/





















































































































Автор: Ластовский Р.П.
Теги: органическая химия химия химическая промышленность химические реакции
Год: 1969




Текст
ВСЕСОЮЗНЫЙ ОРДЕНА ТРУДОВОГО КРАСНОГО ЗНАМЕНИ
> НАУЧНО-ИССЛЕДОВАТЕЛЬСКИЙ ИНСТИТУТ
ХИМИЧЕСКИХ РЕАКТИВОВ И ОСОБО ЧИСТЫХ
ХИМИЧЕСКИХ ВЕЩЕСТВ
МЕТОДЫ ПОЛУЧЕНИЯ
ХИМИЧЕСКИХ РЕАКТИВОВ
И ПРЕПАРАТОВ
ОБЗОРНАЯ ИНФОРМАЦИЯ
Выпуск 20
НИИТЭХИМ
МОСКВА-1969
Редакционная коллегия
Р. II. Л эстонский (гл. редактор), Е. А. Божевольнов,
А. В. Бромберг, В. Г. Брудзь, В. М. Дзиомко,
И. А. Красавин, Г. И. Михайлов, | Г. А. Певцов |
Редактор Б. Г. Козлов
Техн, редактор А. И. Пирожкова
Корректор Н. Р. Казарина
(дано в производство 20.6.69 г.
фрмат бумаги 60 X 90’/ie
Л-52-239 Заказ 1034
Подписано к печати 27.2.1970 г.
Печ. л. 15 Уч.-изд. л. 10,5
Тираж 1000 экз. Цена 84 коя.
Типографии ВАХЗ
СОДЕРЖАНИЕ
Алкилированные ксиленолы. Л. В. Глушкова, В. И. Деревяшаиц,
С. П. Старкова, М. Н. Волкотруб . ..... 7
N- (Р-Алкилмеркаптоэтил)этиленимипы. С. 3. Ивин, В. К- Прожну,
ков, Г. В. Конопатова....................................... 12
Алкилполифосфониты. В. Г. Груздев, К. В. Караванов, С. 3. Ивц 14
2-Алкил-1,3-циклопентандионы. О. В. Иванов, Г. И. Тихона^
В. М. Дзиомко............................................... 16
Бензиловый эфир 4-метилумб§длиферона. Д. А. Дранки-.
В. Г. Брудзь, И. И. Дорошина................................ 19
1-Бензил-3,4,5-триалкилпиразолы. В. М. Дзиомко, О. В. Иванов 21
4,4'-Бис-[2,4-бис-(карбоксиметиламиио)-1,3,5-триазииил-6-амино] -2,!.
толаидисульфокислота. В. Я .Темкина, Г. Ф. Ярошенко, И. Е. Хц.
ченко, Р. П. Ластовский..................................... 24
1,4-Бис-(диэтоксифосфоио)бутен-2.. С. 3. Ивин, В. К. Промоненк^
С. К. Смирнов.............................................. 28
2,2-Бис-(4'-окси-3'-хлор-5'-метилфенил)пропан. А. М. Серебрянц
И. М. Билик, И. М. Миронова................................ 30
2,2-Бис- (4'-окси-3'-хлорфеиил (пропан. А. М. Серебряный, И. М. Б,
лик, И. М. Миронова......................................... 32
1,1-Бис-(3'-хлор-4'-оксифеиил)циклогексап. А. М. Серебрянь.,
И. М. Билик, Н. М. Миронова................................. 35
Бис- (циклогексанон(оксалилдигидразон и бис- (ацетальдегид (оке
лилдигидразон. Г. С. Петрова, А. М. Лукин, И. А. Немировт
Н. С. Фрумина, М. Д. Кофман................................. 38
5-Бром-2-бензоилфураи. 3. И. Назарова, Л. Д. Бабешкина 41
о-Бромфенилдихлорарсип. И. П. Шепилов, К. А. Дунаевой
И. А. Муратова, В. М. Дзиомко............................... 43
1-и-Бутил-3,4,5-триалкилпиразолы. В. М. Дзиомко, О. В. Иванц 45
4-втор-Бутилфенол. И. С. Маркович, Н. В. Круглова, В. М. Л~-
омко........................................................ 48
Гидрохиион-2,6-диметилениминодиуксусная кислота. В. Я- Темкин,
Н. В. Цирульникова, М. Н. Русина, Р. П. Ластовский . 51
Гуанилтиомочевииа. М. Ф. Кондрашова, Е. Я- Яровенко . 54
N-Гуаиилформамидинсульфиновая кислота. М. Ф. Кондрашм,
Е. Я- Яровенко.............................................. 56
N,N- Диалкилацетамиды. Н. А. Егорушкина, В. Я- Темкищ
Р. П. Ластовский............................................ 58
Диалкил(пергалоидизопропеиил)фосфаты. С. 3. Ивин, В. К. Пр
моненков, Е. А. Фокин....................................... 61
Диалкил(гексафторизопропил)фосфаты. С. 3. Ивин, В. К- Прея
ненков, Е. А. Фокин.........................................,64
2,5-Диацетилтиофен. С. В. Цукерман, В. М. Никитченко, Лам В»
Тхием, В. Ф. Лаврушин....................................... 67
3
Ди-[(Рдифенилфосфииил) этил]малоиовая кислота. Т. М. Балашова,
Ю. М. Поликарпов, Т. Я- Медведь..............................70
Диимид нафталин-1,4,5,8-тетракарбоновой кислоты. М. Т. Разумов-
ская, А. И. Белякова, Л. А. Егорова................................................73
2,5-Ди-(оксиметил) тетрагидрофуран. Н. Н. Шмагина, В. И. Федо-
това, А. И. Курашева...............................................................76
5-(Диметиламиио) фурфурол. 3. Н. Назарова, В. С. Пустоваров . 78
(5-ДиметиламиноэтиламиИ. Н. М. Морлян, Ж- Л. Багратуни . 81
2,4-Диметилтиофен. Н. М. Морлян, Ж. Л. Багратуни, Г. В. Бадасян 84
3,4-Диметилтиофен. Н. М. Морлян, Ж. Л. Багратуни, Г. В. Бадасян 87
2,5-Дн(оксиметил)фуран. Н. И. Шмагина, А. И. Курашева, Ф. И. Фе-
дотова ............................................................................90
1,5-Дифеиилкарбогидразид. Г. А. Креймер '..............................................92
Дифенилфосфинилуксусная кислота. Т. М. Балашова, 10. М. По-
ликарпов, Т. Я- Медведь............................................................95
(р-Дифенилфосфинилэтил) малоновая кислота. Л1. Т. Балашова,
Ю. М. Поликарпов, Т. Я. Медведь................................................. 98
Дихлор-сижж-триазиниламинофлуоресцеин I. Ю. Е. Скляр, А. Г. Буб-
нова, Г. И. Михайлов ................................... 101
Дихлораигидрид трихлорметилтиофосфоновой кислоты. Ф. И. Поно-
маренко, С. 3. Ивин, К- В. Караванов..............................................105
Дихлораигидрид трихлорметилфосфоновой кислоты. Ф. Н. Понома-
ренко, С. 3. Ивин, К. В. Караванов................................................107
5-(Диэтиламино) фурфурол. 3. Н. Назарова, В. С. Пустоваров . . 109
Диэтиловый эфир (8-окси-2-хииолил)малоновой кислоты. И. А. Кра-
савин, В. М. Дзиомко, Ю. П. Радин.................................................111
Додецилянтарный ангидрид. Н. М. Морлян, А. Г. Мурадян . . 114
О-Изопропилметилцианфосфонат. И. Д. Шелакова, С. 3, Ивин,
В. К. Промоненков.................................................................116
Калиевая соль n-стнролсульфокислоты. К. М. Ройзен, И. М. Билик 118
n-Карбоксигалланилид. И. М. Морлян, Л. О. Ростомян, Г. А. Егиа-
зарян.............................................................................121
Кислый О-этиловый эфир метилфосфоновой кислоты. И. Д. Шела-
кова, В. К- Промоненков, С. 3. Ивин...............................................124
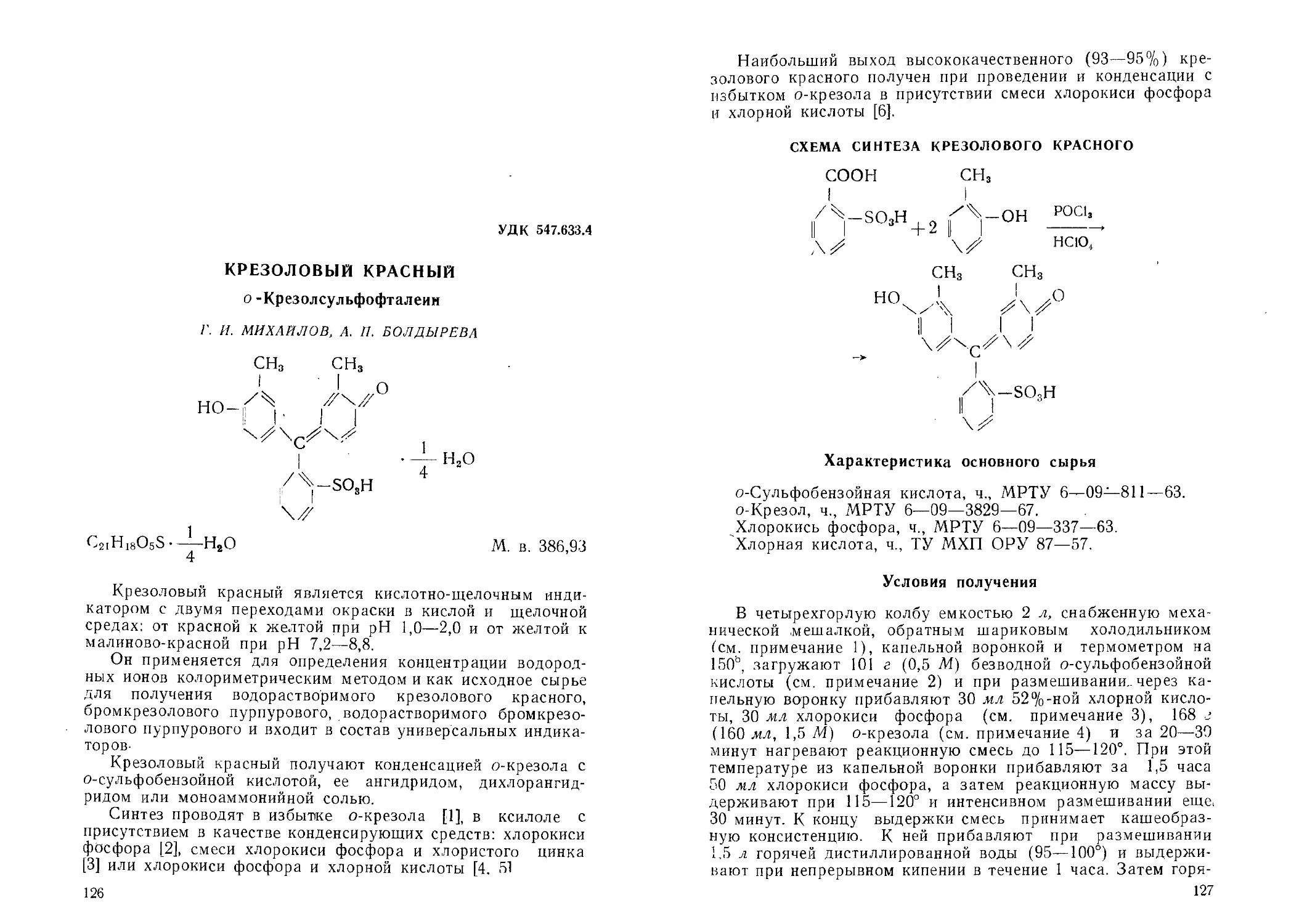
Крезоловый красный. Г. И. Михайлов, А. П. Болдырева 126
Лауриновый эфир метакриловой кислоты. Я. М. Морлян, Л- О. Ро-
стомян ...........................................................................129
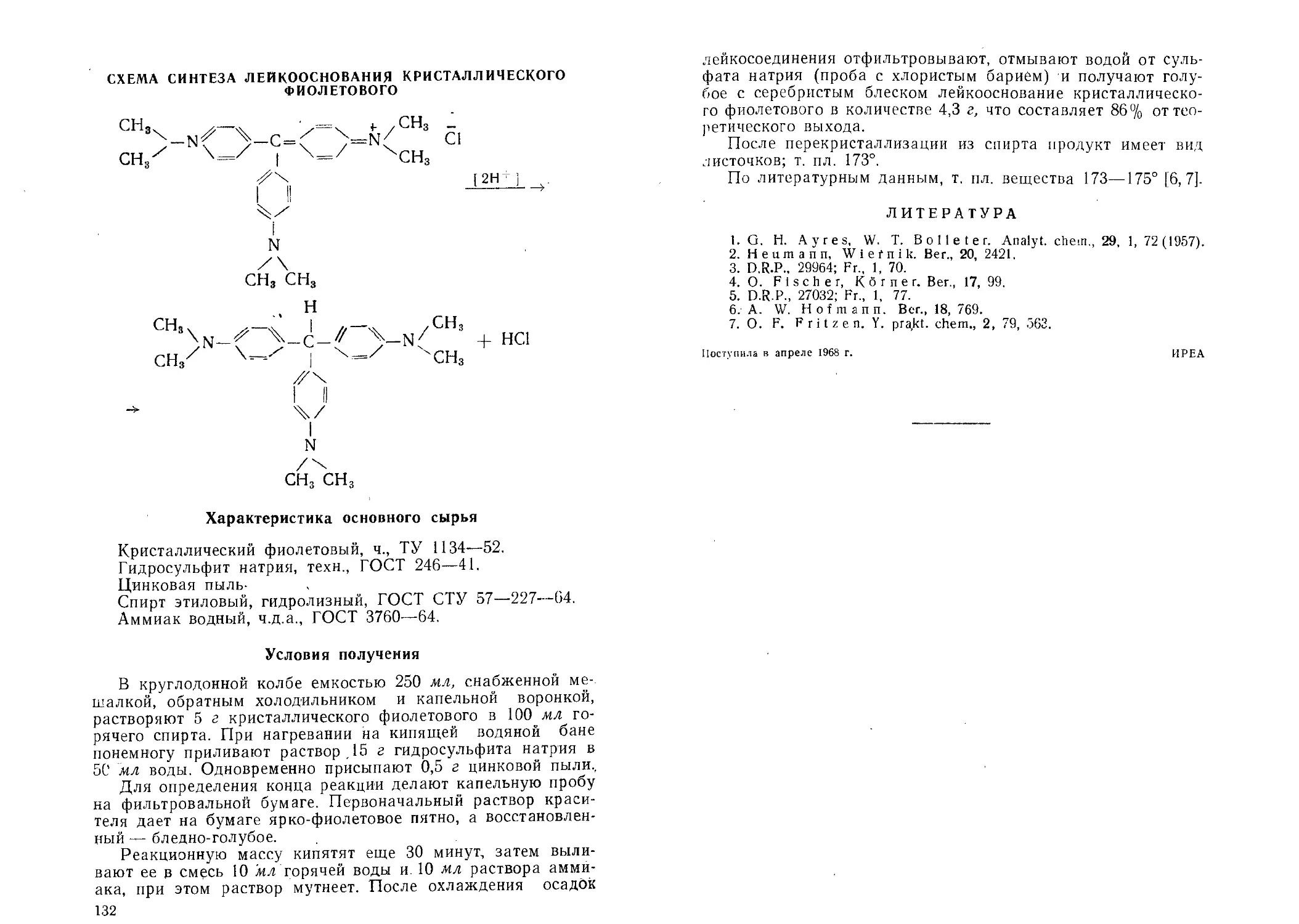
Лейкоосиование кристаллического фиолетового. Г. С. Петрова,
А. М. Лукин, Н. Б. Этинген.......................................................131
1-Метил-З-ацетоксииндол, И. В. Факеева, В. В. Жидкова . . . 134
2-а-Метилбензил-4-алкилфенолы. И. С. Маркович, Н. В. Круглова,
В. М. Дзиомко....................................................................136
1-Метил-3,4,5-триалкилпнразолы. О. В. Иванов, В. М, Дзиомко 139
N-Метилфенилглиции-о-карбоновая кислота. Н. В. Факеева,
В. В. Жидкова.....................................................................142
5-Метокси-З-нитросалициловый альдегид. В. Г. Брудзь, Д. А. Драп-
кина, В. А. Иншакова, И. Е. Бохлунова.............................................144
5-Метоксисалициловый альдегид. В. А. Иншакова, Д. А. Драпкина,
В. Г. Брудзь, Н. Е. Бохлунова.....................................................146
N-о-Метоксифеиилформамидинсульфиновая кислота. Е. Я- Яровен-
ко, Р. П. Ластовский............................................................ 148
2-(п-Метоксифенил)-4Н-3,1-бензоксазои-4. Б. М. Болотин, Д. А. Драп-
кина, В. Г. Брудзь, Р. У. Судиярова...............................................150
о-Нитробензальдегид. Г. А. Креймер....................................................152
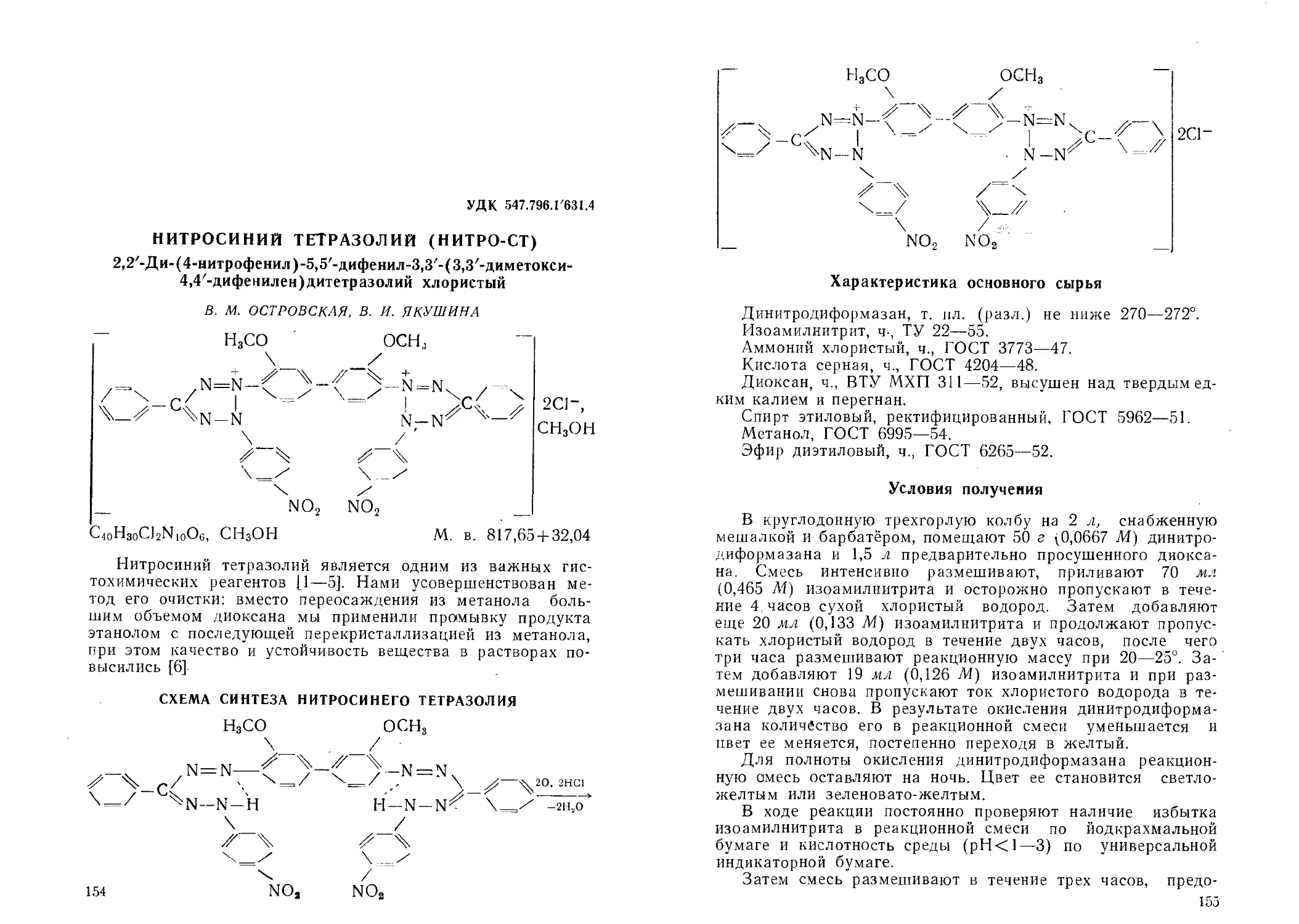
Нитросиний тетразолий (иитро-СТ). В. М. Островская, В. И. Яку-
шина .............................................................................154
5-Оксиметилфурфурол. Н. Н. Шмагина, И. П. Ермакова, В. И. Фе-
дотова, А. И. Курашева............................................................158
N-Оксисукцинимид. Г. Н. Кошелева, 4. К- Салгус.......................................160
4
N-о-Оксифепилформамидинсульфииовая кислота. Е. Я. Яровенко,
Р. П. Ласт.овский............................................. 162
1-(8-Окси-2-хинолил)-2-пропанон. В. М. Дзиомко, И. А. Красавин.
Ю. П. Радин....................................................164
8-Оксихнноли11-2-сульфокислота. И. А. Красавин, В. М. Дзиомко,
И. В. Рубцов................................................167
5-(М-Пиперидино)-2-бензоилфуран. 3. И. Назарова, Л. Д. Бабеш-
кина...........................................................172
5-(1\'-Пиперидино)фурфурол. 3. Н. Назарова, В. С. Пустоваров,
О. Б. Беличенко................................................174
Пропаргиловые эфиры акриловой и метакриловой кислот. Н. М. Мор-
лян, Л. О. Ростомян............................................176
Сополимеры стирола с я-дивинилбензолом. Т. А. Аптова, С. Б. Ма-
карова, Е. В. Егоров...........................................178
Тетраметилдифосфиндисульфид. С. 3. Ивин., В. К. Промоненков 181
Тетрамид иафталнн-1,4,5,8-тстракарбоновой кислоты. М. Т. Разумов-
ская, А. И. Белякова, Л. А. Егорова............................183
3,3', 4,4'-Тетрациандифенилоксид. М. Т. Разумовская, А. И. Беляко-
ва, Г. И. Карельская...........................................186
Тиопиколинанилид. Ю. М. Ютилов, И. А. Свертилова .... 189
3,4,5-Триалкилизоксазолы. О. В. Иванов, В. М. Дзиомко . . 191
3,4,5-Триалкилпиразолы. В. М. Дзиомко, О. В. Иванов, Л. В. Дар-
да ............................................................194
N-Триалкилсилил-, N-алкилмеркапто-, N-алкоксиметилэтиленими-
ны. С. 3. Ивин, В. К- Промоненков, Г. В. Конопатова . 197
2,2,4-Триметил-4- (я-оксифеиил) хромай. ,Г. Г. Кондратьева 199
Трихлорметилдихлорфосфонит. Ф. И. Пономаренко, С. 3. Ивин,
К. В. Караванов................................................202
5-(Трихлорметил) оксазолидон-2. Ф. И. Лукницкий, Б. А. Вовси 204
2,3,5-Триэтокситетратидро.фураи. Л. М. Болотина, Н. И. Куценко 206
о-Фенилбориая кислота. Н. М. Морлян, Л. О. Ростомян 209
Феноловый красный. Е. Я. Яровенко, М. Ф. Кондрашова 212
1-Хлор-4-(алкоксиметил) циклопептены-2. С. 3. Ивин, В. К. Промо-
ненков, Л. А. Зернакова, Ю. Е. Уткин...........................215
2-Хлор-5-аминопиридин. Ю. М. Ютилов, И. А. Свертилова , 218
Хлорангидрид трднс-трихлоркротоновой кислоты. Ф. И. Лукницкий,
Б. А. Вовси ...................................................220
2-Хлор-5-нитропиридин. Ю. М. Ютилов, И. А. Свертилова . 222
5-Хлорфурфурол. 3. И. Назарова, Л. Д. Бабешкина .... 225
Цирконилфосфат в виде сферических гранул. Г. И. Корельскач.
Е. В. Егоров, С. Б. Макарова...................................227
Этиловый эфир (5-аминопропионовой кислоты. С. И. Безуевскаи,
И. М. Билик................................................... 229
Этиловый эфир и>-аминоэпантовой кислоты. С. И. Безуевскач,
И. М. Билик................................................... 231
Этиловый эфир метилфосфонистой кислоты. В. Г. Груздев, К-В. Ка-
раванов, С. 3. Ивин............................................233
0-Этил-0-[р-(триэтилсилил)аллил]метилфосфонат. И. Д. Шелакова.
С. 3. Ивин, В. К- Промоненко...................................235
Алфавитный перечень................................................237
УДК 547.563
АЛКИЛИРОВАННЫЕ КСИЛЕНОЛЫ
Л. В. ГЛУШКОВА, В. И. ДЕРЕВЯНКИНА, С. П. СТАРКОВА,
М. И. ВОЛКОТРУБ
ОН СН8
1 I =
^\—СН—/
I II
Н8С^/ХСН3
3,5-Диметил-2-(а-метилбензил)фенол
С16Н18О М. в. 226,30
Сг4Н26О
СН3 °Нз СН3
3,.5-Диметил-2,6-ди-(а-метилбензил)фенол
М. в. 330,45
2,5-Диметил-4-(а-метил-
бензил)фенол
CieHisO М. в. 226,30
ОН
—СНо
| || '-,г*з
н3с/чу
сн
НгС^^СН,
I I
Н2С ZCH2
сн2
2,5-Диметил-4-циклогексилфенол
СиНгоО
М. в. 204,30
7
Алкилирование ксиленолов осуществляют с применением
в качестве катализаторов концентрированной серной [1] или
фосфорной кислот [2], трехфтористого бора [3], бензолсуль-
фокислоты [4]. При алкилировании 3,5-ксиленола алкилиру-
ющая группа вступает преимущественно во 2-е положение
[5]. При алкилировании изомерных ксиленолов циклогексано-
лом в присутствии фосфорной кислоты при 80° выделены хро-
матографией на окиси алюминия отдельные изомеры цикло-
тексилксиленолов, в том числе и 2,5-диметил-4-циклогексил-
фенол с т. пл. 84° (выходы продуктов не указаны).
Нами предложен способ моно- и диалкилирования 3,4-
ксиленола олефинами в присутствии катионита КУ-2 [6, 7],
что значительно упрощает выделение продуктов реакции.
Алкилированные ксиленолы применяются как антиокси-
данты для светлых резин [8] и бензина {9].
СХЕМА СИНТЕЗА СОЕДИНЕНИИ
ОН
I
н3с/Ч/хсн3
4- сн2=сн-
°н сна
нас/^/Хсна
+ 2СН2=СН ^^
СНа ОН СНз
h8c/Vx'4chs
+ сн2=сн-^
8
он
I сн-сн2
f \-СН3 + НС< )СН2 -*
\ch2-ch2z
/V
н3с
он
I
^\-сн3
I II
Hacz V
-> сн
н2с сн2
I I
Н2С сн2
сн2
Характеристика основного сырья
3,5-Ксиленол (3,5-диметилфенол), ч., т. пл. 62—63°.
2,5-Ксиленол(2,5-диметилфенол), ч., т. пл. 72,5—73,5°.
Стирол, ч. свежеперегнанный, т. кип. 46°/20 мм;
(Д20 — 0,908; п2° —1,5472.
Циклогексен, получен дегидратацией циклогексанола на
катионите КУ-2 по способу [10]; т. кип. 81—82°, п2£ —
1,4455.
КУ-2, обменная емкость 4,9 мг-экв/г, % ДВЕ 6,4.
КУ-1, обменная емкость 4,5 мг-экв!г.
Условия получения
Синтез 3,5-диметил-2-(а-метилбензил)фенола. К смеси
29,32 г (0,24 М) 3,5-ксиленола и 3 а КУ-1 при 90—110° за 30
9
минут приливают 20,83 г (0,2 Л!) стирола. После двухчасо-
вой выдержки при этой же температуре катализат деканти-
руют и перегоняют в вакууме.
Выход 3,5-диметил-2-(а-метилбензил) фенола в виде жел-
товатой маслянистой жидкости равен 25,3 г, что составляет
56% от теоретического; т. кип. 160—170°/3 млг; rf420—1,057;
п%—1,5830.
Найдено, %: С—84,67; Н—8,27; MRD —71,44.
СшНцО. Вычислено, %: С—84,90; Н—7,96; MRD —70,40.
При длительном стоянии продукт закристаллизовывается;
т. пл. 73—73,5° (из н-гексана).
В ИК-спектре имеются очень сильная полоса поглощения
в области 848 см~\ которая характерна для 1,2,3,5-замещен-
ного бензола, а также полосы поглощения в областях 710 и
750—775 см-1, характеризующие монозамещенное бензольное
кольцо [11]. I;
Получение 3,5-диметил-2,6-(а-метилбензил)фенола. Про-
дукт получают аналогично предыдущему, исходя из 24.43 г
(0,2 Л1) 3,5-ксилеиола, 52,07 г (0,5 М) стирола и 0,25 г кон-
центрированной серной кислоты. Реакционную массу нейтра-
лизуют 5%-ным раствором едкого натра, экстрагируют' бен-
золом и экстракт сушат сульфатом натрия. После отгонки
бензола остаток перегоняют в вакууме.
Выход 3,5-диметил-2,6-ди-(а-метилбёнзил) фенола в виде
очень вязкой желтой жидкости, равен 55,5 г, что составляет
84% от теоретического; т. кип. 200—21073 мм; rf420— 1,079;
«д —1,5970.-
Найдено, %: С—87,03; Н—7,99; MR D —104,51.
C24H2GO. Вычислено, %': С—87,10; Н—8,15; MR D —103,75.
Продукт не растворяется в растворе Кляйзена, что указы-
вает на 0,0-замещение [12].
В ИК-спектре имеется полоса поглощения средней интен-
сивности в области 855 см-1, характерная для' пентазамещен-
ного бензола [13] и в областях 705 и 740—770 см-1, харак-
терные для монозамещенных бензольных колец [11].
Получение 2,5-диметил-4-(а-мегилбензил)фенола. Продукт
получают аналогично предыдущему из 12,21 г 2,5-ксиленола,
6 г КУ-2 и 10,41 г стирола.
Выход 2,5-диметил-4-(а-метилбензил) фенола в виде бес-
цветной маслянистой жидкости равен 14,7 г, что составляет
65% от теоретического; т. кип. 170—174°/7 мм. При стоянии
продукт закристаллизовывается; т. пл- 90,5—91° (из петро-
лейного эфира).
Найдено, %: С—84,92; Н—8,07; ОН—7,52.
CjeHigO. Вычислено, %: С—84,90; Н—7,96; ОН—7,16.
В ИК-спектре обнаруживаются полосы поглощения сред-
ней интенсивности в областях 860—870 см~! и 900 см-1, ха-
10
рактерные для 1,2,4,5-замещенного бензола [14], а также в
областях 715 см~': и 780—760 см~\ характеризующие моно-
замещенное бензольное кольцо [И].
Получение 2,5-диметил-4-циклогексилфенола. К смеси
36,64 г (0,3 М) 2,5-ксиленола и 18 г КУ-2 при 125° за 90 ми-
нут прибавляют 16,42 г (0,2 М) циклогексена и выдержива-
ют 20 минут. Алкилат декантируют и перегоняют в вакууме.
Получают 29,4 г (72% от теоретического выхода) 2,5-ди-
метил-4-циклогексилфенола с т. кип. 165—172°/9 мм; т. пл.
73—74° (из петролейного эфира).
По литературным данным, т. пл. продукта, очищенного
хроматографией на окиси алюминия, 84° [2].'
Найдено, %: С—82,02; Н 10,14; ОН—6,58.
С14Н20О. Вычислено, %: С—82,30; Н—9,67; ОН—6,10.
Продукт полностью растворяется в 10%-ном водном рас-
творе едкого натра, что указывает на отсутствие орто-изоме-
ра [2].
ЛИТЕРАТУРА
1. Пат. США, 2900362; РЖхим, 1961, 14П380.
2. С. Parc. Rev. Inst, franc, petrole, 15, 4, 680 (1960).
3. Фр. пат., 1128968, (1957).
4. Пат. США, 2537636 (1946).
5. Ph. Bun-Hoi, М. Sy, М. Nt el. Compt. Rend., 254, 4476 (1962).
6. С. П. Старков, Л. В. Глушкова. Ж- прикл. химии, 40 1
(1967).
7. С. П. Старков, Л. В. Глушкова. Ж- прикл. химии 40, 7
(1967).
8. Англ. пат. 723838; РЖхим, 1965, 37559.
9. Пат. США, 2248827; Zb„ 1942, 2092.
10. И. И. Ш у й к и и, Н. А. Поз ди як, Г. П. Добрынина. Изв.
АН СССР, сер. хим., вып. 9, 1964, стр. 1705.
11. К- Накаииси. Инфракрасные спектры и строение органических
соединений, М., 1965, стр. 32.
12. D. Peppard. Inorg. u. Nucl. chem., 4, 334 (1957).
13. Л. Беллами. Инфракрасные спектоы сложных молекул, М.,
1963.
14. Т. А. Рудольфи. Ж. прикл. спектроскопии, 7, 336 (1965).
Поступила в январе 1968 г. Тамбовск. Гос. педагог, ин-т, НИИ химикатов
для полимерн. материалов
УДК 547.415.3
N-(£ АЛКИЛМЕРКАПТОЭТИЛ)ЭТИЛЕНИМИНЫ
С. 3. ИВИН, В. к. ПРОМОНЕНКОВ, Г. В. КОНОПАТОВА
N-(p-Алкилмеркаптоэтил) этиленимины до сих пор не бы-
ли известны, они могут быть использованы для синтетичес-
ких целей, а также для изучения физиологического действия.
М-(Р-ЭТИЛМЕРКАПТОЭТИЛ)ЭТИЛЕНИМИН
ГН2
C2H5-S-CH2CH,-N( |
ХСН2
C0H13NS М. в. 131,24
СХЕМА СИНТЕЗА М-(₽-ЭТИЛМЕРКАПТОЭТИЛ)ЭТИЛЕНИМИНА
,сн2 .сн,
C2H6-S—CH=CH2+H-N( I C2H6-S—CH2CH2-N^ I
XCH2 xCHa
Характеристика основного сырья
Этиленимин, получен дегидратацией этаноламина серной
кислотой [1]; т. кип. 55—56°; d^°—0,837; п'£ — 1,4136.
Алкилвинилсульфиды, получены дегидратацией щелочью'
соответствующих p-алкилмеркаптоэтанолов [2J.
Этилвинилсульфид, т. кип. 91—93°; Пд—1,4756.
Изобутилвинилсульфид, т. кип. 128—130°; п® —1,4670.
Условия получения
В четырехгорлый реактор с мешалкой, термометром, ка-
пельной воронкой и обратным холодильником помещают
12
10,6 г (0,25 М) этиленимина и 0,5 г металлического натрия.
При температуре кипения этиленимина из капельной ворон-
ки прикапывают 21,75 г (0,25 Л1) этилвинилсульфида, смесь
кипятят в течение 3 часов, после чего перегоняют в вакууме.
Выход N-(р-этилмеркаптоэтил)этиленимина равен 24,0 г.
что составляет 75% от теоретического; т. кип. 85—87° 125 мм;
n'i20 0,9473; /г20 —1,4820.
Найдено, %: N—10,73.
C5H13NS. Вычислено, %: N—10,68.
По описанной методике получают №-(р-изобутилмеркапто-
этил)этиленимин с выходом 60—65%; т. кип. 105—107°/25мм;
d^o—0,9567; п» —1,4779.
№(Р-алкилмеркаптоэтил)этиленимины представляют со-
бой бесцветные подвижные жидкости. При хранении ниже 0°
устойчивы.
ЛИТЕРАТУРА
1. Н. W е n к с г. J. Amer. Chem. Soc., 57, 2328 (1935).
2. J. Arens. Rec. trav. chim., 75 482 (1656).
Поступила в декабре 1967 г.
УДК 678.85
АЛкилПОЛИФОСФОНИТЫ
В. Г. ГРУЗДЕВ, к. В. КАРАВАНОВ, С. 3. ИВИН
До последнего времени способы получения алкилполи-
фосфонитов в литературе не были описаны. Нами разрабо-
тан способ получения этих соединений, основанный на взаи-
модействии алкилдихлорфосфинов с водой или органически-
ми кислотами без растворителя, при температуре—70°
с последующим медленным повышением ее до 100° или 130°.
и с вакуумированием продуктов реакции.
Получить алкилполифосфониты оказалось возможным
лишь в том случае, если все операции проводить в токе
инертного газа (азота или гелия), очищенного от кислорода.
Алкилполифосфониты очень реакционноспособные, белые
кристаллические вещества, энергично окисляющиеся и омы-
ляющиеся; устойчивы при нагревании до 135°. Они легко вза-
имодействуют с хлористой медью (образуя комплексные сое-
динения), со спиртами и многими другими реагентами.
МЕТИЛПОЛИФОСФОНИТ
(СН3РО)„
СХЕМА СИНТЕЗА МЕТИЛ ПОЛИФОСФОНИТА
СН3РС12 J- Н2О.--> (СН3РО)П 4- НС1
Характеристика основного сырья
Метилдихлорфосфин, перегнанный, получают по мето-
дике [1].
Вода дистиллированная, ГОСТ 6709—53.
Условия получения
В трехгорлую колбу с капельной воронкой, трубкой для
подвода инертного газа и трубкой, соединенной со склянкой
14
Тищенко с раствором щелочи, помещают 11,69 г (0,1 Л4) све-
жеперегнанного метилдихлорфосфина. Колбу охлаждают до
—70° (углекислота-ацетон) и приливают 1,8 г (0,1 Л1)
воды (см. примечание 1). Температуру реакционной массы в
течение 40 минут повышают до 10° и затем до 125—130°. (тем-
пература бани) в течение 60 минут так, чтобы выделяющийся
из реакционной смеси хлористый водород проходил через
склянку Тищенко со скоростью 1—2 пузырька в секунду (см.
примечание 2). Содержимое колбы при повышении темпера-
туры постоянно встряхивают. Образование кристаллов ме-
тилполнфосфонита происходит при температуре 125°, их вы-
держивают в вакууме 1—2 мм до постоянного веса при ком-
натной температуре.
Выход равен 6,18 г, что' составляет 99,7% от теоретичес-
кого (см. примечание 3).
Найдено, %: Р—50,0; 49,78; С—19,0; 19,21; Н 4,76, 4,80.
СН3РО. Вычислено, %: Р—49,88; С—19,33; Н—4,83.
По аналогичной методике могут быть получены и другие
алкилполифосфониты.
Примечания:
!. Вместо воды можно использовать безводную муравьиную кислоту.
2. При быстром повышении температуры возможны побочные реак
ции с образованием фосфинов.
3. Количественный анализ вещества проводился после получения
комплексного соединения с хлористой медью.
ЛИТЕРАТУРА
1. И. П. Комков, К. В. Караванов, В. Г. Груздев,
С. 3. Ивин. Сб. «Методы получения хим. реактивов и препаратов», вьш.
12, М„ ИРЕА, 1965, стр. 101.
Поступила в марте 1968 г.
УДК 547.442.3
2-АЛ КИЛ-1,3-ЦИКЛОП ЕНТАНДИОНЫ
О. В. ИВАНОВ, Г. И. ТИХОНОВА, в. м. дзиомко
I К = н-С5Ни
II R= н-С6Н,з
н ХИ
Н-/\/Н
Н_
н
о
III R=h-C7H!3
IV R = н-С8Н17
2-Алкил-1,3-циклопентандионы, применяемые в синтезе
стероидов [1—3], получают конденсацией метилалкилкетонов
с диэтилоксалатом в присутствии металлического натрия или
этилата натрия с последующим каталитическим гидрирова-
нием образующихся 3-алкил-1,2,4-циклопентантрионов [4—5]
или разложением их семикарбазидных производных в присут-
ствии алкоголята натрия [I, 3, '6—9].
Другой метод синтеза 2-алкил-1,3-циклопентандионов за-
ключается во внутримолекулярной конденсации эфиров
у-кетокислот в присутствии этилата натрия или третичного
бутилата калия [10—II].
В разработанный нами ранее метод синтеза 2-алкил-1,3-
циклопентандионов [13], состоящий в ацилировании карбоно-
вых кислот янтарным ангидридом в присутствии безводного
хлорида алюминия, внесены некоторые уточнения и получен
ряд новых 2-алкил-1,3-циклопентандионов.
СХЕМА СИНТЕЗА 2-АЛКИЛ-1.3-ЦИКЛОПЕНТАНДИОНОВ
О
,Н
Н3 0+ rch2coo.h
Н \ /
Н
о
АЮз
c6h6no2
н
н-х\/н
Н— \//XR
н ч
о
Характеристика основного сырья
Алюминий хлористый, безводный, ч., ВТУ МХП 3500—52.
Янтарный ангидрид, ч., ВТУ ГКХ 1439—60.
Нитробензол, ч., ГОСТ 5846—51.
Энантовая кислота, ч., ВТУ ГКХ 1535—61.
Каприловая кислота, ч., ВТУ ГКХ 1531—61.
Пеларгоновая кислота, ч., ВТУ ГКХ 1533—61.
Каприновая кислота, ч., ВТУ ГКХ 1532—61.
Серная кислота, ч., ГОСТ 4204—48.
Хлороформ, ч., ГОСТ 3160—51.
Натр едкий, ч., ГОСТ 4328—48.
Уголь активированный, технический, ГОСТ 6213—53.
Этилацетат, ч., ст. ГОХП 27—1839.
Условия получения
В трехгорлую колбу, снабженную мешалкой, обратным
холодильником с хлоркальциевой трубкой и капельной во-
ронкой помещают 40 г (0,3 М) безводного хлорида алюми-
ния, 10 г (0,1 М) янтарного ангидрида и 70 мл сухого нитро-
бензола. Смесь при постоянном перемешивании выдержива-
ют на глицериновой бане при 70—80° в течение 15—20 минут
и добавляют 0,2 М безводной карбоновой кислоты. Затем
температуру бани поднимают до 135—140° и реакционную
массу при перемешивании выдерживают 7 часов, после чего
ее охлаждают в бане со льдом и понемногу прибавляют
300 мл 15%-ной серной кислоты. Органический слой отделя-
ют, а водный дважды экстрагируют хлороформом (по
50 мл). Экстракты прибавляют к нитробснзольному слою и
подвергают перегонке с паром. Кубовый остаток при охлаж-
дении подщелачивают 5%-ным раствором едкого натра до
pH 9—10, обрабатывают углем на холоду, фильтруют и при
охлаждении нейтрализуют 5—7%-ным раствором соляной
кислоты до pH 5. Выпавший продукт отфильтровывают, су-
шат и кристаллизуют из этилацетата с углем.
Все синтезированные соединения представляют собой бе-
лые кристаллические продукты, легкорастворимые в боль-
шинстве органических растворителей.
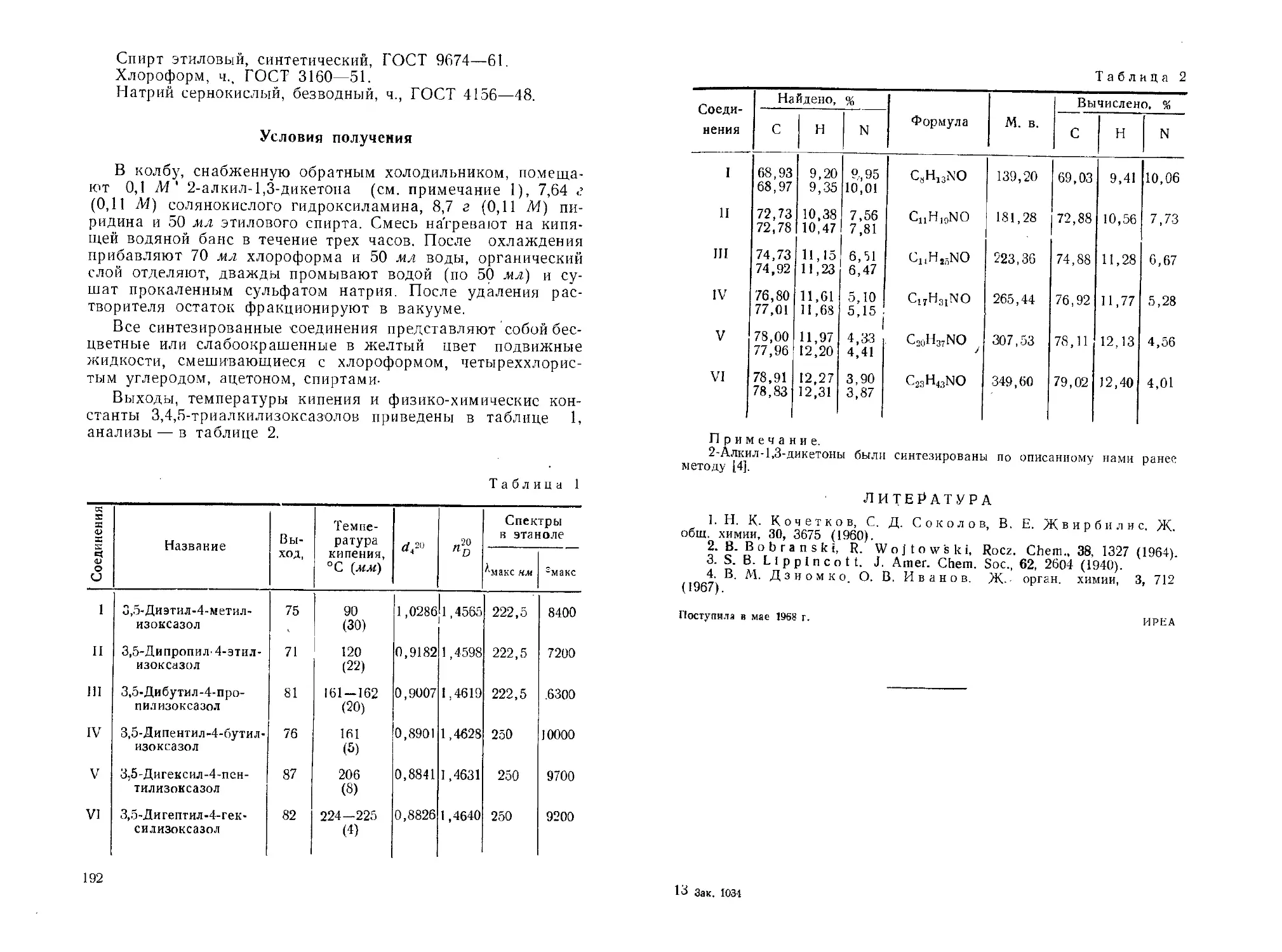
Выходы, температуры плавления и максимумы поглоще-
ния УФ-спектров 2-алкил-1,3-циклопентандионов приведены
в таблице 1, анализы—в таблице 2.
2 Зак. 1034 17
Таблица 1
Соединения! Название Выход, % Температу- ра плавле- ния, °C Спектры (в этаноле) Спектры (в этаноле) 3.10“3 Л1 NaOH)
^•макс НМ £макс ^макс НМ £макс
I 2 -Пентил- 1,3-циклопентан- дион 19,5 147-147,5 250 16800 270 28400
II 2-Гексил-1,3-циклопентан- дион 22,3 141,5-142 250 19000 270 34800
III 2-Гептил-1,3-циклопентан- дион 25,0 137,5-138 255 17600 270 29800
IV 2-Октил-1,3-циклопентан- дион 20,4 138—138,8 255 11600 270 20400
Таблица 2
Соединения Найдено, % Формула М.в. Вычислено, %
С Н С Н
I 71,53 71,62 9,73 9,65 168,238 71,39 9,59
11 72,63 72,69 9,86 9,81 С1ХН18О2 182,265 72,49 9,95
Ill 73,62 73,33 10,15 10,25 С12Н20О2 196,292 73,43 10,27
IV 74,15 73,98 10,60 10,55 С1зН22О2 210,319 74,25 10,54
ЛИТЕРАТУРА
1. Франц, пат. 81978 (1963).
2. Бельг, пат., 632348 (1963).
3. Н. Smith, J. A. Hughes, J. Н. D о u’g 1 a s. J. Chem. Soc.
1964, 4472.
4, М. О г с h i п, L. W. Butz. J. Amer. Chem. Soc., 65, 2296 (1943).
5. J. H. Both e. K. J. Wilkinson. J. Amer. Chem. Soc. 75, 1732,
(1953).
6. J. J. P a n о u s e, C. S a n n 1 e. Bull. Soc. Chim., 1955, 1036.
7. Англ, пат., 776920 (1957).
8. Пат. США, 2811558 (1958).
9. С. В. С. Воусе, J. S. Whitehurst. J. Chem. Soc., 1959,2022.
10. S. Esko la. Ann. Acad. Sci. Fennical, Ser. A, Il Chem, 18,27(1945)
11. Франц, пат., 1363281 (1964).
12. Д. P. Л а г и д з e, С. Н. Ананченко, И. В. Торгов. Изв. АН
СССР ОХН, 10, 1899 (1965).
13. В. М. Дзномко, О. В. Иванов. Ж. орг. химии, 3, 712 (1967).
Поступила в мае 196S г.
18
ИРЕА
УДК 547.587.52'568.1
БЕНЗИЛОВЫЙ ЭФИР 4-МЕТИЛУМБЕЛЛИФЕРОНА
7-Бензилокси-4-метилкумарин
Д. А. ДРАПКИНА, В. Г. БРУДЗЬ, Н. И. ДОРОШИНА
СН3
С,.НГ,СН2ОХ^ХХО/^О
С17Н14О3 М. в. 266,29
Бензиловый эфир 4-метилумбеллиферона представляет ин-
терес как вещество, люминесцирующее в синефиолетовой об-
ласти спектра при воздействии ультрафиолетовой радиации.
Бензиловый эфир 4-метилумбеллиферона был получен
бензилированием 4-метилумбеллиферона бромистым [1] или
хлористым (выход 25%) [2] бензилом в ацетоне в присутст-
вии безводного поташа, а также с выходом 67% бензилиро-
ванием хлористым бензилом в этаноле в присутствии метал-
лического натрия [3]. При воспроизведении условий по [2] мы
получили эфир с выходом сырца 41,2% (т. пл. 112—113°) и
чистого продукта 27%, (т. пл. 415—116°). Значительно повы-
сить выход продукта нам удалось в результате проведения
реакции в метилэтилкетоне вместо ацетона с применением з
качестве катализатора йодистого калия и металлического
йода.
СХЕМА СИНТЕЗА БЕНЗИЛОВОГО ЭФИРА
4-МЕТИЛУМБЕЛЛИФЕРОНА
снэ сн3
I I
Г'Х||//'^ С6Н5СН,С1_
I II I К2СО3(КЛ,.12Г J II I
HO/'^''4o/V) СвН5СН2О/^/\/'^О
2* 19
Характеристика исходного сырья
4-Метилумбеллиферон, получен по методике [4].
Бензил хлористый, ч., ТУ МХП 50-49.
Калий углекислый, ч., ГОСТ 4221-57.
Метилэтилкетон, ч., ВТУ МХП 3024—55.
Калий йодистый, ч., ГОСТ 4232—48.
йод металлический, ч., ГОСТ 4159—48.
Условия получения
В двугорлой колбе емкостью 100 мл, снабженной механи-
ческой мешалкой и обратным холодильником, кипятят в те-
чение 5 часов смесь, состоящую из 30 мл метилэтилкетона,
8,84 г (0,05 М) 4-метилумбеллиферона, 6,96 г (0,055 А4) хло-
ристого бензила, 5 г прокаленного поташа, 0,2 г йодистого
калия и кристаллика йода. Образовавшийся осадок хорошо
промывают 50 мл 2-3%-ного раствора едкого натра, водой и
один раз небольшим количеством .60%-ного этанола.
Получают 11,6—12 г сырого продукта, с т. пл. 112—113э.
После кристаллизации из 300 мл 60—65%-ного этанола вы-
ход эфира равен 9.5—9,7 г, что составляет 72—73% от теоре-
тического, считая на 4-метилумбеллнферон; т. пл. 115—116°.
По литературным данным, температура плавления веще-
ства 117,5° [1]; 118—120° [2]; 112° [3].
ЛИТЕРАТУРА
1. W. Bridge, A. J. Crocker, In. С u Ь i n, A. Robertson.
J. Chem. Soc., 1937, 1530.
2. B. Z. M u 1 la j i, R. C. Shah. Proc. Indian Acad. Sci., 34A, 173
(1951).
3. А. А. Шамшурин, P. А. Ибаду л ин. Tp. Узбекск. гос. ун-та.
сер. хим., 25, вып. 1, I (1941).
4. Синтезы органических препаратов, т. Ill, М., Госхимиздат, 1952,
стр. 218.
Поступила в феврале 1968 г.
ПРЕД
УДК 547.772.1
1-БЕНЗИЛ-3,4,5-ТРИАЛ КИЛ ПИРАЗОЛ Ы
В. М. ДЗИОМКО, О. В. ИВАНОВ
I r=c2h5
II R=h-C3H7
III R = h-C4H9
IV R = h-C5Hh
N-Бензилированные производные пиразола могут быть
получены при взаимодействии бензилгидразина с 1,3-дикето-
нами [1] или посредством алкилирования пиразолов галоид-
ным бензилом [2].
Нами получен ряд новых 1-бензил-3,4,5-триалкилпиразо-
лов алкилированием 3,4.5-триалкилпиразолов хлористым бен-
зилом в присутствии углекислого калия в среде диметилформ-
амида.
СХЕМА СИНТЕЗА 1-БЕНЗЙЛ-3,4,5-ТРИАЛ КИЛ ПИРАЗОЛОВ
R CH,R
„ У—\\ к2со,
RCH2— +СсН5СН2С1 (сн3)2МСНО~>
4N
н
21
CH2R
RCH2-^ \
N /
I
CH2
\/
Характеристика основного сырья
3,4,5-Триалкилпиразолы (см. одноименную статью в на-
стоящем сборнике)-
Диметилформамид, ч., ВТУ РУ—1193—56.
Калий углекислый, ч., ГОСТ 4221—48.
Хлористый бензил, ч., ТУ МХП 50—49.
Натр едкий, ч„ ГОСТ 4328—48.
Хлороформ, ч., ГОСТ 3160—51.
Натрий сернокислый, безводный, ч„ ГОСТ 4156—48.
Условия получения
В трехгорлую колбу, снабженную мешалкой, обратным
холодильником с хлоркальциевой трубкой и капельной во-
ронкой с хлоркальциевой трубкой, помещают 0,1 М 3,4,5-три-
алкилпиразола, 50 мл сухого диметилформамида и 9,7 г
(0,07 Л4) углекислого калия. При постоянном перемешива-
нии за 10 минут прибавляют 13,9 г (0,11 М) хлористого бен-
зила и реакционную смесь выдерживают 3,5 часа на глице-
риновой бане нагретой до 140—150°. После охлаждения к ре-
акционной массе добавляют 70 мл 10%-ного раствора едкого
натра, 100 мл хлороформа и смесь тщательно перемешивают
в течение 15 минут. Органический слой отделяют, дважды
промывают водой (по 50 мл), и сушат прокаленным сульфа-
том натрия. Затем растворители отгоняют, а остаток фрак-
ционируют в вакууме.
Выходы, температуры кипения и физико-химические кон-
станты 1-бензил-3,4,5-триалкилпиразолов приведены в табли-
це 1, анализы — в таблице 2.
22
Таблица 1
1 Соединения! Название Выход, % Температу- ра кипения, °C (мм) rf20 20 «в Спектры в циклогекса- не ^макс 1 £макс
I 1-Бензил-3,5-Дипро- пил-4-этнл пиразол 82 151—152 (6) 0,9699 1,5241 230 7600
11 1 -Бензил-3,5-дибу- тил-4-пропилпиразол 81 185 (4) 0,9437 1,5122 223 8600
III 1-Бензил-3,5-дипен- тил-4-бутнлпиразол 86 218 (2) 0,9315 1,5025 221 7600
IV 1-Бензил-3,5-дигек- сил-4-пентилпиразол 90 232-233 (15) 0,9064 1,4841 227,5 5000
Таблица 2
1 Соединения Найдено, % Формула М.в. Вычислено %
С Н N С Н N
I 79,85 79,99 9,48 9,60 10,30 10,28 c18H2SN2 270,422 79,95 9,69 10,36
11 80,78 80,93 10,24 10,11 8,70 8,74 c2ih32n2 312,503 80,71 10,33 8,96
III 81,59 81,56 10,98 1 к 05 7,81 7,83 Сз<Н38М2 354,584 81,30 10,80 7,90
IV 81,85 81,90 11,02 11,35 7,15 6,97 C3,HUNS 396,665 81,76 11,18 7,06
ЛИТЕРАТУРА
1. И. И. Г р а н д б е р г, А. Н. Кост. Ж. общ. химии, 30, 203 (1960).
2. И. И. Грандберг, ХГС, 279 (1965).
Поступила в мае 1968 г.
ПРЕД
УДК 547.638.3
4,4/-БИС-[2,4-БИС-(КАРБОКСИМЕТИЛАМИНО)-1,3,5-
ТРИАЗИНИЛ-6-АМИНО]-2,2-ТОЛАНДИСУЛЬФО-
КИСЛОТА
В. я. ТЕМКИНА, Г. Ф. ЯРОШЕНКО, Н. Е. ХАВЧЕНКО,
Р. П. ЛАСТОВСКИЙ
s0 н NHCH,COOH
N-C4
/N
| N^Cx-NHCHaCOOH
,nhch2cooh
__ N~ C<
C-^ NH-C7 ~N
z ^N-
SO3H xNHCH2COOH
C28H26N12O14S2 M. в. 818,71
4,4'-Бис-[2,4-бис-(карбоксиметиламино)-1,3,5-триазинил- 6-
амино]-2,2'-толандисульфокислота является аналогом люми-
несцентного реагента для определения Сг3+—триазинилстиль-
бексона [1, 2] в ряду толана. В системе диаминостильбена,
обусловливающей флуоресценцию триазинилстильбексона,
благодаря фотоиндуцировэнной перегруппировке, часть флуо-
ресцирующей транс-формы переходит в нефлуоресцирующую
г(ас-форму, что снижает интенсивность флуоресценции реа-
гента [3]. Отсутствие ч«с-гранс-изомерии в производном то-
лана устраняет этот недостаток реагента.
Синтез комплексона осуществлен нами взаимодействием
диаминотоландисульфокислоты с двумя молями хлористого
24
пиаиура и последующим замещением атомов хлора на ами-
Есоацетатные группы при 45° и 90°.
Исходная диаминотоландисульфокислота получена по ме-
тоду [4] бромированием динитростильбендисульфокислоты,
омылением полученного монобромлактона и восстановлением
динитротоландисульфокислоты двуххлористым оловом.
СХЕМА СИНТЕЗА СОЕДИНЕНИЯ
O2N-^ ^-СН=СН-^ ^-no2->
Х SO3Na NaOaS7
O8N-^_^~CHBr-CH-^ A-NO2 _>
\o3Na .O-O.S
-> O,N-^ ^-C=C-^ ^-NO2->
\o3K icons'7
- H2N--^ ^-C=C-^ ^-NH2->
\sOsH HOsS^
SO3H Cl
N-<
C-^ ^-NH-C^ N
= N—
Cl
/С1
C-^ ^-NH-C<6 N
/ N-Cf
SO3H 4C1
25
SO3H .NHCH^OOH
\_ N-~cZ
C-^ ^-NH-C^ N
==' N=C/
'NHCIECOOH
, nhch2cooh
_ N=c/
C-(f ^-NH-cZ N
/ "" N-C^
SO3H xNHCH2COOH
Характеристика основного сырья
Динитростильбендисульфокислота, ч.
Бром, ч., ГОСТ 4109—64.
Едкое кали, х. ч., ГОСТ 4203—65.
Двухлористое олово, ч., ГОСТ 36—40.
Цианур хлористый, ч., ТУ ЛРЗ 137—65.
Гликокол (аминоуксусная кислота), ч., ГОСТ 5860—51.
Условия получения
Синтез монобромлактона динитростилбендисульфокисло-
ты. Растворяют при нагревании 30 г (0,063 М) динатриевой
соли динитростильбендисульфокислоты в 150 мл горячей во-
ды, затем раствор охлаждают до 55—58° и добавляют 12 г
(0,07 М) брома. Присоединение брома идет при сильном
перемешивании в течение нескольких минут. Выпавший при
охлаждении до 0—3° желто-зеленый осадок монобромлакто-
на отфильтровывают, кристаллизуют из воды, промывают
этанолом и сушат в вакуум-эксикаторе.
Выход 9,4 г (28% от теоретического).
Найдено, %: Вг—15,9; 15,7.
CuHgBrNaNoOioSs. Вычислено, %: Вг—15.
Получение дикалиевой соли динитротоландисульфокисло-
ты. Растворяют 9 г (0,017 Л1) монобромлактона в 150 мл во-
ды при 60—70° и добавляют 30 мл 50п/о-ного раствора едкого
кали. Цвет раствора становится красным.
Выпавший ярко-зеленый осадок отфильтровывают, кри-
сталлизуют из воды и сушат при температуре 50—60°.
Выход 6 г (70% от теоретического).
Получение 4,4'-диаминотолан-2,2'-дисульфокислоты. Рас-
творяют 6 г (0,012 М) калиевой соли динитротоландисульфо-
кислоты в 200 мл горячей воды, нагревают до кипения и к
кипящей реакционной массе порциями добавляют раствор
26
30 г двуххлористого олова в 80 мл воды и 50 мл концентри-
рованной соляной кислоты. После четырехчасового кипяче-
ния охлаждают реакционную смесь и отфильтровывают вы-
павший осадок. Динитротоландисульфокислоту очищают пе-
реосаждением, промывают водой до отсутствия ионов хлора
и сушат при температуре 60—70°.
Выход 3,1 г (70% от теоретического).
Получение 4,4'-бис-[2,4-бис-(карбоксиметиламино)-1,3,5-
1риазинил-6-амино]-2,2'-толандисульфокислоты- К суспензии
3,72 г хлористого цианура в 100 мл воды при размешивании
и температуре 0—5° в течение 10—15 минут добавляют рас-
твор 4,26 г натриевой соли диаминотоландисульфокислоты,
поддерживая pH 6—7 по универсальной индикаторной бу-
маге добавлением 10%-ного раствора углекислого натрия.
После выдержки при 0—5° в течение 30 минут к реакционной
массе добавляют 3 г аминоуксусной кислоты, предваритель-
но нейтрализованной также 10%-ным раствором углекислого
натрия до pH 7. Смесь нагревают при размешивании в тече-
ние 1,5 часа до 97—100°, поддерживая pH 7—8, и оставляют
при этой температуре еще на 5 часов. Охлажденный раствор
подкисляют до pH 2 концентрированной соляной кислотой.
Выпавший желтый осадок натриевой соли комплексона про-
мывают 85%-ным раствором метанола до отсутствия ионов
хлора и переводят в протонированную форму с использова-
нием катионитового сита КРС-5 [5]. Выход продукта равен
5,4 г, что составляет 66% от теоретического.,
Найдено, %: С—40,2; 40,1; Н—4,2; 4,0; N- 19,8; 19,9.
CmH,6N12O,4S2. Вычислено, %: С—40,8; Н—3,4; N—20,4.
ЛИТЕРАТУРА
1. Р. П. Ластовский, Е. А. Божевольнов, В. Я. Темкина,
С. У. Крейнгольд Г. Ф. Ярошенко, В. Н. Антонов. Авт. свид.
186757; Билл, нзобр., № 19 (1966).
2. В. Я. Тем к нн а, Е. А. Божевольнов, Н. М. Дятлова,
С. У. Крейнгольд, Г. Ф. Ярошенко, В. Н. Антонов,
Р. П. Ластовский. Ж. аналит. химии, 22. 1830 (1967).
3. Q. D re fa h I, G. К 0 11 n e r. J. pract. Chem., 31, 5—6, 269 (1966).
4. Р,- R ugg 11, E. Peyer. Helv. Chim. Acta, 9, 929 (1926).
5. P. П. Ластовский, E. Б. Тростяиская, В. Я. Темкина,
И. Д. Колпакова, С. Б. Макарова, Г. Ф. Ярошенко,
Т. А. А п т о в а. Авт. евнд. 178000; Бюлл. изобр.. № 2 (1966).
Поступила в мае 1968 г.
ИРЕА.
УДК 547.341.124
1,4-БИС-(ДИЭТ0КСИФ0СФ0Н0) БУТЕН-2
С. 3. ИВИН, В. к. ПРОМОНЕНКОВ, С. к. СМИРНОВ
(С2Н6О)2РСН,СН=СНСН2Р(ОС2Н6)2
11 II
о о
CiaHaeOePa М. в. 328,28
Известно сравнительно немного алкенильных соединений
фосфора, содержащих несколько замещенных фосфоновых
группировок. К их числу относится неописанный ранее
1,4-бис-(диэтоксифосфоно) бутен-2, способ получения которо-
го приводится ниже. Вещества этого типа могут представить
интерес как мономеры и промежуточные продукты.
СХЕМА СИНТЕЗА 1,4-БИС-(ДИЭТОКСИФОСФОНО)БУТЕНА-2
ВгСН2СН=СНСН2Вг + 2Р(ОС2Н5)3->
(CsH6O)4PCH2CH=CHCH2P(OC»H5)2+2C2H5Rr
II II
О о
Характеристика основного сырья
Триэтилфосфит, получен взаимодействием треххлористо-
го фосфора с абсолютным этиловым спиртом в присутствии
триэтиламина, т. кип. 55715 мм; d420—0,9687; riff —1,4135(1].
1,4-Дибромбутен-2, получен бромированием дивинила по
методике [2]; т. пл. 53° (из петролейного эфира).
Условия получения
В колбу Кляйзена емкостью 150 мл (см. примечание 1)
помещают 21,4 г (0,1 А4) 1,4-дибромбутена-2 и 54 г (0,325 М)
23
триэтилфосфита. Реакционную смесь постепенно нагревают,
доводя температуру до 70—75°, на что уходит 20—30 минут
(см. примечание 2)- При этом начинает отгоняться бромис-
тый этил. Нагрев реакционной массы продолжают еще 2,5—
3 часа, постепенно повышая температуру так, чтобы непре-
рывно отгонялся бромистый этил, и доводя ее в конце этого
периода до 130—140°. При этой температуре смесь выдержи-
вают 30 минут до полного прекращения отгонки бромистого
этила, после чего остаток перегоняют в вакууме.
Получают 27,9 г (85%) 1,4-бис-(диэтоксифосфоно) буте-
на-2; т. кип. 168—170°/10-3 мм; d4w—1,1340; ng — 1,4593.
Найдено, %: С—43,30; Н—7,91; Р—18,34.
С12Н2вО5Р2. Вычислено, %: С—43,90; Н—7,93; Р—18,89.
Примечания:
1. При проведении реакции используют прибор для перегонки в ва-
кууме.
2. В процессе нагрева иногда наблюдается образование кристалли-
ческого осадка, являющегося, по-видимому, промежуточным продуктом
протекающей арбузовской перегруппировки.
ЛИТЕРАТУРА
1. Н. McCom'bie, В. Saunders, G. Stacey. J. Chein. Soc.
1945, 380.
2. L. Owen. J. Chem. Soc., 1949, 243.
Поступила в декабре 1967 г.
УДК 547.638.1
2,2-БИС-(4-ОКСИ-3 -ХЛОР-5 -МЕТИ Л ФЕН ИЛ) ПРОПАН
А. М. СЕРЕБРЯНЫЙ, И. М. БИЛИК, Н. М. МИРОНОВА
С1 СНз
СН, СН’ CH,
C17H1SC12O2 M. в. 325,23
2,2-Бис- (4'-окси-3'-хлор-5'-метилфенил) пропан синтезиро-
ван впервые по разработанному нами методу [1] посредством
хлорирования 2,2-бис- (4'-окси-3'-метилфенил) пропана хлори-
стым сульфурилом.
СХЕМА СИНТЕЗА 2,2-БИС-(4'-ОКСИ-3'-ХЛОР-5'-МЕТИЛФЕНИЛ)-
ПРОПАНА
СН3 —сн3
I ^Н3 I
I /=\
НО-^ ^-С—^OH+2SO2C12 -*
СНз
С1 „„ С1
L । 3 J
НО^ ^-С-4 ^OH+2HC1+2SO2
СНз СНз СНа
Характеристика основного сырья
2,2-Бис- (4'-окси-3'-метилфенил) пропан, получен по мето-
дике [2].
30
Хлористый сульфурил, ч., ТУ НКХП 45—40.
Четыреххлористый углерод, технический, ГОСТ 4—45.
Условия получения
В колбу, снабженную мешалкой, обратным холодильни-
ком, термометром и отводной трубкой (соединенной со склян-
кой Дрекселя, содержащей раствор едкого натра для погло-
щения выделяющихся газов), вносят 3,2 г (0,012 М) 2,2-бис-
(4'-окси-3/-метилфенил) пропана. Добавляют 10 мл четырех-
хлористого углерода и 2,25 мл (0,03 Л4) хлористого сульфу-
рила. Колбу помещают в водяную баню, реакционную массу
размешивают 3 часа при 55°, затем 1 час при 70°, оставля-
ют на ночь при комнатной температуре и затем фильтруют.
Фильтрат промывают водой, водный слой отделяют и отбра-
сывают. Органический слой переносят в чашку Петри и че-
тыреххлористый углерод испаряют в вытяжном шкафу.
Выход 2,2-бис-(4'-окси-3'-хлор-5'-метилфенил)пропана ра-
вен 3,59 г, что составляет 88,4% от теоретического- После
перекристаллизации из 35%-ной уксусной кислоты получают
белое кристаллическое вещество с т. пл. 103—103,5°.
Найдено, %: С—62,85; 62,65; Н—5,60; 5,80; 0—21,39; 21,40.
С18Н18С12О2. Вычислено, %: С—62,77; Н—5,54; С1—21,84.
Дибензоат. Белое кристаллическое вещество с т. пл. 192—
193° (из этилового спирта, затем из уксусной кислоты).
Найдено, %: С—70,42, 70,15; Н—5,23- 5,12; С1—13,30; 13,32.
C3iH26Cl2O4. Вычислено, %: С—69,79; Н—4,88; С1—13,32. \
ЛИТЕРАТУРА
1. И. М. Б и л и к, Н. М. Бондарен, А. М. Серебряный.
Л. С. Рыбкина, Т. М. Чернявская. Англ, пат., 1047058 (1966).
2. И. М. Б и л н к, Н. М. Бондарен, А. М. Серебряный,
Р. Л. Глобус, В. Г. Брудзь. Методы получения химических реакти-
вов и препаратов, вып. 2, М., ИРЕА, 1961, стр. 113.
Поступила в мае 1968 г.
ИРЕА
УДК 547.638.1
2,2-БИС-(4-ОКСИ-З'-ХЛОРФЕНИЛ)ПРОПАН
А. М. СЕРЕБРЯНЫЙ, и. М. БИЛИК, н. М. МИРОНОВА
С1 „„ С1
L : U
“Ос-О™
СНз
С^НиСЬОг М. в. 297,18
В результате конденсации о-хлорфенола с ацетоном с
целью синтеза 2,2-бис- (4/-окси-3/-хлорфенил)пропана было
получено лишь смолообразное вещество [1]. В работах [2, 3]
приведена только температура плавления этого вещества без
описания методики его получения.
При конденсации о-хлорфенола с ацетоном в присутствии
фтористого бора 2,2-бис-(4'-окси-3'-хлорфенил)пропан нами
был синтезирован с выходом 33—36% [4]. Другой разрабо-
танный нами метод получения этого продукта посредством
хлорирования 2,2-бис- (4'-оксифенил) пропана хлористым суль-
фурилом в присутствии сернистого натрия позволяет полу-
чать 2,2-бис- (4'-окси-3'-хлорфенил) пропан хорошего качест-
ва с высоким выходом [5]-
2,2-Бис- (4'-окси-3'-хлорфенил) пропан представляет инте-
рес для получения поликарбонатов, эпоксидных смол и для
проведения ряда других исследовательских работ.
СХЕМА СИНТЕЗА 2,2-БИС-(4'-ОКСИ-3'-ХЛОРФЕНИЛ) ПРОПАНА
сн3
НО—>-OH+2SOsC12
СН3
32
С£ ' СИз J3I
НО-^ ^-С-^ ^-OH4-2HCH-2S02
СН3
Характеристика основного сырья
2,2-Бис-[4'-оксифенил]пропан (дифенилолпропан), ч.,
МРТУ 6-09-7-62.
Хлористый сульфурил, ч., ТУ НКХП 45—40.
Четыреххлористый углерод, технический, ГОСТ 4—65.
Натрий сернистый, плавленый, технический, ГОСТ 596—•
56-
Уксусная кислота, техническая, ГОСТ 7077—56.
Активированный уголь, I сорт, ГОСТ 4453—48.
Условия получения
- В колбу, снабженную мешалкой, обратным холодильни-
ком, термометром и отводной трубкой (соединенной со склян-
кой Дрекселя, содержащей раствор едкого натра для погло-
щения выделяющихся газов), вносят 22,8 г (0,1 М) 2,2-бис-
(Ч'-оксифенил)пропана. Затем добавляют 0,46 г (0,006 М)
растертого в порошок сернистого натрия, 80 мл четыреххло-
ристого углерода и 17,8 мл (0,22 М) хлористого сульфурила.
Колбу помещают в водяную бацю, смесь размешивают 3 ча-
са при 55° и 1 час при 70°, после чего оставляют на 4 часа при
комнатной температуре и затем фильтруют. Фильтрат выли-
вают в воду, перемешивают, органический слой отделяют,
5 раз промывают его водой порциями по 400—500 мл по-
средством декантации. В процессе этой операции выделяется
осадок, который отфильтровывают, отмывают от кислоты до
нейтральной реакции по индикаторной бумажке и сушат при
40°. . '
Выход продукта равен 24,9—25,8 г, что составляет 84—
87% о г теоретическою; т. пл. 84—86°. После двухкратной пе-
рекристаллизации из 38 %-ой уксусной кислоты (10 мл кис-
лоты на 1 г вещества) с применением активированного угля
2,2-бис-(4/-окси-3/-хлорфенил) пропан имеет т. пл. 88—90°
Полученное вещество не дает депрессии температуры пла-
вления в смешанной пробе с образцами, синтезированными
из о-хлорфенола и ацетона в присутствие фтористого бора.
По литературным данным, т. пл. продукта 88,2—88,6° [4];
89—91° [2]; 90,5—91,5° [3].
Найдено, %: С—60,70: 60,60; Н—4,83; 5,02; С1—23,70; 23,90.
CisHhCIjA- Вычислено, %: С—60,61; Н—4,71; С1—23,90.
3 Зак. 1034 33
ЛИТЕРАТУРА
1. Пат. ФРГ, 905977 (1954); Zbl., 1954, 5410.
2. Н. Schnell. Ind. Eng. Chem., 51, 157 (1959).
3. P. В. M a r c h. М. L. В u 11 е г, В. S. С 1 а г с к. Ind. Eng. Chem.,
41, 2176 (1949).
4. И. М. Билик, А. М. Серебряный, Р. Л. Глобус,
В. Г. Брудзь. Ж. общ. химии, 33, 487 (1963).
5. И. М. Билик, Н. М. Бондаре ц, А. М. Серебряные,
Л. С. Рыбкина, Т. М. Чернявская. Англ, пат., 1047058 (1966).
Поступила в мае 1968 г.. И ГЕ А
УДК 547.592.12'562.311
1,1-БИС-(3'-ХЛОР-4-ОКСИФЕНИЛ)ЦИКЛОГЕКСАН
4. М. СЕРЕБРЯНЫЙ, И. М.
БИЛИК, н. М. МИРОНОВА
С18Н18С12О2
М. в. 337,24
Ранее было описано получение 1,1-бис- (3'-хлор-4'-окси-
фенил) циклогексана с выходом 55% из о-хлорфенола и'цик-
логексанона в присутствии щелочи [!]• Продолжительность
этой реакции 210 часов. Имеется также пропись синтеза это-
го вещества из о-хлорфенола и циклогексанона в присутст-
вии катионитов с выходом 20%' [2].
Нами предложена методика получения 1,1-бис- (З'-хлор-
4'-оксифенил) циклогексана посредством хлорирования 1,1'-
бис-(4'-оксифенил) циклогексана хлористым сульфурилом в
присутствии сернистого натрия [3], что позволяет быстро по-
лучать продукт хорошего качества с высоким выходом.
Препарат представляет интерес для получения поликар-
бонатов.
3* 35
СХЕМА СИНТЕЗА 1.1-БИС-(3'-ХЛОР-4'-ОКСИФЕНИЛ)ЦИКЛО-
ГЕКСАНА
ОН он
ОН ОН ||
4 С Х + 2SO2C12 -> 4 с 7 4- 2НС1 + 2SO2
НаС7 'сН, Н2С "сН,
Н2С СН2 Н2С СН,
сн2 Чсн2
Характеристика основного сырья
1,1-Бис(4'-оксифенил)циклогексан, ч„ МРТУ 6-09-3157-66.
Хлористый сульфурил, ч., ТУ НКХП 45—40.
Четыреххлористый углерод, технический, ГОСТ 4—65.
Натрий сернистый, плавленый, технический, ГОСТ 596—
56.
Условия получения
В колбу, снабженную мешалкой, обратным холодильни-
ком, термометром и отводной трубкой (соединенной со склян-
кой Дрекселя, содержащей раствор едкого натра для погло-
щения выделяющихся газов), вносят 15,1 г (0,056 Л1) 1,1-бис-
(4'-оксифенил) циклогексана. Добавляют 0,6 г (0,005 Л1) рас-
тертого в порошок плавленого сернистого натрия, 90 мл че-
тыреххлористого углерода и 10,1 мл (0,13 М) хлористого
сульфурила. Колбу помещают в водяную баню и смесь раз-
мешивают 3 часа при 55° и 1 час при 70°. После этого горя-
чую массу фильтруют через складчатый фильтр на воронке
для горячего фильтрования и фильтрат охлаждают. Выде-
лившийся осадок отфильтровывают, промывают водой от кис-
лоты, ионов хлора и сушат при 50—60°.
Выход продукта равен 13,3 г, что составляет 70% от тео-
ретического; т. пл. 142—144°.
После перекристаллизации из четыреххлористого углеро-
да (10 мл на 1 г) вещество представляет собой белые кри-
сталлы с т. пл- 147,5—148,5°.
По литературным данным [1], температура плавления
продукта 134—141°.
Найдено %: С—64,16; 64,13; Н—5,43; 5,55; С1—20,31; 21,08.
С18Н18С12О2. Вычислено, %: С—64,10; Н—5,34; С1—21,07.
36
ЛИТЕРАТУРА
1. Пат. США, 2858342; С. А., 53, 6165в (1959).
2. Англ, пат., 849965; С. А., 55, 19869h (1961).
3. И. М. Билик, Н. М. Бондарец, А. М. Серебряный.
.1 С. Рыбкина, Т. М. Чернявская. Англ, пат., 1047058 (1966).
Поступила в мае 1968 г.
ИРЕА
УДК 547.447.2
БИС-(ЦИКЛ0ГЕКСАН0Н)0КСАЛИЛДИГИДРА30Н
И БИС-(АЦЕТАЛЬДЕГИД)ОКСАЛИЛДИГИДРАЗОН
Г. С. ПЕТРОВА, А. М. ЛУКИН, И. А. НЕМИРОВСКАЯ,
И. С. ФРУМИНА, М. Д. КОФМАН
C14H22N4O2
0 0 ,
II II |
= N=NH-С—С—NH-N='X
М. в. 278,35
О О
II II
CH3-CH=N-NH-C-C-NH-N=CH-CHS
СвНюТ^Ог М. в. 170,87
Цветные реакции бис-(циклогексанон)оксалилдигидразо-
на и бис-(ацетальдегид)оксалилдигидразона с ионом меди
описаны впервые в работе [1]. В настоящее время первый ре-
актив широко применяется для анализа сталей, чугунов и
сплавов [2]. Имеются также данные о том, что второй реактив
по устойчивости комплексов с медью и интенсивности окрас-
ки превосходит первый [3].
Сведений по синтезу этих реактивов в литературе нами не
найдено. По данным каталога [4], бис-(циклогексанон) окса-
лилдигидразон представляет собой желтоватый порошок с
95%-ным содержанием основного вещества и т. пл. 200—
220° (с разл.).
СХЕМА СИНТЕЗА БИС-(ЦИКЛ ОГЕКСАНОН)-
ОКСАЛ ИЛ ДИГИДРАЗОНА
О
II
c-nh-nh2
c-nh-nh2
II
о
о
II
+ 2 Qi
38
00 /\
"* J--=N-NH-C—C-NH-N='x/! + 2Нз°’
Характеристика основного сырья
Оксалилдигидразид, ч., МРТУ 6-09-2154-65.
Циклогексанон, ч., ТУ МХП 2223—50.
Спирт этиловый, ректифицированный, ГОСТ 9674—61.
Кислота серная, х. ч., ГОСТ 4204—48.
Уксусный альдегид, ч„ ТУ МХП 2633—51.
Условия получения
В колбу с мешалкой загружают 50 мл этилового спирта,
0,2 мл концентрированной серной кислоты, 5,9 г (0,05 Л4) ок-
салилдигидразида и 12 г (0,125 М) циклогексанона. Смесь
размешивают в течение 5 часов при комнатной температуре,
затем осадок отфильтровывают и сушат на воздухе.
Выход бис-(циклогексанон) оксалилдигидразона равен
12,2 г, что составляет 88% от теоретического; т. пл. 210—-213°
(с разл.). По внешнему виду это белый порошок с желтова-
тым оттенком, пригоден для аналитических работ. Чувстви-
тельность реактива к иону меди составляет 0,05 мкг/мл. Для
получения х.ч. соединения производят перекристаллизацию
из спирта.
Найдено, %: N—20,57; 20,56.
CUH22N4O2. Вычислено, %: N 20,12.
СХЕМА СИНТЕЗА БИС-(АЦЕТАЛЬДЕГИД)ОКСАЛ ИЛДИГИДРАЗОНА
О
II
c-nh-nh2
| + 2 СНзСНО
C-NH — NH2
О
О О
II I!
СН3—CH = N—NH—С—С—NH —N=CH—СН3 %- 2 Н2О
Условия получения
В трехгорлую колбу, снабженную мешалкой, обратным
холодильником и термометром, загружают 2,4 г (0,24 Л4) ок-
салилдигидразида и 25 мл спирта. Смесь размешивают при
10° и прибавляют 3 мл (0,05 М) уксусного альдегида. Тем-
39
пература при этом поднимается до 25°, а затем постепенно
снижается. Реакционную массу при 15° размешивают 5 ча-
сов, выпавший белый кристаллический осадок отфильтровы-
вают и сушат на воздухе.
Выход бис-(ацетальдегид)оксалилдигидразона равен 2,6.?,
что составляет 79% от теоретического; т. пл. 230° (с разл.)-
Чувствительность реактива к иону меди составляет
6,02 мкг]мл.
Найдено, %: N—33,07; 33,01.
C6HioN402. Вычислено, %: N—32,91.
ЛИТЕРАТУРА
1, G. N ill son. Acta. Chem. scand , 4, 205 (1950).
2. E. Merck A.Q. Organlshe Reagenzien Fur die anorganishe
Analyse Darmstadt, 1966, стр. 59.
3. E. Jacobsen, F. La ng my hr, A. R. Selmer—Olsen,
Analyt. chim. acta, 24, 579 (1961).
4. Schuchardt. Hauptkatalog, Munchen, 1966/1967, S. 223; Z. Analyt.
Chem., 157, 125 (1957).
Поступила в мае 1968 г.
ИРЕА, Саратовск. Гос. ун-т
УДК 547.727
5-БРОМ-2-БЕНЗОИЛ ФУРАН
5-Бром-2-фурилфенил кетон
3. Я. НАЗАРОВА, Л. Д. БАБЕШДИНА
Вг ' >-СО-
сг
2
М. в. 251,08
5-Бром-2-бензоилфуран впервые был получен бромиро-
ванием 2-фурилфенилкетона с низким выходом [1]. Позже
удалось повысить выход до 30% [2].
Мы применили для получения галоидфурилфенилкетонов
метод Фриделя-Крафтса. При использовании безводного хло-
ристого алюминия в качестве конденсирующего агента для
ацилирования бензола хлорангидридом 5-бром-фуран-2-кар-
боновой кислоты нами получен 5"бром-2-бензоилфуран с вы-
ходом 85%.
СХЕМА СИНТЕЗА 5-БРОМ-2-БЕНЗОЦЛФУРАНА
Вт-/ 5—СОС1 . -» Вг— ^-СО-СвН5+НС1
\q/ А1С13б/в
Характеристика основного сырья
Хлорангидрид 5-бромфуран-2-карбоновой кислоты, т. кип.
110—111718 мм; т. пл. 54—55° [3].
Бензол, х. ч. ГОСТ 5955—51.
Алюминий хлористый, безводный, ГОСТ 4432—46.
Натр едкий, ч.д-а., ГОСТ 4328—48.
Вода дистиллированная, ГОСТ 6709—53.
41
Условия получения
В круглодонную колбу, снабженную обратным холодиль-
ником с хлоркальциевой трубкой, помещают раствор 20,95с
(0,1 Л1) хлорангидрида 5-бромфуран-2-карбоновой кислоты в
107 мл абсолютного бензола. При охлаждении ледяной во-
дой вносят порциями (по 2,5—3 г) 26,7 г (0,2 М) безводного
хлористого алюминия. После прибавления последней порции
реакционную смесь нагревают в течение 5 часов при 50°, а
затем на кипящей водяной бане до прекращения выделения
хлористого водорода (около 30 минут). После охлаждения
содержимое колбы выливают на лед, бензольный слой про-
мывают 10%-ным раствором едкого натра, затем несколько
раз — водой и высушивают над безводным сульфатом нат-
рия. После отгонки бензола под вакуумом водоструйного
насоса кетон перегоняют при 119—122°/15 мм; при охлажде-
нии он закристаллизовывается.
Выход 5-бром-2-бензоилфурана равен 21,3 а, что состав-
ляет 85% от теоретического; т. пл- 43—44°.
По литературным данным, т. пл. продукта 43—44° [2] (см.
примечание).
Примечание.
Аналогично путем ацилирования бензола хлорангидридом 5-хлорфу-
ран-2-карбоновой кислоты получают 5-хлор-2-бензонлфуран с выходом
70%; т. пл. 29,5—30°; т. кип. 190—193727 мм [4].
ЛИТЕРАТУРА
1. J. М. Straley. Iowa State Coll. J. Scl, ll, 115 (1936).
2. R. Grigg, J. A. Knight, M. V. Sargent. J. Chem. Soc.,'
Nov., 6057 (1965).
3. Синтезы гетероциклических соединений, вып. 2, изд. АН Арм. ССР,
Ереван (1957).
4. 3. Н. Назарова, Л. Д. Б а б е ш к и н а. Ж- орган, химии, 2,
вып. 10, 1903 (1966).
Поступила в ноябре 1967 г.
Ростовский н/Д. гос. ун-т
УДК 547.558.2
о-БРОМФЕНИЛДИХЛОРАРСИН
И. П. ШЕПИЛОВ, К. А. ДУНАЕВСКАЯ, Н. А. МУРАТОВА,
В. М. ДЗИОМКО
/Вг
^-AsC12
C6H4AsBrCl2
М. в. 301,83
о-Бромфенилдихлорарсин представляет интерес как реак-
тив для синтеза большого числа мышьякорганических сое-
динений. Его получают восстановлением о-бромфениларсо-
повой кислоты сернистым ангидридом в солянокислой среде
[1, 2]. Нами проверены и уточнены условия получения ука-
занного соединения, причем применявшийся при очистке се-
роуглерод, заменен бензолом.
СХЕМА СИНТЕЗА О-БРОМФЕНИЛДИХЛОРАРСИНА
Вг
7/ ^-As^OH
^ОН
so2
НС1
/Вг
^-AsC12
Характеристика основного сырья
о-Бромфениларсоновая кислота [3], т. пл. 198—199° (с
разл.).
Соляная кислота, ч., ГОСТ 857—57.
Сернистый ангидрид, технический.
Йодистый калий, ч., ГОСТ 4232—48-
Бензол, ч., ГОСТ 8448—61.
Петролейный эфир, т. кип. .40—60°.
43
Условия получения
Суспендируют 34 г (0,122 Л4) о-бромфениларсоновой кис-
лоты в 170 мл соляной кислоты (d=l,12) и добавляют 0,5 г
йодистого калия, при этом выпадает маслообразное вещест-
во красного цвета и раствор становится молочно-белым. В
реакционную смесь при комнатной температуре пропускают
сернистый ангидрид до тех пор, пока солянокислый раствор
не станет полностью прозрачным, а осадок почти не обесцве-
тится, на что обычно требуется 4—6 часов. Затем жидкость
декантируют, маслообразный осадок растворяют в 25—30мл
бензола и сушат безводным хлористым кальцием. После от-
гонки растворителя остаток перегоняют в вакууме, собирая
фракцию, с т. кип. 142—14878—10 мм и перекристаллизовы-
вают из петролейного эфира (около 100 мл).
Выход чистого продукта в виде белого мелкокристалличе-
ского порошка равен 20—21 г, что составляет 54—57% от
теоретического; т. пл. 62—64°.
По литературным данным, т, пл. о-бромфенилдихлорарси-
на 63° [1], 62—64° [2],
ЛИТЕРАТУРА
1. L. Kalb. Ann dev Chemie, 423, 68 (1921).
2. Н. Burton, С. S. Gibson. J. Chem. Soc., 1926, 457.
3. G. J о n s о n. J. Chem. Soc., 1927, 2506.
Поступила в апреле 1968 г.
ПРЕД
УДК 547.772.1
1-н-БУТИЛ-3,4,5-ТРИАЛКИЛПИРА30ЛЫ
В. М. ДЗИОМКО, О. В. ИВАНОВ
Rx CH2R
RCH3—%
Nz
I
CH2(CH2)2CH3
III R=h-C4H9
IV R=h-C5Hh
I r=c2h5
II R = h-C3H7
N-н-Бутильные производные пиразола могут быть полу-
чены при взаимодействии н-бутилгидразина с 1,3-дикетонами
[1 j или при алкилировании незамещенных у азота производ-
ных пиразола галоидным бутилом [2]-
Для получения ряда новых 1-н-бутил-3,4,5-триалкилпира-
золов нами применен метод алкилирования триалкилпиразо-
лов бромистым бутилом в присутствии углекислого калия в
среде диметилформамида.
СХЕМА СИНТЕЗА 1-Н-БУТИЛ-3,4,5-ТРИАЛКИЛПИРАЗОЛОВ
R CH2R
+ н-С.Н9Вг
I
H
R CH,R
—> К Crtg К N
N/
CH2(CH2)2CH3
45
Характеристика основного сырья
3,4,5-Триалкилпиразолы (см. одноименную статью в на-
стоящем сборнике).
Диметилформамид, ч., ВТУ РУ—1193—56.
Калий углекислый, ч., ГОСТ 4221—48.
Бромистый бутил, ч., ВТУ МХП 2783а—51.
Натр едкий, ч., ГОСТ 4328—48.
Хлороформ, ч., ГОСТ 3160—51.
Натрий сернокислый, безводный, ч., ГОСТ 4156—48.
Условия получения
В трехгорлую колбу, снабженную мешалкой, обратным
холодильником с хлоркальциевой трубкой и капельной во-
ронкой с хлоркальциевой трубкой, помещают 0,1 М 3,4,5-три-
алкилпиразоЛа, 50 мл сухого диметилформамида и 9,7 г
(0,07 А4) углекислого калия, а затем при постоянном переме-
шивании за 7—10 минут прибавляют 15,1 г (0,11 М) бромис-
того бутила.
Реакционную массу нагревают на глицериновой бане в
течение 2 часов при 85—90°, затем за 15 минут температуру
поднимают до 120—125° и продолжают нагревание при этой
температуре еще 2 часа. После охлаждения к реакционной
массе добавляют 70 мл 10%-ного раствора едкого натра,
смесь тщательно перемешивают и выделившийся органичес-
кий слой экстрагируют 100 мл хлороформа. Экстракт дваж-
ды промывают водой (по 50 мл) и сушат прокаленным суль-
фатом натрия. Растворители отгоняют, а остаток фракцио-
нируют в вакууме-
Таблица I
Соедине- I НИЯ Название ..Вы- ход, % Темпе- ратура кипения, °C (мм) rf20 „20 nD Спектры в циклогексане
' макс, НМ £макс
I 1-н-Бу тил-3,5-дипропил- 4-этилпиразол 85 143 (11) 0,9046 1,4759 227,5 7100
п 1-н-Бутил-3,5-дибутил-4- пропнлпиразол 82 156 (4) 0,8814 1,4734 227,5 6900
III 1-п-Бутил-3,5-днпен- тил-4-бутил пиразол 86 170—171 (5) 0,8727 1,4722 227,5 5700
IV 1-н-Бутил-3,5-дигексил-4- пентилпиразол 88 174-175 (2) 0,8712 1,4662 227,5 3600
46
Выходы, температуры кипения и физико-химические кон-
станты 1-н-бутил-3,4,5-триалкилпиразолов приведены в таб-
лице 1, анализы в таблице 2.
Таблица 2
Соеди- нения Найдено, % Формула М. в. Вычислено, %
С Н N С Н N
I 76,60 76,35 12,05 12,15 12,01 12,15 C15H28N2 236,40 76,21 11,94 11,85
II 77,81 77,52 12,20 12,17 9,87 9,93 ClgHgiNj 278,48 77,63 12,31 10,06
III 78,48 78,79 12,53 12,50 8,93 8,87 C2iH40N3 320,56 78,68 12,58 8,74
IV 79,62 79,70 12,55 12,68 7,83 7,55 C24H4fiN2 362,64 79,49 12,79 7,72
ЛИТЕРАТУРА
1. L. Knorr. Ann, 279, 232 (1894),
2. И. И. Г р а н д б с р г. ХГС, 1965 (2), 279.
Поступила в мае 1968 г.
ИРКА
УДК 547.563.161
А-втор -БУТИЛФЕНОЛ
И. С. МАРКОВИЧ, И. В. КРУГЛОВА, в. м. дзиомко
он
0
сн
H3CZ хс3нГ1
СюНиО М. в. 150,22
Основным методом получения 4-втор-бутилфенола явля-
ется алкилирование фенола вторичным или н-бутиловым
спиртом [1—4], галоидными алкилами [5], олефинами [6], ме-
тилэтилкетоном [7]. Во всех случаях в результате алкилиро-
вания получается смесь 2- и 4-изомеров. Последний выделя-
ют фракционной перегонкой. Однако фракционированием не
всегда удается выделить чистый 4-втор-бутилфе.нол. В боль-
шинстве работ описано получение 4-втор-бутилфенола в ви-
де жидкости, в то время как чистый продукт представляет
собой кристаллическое вещество с температурой плавления
58—59,5° [4].
Нами алкилирование фенола проведено1 н-бутиловым
спиртом в присутствии 85%-ной фосфорной кислоты по мето-
ду [2]. Для разделения изомеров мы использовали различ-
ную растворимость их кальциевых солей в воде. 4-втор-бу-
тилфенол выделяли в виде кальциевой соли, после разложе-
ния которой получали кристаллический продукт с выходом
20—25%.
СХЕМА СИНТЕЗА 4-втор -БУТИЛ ФЕНОЛА
ОН
\ Y + С4Н9ОН ^-3?--4)->
48
Характеристика основного сырья
Фенол, ч„ ГОСТ 6417—52.
н-Бутанол, ч., ГОСТ 6006—51.
Фосфорная кислота, ч.д.а., ГОСТ 6552—58.
Петролейный эфир, ч., МХТУ 279—59.
Соляная кислота, ч., ГОСТ 3118—46.
Кальций хлористый, кристаллический, ч.д.а., ГОСТ
4141—48.
Условия получения
Алкилирование фенола [2]. В трехгорлую колбу емкостью
250 мл, снабженную мешалкой, обратным холодильником
и капельной воронкой, помещают смесь 46 г (0,5 М) фенола
и 100 мл фосфорной кислоты (уд. вес 1,85) и по каплям при-
бавляют при размешивании 37 г (0,5 Л4) н-бутанола. Смесь
нагревают до 130° и продолжают размешивание в течение
4 часов. После охлаждения отделяют органический слой, про-
мывают его 5%-ным раствором соды до нейтральной реак-
ции, сушат прокаленным сульфатом натрия и перегоняют в
вакууме, собирая фракцию с т. кип. 110—120715 мм; —
1,5192—1,5200.
Выход продукта равен 46 г, что составляет 61,3%, считая
на загруженный фенол.
Выделение 4-втор-бутилфенола. В термостойком стакане
емкостью 250 мл растворяют 15 г полученного бутилфенола
в 120 мл 5 %-кого едкого натра, нагревают раствор до кипе-
ния, добавляют активированный уголь, кипятят в течение
5 минут, отфильтровывают от угля, разбавляют 150 мл воды
и приливают раствор 90 г хлористого кальция в 100 мл воды.
Выпавший зеленоватый осадок отфильтровывают, промыва-
ют водой и переносят в трехгорлую колбу, снабженную хо-
лодильником, мешалкой и капельной воронкой. Приливают
35—40 мл петролейного эфира и' прикапывают 18%-ную со-
ляную кислоту до pH 1—2- Смесь нагревают (размешивая)
при температуре 50° в течение 1 часа, до полного растворения
осадка, затем отделяют органический слой, охлаждают его
4 Зак. 1034 49
примерно до 0° и отфильтровывают выпавшие бесцветные
кристаллы 4-втор-бутилфенола; т. пл. 57°.
Выход равен 3 -ь- 3,4 г, что составляет 20—25%, считая на
загруженный бутилфенол.
После перекристаллизации из петролейиого эфира темпе-
ратура плавления 4-втор-бутилфенола 58—59°.
ЛИТЕРАТУРА
1. Англ. пат. 934282 (1963).
2. В. Тамбовцев а, И. Цукерваник. Ж- общ. химии, 15, 820
(1945).
3. R. С. Huston, R. L. Q u 11 е, D. L. Bailey, J. Amer. Chem.
Soc. 67, 899 (1945).
4. И. Ромадан, Е. А. Кочеткова. Ж. общ. химии, 34, 2767
(1964).
5. О. Б. Кучкаров. Ж. общ. химии, 21, 685 (1951).
6. С. В. Завгородний. Ж- общ. химии, 16, 1495 (1946).
7. Г. Н. Дорофеей ко, С. В. Кривун. Ж- общ. химии, 33, 2970
(1963).
ИРЕА
Поступила в апреле 1968 г.
УДК 547.565.2'292'283.2
ГИДРОХИ НОН-2,6-ДИМЕТИЛЕНИМИ НОД ИУКСУСНАЯ
КИСЛОТА
1,4-Диокси-2,6-бис-[К,Ы'-ди-( карбоксиметил)
аминометилфбензол
В. Я. ТЕМКИНА, Н. В. ЦИРУЛЬНИКОВА, М. Н. РУСИНА,
Р. П. ЛАСТОВСКИИ
ОН
НООСН2С, ! ,СН2СООН
XN — H2C-f >-СН, —N(
НООСН2СХ I J ' ХСН2СООН
он
С16Н2(№О10
М. в. 400,34
Гидрохинон-2,6-диметилениминодиуксусиая кислота яв-
ляется неописанным в литературе комплексоном и образует
хелатные соединения с рядом катионов.
Синтез комплексона осуществлен по реакции Манниха
взаимодействием полученной нами ранее [1] гидрохиионме-
тилеииминодиуксусной кислоты с формальдегидом и имино-
диуксусной кислотой в водно-щелочной среде. Получение его
непосредственно из гидрохинона не дает положительных ре-
зультатов вследствие окисления самого гидрохинона в ще-
лочной среде и вступления только одной иминодиацетатной
группы в кислой среде.
Продукт представляет собой белый порошок, плохо рас-
творимый в воде, нерастворимый в органических раствори-
телях, растворимый в щелочах при образовании мононатрие-
вой соли.
Физико-химическое исследование показало неравноцен-
ность окси-групп при комплексообразовании. На этом осно-
вании предложено указанное строение комплексона.
4* 51
СХЕМА СИНТЕЗА ГИДРОХИНОН-2,6-ДИМЕТИЛЕНИМИНОДИ-
УКСУСНОЙ кислоты
он
сн,соон ,СН3СООН ы „н
+ HN^ +СН3О^
СН2СООН ХСН3СООН НС1
он
ОН
НООСН2С\ 1 ,СН,СООН
>NHsC-f ||-СН2 -Nf +NaCl+H2O
HOOCH2CZ .. . , чснйсоон
он
Характеристика основного сырья
Гидрохинонметилениминодиуксусная кислота [1].
Иминодиуксусная кислота, ч., ВТУ МГУХП 190—58.
Формальдегид, техн., ГОСТ 1625—61.
Натр едкий, ч., ГОСТ 4328—48-
Условия получения
В трехгорлую колбу, снабженную механической мешал-
кой, обратным холодильником и капельной воронкой, поме-
щают 5,1 а (0,02 А!) гидрохинонметилениминодиуксусной ки-
слоты, смоченной 8 мл воды и растворенной-в 3,5 мл 30%-но-
го водного раствора едкого натра и 2,7 г (0,02 М) иминоди-
уксусной кислоты, предварительно нейтрализованной 30%-
ным раствором едкого натра до pH 7—8.
К охлажденной до 10° реакционной смеси добавляют по
каплям при перемешивании 1,6 г (0,02 Л1) 37%-ного раство-
ра формальдегида; размешивают при комнатной температу-
ре в течение 1 часа и при температуре 60—70° в течение 2 ча-
сов. Затем массу охлаждают и подкисляют концентриро-
ванной соляной кислотой до pH 1—2. Выпавший осадок от-
фильтровывают, промывают водой до отсутствия ионов хлора
(проба с AgN'Oa) и сушат в сушильном шкафу при темпера-
туре 50—60°.
Выход продукта равен 4 г, что составляет 50% от теоре-
тического, считая на гидрохинонметилениминодиуксусную
кислоту.
Найдено, %: С—47,3; 47,1; Н—5,2; 5,2; N—7,6; 7,4.
СюНго^Ощ. Вычислено, %: С—48,0; Н—5,0; N—7,0.
52
ЛИТЕРАТУРА
1. Н. В. Цирульников а, В. Я. Темкина, Р. П. Ластов-
< кий. Методы получения химических реактивов и препаратов, вып. 18,
М., ИРЕА, 1969.
Поступила в мае 1968 г.
ИРЕА
УДК 547.496.3
ГУАНИЛТИОМОЧЕВИНА
М. Ф. КОНДРАШОВА, Е. Я. ЯРОВЕНКО
H2N !
^C-NH-C-NH2- 4- (СООН)2
HN^ || 2
S
C2H0N4S-yCaH2O4 M. в. 163,19
По литературным данным, гуанилтиомочевину получают
при многодневном пропускании сероводорода в водный рас-
твор дициандиамида [1] или при нагревании тиофосгена с мо-
чевиной.12].
Нами проверен первый способ, уточнены условия проведе-
ния реакции, выделения и очистки продукта.
СХЕМА СИНТЕЗА ГУАНИЛТИОМОЧЕВИНЫ
H2N. ]
\C-NH-CN + H2S +-^-(СООН)2 ->
HN^ 2
H2N. J , x
>C-NH-C-NH2.^-I соон
HN^ II 2 \ A
s
Характеристика основвого сырья
Дициандиамид, ч., ТУ ГКХ № РУ 1837—62.
Соляная кислота, ч., ГОСТ 3118—46.
Пиридин, ч., ГОСТ 2747—44.
Щавелевая кислота, ч., ГОСТ 5873—51.
Сернистое железо, ч., МРТУ 6-09-1148-64.
54
Условия получения
В цилиндрический сосуд емкостью 200 мл, снабженный
термометром и трубкой для подачи сероводорода, загружают
12,6 г (0,15 Л4) дициандиамида и 100 мл воды. Раствор на-
гревают до 60—70° и при этой температуре пропускают се-
роводород в течение 26 часов при pH 2—3 (по универсальной
индикаторной бумажке); pH среды поддерживают щавеле-
вой кислотой, которую добавляют в реакционную массу в
случае необходимости. Из реакционной массы выпадает оса-
док. Его отфильтровывают, промывают водой и сушат при
температуре 65—70°.
Выход оксалата гуанилтиомочевины равен 10,4 г (42,5%).
После перекристаллизации из 100—150 мл смеси пиридина
и воды (1:1) получают 7,8 г продукта, что составляет 31,9%
от теоретического выхода.
В литературе не указаны выход и температура плавления
оксалатной соли гуанилтиомочевины. По нашим данным,
т. пл. вещества 185,5—186°.
Найдено, %: С—21,63; 21,70; Н—4,79; 4,74;
N—34,79 ; 34,75; S—19,10; 19,21.
C,H6N4S •'/> (COOH)q. Вычислено %: С—22 08; Н—4,29; S—19,63;
N—34,35.
ЛИТЕРАТУРА
1. Е. Bamberger. Вег. 16, 1459 (1883).
2. В. Rathke. Вег., 11, 962 (1880).
Поступили в марте 1968 г.
ИРЕА
УДК 547.496.3
N ГУАНИЛФОРМАМИДИНСУЛЬФИНОВАЯ КИСЛОТА
М. Ф. КОНДРЛШОВЛ, Е. я. ЯРОВЕНКО
H2N-
^C-N=C-NH2-H2O
НЬГ |
SO2H
C2H6O2N'4S-H2O M. b. 168,17
N-Гуанилформамидинсульфиновая кислота не описана в
литературе; она получена окислением щавелевокислой соли
гуанилтиомочевины перекисью водорода при pH 2—3.
Нами разработаны условия получения и выделения этого
соединения-
СХЕМА СИНТЕЗА
n-гуанилформамидинсульфиновои кислоты
H2N.
>C-NH-C-NH2 + 2 Н2О2 ->
HNk II
s
H,N.
)>C-N = C-NH2 +2H2O
HN/Z I
SO2H
Характеристика основного сырья
Перекись водорода, медицинская, ГОСТ 177—55.
Щавелевая соль гуанилтиомочевины, т- пл. 185,5°—186.
Условия получения
В двухгорлую колбу, снабженную механической мешал-
кой и термометром, загружают 0,49 г (0,003 М) щавелево-
•56
кислой соли гуанилтиомочевины и 50 мл воды. Нагревают до
60° и при этой температуре прибавляют 3 мл (0,03 М) 35 % -
ной перекиси водорода. Коней реакции определяют по исчез-
новению перекиси водорода (по йодкрахмальной бумажке).
Затем раствор упаривают досуха и осадок сушат при темпе-
ратуре 65—70°
Получают 0,26 г продукта', что составляет 51% от теоре-
тического выхода.
N-Гуанилформамидинсульфиновая кислота представляет
собой белое кристаллическое вещество с т. пл. 137—138°,
растворимое в воде, нерастворимое в бензоле, ацетоне, эти-
ловом спирте и диоксане.
Найдено, %: С—14,55; 14,22; Н—5,02; 5,05.
C2H6O2N4S.H2O. Вычислено, %: С—14,29; Н—4,80.
Поступила в марте 1968 г. ИРЕА
УДК 547.292'233.375
\,\-ДИЛЛКИЛАЦЕТЛМИДЫ
Н. А. ЕГОРУШКИНА, В. Я. ТЕМКИНА, Р. П. ЛАСТОВСКИИ
N.N-Диалкилацетамиды находят применение в качестве
экстрагентов при разделении ряда элементов [1]. По литера-
турным данным, эти соединения получают взаимодействием
хлорангидридов соответствующих кислот с вторичными ами-
нами [1], дикетонов с аминами [2], калийалкилсульфатов с
натрийалкиламидами [3].
Нами получены Н,М-дилаурилацетамид и N,N-ди (гекса-
децил) ацетамид взаимодействием уксусного ангидрида с
М,№дилауриламином и N.N-ди (гексадецил) амином соответ-
ственно.
N.N-ДИЛАУРИЛАЦЕТАМИД
Gj i Нг3 ч
СцН2з
N— СО СН3
c24h49no
М. в. 367,66
СХЕМА СИНТЕЗА N.N-ДИЛАУРИЛАЦЕТАМИДА
СцНоЗч
)NH+(CH3CO)2O->
СцН2з
СцН23
/N-COCHJ СН3СООН
cnHS3
Характеристика основного сырья
Дилауриламин.
Уксусный ангидрид, ч., ГОСТ 5815—52.
Условия получения
В трехгорлую колбу, снабженную мешалкой, термомет-
ром, обратным холодильником и капельной воронкой, загр-у-
58
жают 50 г (0,154 Л4) дилауриламина и при размешивании за
30 минут прибавляют из капельной воронки 63 ’а (0,61 М)
уксусного ангидрида. Затем реакционную массу нагревают
до кипения (140—143°) и выдерживают при этой температу-
ре в течение 3 часов. После выдержки в течение ночи реак-
ционную смесь переносят в колбу для отгонки и отгоняют
при 27—30°/4—5 мм избыток уксусного ангидрида. Остаток
перегоняют при 204—20672—3 мм и получают 44 г дилаурил-
ацетамида, что составляет 78% от теоретического выхода,
считая на дилауриламин.
Найдено, %: С—78,91; 78,98; Н—14,01; 13,86.
C24H49NO. Вычислено, %: С—78,47; Н—13,35.
N.N-ДИ (ГЕКСАДЕЦИЛ) АЦЕТАМИД
С]вН3з
;n-сосн3
С16Н83
C34H69NO М. в- 507,92
СХЕМА СИНТЕЗА М,\-ДИ(ГЕКСАД ЕЦИЛ)АЦЕТАМИДА
CieH33 Cj6H33 ч
;nh+(ch3co)zo-» )n-coch3+ch8cooh
С1вНЭ8' -
Характеристика основного сырья
Ди (гексадецил) амин.
Уксусный ангидрид, ч., ГОСТ 5815—52.
Ацетон, ч., ГОСТ 2603—63.
Условия получения
В трехгорлую колбу, снабженную мешалкой, термомет-
ром, капельной воронкой и обратным холодильником, загру-
жают 25 г (0,054 АЦ ди (гексадецил) амина и при работаю-
щей мешалке в течение 30 минут прибавляют из капельной
воронки 27,5 г (0,27 /И) уксусного ангидрида. Смесь кипятят
в течение 3 часов и оставляют на ночь.
После отгонки уксусного ангидрида в вакууме водоструй-
ного насоса при температуре не выше 50°, к остатку прибав-
ляют 500 мл ацетона. Полученный осадок отфильтровывают,
сушат в эксикаторе и получают 20 г ди(гексадицил)ацетами-
да, что составляет 72,4% от теоретического выхода, считая
на ди (гексадецил) амин.
Найдено, %: N—2,25; 2,56; С—80,44; 80,45; Н—13,52; 13,49.
C31H69NO. Вычислено, %: .У—2,76; С—80,45; Н—13,61.
59
ЛИТЕРАТУРА
1. Т. Н. Sid dal 1 III. J. Phys. Chem., 64, 12, 1863 (1960).
2. С. И. Бурмистров, E. А. Шилов. Ж- общ. химии, 16, 295
(1946).
3. А. Т i t h е г 1 е у. J. Chem. Soc., 79, 400 (1901).
Поступила в апреле 19§8 г.
ПРЕД
УДК 547.341
ДИ АЛ КИЛ (ПЕР ГАЛОИДИЗОПРОПЕНИ Л)ФОСФАТЫ
С. 3. ИВИН, В. К. ПРОМОНЕНКОВ, Е. А. ФОКИН
Диалкил (пергалоидизопропенил) фосфаты, получаемые
взаимодействием диалкилфосфитов [1] или триалкилфосфитов
[2, 3] с пергалоидацетонами могут быть использованы в син-
тезе фосфорорганических соединений и в качестве пестици-
дов.
ДИЭТИЛ-а-ДИФТОРХЛОРМЕТИЛ-р-
ФТОРХЛОРВИНИЛ ФОСФАТ
(C2H5O),P-OC=CFCI
“II I
О СР2С1
С7Н10С12ЕзО4Р
М. в. 317,27
СХЕМА СИНТЕЗА
ДИЭТИЛ-а-ДИФТОРХЛОРМЕТИЛ-0-ФТОРХЛОРВИНИЛФОСФАТА
(C2H6O),PH+CFsC1CCFC1, -> (C3H6O)2P-OC=CFC1
II II ‘ II I +НС1
О О • О CF2C1
Характеристика основного сырья
Диалкилфосфиты, получены взаимодействием треххлорис-
того фосфора со спиртами [4]:
Диметилфосфит, т. кип. 56—58°/10 мм; —1,1944; п2° —
1,4020;
Диэтилфосфит, т. кип. 71—72°/10 мм; dA°—1,0912; —
1,4080;
Диизопропилфосфит, т. кип. 78—80°/17 мм; riff — 1,4080.
Фторхлорацетоны, получены фторированием гексахлор-
ацетона трехфтористой сурьмой [5]:
61
Дихлортетрафторацетон (симм.), т. кип. 44°; d^20—1,4772,
п2^ —1,3420; а, а, а'-трифтор-а, а7, а'-трихлорацетон, т. кип.
84°; di20—1,5812; л™ —1,3820;
Дифтортетрахлорацетон, т. кип- 122°; %20—1,6349; пг£ —
1,4262.
Условия получения
В четырехгорлую колбу на 250 мл с мешалкой, термомет-
ром, обратным холодильником и капельной воронкой с хлор-
кальциевыми трубками помещают 107,7 г (0,5 А4) а, а, а'-
трифтор-а, а', а'-трихлорацетона. При перемешивании до-
бавляют по каплям 69 г (0,5 А4) диэтилфосфита, поддержи-
вая температуру реакционной массы за счет выделяющегося
тепла реакции в пределах 60—70°. Перемешивание продол-
жают 4 часа при комнатной температуре, после чего реакци-
онную массу перегоняют в вакууме.
Получают 106 г (67%)диэтил-ге-дифторхлорметил-р-фтор-
хлорвинилфосфата, т. кип. 71—72°/1 мм; di20—1,4161; nf® —
1,4172- По внешнему виду продукт представляет собой бес-
цветную, подвижную жидкость, устойчивую при хранении,
водой быстро гидролизуется.
Аналогичным методом могут быть получены и другие про-
дукты. Некоторые из них представлены в таблице.
ЛИТЕРАТУРА
1. С. 3. Ивин, В. К. Промоненков. Авт. свид. 187787 (1966);
Бюлл. изобр., № 21 (1966).
2. W. Perkow. Вег. 87, 755 (1954).
3. J. Allen, О. Johnson. J. Amer. Chem. Soc., 77, 2871 (1955).
4. H. Mocembie, В. Saunders, G. Stacey. J. Chem. Soc.,
1945, 380.
5. B. Saunders, G. Stacey. J. Chem. Soc., 1948, 1773.
Поступила в декабре 1967 г.
УДК 547.341
ДИАЛ КИЛ (ГЕКСАФТОРИЗОПРОПИЛ) ФОСФАТЫ
С. 3. ИВИН, В. к. ПРОМОНЕНКОВ, Е. А. ФОКИН
Ранее неизвестные .диалкил(гексафторизопропил)фосфаты
получены нами взаимодействием диалкилфосфитов с гекса-
фторацетоном [1]. Они могут быть использованы в синтезе
фосфорсодержащих веществ.
ДИЭТИЛ (ГЕКСАФТОРИЗОПРОПИЛ) ФОСФАТ
(C2H5O)2P-OCH(CF3)2
II '
О
C7H11F6O4P М. в. 304,12
СХЕМА СИНТЕЗА
ДИЭТИЛ (ГЕКСАФТОРИЗОПРОП ИЛ )ФОСФАТА
(С2Н5О)2РН + O = C(CF3)2 (C2H6O)2P-OCH(CF3)2
Характеристика основного сырья
Диэтилфосфит, получен взаимодействием треххлористого
фосфора с этиловым спиртом; т. кип. 71—72710 мм; d420—
1,0912; —1,4080 [2].
Гексафторацетон, получен дегидратированием его гидра-
та серной кислотой; т. кип. 28° [3].
Условия получения
В четырехгорлую колбу емкостью 250 мл с мешалкой
термометром, холодильником и барботером помещают 69
(0,5 А1) диэтилфосфита и при перемешивании пропускаю
64
o' с о U-t 5 34,38
И 3 ы вычисл | d 06*8 9,33 1
А и а л о CU 41,47 41,45
X <и 5S СО ж Cl 9,13 9,53 ।
йа с ; 1,3300 1,3513
с- **3 1,5133 1,2996
Т. кип, Смм QO o" CO О Ю 44—45/1
Выход, % 63 ' 61
Соединения Диметил(гексафторизопропил)-фосфат (CH3O)3P-O CH(CF3)2 II 0 C6H7F6O4P M. b. 276,07 Диизопропил(гексафторизопропил)-фосфат ' <M «» p- o X о 1 о 1 d r 4 о s QO oi 00 oo 35 a. 4 П- r J
4L>
X
и
5 Зак. 1034
65
91,4 г (0,55 Л1) гексафторацетона, поддерживая температуру
реакционной массы за счет выделяющегося тепла в пределах
50—60° (см. примечание). Перемешивание продолжают 4 ча-
са при комнатной температуре и затем реакционную массу
перегоняют в вакууме.
Получают 113 г (78%) диэтилгексафторизопропилфосфа-
та с т. кип. 71— 73°/10 мм; d4Z0—1,3233; —1,3585. По
внешнему виду это бесцветная жидкость, устойчивая при хра-
нении-
Найдено, %: Р—10,50; F—37,56.
С7НцР6О4Р. Вычислено, %: Р—10,40; F—37,50.
Аналогичным путем можно получить другие гексафторизо-
пропилфосфаты. Свойства некоторых из них представлены
в таблице.
ЛИТЕРАТУРА
1. С. 3. Ивин, В. К. Пром опенков. Е. А. Фокин.. Ж. общ.
химии, 2511 (1967).
2. Н. М с С о tn b i е, В. С. S a u n d е г s, G. Stacey. J. Chem. Soc.,
1945, 380.
3. T. Brice, J. L a z c r t e, W. Pear Ison. J. Anter. Chem. Soc.
75, 2698 (1953).
Поступила в декабре 1967 г.
УДК 547.733
2,5-ДИАЦЕТИЛТИОФЕН
С. В. ЦУКЕРМАН, В. М. НИКИТЧЕНКО, ЛАМ НГОК ТХИЕМ,
В. Ф. ЛАВРУШИН
НзС-СО-^ Leo-СНз
S7
CgHsOaS М. в. 168,21
При непосредственном ацетилировании тиофена или
2-ацетотиенона выход 2,5-диацетилтиофена, согласно лите-
ратурным данным [1, 2], составляет 5—6%. Однако, как по-
казали наши исследования, в этом случае выход 2,5-диаце-
тилтиофена, как правило, колеблется в пределах от 0,5 до
2%, причем продукт получается загрязненным и дальнейшая
очистка его связана с дополнительными потерями.
Предлагаемая нами методика синтеза 2,5-диацетилтио-
фена окислением 5-этил-2-ацетотиенона в нейтральной среде
позволяет получать 2,5-диацетилтиофен со стабильным выхо-
дом (10%), считая на 2-ацетотиенон.
СХЕМА СИНТЕЗА 2,5-ДИАЦЕТИЛТИОФЕНА
кон
СО—СН8 + NH2-NH»H2O-------
S
i| !| (СНзСО)2О
НеСп \ /
Xsz Mg(C104)2
Н,сД
s
H3C-CO-'< co—сн3
s
5*
67
Характеристика основного сырья
2-Ацетотиенон, т. кип. 89—91°/9 мм, уд. в., 1,168; п'£ —
= 1,5666.
Гидразин-гидрат, ч.д.а., ГОСТ 5832—51.
Уксусный ангидрид, ч.д.а., ГОСТ 5815—52.
Магний хлорнокислый, безводный, ч., ВТУ МХП 3139—57-
Окись магния, ч.д.а-, ГОСТ 4526—48.
Азотная кислота, х.ч., ГОСТ 4461—48.
Условия получения
Синтез этилтиофена. В круглодонную колбу емкостью
6,5 л, снабженную обратным холодильником, помещают 40 г
едкого кали, 280 мл диэтиленгликоля, 100 мл гидразин-гид-
рата, 52 г 2-ацетотиенона и кипятят в течение 45 минут. За-
тем заменяют обратный холодильник на нисходящий и от-
гоняют образовавшийся этилтиофен, собирая фракцию, кипя-
щую при 132—137°. При вторичной перегонке собирают фрак-
цию, кипящую при 133—134°; (<№—0,99; п®—1,5122).
По литературным данным, т. кип. продукта 134—135° [3]
и 133—134,5° [4].
Выход этилтиофена равен 40 г, (85%).
Синтез 5-этил-2-ацетотиенона. В круглодонную колбу> ем-
костью 0,25 л, снабженную обратным холодильником, поме-
щают 11,2 г (0,1 7И) 2-этилтиофена, 15,3 г (0,15 АГ) уксусно-
го ангидрида, 0,45 г (0,02 М) безводного перхлората магния
и кипятят на водяной бане 30—45 минут. После охлаждения
до 50° смесь выливают в 100 мл холодной воды, органический
слой отделяют, а водный экстрагируют эфиром. Органический
слой, объединенный с эфирным экстрактом, тщательно про-
мывают водой, затем насыщенным раствором соды и сушат
безводным сульфатом натрия.
После отгонки растворителя, фракционируют в вакууме,
собирая фракцию, кипящую при 120—122°/12 мм [5] (и;,0 =
= 1,5516).
Выход 5-этил-2-ацетотиенона равен 12,3 г, что составляет
80% от теоретического.
Получение 2,5-диацетилтиофена [6]. В трехгорлую колбу ем-
костью 1 л, снабженную механической мешалкой, термомет-
ром и капельной воронкой, помещают 8 г окиси магния,
240 мл воды, 28 мл азотной кислоты (уд. в. 1,4) (среда
должна быть нейтральной но лакмусу) и 32 г перманганата
калия. Смесь нагревают на водяной бане при 60° до полного
растворения окиси магния и перманганата калия, затем ох-
лаждают до 35° и при энергичном перемешивании из капель-
ной воронки медленно добавляют 16 г 5-этил-2-ацетотиенона.
По окончании добавления продолжают перемешивание еще
68
2,5 часа при той же температуре. Образовавшуюся двуокись
марганца отфильтровывают и осадок на фильтре трижды
промывают горячим этилацетатом (по 30 мл). Органический
слой отделяют, а водный дважды экстрагируют этилацета-
том.
Остаток после отгонки растворителя перекристаллизовы-
вают из этилацетата (1:10) с применением активированного
угля.
Получают бесцветные пластинки с т. пл. 172°. Выход
2,5-диацетилтиофепа равен 2,8 г, что составляет 15% от тео-
ретического-
По литературным данным, т. пл. продукта 172—173° [1].
ЛИТЕРАТУРА
1. Н. D. Н а г t о u g h, А. I. К о z a k, J. Amer. Chem. Soc., 69, 1012
(1947).
2. Е. Н. Hadley, J- Goggin, C..Habertnan. Trans Illinlos
State. Acad. Set, 47, 89 (1955).'
3. N. P. В u u.-H о 1, N. G. H о a n, N. К. К h о i, J. Organ Chem., 15.
959 (1950).
4. W. I. King, F. F. Nord. J. Organ. Chem., 14, 639 (1949).
5. D. Howard, H. D. H a r t oug h. J. Amer. Chem. Soc., 73, 4033
(1951).
6. С. В. Цукерман, В. M. Никитченко, В. Ф. Лаврушин,
Лам Нгок Т х и е м. Авт. свид. 202174; Бюлл. изобр., 19 (1967).
Поступила в ноябре 1967 г.
Харьковск. Гос. ун«г. им. А. М. Горького
УДК 547.558.1'461.3
ДИ [(|3-ДИФЕНИЛФОСФИНИЛ)ЭТИЛ]МАЛОНОВАЯ
КИСЛОТА
Т. М. БАЛАШОВА, Ю. М. ПОЛИКАРПОВ, Т. Я. МЕДВЕДЬ
соон
С6нг I ,свн5
>р-сн2-сн2-с-сн.>-сн2-р(
cchZ|| I ' ||ХС6Н5
о соон о
Сз1НзоОеР2
М. в. 560,52
По литературным данным. ди-{(|3-дифенилфосфинил) -
этил]малоновую кислоту получают омылением ди-[(Р-дифе-
нилфосфинил)этил]малонового эфира раствором едкого кали
[1]. Нами проверен и уточнен этот метод.
СХЕМА СИНТЕЗА
ДИ-КР-ДИФЕН ИЛ ФОСФИНИЛ )ЭТИЛ]МАЛОНОВОй кислоты
2(СвН5)2Р-СН=СН2 + СН2(СООС2Н,5)2
СООС2Н5
I
(СВН6)2Р-СН—СН2-С-СН2-СН2-Р(С|;Н02
I! I II '
О СООС2Н5 о
СООС2Н3
I кон
(С6Н5)гР-СН2- СН2-С-СН2-СН2-Р (С«Н5)2---
II I II
О СООС2Н5 О
70
соон
- (С,,Н5)2Р-СН2-СН2-С-СН2-СНа-Р(С6Н5)3
о соон о
Характеристика основного сырья
Окись винилдифенилфосфина, т. кип. 179—18Г/1 мм, т.
пл. 116—117° (из бензола) [2].
Малоновый эфир, т. кип. 199,3°; d420—1,055; п?£ _[ 4162
ТУ ГКХ ОРУ 126—59.
Натрий металлический, ТУ МХП 1664—50.
Соляная кислота, х. ч., концентрированная, ГОСТ 3118—
46.
Бензол, ч-д.а., ГОСТ 5955—51.
Кали едкое, х.ч., ГОСТ 4203—48.
Этиловый спирт, ректифицированный, ГОСТ 5962—51.
Вода дистиллированная, ГОСТ 6709—53.
Условия получения
Синтез ди-[(^-дифенилфосфинил)этил]малонового эфира.
В четырехгорлую колбу, снабженную мешалкой, термомет-
ром, обратным холодильником и тубусом для подачи азота
•см- примечание), помещают 5,3 г (0,023 М) окиси винилди-
фенилфосфина, 1,8 г (0,011 /И) малонового эфира и 35 м г
бензола. В полученный раствор вносят 0,023 г металлическо-
го натрия и перемешивают 4 часа при кипении бензола. За-
тем растворитель удаляют, остаток перекристаллизовываю г
из бензола и получают 3,3 г ди-[(|3-дифенилфосфинил)этил]
малонового эфира, что составляет 45% от теоретического;
т. пл. 183—184°.
Найдено, %: С -68,2; 68,0; Н—6,3; 6,3; Р—10,2; 10,0.
Сз5Н3вО6Р2. Вычислено. %: С—68.2; Н—6,2; Р—10,0.
Получение ди-[($-дифенилфосфинил)этил\малоновой кис-
лоты. В одногорлую колбу помещают 3,3 г (0,00495 М)
ди-[(0-дифенйлфосфинил)этил]малонового эфира, приливают
раствор 1,7 г (0,03 Af) едкого кали в 8 мл воды, смесь нагре-
вают- на кипящей водяной бане 1 час. По охлаждении при-
бавляют 4 мл Концентрированной соляной кислоты (до кис-
лой реакции по универсальной индикаторной бумаге). Выде-
лившиеся кристаллы отфильтровывают, промывают водой и
перекристаллизовывают из спирта.
Выход ди-[(р-дифенилфосфипил)этил]малоновой кислоты
равен 2,4 г, что составляет 80% от теоретического; т. разл.
71
выше 180°. Эквивалент нейтрализации: найдено — 279; вычи-
слено — 280-
Найдено, %: С—66,9; 66,9; Н—5,6; 5,9; Р—11,0; 11,0.
Сз1Нзо06Рг- Вычислено, %; С—66,4; Н—5,4; Р—11,0.
Примечание.
Синтез [(0-дифенилфосфинил)этил]малоиового эфира проводят в токе
сухого азота.
ЛИТЕРАТУРА
1 .М. И. Кабачник, Т. Я. Медведь, Ю. М. Поликарпов,
К. С. Юдина. Изв. АН СССР, Отд. хим. н„ 1962, 1584.
2 . М. И. Кабачник, Т. Я. Медведь, К). М. Поликарпов,
К. С. Юдина. Изв. АН СССР, Отд. хим. н., 1961, 11, 2029.
Поступила в апреле 1968 г.
ИРЕА
УДК 547.657
ДИ ИМИД НАФТАЛИН-1,4,5,8-ТЕТРАКАРБОНОВОЙ
КИСЛОТЫ
М. Т. РАЗУМОВСКАЯ, А. И. БЕЛЯКОВА, Л. А. ЕГОРОВА
NH
О=С С=О
о-с с=о
NH
CuH6N2O4
М. в. 266,21
Диимид нафталин-1,4,5,8-тетракарбоновой кислоты приме-
няется в органическом синтезе и для получения полимеров.
Он образуется при действии водного раствора аммиака на
диангидрид нафталин-1,4,5,8-тетракарбоновой кислоты [1, 2,
3].
Нами разработан новый метод получения диимида нафта-
лин-1,4,5,8-тетракарбоновой кислоты, основанный на взаимо-
действии нафталин-1,4,5,8-тетракарбоновой кислоты, с моче-
виной. Предложенный метод упрощает синтез и дает высокий
выход продукта.
СХЕМА СИНТЕЗА
ДИИМИДА НАФТАЛИН-1,4,5,8-ТЕТРАКАРБОНОВОЙ КИСЛОТЫ
ноос соон
/NH2
+2 со;
XNH3
ноос соон
NH
o=cf c=o
I II I 4-2C02-|-2H,O
ч/w
I i
O==c c=o
\н
Характеристика основного сырья
Нафталин-1,4,5,8-тетракарбоновая кислота, предваритель-
но очищенная переосаждением технического продукта, 80% -
пая (см. примечание).
Аммиак, ч.д.а., 25%-ный водный раствор, ГОСТ З^бО—64.
Ацетон, ч.д.а., ГОСТ 2603—63.
Мочевина, ч., ГОСТ 6691—53.
Условия получения
В круглодонную, длинногорлую колбу емкостью 250 мл
загружают растертую и тщательно перемешанную в ступке
смесь 20 г (~0,06 Л4) чистой нафталин-1,4,5,8-тетракарбоно-
вой кислоты и 20 г (0,3 М) мочевины, нагревают -ее до 130—
135° (температура бани 140—150°) и выдерживают при этой
температуре в течение одного часа. Затем реакционную мас-
су охлаждают до 18—20°, прибавляют 250 мл 12,5%-ного
водного раствора аммиака и оставляют на ночь. На следую-
щий день содержимое колбы переносят в химический стакан
и перемешивают в течение 1,5 часа. Образовавшийся осадок
отсасывают на фарфоровой воронке через двойной (бумаж-
ный и бязевый) фильтр, промывают на фильтре дистиллиро-
ванной водой до нейтральной реакции по универсальной ин-
дикаторной бумаге, затем ацетоном (30 мл) и сушат при
80° до постоянного веса.
Выход продукта равен 16—16,5 г, что составляет 91,4—
94,2% от теоретического, считая на нафталин-1,4,5,8-тетра-
карбоновую кислоту.
Найдено, %: С—63,48; 63,57; Н—2,73; 2 ,74; N—10,92; 10,98,
Ci4H6N2O4. Вычислено, %: С—63,16; Н—2,63; N—10,52.
Диимид нафталин-1,4,5,8-тетракарбоновой кислоты пред-
ставляет собой мелкокристаллическое вещество желтоватого
цвета, плохо растворимое в органических растворителях и з
воде. При нагревании его выше 270° образуется сублимат из
блестящих желтых иголок.
74
Примечание.
Чистая нафталин- 1,4,5,8-тетракарбоновая кислота получена из техни-
ческого 80%-ного продукта многократным переосаждением, которое про-
изводилось посредством растворения вещества в растворе едкого натра,
обработки углем (марки «Бау»), фильтрования п подкисления фильтрата
< ('рной или соляной кислотой.
ЛИТЕРАТУРА
1. Е. Bamberger, М. Philip. Ann., 240, 147 -188 (1887).
2. М. Frennd. К. Fleischer. Ann., 402, 74 (1913).
3. М. Freund, К. Fleischer. Ann., 399, 224 (1913).
Поступила в феврале 1968 г.
ИРЕА
УДК 547.723
2,5-ДИ-(ОКСИМЕТИЛ)ТЕТРАГИДРОФУРАН
Н. Н. ШМАГИНА, В. И. ФЕДОТОВА, А. И. КУРАШЕВА
Н2С—СН2
i I •
НОН2С-НС СН-СНаОН
СбН12О3 М. в. 132,15
2,5-Ди-(оксиметил) тетрагидрофуран получают каталити-
ческим восстановлением 5-(оксиметил) фурфурола [1]. Нами
уточнены условия получения 2,5-ди (оксиметил)тетрагидро-
фурана из технического 5-(оксиметил) фурфурола.
СХЕМА СИНТЕЗА
2,5-ДИ-(ОКСИМЕТИЛ)ТЕТРАГИДРОФУРАНА
НС—сн „ Н2С-СН3
II II II
НОН8С-С СНО НОН2С-НС СН-СН2ОН
чох xoz
Характеристика основного сырья
5-(Оксиметил)фурфурол (см. одноименную статью в на-
стоящем сборнике).
Водород, ГОСТ 3022—45.
Сплав Ni-AI.
Спирт этиловый, абсолютный, ГОСТ 5962—51.
Натр едкий, ч., ГОСТ 4328—48.
Условия получения
В литровый качающийся автоклав, снабженный карманом
для термопары и манометром, помещают 30 г 88%-ного сы-
76
рого 5-(оксиметил) фурфурола и 12 г никель-скелетного ка-
тализатора (см. примечание). Автоклав двукратно продува-
ют водородом, затем давление водорода доводят до 100 атм.
Нагревают содержимое автоклава до 150—160°, выдержива-
ют при этой температуре 10 часов, автоклав охлаждают до
комнатной температуры, спускают давление, сливают раствор
и отфильтровывают катализатор. От фильтрата отгоняют рас-
творитель и получают 2,5-ди-(оксиметил) тетрагидрофуран с
т. кип. 134—13673 мм, п™ —1,4750-
Выход равен 24 г, что составляет 67% от теоретического.
По литературным данным [1], п-^ —1,4766.
Примечание.
Кусочки сплава Ni-Al размером 1—3 мм обрабатывают 25%-ным рас-
твором едкого натра. Выщелачивание 100 г сплава проводят в трехлиг-
ровой колбе, снабженной холодильником, термометром и капельной во-
ронкой, из которой постепенно к сплаву добавляют раствор щелочи. Че-
рез верхнюю часть холодильника отводится водород. В начале процесса
температура 30—35°, постепенно ее повышают до 90—95°. Катализатор
промывают горячей дистиллированной водой до слабощелочной реакции
по фенолфталеину и хранят под спиртом (ввиду его пирофорности в су-
хом состоянии).
ЛИТЕРАТУРА
1. А. С. Соре, W. N В а х t е г. J. Amer. Chem. Soc., 77,393(1955)
Поступила в сентябре 1967 г.
НИИПластмасс
УДК 547.724.2
5-(ДИМЕТИЛАМИНО)ФУРФУРОЛ
3. Н. НАЗАРОВА, В. С. ПУСТОВАРОВ
(CH3)2N-\ /-СНО
c7h9no2
М. в 139,16
Известные в литературе способы получения 5-(диметил-
амино) фурфурола позволяют синтезировать продукт сравни-
тельно просто и с хорошим выходом (до 80%) [1, 2]. Однако
получаемый при этом альдегид недостаточно чист и потери
его при очистке составляют 30—40%.
Нами разработана методика получения 5- (диметиламп-
но)фурфурола из 5-йодфурфурола. Синтез в 2 стадии с выде-
лением и перекристаллизацией йодида 5-(диметиламино)-
фурфурилидендиметиламмония, обладающего гораздо мень-
шей растворимостью в спирте, чем соответствующий бромид,
позволяет повысить выход продукта на 5—10%. Аминоаль-
дегид, образующийся при разложении йодида, обладает вы-
сокой чистотой и не требует дополнительной очистки.
СХЕМА СИНТЕЗА 5-(ДИМЕТИЛАМИНО)ФУРФУРОЛА
-> (CH3)3N-^ JLcH=N(CH3)2J
(CHs)2N~t J'-CHO
78
Характеристика исходного сырья
5-Йодфурфурол, т. пл. 126°, получают по методике [3].
Диметиламин, 33%-ный водный раствор, ТУ МХП 2645—
51.
Натр едкий, ч., ГОСТ 4328—48-
Спирт этиловый, ректифицированный, ГОСТ 5962—51.
Кали едкое, х.ч., ГОСТ 4203—48.
Кальций хлористый, плавленый. ГОСТ 4460—48.
Вода дистиллированная, ГОСТ 6709—53.
Условия получения
В круглодонную колбу емкостью 200 мл помещают 22,2 г
(0,1 М) 5-йодфурфурола и 30 мл спирта. Затем нагревают
до 50° и порциями (по 3—4 мл) прибавляют 40 мл 50%-ногэ
спиртового раствора диметиламина (приготовленного насы-
щением абсолютного спирта газообразным диметиламином,
полученным нагреванием водного раствора и высушенным
пропусканием над твердым КОН и плавленым хлористым
кальцием), строго следя при этом, чтобы температура смеси
не поднималась выше 55°. После прибавления диметиламина,
на что требуется около 20 минут, раствор выдерживают в те-
чение 10 минут при 55°, далее нагревают до 70° и охлаждают
до минус 10°. Выпавшие кристаллы отделяют, промывают ох-
лажденным спиртом (10 мл) и высушивают.
Выход йодида 5-(диметиламйно)фурфурилидендиметил-
аммония равен 28 г, что составляет 95% от теоретического.
Полученный йодид перекристаллизовывают из минималь-
ного количества этилового спирта, помещают в колбу, до-
бавляют 50 мл воды и прибавляют порциями 25 мл свеже-
приготовленного 40%-ного раствора едкого натра, энергично
встряхивая содержимое колбы. Образующееся при этом мас-
ло отделяют и помещают в чашку Петри. При стоянии (10—
20 минут) масло закристаллизовывается (иногда кристалли-
зация начинается до разделения смеси, в растворе). Кри-
сталлы отделяют, промывают 3—4 раза ледяной водой и вы-
сушивают в эксикаторе над едким кали.
Выход 5-(диметиламино) фурфурола равен 9,3 г, что со-
ставляет 94% от теоретического; продукт представляет белые
кристаллы с т. пл. 75° (из бензола).
Найдено, %: С—60,24; Н—6,64; N—9,87.
C7H9NO3. Вычислено, %: С—60,40; Н—6,52; N- 10,06.
ЛИТЕРАТУРА
1.3. Н. Назарова, В. Н. Новиков. Авт. свид. 170528; Билл,
изобр., 9, (1965).
79
2. 3. Н. Назарова, В. Н. Новиков, В. С. Пустоваров. Ж.
орг. химии, П, 161 (1966),
. 3. 3. Н. Назарова. Ж. общ. химии, 25, 539 (1955).
4. 3. И. Назарова, В. С. Пустоваров. Авт. свид. 185931; Бюлт
изобр., 18 (1966).
Поступила в декабре 1967 г.
Ростовск. н/Д Гос. ун-т
УДК 547.415.1
РДИМЕТИЛАМИНОЭТИЛАМИН
II. М. МОРЛЯН, Ж- л. БАГРАТУНИ
C4H12N2
сн3.
j:NCH2CHaNH2
сн3х
М- в. 86,13
Известны способы получения р-диметиламиноэтиламина
из р-диметиламиноацетонитрила [1—5], из гидробромида
2-бромэтиламина [6], этиленимина [7, 8], диметилглицинонит-
рила [9, Ю] и из N-0-диалкиламиноэтилфталимнда [11, 12].
Й американском патенте [13] приводится способ получе-
ния 0-диметиламиноэтнламина реакцией соли 0-галоидэтил-
амина с водным раствором диметиламина в присутствии ед-
кого натра при 0—50°.
Нами воспроизведен этот метод, причем несколько изме-
нены и усовершенствованы отдельные стадии процесса.
СХЕМА СИНТЕЗА (3-ДИМЕТИЛАМИНОЭТИЛАМИНА
HOCH2CH2NH2+HC1->HOCH2CH2N'H2-HC1
hoch2ch2nh2-hci+soci2—
C1CH2CH2NH2-HC1 + SO2+HC1
ClCH2CHaNH2-HCl + HN(CH3)2
-*(CH3)2NCH2CH2NH24-NaCl + HaO
Характеристика основного сырья
Моноэтаноламин, ч., МРТУ 6-09-3909-67.
Тионилхлористый, ч., МРТУ 6-09-2602-65-
Диметиламин, ч., ТУ МХП 2645—51.
6 Зак. 1034 81'
Этиловый спирт, ректифицированный, ГОСТ 5962—51.
Бензол, ч„ ГОСТ 5955—51.
Натр едкий, ч„ ГОСТ 4328—48.
Условия получения
Синтез гидрохлорида моноэтаноламина. В плоскодонную -
колбу емкостью 250 мл помещают 50 г моноэтаноламина и
100 мл этилового спирта. Содержимое колбы размешивают,
после чего пропускают сильный ток хлористого водорода (см.
примечание 1) в течение 5 часов (см. примечание 2). Образу-
ется осадок моноэтаноламин гидрохлорида в виде белых
игольчатых кристаллов. Фильтруют на воронке Бюхнера,
тщательно отсасывают и сушат в вакуум-эксикаторе 1 (см.
примечание 3).
Выход 50 г (52% теоретического).
Получение гидрохлорида &-хлорэтиламина. В сухую четы-
рехгорлую колбу емкостью 1 л, снабженную мешалкой, тер-
мометром, капельной воронкой и обратным холодильником с
хлоркальциевой трубкой, помещают 97,5 г (1 А4) гидрохло-
рида моноэтаноламина и постепенно через капельную ворон-
ку при охлаждении добавляют раствор 119 г (1 М) хлорис-
того тионила в 100 мл сухого бензола, при этом температура
в колбе не должна превышать 5°. После окончания прибав-
ления хлористого тионила реакционную массу перемешива-
ют 3 часа при 30°, непрореагировавший хлористый тионил и
растворитель удаляют слабым вакуумом. Оставшийся гид-
рохлорид p-хлорэтиламина кристаллизуют из смеси 100 мл
абсолютного этилового спирта и 50 мл диэтилового эфира,
перекристаллизовывают из охлажденного абсолютного эти-
лового спирта и сушат в вакуум-эксикаторе над безводным
хлористым кальцием.
Выход чистого продукта с т. пл. 145° составляет 90 г
(71,4% от теоретического).
Получение fi-диметиламиноэтиламина. К НО г 30—33%-
ного водного раствора диметиламина при 20° и перемешива-
нии добавляют в течение 30 минут 68 г гидрохлорида р-хлор-
этиламина, нагревают 15 минут при 35° и 3 часа при 62—65°.
Затем смесь охлаждают до 20—30° и в течение 30 минут при-
бавляют 125 г едкого натра, перемешивают еще 30 минут и
оставляют на ночь при комнатной температуре. Образовав-
шийся верхний слой отделяют, сушат едким натрием и пере-
гоняют, собирая фракцию с т. кип. 103—108°.
Выход чистого р-диметиламиноэтиламина равен 35 г, что
составляет 75% от теоретического; п'£ —1,4250; df0—0,8165.
По литературным данным температура его кипения
103—105° [1]; 105—108° [2]; 107° [9]; /г® —1,425 [6].
Найдено, %: N—31,58; 31,60.
C4H12N2. Вычислено, %: N—31,76.
82
Примечания:
1. Нельзя допускать сильного нагревания смеси, время от времени
колбу следует охлаждать холодной водой.
2. При более длительном пропускании хлористого водорода выход
гидрохлорида моноэтаноламина практически не увеличивается.
3. Гидрохлорид моноэтаноламина очень гидроскопичен, необходимо
по мере возможности сократить время соприкосновения его с воздухом.
ЛИТЕРАТУРА
I. В. Г. Я ш у н с к и й. Ж- общ. химии 33, 192 (1963).
2. R. A. Turner J. Amer. Chem. Soc.. 68, 1607 (1946).
3. A. Lespagnol a. oth. Bull Soc. chim. Fr., 1960, 833.
4. Hou ben - Weil, V. Xlll, 1957, 563.
5. Пат. ФРГ, 1103937; РЖХим, 1962, 20Л183.
6. H. M. Woodburn, R. С. 0‘. Gel. J. Organ. Chem., 17, 1235
(1952).
7. Г. И. Б p а з, В. А. Скородумов. Докл. АН СССР, 59, 489
(1948).
8. А. Г. Кау и др. Can. J. Chem., 34, 1567 (1956).
9. R. В а 11 z 1 у. J. S. В и с k, W. S. I d е. J. Amer. Chem. Soc., 64,
2232 (1942).
10. Brann. Вег., 40. 3937 (1907).
11. Пат. ГДР, 17068, РЖХим., 1961, 5Л112.
12. W. Voigtlander, Chemische Technik, 11, 324 (1959).
13. Пат. США, 2908714; РЖХим., 1961, 5Л314.
Поступила в мае 1968 г. ЦЗЛ
6*
УДК 547.732
2,4-ДИМЕТИЛТИОФЕН
И. М. МОРЛЯН, ж. л. БАГРАТУНИ, Г. В. БАДАСЯН
Н3С-С-СН
II II
НС С—СН3
V
C6H8S М. в. 112,19
2,4-Диметилтиофен получают взаимодействием а-метил-
левулиновой кислоты с пятисернистым фосфором.
а-Метиллевулиновую кислоту, в свою очередь, можно ПО'
лучить из ацетоуксусного эфира [1—3]-
Нами уточнены и проверены условия получения 2,4-диме-
тилтиофена, исходя из ацетоуксусного эфира.
СХЕМА СИНТЕЗА 2,4-ДИМЕТИЛТИОФЕНА
СН3СОСН2СООС2Н6 ——------> [CH3COCHCOOC,H5]-Na+
j 25 С2Н5ОН 1 ‘
[CH3COCHCOOC2Hs]~Na+ + BrCHCOOC2H5 ->
СНз
— CHSCOCHCOOC2HS
I
CH3CHCOOCaH5
CH3COCHCOOC2H5
I - 6 (НС1)
СН3СНСООС2Н6
— СН3СОСНаСНСООН->2С2Н6ОН+СО2
84 СН3
СН3СОСН2СНСООН —
I
СН3
Н3
3
Характеристика основного сырья
Ацетоуксусный эфир, ч., ГОСТ 9799—61.
Спирт этиловый, абсолютированный, ГОСТ 9674—61.
Натрий металлический, ч., ТУ МХП 1664—50-
Этиловый- эфир а-бромпропионовой кислоты, ч., МРТ-У
6—09—845—63.
Натр едкий, ГОСТ 2263—59. сорт I.
Пятисернистый фосфор, ч„ ГОСТ 3118—46.
Соляная кислота, ч., ГОСТ 3118—46.
-)
Условия получения
Синтез а-метиллевулиновой кислоты. В трехгорлую круг-
лодонную колбу емкостью 2 л, снабженную обратным холо-
дильником, механической мешалкой и капельной воронкой,
помещают 0,5 л абсолютного спирта. При интенсивном пере-
мешивании порциями добавляют 46 г (2 г-атома) металличе-
ского натрия в виде проволоки. После полного растворения
натрия из капельной воронки добавляют по каплям 260 ?
(2 Л4) свежеперегнанного ацетоуксусного эфира. Реакцион-
ную смесь кипятят в течение 4 часов и оставляют на ночь.
На следующий день добавляют 362 г (2 Л4) этилового эфира
а-бромпропионовой кислоты и кипятят на водяной бане в те-
чение 3—4 часов- Затем отгоняют этиловый спирт, и содер-
жимое колбы промывают при перемешивании 1 н. раствором
соляной кислоты. Выделившийся маслянистый слой отделя-
ют, а водный слой несколько раз экстрагируют эфиром. Эфир-
ные вытяжки соединяют с маслянистым слоем и сушат без-
водным сульфатом натрия.
После удаления растворителя остаток, представляющий
собой а-метиллевулиновую кислоту, 'перегоняют в вакууме,
собирая фракцию с т. кип. 135—137°/10 мм.
Выход кислоты равен 200 г, что составляет 66,7% от тео-
ретического, считая на ацетоуксусный эфир.
Получение 2,4-диметилтиофена. Смесь 200 г а-метиллеву-
линовой кислоты и 210 г порошкообразного пятисернистого
фосфора нагревают до 50—60°, при этом начинается экзотер-
мическая реакция и температура реакционной смеси подни-
мается до 150—160°. После окончания реакции продукт от-
гоняют из реакционной смеси и полученный дистиллят про-
мывают холодным раствором едкого натра и перегоняют над
85
металлическим натрием, собирая 2,4-диметилтиофен с т. кип.
136—140°.
После повторной перегонки выход продукта равен 20 г,
что составляет 11,6% теоретического, считая на а-метиллеву-
линовую кислоту с т. кип. 138—139°; d420=0,9 9 6; п^с —1,5010.
2,4- Диметилтиофен представляет собой желтоватую жид-
кость с неприятным запахом, хорошо растворяется во мно-
гих органических растворителях, не растворяется в воде.
Примечание.
По литературным данным, т. кип. 2,4-диметилтиофена 137—138°;
d420=-0,9956 [1, 4].
ЛИТЕРАТУРА
1. Zelinsky. Вег., 20, 2018 (1887).
2. Rinkes. Rec. trav. chim., 52, 1052 (1953).
3. Органические реакции, т. 6, стр. 343.
4. Словарь органических соединений, т. 1, М., ИЛ., 1949, стр. 961.
Поступила в январе 1968 г. Ц.ЗЛ
УДК 547.732
3,4-ДИМЕТИЛТИОФЕН
Н. М. МОРЛЯН, Ж. Л. БАГРАТУНИ, Г. В. БАДАСЯН
Н3С-С—с—сн3
II II
ПС сн
xsz
C6H8S м. в. 112,19
В литературе описан способ получения 3,4-диметилтиофе-
на из диметилянтарной кислоты [1—3], но детальное изложе-
ние синтеза не приведено.
Нами разработаны условия получения 3,4-диметилтиофе-
на из малонового эфира-
СХЕМА СИНТЕЗА 3,4-ДИМЕТИЛТИОФЕНА
СООС2Н5 СН2 + CH3J СООС2Н5 соосан.-, НС-СН3 +ВгСНСООС2Н.-, СООС2Н5 сн3 СООС2Н-, 1 Н3С-СН Н3С-С-СООС2Нг, СООС2Н6 СООС2Н5 1 > нс-сн3 1 СООС2Н5 СООС2Н5 . Н3С-СН 1 Н3С—С-СООС2Н-, 1 СООС2Н5 11С1 87
Н3С-СН-СООС2Н6
| 4-С2Н5ОН + СО2
Н3С-СН-СООС2Н5
н3с—сн-соосан6
I РА
Н3С-СН-СООС2Н-
Н3С С— С-СИ,
II II
НС СН
Характеристика основного сырья
Малоновый эфир, ч., ТУ ГКХ ОРУ 126—59.
Метил йодистый, ч., ТУ МХП 49—51.
Натрий металлический, ч., ТУ МХП 1664—50.
Этиловый эфир а-бромпропионовой кислоты, ч., МРТУ
6—09 845—63.
Пятисернистый фосфор, ч„ ГОСТ 3118—46.
Бензол, ч.д.а., ГОСТ 5955—51.
Диэтиловый эфир, мед., ГОСТ 6265—52.
Натр едкий, ГОСТ 2263—59, сорт Г
Соляная кислота, ч., ГОСТ 3118—46.
Условия получения
Синтез метилмалонового эфира. В двухлитровую кругло-
донную трехгорлую колбу, снабженную обратным холодиль-
ником, мешалкой и капельной воронкой, помещают 1 л абсо-
лютного бензола и 640 г малонового эфира. При перемеши-
вании в течение 2,5—3 часов добавляют рорциями 92 г ме-
таллического натрия в виде проволоки. После растворения
натрия смесь кипятят еще 2 часа. Затем колбу охлаждают
до комнатной температуры и через капельную воронку по-
степенно добавляют 570 г йодистого метила, после чего
смесь промывают холодной водой и 1 н. раствором соляной
кислоты (100 г). Далее верхний масляный слой отделяют, а
водный — экстрагируют эфиром (2 раза порциями по 100 г)
Эфирные экстракты и маслянистый слой соединяют и сушат
безводным сульфатом натрия. После отгонки растворителя
остаток, представляющий собой метилмалоновый эфир, пе-
регоняют в вакууме, собирая фракцию с т. кип. 99°/10 мм.
Выход эфира равен 360 г, что составляет 52% от теоре-
тического.
Получение диэтилового эфира диметилянтарной кислоты.
В двухлитровую круглодонную колбу, снабженную мешал-
кой,. обратным холодильником и капельной воронкой, по-
мещают 1 л абсолютного бензола, 360 г метилмалонового
эфира и при перемешивании небольшими порциями добавля-
ют 46 г металлического натрия в'виде проволоки. Для пол-
88
кого растворения натрия колбу нагревают при 70—80° в те-
чение часа, затем охлаждают, добавляют из капельной во-
ронки 360 г этилового эфира а-бромпропионовой кислоты и
нагревают на водяной бане в течение 2 часов. Затем реакци-
онную смесь, охлаждают и промывают холодной водой и 1 н.
раствором соляной кислоты (100 г).
Выделившийся маслянистый слой отделяют, а водный слой
экстрагируют эфиром (2 раза порциями по 100 мл). Эфир-
ный экстракт и маслянистый слой соединяют и сушат безвод-
ным сульфатом натрия. После отгонки эфира собирают ди-
этиловый эфир диметилянтарной кислоты с т. кип. 162—164°.
Выход продукта равен 320 г, что составляет ~77% от
теоретического, считая на метилмалоновый эфир.
Получение 3,4-дйметилтиофена. Смесь 100 г диэтилового
эфира диметилянтарной кислоты и 70 г порошкообразного
пятисернистого фосфора нагревают до 50.—60°, при этом на-
чинается экзотермическая реакция и температура реакцион-
ной смеси поднимается до 150—170°. После окончания реак-
ции продукт отгоняют из реакционной смеси (140—160°).
Полученный дистиллат промывают холодным раствором ед-
кого натра и перегоняют над металлическим натрием, соби-
рая 3,4-диметилтиофен с т. кип. 144—146°.
Выход продукта равен 13 г, что составляет ~21% оттео-
ретического; =1,4850; (1^° = 0,9950.
ЛИТЕРАТУРА
1. Zelinsky. Вег. 21, 1836 (1888).
2. Steinkopt. Ann., 403, 64 (1914).
3. Shepard Henne, Midglay. J. Amer. Cheni. Soc, 56, 1355
(1934).
Поступила в январе 1968 г.
ЦЗЛ
УДК 547.723
2,5-ДИ(ОКСИМЕТИЛ)ФУРАН
Н. Н. ШМАГИНА, А. И. КУРАШЕВА, В. И. ФЕДОТОВА
НОСН2-Л J'-CH2OH
о
С6Н80з М. в. 128,12
Для синтеза 2,5-ди (оксиметил) фурана использован общий,
метод каталитического гидрирования альдегидов в первич-
ный спирт. Нами подобраны условия получения этого соеди-
нения в присутствии промышленного медно-хромового ката-
лизатора при использовании технического 5-оксиметилфурфу-
рола.
СХЕМА СИНТЕЗА 2,5-Д И(ОКСИМЕТИЛ)ФУРАНА
носн,-'!. )—сно — -* HOCH2-'l J'-CH2OH
о о
Характеристика основного сырья
5-Оксиметилфурфурол (см. одноименную статью в этом
сборнике).
Водород, ГОСТ 3022—45.
Катализатор, медно-хромовый (ГИПХ 105).
Спирт этиловый, абсолютный, ГОСТ 5962—51.
Условия получения
В литровый качающийся автоклав, снабженный карманом
для термопары и манометром, помещают 26,4 г сырого
95%-ного 5-оксиметилфурфурола, 600 мл этилового спирта и
2,6 г медно-хромового катализатора (порошок). Автоклав
90
дважды продувают водородом, после чего, закрыв входной
вентиль, доводят давление водорода до 100 атм, смесь на-
гревают до температуры 140° и в этих условиях выдержива-
ют ее 10 часов; давление в автоклаве поднимается до 150—
160 атм- Затем содержимое автоклава охлаждают до ком-
натной температуры и спускают давление. Автоклав вскры-
вают и сливают раствор, отфильтровывают катализатор и из
фильтрата отгоняют расторитель на водоструйном насосе.
Выход 2,5-ди (оксиметил) фурана равен 25 г, что состав-
ляет 96% от теоретического. После перекристаллизации из
воды т. пл. 75°.
По литературным данным, т. пл- вещества 70—75° [2];
75,5—77° [3].
ЛИТЕРАТУРА
1. А. А. Пономарев, В. В. Зе ленков а. Успехи химии, 20, 589
(1951).
2. Пат. США 3083236; РЖХим., 1965, 1Н73.
3. А. Р. Dunlop, F. N. Peters. The Furans, Nen York, P. 1953,
268.
Поступила в сентябре 1967 г. НИИПластмасс
УДК 547.556.8
1,5-ДИФЕНИЛКАРБОГИДРАЗИД
Дифенил карбазид
Г. А. КРЕЙМЕР
,NH—NH —Э
0=С/
XNH—NH—у /
С|3Н14Н4О М. в. 242,28
1,5-Дифенилкарбогидразид (дифенилкарбазид) является
важным аналитическим реактивом. Применяется он главным
образом для колориметрического определения хрома и как
индикатор при меркуриметрическом определении галоидов.
Дифенилкарбазид обычно получают взаимодействием мо-
чевины с фенилгидразином [1]. По литературным данным [2],
получаемый этим методом препарат плавится при 165—166°,
тогда как в других источниках указываются иные значения
температуры плавления: 169—170° [3]; 172—173° [4]; 175° [51.
Причиной этого является образование наряду с дифенилкар-
базидом 1-фенилсемикарбазида; оба продукта реакции обра-
зуют молекулярное соединение с температурой плавления
около 166° [5]. Для получения чистого препарата с темпера-
турой плавления 170—175° требуется применение труднодо-
ступных исходных веществ: дифенилкарбоната [3] или гвая-
колкарбоната [5].
Нами разработана сравнительно простая методика полу-
чения чистого 1,5-дифенилкарбогидразина из 1-фенилсеми-
карбазида, который легко может быть получен из соляноки-
слого фенилгидразина и мочевины. Впервые этот метод был
применен в работе [2], однако полученный им препарат пла-
вился при 164—165°.
92
СХЕМА СИНТЕЗА ДИФЕНИЛ КАРБОГИДРАЗИДА
/NH,
О=С( +HCl-H2N-NH-f ->
xNH2
nh2
->о =с;
XNH-NH—^q-NH4Cl
zNH2 +H2N-NH-<f
XNH-NH-^
NH—NH—
-> О=С/ +NH3
NH—NH-Z ;
Характеристика основного сырья
Фенилгидразин солянокислый, техн-, ТУ ТСНХ 41—59.
Фенилгидразин-основание. ч., ГОСТ 8750—58.
Аммиачная вода, техн., ГОСТ 9—57.
Мочевина, техн., ГОСТ 2081—57.
Спирт этилбвый, ректифицированный, ГОСТ 5962—51.
Бензол, ч., ГОСТ 8448—57.
Изопропиловый спирт, техн., ГОСТ 9805—61.
Условия получения
Синтез 1-фенилсемикарбазида. В колбу емкостью 0,5 л
помещают пасту солянокислого фенилгидразина, содержа-
щую 72,3 г (0,5 Л4) основного вещества, 42 г (0,7 Л4) моче-
вины, приливают 200 мл дистиллированной воды и 10 мл ам-
миачной воды. Смесь кипятят 5 часов с обратным холодиль-
ником (см. примечание 1), затем выливают в стакан и охла-
ждают до 0—5°. Осадок отжимают и промывают 300 мл дис-
тиллированной воды. Влажный продукт перекристаллизовы-
вают из 700 мл дистиллированной воды, добавляя для освет-
ления 3 г угля. Сушат при 90—100°.
Выход 1-фенилсемикарбазида равен 53 г, что составляет
70% от теоретического; т. пл. 169—173° (в пределах 1°).
Получение Г,5-дифенилкарбогидразида. В трехгорлую кол-
бу емкостью 250 мл, снабженную мешалкой, термометром и
газоотводной трубкой, конец которой выведен в вентиляци-
онный воздуховод, помещают 38 г (0,25 М) 1-фенилсемикар-
93
базида и 47,3 г (0,44 Л4) фенилгидразина (в пересчете на
100%-ный). Колбу помещают в глицериновую баню, нагре-
тую до 170°. Когда масса сделается достаточно подвижной,
включают мешалку и выдерживают массу при температуре
около 165° в течение 2 часов (см. примечание 2). По окончи
нии выдержки колбу вынимают из бани,, массе дают охла-
диться до 120° и осторожно при размешивании приливают
100 мл бензола. Раствор выливают в стакан и колбу сполас-
кивают двумя порциями бензола по 50 мл. Когда масса ох-
ладится до комнатной температуры, выпавший осадок от-
сасывают, промывают четыре раза бензолом порциями по
50 мл и сушат при 50—60°.
Выход технического продукта равен 53 г. Его растворяют
в 430 мл изопропилового спирта при кипячении, раствор
фильтруют и охлаждают до 20° (см. примечание 3). Выпав-
ший осадок отсасывают, промывают спиртом (4 раза по
50 мл), бензолом (4 раза по 50 мл) и сушат при 50—60°.
Выход чистого 1,5-дифенилкарбогидразида равен 40 г, что
составляет 66% от теоретического; т. пл. 171 —175° (в пре-
делах 1°). По внешнему виду полученный продукт бесцветен
или имеет слегка розоватый цвет.
Примечания:
1. В конце выдержки раствор должен иметь pH около 7,5.
2. Реакцию можно вести при температуре 160—175°.
3. Спиртовый раствор нельзя охлаждать быстро. В этом случае оса-
док очень трудно фильтруется.
ЛИТЕРАТУРА
1. К. Н. S 1 о 11 а, К- R. Y а с о b 1. Ztschr. anallt. Chem., 77, 344
(1929).
2. Реактивы и препараты лабораторного назначения, вып. 1. М.—Л ,
ГОНТИ НКТП'СССР, 1938, стр. 111.
3. Р. Caseneuve, Morean. Compt. rend., 129, 1254 (1899).
4. Справочник химика, т. II, М.—Л., ГХИ, 1963, стр. 722.
5. С. R. Nolle г. Journ. Amer. Chem. Soc., 52, 1132 (1930).
Поступила в ноябре 1967 г. ЦЗЛ
94
УДК 547.558.1'292
ДИФЕНИЛФОСФИНИЛУКСУСНАЯ КИСЛОТА
Т. М. БАЛАШОВА, Ю. М. ПОЛИКАРПОВ, Т. Я. МЕДВЕДЬ
С6Н5Х
\р-СН2СООН
С6н/ II
о
СиН^ОзР
М. в. 261,23
Из литературных данных известно, что дифенилфосфинил-
уксусную кислоту получают омылением этилового эфира ди-
фенилфосфинилуксусной кислоты раствором едкого кали с
последующей обработкой концентрированной соляной кис-
лотой [1].
Нами проверен и уточнен указанный метод.
СХЕМА СИНТЕЗА ДИФЕНИЛ ФОСФИНИЛ УКСУСНОЙ КИСЛОТЫ
(СеНБ),РСЦ-С2НБОН -^M^(CeHB)2P(OC,HB)+(C2H6)3N • НС1
(CeHs)2P(OC2H5)+ClCH2COOC2H5 —-
— (С6Н5)2РСН2СООС2Н5+С2Н5С1 t
II
о
(С6Н5)2РСН2СООС2Н5+КОН
о
-> (C6HS)2PCH2COOH+KC1
95
Характеристика основного сырья
Дифенилхлорфосфин, т. кип. 111 —112°/0,3 мм; п'£ —
1,6361 [2].
Спирт этиловый, абсолютированный, ГОСТ 9674—61.
Триэтиламин, ч., МРТУ 6—09—2437—65.
Кали едкое, х.ч., гранулированное, ГОСТ 4203—48.
Гексан, х.ч., МРТУ 6-09-2937-66.
Соляная кислота, х.ч., ГОСТ 3118—46.
Этиловый эфи]э, ч., абсолютный, ГОСТ 6265—52.
Этиловый эфир монохлоруксусной кислоты, ч., МРТУ
6-09-764-63.
Условия получения
Синтез этилового эфира дифенилфосфинистой кислоты. В
четырехгорлую колбу объемом 150 мл, снабженную мешал-
кой, холодильником, капельной воронкой с тубусом для азо-
та (см. примечание 1) и термометром, помещают 51,8 ?
-(0,235 М) дифенилхлорфосфина в 240 мл этилового эфира и
к этому раствору приливают 32 мл триэтиламина в 40 мч
этилового эфира- Полученную смесь охлаждают до 0—5° и
при перемешивании в течение 45 минут прикапывают смесь
14,2 мл этилового спирта и 40 мл этилового эфира, доводят
температуру до комнатной и оставляют на ночь. Образовав
шийся осадок отфильтровывают в токе азота, промывают на
фильтре этиловым эфиром (около 1 л), фильтрат концентри-
руют в вакууме, фракционируют, отбирая фракцию с т. кип.
146—148°/4 мм. Получают 24,2 г, (33%) бесцветной подвиж-
ной жидкости; п2®—1,5891.
По литературным данным, т. кип. продукта 179714 мм;
«“—1,5900 [1]. - . 4J
Получение этилового эфира дифенилфосфинилуксусной
кислоты. В четырехгорлую колбу, снабженную мешалкой,
прямым холодильником с ловушкой для конденсации хлори-
стого этила, капельной воронкой с тубусом для азота и тер-
мометром, помещают 15 мл этилового эфира монохлоруксус-
ной кислоты, нагревают до кипения (143°) и при хорошем
перемешивании прикапывают 6 г этилового эфира дифенил-
фосфинистой кислоты в течение полутора часов (см. приме-
чание 2). После отгонки избытка этилового эфира моно-
хлоруксусной кислоты и охлаждения реакционная масса пол-
ностью закристаллизовывается. Полученный продукт очища-
ют переосаждепием (гексаном из эфира) и получают 3,5 г
(47%) этилового эфира дифенилфосфинилуксусной кислоты
в виде белых иглообразных кристаллов с т. пл. 69—70°.
Найдено, %: С—66,24; Н—6,02, Р—10,42.
CieHiyOsP. Вычислено, %; С—66,5; Н—5,9; Р—10,75.
96
Получение дифенилфосфинилуксусной кислоты. В одно-
горлую колбу емкостью 25 мл помещают 2 г (0,0069 М) эти-
лового эфира дифенилфосфинилуксусной кислоты, прилива-
ют раствор 2,01 г (0,036 Л!) едкого кали в 8,9 мл воды и на-
гревают на кипящей водяной бане с обратным холодильни-
ком в течение 1 часа. По охлаждении к реакционной массе
прибавляют по каплям (при перемешивании стеклянной па-
лочкой) около 8 мл концентрированной соляной кислоты до
кислой реакции по универсальной индикаторной бумаге. К
концу прибавления соляной кислоты появляется желтоватое
масло, которое через 30 минут — 1 час закрйсталлизовывает-
ся в виде белого осадка. После перекристаллизации его из
воды получают 0,99 г (43,5%) дифенилфосфинилуксусной ки-
слоты с т. пл. 145—146°.
По литературным данным, т. пл. продукта 142—144° [3].
Эквивалент нейтрализации: найдено — 260; вычислено—
260.
Примечания:
1. Синтез этилового эфира дифенилфосфинистой и дифенилфосфииил-
уксусной кислоты проводят в токе сухого азота.
2. К концу прибавления этилового эфира дифенилфосфинистой кис-
лоты реакционная масса приобретает желтый оттенок.
ЛИТЕРАТУРА
1. А. Е. Арбузов. ЖРФХО, 42, 395 (1910); А. Е. Арбузов. Ав-
тореферат диссертации. Казань, 1914.
2. Е. Л. Гефтер. Фосфорорганические мономеры и полимеры. М,
АН СССР 1960.
3. К. I s s 1 i е b, G. Thomas. Chem. Вег, 94, 2246 (1961).
Поступила в апреле 1968 г. ИРЕА
7 Зак. 1034
УДК 547.558. Г461.3
(РДИФЕНИЛФОСФИНИЛЭТИЛ)МАЛОНОВАЯ
КИСЛОТА
М Т. БАЛАШОВА, Ю. М. ПОЛИКАРПОВ, Т. Я. МЕДВЕДЬ
СООН
С6Н5 I
,Р-СН2—сн2-сн
С«н/ II I
о соон
С17Н17О5Р М. в. 332,29
Синтез не описанной в литературе (Р-дифенилфосфинил-
этил) малоновой кислоты проводился нами в условиях, близ-
ких к условиям получения ди-(|3-дифенилфосфинилэтил)ма-
лоновой кислоты [1].
СХЕМА СИНТЕЗА (Р-ДИФЕНИЛФОСФИНИЛЭТИЛ)МАЛОНОВОЙ
КИСЛОТЫ
(с6н5)2р-сн=сн2+сн2(соос2н6)2 Ли
о
СООС2Н5
-> (С6Н5)2Р-СН2-СН2-СН
II I
О СООС2Н5
СООС2Н5
(С6Н8)2Р-СН2-СН2-СН
О СООС2Н5
98
соон
-> (CeH5)2P-CH2~CH2-CH
I! I
о соон
Характеристика основного сырья
Окись винилдифенилфосфина, т. кип. 179—18Г/1 мм-, т. пл-
116—117° (из бензола) [2].
Малоновый эфир, ТУ ОРУ 126—59. т. кип. 199,3°; d^0—
1,055; ng1—1,4162.
Натрий металлический, ТУ МХП 1664—50.
Соляная кислота, х.ч., концентрированная, ГОСТ 3118—
46.
Кали едкое, x-ч., ГОСТ 4203—48-
Вода дистиллированная, ГОСТ 6709—53.
Условия получения
Синтез (^-дифенилфосфинилэтил)малонового эфира. В
четырехгорлую колбу, снабженную мешалкой, обратным хо-
лодильником, термометром и тубусом для подачи азота (см.
примечание), помещают 60 мл малонового эфира и при пере-
мешивании добавляют 0,1 г металлического натрия. После
того, как натрий полностью прореагирует, добавляют 15,3 г
(0,067 Л4) окиси винилдифенилфосфина. Реакционную смесь
нагревают до 50° и при этой температуре перемешивают в
течение 7 часов; после охлаждения промывают равным объ-
емом воды и фильтрат сушат прокаленным сульфатом нат-
рия. При стоянии в течение суток из фильтрата выпадают
бесцветные блестящие кристаллы. Осадок отфильтровывают,
промывают этиловым эфиром и маточник концентрируют в
вакууме водоструйного насоса. При охлаждении из маточни-
ка выпадает дополнительное количество кристаллов.
Общее количество полученного (р-дифенилфосфинил-
этил) малонового эфира составляет 18,2 г (70%); т. пл. 97—
98°.
Найдено, %; С—65,4; 65,4; Н—6,7; 6,6; Р—7,9; 7,9.
С21Н25О5Р. Вычислено, %: С—65,1; Н—6,5; Р—8,0.
Получение (fi-дифенилфосфинилэтил)малоновой кислоты.
В одногорлую колбу помещают 9,3 г (0,0244 Л4) (Р-дифенил-
фосфинилэтил) малонового эфира, добавляют раствор 7,2 г
(0,129 Л4) едкого кали в 37 мл воды и нагревают в течение
5 часов на кипящей водяной бане. По охлаждении к реакци-
онной массе добавляют по каплям концентрированную соля-
ную кислоту (до кислой реакции по универсальной бумаге),
при этом выпадает белый осадок, который отфильтровывают,
7* 99
многократно промывают водой, сушат при 50° и получают
7,4 г (99%) (|3-дифенилфосфонилэтил) малоновой кислоты;
т. пл. 146—154° (с разл.).
Полученный продукт перекристаллизовывают из 50%-но-
го водного спирта и промывают кипящим бензолом. Вес очи-
щенного продукта составляет 3 г, (40,3%); т пл. 152—153°
(с разл.).
Эквивалент нейтрализации: найдено—167; вычислено —
166.
Найдено, %: С—62,0; 62,0; Н—5,3; 5,2; Р—9,3; 9,1.
С17Н17О5Р. Вычислено, %: С—61,5; Н—5,12; Р—9,32.
Примечание.
Синтез (Р-дифенилфосфинилэтнл)малоновою эфира проводят в токе
сухого азота.
ЛИТЕРАТУРА
1. М. И. Кабачник, Т. Я. Медведь, Ю. М. Поликарпов,
К. С. Юдина. Изв. АН СССР, Отд. хим. и., 1962, 1584.
2. М. И. Кабачник, Т. Я. Медведь, Ю. М. Поликарпов,
К. С. Юдина. Изв. АН СССР, Отд. хим. и., 1961, 11, 2029.
Поступила в апреле 1968 г.
ИРЕА
УДК 547.633.6
ДИХЛОР-шг.и.и-ТРИАЗИН ИЛАМ И НОФЛУОРЕСЦЕИН I
(ДХТАФ)
Ю. Е. СКЛЯР, А. Г. БУБНОВА, Г. И. МИХАИЛОВ
C23H13CI3N4O5
М. в. 531,76
Дихлор-сшил-триазиниламинофлуоресцеин I, новый флуо-
ресцентный метчик белка с зеленым свечением, впервые син-
тезирован в ИРЕА и нашел применение для качественного и
полуколичественного определения белка (и отдельных функ-
циональных групп) в срезах тканей [1]. ДХТАФ успешно ис-
пользуется также для получения люминесцентных сывороток
взамен флуоресцеинизотиоцианата [2].
Дихлор-силш-триазиниламинофлуоресцеин I представля-
ет собой мелкокристаллический порошок светло-желтого цве-
та, растворимый в этиловом спирте, водных растворах би-
карбоната натрия, соды, двузамещенного фосфата натрия.
Дихлор-силш-триазиниламинофлуоресцеин I получается в
результате реакции аминофлуоресцеина I [3] с цианурхлори-
дом в ацетоне.
101
СХЕМА СИНТЕЗА ДИХЛОР- симм -ТРИАЗИНИЛАМИНО-
ФЛУОРЕСЦЕИНА I
Характеристика основного сырья
Аминофлуоресцеин I, хроматографически чистый, ТУ ВЗ
89—66 (см. примечание 1).
Цианур хлористый, ч., ТУ ЛРЗ 137—65 (см. примеча-
ние 2).
Ацетон, ч.д.а., ГОСТ 2603—51.
Петролейный эфир, ч., МХТУ 279—59, дополнительно пе-
регнанный, т- кип. 40—60°.
Четыреххлористый углерод, ч.д.а., ГОСТ 5827—51.
Условия получения
Растворяют 17,37 г аминофлуоресцеина I в 480 мл аце-
тона, фильтруют (фильтр № 4), фильтр промывают 20 ми
ацетона.
В трехгорлую колбу емкостью 1 л, снабженную мешал-
кой, термометром, капельной воронкой и хлоркальциевой
трубкой, загружают 10,14 г цианурхлорида и 85 мл ацетона.
После растворения цианурхлорида реакционную массу ох-
102
лаждают до 2—4° и при энергичном перемешивании прибав-
ляют по каплям охлажденный до 5° раствор аминофлуорес-
иеина I в течение 2,5 часов. Уже после прибавления первых
порций раствора аминофлуоресцеина начинает выпадать свет-
ло-желтый осадок. Реакционную массу перемешивают еще
в течение 3,5 часов при 2—4°, после чего оставляют на 20 ча-
сов при комнатной температуре. Выпавший светло-желтый
осадок отфильтровывают, промывают ацетоном (2X25 мл) и
петролейным эфиром (2X20 мл) (см. примечание 3). Веще-
ство помещают в круглодонную колбу емкостью 500 мл и
сушат в вакууме (3—5 мм; 20°) до постоянного веса в тече-
ние 6 часов.
Выход вещества равен 24,8—25,5 г, что составляет 93,1 —
95,9% от теоретического, считая на аминофлуоресцеин;
Хмакс 490 нм; ема1[С 82200 (0,1 н. раствор едкого натра); ИК-
спектр: 1700 см1 (СООН), 1640 см' (о-хиноидная структу-
ра), 1607 см~' (бензольное кольцо), 1557 см1, 1132 см~‘
(сшил-триазин).
При хроматографии на бумаге (см. примечание 4) веще-
ство дает одно светло-желтое пятно с Rf 0,46, флуоресци-
рующее зеленым в ультрафиолетовом свете.
Дихлор-сн.им-триазиниламинофлуоресцеин хранят в ва-
куум-эксикаторе над хлористым кальцием. В этих условиях
вещество хранится в течение двух лет без признаков разло-
жения. На воздухе ДХТАФ постепенно гидролизуется, приоб-
ретая оранжевую окраску.
Примечания:
1. Аминофлуоресцеин I анализировался по описанной методике [4].
Использованный препарат содержал 99,1% основного вещества. Хромато-
графия на бумаге показала наличие в препарате следов примесей, флуо-
ресцирующих зеленым в ультрафиолетовом свете (365 нм).
2. Цйанурхлорид обычно содержит некоторое количество циануровой
кислоты и перед реакцией должен быть перекристаллизован. Для этого
75 г цианурхлорида растворяют при 40—50° в 600 мл четыреххлористо-
го углерода, отфильтровывают от нерастворившейся циануровой кисло-
ты (фильтр № 4), осадок на фильтре промывают четыреххлористым уг-
леродом (2 раза по 25 мл), фильтрат упаривают до объема около 150 мл.
затем охлаждают и выпавшие кристаллы отфильтровывают. Выход 58,8 з.
Из маточника после упаривания выделяют дополнительно 5 г. Суммар-
ный выход 63,8 г (85,0%). Перекристаллизованный цианурхлорид можно
хранить в эксикаторе над пятиокисью фосфора до 6 месяцев.
Препарат цианурхлорида, используемый в данной реакции, должен
отвечать следующим требованиям: а) т. пл. 145—146°; б) 0,5 г вещества
должны полностью растворяться в 10 мл четыреххлористого углерода.
3. Промывка петролейным эфиром гидрофобизирует осадок. Вещество,
полученное без такой промывки, очень быстро краснеет и быстрее разла-
гается.
4. Перед хроматографией дихлор-сильи-триазиниламинофлуоресцеин
превращался в нереакционноспособный продукт путем реакции с анили-
ном в ацетоне с водой (4:1) в течение 20 часов при 20°. Для хромато-
графии использовалась бумага «быстрая», Ленинградской бумажной фаб-
103
рики № 2, пропитанная 0,05 Л1 раствором двузамещенного фосфата пат-
рия. В качестве растворителя применялся н-бутанол, насыщенный 0,05 М
раствором фосфата.
ЛИТЕРАТУРА
1. В. Е. Барский, В. Б. Иванов, Ю. Е. Скляр, Г. И. Михай-
л о в. Тезисы докладов Всесоюзного совещания по органическим люмино-
форам, Харьков, 1967, стр. 8.
2. Ю. Е. Скляр, Г. И. Михайлов, К. Л. Шаханина,
В. А. Чибисова. Авт. свид. 194267, Изобретения, пром, образцы, то-
варн. знаки, № 8 (1967).
3. А. Н. Coons, М. М. Н. Kaplan. J. Exp. Med., 91, 1(1950).
4. Ю. Е. С к л я р, А. А. Былннкина, Н. М. Рыбкина,
Г. И. Михайлов. Химические реактивы и препараты, вып. 30, М.,
ИРЕА, 1967, стр. 167.
Поступила в мае 1968 г. ИРЕА
УДК 5.47.241
ДИХЛОРАИГИДРИД
ТРИХЛОРМЕТИЛТИОФОСФОНОВОЙ кислоты
Ф. И. ПОНОМАРЕНКО, С. 3. ИВИН, К. В. КАРАВАНОВ
СС13РС12
II
S
CC1BPS М. в. 252,31
Дихлораигидрид трихлорметилфосфоновой кислоты явля-
ется реакционноспособным соединением, он легко вступает
в реакцию со многими веществами, образуя различные про-
изводные трихлорметилтиофосфоновой кислоты.
До последнего времени способы его получения не были
известны. Лишь в 1967 году был опубликован метод получе-
ния дихлор ангидрида трихлорметилтиофосфоновой кислоты,
основанный на реакции комплексного соединения трихлорме-
тилтетрахлорфосфора и хлористого алюминия с этиленсуль-
фидом или этилмеркаптаном [1].
Нами найден новый способ получения этого препарата,
основанный на взаимодействии комплексного соединения
трихлорметилтетрахлорфосфора и пятихлористого фосфора
[СС13РС1з]+[РС16]“ с сероводородом. По этому методу выход
дихлорангидрида трихлорметилтиофосфоновой кислоты до-
стигает 85—87%.
СХЕМА СИНТЕЗА ДИХЛОРАНГИДРИДА
ТРИХЛОРМЕТИЛТИОФОСФОНОВОЙ КИСЛОТЫ
}CC13PC13']+[PC16]-+H2S -> CC13PC12+PSC13+HC1
s
Характеристика основного сырья
Комплексное соединение трихлорметилтетрахлорфосфора
с пятихлористым фосфором, получают по методике [2].
105
Сероводород, обычный из баллона или получаемый лабо-
раторным способом.
Условия получения
В трехгорлую колбу, снабженную трубкой для пропус-
кания газа и обратным холодильником, помещают , 50 г
(0,1 М) комплексного соединения трихлорметилтетрахлор-
фосфора с пятихлористым фосфором (см. примечание),
200 мл абсолютного бензола и при комнатной температуре
пропускают из предварительно сконденсированного в ло-
вушке 6,8 г (0,2 Л4) сероводорода до полного растворения
комплексного соединения.
Обратный холодильник меняют на прямой и под ваку-
умом водоструйного насоса отгоняют бензол и тиотреххло-
ристый фосфор. Остаток перегоняют в вакууме (10 мм)-
При повторной перегонке получают 22 г дихлорангидри-
да трихлорметилтиофосфоновой кислоты, что составляет 87%
от теоретического выхода; т. кип. 95°/10 мм; т. пл. 120°.
Найдено, %: Р—11,85; 11,76; С1—69,75; 69,83; S—12,48; 12,36.
CC15PS. Вычислено, %: Р—12,28; С1—70,29; S—12,68.
Примечание.
Комплексное соединение трихлорметилтетрахлорфосфора и пятихло
ристого фосфора гигроскопично, поэтому следует избегать излишнего со-
прикосновения его с воздухом.
ЛИТЕРАТУРА
1. Л. Е. Дмитриева, С. 3. Ивин, К. В. Караванов. Методы
получения химических реактивов и препаратов, вып. 15, М., ИРЕА, 1967,
стр. 79.
2. А. В. Кирсанов, Г. К. Федотова, ДоповЩ, АН УССР, 8,
1086 (I960); Ж- общ. химии, 31, 594 (1961).
Поступила в марте 1968 г.
УДК 547.241
ДИХЛОРАНГИДРИД
ТРИХЛОРМЕТИЛФОСФОНОВОЙ кислоты
Ф. Н. ПОНОМАРЕНКО, С. 3. ИВИН, К. В. КАРАВАНОВ
СС13РС12
о
СС15ОР М. в. 236,24
В литературе описан способ получения дихлорангидрида
трихлорметилфосфоновой кислоты, основанный на взаимо-
действии комплексного соединения трихлорметилтетрахлор-
фосфора и хлористого алюминия с водой в четыреххлорис-
том углероде [1].
Основным недостатком этого способа является примене-
ние безводного хлористого алюминия.
Предлагаемый нами способ получения дихлорангидрида
трихлорметилфосфоновой кислоты основан на взаимодейст-
вии комплексного соединения трихлорметилтетрахлорфосфо-
ра и пятихлористого фосфора с водой (метод А) илю серни-
стым ангидридом (метод Б).
СХЕМА СИНТЕЗА ДИХЛОРАНГИДРИДА
ТРИХЛОРМЕТИЛФОСФОНОВОЙ КИСЛОТЫ
СС13РС14-РС15 + Н2О — СС13РС12 + Н3РО4 [Метод А]
н
о
CC1SPC14PC15 + SO, СС13РС12 + SOC12 + РОС13 [Метод Б]
II
О
Характеристика основного сырья
Комплексное соединение трихлорметилтетрахлорфосфора
с пятихлористым фосфором, получают по методике [2].
107
Вода дистиллированная, ГОСТ 6709—53.
Сернистый ангидрид, из баллона.
Условия получения
Метод А. В реактор, снабженный мешалкой, капельной
воронкой и термометром, помещают 50 г (0,1 М) комплекс-
ного соединения трихлорметилтетрахлорфосфора с пятихло-
ристым фосфором (см. примечание) и 150 мл четыреххло-
ристого углерода. К реакционной массе, охлажденной до ми-
нус 20°, при перемешивании из капельной воронки по каплям
прибавляют 9 г (0,5 А1) дистиллированной воды.
После прибавления всего количества воды, не прекращая
перемешивания, реакционную смесь выдерживают 30 минут
при минус 20°. Затем перемешивание прекращают, темпера-
туру доводят до комнатной- Образуется два слоя, которые
разделяют в делительной воронке. Верхний слой—фосфор-
ная кислота. Нижний — дихлорангидрид трихлорметилфос-
фоновой кислоты в четыреххлористом углероде. Раствори-
тель отгоняют, а остаток перегоняют в вакууме.
При повторной перегонке получают 19,9 г дихлорангид-
рида трихлорметилфосфоновой кислоты, что составляет 83%
от теоретического выхода; т. кип. 95°/1 мм; т. пл. 156°.
Найдено, %: С1—75,36; 75,21; 75,21; Р—13,00: 12,89.
СС15ОР. Вычислено, %: С1—75,25; Р—13,08.
Метод Б. В трехгорлую колбу, снабженную трубкой для
пропускания газа и прямым холодильником, помещают 50 г
(0,1 Л4) комплексного соединения трихлорметилтетрахлор-
фосфора с пятихлористым фосфором и при охлаждении ле-
дяной водой пропускают 12,8 г (0,2 М) сернистого ангидри-
да до полного растворения комплексного соединения. Затем
обратный холодильник меняют на прямой и под вакуумом
(водоструйный насос) отгоняют образовавшийся хлористый
тионил и хлорокись фосфора. Остаток перегоняют в вакууме.
При повторной перегонке получают 17,7 г дихлорангид-
рида трихлорметилфосфоновой кислоты, что составляет 75%
от теоретического; т. кип. 95°/1 мм; т. пл. 156°.
Найдено, %: С1—75,35; 75,19; Р—13,00; 12,98.
СС15ОР. Вычислено, %: С1—75,25; Р—13,08.
Примечание.
Комплексное соединение гигроскопично, поэтому следует избегать из-
лишнего соприкосновения его с воздухом.
ЛИТЕРАТУРА
1. А. М. К i п п е а г, R. А. Р е г г е. п. J. Chem. Soc., 1952, 3437.
2. А. В. Кирсанов, Г. К- Федотова. Допов1дь АН УССР. 8,
1086 (1960); Ж- общ. химии, 31, 594 (1961).
Поступила в марте 1968 г.
108
УДК 547.724,2
5-(ДИЭТИЛАМИ НО)ФУРФУРОЛ
3. Н. НАЗАРОВА, В. С. ПУСТОВАРОВ
(C2HS)2N—СНО
C9Hi3NO2 ' М. В 167,20
Известный способ получения 5-(диэтиламино) фурфурола
из 5-бромфурфурола позволяет осуществить его синтез в одну
стадию [1]. Однако полученный таким образом альдегид за-
грязнен примесями и при его очистке потери составляют 40—
50%. Было установлено [2], что целесообразнее проводить
синтез в 2 стадии, исходя из 5-йодфурфурола. В этих усло-
виях промежуточный продукт — йодид 5-(диэтиламино)фур-
фурилидендиэтиламмония легко выделяется из раствора (что
не удается осуществить в случае бромида) и очищается пе-
рекристаллизацией из спирта. При обработке щелочью вод-
ного раствора соли образуется альдегид, который перегоня-
ют в вакууме, хроматографируют в бензоле на колонке с
окисью алюминия и перекристаллизовывают из гексана.
Общий выход альдегида около 70% •
СХЕМА СИНТЕЗА 5-(ДИЭТИЛАМИНО)ФУРФУРОЛА
1-< ;'-сно
XOZ
2HN(C8H5)2
- (C2H6)2N—\о J-CH=N(C2H5)2I- —
-> (c2hs)2n-^o;~cho
109
Характеристика исходного сырья
5-Иодфурфурол [3], т. пл. 126°
Диэтиламин, ч., ВТУ МХП 2938—51.
Вода дистиллированная, ГОСТ 6709—53.
Натр едкий, ч., ГОСТ 4328—48.
Спирт этиловый, ректифицированный, ГОСТ 5962—51.
Калий углекислый, х.ч., ГОСТ 4221—48-
Условия получения
В круглодонную колбу емкостью 200 мл, снабженную об-
ратным холодильником, помещают 22,2 г (0,1 М) 5-йодфур-
фурола, 40 мл диэтиламина, 40 мл спирта и нагревают в те-
чение 1,5 часа при 50°. После этого избыток диэтиламина от-
гоняют, спирт испаряют досуха в чашке Петри. Остаток рас-
творяют в 40 мл спирта, добавляют 0,5 г угля и кипятят
5 минут. Горячий раствор фильтруют, спирт испаряют в чаш-
ке Петри; образующиеся кристаллы растворяют при нагре-
вании в 40 мл спирта, фильтруют, раствор охлаждают до ми-
нус 10°. Выпавший осадок отделяют (если образование осад-
ка не наблюдается, спирт частично испаряют), высушивают,
растворяют в воде и прибавляют 70 мл свежеприготовлен-
ного 40%-ного раствора едкого натра (до разделения смеси
на 2 слоя). Образующееся масло экстрагируют 3 раза бензо-
лом порциями по 30 мл, вытяжки объединяют, сушат без-
водным углекислым калием и перегоняют в вакууме, собирая
фракцию, кипящую при 156—161°/5 мм, растворяют ее в бен-
золе и хроматографируют на окиси алюминия; бензол отго-
няют в вакууме. Полученное масло при охлаждении кристал-
лизуется. Образующийся альдегид перекристаллизовывают
из гексана.
Выход 5-(диэтиламино)фурфурола равен 12 г, что состав-
ляет 70% от теоретического; т. пл. 36°.
По литературным данным [1], т- пл. продукта 35—36°.
Найдено, %: С—64,49; Н—7,99; N—8,56.
C9Hi3NO2. Вычислено, %: С—64,65; Н—7,84; N—8,38.
ЛИТЕРАТУРА
1. 3. Н. Назарова, В. С. Пустоваров. Авт. свид. 185931; Бюлл.
изобр., № 18 (1966).
2. В. С. Пустоваров. Материалы VII и VIII научных конферен-
ций аспирантов РГУ, 1968, стр. 239.
3. 3. Н. Назарова. Ж- общ. химии, 25, 539 (1955).
Поступила в декабре 1968 г.
Ростовск. п/Д гос. ун-т.
УДК 547.831.7'461.3
ДИЭТИЛОВЫЙ ЭФИР
(8-ОКСИ-2-ХИНОЛИЛ)МАЛОНОВОЙ КИСЛОТЫ
2-[Ди(карбэтокси)метил]-8-оксихинолин
И. А. КРАСАВИН, В. М. ДЗИОМКО, Ю. П. РАДИН
C16HI7NO5
4 СН(СООС2Н5)2
ОН
М. в. 303,32
Диэтиловый эфир (8-окси-2-хинолил) малоновой кислоты
в литературе не описан. Он получен нами с выходом 65—
70% при взаимодействии метосульфата 8-окси-1-метоксихино-
линия с натриймалоновым эфиром в абсолютном этаноле.
В результате этой реакции заместитель вступает во 2-ое
положение молекулы, что строго доказано посредством его
расщепления: после гидролиза эфира и декарбоксилирова-
ния образовавшейся кислоты был получен 2-метил-8-оксихи-
нолин.
Диэтиловый эфир (8-окси-2-хинолил) малоновой кислоты
нерастворим в воде, но легко растворяется в разбавленных
растворах щелочей и во многих органических растворителях,
из этанола кристаллизуется в ярко-желтых иглах с т. пл-
112,5—113°. Это вещество принадлежит к числу хелатообра-
зующих соединений.
СХЕМА СИНТЕЗА ДИЭТИЛОВОГО ЭФИРА
(8-ОКСИ-2-ХИНОЛИЛ)МАЛОНОВОй КИСЛОТЫ
| II | + [CH(CC)OC2H6)2]-Na+-
CH3SO4-
ОСН, 111
+ NaCH3SO4+CHsOH
'' l'7 XN- XCH(COOC2H.:)a
OH
Условия получения
В сухой четырехгорлой колбе емкостью 1 —1,5 л, снаб-
женной мешалкой с ртутным затвором, термометром, капель-
ной воронкой и обратным холодильником с хлоркальциевы-
ми трубками, готовят раствор натриймалонового эфира. Для
этого в нее помещают 9,2 г (0,4 г-ат). 'нарезанного металли-
ческого натрия и понемногу прибавляют к нему из капель-
ной воронки 400 мл абсолютного этанола. Когда натрий
полностью растворится (см. примечание 1), раствор охлаж-
дают до 5—7° (см. примечание 2) и добавляют к нему при
размешивании 76,9 г (0,48 М) свежеперегнанного диэтилово-
го эфира малоновой кислоты.
Одновременно в круглодонной колбе, снабженной обрат-
ным холодильником с хлоркальциевой трубкой, растворяют
57,46 г (0,2 /И) метосульфата 8-окси-1-метоксихинолиния (см.
примечание 3) в 380 мл кипящего абсолютного этанола; кол-
бу нагревают на глицериновой бане и перемешивают вруч-
ную ее содержимое, чтобы ускорить растворение вещества.
Полученный оранжево-красный раствор охлаждают до 5°
и прибавляют по каплям за 1 —1,5 часа к раствору натрий-
малонового эфира; добавление проводят при охлаждении
льдом и интенсивном размешивании с такой скоростью, что-
бы температура реакционной массы не превышала 7°.
Образовавшийся раствор перемешивают при охлаждении
в течение 3,5—4 часов, затем добавляют к нему 40 мл ледя-
ной уксусной кислоты и продолжают размешивание еще
1,5 часа.
После этого растворитель отгоняют досуха в вакууме во-
доструйного насоса при нагревании на водяной бане- К еще
теплому остатку добавляют 200 мл хлороформа и 200 мл ди-
стиллированной воды и смесь встряхивают до растворения
твердого вещества. Органический слой отделяют, а водный
экстрагируют еще 50 мл хлороформа. Оба хлороформных
раствора соединяют, промывают водой (50 мл), сушат без-
водным сульфатом натрия в течение нескольких часов, филь-
труют и упаривают досуха в вакууме водоструйного насоса
на водяной бане.
Остаток растворяют при нагревании в 75 мл этанола, ки-
пятят с 1,5 г активированного угля и быстро фильтруют с
отсасыванием; фильтрат оставляют на 10—15 часов для кри-
сталлизации, затем охлаждают льдом в течение 2—3 часов.
112
Выпавшие кристаллы отсасывают, тщательно отжимают
стеклянной пробкой, промывают на фильтре этанолом
(10 мл) и высушивают в вакуум-эксикаторе.
Выход сырого продукта равен 33,5—36,5 г, что составляет
55—60% от теоретического; т. пл. 109,5—111,5°.
Фильтрат упаривают досуха и маслянистый остаток ос-
тавляют в ледяной бане на 5—10 часов. Выпавший осадок
отсасывают, промывают минимальным количеством этанола
и получают дополнительно 6—8 г менее чистого вещества с
т. пл.'105—106°.
Общий выход диэтилового эфира (8-окси-2-хинолил)мало-
новой кислоты составляет 39,5—42,5 г (65—70%).
После перекристаллизации основной порции продукта из
50 мл этанола с активированным углем (1 г) получают 31 —
32 г вещества с т. пл. НО—112°, которая при дальнейшей очи-
стке повышается до 112,5—113° (см. примечание 4).
Примечания:
1. Под конец реакция замедляется и для растворения остатков нат-
рия целесообразно подогреть раствор до кипения.
2. При более низкой температуре начинается выделение осадка натри-
евого производного н образуется густая кашица, которую трудно пере-
мешивать. Если это произошло, необходимо слегка подогреть суспензию
до образования прозрачного раствора и вновь охладить его до 5—7°.
3. Применялся метосульфат 8-окси-1-метоксихинолиния, приготовлен-
ный по описанному нами ранее методу [1]. В данном синтезе можно ис-
пользовать также сырой продукт, полученный нагреванием 32,23 г (0,2 М)
8-ОКСИХННОЛИН-1-оксида [2] и 25,23 г (0,2 Л4) свежеперегнанного нейтраль-
ного днметилсульфата в течение 1—2 часов при 100—103° (температура
бани).
4. Чистый образец диэфнра с т. пл. 112,5—113° был приготовлен по-
вторной перекристаллизацией из этанола и проанализирован.
Найдено, %: С—63,43; 63,77; Н—5,68; 5,73; N—4,67; 4,46.
C,6Hi7NO5. Вычислено, %: С—63,36; Н—5,65; N—4,62.
ЛИТЕРАТУРА
1. И. А. Красавин, В. М. Д з н о м к о, Ю. П. Радин. Методы
получения химических реактивов н препаратов, вып. 13. М., ИРЕА, 1965,
стр. 68.
, 2. К. R a m a i a h, V. R. S г i п i v a s а п. Proc. Indian. Acad. Set., 55A,
360 (1962).
Поступила в мае 1968 г. ИРЕА
8 Зак. 1034
УДК 547.461.2 -
CigHseOs
ДОДЕЦИЛЯНТАРНЫЙ АНГИДРИД
Н. М. МОРЛЯН, А. Г. МУРАДЯН
C12H25-CH-Cf
1 /°
сн2-с<
-о
М. в. 268,39
Алкен-, алкан- и циклоалканянтарные кислоты, их эфиры
и ангидриды применяются в промышленности как пластифи-
каторы, в качестве морозоустойчивых смазок, а также для
вулканизации бутадиенакрилонитрильных каучуков [1—4]-
В американском патенте [5] описан синтез алкан- и цик-
лоалканянтарной кислоты взаимодействием малеинового ан-
гидрида с алканами или циклоалканами в присутствии пере-
кисного инициатора.
Нами проверен и уточнен этот метод, в частности, для по-
лучения додецилянтарного ангидрида в присутствии пере-
киси третичного бутила.
СХЕМА СИНТЕЗА ДОДЕЦИЛ ЯНТАРНОГО АНГИДРИДА
//°
СН С< 135_150о //О
С12Н26+ II )О с^-сн-с77
сн-с< । .о
сн2-с<
Характеристика основного сырья
н-Додекан, ч„ МРТУ 6-09-1269-64.
Малеиновый ангидрид, ч., ГОСТ 5854—51.
Третичный бутил перекись (получение см. [6])-
114
Условия получения
В двухлитровый автоклав, снабженный электрическим
обогревом, загружают 580 г н-додекана, 120 г малеинового
ангидрида и 30 г перекиси третичного бутила. Включают
обогрев и температуру доводят до 135—150°. Содержимое
автоклава нагревают при этой температуре в течение 20—
25 часов, после чего выгружают. Жидкую фазу отделяют от
твердой и подвергают перегонке в вакууме.
Сначала отгоняют невступивший в реакцию н-додекан в
количестве 356 г, а затем основную фракцию с т. кип. 200—
220°/5 мм. Ее повторно перегоняют, собирая чистый доде-
цилянтарный ангидрид с т. кип. 210—212°/1 мм.
Выход равен 50 г, что составляет ~ 14,5% от теоретичес-
кого, счиТая на вступивший в реакцию додекан; «д —
1,4638; Й420 — 0,9661.
Найдено, %: С—71,96; 71,84; Н—10,90; 10,68; MR D —76,65.
С1бН2вОз. Вычислено, %: С—71,60; Н—10,51; MR D—76,09.
ЛИТЕРАТУРА
1. Ямаути, Мацуда, Хирата. J. Cheni. Soc Japan, Industr.
Chem. Sec., 60, 2, 187 (1957); РЖХнм., 1958. 28670.
2. Mika F. Tomas. J. Appl. Chem., 6, 9, 365 (1956); РЖХнм.,
1957, 49405.
3. Мацуда. Амаути, Яманоуэ. J. Chem. Soc. Japan, Industr.
Chem. Sec., 58, 4, 296 (1955); РЖХнм., 1957, 52312.
4. J. Remond. „Rev. prod. chlm.“, 65, 1294, 57, (1962).
5. Пат. США, 3030387 от 17.04.62; РЖХнм., 1963, 18н33п.
6. П. М. Кукарской, Я. Линдеман, Я. Мальчевской,
Т. Р а б е к. Лабораторные работы по химии и технологии полимерных
материалов, перев. с польского. М., «Химия», 1965, стр. 89.
Поступила в январе 1968 г. U3JI
УДК 547.241
О-ИЗОПРОПИЛМЕТИЛЦИАНФОСФОНАТ
И. Д. ШЕЛЛ ДОВА, С. 3. ИВИН, В. Д. ПРОМОНЕНДОВ
ОС3Н7-изо
CHSP^
II XCN
О
C5H10NO2P М. в. 147,11
В литературе сведения о циановых производных алкил-
фосфоновых кислот ограничены. Имеется патент [1], в кото-
ром описывается получение алкиловых эфиров цианфосфоно-
вых кислот перегруппировкой алкилфосфонатов с галоидциа-
нами. При этом наибольший выход указан для реакции с
бромцианом.
Нами разработан простой и надежный способ получения
алкиловых эфиров цианфосфоновых кислот посредством об-
работки алкилфосфонитов хлорцианом. Полученные вещест-
ва могут быть использованы в синтезах различных фосфор-
органических соединений.
СХЕМА СИНТЕЗА О-ИЗОПРОПИЛМЕТИЛЦИАНФОСФОНАТА
/OCsHj-hso
СН3Р(ОС3Н,-изо)г + C1CN — СН3Р< + С3Н7С1
II XCN
О
Характеристика основного сырья
О,О-Диизопропилметилфосфонит, получен по методике [2]
Хлорциан, получен по методике [3] (см. примечание).
116
Условия получения
В четырехгорлую колбу с мешалкой, капельной воронкой,
обратным холодильником и термометром помещают 16,4 г
(0,1 М) О,0-диизопропилметилфосфонита и 250 мл абсолютт
ного диэтилового эфира. При интенсивном перемешивании
к раствору медленно прибавляют 6,25 г (0,1 М) хлорциана
н 25 мл того же эфира. Капельную воронку при этом охлаж-
дают до 5°. Реакция протекает с разогреванием реакционной
массы до температуры 35—40°. По окончании прибавления
хлорциана перемешивание продолжают еще 2 часа, после
чего эфир отгоняют, а остаток перегоняют в вакууме.
Получают 11,8г (80%) О-изопропилцианметилфосфоната;
т. кип. 80°/4 мм; rf420 — 1,0356; п™ —1,4250.
Найдено, %: Р—21,1о.
C5HioN02P. Вычислено, %: Р—21,04.
Примечание.
Хлорциан сильно ядовит, поэтому работа с ним допустима только
при наличии хорошей тяги в вытяжном шкафу.
ЛИТЕРАТУРА
1. G. Schrader. Пат. ФРГ 938187 (1952); С. А„ 10, 106 (1956).
2. F. Hoffmann, N. М о о г е. J. Amer. Chem. Soc., 80, 1152 (1958).
3. А. Н a n t z s с h, L. Mat Ber., 28, 2471 (1895).
Поступила в декабре 1967 г.
УДК 547.538.141
КАЛИЕВАЯ СОЛЬ п-СТИРОЛСУЛЬФОКИСЛОТЫ
К М. РОИЗЕН, И. М. БИЛИК
сн=сн2
V
SOsK
C8H7KO3S
М. в. 222,30
В литературе описаны 3 способа получения калиевой соли
/’-стирол сульфокислоты действием едкого кали* на хлор ангид-
рид п- (а-бромэтил) бензолсульфокислоты [1] хлорангидрид
п- (|3-бромэтил) бензол сульфокислоты [2] или и-толуидиновую
соль и-стиролсульфокислоты [3]-
Нами уточнен второй способ получения этой соли и не-
сколько увеличен ее выход.
СХЕМА СИНТЕЗА КАЛИЕВОЙ СОЛИ л-СТИРОЛСУЛЬФОКИСЛОТЫ
СН2-СН2Вг
| || + 2HSO3C1
Ч/
СН2-СН2Вг
1
I II + H2SO4+HC1
v
SO2C1
СН2-СН,Вг сн=сн,
I I
| II +3 КОН ЯТБн* I I1 + КС1 + КВг+2Н2О
! I
SOsCl SO3K
118
Характеристика основного сырья
(З-Бромэтилбензол, ч., ТУ TCP 1320—63.
Хлорсульфоновая кислота, ч., ТУ НКХП 385—41, пере-
1нанная.
Едкое кали, ч., ГОСТ 4203—65.
Спирт этиловый, абсолютный, ГОСТ 8314—57.
Бикарбонат натрия, ч., ГОСТ 2156—52.
Сульфат натрия, ч., ГОСТ 4166—48.
Вода дистиллированная, ГОСТ 6709—53.
Условия получения
В трехгорлую колбу емкостью 250 мл, снабженную ме-
шалкой, капельной воронкой и трубкой для отвода газов, по-
мещают 176 мл хлорсульфоновой кислоты, охлаждают до 0—
5° и медленно прибавляют 100 г (0,54 М) р-бромэтилбензо-
ла с такой скоростью, чтобы температура не превышала 20°.
Реакционную массу выдерживают 1 час при комнатной тем-
пературе и выливают на мелкораздробленный лед. Смесь
экстрагируют диэтиловым эфиром (400 мл), эфирный экст-
ракт нейтрализуют 4%-ным раствором бикарбоната натрия,
промывают водой и сушат сульфатом натрия, эфир отгоняют.
Полученный хлор ангидрид п-(р-бромэтил) бензолсульфокис-
лоты растворяют в 190 мл охлажденного спирта, переносят
в колбу и, охлаждая раствор ледяной водой, при размеши-
вании медленно добавляют из капельной воронки раствор
121 г едкого кали в 540 мл спирта. Смесь кипятят в течение
1 часа и горячий раствор фильтруют. Остаток на фильтре
обрабатывают 200 мл горячего спирта и вновь фильтруют.
Фильтраты объединяют и охлаждают до минус 20°. Выпав-
шую соль отфильтровывают на воронке Бюхнера и тщатель-
но отжимают. Получают калиевую соль н-стиролсульфокис-
лоты в виде пасты, которую перекристаллизовывают из 60мл
горячей воды, затем из 700 мл спирта с добавлением 2 г ак-
тивированного угля. Перекристаллизованную соль сушат в
вакуум-эксикаторе над хлористым кальцием.
Получают 41—44 г белого кристаллического вещества, что
составляет 35—37% от теоретического выхода, считая на
(Г бромэтилбензол. По литературным данным [2], выход соли
составляет 30% от теоретического.
Если продукт имеет желтоватый оттенок, кристаллизацию
повторяют-
Полученная нами калиевая соль п-стиролсульфокислоты
содержит 95—96% основного вещества по анализу на содер-
жание калия и 96—97% по анализу на двойные связи. По-
следний был проведен по методу [4] с ацетатом ртути (II),
разработанному для стирола и его производных.
119
ЛИТЕРАТУРА
1. G. J. Mora Hi. Bull. Soc. Chirn., 1044 (1953).
2. R. H. Wiley, S. F. Reed. J. Amer. Chem. Soc., 78, 2171(19,56).
3. R. H. Wiley, N. R. Smith, С. C. Ketterer. J. Amer.
Chem. Soc., 76, 720 (1954).
4. R. W. Martin. Anal. Chem., 21, 921 (1949). '
Поступил» в феврале 1968 г. ;14РЕА
УДК 547.587.26'583.5
n-КАРБОКСИГАЛЛАНИЛИД
3,4,5-Триоксибензоил-п-аминобензойная кислота
Н. М. МОРЛЯН, Л. О. РОСТОМЯН, Г. А. ЕГИАЗАРЯН
НО
-.соон
С14Н । |OgN
М. в. 289,25
В литературе [1] указывается, что п-карбоксигалланилид
впервые синтезирован и предложен НИИхимии Харьковского
Государственного Университета для фотоколориметрического
определения малых количеств титана [1, 2, 3].
При неоднократной проверке этой методики нами выясне-
но, что первая стадия этого процесса, т. е. получение хлоран-
гидрида галловой кислоты, указанным способом, не дает по-
ложительных результатов — реакция не идет.
Нами разработана методика плучения хлорайгидрида
галловой кислоты в присутствии триэтиламина в растворе
хлороформа.
СХЕМА СИНТЕЗА п-КАРБОКСИГАЛЛАНИЛ ИДА
НО
но-О-соон--!0^
но
но
121
но
НО-^^—C-C1 + H2N—^_^-соон -►
но7 °
но
-> но-^ С- NH-^-СООН+НС1
\_/ н
но 0
Характеристика основного сырья
Галловая кислота, ч., ТУ МХП 2726—55.
Тионил хлористый, ч.. ВТУ МХП 3591—52.
Эфир диэтиловый, ГОСТ 6265—52.
n-Аминобензойная кислота, ч., ГОСТ 2681—51.
Хлороформ, ч., ГОСТ 3160—51.
Условия получения
Синтез хлорангидрида галловой кислоты- В круглодон-
ную колбу емкостью 1 л, снабженную обратным холодильни-
ком с хлоркальциевой трубкой, механической мешалкой, тер-
мометром и капельной воронкой, помещают 50 г галловой
кислоты, 375 г (250 мл) сухого хлороформа и 2 мл триэтил-
амина. Реакционную массу нагревают на водяной бане при
температуре 45—50° и из капельной воронки в течение 30 ми-
нут добавляют 165 г (100 мл) свежеперегнанного хлорис-
того тионила. После прибавления всего количества хлорис-
того тионила смесь кипятят в течение 6—8 часов при 55—
60°, за это время галловая кислота полностью переходит в
раствор. Растворитель и не вошедший в реакцию хлористый
тионил отгоняют вначале при обычном давлении и затем 2—
3 часа в вакууме при 20—25 мм на кипящей водяной бане.
Остаток в виде вязкой массы янтарного цвета, представляю-
щий собой хлорангидрид галловой кислоты, содержит одну
молекулу сернистого ангидрида.
Выход хлорангидрида галловой кислоты теоретический.
Найдено, %: С1—18,50; 18,55.
С7Н5О4. Вычислено, %: С1 — 18,32.
Получение п-карбоксилгалланилида. В круглодонную кол-
бу емкостью 3 л, снабженную обратным холодильником с
хлоркальциевой трубкой, помещают 50 г хлорангидрида гал-
122
ловой кислоты и 1350 мл абсолютного эфира. После раство-
рения хлорангидрида добавляют 36,1 г п-аминобензойной
кислоты, 74,7 г сухого углекислого калия и кипятят на водя-
ной бане 10—12 часов, периодически перемешивая. Затем
отгоняют растворитель досуха, остаток растворяют в 1500 мл
воды, фильтруют, и фильтрат подкисляют соляной кислотой
до кислой реакции на «конго». Выделившийся осадок филь-
труют, отжимают и промывают водой. После высушивания в
вакуум-сушильном шкафу над серной кислотой выход и-кар-
боксилгалланилида равен 38—40 г, что составляет ~50% от
теоретического; т. пл. 287—289°, что соответствует литератур-
ным данным [1—3].
Сырой продукт перекристаллизовывают 'из воды, насы-
щенной сернистым газом (на 10 г продукта 1 л воды); хоро-
шо растворяется в спирте, ацетоне.
Найдено, %: N—4,58; 4,63.
С14Н11О4. Вычислено, %: N—4,84.
ЛИТЕРАТУРА
1. А. Л. Гер ш у нс, А. Т. Слюсаре в, И. А. Растрепина. Ме-
тоды получения химических реактивов и препаратов, вып. 9. М., ИРЕА,
1964, стр. 75.
2. А. Т. Слюсарев, А. Л. Г е р ш у н с . Укр. хим. ж., 26, 364 (1960).
3. А. Т. Слюсарев, А. Л. Гер ш у нс Х1м1чна промисловкть, 1,
72 (1961).
Поступила в январе 1968 г. ЦЗЛ
УДК 547.241
КИСЛЫЙ О-ЭТИЛОВЫЙ ЭФИР
МЕТИЛФОСФОНОВОЙ кислоты
И. Д. ШЕЛЛКОВА, В. к. ПГ’ОМОНЕНКОВ, С. 3. ИВИН
/ОС2Н5
сн8р/
II ХОН
о
СзНдОзР
М. в. 124,07
О-Алкиловые эфиры метилфосфоновой кислоты находят
широкое применение в лабораторной практике. В литерату-
ре предложен способ их синтеза посредством гидролиза со-
ответствующих хлорэфиров водой [1]. С целью увеличения
выхода нами разработан способ получения указанных про-
дуктов обработкой хлорэфиров 75—85%:hoh муравьиной ки-
слотой.
СХЕМА СИНТЕЗА
КИСЛОГО О-ЭТИЛОВОГО ЭФИРА МЕТИЛФОСФОНОВОЙ кислоты
,ОС2Н8 ОС2Н.,
сн,р( -фнсоон — сн3р; н-нсц-со
II ЧС1 II 4 он
о о
Характеристика основного сырья
Хлорангидрид О-этилового эфира метилфосфоновой кис-
лоты. получен путем прибавления эквимолекулярного коли-
чества спирта в смеси с триэтиламином к дихлорангидриду
метилфосфоновой кислоты; т. кип. 55°/5" мм; d420—1,2280,
я^.—1,4320 [2];
Муравьиная кислота, ч., ГОСТ 5848—60-
124
Условия получения
В четырехгорлую колбу емкостью 100 мл с мешалкой,
обратным холодильником, капельной воронкой и . термомет-
ром помещают 4,6 г (0,1 М) 75—85%-ной муравьиной кис-
лоты и при перемешивании прикапывают 12,4 г (0,1 Л4) хлор-
ангидрида О-этилового эфира метилфосфоновой кислоты в
течение 15 минут. Температуру реакции при этом поддержи-
вают за счет выделяющегося тепла реакции в пределах 65—
70°, затем реакционную смесь нагревают при 100—110° в те-
чение 30 минут и перегоняют в вакууме.
Выход кислого О-этилметилфосфоната равен 8,7 г, что
(.оставляет 70% от теоретического; т. кип. 11571 мм; <7420—
1,1800; п™—1,4245-.
Найдено, %: Р—25.06.
СзНдОдР. Вычислено, %: Р—24,99.
Аналогично получают кислый О-изопропиловый эфир ме-
тилфосфоновой кислоты с т. кип. 118°/1 мм; df0—1,1100;
«20 — 1,4213.
Найдено, %: Р—22,37.
C4II11O3P. Вычислено, %: Р—22,23.
ЛИТЕРАТУРА
1. С. 3. Ивин, И. Д. Ш е л а к о в а. Методы получения химических
реактивов и препаратов, вып. 12. М., ИРЕА, 1965, стр. 82.
2. Н. Разумов, Т. Мухачева. Ж. общ. химии, 27, 2389 (1957).
Поступила в декабре 1967 г.
УДК 547.633.4
КРЕЗОЛОВЫЙ КРАСНЫЙ
о -Крезолсульфофталеин
Г. И. МИХАИЛОВ, А. И. БОЛДЫРЕВА
СН3 СНз
I ' I о
нп /Ч //^//
НО” Р II
A-so3h
\//
• ~г н*°
4
C2IHI8O5S~H«O
М. в. 386,93
Крезоловый красный является кислотно-щелочным инди-
катором с двумя переходами окраски в кислой и щелочной
средах: от красной к желтой при pH 1,0—2,0 и от желтой к
малиново-красной при pH 7,2—8,8.
Он применяется для определения концентрации водород-
ных ионов колориметрическим методом и как исходное сырье
для получения водорастворимого крезолового красного,
бромкрезолового пурпурового, водорастворимого бромкрезо-
лового пурпурового и входит в состав универсальных индика-
торов-
Крезоловый красный получают конденсацией о-крезола с
о-сульфобензойной кислотой, ее ангидридом, дихлорангид-
ридом или моноаммонийной солью.
Синтез проводят в избытке о-крезола [1], в ксилоле с
присутствием в качестве конденсирующих средств: хлорокиси
фосфора [2], смеси хлорокиси фосфора и хлористого цинка
[3] или хлорокиси фосфора и хлорной кислоты [4. 51
126
Наибольший выход высококачественного (93—95%) кре-
золового красного получен при проведении и конденсации с
избытком о-крезола в присутствии смеси хлорокиси фосфора
и хлорной кислоты [6].
СХЕМА СИНТЕЗА КРЕЗОЛОВОГО КРАСНОГО
соон сн3
РОС13
НС1О4
Характеристика основного сырья
о-Сульфобензойная кислота, ч., МРТУ 6—09—811—63.
о-Крезол, ч., МРТУ 6—09—3829—67.
Хлорокись фосфора, ч., МРТУ 6—09—337—63.
Хлорная кислота, ч., ТУ МХП ОРУ 87—57.
Условия получения
В четырехгорлую колбу емкостью 2 л, снабженную меха-
нической мешалкой, обратным шариковым холодильником
(см. примечание 1), капельной воронкой и термометром на
150°, загружают 101 г (0,5 М) безводной о-сульфобензойной
кислоты (см. примечание 2) и при размешивании,, через ка-
пельную воронку прибавляют 30 мл 52%-ной хлорной кисло-
ты, 30 мл хлорокиси фосфора (см. примечание 3), 168 г
(160 мл, 1,5 М) о-крезола (см. примечание 4) и за 20—39
минут нагревают реакционную смесь до 115—120°. При этой
температуре из капельной воронки прибавляют за 1,5 часа
50 мл хлорокиси фосфора, а затем реакционную массу вы-
держивают при 115—120° и интенсивном размешивании еще,
30 минут. К концу выдержки смесь принимает кашеобраз-
ную консистенцию. К ней прибавляют при размешивании
1,5 л горячей дистиллированной воды (95—100°) и выдержи-
вают при непрерывном кипении в течение 1 часа. Затем горя-
127
чий раствор фильтруют и полученный продукт промывают
1 л горячей воды до нейтральной реакции промывных вод и
отсутствия в них ионов хлора (см. примечание 5). Промытую
пасту переносят в колбу емкостью 2 л, добавляют 500 мл дис-
тиллированной воды и отгоняют с водяным паром оставшееся
количество о-крезола. Затем содержимое колбы охлаждают
до 20—30°, отфильтровывают осадок, промывают 200 мл ди-
стиллированной воды, тщательно отжимают, сушат до посто-
янного веса при 100—105° и получают сухой крезоловый кра-
сный в количестве 169—173 г с содержанием основного ве-
щества 93—95%.
Выход в пересчете на 100%-ный индикатор равен 150—
160 г, что составляет 81—83% от теоретического, считая на
о-сульфобензойную кислоту.
Примечания:
1. Выделяющийся в процессе синтеза газообразный хлористый водо-
род улавливают раствором едкого натра в склянке Тищенко, присоеди-
ненной к .обратному холодильнику.
2. Для получения безводной о-сульфобензойной кислоты 258 г кри-
сталлической о-сульфобензойной кислоты нагревают в открытом фарфо-
ровом стакане при размешивании и температуре 140—145° в течение 2—•
2,5 часов, после чего расплавленную кислоту выливают в фарфоровую
чашку и хранят в эксикаторе. Температура плавления безводной кислоты
139—141°. Выход 202 г.
3. Применяют свежеперегианную хлорокись фосфора, кипящую в ин-
тервале 106—108°.
4. Применение избытка о-крезола устраняет к концу синтеза загус-
тевание реакционной массы и обеспечивает хорошее размешивание.
5. Во время промывки реакционного плава .водой наряду с отмыв-
кой кислот удаляется избыток о-крезола, который может быть регенери-
рован.
ЛИТЕРАТУРА
1. П. Н-. Рабинович. Реактивы и препараты лабораторного наз-
начения, вып. 1. М., ИРЕА, 1938, стр. 191.
2. Е. Д. О с е т р о в а, П. Г. М и т ю ш и и а. Авт. свид. 85590; Бюлл.
изобр. 3, 41 (1951).
3. Г. Н. Кошелева, Г. И. Михайлов. Авт. свид. 104125; Бюлл.
изобр. 8, 17 (1956).
4. Е. Я. Я р о в е и к о, М. Ф. Кондрашова, Т. Г. Г а й в о р о н-
с к а я, Г. И. М и х а й л о в. Авт. свид. 187191; Бюлл. изобр., 20, 90 (1966).
5. Е. Я. Я р о в е и к о, М. Ф. Кондрашова. Методы получения
химических реактивов и препаратов, вып. 17. М., ИРЕА, 1967, стр. 140.
6. Г. И. Михайлов, Н. С. Мезенцева, А. П. Болдырева.
Авт. свид. 232277; Изобретения. Промобразцы. Товарные . знаки, 1, 30
(1969).
Поступила в апреле 1968 г.
ИРЕА
УДК 547.391.3'268.15
ЛАУРИНОВЫЙ ЭФИР МЕТАКРИЛОВОЙ КИСЛОТЫ
II. М. МОРЛЯН, Л. О. РОСТОМЯН
СН9
СН2=С-СООСН2(СН2)10СН3
С16Н30О2 М. в. 254,41
Лауриновый эфир метакриловой кислоты (метацен) явля-
ется одним из лучших дезодозирующих средств. Реагируя с
большинством обладающих запахом соединений, он образует
совсем непахучие или слабопахучие сополимеры.
В литературе имеется только одно сообщение о синтезе
лауринового эфира метакриловой кислоты [1]. При проверке
этого метода мы несколько изменили условия получения, в
результате чего увеличился выход чистого продукта.
СХЕМА СИНТЕЗА
ЛАУРИНОВОГО ЭФИРА МЕТАКРИЛОВОЙ КИСЛОТЫ
сн3
СН2 = С-СООН + С]2Н2бОН
сн3
I
-> СН2=С-СООС]2Н25 + Н2О
Характеристика основного сырья
Додециловый спирт^ ч., МРТУ 6—09—3735—67.
Метакриловая кислота, ч., МРТУ 6—09—1768—64.
Бензол, ч„ ГОСТ 5955—51.
Серная кислота, ч., ГОСТ 4204—66.
Гидрохинон, марка Б—635.
Натрий двууглекислый, ч., ГОСТ 2156—52.
9 Зак. 1034 129
Условия получения
В круглодонную колбу емкостью 2 л, снабженную водоот-
делителем и обратным холодильником, помещают 86 г (1 Л4)
метакриловой кислоты, 400 мл бензола, 2 г гидрохинона,
предварительно приготовленный раствор 279 г (1,5 (И) доде-
цилового спирта и 15 г серной кислоты. Смесь нагревают на
водяной бане до прекращения выделения воды, для чего тре-
буется 12—14 часов. Затем отгоняют бензол, остаток нейтра-
лизуют бикарбонатом натрия, фильтруют на воронке Бюх-
нера и подвергают вакуум-перегонке, предварительно добав-
ляя еще 2 г гидрохинона. После выделения первой фракции
в интервале 82—86°/10 мм, собирают лауриновый эфир мета-
криловой кислоты с т. кип. 130—140°/Ю мм.
Выход чистого продукта равен 200 г, что составляет
~80% от теоретического, считая на метакриловую кислоту;
rf420—0,8700; nf —1,4450.
Найдено, %: С—75,27; Н—11,60; MR D —77,80.
С1вНзо02. Вычислено, %: С—75,53; Н—11,80; MR D—77,28.
ЛИТЕРАТУРА
1. Японск. пат. 9198 (1961).
Поступила в мае 3968 г.
ЦЗЛ
УДК 547.632.4
ЛЕЙКООСНОВАНИЕ
КРИСТАЛЛИЧЕСКОГО ФИОЛЕТОВОГО
Г. С. ПЕТРОВА, А. М. ЛУКИН, Н. Б. ЭТИНГЕН
C25H31N3
М. в. 373,54
Лейкооснование кристаллического фиолетового является
чувствительным реагентом для спектрофотометрического оп-
ределения иридия [1].
В литературе проводится ряд методов синтеза этого сое-
динения: его получают нагреванием диметиланилина с гли-
колем и хлористым цинком [2], длительным нагреванием ди-
метиланилина с муравьиной кислотой в присутствии хлори-
стого алюминия [3]; из диметиланилина и этилового эфира
ортомуравьиной кислоты в присутствии хлористого цинка
[4]; нагреванием диметиланилина с гидролом Михлера [5], а
также восстановлением красителя кристаллического фиоле-
тового [6, 7]. Нами выбран метод, заключающийся в восста-
новлении кристаллического фиолетового гидросульфитом нат-
рия в присутствии небольшого количества цинковой пыли.
S* 131
СХЕМА СИНТЕЗА ЛЕИКООСНОВАНИЯ КРИСТАЛЛИЧЕСКОГО
ФИОЛЕТОВОГО
Характеристика основного сырья
Кристаллический фиолетовый, ч., ТУ 1134—52.
Гидросульфит натрия, техн., ГОСТ 246—41.
Цинковая пыль-
Спирт этиловый, гидролизный, ГОСТ СТУ 57—227—64.
Аммиак водный, ч.д.а., ГОСТ 3760—64.
Условия получения
В круглодонной колбе емкостью 250 мл, снабженной ме-
шалкой, обратным холодильником и капельной воронкой,
растворяют 5 г кристаллического фиолетового в 100 мл го-
рячего спирта. При нагревании на кипящей водяной бане
понемногу приливают раствор, 15 г гидросульфита натрия в
50 мл воды. Одновременно присыпают 0,5 г цинковой пыли.
Для определения конца реакции делают капельную пробу
на фильтровальной бумаге. Первоначальный раствор краси-
теля дает на бумаге ярко-фиолетовое пятно, а восстановлен-
ный — бледно-голубое.
Реакционную массу кипятят еще 30 минут, затем выли-
вают ее в смесь 10 лм горячей воды и. 10 мл раствора амми-
ака, при этом раствор мутнеет. После охлаждения осадок
132
лейкосоединения отфильтровывают, отмывают водой от суль-
фата натрия (проба с хлористым барием) и получают голу-
бое с серебристым блеском лейкооснование кристаллическо-
го фиолетового в количестве 4,3 г, что составляет 86% от тео-
ретического выхода.
После перекристаллизации из спирта продукт имеет вид
листочков; т. пл. 173°.
По литературным данным, т. пл. вещества 173—175° [6,7].
ЛИТЕРАТУРА
1. G. Н. Ayres, W. Т. В о 11 е t е г. Analyt. chem., 29, 1, 72 (1957).
2. Heumann, Wiefnik. Вег., 20, 2421.
3. D.R.P., 29964; Fr„ 1, 70.
4. О. Fischer, К б г п e г. Вег., 17, 99.
5. D.R.P., 27032; Fr„ 1, 77.
6; A. W. Hofmann. Вег., 18, 769.
7. О. F. Fritzen. Y. prakt. chem., 2, 79, 563.
Поступила в апреле 1968 г.
ИРЕА
уДК 547.755'292
1-МЕТИЛ-З-АЦЕТОКСИИНДОЛ
N-Метилиндоксилацетат; N-Метил- О -ацетилиндоксил
Н. В. ФАКЕЕВА, В. В. ЖИДКОВА
С11Н P1NO2
М. в. 189,20
1 -Метил-З-ацетоксинол применяется в гистохимии для оп-
ределения эстераз [1].
В литературе описан единственный способ его получения
ацетилированием N-метилфенилглицин-о-карбоновой кислоты
уксусным ангидридом *в присутствии уксуснокислого натрия
[2, 3].
Нами проверен и уточнен этот- способ получения.
СХЕМА СИНТЕЗА 1-МЕТИЛ-З-АЦЕТОКСИИНДОЛА
^,^/СООН
| II +2(СН3СО)2О
Ч/ \N-CH,COOH
I
СН3
\____огпгн
I || || 3+зсн2соон4-со2
V/'nz
снй
134
Характеристика основного сырья
N-Метилфенилглицин-о-карбоновая кислота, ч., (см. одно-
именную статью в настоящем сборнике).
Уксусный ангидрид, ч., свежеперегнанный, ГОСТ 5815—
52.
Натрий уксуснокислый, ч., ГОСТ 199—52.
Спирт этиловый, гидролизный, ТУ 366—65.
Петролейный эфир, ч., МХТУ 279—59.
Уголь активированный, техн., ГОСТ 6213—53-
Вода дистиллированная, ГОСТ 6709—53.
Условия получения
В круглодонную трехгорлую колбу емкостью 0,5 л, снаб-
женную обратным холодильником, мешалкой и термометром,
загружают 249 мл (269,5 г; 2,6 М) уксусного ангидрида и
50 г (0,6 М) уксуснокислого натрия, предварительно прока-
ленного при 120°. При перемешивании реакционную массу
доводят до кипения и добавляют 50 г (0,24 М) N-метилфе-
нилглицин-о-карбоновой кислоты. При температуре 139—140°
смесь выдерживают в течение 1 часа, после чего раствор вы-
ливают в 2,5 л ледяной воды. 1-Метил-З-ацетоксииндол выде-
ляется в виде масла, затвердевающего при дальнейшем пере-
мешивании. Осадок отфильтровывают, промывают 3 л воды
и сушат в эксикаторе над хлористым кальцием.
Выход технического 1-метил-З-ацетоксииндола в виде
светло-коричневого порошка с т. пл. 52,5—54,5° равен 40—
41 г.
Технический продукт растворяют в 180 мл этилового
спирта, нагревают до 60°, добавляют 4 г активированного
угля и размешивают 10—15 минут. Раствор фильтруют, ох-
лаждают до 0°, выпавший осадок отфильтровывают и сушат
в вакууме-эксикаторе.
Для окончательной очистки продукт перекристаллизовы-
вают из петролейного эфира (1:20) и получают 1-метил-З-
ацетоксииндол в виде блестящих белых кристаллов с т. пл.
57—58°.
По литературным данным его т. пл. 57° [2].
Выход чистого продукта равен 28—30 г, что составляет
62—66% от теоретического.
ЛИТЕРАТУРА
1. Э. Пирс. Гистохимия М., ИЛ, 1962, стр. 416.
2. L. Ettinger, Р. Friedlaender. Вег., 45, 2075 (1912).
3. S. Y. Holt, Р. W. Sadler. Proc. Roy. Soc., 148В, 933 (1958).
Поступили в мае 1968 г. ИРЕА
.135
УДК 547.563.1.024
2-а -МЕТИЛБЕНЗИЛ-4-АЛКИЛФЕНОЛЫ
И. С. МАРКОВИЧ, Н. В. КРУГЛОВА, в. м. дзиомко
В литературе описан метод алкилирования 4-трет-бутил-
фенола стиролом в присутствии серной кислоты [1]. Данных
об алкилировании 4-н-бутилфенола и 4-трег-октилфенола не
было найдено и воспроизвести синтез по методу [1] нам не
удалось.
В настоящей работе сообщается об алкилировании 4-н-
бутилфенола, 4-трет’-бутилфенола и 4-трет’-октилфенола сти-
ролом в присутствии КУ-2 по методу [2].
СХЕМА СИНТЕЗА 2-а-МЕТИЛБЕНЗИЛ-4-АЛКИЛФЕНОЛОВ
^\^0Н ^\/°И
I к д- сн2=снс8н5 -- । ||'
R/<W R/^/XCHCeH-
СН3
где R=Tper-C4H9; н-С4Н9; rper-CsHi?
Характеристика основного сырья
4-тре7’-Бутилфенол, т. пл. 97—98° •
4-н-Бутилфенол, ТУ TCP.
4-трет’-Октилфенол, ч., т. пл. 53°.
Стирол, ч„ МРТУ 6—09—4055—67.
Бензол х.ч., ГОСТ 5955—51.
КУ-2.
Условия получения
Синтез 2-а-метилбензил-4-трет-бутилфенола. Смесь 10,5 г
(0,07 Л4) 4-трет-бутилфенола, 5,9 г (0,051 М) стирола (см.
136
примечание 1), 1,1 г КУ-2 (см. примечание 2) нагревают при
130—135° и размешивают в течение 4 часов. По окончании
нагревания к реакционной массе добавляют 30 мл бензола.
Раствор отфильтровывают от катализатора п перегоняют в
вакууме, собирая три фракции:
первую — до 110°/20 мм — следы;
вторую—110°—130720 мм в количестве 4 г (непрореа-
гировавший 4-трет-бутилфенол);
третью — 190—210720 мм в количестве 8,5 г, представля-
ющую собой 2-а-метилбензил-4-трет-бутилфенол.
Выход продукта составляет 77% в расчете на прореаги-
ровавший бутилфенол- После повторной перегонки т. кип. ве-
щества 159—16773—4 мм; d№20—1,0250; п2°—1,5590.
По литературным данным т. кип. 18075 мм [1]; 145—
16072 мм [3].
При хроматографировании на окиси алюминия в тонком
слое по методу [4] получено четкое пятно: Rf =0,57.
Получение 2-а-метилбензил-4-к-бутилфенола. Смесь 10,5 г
(0,07 44) 4-н-бутилфенола, 5,9 г (0,051 44) стирола, 1,1 г
КУ-2 нагревают при 160—165° и размешивании в течение
8 часов. По окончании нагревания к реакционной массе до-
бавляют 30 мл бензола. Раствор отфильтровывают от ката-
лизатора и перегоняют в вакууме, собирая 3 фракции:
первую—до 130725 мм — следы;
вторую — 130—150725 мм в количестве 5,6 г (непрореа-
гировавший 4-н-бутилфенол);
третью — 200—210725 мм в количестве 6,4 г, представля-
ющую собой 2-а-метилбензил-4-н-бутилфенол.
Выход продукта составляет 77%, считая на прореагиро-
вавший бутилфенол.
После повторной перегонки т. кип. вещества 149—15973—
4 мм; <4о20—1,0220; п%—1,5606; 7?/— 0,55.
Найдено, %: С—85,43; 85,23; Н—8,77; 8,38.
CisHssO. Вычислено, %: С—85,00; Н—8,70.
Получение 2-а-метилбензил-4-трет-октилфенола. Смесь 7 г
(0,03 44) 4-грет-октилфенола, 3,5 г (0,03 44) стирола, 1 г
смолы КУ-2 нагревают при 170° и размешивании в течение
8—9 часов. По окончании нагревания к реакционной массе
добавляют 30 мл бензола. Раствор отфильтровывают от ка-
тализатора и перегоняют в вакууме, собирая фракцию, кипя-
щую при 170—20072—4 мм.
Выход продукта равен 9,5 г, что составляет 68% на за-
груженный 4-трет-октилфенол. После повторной перегонки
т. кип- 180—20072—4 мм; п2^—1,5498; А?/ = 0,61.
Найдено, %: С—84,96; 85,04; Н—9,46; 9,57.
С22Н30О. Вычислено, %: С—85,10; Н—9,74.
137
Примечания:
1. Для арилалкилирования используют свежеперегнанный стирол.
2. Катионит выдерживают 3 часа в 2%-ной растворе едкого натра,
отфильтровывают и промывают водой до нейтральной реакции промыв-
ных вод. Затем катализатор выдерживают 18—20 часов в 5%-ной соляной
кислоте, вновь промывают водой до нейтральной реакции и сушат до по-
стоянного веса при температуре 40—50°.
ЛИТЕРАТУРА
1. Пат. США. 2849517; С. А., 52, 21210а.
2. Ф. М. Эгид и с, И. В. Кох а иов а. Хим. промышленность, 1, 23
(1966).
.3. Пат. США, 2276117; С. А, 36, Р46338.
4. Л. А. Хейфиц, Г. И. М о л д о в а н с к а я, Л. М. Шулов. Ж.
аналит. химии, 28, 267 (1963).
Поступила в апреле 1968 г.
ИРЕА
УДК 547,/72,1
1-МЕТИЛ-3,4,5-ТРИАЛКИЛ ПИРАЗОЛ Ы
О. В. ИВАНОВ, в. м. дзиомко
R CH2R
rch2-^
XN-/
сн3
I R=CH3 IV R = h-C4H9
II R = C2H5 V R=h-C5Hh
III R = h-C3H7
N-Метилированные производные пиразола могут быть по-
лучены при взаимодействии метилгидразина с 1,3-дикетона-
ми, при алкилировании пиразолов йодистым метилом в запа-
янной трубке [1] или диметилсульфатом [2].
Нами получен ряд новых 1-метил-3,4,5-триалкилпиразо-
лов алкилированием 3,4,5-триалкилпиразолов йодистым ме-
тилом в присутствии углекислого калия в среде диметил-
формамида.
СХЕМА СИНТЕЗА 1-МЕТИЛ-3.4,5-ТРИАЛ КИЛ ПИРАЗОЛОВ
R CH2R
nri-l К2СОз__
RVHs N (ch3)2ncho->
N /
H
139
R /CH2R
-> RCH2-f
xNz/‘
I
CH3
Характеристика основного сырья
3,4,5-Триалкилпиразолы (см. одноименную статью в на-
стоящем сборнике)-
Диметилформамид, ч., ВТУ РУ—1193—56.
Йодистый метил, ч., ТУ МХП 49—51.
Натр едкий, ч., ГОСТ 4328—48.
Хлороформ, ч., ГОСТ 3160—51.
Натрий сернокислый, безводный, ч., ГОСТ 4156—48.
Условия получения
В трехгорлую колбу, снабженную мешалкой, обратным
холодильником с хлоркальциевой трубкой, помещают 0,1 М
3.4,5-триалкилпиразола, 50 мл сухого диметилформамида и
9,7 г (0,07 М) углекислого калия. Затем при перемешивании
прибавляют за 5 минут 15,6 г (0,11 3'1) йодистого метила.
Реакционную массу при постоянном перемешивании нагрева-
ют на глицериновой бане в течение 2 часов при 75°, затем
температуру в бане за 15 минут поднимают до 120° и выдер-
живают при этой температуре еще 2 часа. После охлаждения
к реакционной массе добавляют 70 мл 10%-ного раствора
едкого натра, тщательно перемешивают и выделившийся ор-
ганический слой экстрагируют 100 мл хлороформа. Экстракт
дважды промывают водой (по 50 мл) и сушат прокаленным
сульфатом .натрия, после чего растворители отгоняют, а ос-
таток фракционируют в вакууме.
Все синтезированные соединения представляют собой
бледно-желтые, легкоподвижные жидкости, смешивающиеся
с органическими растворителями в любых соотношениях и
не смешивающиеся с водой.
Выходы, температуры кипения и физико-химические кон-
станты 1-метил-3,4,5-триалкилпиразолов приведены в табл. 1,
анализы — в табл. 2.
140
Таблица 1
Соединение! Название Выход, % Темпера» тура кипения, °C (мм) Z/20 а4 „2° nD Спектры в циклогексане
Хмакс,ил £макс
I 1-Метил-3,5-ди- этил-4-метилпиразол 88,0 96—97 (20) 0,9358 1,4792 227,5 5400
11 1 -Метил-3,5-дипро- пил-4-этилпиразол 93,5 101—102 (5) 0,9011 1,4784 230 6300
1П 1-Метил-3,5-дибу- тил-4-пропилпиразол 85,0 157 (13) 0,8932 1,4761 230 6500
IV 1-Метил-3,5-дипен- тил-4-бутилпиразол 90,4 164 (3) 0,8805 1,4745 230 6800
V 1 -Метил-3,5-дигек- с ил-4- пентилпиразол 84,7 183-184 (4) 0,8782 1,4732 230 5500
Таблица 2
Соединение Найдено, % Формула М.в. Вычислено, %
С Н N С Н N
I 70,98 71,06 10,48 10,60 18,36 18,30 C9HieN2 152,241 71,01 10,59 18,40
11 74,30 74,20 11,21 11,15 14,50 14,61 Cj-.H^N., 194,324 74,17 11,42 14,41
III 76,45 76,40 12,01 12,20 11,62 11,47 Z 00 СА X о 236,405 76,21 11,94 11,85
IV 77,60 77,58 12,35 12,32 10,02 10,09 C18H34N2 278,486 77,63 12,30 10,07
V 78,93 78,79 12,60 12,77 8,85 8,80 C2iH4ON2 320,565 78,68 12,58 8,74
ЛИТЕРАТУРА
1. L. Knorr. Ann., 279, 232 (1894).
2. И. И. Грандберг, А. Н. Кост. Ж. общ. химии, 30, 203 (1960).
Поступила в мае 1968 г.
ПРЕД
141
УДК 547.586.73
N-МЕТИЛФЕНИЛГЛИЦ.ИН -о-КАРБОНОВАЯ КИСЛОТА
Н. В. ФАКЕЕВА, В. В. ЖИДКОВА
СООН
N—СНаСООН
СН3
С юН11NO4
М. в. 209,20
N-Метилфенилглицин-о-карбоновая кислота применяется
для синтеза N-метилиндоксилацетата [1, 2, 3]. Она получает-
ся метилированием фенилглицин-о-карбоновой кислоты йодис-
тым метилом [3, 4] или кипячением N-метилантраниловой ки-
слоты с небольшим избытком монохлоруксусной кислоты в
концентрированном растворе углекислого натрия [2].
Нами проверен последний способ получения N-метилфе-
нилгл ицин-о-кар боновой кислоты.
СХЕМА СИНТЕЗА N-МЕТИЛФЕНИЛГЛИДИН- о -КАРБОНОВОЙ
КИСЛОТЫ
//\/
СООН
+ ClCH2COOH-|-2Na2CO3
сн3
^/C00Na 2НС. ^\/С00Н
| I! --> | || +2NaCl
N —CH2COONa ^x\n~CH3COOH
142 СНз СН3
Характеристика основного сырья
N-Метилантраниловая кислота, ч., ВТУ РУ 1381.—57.
Монохлоруксусная кислота, ч., ГОСТ 5836—51.
Натрий углекислый, безводный, ч., ГОСТ 83—41.
Соляная кислота, ч., ГОСТ 3118—46.
Активированный уголь, технический, ГОСТ 6213—53.
Условия получения
В круглодонной трехгорлой колбе емкостью 1 л, снабжен-
ной обратным холодильником, мешалкой 'И термометром, рас-
творяют 98 г (0,92 А1) углекислого натрия в 490 мл воды.
При работающей мешалке вносят небольшими порциями
70 г (0,46 А1) N-мстилантраниловой кислоты и 60 г (0,72 М)
монохлоруксусной кислоты. Реакционную массу осторожно
нагревают до температуры кипения (103—104°) и выдержи-
вают при этой температуре в течение 3 часов. Затем подкис-
ляют раствор 100 мл (119 г, 1,2 А4) соляной кислоты до кис-
лой реакции по «конго» и массу охлаждают до 8—10°. Вы-
павший осадок отфильтровывают и сушат при 50—55°.
Выход технического продукта равен 85 г (87,7%); т. пл.
182—184°. Его перекристаллизовывают из 710 мл дистилли-
рованием воды с добавлением 8,5 г активированного угля.
Фильтрат охлаждают до 5—10°, выпавший при этом продукт
отфильтровывают и сушат в сушильном шкафу при 50—55°.
Выход чистой N-метил-фенилглицин-о-карбоновой кисло-
ты равен 70 г, что составляет 72,2%, считая на N-метилант-
оаниловую кислоту; т. пл. 188,5—189° (с разложением).
По литературным данным, т. пл. кислоты 189° (с разл.) [3].
ЛИТЕРАТУРА
1. L. Ettinger, Р. Friedlaender. Вег., 45, 2075 (1912).
2. S. Y. Holt, А. Е. Kellie, D. G. 0‘Sillivan, Р. W. S a n d-
1 е г. J. Chem, Soc., 3, 1217 (1958).
3. D. V о г 1 а п d е г, Е. М u m m е. Вег, 35, 1699 (1902).
4. S. Y. Holt, Р. W. Sadler. Proc. Roy. Soc., 148В, 933 (1958).
Поступила в апреле 1968 г.
ИРЕА
УДК 547.576
5-МЕТОКСИ-З-НИТРОСАЛИЦИЛОВЫЙ АЛЬДЕГИД
5-Метокси-3-нитро-2-оксибенз альдегид
В. Г. БРУДЗЬ, Д. А. ДРАПКИНА, В. А. ИНШАКОВА,
И. Е. БОХЛУНОВА
СНО
НзСО^/^ЫОг
C8H7NO5
М. в. 197,14
5-Метокси-З-нитросалициловый альдегид был получен с
выходом 55% [1] при нитровании 5-метоксисалицилового аль-
дегида азотной кислотой (d—1,42) в ледяной уксусной кис-
лоте при 10—20°. Автор рекомендует применять двенадцати-
кратный избыток азотной кислоты.
Наши попытки воспроизвести синтез в этих условиях ус-
пеха не имели. После добавления примерно третьей части
указанного количества нитрующего агента происходил рез-
кий подъем температуры реакционной массы, сопровождав-
шийся растворением выпавшего до этого нитропродукта.
Альдегид выделить» больше уже не удавалось. Значительно
уменьшив избыток азотной кислоты, мы добились устойчиво-
го выхода продукта, равного 61—65% от теоретического.
СХЕМА СИНТЕЗА
5-МЕТОКСИ-З-НИТРОСАЛИЦИЛОВОГО АЛЬДЕГИДА
СНО
I
ГУ0Н
НзСО-^/11
-ь HNO3
СНзСООН
144
СНО
Н3со/^/ \N0,
Характеристика основного сырья
5-Метоксисалициловый альдегид (получение см. в одно-
именной статье настоящего сборника).
Азотная кислота, ч„ ГОСТ 4461—48.
Уксусная кислота, ледяная, х.ч., ГОСТ 61—51.
Условия получения
В четырехгорлую колбу, снабженную мешалкой, обрат-
ным холодильником, термометром и капельной воронкой, по-
мещают 15,2 г (0,1 М) 5-метоксисалицилового альдегида и
80 мл ледяной уксусной кислоты. Смесь охлаждают до 10° и
в течение 40 минут прибавляют раствор 9,2 мл (0,15 А1)
азотной кислоты (уд. веса 1,42) в 80 мл ледяной уксусной
кислоты, поддерживая температуру 10—18°. Затем дают
30-минутную выдержку при комнатной температуре.
Смесь охлаждают в бане со льдом, осадок отфильтровы-
вают и промывают водой до нейтральной реакции. Получают
11 —12 г альдегида, что составляет 56—61% от теоретическо-
го; т. пл. 129,5—130°. Из реакционного маточника разбавле-
нием водой выделяют дополнительно 0,8—1,5 г с т- пл. 124—
126°. Общий выход продукта равен 12,5—12,8 г. После пере-
кристаллизации из 50 %-ной уксусной кислоты получают
11,6—12 г 5-метокси-З-нитросалицилового альдегида или 59—
61% от теоретического количества; т. пл. 130—131°.
По литературным данным, т. пл- продукта 132° [1].
ЛИТЕРАТУРА
1. L. R u b i n s t е i n. J. Chem. Soc., 127, 2000 (1925).
Поступила в феврале 1968 г. ИРЕА
10 Зак. 10И
УДК 547.576
5-МЕТОКСИСАЛИЦИЛОВЫЙ АЛЬДЕГИД
2-Окси-5-метоксибензальдегид
В. А. ИНШАКОВА, Д. А. ДРАПКИНА, В. Г. БРУДЗЬ,
Н. Е. БОХЛУНОВА
СНО
Г|°н
НзСОЧ/
С8Н8О3 М. в. 152,15
5-Метоксисалиниловый альдегид получен по Реймер-Ти-
ману с выходами 25% [1] и 64,5% [2]. В работе [1] вещество
очищали через бисульфитное соединение.
Альдегид представляет собой масло и охарактеризован
в литературе лишь температурой кипения в вакууме при
разных давлениях.
Нами проверен метод [2], но с предварительным выделе-
нием продукта ’в виде бисульфитного соединения, а также
определен показатель преломления альдегида.
СХЕМА СИНТЕЗА 5-МЕТОКСИСАЛИЦИЛОВОГО АЛЬДЕГИДА
ОН СНО
^\он
| IS + CHCls + NaOH -> | II
X/ Н8С0Х/
OCH8
Характеристика основного сырья
Монометиловый эфир гидрохинона, ч., ВТУХСНХ341—60.
Хлороформ, ч., ГОСТ 3160—51.
Натр едкий, ч., ГОСТ 4328—48.
146
Серная кислота, ч., ГОСТ 4204—48.
Диэтиловый эфир, фарм., Ф.1Х.
Бисульфит натрия, ч., ТУ МХП 1944—49.
Условия получения
В четырсхгорлую колбу, снабженную мешалкой, обратным
холодильником, термометром и капельной воронкой, загру-
жают 125 г (1 Л4) монометилового эфира гидрохинона и
320 г едкого натра, растворенного в 400 мл воды, нагревают
до 70° и при размешивании прибавляют 160 мл хлороформа
за 2,5 часа, поддерживая температуру 70—80°. После 15-ми-
нутной выдержки добавляют 500 мл воды, охлаждают и под-
кисляют 10 н. раствором серной кислоты до слабокислой ре-
акции. От полученной смеси продукт отгоняют с водяным
паром и из дистиллата экстрагируют эфиром. Эфирный рас-
твор встряхивают на механической качалке с 40%-ным рас-
твором бисульфита натрия (500 мл). Бисульфитное соедине-
ние отфильтровывают, промывают- спиртом и эфиром и раз-
лагают 10—20%-ным раствором серной кислоты при нагрева-
нии на водяной бане в течение 20—30 минут. Альдегид извле-
кают эфиром, эфирный экстракт сушат прокаленным сульфа-
том натрия, эфир отгоняют, а остаток перегоняют в вакууме.
Выход 5-метоксисалицилового альдегида равен 62—75 г,
что составляет 41—49% от теоретического; т. кип. 116°/9 мм;
124°/12 мм; п2° =1,5789.
По литературным данным, т. кип- продукта 124°/12 мм [1]:
133715 мм [2].
ЛИТЕРАТУРА
1. L. Rubinstein. J. Chem. Soc., 127, 1998 (1925).
2. Н. В. L i 11 е s р i е. Biochem. Preparat., 3, 79 (1953),
Поступила в феврале 1968 г. ИРЕА.
10*
УДК 547.425.05
N о-МЕТОКСИФЕНИЛФОРМАМИДИНСУЛЬФИНОВАЯ
КИСЛОТА
Е. Я. ЯРОВЕНКО, Р. П. ЛАСТОВСКИЙ
' Н!Ч
V-so2h
ch3oc6h4nhx
CsH)0N2O3S М. в. 214,25
N-о-Метоксифенилформамидинсульфиновая кислота в ли-
тературе не описана. Она, как и другие производные форма-
мидинсульфиновых кислот, может представить интерес в ка-
честве физиологически активного вещества.
СХЕМА СИНТЕЗА
N-о МЕТОКСИФЕНИЛФОРМАМИДИНСУЛЬФИНОВОЙ КИСЛОТЫ
HN.
^С-SH+2H2O2 —
CH3OC6H4NH
HN.
— /С—SO2H4-2H2O
ch3oc6h4nh
Характеристика исходного сырья
N-о-Метоксифенилтиомочевина, т. пл. 144—145°.
Перекись водорода, медицинская, ГОСТ 177-Г-55.
Условия получения
В трехгорлую колбу, снабженную мешалкой, термометром
и капельной воронкой, загружают 1,2 г. (0,007 А1) N-о-меток-
148
сифенилтиомочевины, 0,002 г молибденовокислого аммония и
приливают 24 мл воды. Полученную суспензию нагревают до
50°, при интенсивном размешивании добавляют Змл (0,02 А1)
перекиси водорода, реакционную массу отфильтровывают; из
фильтрата при охлаждении выпадает осадок, который промы-
вают ледяной водой и сушат при 60°. Получают 0,6 г N-o-ме-
токсифенилформамидинсулфиновой кислоты, что составляет
42,8% от теоретического выхода.
Найдено, %: С—41,63; 41,77; Н—4,54; 4,84;
N—11,76; 11,67.
C8HioN203S. Вычислено, %: С—41,37; Н—5,15; N—12,07.
По внешнему виду вещество представляет собой белые
кристаллы с т. пл. 142—143°, растворяется в спирте и воде,
не растворяется в неполярных растворителях.
Поступила в феврале 1968 г. ИРЕА
УДК 547.831.8
2-(п-МЕТОКСИФЕНИЛ)-4Н-3,1-БЕНЗОКСАЗОН-4
Анизоилантранил
Б. М. БОЛОТИН, Д. А. ДРАПКИНА, В. Г. БРУДЗЬ,
Р. У. СУДИЯРОВА
С^НпМОз
М. в. 253,26
2-(n-Метоксифенил)-4Н-3,1-бензоксазон-4 в литературе
описан [1]. Он может быть использован в качестве органичес-
кого люминофора фиолетового свечения. Известен общий спо-
соб получения аналогичных соединений, заключающийся з
обработке антраниловой кислоты соответствующим хлоран-
гидридом в среде пиридина [2].
СХЕМА СИНТЕЗА 2-( га-МЕТ0КСИФЕНИЛ)-4Н-3,1-БЕН30КСА30НА-4
. ч соон.
\ Xliz
сн3о—Z \-coci
C5HfjN
-OCH3
150
Характеристика основного сырья
Антраниловая кислота, ч., ТУ МХП-1713—48.
Анизоилхлорид, ч., СТУ 79—59—X—61.
Пиридин, ч., ГОСТ 2747—44.
Условия получения
В колбу емкостью 150 мл, снабженную мешалкой, термо-
метром и капельной воронкой, помещают раствор 1,7 г
(0,013 Л4) антраниловой кислоты в 50 мл пиридина. При тем-
пературе 10—15° прикапывают раствор 2,2 г (0,013 Л1) ани-
зоилхлорида в 50 мл пиридина. Реакционную массу охлаж-
дают ледяной водой. По окончании прибавления смесь пере-
мешивают еще 15 минут, отфильтровывают выпавшие кри-
сталлы и промывают 10%-ным раствором соды, а затем пере-
кристаллизовывают из спирта.
Выход равен 1,6 г, что составляет 49% от теоретического;
т. пл. 157°.
Найдено, %: С—71,53; 71,41; Н—4,63; 4,67; N—5,24; 5,50.
CisHnNOs. Вычислено, %: С—71,14; Н—4,37; X—5,56.
ЛИТЕРАТУРА
1. С. L. А г reus, R. Е. Marks. J. Chem, Soc., 1956, 1627.
2. G. Heller, G. Fi e s s e 1 tn a n n. Ann., 324, 133 (1902).
Поступила в апреле 1967 г.
И PEA
УДК 547.571
о-НИТРОБЕНЗАЛЬДЕГИД
Г. А. КРЕЙМЕР
no2
C7H5NO3
М. в. 151,12
В лабораторной практике нашел применение метод по-
лучения о-нитробензальдегида посредством окисления ©-ни-
тротолуола хромовым ангидридом в среде уксусного ангид-
рида и последующего гидролиза образующегося о-нитрсбенз-
альдиацетата [1, 2]. Согласно имеющимся в литературе про-
писям, омыление диацетата ведется минеральными кислота-
ми {I, 2] или кальцинированной содой [3]; получаемый при
этом продукт бывает сильно загрязнен смолистыми примеся-
ми и для очистки его необходимо перегонять с водяным па-
ром. Эта операция требует громоздкого аппаратурного офор-
мления, отнимает много времени и ограничивает масштабы
синтеза.
Нами разработана другая методика, позволяющая обой-
тись без перегонки с паром.
СХЕМА СИНТЕЗА о-НИТРОБЕНЗАЛЬДЕГИДА
no2 no,
I I
^-^ососнл + н2о —— |^Х|ГСИО 4-2сн3соон
Ч/ X/
Характеристика основного сырья
о-Нитробензальдиацетат, т. пл. 82—88° [1, 2].
Муравьиная кислота, техн., ГОСТ 1706—42
152
Изопропиловый спирт, техн., ГОСТ 9805—61.
Вода дистиллированная, ГОСТ 6709—53.
Условия получения
В колбу емкостью 0,5 л помещают 100 г о-нитробензаль-
диацетата и 5 г угля, приливают 200 мл муравьиной кисло-
ты и 50 мл дистиллированной воды. Смесь кипятят 2 часа с
обратным холодильником, после чего охлаждают до 20—25°.
раствор отфильтровывают от угля и уголь промывают 25 мл
муравьиной кислоты. Раствор выливают при размешивании
в охлажденную до 2—5° дистиллированную воду (1000 мл).
Реакционную массу выдерживают при 2—5° в течение двух
часов, выпавший о-нитробензальдегид отсасывают, промыва-
ют дистиллированной водой до pH 7 и сушат на воздухе.
Выход светло-желтого цвета продукта равен 52 г, что со-
ставляет 87,2% от теоретического; т. пл. 42—43°.
Для получения чистого препарата полученный о-нитро-
бензальдегид растворяют при нагревании (40—50°) в 210мл
изопропилового спирта, несколько минут размешивают с 3 г
угля, отфильтровывают и уголь промывают 25 мл спирта.
Раствор выливают при размешивании в охлажденную до 2—•
5е дистиллированную воду (1000 мл), выдерживают при 2—
5е два часа, выпавший осадок отжимают и сушат на воздухе.
Выход продукта равен 46 г, что составляет 77,1% °т тео-
ретического; т. пл. 43—44°.
Л ИТЕРАТУРА
1. Синтезы органических препаратов, т. 3. М., ИЛ, 1952, стр. 348.
2. Синтезы органических препаратов, т. 8. М., ИЛ, 1958, стр. 37.
3. Р. М. Гельштейн, Ф. В. Александрова, Г. А. Креймер.
Авт. свид. 164588; Бюлл. изобр., 16 (1964).
Поступила в ноябре 1967 г. ЦЗЛ
УДК 547.796.1'631.4
НИТРОСИНИЙ ТЕТРАЗОЛИЙ (НИТРО-СТ)
2,2/-Ди-(4-нитрофенил)-5,5'- дифенил-3,3/-(3,3/- диметокси-
4,4'-дифенилен)дитетразолий хлористый
В. М. ОСТРОВСКАЯ, В. И. ЯКУШИНА
2С1-
СН3ОН
С40НзоС12К10Об, СНзОН
М. в. 817,65 + 32,04
Нитросиний тетразолий является одним из важных гис-
тохимических реагентов (1—5]. Нами усовершенствован ме-
тод его очистки: вместо переосаждения из метанола боль-
шим объемом диоксана мы применили промывку продукта
этанолом с последующей перекристаллизацией из метанола,
при этом качество и устойчивость вещества в растворах по-
высились [6]
СХЕМА СИНТЕЗА НИТРОСИНЕГО ТЕТРАЗОЛИЯ
Н3СО ОСН3
. N=N y~N = N\
S Ч_с/ ч==/ \
\ = / 4N--N-H H-N-N<
154
Характеристика основного сырья
Динитродиформазан, т. пл. (разл.) не ниже 270—272°.
Изоамилнитрит, ч-, ТУ 22—55.
Аммоний хлористый, ч., ГОСТ 3773—47.
Кислота серная, ч., ГОСТ 4204—48.
Диоксан, ч., ВТУ МХП 311—52, высушен над твердым ед-
ким калием и перегнан.
Спирт этиловый, ректифицированный, ГОСТ 5962—51.
Метанол, ГОСТ 6995—54.
Эфир диэтиловый, ч., ГОСТ 6265—52.
Условия получения
В круглодонную трехгорлую колбу на 2 л, снабженную
мешалкой и барбатёром, помещают 50 г (0,0667 А4) динитро-
диформазана и 1,5 л предварительно просушенного диокса-
на. Смесь интенсивно размешивают, приливают 70 мл
(0,465 М) изоамилнитрита и осторожно пропускают в тече-
ние 4, часов сухой хлористый водород. Затем добавляют
еще 20 мл (0,133 Л4) изоамилнитрита и продолжают пропус-
кать хлористый водород в течение двух часов, после чего
три часа размешивают реакционную массу при 20—25°. За-
тем добавляют 19 мл (0,126 Л4) изоамилнитрита и при раз-
мешивании снова пропускают ток хлористого водорода в те-
чение двух часов. В результате окисления динитродиформа-
зана количество его в реакционной смеси уменьшается и
цвет ее меняется, постепенно переходя в желтый.
Для полноты окисления динитродиформазана реакцион-
ную омесь оставляют на ночь. Цвет ее становится светло-
желтым или зеленовато-желтым.
В ходе реакции постоянно проверяют наличие избытка
изоамилнитрита в реакционной смеси по йодкрахмальной
бумаге и кислотность среды (рН<1—3) по универсальной
индикаторной бумаге.
Затем смесь размешивают в течение трех часов, предо-
155
храняя ее от дневного света (колбу закрывают черной бума-
гой), охлаждают до 5°. образовавшийся осадок отфильтро-
вывают на воронке Бюхнера через плотный бумажный
фильтр, промывают на фильтре 250 мл диэтилового эфира и
сушат над серной кислотой в эксикаторе с открытым краном
в течение 3 дней, предохраняя от действия света.
Выход технического нитросинего тетразолия равен 62 г;
т. пл. 162—190,5° (с разя.).
Для очистки его помещают в коническую колбу емкостью
500 мй, заливают 250 мл этилового спирта, размешивают
в течение 10 минут, охлаждают до 5°, отфильтровывают, про-
мывают на фильтре 100 мл диэтилового эфира, сушат в ва-
куум-эксикаторе и получают 55 г вещества с т. пл. 192—
200° (с разл.).
Для перекристаллизации в коническую колбу емкостью
0,5 л, снабженную обратным холодильником на шлифе, за-
гружают 55 г нитросинего тетразолия и 180 мл метанола н
нагревают на глицериновой бане до растворения. Затем рас-,
твор фильтруют, охлаждают в течение двух часов до темпе-
ратуры— 20° и дают затравку, при этом выпадает осадок,
который отфильтровывают на воронке Бюхнера через плот-
ный бумажный обеззоленный фильтр (см. примечание 2).
Во время фильтрования воронку закрывают чашкой Петри
и черной бумагой. Отфильтрованный продукт быстро перено-
сят во взвешённую чашку Петри и помещают над концентри-
рованной серной кислотой и твердым едким кали в вакуум-
эксикатор, защищенный от света черной бумагой.
Выход нитросинего тетразолия равен 43 г, что соответ-
ствует 76% от теоретического. По внешнему виду вещество
представляет собой мелкие светло-желтые кристаллы, его
оптическая плотность (ФЭК) 0,07, коэффициент молярного
погашения в метаноле 82000, потенциал полуволны второй
волны полярографического восстановления по отношению к
насыщенному каломельному электроду—0,38 'в; т. пл. 189,5—
191,5° (с разл.).
По литературным данным, температура плавления нит-^
росинего тетразолия после перекристаллизации из метанола
равна 180—182° [9]; 184° [7, 8]; 187—188° [6].
Примечания:
1. Хлористый водород получают в колбе Вюрца прн медленном до-
бавлении по каплям 63 м.л (1,2 М) концентрированной серной кислоты
к 150 г (2,76 Д4) хлористого аммония прн подогреве. Пропускание хло-
ристого водорода ведут через склянку Тищенко с концентрированной
серной кислотой. Для улавливания брызг кислоты хлористый водород
пропускают через склянку Дрекселя, наполненную стеклянной ватой.
2. Из метанольного маточника можно осадить днэтиловым эфиром
или диоксаном дополнительно 7 г сырого продукта. -
156
ЛИТЕРАТУРА
1. А. В, Johnson. J. Histochem. and Cytochem, 13, 7, 583 (1965).
2 К. Balogh. J. Histochem. and Cytochem., 14, 1, 77 (1966).
3. I. M. Genis-Gal ver. Nature, 213, 5073, 283 (1967).
4. K. Balogh. J. Histochem., a 13, Cytochem. 7, 533 (1965).
5. J. F. Goggins, H. M. F u 11 m e r. Arch. Oral. Biol., 11. 12, 1365
(1965); РЖбиохим. 15Ф431 (1966).
6. M. H. Малев. Авт. свид. 189105; Изобретения. Промобразцы и
товарные знаки, № 23, (1966).
7. S. S. К а г m а г k а г, J. В а г г п е 11. М. М. N а с h I о s, А. М.
Seligman. J. Amer. Chem. Soc., 81, 3771 (1959).
8. А. А. Прянишников, В. M. Островская, Н. Т. Р а й х л и и.
Химические реактивы и препараты, вып. 25, М., ИРЕА, 1963, стр. 139—
148.
9. В. М. Островская, А. А. Прянишников, О. Т. Лушина.
Методы получения хим. реактивов и препаратов, вып. 8, М., ИРЕА, 1964,
стр. 24.
Поступила в ноябре 1967 г. ИРЕА
УДК 5.47.724.2.
5-ОКСИМЕТ.ИЛ ФУРФУРОЛ
II. II. ШМАГИНА, И. П. ЕРМАКОВА, В. И. ФЕДОТОВА,
А. И. КУРАШЕВА
нс-сн
II II
НОН2С-С С-СНО
С6Н60з
М. в. 126,07
5-Оксиметилфурфурол является сырьем для получения
эфиров, гликолей, диальдегидов, ацеталей, которые могут
быть использованы для производства смол, пластификаторов,
поверхностноактивных веществ.
В литературе описано получение 5-оксиметилфурфурола
дегидратацией сахарозы в присутствии щавелевой кислоты
[1] и иода [2].
Нами проверены литературные данные и уточнены усло-
вия получения и выделения 5-оксиметилфурфурола.
СХЕМА СИНТЕЗА 5-ОКСИМЕТИЛФУРФУРОЛА
Характеристика основного сырья
Сахароза, ч.д.а-, ГОСТ 5833—54.
Щавелевая кислота, ч., ГОСТ 5873—51.
Кальций углекислый, ч., ГОСТ 4530—48-
Свинец уксуснокислый основной, ч., ВТУ МХП 2907—54.
158
Этиловый эфир уксусной кислоты (этилацетат), Ст. ГОХП
27—1839.
Магний сернокислый, ч., ГОСТ 4523—48.
Условия получения
В трехлитровый автоклав, снабженный мешалкой, карма-
ном для термопары и манометром, загружают 1500 мл воды,
500 г сахарозы, 3,5 г щавелевой кислоты, нагревают до 130°
и при этой температуре выдерживают 2,5 часа. После охлаж-
дения смесь выгружают и отфильтровывают от гуминовых
веществ. Фильтрат помещают в круглодонную колбу, снаб-
женную мешалкой и при перемешивании небольшими порци-
ями за 1.5 часа добавляют 90 г углекислого кальция, а за-
тем — 30 г уксуснокислого свинца и перемешивают еще пол-
часа. Осадок отфильтровывают, фильтрат в количестве
1600 мл загружают в трехгорлую колбу, снабженную мешал-
кой, обратным холодильником, термометром, и при перёме-
шивании экстрагируют 1600 мл этилацетата. Экстракцию
повторяют три раза при температуре 40—50°. Экстракт су-
шат сернокислым магнием, отгоняют растворитель в вакууме
водоструйного насоса в токе азота и получают 40—45 г 5-ок-
симетилфурфурола в виде коричневой жидкости (бромное
число 248—243), что составляет 50% от теоретического вы-
хода, считая на фруктозную часть.
5-Оксиметилфурфурол кристаллизуется в виде бесцветных
игл, плавящихся при 31—33°. Он почти не имеет запаха,
очень гигроскопичен, на воздухе быстро расплывается, с во-
дой смешивается во всех отношениях; температура кипения
115—12070,5 мм.
Получен семикарбазон с т. пл. 191° (из этанола).
По литературным данным его т. пл. 192° (1].
Найдено, %: С- -15.9; Н—5.0; N—23,1.
С7Н90з.Мз. Вычислено, %: С—45,8; Н—4,8; N—23,0.
ЛИТЕРАТУРА
1. W. Hawort, W. Jones. J. Chem. Soc., 1944, 667.
2. T. G. Bonner, E. J. В о u r k e, T- Rusz kiewiez, J. Chem.
Soc. 1960, 787.
Поступила в сентябре 1967 г.
Н ИИПластмасс
УДК 5.47.461.4
N-ОКСИСУКЦИНИМИД
Г. Н. КОШЕЛЕВА, А. К. САЛ ГУС
, СН2-СО.
I ;noh
СН2-ССГ
C4H5NO3 М. в. 115,08
N-Оксисукцинимид, применяемый в синтезе пептидов, по-
лучают взаимодействием гидроксиламина и янтарного ангид-
рида [1, 2, 3].
Нами проверен и уточнен один из описанных методов син-
теза, который позволяет получить продукт с выходом 75% от
теоретического.
СХЕМА СИНТЕЗА N-ОКСИСУКЦИНИМИДА
сн2-сок сн,-со.
I ;о + nh,oh -> 1 ’ \noh I Н2о
СНо-СО7 " СНа-СО7
Характеристика основного сырья
Гидроксиламин солянокислый, ч., ГОСТ 5456—51.
Янтарный ангидрид, ч., ВТУ ГКХ 1439—60.
Диоксан, ГОСТ 10455—63.
Натр едкий, ч., ГОСТ 4328—48.
Этиловый эфир (для наркоза), ГОСТ 6205—52.
Вода дистиллированная, ГОСТ 6709—53.
Условия получения
В двухлитровую колбу с мешалкой, термометром и за-
грузочным отверстием помещают 208,5 г (3 М) солянокисло-
го гидроксиламина, 300 мл воды, 150 мл диоксана и 600 мл
160
5 н- раствора едкого натра. Затем при охлаждении добавля-
ют 300 г (3 М) янтарного ангидрида таким образом, чтобы
температура массы не поднималась выше 20°.
Смесь нагревают до 60°, подключают вакуум водоструйно-
го насоса и отгоняют воду и диоксан, при этом температуру
массы постепенно повышают до 70—80°, на что требуется
около трех часов после чего вакуум отключают. Остаток на -
гревают при нормальном давлении до 120° и снова подклю-
чают вакуум водоструйного насоса, медленно повышая тем-
пературу до 160°.
В этих условиях '.массу выдерживают около 1,5 часов до
тех пор, пока не перестанет отгоняться вода (от полноты от-
гонки воды зависят качество и выход продукта), и после ох-
лаждения до 80° добавляют 400 мл высушенного над хлори-
стым кальцием этилацетата. Смесь хорошо перемешивают,
помещают в бязевый патрон и экстрагируют в приборе Сок-
слета в течение 10—12 часов, добавив в колбу прибора
1600 мл этилацетата и 7 г активированного угля.
Горячий раствор отфильтровывают от активированного
угля и охлаждают до комнатной температуры. Выпавшие
кристаллы N-оксисукцинимида отфильтровывают, промывают
400 мл эфира и, сушат при 50° в вакууме водоструйного на-
соса в течение 3 часов.
Выход N-оксисукцинимида равен 260 г, что. составляет
75% от теоретического; т. пл. 95—97°.
N-Оксисукцинимид очень гигроскопичен, поэтому его сле-
дует тщательно оберегать от влаги воздуха.
ЛИТЕРАТУРА
1. G. W. Anderson, J. Е. Zimmerman, F. М. Callahan,
J. Amer. Chem. Soc., 86, 1839(1964).
2. К u n g-T sung Wang, D. N, В r a 11 e s a n i, B. We i nst e i
J. Heterocyclic chetnie, 3, 98 (1966).
3. E. Wiinsc h, E. Jaeger. Zeitschrift fiir physiol chemie, 346
301 (1966).
Поступила в апреле 1968 г. Ин-т. химии приролн.
соединений АН СССР
11 Зак. 1034
УДК 547.542
N-o -ОКСИФЕН ИЛФОРМАМИДИНСУЛЬФИНОВАЯ
КИСЛОТА
Е. Я. ЯРОВЕНКО, Р. П. ЛАСТОВСКИЙ
№
^C~SO8H
HOC6H4NH
C7H8N2.O3S
М. в. 200,22
N-o-Оксифенилформамидинсульфнновая кислота в лите-
ратуре не описана. Она, как и другие производные формами-
динсульфиновых кислот, может представить интерес в каче-
стве физиологически активного вещества.
N- о -ОКСИФЕНИЛФОР^АМИДИНСУЛ3ЬФИНОВОЙ КИСЛОТЫ
HN HN
/С—SH+2H2O2 -> /С-SO2H + 2H2O
hocgh4nh hoc6h4nh
Характеристика исходного сырья
N-о-Оксифенилтиомочевина, т. пл. 150—153°.
Перекись водорода, медицинская, ГОСТ 177—55.
Условия получения
В трехгорлую колбу, снабженную мешалкой, термомет-
ром и капельной воронкой, загружают 2 г (0,01 М) N-о-ок-
сифенилтиомочевины, 0,002 г молибденовокислого' аммония и
приливают 20 мл воды. Полученную суспензию охлаждают
до 5°, по каплям при интенсивном размешивании добавляют
5,6 мл (0,04 М) перекиси водорода, реакционную смесь вы-
162
держивают при этой же температуре в течение 30 минут.
Полученный продукт отфильтровывают и сушат при комнат-
ной температуре.
Получают L4 г N-о-оксифенилформамидинсульфиновой
кислоты, что составляет 60% от теоретического выхода.
Найдено, %: С—42,45; 42,55; Н—4,27; 4,53; N—13,73; 13,68.
CzHeNaOaS. Вычислено, %: С—42,00; Н—4,00; N—14,00.
По внешнему виду вещество представляет собой белые
кристаллы с т- пл. 147—148°; растворяется в спирте, не рас-
творяется в воде и неполярных растворителях.
Поступила в феврале 1968 г. ИРЕА
УДК 547.831.8
1-(8-ОКСИ-2-ХИНОЛИЛ)-2-ПРОПАНОН
2-Ацетонил-8-оксихинолин;
Метил-(8-оксихинальдил) кетон
В. М. ДЗИОМКО, И. А. КРАСАВИН, Ю. П. РАДИН
1 " /
^/'XN^\cH3COCHs
C12HnNO2
М. в. 201,23
В литературе 1-(8-окси-2-хинолил)-2-пропанон не описан.
Нами установлено, что он образуется с 45—47%-ным выхо-
дом при взаимодействии метосульфата 8-окси-1-метоксихино-
линия с натриевым енолятом 2,4-пентандиона в среде абсо-
лютного метанола.
Положение ацетонильной группы определено на основа-
нии результатов ее гидролитического расщепления: прй на-
гревании кетона с концентрированным раствором едкого ка-
ли с 89%-ным выходом образовался 2-метил-8-оксихинолин.
1-(8-Окси-2-хинолил)-2-пропанон почти нерастворим вво-
де, но растворяется в ацетоне, бензоле, спиртах и других ор-
ганических растворителях, из этанола кристаллизуется в зо-
лотисто-желтых иглах с т. пл. 178,5—179,5°.
Являясь производным 8-оксихинолина, 1-(8-окси-2-хино-
лил)-2-пропанон принадлежит к числу хелатообразующих
соединений.
СХЕМА СИНТЕЗА
1-(8-ОКСИ-2-ХИНОЛ ИЛ)-2-ПРОПАНОНА
он осн.
CH3SO4-
I II /I
^/XN/XCH.2COCH3
он
+ NaCH3SO4+CH3COOCH3
Условия цолучения
Сухую чстырехгорлую колбу емкостью 750 мл со шлифами
снабжают мешалкой с ртутным затвором, термометром, ка-
пельной воронкой и обратным холодильником с хлоркальци-
евыми трубками. В колбу помещают 4,60 г (0,2 г-ат) наре-
занного металлического натрия и постепенно прибавляют к
нему из капельной воронки 70 мл абсолютного метанола.
Когда растворение натрия закончится (см. примечание 1),
раствор метилата охлаждают до 5—7° и при размешивании
добавляют к нему 24,0 г (0,24 М) свежеперегнанного 2,4-пеп-
тандиона. Затем колбу погружают в баню со льдом и охлаж-
дают ее содержимое до 2—4°.
Одновременно в круглодонной колбе, снабженной обрат-
ным холодильником с хлоркальциевой трубкой, растворяют
28,73 г (0,1 М) метосульфата 8-окси-1-метоксихинолиния
(см. примечание 2) в 190 мл кипящего абсолютного метанола:
колбу нагревают на глицериновой бане (110—115° в бане) и
помешивают вручную ее содержимое, чтобы ускорить раство-
рение вещества.
Прозрачный оранжево-красный раствор охлаждают до
3—5° и добавляют по каплям за 1 час к раствору натриевого
призводного дикетона при размешивании и охлаждении
льдом. Прибавление ведут с такой скоростью, чтобы темпе-
ратура реакционной массы не превышала 5°, а затем про-
должают перемешивание при 2—4° в течение еще двух часов.
После этого добавляют 40 мл 50 %-ной уксусной кислоты
и размешивают массу при охлаждении два часа. Желтый
осадок отсасывают, тщательно отжимают стеклянной проб-
кой, промывают на'фильтре этанолом (2 раза по 15—20 мл)
и дистиллированной водой (2—3 раза по 30 мл), вновь от-
жимают и высушивают в вакуум-эксикаторе.
Выход 1-(8-окси-2-хинолил)-2-пропанона равен 9,2—9,5 г,
что составляет 45,7—47,2% от теоретического; т. пл. 176,5—
177,5°.
Для очистки вещество, по возможности быстро растворя-
ют в 250—260 мл этанола, заранее нагретого до кипения,
раствор кипятят в течение 5 минут с 2 г активированного
угля и фильтруют с отсасыванием через воронку для горяче-
го фильтрования. Фильтрату дают остыть до комнатной тем-
пературы, затем его помещают на 2—3 часа в баню со
ЛЬДОМ.
165
Выпавшие золотисто-желтые игольчатые кристаллы отса-
сывают, промывают этанолом (15—20 мл), высушивают в
вакуум-эксикаторе и получают 6,2—7,0 г кетона с т. пл.
178,5—179,5° (см. примечание 3).
Примечания:
1. Под конец реакция замедляется и для растворения остатков нат-
рия целесообразно подогреть раствор до кипения.
2. Применялся метосульфат 8-окси-1-метоксихинолиния, приготовлен-
ный по описанному нами ранее методу [1]. Пригоден также сырой про-
дукт, полученный нагреванием 16,12 г (0,1 Л4) 8-окснхинолин-1-оксида
[2] и 12,61 г (0,1 М) свежеперегнанного нейтрального диметилсульфата в
течение 1—2 часов при 100—103° (температура бани).
3. При дальнейшей очистке температура плавления не повышается.
Чистый образец 1-(8-окси-2-хинолил)-2-пропанона был приготовлен по-
вторной перекристаллизацией и проанализирован.
Найдено, %: С—71,52; 71,56; Н—5,58; 5,67; N—6,88; 7,04.
CiaHnNOa. Вычислено, %: С—71,63; И—5,51; N—6,96.
ЛИТЕРАТУРА
1. И. А. Красавин, В. М. Д з и о м к о, Ю. П. Радин. Методы по-
лучения химических реактивов и препаратов, вып. 13, М., ИРЕА, 1965,
стр. 68.
2. К. Ram ala h, V. R. Srinivasan. Proc. Indian. Acad. Scf., 55A
360 (1962).
Поступила в апреле 1968 г.
ИРЕА
УДК 547.831.5
8-ОКСИХИНОЛИН-2-СУЛЬФОКИСЛОТА
И. А. КРАСАВИН, В. М. ДЗИОМКО, И. В. РУБЦОВ
I II I
^S-^sOsH-HsO
он
Cf,H7NO4S-H2O
М. в- 243,24
В литературе 8-оксихинолин-2-сульфокислота не описана,
но ее S-бензилизотиурониевая соль упоминается в моногра-
фии [1]. Однако приведенные там характеристики (т. пл. и
ИК-спектры) приписаны этому веществу ошибочно и принад-
лежат, вероятно, S-бензилизотиурониевой соли 8-оксихино-
лин-5-сульфокислоты [2].
Нами установлено [3], что 8-оксихинолин-2-сульфокислота
может быть получена с 85%-ным выходом в результате ре-
акции неизвестного ранее типа, происходящей при смешива-
нии водных растворов метосульфата 8-окси-1-метоксихиноли-
ния и сульфита натрия.
Положение сульфогруппы доказано превращением про-
дукта реакции в соединения известного строения: при крат-
ковременном нагревании сульфокислоты с гидразин-гидра-
том получен 8-окси-2-гидразинохинолии (выход 89%), а при
кипячении ее водного раствора наблюдалось выделение сер-
нистого ангидрида и образовался 2,8-диоксихинолин (8-окси-
карбостирил) с 90%-ным выходом. Подобные реакции ха-
рактерны для хинолин-2-сульфокислот, отличающихся высо-
кой подвижностью сульфогруппы [4, 5].
Синтез 8-оксихинолин-2-сульфокислоты осуществлен нами
также и по методу, обычно применяемому для получения хи-
нолин-2-сульфокислот из 2-хлорхинолинов [5]. После 12-ча-
сового нагревания 2-хлор-8-оксихинолина с водным раство-
ром бисульфита натрия выход сульфокислоты составил 50%.
1G7
Ооразцы 8-окСихинолин-2-сульфокислоты, полученные
обоими методами, идентифицированы по отсутствию депрес-
сии в пробе смешения их Б-бен^илизотиурониевых солей
(т. пл. 193—194° с разл.).
8-Оксихинолин-2-сульфокислота умеренно растворима в
теплой воде, метаноле и этаноле, значительно хуже раство-
ряется в концентрированной соляной кислоте, нерастворима
в ацетоне, бензоле, диоксане и хлороформе; из разбавленной
соляной кислоты кристаллизуется в светло-желтых мелких
иглах с температурой разложения 270°, содержащих 1 моле-
кулу кристаллизационной воды. Обезвоживание происходит
при нагревании моногидрата до 105—110° или после длитель-
ного высушивания его в вакуум-эксикаторе.
8-Оксихинолин-2-сульфокислота является хелатообразую-
щим соединением и, благодаря легкости обмена сульфогруп-
пы на различные заместители, может служить ценным проме-
жуточным продуктом в синтезе других производных 8-окси-
хинолина.
СХЕМА СИНТЕЗА 8-ОКСИХИНОЛИН-2-СУЛЬФОКИСЛОТЫ ИЗ
МЕТОСУЛЬФАТА 8-ОКСИ-1-МЕТОКСИХИНОЛИНИЯ
J || I + Na,SO3 -» | || ! +
YYw- YXN^4sQ-iNa
0H OCH3 0H
F NaCHsSO. + CHSOH
+ HC1 + H2O -*
+ NaCl
Условия получения
В четырехгорлой колбе емкостью 750 мл, снабженной ме-
шалкой с затвором, термометром, капельной воронкой и от-
водной трубкой, растворяют 25,2 г (0,2 Л4) чистого ' безвод-
ного сернистокислого натрия в 200 мл дистиллированной во-
ды. Раствор охлаждают с помощью ледяной бани и при 2—3°
168
добавляют к нему за 30 минут при размешивании раствор
28,73 г (0,1 М) мётосульфата 8-окси-1-метоксихинолиния (см.
примечание 1) в 60 мл воды. Вскоре начинается выделение
светло-кремового мелкого осадка натриевой соли сульфокис-
лоты.
Реакционную массу перемешивают при 0—2° еще 2,5—
3 часа, затем отводную трубку прибора подсоединяют к ло-
вушке с раствором щелочи и добавляют в колбу из капель-
ной воронки 50 мл 37%-ной соляной кислоты при хорошем
размешивании и охлаждении. Суспензию, цвет которой при
подкислении изменяется от кремового до желтого, переме-
шивают в течение 2—3 часов и оставляют на 10—15 часов в
бане со льдом.
На следующий день холодную (0—2°) суспензию фильт-
руют с отсасыванием, осадок тщательно отжимают, промыва-
ют на фильтре ледяной водой (20—30 мл) и этанолом (15—
20 мл) и высушивают в вакуум-эксикаторе над едким нат-
ром.
Выход сульфокислоты равен 19,2—20.7 г, что составляет
79—85% от теоретического.
Для очистки вещество по возможности быстро растворя-
ют при размешивании в 1200—1400 мл дистиллированной во-
ды, заранее нагретой до 50—55° (см. примечание 2), Раствор
фильтруют с отсасыванием, добавляют к еще теплому филь-
трату 250 мл 37%-ной соляной кислоты и оставляют его на
1,5—2 часа для кристаллизации, а затем охлаждают льдом
в течение 1,5—2 часов. Выпавшие кристаллы отсасывают, от-
жимают, промывают на фильтре ледяной водой (20—30 мл)
и этанолом (15—20 мл) и высушивают в вакуум-эксикаторе
над едким натром.
Выход очищенной 8-оксихинолин-2-сульфокислоты (мо-
ногидрата) равен 15,6—16,3 г (64—67%), температура раз-
ложения в пределах 260—270° (см. примечание 3).
СХЕМА СИНТЕЗА 8-ОКСИХИНОЛИН-2-СУЛЬФОКИСЛОТЫ
ИЗ 2-ХЛОР-8-ОКСИХИНОЛИНА
ОН он
SO3H-H2O
169
Условия получения
Раствор бисульфита натрия, приготовленный пропуска-
нием сернистого ангидрида через раствор 8,0 г (0,2 М) едко-
го натра в 38 мл дистиллированной воды до кислой реакции
(pH 2—3), помещают в круглодонную колбу емкостью
100 мл с пришлифованным обратным холодильником. Доба-
вляют 3,60 г (0,02 М) 2-хлор-8-оксихинолина (см. примеча-
ние 4) и смесь кипятят на масляной бане в течение 2 часов
при 120° (температура бани) и 10 часов при 145—155° (см.
примечание 5).
Затем реакционную массу охлаждают до комнатной тем-
пературы, подкисляют прибавлением 25.мл 37%-ной соляной
кислоты и выдерживают в ледяной бане 1,5—2 часа. Желтый
осадок отсасывают, промывают ледяной водой (3—5 мл) и
этанолом (5 мл) и высушивают в вакуум-эксикаторе под ед-
ким натром.
Получают 2,4—2,5 г (49—51%) сырого продукта с темпе-
ратурой разложения.около 260°, который может быть очищен
по описанному выше способу.
Примечания:
1. Применялся метосульфат 8-окси-1-метоксихинолпния, приготовлен-
ный по описанному' ранее методу [6]. Однако нет необходимости приме-
нять в данном синтезе промытую эфиром кристаллическую соль; вполне
пригоден сырой продукт, полученный нагреванием 16,12 г (0,1 М) 8-ок-
снхинолин-1-оксида и 12,61 г (0,1 Л4) свежеперегнанного нейтрального
диметилсульфата в течение 1—2 часов при 100—103° (температура бани).
2. При растворении сульфокислоты следует строго придерживаться
указанных условий и проводить эту операцию по возможности быстро.
Чтобы ускорить растворение вещества, целесообразно подогревать колбу
в бане с горячей водой, но температура раствора не должна превышать
55—60°, иначе может произойти гидролиз и продукт будет загрязнен
8-оксикарбостирилом.
3. Чистый образец 8-оксихинолин-2-сульфокислоты с температурой
разложения 270° был приготовлен повторной перекристаллизацией и про-
анализирован.
Найдено, %: С—44,76; 44,82; Н—3,36; 3,80; N—5,80;
5,86; S—12,99; 13,13; Н2О—6,91.
C9H7NO4S.H2O. Вычислено, %: С—44,44; Н—3,73; N—5,76; S—13,18;
Н2О—7,41.
4. Применялся 2-хлор-8-оксихинолин с т. пл. 82,5—83,5°, полученный
по описанному ранее методу (7].
5. В процессе нагревания часть 2-хлор-8-оксихинолина возгоняется и
оседает в холодильнике. Время от времени его следует сплавлять назад з
колбу, удаляя ненадолго воду из рубашки холодильника.
ЛИТЕРАТУРА
1. A. Jart. Acta Polytechnics Scandinavica, Chemistry including me-
tallurgy series, 24, Copenhagen, 1963; p. p. 21, 118.
2. A. Jart A. J. В t g 1 e г, V. В 11 s e n Analyt., chim. acta, 31, 472
(1964).
3. И. А. Красавин, В. M. Дзиомко, И. В. Рубцов. Авт.
евнд. 218165; Изобретения. Пром, образцы. Товарные знаки, № 17 (1968).
170
4. Y. Suzuki. Yakugaku Zasshi, 81, 1146 (1961); Chem. Abstrs., 56
3450 c (1962).
5. E. Besthorn, В Qelsselbrecht. Ber., 53, 1017 (1920).
6. И. А. Красавин, В. M. Дзиомко, Ю. П. Радин. Методы
получения химических реактивов и препаратов, вып. 13. М., ИРЕА, 1965,
стр. 68.
7. И. А. К Р а с а в и и, В. М. Дзиомко, Н. И. М и р о ш к и и а,
Ю. П. Радин. Методы получения химических реактивов и препаратов,
вып. 14, М., ИРЕА, 1966, стр. 150.
Поступила в апреле 1968 г. ИРЕА
УДК 547.829
5-(М-ПИПЕРИДИНО)-2-БЕНЗОИЛФУРАН
3. Н. НАЗАРОВА, Л. Д. БАБАШКИНА
О“О-С0~О
CieFliyNOa
М. в. 255,31
В литературе имеется указание о возможности получения
5-(М-пиперидино)-2-бензоил- и 5- (Ы-морфолино)-2-бензоил
Фуранов взаимодействием 5-бром-2-фурилфенилкетона соот-
ветственно с пиперидином и морфолином в спиртовом рас-
творе [1]. Ниже описывается разработанная нами методика
получения 5- (N-пиперидино) -2-бензоилфурана.
СХЕМА СИНТЕЗА 5-(Ы-ПИПЕРИДИНО)-2-БЕНЗОИЛФУРАНА
N Н-НВг
Характеристика основного сырья
5-Бром-2-фурилфенилкетон, т. пл. 43—44° [2].
Пиперидин (для кинопромышленности), ТУ РУ 444—51.
Этиловый спирт, ректифицированный, ГОСТ 5962—51.
Вода дистиллированная, ГОСТ 6709—53.
Условия получения
К 1,7 г (1,97 мл; 0,02 М) 50%-ного водного раствора пи-
перидина добавляют 2,51 г (0,01 М) 5-бром-2-фурилфенилке-
172
тона в 7 мл этилового спирта и смесь нагревают на водяной
бане при 80—85° в течение 4—5 часов. По охлаждении содер-
жимое колбы выливают в 50 мл холодной воды. Через не-
сколько часов выделившиеся желтые пластинчатые кристал-
лы отфильтровывают, промывают холодной водой и пере-
кристаллизовывают из спирта.
Выход 5-(1\т-пиперидино)-2-бензоилфурана равен 2,21 г,
что составляет 90% от теоретического; т. пл. 108,5—109°.
Аналогичным путем получают 5- (М-морфолино)-2-бензо-
илфуран с той лишь разницей, что после нагревания из ре-
акционной смеси отгоняют спирт и остаток выливают в хо-
лодную воду.
Выход 5- (М.-морфолино)-2-бензоилфурана составляет 75%;
т. пл. 87—88° (из спирта).
ЛИТЕРАТУРА
1.3. Н. Назарова Л. Д. Бабешки на. Ж. орг. химии, 2, вып.
10, 1903 (1966).
2. R. Grigg, J. A. Knight, М. V. Sargent. J. Chem. Soc
Nov., 6057 (1965).
Поступила в декабре 1967 г. • Ростовск. н/Д Гос. ун-т
УДК 547.822.3'724.1
- 5-(\-ПИПЕРИДИН0)ФУРФУР0Л
3. И. НАЗАРОВА, В. С. ПУСТОВАРОВ, О. Б. БЕЛИЧЕНКО
\ J-CHO
C10H13NO2
м. в. 179,22
Известный способ получения 5-(N-пиперидино) фурфурола
основан на взаимодействии 5-бромфурфурола с пиперидином
Предлагаемая методика получения 5-(N-пиперидино) -
фурфурола отличается изменением температурных условий
реакции и применением в качестве исходного сырья 5-йод-
ФУРфУР0Ла вместо 5-бромфурфурола. Указанные изменения
упрощают синтез, улучшают выход и качество продукта.
СХЕМА СИНТЕЗА 5-(\-ПИПЕРИДИНО)ФУРФУРОЛА
O_rLln П Гг- - NaOH ________. [i П
-ст —= ----гО'-Ч3>“С’'(7
Характеристика основного сырья
5-Йодфурфурол, т. пл. 126°. получен по методике [2].
Пиперидин, ч., ТУ РУ 444—51-
Натр едкий, ч., ГОСТ 4328—48.
Бензол, ч„ ГОСТ 5955—51.
Калий углекислый, х.ч., ГОСТ 4221—48.
Условия получения
В круглодонную колбу емкостью 200 мл помещают 22,2 г
(0,1 44) 5-йодфурфурола и 40 мл спирта. Смесь нагревают до
174
50е и порциями (по 2—3 мл) при перемешивании добавляют
раствор 20 мл пиперидина в 20 мл спирта. Через 40 минут
раствор нагревают до 65°, охлаждают и прибавляют 50 мл
40%-ного раствора едкого натра. Смесь трижды экстрагиру-
ют бензолом, вытяжки объединяют, сушат безводным угле-
кислым калием, перегоняют в вакууме и получают светло-
желтое масло.
Выход сырого продукта равен 15 г, что составляет 85%
от теоретического. Его растворяют в бензоле и хроматогра-
фируют на окиси алюминия. Растворитель испаряют, 'оста-
ток кристаллизуют из гексана и получают ярко-желтые кри-
сталлы с т. пл. 50—5Г.
Найдено, %: С—67,28; Н—7,18; N—7,98.
CioHiaNOj. Вычислено, %: С—67,02; Н—7,31; N—7,81.
По литературным данным, т. пл. продукта 50,5—51,5°[1].
ЛИТЕРАТУРА
1. В. С. Пустоваров. .Материалы VII и VIII научных конферен-
ций аспирантов, РГУ, 1968, стр. 239.
2. 3. Н. Назарова. Ж. общ. химии, 25, 539 (1955).
Поступила в ноябре [967 г. Ростовск. н/д. Гос. ун-т
УДК 547.391.1'362.3
ПРОПАРГИЛОВЫЕ ЭФИРЫ АКРИЛОВОЙ
И МЕТАКРИЛОВОЙ КИСЛОТ
1-Акрилокси-2-бутин и 1-метакрилокси-2-бутин
Я. М. МОРЛЯН, Л. О. РОСТОМЯН
R
СН2=с! - СООСН2С=СН,
где R = H; СН3
С6Н6О2 М. в. 110,11
С7Н8О2 М. В. 124,14
В литературе почти отсутствуют сведения о получении
этих соединений. Д'Эйлио и Эверс [1] синтезировали их по
методу [2] и исследовали полимеризацию и сополимеризацию
этих соединений.
Нами в основу получения пропаргиловых эфиров акри-
ловой и метакриловой кислот был положен метод [2] с неко
торыми изменениями.
СХЕМА СИНТЕЗА ПРОПАРГИЛОВЫХ ЭФИРОВ
АКРИЛОВОЙ И МЕТАКРИЛОВОЙ КИСЛОТ
R
I , h2so4
сн2=с-соон+носй2с=сн-----------►
R
I
- > СН2=С-СООСН2С=СН
Характеристика основного сырья
Акриловая кислота, ч., ТУ ГКХ № РУ 1730—62.
Метакриловая кислота, ч., МРТУ 6-09-1768-64.
176
Пропаргиловый спирт, имп.
Бензол, ч., ГОСТ 5955—51.
Серная кислота, ч., ГОСТ 4204—66
Гидрохинон, марка Б-635.
Натрий углекислый, ч., ГОСТ 83—63.
Кальций хлористый, плавленый, ГОСТ 4460—48.
Условия получения
В круглодонную колбу емкостью 1 л, снабженную водо-
отделителем и обратным холодильником, помещают 144 г
(2 моля) акриловой кислоты, 250 г бензола, предварительно
приготовленную смесь 140 г (2,5 /И) пропаргилового спирта
и 4 г концентрированной серной кислоты и 0,5 г гидрохино-
на. Смесь кипятят на водяной бане до прекращения выделе-
ния воды. Раствор охлаждают, дважды промывают водой,
нейтрализуют 10%-ным раствором углекислого натрия, сно-
ва промывают водой (pH = 7) и сушат над хлористым каль-
цием в течение ночи. После 'высушивания смесь фильтруют,
переносят в колбу Кляйзена, добавляют 0,5 г гидрохинона,
отгоняют растворитель, а остаток перегоняют в вакууме, со-
бирая фракцию с т.. кип. 52—54°/10 мм.
Выход чистого пропаргилового эфира акриловой кислоты
равен 131 г, что составляет ~60% от теоретического, считая
на акриловую кислоту; <720—1,0040; —1,4445.
Найдено, %: С—65,40; Н—5,50; MRD —29,14.
СбПвОг. Вычислено, %; С—65,45; Н—5,45; MRD—28,52.
Аналогично получают пропаргиловый эфир метакриловой
кислоты. Выход чистого продукта составляет 65% от теоре-
тического, считая на метакриловую кислоту; т. кип. 59—
62°/10 мм; йТ29—0,9850; п™ —1,4497.
Найдено, %: С—67,70; Н- 6,40; MRD —33,73.
СуНвОг, Вычислено, %: С—67,74; Н—6,45; MRD—32,94.
ЛИТЕРАТУРА
1. О. F. D' Aelio, R. С. Evers. J. Polvm. Set, 5, 813-832 (1967)
2. К. Р. Reiss, К. Е. Schulte. Chem." Вег., 87, 964 (.1954).
Поступила в мае 1968 г. . ЦЗЛ
12 Зак. 1034
УДК 678.746.22—136.22.02:66.095.26
СОПОЛИМЕРЫ СТИРОЛА С п-ДИВИНИЛБЕНЗОЛОМ
Т. А. АПТОВА, С. Б. МАКАРОВА, Е. В. ЕГОРОВ
Метод суспензионной сополимеризации стирола с диви-
нилбензолом нашел широкое применение в синтезе различ-
ных ионообменных смол. Однако получение гранул правиль-
ной сферической формы с высокой степенью однородности
по гранулометрическому составу и удельному весу, облада-
ющих высокой механической прочностью, является достаточ-
но трудным.
Иониты аналитического назначения, кроме перечислен-
ных свойств, должны обладать воспроизводимой структурой
и высокой степенью чистоты.
В качестве стабилизатора суспензионной полимеризации
для получения гранул обычно используют крахмал 11], поли-
виниловый спирт [2], карбоксиметилцеллюлозу [3], а также
«мовиол» — поливиниловый спирт, содержащий ацетильные
группы [4].
Поливиниловый спирт не дает устойчивых эмульсий сме-
си стирола и n-дивинилбензола, что приводит к слипанию
гранул и образованию гранул неправильной формы. В связи
с этим на основе поливинилового спирта не удается полу-
чить гранулы сферической формы с размером 50' мк и ниже.
В данной работе в качестве стабилизатора предлагается
использовать сополимер метакриловой кислоты и метилмета-
крилата (ММК)-
В противоположность поливиниловому спирту он образу-
ет более стойкую эмульсию, которая не разрушается даже
при нагревании, что позволяет получить прозрачные стекло-
видные гранулы правильной сферической формы с выходом
около 100%. Преимуществом данного метода является то,
что, используя ММК в качестве стабилизатора эмульсин,
можно получать не только крупные гранулы (0,1 —1,0 мм),
но также, что особенно важно, сферические гранулы сопо-
лимера с размером ниже 20 мк.
178
Характеристика основного сырья
Стирол, ч„ ГОСТ 10003—62 (см. примечание !)•
/г-Дивиннлбензол, ч., РТУ ЛБЗ 49—67, т. пл. 28°.
Сополимер метакриловой кислоты и метилметакрилата,
ВТУ 69—61.
Кальций хлористый плавленный, ч., ГОСТ 4460—66.
Натр едкий, х.ч., ГОСТ 4328—66.
Натрий фосфорнокислый двузамещенный, х.ч, ГОСТ
4172—66..
Бензол, ч.д.а., ГОСТ 5955—51.
Динитрил азоизомасляной кислоты (ДАК), используе-
мый в качестве инициатора, очищают перекристаллизацией
из абсолютного метанола (см. примечание 2).
Условия получения
Стабилизатор готовят следующим образом: в 450 мл ди-
стиллированной воды (см. примечание 3) растворяют 1,6 г
едкого натра, 42 г двузамещенного фосфорнокислого натрия
и 25 г сухого сополимера метакриловой кислоты и метилме-
такрилата. Растворение ведут при 70° в течение 3 часов при
непрерывном перемешивании до образования прозрачного
раствора.
Сополимеризация. Стадию сополимеризации проводят в
полимеризаторе, в качестве которого используют сосуд ци-
линдрической формы, емкостью 600—800 мл (отношение диа-
метра к высоте около 1:2,5), снабженный обратным холо-
дильником и лопастной мешалкой с регулируемым числом
оборотов.
В зависимости от требуемого размера гранул (от 0,75 мм
до 0,01 мм) скорость вращения мешалки составляет от 120
до 1200 об/мин.
В полимеризатор заливают 50 мл раствора стабилизатора
и 250 мл воды, затем при перемешивании добавляют 100 г
смеси мономеров (стирол и п-дивинилбензол), в которой
предварительно растворяют 1,5 г инициатора (смесь моно-
меров готовят в зависимости от заданного состава полимера,
отвешивая n-дивинилбензол и стирол с точностью до 0,01 г).
После того, как капли достигнут определенного желаемо-
го размера (контроль проводят измерением величины капли
под микроскопом) температуру в бане повышают в течение
2 часов от 20 до 60° и выдерживают при этой температуре
еще 2 часа. Затем поднимают температуру до 70° за 30 ми-
нут и выдерживают в течение 30 минут, далее нагревают до
80° за 30 минут и выдерживают при 80° 30 минут. Подъем
температуры производят во всех случаях равномерно, строго
поддерживая скорость подъема, равную 1° за три минуты.
12*
179
Далее полученную смесь охлаждают до 20° в течение
двух часов, равномерно понижая температуру со скоростью
Г за две минуты. Полученные гранулы отфильтровывают на
воронке Бюхнера, промывают несколько раз дистиллирован-
ной водой до обесцвечивания промывных вод и сушат в ва-
куум-шкафу в течение пяти часов при температуре 70±5° и
остаточном давлении 200 мм.
Для удаления из сополимера растворимых продуктов,
его экстрагируют в аппарате Сокслетта в течение четырех
часов кипящим бензолом или дихлорэтаном (растворитель
берут в количестве 800—1500 мл в зависимости от коэффи-
циента набухания сополимера). Затем гранулы отфильтро-
вывают на воронке Бюхнера, промывают бензолом или ди-
хлорэтаном (100—200 мл) и сушат в вакуум-шкафу при
70±5° и остаточном давлении 200 мм в течение трех часов.
Продукт представляет собой прозрачные гранулы пра-
вильной сферической формы; выход около 100 г.
Этим методом можно получать сополимеры с разным со-
держанием сшивающего агента, обладающие различными
коэффициентами набухания.
Полученные сополимеры могут быть использованы для
получения ионообменных смол с различными функциональ-
ными группами.
Примечания:
1. Стирол для освобождения от ингибитора обрабатывают несколько
раз 1 н. раствором щелочи до исчезновения розового окрашивания, про-
мывают водой до нейтральной реакции промывных вод, сушат над про-
каленным хлористым кальцием (не менее двух часов) и перегоняют при
пониженном давлении, отбирая фракцию, кипящую при 46—47720 мм.
d^> — 0,906 г/слЩ п2о —1,5461.
2. Для очистки технического динитрила азоизомасляной кислоты 10 г
его быстро растворяют в 50 мл метанола, подогревая раствор до 40° (не
выше 50°) на водяной бане и фильтруют на воронке горячего фильтрова-
ния с водяным обогревом. Колбу с фильтратом охлаждают холодной во-
дой. Кристаллы, выпавшие при охлаждении, отфильтровывают и сушат в
вакуум-эксикаторе в течение трех часов.
3. Для работы применяют только дистиллированную или деионизо-
ванную воду.
ЛИТЕРАТУРА
1. Ц. Тзйдзи. Реакция получения синтетических полимеров. М.,
Госхимиздат, 1963, стр. 16.
2. К. М. С а л д а д з е, А. Б. Пашков, В. С. Титов. Ионообменные
высокомолекулярные соединения, М., 1960.
3. R. Н. Wiley, J. А 11 е n, S. С h a n g. К. Musselman, Ven-
k a t а с h а|1 a m. J. Phys. Chem., 68,7, 1776 (1964).
4. Н. Н о р f f. Н. L u s s i, Makromolek. chem, 78, 24, 37 (1964);
82, 175, 184, 274, 281 (1965).
Поступила в мае 1968 г.
ИРЕА
УДК 547.241
ТЕТРАМЕТИЛДИФОСФИНДИСУЛЬФИД
С. 3. ИВИН. В. к. ПРОМОНЕНКОВ
(СН()2Р-Р(СН:1)2
S S
C4H12P2S2 М. в. 186,21
Тетраметилдифосфиндисульфид получают взаимодействи-
ем тиотреххлористого фосфора с метилмагниййодидом [1, 2].
Нами подобраны оптимальные условия проведения этой ре-
а кции.
СХЕМА СИНТЕЗА ТЕТРАМЕТИЛДИФОСФИНДИСУЛЬФИДА
6CH3Mgl + 2PSC13 - (CH,j2P-P(CH3)., + 6MglCl |-С.Не
II
S S
Характеристика основного сырья
Тиотреххлористый фосфор [3], т. кип. 125°.
Йодистый метил, ч., т. кип. 42,5°, ТУ МХП 49—51.
Магниевая стружка.
Условия получения
В трехгорлую колбу емкостью 2 л, снабженную мешал-
кой, обратным холодильником (см. примечание 1) и капель-
ной воронкой с хлоркальциевыми трубками, помещают 48,5 г
(2 г-ат). чистых сухих магниевых стружек (см. примеча-
ние 2) и 0,1 г йода. Нагревают колбу пламенем горелки до
появления фиолетовых паров, после чего дают ей остыть до
комнатной температуры и помещают в нее 1000 мл абсолют-
ного диэтилового эфира. При перемешивании и охлаждении
колбы ледяной водой добавляют в нее по каплям 284 г (2 М)
йодистого метила, так чтобы эфир слабо кипел, перемешива-
181
йие при комнатной температуре продолжают 2 часа, затем
при 3—5° за 1,5—2 часа прикапывают 102 г (0,6 М) тиотрех-
хлористого фосфора и колбу нагревают 1 час при 35°. Реак-
ционную смесь, представляющую собой густую белую массу,
по охлаждении выливают в 1000‘ли ледяной воды, подкис-
ленной 50 мл концентрированной серной кислоты. Смесь пе-
ремешивают, фильтруют, промывают на фильтре последова-
тельно дистиллированной водой, спиртом и эфиром.
После сушки в вакууме при 50° в течение 1 часа получа-
ют 39 г тетраметилдифосфиндисульфида, что составляет 69%
от теоретического выхода; т- пл. 222—225°.
Примечания:
1. Холодильник должен обеспечивать эффективное охлаждение при
получении реактива Гриньяра.
2.. Мелкие магниевые стружки промывают ацетоном н эфиром, после
чего высушивают при 50°.
ЛИТЕРАТУРА
1. М. И. Кабачник, И. О. Шепелева. Изв. АН СССР, 56 (1949).
2. Н. Niebergall, В. Langenfeld. Chem. Вег., 95, 64 (1962).
3. Руководство по препаративной неорганической химии, М., ИЛ,
1956, стр. 262.
Поступила в декабре 1967 г.
УДК 547.657
ТЕТРАМИД НАФТАЛИН-1,4,5,8-ТЕТРАКАРБОНОВОЙ
КИСЛОТЫ
М. Т. РАЗУМОВСКАЯ, А. И. БЕЛЯКОВА, Л. А. ЕГОРОВА
h2noc conh2
//\ /Ч
Ч/хЧ
h2noc
CONH3
C14H12N4O4
М. в. 300,27
В литературе мы не нашли описания метода получения й
свойств тетрамида нафталин-1,4,5,8-тетракарбоновой кисло-
ты.
По общеизвестному методу — амидированием нафталин-
1,4,5,8-тетракарбоновой кислоты концентрированным раство-
ром аммиака — получить тетрамид нам не удалось. Как ус-
тановлено, процесс идет только до стадии образования ди-
имида нафталин-1,4,5,8-тетракарбоновой кислоты.
Нами был синтезирован тетрамид нафталин-1,4,5,8-тетра-
карбоновой кислоты взаимодействием нафталин-1,4,5,8-тетра-
карбоновой кислоты с мочевиной в среде трихлорбензола.
СХЕМА СИНТЕЗА
ТЕТРАМИДА НАФТАЛИН-1,4,5,8-ТЕТРАКАРБОНОВОЙ КИСЛОТЫ
Н09С СООН
ХххЧ
| 1! I + 2CO(NH2)2
Ч/ХЧ
ноос соон
183
. h2noc conh2
+ 2H2O (- 2CO2
h2noc conh2
Характеристика основного сырья
Нафталин-1,4,5,8-тетракарбоновая кислота, предвари-
тельно очищенная путем переосаждения технического про-
дукта, 80%-ная (см. примечание 1).
Мочевина, ч., ГОСТ 6691—53
Трихлорбензол, ч., ТУ TCP 443р—61.
Аммиак, ч.д.а., 25%-ный водный раствор, ГОСТ 3760—64.
Ацетон, ч.д.а., ГОСТ 2603—63.
Условия получения
В круглодонную колбу емкостью 1 л, снабженную мешал-
кой, обратным холодильником и термометром, загружают
15 г (~0,049 М) нафталип-1,4,5,8-тетракарбоновой кислоты,
15 г (0,25 М) мочевины и 200 мл трихлорбензола. Смесьпа-
гревают при перемешивании до 130—140° и выдерживают
при этой температуре в течение шести часов, затем темпера-
туру реакционной массы повышают до 155—160° и снова вы-
держивают три часа (см. примечание 2), после чего раствор
охлаждают до 100° и в горячем виде трихлорбензол сливают
с осадка. К осадку прибавляют 200 мл 25%-ного водного
раствора аммиака и перемешивают при комнатной темпе-
ратуре в течение трех часов, затем отфильтровывают с отса-
сыванием. Осадок па фильтре промывают 12,5%-ным водным
раствором аммиака (50 мл), затем водой до нейтральной ре-
акции по универсальной индикаторной бумаге и ацетоном
(20 мл). Сушат продукт при 80° до постоянного веса.
Выход тетрамида нафталин-1,4,5,8-тетракарбоновой кис-
лоты равен 13—14 г, что составляет 86,7—93,3% от теорети-
ческого.
Найдено, %: С—56,10; 56,14; II -3,95; 4,01; N—18,5; 18,44.
C14Ht2N4O4. Вычислено, %: С—56,00; Н—4,02; N -18,65.
Примечания:
1. Чистая нафталин-1,4,5,8-тетракарбоновая кислота получена из тех-
нического 80%-ного продукта многократным переосаждением, которое
производилось посредством растворения вещества в растворе едкого нат-
ра, обработки углем (марки «Вау»), фильтрования и подкисления фнль
трата серной или соляной кислотой.
184
2. Повышение температуры до 175° и увеличение продолжительности
процесса амидирования приводит к образованию диимида нафталин-1,4,
5.8-тетракарбоновой кислоты, что по-видимому, связано с частичным раз-
ложением мочевины в этих условиях.
Поступил» в феврале 1968 г. ИРЕА
УДК 547.562.4'562.1
3,344'ТЕТРАЦИАНДИФЕНИЛОКСИД
3,3'4,4'-Тетрациандифен иловый эфир
М. Т. РАЗУМОВСКАЯ, А. И. БЕЛЯКОВА, Г. И. КОРЕЛЬСКАЯ
CN
CN
C16H6N4O
М. в. 270,25
3,3'4,4'-Тетрациандифенилоксид получают дегидратацией
тетрамида дифенилоксид-3,3', 4, А'- тетракарбоновой кислоты
хлористым тионилом в среде диметилформамида [1].
Нами проверен этот способ и уточнены условия прове-
дения реакции, выделения и очистки продукта.
СХЕМА СИНТЕЗА 3,3',4,4'-ТЕТРАЦИАНДИФЕНИЛ ОКСИДА
conh2
H2NOC -О—CONH2 + 4SOC12 —
г
CONH3
CN
= I
— NC< \-Q_/ X-CN + 4SO2+ 8НС1
CN
186
Характеристика основного сырья
Тетрамид дифенилоксид-3,3',4,4'-тетракарбоновой кислоты,
ч., получен по методу [2].
Тионил хлористый, ч., МРТУ 6—09—2602—65.
Диметилформамид, ч., МРТУ 6—09—2068—65.
Ацетон, ч.д.а., ГОСТ 2603—63.
Условия получения
В четырехгорлую круглодонную колбу емкостью 1 л, снаб-
женную обратным холодильником; мешалкой, капельной
воронкой и термометром, загружают 450 мл диметилформ-
амида и 51 г (0,15 М) тетрамида дифенилоксид-3,3'4,4'-тет-
ракарбоновой кислоты и смесь перемешивают в течение 5—7
минут. Затем, не прекращая перемешивания, прибавляют из
капельной воронки за 25—30 минут 96 мл (158,4 г; 1,32 М)
хлористого тионила, следя за тем, чтобы температура реак-
ционной массы была не выше 50—60°. Реакционную массу
выдерживают при указанной температуре в течение 5,5—6
часов, фильтруют на воронке Бюхнера через двойной бумаж-
ный фильтр и выдерживают раствор при комнатной темпе-
ратуре еще 16—17 часов, причем раствор приобретает свет-
ло-коричневую окраску и из него выпадают кристаллы
3,3',4,4'-тетрациандифенилоксида. Их отфильтровывают, про-
мывают дистиллированной водой, 1%-ным водным раство-
ром едкого натра (2 раза по 25 мл), снова водой до ней-
тральной реакции на лакмус,’ацетоном (25 мл), тщательно
отжимают, сушат сначала в вакуум-эксикаторе над фосфор-
ным ангидридом, а потом при 60—70° до постоянного веса.
Выход технического продукта равен 19 — 20 г; т. пл. 250—
253°
Технический продукт перекристаллизовывают из 140 мл
диметилформамида и получают 14,4—15,2 г чистого продук-
та. Его выход равен 14,4—15,2 г, что составляет 76—80% от
технического; т. пл. 254—258°.
Маточник, состоящий из растворенного 3,3',4,4'-тетраци-
андифенилоксида в диметилформамиде, выливают на 1 кг
измельченного льда. Выпавший желто-коричневый осадок
отфильтровывают, промывают и сушат, как описано выше,
затем дважды перекристаллизовывают из диметилформами-
да с добавкой угля марки «Бау» и получают дополнительно
11 —12 г вещества.
Таким образом, общий выход чистого 3,3',4,4'-тетрациан-
дифенилоксида равен 25—27 г, что составляет 62—67% от
теоретического; т. пл. 254—258° (интервал плавления 2°).
Найдено, %: С—71,05; 71,32; Н—2,54; 2,78; N—20,48;
20,39.
Ci6H6N4O. Вычислено, %: С—71,11; Н—2,22; N—20,73.
По литературным данным, т. ил. продукта 254—256° [1].
187
литература
1. С. S. Marvel, М i с h а е 1, М. М а г t i n. J. Amer. Chem., 80,6600
(1958).
2. M. T. Разумовская, А. И. Белякова, Л. А. Егорова.
Методы получения химических реактивов и препаратов, вып. 18. М., ИРЕА,
1969, стр. 176.
Поступила в апреле I96S г. ИРЕА
УДК 547.828
ТИОПИКОЛИНАНИЛИД
Ю. М. ЮТИЛОВ, И. А. СВЕРТИЛОВА
G12H10N2S
М- в. 214,28
Тиопиколинанилид получают нагреванием смеси а-пико-
лина и серы с анилином [1] или нитробензолом [2].
Нами уточнены условия синтеза тиопиколинанилида по
первому методу, время реакции, температурный режим, оп-
тимальное соотношение компонентов, а также способ очист-
ки продукта.
СХЕМА СИНТЕЗА ТИОПИКОЛИНАНИЛИДА
^N/4c-NH-^
Характеристика основного сырья
а-Пиколин, технический, высушен над едким кали и пе-
регнан, т. кип. 128—129°.
189
Анилин, технический, перегнанный, т- .кип. 183—184°.
Сера, техническая, ГОСТ 127—64.
Спирт этиловый, технический, гидролизный, ГОСТ
8314—57.
Условия получения
В круглодонную колбу емкостью 500 мл, снабженную об-
ратным холодильником, загружают 64 г (2 г-атома) серы,
93 г (1 М) анилина и 93 г (1 Л1) а-пиколина и нагревают
смесь на масляной бане при 160° в течение 10 часов. Непро-
реагировавшие анилин и а-пиколин отгоняют в вакууме.
Светло-коричневый остаток растворяют в 500 мл этилового
спирта, нагревают и отфильтровывают в горячем виде не-
прореагировавшую серу. Спирт отгоняют, а продукт перего-
няют в вакууме, собирая фракцию, кипящую при 198—
20375—6 мм.
Выход тиопиколинанилида равен 135 г, что составляет
62,4% от теоретического.
При стоянии продукт кристаллизуется, т. пл. 41—43°. По-
сле перекристаллизации из смеси гексана с бензолом полу-
чают светло-желтые кристаллы с т. пл. 49—50°.
' По литературным данным [1], т. пл. продукта 50—51°.
ЛИТЕРАТУРА
1. D. Porter. J. Amer. Chem. Soc., 76, 127 (1954).
2. Emmert, Groll. Chem. Bor., 86, 208 (1953).
Поступила в декабре 1967 г.
ВНИИРЕАктивэлектрон
УДК 547.786.1
3,4,5-ТРИАЛ КИЛ ИЗОКСАЗОЛЫ
О. В. ИВАНОВ, в. м. дзиомко
I R = CH3
II r=c2h5
III R=h-C3H7
IV R=h-C4H9
V R = h-C5H,i
VI R = h-C6H|3
Замещенные изоксазолы образуются при взаимодействии
1,3-дикетонов с гидроксиламином в водной [1] или в спирто-
вой среде [2]. 3,4,5-Тризамещенные изоксазолы могут быть по-
лучены конденсацией диоксинов 2-замещенных-1,3-дикетонов
в разбавленной серной кислоте [3].
Нами получен ряд новых 3,4,5-триалкилизоксазолов по
методу [2].
СХЕМА СИНТЕЗА 3,4,5-ТРИАЛКИЛИЗОКСАЗОЛОВ
I
rch2-c-ch-c-ch,r + NH2OH-HC1 - C6H6N
C,H5OH
R,x zCH2R
RCHj—\ ?N
oz
Характеристика основного сырья
2-Алкил-1,3-дикетоны (см. примечание),
Гидроксиламин солянокислый, ч.д.а-, ГОСТ 5456—51.
191
Спирт этиловый, синтетический, ГОСТ 9674—61.
Хлороформ, ч., ГОСТ 3160—51.
Натрий сернокислый, безводный, ч., ГОСТ 4156—48.
Условия получения
В колбу, снабженную обратным холодильником, помеща-
ют 0,1 М ' 2-алкил-1,3-дикетопа (см. примечание 1), 7,64 г
(0,11 А4) солянокислого гидроксиламина, 8,7 г (0,11 Л4) пи-
ридина и 50 мл этилового спирта. Смесь нагревают на кипя-
щей водяной бане в течение трех часов. После охлаждения
прибавляют 70 мл хлороформа и 50 мл воды, органический
слой отделяют, дважды промывают водой (по 50 мл) и су-
шат прокаленным сульфатом натрия. После удаления рас-
творителя остаток фракционируют в вакууме.
Все синтезированные соединения представляют собой бес-
цветные или слабоокрашенные в желтый цвет подвижные
жидкости, смешивающиеся с хлороформом, четыреххлорис-
тым углеродом, ацетоном, спиртами-
Выходы, температуры кипения и физико-химические кон-
станты 3,4,5-триалкилизоксазолов приведены в таблице 1,
анализы — в таблице 2.
Таблица 1
Соединения Название Вы- ход, Темпе- ратура кипения, °C (мм) я20 nD Спектры в этаноле
^макс нм Змакс
1 3,5-Диэтил-4-метил- изоксазол 75 90 (30) 1,0286 1,4565 222,5 8400
II 3,5-Дипропил- 4-этил- изоксазол 71 120 (22) 0,9182 1,4598 222,5 7200
Ill 3,5-Дибутил-4-про- пилизоксазол 81 161—162 (20) 0,9007 1,4619 222,5 .6300
IV 3,5-Дипентил-4-бутил- изоксазол 76 161 (3) 0,8901 1,4628 250 10000
V 3,5-Дигексил-4-пен- тилизоксазол 87 206 (8) 0,8841 1,4631 250 9700
VI 3,5-Дигептил-4-гек- силизоксазол 82 224—225 (4) 0,8826 1,4640 250 9200
192
Таблица 2
Соеди- нения Найдено, % Формула М. в. Вычислено, %
С Н N С Н N
I 68,93 68,97 9,20 9,35 Р,95 10,01 C8H13NO 139,20 69,03 9,41 10,06
и 72,73 72,78 10,38 10,47 7,56 7,81 СцН10ЬЮ 181,28 72,88 10,56 7,73
ш 74,73 74,92 11,15 11,23 6,51 6,47 CltHS5NO 223,36 74,88 11,28 6,67
IV 76,80 77,01 11,61 11,68 5,10 5,15 C17H3lNO 265,44 76,92 11,77 5,28
V 78,00 77,96 11,97 12,20 4,33 4,41 C20H37NO J 307,53 78,11 12,13 4,56
VI 78,91 78,83 12,27 12,31 3,90 3,87 C23H43NO 349,60 79,02 12,40 4,01
Примечание.
2-Алкил-1,3-дикетоны были синтезированы по описанному нами ранее
методу 14].
ЛИТЕРАТУРА
1. Н. К. Кочетков, С. Д. Соколов, В. Е. Жвирбилис. Ж.
общ. химии, 30, 3675 (1960).
2. В. В о b г a n s k i, R. Wo j towski, Rocz. Chem., 38, 1327 (1964).
3. S. В. L I p p I n с о 11. J. Amer. Chem. Soc., 62, 2604 (1940).
4. В. M. Д з и о м к о О. В. Иванов. Ж. орган, химии, 3, 712
(1967).
Поступила в мае 1968 г. ИРЕА
13 Зак. 1031
УДК 547.772.1
3,4,5-ТРИАЛ КИЛ ПИРАЗОЛ Ы
В. М. ДЗИОМКО, О. В. ИВАНОВ, Л. В. ДАРДА
7 ch2r
rch2-
I R = CH3
II r=c2h5
III R = h-C3Hz
IV R = h-C4H9
V R = h-C5Hh
VI R=h-C6H|3
Наиболее распространенный способ получения алкилпи-
разолов состоит во взаимодействии 1,3-дикетонов с серноки-
слым гидразином (или гидразингидратом) в среде спирта
или другого растворителя [1—3]. Разработанный нами ранее
метод [4] сделал легко доступными 2-алкил-1,3-дикетоны.
При взаимодействии их с гидразингидратом в спиртовой сре-
де нами получены, наряду с известными 3,5-диэтил-4-метил-
3,5-дипропил-4-этилпиразолами [5], их высшие гомологи, ра-
нее не описанные в литературе.
СХЕМА СИНТЕЗА 3,4,5-ТРИАЛ КИЛ ПИРАЗОЛОВ
R
RCH2C-CH-C-CH2R + NH2-NH2-H3Q
II II
о о
Rx /CHjR
rch2-/
н
Характеристика основного сырья
2-Алкил-1,3-дикетоны, получение см. [4].
Спирт этиловый, синтетический, ГОСТ 9674—61-
194
Гидразин-гидрат, ч., ГОСТ 5832—51.
Натр едкий, ч., ГОСТ 4328—48.
Хлороформ, ч., ГОСТ 3160—51.
Натрий сернокислый( безводный, ч., ГОСТ 4156—48.
Условия получения
В трехгорлую колбу, снабженную мешалкой, обратным
холодильником и капельной воронкой, помещают 0,1 А1 2-ал-
кил-1,3-дикетона в 70 мл этилового спирта. При перемешива-
нии и охлаждении ледяной водой добавляют по каплям за
15 минут раствор 5,5 г (0,11 М) гидразин-гидрата в 30 мл
этилового спирта. Реакционную смесь нагревают на кипя-
щей водяной бане в течение трех часов, затем добавляют
10 мл 15%-ного раствора едкого натра и нагревание продол-
жают еще 15 минут (см. примечание). После охлаждения к
реакционной массе добавляют 100 мл хлороформа, органи-
ческий слой отделяют и промывают водой до нейтральной
реакции промывной жидкости- Органический слой сушат без-
водным сульфатом натрия, растворители отгоняют, а оста-
ток фракционируют в вакууме.
Выходы, температуры кипения и физико-химические кон-
станты 3,4,5-триалкилпиразолов приведены в таблице 1, ана-
лизы в таблице 2.
Таблица I
Сое- дине- ния Название Вы- ход, % Темпера- тура кипения, °C (мм) rf20 “4 n2fl nD Спектры в циклогексане
^макс нм £макс
I* 3,5-ДиэтиЛ'4-метнл- пиразол 85 199-2(0 (19) — __ 225 4300
11** 3,5-Дипропил-4-этил- пиразол 90 146 (6) — - 225 4500
III 3,5-Дибутил-4-про- пилпиразол 91 157 (2) 0,9018 1,4813 221 5350
IV 3,5-Дипентил-4-бутнл- пиразол 93 189-190 (4) 0,8959 1,4810 222 5600
V 3,5-Дигексил-4-пен- тилпиразол 90 200 (4) 0,8905 1,4795 227 4750
VI 3,5-Дигептил-4-гек- силпиразол 89 225 (1) 0,8765 1,4771 225 4100
* Т. пл. 55°; по литературным данным — 56° [5].
* Т. пл. 64,0—64,5"; по литературным данным — 64—65° [5].
195
Таблица 2
Соеди- нения Найдено, % Формула M. в. Вычислено, %
С Н N C H N
I 69,48 69,46 10,17 10,26 20,21 20,18 c8h14n2 138,21 69,52 10,21 20,27
II 73,16 73,21 11,22 11,20 15,48 15,51 CnH,0N2 180,29 73,28 11,18 15,54
III 75,32 75,34 12,06 11,95 12,28 12,54 C14H26N2 222,37 75,62 11,78 12,60
IV 76,81 77,11 12,49 12,21 10,32 10,50 CnH32N2 264,45 77,21 12,20 10,59
V 78,41 78,16 12,41 12,20 8,83 8,94 306,53 78,37 12,49 9 ,14
VI 79,02 79,15 12,90 12,82 7,90 7,97 348,61 79,24 12,72 8,01
Примечание.
Горячим спиртовым раствором щелочи легко гидролизуются следы
непрореагировавшего 1,3-дикетона.
ЛИТЕРАТУРА
1. Синтезы органических препаратов, вып. 4, М., 1953, стр. 189.
2. L. Knorr, В. Oettinger. Ann, 279 244, (1894).
3. И. И. Грандберг, А. Н. Кост. Ж- общ? химии, 32, 874 (1962).
4. В. М. Дзиомко, О. В. Иванов. Ж- орган, химии, 3, 712
(1967).
5. Н. Musso, К. Figge. Ann., 668, 1 (1963).
Поступила в мае 1968 г.
ИРЕА
УДК 547.415.3
N-ТРИАЛ КИЛСИЛ ИЛ-, N-АЛ КИЛМЕРКАПТО-,
N-АЛ КОКСИМЕТИЛ ЭТИЛ ЕНИМИНЫ
С. 3. ИВИН, В. к. ПРОМОНЕНКОВ, Г. В. КОНОПАТОВА
N-замещенные этиленимины находят разнообразное при-
менение в синтетических исследованиях. До сих пор, однако,
простые методы их получения отсутствуют. Некоторые из
таких веществ получены нами обработкой этиленимина реа-
гентами с активным галоидом.
СН,
| >N-Si(C2H8)s
сн/
СН,.
| )N-SC,H6
сн/
СН,Х
СНа7
N—СН,ОС„Н3
N-Триэтилсилил -
этилснимии
CsH)9NSi М. в. 157,33
N-Этилмеркапго-
этиленимин ,
C4HaNS М. в. 103,13
N-Этоксиметил-
этиленимии
C3HnNO М. в. 101,24
СХЕМА СИНТЕЗА N-ЗАМЕЩЕННЫХ ЭТИЛЕНИМИНОВ
СН,.
2| ;n-h+cix->
сн/
сн2,
| \N-X+
сн/
СН2х
I ;nh-hci
сн/
X = Si(C,Hb)s; SC2Hs; CH2OC2H5
Характеристика основного сырья
Этиленимин, получен дегидратированием |3-аминоэтанола
серной кислотой по методу [1]; т. кип. 55—56°; <Д20—0,837;
—1,4136.
Этилсульфенхлорид, получен хлорированием диэтилди-
сульфида [2].
Триэтилхлорсилан, синтезирован реакцией этилмагний-
бромида с трихлорсиланом [3].
197
Алкилхлорметиловые эфиры, получены из спиртов, пара-
форма и соляной кислоты по методике [4].
Условия получения (общий метод)
В четырехгорлую колбу емкостью 1000 мл помещают 1 At
этиленимина и 500 мл абсолютного эфира. При перемеши-
вании и температуре 8—10° по каплям в течение трех часов
прибавляют 0,5 АЛ галоидопроизводного. Затем реакционную
массу перемешивают при комнатной температуре 2—3 часа,
осадок отфильтровывают, растворитель отгоняют в вакууме
и остаток перегоняют.
Ниже представлена характеристика веществ, полученных
этим методом.
ЛИТЕРАТУРА
1. Н. Wenker. J. Amer. Chem. Soc., 57, 2328 (1935).
2. H. В rint zinger, M, Langhcek. Вег., 84, 557 (1953).
3. L. Sommer, E. Pietrusza, F. Whitmore. J. Amer. Chem.
Soc., 68, 158 (1946).
4. N. Litterscheid. Ann., 330, 122 (1904).
Поступила в декабре 1967 г
УДК 547.563
2,2,4-ТРИМЕТИЛ-4-(/7-ОКСИФЕНИЛ )ХРОМАН
Г. Г. КОНДРАТЬЕВА
С18Нао02
М. в. 268,35
В литературе имеется сообщение о синтезе 2,2,4-триме-
тил-4-(n-оксифенил)хромана конденсацией фенола с окисью
мезитила в присутствии газообразного хлористого водоро-
да [1]. Реакция длится 30 суток, выход 40%.
Повышение температуры до 40° позволило провести реак-
цию за 5 суток с тем же выходом (2].
Нами в качестве катализатора при получении этого про-
дукта использовано молекулярное соединение трехфтористо-
го бора с ортофосфорной кислотой.
СХЕМА СИНТЕЗА 2,2,4-ТРИМЕТИЛ-4-( и-ОКСИФЕНИЛ)ХРОМАНА
ОН „„ °
//\/ СН3, '! H3PO,-BF3
I || .+ ;с = сн-с-снз------------>
Чх сн/
•^Х/ОХ^СНз
I II |^СН,
II |<н
Ч/\
няс/х-( ^он
199
Характеристика основного сырья
Ортофосфорная кислота, ч., ГОСТ 6552—53.
Фенол, ч., ГОСТ 6417—52.
Окись мезитила, ч-, МРТУ 6-09-760-63.
Кали едцое, ч„ ГОСТ 4203—48.
Спирт метиловый, ч.д.а., ГОСТ 6995—54.
Кислота соляная, техн., ГОСТ 1382—42.
Условия получения
Молекулярное соединение фтористого бора с ортофосфор-
ной кислотой. В охлаждаемую ледяной водой склянку с при-
тертой пробкой помещают 500 г. ортофосфорной кислоты и
насыщают ее сухим газообразным трехфтористым бором до
тех пор, пока привес не составит 346 г.
Полученное молекулярное соединение в количестве 846 г,
представляет собой вязкую дымящую жидкость с удельным
весом 1,90; содержание трехфтористого бора 37%.
В герметически закрытой склянке молекулярное соедине-
ние трехфтористого бора сохраняется без изменений в тече-
ние 6 месяцев.
2,2,4-Триметил-4-(п-оксифенил)хроман. В коническую кол-
бу загружают 18,8 г (0,2 М) фенола, 20 г молекулярного
соединения фтористого бора с ортофосфорной кислотой и
4,9 г (0,05 М) окиси мезитила.
Смесь встряхивают до получения однородной массы пос-
тавляют на 5 суток при температуре 18—20°.
По окончанию выдержки частично закристаллизовавшу-
юся массу обрабатывают три раза кипящей водой порциями
по 100 мл.
Водный слой декантируют, твердый остаток обрабатыва-
ют водяным паром для удаления фенола.
Остаток в перегонной колбе представляет собой серый
кристаллический порошок, который растворяют в 50 мл ще-
лочи Кляйзена (см. примечание 1). Щелочной раствор ох-
лаждают до 2—5° и нейтрализуют разбавленной соляной ки-
слотой. Выпавший осадок отфильтровывают, промывают ле-
дяной водой и сушат.
Выход 2,2,4-триметил-4-(л-оксифенил)хромана равен 6,7 г,
что составляет 50% от теоретического, считая на окись мези-
тила; т. пл. 155—156° (см. примечание 2).
Продукт представляет собой белый кристаллический по-
рошок, растворимый в большинстве органических раствори-
телей [2].
Найдено, %: С—80,51; Н—7,33.
CisHzoOa. Вычислено, %: С—80,54; Н—7,46.
200
Примечания:
1. Для приготовления щелочи Кляйзена растворяют 100 г едкого
кали в 125 мл воды и добавляют 375 мл метилового спирта.
2. При перекристаллизации из растворителей 2,2,4-триметил-4-(п-ок-
сифенил)хроман образует с ними прочные продукты присоединения.
Этанольный аддукт состава 4CisH2o02.C2HsOH — белый кристалли-
ческий порошок с т. пл. 163—164°.
Уксусно-кислый аддукт состава 3C1SH20O2.C2H4O2 — белый кристал-
лический порошок с т. пл. 160—161°.
ЛИТЕРАТУРА
1. А. Диании. ЖРФХО, 43, 1310 (1914).
2. W. Baker, А. 1. Floyd, J. F. W. McOmie, J. Pope. J.
Chem. Soc., 1956, 2010.
Поступила в декабре 1967 г.
Тамбовск. Гос. педагогии, ип-т
УДК 547.241
ТРИХЛОРМЕТИЛДИХЛОРФОСФОНИТ
Ф. И. ПОНОМАРЕНКО, С. 3. ИВИН, К. В. КАРАВАНОВ
CCI3PCI2
СС15Р М. в. 220,24
Трихлорметилдихлорфосфонит является промежуточным
реагентом в синтезе разнообразных фосфорганических сое-
динений.
В литературе описано несколько способов получения это-
го соединения. Один из них основан на восстановлении три-
хлорметилтетрахлорфосфора с помощью метилдихлорфосфи-
та. Трихлорметилтетрахлорфосфор получается хлорировани-
ем метилдихлорфосфонита [1].
Второй метод заключается в восстановлении комплексно-
го соединения трихлорметилтетрахлорфосфора и хлористого
алюминия белым фосфором [2].
Трихлорметилдихлорфосфонит получается также при ре-
акции белого фосфора с четыреххлористым углеродом [3] и
действии на комплексное соединение трихлорметилтетра-
хлорфосфора и хлористого алюминия фенилдихлорфосфони-
та в присутствии хлорокиси фосфора [4].
Мы предлагаем получать трихлорметилдихлорфосфонит
взаимодействием комплексного соединения трихлорметилди-
хлорфосфора и пятихлористого фосфора [СС13РС13]+[РС1б]~
с красным фосфором. По этому методу выход трихлорметил-
дихлорфосфонита достигает 70—75%.
СХЕМА СИНТЕЗА ТРИХЛОРМЕТИЛДИХЛОРФОСФОНИТА
[СС13РС13]+[РС16]-+Р -> СС13РС1 +РС18
Характеристика основного сырья
Комплексное соединение трихлорметилтетрахлорфосфора
с пятихлористым фосфором (см. примечание 1), получают по
методике [5].
Красный фосфор, безводный (высушен в вакууме), ч.д.а.
202
Условия получения
В колбу Кляйзена емкостью 0,5 л с прямым холодиль-
ником и приемниками помещают тщательно перемешанную
смесь 50 г (0,1 М) комплексного соединения трихлорметил-
тетрахлорфосфора с пятихлористым фосфором и 49,6 г
(0,4 Л1) красного фосфора и нагревают на водяной бане до
90—100° в течение трех часов. В процессе реакции отгоняет-
ся образовавшийся треххлористый фосфор (75%). Остаток
фракционируют в вакууме 20 мм в токе азота.
При повторной перегонке получают 15,4 г трихлорметил-
дихлорфосфонита, что составляет 70% от теоретического вы-
хода; т. кип. 65—67720 мм; т. пл. 40—41° (см. примечание 2).
Найдено, %: С- 5,21; 5,19; С1—7!9,80; 80,00; Р—13,81; 14,00.
CC15PS. Вычислено, %: С—5,45; С1 — 80,50; Р 14,05.
Примечания:
1. Комплексное соединение трихлорметилтетрахлорфосфора и пяти-
хлористого фосфора гигроскопично, поэтому следует избегать излишнего
соприкосновения его с воздухом.
2. Трихлорметилдихлорфосфонит кристаллический прозрачный про-
дукт, легко окисляется на воздухе.
ЛИТЕРАТУРА
1. L. Quin, Н. Rolston J. Organ. Chem., 23, 1693 (1958).
2.1. L. VanWinkle, S. Bell, G. Morris. Пат. США 2875224;
С. A., 53, 13054 (1958).
3. I. E. Nixon. I. Chem. Soc., 2471 (1964).
4. Л. E. Дмитриева, К. В. Караванов, С. 3. И в и и. Методы
получения химических реактивов и препаратов, вып. 15, М., ИРЕА, 1967,
стр. 148.
5. А. В. Кирсанов, Г. К- Федотова. Допов!д1 АН УССР, 8,
1086 (1960); Ж. общ. химии, 31, 594 (1962).
Поступила в марте 1968 г.
УДК 547.787
5-(ТРИХЛОРМЕТИЛ)ОКСАЗОЛ ИДОН-2
Ф. И. ЛУКНИЦКИЙ, Б. А. ВОВСИ
С13С-СН-СН2
О NH
XCZ
О
C4H4CI3NO2 М. В. 204,44
5-(ТрихлорМетил)оксазолндон-2 не .описан в литературе.
Имеются сведения о высокой антиконвульсантной активно-
сти производных оксалидона-2, а также о седативном их дей-
ствии [1, 2]. Наличие в молекуле оксазолидона трихлорме-
тильной группы позволяет рассчитывать на подобный эффект
этого препарата [3].
СХЕМА СИНТЕЗА 5-(ТРИХЛОРМЕТИЛ)ОКСАЗОЛ ИДОНА-2
н
f NaNO.
C1SC-C-CH2-CO -NH-NH2 —777T-1-*
I П LA
OH
— C1,C-CH-CH,
О NH
xc 7
II
.0
Характеристика основного продукта
.Гидразид 3-окси-4,4,4-трихлормасляной кислоты, получен
из р-трихлорметил-|3-пропиолактона [4] и гидразин-гидрата по
методу [5].
204
Соляная кислота, ч., ГОСТ 3118—46.
Нитрит натрия, ч., ГОСТ 4197—48.
Бензол, ч., ГОСТ 5955—51.
Условия получения
К суспензии 2,88 г гидраЗида 3-окси-4,4,4-трихлормасля-
ной кислоты в 20 мл воды, охлажденной до 2—5°, приливают
23,7 мл 7%-ной соляной кислоты и перемешивают до полно-
го растворения смеси. Затем раствор охлаждают до 0° и к
нему, не прекращая перемешивания, прикапывают раствор
1 г нитрита натрия в 10 мл воды, при этом температуру под-
держивают в пределах 0—5°. Помутневший водный раствор
экстрагируют трижды бензолом (по 10 мл). Экстракт сушат
над прокаленным сульфатом натрия, отделяют сухой бен-
зольный раствор и кипятят его 30 минут с обратным холо-
дильником, после чего бензол отгоняют под вакуумом.
Выход 5-(трихлорметил)оксазолидон-2 равен 1,5 г, что
составляет 59,5% от теоретического; т. пл. 127—128° (бен-
зол +гексан).
ЛИТЕРАТУРА
1. В. Nico la ns. J. Organ. Chem., 26, 2253 (1961).
2. М. Neg we г. Org. Chem. Arzneimittel und ihrc Synonyma, Berlin,
1967, S. 254, 298.
3. R. Bowman. A. Campbell, E. Tanner. J. Chem. Soc.
1963, 692.
4. Ф. И. Лукницкий, Б. А Вовси. Ж- орг. химии, 2. 1895 (1966).
5. Ф. И. Лукницкий, Б. А. Вовси. Ж. орг. химии, 3, 794 (1967).
Поступила в апреле 1968 г.
Ленинград, хим.-фарм. ин-т
УДК 547.722.3.
2,3,5-ТРИЭТОКСИТЕТРАГИДРОФУРАН
Л. М. БОЛОТИНА, Н. И. КУЦЕНКО
Н2С---СН-ОС2Н5
I I
С2Н5О-НС СН-ОС2Н6
'"о7
СюНг'оОд М. в. 204,26
2,3,5-Триэтокситетрагидрофуран — прозрачная, бесцвет-
ная жидкость со слабым эфирным запахом. Он является ис-
ходным продуктом для синтеза производных тропана. Изве-
стный метод получения 2,3,5-триэтокситетрагидрофурана со-
стоит из двух стадий: а) синтез 2,5-диэтокси-2,5-дигидрофу-
рана действием на фуран брома в этиловом спирте [1] или
электролитическим алкоксилированием фурана [2]; б) полу-
чение 2,3,5-триэтокситетрагидрофурана присоединением эти-
лового спирта к 2,5-диэтокси-2,5-дигидрофурану в присутст-
вии кислотного катализатора [3].
Выход 2,3,5-триэтокситетрагидрофурана равен 44%, в ра-
счете па исходный фуран.
Предлагаемый нами метод получения 2,3,5-триэтокситет-
рагидрофурана принципиально отличается от известного ра-
нее метода и дает выход около 60% в расчете на тетрагид-
рофуран.
СХЕМА СИНТЕЗА 2,3,5-ТРИЭТОКСИТЕТРАГИДРОФУРАНА
Н2С-—сн2
I I ЗС12
Н2С СН3-------’
'О7
Н3С- - СН-С1
I I
С1—НС СН-С1
3CaH5ONa
206
НаС----СН-ОС2Н5
I I
C2HSO-HC CH-OC2H6
Характеристика основного сырья
Тетрагидрофуран, х.ч,, перегнанный.
Хлор, ГОСТ 6718—53.
Этиловый спирт, абсолютный, ГОСТ 9674—61.
Натрий, ч., ТУ 1664—50.
Эфир диэтиловый, ГОСТ 6265— 52.
Сульфат натрия, прокаленный, ГОСТ 4166—48.
Условия получения
Синтез 2,3,5-трихлортетрагидрофурана. Прибор для син-
теза представляет собой четырехгорлую грушевидную колбу
(см. примечание 1), снабженную мешалкой, термометром,
обратным холодильником и барботажной трубкой для пода-
чи газа. В колбу помещают 72 г осушенного тетрагидрофу-
рана, охлаждают до температуры 0 + 5° и при этой темпера-
туре пропускают хлор (см. примечание 2) со средней скоро-
стью 12 л!час в течение 4 часов- После этого охлаждающую
баню снимают и нагревают реакционную смесь до 75° (см.
примечание 3). По достижении температуры 75° хлор про-
пускают еще в течение трех часов. Затем продувают азотом
до исчезновения желто-зеленой окраски. Полученную смесь
подвергают вакуумной разгонке.
Выход продукта равен 164—«165 г, что составляет 93—
94% от теоретического.
2,3,5-Трихлортетрагидрофуран имеет следующие кон-
станты: т. кип. 84°/10 мм; п2^ —1,5068; d420—1,5043.
По литературным данным [5], т. кип. 94°/22 мм; п2£ —
1,5068; d420 — 1,5034.
Получение 2,3,5-триэтокситетрагидрофурана. В четырех-
горлой круглодонной колбе, снабженной мешалкой, термо-
метром, капельной воронкой и обратным холодильником,
растворяют 23 г натрия в 500 мл этилового спирта (см. при-
мечание 4). Раствор охлаждают до 10° и при этой темпера-
туре прибавляют 52 г 2,3,5-трихлортетрагидрофурана. По-
следнее следует вести так, чтобы не происходил разогрев
выше 15°. По окончании прибавления трихлорида этиловый
спирт отгоняют при остаточном давлении 100—200 мм, затем
к реакционной смеси прибавляют 150 мл воды. После пере-
мешивания получают прозрачный красновато-коричневый
раствор. Из этого раствора 2,3,5-триэтокситетрагидрофуран
207
экстрагируют бензолом (три раза по 60 мл). Бензольные
вытяжки соединяют вместе и подсушивают сульфатом нат-
рия.
После отгонки бензола и вакуумной перегонки пблучают
40—43 г 2,3,5-триэтокситетрагидрофурана, что составляет
65—70%| от теоретического выхода.
2,3,5- Триэтокситетрагидрофуран имеет следующие кон-
станты: т. кип. 98—100°/10 мм; п20—1,4425; <Ц20—1,0253.
По литературным данным [4], т. кип. продукта 99710 мм.
Примечания:
1. Дно колбы должно быть вытянуто, чтобы столб жидкости в колбе
был как можно выше.
2. Хлор, поступающий из баллона, подвергается осушке в склянке
Дрекселя, наполненной серной кислотой.
3. Все это время подачу хлора не прекращают.
4. Можно вести реакцию со щелочью, но выход 2,3,5-триэтокситетра-
гидрофурана при этом снижается.
ЛИТЕРАТУРА
1.1. A. Fakstorp, D. Raleigh, L. Schniepp. J. Amer. Chem.
Soc., 72, 869 (1950).
2. N. Clauson-Caas, F. Lira bo rg, A. Glens. Acta Chem.
Scand, 6. 531 (1952).
3. N. Clause n—C a a s, J. Nilsen, E. В о s s. Acta Chem. Scand.,
9, 111 (1955).
4. A. S t о 11. A. Lindemann, E. linker. Helv. Chrm, Acta, 39,
1500 (1953).
5. M. Крат ox вил. Coll., 27, 52 (1962).
Поступила в апреле 1968 г.
НИИПластмасс
УДК 547.558.4.
О-ФЕНИЛБОРНАЯ КИСЛОТА
Н. М. МОРЛЯН, Л. О. РОСТОМЯН
^_^В(ОН)2
С6Н7ВО2 М. в. 121,93
Впервые о возможности получения замещенных борных
кислот из реактива Гриньяра и эфиров борной кислоты бы-
ло сообщено в работе [1]. о-Фенилборная кислота может
быть получена при взаимодействии бромистого фенилмаг-
ния с трехфтористым бором [2], фениллития с бутиловым эфи-
ром борной кислоты [3] и дифенилртути с треххлористым бо-
ром [4].
о-Фенилборная кислота применяется в качестве исходно-
го вещества при синтезе фенилбордихлорида [5] и различных
замещенных арилборных и диарилборных кислот и их эфи-
ров [6, 7]-
В описанную ниже методику получения о-фенилборной
кислоты, разработанную на основе прописей [1 и 8], нами
внесены значительные изменения, упрощающие синтез этого
соединения.
СХЕМА СИНТЕЗА о-ФЕНИЛБОРНОЙ КИСЛОТЫ
С6Н3Вг + C6H5MgBr
C6H3MgBr + В(ОСН3)з^[С6Н5В(ОСНз)з]-М§Вг+
[C6HSB (ОСНз) ] 3Mg- Bi+ + 3H2O—
->С6Н5В(ОН)2 + 2СН3ОН + Mg(OH)Br
Характеристика основного сырья
Метиловый эфир борной кислоты, ч., ЕРЗ ТУ 46—67.
Магний металлический.
14 зак. Ю34 209
Бромбензол, ч., МРТУ 6-09-1520-64.
Эфир диэтиловый, ГОСТ 6265—52-
Серная кислота, ч., ГОСТ 4204—48.
Эфир петролсйный, ч„ МРТУ 279-59.
Условия получения
Синтез бромистого фенилмагния. В двухлитровую трех-
горлую колбу, снабженную механической мешалкой, обрат-
ным холодильником и капельной воронкой, помещают 36,5 г
(1,5 г.-атома) магниевых стружек, добавляют 150 мл абсо-
лютного эфира, 17 г бромбензола и несколько кристаллов
йода. Колбу слегка нагревают на водяной бане до начала
реакции, после чего пускают в ход мешалку и добавляют из
капельной воронки 219 г бромбензола в 750 мл абсолютного
эфира с такой скоростью, чтобы реакция шла энергично, но
не очень бурно. После добавления всего бромбензола реак-
ционную массу перемешивают еще 30 минут при температу-
ре 30—35°.
Выход бромистого фенилмагния количественный.
Получение о-фенилборной кислоты. В трехлитровую пя-
тигорлую круглодонную колбу, снабженную механической
мешалкой, двумя градуированными капельными воронками,
обратным холодильником, термометром и трубкой для пода-
чи азота (см. примечание 1), помещают 750 мл абсолютного
эфира. Включают мешалку и пропускают ток сухого азота.
Колбу охлаждают до минус 60° при помощи бани с сухим
льдом и ацетоном и поддерживают указанную температуру
в течение всей реакции (см. примечание 2). В одну капель-
ную воронку помещают 153 г (1,5 М) свежеперегнанного ме-
тилового эфира борной кислоты (см. примечание 3), в дру-
гую раствор 271,5 г (1,5 М) бромистого фенилмагния з
900 мл абсолютного эфира.
При сильном перемешивании из капельных воронок до-
бавляют попеременно, небольшими порциями: сначала
10 мл метилового эфира борной кислоты, затем 30 мл бро-
мистого фенилмагния, причем скорость прибавления должна
быть по возможности большой, но в то же время такой, что-
бы не допускать разогревания смеси выше температуры ми-
нус 60° (см. примечание 4)-
По окончании прибавления всего количества реагентов
перемешивание продолжают еще 20 минут, поддерживая ту
же температуру, при этом смесь становится белой.
Затем температуру реакционной массы повышают до
0—5° и продолжают перемешивание, смесь гидролизуют до-
бавлением 100 мл дистиллированной воды за 5—10 минут,
после чего нейтрализуют 42 мл концентрированной серной
кислоты в 850 мл дистиллированной воды и переносят в де-
210
лйтельную воронку. Отделяют эфирный слой, а водный слой
трижды экстрагируют эфиром порциями по 125 мл.
Эфирный слой и вытяжки соединяют и сушат безводным
сернокислым натрием. После удаления эфира остаток охла-
ждают. о-Фенилборную кислоту, выпавшую в виде мелких
желтоватых кристаллов, отфильтровывают на воронке Бюх-
нера и промывают дважды петролейным эфиром по 250 мл,
который удаляет следы дифенилборной кислоты.
Выход неперекрнсталлизованного продукта равен 130—
140 г.
После перекристаллизации из воды (см. примечание 5)
получают белые игольчатые кристаллы, которые сушат в ва-
кууме при 40—50°/40—50 мм до постоянного веса.
Выход чистой о-фенилборной кислоты равен 70—80 г,
что составляет 40—45% от теоретического; т. пл. 215—217°.
Из маточника после сильного охлаждения добавочно
можно получить еще 10 г вещества.
Примечания:
1. Азот сушат пропусканием через колонку с фосфорным ангидридом.
2. Выход фенилборной кислоты в значительной степени зависит от
температуры реакции.
3. Метиловый эфир борной кислоты (т. кип. 65—67°) с метиловым
спиртом (т. кип. 64") образует азотроппую смесь (1:1), кипящую прн
54,6°. Поэтому необходимо перед опытом перегнать его на хорошей ко-
лонке, чтобы удалить в виде головного погона азеотропную смесь мети-
лового эфира борной кислоты с метиловым спиртом, которая могла обра-
зоваться при хранении в результате гидролиза.
4. При указанной температуре наибольший выход феиилбориой кисло-
ты получается в том случае, когда реагенты берутся в строго стехиомет-
рическом соотношении.
5. Растворимость кислоты в 100 г воды составляет 1,1 г при 0° и 25 г
при 25".
ЛИТЕРАТУРА
1. Khotinsky, Melamed. Вег., 42, 3090 (1909).
2. Krause Nitsch е. Вег., 55 В, 1261 (1922); Герм. пат. 371467
(1923); С. А. 18, 992 (1924).
3. Brinbley, Cerrard, tapper t. J. Chem. Soc., 2956 (1955).
4. Michaelis, Becker. Ber., 15, 180 (1882),
5. Dandegaonkcr, Gerrard, tapper 1. J. Chem. Soc., 2893
(1957).
6. Seaman, Johnson. J. Amer. Chem. Soc., 53, 711 (1931).
7. L a p p e r t. Chem. Revs., 56, 987, 1013 (1956).
8. Синтез органических препаратов, т. 11. М., ЙЛ, стр. 5
Поступила в январе 1968 r. ЦЗЛ
14*
УДК 547.632.3
ФЕНОЛОВЫЙ КРАСНЫЙ
Фенолсульфофталеин
Е. Я- ЯРОВЕНКО, М. Ф. КОН ДР А Ш О ВЛ
О
SO3H
C10H14O5S-~ Н5О М. в. 354,38 + 4,50
Феноловый красный является кислотно-основным индика-
тором с переходом окраски от желтой к красной при pH
6.2—8,4.
Применяется для определения концентрации водородные
гюнов, для приготовления универсальных, комплексометри-
ческих и галоидзаМещенных индикаторов.
Феноловый красный получается конденсацией фенола с
о-сульфобензойной кислотой, с ее дихлорангидридом, ангид-
ридом или моноаммонийной солью в присутствии хлористого
цинка [1], пятиокиси фосфора [2], смеси хлористого цинка
и хлорокиси фосфора [3].
Нами предложен способ конденсации фенола с о-сульфо-
бензойной кислотой в присутствии смеси хлорной кислоты и
хлорокиси фосфора, что дает увеличение выхода индикатора
и улучшает его качество [4].
212
СХЕМА СИНТЕЗА ФЕНОЛОВОГО КРАСНОГО
СООН
/ -SO3H
он
Характеристика основного сырья
о-Сульфобензойная кислота ч., МРТУ 6-09-811-63.
Фенол, ч„ ГОСТ 6417 -52- ’
Хлорокись фосфора, ч.. ВТУ 166—61.
Хлорная кислота, ч., ТУ МХП ОРУ 87—57.
Уксусная кислота, ч., ГОСТ 61 -51-
Соляная кислота, ч., ГОСТ 3118—46.
Аммиак водный, ч., ГОСТ 9—57.
Условия получения
В трехгорлую колбу емкостью 2 л, снабженную мешал-
кой, термометром, капельной воронкой и обратным холо-
дильником, загружают 103 г (0,5 Л1) безводной о-сульфобен-
зойной кислоты, 15 мл 57—58%-ной хлорной кислоты и 15 мл
хлорокиси фосфора. При интенсивном перемешивании нагре-
вают реакционную массу до 60°. Эту температуру поддержи-
вают 30 минут. Затем прибавляют 96 з (1 Л1) фенола, на-
гревают смесь до 125—130° и при этой температуре в течение
часа прибавляют 45 мл хлорокиси фосфора, а затем реак-
ционную массу выдерживают 30 минут при 125—130°.
К полученному плаву прибавляют 200 мл горячей воды,
перемешивают в течение 1 часа при 80—90° и охлаждают до
комнатной температуры, при этом феноловый красный вы-
падает в осадок. Его отфильтровывают, переносят в колбу,
прибавляют 400 мл воды, 83 мл уксусной кислоты и нагрева-
ют, при перемешивании до 80—90°. Затем добавляют 175 мл
аммиака до полного растворения осадка, раствор перемеши-
вают в течение 1,5 часа, охлаждают до 30° и фильтруют.
Фильтрат подкисляют разбавленной Г.1 соляной кислотой до
213
кислой реакции по «конго». Выпавший осадок отфильтровы-
вают, промывают водой до нейтральной реакции в промыв-
ных водах (но универсальной индикаторной бумажке) и су-
шат при 90—100°.
Получают 107—113 г фенолового красного, что составля-
ет 60—63% от теоретического выхода, в расчете на о-сульфо-
бензойную кислоту. Полученный продукт по качеству удов-
летворяет требованиям ГОСТа 4599—61.
Для получения индикатора в химически чистом состоянии
его переосаждают по методу [5].
Найдено, %: С—64,12; 63,8; Н—4,3; 4,44; S—9,2; 9,18.
Ci9Hi4O5S.i/i Н2О. Вычислено, %: С—64,48; Н—3,89; S—9,02.
ЛИТЕРАТУРА
1. W. К. Orndorff, F. W. С h е г w о о d. J. Amer. Chem. Soc.
45, 487, (1923).
2. J. Chr zaszcze wski, M. К о s i n s k i. C. A., 52, 200361, (1958).
3. Г. И. Михайлов, Г. H. Кошелева. Авт. свид. 104125. (1956);
Бюлл. изобр., № 8, 17 (1956).
4. Е. Я. Яровенко, М. Ф. Кондрашова, Т. Г. Гайворон-
с к а я, Г. И. Михайлов. Авт. свид. 187191, Бюлл. изобр. № 20, 90
(1966).
5. Г. Н. Кошелева. Химические реактивы и препараты, вып. 21,
М., ИРЕА, 1956, стр. 57.
Поступила в ноябре 1967 г.
ИРР
УДК 547.514.721.
1-ХЛОР-4-( АЛКОКСИМЕТИЛ) ЦИКЛОПЕНТЕНЫ-2
С. 3. ИВИН, В. К. ПРОМОНЕНКОВ, Л. А. ЗЕРНАКОВА.
Ю. Е. УТКИН
1-Хлор-4-(алкокеиметил) циклопентены-2 ранее были по-
лучены взаимодействием алкилхлорметиловых эфиров с цик-
лопентадиеном в растворителе в присутствии четыреххлори-
стого олова [1].
Нами разработана методика получения указанных ве-
ществ без применения растворителя. Они могут быть исполь-
зованы в разнообразных синтезах-
1-ХЛОР-4- (МЕТОКСИМЕТИЛ) ЦИКЛОПЕНТЕН-2
НС=СН
СН3ОСН2СН/ \CHC1
xchZ
С7НПСЮ
М. в. 146,61
СХЕМА ПОЛУЧЕНИЯ
1-ХЛОР-4-(МЕТОКСИМЕТИЛ)ЦИКЛ О ПЕНТЕНА-2
нс=сн
ch3ocilci + н2с^ ;сн —
хсн/
нс=сн
СН3ОСН8СН/ )СНС1
хсн/
21.г>
o' ж X 58 9 7,54 8,13 со о*
1 Л И 3 ы 1 вычисле. о 54,51 57,61 60,01 ! 58,37
А н а 2Й о” S г 6,91 7,63 8,22 ' 6,37
о =( •-Х га о 54,60 57,38 60,16 58,20
е 1,4680 1,4651 СМ S 1,4830
1,0350 0,9983 0,9797 1,0327
1. кип., °C/ мм 1 S 62-67/4 1 62—68/2 70-74/2
3 CQ 5с 5 2 СП
Соединения 1-Хлор-4-(этоксиметил)циклопентеи-2 1 НС=СН G г и г сч г с? о й1 сн2 С8НЙС1О М.в. 160,64 1-Хлор-4-(изопропоксиметил)циклопентен-2 НС=СН о CQ и X о еч X о о ХУ о о X и о О о г и 1-Хлор-4-(изобутоксиметил)циклопентен-2 НС=СН азо- СДНВОСН2НС^ )СНС1 хснУ С1вНпСЮ М.в. 191,72 1- Хлор-4-(аллилоксиметил)циклопентен-2 Г о П о г и X о о X о г о м г и ч г’ и г и II м г о С8Н13С1О М.В. 172,65
Характеристика основного сырья
Циклопентадиен, получен из дициклопентадиена по мето-
дике [2]; т. кип. 40—42°; </420—0,8016; Пр —1,4446.
Хлорметиловые эфиры, получены из соответствующих
спиртов, параформа и соляной кислоты по способу [3].
Условия получения
В четырехгорлую колбу с мешалкой, термометром, обрат-
ным холодильником и капельной воронкой помещают 1 М
метилхлорметилового эфира и несколько кристаллов свеже-
плавленного хлористого цинка (см. примечание 1). При 5—•
10° и перемешивании прибавляют 72 г (1,2 М) свежеперег-
нанного циклопентадиена (см. примечание 2). По окончании
добавления смесь перемешивают 30 минут при той же темпе-
ратуре, затем один час при комнатной и перегоняют в ваку-
уме.
Выход 1-хлор-4-метоксиметилциклопентена-2 составляет
62 г (42%); т. кип. 62—64°/12 мм; d^0—1,0610; < —1,4714-
По описанной методике можно синтезировать другие сое-
динения этого ряда. В таблице представлена характеристика
некоторых из них.
Примечания:
1. Свежеплавленный цинк обладает лучшей каталитической способно-
стью.
2. Проведение реакции при более дысокой температуре приводит к
понижению выхода продуктов за счет димеризации циклопентадиена.
ЛИТЕРАТУРА
1. R. Dewbenko. J. Organ. Chem., 27, 789 (1962).
2. Синтез органических соединений, М., Гостехиздат, 1950, стр. 20.
3. L. F а г г е п, Т. Fife. J. Amer. Chem. Soc. 47. 2421 (1925).
N. L. Herscheid. Ann., 330, 122 (1904).
Поступила в декабре 1967 г.
216
УДК 547.822.5
2-Х ЛОР-5-АМИНОПИРИДИН
Ю. М. ЮТИЛОВ, И. А. СВЕРТИЛОВА
C5H5C1N2
М. в. 128,5
По литературным данным, 2-хлор-5-аминопиридин полу-
чают восстановлением 2-хлор-5-нитропиридина хлоридом оло-
ва [1] и оловом [2] в среде соляной кислоты; цинковой пылью
в водном растворе серной кислоты [3]; электролитическим
или каталитическим гидрированием в присутствии платино-
вой черни или никелевого катализатора [3]; железом в ук-
сусной кислоте [4] или воде [5].
Практически наибольший интерес представляет последний
метод как более доступный и наименее трудоемкий-
При проверке этого метода нами уточнены условия реак-
ции, способы выделения и очистки продукта. Оказалось, что
максимальный выход продукта достигается при использова-
нии карбонильного железа.
СХЕМА СИНТЕЗА 2-ХЛОР-5-АМИНОПИРИДИНА
Fe,H5O
[Н]
Характеристика основного сырья
2-Хлор-5-нитропиридин, т. пл. 108° [6].
Железо, карбонильное, особо чистое, класса А-2.
Вода дистиллированная, ГОСТ 6709—53.
218
Условия получения
В круглодонную колбу емкостью 500 мл, установленную
в водяной бане и снабженную мешалкой, термометром и об-
ратным холодильником, загружают 100 г (1, 8 М) карбо-
нильного железа и 150 мл воды. Смесь нагревают до 85—
90° и при тщательном перемешивании прибавляют неболь-
шими порциями в течение 1 часа 40 г (0,25 7И) 2-хлор-5-нит-
ропиридина. Затем смесь выдерживают еще два часа при той
же температуре и фильтруют в горячем виде. Непрореаги-
ровавшее железо промывают на фильтре горячей водой три-
жды, порциями по 20 мл, и присоединяют промывные воды
к основному фильтрату. Раствор упаривают в вакууме до
7з объема и охлаждают- Выделившиеся при этом кристаллы
отфильтровывают, сушат на воздухе и получают 25 г про-
дукта с т. пл. 82—83°.
Маточный раствор упаривают в вакууме до !/з объема, ох-
лаждают и получают еще 5,4 г вещества с т. пл. 80—82°, ко-
торое перекристаллизовывают из хлороформа и получают до-
полнительно 5 г бесцветных блестящих кристаллов в т. пл.
82—83°
Таким образом, общий выход 2-хлор-5-аминопиридина ра-
вен 30 г, что составляет 93% от теоретического.
По литературным данным [5], 2-хлор-5-нитропиридин пла-
вится при 83—83,5°.
ЛИТЕРАТУРА
I. А. Чичибабин, Разоренов. ДРФХО, 47, 128G (1915).
2. А. Чичибабин, Былинкин. ДРФХО, 50, 471 (1918).
3. А. В i n z, V. S с h 1 с k h. Вег. 68, 315 (1935).
4. Р 1 е г о 11 i, Н a n р t. С. А., 1, 672, 1926.
.5 . С г a g о е, Н a m 111 о n. J. Amer. Chem. Soc., 67, 53G (1945).
6. А. Чичибабин. ЖРФХО., 46, 1236 (1914).
Поступила в декабре 1967 г. ВНИИРЕЛктивэлектрон
УДК 547.397
ХЛОРАНГИДРИД
mpawc-ТРИХЛОРКРОТОНОВОЙ КИСЛОТЫ
Ф. И. ЛУКНИЦКИЙ, Б. А. вовси
С13С-С=С-С-С1
С4Н2С14О
М. в. 207,87
Хлорангидрид трднс-трихлоркротоновой кислоты может
быть получен при действии на трихлоркротоновую кислоту
тионилхлорида или пятихлористого фосфора [1].
СХЕМА СИНТЕЗА ХЛОРАНГИДРИДА
транс-ТРИХЛОРКРОТОНОВОЙ КИСЛОТЫ
Н
-1-СНг |
I -| pci5 -> а3с-с=с-с-а +
—со | ||
н о
+ РОС13 + НС!
Характеристика основного сырья
(З-Трихлорметил-р-пропиолактон, получен из хлораля, аце-
тилхлорида и триэтиламина [2].
Пятихлористый фосфор, ч„ МРТУ 6-09-604-63.
Условия получения
Смесь, состоящую из 20,6 г пятихлористого фосфора и
18,65 г р-трихлорметил-р-пропиолактона, нагревают на кипя-
220
щей водяной бане, одновременно удаляя в течение 15 минут
часть низкокипящих летучих продуктов при температуре па-
ров не выше 50°.
Затем реакционную смесь разгоняют в вакууме, собирая
фракцию, кипящую при 78—80715 мм.
Выход хлорангидрида гранс-трихлоркротоновой кислоты
равен 15,3 г, что составляет 74,8% от теоретического.
По литературным данным, т. кип. хлорангидрида этой ки-
слоты равна 75711 мм [1]. Транс-Конфигурация подтвержда-
ется ПМР-спектром, где константа спин-спинового взаимодей-
ствия составляет 14,2 гц.
ЛИТЕРАТУРА
1. К- A u w е г s, A. W i s s е b ас h. Вег., 56, 736 (1923).
2. Ф. И. Лукницкий, Б. А. В о в с и. Ж. орган, химии, 2, 1895
(1966).
Поступила в апреле 1968 г. Леничградск. хим-фарм. ин-т
УДК 547.822.5
2-ХЛОР-5-НИТРОП ИРИДИИ
Ю. М. ЮТИЛОВ, И. Л. СВЕРТИЛОВА
C5H3C1NsO2
М. в. 158,54
2-Хлор-5-нитропиридин является ценным исходным веще-
ством для синтеза многих производных пиридинового ряда.
Его получают при обработке 2-амино-5-нитропиридина нит-
ритом натрия (или калия) в разбавленной серной кислоте и
последующем взаимодействии полученного оксипроизводного
с хлорокисью фосфора (или смесью хлорокиси фосфора с
пятихлористым фосфором) [1, 2, 3]. 2-Хлор-5-нитропиридин
может быть также получен непосредственно при действии на
2-амино-5-нитропиридин нитрита натрия в соляной кислоте
[4]
При проверке литературных данных установлено, что хотя
последняя реакция протекает в одну стадию, предпочтение
следует отдать синтезу через оксипроизводное [2], так как в
последнем случае выход продукта значительно выше.
Нами уточнен метод синтеза 2-хлор-5-нитропиридина че-
рез 5-нитропиридон-2 [1] и внесены некоторые изменения в
условия реакции и выделения продукта.
Исходный 2-амино-5-нитропиридин получен нитрованием
2-аминопиридина [5].
СХЕМА СИНТЕЗА 2-ХЛОР-5-НИТРОПИРИДИНА
Н
222
РОС13 ;
Характеристика основного сырья
2-Амино-5-нитропиридин, получен по [5].
Серная кислота, ч., ГОСТ 4204—48.
Азотнокислый калий, х.ч., ГОСТ 4144—48.
Хлорокись фосфора, техническая, перегнанная, т. кип.
105—107°.
Натр едкий, ч., ГОСТ 4328—48.
Сода кальцинированная, ч., ГОСТ 5100—49.
Условия получения
Синтез 5-нитро-2-пиридона. В трехгорлую колбу емко-
стью 3 л, снабженную мешалкой, термометром и воронкой
для сыпучих веществ, помещают 960 мл воды и 128 мл кон-
центрированной серной кислоты. Раствор охлаждают до
комнатной температуры и растворяют в нем 200 г (1,44 М)
2-амино-5-нитропиридина.
Полученную смесь интенсивно перемешивают и прибав-
ляют порциями 132 г (1,55 /И) азотистокислого калия, под-
держивая температуру реакционной массы не выше 25°. При
этом выпадает осадок желтого цвета. Смесь оставляют на
ночь, затем нагревают в течение 30 минут на водяной бане
при 50—60°, охлаждают и нейтрализуют содой до слабоще-
лочной реакции (pH 5—6). Осадок отфильтровывают, про-
мывают несколько раз водой и сушат.
Выход 5-нитро-2-пиридона равен 143 г, что составляет
71% от теоретического, т. пл. 188—189°.
Получение 2-хлор-5-нитропиридина. В одногорлую колбу
емкостью 0,5 л, снабженную обратным холодильником с
хлоркальциевой трубкой, помещают 50 г (0,36 М) 5-нитро-
2-пиридона и 55 мл (92,4 г; 0,6 М) хлорокиси фосфора.
Смесь нагревают при кипении в течение 4 часов, а затем от-
гоняют в вакууме избыток хлорокиси фосфора. Твердый ос-
таток охлаждают ледяной водой и постепенно приливают к
нему небольшое количество воды (~50 мл). Смесь нейтра-
лизуют едким натром до слабощелочной реакции, осадок от-
фильтровывают, промывают несколько раз водой и сушат на
воздухе.
Выход 2-хлор-5-нитропиридина равен 54 г, что составляет
95% от теоретического; т. пл. 108° (из воды).
223
По литературным данным [1, 2, 3], т. пл. вещества 106—
108°.
ЛИТЕРАТУРА
1. А. Чичибабин. ЖРФХО., 46, 1236 (1914).
2. Caldwell, Korn.feld. J. Amer. Chem. Soc., 64, 1696 (1942).
3. Shibasaki. J. Pharm. Soc. Japan., 72, 381 (1952).
4. P h i 1 1 i p s. J. Chem. Soc., 1941, 9.
5. А. Чичибабин. ЖРФХО, 47, 1286 (1915).
Поступила в декабре 1967 г.
ВН ИИРЕАкгивэлсктрон
УДК 547.724.1.
5-ХЛОРФУРФУРОЛ
3. II. НАЗАРОВА, Л. н. БАБЕШКИНА
Cl-<f ^-СНО
C5H3CIO2 М. в. 130,53
Синтез 5-хлорфурфурола с выходом 3—4% был впервые
осуществлен прямым хлорированием диацетата фурфурола
[1]. Хлорированием диацетата фурфурола в сероуглероде
сульфурилхлоридом выход был повышен до 15% [2]. В даль-
нейшем 5-хлорфурфурол с выходом 33% был получен пря-
мым хлорированием фурфурола в сероуглероде в присутст-
вии каталитических количеств серы и перекиси бензоила [3].
Но этот метод неудобен из-за токсичности и огнеопасности
сероуглерода. Попытки перенести условия прямого бромиро-
вания [4] фурфурола на процесс прямого хлорирования при-
вели [5] к снижению выхода до 12—14%.
Было осуществлено [6] прямое хлорирование фурфурола с
использованием тех же катализаторов (серного цвета, пере-
киси бензоила с добавкой в качестве стабилизатора гидрохи-
нона), но без растворителя, при этом выход 5-хлорфурфуро-
ла составил уже 50% (считая на вошедший в реакцию фур-
фурол). Однако этот способ не всегда дает воспроизводимые
результаты.
Нами показано, что для успешного проведения этой ре-
акции необходимо пропускать ток высушенного хлора с такой
скоростью, чтобы температура реакционной смеси поддержи-
валась на уровне 125—130.
В этих условиях можно получить 5-хлорфурфурол с вы-
ходом около 40% (в пересчете на взятый для реакции фурфу-
рол).
15 Зак. 1034 225
СХЕМА СИНТЕЗА 5-ХЛОРФУРФУРОЛА
€ СНО -£1—» С1~^ Э- СНО + НС1
Х(Х xoz
Характеристика основного сырья
Фурфурол, ч., ГОСТ 10930—64, т. кип. 69—72718—20 мм.
Калий марганцевокислый, технический, ГОСТ 5777—51.
Соляная кислота, х.ч., концентрированная, ГОСТ
3118—46.
Перекись бензоила, получена по методике [7].
Серный цвет, ч., ГОСТ 17668—40.
Условия получения
, В трехгорлую колбу, снабженную обратным холодильни-
ком, термометром и барбатером, помещают 0,5 г серного
цвета, 0,9 г свежеполученной перекиси бензоила и 42 мл
(0,5 М) свежеперегнанною фурфурола- В нагретую до 118°
(не выше!) реакционную смесь пропускают в течение 1,5—--2
часов струю хлора, высушенную в склянке Тищенко с серной
кислотой и колонке с безводным хлористым кальцием. Ско-
рость подачи хлора регулируется таким образом, чтобы тем-
пература содержимого колбы была в пределах 125—130°.
О конце реакции судят по понижению температуры смеси до
110—115° даже при увеличении скорости подачи хлора. За-
тем реакционную смесь немедленно подвергают перегонке с
водяным паром. Из отогнанного дистиллята 5-хлорфурфурол
вымораживают, выпавшие кристаллы отфильтровывают и пе-
регоняют либо при нормальном давлении (т. кип. 180°),либо
под вакуумом (водоструйный насос, т. кип- 135—137730—
35 .им).
Выход 5-хлорфурфурола равен 25,2 г, чтб составляет 40%
от теоретического в пересчете на ф^ффурол; т. пл. 35—36°.
Примечание.
При использовании неочищенного фурфурола выход 5-хлорфурфурола
снижается на 12—15%. Реакцию можно проводить без добавления гидро-
хинона.
ЛИТЕРАТУРА
1. Н. Oilman, J. F. Wright. J. Amer. Chem. Soc., 52, 1170 (1930)
2. H. Gilman, J. F. W r 1 g h t. Recuel. Trav. Chim., 50,7-8,833.
(1931).
3. W. J. Chute, J. F. Wright. J. Organ. Chem., 10, 6,541 (1945).
4. 3. H. Назарова. Докл. АН УзССР, 4, 40 (1953).
5. Ф. Т. Пожарский. Уч. зап. Ростовского н/Д ун-та, 60 (1959).
6. 3. Н. Назарова, Ю. А. Бабаев. Ж- общ. химии, 32, 723,
(1962).
7. Л. Г аттерман, Г. Виланд. Практические работы по органи-
ческой химии, 1948, стр. 168.
Поступила в декабре 1967 г. Ростовск. н/Д Гос. ун-т
226
УДК 661.183.12
ЦИРКОНИЛФОСФАТ В ВИДЕ СФЕРИЧЕСКИХ
ГРАНУЛ
Г. И. КОРЕЛЬСКАЯ, Е. в. ЕГОРОВ, С. Б. МАКАРОВА
Неорганический ионит — цирконилфосфат {1, 2] — облада-
ет высокой обменной емкостью, термо- и радиационно-стой-
костью, повышенной селективностью к ряду катионов (в част-
ности, к цезию). Однако цирконилфосфат, полученный осаж-
дением из растворов, имеет вид порошка или гранул непра-
вильной формы, что затрудняет использование его в колон-
ках и не обеспечивает получение четких хроматограмм.
Для приготовления цирконилфосфата в виде сферических
гранул растворы цирконилнитрата и ортофосфорной кисло-
ты быстро смешивают и распыляют в колонке с минераль-
ным маслом [3]. Установлено, что скорость осаждения при
этом можно регулировать посредством комплексообразова-
ния цирконила с цитрат-ионами [4].
Способ получения цирконилфосфата в присутствии цит-
рат-ионов [4] нами был проверен, но вместо цирконилнитра-
та мы применяли циркопилхлорид.
СХЕМА СИНТЕЗА ЦИРКОНИЛ ФОСФАТА
0 НгР03 Нг0 0-H,PD, Лг0 О
I I II
Zr ------0-----h-------О----Р—о
мс(он)с(снгсоон)г он он
Характеристика основного сырья
Цирконилхлорид, х.ч., МРТУ 6-09-2901-66
Лимонная кислота, ч.д.а., ГОСТ 6552—58.
Ортофосфорная кислота, ч.д.а-, ГОСТ 3652—51.
Силиконовое масло, ТУ 2416—54.
15* 227
Бензол, ч.д.а,, ГОСТ 5955—51,
Этиловый спирт, ГОСТ 5962—51.
Условия получения
В химический стакан емкостью 700 мл, снабженный ме-
шалкой с регулируемым числом оборотов, помещают 300 мл
силиконового масла.
В стакан емкостью 100 мл помещают раствор 10 г
(0,031 Л1) цирконилхлорида в 40 мл дистиллированной воды,
к нему приливают раствор 5 г (0,026 /И) лимонной кислоты
в 15 мл дистиллированной воды и перемешивают в течение
5—6 минут до выпадения осадка. Образовавшуюся суспен-
зию выливают в стакан емкостью 150 мл, содержащий 45 мл
(0,055 АГ; уд. вес 1,063) 12%-ной ортофосфорной кислоты;
перемешивают до растворения осадка и полученный раствор
не позднее, чем через 30 секунд выливают в стакан с сили-
коновым маслом при перемешивании (150 об/мин; см. при-
мечание 1). Через 10 минут перемешивание прекращают ' и
приливают 200 мл дистиллированной воды для перевода гра-
нул цирконилфосфата в водную фазу и отделяют слой мас-
ла декантацией (см. примечание 2), Гранулы несколько раз
промывают декантацией дистиллированной водой до pH
4—5, отфильтровывают на воронке Бюхнера, промывают бен-
золом (50 мл) для удаления оставшегося масла, затем—эта-
нолом (50 мл) и хранят под слоем воды.
Выход цирконилфосфата рав4н 10 г (в пересчете на су-
хой продукт). Емкость сорбента равна 4—5 мг-экв/г. .
Примечания:
1. В зависимости от скорости перемешивания можно получить гра-
нулы размером от 0,1 до. 2 лл.
2. Силиконовое масло после отстаивания можно "повторно использо-
вать в процессе.
ЛИТЕРАТУРА
1. Ч. Дм флетт. Неорганические иониты, М., «Мир», 1966.
2. Ионный обмен. Изд-во ЛГУ, 1965.
3. N. Michael, W. D. Fletcher, D. Е. Grouch er, M. J,
Bell. Westinghouse Report CVNA. 135 (1961).
4. С. B. A tn p h 1 e 11, G. H. N а п с о 11 a s, T. Williams. Chem.
Ind., (London), 292 (1959).
Поступила в мае 1968 г.
ИРЕА
УДК 547.466.3'262
ЭТИЛОВЫЙ ЭФИР
р аминопропионовой кислоты
Этиловый эфир (3-аланина
С. И. БЕЗУЕВСКАЯ. И. М. БИЛИК
NHa—CHo CH.j-
ОС2Н-,
C5HiiNO2‘ М. в- 117,14
По литературным данным, этиловый эфир аминопрбпио-
новой кислоты получают действием на его гидрохлорид рас-
творов едких щелочей, карбонатов при сильном охлаждении
[1—3], а также метилата натрия [1] и гидрата окиси свинца
[4].
Нами были проверены и уточнены методы со смесью ед-
ких щелочей и карбонатов, а также с гидратом окиси свинца,
(метод Зелинского [4]). “
СХЕМА СИНТЕЗА ЭТИЛОВОГО ЭФИРА
РАМИНОПРОПИОНОВОИ КИСЛОТЫ
NH2-CH2-CH!-COOH+C2H,,OH+HC1 —
газ
HCl-NH2-CHa-CH2-Cf +н2о
ос2н.,
/О
2HCl-NH2-CH2-CHa-C< +РЬ(ОН)2 —
ос,н5
-> NH2-CH2-CH2-C(f 4-РЬС12+‘2Н2О
ОС2Н?, 229
Характеристика основного сырья
Аминопропионовая кислота (|3-аланин), ч., МРТУ
6-09-94-62.
Свинец, гидрат окиси, ч., МРТУ 6-09-2055-65.
Спирт этиловый, абсолютный, ГОСТ 8314—57.
Условия получения
В трехгорлую колбу, снабженную обратным холодильни-
ком, мешалкой и трубкой для подачи газа, помещают 200 г
р-аланина, добавляют 500 мл этилового спирта и при интен-
сивном перемешивании пропускают через смесь ток хлорис-
того водорода. Через 2—3 часа образуется прозрачный рас-
твор. Спирт отгоняют в вакууме (20—30 мм), после чего об-
разовавшийся гидрохлорид эфира p-аминопропионовой кис-
лоты переносят в эксикатор над серной кислотой и выдер-
живают в вакууме до постоянного веса.
Затем смешивают 23 г полученного гидрохлорида с 17 л
гидрата окиси свинца и помещают в колбу Кляйзена- Нагре-
ванием смеси на масляной бане в вакууме разлагают гидро-
хлорид и одновременно отгоняют этиловый эфир р-амннопро-
пионовой кислоты, собирая фракцию с. т. кип. 58—60°/Ч2—
14 мм.
Выход продукта равен 8—9 г, что составляет 50—60% от
теоретического, считая на гидрохлорид этилового эфира
p-аминопропионовой кислоты.
Найдено, %: N—11,89; 11,95.
C5HhNO2. Вычислено, %: N—11,96.
По литературным данным [3], т. кип. этилового эфира
р-аминопропионовой кислоты 54°/12 мм; 58°/14 мм.
Примечание.
Большее потери и соответственно низкий выход имеют место за счет
разложения этилового эфира p-аминопропионовой кислоты и быстрой по-
лимеризации продуктов разложения.
ЛИТЕРАТУРА
1. I. W i 1 е у, Sons. Organic Syntheses Coll, V. II, 7, 28(1943).
2. Л. Ф и з e p, М. Ф и з е р. Органическая химия, том 1, 1966, стр. 433.
3. Е. Abderhalden, A. Eodor. Hoppe —Seyler's zeit. fur physiol,
chem , 562, 112 (1913). i
4. H. Зелинский, А. Анненков, И. Куликова. ЖРФХО,
XLII1, часть II, 1091 (1869—1900).
Поступила в феврале 1968 г.
ИРЕА
УДК 547.466.3'262
ЭТИЛОВЫЙ ЭФИР
ш-АМИНОЭНАНТОВОЙ КИСЛОТЫ
С. И. БЕЗУЕВСКАЯ, И. М БИЛИК
NH2-CHa-CH,-CH,-CH2-CH2CHa-C<
4 ОС2Н6
C3H20NO2 М. в. 174,26
По литературным данным этиловый эфир ш-аминоэнанто-
ной кислоты получают разложением гидрохлорида этого эфи-
ра щелочами и карбонатами при сильном охлаждении [1—3]
или влажной окисью серебра [4].
Нами предложен метод получения этилового эфира
ы-аминоэнантовой кислоты действием гидрата окиси свинца
на его гидрохлорид по аналогии с получением этилового
эфира |3-аминопропионовой кислоты [5], так как этот метод
прост в выполнении и дает лучший выход.
СХЕМА СИНТЕЗА ЭТИЛОВОГО ЭФИРА «>• АМИНОЭНАНТОВОИ
КИСЛОТЫ
\ГН2 (СН3),;-соон+С2Н6ОН+HCI —
газ
— HCl-NH3-(CH2)G-Cf +Н2О
ОС2Н5
,0
2HC1-NH2-(CH„)6-Cf +РЬ(ОН)2^
ХОС2Н6
,о
-> NH,-(CH2)g-C^ +РЬС1,+2Н2О
хос2н5
231
Характеристика основного сырья
ы-Аминоэнантовая кислота, ч., МРТУ 6-09-3527-67,
Свинец гидрат окиси, ч., МРТУ 6-09-2055-65.
Спирт этиловый, абсолютный, ГОСТ 8314—57.
Условия получения
В трехгорлую колбу, снабженную обратным холодильни-
ком, мешалкой и ^рубкой для подачи газа, помещают 200 г
w-аминоэнантовой кислоты и добавляют 500 мл этилового
спирта. Смесь нагревают до 60° и при этой температуре н
интенсивном перемешивании пропускают через нее ток
хлористого водорода. Через 2—3 часа образуется прозрачный
раствор. Спирт отгоняют в вакууме (20—30 мм), после чего
гидрохлорид этилового эфира <о-аминоэнантовой кислоты пе-
реносят в вакуум-эксикатор и выдерживают над серной кис-
лотой до постоянного веса-
Затем смешивают 20 г гидрохлорида этилового эфира
го-аминоэнаитовой кислоты с 11,5 г гидрата окиси свинца и
помещают в колбу Кляйзена. Нагреванием смеси на масля-
ной бане в вакууме разлагают гидрохлорид и одновременно
отгоняют этиловый эфир <в-аминоэнантовой кислоты, собирая
фракцию с т. кип. 80—82°/2 мм или 92—9573,5 мм.
Выход этилового эфира <о-аминоэнантовой кислоты равен
8—12 г, что составляет 60—90% от теоретического.
Найдено, %: N—8,09; 7,86.
C9H2oN02. Вычислено, %: N—8,05.
По литературным данным [4], т, кип- продукта 80—
85°/2 мм.
ЛИТЕРАТУРА
1. 1. Wiley, Sons. Organic Syntheses Coll, V. 11, 7. 28(1943).
2. Л. Ф и 3 e p, M. Ф и 3 e p. Органическая химия, том 1, 1966, стр. 433.
3. Е. A b d erhalden, A. Fodor. Hoppe — SeyleFs zeit. fur phy-
siol. chem; 562, 112 ((1913).
4. A. H. Несмеянов, E. И. Васильев, P. X. Фреи длина.
Изв. АН СССР, отд. хим. наук, 7, 836 (1958).
5. Н. Зелинский, А. Анненков, И. Куликова. ЖРФХО,
XLIII, часть И, 1091 (1869—1900).
Поступила в феврале 1968 г.
ИРЕА
УДК 547.241
ЭТИЛОВЫЙ ЭФИР
МЕТИЛФОСФОНИСТОЙ кислоты
В. Г. ГРУЗДЕВ, К. В. КАРАВАНОВ, С. 3. ИВИН
СНаР<
II 4
oc2Hs
н
о
С3Н9О2Р
М. в. 108,07
Получение моноэфиров алкилфосфонистых кислот осу-
ществляется взаимодействием алкилдигалоидфосфинов со
спиртами [1, 2] или гидролизом полных эфиров фосфонистых
кислот [3, 4].
Нами разработан другой способ получения моноэфиров
алкилфосфонистой кислоты, заключающийся во взаимодей-
ствии алкилполифосфонитов со спиртами без растворителя.
По этому методу выход моноэфиров алкилфосфонистой кис-
лоты достигает 75%.
СХЕМА СИНТЕЗА ЭТИЛОВОГО ЭФИРА
МЕТИЛФОСФОНИСТОЙ КИСЛОТЫ
ос,н5
(СН3РО)„ + 7iC2HBOH -> nCH8Pf
II ЧН
О
Характеристика основного сырья
Метилполифосфонит, получают по методике, описанной в
статье «Алкилполифосфониты» настоящего сборника.
Этиловый спирт, ректифицированный, абсолютный, ГОСТ
5962—5.
233
Условия получения
К 6,2 г (0,1 ’/И) метилполифосфонита при температуре
100° в один прием приливают 5,7 г (0,1 7И) абсолютного эти-
лового спирта. Реакционную массу нагревают при темпера-
туре 100° в течение 1 часа (см. примечание) и затем фрак-
ционируют.
После Двухкратной перегонки получают 7,9 г этилового
эфира метилфосфонистой кислоты, что составляет 73% от
теоретического; т. кип- 58—5976 мм; d420—1,0531; —
1,4220.
По аналогичной методике могут быть получены и другие
моноэфиры алкилфосфонистой кислоты.
Найдено, %: С—33,00; 33,25; Н—8,15; 8,29; Р—28,19; 28,45.
С2Н9О2Р. Вычислено, %: С—33,33; Н—8,33; Р—28,66.
Примечание.
Все операции проводятся в токе инертного газа, очищенного от кис-
лорода, в противном случае выход моноэфиров алкилфосфонистой кисло-
ты снижается за счет ббразования моноэфиров алкилфосфоновой кислоты.
ЛИТЕРАТУРА
1. Б. А. Арбузов, Н. И. Ризположенскип. Изв. АН СССР,
Отд. хим. н., 5, 956, (1952).
2. А. Н. Пудовик, Д. X. Я р м у х а м е т о в а. Изв. АН СССР,
Отд. хим. н„ 5, '902 (1952).
3. М. И. Кабачник, Е. Н. Цветков. Докл. АН СССР, 117, 817,
(1957).
4. Чжан Жун-юй. Химия и применение фосфорорганических сое-
динений, «Труды II конференции», 310—316 (1962).
Поступила в январе 1968 г.
УДК 547.341.
О-ЭТИЛ-О-[Р-(ТРИЭТИЛСИЛИЛ)АЛЛИЛ]
МЕТИЛФОСФОНАТ
И. Д. ШЕЛАКОВА, С. 3. ИВИН, В. К. ПРОМОНЕНКОВ
/ОС2Н5
СН3Р<
II хоснас=сн2
0 4Si(C2H6)3
Ci2HizO3PSi М. в. 359,19
О-Алкил-О-0-триалкил (алкокси) силилаллилметилфосфона-
ты в литературе не описаны. Нами разработан способ, поз-
воляющий получать указанные соединения из О-алкил-О-про-
паргилметилфосфонатов и триалкил (алкокси)гидридсиланов
в присутствии катализатора Спайера- Продукты интересны
возможностью использования в разнообразных синтезах фос-
форорганических веществ.
СХЕМА СИНТЕЗА
О-ЭТИЛ-О-[р (ТРИЭТИЛСИЛИЛ)АЛЛ ИЛ)МЕТИЛФОСФОНАТА
/ОС2Н5
СНзР< + HSi(CaH5)3 ->
II ХОСН2С^СН
о
ОС2Н5
-> сн3р/
II ХОСН2С=СН2
° 4S1(C2H5)3
Характеристика основного сырья
О-Этил-О-пропаргилметилфосфонат, т. кип. 85°/5 мм;
с/420—1,1181; я'р —14415; получен прибавлением эквимоле-
235
кулярного количества спирта в смеси с триэтиламином к
О-этилметилхлорфосфонату.
Триэтилсилан, т. кип. 108°; d^°—0,7510 [1].
Триэтоксисилан, т. кип. 132—135°; d420—0,8745 [2].
Катализатор Спайера (0,1 М раствор HaPtCle-SHaO в
абсолютном изопропиловом спирте) [3].
Условия получения
В четырехгорлую колбу емкостью 100 мл с мешалкой, ка-
пельной воронкой, термометром и обратным холодильником
помещают 16,2 г (0,1 Л1) О-этил-О-пропаргилметилфосфоната
и 3—4 капли катализатора Спайера. При энергичном пере-
мешивании к смеси прибавляют по каплям 11,6 а (0,1 М)
триэтилсилана- Температура реакционной массы повышается
за счет тепла реакции до 125—130°. По окончании прикапы-
вания перемешивание продолжают в течение 1—2 часов, при
этом температура реакционной смеси самопроизвольно по-
нижается до комнатной. Продукт перегоняют в высоком ва-
кууме.
Выход О-этил-О-[(3- (триэтилсилил) аллил]метилфосфоната
равен 27,1 г, что составляет 75% от теоретического; т. кип.
76—7870,003 мм; d420 — 0,9789; п*> —1,4280.
Найдено. %: Р — 8,73.
C12H17O3PS1. Вычислено, %—8,64.
Аналогично «получаютО-изопропнл-О-[0- (триэтоксисилил)-
аллил]метилфосфонат, т- кип. 125—12770,002 мм; d420 —
1,0455; 1,4278. Выход 72%.
ЛИТЕРАТУРА
1. К. А. Андрианов. Кремнийорганические соединения. М., Гос-
химиздат, 1955, стр. 96.
2. Там же, стр. 147.
3. I. S р е 1 е г, I. Webster, G, Barnes. J. Amer. Chem. Soc.,
79, 974 (1957).
Поступила в декабре 1967 г.
АЛФАВИТНЫЙ ПЕРЕЧЕНЬ
СОЕДИНЕНИЙ, ОПИСАННЫХ В НАСТОЯЩЕМ СБОРНИКЕ
Алкилированные ксиленолы .....................................
N-Алкилмеркаптоэтиленимин.....................................
Ы-(Р-Алкнлмеркаптоэтнл)этиленнмины............................
N-Алкоксиметилэтиленимин......................................
Алкилполифосфониты ...........................................
2-Алкил-1,3-циклопентандионы..................................
Анизоилантранил ..............................................
2-Ацетонил-8-оксихинолин......................................
1 -Бензил-3,5-дибутил-4 пропилпиразол.........................
1 Бензил-3,5-дигексил-4-пентнлпиразол.........................
1 -Бензил-3,5-дипентнл-4-бутилпиразол.........................
1-Бензил-3,5-дипропил-4-этилпнразол....................... .
1-Бензил-3,4,5-триалкилпиразолы . .........................
Бензиловый эфир 4-метнлумбеллиферопа..........................
Бис-.(ацетальдегид)оксалилдигндразон..........................
4,4'-Бис-[2,4-бис- (карбоксилметиламино) - 1,3,5-триазинил-6-амино]
2,2'-толандисульфокислота.................................
1,4-Бис-(диэтокснфосфоио)бутен-2..............................
2,2-Бис-(4'-окси-3'-хлор-5'-метилфенил) пропан................
2,2-Бис-(4'-окси-3'-хлорфенил)пропан . ...............
Бис- (циклогексанон)оксалилдигидразон.........................
1,1-Бис-(3'-хлор-4'-оксифенил)циклогексан ......
5-Бром-2-бензоилфуран .................................
о-Бромфенилдихлорарсин .......................................
5-Бром-2-фурилфенилкетон......................................
1-н-Бутил-3,5-дибутнл 4-пропилпиразол.........................
1-н Бутил-3,5-дигексил-4-пентилпиразол........................
1 -н-Бутил-3,5-дипентнл-4-бутилпиразол........................
1-н-Бутил-3,5-дипропил-4-этилпиразол..........................
1-н-Бутил-3,4,5-триалкилпнразолы..............................
4-вгор-Бутилфеиол.............................................
2-Гексил- 1,3-циклопентаидиои . . . . ,...............
2-Гептил-1,3-циклопентандион
Гидрохинон-2,6-диметилеииминодиуксусиая кислота
Гидрохлорид моио^таноламина ..................................
Гидрохлорид р-хлорэтиламина...................................
Гуаиилтиомочевииа ............................................
N-Гуанилформамидинсульфииовая кислота.........................
7
197
12
197
14
16
150
164
23
23
23
23
21
19
38
24
28
30
32
38
35
41
43
41
46
46
46
46
45
48
17
17
51
82
82
54
56
237
М,М-Диалкилацетамиды........................................
Диалкил(гексафторизопропил) фосфаты.........................
Диалкил (псргалоидизопропени л) фосфаты.....................
4,4'-Диаминотолан-2,2'-дисульфокислота -....................
2,5-Диацетилтиофен..........................................
3,5-Дибутил-4-пропилизокс азол..............................
3,5-Дибутил-4-пропилпиразол.................................
N.N-Ди (гексадецил) ацетамид................................
3,5 Дигсксил-4-пентилизоксазол..............................
3.5-Дигексил-4-пеитилпиразол................................
3,5-Дигептил-4-гексилизоксазол..............................
3,5-Дигептил-4-гексилпиразол..................................
Ди-[(р-дифенилфосфииил)этил]малоиовая кислота ....
Ди-[(р-дифенилфосфинил)этил]малоиовый эфир..................
Диизопропил (гексафторизопропил) фосфат.....................
Диизопропил-а-дифторхлормстилР-фторхлорвинилфосфат
Диимид нафталин-1,4,5,8-тетракарбоновой кислоты ....
Дикалиевая соль динитротоландисульфокислоты ....
2-[Ди(карбэтокси)метил]-8-оксихинолин.......................
N.N-Дилаурилацетамид........................................
5-(Диметиламино)фурфурол....................................
Р-Диметиламиноэтиламин......................................
Диметил (гексафторизопропил (фосфат.........................
3,5-Диметил-2,6-ди- (а-метилбензил)фенол....................
Диметил-а-дифторхлорметил-р-хлорвинилфосфат ....
Диметил-а-дифторхлорметил-р-хлорвинилфосфат ....
3,й-Диметил-2-(а-метилбензил)фенол..........................
2,5-Диметил-4-(а-метилбензил (фенол.........................
2,4-Диметилтиофен...........................................
3,4-Диметилтнофеи...........................................
2,5-Диметил-4-(циклогексил) феиол...........................
2,2'-Ди- (4-нитрофенил) -5,5'-дифенпл-3,3'-(3,3'-диметокси-4,4'- дифени
лен)дитетразолий хлористый .................................
1,4-Диокси-2,6-бис[М,М'- ди- (карбоксиметил)-аминометил]бензол
2,5-Ди- (оксиметил)тетрагидрофураи..........................
2,5-Ди (оксиметил) фураи....................................
3,5-Дипентил-4-бутилизоксазол...............................
3,5-Дипентил-4-бутилпиразол.................................
3,5-Дипдопил-4-этилизоксазол................................
3,й-Дипропил-4-этилпиразол..................................
1,5-Дифенилкарбогидразид....................................
Дифепилфосфнпилуксусная кислота.............................
(Р-Дифенилфосфинйлэтил)малоновая кислота....................
(р-Дифенилфосфинилэтил)малоновый эфир.......................
Дихлорангидрид трихлормети.ттиофосфоновой кислоты
Дихлорангидрид трихлорметилфосфоновой кислоты
Дихлор-сммлг-триазиниламинофлуоресцеин I....................
5-(Диэтиламино)фурфурол.....................................
Диэтил (гексафторизопропил) фосфат..........................
Диэтил-а-дифторхлорметил-Р-фторхлорвииилфосфат
3,5-Диэтил-4-метилизоксазол.................................
3,5-Диэтил-4-метилпиразол . .........................
Диэтил-а-фторднхлорметпл-р-фторхлорвииилфосфат
Диэтиловый эфир димстилянтарной кислоты.....................
Диэтиловый эфир (8-окси-2-хинолил)малоповой кислоты
Додецилярный ангидрид.......................................
М-(Р-Изобутилмеркаптоэтил)этиленимин........................
О-Изопропилметилцианфосфонат................................
О-Изопропил-О-[(р-триэтоксисилил) аллил-метилфосфонат
О-Изопропиловый эфир метилфосфоновой кислоты ....
58
64
61
26
68
192
195
59
192
195
192
195
70
71
65
62
73
26
111
58
78
82
65
7
62
62
7
7
84
87
7
154
51
76
90
192
195
192
195
92
95
98
99
105
107
101
109
64
62
192
195
62
88
111
114
13
116
236
125
238
Калиевая соль п-стиролсульфокислоты.........................
я-Карбоксигалланилид........................................
Кислый О-изопропиловый эфир метилфосфоновой кислоты
Кислый О-этиловый эфир метилфосфоновой кислоты
Крезоловый красный..........................................
о-Крезолсульфоф'галеин......................................
Лауриновый эфир метакриловой кислоты....................
Дейкоосиование кристаллического фиолетового ................
N Метил-О-ацетилиндоксил....................................
1-Метил-З-ацетоксииндол.....................................
2-а-Метилбензил-4-алкилфенолы.......................
2-а-Метилбензил-4-н-бутилфенол..............................
2-а-Метилбензил-4-трет-бутилфенол...........................
1- Метил-3,5-дибутил-4-пропилпиразол........................
1 -Метил-3,5-дигексил-4-пентилпиразол.......................
1-Метил-3,5-дипентил-4-бутИлпиразол.........................
1 -Метил-3,5-дипропил-4-этилпиразол.........................
1-Метил-3,5 диэтил-4-метилпиразол...........................
N-Метилипдоксилапетат.......................................
а-Метиллевулиновая кислота..................................
Метплмалононый эфир . ...............................
Метил- (8-оксихинальдил)кетон...............................
Метилполифосфонит...........................................
1-Метил-3,4,5-трИалкилпира.золы.............................
N-Метилфеиилглицин-о-карбоновая кислота.....................
5-Метокси-3-нитро-2-оксибензальдегид........................
5-Метокси-З-нитросалициловый альдегид.......................
5-Метоксасалициловый альдегид...............................
2-(п-Метоксифенил)-4н-3,1-бензоксазон-4.....................
N-о-Метоксифенилформамидинсульфиновая кислота
о-Нитробензальдегид.........................................
5-Нитро-2-пиридин...........................................
Нитросиний тетразолий.......................................
5-Оксиметилфурфурол.........................................
2-Окси-5-метоксибензальдегид................................
N-Оксисукцинимид............................................
N-о-Оксифенилформамидинсульфиновая кислота ....
1- (8-Окси-2-хинолил) -2-пропанон...........................
8-Оксихиполин-2-сульфокислота...............................
2-Октил-1,3-циклопентандион.................................
2-Пентил-1,3-циклопентанднон................................
5-(П-Морфолино)-2-бензоилфураи..............................
5-(Ы-Пиперидино)-2-бензоилфуран.............................
5-(Ы-Пиперидино)фурфурол....................................
Пропаргиловый эфир акриловой кислоты........................
Пропаргиловый эфир метакриловой кислоты.....................
Сополимеры стирола с п-дивинилбензолом .....................
Тетраметилдифосфиндисульфид.................................
Тетрамид нафталин-1,4,5,8-тетракарбоновой кислоты
З,3',4,4'-Тетрациандифеииловый эфир.........................
3,3',4,4'-Тетрациандифенилоксид.............................
Тиопиколинанилид ...........................................
3,4,5-Триалкилизоксазолы.......................................
118
121
125
124
126
126
129
131
134
134
136
137
136
141
141
141
141
141
134
85
88
164
14
139
142
144
144
146
150
148
162
223
154
158
146
160
162
164
167
17
17
173
172
174
176
176
184
182
178
186
186
189
191
239
3,4,5-Триалкилпиразолы.......................................... 194
N-Триалкилсилилэтиленимин.........................................197
2,2,4-Триметил-4-(п-оксифенил) хромай.............................199
Трихлорметилдихлорфосфонит........................................202
5-(Трихлорметил)оксазолидон-2.....................................204
2,3,5-Трихлормстилтетрагидрофуран.................................209
N-Триэтилсилилэтилеиимин..........................................197
2,3,5-Триэтокситетрагидрофуран....................................207
о-Фенилборная кислота..........................................209
Фенилмагний бромистый..........................................210
1-Фенилсемикарбазид............................................90
Феноловый красный..............................................212
Фенолсульфофталеин.............................................212
1-Хлор-4- (алкоксиметил) циклопентены-2.........................
1-Хлор-4- (аллилоксиметил)циклопентен-2.........................
2-Хлор-5-аминопиридин...........................................
Хлорангидрид галловой кислоты ..................................
Хлорангидрид транс-трихлоркротоновой кислоты....................
5-Хлор-2-бензоилфураи...........................................
1-Хлор-4- (изобутоксиметнл)циклопентен-2........................
1-Хлор-4- (изопропэксиметнл)циклопентеи-2.......................
1-Хлор-4- (метоксиметил)циклопентен-2...........................
2-Хлор-5-нитропиридин...........................................
5-Хлорфурфурол..................................................
1-Хлор-4- (этоксиметил)циклопентен-2............................
215
216
218
122
220
42
216
216
215
222
225
216
Цирконилфосфат в виде сферических гранул...................227
5-Этил-2-ацетотиенои..............................................68
N-Этилмеркаптоэтиленимин........................................198
N-(Этилмеркаптоэтил) этиленнмнн.................................. 12
Этиловый эфир р-аламина......................................229
Этиловый эфир p-аминопропионовой кислоты.......................' 229
Этиловый эфир ш -аминоэнантовой кислоты......................231
Этиловый эфир дифенилфосфинилуксусной кислоты .... 96
Этиловый эфир дифенилфосфинистой кислоты.......................96
Этиловый эфир метилфосфопистой кислоты........................233
Этилтиофен.......................................................68
О-Этил-О-[(Р-триэтилсилил) аллил]метилфоефонат..................235
N-Этоксиметилэтиленимин..........................................198
Замоченные опечатки
Стр. Строка Напечатано Следует читать
100 2 сверху (99%) (₽-дифснплфосфо- (93 %) (“-дифеннлфосфинил)
ннл)
=N\ =ч =-N / =
154 1 формула _N^C%_Z
156 14 снизу электроду— электроду при pH 7,2
3 снизу Дрекселя Тшцецко
157 7 сверху (1966). (1967).
23$ 11 снизу % %: Р-