/




























































Текст
>
л
Г. ЗОНТАГ К. Ш ТРЕН ГЕ
КОАГУ\Я ЦИЯ
И УСТОЙЧИВОСТЬ ДИСПЕРСНЫХ СИСТЕМ
и
т FW’W -ч—.з
«ХИМИЯ» 1973
HANS SONNTAG
KLAUS STRENGE
KOAGULATION
UND STABILITAT
DISPERSER SYSTEME
VEB bEUtSCHER VERLAQ der wissenschaften BERLIN 1970
Г. ЗОНТАГ К. ШТРЕНГЕ
КОАГУЛЯЦИЯ И УСТОЙЧИВОСТЬ ДИСПЕРСНЫХ СИСТЕМ
Перевод с немецкого и редакция О. Г. Усьярова
ЛЕНИНГРАД ИЗДАТЕЛЬСТВО „ХИМИЯ-нградстоа отделение 19 7 3
УДК 541.18.046/.049
~ 3^84
Г. Зонтаг, К. Штренге.
Коагуляция и устойчивость дисперсных систем. Перевод с немецкого и редакция О. Г. Усьярова. Л., «Химия», 1973.
Стр. 152, рис. 60, табл. 23.
В монографии даны основы теории устойчивости и коагуляции дис-персных систем и приведен большой фактический материал, относящийся к исследованию суспензий, эмульсий и пен. Кроме того, на современном уровне рассмотрен ряд вопросов по электрокоагуляции, флотации, очистке сточных вод н другим технологическим процессам.
Книга предназначена для научных работников, инженеров, аспирант тов, занимающихся проблемой устойчивости и коагуляции коллоидных си** стем, а также вопросами физико-химической механики дисперсных струк« тур и материалов. Она может служить пособием для студентов при нзу* чении курса коллоидной химии.
„ 0254—007
3050(01)—73
4—73
Число книг по коллоидной химии весьма ограничено. Если Жучебные пособия, рассчитанные, как правило, на учащихся К* различных специальностей, представлены сравнительно полно, Жто ощущается очевидный недостаток монографий, посвященных ^отдельным наиболее важным разделам науки о коллоидах. Од-|йдам из таких центральных разделов коллоидной химии является Йрдроблема устойчивости и коагуляции дисперсных систем. Можно ||4Жазвать, по-видимому, две монографии, в которых всесторонне ^'обсуждаются вопросы взаимодействия дисперсных частиц и ^устойчивости коллоидных растворов: Е. J. Verwey, J. Th. G. Over-fepeek, Theory of the Stability of Lyophobic Colloids, Amsterdam, ж. 1948 г. (перевода на русский язык нет); Наука о коллоидах, |^лод ред. Г. Р. Кройта, т. 1, ИЛ, 1955.
Последняя монография в настоящее время представляет по-Жчти библиографическую редкость. Оба названных издания прак-Цтически недоступны широкому кругу специалистов, интересую-I? щихся проблемами коллоидной химии. Кроме того, они не Б. включают результатов многочисленных работ, выполненных ^Сравнительно недавно и опубликованных в периодической науч- • чг’цой литературе. Поэтому монография двух известных ученых из Германской демократической республики Г. Зонтага и.К. Штрек-fc'pe, несомненно, привлечет внимание исследователей в области поверхностных явлений и специалистов, работающих в смежных s? научных областях.
£ Книгу характеризуют краткость и порой даже излишняя кон-Спективность изложения. Авторы избегают громоздких матема-тических выводов и преобразований, отсылая читателей к соот-ветствующей литературе. Библиография книги довольно об-1 ширна (около 300 наименований), хотя необходимо отметить, к что труды советских ученых не нашли в ней достаточно пол-I ного отражения. Фактический материал приводится в легко до-> ступной для читателей форме; его распределение по трем основ-I ным разделам (теория взаимодействия частиц дисперсной фазы, теория коагуляции и примеры процессов коагуляции дисперсных „ систем, относящихся к некоторым технологическим операциям) отвечает переходу от простого к сложному и обусловливает
необходимую логическую последовательность развиваемых представлений. Много внимания уделено проблеме устойчивости тонких пленок; ранее эти вопросы, являющиеся предметом собственных исследований авторов монографии, настолько обстоятельно не были рассмотрены.
Книга Г. Зонтага и К. Штренге не лишена ряда недостатков. Так, в монографии встречаются неточные формулировки; давая определения понятиям флокуляции и коалесценции, авторы подчас не соблюдают принятую ими терминологию. Некоторые работы обсуждаются слишком подробно, другие — незаслуженно кратко.
В тех случаях, когда необходимость в исправлениях была очевидной, они вносились без всяких оговорок. Если речь шла о различных точках зрения на один и тот же вопрос, то это указывалось в подстрочных примечаниях. Поскольку в последние годы появилось большое количество работ, относящихся к изучению устойчивости и коагуляции дисперсных систем, не представлялось возможным отметить все подобные исследования и при редактировании было решено ограничиться лишь ссылками на наиболее существенные результаты, опубликованные, в основном, на русском языке.
В заключение пользуемся случаем выразить глубокую благодарность чл.-корр. АН СССР Б. В. Дерягину и профессору И. Ф. Ефремову за ценные советы при переводе и редактировании книги.
О. Усьяров
ПРЕДИСЛОВИЕ АВТОРОВ
Взаимодействие коллоидных частиц друг с другом и с макроповерхностями определяет устойчивость, коагуляцию и реологическое поведение дисперсных систем, а также адгезию микрообъектов к твердым телам в жидкой и газообразной средах; оно оказывает существенное влияние на образование и свойства пространственных структур в суспензиях. Поскольку дисперсные системы широко распространены в природе и в различных отраслях промышленности (например, дисперсии пищевых продуктов, фармацевтических веществ, средств защиты растений, полимеров, строительных материалов, красителей), представлялось необходимым рассмотреть общие закономерности взаимодействия коллоидных частиц, независимо от их агрегатного состояния. При этом мы стремились подчеркнуть отличие процесса флокуляции, связанного с действием молекулярных и ионно-электростатических сил и сопровождающегося сохранением сравнительно толстых жидких прослоек между поверхностями частиц, от процесса коалесценции, который приводит к непосредственному контакту микрообъектов.
Берлин, осень 1969 года
Г. Зонтаг, К.. Штренге
ОСНОВНЫЕ ОБОЗНАЧЕНИЯ, ПРИНЯТЫЕ В ТЕКСТЕ
а — радиус частиц и ионов
А — постоянная Гамакера
с — скорость света; концентрация электролита, моль! л
ес — критическая концентрация
cs — концентрация поверхностно-активного вещества, соответствующая образованию устойчивой пленки
h — расстояние между поверхностями частиц
й0 — расстояние между поверхностями частиц при равновесии сил притяжения и отталкивания
йс — критическая толщина пленки е —• заряд электрона
Е — напряженность электрического поля
F — свободная энергия
^ds — свободная энергия двойного слоя
Fo— площадь, приходящаяся на одну молекулу поверхностно-активного вещества в поверхностном слое
Fs — площадь, приходящаяся на одну молекулу поверхностно-активного вещества в поверхностном слое при концентрации са
fi — постоянная Планка
Is — ионная сила раствора
k — постоянная Больцмана
п — коэффициент преломления
til — число ионов в 1 см3 раствора
N — число коллоидных-частиц в единице объема
Na — число Авогадро
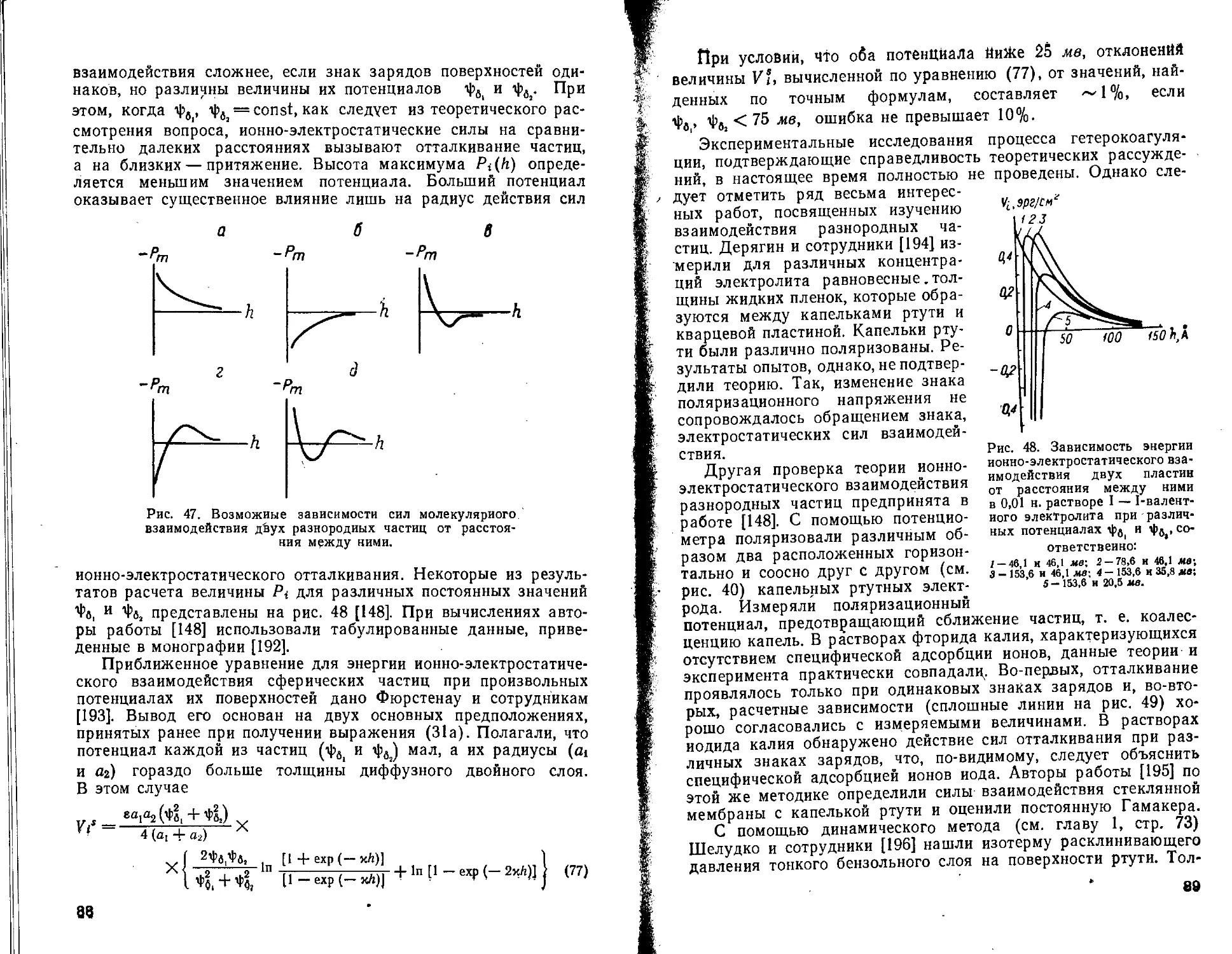
Pi — сила ионно-электростатического взаимодействия частиц
Рт — сила молекулярного взаимодействия частиц
Рп — сила тяжести
Рд — капиллярные силы
q — заряд частицы
г — межатомное расстояние, расстояние между центрами сферических частиц, радиус кривизны, расстояние от плоской поверхности до центра частиц
R ~ радиус плоскопараллельной пленки
t— время
Т — абсолютная .температура
JE— разность потенциалов о — объем 1
V — энергия, отнесенная к единице площади
V{ — энергия ионно-электростатического отталкивания
Vm — энергия молекулярного притяжения
Vd — энергия дисперсионного взаимодействия атомов или молекул
(х) — работа, необходимая для переноса иоиа из бесконечности в некоторую точку, находящуюся на расстоянии х от поверхности И? — фактор замедления коагуляции
и — безразмерный потенциал, равный геф/feT - z — валентность иона
я — поляризуемость
Г — поверхностная концентрация молекул поверхностно-активных веществ
Г3 — поверхностная концентрация молекул поверхностно-активных веществ при объемной концентрации с, б — расстояние между центром тяжести заряда противоиона и гра ницей раздела фаз б„ б2 — толщина адсорбционных слоев е — диэлектрическая постоянная е0 — статическая диэлектрическая постоянная С — электрокииетический потенциал т] — вязкость 0 — краевой угол смачивания х — величина, обратная толщине дебаевской ионной атмосферы А. — длина волны 1-1 — лондоновская длина волны
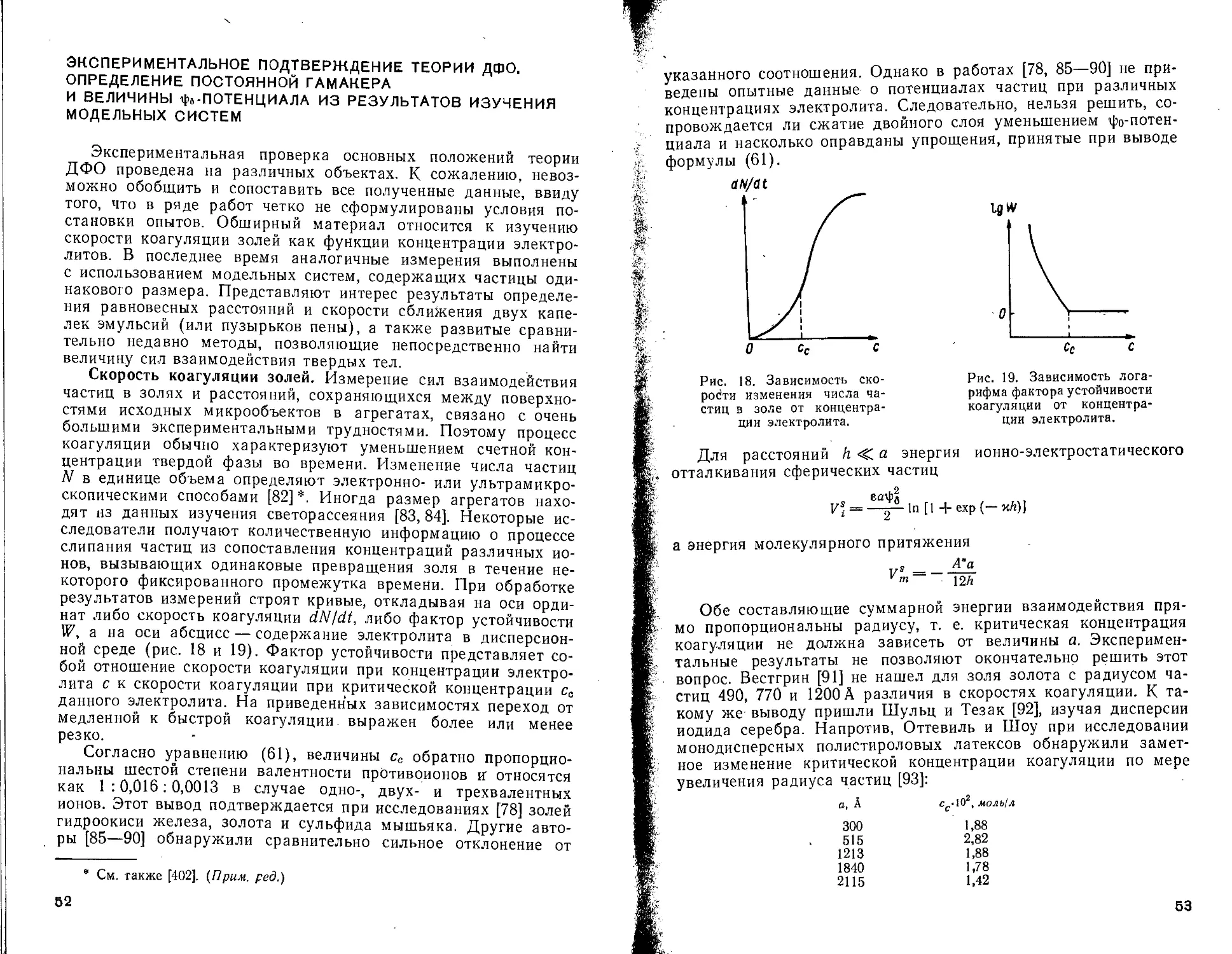
v — частота колебаний атома в основном состоянии vv — характеристическая частота
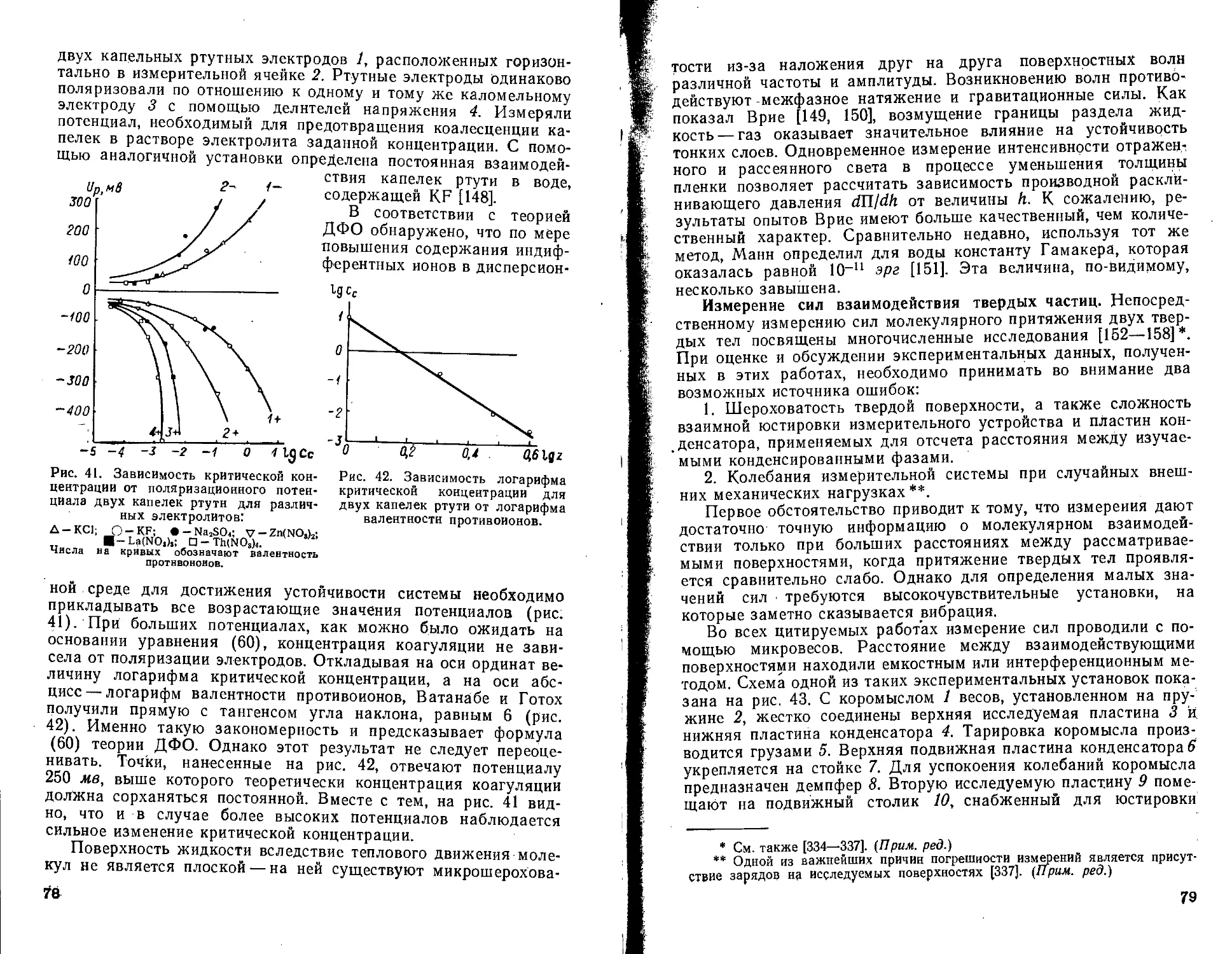
П — суммарное расклинивающее давление
Лт — молекулярная составляющая расклинивающего давления
П( — ионно-электростатическая составляющая расклинивающего дав леиия
Д₽а — лапласовский перепад давления
ДРд — гидростатическое давление р — объемная плотность заряда (То — поверхностная плотность заряда о — поверхностное натяжение Ф — удельный адсорбционный потенциал ф (х) — электрический потенциал на расстоянии х от межфазной гра ницы
фо — потенциал поверхности частиц
фв — потенциал диффузной части двойного слоя ф^ — потенциал в плоскости симметрии между частицами
ВВЕДЕНИЕ
Коллоидные системы в зависимости от состава и структуры частиц можно разделить на три основные группы: дисперсионные коллоиды, ассоциативные или мицеллярные коллоиды и растворы макромолекулярных веществ. Последние получают растворением полимеров в соответствующих средах; процесс сопровождается уменьшением свободной энергии, и, следовательно, возникающая система обладает термодинамической устойчивостью. То же самое относится и к ассоциативным коллоидам, самопроизвольно образующимся в растворах поверхностно-активных веществ (ПАВ) с концентрацией, превышающей критическую концентрацию мицеллообразования. Вопрос об устойчивости имеет наиболее важное значение для дисперсионных коллоидов: аэрозолей, лиозолей, эмульсий и пен. В это состояние можно привести любое вещество либо дроблением компактного материала, либо конденсацией его молекулярного, раствора. Задачи данной монографии ограничиваются рассмотрением дисперсионных коллоидов с жидкой дисперсионной средой.
Особенностью дисперсионных коллоидов является существование между частицами и средой действительной границы раздела фаз с определенной величиной поверхностного натяжения о. Основной вклад в изменение свободной энергии F, вызываемое дроблением вещества, вносят находящиеся на межфазной поверхности атомы, число которых сравнимо с их числом в объеме, т. е. dF ~ csdS (где S— удельная поверхность).
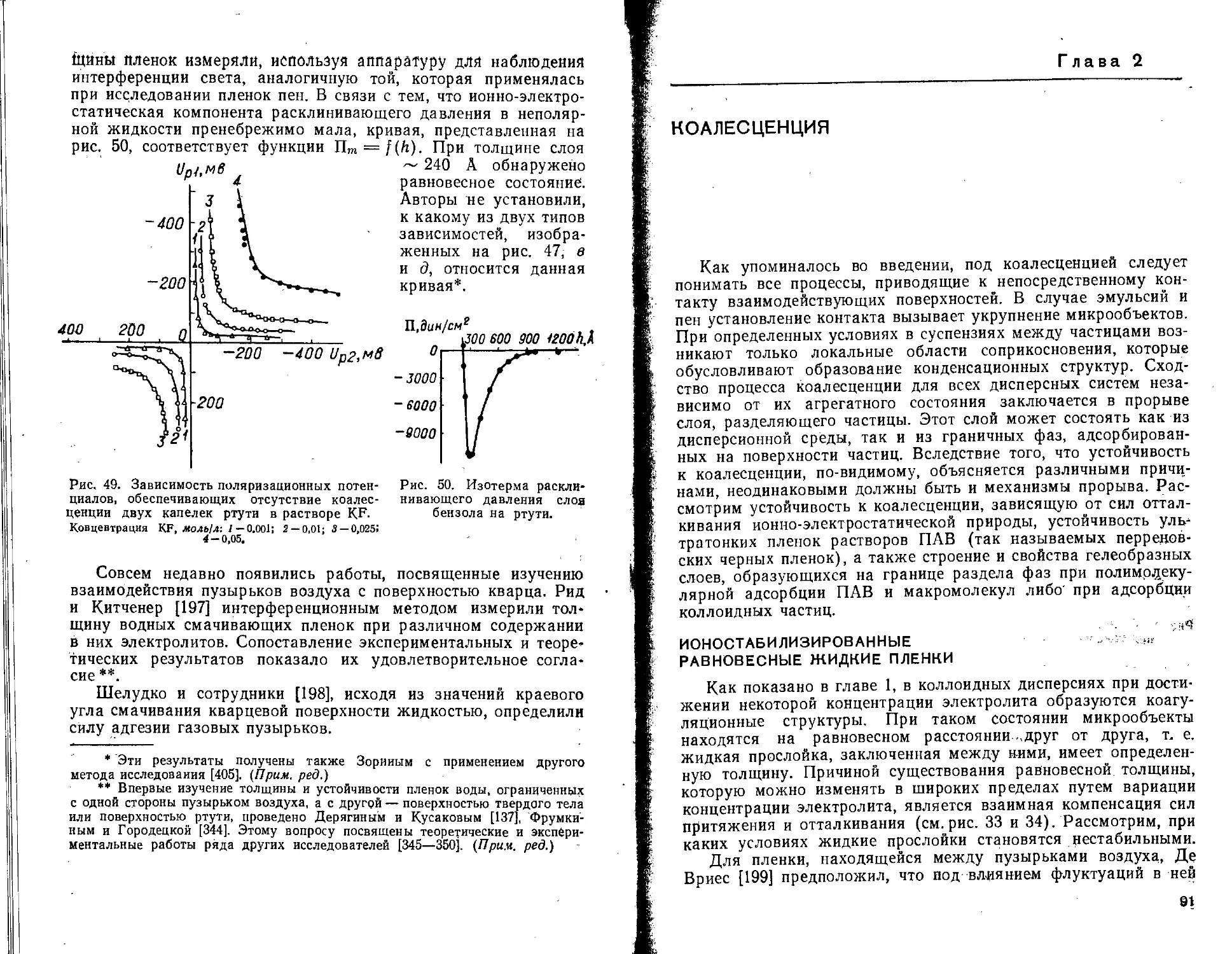
Состояние дисперсионных коллоидов характеризуется избытком свободной энергии, причем укрупнение частиц происходит самопроизвольно, обусловливая уменьшение величины F. Следовательно, дисперсионные коллоиды термодинамически неустойчивы; их временная стабильность может быть связана с наличием энергетического барьера, предотвращающего сближение и взаимную фиксацию частиц на сравнительно малых расстояниях друг от друга (флокуляция) или полное объединение микрообъектов (коалесценция). Исходя из этого, различают дисперсии, устойчивые к флокуляции, и дисперсии, устойчивые к коалесценции. Во флокулированном, но устойчивом к коалесценции состоянии отдельные частицы объединены в очень крупные агрегаты и образуют так называемую коагуляционную структуру. Они сохраняют индивидуальность и разделены тонкими прослойками дисперсионной среды, содержащей в ряде случаев поверхностно-активные и макромолекулярные вещества. Разрушение таких слоев, сопровождающееся либо полным объединением частиц в пенах и эмульсиях, либо возникновением
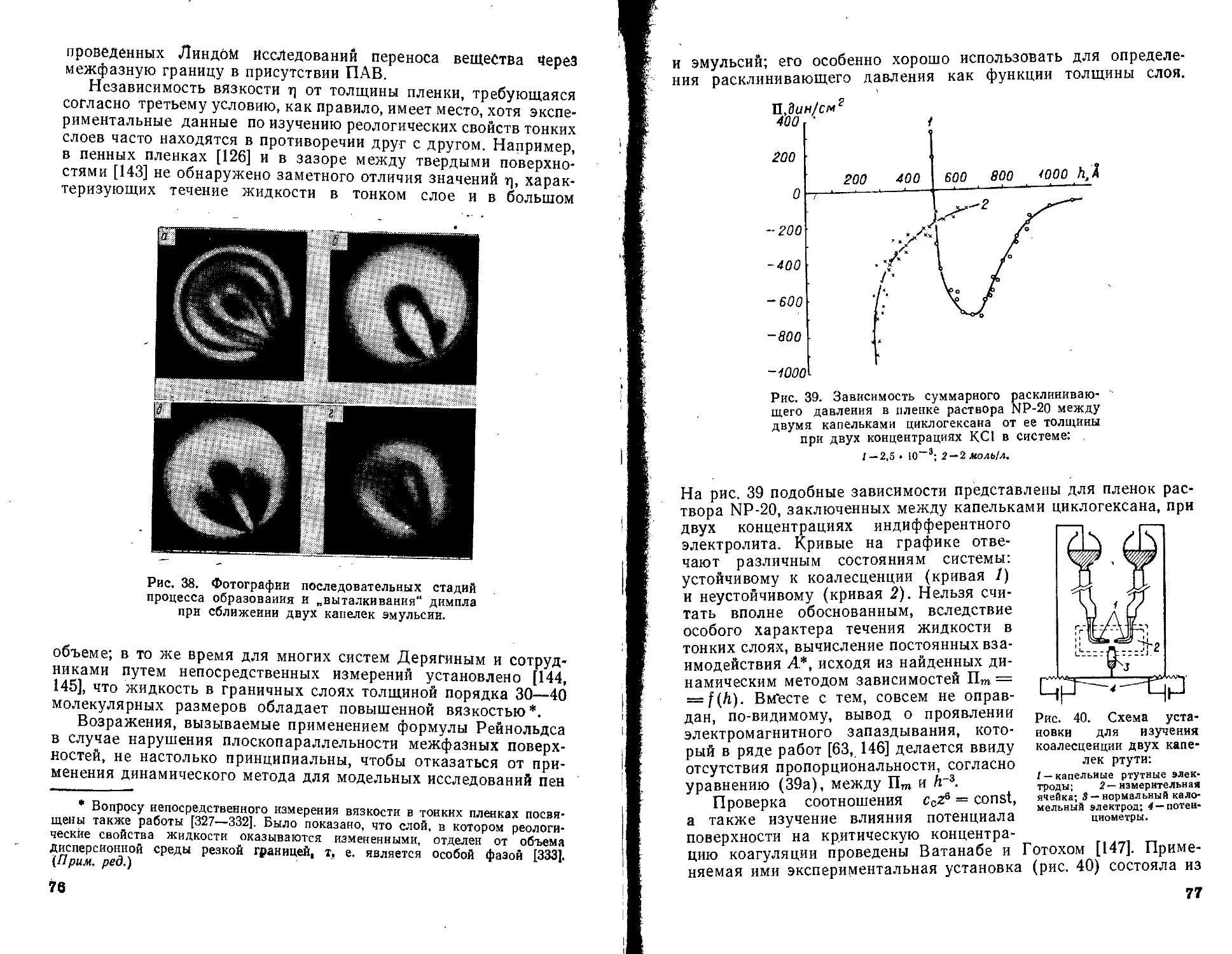
Непосредственных контактов (конденсационные структуры) П случае твердых микрообъектов, мы называем коалесценцией. Термин «коагуляция» означает более общее понятие для коалес-.’ценции и флокуляции*. Характеристика явлений, происходящих при получении и разрушении дисперсионных коллоидов, дана на •рис. 1.
2 Рис. 1. Схематическое изображение процессов, протекаю-
Е’ щих при получении и разрушении дисперсных систем.
Процесс флокуляции зависит от дальнодействующих поверхностных сил. Поэтому для устойчивости к флокуляции необхо-|®мо, чтобы силы отталкивания, обусловленные перекрытием диффузных двойных слоев частиц, превышали на определенном
Е" * Обычно в отечественной и зарубежной литературе термины «флокуля-«аия» и ^коалесценция» используют, вкладывая в них несколько другой смысл. ’^Первый употребляют как синоним понятия «агрегация» или «коагуляция» Й279—282]; под коалесценцией понимают слияние капелек в эмульсиях и туманах, а также объединение пузырьков газа в пенах [283]. Когда необходимо Подчеркнуть отличие между агрегацией, характеризуемой непосредственным соприкосновением частиц, н агрегацией, протекающей при сохранении тонких ^Жидких прослоек, разделяющих отдельные микрообъекты, говорят о «ближней» М «дальней» агрегации или коагуляции [284]. Здесь и в дальнейшем терминология авторов сохраняется. На возможность такой трактовки процессов коалес-••1®нции в дисперсных системах, содержащих твердые частицы, указывал Ребин-’•Дер [283], отмечая аналогию явлений собирательной рекристаллизации и слияния, приводящих в конечном итоге к исчезновению поверхности раздела фаз. (Прим, ред.) ,
расстоянии силы молекулярного притяжения. Особый случай, по-видимому, представляет устойчивость коллоидных систем, у которых поверхностное натяжение на границе дисперсная фаза — среда очень мало или равно нулю [1]*. В этих дисперсиях агрегация не вызывает уменьшения свободной энергии, и они является стабильными в термодинамическом отношении. Иначе говоря, в таких системах практически отсутствует молекулярное взаимодействие частиц.
Силы взаимодействия микрообъектов также оказывают существенное влияние на процесс коалесценции, в частности, когда ей предшествует флокуляция, сопровождающаяся сохране-нйем между частицами расстояний, отвечающих вторичному минимуму (стр. 47). Кроме того, важную роль при получении устойчивых к коалесценции систем играют адсорбционные слои макромолекулярных и поверхностно-активных веществ.
Необходимо отметить, что иногда получение стабильных дисперсий представляет столь же трудную задачу, как и их разрушение. Так, от дисперсий пестицидов, косметических или фармацевтических препаратов требуется, чтобы они были устойчивы к флокуляции. Флокуляция также нежелательна, когда дисперсные системы предназначены для транспортировки, в том числе для перекачки по трубопроводам. Интенсивное образование коагуляционных структур, вызывая сильное повышение вязкости, ухудшает технологические свойства суспензий строительных материалов и промывочных растворов, применяемых при бурении скважин. Напротив, для дисперсий пигментов и латексных красок устойчивость к флокуляции — необязательное требование. Вместе с тем, в емкости, предназначенной для их хранения, не должна происходить коалесценция. Заметное улучшение качества полимерных композиций при введении поверхностноактивных веществ связано с возникновением коагуляционных структур.
Флокуляция и коалесценция имеют большое значение для процессов флотации и очистки различных поверхностей от загрязнений. Диспергированные благодаря механическим воздействиям частицы грязи должны флокулировать под влиянием моющего средства и затем удаляться при промывке. Если же наступает коалесценция, загрязнения вновь прилипают к обрабатываемой поверхности. При флотации флокуляция и коалесценция газовых пузырьков или масляных капель с минеральными частицами являются необходимой предпосылкой эффективности процесса.
В заключение следует еще раз подчеркнуть важность решения задачи о разрушении нежелательных дисперсий (борьба с пенообразованием, деэмульгирование, удаление из различных жидкостей взвешенных частиц и т. д.),
* См. также [285, 286]. {Прим, ред.)
Глава 1
ЗАИМОДЕЙСТВИЕ ДИСПЕРСНЫХ ЧАСТИЦ. РОЦЕСС КОАГУЛЯЦИИ
Электрический двойной слой
Устойчивость дисперсных систем в значительной степени Зависит от распределения ионов вокруг коллоидных частиц или, (Х^даче говоря, вблизи электрода. При соприкосновении двух фаз 'У4. “Ча сто происходит переход носителей электричества. В результате частицы получают избыточный поверхностный заряд в виде 1 'электронов или адсорбированных ионов, который со стороны 'раствора компенсируется равным по величине, но противопо-Ж^ожным по знаку зарядом. Последний образован противоио-
%
Рис. 2. Зависимость потенциала'ф в диффузной части электрического двойного слоя от расстояния х до межфазной границы при низкой (кривая 1) и высокой (кривая 2) концентрациях индифферентного электролита.
нами, а также малым количе-Ж‘1ством коионов, имеющих одина-‘ ковый знак заряда с границей раздела фаз. Концентрацию Кононов и противоионов можно определить раздельно термодина-
мическим методом [2, 3]. Согласно исследованию Ликлемы [3], компенсация поверхностного заряда в золе иодида серебра определяется, главным образом, противоионами. Прэтому в дальней-’шем, исключая особо отмеченные Случаи, только они и будут при--ниматься во внимание.
Вследствие электростатического притяжения противоионы находятся в непосредственной близости от электрода; часть из них расположена в виде неподвижного слоя, аналогичного одной из обкладок плоского конденсатора, а остальные распределены диффузно под действием теплового движения (рис. 2).
Диффузная часть двойного электрического слоя. Распределение противоионов в диффузной части двойного слоя зависит от природы и концентрации электролитов и обычно определяется с помощью трех основных уравнений: уравнения Пуассона . diy [в (*) grad ф (*)] = —4яр (х)
уравнения Больцмана
Г (.*> 1
(х) = «г(°°) ехР[-—j (2)
уравнения для объемной плотности зарядов
р W = 2 zieni W (3)
i
где е—диэлектрическая постоянная в объеме раствора; ф—потенциал; р — объемная плотность заряда; п, — число ионов i-ro сорта в 1 см3; х— расстояние до границы раздела фаз; Wf(x)— работа, необходимая для перемещения иона из объема раствора (х = оо) на расстояние х; k — постоянная Больцмана; Т — абсолютная температура; Zi— валентность иона; е — заряд электрона.
Для аналитического решения уравнения (1) его необходимо упростить. При этом обычно исходят из предположений, сформулированных Гуи [4, 5] и Чепменом [6]:
1. Двойной слой является плоским.
2. Диэлектрическая постоянная раствора не зависит от координаты х.
3. Собственный объем ионов равен нулю, т. е. двойной слой рассматривается как система точечных зарядов.
4. Для перевода иона из объема раствора в двойной слой требуется совершить работу’ только против сил электростатического взаимодействия; т. е. 1У,(х) = 2гвф(х).
Тогда уравнения (1) и (2) приводятся к следующим выражениям:
rf2'!’ (х) 4лр (х) .
------------------ (1а)
и
Г г.еф (х) ]
пг (*) == ni <°°) ехР [--] (2а)
Ниже будет показано, насколько такие предположения выполняются.
Уравнения (1а), (2а) и (3) совместно задают соотношение Пуассона — Больцмана:
d2t[)(x) . 4л v Г г,еф (х)1
2и2^(оо)ехр[—аг—J <4)
i
Проводя первое интегрирование выражения (4) при условии х->оо, ф = 0 и dty/dx = 0, для симметричного электролита с валентностью ионов z имеем:
(х) Г 8л«г (оо) kT Т/г Г [ zety \ / геф \1
dx = ~ [ в J [ехр “ ехр -
1
1
F . л
г После второго интегрирования, принимая, что при х = О л = ф01 получим формулу, характеризующую зависимость потенциала от расстояния до границы раздела фаз
|Де х — величина, обратная толщине диффузного электрического двойного слоя
4ле2 У п^2 i
В Поскольку значение параметра н прямо пропорционально Ьорню квадратному из концентрации электролита, содержание Монов в растворе, как видно из уравнения (6), определяет изменение потенциала в двойном слое. Введение индифферентных электролитов в дисперсионную среду вызывает более сильное Впадение потенциала, что имеет существенное значение для коагуляции коллоидных систем.
К ' Ограничиваясь для низких потенциалов первыми двумя членами разложения в ряд показательных функций, из выражения (6) найдем
Фо
V.X = In Ф
(8)
|т. е. по мере увеличения расстояния до границы раздела потенциал стремится к нулю по экспоненциальному закону. При [х = 1/х значение ф уменьшается в е раз по сравнению с фо:
С другой стороны, для высоких потенциалов и больших расстояний до границы раздела, когда г<?ф0/£7’ 55? 1 и ze^jkT 1
Используя уравнение (5) и принимая во внимание, что
£ получим соотношение между поверхностной плотностью зарядов
* Со и потенциалом:
erijkT ТА Г / геф0 \ / геф0
ff° ~ НН Lexp \~йП “ехр V '2AF/J -(,Оа)
15
Коррекция уравнения Пуассона — Больцмана. В последнее время опубликованы многочисленные теоретические и экспериментальные работы, в которых изучены границы применимости соотношения Пуассона — Больцмана и проверены предположения, принятые Гуи и Чепменом. Из результатов измерения емкости двойного слоя следует, что диэлектрическая постоянная в во внутренней области двойного слоя понижена вследствие высокой напряженности поля Е (диэлектрическое насыщение) и структурирующего влияния фазы, граничащей с объемом дисперсионной среды. Зависимость е(£) впервые теоретически рассмотрена Бусом [7], а несколько позднее и более строго — Букингемом [8]. Связь между е и концентрацией ионов экспериментально исследована в работе [9], причем установлено, что в уменьшается при увеличении содержания ионов. Величины Е, щ и, следовательно, е изменяются по мере удаления от границы раздела фаз. Поэтому уравнение (1а), предполагающее постоянство значений е, необходимо модифицировать [10—14].
Спарней [12] представил диэлектрическую постоянную в виде суммы двух величин; ео и е (где е0 — статическая постоянная); первая из них не зависит, а вторая зависит от расстояния х. Он показал, что при этом уравнение Пуассона может быть записано следующим образом:
d2if (х) 4лр (х) I Г, . d2i|> (х) ., 7
------[(e-eo)-^- + gradtgrade] (П)
Здесь е= е(Е) — функция от напряженности поля и концентрации ионов
п, п
8 = 8o+a£2 + 6+^_L+S_^
02)
где Na — число Авогадро.
Коэффициент а < 0 и, согласно Малшу [15], приблизительно равен —3-10'6 (см-се№)/г, если для значения Е принята электростатическая система измерения. Константы б+ и б_ зависят от гидратации ионов и варьируют в пределах от —3 до —20 смР/моль. В связи с тем, что катионы, как правило, сильнее гидратированы, чем анионы, для большинства случаев 1б+1 > Iб_|. Подставляя выражение (12) в уравнение (11), получим модифицированное уравнение Пуассона:
«з>
Здесь
16 . .. -
НШИ.
к явно завышенным
еЧ/кТ
Ю
8
6
л
lai
г
Рис. 3. Зависимость безразмерного потенциала диффузной части электрического двойного слоя от радиуса ионов [22].
В отличие от уравнения Пуассона выражение (13) имеет: Ц) поправочный член х отрицательным коэффициентом а, обусловливающий увеличение производной, т. е. более сильное изменение напряженности поля по мере удаления от поверхности раздела фаз; 2) поправочный член с коэффициентом Ь, величина которого зависит от гидратации анионов и катионов.
' В соответствии с третьим предположением Гуи и Чепмена gfcM. стр. 14) ионы рассматривают как точечные заряды, что приводит при больших потенциалах поверхности и высоком содержании электролита в дисперсионной среде ачениям концентрации ионов вблизи ^границы раздела фаз. Так, вычисляя по уравнению (2а) поверхностную концентрацию для 1 -10-2 н. раствора симметричного электролита при z~l и фо=ЗОО мв, ВйЪлучим примерно 1500 г-ион!д [16]. #то затруднение можно устранить, если уравнение Больцмана ввести член, ’Считывающий объем гидратированных ’Ионов [12, 13, 16—21]*. Согласно Айвену и Вике [18], гидратные оболочки являются непроницаемыми в случае одинаково заряженных ионов; наоборот, ^Противоположно заряженные ионы способны взаимно проникать через гидратные атмосферы. Введение в уравнение ^Больцмана поправки на объем ионов /способствует уменьшению абсолютной величины плотности заряда у межфазной границы. Представленная на рис. 3 кривая зависимости потенциала от радиуса иона рассчитана, ; исходя из предположения о жесткой гидратной оболоч-ке [22].
Другая коррекция уравнения Больцмана связана с учетом /Эффектов поляризации ионов в электрическом поле диффузной части двойного слоя. Поляризация обусловливает введение особого энергетического члена в уравнение Больцмана [23]. Полагая коэффициенты активности катионов и анионов равными
* Изучение влияния собственного объема ионов на распределение потен-£ циала в двойном слое проведено Мартыновым. В работе [287] на основании .уравнения Боголюбова, являющегося следствием канонического распределения Тиббса, получены, согласно исследованиям Левина и Кирьянова [288], исходные формулы теории Гуи —Штерна и определены границы их применимости. ^'Показано, что теория Гуи —Штерна справедлива в случае очень малых потенциалов поверхностей и низких концентраций электролита (до 10"’—Ю~2 моль/л), -причем при более высоком содержании ионов она не только количественно, но и качественно не описывает свойства двойного слоя. Строгий учет объема 'Гидратированных ионов существенно улучшает сходимость теоретических и экспериментальных данных [289]. {Прим, ред.) _
йсшвшвй фнлиап
НАУЧНАЯ
единице [12] и принимая, что условие
<kT
8я \дп1 1Т.1.П.Е
выполнено, можно получить уравнение Больцмана в виде:
Г / еф \ £"2б, / еф
п+ =.п+ (оо) |_ехр j + 8яйГлГв exp (14)
Или для анионов:
га-— я-(оо) Гехр f—4~ 8дЛъ (14а)
L \ я/ / оТСЯл /V д у к1 j J
Поскольку знак постоянных 6+ и 6- отрицательный, учет поляризации приводит к уменьшению содержания ионов в двойном слое. Принятые предположения ограничивают применимость этих формул, которые справедливы только для низких концентраций электролита и малых потенциалов поверхности.
Еще одно уточнение уравнения Больцмана состоит в том, что при перенесении иона из объема раствора в определенную точку двойного слоя необходимо совершить работу против собственной ионной атмосферы. Соответствующие расчеты выполнены Вильямсом [24]. Как показано, влияние этого фактора становится меньше при увеличении содержания ионов в дисперсионной среде; для потенциала порядка 100 мв и их—>0 погрешность, связанная с использованием уравнения Больцмана *, составляет приблизительно 5%, а при фо — 200 мв — около 8%. Существенный вклад в распределение потенциала вблизи заряженной поверхности, согласно исследованиям Левина и Белла [25], вносят так называемые полостные (cavity) эффекты. Они вызваны тем, что гидратированный ион, входящий в диффузную часть электрического двойного слоя, должен вытеснять заряд, который занимает конечный объем вблизи границы раздела фаз. Такой переход приводит к изменению потенциала в точке, отвечающей центру иона, и является причиной возникновения потенциалов полостей [17]. Обе составляющих суммарного изменения потенциала имеют различные знаки.
Численный анализ модифицированного уравнения Пуассона— Больцмана, выведенного на основании статистического рассмотрения раствора электролита и предположения, что гид-’ ратированные ионы представляют собой твердые шары, распределенные в структурированной среде, проведен Левиным и Беллом. [25]. Это позволяет оценить, какой вклад вносит каждый отдельный фактор в суммарный коррекционный коэффициент
* Наряду с учетом ионных атмосфер внутри двойного слоя необходимо принимать во внимание влияние сил изображения на распределение заряда вблизи границы раздела фаз. Показано [290], что при сравнительно больших концентрациях и особенно в случае противоионов с высокой валентностью роль сил изображения становится весьма существенной. (Прим, ред.)
л. «Ч .
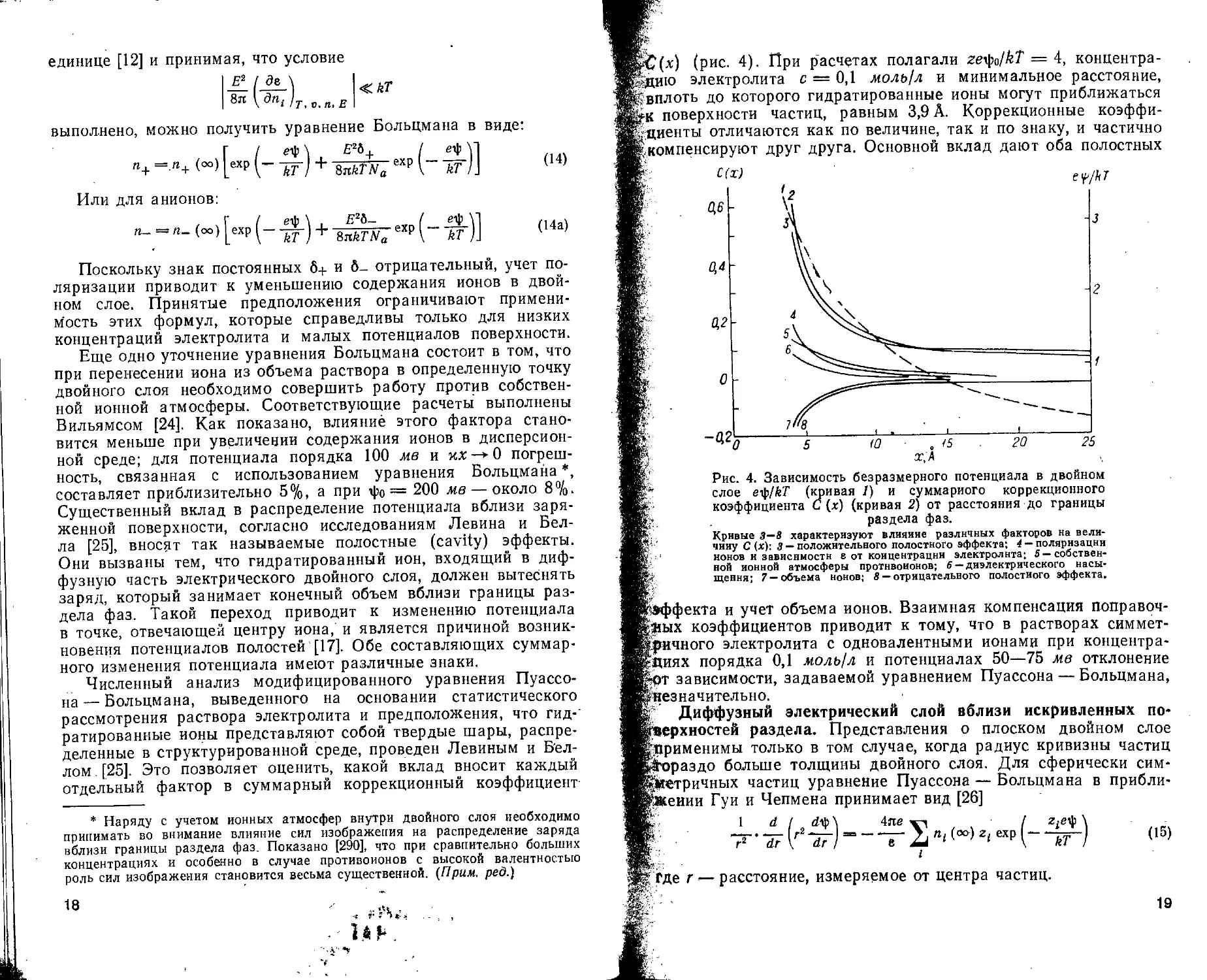
(рис. 4). При расчетах полагали ze^a/kT — 4, концентра-|дцию электролита с = 0,1 моль/л и минимальное расстояние, I вплоть до которого гидратированные ионы могут приближаться Ь-к поверхности частиц, равным 3,9 А. Коррекционные коэффи-F диенты отличаются как по величине, так и по знаку, и частично к компенсируют друг друга. Основной вклад дают оба полостных
Ми Рис. 4. Зависимость безразмерного потенциала в двойном
Ию’ слое ety/kT (кривая 1) и суммарного коррекционного
- коэффициента С (х) (кривая 2) от расстояния до границы
Б’ раздела фаз.
ЗИИ Кривые 3—8 характеризуют влияние различных факторов на вели-
ШК чину С (х): 3 — положительного полостного эффекта; 4 — поляризации
Мк ионов и зависимости в от концентрации электролита; 5 — собственник ной нонной атмосферы противоионов; б— диэлектрического насы-
Mgv- щення; 7 —объема ионов; 8 — отрицательного полостного эффекта.
Игэффекта и учет объема ионов. Взаимная компенсация поправоч-Нкяых коэффициентов приводит к тому, что в растворах симмет-иричного электролита с одновалентными ионами при концентра-ИКДиях порядка 0,1 моль!л и потенциалах 50—75 мв отклонение Йт зависимости, задаваемой уравнением Пуассона — Больцмана, НВнезначителыю.
Вк' Диффузный электрический слой вблизи искривленных по* Ийгверхностей раздела. Представления о плоском двойном слое ^Кприменимы только в том случае, когда радиус кривизны частиц РГораздо больше толщины двойного слоя. Для сферически сим-Ящцетричных частиц уравнение Пуассона — Больцмана в прибли-Я^'Жеиии Гуи и Чепмена принимает вид [26]
SSL’ 1 d / dty\ 4ле хт / \
F ------—kT~) <15>
як i
Где г — расстояние, измеряемое от центра частиц.
Решение этого уравнения возможно только с помощью электронной вычислительной машины. Результаты расчетов табулированы в работе [27]. Для малых потенциалов можно пренебречь членами разложения"экспоненты в ряд, начиная с третьего, и получить, по аналогии с выводом Дебая и Гюккеля для
сильных электролитов, следующие
Рис. 5. Схема распределения противоионов вблизи заряженной плоской поверхности (а) н зависимость потенциала от расстояния до межфазной границы (б).
выражения для потенциала
ф = фоу ехр [х(а —г)] (16)
и для плотности поверхностного заряда
* Ж ==__£2k(I + xa)(I7)
4л \ dr /г—о 4ла ' ’
где а — радиус сферической частицы *.
Плоская часть электрического двойного слоя — слой Штерна. Различие в зависимостях потенциала двойного слоя ф от расстояния до границы раздела фаз, вычисленных на основании модели Гуи — Чепмена и найденных из экспериментальных данных, впервые объяснено Штерном [28] и несколько позднее Грэмом [29, 30]. Авторы работ [28— 30] приняли во внимание, что ионы имеют определенные размеры и поэтому могут приближаться только на конечное расстояние S к поверхности раздела (рис. 5). Следовательно, в уравнение Гуи — Чепмена необходимо подставить вместо фо
величину ф6. Кроме того, как показано, в уравнении Больцмана
следует учесть энергию адсорбции ионов на поверхности. Соот-
ветствующие постоянные, входящие в показатели степени основания натуральных логарифмов, имеют большое значение при изучении специфического влияния ионов.
Согласно развитым представлениям, двойной слой можно разделить на три части:
1. Непосредственно на межфазной границе находится слой, образованный специфически адсорбированными ионами (внутренний слой ГельмгоЛьца). Эти ионы адсорбируются без своих
* Сравнительно недавно Духиным и сотрудниками [291] получены приближенные формулы распределения потенциала вокруг сферической коллоидной частицы, обеспечивающие достаточно высокую точность расчета, при произвольных потенциалах фо и параметрах на. (Прим, ред.)
гидратных оболочек. В связи с тем, что катионы, как правило, гидратированы сильнее анионов (табл. 1), последние, за исключением иона фтора, более склонны к специфической адсорбции
независимо
от знака
заряда поверхности.
Таблица 1
Числа
гидратации
ионов [31]
Катион Число гидратации
Li+ 5
Na+ 5
К+ 4
Rb+ 3
Анион Число гидратации
F" 4
СГ 1
Вг' 1
Г 1
2. Противоионы расположены во внешней части слоя Гельмгольца на расстоянии 6 от межфазной границы. В этом слое потенциал падает до величины фб.
3. К внешней части слоя Гельмгольца примыкает диффузный электрический слой.
Понимая под штерновским двойным слоем внутреннюю и внешнюю обкладку слоя Гельмгольца, для суммарной поверхностной плотности заряда имеем
где as — заряд слоя Штерна; <та — заряд диффузной части двой-
ного слоя.
Исходя из того, что процесс адсорбции противоионов характеризуется изотермой адсорбции Лангмюра, при отсутствии спе-
цифической адсорбции величину <js можно определить из уравнения [26]
as = —
1-+
Na / г еФб + Ф — еХр. -------------
п?М \ kT
'(18)
где АЛ — число адсорбционных центров, отнесенное к единице площади; М — молекулярный вес растворителя; <р— удельный химический адсорбционный потенциал противоионов.
При рассмотрении структуры двойного слоя мы предполагали равномерное распределение зарядов на поверхности. Есин и Марков [32] ввели понятие дискретности заряда, учитывающее тот факт, что адсорбция сопровождается вытеснением имеющегося на поверхности иона. Иначе говоря, сначала должно возникать вакантное место, на которое затем адсорбируется новый ион. Поэтому дискретность расположения ионов
обусловливает более сильное уменьшение потенциала по сравнению со случаем равномерно распределенного поверхностного заряда.
ЭЛЕКТРОСТАТИЧЕСКИЕ СИЛЫ
ОТТАЛКИВАНИЯ ДИСПЕРСНЫХ ЧАСТИЦ
Электростатические силы отталкивания дисперсных частиц являются следствием их одинакового заряда. Однако нельзя вычислять силы отталкивания непосредственно по закону Кулона в связи с тем, что частицы окружены противоионами, компенсирующими их заряд, и за пределами двойного слоя напряженность электрического поля равна нулю. Взаимодействие двух частиц обнаруживается только тогда, когда перекрываются их диффузные электрические слои. Вычисление энергии электростатического отталкивания впервые проведено Дерягиным [33—36]*, а позднее Фервеем и Овербеком [26].
При условии сохранения термодинамического равновесия во время сближения частиц, сопровождающегося перекрытием двойных сдоев, не происходит изменения потенциала на межфазных границах; напротив, заряд сто поверхностей уменьшается **. Такое уменьшение заряда вызывает отталкивание двух частиц***.
С другой стороны, мож носила гать, что иногда за время сближения частиц, происходящего в результате теплового движения, не успевает устанавливаться состояние адсорбционного равновесия. Тогда перекрытие двойных слоев необходимо рассматривать как процесс, протекающий при постоянном заряде и увеличении потенциала .поверхностей. Поскольку зависимости энергии отталкивания от расстояния между частицами в обоих случаях незначительно отличаются друг от друга [37] — энергия ионно-электростатического взаимодействия при малых расстоя
* См. также [38, 294, 303], (Прим., ред.)
** В общем случае при условии термодинамического равновесия в процессе сближения частиц как потенциал, так и заряд поверхностей изменяются; вместе с тем вплоть до расстояний, сравнимых с радиусом действия сил специфической адсорбции, сохраняется постоянной энергия адсорбции потенциал-определяющих ионов. (Прим, ред.)
*** Это утверждение авторов является неточным. Анализ причин отталкивания дан в работах [33—36], а также в обзорной статье Дерягина [292]. При перекрытии двойных слоев силы, действующие со стороны внутренних обкладок обеих коллоидных частиц на ионы, находящиеся между сближаемыми поверхностями, не будут полностью экранированы наружными обкладками. Такое изменение сил нарушает статистическое равновесие, которое имелось до наложения друг на друга ионных атмосфер, и вызывает перераспределение зарядов. В результате концентрация электролита в пленке жидкости, разделяющей частицы, оказывается отличной от концентрации электролита в не-перекрытом двойном слое. Следовательно, с перекрытием двойных слоев могут быть связаны силы как осмотической, так и электростатической природы, причем их одновременный учет представляет необходимую предпосылку для правильной оценки ионно-электростатического взаимодействия. (Прим, ред.)
ниях для постоянного заряда выше, чем для постоянного потенциала — в последующем изложении будем исходить из условия постоянства потенциала *.
Существуют два пути расчета энергии ионно-электростатического взаимодействия: либо находят изменение свободной энергии двойного слоя при переходе от бесконечно удаленных частиц к частицам с перекрытыми двойными слоями, либо получают формулу для энергии, проводя интегрирование выражения для силы отталкивания двух одинаково заряженных поверхностей как функции расстояния между ними. Более подробно оба способа вычисления исследованы в монографии Фервея и Овер-бека [26].
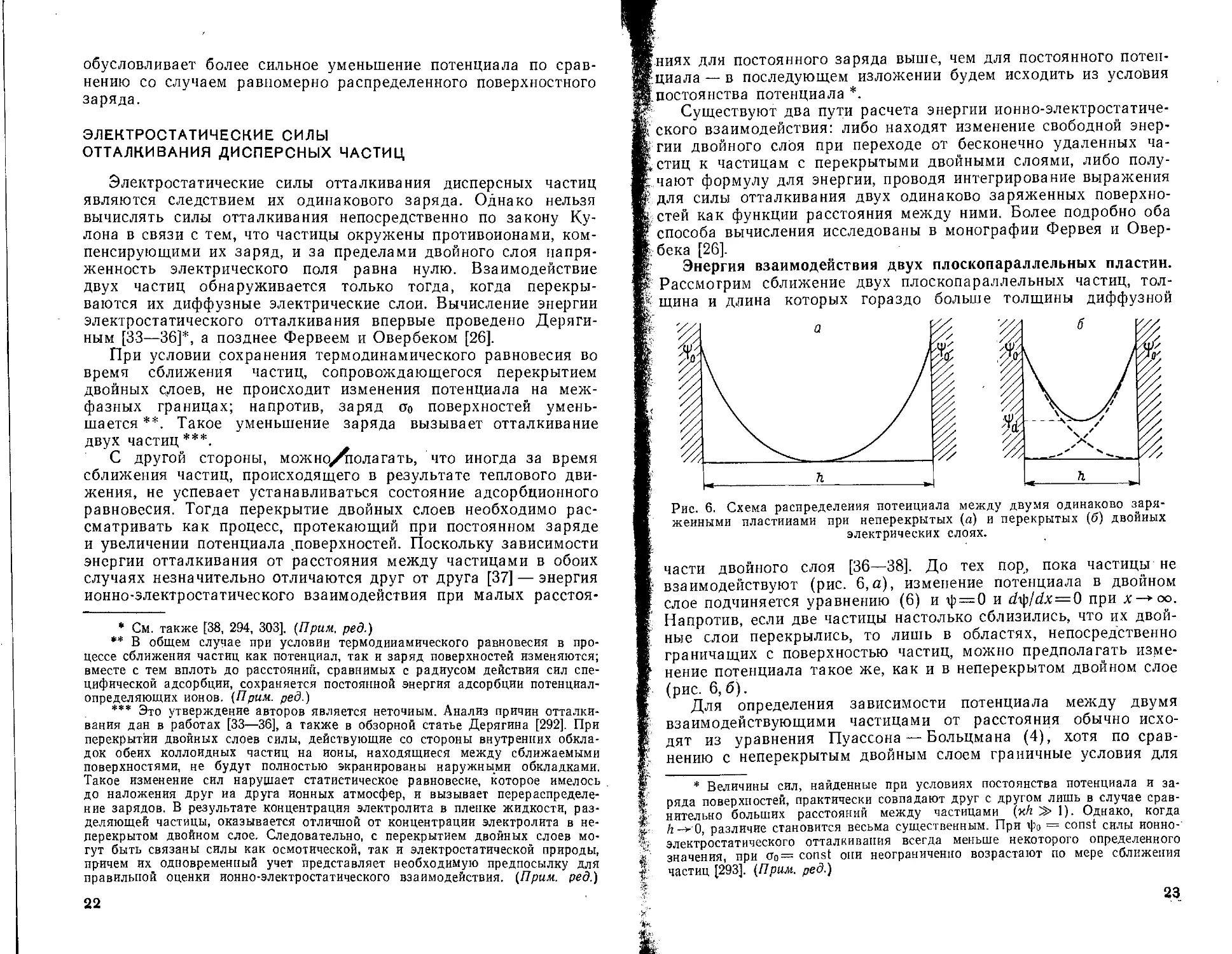
Энергия взаимодействия двух плоскопараллельных пластин. Рассмотрим сближение двух плоскопараллельных частиц, толщина и длина которых гораздо больше толщины диффузной
Рис. 6. Схема распределения потенциала между двумя одинаково заряженными пластинами при неперекрытых (а) и перекрытых (б) двойных электрических слоях.
части двойного слоя [36—38]. До тех пор, пока частицы не взаимодействуют (рис. 6,а), изменение потенциала в двойном слое подчиняется уравнению (6) и ф = 0 и dty/dx=0 при х-*оо. Напротив, если две частицы настолько сблизились, что их двойные слои перекрылись, то лишь в областях, непосредственно граничащих с поверхностью частиц, можно предполагать изменение потенциала такое же, как и в неперекрытом двойном слое (рис. 6, б).
Для определения зависимости потенциала между двумя взаимодействующими частицами от расстояния обычно исходят из уравнения Пуассона — Больцмана (4), хотя по сравнению с неперекрытым двойным слоем граничные условия для
* Величины сил, найденные при условиях постоянства потенциала и заряда поверхностей, практически совпадают друг с другом лишь в случае сравнительно больших расстояний между частицами (x/z 3> 1). Однако, когда й-> 0, различие становится весьма существенным. При фо = const силы ионноэлектростатического отталкивания всегда меньше некоторого определенного значения, при Оо= const они неограниченно возрастают по мере сближения частиц [293]. (Прим, ред.)
интегрирования будут другими: гр = фо, когда х = 0, и гр = гр,/ при х = h!?,. Так как грй представляет минимальное значение потенциала (см. рис. 6,6), то для х = h/2 производная потенциала по расстоянию dtyldx = 0.
Первое интегрирование уравнения (4) при измененных граничных условиях дает:
йгр / 8nkT \'1г Г _ . I г~егр ) + ,
\П‘ (°°)еХр1Лт / + (°°)еХр
г+егр \ kT /
_ (г егрД I г
— nt (оо) ехр —I — «, (°°) ехру----------
(19)
Второе интегрирование аналитически провести трудно. Эту
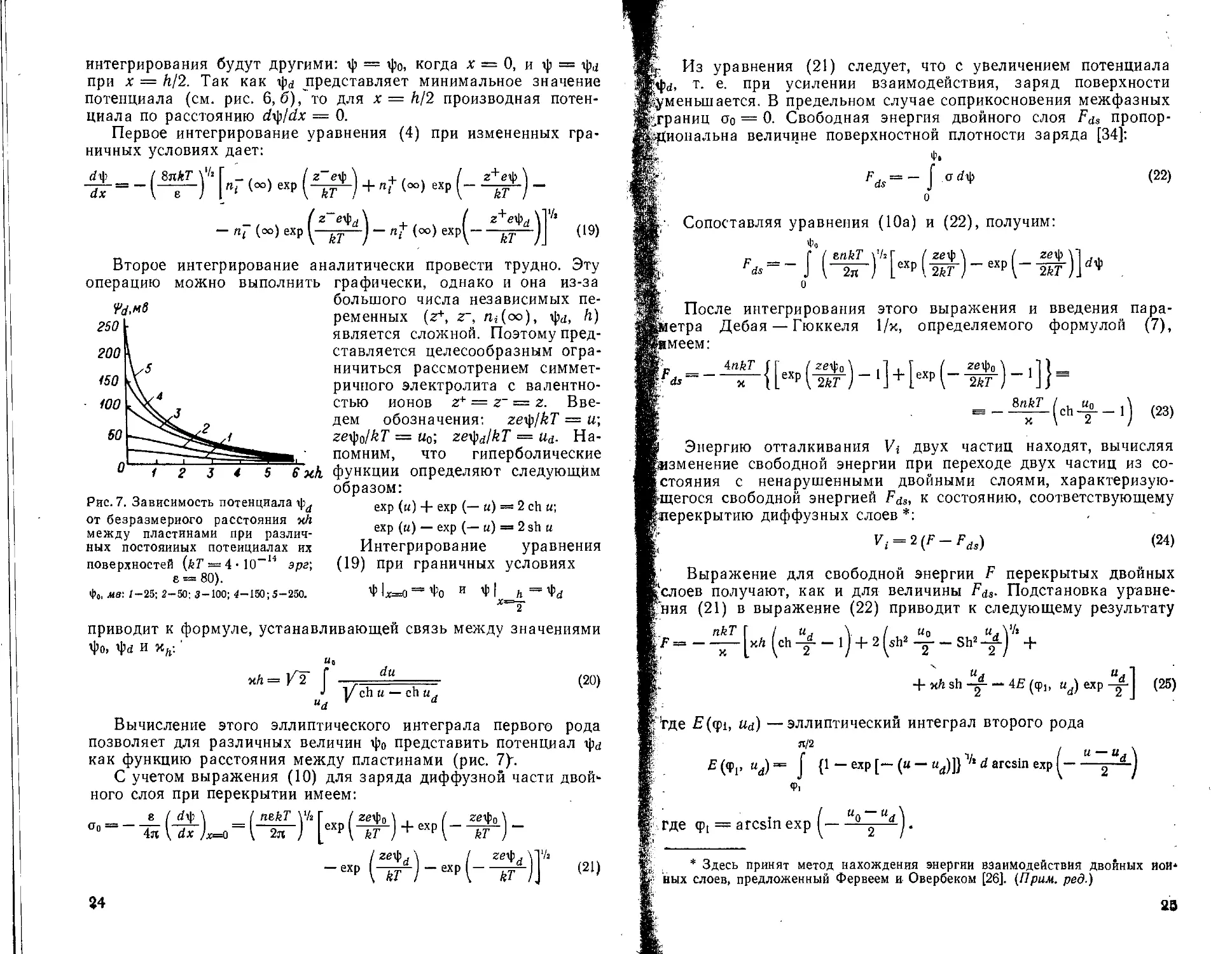
операцию можно выполнить
Рис. 7. Зависимость потенциала от безразмерного расстояния мЛ между пластинами при различных постоянных потенциалах их поверхностей (kT = 4 • 10-14 эрг\ е = 80).
Фо, мв: 1-25; 2-50: 3-100; 4-150; 5-250.
графически, однако и она из-за большого числа независимых переменных (z+, Z-, п;(оо), грй, h) является сложной. Поэтому представляется целесообразным ограничиться рассмотрением симметричного электролита с валентностью ионов z+ = z~ = z. Введем обозначения: zetyjkT = м; zetyojkT = ы0; zetydlkT = Ud- Напомним, что гиперболические функции определяют следующим образом:
ехр (и) + ехр (— и} = 2 ch и;
ехр (и) — ехр (— и) = 2 sh и Интегрирование уравнения (19) при граничных условиях
гр |х=о — гр0 и г|> | h = грй
Х~2
приводит к формуле, устанавливающей связь между значениями гро, и хд:'
«0
и/г = /Г f ,, du — (20)
J |ЛсЬ и — ch ud
Вычисление этого эллиптического интеграла первого рода позволяет для различных величин фо представить потенциал ф<; как функцию расстояния между пластинами (рис. 7)".
С учетом выражения (10) для заряда диффузной части двойного слоя при перекрытии имеем:
е ( rf-ф \ I nzkT \‘/г Г / гегЬ0 ) , ( геф0 \
а°— 4л \ dx )х=0— I 2л ) [еХр(“Й^) + еХр1 ~kf~)
/ геф . \ / гегр . \Т/а
-ехр ^-ехр^------------
(21)
К. Из уравнения (21) следует, что с увеличением потенциала т. е. при усилении взаимодействия, заряд поверхности ^уменьшается. В предельном случае соприкосновения межфазных । границ оо = 0- Свободная энергия двойного слоя FdS пропор-&|Хиональна величине поверхностной плотности заряда [34]:
V (22)
о
Ж,- Сопоставляя уравнения (10а) и (22), получим:
К „ f / enkT x'h Г / zeib \ { аеф \1 ,
№. Fds J ( 2л ) [еХр I. 2kT ) еХр\ 2fcT .
Я 0
ж После интегрирования этого выражения и введения пара-Миетра Дебая — Гюккеля 1/х, определяемого формулой (7), Внмеем:
— + Ь (-»->]}"
К <»>
К Энергию отталкивания V, двух частиц находят, вычисляя ^изменение свободной энергии при переходе двух частиц из со-Встояния с ненарушенными двойными слоями, характеризую--щегося свободной энергией FdS, к состоянию, соответствующему «перекрытию диффузных слоев *: -
К 7Z = 2(F-Fds) (24)
В' Выражение для свободной энергии F перекрытых двойных «слоев получают, как и для величины FdS- Подстановка уравне-№ния (21) в выражение (22) приводит к следующему результату Н nkT Г / и. \ / и0 uj\‘^
~Sh^) +
Ц. + x/i sh -у — 4Е (Ф1, ud) ехр -y-j (25)
||!где Е (ф1, Ud) —эллиптический интеграл второго рода
р Я/2
Ж г v u^ud\
К Е (фр Ud) = j {1 - ехр [- (u - Ud)]} 1г d arcsin ехр -J
В - ф’
К, ' I ип —и.\
в где <pt = arcsin ехр I-- ).
j . * Здесь принят метод нахождения энергии взаимодействия двойных иои*
к ных слоев, предложенный Фервеем и Овербеком [26]. (Прим, ред.)
Определяя разность 2(f — FdB), находят величину энергий отталкивания двух частиц. .
Вычисления с использованием этих достаточно сложных выражений для различных потенциалов ф0 и грй проведены Фер-веем и Овербеком [26]*. Зависимость энергии отталкивания от параметра Дебая — Гюккеля 1/х, концентрации ионов п, потенциала поверхности частиц ф0 и потенциала в плоскости симметрии фц можно выразить с помощью формулы:
vi = 7 f (to- td) = -^2 f i (to- td) (26)
Последнее равенство справедливо, поскольку величина п прямо пропорциональна отношению х2/г2. Необходимо учиты-
Рис. 8. Зависимость энергии ионно-электростатического отталкивания двух пластин в растворе симметричного электролита (z= 1, с — 9,5 10-2 моль/л} от безразмерного расстояния между ними при постоянных потенциалах поверхностей ф0, равных 100 мв (кривая /) и 250 мв (кривая 2), а также при постоянном заряде диффузного двойного слоя 1,33 • 10“5 к./смг (кривая 3).
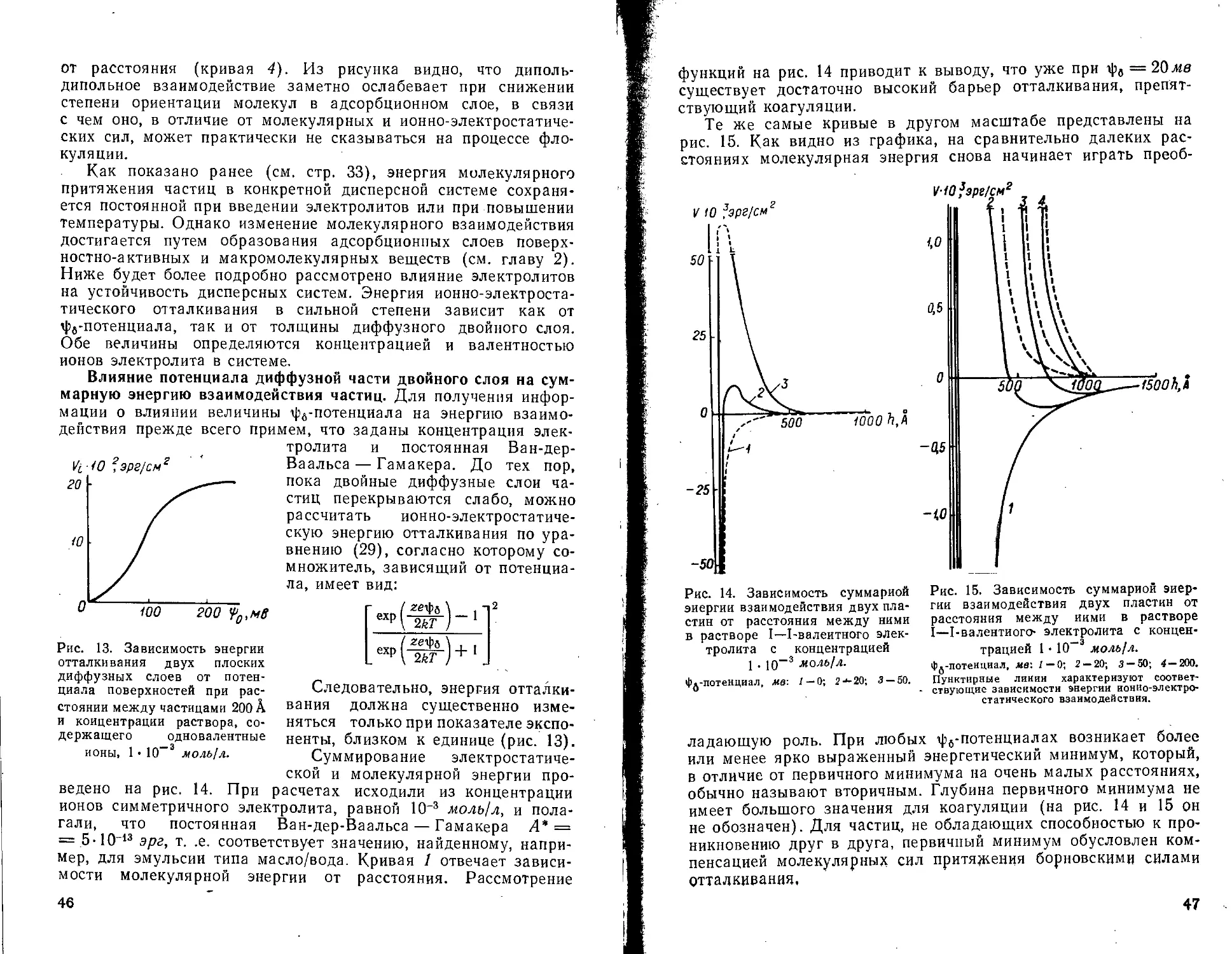
вать, что произведение x/i, в соответствии с уравнением (20), также является функцией потенциалов фо и фа. Таким образом, энергия ионно-электростатического отталкивания при некоторых заданных значениях ф0 и фу и, следовательно, при фиксированной величине х/i определяется еще валентностью противоионов г и их концентрацией п.
На рис. 8 представлена зави-
симость энергии отталкивания от расстояния x/i для различных потенциалов ф0. В связи с тем, что энергия отталкивания зависит от дебаевской толщины двойного слоя и валентности ионов, значения ординат должны быть умножены на соответствующие коэффициенты, чтобы получить Vi в эрг/см2. Этот коэффициент в случае симметричного электролита (г=1) равен 1 для х — = 1-Ю7 саг1; при х == 1- 10е ел/-1
При расчетах полагали kT*=4 10“14 э?г.
коэффициент равен 0,1 и т. д. Для перехода от z — 1 к г = 2 требуется деление значений ординат на 4 и т. д.
Как следует из рис. 8, величина Vi в сильной степени зависит от фо-потенциала (кривые 1, 2). По мере увеличения значений x/i это влияние становится слабее. Сравнение кривой 3, полученной при условии постоянства заряда диффузной части
* Аналитическим путем выражение для энергии ионно-электростатическогО взаимодействия плоских частиц получено Муллером [293]. (Прим, ред.)
^'двойного слоя * [37], и кривой 2 указывает на то, что существен-гное различие между ними обнаруживается только при малых (расстояниях h.
; Для определения силы электростатического отталкивания Г Pi, отнесенной к единице площади, или электростатической [[.компоненты расклинивающего давления слоя Пг можно предложить различные пути [26,34,36,38]**. Так, при условии посто-^янства потенциала ф0 величину Р, находят дифференцированием формулы (25):
I dF I
В Pi — -~п~ == — nkT
к * dh H’Q=const
ze^d\ п] kT I J
as — 2nkT (ch ud — 1) (27)
К Из этого простого на вид уравнения вычислить значение Р, дГлля некоторого расстояния h весьма затруднительно, так как щпотенциал ф<; и произведение x/i связаны друг с другом слож-Шйым выражением (20).
Ж Приближенное уравнение для больших расстояний между Жчастицами. Приближенно зависимость между величинами ф0, Ж-^d и x/i можно найти при условии слабого перекрытия двойных Желоев, т. е. когда ф0 фа- В этом случае значение ф<г представ-Шдяется в виде суперпозиции потенциалов ф(й/2), соответствую-Жптих ненарушенному двойному слою [26, 36]. Связь между вели-Чинами ф(й/2) и фо для ненарушенного двойного слоя опреде-Жйяется при больших расстояниях уравнением (9):
{ гефо ) ,
\ 2kT )
^=4ехр(^А)
L Тогда для потенциала на середине расстояния между части-Йами имеем
td “ 2ф W2)
[ЛИ
/ гефо \ exMwd-
zeib .
-^- = 8еХр
Ч^)+1
(28)
% Раскладывая экспоненциальные функции формулы (27) (в ряд и ограничиваясь первыми тремя членами разложения,
X * Теория ионно-электростатического взаимодействия при постоянстве за-2 ряда поверхностей развита в работе [293]. (Прим, ред.)
* ** Согласно определению, данному Дерягиным [295], «расклинивающее
4 Давление тонкого плоскопараллельного слоя жидкости, разделяющего два '•’тела, равно давлению П, с которым (в дополнение к «нормальному» гидростатическому давлению в слое) действует в состоянии равновесия слой Жидкости на ограничивающие его тела, стремясь их раздвинуть». (Прим, ред.)
для силы ионно-электростатического взаимодействия получим:
(28а)
Сопоставляя уравнения (28) и (28а), найдем
Pt = — 64пйГ ехр (— и/;)
(286)
При тех же предположениях энергию электростатического взаимодействия можно вычислить из выражения:
л
TZ f п Л GiflkT
Vi— I P(dh——-—exp (— nh)
CO
(29)
Энергия взаимодействия сферических частиц. Расчет энергии отталкивания сферических частиц радиуса а при малых расстояниях между поверхностями проводят обычно так же, как и в случае параллельных пластин [20, 36]*. Рассмотрим общее уравнение:
- (30)
Для вычислений V* по уравнению (30), как правило, используют функции, приведенные в монографии [39]. Сложное точное выражение для энергии отталкивания как функции расстояния дано в работе [40]. Оно найдено разложением в ряд функции Vi (h). Ограничиваясь в разложении первыми двумя членами, нетрудно найти часто применяемое приближенное решение [41] для больших расстояний между частицами:
’ехо
s 8 (fe?)2 еа ехр ( — xfe) Р \ 2fe7 ]
1 e2z2 / zeib01 , , ' '
1ехрЫг)+1-1
Необходимо отметить, что энергия отталкивания сферических частиц прямо пропорциональна их радиусу. Поэтому понятны трудности, с которыми приходится сталкиваться при получении устойчивых тонкодисперсных систем. Так, если величина а мала, то энергия отталкивания может оказаться одного порядка с энергией теплового дви&ения.
Другое приближенное решение уравнения (30) получено для частиц с низким значением пртенциала фо при условии, что их
* См. также [294]. Прим, ред.
’ радиус гораздо больше толщины диффузного двойного слоя [36]’.
; V] = -^-In [1 + ехр(-хЛ)] (31а)
Учет коррекции уравнения Пуассона — Больцмана. Вычисле-1 ние распределения потенциала между двумя взаимодействую-s' щими плоскими частицами и свободной энергии двойных слоев. J с учетом объема ионов, зависимости диэлектрической постоян-? ной от напряженности поля и концентрации электролита, поля-[ ризации ионов электрическим полем двойного слоя, собственной s ионной атмосферы ионов и полостных эффектов предпринято ; Левиным и Беллом [25]. Численный анализ сложного интеграла авторами еще не завершен. Однако, принимая во внимание влияние различных факторов на распределение потенциала в двойном слое, следует ожидать более сильного уменьшения *•- электростатических сил отталкивания с расстоянием по сравне-f нию с закономерностью, предсказываемой уравнением Пуас-! сона — Больцмана. Вместе с тем, ниже будет показано, что в связи с противоположным действием ряда факторов, по край-: ней мере, для симметричного электролита, содержащего одно-! валентные ионы, коррекция уравнения Пуассона — Больцмана не вносит существенных изменений в теорию устойчивости лио-: фобных коллоидов.
Учет слоя Штерна. Полная интерпретация структуры двой-; ного слоя требует учета объема противоионов (минимального У расстояния 6 от границы раздела фаз до центра тяжести заря-? дов противоионов) и их специфической адсорбции. Поэтому f плоскость, от которой начинается двойной слой и тем самым об-;• ласть действия сил отталкивания, должна быть сдвинута на рас-: стояние х = 6. Таким образом, для расчета электростатического ' отталкивания имеет значение не потенциал фо, а потенциал слоя . Штерна фв.
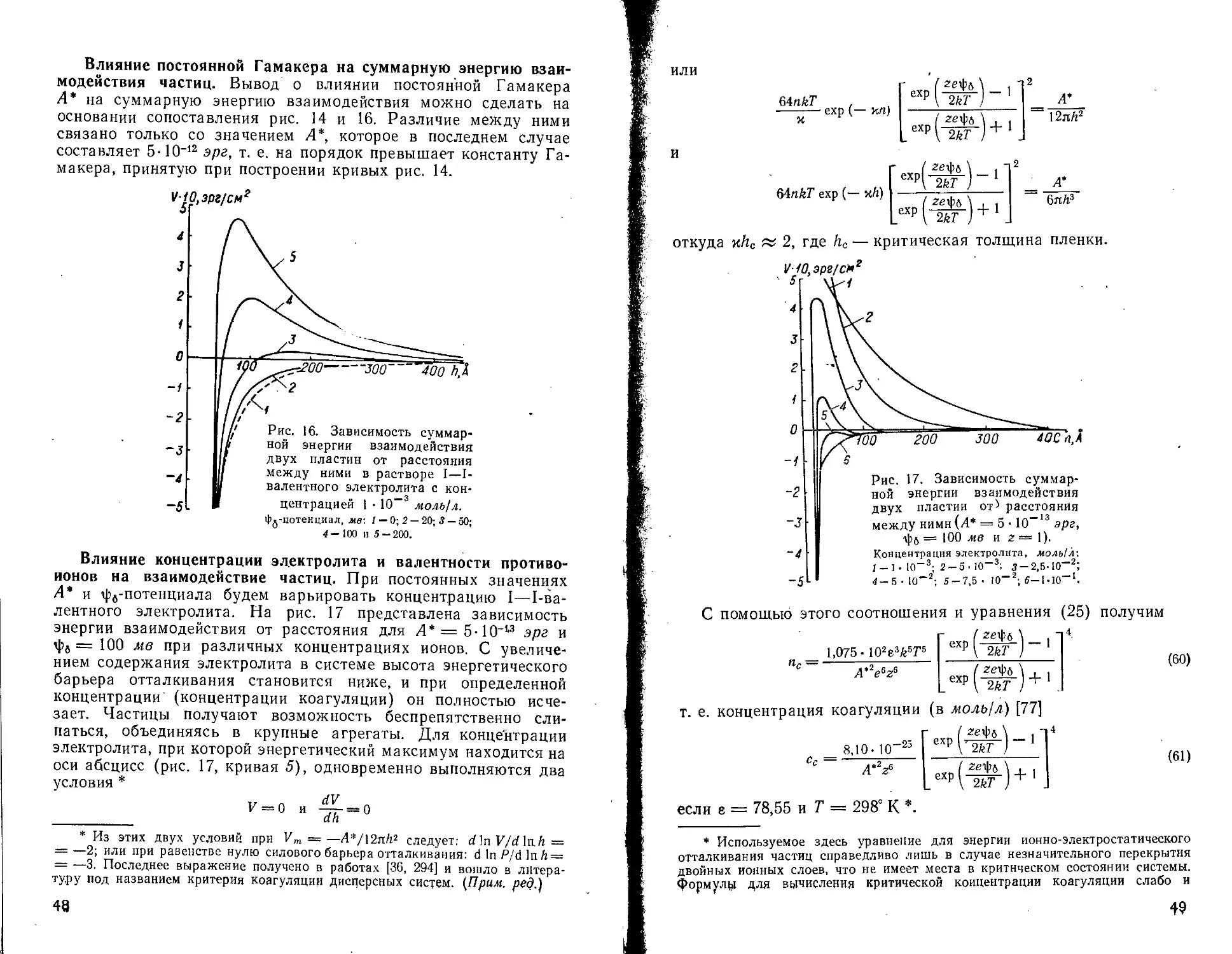
; Не рассматривая особый случай специфической адсорбции ’ коионов, заметим, что ввиду неравенства фо Ф« наличие слоя [ Штерна приводит к уменьшению сил электростатического от-= талкивания. Вместе с тем, при возникновении слоя Штерна про-f является специфическое действие ионов; оно находит выражение в так называемых лиотропных рядах и обнаруживается, например, при’изучении коагуляции золя иодида серебра, вызываемой - добавлением различных нитратов щелочных металлов [42, 43]. ’’ Поскольку потенциал фв в противоположность потенциалу ф0 подвержен сильному влиянию электролитов и уменьшается при возрастании содержания ионов, уравнение для энергии отталки- вания содержит наряду с величиной концентрации еще параметр, зависящий ОТ п:
Vi ~ ~~ i ('Ъб, tyd) Л
Рис. 9. Зависимость энергии ионно-электростатического отталкивания двух пластин с потенциалом поверхностей 50 мв от расстояния h. Концентрация электролита в дисперсионной ' среде, моль/л: г _ 1-10—3- 2-5- 10~3;
3 — 2,5 10~2.
То же самое относится и к приближенным уравнениям, в которых гр0 необходимо заменить на *.
Влияние концентрации электролитов на энергию отталкивания. Устойчивость лиофобных коллоидов к флокуляции в значительной степени определяется концентрацией ионов1в дисперсионной среде. Из приведенных выше теоретических представлений следует, что дестабилизация может быть вызвана различными причинами: 1) уменьшением толщины двойного слоя при увеличении содержания электролита; 2) сдвигом равновесия диссоциации гомогенных групп на поверхности частиц, приводящим непосредственно к изменению потенциала ф0; 3) образованием при высоких концентрациях электролита штерновского слоя, в котором происходит падение потенциала до величины фв.
Толщина двойного слоя и радиус действия ионно-электростатических сил всегда уменьшаются с ростом концентрации, так как, если исключить из рассмотрения слой Штерна, значение х прямо пропорционально Уп. Вместе с тем, согласно уравнению (26), вывод которого основан на предположении о постоянстве потенциала, по мере увеличения содержания ионов энергия отталкивания изменяется бо
лее резко (рис. 9). В достаточно концентрированных растворах электролитов заполнение штерновского слоя и падение потенциала между плоскостями х == 0 и х — 6 возрастают. Поэтому следует принять, что формирование штерновского слоя приводит к заметному уменьшению энергии ионно-электростатического отталкивания.
МОЛЕКУЛЯРНОЕ ВЗАИМОДЕЙСТВИЕ
ДИСПЕРСНЫХ ЧАСТИЦ
Силы притяжения, которые действуют между отдельными атомами и молекулами, называются силами Ван-дер-Ваальса. Они много слабее сил химической связи, не могут быть насыщены и в первом приближении аддитивны. Благодаря суммированию по всем атомам, содержащимся в объеме коллоидных и
* Влияние слоя Штерна на взаимодействие в коагуляцию дисперсных частиц изучено в работе [401]. (Прим, ред.)
макроскопических чаСтиЦ, МолёкуЛярйые силы, обладая большим радиусом действия, играют важную роль в процессах коагуляции дисперсных систем и адгезии.
Взаимодействие атомов и молекул. Силы Ван-дер-Ваальса могут слагаться из трех компонент: 1) диполь-дипольного взаимодействия (силы Кеезома); 2) индукционного взаимодействия (силы Дебая); 3) дисперсионного взаимодействия (силы Лондона). Существование первых двух типов взаимодействий предполагает наличие по крайней мере наведенного дипольного момента у обеих молекул. Между неполярными молекулами действуют только дисперсионные силы, которые обусловлены флуктуациями зарядов, возникающими вследствие движения электронов. Электронные флуктуации в атомах или молекулах приводят к появлению изменяющихся во времени диполей. Взаимное влияние флуктуационных диполей вызывает фазовый сдвиг колебаний (при малых расстояниях он составляет 0°) и поэтому две неполярные молекулы всегда притягиваются друг к другу.
Для расчета вклада дисперсионного взаимодействия в суммарную энергию молекулярного притяжения рассматривают два атома как связанные изотропные гармонические осцилляторы, имеющие три измерения [44—48]. Для двух атомов, обладающих поляризуемостями aoi и ссог и частотами voi и V02 в основном состоянии, получено приближенное уравнение:
V, , 3ЛЙ 'Vol'Voz
-6 । ао1^о2 Vj2)
• Vol + V02
Здесь Vd —энергия дисперсионного взаимодействия; г — Межатомное расстояние; fi —постоянная Планка: h.— h/2n, для двух одинаковых атомов формула (32) упрощается
Vd =* - voao (32а)
а для Двух идентичных молекул, каждая из которых имеет з электронных осцилляторов, согласно [49], имеем
*zd=s:-'2^~'voaol/s (326)
где ==vv— характеристическая частота. Частота в основном состоянии атомов зависит от заряда электрона е и его массы те:
v0 = л/(33) 2л у теа0
Следовательно
Такая форма Записи наиболее удобна, поскольку в формуле (32в) отсутствует трудно определяемая величина vo.
Статическую поляризуемость можно получить из результатов оптических наблюдений
3 «о ~ 1 М а0 —------- —г,—--- (34)
2лУй Пд + 2 р
где По — коэффициент преломления, который находят путем экстраполяции на область больших длин волн; М и р — молекулярный вес и плотность вещества, соответственно.
При вычислениях часто делается предположение, что произведение 2nfivo равно величине энергии ионизации. Из сравнения значений 2nfivo, найденных оптическим методом [48], и энергии ионизации / [50] видно (табл. 2), что это предположение не всегда оправдывается.
Таблица 2
Сопоставление величин энергии ионизации I и произведения 2nfiv0, найденного оптическим методом, для некоторых простых молекул
Молекулы глйуо-Ю11, эрг * 2.10й, эрг 7/2nftv0, %
Не 4,10 3,84 94
о2 2,38 2,00 84
Na 2,73 2,46 90
Н2О 2,16 2,01 93
ecu 2,50 1,76 71
с2н4 1,63 1,68 103
с2нв 2,06 1,88 91
с6нв 1,68 1,54 92
* Величины Vo определены по уравнениям (33) и (34).
При вычислении частоты колебаний vo сложных молекул из опытных данных измерения молекулярной рефракции Ra в области видимого света получены результаты [50], представленные в табл. 3. Как видно, такой метод приводит к еще большему
Таблиц а 3
Сопоставление величин энергии ионизации I
и произведения 2«5v0, полученного измерением молекулярной рефракции, для некоторых сложных веществ
Молекулы До, СмЗ/моЛЬ vo.IO“16, сек*"1 гяйуо'Ю11, эре Z-1011, эре 2/2nftvo, %
Бензол ..... 26,6 6,785 0,515 1,54 300
Октан ...... 39,0 0,645 0,422 0,425 1,64 388
л-Ксилол .... 38,6 0,650 1,44 340
Толуол 33,6 0,695 ' 0,455 1,43 315
Циклогексан . . 27,5 0,767 0,503 1,71 1,36 340
Хлорбензол . . . 62,0 0,510 0,334 407
различию между произведением 2nfivo и величиной энергии ионизации I.
Справедливость уравнения (32) ограничена; оно выполняется от значений расстояний, соизмеримых с размерами диполя (2—3 А), до значений г, меньших, чем лондоновские длины волн А,, = cv“],где с — скорость света.
При расстояниях, превышающих лондоновскую длину волны, наблюдается эффект запаздывания, связанный с конечной скоростью распространения электромагнитных волн: для двух взаимодействующих диполей изменяется сдвиг фаз — он перестает быть равным 0° — и наступает ослабление энергии взаимодействия.
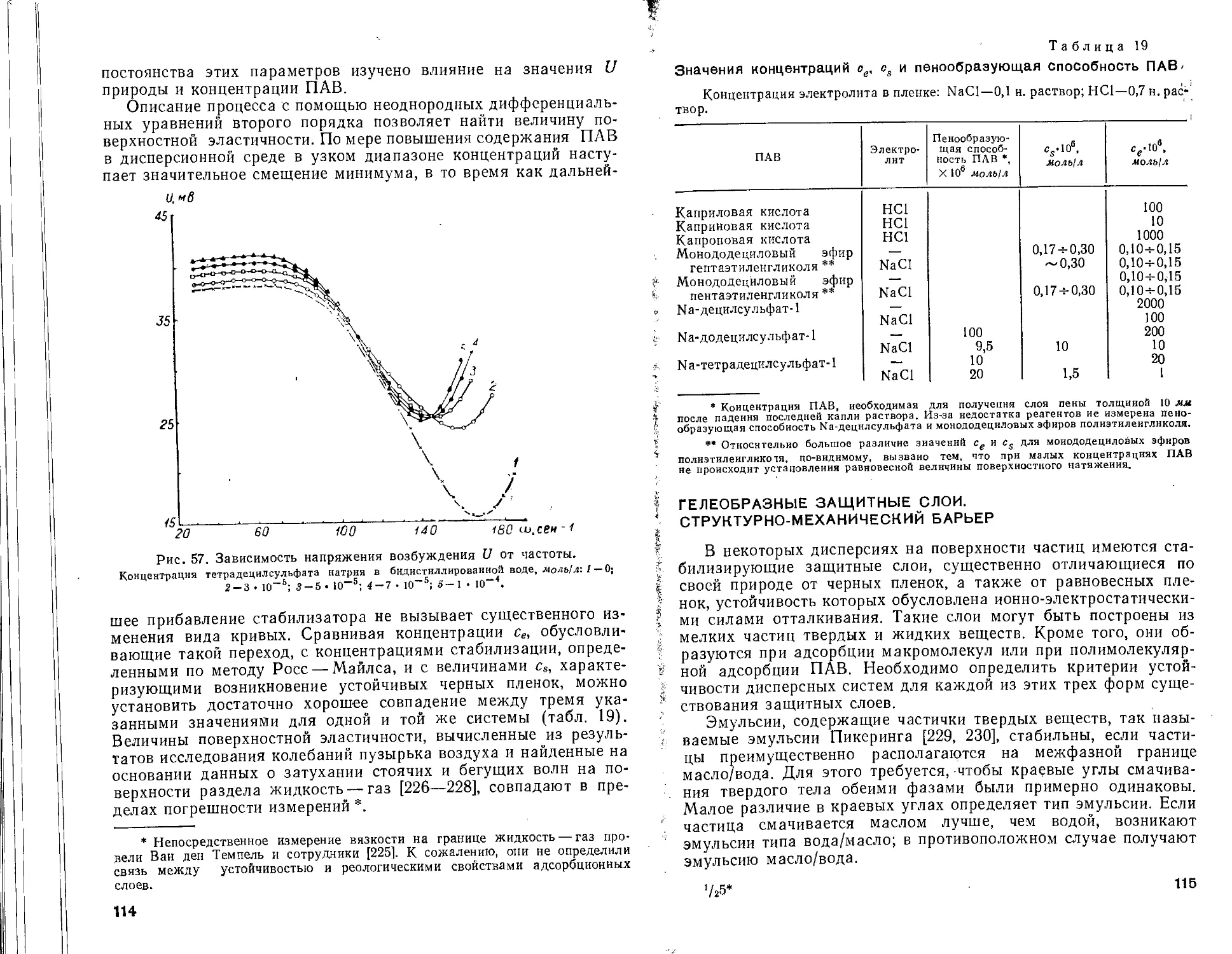
Силы притяжения макроскопических частиц. Уже в 1932 г. Кальман и Вильштеттер [51] указали на то, что благодаря суммированию межатомных взаимодействий силы притяжения макроскопических частиц должны быть существенно больше сил притяжения отдельных атомов или молекул. Могут быть предложены два пути для вычисления энергии взаимодействия конденсированных фаз. Де Бер [52] и Гамакер [53] исходили из аддитивности атомных сил взаимодействия (микроскопическая теория). Лифшиц [54] избрал макроскопический способ рассмотрения, при котором не предполагается аддитивность дисперсионных сил, справедливая только для разреженного газа. Ренне и Ниибер [55] недавно установили, что с помощью микроскопической теории также могут быть получены результаты, согласующиеся с данными макроскопической теории, если неаддитивность учитывается введением соответствующих поправочных членов. В последнее время Лангбейном [56] на основе микроскопической теории и учета макроскопической экранировки флуктуационного поля выведено общее уравнение для энергии взаимодействия частиц.
Микроскопическая теория. Сферические частицы. Согласно работе [53], для энергии молекулярного притяжения двух сферических частиц радиусами «1 и а2, каждая из которых содержит q атомов в 1 см3, имеем ,
Vm = — j j (q'^lr6) dv2 (35)
«2
где Vt и v2 — объемы обеих частиц; dvi и dv2— элементарные объемы,' расстояние между которыми равно г; р— постоянная Лондона:
₽ = й^оао (36)
Интегрирование формулы (35) приводит к уравнению:
.. At 2^2___________________________2а1а2________
Vm = 6 \ А2 + 2a(A + 2л2Л "* h2 -f- 2ath + 2a2h + 2Й1а2 +
in h2 + 2а,/г + 2a~h
h2 + 2aih + 2a2h + 2eJa2 ) ' *
Здесь А — постоянная Ван-дер-Ваальса — Гамакера (Д = == рзт2<72); h — кратчайшее расстояние между поверхностями частиц.
Для сфер одинакового радиуса (ai = а2) уравнение (37) принимает вид:
У = _ Д Г 2а2 .______2а1_____, , ft2 + 4aft 1 .
т 6 L Л2 + iah h2 + 4ah + 2а2 + п h2 + 4ah + 2а2 J 1 '
Дальнейшее упрощение возможно, если радиус частиц гораздо больше, чем расстояние h между ними:
у = — -Аа-т 12/г
(38)
Плоскопараллельные пластины. Формула для энергии молекулярного взаимодействия, отнесенной к единице площади двух бесконечных плоскопараллельных пластин толщиной б, выведена Фервеем и Овербеком [26]:
(39)
При некоторых предположениях уравнение (39 )можно упростить.
Рассмотрим прежде всего толстые пластины, когда б h. В этом случае
и сила молекулярного притяжения
р - А
т dh &ths
Напротив, если расстояние между пластинами значительно больше их толщины, т. е. б h
ДХ2
Как уравнения для сферических частиц, так и уравнения для плоскопараллельных пластин относятся к взаимодействию в вакууме и применимы только для малых расстояний, при которых можно пренебречь эффектом электромагнитного запаздывания.
Влияние дисперсионной среды на энергию молекулярного взаимодействия. В суспензиях и эмульсиях микрообъекты разделяет более или менее толстая прослойка конденсированной среды. Этот факт учтен уже при вычислениях, проведенных Га-макером [58], причем, как и ранее, предполагалась аддитивность действия дисперсионных сил. Хотя такое предположение для конденсированных систем не выполняется, полученные результаты по крайней мере для малых расстояний можно рассматри
вать в качестве рабочей гипотезы при анализе взаимодействия дисперсных частиц *.
Пусть две частицы сначала удалены на бесконечно большое расстояние друг от друга, на котором не проявляется их взаимодействие. Приближая частицу 2 на расстояние h, получим изменение ее энергии взаимодействия
И12- V02 '
где V12 — энергия взаимодействия двух частиц; V02 — энергия взаимодействия частицы 2 с объемом дисперсионной среды, находящимся перед сближением на расстоянии h от частицы 1.
Этот объем среды необходимо перевести от частицы 1 на бесконечное расстояние, для чего совершить работу
Voo — V01
где Vol — энергия взаимодействия частицы 1 с объемом дисперсионной среды, расположенным на расстоянии h от ее поверхности: Voo — энергия взаимодействия переведенного объема с дисперсионной средой.
Тогда общее изменение энергии взаимодействия составит
V — V[2 + Too— Т02— Vol (40)
а для двух одинаковых частиц
V = Уц + Voo — 2Voi (40а)
При лаком выводе предполагается, что взаимодействие одного вещества не изменяется в присутствии второго вещества.
Сравнивая изменение энергии V с энергией молекулярного взаимодействия, получим аналогично уравнению (39а) для двух пластин бесконечной толщины
у = _ m 12лЛ2
где А* = Ai 4- До — 2До1 — сложная константа Ван-дер-Ваальса— Гамакера; Ai и До — постоянные взаимодействия Ван-дер-Ваальса— Гамакера для дисперсной фазы, состоящей из частиц 1, и дисперсионной среды, соответственно; A»i — постоянная взаимодействия фазы и среды.
В связи с тем, что величины vo для отдельных веществ слабо различаются (см. табл. 2 и 3), в соответствии с уравнениями (32), (36) и (38) можно записать
Aoi-PMo)*'*
откуда
А* = (л’^-Л’/2)2 (41)
* Более строго задача о молекулярном взаимодействии коллоидных частиц в жидкой дисперсионной среде изучена Лифшицем, Дзялошикским и Питаевским [67], а также Русановым и сотрудниками [296—299]. (Прим, ред.)
Следует еще раз подчеркнуть, что определение сложной константы Ван-дер-Ваальса — Гамакера предполагает аддитивность постоянных для отдельных компонентов.
Влияние гомогенных адсорбционных слоев на энергию молекулярного взаимодействия. Взаимодействие двух сферических частиц радиуса а, окруженных гомогенными слоями третьего вещества толщиной рассмотрено Волд [57], а также Нинхе-мом и Парсегианом [58—60]. Для энергии молекулярного притяжения получено уравнение
Vm = ~ [(4 - А*)2 Н33 (х, у) + (Л* - Л'/2)2 (х, у) +
+ 2(ЛУ* - Лз/2)(Л‘'2 - Л1/’) Н13 (х, у)] (42)
где Аз — постоянная Ван-дер-Ваальса — Гамакера для адсорбционных слоев 3; Н(х, у)—функция Гамакера [выражение в скобках в уравнении (37)]
Н (х - У . I - - У_____________l 2 и А-ху + х
( ’ У) хг _|_ Ху 4. х + хг _|_ Ху х _|_ у + х2 + ху -f- х + у
Символы х и у введены для упрощения записи, причем х = й/2щ; у = а2/аг, Oi < а2; h — минимальное расстояние между поверхностями частиц (рис. 10).
Рис. 10. Схема к расчету молекулярного взаимодействия пластин в неполярной (а) и полярной (б) дисперсионной среде при наличии на их поверхности ориентированных адсорбционных слоев:
О — дисперсионная среда; / — дисперсная фаза; 3 — адсорбционный слой толщиной б/, 4 — слой толщиной ба, образованный полярными группами вокруг коллоидных частиц.
Для случая х < 1
Л (х, у) = —
х О + У)
В уравнение (43) вместо х и у необходимо подставить следующие величины: х = (h — 26f)/2(a + di) и у — I для Н33; х — h/2a и (/=1 для #и; х=(й —6j)/2a и у = 1 -фbja для Д1з.
При этом выражение (42) принимает вид:
- -нН - Йяг+«' - +
+ 4 W’ - w Wf - 4‘) ТГ-4‘+<./«>] <42а>
Аналогично, энергия молекулярного взаимодействия двух, плоскопараллельных пластин бесконечной толщины вычисляется по формуле*:
1 [(4Л-4)2
12л (A-2S,)2
(4 - а'(^ 2(4 - 4) (4 - л'/*)
Л2 + (Л — д,)2
(44)
Для дисперсных систем с неполярными дисперсионными средами (см. рис. 10, а), например, для эмульсий типа вода/ма-сло, можно положить Ао ж А3, так как молекулярное взаимодействие углеводородных Цепей поверхностно-активных веществ друг с другом и с масляной средой практически одинаково**. Тогда
vw = -72-(4-^)2t (45)
или
и _ А*а Ч
Vm — ~ Д2Л (45а)
а для параллельных пластин
В уравнения (45) и (456) входит величина расстояния между поверхностями частиц, которую обычно определяют с помощью оптических и электрических измерений, поскольку граница сильно отличных значений коэффициентов преломления или диэлектрических постоянных совпадает с поверхностью дисперсных частиц.
При условии Аз ~ Ai, выполняющемся, например, в случае эмульсий типа масло/вода, суспензий парафина, полистирола (см. рис. 10,6), для сферических частиц
v______1 (A'l, .A'ltf а + ^ A"(a + 6t) (45 .
Vm~ 12 Ио Аз) Д —2d, 12 (й —2d,) 14&В)
и для пластин
А*
Vm = 12л (й-2d,)2' <45г)
* Эта задача решена Нерпиным [300, 301]. (Прим, ред.)
** Переход от сравнительно толстого адсорбционного слоя к моиомоле-кулярному является некорректным, поскольку микроскопическая теория учитывает парные взаимодействия путем интегрирования в пределах от оо до od, где б0 — некоторая толщина порядка 2—3 молекулярных размеров. (Прим, ред.)
Как правило, радиус частиц существенно больше толщины адсорбционного слоя и можно пренебречь величиной Si в числителе. Необходимо, однако, принимать во внимание, что значение h отсчитывается от поверхностей частиц и равно расстоянию, определяемому оптическим методом. Заметные отклонения от зависимости, задаваемой простым уравнением (38), по-видимому, проявляются, если Ао A i ¥= А3. Следует поэтому, исходя из выражения (42а), проверить, насколько сильно сказывается влияние адсорбционных слоев на энергию Vm.
При рассмотрении молекулярного взаимодействия частиц, имеющих адсорбционные слои, предполагалось слабое различие во взаимодействии полярных групп молекул поверхностноактивных веществ и молекул среды. Такое предположение нельзя считать обоснованным. Например, взаимодействие сульфонатных групп друг с другом должно сильно отличаться от взаимодействия их с молекулами1 воды. Принимая, что полярные группы образуют вокруг коллоидных частиц слой 4 толщиной (см. рис. 10), характеризуемый постоянной At, для энергии молекулярного взаимодействия получают -более сложное выражение, в котором учтена ориентация адсорбированных молекул поверхностно-активных веществ [57]. Так, для сферических частиц, схематически изображенных на рис. 10, а, имеем
- ТТ ~ 42)2 3 (х, у) + (л* _ 4)2 4 (х> у} +
+ (Л*/1 - Л^)2 Н1 1 А + 2 И2 - 4'*) И’ - 4 (х, у) +
+2(4 - Л*'») (Л7’ - А*) Н{ 4 (х, у) + 2 (Л^!—Л^2)(л'/!—Л4г) н\ з $ (45д)
где значения аргументов х и у для различных Hi, выбирают
следующим образом:
h — 26] — 2б2
з Х 2 (а + б, + б2) ’ г/== 1
- h - 2б2
/Г 4 4 1 2 (а + б2) ’ г/= 1
R . h — б] — 2б2 о + б| + б2
2(а + 62) ’ а + б2
h
1 Х~ 27*
^14 й-б2 Х ’ 2а ’ CL 4- &2 г/ = а
Я13 h — б] — б2 Х 2а g-4-б] +бг v а
При ориентации полярных групп в сторону объема дисперсионной среды (рис. 10,6) в уравнении (45д) необходимо поменять местами На и Htt, а также Si и 6г- Поскольку постоянные Гамакера в формулы (42а) и (45д) входят в виде разно-
Рис. 11. Схема к расчету взаимодействия одной молекулы с шаровым сегментом конденсированной фазы.
Стей, очевидно, что поверхностно-активные вещества или I макромолекулярные защитные слои должны оказывать сильное : влияние на взаимодействие коллоидных частиц. К сожалению, ; Точная экспериментальная проверка этих уравнений в настоящее время невозможна из-за отсутствия данных о значениях i постоянных А для отдельных веществ.
Влияние электромагнитного запаздывания на энергию моле-< кулярного взаимодействия. Ввиду конечной скорости распрост-j ранения электромагнитных волн при расстоянии между двумя : атомами, сравнимом по порядку величины с лондоновской дли-ной волны, фазовый сдвиг флуктуи-J рующих диполей отличается от 0° и f дисперсионные силы уменьшаются быстрее, чем это следует из уравне-; ния (32). Казимир и Польдер [61] вы-числили, что для межатомных рас-> стояний г Ki дисперсионная энергия изменяется пропорционально 1/г7, а не 1/т6, причем поправочный коэффи-• циент, который необходимо ввести • в уравнение (32), является функцией межатомного расстояния:
3nfivoa§ /2Яг\
Vd~ 2r* '\Ьг)
Согласно работе Овербека [39], функцию f(2nr/X;j для различных расстояний можно представить в виде выражений:
и
2л г
2,45Аг 2.04Л/
2лг 4я2г2
при
2лг Хг
(46)
при
3<
2лг Лг
(47)
На основе этих формул выведены уравнения для определения энергии молекулярного взаимодействия двух плоскопараллельных пластин [62] и молекулярной составляющей расклинивающего давления тонких пленок [63]. Исходя из аддитивности взаимодействий, Шелудко [63] получил выражение для энергии, суммируя взаимодействие одной молекулы со всеми молекулами, находящимися в шаровом сегменте конденсированной фазы большого объема (рис. 11).
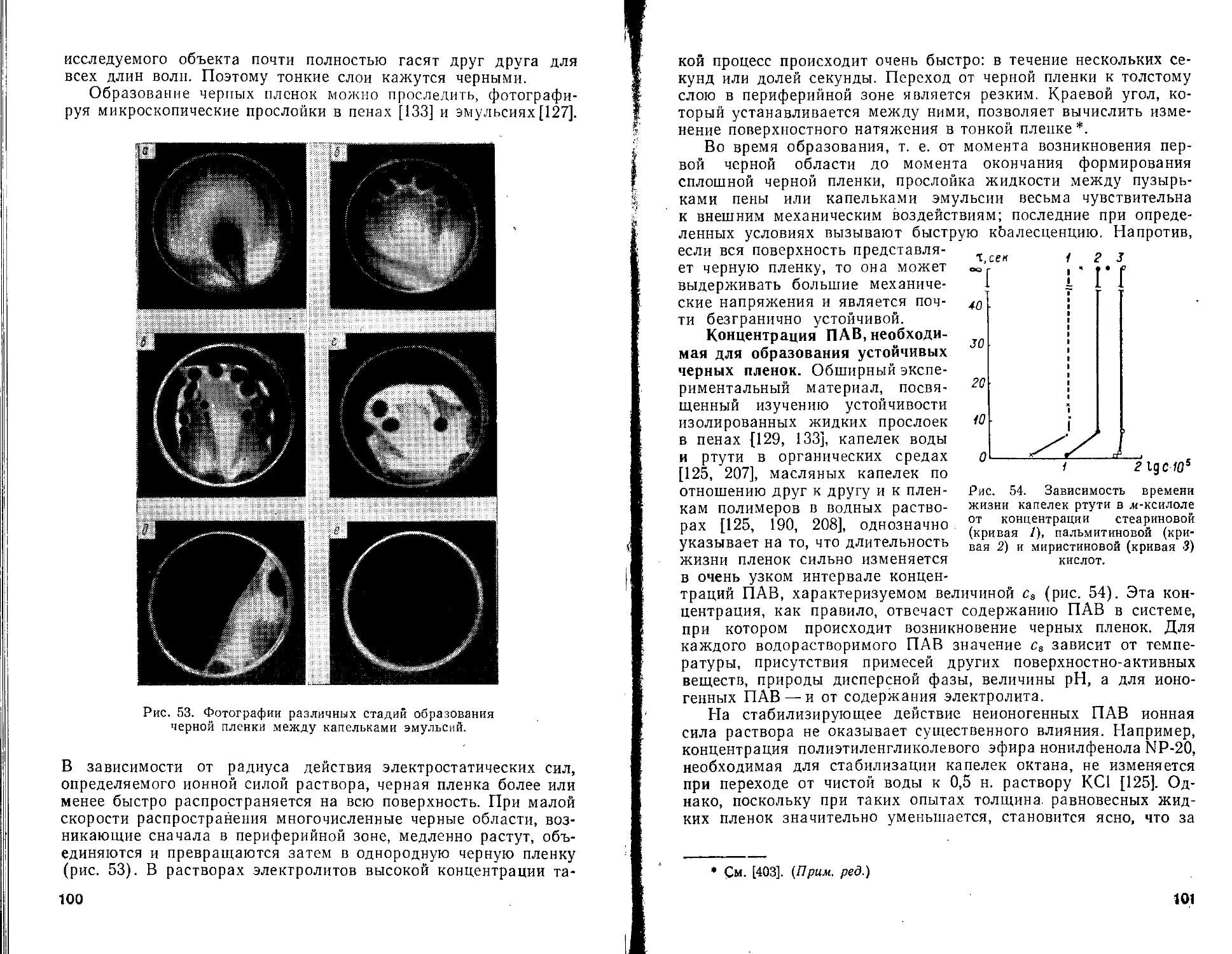
В соответствии с уравнениями (46) и (47) рассматривают две области расстояний
ЗХ;
2л
ЗХ; 2п
В первом случае интегрирование проводят в пределах г = h аг — ЗХ//2л, во втором — от г = 3?ч/2л до г = оо. В результате
имеем для 0 < h < 3A;/2jt
- - тя? [1 »' - °'28 Т + »•1735 (тг)’ -
- 0,028 (~У + 0,00193 (^-)4] (48)
и для ЗЛ(/2л < h < оо
_ А 0.098Л?
Vm==~ 72ЙЛ2 ' (2лЛ)2 ^49)
Дифференцируя выражения (48) и (49) по расстоянию, получим формулу для силы притяжения, отнесенной к единице площади двух плоскопараллельных частиц
P"!=='6^-f1’01 “0’28^ + 0>0143('Т^У “°’00196('Т^У] (50)
6л/^ L м \ Л/ ) \ А./ / j
и соответственно
= л4з-[1,47-А--0,811б(-г^гУ] (51)
бл/г1 L 2л/г \ 2л/г ) j ' '
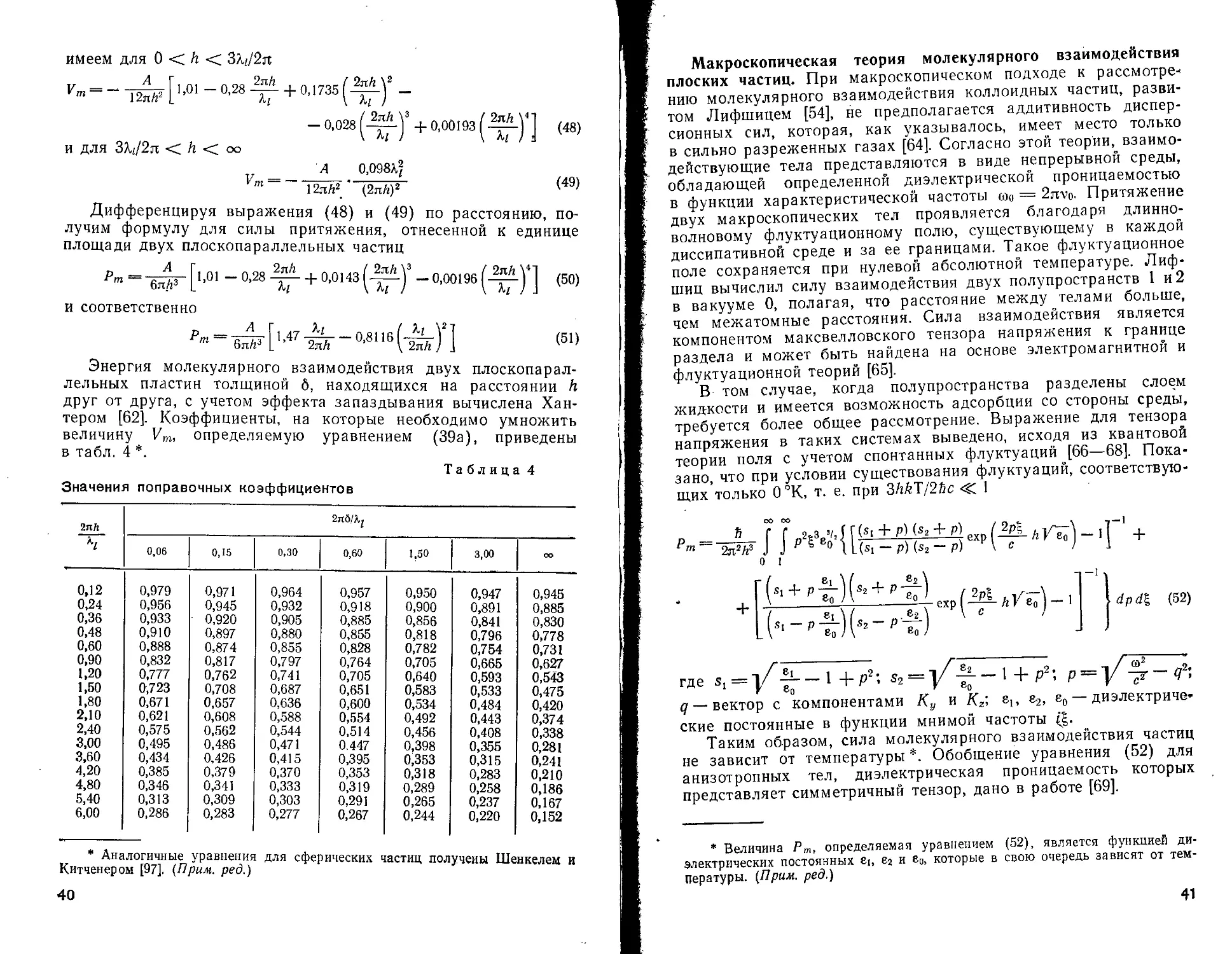
Энергия молекулярного взаимодействия двух плоскопараллельных пластин толщиной б, находящихся на расстоянии h друг от друга, с учетом эффекта запаздывания вычислена Хантером [62]. Коэффициенты, на которые необходимо умножить величину Vm, определяемую уравнением (39а), приведены в табл. 4 *.
Таблица 4
Значения поправочных коэффициентов
2яЛ h 2it6az
0,06 0,15 0,30 0,60 1,50 3,00 СО
0,12 0,979 0,971 0,964 0,957 0,950 0,947 0,945
0,24 0,956 0,945 0,932 0,918 0,900 0,891 0,885
0,36 0,933 0,920 0,905 0,885 0,856 0,841 0,830
0,48 0,910 0,897 0,880 0,855 0,818 0,796 0,778
0,60 0,888 0,874 0,855 0,828 0,782 0,754 0,731
0,90 0,832 0,817 0,797 0,764 0,705 0,665 0,627
1,20 0,777 0,762 0,741 0,705 0,640 0,593 0,543
1,50 0,723 0,708 0,687 0,651 0,583 0,533 0,475
1,80 0,671 0,657 0,636 0,600 0,534 0,484 0,420
2,10 0,621 0,608 0,588 0,554 0,492 0,443 0,374
2,40 0,575 0,562 0,544 0,514 0,456 0,408 0,338
3,00 0,495 0,486 0,471 0.447 0,398 0,355 0,281
3,60 0,434 0,426 0,415 0,395 0,353 0,315 0,241
4,20 0,385 0,379 0,370 0,353 0,318 0,283 0,210
4,80 0,346 0,341 0,333 0,319 0,289 0,258 0,186
5,40 0,313 0,309 0,303 0,291 0,265 0,237 0,167
6,00 0,286 0,283 0,277 0,267 0,244 0,220 0,152
для сферических частиц получены Шенкелем и
* Аналогичные уравнения Китченером [97]. (Прим, ред.)
Макроскопическая теория молекулярного взаимодействия плоских частиц. При макроскопическом подходе к рассмотрев нию молекулярного взаимодействия коллоидных частиц, развитом Лифшицем [54], не предполагается аддитивность дисперсионных сил, которая, как указывалось, имеет место только в сильно разреженных газах [64]. Согласно этой теории, взаимодействующие тела представляются в виде непрерывной среды, обладающей определенной диэлектрической проницаемостью в функции характеристической частоты <оо = 2jivo. Притяжение двух макроскопических тел проявляется благодаря длинноволновому флуктуационному полю, существующему в каждой диссипативной среде и за ее границами. Такое флуктуационное поле сохраняется при нулевой абсолютной температуре. Лифшиц вычислил силу взаимодействия двух полупространств 1 и2 в вакууме 0, полагая, что расстояние между телами больше, чем межатомные расстояния. Сила взаимодействия является компонентом максвелловского тензора напряжения к границе раздела и может быть найдена на основе электромагнитной и флуктуационной теорий [65].
В том случае, когда полупространства разделены слоем жидкости и имеется возможность адсорбции со стороны среды, требуется более общее рассмотрение. Выражение для тензора напряжения в таких системах выведено, исходя из квантовой теории поля с учетом спонтанных флуктуаций [66—68]. Показано, что при условии существования флуктуаций, соответствующих только О °К, т. е. при ЗЛАТ/2ЙС С 1
Р = Ъ- - |
О
exp
dpdl (52)
где 3.=]/ Т--1 +р2-. з2 = у ^-1 + р2; Р = 1/^-9г;
V С'О Т CQ v L
q— вектор с компонентами Ку и /Сг; eIt е2, е0— диэлектрические постоянные в функции мнимой частоты
Таким образом, сила молекулярного взаимодействия частиц не зависит от температуры*. Обобщение уравнения (52) для анизотропных тел, диэлектрическая проницаемость которых представляет симметричный тензор, дано в работе [69].
* Величина Рт, определяемая уравнением (52), является функцией диэлектрических постоянных в], е2 и е0, которые в свою очередь зависят от температуры. (Прим, ред.)
Упрощение выражения (52) возможно, если рассматривать либо очень малые, либо очень большие расстояния по сравнению с длинами волн, соответствующими последней линии в спектре поглощения каждого вещества (лондоновская длина волны). Для h С X; получим
Рт = .А.з Г Гх2 Г+ ео) + е°Ьехр x-lldxdl (53)
т I6n2/i3 J J L (61 — е0) (е2 — е0) 1
о о
а для расстояний h А.;
he
m 32л2/;4 Ve00 J
i I Г (SO1 + p) (sos+ p) pyn Y
>2 L («01 - p) («02 - P) P
„ 1 _ 801 ) I „ I g°2 )
So a + P ~— «02 + p ~—
---------е<юА-------------г™/ ex₽ x _ ( eO2 \
«02 -- P ~Z I
800 /
dp dx (54)
soi
Здесь s01 — у g°° 1 p2; s02 — у 1 -|-p2; e01, e02, —
статические диэлектрические проницаемости.
Для двух одинаковых частиц eOi = еог и уравнение (54) еще более упрощается:
Рт = ф М = А (55)
240/г4 КеОо \ е01 + еОо / \ е00 / /г4
Функция q)(eoi/eoo) табулирована Лифшицем [54], а расчеты при eoi е02 проведены Деверо и Дебройном [70]. Для взаимодействия двух металлических пластин в среде с диэлектрической проницаемостью ем функция <р(оо) = 1, так как eoi = = еог = оо. Тогда
hen2 240/г4
(55а)
Такое же выражение найдено Казимиром [61], исходя из микроскопической теории, при еоо = 1- Для практического использования целесообразно записать формулу (53) в ином виде. Считая единицу в квадратных скобках малой величиной по сравнению с членом, содержащим ехр х и, кроме того, проводя интегрирование по х, получим интеграл по £ [или в уравнении (54) по р]:
т UnW J (ei + eo)(e2 + eo) <56)
0
Микроскопическая теория Ренне и Ниибера. Ренне и Ниибер [55] дали для силы молекулярного взаимодействия атома и конденсированной фазы, ограниченной с одной стороны, точный
вывод формулы, в котором путем введения поправочных коэффициентов учитываются взаимодействия трех, четырех и т. д. частиц. Это уравнение соответствует выражению (53) в случае сильно разреженной среды. Таким образом, микроскопическая и макроскопическая теория равноценны, если в первой принимается во внимание неаддитивность взаимодействий. Для моделей атом — стенка и двух плоскопараллельных пластин Ренне и Ниибер вывели простые приближенные формулы, исходя из парных и тройных корреляций.
Для первой модели зависимость молекулярной силы от расстояния при h < Ki характеризуется уравнением
/3 е0,+ 1 3X1
4р-~ [1 ~ “ ТЛ (57)
а для второй модели
n2fivft<72an Г / е01 + 1 3 \1
[1 " - 2 )J (58)
Величина, вычитаемая из единицы в квадратных скобках, отвечает поправочному коэффициенту неаддитивности взаимодействий.
В табл. 5 сопоставлены приближенные уравнения для силы молекулярного взаимодействия пластин при больших и малых расстояниях h.
Из уравнений макроскопической теории, соответствующих малым расстояниям между поверхностями, следует, что в обратных системах 1—0—1 и 0—1—0 молекулярные силы практи-
СО
чески равны друг другу при равенстве интегралов J для о
частиц 1 и 2. Последнее наблюдается в случае веществ, имеющих во всем интервале частот одинаковые зависимости диэлектрических постоянных. Величина [(ei — ео)/(еГ-ф ео)]2 в уравнении (56) принимает значение от 0 до 1 и изменяется для многих дисперсных систем в узком интервале от 0,9 до 1. Поскольку частота vo совпадает по порядку величин у различных веществ (2-10'5 сек.~1 для воды; 1,3-1015 сект1 для бензола; 1,5-10!5 сект1 для ртути), можно оценить значение К = fivo/Зл2. Отсюда, для постоянной Ван-дер-Ваальса — Гамакера А = блК получим среднее значение, равное 4-10~13 эрг.
Переход от энергии взаимодействия пластин к энергии взаимодействия искривленных поверхностей производится с помощью соотношения, выведенного Дерягиным [71]*
ОО
Vsm = G $Vm(h)dh (59)
л
* См. также [302]. (Прим, ред.)
где G — так называемый коэффициент формы, который для взаимодействия пластины с шаром радиуса а равен 2ла, а для двух шаров составляет ла.
Таблица 5 формулы для вычисления сил молекулярного взаимодействия двух пластин
Соотношения между диэлектрическими постоянными
Литература
е01 — е02 ИЛИ
8i = е2
h
h
8л2Л3
801 — 8q2 ““ 00
ir2fiv0<72a§
W
El + 1
e, — 1
54
55
8л2/г3 f 0 n2fiv0<72a2 / 5 4/P 3n<?ao
(56)
54
55
®01 — ®02 ИЛИ
81 — 82
Bqi — 802 = OO
n2hc / e01 — e00 \2 / Bui
240 h4 У~ё^ \ Eoi + 800 / \ e00
л2 he
240 h4 /^7
(55а)
СУММАРНЫЕ СИЛЫ ВЗАИМОДЕЙСТВИЯ ЧАСТИЦ.
УСТОЙЧИВОЕ СОСТОЯНИЕ ДИСПЕРСИЙ
Устойчивость дисперсных систем или, иначе говоря, скорость их коагуляции зависит от знака и величины суммарной энергии взаимодействия, обусловленной сложением ионно-электростатической энергии отталкивания и энергии притяжения Ван-дер-Ваальса — Лондона.
Такое объяснение стабильности дисперсных систем впервые дано Дерягиным, а затем Фервеем и Овербеком и вошло в научную литературу как теория ДФО *. Рассматривая радиус
* Часто эту теорию называют теорией Дерягина — Ландау — Фервея — Овербека (ДЛФО), (Прим, ред.)
а промежуточных расстоя-
Рис. 12. Зависимость энергии диполь-дипольного взаимодействия от расстояния.
Степень ориентации диполей воды, %; I — 100; 2 — 55; 3 — 35. Кривая 4 характеризует энергию молекулярного взаимодействия двух пластин.
действия двух составляющих суммарной энергии взаимодействия плоских частиц бесконечной толщины, можно заметить, что Vm изменяется пропорционально \/h2 [уравнение (39а)], в то время как Vt уменьшается при увеличении расстояния по экспоненциальному закону. Из этого следует, что па малых и больших расстояниях между взаимодействующими поверхностями преобладает энергия притяжения, а ниях — ионно-электростатическая энергия отталкивания.
•Некоторые авторы [72—75] считают диполь-дипольное взаимодействие адсорбированных молекул воды или других полярных молекул, ориентированных в определенном направлении, существенным фактором флокуляции. Однако вычисление этого взаимодействия в настоящее время связано с принципиальными трудностями. В зависимости от того, на основании каких моделей проводится расчет, варьирует знак диполь-дипольной компоненты взаимодействия. Так, Мартынов и Смилга [76] полагают, что расположение двух частиц с ориентированными адсорбционными слоями соответствует минимуму энергии, если происходит соприкосновение последних. Строго говоря, предложенный в работе [76] метод учета взаимодействия адсорбционных слоев справедлив, когда расстояние между частицами сравнимо с плечом
диполя *. В противном случае должно проявляться отталкивание.
Силы диполь-дипольного взаимодействия, главным образом, определяются числом адсорбированных диполей, приходящихся на единицу площади; для двух пластин, окруженных одним или многими слоями ориентированных молекул воды, они оценены Хавеманом. Вследствие теплового движения не все диполи располагаются перпендикулярно поверхности. В целях упрощения предполагалось, что соседние молекулы адсорбционного слоя не оказывают влияния друг на друга и дипольные силы подчиняются правилу аддитивности.
Результаты расчета представлены на рис. 12. Для сравнения на график нанесена кривая зависимости молекулярной энергии
* Такое ограничение в работе [76] отсутствует. (Прим, ред.)
от расстояния (кривая 4). Из рисунка видно, что диполь-дипольное взаимодействие заметно ослабевает при снижении степени ориентации молекул в адсорбционном слое, в связи с чем оно, в отличие от молекулярных и ионно-электростатических сил, может практически не сказываться на процессе фло
куляции.
Как показано ранее (см. стр. 33), энергия молекулярного притяжения частиц в конкретной дисперсной системе сохраняется постоянной при введении электролитов или при повышении температуры. Однако изменение молекулярного взаимодействия достигается путем образования адсорбционных слоев поверхностно-активных и макромолекулярных веществ (см. главу 2). Ниже будет более подробно рассмотрено влияние электролитов на устойчивость дисперсных систем. Энергия ионно-электростатического отталкивания в сильной степени зависит как от фд-потенциала, так и от толщины диффузного двойного слоя. Обе величины определяются концентрацией и валентностью
ионов электролита в системе.
Влияние потенциала диффузной части двойного слоя на суммарную энергию взаимодействия частиц. Для получения информации о влиянии величины фб-потенциала на энергию взаимодействия прежде всего примем, что заданы концентрация электролита и постоянная Ван-дер-Ваальса— Гамакера. До тех пор, пока двойные диффузные слои частиц перекрываются слабо, можно рассчитать ионно-электростатическую энергию отталкивания по уравнению (29), согласно которому сомножитель, зависящий от потенциала, имеет вид:
Рис. 13. Зависимость энергии отталкивания двух плоских диффузных слоев от потенциала поверхностей при расстоянии между частицами 200 А и концентрации раствора, содержащего одновалентные ионы, 1 • 10-3 моль/л.
2
ехр
ехр
Следовательно, энергия отталкивания должна существенно изменяться только при показателе экспоненты, близком к единице (рис. 13).
Суммирование электростатической и молекулярной энергии прорасчетах исходили из концентрации
ведено на рис. 14. При
ионов симметричного электролита, равной 10s моль!л, и полагали, что постоянная Ван-дер-Ваальса — Гамакера А* = = 5-1013 эрг, т. .е. соответствует значению, найденному, например, для эмульсии типа масло/вода. Кривая 1 отвечает зависимости молекулярной энергии от расстояния. Рассмотрение
функций на рис. 14 приводит к выводу, что уже при фв = 20л«в существует достаточно высокий барьер отталкивания, препятствующий коагуляции.
Те же самые кривые в другом масштабе представлены на рис. 15. Как видно из графика, на сравнительно далеких расстояниях молекулярная энергия снова начинает играть преоб-
Рис. 15. Зависимость суммарной энергии взаимодействия двух пластин от расстояния между ними в растворе I—I-валентиого электролита с концентрацией 1 • 10-3 моль/л.
фупотеициал, мв'. / —0; 2 — 20; 3 — 50; 4 — 200. Пунктирные линии характеризуют соответствующие зависимости энергии ионио-электро-статического взаимодействия.
Рис. 14. Зависимость суммарной энергии взаимодействия двух пластин от расстояния между ними в растворе I—I-валеитного электролита с концентрацией 1 . io-3 моль!л.
фупотенциал, лв: Г —0; 2-^20; 3 — 50.
ладающую роль. При любых фб-потенциалах возникает более или менее ярко выраженный энергетический минимум, который, в отличие от первичного минимума на очень малых расстояниях, обычно называют вторичным. Глубина первичного минимума не имеет большого значения для коагуляции (на рис. 14 и 15 он не обозначен). Для частиц, не обладающих способностью к проникновению друг в друга, первичный минимум обусловлен компенсацией молекулярных сил притяжения борновскими силами отталкивания,
Влияние постоянной Гамакера на суммарную энергию взаимодействия частиц. Вывод о влиянии постоянной Гамакера Д* на суммарную энергию взаимодействия можно сделать на основании сопоставления рис. 14 и 16. Различие между ними связано только со значением А*, которое в последнем случае составляет 5-Ю-12 эрг, т. е. на порядок превышает константу Гамакера, принятую при построении кривых рис. 14.
У10,эрг1смг
О
4
3 г
1
о
-1
-2
~3
-л
-5
Рис. 16. Зависимость суммарной энергии взаимодействия двух пластин от расстояния между ними в растворе I—I-валентного электролита с концентрацией 1 • 10-3 моль/л.
ф ^-потенциал, мв-. I — 0; 2 — 20; 3 — 50;
4— 100 и 5-200.
Влияние концентрации электролита и валентности противоионов на взаимодействие частиц. При постоянных значениях А* и ^-потенциала будем варьировать концентрацию I—1-ва-лентного электролита. На рис. 17 представлена зависимость энергии взаимодействия от расстояния для /4* = 5-10~13 эрг и = 100 мв при различных концентрациях ионов. С увеличением содержания электролита в системе высота энергетического барьера отталкивания становится ниже, и при определенной концентрации (концентрации коагуляции) он полностью исчезает. Частицы получают возможность беспрепятственно слипаться, объединяясь в крупные агрегаты. Для концентрации электролита, при которой энергетический максимум находится на оси абсцисс (рис. 17, кривая 5), одновременно выполняются два условия *
* Из этих двух условий при = — А*/12л/г2 следует: dlnV/dln,ft = = —2; или при равенстве нулю силового барьера отталкивания: d In Р/д. 1п/г = = —3. Последнее выражение получено в работах [36, 294] и вошло в литературу под названием критерия коагуляции дисперсных систем. (Прим, ред.)
или
и
MnkT .
-----exp (— wi)
4*
12лйа
64nkT exp (— х/г)
4*
6лЛ3
откуда х/гс » 2, где hc — критическая толщина пленки.
-2
-S
-4
-5
Рис. 17. Зависимость суммарной энергии взаимодействия двух пластин от^ расстояния между ними (4* — 5 10“13 эрг, фб = 100 мв и 2=1).
Концентрация электролита, моль/л\ 1-1. 10~3; 2-5-10-3; ,3-2,5-Ю-2; 4 —5 • 10~2; 5-7,5- 10-2; 5-1-10-1.
С помощью этого соотношения и уравнения (25) получим
пс =
1,075 • 102е3Ф57'5
4*2e6z6
(60)
т. е. концентрация коагуляции (в моль/л) [77]
8,10- 10~25
4*2z6
ехр
2kT ,) +
(61)
если е = 78,55 и Т = 298° К *.
* Используемое здесь уравнение для энергии ионно-электростатического отталкивания частиц справедливо лишь в случае незначительного перекрытия двойных ионных слоев, что не имеет места в критическом состоянии системы, формулу для вычисления критической концентрации коагуляции слабо и
В коллоидных дисперсиях, для которых потенциалы фо и соответственно не изменяются при добавлении электролита или настолько велики, что выражение в квадратных скобках с достаточной степенью точности можно положить равным единице, ионно-электростатическое взаимодействие определяется практически только зарядом противоионов; концентрации коагуляции таких систем одно-, двух- и трехвалентными ионами относятся как 1:0,016:0,0013. Подобная закономерность часто наблюдается при экспериментальных исследованиях и известна под названием правила Шульце — Гарди [78, 79].
Основные результаты, полученные выше для бесконечных пластин, могут быть распространены также на пластины конечного размера и сферические частицы. При этом необходимо, однако, принимать во внимание, что частицы благодаря тепловому движению обладают определенной кинетической энергией. В том случае, когда тепловая энергия больше энергетического барьера, они коагулируют, несмотря на действие сил отталкивания.
Общее рассмотрение вопроса устойчивости. Анализируя приведенный выше теоретический и экспериментальный материал, можно прийти к выводу о существовании трех видов кривых зависимости энергии взаимодействия от расстояния, каждая из которых характеризует определенное состояние устойчивости системы.
Первое состояние отличается тем, что либо на всех расстояниях между частицами силы притяжения преобладают над силами отталкивания, либо энергетический барьер меньше энергии теплового движения. Такие дисперсии подвержены быстрой коагуляции, вызывающей образование крупных агрегатов. Отдельные частицы в ходе этого процесса сближаются вплоть до непосредственного контакта, а затем укрупняются вследствие слияния (в случае пузырьков пены или капелек эмульсий) или благодаря перекристаллизации (в случае твердых тел). Быстрая коагуляция, однако, не всегда приводит к увеличению размеров частиц: между взаимодействующими поверхностями могут сохраняться очень тонкие прослойки жидкости или адсорбционные слои толщиной несколько ангстрем, предотвращающие непосредственный контакт и, тем самым, укрупнение частиц [80,81]*. Такие системы называют устойчивыми к коалесценции. Они отличаются незначительной энергией разрушения связей (редиспергирования).
сильно заряженных золей впервые даны в работах [294, 303] Дерягиным и Ландау, а при произвольных постоянных потенциалах поверхности частиц — Барбоем [304—306]. Рассмотрение вопросов коагуляции дисперсных систем, исходя из условия равенства нулю потенциального барьера отталкивания двух частиц, проведено Муллером [293]. (Прим, ред.)
* См. также [307]. (Прим, ред.)
Об .устойчивости к коалесценции пен и эмульсий обычно судят по отсутствию заметного их расслаивания. Для твердых частиц, обладающих постоянством формы, часто нельзя однозначно установить, идет ли речь о коагуляционных структурах — агрегированных первичных частицах или о конденсационных структурах — микрообъектах, «спаянных» друг с другом в отдельных областях поверхности соприкосновения. Быстрая коагуляция дисперсий достигается добавлением электролитов. Эффективность действия ионов может быть связана как со сжатием диффузной части двойного слоя (концентрационная коагуляция), так и с уменьшением фо- или фв-потенциалов (нейтрализационная коагуляция). Как правило, концентрация электролита порядка 10-1 моль/л оказывается достаточной, чтобы вызвать быструю коагуляцию.
При сравнительно глубоком вторичном энергетическом минимуме и высоком барьере отталкивания частицы быстро флокулируют, обусловливая образование коагуляционных структур. Последние при низких напряжениях сдвига обнаруживают ползучесть. Под влиянием интенсивных механических воздействий они разрушаются, переходя в легкотекучее состояние, характеризующееся постоянной вязкостью. После снятия внешней нагрузки происходит восстановление связей между частицами системы. Таким образом, коагуляционные структуры могут проявлять склонность к тиксотропным превращениям. Поскольку прочность связи частиц определяется глубиной и координатой вторичного минимума, свойства таких систем существенно зависят от концентрации электролита в дисперсионной среде. Уменьшением содержания ионов можно вызвать пептизацию, т. е. переход геля в золь*.
Третье состояние устойчивости характеризуется тем, что кинетическая энергия частиц kT больше глубины вторичного энергетического минимума, но меньше барьера отталкивания. При этом соударения микрообъектов, участвующих в броуновском движении, очень редко приводят к возникновению агрегатов: Дисперсные системы, находящиеся в таком состоянии, являются стабильными; они обычно содержат малые количества электролита: ~ 10~3 моль)л.
* Вопрос о возможности фиксации частиц на сравнительно далеких расстояниях, отвечающих координате вторичного минимума, впервые подробно рассмотрен Ефремовым и Нерпиным [284, 308—312]. Влияние электролитов на процесс дальней агрегации и зависимость физико-мехапических свойств систем, возникающих в результате его протекания, от концентрации и валентности противоионов в дисперсионной среде изучены в работах [313, 314]. Показано, что в отличие от соотношения ccz? = const, установленного при исследовании коагуляции лиофобных коллоидов, в случае фиксации частиц относительно друг друга на далеких расстояниях должно выполняться равенство ecz2-s = = const, когда фо 75 мв. (Прим, ред.)
ЭКСПЕРИМЕНТАЛЬНОЕ ПОДТВЕРЖДЕНИЕ ТЕОРИИ ДФО. ОПРЕДЕЛЕНИЕ ПОСТОЯННОЙ ГАМАКЕРА
И ВЕЛИЧИНЫ Пи-ПОТЕНЦИАЛА ИЗ РЕЗУЛЬТАТОВ ИЗУЧЕНИЯ МОДЕЛЬНЫХ СИСТЕМ
Экспериментальная проверка основных положений теории ДФО проведена на различных объектах. К сожалению, невозможно обобщить и сопоставить все полученные данные, ввиду того, что в ряде работ четко не сформулированы условия постановки опытов. Обширный материал относится к изучению скорости коагуляции золей как функции концентрации электролитов. В последнее время аналогичные измерения выполнены с использованием модельных систем, содержащих частицы одинакового размера. Представляют интерес результаты определения равновесных расстояний и скорости сближения двух капелек эмульсий (или пузырьков пены), а также развитые сравнительно недавно методы, позволяющие непосредственно найти величину сил взаимодействия твердых тел.
Скорость коагуляции золей. Измерение сил взаимодействия частиц в золях и расстояний, сохраняющихся между поверхностями исходных микрообъектов в агрегатах, связано с очень большими экспериментальными трудностями. Поэтому процесс коагуляции обычно характеризуют уменьшением счетной концентрации твердой фазы во времени. Изменение числа частиц N в единице объема определяют электронно- или ультрамикро-скопическими способами [82] *. Иногда размер агрегатов находят нз данных изучения светорассеяния [83,84]. Некоторые исследователи получают количественную информацию о процессе слипания частиц из сопоставления концентраций различных ионов, вызывающих одинаковые превращения золя в течение некоторого фиксированного промежутка времени. При обработке результатов измерений строят кривые, откладывая на оси ординат либо скорость коагуляции dN/dt, либо фактор устойчивости W, а на оси абсцисс — содержание электролита в дисперсионной среде (рис. 18 и 19). Фактор устойчивости представляет собой отношение скорости коагуляции при концентрации электролита с к скорости коагуляции при критической концентрации са данного электролита. На приведенных зависимостях переход от медленной к быстрой коагуляции выражен более или менее резко.
Согласно уравнению (61), величины се обратно пропорциональны шестой степени валентности противоионов и относятся как 1 :0,016:0,0013 в случае одно-, двух- и трехвалентных ионов. Этот вывод подтверждается при исследованиях [78] золей гидроокиси железа, золота и сульфида мышьяка. Другие авторы [85—90] обнаружили сравнительно сильное отклонение от
* См. также [402]. (Прим, ред.)
указанного соотношения. Однако в работах [78, 85—90] не приведены опытные данные о потенциалах частиц при различных концентрациях электролита. Следовательно, нельзя решить, сопровождается ли сжатие двойного слоя уменьшением фо-потен-
циала и насколько оправданы упрощения, принятые при выводе формулы (61).
Рис. 18. Зависимость ско-роёти изменения числа частиц в золе от концентрации электролита.
Рис. 19. Зависимость логарифма фактора устойчивости коагуляции от концентрации электролита.
Для расстояний h <С а энергия ионно-электростатического отталкивания сферических частиц
V® = —2~ In [ 1 + ехр (— х/г)]
а энергия молекулярного притяжения у® =__d2£ т 12ft
Обе составляющие суммарной энергии взаимодействия прямо пропорциональны радиусу, т. е. критическая концентрация коагуляции не должна зависеть от величины а. Экспериментальные результаты не позволяют окончательно решить этот вопрос. Вестгрин [91] не нашел для золя золота с радиусом частиц 490, 770 и 1200 А различия в скоростях коагуляции. К такому же выводу пришли Шульц и Тезак [92], изучая дисперсии иодида серебра. Напротив, Оттевиль и Шоу при исследовании монодисперсных полистироловых латексов обнаружили заметное изменение критической концентрации коагуляции по мере увеличения радиуса частиц [93]:
а, А Сс-102, моль/л
300 1,88
515 2,82
1213 1,88
1840 1,78 1.42
2115
Очевидно, независимость критической концентрации от радиуса частиц должна проявляться только в тех случаях, когда справедливы приближенные уравнения (31а) и (38) для ионноэлектростатического и молекулярного взаимодействий.
Дерягин и Муллер [94] получили другую формулу
—4(1+1+1п4) (б2)
на основании которой нельзя установить в явном виде функциональное соотношение между критической концентрацией и зна-
18*
Рис. 20. Зависимость лога-
рифма фактора устойчивости от логарифма концентрации электролита для золей иодида серебра [91] (кривые 1 и 2) и латекса полистирола [93] (кривая 3) в присутствии нитратов: 1, 3-Ba(NOs)2; 2-KNOs.
чением а. Однако, исходя из уравнения (62), можно показать, что увеличение радиуса частиц приводит к уменьшению содержания ионов в дисперсионной среде, необходимого для исчезновения барьера отталкивания *.
Этот вопрос изучен также Ририн-ком и Овербеком [91]. Они использовали выражение для фактора устойчивости, выведенное Фуксом [95] для' аэрозолей. В работе [95] медленная коагуляция рассматривается как процесс диффузии частиц в силовом поле, причем
du
(« + 2)2
(63)
где и = h[a.
Из результатов экспериментальных исследований следует, что между логарифмом фактора устойчивости и ло-
гарифмом концентрации имеется линейная зависимость (рис. 20). Такую же закономерность предсказывает теория ДФ.О:
г (гефа \ , I2 ' ‘4*_______,1S. 10, “"I ar )
Уравнение (64) позволяет определить тангенс угла наклона прямых. Величины d)gW/d\gc, найденные Риринком и Овербеком и приведенные в табл. 6, находятся в неудовлетворительном согласии с формулой (64).
* Вместе с тем высота потенциального барьера отталкивания становится ниже по мере уменьшения радиуса частиц, а поэтому на кривой зависимости се = /(а) должен существовать максимум, так как lim се = о [315]. (Прим. а->0
ред.)
Таблица 6
Значения фб и А* для золей иодида серебра с различным радиусом частиц [91]
Электролит а, А dig IT dig с Фд, -ив Л*-1013, эрг
ККЮз 250 — 10,6 -48 2
520 -5,9 -24 0,5
2000 —7,3 — 14 0.2
Ba(NO3)2 250 -8,0 -53 9
520 -8,0 -30 4
2000 -11,0 -12 2
La(NO3)3 520 -5,8 -28 10
Авторы предприняли попытку, используя эти значения dlgWVdlgc, из выражения (64) вычислить фв-потенциал, а затем оценить константу Гамакера А*. Производя построение кривых, характеризующих энергию взаимодействия частиц как функцию расстояния для различных постоянных А* при фиксированном фб-потенциале, и рассчитывая фактор устойчивости W по уравнению (63), путем экстраполяции Риринк и Овербек получили величину А*, которая соответствовала опытным данным. Не удивительно, что вычисленные таким образом постоянные для одного и того же вещества различаются более чем на порядок (см. табл. 6).
Другая возможность определения фв-потенциала или константы А* состоит в измерении одной из этих величин независимым способом. Поскольку непосредственно измерить ^-потенциал невозможно, обычно принимают его равным электрокине-тическому потенциалу £; последний находят, например, исходя из скорости перемещения частиц в электрическом поле [84, 93, 96, 101]. Постоянные Гамакера, вычисленные при условии фб=£, представлены в табл. 7.
Таблица 7
Постоянные Ван-дер-Ваальса—гамакера для частиц различных золей в водной дисперсионной среде
Дисперсная фаза А ’-Ю13, эрг Литература
Арахиновая кислота 0,03 101
Двуокись кремния 0,5-0,2 105
Золото 6 91
Иодид серебра 0,5-9 91
Парафин 1,5 98
Полистирол 0,1-1 93
Селен 0,5-5 91
Предположение равенства значений фв и £ нельзя считать обоснованным. Во-первых, это фактически означает совпадение слоя Штерна с границей скольжения жидкости относительно
частицы в электрическом поле, что, по-видимому, мало вероятно. Во-вторых, вычисление ^-потенциала из скорости перемещения частиц требует учета ряда явлений (эффектов поляризации двойного слоя и т. д.), теория которых развита только для частиц простой формы [102].
Надежные величины постоянной А* найдены Ликлемой для
золей Agl [103]. Он измерял поверхностную плотность заряда на частицах дисперсий иодида серебра с известной удельной поверхностью методом потенциометрического титрования. Иссле-
дование емкости двойного слоя в растворах нитратов щелочных металлов показало, что катионы специфически адсорбируются только при сравнительно низких температурах; напротив, двойной слой при 85 °C имеет диффузное строение. Зная плотность поверхностных зарядов, Ликлема по формуле (Юа) вычислил потенциал поверхности, а затем, определяя критическую концентрацию, по уравнению (61) оценил значение А*.
Влияние на процесс флокуляции адсорбционных слоев поверхностно-активных и макромолекулярных веществ. Добавление неионогенных [73, 106] и ионогенных [107] ПАВ в достаточной концентрации
Рис. 21. Зависимость логарифма критической концентрации La (NO3)3 для золей иодида серебра от содержания в системе неионогенных ПАВ: / — октиловый эфир полиэтилепгликоля (6); 2 — дециловый эфир полиэтилеигликоля (6); 3 — додециловый эфир полиэтиленгликоля (6); 4 — гексадециловый эфир поли-этиленгликоля (9); 5—гексадециловый эфир полиэтиленгликоля (6) [106].
существенно уменьшает скорость флокуляции золей иодида серебра * (рис. 21). Еще сильнее выражено стабилизирующее действие водорастворимых полиэлектролитов, так называемых защитных коллоидов, — белковых веществ, альгинатов, карбок-симетилцеллюлозы, алкилполиамина и т. д. [78].
Согласно результатам многих исследований, вокруг частиц золя должен существовать избыток макромолекул, необходимый для образования насыщенного монослоя или даже полислоя. Электронномикроскопические снимки непосредственно доказали наличие таких защитных оболочек [108]. Например, адсорбционные слои метилцеллюлозы па частицах полистирола имеют толщину 70—100А. Для характеристики влияния искус
* См. также [316—324] и обзорную статью Воюцкого [325]. (Прим, ред.)
ственных и природных полимеров на устойчивость золей используют различные эмпирические методы: определение золотого [109] или рубинового [110] чисел, показателя защитного действия [111] и т. д. Однако значение этих методов невелико, так как стабильность дисперсных систем в сильной степени зависит от специфического взаимодействия макромолекул с поверхностью коллоидных частиц.
Следует отметить, что наряду с защитным действием в некоторых случаях наблюдается уменьшение устойчивости по мере добавления макромолекулярных веществ, а иногда-—выпадение флокул. Такое явление называют сенсибилизацией [78]*. Оно, как правило, обнаруживается при малом содержании макромолекул в дисперсионной.среде и объясняется образованием между отдельными частицами мостиков стабилизатора. В суспензиях каолина и полистирола возникновение мостиков доказано электронномикроскопическими исследованиями [108, 112—114]. Концентрациям метилцеллюлозы до 1—2% от веса твердой фазы обычно отвечает неустойчивое, а выше 4%—устойчивое состояние Дисперсной системы. Решающее влияние на защитное действие макромолекул оказывает соотношение между количеством полимера и удельной поверхностью частиц. Для стабилизации суспензий полистирола, например, необходима поверхностная концентрация метилцеллюлозы ~6-10 ‘1 г/м2. Аналогичные соотношения установлены и для ряда других макромолекулярных веществ.
По мнению некоторых исследователей, причиной стабилизирующего действия ПАВ и макромолекулярных веществ является ослабление молекулярного притяжения частиц дисперсной фазы при наличии на их поверхности адсорбционных слоев. Так, в работе [106], исходя из критических концентраций Ba(NO3)2 для золей иодида серебра, стабилизированных додециловым эфиром полиэтиленгликоля (6), вычислены значения сложной постоянной Гамакера А*, которые уменьшаются при увеличении содержания полимера в системе:
Концентрация додецилового
эфира полиэтилеигликоля А • 1м ।
(6), модь/л эра
0 7 3,1
5-10"' 1,6
1 • 10"в 0,9
5-10“’ 0,72
1 • 10_6 0,32
Воспользуемся представлениями Волд [57] для оценки влияния адсорбционных слоев ПАВ и макромолекулярных веществ на устойчивость дисперсных систем. Пусть коллоидные частицы радиуса а имеют постоянную молекулярного взаимодействия
* Уменьшение устойчивости золей при введении неионогенных ПАВ наблюдали во многих работах [317—320]. (Прим, ред.)
Лj. Кроме того, примем, что константы Гамакера для адсорбционных слоев и дисперсионной среды равны соответственно А3 и До. В этом случае:
V. — -Г [ W- - + (л? - +
+ • W - л?) (л’ - л?.)---+ '
(h -fij) (2 + -^-)
Макромолекулы в поверхностном слое сильно сольватированы, т. е. можно положить Аз = Ао. Тогда первый и последний член обращаются в ноль, и формула для молекулярной энергии получает следующий вид:
При условии Аз = Ао молекулярное взаимодействие коллоидных частиц в присутствии адсорбционных слоев равно их молекулярному взаимодействию в чистой дисперсионной среде. Знаменатель последней формулы содержит величину расстояния между поверхностями частиц, а не между адсорбционными слоями. Поэтому защитное действие высокомолекулярных веществ проявляется, если адсорбционные слои достаточно прочные и настолько толстые, что при h = 26t энергия молекулярного притяжения частиц сравнима с кинетической энергией, т. е. составляет ~ 4 • 10-14 эрг.
С целью проверки этих теоретических представлений исследованы суспензии полистирола, стабилизированные защитными коллоидами. Константу Гамакера для воды Ао на основании изучения разновесных толщин пленок принимали равной 3,5-10~13 эрг [115]. Величину At = 8,5-10~13 эрг находили, исходя из значения А* = 1,1 -К)-13 эрг для латекса полистирола [85, 93], согласно формуле А = (А|/х — Aq‘)2. Результаты расчета энергии молекулярного притяжения двух сферических частиц радиуса 1000 А для толщин 61 = 24 А и 61 = 50 А при различных постоянных А3 представлены в табл. 8.
Для расстояний между поверхностями частиц ~200А энергия молекулярного притяжения Vsm кТ, т. е. толщина защитных слоев ~100 А может быть достаточна для предотвращения процесса флокуляции. Из экспериментальных данных работы [Ю8] следует, что устойчивость дисперсной системы достигается, когда 61 ~ 70 4- 100 А. Таким образом, опытные и расчетные значения 64, соответствующие стабильному состоянию суспензии, хорошо согласуются друг с другом.
* См. стр. 37. (Прим, ред.)
Таблица 8
Энергия молекулярного взаимодействия сферических частиц полистирола радиусом 1000 А в воде и в водных растворах, содержащих защитные коллоиды
h, А -V^-1014. spa
«, = 0 А3=2.10-13 эрг А3— 5*10 13 эрг А3 = 8.10“13 эрг
«,=24 А «,=50 А «, = 24 А «, = 50 А «, = 24 А «,=50 А
50 18,2 82,1 80,7 397,0
75 12,1 8,9 17,6 31,8
100 9,0 7,0 11,5 17,0
' 105 8,6 6,7 31,1 10,8 . 34,7 15,5 163,0
125 7,2 5,9 6,7 8,7 13,4 11,6 8,8 34,0
150 6,1 5,1 4,5 7,0 9,1 17,5
175 5,2 4,5 3,8 5,8 7,1 7,1 11,9
200 4,5 3,9 3,4 5,0 5,9 6,0 9,0
250 3,6 3,2 2,8 3,9 4,4 4,5 6,1
500 1,8 1,7 1,6 1,9 2,0 2,0 2,3
1000 0,9 0,9 0,8 0,9 1,0 1,0 1,1
Влияние адсорбционных сольватированных слоев макромолекулярных веществ на устойчивость дисперсных систем, по-види-^мому, объясняется двумя причинами. Во-первых, при исчезающем малом взаимодействии адсорбционных слоев друг с другом они являются барьером, препятствующим сближению частиц на расстояния, при которых силы молекулярного притяжения становятся значительными (см. главу 2, стр. 115). Во-вторых, наличие макромолекул на межфазной границе может существенно ^ослабить силы притяжения частиц. Вероятно, первый фактор ‘стабилизации играет решающую роль*. Поскольку энергия молекулярного притяжения прямо пропорциональна радиусу взаимодействующих поверхностей, для получения устойчивых золей, в отличие от суспензий, требуется меньшее количество полимера на единицу площади границы раздела фаз. Устойчивость грубодисперсных суспензий наступает только в том случае, когда вследствие повышения концентрации макромолекулярного раствора в нем происходит образование геля и твердые частицы прочно удерживаются в его решетке**.
* По мнению ряда исследователей [318—321], в результате адсорбции молекул ПАВ на коллоидных частицах лиофобных золей происходит лиофилизация их поверхности и образуются системы, агрегативная устойчивость которых обеспечивается благодаря возникновению сольватных слоев; перекрытие последних связано с проявлением сил отталкивания. (Прим, ред.)
** В этом случае, очевидно, можно говорить лишь об отсутствии слипания частиц, а не об устойчивости дисперсной системы в целом. (Прим, ред.)
Непосредственное ослабление молекулярного взаимодействия часгиц в присутствии адсорбционных слоев полимеров нетрудно оценить на основании приведенных выше уравнений. При А3 Ф До величина первого члена в правой части формулы (42а) становится достаточно большой, так как его знаменатель содержит разность h — 261. Следовательно, ослабление молекулярного взаимодействия возможно только в случае близких значений постоянных Д3 и Ао. Как показывает сопоставление расчетных данных, относящихся к энергии притяжения частиц полистирола в водных растворах полимеров (см. табл. 8), для ряда параметров действительно обнаруживается ослабление взаимодействия при наличии адсорбционных слоев. Такое изменение наблюдается для расстояний h > 26i потому что, когда h -*261, первый член в правой части выражения (42а) стремится к бесконечности. Вместе с тем, при достаточно больших значениях постоянных А3 благодаря адсорбции наступает существенное увеличение сил молекулярного притяжения частиц.
Некоторые исследователи [73, 106] проводят сравнение величин Vm, относящихся к взаимодействию частиц в чистой дисперсионной среде и в растворах полимеров, отсчитывая соответственно расстояние либо от твердой поверхности, либо от внешней границы адсорбционного слоя. При этом, естественно, нельзя сделать вывод об ослаблении (или усилении) молекулярного притяжения за счет образования полимолекулярной пленки защитного коллоида. Кроме того, если А3 < At и расстояние отсчитывается от поверхности адсорбционного слоя, находят очень низкие значения энергии взаимодействия. Поэтому избранный нами путь рассмотрения вопроса о молекулярном притяжении дисперсных частиц в растворах защитных коллоидов, по-видимому, более приемлем для получения информации о влиянии высокомолекулярных соединений на устойчивость дисперсных систем.
Аналогично полимерам ПАВ при введении в дисперсионную среду образуют на границе раздела фаз адсорбционные слои, которые в первом приближении гомогенны и характеризуются определенной константой Гамакера А3 *. На рис. 22 представлена зависимость энергии притяжения частиц иодида серебра от расстояния h. в растворе ПАВ. При расчетах полагали Ао = = 3,5- 1СГ1Я эрг и 61 = 15 А. Постоянную взаимодействия At = =?= 1,3-10-’2 эрг находили на основании экспериментальных данных [ЮЗ], приведенных в табл. 7. Из графика видно, что влияние адсорбционных слоев на энергию притяжения частиц иодида серебра незначительно и ограничено областью малых расстояний между частицами.
Существенный вклад в суммарную силу взаимодействия частиц, вероятно, должны вносить полярные группы адсорбиро-
* См. стр. 37. (Прим, ред.}
ванных молекул ПАВ. В качестве постулата примем, что полярные группы образуют слой очень малой толщины б2 и постоянная Гамакера для таких слоев равна Д4. Исходя из этих предположений, по уравнению (45д) произведена оценка энергии
-5 - I Рис. 22. Зависимость энергии взаимо
2 действия частиц иодида серебра радиу
-6 - сом1000 А от расстояния между их поверх-
ностями в присутствии адсорбционных
- 7 - слоев ПАВ.
1 * „ Л3, эрг: /-2 10-13; 2-0; 3-9 • Ю-13.'
Ут-10,зрг
400 h,A
О
200
-10
J
•Рис. 23. Зависимость энергии взаимодействия частиц иодида серебра радиусом 1000 А от расстояния между их поверхностями в отсутствие (кривая 7) и при наличии адсорбционных слоев полярных молекул (кривые 2—4).
At. эрг-. 2-0; 3-3.5 • 10~13; 4-5 • 10-12.
притяжения частиц иодида серебра радиусом 1000 А как функции расстояния h между ними (рис. 23). Как и ранее, полагали Аг) = 3,5-10’13 эрг, А^ГЗХ X 10-12 эрг, Аз = 9-1013 эрг и б! — 15А; значение б2 составляло 3 А. Сопоставление зависимостей, рассчитанных для различных постоянных Д4, приводит к выводу, что заметное усиление молекулярного притяжения частиц происходит только при расстояниях, не превышающих 250 А. Абсолютные величины таких изменений обусловлены выбором константы Гамакера Д4, которая, строго говоря, является гипотетической.
К сожалению, в настоящее время отсутствует экспериментальное подтверждение развиваемых здесь представлений.
О сильном влиянии полярных групп в молекулах ПАВ на взаимодействие частиц можно судить лишь на основании измерений равновесных толщин h0 пленок воды, содержащей добавки стабилизатора, между двумя капельками масла при одинаковой
4
-f5 <з
Vm10^
концентрации индифферентного электролита *. Отличие значений ho, соответствующих, например, приблизительно равной плотности адсорбционных слоев алкен- и алкансульфонатов, нельзя объяснить, предполагая, что потенциал диффузного двойного слоя варьирует от нуля до бесконечности (табл. 9).
Таблица 9
Влияние ПАВ на равновесные расстояния ha
между капельками циклогексана в б-10~2н. растворе KCI
ПАВ Площадь, занимаемая молекулой в поверхностном слое, А2 Равновесная толщина пленки, А
Цолиэтиленгликолевый эфир ионилфенола
NP-12.................................
NP-20 *...............................
NP-30.................................
Na-додекансульфонат-!.....................
Na-3-гидроксидодекансульфонат-1...........
Na-2-гидроксидодекансульфонат-!...........
Na-3-оксододекансульфонат-!...............
Na-2-додеценсульфонат-! . '...............
Na-додецилсульфонат-!.....................
Na-лаурилсаркозид.........................
1110
52
52
57
55
53
46
69
105
125
145
175
155
170
165
150
170
185
• Препарат NP-23 по строению и свойствам аналогичен вспомогательному веществу ОП-20, выпускаемому отечественной промышленностью. Здесь и далее .цифры индекса ПАВ соответствуют среднему содержанию этиленоксндных звеньев в молекуле. (Прим, ред.)
Проведем краткое обобщение результатов, полученных в этом разделе. Внесение двух частиц из газовой фазы в жидкость вызывает существенное изменение характера их взаимодействия, причем влияние адсорбционных слоев, как правило, невелико и сказывается только на сравнительно малых расстояниях. Поэтому причина увеличения устойчивости дисперсных систем по мере повышения содержания ПАВ заключается, по-видимому, не только в молекулярном взаимодействии микрообъектов. Очевидно, от свойств адсорбционных слоев, главным образом, должны зависеть силы ионно-электростатического отталкивания частиц. Такая зависимость обусловлена рядом явлений: 1) изменениями фо-потенциала, связанными либо непосредственно с адсорбцией ионогенного ПАВ, либо с вытеснением потенциал-определяющих ионов с границы раздела фаз; 2) нарушением исходного распределения зарядов в двойном слое. В частности,
* На это указывают также результаты изучения устойчивости золей сульфида мышьяка в присутствии алкиловых эфиров полиэтиленгликоля, содержащих 18 и 98 оксиэтильных групп [309, 310]. Как правило, все производные, отвечающие большей длине оксиэтильиой цепи, обладали ярко выраженным стабилизирующим действием. (Прим, ред.)
0 -W -8 -6 -4lgc
Рис. 24. Зависимость потенциала диффузной части двойного слоя в прослойке пены от концентрации Na-додецилсульфата-1 в растворе КС1 при постоянной ионной силе 1 • 10-* моль!л.
благодаря адсорбции сдвигается плоскость, отвечающая потен-Ж циалу диффузного двойного слоя, на расстояние, равное бь Ц- Оценка и учет всех этих явлений на современном этапе разви-Ж тия теории взаимодействия дисперсных частиц не представ-Ж ляются возможными.
й’- Измерение потенциала двойного диффузного слоя в пенных пленках, стабилизированных ионогенными ПАВ, проведены в pair боте [117]. Исходя из равновесных толщин h0, на основании тео-Ц*- рии ДФО рассчитаны значения фб, которые, как и следовало Ш ожидать, возрастают вплоть до насы-Ж щения при увеличении концентрации Ж* ПАВ (рис. 24). Аналогичные законо-Цг мерности должны наблюдаться и для fe эмульсий. Более сложные соотноше-ж ния между фа-потенциалом. и концен-® трацией ПАВ, по-видимому, суще-Ж' ствуют в случае золей, заряд которых Н; зависит от поверхностной диссоциа-К ции или от предпочтительной адсорб-Як ции каких-либо ионов, имеющихся в ЯЖ растворе. В таких системах может об-наруживаться как увеличение, так и
Ж уменьшение фа-потенциала, причем К его значение будет определяться на-правлением ориентации полярных Яе: групп молекул стабилизатора на гра-Щ нице раздела фаз.
Ж Исследования моделей пен и эмульсий. Жидкие прослойки Иг в пенах и эмульсиях являются хорошими объектами для коли-Щ;чественного изучения взаимодействия дисперсных частиц, по-скольку плоскопараллельные межфазные границы в первом при-Мг ближении эквипотенциальны. Кроме того, весьма ценно, что про-И| ведение измерений в таких системах не вызывает принципиальных затруднений. Во всех исследованиях информацию о поверхност-Як пых силах, действующих в тонких пленках, получали, определяя Ии либо равновесную толщину последних, либо скорость вытекания раствора из прослойки, иначе говоря, скорость сближения частиц. ИВ Впервые подобные измерения проведены на микроскопиче-К ских горизонтальных пенных пленках Дерягиным и Титиевской В- [118—120]*. Применяемая в этих работах аппаратура схемати-чески изображена на рис. 25. Два воздушных пузырька 1 и 2 К посредством стеклянных рамок 3 и 4 приводили в соприкоснове-К ние друг с другом в исследуемом растворе 5. Равновесные тол-К; щины измеряли методом интерференции света. Расклинивающее Я| давление пленки компенсировали внешним давлением, которое к’ отсчитывали по манометру 6.
* См. также [326]. (Прим, ред.)
Несколько видоизмененная аппаратура использована Дейвисом для пен [121] и Ван ден Темпелем для эмульсий [122]. Дальнейшее усовершенствование методики исследования выполнено Шелудко и сотрудниками [63, 123]. Сконструированная ими установка (рис. 26)чсравнительно проста и позволяет измерять равновесную толщину горизонтальных пленок пены, а также скорость ее изменения при отсутствии равновесия. Образование пенных пленок производили в стеклянной рамке / путем отсасывания
Рис. 25. Схема установки для исследования пленок между двумя воздушными пузырьками [ 118]:
1,2 —пузырьки воздуха; 3, 4 — стеклянные рамки; 5 — исследуемый раствор; 6— манометр.
Рис. 26. Схема установки для исследования пенных пленок [63]:
1 — стеклянная рамка; 2 — микрометрическая пипетка.
жидкости микрометрической пипеткой 2. Наступающее самопроизвольное изменение толщины пленок регистрировали интерференционным методом. Аналогичная установка применена Ми-зелсом и Джонсом для изучения поведения жидких прослоек при различных внешних давлениях [124].
Аппаратура для определения скорости сближения капелек эмульсий предложена Зонтагом и сотрудниками [125]; схема ее представлена на рис. 27. Капельки получали с помощью микрометрических винтов 1 и 2 на концах двух соосных капилляров 3 и 4, находящихся в дисперсионной среде. Нижний капилляр имел расширение для сведения к минимуму помех, связанных с отражением световых лучей от стеклянных стенок при проведении интерференционных измерений. Наружные концы капилляров закрывали, чтобы случайные колебания давления воздуха не вызывали изменения площади соприкосновения капелек.
Результаты исследования вертикальных пенных пленок изложены в работе [126]. Подобные пленки образуются при медлен-
ном подъеме из раствора ПАВ проволочной рамки /, соединен* ной с микрометрическим винтом 2 (рис. 28). Скорость подъема составляет 4-Ю-7 — 5-10—2 см!сек. Изменение толщины пленки вследствие испарения исключают, покрывая дно измерительного сосуда 3 раствором ПАВ и создавая в ячейке малое избыточное давление паров азота.
Как показало изучение картин интерференции света (рис. 29,6), уже при сравнительно больших расстояниях между поверхностями газовых пузырьков или капелек эмульсий на-
Рис. 27. Схема ячейки для определения скорости сближения капелек эмульсий и равновесных расстояний между ними:
/> 2 — микрометрические винты; 3 — верхний капилляр диаметром 2 мМ; 4 —иижинй капилляр с расширением.
ступает деформация межфазных границ, приводящая к возникновению почти плоскопараллельной зоны соприкосновения радиуса R (рис. 29,а). (К вопросу о плоскопараллельное™ промежуточного слоя мы еще вернемся ниже при рассмотрении процесса сближения частиц дисперсной фазы.) В периферийной части зоны соприкосновения действует капиллярное давление, связанное с кривизной поверхности и обусловливающее вытекание раствора из центра пленки и тем самым уменьшение ее толщины h. Утоньшение вертикальных пленок происходит благодаря действию гидростатического давления.
Для вычисления толщины слоя измеряют интенсивность [° отраженного света. Кроме того, находят интенсивности при минимальном /°min и максимальном /°тах отражении [123, 125]. Последняя величина соответствует оптической толщине пленки, равной четверти длины волны. Блок-схема применяемой для этого установки изображена на рис. 30. Поскольку в формулу для определения толщины слоя входит отношение интенсивностей, не требуется знать их абсолютные величины. К сожалению, расчеты, основанные на опытных данных интерференционных
измерений, приводя! к падежным результатам только в случае прямых эмульсий, когда неполярные радикалы, обладающие Коэффициентом преломления, свойственным маслу, расположены
Рис. 28. Схема аппаратуры для получения равновесных вертикальных пленок пены:
1 — проволочная рамка; 2 — микрометрический -виит; 3 — измерительный сосуд.
Рнс. 29. Схема образования плоскопараллельной зоны соприкосновения между капельками эмульсий (а) и картина интерференции для разделяющего их слоя (б) [127].
в дисперсной фазе и справедлива однослойная модель (рис. 31,а). Тогда
/ /°-Zmln \'f>
_ /0 /
V max ' mjn/
где tii — коэффициент преломления воды; п — коэффициент пре* ломления дисперсной фазы,
Пенные пленки представляют трехслойную систему (рис. 31,6). Их структура обусловливает сложное, хотя и симметричное изменение коэффициента преломления. Можно, однако, приближенно считать, что у границы с воздухом имеется два тонких внешних слоя постоянной толщины Л2 с коэффициентом преломления п2, содержащих между собой водное ядро 1
Рис. 30. Блок-схема установки для измерения толщин тонких жидких пленок методом интерференции световых лучей.
толщиной hi с коэффициентом преломления П] [121, 126]*. Определение значений h для пенной пленки на основании однослойной модели приводит к слишком высоким величинам. Зависимость абсолютной погрешности расчета от толщины адсорбционных слоев й2 представлена на рис. 32 [121]. При построении графика полагали п2 = 1,5; nt = 1,33 и Ai = 90 А. Абсолютная погрешность расчета практически не зависит от толщины водного ядра.
Равновесное состояние слоя устанавливается, когда силы притяжения и отталкивания взаимно компенсируют друг друга, т. е. SP = 0. Величина SP характеризует суммарную силу
* О преломлении света можно говорить только в том случае, когда размеры неоднородностей среды значительно больше длины волны. Поэтому нельзя приписывать мономолекулярному слою какую-либо величину коэффициента преломления, характеризующего объемное свойство веществ. (Прим, ред.)
взаимодействия дисперсных частиц, отнесенную к единице площади зоны соприкосновения. В горизонтальных пенных пленках или в прослойках жидкости между двумя капельками эмульсий наряду с ионно-электростатическими Pi и молекулярными Рт силами проявляются капиллярные силы Ра, связанные с кривизной поверхности. Для вертикальных пенных пленок необходимо
Рис. 31. Схемы однослойной (а) и трехслойнон (б) пленки:
1 — водная фаза; 2 — масляная фаза;
3 — воздух.
Рис. 32. Зависимость поправки к вычисленному, исходя из однослойной модели, значению h пенной пленки от толщины адсорбционного слоя.
учитывать также силу тяжести. При условии равновесия с учетом определения понятия расклинивающего давления *
П, = Пт + ДРа 4- ДРд
где Пт — молекулярная составляющая расклинивающего давления; ДРа— лапласовский перепад давления; Д/\— гидростатическое давление.
Величина ДРа не зависит от толщины прослойки и известна, если заданы поверхностное натяжение и главные радиусы кривизны. Значение ДРа, кроме того, можно получить, измеряя скорость сближения капель или пузырьков для расстояний h, превышающих 1000 А. При вычислении лапласовского перепада давления необходимо учитывать деформацию межфазных границ (рис. 29,а), т.е. принимать во внимание радиус R плоскопараллельной зоны соприкосновения. Справедливо следующее уравнение [128]
A^=T?^r (67)
* См. стр. 27. (Прим, ред.)
которое при г > /? переходит в известную формулу:
Вследствие образования плоскопараллельной зоны соприкосновения, вычисление расклинивающего давления производится по уравнениям, полученным для плоских частиц. Подставляя в выражение (66) соответствующие формулы для лапласовского перепада давления, молекулярной и ионно-электростатической компонент расклинивающего давления, имеем:
64nkT exp (— хйо)
Л* 2ргг блйо г2 — R2
(68)
При Т = 298 °К и е = 78,5 получим
1,59 • 1О»сГ ехр ехр (- 0,329 • 1О»йо /7?) =
L ехр (19,83гфб) + 1 J г
где/8—ионная сила, моль!л.
( В уравнении (68а) неизвестны величины А* и фв. Однако, задавая концентрацию с (в моль/л) электролита настолько низ-г кой, чтобы равновесные толщины йо пленок превышали 6- 10~б см, можно пренебречь молекулярной составляющей расклинивающего давления по сравнению с лапласовским перепадом давления и определить значение фв. Так, для пенных пленок, стабилизированных лауратом натрия [118], сапонином [128], полигли-колевым эфиром октилфенола, содержащим 20 этиленоксидных групп [129], тетрадецилсульфатом натрия [129] и додецилсульфатом натрия [130], фв соответственно составляет 50, 90, 35, 115 и 39 мв, Для эмульсий типа масло/вода, в дисперсионной среде которых растворены полигликолевые эфиры нонилфенола NP-20 или NP-30 получена величина ф6, равная 50 мв [127]. Эти вычисления основываются обычно на упрощенном уравнении (286) для электростатической компоненты расклинивающего давления П,, т. е. на предположении, что потенциал в плоскости симметрии между поверхностями ф<г мал по сравнению с потенциалом диффузной части электрического двойного слоя. Недавно Ексеровой [117] предложен другой путь определения фб-потенциала. Она исходила из точного уравнения (27), устанавливающего связь между величинами Пг и фа
П,= 2^7’(сЬ-^--1) \ Ki /
а также уравнения (20) для зависимости величин от фв и расстоянии h
иб
хй = V2 f-------—-----п-
J (ch и — ch /а ud
где u6 = ze^JkT и ua — ze^alkT.
•Заменяя переменную интегрирования и вводя параметр 9, обе формулы записываются в виде*:
П; = 4nkT th2 0 (69)
и
х/г = 2 sin QF (<pj, sin 0) (70)
Здесь f(<p6, sin9) — эллиптический интеграл первого рода с ймплитудой <рв и модулем sin 0, причем
shJ^=tfgO_
2 cos <pj 4 '
И
Ch-T = ‘sin0'
Зная величину П<, из уравнения (69) находят параметр 0, а затем из выражения (70) при известной толщине й0— значение эллиптического интеграла. Используя таблицы интегралов [131], нетрудно определить при некоторой фиксированной величине 77(<p6sin0) амплитуду <р6 и по формуле (71) ф6-потенциал. Таким путем для ряда ПАВ вычислены значения ф6-потенциалов пенных пленок, соответствующие максимальной адсорбции (см. рис. 24); полученные результаты представлены в табл. 10.
Таблица 10
Значения ^-потенциала, соответствующие
максимуму адсорбции некоторых ПАВ
ПАВ с-106, моль! л Фй,- J МВ
Монодециловый эфир гексаоксиэтиленгликоля . . . 12,0 54 '
Na-октадецилсульфат-! . 60,0 150
Na-тетрадецилсульфат-! Полиэтиленгликолевый эфир октилфенол.а, содер- 32,0 115
жащий 20 этиленоксидных групп 4,6 36
Сукрозеддодецилуретан 35,0 26
Цетилпиридиний хлорид 14,2 100
По мере увеличения концентрации ионов в дисперсионной среде становится все заметнее влияние молекулярных сил на
* Уравнение (70) впервые получено Барбоем [305]. {Прим, ред.)
взаимодействие частиц. Измеряя равновесные толщины пленок И постулируя, что ф6-потенциал не зависит от содержания электролита (предположение, которое для неионогенных ПАВ в первом приближении выполняется [132]), можно оценить постоянную Ван-дер-Ваальса — Гамакера. Как показано, для пенных пленок А* = 3,5-10~13 эрг [115], а для капелек воды в циклогексане А* = 1,5-1013 эрг [127]. Приведенные выше значения ^-потенциала и константы А*, очевидно, являются точными только в том случае, если справедлива теория ДФО.
Для экспериментальной проверки теории необходимо знать ионную силу раствора, потенциал диффузного слоя, постоянную А*, толщину слоя йо и лапласовский перепад давления в пленке. Ионную силу раствора сравнительно просто вычислить, исходя из концентраций электролита и концентрации ПАВ при условии, что последняя не превышает критической концентрации мицелло-образования. Лапласовский перепад давления определяют независимым методом. Часто поверхностный потенциал фо находят, полагая 100%-ную диссоциацию молекул ионогенного ПАВ, адсорбированных на межфазной границе. При этом его значение, как правило, оказывается сильно завышенным по сравнению с величиной ф6-потенциала, полученной на основании опытных данных о равновесной толщине пленки. Например, для пейных пленок, стабилизированных Na-додецилсульфатом (площадь, приходящаяся на одну молекулу в поверхностном слое, равна 52 А2), при ионной силе 5-10-3 моль!л фо = 224 мв, в то время как из результатов измерения равновесной толщины слоя следует, что ф6 = 39 мв [130]. Однако такое различие не вызывает существенного изменения расчетной величины постоянной взаимодействия А*, поскольку влияние потенциала на толщину равновесных пленок не очень велико.
На рис. 33 приведены полученные различными авторами зависимости равновесных толщин пенных пленок.от ионной силы раствора. Равновесные толщины, найденные Шелудко при ДР0 == 760 дин/см2, относятся к потенциалу диффузного слоя 90 мв. Аналогичные закономерности установлены для тонких водных слоев, разделяющих две капли углеводородных жидкостей, в присутствии различных ПАВ (рис. 34).
Таким образом, результаты исследования равновесных тонких пленок подтверждают основные положения теории ДФО. Особенно хорошее соответствие между теорией и экспериментом наблюдается в том случае, когда при вычислениях принимают сравнительно низкие значения ф6-потенциалов.
.Непосредственное измерение сил отталкивания в тонкой жидкой прослойке, связанных с существованием двойного слоя, предпринято Мизелсом [124] путем определения изменения равновесных толщин горизонтальных пенных пленок под действием внешнего давления и при сохранении постоянной ионной силы
раствора (рис. 35) *. Для вычисления теоретических зависимостей полагали А* = 6-10-13 эрг и ^-потенциал, равным 100 мв
Рис. 33. Зависимость равновесной толщины пенных пленок от ионной силы раствора для различных ПАВ (дрст==300дин!см2-, А*=3,5 • Ю-13эрг): Д —олеат натрия [119]; ф —олеат натрия 7133]; X —сапо* ннн [115, 128]; О — Додецилсульфат натрия (130); И-доде-цнлсульфат натрия [134]. Сплошные линии соответствуют зависимостям, рассчитанным согласно теории ДФО для ф^=50л8 (кривая J) и ффясо (кривая 2).
(прямая /) и <ю (прямая 2). Сопоставление расчетных и экспериментальных данных указывает на их достаточно хорошее каче-
Рис. 34. Зависимость толщины водных равновесных пленок между капельками масла от ионной силы раствора для различных стабилизаторов н углеводородов (ДРа = 300 дин!см?\ А*=» = 1,5- 10-13 эрг):
О —сульфат натрия — парафиновое масло [122]; ♦ —полиэтнленгликоле&ый эфир нонилфенола (NP-20) — циклогексан [127]; X — полиэтнлеигликолевый эфир ноннлфенола (NP-30) — циклогексан [127]); Д — Na-3-гндроксидодекаисуль-фонат —циклогексан (по данным Зонтага). Сплошные лнннн соответствуют зависимостям, рассчитанным на основаинн теории ДФО при
(кривая /) и Фд = °о (кривая 2).
СТВенное (но не количественное) согласие. Измеренные толщины слоев h все без исключения очень велики. Даже принимая бес-
* Подобные измерения впервые проведены Дерягиным н Титиевской [118—119]. (Прим., ред.)
конечно большой потенциал диффузного слоя, не удается добиться удовлетворительного согласия. По мнению Мизелса, причиной этого является слишком малая поправка к толщине слоя, вводимая на основе трехслойной модели. Соответствие между
опытными и теоретическими данными существенно улучшается, если вычисления проводить, исходя из другого значения площади Fo, занимаемой одной молекулой ПАВ в поверхностном слое. Согласно исследованиям Нильсона [135], для Na-тетраде-цилсульфата-1 величина /^составляет 36 А2, а не 52 А2, как предполагалось при обработке результатов изучения интерференции света. Тогда абсциссы точек на рис. 35 должны быть уменьшены на 12 А. Однако более удобно сместить прямые 2 вправо на расстояние, соответствующее Й4-12А (прямые 3). Это дает возможность убедиться, что при введении такой поправки, хотя и для бесконечно больших потенциалов диффузного слоя, достигается почти полное совпадение теоретических и экспериментально установленных зависимостей.
Интересный метод определения молекулярных сил из данных об уменьшении толщины пенной пленки предложен Шелудко [123]. Метод основан на рассмотрении пенной пленки как плоскопараллельного диска с расширением у краевой зоны (см. рис. 29,а). Скорость вытекания, выражае-
Рис. 35. Зависимость суммарного расклинивающего давления в пенной пленке от ее толщины прн 0,0012 (а) и 0,0094 моль/л (б).
Сплошные линии — теоретические зависимости, рассчитанные прн ф^» 100 мв (прямые /) и Ф^==со (Прямые 2}. Пунктирные линии 3 построены для ф^ = с» с учетом поправки к толщине пленки.
мая через изменение толщины как функции времени, может быть найдена из уравнения Рейнольдса
di
ЗП 3/?2п
(72)
где ht и h — толщины пленки при времени t и при t = 0, соответственно; R—радиус плоскопараллельной зоны соприкосновения; т) — динамическая вязкость раствора в прослойке.
Для достаточно толстых пленок (ft > 1000 А) значение IJ определяется только лапласовским перепадом давления ДР0. Поэтому функция 1/Л? — I/ft2 = f(0 должна характеризоваться прямой линией (рис. 36). При малых толщинах слоя оказыва-
Рис. 36. Зависимость величины (i/^-1/л2) от времени для пенных пленок, стабилизированных ОП-Ю [136].
ются существенными молекулярная составляющая расклинивающего давления (Пт) и, в случае низкого содержания электролита в дисперсионной среде, ионно-электростатическая компонента (П,), которые обусловливают отклонение экспериментальных точек от указанной зависимости. Из величины этого от-
клонения, соответствующего высокой концентрации ионов в пленке, можно вычислить Пт, а затем и константу А*. Таким образом Шелудко
нашел, что для водных пенных пленок А* — 7-10~134-11 • 10~13 эрг; она приблизительно в три раза превышает значение А*, определенное в работе [115] из опытных данных о равновесной толщине тонких жидких слоев. Еще большее различие постоянных 4* (на порядок) получают, изучая модель эмульсии динамическим и статическим методами [127]. Поэтому представляется необходимым проверить, выполняются
ли для течения жидкости в пленке основные предположения, на
которых основан вывод уравнения Рейнольдса. Они заключаются в следующем:
1. Течение жидкости происходит между плоскопараллельными стенками.
2. Адсорбционный слой является прочным и не участвует в движении.
3. Вязкость жидкости в пленке равна вязкости жидкости в объ
Рис. 37. Схема утолшения водной пленки 2 между двумя капельками масла /.
еме.
При экспериментальных исследованиях обнаружено, что по мере приближения пузырьков газа или капелек эмульсий к плоской твердой поверхности [137—141] или при сближении капелек эмульсий друг с другом [127, 132, 138, 141] жидкость из центральной части зоны соприкосновения вытекает медленнее, чем из периферийной зоны. Такой характер течения приводит к возникновению в середине пленки линзоподобного утолщения, так называемого «димпла» (рис. 37). Толщину слоя вдоль оси симметрии и в периферийной зоне, исходя из гидродинамических соображений, можно рассчитывать по уравнениям, учитывающим только лапласовский перепад давления как фактор, кото
рый определяет процесс течения [142]
0,096г6ч<7 V * aRt )
(73)
и
Лтщ = 3.05/12тах4 (74)
где q — число граничных поверхностей, на которых скорость течения равна нулю.
Экспериментальная проверка [140] указывает на то, что удовлетворительное согласие опытных данных с теорией имеется только в случае газовых пузырьков на твердых подложках.
Вероятность образования утолщений в тонких слоях жидкости, разделяющих два пузырька газа или две капельки эмульсии, в сильной степени зависит от радиуса зоны соприкосновения. Как установили Шелудко и сотрудники, утолщение в пленке не наблюдается, когда величина R не превышает 10-2 см. При больших радиусах димплы самопроизвольно исчезают в процессе утоньшения прослойки примерно до 1000 А; в дальнейшем сохраняется лишь плоскопараллельная пленка, для которой справедливо уравнение Рейнольдса.
Иначе происходит сближение капелек эмульсий. При незначительной площади зоны их соприкосновения (/? •< 10-2 см) обнаруживается возникновение постепенно уменьшающегося в объеме димпла — для расстояний ~1000 А его уже невозможно заметить визуально. В случае R > 10-2 см утолщение как бы «выталкивается» в том или ином случайном направлении (рис. 38). Однако при толщине пленок h < 1000 А димпл снова образуется, по-видимому, вследствие действия молекулярной составляющей расклинивающего давления.
Различие между величинами /гтах и /imin определяется концентрацией электролита в дисперсионной среде. Оно может быть настолько мало, что утолщение незаметно при наблюдении пленки в целом. Только проводя измерения толщины в отдельных областях слоя с по'мощью фотоумножителя, можно однозначно доказать существование димпла. Вторичный димпл выравнивается, когда расстояние между капельками эмульсий достигает ~300А.
Специфика поведения пленок в пенах и эмульсиях, вероятно, связана С величинами поверхностных натяжений, которые составляют 60 и 5—15 дин/см для границ раздела жидкость — газ и жидкость — жидкость, соответственно. Таким образом, первое условие применимости формулы Рейнольдса соблюдается только при определенных толщинах и размерах тонких слоев.
Предположение о наличии неподвижных адсорбционных слоев, принятое при выводе уравнения (72), по-видимому, обычно выполняется. На это, в частности, указывают результаты
проведенных Линдом исследований переноса вещества Через межфазную границу в присутствии ПАВ.
Независимость вязкости т] от толщины пленки, требующаяся согласно третьему условию, как правило, имеет место, хотя экспериментальные данные по изучению реологических свойств тонких слоев часто находятся в противоречии друг с другом. Например, в пенных пленках [126] и в зазоре между твердыми поверхностями [143] не обнаружено заметного отличия значений т], характеризующих течение жидкости в тонком слое и в большом
Рис. 38. Фотографии последовательных стадий процесса образования и „выталкивания" димпла при сближении двух капелек эмульсии.
объеме; в то же время для многих систем Дерягиным и сотрудниками путем непосредственных измерений установлено [144, 145], что жидкость в граничных слоях толщиной порядка 30—40 молекулярных размеров обладает повышенной вязкостью*.
Возражения, вызываемые применением формулы Рейнольдса в случае нарушения плоскопараллельности межфазных поверхностей, не настолько принципиальны, чтобы отказаться от применения динамического метода для модельных исследований пен
* Вопросу непосредственного измерения вязкости в тонких пленках посвящены также работы [327—332]. Было показано, что слой, в котором реологические свойства жидкости оказываются измененными, отделен от объема дисперсионной среды резкой границей, т, е. является особой фазой [333]. (Прим, ред.)
и эмульсий; его особенно хорошо использовать для определения расклинивающего давления как функции толщины слоя.
Рис. 39. Зависимость суммарного расклинивающего давления в пленке раствора NP-20 между двумя капельками циклогексана от ее толщины при двух концентрациях КО в системе: /-2.5 . 10~3; 2-2моль/л,
На рис. 39 подобные зависимости представлены для пленок раствора NP-20, заключенных между капельками циклогексана, при
двух концентрациях индифферентного электролита. Кривые на графике отвечают различным состояниям системы: устойчивому к коалесценции (кривая 1) и неустойчивому (кривая 2). Нельзя считать вполне обоснованным, вследствие особого характера течения жидкости в тонких слоях, вычисление постоянных взаимодействия А*, исходя из найденных динамическим методом зависимостей Пт = — f(h). Вм'есте с тем, совсем не оправ
Рис. 40. Схема установки для изучения коалесценции двух капелек ртути:
1 — капельные ртутные электроды; 2~ измерительная ячейка; 3 — нормальный каломельный электрод; 4 — потенциометры.
дан, по-видимому, вывод о проявлении электромагнитного запаздывания, который в ряде работ [63, 146] делается ввиду отсутствия пропорциональности, согласно уравнению (39а), между Пт и h~\
Проверка соотношения саг? = const, а также изучение влияния потенциала
поверхности на критическую концентра-
цию коагуляции проведены Ватанабе и Готохом [147]. Применяемая ими экспериментальная установка (рис. 40) состояла из
двух капельных ртутных электродов 1, расположенных горизонтально в измерительной ячейке 2. Ртутные электроды одинаково поляризовали по отношению к одному и тому же каломельному электроду 3 с помощью делителей напряжения 4. Измеряли потенциал, необходимый для предотвращения коалесценции капелек в растворе электролита заданной концентрации. С помощью аналогичной установки определена постоянная взаимодей-
-5 -4 ^3 ^2 О ТLg Сс
ствия капелек ртути в воде, содержащей KF [148].
В соответствии с теорией ДФО обнаружено, что по мере повышения содержания индифферентных ионов в дисперсион
Рис. 41. Зависимость критической концентрации от поляризационного потенциала двух капелек ртутн для различных электролитов:
Д - KCJ; о - KF; • - Na2SO4; V - Zn(NO8)2; И - La(NO|)3; □ - Th(NOs)4.
Числа на кривых обозначают валентность противоионов.
Рис. 42. Зависимость логарифма критической концентрации для двух капелек ртути от логарифма валентности противоионов.
ной среде для достижения устойчивости системы необходимо прикладывать все возрастающие значения потенциалов (рис. 41). При больших потенциалах, как можно было ожидать на основании уравнения (60), концентрация коагуляции не зависела от поляризации электродов. Откладывая на оси ординат величину логарифма критической концентрации, а на оси абсцисс— логарифм валентности противоионов, Ватанабе и Готох получили прямую с тангенсом угла наклона, равным 6 (рис. 42). Именно такую закономерность и предсказывает формула (60) теории ДФО. Однако этот результат не следует переоценивать. Точки, нанесенные на рис. 42, отвечают потенциалу 250 же, выше которого теоретически концентрация коагуляции должна сорханяться постоянной. Вместе с тем, на рис. 41 видно, что и в случае более высоких потенциалов наблюдается сильное изменение критической концентрации.
Поверхность жидкости вследствие теплового движения молекул не является плоской — на ней существуют микрошерохова
тости из-за наложения друг на друга поверхностных волн различной частоты и амплитуды. Возникновению волн противодействуют межфазное натяжение и гравитационные силы. Как показал Врие [149, 150], возмущение границы раздела жидкость— газ оказывает значительное влияние на устойчивость тонких слоев. Одновременное измерение интенсивности отражен,? ного и рассеянного света в процессе уменьшения толщины пленки позволяет рассчитать зависимость производной расклинивающего давления dTl/dh от величины h. К сожалению, результаты опытов Врие имеют больше качественный, чем количественный характер. Сравнительно недавно, используя тот же метод, Манн определил для воды константу Гамакера, которая оказалась равной 10-11 эрг [151]. Эта величина, по-видимому, несколько завышена.
Измерение сил взаимодействия твердых частиц. Непосредственному измерению сил молекулярного притяжения двух твердых тел посвящены многочисленные исследования [152—158]*. При оценке и обсуждении экспериментальных данных, полученных в этих работах, необходимо принимать во внимание два возможных источника ошибок:
1. Шероховатость твердой поверхности, а также сложность взаимной юстировки измерительного устройства и пластин конденсатора, применяемых для отсчета расстояния между изучаемыми конденсированными фазами.
2. Колебания измерительной системы при случайных внешних механических нагрузках**.
Первое обстоятельство приводит к тому, что измерения дают достаточно точную информацию о молекулярном взаимодействии только при больших расстояниях между рассматриваемыми поверхностями, когда притяжение твердых тел проявляется сравнительно слабо. Однако для определения малых значений сил требуются высокочувствительные установки, на которые заметно сказывается вибрация.
Во всех цитируемых работах измерение сил проводили с помощью микровесов. Расстояние между взаимодействующими поверхностями находили емкостным или интерференционным методом. Схема одной из таких экспериментальных установок показана на рис. 43. С коромыслом / весов, установленном на пружине 2, жестко соединены верхняя исследуемая пластина 3 и нижняя пластина конденсатора 4. Тарировка коромысла производится грузами 5. Верхняя подвижная пластина конденсатора^ укрепляется на стойке 7. Для успокоения колебаний коромысла предназначен демпфер 8. Вторую исследуемую пластину 9 помещают на подвижный столик 10, снабженный для юстировки
* См. также [334—337]. (Прим, ред.)
** Одной из важнейших причин погрешности измерений является присутствие зарядов ид исследуемых поверхностях [337]. (Прим, ред.)
тремя микрометрическими винтами И и двумя мембранами 12\ управление последними осуществляется посредством создания давления в камере 13, заполненной силиконовым маслом. Повышение чувствительности микровесов достигают компенсацией веса коромысла с помощью специального рычага (на рис. не показано). Вся установка плавает на плоту, что сводит к минимуму влияние случайных сотрясений.
Рис. 43. Схема установки для измерения сил молекулярного взаимодействия твердых тел [157]'.
/ — коромысло; 2—пружина; 3 —верхняя исследуемая пластина;
4 — нижняя пластина конденсатора; 5 — тарнровочиые грузы;
6 —верхняя пластина конденсатора; 7 —стойка; 3 —демпфер;
9— нижняя исследуемая пластина; 10—подвижный столик; И—микрометрические винты; 12 — мембраны; 13 — камеры давления.
Результаты экспериментального изучения взаимодействия твердых тел в вакууме подтверждают в пределах погрешности измерений макроскопическую теорию Лифшица [54] *. Найденные для плавленого кварца двумя различными авторами зависимости сил молекулярного притяжения от расстояния представлены на рис. 44 и 45. Постоянные взаимодействия А, вычисленные из опытных данных, а также на основании макроскопической и микроскопической теорий, сопоставлены в табл. 11. Для сравнения, исходя из ранее табулированных значений А, с учетом уравнений (51) и (55), формально рассчитывали величину В, причем лондоновскую длину волны полагали равной 10~5 см. Из таблицы видно, что имеется достаточно хорошее сорлдси?
* См. [329]. (Прим, ред.)
между экспериментально найденными и предсказанными теорией значениями В, хотя нельзя не заметить сильного отличия постоянных взаимодействия, определенных различными авторами для одного и того же материала твердых тел. Непонятной остается также зависимость сил молекулярного притяжения от радиуса кривизны.
Рис. 45. Зависимость логарифма величины сил молекулярного притяжения плоской пластины и линзы с радиусом кривизны 715,2 см, изготовленных из плавленого кварца, от логарифма кратчайшего расстояния между ними
Тангенс угла наклона равен — 3 В—0,66 • 10~'9 эрг-см [158].
Рис. 44. Зависимость логарифма величины сил молекулярного притяжения плоской пластины и шара радиусом 11,1 см, изготовленных из плавленого кварца, от логарифма кратчайшего расстояния между ними.
Тавгенс угла наклона прямой равен — 3, В=0,81 • Ю—19 эрг-см [153].
В связи с рассмотрением вопроса о взаимодействии твердых тел необходимо указать еще на модельные исследования Дерягина и его сотрудников [160—163]. В этих работах измеряли силовой барьер, препятствующий сближению скрещенных платиновых или золотых нитей в растворах электролитов, и затем с помощью формулы (59) рассчитывали энергию отталкивания двух пластин. Проволочки поляризовали до определенного одинакового потенциала. Из результатов измерений вычислены постоянные Гамакера А*, которые в случае платины и золота оказались равными 10-10~13 и 44-10'13 эрг, соответственно*.
* В цитируемых работах (см., кроме того, [338, 339]), а также в ряде статей [340—342], посвященных изучению сил прилипания пластин и нитей в растворах электролитов, показано, что отсутствие непосредственного контакта поверхностей связано в некоторых случаях с действием сил отталкивания молекулярной природы, обусловленных перекрытием граничных фаз. По-видимому, не принимая во внимание эту компоненту взаимодействия, представляется невозможным объяснить совокупность экспериментальных данных, установленных при исследовании процессов адгезии частиц В жидких средах [343]. [Прим, ред.)
Таблица 11
Теоретические и экспериментальные значения постоянных молекулярного взаимодействия двух твердых тел
Вещество Снс-те* ма * Радиус кривизны, см эрг.см А-1013, эрг Литература
макроскопическая теория микроскопическая теория экспериментальное значение
Плавленый кварц |П— л 10,0 0,57 0,75 1,21 9,8
п—л 11,1 0,57 0,75 0,81 6,5 | 152
п—л 25,4 0,57 0,75 1,37 11,1
п—п 0,57 0,75 2,00 11,6 155-
П—л 715,2 0,57 0,75 1,15 9,3
п—л 715.2 0,57 0,75 0,66 5,3
Кристаллический п—л 715,2 0,70 0,96 0,74 6,0 158
кварц п—л 400,0 0,70 0,96 0,84 6,8
Боросиликатное п—п 0,68 0,90 1,12 9,0 157
стекло
Галогенид талия п—л 5,2 2,25 3,50 4,96 40,0
п—л 12,5 2,25 3,50 3,51 28,3
Плавленый п—л 5,0 2,18 — 3,60 29,0 153
кварц — хром п—л 10,0 2,18 — 3,00 24,2
Кристаллический п—л 715,2 2,40 2,90 23,4
кварц — хром Пленки хрома тол- п—л 715,2 7,9-11,7 8,2—18,0 66-145
щиной 380 А на кварце 158
Пленки хрома тол- п—л 715,2 2,4—10,8 —и. 4-9 32-73
щиной 80 А на кварце
Золото—германий п—л 4-l°-3S 8,3 — 1-10 8-80 156
Золото — стекло п-л 4- 10 2,3 — 1-10 8-80
Слюда — слюда Ц-Ц — — 0,87 10** 159
* П —пластины; Л — линзы; Ц — цилиндры, *♦ Значение, найденное экспериментально.
КОАГУЛЯЦИЯ В НЕПОЛЯРНЫХ СРЕДАХ
Дальнее взаимодействие частиц в дисперсиях определяется величинами молекулярных и ионно-электростатических сил. В то время как для молекулярного взаимодействия частиц в неполярных средах справедливы установленные ранее закономерности (см. главу I, стр. 30), для расчета энергии ионно-электростатического отталкивания необходимо получить другие формулы. В жидкости с низкой диэлектрической постоянной диссоциация 32
незначительна, и, следовательно, мала концентрация ионов. Последняя составляет по порядку величины 10-10.моль/л. Поэтому противоионы вокруг коллоидных частиц располагаются в виде сильно размытого диффузного двойного слоя (1/х та 10~4 см). Можно рассматривать вычисление энергии отталкивания двух сферических частиц с радиусом а и потенциалом поверхности фо как простую электростатическую проблему, • пренебрегая экранировкой заряда поверхности противоионами, располагающимися в диффузном слое. Однако это решение сложно, так как в интересующем нас случае радиус частиц велик по сравнению с расстоянием.
С целью упрощения задачи предположим, что заряд q частиц концентрируется в центре. Тогда емкость шара равна его радиусу и для энергии отталкивания получим:
2 . 2 _ q
Vi " е (Л + 2а) Л + 2а
Аналогично, для энергии отталкивания пластин с площадью F имеем:
(76)
В отличие от растворов электролитов, в неполярных средах силы ионно-электростатического взаимодействия уменьшаются очень медленно при увеличении толщины жидкой прослойки, разделяющей частицы. Это, в частности, видно из табл. 12, где
Таблица 12
Силы притяжения и отталкивания двух сферических частиц в неполярной среде
л, А Н?-1013, Эрг vsm -10», эрг
Фс=25 Мв Фо=50 мв фй= 100 мв
а — 1,10-4сл
50 6,94 27,7 111 -801
100 6,91 27,7 111 -39,1
250 6,86 27,4 ПО -14,7
500 6,79 27,1 108 - 6,99
1000 6,61 ' 26,4 106 - 3,28
1=1- 10—5 см
50 0,679 2,71 10,8 -6,99
100 0,661 2,64 10,6 —3,28
250 0,618 2,48 9,65 -1,23
500 0,555 2,22 8,88 —0.602
1000 0,462 1,85 7,41 -0,291
приведены значения энергии молекулярного притяжения и ионно-
электростатического отталкивания для различных радиусов а, расстояний h и фо-потенциалов при е = 2 и Л* = 5-10~13 эрг. Сопоставление величин Vsm и позволяет сделать вывод о существовании барьера отталкивания для частиц с радиусом а — 1-Ю-5 см и потенциалом ф0 = 25 мв. Однако высота барьера меньше энергии теплового движения микрообъектов, и поэтому он практически не предотвращает флокуляцию. Поскольку в неполярных средах потенциал поверхности обычно не превышает 100 мв, устойчивость таких систем, очевидно, может до-
стигаться только в случае достаточно крупных частиц с размером более 10’5 см. Вместе с тем, необходимо принимать во внимание, что в зависимости от разности плотностей происходит либо седиментация, либо всплывание столь больших частиц.
По виду седиментационного осадка часто трудно определить, идет ли речь о стабильной или нестабильной системе. Оценку можно произвести, зная энергию, требующуюся для проведения редиспергирования. Известное различие проявляется также в скорости седиментации. Стабильные
Рис. 46. Зависимость расклинивающего давления от толщины пленок октана, находящихся между капельками воды и стабилизированных span-80.
дисперсии седиментируют медленно, нестабильные — быстро и характеризуются резкой границей раздела осадок —дисперсионная среда.
Работы, посвященные исследованию дисперсных систем с неполярной средой, немногочисленны и содержат порой противоречивые результаты. Обычно стабильные дисперсии в слабо-проводящих жидкостях получают при добавлении макромолекулярных и поверхностно-активных веществ. Как однозначно показано при изучении пенных углеводородных пленок [63, 146] и эмульсий типа вода/масло, их устойчивость не связана с ионноэлектростатической компонентой расклинивающего давления. Величина суммарного расклинивающего давления как функция толщины тонкого слоя представлена на рис. 46. Кривая по
строена на основании опытных данных измерения скорости утоньшейия пленки. Определение равновесных толщин углеводородных пленок всегда приводит к значению, приблизительно соответствующему удвоенному размеру молекул ПАВ. Этот вопрос подробнее рассмотрен в главе 2.
Измерение -скорости утоньшения жидких слоев интерференционным методом дает информацию для расчета константы Гамакера. Так, для пленок бензола, стабилизированных силиконовым маслом, А = 2,3-10-12 эрг, для пленок хлорбензола в при-
Сутствии «Аэрозоля МА» и для анилина с добавками Додецилового спирта — А = 5,8-10~12 эрг. Эти значения, вероятно, являются завышенными, поскольку при использовании аналогичной методики в эмульсиях типа масло/вода для октана найдено: Д = 9.10~13 эрг. Еще меньшая величина константы Гамакера (8,5-10-13 эрг) характеризует взаимодействие частиц полистирола, причем оба последних значения достаточно хорошо согласуются друг с другом.
Измерение толщины масляных пленок между капельками ртути или воды удобно проводить емкостным методом [125, 167, 170, 171]. Обычно тонкие слои неполярных жидкостей получают либо в ячейке, схема которой приведена на рис. 27, либо с помощью проволочных рамок [167, 168]. Емкостной метод имеет перед оптическим то преимущество, что он дает возможность с высокой точностью определять малые значения толщин. Масляные пленки между капельками воды представляют однослойную систему, потому что неполярные радикалы молекул ПАВ, расположенные в дисперсионной среде, обладают диэлектрической постоянной, приблизительно равной диэлектрической постоянной углеводородов*. Вместе с тем, измерения емкости должны сопровождаться контролем плоскопараллельности тонкого слоя оптическим методом. Как обнаружено при наблюдении сближения капелек воды в октане и ксилоле, неравномерность пленки жидкости по толщине выражена сильнее по сравнению со случаем эмульсий типа масло/вода (см. стр. 75) и, следовательно, уравнение Рейнольдса (72) позволяет произвести лишь грубую оценку постоянной взаимодействия А *.
В соответствии с результатами исследований единичных тонких слоев жидкости, характеризующейся низкой диэлектрической постоянной, стабилизация обратных эмульсий наступает только при добавлении ПАВ [172, 173]. На основании изучения реологических свойств этих эмульсий при известных условиях можно сделать вывод о силах молекулярного взаимодействия отдельных частиц и вычислить константу Гамакера. Таким путем в работе [172] для капелек воды, стабилизированных Span-80** в парафиновом масле найдеца величина А*, равная 5-Ю-13 эрг.
Устойчивость к флокуляции суспензий и золей с неполярной средой не обязательно связана с присутствием макромолекулярных и поверхностно-активных веществ, хотя наиболее стабильные системы образуются в растворах ионогенных ПАВ. По-видимому, при адсорбции последних важную роль играют силы ионно-электростатического отталкивания частиц. Имеются многочисленные опытные данные, относящиеся к получению
* Диэлектрическая проницаемость характеризует объемные свойства материала и поэтому приписывать радикалам некоторую величину е, строго говоря, некорректно. (Прим, ред.)
** Моиоолеат сорбитана —- технический продукт. (Прим, ред.}
устойчивых суспензий и золей с неполярной средой. Так, известны суспензии сажи в бензоле (а = 2,5 • 10~5 см, £ — 25 мв) [174], суспензии различных окислов, карбоната кальция и сульфата бария в ксилоле (а = 5-10-5 см, £ = 40 мв) [175], дисперсии окиси и гидроокиси алюминия в толуоле [176] с радиусом частиц 4-10-5 см и 5-10~5 см, соответственно (£ = 40 мв). Двойной слой в слабопроводящих жидкостях довольно толстый. Поэтому с достаточной степенью точности можно полагать £ = ф6.
Теория взаимодействия дисперсных частиц подтверждается также результатами работы [177], в которой обнаружено, что дисперсии сажи в парафиновых и ароматических углеводородах при размере частиц 0,1-10~5 — 3-10~5 см являются неустойчивыми. Напротив, стабилизация системы наблюдается в алкил-бензоле, имеющем одну или две углеводородных цепи [177], а также в гептане, содержащем добавки алкилбензола [178]. В обоих случаях установлено отсутствие заметного электрофоретического перемещения частиц.
По сравнению с полярными дисперсионными средами, в неполярных средах молекулярное взаимодействие частиц проявляется слабее*. Это можно показать, непосредственно вычисляя сложную константу Гамакера А*. Например, для воды она составляет 3,5-10~13 эрг, а для масляной фазы, такой как октан,-9-10-13 эрг. Поскольку А* = (Дг* — До2), при Д1 = const молекулярное притяжение частиц в неполярной среде должно быть меньше, если Д1 > До.
Величина Д* = 5-10~13 эрг, принятая при расчете табл. 12, соответствует А{ = 2,75-10~12 эрг. Проводя аналогичные вычисления для частиц иодида серебра (Д1 = 1,3-10-12 эрг), получим Vm= 6 • 10-14 эрг при а=1-10'5 см и h = 50 А. Следовательно, адсорбционные слои Si толщиной 25 А вполне достаточны для стабилизации по отношению к флокуляции, если предполагать, что алкильные цепи расположены в органической среде и Дз — До (см. стр. 37). Существование полимолекуляр-ных слоев ненасыщенных жирных кислот на частицах двуокиси титана доказано адсорбционным методом [179]. Полислои олеиновой кислоты, образующиеся из ее ^растворов в октане на капельках ртути, обнаружены при измерении емкости тонких пленок [180]. Такой механизм стабилизации, вероятно, определяет устойчивость дисперсий, содержащих частицы размером НО'3 — 5-Ю-5 см [181 —185], хотя для некоторых из рассмотренных систем наблюдалась зависимость между интенсивностью коагуляции и электрокинетическим потенциалом частиц.
Защитное действие макромолекулярных добавок в неполярных средах подчиняется основным закономерностям, установ
* Этот вывод нельзя считать вполне обоснованным ввиду того, что он получен на основании предположения об аддитивности межмолекулярных взаимодействий. (Прим, ред.)
ленным в случае водных дисперсий. Адсорбционные слои только тогда обеспечивают устойчивость дисперсной системы, когда их толщина достаточна и на расстояниях h = 26i величина энергии молекулярного притяжения частиц уменьшается приблизительно до kT. Вместе с тем, необходимо, чтобы взаимодействие между адсорбционными слоями было незначительно. Как показано при эллипсометрических измерениях [186, 187], толщины поверхностных пленок на частицах двуокиси титана в бензольных растворах полиэфиров и алкидных смол достигают 40—60 и 100 А, соответственно. Очевидно, что столь толстые адсорбционные слои обусловливают получение устойчивых к флокуляции суспензий.
ВЗАИМОДЕЙСТВИЕ РАЗНОРОДНЫХ ЧАСТИЦ. ГЕТЕРОКОАГУЛЯЦИЯ
Изучение взаимодействия разнородных частиц имеет исключительно большое значение для многих технологических процессов. К таким процессам следует отнести слипание газовых пузырьков и масляных капель с минеральными частицами при флотации [188, 189], предотвращение осаждения частиц грязи на твердых подложках в промывочной ванне [190], повышение прочности веществ при введении активных наполнителей, склеивание различных материалов и т. д.
Взаимодействие разнородных и однородных частиц подчиняется одним и тем же закономерностям или, точнее говоря, определяется величиной молекулярных и ионно-электростатических сил, проявляющихся при сближении поверхностей [191]. Сила молекулярного взаимодействия двух пластин в дисперсионной среде, согласно макроскопической теории Лифшица [54], для расстояний h, меньших лондоновской длины волны Л; вычисляется из уравнения (53) или из приближенной формулы (56). Знак силы молекулярного взаимодействия определяется значениями комплексных диэлектрических постоянных [66]. Очевидно, при произвольных ео, Bi и ег величина интеграла может быть как положительной, так и отрицательной, причем в первом случае частицы должны испытывать притяжение, а во втором— отталкивание. При больших расстояниях зависимость Рт от h представляет знакопостоянную функцию (рис. 47, а, б). По мере сближения взаимодействующих поверхностей возможна инверсия знака величины Рт из-за различного изменения комплексной диэлектрической проницаемости жидкости в тонких слоях (рис. 47, в—д).
Теория ионно-электростатического взаимодействия разнородно заряженных частиц и их коагуляция разработана Дерягиным [191]; она получила дальнейшее развитие в работе Деверо и де Бройна [192]. Показано, что противоположно заряженные Частицы всегда притягиваются друг к другу. Однако характер
взаимодействия сложнее, если знак зарядов поверхностей одинаков, но различны величины их потенциалов и "Фе,- При этом, когда фв, 1]^ = const, как следует из теоретического рассмотрения вопроса, ионно-электростатические силы на сравнительно далеких расстояниях вызывают отталкивание частиц, а на близких — притяжение. Высота максимума Pi(h) определяется меньшим значением потенциала. Больший потенциал оказывает существенное влияние лишь на радиус действия сил
Рис. 47. Возможные зависимости сил молекулярного взаимодействия двух разнородных частиц от расстояния между ними.
ионно-электростатического отталкивания. Некоторые из результатов расчета величины Pi для различных постоянных значений %, и 'Фа,, представлены на рис. 48 [148]. При вычислениях авторы работы [148] использовали табулированные данные, приведенные в монографии [192].
Приближенное уравнение для энергии ионно-электростатического взаимодействия сферических частиц при произвольных потенциалах их поверхностей дано Фюрстенау и сотрудникам [193]. Вывод его основан на двух основных предположениях, принятых ранее при получении выражения (31а). Полагали, что потенциал каждой из частиц (ф41 и мал, а их радиусы (щ и аг) гораздо больше толщины диффузного двойного слоя. В этом случае
П 4(а,4-а2) А
,,( 2te,te, , [1+ехр(-хй)] ]
X 1 , „2 1п Ti-----1--мГ + In [1 - ехр (- 2хй)] } (77)
I + Н [1 - ехр (- хЛ) ’ V г > М J )
не
проведены. Однако сле-
Рис. 48. Зависимость энергии ионно-электростатического взаимодействия двух пластин от расстояния между ними в 0,01 н. растворе I — 1-валент-иого электролита при различных потенциалах и соответственно:
/ — 46,1 и 46,1 мв; 2 — 78,6 и 46,1 мв; 3 - 153,6 и 46,1 мв; 4 — 153,6 и 35,8 мв; 5-153,6 и 20,5 мв.
При условии, что оба потенциала Ниже 25 мв, отклонений величины V’, вычисленной по уравнению (77), от значений, найденных по точным формулам, составляет ~1%, если фв1, фбг < 75 мв, ошибка не превышает 10%.
Экспериментальные исследования процесса гетерокоагуля-ции, подтверждающие справедливость теоретических рассуждений, в настоящее время полностью "
/ дует отметить ряд весьма интересных работ, посвященных изучению взаимодействия разнородных частиц. Дерягин и сотрудники [194] измерили для различных концентраций электролита равновесные. толщины жидких пленок, которые образуются между капельками ртути и кварцевой пластиной. Капельки ртути были различно поляризованы. Результаты опытов, однако, не подтвердили теорию. Так, изменение знака поляризационного напряжения не сопровождалось обращением знака, электростатических сил взаимодействия.
Другая проверка теории ионно-электростатического взаимодействия разнородных частиц предпринята в работе [148]. С помощью потенциометра поляризовали различным образом два расположенных горизонтально и соосно друг с другом (см. рис. 40) капельных ртутных электрода. Измеряли поляризационный потенциал, предотвращающий сближение частиц, т. е. коалесценцию капель. В растворах фторида калия, характеризующихся отсутствием специфической адсорбции ионов, данные теории и эксперимента практически совпадали,. Во-первых, отталкивание проявлялось только при одинаковых знаках зарядов и, во-вторых, расчетные зависимости (сплошные линии на рис. 49) хорошо согласовались с измеряемыми величинами. В растворах иодида калия обнаружено действие сил отталкивания при различных знаках зарядов, что, по-видимому, следует объяснить специфической адсорбцией ионов иода. Авторы работы [195] по этой же методике определили силы взаимодействия стеклянной мембраны с капелькой ртути и оценили постоянную Гамакера.
С помощью динамического метода (см. главу 1, стр. 73) Шелудко и сотрудники [196] нашли изотерму расклинивающего давления тонкого бензольного слоя на поверхности ртути. Тол-
Шины пленок измеряли, используя аппаратуру для наблюдения интерференции света, аналогичную той, которая применялась при исследовании пленок пен. В связи с тем, что ионно-электро-
статическая компонента расклинивающего давления в неполярной жидкости пренебрежимо мала, кривая, представленная на
50, соответствует функции Hm = f(h). При толщине слоя
рис.
400
Рис. 49. Зависимость поляризационных потенциалов, обеспечивающих отсутствие коалесценции двух капелек ртути в растворе K.F. Ковцевтрация KF, моль!л: 7 — 0,001; 2 — 0,01; 3 — 0,025;
4-0,05.
~ 240 А обнаружено равновесное состояние. Авторы не установили, к какому из двух типов зависимостей, изображенных на рис. 47, в и д, относится данная кривая*.
Рис. 50. Изотерма расклинивающего давления слоя бензола на ртути.
Совсем недавно появились работы, посвященные изучению взаимодействия пузырьков воздуха с поверхностью кварца. Рид и Китченер [197] интерференционным методом измерили тол* щину водных смачивающих пленок при различном содержании в них электролитов. Сопоставление экспериментальных и теоретических результатов показало их удовлетворительное согласие **.
Шелудко и сотрудники [198], исходя из значений краевого угла смачивания кварцевой поверхности жидкостью, определили силу адгезии газовых пузырьков.
* Эти результаты получены также Зориным с применением другого метода исследования [405]. (Прим, ред.)
** Впервые изучение толщины и устойчивости пленок воды, ограниченных с одной стороны пузырьком воздуха, а с другой — поверхностью твердого тела или поверхностью ртути, проведено Дерягиным и Кусаковым [137], Фрумкиным и Городецкой [344]. Этому вопросу посвящены теоретические и экспериментальные работы ряда других исследователей [345—350]. (Прим, ред.)
Г лава 2
КОАЛЕСЦЕНЦИЯ
Как упоминалось во введении, под коалесценцией следует понимать все процессы, приводящие к непосредственному контакту взаимодействующих поверхностей. В случае эмульсий и пен установление контакта вызывает укрупнение микрообъектов. При определенных условиях в суспензиях между частицами возникают только локальные области соприкосновения, которые обусловливают образование конденсационных структур. Сходство процесса коалесценции для всех дисперсных систем независимо от их агрегатного состояния заключается в прорыве слоя, разделяющего частицы. Этот слой может состоять как из дисперсионной среды, так и из граничных фаз, адсорбированных на поверхности частиц. Вследствие того, что устойчивость к коалесценции, по-видимому, объясняется различными причинами, неодинаковыми должны быть и механизмы прорыва. Рассмотрим устойчивость к коалесценции, зависящую от сил отталкивания ионно-электростатической природы, устойчивость уль-тратонких пленок растворов ПАВ (так называемых перрецов-ских черных пленок), а также строение и свойства гелеобразных слоев, образующихся на границе раздела фаз при полимолеку-лярной адсорбции ПАВ и макромолекул либо' при адсорбции коллоидных частиц.
'• - -э"?
ИОНОСТАБИЛИЗИРОВАННЫЕ .
РАВНОВЕСНЫЕ ЖИДКИЕ ПЛЕНКИ
Как показано в главе 1, в коллоидных дисперсиях при достижении некоторой концентрации электролита образуются коагуляционные структуры. При таком состоянии микрообъекты находятся на равновесном расстоянии ..друг от друга, т. е. жидкая прослойка, заключенная между ними, имеет определенную толщину. Причиной существования равновесной толщины, которую можно изменять в широких пределах путем вариации концентрации электролита, является взаимная компенсация сил притяжения и отталкивания (см. рис. 33 и 34). Рассмотрим, при каких условиях жидкие прослойки становятся нестабильными.
Для пленки, находящейся между пузырьками воздуха, Де Вриес [199] предположил, что под влиянием флуктуаций в ней
самопроизвольно возникают утоньшения. Если дальнейшее расширение области утоньшения связано с уменьшением свободной энергии, то наступает прорыв слоя. Снижение энергии системы происходит вследствие исчезновения метастабильного участка пленки при образовании утоньшения, а увеличение определяется возрастанием межфазной поверхности раздела *. Суммарное изменение энергетического состояния жидкого слоя, вызванное колебаниями толщины, обусловливает интенсивность процесса прорыва. При переходе от толстых пленок к тонким
возрастает вероятность их разрушения, так как при этом уменьшается энергия образования поверхности раздела фаз и увеличивается энергия, выделяющаяся благодаря возникновению флуктуационного утоньшения. Однако представления Де Вриеса находятся в противоречии с экспериментальными данными. Как следует из проведенных им вычислений, энергия активации, необходимая для возникновения утоньшения, уже для пленок толщиной 105А настолько высока, что их прорыв не должен наблюдаться.
Рассмотрим сначала две частицы золя, которые в ходе процесса флокуляции сблизились до равновесного расстояния. В результате теплового движения в некоторый момент времени частицы выходят из состояния равновесия и приближаются на меньшие расстояния. Это мгновенное нарушение равновесия может вызвать коалесценцию, если производная сил притяжения по расстоянию изменяется сильнее, чем производная сил
Рис. 51. Схема локального изменения толщины пленки.
отталкивания, т. е. при выполнении неравенств
dh dPm . dh ' dPi dh (78)
или dHm . dh ' dn.[ > dh (78a)
В случае межфазных границ,
характеризующихся возможностью изменения своей формы, во флокулированном состоянии образуется почти плоскопараллельная зона соприкосновения, как это наблюдается, например, в пенах и эмульсиях (рис. 29а). Для таких систем разрыв пленок связан с преобразованием
формы и увеличением поверхности соприкосновения фаз (рис. 51). Поэтому неравенство (78а), отвечающее переходу пленки в неустойчивое состояние, должно содержать еще одну
* Потеря устойчивости связана с течением внутри слоя в сторону от области, где намечается его разрыв, а сохранение устойчивости, наоборот, должно обеспечиваться подтеканием жидкости к этой области. Иными словами, критерием устойчивости является направление потока в пленке при наличии градиента ее толщины. Следовательно, устойчивости пленок может быть дана наглядная гидромеханическая трактовка 1351]. (Прим, ред.}
(79)
компоненту расклинивающего давления определяемую
изменением границы раздела при возникновении утоньшения:
dnm dnf , d dh > dh + dh
Величину ДР* не следует отождествлять с капиллярным давлением в пенах и эмульсиях, обусловленным кривизной поверхности дисперсных частиц: последнее не зависит от толщины пленки и всегда способствует притяжению *.
Чтобы можно было оперировать с неравенством (79), необходимо знать величины всех составляющих расклинивающего давления тонкого слоя как функции его толщины h. Для молекулярной составляющей обычно исходят из уравнения (39а), дифференцируя его по h, или из выражения (51). Ионно-электростатическая компонента вычисляется по формуле (286). Однако очень трудно получить данные о значении ДР’, так как в настоящее время не существует единой точки зрения о размерах областей, на которые распространяются колебания толщины пленок, приводящие к прорыву.
Теоретические представления о механизме разрыва неустой-; чивых плоскопараллельных пленок пены, имеющих форму диска, без учета электростатической составляющей расклинивающего давления развиты в работах Шелудко [133, 200, 201]** и Врие [202].
i Согласно работам Шелудко, периодические колебания тол-; щины в пленке возникают благодаря стоячим цилиндрическим : волнам, причем предполагается, что длина волны % находится !. в определенном соотношении с радиусом R диска. Пленка жид-; кости нестабильна, когда флуктуации толщины, характеризуемые амплитудой волны, обеспечивают выполнение неравенства (79). В случае предельной толщины слоя, для которой возникновение утоньшения еще не приводит к прорыву, пренебрегая ионно-электростатической компонентой, имеем:
d ДР'
= ——° (80)
dh dh '
Лапласовский перепад давления ДР', способствующий стабилизации толщины пленки, зависит от поверхностного натяже-
* Значение ДРО характеризует локальный лапласовский перепад давления в плоскопараллельной пленке, связанной с временным изменением ее толщины. В общем случае
где о — поверхностное натяжение на границе раздела фаз; у—координата, отсчитываемая вдоль горизонтальной оси. (Прим, ред.)
* * См. также [352]. (Прим, ред.)
ния ст и радиуса кривизны г* элемента объема, деформированного волнами. Для малого изменения толщины Д/i > 0 запишем уравнение, связывающее величину г* и длину поверхностной волны Л
откуда для ДР* следует:
d ЬРа 64<т dh = T
(82)
Данные о длине волны X в настоящее время отсутствуют, однако можно предполагать, что она пропорциональна радиусу пленки.
Дважды дифференцируя по h уравнение для молекулярного взаимодействия (39а), получим:
dHm = А dh = 2лЛ4
(83)
Приравнивая правые части формул (82) и (83), для критической толщины имеем: . / лл2
с = \ 128 ла
(84)
Чтобы учесть влияние эффекта электромагнитного запаздывания молекулярных сил, необходимо продифференцировать по h уравнение (51) . Тогда для критической толщины получим значительно более сложное выражение:
0,49ЛЬ 0,171 ЛХ? 64<т
ТС <1 с fl q ht
Пренебрегая членом, пропорциональным l/ft®, ближенное решение в виде:
_ ( 0Л9ЛЛ/Л2 \’/s
с~ ( 64л2а )
запишем при-
(86)
Врие, напротив, полагает хаотическое распределение флуктуаций толщины. Исходя из вероятности возникновения флуктуаций, он [202] вывел уравнение для критической толщины, содержащее только измеряемые величины. Колебания толщины пленки происходят в результате наложения всех независимых стационарных синусоидальных волн, амплитуды которых являются показательными функциями времени. Вывод уравнения для hc основан на сравнении скорости распространения флуктуации, приводящей к прорыву, и скорости уменьшения толщины при вытекании раствора из пленки.
Первый параметр характеризуется временем 24faty dn (87)
h4h
где f — коэффициент, слабо зависящий от толщины и изменяющийся от 4 до 7 (см2-сек) 1г.
Второй параметр может быть вычислен из уравнения Рейнольдса (72):
1 ) а \ ДА2 ) 4_. П
dt = 3 ‘ п/?2
Найдены два приближенных решения для критической толщины: для больших толщин, когда скорость вытекания определяется только капиллярным давлением искривленной поверхности, т. е. при Пт -С
/ Л2Л>2 \'h
(88)
и для расстояний, при которых Пт ДЛт
hc = 0,22 ' (89)
Для малых.толщин пленок уравнения Щелудко и Врие совпадают с точностью до постоянного числового множителя. Так, согласно работам [.199] и [133, 200, 201], критическая толщина зависит от молекулярной составляющей расклинивающего давления (Л'л), радиуса плоскопараллельной Пленки (R'11) и удельной поверхностной энергии (о|/4)- При рассмотрении сравнительно толстых пленок нельзя не учитывать эффекта электро-s магнитного запаздывания дисперсионных сил. В этом случае следует использовать уравнение (86). Однако это уравнение соответствует состоянию, реализующемуся, по-видимому, очень редко. Необходимо еще раз подчеркнуть, что выражения (80), (82) — (89) относятся к нестабильным пленкам, в которых ионно-электростатические силы не проявляют своего действия'. Экспериментальная проверка теории проведена на мыльных пленках [201, 203] и капельках эмульсий [204, 205]*. Подтверждение, естественно, можно ожидать при выполнении условий плоскопараллельности и равенства нулю скорости течения на межфазной границе. Кроме того, должна быть известна зависимость расклинивающего давления от толщины при постоянйой температуре. Прежде всего рассмотрим, как изменяются значения /ic, когда радиус частиц или величина поверхностного натяжения стремятся к нулю. Уменьшение величины о в соответ-
* Экспериментальная проверка теории впервые проведена в работах [118—120]. (Прим, ред.)
СТййй с уравнениями (86), (88) и (89) должно приводить к ро-сту критической толщины вплоть до бесконечности, т. е. обусловливать увеличение неустойчивости. Однако известно, что
именно при равенстве нулю поверхностного натяжения система является неограниченно устойчивой. При R—>0, как следует из уравнений для критической толщины, стабильность очень ма-
леньких пузырьков пен и капелек эмульсий беспредельно воз-
растает. Вместе с тем, результаты экспериментальных исследо-
ваний водных прослоек пен, пленок хлорбензола и анилина не
закономерность. Экстраполяция зависимо-
подтверждают такую
Рис. 52. Зависимость лога-
рифма критической толщины от логарифма радиуса пленок:
7 —вода: 5~авилии; J —хлорбензол»
сти критической толщины от радиуса пленки всегда дает для R — 0 конечную величину hc.
Представляя зависимость hc от R в логарифмическом масштабе (рис. 52) и определяя из угла наклона показатель степени при значении R, для хлорбензола, анилина и воды найдем величину, близкую к 2/7. На первый взгляд это говорит о справедливости уравнения (88) для хлорбензола и анилина. Однако уравнение (88) обычно не соответствовало условиям проведения опытов, при которых, как правило, выполнялось соотношение Пт 3* &Ра-
Измерения в случае тонких слоев между капельками эмульсий дали совершенно иные результаты [205]. Так, не обнаружена зависимость критической толщины пленки от величины по-
верхности соприкосновения. По-видимому, причина состоит в том, что подобные пленки нельзя рассматривать как плоскопараллельные. В периферийной зоне пленок всегда имеется утоньшение, а в центре — утолщение (см. рис. 37) *. Эта неоднородность увеличивается с возрастанием радиуса. При измерении толщины интерференционным методом путем фотометри-рования всей пленки влияние радиуса изучается лишь формально. Напротив, если измеряют толщину малых сегментов пленки и направляют фотоумножитель в место с наименьшей толщиной, то значение hc оказывается независимым от радиуса.
Неравномерность толщины пленки между двумя капельками не является случайной, а возникает при вытекании раствора из прослойки, т. е. она связана с гидродинамикой течения. Поэтому нё удивительно, что при исследованиях эмульсий исчезает влияние поверхностного натяжения на устойчивость к коалесценции.
♦ Аналогичную форму могут иметь также пленки, заключенные между пузырьком газа и твердой поверхностью [137]. (Прим, ред.)
Отсюда следует заключить, что йрорыв пленки, разделяющей капельки эмульсий, не зависит от соотношения энергий, определяемых исчезновением метастабильного участка слоя и образованием новой поверхности раздела фаз; он наступает, когда в процессе сближения деформированные периферийные зоны становятся очень тонкими, в результате чего может произойти значительное увеличение молекулярной компоненты расклинивающего давления. Исследование капелек эмульсии позволяет установить влияние поверхностного натяжения на устойчивость тонкого жидкого слоя, не повышая концентрации ПАВ в пленке. С малыми количествами ПАВ, которые растворимы только в дисперсной фазе, можно достичь большей степени заполнения адсорбционного слоя исходного эмульгатора и, таким образом, варьировать величину поверхностного натяжения.
Рассмотрим, например, водные пленки, стабилизированные полиэтиленгликолевым эфиром нонилфенола NP-20 и разделяющие капельки октана. В качестве маслорастворимого ПАВ применен моноглицерид олеиновой кислоты. При обсуждении результатов, представленных в табл. 13, необходимо принимать во внимание, что моноглицерид олеиновой кислоты имеет поверхностную активность на два-три порядка меньшую, чем NP-20. Поэтому равной концентрации в объеме раствора соответствуют различные поверхностные концентрации. Приведенные данные однозначно подтверждают отсутствие зависимости критической толщины от поверхностного натяжения.
Таблица 13
Влияние поверхностного натяжения на критическую толщину пленок 0,1 М раствора КС1 между двумя капельками октана, содержащего различные количества моноглицерида олеиновой кислоты
Концентрация раствора NP-20 составляет 3 • 10“5 моль/л.
Концентрация моноглнце-рида олеиновой кислоты молъ/л Поверхностное ватяженяе, дин/см Лс, А
0 16,6 175
2,8 • ГО-5 12,5 185
1.4-10“] 9.1 180
2,1 • 10“4 4,0 185
Вместе с тем, опыты, проведенные с пенными пленками хлорбензола и анилина, показали, что поверхностное натяжение может оказывать существенное влияние на устойчивость к коалесценции [201]*. Так, при одинаковых радиусах пленок их
* См. также [119]. (Прим, ред.)
критическая толщина для обеих жидкостей значительно различается (табл, 14), Согласно исследованиям Манева и Боуле-вой [125], постоянные Гамакера и лондоновские длины волн для обеих органических сред равны и, по-видимому, различие в свойствах пленок может быть связано только с величинами поверхностного натяжения. Для хлорбензола значение а составляет 32,6 дин!см, а для анилина о = 42,6 дин!см.
Таблица 14
Зависимость критической толщины от радиуса пленок хлорбензола и анилина
Радиус пленки, мм А
хлорбензол аиилин
0,036 300 278
0,120 406 378
0,154 435 411
0,200 469 437
При модельных исследованиях эмульсий показано [204], что критическая толщина пленок изменяется в зависимости от молекулярного взаимодействия, проявляющегося в системе. В частности, такие изменения достигаются путем подбора соответствующих ПАВ и органической среды. На вопрос, оказывают ли добавки электролитов влияние на молекулярное взаимодействие, следует ответить отрицательно, поскольку в растворах КС1, K2SO4 и MgSO4 в области концентраций от 5-10~2 до 2 моль/л не наступает заметного изменения критической толщины пленок.
Молекулярное взаимодействие капелек эмульсий определяется постоянной Ван-дер-Ваальса — Гамакера А*, учитывающей наличие жидкой прослойки между рассматриваемыми микрообъектами [см. уравнение (41)]. Вероятно, нельзя ожидать слишком сильного изменения критической толщины для различных ПАВ и неполярных органических жидкостей. Так, критическая толщина водной пленки, стабилизированной NP-20, практически не зависит от природы дисперсной фазы:
А
Октан.......................... 185
Хлорбензол.......................195
Циклогексан .....................210
Декалин .........................215
Аналогично не обнаружено значительного влияния на критическую толщину водных прослоек, заключенных между капельками октана, длины углеводородной цепи в ряду полиэтиленгли-
Колевых эфиров нонилфенола, а также молекулярной структуры применяемых ПАВ:
NP-12.............................210
NP-20............................ 185
NP-30 . .........................175
Na-3-гидрокситетрадекансульфонат 232
Из приведенного материала видно, что результаты изучения критической толщины и ее зависимости от поверхностной энергии, молекулярной составляющей расклинивающего давления и радиуса соприкосновения содержат много неясностей и противоречий. При достижении величины hc происходит либо прорыв пленки, либо, если на границе раздела фаз существуют адсорбционные слои ПАВ с оптимальной поверхностной концентрацией, наблюдается скачкообразное изменение толщины, приводящее к возникновению нового метастабильного состояния — так называемых перреновских черных пленок*.
УСТОЙЧИВОСТЬ ПЛЕНОК,
СТАБИЛИЗИРОВАННЫХ МОНОМОЛЕКУЛЯРНЫМИ АДСОРБЦИОННЫМИ СЛОЯМИ ПАВ.
ЧЕРНЫЕ ПЛЕНКИ
Как показано выше, слой жидкости между двумя дисперсными частицами при критической толщине становится неустойчивым. Этот процесс не обязательно приводит к коалесценции. Когда на межфазной границе имеются адсорбционные слои с определенной поверхностной концентрацией, возникает новое квазиравновесное состояние. Оно характеризуется сохранением относительно тонкой пленки, предотвращающей коалесценцию.
Явление скачкообразного перехода от толстых пленок к тонким известно сравнительно давно. Еще Ньютон [206] наблюдал в мыльных пузырях черные «дыры», которые в действительности представляли очень тонкие пленки. Существование черных пленок обнаружено не только в пене. В последние годы доказано, что они обусловливают устойчивость к коалесценции эмульсий вода/масло и масло/вода, а также капелек ртути и частиц полимеров, находящихся в водных растворах ПАВ и макромолекул. Термин «черная пленка» впервые введен при обсуждении результатов наблюдения мыльных пузырей в отраженном свете. Свет отражается от обеих фазовых границ и, так как при переходе от оптически более плотной среды к менее плотной наступает фазовый сдвиг на 180°, оба луча при малой толщине
* Теория флуктуационного прорыва перреновских черных пленок развита Дерягиным и Гутопом [353]. Изучению устойчивости тонких жидких слоев посвящены также работы [354—358]. (Прим, ред.)
исследуемого объекта почти полностью гасят друг друга для всех длин волн. Поэтому тонкие слои кажутся черными.
Образование черных пленок можно проследить, фотографируя микроскопические прослойки в пенах [133] и эмульсиях [127].
Рис. 53. Фотографии различных стадий образования черной пленки между капельками эмульсий.
В зависимости от радиуса действия электростатических сил, определяемого ионной силой раствора, черная пленка более или менее быстро распространяется на всю поверхность. При малой скорости распространения многочисленные черные области, возникающие сначала в периферийной зоне, медленно растут, объединяются и превращаются затем в однородную черную пленку (рис. 53). В растворах электролитов высокой концентрации та-
жизни капелек ртути в л-ксилоле от концентрации стеариновой (кривая /), пальмитиновой (кривая 2) и миристиновой (кривая 3) кислот.
кой процесс происходит очень быстро: в течение нескольких секунд или долей секунды. Переход от черной пленки к толстому слою в периферийной зоне является резким. Краевой угол, который устанавливается между ними, позволяет вычислить изменение поверхностного натяжения в тонкой пленке *.
Во время образования, т. е. от момента возникновения первой черной области до момента окончания формирования сплошной черной пленки, прослойка жидкости между пузырьками пены или капельками эмульсии весьма чувствительна к внешним механическим воздействиям; последние при определенных условиях вызывают быструю кбалесценцию. Напротив, если вся поверхность представляет черную пленку, то она может выдерживать большие механические напряжения и является почти безгранично устойчивой.
Концентрация ПАВ, необходимая для образования устойчивых черных пленок. Обширный экспериментальный материал, посвященный изучению устойчивости изолированных жидких прослоек в пенах {129, 133], капелек воды и ртути в органических средах [125, 207], масляных капелек по отношению друг к другу и к пленкам полимеров в водных растворах [125, 190, 208], однозначно указывает на то, что длительность жизни пленок сильно изменяется в очень узком интервале концен
траций ПАВ, характеризуемом величиной cs (рис. 54). Эта концентрация, как правило, отвечает содержанию ПАВ в системе, при котором происходит возникновение черных пленок. Для каждого водорастворимого ПАВ значение cs зависит от температуры, присутствия примесей других поверхностно-активных веществ, природы дисперсной фазы, величины pH, а для ионогенных ПАВ — и от содержания электролита.
На стабилизирующее действие неионогенных ПАВ ионная сила раствора не оказывает существенного влияния. Например, концентрация полиэтиленгликолевого эфира нонилфенола NP-20, необходимая для стабилизации капелек октана, не изменяется при переходе от чистой воды к 0,5 н. раствору КС1 [125]. Однако, поскольку при таких опытах толщина, равновесных жидких пленок значительно уменьшается, становится ясно, что за
* См. [403]. (Прим, ред.)
устойчивость ответственны в первую очередь физико-механические свойства адсорбционных слоев.
Концентрации стабилизации cs, определенные при модельных исследованиях, позволяют указать благоприятные условия получения устойчивых дисперсий. При этом, конечно, необходимо принимать во внимание, что решающее значение для стабилизации имеет концентрация ПАВ не в объеме, раствора, а на межфазной границе. В модельных системах поверхность жидкой прослойки, заключенной между двумя капельками или пузырьками, мала, и адсорбция не приводит к заметному изменению содержания ПАВ в объеме. При изготовлении эмульсий путем смешения определенных количеств воды и масла из-за большой удельной поверхности частиц дисперсной фазы нельзя не учитывать связанное с адсорбцией снижение концентрации ПАВ в объеме дисперсионной среды. Поэтому для образования адсорбционных слоев с поверхностной концентрацией Г,, обеспечивающей устойчивость эмульсии, требуется увеличить содержание ПАВ в растворе по сравнению с cs. Необходимый избыток ПАВ зависит от степени дисперсности и объемного соотношения фаз вода/масло. Следовательно, стабилизирующее действие ПАВ характеризует величина Г5, а не cs. К такому же выводу пришли авторы работы [209].
Ниже приведены значения cs для различных ПАВ в 0,1 М растворе КС1, соответствующие образованию устойчивых микроскопических пленок пен [129]:
Полиэтиленгликолевый эфир октилфенола*
п = 7..............................
« = 9..............................
«=15...............................
« = 20.............................
Na-гексилсульфат-1....................
Na-октилсульфат-! ....................
Na-децилсульфат-!.....................
Na-ундецилеульфат-!...................
Na-додецнлсульфат-!...................
Na-тетрадецилсульфат-!................
Ыа-октилсульфат-2.....................
Na-октилсульфонат-!...................
Na-лаурат.............................
с$,моль;л
1,36- 10“ ® 1,08- 10"5 5,2- 10"’ 4,6- 10"° 1,2- io-г.2. 10~4 2,0- 10" ® 9,0 • 10" 7,0-10" 1,5- 10~ ’ 8,6 10“’ 6,5-10~4
1,3- 10~в
• Величина п соответствует среднему содержанию этилен* оксидных звеньев в молекуле ПАВ.
В табл. 15 сопоставлены концентрации ПАВ, необходимые для стабилизации пленки 0,05 М раствора КС1 между двумя ка-пельками циклогексана, а также соответствующие значения поверхностной концентрации и площади Fs, занимаемой молекулой ПАВ в поверхностном слое [208]. Измерения проводили по методике, изложенной в главе I (стр. 65). Из сравнения способности ПАВ образовывать пены и эмульсии видно, что для достижения
устойчивых к коалесценции пен требуются значительно меньшие количества ПАВ, чем для эмульсий. К сожалению, в работе [129] не определены поверхностные концентрации, так что нельзя установить, наступает ли стабилизация для обоих типов систем при одинаковой степени покрытия поверхности.
Таблица 15
Концентрация поверхностно-активных веществ cs, необходимая для образования устойчивых к коалесценции пленок 0,05 М раствора КС1 между капельками циклогексана
ПАВ cs* молъ1я моль) см1 ps> ' А2
Na-додекансульфонат-! Na-3-гидроксидодекансульфонат-! оо СЛ СО о о L L 1 3.2-10-;” 52
Na-2-гидроксидодекансульфонат-! 2- 10 “ 2,9- 10 57
№-1-гидроксидодекансульфоиат-2 (насыщенный раствор) Отсутс 9 • 10“3 гвие черных 3,0- 10"'” пленок
Na-3-оксододекансульфонат-! . . . 55
Na-2-оксододекансульфонат-! . . . Отсутствие черных пленок
Na-2-додецеисульфонат-! 6-10“’ 3,1-10"“' 53
Na-2-тетрадеценсульфонат-! .... Na-3-гидроксиоктаисульфонат-! . . 2-10 | Отсутствие черных пленок
Na-3-гидроксигексадекансульфоиат-! 1- IO” 3,6-10",° 46
Na-додецилсульфат-! 4-10 ’ 2,4 10 10 69
Na-лаурилсаркозид Цетилтриметиламмонийбромид . . . Полиэтиленгликолевый эфир ионил-фенола NP-12 NP-20 NP-30 * NW- (О к; ООО О о III 1 , -4 ел ел сл { 1,4 • 10“10 111 , ;
• Технический продукт.
Для оценки эмульгирующей способности различных ПАВ необходимо подробнее изучить данные табл. 15. Рассмотрим прежде всего ряды алкан- и алкенсульфонатов натрия. Na-додекаи-сульфонат вызывает образование устойчивых пленок между двумя капельками циклогексана только в том случае, когда эмульгатор трудно применять, так как его граница растворимости находится около 10~3 моль/л, т. е. практически совпадает с cs. Поэтому количество ПАВ, необходимое для приготовления устойчивых эмульсий, не может быть растворено в дисперсионной среде. Введение во второе положение молекулы до-де.кансульфоната гидроксильной группы или оксогруппы незначительно изменяет его растворимость. Так как одновременно снижается поверхностная активность, то значение сдвигается в сторону больших величин или вообще .не достигается.
Гидроксильная группа или оксогруппа в третьем положении увеличивает растворимость продукта почти на два порядка и уменьшает его поверхностную активность, приводя к дальнейшему возрастанию значения cs. В связи с тем, что изменение cs несущественно по сравнению с повышением растворимости, такие вещества можно применять в качестве эмульгаторов.
Только наличие в молекуле двойной связи дает желаемый результат — увеличение растворимости и поверхностной активности. Оба эти параметра определяют также влияние длины алкильной цепи на эмульгирующую способность. Например, увеличивая длину цепи алкенсульфатов при переходе от Na-додецен-сульфоната к Na-тетрадеценсульфонату, вызывают усиление эмульгирующей способности, поскольку при относительно неизменной растворимости повышается поверхностная активность. В случае Na-гексадеценсульфоната и Na-октадеценсульфоната начинает проявляться уменьшение растворимости стабилизаторов.
Сравнение значений Гг, соответствующих концентрациям cs, а также площадей, занимаемых одной молекулой различных ПАВ в поверхностном слое и составляющих около 55 А2, приводит к выводу, что для всех продуктов устойчивые пленки образуются при одной и той же степени заполнения поверхности. Последнее обстоятельство, по-видимому, позволяет сделать заключение о независимости структуры пленок, полученных из растворов сульфонатов, от химической природы заместителей в молекулах ПАВ. Как следует из экспериментальных данных, устойчивость к коалесценции не связана с образованием насыщенного монослоя, так как минимальная площадь, приходящаяся на одну молекулу алкилсульфонатов, приблизительно равна 42 А2. Наряду с сульфонатами аналогичным образом, изучены другие ПАВ. При этом установлено, что очень хорошими стабилизаторами являются полиэтиленгликолевые эфиры нонилфенола с различным содержанием этиленоксидных звеньев, а также цетилтриметиламмонийбромид.
Стабилизирующее действие некоторых промышленных ПАВ на пленки октана, находящиеся между двумя капельками воды, можно характеризовать следующими данными:
«/л
Span-65*........................... 1,0
Span-80 ......................... 0,1
Span-85** ......................... 1,0
Моноглицерид олеиновой кислоты 0,42
Холестерин....................... 3,7
Концентрация ПАВ, необходимая для получения устойчивых тонких жидких прослоек, в значительной степени зависит от природы масляной фазы [125]. Так, стабилизация пленок 0,5 н.
* Тристеарат сорбитана — технический продукт. (Прим, ред.)
' **Тряолеат сорбнтяна — технический продукт» (Прим. реф.)
раствора КС1 между капельками ряда углеводородных жидкостей и их производных достигается при различном содержании эмульгатора — промышленного образца NP-20:
Октан.......................
Декалин.....................
Циклогексан.................
Бензол .....................
Хлорбензол .................
Аналогичные результаты получены при модельных исследованиях обратных эмульсий. Обнаружено, что возникновение устойчивых углеводородных пленок между капельками 0,05 н. водного раствора КС1 в зависимости от природы дисперсионной
среды происходит в широком диапазоне концентраций моногли-
церида олеиновой кислоты:
Октан............................
Додекан..........................
Декалин.......................;
Циклогексан......................
Ксилол ..........................
Тетралин.........................
Хлорциклогексаи .................
Хлорбензол ......................
cs, моль!л
5,5- 10~*
2,8- 10
8,4- 10~*
9,0- 10~*
5,0- 10 , 1,4- 10~2
Отсутствие устойчивости
То же
Изучению стабилизирующего действия ПАВ на пленки, находящиеся между разнородными поверхностями, насколько нам известно, посвящена только одна работа [190]*. В этой работе измерены величины концентраций, обусловливающие устойчивость водных прослоек между каплями олеиновой кислоты и циклогексана, с одной стороны, и подложками, изготовленными из ряда полимерных материалов, с другой. Некоторые сведения содержатся в табл. 16.
Полученные результаты приводят к выводу о существовании на обеих межфазных границах определенных поверхностных концентраций ПАВ, обеспечивающих устойчивость к коалесценции. Из сопоставления данных табл. 15 и 16 видно, что концентрация стабилизации в системе циклогексан — пленка — полимер, зависящая от химической природы макромолекул, всегда больше концентрации ПАВ, необходимой для получения устойчивой водной прослойки между капельками циклогексана. Более подробно мы остановимся на этих опытах в главе 3 при рассмотре-. нии процесса стирки.
* См. также работы [345, 346], относящиеся к исследованию устойчивости тонкйх пленок воды между поверхностью кварца и капельками нефти в присутствии ПАВ. (Прим, ред.) .
Таблица 16
Концентрация ПАВ, необходимая для стабилизации жидкой прослойки между капелькой циклогексана и полимерной пленкой
Материал полимерной подложки cs, моль!Л
Na-олеат Na-додецил-сульфат цетилгр име-тиламмоний-бромид NP-20
Полиамид Политетрафторэтилен Полиэтилен Полиэфир •е> « w со III 1 О о О' о — ь. со 5- 10"3 7-10~3 Отсутствие устойчивости 6- 10"3 5- 10"’ 7-10"’ 7-10" 7-10"’ 1,4- 10"’ 1,5- 10 Отсутствие устойчивости 1,4-10"’
Толщина черных пленок. Стабилизирующее действие ПАВ проявляется также в очень тонких черных пленках. Поэтому желательно получить более подробную информацию об их толщине и структуре. Измерения толщины мыльных пленок и пленок в эмульсиях типа масло/вода могут быть проведены только ин-терферометрически (см. главу I, стр. 67).
К сожалению, методика определения малых толщин недостаточно надежна. Вследствие того, что прослойки пены представляют трехслойные системы (см. рис. 31,6)-, необходимо знать коэффициент преломления адсорбционных слоев, чтобы получить данные о размере водного ядра, заключенного между ними. В качестве первого приближения можно положить равными коэффициенты преломления адсорбционных слоев и объемной фазы и оценить толщину адсорбционного слоя, исходя из площади, занимаемой одной молекулой [126]. Напротив, весьма спорно использовать при расчетах вместо коэффициента преломления .родного ядра соответствующую величину для объемной фазы, так как в результате структурообразования молекул воды в тонких пленках, очевидно, происходит изменение коэффициента .преломления. При модельных исследованиях эмульсий положение несколько лучше в связи с тем, что пленка между двумя Капельками, масла, вероятно, является однослойной (рис. 31, а). Однако и тогда остается неизвестным коэффициент преломления, водного ядра.
Более точные результаты для эмульсий вода/масло можно •получить при,совместных интерферометрических и емкостных измерениях-. [21Q], Преимущество последнего метода обусловлено сильной, зависимостью емкости от толщины прослойки масла. Углеводородные цепочки адсорбционных слоев, «растворенные» в органической фазе, имеют диэлектрическую постоянную, практически совпадающую с диэлектрической постоянной предельных и циклических углеводородов. Поэтому для вычисления величины h можно' использовать значение 8, характеризующее дисперсионную среду, причем сольватированные полярные
Т06
группы в водных капельках следует рассматривать в виде последовательно включенных емкостей *. Очевидно, измерения толщины черной пленки между твердыми поверхностями трудно осуществить ввиду их шероховатости**.
Изучение моделей пен [133, 134, 211, 212] и эмульсий типа масло/вода [132] показало, что устойчивость черных пленок с толщиной, превышающей 60 А, определяется действием вандер-вдальсовых и электростатических сил3*. Эти пленки, согласно данным работы [211], называют первичными пленками. При определенных условиях они самопроизвольно переходят в еще более тонкие пленки, которые впервые обнаружены Перреном [213]; их называют вторичными или перреновскими черными пленками. Переход .от первичных ко вторичным черным пленкам в пенах происходит при испарении жидкости [211, 213] или при добавлении определенных электролитов [134], а в слоях, заключенных между капельками масла, — только при введении электролитов [205]. Расклинивающее давление вторичных черных пленок должно быть очень велико, так как превращение первичных черных пленок во вторичные сопровождается сильной деформацией газовых пузырьков пены или капелек эмульсии.
Мизелс с сотрудниками [134] изучил устойчивое состояние двух типов черных пленок в водных растворах Na-додецилсуль-фата в присутствии электролитов и неэлектролитов. Например, показано, что при 0,26 моль/л NaCl образуются вторичные пленки, в то время как для меньшего содержания ионов в дисперсионной среде являются устойчивыми первичные пленки. Существование двух различных областей равновесной толщины полимолекулярного жидкого слоя может быть доказано экспериментально. Так, если в течение короткого промежутка времени нагревают в некоторых участках первичную черную пленку, то в этих местах вследствие испарения растворителя возникает вторичная черная пленка; однако после прекращения нагревания она снова переходит в первичную пленку. Следовательно, происхождение вторичной черной пленки не связано с кинетикой процесса (малой скоростью образования при незначительных концентрациях электролита), а определяется равновесными термодинамическими факторами.
* Обоснование методов интерферометрических и емкостных измерений толщины пленок является некорректным, поскольку коэффициент преломления и диэлектрическая проницаемость относятся к объемным свойствам веществ (см. стр. 67 и 85). {Прим, ред.)
** Явления скачкообразного изменения толщины водной прослойки между твердыми поверхностями, аналогичные процессу образования черных пленок в пенах, обнаружил Норриш [359], изучая набухание монтмориллонита в растворах электролитов методом рассеяния рентгеновских лучей под малыми углами. Измеренные при этом межплоскостные расстояния составляли, как правило, 20—100 А, т. е. соответствовали толщинам черных пленок. {Прим, ред.)
3* См. также [360]. {Прим, ред.) . _
Когда помимо NaCl добавляют еще другие электролиты, то они либо способствуют, либо препятствуют получению вторичных черных пленок. Из табл. 17 видно, что при введении некоторых солей изменяются концентрации NaCl, приводящие к образованию вторичных пленок. Нейтральные соединения, такие как додециловый спирт или мочевина, не оказывают на этот процесс заметного влияния. Ионы калия и кальция способствуют, а ионы лития, водорода, алкиламмония препятствуют возникновению вторичных черных пленок.
Таблица 17
Влияние состава электролита на образование и равновесную' толщину вторичных черных пленок, стабилизированных Na-додецилсульфатом [134]
Концентрация NaCl, моль! л Концентрация второй соли, молъ/л Толщина вторичной пленки, А
0,246 Пленка не образуется
0,260 — 43,0
0,300 44,8
0,350 — 44,6
0,450 — 45,3
L1C1
0,300 0,050 43,2
0,300 0,100 Пленка не образуется
0,350 0,200 44,9
0,350 0,400 50,2
0,350 0,600 48,6
КС1 ч
0,200 0,100 44,9
НС1
0,303 0,016 41,5
0,300 0,040 Пленка не образуется
0,350 0,100 То же
(CH3)4NBr
0,270 0,040 Пленка не образуется
0,300 0,010 44,3
0,370 0,060 Пленка не образуется
Толщина вторичных черных пленок слабо зависит от состава раствора электролита и размеров молекул ПАВ. В ряду Na-децилсульфат — Na-тетрадецилсульфат она увеличивается от 39,8 до 48 А. Для вторичных черных пленок 0,1 М раствора NaCl, стабилизированных Na-додецилсульфатом и полученных путем испарения растворителя, значение й, составляет 43 А [214]. Произведя оценку толщины монослоя с учетом площади, занимаемой одной адсорбированной молекулой на границе раздела фаз, для Na-децилсульфата, Na-додецилсульфата и Na-тетрадецилсуль-фата соответственно найдем 7, 8,5 и 10 А. Отсюда следует, что во вторичной черной пленке должна содержаться водная прослойка с толщиной 26—28 А.
Аналогично со случаем пенных пленок, образование вторичных черных пленок в тонких слоях жидкости между капельками эмульсий обнаружено не для всех электролитов; оно наступает при концентрации ионов, превышающей некоторое определенное значение [205]. К сожалению, точные измерения в модельных эмульсиях не могут быть проведены, так как при этом очень низка интенсивность отраженного света.
Равновесие между первичными и вторичными черными пленками изучено для целого ряда ПАВ. Применяя в качестве стабилизатора полиэтиленгликолевый эфир нонилфенола NP-20, вторичные черные пленки получают при добавлении Кг5О4(с> >0,60 моль/л) и MgSO4(c > 0,26 моль/л). В растворах КС], NaC104 и Са(МОз)2 вплоть до концентраций, соответствующих насыщенным растворам, образуются только первичные черные пленки. В случае анионактивного вещества, например, Na-тетра-деценсульфоната, вторичные черные пленки возникают при введении MgSO4 (с > 0,125 моль/л), однако они не обнаружены в растворах КС1 и Кг5О4. Цетилтриметиламмонийбромид не вызывает образования вторичных пленок растворов КС1, Кг$О4 и MgSO4*.
Причины перехода от первичных ко вторичным черным пленкам до настоящего времени не объяснены. Мизелс [134] предполагает, что, начиная с некоторой достаточно высокой концентрации электролита, ионы, специфически адсорбирующиеся на межфазной границе, располагаются в виде штерновского слоя. Поэтому электростатическое отталкивание, обусловленное перекрытием диффузных атмосфер, практически не проявляется **. Следовательно, переход от первичных пленок ко вторичным объясняется значительным увеличением сил притяжения.
Результаты исследований, выполненных на модельных эмульсиях, не согласуются с таким объяснением. Например, для неионогенного раствора NP-20 отсутствует специфическая адсорбция ионов и, несмотря на это, наблюдается образование вторичноц черной пленки. С другой стороны, эти пленки не возникают, когда в качестве стабилизатора применяют цетилтриметиламмонийбромид, хотя при высокой концентрации ПАВ на межфазной границе должна происходить специфическая адсорбция анионов, так как их способность к адсорбции выражена сильнее, чем у катионов.
В заключение еще раз сопоставим имеющийся в настоящее время экспериментальный материал, относящийся к образованию и свойствам вторичных черных пленок. Подобные пленки
* Влияние валентности ионов иа переход от первичных черных пленок к вторичным изучено в работе [361]. (Прим, ред.)
** Для пленок, стабилизированных ионогенпыми ПАВ, учет ионно-электростатического взаимодействия, по-видимому, следует проводить на основе модели двойного слоя с постоянной плотностью поверхностного заряда [362]. (Прим, ред.)
являются очень тонкими, однако, межплоскостное расстояние в них больше, чем удвоенная толщина адсорбционного слоя. Таким образом, они содержат в себе водное ядро. При их возникновении происходит деформация капелек или пузырьков, обус-
ловленная изменением сил, которые действуют в тонком слое и приводят к увеличению контактной поверхности. Силы взаимо-
действия можно оценить, измеряя площади первичной и вторич-
Рис. 55. Схема определения краевого угла смачивания черной пленки объемной фазой.
ной черных пленок [215, 216], а также краевой угол, существующий между, пленкой и объемной фазой (рис. 55).
Указанные величины обычно определяют интерференционным методом. Краевой угол 0 связан с поверхностным натяжением объемной фазы о и с поверхностным натяжением пленки
од следующим уравнением:
cos 0 = —
<т
(90)
Для тонких пленок давление пара, парциальный мольный объем и расклинивающее давление зависят от толщины. Разность поверхностных натяжений объемной фазы и пленки можно выразить через расклинивающее давление П [215]*:
оо
<т — afi = — ПА — J П dh h
(91)
В случае вторичных черных пленок толщиной 44 А Мизелс и сотрудники [217, 218] нашли, что краевой угол 0 = 8°50', а поверхностное натяжение 0^ = 29,15 дин!см. Отсюда с учетом значения о = 29,5 дин{см имеем П=1,5-10в дин/см2. Если на расстояниях порядка 44 А действуют только молекулярные силы, то константа Гамакера будет равна 1,5-10—12 эрг. Она превышает на порядок величину постоянной, вычисленную из экспериментальных данных изучения равновесной толщины. Нельзя однозначно установить, объясняется ли такое расхождение влиянием полярных групп на молекулярное притяжение или возникновением дополнительных дипольных сил взаимодействия адсорбированных молекул воды, ориентированных благодаря образованию водородных связей. Исследование поведения углеводородных черных пленок между капельками воды может содействовать решению поставленного вопроса.
Работы в этой области немногочисленны. Так, некоторыми авторами определена толщина черных углеводородных пленок, стабилизированных маслорастворимыми ПАВ. Результаты исследований представлены в табл. 18. Из таблицы видно, что
* Вывод этого уравнения дан в работах [403, 404]. (Прим, ред.)
Таблица 18
Толщина черных углеводородных пленок, находящихся между капельками воды и стабилизированных различными ПАВ
Концентрация электролита в капельках: КС1—0,05 н. раствор;
NaCl — 0,1 н. раствор.
ПАВ Углеводородная среда Электролит Толщина черной пленки, A Метод исследования Литература
Диизотридецилцитрат Октан KCI 30 Емкостной 125
Лецитин Декан NaCl 70 Интерференцией- 220
ный
Монобрассидат сорбита > NaCl 56 Емкостной 168
Моноглицерид олеиновой Октан KCI 41 Интерференцион- 219
кислоты Ксилол КС1 30 ный и емкост-
Моноглицерид стеа- Гексан NaCl 50 Интерференцион- 169
рнновой КИСЛОТЫ НЫЙ
Монолаурат сорбита Декан NaCl 31 Емкостной 168
Моноолеат сорбита Октан KCI 39 Интерференцион- 219
Кснлол KCI 28 ный н емкост-
ной
Декан NaCl 46 Емкостной 168
Монопальмитат сорбита » NaCl 42 » 168
Холестерин Октан KCI 23 » 125
толщина пленок приблизительно соответствует удвоенной длине молекулы ПАВ. Различие в толщине пленок для растворов одних и тех же ПАВ в октане и ксилоле следует объяснить неодинаковой поверхностной концентрацией адсорбированных молекул. Например, площадь Fs, занимаемая одной молекулой моноглицерида масляной кислоты в поверхностном слое, составляет для растворов в октане и ксилоле соответственно 25 и 61,5 А2, а для растворов моноолеата сорбита—40,8 и 123 А2. В адсорбционные слои входят молекулы растворителя и, следовательно, молекулы ПАВ расположены под углом к границе раздела фаз. Проникновение бензола в адсорбционные слои мойоглицерида пальмитиновой кислоты доказано спектроскопическим методом Ределем.
В отличие от водных черных пленок, по данным работы Штеммера и Зонтага, образование пленок из углеводородных жидкостей не связано со значительным увеличением поверхности. В частности, это подтверждено Хейдоном и Тейлором [221 ] путем определения краевых углов. Необходимо отметить, что найденное таким образом значение разности поверхностных натяжений о — Gh, составляющее 0,00197 дин/см, очень мало. Вычисляя по уравнению (91) расклинивающее давление при h = 42 А, имеем П = 9-103 дин/см2. Тогда, полагая, что в пленке действует только молекулярная составляющая расклинивающего
давления, ДЛЯ Постоянной Гамакера получим значение 7-10-15 эрг. По-видимому, ошибка заключается в методике, так как в условиях проведения опыта не устанавливалось истинное равновесие системы. Измерения, проведенные на пленках неполярных веществ, позволяют сделать вывод, что полное подавление ионно-электростатических сил не является основной причиной возникновения вторичных черных пленок.
В ряде работ [222, 223] для изучения структуры черных пленок использовали метод инфракрасной спектроскопии. Однако, так как вплоть до настоящего времени объектами исследования являлись только первичные черные пленки, имеющие, как известно, сравнительно толстое водное ядро, полученные результаты содержат мало информации о строении адсорбционных слоев ПАВ.
Механическая прочность пленок. Как показано ранее (стр. 101) по мере увеличения содержания в системе ПАВ в узком интервале концентраций устойчивость тонких жидких слоев значительно возрастает. Можно предполагать, что в той же самой области наступает изменение механических свойств пленок. Попытка оценить влияние на устойчивость механических свойств адсорбционного слоя, определяемых путем измерения поверхностной вязкости и прочности на межфазных границах больших размеров, не привела к положительному результату, поскольку измеряемые эффекты слишком малы и деформация под действием сдвиговых напряжений не соответствует элементарному акту процесса разрыва. Разрушению тонкого жидкого слоя всегда должно предшествовать появление более тонкой локальной области— слабого места, скорость «залечивания» которого меньше, чем скорость разрыва пленки. Для устойчивости решающее значение имеют физико-механические свойства, проявляющиеся при сжатии и растяжении адсорбционных слоев — поверхностная эластичность (модуль поверхностного сжатия) и поверхностная вязкость. Последняя величина, зависящая от скорости деформации, характеризует процесс релаксации молекул ПАВ в адсорбционном слое, а также диффузионный обмен, происходящий в пленке при изменении ее толщины под действием внешней нагрузки *.
Периодическое растяжение и сжатие адсорбционных слоев можно наблюдать при возникновении в системе поперечных волн. Однако более целесообразно для изучения структуры пле-
* Большой вклад в изучение физико-механических свойств тонких пленок и адсорбционных слоев внесли работы Трапезникова и его сотрудников [363—371]. Непосредственное измерение вязкости жидкости в тонких пленках, находящихся на твердых поверхностях, осуществлено Дерягиным, Кусаковым и рядом других исследователей [144, 145, 327—332]. Как установлено [333], адсорбция молекул ПАВ на границе раздела фаз обусловливает возникновение сольватных слоев, вязкость которых в два-три раза превышает вязкость жидкости в объеме. (Прим, ред.)
Рис. 56. Зависимость коэффициента затухания поперечных колебаний на межфазной границе вода — циклогексан от частоты.
Концентрация раствора NP-20, молъ1л: 2 -0; 2 — I • 10~6; 3-1- 10~°; 4-9 • 10~°; 5-2 • Ю~5.
нок использовать продольные колебания, распространяющиеся в адсорбционных слоях. Подобные исследования выполнены Лу-кассеном [224]. Трудность экспериментальной работы состоит в том, что для высокоэластичных тел, как и для адсорбционных слоев, характерно более сильное затухание продольных волн. Измерение затухания колебаний проведено, в основном, в тех случаях, когда адсорбционные слои являлись нерастворимыми. Поэтому не представляется возможным сопоставить эти данные с результатами исследований устойчивости пленок.
Определенная связь между устойчивостью пленок и механи-; ческими свойствами адсорбционных слоев ПАВ обнаружена Я Фрайзом. Он нашел частотную за-J висимость затухания поперечных ’ волн на межфазной границе вода — f циклогексан в присутствии различ-? ных количеств полиэтиленгликолевого эфира нонилфенола NP-20, применяемого в качестве стабилизатора. Показано, что при концентрации ПАВ, равной с3, коэффициенты затухания а, соответствующие фикси-' рованной частоте, резко возрастают (рис. 56). В настоящее время теоретическая обработка результатов измерений еще не завершена и отмеченное явление не получило достаточно полного объяснения.
Кретцшмаром и Лункенхайме- ром для изучения механических свойств адсорбционных слоев предложен новый метод. Принцип его
заключается в следующем. В капилляр, заполненный раствором ; ПАВ, вводили пузырек воздуха определенного размера. В системе возбуждали колебания различной частоты и амплиту-V ды, обусловливающие периодические изменения объема воздуха. ’ Пузырек проектировали на фотоэлемент, с помощью которого происходящие синусоидальные изменения объема преобразовы-; вались в синусоидальные электрические колебания. В зависимости от частоты измеряли напряжение U, необходимое для воз- буждения колебаний некоторой постоянной амплитуды, регистри-руемой осциллографом. Напряжение возбуждения прямо про-? порционально изменению объема и, следовательно, изменению ё давления в пузырьке. Таким образом получены кривые U = ». =/(и), на которых при определенных условиях обнаружен ми-| нимум (рис. 57).
I Характер кривых зависит от различных параметров: радиуса капилляров, глубины их погружения в исследуемую жидкость, объема пузырька, а также степени возбуждения. В условиях
постоянства этих параметров изучено влияние на значения U природы и концентрации ПАВ.
Описание процесса с помощью неоднородных дифференциальных уравнений второго порядка позволяет найти величину поверхностной эластичности. По мере повышения содержания ПАВ в дисперсионной среде в узком диапазоне концентраций наступает значительное смещение минимума, в то время как дальней-ц мб 45 г
Рис. 57, Зависимость напряжения возбуждения U от частоты. Концентрация тетрадецилсульфата натрия в бидистиллироваииой воде, моль!л'. I — 0;
2 — 3 . Ю~5; 3-5 • 10-s; 4-1 • 10~5; 5-1 • 10-4.
шее прибавление стабилизатора не вызывает существенного изменения вида кривых. Сравнивая концентрации се, обусловливающие такой переход, с концентрациями стабилизации, определенными по методу Росс — Майлса, и с величинами щ, характеризующими возникновение устойчивых черных пленок, можно установить достаточно хорошее совпадение между тремя указанными значениями для одной и той же системы (табл. 19). Величины поверхностной эластичности, вычисленные из результатов исследования колебаний пузырька воздуха и найденные на основании данных о затухании стоячих и бегущих волн на поверхности раздела жидкость — газ [226—228], совпадают в пределах погрешности измерений *.
* Непосредственное измерение вязкости на границе жидкость — газ провели Ван деп Темпель и сотрудники [225]. К сожалению, они не определили связь между устойчивостью и реологическими свойствами адсорбционных слоев.
Таблица 19
Значения концентраций се, cs и пенообразующая способность ПАВ-
Концентрация электролита в пленке: NaCl—0,1 н. раствор; НС1—0,7 н. рас-' твор.
ПАВ Электролит Пенообразующая способность ПАВ *, X 106 моль/л cs-10°, моль/л се-10в. МОЛЬ/Л
Каприловая кислота НС1 100
Каприновая кислота НС1 10
Капроновая кислота НС1 1000
Монододециловый эфир — 0,17 ч-0,30 0,10ч- 0,15
гептаэтиленгликоля ** NaCl ~0,30 О.Юч-0,15
Монододециловый эфир — 0,10ч-0,15
пентаэтиленгликоля** NaCl 0,17 ч-0,30 0,104-0,15
с Na-децилсульфат-! — 2000
NaCl 100
ь Na-додецилсульфат-1 — 100 200
NaCl 9,5 10 10
? Na-тетрадецилсульфат-! — 10 20
NaCl 20 1,5 1
i' ♦ Концентрация ПАВ, необходимая для получения слоя пены толщиной 10 мм
после падения последней капли раствора. Из-за недостатка реагентов ие измерена пенообразующая способность Na-децнлсульфата и монододециловых эфиров полиэтилеигликоля.
** Относительно большое различие значений се и cs для монододециловых эфиров полиэтилеигликотя, по-видимому, вызвано тем, что при малых концентрациях ПАВ не происходит установления равновесной величины поверхностного натяжения.
| ГЕЛЕОБРАЗНЫЕ ЗАЩИТНЫЕ СЛОИ.
< СТРУКТУРНО-МЕХАНИЧЕСКИЙ БАРЬЕР
В некоторых дисперсиях на поверхности частиц имеются ста-J билизирующие защитные слои, существенно отличающиеся по | своей природе от черных пленок, а также от равновесных пле-J нок, устойчивость которых обусловлена ионно-электростатически-I ми силами отталкивания. Такие слои могут быть построены из мелких частиц твердых и жидких веществ. Кроме того, они об-; разуются при адсорбции макромолекул или при полимолекуляр-S ной адсорбции ПАВ. Необходимо определить критерии устой-? чивости дисперсных систем для каждой из этих трех форм суще-‘ ствования защитных слоев.
Эмульсии, содержащие частички твердых веществ, так назы-; ваемые эмульсии Пикеринга [229, 230], стабильны, если частицы преимущественно располагаются на межфазной границе масло/вода. Для этого требуется, -чтобы краевые углы смачивания твердого тела обеими фазами были примерно одинаковы. Малое различие в краевых углах определяет тип эмульсии. Если частица смачивается маслом лучше, чем водой, возникают эмульсии типа вода/масло; в противоположном случае получают эмульсию масло/вода.
ПАВ могут оказывать сильное влияние на краевой угол и тем самым создавать оптимальные условия смачивания. Частицы на межфазной границе образуют барьер, препятствующий сближению двух капель до расстояний, при которых наступает коалесценция.
К системам типа Пикеринга относят также эмульсии, стабилизированные продуктами гидролиза мыл многовалентных металлов. В частности, к указанному классу принадлежат эмульсии масло/вода в присутствии олеата алюминия при значении pH, отвечающем образованию в системе полимерной гидроокиси алюминия; их исследование проведено Корецким и Таубманом [231].
Эмульсии Пикеринга устойчивы лишь по отношению к коалесценции, когда заряд твердых частиц недостаточен для появления барьера отталкивания против флокуляции; их коалесценция достигается добавлением ПАВ, которые не являются, стабилизаторами и одновременно сильно уменьшают краевой угол смачивания твердой поверхности одной из жидких фаз. Поэтому твердые частицы переходят с межфазной границы в объем и капли могут объединяться. Вероятно, особый случай структурномеханического барьера, обусловленного капельками микроэмульсий, наблюдала Никитина [232, 233]. Его возникновение связано с турбулентным массообменом через межфазную границу, причем направление переноса вещества определяло тип эмульсий. Разрушение последних проводилось обычными способами.
Большой интерес вызывают относительно мало изученные адсорбционные слои макромолекул. Действие защитных коллоидов в водных растворах известно уже сравнительно давно, и данной проблеме посвящен ряд феноменологических исследований [78, 109, 234]. Аналогичное влияние на устойчивость эмульсий и суспензий оказывают полимолекулярные слои ПАВ, возникающие при адсорбции из растворов ПАВ достаточно высокой концентрации. Несмотря на подробное экспериментальное исследование, причины стабилизации при введении в дисперсную систему защитных коллоидов до сих пор еще окончательно не выяснены. Фрейндлих [78] постулировал, что для достижения устойчивости необходимы особые свойства адсорбционных слоев: прочность, наличие ориентации молекул и их достаточно высокая энергия связи с подложкой.
Ребиндер [235] впервые ввел понятие структурно-механического барьера. Этот барьер обусловлен адсорбционно-сольватными слоями, обладающими вязко-эластичными свойствами, и препятствует коалесценции при соударении двух частиц; его существование возможно как в полярных, так и в неполярных дисперсионных средах.
Толщина и механические свойства защитных слоев изучены недостаточно полно. Можно, однако, полагать, что для некоторых систем стабилизация достигается при сохранении между 116,
взаимодействующими поверхностями равновесных расстояний, превышающих десятки и сотни ангстрем. Так, по данным работы [180], капельки циклогексана в водном растворе поливинилового спирта отделены друг от друга прослойкой дисперсионной среды толщиной 800 А. Молекулы сложного полиэфира образуют на двуокиси титана слои толщиной 40—60 А [236], а на металлических поверхностях — 70 А [237]. Для растворов метилцеллюлозы, стабилизирующих частицы сложного полиэфира, Фишер [108] определил равновесные расстояния, равные 140—200 А. Как обнаружено при экспериментальном исследовании устойчивости капелек ртути в углеводородных жидкостях, содержащих добавки стабилизаторов — олеиновой кислоты и диизононилцитрата, толщина защитных слоев составляет 450 А для первого и 350— 750 А для второго вещества [238].
Если приложить к слипшимся капелькам ртути различные по
знаку электрические потенциалы, то можно, измеряя величину
деформации сжатия пленок под действием электростатических сил, оценить эластичность адсорбционных слоев в направлении, нормальном к межфазной границе. Эти слои обладают свойствами высокоэластичных тел с модулем эластичности 2- 105—7 • 105 дин/см2 и препятствуют сближению на расстояния, при которых может произойти коалесценция. Сравнение модуля эластичности и мо-
лекулярных сил проведено на рис. 58. Из графика видно, что для расстояний, превышающих 200 А, эластичность тонких пле-
Рис. 58. Зависимость сил молекулярного взаимодействия двух металлических пластин от расстояния между их поверхностями (е = 2;
А* =7,5- 10“12 эрг).
Пунктиром ограничен диапазон изменения высокоэластнчных напряжений, которые возникают при сближении частиц, имеющих на своей поверхности защитные слои.
нок достаточна, чтобы противодействовать непосредственному
слипанию частиц.
Адсорбция жирных кислот на поверхности металлических частиц, по-видимому, сопровождается химическими реакциями [239, 240]. Образующиеся при такой стабилизации пленки различным образом воспринимают нормальные и тангенциальные нагрузки. В то время как эластичность слоя в направлении нор'1' мали относительно велика, он легко разрушается под действием касательных напряжений. Вероятно, поэтому жирные кислоты не оказывают сильного влияния на устойчивость против слипания тонкодисперсных металлических частиц, при столкновении которых могут возникать значительные сдвиговые напряжения в пленке. Следовательно, тонкие слои, предотвращающие коалесценцию, должны иметь гелеобразную изотропную структуру, чтобы эластично воспринимать напряжения во всех направлениях.
Разрушение рассмотренных выше слоев достигается термическим, электрическим и сорбционным методами.
В заключение отметим, что крупный вклад в изучение физико-механических свойств полимолекулярных адсорбционных слоев и объяснение вопросов устойчивости тонких пленок растворов ПАВ внесли Ребиндер и его сотрудники [241—243].
РАЗРУШЕНИЕ УСТОЙЧИВЫХ ЖИДКИХ ПЛЕНОК
Данные о разрушении устойчивых слоев получены при изучении пен и эмульсий, так как для этих дисперсных систем возможно непосредственное наблюдение процесса прорыва пленок, сопровождающегося объединением отдельных частиц и образованием компактных фаз. Очевидно, что флокуляция является необходимой для наступления коалесценции в пенах и эмульсиях, характеризующихся существованием устойчивых тонких жидких пленок между дисперсными частицами. Разрушение адсорбционных слоев может происходить при десорбции стабилизатора с межфазной границы вследствие изменения состояния системы (температуры, концентрации различных компонентов в пленке и др.), благодаря химическому модифицированию молекул ПАВ и при действии внешней нагрузки, способной преодолеть их механическую прочность. Однако разрушение некоторых дисперсных систем может быть вызвано только при использовании специфических приемов.
Коалесценция при адсорбционном замещении. Этот метод широко распространен и применяется, главным образом, для пен и эмульсий типа вода/масло. Разрушение дисперсий происходит при добавлении ПАВ, которое адсорбируется сильнее, чем исходный стабилизатор, не способствуя образованию устойчивых слоев. Выбор таких веществ для пен (см. главу 3) или дисперсий твердых веществ сильно ограничен, так как дестабилизатор должен быть растворим в той же дисперсионной среде, что и стабилизатор. Для разрушения эмульсий типа вода/масло, напротив, имеется широкий круг ПАВ, которые растворяются в дисперсной фазе.
Ввиду того, что поверхностная активность маслорастворимых эмульгаторов на порядок ниже соответствующей величины для водорастворимых ПАВ, последние оказывают значительное влияние на устойчивость эмульсий вода/масло. Необходимо, конечно, принимать во внимание особенности действия эмульгаторов на дисперсную систему. При низких концентрациях водорастворимых ПАВ, применяемых для разрушения эмульсий, наблюдается весьма существенное повышение ее устойчивости, связанное, очевидно, с увеличением степени заполнения адсорбционных слоев [219]. Разрушение жидких пленок, разделяющих отдельные капельки дисперсной фазы, происходит только при определенном оптимальном содержании эмульгатора в воде.
Возможность дополнительной стабилизации, обусловленной введением деэмульгатора, доказана при модельных исследованиях. Растворяя в масле некоторое количество ПАВ, недостаточное для устойчивости слоя, а в дисперсной фазе возрастающие количества деэмульгатора, получают пленки, сохраняющиеся без изменения своей толщины в течение длительного промежутка времени (табл. 20). Из таблицы видно, что по мере увеличения содержания раствора NP-12 в системе сначала возникают стабильные пленки масла и лишь при еще больших добавках происходит их разрушение.
Таблица 20
Влияние концентрации деэмульгатора NP-12 на стабилизацию пленок раствора моноглицерида олеиновой кислоты в октане, . находящихся между капельками воды
Обозначения: + устойчивость; — отсутствие устойчивости.
Концентрация деэмульгатора, г!л Концентрация моноглицерида олеиновой кислоты, г1л
0,08 0,10 0,125 0,30
0 —
1•io~! — .— +
5 • 10~® — —• — +
7-10, —— —- +
1 • — + +
2-Ю — + + +
5-10~* + + + +
8 10~ •—- — +
1 • ’°2 — —« +
1-10 — — — —,
Взаимное усиление стабилизирующего действия водо- и маслорастворимых ПАВ не ограничивается системами типа вода/масло. Так, при полимеризации винилацетата получается тонкодисперсный латекс только в том случае, когда исходный винилацетат содержит маслорастворимые ПАВ, благодаря которому повышается устойчивость мельчайших частиц.
Другое ограничение в процессе разрушения эмульсий — использование больших количеств деэмульгаторов. В этом случае водорастворимые ПАВ, вызывая обращение фаз при достаточно высоких концентрациях, ведут себя как стабилизаторы [244]. Явление обращения фаз изучено в эмульсиях раствор электро-лита/октан (табл. 21).
При выборе водорастворимых ПАВ для деэмульгирования необходимо найти такое вещество, которое, несмотря на высокую поверхностную активность, обладало бы незначительной эмульгирующей способностью для систем типа масдо/вода. Таким требованиям отвечают, например, сополимеры этилен- и пропи-ленгликоля.
Влияние деэмульгатора NP-12 на устойчивость и тип дисперсной системы раствор MgSO4 — октан, стабилизированной препаратом span-80
Обозначения: + устойчивость; — отсутствие устойчивости; В/М и М/В — эмульсии вода/масло и масло/вода, соответственно.
Концентрация MgSO*. моль/л Концентрация NP-12, г/л
0 0,001 0,003 0,095 0,01. 0,03 0,05 0.1
2,5- 10-3 + + + + + +
В/М В/М В/М В/М В/М В/М. В/М М/В
4- 10 2 + + + + + + — —
В/М В/М В/М В/М В/М В/М М/В М/В
Вытеснение маслорастворимых ПАВ из адсорбционного слоя часто достигают при повышении концентрации электролита. Увеличение содержания ионов приводит к дегидратации полярных групп молекул ПАВ и вызывает снижение энергии адсорбции. Связанный с этим переход молекул ПАВ из поверхностных слоев в объем пленки создает условия для коалесценции капелек [245].
Независимо от типа, эмульсии, стабилизированные твердыми частицами, разрушают, изменяя смачиваемость последних путем добавления масло- или водорастворимых ПАВ. В данном случае также необходимо учитывать, что при передозировке количества деэмульгаторов происходит обращение фаз [231].
Адсорбция стабилизатора на межфазных поверхностях в значительной степени определяется температурой. Поэтому нагревание или охлаждение пленок может приводить к их разрушению. Как правило, с повышением температуры адсорбция ПАВ уменьшается. Однако в ряде систем отмечена обратная зависимость, ее следует объяснить скачкообразным увеличением молекулярной растворимости стабилизатора при некоторой температуре [246]. Вероятно, в таких системах коалесценция должна наступать при охлаждении.
Кларе [210] измерил адсорбцию span-80 на межфазной границе раствор электролита/октан как функцию температуры и сопоставил величины адсорбции с устойчивостью водных капелек в том же растворе ПАВ (рис. 59). Зависимости поверхностного натяжения от температуры для различных концентраций имеют минимум; его возникновение связано как с процессом десорбции, так и с уменьшением свободной энергии межфазной поверхности при нагревании. По мере увеличения концентрации минимум сдвигается в сторону больших температур. Аналогичным образом изменяется устойчивость пленок. В концентрированных растворах ПАВ или в случае ПАВ, характеризующихся высокой
поверхностной активностью, коалесценция не наблюдается при достаточно сильном нагревании (рис. 59, кривая 4).
Изучение стабильных углеводородных черных пленок толщиной 39 А, полученных с помощью эмульгатора span-80 между капельками воды, показало [219], что температура, по-видимому,
оказывает влияние не только на десорбцию ПАВ, но и на механические свойства адсорбционных слоев. Однако для решения данной проблемы необходимы дальнейшие исследования.
Химическое разрушение адсорбционных слоев. Химический метод, в принципе, применим для любых дисперсных систем при условии, что используемые для стабилизации молекулы ПАВ в
Рис. 59. Зависимость поверхностного натяжения в системе 0,05 н КС1/раствор span-80 в октане от температуры.
Концентрация ПАВ, молъ!л'. / — 0,01; 2 — 0,05; 3 — 0,1; 4 — 0,5. Стрелки указывают температуру, при которой наступает коалесценция.
результате химических реак-ций замещения, расщепления или ионного обмена теряют способность образовывать устойчивые пленки. На
пример, если подкисляют эмульсии масло/вода, стабилизированные солями щелочных металлов карбоновых кислот, то возникают свободные жирные кислоты, не обладающие эмульгирующими свойствами. В частности, такой прием нашел широкое
распространение в технологии изготовления пенопластов из латексов [247]. Разрушение дисперсных систем производят также введением в дисперсионную среду катионов щелочноземельных металлов. В эмульсиях типа масло/вода Са2+, Sr2+ и Ва2+ при известных обстоятельствах вызывают обращение фаз.
Подкисление растворов алкилсульфатов не приводит к прорыву пленок в связи с тем, что соответствующие кислоты хорошо диссоциируют и имеют высокую поверхностную активность. В этом случае добавляют алкиламмониевые соединения или соли тяжелых металлов.
Влияние электрического поля на коалесценцию. Электрическое деэмульгирование. Известно, что приложение разности потенциалов существенно ускоряет разрушение эмульсий. Это явление используют в процессе удаления воды из нефти [248].
Изучению механизма прорыва пленок во внешнем электрическом поле посвящена работа [219]. Обнаружено, что при малой разности потенциалов U между двумя капельками воды, разделенными прослойкой углеводородной жидкости, возникают дополнительные силы притяжения. При увеличении напряжения устойчивые пленки в определенном интервале значений U
становятся нестабильными и разрушаются. Например, среднее постоянное напряжение, при котором стабилизированная моно-глицеридом олеиновой кислоты пленка октана толщиной 42 А становится неустойчивой, близко к 375 мв и только незначительно зависит от концентрации ПАВ. В табл. 22 для двух эмульгаторов, растворенных в октане и ксилоле, приведены значения постоянных электрических напряжений, вызывающих разрушение черной пленки.
Таблица 22
Постоянные электрические напряжения, обусловливающие разрушение черных углеводородных пленок между капельками воды
Стабилизатор Дисперсионная среда и, Мв £, в/см л, А
Моиоглицерид олеиновой Октан 375 8,9 • 105 42
кислоты Ксилол 235 7,8- 105 30
Span-80 Октан 330 8,5- Ю5 39
Ксилол 225 8,1 • 105 28
AN/N
О'------------------- -
J50 450 U,мд
Рис. 60. Плотность распределения напряжения U, приводящего к прорыву черных углеводородных пленок в системе 0,05 и. КС1/рас-твор моноглицерида олеиновой кислоты в октане с концентрацией 5 г/л.
Вычисленные значения напряженности, характеризующие устойчивость пленок в электрическом поле, по своему порядку совпадают с величинами напряженности пробоя диэлектриков (например, для полиэтилена Е == = 6- 10s в/см). На основании полученных данных можно предположить, что деэмульгирование в системе вода/масло наступает, когда создаются условия, необходимые для пробоя углеводородных прослоек. Так как физико-механические свойства и, следовательно, устойчивость пленок одинакового состава несколько отличаются друг от друга, разрушение эмульсий соответствует некоторому интервалу значений V (рис. 60). Вид представленной на рисунке
зависимости напоминает функцию распределения времени жизни исследованных черных пленок [165].
Уже при сравнительно невысоких напряженностях поля в тонкой углеводородной прослойке между капельками раствора электролита образуются слабые места, обусловливающие в конечном итоге коалесценцию капелек. Экспериментальные данные, относящиеся к такому «статическому» разрушению черных пленок, сопоставлены для двух различных концентраций стабилизатора в табл. 23.
Влияние переменного электрического поля на устойчивость углеводородных пленок
Дисперсная система U, мв ф — 1 N —% N '
0,05 н. КС1/октан + 0,5 г/л моногли- 0 0 100
церид олеиновой кислоты 100 0 100
120 14 32
140 14 16
180 18 14
190 18 0
200 22 0
220 40 0
0,05 и. КС1/октан + 5 г/л моногли- 0 0 100
церид олеиновой кислоты 100 0 100
200 0 100
230 0 13
260 2 0
280 10 0
Из таблицы видно, что для изученных концентраций ПАВ черные пленки в отсутствие поля и при низких значениях его напряженности являются устойчивыми. По мере повышения напряженности количество Л/» слоев, сохраняющих стабильность в течение длительного промежутка времени, становится все меньше. В то же время количество No черных пленок, разрушающихся сразу же после приложения электрического поля, значительно увеличивается. При Е — const значения No и М» зависят от содержания ПАВ в системе.
Часто проводимое сравнение электрического деэмульгирования и электрического обеспыливания оправдано только с точки зрения аппаратурного оформления. Частицы пыли, обладающие, как правило, избыточным зарядом какого-либо знака, перемещаются вдоль силовых линий внешнего электрического поля. Капельки эмульсии, напротив, практически не осаждаются на электродах— ее коалесценция достигается раньше в результате интенсивного разрушения пленок. Причина коалесценции во всем объеме дисперсной системы, имеющей агрегаты — относительно большая разность потенциалов между поверхностями двух капелек водного раствора электролита. Последняя возникает вследствие существенного различия величин удельной электропроводности фаз *.
* Изучению поведения эмульсий в электрическом поле посвящена монография [372]. Необходимо отметить, что причиной разрушения эмульсий и пен под действием внешней разности потенциалов является не только электрический пробой жидких пленок, разделяющих отдельные микрообъекты. Значительное влияние оказывают силы притяжения дипольной природы, действующие между частицами и возникающие благодаря эффектам поляризации двойного ионного слоя, а также материала дисперсной фазы [373]. (Прим, ред.)
Коалесценция при механических нагрузках. Подвергая устойчивую к коалесценции систему действию сдвиговых напряжений, вызывают деформацию частиц дисперсной фазы. Появляющиеся при этом «слабые» места в адсорбционных слоях могут быть не «залечены» из-за сравнительно малой подвижности адсорбированных молекул. Разрушение дисперсий происходит также в том случае, когда внешняя нагрузка достаточна для преодоления прочности адсорбционных слоев. Возможность изменения устойчивости пленок под влиянием механических напряжений имеет важное значение для процессов переработки латексов [249]. Сдвиг эмульсии обычно не вызывает коалесценцию а, наоборот, приводит к диспергированию, так как затраты энергии на разрушение слоев превышают работу образования новой поверхности.
Глава 3
ФЛОКУЛЯЦИЯ И КОАЛЕСЦЕНЦИЯ
В НЕКОТОРЫХ ТЕХНОЛОГИЧЕСКИХ ПРОЦЕССАХ
Коагуляционные процессы, широко распространенные в природе и технике, играют решающую роль в различных отраслях промышленности и сельского хозяйства. Они определяют в ряде случаев протекание полезных, а иногда нежелательных побочных явлений. Для специалиста в конкретной области знаний общие закономерности флокуляции и коалесценции частиц в дисперсных системах, цак правило, неизвестны. Поэтому будет полезным показать на более или менее произвольно выбранных примерах, что Изучение устойчивости и дестабилизации дисперсий представляет интерес не только для химиков-коллридников.
ФЛОТАЦИЯ
При флотационной подготовке руды и флокуляция, и коалесценция проявляются достаточно четко. Первая ступень — прилипание пузырьков газа к частицам руды при сохранении жидкой прослойки, разделяющей разнородные поверхности,— представляет классический пример гетерофлокуляции (см. главу 1, стр. 87).
Вторая ступень — установление непосредственного контакта частиц и газовых пузырьков — соответствует гетерокоалесценции. В принципе, флокуляции было бы достаточно для транспортировки маленьких частиц с помощью пузырьков газа на поверхность. Однако «флокуляционная флотация» невозможна по следующим причинам. Во-первых, сохранение относительно больших расстояний между взаимодействующими поверхностями определяет сравнительно слабую связь частиц и пузырьков воздуха. Поэтому крупные микрообъекты, если не произошла коалесценция, отрываются от всплывающих пузырьков и. осаждаются на дно флотационной ванны. Во-вторых, флокуляция практически не зависит от природы поверхности, т. е. не специфична. Коалесценцию вызывают введением в систему так называемого коллектора, который позволяет использовать флотационный процесс для разделения различных материалов.
Коалесценция наблюдается, когда случайное соударение пузырьков и частиц в результате направленного или броуновского
движения сопровождается их сближением благодаря преобладанию сил притяжения на всех расстояниях между поверхностями. Для сил ионно-электростатического взаимодействия справедливы закономерности, установленные для слабо заряженных частиц, так как при одинаковом знаке зарядов частиц руды и пузырьков воздуха высота барьера отталкивания зависит от меньшей величины фв-потенциала. Отсюда следует вывод, что для протекания коалесценции при флотации необходимо обеспечить низкий потенциал на одной из двух межфазных границ — жидкость/газ или твердое тело/жидкость *.
Однако, снижая потенциал поверхности частиц руды путем специфической адсорбции ионов, можно достичь такого состояния, при котором легко наступает не только прочное прилипание частиц к газовому пузырьку, но и их флокуляция друг с другом, приводящая к возникновению труднофлотируемых крупных агрегатов. Это объясняет, в частности, симбатное изменение элек-трокинетического потенциала и эффективности флодации при увеличении концентрации электролита в дисперсионной среде [189]. Для самого флотационного акта — коалесценции пузырьков газа и частиц руды — электрокинетический потенциал не имеет большого значения **. С целью создания оптимальных условий процесса необходимо снизить потенциал на границе жидкость/газ или, еще лучше, подбирая соответствующие ПАВ, добиться такого положения, при котором знаки зарядов разнородных поверхностей были бы различны.
При изучении вопроса о молекулярном взаимодействии микрообъектов в процессе флотации можно исходить в первом приближении из представлений Гамакера (см. главу 1, стр. 34). Для частицы руды 1 и пузырька воздуха 2, разделенных прослойкой жидкости 0, имеем:
V (Л) « Г12 (Л) + Voo (Л) - Г02 (Л) - Г01 (Л)
Пренебрегая величинами V12 и Г02 ввиду их малости, получим:
Г(/г) = У00(А)-701(А) (92)
Отсюда следует, что сближение газового пузырька и частицы руды происходит, когда энергия взаимодействия молекул воды
* Согласно теории взаимодействия и коагуляции разнородных частиц [191], высота барьера отталкивания, определяемая суммой ионно-электростатических и молекулярных сил, зависит от потенциалов обеих поверхностей, хотя значение функции Pt(h) в экстремальной точке (см. стр. 89) задает величина фб = min (фбь фа2}. (Прим, ред.)
** Элементарный акт флотации является частным случаем гетерокоагуляции, которая в свою очередь существенно зависит от электрических свойств взаимодействующих поверхностей. Поэтому ^-потенциал, отличный по величине от потенциала поверхности фо, хотя и не определяет количественно параметров системы, отвечающих слипанию пузырьков воздуха и частиц твердой фазы, в известной мере может характеризовать эффективность этого процесса. (Прим, ред.)
Друг С другом Voo больше энергии взаимодействия жидкой и твердой фаз Voi *. Такое сближение после достижения критической толщины пленки обусловливает коалесценцию. Наоборот, если Voi > Voo непосредственного слипания пузырька газа и частицы руды не наблюдается. Поскольку обе составляющие Voi и V0o суммарной энергии взаимодействия изменяются различным образом по мере уменьшения расстояния h между поверхностями, зависимость P(h) характеризуется одной из кривых, представленных на рис. 47. Для флотационного разделения веществ важно, чтобы функция P(h) соответствовала кривой рис. 47,6. Сформулируем условия, выполнение которых обеспечит подобную зависимость суммарного расклинивающего давления от толщины пленки.
Рассмотрим два случая:
1. Расстояние между пузырьком воздуха и частицей руды велико по сравнению с толщиной слоя б молекул воды или коллектора, адсорбированных на границе раздела твердое тело— жидкость.
2. Значения h и б сопоставимы друг с другом, однако h > 26.
При больших расстояниях между пузырьком воздуха и частицей руды, как показано ранее (см. главу 1, стр. 62), влияние адсорбционных слоев на молекулярн.ое притяжение сказывается очень слабо и справедлива формула (92). Поэтому зависимость П(й) является монотонно убывающей, отрицательно определенной функцией, когда разности статических или комплексных диэлектрических проницаемостей, входящие в уравнения (53) и (54), имеют одинаковый знак. Для воздуха 8г — еог = 1, т. е. должны совпадать знаки выражений ei — 80 и 1 — 80 или 80i — — еоо и 1 — 8оо- Таким образом, сближение частиц руды с газовым пузырьком будет наступать, если их статические и комплексные диэлектрические проницаемости меньше соответствующих величин для воды **.
Существование адсорбционных слоев может заметно изменять характер взаимодействия микрообъектов на достаточно близких
* Авторы не учитывают значительного влияния на флотацию электрического поля, которое возникает вокруг пузырька воздуха, всплывающего в жидкости. Теория эффекта Дорна применительно к процессу разделения минеральных частиц развита Дерягиным и Духиным [374—380]. (Прим, ред.)
** Известно, что устойчивость жидких слоев между пузырьком воздуха и Частицей минерала в сильной степени зависит от граничных фаз, существующих в ряде случаев на поверхности твердых тел [333] и заметно изменяющих характер молекулярного взаимодействия в системе [300, 301]. Например, изотерма молекулярной составляющей расклинивающего давления смачивающей пленки на кварце при наличии сольватного слоя имеет вид кривой, изображенной на рис. 47, д. Функция П(Л), очевидно, определяется, кроме того, знаком заряда и потенциалом поверхностей твердое тело — жидкость и Жидкость — газ, а также концентрацией и валентностью ионов электролита. Условие устойчивости смачивающих пленок, основанное на предположении равенства нулю заряда на границе жидкость — газ, сформулировано в работе [348]. (Прим, ред,)
расстояниях. Например, хемосорбция воды на поверхности частиц руды вызывает сильное увеличение значений VOi и тем самым определяет возникновение устойчивой пленки жидкости между твердой и газообразной фазами. Флотация в этих системах осуществляется только с помощью коллектора 3. Последний способен вытеснить с твердой поверхности хемосорбированные молекулы, причем его энергия взаимодействия со средой К0з меньше, чем Уоо. Тогда для расстояний, сравнимых с толщиной слоя коллектора (Л > 26), уравнение (92) принимает вид:
т = И00(й-6)-У0з(й-6) (93)
Формула (93) позволяет сделать заключение, что знак суммарной энергии зависит как от расстояния h, так и от взаимодействия молекул среды с молекулами ПАВ. Введение коллектора в систему приводит к гидрофобизации поверхности минеральных частиц (кварца, окисей металлов и т. д.). Иначе говоря, он обусловливает селективность флотационного процесса. Следовательно, коллектор должен отвечать двум основным требованиям: специфически адсорбироваться и иметь гидрофобные радикалы, обращенные в сторону жидкой фазы.
В ряде случаев коалесценции пузырьков воздуха и частиц руды, по-видимому, препятствует образование устойчивых черных пленок (см. главу 2, стр. 99). В настоящее время, насколько нам известно, изучение данного вопроса еще не проведено. Однако вполне обоснованно предполагать, что стабильные черные пленки будут возникать лишь при высоких концентрациях коллектора *.
Для флотации мелкодисперсных коллоидных частиц необходима *йх флокуляция. Сопровождаемое гидрофобизацией слипание микрообъектов в золе вызывается уменьшением заряда поверхности твердой фазы при введении коллектора. Таким способом, добавляя алкилсульфаты, производят, например, флотацию частиц гидрозоля окиси железа [250]. Аналогичным образом поступают, когда подлежащее выделению вещество получают в виде осадка при химической реакции. Частицы in statu nascendt сначала флокулируют и только потом флотируют [251]. Коллектор, применяемый для извлечения ионов, должен образовывать с ними либо нерастворимое, либо комплексное соединение. Метод ионной флотации развит, в частности, для Си2+ [251] и СггО? [252]. Коалесценция газовых пузырьков при наличии на их поверхности слоя частиц и переход к стабильным пенам, характеризующимся высокой концентрацией минерала, подробно рассмотрены в работе [253].
* При высоком содержании коллектора может наблюдаться так называемое явление «перемасливаиия» [381], заключающееся в гидрофилизации поверхности при образовании полимолекулярного адсорбционного слоя мыл. (Прим, ред.)
Устойчивость дисперсий играет важную роль при агломерационной флотации [254]. Для осуществления ее необходимы прежде всего стабильные эмульсии типа масло/вода. Извлечение частиц руды из суспензии происходит более полно при меньших размерах капелек и наступает в результате гетерокоалесценции. Капельки масла, содержащие минерал, в дальнейшем флотируются обычным способом с помощью пузырьков воздуха. Эти процессы еще мало изучены и до сих пор проводятся на основе эмпирических данных. Теоретическое обоснование всех технологических операций возможно только в том случае, если достаточно хорошо проведено исследование флотации и коалесценции капелек масла друг с другом, а также с частицами минералов и пузырьками пены *.
РАЗРУШЕНИЕ ПЕН
Можно привести много примеров, относящихся к различным областям химической технологии и показывающих, насколько важна и сложна проблема борьбы с пенообразованием. В связи с тем, что разрушение пен представляет не что иное как процесс коалесценции, необходимо принимать во внимание основные факторы, обусловливающие устойчивость тонких жидких слоев и причины, вызывающие разрыв пленок (см. главу 2, стр. 99).
Чаще всего для борьбы с пенообразованием применяют метод адсорбционного замещения: в дисперсионную среду вводят специальные вещества — пеногасители. В качестве последних используют ПАВ, которые вытесняют молекулы стабилизаторов с межфазной границы и не способствуют возникновению устойчивой пленки. Большинство из этих веществ слабо растворимы в дисперсионной среде. Широкое распространение получили, например, силикон, трибутилфосфат [255], высшие алифатические спирты и, главным образом, октанол, а также продукты присоединения этиленоксида к пропиленоксиду [256]. Все они образуют мономолекулярный адсорбционный слой с механической прочностью, недостаточной для предотвращения коалесценции.
Разрушение пен сравнительно просто достигается при охлаждении или нагревании. В первом случае разрыв пленок осуществляется благодаря кристаллизации воды, во втором — в результате десорбции стабилизатора и испарения растворителя из жидких прослоек.
Разнообразны механические методы борьбы с пенообразованием. Их преимущество состоит в том, что они не связаны с внесением в систему посторонних веществ и экономически выгодны. Эти методы применяют, когда требуется регенерация продуктов, содержащихся в пене. Малые количества пены можно
* Физико-химические основы процесса флотации подробно рассмотрены В работах [300, 382]. (Прим, ред.)
разрушить с помощью звуковых колебаний определенной частоты и интенсивности (II кгц, 150 дб). Изготовление соответствующих установок с большой производительностью слишком дорого. Подробные исследования [257] разрушения пены быстро вращающимся диском, расположенным перпендикулярно к ее поверхности, позволили сделать вывод, что механические воздействия (например, центрифугирование, продувание через сопло) обусловливают возникновение в жидких прослойках сдвиговых напряжений. Последние, как известно, приводят к прорыву тонких пленок (см. главу 2, стр. 124).
Опрыскивание горячей пены холодной Водой вызывает, наряду с напряжением сжатия и сдвига, десорбцию молекул стабилизатора вследствие разбавления раствора. Такой метод разрушения пен следует рассматривать как механико-адсорбционный.
ДЕЭМУЛЬГИРОВАНИЕ НА ПРИМЕРЕ
УДАЛЕНИЯ ВОДЫ ИЗ НЕФТИ
Для удаления капелек воды из сырой нефти имеют большое значение как процесс коалесценции, так и процесс флокуляции, хотя, очевидно, первый является определяющим. Можно полагать, что в связи с очень малой концентрацией ионов в углеводородной среде, силы ионно-электростатического отталкивания частиц дисперсной фазы малы и случайные соударения микрообъектов приводят к их слипанию. Иначе говоря, в сырой нефти должны присутствовать агрегаты капелек. Для их удаления необходимо провести флокуляцию, создавая в системе другие капли, содержащие деэмульгатор, а затем коалесценцию. Поскольку скорость коагуляции в значительной степени зависит от вязкости дисперсионной среды, желательно осуществлять деэмульгирование при повышенной температуре.
Исходная флокуляция капелек играет большую роль также при использовании внешнего электрического поля для разрушения эмульсий. Поэтому целесообразно перед электрическим деэмульгированием диспергировать в сырой нефти воду или водный раствор дестабилизатора. В результате такой обработки существенно увеличивается счетная концентрация частиц дисперсной фазы и тем самым количество и размер образующихся агрегатов.
Капельки воды в сырой нефти стабилизированы коллоидными частицами, создающими вокруг них структурно-механический барьер (см. главу 3, стр. 115). Путем фракционной фильтрации, применяя фильтры с различным радиусом пор, Нейман [258] нашел для пяти масел, взятых из ряда месторождений, что самые крупные частицы остаются в виде осадка на фильтрах со средним диаметром капилляров 200—300 А. Таким образом, расстоя-130
ние, на которое приближаются друг к другу две капельки воды в процессе флокуляции, составляет ~ 500 А. Эта же величина найдена при исследованиях модельных эмульсий [244].
Коалесценцию можно вызвать двумя способами:
I) вытеснением стабилизирующих частиц с межфазной границы путем изменения их смачиваемости (иногда такой метод неправильно называют химическим деэмульгированием);
2) непосредственным разрушением адсорбционных слоев. Оба способа практически не отличаются по эффективности [259].
Для вытеснения коллоидных частиц с межфазной поверхности обычно используют как водорастворимые, так и маслорастворимые ПАВ. Однако на практике чаще применяют первые (например, этоксилированные соединения). При выборе деэмульгаторов и определении их количества, необходимого для разрушения эмульсий, следует принимать во внимание явление обращения фаз, которое может существенно осложнить процесс [260].
Электрическое деэмульгирование подробно рассмотрено в главе 2 (стр. 121). Приложение внешней разности потенциалов обусловливает сильную поляризацию хорошо проводящих капелек воды и при условии их флокуляции — электрический пробой промежуточной углеводородной пленки. Если толщина адсорбционных слоев составляет 500 А, падения напряжения в зазоре между частицами около 400 мв вполне достаточно для разрушения эмульсий [219].
ПРОЦЕСС СТИРКИ
. При разработке большинства моющих средств, а также ряда составов, применяемых в текстильной промышленности, для проверки их эффективности проводят тест «загрязнение — стирка». При этом, как правило, в качестве меры чистоты принимают показатель белизны — отношение ремиссии света при какой-либо степени очистки к стандартной ремиссии света. Многие полученные таким образом эмпирические данные, очень важные для практики, приобретают еще большую ценность, когда раскрывается механизм явлений, сопровождающих стирку. Например, известный факт, что стирать грязное белье после долгого хранения труднее, чем белье, не подверженное длительной выдержке, объясняется молекулярным притяжением частиц загрязнений и волокна (см. главу 2, стр. 87). Как показали результаты измерения сил отрыва мелких частиц от плоской твердой поверхности [261, 262], прочность связи микрообъектов с подложкой существенно возрастает во времени. Такое изменение наступает вследствие деформации подложки в зоне соприкосновения и вытеснения воздуха, воды или жира из зазора между взаимодействующими поверхностями. Кроме того, в работах [261, 262] обнаружено, что пигментная защита, образующаяся в ванне,
предназначенной для окраски, удаляется значительно легче, чем частицы, нанесенные на сухой материал *.
Поведение в процессе стирки загрязнений, представленных полярными и неполярными органическими веществами, изучали в модельных опытах. Исследовали коалесценцию капелек и пленок, изготовленных из различных полимеров. Полученные экспериментальные данные позволяют сделать вывод, что специфическое влияние моющих средств на стирку связано с возникновением на оторвавшихся частицах и поверхности подложки адсорбционных слоев. Эти слои после выключения стиральной машины способны предотвратить повторную коалесценцию. Иными словами, стирка полимеров может осуществляться обычным способом, поскольку при полоскании, благодаря уменьшению концентрации электролита, происходит редиспергация (пептизация) частиц грязи и их удаление от очищаемой поверхности. В случае полимеров, обладающих низкой поверхностной энергией, таких как полиэтилен и политетрафторэтилен, не удается избежать повторной коалесценции с капельками полярных органических веществ (жирных кислот); для удаления неполярных масел требуются специальные ПАВ или высокие концентрации моющего средства. Материалы из полиэтилена и политетрафторэтилена, как правило, очищают, создавая сильный поток жидкости, транспортирующий механически отделенные от подложки частицы жира.
Изложенные результаты подтверждены Новаком и сотрудниками, которые’ радиохимическим методом исследовали способность различных ПАВ к удалению загрязнений. В работе [263] показано, что эффективность действия ПАВ в значительной степени определяется природой подложки. Так, пленки из меламинформальдегидной смолы легче очистить, чем пленки слабополярных полимеров (полиэтилена, полистирола), но тяжелее, чем поверхность металлов и стекол.
При очистке материалов, характеризующихся низкой величиной поверхностной энергии, коэффициент растекания не имеет решающего значения, поскольку в процессе стирки не наступает самопроизвольного смачивания, как это наблюдается для некоторых гидрофильных волокон. Иначе говоря, для гидрофобных поверхностей специфическое влияние ПАВ сказывается, главным образом, на коалесценции, потому что отрыв частиц загрязнений происходит благодаря механическим воздействиям. Такое отделение капелек масла от подложки требует сравнительно малых затрат энергии, равных приблизительно работе образования новой поверхности жидкости (^ 30 эрг!см2).
* Зависимость' сил адгезии от длительности контакта кварцевых нитей обнаружена в работах [340, 341]. В дальнейшем этот вопрос более подробно рассмотрен рядом исследователей [342, 343]. Различие между прочностью прилипания частиц, нанесенных на плоскую поверхность из воздушной и из жидкой сред, отмечал Зимон [343]. (Прим, ред.)
Считавшаяся ранее весьма важной роль пены как носителя загрязнений на самом деле не столь существенна. Несмотря на то, что в отдельных случаях флотация частиц грязи, несомненно, проявляется, пена не вносит заметного вклада в действие многих препаратов (предназначенных, например, для мойки машин). Вообще, для моющего действия справедливы основные закономерности, установленные при изучении устойчивости дисперсных систем.
В ванне для стирки содержание электролита достаточно велико и, следовательно, необходимо позаботиться о том, чтобы защитить удаленные от очищаемой поверхности диспергированные частицы загрязнений от повторной коалесценции. В качестве стабилизаторов можно использовать защитные коллоиды (например, карбоксиметилцеллюлозу) или специфически адсорбирующиеся носители зарядов — ионогенные ПАВ и полифосфаты.
СТРУКТУР00БРА30ВАНИЕ
В КОНЦЕНТРИРОВАННЫХ ДИСПЕРСИЯХ
ТВЕРДЫХ ВЕЩЕСТВ
Изучение проблемы.образования пространственных структур в суспензиях привело к возникновению самостоятельной области науки — физико-химической механики дисперсных систем [264]. Основное содержание этого научного направления—исследование и нахождение условий получения твердых тел с определенными механическими свойствами. Свойства пространственных структур зависят, главным образом, от того, существуют или отсутствуют между частицами тонкие слои дисперсионной среды; В первом случае речь идет о флокулированных системах (коагуляционные структуры), во втором — о коалесцированных системах (конденсационные структуры) [265,266]*.
Реологические параметры коагуляционных структур находятся в непосредственной связи с толщиной пленки, препятствующей коалесценции. Относительно толстые слои, соответствующие равенству дальнодействующих сил отталкивания и притяжения (см. главу 1, стр. 50), обусловливают меньшую прочность дисперсных систем, чем тонкие [267]. Существенное изменение реологических свойств дисперсий наступает, когда исчезает пленка, разделяющая твердые частицы. Оно, в частности, проявляется в увеличении прочности пространственной структуры на порядок и более.
Под влиянием внешних сил коагуляционные структуры разрушаются и после снятия нагрузки вновь восстанавливаются в
* Основные положения физико-химической механики, развитые Ребиндером и его сотрудниками, приведены в ряде монографий [264, 383—386]. (Прим, ред.)
течение некоторого промежутка времени. Такой обратимый переход из твердого в жидкотекучее состояние называют тиксотропным превращением. Восстановления исходной прочности в системах, характеризующихся кристаллизационной связью между частицами и подвергнутых разрушению, как правило, не происходит. При действии сдвигающей нагрузки, практически не вызывающей разрушения, коагуляционные структуры обнаруживают два вида высокоэластичной деформации: быструю (время релаксации 10-3—10-2 сек) и медленную (время релаксации 102—103 сек). Первая соответствует процессу переориентации, вторая — сдвигу частиц относительно друг друга без изменения равновесных расстояний [268] *.
Коагуляционные структуры широко распространены в природе и технике. Они играют значительную роль при транспортировке и перемешивании концентрированных суспензий (приготовление бетона и асфальта, производство красителей и пищевых продуктов: майонеза, сбитых сливок и т. д.). Тонкие жидкие прослойки дисперсионной среды разделяют микрообъекты в гелях. Влияние наполнителей на свойства различных композиций, полученных на основе высокомолекулярных соединений, объясняется образованием пространственной связи между полимерами и частицами вносимого вещества.
Можно считать, что коагуляционные структуры являются переходной ступенью для возникновения кристаллизационных структур. Например, «сплавление» частиц достигают при нагревании. В ряде случаев непосредственная связь между отдельными микрообъектами системы наступает благодаря процессам перекристаллизации, как это имеет место при твердении вяжущих материалов [269]**. Роль взаимодействия твердых частиц при брикетировании, таблетировании и грануляции обсуждена в работах [270, 271].
ЭЛЕКТРОКОАГУЛЯЦИЯ. ЭЛЕКТРОЛАКИРОВКА
В последние годы находит все большее распространение метод отложения растворенных или диспергированных частиц на металлических поверхностях под воздействием внешнего электрического поля. Хотя уже сравнительно давно известно [78, 272], что осаждение наступает вследствие коагуляции, связанной с повышением концентрации продуктов электрохимической реакции на электроде, до настоящего времени сохранилось неоправданное предположение о непосредственном разряде частиц [273;
* Реологические свойства коагуляционных дисперсных структур и тиксотропные превращения гелей подробно изучены в работах [387—396]. (Прим. Ред.)
** См. также [397—398]. (Прим, ред.)
274] *. В действительности процесс электрофоретического получения покрытий представляет последовательность взаимосвязанных сложных явлений, в которой важную роль играют как флокуляция, так и коалесценция [275, 276]. Транспортировка частиц дисперсной фазы в ванне происходит почти полностью только за счет конвекции и лишь в тонком диффузионном граничном слое заметно сказывается их электрофоретическое перемещение [276].
На первом этапе формирование покрытий из водных дисперсий объясняется флокуляцией под действием электролитов частиц лака, окисей и т. д. На катоде наблюдается накопление ОН-, а на аноде возникают ионы водорода. Кроме того, в ряде случаев отмечается анодное растворение металла. Такое повышение содержания ионов способствует образованию вблизи электрода коагуляционной структуры (см. главу 3, стр. 133). Она не препятствует переносу ионов от поверхности металла к границе раздела покрытие — объем дисперсии и поэтому толщина слоя может увеличиваться. Коалесценция частиц лака на первом этапе нежелательна, поскольку создание компактного изоляционного осадка фактически означает прекращение процесса. Непосредственное слипание микрообъектов должно происходить, когда достигается оптимальная толщина покрытия.
Поскольку речь идет не об электрохимическом методе осаждения, сцепление частиц дисперсной фазы с электродом также осуществляется благодаря гетерокоалесценции. Следовательно, на качество покрытия существенное влияние, по-видимому, оказывает модификация металлической поверхности. Однако эта проблема, по нашему мнению, практически не изучена.
ПОДГОТОВКА СТОЧНЫХ вод
При подготовке сточных вод различного происхождения, наряду с некоторыми другими операциями, проводят флокуляцию, а в ряде случаев и коалесценцию. Отделение.твердых взвешенных частиц обычно осуществляют путем флокуляции.
Чтобы избежать нового загрязнения из-за введения в систему электролита, принимают во внимание, что гидролизованные ионы многовалентных металлов, находящиеся в растворе в виде гидроксильных комплексов (изополикатионы), благодаря' высокому заряду и специфической адсорбции на границе твердое тело — жидкость гораздо легче вызывают коагуляцию, чем соответствующие простые ионы [277]. Так, при использовании
* Влияние электрохимических процессов на электрофоретическое осаждение частиц на электроды из суспензий подробно рассмотрено Коелмаисом [399]. Однако нельзя не принимать во внимание, что при наложении внешнего электрического поля в объеме дисперсной системы вследствие поляризационного взаимодействия частиц происходит их коагуляция [366]. Следовательно, на электроде осаждаются, как правило, не отдельные микрообъекты, а цепочечные агрегаты, ориентированны? вдодь силовых линий поля [400]. (Прим, ред.)
А1+3 в среде, характеризующейся значением pH 3,5, для полной флокуляции тонкодисперсной суспензии кварца достаточно концентрации порядка 10-6—10~5 г-ион!л. Суспензия кварца может рассматриваться как модельная система для сточных вод, содержащих взвешенные частицы загрязнений.
В заключение отметим, что этот обзор явлений флокуляции и коалесценции, протекающих в некоторых отраслях химической технологии, несмотря на краткость, показывает важность изучения коллоидно-химических процессов в дисперсных системах.
ЛИТЕРАТУРА
1. П. А. Р е б и н д е р, Коллоидн. ж., 20, 528 (1958).
2. D. С. Grahame, В. A. Soderberg, J. Chem. Phys., 22, 449 (1954).
3. J. L у k 1 e m a, Trans. Faraday Soc., 59, 418 (1963).
4. G. Gouy, J. Phys. Radium, 9, 457 (1910).
5. G. Gouy, Ann. Phys., 7, 129 (1917).
6. D. L. Chapman, Philos. Mag., 25, 475 (1913).
, 7. F. Booth, J. Chem. Phys., 19, 391 (1951); 19, 1327 (1951); 19, 1615 (1951).
8. A. D. Buckingham, ibid., 25, 428 (1956).
9. J. B. Hasted, D. M. Ritson, С. H. Collie, ibid., 15, 1 (1948).
10. D. C. G r a h a m e, ibid., 18, 903 (1950).
11. В. E. Conway, J. O’M. В о c k r i s, J. A. Ammar, Trans. Faraday Soc., 47, 756 (1951).
12. M. J. S p a r n a a y, Rec. trav. chim., 77, 872 (1958).
13. H. Brod owsky, H. Strehlow, Z. Elektrochem., Ber, Bunsenges. phys. Chem., 63, 262 (1959).
14. A. Steinchen - Sanield, A. Santel d, H. Hurwitz, Kollogium uber Grenzflachenerscheinungen, Kpnigl. Flamisch. Akad. d. Wissenschaften, 1965. uber Grenzflachenerscheinungen, Kbnigl. Flamisch. Akad. d. 'Wissenschaften, 1965.
15. J. Malsch, Phys. Z., 29, 770 (1928).
16. J. Lyklema, Med. Electron, Biol. Eng., 2, 265 (1964).
17. E. H u c k e 1, G. К r a f f t, Z. phys. Chem., 3, 135 (1955).
18. M. Eigen, E. Wicke, Z. Elektrochem., Ber. Bunsenges. phys. Chem., 56, 551 (1952).
19. H. D. О h 1 e n b u s c h, ibid., 60, 607 (1956).
20. S. Levine, G. M. Bell, Trans. Faraday Soc., 53, 143 (1957).
21. S. Levi ne, G. M. Bel 1, J. Phys. Chem., 64, 1188 (1960).
22. D. A. Haydon, F. H. Taylor, Philos. Trans. Roy. Soc. (London), Ser. A, 253, 255 (1960); цит. no D. Haydon, Recent Progress in Surface Science, vol. 1, New York — London, 1964.
23. I. Prigogine, P. Mazur, R. D e f a y, J. Chem. Phys., 50, 146 (1953).
24. W. E. W i 1 1 i a m s, Proc. Phys. Soc., A66, 372 (1953).
25. S. Levine, G. M. Bell, Discuss. Faraday Soc., 42, 69 (1966).
26. E. J. V e r w e y, J. T h. G. О v e rb e e k, Theory of the Stability of Lyophobic Colloids, Amsterdam, 1948.
27. A. L о e b, P. H. W i e г s e m a, J. Th. G. О v e r b e e k, The Electrical Double Layer around a Spherical Colloid Particle, Mass. Inst. Techn. Press, 1960.
28. O. Stern, Z. Elektrochem., 30, 508 (1924).
29. R. B. Whitney, D. C. Grahame, J. Chem. Phys., 9, 827 (1941).
30. D. C. Grahame, Z. Elektrochem., Ber. Bunsenges. phys. Chem., 62, 264 (1958).
31. J. O’M. Bockris, M. A. V. Devanathan, K. Muller, Proc. Roy. Soc. (London), Ser. A, 274, 55 (1963).
32. O. A. Esin, B. F. Markov, Acta physicochim. URSS, 10, 353 (1939).
33. В. V. Derjaguin, ibid., 10, 333 (1939).
34. Б. В. Д e p я г и н, Коллоиди. ж., 6, 291 (1940).
3$. Б. В. Д е р я г и и, Там же, 7, 285 (1941).
36. В. V. Derjaguin, L. D. Landau, Acta physicochim. URSS, 14, 633 (1941).
37. G. Fren S, Dissertation, Utrecht, 1968.
38. Б. В. Дерягин, Изв. АН СССР. Сер. хим., Xs 5, 1153 (1937).
39. Г. Р. Кройт, Наука о коллоидах, ИЛ, 1955.
40. S. Levine, Discuss. Faraday Soc., 18, 202 (1954).
41. H. R e e r i n k, J. Th. G. Overbee k, ibid., 18, 74 (1954).
42. J. L у k 1 e m a, Abh. deutsch. Akad. Wiss., KL Chem., Biol., Geol., 6a, 542 (1966).
43. J. Lyklema, Discuss. Faraday Soc., 42, 81 (1966).
44. F. L О n d о n, Z. Phys., 63, 245 (1930).
45. F. London, Trans. Faraday Soc., 33, 8 (1937).
46. H. Margenau, Rev. mod. Phys., 11, 1 (1939).
47. W. Kauzmann, Quantum Chemistry, An Introduction, New York, 1957.
48. Э. А. Мелвин-Хьюз, Физическая химия, т. 1, ИЛ, 1962.
49. J. С. Slater, J. G. Kirkwood, Phys. Rev., 37, 682 (1931).
50. Landolt-Bornstein, Zahlenwerte und Funktionen, Berlin — Gottingen— Heidelberg, 1950.
51. H. Kai Iman, M. Wil 1st fit ter, Naturwiss., 20, 952 (1932).
52. J. H. De Boer, Trans. Faraday Soc., 32, 21 (1936).
53. H. С. H a m a k e r, Physika, 4, 1058 (1937).
54. E. M. Л и ф ш и ц, ЖЭТФ, 29, 94 (1955).
55. М. J. Renne, В. R. A. Nijboer, Chem. Phys. Letters, 1, 317 (1967).
56. D. L a n g b e i n, Proceedings of the Conference «Physics of Adhesion», Karlsruhe, 1969, p. 55.
57. M. J. V о 1 d, J. Colloid Sci., 16, 1 (1961).
58. B. W. N i n h a m, V. В. P a r s e g i a n, Nature, 224, 1197 (1969).
59. B. W. N i n h a m, V. B. Parsegian, J. chem. Phys., 52, 4578 (1970).
60. B. W. N i n h a m, V. В. P a r s e g i a n, ibid., 53, 3398 (1970).
61. H. B. G. C a s i m i r, D. P о 1 d er, Physic. Rev., 73, 360 (1948); H. B. G. C a-
s i m i r, Proc. Kon. nederl. Acad. Wetensch., Ser. B, 51, 793 (1948).
62. R. J. Hunter, Austral. J. Chem., 16, 774 (1963).
63. A. Scheludko, D. Platikanov, E. Manev, Discuss. Faraday Soc., 40, 253 (1965).
64. T. К i h a г a, Adv. Chem. Phys., 1, 261 (1958).
65. С. M. P ы т о в, Теория электрических флуктуаций и теплового излучения, Изд. АН СССР, 1953.
66. И. Е. Дзялошинский, Е. М. Лифшиц, Л. П. Питаевский, ЖЭТФ, 36, 1797 (1959).
67. И. Е. Дзялошинский, Е. М. Лифшиц, Л. П. Питаевский, Там же, 37, 229 (1959).
68. I. Е. Dzjaloschinski, Е. М. Lifschitz, L. Р. Pitajewski, Adv. in Phys., 10, 165 (1961).
69. T. К i h a r a, N. H о n d a, J. Phys. Soc. Japan, 20, 15 (1965).
70. O. F. Devereux, P. L. de Bruyn, J. Chem. Phys., 37, 2147 (1962).
71. В. V. Derjaguin, Kolloid. Z., 69, 155 (1934).
72. G. A. Johnson a. oth., Discuss. Faraday Soc., 42, 120 (1966).
73. Ju. M. Glazman, ibid., 42, 255 (1966).
74. В. M. Барбой, Ю. M. Г л а з м а н, Г. И. Фукс, Тезисы докладов на VI Всесоюзной конференции по коллоидной химии, Изд. Воронежск. уи-та, Воронеж, 1968, стр. 17.
75. Р. Э. Нейман, Коагуляции синтетических ленестков, Изд. Воронежск. ун-та, Воронеж, 1967.
76. Г. А. Мартынов, В. П. С ми л г а, Коллоидн. ж., 27, 250 (Г965).
77. В. V. Derjaguin, L. D. Landau, Acta physicochim. URSS, 14, 633 (1941).
78. H. Freundlich, Kapillarchemie, Bd. 2, Leipzig, 1932.
79. H. Schulze, J. prakt. Chem., 25, 431 (1882); 27, 320 (1883) (цит. no [78]).
80. P. A. Rebinder, N. W. Michailov, Rheol. acta (Erganzungs. zur Kolloid. Z.), 1, 361 (1961). *
81. P. A. R e b i n d е г, J. Pure a. Applied Chem., 10, 337 (1965).
82. R. H. Otte will, J. Wilkins, J. Colloid Sci., 15, 512 (1960).
83. R. H. Otte will, A. Sirs, Bull. Photometric Spectrometry Group, 10, 262 (1957).
84. A. W a t i 11 о n, A. M. J о s e p h • P e t i t, Discuss. Faraday Soc., 42, 143 (1966).
85. P. 9. Нейман, О. А. Ляшенко, Коллоидн. ж., 24, 494 (1962).
86. W. О s t w а 1 d, Kolloid-Beihefte, 10, 209 (1919).
87. N. J s h i z a k a, Z. phys. Chem., 83, 97 (1913).
88. A. Kumar, Amal K. Bhattacharya, Albani K. Bhattacha-rya, J. Indian Chem. Soc., 39, 361 (1962).
89. M. Mirnik, Nature (London), 199, 555 (1963).
90. M. Sing, О. P. Bansal, J. Indian Chem. Soc., 41, 85 (1964).
91. H. Reerink, J. Th. G. Overbeek, Discuss. Faraday Soc., 18, 74 (1954).
92. К. H. Schulz, B. Tez a k, Ark. Kemi, 26, 187 (1954).
93. R. H. Otte will, J. N. Shaw, Discuss. Faraday Soc., 42, 154 (1966).
94. В. M. Муллео, Б. В. Дерягин, ДАН СССР, 176, 1111 (1967).
95. N. A. Fu ch s, Z. Phys., 89, 736 (1934).
96. G. A. Johnson, J. Goldfarb, B. A. Pet hie a, Trans. Faraday Soc., 61, 2321 (1965).
97. J. H. S c h e n k e 1, J. A. Kitchener, ibid., 56, 161 (1960).
98. S. N. Srivastava, D. A. Haydon, ibid., 60, 971 (1964).
99. S. N. S r i v a s t a v a, Indian J. Chem., 3, 376 (1965).
100. R. H. Otte will, M. C. Rastogi, A. Watanabe, Trans. Faraday Soc., 56, 880 (1960).
101. R. H. О 11 e w i 11, D. J. W i 1 k i n s, ibid., 58, 608, (1962).
102. P. H. W i e г s e m a, Dissertation, Utrecht, 1964.
103. J. L у k 1 e m a, Abh. dtsch. Akad. Wiss. Berlin, KL Chem., Geol., Biol., 6a, 542 (1966).
104. A. Wat ill on, A. M. Joseph, Proceedings of the 3d International Congress on Surface Activity, Sec. A, London, 1960, p. 145.
105. A. W a t i 11 о n, Ph. G e г a r t, Proceedings of the 4th International Congress on Surface Activity, Sec. B, Brussel, Vol. VI, 1964, p. 28.
106. R. H. Otte will, K. G. Mathai, Trans. Faraday Soc., 62, 759 (1966).
107. M. Mirnik, R. D e s p о t о v i с, M. J. H e г a k, Abh. dtsch. Akad. Wiss.
Berlin, KL Chem., Geol., Biol., 6a, 944 (1966).
108. E. W. Fischer, Kolloid. Z., 160, 120 (1958).
109. R. Zsigmondy, Kolloidchemie, Leipzig, 1923.
110. Wo. Ostwald, Kolloid-Beihefte, 10, 179 (1919).
111. H. Thiele, H. S. V. Levern, J. Colloid Sci., 20, 679 (1965).
112. A. Audsley, A. Fursey, Nature (London), 208, 753 (1965).
113. V. K- L a M e r, Discuss. Faraday Soc., 42, 248 (1966).
114. R. W. Slater, J. A. Kitchener, ibid., 42, 267 (1966).
115. A. Scheludko. D. Ex e row a, Kolloid. Z., 168, 24 (1960).
116. H. Sonntag, Tenside, 5, 188 (1968).
117. D. Exerowa, Kolloid. Z. u. Z. Polymere, 232, 703 (1969).
118. Б. В. Дерягин, А. С. Титиевская, Коллоидн. ж., 15, 16 (1953).
119. В. V. Derjaguin, A. S. Т i t i j е w s k a j a, Proceedings of the 2 International Congress on Surface Activity, vol. 1, London, 1957, p. 211.
120. Б. В. Дерягин, А. С. Титиевская, В. X. Выборнова, Коллоидн. ж, 22, 396 (1960).
121. Е. М. Duyvis, Dissertation, Utrecht, 1962.
122. М. van den Tempel, J. Colloid Sci., 13, 125 (1958).
123. A. Scheludko, Kolloid. Z„ 155, 39 (1957).
124. K. J. M у s e 1 s, M. N. Jones, Discuss. Faraday Soc., 42, 42 (1966).
125. H. S о n n t a g, J. N e t z e 1, H. К1 a r e, Kolloid. Z. u Z. Polymere, 211, 121 (1966).
126. J. L у k 1 e m a, P, C. Scholten, K. J. M у s e 1 s, J. Phys. Chem., 69, 116 (1965).
127. J. Netzel, Dissertation, Humbolt—Universitat, Berlin, 1968,
128. A. Scheludko, D. Exerowa, Kolloid. Z., 165, 148 (1959); D. Exerowa, J. I w a n о w, A. Scheludko, Ann. Univ. Sofia, 56, 157 (1962).
129. D. Exerowa, A. Scheludko, 4 International Kongress fur grenzfla-chenactive Stoffe, Brussel, Bd. 2, 1964, S. 1097.
130. J. .Lyklema, K. J- M у s e 1 s, J. Am. Chem. Soc., 87, 2539 (1965).
131. В. M. Беляков, P. И. Кравцова, M. Г. Раппопорт, Таблицы эллиптических интегралов, т. 1, 2, Изд. АН СССР, 1962, 1963.
132. J. Netzel, Н. Sonntag, Abh. dtsch. Akad. Wiss., Berlin, KI. Chem., GeoL, Biol., 6a, 589 (19’66).
133. A. Scheludko, Proc, nederl. Acad. Wetensch., ser. B65, 87 (1962).
134. M. N. Jones, K. J. M у s e 1 s, P. C. Scholten, Trans. Faraday Soc., 62, 1336 (1966).
135. G. Nilsson, J. Phys. Chem., 61, 1135 (1957).
136. А. Шелудко, ДАН СССР, 123, 1074 (1958).
137. Б. В. Дерягин, М. М. Ку саков, Изв. АН СССР. Сер. хим., № 5, 1119 (1937).
138. Р. S. Prokhorov, Discuss. Faraday Soc., 18, 41 (1954) .
139. G. A. H. Elton, Proc. Roy. Soc. (London), Ser. A, 194, 275 (1948).
140. R. S. Allan, G. E. Charles, S. G. Mason, J. Colloid. Sci., 16, 150
(1961).
141. D. P 1 a 11 k а п о w, J. Phys. Chem., 68, 3619 (1964).
142. S. P. F r a n k e 1, K. J- M у s e 1 s, ibid., 66, 190 (1962).
143. F. P. Bowden, S. H. В a s t о w, Nature (London), 18, 828 (1935).
144. Б. В. Дерягин и др., Коллоидн. ж., 24, 289 (1962).
145. В. V. D е г j a g u i n, Discuss. Faraday Soc., 42, 109 (1966).
146. E. M a n e w, M. В о u 1 e w a, Abh. dtsch. Akad. Wiss., Berlin, KI. Chem., GeoL, Biol., 6a, 557 (1966).
147. A. Watanabe, R. Gotoh, Kolloid. Z. u. Z. Polymere, 191, 36 (1963).
148. Sh. Usui, T. Yamasaki, J. Phys. Chem., 71, 3195 (1967).
149. A. Vrij, J. Colloid Sci., 19, 1 (1964).
150. A. Vrij, Adv. Colloid, a. Interface Sci., 2, 39 (1968).
.151. J. A. M a n n, J. Colloid a. Interface Sci., 25, 437 (1967).
152. Б. В. Дерягин, И. И. Абрикосова, ЖЭТФ, 21, 945 (1951).
153. В. V. Derjaguin, I. I. Abrik os sow a, J. Phys. Chem. Solids, 5, 1 (1958).
154. В. V. Derjaguin a. oth., Discuss. Faraday Soc., 18, 24 (1954).
155. W. Black a. oth., Trans. Faraday Soc., 56, 1597 (1960).
156. M. J. S p a r n а а у, P. W. J. Jochems, 3 International Kongress fiir grenzflachenaktive Stoffe, Koln, B, I960, S. 375.
157. J. A. Kitchener, A. P. Prosser, Proc. Roy. Soc. (London), Ser. A, 242, 403 (1957).
158. A. v a n Silfhout, Proc. Kon. nederl. Acad. Wetensch., 69, 501 (1966).
159. D. Tabor, R. H. S. W i n t e r t о n, Nature (London), 219, 1120 (1968).
160. В. V. D e r j a g u i n a. oth., J. Colloid Sci., 19, 113 (1964).
161. Б. В. Дерягин, Я. И. Рабинович, Коллоидн. ж., 31, 42 (1969).
162. Б. В. Дерягин, В. М. Муллер, Я. И. Рабинович, Там же, 31, 304 (1969).
163. Б. В. Дерягин, Я. И. Рабинович, Там же, 31, 42 (1969).
164. Т. Н. Воропаева, Б. В. Дерягин, Б. Н. Кабанов, Там же, 24, 396 (1962). .
165. Н. Sonntag, Z. phys. Chem., 221, 373 (1962).
166. Н. Sonntag, 4' International Kongress fiir grenzflachenaktive Stoffe, Brussel, Bd. 2, 1964, S. 1089.
167. T. H a n a i, D. A. Haydon, J. Taylor, Proc. Roy. Soc. (London), Ser. A, 281, 377 (1964).
168. J. Taylor, D. A. Haydon, Discuss. Faraday Soc., 42, 51 (1966).
169, H. T. Tien, E. A. Dawidowicz, J. Colloid a. Interface Sci., 22, 438 (1966). ...
170. H. Sonntag, Mbr. dtsch. Akad. Wiss., Berlin, 4, 330 (1962),
171. J. J. C. Oo men, Philips Res. Rep., Suppl,, 4, I (1967),
172. P. S h е г m а п, J. Phys. Chem., 67, 2531 (1963).
173. W. Albers, J. Th. G. Overbee k, J. Colloid Sci., 14, 561 (1959).
174. J. L. v a n der Minne, P. H. Hermanie, ibid., 7, 600 (1952).
175. H. К о e 1 m a n s, Philips Res. Rep., Suppl., 10, 161 (1955).
176. L. A. Romo, 4 International Kongress fiir grenzflachenaktive Stoffe, Briis-sel, Bd. 2, 1964, S. 1279.
177. M. van der Waarden, J. Colloid Sci., 5, 317 (1950).
178. G. D. Parfitt, E. Willis, ibid., 22, 100 (1966).
179. R. H. Ottewill, J. M. Tiffany, J. Oil Colour Chemist’s Assoc., 50, 844 (1967).
180. K. Strenge, H. Sonntag, Tenside, 6, 61 (1969).
181. K. Strenge, Diplomarbeit, TU, Dresden, 1962.
182. D. N. L. McGown, G. D. Parfitt, E. Willis, J. Colloid Sci., 20,
650 (1965).
183. К. E. Lewis, G. D. Parfitt, Trans. Faraday Soc., 62, 1652 (1962).
184. D. N. L. McGown, G. D. Parfitt, Discuss. Faraday Soc., 42, 225 (1966).
185. P. T. Dawson, D. A. Haydon, Kolloid. Z. u. Z. Polymere, 203, 133 (1965).
186. V. T. Crowl, M. A. M a 1 a t i, Discuss. Faraday Soc., 42, 267 (1966).
187. R. A. Stromberg, L. E. Smith, J. phys. Chem., 71, 2470 (1967).
188. A. H. Ф p у м к и n, ЖФХ, 12, 1 (1938).
189. A. Scheludko, Kolloid. Z„ 191, 52 (1963).
190. H. Sonntag, K. S t r e n g e, Tenside, 4, 129 (1967).
191. Б. В. Дерягин, Коллоидн. ж., 16, 425 (1954); Discuss. Faraday Soc., 18, 85 (1954).
192. O. F. Devereux, P. L. de Bruyn, Interaction of Plane-parallel Double Layers, Cambridg, Mass., 1963.
193. R. Hogg, T. W. Healy, D. W. F u e r s t e n a u, Trans. Faraday Soc., 62, 1638 (1966).
194. Б. В. Дерягин и др., Коллоидн. ж., 23, 535 (1961).
195. S h. Usui, Т. Yamasaki, J. Colloid Interface Sci., 29, 629 (1969).
196. A. Scheludko, D. Platikanow, Kolloid. Z. u Z. Polymere, 175, 150 (1961).
197. A. D. R e a d, J. А. К 11 c h e n e r, J. Colloid, a. Interface Sci., 30, 391 (1969).
198. A. Scheludko,. B. Radoev, A. Fabrikant, Годишник Соф. ун-та, хим. фак., 63, 1 (1968/1969).
199. A. J. d е Vries, Rec. trav. chim., 77, 338 (1958).
200. A. Scheludko, Adv. Colloid a. Interface Sci., 1, 391 (1967).
201. A. Scheludko, E. Ma new, Trans. Faraday Soc., 64, 1123 (1968).
202. A. Vrij, Discuss. Faraday Soc., 42, 23 (I960).
203. Д. Ексерова, И. T. Коларов, Годишник Соф. ун-та, хим. фак., 59, 207 (1966).
204. Н. Sonntag, J. Netzel, Tenside, 3, 296 (1966).
205. X. 3 о и т а г, Коллоидн. ж., 33, 529 (1971).
206. И. Ньютон, Оптика, Гос. изд., 1927.
207. Н. S о n n t a g, Z. phys. Chem., 222, 365 (1962).
208. Н. Sonntag, F. P use he 1, B. Strobel, Tenside, 4, 349 (1967).
209. R. D. Void, R. C. Groot, J. Colloid Sci., 19, 384 (1964).
210. H. К 1 a r e, j r., Dissertation, Berlin, 1967.
211. K. J. M у s e 1 s, K- S h i n о d a, S. Frankel, Soap Films, New York, 1959.
212. E. M. D u у v 1 s, Thesis, Utrecht, 1962.
213. J. Perrin, Ann. Phys., 10, 165 (1918).
214. P. R. Muss el white, J. A. Kitchener, J. Colloid a. Interface Sci., 24, 80 (1967).
215. A. Scheludko, B. Radoev, T. Kolarow, Trans. Faraday Soc. 64, 2213 (1968).
216. T. Kolarow, A. Scheludko, D. Exerowa, ibid., 64, 2864 (1968).
217. K. J. My seis, H. F, Huisman, R. H, Razouk, J. Phys. Chem., 70, 1339 (1966),
218. F. Huisman, К. M у s e 1 s, ibid., 73, 489 (1969')’.
219. H. S о n n t a g, H. К 1 а г e, Tenside, 4, 104 (1967).
220. H. Tien, S. Carbone, E. A. D a vi d owicz, Kolloid. Z. u. Z. Polymere, 212, 165 (1968).
221. D. A. Haydon, J. Taylor, Nature (London), Ser. A, 217, 739 (1968).
222. J. M. Cor kill a. oth., Proc. Roy. Soc. (London), Ser. A, 273, 84 (1963).
223. J. M. Corkill, J. F. Goodman, С. P. Ogden, Trans. Faraday Soc., 61, 583 (1965).
224. J. Lucassen, ibid., 64, 2221 (1968).
225. F. van Voorst Vader, Th. F. Erkens, M. van den Tempel, ibid., 60, 1170 (1964).
226. J. Lucassen, R. S. Hansen, J. Colloid a. Interface Sci., 22, 32 (1966).
227. J. Lucassen, E. H. Lucassen-Reynders, ibid., 25, 496 (1967).
228. D. Thiessen, P. Schwartz, Z. phys. Chem. (Leipzig), 236, 363 (1967).
229. S. U. Pickering, J. Chem. Soc. (London), 91, 2001 (1907).
230. S. U. Pickering, Kolloid. Z., 7, 11 (1910).
231. A. F. Koretzki, A. B. T.aubman, Abh. dtsch. Akad. Wiss., KI. Chem., Biol., Geol., 6a, 576 (1966).
232. S. A. Nikitina, Abh. dtsch. Akad. Wiss., Kl- Chem., Biol., Geol., 6a, 609 (1966).
233. С. А. Никитина, О. С. Мочалова, Коллоидн. ж., 30, 264 (1968).
234. R. Zsigmondy, Р-. A. Thiessen, Das kolloide Gold, Leipzig, 1925.
235. П. А. Ре бин дер, Предисловие к русскому изданию книги В. Клейтона «Эмульсии», ИЛ, 1950.
236. V. Т. С г Owl, М. А. М а 1 a t i, Discuss. Faraday Soc., 42, 301 (1966).
237. P. Peyser, D. J. T u t a s, R. R. Stromberg, J. Polymer Sci., 5, 651 (1967).
238. K. S t г e n g e, Dissertation, Humbolt — Universitat, Berlin, 1969.
239. S. Tam ay, J. Phys. Chem., 63, 1283 (1959).
240. M. J. D. Low, К- H. В г о w n, H. I n о n e, J. Colloid a. Interface Sci., 24, 252 (1967).
241. P. A. Rebinder, A. A. Trapeznikow, Acta physicochim. URSS, 9, 257 (1938).
242. П. А. Ребиидер, Коллоидн. ж., 20, 527 (1958).
243. П. А. Ребиндер, А. Б. Таубман, Там же, 23, 359 (1961).
244. Н. Sonntag, Н. К 1 а г е, jr., Tenside, 2, 365 (1965).
245. Н. Sonntag, Н. К 1 а г е, jr., ibid., 2, 33 (1965).
246. F. К г a f t, Н. W i g 1 о w, Ber., 28, 2566 (1895).
247. E. W. Mad ge, Latex Foam Rubber, London, 1962.
248. W. Clayton, C. G. Summer, The Theory of Emulsions and their Technical Treatment, London, 1954.
249. W. Mui steph, W. Poge, Anwendungstechnik der Plast- und Elastdis-persionen, Leipzig, 1964.
250. R. B. Grieves, D. Bhattacharyya, С. I. Crandall, J. Appl.
Chem., 17, 163 (1967).
251. A. J. R u b i n, J. D. J о h n s о n, J. C. L a m b, Ind. Eng. Chem., Proc. Desn.
Develop., 5, 368 (1966).
252. R. B. Grieves, T. E. Wilson, Nature (London), 205, 1066 (1965).
253. A. M. G a u d i n, Flotation, New York, 1957.
254. H. Schubert, J. Schmidt, Bergakademie Freiberg, 15, 850 (1963).
255. S. Ross, J. N. Butler, J. Phys. Chem., 60, 1255 (1956).
256. Patentrundschau, Tenside, 4, 104 (1967).
257. M. Goldberg, E. Rubin, Ind. Eng. Chem., Proc. Desn. Develop., 6, 195 (1967).
258. H. J. Neumann, Erdol u. Kohle, 18, 865 (1965).
259. E. С. H о u s a n, R. D. W i 1 s d о n, ibid., 19, 401 (1966).
260. H. Sonntag, H. Klare, jr., Kolloid. Z. u. Z. Polymere, 195, 35 (1964).
261. G. Bohme, W. Kling, H. Krupp, H. Lange, G. Sandstede, Z. angew. Phys., 16, 486 (1964).
262. G. В oh me u. a., Z. angew. Phys., 19f 265 (1965).
263. H. Uhlich, К. Schumann, W. Nowak, Tenside, 4, 133 (1967).
264. Сб. «Физико-химическая механика дисперсных структур», под ред. П. А. Ребиндера, Изд. «Наука», 1963.
265. П. А. Р е б и н д е р, Коллоидн. ж., 20, 527 (1958).
266. Р. A. R е b i n d е г, J. Pure. a. Applied Chem., 10, 337 (1965).
267. А. Ф. Полак, Коллоидн. ж., 24, 206 (1962).
268. Е. Д. Щукин, Тезисы 5 Всесоюзной конференции по коллоидной химии, Одесса, 1962.
269. Е. Е. С е г а л о в а, П. А. Ре бин дер, Строит, материалы, 1, 20 (1960).
270. Н. Rumpf, Chem.-Ing.-Tech., 30, 144 (1958).
271. W. Pietsch, Staub-Reinhaltung der Luft, 27, 20 (1967).
272. B. Schmauss, Ann. Phys., 18, 628 (1905).
273. К. E d e 1 m a n n, Lerbuch der Kolloidchemie, Bd. 1, Berlin, 1968.
274. К- H. Fr a n ge n, Farbe u. Lack, 70, 271 (1964).
275. F. В eck, ibid., 72, 218 (1966).
276. F. Beck, H. Pohlemann, H. Spoor, ibid., 73, 298 (1967).
277. E. M a t i j v i c, L. J. Stryker, J. Colloid a. Interface Sci., 22, 68 (1966).
278. H. Hahn, W. Stumm, Proceedings ot the 154th Conference of American Chemical Society, Chicago, 1967.
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
279. С. С. В о ю ц к и й, Курс коллоидной химии, Изд. «Химия», 1964, стр. 290.
280. Е. Mane go Id, Grundriss der Kolloidkunde, Dresden — Leipzig, 1959, S. 75.
281. A. Kuhn, Kolloidchemisches Taschenbuch, Leipzig, 1960, S. 113.
282. V. K. La Mer, J. Colloid Sci., 19, 291 (1964).
283. Краткая химическая энциклопедия, т. 2, Изд. «Советская энциклопедия», 1963, стр. 607 и 610.
284. И. Ф. Ефремов, Периодические коллоидные структуры, Изд. «Химия», 1971, стр. 5.
285. В. М. Барбой, Ю. М. Г л а з м а н, Г. И. Фукс, Коллоидн. ж., 32, 312 (1970).
286. В. М. Барбой и др., Там же, 32, 480 (1970).
287. Г. А. Ма р т ы н о в, сб. «Исследования в области поверхностных сил», Изд. «Наука», 1964, стр. 79.
288. В. Г. Л е в и ч, В. А. Кирьянов, ДАН СССР, 131, 1134 (1960).
289. Г. А. Мартынов, сб. «Исследования в области поверхностных сил», Изд. «Наука», 1964, стр. 102.
290. Г. А. М а р т ы н о в, Там же, стр. 90.
291. С. С. Духин, Н. М. Семенихин, Л. П. Ш а пин с кая, ДАН СССР, 193, 395, 1970.
292. Б. В. Дерягин, Труды третьей Всесоюзной конференции по коллоидной химии, Изд. АН СССР, 1956, стр. 225.
293. В. М. Муллер, сб. «Исследования в области поверхностных сил», Изд. «Наука», 1967, стр. 270.
294. Б. В. Дерягин, Л. Д. Ландау, ЖЭТФ, 11, 802 (1941); 15, 662 (1945).
295. Б. В. Дерягин, Коллоидн. ж., 17, 207 (1955).
296. Ф. М. К у н и, А. И. Р у с а н о в, Там же, 32, 232 (1970).
297. А. И. Русанов, Ф. М. Куни, Там же, 33, 121 (1971).
298. Е. Н. Трофимова, Ф. М. Куни, А. И. Русанов, Там же, 31, 578 (1969):
299. Ф. М. Куни, А. И. Русанов, Е. Н. Бродская, Там же, 31, 860 (1969).
300. С. В. Н е р п и н, Автореф. докт. дисс., ЛИИВТ, 1956.
301. С. В. Нерп ин, А. Ф. Ч уд нов с кий, Физика почвы, Изд. «Наука», 1967, стр. 94.
3Q2. Б- ₽. Д е р я г и н, ЖФХ, 6, 1306 (1935).
303. В. V. Derjaguin, Trans. Faraday Soc., 36, 203, 1940.
304. В. M. Барбой, Ю. M. Г л а з м а н, Коллоидн. ж., 25, 282 (1963).
305. В. М. Барбой, Там же, 25, 385 (1963).
306. В. И. Барбой, Там же, 26, 409 (1964).
307. Н. В. Михайлов, П. А. Р е б и и д е р, Там же, 17, 107 (1955).
308. И. Ф. Ефремов, Там же, 18, 276 (1956).
309. И. Ф. Ефремов, Труды Ленинградск. ин-та советской торговли, Гос-торгиздат, 1950, стр. 36.
310. И. Ф. Ефремов, Автореф. докт. дисс., ЛТИ им. Ленсовета, 1967.
311. И. Ф. Ефремов, С. В. Нерпин, Коллоидн. ж., 19, 757 (1957).
312. И. Ф. Ефремов, С. В. Нерпин, ДАН СССР, 113, 846 (1957).
313. Г. М. Лукашенко и др., Коллоидн. ж., 33, 106 (1971).
314. Г. М. Лукашенко, О. Г. У с ь я р о в, И. Ф. Ефремов, сб. «Поверхностные силы в тонких пленках и дисперсных сист.емах», Изд. «Наука», 1972, стр. 35.
315. В. М. Муллер, Автореф. канд. дисс., ин-т физической химии АН СССР, 1967, сто. 71.
316. Ю. М. Гл азман, М. Е. Краснокутская, Коллоидн. ж., 27, 815 (1865).
317. М. Е. Краснокутская, Ю. М. Г л а з м а н, сб. «Исследования в области поверхностных снл», Изд. «Наука», 1967, стр. 219.
318. М. Е. Краснокутская, Ю. М Глазман, Коллоидн. ж., 28, 847 (1966).
319. И. П. С а п о н, Ю. М. Глазман, Там же, 27, 601 (1965).
320. Ю. М. Глазман, И. П. С а п о н, сб. «Исследования в области поверхностных сил», Изд. «Наука», 1964, стр. 258.
321. И. П. Сапон, Ю. М. Глазман, Коллоидн. ж.,. 30, 274 (1968).
322. Ю. М. Глазман, Ж. Г. Блащук, Там же, 31, 809 (1969).
323. Ю. М. Глазман, Г. М. К а б ы ш, Там же, 31, 27 (1969).
324. Р. М. П а н и ч и др., Там же, 24, 733 (1962).
325. С. С. Воюцкий, Усп. хим., 30, 1237 (1961).
326. Б. В. Дерягин, А. Т. Титиевская, ДАН СССР, 89, 1041 (1953).
327. Б. В. Дерягин, Г. М. Страховский, Д. С. Малышева, ЖЭТФ, 16, 172 (1946).
328. Б. В. Дерягин, М. М. К У с ак о в, К. С. Крым, Там же, 16, 179 (1946).
329. М. М. К у с а к о в, Там же, 16, 451 (1946).
330. Б. В. Дерягин, Е. Ф. Пичугин, ДАН СССР, 63, 53 (1948).
331. М. М. Кусаков, К. С. Крым, ЖЭТФ, 16, 266 (1946).
332. Б. В. Дерягин, В. В. Ка,расев, Коллоидн. ж., 15, 365 (1953).
333. Б. В. Дерягин, Труды Всесоюзной конференции по коллоидной химии Изд.. АН УССР, Киев, 1952, стр. 26.
334. Б. В. Дерягин, И. И. Абрикосова, ДАН СССР, 90, 1055 (1953).
335. Б. В. Дерягин, И. И. Абрикосова, Там же, 108, 214 (1956).
336. И. И. Абрикосова, Б. В. Дерягин, ЖФХ, 32, 442 (1958).
337. Б. В. Дерягин, И. И. Абрикосова, Е. М. Лифшиц, Усп. физ. наук, 64, 493 (1958).
338. Т. Н. Воропаева, Б. В. Дерягин, Б. Н. Кабанов, сб. «Исследования в области поверхностных сил», Изд. АН СССР, 1961, стр. 143.
339. Т. Н. Воропаева, Б. В. Дерягин, Б. Н. Кабанов, ДАН СССР, 128, 981 (1959).
340. А. Д. Малкина, Б. В. Дерягин, Коллоидн. ж., 12, 431 (1950).
341. Б. В. Дерягин, Н. И. Моск в и тин, М. Ф. Фут ран, Труды Третьей Всесоюзной конференции по коллоидной химии, Изд. АН СССР, 1956, стр. 285.
342. Г. И. Ф у кс, Там же, стр. 301.
343. А. Д. Зим он, Адгезия пыли и порошков, Изд. «Химия», 1967, стр. 108.
344. А. Н. Фрумкин, А. В. Городецкая, ЖФХ, 12, 512 (1938).
345. М. М. Кусаков, Л. И. Мекеницкая, Доклад на IV Международном нефтяном конгрессе в Риме, Изд. АН СССР, 1955.
346. М. М. Кусаков, Л. И. Мекеницкая, сб. «Исследования в области поверхностных сил», Изд. АН СССР, 1961, стр. 17,
347. В. Д. Соболев, 3. М. Зорин, сб. «Исследования в области поверхностных сил», Изд. «Наука», 1967, стр. 36.
348. С. В. Нерпин, Б.. В. Дерягин, ДАН СССР, 100, 17 (1955).
349. Б. В. Дерягин, Н. В. Ч у р а е в, И. Г. Ершова, Там же, 182, 368 (1968).
350. В. Я. Волкова, С. В. Нерпин, О. Г. У с ь я р о в, сб. «Вопросы энерго- и массообмена в системе почва — растение — атмосфера», Гидрометео-издат, 1971, стр. 184.
351. Б. В. Дерягин, С. В. Нерпин, ДАН СССР, 99, 1029 (1954).
352. А. Шелудко, Д. Ексерова, Д. Платиканов, Коллоидн. ж., 25, 606 (1963).
353. Б. В. Дерягин, Ю. В. Г утоп, Там же, 24, 431 (1962).
354. Б. В. Дерягин, Ю. В. Г утоп, Там же, 30, 19 (1968).
355. G. A. Marrucci, Chem. Eng. Sci., 24, 975 (1969).
356. W. R. McEntee, K. J. M у s e 1 s, J. Phys. Chem., 73, 3018 (1969).
357. S. F r a n k e 1, K. J. M у s e 1 s, ibid., 73, 3028 (1969).
358. А. И. Русанов, Фазовые равновесия и поверхностные явления, Изд. «Химия», 1967, стр. 259.
359. К. Norrish, Discuss. Faraday Soc., 18, 120 (1954).
360. А. Шелудко, Д. Ексерова, ДАН СССР, 127, 149 (1959).
361. М. N. J о п е s, Kolloid. Z. u. Z. Polymere, 230, 236 (1969).
362. G. Ibbotson, М. N. Jones, Trans. Faraday Soc., 65, 1146 (1969).
363. А. А. Трапезников, В. А. Огарев, Коллоидн. ж., 24, 97 (1962).
364. Л. А. Ш и ц, А. Трапезников, Там же, 25, 613 (1963).
365. А. А. Трапезников, Е. С. Докукина, ЖФХ, 45, 717 (1971).
366. А. А. Трапезников, Е. С. Докукина, Коллоидн. ж., 32, 272 (1970).
367. А. А. Трапезников, К. В. Зотова, Н. В. Шамрова, Там же, 32, 437 (1970).
368. К. В. Зотова, Н. В. Шамрова, А. А. Трапезников, Там же, 32, 369 (1970).
369. А. А. Трапезников, Л. С. Чуракова, Е. С. Докукина, Там же, 33, 603 (1971).
370. Е. С. Докукина, А. А. Трапезников, Там же, 33, 674 (1971).
371. Н. В. Шамрова, К. В. Зотова, А. А. Трапезников, Там же, 33, 618 (1971).
372. Г. М. П а н ч е н к о в, Л. К. Ц а б е к, Поведение эмульсий во внешнем электрическом поле, Изд. «Химия», 1969.
373. J. S t a u f f, Kolloid. Z„ 143, 162 (1955).
374. Б; В. Дерягин, С. С. Д у х и н, Изв. АН СССР. ОТН, Металлургия и топливо, № 1, 82 (1959).
375. Б. В. Дерягин, С. С. Ду хин, В. А. Л и си чей ко, ЖФХ, 33, 2280 (1959).
376. Б. В. Дерягин, С. С. Духин, Коллоидн. ж., 21, 37 (1959).
377. С. С. Духин, Б. В. Дерягин, В. А. Лисиченко, ЖФХ, 34, 524 (I960).
378. С. С. Духин, Б. В. Д е р я г и и, Коллоидн. ж., 22, 587 (1960).
379. С. С. Духин, ЖФХ, 34, 1053 (1960).
380. С. С. Духин, Б. В. Дерягин, Там же, 35, 1453 (1961).
381. П. А'. Ребиндер, И. Солдатов, А. Чертавских, В. Шнеер-сон, М. Римская, Н. Лубман, М. Липец, Исследование в области поверхностных явлений, ОНТИ, 1936.
382. В. И. Классен, В. А. Мокроусова, Введение в теорию флотации, Госметаллургиздат, 1953.
383. П. А. Р е б и н д е р, Л. А. Ш р е й н е р, К. Ф. Ж и г а ч, Понизители твердости в бурении, Изд. АН СССР, 1944.
384. П. А. Р е б и н д е р, В. И. Л и х т м а н, Г. В. Карпенко, Влияние поверхностно-активной среды на процессы деформации металлов. Изд. АН СССР, 1954.
385. П. А. Ребиндер, Физико-химическая механика г—новая область науки, Изд. «Знание», 1958,
386. В. И. Л и х t М а н, В. Д. Щукин, П. А. Ре б ин дер, Физико-хйМИче-ская механика металлов. Изд. АН СССР, 1962.
387. Л. А. Абдурагимова, П. А. Ребиндер, Н. Н. Серб-Сербина, Коллоидн. ж., 17, 184 (1955).
388. Л. В. Иванова-Чумакова, П. А. Ребиндер, Коллоидн. ж., 18, 429 (1956).
389. Л. В. Иванова-Чумакова, П. А. Ребиндер, Там же, 18, 540 (1956).
390. П. А. Ре б ин дер, Г. И. Крус, Там же, 18, 682 (1956).
394. В. А. Федотова, П. И. Па раска, П. А. Ре бин дер, Там же, 32,
794 (1970).
392. Э. А. Александрова н др., Там же, 33, 6 (1971).
393. Э. А. Александрова и др., Там же, 33, 306 (1971).
394. О. И. Лукьянова н др., Там же, 33, 415 (1971).
395. В. А. Федотова, П. И. П а р а с к а, П. А. Р е б н н д е р, Там же, 33,
277 (1971).
396. Е. Д. Щукин, П. А. Ребиндер, Там же, 33, 450 (1971).
397. А. Ф. П о л а к, Там же, 25, 359 (1963).
398. В. М. Быков, Н. В. Михайлов, сб. «Физико-химическая механика дисперсных структур», Изд. «Наука», 1966, стр. 323.
399. Н. Koelmans, Philips Res. Rep., 10, 161 (1955).
400. О. Г. У с ь я р о в, И. С. Лавров, И. Ф. Ефремов, Коллоидн. ж., 28, 596 (1966).
401. Г. А. Мартынов, В. М. Муллер, сб. «Поверхностные силы в тонких пленках и дисперсных системах», Изд. «Наука», 1972, стр. 7.
402. Б. В. Дерягин, Н. М. Кудрявцева, сб. «Исследования в области поверхностных сил», Изд. АН СССР, 1961, стр. 183.
403. Г. А. Мартынов, Б. В. Дерягин, Коллоидн. ж., 24, 480 (1962).
404. A. N. Frumkin, Acta physicochim. URSS, 9, 313, 1938; Б. В. Дерягин, Коллоидн. ж., 14, 137 (1940).
405. 3. М. Зорин, сб. «Исследования в области поверхностных сил», ЙЗд, «Наука», 1964, стр. 146.
предметный указатель
Адсорбция
анионов 20
катионов 21, 63
специфическая 56, 63, 89, 135 макромолекул 56, 87, 91
поверхностно-активных веществ
36, 60, 91
Адсорбционные слои 56, 86, 116
влияние
иа молекулярное взаимодействие частиц 36, 60
иоино-электростатнческие силы 62
вязкость 112
ориентация молекул 36, 45, 63
площадь, приходящаяся на одну
молекулу ПАВ 73
разрушение 121
адсорбционным замещением 120
при изменении температуры 121
химическое 121
сдвиговая прочность 58, 117
стабилизирующее действие 87
толщина 56, 58, 117
эластичность 112, 114, 117
Аэрозоли 54
Ван-дер-Ваальса — Г амакера посто-
янная (см. Постоянная молекулярного взаимодействия)
Взаимодействие коллоидных частиц И, 13
барьер отталкивания 47 сл., 59,
влияние концентрации и валентности ионов 48
вторичный минимум 47, 51
ионно-электростатическое 22
молекулярное 30
первичный минимум 47
разнородных 87
нонио-электростатическое 87 молекулярное 87
суммарные силы 44
Взаимодействие ориентированных диполей 45
Водоподготовка 135
Волны поверхностные 79
затухание 114
поперечные 113
продольные 113
Вязкость жидкости
в пленке 73, 76
вблизи границы раздела фаз 76,
112
Гамакера функции 36
Гетерокоагуляция 87
теоретическое рассмотрение 87
экспериментальное исследование
89
при флотации 125
при электрофорезе 134
Гетерофлокуляция 90, 106, 125
Давление
гидростатическое 65, 68
капиллярное 65, 68
Двойной слой
вблизи сферических частиц 19
влияние температуры 56
дискретность зарядов 18, 19
диффузная часть 13, 21
диэлектрическая постоянная 16,
29
емкость 56
Двойной слой
плоский 14
поляризация 56, 135
распределение потенциала 13, 15,
19, 46
свободная энергия 25
слой Гельмгольца 20
Штерна 20, 29
толщина 15
Деэмульгирование 121, 130 адсорбционным замещением 121 в электрическом поле 122 нефтепродуктов 130 подкислением 121 при действии электролитов 121 при механических нагрузках 124
Димпл 74
Диполь флуктуирующий 31
Диссоциация поверхностная 30, 63
Диффузия частиц 54
Диэлектрическая проницаемость 13, 41 адсорбционных слоев 106 комплексная 41 среды в пленке 106 статическая 42
Заряд ионов
влияние на взаимодействие коллоидных частиц 26
коагуляцию 54, 77
равновесную толщину пленок 90, 108
переход от первичных черных пленок ко вторичным 109
объемная плотность в двойном слое 15, 17
поверхностный плоской границы раздела фаз
15
дискретность 21, 29
измерение 13, 15, 20, 24, 56 сферических частиц 20, 93
Защитное действие 57
золотое число 57
рубиновое число 57
Защитные слои 56
альгинатов 56
белковых веществ 56
в неполярных средах 86 карбоксиметилцеллюлозы 56 прочность 58
толщина 59, 87
Ионы
концентрация в двойном слое 14, 18
многовалентные, гидролизованные 135
поляризация 17, 29
потенциалопределяющие 21, 62
размер 17, 19, 29
Коагуляция 11, 13, 47, 50 быстрая 50 в неполярных средах 82 зависимость от размера частиц
55
концентрационная 51
Коагуляция
медленная 54
нейтрализационная 51
определение 11
скорость 52
Коалесценция 10, 50, 89, 91, 124
вероятность 122
влияние валентности ионов 78
при разрушении пен 129
при стирке 132
при удалении воды из нефти 130
при электрофорезе 135
экспериментальное изучение 77 Коллектор 125 Коллоиды
дисперсионные 10
мицеллярные 10
Концентрация частиц нечетная 52
Критическая концентрация 48, 52, 77 влияние ф^-потенциала 50, 77 в присутствии ПАВ 56 зависимость от радиуса частиц 53
Коэффициент преломления адсорбционных слоев 67, 106 жидкости в пленке 67, 106 Краевой угол между черной пленкой и объемной фазой 110
Критерий коагуляции 48
Модельные системы 63
Пенообразующая способность ПАВ 114
Пены 63, 114, 129
разрушение 129
Пептизация 51
при стирке 132
Пленки 63
анилина 85, 96, 97
критическая толщина 98
воды 97, 98
влияние органической фазы эмульсий 98
интенсивность отраженного света 65
интерференция света 63
ионностабилизированные 91 критерий устойчивости 92 механические свойства 112 пенные
вертикальные 64
горизонтальные 63
равновесное состояние 63, 67, 91 стабилизированные мономолеку-лярными адсорбционными слоями 99
теория прорыва 92
толщина 68, 72, 73, 77, 84
влияние фй-потенциала 71 измерение 63, 64, 85
Пленки
толщина
критическая 49, 94, 96
углеводородные 84 бензола 84
измерение толщины 85
ксилола 85
октана 85
хлорбензола 84, 96, 97
устойчивость 91, 95, 96
флуктуации толщины 92
форма 73, 74
черные (см. Черные пленки) Поверхностно-активные вещества 36, 56
диссоциация молекул в растворе 71
' мицеллообразование 71 поверхностная активность 103 связь строения и эмульгирующей способности 103
стабилизирующее действие' 101 Поверхностное натяжение 68, 11-0 Полиэлектролиты (см. Защитные коллоиды)
Поляризуемость атомов и молекул 30 Постоянная молекулярного взаимодействия 34
кварца 82.
определение 52
сложная 35
слюды 82
сравнение найденных различными
методами 82
стекла 82
Потенциал поверхности 13, 26, 84
полости 18
поляризационный 89
слоя Штерна 28
экспериментальное определение 52
электрокинетический 55, 56, 86 Правило Шульце — Гарди 50
Расклинивающее давление 63, 77
ионно-электростатическая составляющая 27
молекулярная составляющая 39, 68
связь с поверхностным натяжением пленки 110
экспериментальное определение 73, 77, 81, 84, 89
Редиспергирование 84
Рефракция молекулярная 32
Свободная энергия дисперсных систем 10
Сенсибилизация 57
Силы взаимодействия
дисперсионные атомов и молекул
изображения 18
индукционные 31
ионно-электростатические 22
влияние концентрации и валентности ионов 30
в неполярных средах 82
плоскопараллельных частиц 23,
27
разнородных частиц 87
сферических частиц 28
учет слоя Штерна 29
экспериментальное исследование 81
капиллярные 92
молекулярные, влияние 33
адсорбционных слоев 36
дисперсионной среды 34
постоянной Ван-дер-Ваальса —
Гамакера 48
эффекта электромагнитного запаздывания 39
молекулярные
макроскопическая теория 41
микроскопическая теория 33, 42
плоскопараллельные частицы 34
разнородные частицы 87
сферические частицы 33
измерение 73, 79
Скорость сближения частиц 52, 63
седиментации 84
Структурирование в суспензиях 133
при электрофорезе 135
Структурно-механический барьер 115
Структуры
коагуляционные 51, 91, 133
деформация 134
конденсационные 51, 91, 133
кристаллизационные 134
Суспензии 37
двуокиси титана 86, 117
‘ гидроокиси алюминия 86
каолина 57
карбоната кальция 86
кварца 136
окиси алюминия 86
полистирола 53, 56, 57, 58, 59, 85
сажи 86
сульфата бария 86
Тиксотропия 51, 134
Уравнение
Больцмана 14, 18
Пуассона 13, 16
Пуассона — Больцмана 14, 23, 29
для плоских частиц 14, 18
для сферических частиц 20
Уравнение
Пуассона — Больцмана коррекция 16, 29
Рейнольдса 73, 95
Устойчивость дисперсных систем 44, 50, 52
Флокуляция 10, 30, 84, 86, 92
влияние ПАВ 56
при удалении воды из нефти 130
при электрофорезе 135
Флотация 87, 125
агломерационная 129
ионов 128
окиси железа 128
Флуктуации 41, 91
вероятность образования 94
распространение 93, 95
Частота колебаний
атома в основном состоянии 31
характеристическая 31
Черные пленки 91, 99
влияние механических воздействий 124
электролитов 107, 109, 121
вторичные 107
Черные пленки
концентрация ПАВ, необходимая для их образования 101
краевой угол между пленкой и объемной фазой НО
первичные 107
переход первичных пленок во вто-
ричные 109
толщина 106
измерение 106
углеводородов 104
Электрокоагуляция 134
Электролакировка 134
Электрофорез суспензий 134
Эмульгирующая способность ПАВ 103
Эмульсии 37, 46, 92
вода/масло 84, 99
масло/вода 85, 99
Пикеринга 115
реологические свойства 85
Энергия ионизации атомов и молекул
Эффект полостной (cavity) 18, 29
электромагнитного запаздывания 33, 39
ОГЛАВЛЕНИЕ
Предисловие редактора перевода .................................. 5
Предисловие авторов...............................................7
Основные обозначения, принятые в тексте...................8
Введение ............................ '.......................Ю
Глава 1. Взаимодействие дисперсных частиц. Процесс коагуляции 13
Электрический двойной слой . . .............................13
Электростатические силы отталкивания дисперсных частиц .... 22
Молекулярное взаимодействие дисперсных частиц...............30
Суммарные силы взаимодействия частиц. Устойчивое состояние дисперсий ................................................... 44
Экспериментальное подтверждение теории ДФО. Определение постоянной Гамакера и величины фа-потенциала из результатов изучения модельных систем.....................................52
Коагуляция в неполярных средах .............................82
Взаимодействие разнородных частиц. Гетерокоагуляция ..... 87
Глава 2. Коалесценция.......................................... 91
Ионостабилизированные равновесные жидкие пленки...........91
Устойчивость пленок, стабилизированных мономолекулярными адсорбционными слоями ПА.В. Черные пленки....................99
Гелеобразные защитные слои. Структурно-механический барьер . .115
Разрушение устойчивых жидких пленок......................118
Глава 3. Флокуляция и коалесценция в некоторых технологических процессах ... ................ 125
Флотация ................................................. 125
Разрушение пен.............................................129
Деэмульгирование на примере удаления воды из нефти.........130
Процесс стирки.............................................131
Структурообразование в концентрированных дисперсиях твердых веществ .................................................. 133
Электрокоагуляция. Электролакировка........................134
Подготовка сточных вод.....................................135
Литература .................................................... 137
Предметный указатель .... ...............................147
Ганс Зонтаг
Клаус Штренге
КОАГУЛЯЦИЯ И УСТОЙЧИВОСТЬ
ДИСПЕРСНЫХ СИСТЕМ
Редактор Л. Ф. Травина
Технический редактор Ф. Т. Черкасская
Обложка художника Л. А. Я Ц е и к о
Корректор Г. П. Батракова
Сдано в набор 16/III 1972 г. Подписано к печати 11/XII 1972 г.
формат бумаги 60 X 90l/ie. Бумага тнпогр. № 2. Усл. печ. л. 9,5. Уч.-нзд. л.
10,05. Тираж 5000 экз. Заказ № 291, Цена 1 руб.
Ленинград, 191186. Издательство „Химия",
Ленинградское отделение, Д-186, Невский пр., 28
Ордена Трудового Красного Знамени
Ленинградская типография № 2 имени Евгении Соколовой <кСоюзполиграфпрома> прн Государственном комитете Совета Министров СССР по делам издательств, полиграфии и книжной торговли.
Измайловский проспект, 29