130
200
640
Симметрия
Aig
Eg
^20
Т 2U
*1U
Т\и
Вырождение
1
2
3
3
3
3
Активность
Активна в спектре
комбинационного
рассеяния
То же
» »
Неактивна
Активна в инфракрасном
спектре
То же
Наблюдавшаяся
частота, см-1
656±3
511±3
202±3
—
—
623 или
больше
Электрические и магнитные свойства.
Диэлектрическая постоянная. Мартин и Амфлетт вначале получили для диэлектрической
постоянной газообразного гексафторида урана при комнатной температуре
в атмосферном давлении величину 1,004,[79], позже они уточнили это
значение в 1,0043±0,0001 [46]. Совсем недавно Амфлетт, Маллингср и Томас [281
определили, что диэлектрическая постоянная для газообразного гексафторида
Глава 13. Гексафторид урана
351
Таблица 153
РАССЧИТАННЫЕ И НАБЛЮДАВШИЕСЯ ОБЕРТОНЫ
В ИНФРАКРАСНОМ СПЕКТРЕ [55]
Обозначение
^5
^4 + ^5
^5 + ^3
^6
^4 + ^2
^5 + ^2
^6 + Ъ
^5 + ^1
^0 + ^2
^6 + ^1
Частота. см~1
по расчету
200
300
400
640
641
т.
840
856
1 151
1296
по
наблюдению
_
__
—
623
675
713-719
825
850
1 163
1295
2 053
Интенсивность
1 000
10
0,5
2
0,25
4
3
0,5
равна 1,0038±0,0001 при 19,6° и 760 мм рт. ст. Вычисленная из этих данных
молярная поляризация равна 30,7±1,0сл*3 при тех же температуре и давлении.
В табл. 154 представлены результаты тщательных определений, проведенных
в Принстонском университете [85]. Зная диэлектрическую постоянную е
газа, можно вычислить молярную поляризацию Р, а из нее в свою очередь на
основании уравнения Клаузиуса—Мозотти
и диэлектрическую постоянную жидкости. Принстонской группой выведены
значения Р=27,1 см3 и е=2,18 при 65°, что является типичным для
неполярных жидкостей.
Дипольный момент. Из диэлектрической постоянной были вычислены
дипольные моменты газообразного гексафторида при различных
температурах [85]. Найдено (см. табл. 154), что молярная поляризация не изменяется
с температурой. Р можно определить из хорошо известного уравнения Дебая:
Отсутствие заметной зависимости Р от температуры показывает, что
дипольный момент, вероятно, равен нулю. На основании обычных критериев
выводится, что хотя дипольный момент гексафторида может иметь величину
меньше 0,5-10~18, логичнее предполагать, что он равен нулю.
Магнитная восприимчивость. Гексафторид урана, повидимому,
парамагнитен. Удельная восприимчивость твердого гексафторида определена в
+0,12-10"6, а молярная—в +43-10"6 [6]. После внесения в значение молярной
восприимчивости поправки на диамагнетизм (U=23, F=7,25) [86] получается
величина +106-10~в. Парамагнетизм, повидимому, не зависит от температуры.
Ионизационный потенциал. Определены [87, 88] критические
ионизационные потенциалы одновалентных ионов, образованных бомбардировкой
гексафторида медленными электронами. Они приведены в табл. 155. Вероятный
интервал погрешностей этих данных составляет от 5% для иона UF5+ до
15% для иона U+.
352 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Таблица 155
КРИТИЧЕСКИЕ ИОНИЗАЦИОННЫЕ
ПОТЕНЦИАЛЫ ГАЗООБРАЗНОГО
ГЕКСАФТОРИДА УРАНА [87]
Ион
UFJ
UF4+
UFJ
UF*
UF+
U*
Потенциал, в
15,5
20,1
S3,5
29,9
* 37,9
50,3
Структура молекулы гексафторида урана. Для
установления структуры гексафторида применялись методы электронографии
и рентгеновской кристаллографии, изучение спектров инфракрасного и
комбинационного рассеяния света и определение дипольного момента.
Имеющийся материал говорит в пользу неискаженной октаэдрической структуры
гексафторида.
Электронография. Впервые электронографические исследования структуры
гексафторида были проведены Брауне и Пинноу [89]. Обнаружено, что гекса-
фториды урана, вольфрама и молибдена обладают неправильной структурой
и асимметрией ромбической голоэдрии; расстояния между металлом и фтором
для каждой из трех пар атомов фтора относятся как 1 : 1,12 : 1,22. В случае
гексафторида это дает для расстояний U—F значения 1,78; 1,99 и 2,17 А. Бауэр
[90] исследовал диффракцию электронов с энергией 42 кв парообразным гек-
сафторидом и пришел к заключению, что в молекуле гексафторида все шесть
расстояний U—F не могут быть одинаковыми. Его данным соответствуют две
модели (симметричная октаэдрическая структура, так же как и некоторые
другие возможные структуры, была исключена). Бауэр выбрал как наиболее
вероятную структуру искаженный октаэдр с атомами фтора в шести вершинах
и атомом урана в плоскости, образованной четырьмя атомами фтора.
Межатомные расстояния для этой модели следующие:
два смежных U—F = 1,87±0,02 А;
два противоположных . . . U—F = 2,12^0,03 А;
два смежных U—F = 2,22^0,03 А.
Искажение октаэдра такое, что все расстояния F—F приблизительно
одинаковы (около 2,9 А). Эта структура сходна со структурой Брауне и Пинноу,
однако абсолютные значения межатомных расстояний примерно на 5% больше.
Модель, предложенная Бауэром, не имеет центра симметрии и должна
поэтому иметь небольшой дипольный момент. Аналогия соструктурой
металлического урана, найденная Джэкобом и Уорреном [91], вполне очевидна.
Дипольный момент. Смит [85] показал, что структура, предложенная
Бауэром, должна иметь дипольный момент, по меньшей мере равный 1,5-10~18,
вероятно даже больший. Измерения диэлектрической постоянной показали,
что момент не может быть более 0,5 • 10"18, а скорее всего он равен нулю.
Вследствие этого структура, предложенная Бауэром, неприемлема. Смит отметил
также, что одна из моделей, отвергнутых Бауэром, вполне соответствует данным
электронографии. Эта структура представляет собой октаэдр с неискаженными
Таблица 154
ДИЭЛЕКТРИЧЕСКАЯ ПОСТОЯННАЯ ГАЗООБРАЗНОГО
ГЕКСАФТОРИДА УРАНА
Температура,
°С
59,5
67,4
89,0
Диэлектрическая
постоянная, е
1,00297±0,00006
1,00292±0,000003
1,00273±0,00005
Молярная
поляризация Р,
см*
27,0±0,2
27,2±0,3
27,0±0,2
Глава 13. Гексафторид урана
353
углами и следующими межатомными расстояниями:
два противоположных .... U—F = 1,87 А;
два противоположных. . . . U—F = 2,12 А;
два противоположных .... U—F = 2,22 A.
Такая структура должна иметь три пары взаимно противоположных и
компенсирующих друг друга диполей, которые дают для молекулы в целом момент,
равный нулю.
Данные по спектрам инфракрасному и комбинационного рассеяния [81—
84]. Наличие только трех линий комбинационного рассеяния является
хорошим доказательством в пользу симметричной октаэдр и чес кой структуры
молекулы гексафторида; для любой другой модели число линий должно бы быть
больше.* Если молекула гексафторида представляет правильный октаэдр, она
должна принадлежать к группе симметрии 0^ Правило отбора показывает,
что в спектре комбинационного рассеяния таких молекул можно ожидать
только три линии; тетрагональный октаэдр ф^или С^) дал бы гораздо большее
число линий. Только плоский шестиугольник (Об/О может дать такое же
небольшое число линий, как и правильный октаэдр. Отмечено, что если бы даже
только две противоположные связи уран—фтор были удлиненными, линии 511
и 202 см'1 в спектре комбинационного рассеяния были бы расщепленными.
Между тем подобного расщепления не наблюдалось. Инфракрасный спектр
показывает наличие только одной основной частоты между 600 и 2100 смх,
и это также может быть объяснено на основании предположения о полной
октаэдр и чес кой симметрии.
Рентгенографические данные для кристалла гексафторида [65].
Рентгенографические анализы цилиндрических монокристаллов показывают, что
твердый гексафторид принадлежит к ромбической голоэдрии; пространственная
группа D]>i—Рпти. При 24° константы решетки следующие:
а = 9,900 ± 0,002А; Ь = 8,962 ± 0,002А; с - 5,207 ± 0,002 А.
Элементарная ячейка содержит четыре молекулы, и плотность твердого
гексафторида при 25°, вычисленная из рентгенографических данных, равна
5,060±0,005 г/см3. В табл. 156 сопоставлены значения экспериментально най-
Таблица 156
СРАВНЕНИЕ ЭКСПЕРИМЕНТАЛЬНО НАЙДЕННЫХ И ИДЕАЛЬНЫХ
ПАРАМЕТРОВ КРИСТАЛЛИЧЕСКОЙ РЕШЕТКИ ГЕКСАФТОРИДА УРАНА
Атом
и
Fi
F„
Fin
F1V
X
0,1295
0,003
0,250
0,014
0,246
найденные
У
1
т
1
т
1
7
0,093
0,093
Параметры
z
0,081
-0,250
0,417
0,250
-0,083
X
1
"8
0
1
7
0
1
4
идеальные
У I
1
4
1
4
1
7
1
12
1
72
z
1
2
1
~4
5
12
1
4
1
12
23 Химия урячз
354 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
денных и «идеальных» параметров для этой структуры. Рентгенографические
данные не свидетельствуют о совершенно правильной октаэдрической
конфигурации молекулы в кристалле. Атомы фтора приблизительно расположены по
принципу плотнейшей упаковки. Имеется, повидимому, действительное, хотя
и небольшое отклонение от идеальной симметрии, выражающееся в небольшом
сплющивании молекулы вдоль нормали к плоскости симметрии. Это небольшое
искажение может быть результатом анизотропных колебаний в кристалле.
Оно совместимо с совершенно правильной конфигурацией молекулы в парах.
Конфигурации, предложенные Бауэром для пара, определенно исключаются
в случае кристалла.
ХИМИЧЕСКИЕ^СВОЙСТВА ГЕКСАФТОРИДА УРАНА
Гексафторид является весьма реакционноспособным веществом. Его
химические свойства будут рассмотрены в следующих трех разделах: 1) общие
химические свойства, 2) растворы гексафторида в различных растворителях
и 3) коррозионное действие на металлы и другие аппаратостроительные
материалы.
Общие химические свойства. Отношение к воде.
Гексафторид урана энергично реагирует с водой с образованием главным образом
U02F2 и фтористого водорода. Эта реакция сопровождается выделением
значительного количества тепла (см. стр. 334). При проведении этой операции
в большом масштабе целесообразно вводить гексафторид в разбавленную
щелочь либо в виде пара, либо по каплям после сжижения. Ориентировочные
расчеты показывают, что теплота растворения гексафторида в разбавленном
растворе едкого натра равна примерно—118ккал/моль [92]. Реакция
гексафторида со щелочью протекает следующим образом:
UF6 + 40H: -> U02+ + 6F" + 2Н20, рН = 7,0 - 7,5; (64)
2U05+ + 60Н- + 2Na+ -> Na2U207 + ЗН20, рН = 10,0 - 10,5. (65)
При парциальном давлении гексафторида в 15 мм рт. ст. он реагирует с
водяным паром с выделением густого белого быстро оседающего дыма. При
давлении 0,4—0,5 мм рт. ст. гексафторид заметно не дымит [93].
Рентгенографические исследования показали, что при взаимодействии гексафторида с парами
воды образуется не чистый U02F2, а комплексное соединение последнего
с фтористым водородом и водой, из которого может быть получен чистый
U02F2 нагреванием до 180° или выше [94]. Эти результаты, вероятно,
требуют еще проверки.
Отношение к кислороду, азоту и двуокиси углерода [3]. Гексафторид вполне
устойчив по отношению к кислороду, азоту и сухому воздуху. Он не реагирует
с двуокисью углерода, однако при фторировании в присутствии последней
возможно образование карбонилфторида с последующим взрывом, что требует
принятия соответствующих мер предосторожности.
Отношение к галогенам [3]. Гексафторид урана заметно растворяется
в жидком хлоре или броме. С газообразными хлором или бромом он не
реагирует ни на холоду, ни при нагревании. Отношение к иоду не изучено.
Восстановление гексафторида урана неметаллами [95]. Руфф и Хайн-
цельман утверждают, что гексафторид мгновенно реагирует с водородом при
комнатной температуре. Однако недавно сделанные многократные попытки
восстановить гексафторид водородом окончились неудачей. Так, было
показано [96], что при температуре ниже 390° взаимодействия вообще не
происходит. Даже при температуре выше 500° реакция протекает довольно
медленно и неполностью и дает сложную смесь различных продуктов. Добавление
небольших количеств хлора не ускоряло реакцию. Подобные же наблюдения
сделаны и в Англии; отмечено, что реакция между гексафторидом и
водородом требует значительной энергии активации. Она может быть возбуж-
Глава 13. Гексафторид урана
355
дена, например, действием ультрафиолетового света или электрического
разряда. Были испытаны различные вещества в качестве катализаторов, в
частности вода, тетрафторид и тетрахлорид урана, хлор, бром, иод, динитрид урана,
никель, хлористый водород, монохлорид ртути и трихлорид железа. Заметное
действие оказывали лишь хлориды, но даже в их присутствии реакция шла
медленно и в результате ее получались загрязненные продукты [77, 94].
При пропускании сернистого газа через гексафторид при комнатной
температуре или при нагревании смеси его с газообразным гексафторидом в
никелевой трубке до 150° восстановления гексафторида не наблюдалось [97]. Этилен
при тех же условиях, повидимому, восстанавливает гексафторид, однако в
продуктах реакции содержится значительное количество углеродистых веществ
[97]. Найдено, что при использовании горелки (типа ацетиленовой) можно
восстановить гексафторид пропаном до тетрафторида [98]. Смесь водорода
и арсина при комнатной температуре не восстанавливает гексафторида.
Гексафторид реагируете хлористым водородом при 200°, реакция протекает
быстрее в интервале температур 250—300° [99]. При 200° получается смесь
коричневого, зеленого и черного продуктов, которая после обезгаживания в
вакууме становится зеленой (ср. U2F9 и U4F17). При 250° получается
преимущественно коричневый продукт с небольшой примесью зеленого и черного.
Реакцию лучше проводить в цельнометаллической никелевой аппаратуре. Пары
гексафторида уносятся током азота в камеру, где они смешиваются с хлористым
водородом при температуре 225°. Продукт реакции состоит из белых и голубых
игл, которые на воздухе быстро становятся зелеными. Для того чтобы получить
чистый тетрафторид урана, кристаллы необходимо нагревать в вакууме.
Повидимому, это наилучший метод восстановления гексафторида до тетрафторида.
С бромистым водородом реакция спокойно протекает при 80° с
образованием светлозеленого рыхлого порошка. Путем анализа установлено, что в
продукте реакции в качестве примеси содержится оксифторид. Его образование
приписывается наличию влаги в броме, применявшемся для получения НВг.
Для реакций восстановления гексафторида водородом и двумя
упомянутыми галогеноводородами (при 25°) были выведены следующие термодинамические
величины:
UF6TB + Н2 газ -» UF4 тв + 2HFras, (66)
AF°=-68900 кал;
UFe TB + 2НВггаз -+ UF4 Газ + 2HFra3 + Br2, (67)
AF°= -43500 кал;
UF6 тв + 2НС1газ -* UF4 тв + 2HFra3 + С1а газ, (6 8
AF° = -23400 кал.
Реакция восстановления гексафторида водородом должна иметь очень
большую энергию активации, так как даже при 600° она идет недостаточно
быстро, тогда как две другие реакции протекают гораздо легче.
Изучено также восстановление гексафторида аммиаком [100].
Газообразный аммиак реагирует с гексафторидом при температуре сухого льда. Эта
реакция проводилась также в газовой фазе при 300° в никелевом реакторе.
Получающийся при этом продукт не является чистым тетрафторидом.
Рентгенографические исследования показали присутствие в нем фторида аммония.
В спектре продукта восстановления было обнаружено также несколько новых
не идентифицированных линий, однако линий, принадлежащих тетрафториду,
не было. Химический анализ показал наличие двойной соли, соответствующей
смеси 2% NH4F и 98% NH4UF6.
При сплавлении тетрафторида урана с избытком бифторида аммония при
450° до вытеснения всего фтористого водорода получался продукт, который
23*
356 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
давал рентгенограмму, практически идентичную с рентгенограммой продукта
восстановления гексафторида аммиаком. Аналитическим путем найдено, что
эти продукты практически идентичны и различаются лишь по содержанию
фторида аммония. Рентгенографически показано, что расположение атомов урана
в этой двойной соли сильно отличается от расположения их в тетрафториде.
Исследовано также восстановление в жидкой фазе гексафторида тетрафтори-
дом кремния, трихлоридом фосфора и тионил хлоридом SOCl2. Все эти реагенты
восстанавливают гексафторид при комнатной температуре, причем тионилхло-
рид, очевидно, дает количественный выход тетрафторида урана [101].
Аморфный углерод при нагревании восстанавливает гексафторид урана до
тетрафторида, при этом образуются еще четырехфтористый углерод и другие
фторированные углеводороды. Кремний и мышьяк при нагревании дают
соответственно тетрафторид кремния и трифторид мышьяка. Фосфор реагирует
подобно кремнию и при умеренном нагревании дает трифторид фосфора.
Гексафторид урана реагирует также с серой с образованием тетрафторида и дисульфида
урана и газообразного фтористого производного серы (т. пл. —135°, т. кип.
—40°), повидимому тетрафторида серы [3].
Изучена реакция между гексафторидом урана и дицианидом ртути с целью
получить летучие соединения урана, содержащие группу —NC (которая изо-
стерична СО). Гексафторид урана перегоняли в сосуд с сухим дицианидом
ртути и смесь оставляли на 24 часа. Однако летучих продуктов (кроме
гексафторида) не было обнаружено, и цианид оставался неизменным [1021.
Растворы гексафторида в различных
растворителях. Различные органические соединения [3, 19]. При растворении
гексафторида в некоторых органических растворителях образуются практически
идеальные растворы. Ввиду того что гексафторид является сильным
фторирующим агентом, он реагирует с большинством органических соединений с
образованием фтористого водорода и фторуглеродов. Однако растворы
гексафторида в некоторых хлорированных углеводородах вполне устойчивы в течение
периодов времени до нескольких недель.
Спирт и эфир быстро реагируют с гексафторидом при комнатной
температуре, при этом получаются фтористый водород, U02F2 и углеродистое вещество.
Бензол, толуол и ксилол также легко реагируют. В нитробензоле гексафторид
растворяется с образованием тёмнокрасного раствора, дымящего на воздухе.
Парафиновые углеводороды (например, я-цетан С16Н34) не растворяют
гексафторид, но быстро реагируют с ним при комнатной температуре с выделением
фтористого водорода и обугливанием. Масла, содержащие непредельные
углеводороды, реагируют еще быстрее. В сероуглероде гексафторид не растворяется.
С сухим сероуглеродом реакция происходит медленно; при взаимодействии же
с влажным сероуглеродом образуются фториды серы, подобные (возможно,
даже идентичные) получающимся при действии гексафторида урана на серу.
Галогенированные углеводороды [3, 19]. Гексафторид растворим в четырех-
хлористом углероде, хлороформе и симметричном тетрахлорэтане. Из них
последний, С12СН—CHCU, образует наиболее устойчивый раствор; реакция
гексафторида с растворителем становится заметной при комнатной температуре
лишь через несколько дней. При кипячении желтый раствор гексафторида
в тетрахлорэтане обесцвечивается. По охлаждении желтая окраска опять
восстанавливается; это, возможно, указывает на то, что окраска обусловлена
образованием комплексного соединения. Изучено несколько реакций гексафторида,
растворенного в тетрахлорэтане. Окись азота NO окрашивает такие растворы
в сине-зеленый цвет; газообразный аммиак сообщает им зеленую окраску,
причем одновременно выделяется хлопьевидное вещество, содержащее фтор,
группу NH4 и четырехвалентный уран. Это вещество легко растворяется в
разбавленной серной кислоте, что отличает его от тетрафторида урана. Состав этого
вещества неизвестен. Трихлооид мышьяка осаждает вещество цвета бурой ржав-
Глава 13. Гексаф'торид урана
357
чины, растворимое в избытке реагента. Эти реакции гексафторида в растворе
заслуживают более детального изучения. Пентахлорэтан СС13СНС12 также
образует с гексафторидом урана желтый раствор, который при кипячении
становится бесцветным. При охлаждении такого раствора из пего выпадают
кристаллы гексафторида. Растворы в этом растворителе более устойчивы, чем
растворы в тетрахлорэтане; при стоянии в течение нескольких недель они
разлагаются лишь в незначительной степени. 1,2-дифтор-1,1,2, 2-тетрахлор-
этан FC12CCC12F реагирует с гексафторидом довольно быстро. При этом
происходит выделение газов (вероятно, дихлордифторметана) и в конечном
счете весь гексафторид восстанавливается до тетрафторида. Трихлорэтилен
С12С = СС1Н мгновенно восстанавливает гексафторид до тетрафторида.
Фтор угле роды. Полностью фторированные углеводороды чрезвычайно
устойчивы по отношению к большинству реагентов. Единственным известным
веществом, реагирующим с ними при обычных температурах, является фтор,
который превращает их в четырехфтористый углерод. Металлический натрий
или калий реагирует с фторуглеродами при температуре выше 400°. Фтор-
углероды термически весьма устойчивы и не изменяются при температурах
400—500°. Азотная кислота (96%), дымящая серная кислота, подкисленный
хромат и растворы перманганата на них не действуют. Бром и иод также не
реагирук*г, под действием водорода (100 am, 450°) часть фтора превращается
в фтористый водород. Разбавленные и концентрированные щелочи при
температурах ниже 100° не действуют на фторуглероды. Жидкие насыщенные фтор-
углероды нерастворимы в воде, спиртах и углеводородах, в хлорированных
углеводородах они заметно растворяются, а при повышенных температурах
смешиваются с ними во всех отношениях. Фторуглероды смешиваются во всех
отношениях и с этиловым эфиром и с частично фторированными углеводородами,
например с бензотрифторидом C6H5-CFV Более подробные сведения о
получении и других свойствах этих интересных соединений читатель может
почерпнуть из сообщений по фторуглеродам [103].
Физические свойства некоторых фторуглеродов приведены в табл. 157.
Растворы гексафторида урана во фторуглеродах чрезвычайно устойчивы.
Наблюдающееся иногда восстановление обычно можно объяснить реакцией
с веществом, из которого изготовлен сосуд, или с неполностью фторированными
Таблица 157
ФИЗИЧЕСКИЕ СВОЙСТВА НЕКОТОРЫХ ФТОРУГЛЕРОДОВ
Эмпирическая
формула
CF4
QFg
C3F8
QFio
QFi2
C7F14
C7F16
C7Fie
C8F16
CgFjg
QF18
QF20
Q0F18
^"12^22
QeFs4
Т. пл., °C
-183,5
-100,6
-183
-12
50
—
—
-60
—
—
—
—
-75
75
114
Т. кип., °C
-128,0
-78,2
-38
22
52
76
83
82,2
100
104
123
123
138
90 (90 мм рт. ст.)
240
ж
1,91 (-183°)
1,70 (-100°)
1,45(0,2°)
1,67
1,714
1,789
1,730
1,721
1,85
1,73
1,89
1,80
1,92
—
358 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
углеводородами, которые могут присутствовать в растворителе. С чистыми
фторуглеродами, как, например, с перфторциклогексаном или перфтор-
гептаном, реакции не наблюдается.
Растворы гексафторида урана в неорганических растворителях. Поведение
гексафторида в большом числе различных неорганических растворителей
изучено лишь с качественной стороны [104], и лишь для системы гексафторид
урана—трифторид бора были получены количественные данные. Для этой системы
разработана также диаграмма температура плавления—состав (рис. 56) [105].
При —125° гексафторид урана лишь умеренно растворим в трифториде бора.
19,00
I
| 20,00
В
^ 21,00
82,00 85,00 90,00 95,00 100,00
BF3 , % (мол. кажущийся)
Рис. 56. Диаграмма плавкости системы трифторид
бора—гексафторид урана.
О—экспериментально определенные точки. 0,16 лв=1°С (для многоспайной
термопары).
Реакции гексафторида урана с металлами и
другими веществами, из которых изготовлена
аппаратура. Гексафторид урана реагирует с большинством металлов и других
веществ, из которых изготовляется аппаратура. Поэтому проблема отыскания
материала, инертного к гексафториду, подвергалась тщательному изучению.
Металлы и сплавы. Для повышения коррозионной устойчивости
различных материалов применяются разнообразные методы. Руфф и Хайнцельман [3]
получили некоторые качественные результаты по коррозионной стойкости путем
обработки различных веществ парами гексафторида. Они установили, что
золото и платина не реагируют с гексафторидом на холоду, при нагревании же
эти металлы несколько тускнеют. Ртуть реагирует уже на холоду. На
медь и серебро гексафторид действует слабо при нагревании. Свинец, олово,
цинк и железо подвергаются более сильному воздействию, чем медь и серебро.
Алюминий покрывается белым налетом. Натрий быстро реагирует с
гексафторидом и при нагревании даже загорается.
Имеются указания, что никель, медь и алюминий устойчивы по
отношению к гексафториду. Для изготовления трубопроводов медь, вероятно, более
подходит, чем алюминий, так как медным трубам легче сообщить герметичность
в условиях вакуума. Эти три металла значительно превосходят все другие.
Пригодны также и сплавы с высоким содержанием никеля, меди или алюминия.
Вещества, которые образуют летучие фториды и легко переходят в состояния
с различной валентностью, непригодны для работы с гексафторидом.
Необходимо, однако, подчеркнуть, что эти указания справедливы лишь для очень
чистого вещества; в присутствии даже небольших количеств фтористого
водорода коррозионное действие гексафторида резко увеличивается.
к
У-
[о
Г^>'"' '
1 1
о
—о ■•■— ■
1
1 1
о о__Д-
1 1
1 1
-;27\Л
1 1
Глава 13. Гексафторид урана
359
ЛИТЕРАТУРА
1. Ruff 0.,Heinzelmann А., Вег., 42, 495 (1909).
2. Moissan H., L e Fluor, S t e i n h e i 1 G., Paris (1900).
3. Ruff O., H e i n z e 1 m a n n A., Z. anorg. Chem., 72, 63—84 (1911).
4. G г о s se A. V., К г о n e n b e г g P., Z. anorg. u. allgem. Chem., 204, 184 (1932).
5. A s t о n F. W., Nature, 128, 725 (1931).
6. H e n к el R., К 1 e m m W., Z. anorg. u. allgem. Chem., 222, 70 (1935).
7. В г о w n H. S., Report CC-408, Jan. 8, 1943 (MP Chicago 1).
8. Cong rev e W. L., Report BR-436, June 2, 1944 (British 1).
9. Abel son P., Report A-58 (Natl. Bur. Standards 1).
10. S к i n n e r H. A., Report BR-423, May 8, 1944 (British 2).
11. Smith F„Grandy K.,Tober F. W., Report CD-4101, Jan. 16, 1945 (CEW-
TEC 1).
12. Fowler R. D., The Preparation of Uranium Hexafluoride from Oxides, in
National Nuclear Energy Series, Division VIII, vol. 6 (Johns Hopkins 1).
13. McClure D. S., Report A-1224, Oct. 28, 1943 (SAM Columbia 1).
14. H a p p G. P., С a m e г о n A. E., Report CD-569, Mar. 12, 1945 (CEW-TEC 2).
15. W e i n s t о с к В., Report A-569, Mar. 15, 1943 (SAM Columbia 2).
16. Fytelson M., Lowenhaupt H. S., Wasserman D., Report A-540,
Feb. 11, 1943 (Yale 1).
17. M i 11 e r A. J., Brown К. В., G г a d у H. R., Y о u n g H. A., Report CD-
453, July 19, 1944; Whitney J., Smith F.,Mi 1 1 er A. J., Report CD-2.355.1,
Oct. 15, 1945 (CEW-TEC 3).
18. D о w n i n g F. В., В e n n i n g A. F., L i n с h A. L., Report A-728, May 17,
1943 (Du Pont 1).
19. G г о s s e A. V., Report A-83, June 25, 1941 (SAM Columbia 3).
20. G г о s s e A. V., Report A-3389 (SAM Carbide and Carbon 1).
21. F г i e d S., D a v i d s о n N. R., Report N-1722, Nov. 14, 1944; The Reaction of
UF4 with Dry 02; a New Synthesis of UF6, in National Nuclear Energy Series, Division
VIII, vol. 6 (MP Chicago 2).
22. К г a u s С A., Report CC-1717, July 29, 1944, p. 4 (Brown 1).
23. E r g e n W. K., Report A-3387, July 29, 1942 (SAM Carbide and Carbon 2).
24. H e n n e A. L, Renoll M. W., J. Am. Chem. Soc., 60, 1060 (1938).
25. В a h 1 a u G., Dissertation, University of Danzig (1919).
26. P e r r i n M. W., Report B-33, Sept. 1941 (British 3).
27. Davidson N. R., L о n g E. A., Report A-154, Apr. 13, 1942 (SAM Columbia 4).
28. A m p h 1 e t t С. В., M u 1 1 i n g e r L. W., T h о m a s L. F., Trans. Faraday
Soc., 44, 927 (1948).
29. P г i e s t H. F., Report A-121, Feb. 20, 1942 (SAM Columbia 5).
30. Dowson A. G., Report BR-287, Appendix 1, Aug. 26, 1943 (British 4).
31. S к i n n e r W. J., Report BR-347, Nov. 18, 1943 (British 5).
32. T u г к e v i с h A., M e t г о р о 1 i s N., L i b b у W. F., Report A-180, June
1942 (SAM Columbia 6).
33. H i с к s J. F. G., Wei 1 L., Co 1 e m a n C, Y u r m a n Т., Report [A] M-1136,
Aug. 8, 1944 (SAM Columbia 7).
34. H о d g s о n M. A. E., Report BR-633, July 18, 1945 (British 6).
35. К i r s h e n b a u m I., Report A-753, July 1, 1943 (SAM Columbia 8).
36. Brickwedde F. G., H о g e H. J., S с о t t R. В., J. Chem. Phys., 16, 429
(1948).
37. Brickwedde F. G., H о g e H. J., S с о t t R. В., Report A-607 (Natl. Bur.
Standards 2).
38. S i m о n F., F e r g u s о n J., Report B-37, Jan. 1942 (British 7).
39. С r i s t R. H., W e i n s t о с к В., Report A-571, Apr. 10, 1943 (SAM Columbia 9).
40. W e i n s t о с к В., С г i s t R. H., J. Chem. Phys., 16, 436 (1948).
41. W i 1 1 i a m s о n А. Т., L у n с h, Report B-18, July 9, 1941 (British 11).
42. H a w о г t h W. N.. Report B-79, June 1942 (British 8).
43. A m p h 1 e t t С. В., T h о m a s L. F., Report B-18, Aug. 26, 1941 (British 12).
44. A m p h 1 e t t С. В., T h о m a s L. F., Report B-27, Oct. 21, 1941 (British 13).
45. Simon F., Report B-3, May 1942 (British 9).
46. Directorate of Tube Alloys [B] LRG-64, Mar. 13, 1944 (British 10).
47. Directorate of Tube Alloys, [B]LRG-19, Apr. 1943 (British 14).
48. H о g e H. J., W e с h s 1 e г М. Т., Report A-1591, Dec. 18, 1943 (Natl. Bur.
Standards 3).
49. Awbery J. H., Report BR-290, Aug. 1943 (British 15).
50. Extracts from Report A-763, July 19, 1943 (SAM Columbia 10).
51. L о n g E. A., D a v i d s о n N. Rr, quoted by L i b b у W. F., Report A-1228x,
Aug. 29, 1944 (SAM Columbia 11).
360 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
52. Arms, Report B-36, Dec. 1941 (British 16).
53. Simon F., Report B-55, Mar. 1942 (British 17).
54. К i r s h e n b a u m I., Report A-753, Addendum, Oct. 26, 1943 (SAM Columbia 12).
55. Bigeleisen J., Mayer M. G., Stevenson P. C, Turkevich J.,
J. Chem. Phys., 16, 442 (1948).
56. M a s i J. F., J. Chem. Phys., 17, 755 (1949).
57. Ha worth W. N.. Report B-38, Feb. 1942 (British 18).
58. В о о t h E. Т., С a 1 1 i h a n D., Haggsstrom E.,Nordsick N.. Report
A-87, Dec. 27, 1941 (SAM Columbia 13).
59. Report B-32, May, 1942 (British 19).
60. T а у 1 о г A. H., A g г о n P. A., to be published (SAA1 Carbide and Carbon 3).
61. L 1 e w e 1 1 у n D. R., S w a i n e, Report B-100, Sept. 1942 (British 20).
62. L 1 e w e 1 1 у n D. R., Swaine, Report BR-427, 1944; Roberts L. E. J.,
Report BR-468, 1944; cited in Report [A]M-2511 (British 21.)
63. Present R. D., Report [A]M-2511, June 8, 1945 (SAM Carbide and Carbon 4).
64. P r i e s t H. F., Report A-2142, Oct. 1942 (SAM Columbia 15).
65. Hoard J. L.Stroupe J. D., Reports A-1242, Mar. 1, 1944; A-1296, June 30,
1944 (Cornell 1).
66. H о g e H. J., W e с h s I e г М. Т., J. Chem. Phys., 17, 617 (1949).
67. Priest H. F., Report A-139, Mar. 29, 1942 (SAM Columbia 16).
68. Wechsler M. T.,Hoge H. J., Report A-456, Feb. 25, 1943 (Natl. Bur.
Standards 4).
69. Johnson С. Н., Report B-I9, July 7, 1941 (British 22).
70. J о h n s о п С. Н., Report B-97, July 1942 (British 23).
71. N e у E. P., A r m i s t e a d F. C, quoted by M u 1 1 i к e n R. S., Report CC-567,
Mar. 25, 1943 (Virginia 1).
72. N e у E. P., A r m i s t e a d F. C, Phys. Rev., 71, 14 (1947).
73. К i r s h e n b a u m A. D., Report A-732, June 10, 1943 (SAM Columbia 17).
74. R о b e r t s L. E. J., Report BR-468, July 1944 (British 24).
75. F о w 1 e r R. D„Anderson H. C.Weber С E., Report A-1398, Nov. 15,
1942 (SAM Columbia 18).
76. M e у е г s о n A. L., E i с к e r J. H., Report A-3825, Dec. 26, 1945 (SAM Carbide
and Carbon 5).
77. Directorate of Tube Alloys, [B]LRG-20, May 1943 (British 25).
78. L i p к i n D., Weisman S., Report A-520, Dec. 17, 1942 (UCRL I).
79. M a r t i n A. E., Amphlett С. В., Report B-117, Nov. 1942 (British 26).
80. Duncan A. B. F., Report A-565, Mar. 15, 1943 (SAM Columbia 19).
81. D u n с a n A. B. F., Report A-584, Apr. 15, 1943 (SAM Columbia 20).
82. U г е у H. C, Report A-750, July 10, 1943 (SAM Columbia 21).
83. Duncan A. B. F., Murphy G. M., Report A-580, Apr. 5, 1943 (SAM Columbia 22).
84. Mayer M. G., В i g e 1 e i s e n J., Report A-1269, May 4, 1944 (SAM Columbia 14).
85. S m у t h С P., H a n n а у N. В., Report A-2130, Oct. 1, 1944 (Princeton 1).
86. T i 1 к W., Klemm W., Z. anorg. u. allgem. Chem., 240, 355 (1939).
87. W h i t e J. R., С a m e г о n A. E., Phys. Rev., 71, 907 (1947).
88. Cameron A. E., W h i t e J. R., Report CD-B-6.460.9, Jan. 17, 1946 (CEW-TEC 4).
89. В г a u n e H., P i n n о w P., Z. physik. Chem., B-35, 239 (1937).
90. Bauer S. H., Report A-1209, Aug. 10, 1943 (SAM Columbia 23).
91. J а с о b G. W., W a r г с n В., J. Am. Chem. Soc, 59, 2588 (1937).
92. Klein D. X., Reports A-1055, June 3, 1944; A-1058, Sept. 16, 1944 (Du Pont 2).
93. Gordon, Report B-99, Aug. 1942 (British 27).
94. Directorate of Tube Alloys, [B]LRG-22, July 1943 (British 28).
95. Jons I. В., T e v с b a ug h A. D., G 1 a d г о v E., W a 1 s h K.,C h i о t t i P.,
А у с r s В., V a s 1 о w F., Fisher R. W., The Reduction of Uranium Hexa-
fluorideto Uranium Tetrafluoride, in National Nuclear Energy Series, Division VIII,
vol. 6 (MP Ames 1).
96. T e v e b a u g h A. D., W a 1 s h K., J о h n s I. B., Report CC-1245, Jan. 8, 1944
(MP Ames 2).
97. T e v e b a u g h A. D., V a s 1 о w F., F i s h e r R. W., Report CK-1516, Mar. 10,
1944, p. 6 (MP Ames 3).
98. Bernhardt H. A., V о s s D. J., В r u s i e J. P., Report A-3618, Dec. 6, 1945
(SAM Carbide and Carbon 6).
99. Gladrow E. M., С h i о t t i P., Report CK-1498, May 10, 1944, p. 11 (MP
Ames 4).
100. А у e r s В., Report CC-1504, Aug. 10, 1944, p. 6 (MP Ames 5).
101. Calkins V. P., Report CD-0.350.4, Sept. 25, 1945 (CEW-TEC 5).
102. Directorate of Tube Alloys, [B]LRG-17, Feb. 1943 (British 29).
103. National Nuclear Energy Series, Division VII, vol. 1, etc.
104. Kirshenbaum I., Report A-711, May 14, 1944 (SAM Columbia 24).
105 Kirshenbaum 1., Report [AJM-709, Febr. 16, 1944 (SAM Columbia 25).
Глава 14
СОЕДИНЕНИЯ УРАНА С ХЛОРОМ
Известны четыре соединения урана с хлором: трихлорид, тетрахлорид,
пентахлорид и гексахлорид. Первые три получены и исследованы еще в
первоначальных работах по химии урана. Гексахлорид же получен лишь
недавно в радиационной лаборатории Калифорнийского университета. Это
соединение, а также гексафторид урана представляют особый интерес, так как они
являются единственными соединениями шестивалентного урана, не
содержащими кислорода. Начиная с 1942 г. было получено много новых сведений о
хлоридах урана, этот материал и составляет большую часть настоящей главы.
Для удобства в табл. 158 сопоставлены некоторые физические свойства
хлоридов урана.
Таблица 158
НЕКОТОРЫЕ ФИЗИЧЕСКИЕ СВОЙСТВА СОЕДИНЕНИЙ УРАНА С ХЛОРОМ
Формула
соединения
иС13
исц
иаб
UCie
Т. пл., °С
842+5
590+1
300 б)
177,5 б)
Плотность а),
5,51
4,87
3,81 в>
3,59
Давление пара
1 А В
'£ Рмм рт. ст-~А j
А
10,0
13,2995
6,634
В
12 000
10 427
2 422
дявозг,
ккал -моль-1
55,0
47,7
11,12
о
F
ккал'Моль— 1
-196,2
-229,0
-235,7
-241,4
' 5298.
энтропийные
единицы
40,0
47,14
57,0 б)
68,26
а) На основании рентгенографических данных.
б) Значения, найденные расчетом.
в) Значение, полученное иммерсионным методом (с бензолом).
ТРИХЛОРИД УРАНА
Впервые трихлорид урана был получен восстановлением тетрахлорида
водородом [1]. Этот метод и до сих пор является наиболее удобным, особенно
в случае отсутствия чистого металлического урана. Необыкновенная
устойчивость трихлорида на воздухе обусловила значительный интерес к этому
соединению.
Получение трихлорида урана. Помимо восстановления
тетрахлорида урана водородом, трихлорид может быть также получен путем
взаимодействия гидрида урана с хлористым водородом. Некоторые другие
реакции, также приводящие к образованию трихлорида, практического
значения не имеют.
362 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Тетрахлорид+водород [2—8]. При температуре ниже 500° реакция
восстановления тетрахлорида водородом
UCI4 + yH2^UCl3 + HCl (1)
протекает медленно. При 525—550° скорость ее еще настолько мала, что для
доведения реакции до конца требуется около 5 час. (см. стр. 390). Однако чтобы
избежать плавления тетрахлорида, необходимо поддерживать температуру
ниже 590°, так как расплав покрывается слоем трихлорида, который
препятствует дальнейшему восстановлению. Поэтому можно рекомендовать большую
часть тетрахлорида превращать в трихлорид при температуре ниже 575°, а
заканчивать восстановление при температурах до 650°. Излишне говорить,
насколько важна тщательная очистка применяемого водорода.
Восстановление водородом под давлением протекает гораздо быстрее [5, 71.
При 7 am и 525—550° реакция проходит в четыре раза быстрее, чем при
атмосферном давлении. Для предотвращения спекания реакционной смеси ее
выдерживают 30 мин. при 525° и затем медленно нагревают до 550°. Реакцию можно
проводить в сосуде из нержавеющей стали, снабженном отводными трубками
для медленного выпуска газов. Полученный 'таким путем трихлорид
представляет собой темнозеленое кристаллическое вещество.
Гидрид водорода+хлористый водород [9, 10]. При нагревании тонкоизмель-
ченного металлического урана или его гидрида с хлористым водородом в
интервале температур 250—300° получается трихлорид оливково-зеленого цвета:
UH3 + ЗНС1 -^ UC13 + ЗН2. (2)
Реакция легко протекает и является очень удобным методом получения
трихлорида при наличии металлического урана. Для проведения этой
реакции пригоден прибор, применяемый для получения трибромида урана [11]
(см. стр. 412). Хлористый водород сильно адсорбируется трихлоридом урана
и может быть удален лишь при 150° в вакууме.
Различные способы получения трихлорида урана. Тетрахлорид урана может
быть восстановлен при повышенной температуре аммиаком, но получающийся
продукт содержит азот [12]. Можно в качестве восстановителя применять
также йодистый водород:
UC14 + HJ 30Q-350:, UC13 + HC1 +1J2. (3)
Продукт реакции можно освободить от иода током сухого азота при 200—
300° [13].
Наблюдения показали, что пары тетрахлорида урана реагируют с
нагретыми металлами согласно уравнению
*UCl4 + M-*MClx + xUCl3. . (4)
В том случае, если металл М образует летучий хлорид, наблюдается очень
хороший выход кристаллического трихлорида. Особенно пригоден для этой
цели металлический циьк [14, 15].
При нагревании мононитрида урана UN в парах тетрахлорида в течение
нескольких часов при 500° получается, повидимому, трихлорид. Реакционная
масса растворяется в воде с выделением значительного количества газов и
образованием раствора пурпурного цвета [16]. Как препаративный метод получения
трихлорида эта реакция не представляет интереса.
Растворы трихлорида в концентрированной соляной кислоте более
устойчивы, чем растворы в чистой воде. Такие растворы могут быть получены
электролитическим восстановлением 15%-ного раствора трехокиси урана в соляной
кислоте (уд. вес 1,12) [17]. Электролиз проводится в атмосфере углекислого
Глава 14. Соединения урана с хлором
363
газа в ванне, снабженной пористой диафрагмой и ртутным катодом, при
напряжении 110 б и силе тока 1,5—2,0 а. Анолитом служит концентрированная
соляная кислота. После того как раствор, который сначала быстро приобретает
зеленую окраску, становится грязного коричнево-зеленого цвета, силу тока
уменьшают до 0,75—1,0 а. В дальнейшем электролиз проводят при 0° с
периодической добавкой соляной кислоты до тех пор, пока не будет достигнут
характерный для урана (III) красный цвет раствора. Растворы урана (III) можно
получить также восстановлением уранилхлорида U02C12 в соляной кислоте
цинком [18].
Очистка трихлорида урана. Трихлорид урана может быть очищен от
летучих примесей (например, от тетрахлорида) нагреванием в течение нескольких
часов в вакууме при 770° [19]. Предложен остроумный метод очистки,
основанный на превращении трихлорида урана в трихлориодид UC13J перегонкой в токе
паров иода:
UCl3 + lj2^UCl3J. (5)
Трихлориодид урана летуч; при охлаждении он разлагается на исходные
трихлорид и иод. В результате этого достигается перегонка трихлорида при
гораздо более низких температурах, чем соответствующие его собственной
летучести [20].
Физические свойства трихлорида урана. При комнатной
температуре трихлорид окрашен в оливково-зеленый цвет. При 300° он
становится красновато-коричневым, при температуре выше 450°—темнопурпур-
ным. При охлаждении трихлорида до комнатной температуры его оливково-
зеленая окраска восстанавливается. Препарат, получающийся восстановлением
тетрахлорида водородом, имеет обычно красный цвет.
Температура плавления [6,21]. Как установлено при помощи
термического анализа, трихлорид плавится при 842 ± 5°.
Летучесть* Упругость пара трихлорида урана измерена по методу
молекулярного истечения. Верхний предел летучести соответствует
приблизительно Ы0~2лш рт. ст. при 820° и 3,5-Ю"3жж рт. ст. при 750° [22].
Было выведено эмпирическое уравнение, описывающее данные по упругости
пара в интервале температур 600—1000° [23]:
\gPMM рт. ст.= - ^~^+ Ю,00. (6)
Найденная из этого уравнения теплота возгонки равна 55 000 кал-моль'1.
Кристаллическая структура трихлорида урана [24, 25]. Трихлорид
имеет гексагональную элементарную ячейку, содержащую две молекулы.
Константы решетки имеют следующие значения:
^ = 7,428 ± 0,003 А; а3 = 4,312 ± 0,003 А.
Пространственная группа С63/т(С1л). Положения атомов в решетке
следующие:
«■±G4t>
6С1в±( xyj^^x-y.^Qy-x, *. -J")»
где * = 0,375 ± 0,014, # = 0,292 ± 0,014.
Таким образом, каждый атом металла окружен девятью атомами галогена:
из них три находятся на расстоянии 2,95 А и шесть—на расстоянии 2,96 А.
Ближайшее расстояние между двумя атомами хлора равно 3,45 А. Структура
364
Таблица 159
УДЕЛЬНАЯ ТЕПЛОЕМКОСТЬ, ЭНТРОПИЯ И ЭНТАЛЬПИЯ UC13 ОТ О ДО 300° К
Температура,
°К
о
5
10 !
15
20
25 1
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
105
ПО
115
120
125
130
135
140
145
150
155
160
165
170
175
180
185
190
195
200
205
210
215
220
225
230
235
ср ,
межд.дж-моАЬ-1 -град-*
0,0
0,476 а)
3,15а)
6,64
9,30
12,38
15,51
18,94
23,07
27,2
31,6
36,7
41,8
46,4
50,9
55,2
59,1
62,9
66,6
70,3
73,1
75,4
77,8
79/9
82,1
84,0
85,6
87,2
88,6
89,9
91,2
92,4
93,4
94,3
95,1
95,8
96,4
97,0
97,4
97,8
98,1
98,4
98,8
99,2
99,6
1 100,1
100,5
100,8
5,
межд. дж-мол ь-1-град-!
0,0
0,159
1,181
3,13
5,43
7,84
10,37
13,01
15,80
18,76
21,85
25,10
28,51
32,04
35,64
39,30
42,99
46,69
, 50,39
! 54,10
| 57,78
61,40
64,96
! 68,46
71,91
75,30
78,63
, 81,89
85,09
88,22 .,
91,29
94,30
97,25
100,14
102,97
105,73
108,44
111,09
113,68
116,22
118,70
121,12
123,50
125,83
128,11
130,35
132,56
134,72
я-4.
межд. дж-моль-^
0,0
0,597
8,73
33,39
73,67
127,8
197,6
283,4
388,1
513,8
660,8
831,6
1028
1 248
1492
1757
2 043
2 348
2 672
3 014
3 373
3 744
4 128
4 522
4 927
5 342
5 766
6 198
6 638
7 084
7 537
7 996
8 460
8930
9 403
9 881
10361
10 845
11331
11819
12 309
12 800
13 293
13 788
! 14 285
! 14 784
| 15 286
15 789
Глава 14. Соединения урана с хлором
365
Температура,
°К
240
245
250
255
260
265
270
275
280 .
285
290
295
298,16
300
305
310
315
320
325
330
335
3*0
345
350
355
360
365
370
375
380
межд.дж-моль-1-град-1
101,1
101,4 |
101,7 ;
102,0
102,2
102,6
102,7
102,8
102,9
103
103
103
103
103
103
103
103
103
103
103
103
103
103
103
103
103
103
103
103
103
Продолжение табл. 159
s, 1
межд.дж-моль-1-град~1
136,85
138,94
140,49
143,01
144,99
146,94
148,85
150,73
152,58
154,40
156,19
157,95
159,04
159,68
161,38
163,06
164,71
166,33
167,93
169,50
171,05
| 172,57
174,08
175,56
177,02
178,46
179,88
181,28
182,67
184,03
межд .дж • мол 6-1
16 294
16 800
17 308
17817
18 328
18 839
19 352
19 865
20 379
20 893
21 408
21923
22 248
22 438
22 953
23 468
23 983
24 498
25013
25 528
26 043
26 558
27 073
27 588
28 103
28 618
29 133
29 648
30 163
30 678
а) Экстраполировано с применением функции Девая1 15,665 D ( J -).
трихлорида координационного типа и связи между атомами урана и хлора
имеют, вероятно, преимущественно ионный характер. Трихлорид урана
изоморфен трихлориду церия, трибромидам лантана и церия и ряду гидроокисей
редкоземельных элементов.
Вычисленная из рентгенографических данных плотность трихлорида урана
равна 5,51 г/см3. Гидростатическим взвешиванием в четыреххлористом
углероде было получено значение 5,35 г/см'г. Из этих данных видно, что
плотность трихлорида больше плотности тетрахлорида [8].
Термодинамические данные [26]. Были определены удельная
теплоемкость, энтропия и энтальпия трихлорида в температурном интервале от 15
до 380° К [26] и от 273 до 1000° К [27, 28]. Точность полученных
результатов, которые приведены в табл. 159 и 160, лежит, вероятно, в пределах
нескольких десятых долей процента. Значения, приводимые в табл. 160»
более надежны.
Збб Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Таблица 160
УДЕЛЬНАЯ ТЕПЛОЕМКОСТЬ, ЭНТРОПИЯ И ЭНТАЛЬПИЯ
ТРИХЛОРИДА УРАНА ОТ 0 ДО 727е а)
Температура,
°С
б
50
100
150
200
250
300
350
400
450
500
550
600
650
700
726,84(1000° К)
Ср,
межд. дж*моль~1-град~1
101,7
102,5
103,3
104,1
104,9
105,9
106,8
107,9
109,0
110,3
111,7
113,5
115,8
119,0
123,5
126,7
S,
межд. дж-моль^1'град"1
149,99
167,15
181,95
194,99
206,66
217,23
226,93
235,90
244,28
252,13
259,55
266,61
273,38
279,92
286,29
289,69
H-El
0
межд. дж-моль-1
19 674
24 778
29 921
35 103
40 329
45 601
50 921
56 290
61714
67 194
72 741
78 369
84 101
89 970
96 028
99 379
а) 4,1833 межд. дж~\ кал; 0° С=273,16в К.
Теплота образования трихлорида была недавно вновь определена в
радиационной лаборатории Калифорнийского университета [29] по методу
Бильца и Фендиуса [30] (см. стр. 388). Для реакции
UTB ~Ь "о" ^2 газ —* ^^3 тв (' )
получено следующее значение:
ДЯ273= —212,1 ±0,6 ккал-моль'1.
Подробности расчета читатель может найти в оригинальной работе.
В той же лаборатории было изучено равновесие (1)
UC14TB+| H2ri3 7± иС13тв + НС1газ. (1)
Результаты этого исследования были критически обработаны Маквудом
[31—33]. Значения константы равновесия этой реакции приведены в табл. 161.
Изменение теплоемкости для реакции (1) может быть вычислено следующим
образом:
иС1зтв: Ср = 23,42 + 4,132-10-3Г
НС1газ: Ср-6,7+ 0,84-10-3 Г
-UCUtb: -Ср= -27,56-4,412-10~3 Т
-|н2газ: -уСр= -3,31 -0,405- КГ3 Т
ДСр=-0,75+1,55.10-4Г
Отсюда получается выражение для теплоты реакции:
ДЯГ= 17,270 клад-0,75 Г + 7,79-10~5 Г2.
Глава 14. Соединения урана с хлором 367
Таблица 161
КОНСТАНТА РАВНОВЕСИЯ ДЛЯ РЕАКЦИИ ВОССТАНОВЛЕНИЯ
ТЕТРАХЛОРИДА УРАНА а)
•
Температура
°С
400
420
453
477
502
°К
673
693
726
750
775
1 i лЗ
— кг
1,486
1,443
1,377
1,333
1,290
К [32]
0,295
0,438
0,789
1,100
1,550
Я[31]
0,386
0,549 (425° С)
0,762 (450° С)
1,014 (475° С)
1,277 (500° С)
') к-р-22 .
Значение AS2e8, вычисленное из констант равновесия, равно 23,00.
К сожалению, это значение отличается на 3,1 энтропийной единицы от
полученного на основании третьего закона термодинамики (AS298= 19,92).
Причина этого расхождения неизвестна. Если предположить, что значение
энтропии для тетрахлорида урана при 298,16° К, равное 47,14 энтропийной
единицы, правильно, то энтропия трихлорида при 298,16° К должна быть равна
40,5 энтропийной единицы.
Используя последнее значение, Маквуд сделал вывод, что наиболее
достоверными данными для реакции
UTB +"2 СЬ газ —> UCI3 тв (7)
являются следующие:
Д#298 = —212,0 ккал-мо/иг1;
д^298= —196>7 ккал-моль"1;
Д5298= —51,5 энтропийной единицы;
Д# = —213700 + 5,70 Г-7,79-10"4 Г2; (8)
Af°=_2i3700-13,131gr + 7,79.10-4r2 + 89,237\ (9)
Химические свойства трихлорида урана. В этом
разделе будут рассмотрены в основном реакции твердого трихлорида. Послед^
ний является типичным трехвалентным галогенидом; по своим физическим
и химическим свойствам он напоминает галогениды трехвалентных
редкоземельных элементов. Главное отличие состоит лишь в том, что трихлорид урана
как твердый, так и в растворе является сильным восстановителем.
Отношение к воде. Трихлорид урана гигроскопичен, но в гораздо меньшей
степени, чем другие хлориды урана. Если парциальное давление паров
воды /?н2о равно 2,4 мм рт. ст., то поглощения воды не происходит; при рн2о =
= 3,4 мм рт. ст. наблюдается слабое поглощение. В токе азота, содержащем
пары воды с парциальным давлением рн2о = 9 мм рт. ст., трихлорид
расплывается, образуя пурпурный раствор, из которого медленно выделяется водород.
Во влажном воздухе (рн2о = 9 мм рт. ст.) трихлорид не расплывается, но быстро
подвергается гидролизу с одновременным окислением до UOCl2. Получающийся
368 Часть IV. Галогениды, оксигалогениды, бореидриды и карбонил урана
продукт представляет собой сухую спекшуюся массу [34]. Во влажном
воздухе трихлорид увеличивается в весе приблизительно на 1% в минуту [22].
При растворении трихлорида в воде образуется раствор нестойкого
пурпурного цвета, который быстро изменяется на грязно-зеленый, что указывает
на присутствие урана (IV). Растворимость трихлорида в воде при 0°
составляет 3—4,8моль1л (рН2 М водного раствора трихлорида равен 2,4) [35]. Хотя
это значение и является весьма приближенным, оно дает понятие о степени
гидролиза.
При растворении трихлорида в воде количество выделяющегося водорода
соответствует с точностью до нескольких процентов вычисленному для
окисления трехвалентного урана в четырехвалентный [35]. При 24° выделение
водорода идет в четыре раза быстрее, чем при 0°. При 0° трихлорид может находиться
в насыщенном растворе над твердой солью в течение значительного
промежутка времени без существенного окисления. Процесс окисления был изучен
по изменению спектра поглощения. По мере того как происходит окисление,
полосы, характерные для иона трехвалентного урана (широкая полоса в
зеленой и две смежные четкие полосы в красной части спектра), постепенно
ослабевают, однако интенсивная полоса в желто-зеленой части
спектра,-характерная для урана (IV), появляется не сразу.
Скорость растворения трихлорида в соляной кислоте уменьшается с
увеличением концентрации последней; в 6 М соляной кислоте растворяется за час
при0° меньше трихлорида, чем в I M кислоте за полчаса. Растворы трихлорида
в концентрированной соляной кислоте значительно устойчивее, чем в воде.
Теплота растворения трихлорида в соляной кислоте (1 моль НС1 на 8,808 моля
воды) равна 40,6ккал-моль"1 [30]. В литературе указывается, что при прили-
вании серной кислоты к водному раствору трихлорида выпадает сульфат
урана (III). Действие же аммиака, фосфорной, щавелевой или
плавиковой кислот приводит к образованию солей четырехвалентного урана [17]
(см. гл. 12).
Отношение к другим растворителям [36—38]. Трихлорид урана
растворяется в ледяной уксусной кислоте с образованием раствора красного цвета.
Метиловый спирт и формамид реагируют с трихлоридом. В четыреххлористом
углероде, ацетоне, хлороформе, пиридине, углеводородах и неполярных
растворителях трихлорид нерастворим. Растворимость его в полярных
растворителях меньше, чем растворимость тетрахлорида и тетрабромида урана. В
неорганических растворителях, например трихлориде фосфора и тетрахлориде
олова, трихлорид урана нерастворим. С трихлоридом фосфора образуется
эмульсия зеленого цвета.
Термическое разложение трихлорида урана. При нагревании трихлорида
в вакууме до 840° происходит возгонка различных веществ, которые оседают
на холодных частях аппаратуры. Дальше всего от нагреваемых частей прибора
оседает коричневый пентахлорид урана, ближе—зеленый тетрахлорид, еще
ближе к месту нагрева—рыжевато-коричневый трихлорид. Налет тетрахлорида
образуется впервые, когда температура достигает 500°, налет пентахлорида
появляется при 720° и при 840° начинает возгоняться неизмененный трихлорид.
Остаток от возгонки представляет блестящие светлые бесформенные куски
металлического вида [22]. Эти наблюдения объясняются диспропорциониро-
ванием трихлорида
2UC13^UC12H-UC14. (10)
Однако образование дихлорида далеко еще нельзя считать доказанным [10,
39] (см. гл. 15).
Отношение к окислителям. При нагревании трихлорида на воздухе до
700° и последующей перегонке в вакууме возгоняется твердое вещество желтого
цвета, вероятно уранилхлорид. Возможно, что реакция идет следующим
Глава 14, Соединения урана с хлором
369
образом [22]:
2UC13 +02 -* U02C12 + UC14. (11)
С чистым кислородом трихлорид урана начинает реагировать при температуре
около 150°. Получающийся при этом продукт представляет, повидимому,
уранилхлорид с примесью некоторого количества двуокиси урана и не-
прореагировавшего трихлорида [40]. Трихлорид легко реагирует с хлором
при 250° с образованием тетрахлорида:
иС13 + |с12~-+иС14. (12)
При этом выделение тепла очень невелико и плавления не происходит [9].
При более высоких температурах, однако, реакция идет весьма бурно. Так,
при действии хлора при 300° трихлорид загорается и температура реакционной
смеси доходит примерно до 800°. При этом выделяются бурые пары. Если
дополнительно пропускать хлор над уже затвердевшей реакционной массой
при 420° в течение 6 час, образуется второй летучий коричневый продукт.
Первый коричневый возгон имеет состав UC13(67; состав второго соответствует
формуле UCl5t33- Наряду с упомянутыми хлоридами образуется также
небольшое количество зеленых кристаллов, которые конденсируются в
непосредственной близости от реакционной печи и имеют состав UC14,83. Хотя
результаты соответствующих анализов и не очень надежны, они все же здесь
приводятся ввиду того, что они могут иметь значение для решения вопроса
о существовании еще других хлористых соединений урана, аналогичных
U2Fe и U4F17[8] (см. стр. 316).
Таблица 162
ФАЗОВЫЕ СООТНОШЕНИЯ В НЕКОТОРЫХ СИСТЕМАХ ТРИХЛОРИД
УРАНА-ГАЛОГЕНИД МЕТАЛЛА
Система
UC13-KC1
UC13—NaCl
UCl3-BaCla
UC13-UC14
Формула соединения
2KCbUCl3
3KC1.UC13
Отсутствует
»
»
Т. пл. соединения,
°С
625±5
При 530±5
превращается в
2KC1.UCI3 и
КС1
«
Эвтектика
КС1-К2иС1б
[20% (мол.) UC13]
2K2UC15—UC13
[500^ (мол.) UC13]
NaCl-UCl3
[33% (мол.) иС13]
Одна эвтектическая
смесь, состав ее не
определен
Взятые вещества не
смешиваются в жидкой
фазе и лишь слабо
растворимы в твердой
фазе
Т. пл.
эвтектики,
•С
590J-5
545±5
520±5
635—640
24 Химия ураиа
370 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Бром и иод реагируют с трихлоридом с образованием смешанных галоге-
нидов [41] (см. стр. 434). Сухой фтористый водород окисляет трихлорид при
повышенных температурах до тетрафторида [13]:
UC13 + 4HF— -+UF4 + 3HCl+i-H2. О3)
Жидкий безводный фтористый водород реагирует с трихлоридом при 25°
с выделением хлористого водорода; предполагают, что получающийся при этом
продукт имеет состав UF3 [42].
Отношение к восстановителям. Сообщается, что водород восстанавливает
трихлорид с образованием дихлорида и хлористого водорода:
иС1я + уНа->иС12 + НС1. (14)
Однако в литературе не приводится экспериментальных данных или
результатов анализов, которые могли бы подтвердить это заключение [39].
Трихлорид урана не реагирует с атомарным водородом при 700° [43].
Комплексные соединения трихлорида урана. При помощи термического
анализа исследован ряд комплексных соединений, содержащих трихлориды
и галогениды других металлов [21 ]. Результаты этого исследования
представлены в табл. 162 (стр. 369).
ТЕТРАХЛОРИД УРАНА
Впервые тетрахлорид урана был получен в 1842 г. [1 ]. С тех пор это
соединение изучалось многими исследователями.
В связи с разработкой способа получения тетрахлорида было изучено
несколько реакций галогенирования. Теплота и свободные энергии некоторых
из них приведены в табл. 163. Энтропии реакций галогенирования довольно
велики. Вследствие этого при выборе хлорирующего агента целесообразно
исходить не из теплот реакции, а из свободных энергий.
Таблица 163
СВОБОДНЫЕ ЭНЕРГИИ НЕКОТОРЫХ РЕАКЦИЙ ОБРАЗОВАНИЯ ТЕТРАХЛОРИДА УРАНА
Реакция а)
U02 + 4HC1 -> UC14 + 2H20
U02 + C + 2CI2 -» UC14 + C02
UO2 + CCI4 -> UQ4+CO0
U02 + 2CC14 -» иС14 + 2СОС12
U02 + 2COCla -> UC14 + 2C02
U02 + 2SOCla ~> UC14 + 2S02
U02 + yS2ra3 + 2Cl2 -> UCI4 + SO2
UO2 + 2PCI5 —> UCI4 + 2POCI3
и02 + 2РС1з + 2С12 -> иС14 + 2РОС1з
А#298*
ккал б)
-8,0
-75,1
-49,4
-36,2
-62,5
-37,0
-51,5
-75,4
-117,4
AS298.
энтропийные
единицы б)
-59,9
-28,4
+ 5,4
+ 14,5
-3,7
-0,1 в>
-46,1
+ 8,3В>
-72,3 в>
L
Д^зоо.
ккал
+ 10,0
-66,5
-51,0
-40,6
-61,4
-36,7
-77,9
4^800.
ккал
+39,9
-52,3
-53,7
-47,8
-59,5
-36,9
-14,7
-59,5
Уравнение
№
(15)
(16)
(17)
(18)
(19)
(20)
(21)г)
(22)
(23)
а) Все вещества, за исключением тетрахлорида урана, двуокиси его и углерода, взяты в парообразном
состоянии. Для двуокиси урана S298—18,63 энтропийной единицы; Д#298=_270,4 ккал (см. гл. 11).
Для тетрахлорида урана 5298 = 47,14 энтропийной единицы; ДЯ2 8е-251,0 ккал (см. стр. 386).
б) Значения теплот образования взяты из книги Быховского и Россини [44]; значения энтропии—
из работы Келли [45].
в) Энтропии SOCl2 и РОС13 взяты из данных Иоста и Рассела [46].
г) Это уравнение представляет реакцию с применением S2CI2 в качестве хлорирующего агента.
Глава 14. Соединения урана с хлором
371
Большинство реакций галогенирования протекает с практически
приемлемой скоростью лишь при температурах порядка 800° К, поэтому в табл. 163
приведены значения свободной энергии и для 300 и для 800° К.
Для ряда аналогичных реакций часто наблюдается параллелизм между
количеством освобождающейся свободной энергии и скоростью установления
равновесия, но это, конечно, не является общим правилом. Поэтому нельзя
считать, что относительная легкость, с которой различные реагенты,
перечисленные в табл. 163, взаимодействуют с двуокисью урана, будет изменяться
точно в том же порядке, что и AF реакции. Однако можно заранее сказать,
что реакции галогенирования, протекающие без заметного уменьшения стан*
дартной свободной энергии, должны быть лишены практического значения.
Даже если в подобного рода реакциях (например, в реакции с хлористым
водородом) равновесие будет достигаться быстро, малые равновесные
концентрации продуктов реакции не дадут возможности удалять их со скоростью,
обеспечивающей завершение реакции за приемлемый промежуток времени.
Необходимо помнить, что значения свободной энергии, приведенные
в табл. 163, являются средними значениями для двух стадий хлорирования,
например:
U02 + SOCl2 -> UOCl2 + S02; (24)
UOCl2 + SOCl2 -> UC14 + S02. (25)
По всей вероятности, в первой стадии выделяется больше энергии, чем во
второй, поэтому, если хлорирующий агент не очень активен, реакция может
остановиться на стадии образования UOCl2.
Все другие перечисленные реагенты должны, повидимому, более или
менее легко превращать двуокись урана в тетрахлорид. Четыреххлористый
углерод и фосген [уравнение (19)] являются, повидимому, энергичными
хлорирующими агентами, как и SOCl2 [уравнение (20)]. Пентахлорид фосфора
особенно применим при низких температурах, при которых он не диссоциирует
на трихлорид фосфора и хлор.
При выборе реагента необходимо принимать в расчет и другие соображе-
ния помимо термодинамических. Существенно важно, чтобы получался чистый
тетрахлорид, свободный от хлорирующего реагента. Это требование снижает
применимость, например, S2C12 [уравнение (21)]. Другими факторами, влияю-
щими на выбор реагента, являются легкость обращения с ним, коррозионная
стойкость, а также природа продуктов реакции.
Наиболее важными исходными материалами для получения тетрахлорида
являются окислы урана. Можно также применять свободный металл, гидрид,
карбид, нитрид и сульфид урана, но они менее доступны. С другой стороны,
соединения уранила весьма доступны, но они недостаточно реакционноспособ-
ны для того, чтобы приобрести существенное значение.
Важным фактором во всех реакциях хлорирования с использованием
окислов урана в качестве исходных материалов является реакционная
способность применяемого окисла. Реакционноспособная двуокись урана может быть
получена, например, восстановлением высших окислов метаном, аммиаком
или этиловым спиртом при температуре ниже 750° или термическим
разложением таких соединений урана (IV), как сульфат, оксалат или бензоат.
Полученная таким путем двуокись урана представляет собой серо-черный пирофор*
ный порошок, легко окисляющийся на воздухе. Двуокись урана кофейно-корич-
невого цвета, получающаяся восстановлением или термическим разложением
при температуре выше 800°, гораздо более инертна.
Причина различной активности окислов урана в настоящее время еще не
совсем выяснена, особенно в отношении двуокиси. Современные исследования
показали, что трехокись урана может существовать по крайней мере в четырех
24*
372 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
различных кристаллических формах, которые значительно различаются между
собой по термической устойчивости (см. гл. 11). Возможно, что более
подробное изучение приведет к открытию полиморфизма и у двуокиси. Реакционная
способность окислов может также зависеть от точного отношения U : О в
продукте (согласно изложенному в гл. 11). В случае низшего окисла U02 это
отношение может изменяться от 1 : 1,75 до 1 : 2,30 без изменения
кристаллической структуры.
Реакции галогенирования могут проводиться как в газовой фазе (для
чего парообразный реагент пропускается над соединением урана при
повышенных температурах), так и в жидкой фазе при более умеренных температурах.
Большое число галогенирующих агентов может быть использовано и в жидкой
и в газовой фазах.
Критический обзор препаративных методов получения тетрахлорида был
дан Вагнером [47]; настоящее обсуждение во многих отношениях следует
обзору Вагнера.
Препаративное получение тетрахлорида урана.
Двуокись урана и хлористый водород. Термодинамические данные (ср. табл. 163)
указывают, что при повышенных температурах равновесие реакции
превращения двуокиси урана в тетрахлорид действием хлористого водорода не
благоприятствует галогенированию. Равновесные концентрации тетрахлорида
и воды, рассчитанные из соотношения —&F=RT\nK, настолько малы,
что эти продукты не могут быть удалены со скоростью, достаточной для того,
чтобы обеспечить приемлемую скорость превращения. Действительно, было
найдено [48], что при обработке двуокиси урана (полученной разложением
оксалата четырехвалентного урана) хлористым водородом в газообразной фазе
при 400° реакция практически не идет. При низких температурах
термодинамические соотношения становятся более благоприятными. Опыты показали,
что, если применять растворитель типа воды или водного раствора спирта,
хлорирование происходит; однако, как упоминалось выше, двуокись
превращается не в тетрахлорид урана, а только в UOCl2 [49]. Трехокись урана
также реагирует с хлористым водородом в этиловом спирте или четырех-
хлористом углероде, но и при этом образуется не тетрахлорид, a U02C12.
Сообщается, что активная двуокись урана реагирует с хлористым водородом
в абсолютном этиловом спирте, но образование тетрахлорида в таких
растворах не было доказано [48].
Двуокись урана, углерод и хлор. Впервые тетрахлорид урана был получен
обработкой хлором тесной смеси двуокиси урана с углем [1]. С тех пор эта
реакция использовалась многими исследователями [18, 50—52]. Вероятно,
здесь фактически хлорирующим агентом является четыреххлористый углерод.
Для получения тесной и реакционноспособнои смеси с углем можно смешать
сахар с двуокисью и прокалить смесь. Колани [53] считает, что особенно
пригоден для проведения процесса сахарный уголь или ламповая сажа, но другие
исследователи нашли, что уголь, приготовленный различными другими способами,
также вполне удовлетворителен. В условиях опыта, требующего пропускания
большого избытка хлора, образуется, помимо тетрахлорида, также
значительное количество пентахлорида [54]. Сделаны попытки [55] свести к минимуму
загрязнение тетрахлорида пентахлоридом путем применения смеси с большим
избытком угля, но эти попытки, повидимому, не дали ожидаемых результатов.
Наилучшее соотношение между двуокисью урана и графитом равно примерно
0,6 [56]. Вместо двуокиси может быть использован любой окисел или оксига-
логенид урана, однако при прочих равных условиях двуокись дает
наименьшее количество пентахлорида. Со смесью уранат натрия—ламповая сажа
реакция протекает удовлетворительно при температуре 600° [57].
Основным преимуществом этого метода является доступность исходных
материалов. Недостатками, которые, повидимому, перевешивают преимущества,
Глава 14. Соединения урана с хлором
373
нужно считать образование сравнительно больших количеств пентахлорида
(от которого необходимо избавляться последующей обработкой), высокая
температура процесса, что предъявляет жесткие требования к аппаратуре,
и присутствие значительных количеств фосгена в отходящих газах.
Недавно этот метод был заново изучен [58]. Таблетки, приготовленные
прессованием смеси трехокиси урана с углем, загружались в вертикальный
реактор, в нижней части которого был помещен дырчатый графитовый диск.
Хлор вводился в смесь снизу. Температура реакционной зоны поддерживалась
на уровне 850°. При этой температуре тетрахлорид является жидкостью,
поэтому он просачивается через слой таблеток и собирается на дне реактора.
Однако и в этом случае наблюдалось образование больших количеств
пентахлорида, который уносился в виде пыли током газа. В общем эти результаты
не являются обнадеживающими.
Двуокись урана и пары четыреххлористого углерода. Впервые четырех-*
хлористый углерод был применен в качестве хлорирующего агента для
превращения окислов металлов в хлориды Уаттсом и Беллом [59]. Возможность
использования этого реагента была подтверждена впоследствии другими
авторами [60, 61], которые изучали реакции взаимодействия окислов многих
металлов с этим реагентом. Для хлорирования окислов урана четыреххлори-
стый углерод был впервые применен Колани [53], установившим, что и
двуокись и закись-окись урана U308 при температуре красного каления реагируют
с газообразным четыреххлористым углеродом. Им было замечено, что
количество пентахлорида, образующегося одновременно с тетрахлоридом, гораздо
меньше в случае двуокиси урана, чем при закиси-окиси. В других опытах [62],
проведенных примерно с тридцатью окислами металлов, которые
обрабатывали парами четыреххлористого углерода, также было отмечено, что закись-
окись урана при 300° дает смесь тетра- и пентахлорида. Во всех этих старых
исследованиях применялся газообразный четыреххлористый углерод при
повышенных температурах. Майкл и Мэрфи [63] были первыми, изучавшими
галогенирование с четыреххлористым углеродом в жидкой фазе. Они
использовали как жидкий реагент, так и растворы хлора в последнем. Их работа
лежит в основе современных методов превращения окислов урана в тетра-,
пента- и гексахлорид путем хлорирования в жидкой фазе.
Течение реакции между двуокисью урана и четыреххлористым углеродом
было изучено путем измерения объема и анализа газообразных продуктов. При
450±20° (оптимальная температура для реакции с «неактивной» двуокисью
урана) основным процессом является следующий [64] [ср. уравнение (17)]:
U02 + CCI4-*UCI4-bC02.
В отходящих газах присутствуют также небольшие количества фосгена, окиси
углерода и хлора [65], что указывает на одновременное протекание ряда
параллельных реакций, как, например:
U02 + СС14 -> UOCl2 + СОС12; (26)
UOCl2 + СС14 -> UC14 + СОС12; (27)
U02 -t 2СОС12 -+ UCI4 + 2С02; (19)
СОС12->СО + С12; (28)
UCI4 + i-CI2-->UCl5. (29)
Основная реакция (17) является экзотермической и протекает с выделением
50 ккал тепла на 1 моль; местный перегрев может повлечь за собой частичное
сплавление и таким образом стать причиной неполного превращения.
Делались попытки проводить эту реакцию при температурах, достаточных для
того, чтобы тетрахлорид возгонялся из зоны реакции, однако этот метод
374 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
оказался неудовлетворительным из-за образования значительных количеств
пентахлорида урана и гексахлорэтана.
Интенсивному изучению подверглась также реакция между трехокисью
урана и парами четыреххлористого углерода, так как часто желательно
осуществление непосредственного превращения трехокиси в тетрахлорид без
предварительного получения двуокиси. Реакция газообразного четыреххлористого
углерода с трехокисью урана протекает почти при тех же самых условиях, что
и с двуокисью, но ввиду более высокой валентности металла в окисле пента-
хлорид образуется в большем количестве. При повышении температуры с 300
до 350° количество тетрахлорида возрастает быстрее, чем количество
пентахлорида [66]; однако относительное содержание последнего в тетрахлориде также
увеличивается (см. табл. 164). В случае применения «активной» трехокиси урана,
полученной прокаливанием дигидрата перекиси, хлорирование при 350°доходит
до конца.
Таблица 164
ОБРАЗОВАНИЕ ПЕНТАХЛОРИДА УРАНА ИЗ ТРЕХОКИСИ УРАНА
И ЧЕТЫРЕХХЛОРИСТОГО УГЛЕРОДА (ПАРЫ)
Температура,
°С
300
350
Пре вращение,
%
79,6
92,2
Образование
UCl5l
%
8,3
11,7
Образование
UC14,
%
71,3
80,5
Содержание
uci5 в исц,
о/
/О
11,7
14,5
Механизм этой реакции был изучен методами, сходными с теми, которые
применялись для изучения реакции хлорирования двуокиси урана. Реакция
состоит из быстрой начальной стадии, в результате которой получаются
двуокись углерода, хлор и уранилхлорид [67], и второй стадии—превращения
уранилхлорида в тетрахлорид урана, которое идет медленно при 400°.
Главным стехиометрическим уравнением реакции при 400° можно считать следующее
[67, 68]:
2U03 + ЗСС14 -» 2UC14 + ЗС02 + 2С12. (30)
Для объяснения присутствия фосгена, окиси углерода, хлора и пентахлорида
урана в продуктах реакции было сделано предположение о существовании
следующих параллельных реакций:
U03 + CC14 -» U02C!2 + COCI2; (31)
U03 + СОС12 -> U02C12 + С02; (32)
2U02C12 + 2СС14 -* 2UC16 + 2C02; (33)
2UCI6-^2UC15 + C12; (34)
2UC15*-»2UC14 + C12; (35)
COCla-*CO + CI2. (28)
Результаты, достигнутые в случае применения закиси-окиси урана, являются
промежуточными между теми, которые получены с двуокисью и трехокисью.
Хлорирование может быть проведено при температуре около 450°, причем
количество образующегося пентахлорида составляет 8—12%.
Сделаны попытки уменьшить количество образующегося пентахлорида
(при хлорировании трехокиси урана) введением окиси углерода, хлорос{юрма
или других соединений, способных реагировать со свободным хлором:
СО + С12-->СОС12; (36)
CHCIa + Cla-^CCI.-hHCI, (37)
Глава 14. Соединения урана с хлором
375
и предотвращающих, таким образом, образование пентахлорида урана [69, 70].
Сообщается [71 ], что, действительно, присутствие окиси углерода сильно
уменьшает образование пентахлорида, но получающийся результат в общем
неудовлетворителен. Добавление метана, двуокиси серы, хлороформа и различных
других хлорированных углеводородов оказалось неэффективным. Применение
активной двуокиси урана и проведение реакции при низкой температуре
остаются лучшими факторами, способствующими получению тетрахлорида
урана, свободного от пентахлорида.
Реакция между трехокисью урана и четыреххлористым углеродом в жидкой
фазе* Разложение пентахлорида. Установлено [3J, что двуокись или трехокись
урана медленно реагирует с жидким четыреххлористым углеродом при
температуре его кипения (77°) с образованием тетрахлорида. Повышение
температуры и давления сильно ускоряет реакцию в жидкой фазе. Так, при 250°
двуокись урана быстро реагирует с жидким четыреххлористым углеродом.
Высшие окислы урана реагируют значительно легче, чем двуокись, т. е. они
требуют меньших температур и давлений. Поэтому наибольшее внимание было
уделено реакции между жидким четыреххлористым углеродом и трехокисью
урана.
При температурах 115—185° основным продуктом этой реакции является,
вероятно, пентахлорид урана:
2U03 + 6СС14 ^ 2UCI5 + 6СОС12 + С12. (38)
Путем подбора подходящих условий можно по этой реакции получать и пента-
и тетрахлорид.
При нагревании в токе азота, двуокиси углерода или даже хлора
пентахлорид разлагается с переходом в тетрахлорид [4, 5, 7, 72—76]:
2UC]5^2UC14 + C12. (35)
Разложение начинается при температуре ниже 100° и завершается при 250°.
Летучесть и плохая теплопроводность пентахлорида несколько осложняют
практическое осуществление дехлорирования. Последнее легко достигается
следующим образом: пентахлорид помещают в узкий вертикальный
стеклянный цилиндр, который затем медленно опускают в печь, нагретую до 500°.
На поверхность пентахлорида направляют ток углекислого газа или азота для
того, чтобы способствовать конденсации вещества, которое в противном случае
могло бы теряться вследствие сублимации.
Дехлорирование может быть также выполнено кипячением растворов
пентахлорида с обратным холодильником в таких жидкостях, как монохлорид
серы, тетрахлорид олова, гексахлорпропен и др. [77]. Однако очистка
образующегося продукта от растворителя представляет некоторые трудности. Лучшие
результаты получаются при применении тионилхлорида; в этом случае
дехлорирование и освобождение от растворителя достигаются без особых
трудностей [78].
Хлорирование в газообразной фазе другими органическими галогенопро-
изводными. В качестве реагентов для превращения окислов урана в тетрахлорид
при низких температурах без образования фосгена или пентахлорида было
проверено большое число галогенированных органических соединений. Эти
соединения перечислены в табл. 165. В общем соединения с количеством атомов
углерода большим четырех неудовлетворительны ввиду образования
слишком большого количества углерода. В случае применения хлороформа или
смеси его с четыреххлористым углеродом образуется значительное количество
углерода. Чистый хлороформ реагирует медленно, превращение протекает
неполностью, и получающийся пентахлорид загрязнен продуктами пиролиза [70].
Гексахлорпропен и гексахлорциклопентадиен, ненасыщенные соединения,
имеющие атомы хлора в альфа-положении к двойной связи, являются
376
Таблица 165
НЕКОТОРЫЕ ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ, ИССЛЕДОВАННЫЕ В КАЧЕСТВЕ
ГАЛОГЕНИРУЮЩИХ АГЕНТОВ ДЛЯ ТРЕХОКИСИ УРАНА [79-82]
Соединение
Формула
Т. пл., вС
-22,6
186,9-187,4
-19
39
32
Четыреххлористый углерод
Гексахлорэтан
Тетрахлорэтилен
Хлораигидрнд трихлоракриловой
кислоты
Гексахлорбутадиен
Хлорангндрид трихлоруксусной
кислоты
Гексахлорпропен
Гексахлорциклопентадиен
Днхлордибромметан . . .
Трихлорбромметан . . .
Трихлорацетоннтрил . .
Хлористый метил ....
Хлористый метилен . . .
Хлороформ
1,1,1-Трихлорэтан ....
снле-Тетрахлорэтан . . .
1,2,3-Трихлорпропан . . .
Трихлорэтилен
Трихлоркумол
Хлораль
Хлористый аллил ....
2,3-Дихлорпрог!ен-1 . . .
Бензотрихлорид
3,3-Дихлорпентадион-2,4
1,1-Дихлорэтан . .
1,1,2-Трихлорэтан .
Этилтрихлорацетат
2,2-Дихлорпропан .
СС14
СС13СС13
СС12-СС1а
С12С = СС1-СОС1
СС!2 = СС1-СС1 = СС12
СС13СОС1
СС13 = СС1СС13
С5С1в
СВг2С1а
СВгС13
CC13CN
СН3С1
CH2Cia
СНС13
СН3СС13
СНС12СНС13
СН2С1СНС1СН2С1
СС12 = СНС1
(CH3)2CHQH2Ci3
CC13CHO
СН2=-СНСН2С1
Н2С = СС1-СН2С1
С,Н5СС13
СН3СОСС12СОСН3
СН3СНС12
СН2С1СНС12
СС13СООСН2СН3
СН3СС12СН3
-70
22
-21
-97,7
-96,7
-63,5
-36
-14,7
-73
-57
-136,4
-4,75
-96,7
-36,7
-34,6
а) Числа в круглых скобках указывают давление, мм рт.ст.
Глава 14. Соединения урана с хлором
377
удовлетворительными реагентами. Однако ни одно из этих соединений не
имеет каких бы то ни было преимуществ перед четыреххлористым углеродом
при хлорировании в газообразной фазе.
Другие способы галогенирования в жидкой фазе [81, 83—85]. Испытана
большое число органических галогенсодержащих соединений [80, 81] для
галогенирования в жидкой фазе (см. табл. 165). Насыщенные хлористые
соединения дают заметное превращение только в том случае, если они обладают
гораздо более высокой температурой кипения, чем четыреххлористый углерод;
они не очень эффективны. Соединения, содержащие винил ьный хлор (т. е. с
группировкой С = С—С1), обладают небольшой хлорирующей способностью, как
и следовало ожидать, на основании общей инертности такого атома хлора
в реакциях замещения. Однако хлорангидрид трихлоракриловой кислоты
реагирует с трехокисью урана, образуя нерастворимое в воде, но растворимое в
ацетоне и бензоле соединение, которое является, повидимому, частично
хлорированным окислом. Соединения с аллильным хлором (группировкой С=С—СС1)
более эффективны. Так, гексахлорпропен, гексахлорциклопентадиен С5С1в и
бензотрихлорид СвН5СС13 энергично реагируют со многими окислами. Однако
выделить чистые продукты из реакционной смеси не всегда возможно.
Найдено, например, что кипячением с обратным холодильником
соответствующей окиси и таких галогенированных углеводородов, как
гексахлорпропен, можно получить хлориды молибдена (IV), вольфрама (VI), кобальта (III),-
ниобия, тантала, ванадия (III) и ртути (II) и оксихлорид вольфрама (VI) [86].
В общем с окислами элементов первых четырех групп периодической системы
реакция или совсем не идет, или протекает в незначительной степени. Такие
вещества, как трехокись урана, закись-окись урана, U02+2U03> U04-2H20,
уранилхлорид, уранилсульфат, уранилнитрат и другие соединения уранила,.
при кипячении в гексахлорпропене с обратным холодильником превращаются
в тетрахлорид. Чистые двуокись урана и уранат аммония не вступают в
подобную реакцию [77, 81, 87, 88]. С трехокисью урана быстрая и экзотермическая
реакция начинается при температуре ниже 100°. Сначала образуется пента-
хлорид, легко растворимый в реакционной смеси; при дальнейшем нагревании
до 165° он разлагается. Если отношение числа молей гексахлорпропена к числу
молей трехокиси ниже 5:1, реакция проходит неполностью. После отфиль-
тровывания тетрахлорида последний промывают четыреххлористым углеродом
для удаления следов органического вещества. Основным органическим
продуктом этой реакции является хлорангидрид трихлоракриловой кислоты
СС12=СС1СОС1 (т. кип. 158°) [81,88,89]. Так как это соединение способно
растворять производные урана, то, прежде чем пытаться выделить продукты
реакции, необходимо хлорангидрид отогнать.
Изучены и другие хлорированные углеводороды в качестве галогенирую-
щих агентов для трехокиси урана с использованием запаянных стеклянных
трубок [90]. В случае применения дихлордибромметана и трихлорбромметана
получался чистый тетрахлорид. При 130° соединения, перечисленные ниже,,
давали менее удовлетворительные результаты:
СН3СОСС12СОСН3, СС13СНО, СС13СОС1, СН3СНС12,
СС12 = СС12, СН2С1СНС12, СС13СООСН2СН3,
СНС1 = СС]2, CC13CN, CH3CCI3, СН3СС12СН3.
Эти соединения расположены приблизительно в порядке убывания реакци-
онноспособности. Хлороформ не реагирует даже в присутствии заметных
количеств пентахлорида.
Двуокись урана и фосген. В качестве галогенирующего агента фосген был
впервые применен Шовене [91], который нашел, что он является сильным га-
логенирующим средством с широкой областью применения. Он реагирует не
378 Часть IV, Галогениды, оксигалогениды, боргидриды и карбонил урана
только с окислами урана, но также с сульфидами и фосфатами урана. Так,
минерал отунит реагирует при 800° следующим образом:
2U02 • СаО • Р205 + 8СОС!2 -> 2UCI4 + 2РОС13 + 8С02 + СаС!2. (39)
Шовене установил, что закись-окись урана реагирует с фосгеном при 450°;
Розенгейм иКелми [92] подтвердили это сообщение. Активная двуокись урана
реагирует при несколько более низких температурах. В современных работах
фосген не пользуется широким применением главным образом из-за его
отравляющего действия. Английские исследователи отметили полное превращение
закиси-окиси урана в тетрахлорид урана под действием фосгена при 900—940°[93].
Фосген применяется в промышленном масштабе для превращения двуокиси
тория в безводный хлорид. В возможности применения его для приготовления
любых безводных хлоридов не приходится сомневаться.
Окислы урана и тионилхлорид [48, 79, 82, 87, 94]. Тионилхлорид является
эффективным средством для превращения окислов урана в тетрахлорид.
Трехокись урана (полученная разложением перекиси) при действии паров
тионилхлорида в условиях температуры 350° быстро превращается в
тетрахлорид. Закись-окись урана реагирует медленнее. Дигидрат перекиси урана
и уранат аммония могут быть прохлорированы при 350°. Для получения
продукта, наименее загрязненного пентахлоридом урана, наиболее подходящим
исходным веществом является активная двуокись урана (полученная из
U04-2H20 или из трехокиси восстановлением парами спирта) [31].
В жидкой фазе трехокись урана может быть переведена в пентахлорид
кипячением с обратным холодильником с тионилхлоридом. Этой реакции
способствует добавка тетрахлорида. Выделение пентахлорида из реакционной смеси
наталкивается на некоторые трудности ввиду высокой растворимости его в тио-
нилхлориде (табл. 166). Из таких растворов кристаллизуется соединение
Таблица 166
РАСТВОРИМОСТЬ НЕКОТОРЫХ СОЕДИНЕНИЙ УРАНА В ЖИДКОМ
ТИОНИЛХЛОРИДЕ [95]
Соединение
UC15
Растворимость,
г 1мл
3-10-*
1,29
Соединение
UC16
U02C12
Растворимость,
г} мл
0,17
3-10-4
UCl5-xSOCl2 красноватого цвета; этот комплекс может быть разложен
нагреванием в вакууме до 100°. Растворы пентахлорида урана в тионилхлориде
могут быть одновременно дехлорированы и десольватированы распылительной
сушкой при 300° [78].
Хлорирование трехокиси урана жидким тионилхлоридом проводится
при температуре 100—130° и давлении 5,3—10,5 am [87, 96]. Реакция может
быть направлена таким образом, что получается тетра- или пентахлорид,
однако результат довольно трудно предугадать.
Сульфурилхлорид S02C12 не хлорирует ни трехокиси урана, ни закиси-
окиси его.
Двуокись урана и монохлорид серы. Монохлорид серы S2C12 был предложен
в качестве хлорирующего средства [97]. Затем рядом исследователей эта
реакция была применена к окислам урана. Установлено [53], что при температуре
красного каления смесь хлора и монохлорида серы реагирует с закисью-окисью
урана; однако без возгонки тетрахлорида реакция не доходит до конца.
Одновременно образуются значительные количества пентахлорида. Позже было
показано, что образование пентахлорида невелико, если применять монохлорид
Глава 14. Соединения урана с хлором
379
серы без хлора [98, 99]. Опубликована [100] подробная методика получения
тетрахлорида путем введения закиси-окиси урана в реакцию с
монохлоридом серы. Изучена [101] реакция закиси-окиси урана с монохлоридом серы
при 900°. Опыты были поставлены таким образом, что реакция и возгонка могли
протекать одновременно.
«Активные» окислы начинают реагировать с монохлоридом серы при
температуре около 250°, а при 450° или даже ниже реакция обычно доходит до
конца. При повышенных температурах монохлорид серы фактически
диссоциирует на серу и хлор.
С точки зрения термодинамики, процесс
U02 + 2S2C12 -> UC14 + S02 + 3S (40)
более выгоден, чем
2U02 + S2C12 + 3C12 —> 2UC14 + 2S02. (41)
Тем не менее можно рекомендовать использование хлора в качестве
газоносителя, так как при этом уменьшается загрязнение получающегося
продукта серой. В жидкой фазе реакция протекает при более низких температурах.
Кипячение закиси-окиси урана с обратным холодильником(138°)с монохлоридом
серы при атмосферном давлении в течение 7 час. способствует 95%-ному
превращению в тетрахлорид [3, 72, 79]. В подобную реакцию вступают все окислы
урана. Повышенное давление способствует более быстрому превращению.
Получающийся продукт обычно сильно загрязнен серой, которую трудно
удалить. Возгонкой в вакууме практически удалить ее не удается [93, 102, 103].
Предложено очищать тетрахлорид, содержащий серу, обработкой хлором
с целью превращения серы в летучий тетрахлорид серы [104], однако при этих
условиях могут образоваться большие количества пентахлорида урана.
Хлорирование другими безводными неорганическими хлоридами [104—106].
Изучено хлорирование двуокиси урана с помощью большого числа
неорганических галогенидов. С точки зрения термодинамики, реакция может идти лишь
при применении галогенидов металлов, которые образуют очень устойчивые
окислы, так как двуокись урана сама по себе весьма стойка. Ни один из
изученных реагентов не оказался особенно пригодным, за исключением, пожалуй,
хлорида алюминия. Хлорид аммония, который широко применяется для
превращения окислов редкоземельных элементов в хлориды, в случае урана
оказался неудовлетворительным [107]. Результаты изучения реакций двуокиси
урана с галогенидами металлов представлены в табл. 167.
Изучено также отношение тетрафторида урана ко многим из
вышеперечисленных реагентов. Тетрафторид легко реагируете газовой фазе с хлоридами
алюминия и бора с переходом в тетрахлорид [108]. В жидкой фазе
тетрафторид урана превращается в тетрахлорид при кипячении с обратным
холодильником со следующими соединениями: с тетрахлоридом кремния (1 час) на 5%,
с трихлоридом сурьмы (2 часа) на 10—20%, с тетрахлоридом олова (2 часа)
на 80% и с пентахлоридом тантала (1 час) на 90%. Как и можно было ожидать
на основании теплот образования фторидов, сплавление тетрафторида кремния
с хлоридами натрия, калия, меди (I), никеля (II), марганца (II), свинца (II)
и олова (II) не приводит к образованию тетрахлорида урана; такие же
соединения, как хлориды бериллия, магния, кальция, бария и NaAlCl4, превращают
тетрафторид урана при 700—800° в тетрахлорид [109]. Выделить последний
из реакционной смеси в чистом виде, без примеси хлорирующего агента, трудно.
При нагревании в вакууме до 750° смеси сульфата урана (IV) с
эвтектической смесью NaCl+CaCl2(51,5% последнего) образуется сублимат
тетрахлорида; за несколько часов превращение достигает 10%. В случае взаимодействия
с хлоридом бария при 580° в течение 13 час. образуется лишь небольшое
количество зеленого возгона [48].
380 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Таблица 167
РЕАКЦИЯ ДВУОКИСИ УРАНА а) С НЕОРГАНИЧЕСКИМИ ГАЛОГЕНИДАМИ
NH4Clf
СаС!3
ВеС13
MgCI3
SnCl2,
NiCl.,
CdCl
Галогенид
CuCl,
ZnCl3
CuCl2,
2, PbCl
MnCl3
AICI3
SbG3
PCI3
FeCi3
SiCI4
SnCl4
PC15
TaCl5
KCI,
FeCl2
Hg2Cl2
, CoCl2,
2, HgCl3, BaC!2,
Условия реакции б)
Сплавление при температуре
плавления соли
700-800°
500°
700-800°
500°
500°
250°
Кипячение с обратным
холодильником
То же
300° !
Кипяченяе с обратным
холодильником
То же
300°
Кипячение с обратным
холодильником
Результаты
Реакция не идет
Образуется 5% UCI4
Происходит полное
превращение в UC14 в течение 4 час.
Образуется 15% UC14
Получается небольшое
количество зеленого возгона
Реакция не идет
Реакция протекает быстро
Реакция не идет
Образуется 5% UC14
Образуется неидентифициро-
ванное соединение урана
(не UC14)
Реакция не идет
Реакция не идет
Происходит превращение
на 20%
Образуется 10-20%'UC14
а) В этих опытах пользовались весьма нереакционноспособной окисью Маллинкродта.
б) В тех случаях, когда температура была выше точки плавления галогенида, реакцию проводили
в запаянной трубке.
Получение тетрахлорида урана в водных растворах. Водные растворы
тетрахлорида легко получить путем восстановления растворов уранилхлорида.
Восстановление может быть выполнено электролитически, фотохимически,
амальгамой серебра, кадмия или висмута, металлической медью или
гидросульфитом натрия Na2S204[18, 110—112]. Может потребоваться обработка
реакционной смеси воздухом для окисления урана (III) в случае его
образования.
Попытки выделить из таких растворов безводный тетрахлорид лишь
отчасти увенчались успехом. Розенгейм и Келми [92] сообщают, что при
выпаривании водного раствора до сиропообразной консистенции с последующей
обработкой абсолютным спиртом, насыщенным хлористым водородом, получается
декагидрат UCl4-10H2O. Однако работы исследовательской группы из Эймса
не подтвердили этого наблюдения. При дегидратации в токе хлористого
водорода этими исследователями [113] была получена смесь двуокиси урана и
UOCl2. Эти результаты были впоследствии подтверждены другими авторами.
Прямая дегидратация растворов урана (IV) по методу Клейнхекселя и
Кремерса [114], являющемуся весьма эффективным в случае редкоземельных
Глава 14. Соединения урана с хлором
381
элементов, не приводит к образованию безводного тетрахлорида. Однако
получающийся таким путем оксидихлорид может быть превращен, по крайней
мере частично, в тетрахлорид термическим диспропорционированием [49]
(см. стр. 473). /
Многообещающие результаты были получены при дегидратации водных
растворов тетрахлорида с помощью азеотропной перегонки [115]. К водному
раствору тетрахлорида добавляли бензол, четыреххлористый углерод,
хлороформ и т. д. и отгоняли азеотропную смесь до удаления всей воды. После
отгонки азеотропообразователя остается твердый зеленый продукт, из которого
возгонкой может быть получено от 10 до 70% тетрахлорида урана.
Разные способы получения тетрахлорида урана. Галогенирование
различных соединений урана. Металлический уран легко реагирует со многими гало-
генирующими агентами с образованием тетрахлорида [1, 18, 116]:
U + 2Cla-^-UCl4. (42)
Вместо металла может быть использован его гидрид (см. гл. 8):
UH3 + 2C0C12 ^VUC14 + 2C0 + -|H2; (43)
UH3 + \ С1а -* UC14 + 3HC1. . (44)
При более высоких температурах металлический уран реагирует также с
хлороформом или хлористым водородом. Прямая реакция между гидридом урана
и хлором не подходит для использования в лаборатории, так как, раз
начавшись при 200°, она уже не поддается регулированию. Гораздо удобнее при
получении тетрахлорида из металлического урана использовать последователь-:
но следующие реакции [9]:
U+|-H2->UH3; (45)
UH3 + 3HC1 -* UC13 + ЗН2; (2
UCl3 + yCl2-*UCl4. (12)
Сульфид [104] и карбид [117] урана можно прохлорировать газообразным
хлором при 400°; карбид при повышенных температурах также реагирует с
хлористым водородом. Нитриды урана UN и UNi>75 легко реагируют с хлористым
водородом при 400—500° с образованием смеси хлоридов аммония и урана [118]
(см, гл. 10). Большинство солей уранила можно хлорировать в паровой фазе,
однако они реагируют медленнее, чем окислы. Уранаты и диуранаты частично
хлорируются в жидкой фазе четыреххлористым углеродом, но продукты
реакции пока еще недостаточно хорошо изучены [109].
Сделаны попытки превратить органические соединения урана (IV) в
тетрахлорид обменным разложением. Так, ацетилацетонат урана (IV) был
обработан в бензольном или эфирном растворе безводным хлористым водородом.
В результате реакции образовалась темная смола, и тетрахлорид выделить не
удалось [119]. Как и в случае тория, белый твердый осадок, который сначала
выпадает при пропускании хлористого водорода, еще содержит органические
вещества. При обработке безводным хлористым водородом бензоата урана (IV)
U(OOCC6H5)4-H20, взвешенного в этиловом эфире, образуются зеленые
соединения с отношением хлора к урану от 2,5 до 3,8 и содержанием
органических веществ от 20 до 40% [120]. Зеленый продукт растворяется в воде с
образованием зеленого раствора, однако тетрахлорид урана из такого продукта не
удалось выделить возгонкой.
Очистка тетрахлорида урана. Тетрахлорид может быть очищен непог
•средственной возгонкой в вакууме или возгонкой в токе хлора (275—700°) в виде
382 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
высшего хлорида с последующим обратным превращением в тетрахлорид [72].
Возгонка может быть выполнена также в токе инертного газа, как, например,
азота или гелия. В азоте возгонка быстро протекает при температуре 600—650°
и давлении 3—5 мм рт. ст.; тетрахлорид возгоняется не плавясь. Если возгонку
вести при атмосферном давлении, то количество нелетучего остатка может быть
сильно уменьшено добавлением к азоту четыреххлористого углерода [4].
Для этих же целей применяется гелий с очень небольшой примесью хлора.
Качество очищенного тетрахлорида может быть определено с помощью
разных критериев, наиболее важными из которых являются растворимость
в некоторых подходящих растворителях и летучесть [121—123].
Физические свойства тетрахлорида урана.
Температура плавления. Наиболее надежным из всех определенных значений
температуры плавления тетрахлорида является 590±Г [124, 125]. Средним (не-
взвешенным) из нескольких определений, выполненных различными другими
исследователями, является 589° [4, 6, 21, 126—129]. Приведенное в литературе
значение 564° [51], несомненно, является заниженным.
Точки превращения. Изучение кривой охлаждения тетрахлорида
показывает, что при температурах 212, 506—522 и 543—565° твердый тетрахлорид
претерпевает фазовые превращения. Тепловые эффекты этих превращений
невелики (приблизительно 1 икал-моль'1) [130]. Проверка в Национальном бюро
стандартов [131] не подтвердила наличие фазового превращения при 211°.
Поэтому действительное существование упоминавшихся фазовых превращений
остается сомнительным.
Температура кипения. Наилучшее прямое определение температуры
кипения тетрахлорида дает значение 792° при давлении 760 мм рт. ст. [124, 125].
Заметного разложения при этой температуре не наблюдалось. Несколько
других определений температуры кипения обладают сомнительной точностью
[127, 132, 133].
Давление пара. Давление пара твердого тетрахлорида было определено
двумя методами—методом истечения (в Беркли) и методом испарения (в Эймсе).
Полученные результаты не согласуются друг с другом. Причина
расхождения не ясна, однако данные, полученные первым методом, повидимому, более
надежны. Согласно критическому обзору измерений по методу истечения [134],
давление пара твердого тетрахлорида может быть выражено следующим
уравнением:
lgp**PT.cx.= -^+13,2995 (350-505°). (46)
Значения, рассчитанные по этому уравнению, приведены в табл. 168. Теплота
возгонки тетрахлорида равна 47,7 ккал-моль'1', свободная энергия возгонки
Таблица 168
ДАВЛЕНИЕ ПАРА ТВЕРДОГО ТЕТРАХЛОРИДА УРАНА
Температура, °С
350
360
370
380
390
400
410
420
Давление пара,
мм рт. ст.
0,00037
0,00067
0,0012
0,0022
0,0038
0,0064
0,011
0,018
Температура, °С
430
440
450
460
470
480
490
500
505
Давлерше пара,
мм рт. ст.
0,030
0,047
0,076
0,12
0,18
0,28
0,43
0,64
0,80
Глава 14. Соединения урана с хлором
383-
определяется по следующей формуле:
AF0 = 60606 + 36,85 Т]gГ- 170,9 Т кал-моль1.
(47)
Произведено повторное определение давления пара жидкого тетрахлорида
[124, 125]. Полученные результаты точнее ранее полученных значений [127].
Они могут быть выражены уравнением
IgAwA/pT.cT. = —^- + 9,65
(590 4-790°).
(48)
Экспериментальные значения приведены в табл. 169 и нанесены на график
(рис. 57). Рассчитанная из этих данных теплота парообразования равна.
W00
100
500
400
300
Ё
ъ.200
с:
575 600
Температурау °С
650 700 750
800 850
100
70
50
40
30 \-
20
/
/
Iff Рммртст =-7205r'+9,65j
/
15
1,20
V5
VO
105
1 Ю3
100
Q95
Q90
Рис. 57. Давление пара жидкого тетрахлорида урана.
X, П. О — точки для разных препаратов. Температура кипения при
атмосферном давлении 792°.
33 ккал-моль'1. Скрытая теплота плавления составляет, таким образом,
47,7—33 = 14,7 ккал-моль"1, и константа Трутона равна 31. Следовательно,
жидкий тетрахлорид является сильно ассоциированной жидкостью.
384 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Экстраполяция кривой давления пара жидкого тетрахлорида приводит
к значению давления пара в точке плавления (590±1°) р=19,5±1 ммрт. ст. Это
наиболее надежная из предложенных величин для координат тройной точки.
Измерения давления пара, сделанные в Эймсе методом испарения [133],
дали результаты, совершенно отличающиеся по порядку величины (гораздо
более высокие, чем приведенные в табл. 169). Они, по всей вероятности,
содержат систематическую ошибку и в дальнейшем нами обсуждаться здесь не
будут.
Таблица 169
ДАВЛЕНИЕ ПАРА ЖИДКОГО ТЕТРАХЛОРИДА УРАНА
Температура,
°С
789,5
788
791
769
770
763,5
761
757
747
742
733
727,5
Давление пара.
мм рт. ст.
1
74,63
74,55
74,26
54,6
54,1 '
54,0
47,2
46,0
39,5
37,4
30,9
28,0
Температура,
°С
714
697,5
692
660
648
639
632
628
611
| 603
597
591
Давление пара,
мм рт. ст.
23,0
17,2
15,0
9,0
6,1
6,0
4,7
4,6
3,2
2,7
2,3
1,9
Кристаллическая структура и плотность. Тетрахлорид урана образует
темнозеленые октаэдрические кристаллы с металлическим блеском.
Рентгенографическое исследование [135—137] показывает, что они имеют
тетрагональную симметрию с константами решетки
ах = 8,296 ± 0,009 А; а3 = 7,№ ± 0,009А.
Элементарная ячейка содержит четыре молекулы тетрахлорида; рассчитанная
плотность равна 4,87 г/см2. Группа симметрии 14 /amd, положения атомов
следующие:
4U в ЦЬ);
16С1 в 16(A);
•с х=0,281 и 2=0,398. Эти результаты позже были подтверждены и другими
исследователями [138—140]. Плотность была определена также прямыми
измерениями. Значения, полученные при ранних исследованиях, равны 4,854 [57]
и 4,725 [52]. Плотность определяли [99] по методу гидростатического
взвешивания в водороде в интервале температур от комнатной до температуры
жидкого воздуха. Полученные величины составляют 4,950 при —193° и 4,890 при
—78°. Значение, найденное для плотности при 19°, хорошо согласуется с
найденным рентгенографически. В Эймсе была найдена величина 4,88 г/см3 при 24°,
что также хорошо согласуется с рентгенографическими данными [76].
Измерена также плотность парообразного тетрахлорида [141]. Средняя
из полученных величин составляет 13,31 (по воздуху); из этого значения
вытекает отсутствие ассоциации в парах тетрахлорида. Эбулиоскопиче-
ское определение молекулярного веса в трихлориде висмута в качестве
растворителя показывает, что в растворах, по крайней мере в разбавленных
Таблица 170
УДЕЛЬНАЯ ТЕПЛОЕМКОСТЬ, ЭНТРОПИЯ И ЭНТАЛЬПИЯ ТВЕРДОГО
ТЕТРАХЛОРИДА УРАНА ОТ 0 ДО 355° К
Температура,
°К
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
105
ПО
115
120
125
130
135
140
145
150
155
160
165
170
175
180
185
190
195
200
205
210
215
220
225
Ср,
межд. дж • моль~^ . град~1
0,0
0,255а>
2,035а>
6,473
13,01
20,50
28,17
35,27
41,64
47,26
52,24
56,68
60,78
64,73
68,34
71,72 #
74,88
77,80
80,61
83,12
85,48
87,72
89,85
91,82
93,72
95,47
97,10
98,66
100,1
101,5
102,8
103,9
105,0
106,1
107,1
108,0
108,9
109,7
110,5
111,2
111,9
112,6
113,2
113,8
114,4
115,0
5,
межд. дж - моль~1 • град~1
0,0
0,0843
0,6793
2,251
4,977
8,673
13,09
17,98
23,11
28,35
33,59
38,79
43,88
48,93
53,83
58,66
63,40
68,04
72,56
76,97
81,30
85,55
89,66
93,72
97,63
101,5
105,3
109,0
112,6
116,1
119,6
123,0
126,3
129,5
132,7
135,9
138,9
141,9
144,9
147,7
150,6
153,3
156,0
158,7
161,3
163,9
межд. дж • моль~ъ
0,0
0,318
1 5,098
25,22
73,55
156,9
278,8
437,6
| 630,2
852,5
1 102,0
1374
1668
1982
2 314
2 665
3 032
3413
3 809
4219
4 640
5 074
5517
5 972
6 435
1 6 908
1 7 390
7 879
8 376
8 880
9 391
9 908
10 430
10 959
11491
12 029
12 571
13 118
13 668
14 223
14 781
15 342
15 906
16 474
17 045
17618
25 Химия урана
386 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Продолжение табл. 170
Температура,
°К
230
235
240
245
250
255
260
265
270
275
280
285
290
295
298,16
300
305
310
315
320
325
330
335
340
345
350
355
Ср.
межд. дж ♦ моль~1 - град"1
115,5
116,1
116,6
117,0
117,5
118,0
118,5
118,9
119,4
119,8
120,3 |
120,7
121,2
121,6 !
121,8
122,0 1
122,4
122,7
123,0
123,4
123,7
124,0
124,3 I
124,5
124,8 1
125,1 |
125,3 |
S,
межд. дж ♦ моаь~1 • град"*
166,4
168,9
171,4
173,8
176,2
178,5
180,8
183,0
185,3
187,5
189,6
191,8
193,9
196,0
197,2
198,0
200,0 "
202,0
204,0
205,9
207,8
209,7
211,6
213,4
215,2
217,0
218,8
H-Es,
1 межд. дж • моль~1
18 194
18 773
19 355
19 939
20525
21 114
21705
22 298
22 894
23 492
24 092
24 695
25 299
25 906
26 291
26515
27 126
27 739
28 353
28 970
29 587
30 206
30 827
31449
32 072
32 697
33 323
а) Экстраполировано с применением функции Дебая 0,15396 D
растворах при высоких температурах, тетрахлорид существует в виде простых
молекул [142].
Термохимические данные. Сотрудниками Национального бюро
стандартов [143] были определены теплоемкость*, энтропия и энтальпия твердого
тетрахлорида в интервале температур 15—355° К (табл. 170). Во избежание
окисления и гидролиза измерения проводились в атмосфере сухого гелия.
Значения S и функции Н—Ejj} были получены с помощью интегрирования по
таблицам с использованием правила Симпсона. При 298,16° К энтропия
твердого тетрахлорида равна 47,14 энтропийной единицы. Эти результаты были
расширены во второй серии опытов [28, 131J, охватившей температурный
интервал 0—427° (табл. 171). Разница между двумя рядами значений в области,
перекрываемой ими, составляет около 0,7%; вероятно, значения, полученные
в более высокотемпературной области (табл. 171), более точны ввиду лучшего
качества использованного тетрахлорида.
* Использованный образец содержал менее 0,02% нелетучих примесей.
Глава 14. Соединения урана с хлором
387
Таблица 171
УДЕЛЬНАЯ ТЕПЛОЕМКОСТЬ, ЭНТАЛЬПИЯ И ЭНТРОПИЯ ТВЕРДОГО
ТЕТРАХЛОРИДА УРАНА ПРИ ТЕМПЕРАТУРАХ ОТ О ДО 427е [131]а>
Температура,
°С
0
50
100
150
200
250
300
350
400
426,84
(700° К)
Ср,
кал • г""1 ♦ град~1
0,07516
0,07735
0,07917
0,08063
0,08196
0,08328
0,08472
0,08616
0,08760
0,08838
Н-Но.
кал • г~1
0
3,815
7,728
11,726
15,790
19,920
24,120
28,392
32,736
35,099
S-So.
кал ♦ г~1 • град-1
0
0,012830
0,024083
0,034132
0,043204
0,051501
0,059164
0,066309
0,073011
0,076450
а) В другой работе [28] те же авторы сосбщают несколько другие
пересчитанные значения.
Экспериментальные данные, приведенные в табл. 171, описываются
следующими уравнениями* (с точностью, указанной в скобках; для температур
ниже 0° эти уравнения неприменимы):
Ср = 0,07608 + 2,88- Ю"5*- тгг^2 кал -г"1 -град'1
(* + 41)2
(точность ±0,35%; 0-427°),
где /—температура, °С;
Н- Н0* = 0,07608/ + 1,44- Ю-5/2- 0,032-
—— кал • г 1
t + 4l
(49)
(50)
(точность ±0,2%; 0-427°),
где / — также температура, °С;
0,0056
S-So = 0,15706 lgТ+2,88-10-Б Т- 0,39062 + т^_ш
+ 5,6.10-5lg(£^?.) кал-г-1-град~1
(51)
(точность ±0,2%; 0-427°),
где Т=/+273,16.
При температурах выше 100° последние члены в уравнениях (49) и (50)
и последние два члена в уравнении (51) ничтожно малы.
Для того чтобы получить абсолютные значения энтальпии и энтропии,
необходимо к величинам, которые приведены в табл. 171 или выведены из
уравнений (50) и (51), прибавить 14,644 кал-г'1 и 0,П748 кал-г'1 •град'1
соответственно (эти значения взяты из табл. 170).
Теплота образования тетрахлорида урана впервые была определена Биль-
цем и Фендиусом [30]; это определение было затем повторено в Беркли [ЗЗК
* Джиннингсом и Корруцини [28] для этих же данных предложены другие
уравнения.
25*
388 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Метод определения состоял в растворении металлического урана и
тетрахлорида урана по отдельности в соляной кислоте, содержащей 10% хлорида
железа (III), и в сравнении этих теплот (измерения проводились в ледяном цель-
ностеклянном калориметре типа бунзеновского). Кислоту и хлорид железа
брали в столь большом избытке, что тепловые эффекты, вызванные изменением
концентрации, были ничтожно малы. Водород, выделяющийся при
взаимодействии металлического ураиа с кислотой, может восстановить часть ионов
железа (III); при расчетах это учитывалось путем определения содержания
двухвалентного железа с помощью сульфата церия (IV) после завершения
растворения.
Из теплот растворения в сочетании с другими известными
термохимическими данными можно рассчитать теплоту образования
UTB + 2Cl2ra3->UCl4TB, (42)
Д#273= —250,9 ±0,6 ккал • моль"1.
Более ранние определения Бильца и Фендиуса [30] дали очень близкий к этому
результат.
В начале этой главы [ср. уравнение (1)] рассматривалась равновесная
реакция восстановления тетрахлорида урана
UC14 + JH2 ^UC1, + HC1.
Из экспериментальных результатов, приведенных там, были выведены
следующие значения для теплоты и свободной энергии образования тетрахлорида:
Д#208=— 251,0 ккал-моль'1;
AF°298= —229,65 ккал-моль"1;
AS208= —71,5 энтропийной единицы;
д#= -252600 + 5,70Г-7,79-10-4Г2;
AF* = -252600~13,13rigr + 7,79.10-4r2+109,257\
Найденное таким образом значение для теплоты образования тетрахлорида
хорошо согласуется с полученным по методу Бильца и Фендиуса. Реакция
образования тетрахлорида из трихлорида и хлора является эндотермической,
поэтому с ростом температуры возрастает термическая устойчивость тетра-хло-
рида с точки зрения диссоциации натрихлорид и хлор. Расчеты показывают, что
при давлении ниже атмосферного и любой достижимой температуре
газообразный тетрахлорид не может быть превращен в трихлорид путем разложения.
Оптические свойства. Красные пары тетрахлорида поглощают свет с
длиной волны между 500 и 220 тр.. Поглощение становится интенсивным вблизи
310 тр. и растет в направлении более коротких волн. По мере роста
концентрации паров (t=430°HBbiuie) поглощение распространяется на область видимого
спектра. Можно было открыть оптическим путем в 37,5-сантиметровой трубке
пары тетрахлорида под давлением всего в 3-Ю"3 мм рт. ст. [144].
Был также изучен спектр поглощения водных растворов тетрахлорида [145].
Максимумы наблюдались при длинах волн 429,5, 485, 495, 549, 649 и 672тр..
При длине волны 649 тр. закон Бэра справедлив в интервале концентраций от
0,007 до 0,020 М.
Электрические и магнитные свойства. Магнитная восприимчивость.
Определена [146] молярная магнитная восприимчивость твердого тетрахлорида в
виде функции температуры (табл. 172). Закон Кюри справедлив при низких
температурах.
Поскольку отмечается, что тетрахлорид неустойчив при температуре выше
комнатной, необходимо сделать вывод, что не было принято соответствующих
Глава 14. Соединения урана с хлором
389
Таблица172 Таблица173
МОЛЯРНАЯ МАГНИТНАЯ ВОСПРИИМЧИВОСТЬ ЭЛЕКТРОПРОВОДНОСТЬ
ТЕТРАХЛОРИДА УРАНА [146] ТЕТРАХЛОРИДА УРАНА
Температура,
. °к
90
198
273
289
х (мол.) • 1 0^>
1058
488
413
410
х (мол.) • Т
0,952
0,965
1,130
1,185
Температура,
°С
570
598
620
Удельная
электропроводность, омг*
0,34
0,42
0,48
мер против попадания влаги и воздуха в продолжение эксперимента.
Следовательно, эти результаты довольно сомнительны и цитируются здесь лишь
ввиду отсутствия более точных данных.
Электропроводность^. Расплавленный тетрахлорид является хорошим
проводником электричества [147]. Изучена [51] удельная электропроводность
такого расплава как функция температуры. Хотя их измерения, повидимому,
выполнены с достаточной тщательностью, можно сомневаться в правильности
результатов ввиду того, что температура кипения тетрахлорида, приведенная
авторами, значительно отличается от общепринятой для чистого вещества.
Несмотря на это, в табл. 173 приводятся эти данные вследствие отсутствия
других измерений. Электролитическое восстановление расплавленного
тетрахлорида урана было использовано для получения металлического урана
(см. гл. 4).
Химические свойства тетрахлорида урана.
Тетрахлорид притягивает воду и окисляется на воздухе, поэтому все операции
с ним должны проводиться в атмосфере сухого инертного газа.
Отношение к воде. Тетрахлорид урана очень гигроскопичен. Хотя он
и менее чувствителен к влаге, чем пентахлорид или гексахлорид, все же он
реагируете парами воды, если их парциальное давление превышает2мм рт. ст.
Поглощение воды не ограничивается образованием гидрата тетрахлорида,
а протекает дальше в виде химической реакции с образованием UOCl2 и
хлористого водорода. Этот процесс длится до полного растворения и разложения
тетрахлорида [148]. Однако установлено, что выделение хлористого водорода
протекает очень слабо до тех пор, пока не будет поглощена вода в количестве,
достаточном для образования дигидрата тетрахлорида [21].
Тетрахлорид урана растворяется в воде с выделением тепла и образованием
зеленого раствора, который проявляет характерные свойства иона урана (IV)
[149]. Теплота растворения в растворе соляной кислоты (1 моль хлористого
водорода на 8,808 моля воды) была определена в 39,4 + 0,2 ккал-моль"1 [30].
Поскольку водные растворы тетрахлорида всегда имеют кислую реакцию,
очевидно, они претерпевают сильный гидролиз. При попытках сконцентрировать
водные растворы тетрахлорида выпариванием выделяется хлористый водород
и выпадает сложная смесь гидратированных окислов урана (ионный состав
водных растворов тетрахлорида будет рассмотрен во второй книге, где
всесторонне будут обсуждены свойства растворов четырехвалентного
урана).
Под действием водяного пара при 600° тетрахлорид превращается
в закись-окись урана U308.
Отношение к неводным растворителям. В общем тетрахлорид растворим
в сильно полярных растворителях и нерастворим в неполярных, например
в таких углеводородах, как бензол, хлороформ и эфир. Он растворим в
некоторых кислородсодержащих органических растворителях, однако вследствие
390 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
сольволиза регенерировать из таких растворов тетрахлорид нельзя. Например,
при растворении последнего в метиловом или этиловом спирте имеет место алко-
голиз [50]. Небольшие количества воды могут, очевидно, присутствовать
в спиртовых растворах, не вызывая гидролиза [82]. Растворы тетрахлорида
в спирте проводят электрический ток.
В ацетоне тетрахлорид растворяется (вероятно, это растворение
сопровождается химической реакцией) с образованием зеленовато-желтого раствора
[150]. Пирле [151] нашел, что такой раствор обладает значительной
электропроводностью. Тетрахлорид растворяется и в пиридине, этилацетате и этил-
бензоате с химической реакцией.
При температурах ниже 70° тетрахлорид нерастворим в таких
неорганических растворителях, так РС13, SnCl4 и РОС13 [152], а также в жидком
безводном фтористом водороде [153].
Отношение к окислителям. Реакция между тетрахлоридом и кислородом
при повышенных температурах изучена в лабораториях Теннесси—Истмен
[154—155]. Результаты, полученные в этих лабораториях, отличаются от
найденных в более ранней работе, выполненной в Беркли [156]. По первым
данным [155], реакция тетрахлорида с кислородом протекает в две стадии:
UC14 + 02 ^ Ш2С12 + С12; (52)
3U02Cl2 + 02 ^U308 + 3C12. (53)
Наинизшая температура, при которой реакция протекает с измеримой
скоростью, равна 230±5° для реакции (52) и 250+10° для реакции (53).
Авторы считают, что образующееся при этом «черное вещество» является
закисью-окисью урана, однако это не было подтверждено аналитическими
данными. Энергии активации для обеих реакций были определены в 12 и 45 ккал
соответственно. Кинетические исследования показывают, что стадией,
определяющей скорость реакции, является собственно окисление, а не диффузия
газа через твердое вещество.
Хлор реагирует с тетрахлоридом при повышенных температурах с
образованием смесей пента- и гексахлорида (эта реакция рассматривается на стр. 393
при обсуждении получения пентахлорида). Можно тетрахлорид урана прохло-
рировать до смеси пента- и гексахлорида при температуре 115—125° с помощью
раствора хлора в четыреххлористом углероде. Фтор превращает тетрахлорид,
как и другие соединения урана, в гексахлорид. Эта реакция может
представлять опасность из-за образования взрывчатой смеси хлора с фтором.
Отношение к восстановителям. При атмосферном давлении и температуре
525—550° водород восстанавливает тетрахлорид до трихлорида [6] (см. стр. 362).
В противоположность утверждениям, имеющимся в более старой литературе,
бромистый и йодистый водород также могут восстанавливать тетрахлорид до
трехвалентного состояния [13] (вместо простой замены атомов галогена):
UCI, + 4HBr i^zii0^ UBr3 + 4HC1 + у Вг2; (54)
»
uci4 + hj ii^zii0^ uci3 + Hci+i-j2. (3)
Однако при более высоких температурах бромистый водород дает тетрабромид
урана, а йодистый водород—смешанные иодхлориды. При действии
фтористого водорода всегда образуется тетрафторид урана. Активные металлы (литий,
натрий, калий, кальций, магний и алюминий) восстанавливают тетрахлорид
урана до металлического урана (см. гл. 4).
Реакции обменного разложения. Сульфиды и селениды реагируют с
тетрахлоридом с образованием соответствующего сульфида или селенида
Глава 14. Соединения урана с хлором
391
урана (IV) [53]:
UC14 + 2H2S —LlJS2 + 4HCl; (55)
UCl4+2H2Se ^_-.USe2 + 4HCl. (56)
При,повышенных температурах тетрахлорид реагирует в общем с
большинством кислородсодержащих соединений с образованием двуокиси урана.
Сообщается, что двуокись кремния (кварц) не реагирует с расплавленным тетрахло-
ридом урана [52], однако стекло подвергается сильному разрушению [55].
Двуокись урана взаимодействует с расплавленным тетрахлоридом урана с
образованием оксидихлорида.
Комплексные соединения тетрахлорида урана. Тетрахлорид урана является
координационно ненасыщенным соединением и поэтому образует большое
количество комплексных соединений. При умеренных температурах он
поглощает газообразный аммиак с образованием соединений, содержащих одну,
две или три молекулы аммиака. Часть поглощенного таким образом аммиака
остается связанной даже в вакууме. С жидким аммиаком образуется зеленовато-
белое вещество состава UC14-12NH3. •
UC14TB+12NH3~>UC14.12NH3+183 ккал. (57)
При нагревании этого соединения до 100° выделяется столько аммиака, что
в остатке образуется UC14-8NH3. Оба эти соединения на воздухе неустойчивы
и реагируют с водой с образованием гидратированной гидроокиси урана (IV).
В условиях высоких температур получается смесь окиси и нитрида [157].
Сходные продукты присоединения тетрахлорида образуются при действии
хинолина и пиридина в ацетоновом растворе. Эти комплексные соединения
выделяются в виде желтых кристаллов и могут быть перекристаллизованы из
кипящего абсолютного спирта [150].
Получены также двойные соли типа M2UC16, где М представляет литий,
натрий, калий или R4N. Некоторые из этих соединений нашли применение для
электрохимического получения металлического урана (см. гл. 4). Ввиду
большей устойчивости на воздухе они иногда применяются при химической
препаративной работе вместо безводного тетрахлорида. Соединения такого типа
не могут быть получены осаждением из водных растворов. Производное калия,
K2UC16, может быть приготовлено путем реакции между парами тетрахлорида
урана и хлоридом калия при температуре красного каления [158]. Оно
представляет темнозеленые кристаллы, слабо гигроскопичные на воздухе,
плавящиеся при 350°. Это соединение может, быть нагрето до 550° без выделения
паров. Оно растворяется в воде с разложением, растворимо в спирте и в этил-
ацетате и практически нерастворимо в эфире. Тем же путем могут быть
получены производные натрия и рубидия, которые обладают такими же свойствами,
как и калиевое. Существование соединения лития этого типа пока еще точно не
установлено (см. табл. 174).
Хлориды щелочноземельных элементов также образуют подобные двойные
соли с тетрахлоридом урана. Было получено два таких соединения, а именно
BaUCl6 и SrUCl6, которые близко напоминают калийную двойную соль.
Известны также соответствующие пиридиновые и хинолиновые соли, (C5HGN)2UC16
и (C9H8N)2UC16. Они могут быть получены путем приливания пиридина
или хинолина к раствору тетрахлорида в абсолютном спирте, насыщенному
хлористым водородом [92].
Изучены фазовые соотношения в системах, содержащих тетрахлорид урана
и тот или иной другой галогенид, с помощью термического анализа [21].
Результаты этих исследований приведены в табл. 174. Рентгенографического
исследования этих систем пока еще не проведено (см. гл. 12).
392 Часть IV. Галогениды, оксигалогениды, боргидриды и карбон ил урана
Таблица 174
ФАЗОВЫЕ СООТНОШЕНИЯ В СИСТЕМАХ ТЕТРАХЛОРИД УРАНА-ГА ЛОГЕ НИД МЕТАЛЛА
Система
UC14-KC1
UC14—NaCl
UC14—LiCl
UCI4—СаС12
UCl4-BaCl2
UC14-UC136)
Формула
соединения
к2ис1в
Na2UCle
Отсутствует
»
Ba2UCl8
Отсутствует
Т. пл. соединения,
°С
650±5а>
При 430+5
превращается в
NaCl+UCl4
Эвтектика
KCl-K2UCle [25%
(мол.) UC14];
K2UCle—UC14
[53% (мол.) UC14]
2Na2UCle—UC14
[50% (мол.) UC14]
?
?
?
Компоненты
нерастворимы в жидкой
фазе; слабо
растворимы в твердой
фазе
Т. пл.
эвтектики,
°С
560+5;
350+5
370+5
410 и
430+5
485+5
515 и
425+5
а) Объяснить расхождение между этим значением и приведенным выше на стр. 391 невозможно.
б) Группа, работавшая в Эймсе, сообщает, что температура плавления системы UCI3—ЬТС14„
содержащей 11,7% (мол.) трихлорида урана, лежит иа 14° ниже точки плавления тетрахлорнда.
ПЕНТАХЛОРИД УРАНА
Пентахлорид урана был открыт Роско в 1874 году в качестве побочного
продукта при получении тетрахлорида. Впоследствии многие исследователи
отметили образование высших хлоридов урана в том случае, когда
тетрахлорид в процессе получения вводится в соприкосновение с хлором при
повышенных температурах. Однако этим побочным продуктам уделялось мало внимания.
Руфф и Хайнцельман [159] безуспешно пытались использовать пентахлорид
в качестве промежуточного продукта при получении гексафторида урана;
однако в общем трудности в обращении с пентахлоридом препятствовали
дальнейшему исследованию этого соединения.
Интерес к нему снова возник в 1943 г., когда было найдено, что этот
хлорид подвергается термическому диспропорционированию на тетрахлорид
и ранее неизвестный гексахлорид [160].
2UCl5-*UCl4 + UCle. (58)
Для анализа пентахлорид обычно подвергают гидролизу и определяют
уран (IV) и уран (VI) в образующемся растворе. Если в анализируемой пробе
присутствуют UOCl4 или уранилхлорид, значения, получающиеся для
содержания урана (VI), оказываются завышенными.
Глава 14. Соединения урана с хлором
393
Получение пентахлорид а. Для получения пентахлорида
имеются два практических метода. Первый состоит в обработке низших
хлоридов урана газообразным хлором при высоких температурах; второй,
основанный на работе Майкла и Мэрфи [63], заключается в хлорировании трехокиси
урана четыреххлористым углеродом в жидкой фазе.
Реакция тетрахлорида урана с парами четыреххлористого углерода.
Действием хлора при 520—555° тетрахлорид может быть превращен в пентахлорид
[161]:
2UC14 + CI2->2UC15. (59)
Предложено добавлять к хлору небольшие количества четыреххлористого
углерода, однако это, повидимому, не имеет особого значения, особенно если
исходный материал (тетрахлорид урана) достаточно чист [162].
Отношение CI : U в продукте реакции (59) зависит от скорости охлаждения
и может доходить до 5,5, что указывает на значительно большее содержание
хлора, чем это соответствует формуле UC15. Сделано предположение, что при
реакции образуются соединения #UCl6-£/UCl4, причем отношение х : у может
принимать значения больше единицы. Однако более вероятно, что в этих
промежуточных соединениях^ так же как и в пентахлориде, все атомы урана
равнозначны и несут одинаковый заряд [ср. структуры фторидов урана (гл. 12
и 13) и U205 (гл. 11)].
Смеси тетра- и пентахлорида могут быть превращены в чистый
пентахлорид действием жидкого хлора при повышенных температуре и давлении
[159]. При пропускании хлора через суспензию тетрахлорида в кипящем
четыреххлористом углероде (76°) пентахлорида не образуется [6].
Сделаны попытки получить пентахлорид введением двуокиси урана в
реакцию с парами четыреххлористого углерода в токе воздуха при температуре
550°, однако анализ показал, что получающийся продукт содержит кислород
(уранилхлорид или UOCl4) [163]. В пользу существования достаточно
устойчивого UOCl4 говорят результаты опытов по хлорированию уранилхлорида
четыреххлористым углеродом при низких температурах (ср. [164]). Этот вопрос
будет рассмотрен подробнее ниже.
Реакция между окислами урана и четыреххлористым углеродом в жидкой
фазе- Как уже указывалось, в разделе о получении тетрахлорида, галогениро-
вание окислов урана четыреххлористым углеродом в жидкой фазе может
быть проведено таким образом, чтобы получить или тетрахлорид, или
пентахлорид. Здесь мы рассмотрим специфические условия, при которых образуется
пентахлорид.
Впервые реакция окислов урана с жидким четыреххлористым углеродом
была изучена Майклом и Мэрфи [63], которые установили, что при 250° трех-
окись или закись-окись урана быстро взаимодействует с этим реагентом
в запаянной трубке с образованием пентахлорида урана (ср. [2]). Недавно
исследовано также поведение кипящего четыреххлористого углерода при
атмосферном давлении. В этих условиях реакция идет гораздо медленнее:
после 24-часового кипячения с обратным холодильником превращение
составляло лишь 10—30%. В случае предварительной добавки даже небольшого
количества пентахлорида урана требуется очень небольшое время нагрева
трехокиси или закиси-окиси, чтобы получить полностью растворимые в воде
продукты. Однако эти продукты представляют собой главным образом окси-
хлориды, и их превращение в пентахлорид протекает лишь очень медленно.
Если взять большой избыток четыреххлористого углерода, то после
многочасового кипячения с трехокисью урана последняя полностью превращается
в пентахлорид; при этом требуется заранее добавить большие количества
последнего. Через кипящую смесь необходимо все время пропускать хлор
[5,6,165]. Трудность проведения реакции в жидкой фазе при атмосферном
394 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
давлении состоит еще в том, что малейшие следы кислоты, повидимому, очень
сильно подавляют реакцию [166].
Механизм превращения [167, 168]. При интерпретации механизма реакции
галогенирования окислов урана четыреххлористым углеродом в жидкой фазе
должны быть объяснены следующие три факта: 1) реакция может быть
направлена в сторону образования тетра-, пента- или гексахлорида урана; 2) пента-
хлорид действует как катализатор; 3) в продуктах реакции содержатся хлор
и фосген.
Суммарные реакции образования пентахлорида при взаимодействии
трехокиси или закиси-окиси урана с четыреххлористым углеродом с выделением
хлора и фосгена могут быть представлены следующими уравнениями:
2U03 + 6CCI4 -» 2UC15 + 6COCI2 + С12; (38)
2U308+ 16СС14 —> 6UC15+ 16СОС12 + С12. (60)
При более высоких температурах пентахлорид разлагается на тетрахлорид
и хлор:
2UCI5^ 2UC14 + CI2. (61)
При этих условиях продукт реакции может содержать много тетрахлорида
или даже представлять чистый тетрахлорид (см. стр. 373). При добавлении
в систему избытка хлора равновесие в уравнении (61) смещается влево и, кроме
того, может идти дальнейшее хлорирование, приводящее к образованию
гексахлорида урана:
2UC15 + C12 ^ 2UC16. (62)
Даже если не добавлять избытка хлора и удалять растворитель при
температуре ниже его температуры кипения, в получающемся пентахлориде урана
всегда присутствует несколько процентов гексахлорида. Это может быть
объяснено тем, что в результате протекания реакции (61) выделяется достаточное
количество хлора для образования небольшого количества гексахлорида-по
реакции (62). Равным образом, это может быть просто результатом прямого
диспропорционирования двух молекул пентахлорида на тетра- и гексахлорид.
По уравнениям (38) и (60) предполагается, что первичным продуктом
реакции является пентахлорид. Однако более вероятно предположение, что
первичный продукт (по крайней мере при трехокиси урана) представляет собой
гексахлорид:
и03 + ЗСС14 -► UC]6 + ЗСОС12. (63)
В Браунском университете было проведено подробное исследование с целью
выяснения механизма автокаталитического действия пентахлорида на реакцию
трехокиси урана с жидким четыреххлористым углеродом. Одни эти вещества
заметно не реагируют друге другом при температуре 65° даже при выдерживании
в течение 10час, но при добавлении пентахлорида урана (2UC15: 1U03) за 3 часа
образуется растворимый в воде продукт. Исследование продуктов реакции,
выделенных через промежутки времени от 2 до 16 час, указывает на
медленное, постепенное хлорирование урана. Полностью растворимое в воде вещество,
полученное по удалении четыреххлористого углерода после 15 час
взаимодействия, было разделено на четыре фракции на основании убывающей
растворимости в четыреххлористом углероде; каждую фракцию анализировали на
отношение CI : U и содержание кислорода (при возгонке в вакууме при 750°
получается остаток двуокиси урана, количество которого пропорционально
содержанию кислорода в исследуемой фракции). Результаты анализов довольно
убедительно показали, что растворимое в воде вещество является смесью UC15
Глава 14. Соединения урана с хлором
395
(растворимого в четыреххлористом углероде), UOCl4 (немного растворимого),
UOCl3 (еще менее растворимого) и U02C12 (нерастворимого).
Установив, таким образом, промежуточное образование оксихлоридов,
можно объяснить автокаталитическое действие пентахлорида урана, если
предположить, что сначала реакция идет между трехокисью урана и пентахло-
ридом с образованием оксихлоридов. Последние подвергаются затем
хлорированию четыреххлористым углеродом, которое происходит легче, чем
хлорирование трехокиси урана, Вероятным механизмом первой стадии процесса
является следующий:
U08 + UC]5 ->UOCl3+U02Cl2 (64)
U02C12 + UC15 -> UOCl3 + UOCl4 (65)
1Ю8 + 2UC15 -> 2U0C13 + UOCl4 (66)
Дальнейшее хлорирование промежуточных оксихлоридов может протекать
следующим образом:
2U0C13 + 2СС14 _> 2UC16 + 2СОС12; (67)
UOCI4 + СС14 -» UC16 + СОС12. (68)
Суммарная реакция может быть представлена следующим уравнением:
U03 + 2UC15 + ЗСС14 -+ 2UC15 + UC16 + ЗСОС12. (69)
Таким образом, на каждые 2 моля добавленного пентахлорида в конечном
продукте должен содержаться 1 моль гексахлорида, если только условия
таковы, что последний не разлагается на пентахлорид и хлор. Методика
получения пентахлорида обычно обеспечивает полное разложение гексахлорида
(или предшествующего ему UOCl4). Условия получения гексахлорида в
качестве конечного продукта будут рассмотрены ниже. Приведенный механизм
реакции объясняет, почему для полного превращения с максимально
возможной скоростью необходимо соотношение 2UC15:1U03.
Термическая устойчивость вышеупомянутых промежуточных
оксихлоридов изучена только с качественной стороны. Найдено, что при нагревании
смеси растворимых в воде промежуточных продуктов реакции до 130° легко
выделяется хлор, и даже при столь низкой температуре, как 88°, были получены
заметные количества хлора. Ввиду того, что из пентахлорида, так же как и из
гексахлорида, при 130° получаются лишь следы хлора (при атмосферном давлении),
источником хлора может являться только разложение промежуточных
оксихлоридов. Однако фракция 1ЮС13, полученная из сырой смеси ступенчатым
растворением в четыреххлористом углероде, оказалась необычайно
устойчивой; ее разложение наблюдалось только при температуре выше 375°.
Разложение при низкой температуре объясняется поэтому лучше всего реакцией
UOCl4 -^ UOCI3 +± С12. (70)
Благодаря высокой термической устойчивости 1ЮС13 разложение UOCl4
останавливается на стадии образования UOCl3 и не доходит до UOCl2.
Хлорирование трехокиси урана с помощью жидкого тионилхлорида
и пентахлорида урана, вероятно, протекает таким же путем, как и реакция
с четыреххлористым углеродом.
Физические свойства пентахлорида урана. Физические
свойства этого соединения изучены мало. Приготовленное хлорированием тетрахло-
рида, оно обычно представляет собой красно-коричневый микрокристаллический
порошок. При высоком давлении в автоклаве пентахлорид получается в виде
красновато-черных кристаллов с металлическим блеском. Были сделаны
396 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
попытки перекристаллизовать в автоклаве красно-коричневые микрокристаллы
пентахлорида из раствора в четыреххлористом углероде, однако эти попытки
не увенчались успехом [169].
Плотность. Определения плотности пентахлорида прямым взвешиванием
в бензоле дали значение 3,81 г/см3 [170]. Насыпной вес пентахлорида,
полученного в актоклаве, равен 1,95 г/см3 [171].
Температура плавления и летучесть. Пентахлорид подвергается и
термическому разложению и диспропорционированию, а поэтому точные значения
его температуры плавления или летучести отсутствуют. Упругость пара его
при 50° была определена в 10"'7 мм рт. ст.; ввиду того что он подвержен
диспропорционированию, высказываются сомнения в возможности его
существования в виде пара при температурах выше 60° [172]. Разделение пента- и гек-
сахлоридов возгонкой осуществить трудно или даже невозможно, что
принимается за доказательство довольно высокой летучести пентахлорида.
Последний может быть перегнан в атмосфере хлора с небольшой примесью четырех-
хлористого углерода, однако сублимат оказывается обогащенным гексахло-
ридом [173].
Молекулярный вес [174]. Молекулярный вес пентахлорида урана был
определен эбулиоскопически в растворе четыреххлор истого углерода и
оказался очень близким к вычисленному для димерной молекулы (UC15)2. Таким
образом, можно предполагать, что молекулы пентахлорида урана существуют
в растворе в виде U2Cllrt.
Термохимия [29]. Теплота образования пентахлорида урана была
определена в Беркли измерением разности теплот растворения тетра- и пентахлорида
в воде, содержащей избыток хлорида железа (III):
UC14 тв + 2FeCl3 щ + 2Н2Ож -> Ш2С!2а, + 4НС1ад + 2FeCI2 aq, (71)
А# = —24,1 ккал;
UC15TB + FeCl3 aq + 2Н2Ож -► U02C12 aq + 4НС1а, + FeCl2 ^ (72)
А Н — —33,9 ккал;
FeCl2 aq + у Cl2 aq -> FeCl3 aqy (73)
ДЯ= -21,0 ккал.
Отсюда
UCl4TB + yCI2
aq * Г, тв> (74а)
Д#= — 71,2 ккал-моль'1.
Если использовать еще уравнение (42), получим
итв + 4с12газ —UC15TB, (746)
А#273= - 262,1 ± 0,6 ккал - моль"1.
Энтропия пентахлорида была определена в i?298 =^ 57,0 энтропийных единиц;
в этом случае Д£ для уравнения (74а) равно —88,3 энтропийной единицы и
AF°298= -235,7±2,0 ккал*мольх [175].
Диэлектрическая постоянная. Диэлектрическая постоянная твердого
пентахлорида равна 1,9 и в заметной степени с частотой не изменяется [176].
Спектр поглощения растворов пентахлорида урана. Получен спектр
поглощения растворов пентахлорида в четыреххлор истом углероде и тионилхло-
риде [177]. Пентахлорид имеет довольно широкий максимум при 850 т\*. в обоих
растворителях и признаки еще одного максимума при 650 т\х в четы-
Глава 14. Соединения урана с хлором
397
реххлористом углероде и при 950 m4u в тионилхлориде. В спектре гексахлорида
обнаружены [177] максимумы при тех же самых частотах, в спектре же тетра-
хлорида поглощения в этой области спектра не наблюдается. Хотя спектры
пента- и гексахлорида определенно отличаются друг от друга, на основании
того факта, что максимумы в спектрах поглощения этих соединений
наблюдаются примерно при одних и тех же частотах, сделан вывод, что пентахлорид
урана должен быть представлен как UC14-UC16. Доказательство в пользу этого
заключения довольно шатко. Однако окончательное решение должно быть
отложено до тех пор, пока не будет снята рентгенограмма твердого
пентахлорида.
Химические свойства пентахлорида урана.
Отношение к растворителям. Пентахлорид урана очень гигроскопичен. Он
чувствителен к влаге при парциальном давлении паров воды в 0,007 мм рт. ст. и выше
[178]. Вода немедленно разлагает его.
2UC15 + 2Н20 -> UC14 + U02C12 + 4HC1. (75)
Во избежание потери хлористого водорода в том случае, когда эта реакция
используется для аналитических целей, рекомендуется вносить пентахлорид
в разбавленную серную кислоту, охлажденную до—78°, и затем давать
реагентам постепенно нагреваться [179].
Изучена растворимость пентахлорида в большом числе органических
растворителей. Данные по растворимости в четырех хлор истом углероде в
зависимости от температуры представлены в табл. 175. Они показывают, что
Таблица J 75
РАСТВОРИМОСТЬ ПЕНТАХЛОРИДА УРАНА
В ЧЕТЫРЕХХЛОРИСТОМ УГЛЕРОДЕ [34, 165, 168]
Температура,
°С
-20
-16
0,0
22,8
25,0
70
160
Количество
UCU на 100 г
СС14, г
0,29
0,45
0,94
1,04
5,3
20
(ориентировочно)
UCU на 100 г
СС14 + С12а), г
0,26
—
0,43
—
0,82
а) 87,5% СС14+12,5% С12.
добавление хлора уменьшает растворимость пентахлорида урана в этом
растворителе.
Несколько различных наблюдений по растворимости представлено
в табл. 176.
Диметиловый эфир энергично реагирует с пентахлоридом с образованием
вещества оранжевого цвета, растворимого в воде. Диэтиловый эфир, ацетон,
этилацетат, формамид, диоксан и спирты также немедленно реагируют с ним;
хлороформ, ацетофенон и хлорбензол реагируют медленнее. С топропиловым
эфиром реакция не идет [8, 168]. Пентахлорид урана хорошо растворим в
сероуглероде и тионилхлориде.
398 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Таблица 176
РАСТВОРИМОСТЬ ПЕНТАХЛОРИДА УРАНА В РАЗЛИЧНЫХ
РАСТВОРИТЕЛЯХ [180]
Растворитель
Тионилхлорид
Пиридин
Бензотрихлорид
Р,Р',-Дихлорэтнловый
эфир
Тетралнн
Тетрахлорэтан
Тетрахлорэтилен
Бензол, ксилол
Фреон 11, фреон 21
Фреон 113
Растворимость
Растворим
Мало растворим
Растворим
Немного растворим
Мало растворим
Растворим
Растворим: 9% при 22°,
25% при 120°
Нерастворим
Быстрая реакция с
выделением С12
Растворим
Примечание
Красновато-коричневый
раствор
Повндимому, образуется
комплексное
соединение
При стоянии или
кипячении с обратным
холодильником
образуется темное твердое
вещество
Зеленый раствор
Прн кипячении с
обратным холодильником
выпадает осадок
Темнокорнчневый раствор
Химически не реагирует
При кипячении
образуется осадок
Реакции окисления. Пентахлорид реагирует с кислородом с образованием
смеси оксихлоридов. При взаимодействии с хлором при высокой температуре
пентахлорид частично превращается в гексахлорид:
2UC15 + C12 ^2UCIe. (62)
Реакция обратима, однако обработкой пентахлорида хлором в парах четырех -
хлористого углерода можно достигнуть практически полного его превращения
в гексахлорид [7]. Эта реакция будет обсуждаться ниже. Газообразный фтор
превращает пентахлорид урана в гексафторид.
Реакция обменного разложения. При действии жидкого безводного
фтористого водорода пентахлорид превращается в пентафторид (см. гл. 12):
UC15 + 5HF->UF5 + 5HC1. (76)
Реакции восстановления. Металлический натрий восстанавливает
пентахлорид до металлического урана. Однако пентахлорид урана не является
особенно подходящим исходным веществом для получения металлического урана
[181]. Хлороформ восстанавливает пентахлорид до тетрахлорида [182].
Восстановление может быть проведено в автоклаве при 140°, однако эта реакция
не имеет большого практического значения, так как получающийся тетра-
хлорид загрязнен гексахлорэтаном. Пентахлорид урана может быть
дехлорирован кипячением с обратным холодильником в монохлориде серы, хлориде
Глава 14. Соединения урана с хлором
399
олова (IV), гексахлорпропене и т. п., но очистить получающийся продукт от
растворителя обычно трудно.
Диспропорционирование и термическое разложение- При нагревании
пентахлорид подвергается распаду [54]:
2UC15^2UC14 + C12. (61)
Некоторое разложение наблюдается уже при температуре ниже 100°; при 250°
оно становится быстрым, причем его скорость примерно одинакова в атмосфере
хлора и в атмосфере чистого азота [178], что указывает на то, что реакция
заметно необратима при этих условиях. Сообщается, что при 75° пентахлорид
выделяет заметные количества хлора и что скорость разложения быстро
увеличивается при 175° [183].
Термическая устойчивость пентахлорида изучалась также Мартином
и Элдау [184]. Эти исследователи, очевидно, не знали о существовании гексахло-
рида; проверка их экспериментальной методики показывает, что результаты,
вероятно, относятся к смеси пента- и гексахлорида, а не к чистому пентахло-
риду. В Беркли была изучена кинетика процесса термического разложения
пентахлорида [172]. Эта реакция по отношению к пентахлориду, повидимому,
первого порядка. Константа скорости ее представляет величину порядка
1-Ю"3 мин."1 для интервала температур 100—152°; энергия активации была
вычислена в 28 ккал-моль'1.
Помимо термического разложения пентахлорид подвергается еще диспро-
порционированию при температуре 100—175° в высоком вакууме:
2UC15->UC14 + UC16. (58)
Однако эта реакция еще не дает нам права рассматривать пентахлорид
как UC14-UC]G. Весьма вероятно, что, как и в случае пентафторида, все атомы
урана равнозначны. Подробности термического диспропорционирования
пентахлорида на тетра- и гексахлорид будут рассмотрены ниже.
Комплексные соединения пентахлорида урана. Сообщается, что при
взаимодействии трехокиси урана с пентахлоридом фосфора при температуре 180—
190° в запаянной трубке образуется двойная соль UC15-PC15 [185]. После
удаления РОС13 эта двойная соль остается в виде желто-красной аморфной массы,
которая плавится без разложения. Водородом она восстанавливается до
хлористого водорода и фосфина; сплавление с хлоридом калия дает тетрахлорид
урана, пентахлорид фосфора и хлор. В воде осаждается фосфат урана (IV)
и образуется UOif. Желательно было бы более подробно изучить этот комплекс.
ГЕКСАХЛОРИД УРАНА
До получения гексахлорида урана единственным известным
бескислородным соединением шестивалентного урана был гексафторид. Гексахлорид урана
был открыт в результате сделанного наблюдения, что чистый в первоначальном
виде пентахлорид не может быть полностью возогнан, если температура
недостаточно высока для того, чтобы возогнался также и тетрахлорид. При
возгонке пентахлорида при температуре ниже 550° всегда получался остаток,
близкий по составу к тетрахлориду [160]. Сублимат можно было уже вновь
возогнать в вакууме ил и в атмосфере азота или хлора без вторичного образования
остатка. Анализ сублимата показал, что он представляет собой
гексахлорид; было совершенно очевидно, что он образовался в результате
диспропорционирования UC15:
2UC15->UC14 + UC16. (58)
Довольно неожиданная устойчивость гексахлорида в сочетании сего заметной
летучестью вызвала значительный интерес к этому соединению. Сталя усиленно
400 Часть /V. Галогениды, оксигалогениды, боргидриды и карбонил урана
изучаться методы получения гексахлорида, и были сделаны известные
успехи в изучении физических и химических свойств.
Получение гексахлорида урана. Для получения
гексахлорида известны три метода: 1. Вышеупомянутое диспропорционирование
пентахлорида в вакууме. Максимальное количество гексахлорида,
получающееся таким образом, составляет 54,6% отвеса пентахлорида. 2. Введениететра-
хлорида или пентахлорида в реакцию с хлором при повышенных
температурах. Этим путем может быть получен продукт с отношением CI : U, равным
5,8 : 1. Он может быть использован для получения чистого гексахлорида с
помощью фракционирования. 3. Обработка низшего хлорида (обычно
пентахлорида) или трехокиси урана смесью хлора и четыреххлористого углерода в
жидкой фазе. Последний метод также не дает сразу чистого гексахлорида.
Необходима дальнейшая обработка для удаления пентахлорида,
примешанного к продукту.
Получение гексахлорида урана диспропорционированием UC15 [11].
Получение гексахлорида возгонкой пентахлорида в вакууме может быть проведено в
сосуде из стекла или нержавеющей стали, снабженном конденсатором,
охлаждаемым твердой углекислотой. Чистота образующегося гексахлорида в
значительной мере зависит от состава исходного вещества. Чем выше отношение CI : U
в последнем, тем меньше фракционированных возгонок требуется для
получения чистого гексахлорида [8, 186]. Необходимость многократного
фракционирования вытекает из того факта, что пентахлорид сам по себе обладает заметной
упругостью пара [187—190].
Различные исследователи использовали для проведения диспропорциони-
рования температуры в интервале от 80 до 240° [171]. Хотя скорость возгонки
растет с увеличением температуры исходного вещества, все же лучше избегать
температур, превышающих 150°, чтобы свести к минимуму одновременную
возгонку пентахлорида. Диспропорционирование лучше всего проводить при
температуре 120—150° и давлении порядка Ы0"4 мм рт. ст. [191]. Чем ближе
состав исходного вещества к пентахлориду, тем ниже должна быть
применяемая температура, чтобы избежать потерь пентахлорида. Низкое давление
способствует диспропорционированию, а поэтому существенно важно создание
хорошего вакуума. Обязательно применение насосной системы высокой
мощности, чтобы обеспечить высокий вакуум даже в условиях непрерывного
выделения газов из реакционной массы.
Получение смесей пента- и гексахлорида урана высокотемпературным
хлорированием в газообразной фазе. При действии хлора на тетрахлорид при
повышенных температурах образуются смеси пентахлорида и гексахлорида:
UC14 + IC12-*UC15 (29)
UCl5+iC!2^UCle (62)
UCI4 + Cl2->UCle. (77)
Эти реакции протекают при температуре выше 350°; состав образующегося
продукта в значительной степени зависит от скорости, с которой охлаждаются
продукты реакции [178]. Чем больше эта скорость, тем выше содержание
гексахлорида. При 350° отношение CI : U в получающемся продукте выше, чем при
450° [192]. Опыты показали, что предлагаемое иногда добавление паров
четыреххлористого углерода к хлору не имеет особенно существенного значения.
При указанных выше температурах может быть получен продукт, содержащий
30% гекса- и 70% пентахлорида.
Глава 14. Соединения урана с хлором
401
Очистка гексахлорида урана. Чистый безводный гексахлорид может быть
возогнан при температуре 75—100° в вакууме порядка 1 • \0~*мм рт. ст. Обычно
гексахлорид очищают многократной возгонкой в вакууме, однако он может
быть очищен и перегонкой в токе инертного газа под уменьшенным давлением.
Продукт, получающийся возгонкой гексахлорида в токе аргона при давлении
1—2 см рт. ст., является кристаллическим, и, по имеющимся данным, он более
чист, чем получаемый возгонкой в вакууме [76J. Возгонка же в хлоре или азоте,
повидимому, неудовлетворительна. В случае применения хлора абсорбируются
большие количества его, которые не могут быть вытеснены даже путем
продолжительного пропускания инертного газа над продуктом [193]. Было изучено
также влияние следов четыреххлористого углерода на процесс возгонки
гексахлорида урана [194]. После растворения в четыреххлористом углероде или
выдерживания в его парах гексахлорид никогда не возгоняется полностью;
всегда получается остаток, который не может быть возогнан при 100°.
Физические свойства гексахлорида урана.
Гексахлорид урана образует прекрасные черные или темнозеленые кристаллы.
Тот или иной внешний вид зависит от метода очистки и от скорости роста
кристаллов.
Температура плавления [195]. Несмотря на термическую неустойчивость
гексахлорида, оказалось возможным определить его температуру плавления.
Измерение было выполнено путем погружения тонкостенных трубочек с гекса-
хлоридом в масляную баню, заранее нагретую до различных температур.
При атмосферном давлении и 177,5±2,5° мгновенно образуется жидкая
фаза. При этой температуре равновесное давление хлора, вызванное
разложением,
UC16-^UCI4 + C12, (78)
должно быть достаточно велико, о чем можно судить по быстрому исчезновению
жидкого гексахлорида.
Кристаллическая структура и плотность [136, 196, 197].
Кристаллический гексахлорид был изучен рентгенографически; было найдено, что он
имеет гексагональную элементарную ячейку с константами
а^ 10,90 ± 0,02 А; а3 = 6,03±0,01 А.
Элементарная ячейка содержит три молекулы гексахлорида урана;
пространственная группа C3m(Dld)-
Положения атомов следующие:
lUj в (0 0 0),
2U„ в ±(уу*), гдегЦ.
Имеются три группы атомов хлора в положениях ±(x2xz) ( Txxz) (xxz) с
параметрами:
X 2
Cli 0,10 0,25
С1ц 0,43 0,25
Clin 0,77 0,25
Кристаллы гексахлорида обладают типичной молекулярной
пространственной решеткой, построенной из отдельных молекул гексахлорида
что вполне совместимо с наблюдающейся высокой летучестью этого соединения.
Шесть атомов хлора вокруг каждого атома урана образуют почти правильный
октаэдр с расстоянием U—С1, равным 2,42А. Поскольку нормальный
эффективный радиус атома хлора равен 0,99А, эффективный радиус атома урана
26 Химия урана
402 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
составляет 1,43А. Наименьшее расстояние между атомами хлора из разных
молекул гексахлорида равно 3,85А.
Плотность, рассчитанная из рентгенографических данных, равна 3,59 г/см*.
Прямым взвешиванием в бензоле найдено значение 1,56 г/см3 [1861, однако
измерения в тетрагидронафталине дали величину 3,36±0,17 г/см? [198].
Насыпной вес равен примерно 1,75 г/см? [199].
Давление пара. Давление пара гексахлорида определено независимо двумя
методами. Полученные результаты плохо совпадают. Одна группа измерений
была выполнена с цельностеклянным эффузионным манометром [200].
Давление пара и свободная энергия возгонки могут быть выражены с точностью до
1% или даже большей следующими уравнениями:
lgP:
2422
f 6,6337 (0-200°),
Д#ВОзг= 11120 кал-моль \
A F0 = 17500 + 36,85 Tig Г- 129,0 Т.
(79)
(80)
(81)
В табл. 177 приведены значения давления пара, рассчитанные из уравнения
(79).
Давление пара гексахлорида урана было также определено по методу
испарения с применением очищенного гелия [11, 201]. В результате была
введена поправка на выделение хлора, образующегося при разложении
гексахлорида. Полученные значения давления пара значительно ниже определенных
методом молекулярного истечения (табл. 178).
Т
а б л и ц а 177
ДАВЛЕНИЕ ПАРА ГЕКСАХЛОРИДА УРАНА
(МЕТОД МОЛЕКУЛЯРНОГО ИСТЕЧЕНИЯ)
Температура,
°С
0
20
40
60
80
100
Давление
пара,
ММ рТ. СТ.
0,005
0,023
0,08
0,22
0,59
1,35
Температура,
120
140
160
180
200
Давление
пара,
мм рт. ст
2,95
5,75
11,07
18,60
32,35
Та
i б л и ц а 178
ДАВЛЕНИЕ ПАРА
ГЕКСАХЛОРИДА УРАНА
(МЕТОД ИСПАРЕНИЯ)
Температура,
°С
75,5
98,5
120,0
138,0
Давление
пара,
мм рт. ст.
0,06
0,24
0,89
1,75
Экспериментальные данные, полученные методом испарения, могут быть
представлены следующим уравнением:
3788
1 О/ОО . л -л
lgP= -— + 9,52,
Д#во?,г= — 17300 кал-моль'1.
(82)
(83)
Значения, найденные методом испарения, вероятно, в принципе более точны,
чем найденные путем измерения молекулярного истечения. В последние не
внесены поправки на термическое разложение гексахлорида, что неизбежно
приводит к завышенным результатам. Однако исследователи, работавшие по
методу истечения, отметили лишь ничтожно малое разложение при
температурах ниже" 130°, в то время как другие, пользовавшиеся методом испарения,
нашли, что разложение имеет место практически при всех температурах
Таблица 179
УДЕЛЬНАЯ ТЕПЛОЕМКОСТЬ, ЭНТРОПИЯ И ЭНТАЛЬПИЯ ГЕКСАХЛОРИДА
УРАНАа) ОТ О ДО 350° К
Температура,
°К
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
105
110
115
120
125
130
135
140
145
150
155
160
165
170
175
180
185
190
195
200
205
210
215
220
225
230
ср,
межд. дж • моль 1 • град~1
0,0
0,793б>
6,01
15,66
25,18
34,35
43,51
52,18
60,33
67,73
74,53
80,74
86,37
91,46
96,01
100,2
104,6
108,6
112,0
115,3
118,4
121,3
124,2
126,9
129,6
132,1
134,4
136,8
139,0
141,0
143,0
144,8
146,5
148,1
149,6
151,1
152,5
153,8
155,1
156,4
157,6
158,7
159,8
160,9
161,9
163,0
164,0
\S\
межд. дж - м<ы(>~'1 • град~*
0,0
0,264
2,078
6,26
12,08
18,68
25,76
33,12
40,63
48,17
55,66
63,06
70,33
77,45
84,40
91,16
97,77
104,23
110,54
116,69
122,68
128,53
134,24
139,82
145,28
150,62
155,85
160,96
165,98
170,89
175,71
180,42
185,05
189,58
194,03
198,39
202,66
206,86
210,98
215,02
219,00
222,90
226,74
230,51
234,23
237,88
241,47
И~Е1'
' межд. дж • моль-1
0,0
0,991
15,52
68,78
171,0
319,9
514,6
754,1
1 036
1 356
1712
2 100
2518
2 963
3 432
3 923
4 435
4 968
5519
6 088
6 672
7 271
7 885
8513
9 154
9808
10 475
11 153
11842
12 542
13 252
13 972
14 700
15 437
16 181
16 933
17 692
18 458
19 230
20 009
20 794
21585
22 381
23 183
23 990
24 802
25 619
26*
404 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Продолжение табл. 179
Температура,
°К
235
240
245
250
255
260
235
270
275
280
285
290
295
298,16
300
305
310
315
320
325
330
335
340
345
350
ср.
межд. дж • моль~1 • град~1
164,9
165,9
166,8
167,7
168,6
169,5
170,4
171,2
172,1
172,9
173,7
174,5
175,2
175,7
176,0
176,7
177,5
178,2
178,9
179,7
180,5
181,3
182,1
182,9
183,8
S,
межд. дж*моль~1*град-1
245,01
248,49
251,92
255,30
258,62
261,91
265,14
268,34
271,48
274,59
277,66
280,69
283,68
285,54
286,63
289,54
292,42
295,27
298,08
300,86
303,61
306,33
309,02
311,69
314,33
Я-£*,
межд. дж-моль"1
26 442
27 269
28 100
28 936
29 777
30 622
31472
32 326
33 184
34 046
34 912
35 783
36 657
37 212
37 535
38 417
39 303
40 192
41084
41981
42 882
43 786
44 694
45 607
46 524
а) Использованный UCle содержал 6,3% иСЦ; были сделаны соответствующие поправки.
б) Экстраполировано с применением функции Дебая 58.17D (—-^—) .
Раньше чем сделать окончательные выводы, необходимо провести
сравнительные измерения над одной и той же пробой.
Термохимические данные. Удельная теплоемкость, энтропия и энтальпия
гексахлорида в интервале от 0 до 350° К были определены в Национальном
бюро стандартов [202] (табл. 179). Энтропия при 298° К определена в 68,28
энтропийной единицы.
Теплота образования гексахлорида была определена в радиационной
лаборатории Калифорнийского университета [29].
UC14TB f 2FeCI3 ац + 2Н2Ож -» U02C12 aq + 4НС1а, + 2FeCl2 aq, (84)
А Н = — 24,1 ккал;
UC16TB + 2Н2Ож -* U02C12 aq + 4НС1а„ (85)
Д#= —44,7 ккал;
2FeCl2 aq + Cl2 ra3 -» 2FeCl3, (86)
Д#= -42,0 ккал.
Глава 14. Соединения урана с хлором
405
(87)
Отсюда
UTB + оЫ2 газ > UCJ6 TB,
АЯ273= —272,3 ± 0,7 ккал-моль'1,
&F°29B= —241,4 ± 1,0 ккал-момг1.
Оптические свойства [203, 204]. Изучение спектра поглощения паров
гексахлорида представляет трудности ввиду низкого давления пара. Спектр
поглощения был исследован при температурах 76,5 и 100° с применением трубки
7,5,
7000 6000 5000
Длина волны, А
4000
3000
Рис. 58. Спектр поглощения раствора гексахлорида урана в
перфтор гептане.
длиной 50 см. При этом была обнаружена слабая непрерывная полоса
поглощения при 20 000 см'1. Гораздо более интенсивная полоса наблюдалась при
27 000 см~1г, она, очевидно, соответствует полосе поглощения гексафторида
урана. Был также изучен спектр поглощения пленок твердого гексахлорида,
снятый при 77°К. Как и следовало ожидать, при низкой температуре полосатая
структура более четкая. При 20 000 см'1 был обнаружен ряд довольно
размытых полос; ниже 17 000 см'1 полосы становились слабее, но делались более
четкими. Ультрафиолетовый спектр ниже 30 000 см'1 не был исследован.
На рис. 58 представлен спектр поглощения раствора гексахлорида в одном
из фторуглеродов, качественно напоминающий спектр поглощения этого
хлорида в парах.
Химические свойства гексахлорида урана. О хи:
мических свойствах гексахлорида известно мало. Он крайне неустойчив в
присутствии влажного воздуха, поэтому манипуляции с ним должны проводиться
в вакуум-аппарате или сухой камере. С водой гексахлорид бурно реагирует
с образованием уранилхлорида.
Растворимость. Гексахлорид урана растворим в четыреххлористом
углероде с образованием устойчивого коричневого раствора. Определена
растворимость его в этом и в нескольких других растворителях (табл. 180).
Повидимому, гексахлорид реагирует с тетрахлорэтиленом [180] и
нафтеновыми углеводородами [203]. Во фторуглероде QF^ и изобутилбромиде он
немного растворим.
Термическое разложение. Точных данных о термическом разложения
гексахлорида урана в настоящее время привести нельзя. Вероятно, он устойчив
406 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Таблица 180
РАСТВОРИМОСТЬ ГЕКСАХЛОРИДА УРАНА
В РАЗЛИЧНЫХ РАСТВОРИТЕЛЯХ [168]
Растворитель
СС14
СС14
ecu
6,6% С12—93,4% СС14 . . .
!2,5о/0С12—87,5о/0 СС14 . . .
12,5% С12—87,5% СС14 . . .
Бензол
СН3С1
Фреон 113
Температура,
°с
-18
0
20
-20
-20
0
-33
80
j -24
1 45
На 100 г
раствора, г
2,64
4,9
7,8
2,4
2,23
3,98
2,20
Нерастворим
1 1>16а>
1,83а)
а) Заметной реакции между раст ворителем и гексахлоридом ие
наблюдалось.
до температур 120—150°. Изучена кинетика процесса термического
разложения гексахлорида, оказавшаяся довольно сложной [172]. Кривая скорость
распада—время имеет точку перегиба; это принимается как доказательство
фазового превращения одной кристаллической формы в другую, более
устойчивую. Наблюдался вполне определенный индукционный период, который
с ростом температуры от 140 до 170° уменьшался с 30 до 5 мин. Длительность
индукционного периода, весьма возможно, зависит от всей предыдущей истории
образца. На скорость реакции разложения гексахлорида заметно влияет также
наличие катализатора; активными катализаторами для термического
разложения гексахлорида являются нержавеющая сталь и монель-металл;
позолоченная или никелированная медная проволока является совсем инертной.
Скорость разложения гексахлорида в газообразной фазе ничтожно мала
по сравнению со скоростью в твердой фазе. В интервале температур 130—180°
значение константы скорости k для реакции
2UCL
2UC15TB + C1
2 газ
(88)
лежит между МО"3 и 1 • 10"1 мин."1 Эти широкие пределы объясняются
отсутствием точных данных о давлении пара гексахлорида. Для реакции (88)
требуется энергия активации около 40 ккал-моль1.
Реакция гексахлорида урана с фтористым водородом [188, 189]. При
обработке гексахлорида очищенным безводным фтористым водородом при
комнатной температуре в реакторе с платиновой обкладкой получается пентафторид
урана, а не гексафторид:
2UC16 + 10HF -> 2UF5 + 10НС1 + Cl2
(89)
ЛИТЕРАТУРА
Pel i got E., Ann. chim. et phys., [3] 5, 5—20 (1842).
Kraus C. A., Report [A]M-I060, Nov. 2, 1942 (Brown 1).
Kraus C. A., Report CC-342, Nov. 15, 1942 (Brown 2).
Kraus C. A., Report CC-365, Nov. 17, 1942 (Brown 3).
Kraus C. A., Report A-505, Jan. 9, 1943 (Brown 4).
Kraus C. A., Report A-522, Feb. 1, 1943 (Brown 5).
Глава 14. Соединения урана с хлором
407
7. К г a u s С. A., Report A-726, June 1, 1943 (Brown 6).
8. J о h n s о п О., В u t 1 е г Т., Report CC-1496, May 11, 1944 (MP Ames 1).
9. Newton A. S., Johnson О., К а и t A., N о t t о r f R. W., Report CC-705,
June 7, 1943 (MP Ames 2).
10. В u t 1 e г Т., Report CC-1500, June 17, 1944 (MP Ames 3).
11. Johnson O., Butler Т., Newton A. S., The Preparation, Purification,
and Properties of Anhydrous Uranium Chlorides, in National Nuclear Energy Series,
Division VIII, vol. 6 (MP Ames 4).
12. R a m m e 1 s b e r g C, Pogg. Ann., 55, 322; 56, 129 (1842).
13. G a t e s J. W., A n d г e w s L. J., Report CD-477, Nov. 9, 1944 (CEW-TEC 1).
14. Y о u n g H. S., W a g n e г E. L.f M i 1 1 e r A. J., Report CD-2.350.5, Nov. 23,
1945 (CEW-TEC 2).
15. Young H. S., Preparation of Uranium Trichloride by Reduction of the
Tetrachloride with Metals, in National Nuclear Energy Series, Division VIII, vol. 6 (CEW-TEC
32).
16. J о h n s о n O., Report A-3190, Nov. 25, 1944 (MP Ames 5).
17. R о s e n h e i m A., L о e b e 1 H., Z. anorg. Chem., 57, 235 (1908).
18. Z i m m e г m a n n C, Ann., 213, 301, 304 (1882); Ber., 15, 849 (1882).
19. К г a u s C. A., Report CC-1715, July 6, 1944 (Brown 7).
20. Barkelew C. H., Report RL-4.6.906, Jan. 18, 1945 (UCRL 1).
21. Kraus C. A., Report [A]M-251, July 1, 1943 (Brown 8).
22. A 1 t m a n D., Report RL-4.6.156, June 11, 1943 (UCRL 2).
23. M а с W о о d G. L.Altman D., Report RL-4.7.600, Oct. 24, 1944 (UCRL 3).
24. Z а с h а г i a s e n W. H., Report CK-1487, Mar. 27, 1944 (MP Chicago 1).
25. Z а с h a r i a s e n W. H., J. Chem. Phys., 16, 254 (1948).
26. F e r g u s о n W, J., P r a t n e r J. L., Report A-3143, Nov. 18, 1944 (Natl. Bur.
Standards 1).
27. G i n n i ngs D. C, Cor rifcci n i R. J., Report A-4120, Nov. 20, 1946 (Natl. Bur.
Standards 6).
28. G u n n i n g s D. C.Corruccini R. J., J. Research Natl. Bur. Standards, 39,
309—316 (1947).
29. Barkelew С H., Report RL-4.6.929, Aug. 28, 1945 (UCRL 4).
30. В i 1 t z W., Fendius C, Z. anorg. u. allgem. Chem., 176, 49—63 (1928).
31. A 1 t m a n D., Report RL-4.6.276, Aug. 14, 1945 (UCRL 5).
32. Gregory N. W., Report RL-4.6.928, Aug. 24, 1945 (UCRL 6).
33. M а с W о о d G. E., Thermodynamic Properties of Some Uranium Compounds, in
National Nuclear Energy Series, Division VIII, vol. 6 (UCRL 7).
34. Kraus C. A., Report N-29, Apr. 15, 1943 (Brown 9).
35. A 1 t m a n D., Report RL-4.6.166, June 25, 1943 (UCRL 8).
36. J о h n s о n O., Butler Т., Report CC-1974, Nov. 18, 1944 (MP Ames 6).
37. С a 1 к i n s V. P., Report CC-1500, June 17, 1944 (MP Ames 7).
38. E p h r a i m F., M e z e n e r M., Helv. Chim. Acta, 16, 1259 (1933).
39. Sea b org С Т., Report N-930, Apr. 5, 1944 (MP Chicago 2).
40. J о h n s о n O., Report CC-1500, June 17, 1944 (MP Ames 8).
41. Edwards F., W а г f J. C, Report CC-1496, May 11, 1944 (MP Ames 9).
42. Kraus C. A., Report CC-1772, Feb. 1944 (Brown 10).
43. К a r 1 e J., Report CK-I512, Apr. 14, 1944 (MP Chicago 3).
44. Bichowsky F. R., R ossi ni F. D., The Thermochemistry of Chemical
Substances, Reinhold Publishing Corp., N. Y., 1936.
45. Kelly K. K., IX. The Entropies of Inorganic Substances. U. S. Bureau Mines Bull.,
434 (1941).
46. Yost D. M., Russell H., Systematic Inorganic Chemistry, Prentice-Hall, Inc.,
N. Y., 1944.
47. Wagner E . L., Report CD-0.350.9, Feb. 1946 (CEW-TEC 4).
48. Mc Bee E. Т., Report [A]M-2102, Mar. 21, 1945 (Purdue 1).
49. Davidson P. H., M u e 1 1 e г M. E., Report RL-4.6.250, Feb. 26, 1944(UCRL9).
50. Fisher A., Z. anorg. Chem., 81, 177, 190 (1913).
51. Voigt A., Biltz W., Z. anorg. u. allgem. Chem., 133, 277^305 (1924).
52. H о n i g s с h m i d O., S с h i 1 z W. E., Z. anorg. u. allgem. Chem., 170, 145—160
(1928).
53. Co 1 a n i A., Ann. chim. et phys. [8] 12, 66—80 (1907).
54. Roscoe H., Ber., 7, 1131—1133 (1874).
55. Biltz W., Fendius C, Z. anorg. u. allgem. Chem., 172, 386-391 (1928).
56. W a g n e г E. L., G о t t s с h a 1 1 H. A., Report CD-MPR,13, Feb. 1945 (CEW-
TEC 5).
57. S t e d m a n D. F., В г о w n A. G., Report A-7, Apr. 1, 1941 (Brown 11).
58. Parsons G. S., M с С a b e C. L., E n g 1 e G. F.,LarsonC. E., Reports
CD-564, Apr. 2, 1945; CD-541 and CD-5701, May, 1945; Parsons G. S., L a r-
son С. Е., Reports CD-5715, June 1945; CD-5718, June, 1945 (CEW-TEC 6).
408 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
59. Watts С. W., Bell С. A., J. Chem. Soc., 33, 442 (1878).
60. Demarcay E., Compt. rend., 104, 111 (1887).
61. Meyer L., Ber., 20, 681 (1887).
62. С a m b о u 1 i v e s P., Compt. rend., 150, 175 (1910).
63. M i с h a e 1 A., Murphy A., Jr., Am. Chem., J., 44, 365—384 (1910).
64. R e i b e r H. G., Y о u n g H. A., Report RL-4.6.54 (UCRL 16).
65. R ei b er H. G., Report RL-4.7.600, Sec. D, Oct. 24, 1944 (UCRL 14).
66. Steel e G., Report RL-4.6.113, Apr. 13, 1943 (UCRL 17).
67. R ei b er H. G., Report RL-4.6.70, Mar. 13, 1943 (UCRL 18).
68. R ei b er H. G., Report RL-4.6.105, Apr. 10, 1943 (UCRL 19).
69. R ei ber H. G., Report RL-4.6.67 (UCRL 20).
70. R ose nf el d S., Report RL-4.6.119, Aug. 28, 1944 (UCRL 21).
71. В a b с о с к S. H., Report RL-4.6.61, July 13, 1943 (UCRL 22).
72. Kraus С A., Report [A]M-885, Oct. 7, 1942 (Brown 14).
73. Kraus С A., Report CC-402, Dec. 1, 1942 (Brown 15).
74. Kraus C. A., Report A-1088, Nov. 3, 1943 (Brown 16).
75. Kraus С A., Report A-1089, Nov. 1, 1943 (Brown 17).
76. J о h n s о n O., Butler Т., Report CC-1524, Mar. 1944 (MP Ames 10).
77. Pitt B. M.,Curran W. F.,Schmitt J. M., Wagner E. L., Miller
A. J., Report CD-2.350.3, July, 1945 (CEW-TEC 9).
78. P a r s о n s G. S., M с С a b e C. L., E n g 1 e G. F., L а г s о n С. Е., Report
CD-5701, May 1945 (CEW-TEC 10).
79. Kamen M., Report RL-4.6.8, Nov. 30, 1942 (UCRL 24).
80. D e a n L. В., W a g n e r E. L.Miller A. J., Report CD-4107, June 12, 1945;
Wagner E. L., P i t t B. M.,Curran W. F., Schmitt J. M.,
Miller A. J., Report CD-2.350.1, July 10, 1945 (CEW-TEC 11).
81. Miller A. J., D e a n L. В., Preparation of Uranium Tetrachloride with Hexachlo-
ropropylene and Other Chlorinated Organic Compounds, in National Nuclear Energy
Series, Division VIII, vol. 6 (CEW-TEC 33).
82. Mc Bee E. Т., Report [A]M-2101, Feb. 21, 1945 (Purdue 2).
83. Wagner E. L., P i t t В. М., Report CD-MPR-14, Mar. 1945, reported by
Miller A. J., Y о u n g H. A., CEW-TEC, Part II, p. 12 (CEW-TEC 12).
84. G о t t s с h a 1 1 H. A., Manneschmidt J. F., Schmitt J. M.,
Wagner E. L., M i 1 1 e г A. J., Report CD-2. 350. 6, Jan. 29, 1946 (CEW-TEC 15).
85. McBee E. Т., Report [AlM-2113, Jan. 1946 (Purdue 3).
86. Pitt B. M., Wagner E. L., Miller A. J., Report CD-2.355.3, Jan.
14, 1946 (CEW-TEC 14).
87. M с В е е Е. Т., Report [AlM-2103, Mar. 1945 (Purdue 4).
88. W a g n e r E. L., Pitt B. M., D e a n L. В., M i 1 I e r A. J., Report CD-4106,
Apr. 1945 (CEW-TEC 13).
89. M с В e e E. Т., Evans L. R., Report A-2704, Dec. 14, 1945 (Purdue 5).
90. G a v 1 i n G., H u b b a r d J. V„ Report CD-5. 350.5, Dec. 12, 1945 (CEW-TEC 16).
91. Chauvenet E., Compt. rend., 152,87, 1250(1911).
92. R о s e n h e i m А., К e 1 m у М., Z. anorg. u. allgem. Chem., 206, 31, 32 (1932).
93. Wood T. K., Report [B]LRG-4, Dec. 22, 1942 (British 1).
94. McBee E. Т., Pea re e D. W., Mezey R., Report A-2706, Feb. 5, 1946
(Purdue 6).
95. St ere tt С. С, Van Wazer J. R., Repotr CD-3.360. 4, Sept. 21, 1945
(CEW-TEC 17).
96. Hubbard J. V., Perlmutter H. A., G о г e n H. L., С 1 e w e t t G. H.,
Report CD-5.-350.3, Aug. 28, 1945; L о w r i e R. S., Hubbard J. V., С 1 e-
w e t t G. H„ Report CD-5.350.6, Dec. 20, 1945; Perlmutter H. A.,Coobs
J. H., Lowrie R. S., Miller A. J., Report CD-2.350.9, Mar.12, 1946
(CEW-TEC 8).
97. M a t i g n о n С, В о u r i о n F., Ann. chim. et phys., [8] 5, 127—136 (1905).
98. L e 1 у О., Jr., Hamburger L., Z. anogr. Chem., 87, 209—228 (1914).
99. H u 1 s m a n n О., В i 1 t z W., Z. anogr. u. allgem. Chem., 219, 357—366 (1934).
100. Moore R. W., Trans. Am. Electrochem. Soc, 43, 317—323 (1923).
101. G о g g i n J. F., С г о m i n J. J., Fogg H. C, JamesC, Ind. Eng. Chem.,
18, 114—116 (1926).
102. H a w о r t h W., Peport B-20 (British 2).
103. R о d d e n С J., Report A-36, Mar. 1942 (Natl. Bur. Standards 2).
104. Jenkins F. A., Report RL-4.6.232, Dec. 15, 1943 (UCRL 26).
105. CalkinsV. P., LarsonC. E.f Report CD-0.350.4, Sept. 1945 (CEW-TEC 19).
106. Miller A. J., WagnerE. L, Report CD-0.100.25, Jan. 1946 (CEW-TEC 20).
107. Booth H. S., editor, Inorganic Syntheses, vol. I, McGraw-Hill Book Company
Inc., New York, 1939, p. 28.
108. Calkins V. P., Larson С. Е., Report CD-0.350.5, Sept. 1945 (CEW-
TEC 21).
Глава 14. Соединения урана с хлором
409
109. Wagner E. L., Gottschall H. A., Report CD-0.100.25, Jan. 1946 (CEW-
ТЕС 22).
ПО. Kohlschutter V/, Rossi M., Ber., 34, 1473(1900).
111. Giolitti R, AgamennoneG., Atti accad. nazl. Lincei, [5] 14, 165
(1905).
112. A 1 о у J., R о d i e г Е., Bull. soc. chim. France, [4] 31, 247 (1922).
113. Neher C, Lipkind H., Lyon W., Sleight W., Report CT-1780,
Aug. 9, 1944; Spedding F., Report CT-1780, July 10, 1944 (MP Ames 11).
114. Kleinheksel J., Kremers H., J. Am. Chem. Soc, 50, 959 (1928).
115. Gregory W. A., Grogg H. J., Grieger P. F., Gates J. W.,
Report CD-I.350.1, Aug. 1, 1945 (CEW-TEC 23).
116. Moissan H., Compt. rend., 122, 1092 (1896).
117. M о i s s a n H„ Bull. soc. chim. Paris., (3) 17, 266 (1897).
118. S p e d d i n g F., Report CC-1504, June 10, 1944 (MP Ames 12).
119. D о г о u g h G. D., Report RL-4.6.283, Aug. 18, 1944 (UCRL 27).
120. Wagner E. L., Pitt B. M., Report CD-MPR-13, Sec. Ill, Feb. 1945 (CEW-
TEC 24).
121. Burns R. E., DeVries Т., Report A-2714, Mar. 12, 1946 (Purdue 7).
122. M с В e e E. Т., D e V r i e s Т., R о t h г о с к G. M., Report A-2705, Dec. 14,
1944 (Purdue 8).
123. Sterett С С, Van Wazer J. R.t Report CD-GS-37, Apr. 26, 1945 (CEW-
TEC 26).
124. Wagner E. L., Grady H. F., Miller A. J., Report CD-2.350.4, Sept.
18, 1945 (CEW-TEC 27).
125. Young H. S., Grady H. F., Physical Constants of Uranium Tetrachloride,
in National Nuclear Energy Series, Division VIII, vol. 6 (CEW-TEC 35).
126. Butler Т., Newton A. S., Report CC-1500, June 17, 1944 (MP Ames 13).
127. Jenkins F. A., Report RL-4.6.3 (UCRL 32).
128. Polissar M. J., Report RL-4.6.36, Oct. 23, 1942 (UCRL 33).
129. A 1 t m a n D., Report RL-4.6.276., Aug. 14, 1944 (UCRL 34).
130. Gregory N. W., Report RL-4.6.923, Aug. 1, 1943 (UCRL 31).
431. С i n n i n g s D. С, С о г г и с с i n i R. J., Report A-3948, July 1946 (Natl.
Bur. Standards 3).
132. В u t 1 e г Т., N о t t о r f R. W., Report CC-1975, Sept. 10, 1944, p. 7 (MP Ames 14).
133. Johnson O., Butler Т., Report CC-1778, Aug. 18, 1944 (MP Ames 15).
134. Mueller M. E., Report RL-4.6.934, Sept. 18, 1945 (UCRL 35).
135. Mooney R. С L., Reports CP-1533, Mar. 31, 1944; CP-1507, Apr. 3, 1944 (MP
Chicago 4).
136. Z а с h a r i a s e n W. H., Report CC-2768, Mar. 12, 1945 (MP Chicago 5).
137. Mooney R. C. L., Acta Crystailographica, 2, 189—191 (1949).
138. Burford W. В., Ill, Report [A]M-2121, Feb. 1945 (Johns Hopkins 1).
139. Burford W: В., Ill, Report [A]M-2122, Mar. 1945 (Johns Hopkins 2).
140. Baenziger N. C, Rundle R. E., Report CC-1500, June 17, 1944 (MP
Ames 16).
141. Z i m m e г m a n n C, Ber., 14, 1934—1939 (1881).
142. Rugheimer L., Gonder L., Ann., 364, 45 (1908).
143. Ferguson W. J., P r a t h e г J. L.Scott R. В., Report A-1920, May 3,
1944 (Natl. Bur. Standards 4).
144. Reynolds F. L., Davidson P. H., Reports RL-4.6.231, Nov. 30, 1943;
RL-4.5.236, Dec. 28, 1943 (UCRL 36).
145. Howland J. J., Jr., Absorption Spectra of Uranium (III) and Uranium (IV) in
Molar Hydrochloric Acid, in National Nuclear Energy Series, Division VIII, vol. 6
(MP Chicago 6).
146. S u с к s m i t h W., Phil. Mag., [7] 14, 1121 (1932).
147. Hampe W., Chem. Ztg., 12, 106 (1888).
148. Mueller M. E., Report RL-4.6.191, Aug. 19, 1943 (UCRL 37).
149. Zimmermann C, Ann., 216, 9, 10 (1883).
150. Renz C, Z. anorg. Chem., 36, 110 (1903).
151. P i e г 1 ё С. A., J. Phys. Chem., 23, 541 (1919).
152. Calkins V. P., Reports CC-1496, May 11, CC-1500, June 17, 1944 (MP Ames 17),
153. Directorate of Tube Alloys, Report B-55, Mar. 1942 (British 3).
154. J oh n G. S., V a n W a zer J. R., Report CD-3.385.4, Aug. 22, 1945 (CEW-TEC 28).
155. Van Wazer J. R., J о h n G. S., J. Am. Chem. Soc, 70, 1207 (1948).
156. Booker J., DeHaan A., Report RL-4.6.6, Nov. 22, 1942 (UCRL 38).
157. Beck G., Z. anorg. u. allgem. Chem., 206, 416, 421 (1932).
158. Alloy J., Bull. soc. chim. Paris, [3] 21, 264 (1899).
159. Ruff О., Н ei n z e 1 m a n n A., Z. anorg. Chem., 72, 65 (1911).
160. Carter J. F., Jenkins F. A., Kamen M., Larson С. Е., Lipkind D,,
Weismann S., Report RL-4.6.13, Jan. 17, 1943 (UCRL 42).
410 Часть IV. Галогсниды, оксигалогениды, боргидриды и карбонил урана
161. Webb A. D.f Reports RL-4.6.102 and RL-4.6.87, Маг. 25, 1943; Kyle H. P.,
Report RL-4.6.50, Aug. 7, 1944; We b b A. D.,Kyle H. P., Report RL-4.6.51,
Mar. 1943 (UCRL 43).
162. Reiber H. G., Report RL-4.6.116, Apr. 23, 1943 (UCRL 44).
163. Wilder C. D., Reports RL-4.6.65, RL-4.6.68, RL-4.6.73, RL-4.6.103, RL-4.6.106,
RL-4.6.129, Apr. 26, 1943; Young H. A., Report RL-4.6.Ill, Apr. 20, 1943
(UCRL 45).
164. Mueller M. E., Report RL-4.6.88, Mar. 28, 1943 (UCRL 46).
165. Kraus C. A., Report N-12, Dec. 15, 1942 (Brown 19).
166. Reynolds F. L., Report RL-4.6.80, Feb. 13, 1943 (UCRL 47).
167. Kraus C. A., Report A-1085, Aug. 1, 1943 (Brown 22).
168. Kraus C. A., Report A-1087, Oct. 1, 1943 (Brown 23).
169. Steele G. D., Report RL-4.6.99, Mar. 26, 1943 (UCRL 53).
170. Johnson O., Butler Т., Report CC-1500, June 17, 1944 (MP Ames 18).
171. Jenkins F. A., Reports RL-4.6.85A, Feb. 22, 1943, RL-4.6.47, Mar. 9, 1943
(UCRL 54).
172. A 1 t m a n D., Report RL-4.6.161, June 21, 1943 (UCRL 55).
173. Reiber H. G., Report RL-4.6.60 (UCRL 56).
174. G о r e n H. L., L о w r i e R. S., H u b b a r d J. V., Report CD-5.350. 8, Feb. 28,
1946 (CEW-TEC 29).
175. MacWood G. E., Report RL-4.7.602, Dec. 18, 1945 (UCRL 57).
176. Sterett С. С, Halpern I., Van Wazer J. R., Report CD-GS-28, Mar. 16,
1945 (CEW-TEC 30).
177. Sterett С. С, Calkins V. P., Report AECD-2443 (1949).
178. Kraus С A., Report A-557, Mar. 1, 1943 (Brown 18).
179. Kraus С A., Report [AJ-BM-4, 4, Jan. 3, 1944 (Brown 24).
180. Allen M. В., Report RL-4.6.19, Feb. 13, 1943 (UCRL 58).
181. Hunter M. A., J о n e s A., Trans. Am. Electrochem. Soc., 44, 27 (1923).
182. Wood W. C, Bradley H., Report RL-4.6.288, Sept. 2, 1944 (UCRL 59).
.183. Gavlin C.Hubbard J. V., С 1 e w e t t G. H., Report CD-PPO-14, Apr. 3,
1945 (CEW-TEC 31).
184. Martin H., Eldau K. H., Z. anorg. u. allgem. Chem., 251, 295—304 (1943).
185. Cronander W., Bull. soc. chim. Paris, [2] 19, 500 (1873).
186. Butler Т., Report CC-1778, Aug. 18, 1944 (MP Ames 19).
187. Kraus С A., Report CC-1685, Mar. 10, 1944 (Brown 25).
188. Kra us С A., Report A-1090, Dec. 1, 1943 (Brown 26).
189. Kraus С A., Report A-1091, Dec. 1943, (Brown 27).
190. К г о h n R., Report RL-4.6.238, Jan. 4, 1944 (UCRL 60).
191. Davidson P. H., Report RL-4.6.86, Mar. 23, 1943 (UCRL 61).
192. Kraus С A., Report N-18, Apr. I, 1943 (Brown 20).
193. Allen M. В., Report RL-4.6.77, Feb. 6, 1943 (UCRL 64).
194. Reynolds F. L., Report RL-4.6.78, Feb. 7, 1943 (UCRL 65).
195. Reynolds F. L., Report RL-4.6.188, May 2, 1943 (UCRL 66).
196. Z а с h a r i a s e n W. H., Reports N-1574, Feb. 28, 1945; CF-2796, Mar. 15, 1945
(MP Chicago 8).
197. Z а с h a r i a s e n W. H., Acta Crystallographica, 1, 285—287 (1948).
198. Davidson P. H., H о 1 m e s J. A., Report RL-4.6.182, July 29, 1943 (UCRL 67).
199. Jenkins F. A., Report RL-4.6.47, Mar. 9, 1943 (UCRL 68).
200. A 1 t m a n D., Lipkin D., W e i s s m a n S., Report RL-4.6.22, Feb. 27, 1943
(UCRL 69).
201. В u г 1 e г Т., J о h n s о n O., Report CC-1975, Sept. 10, 1944 (MP Ames 20).
202. Ferguson W. J., Rand R. D., Report A-3357, Mar. 28, 1945 (Natl. Bur.
Standards 5).
203. U re у H. C, Report A-750, July 10, 1943 (SAM Columbia 1).
204. Duncan A. B. F., Report A-584, Apr. 15, 1943 (SAM Columbia 2).
Глава 15
БРОМИДЫ, ИОДИДЫ И ПСЕВДОГАЛОГЕНИДЫ
УРАНА
ТРИБРОМИД УРАНА [1]
Трибромид урана UBr3 во многих отношениях напоминает трихлорид.
Он немного более гигроскопичен, но в общем вступает в те же реакции, что
и трихлорид, и обладает также сходными с ним физическими свойствами.
Методы получения трибромида и трихлорида также очень близки между собой.
Сходство между трибромидом и трихлоридом урана подчеркивается здесь ввиду
того, что химия трихлорида изучена гораздо лучше и много качественных
данных относительно трибромида может быть приписано ему по аналогии
с трихлоридом.
Получение трибромида урана. Наиболее старый метод
получения трибромида заключается в восстановлении тетрабромида
водородом [2]. Однако эта реакция дает довольно неудовлетворительные результаты,
поэтому была разработана методика, по которой получается более чистый
продукт.
Получение трибромида урана из гидрида и бромистого водорода [3—9].
Гидрид урана легко и спокойно реагирует с бромистым водородом при 300° с
образованием трибромида урана:
250°
2U + 3H2 >2Ш3; (1)
300°
UH3 + ЗНВг > UBr3 + ЗН2. (2)
Аппарат, подходящий для проведения этого процесса, показан на рис. 59
(см. также [1]). Необходимо пользоваться чистыми реагентами, водород
должен быть тщательно очищен от следов кислорода, а бром, применяемый для
получения бромистого водорода, должен быть высушен над фосфорным
ангидридом. При наличии металлического урана этот метод очень удобен.
Реакция между металлическим ураном и бромом [10—12]. В результате
взаимодействия металлического урана с парами брома образуется трибромид
урана
2U + 3Br2-^2UBr3. (3)
При этом необходимо брать стехиометрические количества реагентов.
Подходящим реактором для бромирования урана может служить большая трубка
из стекла пирекс с боковым отводом для брома. Пока металл нагревается до
300—500°, бром в резервуаре поддерживается в замороженном состоянии,
затем его расплавляют и дают парам реагировать с металлом. Целесообразно
предварительно измельчить уран, что можно сделать путем превращения его
в гидрид с последующим разложением этого гидрида. Когда весь бром
оказывается израсходованным, систему нагревают до 500—550°. Этим путем, как
показывает анализ, может быть получен продукт, близкий по составу к три-
бромиду урана.
Восстановление тетрабромида урана водородом. История показывает,
что получение трибромида впервые было выполнено восстановлением
412 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
тетрабромида урана водородом при температурах, близких к точке'"плавлечия
тетрабромида [2, 13]:
2UBr4 + Н2 -* 2UBr3 + 2НВг. (4)
Однако оказалось, что восстановление не обязательно останавливается на
стадии трибромида и что иногда образуются продукты, в которых: отношение
Рис. 59. Прибор для получения безводного трибромида урана,
1 — реакционная камера; 2 —газопромывная камера; 3 —диск из пористого
стекла; 4—выпускное отверстие для системы продувки прибора; 5 —краны;
6 — платинированный асбест; 7—барботер с бромом; 8 —капельная
воронка; 9 — вход газа; 10—трехходовый кран; 11 — электронагревательная
печь сопротивления; 12 — стандартный конический шлиф; 13 — резиновая
трубка; /4 —выходное отверстие (к тяге); /5—промывная склянка с
серной кислотой и ловушка; 16—манометр; 17 — пробка из стеклянной ваты.
бром : уран заметно меньше трех [7,14,15]. Это установлено как путем анализа
получавшегося продукта, так и измерением количества выделявшегося
бромистого водорода. Не совсем ясно, происходит ли при этом восстановление
до низшего бромида урана; неизвестно также, вызывается ли это явление
присутствием следов влаги или кислорода. Реакция восстановления начинается
при 430°, однако до 550° скорость ее чрезвычайно мала. При 700° она идет
быстро, но при этой температуре наблюдается значительное воздействие реагентов
Глава 15. Бромиды, иодиды и псевдогалогениды урана 413
на вещество, из которого изготовлен реактор (кварц). В связи с
неопределенностью состава продукта и малой скоростью реакции метод восстановления
водородом имеет сомнительную ценность.
2^Ф изические свойства трибромида урана. По общим
физическим свойствам трибромид урана напоминает трихлорид урана и три-
бромиды редкоземельных элементов лантана и неодима. Подобно им, он
является тугоплавким нелетучим кристаллическим веществом.
Температура плавления. Было проведено несколько определений
температуры плавления трибромида урана [9, 11, 16—19]. Значение 755° [16]
получено не прямым определением точки плавления, а путем изучения систем
U—UBr3 и UBr3—UBr4 с точки зрения правила фаз. Вероятно,
значение 730°, найденное в радиационной лаборатории Калифорнийского
университета путем прямых измерений, является более точным.
Кристаллическая структура и плотность [20, 21]. Рентгенографическими
методами показано, что трибромид урана имеет гексагональную элементарную
ячейку с константами
ах = 7,926 ± 0,002А; а3 = 4,432 ± 0,002А.
Элементарная ячейка содержит две молекулы; вокруг каждого атома урана
находится девять атомов брома на среднем расстоянии U—Вг в 3,12 А.
Трибромид урана изоморфен трихлоридам урана, лантана и неодима, три-
бромидам лантана и празеодима и гидроокисям лантана и неодима [22—24].
Пространственная группа С63//л (Ci/J. Подробно об этом структурном типе см.
гл. 14, стр. 363.
Плотность трибромида, найденная из рентгенографических данных, равна
6,53 г/см3. Прямое пикнометрическое определение (в ксилоле) дало среднее
значение 5,98 г/см3 [25].
Давление пара трибромида урана. Определение давления пара трибромида
урана представляет некоторые трудности. Измерения должны проводиться
в условиях повышенных температур, при которых воздействие на материал
аппаратуры довольно значительно. Далее, состав остатка медленно меняется от
отношения U : Вг= 1 : 3 до значений, приближающихся к 1 : 2; менялось это
отношение и в дестиллате(от 1 :3,2 до 1 : 3,8). Хотя заметная часть последнего,
повидимому, представляет собой трибромид, в нем почти несомненно
присутствует и некоторое количество тетрабромида, особенно при температурах
выше 900°. Давление пара трибромида измерено по методу истечения с
применением сосуда из молибдена [91. Данные, приведенные в табл. 181, являются,
Таблица 181
ДАВЛЕНИЕ ПАРА ТРИБРОМИДА УРАНА, ОПРЕДЕЛЕННОЕ МЕТОДОМ
ИСТЕЧЕНИЯ В СОСУДЕ ИЗ МОЛИБДЕНА
Температура,
°С
600
650
700
750
800
Давление пара,
мм рт. ст.
2,0- 10-5
1,6. Ю-4
1,3. 10-3
8,0. Ю-3
3,2- 10~2
Температура,
°С
850
900
950
1000
Давление пара,
мм рт. ст.
1,6- Ю-1
5,0 . Ю-1
1,6
5,0
вероятно, наилучшими из всех известных для трибромида. Они могут быть
представлены уравнением
lg рмм рт. ст. = f Ь 12,5. (5)
414 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Таблица 182
ДАВЛЕНИЕ ПАРА ТРИБРОМИДА УРАНА,
ОПРЕДЕЛЕННОЕ МЕТОДОМ ИСПАРЕНИЯ
В ТОКЕ ИНЕРТНОГО ГАЗА
На основании уравнения (5) вычислена теплота испарения, равная
68 ккал-моль'1.
Применен также метод насыщения газа (метод испарения) [10]. Для
предотвращения диспропорционирования применялась смесь трибромида с
металлическим ураном. Жидкая фаза приближалась по составу к дибромиду,
в то время как состав дестиллата соответствовал трибромиду (табл. 182).
Согласованность этих двух рядов
измерений неудовлетворительная, и
сомнительно, чтобы в обоих случаях измерялась
одна и та же величина. Возможно, что
один или оба ряда значений давления
пара в действительности отвечают
давлению диспропорционирования, а не
подлинному давлению пара. Все это
указывает на необходимость дальнейшей
работы в этом направлении.
Термохимические данные [26].
Теплота образования трибромида урана
определена путем измерения теплоты
растворения его в растворе хлорида
железа (III). Исходя из этой теплоты
растворения в сочетании с термохимическими
данными для других галогенидов урана и соответствующими литературными
данными можно вычислить теплоту и свободную энергию образования
трибромида урана:
Температура,
°С
915
1014
1062
1124
Д
явление пара,
мм рт. ст.
0,30
1,9
3,1
6,6
икрист Ч о" -^Г
2газ
UBr.
зкрист,
(6)
А#27з= — 181,6 ± 0,6 ккал • моль'1.
Для энтропии трибромида при 298° К дается значение 50,6 энтропийной
единицы [27]. Отсюда выводится энтропия образования (—49,4 энтропийной
единицы) иА/7°298-=—166, 9±1,5. Происхождение этих значений для энтропии
неясно, и они, без сомнения (по крайней мере частично), являются
ориентировочными.
Данных по теплоемкости трибромида не имеется.
В интервале температур 100—500° фазовых превращений не
обнаружено [28].
Химические свойства трибромида урана. Трибромид
представляет собой темнокоричневое кристаллическое вещество. Он очень
гигроскопичен, гораздо больше, чем трихлорид. При комнатной температуре
он заметно реагирует с кислородом воздуха.
Отношение к воде. Трибромид урана растворяется в воде с бурным
выделением газа и образованием прозрачного темного красно-фиолетового раствора,
который за 30—60 сек. изменяет свою окраску на яркозеленую, характерную
для растворов солей урана (IV) [14]. Измерено количество выделившегося
водорода; оно соответствовало (с точностью до нескольких процентов)
вычисленному для окисления урана (III) до урана (IV) [29|.
Отношение к другим растворителям [14, 15, 30]. Трибромид урана,
подобно тетрабромиду, почти нерастворим в неполярных растворителях.
Например, в 100 мл бензола или этилбромида растворяется менее чем 0,5 г
трибромида. Кристаллы безводного трибромида не меняют окраски в тг-кси-
лоле. В полярных растворителях трибромид, как правило, менее растворим,
чем тетрабромид. Растворение трибромида в полярных растворителях
сопровождается обычно химической реакцией. Так, при растворении трибромида
Глава 15. Бромиды, иодиды и псевдогалогениды урана
415
в формамиде образуется тёмнокрасный раствор, который через несколько минут
превращается в зеленый с выделением газа. В растворе бромистого водорода
в формамиде (32 г НВг на 100 мл формамида) трибромид немедленно разлагается
с бурным выделением газа и образованием зеленого раствора урана (IV).
Прибавление трибромида к ацетамиду при температуре 110—115° приводит
к образованию весьма тёмнокрасного раствора, который в течение нескольких
секунд становится зеленым, причем одновременно наблюдается выделение газа.
Трибромид быстро разлагается этиловым спиртом, безразлично, присутствует
ли кислород воздуха или нет; при этом одновременно выделяется водород.
В неорганических растворителях, как, например, в трихлориде фосфора и тет-
рахлориде олова, трибромид урана нерастворим [31J.
Восстановление трибромида урана (см. ниже). Трибромид
восстанавливается металлическим кальцием до металлического урана с хорошим выходом.
Для проведения этой реакции может быть использована обычная
методика [5, 32] (см. также гл. 4). Было изучено электрохимическое восстановление
растворов трибромида в расплавленных солях [10, 33, 34]. Электролиз раствора
трибромида в бромиде бария (4—10% UBr3) при 870—910° дал
неудовлетворительный результат—металлический уран отлагался в виде губчатой массы,
плохо пристававшей к катоду. Для получения слоя урана на железном,
молибденовом или урановом катоде может быть использован раствор трибромида
урана в бромиде стронция [10—20% (мол.) UBr3]. Выход по току составляет
около 50%. Чистый расплавленный трибромид не является подходящим
электролитом для осаждения урана.
Окисление трибромида. Окисление трибромида до тетрабромида бромом
будет рассмотрено на стр. 418. Кислород реагирует с трибромидом даже при
комнатной температуре, однако продукты окисления не идентифицированы.
С качественной стороны трибромид окисляется, повидимому, быстрее, чем
трихлорид.
Диспропорционирование трибромида урана. Фазовые соотношения. При
обсуждении данных по давлению пара трибромида было отмечено, что состав
дестиллата может рассматриваться как результат частичного диспропорциониро-
вания при температуре выше 900°:
2UBr3 ->UBr2 + UBr4. (7)
Вопрос о существовании галогенида двухвалентного урана вызвал,
естественно, интерес, особенно после того, как было сделано наблюдение, что
металлический уран растворяется в расплавленном трибромиде и образует при
охлаждении твердые продукты с эмпирической формулой, близкой к UBr2.
Проведено исследование системы U—UBr3 с точки зрения правила фаз [111.
Результаты ясно показали, что при растворении металлического урана в
расплавленном трибромиде никакого соединения не образуется. При соотношении
бром : уран, равном 2,85—2,9 (что соответствует молярной доле трибромида
0,98—0,95), образуется эвтектика. По охлаждении образуется смесь
трибромида с ураном. Эти результаты определенно исключают возможность
существования дибромида как стабильного при комнатной температуре соединения.
Поэтому образование тетрабромида в исследованиях по давлению пара
трибромида должно быть объяснено не простым диспропорционированием, а как-то
иначе. Повидимому, приемлемым является предположение, что тетрабромид
образуется за счет реакции трибромида со следами влаги или кислорода.
Составлена фазовая диаграмма системы UBr3—UBr4 [16] (рис. 60). Данные
показывают, что это простая система с эвтектикой, характеризующаяся
ничтожной взаимной растворимостью компонентов в жидкой фазе и значительной
растворимостью трибромида в твердом тетрабромиде. Эвтектика содержит
76% (мол.) трибромида и имеет точку плавления 490°.
416 Часть IV, Галогениды, оксигалогениды, борг ид р иды и карбонил урана
Изучены также системы SrJ2—UBr3 и SrBr2—UBr3 [18]. Полученные
предварительные данные указывают на отсутствие соединений в этих системах
(рис. 61).
Комплексные соединения. Различные реакции. При действии газообразного
аммиака на трибромид урана наблюдается химическая реакция. Характерный
коричневый цвет трибромида исчезает и образуется весьма объемистое зеленое
вещество, которое имеет состав UBr3-6NH3. При обработке трибромида жидким
7до г
40 60
UBr4 , %(мпл.)
Р*и с. 60. Фазовая диаграмма системы UBr3—UBr4.
аммиаком с последующим высушиванием в вакууме получается твердое
вещество, содержащее от трех до четырех молекул NH3 на одну молекулу
трибромида [30 h
При нагревании трибромида в кварцевом сосуде до прекращения
образования дестиллата получается остаток, свободный от брома. Исследование
показывает, что этот остаток состоит из растворимой в воде части и нерастворимой
части черного цвета. Для объяснения этого явления была предложена
следующая реакция:
12UBr3 + 2Si02 -> USi2 + 2U02 + 9UBr4. (8)
Этот факт, между прочим, указывает на то, что длительное нагревание
трибромида в кварцевом или стеклянном сосуде при повышенных температурах
является нежелательным.
Глава 15. Бромиды, иодиды и псевдогалогениды урана 417
При проверке коррозионного действия трибромида на некоторые металлы
установлено, что молибден вполне устойчив и является более подходящим
материалом для работы с трибромидом, чем нержавеющая сталь [9].
1—1 i г—ц
/
/ Л
/
/
/ н
/
/
/ Н
/
-V -J
^.^^ ~|
о I
I I I
™~0 0,25 0,50 Q75 1,00 0 0,25 0,50 Q75 1,00
Молярная доля U Вг3 Молярная доля (J В г3
а б
Рис. 61. Фазовая диаграмма систем:
a- SrBr2-UBr3; б - SrJ2-UBr3.
ТЕТРАБРОМИД УРАНА [1]
По своим физическим и химическим свойствам тетрабромид урана UBr4
близко напоминает тетрахлорид. Он является менее устойчивым соединением,
что видно из меньшей величины его свободной энергии образования.
икрист ~т~ ^^--'ггаз * UL.I4KpIICTJ
д f°298= —229,0 ± 1,0 ккал - моль'1;
икрист Н~ ^£>Г2газ ^ иг>Г4КрИст^
д^°298= — 190,2 ± 1,5 ккал • моль'1.
Методы получения тетрабромида менее разнообразны, чем в случае тет-
рахлорида. Единственным известным реагентом, непосредственно
превращающим окислы урана в тетрабромид, является смесь угля и брома при
повышенной температуре. В противоположность четыреххлористому углероду, который
находит столь значительное применение при получении тетрахлорида урана,
четырехбромистый углерод не может быть использован в качестве бромирую-
щего агента, так как при высокой температуре, требуемой для превращения,
он полностью разлагается. При действии бромистого водорода в условиях
любых температур окислы урана не превращаются с достаточно высокой
скоростью в тетрабромид, несмотря на то, что термодинамические условия этой
реакции более благоприятны, чем условия реакции хлорирования окислов
урана хлористым водородом. Ввиду того что бромирование окислов урана
бромом в присутствии угля трудно осуществить на практике, приходится
получать тетрабромид из других соединений урана. Свободный бром является
27 Хиуия урана
750 Ь
*> Г
I
15
650
КПП
L 1 Т~
Г /
/
1 у
L_ '
г /
1 /
К /
LV
IV i i
—1—и
J
л
/
/
/ Ч
/
/
/ J
i I, _..i.
.„j
700
**
Н
W600]
о
°-
4)
с
*
Й
500\
апп\
(9)
(10)
418 Часть IV. Га/югениды, оксигалогениды, боргидриды и карбонил урана
достаточно сильным бромирующим агентом для превращения металлического
урана или трибромида урана, а также его карбида, нитрида или сульфида
в тетрабрсмид.
Получение тетрабромида урана. Поскольку и бром и
тетрабромид являются сильно корродирующими агентами, получение
тетрабромида проводят обычно в стеклянном или кварцевом сосуде. Никель и золото
обладают удовлетворительной стойкостью; нержавеющая сталь, монель-
металл и медь непригодны для этих целей [7].
Двуокись урана, уголь и бром. Как упоминалось выше, этот метод был
первым, предложенным для получения тетрабромида урана, но он неудобен.
Впервые он был применен Германом [35] и в принципе использовался почти
всеми исследователями до самого последнего времени [13]. Попытки Колани [361
усовершенствовать эту методику не дали удовлетворительных результатов.
Этот автор утверждает также, что тетрахлорид может быть превращен в
тетрабромид действием бромистого водорода при 590°, что, однако, сомнительно.
Получение тетрабромида путем введения в реакцию двуокиси или закиси-
окиси урана с бромом и углем и его очистка достаточно подробно изучены [37,
38], причем для определения атомных весов пользовались очищенным тетра-
бромидом.
Реакция проводится обработкой бромом смеси двуокиси или закиси-
окиси урана с избытком угля при температуре яркокрасного каления. Бром
лучше вводить в токе инертного газа, например азота или двуокиси углерода.
Тетрабромид конденсируется на более холодных частях аппаратуры.
Металлический уран ибром[\3, 39, 40]. Эта реакция является очень
удобной для получения тетрабромида. Бром (вводимый в токе гелия) легко
реагирует с металлическим ураном в виде стружек при 650°. Процесс проводят
в кварцевом приборе такой конструкции, что получающийся тетрабромид
отгоняется по мере образования (рис. 62 [39]).
Металлический уран и бром (через гидрид урана) [3]. Превращение
металлического урана в тетрабромид может быть также выполнено с
промежуточным образованием гидрида урана:
2U + 3H2 -> 2UH3; (1)
2UH3 + 7Br2 —> 2UBr4 + 6HBr. (11)
Этот процесс менее удовлетворителен, чем прямое превращение металла в
тетрабромид. Неудобства заключаются в трудности регулирования реакции и в
местных перегревах, которые могут привести к закупориванию прибора.
Трибромид урана и бром [3, 5, 25, 41—43]. Эта реакция является отличным
способом получения тетрабромида:
2UBr3 + Br2 300° -. 2UBr4. (12)
Бром вводится в реакцию в токе азота, который предварительно тщательно
очищается пропусканием над окисью меди при 600° аскаритом, ангидроном
и, наконец, нитридом урана при 600°. Реакция легко протекает при 300°,
однако время от времени требуется покачивание прибора во избежание
спекания и образования каналов, по которым пары брома могут проходить не
прореагировав. При температуре ниже 200° реакция проходит неполно. Можно
проводить реакцию без отгона образующегося тетрабромида. В некоторых
случаях это может быть целесообразно.
Нитрид урана и бром [44]. По существу этот метод основан на применении
металлического урана, так как до сих пор нитрид получают только из металла.
Реакция
UN + 2Bra-->UBr4 + 4-N2 (13)
Глава 15. Бромиды, иодиды и псевдогалогениды урана
419
протекает весьма спокойно при 600°. Бром целесообразно вводить в токе
очищенного азота. .
Карбид урана и бром [45—47]. Реакция между бромом и карбидом урана
является одним из методов превращения двуокиси урана в тетрабромид:
U02 + 3C->UC2 + C02; (14)
UC2 + 2Br2 -> UBr4 + 2C. (15)
Взаимодействие брома с карбидом быстро протекает при 800°. Требуемая
высокая температура (см. стр. 418) является серьезным неудобством, ввиду того
Рис. 62. Прибор для получения безводного тетрабромида урана.
I — кварцевая реакционная колба, содержащая трибромид урана или металлический
уран; 2 — кварцевая колба емкостью 1 л, используемая в качестве конденсатора;
8 — рефлектор из алюминиевой фольги для теплоизоляции; 4 — электронагревательная
печь сопротивления; 5 —колба из стекла пирекс емкостью 1 .1. используемая в качестве
приемника; б —соединение кварца с пирексом. снабженное смазкой апьезон W; 7 — ре-
виноваи трубка; 5 —краны; 9 — промывалка с серной кислотой и ловушка; 10 — колба
с бромом; //—стандартный конический шлиф; /2 —вход газа; 13 — выходное отверстие
(к тяге); 14 — отверстие (которое иногда забивается); /5 —пробка из стеклянной ваты
для удерживания дисперсного продукта; 16 — отодвигающаяся крышка для наблюдений;
17—соединение кварца со стеклом пирекс.
что при этой температуре возможна сильная коррозия. Другой недостаток
этого метода состоит в том, что часть образующегося тонкодисперсного
углерода уносится током газа и загрязняет продукт.
Сульфид урана и бром или бромистый водород [47, 48]. Тетрабромид урана
может быть получен действием брома или бромистого водорода на сульфид
урана:
US2+ ЗВг2 -* UBr4 + S2Br2; (16)
US2 + 4HBr -> UBr4 + 2H2S. (17)
Реакция с бромом протекает при комнатной температуре; бромирование с
помощью бромистого водорода требует несколько более высокой температуры.
Монобромид серы может быть отогнан при температуре ниже 450°; реакцию
обычно заканчивают возгонкой тетрабромида при 600°. Эти реакции не имеют
практического значения из-за трудностей, связанных с получением сульфида
урана.
Двуокись урана, сероуглерод и бром (U;i02S4+Br2) [39, 42, 44]. Как
хорошо известно, многие окислы (особенно двух- и трехвалентных металлов) v
27*
420 Часть IV. Галогениды, оксигалогениды, боргидриди и карбонил урана
превращаются в сульфиды при обработке сероуглеродом при повышенных
температурах.
В случае двуокиси урана, однако, реакция протекает с образованием
смешанного оксисульфида урана (IV) (см. гл. 11):
3U02 + 4CS2 900° ^ U30,S4 -f 4COS. (18)
При обработке этого соединения бромом наблюдается реакция уже при 50—100°;
образующийся монобромид серы может быть отогнан при 400°. Остаток,
представляющий светлокоричневый порошок, разлагается при 600° с образованием
тетрабромида урана. Этот остаток представляет оксибромид урана (IV),
который в свою очередь разлагается на двуокись урана и тетрабромид при 800°:
U302S4 + 6Br2 50-100° , 2UOBr24 UBr4 + 2S2Br2 (19)
2UOBr2 800-!000° , UBr4+UOe (20)
U302S4 + 6Br2 800° , 2UBr4 4 U02 + 2S2Bra (21)
Если реакцию заканчивают при 800°, то в конечном результате из 3 молей
двуокиси урана образуется 2 моля тетрабромида. Для оставшегося
неизмененного моля двуокиси требуется повторная обработка. Этот интересный цикл
заслуживает дальнейшего исследования.
Двуокись урана, сера и бром. Тетрабромид урана был получен [49] путем
реакции в жидкой фазе между двуокисью урана и монобромидом серы или
смесью серы и брома. Двуокись урана в избытке серы и брома нагревали с
обратным холодильником в течение 12 час. при 170°; затем избыток реагента
отгоняли, а продукт промывали четыреххлор истым углеродом. Можно заранее
предсказать, что получение свободного от серы продукта по этому методу
натолкнется на значительные трудности.
Общий обзор методов получения. В табл. 183 указаны обсуждавшиеся выше
реакции, по которым может быть получен тетрабромид урана из различных
соединений урана и свободного брома.
Таблица 183
ПОЛУЧЕНИЕ ТЕТРАБРОМИДА УРАНА
Реагенты
(+ бром)
ио2, с
и3о8, с
и
иН3
UBr,
UN
ис,
us2
u3o2s4
uo2,s
Температура, eC
700-900
700-900
650
?
300
600
800
450
800
170
Газ, несущий
бром
N.. СО,
N„ СО,
Не
Не
N,
N,
N,
N,
N,
Побочные
продукты
' СО
со
Отсутствуют
НВг
Отсутствуют
N,
с
S2Br2
(Ю2, S2Bra
Уравнение
(22)а)
(23)
(10)а)
(11)
(12)а)
(13)
(15)
(16)
(21)
а) Эти реакции являются практически наилучшими с точки зрения доступности
исходных материалов, а также простоты операций.
Получение тетрабромида урана из раствора. Водные растворы
тетрабромида могут быть получены легко, а поэтому сделаны попытки приготовить из
Глава 15, Бромиды, иодиды и псевдогалогениды урана
42J
них безводную соль. Растворы тетрабромида можно получить
электрохимическим восстановлением растворов уранилбромида, химическим
восстановлением такими агентами, как металлический уран, или растворением гидратиро-
ванной гидроокиси урана (IV) в бромистоводородной кислоте. Установлено,
что такие растворы не могут быть обезвожены без весьма значительного
гидролиза. В этом отношении растворы тетрабромида в воде напоминают водные
растворы тетрахлорида с той разницей, что в случае тетрабромида гидролиз
идет даже легче. Гидролиз может иметь место даже в токе бромистого водорода.
Это неудивительно, если вспомнить, что бромистый водород не способен
превращать двуокись урана в тетрабромид. При концентрировании растворов
тетрабромида под пониженным давлением как в условиях комнатной
температуры, так и при 180° даже в атмосфере бромистого водорода наблюдается
значительное окисление [50]. Имеются указания, что тетрабромид образуется при
фотохимическом восстановлении раствора трехокиси урана в спирте,
насыщенном бромистым водородом, однако это сообщение требует проверки [51].
Очистка тетрабромида урана [7, 42, 52, 53]. Тетрабромид урана лучше
всего может быть очищен перегонкой в вакууме или возгонкой в токе инертного
газа, содержащего небольшое количество брома. В качестве несущего газа
применимы тщательно очищенные азот или гелий.
Физические свойства тетрабромида урана.
Физические свойства тетрабромида не изучены столь систематически, как
свойства тетрахлорида, и имеющиеся сведения весьма отрывочны.
Температура плавления [44, 54, 55]. При определении температуры
плавления тетрабромида получено значение 519±2°. Из данных термического
анализа вычислена температура 516° [28].
Температура кипения[44, 56]. Прямым определением при давлении 740мм
рт. ст. установлена температура кипения тетрабромида, равная 765° (с
разложением). Экстраполяцией данных по давлению пара тетрабромида (применялся
метод испарения) получено значение 761° при 760 мм рт. ст., что
удовлетворительно согласуется с величиной, найденной непосредственным измерением.
Плотность. Старые значения [37] для плотности тетрабромида, вероятно,
ошибочны. Прямым определением с бензолом и ксилолом в качестве пикнометри-
ческих жидкостей получено значение 5,35 г/см3 при 26° [25].
Рентгенографические данные, из которых могла бы быть рассчитана плотность тетрабромида,
отсутствуют. Величина плотности пара, найденная Циммерманом [13],
показывает, что тетрабромид в парах не полимеризован.
Давление пара. Давление пара тетрабромида изучено по методу молеку*
лярного истечения и по методу испарения. Исследования проводились в
разных температурйых интервалах, а поэтому результаты трудно сравнимы, за
исключением одной точки (450°), для которой совпадение является
удовлетворительным. Результаты, полученные методом истечения, приведены в табл. 184.
Результаты определений давления пара тетрабромида методом испарения
приведены в табл. 185 [1].
Из этих данных выводятся следующие уравнения:
для возгонки: lgp**pT.c*.= f f-14,56; (24)
для испарения: lgрмм рт. ст. «= ^- + 9,71; (25)
ДЯвозг = 50 000 кал -моль"1;
Д#исп - 32 000 кал-моль"1;
ДЯолавл == *8 000 кал • моль~1.
' 422 Часть IV. Галогениды, оксигалогениды, боргидриды и карбон ил урана
Таблица 184
ДАВЛЕНИЕ ПАРА ТЕТРАБРОМИДА УРАНА,
ОПРЕДЕЛЕННОЕ МЕТОДОМ ИСТЕЧЕНИЯ
[53,57]
Температура,
°С
300
330
350
365
375
400
450
Давление пара, мм рт. ст.
1,08.10-*
8,45-10-*; 9,55-10-*
3,08-10-3; 3,24-10-3;
3,33- Ю~3
5,б1.10-3
1,27.10-а
2,25-10-а; 6,23.10-*
2,9.10-1
Таблица 185
ДАВЛЕНИЕ ПАРА
ТЕТРАБРОМИДА УРАНА, ОПРЕДЕЛЕННОЕ
МЕТОДОМ ИСПАРЕНИЯ В ТОКЕ
ИНЕРТНОГО ГАЗА 144]
Температура,
°С
450
475
500
525
550
575
600
625
Давление пара,
мм рт. ст.
0,30
0,95
2,85
9,15
13,5
24,0
40,8
70,1
Данные табл. 184 приводят к значению ДНВ0зг> равному 48 700 кал моль'1.
Вычисленная константа Трутона (SH/T) равна 31,0, а константа Гильдебранда
32,8. Эти величины указывают на значительную ассоциацию в жидком
состоянии. Величины, полученные методом испарения, повидимому, более надежны;
поэтому термодинамические данные, основанные на значениях давления пара,
полученных методом испарения, вероятно, являются более точными.
Термохимические данные. Теплота растворения тетрабромида в воде, по
определению, равна—33,1 ±0,1 ккалмоль'1. В сочетании с другими
соответствующими данными отсюда может быть вычислена теплота образования
тетрабромида [26]:
Ц
крист
+ 2Вг
2 газ
UBr,
4крист>
(Ю)
д#°298= — 211,3 ± 0,6 ккал-моль'1.
Маквуд [27] дает для энтропии тетрабромида значение S298 = 58,5
энтропийной единицы, откуда можно вывести:
Д5Я98=—70,8 энтропийной единицы;
д^7°298==г — 190,2 ± 1,5 ккал-моль~х.
В радиационной лаборатории Калифорнийского университета [55, 58, 59]
определена константа равновесия реакции
UBr
4крист
+4-н
2 газ
UBr
3 крист
+ НВгг
(26)
в интервале температур 375—525° (табл. 186).
Вероятно, более поздние результаты, полученные Грегори [60], являются
более точными. Если принять для реакции (26) значение ДСР, равное —7,59
при 500°, то АЯ0 составит 19,913 ккал-моль'1. Данные табл. 186 приводят (для
этой же реакции) к значению AS29ft = 23,96 энтропийной единицы.
Химические свойства тетрабромида урана. Тет-
рабромид урана представляет кристаллическое вещество, окраска которого
меняется от светлой рыжевато-красной до темнокоричневои в зависимости от
размера кристаллов. Он очень гигроскопичен и быстро окисляется во влажном
воздухе до уранилбромида. Отсюда следует, что все манипуляции с тетрабро-
мидом должны проводиться в атмосфере сухого инертного газа.
Растворимость [15, 25, 30, 61, 62]. Тетрабромид урана легко растворяется
в воде с образованием зеленого раствора, обладающего характерными
свойствами растворов урана (IV). Растворенный тетрабромид заметно гидролизован.
Глава 15. Бромиды, иодиды и псевдогалогениды урана
423
Таблица 186
КОНСТАНТЫ РАВНОВЕСИЯ РЕАКЦИИ ВОССТАНОВЛЕНИЯ ТЕТРАБРОМИДА УРАНА
ВОДОРОДОМ3 )•
Температура» °С
375
400
425
450
475
500
525
Температура, °К
648
673
698
723
748
773
798
~г- 108
1,543
1,486
1,433
1,383
1,337
1,294
1,253
К [59]
0,080
0,124
0,188
0,256
0,365
—
—
К [551
0,141
0,170 (420° С)
0,250
0,320
0,440
0,570
Изучено поведение тетрабромида в большом числе органических
растворителей. Неполярные растворители, как «-гептан, бензол, толуол, тг-ксилол,
бромбензол, тетралин и четыреххлористый углерод, не растворяют
тетрабромида и не реагируют с ним. Неполярные неорганические жидкости, как три-
хлорид и оксихлорид фосфора, бром и тетрахлорид олова, также не
растворяют тетрабромид при температурах ниже 70°. Наоборот, в полярных
растворителях тетрабромид растворим. Такие растворители, как уксусная кислота,
метиловый спирт, этиловый спирт, фенол и анилин, реагируют с тетрабромидом
с выделением бромистого водорода. При действии полярных растворителей,
обладающих окислительными свойствами, например нитробензола, нитро-
метана или бензальдегида, тетрабромид превращается в уранилбромид.
Растворители, содержащие основную кислородную группу, как, например,
диоксан, ацетон, диэтиловый эфир, этилацетат и амилацетат, реагируют
с тетрабромидом с образованием устойчивых сольватов, из которых тетрабромид
трудно регенерировать. Эти последние растворы имеют зеленую окраску
в отраженном свете и красную в проходящем. Соединения, содержащие
карбонильную группу, являются лучшими растворителями, чем те, которые
содержат только эфирную связь. Нитрилы, например ацетонитрил и бен-
зонитрил, растворяют тетрабромид в количестве до 5 г на 100 мл
растворителя, причем при стоянии таких растворов выделяются нерастворимые
сольваты.
Тетрабромид нерастворим в жидком аммиаке, но образует с ним
нерастворимые аммиакаты. Другие амины, как анилин, бутил амин, триэтиламин,
диэтиланилин и пиридин, лишь слабо растворяют тетрабромид урана, но
образуют с ним нерастворимые продукты присоединения. В формамиде тетрабромид
растворяется с выделением значительного количества тепла. Растворы
тетрабромида в формамиде устойчивы при комнатной температуре, при температурах
же выше 100° наблюдается некоторая реакция, о чем можно судить по
образованию осадка. Очень хорошо растворим тетрабромид в расплавленном ацет-
амиде (50 г на 100 мл ацетамида). Раствор, повидимому, совершенно устойчив
при 145° в течение нескольких дней и имеет такую же окраску и спектр
поглощения, как и раствор тетрабромида в формамиде. Некоторые
количественные данные по растворимости тетрабромида в различных растворителях
сведены в табл. 187 и 188.
Изучена также растворимость тетрабромида в некоторых расплавленных
солях. При 900° может быть приготовлен 10%-ный раствор тетрабромида в
бромиде бария. Кроме того, тетрабромид урана растворим (в неизвестном
количестве) в расплавленной смеси NaCl и КС1 при 725° [33].
424 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Таблица 187
РАСТВОРИМОСТЬ ТЕТРАБРОМИДА УРАНА В ОРГАНИЧЕСКИХ
РАСТВОРИТЕЛЯХ ПРИ 25° [61]
Растворитель
Диоксан
Нитрометан
Бензола<
Четыреххлористый углерод . .
я-Гептан
Плотность
ненасыщенного раствора,
г /мл
1,573
1,841
1,523
1,126
1,057
1,271
0,869
1,583
0,678
Растворимость
в 100 г
растворителя (по
анализу на уран), г
64,81
185,2
91,3
35,3
2,84
17,86
14,5
0,0
0,0
0,0
а) Тетрабромид нерастворим также в толуоле, л-ксилоле и тетралине.
Таблица 188
РАСТВОРИМОСТЬ ТЕТРАБРОМИДА УРАНА В РАЗЛИЧНЫХ РАСТВОРИТЕЛЯХ [621
Растворитель а)
Растворимость в 100 мл
растворителя, г
Этилбромид б) . . .
Этилбромид (S02) б)
Бромбензол . . . .
Дмэтиловый эфир .
Диоксан
Ацетон
Ацетон (S02) . . .
Уксусная кислота .
Нитробензол . . . .
Формамид . . . .
Формамид (НВг) . .
Очень мало растворим
ь » »
» т> »
Мало растворим
» »
15
30
55
3
55
5
Цвет
I
Сильно окрашен
» »
» »
Красновато-оранжевый
Тёмнокрасно-коричневый
Темнофиолетово-красный
Очень темнозеленый
Тёмнокрасно-коричневый
а) Формула в скобках (SO2) указывает, что растворитель был насыщен сернистым
газом. (НВг) указывает на присутствие примерно 40 г НВг в 100 мл формамида.
б) Добавка бензола в количестве 5% (объемн.).
Реакции окисления. Под действием кислорода тетрабромид превращается
в уранилбромид. В результате реакции с хлором образуется тетрахлорид.
Жидкий бром не реагирует с тетрабромидом даже при 230° [13]. В процессе
перегонки тетрабромида с парами брома при 750° признаков образования
пентабромида или других высших бромидов не обнаружено.
Реакция восстановления. При температурах 470—700° водород
восстанавливает тетрабромид до трибромида:
UBr4 + ^H2-* иВГз + НВг.
(26)
Глава 15. Бромиды, иодиды и псевдогалогениды урана
425
Когда восстановление достигает стадии трибромида, выделение бромистого
водорода резко падает [15] (см. получение трибромида, стр. 412).
Кальций и магний восстанавливают тетрабромид до металлического урана.
Из-за летучести тетрабромида восстановление приходится проводить в
автоклаве. Тетрабромид урана может быть восстановлен: 1) электрохимическим
путем; 2) из раствора в расплавленных солях [33]; 3) из растворов в некоторых
органических растворителях [15, 30, 62]. По последнему способу в Беркли
проведены обширные исследования, их результаты, однако, нельзя считать
убедительными. При электролизе растворов тетрабромида в бензоле, диэти-
ловом эфире, диоксане, ацетоне, уксусной кислоте и нитробензоле на катоде
образовывался сложный неметаллический осадок. При электролизе растворов
тетрабромида в ацетонитриле, бензонитриле, воде и жидком аммиаке
металлический уран также не получался. Из растворов же в формамиде, ацетамиде
и этиловом спирте выделен осадок (на платиновых катодах), который
представлял собой, по мнению исследователей, металлический уран. Идентификация
осадка была произведена на основании нерастворимости его в воде и
растворимости в разбавленной соляной кислоте с выделением газа. К сожалению,
некоторые соединения урана ведут себя аналогичным образом, и, следовательно,
идентификация этого осадка остается под сомнением. Выход по току при
электролизе очень мал (около 5%), даже при наиболее благоприятных условиях.
Поэтому этот процесс не имеет или почти не имеет практического значения.
Несмотря на это, дальнейшие исследования сложных явлений, связанных
с электролизом этих органических растворов, могут представлять интерес.
Термическое разложение тетрабромида урана. Перегонка тетрабромида
в токе азота приводит к его частичному разложению [38]:
UBr4 -*UBr3 + 4Br2' (27)
В эвакуированной трубке с раскаленной нитью пары тетрабромида
разлагаются на бром и металлический уран, оседающий на нити (см. на стр. 111
о получении металлического урана по методу термического разложения).
Координационные соединения. При комнатной температуре и атмосферном
давлении тетрабромид урана поглощает аммиак с небольшим выделением тепла.
При такой обработке тетрабромида в течение 1 часа с последующим
выдерживанием в вакууме в течение 5 мин. образуется твердое вещество UBr4-4NH3
серого цвета. Жидкий аммиак дает твердые вещества с составом, меняющимся
от UBr4-5NH3 до UBr4-6NH3. Последнее имеет белый цвет с зеленоватым
оттенком [30].
Сообщается [63], что тетрабромид урана в парах при повышенных
температурах реагирует с бромидами щелочных металлов с образованием двойных
солей типа M2UBr6. Реакция проходит таким же образом, как и в случае
соответствующих хлоридов, но не так легко. Продукт, получающийся при
взаимодействии бромида калия с парами тетрабромида урана, представляет вещество
зеленого цвета, легко растворимое в воде. Получено также аналогичное
натриевое производное. Свойства этих соединений изучены недостаточно, и их
существование и состав требуют проверки.
трииодид и; тетраиодид урана
Три- и тетраиодид урана удобнее рассматривать вместе, потому что они
легко превращаются друг в друга. Тетраиодид отличается от других галогенидов
урана (IV) своей термической неустойчивостью. Напомним, что тетрахлорцд не
разлагается с образованием трихлорида ни при каких легко достигаемых
температурах (при атмосферном давлении), а тетрабромид лишь слабо диссоцирует
426 Часть IV, Галогениды, оксигалогениды, боргидриды и карбонил урана
при температурах порядка 700—800°. Напротив, тетраиодид очень легко
распадается на трииодид и иод даже при сравнительно низких температурах.
Поэтому для определения стабильности того или иного иодида в системе
UJ4^UJ3 + 4"J2 (28)
нужно знать парциальное давление иода.
В старой литературе единственным упоминавшимся иодидом урана был
тетраиодид [64]. Гишар [65] в результате исследований, основанных на
наблюдениях Муассана [66], установил, что при обработке металлического урана
в запаянной трубке иодом при температуре 550° и давлении 1 атм получается
продукт, состав которого соответствует тетраиодиду. Однако Гишар сообщает,
что это соединение плавится при температуре выше 500° и практически не
летуче, что характерно для трииодида, а не для тетраиодида.
Карбид урана также, повидимому, реагирует с иодом, но получить таким
путем чистый продукт не удалось 167]; однако, судя по более поздним работам
[68], эта реакция имеет, вероятно, некоторые преимущества.
Ранние попытки получить тетраиодид действием иода на смесь двуокиси
урана с углем окончились неудачей [35]. Исследование соответствующих
термодинамических величин показывает, что состояние равновесия этой реакции
невыгодно для иодирования. Попытки получить иодиды урана иодированием
двуокиси урана действием трииодидов алюминия, сурьмы и висмута пока не
дали результатов, хотя известно, что какая-то реакция в этом случае
протекает [68]. При действии йодистого водорода на тетрахлорид образуется,
повидимому, некоторое количество тетраиодида, однако получающийся продукт
сильно загрязнен хлоридами [36]. Исследованы также способы получения
тетраиодида урана в водных растворах. Установлено, что такие водные
растворы могут быть получены растворением гидратированной гидроокиси урана (IV)
в иодистоводородной кислоте. Попытки обезвоживать такой раствор были
безуспешными. После удаления растворителя получался продукт, содержащий
значительное количество свободного иода [69]. Недавно эта работа была
повторена в Металлургической лаборатории [70, 71]. Обезвоживание
проводилось в вакууме или в токе смеси водорода с йодистым водородом;
анализ продукта дал для него отношение урана к иоду, равное 2,70—2,73.
Ввиду трудностей, возникающих при обезвоживании растворов других
галогенидов урана (IV) (стр. 421), образование продукта с таким низким
содержанием иода (содержащего, повидимому, двуоокись урана)
неудивительно.
Получение три- и тетраиодида урана
взаимодействием урана с иодом. Наиболее удобным методом получения
обоих иодидов является прямое соединение элементов. Трииодид урана получают
медленной перегонкой стехиометрического количества иода в эвакуированную
колбу, содержащую тонкоизмельченный уран при 350°. После поглощения всего
иода реакционный сосуд герметически закрывают и нагревают сначала при
130° в течение нескольких часов и затем 15—20 час. при 570°. Получающийся
продукт представляет практически чистый трииодид с очень малой примесью
тетраиодида [34]. Такой же способ может быть применен и для получения
тетраиодида, однако в этом случае желательно к системе присоединить резервуар
с иодом, чтобы в течение всего хода реакции можно было поддерживать
парциальное давление иода около 1 атм.
Реакция может быть также проведена поточным методом, что удобнее
в тех случаях, когда требуется получать большие количества иодида. На
рис. 63 показан прибор, применявшийся в радиационной лаборатории
Калифорнийского университета. В этом случае пользовались горизонтальным
реактором, соединенным с линией вакуума. К входному отверстию реактора
Глава 15. Бромиды, иод иды и псевдогалогениды урана
427
присоединяли резервуар с иодом, и пары иода пропускали через металлический
уран, нагретый до 525°. Приемник отделялся от реактора пористым стеклянным
диском, температура в приемнике поддерживалась на уровне 400°. Нужное
парциальное давление иода достигалось погружением резервуара с иодом
в баню, нагретую до 30°. Реакция идет довольно медленно, но с хорошим
выходом. Чистота получающегося трииодида составляет 98—99% [68].
К вакуум -линии через ловушку
с ншдним воздухом
ЗЕЕ^_
Рис. 63. Прибор для получения трииодида ураиа.
/—реакционная зона печи. 525° (длиной примерно 25 см); 2—конденсационная зона печи, 400° (длиной
примерно 25 см); 3 — конденсационная зона печи, 350° (длиной примерно 25 см); 4 — перфорированный
диск из алунда; 5 —пришлифованный стеклянный диск, посаженный на сургуче: 6 — иод; 7 —
приемник для трииодида урана; 8 — кран для откачивания приемника после отключения его от трубки;
5>—порция металлического урана, внесенная через отверстие, закрытое стеклянным диском 5; 10 —
место максимального осаждения трииодида урана; У/ —место осаждения тетраиодида урана; /2
—крышка, предохраняющая приемник от загрязнения; 13 —скребок из нержавеющей стали для снятия осадка
трииодида урана; 14— уплотнение Вильсона с сильфоном; 15 — пришлифованное стекло, посаженное
на сургуче; 16—шлифы.
Тот же прибор может быть применен для получения тетраиодида.
Единственная разница заключается в том, что парциальное давление иода должно
быть достаточно высоким для того, чтобы подавить диссоциацию тетраиодида:
1
Шд
1Д. + -Ы,
(28)
Таблица 189
ОТНОШЕНИЕ ИОД : УРАН КАК
ФУНКЦИЯ ТЕМПЕРАТУРЫ КОНДЕНСАЦИИ
Для этого достаточно давления в 100—200 мм рт. ст. (что соответствует
температуре резервуара с иодом 120—140°). Чтобы иод не проходил через систему
слишком быстро, реактор присоединяется к линии вакуума через капилляр.
Рекомендуемая температура приемника 300°.
Давление иода в системе нельзя понижать
до тех пор, пока приемник не охладится.
В предварительных опытах,
проведенных по этому методу, пары иода пропускали
над металлическим ураном при 500—530° и
получающиеся продукты осаждали на стенках
приемника, имевшего температурный градиент.
Затем приемник разрезали на части и состав
возгона определяли как функцию
температуры поверхности [72] (табл. 189).
Приведенные результаты дают указания
относительно оптимального температурного
интервала конденсации, а также
свидетельствуют о существовании какого-то высшего иодида. Дальнейшее
исследование этого вопроса представляет определенный интерес.
Температурный
интервал, °С
310—386
278—354
250—290
235—253
Отношение
J: U
3,05-2,99
3,65-3,19
4,54—4,20
5,69 80
428 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Удобным методом получения тетраиодида является иодирование трииодида.
Для этого применяется вертикальный реактор, на дне которого находится иод
в жидком состоянии, а в верхней, суженной, части помещается трииодид.
Образующийся тетраиодид плавится и стекает на дно в избыток иода.
Другая исследованная реакция иодирования заключалась в обработке
гидрида урана метилиодидом при температуре 275—300°. Реакция протекает
быстро и сопровождается образованием трииодида, однако сведений о чистоте
получающегося продукта не имеется [73].
Физические свойства три- и тетраиодида.
Отсутствие надежных данных о физических свойствах иодидов является серьезным
пробелом в наших современных знаниях по химии галогенидов урана.
Температуры плавления и кипения [60]. Точка плавления трииодида равна
примерно 680°. Температура плавления тетраиодида 506° (при измерении
в атмосфере иода); температура кипения приблизительно 762°.
Кристаллическая структура [74]. Три- и тетраиодид урана образуют
черные игольчатые кристаллы. Трииодид кристаллизуется в ромбической
системе, элементарная ячейка его содержит четыре молекулы:
аг= 13,98 ± 0,02А; а2-4,33 ± 0,02А; а3 = 9,99 ± 0,02А.
Рассчитанная плотность трииодида равна 6,76 г/см3. Трииодид урана изоморфен
иодиду лантана и бромидам неодима и самария, он принадлежит к
пространственной группе симметрии Сстт (Djh). Положения атомов следующие:
4U в
4J в ±(*20-i-);
хг = 0,250;
х2 = 0,069;
8J
в ±(*3 0г3)(*з 0-g—*8); *s = 0,361; г3=-0,050.
Каждый атом урана связан с восемью атомами иода со средним межатомным
расстоянием U—J, равным 3,35 А.
Кристаллографических данных для тетраиодида не имеется.
Давление пара тетраиодида [53]. Трииодид урана сравнительно не
летуч и напоминает в этом отношении трихлорид. Тетраиодид более летуч.
Сделано несколько определений давления пара. Эти измерения осложняются
диссоциацией тетраиодида на трииодид и иод, в связи с чем оценка точности
результатов довольно затруднительна. Данные, приведенные в табл. 190,
Таблица 190
ДАВЛЕНИЕ ПАРА ТЕТРАИОДИДА
(МЕТОД ИСТЕЧЕНИЯ)
Таблица 191
Температура, °С
300
330
350
360
380
410
Давление пара, мм
рт. ст
2,4 .10-5
2,32-Ю-4
9,48. Ю-4
1,98-Ю-3
7,04-Ю"3
4,25-Ю-2
ДАВЛЕНИЕ
С ТРИ-
Температура,
523
588
625
644,2
666
ИОДА В РАВНОВЕСИИ
И ТЕТРАИОДИДОМ
°К
Давление
рт.
8,82
3,82
1,84
4,17
8,05
иода, мм
ст.
ю-8
ю-«
ю-6
ю-6
ю-*
пригодны, вероятно, для грубой оценки летучести тетраиодида. Из этих
данных вычисляется теплота возгонки ДЯВОЗг=52 800 кал-моль*1, это значение
кажется завышенным при сравнении с данными для других галогенидов
Глава 15. Бромиды, иодиды и псевдогалогениды урана
429
урана (IV). В более поздней работе, выполненной в радиационной лаборатории
Калифорнийского университета, приведенные значения были приблизительно
подтверждены. Полученные в этой лаборатории данные по давлению пара
тетраиодида соответствуют следующему уравнению (ср. [73, 75]):
lgp= 15,53- 11'5^103 . (29)
Термохимические данные [27]. В радиационной лаборатории
Калифорнийского университета определены следующие значения для теплот и свободных
энергий образования три- и тетраиодида:
з
д#298= — 136,9 ± 0,6 ккал'М0ль~\
UTB + 2J2ra3 —» Ш4ТВ; (31)
Д#298= — 156,7 ± 0,6 ккал • моль'1.
Энтропия 5298 твердых трииодида и тетраиодида определена приблизительно
равной 59,5 и 68,0 энтропийных единиц соответственно. Отсюда вычислены
свободные энергии: д7°298 =—123,2± 1,5 ккал • моль~{ для трииодида и
^F°298=—136,2± 1,5 ккал • моль~1 для тетраиодида.
Химические свойства три- и тетраиодида урана.
Отношение к воде. Иодиды урана очень гигроскопичны и легко растворяются
в воде. Трииодид урана образует красивый тёмнокрасный раствор; растворы
тетраиодида имеют характерный зеленый цвет иона урана (IV) [72]. В водном
растворе тетраиодид сильно гидролизован, как это видно из сильнокислой
реакции раствора.
Растворение трииодида в воде часто сопровождается энергичной реакцией.
В свежеприготовленном виде трииодид очень бурно реагирует с водой (или даже
с влагой воздуха) с образованием густых облаков иода. Предварительное
нагревание трииодида до 800° сильно замедляет эти реакции [73].
Восстановление иодидов. Водород при умеренно повышенных температурах
восстанавливает тетраиодид до трииодида и йодистого водорода. Трииодид
может быть восстановлен электрохимическим путем. Так, при электролизе
раствора трииодида в расплавленном иодиде стронция получается
металлический уран [34]. Для электролиза можно применять вольфрамовый анод и
молибденовый катод; температура ванны поддерживается на уровне 540°.
Окисление иодидов. Трииодид урана умеренно устойчив в растворах
бескислородных кислот, но легко окисляется в щелочном растворе [76, 77].
Тетраиодид урана реагирует с хлором при комнатной температуре с выделением тепла.
При действии кислорода или сухого воздуха при повышенных температурах
трииодид и тетраиодид превращаются в закись-окись урана, при комнатной
температуре—в уранилиодид. В последнем случае предполагают образование
промежуточных продуктов, например оксидииодида1ЮЛ2, однако определенные
данные, касающиеся этого вопроса, отсутствуют [70].
Термическое разложение тетраиодида урана. Как упоминалось выше ^
[ср. уравнение (28)], между тетраиодидом, трииодидом и иодом существует
равновесие. Были измерены давления иода в равновесии с твердыми трииодидом
и тетраиодидом [78] (табл. 191).
Если для ДСР в уравнении (28) принять значение —1,54, можно рассчитать
следующие соотношения [27]:
ДЯ= 18 720-1,54 7;
Д#298=18,3 ккал-моль'1', AF°298=--11,51 ккал-моль'1;
Д5298 = 22,64 энтропийной единицы.
430 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Эти данные могут быть использованы для расчета парциального давления иода,
требующегося для получения три- и тетраиодида из металлического урана
и иода.
Термическое разложение тетраиодида урана находит практическое
применение при получении металлического урана методом раскаленной проволоки
[79]. Этот метод основан на термической диссоциации тетраиодида при
достаточно высоких температурах:
UJ4ra3 ^ ^крист "Т ^2 газ' ("U
Разложение осуществляется на накаленной добела нити в эвакуированной
системе. При температурах выше 1300°К тетраиодид термодинамически
неустойчив в отношении составляющих его элементов. Затруднения вызывает
распад тетраиодида на иод и нелетучий трииодид [согласно уравнению (28)].
Для того чтобы достигнуть благоприятного соотношения между желательным
[уравнение (31)] и нежелательным [уравнение (28)] направлением процесса,
необходимо тщательно регулировать количество газообразного иода. Поэтому
эти два равновесия распада изучены в некоторых деталях (ср. стр. 112,
«Получение металлического урана»).
Разные реакции. При повышенных температурах трииодид является
довольно коррозионно активным веществом. При 800° стекло и фарфор
разъедаются им с образованием кремния. Из металлов никель и серебро более
устойчивы, чем платина [731.
CMFllIAHHblE ГАЛОГЕНИДЫ ТРЕХ- И ЧЕТЫРЕХВАЛЕНТНОГО УРАНА [60, 80]
Кристаллические структуры галогенидов трех- и четырехвалентного урана
являются такими, что в любом из этих соединений возможно замещение одних
атомов галогена другими в широком интервале составов. При некоторых сте-
хиометрических соотношениях твердые растворы обладают максимальной
устойчивостью и ведут себя как истинные соединения. Возможно существование
шести классов соединений, содержащих 18 отдельных веществ типа UX^Y^n,
где X и Y—два различных галогена. Получено по крайней мере по одному
соединению каждого класса. Известно также несколько смешанных
галогенидов трехвалентного урана.
Способы получения смешанных галогенидов не могут быть основаны на
замещении одного атома галогена другим, так как такие реакции не идут.
Для получения смешанных галогенидов урана (IV) имеются два метода:
1) действие на галогенид трехвалентного урана галогеном с более высоким
атомным номером и 2) введение в реакцию двух разных тетрагалогенидов при
высоких температурах. Смешанные галогениды урана (III) могут быть
получены следующими способами: 1) термическим разложением смешанных
галогенидов урана (IV), 2) восстановлением смешанных соединений урана (IV)
водородом и 3) сплавлением двух тригалогенидов.
В табл. 192 перечислены все известные в настоящее время смешанные
галогениды урана. Методы получения и большинство их свойств будут
рассмотрены отдельно в том порядке, в котором эти соединения приведены в таблице.
Однако для удобства сведения о давлении пара и термохимические данные для
всех смешанных галогенидов будут представлены в форме обзора на
стр. 438, 439.
Наиболее обширные исследования в этой области были проведены в
радиационной лаборатории Калифорнийского университета. Для более подробного-
ознакомления с данным вопросом следует рекомендовать статьи Грегори [811.
Хлор-, бром- и иодфториды урана (IV). Монохлортри-
фторид урана UC1F3 [82, 831. Это соединение было получено по реакции
UF3 + 4-Cla _>UCIF3. (32)
Таблица 192
СМЕШАННЫЕ ГАЛОГЕНИДЫ ТРЕХ- И ЧЕТЫРЕХВАЛЕНТНОГО УРАНА
Соединение
UC1F,
UC13F2
UBrF,
UJF3
UBrCl3
UBr2CI2
UBr3Cl
UJC13
UJ2C12
UJ3CI
Метод получения
UF3+i-Cl2
U02F2+2CC14
UF3 + -iBr2
UF3+Ij3
UC!3+i-Br2
UCI4 + UBr4
UCl4 + 3UBr4
UCl3+i-J2
UJ4 + UC14
UJ3 + UC14
Т. пл.,
oCa)
(460)
521
510
502
<490
<500
< 500
Т. кип.,
°C
784
780
771
Устойчивость
Устойчив, но не может
быть возогнан
Диспропорционирует на
UQ4 и UF4
Устойчив до 450°
9 ниже 100°
Возгоняется без разложения
» ь ъ
ь ь »
Выше 250° разлагается с
выделением иода
То же
Возгоняется без
разложения при низком давлении
иода в системе
Температура, при
которой лавление
пара равно
1 • Ю-з мм, рт.
ст.а), °С
356
340
341
(340)
(340)
(340)
ккал -моль~1
-220,3± 1,5
-210,5+1,5
-200,5± 1,5
-206,4± 1,5
ЛЯ298»
ккал 'М0ль~^
-241,6
-230,6
-220,1
-227,3
5298-
энтропийные
единицы
50,0
57,7
60,7
53,1
Продолжение табл. 192
Соединение
ШВг3
Ш2Вг2
Ш3Вг
UBrCl2
UBr2Cl
UJC12
ШаС1
ШВг2
Ш2Вг
Метод получения
UBr3 + yJ2
UJ4 + UBr4
3UJ4+UBr4
2UC!3 + UBr3
UBr3Cl + H2
UCI2J2->UJCi2 + yJ2
UCI3 + 2UJ3
Ш2Вг2 -> UJBr2 + yJ2
UJ3Br -* Ш2Вг + i- J2
Т. пл.,
оСа)
478
<500
<500
(800)
(775)
(750)
(725)
(700)
(690)
Т. кип.,
°С
Устойчивость
Возгоняется без
разложения при низком давлении
иода в системе
При 300° разлагается с
выделением иода
При 250° разлагается с
выделением иода; менее
устойчив, чем Ш3С1
Устойчив
»
»
»
»
»
Температура, при
которой давление
пара равно
1 • Ю-з мм. рт.
ст.а), °С
330
(330)
(330)
(700)
(700)
(700)
(700)
(700)
(7С0)
А^2 98»
ккал'моль"!
-177,5± 1,5
-187,2± 1,5
-178,0± 1,5
д#298»
ккал *моль~1
-195,4
-202,1
-191,9
52 в»
энтропийные
единицы
70,9
44,8
50,6
О Числа в скобках являются ориентировочными значениями.
Глава 15. Бромиды, иодиды и псевдогалогениды урана
433
При 315° реакция идет медленно и для ее завершения требуется много часов.
Обработка трифторида хлористым водородом не дает монохлортрифторида.
Рентгенографическое исследование загрязненного образца
монохлортрифторида показало, что он имеет кубическую структуру с ах=8,64 А. Это
соединение довольно устойчиво и не разлагается при нагревании до 400° в атмосфере
аргона с выделением хлора. Однако летучестью оно не обладает.
Дихлордифторид урана UC12F2. Дихлордифторид урана был впервые
лолучен [84, 85] по реакции
U02F2 + 2СС14 -+ UC12F2 + 2СОС12 + С12. (33)
Реакция может быть проведена либо в жидкой фазе при температуре 130° под
давлением, либо в газообразной фазе при 450°. Второй способ более удобен.
Интересно отметить, что при осуществлении реакции в жидкой фазе получается
небольшое количество вещества, растворимого в четыреххлористом углероде,
которое, как предполагают^ является смешанным галогенидом пятивалентного
урана, например UC13F2 или UC14F. Однако соединения такого типа пока еще
точно не идентифицированы. При растворении в воде дихлордифторид дает ионы
хлора и фтора, что указывает на то, что это вещество не является механической
смесью тетрахлорида и тетрафторида, так как последний нерастворим в воде.
При нагревании дихлордифторид подвергается диспропорционированию:
2UC12F2 -> UF4 + UC14. (34)
Тетрахлорид может быть удален возгонкой в вакууме. Смешанный галогенид
в таком виде не может быть перегнан [84, 85].
Сделано несколько попыток [83, 86] приготовить дихлордифторид по
реакции
UC14 + UF4 -> 2UC12F2. (35)
На основании отдельных наблюдений можно, повидимому, сделать вывод, что
дихлордифторид в этом случае действительно образуется и плавится при
температуре около 460°. Однако приготовленный таким образом продукт менее
чист, чем тот, который получается при взаимодействии уранилфторида с четы-
реххлористым углеродом. Это, вероятно, объясняется более высокой
температурой реакции.
Монобромтрифторид урана UBrF3 [83]. Указанный галогенид может
быть получен тем же путем, что и соответствующее хлоропроизводное:
UF3 + 4Br2 -> UBrF8- (36)
Реакцию можно осуществить путем пропускания паров брома в токе гелия^над
трифторидом при 250°. Обычно, если только не проводить реакцию в течение
очень длительного времени, получается смесь бромтрифторида и трифторида
урана. При температуре 450° в атмосфере гелия бромтрифторид менее
устойчив; водород при 200—400° медленно восстанавливает его до трифторида.
Моноиодтрифторид урана UJF3 [83]. Пары иода (в токе гелия) реагируют
с трифторидом при 180° с образованием смеси трифторида и иодтрифторида.
Эта смесь имеет коричневато-черный цвет, гигроскопична, на воздухе при
комнатной температуре из нее выделяется иод. В отсутствие воздуха и влаги
моноиодтрифторид, повидимому, устойчив до 100°. Это соединение еще не
вполне идентифицировано.
Ни хлор-, ни бром-, ни иодфторид нельзя выпарить без разложения.
Бром- и иодхлориды урана (IV). Эти соединения были
изучены лучше всех остальных смешанных галогенидов урана. В настоящее
время имеются данные по упругостям пара, теплотам образования и
кристаллохимии нескольких членов этой группы. Они вполне точно идентифицированы,
Химия урана
434 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
однако желательно было бы иметь по ним более обширные рентгенографические
данные.
Монобромтрихлорид урана UBrCl3 [86—88]. Лучше всего получать это
соединение бромированием трихлорида урана:
UCl3 + -^-Br2-^UBrCl3. (37)
Реакция проводится нагреванием трихлорида с парами брома в закрытой системе
до 500°. Ввиду того что скорость бромирования сильно зависит от
парциального давления брома, последнее должно поддерживаться на уровне,
соответствующем давлению пара над жидким бромом при комнатной температуре.
Получающийся монобромтрихлорид очищается возгонкой.
Скорость реакции (37) с жидким бромом при 125° слишком мала для того,
чтобы этот вариант имел практическое значение. J
Изучено также несколько других методов получения монобромтрихлорида.
Достаточно хорошие результаты могут быть получены сплавлещгем тетрабро-
мида с тетрахлоридом:
UBr4 + 3UCl4 -> 4UBrCl3. (38)
Однако,продукт возгонки содержит до 20% тетрахлорида. Аналогичная
реакция проведена с трихлоридом и тетрабромидом:
UCl3 + UBr4 ^1 UBrCl3 + UBr3. (39)
Реакция идет при 550°, но при этом, повидимому, устанавливается равновесие.
Продукт, получающийся в результате возгонки реакционной массы,
представляет собой смесь монобромтрихлорида и тетрабромида. Поэтому эта реакция
непригодна для получения чистого монобромтрихлорида.
Другой путь получения последнего, изученный, правда, лишь частично,
заключается в разложении комплексных соединений тетрахлорида урана с
бромидом натрия. При нагревании этих веществ до 500° образуется соединение,
имеющее, повидимому, состав Na2UCl4Br2. При возгонке этого соединения
в вакууме получается смесь тетрахлорида (25%) и монобромтрихлорида (75%).
При сплавлении тетрахлорида урана с бромидом натрия при 700° и возгонке
в вакууме получается смесь, состоящая из 73% монобромтрихлорида и 27%
дибромдихлорида урана. Для объяснения этих результатов были предложены
следующие реакции:
UCl4 + 2NaBr ^l Na2UBr2Cl4; (40)
Na2UBr2Cl4 -> UBrCl3 + NaBr + NaCl; (41)
UBrCl8 + 2NaBr ^ Na2UBr3CI3; (42)
Na2UBr3Cl3 -* UBr2Cl2 + NaBr + NaCl. (43)
Подобным же образом можно получать соединение Na2UBr4Cl2, однако при
возгонке последнего получается только смесь триброммонохлорида и
дибромдихлорида.
Монобромтрихлорид урана представляет гигроскопичное темнозеленое
кристаллическое вещество, которое во многих отношениях напоминает тетра-
хлорид. Оно легко растворяется в воде с образованием зеленого раствора, из
которого при действии азотной кислоты может быть вытеснен бром.
Температура плавления монобромтрихлорида равна 521°; ориентировочное значение
теплоты плавления составляет 11,6 ккал/моль. При действии водорода при
400—500° образуется смесь, состоящая приблизительно из 70% (мол.) моно-
бромдихлорида и 30% (мол.) трихлорида. Термодинамика этого равновесия
будет обсуждаться ниже.
Глава 15. Бромиды, иод иди и псевдоеалогениды урана 435
Монобромтрихлорид термически устойчив и может быть возогнан в
вакууме без разложения. В сухом воздухе он не изменяется, но с парами воды
быстро реагирует с образованием оксигалогенидов.
Проведено предварительное исследование кристаллической структуры
монобромтрихлорида [23, 89]; к сожалению, исследованию подвергали только
вещества довольно неопределенного состава. Повидимому, монобромтрихлорид
имеет тетрагональную структуру, очень сходную со структурой тетрахлорида.
Константы решетки его следующие: ах =8,434 А, а3 =7,690 А, и, таким
образом, размеры элементарной ячейки его немного превосходят размеры ячейки
тетрахлорида. Из рентгенограммы видно, что решетка смешанного галогенида
не является точно объемноцентрированной и что замещение хлора бромом не
является неупорядоченным.
Дибромдихлорид урана (IV) UBr2Cl2 188]. Лучше всего получать это
соединение сплавлением (при 590°) эквимолекулярной смеси тетрахлорида и тетра-
бромида в кварцевой трубке приблизительно в течение трех дней. Реакцию
проводят в атмосфере гелия. По окончании нагревания обычно около трех
четвертей всей исходной смеси получается в виде возгона. Образующийся
продукт представляет собой темнозеленое кристаллическое вещество и имеет состав
UBr2Cl2 (с содержанием приблизительно 1 % избытка тетрахлорида). Состав
возгона указывает, что последний является химическим соединением, а не смесью
тетрахлорида и тетрабромида, так как эти вещества сильно различаются по
давлению пара, и содержание брома в возгоне, полученном из
эквимолекулярной смеси, было бы намного больше, чем это имеет место на самом деле.
По внешнему виду дибромдихлорид больше напоминает тетрахлорид, чем
тетрабромид. Его температура плавления равна 510°; теплота плавления состав-^
ляет ориентировочно 12,3 ккал-моль'1. Температура кипения (рассчитанная из
приводимых ниже данных по давлению пара) равна 780°. Восстановление его
водородом (350—450°) дает смесь примерно 60% диброммонохлорида и 40% мо-
нобромдихлорида. Термодинамика процесса восстановления будет обсуждаться
ниже (стр. 439). Дибромдихлорид термически устойчив и может быть возогнан
в закууме без разложения. В сухом воздухе он не изменяется, но его необходимо
защищать от следов влаги, так как он явлйется очень гигроскопичным.
Триброммонохлорид урана (IV) UBr3Cl. Продукт состава UBr3Cl получается
в виде возгона при нагревании смеси тетрахлорида с тетрабромидом (с
молярным соотношением 1 : 3) до 590° в течение одного дня в атмосфере гелия.
Прямое хлорирование трибромида дает лишь тетрахлорид. Как и можно было
ожидать, триброммонохлорид напоминает скорее тетрабромид, чем тетрахлорид.
Триброммонохлорид представляет собой светлокоричневое вещество,
плавящееся при 502°; теплота плавления его составляет 13,6 ккал-моль*1 160]. Он
может быть возогнан в вакууме без разложения. При реакции с водородом
(350—450°) образуется смесь, состоящая из 40% трибромида и 60%
диброммонохлорида урана.
Моноиодтрихлорид урана (IV) ШС13 [87]. Ввиду термической
неустойчивости этого соединения получить его в чистом виде довольно трудно. Оно
может быть приготовлено иодированием трихлорида:
UCl, + 4-J«^=uJC,3- (44)'
В газообразной фазе моноиодтрихлорид, повидимому, устойчив даже при 500°
Однако при осаждении твердого вещества наблюдается разложение. Поэтому
существенно важно поддерживать парциальное давление иода в системе выше
того, которое соответствует давлению разложения моноиодтрихлорида.
Это соединение представляет собой вещество цвета от
красновато-коричневого до черного, отдающее иод даже при 225° в вакууме. Давление
разложения составляет 5« 10~4 мм рт. ст. при 200° и 1,34-10"1 мм рт. ст. при 302°.
28*
436 Часть IV. Галогениды, оксигалогенидЫу боргидриды и карбонил урана
Разложение протекает настолько интенсивно, что делает невозможным
измерение давления пара моноиодтрихлорида; однако имеются указания, что давление
пара этого соединения больше, чем у тетрахлорида, и, вероятно, весьма близко
к давлению пара тетраиодида.
Моноиодтрихлорид чрезвычайно гигроскопичен. Во влажном воздухе он
отдает иод с образованием оксигалогенида. Поэтому его нельзя вводить
в соприкосновение с воздухом или влагой.
Дииоддихлорид урана (IV) Ш2С12. Это соединение может быть получено
тем же путем, что и дибромдихлорид, т. е. сплавлением эквимолекулярной смеси
тетрахлорида с тетраиодидом (в присутствии небольшого количества иода для
уменьшения разложения тетраиодида) [60]:
UC14+UJ4
2Ш2С12.
(45)
Дииоддихлорид представляет собой черное кристаллическое вещество. Он
определенно более устойчив, чем моноиодтрихлорид. Упругости пара и
температуры плавления обоих этих соединений, повидимому, весьма близки.
Трииодмонохлорид урана (IV) Ш3С1. Это соединение было получено
сплавлением тетрахлорида урана с трииодидом при температуре, превышающей
точку плавления тетрахлорида [86]:
Шз + UClj
G00°
ULC1 + UCL
(46)
После возгонки получается продукт, содержащий свыше 90% трииодмонохлори-
да и примесь трихлорида в качестве наиболее стойкого загрязнения.
Присутствие последнего в сублимате вызвано небольшими количествами свободного
иода, который неизбежно образуется при действии воздуха и влаги на исходный
трииодид. Под действием этого иода трихлорид превращается в
моноиодтрихлорид, который возгоняется вместе с трииодмонохлоридом, но разлагается
вновь на трихлорид и иод после конденсирования.
Трииодмонохлорид представляет собой черное кристаллическое вещество
с красным оттенком в тонкоизмельченном состоянии. Он гораздо более
устойчив, чем моноиодтрихлорид или любой другой иодхлорид, и может быть возо-
гнан без заметного разложения при низком парциальном давлении (0,1 мм
рт. ст.). Он немного более летуч, чем тетрабромид урана.
Иодбромиды урана (IV). Известны все три возможных члена
этого класса. Они представляют собой сравнительно неустойчивые соединения,
которые легко теряют иод.
Единственным членом этой группы, который
может быть возогнан без разложения,
является моноиодтрибромид.
Представляет интерес сравнение термической
устойчивости иодбромидов и иодхло-
ридов (табл. 193).
Все иодбромиды чрезвычайно
гигроскопичны, и их необходимо
получать и хранить без доступа воздуха
и влаги. Все они представляют собой
черные кристаллические вещества.
Моноиодтрибромид урана (IV) ШВг3. Лучше всего получать это
соединение по реакции
Таблица 193
ТЕРМИЧЕСКАЯ УСТОЙЧИВОСТЬ
ИОДХЛОРИДОВ И ИОДБРОМИДОВ
Устойчивость
Наибольшая
Средняя
Наименьшая
Исдхлориди
Ш3С1
Ш2С12
ШС13
Иодбромиды
ШВгз
Ш2Вг2
Ш3Вг
ивгз+4-J
500°
ШВгч
(47)
При охлаждении необходимо поддерживать парциальное давление иода
в 0,5 мм рт. ст. Этот метод вполне пригоден в данном случае ввиду того, что
Глава 15. Бромиды, иод иды и псевдогалоген иды урана 437
моноиодтрибромид гораздо более устойчив, чем моноиодтрихлорид (для
приготовления которого этот метод менее подходит).
Моноиодтрибромид летуч и обладает заметным давлением пара (Ы0~2 мм
рт. ст. при 375°). Он может быть возогнан без заметного разложения при
давлении иода в 0,1 мм рт. ст. При 400° в вакууме он разлагается на трибромид
и иод. Температура плавления его равна 478°.
Дииоддибромид урана (IV) Ш2Вг2. Наиболее удобным методом получения
этого соединения является сплавление эквивалентных количеств тетраиодида
и тетрабромида:
UJ4 + UBr4 -> 2Ш2Вг2. (48)
Качественно это соединение близко напоминает моноиодтрибромид, за
исключением того, что последний термически гораздо более устойчив.
Трииодмонобромид урана (IV) Ш3Вг. Сплавление смеси тетраиодида и
тетрабромида в соответствующих соотношениях приводит к образованию
трииодмонобромида:
UBr4 + 3UJ4 -> 4UJ3Br. (49)
Продукт реакции возгоняется из зоны реакции. Это соединение термически
неустойчиво. При 300° оно легко превращается в дииодмонобромид и иод.
Термическая устойчивость его сравнима с устойчивостью дииоддибромида.
Галогениды урана (IV), содержащие три
различных галогена. В чистом виде соединения иодбромдихлорид и иодди-
бромхлорид не были получены. Однако были получены их смеси по следующим
реакциям:
2UBr2Cl2 + H2 -* UBrCl2 + UBr2Cl4- HBr + HCl. (50)
Последующее иодирование продуктов реакции дает
UBrCl2 + UBr2Cl + J2 -> UJBrCl2 + UJBr2Cl. (51)
Имеются основания полагать, что иоддибромхлорид более устойчив в
отношении потери иода, чем иодбромдихлорид.
Смешанные галогениды урана (III). Смешанные
галогениды трехвалентного урана могут быть получены следующими способами:
1) термическим разложением смешанных галогенидов урана (IV); 2)
восстановлением смешанных галогенидов урана (IV) водородом и 3) сплавлением смеси
двух галогенидов урана (III).
Все смешанные галогениды урана (III) представляют собой черные
кристаллические вещества, напоминающие по своей термической устойчивости,
летучести и т. п. трихлорид и трииодид. Их физические и химические свойства
изучены слабо. Однако восстановление водородом галогенидов урана (IV)
позволило рассчитать термодинамические константы для монобромдихлорида
и диброммонохлорида урана.
Монобромдихлорид урана (HI) UBrCl2. При восстановлении монобром-
трихлорида водородом получается смесь монобромдихлорида и трихлорида.
Иодированием смеси можно удалить трихлорид в виде летучего моноиодтри-
хлорида. Однако наиболее удобным методом получения монобромдихлорида
является сплавление трихлорида с трибромидом:
2UCl3 + UBr3 850° -» 3UBrCl2. (52)
Продукт реакции может быть очищен от непрореагировавшего трихлорида
обработкой парами иода и отгонкой образующегося моноиодтрихлорида.
Диброммонохлорид урана (III) UBr2Cl. Это соединение приготовлено
восстановлением триброммонохлорида водородом. Получавшуюся этим путем
смесь диброммонохлорида и трибромида обрабатывали иодом; пары
образовавшихся моноиодтрибромида и иоддибромхлорида пропускали в токе паров иода
438 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
(0,1 мм рт. ст. ) через трубку, нагретую до 385°. При этих условиях иодди-
бромхлорид разлагается с образованием дибромхлорида, более же
устойчивый моноиодтрибромид отгоняется.
Сплавление трибромида и трихлорида (1 : 2) также должно приводить
к образованию диброммонохлорида урана.
Моноиоддихлорид урана (III) UJC12. Данное соединение в чистом виде
получается при термическом разложении дииоддихлорида урана в вакууме.
Моноиоддибромид урана (III) UJBr2. Этот галогенид получается
термическим разложением дииоддибромида в вакууме.
Дииодмонобромид урана (III) Ш2Вг. Это соединение может быть легко
получено термическим разложением трииодмонобромида в вакууме.
Все эти соединения растворяются в воде с образованием растворов красного
цвета. Они очень гигроскопичны, однако в меньшей степени, чем соединения
урана (IV).
Измерения давления пара. Значения давления пара моно-
бромтрихлорида [90], триброммонохлорида [91], дибромдихлорида [92] и моно-
иодтрибромида урана [93] были определены методом молекулярного истечения.
Полученные экспериментальные данные сведены в табл. 194 и 195. Теплоты
и свободные энергии возгонки даны в табл. 196.
Таблица 194
УРАВНЕНИЯ а), ОПИСЫВАЮЩИЕ ВЕЛИЧИНЫ ДАВЛЕНИЯ ПАРА
НЕКОТОРЫХ СМЕШАННЫХ ГАЛОГЕНИДОВ УРАНА (IV) [27]
Соединение
иВгС13
UBr2Cl2
UBr3Cl
ШВг3
А
13,852
13,149
13,280
13,416
в
10 526
9 901
10 000
9 901
Температурный
интервал, °С
320-430
330-404
320—420
315-382
а) 1*Рмм
рт. ст.'
-А*
Т
Таблица 195
ИНТЕРПОЛИРОВАННЫЕ ЗНАЧЕНИЯ ДАВЛЕНИЯ ПАРА ДЛЯ НЕКОТОРЫХ
СМЕШАННЫХ ГАЛОГЕНИДОВ УРАНА (IV), мм рт. ст.
Температура,
°С
320
330
340
350
360
370
380
390
400
410
420
430
UBrCl3
1,25-Ю-4
2,48
4,75
9,00
1,66
3,00
5,40
9,50
1,64
2,80
4,70
7,90
ю-4
ю-4
ю-4
Ю-3
ю-3
ю-3
ю-3
Ю-2
10-2
ю-2
Ю-2
UBr2Cl2
2,85-Ю-4
5,42-Ю-4
1,00-10~3
1,82-Ю-3
3,23. 10~3
5,70-10-а
9,80. Ю-3
1,67.10-2
2,77-Ю-2
4,62-Ю-2
иВг3С1
2,57.Ю-4
4,95. Ю-4
9,30-Ю-4
1,70-Ю-3
3,02-Ю-3
5,40-Ю-3
9,40-Ю-3
1.6Ы0-2
2,69-10-2
4,50-10-2
7,30-10-2
ШВг3
5,25-Ю-4
9,93-10"4
1,84.10"»
3,34-10"3
5,96-10-
1,04.10-2
1,79-Ю-2
Глава 15. Бромиды, иод иды и псевдогалоеениды урана 439
Таблица 196
ТЕПЛОТЫ И СВОБОДНЫЕ ЭНЕРГИИ ВОЗГОНКИ НЕКОТОРЫХ СМЕШАННЫХ
ГАЛОГЕНИДОВ УРАНА (IV)
Соединение
UBrCIa
UBr2Cl2
ивгзсГ
ШВг3
Д^В03Г,
ккал-моль~1
48,5
45,5
• 46,3
43,0
Д/гвозга), кал-моль 1
ДЯо
57 692
56 000
54 474
51 417
А
36,85
36,85
34,55
34,55
в
168,5
167,1
158,2
154,7
а) д^возг - U4±AT\Qr-BT.
Термохимия. Для семи смешанных галогенидов измерены теплоты
растворения, упругости диссоциации и равновесия восстановления водородом;
результаты этих измерений дают возможность рассчитать теплоты и свободные
энергии образования, а также энтропии этих соединений. Точные данные для
теплоемкости отсутствуют, и вытекающая отсюда неточность в значениях ДСР
для равновесий реакции восстановления отражается на конечных значениях.
Равновесия восстановления [94]. Результаты исследований равновесий
восстановления приведены в табл. 197.
Таблица 197
РАВНОВЕСИЕ РЕАКЦИИ ВОССТАНОВЛЕНИЯ СМЕШАННЫХ ГАЛОГЕНИДОВ УРАНА
ВОДОРОДОМ
Реакция
иВгС13тв + \ Н2газ 2 иВгС12тв + НС1газ
ивгС13тв+1н2газ г ис13тв+нвггаз
ивг2с12тв+-1 н2газ г ивггатв + нс1газ
UBr2Cl2TB + l Н2газ - UBrCI2TB + HBrra3
иВг3С1тв + 1н2газ - иВг3тв + НС1газ
UBr3ClTB + i-H2ra3 - UBr2C!TB + HBrra3
ДЯ?оо.
ккал-моль-1
16,2
16,3
17,6
16,9
17,1
16,9
д^° .
700'
ккаЛ'МОЛЬ~~±
1,73
2,98
2,68
3,18
3,61
2,98
Д57о<ь
энтропийные
единицы
20,67
20,03
21,62
20,03
19,84
20,47
Уравнение
(53)
(54)
(55)
(56)
(57)
(58)
Давление паров иода над моноиодтрибромидом [95] и моноиодтрихлоридом
урана [93]. Измерены давления разложения этих соединений как функции
температуры. Полученные результаты приведены в табл. 198.
Термодинамические константы. Эти константы определены для семи
галогенидов. Полученные результаты приведены в табл. 192. Относительно
подробностей вычислений мы отсылаем к статье Маквуда [96].
440 Часть IV. Галогениды, оксигалогениды, боргидриды и карбснил урана
Таблица 198
ЗНАЧЕНИЯ ДАВЛЕНИЯ ИОДА НАД ШС13 И ШВг3
ШС13 II
температура,
"С
198,6
221,3
241,0
261,7
280,0
302,0
давление J2.
мм рт. ст.
4,73-Ю-4
2,09.10-3
6,64-Ю-з
1,70.10-2
5,07-10-2
1,35.10-!
ШВг3
температура,
°С
277,3
293,2
314,9
331,1
349,7
368,2
381,4
давление J2,
мм рт. ст.
5,44-Ю-4
1,20.10-з
3,05-Ю-з
6,18-Ю-з
1,49-10-2
2,44.10-2
3,75-10-2
БОРГИДРИД УРАНА (IV) U(BH4)4
Боргидриды металлов [97, 98] являются сравнительно недавно
открытым классом соединений. Их можно рассматривать как «псевдогалогениды»„
где вместо галогена стоит радикал ВН4; такое представление оправдывает
помещение их в эту главу.
До открытия боргидрида урана были известны боргидриды лития,
бериллия и алюминия [99— 101]. Они получены действием диборана на
соответствующие металлалкилы:
2LiC2H5 + 2В2Н6 -> 2LiBH4 + (C2H5)2B2H4; (59)
Ве(С2Н5)2 + 2В2Н6 -» Ве(ВН4)2 + (С2Н5)2В2Н4; (60)
2А1(СЯН5)8 + 6В2Н6 -> 2А!(ВН4)3 + 3(С2Н5)2В2Н4. (61)
В действительности эти реакции идут гораздо сложнее, чем это
представлено уравнениями, так как в начальной стадии реакции образуются триалкил-
производные бора, а на последующих стадиях—сложные смеси алкилдибо-
ранов. Боргидриды лития и бериллия—солеобразные соединения, тогда как
боргидрид алюминия является типичным неполярным соединением.
Это—'летучая жидкость (т. кип. 44,5°), растворимая в бензоле. Ввиду того что
боргидриды бериллия и алюминия являются наиболее летучими соединениями
этих металлов, казалось заманчивым попытаться получить соответствующее
соединение урана.
Получение промежуточных продуктов. В отличие от бор-
гидридов других металлов боргидрид урана (IV) нельзя было получить
реакцией между металлалкилом и дибораном, так как ураналкилы неизвестны.
Наилучший из известных до сих пор методов получения боргидрида урана
основан на реакции боргидрида алюминия с тетрафторидом урана.
Описанный в литературе метод приготовления боргидрида алюминия является
очень длительным и пригоден лишь для получения небольших количеств этого-
вещества. Необходимо было поэтому разработать более практичные методы
получения боргидрида алюминия. Подробности этой интересной работы будут
опубликованы в другом месте.
Для получения боргидрида алюминия могут быть использованы
следующие реакции:
NaH + В(ОСН3)з -* NaBH(OCH3)3; (62>
2NaBH(OCH3)3 + B2He -> 2NaBH4 + 2В(ОСН3)3; (63)
3NaBH4 + AICI3 -> А1(ВН4)з + 3NaCl. (64)
Глава 15. Бромиды, иод иды и псевдогалоген иды урана 441
Диборан может быть получен [102] по одной из следующих двух реакций:
6LiH + 8(C2H5)20:BF3 -> В2Н6 + 6LiBF4 + 8(C2H5)20 (65)
или
6NaBH(OCH3)3+ 8(C2H5)20:BF3 ->
-* В2Ы6 + 6NaBF4 + 6В(ОСН3)3 + 8(С2Н5)20. (66)
Получение боргидрида урана (IV). Боргидрид урана (IV)
образуется при действии боргидрида алюминия на тетрафторид урана.
Реакция экзотермична и происходит самопроизвольно при комнатной температуре.
Ход ее выражается следующим уравнением:
UF4 + 2А1(ВН4)3 -* U(BH4)4 + 2A1(BH4)F2. (67)
Тетрафторид урана для этой реакции лучше всего получать дегидратацией
2UF4-5H20 (ср. гл. 12). Окончательно дегидратацию завершают перед самым
употреблением: реакционную трубку с моногидратом тетрафторида урана
присоединяют к линии вакуума и проводят дегидратацию при 350° и 1 • Ю-5 мм
рт. ст.; затем перегоняют боргидрид алюминия в реакционную трубку и
отключают вакуум. Ввиду того что при реакции (67) выделяется значительное
количество тепла, рекомендуется помещать реакционную трубку на несколько
часов в охлаждающую смесь из льда с солью, а затем давать реакции
заканчиваться при комнатной температуре. По окончании реакции трубку
снова соединяют с вакуумной линией через магнитный затвор и отгоняют
боргидрид урана и избыток боргидрида алюминия. Боргидрид урана
улавливается в U-образной трубке, охлажденной до —20°, а боргидрид
алюминия—в такой же трубке, погруженной в жидкий азот. Для возгонки
больших количеств (25 г) может потребоваться несколько часов, причем
необходимое время сильно зависит от. диаметра трубок*. Таким путем боргидрид
урана (IV) может быть получен с выходом 80—90% (считая на
использованный тетрафторид).
Сделаны попытки найти способ получения боргидрида урана, который
не требовал бы применения чрезвычайно легко самовоспламеняющегося
боргидрида алюминия. Единственной такой перспективной реакцией является
взаимодействие тетрафторида урана с боргидридом лития в эфирном
растворе. При этом образуется боргидрид урана, однако выход и чистота
получаемого продукта неудовлетворительны. Требуется дальнейшее исследование
этой реакции. Изучено несколько других процессов, однако все они не дали
желаемых результатов. Несколько подобных реакций перечислено ниже.
Действие боргидрида алюминия на тетрахлорид урана. Этим способом
получается немного боргидрида урана (IV), однако большая часть урана
восстанавливается до трехвалентного состояния.
Действие боргидрида лития, натрия или калия на тетрафторид или
тетрахлорид урана при температурах до 125° в отсутствие растворителей.
Боргидрид урана таким путем вообще не получался.
Действие диборана или карбонилборана ВН3-СО [103] на металлический
уран или его гидрид. Никакой реакции при этом не наблюдалось в широком
интервале температур и давлений. Однако известно, что взятые металлический
уран и гидрид были загрязнены, так что желательно повторное исследование
этой реакции.
Формула боргидрида урана установлена путем его гидролиза:
U(BHt)4 + 16H20->U(OH)4 +4H3B03 + 16Н2. (68)
* Нелетучий остаток после отгонки самопроизвольно и бурно воспламеняется на
воздухе; поэтому перед тем как впускать воздух в реакционный сосуд, необходимо
подвергнуть этот остаток гидролизу медленным впуском водяного пара.
-142 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Количество водорода измеряли с помощью насоса Теплёра; бор определяли
путем отгонки в виде метилбората и титрования гидроокисью бария с маннитом;
уран определяли в виде закиси-окиси. Из полученных данных вытекает формула
UlloB4>oH16tl или U(BH4)4.
Получение алкильных производных боргидрида
урана (IV). Обработка боргидрида урана триметилбором в эвакуированном
запаянном сосуде при 50—70° приводит к образованию зеленой умеренно
вязкой жидкости и небольшого количества нелетучего коричневого твердого
вещества. При температуре ниже 50° реакция протекает медленно.
Из этой смеси были выделены два летучих соединения урана. Менее летучее
представляет собой твердое кристаллическое вещество, бесцветное в тонких
слоях и от сиреневого до почти черного цвета в крупных кристаллах. Более
летучее соединение после очистки путем дробной возгонки является зеленым
твердым веществом. Разница в летучести этих двух соединений достаточно
велика для того, чтобы их можно было частично разделить дробной перегонкой
и возгонкой; зеленое вещество проходит через ловушку, охлажденную до —10°,
сиреневое—конденсируется в ней. Достигаемое таким образом разделение
неполно, однако обработкой сиреневого вещества избытком триметилбора можно его
очистить от последних следов зеленого соединения. Последнее вещество может
быть получено в чистом виде обработкой загрязненного препарата избытком
диборана при 30—40°. Выбор этих операций был продиктован строением
данных соединений, которое было установлено путем анализа, а также по
реакциям с водой и хлористым водородом (см. стр. 444). При взаимодействии зеленого
вещества с хлористым водородом получаются диборан и метилборхлориды
(главным образом СН3ВС12); в случае действия хлористого водорода на
соединение сиреневого цвета диборана не образуется. Ни в том, ни в другом случае
не выделяется метана. Ясно поэтому, что эти соединения содержат углерод
в виде метальных групп. Есть основания полагать, что в обоих соединениях
метальные группы присоединены к бору, а не к урану, что в сиреневом
соединении группы ВН4 отсутствуют и что каждый атом углерода присоединен
к отдельному атому бора. Вместе с данными анализа и определениями
молекулярного веса результаты изучения приведенных выше реакций дают
возможность утверждать, что зеленое соединение является монометилборгид-
ридом урана U(BH4)3(BH3CH3), сиреневое же—тетраметилборгидридом урана
U(BH3CH3)4. Упомянутые выше методы очистки этих соединений основаны на
том, что триметилбор должен алкилировать монометилборгидрид,
находящийся в виде следов в тетраметилборгидриде, а диборан—деалкилировать
тетраметилборгидрид, следы которого присутствуют в монометилборгид-
риде.
Триэтилбор реагирует с боргидридом урана так же, как и триметилбор,
однако из реакционной смеси невозможно выделить чистые соединения.
Это справедливо и для высших алкильных производных бора, которые также
взаимодействуют с боргидридом урана, но дают реакционные смеси, которые
пока не удалось разделить. Летучесть этильных и более высоких алкильных
производных боргидрида урана, очевидно, гораздо меньше, чем
соответствующих метальных соединений.
Физические свойства боргидридов урана (IV).
Величины давления пара определены с помощью стеклянного манометра
Бурдона в цельностеклянной аппаратуре. Полученные данные сведены в
табл. 199 и 200.
Тот же прибор, который применялся для измерения упругости пара, был
использован и для определений молекулярных весов. Эти определения
показали, что данные соединения представляют собой мономеры.
Глава 15. Бромиды, иодиды и псевдогалогениды урана 443
Таблица 199
ЗНАЧБНИЯ КОНСТАНТ В УРАВНЕНИЯХ а)
ДЛЯ ЗАВИСИМОСТИ ДАВЛЕНИЯ ПАРА БОРГИДРИДОВ
ОТ ТЕМПЕРАТУРЫ (ПО ОПЫТНЫМ ДАННЫМ)
Формула соединения
U(BH4)4
и(ВН4)з(ВН3СНя)
U(BH3CH3)4
А
4264,6
3150,0
2970,0
а) *8Рмм рт. <n. = R-f ■
ИНТЕРПОЛИРОВАННЫЕ ЗНАЧЕНИЯ ДАВЛЕНИЯ IIAI
ММ РТ. СТ.
Температуря, СС
30
35
40
45
50
55
60
65
70а)
75й)
U(BH4)4
в
13,354
10,679
8,820
Таблиц л 200
\\ ПОРГИДРИДОВ УРАНА.
1т(ВН|)з(ВНяСН3)
1 !
0,19 | 1,92
0,33
0,54
0,89
1,43
2,27
3,56
5,51
8,40
12,68
2,83
4,12
5,93
8,45
11,89
16,48
22,91
31,26
42,36
1ДВН3С11з)|
0,10
0,15
0,21
0,30
0,42
0,58
0,80
1,08
1,45
1,93
а) Значения при этих температурах получены при помощи значительной экстраполяции.
Боргидрид урана возгоняется без плавления; он плавится при 126° с
интенсивным разложением. Температура плавления U(BH3CH3)4 равна примерно
72—74°. Точность данных по температуре плавления монометильного
производного сомнительна. Экспериментально определенная температура равнялась 95°,
но она относится к образцу, возможно, подвергшемуся диспропорционированию
на U(BH4)4 и более полно метилированные производные.
Фотохимические свойства [104, 105]. В парообразном состоянии
боргидрид урана разлагается и фотохимически и термически. Поэтому спектр
поглощения его мог быть изучен лишь при очень низких давлениях. Найдена
сплошная полоса поглощения при 21 000 см'1 (р=4 мм рт. ст., d=50 см).
Химические свойства бор гидрида урана и его
алкильных производных. Боргидрид урана (IV) представляет
собой блестящее зеленое кристаллическое вещество, которое в крупных кусках
выглядит почти черным. Как упоминалось выше, монометильное производное
также окрашено в зеленый цвет, а тетраметильное образует сиреневые
кристаллы.
Растворимость. Вода и спирт энергично реагируют с боргидридом урана.
В этиловом эфире боргидрид растворим в количестве 2 г на 100 мл при
комнатной температуре. Эфир не может быть полностью удален испарением без
разложения боргидрида; при —80° образуется устойчивый моноэфират,
444 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
U(BH4)4-(C2H5)20. Неполярные растворители, как «-гептан, растворяют бор-
гидрид медленно и лишь в небольшой степени; из таких растворов последний
может быть снова выделен в неизмененном виде. Хорошо очищенный бензол не
реагирует с боргидридом, однако удалить его без разложения .последнего
трудно. Для практических целей растворимость боргидрида в бензоле слишком
мала. Растворимость алкилборгидридов не определялась.
Отношение к Ьоздуху. С сухим воздухом боргидрид урана реагирует
медленно. При этом образуются желтые нелетучие продукты, далее не
исследованные. Это вещество можно выставлять на короткое время на сухой
воздух без опасения разложения.
Реакция гидролиза.
U(BH4)4 + 16H20 -> U(OH).1 + 4H3R03+ 16Н2; (69)
U(BH4)4 + 4HCl-> UCl4 + 2B2He + 4H2; (70)
U(BH4)4+ I6CH3OH ^U(OCH3)4 + 4B(OCH3)3 + 16H2. (71)
Подобным же образом реагируют и алкилированные боргидриды:
U(ВН4)3(ВН3СН 3) -Ь 6НС1 —> UC14 + I ВаНв + СН3ВС12 + 6Н2; (72)
U(BH3CH3)4+ 12HC1 ->UC11 + 4CH3BCI2+ 12H2. (73)
Отношение к диборану. При комнатной температуре боргидрид урана,
очевидно, не реагирует с дибораном:
U(BH4)4 + B2H6 -^ реакция не идет. (74)
При действии диборана на алкилборгидриды образуются простые
боргидриды:
U(BH4)3(BH3CH3) + В2НС -> U(BH4)4 + CH3BH2BH3; (75)
U(BH3CH3)4-]-B2H6 -> U(BH4)4 + (CH3)4B2H2. (76)
Отношение к метилборату. В результате взаимодействия триметокси-
бора В(ОСН3)3 с боргидридом урана при 70° в течение 1 часа получаются
продукты, значительно менее летучие, чем боргидрид. Механизм этой реакции
сложен, так как помимо основного твердого продукта образуются водород,
диборан и диметоксиборан ВН(ОСН3)2. Исходя из данных анализа, можно
полагать, что белое твердое вещество, образующееся при этой реакции,
является монометоксизамещенным боргидрида урана U(BH4)3(BH3OCH3).
Отношение к боралкилам и другим металлалкилам. Реакция между
боргидридом урана и триметилбором, дающая метилборгидриды урана, уже
была рассмотрена. Цинкдиметил и алюминийтриметил вызывают
интенсивное разложение боргидрида урана при 50°. Получающееся при этом
твердое вещество черного цвета не было подвергнуто дальнейшему исследованию.
Отношение к литийэтилу. Поскольку известно, что реакция
3LiC2H5 + А 1(ВН4)3 -> 3LiBH4 + А1(С2Н5)3 (77)
протекает легко, представлялось целесообразным применить этот процесс
для получения алкильных производных урана.
U(BH4)4 + *LiC2H5 -> U(BH4)4-*(CaHB), + х LiBH4. (78)
Однако при выполнении этой реакции в бензольном растворе образуется
черное нелетучее вещество, являющееся, очевидно, продуктом
восстановления; доказательств образования связи U—С не получено.
Отношение к окиси углерода [106]. При 50° окись углерода не
реагирует с боргидридом урана. При 80° наблюдается реакция, в результате
Глава 15. Бромиды, иодиды и псевдогалоген иды урана
445
которой образуется неизвестный продукт, значительно менее летучий, чем
исходный боргидрид.
Термическая устойчивость боргидридов. Разложение боргидридов урана
очень легко протекает в газообразной фазе, особенно при соприкосновении
ларов с твердыми поверхностями. Стенки сосуда из стекла пирекс заметно
катализируют разложение. Такие металлы, как медь и никель, также очень
эффективны в отношении ускорения разложения, в то время как алюминий
и серебро, вероятно, менее активны, чем стекло пирекс.
Изучена кинетика разложения на меди [107]. В присутствии свежевос-
становленной меди разложение идет со скоростью 7,6- 10е моль - см'2 • час"1
при 55° и 10"4 мм рт. ст. В этом случае могут протекать следующие реакции:
U(BH4)4 ^и(ВН4)3+уВ2Н6 + 4н?; (79)
U(BH4)4 -* U+2B2H6 + 2H2. (80)
Количества водорода и диборана равны, но этот факт, конечно, не
позволяет определить, какая именно из указанных реакций действительно имеет
место. При пропускании паров боргидрида урана через нагретую трубку
происходит разложение с выделением борида урана, образующего на стекле
прекрасное зеркало.
Попытки получения боргидридов урана (VI). При действии
диборана, карбонила бора или боргидрида лития на гексафторид, гекса-
хлорид, пента- и гексаметилат, пента- и гексаэтилат и пентаметилатмоно-
этилат урана боргидрида урана (VI) не получалось. Вместо него всегда
происходило образование боргидрида урана (IV) в смеси с другими
соединениями четырехвалентного урана. Этот результат можно было
предвидеть, учитывая сильные восстановительные свойства этих соединений
бора. Трехокись урана не реагировала ни с одним из вышеприведенных
реагентов.
ПОПЫТКИ ПОЛУЧЕНИЯ КАРБОНИЛА УРАНА
По аналогии с хромом, молибденом и вольфрамом, которые дают
летучие соединения состава Сг(СО)6, Мо(СО)б и W (CO)G, можно было
предполагать, что уран, являющийся членом той же группы периодической
системы, также может образовать соединение U (СО)б. Гексакарбонилы хрома,
молибдена и вольфрама легко получаются с хорошими выходами
разнообразными методами. Эти карбонилы летучи и весьма устойчивы.
При рассмотрении возможности существования гексакарбонила урана
необходимо учитывать правило Сиджвика и Бэйли [108]. Это правило
основано на гипотезе, что каждая группа СО в карбониле металла отдает
два электрона центральному атому металла и что только те карбонилы
устойчивы, в которых центральный атом металла приобретает таким образом
электронную конфигурацию благородного газа. Нужно, чтобы по крайней
мере 26 электронов вошло в электронную оболочку урана для приобретения
последним электронной конфигурации гипотетического благородного газа,
следующего за ним в периодической системе. Уже одни лишь
пространственные соображения, повидимому, должны исключить возможность
существования тридекакарбонила урана U(CO)13- Приведенное рассуждение является,
таким образом, теоретическим доказательством невозможности
существования карбонила урана. Однако правило Сиджвика и Бэйли само лишено
теоретического обоснования. Приобретение электронной конфигурации
благородного газа можно рассматривать как решающий фактор лишь при
определении стабильности таких молекул, которые построены из элементов,
непосредственно предшествующих в периодической системе благородному
газу или непосредственно следующих за ним.
446 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Имеются частичные обзоры литературы [109—111], относящейся к карбо-
нилам металлов. Эти обзоры можно рекомендовать для ознакомления
с подробностями методов получения, строения и свойств карбонилов
металлов.
Действие окиси углерода на металлический
уран [106]. Все известные карбонилы металлов (за исключением карбонила
хрома) получены непосредственным соединением металла с окисью углерода.
Поэтому делались многократные попытки при самых разнообразных условиях
вызвать реакцию между металлическим ураном и окисью углерода. Согласно
отчетам, проведены следующие опыты получения карбонила урана:
1. Порошок металлического урана (98,6%-ный) нагревали с окисью
углерода при давлении 207 ати и температуре 165° в сухом гексане.
2. Ту же реакционную смесь нагревали до 30° при давлении 169 ати.
3. «Активированный» металлический уран (полученный разложением
гидрида) обрабатывали окисью углерода при максимальной температуре 114°
и максимальном давлении 500 ати.
4. «Активированный» металл (полученный нагреванием металла до 250°
в вакууме) обрабатывали окисью углерода при температуре 266° и давлении
608 ати.
5. Металлический уран (96%) нагревали в омедненном стальном цилиндре
при температурах до 200° и давлениях до 250 am [112].
Ни в одном из этих опытов образования летучего карбонила урана не
наблюдалось. Необходимо отметить, что использованный металл не был очень
чистым. Тонкоизмельченный металл, применявшийся в опытах 1—4, почти
наверняка был покрыт поверхностной пленкой, которая, весьма вероятно,
могла мешать реакции. Окись углерода содержала 0,5% водорода и 2,4%
азота. Вполне возможно, что эти загрязнения, особенно при повышенных
температурах, препятствовали желаемой реакции.
Действие окиси углерода [106] и карбонила никеля
на гидрид урана. Гидрид урана, применявшийся для этих опытов, был
получен из металлического урана. Давления разложения гидрида (3 мм рт. ст.
при 225°; 150 мм при 360°), сообщенные группой исследователей, работавшей
с этим гидридом, значительно выше значений, приведенных другими
исследователями (ср. гл. 8). Это вызывает сомнения относительно идентичности
применявшихся веществ. При обработке гидрида окисью углерода в интервале
температур от —180 до 300° и давлениях до 600 am не наблюдалось никаких
признаков реакции. Действие карбонила никеля Ni(CO)4 на гидрид урана
(в присутствии трихлорида или диметилхлорида алюминия) также не привело
к образованию какого-либо летучего соединения урана.
Действие окиси углерода и восстановителей на
галогениды урана. В 1927 г. открыт новый способ получения
карбонилов металлов [113]. Он состоит в действии восстановителей в присутствии
окиси углерода на галогенид металла. В качестве восстановителей сначала
применяли магнийорганические соединения. Затем для этой цели стали
применять тонкодисперсные металлы, такие, как серебро, медь, кадмий или цинк
[114, 115]. Предпринимались многократные попытки получить летучий
карбонил урана с помощью этих методов.
Тетрахлорид урана обрабатывали этилмагнийбромидом и окисью углерода
в эфирно-бензольном растворе [116]. Аналогичная реакция была выполнена
с цинковой пылью вместо магнийорганических соединений. В обоих случаях
поглощалось некоторое количество окиси углерода, однако никакого летучего
соединения урана выделить не удалось. Интересно отметить, что в обоих
случаях реакционную смесь подвергали гидролизу (в воде или разбавленной
серной кислоте); при этом, очевидно, предполагалось, что карбонил урана, если
только он вообще существует, не должен разлагаться водой.
Глава 15. Бромиды, иодиды и псевдогалоген иды урана
447
Вышеприведенная реакция была применена к уранилхлориду, гекса-,
тетра- и трихлориду урана, смеси тетра- и пентахлорида и ацетилацетонату
и(СН3СОСНСОСН3)4 [112]; в качестве магнийорганических соединений
пользовались метилмагнийиодидом, этилмагнийбромидом, фенилмагнийбромидом,
а-нафтилмагнийбромидом и фениллитием. Никакого летучего соединения
урана не обнаружено. Так же безуспешно было применение диметил-, диэтил-
цинка и бутиллития [106].
Кроме того, проведено несколько опытов обработки тетрахлорида и тетра-
бромида урана в ацетоновых растворах окисью углерода и цинковой пылью
[106]. Этот способ дает отличные результаты с пентахлоридом молибдена,
однако в случае урана он оказывается безуспешным. Повидимому, единственным
результатом реакции является восстановление урана до трехвалентного
состояния.
Действие окиси углерода на галогениды урана
в присутствии переносчиков [106]. Удобным способом
получения карбонилов металлов является действие окиси углерода на водные
растворы галогенидов металлов в присутствии щелочного раствора цианида
калия или сульфида натрия [117—120]. Проведено несколько опытов с гало-
генидами урана, в которых в качестве «переносчика»окиси углерода применялся
цианид калия, однако реакции не наблюдалось. Как переносчик был
испробован и цистеин; исходным продуктом являлся трихлорид урана [121,122].
При этом происходила какая-то реакция; однако продукты ее не были
идентифицированы. Гидросульфиды щелочных металлов и соли уранила в
присутствии окиси углерода также не дают летучего соединения урана [112].
Выводы. Из приведенного материала видно, что успешное
приготовление летучего карбонила урана или даже доказательство существования
такого соединения является нелегкой задачей. Обзор последних работ показывает,
что было проверено большинство возможных методов получения карбонилов
металлов, если не все эти методы. Что касается работ, сделанных с
металлическим ураном, то весьма желательно повторить эти эксперименты, используя
имеющийся теперь очень чистый металлический уран и чистую окись углерода.
Предложено (и, повидимому, эта мысль заслуживает внимания) применять
сплавы, например сплав никеля с ураном [123]. Во всех опытах,
выполненных до сих пор, в которых наблюдалась реакция (как это часто бывало), не
предпринималось попыток выяснить ее течение. Все, что делалось,
заключалось в быстром определении, образуется или не образуется летучее соединение
урана. Понимание того, что происходит с ураном в случае некоторых таких
реакций, могло бы помочь в планировании дальнейших экспериментов.
Методы переработки реакционной смеси были основаны на аналогии со
способом получения гексакарбонила хрома, нечувствительного к влаге.
Возможно, что<и(СО)6, если он существует, чувствителен к воде. При выборе
растворителей не уделялось достаточного внимания способности урана (IV)
функционировать в качестве окислителя или восстановителя. Наконец,
имело бы, пожалуй, смысл снова исследовать некоторые из ранее изученных
реакций с заменой тетрахлорида урана пента- и гексахлоридом.
На основании того, что попытки получить карбонил урана оканчивались до
сих пор неудачей, нельзя сделать еще окончательного вывода относительно
устойчивости этого соединения. Необходимо, однако, отметить, что уран
является сильно электроположительным элементом, а карбонилы
элементов этого типа, например бериллия или магния, неизвестны.
ЛИТЕРАТУРА
1. Spedding F. Н., N е w t о n A. S., N о t t о г f R. W., Р о w е 1 1 J., С а !-
kins V. P., The Preparation and Some Properties of UBr3, UBr4, UOBr.,, ard
U02Br2, in National Nuclear Energy Series, Division VIII, vol. 6 (MP Ames 1).
448 Часть IV, Галогениды, оксигалогсниды, боргидриды и карбонил урана
2. A i b eg of f G., Ann., 233, 119 (1886).
3. Newton A. S., Johnson D., Kant A., Nottori R. W., Report CC-705
June 7, 1943 (MP Ames 2).
4. Newton A. S., J о h n s о n D., К a n t A., N о t t о г f R. W.f Report CC-725,
June 15, 1943 (MP Ames 3).
5. Lyon W., 111 i f J., L i p к i n d H., Report CK-1526, Apr. 15, 1944 (MP Ames 4).
6. Lyon W., I 11 i f J., L i p к i n d H., Report CK-1494, May 4, 1944 (MP Ames 5).
7. Nottorf R. W., Powell J., Report CC-1524, Mar. 10, 1944 (MP Ames 6).
8. К a t z J. J., D a v i d s о n N. R., Report CK-1221, Jan. 5, 1944 (MP Chicago 1).
9. A 1 t m a n D., Report RL-4.6.202, Sept. 18, 1943 (UCRL 1).
10. Webster R. A., Report CK-873, Aug. 20, 1943 (MP Berkeley 1).
11. Thurmond С D., Report CC-2522, Jan. 15, 1945 (MP Berkeley 2).
12. E a s t m a n E. D., F о n t a n a B. J., W e b s t e r R. A., Observations on the
Preparation of Tribromide and Triiodide of Uranium, in National Nuclear Energy
Series, Division VIII, vol. 6 (MP Berkeley 3).
13. Z i m m e г m a n n C, Ann., 216, 2—7 (1882).
14. F о n t a n a B. J., Report CC-586, Apr. 21, 1943 (MP Berkeley 4).
15. Fo nt a n a B. J., Report CK-677, May 15, 1943 (MP Berkeley 5).
16. С a 1 к i n s V. P., N о t t о г f R. W., Report CC-1975, Oct. 24, 1944 (MP Ames 7).
17. S t i с к 1 a n d A. E., Report CC-939, Sept. 1943 (MP Berkeley 6).
18. T h u г m о n d С D., S t i с к 1 a n d A. E., Report CC-1739, June I, 1944 (MP
Berkeley 7).
19. Warner J. C, Report CK-1396, Feb. 17, 1944 (MP Chicago 2).
20. Z а с h a r i a s e n W. H., The Crystal Structure of Trichlorides, Tribromides, and
Trihydroxides of Uranium and of Rare Earth Elements, in National Nuclear Energy
Series, Division VIII, vol. 6 (MP Chicago 3).
21. Z а с h a r i a s e n W. H., J. Chem. Phys., 16, 254 (1948).
22. Bae'nziger N. C, R u n d 1 e R. E., Report CC-1524, Apr. 14, 1944 (MP Ames 8).
23. В a e n z i ger N. C, R u n d 1 e R. E., Report CN-1495, Apr. 15, 1944 (MP Ames 9).
24. Z a ch a r i a s e n W. H., Reports CP-1576, Apr. 27, 1944; CC-2768, Mar. 12, 1945
(MP Chicago 4).
25. N о t t о г f R. W., Powell J., С a 1 к i n s V. P., Report CC-1524, Apr. 14,
1944 (MP Ames 10).
26. В а г к e 1 e w С H., Report RL-4.6.329, Aug. 28, 1945 (UCRL 2).
27. M а с W о о d G. E., Report RL-4.7.602, Dec. 18, 1945 (UCRL 3).
28. G r e g о г у N. W., Report RL-4.6.923, Aug. 1, 1945 (UCRL 4).
29. T h u г m о n d С D., Report CC-938, Sept. 1943 (MP Berkeley 8).
30. F о n t a n a B. J., Report CK-810, July 20, 1943 (MP Berkeley 9).
31. Calkins V. P., Report CC-1500, June 17, 1944 (MP Ames 11).
32. Lyon W., I 1 1 i f J.,L i p к i n d H., Report CT-1180, Dec. 25, 1943 (MP Ames 12).
33. W e b s t e г R. A., Report CK-671, May 15, 1943 (MP Berkeley 10).
34. Webster R. A., Report CC-2105, Aug. 15, 1944 (MP Berkeley 11).
35. Hermann H., Dissertation, University of Gottingen, p. 29 (1861).
36. С о 1 a n i A., Ann. chim. et phys., [8] 12, 59—74 (1907).
37. R i с h а г d s T. W., Merigold B. S., Z. anorg. Chem., 31, 235 (1902).
38. H 6 n i g sc h m i d O., Monatsh., 36, 51—68 (1915).
39. N о t t о г f R. W.,Powell J., Report CC-1781, Aug. 10, 1944 (MP Ames 13).
40. S m i t h C, Cunningham В. В., Report CK-932, Sept. 11, 1943 (MP
Chicago 5).
41. N о t t о г f R. W., Powell J., Report CC-1496, Apr. 10, 1944 (MP Ames 14).
42. N о t t о г f R. W., Powell J., Report CC-1778, July 10, 1944 (MP Ames 15).
43. К e 1 1 e r W. H., Report CT-1270B, Jan. 29, 1944 (MP Ames 16).
44. N о t t о г f R. W., P о w e 1 1 J., Report CC-1504, June 10, 1944 (MP Ames 17).
45. A h m a n n D. H.f Report CC-298, Oct. 15, 1942 (MP Ames 18).
46. Lyon W., I 1 1 i f J., L i p к i n d H., N e h e г С, Report CK-1498, June 17, 1944
(MP Ames 19).
47. Powell J., Report CC-1778, Aug. 18, 1944 (MP Ames 20).
48. P о w e 1 1 J., Report CC-1504, Aug. 10, 1944 (MP Ames 21).
49. Carter J. M., пат. США 2469916, May 10 (1949).
50. N e h e r C.Lipkind H., L у о n W\, S 1 e i g h t N., Report CT-1780, Aug. 9,
1944 (MP Ames 22).
51. A 1 о у J., R о d i e г Е., Bull. soc. chim. France, [4] 31, 247 (1922).
52. Powell J., Report CC-1781, Oct. 18, 1944 (MP Ames 23).
53. S с h e 1 b e r g A., T h о m p s о n R. W., Report A-809, Oct. 10, 1942 (Princeton 1).
54. N о t t о г f R. W., Powell J., N e w t о п A. S., Report CC-1500, June 17,
1944 (MP Ames 24).
J55. A 1 t m a n D., Report LR-4.6.276, Aug. 14, 1944 (UCRL 5).
Г)0. N о t t о г f R. W., В u t 1 e г Т. A., Report CC-1975, Oct. 24, 1944 (MP Ames'25).
'7. Thompson R. W., Schel b erg A, Report A-179, May 4, 1942 (Princeton 2)
Глава 15. Бромиды, иодиды и псевдогалоген иды урана 449
58. М а с W о о d G. E.,Altman D., Report RL-4.6.600 (A-l) (UCRL 6).
59. G г e g о г у N. W.f Report RL-4.6.928, Aug. 24, 1945 (UCRL 7).
60. G г e g о г у N. W., Preparation and Properties of the Uranium Halides, in National
Nuclear Energy Series, Division VIII, vol. 6 (UCRL 13).
61. Galkins V. P., Report CC-1496, May 11, 1944 (MP Ames 26).
62. F о n t a n a B. J., Report CC-585, Apr. 21, 1943 (MP Berkeley 12).
63. A 1 oy J., Bull. soc. chim. Paris, [3j 21, 265 (1899).
64. Gmelins Handbuch der anorganischen Chemie, System No 55, p. 140, Verlag, Chemie,
Berlin (1936).
65. G u be h а г d M.f Compt. rend., 145, 921 (1907); or Bull. soc. chim. Paris, [41 3 (1908).
66. M о i s s a n H., Compt. rend., 122, 1092 (1896).
67. D a a n e A. H., Report CC-298, Oct. 15, 1942 (MP Ames 27).
68. G г e g о г у N. W., Report RL-4.6.272, July 20, 1944 (UCRL 8).
69. S e n d t n e г R., Ann., 195, 331 (1879).
70. Foster L. S., T v г z i с к у J., Report CK-897, Aug. 28, 1943 (MP Chicago 6).
71. Foster L. S.,Tvrzicky J., Memorandum 123, Sept. 18, 1943; Report CT -963,
Oct. 11, 1943 (MP Chicago 7).
72. Holmes J. A., Report RL-4.6.252, Mar. 7, 1944 (UCRL 9).
73. А у г e s J. A., Report CN-1243, Jan. 8, 1944 (MP Ames 28),
74. Z а с h а г i a s e n W. H., Reports CP-1811, July 4, 1944; CP-1954, Aug. 10, 1944
(MP Chicago 8).
75. A 1 t m a n D., Mueller M. E., Prescott С. Н., Report RL-4.6.273,
July 27, 1944 (UCRL 10).
76. S e a b о г g G. Т., Report N-930, Apr. 5, 1944 (MP Chicago 9).
77. M a g e 1 Т., F о s t e г L. S., Report CT-963, Oct. 11, 1943 (MP Chicago 10).
78. M а с W о о d G. E., M u e 1 1 e г M. E., A 1 t m a n D., Report RL-4.6.277, Dec. 8,
1944 (UCRL 11).
79. Prescott С H., Jr., Reynolds F. L., Holmes J, A., The Preparation
of Uranium Metal by Thermal Dissociation of the Iodide, in National Nuclear Energy
Series, Division VIII, vol. 6 (UCRL 12).
80. "W a r f J. С, В a e n z i g e r N. C., Preparation and Properties of Some Mixed
Uranium Halides, in National Nuclear Energy Series, Division VIII, vol. 6 (MP Ames 29).
81. Gregory, National Nuclear Energy Series, Division VIII, vol. 6.
82. War f J. C, E d w a r d s F., Report CC-1496, Apr. 10, 1944 (MP Ames 30).
83. W a r f J. C, et al., Report CC-1785, Sept. 10, 1944 (MP Ames 31).
84. G a t e s J. W.,Jr1( Andrews L. J., В I о с к В. Р., Y о u n g H. A., Report
CD-460, Aug. 26, 1944 (CEW-TEC I).
85. G a t es J. W., CI e wet t G. H., Y о u n g H. A., Report CD-454, July 20, 1945
(CEW-TEC 2).
86. G r e g о г у N. W., Report RL-4.6.905, Jan. 8, 1945 (UCRL 14).
87. G г e g о г у N. W., Report RL-4.6.275, Aug. 7, 1944 (UCRL 15).
88. Gregory N. W., Report RL-4.6.295, Oct. 7, 1944 (UCRL 16).
89. R u n d 1 e R. E., В a e n z i g e r N. C, Report CC-1500, May 10, 1944 (MP Ames 32).
90. E s t a b г о о к M„ Davidson P. H., Streeter I., Report RL-4.6.902,
Dec. 6, 1944 (UCRL 17). '
91. E s t a b г о о к MM Davidson P. H., S t r e e t e r I., Report RL-4.6.903,
Dec. 29, 1944 (UCRL 18).
92. D a v i d s о n P. H., Street er I.r Est abrook M., Report RL-4.6.908,
Jan. 23, 1945 (UCRL 19).
93. D a v i d s о n P. H., M а с W о о d G. E., Streeter I., Report RL-4.6.932,
Aug. 30, 1945 (UCRL 20).
94. G r e g о г у N. W., Report RL-4.6.936, Sept. 9, 1945 (UCRL 21).
95. D a v i d s о n P. H., M а с W о о d G. E., Streeter I., Report RL-4.6.933,
Aug. 31, 1945 (UCRL 22).
96. M а с W о о d, National Nuclear Energy Series, Division VIII, vol. 6.
97. Sc h 1 e s i n g e r H. I., Brown H. C, Report A-1221, July 29, 1943 (Univ.
Chicago Dept. Chem. 1).
98. Schlesinger H. I., В г о w n H. C, Uranium Borohydride and Its Alkyl
Derivatives, in National Nuclear Energy Series, Division VIII, vol. 6 (Univ. Chicago
Dept. Chem. 2).
99. S с h 1 e s i n g e г H. I., S a n d e г s о n R. Т., В u г g А. В., J. Am. Chem. Soc.,
61, 536 (1939); 62, 3421 (1940).
100. Schlesinger H. I., В г о w n H. C, J. Am. Chem. Soc, 62, 3429 (1940).
101. Burg А. В., S с h 1 e s i n g e г H. I., J. Am. Chem. Soc, 62, 3425 (1940).
102. Schlesinger H. I., SchaefferG. W., Barbaras G. D., Report»
CP-I558, May 24, 1944; CK-2737, Mar. 5, 1945 (MP Chicago 11).
103. Burg А В., S с h 1 e s i n g e г Н. I., J. Am. Chem. Soc, 59, 780 (1937).
104. U г е у H. C, Report A-750, July 10, 1943 (SAM Columbia 1).
105. Duncan A. B. F., Report A-584, Apr.' 15, 1943 (SAM Columbia 2).
29 Химия урана
450 Часть IV. Галогениды, оксигалогенидьц боргидриды и карбонил урана
106. Calingaert G., Soroos HM Dykstra F. J., HnizdaV., Capin-
j ol a J. V., Report A-542, Feb. 16, 1943 (Ethyl Corp. 1).
107. T u г к e v i с h А., В г о w n H. C.Lewinsohn V., L i b b у W. F., Report
A-383, Oct. 19, 1942 (SAM Columbia 3).
108. S i d g w i с к N. V., Bailey R. W., Proc. Roy. Soc. London, A144, 521 (1934).
109. H i e b e г W., Die Chemie, 55, 7, 24 (1942).
ПО. В 1 a n с h a r d A. A., Chem. Revs., 26, 409 (1940); 21, 3 (1937).
111. Trout W. E., J. Chem. Education, 14, 453, 575 (1937); 15, 77, 113 (1938).
112. Report B-22 (British 1).
113. J о b А., С a s s a 1 A., Bull. soc. chim. France, 41, 1041 (1927).
114. Hieber W., Romberg E., Z. anorg. u. allgem. Chem., 221, 322 (1935).
115. Кочешков К. А., Несмеянов А. Н., Н а д М. M., Российская
И. М., Б о р и с о в а Л. М., ДАН СССР, 26, 54—59 (1940).
116. М с D u f f i e M., Report A-806, Oct. 5, 1942 (Princeton 3).
117. M a n с h о t W., Gall H., Ber., 62, 678 (1929).
118. W i n d sor M. M., В 1 a nc h a r d A. A., J. Am. Chem. Soc, 55, 1877 (1933).
119. В 1 a n с h a r d A. A., R a f t e г J. R., A d a m s W. В., Jr., J. Am. Chem. Soc.,
56, 18 (1934).
120. Hieber W., S с h u 1 t e n H.,Marin R., Z. anorg. u. allgem. Chem., 240, 264
(1939).
121. Schubert M., J. Am. Chem. Soc., 55, 4563 (1933).
122. Coleman G. W., В 1 a n с h а г d A. A., J. Am. Chem. Soc, 58, 2160 (1936).
123. S u g ar ma n N.. Elliott N.. С or у ell C. D., Report CC-144 (MP Chicago 12).
Глава 16
ОКСИГАЛОГЕНИДЫ УРАНА
Оксигалогениды шестивалентного урана типа U02X2 (где X—галоген)
были одними из первых изученных соединений урана. Несмотря на это, до
сих пор определено лишь небольшое количество физических констант этих
веществ с большей или меньшей точностью, и химия этих соединений все еще
носит отрывочный характер. Оксигалогениды урана (IV) до 1940 г. не были
известны, однако в настоящее время оксихлорид и оксибромид легко
доступны и относительно их накоплен значительный фактический материал.
Доказательства вероятности существования оксигалогенидов урана (VI) типа
UOX4 и урана (V) типа UOX3 были уже рассмотрены в гл. 14.
Здесь будет обращено внимание на галогениды уранила и на
оксигалогениды урана (IV). Вопросы, связанные с ионными равновесиями и физическими
свойствами водных растворов этих соединений, будут рассматриваться во
второй книге этого тома.
УРАНИЛФТОРИД U02Fa
Уранилфторид был впервые получен путем реакции между плавиковой
кислотой и двуокисью урана. Берцелиус [11 обрабатывал трехокись урана
плавиковой кислотой и после выпаривания получил аморфное вещество белого
цвета. Болтон [2] приготовил растворы уранилфторида, действуя плавиковой
кислотой на закись-окись урана:
U308 + 8HF -> UF4 + 2U02Fa + 4HaO. (1)
Выделить кристаллический уранилфторид из этого раствора ему не
удалось. Смительс [3] при повторении работы Болтона нашел, что путем
выпаривания водного раствора уранилфторида можно выделить это соединение в виде
яркожелтого мылообразного вещества. В одном случае им было получено
частично кристаллическое вещество с перламутровым блеском. Гидратирован-
ное аморфное вещество, полученное выпариванием водного раствора
уранилфторида, Смительс назвал р-формой в отличие от вещества, полученного в очень
малых количествах путем нагревания тетрафторида урана в закрытом тигле
и названного им а-формой. Последнее соединение является, вероятно,
безводным уранилфторидом и образуется по реакции
2UF4 + 02 ->UFe + U02Fa. (2>
Дитте [4] ошибочно принял, что соединение, получаемое прокаливанием
тетрафторида урана при ограниченном доступе воздуха, является оксифтори-
дом состава UOF4.
Получение уранилфторида из водных
растворов. Уранилфторид способен образовывать кислые и основные соли
неопределенного состава. Для получения чистого нейтрального уранилфторида [51
известное количество окисла (закиси-окиси или гидрата перекиси) растворяют
в рассчитанном количестве плавиковой кислоты и полученный раствор выпари-
29*
452 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
вают затем досуха. Получаемый продукт обычно имеет более высокое
содержание урана, чем вычисленное для нейтральной соли. К нему добавляют
плавиковую кислоту в количестве, эквивалентном избытку урана, и воду в количестве,
достаточном для растворения соли, и дают затем соли выкристаллизоваться.
Полученные таким путем кристаллы представляют собой, повидимому, дигид*
paTU02F2.2H20.
Уранилфторид трудно кристаллизуется, образуя при концентрировании
раствора вязкую сиропообразную жидкость. Даже если вызвать
кристаллизацию дигидрата уранилфторида, при отделении кристаллов от маточного
раствора возникают обычно большие трудности. Эти кристаллы представляют собой
мягкие тонкие пластинки светложелтого цвета. Они настолько гигроскопичны,
что их трудно высушить. Поэтому часто выпаривают растворы досуха при 150—
200° без попытки достигнуть кристаллизации. При 200° получается продукт,
соответствующий по составу нейтральной соли, и содержащий менее 0,1 % воды.
Безводный уранилфторид может быть получен также путем реакции между
уранилацетатом и фтористоводородной кислотой 16]. Для полного удаления
уксусной кислоты раствор несколько раз выпаривают с фтористоводородной
кислотой в платиновом тигле на водяной бане. Остаток затем выпаривают
досуха для удаления воды и избытка фтористоводородной кислоты. При
продолжительной сушке полученного продукта в вакуум-эксикаторе образуется
безводный уранилфторид. Этот метод применялся для получения уранилфторида
английскими исследователями [71.
Получение уранилфторида
высокотемпературным гидрофторированием. Безводный уранилфторид легко
получить путем реакции между трехокисью урана и газообразным фтористым
водородом:
U03 -f- 2HF -> U02F2 + Н20. (3)
Подходящей температурой для этой реакции является, повидимому, интервал
350—500° [8]. Для промышленного производства рекомендуются температуры
порядка 550° [9]. Другие исследователи, чтобы избежать образования закиси-
окиси урана изтрехокиси и последующего загрязнения продукта тетрафторидом,
предпочитают температуры, близкие к 400° [10, 11]. Свойства применяемой
трехокиси урана определяют оптимальную температуру. Вероятно, это
наиболее удобный метод получения безводного уранилфторида.
Можно упомянуть также и следующую реакцию получения безводного
уранилфторида (хотя она и не имеет особенного значения как препаративный
метод):
U02-HP04-xH20 + 2HF Э50-Б00\ U02F2 + H3P04 + xH20. (4)
Выше 500° наблюдается восстановление и начинается образование тетрафто-
рида урана [12].
' Уранилфторид может быть легко получен взаимодействием безводного
уранилхлорида с жидким безводным фтористым водородом при комнатной
температуре [13]. Лучше всего проводить эту реакцию в платинированном
никелевом реакторе; реакционную смесь выдерживают в течение ночи и затем отгоняют
хлористый и фтористый водород в вакууме при 450°. При этом получается
продукт, свободный от хлора и полностью растворимый в воде.
Различные реакции, при которых образуется
уранилфторид. Уранилфторид образуется в результате некоторых
реакций, которые, однако, не могут быть рекомендованы в качестве удобных
препаративных методов получения этого соединения [8]:
UO, + 2F2 зб0' , U02F2 + OF, (?); (5)
U(VFt 350' , UOJV, (6)
U308 + 5Ft 8б0'-> 3U02F2 + 20F2 (?). (7)
Глава 16. Оксигалогениды урана
453
Уранилфторид является одним из продуктов следующей реакции [14]:
2UF4 + Oa -> L'Fe + U02Fa. (8)
В имеющихся отчетах указывается, что при действии паров воды на
гексафторид урана образуется комплексное соединение уранилфторида,
фтористого водорода и воды, из которого нагреванием до 180° может быть
получен уранилфторид [15). Существование этого комплекса требует
подтверждения.
Физические свойства уранилфторида. Безводный
уранилфторид представляет собой вещество светложелтого цвета; никаких других
данных о его физических свойствах нет. Он подвергается термическому
разложению, а поэтому его температура плавления неизвестна.
Кристаллическая структура безводного уранилфторида.
Кристаллическая структура уранилфторида была изучена Захариазеном [16, 17].
Идеальная структура его ромбоэдрическая с одной молекулой в элементарной
ячейке, имеющей следующие константы:
а = 5,755±0,003А; а-42°47'±3'.
Пространственная группа RZm\ положения атомов следующие:
Ш в (0 0 0);
20 в ±{иии), где н = 0,122;
2F в ±(vvv)f где t> = 0,294.
Каждый атом урана связан с двумя атомами кислорода, находящимися
на расстоянии U — О, равном 1,91 А, и с шестью атомами фтора на
расстоянии U — F, равном 2,50А. Структура состоит из слоев атомов урана,
01стоящих друг от друга на 5,22 А: оси групп уранила перпендикулярны
к этим слоям. Атомы фтора расположены на расстоянии 0,61 А выше и ниже
каждого слоя атомов урана, слои соединены друг с другом слабыми связями
О —О и О —F. Фактически образцы уранилфторида всегда обладают более
или менее неупорядоченными слоями из-за смещения плоскостей их
относительно друг друга.
Плотность уранилфторида, рассчитанная из рентгенографических данных,
равна 6,37 г/см3. Прямое измерение путем гидростатического взвешивания
в бензоле дало значение 5,8 г/см3, однако эта величина, повидимому,
занижена вследствие неполного вытеснения воздуха при определении [18].
Английские исследователи дают для насыпного веса значение 2,95 г/см99
а для утрясенного—2,55 г/см3 [10]*.
Оптические и фотохимические свсйства уранилфторида. Спектр
поглощения и спектр флюоресценции уранилфторида будут рассмотрены во
второй книге. '
Удельная теплоемкость, энтальпия и энтропия уранилфторида [19, 20].
Теплоемкость уранилфторида измерена в интервале температур 13 —418°К.
Вычисленная энтальпия Н°—Н° составляет 63,96; 77,62 и 108,15 межд. дж-г'1
при температурах 298,16; 338,16 и 423,16° К соответственно, а энтропия
при тех же температурах 0,4400; 0,4830 и 0,5635 межд. дж-г'1-град'1.
Фазовых превращений не обнаружено. В табл. 201 приведены значения
удельной теплоемкости, энтальпии, энтропии и свободной энергии
уранилфторида для температур с интервалом через 5° К. Вероятная ошибка
—порядка нескольких десятых долей процента.
Химические свойства уранилфторида. Отнсшение к воде»
Безводный уранилфторид растворим в' воде, метиловом и этиловом слцрте}
* Очевидно, здесь в оригинале ошибка.—Прим. пере в.
о ^
см <
с*
* е
го S
<
Си
К
и
(X
W
Ж
с*
<
Ж
О
о
ей
О
5
л 1
ее4
^
i *
<-•
•-•
«'1
5э *
1
51
О © Л
1
1 о <ъ
*8
1 »н
1
'О
1 ^
О •
*
1 *°
1 31
Температура,
о
о
о
о
о
—г СЛ
см со
О СО
о о
о о
о о
г» ю
— СО
о —•
о о
о о
о о
т*« ч*
о о
о о
о о
—| СО
ю о
О "f
88
о о
ю о
о
CD
со
о
о
о
чГ
Ч*
8
CD
CM
CD
ч*«
О
О
со
CM
—*
О
О
ю
00
о
ю
о
о
СО
4f
СЛ
о
о
о
R
СО
о
СО
со
ЯР
см
о
о
о
см
8
~ч
о
m
n-
ю
о
о
СО
см
о
со
00
я
о
о
8
<0
8
см
о
во
00
см
см
о
о
во
со
N-
ч*
о
во
N.
4f
4f
о
о
8
^^
СО
ч*
СО
о
о
S
СО
о
о
о
оо
см
n-
о
N.
со
1С
in
о
о
3
СО
со
Ш
о
ч*
N.
о
о
СО
S
РЧ
N.
СО
со
со
о
о
о
4J»
о ы о
СО 00 СП
г*- ел см
о о ~
СЛ Ю СО
« О) О
N Ю Ю
ч* in со
о о о
CD <D CD
СМ 00 Ш
ел о оо
со во см
«« W
N. СЛ —|
ч* О СЛ
Is- СЛ —
£82
О О О
Ю О Ш
тг щ ю
со
ел
со
со
00
тг
ч*
N»
О
О
см
ел
см
00
см
ел
ч*
ю
—
~"
О
о
со
СО N-
с§ 00
О "*•
см см
о ю
см ел
Tf СО
S8
о о
г^ ю
со ел
т»« О
СО «*
СО СО
— N.
r^ in
см со
—< -^
О О
m о
СО N»
со
ю
N-
сл
см
&
со
О
о
СО
СО
ел
N.
ч*
см
$
ч*
"""■
о
in
N-
ч*
N-
ю
СО
СО
СО
о
о
N.
О
•""•
ч*
ел
к
см
о
Я8
8
ю
со
см
ю
*~*
о
о
00
см
СО
со
о
«—»
СО
со
~*
о
in
00
со
ч*
ч*
N.
ч*
см
СО
см
СО
о
N.
ч*
со
—
N.
00
N.
со
N.
«-*
о
SS3
СО СО
ч* —•
Ю СО
ел -^
ч* 1П
о о
О ч*
СО СО
ю об
О СЛ
00 00
СО N-
см ч*
СМ 00
СО 00
.—1 —е
о о
SS о
СО СО
Tf —
Tf N-
ел n«
СО N.
S °*
00 СЛ
о ел
со со
о о
о оо
ч* СМ
СП СЛ
СЛ О
СЛ
СО N.
4f О
СЛ О
—. см
о о
ю о
о —•
о
ч*
ч*
со
00
in
о
ел
N.
о
N.
ч^
СЛ
-щ
CD
N.
о
см
о
ю
в
см
со
ю
СЛ
о
о
00
00
о
S
СЛ
см
см
СО
см
о
о
см
ч* О СЛ ~* СО —* 00
СМ СО N. N. О СО СЛ
in in in со оо en «-«
О —* СМ СО ч* in N.
СМ т»« in т»« СМ 00 СО
00 1П — СО О СМ ч^
СО 1П ч^ СМ — СЛ N.
СЛ О — СМ СО СО 4f
О О О О О О О
СЛ О —* 00 СМ —* ч*
N. СЛ СО СЛ СЛ — 1П
О — СО 4f СО СЛ —
Tf to CO N 00 О «
со см оо см со со о
СЛ in О СО — СО —*
—« СМ СО СО ч*« т^ ю
СМ СМ СМ СМ СМ СМ СМ
о о о о о о о
in о in о in о in
СМ СО СО ч* ^f in 1П
m
in
4f
00
N.
ч#
m
in
CM
о
CM
ч*
CM
CM
s
lO
CM
о
о
со
CM
in
N.
ел
о
ч*
СО
со
см
о
о
N-
СО
см
00
гл
in
см
о
in
со
СЛ ч*
оо со
О ч*«
-н СМ
см см
см со
см ел
— 00
h^ N.
см см
о о
СЛ СЛ
— TJ.
О СО
in со
см см
о ел
ч* N.
со со
см см
о о
о ю
N. N-
00
N-
оо
СО
см
СО
ю
СО
оо
см
о
00
СЛ
со
N»
см
N.
N.
см
о
3
О 00
со -1
СО 00
m со
CM CM
CO —*
О ч*
^ —
СЛ О
см со
О О
со -*
s%
СЛ О
см со
СО 00
in оо
N. N.
см см
О О
ш о
со ел
СО ср
4f О
со ел
00 СЛ
см см
О СЛ
N- 00
S2
СО со
О О
ч* СО
1П N.
оо см
— СО
СО со
СМ ч*
см in
оо оо
см см
О О
in о
см
о
1П
СО
N.
m
см
см
СО
о
я
N.
ч*
со
со
8R
см
о
■
in
ГО
со
СО
N.
ж
см
СО
о
гл
\п
со
СО
N.
гл
см
о
о
см
см
о
оо
ч*
СО
N.
00
<П
ГО
СО
о
со
см
со
N.
СО
гл
4f
СЛ
см
о
in
см
m
о
ю
со
СО
во
СП
го
4f
СО
о
N.
о
СЛ
СО
гл
N.
гл
см
о
^8
СМ О
00 О
со 4f
—« in
4f О
S in
CO CO
О О
ч* СО
О -ч
со —1
о см
Ч* Tf
CO N-
88
СО СО
о о
О 4П О
см см со
см см см
о as
оо to о
tT О н
о оп л
w -r 2
S a: ro
» 2 § ;,
2 аз s о
§ — Й
ГР *^
ь 1з 3
^ Сз X
я -» • тз £ н ^
кс Й э 5 % о ^
О ъэ
S Ь 5 Я я
2 л н тз g
хз ь S s v; ^ о
Со § Е За ^ m ^
н-5 п> w 2 2
Н 43
~ S
я го Е
го
го
го п> о
го
ГО )а
8 s
ас ^
^ Вэ w О
— - - д
S §
Я g
2 ь п> »
2 чЭ-ja ь»
Ь ч Г) Со
о о н о
со я го
<т> ^ ас
со сг
ж го ~
р:
со
я 3
я Ь чз
2 ' ^
2 о?
to со чз
О- из
К ГО
8ё
ГО
ч
со
го
тз
го
н
О)
СО W
4Ь 4*.
tO tO
ел о
о о
со Ъо
аз ст>
ОО -vj
00 00
SS
Ъо \о
СО 00
— со
о о
$£ сл
аз стз
ел о
О -vi
00 СО
со to
— 00
со ел
; со ~
О 00
4*.
сл
о
со
оз
CJ3
00
о
о»
—
о»
со
о
СЛ
сл
CJ3
со
со
to
сл
-4
to
сл
4*
о
о
со
оз
ат
оо
о
со
со
to
о
сл
сл
00
со
to
to
со
Oi
4*
4*
о
сл
о
со
аз
4*.
00
о
4*.
со
сл
о
сл
4*.
-vl
4*.
to
о
to
S?
4*
8
о
со
аз
&
со
со
аз
-vj
>£»
О
se
ю
оо
00
-vl
а^
00
О
СО
со
СЛ
О
СО
аз
to
-vl
СО
-vl
00
£
О
сл
СО
00
со
4^»
-vj
^1
аз
со
со
о
о
со
аз
-vl
СО
аз
о
4^
аз
о
сл
со
со
-vl
о
to
о
со
00
со
00
сл
о
со
аз
о
*n|
со
4^
to
4^
О
СЛ
Ю
СО
О
4*.
О
СО
4^
4^
о
%
О
О
СО
сл
СО
аз
СО
N3
4^
4^
О
О
сл
го
4^
со
со
о
аз
оо
о
аз
со со
-4 -О
сл о
о о
со со
сл ел
оо -о
4*. tO
СО 00
О ОС
аз оо
4^ СЛ
СЛ СЛ
о о
СЛ СЛ
<Х> 4*.
сл -ч
-J -4
О О
4*. —
— аз
^о ~
аз —
со
аз
сл
о
со
сл
аз
о
00
-v]
о
-V)
to
о
сл
S?
СО
to
со
со
о
сл
о
со
а>
о
о
со
сл
4*
аз
00
сл
to
СО
аз
о
сл
о
сл
о
to
СО
аз
сл
to
СО СО
сл сл
сл о
о о
со со
сл сл
со ~
to -<i
00 00
со —
СЛ -vl
to аз
аз 4=*
о о
СЛ 4*.
О СО
о сл
о о
•ч -4
СО СО
4* 1—
о сл
о —
о to
со
4^
сл
о
со
сл
о
to
00
о
о
S
о
4*.
СО
о
о
to
оо
со
о
4*.
со
со
4*
о
о
со
4*
00
аз
-4
00
to
аз
ю
о
4^
00
4^
со
to
00
аз
аз
to
со
С>й
сл
о
со
4^
-vj
о
^J
аз
сл
to
со
о
4^
-vl
СО
-vl
аз
00
£ь
_
со
со
СО
со
о
о
со
4^
сл
4ь>
-4
4*
-<1
СО
ю
о
4^
-4
4^
СЛ
аз
00
к-
00
4^-
СО СО
to to
сл о
о о
со со
4^ 4^
со to
оо to
-4 -4
СО —
88
СО 4^
О О
4^ 4^
аз аз
со со
со со
о оо
-4 -4
СО -4
4*. —
ел to
4* »—
СО
СЛ
о
со
4^.
СО
о
о
со
со
Q СО
аз
аз
СО
а>
4^
^i
о
4^
88
аз
О
"vl
4^
00
4^
аз
-v)
СО
4^
-4
О
4».
СЛ
со
-4
-4
to
СЛ
со
а>
со
о
сл
о
со
со
-vj
4^
аз
аз
to
сл
аз
о
4*
4^
■^4
аз
-4
^ч1
О
to
00
4>>
со
8
О
СО
со
сл
-4
аз
4^
сл
-vl
4»>
О
4^
4^
Ю
О
аз
00
гг
сл
00
ю
со
сл
о
со
со
со
со
аз
to
СО
О
о
о
4^
со
аз
4^
00
(Т)
сл
00
аз
to
to
СО
о
о
со
со
to
о
аз
~
to
со
сл
о
4*
СО
о
^1
СО
аз
со
аз
со
4*.
to
00
сл
о
со
со
о
о
сл
со
сл
-vl
со
о
4*
to
сл
о
со
аз
сл
сл
сл
to
00
о
о
со
ю
00
о
сл
-vl
со
со
4^
о
4ь
СО
to
о
сл
СО
4^
4*
со
to
-vl
сл
о
со
to
сл
СО
сл
аз
со
о
о
о
4^
со
со
сл
-4
со
аз
to
to
-4
о
о
со
to
со
-v]
сл
4ь
аз
^1
аз
о
4*
о
-vl
со
сл
сл
сл
со
о
to
оз
сл
о
со
to
4*
сл
со
о
аз
со
о
4*
о
со
со
сл
со
to
СО
о
to
а'з
о
о
со
СО
о
сл
4*
аз
to
о
со
СО
сл
to
со
сл
—*
IO
СО
00
to
сл
сл
о
со
аз
аз
4*
со
00
-v|
ю
о
со
00
СО
о
сл
£ь
СО
со
со
-4
to
сл
о
о
со
4^
to
4*.
00
to
со
сл
о
со
00
to
оо
£ь
-4
4*
О
00
to
£>.
СЛ
о
со
аз
4*
аз
-4
со
о
со
-vl
оз
4*
СО
4*.
UI
сл
о
ю
4ь.
О
О
СО
о
СО
о
4*
СЛ
«
-vl
со
о
со
-vl
о
о
со
4*
со
аз
4*
СО
to
со
сл
о
со
о
аз
4=*
4*.
со
аз
4*
О
СО
аз
со
аз
4*
*"*
00
8
00
i
Темпера
tVl - °
1
1
то
1*
о
Oj •
^ ^
• О О
Си -
1
58
CD
0j О
1 -
а.
1
; ^
1 vt"
456 Часть IV. Галогениды, оксигалогениды, борг ид р иды и карбонил урана
Таблица 202
РАСТВОРИМОСТЬ УРАНИЛФТОРИДА В ВОДЕ
Температура,
•с
25,0
75,0
99,9
Растворимость UO2F2
% (вес.)
67,3
69,6
72,4
моли\л
5,18
5,59
6,08
Плотность,
г\см*
2,405
2,472
2,588
Таблица 203
РАСТВОРИМОСТЬ УРАНИЛФТОРИДА
ПРИ РАЗЛИЧНЫХ ТЕМПЕРАТУРАХ
Температура,
°С
1
25
60
100
Растворимость U02F2
% (вес.)
61,4
65,6
71,0
| 74,1
молярные
доли
0,0862
0,100
0,125
0,143
оказавшаяся равной 1,0 ккал-моль1. Растворимость уранилфторида в воде
с температурой возрастает, а в плавиковой кислоте падает, в результате
чего при нагревании наблюдается кристаллизация уранилфторида.
Выделенный при низких температурах уранилфторид очень
гигроскопичен; полученный же высокотемпературным гидрофторированием не
расплывается даже при длительном стоянии на воздухе. Гидра тированный
уранилфторид может быть дегидратирован при 120° без заметного
разложения [10, 22]. Гидраты уранилфторида, как правило, неустойчивы при
температурах выше 100°. Для полной дегидратации уранилфторида,
содержащего избыток трехокиси урана, требуется более высокая температура
(до 250°). При 250° уранилфторид меняет окраску и становится розовым,
однако химически, повидимому, он при этом не меняется [5].
Водные растворы нейтрального уранилфторида не действуют на стекло
даже при температура кипения. При нагревании 2УИ водных растворов
уранилфторида в трубках из стекла пирекс до 200° в течение 10 суток
образования осадка не замечено [5]. В результате обработки перегретым
водяным паром при 900° уранилфторид теряет весь фтор и превращается
в закись-окись урана. В случае проведения этой операции при 450°
образуется в основном трехокись урана [23].
Система U02F2— HF— H20 [21]. Фазовый анализ этой системы проведен
по методу определения растворимости (применявшийся уранилфторид
получали реакцией трехокиси урана с фтористым водородом). Различные смеси
выдерживали при 25° в течение двух-трех недель до достижения равновесия.
Полученные результаты представлены в табл. 204.
Таблица 204
РАВНОВЕСНЫЙ СОСТАВ ЖИДКОЙ И ТВЕРДОЙ ФАЗ В СИСТЕМЕ
HF
0,00
11,88
20,70
25,75
32,51
41,70
U02F2
Жидкая фаза
состав. %
U02F2
65,55
31,88
22,99
18,19
1,35
6,10
н20
34,45
56,24
57,01
56,01
56,14
52,20
— HF—Н20 при 25е
.25
«25
2,224
1,440
—
—
1,231
1 —
Твердая фаза
состав, %
HF
.
8,52
18,20
14,51
12,06
I 3,50а)
| 8,77
U02F2
51,95
32,50
51,25
64,47
83,76а)
75,2
н2о
__
39,53
49,30
34,24
23,47
12,74а)
16,03
а) Частично высушена.
Глава 16. Оке иго л о ген иды урана
457
Приведенные данные показывают заметное уменьшение растворимости
с ростом концентрации плавиковой кислоты. В исследованной области
имеется лишь одна твердая фаза. При экстраполяции конод между точками
соответствующих составов твердой и жидкой фаз получается след>ющий
состав равновесной твердой фазы: t702F2-H20 (рис. 64).
Термическое разложение уранилфторида. Уранилфторид, повидимому,
устойчив на воздухе до 300°. При более высокой температуре наблюдается
разложение с образованием закиси-окиси урана [24, 25J. В результате
iOO°/o
Н20 50% HF
100% Ю0%
Рис. 64. Система U02F2—HF—Н20 при 25Ь.
подробного изучения [26] термического разложения уранилфторида
установлено, что оно наблюдается даже при 200°, однако разложение в
основном протекает при 850-900° (в вакууме). Уранилфторид при этом не
плавится. В печи остается черный коксоподобный остаток закиси-окиси;
одновременно отгоняются желтое вещество L02F2 (?) и зеленое вещество—
загрязненный тетрафторид. Имеются также доказательства в пользу
выделения фтора (или, может быть, гексафторида урана), так как обычно
в ртутном диффузионном насосе образуется налет фторида ртути (I). Для
объяснения происхождения различных продуктов термического разложения
уранилфторида предполагается одновременное протекание нескольких
реакций:
2U02F2 -* U02 + UF4 + 02; (10)
U02F2 ^U02 + F2; (И)
4U02F2 -» U308 + UF4 + 2F2. (12)
Однако имеющиеся доказательства слишком скудны для того, чтобы можно
было решить, какая из этих реакций преобладает. Первичной реакцией,
которой можно, вероятно, объяснить наблюдаемые явления (по крайней
458 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
мере частично), является следующая:
3U02F2 ~> UF6 + 4 U308+ |<Э2. (13)
Фрид и Дэвидсон вычислили ориентировочно свободную энергию этой
реакции; из их расчетов следует, что для ее протекания необходима температура
по крайней мере 1300° К [14]. Однако при этом расчете было сделано
несколько произвольных допущений, поэтому нельзя исключить возможность
образования гексафторида урана по этой реакции. Желательна дальнейшая
работа над этим вопросом, так как образование гексафторида путем
разложения уранилфторида представляет некоторый интерес.
Некоторые химические реакции уранилфторида, Уранилфторид может
быть восстановлен водородом:
U02F2 + H2 -> U02 + 2HF. (14)
Эта реакция быстро протекает при 600° й довольно медленно при 500°
[8, 27]. Получаемая таким образом двуокись урана весьма реакционноспо-
собна. При действии фтора уранилфторид превращается в гексафторид:
U02F2 + 4F2 -> UF6 + 2F20. (15)
Типичной реакцией для всех растворов, содержащих ион U02+, но
особенно изученной в отношении уранилфторида, является восстановление
хлоридом олова (II) (см. гл. 12).
U02F2 + 4HF+ SnCl2 -> UF4 + SnCl2F2 + 2H20. (16)
Растворы уранилфторида, содержащие глюкозу или спирт,
восстанавливаются на прямом солнечном свету до тетргфторида. При сплавлении
со щелочью, например с едким натром или окисью кальция, уранилфторид
превращается в смесь ураната и фторида натрия (или кальция) [28].
Комплексные соли уранилфторида, Уранилфторид является
координационно ненасыщенным соединением и образует большое число двойных солей.
С фторидами металлов образуются двойные соединения следующих типов:
Л. MU02F3 или MF-U02F2 (M--Na);
Б. M3U02F5 или 3MF-U02F2 (M = K, NH4,~Ba);
В. M3(U02)2F7 или 3MF-2U02F2 (M=K);
Л M5(U02)2F9 или 5MF-2U02F2 (M = K).
В 1884 г. Дитте [4] описал соединения ряда М41Ю2Р6 (M = Li, Na, К,
Rb, T1), которые он получил сплавлением закиси-окиси урана с фторидом
калия и небольшим количеством карбоната калия. Он описал также соли
типа M4UOF8-flH20 (М = К, Rb). Смительсом [3], однако, показано, что
последние соединения относятся, по всей вероятности, к типу £, в то время
как первые являются просто уранатами. Сообщается о получении двух
соединений Cs4U02Fc и K4U02Fc [29J, однако подробности получения и данные
анализа не опубликованы. Соединение цезия образует моноклинные
призматические кристаллы. В связи с этим нельзя, повидимому, считать
исключенным существование комплексных соединений типа 4MF-U02F2; они,
вероятно, могут быть получены, хотя скорее всего и не по методам Дитте.
Хотя комплексные соединения уранилфторида были открыты и изучены
Болтоном [30], первым, кто разъяснил их взаимоотношения, был Бэкер [31].
При добавлении фторида калия к раствору уранилнитрата или
уранилфторида выпадает КзЬ'О^. Если эту соль перекристаллизовать из чистой
воды (или из водного раствора, содержащего менее 13% дифторида калия),
Глава 16. Оксигалогениды урана
459
образуется K5(U02)2F9. Далее, если K3U02F5 или K5(U02)2F9
перекристаллизовать из раствора уранилнитрата, образуется K3(UCL)2F7.
Из этих солей довольно подробно была изучена соль K3tj02F5. Она плавится
с разложением при температуре красного каления. При нагревании на
воздухе имеет место потеря фтора и соль превращается в уранат. Теплой
концентрированной серной кислотой она полностью разлагается. Добавление
к раствору K3U02F6 аммиака или едкого натра вызывает осаждение диура-
ната; карбонаты же аммония и натрия дают при этом растворимые в воде
комплексные соединения. Последние реакции доказывают, что связь урана
в комплексе K3U02F- не является очень прочной. При действии на раствор
этого комплекса солями меди, серебра, цинка, ртути, железа или платины
осадка не образуется, при введении же ионов бария, кальция и свинца
выпадают нерастворимые комплексные соли типа Ba2(UO2)2F10-2H2O. В
результате фотохимического восстановления растворов K3L'02F5 щавелевой
или муравьиной кислотой выпадает K2UF6. Все комплексные соли при
нагревании на воздухе или сплавлении с содой превращаются в уранаты;
при прокаливании аммонийной соли образуется закись-окись урана и
улетучивается фторид аммония.
Перекись водорода окисляет NaU02F3 или K3U02F6 в водном растворе
до нерастворимых надсоединений, для которых был предложен следующий
состав: NaU04F-5H20 и K4U4015F6-4H20. Эти формулы нельзя считать
окончательно установленными. При температуре выше 100° надсоединения
разлагаются с выделением кислорода [32]. Изучена электропроводность
водных растворов K3U02F5 и (NH4)3U02F6 [33]; полученные результаты
доказывают существование устойчивого иона (UOgF^)3" (ср. с
кристаллографическим доказательством, приводимым ниже). В табл. 205 приведены
некоторые данные, относящиеся к получению и кристаллографическим
свойствам этих комплексных солей.
Недавно изучена кристаллическая структура K3U02F5 [36]. Вещество,
полученное при добавке фторида калия к концентрированному раствору
уранилнитрата, состояло из одной фазы — безводного K3U02F6. Эта соль
имеет тетрагональную структуру и принадлежит к объемноцеитрированной
трансляционной группе. Элементарная ячейка имеет следующие константы:
Да-9,05 ±0,05 А; а3 = 18,10 ± 0,10 А.
Она содержит восемь молекул. Вычисленная плотность равна 4,29 г/см3;
пространственная группа симметрии, вероятно, 14/amd. Выведена
приближенная структура этого соединения; интересной особенностью является
существование в решетке ионов (U02F4)2~, тогда как пятый атом фтора связан
только с калием.
Уранилфторид образует ряд продуктов присоединения с аммиаком [6].
При действии жидкого аммиака на уранилфторид (или аммиакаты с меньшим
содержанием аммиака) образуется аммиакат состава U02F2-4NH3. Это
соединение представляет собой вещество темного оранжево-красного цвета, более
устойчивое, чем соответствующие производные уранилбромида и уранилхло-
рида. Газообразный аммиак реагирует с уранилфторидом с образованием
оранжево-красного аммиаката U02Fa-3NH3, который при нагревании превращается
в желтый UOoF2-2NH3.
Приготовлены также комплексные соединения уранилфторида с
органическими основаниями [37]. Раствор соответствующего основания (в разбавленной
плавиковой кислоте) приливают к раствору уранилнитрата (содержащему ионы
фтора). Выпавший осадок отделяют, промывают сначала разбавленной
плавиковой кислотой, затем водой и наконец высушивают между листами
фильтровальной бумаги. Большинство этих соединений устойчиво на воздухе в
течение длительного времени; при нагревании они превращаются в закись-окись
Таблица 205
ПОЛУЧЕНИЕ И СВОЙСТВА КОМПЛЕКСНЫХ СОЕДИНЕНИЙ УРАНИЛФТОРИДА И ФТОРИДОВ МЕТАЛЛОВ
Формула соединения
NaU02F3.4H20
K3U02F5
(NH4)3U02Fe
Отношение
фторида
металла
к уранил-
фторнду
1 :1
3:1
3:1
1
Кристаллический
габитус L31]
Моноклинные
Тетрагональные; заметно не
флюоресцируют
Тетрагональные; сильно
флюоресцируют под
действием
рентгеновских или
ультрафиолетовых лучей
Кристаллографические
данные [31]
а: Ь: с= 1,0270:1:0,5222;
Р = 94°51'
а: г=0,992
Показатель преломления
равен 1,495 [34]
Плотность при
20°. г\см*
[31J
4,263
3,186
Растворимость
12,5 г на 100 г
воды при 21° [2];
осаждается при
добавлении спирта
10,11 г на 100 г
раствора при 27е;
20,70 г на 100 г
раствора при 81,3°
[35]; нерастворим
в спирте
Получение
Кристаллизуется при медленном
выпаривании растворов уранил-
нитрата и фторида натрия или
растворов ураната натрия во
фтористоводородной кислоте; точные
условия неизвестны; при
перекристаллизации из воды
получается дигидрат [2]
Выпадает в виде желтого
кристаллического вещества при
добавлении небольшого избытка
фторида калия к раствору ура-
нилнитрата; является первичным
продуктом реакции между
фторидом калия и уранилфторидом или
уранилнитратом [2, 3, 31]
Получается при добавлении
фторида аммония к раствору ура-
иилнитрата [2, 31]
Продолжение табл. 205
Формула соединения
Ва3(иог),Р„-2Н,0
Ks(UOa)2F,-2H80
K5(UOa)2F,
1
Отношение
фторида
металла
к уранил-
фториду
3:1
3:2
5:2
Кристаллический
габитус [31J
Моноклинные;
обладают
отчетливой зеленой
флюоресценцией
Триклинные;
большие
кристаллы, обладают
отчетливой
флюоресценцией
, Кристаллографически*
ные 131J
а:6:с = 0,918: 1 :
p=IJ4°0'
а:6:с = 0,6222: 1
о= 72°38';
р=11бс23';
1 = 11Г57'
дан-
0,978;
0,568;
Плотность при
20°. <?/с*з
[31]
4,108
4,379
растворимость
Весьма слабо
растворим в
горячей воде
Растворим
в теплой воде
Получение
Получается высушиванием при
100° продукта, образующегося
при сливании растворов ВаС12 и
K3U02F5; тем же способом
получаются соединения кальция
и свинца [2]
Получается при добавлении
к раствору уранилфторида
фторида калия в количестве,
недостаточном для образования
стойкого осадка, с последующим
выпариванием или путем
кристаллизации K?U02F5 или K5(U02)2F9
из растсора уранилнитрата или
уранилфторида [31]
Получается при
перекристаллизации K3U02F5 из воды или из
раствора бифторида калия с
концентрацией менее 13% 131]
462 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
урана. Они немного растворимы в воде, причем обычно их растворимость
возрастает с температурой. Эти комплексы растворяются в кислотах;
основания, например едкий натр и аммиак, осаждают из этих растворов уранаты, при
действии карбонатов образуются растворимые комплексы. В органических
растворителях, как, например, в ледяной уксусной кислоте, спирте, эфире
и ацетоне, комплексы с органическими основаниями не растворимы. В табл. 206
перечислено несколько таких соединений и даны их растворимости в воде.
Таблица 206
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ УРАНИЛФТОРИДА С ОРГАНИЧЕСКИМИ ОСНОВАНИЯМИ
Основание
Пиридиний
Пиридиний
Хинолиний
Хинолиний
Тетраметиламмоний
Тетраэтиламмоний
Тетраэтиламмони й
Триметил-л-толиламмоний
Триметил-тг-толиламмоний
Тетраметилпиридиний
Фени лдиэтиламмони й
Фенилдиэтиламмоний
Триэтилсульфоний
Триметиламмоний
Пропиламмоний
Пропиламмоний
Тетрапропиламмоиий
Метилэтилпропилфеииламмоний
Фениламмоний
Фенилдиметиламмоний
Соединение
C6HeNHU02F3-H20
C5HeNH(U02)2F5.3HaO
C9H8NHU02F3.H20
C9H8NHU02F5.2H20
(CH3)4N(U02)2F5.2H20
(C2H5)4NU02F3
(C2H5)4NU02F3.2H20
(CH3CeH4) (CH3)3NU02F3-2H20
(CHaCGH4) (CH3)3N(U02)2F5.H20
(QH2) (CK3)4NH (U02)3F7.6HaO
C6H5N (C2H5)2HU02F3.2H20
CeH5N (C2H5)2H (U02)2F5.2H20
(C2H5)3S(U02)2F5.2H20
(CH3)3NH(U02)2F5.2H20
C3H7NH3U02F3-2H20
C3H7NH3(U02)3F7.6H20
(C3H7)4N(U02)3F7-2H20
(CH3) (CaH5) (C3H7) (CeH5) N (UOa)3F7 - 6HaO
CeH5NH3UOaF3.3H20
CeH5N (CH3)2H(U02)2F5-H20
Растворимость в воде
при 20°,
а/100 мл
1,289
1,952
0,126
0,979
0,143
0,716-
0,771
1,645
3,091
0,708
1,759
3,896
0,897
Все эти соединения можно подразделить на три типа:
Л. MU02F3-ttH20 или MF-U02F2;
£. M(U02)2F5->zH20 или MF.2U02F2;
В. M(U02)3F7-azH20 или MF-3U02F2,
где М~одновалентное органическое основание.
ОКСИФТОРИД УРАНА (IV) LOFa
В 1905 г. Джиолитти и Агаменнон [38] сообщили, что ими якобы было
получено соединение UOF2-2H20. В 1904 г. Джиолитти [39] разработал метод
определения урана, основанный на осаждении его в виде тетрафторида.
Результаты, которые он при этом получал, были весьма непостоянными.
В поисках причины этого он пришел к неожиданному выводу, что тетрафторида
урана вообще не существует, при приливании же плавиковой кислоты к
раствору солей урана (IV) выпадает оксифторид урана (IV).
Вряд ли можно сомневаться в том, что причиной таких выводов Джиолитти
были весьма несовершенные методы анализа. Его определения фтора могли
содержать ошибку порядка нескольких сотых процентов.
Глава 16. Оксигалогениды урана
463
В процессе этой работы Джиолитти и Агаменнон сделали несколько
наблюдений над реакцией между плавиковой кислотой и закисью-окисью урана,
которые были подтверждены современными исследованиями [40, 41]. При
обработке закиси-окиси урана плавиковой кислотой образуются две твердые
фазы, которые ввиду разницы в плотностях легко могут быть разделены в
жидкости с промежуточным удельным весом. Менее плотное вещество имеет
сике-зеленую окраску; более плотное окрашено в зеленый цвет. Природа этих
двух веществ пока не известна.
Были сделаны попытки получить оксифторид урана (IV) обработкой окси-
хлорида жидким безводным фтористым водородом при комнатной температуре,
однако единственным продуктом реакции был тетрафторид [13]. Попытки
приготовить оксифторид сплавлением двуокиси урана с тетрафторидом также
окончились неудачей [42].
УРАНИЛХЛОРИД U02C!2
Безводный уранилхлорид можно приготовить только при высокой
температуре в газообразной фазе. Водные растворы уранилхлорида получаются легко,
однако пока еще не решена задача обезвоживания их без образования основных
солей.
Приготовление безводного уранилхлорида.
Пелиго [43, 44] для получения безводного уранилхлорида хлорировал двуокись
урана сухим хлором при температуре красного каления:
U02 + C12 -> 1_Ю2С12. (17)
Реакция обычно не доходит до конца. Регельсбергер [45] отделял
образовавшийся уранилхлорид от непрореагировавшей двуокиси экстрагированием его
эфиром. Недостатком данного метода является невозможность последующего
полного отделения эфира от уранилхлорида. Позже группой исследователей [46]
было установлено, что реакция между двуокисью урана и техническим сухим
хлором идет при 500° и дает продукт, содержащий некоторое количество закиси-
окиси урана. С трехокисью урана хлор не реагирует при 400°; при более
высокой температуре образуется закись-окись [47].
При обработке хлором активной двуокиси урана, приготовленной путем
восстановления трехокиси метаном (см. гл. 11), образуется уранилхлорид [48].
U302S2(U02-^US) реагируете хлором при 60°. По окончании реакции
образовавшийся тетрахлорид урана удаляют возгонкой в токе хлора при 600°, причем
остается желтый безводный уранилхлорид [49].
Кроме того, известны и многие другие реакции, в которых образуется
безводный уранйхлорид; например, при реакции четыреххлористого углерода
с различными окислами урана образуется некоторое количество уранилхлорида,
который, повидимому, может быть отделен от одновременно образующегося
тетрахлорида урана возгонкой в вакууме при повышенных температурах или
возгонкой в токе хлора (см. гл. 14). Если трехокись урана обработать парами
четыреххлористого углерода при 290°, продукт реакции содержит 23%
уранилхлорида и 77% тетрахлорида урана [50]. Если вместе с четыреххлористым
углеродом вводится окись углерода или, что еще лучше, хлороформ, то
выход уранилхлорида возрастает [51]. Но это не особенно удобные
препаративные методы.
Хлористый водород легко реагирует с трехокисью урана с образованием
уранилхлорида. Реакция является экзотермической и самопроизвольно
протекает при комнатной температуре. Необходимым условием является присутствие
влаги,так как при использовании безводной трехокиси урана реакция идет
крайне медленно. Закрытую систему, содержащую частично гидратированную
трехокись урана, эвакуируют без нагревания для удаления воздуха, но с
оставлением возможно большего количества воды. Затем вводится хлористый водород со
464 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
скоростью, достаточной для поддержания в реакционной системе давления
в 1 атм. Для окончания реакции при комнатной температуре требуется
приблизительно от 18 до 24 час. Приготовленное таким образом вещество имеет
состав, соответствующий формуле U02C12 • Н20. Гидратированный уранилхлорид
может быть высушен без разложения в токе сухого хлористого водорода при
300° [52]. Опубликовано краткое сообщение (лишенное подробностей), что при
обработке хлористым водородом суспензии трехокиси урана в этиловом спирте
или четыреххлористом углероде образуется уранилхлорид [53]. Ценность этой
реакции в качестве препаративного метода неизвестна.
Наилучшим методом получения безводного уранилхлорида является,
вероятно, реакция между тетрахлоридом урана и кислородом при 300—350°:
UC!4 + 02 -* Ш2С!2 + С12. (18)
Реакционная масса имеет тенденцию к спеканию, что мешает завершению
реакции; спекание можно предотвратить встряхиванием реакционной трубки. Смеси
пента-, гекса- и трихлорида урана также реагируют с кислородом с
образованием уранилхлорида, но тетрахлорид все-таки является, повидимому,
наилучшим исходным материалом [47].
Ацетил хлор ид СН3СОС1 реагируете различными окислами с образованием
галогенидов или оксигалогенидов [54]. При реакции между трехокисью урана
и жидким ацетилхлоридом при комнатной температуре получается светложел-
тый кристаллический порошок, имеющий состав 1Ю2С12(СН3СО)20. О
возможности получения чистого уранилхлорида из этого комплекса ничего не
сообщается. Представляла бы интерес дальнейшая работа в этой области.
Получение гидратов уранилхлорида. Водные рас-
* творы уранилхлорида получены осторожным окислением раствора тетрахлорида
урана азотной кислотой [55], растворением моногидрата трехокиси урана
в эквивалентном количестве соляной кислоты [56], а также введением стехио-
метрического количества хлорида бария в раствор уранилсульфата [57]. Путем
выпаривания таких растворов можно получить твердые гидраты. Обычно
получаются аморфные продукты, но при медленном испарении в эксикаторе
образуются флюоресцирующие желтовато-зеленые кристаллы U02C12-3H20,
обладающие двойным лучепреломлением и легко разлагающиеся [56]. Кристаллизацию
можно иногда вызвать введением в концентрированный раствор уранилхлорида
затравки из небольшого количества кристаллов, полученных обработкой
некоторой части полуаморфного вещества концентрированной соляной кислотой
и спонтанной кристаллизацией путем медленного испарения в эксикаторе.
Моногидрат U02C12'H20 получается медленным выпариванием водного
раствора уранилхлорида в сухом воздухе [57]. Этот моногидрат получается
также выпариванием раствора уранилхлорида при 120° досуха [58]. При
насыщении хлористым водородом насыщенного водного раствора уранилхлорида
при—10° происходит образование 1Ю2С12-НСЬ2Н20 в виде желтых, весьма
неустойчивых кристаллов [59].
Во всех вышеупомянутых работах принималось, что гидраты
уранилхлорида не могут быть дегидратированы без значительного разложения. Так,
сообщается [56], что во время испарения выделяется хлористый водород, притом
даже при комнатной температуре. Склонность к образованию основной соли
очень велика. Из сиропообразного продукта выпариванием раствора
уранилхлорида выделено соединение U02(0H)C1-2H20 в форме небольших желтых
игл [56]. Это вещество, вероятно, идентично с веществом, описанным гораздо
раньше Лекану [60]. Оно устойчивее гидрата уранилхлорида;
кристаллизационную воду оно теряет при 150° без дальнейшего разложения.
Физические свойства уранилхлорида.
Кристаллическая структура [61]. Рентгенограммы порошка безводного уранилхлорида
и игольчатых кристаллов, полученных конденсацией его из паров (500°), ока-
Глава 16. Оксигалогениды урана
465
зались различными. Рентгенограммы игольчатых кристаллов обнаружили много
максимумов, отсутствовавших в порошкограммах. Кристалл относится к
ромбической сингонии. Элементарная ячейка его содержит четыре молекулы.
Константы решетки имеют следующие значения:
Иглы: а0=8,71 ±[0,01А; &0 = 8,39 ± 0,01А; с0 = 5,72 ± ОДНА.
Порошок: а0=8,69 ±Ь0,01А; 60 = 8,39 ± 0,01 А; с0 = 5,70 ± 0.01А.
Рассчитанная из рентгеновских данных плотность равна 5,426 г/см3.
Прямое измерение плотности путем гидростатического взвешивания в бензоле
дало 5,28 г/см* [49] в соответствии с прежними литературными данными.
Летучесть. Уранилхлорид обладает некоторой летучестью в токе хлора
или кислорода при температуре выше 500°. Найдено, однако, что он заметно
не улетучивается при нагревании в течение 7 час. в атмосфере хлора при 630° [62].
Результаты, полученные при изучении летучести с применением меченых
атомов, показали, что летучесть при температуре ниже 775° незначительна.
Но эти выводы требуют подтверждения [63]. Разложение уранилхлорида
затрудняет анализ данных, полученных при изучении летучести. Сообщается, что
безводный уранилхлорид плавится при относительно низкой температуре
(температура красного каления). Пары имеют оранжево-желтый цвет [43,44].
Другие физические свойства. Кристаллы безводного уранилхлорида не
обладают триболюминесценцией [64]. Расплавленный уранилхлорид проводит
электрический ток; при этом выделяется хлор и из расплава осаждается
двуокись урана [65].
Химические свойства уранилхлорида. Безводный
уранилхлорид является яркожелтым кристаллическим веществом. Гидраты
имеют зеленоватый оттенок и, гювидимому, флюоресцируют. Как безводная,
так и гидратированные формы уранилхлорида очень гигроскопичны и на
воздухе быстро образуют вязкие растворы. В сухом воздухе соединения
устойчивы в течение неограниченно долгого времени.
Растворимость в воде и устойчивость водных растворов. Уранилхлорид,
его гидраты и основные соли хорошо растворимы в воде. Растворимость три-
гидрата составляет 746 частей па 100 частей воды при 18°; она еще выше при
более высоких температурах [56], что кажется, однако, странным.
Растворимость больше в растворе соляной кислоты [66]. Плотность насыщенного водного
раствора при 18° равна 2,740 г/см?\ таким образом, стекло или кварц будут
всплывать в такой жидкости. Плотность растворов, содержащих 1—10%
уранилхлорида при 13—16°, равна от 1,0056 до 1,0517 г/см* [67]. Молярная
теплота растворения U02C12H20 (в 2500 молях воды), по определению,
равна 6,05 ккал (при 18—20°) 168]. Но это значение неточно вследствие
неопределенности состава использованной соли.
Водные растворы уранилхлорида термически и фотохимически
неустойчивы [56]. Растворы обычно дают кислую реакцию на лакмус, что указывает
на заметный гидролиз. Хотя де-Коник [69] и оспаривает существование
соединения U02(0H)C1-2H20, найденного другими исследователями, но
образование этой основной соли в результате гидролиза можно считать, повидимому,
довольно точно установленным.
Неводные растворы уранилхлорида. Уранилхлорид растворим в метил-
ацетате, этилацетате, ацетоне и пиридине, но о наличии реакции между этими
соединениями и уранилхлоридом ничего не сообщается [70, 71]. Безводный
уранилхлорид нерастворим в четыреххлористом углероде, ксилоле и бензоле.
Он нерастворим в простых эфирах и хлороформе и не реагирует с ними.
Уранилхлорид растворим в спиртах, ацетофеноне, пиридине, диоксане и реагирует
со всеми этими растворителями [47]. Тригидрат уранилхлорида растворим
в спирте и эфире.
30 Химия урана
466 Часть IV. Галогениды, оксиеалогениды, боргидриды и карбонил урана
Унру [6] приготовил растворы безводного уранилхлорида в амиловом
спирте путем повторного выпаривания уранилацетата сначала с соляной
кислотой, затем с водой, с последующим растворением гидратированного
уранилхлорида в амиловом спирте и отгонкой воды и части спирта.
Термическая устойчивость. Уранилхлорид и его гидраты легко разлагаются
при повышенных температурах. При прокаливании на воздухе уранилхлорид
превращается в закись-окись урана. Сообщается, что уранилхлорид при
температурах, превышающих 450°, разлагается в вакууме с выделением хлора
и образованием смеси двуокиси и закиси-окиси урана [72]. Исследование этого
вопроса привело к следующим результатам [49]. В токе азота разложение
до двуокиси урана и хлора происходит при температуре выше 400°. В вакууме
оно начинается при 300°. В токе хлора уранилхлорид плавится примерно при
500°, превращаясь в красновато-бурую жидкость. При повышении температуры
до 900° происходит разложение до двуокиси урана и хлора с частичной
возгонкой уранилхлорида и пентахлорида урана, образующегося за счет реакции
с хлором. Большая часть уранилхлорида превращается в черную
кристаллическую двуокись. Рентгенографическое исследование показало, что двуокись,
полученная этим путем, немного отличается от обычной бурой двуокиси урана.
Отношение к восстановителям. Безводный уранилхлорид
восстанавливается металлическим калием до двуокиси урана и хлорида калия [43, 44]. Магний
при температуре красного каления частично восстанавливает уранилхлорид
до металлического урана [73]. Водород, цинк, медные стружки не оказывают
восстанавливающего действия [67, 74]. Сероводород восстанавливает
уранилхлорид до двуокиси урана, с выделением серы и хлористого водорода.
Отношение к щелочам [57, 67, 74]. Сплавление с едким кали или едким
натром на воздухе дает смесь диураната с небольшим количеством ураната.
С гидроокисью кальция образуется закись-окись урана и некоторое
количество ураната кальция; если же сплавление ведется без доступа воздуха, то
вместо закиси-окиси урана образуется двуокись. Гидроокись бария действует
подобным же образом. Сплавление с окисью кальция или бария на воздухе
дает закись-окись урана; при сплавлении с СаО получается уранат и диура-
нат кальция, в случае ВаО образуется в основном диуранат бария.
Гидроокись и окись стронция ведут себя подобно соединениям бария.
Отношение к кислотам [67, 75]. При нагревании уранилхлорида с азотной
кислотой выделяются хлор и окислы азота. Концентрированная серная кислота
превращает уранилхлорид в уранилсульфатс выделением хлористого водорода.
Уранилхлорид растворяется в селеновой кислоте; при нагревании выделяется
хлор и образуется раствор селенита натрия.
Реакции в водных растворах. Уранилхлорид в водных растворах вступает
во все реакции, характерные для уранил-иона.
Комплексные соединения уранилхлорида с аммиаком и органическими
основаниями. Известен обширный ряд аммиакатов уранилхлорида. Высушенный в
вакууме уранилхлорид поглощает две молекулы аммиака, образуя оранжевое
соединение U02C12-2NH3; из двух молекул аммиака одна связана так
прочно, что не улетучивается в вакууме [76, 77]. Если эфирный раствор
уранилхлорида при нагревании обработать аммиаком,то образуется осадок
U02C12-2NH3-(C2H5)20, из которого эфир может быть отогнан в вакууме [45].
Этот же эфират может быть получен из раствора хлорида в
амиловом спирте обработкой раствора аммиаком; осадок высушивают эфиром [6].
Диаммиакат разлагается при нагревании до 100° с образованием хлорида
аммония; при прокаливании на воздухе он превращается в закись-окись
урана. При повышенных температурах он восстанавливается водородом или
аммиаком до двуокиси урана [45]. Известен и триаммиакат, но только
в форме эфирата U02C12-3NH3-(C2H5)20. Это соединение получпется
обработкой эфирата диаммиаката газообразным аммиаком; оно представляет
Глава 16. Оксигалогениды урана
467
собой вещество оранжевого цвета, устойчивое на воздухе при комнатной
температуре, но теряющее аммиак при нагревании. При взаимодействии
уранилхлорида с жидким аммиаком при 5° образуется неустойчивый тетра-
аммиакат. Это аморфное твердое вещество оранжево-красного цвета,
которое начинает разлагаться при температуре 10° [6]. При обработке
уранилхлорида жидким аммиаком также наблюдалось образование
серовато-зеленого осадка, но состав получающегося при этом соединения не
установлен [78].
Наиболее точная работа по изучению системы уранилхлорид — аммиак
была проведена Спаку [79], исследовавшим реакцию между жидким
аммиаком и уранилхлоридом при —78°. В этих условиях уранилхлорид образует
декааммиакат U02C12- 10NH3. По мере возрастания температуры аммиак
отгоняется. Измерения давления паров указывают на существование следующих
12
Ю 8 6 4
Аммиак, моли
Рис. 65. Система U02C12—NH3[79].
комплексных соединений: U02Cl2. 5NH3, U02C12. 4NH3, U02C12.3NH3,
U02C12-2NH3 и U02Cl2-NH3. Рис 65 иллюстрирует полученные результаты.
Температуры на изотермах указывают области термической устойчивости
различных соединений. Пентааммиакат отчасти неустойчив даже при — 44°,
в то время как моноаммиакат вполне устойчив до 130°.
Теплота образования аммиакатов изменяется от — 9,63 ккал на 1 моль
для пентааммиаката до — 18,03 ккал на 1 моль для моноаммиаката.
Многие органические основания также образуют комплексные соединения
с уранилхлоридом. Хотя в координационной сфере урана в уранилхлориде,
повидимому, максимально могут разместиться четыре молекулы аммиака,
число органических групп вследствие пространственных затруднений обычно
равно двум. Способы получения и свойства многих подобных комплексов
даны в табл. 207. Комплексообразующие группы помимо основных азотистых
соединений включают и соединения, главнейшие свойства которых
обусловлены наличием кислорода или серы.
Комплексные соединения уранилхлорида и галогенидов металлов.
Уранилхлорид образует ряд двойных солей типа M2U02C14, где М —
одновалентный металл или эквивалентный ион. Безводные К2Ш2С14 и Na2U02Cl4
получены обработкой галогенида соответствующего щелочного металла парами
уранилхлорида при температуре красного каления [66]. Двойные соли
представляют твердые вещества золотисто-желтого цвета, растворимые в
воде и плавящиеся при температуре красного каления без выделения
паров,
»•*
468
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ УРАНИЛ
Органическое
основание
Комплексное соединение
Метод приготовления
Этиловый
[45]
эфир
2,3-Диметилхромон
[80]
2,3-Диметилтио-
хромон [81]
Анилин [82]
л-Толуидии [82]
Пиридин [83]
л-Нитрозодиметил-
анилин [84]
л-Нитрозодиэтил-
анилии [85]
Дикетопиперазин
[86]
Ацето-л-фенетидин
[87]
Метилацетани-
лид [88]
Феннлдиметилпира-
золон
(антипирин) [89]
Бромантипирин [90]
Диметиламиноанти-
пирин
(пирамидон) [91]
U02C12-2(C2H6)20
UO2CI2-2CHH10O2
UO2Cl2-2C11H10OS
U02Cl2.2CeH5NH2
U02C12.2C7H7NH2
U02C12.2C6H5N
U02Cl22(CH3)2NCeH4NO
U02Cl2-2(C2H5)2NCeH4NO
U02C12C4H6N202-1,5H20
UO2Cl2.2C10H13O2N
UO2Cl2.3C10HI3O2N
U02C12 2C0H5N (СНз)СОСНз
U02Cl2-2CnH12N20
иОзСи-гСцНцВгЫзО
U02CI2-CnHuN(CH3)2N20
U02Ct2.2CuHnN(CHg)2NaO
Выпаривание раствора уранилхло-
рида в эфире
Из концентрированного
солянокислого раствора компонентов;
перекристаллизация из
концентрированной соляной кислоты
Выпаривание концентрированного
солянокислого раствора
компонентов
Из спиртового раствора
компонентов; перекристаллизация из
спирта
Выпаривание спиртового раствора
компонентов
Из раствора U02C12-a;H20 в
амиловом спирте и раствора
пиридина в хлороформе при
охлаждении
Из теплого спиртового раствора
компонентов
То же
Из теплого спиртового раствора
Из теплого раствора в амиловом
спирте
Из раствора компонентов в
этиловом или амиловом спирте
Из водного раствора компонентов
Из спиртовых растворов
компонентов при повышенных
температурах
То же
Из спиртовых растворов
компонентов иа холоду
а) Все ссылки иа работы Раскаиу взяты по Гмелину [92]
469
Таблица 207
ХЛОРИДА С ОРГАНИЧЕСКИМИ ОСНОВАНИЯМИ *
Свойства
Желтые иглы;
разлагается во
влажном воздухе; эфир
не может быть
отогнан
Длинные
блестящие яркожелтые
призмы;
разлагается без
плавления
—
Небольшие желтые
иглы
Желто-зеленые
ромбические
кристаллы
Желтое соединение
со слабой зеленой
флюоресценцией;
очень
гигроскопично
Кирпично-красное;
устойчиво на
воздухе
Оранжевого цвета;
аморфное
Желтые кристаллы;
устойчивы на
воздухе
Желтые
кристаллы с зеленой
флюоресценцией;
устойчивы на
воздухе
Блестящие
пластинчатые
желтые кристаллы
Устойчиво на
воздухе даже при
нагревании
—
Аморфное; желтое
0
Растворители
вода
—
—
—
—
творимо
Трудно
творимо
То же
творимо
То же
»
»
»
этиловый
спирт
—
Слабо
растворимо
То же
Растворимо
—
Растворимо при
нагревании
—
—
Рас
творимо при
нагревании
Растворимо
Трудно
растворимо
Растворимо при
нагревании
Растворимо
амиловый
спирт
—
—
—
—
—
Нерастворимо
То же
Растворимо при
нагревании
Слабо
растворимо
—
Слабо
растворимо
эфир
—
—■
—
—
Нерастворимо
Слабо
растворимо
То же
Очень
слабо
растворимо
Слабо
растворимо
Растворимо
Нерастворимо
То же
этил-
ацетат
—
—
—
—
—
—
—
—
—
Очень
слабо
творимо
ацетон
—
Слабо
растворимо
То же
—
—
Нерастворимо
То же
»
Растворимо
—
Очень
слабо
растворимо
Слабо
растворимо
хлороформ
—
—
—
—
Нерастворимо
Слаборастворимо
То же
Очень
слабо
растворимо
_
Растворимо
Нерастворимо
Слабо
растворимо
бензол
—
"
—
—~"
творимо
—
—
—
470 Часть IV. Галогениды, оксиеалогениды, боргидриды и карбонил урана
Подобные соединения могут быть приготовлены также из водных
растворов, причем в этом случае обычно получается дигидрат. Соединения
K2U02C14 • 2Н20 и (NH4)2U02C14-2H20 были впервые приготовлены
растворением диураната калия или аммония в концентрированной соляной кислоте
с последующим выпариванием раствора до начала кристаллизации [44].
Образование этих солей происходит также при кристаллизации из раствора,
содержащего уранилхлорид и соответствующий галогенид щелочного метала.
Если М=К, то для предотвращения выпадения хлорида калия необходимо
применять избыток уранилхлорида или работать с концентрированными
растворами соляной кислоты. Так, соединение K2U02C14 • 2Н20 было
получено путем кристаллизации водного раствора, содержавшего экви-
молярные количества компонентов и, кроме того, по крайней мере 15%
соляной кислоты [93]. Соль представляет желтые триклинные кристаллы
с отношением осей а:Ь\с = 0,607:1:0,560, а = 80°4Г, р = 77°42' и 7=9Г18'.
Она очень хорошо растворима в воде. При температуре ниже 60°
растворение происходит с разложением, нерастворившийся остаток состоит главным
образом из хлорида калия; при температуре выше 60° раствор и донная
фаза имеют одинаковый состав. Теплота растворения в бесконечно
разбавленном растворе составляет около 2 ккал-моль'1 при 18° (1 моль на 2500 молей
воды) [68]. При 100° соединение дегидратируется с частичным разложением.
Оно плавится при температуре красного каления с выделением хлора;
восстанавливается водородом. В противоположность K3U02F5 соединение
K2U02C14 • 2Н20 не восстанавливается на солнечном свету растворами
муравьиной или щавелевой кислоты [30].
Соли рубидия Rb2U02Cl4-2H20 и цезия Cs2U02Cl4 получаются
аналогично K2U02C14 [93]. Соль рубидия изоморфна солям калия и аммония. Соли
рубидия и цезия растворяются в воде без разложения. Таким образом,
они устойчивее комплекса с хлоридом калия (а также с хлоридом аммония,
ср. ниже), который диссоциирует при растворении в условиях комнатной
температуры. Повидимому, существует определенная зависимость между
устойчивостью комплекса и размером катиона; чем последний больше, тем
комплекс прочнее. Соль цезия кристаллизуется без воды, 'соль рубидия
иногда также образуется в безводной форме. Уэллс и Болтвуд [94], первые
изучившие эти соединения, сообщают, что цезиевый комплекс кристаллизуется
в виде блестящих ромбических кристаллов, однако по другим данным [95]
эти кристаллы относятся к триклинной сингонии. Проведены опыты по
выращиванию крупных кристаллов из солей калия, цезия и аммония; изучены
спектры поглощения и флюоресценции этих кристаллов [29, 96].
Четвертичные аммониевые соли образуют аналогичный ряд соединений.
Они отличаются от комплексов с аммиаком и органическими основаниями
(стр. 467) в том отношении, что комплексообразователь является солью, а не
свободным основанием. В литературе [93] описаны продукты присоединения
хлоридов аммония, триметиламмония, тетраметиламмония и тетраэтиламмония;
попытки приготовить производные гидроксиламина и гидразина не
увенчались успехом. Аммоний уранилхлорид (NH4)2U02C14-2H20 может быть получен
из раствора уранилхлорида и хлорида аммония в концентрированной соляной
кислоте. Он образует очень неустойчивые кристаллы, изоморфные калиевой
соли. В воде при температуре ниже 70° растворяется с разложением.
Солянокислые моно-, ди- и триметиламины образуют неустойчивые соединения типа
[(CH3)3N]2U02C14. Тетраметиламмонийуранилхлорид [(СН3)4 N]2U02C14 может
быть выделен из водных растворов компонентов в виде зеленовато-желтых,
сильно флюоресцирующих тетрагональных кристаллов (а : с= 1 : 0,9057),
растворяющихся в воде без разложения. Таким же путем, а именно
медленной кристаллизацией из водных растворов, содержащих эквивалентные
количества компонентов, образуется и тетраэтиламмонийуранилхлорид
Глава 16. Оксигалогениды урана
471
[(C2H5)4N]2U02CI4. Он представляет желтые тетрагональные кристаллы
(а : с= 1 : 0,9094), изоморфные соответствующему тетраметильному
соединению и подобно последнему растворяющиеся в воде без разложения. Этилен-
диаммонийуранилхлорид C2H4(NH3)2U02C14 [97] лучше всего получить путем
приливания избытка соляной кислоты к водному раствору этилендиамина
и последующего добавления эквимолярного количества уранилхлорида. Это
соединение образует весьма гигроскопичные желтые призматические
кристаллы, плавящиеся при температуре 219° (не резко). Подобным же
образом были приготовлены следующие соединения: пиридиыийуранилхлорид
(C5HeN)2U02Cl4, желтый кристаллический порошок, растворимый в воде
и спирте [98], р-лутидинийуранилхлорид (C7H10N)2UO2Cl4 [99J и хиноли-
нийуранилхлорид (C9H8N)2 U02C14 [100]. Известна также аналогичная оксо-
ниевая соль, ксантилийуранилхлорид (C13H90)2 U02C14 [101].
ОКСИХЛОРИД УРАНА (IV) 1ЮС12
В старой литературе имеется несколько сообщений об этом веществе.
Указывается [102], что безводный оксихлорид урана (IV) получается при
фотохимическом восстановлении эфирного раствора уранилхлорида на прямом
солнечном свету. Образующийся продукт имеет светлозеленую окраску. При
восстановлении уранилхлорида в спирто-эфирных растворах получаются
сложные смеси основных солей; в водном растворе образуется лишь гидра-
тированная двуокись урана. Доверять этим результатам трудно, так как
подробности эксперимента не даны, а описание продуктов вызывает сомнение
в правильной их идентификации.
Указывается также [59, 66], что при выпаривании раствора тетрахло-
рида урана при низких температурах в вакууме получается аморфный осадок.
Повторное растворение последнего в этиловом спирте и высаживание эфиром
приводит к образованию продукта, который после тщательной промывки
эфиром и вьхушивания в эксикаторе имеет состав UOCl2-0,5H2O. Это
кристаллическое вещество яркозеленого цвета, хорошо растворимое в воде.
При нагревании водных растворов указанного соединения происходит
выделение гидратированной двуокиси урана. Эти данные были подтверждены
наблюдениями, сделанными в радиационной лаборатории Калифорнийского
университета: при высушивании водного раствора тетрахлорида при 100°
на воздухе или при 120° в сухом хлористом водороде получались
соединения состава UOi,iClli9- 1,2H20 и UOCl2t02- 1,25H20 [103] (см. также гл. 14).
Наиболее простой и в общем удовлетворительный метод получения
оксихлорида урана состоит в растворении двуокиси урана в избытке
расплавленного тетрахлорида (600°) [13, 104]. При этих условиях
устанавливается равновесие
U02 + UCI4 ^ 2UOCl2. (19)
После охлаждения и измельчения избыток тетрахлорида возгоняют при 450°
в вакууме; в этих условиях диспропорционирование оксихлорида ничтожно
мало.
В радиационной лаборатории Калифорнийского университета оксихлорид
урана (IV) был приготовлен действием паров тетрахлорида урана на его
двуокись [103]. Получаемый таким образом оксихлорид урана менее чист,
чем тот, который образуется при реакции в жидкой фазе.
Оксихлорид урана (IV) пытались получить следующими двумя
методами, не давшими, однако, удовлетворительных результатов. В одном случае
действовали хлором на U302S4 (по аналогии с реакцией, успешно
примененной для получения оксибромида урана), но полученный продукт пред-
472 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
ставлял смесь уранилхлорида и высших хлоридов урана. В другом случае
над тетрахлоридом пропускали перегретый водяной пар в токе инертного
газа; однако и при этом не получили оксихлорида урана
удовлетворительной чистоты [49].
Исследователи Эймс кого университета описывают оксихлорид
урана (IV) как желтое вещество с перьевидными кристаллами, сотрудниками же
Браунского университета цвет этих кристаллов был определен как зеленый.
Желтые кристаллы устойчивы на воздухе и растворяются в воде
с образованием зеленого раствора. Получены предварительные
рентгенографические данные для монокристаллов [105]. Повидимому, они принадлежат
к тетрагональной или гексагональной сингонии. Межплоскостные расстояния
вдоль оси игольчатых кристаллов равны 3,32 А, как это выведено из
расстояний между слоевыми линиями. Расстояния между слоевыми линиями
(не отчетливо выраженными) нормально к оси игл составляют около 40 А. Были
проведены и некоторые другие предварительные рентгенографические
исследования оксихлорида, однако выводов относительно структуры кристаллов
пока еще не сделано [106]. Для диэлектрической постоянной твердого
оксихлорида сообщается значение 2,4 [107].
Теплота образования оксихлорида урана (IV) [108]. По
определению теплота растворения оксихлорида урана (IV) Д// =
= — 16,7 ± 0,2ккал-моль'1. Отсюда может быть рассчитана теплота его
образования:
UTB + С12 газ + 4 °2 газ ^ UOCl2 тв; (20)
А#296= —261,7 ккал-моль1.
Равновесное давление паров тетрахлорида урана над
оксихлоридом [109]. Как уже упоминалось, реакция
U02 тв + UC14 газ Z- 2UOC!2 TB (21)
является обратимой. Измерено равновесное давление паров тетрахлорида
над оксихлоридом в интервале 460 — 540°; полученные результаты приведены
в табл. 208. Вычисленное из этих данных значение АЯ для реакции (21)
равно 55,1 ккал.
Таблица 208
РАВНОВЕСНОЕ ДАВЛЕНИЕ ПАРОВ ТЕТРАХЛОРИДА УРАНА НАД ЧИСТЫМ
ОКСИХЛОРИДОМ
Температура,
°С
460
470
480
490
500
^г-юз
1,364
1,346
1,328
1,311
1,294
Давление
UC14, мм
рт. ст.
7,МО-4
1,18-Ю-3
1,95ЛО-3
3,13-Ю-3
5,0. Ю-3
Температура,
' °С
510
520
530
540
_1.10з
1,277
1,261
1,245
1,230
Давление
UCU, мм
рт. ст.
8,Ы0-3
1,28.10-2
1,97-10-2
з,оз.ю-2
Равновесное давление паров тетрахлорида над оксихлоридом при 500°
приблизительно в 100 раз меньше давления пара тетрахлорида при той же
v температуре.
Равновесие реакции восстановления тетрахлорида
водородом [НО]. Хотя продукты восстановления оксихлорида
водородом точно не идентифицированы, вероятнее всего, что при этом образуется
Глава 16. Оксигалогениды урана
473
оксимонохлорид UOCI. Следовательно, все расчеты должны относиться
к реакции
UOCI2 тв + ±На газ Z- UOCI тв + НС1газ. (22)
В табл. 209 даны опытные значения констант равновесия этой реакции.
Таблица 209
КОНСТАНТЫ РАВНОВЕСИЯ ДЛЯ РЕАКЦИИ ВОССТАНОВЛЕНИЯ
ОКСИДИХЛОРИДА ВОДОРОДОМ3)
Температура,
°С
зсо
350
385
400
450
Температура,
0 К
573
623
658
673
723
Л. юз
1,745
1,605
1,520
1,486
1,383
А'
<0,004
0,0061
0,0095
0,011
0,020
Я—PHCl/pi/2 (р—давление, am).
Из этих данных вычисляется: Д#673= 10,6 ккал-моль"1 (±10—15%);
Afg73 = 6,03 ккал-жоль'1 и AS673 = 6,79 энтропийной единицы.
Термодинамические константы. Для термодинамических
констант оксихлорида предложены следующие значения [111]:
ДЯ298= —261,7 ккал-моль"1;
AF298= — 246,3 ± 1,5 ккал-моль'1;
S298=^38,l энтропийной единицы.
Подробности расчетов и допущенных предположений см. в
оригинале [111].
Химические свойства. Химические свойства оксихлорида
урана (IV) мало изучены. При 170° он реагирует с четыреххлористым
углеродом с образованием тетрахлорида. С жидким фтористым водородом при
комнатной температуре образуется тетрафторид урана, а не оксифторид.
УРАНИЛБРОМИД U02Br2
Уранилбромид заметно менее устойчив, чем уранилфторид или уранил-
хлорид. По Герману [112], уранилбромид может быть получен
пропусканием паров брома при повышенной температуре над смесью двуокиси урана
с древесным углем; при этом образуется смесь тетрабромида урана и ура-
нилбромида. Последний может быть отделен растворением продукта реакции
в смеси спирта с эфиром [6]. Из этого раствора можно выделить
гигроскопичные флюоресцирующие красные игольчатые кристаллы эфирата
уранилбромида U02Br2-2(C2H5)20. В вакууме большая часть эфира теряется.
При обработке закиси-окиси урана бромом или бромистым водородом
реакции не наблюдается [ИЗ]. Ряд исследователей получил безводный
уранилбромид дегидратацией гидрата (см. стр. 474).
Получение безводного уранилбромида [114]. Безводный
уранилбромид получен по реакции между тетрабромидом урана и кислородом:
UBr4 + 02->U02Br2 + Br2. (23)
474 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Для ее проведения очень важно регулирование температуры. При
температуре ниже 140° реакция идет чрезвычайно медленно; при 200° образуются
значительные количества закиси-окиси урана. Наиболее подходящими
являются температуры в интервале 150—160°; в этих условиях легко может
быть достигнут 96%-ный выход уранилбромида. Рентгенограмма полученного
продукта показывает отсутствие двуокиси, закиси-окиси, трехокиси и тетра-
бромида урана.
Трибромид урана интенсивно горит в атмосфере кислорода при комнатной
температуре. Поэтому эта реакция не является удобным препаративным
методом для получения уранилбромида. Замечено [115], что двуокись урана не
реагирует с бромом даже при 720°. Сухой бромистый водород реагирует при 100°
с сухим диуранатом аммония (NH4)2U207 с образованием в числе прочих
продуктов реакции растворимого в воде вещества, которое, повидимому, является
двойным бромидом аммония и уранила.
Получение гидратированного уранилбромида.
Приготовлены [113] растворы уранилбромида нагреванием суспензии двуокиси
урана в воде с бромом по старому методу Бертемо [116]. После удаления
избытка брома выпариванием оставался раствор уранилбромида, который
можно было затем упарить до сиропообразного состояния. Выход кристаллов
из сиропа мал, а их растворимость в воде и спирте настолько велика, что
отмыть их от маточного раствора почти невозможно.
Растворением гидратированной двуокиси урана в водном растворе броми-
стоводородной кислоты был получен кристаллогидрат уранилбромида [117].
Желтый раствор концентрировали до сиропообразной консистенции и затем
обезвоживали в эксикаторе. Полученные таким путем гигроскопичные
неустойчивые кристаллы имели состав U02Br2-7H20. Повторное выпаривание уранил-
ацетата сначала с бромистоводородной кислотой, а затем с водой также приводит
к образованию водного раствора уранилбромида [6]. Из этого раствора в
эксикаторе образуются большие желто-зеленые кристаллы. Эти кристаллы
растворимы в амиловом спирте; кристаллизационная вода собирается отдельным
слоем, и спиртовый раствор может быть слит. Еще более полно раствор может
быть обезвожен с помощью азеотропной перегонки. Для обезвоживания
предложено также применение эфира, однако показано [113], что последний
реагирует с уранилбромидом. Сомнительно, чтобы из спиртовых растворов
можно было получать уранилбромид, свободный от спирта.
Свойства уранилбромида. Уранилбромид представляет
собой яркокрасное, весьма гигроскопичное твердое вещество, которое желтеет
в присутствии паров воды. Оно очень легко растворяется в воде с образованием
желтого раствора. Растворы уранилбромида в этиловом спирте довольно
устойчивы; в присутствии влаги и света наблюдается частичное восстановление соли
уранила до урана (IV) [118]. Уранилбромид, как уже отмечалось выше,
растворим в эфире и амиловом спирте.
Гидрат уранилбромида U02Br2-7H20 разлагается во влажном воздухе
с выделением бромистого водорода и образованием гидратированной двуокиси
урана. В воде кристаллогидрат очень хорошо растворим и проявляет свойства,
характерные для иона UO^. При кипячении водные растворы уранилбромида
подвергаются сильному гидролизу [119].
Уранилбромид термически неустойчив, даже при комнатной температуре
наблюдается медленное разложение его с выделением брома; в атмосфере гелия
при 250° бром выделяется еще быстрее. Однако даже при 350° для полного
разложения необходимо 48 час. Ввиду того что даже при 720° отсутствует обратная
реакция, реакция разложения уранилбромида необратима и ее скорость
является функцией только температуры, а не парциального давления брома [120].
Прокаливание гептагидрата уранилбромида без доступа воздуха приводит
в конце концов к образованию двуокиси урана с одновременным выделением
Глава 16. Оксигалогениды урана
475
брома и бромистого водорода. Получаемая двуокись урана красного цвета
и является, повидимому, еще одной из разновидностей этого окисла (см. гл. 11).
Подобно уранилфториду и уранилхлориду, уранилбромид легко
образует двойные соли. Действием аммиака на эфирные или спиртовые растворы
уранилбромида могут быть получены аммиакаты состава U02Biy2NH3,
U02Br2-3NH3H U02Br2-4NH3 [6]. При обработке диаммиаката жидким"
аммиаком образуется тетрааммиакат, окрашенный в глубокий оранжево-красный цвет
..и быстро разлагающийся при комнатной температуре.
Были также приготовлены соединения типа MgUOoBrj • 2Н20 [117]. При
растворении диураната аммония или калия в бромистоводородной кислоте
и последующем выпаривании раствора на водяной бане до кристаллизации
получается соответственно (NH4)2U02Br4 • 2Н20 или K2U02Br4 • 2H20; эти
соединения представляют большие желтые ромбические кристаллы, очень хорошо
растворимые в воде; они образуются только в присутствии большого избытка
бромистоводородной кислоты. Чистая соль, растворенная в воде, не может быть
вновь выделена. Прокаливание калийного комплекса приводит к образованию
смеси двуокиси урана и солей калия. Двойные соли уранилбромида менее
устойчивы, чем соответствующие хлористые соединения. Путем приливания
Таблица 210
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ УРАНИЛПРОМНДА С ОРГАНИЧЕСКИМИ ОСНОВАНИЯМИ
Органическое
основание
л-Нитрозодиме-
тиланилин
л-Нитрозоди-
этиланилин
Метилацета-
нилид
Ацето-я-фенети-
дин
Антипирин
Бромантипирин
Комплексное соединение
UO;>Br2.2(CH3)2NCGH4NO
UOoBr,. 2(C2H5)2NCGH4NO
UOoBr2. 2C6H5N(CH;t)(C2H30)
UOoBr3-4C10HJ3OaN
UO,Biv2CnHJ2N20
иОгВга-гСцН^ВгЫзО
1
Внешний вид
Устойчиво на
воздухе; порошок
кирпично-красно-
го цвета
Темный кнрпично-
красный
порошок
Устойчиво на
воздухе; блестящие
желтые
кристаллы
Устойчиво на
воздухе; оранжево-
желтого цвета
Устойчиво на
воздухе
Желтые иглы
Растворимость
Мало растворимо в воде;
нерастворимо в спирте,
эфире, ацетоне и
хлороформе
То же
Растворимо в воде и
спирте; нерастворимо
в эфире
Нерастворимо в воде;
заметно растворимо в
| спирте, ацетоне и
хлороформе; нерастворимо
в эфире и амиловом
1 спирте; полностью
растворимо в амиловом
спирте при нагревании
Растворимо в воде;
слабо растворимо в
спирте; нерастворимо в
эфире и хлороформе
Растворимо в теплой воде
или спирте; слабо
растворимо в кипящем
ацетоне; нерастворимо
в эфире и хлороформе;
растворимо в ссляной
кислоте
476 Часть 1V. Галоген иды, оксигалогсниды, борг ид р иды и карбонил урана
пиридина в кипящий раствор трехокиси урана в избытке спиртового раствора
бромистого водорода получено соответствующее пиридиновое производное
(C5H6N)2UOoBr4. По охлаждении выпадают желтые кристаллы комплексной
соли [121]. -
Уранилбромид с различными органическими основаниями образует ряд
продуктов присоединения. Описан эфират состава U02Br2-2(C2H5)20 [6]. Он
представляетсобой желто-зеленое флюоресцирующее кристаллическое вещество,
очень гигроскопичное и быстро разлагающееся на воздухе с выделением брома.
Изучен ряд продуктов присоединения органических азотистых оснований к
уранилбромиду [87, 89, 90, 122]. Получаются они таким же путем, как и
соответствующие производные уранилхлорида (ср. стр. 468). По
растворимости уранилбромидные комплексы отличаются от уранилхлоридных. Данные
по этой растворимости уранилбромида приведены в табл. 210.
Получено также ксантилиевое производное UOoBr2-2C13H9OBr желтые
кристаллы [101].
ОКСИБРОМИД УРАНА (IV) UOBr2
Работы по синтезу этого соединения были предприняты потому, что при
возгонке тетрабромида урана часто оставалось какое-то желтое вещество
[115]. Его анализ показал, что это оксибромид урана (IV). Был разработан метод
[123] его получения, основанный на следующих реакциях:
3U02 + 2CS2 ^ U302S4 + 2C02; (24)
UAS4 + 6Br2 IT 2UOBr2 + UBr4 + 2S2Br2. (25)
Пары брома в токе азота пропускают над U302S4 при 600° до
прекращения отгонкд тетрабромида урана и монобромида серы. Нелетучий
оксибромид урана (IV) представляет порошок цвета от зеленовато-желтого до
желтого. Анализ дает состав, близкий к UOBr2, а рентгенографические
исследования показывают, что он является химическим соединением, а не
механической смесью двуокиси и тетрабромида урана.
Оксибромид урана (IV) не особенно гигроскопичен, но в воде легко
растворяется с образованием зеленого раствора, устойчивого в течение
нескольких часов, после чего из него начинает выпадать черный осадок,
вероятно, гидратироваиной двуокиси урана. Этот осадок сходен с тем,
который получается при действии щелочи па водный раствор тетрабромида
урана. Предполагают [124], что в растворе оксибромида урана (IV)
содержатся устойчивые ионы U02f; такая интерпретация подтверждается тем,
что при электрометрическом титровании растворов тетрабромида урана
требуется для полного осаждения четыре эквивалента щелочи, а при
титровании оксибромида — только два. Этот вопрос будет подробнее
рассматриваться во второй книге.
В атмосфере инертного газа оксибромид урана устойчив и нелетуч
при 600°, при температуре 800°, однако, наблюдается диспропорциониро-
вание его на двуокись и тетрабромид урана. При прокаливании на воздухе
он переходит в закись-окись урана.
Равновесие реакции восстановления оксибромида
урана (IV) водородом [125]. Равновесие восстановления оксибромида
урана водородом изучено в интервале 300 — 400°. Реакция протекает с
образованием бромистого водорода. Анализ полученных данных сделан на
основании предположения, что другим продуктом реакции является оксимонобро-
мид урана UOBr:
UOBr2TD + ± Н2Газ -> ШВгтв + НВггаз. (26)
Опытные значения константы равновесия приведены в табл. 211.
Глаза 16. Оксигалогениды урана
477
Таблица 211
КОНСТАНТЫ РАВНОВЕСИЯ ДЛЯ РЕАКЦИИ ВОССТАНОВЛЕНИЯ
ОКСИБРОМИДЛ УРАНА (IV) ВОДОРОДОМ3)
Температура,
°С
300
350
375
400
425
440
Температура,
°К
573
623
648
673
698
713
^г-юз
1,745
1,605
1,543
1,486
1,433
1,403
К
< 0,004
0,0061
0,0078
0,0108
0,0139
0,0162
"PHBi7PHo (P—давление, am).
На основании данных табл. 211 вычисляются следующие величины для
реакции (26):
д#ь93=10,1 ккал-моль'1;
AF673 = 6,06 ккал-моль'1;
AS673 = 6,01 энтропийной единицы.
Термодинамические константы. Для оксидибромида были
предложены следующие значения термодинамических величин [111]:
Д#298= —246,9 ± 0,7 ккал-моль'1;
&F298= —231,3 ± 2,0 ккал-моль'1;
Д5298 = 42,9 энтропийной единицы (ориентировочно).
Приведенное значение для теплоты образования основано на
калориметрическом измерении теплоты растворения оксидибромида, давшем
Д#=-16,3±0,2 ккал-моль'1 [108].
УРАНИЛИОДИД U02J2
Сомнительно, чтобы чистый уранилиодид был когда-нибудь приготовлен
в твердом состоянии. Из того немногого, что о нем известно, можно с
вероятностью заключить, что он менее устойчив, чем уранилбромид, уже
обладающий определенной тенденцией к потере галогена. Различные исследователи
безуспешно пытались получить уранилиодид путем реакции между парами
иода, йодистым водородом и смесью двуокиси урана с углеродом [6].
Однако водные растворы уранилиодида можно легко получить действием
иодистоводородной кислоты на у ранил ацетат [61. Концентрирование такого
раствора путем нагревания приводит к значительному разложению
уранилиодида. При обезвоживании в вакууме получаются желто-зеленые
флюоресцирующие игольчатые кристаллы, быстро разлагающиеся на воздухе с выделением
иода.Это вещество не было проанализировано. Ранее сделанные попытки [117]
сконцентрировать растворы U03-H20 в иодистоводородной кислоте также
всегда приводили к выделению больших количеств иода; получавшийся продукт
был сильно загрязнен свободным иодом. Повидимому, растворы уранилиодида
не могут быть обезвожены без значительного разложения последнего.
478 Часть IV. Галогениды, оксигалогениды, боргидриды и карбонил урана
Растворы уранилиодида в воде или в органических растворителях могут
быть легко приготовлены путем реакции обменного разложения. Реагенты
подбирают таким образом, чтобы одним из продуктов был уранилиодид, а
другим—соль, нерастворимая в применяемом растворителе. Так, в одном случае
к эфирному раствору частично дегидратированного гексагидрата уранилни-
трата был добавлен небольшой избыток иодида бария. После отделения
нерастворимого в эфире нитрата бария и концентрирования красного раствора
в вакууме было получено неустойчивое, легко расплывающееся
кристаллическое вещество красного цвета [66]. Раствор уранилиодида был получен также
при действии на раствор уранилнитрата в метаноле йодистым натрием. Кроме
того, неводные растворы уранилиодида были приготовлены путем растворения
в эфире сухого остатка от выпаривания водного раствора с последующей
обработкой эфирного раствора хлоридом кальция и металлическим натрием для
отнятия воды и свободного иода [6]. Выпариванием эфирного раствора в
вакууме или в токе сухого воздуха можно получить кристаллы предполагаемого
состава U02J2-2(C2H5)20, загрязненные иодом. Для получения водных растворов
уранилиодида также можно использовать реакции обменного разложения.
Так, например, можно обработать раствор уранилсульфата эквивалентным
количеством иодида бария или кальция [126].
В литературе указывается, что уранилиодид растворим в метилацетате,
этилацетате, ацетоне и пиридине [70, 71 ], а также в воде, эфире и метиловом,
этиловом или амиловом спиртах.
С аммиаком уранилиодид образует ряд продуктов присоединения [6].
При пропускании аммиака в раствор уранилиодида в эфире или амиловом
спирте получается диаммиакат U02J2-2NH3, более продолжительное
пропускание аммиака приводит к образованию золотисто-желтого аморфного
вещества состава U02J2-3NH3. Обработка диаммиаката жидким аммиаком при 0°
приводит к образованию неустойчивого соединения состава U02J2-4NH3, быстро
разлагающегося при температуре выше 5°.
Водные или этанольные растворы уранилиодида обладают способностью
растворять обычно нерастворимые иодиды тяжелых металлов, как висмута (III)
или ртути (II). Сообщается, что из подобных растворов были выделены
соединения, имеющие, например, состав U02BiJ5[126].
ЛИТЕРАТУРА
1. Berzelius J. J,, Pogg. Ann., 1, 34 (1824).
2. В ol t on H. C..Z. Chem., [2] 2, 353 (1866).
3. S m i t h e 1 1 s A., J. Chem. Soc., 43, 130 (1883).^
4. D i t t e A., Ann. Chim. et phys., [6] 1, 341 (1884).
5. H e b e r t J., M a d d о с k A. G., Report MC-43, Mar. 13, 1944 (Montreal 1).
6. U n r u h A., Dissertation, University of Rostock, p. 37 (1909).
7. P e i s e r H. S., Alcock T. C, Report BR-589, Mar. 19, 1945 (British 1).
8. Brown H. S., Hill O. F., Jaffey A. H., Report CN-343, Nov. 15, 1942
(MP Chicago 1).
9. H о 1 b г о с k С. E., Report N-25, Apr. 17, 1943 (Du Pont 1).
10. Report BR-442, date received July 6, 1944 (British 2).
11. H i s k e у С. F., Report N-673, Feb. 17, 1944 (SAM, Columbia 1).
12. Gates J. W., Andrews L. J., P i t t R. В., Report CD-4001, Jan. 30,
1945 (CEW-TEC 1).
13. К r a u s C. A., Report CC-1717, July 29, 1944 (Brown 1).
14. Fried S., Davidson N. R., Report N-1722, Nov. 14, 1944 (MP Chicago 2).
15. Report [B] LRG-21, June 1943 (British 3).
16. Z а с h a r i a s e n W. H., Reports CP-1249, Jan. 24, 1944; CK-1367, Feb. 15, 1944,
and CC-2768, Mar. 12, 1945 (MP Chicago 3).
17. Z а с h a r i a s e n W. H., Acta Crystallographica, 1, 277 (1948).
18. Katz M. S., Report CC-1744, June 17, 1944 (SAM Columbia 2).
19. Wacker P. F., Cheney R. K., Report MDDC-1096, July 9, 1947 (Natl. Bur.
Standards 1).
20. Wacker P. F., С h e n e у R. K., J. Research Natl. Bur. Standards, 39, 317 (947).
Глава 16. Оксигалогениды урана
479
21. Kunin R., Report A-3255, May 8, 1945 (SAM Carbide and Carbon 1).
22. D e a n G. R., Bradt R., К a t z M. S., Report CC-1382, Mar. 6, 1944 (SAM
Columbia 3).
23. С 1 i n e W. D., Report CC-1983, Dec. 9, 1944 (MP Ames 1).
24. G a t e s J. W., С 1 e w e t t G. H., A n d r e w s L. J., Pitt R. В., Report
CD-457, Aug. 4, 1944 (GEW-TEC 2).
25. Report [B1LRG-22, July 1943 (British 4).
26. V a s 1 о w K.Newton A. S., Report CK-1498, June 17, 1944, p. 10 (MP Ames 2).
27. D о b r a t z I. W., Downing R. С, М а у n а г d C. W., S m i t h S. В.,
S p i e g 1 e r L., Report N-33, May 13, 1943 (Du Pont 2).
28. Aloy J., Rodier E., Bull. Soc. Chim. France, [4] 31, 247 (1922).
29. Freed S., Report [A] M-144, Sept. 2, 1943 (SAM Columbia 4).
30. В о 1 t о n H. C, Z. Chem., [2] 2 356 (1866).
31. Baker H., J. Chem. Soc, 35, 760 (1879).
32. Лордкипанидзе С, ЖРФХО, 32, 286 (1900).
33. M i о 1 a t i A., A 1 v i s i U., Atti accad. nazl. Lincei, [5] 6, II, 380 (1897).
34. В о 1 1 a n d A., Monatsh., 31, 387, 403 (1910).
35. Burger H., Dissertation, University of Bonn, p. 49 (1904).
36. Z а с h a r i a s e n W. H., Report CK-2737, Mar. 5, 1945, p. 12 (MP Chicago 4).
37. О 1 s s о n F., Z. anorg. u. allg. Chemie, 187, 112 (1930).
38. G i о 1 i t t i F., A g a m e n n о n e G., Atti accad. nazl. Lincei, [5] 14, I, 114, 165
(1905).
39. G i о 1 i t t i F., Gazz. chim. ital., 34, II, 166 (1904).
40. В a e n z i g e r N. C.Tevebaugh A. D., Report CC-1504, Aug. 10, 1944 (MP
Ames 3).
41. L i v i n g s t о n R., Personal communication (MP Chicago 5).
42. Sped ding F. H., Wilhelm H. A., Report CC-1517, Mar. 10, 1944 (MP Ames 13)
43. P e 1 i g о t E., Ann., 43, 278 (1842).
44. P e 1 i g о t E., Ann. chim. et phys., [3] 5, 36 (1842).
45. R eg el s b er ger F. F., Ann., 227, 119 (1885).
46. Kraus С A., Report A-256, Aug. 14, 1942 (Brown 2).
47. J о h n s о n O., Report CC-1500, June 17, 1944 (MP Ames 4).
48. К i 1 n e r S., P о 1 i s s a r M. J., Report RL-4.6.42, Nov. 7, 1942 (UCRL 1).
49. J о h n s о n O., Report CC-1781, Oct. 18, 1944 (MP Ames 5).
50. В a b с о с k S. H., Report RL-4.6.57, Jan. 9, 1943 (UCRL 2).
51. R о s e n f e 1 d S., Report RL-4.6.84, Febr. 20, 1943; Reiber H. G., Report
RL-4.6.70, Mar. 13, 1943 (UCRL 3).
52. Kraus С A., Report A-360, Oct. 26, 1942, p. 9 (Brown 3).
53. Mc В ее Е. Т., Report [A] M-2102, Mar. 21, 1945 (Purdue I).
54. С h г ё t i e n А., О е с h s e 1 G., Compt. rend., 206, 254—256 (1938).
55. А г f v e d s о n J. A., Pogg. Ann., 1, 269 (1824).
56. M у 1 i u s F., Dietz R., Ber., 34, 2774 (1901).
57. d e Conick W. O., Compt. rend., 148, 1769 (1909).
58. S p e v а с k J., G г о s s e A. V., Report A-38, Aug. 21, 1941 (SAM Columbia 5).
59. Aloy J., Bull. Soc. chim. Paris, [3], 25, 154 (1901).
60. L e с a n u L. R., J. pharm. chim., 11, 285 (1825).
61. В a e n z i g e r N. C, R u n d 1 e R. E., Reports CC-1504, Aug. 10, 1944; CC-1778,
Aug. 18, 1944 (MP Ames 6).
62. M с В ее Е. Т., Report [A] M-2103, Apr. 21, 1945 (Purdue 2).
63. Garner С S., Kent J. W., Report CN-343, Nov. 15, 1942 (MP Berkeley 1).
64. T r a u t z M., Z. phys. Chem., 53, 48 (1905).
65. H a m p e H., Chem. Ztg., 12, 106 (1888).
66. Aloy J., Ann. chim. et phys., [7] 24, 417 (1901).
67. de Conick W. O., Ann. chim. et phys., [8] 3, 500 (1904).
68. Aloy J., Compt. rend., 122, 1541 (1896).
69. de Conick W. O., Bull, classe sci. Acad. roy. Belg., 836 (1909).
70. N a u m a n n A., Ber., 37, 3601, 4328, 4609 (1904).
71. Naumann A., Ber., 42, 3790 (1909).
72. Kraus С A., Report N-1789, July 1, 1943 (Brown 4).
73. Seubert K., Schmidt A., Ann., 267, 239 (1892).
74. d e С о n i с k W. O., Bull, classe sci. Acad. roy. Belg., p. 173 (1909).
75. de Conick W. O., Bull, classe sci. Acad. roy. Belg., 709 (1903).
76. Peters W., Ber., 42, 4833 (1909).
77. Peters W., Z. anorg. Chem., 78, 159 (1912).
78. Rosenheim A., J а с о b s о h n F., Z. anorg. Chem., 50, 306 (1906).
79. S p а с u P., Z. anorg. u. allg. Chem., 230, 181 (1936).
80. S i m о n i s H., E 1 i a s A., Ber., 48, 1513 (1915).
81. S i m о n i s H., E 1 i a s A., Ber., 49, 777 (1916).
82. L e e d s A. R., J. Amer. Chem. Soc, 3, 145 (1881).
480 Часть IV. Галогениды, оксигалогенидыу боргидриды и карбонил урана
83. R a s с a n u R., Ann. sci. univ. Jassy, 16, 461 (1930—1931).
84. Rase a nu R., Ann. sci. univ. Jassy, 17, 133 (1931—1932).
85. Rase a nu R., Ann. sci. univ. Jassy, 17, 143 (1931—1932).
86. A s a h i n а Т., D 6 n о Т., Z. physiolog. Chem., 186, 134 (1930).
87. R a sc a n u R., Ann. sci. univ. Jassy, 16, 480, 481, 484, 486, 489 (1930—1931).
88. R й s с a n u R., Ann. sci. univ. Jassy, 17, 75 (1931—1932).
89. Rascanu R., Ann. sci. univ. Jassy, 16, 54 (1930—1931).
90. Rascanu R., Ann. sci. univ. Jassy, 18, 94 (1932—1933).
91. Rase a nu R., Ann. sci. univ. Jassy, 16, 465, 466, 467, 470, 473 (1930—1931).
92. Gmelins Handbuch der anorg. Chemie, System No 55, 135—136, Verlag Chemie, Berlin.
93. R i m b а с h E., Ber., 37, 461 (1904).
94. W e 1 1 s H. L., В о 1 t w о о d В. В., Z. anorg. Chem., 10, 183 (1895).
95. N i с h о 1 s E. L, HowesH. L, Carnegie Inst. Wash. Pub., 298, 218 (1919).
96. D i e к e G. H., D u n с a n A. B. F., Spectroscopic Properties of Uranium Compounds.
National Nuclear Energy Series, Division III, vol. 2; McGraw-Hill Book Co. Inc.
N. Y., 1949.
97. G г о s s m a n n H., Schuck В., Z. anorg. Chem., 50, 26 (1906).
98. Kalischer В., Dissertation, Univ. of Berlin, 49 (1902).
99. W i 1 1 i a m s С G., Chem. News., 44, 308 (1881).
100. Williams G., J. prakt. Chem. 69, 358 (1856).
101. Fosse R., L e s a g e L., Compt. rend., 142, 1544 (1906).
102. В e n г a t h A., Z. wiss. Photogr., 16, 258 (1917).
103. Davidson P. H.,MullerM. E„ Report RL-4.6.259, Febr. 26, 1944 (UCRL 4).
104. Kraus С A., Report CC-342, Nov. 15, 1942 (Brown 5).
105. В a e n z i g e r N. C, Report CC-1778, Aug. 18, 1944 (MP Ames 7).
106. Burford W. В., Ill, Report [A] M-2121, Feb. 1945 (Johns Hopkins 1).
107. S t e г e t t C, H a 1 p e r n I., V a n V e z e r J., Y о u n g H. A., Report CD-GS-28,
Mar. 16, 1945 (CEW—TEC 3).
108. В а г к e 1 e w С H., Report RL-4.6.937, Oct. 11, 1945 (UCRL 5).
109. Davidson P. H., Streeter I., Report RL-4.6.920, June 5, 1945 (UCRL 6).
110. Gregory N. W., Report RL-4.6.938, Oct. 13, 1945 (UCRL 7).
111. M а с W о о d G. E., Report RL-4.7.602, Dec. 18, 1945 (UCRL 8).
112. Hermann H., Dissertation, University of Gottingen, 31 (1861).
113. R i с h a r d s T. W., M e г i g о 1 d B. S., Chem. News, 85, 187 (1902); Z. anorg.
Chem., 31, 244 (1902).
114. Powell J.,Nottorf R. W., Report CC-1500, June 1944; Powell J.,
Report CC-1504, Aug. 10, 1944 (MP Ames 8).
115. Powell J., N ot tor! R. W., Report CC-1496, May 11, 1944 (MP Ames 9).
116. В е г t h e m о t J. В., Ann. chim. et phys., [2] 44, 387 (1830).
117. S e n d t n e г R., Ann., 195, 325 (1879).
118. Font ana B. J., Report CK-810, July 20, 1943 (MP Berkeley 2).
119. de Conic к W. О., Bull, classe sci. Acad. roy. Belg., 1025 (1902).
120. N о t t or f R. W., Report CC-1778, Aug. 18, 1944 (MP Ames 10).
121. L о e b e 1 H., Dissertation, University of Berlin, p. 42 (1907).
122. Rascanu R., Ann. sci. univ. Jassy, 16, 54, 496 (1930—1931).
123. Powell J., Report CC-1778, Aug. 18, 1944 (MP Ames 11).
124. Powell J., N e w t о n A. S., Report CC-1778, Aug. 18, 1944 (MP Ames 12).
125. Gregory N. W., Report RL-4.6.940, Dec. 13, 1945 (UCRL 9).
126. Truttwin H., Герм. пат. D. R. P. 420391 (from A. Brauer and J, D'Ans.,
Fortschritte in der anorganisch-chemischen Industrie III, 1327 (1924—1927).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Алланит, урансодержащий минерал 81
Ампангабеит, состав, свойства и
месторождения 72, 86, 91
Атмосфера, распределение элементов 63
Бассетит, состав, свойства и
месторождения 82, 86, 87
Беккерелит, состав, свойства и
месторождения 75, 86
Бетафит, извлечение урана 107
— состав, свойства и месторождения 68,
72, 86, 87, 91
Биллиетит, урансодержащий минерал 74
Бломстрандин, состав, свойства и
месторождения 71, 86
Бломстрандит, состав, свойства и
месторождения 72, 86, 91
Браннерит, состав, свойства и
месторождения 70, 86
Брёггерит, состав, свойства и
месторождения 66, 68, 86
Вальпургит, состав, свойства и
месторождения 84, 86, 87
Ванденбрандит, состав, свойства и
месторождения 77, 86
Вандендрисшеит, урансодержащий
минерал 74
Виикит, состав, свойства и месторождения
71, 86
Гадолинит, урансодержащий минерал 81
Гатчеттолит, состав, свойства и
месторождения 69, 86, 87
Гильпинит, состав, свойства и
месторождения 80, 86, 87
Гуммит, состав, свойства и месторождения
74, 75, 86, 91
Дакеит, состав, свойства и месторождения
79, 86
Девиндтит, состав, свойства и
месторождения 83, 86
Делоренцит, состав, свойства и
месторождения 72, 86
Джалмаит, состав, свойства и
месторождения 72, 86
Дидерихит, урансодержащий минерал 74
Дюмонтит, состав, свойства и
месторождения 83, 86
Иоганнит, состав, свойства и
месторождения 80, 86, 87
Иттриалит, урансодержащий минерал 81
Иттрокразит, состав, свойства и
месторождения 71, 86
31 Химия ураиа
Иттротанталит, состав, свойства и
месторождения 70, 86
Ишикаваит, состав, свойства и
месторождения 70, 86
Казолит, состав, свойства и
месторождения 78, 86—88
Карнотит, извлечение урана 103 ел., 107
— обработка 103
— состав, свойства, месторождения и
добыча 63, 85, 86, 89
Кларкеит, состав, свойства и
месторождения 76, 86
Клевеит, состав, свойства и
месторождения 66, 68, 86
— структура и состав 206
Кольм, состав, свойства, месторождения
и добыча 63, 81, 86, 91
Корвузит, урансодержащий минерал 81
Ксенотим, урансодержащий минерал 81
Купросклодовскит, флюоресценция 87
Кюрит, состав, свойства и месторождения
76, 86, 87
Ламбертит, состав, свойства и
месторождения 77, 86
Либигит, состав, свойства и
месторождения 79, 86
Линдокит, состав, свойства и
месторождения 71, 86
Макиитошит, урансодержащий минерал 78,
86
Мезуит, урансодержащий минерал 74
Менделеевит, состав, свойства и
месторождения 72, 86
Метаторбернит, состав, свойства и
месторождения 81, 83, 86
Метландит, урансодержащий минерал 78,
86
Микролит, состав, свойства и
месторождения 69, 86
Монацит, урансодержащий минерал 81
Наегит, урансодержащий минерал 81
Нивенит, состав, свойства и
месторождения 66, 68, 86
Николаит, урансодержащий минерал 78, 86
Отунит, переработка 105
— реакция с фосгеном 378
— состав, свойства и месторождения 81,
82, 86, 90, 91
Параскупит, урансодержащий минерал 74
Парсонсит, состав, свойства и месторожде»
ния 83, 86
482
Предметный указатель
Пегматит, месторождения урановых
минералов в нем 63, 69, 86 ел.
Пильбарит, состав, свойства и
месторождения 78, 86, 91
Пирохлор, состав, свойства и
месторождения 69, 86
Приорит, состав, свойства и
месторождения 71, 86
Рандит, состав, свойства и месторождения
85,86
Раувит, состав, свойства и месторождения
85, 86, 89
Резерфордин, состав, свойства и
месторождения 79, 86
Ренардит, состав, свойства и
месторождения 83, 86
Ретзерфордит, состав, свойства и
месторождения 70, 86
Рихетит, урансодержащий минерал 74
Роуландит, урансодержащий минерал 81
Салеит, состав, свойства и месторождения
82, 86
Самарскит, состав,"свойства и
месторождения 71, 86, 91
Самирезит, состав, свойства и
месторождения 72, 86
Склодовскит, состав, свойства и
месторождения 74, 78, 86, 88
Скупит, состав, свойства и месторождения
74, 75, 86
Соддиит, состав, свойства и
месторождения 77, 86
Стасит, состав, свойства и месторождения
84, 86
Студтит, урансодержащий минерал 74
Торбернит, состав, свойства и
месторождения 81, 83, 86-88, 91
Торианит, состав, свойства и
месторождения 66, 86
Трёгерит, состав, свойства и
месторождения 81, 84, 86, 87
Тюямунит, состав, свойства и
месторождения 85—87, 89
Уванит, состав, свойства и месторождения
85, 86, 89
Ураконит, состав, свойства и
месторождения 80, 86
Уран, атомный вес 11 ел.
— в воде 61 ел.
живой материи 62
— — изверженных породах 59 ел.
космическом пространстве 62
— — осадочных породах 61
— извлечение из руд 99 ел.
— изотопный состав 7 ел.
— отношение U238: U235 8 ел.
— первоначальное содержание U235 в
земле 9, 10
— распространенность 59 ел.
— руды и соли, статистика 91
— системы с бором 178
__ — ___ водородом 155 ел.
— — — кислородом 202 ел.
— — — кремнием 187 ел.
Уран, системы с мышьяком 199
серой 270
углеродом 178 ел.
Уран-атом, Зеемана эффект 39 ел.
— комбинации рентгеновских термов 18
— константы экранирования 25
— оптические термы 41 ел.
— оптический спектр 26 ел.
U1 42 ел.
UH 47 ел.
— наиболее интенсивные линии
28 ел.
устойчивые линии 38
— размеры 117
— рентгеновские термы 22 ел.
— рентгеновский спектр 14 ел.
— сверхтонкая структура оптического
спектра 39
— смещение линий оптического спектра
27 ел.
— тонкая структура границ поглощения
15 ел.
— упаковочный коэффициент (см. также
эффект упаковки) 12
— флюоресценция в рентгеновском
спектре 21
— фотоэлектрический эффект при
облучении рентгеновскими лучами 21 ел.
— характеристический рентгеновский
спектр 16 ел.
/(-серия 18
L-серия 18 ел.
М-серия 20 ел.
Л/-серия 21
— Штарка эффект 39
— электронные конфигурации 49 ел.
— эффект упаковки (см. также
упаковочный коэффициент) 12
Уран металлический, амальгамирование
149 ел.
— — давление паров 129 ел.
диффузия водорода 155
— — интерметаллические соединения 150
ел., 172
коэффициент Пуассона 121
— — кристаллическая структура 116 ел.
— — магнитная восприимчивость 137
модуль сдвига 120
— — модуль упругости 120, 121
оптическая излучательная
способность 137
■ плотность 118
— — поглощение рентгеновских лучей 16
получение восстановлением галоге-
нидов 109 ел.
— окислов 108 ел.
— — — разложением галогенидов 111 ел.
— — — электролитическим
восстановлением ПО ел.
— — предел текучести 121 ел.
— — проникание водорода 162
— — прочность 122 ел.
— — растворимость в металлах 148 ел.
азота 192
— — — водорода 155 ел.
— кислорода 202
— — — металлов 148 ел.
— — — углерода 179
— — реакция с азотом 142, 196 ел.
Предметный указатель
483
Уран металлический, реакция с аммиаком
144, 196 ел.
— — инертными газами 143
— __ — — борной кислотой 146
— — бромистым водородом 143
бромом 142, 411
— — водой 143
— — —■ — водородом 141, 157 ел.
— — двуокисью углерода 144
„ двуокисью урана 146, 244
— — — — дейтерием 141, 173 ел.
— — йодистым водородом 143
— — — — иодом 143
— _ — _ кислородом 142, 243 ел.
— — — — кислотами 144 ел.
— — кремнеземом 146
— кремнием 142, 187
— — — — метаном 144, 184
— — — — метиловым спиртом 146
— — — — мышьяком 142, 199
— — — — окисью азота 144, 185
окисью углерода 144, 185
— — — — персульфатом 146
■ селеном 142, 275
серой 142, 272 ел.
— — — — солями 145 ел.
— — тетрафторидом урана 146, 287
— — углеродом 141, 184
фосфором 142, 199
— фтористым водородом 143,
296
фтором 142, 322
— — хлорированными
углеводородами 146
хлористым водородом 362
хлором 142, 381
щелочами 145
сплавы 152
— — твердость 119 ел.
твердые растворы 148 ел.
— — температура плавления 129
теплоемкость 124 ел.
• теплопроводность 136 ел.
— — теплосодержание 125 ел.
— — теплота образования 112
парообразования 129 ел.
— плавления 129
— — термическое расширение 118 ел.
— — термоэлектродвижущая сила 137
— — ударная вязкость 123 ел.
фазовые превращения 127 ел.
электропроводность 131 ел.
энтропия 125 ел.
■Урана антимониды, получение и свойства
199 ел.
Урана арсениды/пол учение и свойства 199
Урана боргидрид, алкилирование 442
— —- получение 441 ел.
— — растворимость 443 ел.
реакции 444 ел.
— — физические свойства 442 ел.
— — химические свойства 443 ел.
Урана бориды, получение и структура 178
Урана гексафторид, вязкость 347
— — гидролиз 354
давление пара 328 ел., 332 ел.
коэффициент самодиффузии 346
кристаллическая структура 353 ел.
критические константы 334
Урана гексафторид, магнитные свойства
351 ел.
— — обращение с ним 326 ел.
— — очистка 326 ел.
— — плотность 343 ел., 353
поверхностное натяжение 347
показатель преломления 347 ел.
— — получение без применения
элементарного фтора 325 ел.
фторированием урана и его
соединений 322 ел.
растворы 356 ел.
— — реакция с аппаратостроительными
материалами 358 ел.
— — свободный пробег молекулы 346
скорость звука 339
— — спектральные свойства 348 ел.
структура 352 ел.
температура кипения 333
температура плавления 328
— — теплопроводность 342
— — теплота возгонки 340 ел.
парообразования 340 ел.
плавления 342
— — термические и термодинамические
свойства 334 ел.
тройная точка 333
— — химические свойства 354 ел.
— — электрические свойства 350 ел.
Урана гексахлорид, давление пара 402
— — кристаллическая структура 401 ел.
механизм получения 394 ел.
— — оптические свойства 405
— — очистка 401
плотность 402
получение 400
— — растворимость 405
температура плавления 401
термическое разложение 405 ел.
термодинамические свойства 403 ел.
фторирование 406
Ураиа гептадекафторид U4F17,
кристаллическая структура 319
— — — получение 312 ел.
термодинамические функции 316
ел.
— химические свойства 319
Урана гидрид, амальгама 171 ел.
— — давление разложения 160 ел.
изотермы разложения и образования
157 ел.
— — квази-гидрид 171
кинетика образования 162 ел.
— разложения 165
—• — кристаллическая структура 166
— — механизм образования 164 ел.
окисление 169 ел.
— — плотность 167
получение 157, 165 ел.
— — применение 172 ел.
— — природа 157
реакции с водой 170
— — — — газами 168 ел.
— — — — кислотами 170
— — основаниями 170
— растворителями 170
— — состав 158 ел.
теплота разложения 160 ел.
— — термодинамические константы 162
31*
484
Предметный указатель
Урана гидрид, электропроводность 167
Урана двуокись, гидраты 232 ел.
восстановление до металла 247
— — галогенирование 264 ел.
■ ■ гидрозоль 242
давление пара 216
кристаллическая структура 215
■ кристаллический габитус 215 ел.
магнитные свойства 221 ел.
— — область существования 205 ел.
образование из высших окислов
249 ел.
образование из урана 248
окисление в гидраты перекиси 258
трехокиси 257 ел.
трехокись 256
плотность 208, 216
получение из других соединений
урана 259 ел.
превращение в закись-окись 252 ел.
■ — карбиды 265 ел.
— — перекись 258 ел.
— — — — соли урана (IV) и уранила
268 ел.
сульфиды 265
уранаты и перуранаты 266 ел.
растворение в кислотах 268
твердость 216
температура плавления 216
— — теплоемкость 218 ел.
теплопроводность 219
— — теплосодержание 218 ел.
— — теплота парообразования 216
— — термодинамика образования и
превращения окислов 243 ел.
цвет 222 ел.
электрические свойства 219 ел.
энтропия 218 ел.
Урана дейтерид, давление разложения 173
— — константа решетки 175
плотность 175
■ получение 173
— — скорость и теплота образования
173 ел.
Урана дибромдихлорид, давление пара 438
— — получение и свойства 431, 435
— — термодинамика 439
Урана диброммонохлорид, получение и
свойства 432, 437 ел.
Урана дииоддибромнд, получение и
свойства 432, 437
Урана дииоддихлорид, получение и
свойства 431, 436
Урана дииодмонобромид, получение 432,
438
Урана дииодмонохлорид, получение и
свойства 432
Урана дикарбид, кристаллическая
структура и плотность 182
— — получение 182 ел.
— — температура кипения 181
плавления 181
— — тепловые и термодинамические
свойства 182 ел.
— — химические реакции 185 ел.
Ураиа динитрид, кристаллическая
структура и плотность 194
— — теплота образования 198
химические реакции 198
Урана дисульфид, кристаллическая
структура и плотность 271
— — магнитные свойства 271
получение 273
— — превращение в низшие сульфиды
272 ел.
— — температура плавления 271
—'— химические реакции 274
Урана дихлордифторид,получение и
свойства 431, 433
Урана закиси-окиси гидраты 233
Урана закись-окись, восстановление до
U02 250 ел.
— металла 247
галогенирование 264 ел.
— — излучательная способность 227 ел.
кристаллическая структура 223 ел.
— -— магнитные свойства 226 ел.
— — область существования 208 ел.
— — окисление в гидраты перекиси 258
— — гидраты трехокиси 257 ел.
— — трехокись 256 ел.
плотность 208, 225
■— — получение из двуокиси 252 ел.
— — — — других урановых соединений
260 ел.
— трехокиси 254 ел.
— — превращение в соли урана (IV) и
уранила 268 ел.
сульфиды 265
— — — — уранаты и перуранаты 266 ел.
— — разложение до двуокиси 249 ел.
— — растворение в кислотах 268
твердость 226
теплоемкость 226
термодинамика образования и
превращения в другие окислы 243 ел.
— — термоэлектродвижущая сила 226
цвет 227
— — электрические свойства 226
— -— энтропия 226
Урана иодбромдихлорид, получение 437
Урана иоддибромхлорид, получение 437
Ураиа карбонил, попытки получения 445 ел.
Урана монобромдихлорид, получение и
свойства 432, 437
Урана монобромтрифторид, получение и
свойства 431, 433
Урана монобромтрихлорид давление
пара 438
кристаллическая структура 435
—■ — получение и свойства 431, 434 ел.
— — термодинамика 439
Урана моноиоддибромид, получение 432, 438
Урана моноиоддихлорид, получение 432, 438
Урана моноиодтрибромид, давление иода
439
давление пара 438
получение и свойства 432, 436 ел.
— — термодинамика 439
Урана моноиодтрифторид, получение и
свойства 431, 433
Ураиа моноиодтрихлорид, давление иода
440
— — получение и свойства 431, 435 ел.
Урана монокарбид, кристаллическая
структура и плотность 179
— — получение 184 ел.
— — смесь с ураном 181
Предметный указатель
485»
Урана монокарбид, твердые растворы 180
температура плавления 179
термодинамика образования 182
Урана монометнлборгидрид, получение и
свойства 442 ел.
Урана мононитрид, кристаллическая
структура и плотность 192
— — теплота образования 197
— — химические реакции 198
Ураиа моноокись, давление пара 215
— — кристаллическая структура 205,
215
область существования 204
плотность 208
получение из урана и двуокиси
урана 248
Ураиа моносульфид, кристаллическая
структура и плотность 270
— — магнитные свойства 271
получение 271 ел.
' температура плавления 271
химические реакции 273 ел.
Урана монохлортрифторид,
кристаллическая структура 433
получение и свойства 430, 431
Урана нитриды, давление разложения 195
получение 196 ел.
теплота образования и разложения
197 ел.
фазовые отношения (см. также Урана
динитрид, моионитрид, полуторный
нитрид) 192 ел.
Урана нонафторид U2Fg,
кристаллическая структура 318 ел.
— — — получение 314 ел.
— термодинамические функции 316
ел.
— химические реакции 319
Урана окислы, восстановление до
металла 247 ел.
общий обзор 202 ел.
термодинамика образования и
взаимных превращений 243 ел.
— — фазовые отношения: область U—UO
204 ел.
UO—UOo.g 205 ел.
U02 з—UO2,e7208 ел.
Ш2,67иО3|0 211 ел.
— — — — — выше U03 (см. также
Урана двуокись, закись-окись,
перекись, трехокись) 214
Урана оксибромид, получение и свойства
476 ел.
Урана оксисульфиды, кристаллическая
структура и плотность 274
получение и реакции 274 ел.
Ураиа оксифторид, попытки получения 462
Урана оксихлорид, восстановление
водородом 472 ел.
давление пара 472
получение 471 ел.
— — термодинамика 472 ел.
— — химические свойства 473
Урана пентафторид, кристаллическая
структура 317 ел.
получение 314 ел.
превращение а^±$ 318
температура плавления и летучесть
319
Урана пентафторид, термодинамические
функции 316 ел.
— — химические реакции 319
Урана пентахлорид, гидролиз 397
— — давление пара 396
— — двойные соли 399
— •— диэлектрическая постоянная 396
— — механизм получения 394 ел.
— — молекулярный вес 396
— — окисление и восстановление 398
— — плотность 396
— — получение 393 ел.
— — растворимость 397
— — спектральные свойства 396
—• — температура плавления 396
— — термическое разложение 399
— — термодинамические свойства 396
Урана перекиси гидраты, дегидратация'
239 ел., 258
— кристаллическая структура 241
— — — образование из низших окислов
258
— — — оптические свойства 242
— — — парамагнетизм 241 ел.
— — — плотность 241 ел.
— превращение в трехокись и
гидраты ее 258
— состав 239 ел.
— — — твердость 241
Урана перекись, получение 258, 263
— — существование 214, 232
Ураиа полуторный карбид, природа 181
— — — химические реакции 185 ел.
Урана полуторный нитрид,
кристаллическая структура 193 ел.
— — — плотность 194
— — — теплота образования 198
— — — химические реакции 198
Урана полуторный сульфид,
кристаллическая структура 271
— — — магнитные свойства 271
— — — получение 272 ел.
— превращение в моносульфид 274
— температура плавления 271
— химические реакции 274
Урана пятиокись, кристаллическая
структура 223
плотность 208, 223
— — существование ее 209
Урана селениды 275
Урана силициды, кристаллические
структуры 188 ел.
— — получение 187 ел.
— — фазовая диаграмма 187 ел.
Урана теллуриды 275 ел.
Урана тетрабромид, давление пара 421
— — координационные соединения 425
— — окисление и восстановление 424
— — очистка 421
— — плотность 421
— — получение 418 ел.
— — — из раствора 420 ел.
— — растворимость 422 ел.
— — температуры кипения и плавления*
421
— — термическое разложение 425
— — термодинамика 422
Урана тетраиодид, гидролиз 429
— — давление пара 428 ел.
486
Предме тный указатель
Урана тетраиодид, окисление 429
— ■— получение 427 ел.
— — температура плавления и кипения 428
термическое разложение 429 ел.
— — термодинамические свойства 429
Урана тетраметилборгидрид, получение и
свойства 442 ел.
Урана тетрафторид, гидролиз 303 ел.
— — давление пара 298
— — кристаллическая структура 298 ел.
— — магнитная восприимчивость 302 ел.
— — окисление 305 ел.
— — оптические свойства 298
— — плотность 299
получение 293—297
— — растворимость в воде 303
— — реакции без изменения валентности
306
— — реакции с гексафторидом 314
— — сплавление с солями 306
— — температура плавления 298
— — термодинамические данные 299 ел.
— — фазовый состав системы с NaF
и KF 307 ел.
Ураиа тетрафторида гидраты,
дегидратация 304 ел.
— — — кристаллическая структура и
плотность 299
— — — получение 292 ел.
Урана тетрафторида комплексы,
кристаллическая структура и плотность 308 ел.
Урана тетрахлорид, аммиакаты 391
— — восстановление 390
— — гидрат 380
— — гидролиз 389
— — давление пара 382 ел.
— — двойные хлориды 391 ел.
— — кристаллическая структура 384
— — магнитная восприимчивость 388 ел.
— — механизм получения 374, 394
— — окисление 390
— — оптические свойства 388
— — очистка 381 ел.
— — плотность 384
получение 372, 380
— — — в растворе 380 ел.
— — растворимость 389 ел.
— — свободная энергия образования
370 ел.
— — температуры плавления,
превращения и кипения 382
— — теплота возгонки 382
— — теплота образования 387 ел.
— — теплота плавления и
парообразования 383
— — термодинамические данные 385 ел.
— — электропроводность 389
Урана трехокиси гидраты, дегидратация
237 ел.
— — — кристаллические структуры и
модификации 235 ел.
— — — плотность 236
__ „ — получение из других соединений
урана 262 ел.
— — — — — низших окислов 257 ел.
— — — превращение в диураиаты 267
— — — раскисление 255
— — — твердость 236
— — — теплоты нейтрализации 268
Урана трехокиси гидраты, термодинамические
свойства 235
— — — фазовые отношения в системе
и03—Н20 233 ел.
— — — цвет 236
— — — флюоресценция 236
Урана трехокись, восстановление до
двуокиси 250 ел.
— — галогенирование 264 ел.
— — гидратация 237 ел.
— — гидрозоль 242
— — давление пара 229 ел.
— — кристаллизация 228
— — кристаллическая структура 228 ел.
— — магнитные свойства 231 ел.
— — область существования 211 ел.
— — окисление в гидрат перекиси 258 ел.
плотность 208, 229
— — получение из дигидрата перекиси
258
— — — — других урановых соединений
261 ел.
— „ — — низших окислов урана 256 ел.
— — превращение в низшие окислы
254 ел.
— — — — соли урана и уранила
268 ел.
— — — уранаты и перуранаты 266 ел.
— — разложение 254
— — растворимость в кислотах 268 ел.
— — — — растворах солей 270
— — теплоемкость 230
— — теплопроводность 231
— — теплосодержание 230
— — теплота нейтрализации 268
— — термодинамика образования и
превращения в другие окислы 243 ел.
цвет 232
— — электрические свойства 231
— — энтропия 231
Урана трибромид,давление пара 413 ел.
комплексные соединения 416
— — коррозийное действие 417
— — кристаллическая структура и
плотность 413
— — растворимость 414 ел.
— — реакции 415
— — температура плавления 413
— — термодинамические свойства 414
— — фазовые отношения 415 ел.
Урана триброммонохлорид, давление
пара 438
— — получение и свойства 431, 435
— — термодинамика 4391
Урана трииодид, коррозийное действие
430
— — кристаллическая структура 428
— — окисление 429
— — получение 426 ел.
— — температура плавления 428
— — термодинамические свойства 429
Урана трииодмонобромид, получение и
свойства 432, 437
Урана трииодмонохлорид, получение и
свойства 431, 436
Урана трифторид, получение 286 ел.
— — температура плавления 287
—■■— химические реакции 288 ел.
Урана трихлорид, восстановление 370
Предметный указатель
487
Урана трихлорид, гидролиз 367 ел.
— — давление пара 363
— — комплексные соединения 370
• кристаллическая структура 363
— — окисление 368 ел.
очистка 363 ел.
плотность 365
получение 361 ел.
растворение 367 ел.
температура плавления 363
— — термическое разложение 368
термодинамические данные 364 ел.
Урана фосфиды, получение и свойства 199
Урана хлориды, сопоставление свойств
(см. также урана гексахлорид, пентахло-
рид, тетрахлорид, трихлорид) 361
Уранилбромид, комплексные соединения
475 ел.
— получение безводной соли 473 ел.
гидратов 474
— свойства 474 ел.
Уранилиодид, получение растворов 477 ел.
Уранилфторид, комплексные соединения
458 ел.
— кристаллическая структура и
плотность 453
— растворимость 455
— система с HF и водой 456 ел.
— термическое разложение 457 ел.
— термодинамические свойства 453 ел.
— химические реакции 453
Уранилхлорид, комплексные соединения
466 ел.
— кристаллическая структура 464 ел.
— летучесть 465
— получение безводной соли 463 ел.
— — гидрата 464
— растворы 465 ел.
— термическая устойчивость 466
— химические реакции 465
Уранинит, состав, свойства, месторождения
и добыча 64—67, 86, 88
Урановая смоляная руда, извлечение
урана 99 ел.
обработка 261
— — — состав, свойства и добыча 64, 73,
86, 87 ел.
Урановые минералы, классификация 64 ел.
— — месторождения 87 ел.
распространение 62 ел.
— — флюоресценция 86 ел.
Урановые низкосортные руды, извлечение
урана 107
Уранолепидит, состав, свойства и
месторождения 77, 86
Уранопилит, состав, свойства и
месторождения 74, 80, 86
Ураноспинит, состав, свойства и
месторождения 81, 84, 86
Ураносферит, состав, свойства и
месторождения 77, 86, 87
Ураноталлит, состав, свойства и
месторождения 74, 79, 86
Уранотил, состав, свойства и
месторождения 77, 86
Уранофан, состав, свойства и
месторождения 77, 86, 91
Уранохальцит, состав, свойства и
месторождения 80, 86
Ураноцирцит, состав, свойства и
месторождения 81, 86
Ураношпатит, состав, свойства и
месторождения 83, 86
Фергусонит, состав, свойства и
месторождения 70, 86, 91
Фоглианит, состав, свойства и
месторождения 80, 86
Фоглит, состав, свойства и месторождения
79, 86
Фосфуранилит, состав, свойства и
месторождения 82, 86, 87
Фритчеит, состав, свойства и
месторождения 82, 86, 87
Фурмарьерит, состав, свойства и
месторождения 76, 86, 87
Хьельмит, состав, свойства и
месторождения 71, 86
Цейнерит, состав, свойства и
месторождения 81, 84, 86—89
Циппеит, состав, свойства и
месторождения 80, 86
Циркелит, состав, свойства и
месторождения 72, 86
Циркон, урансодержащий минерал 81
Циртолит, урансодержащий минерал 81
Чевкинит, урансодержащий минерал 81
Шарпит, состав, свойства и месторождения
79, 86
Шинколобвит, состав, свойства и
месторождения 78, 86
Шрёкингерит, состав, свойства и
месторождения 79, 86
Эвксенит-поликраз, состав, свойства и
месторождения 68, 71, 86, 91
Эльсвортит, состав, свойства и
месторождения 69, 86, 87
Эпиянтинит, урансодержащий минерал 74
Эшвегит, состав, свойства и
месторождения 71, 86
Эшинит, состав, свойства и месторождения
71, 86
Элементы, распределение по различным
зонам земли 63
Янтинит, состав, свойства и
месторождения 74, 75, 86, 87
СОДЕРЖАНИЕ
О т р е д а к ц и и 3
Часть I
УРАН КАК ЭЛЕМЕНТ
Глава 1. Изотопный состав и атомный вес природного урана 7
Изотопный состав 7
Атомный вес И
Литература 13
Глава 2. Свойства атома урана 14
Рентгеновский спектр Н
Оптический спектр 26
Литература 56
Глава 3. Уран в природе 59
Общий обзор 59
Распространение и состав минералов, содержащих уран 62
Промышленные месторождения урановых минералов 87
Литература 92
Часть II
МЕТАЛЛИЧЕСКИЙ УРАН
Глава 4. Извлечение урана из руд и получение металлического урана 99
Извлечение урана из руд 99
Получение металла 107
Литература 113
Глава 5. Физические свойства металлического урана ИЗ
Структура и механические свойства твердого урана 116
Механические свойства урана 119
Термодинамические свойства урана 124
Электронные свойства урана 131
Литература 138
Глава 6. Химические свойства металлического урана 141
Реакции с неметаллами • 141
Реакции с соединениями неметаллов 143-
Реакции с водными растворами кислот 144
Реакции с водными растворами щелочей 145
Реакции с растворами солей тяжелых металлов 145
Другие реакции 146
Литература 147
Содержание 489
Глава 7. Интерметаллические соединения и сплавы урана 148
Получение сплавов и интерметаллических соединений 148
Взаимная растворимость урана и различных металлов • 148
Интерметаллические соединения 151
Литература - 152
Часть III
БИНАРНЫЕ СОЕДИНЕНИЯ УРАНА
(за исключением галогенидов)
Глава 8. Система уран—водород 155
Растворимость водорода в твердом и жидком металлическом уране 155
Гидрид урана 157
Дойтерид урана 173
Литература 175
Глава 9. Бориды, карбиды и силициды урана 178
Система уран—бор 178
Система уран—углерод 178
Система уран—кремний 187
Литература 190
Глава 10. Соединения урана с элементами подгруппы Va 192
Нитриды урана 192
Фосфиды урана 199
Арсениды и антимониды урана 199
Литература 200
Глава 11. Окислы, сульфиды, селениды и теллуриды урана 202
Окислы и гидроокиси урана 202
Фазовые отношения в системе уран—кислород 204
Физические свойства безводных окислов урана 214
Гидраты окислов урана 232
Окисление и восстановление урана и его окислов 243
Переход других соединений урана в окислы 259
Превращение окислов в другие соединения урана 264
Сульфиды урана 270
Система уран—сера 270
Селениды и теллуриды урана 275
Литература 276
Часть IV
ГАЛОГЕНИДЫ, ОКСИГАЛОГЕНИДЫ, БОРГИДРИДЫ И КАРБОНИЛ УРАНА
Глава 12. Нелетучие фториды урана 285
Трифторид урана 285
Тетрафторид урана 289
Пентафторид урана и промежуточные фториды U2F9 и U4F17 312
Литература . 319
Глава 13. Гексафторид урана 322
Получение гексафторида ураиа 322
Физические свойства гексафторида урана 328
Химические свойства гексафторида урана 354
490 Содержание
Литература 359
Глава 14. Соединения урана с хлором 361
Трихлорид урана 361
Тетрахлорид урана 370
Пентахлорид урана 392
Гексахлорид урана 399
Литература 466
Глава 15. Бромиды, иодиды и псевдогалогениды урана 411
Трибромид урана 411
Тетрабромид урана 417
Трииодид и тетраиодид урана 425
Смешанные галогениды трех- и четырехвалентного урана 430
Боргидрид урана (IV) U(BH4)4 440
Попытки получения карбонила урана 445
Литература 447
Глава 76. Оксигалогеииды урана 451
Уранилфторид U02F2 451
Оксифторид урана (IV) UOF2 " 462
Уранилхлорид U02CI2 463
Оксихлорид урана (IV) UOCL 471
Уранилбромид U02Br2 473
Оксибромид урана (IV) U02Br2 476
Уранилиодид U02J2 477
Литература 478
Предметный указатель 481
Д ж. к а ц и Е. Р а б и н о в и ч
ХИМИЯ УРАНА
Редактор В. В. АРНОЛЬДОВ
Технический редактор
Е. С, Герасимова
Корректор Г. А. Скуратова
Сдано в производство ЗО/ХП 1953 г.
Подписано к печати 10/V 1954 г.
Т-02886. Бумага 70xi08i/i6 =
= 15,4 бум. л. 42,1 печ. л.
Уч.-издат. л. 42.10. Изд. № 3/2080.
Цена 31 р. 45 к. Заказ № 1424
Издательство иностранной
литературы
Москва, Ново-Алексеевская, 52
16-н тип. Союзполиграфпрома Глав-
издата Министерства культуры СССР.
Москва, Трехпрудный пер., 9.
книги по химии,
выпущенные
ИЗДАТЕЛЬСТВОМ ИНОСТРАННОЙ ЛИТЕРАТУРЫ
Дж. Митчел и Д. Смит. Акваметрия. Перевод с английского,
1952, 426 стр., ц. 22 р. 50 к.
А. Гровс. Анализ силикатов. Перевод с английского, 1953, 304 стр.,
ц. 15 р. 70 к.
Фотографическая регистрация ионизирующих излучений. Сборник
статей. Перевод с английского, итальянского, немецкого и
французского, 1953, 400 стр., ц. 19 р. 65 к.
A. Шварц и Дж. Перри. Поверхностноактивные вещества.
Перевод с английского, 1953, 544 стр., ц. 29 р. 50 к.
Радиационная химия. Сборник статей. Перевод с английского, 1953,
332 стр., ц. 15 р. 70 к.
ГОТОВЯТСЯ К ПЕЧАТИ
B. М. Лат и мер. Окислительные состояния элементов и их
потенциалы в водных растворах. Перевод с английского, объем 28 авт.
листов.
Физическая химия фотографических процессов. Сборник статей.
Перевод с английского, немецкого и французского, объем 35 авт.
листов.
Книги продаются в магазинах книготоргов и высылаются почтой
наложенным платежом (без задатка) республиканскими, краевыми
и областными отделами «Книга—почтой».
ОПЕЧАТКИ
Страница
38
134
169
184
330
364
370
Строка
25 снизу
табл. 53, 2 графа,
6 сверху
табл. 62, 3 графа,
8 сверху
табл. 66, 2 графа,
4, 5 сверху
табл. J31, 6 графа,
11 сверху
2 сверху
5 снизу
Напечатано
различными
0,896
Ш(?)
ккал*г-атом U"1
1,072
от 0 до 300° К
ДН28
Следует читать
с различными
0,086
Ш3(?)
ккал-г-атомг1 U
1072
от 0 до 380° К
ДН208
Зак. 1424