





























































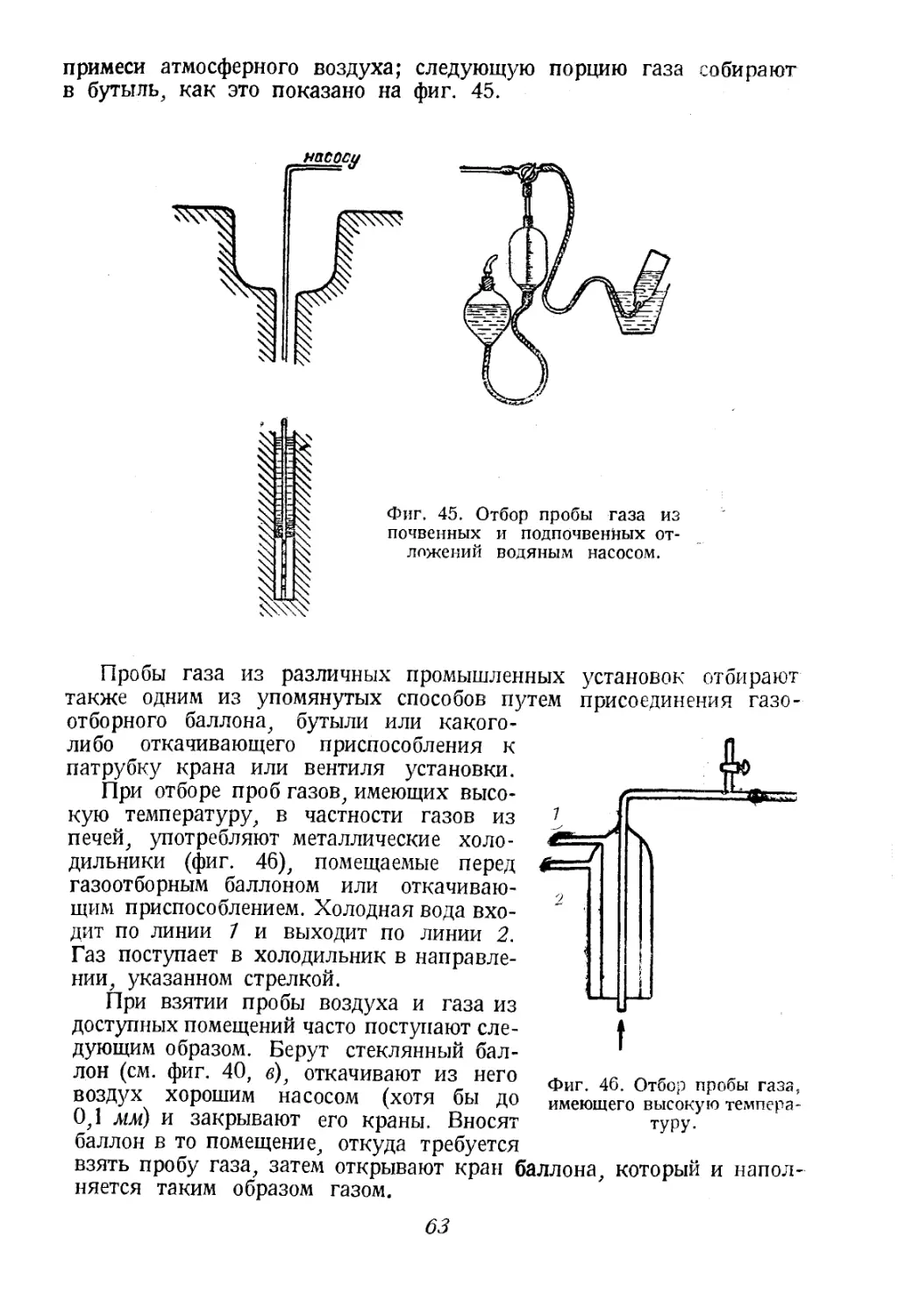
























































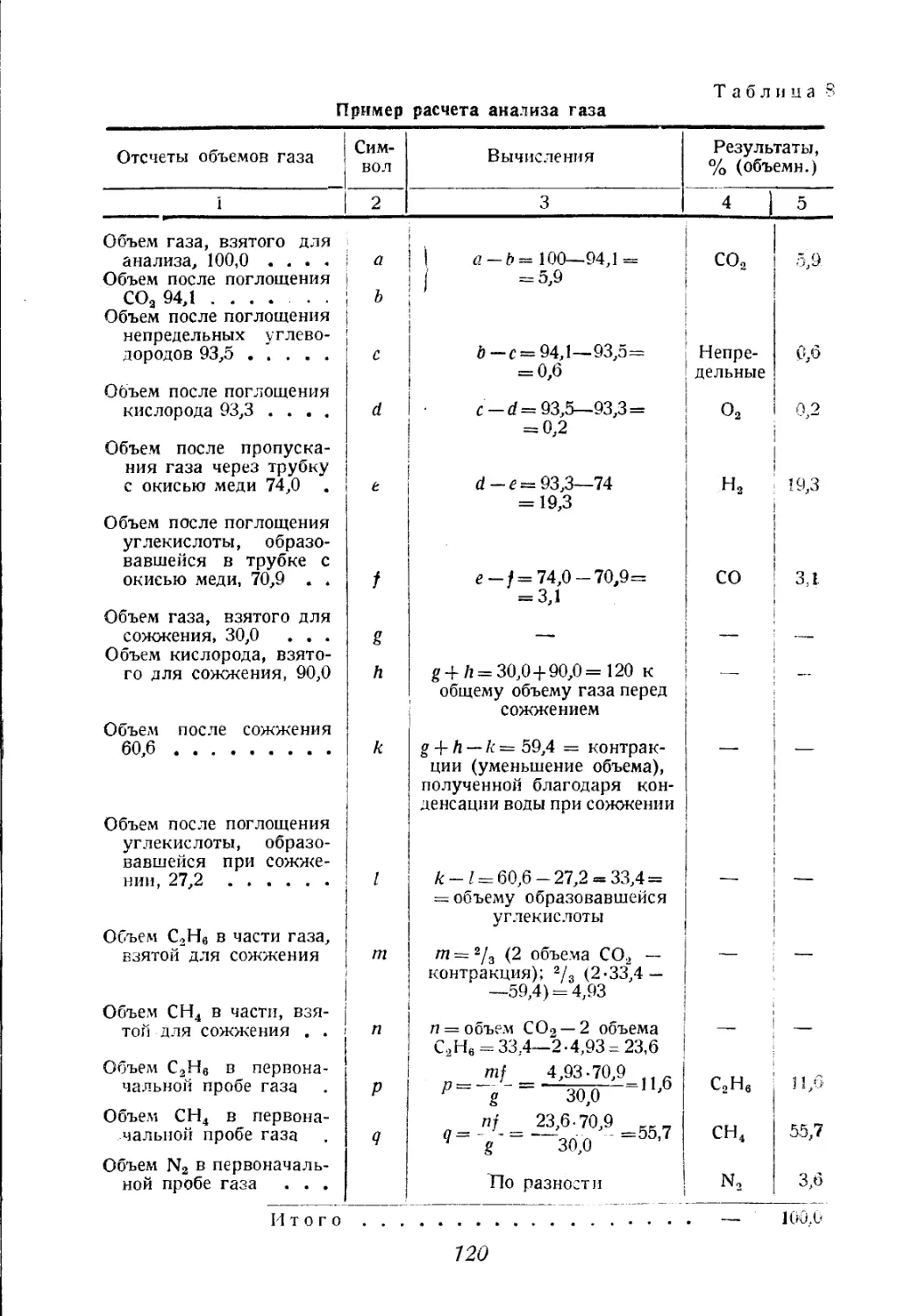






















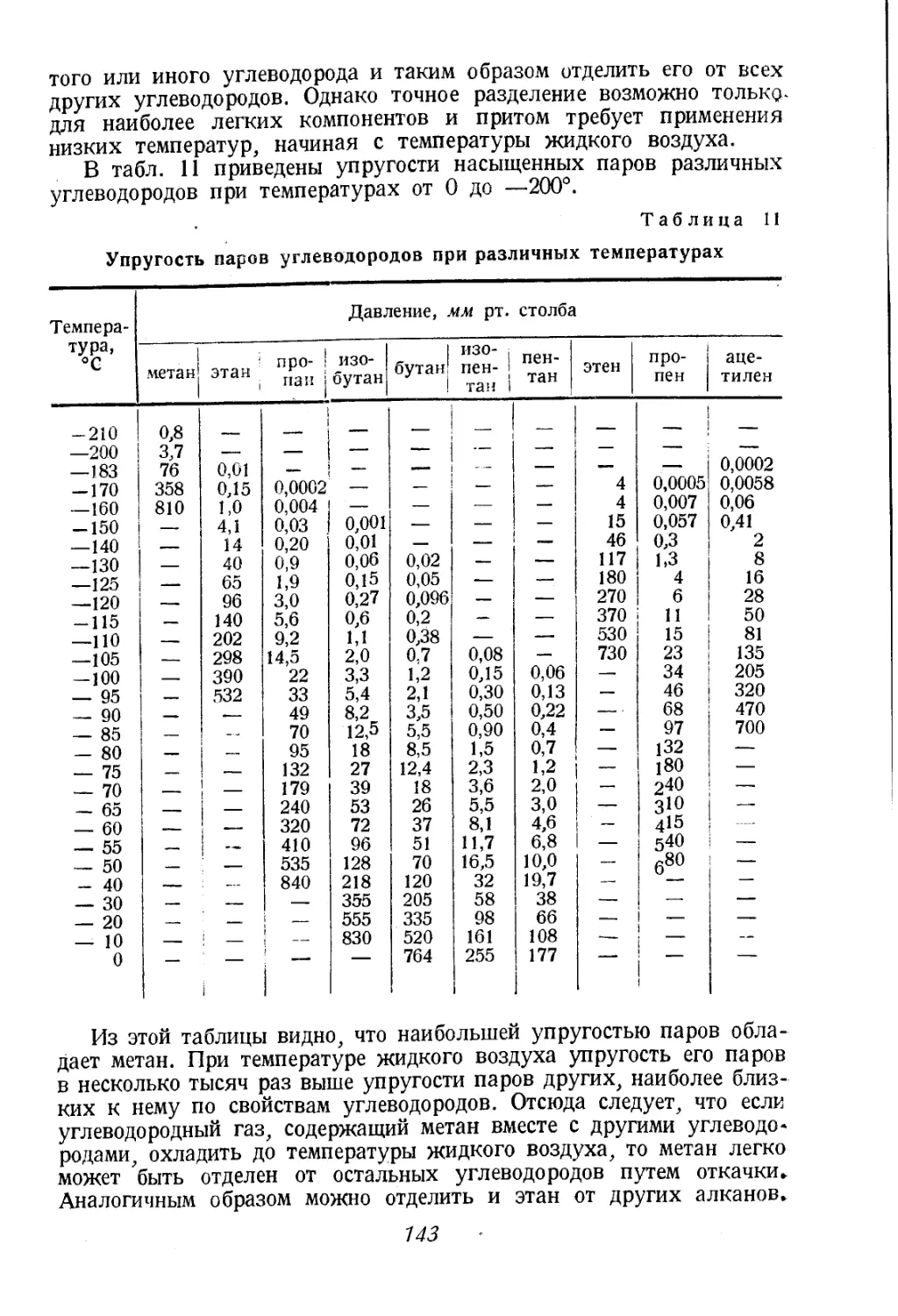















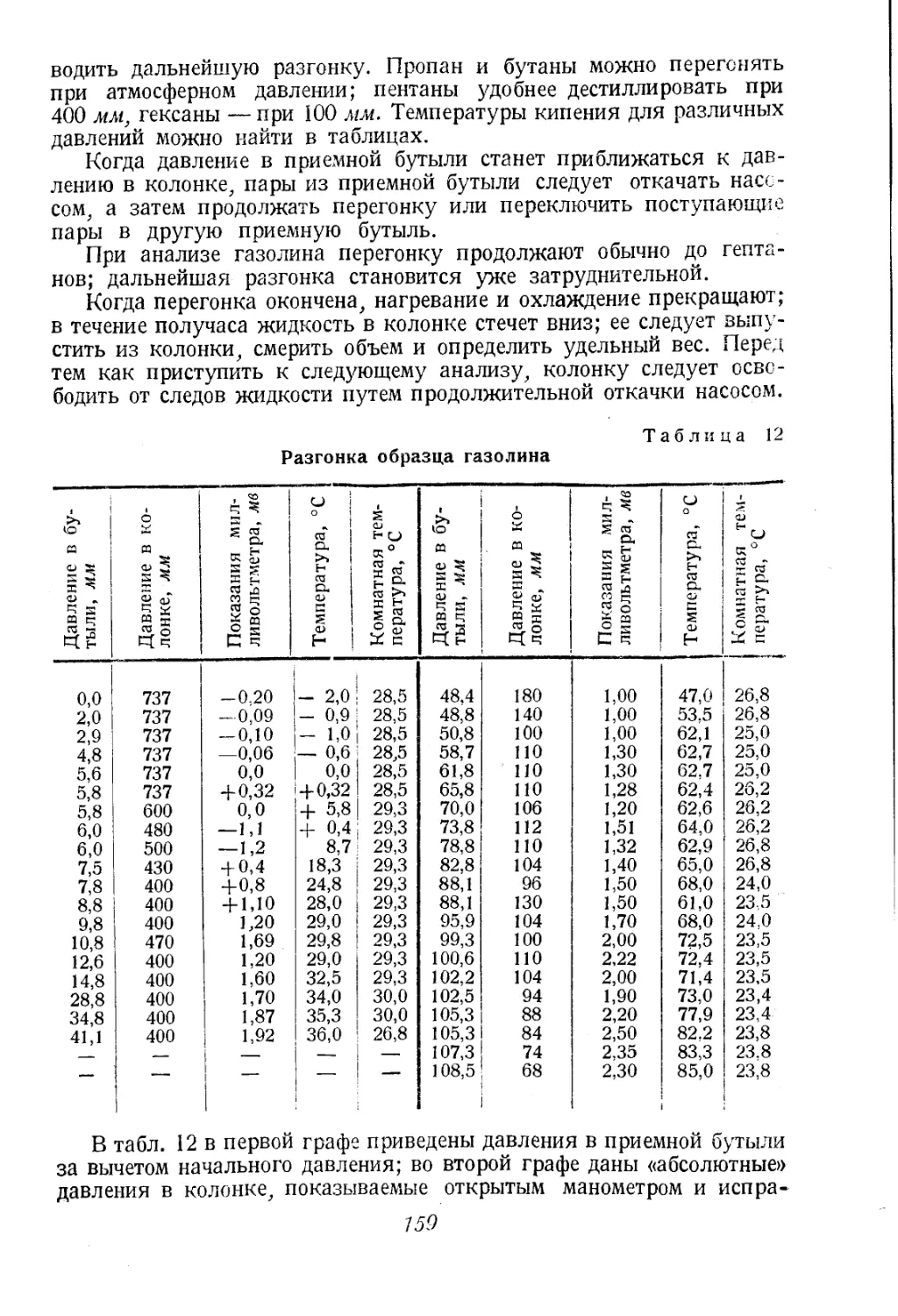


























































































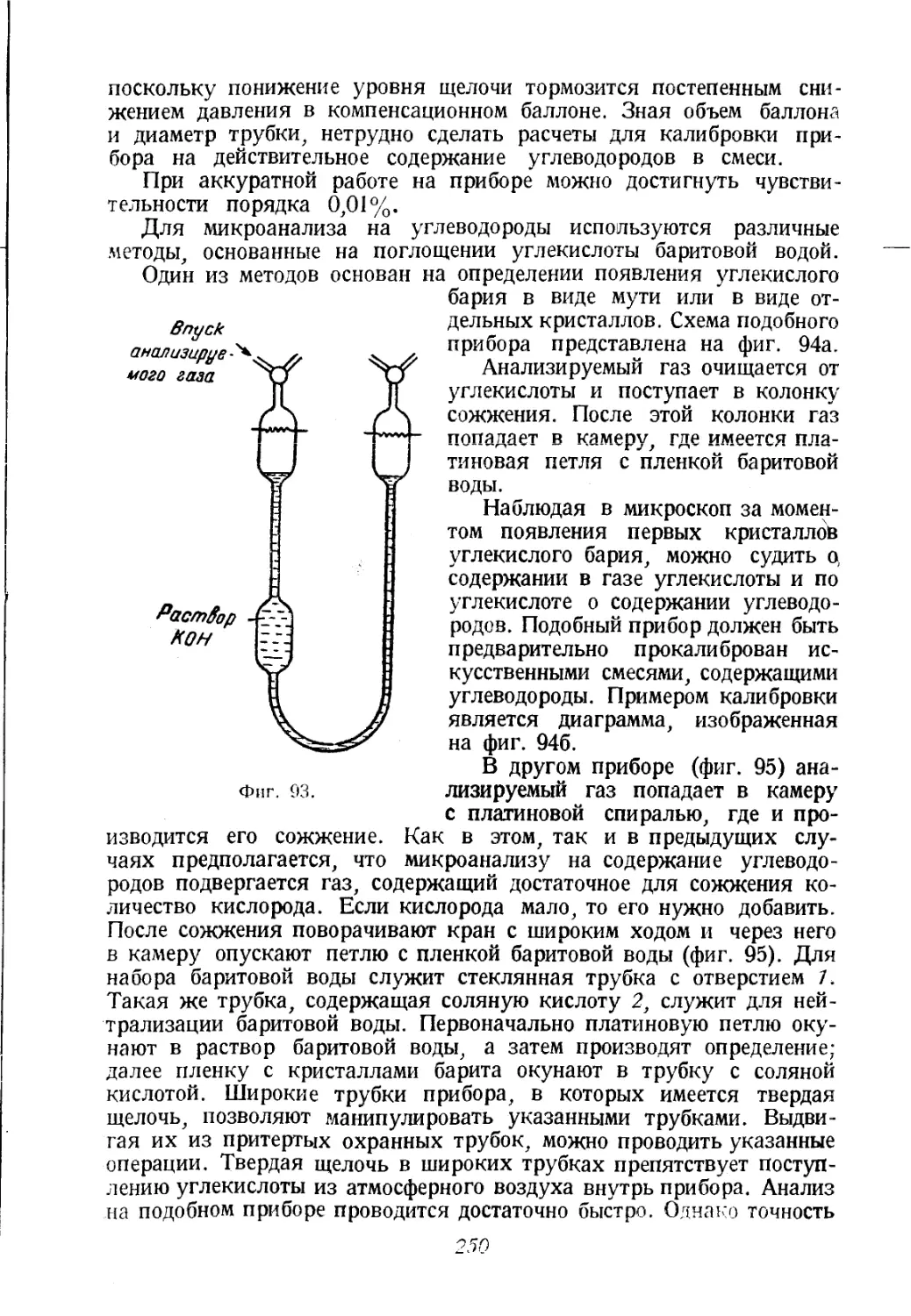























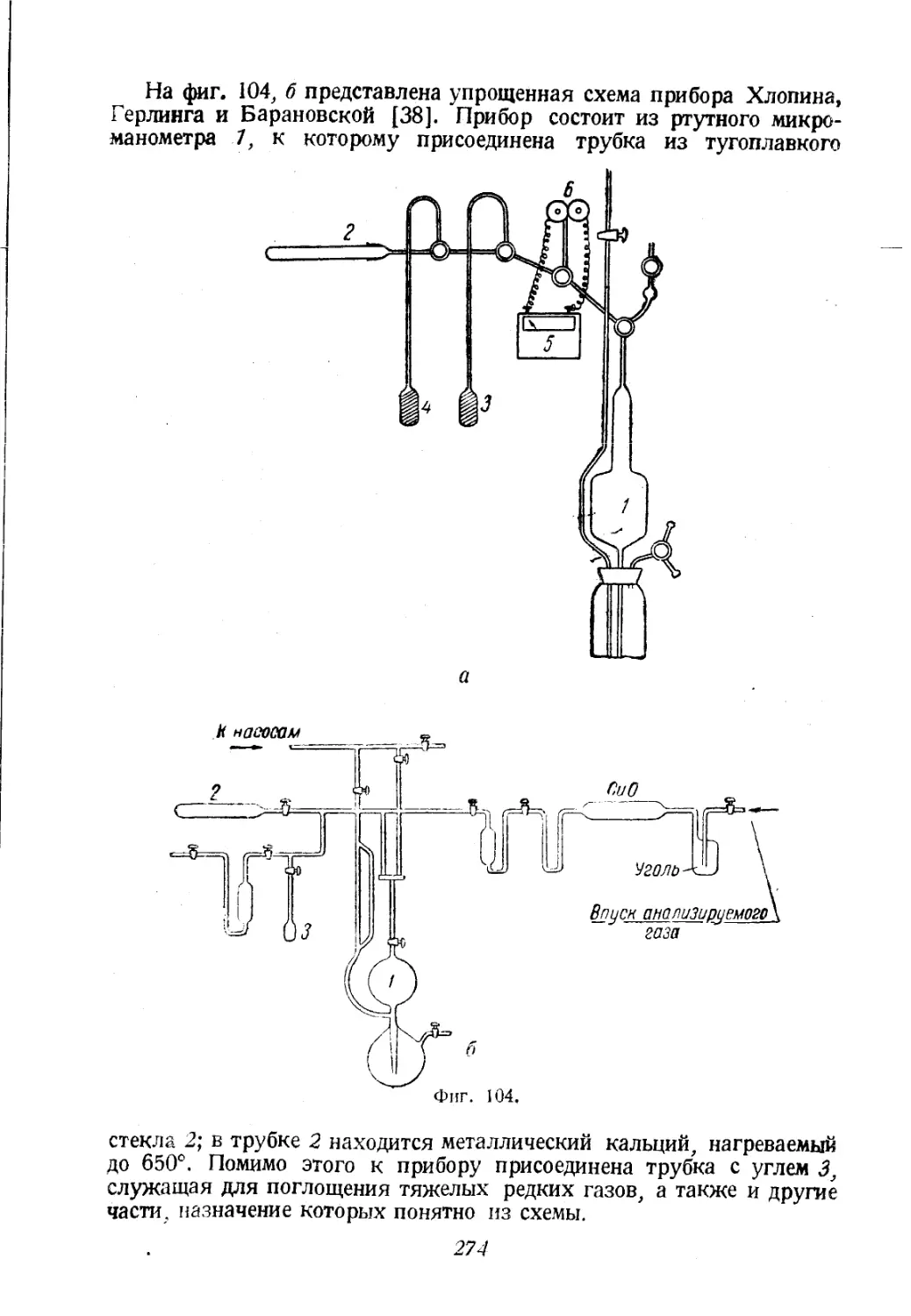








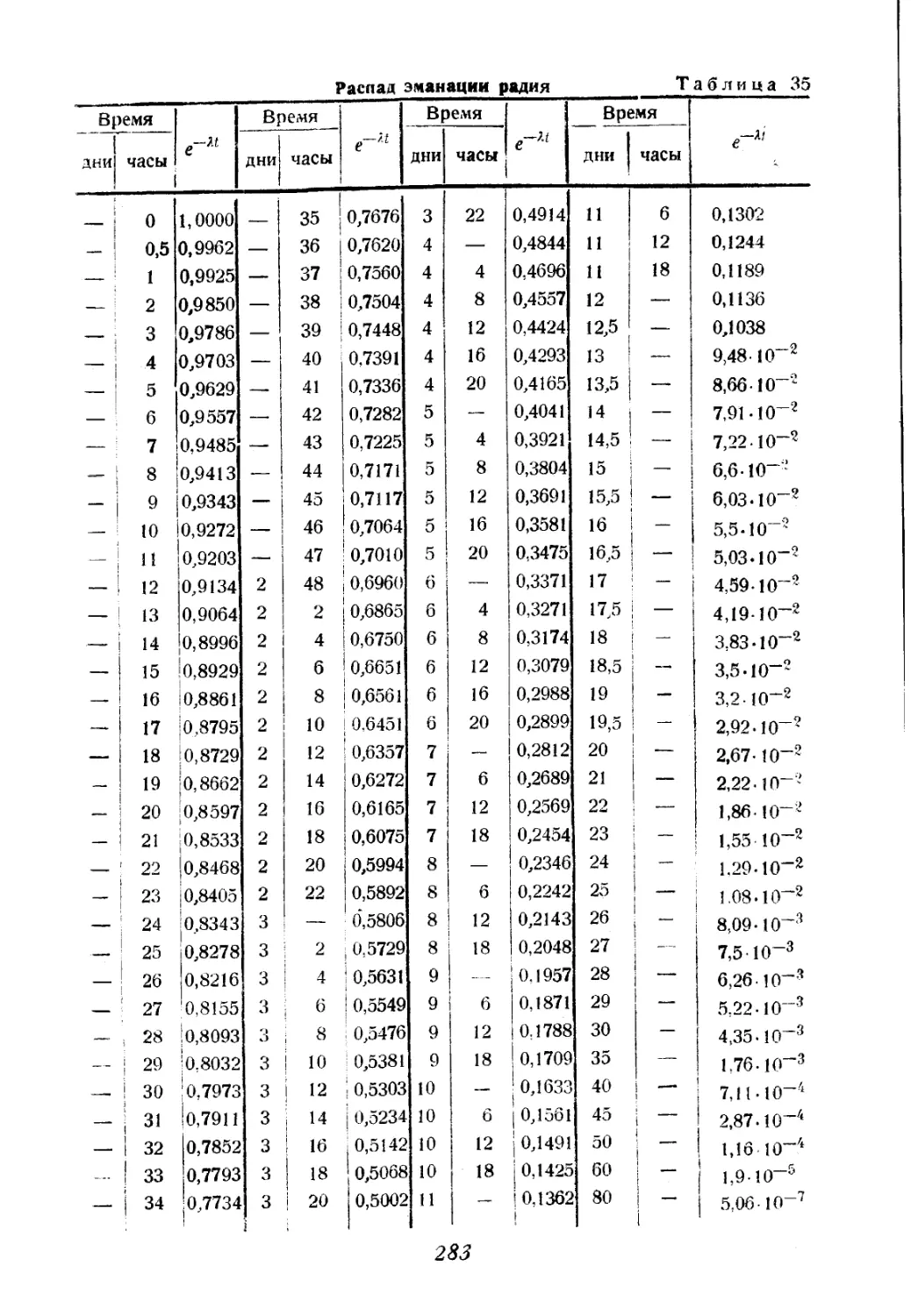





















































Автор: Соколов В.А.
Теги: физика анализ госиздат издательство москва инертные газы газы анализ газов
Год: 1950




Текст
В. А. СОКОЛОВ
I,
Г n.’OBEi’tiiJ
1960
АНАЛИЗ ГАЗОВ
ГОСУДАРСТВЕННОЕ НАУЧНО-ТЕХНИЧЕСКОЕ ИЗДАТЕЛЬСТВО
НЕФТЯНОЙ И ГОРНО-ТОПЛИВНОЙ ЛИТЕРАТУРЫ
Москва 1950 Ленинград
13-5-3
Л
АННОТАЦИЯ
Настоящая книга представляет собой руководство по анализу
газов.
В кнцге описаны разнообразные методы и приборы, применяе-
мые для анализа природных, а также промышленных газов и в пер-
вую очередь газов нефтяных месторождений и газов нефтеперера-
батывающих заводов. Описанные методы и приборы могут быть
также использованы для нужд других отраслей промышленности
и для различных научно-исследовательских целей.
Первый раздел книги касается методики и техники газоанали-
тических определений, затем описываются методы и приборы для
общего газового анализа, для анализа углеводородных газов и
для анализа сернистых, азотистых и других газов неорганического
характера. Значительное место в книге занимают современные ме-
тоды микроанализа газов, именно углеводородных, редких и др.
Последние главы книги содержат описание физических мето-
дов газового анализа и методов непрерывного анализа с автомати-
ческой или полуавтоматической регистрацией показаний приборов.
Книга предназначена для техников, инженеров и научных
сотрудников, работающих в области исследования и использования
природных и промышленных газов.
ГЛАВА ПЕРВАЯ
ОБЩИЕ СВЕДЕНИЯ И ТЕХНИКА
ГАЗОАНАЛИТИЧЕСКИХ ОПРЕДЕЛЕНИЙ
§ 1. Понятие о газовом анализе и его подразделениях.
Законы газового состояния
Анализ газов представляет собой специфическую отрасль ана-
литической химии, поскольку методы газового анализа резко
отличаются от обычных методов анализа жидких и твердых веществ.
Особенности газового состояния вещества обусловливают широкое
применение при анализе газов физических методов. Во многих слу-
чаях анализ газов вообще производится исключительно с помощью
только физических методов. В других случаях физические методы
исследования газов сочетаются с химическими определениями.
В анализируемой газовой смеси могут встречаться как газообраз-
ные элементы, так и различные газообразные их соединения.
При анализе природных газов приходится встречаться с газо-
образными элементами и соединениями, к числу которых в первую
очередь относятся кислород, водород, азот, углекислый газ, окись
углерода, сероводород, сернистый ангидрид, метан, этан, пропан,
бутан и другие высшие парафиновые углеводороды, редкие газы
(гелий, неон, аргон, криптон, ксенон). В промышленных га-
зах главным образом встречаются окислы азота, сернистый и сер-
ный ангидрид, аммиак, водород, окись углерода, предельные и не-
предельные углеводороды, галоиды и их производные, пары разно-
образных органических соединений.
Не все перечисленные газы встречаются одновременно. В при-
родных газах, например, не приходится ожидать примесей некото-
рых газов, характерных для промышленных установок. С другой
стороны, в различных газах промышленного характера часто нельзя
ожидать наличия некоторых компонентов, встречающихся в при-
родных газах. В зависимости от целей анализа, состава и количе-
ства исследуемого газа методика применяемого анализа бывает раз-
личной. Можно указать на следующие основные подразделения га-
зового анализа.
1. Общий анализ.
2. Анализ углеводородных газов.
1*
3
3. Методы определения сернистых, азотистых, галоидных и дру-
гих неорганических газов.
4. Методы микроанализа на различные газообразные элементы
и соединения.
5. Анализ на редкие газы.
б. Специальные физические методы газового анализа.
7. Методы непрерывного газового анализа с автоматической ре-
гистрацией.
Общий газовый анализ заключается в определении наиболее
известных газообразных элементов и соединений, причем те из них,
которые характеризуются схожестью своих химических свойств,
определяют суммарно. При этом виде анализа кислотные газы (СО2
и H2S) поглощают щелочью и определяют их в сумме. Отдельно опре-
деляют кислород, водород и окись углерода, последние два газа —•
обычно путем сожжения. Определяют с помощью сожжения также
суммарное содержание углеводородов, а с помощью поглощения —
ненасыщенные углеводороды. Кроме того, по разности определяют
азот вместе с редкими газами.
Газы щелочного характера (NH3) поглощают раствором кислоты.
Подобные определения не позволяют судить о содержании от-
дельных индивидуальных углеводородов. Между тем надобность
в определении индивидуальных углеводородов очень велика, осо-
бенно в нефтяной промышленности и при различных исследованиях
природных газов. Для этих целей употребляют специальные методы
анализа углеводородных газов, основанные на разгонке газовой
смеси на отдельные компоненты при низких температурах, а также
и иные методы.
При общем газовом анализе азот, как это упомянуто выше, опре-
деляется вместе с редкими газами. Чтобы определить отдельные
индивидуальные редкие газы (гелий, неон и т. д.), применяют спе-
циальные методы, большинство которых основано на сорбции газов
углем при низких температурах.
Для определения сернистых и азотистых газообразных соеди-
нений, как и для определения галоидов и их производных и многих
других неорганических газов, а также паров многих органических
веществ, применяют разнообразные специальные, преимущественно
химические, аналитические методы.
Во многих случаях требуется применять методы анализа, обла-
дающие очень высокой чувствительностью и позволяющие прово-
дить анализ очень малых количеств газа или определять ма-
лые концентрации интересующих нас газообразных компонентов в
анализируемой газовой смеси. Для этой цели существуют специ-
альные разнообразные методы микроанализа газов.
Многие аналитические исследования могут быть проведены с
помощью физических методов. В частности, сюда относятся методы
спектрального газового анализа, а также и другие оптические ме-
тоды.
Для ряда производственных, а также научных целей требуется
непрерывный газовый анализ, при котором содержание интересу-
4
ющих нас компонентов автоматически записывается. Для этой цели
разработаны разнообразные методы автоматического газового ана-
лиза, основанные на сравнении теплопроводности газов и на их сож-
жении с применением в обоих случаях электрических измерений,
а также на иных принципах.
При исследовании газов и изучении их состава большую помощь
может принести определение таких свойств газа, как удельный вес,
теплотворная способность и т. п., которые являются косвенными
показателями газового состава и в отдельных случаях могут дать
довольно точное представление о содержании в газах тех или иных
компонентов.
Законы газового состояния
При работе с тазами в первую очередь приходится сталкиваться
с физическими законами, которым газы подчиняются. Эти физи-
ческие законы не являются безукоризненно точными. При деталь-
ных исследованиях все газы обнаруживают те или иные уклонения
от законов, с чем и приходится считаться при точной работе. Вообра-
жаемый газ, который совершенно точно подчиняется законам газо-
вого состояния, называется идеальным. По своим свойствам к иде-
альному газу приближаются водород и гелий, находящиеся в со-
стоянии большого разрежения. Основные законы газового состояния
следующие.
1. При постоянной температуре объем данной массы газа об-
ратно пропорционален давлению или, иначе говоря, произведение
объема V на давление Р при неизменной температуре есть величина
постоянная:
VP — const,
откуда следует
VP = = V2P2 = const,
или
v2 Рг ’
Следовательно, на основании этого закона, зная объем и давле-
ние данной массы газа, можно вычислить его объем при другом дав-
лении или вычислить давление при изменившемся объеме. Как уже
было упомянуто, все реальные газы более или менее уклоняются
от этого закона. Эти уклонения особенно заметны при применении
больших давлений; например, метан при давлении в 40 ат сжи-
мается на 9% больше, чем это следует по данному закону. При ана-
лизе газа, при определении его удельного веса и при других иссле-
дованиях не приходится иметь дела с большими давлениями. Обычно
все измерения объема газа производят при давлении, равном 1 ат,
и только в тех случаях, когда количество газа мало, его объем из-
меряют при низком давлении. Работами многих исследователей уста-
новлено, что при низких давлениях тазы вполне удовлетворительно
подчиняются этому закону. Уклонения становятся слегка замет-
ными при применении лишь давлений, начиная от 100 мм рт. столба
5
и выше. Для давлений от 0,5 до 1 ат указывалась следующая фор-
мула [1], выражающая зависимость между объемом и давлением газа:
РУ = Р<Уо [ 1 -Ь 2 (1 - В) Р] = P0V0 (1 + аР),
где Ро и Уо— давление и объем при крайнем разрежении; Р выра-
жено в атмосферах.
Величина коэфициента а для различных газов такова:
Н2 ................—0,00052
О2 ................—0,00057
N2 ................—0,00043
Не . ..............-0,0006
СО2 ................ -0,006
СО ... .............—0,0005
NH3.................+0,015
SO,..................+0,023
Для газов, температура которых далека от температуры их кипе-
ния, эти отклонения невелики—около 0,5%; для прочих же га-
зов эти отклонения уже значительны и при точной работе их при-
ходится учитывать.
В табл. 1 приведены величины PV при разных давлениях по отно-
шению к величине PV при I ат и 0°, принятой условно за единицу.
Таблица 1
Значения PV для некоторых газов при различных давлениях
Давление, ат Величина PV для
кислорода азота углекислоты водорода
1 1,0000 1,0000 1,0000 1,0000
100 0,9265 0,9910 0,2020 1,0690
200 0,9140 1,0390 0,3850 1,1380
500 1,1570 1,3900 0,8910 1,3565
1000 1,7360 2,0700 1,6560 1,7250
2500 3,3238 3,9200 2,6850
Из углеводородных газов метан дает наименьшие отклонения.
При давлении около 30 ат отклонение меньше 1%. Для более тя-
желых углеводородов (начиная с пропана) это отклонение дости-
гает 2—3% при обычной температуре даже в пределах атмосфер-
ного давления.
2. При постоянном давлении при повышении температуры все
газы расширяются одинаково, а именно, при повышении температу-
ры на 1° газ расширяется на своего объема при 0°.
Коэфициент расширения
а = ^+- = 0,003663.
л id
Этот закон можно выразить следующей формулой:
V = V0(\ + at),
где 1%— объем газа при 0°;
V—объем газа при температуре ГС.
6
Можно написать:
V2 = V0 (1 4- aQ
и т. д., откуда следует, что
273 + и
Vi _ V0(l+aG) 273 273-Hi _ Л
V2 VO(1+«G) 273 + f2 “ 273 + ta ” T2 >
273
т. e. при постоянном давлении объемы одного и того же количества
газа прямо пропорциональны абсолютным температурам. Этот закон
позволяет, зная объем газа и температуру, вычислить объем этого
газа при другой температуре или вычислить температуру, зная но-
вый объем газа.
Оба вышеприведенных закона не являются абсолютно точными,
так как коэфициент расширения не строго одинаков для всех газов
и, кроме того, он изменяется в зависимости от температуры и давле-
ния.
3. Объединение этих двух законов дает известное уравнение:
PV = RT,
р v
где R —газовая постоянная, равная -~^3° ;
Т — абсолютная температура, равная 273 4-1.
4. Предыдущее уравнение абсолютно справедливо для идеального
газа, а все реальные газы более или менее уклоняются от него.
Одной из причин является то обстоятельство, что молекулы газа
сами по себе занимают некоторый объем, в теоретическом же выводе
уравнения PV = RT это не учитывается. Таким образом, если мы
будем точно рассчитывать изменение объема газа в зависимости от
давления, то мы должны принимать во внимание не весь объем газа,
а объем газа без объема самих молекул. Обозначим этот объем мо-
лекул газа через Ь.
В уравнении PV = RT не учитывается также взаимное притя-
жение молекул друг к другу. Пр мере увеличения давления и, сле-
довательно, уменьшения объема расстояния между молекулами газа
уменьшаются, а сила притяжения увеличивается, что и ведет к на-
рушению этого уравнения. Обозначим это дополнительное давление,
образующееся вследствие увеличения силы притяжения между мо-
лекулами, через £. Величина а, как и величина V, есть постоянная
для данного газа. Дополнительное давление зависит от объема: чем
меньше объем (вследствие увеличения давления), тем больше давле-
ние. Если мы условно разобьем весь газ на слои, то эти слои будут
притягиваться друг к другу вследствие молекулярного взаимодей-
ствия С силой, пропорциональной плотности как притягивающего
слоя, так и притягиваемого, иначе говоря, с силой, пропорциональ-
ной квадрату плотности; квадрат плотности, как известно, обратно
?
пропорционален квадрату объема, поэтому в знаменатель величины а
и поставлена величина V2.
С введением этих поправок уравнение PV = RT переходит о
уравнение
(P + yr)(V-b) = RT.
Таблица 2
Значения констант а и Ь, вычисленных из
критических условий [2]
Газ а b t °C ‘кр’ Ркр, ата Укр, см3]г
Водород 0,245-105 6 26,6 289,9 12,8 64,5
Азот 1,347-106 38,6 -147,1 33,5 90
Кислород .... 1,36 -106 31,9 — 118,8 49.7 74
Воздух .... 1,33 -10е 36,6 —140,7 37,2 87
Водяной пар . . 5,48 -10е 30,6 374,0 217,7 45
Окись углерода . 3,6 -10е 42,8 31,1 73,0 95,5
Углекислота . . 1,46 -10е 39,4 —139,0 72,9 90,0
Метан ..... 2,25 -10е 42,8 — 82,5 45,8 99
Ацетилен .... 4,37 -10е 51,2 36,0 62 113
Этан 5,38 -10е 62,8 32,1 48,3 143
Этен 4,48 -10е 57,2 9,7 50,9 127
Пропан 8,48 -10е 84,5 96 43 190
н-Бутан 14,57 -10" — 152 36 250
н-Пентан .... 19,6 -10е 197 33 310
н-Гексан .... 24,6 -10® — 234,8 29,6 367
П римечание. Величины а и Ь даны для моля вещества. Давление выражено
в ата, объем —в ем3, масса—в г. Если взять массу газа в 1 см3 при 0° и
1 ата, то табличные значения а надо умножить на 1,99-10~“9, ab~ на
4,46-10-5.
5. Всякий газ распространяется в другом газе, кек в пустоте.
Отсюда следует, что при наличии смеси газов каждый газ занимает
объем, равный объему всей смеси. Давление, которое испытывает
каждый отдельный газ, входящий в состав смеси, пропорционально
его процентному содержанию.
6. Вес газа, растворенного в жидкости, пропорционален давле-
нию, производимому этим газом на жидкость. Газы, входящие в
состав смеси, растворяются в жидкости пропорционально той доле
давления, которая приходится на каждый газ в отдельности.
Этот закон может быть выражен следующей формулой:
Q=kP,
где О —вес газа в единице объема растворителя;
Р — давление газа над растворителем;
—постоянная величина.
Поскольку объем газа обратно пропорционален давлению (PV ~
= const), объем, растворенного газа не зависит от давления.
8
7. При одинаковых температурах и одинаковых давлениях в
равных объемах различных газов содержится равное число молекул.
Таким образом, если взять одинаковые объемы двух различных
газов, то чем больше молекулярный вес газа, тем он тяжелее. Иначе
говоря, удельные веса газов при одинаковых температуре и давлении
прямо пропорциональны молекулярным весам:
71 = Мг
7-2 М2 >
где У1 и у2— удельные веса двух разных газов;
Mi и М2 — их молекулярные веса.
§ 2. Приборы и приспособления общего назначения, применяемые
в газовом анализе
Образец анализируемого газа должен находиться в герметически
закрытом сосуде или резервуаре. При наличии даже очень неболь-
шой негерметичности в этой емкости газ может уйти и смешаться с
атмосферным воздухом. Даже если газ уйдет только частично, то
это явление будет сопровождаться поступлением внутрь содержа-
щей газ емкости атмосферного воздуха и, таким образом, анализ
не даст правильного представления о составе исследуемого газа.
Поэтому анализируемый газ, если речь идет об анализе изолирован-
ных образцов этого газа, помещают обычно в какую-либо стеклян-
ную бутыль, бюретку, пипетку или иную емкость. Когда газ соби-
рают под небольшим давлением, то предпочитают иметь стеклян-
ную тару, так как стекло имеет большие преимущества перед дру-
гими материалами. Чистота и прозрачность стекла позволяют су-
дить о состоянии газовой пробы и о количестве газа и запорной жид-
кости, содержащихся в той или иной емкости.
Большие количества газа можно собирать под давлением в метал-
лические баллоны. Следует, однако, учесть, что в этом случае не-
которые газы и пары (пропан, бутан и др.) будут конденсироваться.
Краны. Одним из важнейших моментов газоаналитических опре-
делений является перевод газа из одной емкости в другую. Если
делается анализ твердого или жидкого вещества, то перевод части
этого вещества в другой сосуд не представляет обычно затруднений
и производится путем переливания или пересыпания вещества, на-
пример, из одной колбы в другую или из колбы в стакан и т. д. Когда
анализируется образец газа, то перевод этого газа из одной емкости
в другую требует применения ряда предосторожностей и специфи-
ческого устройства аппаратуры. При переводе газа из одной емкости
в другую важнейшим элементом применяемой аппаратуры явля-
ются газовые краны. Иногда краны заменяют металлическими за-
жимами, которые позволяют пережимать каучуковые трубки. Во
всех этих случаях образец газа, содержавшийся первоначально в
емкости с закрытым краном или с пережатым зажимом каучуком,
вытесняется из этой емкости или откачивается из нее, для чего кран
Р
или зажим открывают. Чтобы газ при этой операции никуда не ушел,
кран и каучуковые соединения должны быть герметичными.
На фиг. 1—8 даны схемы разнообразных кранов, применяемых
в газоаналитических установках. Обычный двухходовый газовый
кран (фиг. 1) по своему устройству ничем не отличается от кранов,
применяемых в аналитической химии. Однако газовые краны должны
быть более герметичными, поэтому они, с одной стороны, лучше при-
тираются, чем обычные химические краны, а с другой стороны, для
повышения герметичности крана его муфту и пробку делают несколько
длиннее и шире, чем у обычных кранов (муфтой называется на-
ружная, обычно неподвиж-
ная часть крана, а вра-
щающаяся часть с просвер-
ленным ходом называется
пробкой). Для получения
хорошей герметизации га-
зовый кран должен смазы-
ваться так называемой ва-
куумной смазкой. Когда
давление газа внутри при-
бора близко к атмосфер-
Фиг. 1. Газовый кран.
ному, вопрос о герметичности кранов не является столь острым.
В этих случаях можно применять смазку из вазелина и даже
глицерина. Однако, когда внутри прибора хотя бы даже на некото-
рых стадиях его работы получается разрежение, опасность поступ-
ления внутрь прибора атмосферного воздуха сильно возрастает,
поэтому применяемые краны и следует смазывать вакуумной смаз-
кой, рецепт которой приводится ниже.
Перед смазкой кран надо тщательно вымыть и просушить. Для
удаления имеющихся на пробке и муфте жировых веществ их сле-
дует смыть бензолом, можно и чистым спиртом. После того как кран
будет вымыт и высушен, на его пробку наносят некоторое количество
смазки и вращают в каком-либо направлении до тех пор, пока не
исчезнут полоски и шлиф не станет совершенно прозрачным. Не
следует наносить на пробку слишком много смазки, иначе эта смазка
может забить ход в пробке.
Особенно большие предосторожности приходится принимать, ко-
гда внутри газоаналитической установки имеется высокий вакуум.
Обычные химические краны, а также небольшие газовые краны в
этих случаях часто бывают неудовлетворительными и пропускают
в прибор некоторое количество атмосферного воздуха. Однако даже
небольшое количество атмосферного воздуха, содержащего, кроме
того, углекислоту и пары воды, попадающее внутрь прибора, мо-
жет при точных определениях вызвать большие погрешности. По-
этому в высоковакуумных установках применяют специальные вы-
соковакуумные краны, очень тщательно пришлифованные, зна-
чительно больших размеров, чем обычные газовые краны, и имеющие
иногда специальные приспособления, снижающие возможность про-
никновения внутрь атмосферного воздуха.
10
На фиг. 2 представлен высоковакуумный кран. Нижняя часть
муфты этого крана несколько расширена и далее переходит в стек-
лянную трубку, сообщающуюся с вакуумной установкой. Эта уста-
новка сообщается с трубкой, которая подходит к крану слева, через
отверстие, имеющееся в пробке крана. Вследствие подобного уст-
ройства нижняя часть муфты не мо-
жет служить местом проникновения
атмосферного воздуха, что возможно,
например, у крана, изображенного
на фиг. 1. Верхняя часть муфты крана
(фиг. 2) устроена таким образом, что
образуется углубление, которое может
быть залито ртутью или смазоч-
ным веществом (вазелин, вакуумная
Фиг. 2. Высоковакуумные краны.
смазка).
Применение в этом случае сма-
зочных веществ имеет преимуще-
ство перед ртутью, поскольку пары ртути ядовиты, а попадание их
в атмосферу при указанном устройстве неизбежно. В этих случаях,
Фиг. 3. Трехходовые и четырехходовые краны.
правда, применяют резиновые баллоны, которыми прикрывают
верхнюю часть крана вместе с налитой ртутью. Однако это средство
не устраняет возможности попадания ртутных паров в атмосферу,
поскольку наливание ртути и ее удаление неизбежно приводят к
разбрызгиванию и попаданию в атмосферу.
Во многих случаях краны, присоединенные к каким-либо емко-
стям, содержащим газ, имеют не два хода, а три. Подобное устроЙ-
11
ство кранов имеет ряд преимуществ, позволяя направлять газ^по
той или иной трубке. На фиг. 3, а представлена схема Т-образного
трехходового крана. Пробка крана имеет Т-образный ход, причем в
зависимости от поворота или все три отвода крана могут быть соеди-
нены между собой, или два из них. На фиг. 3, б представлен трех-
ходовый кран иной конструкции: пробка крана имеет два косых
параллельных хода. В этом кране путем соответствующих поворо-
тов пробки имеется возможность попеременно соединять правую
трубку с той или иной из левых. На фиг. 3, в представлена схема
трехходового крана с так называемым полумесячным ходом в пробке.
Отводные трубки этого крана расположены на равном расстоянии
друг от друга по окружности муфты. Имеющийся в пробке изогну-
тый ход позволяет соединять любую пару этих трубок. На фиг. 3, г
представлены четырехходовые краны с одним и двумя ходами в^проб-
ке. На фиг. 3, д приведена еще одна конструкция трехходового крана,
в котором третий ход расположен в пробке крана. Существуют’краны
и более сложной конструкции. Так, например, на фиг. 4 представ-
Фиг. 5. Кран для приборов с повы-
шенным газовым давлением.
лены двухъярусный и трехъярусный краны, позволяющие одно-
временно соединять несколько параллельных ходов.
Все эти краны могут быть сделаны таким образом, чтобы нижняя
часть муфты была закрыта и заполнена ртутью или каким-либо сма-
зочным веществом, а в верхней части имелось расширение, куда
также может быть налита ртуть или смазочное вещество для предо-
хранения от попадания внутрь прибора атмосферного воздуха
(фиг. 5, а). Если в приборе имеется повышенное по сравнению с атмо-
сферным давление, то некоторые авторы рекомендуют применять
кран, схематически представленный на фиг. 5, б. Воздушная ка-
мера, расположенная ниже пробки, до начала опыта может быть
наполнена газом под повышенным давлением, так что потом пробка
всегда прижимается к муфте и не пропускает газ наружу.
В некоторых случаях смазка крана оказывает нежелательное
действие на анализируемые газообразные вещества, поэтому были
разработаны различные конструкции кранов, которые держат ва-
куум и без смазки. Один из подобных кранов представлен на фиг. 6.
В этом кране всякому нежелательному соединению препятствуют
ртутные затворы [3]; им можно пользоваться только в горизонталь-
ном положении. Чтобы избежать заедания, рекомендуется изгото-
влять пробки и муфты из разных сортов стекла. На фиг. 7 изображены
краны, действующие без смазки, сконструированные Б. М. Нака-
шидзе. .
Кроме стеклянных кранов, в некоторых случаях пользуются
вентилями, сделанными целиком из металла. Обычные вентили на
Фиг. 6. Газовый бессма-
зочный кран с ртутным
запором.
Фиг. 7. Газовые бессмазочные краны.
7 — канавка с ртутью; 2 — смазываемая часть.
резьбе с тем или иным уплотнением могут применяться только тогда,
когда внутри соответствующей установки, где расположены эти
вентили, имеется повышенное давление. Небольшая течь через на-
резку и уплотнение не влияет на состав со-
держащихся в установке газов. Если же
внутри установки имеется вакуум, то при-
менение подобных вентилей недопустимо.
В этих случаях можно применять краны,
аналогичные стеклянным, но сделанные из
металла, а также краны мембранного типа.
Подобный кран с мембраной представлен на
фиг. 8. Гофрированная или плоская мембрана
из меди, латуни или томпака припаивается
к креплению подобного крана, а соответ-
ствующая рукоятка с винтовым ходом позво-
ляет закрывать или открывать отверстие,
расположенное под центральной частью мем-
браны. При таком устройстве подтекание
атмосферного воздуха внутрь установки совер-
шенно исключается. Чтобы указанное отвер-
стие было вполне надежно и крепко закрыто,
Фиг. 8. Мембранные
металлические газовые
в центральной части мембраны имеется свин- краны.
цовая или иная прокладка. При соот-
ветствующем нажиме мембрана может крепко прижиматься к вход-
ной трубке, так что переход газа из одной части прибора в другую
прекращается. Применяются в некоторых случаях и гофрирован-
ные металлические трубки из специальных сплавов. Эти трубки мо-
гут быть сжаты почти в 1,5—2 раза по своей длине, поэтому возможно
опять-таки закрывать отверстие одной из трубок, как это показано
на фиг. 8, и воспрепятствовать прохождению газа.
Когда требуется исключительно высокий вакуум и приходится
работать с очень малыми количествами газа, наличие кранов ведет
13
к различным погрешностям. В этих случаях пользуются обычно
ртутными затворами, устройство которых описано ниже, и приме-
няют стеклянные установки, целиком спаянные из стекла;
Соединения. Отдельные части приборов, как стеклянных, так
и из иных материалов, при газоаналитических работах часто при-
ходится соединять. Наиболее совершенным способом соединения
является спайка стеклянных частей. Однако иногда спаивать стек-
лянные трубки не представляется возможным, иногда же соединяе-
мые части изготовлены из разнородного материала. Часто, когда
внутри установки нет высокого вакуума, спайка не требуется.
В этих случаях две стеклянные трубки обычно соединяются с помощью
каучуков. Для этой цели на концах соединяемых частей рекомен-
Фиг. 9. Соединения трубок.
1 — каучук; 2 — замазка.
дуется делать так называемые оливки, т. е. расширения, как это
показано на фиг. 9, а. Трубки соединяют каучуком, как говорят,
встык. Каучук для таких соединений желательно употреблять наи-
более эластичный. Внутренний диаметр каучуковой трубки должен
быть меньше наружного диаметра соединяемых частей. Для боль-
шей герметичности рекомендуется конец каучука слегка смазывать
вакуумной смазкой. При несколько повышенном давлении внутри
трубок необходимо каучук перевязывать проволокой.
Соединения встык делают для того, чтобы газ, содержащийся
внутри прибора, насколько возможно меньше соприкасался с кау-
чуком, так как каучук может оказать вредное влияние на состав
газа. Как известно, всякий газ обладает способностью диффузйи
через самые разнообразные материалы. Каучук в некоторой степени
проницаем для газов. Кроме того, каучук является хорошим сор-
бентом для некоторых газообразных соединений, в частности для
U
паров различных органических веществ. Углекислый газ и водя-
ные пары хорошо диффундируют через каучук. Если анализируе-
мый газ проходит через длинную каучуковую трубку, то таким об-
разом возможно изменение состава газа вследствие диффузии газа
через каучук и поглощения каучуком некоторых из его компонентов.
насколько хорошо каучук может поглощать некоторые составные
части газа, можно видеть из следующего примера. Автору настоящей
монографии было необходима впустить в один микроаналитический
прибор некоторое количество паров октана. Для этой цели через
барботер с октаном продувался воздух, который затем через отре-
зок каучуковой трубки попадал в аналитический прибор. Несмотря
на очень высокую чувствительность прибора, в 1000 раз превосходив-
шую концентрацию впускаемых паров октана, последний совер-
шенно не попадал в прибор, целиком задерживаясь каучуковой труб-
кой. Таким образом, если газ содержит компоненты, хорошо погло-
щаемые каучуком или сравнительно хорошо диффундирующие через
него, то состав газа может существенно измениться. Следует также
учесть, что каучук, уже содержащий сорбированные газы или водя-
ные пары, будет отдавать их в тот газ, который проходит мимо
него.
Все эти обстоятельства заставляют сделать вывод, что примене-
ние каучуковых соединений нежелательно при точных газоаналити-
ческих работах, особенно когда приходится иметь дело с малыми ко-
личествами анализируемого газа. Если нельзя избежать употреб-
ления каучука, то рекомендуется применять определенные сорта
его, обладающие сравнительно меньшей сорбционной способностью.
Так, например, имеется указание, что трубки, изготовленные из не-
которых видов искусственного каучука, обладают меньшей способ-
ностью поглощать многие газообразные компоненты, чем трубки
из натурального каучука. При работах с высоким вакуумом приме-
нение каучуковых соединений совершенно недопустимо. Во многих
случаях при газоаналитических работах, когда применение кау-
чука недопустимо, спаять отдельные части прибора почему-либо
также невозможно вследствие отсутствия газа или стеклодува или
вследствие разнородности материалов соединяемых частей. В этих
случаях применяют различные замазки, которыми герметизируют
места соединения отдельных частей. Чтобы обеспечить герметич-
ность подобного соединения, рекомендуются устройства, схемати-
чески представленные на фиг. 9, б и 9, в. Обычно делают это таким
образом, чтобы одна из соединяемых трубочек была уже другой и
входила в нее на довольно значительное расстояние (2—3 см). Соеди-
няемые трубки нагревают, покрывают соответствующей распла-
вленной замазкой и вставляют одну в другую. Выступающую замаз-
ку оплавляют так, чтобы не было нигде трещин или отверстий. Для
большей уверенности в герметичности соединяемые части иногда
изгибают, как это видно на фиг. 9, г, и погружают в стеклянную
чашечку, которую целиком заливают замазкой. Следует учесть,
что высокий вакуум могут держать только специальные замазки,
например пицеин и др. (о различных типах замазок см. ниже).
15
Когда соединяемые части должны легко отъединяться друг от
друга или вращаться, применяют шлифы (фиг. 10). Для повы-
шения герметичности устраивают шлифы с ртутными затворами.
Следует еще раз отметить, что применение ртути вследствие ядови-
тости ее паров не рекомендуется и лучше подобные затворы запол-
нять смазочным материалом.
Если соединение должно быть до известной степени гибким, не-
которые авторы рекомендуют применять каучуковые шланги и труб-
ки, покрытые изнутри и снаружи тонким слоем пицеина. Это покры-
тие легко наносится путем нагрева каучуковой трубки и впускания
в нее расплавленного пицеина. Если трубка нагрета, например, до
70—80°, то расплавленный пицеин легко растекается по внутренней
поверхности трубки, обеспечивая гер-
метичность каучукового соединения.
Соединение кварца со стеклом
может производиться при помощи
шлифов или применением замазки.
Точно так же на шлифах могут соеди-
няться стеклянные трубки с фарфо-
ровыми или с металлическими.
Затворы. Описанные выше газо-
вые краны, как стеклянные, так и
металлические, не всегда могут при-
Фиг. ю. Шлифы. меняться. Стеклянные краны с вра-
щающейся пробкой на смазке могут
пропускать через себя хотя и очень небольшое, но измеримое коли-
чество газа. Кроме того, смазка крана может вредно влиять на состав
газа, поглощая из него те или иные компоненты. Металлические
краны не всегда удобны для применения, так как часто бывают
затруднения при соединении их со стеклянными частями. В связи
с этим в различных газоаналитических аппаратах применяют иные
устройства, препятствующие переходу газа из одной части прибора
в другую и позволяющие переводить газ из одной части в другую.
На фиг. 11 представлены ртутные затворы, часто применяемые
в приборах и установках вакуумного типа [4]. Если разница давле-
ний в соединяемых частях прибора невелика, то пользуются ртут-
ным затвором, представленным на фиг. 11, а. Поднимая ртуть выше
спая трубок, отъединяют правую трубку от левой. Опускание ртути
ниже спая ведет к соединению частей прибора, и газ из трубки А
может перейти в трубку В. В тех случаях, когда в процессе анализа
или действия вакуумной установки перепад давления достигает
больше 5—10 '-мм, изображенный на фиг. 11, а ртутный затвор
не всегда Сбудет ^удовлетворительно работать, так как при опу-
скании ртути пузырьки газа могут перебросить ртуть в другую часть
прибора, что не всегда желательно. Во избежание этого в той или
иной трубке или устраивают расширение (11,0), или помещают предо-
хранитель, представляющий собой полый шарообразный или цилин-
дрический запаянный и пришлифованный отрезок трубки, который,
16
плавая на ртути, при ее поднятии закрывает отверстие, ведущее к
трубке В (см. фиг. 11, в).
На фиг. 11, г представлен затвор с двумя подобными клапанами,
снабженный также кранами, регулирующими высоту поднятия ртути.
В некоторых случаях, чтобы воспрепятствовать перебросу ртути,
ставят предохранитель иного типа, а именно, с пластинкой из пори-
стого стекла (фиг. 11, г). Опускание ртути при наличии такого предо-
хранителя не ведет к перебросу ее в другую часть прибора1. Созда-
ние очень высокого вакуума через такую пластинку происходит
медленно. Для ускорения создания вакуума ставится второй за-
твор-, состоящий из широких трубок, способствующих более скорому
и более полному созданию вакуума (11, д). Второй [затвор откры-
вают после откачки через первый затвор.
Фиг. 11. Ртутные затворы.
На фиг. 12, а изображена другая конструкция ртутного затвора.
Внутри широкой трубки затвора диаметром 2—3 см помещается
более узкая. Подняв уровень ртути выше конца этой трубки, произ-
водят перекрытие частей прибора А и В. При опускании ртути в
широкой трубке пузырьки газа проходят через ртуть и переброса
не будет.
В аналитической практике встречаются случаи, когда внутрь
прибора, где имеется вакуум, приходится впускать газ, находящийся
под атмосферным давлением. Если применение какого-либо крана
нежелательно, то берут ртутный впускной затвор, схематически
изображенный на фиг. 12, б. Внутри трубки диаметром 15—20 мм
проходит тонкий длинный капилляр; длина трубки превышает 76 см,
и даже при вакууме внутри прибора атмосферный воздух или газ,
находящийся под атмос ым давлении, не моя^г войти внутрь 1 2 *
1 L Q ' «СА )
1 Пред поставлен или в однолсили в обоих коленах
затвора в зависимое откачки^
Курсов ----
2 в- A-l№W?re 17 ЦЦЧ*
I К лфтяясй Пр
прибора. При понижении уровня ртути в затворе газ мелкими пу-
зырьками идет через капилляр и поднимается вдоль него. Практика
показала, что перебрасывания ртути в этом случае не происходит.
Эти затворы применяют в вакуумных приборах для микроанализа.
Чтобы воспрепятствовать перебросу ртути в затворе, изображен-
ном на фиг. 12, а} можно употреблять трубки, по окружности кото-
Фиг. 12. Ртутные затворы.
рых имеются мелкие отверстия (12; в). Газ будет проходить по этим
отверстиям мелкими пузырьками, и переброса ртути не произойдет.
В то же время следует отметить, что создание вакуума через подоб-
ные затворы представляет некоторые затруднения вследствие
незначительного диаметра отверстий. Чтобы избежать этих затрудне-
ний, рекомендуется иметь двойной затвор, аналогичный схемати-
чески представленному на фиг. 11, д.
Получение вакуума. Во многих случаях при газовом анализе
в тех или иных установках, баллонах или трубках приходится созда-
вать вакуум, т. е. откачивать их до небольшого давления. Подобная
откачка необходима, когда требуется полностью удалить тот или
иной газ и когда вытеснение этого газа жидкостью, например водой
или ртутью, по условиям анализа невозможно или не может дать
требуемой степени разрежения.
Следует учесть, что при вытеснении газа из трубки или сосуда
путем заполнения ртутью содержащийся в этой трубке газ не будет
вытеснен целиком. Если после вытеснения газа уровень ртути опу-
стить, то в пространстве над ртутью все-таки будет оставаться неко-
торое, хотя и очень незначительное количество газа.
J 18
При заполнении ртутью сосуда или трубки некоторое количество
газа в виде мелких пузырьков, прилипших к стенкам сосуда, не-
избежно останется. Если поднять уровень ртути второй раз и затем
третий, то, конечно, степень вакуума повысится. Однако при выпуске
этого газа через кран полностью удалить его опять-таки крайне за-
труднительно. Таким образом, даже в тех случаях, когда в качестве
вытесняющей жидкости используется ртуть, полное удаление газа
затруднительно. Тем более нельзя достигнуть полного удаления
газа путем вытеснения его водой, так как сама вода всегда имеет
значительную упругость паров, а кроме того, содержит в себе рас-
творенный воздух и другие газы, которые при понижении давления
выделяются из воды. В связи с этим при газоаналитических работах
очень часто пользуются для удаления газа различными вакуумными
насосами.
Для получения сравнительно небольшого вакуума, а также для
непрерывного тока воздуха часто применяют водоструйные насосы.
Существует много типов этих насосов.
Наиболее употребительные типы представлены на фиг. 13, 1 и 2. Во-
доструйный насос присоединяют к водопроводному крану и через него
пускают ток воды. Двигаясь с большой скоростью и под значитель-
ным напором через узкие трубки водяная струя увлекает окружа-
ющий ее газ, который проталкивается через нижнюю часть насоса
в атмосферный воздух. Внутри самого насоса и в любом аппарате,
присоединенном к трубке насоса, создается таким образом вакуум,
степень которого зависит, с одной стороны, от напора воды, а с дру-
гой стороны, от качества самого насоса (диаметр трубок, величина
зазора и т. д.). Совершенно очевидно, что предельный вакуум, кото-
рый может быть достигнут с помощью водоструйного насоса, не мо-
жет быть ниже упругости паров воды. Для улучшения вакуума ино-
гда применяют осушительные устройства в виде трубок, содержащих
хлористый кальций или фосфорный ангидрид, благодаря чему сте-
пень вакуума может быть повышена.
2* 19
Необходимо учесть, что всякое уменьшение напора воды при
действующем водоструйном насосе может повести к большим непри-
ятностям, если между насосом и присоединенными к нему приборами
не поставить предохранительных клапанов. Ослабление напора воды
может привести к тому, что вода, содержащаяся в нижней части
насоса, атмосферным давлением будет переброшена в прибор. В каче-
стве предохранительных клапанов применяют различные устройства.
Один из типов этих клапанов представляет собой поплавок, кото-
рый закрывает трубку насоса при повышении уровня воды.
В качестве предохранителя используют часто резиновую пробку,
нижняя часть которой надрезана, как это показано на фиг. 13, а.
При приближении вода прижимает надрезанную часть к пробке,
вследствие чего дальнейшего проникновения воды в вакуумную
линию не
Фиг. 14. При-
способления для
поддержания по-
стоянного давле-
ния.
будет [5]. Когда необходимо прекратить действие водо-
струйного насоса, следует перекрыть прежде всего
кран откачиваемой установки, затем соединить ва-
куумную линию с атмосферной, а затем уже закрыть
кран, подающий воду.
Водоструйными насосами пользуются также для
получения некоторого избыточного давления; в этом
случае применяют нагнетающие водоструйные насосы.
Схема подобного насоса представлена на фиг. 13, 3.
Отводная трубка водоструйного насоса открыта
и соединена с атмосферой. Струя воды увлекает
атмосферный воздух в ту или иную емкость, вслед-
ствие чего в ней создается некоторое повышенное
давление, которое и гонит воздух в указанном на
фиг. 13 направлении. Следует, конечно, учесть, что
давление, создаваемое таким водоструйным насосом,
будет невелико. Однако для многих целей подобная
струя воздуха вполне достаточна. Так, например,
с помощью подобного нагнетающего водоструйного
насоса при надлежащем напоре воды можно подавать воздух для
ручной стеклодувной горелки, для промывки воздухом тех или
иных емкостей и т. п.
Часто при газоаналитических работах бывает необходимо под-
держивать постоянное давление или постоянный вакуум. С помощью
сосуда, изображенного на фиг. 14, вакуум, создаваемый в установке,
присоединенной к трубке Д, не может быть больше высоты столба
жидкости, считая от конца трубки Б. Если вакуум станет больше
этой величины, то через трубку Б внутрь системы начнут проходить
пузырьки воздуха.
Как уже было упомянуто выше, степень вакуума, даваемая водо-
струйным насосом, сравнительно невелика, поэтому, если тре-
буется вакуум меньше 10—20 мм рт. столба, применяют различные
масляные насосы. Одним из наиболее употребительных ручных мас-
ляных насосов является насос Комовского. Вращая рукоятку махо-
вика, имеющуюся в этом насосе, можно быстро получить довольно
хороший вакуум.
20
Для постоянного получения вакуума или откачки значительного
количества газа пользуются обычно вращающимся масляным насо-
сом, присоединенным к электромотору. Имеется очень много типов
этих масляных насосов.
С помощью масляного насоса можно получить вакуум, меньший
I мм рт. столба. При употреблении высокосортного масла удается
получить вакуум, достигающий 0,01 мм рт. столба. При этом сле-
дует учесть, что получение подобного вакуума возможно лишь при
хорошей герметичности всей установки и соединительных трубок.
Как уже было упомянуто выше, каучуковые трубки, особенно места
их соединения со стеклянными или металлическими трубками, явля-
ются всегда источниками той или иной течи, вследствие чего полу-
чение даже такого вакуума, как 0,01 мм рт. столба, представляет
значительные затруднения.
Между приборами, в которых создается вакуум, и масляным
насосом рекомендуется ставить ловушку, чтобы избежать перебра-
сывания масла в откачиваемый прибор при остановках мотора масля-
ного насоса. При остановке мотора масляного насоса необходимо
перекрыть соответствующие краны и соединить вакуумную линию
с атмосферой.
Если требуется получить еще более высокий вакуум, то приме-
няют ртутные насосы. На фиг. 15 представлен ртутный насос, дей-
ствие которого заключается в том, что при поднятии склянки 1 с
ртутью последняя заполняет баллон 2 и вытесняет содержащийся
в нем воздух через капиллярную трубку 3. При опускании ртути в
баллоне 2 создается вакуум. Атмосферный воздух давит на ртуть,
содержащуюся в баллончике 4. Однако попасть внутрь баллона 2
воздух не может, если высота трубки 3 превышает 760 мм. Эта трубка
при опускании ртути в баллоне 2 заполняется ртутью из баллончика
4, что и препятствует проскакиванию атмосферного воздуха
внутрь прибора. При опускании ртути ниже соединения трубки 5
с баллоном 2 газ или воздух, содержащиеся в установках и при-
борах, присоединенных к насосу, поступают в баллон 2. Проводя
последовательно несколько раз поднятие и опускание ртути, можно
откачать газ из присоединенного к насосу прибора. В верхней
части трубки 5 следует поместить предохранитель, который пре-
пятствует проникновению ртути при поднятии сосуда 7 в откачи-
ваемую установку. С помощью указанных манипуляций можно
получить сравнительно высокий вакуум. Предел вакуума зависит
от течи через соединения и краны откачиваемой установки, а также
лимитируется выделением сорбированных газов поверхностью
стекла и, наконец, упругостью паров самой ртути, составляющей
при комнатной температуре величину порядка 0,001 мм рт. столба.
Для устранения течи, обусловливаемой негерметичностью соеди-
нений, рекомендуется не применять каучуковые или иные соеди-
нения, а целиком припаивать откачиваемую установку непосред-
ственно к насосу. Для устранения выделения сорбированного газа
из стекла рекомендуется предварительный прогрев как самого насоса,
так и соединительных трубок и установки до температуры 200—300°.
27
При непрерывной работе насоса, когда из откачиваемого сосуда
необходимо удалить также и пары ртути, между насосом и откачи-
ваемым сосудом помещают ловушки, погружаемые в жидкий воз-
дух. Схема подобной ловушки представлена на фиг. 15, а. Ловушка
представляет собой одну или две широкие трубки со впаянными в них
более узкими трубками, которые погружаются
Фиг. 15. Ртутный насос. Фиг. 16. Ртутный насос.
а — дьюаровский сосуд с жидким воздухом.
На фиг. 16 представлена схема ртутного насоса другого типа.
В этом насосе ртуть, стекая каплями по капиллярной трубке, увле-
кает за собой воздух, содержащийся в промежутке между капель-
ками, и таким образом в присоединенном к насосу сосуде постепен-
но создается высокий вакуум. Чтобы ртуть могла опять подняться
в верхнюю часть насоса, баллон 7 делают закрытым и в нем создают
некоторый вакуум. В то же время через трубку 2 непрерывно впу-
скают атмосферный воздух, вследствие чего капельки ртути посте-
пенно двигаются вверх и попадают, наконец, в верхний баллон.
При достаточно продолжительном действии ртутных насосов и
при применении упомянутых предосторожностей, т. е. спайки всех
частей, вымораживания ртути, прогрева трубок, можно получить
вакуум порядка 10~4 — 10-5 мм рт. столба.
Для получения высокого вакуума часто употребляют насосы
ртутно-конденсационного типа.
Устройство подобного насоса схематически представлено на фиг. 17
и основано на том же принципе, что и устройство водоструйного
насоса. Струя ртутных паров, образовавшихся вследствие нагрева
22
колбы с ртутью газовой или электрической горелкой, увлекает воз-
дух, заключающийся в присоединенных к насосу трубках, и напра-
вляет его в так называемую форвакуумную часть насоса. Из этого
форвакуума воздух или газ откачивается масляным или иным насо-
сом. Чтобы указанный насос действовал, в форвакуумной части не-
обходимо давление не больше 10—15 мм рт. столба. Для повышения
степени вакуума применяют иногда два последовательно расположен-
ных ртутных конденсационных насоса. Первый насос создает фор-
вакуум для второго насоса. Эти насосы позволяют создавать весьма
высокий вакуум (при соблюдении вышеупомянутых предосторож-
ностей) и обладают очень большой скоростью откачки.
Во многих случаях для получения высокого вакуума весьма удоб-
ным является уголь, охлаждаемый до температуры жидкого воз-
духа (или жидкого кислорода или жидкого азота). Стеклянный
баллон с углем сообщается через трехходовый кран с откачиваемым
прибором. Третий ход крана служит по мере надобности для откачки
газа, поглощенного углем. Предварительно уголь должен прокали-
Фиг. 17. Ртутно-
конденсационный
насос.
ваться в течение нескольких часов до температуры 300—350°, а выде-
ляющиеся из угля газы должны хорошо откачиваться. При погру-
жении трубки с углем в дьюаровский сосуд с жидким воздухом уголь
сорбирует содержащийся над ним газ. При соответствующем пово-
роте крана газ, содержащийся в той или иной присоединенной к углю
установке, попадает в пространство, расположенное над углем, и
уголь, поглощая газ, создает в установке вакуум (фиг. 18, а).
23
В небольших по объему установках вакуум получается в этом
случае довольно быстро. Следует, однако, отметить, что чем больше
газа поглощает уголь, тем меньший вакуум он способен создать.
В конце концов, при большом количестве поглощаемого газа может
наступить такое положение, что уголь' перестанет создавать нуж-
ный вакуум. Поэтому для получения высокого вакуума через упо-
мянутый трехходовый кран предварительно откачивают газ масля-
ным насосом, а затем уже переключают кран таким образом, чтобы
к откачиваемой установке был присоединен уголь. Если в процессе
создания вакуума уголь уже поглотил значительное количество
газа, то удаляют дьюаровский сосуд, подогревают уголь до комнат-
ной температуры или несколько выше пламенем газовой горелки и
производят откачку угля, после чего он снова может служить для
создания вакуума.
Необходимо заметить, что свежий уголь, даже прокаленный упо-
мянутым способом, обычно не обладает полной способностью к созда-
нию вакуума. По мере того как уголь поглощает газ, а затем этот
газ из него откачивают, уголь, как говорят, активируется и его спо-
собность к созданию вакуума повышается. Для получения весьма
высокого вакуума можно Потреблять два или три последовательно
соединенных угольных поглотителя, как это представлено на
фиг. 18, б. Так как при весьма высоком вакууме краны нежелатель-
ны, то применяют ртутные затворы.
Первоначально в установке создается вакуум с помощью масля-
ного насоса или путем вытеснения газа ртутью. Затем кран или труб-
ку, ведущие к насосу, закрывают ртутным затвором и прибор при-
соединяют к одному из изображенных на фиг. 18 угольных погло-
тителей, который и создает некоторый вакуум. После этого ход к
первому угольному поглотителю С\ (фиг. 18, в) закрывают ртут-
ным затвором и всю установку переключают на второй угольный
поглотитель С2. Таким путем можно получить весьма высокий ва-
куум внутри установки. Чем больше количество угля, которое
охлаждается жидким воздухом и присоединено к откачиваемой уста-
новке, и чем меньше газа осталось в установке, тем выше в ней
вакуум после откачки углем.
Следует также учесть, что угольный поглотитель, охлаждаемый
жидким воздухом, поглощает не только газ, но и ртутные и иные
пары. При получении весьма высокого вакуума и в этом случае тре-
буется применять все упомянутые выше мероприятия, а именно:
спайку всех частей, замену кранов ртутными затворами и прогрев
как откачиваемой установки, так и соединительных трубок.
Бюретки, аспираторы. Если требуется взять некоторое коли-
чество газа для последующего его анализа и провести иные опреде-
ления, употребляют различные приспособления, которые предста-
вляют собой или один из типов газовых бюреток, или при большем
количестве образца газа один из типов газометров или аспираторов.
Наиболее простой тип газовой бюретки представлен на фиг. 19, а,
В верхней части бюретки имеется кран, а к нижней части на каучуке
24
присоединен уравнительный сосуд. Вся бюретка проградуирована
по объему,, начиная от крана. Путем поднятия уравнительного сосуда
с водой находившийся ранее в бюретке воздух или газ вытесняется
через кран, после чего кран
Опуская уравнительный сосуд,
забирают в бюретку то или иное
количество газа, после чего кран
закрывают.
Когда требуется отобрать
большое количество газа, при-
меняют газометр или аспиратор.
На фиг. 19, б представлен один
из типов подобных аспираторов,
который в то же время может
играть роль водяного насоса для
откачки газа. Верхний баллон 7
имеет емкость 0,5—1 л и при-
соединяется через каучук к урав-
нительному сосуду 2 несколько
большей емкости. Опусканием
уравнительного сосуда можно
забрать в баллон 7 соответ-
ствующее количество газа. При-
меняя в верхней части баллона
присоединяют к источнику газа.
Фиг. 19. Газовая бюретка и аспиратор.
трехходовый кран, можно производить откачку газа из какой-либо
емкости, попеременно поднимая и опуская уравнительный сосуд при
соответствующем повороте трехходового крана. Совершенно очевидно,
что разрежение, достигаемое подобным насосом, очень невелико и не
может быть больше разности уровней в уравнительном сосуде и
в баллоне 7.
На фиг. 20, а представлен один из типов газометров, имеющий
емкость несколько литров. Газ впускают в газометр через кран. Газ
в подобном газометре находится всегда под некоторым давлением,
вследствие чего даже при небольшой течи крана можно не опасаться
попадания внутрь его атмосферного воздуха. При наличии такой
течи будет иметь место только некоторое выделение газа из газо-
метра наружу. На фиг. 20, би 20, в представлены газометры зна-
чительно большего размера. Подобные газометры обычно делают
металлическими. За уровнем жидкости в этом газометре следят ло-
ее положению в уравнительной трубке.
Промывалки, поглотители. Весьма часто при работе газы при-
ходится пропускать через раствор для поглощения тех или иных
компонентов.
Для этой цели применяют различные склянки, промывалки,
схематически изображенные на фиг. 21. На фиг. 21, а представлена
одна из таких склянок. Одна из трубок верхней пришлифованной
части склянки длинная и доходит до дна основного сосуда, в кото-
ром налит тот или иной раствор. С помощью водоструйного насоса
25
или другого приспособления газ просасывается через данную склян-
ку. Газ проходит пузырьками через раствор и выходит из склянки.
На фиг. 21, б представлена склянка Тищенко. Эта склянка устроена
таким образом, что по всей ее высоте по ее середине проходит сте-
клянная перегородка, имеющая в нижней части небольшое отвер-
стие. Газ впускается с одной стороны склянки и пробулькивает через
раствор, который при этом переходит целиком в другую половину
колонки. Эта склянка имеет то преимущество, что может быть ис-
пользована независимо от того, с какой стороны впускается газ.
Имеются промывалки, в которых газ для лучшего поглощения
проходит по спиралеобразным ходам, чем удлиняется путь газа
по раствору (фиг. 21, в).
На фиг. 21, г представлены склянки, в которых используются
стеклянные фильтры. Подобные фильтры обусловливают образова-
ние очень мелких пузырьков газа, вследствие чего достигается очень
полное и быстрое поглощение или растворение газа. Следует учесть,
что в упомянутых выше склянках имеется довольно большое про-
странство над раствором, поэтому они удобны только в тех слу-
чаях, когда приходится иметь дело с большими количествами газа.
Если приходится иметь дело со сравнительно небольшими коли-
чествами газа, применяют другие типы промывалок. На фиг. 22, а
представлен простой барботер, состоящий из стеклянной трубки со
впаянной в нее узенькой трубкой, заканчивающейся обычно капил-
ляром. Объем пространства, которое находится над раствором, может
быть очень небольшим. Следует учесть, что при работе с подобным
простым барботером не всегда достигается необходимая степень
26
Фиг. 22. Барботеры.
27
в
поглощения раствором того или иного компонента газовой смеси.
В этих случаях стараются так построить барботер, чтобы пузырьки
газа не сразу поднимались на поверхность раствора, а проходили
в жидкости более длинный путь. Для этого внутрь барботера поме-
щают спиральную стеклянную трубку, как это показано на фиг. 22, в,
в которой газ соприкасается с раствором значительно большее время.
Для еще более полного поглощения ставят последовательно два, а
когда необходимо, то и три барботера. Несколько иная конструкция
барботера представлена на фиг. 22, б, где газ медленно движется по
горизонтальной трубке, способствуя в то же время и циркуляции
раствора.
Очень часто при газовом анализе приходится употреблять твер-
дые поглотители (например, при поглощении из газа водяных паров),
Фиг. 23. Трубки для твердых поглотителей.
для чего применяют хло-
ристый кальций, фосфор-
ный ангидрид, натрон-
ную известь, натронный
асбест и некоторые дру-
гие поглотители. На
фиг. 23, а представлена
обычная так называемая
хлоркальциевая трубка
с притертыми пробками.
Эту трубку можно за-
полнять также фосфор-
ным ангидридом. Сле-
дует учесть, что хлори-
стый кальций хотя и
является неплохим осу-
шителем, но не может
дать такой степени поглощения, как фосфорный ангидрид. Поэтому
в высоковакуумных установках и в других приборах, где требуется
наиболее полное удаление влаги из газа, как правило, применяют
фосфорный ангидрид. Хорошим осушающим средством является
натронный асбест. Указанные реактивы помещают или в хлоркаль-
циевую трубку, или в иную стеклянную трубку, которая непосред-
ственно припаивается к тому прибору, где требуется производить
осушку газа (см. фиг. 23, б).
Так как фосфорный ангидрид представляет собой порошок, то,
чтобы избежать его выдувания током газа, а следовательно, и попа-
дания его внутрь установки, рекомендуется с обеих сторон трубки
с фосфорным ангидридом поместить стеклянную вату.
«а
Реометры. Для замера скорости протекания газа применяют так
называемые реометры. Реометры бывают открытые и закрытые. От-
крытый реометр представлен на фиг. 24, а. Разница в высотах жид-
кости колена реометра характеризует скорость течения газа,
а следовательно, и его количество, протекающее в единицу
времени.
28
Закрытый реометр изображен на фиг. 24, б. Между трубками
/ и 2 находится суженная часть, создающая перепад давления в ко-
ленах реометра. В частности, этот перепад определяет скорость те-
чения газа. Имея набор трубок и капилляров различного диаметра,
которые можно помещать между трубками 7 и 2, можно один и тот
же реометр употреблять для измерения весьма разнообразных ско-
ростей протекания газа.
В последнее время получили распространение безжидкостные
реометры (фиг. 24, в). В этих реометрах применяют стеклянные
трубки, внутренний диаметр которых в верхней и
нижней частях неодинаков.
Диаметр нижней части трубки реометра несколько
меньше, чем верхней. Внутри трубки имеется попла-
вок из "пластмассы, фарфора или иного материала.
Поплавок имеет спиралевидную канавку. Струя
газа, двигающаяся по трубке реометра, поднимает
поплавок до некоторого
уровня, при котором обес-
печивается прохождение
газа через зазор между по-
фиг. 24. Реометры.
плавком и стенками трубки. Чем больше скорость газа, тем выше
поднимается поплавок, вращающийся вокруг своей оси и служащий
указателем скорости газа по шкале, нанесенной на .трубке.
Подобные реометры имеют, в частности, то преимущество, что
могут применяться при низких температурах, когда водяные" рео-
метры не могут быть использованы, а применение в реометре других
жидкостей по тем или иным причинам нежелательно. Следует также
учитывать, что указанные безжидкостные реометры могут быть
сделаны весьма чувствительными.
Замазки. При соединении отдельных частей прибора, как сте-
клянных, так и металлических, приходится иногда применять раз-
личные замазки. Чтобы обеспечить герметичность соединения, соеди-
няемые части должны тщательно высушиваться, разогреваться и в
разогретом состоянии на них должна наноситься та или иная рас-
плавленная замазка. После соединения частей следует пламенем
29
горелки оплавить замазку так, чтобы она приобрела ровный вид
и растеклась по поверхности соединяемой трубки или частей при-
бора. В качестве замазки употребляют различные вещества, обла-
дающие способностью хорошо приставать к стеклянным или металли-
ческим частям. Одной из лучших замазок является пицеин, изго-
товляемый из каучука и асфальта. Применяется также менделеев-
ская замазка, представляющая собой сплав из канифоли и пчели-
ного воска с добавлением минеральных веществ.
Следует отметить, что как пицеин, так и менделеевская замазка
не могут выдерживать температуры выше 40—50°. Чем выше темпе-
ратура, тем труднее подобрать условия, обеспечивающие герметич-
ность соединения, сделанного с помощью какой-либо замазки или
клея. Весьма устойчивы и механически прочны замазки, изгото-
вленные из окиси свинца и льняной олифы, а также из смеси глета
и глицерина.
Чтобы обеспечить газонепроницаемость подобных замазок, их
можно покрыть тем же пицеином или менделеевской замазкой. По-
добные соединения могут держать вакуум и при температурах, зна-
чительно превышающих температуру расплавления пицеина или
менделеевской замазки. Мелкие поры указанных твердых замазок
будут закрыты расплавленным пицеином или менделеевской замаз-
кой и таким образом будет обеспечена газонепроницаемость соеди-
нения. Для обеспечения герметичности соединения иногда приме-
няют легкоплавкие сплавы. Однако эти легкоплавкие сплавы не
всегда хорошо пристают к стеклу и сами по себе иногда имеют не-
которую пористость, вследствие чего они не всегда обеспечивают
газонепроницаемость соединения.
Некоторые авторы рекомендуют сплав, состоящий из 40% вис-
мута, 25% свинца, 10% олова, 10% кадмия и 15% ртути. Если по-
добную металлическую замазку покрыть слоем смоляной замазки,
то можно придать соединению высокую газонепроницаемость. Если
соединяемые части должны по возможности противостоять кислот-
ным парам, хлору и т. п., то применяют сплав асфальта и каучука.
Состав, называемый твердым морским клеем, приготовляют про-
должительным нагреванием на парафиновой бане двух частей ас-
фальта и одной части каучука в керосине. Во многих случаях приме-
няют также жидкое стекло. Замазки на жидком стекле приготовляют
размешиванием жидкого стекла с известью, мелом, безводным гип-
сом, окисью цинка, каолином и т. п.; эти замазки затвердевают через
2—3 часа. В некоторых случаях для герметичных соединений при-
годны расплавленное хлористое серебро и кремнекислый свинец.
В качестве замазки и склеивающего вещества часто применяют
шеллак как в чистом виде, так и в смеси с некоторыми другими ве-
ществами. При смешивании шеллака с 20—40% древесного дегтя
получают замазку, напоминающую по своим свойствам известный
цемент Котинского.
При смешивании шеллака с бутилфталатом можно получить за-
мазку с очень малым давлением паров, рекомендуемую при работе
с высокий! вакуумом.
30
Смесь 32 г амилацетата со 100 г шеллака дает замазку,, облада-
ющую высокой прочностью на разрыв.
В качестве замазок, обладающих хорошей склеивающей спо-
собностью и крайне незначительной упругостью паров, применяют
также бакелит и органическое стекло (люцит, плексиглас). Эти ве-
щества характерны тем, что при соответствующем нагревании они
полимеризуются и затвердевают. Бакелитовые лаки обычно состоят
из растворов еще не полимеризованных видов бакелита. Бакелито-
вую замазку изготовляют как в виде твердых веществ, так и в виде
вязких жидкостей.
В твердол! виде замазка плавится при температуре около 80°,
а при нагревании до более высокой температуры полимеризуется,
переходя в такую модификацию, которая не плавится даже при 200—
250° и лишь обугливается при температуре выше 285°.
К числу первоклассных замазок относятся алкидные смолы, полу-
чающиеся действием фталевого ангидрида на гликоль, глицерин и
другие полигидридные алкоголи. Гликольфталат особенно реко-
мендуется как вакуумная смазка благодаря низкому давлению его
паров, липкости, смачивающей способности в расплавленном со-
стоянии и прозрачности. Гликольфталат хорошо приклеивается к
металлам, особенно к алюминию.
Высокими качествами обладают апиезоновые замазки, получае-
мые из рафинированных остатков после перегонки парафиновых
масел и очистки этих остатков от примесей, обладающих значитель-
ным давлением паров.
Очень высокими качествами как вакуумная замазка обладает
следующий состав: 148 г фталевого ангидрида смешивают с 62 г эти-
ленгликоля, смесь нагревают в течение 1—1,5 часа при температуре
130°, при этом она превращается в однородную сиропообразную мас-
су; затем температуру поднимают до 180°. Через 3—4 часа замазка
готова.
Вакуумная смазка. Для приготовления вакуумной
смазки берут 30 частей натурального каучука, 25 частей белого ва-
зелина и 5 частей парафина. Каучук нарезают мелкими кусочками
и всю смесь держат в течение 180—200 час. при температуре 150—
160° при постоянном перемешивании.
Для получения более подвижной смазки увеличивают в смеси
долю вазелина.
Очистка ртути. Проверку и градуировку бюреток следует про-
изводить чистой ртутью, для чего ее фильтруют и сушат при ПО—
130°. При необходимости иметь очень чистую ртуть ее перегоняют
один или лучше два раза.
Для предварительной очистки ртути употребляют прибор, изо-
браженный на фиг. 25, представляющий собой стеклянный баллон
длиной около 70 см, в верхнюю часть которого впаяны две трубки —
одна с краном, ведущая к вакуумному насосу, другая—закрыва-
ющаяся каучуковой пробкой и служащая для наполнения всего
прибора 5—8%-ным раствором азотной кислоты. К верхней части
31
трубки при помощи менделеевской замазки прикреплена деревян-
ная воронка, в которую и наливается ртуть. Если трубку 1 закрыть
каучуковой пробкой, а через кран 2 выкачивать воздух, хотя бы
даже водоструйным насосом, то ртуть под действием атмосферного
давления начнет фильтроваться сквозь дерево и очень мелкими ка-
пельками падать в раствор азотной кислоты, а затем опускаться
на дно баллона 1. Проходя через раствор азотной кислоты, ртуть очи-
щается от различных растворенных в ней металлов, дерево же не
пропускает никаких механических
примесей. Ртуть, выпускаемая
через нижний кран, будет уже
довольно чистой и пригодной для
Фиг. 25. Прибор
для очистки
ртути.
Фиг. 26. Приборы для перегонки
ртути.
перегонки. Чтобы ртуть хорошо фильтровалась через дерево, сна-
ружи всю деревянную воронку необходимо обмазать горячей мен-
делеевской замазкой, чтобы не было течи воздуха сквозь дерево,
при наличии которой и при слабом насосе получающийся вакуум
может оказаться недостаточным, чтобы ртуть стала фильтроваться
через дерево. Толщина слоя дерева, через которое фильтруется ртуть,
должна быть 1—1,5 см. Кроме дерева, для этой же цели употре-
бляют также замшу.
Ртуть после выпуска через нижний кран следует промыть не-
сколько раз дестиллированной водой, чтобы очистить от следов азот-
ной кислоты, а затем нагреть до 120—130°, чтобы удалить воду. Со-
вершенно сухую ртуть для окончательной очистки следует пере-
гнать на каком-либо из приборов, изображенных на фиг. 26.
32
Прибор, изображенный на фиг. 26, а, устроен следующим обра-
зом: широкая трубка 1 диаметром 10—12 мм опирается своим кон-
цом на каучуковую пробку сосуда 2; на другом конце трубка 1 имеет
расширение 3 с 70—80 мм в поперечнике. Сквозь трубку сосуда 2
и по всей длине трубки 7 проходит трубка 4, которая в верхней части
(выше крана 5) имеет диаметр 5—7 мм, а в нижней части (ниже кра-
на 5) имеет внутренний диаметр не более 1,5 мм. Резервуар 6 напол-
няется ртутью, которую можно по каучуковой трубке перелить в
сосуд 2. От дна сосуда 2 до открытого конца трубки 4 должно быть
82—85 см, от места впайки крана 5 до сосуда 7 — около 90—95 см.
Под расширением 3 помещается кольцевая газовая горелка, а над
ним асбестовый колпак. Трубка 7 в месте прикосновения к пробке
сосуда 2 имеет выемку, позволяющую ртути свободно входить
внутрь.
Для того чтобы прибор пустить в ход, резервуар 6 наполняют
ртутью, которую требуется перегнать, а сосуд 7 наполняют дестил-
лированной ртутью. Если дести л лированной ртути нет, то в сосуд 7
наливают той же ртути, которую дестиллируют, но в этом случае
первые порции дестиллированной ртути, получившиеся через боко-
вую трубку сосуда 6, вливают обратно в резервуар 6 до тех пор, пока
не пройдет по крайней мере восьми- или десятикратный объем того
количества ртути, которое было помещено в сосуд 7.
Затем через кран 5 производят возможно полную откачку воз-
духа из всего прибора (можно обойтись и водоструйным насосом).
При откачке ртуть из сосуда 7 поднимется по трубке 4, а из сосуда 2
по трубке 7 и заполнит часть сосуда 3. Необходимо все время смот-
реть, чтобы ртуть из сосуда 6 не перелилась через открытый конец
трубки 4. По мере нагревания ртути ее пары будут оседать на внут-
ренней поверхности трубки 4 в той ее части, которая проходит внутри
трубки 7, служащей как бы холодильником. Постепенно капельки
ртути начнут падать в капиллярную часть трубки 4.
При диаметре капилляра не более 1,5—2 мм капельки ртути бу-
дут давать цепочку, т. е. капли ртути будут чередоваться с пузырь-
ками воздуха. Как только начнет образовываться цепочка, кран 5 сле-
дует закрыть, так как дальше ртуть, образуя цепочку, будет увле-
кать пузырьки воздуха вниз и производить откачку автомати-
чески.
Прибор, изображенный на фиг. 26, б, представляет собой колбу,
подогреваемую электрическим током, в которую ртуть поступает
из стакана 7. Колба и широкая трубка 2 покрыты толстым слоем
асбеста. Пары ртути из колбы попадают в широкую трубку (холо-
дильник) 3 и дальше в капиллярную трубку 4. Через кран 5 про-
изводится откачка воздуха.
Для большей чистоты ртуть лучше перегнать два раза, для чего
следует иметь два прибора для перегонки — на одном перегонять
ртуть, только профильтрованную и пропущенную через азотную
кислоту, а на другом работать только с ртутью, перегнанной уже
один раз.
3 В. А. Соколов
33
§ 3. Измерения объема и давления газа
Измерение объема
Для измерения объема газа при анализе обычно употребляют
градуированные стеклянные трубки того или иного размера и той
или иной формы в зависимости от количества газа, требуемой точ-
ности измерения и условий анализа.
Наиболее употребительные формы газовых бюреток изображены
на фиг. 27. Обычная газовая бюретка представляет собой градуиро-
ванную стеклянную трубку/ на одном конце которой имеется кран,
а на другой конец надевается каучуковая трубка, соединяющая
бюретку с уравнительным сосудом.
В уравнительный сосуд и бюретку наливают ртуть или водный
раствор чистой поваренной или глауберовой соли. Употребляют
также и дестиллированную воду.
Между бюреткой и уравнительным сосудом иногда помещают
кран, назначение которого не допускать, когда это требуется, под-
нятия и опускания ртути или воды в бюретке при перемещении ура-
внительного сосуда. Отличие газовой бюретки от химической заклю-
чается в том, что у газовой бюретки нуль находится у самого крана,
а у химической — на другом конце бюретки; кроме того, обычно
<34
у химической бюретки градуировка не доведена до крана, а потому
химической бюреткой для измерения объема газа пользоваться нельзя.
Газовые бюретки изготовляют емкостью 50, 100 или 200 см3', встре-
чаются также специальные бюретки емкостью меньше 50 см3 и больше
200 о*3. Наиболее употребительны бюретки емкостью 100 ел/3; они
удобны тем, что отсчеты по бюретке при анализе дают прямо про-
центное содержание газов.
Бюретка I (фиг. 27) представляет собой цилиндрическую трубку
длиной обычно от 60 до 80 см и диаметром от 13 до 15 мм. Бюретка
проградуирована на кубические сантиметры, а каждый куби-
ческий сантиметр обычно разделен на 5 или 10 частей. Таким обра-
зом, каждое маленькое деление на бюретке емкостью 100 см3 соот-
ветствует 0,2 или 0,1 см3, или, иначе говоря, 0,2 или 0,1% всего объ-
ема бюретки. Бюретка II отличается от бюретки I тем, что идущая
от крана градуированная узкая трубка имеет расширение на конце.
Емкость широкой и узкой частей вместе равна обычно 100 см3. Черта,
соответствующая емкости 100 см3, ставится за широкой частью бю-
ретки. Диаметр узкой трубки берут в зависимости от желаемой точ-
ности измерений. При внутреннем диаметре трубки 4—5 мм можно
каждый кубический сантиметр проградуировать до 0,01 см3. Широ-
кая часть бюретки в большинстве случаев не градуируется. Бюретки
типа II употребляют при анализе газа в тех случаях, когда про-
центное содержание интересующей нас составной части газа очень
мало, а главная составная часть газа поглощается в первых стадиях
его анализа.
Представим себе, что в газе содержится 97% углекислого газа,
в остатке же требуется с большой точностью определить содержа-
ние кислорода или окиси углерода. Бюретка типа II является един-
ственной подходящей для решения такой задачи. Если же мы имеем
противоположный случай, а именно, газ, содержание которого тре-
буется узнать с большой точностью, приходится определять пер-
вым, тогда наиболее подходящей будет бюретка типа III.
Краны в верхнем конце газовых бюреток бывают различных кон-
струкций. Ставятся простые двухходовые краны, как у бюреток
I и II, ставятся краны и трехходовые и еще более сложные.
Для отсчета объема газа в бюретке поступают так. При закры-
том верхнем кране и открытом нижнем уравнительный сосуд под-
носят вплотную к бюретке и устанавливают на такой высоте, чтобы
ртуть или вода в бюретке и в уравнительном сосуде была на одном
уровне. При таком положении производят отсчет, который и будет
соответствовать объему газа при температуре и барометрическом
давлении в момент измерения.
Так как вода и упомянутые водные растворы солей имеют при
комнатной температуре значительную упругость паров, то для полу-
чения точных результатов необходимо вводить поправки на присут-
ствие в газе водяных паров.
Кроме того, вода в значительной степени растворяет некоторые
газы, что при анализе может повести к искажению результатов. Этих
недостатков нет у ртути, упругость паров которой при комнатной
35
температуре настолько мала, что никаких поправок вводить не по-
требуется (табл. 3).
Таблица 3
Давление насыщенных паров ртути при
различных температурах
Темпе- ратура, °C Давление насыщенных паров, мм Темпе- ратура, °C Давление насыщенных паров, мм
0 0,0002 24 0,0018
10 0,0005 26 0,0022
16 0,0009 28 0,0025
18 0,0011 30 0,0030
20 0,0013 34 0,0041
22 0,0016 40 0,0065
Растворимость газов в ртути ничтожна. По этим причинам при
точной работе в качестве вытесняющей жидкости в бюретке сле-
дует употреблять ртуть.
Ртуть как жидкость, не смачивающая стекло, образует в бюрет-
ках выпуклый мениск, что и приходится принимать во внимание
при измерении объема газов. Ни то деление, которое приходится
против верхнего края мениска, ни то, которое приходится против
нижнего, не соответствуют истинному объему части бюретки, нахо-
дящемуся над ртутью и ограниченному плоскостью, проходящей
через деление, но во всяком случае верхняя граница мениска в ши-
роких трубках ближе подходит к истинному объему и при отсчетах
за объем газа принимают то деление, которое приходится против
верхней границы (фиг. 27; а, линия а—а). Чтобы не вводить никаких
поправок на мениск, газовые бюретки следует градуировать таким
образом, чтобы деления, приходящиеся против верхней границы
мениска, соответствовали истинному объему газа и уже включали
в себя поправку на мениск.
В табл. 4 даны объемы мениска для трубок различного диаметра.
Колебания температуры могут значительно влиять на объем газа
в бюретке. Если взять в руку бюретку за то место, где находится
газ, и подержать в течение нескольких секунд, то уже можно заме-
тить увеличение объема. Точно так же быстро действуют всякие из-
менения температуры воздуха в помещении, где находится бюретка.
При открывании дверей, окон, форточек холодный или теплый поток
воздуха всегда может изменить объем газа в бюретке в ту или другую
сторону. Чтобы предохранить бюретку от колебаний температуры,
ее помещают в широкую стеклянную трубку (фиг. 28, а), которая
держится на резиновых пробках, плотно надетых на бюретку. В эту
широкую охранную трубку наливают воду.
36
Поправка на мениск
Таблица 4
Диаметр трубки, мм i Двойной объем мениска, см3 Диаметр трубки, мм Двойной объем мениска, ем3
ртуть вода ртуть вода
1 0,0026 0,0021 14 0,118 0,291
2 0,0052 0,0043 15 0,140 0,343
3 0.0090 0,0093 16 0,162 0,395
4 0,0128 0,0144 17 0,174 0,443
5 0,0200 0,026 18 0,185 0,492
6 0,027 0,039 19 0,181 0,520
7 0,035 0,062 20 0,177 0,548
8 0,043 0,084 21 0.180 0,605
9 0,056 0,114 22 0,182 0,663
10 0,068 0,144 23 0,179 0,708
11 0,080 0,190 24 0,176 0,754
12 0,091 0,236 25 0,173 0,800
13 0,104 0,263
Часто применяют бюретки, в которых всякие изменения темпе-
ратуры и давления, происходящие во время работы, автоматически
компенсируются. Подобная бюретка изображена на фиг. 28, б.
В широкой охранной трубке рядом с бюреткой помещена стеклянная
компенсационная трубка, запаянная с одного конца и сообщающая-
ся другим концом с манометром 7. который соединен с левой труб-
кой трехходового крана. В манометр наливают подкрашенную воду.
При первом измерении объема газа поступают следующим образом.
Газ забирают в бюретку обычным способом, опуская уравнитель-
ный сосуд. Затем кран бюретки закрывают, а кран 2 открывают,
уравнительный сосуд поднимают на такую высоту, чтобы ртуть в
нем и в бюретке стояла приблизительно на одной высоте. После этого
поворачивают кран, чтобы сообщить бюретку с манометром. Слегка
приподнимая и опуская уравнительный сосуд, добиваются того,
что уровень воды в обоих коленах манометра будет на одной высоте,
и в этот момент закрывают кран 3, сообщающий бюретку с уравни-
тельным сосудом. Закрыв верхний кран бюретки и кран 2, произ-
водят отсчет объема газа. Обычно бюретка присоединена к какому-
либо прибору для анализа, а заключающийся в бюретке газ во время
анализа приводится в соприкосновение с тем или иным поглотите-
лем, отчего объем газа все уменьшается. После каждого поглотителя
приходится измерять оставшийся объем газа. Эти измерения объема
производят так же, как было описано, с той только разницей, что
кран все время остается закрытым. Производя все отсчеты объема
газа при одинаковых высотах воды в манометре, мы исключаем влия-
ние изменений атмосферного давления и температуры. Предположим,
что за время испытания газа атмосферное давление изменилось. Так
как компенсационная трубка не сообщалась за время испытания с
37
атмосферным воздухом, то в ней сохранилось то же давление, какое
было и при первом измерении. Таким образом, все измерения объема
газа в описанной бюретке производятся при постоянном давлении.
Точно так же всякое изменение объема газа в бюретке в зависимости
от колебаний температуры компенсируется изменением упругости
воздуха в компенсационной трубке.
При отсчете объема газа необходимо, чтобы уровни ртути в бю-
ретке и уравнительном сосуде были
Фиг. 28. Бюретка с компенсатором и бю-
ретка с манометрической трубкой.
одинаковы. Наличие широкой
охранной трубки делает за-
труднительным определение
на-глаз момента нахождения
уровней ртути в одной пло-
скости. Это облегчается мано-
метром, вынесенным за пре-
делы охранной трубки, по
которому довольно легко уло-
вить требуемый момент от-
счета. Применение подобных
манометров имеет один недо-
статок: установить манометр
так, чтобы уровень воды в
обоих коленах был одинаков,
можно только при открытом
кране бюретки, причем эта
установка совершается не
сразу, а после нескольких
колебаний манометра в ту и
другую сторону. При этих
колебаниях манометра часть
газа или воздуха, находяще-
гося над левым коленом манометра (фиг. 28, б), будет, безусловно,
попадать в бюретку и таким образом влиять на состав взятого для
испытания газа. Количество газа, попадающего в бюретку, обычно
невелико, но при очень точной работе это изменение заметно.
Чтобы достаточно точно вести отсчеты объема газа, применяют
бюретку, изображенную на фиг. 28, а. Внутри охранной трубки
рядом с бюреткой идет манометрическая стеклянная трубка 8—10 мм
диаметром, сообщающаяся одним концом с уравнительным сосудом
и бюреткой, а другим концом — с атмосферным воздухом. Так как
ртуть в уравнительном сосуде и в трубке будет всегда на одной и
той же высоте, то закрывание крана для отсчета объема следует про-
изводить в тот момент, когда уровни ртути в бюретке и в трубке бу-
дут в одной плоскости. Так как бюретка и трубка находятся рядом,
то этот момент заметить легко.
Необходимо заметить, что упомянутые компенсационные устрой-
ства (фиг. 28, б) применяются не всегда. Колебания атмосферного
давления за время анализа обычно настолько невелики, что суще-
ственно не влияют на измерения объема газа. Колебания же темпе-
ратуры при наличии охранной трубки с водой зависят в основном
38
Фиг. 30.
градуировки
ретки.
Фиг. 29. Бю-
ретка для точ-
ного измерения
объема газа.
от температуры воды, которая замеряется термометром, на осно-
вании чего и вводятся соответствующие поправки.
Если требуется измерять объемы газов с большой точностью,
употребляют бюретку особого устройства, изображенную на фиг. 29.
Бюретка состоит из трех частей — узкой трубки 1 емкостью 20 см3,
трубки 2 с расширениями, каждое емкостью 20 см3, и трубки 3, у
которой каждое расширение имеет емкость 100 см3. Трубка 1 гра-
дуируется до 0,05 см3 или до еще более мелких объемов; емкость
трубок 7 и 2 вместе должна быть не меньше емкости одного расши-
рения трубки 3. Применяют чаще всего бюретки с двумя трубками
7 и 2, причем трубка 2 состоит из
пяти расширений по 20 см3 или
из десяти расширений по Ю см3.
Число расширений трубки 3
может быть любое. Все расшире-
ния соединены между собой капил-
лярами, на которых и нанесены
метки, соответствующие целым сот-
ням или десяткам куб. сантиме-
тров. Каждая из трубок 7, 2 и 3
имеет свой кран, сообщающий ее
уравнительным сосудом; к с крану
бюретки можно присоединить ма-
нометр 4, который другим концом
сообщается с компенсатором.
Продажные бюретки не всегда
бывают точно проградуированы,
поэтому, приступая к работе с но-
вой бюреткой, ее следует проверить.
Проверка бюреток. При про-
верке бюреток, а также и при
их первоначальной градуировке поступают так. Бюретку устанав-
ливают вертикально и к нижнему концу на толстостенном кау-
чуке присоединяют кран с капиллярными ходами (1—2 мм). Ниж-
няя оттянутая трубка крана должна иметь на выходе очень узкое
отверстие, не больше 0,5 мм (фиг. 30, 7). На капиллярный кран на-
девают каучук уравнительного сосуда. Соединение капиллярного
крана с бюреткой должно делаться особенно тщательно: кран и бю-
ретка должны иметь расширения на стекле, так называемые оливки,
служащие для более плотного прижимания каучука; каучук обя-
зательно должен быть толстостенным; кран и бюретка должны соеди-
няться встык, а весь соединяющий каучук должен обматываться
железной проволокой.
В уравнительный сосуд наливают чистую сухую ртуть в коли-
честве, достаточном для наполнения всей бюретки. Когда ртуть за-
полнит нижний капиллярный кран, его закрывают. Затем верхний
кран бюретки соединяют с ртутным или масляным насосом и из бю-
ретки откачивают воздух. Когда воздух будет выкачан хотя бы до
Проверка
бю-
39
вакуума в несколько миллиметров, капиллярный кран открывают
и ртуть постепенно заполняет всю бюретку. Когда ртуть подойдет
к верхнему крану на расстояние 2—3 мм, нижний капиллярный
кран закрывают, а откачку продолжают до вакуума 1—2 мм, после
чего верхний кран закрывают, а бюретку отъединяют от насоса.
Затем, „подняв уравнительный сосуд до верхнего крана, открывают
нижний кран; при этом ртуть заполнит оставшееся в бюретке про-
странство до верхнего крана. После этого нижний капиллярный
кран закрывают и с него осторожно снимают каучук уравнительного
сосуда.
Наполнять бюретку ртутью под вакуумом желательно потому,
что в противном случае пузырьки воздуха прилипают к внутренним
стенкам бюретки, что ведет к значительный! погрешностям при гра-
дуировке. Так как верхний кран закрывают уже тогда, когда объем
над ртутью до крана не превышает десятой доли кубического санти-
метра, то при вакууме в 1—2 мм объем остающегося в верхней части
бюретки воздуха будет чрезвычайно мал и при поднятии уравни-
тельного сосуда ртуть целиком заполнит весь объем без каких-либо
видимых глазом пузырьков воздуха.
При снятии каучука с капиллярного крана необходимо следить,
чтобы весь нижний отросток крана остался заполненным ртутью.
Бюретка готова теперь для проверки или градуировки. Под капил-
лярный кран подставляют стакан, совершенно чистый, сухой и тща-
тельно взвешенный на аналитических весах. Открыв верхний кран,
осторожно (через нижний кран) выпускают ртуть в стакан, пока
верхняя граница мениска не дойдет до метки 1 см3 (или до любой
другой метки). Совершенно очевидно, что объем вытекшей ртути в
точности равен объему вошедшего в бюретку воздуха. Стаканчик
с вытекшей ртутью взвешивают, полученный вес ртути делят на вес
1 см3 ртути при данной температуре и таким образом получают объем,
соответствующий метке, до которой дошел мениск. Как это совер-
шенно очевидно, никаких поправок на мениск вводить в этом случае
не требуется. Проверку первых кубических сантиметров бюретки
можно вести до 0,1 см3, дальше же проверку можно вести через 1 гл*3.
Пока вес невелик, стаканчик с ртутью можно взвешивать на анали-
тических весах, в дальнейшем же следует пользоваться весами с
точностью до 0,01—0,02 г и допускающими нужную нагрузку. При
взвешивании 1—2 кг достаточна точность 0,1 г. При всех этих опе-
рациях необходимо следить, чтобы ртуть не разбрызгивалась при
выпускании ее в стакан, поэтому стакан следует ставить настолько
высоко, чтобы между уровнем ртути и концом крана был промежуток
всего лишь в несколько миллиметров.
Известно, что ртуть при поднятии образует более выпуклый ме-
ниск, а при опускании более плоский. Но если ртути дать некоторое
время постоять спокойно и при этом постукивать пальцами по стен-
кам бюретки в том месте, где находится мениск, то последний посте-
пенно примет свою нормальную форму. Все отсчеты при проверке и
градуировке бюреток следует делать только после того, как мениск
примет нормальную форму.
40
Когда бюретка градуируется впервые, следует поступать так: выпу-
скать ртуть надо не в стаканчик, а в специальную узкую мензурку,
правильно и тщательно проградуированную, или присоединив гра-
дуированную трубку 2 (фиг. 30). Как только натечет 1 со3. на бю-
ретке ставят метку (острым графчиком процарапывают парафин или
воск, которым предварительно обливают бюретку, а затем по окон-
чании всей операции ставят цифры и травят фтористым водородом).
Для градуировки мензурки отвешивают такое количество ртути,
которое соответствовало бы 1 см3, наливают его в мензурку и ставят
метку, затем отвешивают и наливают второй кубический сантиметр,
третий и т. д.
Если требуется большая точность, в мензурку наливают по 0,1 см3
ртути, пока не наберется 1 см3. Мензурка емкостью в несколько
кубических сантиметров вполне достаточна, так как, когда она
наполнится, ртуть из нее выливают и продолжают работу. Куби-
ческие сантиметры, нанесенные на бюретку, следует делить на) 5,
10 и 20 частей при помощи делительной машины, которой пользу-
ются градуировщики и которая очень просто устроена. Если куби-
ческие сантиметры нанесены на бюретку, промежуточные деления
можно нанести хотя и с меньшей точностью, чем делительной маши-
ной, от руки, измерив расстояние между делениями, соответству-
ющими целым кубическим сантиметрам, и разделив это расстояние
на необходимое число частей.
Объем, занимаемый единицей веса ртути, зависит от ее удель-
ного веса, который меняется в зависимости от температуры. По этой
причине следует при проверке и градуировке надевать на бюретку
охранную трубку и наполнять ее водой, за температурой которой
Таблица 5
Вес 1 см3 чистой ртути при различных температурах в безвоздушном
пространстве
(в воздухе вес ртути меньше, чем приведенный в таблице, на вес 1 см3 воздуха
т. е. приблизительно на 0,0013 г)
Темпера- тура, °C Вес ртути, г Темпера- тура, °C Вес ртути, г Темпера- тура, °C Вес ртути, г
15,0 13,5593 20,0 13,5489 25,0 13,5385
15,5 13,5583 20,5 13,5479 25,5 13.5374
16,0 13,5573 21,0 13,5468 26,0 13,5364
16,5 13,5562 21,5 13,5458 26,5 13,5353
17,0 13,5552 22,0 13,5447 27,0 13,5343
17,5 13,5541 22,5 13,5437 27,5 13,5332
18,0 13,5531 23,0 13,5426 28,0 13,5322
18,5 13,5520 23,5 13,5416 28,5 13,5312
19,0 13,5510 24,0 13,5405 29,0 13.5301
19,5 13,5499 24,5 13,5395 29,5 13,5291
30,0 13.5280
Л
необходимо следить. Желательно, чтобы во время проверки темпе-
ратура воды по всей высоте была одинакова, чего можно достигнуть
легким перемешиванием ее стеклянной палочкой или пропусканием
слабого тока воздуха через трубку, опущенную до дна охранной
трубки: поднимающиеся пузырьки воздуха произведут достаточное
перемешивание.
В табл. 5 приведены веса 1 см3 совершенно чистой ртути при раз-
личных температурах.
Для измерения очень малых объемов газа пользуются градуиро-
ванными капиллярами. Измерять объем газа можно также при дав-
лениях, во много раз меньших атмосферного; это позволяет еще бо-
лее повысить чувствительность измерений. Различные приспособле-
ния для замера малых количеств газа описаны ниже в разделе, каса-
ющемся измерения давления (фиг. 33, 34) и в главе о микроанализе
газов.
Измерение давления
Большинство измерений объемов производится при атмосферном
давлении, но атмосферное давление не остается постоянным, а может
колебаться в разные дни в пределах не-
Фиг. 31. Ртутные баро-
метры.
скольких сантиметров. По этой причине
для точного определения количества газа
следует измерять также и атмосферное
давление по точному барометру. Ртутные
барометры (наиболее точные) бывают или
чашечные или сифонные; схематическое
устройство их видно на фиг. 31. В неко-
торых чашечных барометрах дно чашки
с ртутью сделано из замши (фиг. 31). Поль-
зуясь винтом, прикрепленным к замшевому
мешку, можно подвести уровень ртути к
самому концу штифта. Крайняя оконеч-
ность штифта является нулем миллиметро-
вой шкалы, идущей вверх вдоль баро-
метра.
Так как плотность ртути и длина деле-
ний шкалы зависят от температуры, то не-
обходимо относить высоту ртутного столба
и длину делений шкалы барометра к какой-
либо определенной температуре. За такую
температуру обычно принимают 0°.
Пусть а— коэфициент кубического рас-
ширения ртути и — коэфициент линей-
ного расширения материала шкалы.
Обозначим через h отсчитанную высоту барометра при темпе-
ратуре /°, а через /Уо высоту барометра, приведенную к 0°. Если
деления шкалы верны при 0°, то отсчитываемая по шкале длина /,?
делений при /° в действительности будет равна h (1 + /Ю- Так как
высоты уравновешивающихся столбов ртути при различных темпе-
42
ратурах обратно пропорциональны ее плотностям при этих темпе-
ратурах и прямо пропорциональны удельным объемам ее, то
н0 =Dt = 1
Л (1 4-/5Г) Do 1 4- at >
откуда
или приближенно
Но== /2 [!-(«-/?) /].
Поправка на капиллярную депрессию (табл. 6) и на несовпа-
дение острия с нулем шкалы устанавливается сравнением показания
барометра с показаниями нормального барометра и дается в виде
некоторой постоянной при каждом барометре. Кроме ртутных баро-
метров, пользуются еще металлическими анероидами, но они дают
меньшую точность.
Таблица 6
Капиллярная депрессия ртути (мм)
Радиус трубки, мм Высота мениска, мм
0,2 0,4 0,6 0,8 1,0 1,2 1,4 1,6 1,8
1,0 2,46 4,40 • - - , — —
1,4 1,26 2,36 3,22 —. — — —
1,8 0,75 1,44 2,02 2,48 —— — — — —
2,2 0,49 0,95 1,36 1,70 1,98 — — — —
2,6 0,34 0,66 0,96 1,22 1,44 1,61 — ——
3,0 0,24 0,48 0,70 0,90 1,07 1,21 1,32 — —
3,5 0,17 0,34 0,49 0,64 0,76 0,87 0,96 1,04 —
4,0 0,12 0,24 0,35 0,46 0,56 0,64 0,71 0,77 0,82
4,5 0,09 0,18 0,26 0,34 0,41 0,47 0,53 0,58 0,62
5,0 0,07 0,13 0,19 0,25 0,30 0,35 0,40 0,44 0,47
5,5 0,05 0,10 0,14 0,19 0,23 0,27 0,30 0,33 0,36
6,0 0,04 0,07 0,11 0,14 0,18 0,20 0,23 0,25 0,27
6,5 0,03 0,06 0,09 0,11 0,14 0,16 0,18 0,20 0,21
7,0 0,02 0,04 0,06 0,08 0,10 0,12 0,14 0,15 0,16
Если требуется измерить давление какого-либо газа, пользуются
сифонными манометрами с одним открытым концом или укорочен-
ными манометрами с запаянным концом (фиг. 32).
Измерение малых давлений. Для измерения малых давлений
применяют разнообразные типы манометров в зависимости от коли-
чества имеющегося газа и степени разрежения. На фиг. 32 пред-
ставлены обычные ртутные укороченные манометры, позволяющие
определять давления газа, начиная от 1—2 мм рт. столба и до давле-
ний, исчисляемых десятками сантиметров ртутного столба.
Если не приходится опасаться вредного влияния водяных паров,
применяют водяные манометры. Чувствительность водяного мано-
43
метра, естественно, значительно больше ртутного в соответствии с
разницей удельных весов воды и ртути. Для получения еще более
чувствительного замера употребляют наклонные манометры, ртут-
ные или водяные. При небольшом угле наклона чувствительность
водяного манометра может быть очень высокой. В тех случаях, когда
нежелательно применение воды, применяют различные масла или
керосин. Подобные чувствительные наклонные манометры приме-
няются главным образом при измерении небольшого перепада дав-
лений. Давление, соот-
ветствующее высокому
вакууму, измеряют спе-
циальными маномет-
рами.
На фиг. 33 (а и б)
представлен один из наи-
более употребительных
манометров, применяе-
мых в вакуумных уста-
новках. Он представляет
собой стеклянный бал-
лон, нижняя трубка
которого сообщается с
Фиг. 33. Манометр, применяемый в вакуумной
установке.
4 Измеряемое
I давление
Фиг. 32. Ртутные
манометры.
уравнительным сосудом, а кроме того, имеет отвод, сообщающийся
с той установкой, в которой определяется давление.
Верхняя часть манометра — капилляр очень маленького диа-
метра, а следовательно, и объема. Параллельно с капилляром идет
манометрическая трубка, другой конец которой присоединен к
вакууму, создаваемому отдельно насосом или углем, или к той же
самой откачиваемой установке, где уже имеется достаточно большой
вакуум.
Следует учесть, что для манометрической трубки нет надобности
добиваться вакуума, большего 0,1—0,2 мм рт. столба, т. е. той вели-
чины, которая может быть отсчитана на-глаз.
Совершенно очевидно, что давление в баллоне манометра при
достаточном диаметре соединительных трубок будет такое же, как
и в установке. Поднимая ртуть из уравнительного сосуда и запол-
няя ею баллон манометра, производят сжатие газа.
44
Разница уровня ртути на манометрической труоке и в капилляре
показывает давление газа внутри капилляра. Зная объем баллона
манометра и объем капилляра, которые градуируются заранее, можно
вычислить то давление, которое первоначально существовало в бал-
лоне до поднятия ртути, а следовательно, и давление, имеющееся
в присоединенной к манометру установке. Подобным манометром
можно измерять давления до 10~4—10~5 мм рт. столба. Для того чтобы
имелась возможность измерять как очень малые давления, так и
давления порядка I0-2—Ю-1 мм рт. столба, измерительный капил-
ляр делают ступенчатым. Верхняя его часть представляет собой
узкий капилляр, ниже распола-
гается более широкий, а еще
ниже трубка диаметром в не-
сколько миллиметров. Соответ-
ственно этому имеется и не-
сколько манометрических трубок
таких же диаметров, как и из-
мерительные трубки (фиг. 33).
На фиг. 34 представлена
схема манометра, построенная
на аналогичном же принципе, но
в сочетании с ртутным затвором.
Подобные манометры приме-
нялись автором настоящей книги
в различных аппаратах для
углеводородного микроанализа.
Преимущество представлен-
ного на фиг. 34 устройства за-
ключается в том, что манометр
одновременно может играть и
роль вакуумного ртутного на-
соса [4]. Поднимая ртуть в ма-
нометре и опуская ртуть в за-
творе, можно производить от-
качку той установки, которая
присоединена к упомянутому
устройству. Если же ртуть в
затворе будет поднята до самого
верха, то этот насос может быть
ИСПОЛЬЗОВан В качестве мано- Фиг. 34. Микроманометр,
метра.
Верхняя часть насоса представляет собой изогнутый капилляр,
проградуированный по своей длине по объему. Устанавливая ртуть
на определенной метке и замеряя показания манометрической трубки,
можно определить очень малые количества газа, а отсюда при не-
обходимости можно вычислить и давление в присоединенной к мано-
метру установке.
Подобная комбинация насоса и манометра удобна для откачки
и замера малых количеств газа.
45
Фиг.. 35. Определение вакуума.
а — тепловой манометр; б — схема включения теплового
манометра.
Для определения малых давлений’применяют и многие другие
типы микроманометров, например манометры мембранного типа, в
которых небольшие давления передаются на мембрану (стеклянную,
резиновую, металлическую); смещение этой мембраны тем или иным
путем передается на ось или нить с прикрепленным к ней зеркаль-
цем. Поворот зеркальца,
регистрируемый по шка-
ле, позволяет судить о
величине давления.
Следует также упо-
мянуть о манометре,
устройство которого ос-
новано на измерении те-
плопроводности. В со-
суде, где производится
измерение давления,
имеется натянутая пла-
тиновая нить, подогре-
ваемая электрическим
током. Электрическое
сопротивление этой нити
замеряется с помощью
мостика Уитстона. В за-
висимости от давления
газа меняется и его
теплопроводность, что
позволяет по температуре накаливаемой нити, а следовательно, и
по ее сопротивлению судить о степени вакуума (фиг. 35).
Известны также и другие многочисленные типы манометров для
измерений малых давлений газов.
§ 4. Стеклодувные вспомогательные работы
Большинство газоаналитических приборов представляет собой
установки, спаянные или собранные из стеклянных частей. При
работе с этими приборами очень желательно обладать навыком в
обращении со стеклом и в пайке стеклянных частей [3].
Газовая паяльная горелка устраивается так, чтобы было возможно
продольное передвижение внутренней трубки, подводящей воздух,
в наружной гильзе, по которой идет газ. Если концы обеих трубок
сближены, то получают остроконечное пламя; если наружная трубка
оттянута назад или внутренняя выдвинута вперед, то получается
более широкое пламя. В зависимости от величины обрабатываемого
предмета применяют то или другое пламя. При обработке тугоплав-
ких сортов стекла нагрев удается повысить, если на пути пламени
за обрабатываемым предметом поместить перпендикулярно к пла-
мени пластинку из древесного угля или лучше из огнеупорного ша-
46
мотного кирпича. Для получения еще более высокой температуры
рекомендуется питание горелки кислородом вместо воздуха.
Воздух подается обычно или кожаным мехом с ножной педалью,
или водоструйным насосом, имеющим приспособление для подачи
воздуха под давлением, или же ротационным насосом, приводимым
в движение электромотором.
При спайке сложных установок, когда нет возможности собирать
или ремонтировать установку на стеклодувном столе, применяют
ручные паяльные горелки.
•Стекло употребляют почти исключительно в виде трубок; запас
трубок различных диаметров и разной толщины стенок необходимо
иметь под рукой. Следует иметь в виду, что стекла различного состава
трудно спаиваются. Поэтому нужно всегда иметь возможно полный
ассортимент различных трубок одного сорта.
Нагревать стеклянные трубки следует медленно и тем медленнее
и осторожнее, чем шире трубка и чем толще ее стенки. Прежде всего
подогревают трубку в коптящем пламени, чтобы повышение темпе-
ратуры трубки происходило не очень быстро. Трубка, нагреваемая
с конца, лопается гораздо чаще, чем нагреваемая на некотором рас-
стоянии от конца. При подогревании и в дальнейшем при размягче-
нии стекла обрабатываемый предмет нужно равномерно и непре-
рывно двигать в пламени, чтобы в нем не возникло заметных раз-
ностей температур и связанных с этим внутренних напряжений.
Трубки, например, при нагреве все время вращают. Когда пламя
окрасится в желтый цвет, опасность появления трещин мино-
вала.
По окончании обработки предмета, по удалении его из пламени,
необходимо еще раз вдунуть в него ртом воздух, чтобы в момент за-
твердения внутри предмета существовало избыточное давление. Обра-
ботанный таким образом предмет лопается впоследствии гораздо
реже.
При работе с тонкими, быстро остывающими трубками для удоб-
ства вдувания на них надевают длинную и легкую резиновую трубку,
другой конец которой берут в рот. Для’ задержания водяных паров
и воды в резиновую трубку вставляют отрезок стеклянной трубки,
наполненный ватой и хлористым кальцием. Если готовое изделие
спаяно из отдельных частей, то места соединения должны быть надле-
жащим образом отожжены, или, как принято говорить, закалены.
Для этого двигают изделие взад и вперед в коптящем пламени
горелки или, если опасаются восстанавливающего действия коптя-
щего пламени, в горячем воздухе от бунзеновской горелки. Охлаж-
дение должно быть тем медленнее, чем больше разница в толщине
спаянных между собой трубок. Все обрабатываемые трубки должны
быть сухими и свободными от пыли.
Резка трубок. Трубки диаметром до 8 мм разрезают следующим
образом. В избранном месте проводят надрез ножом для стекла,
затем берут трубку обеими руками так, чтобы большие пальцы при-
ходились по другую сторону надреза. Энергичное сгибание и одно-
47
временное растягивание трубки вызывают появление аккуратной
круговой трещины (очень толстостенные трубки разрезать таким
образом не удается).
В качестве ножа для стекла лучше всего пользоваться трехгран-
ным напильником или специальным ножом из твердой стали или по-
бедита. Назначение ножа заключается в нанесении трещины в стекле,
поэтому им нужно нажимать на трубку, осторожно ее вращая.
Иногда необходимо разрезать трубку в таких условиях, что ее
неудобно ломать. Тогда поступают следующим образом. Делают,
как обычно, надрез ножом, затем оплавляют конец тонкой стеклян-
ной палочки в пламени горелки и этим концом, еще раскаленным,
прикасаются к одному из концов надреза. При этом появляется кру-
говая трещина и трубка лопается.
Способ резки толстостенных или широких трубок состоит в том,
что на желаемом месте делают ножом возможно глубокий надрез.
Для быстрого местного разогревания применяют изогнутые полу-
кругом железные прутья толщиной около 3 мм, рукоятку которых
берут в руку, а изогнутую часть нагревают возможно сильнее на
паяльной горелке. На эту раскаленную изогнутую часть помещают
трубку надрезанным местом и медленно вращают; через некоторое
время трубка разламывается по надрезу. Тонкостенные трубки раз-
резают также при помощи остроконечного пламени. Сделанный за-
ранее надрез удлиняют, постепенно подставляя пламени все новые
участки окружности трубки. Образуется первоначально небольшая
трещина; трубку нужно слегка двигать вдоль пламени взад и вперед
в желательном направлении трещины; распространение трещины
можно ускорить, прикасаясь к накаленному участку слегка смочен-
ным пальцем. Если трубка очень широка, полезно предварительно
наметить направление трещины чернилами или мелом, так как в
противном случае легко повести трещину вкось.
Отрезанные края трубки должны оплавляться в пламени в тех
случаях, когда^они предназначены для сборки приборов.
Нагревание трубки. Вращение. При нагревании трубки необхо-
димо непрерывно и равномерно вращать ее вокруг оси в обе стороны.
Вращение трубки только в одну сторону требует попеременного
перехватывания трубки с обеих сторон, вследствие чего (если сере-
дина трубки уже размягчилась) могут появиться сдвиги стекла по
винтовой линии. Но при некоторой опытности вращение в одну
сторону предпочтительнее.
При вращении трубки руки располагают так, чтобы или оба локтя
опирались на плоскость стола, или предплечья опирались на перед-
ний край стола. В соответствии с этим подбирают высоту стола и
стула. Трубку берут с обеих сторон на таком расстоянии от обра-
батываемого места, чтобы и после окончательного размягчения стекла
можно было удерживать ее в правильном положении.
Если работают без резиновой трубки, как это практикуют стекло-
дувы-профессионалы, то левой рукой захватывают трубку сверху
так, чтобы указательный и средний пальцы оказались с одной сто-
48
роны, а большой — с другой. Мизинец и безымянный пальцы слу-
жат в качестве направляющих. Правую сторону трубки захваты-
вают снизу таким образом, чтобы указательный и большой пальцы
перекатывали трубку по концевым суставам остальных пальцев.
Вращают медленно и равномерно (примерно один оборот в две
секунды) и при каждой перемене направления движения на мгно-
вение останавливаются. Угол поворота должен быть не меньше 360°.
Время от времени трубку вынимают из пламени, не прерывая вра-
щения, чтобы дать время выравняться температуре. Равномерность
температуры можно контролировать и регулировать также по по-
стоянству окраски пламени и по равномерности свечения изделия
при полном его повороте в пламени.
Оттягивание трубок. Трубка нагревается в желаемом месте
при непрерывном вращении до размягчения, затем удаляется
из пламени и оттягивается. В зависимости от степени размяг-
чения и скорости растягивания получается та или иная толщина
стенок. Обычной ошибкой начинающих является недостаточное на-
гревание трубки; кроме того, они не дают стенкам трубки достаточно
утолститься («стопиться»), вследствие чего оттянутый конец полу-
чается чересчур тонкостенным. Если нужно оттянуть конец как у
пипетки, то нагревают трубку до образования значительного утол-
щения, стенки трубки медленно растягивают, затем дают охладиться
и отрезают. Если же трубку необходимо запаять, то оттягивают быст-
ро, чтобы на конце не собралось слишком много стекла, которое
потом все равно придется удалять.
Запаивание трубок. У узких трубок просто нагревают конец
при постоянном вращении до сплавления краев. Более широкие
трубки приходится перед запаиванием не
отрезать, а оттягивать. Оттянутый конец
участком а (фиг. 36) снова вносят в пла- । iu» < j
мя, правую часть снова оттягивают при I -— |\^
вращении. Затем оставшуюся часть вносят \ |
в пламя, время от времени вынимают и |\ --- ----- ц-/7
поддувают до тех пор, пока не образуется |
полушарообразное донышко той же тол- i
щины, что и стенки трубки.
Гораздо труднее сделать плоское дно. фиг- 36- Запаивание трубки.
В этом случае поступают по предыдущему, И й
но несколько больше нагревают утонченное дно до участка 6, при-
чем дно утолщается. Главное условие при этом — равномерное вра-
щение, иначе дно получается косым. В заключение донышко при-
жимают к плоскому древесному углю и поддувают внутрь. Затем
тщательно обогревают на пламени без дутья. Несмотря на эту предо-
сторожность, такое дно часто растрескивается.
Для временного затыкания трубок, которые потом будут разду-
ваться, подбирают ассортимент обрезков резиновых трубок, с одного
конца закрытых отрезком стеклянной палочки подходящего диа-
4 В. А. Соколов. 49
метра/ или набор корковых пробок различных диаметров, причем
очень пригодны вырезки, остающиеся после сверления пробок трубоч-
ными сверлами; часто пользуются также асбестом.
Выдувание шаров. Чтобы получить шар на конце трубки, ее за-
плавляют, нагревают необходимый участок и осторожно раздувают.
Раздувать нужно вне пламени, при постоянном вращении. Для полу-
чения больших шаров раздувают сначала небольшой толстостенный
шар, снова нагревают его по возможности равномерно и затем уже
окончательно раздувают до нужного диаметра.
Если нужно выдуть шар посреди трубки, то ее с одного конца
затыкают или запаивают, нагревают нужное место и, сжимая трубку
вдоль оси, утолщают при легком поддувании стенки трубки на нуж-
ную для образования шара величину, затем выдувают его вне пла-
мени при непрерывном вращении. В этом случае целесообразно
раздувать шар постепенно.
Спаивание трубок. Нужно иметь в виду, что края трубок хорошо
спаиваются между собой, если они изготовлены из одинаковых сор-
тов стекла.
Упражнения начинают со спаивания трубок одинакового диа-
метра, причем одну из них с одного конца затыкают, затем спаи-
ваемые концы хорошенько размягчают, удаляют из пламени, уве-
ренным движением соединяют уже вне пламени и при поддувании
слегка растягивают. При некотором опыте трубки спаиваются та-
ким образом с одного нагрева. Начинающему обыкновенно необхо-
димо снова нагреть место соединения и снова растянуть при легком
поддувании. Если спай оказался чересчур тонкостенным, то его на-
гревают и, слегка поддувая, осаживают. При всех этих манипуля-
циях необходимо не прекращать вращения трубки.
Широкие трубки трудно спаиваются с одного нагрева. В этом
случае поступают таким образом. Обогревают весь край, затем сильно
нагревают край в одном месте, соединяют с другой трубкой и зател!
шаг за шагом нагревают и сжимают трубки по всей окружности.
Если в спае окажутся отверстия, приготовляют в запас стеклянные
нити, которые можно быстро сплавить в каплю; этими каплями за-
клеивают отверстия. В заключение весь спай сильно нагревают и
слегка растягивают при слабом поддувании.
При спаивании трубок различных диаметров края более широкой
стапливают до тех пор, пока отверстие ее не будет соответствовать
просвету более узкой трубки, и затем соединяют размягченные концы,
как указано выше, при поддувании и растягивании. Таким обра-
зом спаивают трубки в том случае, если более широкая трубка —
тонкостенная, а узкая — толстостенная, как, например, у пипеток.
Если узкая трубка имеет очень малый просвет и толстые стенки,
то край ее расширяют следующим способом; на конце трубки выду-
вают толстостенный шарик, затем наружную половину его разогре-
вают, после чего при сильном вдувании наружная стенка шарика
лопается или ломается ножом и конец трубки приобретает вид, по-
казанный на фиг. 37.
50
Если более широкая трубка много шире или имеет толстые стенки,
то ее оттягивают до ширины более узкой трубки, следя за тем, чтобы
толщина стенок оттягиваемой части соответствовала припаиваемой
трубке. После этого оттянутый конец разре-
зают в подходящем месте и спаивают, как
указано выше.
Капилляры, которые необходимо спаять
с трубками другого диаметра, должны быть
раздуты на конце в шарик, который разла-
мывают так, чтобы оставался более или
менее правильный рант, как показано в раз-
резе на фиг. 37.
Фиг. 37. Расширение
капилляра.
Т-образные трубки (тройники). Прежде
всего нагревают на остроконечном пла-
мени то место трубки, к которому должен быть припаян отросток.
При этом в трубку сильно дуют, так что к моменту размягчения
в этом участке образуется отверстие. Размер пламени, а также
силу вдувания регулируют в зависимости от того, какого диаметра
должен быть отросток: чем толще отросток, тем больше пламя и
тем сильнее вдувание. Отверстие
Фиг. 38. Тройник.
должно быть несколько меньше диа-
метра припаиваемого отростка.
Затем оба спаиваемых участка
одновременно размягчают, соеди-
няют кратковременным прижима-
нием, а затем (при вдувании в них
воздуха) их слегка растягивают.
Если есть уже некоторый навык,
то спаивание на этом заканчи-
вается; для этого необходимо, чтобы
стекло было сильно прогрето и чтобы соединение производилось быстро
и уверенно. У начинающего обычно получаются наплывы и складки.
Их уничтожают, направляя на дефектное место остроконечное пламя,
нагревая, поддувая и снова нагревая до спадения до желаемых раз-
меров. Таким образом поступают до тех пор, пока не будут уничто-
жены все наплывы и складки; обыкновенно приходится размягчать
и раздувать главным образом оба входящих угла. Нафиг. 38,а пока-
заны правильная а и неправильная б формы спая; последняя рано
или поздно даст трещину. Все эти операции следует производить
с одного нагрева, стараясь не давать концам охладиться, так как
при вторичном разогревании они могут дать трещину. В заключение
все место спая и та часть трубки, которая лежит по другую сторону
отверстия, должны быть разогреты и слегка раздуты, а затем мед-
ленно охлаждены.
Сгибание трубок. Трубки до 5 мм лучше всего сгибаются на
горелке с плоским пламенем, где их и разогревают при постоян-
ном вращении до сгибания их под влиянием собственного веса.
4*
51
Если требуется согнуть под острым углом, то нагревают в пламени
паяльной горелки и сгибают при одновременном поддувании.
Так же поступают с широкими трубками. Начинающему довольно
трудно привыкнуть к сгибанию широких тонкостенных трубок; для
этого требуется большое пламя, на котором нагревают осторожно
и равномерно довольно длинный участок трубки; последний затем
сгибают при одновременном поддувании/ Легко образующиеся при
этом складки раздувают по окончании сгибания.
Пламя бунзеновской горелки совершенно непригодно для сги-
бания трубок: так как его внутренний конус значительно холоднее
наружного, то трубка размягчается на краях пламени сильнее, чем
в середине, что придает сгибу очень неправильную форму.
Отверстия в стекле. Отверстия в стеклянных изделиях делают
на стеклодувной горелке, для чего нужное место разогревают и
продувают. Если необходимо сделать очень маленькое отверстие, то
соответствующее место нагревают и продувают, не вынимая изде-
лие из пламени. Для получения больших отверстий продувание
производят вне пламени.
Отверстия в тонкостенном стекле делают с помощью заострен-
ного и раскаленного добела угля от дуговой лампы, которым и про-
тыкают изделие в желаемом месте.
Толстое стекло сверлят на токарном станке, или трехгранным
напильником, или, что лучше, заостренным стальным закаленным
прутиком, постоянно смачиваемым раствором камфоры в скипидаре,
или же латунной или медной трубкой, смазываемой кашицей из
наждака и воды.
Особенно нужно следить, чтобы при сверлении стекло не разогре-
валось, так как это служит причиной появления трещин.
Впаивание платиновой проволоки. В большинстве случаев пла-
тина вводится в кончик трубки. В пламени выдувают отверстие
такой величины, чтобы в него входила проволока (фиг. 39, а).
Затем вводят эту проволоку на место и нагревают спаиваемое
место и проволоку до окончательного оплавления отверстия. По
окончании этой работы раздувают кончик, как обычно. Чтобы
удобнее манипулировать с платиновой проволокой, припаивают ее
временно к отрезку стеклянной трубки, служащему рукояткой.
Лишние кусочки стекла удаляют с платиновой проволоки раздавли-
ванием плоскогубцами.
Важно следить, чтобы платина и стекло всюду давали прямой
угол; это дает уверенность в том, что впоследствии не появятся тре-
щины. В спайках, подобных изображенной на фиг. 39, б, часто по-
являются трещины.
Часто впаивают платину посредством особого («промежуточ-
ного») стекла. Это стекло, обычно содержащее свинец, отли-
чается вязкостью и способностью спаиваться с платиной и другими
сортами стекла. Содержащее свинец стекло нельзя обрабатывать в
восстановительном пламени. Охлаждение нужно вести над пла-
52
менем. Для впаивания меди, серебра, никеля употребляют эмаль
или синее промежуточное стекло, но прочность такого спая меньше,
чем спая с платиной. При употреблении эмали поступают по преды-
дущему, но оставляют отверстие диаметром 1—2 мм, проволоку в
надлежащем месте окружают каплей расплавленного промежуточ-
ного стекла, вдвигают ее в отверстие и нагревают до сплавления
(фиг. 39, в). Затем поддувают и медленно охлаждают.
Непосредственно с платиной спаиваются стекло № 23 завода
«Дружная горка», молибденовое стекло, а также йенское стекло
16-Ш.
Несколько труднее впаять платиновую проволоку сбоку трубки.
В этом случае нагревают избранное место добела, продувают, держа
Фиг. 39. Впаивание платиновой
г
проволоки.
в пламени, отверстие и дальше поступают, как описано выше
(фиг. 30, г).
Обработка кварца. Способы обработки кварца сходны со спо-
собами обработки стекла, но работу ведут в более горячем пла-
мени (с применением кислородного дутья или гремучего газа).
При этой работе глаза должны защищаться темнооранжевыми или
коричнево-черными очками. Поддувание ведут в пламени. Нагре-
вать можно быстро, медленного охлаждения не требуется. Платина
в кварц плотно не впаивается.
Стеклодувный монтаж и ремонт приборов с помощью ручной паяль-
ной горелки. Первоначальный монтаж, а также и ремонт больших га-
зоаналитических и главным образом вакуумных установок обычно про-
изводят с помощью ручных паяльных газовых горелок. При первона-
чальном стеклодувном монтаже отдельные стеклянные части укрепляют
на соответствующей раме или подставке, после чего эти части соеди-
няют между собой стеклянными трубками, производя спайку с по-
мощью ручной газовой горелки. При монтаже следует учитывать,
что деревянные штативы и подставки, особенно изготовленные из
сырого материала, в дальнейшем деформируются вследствие высыха-
ния, что часто ведет к поломке укрепленных на них стеклянных
частей. Особое внимание при монтаже следует обращать на установку
стеклянных баллонов с ртутью, обычно имеющихся почти во вся-
53
кой вакуумной газоаналитической установке. Подобные баллоны
должны укрепляться неподвижно, лучше всего непосредственно
на деревянных подставках, без каких-либо мягких прокладок.
Стеклянные части, расположенные выше баллона с ртутью и
спаянные с ним, не рекомендуется укреплять, как говорят, намертво.
В противном случае, т. е. при наличии мягкой прокладки и укреп-
ления намертво вышерасположенных частей, может впоследствии
произойти разрыв или поломка этих частей вследствие оседания
тяжелого, наполненного ртутью баллона по мере уплотнения про-
кладки или деформации всей подставки.
При монтаже или при ремонте установки отдельные части ее сле-
дует изготовлять обычным образом на стеклодувном столе, поль-
зуясь ручной горелкой только при соединении этих частей или для
запаивания трещин и отверстий. Для соединения двух частей уста-
новки эти части укрепляют на раме. Предварительно к одной из этих
частей припаивают стеклянную трубку необходимого диаметра и
такой длины, чтобы при соответствующем изгибании эта трубка до-
стигла другой спаиваемой части.
Удлиненная трубка одной из частей изгибается в пламени руч-
ной горелки так, чтобы ее конец вплотную подошел к трубке другой
спаиваемой части. Для изгиба трубки прогревают место изгиба по-
переменно то с одной, то с другой стороны, направляя соответству-
ющим образом пламя ручной горелки. Когда место изгиба доста-
точно размягчится, производят изгиб, раздувая в то же время это
место, чтобы не было наплывов и складок в стекле.
Если изгибаемая трубка слишком длинна, ее обрезают описан-
ным выше образом, а затем уже изгибают так, чтобы трубки соеди-
нились встык. Если изгибаемая трубка немного коротка, то, соот-
ветствующим образом прогревая и размягчая места изгиба или дру-
гих частей, трубку опять-таки подводят встык к другой спаи-
ваемой трубке.
Для спайки трубок на место их стыка направляют пламя ручной
горелки, попеременно нагревая место стыка с разных сторон. Когда
начнется размягчение концов трубок, их стараются еще более сбли-
зить так, чтобы заплавились все промежутки между концами тру-
бок. Это сближение и сжатие концов трубок можно производить
путем смещения той или иной спаиваемой части, которые для этой
цели не должны быть крепко соединены с рамой прибора или соот-
ветствующими держателями.
Часто случается, что сблизить и сжать размягченные концы тру-
бок не удается и между ними образуется или круговой просвет, или
отверстие той или иной формы. В этих случаях поступают следу-
ющим образом. Перед началом монтажа или ремонта изготовляют
(хотя бы в пламени той же ручной горелки) несколько сплошных
стерженьков стекла различных диаметров (1—3 мм). Когда между
спаиваемыми трубками образовалась щель или появилось отвер-
стие, берут один из таких стеклянных стерженьков и разогревают
его конец до расплавления, держа пламя горелки таким образом,
чтобы разогревалось одновременно и спаиваемое место. Это спаи-
54
ваемое место не следует греть слишком сильно. Нужно нагревать
его лишь настолько, чтобы края щели или отверстия не растека-
лись и не опадали, иначе щель и отверстие будут увеличи-
ваться. При подобном нагреве спаиваемого места щель или отвер-
стие замазывают расплавленным стеклом конца стерженька. Когда
вся щель или отверстие будут замазаны (хотя бы и наложением боль-
ших порций стекла стерженьков), все спаиваемое место постепенно
нагревают более сильно до полного размягчения и раздувают спай
так, чтобы не осталось в стекле ни наплывов, ни складок. При раз-
дувании спая поступают таким образом, чтобы несколько раз вду-
вание воздуха в спаиваемую трубку чередовалось с некоторым вса-
сыванием воздуха. Размягченное место спая попеременно то разду-
вается, то сжимается, что способствует слипанию мелких отверстий
и более равномерному распределению расплавленного стекла. Боль-
шие наплывы стекла следует расплавлять и удалять с помощью стек-
лянного стерженька, слегка разогретого во избежание его растре-
скивания в пламени горелки. Спаиваемое место после окончания
пайки следует отжечь, нагревая его все более и более слабо паяль-
ным пламенем горелки или пользуясь коптящим пламенем.
Аналогичным же образом запаивают щели, трещины и отверстия,
образовавшиеся в спаиваемых частях прибора. Если щель или от-
верстие невелики, их замазывают расплавленным стеклом, как это
описано выше. Если же щель или отверстие слишком велики, выре-
зают кусок трубки, припаивают новый, соединяют его встык и посту-
пают дальше вышеописанным образом.
ГЛАВА ВТОРАЯ
ОТБОР ГАЗОВЫХ ПРОБ.
ИЗВЛЕЧЕНИЕ РАСТВОРЕННЫХ И СОРБИРОВАННЫХ ГАЗОВ
§ 1. Отбор проб природных и промышленных газов
При отборе проб природных и промышленных газов могут встре-
титься следующие случаи:
1) газ каптирован, т. е. находится в трубопроводе, который имеет
манометрические или иные патрубки небольшого сечения с кра-
нами;
2) газ каптирован, но каптажная труба у выхода газа открыта;
3) газ не каптирован и свободно выделяется с поверхности земли;
4) газ не каптирован и свободно выделяется из воды минераль-
ного источника, озера, реки и т. п.;
5) газ каптирован, но находится в трубопроводе под вакуумом.
Пробу газа можно брать в стеклянные трубки (фиг. 40), в бу-
тылки и склянки (фиг. 40, д), аспираторы, газгольдеры или же в
металлические баллоны [6]. Отбор проб газа в какие-либо баллоны
из прорезиненной материи или из каучука совершенно недопустим
при условии пересылки пробы. Брать пробы газа в каучуковые бал-
лоны можно только, если исследование газа будет произведено вскоре
же после взятия пробы, если большой точности в результатах иссле-
дования не требуется и газ не содержит легко поглощаемых каучу-
ком соединений. Надежным способом хранения газовой пробы яв-
ляется запаивание газа в стеклянные баллоны (фиг. 40, в). Однако
запаивание баллона с газом часто не может быть применено. При
наличии в газе горючих компонентов и необходимости точного их
определения запайка нежелательна.
Если исследование газа будет произведено после недолгого хра-
нения пробы, вместо запаивания можно закрыть концы трубки с
газом толстостенным каучуком со вставленными в него оплавленными
стеклянными палочками (фиг. 40, б). Как на палочках, так и на стек-
лянной трубке должны быть оливки. Если газ будет исследоваться
не скоро и должен храниться продолжительное время, то трубки, за-
крытые каучуком, лучше не применять, так как через каучук хотя
и медленно, но всегда происходит диффузия газа.
Если требуется хранить газ продолжительное время, то пробу
следует или запаять в стеклянный баллон или набрать в стеклянную
56
трубку, имеющую на концах краны (фиг. 40, а), которые должны
быть хорошо притерты и хорошо закрыты при хранении газа, или
набрать в стеклянную бутыль и хранить ее в перевернутом состоя-
нии, закрыв пробкой, залив замазкой и оставив внутри некоторое
количество воды, служащее водяным запором. Можно хранить газ
под давлением и в металлических баллонах с хорошими вентилями.
На фиг. 40, г представлен тип газовых баллонов для длитель-
ного хранения газа и при необходимости очень точного анализа.
Баллон концом 7 присоединяется к источнику газа. В баллоне пред-
варительно создается высокий вакуум, а затем он заполняется га-
зом и соединительная трубка в суженной капиллярной части пере-
паивается. На другом конце баллона имеется шарик 2 с тонкими стен-
ками внутри более широкой трубки, имеющей боковой отвод. Чтобы
взять газ из баллона, этот боковой отвод припаивают к аналитиче-
скому прибору, а в трубку осторожно помещают железный цилинд-
рик с острым концом. Затем запаивают конец трубки и создают внутри
нее вакуум. С помощью магнита, подносимого снаружи к трубке,
поднимают железный цилиндрик и удаляют магнит. Цилиндрик
падает, разбивает стеклянный шарик, и газ из баллона без загряз-
нений может быть переведен в аналитический прибор.
В первом из перечисленных выше случаев, т. е. когда газ
каптирован, имеется патрубок с вентилем и в трубопроводе доста-
57
точное давление, чтобы из патрубка шла струя газа, набор пробы
производят следующим образом.
Если газ требуется набрать сухим способом, то на патрубок на-
девают один конец каучука, а другой конец сообщают с трубкой,
приведенной на фиг. 40 (я, б, в или г). Открыв вентиль, пускают
струю газа через трубку. Спустя некоторое время, когда весь воздух
будет уже вытеснен и в трубке будет находиться только газ из трубо-
провода, закрывают прежде всего вентиль трубопровода, затем за-
крывают краны трубки (а) или закрывают концы каучуков стеклян-
ными палочками (б) и т. п. Если через трубку прошел 20-кратный
объем газа, то можно считать, что в труб-
Фиг. 41. Отбор проб газа.
ке практически не осталось воздуха.
Когда нежелательно выпускать много
газа на воздух или когда давление в тру-
бопроводе очень мало, можно пользовать-
ся прибором, изображенным на фиг. 41.
После вентиля патрубка ставят трехходо-
вый кран и стеклянную трубку 7 одним
концом присоединяют к этому крану, а
другим соединяют с уравнительным сосу-
дом, в который налита ртуть или вода.
Поднимая уравнительный сосуд, заполняют
ртутью или водой всю трубку 7 до трех-
ходового крана. Поворачивая трехходовый
кран так, чтобы каучук, идущий к патруб-
ку, сообщался с атмосферным воздухом,
открывают вентиль и промывают газом
патрубок и соединительный каучук, затем,
повернув трехходовый кран, пускают газ
в трубку 7, понижая уравнительный сосуд.
После того как вся трубка 7 наполнится
газом, закрывают вентиль, а затем краны
трубки. Первую пробу газа рекомендуется, если требуется большая
точность, выпустить на воздух и забирать на анализ в трубку 7
лишь вторую или третью пробу. Если давление газа в трубопроводе
равно атмосферному или немного меньше его, то сосудом 7 пользуются
как насосом и перед набором пробы откачивают воздух из каучука,
идущего к патрубку.
При подобных операциях ртуть имеет то преимущество перед
водой, что она не растворяет газов в заметных количествах. Так как
растворимость различных газов в воде не одинакова, то при поль-
зовании водой для взятия пробы может измениться процентный хи-
мический состав газа вследствие большей растворимости одних га-
зов и меньшей растворимости других.
Если пробу газа берут в стеклянную бутыль с водяным запо-
ром, то на патрубок надевают каучук, другой конец которого сво-
боден или присоединен к изогнутой стеклянной трубке. Открыв вен-
тиль и промыв газом каучук, вставляют стеклянную трубку в горло
бутыли, как это указано на фиг. 42. Бутыль наполняют водой и опу-
58
скают вверх дном в какое-либо ведро или бадью. Вода, наполня-
ющая бутыль и ведро, должна быть предварительно насыщена тем
газом, проба которого берется, для чего газ некоторое время про-
пускают через воду. Если нельзя приготовить насыщенную газом
воду, следует пользоваться прокипяченной водой. Как только бу-
тыль наполнится газом, но в ней еще останется небольшое коли-
чество воды, ее плотно закрывают под водой резиновой или хорошей
корковой пробкой, вынимают из воды, не переворачивая, обтирают,
сушат и наклеивают этикетку.
При длительном хранении проб в большинстве инструкций реко-
мендуется заливка пробок менделеевской замазкой. Следует заме-
тить, что при наличии водяного затвора и хорошей пробки, не допу-
скающей утечки воды, это мероприятие не оказывает существенного
влияния на сохранность газа. Для макроанализа состав газа оста-
нется неизменным и без применения замаз-
ки. Что же касается микроаналитических
определений, то вообще по ряду причин
не рекомендуется длительное хранение
пробы до анализа. Заливка пробки замаз-
кой, обвязывание марлей и другие анало-
гичные мероприятия могут принести поль-
зу главным образом в отношении сохран-
ности самой пробки во время пересылки
и перевозки пробы.
Бутыль с газом следует пересылать и
перевозить в перевернутом виде так, чтобы
между газом и пробкой всегда был слой во-
Фиг. 42. Отбор пробы газа.
ды. Следует заметить, что в большинстве случаев в течение некоторого
времени (1—2 месяца) можно совершенно безопасно держать бутыль
и в нормальном положении, так как хорошо смоченная пробка, плотно
прилегающая к стенкам, действует так же, как и водяной запор.
Автору этой книги пришлось однажды анализировать пробы газов,
которые были набраны в бутыли с водяными запорами, но бутыли
в течение двух месяцев стояли в нормальном положении. Сравне-
ние анализов этих проб с анализом контрольных проб, сохранив-
шихся в перевернутом состоянии, показало, что практически нет
никакого различия в составе газа первых и вторых. При пользо-
вании каучуковыми пробками их следует вдавливать неглубоко,
чтобы при вскрытии можно было взяться за пробку руками.
Во всех случаях перед взятием пробы газа следует удостовериться
в отсутствии течи в применяемых приспособлениях (трубках, на-
сосе и т. д.).
Если при опущенном неподвижном уравнительном сосуде уро-
вень воды или ртути в насосе не понижается, значит течи нет.
Пробы газа можно брать также в стальные баллоны (для кисло-
рода, углекислоты, хлора), позволяющие взять большое количество
газа. Для наполнения таких баллонов употребляют компрессор,
но до давления 7—8 ат баллон можно наполнять и при помощи руч-
ного насоса. Газ несколько раз накачивают в баллон, а затем вы-
59
пускают, чтобы такой промывкой удалить из баллона воздух, а за-
тем уже окончательно наполняют и тщательно закрывают. При дав-
лении 7—8 ат из газа может выпасть газолин, поэтому проба газа,
взятая из баллона, не всегда будет одинакова по своему составу с
тем газом, которым наполнялся баллон.
Если у нас имеется второй из перечисленных выше случаев, т. е.
газ каптирован, но каптажная труба у выхода газа открыта, то для
взятия пробы газа лучше всего конец трубы закрыть деревянной
или иной пробкой, через которую пропустить стеклянную или метал-
лическую трубку (фиг. 43, а). На трубку надевают каучук, через
который и набирают пробу газа каким-либо из вышеописанных спо-
собов. Перед взятием пробы газ следует пропускать некоторое время
вхолостую для удаления воздуха, который может быть примешан
Фиг. 43< Отбор проб газа.
к газу в верхней части трубы. Выпускаемый вхолостую газ может
быть использован для насыщения воды, употребляемой при взятии
проб. При отсутствии пробки или при невозможности закрыть отвер-
стие вследствие большого давления или по каким-либо другим при-
чинам в скважину опускают воронку с надетым на нее каучуком
(фиг. 43, б), через который и набирают пробу. При сильном давлении
воронку" будет выкидывать; тогда вместо воронки берут стеклянную
60
Фиг. 44. Отбор пробы газа, находящегося
в трубопроводе под вакуумом. Отбор пробы
газа с запайкой в стеклянный баллон.
или металлическую трубку. Воронку или трубку следует опускать
возможно глубже.
Если газ не каптирован и свободно выделяется с поверхности
земли, то место выхода газа прикрывают воронкой, которую вдавли-
вают в землю и землей же засыпают с боков (фиг. 43, в). В этом слу-
чае, прежде чем набирать пробу, газ пропускают вхолостую для
промывки воронки и трубки от воздуха (20-кратный объем воронки).
Во всех случаях выпускать
газ вхолостую следует, опу-
стив конец каучуковой труб-
ки в воду.
При выделении газа с по-
верхности воды поступают
так, как изображено на фиг.
43, г. Стеклянную бутыль на-
полняют той водой, из кото-
рой выделяется газ, вставля-
ют в нее под водой воронку и
помещают над тем местом, от-
куда идут пузырьки газа. По-
сле наполнения бутыль с га-
зом под водой закрывают проб-
кой, оставив водяной запор.
Когда пробы газов берут
из газопроводов, газ иногда
находится под некоторым ва-
куумом. Схема отбора пробы
для этого случая изображена
на фиг. 44, а. Газ, находя-
щийся под вакуумом, посту-
пает через вентиль 3. Стек-
лянный насос 1 снабжен трех-
ходовым краном 2, попереч-
ный разрез которого изобра-
жен справа. Кран 2 соединен
одним концом каучуком с вентилем 3, через другой конец стеклян-
ные сосуды наполняют газом. Насос наполняют ртутью, а при не-
большом вакууме можно пользоваться водой. Открыв вентиль 3
и повернув кран 2 так, чтобы насос 7 сообщался с вентилем 3, опу-
скают уравнительный сосуд 4. Газ под вакуумом наполняет насос 7,
затем кран 2 закрывают. При поднимании уравнительного сосуда 4
газ будет выходить по трубке 5, конец которой следует погрузить в
роду. Кран 2 следует открывать, соединяя насос 7 и трубку 5 лишь
после того, как давление в насосе 7 превысит атмосферное. Повторяя
эту операцию несколько раз, промывают газом все соединительные
трубки, после чего приступают к наполнению газом склянки 6, для
чего ее помещают над концом трубки 5.
Если желательно набрать пробу газа сухим способом и запаять
ее в стеклянный баллон, то поступают так, как это изображено на
61
фиг. 44, б. Трехходовый кран 7 соединяют с сосудом 2, в который
набирают газ. Сосуд 2 должен иметь перетяжки для последующей
запайки. Предварительно поворотом крана 7 соединяют сосуд 2 с
атмосферным воздухом; подняв уравнительный сосуд 3 так, чтобы
ртуть вошла в канал крана 7, вытесняют таким образом из сосуда 2
весь воздух. Повернув кран 7 так, чтобы сосуд 2 сообщался с каучу-
ком, ведущим к вентилю 5, опускают сосуд 3 и засасывают в сосуд 2
воздух, находившийся в трубках, соединяющих кран 7 с вентилем.
Закрыв кран 7, поднимают сосуд и вытесняют откачанный воздух
наружу. Повторив эту операцию 2—3 раза, можно считать, что воз-
дух из соединительных трубок откачан. Открыв вентиль 5 и сооб-
щив его через кран 7 с сосудом 2, опускают сосуд 3 до тех пор, пока
ртуть не дойдет до нижнего края насоса 4. Затем закрывают кран 7
и, поднимая сосуд 3, вытесняют газ из насоса 4 в сосуд 2, после чего
производят запайку. Первую порцию газа, попавшую в баллон 2,
следует выпустить, промыв таким образом баллон 2 от следов воз-
духа, который может остаться в баллоне 2 при заполнении его ртутью
в виде пузырьков, приставших к стенкам. Запайку можно произво-
дить небольшим пламенем газовой или спиртовой горелки. При на-
личии в трубопроводе повышенного давления через трубку про-
пускают 20-кратный объем газа, вентиль трубопровода закрывают
и запаивают трубку: сначала дальний ее конец, а затем ближайший
к трубопроводу.
При проведении запайки следует помнить, что внутри запаивае-
мого баллона должно быть давление несколько меньшее, чем атмо-
сферное, тогда при нагревании перетяжка сама слипается и запайка
происходит легко. В противном случае, если давление в баллоне
превышает атмосферное, газ при нагревании прорывает расплавлен-
ное стекло и образуется отверстие, которое трудно запаять. Чтобы
атмосферным давлением не прорывало расплавленное стекло внутрь,
стенки перетяжки необходимо делать толщиной около 1 мм при диа-
метре отверстия, равном 0,5—0,7 мм, длина перетяжки не должна
быть менее 2—3 см.
Для откачки газа, находящегося под вакуумом, можно употреб-
лять насосы, имеющие емкость 300, 400 и 500 см3, т. е. требующие
для работы от 4 до 7 кг ртути. Применение воды в качестве вытесня-
ющей жидкости в насосе возможно только в тех случаях, когда дав-
ление газа немногим меньше атмосферного. Вакуум 0,5 ат соот-
ветствует уже 5 м вод. столба.
В некоторых случаях приходится отбирать пробы газа непосред-
ственно из осадочных горных пород, из почвенных или подпочвен-
ных отложений [7].
В этих случаях бурят мелкие скважины с помощью ручного бура
до глубины 1—3 м. После этого в скважину вставляют пробоотбор-
ник той или иной конструкции, над которым создается герметиза-
ция путем засыпки породой и заливания глинистым раствором. За-
тем с помощью водяного насоса, как это видно на фиг. 45, произво-
дится откачка почвенного или подпочвенного газа. Первая порция
откачиваемого газа отбрасывается, так как она .может содержать
62
примеси атмосферного воздуха; следующую порцию газа собирают
в бутыль, как это показано на фиг. 45.
Пробы газа из различных промышленных установок отбирают
также одним из упомянутых способов путем
отборного баллона, бутыли или какого-
либо откачивающего приспособления к
патрубку крана или вентиля установки.
При отборе проб газов, имеющих высо-
кую температуру, в частности газов из
печей, употребляют металлические холо-
дильники (фиг. 46), помещаемые перед
газоотборным баллоном или откачиваю-
щим приспособлением. Холодная вода вхо-
дит по линии 1 и выходит по линии 2.
Газ поступает в холодильник в направле-
нии, указанном стрелкой.
При взятии пробы воздуха и газа из
присоединения газо-
доступных помещений часто поступают еле- ♦
дующим образом. Берут стеклянный бал- ’
лон (см. фиг. 40, в), откачивают из него фиг 4б Отбор пробь, газз>
ВОЗДух хорошим насосом (хотя бы ДО имеющего высокую темпера-
0,1 лш) и закрывают его краны. Вносят туру.
баллон в то помещение, откуда требуется
взять пробу газа, затем открывают кран баллона, который и напол-
няется таким образом газом.
63
Пробу газа в доступном помещении можно также взять, напол-
нив предварительно баллон или бутыль водой и вылив воду в ис-
следуемом помещении.
Перевод пробы газа в бюретку. При переводе
газа из бутыли с водяным запором в бюретку прежде всего соскабли-
вают с пробки замазку, а затем пробку под водой вытаскивают што-
пором. Если в бюретке в качестве вытесняющей жидкости употреб-
ляется вода, то поступают так, как изображено на фиг. 47, а. Берут
каучук и один конец его надевают на трубку крана бюретки. Подня-
тием уравнительного сосуда заполняют водой всю бюретку и трубку,
идущую от крана. Эти операции необходимо проделать так, чтобы
в каучуке и трубках не осталось пузырьков воздуха. Для напол-
нения каучука водой без пузырьков применяют еще следующий при-
ем: каучук надевают на бюретку, а затем поднятием уравнительного
сосуда наполняют водой бюретку и каучук. При этом конец кау-
чука следует держать выше крана бюретки, в противном случае вода,
спускаясь по каучуку, всегда оставляет пузырьки воздуха. Если
уравнительный сосуд нельзя поднять достаточно высоко, то воду
засасывают ртом через конец каучука, причем трубку и в этом случае
следует держать выше крана, чтобы в направлении от крана к сво-
бодному концу каучук нигде не опускался ниже. Вставив конец кау-
чука или имеющуюся на каучуке стеклянную трубку в бутыль, от-
крывают кран бюретки и опускают уравнительный сосуд. В бюретку
первоначально попадет вода из каучука, а затем уже войдет газ.
64
Если в бюретке употребляется ртуть и необходимо избежать попа-
дания воды в бюретку, то между нею и каучуком, идущим к бутыли
с газом, следует поставить трехходовый кран, припаяв его к бюретке,
как это показано на фиг. 47, б, или присоединив на каучуке. Сво-
бодный конец трехходового крана присоединяют к водоструйному
или какому-либо иному насосу. Каучук и стеклянную трубку напол-
няют водой, трубку вставляют в бутыль с газом, а затем каучук на-
девают на один из концов трехходового крана. Предварительно под-
нятием уравнительного сосуда заполняют • ртутью всю бюретку и
трубку, идущую к трехходовому крану 7. Закрыв кран бюретки,
поворотом крана 7 соединяют водоструйный насос с трубкой, иду-
щей к бюретке, и откачивают из нее остатки воздуха. После этого
осторожно открывают кран так, чтобы водоструйный насос сооб-
щался с каучуком, идущим к бутыли с газом. Кран следует открыть
только немного, чтобы вода из каучука медленно начала двигаться
к крану 7, а затем по трубке, идущей к насосу. Когда в трубке, иду-
щей к насосу, около крана будет уже не вода, а газ, кран 7 повора-
чивают так, чтобы трубка, идущая к бюретке, была соединена с труб-
кой, идущей к бутыли. Медленно открывая кран бюретки, засасы-
вают в нее газ.
При взятии газа в бюретку в бутыль входит вода из ванны, кото-
рая до некоторой степени может изменить химический состав оста-
ющегося газа вследствие растворения отдельных компонентов этого
газа. Чтобы избежать поступления свежей воды в бутыль, стеклян-
ную трубку можно вводить в бутыль на каучуковой пробке. Вода из
ванны при этом не будет входить в бутыль, газ же из бутыли при-
дется откачивать.
Если проба газа набрана в баллон и закрыта каучуками со стек-
лянными палочками, то поступают так. Из каучука, идущего от бю-
ретки, имеющего на конце кран, выкачивают воздух, а канал и
трубку крана заполняют ртутью или водой. Затем под ртутью или
под водой вынимают стеклянную палочку, которой закрыт каучук
баллона, и присоединяют баллон к крану на конце каучуковой труб-
ки, идущей к бюретке. Открыв все краны, опусканием уравнитель-
ного сосуда забирают газ в бюретку. Второй конец баллона можно
оставить закрытым, тогда газ из него придется откачивать, или же
его можно открыть, тогда по мере выхода газа баллон будет напол-
няться ртутью или водой.
Когда газ заключен в баллон с кранами на концах, то его пере-
водят в бюретку способом, изображенным на фиг. 47, в. При помощи
трехходового крана откачивают воздух из соединительных трубок,
а затем, повернув кран баллона, забирают газ в бюретку. Другой
конец баллона можно оставить закрытым или опустить в воду или
ртуть, но при этом конец трубки, идущий от крана, тоже следует
заполнить водой или ртутью. Если кран свободного конца баллона
закрыт, то газ придется откачивать, если же свободный конец бал-
лона опущен в воду или ртуть или присоединен к уравнительному
сосуду, то весь газ за один прием можно перевести из баллона в бю-
ретку. Газ, заключенный в стеклянный, запаянный с обоих кон-
5 В. А. Соколов.
цов баллон, можно перевести в бюретку следующим образом. На
запаянных концах баллона делают напильником надрезы, а затем
на эти концы надевают каучуки (фиг. 47, г). Уравнительный сосуд 1
наполняют ртутью и, пользуясь трехходовыми кранами 2 и 3, отка-
чивают из соединительных трубок воздух, после чего обламывают
находящиеся в каучуках концы баллона. Открыв кран бюретки и
поднимая уравнительный сосуд 7, переводят газ из баллона в бю-
ретку.
§ 2. Извлечение растворенных газов
В аналитической практике часто требуется определить состав
газа, находящегося в растворенном состоянии в воде или иной жид-
кости. Количество этого газа в свободном состоянии, находящееся
над жидкостью, иногда бывает очень невелико, а потому для прове-
дения анализа необходимо прежде всего извлечь газ из того или
иного раствора.
Состав природных газов приходится изучать при исследовании
газов, растворенных в водах различных минеральных источников,
а также в буровых водах и в глинистом растворе буровых скважин.
При изучении газов, растворенных в водах минеральных источ-
ников, а также в буровых водах, при невозможности проведения
анализа на месте приходится отбирать пробу воды или раствора, а
затем перевозить эту пробу в лабораторию для извлечения газа из
раствора и анализа. При отборе проб воды минеральных источни-
ков, содержащих газ, а также проб буровых вод желательно этот
отбор производить таким образом, чтобы упругость газа соответство-
вала той его упругости, которая имеется на глубине залегания этих
вод. Для тщательного соблюдения этого условия применяют приспо-
собления, описываемые в специальной литературе [8].
Во всяком случае отбор пробы воды рекомендуется проводить
таким образом, чтобы растворенные газы не выделялись в атмосферу.
Для этого естественную струю воды или получаемую путем откачки
пропускают через бутыль для отбора пробы. Желательно, чтобы
через эту бутыль прошел объем воды не менее чем 4—5-кратный.
После этого бутыль будет содержать воду с тем количеством газа,
которое примерно соответствует его упругости в воде. После запол-
нения бутыли водой ее следует закрыть. Если проба не подвергается
немедленно дегазации, а пересылается в лабораторию и хранится
сравнительно долгое время, то необходимо принять меры, чтобы
повышение или понижение температуры не привело к порче пробы.
Расширение воды может выбить пробку, и таким образом проба будет
испорчена; с другой стороны, замерзание воды в бутыли точно так
же приведет к разрушению бутыли и порче пробы.
Для устранения этого рекомендуется в пробку бутыли вставлять
стеклянную трубку с резиновым наконечником, который также за-
полняют исследуемой водой. Резиновый наконечник закрывают стек-
лянной заглушкой. Небольшое расширение воды при повышении
температуры вызовет расширение резинового наконечника, но не при-
ведет к выталкиванию пробки. Чтобы устранить замерзание пробы,
66
Фиг. 48.
Откачка га-
за, раство-
ренного в
воде.
необходимо как ее пересылку, так и хранение проводить в помеще-
нии, имеющем температуру выше 0°.
После получения пробы в лаборатории требуется извлечь газ из
содержащейся в бутыли воды. Для наиболее полного извлечения
рекомендуется поступать следующим образом.
Стеклянную колбу откачивают хорошим насосом до достаточно
высокого вакуума. Резиновую трубку, присоединенную к бутыли,
пережимают зажимом. Стеклянную палочку (заглушку) вынимают
и соединяют резиновый отросток с колбой через трехходовый кран,
как это показано на фиг. 48. Соответствующим пово-
ротом крана, присоединенного одним концом к вакуум-
ному насосу, удаляют воздух из соединительного тол-
стостенного каучука. После этого открывают зажим и
имеющаяся в бутыли вода переливается в колбу. Не-
обходимо, чтобы объем колбы был больше объема бу-
тыли и чтобы таким образом вся вода могла попасть
в колбу. После того как вода попадает в колбу, ее не-
сколько подогревают и при этом откачивают газ через
кран с помощью ртутного насоса. Когда вся вода переве-
дена в колбу, то газ, разумеется, откачивают и из бутыли.
Откачиваемый газ направляется обычно в бюретку
для сохранения, замера и последующего анализа. Отка-
чивать газ из воды или раствора можно ртутным на-
сосом и без подогрева. Понижение давления при опу-
скании ртути вызывает вскипание жидкости, вследствие
чего происходит довольно быстрая и полная дегазация
раствора. Легкий подогрев ускоряет этот процесс.
Если не требуется точного определения количества
содержащегося в растворе газа, то поступают таким
образом. Пробу воды отбирают в бутыль, несколько раз
заполнив ее водой. Окончательно заполняют бутыль
водой не до самого верха, а лишь до горлышка, после
чего ее закрывают обыкновенной резиновой или хоро-
шей корковой пробкой. Желательно, чтобы в пробке
была стеклянная (или металлическая) трубка с каучу-
ком и заглушкой. В крайнем случае подобную пробку л
вить уже в лаборатории, хотя при этом небольшая часть газа может
потеряться. Пробу воды переливают затем в колбу и откачивают
как было описано выше.
Для извлечения газов из глинистых растворов, употребляемых
при бурении глубоких скважин, пробу раствора можно дегазировать
также вышеописанным путем. Следует только учесть, что глинистый
раствор может быть очень густым, поэтому перевод его в колбу и
извлечение газа сопряжены с некоторыми трудностями. Рекомен-
дуется применять в этом случае более широкие соединения. При
проведении подобных исследований глинистого раствора на присут-
ствие тех или иных газов наиболее рационально непрерывное извле-
чение газов из вытекающего при бурении глинистого раствора и ана-
лиз их (газовый кароттаж).
вста-
5*
67
Для наиболее полного извлечения газа глинистый раствор про-
пускают через металлический или стеклянный сосуд, нагреваемый
электрическим током через обмотку; выделяющийся газ откачивают.
Во многих случаях при газовом кароттаже не требуется знать пол-
ного количества растворенного газа; желательно извлекать и ана-
лизировать только тот газ, который свободно выделяется из глини-
стого раствора при его выходе на дневную поверхность из буровой
скважины.
Для этой цели применяют различные дегазаторы. Глинистый рас-
твор заставляют стекать по наклонно поставленным плоскостям,
при этом содержащиеся в нем пузырьки газа, а также частично рас-
творенные газы выделяются и увлекаются током воздуха в аналити-
ческий прибор, непрерывно регистрирующий содержание углеводо-
родных газов. Методы извлечения газа из глинистого раствора под-
робно описываются в специальной литературе по газовому карот-
тажу.
§ 3. Извлечение газов, сорбированных твердыми телами
Для извлечения газов, сорбированных твердыми телами, приме-
няют различные приспособления в зависимости от характера сорб-
ции и необходимой степени извлечения.
Для извлечения газа, который сравнительно слабо удерживается
на поверхности твердого тела, помещают это твердое тело (например,
горную породу) в стакан, изображенный на фиг. 49. Стакан закры-
Фиг. 49. Стакан для откачки газа из горной
породы.
вают герметичной крыш-
кой и через патрубок
крышки откачивают газ с
помощью ртутного насоса.
При этом применяют раз-
личной степени подогрев.
Следует учесть, что
очень сильный подогрев
осадочных пород, напри-
мер выше 100—120°, мо-
жет вызвать, как показали
проведенные опыты, разло-
жение некоторых соедине-
ний.
Необходимо также учесть, что при подобном способе дегазации
в откачиваемом газе будет находиться и некоторое количество атмо-
сферного воздуха,
Чтобы в горной породе сохранился этот легко выделяющийся
газ, требуется и соответствующая методика отбора проб. Поэтому
при отборе образец горной породы немедленно помещают в упомяну-
тый герметический стакан. Эти стаканы делают или металлические
с крышкой и прокладкой, или стеклянные с пришлифованной проб-
кой.
Если же необходимо извлечь газ, глубоко сорбированный, ука-
занных мероприятий (вакуума и небольшого подогрева) часто недо-
65
статочно для полного и быстрого извлечения газа. В этих случаях
исследуемую пробу сначала измельчают, а затем уже производят
основательный подогрев и откачку. При измельчении некоторая
часть газа может потеряться, поэтому многие авторы рекомендуют
производить измельчение в герметичном приспособлении (мельнице,
ступке). Как показали опыты, раздробление, измельчение и расти-
рание твердой породы или какого-либо другого материала сильно
способствуют более полному извлечению газа.
Для этой цели в герметически закрытую мельницу помещают обра-
зец породы или другого твердого 'тела и металлические шары или
куски металла неправильной
формы [9].
При вращении мельницы эти
металлические предметы ударя-
ются о твердые частицы поро-
ды, что способствует более пол-
ному извлечению газа. Выде-
ляющиеся газы по остановке
мельницы откачивают ртутным
насосом.
Вакуумная мельница для из-
влечения газов, разработанная
М. М. Элинсон и Ф. М. Чистя-
ковым, представлена схематиче-
ски на фиг. 50. Мельница имеет
чечевицеобразную форму и вра-
щается на двух подшипниках
вокруг своей оси. Мельница
Фиг. 50. Шаровая вакуумная мельница.
имеет крышку, которая с по-
мощью фланцев и резиновой прокладки соединена с основным чече-
вицеобразным корпусом. Крышка может наглухо привинчиваться
к корпусу, прокладка же обеспечивает полную герметичность мель-
ницы.
Твердые тела, из которых требуется извлечь газы, помещают
внутрь корпуса мельницы, куда кладут также стальные шары диа-
метром 40 мм. Внутрипериферическая часть мельницы выточена по
радиусу измельчающих шаров.
При определенной скорости вращения мельницы (80—100 об (мин)
измельчающие шары поднимаются под действием центробежной
силы в верхнюю часть мельницы, а затем падают, разбивая иссле-
дуемое твердо'е тело и ударяя по его кускам и обломкам. Выде-
ляющиеся при этом газы откачиваются из мельницы через газоот-
водную трубку, соединяющуюся с внутренней частью мельницы
через специальный сальник.
Как показали опыты, извлечение газа из твердого тела путем
ударов его стальными шарами весьма эффективно.
Для получения наиболее точных данных о содержании газа в
твердых телах рекомендуется поступать следующим образом. Ис-
следуемые образцы твердого тела, в частности образцы горной по-
того газа, который имеется
Фиг. 51. Извлечение газа из
твердого тела путем раство-
рения.
метичность. Точно так же
роды, уже с момента их отбора помещают в стеклянные баллоны с
притертой крышкой. Шлиф смазывают или густой высоковакуумной
смазкой или жидким стеклом, которое, застывая, обеспечивает гер-
метичность и прочность соединения. Подобная укупорка пробы де-
лается для того, чтобы в процессе переноски, перевозки и хранения
пробы не происходила ее дегазация.
Стеклянный герметично закрытый баллон с исследуемым веще-
ством помещают внутрь мельницы вместе со стальными шарами,
после чего мельницу закрывают и в ней через газоотводную трубку
создают вакуум. Это обеспечивает удаление практически всего вред-
ного объема воздуха, имеющегося внутри мельницы, не затрагивая
в породе, что позволяет в дальнейшем
более точно определить содержание
газа в исследуемом веществе и точнее
анализировать выделяющийся газ.
После создания вакуума мельниц}/
начинают вращать, для чего приме-
няют небольшой мотор с соответствую-
щей передачей. Стальные шары разби-
вают стеклянный сосуд и измельчают
при вращении мельницы исследуемые
образцы твердого тела. Выделяющиеся
газы откачивают ртутным насосом, объ-
ем их замеряют и направляют их в ана-
литический прибор. Сравнительно не-
большое количество воздуха, которое
имеется внутри стеклянного баллона
между кусками исследуемого твердого
тела, может быть учтено при анализе
газа по содержанию кислорода.
Описанная мельница до своего при-
менения должна быть испытана на гер-
ледует предварительно проверить мето-
дику дегазации путем проведения холостого опыта с пустым сте-
клянным баллоном.
В других случаях для полного извлечения рекомендуется раство-
рение твердого вещества. Если это вещество сравнительно легко
растворяется, то выделение газа из раствора под вакуумом при на-
греве является сравнительно легкой операцией и может быть произ-
ведено очень полно.
Для растворения применяют различные кислоты; вещества, ко-
торые в кислотах плохо растворяются, предварительно сплавляют
с содой и образовавшиеся карбонаты растворяют в соляной кис-
лоте.
Схема одного из подобных приборов ияобпажлна .на фшч
В одну ампулку 1 помещают твердое истолченное вещество, в ко-
тором требуется определить содержание газа; в другую ампулку 2
помещают раствор кислоты. С помощью соответствующих кранов
можно предварительно откачать из всей системы воздух, а затем,
70
повернув аппарат, присоединенный к аналитической установке на
шлифе 3, можно перелить жидкость из ампулки 2 в ампулку 7, где
и происходит растворение твердого тела. Баллончик 7 можно при
этом подогреть, выделяющийся газ частично сконденсировать в ло-
вушке с помощью глубокого охлаждения, а неконденсирующиеся
газы откачать ртутным насосом. Можно произвести откачку и без
глубокого охлаждения, но поставив по пути реагенты для погло-
щения воды и кислотных паров.
ГЛАВА ТРЕТЬЯ
ОБЩИЙ ГАЗОВЫЙ АНАЛИЗ
§ 1. Назначение общего газового анализа. Основные физико-
химические свойства газов, определяемых при общем анализе
(СО2, О2, Н2, СО, N2, простейшие углеводороды)
Общий газовый анализ предназначен для определения в газо-
вых смесях наиболее часто встречающихся при анализах газо-
образных элементов и соединений, а именно: углекислоты, кисло-
рода, окиси углерода, водорода, азота и простейших углеводород-
ных газов [10, 11].
Углекислоту, кислород, а иногда и окись углерода определяют
поглощением этих газов соответствующими реагентами. Водород,
окись углерода и углеводороды определяют обычно путем сожжения
с последующим замером изменения объема газа и продуктов сожже-
ния (СО2).
Азот определяют по разнице после удаления других упомянутых
выше газов. В некоторых случаях при общем анализе применяют
также раздельное определение непредельных и предельных угле-
водородов с помощью соответствующих реагентов.
Необходимо учитывать, что при общем анализе можно определять
только простейшие углеводороды, как метан, этан или этен. Если
в газе присутствуют также и более тяжелые углеводороды, как пре-
дельные, так и непредельные, указанная методика является неудо-
влетворительной. В этих случаях более или менее надежно можно
определять при общем анализе только суммарное содержание угле-
водорода путем сожжения.
При общем анализе газ забирают в бюретку, а затем направляют
в пипетку, содержащую реактив, поглощающий те или иные компо-
ненты газовой смеси.
После того как поглощение какого-либо компонента закончено,
газ забирают обратно в бюретку и оставшийся объем замеряют. По
разнице судят о содержании поглощенного компонента.
Методы сожжения, применяемые при общем анализе, заключа-
ются в том, что анализируемый газ в смеси с кислородом приводится
в контакт с раскаленной платиной, где и происходит полное сож-
жение газа с образованием в качестве конечных продуктов воды и
углекислоты. В других случаях газ пропускают над нагретой окисью
72
меди. При температуре около 300° сгорает окись углерода и водород,
а при температуре 700—800° происходит сожжение всех углеводо-
родов.
Существуют многочисленные приборы для общего газового ана-
лиза. Некоторые из них позволяют определить все упомянутые выше
компоненты, именно СО2, О2, СО, Н2, N2, СН4, С2Н6, С2Н4. Дру-
гие же приборы устроены таким образом, что позволяют определять
только некоторые компоненты. Эти приборы не имеют приспособле-
ний для сожжения и служат для определения таких газов, как кисло-
род, углекислота и окись углерода методом поглощения. Наконец,
есть приборы, в которых определяется только один какой-либо ком-
понент, например углекислота или окись углерода. Имеются также
приборы, в которых определяют только горючие газы, содержащиеся,
например, в воздухе, путем сожжения и поглощения образовавшихся
продуктов сожжения.
Ниже приводится описание основных химических и физических
свойств газов, определяемых при общем газовом анализе.
Свойства отдельных газов
Углекислый газ (СО2). Удельный вес углекислого газа
или углекислоты по отношению к воздуху равен 1,529. Вес 1 л при
0° и 760 мм рт. столба равен 1,9767 г. Углекислый газ в значитель-
ной степени поглощается водой, а именно: 1 л воды при 760 мм
рт. столба растворяет СО2 в л:
при 0°
» 5°
» 10°
» 15°
................. 1,713
..................1,424
................. 1,194
................. 1,019
при 20° 0,874
» 25°.................; . 0,759
>, 30°................ . 0,665
1 см1 * 3 серной кислоты (удельный вес 1,78) растворяет при 14° и
816 мм рт. столба 1,16 см3 углекислого газа. При температуре —78,5е
твердая углекислота испаряется, непосредственно переходя в газо-
образное состояние. При температуре —56,6° и 5,2 ат углекислота
переходит в жидкое состояние.
Углекислый газ получается в лабораториях обычно действием
соляной кислоты на мрамор (соляную кислоту берут разбавленной
в отношении 1 : 1).
Углекислота бывает загрязнена следами хлористого водорода,
сероводорода, кислорода и азота. Для очистки углекислого газа
его пропускают через трубку, наполненную кусками двууглекислого
калия или натрия, далее через трубку с кусками пемзы, пропитан-
ной раствором медного купороса, и затем для удаления кислорода
через нагретую докрасна кварцевую трубку с металлической медью.
Для очистки углекислого газа от азота его медленно пропускают
через сосуд, погруженный в жидкий воздух. Углекислый газ пере-
ходит в твердое состояние. После откачки хорошим насосом из со-
суда и соединительных трубок азота будем иметь чистый углекис-
лый газ, вынув сосуд из жидкого воздуха и дав ему нагреться.
73
Для объемного определения углекислоты в газовых смесях упо-
требляют раствор едкого кали. Для этой цели растворяют одну часть
твердого едкого кали в двух частях воды. 1 см3 раствора такой едкой
щелочи поглощает 40 см3 углекислого газа согласно реакции
СО2 + 2 КОН = К2СО3 + Н2О
При небольших количествах СО2 поглощение производят при
помощи титрованной баритовой воды и оттитровывают избыток
Ва(ОН)2 щавелевой или соляной кислотой. Индикатором при титро-
вании служит розоловая кислота или спиртовой раствор фенолфта-
леина.
Кислород (О2). Удельный вес кислорода по отношению к
воздуху равен 1,1053. При 0° и 760 мм рт. столба вес 1 л кислорода
равен 1,429 г. Температура кипения кислорода равна —183°, тем-
пература плавления —218°.
Кислород мало растворим в воде. 1 л воды растворяет следующие
количества (в см3) чистого кислорода, находящегося под давлением
760 мм рт. столба:
при 0°
о 5°
> 10°
> 15°
48,89 при 203........................31,03
........... 42,87 » 25°.................28,31
................................................. 38,02 » 303.26,08
.......... 34,15
Приведенные количества кислорода относятся к тому случаю,
когда вода соприкасается с чистым кислородом при 760 мм рт. столба.
Если же кислород является лишь составной частью того газа, кото-
рый находится над водой, то количества его, растворимые в воде,
будут меньше в соответствии с его парциальным давлением. Напри-
мер, если мы имеем над водой воздух при 760 мм рт. столба, то пар-
циальное давление кислорода будет равно 0,209 ат, так как содер-
жание кислорода в воздухе равно 20,9%. Чтобы узнать, какое коли-
чество кислорода из воздуха растворится в 1 л воды, необходимо
соответствующую величину для чистого кислорода умножить на
его парциальное давление. При 0° чистый кислород растворяется в
1 л воды в количестве 48,9 см3. Кислород воздуха растворится в 1 л
воды в количестве 48,9 х 0,209 = 10,2 см3.
1 л воды растворяет*следующие количества (в см3) кислорода
из воздуха:
при 0° 10,24
» 5° 8,98
» 10° 7,97
» 15° 7,16
при 20° 6,50
» 25’ 5,93
» 30° 5,47
Кислород в чистол! виде получают нагреванием марганцевокалие-
вой или бертолетовой соли или путем электролитического разложения
воды. При электролитическом получении кислорода берут дестил-
лированную воду, кипятят ее для удаления растворенного воздуха,
добавляют несколько процентов (10—15%) чистой серной кислоты
и наливают ее в прибор для электролиза. Сила тока не должна пре-
вышать 0,1 «на 1 см3 электрода. 1 а дает в течение часа около 2 л
74
кислорода. Электролитический кислород обычно содержит неболь-
шую примесь водорода, от которого можно освободиться, пропуская
газ через накаленную докрасна кварцевую трубку, наполненную
окисью меди. Водород при этом сгорает, проходящий же чистый кис-
лород следует высушить фосфорным ангидридом.
Содержание кислорода в газе можно определять при помощи
различных поглотителей, а именно: 1) щелочного раствора пирогал-
лола, 2) желтого фосфора, 3) гидросернистого натрия, 4) хлористого
хрома и 5) меди (при красном калении). Наиболее употребитель-
ными являются щелочный раствор пирогаллола и гидросернистый
натрий.
Для приготовления щелочного раствора пирогаллола берут 10—15 г
пирогаллола, растворяют в 30 см3 воды и смешивают с раствором
100 г едкого кали в 80 см3 воды. Скорость поглощения кислорода
этим раствором зависит от температуры. Содержащийся в 100 см3
воздуха кислород поглощается при температуре 20—25° в течение
нескольких минут. При низкой температуре поглощение идет зна-
чительно медленнее и при 0° часто не заканчивается даже после полу-
часового воздействия поглотителя. Щелочный раствор пирогаллола
поглощает также углекислоту, поэтому при определении кислорода
углекислота должна предварительно удаляться едким кали.
Поглощение кислорода желтым фосфором производят пропуска-
нием газа через влажный фосфор. Присутствие кислорода узнается
по образованию белого облака, исчезновение которого служит при-
знаком окончания поглощения. Поглощение кислорода фосфором
лучше всего протекает при 15—20°. При более низких температурах
для поглощения необходимо больше времени, при 0° оно почти пре-
кращается. Применение фосфора в качестве поглотителя кислорода
не всегда возможно, так как при некоторых условиях фосфор пере-
стает поглощать кислород. При содержании кислорода выше 60%
поглощения его фосфором не происходит. Если газ содержит следы
тяжелых углеводородов, эфирных масел, спирта, аммиака и т. п., то
поглощения кислорода фосфором также не происходит. Эти недо-
статки ограничивают применение фосфора в качестве поглотителя
для кислорода. По этой причине не рекомендуется применять фос-
фор для определения кислорода в природных газах, содержащих
углеводороды.
Например, смесь природного газа с воздухом, содержавшая около
40% метана, 37% этана и 23% воздуха, была введена в пипетку с
фосфором. В газе присутствовало также небольшое количество более
тяжелых углеводородов; никакого уменьшения в объеме смеси не
последовало даже после часового контакта с фосфором при 20°.
С течением времени желтый фосфор под действием света покры-
вается тонким слоем красного фосфора, который препятствует погло-
щению кислорода. Палочки желтого фосфора, покрывшиеся крас-
ным фосфором, должны удаляться из пипетки, расплавляться под
водой и переливаться в новые палочки.
Для приготовления раствора гидросернистого натрия берут 50 г
соли и растворяют в 250 см3 воды; сюда же добавляют 40 см3 рас-
75
твора едкого кали (5КОН 7Н2О). Преимуществом этого раствора
является то, что его можно употреблять как при низкой, так и при
высокой температуре; при всех условиях поглощение протекает
количественно в течение нескольких минут.
На поглощение кислорода гидросернистым натрием не влияет
присутствие тех газов, которые препятствуют подобной же реакции
кислорода с фосфором.
Реакция кислорода с гидросернистым натрием происходит по
уравнению
2 Na2S2O4+ 2Н2О О2 = 4NaHSO3
откуда следует, что 1 г Na2S2O4 может поглотить 64 см3 кислорода.
Кислород можно также определять путем сожжения или взрыва
его смеси с водородом. Сожжение протекает согласно уравнению
О2 + 2Н2 = 2Н2О
Таким образом, при сожжении одного объема кислорода исче-
зают три объема газа, так как образующиеся пары воды конденси-
руются и их объем ничтожен по сравнению с объемом газа, из кото-
рого они образовались.
Окись углерода (СО). Окись углерода имеет удельный
вес по отношению к воздуху 0,9672. При 0° и 760 мм рт. столба 1 л
СО весит 1,2504 г. Температура кипения окиси углерода равна —190е,
температура плавления —207°.
1 л воды растворяет следующие количества (в см3) окиси угле-
рода при давлении 760 мм рт. столба:
при 0°................35,3 при 20°...........23,2
» 5°................31,5 » 25°............21,4
» 10°...............281 » 30°............ . 20,0
» 15°................25,4
Окись углерода можно получить приливанием по каплям муравьи-
ной кислоты к нагретой до 150° серной кислоте. Серная кислота
отнимает от муравьиной воду, а остаток, представляющий собой
окись углерода, выделяется согласно уравнению
НСООН?— Н2О = СО
Для освобождения выделяющейся окиси углерода от паров воды
и от кислых паров ее пропускают через холодильник, конец кото-
рого входит в пустую склянку, служащую для собирания конденси-
рующихся паров, затем через раствор едкого кали и, наконец, для
окончательной осушки через трубку с фосфорным ангидридом.
Аналитическое определение окиси углерода в газе производится
или поглощением или сожжением.
Для абсорбции окиси углерода употребляют аммиачный раствор
однохлористсй меди, который приготовляют следующим образом:
растворяют 250 г хлористого аммония в 750 см3 воды и прибавляют
туда 200 г однохлористой меди (Сп2С12). Перелив раствор в склянку,
в него опускают несколько медных спиралей, закрывают пробкой
и после обесцвечивания раствора добавляют ]/з его объема аммиака
76
удельного веса 0,910. Аммиачный раствор следует хорошо взбол-
тать и дать постоять в течение нескольких часов, после чего он готов
к употреблению.
В поглотительную пипетку, содержащую аммиачный раствор
однохлористой меди, следует также опустить медную спираль для
сохранения реагирующей способности раствора.
Раньше для поглощения окиси углерода употребляли соляно-
кислый раствор однохлористой меди, но затем от него отказались,
так как поглощение окиси углерода солянокислым раствором не
может быть количественным.
Поглощение СО однохлористой медью происходит по уравнению
Сн2С12 + 2СО = Си2С12 2СО
Соединение Си2С12 • 2СО неустойчиво, и раствор, поглотивший
окись углерода, выделяет обратно некоторое количество ее при взбал-
тывании.
Поглощение окиси углерода аммиачным раствором происходит
количественно и необратимо по той причине, что образующееся со-
единение Си2С12 • 2СО реагирует дальше по уравнению
Си2С12 • 2СО 4- 4NH3 + 2НаО = Cu2 + 2COONH4 + 2NH4C1
Аммиачный раствор, бывший долго в употреблении, значи-
тельно менее энергично поглощает СО, чем свежий раствор, и может
даже выделять СО обратно, хотя и не так легко, как солянокислый
раствор. По этой причине при точном анализе рекомендуется сна-
чала поглотить большую часть СО старым раствором, а остатки свеже-
приготовленным или мало употреблявшимся.
Аммиачный раствор однохлористой меди помимо окиси угле-
рода поглощает также кислород, углекислоту, ацетилен, этен и фос-
фористый водород, которые должны быть удалены до определения
окиси углерода.
Окись углерода может также определяться пропусканием через
йодноватый ангидрид при температуре около 150°. Реакция проте-
кает по уравнению
J2O5 + 5CO = J2 + 6CO2
Выделяющийся иод оттитровывают 0,001 N раствором серно-
ватистонатриевой соли или же определяют количество образова-
вшейся углекислоты, объем которой равен объему бывшей в газе
окиси углерода. Этот метод особенно пригоден для определения ми-
нимальных количеств окиси углерода.
Йодноватый ангидрид реагирует с углеводородами, сероводоро-
дом, сернистым газом и другими газами, обладающими восстанови-
тельными свойствами. Эти газы должны удаляться до определения СО.
Сожжение окиси углерода может производиться или путем взрыва
или пропускание.м через раскаленные палладий, платину или окись
.меди.
77
При сожжении с окисью меди СО окисляется до СО2 за счет кис-
лорода окиси меди, во всех же остальных случаях для сожжения
прибавляют к СО кислород или воздух. Сожжение происходит по
уравнению
2СО + О2 = 2СО2
Следовательно, из трех объемов смеси СО и О2 получаются два
объема углекислоты. Объем образовавшейся углекислоты равен
объему первоначально имевшейся окиси углерода.
Для качественного обнаружения незначительных количеств окиси
углерода применяют бумажки, пропитанные раствором хлористого
палладия. В присутствии окиси углерода бумажка чернеет вслед-
ствие выделения металлического палладия. Этот способ нельзя при-
менять в присутствии водорода, ненасыщенных углеводородов и
сероводорода, от которых бумажка также чернеет.
Водород (Н2). Удельный вес водорода по отношению к воз-
духу равен 0,06962. При 0° и 760 мм рт. столба вес 1 л равен 0,08987 г.
Температура кипения водорода равна —253°, температура плавле-
ния —259°.
1 л воды растворяет при 760 мм рт. столба следующие количе-
ства (в о*3) водорода:
при 0°................. . 21,48
» 5°....................20,44
» 10°....................19,55
при 15°.....................18,83
» 20° .....................18,19
» 25°......................17,54
Чистый водород лучше всего получать электролизом дестилли-
рованной воды, подкисленной серной кислотой. Водород при этом
выделяется на катоде. Для очистки получающегося водорода от сле-
дов кислорода его пропускают через накаленную докрасна кварце-
вую трубку с платинированным асбестом.
Водород можно получить также действием разбавленной (1:6)
серной кислоты на цинк. Если цинк не вполне чист, то водород будет
загрязнен сероводородом, фосфористым и мышьяковистым водо-
родом. Для очистки водород пропускают через раствор едкого кали,
через раствор марганцовокислого калия, через нагретую докрасна
трубку с платинированным асбестом и, наконец, для осушки через
фосфорный ангидрид.
Водород можно определять поглощением или сожжением.
Поглотителем для водорода является раствор коллоидального
палладия и пикринонатриевой соли, к которому прибавлено немного
протальбиновокислого натрия в качестве защитного коллоида [14].
Водород, поглощаемый палладием, восстанавливает пикриновую ки-
слоту в триамидофенол
С6Н2 (NO2)3 ОН + 18Н = С6Н2 (NH2)3 ОН + 6Н2О
Для абсорбции водорода применяют также раствор 2 г коллои-
дального палладия и 5 г пикриновой кислоты, нейтрализованной
22 см3 1 N раствора NaOH. Раствор разбавляют водой до 100—
ПО ем3. Продолжительность процесса поглощения водорода рас-
75
твором коллоидального палладия составляет 10—30 мин. Предвари-
тельно из анализируемого газа должны удаляться углекислота, не-
насыщенные углеводороды, кислород и окись углерода. Этот метод
дает возможность определять водород в присутствии насыщенных
углеводородов. Раствор приведенной выше концентрации в состоянии
поглотить около 4 л водорода. Раствор коллоидального палладия
стоит сравнительно дорого, но зато он может служить очень долго,
так как его можно регенерировать.
Как только поглотитель начинает ослабевать, его выливают из
пипетки, споласкивают пипетку несколько раз водой и эту воду при-
бавляют к поглотителю. К этому разбавленному раствору погло-
тителя прибавляют по каплям разбавленную серную кислоту до тех
пор, пока не прекратится выделение осадка. Избытка кислоты сле-
дует избегать, в противном случае палладиевый золь под влиянием
кислорода воздуха перейдет в раствор в виде сернокислого палла-
дия. Осадок, который, кроме твердого палладия и протальбиновой
кислоты, содержит еще не прореагировавшую пикриновую кислоту,
промывают водой, причем в раствор переходит немного пикриновой
кислоты, палладий же не растворяется. После этого осадок, суспен-
дированный в небольшом количестве воды, растворяют, прибавляя
по каплям раствор едкого натра. После прибавления соответству-
ющего количества пикрата натрия получается раствор, опять год-
ный к употреблению.
Смешивая газ, содержащий водород, с воздухом или с кислоро-
дом, можно произвести сожжение его несколькими способами, а
именно: путем взрыва, путем пропускания через нагретый палла-
дий или накаленную платину и путем пропускания через накален-
ную трубку с окисью меди. Во всех этих случаях сожжение проис-
ходит согласно уравнению
2На + О2«2НаО
т. е. при сгорании 2 объемов Н2 уменьшение объема смеси равно 3/2
объема Н2, так как образующаяся вода практически не занимает ни
какого объема по сравнению с объемом газа. При сожжении с окисью
меди исчезнувший объем газа равен объему водорода. При взрыве,
при пропускании газа через накаленную платину и т. п., кроме
водорода, будут сгорать также окись углерода и углеводороды.
Для поглощения водорода употребляют также губчатый палла-
дий и палладиеву чернь.
Если желательно определить весь содержащийся в газовой смеси
водород, как свободный, так и связанный, можно пропустить газ
через накаленный магний. Углерод, кислород, азот и другие эле-
менты фиксируются магнием, а водород выделяется в свободном сот
стоянии.
Азот (N2). Удельный вес азота по отношению к воздуху
равен 0,9673. При 0° и 760 мм рт. столба вес 1 л равен 1,2505 г.
Температура кипения азота равна —195,8°, температура плавле-
ния —209,8°.
79
1 л воды при 760 мм рт. столба растворяет азота (в см3):
при 0°
» 5°
» 10°
- 15°
23,59 пои 20°...................15,98
21,03 » 25°...................14,89
18,95 » 30°...................13,98
17,31
Чистый азот лучше всего получать нагреванием концентриро-
ванного водного раствора азотистокислого калия и хлористого
аммония в молекулярном отношении и пропусканием выделяюще-
гося азота через накаленную медь для удаления следов окислов азота.
В большинстве случаев при газовом анализе азот определяют по
разности. После поглощения отдельных газов различными реаген-
тами и после сожжения горючих газов остаток принимается за азот.
В действительности же, кроме азота, в остатке могут быть редкие
газы: гелий, аргон, неон, ксенон и криптон.
Наилучшим поглотителем для азота является металлический каль-
ций при температуре около 700° и металлический литий при 400°.
Метан (СН4). Удельный вес метана по отношению к воздуху
равен 0,5545. При 0° и 760 мм рт. столба вес 1 л равен 0,7168 г.
Температура кипения метана равна —161,4°, температура плавле-
ния —184°. 1 л воды при 760 мм рт. столба растворяет метана (в см3):
при 0° 55,63
» 5° 48,05
» 10° 41,77
при 20°...................30,08
» 25°....................30,06
» 30°....................27,62
Искусственным образом метан получается из йодистого метила,
на спиртовый раствор которого действуют цинком с осажденной на
нем медью. Реакция протекает по уравнению
2CH3J + 2Zn + 2НОН = ZnJ2 + Zn (OH)2 + 2CH4
Для получения цинка, покрытого медью, гранулированный цинк
обливают несколько раз разбавленной серной кислотой, а затем
.2%-ным раствором медного купороса, после чего цинк промывают
водой и спиртом.
Для очистки метана, получаемого из йодистого метила, его про-
пускают через дымящую серную кислоту и через едкое кали, а для
окончательной осушки — через фосфорный ангидрид.
Так как для метана неизвестны никакие поглотители, то его опре-
деляют или путем сожжения или разгонкой при низких температурах.
Сгорание метана происходит по уравнению
CH4-j-2O2 = СО2 + 2Н2О
т. е. на один объем сгоревшего метана идет два объема кислорода и
образуется один объем углекислоты. Помимо метана при сожжении
газа будут сгорать также водород, окись углерода и все имеющиеся
в газе углеводороды. Когда в газе имеется более двух углеводоро-
дов, то эти углеводороды не могут быть определены путем сожже-
ния. В этом случае при анализе обычно применяют разгонку при
низких температурах.
80
Этан (С2Н6). Удельный вес этана по отношению к воздуху
равен 1,0491. При 0° и 760 мм рт. столба 1 л этана весит 1,3562 г.
Температура кипения этана —88,3°, температура плавления —172°.
В 1 л воды при 0° и 760 мм рт. столба растворяется 47 см3 этана.
В лаборатории этан можно получить или электролизом уксусно-
калиевой соли, или действием металлов на органические галоидные
соединения
2СН3 J + 2Na = 2Na J + С2Н6
В наиболее чистом виде этан может получаться путем разгонки
содержащего этан природного газа при низких температурах.
Пропан (C3Hg). Удельный вес пропана по отношению к воз-
духу равен 1,562. При 0° и 760 мм рт. столба 1 л пропана весит 2,02 г.
Температура кипения пропана равна —42,2°, температура плавле-
ния —191°.
Пропан может получаться или действием металлов на галоид-
ные соединения, или (при желании получить пропан в наиболее чи-
стом состоянии) разгонкой природного газа при низкой температуре.
Последним же способом производится определение пропана.
Бутан (С4Н10). Удельный вес бутана по отношению к воз-
духу равен 2,07. При 0° и 760 мм рт. столба 1 л бутана весит 2,6726 г.
Температура кипения бутана —0,6°, температура плавления —135°.
Бутан получается или электролизом соли пропионовой кислоты,
или действием металлов на галоидные соединения, или лучше всего
разгонкой природного газа при низких температурах.
У бутана имеется изомер — изобутан. Температура кипения изо-
бутана —12,2°, температура плавления —145°.
Бутан и изобутан определяются в газе путем разгонки при низ-
ких температурах.
Этен (С2Н4). Уд. вес этена по отношению к воздуху равен 0,9748.
При 0° и 760 мм рт. столба 1 л этена весит 1,2603 г. Температура ки-
пения этена —103,9°, температура плавления —169,4. 1 л воды рас-
творяет этена (в см3):
при 0° .............. 226 при 15°..............139
» 10° ...............162 » 20°................122
Абсорбирующими веществами для этена являются дымящая сер-
ная кислота (содержащая 20—25% SO3) и бромная вода. Оба эти
реактива обычно употребляют для поглощения всех непредельных
углеводородов. При желании определить отдельно газы группы этена
следует пользоваться разведенной бромной водой (0,1 N). Для опре-
деления этена в присутствии ацетилена можно пользоваться аммиач-
ным раствором азотнокислого серебра, который поглощает ацетилен
целиком, а этен в очень незначительной степени. Этен можно опре-
делять также путем сожжения.
Ацетилен (С2Н2). Удельный вес ацетилена по отношению к
воздуху равен 0,912. При 0° и 760 мм рт. столба 1 л ацетилена весит
1,1791 г. При температуре —83,6° твердый ацетилен возгоняется,
непосредственно переходя в газообразное состояние. При темпе-
6 В. А. Соколов 81
5 °'
ратуре —81,8° и при давлении 760 мм рт. столба ацетилен переходит
в жидкое состояние. 1 л воды при нормальных условиях растворяет
1 л ацетилена.
Абсорбирующим веществом для ацетилена являются дымящая
серная кислота и насыщенная бромная вода. Разведенная же бром-
ная вода (OJ N) в присутствии соляной кислоты действует очень
медленно и дает возможность определять ацетилен объемным путем
в присутствии этена.
Ацетилен поглощается аммиачным раствором азотнокислого сере-
бра и аммиачным раствором однохлористой меди.
Аммиачный раствор азотнокислого серебра приготовляют рас-
творением 10 г AgNO3 в 500 см3 дестиллированной воды, куда при-
бавляют НС1 до слабокислой реакции, а затем аммиак до щелоч-
ной реакции.
В качестве поглотителя ацетилена и его гомологов в присутствии
этена и предельных углеводородов применяют иногда раствор
иода, содержащий 25 г иодистой ртути и 30 г йодистого калия
в 100 см3 воды, к которому прибавлено немного едкого кали; 1 см3
этого раствора может поглотить 20 см3 ацетилена.
§ 2. Методика общего газового анализа
Общий анализ газа заключается в последовательном поглощении
его компонентов и в измерении оставшегося объема.
После этого производят сожжение газа, определение образо-
вавшейся углекислоты и измерение негорючего остатка.
Методика общего анализа в первоначальном виде заключалась
в том, что бюретка, наполненная анализируемым газом, присоеди-
нялась к пипетке, содержащей тот или иной реагент. Газ впускался
в пипетку, а после окончания поглощения переводился опять в бю-
ретку, где и измерялся оставшийся объем. Разница между перво-
начально взятым объемом газа и получившимся после поглощения
соответствует содержанию той составной части газа, которая погло-
тилась в данной пипетке.
Пипетки, применяемые для поглощения, имеют разную форму
в зависимости от свойств поглотителя (фиг. 52). Пипетки с двумя
шарами (фиг. 52, а) применяются в тех случаях, когда наливаемый
в них раствор не реагирует с атмосферным воздухом. Во всех пипет-
ках от нижнего шара идет капиллярная трубка, внутренний диа-
метр которой 1—2 мм. Простая пипетка, изображенная на фиг. 52, а}
применяется главным образом для определения углекислого газа,
для чего в нее наливают раствор едкого кали. Раствор должен запол-
нить весь нижний шар, капилляр и очень небольшую часть верх-
него шара. Емкость нижнего шара должна быть несколько больше
емкости бюретки с анализируемым газом. Емкость верхнего шара
также должна быть больше емкости бюретки. На оба конца пипетки
надевают каучуковые трубки с зажимами, которые должны быть
закрыты, когда пипетка не употребляется.
Пипетки, имеющие четыре шара, употребляются для таких рас-
творов, как пирогаллол, гидро сернистый натрий и аммиачный рас-
52
твор однохлористой меди, которые поглощают кислород из воздуха.
Раствор, имеющийся в шаре 7 и в шаре 2 (фиг. 52, б), служит для
поглощения газа, а тот же самый раствор, находящийся в осталь-
ных двух шарах, служит гидравлическим запором и не допускает
соприкосновения первого раствора с воздухом. При анализе трубка,
идущая вверх из последнего шара, должна быть открыта, поэтому
при каждом наполнении пипетки анализируемым газом в послед-
ний шар поступает новая порция воздуха. Пирогаллол и другие
растворы можно, конечно, употреблять и в простых пипетках, но в
этом случае продолжительность службы этих растворов будет не-
велика вследствие постоянного поглощения кислорода воздуха.
• Для твердых поглотителей, как фосфор, применяют пипетку,
изображенную на фиг. 52, в. Первый сосуд, имеющий цилиндриче-
скую форму, оканчивается внизу отверстием, через которое в сосуд
помещают палочки фосфора. Отверстие затем закрывают каучуковой
пробкой и наполняют пипетку водой.
Фиг. 52. Пипетки, применяемые при общем газовом анализе, и пи-
петка для твердых поглотителей.
Для производства анализа испытуемый газ набирают в бюретку
той или иной конструкции. В качестве вытесняющей жидкости в
бюретке при анализе, не требующем большой точности, употребляют
воду. Так как в результате анализа должно определяться процент-
ное содержание в газе каждого компонента, то для упрощения рас-
четов удобнее всего начинать анализ, имея в бюретке ровно 100 см3
газа при атмосферном давлении. Чтобы набрать в бюретку ровно
100 см3 газа, поступают следующим образом. До присоединения
бюретки к пипетке (фиг. 53) поднятием уравнительного сосуда вытес-
няют из бюретки весь воздух или какой-либо газ, после чего напол-
няют бюретку исследуемым газом, пользуясь для этого приемами,
указанными в предыдущей главе. Следует взять газа несколько боль-
ше 100 см3, именно 102—103 см3, при атмосферном давлении. Закрыв
кран 7, медленно поднимают уравнительный сосуд до тех пор, пока
вода в бюретке не дойдет до метки 100 см3, в этот момент закрывают
кран 2. Таким образом, в бюретке будет ровно 100 см3 газа, но под
давлением, немного большим атмосферного. Открыв кран 7, сооб-
щают бюретку на секунду с воздухом; при этом весь излишек газа
(свыше 100 см3) выйдет наружу. После закрытия крана 7 будем иметь
б* 83
в бюретке ровно 100 см3 газа при атмосферном давлении, что и про-
веряют, открыв кран и измеряя объем газа обычным способом.
Перед присоединением к бюретке уровень поглотителя в пипетке
следует довести до каучука на капиллярной трубке или до какой-
либо метки.
После наполнения бюретки газом
как это изображено на фиг. 53, при
Фиг. 53. Соединение пипетки и бюретки
при общем газовом анализе.
ее присоединяют к пипетке,
помощи отрезков каучуков и
капиллярной трубки (фиг.
53, а). Капиллярная трубка
должна присоединяться к бю-
ретке и к пипетке встык и
каучуки должны надеваться
плотно. Присоединив трубку,
изображенную на фиг. 53, а,
к пипетке, сжимают каучук,
находящийся на другом кон-
це соединительной трубки
(или бюретки), пальцами так,
чтобы из него вытеснить воз-
дух, и делают второе кау-
чуковое соединение.
Заметив положение рас-
твора в капилляре, переводят
газ из бюретки в пипетку,
для чего, подняв уравнитель-
ный сосуд, открывают кран 7.
Газ входит в нижний шар
пипетки, вытесняя поглоти-
тель в верхний шар. Когда
уровень воды в бюретке дой-
дет до крана 7, кран 2 (или 7)
закрывают и оставляют газ
в пипетке на некоторое вре-
мя. После этого, открыв
кран 7 или кран 2 и опуская
уравнительный сосуд, перево-
дят газ обратно из пипетки
в бюретку, доводя раствор
пипетки до первоначального
положения в капилляре. Эту
операцию перепускания газа из бюретки в пипетку и обратно следует
повторить несколько раз, отмечая остающийся объем газа после
каждой операции. Если при двух следующих одна за другой опера-
циях не будет уменьшения объема, то поглощение можно считать
оконченным. Опустив уравнительный сосуд, газ окончательно пере-
водят в бюретку и, закрыв кран 7, измеряют оставшийся объем. Так
как первоначально газа было взято 100 см3, то число поглощенных
кубических сантиметров будет равно процентному содержанию ком-
понента, поглощаемого в данной пипетке.
84
Сначала газ пускают в пипетку с раствором едкого кали, кото-
рый поглощает углекислый газ. Это поглощение происходит очень
быстро и целиком заканчивается после 2—3 операций впускания
газа в пипетку.
Следует учесть, что одновременно с углекислотой раствором
едкого кали будут поглощаться и другие газы кислотного харак-
тера (H2S, SO2 и др.).
После определения содержания углекислоты в газе пипетку с
едким кали отъединяют от бюретки и на ее место ставят другую с
бромной водой или дымящей серной кислотой для определения не-
предельных углеводородов (при наличии в газе пропана, бутана и
других это определение будет неточным). Непредельные углеводо-
роды поглощаются бромной водой или дымящей серной кислотой
довольно быстро, а именно, в течение 3—4 мин., при легком взбал-
тывании раствора в пипетке. Перед окончательным отсчетом остав-
шегося объема бюретку присоединяют опять к пипетке с едким кали,
газ переводят на одну минуту в пипетку, а затем опять в бюретку,
где и определяют оставшийся объем. Газ впускают в пипетку с ед-
ким кали для удаления паров серной кислоты или брома, которые
вызывают увеличение объема газа.
После непредельных углеводородов в газе определяют кислород,
присоединяя к бюретке пипетку с раствором пирогаллола, гидро-
сернистого натрия, фосфора или другого поглотителя. Поглощение
кислорода пирогаллолом происходит сравнительно медленно, и чем
ниже температура, тем больше времени требуется для полного погло-
щения. Газ в пипетке с пирогаллолом оставляют в течение 5 мин.
и затем, переведя в бюретку, замечают оставшийся объем; далее
опять впускают газ в пипетку и оставляют его там в течение
3 мин. Если произошло дальнейшее уменьшение объема, газ
опять вводят в пипетку и продолжают это делать до тех пор,
пока два последовательных измерения объема не окажутся оди-
наковыми.
При невысокой температуре поглощение происходит очень мед-
ленно. В этих случаях рекомендуется помещать пипетку (стеклян-
ную ее часть) в ванну с теплой водой.
Гидросернистый натрий также применяют для поглощения кисло-
рода; скорость поглощения в меньшей степени зависит от темпера-
туры.
Поглощение кислорода можно производить влажным фосфором
(для чего пипетку с фосфором заполняют водой) только в том случае,
если в газе нет тяжелых углеводородов. Присутствие кислорода в
газе узнают по образованию белого облака, а исчезновение послед-
него служит признаком окончания поглощения. Если в газе слишком
много кислорода (свыше 60%), то он не поглощается фосфором. Не
рекомендуется также употребление фосфора в том случае, если в
газе имеется и кислород и значительное количество водорода, так
как при этом бывают взрывы.
После определения кислорода к бюретке присоединяют пипетку
с аммиачным раствором однохлористой меди для определения СО,
85
а затем производят сожжение газа, присоединяя специальные пипетки
для сожжения1.
При переведении газа из пипетки в бюретку следует соблюдать
осторожность и опускать уравнительный сосуд медленно, чтобы по-
глотительный раствор из пипетки ни в коем случае не попал в соеди-
нительную трубку, ни тем более в бюретку. Если поглотитель в ка-
пилляре пипетки разделен пузырьками газа, то, чтобы перевести
эти пузырьки газа в бюретку, следует, слегка приподнимая и опуская
уравнительный сосуд, заставлять поглотитель подниматься и опу-
скаться по всему вертикальному отрезку капилляра пипетки. Пузырь-
ки газа при этом всплывают наверх.
Если при всех манипуляциях какой-либо из поглотителей попал
в соединительную трубку или в бюретку, то перед следующим ана-
лизом трубку и бюретку необходимо тщательно вымыть и наполнить
бюретку чистой водой. В противном случае поглотитель, попавший
в воду бюретки, будет реагировать с газом раньше, чем газ будет
впущен в пипетку с тем же поглотителем, что может повести к очень
большим ошибкам.
Все пипетки после разъединения с бюреткой должны быть за-
крыты каучуком со стеклянными палочками или зажимами.
Если за время анализа температура газа и атмосферное давле-
ние оставались постоянными, то нет надобности делать приведение
газа к 0° и 760 мм рт. столба. Если же температура и давление меня-
лись, то как начальный объем, так и объем газа, остающийся после
каждого поглощения, следует привести к 0° и 760. мм рт. столба по
формуле
V V(B-W)
760(1 +at) 3
где Vo — объем газа при 0° и 760 мм рт. столба;
V — наблюдаемый объем при барометрическом давлении В
и температуре /;
а —коэфициент расширения газа, равный .
Так как бюретка наполнена водой и газ всегда насыщен водяными
парами, то для получения действительных объемов отдельных со-
ставных частей газа (без водяных паров) из барометрического дав-
ления следует вычитать величину W, представляющую собой упру-
гость водяных паров при температуре /. Упругость паров зависит
от температуры.
Применение воды в качестве вытесняющей жидкости в бюретке
имеет свои недостатки, поэтому анализ, произведенный над водой,
нельзя считать точным^ хотя для большинства технических целей он
является вполне удовлетворительным.
Все газы до некоторой степени растворимы в воде, что ведет к
значительным неточностям в анализах газовых смесей. Как только
газ соприкасается с водой, все составные части начинают растворять-
ся в воде в соответствии с коэфициентом растворимости и с парци-
1 Способы сожжения описаны ниже.
альным давлением; в то же время газы, находившиеся в воде, будут
стремиться выделиться из нее. Таким образом, происходит газовый
обмен между водой и газовой смесью. Влияние растворимости газов
особенно заметно по отношению к углекислоте и непредельным угле-
водородам. Эти газы растворяются значительно больше азота, водо-
рода, метана и др. Для устранения погрешностей, связанных с рас-
творимостью газа, в бюретке вместо воды употребляют раствор пова-
ренной соли или сернокислого натрия. Растворимость газов в этих
растворах меньше, чем в воде.
Помимо растворения в воде газы растворяются и в абсорбентах.
Непредельные углеводороды, кислород, азот и метан растворяются
до некоторой степени в едком кали и т. д. Чтобы по возможности,
уменьшить ошибки, происходящие от этого растворения, рекомен-
дуется сделать три или четыре анализа газа для насыщения абсор-
бентов. Результаты четвертого или пятого анализов того же газа
будут наиболее точными.
Несмотря на все эти предосторожности, употребление жидких
реагентов и воды в бюретке ведет к неизбежным ошибкам. Насыще-
ние абсорбентов газом может практически исключить ошибки, про-
исходящие вследствие растворения в них газов, но это очень услож-
няет анализ. Насыщение же воды (или раствора NaCl) в бюретке
анализируемым газом не достигает своей цели, так как хотя оно и
дает возможность сделать правильный первый отсчет объема газа,
но по мере поглощения компонентов состав остающейся газовой
смеси будет меняться, а поэтому первоначальный газ, растворенный
в воде, частично перейдет в оставшийся после поглощения и про-
исходящий газовый обмен поведет к дальнейшим ошибкам.
К недостаткам воды, применяемой в качестве вытесняющей жид-
кости в бюретке, относится также ее свойство смачивать стенки бю-
ретки. Если мы закроем верхний кран бюретки и сделаем два от-
счета объема газа: один сейчас же после закрытия, другой через
3—4 мин., то заметим между ними разницу, происходящую от того,
что за время стояния вода, бывшая на стенках бюретки, стекла вниз.
Таким образом, правильный отсчет объема газа может быть сделан
лишь спустя несколько минут после переведения газа в бюретку.
Из всего вышеприведенного следует, что очень точный анализ
газа не может быть сделан, если в бюретке употребляется вода.
При более точном анализе в качестве вытесняющей жидкости
в бюретке применяется ртуть. Точно так же для уменьшения погреш-
ностей, происходящих от растворения газов в абсорбентах, в точном
анализе употребляют пипетки, в которые наливают ртуть, а на по-
верхность ртути — небольшое количество абсорбента. При точном
анализе применяют также бюретки с компенсационным приспособле-
нием для автоматической поправки на изменение температуры и
давления. Поскольку анализируемый газ приводится в соприкосно-
вение с растворами, имеющими различную упругость паров, для
получения более точных результатов все измерения объема газа сле-
дует проводить при насыщении газа водяными парами. Для этой
цели в бюретку с ртутью вводят 1—2 капли дестиллированной воды,
87
которая и насытит своими парами газ. Количество введенной воды
будет слишком мало, чтобы растворить в себе заметное количество
газа.
Описанная выше методика применяется в разнообразных приборах
для общего анализа. В приборах, употребляемых в настоящее время,
поглотительные пипетки имеют несколько иное, более совершенное
устройство. Кроме того, ряд таких пипеток соединяют между собой
и с бюреткой с помощью стеклянной трубки с кранами, что предста-
вляет значительное удобство при анализе. Описание этих приборов
дано в § 3.
Сожжение газа
Сожжение газа применяется для определения водорода, окиси
углерода и углеводородов группы метана.
Сожжение этих газов происходит по следующим уравнениям:
2Н24-О2 = 2Н2О
2СО 4-О2 = 2СО2
СН4+2О2 = СО2+ 2Н2О
2СаНв + 7О2 = 4СО2 + 6Н2О
С3Н8 4- 5О2 = ЗСО2 + 4Н2О
Иногда сожжением определяют также и непредельные углеводо-
роды, которые сгорают согласно уравнениям
2С2Н2 4- 5О2 = 4СО2 4- 2Н2О
С2Н4 4- ЗО2 = 2СО2 4- 2Н2О
При сожжении газа всеми способами, кроме сожжения с окисью
меди, к нему прибавляют кислород или воздух. Как видно из при-
веденных уравнений, продуктами горения являются углекислый
газ и вода. Объем воды, образовавшейся при горении, практически
ничтожен по сравнению с объемом газа, поэтому его обычно не при-
нимают во внимание. Объем же образовавшейся углекислоты всегда
меньше объема сгоревшего газа вместе с израсходованным на это
горение кислородом. Поэтому после сожжения всегда наблюдается
уменьшение объема газовой смеси (газ 4- кислород) или так назы-
ваемая «контракция». Объем образовавшейся углекислоты, объем
пошедшего на сожжение кислорода и контракция являются харак-
терными величинами для каждого газа. Знание этих величин дает
возможность определить содержание составных частей сжигаемого
газа.
При сгорании водорода объем его равен двум третям наблюден-
ной контракции, так как на два объема водорода требуется один
объем кислорода.
При сгорании окиси углерода ее объем в два раза больше кон-
тракции, так как на два объема СО требуется один объем О2, в ре-
зультате же сожжения образуются два объема углекислого газа.
55
Объел! образовавшегося углекислого газа равен объему сгоревшей
окиси углерода.
При сгорании метана его объем равен половине контракции, а*
объем образовавшейся СО2 равен объему метана.
При сгорании этана его объем равен двум пятым контракции, а
объем образовавшейся СО2 в два раза больше объема сгоревшего
этана.
По приведенным уравнениям подобным же образом легко вычис-
лить зависимость между объемол! сгоревшего газа, контракцией,
количеством образовавшейся углекислоты и количеством истрачен-
ного на сожжение кислорода.
По этой зависимости вычисляют процентное содержание горю-
чего газа в смеси.
Если мы имеем смесь углеводородов, то определить отдельные
составные части не всегда возможно при помощи сожжения. При
сожжении газа известны обыкновенно следующие величины:
1) объем газа, равный сумме объемов отдельных составных ча-
стей;
2) количество пошедшего на сожжение кислорода;
3) количество образовавшейся углекислоты;
4) контракция.
Приведенные величины не являются независимыми. Контракция
есть функция количества кислорода и углекислоты, а количество
углекислоты находится в функциональной зависимости от коли-
чества кислорода. Таким образом, сожжение дает только два неза-
висимых уравнения, а потому при помощи сожжения в этол! случае
можно определить только два углеводорода. Следовательно, можно
определить при помощи сожжения процентный состав газовой смеси,
содержащей не более двух углеводородов, при условии, что формула
каждого компонента известна.
Предположим, что имеется смесь метана и этана. Обозначим про-
центное содержание метана через х, процентное содержание этана
через у, контракцию через Ук, а объем образовавшейся углекислоты
через С. Будем иметь
VK = 2x+-|-y,
G = x + 2y,
откуда легко вычисляются х и у.
Если, кроме горючих газов, в смеси есть и негорючие, например
азот, то они определяются по разности после сожжения и после по-
глощения углекислоты и оставшегося кислорода.
При сожжении с окисью меди построение уравнений несколько
иное, так как в этом случае к газовой смеси не прибавляют ни
кислорода, ни воздуха. При сожжении водорода с окисью меди кон-
тракция будет равна объему сгоревшего водорода. При сожжении
метана объем газа до и после сожжения останется одинаковым, так
как вместо сгоревшего метана образуется такой же объем углскнс-
89
лого газа. Соответствующим образом меняется уравнение контрак-
ции и для других горючих газов.
Для сожжения газа употребляют следующие приборы: 1) пи-
петку для взрыва, 2) пипетку для сожжения, 3) платиновый капил-
ляр, 4) трубку с палладиевым асбестом или палладиевой чернью
и 5) трубку с окисью меди.
Фиг. 54. Пипетка для взрыва газа и пипетка для сожжения с платиновой
проволокой.
Пипетка для взрыва изображена на фиг. 54, а. Она состоит из
толстостенного стеклянного шара со впаянными в него двумя пла-
тиновыми электродами. Верхняя часть шара заканчивается капил-
ляром. Нижняя часть при помощи толстостенного каучука соеди-
няется с уравнительным сосудом. Чтобы произвести сожжение газа,
к нему прибавляют избыточное количество кислорода (или соответ-
ствующее количество воздуха), измеряют в бюретке объем полу-
чившейся смеси, а затем вводят в пипетку для взрыва, у которой
шар и капилляр предварительно заполнены ртутью. К электродам,
впаянным в пипетку, присоединяют провода от вторичной обмотки
индукционной катушки; при пускании тока в первичную обмотку
между электродами проскакивают искры и происходит взрыв газа.
Если газ слишком разбавлен негорючими составными частями, то
при взрыве полного сгорания не происходит; полное сгорание про-
исходит, когда на 30 частей горючего газа приходится не более 100
частей негорючего газа. При употреблении для взрыва газов кисло-
рода может образоваться значительное количество окислов азота,
что поведет к заметной ошибке.
После охлаждения газ из пипетки переводят в бюретку, изме-
ряют получившийся объем и определяют образовавшееся количе-
ство углекислоты. По этим данным вычисляют процентное содержа-
ние составных частей газа по вышеприведенным уравнениям.
Пипетка для сожжения изображена на фиг. 54, б. Через каучу-
ковую пробку в нижней части пипетки проходят две стеклянные
трубки, целиком заполненные ртутью. В верхние концы трубок
90
впаяна платиновая проволока, идущая спиралью; концы платино-
вой проволоки погружены в ртуть, находящуюся в трубках. В ниж-
ние концы стеклянных трубок также впаяны проволоки, через ко-
торые подводится ток. При сожжении газа в пипетку первоначально
вводят измеренное количество кислорода. Объем сжигаемого газа
измеряют в бюретке. Пускают ток по платиновой спирали, доводя
ее до красного каления и регулируя накал специальным реостатом,
и впускают постепенно в пипетку сжигаемый газ. Когда весь газ
впущен, оставляют его при накаленной спирали в пипетке несколько
минут. После прекращения тока и охлаждения газа его переводят в
бюретку и поступают так же, как и в предыдущем случае.
Сожжение газа при помощи платинового капилляра изображено
на фиг. 55. Капилляр представляет собой толстостенную платиновую
трубку с очень узким каналом. На обоих концах платинового капил-
ляра находятся медные холодильники, наполненные водой. Сжигае-
мый газ вместе с кислородом забирают в бюретку 7. Платиновый
капилляр накаливают несветящейся частью газовой горелки. Если
после многократного пропускания газа из бюретки в пипетку 2 че-
рез накаленный капилляр не будет больше уменьшения объема,
то сожжение можно считать оконченным.
В некоторых случаях для сожжения газа применяют кварцевукг
трубку, содержащую много витков платиновой проволоки, нака-
ливаемой электрическим током.
Фиг. 55. Платиновый капилляр для сожжения газа.
Для сожжения газа употребляют также палладиевый асбест.
Этот способ интересен тем, что он дает возможность сжечь водород
отдельно от метана. Смесь газов пропускают через накаливаемую
стеклянную трубку, в которой находится палладиевый асбест. При
нагревании палладиевого асбеста до 200—300° водород количественно
сгорает в воду, метан же не изменяется Ч Сожжение производится,
1 Разные авторы указывают различные температуры начала окисления ме-
тана (от 150 до 400°), поэтому действие катализатора, зависящее от условий его
приготовления, должно предварительно проверяться.
91
как изображено на фиг. 55, с той только разницей, что вместо пла-
тинового капилляра помещают капиллярную стеклянную трубку с
палладиевым асбестом. При этом способе сжигаемый газ следует
смешивать не с кислородом, а с воздухом, в противном случае часть
метана обязательно сгорит.
Для приготовления палладиевого асбеста растворяют 3 г хлори-
стого палладия в возможно малом количестве воды, приба-
вляют 3 см3 насыщенного на холоду раствора муравьинокислого
натрия и соды до сильно щелочной реакции, а затем 1 г длинных
волокон мягкого асбеста, которые и всасывают всю жидкость. Во-
локна, пропитанные жидкостью, сушат на водяной бане, причем на
асбесте выделяется равномерно черный, мелко раздробленный пал-
ладий :
Na2PdCl4-b HCOONa = 3NaCl + НС1 + CO2+Pd
Соляная кислота нейтрализуется углекислым натрием; после
полной просушки на водяной бане полученную массу размягчают
теплой водой, переносят на воронку и промывают теплой водой до
удаления приставших к асбесту солей. Полученный палладиевый
асбест затем сушат и сохраняют в хорошо закрывающейся банке.
В капиллярную трубку вставляют лишь небольшое волокно пал-
ладиевого асбеста.
Вместо палладиевого асбеста можно употреблять палладиевую
проволоку длиной 50—60 см, сложенную три раза. При нагревании
трубки с палладиевой проволокой до 400° происходит полное сгора-
ние водорода и окиси углерода, а метан остается неизменным.
Отдельное сожжение водорода в газовой смеси можно произвести
также при помощи палладиевой черни. Если пропускать смесь водо-
рода, метана и воздуха с избытком кислорода над палладиевой чернью
при температуре не выше 100°, то сгорает только водород.
Для сожжения газа применяют также пористые материалы, на
которые нанесен платиновый катализатор (см. ниже).
Сожжение газа может также производиться при помощи окиси
меди. Трубку из тугоплавкого стекла или кварца внутренним диа-
метром 5—6 мм и длиной около 20 см наполняют кусочками медной
тонкой проволоки, покрытой окисью меди. При прохождении горю-
чего газа через нагретую трубку окись меди отдает свой кислород,
за счет которого и происходит сожжение. Таким образом, в этом спо-
собе нет надобности прибавлять к газу кислород или воздух, что во
многих случаях является большим преимуществом. При нагревании
окиси меди до 280—300° сгорают только водород и окись углерода.
Сгорание предельных углеводородов производят при 800°.
При сожжении газа путем взрыва, накала платиновой проволоки
и в платиновом капилляре сжигаются нацело все горючие газы.
Остальные способы позволяют вести дробное сожжение, именно сжи-
гать водород и окись углерода отдельно от метана. Следует заметить,
что для анализа природного горючего газа методом дробного сож-
жения наиболее пригодна окись меди при температуре 280—300°.
Способы сожжения с палладиевым асбестом, палладиевой прове-
рь
локой и т. д. не применимы для анализа природного горючего газа.
Присутствие в природном газе тяжелых углеводородов меняет ха-
рактер сожжения. При температуре сгорания водорода и окиси угле-
рода всегда сгорают частично и тяжелые углеводороды.
Если природный горючий газ желательно сжечь целиком, то
лучше всего газ сжигать в пипетке для сожжения или в трубке с
окисью меди при температуре 800—900°. При избытке кислорода в
пипетке газ сгорает нацело, без образования окислов азота. При
пропускании газа несколько раз через накаленную окись меди сго-
рание происходит практически также нацело. Преимуществом сож-
жения с окисью меди является то обстоятельство, что при этом спо-
собе нет надобности смешивать газ с кислородом или воздухом, так
как это смешивание служит источником ошибок: кислород не всегда
обладает 100%-ной чистотой, а воздух не всегда содержит точно
полагающееся количество кислорода (20,9%). Недостаток полного
сожжения с окисью меди заключается в том, что при этом способе
требуется продолжительное пропускание газов через окись меди
и трубка с окисью меди должна быть из кварца или из очень туго-
плавкого стекла, трудно поддающихся обработке в обычных лабо-
раторных условиях. Недостатком пипетки для взрыва является
неполное сгорание, если горючий газ сильно разбавлен негорючим.
Кроме того, при большом избытке кислорода образуются окислы
азота.
Платиновый капилляр не всегда удобен при сожжении угле-
водородного природного газа. Сожжение углеводородов в капил-
ляре происходит не полностью. Капилляр засоряется углеродом,
выделяющимся в результате неполного сгорания газа.
Окись меди при сожжении газа частично восстанавливается до
металлической меди. Обратный перевод меди в окись меди дости-
гается пропусканием воздуха через накаленную докрасна трубку.
При анализе газов методом сожжения, если требуется максималь-
ная точность, необходимо вводить поправки- на неравенство моле-
кулярных объемов различных газов. Граммолекулярный объем или,
иначе говоря, объем, занимаемый молекулярным весом газа, выра-
женным в граммах, для идеального газа при нормальных условиях
равен 22,41 л. Газы, с которыми приходится иметь дело при сож-
жении, дают некоторые отклонения от этой величины. Например,
молекулярный объем углекислого газа составляет 99,4% молеку-
лярного объема окиси углерода. Уравнение сгорания окиси угле-
рода может быть представлено в следующем виде:
2СО + О2 = 2СО2
т. е. 2 молекулярных объема СО -h 1 молекулярный объем О2 дают
2 молекулярных объема углекислого газа. Так как молекулярный
объем СО2 составляет только 99,4% от молекулярного объема СО,
то в приведенное уравнение должна быть введена поправка, а именно,
при приведенной реакции образуются не 2 объема СО2, а (2 х 0,994) =
= 1,988. Исправленное уравнение будет иметь вид:
2СО + О2= 1,988 СО2
93
Подобным же образом составляют уравнения и для других слу-
чаев сожжения газа, причем следует пользоваться величинами моле-
кулярных объемов газов. Если парциальное давление газа меньше
одной атмосферы, то газ по своим свойствам ближе к идеальному.
Поэтому молекулярный объем газа меняется в зависимости от пар-
циального давления смеси.
В табл. 7 приведены молекулярные объемы углекислого газа и
этана в зависимости от парциального давления при 20°. Приведен-
ные в табл. 5 величины даны по отношению к молекулярному объему
идеального газа. Молекулярные объемы метана при давлении около
1 ат и ниже практически мало отличаются от молекулярного объема
идеального газа.
Таблица 7
Молекулярные объемы СОа и СаНв
при 20°
Парциаль- ное давле- ние, мм рт. столба Молекулярные объемы
со2 С2Нв
100 0,9993 0,999
200 0,9986 0,997
300 0,9980 0,996
400 0,9972 0,995
500 0,9965 0,993
600 0,9958 0,992
700 0,9951 0,991
760 0,9947 0,990
Приготовление платинового катализатора [12]
Определение пористости носителя. Платина
наносится на керамическую массу в виде кусочков обычных фарфо-
ровых пористых тарелок или пластинок, применяемых в органиче-
ских лабораториях для отжигания кристаллических препаратов.
Можно пользоваться керамической массой из каолина. Для нане-
сения определенного количества процентов платины необходимо
знать пористость носителя. Пористость определяют обычным техни-
ческим методом. Навеску кусочков в 10—15 г погружают в воду на
24 часа, после чего их вынимают из воды, быстро и осторожно обсу-
шивают с помощью фильтровальной бумаги и взвешивают. Привес
показывает, сколько кубических сантиметров воды (или соответ-
ственного водного раствора) может быть впитано определенным ко-
личеством носителя. Все взвешивания производят на технико-хими-
ческих весах. Обычно работают с носителями, обладающими пори-
стостью около 30%.
Приготовление катализатора. Носители про-
питывают раствором платинохлористоводородной кислоты H2PtCl6,
94
получаемой путем растворения платины в царской водке. После
растворения металла раствор несколько раз выпаривают досуха с
соляной кислотой, пока не разложится вся бывшая в растворе азот-
ная кислота. Затем перед пропиткой носителя платинохлористо-
водородной кислотой раствор разбавляют водой до определенного
объема в зависимости от пористости носителя. Раствор разбавляют
так, чтобы впитываемый носителем объем его содержал то коли-
чество платины, которое требуется нанести на носитель.
Пример. При изготовлении 10%-ного платинового катализатора и при пори-
стости носителя в 30% следует считать, что 100 г носителя поглотят 30 см* рас-
твора H2PtClfi. Поэтому крепость раствора должна быть такова, чтобы в каж-
дых 30 см* его содержалось 10 г платины (считая на металл).
Перед пропиткой носитель дробят на куски желаемого размера. Пропитку
ведут в течение 12—14 час. Носители погружают в горячий раствор H2PtCl6r
которому затем дают остыть. После пропитки раствор H2PtCle сливают, про-
питанные носители сушат на водяной бане и затем восстанавливают. Восста-
новление нужно вести в струе электролитического водорода, поместив носители
в стеклянную трубку, обогреваемую трубчатой электропечью.
Температура должна медленно повышаться до 120—140°. Процесс восста-
новления длится 8 час. и больше. Конец реакции узнается по прекращению выде-
ления хлористого водорода; после охлаждения в атмосфере водорода катализа-
тор можно считать готовым.
§ 3. Приборы, применяемые при общем газовом анализе
пипетки для сожжения.
J 2 /
Фиг.
общего
56. Прибор для
анализа.
Чтобы произвести полный общий анализ газа вышеописанным
образом, необходимо иметь ряд отдельных приборов, а именно: бю-
ретку, набор пипеток для поглощения и
При анализе приходится много раз
присоединять и отъединять бюретку
от пипеток, что представляет боль-
шие неудобства, так как отни-
мает время и требует большой осто-
рожности. При неаккуратной работе
в каучуки и капилляры можно на-
брать некоторое количество воздуха,
что поведет к неточным результатам
анализа.
Современные приборы для общего
анализа основаны на конструкции,
данной Орса. С течением времени в
этот прибор многими исследователями
были внесены различные усовершен-
ствования, и в настоящее время из-
вестно большое количество приборов,
основанных на этом принципе, но отличающихся друг от .друга в дета-
лях. На фиг. 56 изображен простой прибор с тремя поглотительными
пипетками. Бюретку 5 присоединяют к стеклянному капилляру, от
которого отходят также три капиллярные трубки с кранами (так назы-
ваемая «гребенка»). Идущие вниз от кранов капилляры при помощи
каучуков соединены с пипетками, наполненными стеклянными тру-
95
бочками, служащими для увеличения поверхности поглотителя,
что ускоряет процесс поглощения. К прибору присоединен на кау-
чуке капиллярный трехходовый кран, разрез которого изображен
на фиг. 56 отдельно. Пипетка, служащая для поглощения, состоит
из двух соединенных между собой цилиндрических сосудов; к крану
присоединен тот сосуд, который наполнен стеклянными трубочками.
Другой сосуд закрыт каучуковой пробкой с проходящей через нее
стеклянной трубкой. На трубку надет каучук, оканчивающийся
каучуковым же мешочком.
Перед проведением анализа в уравнительный сосуд бюретки нали-
вают воду или ртуть (ртуть наливают, если требуется большая точ-
ность, причем на поверхности ртути в бюретке должно быть немного
воды). В пипетки наливают соответствующие растворы. В пипетку 7
наливают раствор едкого кали, в пипетку 2 — раствор пирогаллола,
в пипетку 3 — аммиачный раствор однохлористой меди. Эти погло-
тители наливают в том случае, если в газе определяют СО2, О2 и СО.
Если определяют иные составные части газа, то берут и другие по-
глотители. Растворы наливают в пипетки через горлышко при от-
крытом кране 4 и открытом кране пипетки. Когда уровень раствора
в пипетке будет стоять немного выше середины, то, закрыв кран 4
так, чтобы не было сообщения атмосферного воздуха с капиллярами,
и открыв кран бюретки, опускают уравнительный сосуд (предвари-
тельно уровень воды в бюретке следует поднять до крана). Опуска-
ние уровня жидкости в бюретке вызовет поднятие уровня поглоти-
теля в пипетке. Уровень поглотителя в пипетке поднимают до тех
пор, пока поглотитель не дойдет до метки на капилляре, имеющемся
в верхней части пипетки. В этот момент кран закрывают и в гор-
лышко заднего сосуда пипетки вставляют пробку с каучуковым меш-
ком. Воздух, перешедший из пипетки в бюретку, вытесняют затем
поднятием уравнительного сосуда при открытых кране бюретки
и кране 4. Подобным же образом наполняют и другие пипетки. При
манипуляциях с одной пипеткой краны остальных должны быть,
естественно, закрыты.
После наполнения пипеток поглотителем прибор следует про-
верить на герметичность всех каучуковых соединений и кранов.
Каучуки, соединяющие краны с пипетками, должны быть плотно
надеты на капилляры; рекомендуется каждый каучук обвязать вверху
и внизу мягкой проволокой. Капилляр крана и капилляр пипетки
должны соединяться встык. Плотность каучуковых соединений кра-
нов с пипетками определяют по положению уровней поглотителей
в капиллярах. Если уровень поглотителя в капилляре за 1—2 часа
опускается на заметную величину, это означает, что имеется течь
или через каучуковое соединение или через кран пипетки. При нали-
чии течи начинать анализ газа ни в коем случае не следует. Для ее
устранения следует или сменить каучук или смазать кран (если кран
плохо притерт, то никакая смазка не поможет и кран следует сме-
нить). Плотность соединений и кранов других частей прибора про-
веряют путем наблюдения за положением уровня жидкости в бю-
ретке. Если при опущенном уравнительном сосуде уровень жидкости
96
в бюретке не понижается, то испытание можно считать закончен-
ным удовлетворительно, в противном случае может потребоваться
как смена каучуков, так и смена крана 4 или бюретки.
Анализ газа на описанном приборе производят следующим обра-
зом: поглотители в пипетках доводят до меток на капиллярах; подня-
тием уравнительного сосуда заполняют ртутью или водой всю бю-
ретку до крана, после чего кран бюретки закрывают. Каучуковую
трубку, по которой поступает анализируемый газ, присоединяют к
свободному концу крана 4; кран 4 поворачивают сначала таким об-
разом, чтобы его третий ход, идущий через пробку крана, был при-
соединен к той трубке, через которую идет анализируемый газ. Если
испытуемый газ находится под некоторым давлением, то его выпу-
скают через кран 4 в течение некоторого времени, чтобы вытеснить
воздух из подводящего каучука. Если газ под собственным давле-
нием не может выходить через кран 4 (например, проба газа находится
в стеклянной бутыли с водяным запором), то третий ход крана, име-
ющий вывод через пробку крана, присоединяют при помощи кау-
чука к водоструйному насосу, которым выкачивают воздух из под-
водящего каучука и воду, которая туда может попасть, если газ
берут из бутыли, находящейся в водяной ванне1. Когда воздух из
подводящего каучука будет весь удален, кран 4 поворачивают так,
чтобы источник газа сообщался с прибором. При открывании крана
бюретки уровень ртути в ней будет понижаться и бюретка будет
наполняться газом. Когда будет набрано достаточное количество
газа (для упрощения расчетов рекомендуется брать 100 см1 2 спо-
собом, описанным ниже), кран 4 поворачивают так, чтобы прибор
не сообщался ни с атмосферным воздухом, ни с источником газа.
Перед анализом гребенку сообщают на мгновение с атмосферным
воздухом, чтобы давление в капиллярах гребенки было равно дав-
лению в бюретке. Затем поднимают уравнительный сосуд и откры-
вают кран бюретки и первой пипетки. Газ из бюретки будет пере-
ходить в пипетку, и когда уровень ртути поднимется до крана, его
следует закрыть и оставить газ на некоторое время в пипетке, после
чего газ переводят обратно в бюретку. Газ из бюретки в пипетку и
обратно переводят несколько раз до тех пор, пока объем газа не пере-
станет уменьшаться. Для поглощения СО2 требуется обычно 2—3 мин.,
для поглощения кислорода пирогаллолом — 5—10—15 мин. в зависи-
мости от температуры (при 0° поглощение кислорода пирогаллолом
иногда не заканчивается и в течение 30 мин.). Когда поглощение
считается законченным, то окончательно измеряют оставшийся
объем газа, для чего, доведя уровень поглотителя в капилляре до
метки, закрывают кран пипетки. Делают отсчет объема газа в
бюретке в тот момент, когда жидкость в бюретке и уравни-
тельном сосуде будет стоять на одной высоте. Подобным же обра-
зом производят поглощение и следующих составных частей газа,
переводя газ в пипетку 2, а затем в пипетку J2. Все измерения объема
1 Способы переведения проб газа в бюретки описаны в главе II.
2 Рецепты растворов даны ниже.
97
7 В. А Соколов.
Фиг. 57. Прибор для общего анализа
газа с приспособлениями для сож-
жения.
газа после поглощений следует производить при открытом кране
бюретки. Если в бюретке в качестве вытесняющей жидкости приме-
няют воду, то этот кран бюретки вообще не является необходимым.
В этом случае верхний свободный конец бюретки, не имеющий крана,
присоединяют каучуком к гребенке; отсчеты объемов взятого газа
будут не совсем точны, но для большинства технических анализов
эти погрешности не имеют значения. Чтобы вытеснить воздух из
бюретки, не имеющей крана, перед
набиранием газа можно ставить
уравнительный сосуд с водой на
верхнюю крышку футляра прибо-
ра. Давление в этом случае еще
настолько мало, что вода из бю-
ретки не попадет в капилляры гре-
бенки. Для более точных анализов
следует употреблять бюретку с
краном.
Необходимо следить, чтобы ни
один раствор поглотителя не по-
падал ни в капилляр гребенки, ни
в бюретку. Если же это случилось,
прибор следует разобрать и вы-
мыть.
Резиновые мешочки присоеди-
няют к пипеткам для того, чтобы
предохранить растворы от сопри-
косновения с окружающим возду-
хом.
Прибор, изображенный на фиг.
56, имеет три пипетки для погло-
тителей. Существуют приборы,
имеющие большее число пипеток
для поглощения и пипетки для
сожжения газа различными спосо-
бами. На фиг. 57 изображен при-
бор с тремя пипетками для поглощения, пипеткой для сожжения
и трубкой с окисью меди, нагреваемой электрической печью. У этого
прибора бюретка имеет компенсационное устройство для автомати-
ческой поправки на измерение температуры и давления.
Прибор ВТИ
На фиг. 58 изображена схема прибора, разработанного Всесоюз-
ным теплотехническил! институтом (ВТИ). Этот прибор имеет бю-
ретку 1 с расширениями и с вертикальной градуированной трубкой
объемом 20 см3. Бюретка помещена в охранной трубке с водой; пи-
петки 2—7 — разъемные. Верхняя часть, содержащая стеклянные
трубочки, имеет внизу длинную трубку, которая на шлифе вста-
вляется в уравнительный сосуд.
98
Через трехходовые краны гребенки присоединена кварцевая
трубка 8 с окисью меди. Для полного сожжения газа трубку нагре-
вают пламенем газовой горелки или проволокой с высоким сопро-
тивлением, накаливаемой электрическим током. Для дробного сож-
жения применяют тот или иной обогрев, но с обязательным контро-
лем за температурой трубки. Помимо трубки с окисью меди в при-
боре имеется и пипетка для сожжения 9.
Имеется большое число модификаций этого прибора, в которых
комбинируется различное число пипеток с теми или иными приспо-
соблениями для сожжения газа (пипетка для сожжения, трубка с
окисью меди, трубка с палладизированным асбестом и т. д.). Пипетки
для поглощения применяются различного устройства, хотя и оди-
Фиг. 58. Схема прибора ВТИ.
наковые по принципу действия. Краны гребенки делают трехходо-
выми, как показано на фиг. 58.
Применяют также простые гребенки с отводами и с простыми
двухходовыми кранами. Гребенка представляет собой толстостен-
ный капилляр с внутренним диаметром 1,5—2 мм.
Анализ газа на приборе ВТИ производят следующим образом.
Предварительно доводят уровни поглотителей во всех пипетках
до соответствующих меток на капиллярах, после чего пипетки для
взрыва и сожжения наполняют измеренным количеством воздуха
или кислорода. Затем наполняют гребенку (состоящую в этом слу-
чае из ряда трехходовых кранов), трубку с окисью меди и все соеди-
нения азотом. Азот проще всего приготовить на этом же приборе,
взяв 100 см3 воздуха и поглотив из него при помощи пирогаллола
кислород. Для наполнения гребенки и трубок азотом из них отка-
чивают воздух при помощи водоструйного насоса, а затем выпускают
из бюретки приготовленный азот, закрыв, разумеется, кран, веду-
щий к насосу. Затем, закрыв кран бюретки, опять откачивают газ
из гребенки, после чего, промыв трубки азотом и окончательно за-
7* 99
крыв кран, ведущий к насосу, впускают опять из бюретки в трубки
азот. Поглотители в пипетках никогда не следует подводить вплот-
ную к крану, поэтому между поглотителями и краном всегда оста-
нется небольшое количество воздуха (или газа от предыдущего ана-
лиза). Этот воздух также можно заменить азотом, для чего в пипетки
впускают по 5—б см3 азота из бюретки. Закрыв кран бюретки, при
помощи водоструйного насоса поднимают уровни поглотителей до
требуемой высоты, а затем, повернув соответствующим образом кран,
откачивают азот из гребенки. Закрыв кран, ведущий к насосу, впу-
скают в капилляры гребенки последнюю порцию азота. Давление
газа в гребенке доводят до атмосферного, для чего пользуются мано-
метром, присоединенным к бюретке. При откачке и при наполнении
азотом гребенки необходимо следить за тем, чтобы трехходовые кра-
ны, ведущие к трубке с СнО, соединяли ее с гребенкой (метки на
рукоятках кранов должны быть обращены кверху).
Испытуемым газом наполняют бюретку, как и на простом при-
боре для анализа, описанном выше. Бюретка имеет обычно трех-
ходовый кран. К одному из концов этого крана присоединяют источ-
ник газа. Из подводящего каучука вытесняют или выкачивают воз-
дух, после чего газ забирают в бюретку. Чтобы перевести газ из бю-
ретки в пипетку,, поднимают уравнительный сосуд, открывают кран
бюретки (находящийся над бюреткой кран после взятия газа в бю-
ретку повертывают так, чтобы бюретка соединялась с гребенкой)
и кран соответствующей пипетки. Кран пипетки при этом поверты-
вают так, чтобы рукоятка крана была направлена вниз, а метка на
кране обращена в сторону бюретки. Газ перепускают из бюретки
в пипетку несколько раз до тех пор, пока объем газа не перестанет
уменьшаться.
В пипетки прибора наливают различные поглотители. В большин-
стве случаев в первую пипетку наливают раствор едкого кали, во
вторую — бромную воду, в третью — дымящую серную кислоту,
в четвертую — щелочной раствор пирогаллола, в пятую — аммиач-
ный раствор однохлористой меди, шестую наполняют также рас-
твором однохлористой меди, но свежеприготовленным.
Если в газе содержится значительное количество сероводорода,
для его поглощения рекомендуют употреблять раствор сернокислой
меди. В этом случае газ в первую очередь вводят в пипетку с этим
раствором, а затем уже определяют СО2 при помощи КОН и т. д.
Вместо раствора пирогаллола можно употреблять гидросернистый
натрий или другие поглотители. Для поглощения СО можно взять,
конечно, и одну пипетку с аммиачным раствором однохлористой
меди, однако применение двух пипеток даст лучшие результаты.
После поглощения H2S, СО2, непредельных углеводородов, О2, СО
в газе остаются Н2, N2 и углеводороды предельного ряда. Окись
углерода и водород можно определять не поглощением, а сожже-
нием, как это описано выше.
Для полного сожжения газа поступают так же, как описано в
предыдущем параграфе, пользуясь пипеткой для сожжения. Если
газ содержит несколько горючих компонентов, то при помощи сож-
100
жения можно определить только их сумму. После окончания сож-
жения газу дают охладиться до комнатной температуры в пипетке,
а затем переводят его в бюретку для измерения объема. Для более
быстрого охлаждения газа на пипетку направляют насосом струю
воздуха. После поглощения образовавшейся при сожжении СО2 и
не вступившего в реакцию О2 определяют остаток, представляющий
собой сумму азота и редких газов. Так как содержание редких га-
зов в большинстве случаев невелико, то этот негорючий остаток при-
нимают за азот. Ниже приводится примерный расчет при анализе
газа методом сожжения.
Предположим, газ состоит из Н2, СО, СН4 и N2.
Обозначим объем Н2 в газе через х, СО — через у и СН4 — через z.
Наблюденная после сожжения контракция будет равна:
3 1
К = -2-х+ —у + 2z.
Объем образовавшейся углекислоты С будет равен:
с = у + z.
Объем израсходованного на сожжение кислорода S (определяемый по раз-
ности между взятым кислородом и оставшимся после сожжения) будет равен:
S = 4-х + -у У + 2z.
Из этих уравнений определяем величины %, у и z:
x = K — S;
4 1
у= ~С
О о
г = 8-4-к-4- с‘
О *3
или
z = С — у.
При помощи окиси меди можно провести дробное сожжение, а
именно, сжечь Н2 и СО отдельно. Это сожжение производят при
температуре 280—300°, температуру контролируют по термометру,
вставляемому в электрическую печь. Газ пропускают несколько
раз через СнО, переводя его из бюретки в пипетку для сожжения
или для взрыва (газ необходимо переводить в пипетку с ртутью,
чтобы избежать поглощения образующейся СО2). Газ через СиО
рекомендуется пропускать со скоростью 8—10 см3]мин. При сож-
жении с СиО расчет очень прост: объем образовавшейся СО2 равен
объему сгоревшей СО. Недостающий объем за вычетом объема
СО из первоначального объема представляет собой водород.
Объем после сожжения следует измерять лишь после охлаждения
трубки с СиО до комнатной температуры. Для ускорения процесса
охлаждения на трубку с СиО направляют струю воздуха.
Все измерения объема рекомендуется производить следующим
образом: опускают уравнительный сосуд до тех пор, пока ртуть в
101
нем и в бюретке не будет на одной высоте (на-глаз); только после
этого кран бюретки закрывают и при помощи манометра точно изме-
ряют объем.
Если требуется произвести сожжение наиболее точным обра-
зом, в уравнения вводят поправки на молекулярный объем.
Для многих технических целей в природном горючем газе доста-
точно определить содержание углекислоты и кислорода, причем по
содержанию кислорода заключают о количестве воздуха, попавшего
в газ, так как свободный кислород в газе встречается крайне редко.
Если газ содержит тяжелые углеводороды (этан, пропан, бутан
и т. д.), то нет смысла определять поглощением СО и непредельные
углеводороды.
Для общего анализа природного горючего газа, содержащего
тяжелые углеводороды, пригоден прибор с двумя пипетками для
поглощения и пипеткой для сожжения. На подобном приборе опре-
деляют СО2, О2, сумму углеводородов и N2. Полный же анализ этого
газа можно сделать только при помощи других методов.
При работе в лаборатории желательно всегда иметь прибор с боль-
шим числом пипеток для поглощения и различными приспособле-
ниями для сожжения газа, что позволит анализировать не только
природные, но и промышленные газы. Кроме того, хотя природный
газ обычно и не содержит СО, Н2 и непредельных углеводородов,
это не исключает возможности их обнаружения в отдельных случаях.
Иногда при анализе отдельно определяют алкены путем погло-
щения серной кислотой различной крепости, которой и наполняют
несколько пипеток прибора. Для этих целей применяют и другие
поглотители. В некоторых случаях в одну из пипеток наливают рас-
твор, поглощающий водород. Если в газе предполагается присут-
ствие аммиака, то для его удаления из газа перед прибором ставят
промывалку со слабым раствором серной кислоты.
Способы приготовления различных реагентов, применяемых в
приборах для общего анализа, и действие этих реагентов на раз-
личные газы описаны в § 4 настоящей главы.
Для газового анализа применяют также приборы типа Норзе
(фиг. 59), видоизмененные и усовершенствованные за последнее вре-
мя Кнорре и Зиминым [12].
Газоанализатор Кнорре — Зимина — Норзе
Устройство аппарата (фиг. 59, а). Измерительная
бюретка 3 посредством распределительной трубки 11, укрепленной
на планке 23, и крановых соединений 14 сообщается с поглотитель-
ными сосудами 77 и с фильтром 13, через который ножное разрежаю-
щее приспособление 20 просасывает анализируемый газ, частично
поглощаемый реактивами в сосудах 77, частично сжигаемый в
петле 7 бензиновой горелкой 22 или электропечью. Измеритель-
ная бюретка 3, градуированная на 100 см*, резиновой трубкой
соединяется со склянкой 7. Чтобы избежать колебания темпе-
ратуры газа, бюретку с газом помещают в наполненный водой
колокол 4, который устанавливают в стакане 5, причем нижний,
102
открытый кран колокола не доходит до дна стакана. Верхняя часть
колокола трубочкой 6 сообщается со сжигательной петлей 7, вслед-
ствие чего сжигаемый в последней газ может перепускаться под
колокол 4, из которого в этом случае вода переходит в сообщающийся
с ним стакан 5. Крановые соединения 14 тремя ветвями соединяют
/4/J
Фиг. 59. Прибор Кнорре — Зимина — Норзе.
распределительную трубку 11 с поглотительными сосудами 77. Каж-
дый из этих сосудов состоит из двух сообщающихся камер, из кото-
рых рабочая камера 18 своей нижней частью, имеющей сужения,
входит в вспомогательную камеру. В рабочую камеру вставлена
капиллярная трубка (форсунка) 15, по которой газ поступает в ниж-
нюю часть камеры 18. Загнутый конец распределительной трубки 77
сообщается с фильтром, соединенным 'с газопроводом и с ножным
разрежающим приспособлением. Сжигательная петля 7 (кварце-
вая или платиновая) с помощью штепселя 8 соединяет распредели-
тельную трубку 77 с колоколом 4.
При засасывании очень пыльного или содержащего много сажи
газа желательно пользоваться добавочным фильтром (стеклянная
трубка с уширением, набитым стеклянной или обыкновенной ватой).
Такой фильтр следует устанавливать у самой заборной трубки, чтобы
избежать засорения резиновой трубки и прибора.
Краны прибора должны быть хорошо смазаны во избежание про-
пусков или заеданий. Последнее особенно легко может случиться
на среднем кране у склянки с бромом (или серной кислотой).
103
Перед анализом необходимо осмотреть все краны и опробовать
аппарат на отсутствие пропусков как в кранах, резиновых трубках
и пробках, так и у сжигательной петли, чтобы,, в дальнейшем не
испортить анализа.
Если для сжигания пользуются кварцевой петлей, то после
присоединения ее необходимо проверить на плотность в холодном
и горячем состояниях (при раскаленном кварце) прокачиванием НО см
воздуха.
Охлаждать кварц смачиванием можно, но не рекомендуется,
так как это отражается на его прочности и долговечности. Кварце-
вая трубка заполняется платинированным асбестом. Можно реко-
мендовать применение кварцевой или стеклянной (пирекс) капил-
лярной трубки с впаянной в нее платиновой проволочкой, нагре-
ваемой электрическим током. При этом возможен контроль над тем-
пературой дожигания.
После сжигания необходимо охладить газ до нормальной тем-
пературы, для чего следует на некоторое время оставить пробу
в бюретке и убедиться, что при продолжающемся остывании не про-
исходит сокращения объема.
Для сжигания на бензиновой горелке достаточно трех-четырех
прокачек (туда и обратно) через раскаленную петлю.
Зарядка прибора. Приступая к зарядке прибора, сни-
мают крышку ящика, откидывают боковую полку 27 и вынимают
из гнезд и держателей 30, 31 наполнительную склянку 7, горелку 22
и штепсель с кварцевой петлей 7. Откинув затем рамку пюпитра,
вынимают ножное разрежающее приспособление 20 и соединяют
его с трубкой фильтра 13. Кварцевую петлю 7, держа ее во избе-
жание поломки за деревянную оправу (штепсель) 8, вставляют на
место, для чего, надвигая оправу правой рукой, левой заводят
в нижнюю пробку конец соединительной трубки 6, а затем,
придерживая распределитель 77, насаживают верхнюю трубку 6,
придерживая все время оправу 8 петли 7. Перед установкой петли 7
полезно смочить отверстие пробок 26 водой для лучшего скольже-
ния стерла. На полку 27 ставят горелку 22.
В склянку 7 наливают кипяченую воду, которую желательно
предварительно насытить газом во избежание поглощения пробы
при анализе. Затем наполняют наружный стакан 5 кипяченой водой
(сырая вода дает помутнение стекла от выделения пузырьков газа)
и подтягивают ее во внутреннем колоколе 4 до капилляра 32, мани-
пулируя склянкой 7 и кранами 9 и 10. Когда вода в колоколе 4 под-
тянута доверху, уровень воды в стакане 5 должен стоять около
деления 30 бюретки.
Наливают немного воды в фильтр 13 для образования гидравли-
ческого затвора и приступают к зарядке поглотительных сосудов 77,
для чего кран 14 каждого из них устанавливают на соединение внут-
ренней камеры 18 через капилляр 16 с распределителем 77, который
соответствующей установкой кранов 10 и 9 сообщается с атмосфе-
рой. Затем надевают резиновую трубку (длиной около 300 мм) с
воронкой на задний тубулусик сосуда 77, через который и наполняют
104
сосуд реактивом. Последний при некотором сжатии воздуха в на-
ружной камере 19 поглотителя 17 начинает подниматься во внутрен-
ней камере 18. Воздух удаляют через открытые заранее краны 14,
10 и 9. Доливанием реактива из подъемной воронки подводят его
в капилляр и закрывают кран 14. Тогда вся внутренняя камера 18
поглотителя окажется заполненной реактивом (требуется около ПО—
120 см*). Нажатием резиновой трубки воронки, наполненной реак-
тивом, вбрызгивают в наружную камеру 19 еще несколько кубиков
раствора для большей надежности гидравлического затвора, сли-
вают из воронки оставшийся реактив, снимают воронку с трубкой;
и надевают на тубулусик резиновый мешок для разобщения с воз-
духом (кроме склянки со щелочью, тубулусик которой остается от-
крытым). В качестве реактивов употребляют:
в первой (от бюретки) склянке для поглощения СО2 — водный
раствор едкого кали (1 : 2);
во второй (от бюретки) склянке для поглощения С2Н4 и др. —
водный раствор брома или дымящую серную кислоту;
в третьей (от бюретки) склянке для поглощения О2—раствор
18 г пирогаллола в 40 гл/3 горячей воды, залитый 70 см3 раствора
едкого кали (1:2).
Бром берут в крепком, но не пересыщенном растворе (светло-
красного цвета). При пользовании в качестве реактива для погло-
щения С2Н4 и С2Н2 дымящей серной кислотой, разъедающей ре-
зину, надо поглотительный сосуд с резиновой пробкой заменить со-
судом с впаянной форсункой.
Анализ. Перед анализом пробы желательно сделать анализ
окружающего (в данном месте) аппарат воздуха для определения
содержания в нем кислорода, так как количество последнего, входя-
щее в формулы для подсчетов испытуемого газа, может отличаться
от принятых 20,9%, что отразится на точности результата. То же
относится и к возможному содержанию СО2 в воздухе.
Соединив трубку фильтра с газопроводом, приступают к произ-
водству анализа, продувая газопровод как перед анализом, так и
во время анализа (для следующей пробы), действуя ногой на раз-
режающее приспособление 20 при постоянном токе газа к прибору.
Продувка аппарата производится поднятием и опусканием склянки 7
через тройник 27 с краником 9 бюретки 3 и краны 12 и 10 распреде-
лителя или специальной грушей, устанавливаемой на склянке 7.
Можно пользоваться и обычной грушей.
Подведя уровень воды в бюретке до бокового капиллярного кра-
ника 9, открывают кран 12 и забирают в бюретку, опуская склянку 7,
пробу газа, несколько большую 100 см3. Закрыв кран 12 и сжав
газ в бюретке до 100 см3 поднятием склянки 7, зажимают соединя-
ющую склянку с бюреткой резиновую трубку 2 и поворотом кра-
ника 9 выбрасывают излишек газа для приведения его к атмосфер-
ному давлению.
Поставив склянку 7 на верх прибора, поворачивают кран 14 справа
налево на соединение с форсункой 15 первого поглотителя и пере-
705
качивают газ в первый сосуд (поглощение СО2). Перед окончанием
перекачки, подтормаживая воду зажатием левой рукой трубки 2,
не снимая склянки 1, чтобы не затянуть реактива в форсунку 15
и далее в кран, переводят кран 14 направо на соединение с капил-
ляром 16 и затем уже, опуская склянку 7, поднимают уровень реак-
тива. Если нужно, повторяют операцию и, подведя реактив в капил-
ляр 16 и закрыв кран 14, по уменьшению объема газа в бюретке 3
судят о количестве поглощенной СО3. Затем повторяют то же со
второй склянкой; выделяющиеся при этом пары брома (или
серной кислоты) наполняют бюретку и увеличивают объем пробы.
После двух-трех прокачек реактив подводят в капилляр и кран за-
крывают, а оставшийся в бюретке газ перегоняют опять в раствор
едкого кали до полного поглощения паров брома (или серной кис-
лоты). Затем уже по уменьшению объема против первого отсчета
судят о поглощенных С2Н4, С2На и др., после чего подобным же обра-
зом перекачкой в третий сосуд определяют содержание кислорода,
для полного поглощения которого требуется при свежем реактиве
ют двух до четырех перекачиваний. Число перекачиваний при све-
жих реактивах надо считать нормальным для поглощения: одно —
для СО2, два — для С2Н4 и три — для О2. Крановая часть 14 третьей
склянки имеет на правом колене уширение для улавливания
пузырьков раствора пирогаллола в случае попадания их в капил-
ляр.
Сжигание. Закончив поглощение СО2, С2Н4 и О2, при-
ступают к определению СО, СН4 и других горючих газов сжига-
нием в кварцевой трубке 7, наполненной платинированным асбе-
стом, для чего предварительно через кран 9, поднимая склянку 7,
переводят в нее большую часть пробы, оставляя 15—30 см3 в зави-
симости от анализируемого газа. Чем богаче горючими газ, тем
меньше оставляемая часть пробы. Уменьшив пробу, добавляют
в нее воздух, опуская склянку 7 так, чтобы весь объем был около
95—98 см3, после чего закрывают кран 9. Доводить до 100 см3 со-
держание смеси не рекомендуется, так как легко перейти за 100 см3
и анализ не будет достаточно точным.
Установив кран 10 на соединение бюретки с петлей 7, зажигают
горелку 22 и нагревают петлю, медленно пропуская 3—4 раза под-
нятием и опусканием склянки 7 всю пробу через раскаленную трубку.
По окончании сжигания гасят горелку и несколькими перекачи-
ваниями пробы из бюретки 3 в колокол 4 охлаждают пробу.
Охлаждение должно производиться до постоянства уровня в бю-
ретке при подтянутой в капилляр 32 воде в колоколе. От пол-
ноты охлаждения в значительной степени зависит точность ана-
лиза.
Охладив пробу, подводят воду в колоколе 4 в капилляр 32 и,
переключив кран Ю на соединение бюретки с распределителем 1l’>
устанавливают уменьшение объема газа после сожжения. Затем
перекачкой газа в соответствующие поглотители дополнительно
определяют содержание СО2, образовавшейся из сгоревших газов,
.и оставшегося неизрасходованным кислорода воздуха.
106
Г азоанализатор с микробюреткой
Бюретки. Основной частью газоанализатора является изме-
рительная часть, т. е. бюретка, представляющая сдвоенную бю-
ретку сложного вида, а именно рабочую бюретку 7 и микробюретку 2
(фиг. 59, б).
Рабочая бюретка 7 —это сосуд объемом в 100 см3, разделенный
тремя пережимами на четыре равные части емкостью 25 см3 каж-
дая. Деления в 25, 50, 75 и 100 см3 нанесены для точности отсчета
на узких местах бюретки, т. е. на пережимах. Нижняя часть рабо-
чей бюретки до 25 см3 градуирована делениями по 0,2 см3. В верх-
ней части рабочей бюретки имеется карман для термометра, а на
капиллярном конце припая одноходовый кран, служащий для соеди-
нения с внешним воздухом (атмосферой). В нижней части бюретка
заканчивается оттянутым концом с оливкой.
Микробюретка 2 представляет прямую трубку емкостью 25 см3,
отградуированную по всей длине с ценой деления 0,05 см3. В верхней
части микробюретка 2 кончается капиллярной трубкой, а в ниж-
ней — оттянутым концом с оливкой. Эта микробюретка и является
основной измерительной частью прибора.
Микробюретка 2 и рабочая бюретка 7 соединены в верхней части
тройником 3 посредством резиновых муфт, а в нижней части —трой-
ником 4.
Тройник 4 имеет угловой кран 5 и соединен шлангом <5 с напорной
склянкой 7.
Колокол рабочей бюретки. Рабочая бюретка 7
вставляется в колокол 8 (показан на схеме в верхней части), пред-
ставляющий стакан с двойными стенками, спаянными вверху и от-
тянутыми в одном месте в узкую и короткую (1—2 см) трубочку 9.
В нижней части наружные стенки колокола оттянуты в трубку, в
которую плотно вставляется нижний конец рабочей бюретки 7 с
помощью резиновых прокладок, а внутренние стенки прямые и не
доходят до конца наружных на 3—4 см. Внизу к наружным стенкам
колокола припаян короткий отросток для слива воды.
Назначение колокола — создать так называемую водяную ру-
башку для охлаждения газа до температуры воздуха, окружающего
прибор. Кроме того, колокол заменяет сосуд для прокачки газа во
время [сжигания.]
Стакан микробюретки. Микробюретка 2 встав-
ляется в особый стакан (не показан на схеме), представляющий стек-
лянную трубку, обрезанную с обоих концов. Нижний конец
стакана плотно закрывают резиновой пробкой, чтобы не протекала
вода; верхний же можно закрыть неплотно резиновым кольцом.
Стакан служит водяной рубашкой для микро бюретки.
Манометр. Рядом с микробюреткой устанавливают мано-
метр 10 в виде длинной трубки, доходящей до верхней крышки ящика
прибора. Вверху манометр имеет одноходовый кран 77. Посредством
резинового соединения манометр наращивается дополнительной тру-
бочкой выше ящика прибора, а внизу он соединен с микробюреткой.
107
Назначение манометра — уточнить взятие отсчета газа по микро-
бюретке. Отсчет берут при совпадении уровней менисков жидкостей
в микробюретке и манометре, а не в напорной склянке, как обычно
в других газоанализаторах.
Совпадение уровней в микробюретке и манометре показывает,
что в микробюретке (а равно и в рабочей бюретке) в момент отсчета
имеет место атмосферное давление.
Зеркало. Для большего уточнения глазного отсчета по микро-
бюретке позади нее и манометра ставится зеркало 12. Отсчет берут
при совпадении зеркальных изображений и фактических менисков
жидкостей в микробюретке и манометре.
Распределительная трубка—гребенка. Гре-
бенка 13 представляет собой капиллярную трубку с внутренним
диаметром 1,5 мм, внешним б—8 мм, четырьмя отростками для
присоединения бюретки и поглотительных сосудов. Левый конец
гребенки отогнут вниз и служит для соединения прибора с источ-
ником газа через фильтр.
Гребенка имеет два крана. Левый кран 14 простой, одноходовый
и служит для забора пробы газа. Правый кран 15 угловой и слу-
жит для соединения бюретки или с поглотительными сосудами (во-
обще с левой частью гребенки), или с колоколом рабочей бюретки —
в зависимости от того, как он установлен.
Назначение гребенки — соединить все части прибора в одну
целую систему и дать возможность сообщать бюретку по очереди с
источником газа, поглотительными сосудами и дожигательным
устройством.
Поглотительный сосуд. Поглотительный сосуд 16
состоит из двух сообщающихся камер, из которых рабочая своей
нижней суженной частью входит во вспомогательную 18. В рабочую
камеру вверху в середине впаяна капиллярная трубка (форсунка)
19, а сбоку припаян еще капиллярный отросток. По форсунке
газ поступает в нижнюю часть рабочей камеры и, пройдя вверх
через реактив, вытесняет последний во вспомогательную камеру 18.
К задней стороне вспомогательной камеры 18 припаян тубулусик,
на который надевается резиновый мешок.
Фильтр. Фильтр 22 состоит из стеклянной пробирки, не-
сколько раздутой у верха в шарик, приплюснутый с двух сторон.
Внутрь шарика впаяна доходящая почти до дна узкая трубочка с
наружный! отростком, на который надевается подводящая газ рези-
новая трубка. Горловина фильтра закрывается плотно резиновой
пробкой, через которую с помощью трубки 23 фильтр присоединяется
к гребенке прибора.
Назначение фильтра — очищать забираемый для анализа газ от
механических примесей, для чего в шаровидную часть кладется стек-
лянная вата, а в нижнюю часть фильтра заливается небольшое коли-
чество воды.
У гл свой кран. Бюретка и поглотительные сосуды соеди-
няются с гребенкой через угловые краны 15 и 24. Муфта углового
крана имеет три капиллярных отростка-конца. Пробка крана имеет
108
два просверленных под прямым углом отверстия, соединяющиеся
между собой. Угловые отверстия в пробке дают возможность при
надобности соединять гребенку с поглотительным сосудом через
форсунку или через капиллярный отросток, что необходимо делать
при анализе. Угловой кран является закрытым в любом положении,
когда он не соединяет какие-либо капиллярные концы муфты.
Сжигательная петля. Сжигательная петля для
горючих составляющих газа (СО, СН4 и др.) представляет со-
гнутую стеклянную трубочку с капиллярными концами. Петля
наполнена платиновым катализатором1. Капиллярные концы петли
проходят через стеклянный холодильник. Нижний конец холо-
дильника посредством резинового шланга присоединяется к водо-
проводу, и в холодильник во время дожигания газа подается про-
точная вода. Из холодильника вода отводится через верхний конец
также резиновым шлангом. Одним концом своим петля присоеди-
няется к колоколу рабочей бюретки посредством соединительного
отростка 9, а другим — к правому концу гребенки 13. К прибору
петля присоединяется посредством резиновых муфточек. Поддержи-
вается петля специальным металлическим бандажником.
Сжигательные печи. Обогревается сжигательная
петля электрическими печами. Прибор имеет две печи, одна из ко-
торых рассчитана на температуру 200° для сжигания СО и Н2 и
вторая — на температуру 450° для сжигания СН4.
Для получения необходимой и устойчивой температуры обогрева
петли печи питают током от сети через 12-е трансформатор. Укреп-
ляют печи на металлическом стержне, который в свою очередь при-
крепляют к стенке ящика прибора. Печи могут быть повернуты во-
круг стержня, что позволяет обогревать петлю поочередно обеими
печами.
Сборка прибора. Сборку прибора начинают со сборки несколь-
ких деталей в одну сложную, после чего сложные детали соединяют
между собой. Перед сборкой прибора все части его должны быть
тщательно вымыты, особенно измерительные—микробюретка, ра-
бочая бюретка и манометр. На чисто вымытом стекле не должно оста-
ваться капель после стекания жидкости.
Сборка бюретки. Сначала рабочую бюретку и микро-
бюретку вставляют в охладительные рубашки (колокол и стакан)
и бюретка нижним концом вводится в колокол примерно на 4—б см
так, чтобы резиновая трубка плотно прилегала к стенкам колокола.
На боковой отросток колокола 9 надевают резиновую трубочку дли-
ной б—8 см, закрытую стеклянной палочкой. После этого наливают
в колокол немного воды и испытывают плотность сборки. Затем вверху
колокола, между его внутренними стенками и бюреткой, неплотно
кладут разрезное резиновое кольцо, чтобы предохранить жидкость в
колоколе от пыли и центрировать бюретку в колоколе. Далее ра-
1 Приготовление платинового катализатора описано выше в § 2.
109
бочую бюретку вставляют в свое гнездо в ящике прибора и присоеди-
няют к гребенке через тройник 3, ранее присоединенный к гребенке.
Микробюретку укрепляют в стакане посредством резиновых про-
бок, из которых нижняя служит дном стакана и потому должна
плотно прилегать. Микробюретку вставляют в гнездо в ящике при-
бора и также присоединяют к гребенке через тройник 3.
Соединения рабочей бюретки и микробюретки с тройником 3, а
последнего с гребенкой должны быть выполнены встык. Резиновые
муфточки должны плотно охватывать стекло, чтобы в местах соеди-
нений не проходил воздух. Если же будут неплотности соединений,
что может быть обнаружено изложенным ниже способом, то до устра-
нения их прибор к употреблению не годится.
Для более легкого надевания и для лучшего прилегания к стеклу
резиновые муфточки перед надеванием следует смочить водой. Бю-
ретку соединяют резиновой трубкой 6 длиной примерно 1 м с напор-
ной склянкой 7. Резиновая трубка 6 должна быть эластичной и плотно
охватывать конец тройника и тубулус склянки 7. На этом закан-
чивается монтаж измерительной части.
Сборка поглотительных сосудов. К погло^-
тительным сосудам сначала присоединяют угловые краны 24 и за-
тем уже сосуды присоединяют к гребенке. Все соединения угловых
кранов как с сосудами, так и с гребенкой должны быть смонтиро-
ваны встык (прилегание стекла к стеклу), а соединительные муф-
точки должны плотно прилегать к стеклу и не пропускать воздуха.
Резиновые муфточки обычно нарезаются из вакуумных (толстостен-
ных) резиновых трубочек. На задние тубулусики второго и третьего
сосудов (считая первый от бюретки) надевают резиновые мешки.
Сборка фильтра. Горловину фильтра 22 закрывают
плотно резиновой пробкой, в которую иногда вставляют тройник.
Вторым концом тройника фильтр присоединяют к левому изогнутому
концу гребенки. На свободный боковой конец тройника надевают
всасывающую грушу. Боковой отросток фильтра соединяют резино-
вой трубкой с газопроводом.
Очень часто фильтр соединяют с гребенкой не тройником, а пря-
мой соединительной трубочкой (как показано на схеме). В этих слу-
чаях газ просасывают через газопровод всасывающей грушей, вста-
вленной плотно в горло напорной склянки 7.
При заборе газа для анализа из аспиратора, что чаще имеет место
при работе на описываемом приборе, фильтр не ставят, а левый
конец гребенки непосредственно присоединяют к аспиратору по-
средством резиновой узкой трубочки или лучше через соединитель-
ный стеклянный капилляр. Засасывают газ в прибор разрежением
в бюретке, создаваемым наполнительной склянкой 7.
Сборка сжигательного устройства. Сжигатель-
ная петля обычно прилагается к прибору в готовом смонтированном
виде, так что остается только соединить ее с прибором, что выпол-
няется посредством резиновых муфточек. Один конец петли при-
соединяют к правому концу гребенки, второй—к колоколу рабо-
чей бюретки посредством стеклянного капиллярного отростка Р.
110
Соединение петли с гребенкой и отростком должно быть встык, а
муфточки не должны пропускать воздуха.
На конец холодильника надевают резиновые шланги и нижний
конец холодильника соединяют с водопроводом. Для контроля тем-
пературы сжигания к петле присоединяют термопару, головку ко-
торой укрепляют в середине сгиба петли. Термопару обычно привя-
зывают асбестовым шнуром, а свободные концы ее присоединяют
к милливольтметру, устанавливаемому на столе рядом с прибо-
ром.
Сжигательные печи укрепляют на металлическом стержне, при-
соединенном к стенке ящика прибора. Печи могут перемещаться
вдоль и вокруг стержня, благодаря чему могут поочередно надви-
гаться на петлю. Печи обогреваются от сети через трансформатор.
Зарядка прибора и проверка на плотность. Перед зарядкой при-
бора все краны тщательно смазывают специальной вакуумной смаз-
кой, сваренной из вазелина, парафина и сырой резины. Смазывать
краны необходимо для плотности прилегания и плавного поворачи-
вания пробки крана в муфте и для предотвращения «замерзания
крана» от действия реактива, который может попасть в него по
неопытности или неосторожности работника, выполняющего анализ
газа. Хорошо смазанный кран должен быть прозрачен и не иметь
матовых полосок. Перед смазываниел! необходимо снять старую
смазку, протерев для этого пробку и муфту крана ватой, смоченной
бензином. Смазывать нужно только пробку крана, наложив тонкий
слой вакуумной смазки и растерев ее равномерно пальцем на
пробке. Муфту крана смазывают, поворачивая в ней несколько раз
пробку. Краны, которые после смазки не становятся прозрачными,
не держат вакуума.
Смазав краны, можно приступать к зарядке прибора и проверке
на плотность.
Измерительную часть прибора, т. е. рабочую бюретку, микро-
бюретку и манометр, заливают насыщенным раствором поваренной
соли посредством напорной склянки 7. Для этого в нее наливают
200—250 см3 раствора соли, открывают нижний кран 5 на сообще-
ние рабочей бюретки, микробюретки и манометра с напорной склян-
кой 7, а кран манометра 77 и воздушный кран рабочей бюретки —
на сообщение с воздухом. Кран 75 повертывают влево на гребенку,
кран 74 и все три крана 24 закрывают. Затем, подняв вверх напор-
ную склянку 7, заливают измерительную часть раствором соли до
верхних рисок, выталкивая наружу имеющийся там воздух. Не сле-
дует заливать раствором соли соединительный капилляр 5 и тем
более гребенку.
Залив раствором соли манометр и бюретки, закрывают краны 77
и воздушный, все же другие краны остаются в указанном выше по-
ложении. Напорную склянку 7 ставят на стол. Если после этого
уровни жидкости в микробюретке, рабочей бюретке и манометре
не спускаются вниз в течение 1—2 мин., то можно считать, что все
резиновые соединения и краны в порядке.
’ 777
В склянке 7 после заливки измерительной части должно остаться
столько соляного раствора, чтобы был закрыт верхний край тубу-
луса, иначе через тубулус будут проскакивать пузырьки воздуха
в бюретку.
Далее заливают колокол рабочей бюретки также раствором по-
варенной соли. Удобнее всего залить колокол через боковую оття-
нутую трубочку 9 посредством воронки. Раствор поваренной соли
наливают немного выше риски 50 см на рабочей бюретке. Затем снова
присоединяют трубочку 9 к сжигательной петле и соединения эти
испытывают на плотность, когда они уже присоединены к прибору.
Для этого на задний отросток сосуда 21 надевают резиновую тру-
бочку длиной 15—20 см.
Поглотительные сосуды заливают соответствующими реактивами.
Так, первый сосуд (считая от бюретки) заливают раствором КОН
для поглощения СО2, второй сосуд (средний) — бромной водой для
поглощения С2Н4 и третий — щелочным раствором пирогаллола
для поглощения О2 \
Залить поглотительные сосуды реактивами можно до присоеди-
нения их к прибору или после присоединения. Удобнее заливать
поглотительные сосуды (кроме бромного), когда они уже присоеди-
нены к прибору. Для этого на задний отросток сосуда 21 надевают
резиновую трубочку длиной 15—20 см с небольшой воронкой. Угло-
вой кран 24- заливаемого сосуда ставят на сообщение капилляра
с гребенкой и бюреткой, два других крана 21 и 14 должны быть
закрыты, кран 15 должен быть повернут влево на гребенку и бю-
ретку, рабочая бюретка должна быть заполнена доверху раствором
соли, кран 5 закрыт. Реактив наливают в сосуд через воронку, а
воздух из сосуда засасывают в бюретку, для чего опускают вниз
напорную склянку и открывают кран 5. Реактивом должна запол-
няться вся камера, а также форсунка и капилляр до резино-
вых муфт. Поднятия реактива в форсунке достигают поворотом кра-
на 24 на форсунку, что нужно делать очень осторожно, так как по
форсунке очень легко засосать реактив в гребенку и даже в бюретку,
после чего необходимо снова мыть весь прибор. При повороте крана
24 на форсунку следует сначала поднять напорную склянку вверх
и ввести воздух через форсунку в сосуд, а затем уже начать осто-
рожно подтягивать жидкость, опуская для этого напорную склянку.
После этого кран 24 закрывают. Во вспомогательной камере 18 дол-
жен оставаться гидравлический затвор.
При заливке поглотительного сосуда пирогаллолом водный рас-
твор пирогаллола с водным раствором щелочи лучше смешивать в
самом сосуде (во избежание порчи пирогаллола), т. е. влить отдельно
в сосуд водный пирогаллол и водную щелочь и уже в самом сосуде
перемешать их.
Заливать поглотительный сосуд бромной водой необходимо в
вытяжном шкафу, отсоединив сосуд от прибора. Сначала в сосуд
1 В зависимости от целей анализа в поглотительные сосуды могут быть
налиты и другие реактивы.
772
наливают дестиллированную или кипяченую воду через задний тубу-
лусик 21 или через отросток 30 и затем уже через отросток посред-
ством маленькой воронки, соединенной с ним встык, вливают при-
мерно 0,5 см3 брома. После добавки брома надо быстро закрыть кран
и надеть резиновый мешок, надутый воздухом, и с помощью мешка
поднять воду в форсунке и в капилляре. Подтянуть уровни можно
также бюреткой после присоединения сосуда к прибору. Бром дает
очень сильные ожоги кожи и, испаряясь, вредно влияет на дыха-
тельные пути, поэтому, наливая бром, необходимо принять все меры
предосторожности.
Если уровни реактивов в капиллярах и форсунках 79, про-
стояв 1—4 мин., не снижаются, можно считать, что все резиновые
соединения и краны 24 не пропускают воздуха.
Анализ газа. Анализ газа производят на приборе, заранее со-
бранном и проверенном на плотность. Перед забором пробы газа для
анализа прибор приводят в рабочее состояние, т. е. все уровни жид-
костей подводят вверх так, чтобы их мениски были хорошо видны
и не входили в резиновые соединения. Мениски в бюретках устана-
вливают на риски 25 (микробюретки) и 100 (рабочая бюретка).
В манометре мениск может занимать произвольное положение. Если
забираемый газ находится под давлением, удобнее мениск в мано-
метре установить на делении 2—3 см3 по микробюретке. Мениск
в манометре можно установить в любом положении при открытых
кранах 77 и 5, манипулируя напорной склянкой 7. В колоколе мениск
доводят до конца трубочки 9, а мениски в поглотительных сосудах
в форсунке 79 и отростке —до соединительных резиновых муф-
точек. Все мениски, кроме микробюретки и манометра, подтягивают
посредством рабочей бюретки и напорной склянки 7.
Сначала подтягивают мениск в колоколе. Для этого кран 15 по-
вертывают вправо на петлю и бюретку, после чего рабочая бюретка
и колокол представляют два сообщающихся вверху сосуда. Микро-
бюретку и манометр нужно отключить от напорной склянки, кран 5
следует повернуть на рабочую бюретку и напорную склянку, кран
манометра 14 закрыть. Затем открывают воздушный кран бюретки
на воздух и, поднимая напорную склянку, выталкивают воздух из
рабочей бюретки и заполняют ее раствором соли. После этого воз-
душный кран закрывают. Опуская затем осторожно напорную склян-
ку, засасывают воздух в рабочую бюретку из колокола и вместе с
тем поднимают в колоколе уровень жидкости до конца отростка 9.
Во время поднятия уровня мениска в колоколе наблюдается дви-
жение трех уровней, из которых первый — уровень жидкости в
рабочей бюретке — и второй — уровень жидкости между бюрет-
кой и внутренними стенками колокола — поднимают. Подняв уро-
вень мениска в колоколе, кран 15 повертывают влево на гребенку,
отключая этим колокол от бюретки.
Далее также посредством рабочей бюретки подтягивают пооче-
редно мениски в поглотительных сосудах. Для этого сначала опять
выталкивают воздух из рабочей бюретки через воздушный кран.
8 В. А. Соколов
113
Затем кран 24 подтягиваемого сосуда повертывают на капилляр
и на гребенку. Два других крана 24 и кран 14 должны быть закрыты.
Опуская напорную склянку, засасывают воздух из поглотитель-
ного сосуда в рабочую бюретку и вместе с тем поднимают уровень
жидкости в капилляр. Переключив кран 24 на форсунку, подтя-
гивают уровень жидкости в форсунке. Подтягивать жидкость в фор-
сунке, как уже указывалось выше, нужно очень осторожно, соблю-
дая при этом все указанные выше правила. При этих условиях жид-
кость в форсунке подтягивается плавно. Подтянув уровни жидкости
в форсунке 19 и капилляре, кран 24 закрывают.
При подтягивании уровней рекомендуется зажимать пальцами
левой руки резиновую трубку, соединяющую напорную склянку
и бюретку, чтобы задержать быстрый подъем жидкости. Подтягива-
ние уровней жидкости как в колоколе, так и в поглотительных сосу-
дах происходит вследствие разрежения, создаваемого в рабочей
бюретке при опускании напорной склянки, благодаря чему атмосфер-
ное давление, передающееся через тубулусик сосуда реактиву
последнего, значительно превышает давление в бюретке и, следо-
вательно, заставляет реактив сосуда подниматься в его рабочей части.
В свою очередь реактив производит давление на воздух, имеющийся
в поглотительном сосуде, который под этим давлением переходит
в бюретку, давит на имеющуюся там жидкость и заставляет послед-
нюю опускаться.
Взятие пробы газа и отсчет. Установив уровни
жидкости в колоколе и поглотительных сосудах, поднимают уровни
в микробюретке и рабочей бюретке, выталкивая воздух через воз-
душный кран. После этого кран 5 лучше закрыть, т. е. разобщить
бюретки с напорной склянкой. Кран 15 должен быть открыт на гре-
бенку, а кран 14 закрыт. Далее прибор соединяют с аспиратором или
с газопроводом.
Если пробу газа берут из аспиратора, то левый свободный конец
гребенки непосредственно соединяют с ним стеклянным капилля-
ром; если же пробу газа берут из газопровода, то к прибору необхо-
димо присоединить фильтр со стеклянной ватой и через фильтр соеди-
нить с газопроводом.
Чтобы засосать пробу газа из аспиратора, надо напорную склянку
аспиратора поставить вверх, напорную склянку прибора спустить
и затем открыть кран аспиратора и краны 14 и 5 прибора.
Сначала забирают 25—50 см3 газа и кран 14 закрывают. Забран-
ный газ выбрасывают наружу через воздушный кран, делая это с
целью промывки соединительного капилляра и гребенки. Затем уже
забирают пробу газа для анализа. Снова закрывают кран 14 и на-
бирают в рабочую бюретку примерно 75 см3 газа (до риски 25), после
чего кран 5 переключают на микробюретку и набирают еще в микро-
бюретку около 25 см3 газа (лучше меньше). После этого кран 14 за-
крывают. Кран манометра 77 во время забора пробы газа должен
быть закрыт. Не следует стремиться взять точно 75 см3 газа но ра-
бочей бюретке и 25 см3 по микробюретке.
114
Чтобы отсчитать объем взятого газа, сначала устанавливают ме-
ниск в рабочей бюретке точно на риске 25 посредством напорной
склянки при открытом кране 5; установив мениск в рабочей бюретке,
кран 5 переключают на микробюретку и устанавливают мениски в
микробюретке и манометре. Для зтого открывают кран 77 и, мани-
пулируя напорной склянкой 7, устанавливают на одной горизон-
тали уровни менисков в Микробюретке и манометре. После этого
кран 5 закрывают, напорную склянку ставят на стол и берут отсчет
газа по шкале микробюретки. Отсчет берут в том случае, если уста-
новленные мениски в микробюретке и манометре и зеркальные изо-
бражения их в зеркале 12 находятся на одной горизонтали. Запись
отсчета принято вести по водяной шкале (микробюретка обычно
имеет одну шкалу). Взяв отсчет, кран 77 манометра закрывают и
открывают лишь тогда, когда снова берут отсчет. Открывая кран 77
манометра, нужно всегда поднимать напорную склянку, так как
через манометр возможно проскакивание пузырьков воздуха в микро-
бюретку.
Если забранная проба газа превышает 100 см3, часть газа выбра-
сывают через воздушный кран. Выбрасывать избыток газа лучше
из микробюретки при закрытом кране 77, поднимая напорную склян-
ку 7.
Поглощение. После записи отсчета взятой пробы газа
ведут анализ путем поглощения составных частей газа.
Чтобы поглотить СО2, газ из бюретки прокачивают через пер-
вый сосуд, наполненный раствором КОН. Для этого поднимают
напорную склянку, ставят угловой кран 24 поглотительного сосуда
в положение, соединяющее бюретку (через гребенку) с форсункой
(риски крана направлены вверх и влево), и, открыв кран 5 микро-
бюретки, переводят сначала весь газ из микробюретки в сосуд, а
затем, не опуская напорной склянки, кран 5 переключают на ра-
бочую бюретку и переводят газ из рабочей бюретки также в сосуд.
Газ выходит из форсунки отдельными пузырьками внизу рабочей
камеры и, пройдя через реактив, поднимается в верх камеры.
Собираясь здесь, газ давит на реактив, который под этим давлением
начинает опускаться и входит во вспомогательную камеру 18, вы-
талкивая оттуда воздух через тубулусик сосуда наружу или в ре-
зиновый мешочек. Таким образом, газ будет соприкасаться с реак-
тивом, который отберет от него ту составную часть, которую данный
реактив поглощает.
Переведя весь газ из рабочей бюретки в сосуд и не опуская напор-
ной склянки, переключают угловой кран 24 на соединение бюретки
с капиллярным отростком. Теперь, опустив напорную склянку,
перекачивают весь газ обратно в рабочую бюретку, а потом опять
в сосуд через форсунку и т. д. Газ через КОН прокачивают три раза,
пользуясь при этом только рабочей бюреткой. Вводят газ в погло-
тительный сосуд всегда через форсунку 19, а вытягивают из сосуда
в бюретку через капилляр. После третьего прокачивания часть
газа, примерно 75 см3, забирают в рабочую бюретку и затем, пере-
ключив кран 5, остаток газа забирают в микробюретку. Далее
8*
775
делают отсчет газа, как это было описано выше при заборе пробы
газа.
Во время прокачки газа через поглотительные сосуды, да и вообще
во время анализа, нужно твердо помнить правило: «следи за подни-
мающимся уровнем», во избежание засасывания реактива или рас-
твора соли в гребенку. Также нужно помнить, что кран 77 мано-
метра во время анализа всегда должен быть закрыт и открывать
его надо только при взятии отсчета.
Если КОН долгое время был в употреблении или содержание
СО2 в газе большое, необходимо прокачать газ через тот же реактив
еще 2—3 раза для контрольного отсчета.
Если показание после этой контрольной прокачки и показание,
записанное ранее, совпадут, то прокачку пробы газа через КОН
можно кончить и приступить к дальнейшему ведению анализа. Если
же эти показания не совпадут, то нужно продолжать прокачку до
тех пор, пока не совпадут какие-либо два рядом стоящие показания.
Чтобы поглотить С2Н4, газ прокачивают 3—4 раза через сосуд
с бромной водой. После этого, не делая отсчета, производят 3—4
раза прокачку газа опять через КОН для поглощения выделившихся
паров брома, без поглощения которых данные анализа будут оши-
бочны, после чего уже берут отсчет. При анализе газа на содержание
С2Н4 на дне бромного сосуда должна быть свободная капля брома
(бурая тяжелая капля), иначе раствор бромной воды может оказаться
слабым и неспособным поглощать.
Чтобы поглотить О2, газы прокачивают через сосуд с пирогалло-
лом 5—6 раз.
При поглощении О2 необходимо сделать контрольный отсчет.
Пирогаллол очень скоро портится при хранении, поэтому его не-
обходимо проверить перед анализом газа. Проверяют пирогаллол
прокачиванием определенного количества забранного воздуха. При
хорошем качестве пирогаллола для полного поглощения кислорода
из 100 см3 воздуха достаточно 6—7 прокачиваний. Содержание кисло-
рода в воздухе принимается равным 20,9%.
Сжигание. СО, Н2 и СН4 в газе на описываемом приборе
определяют методом сжигания над высокоактивным платиновым
катализатором. Данный катализатор дает возможность определить
СО, Н2 и СН4 в газе двумя методами: 1) методом раздельного сжи-
гания, дающим прямое определение СО, Н2 и СН4; 2) методом обыч-
ного суммарного сжигания, когда содержание СО, Н2 и СН4 опре-
деляется путем косвенных подсчетов.
Анализ с помощью раздельного сжига-
ния (по Г. Ф. Кнорре). После поглощения СО2, С2Н4 и О2 запи-
сывают весь остаток газа по газовой шкале. Затем часть газа выбра-
сывают из бюретки, так как для сжигания необходимо добавить
к нему воздух.
Количество газа, оставляемого для сжигания, зависит от со-
держания в нем СО, Н2 и СН4. При анализе топочных газов для
сжигания газа можно оставить 45—55 см3. Выбрасывают часть газа
через воздушный кран, для чего напорную склянку поднимают и,
116
открыв воздушный кран (кран 77 должен быть закрыт), поднимают
мениск жидкости в рабочей бюретке примерно до половины второго
раздутия, т. е. выше риски 50, и затем, не спуская напорной склянки
(во избежание подсоса воздуха), закрывают воздушный кран. Далее
делают отсчет оставшегося газа, как было описано выше.
После этого добавляют в бюретку воздух. Для этого напорную
склянку ставят на стол, кран 5 повертывают на сообщение бюреток
с напорной склянкой (кран 77 закрыт). Затем открывают воздуш-
ный кран и засасывают воздух в рабочую бюретку примерно до риски
25 и в микробюретку до деления 3—4 слА Далее воздушный кран
закрывают, делают отсчет и записывают объем.
Добавив воздух, приступают к сжиганию газа. Сначала на сжи-
гательную петлю надвигают обогревательную печь, дающую темпе-
ратуру 150—200°, и переключают ее к источнику тока. Концы термо-
пары петли присоединяют к милливольтметру. В холодильник петли
пускают слабый ток воды. Когда петля нагреется до 150 — 200°,
начинают сжигание газа. Для этого, подняв напорную склянку,
повертывают кран 75 вправо (на петлю и бюретку) и начинают
перекачивать газ из бюретки через петлю в колокол и обратно.
Перекачивают газ 3 — 4 раза. После этого обогревательную печь
отодвигают от петли и начинают охлаждать газ и петлю, пере-
качивая опять газ в колокол и бюретку. Когда стрелка милливольт-
метра покажет 0, подтягивают уровень жидкости в колоколе до кон-
ца отростка 9 и переключают кран 15 влево, т. е. снова на гребенку
и бюретку. Если газ и петля совсем остыли, то уровень жидкости
в отростке 9 не должен подниматься, в противном случае необходимо
еще несколько раз перекачать газ через петлю в колокол и обратно.
Для контроля температуры газа в рабочую бюретку вставляют термо-
метр. Убедившись в полном остывании газа, делают отсчет дого-
ревшего газа и записывают объем.
Во время перекачивания газа через петлю, как при перекачи-
вании газа через поглотительные сосуды, пользуются только рабо-
чей бюреткой и лишь после последнего прокачивания забирают газ
в рабочую бюретку и микробюретку.
При температуре обогрева петли до 200° в газе сгорают только
СО и Н2, метан же остается несгоревшим. Обогревать петлю выше
200° не следует, так как это может повлечь частичное сжигание
метана. Нельзя также сжигать СО и Н2 при температуре ниже 150°,
так как в этом случае можно не получить полного сжигания
СО и Н2.
Соотношения объемов при сжигании будут следующие:
1. Н2 4- 0,50*-------> Н2О
1 объем 0‘5 объема 0 объемов
2. СО + 0,5О2--------> СО2
1 объем 0,5 объема 1 объем
При реакции горения видно, что объем газа после сжигания
будет меньше, чем до сжигания, и что в сожженном газе будет нахо-
диться СО2, объемное количество которой будет равно содержа-
вшемуся в газе количеству СО.
777
После поглощения СО2 переходят к сжиганию СН4. Тех-
ника сжигания СН4 та же, что и при сжигании СО 4- Н2, с той
лишь разницей, что сжигательную петлю обогревают до 450°, для
чего на петлю надвигают печь, дающую температуру 450°. Добавле-
ния воздуха перед сжиганием СН4 не требуется, так как воздух
сразу добавляют в избытке.
Для сжигания метана также достаточно трех прокачиваний.
Запись отсчета после сжигания СН4 делается только после полного
остывания газа. При горении СН4 образуются вода и СО2:
СН4Ч-2О2->2Н2О4-СО2
1 объем4-2 объема-»0 объемов4-1 объем
поэтому объем газа после сжигания метана будет меньше, чем до
сжигания, и в сожженном газе будет находиться СО2. Для опре-
деления СО2, оставшегося после сжигания метана, газ опять про-
качивают через поглотительный сосуд с КОН и данные отсчета запи-
сывают. Этим заканчивается анализ по методу раздельного сжи-
гания.
Подсчет метана по данным анализа возможен двумя способами.
Так, из уравнения горения метана получим
объем СН4 = объему СО2 (сжигание при 450° С)
или
СН4 = 0,5К,
где К — сокращение объема после сжигания при 450° С.
При правильном ведении анализа и при отсутствии в газе дру-
гих предельных углеводородов (С2Нв и т. д.) оба определения долж-
ны дать одну и ту же величину. Данные подсчета метана по первой
и по второй формулам могут резко разойтись, если отсчет СН4 при
450° был взят при неполном остывании газа. Поэтому температур-
ный контроль должен быть строгим как при сжигании газа, так
и во все время ведения анализа. Для получения точных результатов
необходимо, чтобы все отсчеты газа от начала до конца анализа были
взяты при одной и той же температуре.
На точность анализа газа сильно влияет также стекание жид-
кости в бюретке и манометре. При плохом стекании на стенке бю-
ретки остаются капли жидкости, количество которых может быть
различно при взятии различных отсчетов, что отражается на вычи-
слениях. Поэтому необходимо тщательно мыть измерительную часть
прибора.
Из приведенных выше формул для подсчета СО, Н2 и СН4 видно,
что при раздельном методе сжигания мы имеем прямое определе-
ние СО и СН4 и только определение Н2 зависит от определения СО,
но возможная при этом ошибка весьма незначительна, так как в
формулу подсчета Н2 количество СО входит в половинном значении.
Анализ по метод у. суммарного сжигания.
Метод суммарного сжигания состоит в том, что СО, Н2 и СН4 сжи-
гаются совместно, как это имеет место при сжигании в платиновом
118
капилляре, или над раскаленной платиновой проволокой, или в
кварцевой петле. Поэтому после поглощения СО2, С2Н4 и О2 и доба-
вления воздуха к сжигаемому газу на петлю сразу надевают печь,
дающую температуру 450°, и газ прокачивают через петлю 3—4 раза.
При этом СО, Н2 и СН4 сгорают одновременно. После остывания
газа делают отсчет.
Далее газ прокачивают через сосуд с КОН для определения в
сожженном газе СО2, образовавшейся от горения СО и СН4, и за-
тем газ прокачивают через сосуд с пирогаллолом для определения
оставшегося после сжигания О2. Поглощением О2 анализ закан-
чивается.
Вычисления при газовом анализе
Вышеприведенные расчеты основаны на том предположении,
что молекулярные объемы всех газов равны. Углекислота и этан
уклоняются от этой закономерности. Поэтому, когда парциальные
давления этих двух газов высоки в газовых смесях, при определе-
нии парафиновых углеводородов с помощью сожжения должны быть
введены поправки.
Следующие уравнения позволяют произвести точные вычисления
в газовом анализе:
Р и d3 С — (di + 2 — dp СО„
26 d3(d2—0,5 —2dx) ’
rij СО2 лр тт
Сп4 — —2С2п6,
“з
где С2Н6—объем этана в части газа, взятой для сожжения (теоре-
тический объем, который занял бы этан, если бы он был
идеальным газом);
СН4 —объем метана в части газа, взятой для сожжения;
^—молекулярный объем метана (объем идеального газа
равен единице) при парциальном давлении, под которым
он находится в части газа, взятой на сожжение;
d2 —молекулярный объем этана (объем идеального газа равен
единице) при парциальном давлении, под которым он
находится в части газа, взятой на сожжение;
d3—молекулярный объем углекислоты при парциальном да-
влении, под которым она находится в газовой смеси,
образовавшейся после сожжения;
СО2 — наблюденная контракция (уменьшение объема) вследствие
поглощения углекислоты;
С— наблюденная контракция вследствие конденсации водя-
ных паров.
Значения молекулярных объемов были приведены ранее (§ 2).
Если парциальные давления метана и углекислоты во время ана-
лиза ниже 30%, то ошибки, полученные вследствие отклонения моле-
кулярных объемов от молекулярного объема идеального газа, могут
не приниматься во внимание.
119
Пример расчета анализа газа
Таблица -8
Отсчеты объемов газа Сим- вол Вычисления Результаты, % (объемн.)
i 2 3 4 5
Объем газа, взятого для анализа, 100,0 .... а | а — Ь = 100—94,1 = со2 5,9
Объем после поглощения СО2 94,1 ...... Объем после поглощения непредельных углево- дородов 93,5 ъ с 1 = 5,9 Ь —с = 94,1—93,5 = Непре- 0,6
Объем после поглощения кислорода 93,3 .... d = 0,6 с -d = 93,5—93,3= дельные о2 0,2
Объем после пропуска- ния газа через трубку с окисью меди 74,0 е = 0,2 d—e~ 93,3—74 н2 19,3
Объем после поглощения углекислоты, образо- вавшейся в трубке с окисью меди, 70,9 . . f = 19,3 е—/= 74,0 —70,9= СО 3,1
Объем газа, взятого для сожжения, 30,0 . . . g = 3,1
Объем кислорода, взято- го для сожжения, 90,0 Л g + /2 = 30,0+90,0= 120 к —- —
Объем после сожжения 60,6 к общему объему газа перед сожжением g 4- h — к = 59,4 = контрак-
Объем после поглощения углекислоты, образо- вавшейся при сожже- нии, 27,2 1 ции (уменьшение объема), полученной благодаря кон- денсации воды при сожжении к — / = 60,6 — 27,2 = 33,4 = —
Объем С2Н6 в части газа, взятой для сожжения т = объему образовавшейся углекислоты т = 2/3 (2 объема СО2 —
Объем СН4 в части, взя- той для сожжения . . п контракция); 2/3 (2-33,4 — —59,4) = 4,93 п = объем СО2 —2 объема
Объем С2Н6 в первона- чальной пробе газа р С2Нв = 33.4—2-4,93 = 23,6 mf 4,93-70,9 Д р- 1 30,0 -"'6 с2н6 11 fi
Объем СН4 в первона- чальной пробе газа Ч nf 23,6-70,9 30,0 =ss’7 сн4 55,7
Объем N2 в первоначаль- ной пробе газа . . . По разности n2 3,6
Итого........................... — 100.G
120
§ 4. Реагенты, применяемые при общем газовом анализе, и их
действие на различные газы
Поглотители для углекислоты (СО2). Едкий кали и едкий
натр. Для определения углекислоты употребляют растворы едкого
кали и едкого натра, причем одни аналитики отдают предпочтение
первому, другие — второму. Различные авторы рекомендуют кон-
центрации едкого кали и едкого натра, значительно различающиеся
между собой. Для обычных целей газового анализа приготовляют
растворы в следующих пропорциях:
Раствор едкого кали
Едкий кали (чистый) ............................ 360 г
Дестиллированная вода в количестве, необходимом для
доведения раствора до ........................ 1000 см3
Раствор едкого натра
Едкий натр (чистый).............................315г
Дестиллированная вода в количестве, необходимом для
доведения раствора до....................... 1000 см3
Эти растворы одинаково пригодны для поглощения углекислоты.
Существует мнение, что раствор едкого кали обладает несколько
большей скоростью поглощения, чем раствор едкого натра. В обоих
случаях рекомендуется применять КОН и NaOH, очищенные элек-
тролизом, так как эти же щелочи употребляют и при изготовлении
растворов пирогаллола для определения кислорода. Плохо очи-
щенные едкие щелочи могут обусловить выделение этими растворами
окиси углерода.
Если газовая смесь содержит компоненты кислотного характера,
как-то: двуокись серы, сероводород или циан, то эти компоненты
должны быть удалены раньше, чем будет определена углекислота,
иначе они будут поглощены едкой щелочью и таким образом будут
приняты за углекислоту. Едкие щелочи также поглощают некото-
рые углеводороды (в частности, бензол).
Едкий барий. Когда количество углекислоты, содержа-
щейся в анализируемом газе, очень невелико и приближается к со-
держанию углекислоты в воздухе, некоторые аналитики предпочи-
тают поглощение углекислоты баритовой водой вместо поглощения
более сильными щелочами. Этот поглотитель обычно употребляют,
когда определяют углекислоту титрованием. В большинстве случаев
этот метод позволяет определять СО2 с большой точностью. Употре-
бляющийся для этих целей раствор содержит в литре 14 г гидрата
окиси бария и 1 г хлористого бария; последний вводят для обеспе-
чения лучшей конечной точки анализа. 1 ем3 этого раствора равно-
значен приблизительно 1 см3 углекислоты, измеренной при нор-
мальных условиях. Раствор стандартизируется щавелевой кислотой,
приготовляемой путем растворения 5,3653 г чистой кристалличе-
ской щавелевой кислоты в 1 л кипящей, только что приготовленной
дестиллированной воды. В качестве индикатора употребляют фенол-
фталеин. В сосуд, содержащий 1 л (или больше) газа, вводят через
121
капельную воронку точно измеренное количество гидрата окиси
бария и хорошо встряхивают. Избыток гидрата окиси бария от-
титровывают щавелевой кислотой.
Поглотители для непредельных углеводородов (этен, пропен,
ацетилен, бутен, бензол и более тяжелые углеводороды). Д ы м я-
щ а я серная кислота. Если нет необходимости в отдель-
ном определении каждого из компонентов, то употребляют дымя-
щую серную кислоту. Для большей надежности определений свежая
кислота должна содержать избыток в 15—20% SO3.
Кислота может быть покупная или приготовляться нагреванием
концентрированной кислоты в реторте, соединенной с охлаждаемым
приемником, содержащим чистую концентрированную серную кис-
лоту. Таким образом, SO3, поступающий из реторты в приемник,
абсорбируется находящейся в нем серной кислотой. Реагент годится
для употребления только до тех пор, пока в нем имеется избыток
SO3, показателем чего служит сильное выделение дыма.
Реагент поглощает также и высшие углеводороды алканового
ряда, как-то: пентан С5Н12 и гексан С6Н14, так как последние рас-
творяются. Алкены, как этен С2Н4 и пропен С3Н6, ароматические
углеводороды, как бензол С2Н6, а также ацетилен С2Н2, поглощаются
вследствие химической реакции. При этом этен переходит в C2H6S2O7,
бензол — в C6H4SO3, ацетилен — в C2H4SO4. При продолжитель-
ном соприкосновении кислота поглощает также пропан и бутан,
а частично и этан, но в обычных условиях количество поглощенного
этана незначительно, за исключением тех случаев, когда процент-
ное содержание этана велико. При прохождении газа через пипетку
с дымящей серной кислотой некоторое количество SO3 уносится
вместе с проходящим газом. Поэтому после этой операции необхо-
димо пропустить газ через пипетку с едкой щелочью.
Бро м. Для поглощения непредельных углеводородов некото-
рые аналитики применяют бром. Его приготовляют растворением
5 г бромистого калия в 100 см3 дестиллированной воды и добавле-
нием брома до полного насыщения раствора. Бром поглощает этен,
переходящий в бромистый этен С2Н4Вг2, и пропен, переходящий в
бромистый пропен С3Н3Вг2. Бром не поглощает бензола полностью
подобно дымящей серной кислоте. Как и при дымящей серной кис-
лоте, некоторое количество брома увлекается вместе с газом из пи-
петки. Поэтому после пипетки с бромом газ необходимо пропустить
через пипетку со щелочью перед его измерением.
Бром частично поглощает и тяжелые парафиновые углеводороды.
Однако этан и пропан поглощаются бромом незначительно.
Хлористый палладий. В разбавленном растворе —
от 0,5 до 1%—PdCl2 восстанавливают до палладия окисью угле-
рода этеном и его гомологами по реакциям
PdC 12 + СО + Н2О = 2НС1 + Pd 4- СО2
PdCl2 + С2Н4 = С2Н4С12 +Pd
122
Осажденный палладий — черного цвета и легко определяется.
Эти реакции применяют для обнаружения малых количеств окиси
углерода или алкенов и дают возможность заметить 0,2% любого
из этих соединений при медленном пропускании 100 см3 таза через
30—40 см3 раствора. Хлористый палладий медленно поглощает также
водород, поэтому пользоваться им можно только для качественных
определений водорода.
Растворы для разделения алкенов. Для разделения этена, про-
пена и бутена на фракции при отсутствии окиси углерода используют
достаточно удовлетворительно растворы серной кислоты различ-
ной крепости в присутствии сернокислого серебра и сернокислого
никеля в качестве катализатора. Эти растворы следующие: серная
кислота плюс 10% воды, серная кислота обычной концентрации
(удельный вес 1,84) и серная кислота обычной концентрации, содер-
жащая 10% дымящей серной кислоты (20% избытка SO3). Все эти
растворы насыщены сернокислым никелем и сернокислым серебром.
Первая смесь служит для поглощения бутена, вторая — пропена
и третья — этена. Эти растворы частично поглощают высшие ал-
каны, пары бензола и некоторое количество окиси углерода. Отсюда
следует, что’ эти смеси не пригодны для употребления в обычных
условиях в поглотительной аппаратуре. При наличии окиси угле-
рода удовлетворительные результаты были получены при пропуска-
нии газа через серную кислоту удельным весом 1,62 для поглощения
бутена, через серную кислоту удельным весом 1,70— для погло-
щения пропена и через обычную кислоту, содержащую 10% дымя-
щей серной кислоты, —для поглощения этена. Действие этих кислот
несколько замедленно, и разделение бутена и пропена происходит
не очень точно.
Поглотители для кислорода. Для поглощения кислорода наи-
более часто употребляют сильно щелочные калиевые или натриевые
растворы пирогаллола. Однако наиболее быстрым способом погло-
щения, а в некоторых случаях и наиболее точным является, по мне-
нию многих исследователей, поглощение кислорода хлористым хро-
мом. Существует много рецептов для приготовления раствора пиро-
галлола.
Раствор пирогаллола в едком кали. Раствор
едкого кали приготовляют растворением 600 г электролитического
едкого кали в воде с последующим прибавлением воды до 1000 см3.
Точно так же раствор пирогалловой кислоты готовят растворением
300 г чистой пирогалловой кислоты в 800 см3 воды с последующим
добавлением воды до 1000 см3. Для приготовления пирогаллового
раствора, идущего на анализ, берут (в зависимости от емкости пи-
петки) 40—50 см3 раствора пирогалловой кислоты, помещают в по-
глотительную пипетку и доливают в нее 3 объема (140—175 см3)
раствора едкого кали. Пипетку закрывают резиновой пробкой для
предохранения от влияния воздуха, после чего оба раствора тща-
тельно перемешивают.
123
Раствор пирогаллола в едком натре. Рас-
твор готовят растворением 1 части чистого едкого натра в 1 части
воды и прибавлением 2,5 объемов (150—160 см3 в зависимости от
емкости пипетки) этого раствора к 1 объему (60—65 см3) раствора
пирогалловой кислоты, приготовленного по вышеописанному спо-
собу.
Оба указанных раствора пирогаллола — хорошие поглотители
кислорода, и со свежеприготовленными растворами процесс погло-
щения идет весьма быстро. После поглощения 8—10 объемов кисло-
рода действие их ослабевает. Их активность зависит также от тем-
пературы. При температуре около 0° поглощение идет очень медленно.
Наиболее благоприятна для поглощения температура приблизительно
20 — 30°. Употребление едкого натра или едкого кали, очищенных
спиртом, не рекомендуется, так как возможно выделение растворами
окиси углерода. Некоторые считают, что окись углерода выде-
ляется только после того, как раствор поглотит 8—10 объемов
кислорода, другие же полагают, что если раствор достаточно кон-
центрирован, т. е. насыщен, то он не выделяет окиси углерода. Эти
растворы, будучи сильно щелочными, поглощают все те газы, кото-
рые поглощаются растворами едких щелочей, а также небольшие
количества непредельных углеводородов.
Желтый фосфор применяется для поглощения кисло-
рода в некоторых газах. Для анализа его отливают небольшими
палочками диаметром 4—6 мм. Приготовляют палочки следующим
образом. Желтый фосфор помещают под водой в пробирку, кото-
рую затем опускают в водяную баню с температурой около 50°. Стек-
лянную трубку требующегося внутреннего диаметра погружают в
расплавленный фосфор, который втягивают в трубку всасыванием
до нужной высоты, после чего трубку вынимают и немедленно погру-
жают в сосуд с холодной водой. Затвердевший фосфор выталкивают
стеклянной палочкой в пипетку, наполненную водой. Необходимо
следить, чтобы фосфор не приставал ко дну; при этом следует менять
воду в пипетке для удаления окислов фосфора. Это необходимо де-
лать всякий раз, когда дым в пипетке поглощается медленно или не
полностью в течение одной-двух минут. Пипетка, содержащая фос-
фор, закрывается черной бумагой, чтобы избежать перехода желтого
фосфора в красный под влиянием света. Помимо того, что желтый
фосфор опасен в обращении, он не может употребляться для опре-
деления кислорода, если его много содержится в газе, например в
воздухе, так как в этом случае фосфор выделяет такое большое коли-
чество тепла, что расплавляется. Желтый фосфор не может быть
использован для анализа газов, содержащих следы этена, бензола,
спиртов, нефтяных паров или аммиака, так как они препятствуют
действию фосфора на кислород. Действие желтого фосфора замед-
ляется при падении температуры ниже 15°. Замедление реакции
наступает также при содержании в анализируемом газе больших
количеств метана и этана.
Хлористый хром. Несмотря на то, что хлористый хром
является наиболее быстро действующим поглотителем кислорода и
124
применяется для этого уже давно, он все же не может быть широко
использован для поглощения кислорода, так как обладает свой-
ством быстро восстанавливаться и приготовление, а также хранение
его в восстановленном виде представляют большие трудности. При-
готовление хлористого хрома можно производить следующим обра-
зом. Хромовую кислоту обрабатывают соляной кислотой или же хлор-
ный хром растворяют в воде и подкисляют соляной кислотой. Кис-
лотный раствор переводят в колбу, откуда воздух вытесняют свободным
от кислорода газом; колба должна закрываться пробкой с двумя отвер-
стиями для крана и соединительной трубки. Хлорный хром восстана-
вливают прибавлением металлического цинка. Когда вся соль вос-
становится до хлористого хрома, раствор переводят в насыщенный
раствор уксуснокислого натрия, причем выпадает красноватый оса-
док нерастворимого уксуснокислого хрома, являющегося более устой-
чивым. После промывания уксуснокислого хрома разбавленной
уксусной кислотой и водой он при прибавлении соляной кислоты
переходит в хлористый хром. Приготовленный таким образом хло-
ристый хром поглощает только кислород. Сероводород, углекислота
и другие газы проходят через раствор хлористого хрома без измене-
ния их объемов.
Более простой метод приготовления хлористого хрома состоит
в том, что в раствор хлорного хрома, находящийся в пипетке, до-
бавляют соляную кислоту и восстанавливают до хлористого хрома
цинком в атмосфере, лишенной кислорода.
Приготовленный таким путем раствор быстро поглощает кисло-
род и в присутствии избытка соляной кислоты не поглощает серо-
водород и углекислоту, но медленно выделяет свободный водород.
Если избыток кислоты невелик и температура держится около 20°,
то количество выделяющегося свободного водорода невелико и не
может быть замерено в бюретках обычного типа.
При нейтрализации раствора избытком цинка раствор может по-
глощать небольшие количества сероводорода и углекислоты. Но
это обстоятельство не имеет значения, так как сероводород и угле-
кислоту определяют перед определением кислорода. Устойчивые
растворы в количествах, достаточных для заполнения пипетки, лучше
всего приготовлять восстановлением амальгамированным цин-
ком. Для приготовления восстановителя 5 г ртути растворяют
сначала в разбавленной азотной кислоте (1 : 1), полученный раствор
доводят до 250 см3 и помещают в литровый сосуд. 500 г гранулиро-
ванного цинка помещают в сосуд, взбалтывают несколько минут и
оставляют стоять 10—15 мин., после чего цинк промывают деканта-
цией водой. В трубку редуктора насыпают слой стеклянных
бус, которые покрывают стеклянной ватой, а поверх ваты кладут
тонкий слой асбеста.
Держа амальгамированный цинк под водой, помещают его
на асбестовый слой, промывают 57%-ным раствором серной кислоты
и затем водой. Потом раствор из 75 г хлорного хрома и 190 см3
воды, подкисленной 10 см3 соляной кислоты, приливают к восста-
новителю до окрашивания воды на дне редуктора. После этого
725
редуктор через небольшой кран соединяют с задней камерой пипетки,
наполненной коксовым или другим газом, свободным от кислорода,
куда по каплям и спускают остаток раствора, проходящий при этом
через восстановитель. Пипетку наполняют таким способом до соответ-
ствующего уровня. Далее пипетку немедленно соединяют с приборол!
и раствор надежно изолируют от воздуха. Этот раствор не выделяет
водород и поглощает весь кислород из воздуха при 2—4 подачах
воздуха через барботирующую пипетку.
Гидрат закиси марганца. Гидрат закиси марганца
применяют для определения малых количеств кислорода, которые
не могут определяться ни фосфором, ни другими обычными спосо-
бами. Свежеприготовленный гидрат закиси марганца в виде бе-
лого осадка в водном растворе прекрасно поглощает кислород, окис-
ляясь до гидрата окиси марганца, имеющей коричневый цвет. При
обработке гидроокиси иодистым калием и серной кислотой освобо-
ждается иод и раствор окрашивается в розовый цвет. При взбалтывании
раствора с 100 см3 газа легко определяются следы кислорода. Опре-
деление может быть не только качественным, но и количественным.
Для этого свободный иод оттитровывают тиосульфатом натрия. Вслед-
ствие нерастворимости этот реагент не годится для пипеточного ана-
лиза
Поглотители для окиси углерода. Для определения окиси угле-
рода применялись до сих пор аммиачный раствор однохлористой
меди и солянокислый раствор однохлористой меди. Но в последнее
время было установлено, что Си2О, взвешенная в серной кислоте,
также поглощает окись углерода.
Добавление фенола еще более повышает качество поглотителя.
Приводим способы приготовления и свойства реагентов для по-
глощения окиси углерода.
Аммиачный раствор одг.охлористой меди
Хлористый аммоний (химически чистый)................... 300 г
Однохлористая медь..........«... ............• • 270 г
Вода .............................................• . . 1000 см3
Чтобы приготовить раствор для поглощения СО, требуется 1 объем
NH4OH (уд. вес 0,90) на 3 объема раствора однохлористой меди.
Аммиачный раствор поглощает кислород из воздуха. Поэтому
лучше всего растворить соль в воде, а затем в пипетке приливать к
раствору аммиак в соответствующей пропорции. Закрыв поглоти-
тельную часть пипетки, заполняют компенсационную ее часть рас-
твором и приливают аммиак до тех пор, пока выпавший белый оса-
док не начнет исчезать при взбалтывании. Избыток аммиака в виде
паров легко уходит вместе с газом после поглощения, поэтому не-
обходимо после поглощения СО пропускать газ через пипетку с
кислотой для поглощения паров аммиака. Поэтому многие аналитики
предпочитают употреблять кислотный раствор вместо аммиачного.
1 Приготовление раствора гидросернистого натрия см. стр. 75.
126
Кислотный раствор однохлористой меди приготовляют следу-
ющим образом.
Кислотный раствор однохлористой меди
Однохлористая медь (химически чистая)............. 75 г
Соляная кислота (химически чистая, концентрированная) . 600 см3
Вода в количестве, необходимом для доведения раствора
до.............................................. 1000 см3
Упругость паров этого раствора не превышает упругости паров
воды, но для предохранения газов от паров соляной кислоты, выде-
ляющихся в поглотительной пипетке, необходимо после поглощения
окиси углерода пропустить газ через пипетку со щелочью. Кислот-
ный раствор однохлористой меди также поглощает кислород, по-
этому необходимо предохранять его от влияния атмосферного воз-
духа.
Оба раствора однохлористой меди — кислотный и аммиачный —
обладают большой способностью к окислению, поэтому необходимо
хранить их в восстановительной среде. Несколько кусочков медной
проволоки или медной стружки, положенных в раствор в компен-
сационную часть пипетки, предохраняют однохлористую медь от
окисления. Кислотный раствор должен иметь желто-зеленый или
соломенный цвет, и несколько капель раствора, влитые в воду, долж-
ны дать белый осадок. В качестве восстановителя кислотного рас-
твора можно употреблять вместо меди раствор хлористого олова.
Раствор хлористого олова готовят растворением 25 г SnCl2 в 100 см3
разведенной соляной кислоты (1 : 1). Полученный раствор медленно
приливают к раствору однохлористой меди, который необходимо
при этом встряхивать до приобретения им соломенного цвета. Это
условие обязательно, потому что если раствор окрашен в зелено-
ватый цвет, то он будет поглощать, кроме окиси углерода, и другие
газы.
Действие этих растворов обусловлено образованием нестойкого
соединения, имеющего формулу Си2С12 • 2СО. Свежеприготовлен-
ные растворы поглощают нацело окись углерода. Однако после по-
глощения некоторого количества окиси углерода соединение ста-
новится весьма нестойким и отдает окись углерода при малом содер-
жании ее в газе. Это заставляет пропускать анализируемый газ через
2—3 пипетки с поглотителем. Когда последняя пипетка поглотит
10 см3 окиси углерода, необходимо заменить ее пипеткой со свежим
раствором, а остальные пипетки передвинуть на одну. Кроме кис-
лорода и окиси углерода, раствор поглощает также ацетилен, этен
и другие газы. Поэтому упомянутые газы должны поглощаться до
определения окиси углерода, необходимо также сделать несколько
контрольных предварительных анализов. Это правило обязательно'
для обоих растворов (кислотного и аммиачного), как свежепригото-
вленных, так и бывших в употреблении. Вместо двух или трех пипе-
ток с однохлористой медью можно употреблять одну пипетку с одно-
хлористой медью вместе с пипеткой, содержащей смесь сульфата
меди (Cu2SO4) и /1-нафтола. Газ пропускают сначала через пипетку
127
с однохлористой медью, где поглощается большая часть окиси угле-
рода; остальная часть поглощается во второй пипетке, содержащей
указанную выше смесь сульфата меди с /7-нафтолом. Достаточное
для помещения в пипетку количество этой смеси приготовляют сле-
дующим образом:
Закись меди (химически чистая, приготовленная мокрым
способом)............................................ 20 г
Серная кислота (химически чистая, удельный вес 1,84) . . 200 см3
Дестиллированная вода....................................25 слг3
Д-нафтол (химически чистый) .............................25 см3
Кислоту приливают к воде и дают ей остыть. Закись меди поме-
щают в фарфоровую ступку и, приливая понемногу разбавленную
кислоту, быстро и хорошо смешивают их, растирая пестиком до тех
пор, пока вся закись не будет взвешена. Тогда приливают Д-нафтол
и смешивают обе жидкости. После фильтрации смеси через стек-
лянную вату для удаления несмешавшейся части Д-нафтола
смесь готова к употреблению. Ее необходимо тотчас же пере-
вести в пипетку и защитить от действия воздуха, так как она посте-
пенно поглощает из него кислород. Смесь нужно держать при темпе-
ратуре выше 15°, потому что нафтол при более низкой темпера-
туре отделяется от смеси.
Для получения наилучших результатов закись меди следует
готовить мокрым способом, так как при приготовлении сухим спо-
собом она дает смесь, медленно поглощающую окись углерода. Про-
дажная закись меди в большинстве случаев приготовляется сухим
способом, поэтому желательно приготовлять ее в лаборатории. Наи-
более удобно готовить закись меди путем обработки ацетата меди
глюкозой. К 1000 см3 воды, налитой в 2-л сосуд, добавляют 100 г
.ацетата меди, нагревают до растворения ацетата в воде и, если не-
обходимо, отфильтровывают нерастворившиеся частицы. В то же
время растворяют 60 г глюкозы в 400 см3 воды и добавляют этот рас-
твор в кипящий раствор ацетата меди. Кипятят до тех пор, пока
голубая окраска несколько не побледнеет, после чего раствор оста-
вляют стоять до выпадения осадка красной закиси меди. Осадок
декантируют, собирают на фильтр, промывают декантацией несколько
раз водой, а потом один раз спиртом. Приготовленную таким обра-
зом закись меди высушивают при 90—100°, лучше всего в вакуум-
эксикаторе. Готовую смесь хранят в закрытом сосуде.
Необходимое количество глюкозы можно получить из тростни-
кового сахара растворением 60 г его в 400 см3 воды; затем к раствору
добавляют 3 капли серной кислоты и кипятят его в течение 5 мин.
Избыток кислоты нейтрализуют углекислым кальцием, после чего
отфильтровывают раствор.
Закись меди образует сульфат меди (Cu2SO4), слабо раствори-
мый и очень устойчивый по отношению к серной кислоте. С окисью
углерода сульфат образует устойчивое соединение. Эта смесь не
выделяет свободной окиси углерода, подобно описанному раствору
однохлористой меди, и предел насыщения зависит от количества
присутствующего сульфата меди. Вышеописанная смесь может по-
128
глотить объем окиси углерода, в 18 раз превышающий ее собствен-
ный. Ее действие несколько медленнее действия однохлористой меди;
после того как она будет насыщена окисью углерода наполовину,
действие раствора еще более замедлится, однако поглощение окиси
углерода останется достаточно полным до предела насыщения. Для
быстрого анализа можно применить две пипетки: одна из них содер-
жит однохлористую медь, а вторая — сернокислую закись меди.
Газы с большим содержанием окиси углерода предварительно
обрабатывают однохлористой медью, а затем пропускают через пи-
петку с указанной смесью. Эта смесь поглощает также этен, про-
пен, бутен, небольшое количество ацетилена и немного кислорода,
но не поглощает метан, другие алканы, водород и азот.
§ 5. О результатах и точности общего газового анализа
Приборы и реагенты, применяемые при общем анализе, исполь-
зуют по-разному в зависимости от целей анализа и состава газа.
Когда компоненты газа известны и число этих компонентов невелико,
можно подобрать такие условия анализа, которые обеспечат высо-
кую точность определений. При подобных наиболее благоприятных
условиях точность определений может быть доведена до величин,
лежащих в пределах колебаний объема газа от изменений темпера-
туры и атмосферного давления за время анализа. При очень тща-
тельном проведении анализа можно добиться точности определений
в 0,1% по отношению к объему газа.
Менее точными становятся аналитические определения, когда
компоненты газа неизвестны и их число велико. Следует учесть,
что почти каждый реагент, применяемый в приборах для общего
анализа, в некоторой степени растворяет любой газообразный компо-
нент. Присутствие хорошо растворимых газов плохо отражается на
точности анализа, поэтому хорошо растворимые газы желательно
определять и удалять в начале анализа. В данном случае речь идет
о физическом растворении газа в жидкости, а не о химическом по-
глощении в результате той или иной реакции. Многие реагенты,
химически связывая тот или иной компонент, оказывают в то же
время большее или меньшее химическое действие и на некоторые
другие компоненты. Все эти обстоятельства при наличии сложной
смеси газов снижают точность и надежность анализа.
Одной из реакций, дающих наиболее надежные и точные опре-
деления, является поглощение углекислого газа щелочью. Эта реак-
ция происходит быстро и количественно и почти не затрагивает дру-
гих газов, за исключением газов, имеющих кислотный характер,
как H2S и др. Эти кислотные газы при поглощении щелочью будут
определяться суммарно с СО2. Сульфат меди или другие поглоти-
тели H2S растворяют также и СО2, что может частично повлиять
и на содержание других газов. Если в газе помимо СО2 присутствует,
например, H2S или другие кислотные газы, то проведение анализа
по описанной выше методике становится более затруднительным и
для многих целей точность подобного анализа будет неудовлетвори-
9 в. А. Соколов.
129
тельной. Реагенты, применяющиеся для поглощения H2S, будут
влиять как на содержание в газе СО2, так и некоторых других ком-
понентов. Поэтому сложные смеси кислотных газов не могут быть
точно проанализированы с помощью вышеописанных методов общего
анализа и для подобных случаев применяют другие методы, которые
описываются в одной из следующих глав.
Следует указать еще на один газ, который методами общего ана-
лиза определить затруднительно и который может исказить резуль-
таты анализа других компонентов. Это — закись азота N2O, кото-
рая довольно хорошо растворяется в воде, но химически не связы-
вается и не поглощается ни одним из применяемых в общеА! анализе
реагентов. В природных условиях закись азота была обнаружена
впервые в почвенных и подпочвенных газах при газосъемочных ра-
ботах. Концентрация ее была невелика; закись азота была обна-
ружена с помощью методов микроанализа (см. главу VI). Однако
не исключена возможность наличия закиси азота в некоторых слу-
чаях в природных газах и в значительно больших концентрациях.
Закись азота присутствует в некоторых промышленных газах. Зна-
чительная растворимость закиси азота в воде и водных растворах
обусловит, так сказать, ее размазывание по всем реагентам в про-
цессе анализа, что поведет к неточности всех определений. При сож-
жении с окисью меди при 300° закись азота остается практически
неизменной. При сожжении с платиной при высокой температуре
закись азота разлагается на азот и кислород. Если температура или
время контакта недостаточны, то часть закиси азота останется и при
замере после сожжения и удаления СО2 будет принята за азот. Для
точного определения закиси азота и ее отделения от других газов
необходимо применять специальные методы, описываемые в главах
V и VI.
Определение индивидуальных газообразных углеводородов с по-
мощью методов общего анализа представляет очень трудную, а в
большинстве случаев невыполнимую задачу. Эта задача может быть
удовлетворительно разрешена лишь при наличии в газе только од-
ного или двух наиболее легких газообразных углеводородов (метан
и этан; метан и этен или ацетилен и т. п.). Присутствие углеводоро-
дов, более тяжелых, чем этан и этен, уже заметно искажает резуль-
таты анализа. Определение индивидуальных углеводородов при
значительном их числе производится специальными методами, опи-
сываемыми в главах IV и VI. В этих случаях при общем анализе
более или менее точно может быть определено путем сожжения только
суммарное содержание углеводородов. Однако при одновременном
присутствии Н2, СО и непредельных снижается и точность суммар-
ного определения углеводородов путем сожжения. Водород и окись
углерода могут определяться с помощью окиси меди при 300°; однако
при этом окисляется ацетилен, а частично и другие непредельные
углеводороды.
Азот определяется при общем анализе по разности. В этом азоте,
как было упомянуто выше, может присутствовать частично закись
азота. Кроме того, в азоте присутствуют редкие газы — гелий, неон,
130
аргон, криптон и ксенон. Для определения индивидуальных редких
газов применяют специальные методы, описываемые в главе VII.
Все вышеуказанное заставляет при постановке общего анализа
и для получения наиболее точных результатов тщательно выбирать
реагенты и методы определения отдельных компонентов.
§ 6. Некоторые методы определения СО2, СО и Н2
Определение СО2. Для многих практических случаев
нет необходимости проводить более или менее полный общий ана-
лиз, а требуется знать содержание в анализируемом газе только
одного или двух компонентов.
Наиболее часто содержание од-
ного или двух компонентов тре-
буется определить при контроле
за газами, являющимися ^про-
дуктами горения. Так как го-
рение происходит с помощью
воздуха, то основное внимание
часто обращают только на ^со-
держание СО2 и СО, а в некото-
Фиг. 60. Приборы для определения СО2.
рых случаях и на содержание кислорода. В этих случаях чаще всего
применяют автоматические газоанализаторы, описываемые в IX главе.
Однако иногда определяют СО2 путем эпизодических замеров с по-
мощью приборов, содержащих раствор щелочи. Замеряя объем газа
до и после его поглощения щелочью, определяют содержание дву-
окиси углерода. Для этой цели можно пользоваться бюреткой с при-
соединенной к ней единственной пипеткой, содержащей раствор
щелочи.
9*
131
Имеются и более простые приборы, применяемые в тех случаях,
когда не требуется очень большая точность определений. На
фиг. 60, а представлена схема прибора, в котором проводятся подоб-
ные определения; он применяется для анализа дымовых газов [13].
Этот газ в количестве нескольких кубических сантиметров засасы-
вается ручным поршневым насосом и попадает в сосуд, наполненный
сухим поглотителем (например, гашеной известью). Сосуд, содер-
жащий щелочь, соединен с манометром, другой конец которого сооб-
щается с воздухом. Поглощение углекислоты щелочью после закры-
тия прибора вызовет уменьшение давления в камере, что и будет
обнаружено по смещению уровня жидкости, содержащейся в мано-
метре. Шкалу манометра можно предварительно проградуировать
на процентное содержание СО2.
Преимуществом подобного прибора является его крайняя про-
стота и быстрота определений; точность показаний прибора порядка
0,5-1%.
Схема другого аналогичного прибора изображена на фиг. 60, б.
Анализируемый газ проходит через фильтр, где задерживаются твер-
дые дымовые частицы, и просасывается резиновой грушей через
верхнюю камеру прибора при условии, что верхняя камера будет
отъединена от нижней с помощью пробки. Нижняя камера запол-
нена щелочью. Внизу этой камеры имеется отвод, к которому при-
соединена сбоку стеклянная трубка, имеющая вверху закрывающий
ее клапан.
Перед анализом прибор устанавливают так, чтобы уровень ще-
лочи стоял на нуле при открытом клапане и кране. После впуска
газа, что производится путем просасывания, открывается пробка,
газ при этом входит в контакт со щелочью и начинается его погло-
щение. По понижению уровня щелочи в манометрической трубке
можно судить о содержании СО2. Эту трубку можно проградуировать
на процентное содержание углекислоты. Клапан, закрывающий
манометрическую трубку, во время измерения должен быть открыт.
Определение окиси углерода с применением пятиокйси иода
Описываемый ниже аппарат позволяет производить анализы на
СО достаточно быстро с точностью до 0,002% в обычных условиях.
Комплект аппаратуры, содержащий приспособление для очистки
азотом, показан на фиг. 61. Образец газа пропускают через аппарат
из склянки 7 с помощью вакуумной установки, присоединенной к
капиллярной трубке 2. Образец последовательно проходит через
трубку 3, сосуды с хромовой кислотой 4 и 5, V-образную трубку 6,
трубку с пятиокисью иода 7, трубку 8, содержащую иодистый калий,
и ртутный затвор 9 перед вакуумной линией, присоединенной к ка-
пиллярной трубке 2. В то время как образец газа проходит по ап-
парату, азот выпускается через трубку 10 при закрытом кране 77.
После наролнения аппарата закрывают газовый кран 72 и откры-
вают кран 77. Вследствие этого азот проходит через ртутный затвор
13 и через систему так же, как и образец газа. Скорость прохожде-
132
ния азота регулируют так, чтобы он понемногу выходил и из
трубки 10.
Склянка с образцом газа 7 сделана из стекла и имеет объем около
500 см3. Точный ее объем должен тщательно определяться, и содер-
жащийся в ней газ должен приводиться вычислениями к нормаль-
ным условиям.
Для подогрева пятиокиси иода применяют воздушную баню с
электрическим нагревом. Баня состоит из двух латунных коробок
Фиг. 61. Прибор для определения СО с применением J2OS.
14 и 15. На внутреннюю коробку поверх изоляции наматывают спи-
раль хромовой (хромель А) проволоки 22-го калибра длиной 18,3 м.
Спираль удерживается на месте асбестовым цементом. Промежутки
между стенками 16 заполнены асбесто-фибровой изоляцией. Асбе-
стовая пластинка 17 закрывает баню сверху. Температура бани ре-
гулируется реостатом 18 (100 ом и 1,5 а) с несколькими сериями
витков.
Сосуды с хромовой кислотой наполняют через воронки 19 и 20.
Опорожнивают их с помощью вытяжных склянок, присоединяемых
к стеклянным трубкам с кранами 21 и 22; V-образная трубка 6 со-
держит в верхней части слой стеклянной ваты, под ней слой твер-
дого едкого кали, затем опять слой стеклянной ваты; остальное про-
133
странство трубки заполнено фосфорным ангидридом. Стеклянные
пробки 23, 24 и 25 заливают парафином.
В трубке 7 помещены перемежающимися слоями стеклянная вата
и пятиокись иода.
Трубка имеет три отделения, наполненные йодистым калием,
что обеспечивает достаточную поверхность для поглощения всего
иода, выделяющегося в приборе.
Рама 26, поддерживающая стеклянные части, сделана из I" угло-
вого железа.
Приготовление и нормализация растворов. Для удаления не-
предельных углеводородов, альдегидов и некоторого количества
водяных паров служат две колонки с хромовой кислотой (пригото-
вленной насыщением концентрированной серной кислоты двухро-
мовокислым калием). Обычно колонки с хромовой кислотой сохра-
няют при комнатной температуре; однако если известно, что в газе
присутствует небольшое количество альдегидов или этена или хотя
бы предполагается их присутствие, то первую хромовую колонку
нагревают до 100°. Для удаления следов кислоты и небольших коли-
честв углекислоты насыпают сверху в V-образную трубку 6 немного
едкого кали и остаток трубки наполняют фосфорным ангидридом,
который служит для полного осушения газа перед его впуском в
трубку с пятиокисью иода.
Пятиокись иода. Трубка с пятиокисью иода имеет при
анализе температуру 150°. При этой температуре реакция с окисью
углерода происходит достаточно быстро и полно. Трубки, иду-
щие от колонки с пятиокисью иода, должны нагреваться; благо-
даря этому иод в них не конденсируется. Для анализа употребляет-
ся пятиокись иода высшей чистоты, известная в продаже как ^ан-
гидрид иодной кислоты».
Чистый иодный ангидрид должен иметь температуру распада
не ниже 260°. Однако ангидрид, имеющийся в продаже, обычно со-
держит низшие окислы иода, иодную кислоту и, возможно, другие
компоненты, которые имеют более низкую температуру распада,
чем иодный ангидрид. Для приготовления иодного ангидрида, при-
годного для определения окиси углерода, необходимо держать его
при температуре 205—215° около двух дней при пропускании через
него достаточного количества воздуха или кислорода. При этой тем-
пературе разложатся и уйдут все нестойкие компоненты. Затем тем-
пературу снижают до 150° и очистку продолжают еще два дня. После
этого можно считать иодный ангидрид годным для анализа. После
пропускания азота в течение 2—3 час. через ангидрид крахмал, при-
бавленный в иодистый калий, не должен обнаружить следов иода.
Однажды приготовленный иодный ангидрид может служить в те-
чение 6 месяцев при ежедневной работе. Анализ производится в те-
чение 15 мин. (приблизительно).
Азот, употребляемый для очистки аппа-
ратур ы. Для очистки аппарата, а также для удаления из трубки
с иодным ангидридом свободного иода необходимо, чтобы азот абсо-
131
лютно не содержал окиси углерода. Воздух в комнате, где сжи-
гается какое-нибудь топливо, и воздух промышленных центров всегда
содержит небольшие количества окиси углерода. Поэтому при при-
менении воздуха для очистки необходимо предварительно удалить
из него окись углерода. Практически для этого нужно иметь второй
аппарат, подобный описанному выше. Применение второго аппа-
рата специально для очистки азота потребовало бы дополнительной
затраты труда и времени. Чтобы избежать этого, рекомендуется
пользоваться чистым азотом в баллонах. Азот, приготовленный спо-
собом глубокого охлаждения, совершенно свободен от окиси угле-
рода и не вступает в реакцию с иодным ангидридом.
Раствор йодистого калия. Употребляется раствор
из 10% (по весу) йодистого калия и 90% (по весу) дестиллироваиной
воды. Его нужно хранить в темном месте; при надлежащем приго-
товлении он не утрачивает своих качеств в течение нескольких не-
дель. При освещении солнечным светом раствор медленно разла-
гается. Его необходимо проверять, применяя в качестве индикатора
крахмал. Свежий раствор не должен вызывать появления синей
окраски.
Крахмальный индикатор. Картофельный крахмал
является лучшим индикатором для определения свободного иода.
Он чувствителен к двум каплям 0,001 N раствора иода в 20 см3 воды.
Пшеничный крахмал чувствителен только к трем каплям. Для при-
готовления раствора 2—3 г крахмала разбавляют несколькими ку-
бическими сантиметрами холодной воды, а затем обваривают кипя-
щей дестиллироваиной водой в количестве до 200 см3. Кипячение
продолжают в течение 2—3 мин., а затем охлаждают до комнатной
температуры. Каждый день необходимо приготовлять свежий рас-
твор, иначе при титровании иода не может быть ясного перехода
от синего до белого цвета. Кроме того, при употреблении старого
раствора вместо синего цвета получается розовый, который пере-
ходит в белый цвет очень медленно, и, таким образом, конечная точка
титрования становится неточной.
Если титруется концентрированный иодный раствор, то крах-
мал не следует приливать до тех пор, пока большая часть не обес-
цветится тиосульфатом. Если прилить к концентрированному рас-
твору иода крахмал, то выпадающий при этом хлопьевидный осадок
затруднит определение конечной точки реакции.
Раствор тиосульфата натрия. Раствор Na2S2O3
должен быть 0,001 N или, другими словами, содержать 0,24332 г
в 1 л. Перед титрованием нужно оставить раствор на один месяц
для установления равновесия. По прошествии этого времени рас-
твор остается весьма устойчивым в течение нескольких месяцев.
Рекомендуется приготовлять свежий раствор ежемесячно и титро-
вать его только по истечении месяца. Раствор тиосульфата натрия
титруют чистым иодным раствором. Имеющийся в продаже иод пред-
варительно очищают возгонкой одним из известных способов.
Навеска очищенного двойной возгонкой иода для получения
(приблизительно) 0,001 N раствора должна быть очень точно взве-
135
Шена, и полученный из нее раствор считается стандартным. Иод
очень гигроскопичен и легко испаряется. Поэтому при взвешива-
нии его нужно принимать специальные предосторожности. В стакан-
чик для взвешивания помещают около 2 г KJ и несколько капель
воды и взвешивают, затем прибавляют около 2 г J и снова взвеши-
вают. Разница в весе указывает на количество чистого иода. После
взвешивания в раствор добавляют дестиллированную воду в коли-
честве до 1 л.
Нормальность раствора может быть проверена окисью мышьяка.
Для обычных целей вес иода и его нормальность могут не прове-
ряться.
Раствор тиосульфата натрия титруется полученным раствором
иода с применением крахмала в качестве индикатора.
Для расчетов применяется следующая схема вычислений.
Если 0,0974 г J взвешены в 1000 см3 воды, то в 1 см3 раствора
содержится 0,0000974 г J2; при титровании 10 см3 J2 требуется
8,37 см3 Na2S2O3.
I см3 Na2S2O3--Hy-= 1,195 см3 J2,
или
1 см3 Na2S2O3= 1,195-0,0000974 = 0 0001164 г J2;
1 см3 Na2S2O3 =• 0,0001164 = 0,0518 см3 СО
при 0° и 760 мм рт. столба.
Коэфициент к для перевода кубических сантиметров Na2S2O3
в кубические сантиметры СО выводится следующим образом.
В приготовленном растворе тиосульфата натрия 1 см3 равен
0,000127 г иода; из реакции J2O54-5CO = Ja4-5CO2 эквивалент
0,00007 г СО получается из уравнения
140 : 253,8 = х: 0,000127,
откуда х = 0,00007.
1 см3 окиси углерода при 0° и 760 мм рт. столба весит 0,00125 г;
0,00007 г окиси углерода равны 0,056 см3 при 0° и 760 мм рт. столба.
Приводя результаты к 15° и 760 мм рт. столба, имеем:
0,056 :х= 273 : 288,
откуда х = 0,05608 см3 СО при 15° и 760 мм рт. столба (сухой).
Так как взятая проба всегда насыщена водяными парами, то это
число должно быть приведено от сухого состояния к насыщенному.
Упругость водяных паров воды при 15° равна 12,79 мирт. столба:
PV (Р'~ p)V'
Т — т> ,
где р—упругость водяных паров при 15°.
136
Тогда, подставляя числовые значения, имеем:
760-0,05908 _ 747,21 V'
288 ~ 288 ’
откуда V'= 0,060091 см3 СО при 15° и 760 мм рт. столба (насы-
щенный).
Общий множитель из вышеописаш ого определится:
140-288-760-х
“ 253,8-0,00125-273-747,21 ’
где х равно содержанию иода в а на 1 см3 тиосульфата;
_ 30643200
64715297 х‘
Так как взятая проба всегда насыщена водяными парами, то
полученное число должно быть преобразовано с помощью уравнения
ру (Р.-р^У'
т т >
где /г—давление паров воды, при 0' равное 4,579 мм рт. столба.
Тогда
760-0,056 _ 755,42 V'
273 — 273 *
где V —объем окиси углерода, насыщенной парами воды при (F
и 760 мм рт. столба, см3.
Исправленная формула для коэфициента к:
, 140-760-х
К~ 253,8-0,00125-755,42 ’
где х —содержание иода в г на 1 см3 тиосульфата;
238,9
Описанный метод и аппаратура могут применяться для обычных
анализов продуктов горения при содержании окиси углерода до
0,002%. Азот, кислород и углекислота, содержащиеся в продуктах
горения, или малые количества водорода, метана, сероводорода,
сернистого ангидрида и окисей азота практически не влияют на точ-
ность анализа. Если присутствует формальдегид в количестве свыше
1%, то первая колонка с хромовой кислотой должна быть нагрета
до 100°. Если же присутствует этен, хотя бы в малых количествах,
то анализ становится неточным.
Определение СО с помощью гопкалита
Для определения окиси углерода применяют также приборы,
основанные на поглощении СО гопкалитом. Гопкалит представляет
собой катализатор, состоящий из окисей некоторых металлов
(Си, Мп и др.).
137
Этот катализатор окисляет СО до СО2 за очень короткий про-
межуток времени при невысокой температуре. В результате этого
окисления образуется углекислый газ, который поглощается и опре-
деляется каким-либо из вышеописанных способов. Для определения
окиси углерода этим путем требуется предварительно удалить из газа
углекислоту.
Определение Н3
Для определения водорода путем поглощения применяют в неко-
торых случаях раствор из коллоидального палладия. Этот раствор
обладает способностью поглощать водород при температурах порядка
20—30°. Способ приготовления этого раствора описан Вейнбергом
[14]. Помимо поглощения водорода указанным жидким раствором в
некоторых случаях применяют поглощение его губчатым палладием.
ГЛАВА ЧЕТВЕРТАЯ
АНАЛИЗ УГЛЕВОДОРОДНЫХ ГАЗОВ
§ 1. Углеводородные газы и их свойства
Углеводородные газы являются важнейшим объектом эксплоатации
при использовании как природных, так и промышленных газов.
Большое значение углеводородных газов в промышленности и
специфические особенности этих газов привели к тому, что для их
анализа разработаны различные специальные методы и приборы.
Одной из причин специфичности анализа углеводородных газов
является то, что эти газы по своим химическим свойствам мало отли-
чаются друг от друга. Можно говорить только о том, что определен-
ные группы углеводородных газов, принадлежащие к одному и тому
же гомологическому ряду, обладают некоторой схожестью хими-
ческих свойств. В то же время между членами одного и того же гомо-
логического ряда различия в химических свойствах очень незна-
чительны. Во всяком случае они не настолько велики, чтобы ими
можно было пользоваться для разделения или определения индиви-
дуальных углеводородов.
Например, газообразные алканы, начиная с метана и кончая
парами гептана и еще более тяжелых углеводородов, обладают весьма
близкими химическими свойствами. Эти углеводороды весьма инерт-
ны при действии кислорода и других окисляющих веществ при обыч-
ных температурах, химически нейтральны и не поглощаются щело-
чами и слабыми кислотами. Более тяжелые углеводороды, начиная
с пропана, обладают только большей способностью растворяться
в крепкой серной кислоте.
Более тяжелые углеводороды лучше растворяются в органиче-
ских растворителях. Например, если коэфициент растворимости
метана в бензине равен 0,6, то для пропана эта величина составляет
уже более 5, а пентан смешивается с бензином в любой пропорции.
Алкены отличаются от алканов значительно большей реакцион-
ной способностью. По месту двойной связи эти углеводороды срав-
нительно легко присоединяют галоиды. Кроме того, они вообще
гораздо лучше алканов окисляются и сравнительно легко полимери-
зуются. Однако между алкенами точно так же нет большой разницы
в их реакциях с теми или иными реагентами, чтобы можно было пол-
ностью отделить индивидуальные углеводороды друг от друга.
133
Имеется, однако, несколько способов разделения простейших алкенов
с помощью брома и серной кислоты определенной концентрации.
Цикланы (циклопропан, циклобутан, циклопентан) и аромати-
ческие углеводороды имеют много общего в отношении химических
свойств с алканами. Они также обладают сравнительно большой
стойкостью к окислению при невысоких температурах и ко многим
другим реакциям. Многие жидкие при обычных условиях углеводо-
роды обладают при этом значительной упругостью пара и, присут-
ствуя в газовой фазе, анализируются как газообразные компоненты.
Некоторые из описанных ниже приборов, получивших широкое
применение, специально предназначены для анализа смесей жидких
углеводородов путем подогрева их и перевода в газовую фазу.
Следует отметить, что общее количество известных индивидуаль-
ных газообразных и жидких углеводородов со значительной упру-
гостью пара очень велико. Оно намного превышает количество всех
других компонентов, могущих встречаться как в природных, так и
в промышленных газах. В то же время для многих практических
целей желательно знать не суммарное содержание углеводородов
и не содержание отдельных групп углеводородов (алканов, алкенов
или цикланов), а содержание индивидуальных углеводородных со-
единений.
В табл. 9 и 10 дан перечень наиболее простых углеводородов и
приведены температуры их кипения.
Многочисленность индивидуальных газообразных и жидких
углеводородов и схожесть их химических свойств привели к
тому, что для точного анализа смесей углеводородных газов разра-
Таблица 9
Температура кипения алкановых углеводородов
Наименование Температура кипения, °C Наименование Температура кипения, °C i
Метан СН4 —161,4 2,2-диметилпентан C7Hie + 78,6
Этан С2Нв — 88,6 2,4-диметилпентан C,Hie + 83,9
Пропан С3Н8 — 42,1 3,3-диметилпентан C7Hie + 87.0
Изобутан С4Н10 .... — 117 2-метилгексан C7Hi6 . . + 90,4
н-Бутан С4Н10 ..... — 0,5 3-метилгексан С7Н16 . . + 92,0
2,2-диметилпропан С5Ню + 9,5 3-этилпентан C7Hie . . -t- 93,8
Изопентан С5Н12 + 28,0 н-Гептан С7Н1в .... 4- 98,6
н-Пентан С6Н12 .... 4- 36,1 2,5-диизобутил С8Н1ь . . 4-109,2
2,2-диметилбутан CeHu , 4- 49,6 н-Октан C8Hi8 4-125,5
Диизопропил СвН14 . . 4- 58,1 Нонан С9Н20 4-149,5
Изогексан CeHi4 ...... . + 62,0 Декан Ci0H22 .... 4-173,0
3-метилпентан С6Н14 . . ! + 64,0 Ундекан СцЩ .... 4-197,0
н-Гексан СвН14 ... ; + 69.0
UO
Таблица 10
Температура кипения алкенов и цикланов
Наименование Температура кипения,СС Наименование Температура кипения, °C
Алкены Диметилацетилен С4Н6 । i +28,9
Этен С2Н4 Пропен С3Н8 —103,8 —47,7 Аллен С3Н4 1,3-бутадиен С4Н6 . . . 1,2-бутадиен С4Н6 . . . —34,3 —2,6 + 19,0
Бутены Цикланы
1,1-диметилэтен С4Н8 (изобутен) ..... Этилстен С4Н8 (бутен) . 1,2-диметилэтен С4Н8 (изобутен) • -7,0 —6,0 + 1,4 Циклопропан С3Н8 . . . Метилциклопропан С4Н8 Циклобутан С4Н8 . . . 1,1 -диметилтриметилен с3н10 —34,4 +5,0 + 12,6 +21,0
н-Пентен С5Н10 .... +40,0 Метилциклобутан С3Н10 +42,0
Ацетилен С2Н2 .... —83,6 Циклопентан С5НТ8 . . + 49,5
Аллилен С3Н4 —23,2 Циклогексан C6Hi2 . . + 31,4
Этилацетилен С4Н6 + 18,5 Бензол С8 Н6 +79,6
ботаны и применяются различные специфические методы. Эти методы
вследствие вышеуказанных обстоятельств основаны преимуще-
ственно на различии физических свойств углеводородов.
К числу реакций, которым легко подвергаются углеводороды
при высоких температурах, принадлежит их сожжение. Начиная
с температуры красного каления, в присутствии кислорода все угле-
водородные газы сгорают с образованием в качестве конечных про-
дуктов окисления углекислоты и паров воды. Однако одним
сожжением нельзя дать полного анализа сложных углеводородных
смесей. Как было описано в главе III, путем сожжения можно опре-
делить содержание одного или двух простейших углеводородов. Иден-
тификация углеводородов может производиться путем учета коли-
чества сгоревшего газа и количества образовавшейся углекислоты.
Например, один объем метана при сгорании дает один объем угле-
кислоты. Один объем этана дает при сожжении два объема угле-
кислоты. Однако такое же двойное количество углекислоты дает
и сожжение этена, а также и ацетилена. Поэтому, получая, напри-
мер, двойной объем углекислоты после сожжения и не зная, что пред-
ставляют собой эти углеводороды, нельзя решить вопрос, являются
ли сгоревшие углеводороды этаном, этеном или ацетиленом. Про-
водя сожжение не с окисью меди, а над накаленной платиной с до-
бавкой кислорода, можно по количеству истраченного на сожжение
кислорода судить, является ли сгоревший газ этаном или этеном и
каково содержание каждого из этих компонентов. Однако сожже-
ние смеси из трех и более углеводородных компонентов вообще не
позволяет решить задачи о составе газа, поскольку число неизвест-
ных превышает число независимых уравнений, которые могут быть
построены для этих случаев.
141
Комбинированное сожжение с поглощение?,! алкенов бромной
водой или серной кислотой может помочь в разрешении задачи ана-
лиза лишь для наиболее легких компонентов., поскольку в указан-
ных реагентах растворяются не только алкены, но и пары алканов.
Если требуется определить суммарное содержание углеводородов,
то задача решается сравнительно легко' путем сожжения. Задача
становится еще легче, если мы имеем дело с природными газами,
так как в природных газах алкены обычно не встречаются. Присут-
ствие в газе только алканов, даже если при этом присутствуют водо-
род и окись углерода, позволяет полностью определить суммарное
содержание углеводородов путем сожжения. Окись углерода и водо-
род могут сжигаться с помощью окиси меди при температуре 300°,
а все углеводороды могут определяться путем сожжения при более
высоких температурах.
Иногда при анализе газов наибольший интерес представляет
суммарное содержание только тяжелых углеводородов, начиная с
этана. В этих случаях задача может решаться путем конденсации
газа при температуре жидкого воздуха с предварительным удале-
нием из газа углекислоты и паров воды. Метан при температуре жид-
кого воздуха откачивается, а тяжелые углеводороды могут быть
определены после удаления жидкого воздуха путем откачки и изме-
рения конденсата.
Для анализа углеводородных газов предложены следующие ме-
тоды:
1) разгонка смесей углеводородов при низких температурах;
пользуясь значительной разницей в температурах кипения легких
углеводородов, можно разделить смесь на соответствующие компо-
ненты; однако этот способ трудно применять для анализа и раздель-
ного определения углеводородов, температуры кипения которых
близки друг к другу;
2) разгонка углеводородных смесей с помощью эффективно дей-
ствующих ректификационных колонок; в настоящее время этот спо-
соб является наиболее разработанным;
3) разделение смесей углеводородов на отдельные компоненты
с помощью сорбентов;
4) анализ с помощью масс-спектрографа, позволяющий про-
водить детальный анализ углеводородов; однако этот способ весьма
сложен (см. главу VIII).
§ 2. Анализ углеводородных газов с применением разгонки при
низких температурах
Одним из свойств, которым можно воспользоваться для разде-
ления и определения индивидуальных углеводородов, является раз-
личие их упругости паров при различных температурах. Поэтому
для легких углеводородов можно подобрать такие условия, когда
упругость паров одного углеводорода во много раз выше упругости
паров другого, более тяжелого углеводорода. Вследствие этого воз-
можно при соответствующих температурах производить откачку
U2
того или иного углеводорода и таким образом отделить его от всех
других углеводородов. Однако точное разделение возможно только*
для наиболее легких компонентов и притом требует применения
низких температур, начиная с температуры жидкого воздуха.
В табл. 11 приведены упругости насыщенных паров различных
углеводородов при температурах от 0 до —200°.
Таблица 11
Упругость паров углеводородов при различных температурах
Темпера- тура, °C Давление, мм рт. столба
метан этан про- ! пан j изо- бутан бутан ИЗО- ; пен- 1 тан 1 пен- тан этен про- пен аце- тилен
-210 0,8 — — — —-
—200 3,7 — — — — —• — — ——
—183 76 0,01 — —- -- — — —— 0,0002
-170 358 0,15 0,0002 — — — — 4 0,0005 0,0058
—160 810 1,0 0,004 — — — — 4 0,007 0,06
-150 —— 4,1 0,03 0,001 — — — 15 0,057 0,41
—140 14 0,20 0,01 — — 46 0,3 2
—130 40 0,9 0,06 0,02 — 117 1,3 8
—125 —- 65 1,9 0,15 0,05 — — 180 4 16
—120 - - 96 3,0 0,27 0,096 — — 270 6 28
-115 140 5,6 0,6 0,2 — — 370 И 50
—110 202 9,2 1,1 0,38 — — 530 15 81
—105 298 14,5 2,0 0,7 0,08 — 730 23 135
-100 390 22 3,3 1,2 0,15 0,06 — 34 205
— 95 532 33 5,4 2,1 0,30 0,13 — 46 320
— 90 — 49 8,2 3,5 0,50 0,22 68 470
— 85 — W 70 12,5 5,5 0,90 0,4 — 97 700
- 80 — — 95 18 8,5 1,5 0,7 132 —
— 75 —- 132 27 12,4 2,3 1,2 180 —
— 70 — 179 39 18 3,6 2,0 — 240 - —
- 65 — 240 53 26 5,5 3,0 — зю —
— 60 — 320 72 37 8,1 4,6 — 415
— 55 —* 410 96 51 11,7 6,8 — 540 —
— 50 — 535 128 70 16,5 10,0 — б80 —
- 40 КМ* —— 840 218 120 32 19,7 — —
— 30 — — — 355 205 58 38 —• —. —
— 20 555 335 98 66 — — —
— 10 —— 830 520 161 108 *• —
0 — — — 764 255 177 —
Из этой таблицы видно, что наибольшей упругостью паров обла-
дает метан. При температуре жидкого воздуха упругость его паров
в несколько тысяч раз выше упругости паров других, наиболее близ-
ких к нему по свойствам углеводородов. Отсюда следует, что если
углеводородный газ, содержащий метан вместе с другими углеводе*
родами, охладить до температуры жидкого воздуха, то метан легко
может "быть отделен от остальных углеводородов путем откачки.
Аналогичным образом можно отделить и этан от других алканов.
143
Однако чем тяжелее углеводороды, тем меньше разница упругости
паров. Поэтому разделение тяжелых углеводородов подобным пу-
тем затруднительно, особенно если в газе помимо алканов присут-
ствуют алкены и цикланы.
На фиг. 62 изображен прибор, применяемый для подобного про-
стого разделения углеводородов при различных низких темпера-
турах. Этот прибор предназначен для анализа смесей углеводород-
ных газов [15, 16]. Такие составные части газа, как углекислота
и пары воды, должны удаляться перед анализом. В данный прибор
включены и приспособления для общего анализа. Это мероприятие
Фиг. 62. Прибор для полного анализа углеводородных природных
газов.
позволяет проводить более точно весь анализ и определять все основ-
ные компоненты, содержащиеся в газовой смеси, иначе говоря,
провести наиболее полный газовый анализ.
Прибор (фиг. 62) состоит из бюретки с охранной трубой, име-
ющей вверху трехходовый кран 5, ртутного насоса 77 с уравнитель-
ным сосудом, трех или четырех конденсационных трубок 72, 13, 14,
баллончика с углем 15. В приборе имеется несколько кранов 2—4,
разъединяющих конденсационные трубки, а также краны, распо-
ложенные выше бюретки, позволяющей направлять газ обратно в
конденсационную систему или в прибор для общего анализа.
Разгоночная часть прибора, изображенного на фиг. 62, может быть
использована и отдельно в тех случаях, когда нет надобности про-
водить полный анализ, а требуется определить только содержание
тех или иных углеводородных газов.
144
Конденсационная трубка представляет собой обычную стеклян-
ную трубку диаметром 12—15 мм, в которую впаяна трубка более
узкого диаметра 5—7 мм. В верхней части конденсационной трубки
имеется ртутный манометр, позволяющий следить за упругостью
паров жидкого конденсата. Баллончик с углем, присоединенный
к прибору через трехходовый кран, служит для получения вакуума
в приборе и откачки тех или иных компонентов, для чего баллон-
чик с углем погружается в дьюаровский сосуд с жидким воздухом.
Чтобы избежать переброса ртути из насоса в конденсационные труб-
ки, применяют предохранитель, закрывающий ход при поднятии
ртути в насосе. Уровень ртути в насосе поднимается путем впуска
или откачки воздуха из уравнительного сосуда через кран.
Анализ производится следующим образом. Первоначально в при-
боре создается вакуум, что достигается путем откачки масляным
насосом через краны 1 и 8. Во время этой откачки производят от-
качку и из уравнительного сосуда через отвод 9. После того как в
приборе создастся вакуум, что видно по неизменному положению
уровня ртути в манометрах, краны 7, 6 и 8 закрывают. Более пол-
ный вакуум получают, присоединив весь прибор к трубке с углем 75,
которую погружают в дьюаровский сосуд с жидким воздухом. Уро-
вень ртути в насосе при этом должен быть опущен ниже трубки,
идущей к углю. При создании более полного вакуума в дальнейшем
необходимо заполнить ртутью весь насос и, сжав находившийся
там газ, пустить его через кран и бюретку в конденсационную си-
стему или откачать через кран 8. Бюретка точно так же должна запол-
няться ртутью. При всех этих манипуляциях следует соблюдать
правила, изложенные в главе I.
Для проверки степени вакуума ртуть в насосе опускают, затем
поднимают и, собрав таким образом газ, направляют его в бюретку
или в капилляр 10, где и производят замер. Если количество отка-
чанного газа очень невелико, то можно начинать анализ. Практи-
чески, даже если остаток газа или воздуха в приборе составляет вели-
чину порядка 0,1 см3, можно уже начинать технический анализ.
Для более точного анализа замер производят в капиллярной дуге
насоса 10, пользуясь соседней трубкой 77 как манометром.
Анализируемый газ впускается через барботер со щелочью и
трубку с фосфорным ангидридом. Как углекислый газ, так и пары
воды должны быть удалены из газа, прежде чем он войдет в прибор.
Впуск газа производится через кран 8. Содержание углекислого
газа можно определить в отдельной пробе. Возможно также трубку
с осушающим веществом (Р2О5) поместить между кранами 7 и 4.
В этом случае проба газа забирается через кран 8 без применения бар-
ботера со щелочью и осушителя. В газе определяют углекислоту и
кислород, после чего его направляют в конденсационные трубки
через кран 7 и осушитель 19.
Конденсационные трубки 12, 13, 14 погружаются в дьюаровский
сосуд с жидким воздухом, жидким кислородом или жидким азотом.
Анализируемый газ забирается в бюретку в количестве, соответ-
ствующем требуемой точности анализа, и направляется в конден-
10 ЕЦ А. Соколов. 145
сационные трубки. Для анализа берут 100 см3. При необходимости
повысить точность определений берут большее количество газа,
последовательно забирая несколько порций его в бюретку и напра-
вляя их в конденсационные трубки. Газ, содержащий углеводороды,
попадая в первую же трубку, конденсируется, переходя в жидкое
состояние. Если в газе имеется значительное количество неконден-
сирующихся компонентов, например в нем присутствуют азот, кисло-
род, водород, то газ заставляют несколько раз циркулировать по
всей системе, пользуясь насосом и бюреткой. При этом все угле-
водороды переходят в жидкое и твердое состояния с ничтожной упру-
гостью пара, кроме метана, который в некоторой своей части будет
находиться в газообразном состоянии.
После окончания впуска необходимого количества газа отка-
чивают неконденсирующиеся газы вместе с метаном. Для этой цели
опускают ртуть в насосе, а затем поднимают. Попавший в баллон
насоса газ направляется при этом в бюретку. Сделав последовательно
ряд откачек, собирают газ в бюретке. Эту откачку нужно вести до
тех пор, пока манометры не покажут нулевого давления. Для более
полного извлечения метана из конденсата рекомендуется, перекрыв
краны, удалить дьюаровский сосуд и дать конденсату испариться,
что будет видно по поднятию ртути в манометре. Когда ртуть подни-
мется на 10—20 см, опять опускают конденсационную трубку в жид-
кий воздух. При этом освобождается некоторое количество ранее
растворенных неконденсированных газов и метана, которые и отка-
чиваются ртутным насосом. Необходимо все эти газы соединить вместе
в бюретке и замерить. Когда количество откачанных газов ве-
лико и емкость бюретки недостаточна, то определенную порцию
газов направляют в одну из пипеток, содержащую ртуть, на поверх-
ности которой имеется небольшое количество воды для увлажнения
газа. Вообще для наиболее полной откачки газа, если его количество
составляет значительную часть бюретки, рекомендуется основную
массу газа направить для сохранения в пипетку, а после этого, поль-
зуясь насосом и бюреткой, собрать последние порции откачиваемых
газов. При малом количестве газа можно обойтись и без перевода
газа в пипетку. В приборе для общего анализа, присоединенном к
прибору для разгонки, необходимо, следовательно, иметь пипетку
с ртутью для сохранения газа. Эта пипетка 16 служит также для сож-
жения газа. Если смесь неконденсирующихся газов и метана не под-
вергается анализу, эти газы выпускаются из прибора через кран 8.
Откачанный при вышеописанных условиях газ представляет
собой смесь водорода, окиси углерода, кислорода, азота, метана и
редких газов. Если можно не опасаться присутствия водорода и
окиси углерода, то в откачанной смеси имеются только азот, кисло-
род, метан и редкие газы. Для удаления кислорода можно восполь-
зоваться одним из растворов, применяемых при общем анализе, после
чего газ будет состоять из азота с редкими газами и метана.
Для проведения анализа откачанного газа рекомендуется прежде
всего заполнить капилляр гребенки прибора чистым азотом, как
это описано в главе III.
146
После удаления кислорода производится сожжение метана и
определение по разности азота. Если в газе присутствуют водород
и окись углерода, то можно провести их сожжение в трубке с окисью
меди 18 при 300°.
Газы, определяемые при общем анализе, будут проанализированы
таким образом достаточно точно, так как компоненты, мешающие
определению СО и СН,, удалены из газовой смеси вследствие кон-
денсации в трубках 72, 13 и 14. Эта сконденсированная часть газа
будет состоять из алканов, алкенов и цикланов. Кроме того, в кон-
денсационных трубках будет находиться закись азота. При темпе-
ратуре жидкого воздуха закись азота обладает очень незначительной
упругостью паров и подавляющая ее часть остается в конденсацион-
ных трубках. Упругость паров закиси азота при низких темпера-
турах близка к упругости паров этана.
Соединение прибора для разгонки с прибором для общего анализа
позволяет полно и точно проводить всякий анализ газа. Это особенно
важно при анализе природных газов, когда мы имеем дело с неиз-
вестными до сих пор выходами газа или с газами из новых, вскрытых
бурением пластов. Состав газа в этих случаях совершенно неизве-
стен, поэтому всегда желательно провести наиболее полное его ис-
следование. Откачанный газ после удаления кислорода направля-
ют в трубку для сожжения с окисью меди, где сжигаются водород
и окись углерода при 300°. Кислород можно определить в газе и до
конденсации, хотя это и не обязательно. Можно кислород опреде-
лить и удалить после откачки. Однако это удаление кислорода не-
обходимо провести до сожжения с окисью меди. Остаток после
сожжения метана и определения углекислого газа состоит из азота
и редких газов. При необходимости определения редких газов
остаток надлежит направить в пипетку с ртутью, для того чтобы
в дальнейшем провести на этом же разгоночном приборе также и опре-
деление гелия.
Для определения гелия поступают следующим образом. Газ,
состоящий из смеси азота с редкими газами, направляют в трубку
е углем 15. Азот и тяжелые редкие газы при этом поглощаются, а
гелий и неон откачиваются ртутным насосом в капилляр/ где и за-
меряются. Чистоту гелия и полноту поглощения наблюдают по
спектру в разрядной трубке 20, присоединенной к трубке с углем, и
манометру. Откачку гелия из угля следует проводить при закры-
том кране 1. Измерять количество откачанного гелия следует так,
как описано в главе I, причем манометром будет служить та трубка,
которая идет от насоса к конденсационным трубкам. Более подробно
методика определения редких газов описывается в главе VII.
Если требуется провести еще более детальный анализ на ред-
кие газы с определением аргона, то в систему включают тугоплавкую
трубку, куда помещают металлический кальций или литий для по-
глощения азота (см. главу VII).
Если в откачиваемых газах не требуется определять содержа-
ние водорода, окиси углерода, метана и азота, т. е. если не тре-
буется проводить общий анализ (например, если таковой уже прове-
10* U7
ден в отдельной пробе), то гелий определяют следующим образом.
После впуска газа в конденсационные трубки присоединяют к ним
уголь через кран 7, который потом закрывают. Затем с помощью
ртутного насоса собирают остатки газа в трубках и также напра-
вляют его в уголь. Проверив чистоту спектра гелия, последний от-
качивают в капилляр и замеряют.
Применяя описанную установку, мы можем, следовательно, раз-
делить газ на две части. Первая часть — это углеводороды, более
тяжелые, чем метан, с примесью закиси азота. Эти углеводороды
могут замеряться суммарно или в дальнейшем может
_______ производиться их разгонка с определением индиви-
дуальных углеводородов. Вторая часть — это газы,
Г______4 не конденсирующиеся и откачивающиеся при темпера-
V )) тУРе жидкого воздуха, куда входят метан, азот, редкие
газы, водород, кислород, окись углерода. Эти газы
ca_w____> после откачки анализируют на приборе для общего
т анализа, где и определяют содержание указанных ком-
р L понентов.
Углекислый газ с сероводородом определяются сум-
§ марно или в начале анализа или удаляются из газа
& ЭД до впуска в прибор.
$ Сконденсированный в трубках 72, 13 и 14 остаток,
II в котором находятся более тяжелые углеводороды, чем
Sj эд метан, а также закись азота, если она присутствует
в газе, подвергаются на приборе дальнейшему анализу.
Для этой цели служат имеющиеся в приборе 3—4 кон-
денсационные трубки, позволяющие более тщательно
Фаллический 0ТДелять углеводороды друг от друга.
блок для Как это видно из табл. 11, при температуре жид-
охлаждения кого воздуха из конденсационной трубки может быть
конденсацион- откачан только метан, остальные углеводороды имеют
нои трубки. слишком низкую упругость пара.
Чтобы выделить этан, необходимо повысить тем-
пературу. Для получения температур порядка минус
120—160° применяют специальные приемы. Наиболее удобным
является употребление металлических блоков, изображенных на
фиг. 62а. Эти блоки помещают в дьюаровский сосуд и
в них постепенно подкачивают жидкий воздух с помощью
груши.
Блок представляет собой металлическую толстостенную трубку,
в которую впаян патрубок; внутрь патрубка входит конденсацион-
ная трубка. Пространство между стенками этих металлических тру-
бок заполнено медными или латунными опилками или стружками.
Нижняя стенка блока имеет ряд отверстий. Кроме того, в блоке
имеется еще одна латунная трубка, куда вставляется термометр
или термопара.
Для достижения желаемой температуры блок вместе с дьюаров-
ским сосудом устанавливают таким образом, чтобы конденсацион-
ная трубка вошла в центральную часть блока. Затем начинают под-
745
качивать жидкий воздух. Избыток жидкого воздуха стекает на дно
дьюаровского сосуда.
Подобное устройство сделано для того, чтобы по достижении
даже сравнительно низких температур жидкий воздух не накапли-
вался в стаканчике блока, иначе в нижней части блока обязательно
будет температура жидкого воздуха, а в верхней части более высокая.
Чтобы обеспечить равномерную температуру по всему блоку,
дают избытку жидкого воздуха стекать на дно дьюаровского сосуда.
По мере охлаждения блока температура понижается; за темпера-
турой следят по пентановому термометру или термопаре. Не реко-
мендуется употреблять для указанной цели какие-либо охлажденные
жидким воздухом ванны из пентана или других органических жид-
костей вследствие опасности воспламенения их при соприкосновении
с жидким кислородом.
Для проведения разгонки в первой конденсационной трубке 12
(фиг. 62) создают температуру —125°, а вторую трубку погружают
в жидкий воздух. При этой температуре значительная часть этана
перейдет во вторую трубку вместе с некоторым количеством про-
пана. После этого делают вторую перегонку, создают в трубке 13
температуру —140° и трубку 14 погружают в жидкий воздух. При
этом условии происходит дальнейшая очистка этана от пропана и в
трубке 14 конденсируется довольно чистый этан.
Следует учесть, что вместе с этаном будет отгоняться и закись
азота. Таким образом, в трубке 14 будем иметь смесь этана с закисью
азота. Если в газе присутствует этен, то он также будет отгоняться
вместе с этаном. Как это видно из таблицы упругости паров, этен
обладает даже большим давлением, чем этан, поэтому он еще более
полно и раньше этана начнет отгоняться.
Указанная фракция, состоящая из этана, этена и закиси азота,
откачивается насосом и замеряется.
Для определения в этой смеси отдельно этена необходимо пустить
газ в пипетку с бромной водой, где произойдет поглощение этена,
после чего в газе останутся этан и закись азота. Раздельное опреде-
ление этана и закиси азота может быть произведено различными
путями.
Газ весь целиком может быть сожжен при высокой температуре
порядка 900° с помощью окиси меди, содержащей в то же время не-
которое количество и красной меди. Этан при этом весь сгорает, а
закись азота разлагается на элементы, причем кислород окисляет
медь и, таким образом, остаток после поглощения углекислоты будет
представлять собой азот. Объем азота будет равен объему имевшейся
ранее закиси азота. Можно определить содержание этана и закиси
азота, если определить их удельный вес.
Поскольку в данном случае мы имеем дело с двумя компонентами,
то по удельному весу смеси можно судить о составе каждого из ком-
понентов. Однако для этой цели требуется довольно значительное
количество газа, так как разница их удельных весов не очень велика.
Можно провести также отдельно сожжение этана с помощью на-
каленной платины при сравнительно слабом накале. Этан при этом
149
сгорает, а закись азота остается неизменной. Если произойдет не-
которое разложение закиси азота, то об этом можно судить по коли-
честву образовавшегося кислорода. Кислород может быть определен
в одной из пипеток.
После выделения этана, этена и закиси азота в конденсационной
трубке остается остаток, содержащий более тяжелые углеводороды.
Необходимо также учесть, что при указанных выше температурах
частично вместе с этаном будет откачиваться и ацетилен, если он
присутствует в газе. При повышении температуры конденсационной
трубки 72 до —120° в трубку 13 будет перегоняться пропан и частично
изобутан. Вторая перегонка может быть произведена при температуре
—135°. Если требуется большая точность разделения, число пере-
гонок желательно увеличить.
Необходимо учесть, что во фракцию пропана попадут также про-
пен и ацетилен; частично возможна и примесь аллена. Эту фракцию
после откачки можно анализировать тем же путем, как описано выше.
Пропен и ацетилен могут поглощаться соответствующими реаген-
тами, пропан же останется непоглощенным.
После удаления пропана и указанных алкенов в конденсационной
трубке остается смесь, состоящая из изобутана, бутана, а также из
различных более тяжелых алканов и алкенов.
Дальнейшая разгонка на указанном приборе мало эффективна
вследствие слишком незначительной разницы в упругости паров более
тяжелых углеводородов.
Вопрос о применении тех или иных вариантов зависит от целей
анализа и требований, предъявляемых к нему.
Если мы имеем дело с неизвестными ранее природными газами,
то целесообразно проводить наиболее полный анализ с определением
различных газов неорганического характера, раздельным определе-
нием индивидуальных углеводородов, определением редких газов
и т. д. В других случаях нет надобности проводить подобный анализ.
Во многих промышленных установках, например на газолиновом
заводе, требуется знать для контроля за производством только состав
газа, поступающего на завод, и состав получаемого газолина. В этих
случаях нет надобности проводить такие определения, как опреде-
ление редких газов, водорода, окиси углерода и т. д. В других слу-
чаях, например на нефтеперерабатывающих заводах, помимо угле-
водородного состава, несомненно, интересно знать и содержание водо-
рода, окиси углерода и наличие различных непредельных индиви-
дуальных углеводородов.
В промышленных условиях могут встретиться различные положе-
ния, когда требуется проводить контроль только за одним или не-
большим числом наиболее интересующих нас компонентов газовой
смеси, поэтому очень часто в промышленных условиях аналитиче-
ские приборы могут представлять собой установки, определяющие
небольшое число компонентов, но в то же время проводящие эти
определения возможно часто, а в лучшем случае автоматически.
При проведении упомянутого выше анализа природного
газа не всегда требуется детальное определение тяжелых индиви-
150
Фиг. 63. Упрощен-
ный прибор для
суммарного опре-
деления тяжелых
газообразных угле-
водородов.
на воздух в за-
откачки удаляют
дуальных углеводородов. Для многих целей представляют интерес
только суммарное определение тяжелых углеводородов и отдельное
определение метана. В этих случаях прибор для анализа весьма
упрощается. В приборе необходим ртутный насос, аналогичный опи-
санному выше, одна конденсационная трубка для сжижения тяжелых
углеводородов и трубка с углем, служащая для создания вакуума
и определения редких газов. К прибору присо-
единяется установка для общего анализа с трубкой
для сожжения с помощью окиси меди и несколь-
кими пипетками для поглощения различных газов.
Иногда, особенно при изучении горючих при-
родных газов, наибольший интерес представляет
именно содержание тяжелых углеводородов. Для
этих целей после конденсации тяжелых углево-
дородов в приборе создается вакуум. После уда-
ления жидкого воздуха откачиваются все сконден-
сированные тяжелые углеводороды и замеряются
их количества в бюретке. Для этих целей может
быть применен и более упрощенный прибор, схе-
матически изображенный на фиг. 63.
Прибор предназначен для определения в газе
только тяжелых углеводородов. Анализируемый
газ очищается от углекислоты и паров воды,
проходя через щелочь и фосфорный ангидрид,
затем попадает в градуированную бюретку, имею-
щую внизу расширение и могущую выполнять
также роль ртутного насоса. Газ забирается в
бюретку и по давлению в манометрической трубке
вычисляется его количество.
После замера газ направляется в конденсацион-
ную трубку, где тяжелые компоненты переходят
в жидкое или твердое состояние. После оконча-
ния конденсации оставшийся газ откачивают и
удаляют из прибора. Этот газ может быть напра-
влен в прибор для общего анализа или выпущен
висимости от целей определения. После окончания
дьюаровский сосуд, дают тяжелым углеводородам испариться и отка-
чивают их насосом, опуская ртуть в баллоне при открытом кране.
Закрыв кран, поднимают ртуть в баллоне и делают замер количе-
ства газа, отмечая объем и соответствующую разницу давлений.
Для точного определения этой разницы манометрическая трубка
должна быть проградуирована, чтобы при вакууме и в бюретке и
в манометрической трубке было определено, какому делению мано-
метра соответствует определенное деление бюретки. Откачку и замер
следует последовательно произвести несколько раз, выпуская замерен-
ные порции газа из установки или направляя их в прибор для
общего анализа. Подобным способом можно определять в природном
газе сравнительно небольшие примеси углеводородов, более тяже-
лых, чем метан.
151
§ 3. Приборы низкотемпературной разгонки с применением
ректификационной колонки
Подобные приборы служат для детального анализа тяжелых угле-
водородных газов, а также для анализа легких жидких углеводо-
родов [17].
Действие приборов основано на применении очень эффективно
действующей ректификационной колонки, в нижней части которой
Фиг. 64. Прибор с ректификационной
колонкой.
7 — милливольтметр; 2 — термос; 3 — ввод паров;
4 — ввод жидкого воздуха; 5 — термопара; 6 —
жидкий воздух; 7 — термос; 8 — К вакуумному
насосу; 9 — разрез трубы с насадкой; 10 — мано-
метр установки; 11 — манометр приемника; 12 —
термометр; 13 — холодное соединение; 14—16 —
схема действия термопары; 17 — линия подачи газа;
18 — колонка; 19 — вакуумный кожух; 20—21 —
осушитель; 22— сосуд с ртутью; 23—трансформатор;
24 — приемник; 25 — схема наружного нагрева;
Sx — S-j — краны.
имеется приспособление
для подогревания, а в’верх-
ней — для охлаждения.
Ректификационная ко-
лонка представляет собой
трубку того или иного
диаметра, содержащую на-
бивку в виде специально
свернутой проволоки или
металлической соответст-
вующим образом изогну-
той сетки или ленты, по-
зволяющей жидкой флегме
стекать по ней в нижнюю
часть колонки. Эффектив-
ность действия колонки
обусловлена применение.м
вакуумной рубашки, в ко-
торую вставляется колон-
ка. Вакуумную рубашку
изготовляют из хорошего
стекла, она имеет двой-
ные стенки, между кото-
рыми имеется высокий ва-
куум. Стенки посеребрены
для наилучшей эффектив-
ности действия и наимень-
шей потери тепла. В не-
скольких местах вакуум-
ной рубашки имеются не-
посеребренные полоски или
кружочки, позволяющие
следить за тем, что проис-
ходит в самой колонке.
Внутренняя труба вакуум-
ной рубашки бывает ино-
гда окружена полирован-
ной металлической трубкой (рефлектором), имеющей ряд отверстий.
Для разделения легких углеводородных газов (этана, пропана,
изобутана, этена и др.) применяют колонку^диаметром 3—5 мм. Ниж-
няя часть самой колонки, которую вставляют в вакуумную рубашку.
152
представляет собой баллон того или иного размера, куда первона-
чально вводят жидкость, подвергающуюся анализу. При анализе
газа его конденсируют в этом баллоне при температуре жидкого
воздуха. Для подогрева баллона применяют проволочную обмотку
или иной тип электрического подогревателя. Рекомендуется иметь
набор колонок с баллонами различной емкости, употребляемых в за-
висимости от количества анализируемого газа. Для разделения лег-
ких жидких углеводородов применяют более широкие трубки диа-
метром 8 мм и шире. К этим колонкам присоединяют баллоны раз-
личных размеров, куда помещают анализируемую жидкость.
Фиг. 65. Прибор с ректификационной колонкой.
В первоначальных моделях прибора вся система колонки и ва-
куумной рубашки была спаяна, как это показано на фиг. 64. Однако
в дальнейшем оказалось более удобным вакуумную рубашку делать
отдельно, чтобы в нее можно было вставлять различные колонки.
Прибор с ректификационной колонкой предназначается для ана-
лиза газов или жидкостей. В зависимости от этого несколько изме-
няется и устройство прибора. Схема прибора для анализа газа пред-
ставлена на фиг. 65.
Исследуемый образец газа предварительно освобождается от
углекислоты и паров воды и впускается через кран в нижнюю часть
колонки, охлаждаемую жидким воздухом. Если в газе присутствуют
непредельные углеводороды, то не рекомендуется применять пяти-
окись фосфора, которая их поглощает; лучше применять безводный
153
перхлорат магния. При поднятии уравнительного сосуда с ртутью,
присоединенного к крану, ртуть вводится в нижнюю часть колонки
и получается, таким образом, ртутный затвор, который не позволяет
впущенному в колонку и сжижаемому газу каким-либо образом
выйти назад через кран. В других конструкциях прибора ограничи-
ваются перекрытием крана. Колонка в верхней части проходит через
открытый сосуд с двойными стенками. В этот сосуд, когда дьюаровский
сосуд спаян с рубашкой, наливают легкокипящий газолин, служа-
щий охладителем. Для охлаждения газолина служит металлический
сосудик, который со всех сторон наглухо запаян и имеет две ввод-
ные металлические трубки. Через одну трубку в него подкачивается
жидкий воздух из дьюаровского сосуда, другая же остается открытой
и служит для выхода воздуха. Колба с жидким воздухом показана
на фиг. 65 справа от колонки. Металлический сосуд погружается
в газолин, и жидкий воздух накачивается при помощи резиновой
груши. Таким образом, верхняя часть колонки проходит через
охлажденный газолин. Верхняя часть колонки называется конден-
сатором. Верхушка колонки закрывается каучуковой пробкой, через
которую проходит трубка, соединяющая колонку с остальными
частями прибора. Употребление газолина в этом приборе имеет ряд
неудобств. Как упомянуто выше, применение газолина в присутствии
жидкого кислорода представляет большую опасность. Известны слу-
чаи взрыва и пожара при его применении. Более удобно применение
охладителя, представляющего собой металлический блок, который
охлаждается жидким воздухом.
Через пробку в верхней части колонки проходит термопара медь—
константан, при помощи которой по милливольтметру или потен-
циометру измеряется температура внутри колонки, иначе говоря,
температура отгоняемого углеводорода. Схема термопары и способ
применения потенциометра описаны ниже.
Проходящая через пробку колонки трубка соединяется каучуком
со стеклянным капилляром, имеющим несколько кранов. От одного
из кранов идет манометр, заполненный ртутью и открытый с другого
конца, служащий для измерения давления в колонке. Другой ртутный
манометр, но уже закрытого типа, служит для измерения давления
газа в приемных бутылях, сосудах и бюретках. Один из кранов
(фиг. 65) служит для присоединения всего прибора к вакуумному
насосу.
Сжиженные газы после удаления дьюара заставляют кипеть в
нижней части колонки, подогреваемой током. Благодаря большой
эффективности действия колонки углеводороды разделяются и один
за другим поступают в бутыли или баллоны. Количество поступив-
шего газа определяют по манометру, так как объел! бутыли или бал-
лона определяется заранее. За температурой отгоняемого углеводо-
рода наблюдают по милливольтметру. В тот момент, когда один угле-
водород целиком перейдет в бутыль и начнет перегоняться другой,
температура пара резко меняется. По этил! наблюдениям можно по-
строить кривую зависимости количества газа, попавшего в приемную
бутыль, от температуры. По этой кривой можно определить содержа-
154
ние отдельных компонентов. Для действительного изолирования
одних отгоняемых углеводородов от других требуется поставить
несколько баллонов и переключать в них газ при изменении темпе-
ратуры. Кривая зависимости температуры и объема отогнанного газа
не имеет вида ломаной линии с резко выраженными углами (что
было бы, если бы разделение было идеальным), а имеет вид кри-
вой со сглаженными углами. Но на той части кривой, которая
сглаживает угол, можно найти среднюю точку, которая определит
содержание соседних компонентов.
Перед анализом прибор следует проверить на отсутствие течи.
Для этого воздух из прибора откачивают масляным насосом, а затем
закрывают краны. После того как прибор постоял несколько часов, от-
крывают краны и присоединяют поочередно отдельные части к мано-
метру. Если давление в манометре падает хотя бы на 1—2 мм, это
говорит о наличии течи, которая может происходить через каучук,
через пробку или через кран. Для устранения течи кран следует лучше
смазать, каучуки промазать пицеином и залить пицеином пробку.
Перед анализом необходимо прежде всего охладить конденсатор до
такой температуры, чтобы сконденсировать наиболее летучие ком-
поненты анализируемого газа. Если в газе присутствует метан, то
температура газолина или блока должна быть около минус 165—
170°, т. е. несколько ниже температуры кипения метана. После этого
поворачивают кран так, чтобы отъединить колонку от остальных
частей приборами присоединяют бутыль для приема газа. Убедив-
шись, что манометр показывает постоянное давление внутри при-
бора, приступают к вводу газа. Газ, который вводится через осуши-
тельную трубку, дойдя до верхней части колонки, будет конденси-
роваться и конденсат будет стекать вниз. Впуск подобным образом
большого количества газа требует много времени, поэтому рекомен-
дуется газ предварительно охлаждать, что значительно ускоряет
процесс ввода газа.
Для разгонки обычно берут несколько литров газа. Если он очень
сухой (т. е. содержит мало тяжелых углеводородов), то, чтобы опре-
делить состав жидких углеводородов, естественно, требуется боль-
шое количество газа. Присутствие в газе азота, кислорода и дру-
гих конденсирующихся в колонке газов вызывает повышение давле-
ния до атмосферного и выше. Если это наблюдается и давление в
колонке поднимается выше 900 мм, то неконденсирующиеся газы
следует переводить в приемную бутыль. Скорость впуска газа регу-
лируется вентилем и краном (фиг. 65). После того как требуемое
количество газа введено в колонку, кран закрывают, введя предва-
рительно ртуть в трубку, погруженную в дьюар. Ртуть, войдя в
охлажденную часть, замерзает и образует затвор, после чего дьюар
удаляют и начинают дестилляцию.
Наблюдатель должен регистрировать показания милливольтметра
и одновременно манометра приемных сосудов, следя за тем, чтобы
показания открытого манометра оставались одинаковыми, т. е. чтобы
кипение происходило при атмосферном давлении. Образующиеся
пары доходят до верхней части колонки, где конденсируются, и сте-
155
кают вниз. Чтобы поддержать ток вниз, необходимо отнимать тепло
от конденсатора путем добавления жидкого воздуха в охлажденный
металлический сосуд. Тепло, доставляемое током, и тепло, отнимае-
мое от конденсатора, должно быть сбалансировано, чтобы давление
в колонке оставалось практически постоянным, а проволока, про-
ходящая по всей системе колонки, должна быть везде покрыта тон-
ким слоем жидкости, но количество этой жидкости должно быть
минимальным. При отсчете показаний на манометре следует пово-
рачивать кран 4 так, чтобы манометр и бутыль не сообщались с ко-
лонкой. Это нужно для того, чтобы давление в бутыли пришло в равно-
весие с давлением в манометре, так как при больших скоростях при
перегонке наблюдается отставание давления в бутыли от показаний в
манометре. Первое время милливольтметр будет показывать тем-
Фиг. бб. График анализа газа.
метана. Если температура паров долгое время остается постоянной,
это значит, что дестилляция идет еще хорошо. В этом случае ско-
рость перегонки можно увеличить, усилив нагрев и приоткрыв кран,
однако следя за тем, чтобы температура не повышалась. Когда тем-
пература паров начнет слегка повышаться, следует уменьшить ско-
рость перегонки. Когда большая часть одного из углеводородов будет
удалена, температура паров начнет повышаться, пока не дойдет до
температуры кипения следующего углеводорода. Если при повышении
температуры давление в колонке упадет, следует закрыть кран, пока
давление не поднимется. Перегонку кончают, когда при атмосферном
давлении в колонке уже не будет жидкости. Оставшиеся пары пере-
водят в эвакуированную бутыль и по давлению в ней определяют их
количество. В результате наблюдения получают кривую зависимости
температуры от давления в бутыли, по которой можно судить о.
составе газа (фиг. 66).
156
Анализ газолина. Схема прибора для анализа газолина, бен-
зина и т. п. представлена на фиг. 67. Соответствующий образец вво-
дится через кран. Конденсатор колонны должен предварительно
охлаждаться до температуры, более низкой, чем температура кипе-
ния наиболее летучего компонента. После того как достаточное коли-
Фиг. 67. Прибор для анализа смесей
жидких углеводородов.
чество’газолина будет введено в колонку (обычно достаточно 60 см3),
впускной кран закрывают, отъединяют тот сосуд, из которого была
взята проба, присоединяют бутыль с ртутью и, поднимая ее, создают
ртутный запор, следя, чтобы в колонку не попал воздух (воздух из
соединений может быть вытеснен через боковую трубку крана).
В приборы других конструкций образец бензина может наливаться
157
непосредственно в колбочку, которая и присоединяется к колонке,
как это показано на фиг. 67.
Прибор для анализа жидких углеводородов содержит еще не-
сколько деталей, которые не требуются при анализе газа: подогре-
ватель, дополнительная обмотка для подогрева колонки и ряд дру-
гих приспособлений, показанных на фиг. 67.
Объем взятого образца можно вычислять по количеству получен-
ных паров и по объему остатка, который по окончании перегонки
может быть выпущен из прибора и тщательно измерен.
При введении газолина в прибор предварительного охлаждения
не требуется: в этом и заключается разница между этим прибором
и прибором для анализа газа. В двойном приборе один из них имеет
приспособление для предварительного охлаждения, а другой —
нет, поэтому на двойном приборе можно одновременно производить
и анализ газа и анализ газолина. При анализе газолина необходимо
особенно следить за тем, чтобы вводный кран был герметичным. Этот
кран рекомендуется смазывать патокой, смешанной с глицерином,
касторовым маслом или другим подобным веществом, не взаимодей-
ствующим с газолином.
Проверив милливольтметр, указатель которого должен быть на
нуле при отъединении термопары, приступают к перегонке.
Пустив ток в нагреватель, доводят давление в колонке до атмо-
сферного. Если температура паров приближается к комнатной до
того, как давление в колонке сделается атмосферным, необходимо
начинать перегонку не при атмосферном, а при низком давлении,
иначе пары из колонки будут конденсироваться в капиллярных соеди-
нениях.
Сначала открывают кран, соединяющий колонку с приемными
баллонами. Замечают показания милливольтметра и одновременно
показания манометра приемной бутыли, следя за тем, чтобы показа-
ния открытого манометра оставались постоянными. Эти данные необ-
ходимы для построения кривой зависимости количества отогнанных
паров от температуры.
Если температура паров долгое время остается постоянной, это
означает, что перегонка идет хорошо. В этом случае скорость пере-
гонки можно увеличить, следя, однако, чтобы температура не повы-
шалась. Когда температура паров начнет слегка повышаться, сле-
дует насколько возможно уменьшить скорость перегонки. Когда
один из углеводородов практически будет удален, температура паров
начнет повышаться, а давление в колонке может начать уменьшаться,
так что потребуется пустить в нагреватель побольше тока, чтобы
довести давление опять до атмосферного. Пока не будет достигнуто
атмосферное давление и колонка не придет в равновесие, рекомен-
дуется отъединять колонку от приемной бутыли, закрыв соответ-
ствующий кран. Чем эффективнее процесс фракционировки, тем
быстрее поднимается температура до температуры кипения следую-
щего углеводорода.
Для приближения температуры паров к комнатной температуре
следует уменьшить давление в колонке и при этом давлении произ-
158
водить дальнейшую разгонку. Пропан и бутаны можно перегонять
при атмосферном давлении; пентаны удобнее дестиллировать при
400 мм, гексаны — при 100 мм. Температуры кипения для различных
давлений можно найти в таблицах.
Когда давление в приемной бутыли станет приближаться к дав-
лению в колонке, пары из приемной бутыли следует откачать насо-
сом, а затем продолжать перегонку или переключить поступающие
пары в другую приемную бутыль.
При анализе газолина перегонку продолжают обычно до гепта-
нов; дальнейшая разгонка становится уже затруднительной.
Когда перегонка окончена, нагревание и охлаждение прекращают;
в течение получаса жидкость в колонке стечет вниз; ее следует выпу-
стить из колонки, смерить объем и определить удельный вес. Перед
тем как приступить к следующему анализу, колонку следует осво-
бодить от следов жидкости путем продолжительной откачки насосом.
Таблица 12
Разгонка образца газолина
Давление в бу- тыли, льи 1 Давление в ко- лонке, мм Показания мил- ливольтметра, мв Температура, °C | Комнатная тем- пература, °C Давление в бу- тыли, мм Давление в ко- лонке, мм . Показания мил- ливольтметра, мв Температура, °C Комнатная тем- пература, °C
0,0 737 -0.20 - 2,0 : 28,5 48.4 180 1,00 47,0 26,8
2,0 737 -0,09 - 0,9 ; 28,5 48,8 140 1,00 53.5 26,8
2,9 737 — 0,10 - 1,0 1 28,5 50,8 100 1,00 62,1 25,0
4,8 737 —0,06 — 0,6 28,5 58,7 НО 1,30 62,7 25,0
5,6 737 0,0 0,0 28,5 61,8 ПО 1,30 62.7 25,0
5,8 737 + 0,32 + 0,32 28,5 65,8 ПО 1,28 62,4 26,2
5,8 600 0,0 + 5,8 29,3 70,0 106 1,20 62,6 26,2
6,0 480 — 1,1 + 0,4 : 29,3 73,8 112 1,51 64,0 26,2
6,0 500 — 1,2 8,7 ' 29,3 78,8 ПО 1,32 62,9 26,8
7,5 430 + 0,4 18,3 29,3 82,8 104 1,40 65,0 26,8
7,8 400 +0,8 24,8 29,3 88,1 96 1,50 68,0 24,0
8,8 400 + 1,10 28,0 29,3 88,1 130 1,50 61,0 23,5
9,8 400 1,20 29,0 29,3 95,9 104 1,70 68,0 24.0
10,8 470 1,69 29,8 29,3 99,3 100 2,00 72,5 23,5
12,6 400 1,20 29,0 29,3 100,6 ПО 2,22 72,4 23,5
14,8 400 1,60 32,5 29,3 102,2 104 2,00 71,4 23,5
28,8 400 1,70 34,0 30,0 102,5 94 1,90 73,0 23,4
34,8 400 1,87 35,3 30,0 105,3 88 2,20 77,9 23,4
41,1 400 1,92 36,0 26,8 105,3 84 2,50 82.2 23,8
— — — — 107,3 74 2.35 83,3 23.8
— — — — — 108,5 68 2,30 85,0 23,8
В табл. 12 в первой графе приведены давления в приемной бутыли
за вычетом начального давления; во второй графе даны «абсолютные»
давления в колонке, показываемые открытым манометром и испра-
159
вленные по барометру; в третьей графе даны показания милливольт-
метра. По данным второй и третьей граф вычисляется четвертая
графа; она получается переводом показаний милливольтметра в гра-
дусы Цельсия по соответствующей
Фиг. 68. График анализа жидких угле-
водородов.
1 — гексены; 2 — пентаны и пентены; 3 —
бутаны и бутены; 4 — пропан и пропен;
5 — этан и этен; 6 — метан, кислород и
воздух.
калибровке и приведением этих
температур к атмосферному да-
влению с помощью соответствую-
щих кривых. Последняя гра-
фа дает комнатную температуру.
На основании давления в бу-
тыли и температур (т. е. данных
первой и четвертой граф) строит-
ся диаграмма (фиг. 68). На каж-
дой кривой поднятия темпера-
туры берется средняя точка по
методу «равных площадей». Для
правильного определения сред-
ней точки берется не половина
длины кривой поднятия темпе-
ратуры, а точка пересечения
кривой с такой вертикальной
прямой, которая образовала бы
две равные площади. Замечая,
какому давлению она соответст-
вует, получаем ряд значений,
откуда вычисляем давления, соответствующие различным углеводо-
родам, путем вычитания из давления для данного углеводорода
давления для предыдущего. В результате имеем:
н-Бутан..........................7,1 мм
Изопентан........................6,9 »
н-Пентан .......................34,8 »
Диизопропил ................... 35,2 »
н-Гексан........................24,5 »
Отсюда, зная объем бутыли и ее температуру, можно вычислить
объем и вес каждого углеводорода. Подобным же образом производят
вычисления и в тех случаях, когда анализируется газ.
Прибор с ректификационной колонкой дает возможность произ-
вести анализ углеводородного газа, при котором могут быть опре-
делены не только газообразные компоненты, но и компоненты жид-
кой фракции, а именно, пентан, гексан и т. д. Точно так же на приборе
могут определяться и некоторые непредельные углеводороды, как это
видно на фиг. 68.
Изготовление подобной установки для анализа газа с помощью
ректификационной колонки собственными силами вполне возможно
в исследовательской лаборатории.
Наиболее трудно изготовить колонку. При изготовлении колонки
весьма желательно применение стекла пирекс, но можно сделать
колонку и из хорошего, например, молибденового стекла, принимая
только некоторые меры предосторожности при работе.
160
Способ откачки воздуха из пространства между стенками ва-
куумной рубашки и способ серебрения стенок такой же, как и для
дьюаровских сосудов.
Столь же ответственной частью, как колонка, является и милли-
вольтметр. От милливольтметра зависит правильность показаний
температуры. Изготовлять подобный милливольтметр с правильно
проградуированной шкалой можно только при наличии соответствую-
щих материалов и оборудования, приспособленного для изготовле-
ния мелких и точных деталей. Очень часто милливольтметры имеют
дефекты, не позволяющие считать их точными приборами для изме-
рения электродвижущей силы; например, стрелка милливольтметра
не становится на нуль, чувствительность милливольтметра падает,
вследствие чего изменяется градуировка. Более точно электродвижу-
щую силу можно измерить при помощи потенциометра. Измерения
низких температур потенциометром и другими способами подробно
описаны ниже.
На фиг. 69 представлена схема полуавтоматического прибора, раз-
работанного Б. Б. Каминероми Л. А. Потоловским [50]. Этот прибор
по принципу своего действия не отличается от прибора, описанного
выше. Анализируемый газ поступает через кран 5, освобождается
от паров воды и углекислоты в трубках 7 и 2 и попадает в колбочку 6
ректификационной колонки. Давление замеряется манометром 3,
включаемым краном 4. Разгонка проводится обычным путем, а от-
гоняемые газы направляются в приемники 7. Нововведением в этом
приборе является запись количества газа, поступи ов какой-
либо из приемников, с помощью потенциометра 8, регистрирующего
температуру.
Газ, поступающий в приемник, вытесняет раствор поваренной соли,
который поступает в цилиндрический сосуд 9. При замыкании по-
движного контакта 10 включается мотор 77, который через червячную
передачу 12 поднимает контакт и одновременно вращает вал с бума-
гой для записи в потенциометре. Таким образом, получается запись
количества газа и температур, при которых этот газ выходит из ко-
лонки. Подобная запись может быть проведена для фракций, отго-
няемых при атмосферном давлении, до С4 включительно.
Электропитание прибор получает от сети напряжением 110 или
220 в. Напряжение подведено к потенциометру 8, к мотору 77, к ка-
тушке электромагнитного клапана 75 и к трансформаторам 13 и 14.
От трансформатора 74 ток напряжением 12 в используется для пита-
ния цепей, включающих реле 77 и контакты 17 в открытом мано-
метре 18, а также реле 19 мотора и контакты 10 в цилиндре 9
(фиг. 69," б).
Способы измерения низких температур
При анализе углеводородных газов во всех описанных выше мето-
дах приходится измерять низкие температуры.
Для измерения температур ниже —30° или —40° можно пользо-
ваться следующими способами: 1) газовым или пентановым термо-
метром, 2) термопарой и 3) термометром электрического сопротивления.
11 В. А. Соколов. 161
Фиг. 69. Полуавтоматизированный прибор.
162
Газовый термометр, наполненный водородом или гелием, является
наиболее точным прибором для измерения температуры, но для из-
мерения температур в приборах для анализа газа он неудобен при
большом размере баллона с газом. Проще всего низкие температуры
определяются пентановым термометром, позволяющим производить
измерения до —200°. Некоторые сорта технического пентана обла-
дают способностью не затвердевать даже до —200°; обыкновенный
термометр, наполненный таким пентаном, служит для измерения
низких температур. Продажные пентановые термометры обычно дают
показания с точностью до 1—2°. Пентановые термометры не приме-
няются лишь в тех случаях, когда требуется измерить температуру
внутри дестилляционной трубки или колонки.
Термопара обладает теми преимуществами, что имеет незначи-
тельный объем и малую тепловую инерцию. Благодаря этому при
помощи термопары точнее, чем какими-либо другими способами
можно определить истинную температуру в данной точке простран-
ства. Схема термопары дана на фиг. 70, а. Спай двух проволок, напри-
Фиг. 70. Схема включения термопары.
мер медной 5 с константановой 4, помещается в то место, температуру
которого требуется измерить; 5 — медные проволоки, 4- — констан-
тановая, 3 — милливольтметр, показывающий электродвижущую
силу, даваемую термопарой. Если оба спая 7 и 2 будем держать при
одной и той же температуре, то милливольтметр 3 не покажет ника-
кого отклонения; если температуры спаев будут меняться, то откло-
нения милливольтметра будут зависеть от температур одного и дру-
гого спаев. Поэтому при пользовании термопарой один спай следует
поместить в то место, температуру которого измеряют, а другой все
время держать при постоянной температуре; в этом случае электро-
движущая сила будет зависеть только от температуры первого спая 7.
Обычно второй спай 2 держат во время измерения при температуре 0°
(смесь воды и льда). Шкала милливольтметра, которым производится
измерение электродвижущей силы термопары, может непосредственно
разделяться на градусы. Измерение низких температур при помощи
стрелочного милливольтметра или гальванометра со шкалой, деленной
на градусы, является наиболее простым и удобным способом. К сожа-
лению, электродвижущая сила, даваемая термопарой, очень не-
велика, а чувствительность стрелочных гальванометров ограничена,
поэтому, когда требуется измерить низкие температуры очень точно,
следует измерять электродвижущую силу термопары потенциометром.
И* тбз
Чтобы увеличить электродвижущую силу, соединяют несколько
термопар последовательно, как это изображено на фиг. 70, б, где
7 — медные проволоки, 2 — константановые. Четные спаи поме-
щают в то место, температуру которого измеряют, а нечетные —
в сосуд с тающим льдом. Соединив последовательно три термопары,
получим втрое большую электродвижущую силу, что позволит изме-
рять ее с большей точностью на стрелочном гальванометре. При изме-
рении температуры таким способом необходимо следить, чтобы про-
волоки термопары касались одна другой только в месте спаев; если
они должны быть близко одна к другой, то должны быть изолированы,
чтобы при касании не происходило короткого замыкания. Те концы
термопар, которые помещаются в смесь воды со льдом, должны быть
также изолированы, чтобы не происходило замыкания через воду;
надежнее всего поместить покрытые изоляционным лаком концы термо-
пар в металлическую трубку с дном, а эту трубку уже поместить
в тающий лед. Когда требуется небольшая точность, можно обой-
тись и без тающего льда; в этом случае гальванометр будет показы-
вать разницу между температурами спаев.
При помощи потенциометра, т. е. при помощи сравнения термо-
электродвижущей силы с нормальным элементом, можно достичь
гораздо большей степени точности в измерении низких температур,
чем при помощи стрелочного милливольтметра или гальванометра.
Схема простого потенциометра изображена на фиг. 71, а, где 7 —
аккумулятор, 2 — нормальный элемент, электродвижущая сила ко-
торого равна 1,0183 в, 3 — чувствительный гальванометр (нулевой),
4 — калиброванная проволока из металла с большим удельным со-
противлением и малым температурным коэфициентом, около которой
имеется линейка с миллиметровыми делениями. Электродвижущие
силы аккумулятора и нормального элемента направлены в противо-
положные стороны. Скользящий контакт устанавливается на пол-
ное отсутствие тока в гальванометре; обозначим расстояние от точки 5
до скользящего контакта через ег Поместив вместо нормального
элемента замеряемую электродвижущую силу, находят и для нее
на калиброванной проволоке такую точку, где через гальванометр
не идет ток; обозначим новое расстояние от 5 до скользящего кон-
такта через е2. Так как проволока 4 калиброванная, то потенциал
аккумулятора распределяется по ней пропорционально длине. Через
гальванометр не будет итти тока тогда, когда скользящий контакт
попадет в точку, потенциал которой равен включенному измеряемому
потенциалу. Так как потенциал нормального элемента нам известен,
то измеряемый потенциал х будет равен:
x = _eL.i 0183.
Электродвижущая сила термопары очень невелика; например,
для термопары медь — константан при —185° она равна прибли-
зительно 5 мв. Если длина проволоки 4 будет равна 1 м, то все изме-
рения низких температур будут происходить на протяжении пер-
вых 5 мм калиброванной проволоки. Отсюда совершенно очевидно,
164
что, измеряя низкие температуры на простом потенциометре, изо-
браженном на фиг. 71, мы получим слишком неточные величины.
Для точного измерения низких температур следует пользоваться
потенциометром, схематически изображенным на фиг. 71, б\ 1 —ак-
кумулятор, 2 — нормальный элемент, 3 — гальванометр (нулевой),
4—термопара, ас — калиброванная проволока из манганина или
никелина, 5 и б — сопротивления, 7 и 8 — скользящие контакты.
При термопаре медь — константан и нижнем пределе измеряемой
температуры —210° необходимо иметь возможность измерять электро-
движущую силу приблизительно до б мв. Так как электродви-
жущая сила нормального элемента равна 1,018 в, то сопротивле-
ние 5 должно быть больше сопротивления проволоки ас в 160—
170 раз. При этом условии 6 мв будут приходиться на проволоку ас
Фиг. 71. Схема потенциометра.
и будут распределены пропорционально ее длине, которую можно
взять равной 0,5 или 1 м. На линейке, находящейся около ас, можно
непосредственно нанести градусы температуры. Таким образом,
шкала от 0° приблизительно до —200° будет нанесена на протяжении
0,5 или 1 м, откуда видно, что измерять температуру можно с боль-
шой точностью. Один из полюсов нормального элемента присоединен
к концу сопротивления 5; сопротивление 6 должно быть в 1,2 или
1,3 раза больше сопротивления 5 (так как электродвижущая сила
аккумулятора может доходить до 2,2—2,3 в). Порядок измерения
на потенциометре следующий; соединив переключателем 9 гальвано-
метр с нормальным элементом, передвигают скользящий контакт 8
до тех пор, пока гальванометр не покажет отсутствия тока. Замыка-
ние цепи нормального элемента производят ключом 10. Отсутствие
тока в цепи нормального элемента показывает, что падение потен-
циала от точки a j\o конца 5 равно 1,018 в. Так как эта установка
на нормальный элемент производится перед каждым измерением
температуры, то мы всегда имеем от а до конца 5 потенциал 1,018 в,
а следовательно, градуировка ас на градусы остается постоянной.
После установки на нормальный элемент переключателем 9 соеди-
няют гальванометр с термопарой, передвигают контакт 7 до той точки,
в которой гальванометр опять покажет отсутствие тока, и отсчи-
165
тывают температуру, обозначенную против этой точки. Потенциал,
приходящийся на проволоку ас, постоянен во время измерения по-
стольку, поскольку постоянен потенциал аккумулятора. Рекомен-
дуется, включив аккумулятор, начинать измерения только спустя
5—10 мин., так как в первые минуты потенциал аккумулятора
несколько падает, а затем останавливается на постоянном уровне;
по этой именно причине в цепь аккумулятора не введен ключ для
замыкания; аккумулятор следует присоединить к клеммам и оста-
вить в таком положении во все время измерения. Для получения
точных результатов необходимо употреблять хорошо заряженный
аккумулятор; если потенциал аккумулятора начинает падать, его
следует отправить в зарядку. Шкалу проволоки ас потенциометра
следует градуировать на градусы и эту градуировку время от
времени проверять, погружая термопару в вещества с известной
температурой кипения. Изменения, происходящие в градуировке,
обусловлены главным образом термопарой, электродвижущая сила
которой несколько меняется с течением времени. Точно так же
термопары из одних и тех же металлов имеют несколько отличные
электродвижущие силы. Поэтому при точных измерениях каждая
термопара должна иметь свою градуировку. Сам потенциометр
дает правильные показания в милливольтах до тех пор, пока про-
волока ас остается действительно калиброванной, т. е. сохраняет
по всей длине постоянство площади поперечного сечения, поэтому
рекомендуется пользоваться скользящим контактом с осторожно-
стью: не нажимать при передвижении контакта и вообще избегать
всяких механических повреждений калиброванной проволоки. Весьма
удобны для точных измерений потенциометры с несколькими де-
кадными наборами сопротивлений, начиная от набора по 0,1 ом и
кончая набором по 1000 ом.
Для измерения термоэлектродвижущей силы можно пользоваться
также схемой, изображенной на фиг. 71, в; здесь 7 —точно измерен-
ное сопротивление, равное 0,01 ом, 2 — переменное сопротивление
порядка 2000 ом. Термоэлемент 3 присоединен параллельно. Изменяя
сопротивление 2, добиваются полного отсутствия тока в гальвано-
метре 4 и замечают силу тока, показываемую миллиамперметром 5.
Обозначим силу тока через 1, тогда электродвижущая сила термо-
пары е будет равна:
fe = IR.
При сопротивлении R = 0,01 ом один миллиампер соответствует
0,01 мв.
Наиболее употребительными термопарами для измерения низ-
ких температур являются медь — константан и железо—констан-
тан. Термопара медь—константан обладает несколько меньшей чув-
ствительностью, чем термопара железо—константан, но зато обла-
дает большим постоянством; поэтому термопару медь—константан
следует предпочесть термопаре железо—константан. Табл. 13 харак-
теризует зависимость между температурой и электродвижущей силой
для термопары медь—константан.
166
Таблица 13
Зависимость между электродвижущей силой (мв) и температурой (°C) для
термопары медь —константан
мв °C Мв °C Мв. °C Мв СС мв i °C мв °C
0 0,6 1,0 —26,82 2,0 —55,81 3,0 - 87,86 4,0 — 124,46 5,0 -169,14
о,1 —2,60 1,1 —29,61 2Д —58,86 3,1 — 91,23 4,1 —128,47 5,1 —174,34
0,2 —5,22 1,2 —32,42 2,2 —61,94 3,2 — 94,74 4,2 —132,56 5,2 — 179,74
0,3 —7,85 1,3 -35,26 2,3 —65,05 3,3 - 98,25 4,3 —136,74 5,3 —185,38
0,4 —10,50 1,4 —38,12 2,4 —68,20 3,4 —101,82 4,4 —141,02 5,4 — 191,27
0,5 -13,17 1,5 -41,01 2,5 —71,39 3,5 —105,45 4,5 —145,41 5.5 — 197,44
0,6 -15,86 1,6 -43,93 2,6 —74,61 3,6 —109,13 4,6 —149,91 5,6 —203,95
0.7 -18,57 1,7 —46,84 2,7 —77,87 3,7 —112,87 4,7 —154,52 5,7 —210,92
0.8 —21,30 1,8 -49,80 2,8 —81,16 3,8 —116,67 4,8 —159,25 5,8 —218,47
0.9 —24,05 1,9 —52,79 2,9 —84,49 3,9 -120,53 4,9 —164,12 — —
Как уже сказано выше., электродвижущие силы отдельных термо-
пар несколько отличаются друг от друга; поэтому данные, приведен-
ные в табл. 13, являются некоторыми средними величинами.
Термоэлектродвижущие силы различных пар в области комнат-
ных температур приведены в табл. 14.
Таблица 14
Электродвижущая сила различных термопар
Наименование на 1° С Наименование /г? на 1° С
Платина- платина + 4-10% радия ... • Константан—серебро . . Константан—медь . . . Константан—железо . . Константан—хромони- кель Константан—хромин . . 6,4 40 41 53 56 59 Висмут—железо . . . Висмут—сурьма . . . 95Bi, 5Sn--97 Bi, 3Sn . Теллур—никель . . . Теллур—платина . . . Теллур—висмут . . . 92 100 120 330—520 500 500—550
Термопары из теллура обладают наибольшей электродвижущей
силой, и применение их может облегчить измерения. Небольшие
проволочки из таких металлов, как теллур или висмут, можно полу-
чить, разбрызгав расплавленный металл на пластинке зеркального
стекла; при этом всегда можно найти несколько полосок металла
подходящей длины и формы.
Помимо пентановых термометров и термопар низкие температуры
можно измерять при помощи термометров сопротивления. За исклю-
чением специальных сплавов, как манганин, константан и т. д., со-
противление всех металлов меняется в зависимости от температуры.
767
На этом основан способ определения температуры по изменению
сопротивления проволоки, помещенной в то место, температуру
которого требуется определить. В качестве термометра сопротивле-
ния обычно употребляют платиновую проволоку диаметром 0,05—
0,15 мм. 1 м платиновой проволоки диаметром 0,05 мм имеет сопро-
тивление около 45 ом, а диаметром 0,15 мм — около 5 ом. Изменение
сопротивления платиновой проволоки с температурой видно из
табл. 15, где — сопротивление при данной температуре и /?0 —
сопротивление при 0°.
Для измерения низких температур можно пользоваться также
чистой свинцовой проволокой (табл. 16).
Таблица 15
Зависимость сопротивления
платины от температуры
Таблица 16
Зависимость сопротивления
свинца от температуры
=G Rt Ro °C К. °C °C я, я.
Ro
—253 0,008 0 1,000 -193 -173 0,259 0,333 —73 —53 0,709 0,788
— 183 0,247 + 100 1,396 —153 -133 0,407 0,482 -33 — 13 0.867 0.955
— 76 0,695 — —— —113 — 93 0,557 0,633 - 3 4 100 1,000 1,422
водить по схеме мостика
Фиг. 72. Схема измерения
температуры термометром со-
противления.
Как видно из приведенных таблиц, сопротивление платины и
свинца падает приблизительно в 4 раза при изменении температуры
от 0° до температуры жидкого воздуха.
Измерение температуры термометром сопротивления следует произ-
Уитстона, изображенной на фиг. 72, где
7 — аккумулятор, М — М — калиброван-
ная проволока, 2 — гальванометр, 4 —
термометр сопротивления, 3 — скользящий
контакт, 5 — постоянное сопротивление
приблизительно такой же величины, как
и сопротивление термометра при тех тем-
пературах, которые желательно измерять
с наибольшей точностью. Шкалу мостика
можно проградуировать на градусы.
Платиновую проволоку, служащую тер-
мометром сопротивления, можно свернуть
спиралью или намотать на крестообразно
скрепленные пластинки слюды. Спираль
или обмотку обязательно следует делать
двойной. К концам проволоки следует приварить или припаять
толстые провода. При измерениях термометром сопротивления
пропускают по возможности слабый и кратковременный ток, чтобы
168
не нагревалась сама проволока. Как у пентановых термометров, так
и у термопар и термометров сопротивления следует время от времени
проверять калибровку. Проверка производится путем помещения
термопары или термометра в различные химически чистые вещества,
находящиеся в состоянии кипения или плавления. Температуры
кипения и плавления некоторых веществ очень точно известны, л
по этим температурам производят калибровку. В табл. Г7 приведены
температуры кипения и плавления этих веществ.
Табл ицa 1 7
Температуры кипения и плавления веществ, применяемых для
калибровки термометров для низких температур
Вешество Темпера- тура, С Состояние
Азот -195,8 Кипение
Кислород -183,0 »
Изопентан ... ... .... —159,'6 Отвердение
Метилциклогексан -126,3 »
Эфир — 116,3 Быстрое отвердение или
медленное таяние
Сероуглерод -111,6 Отвердение
Толуол — 95,1 )>
Углекислота (твердая) - 78,5 Испарение
Хлороформ — 63,5 Отвердение
Четыреххлористый углерод .... — 22,9 »
§ 4. Анализ углеводородных газов с помощью адсорбентов
Помимо описанных выше методов разделения углеводородов
с помощью низких температур для этих же целей может быть исполь-
зована сорбция углеводородных газов различными твердыми адсор-
бентами. В качестве сорбента чаще всего применяют уголь. Уголь
при низкой температуре, как уже было упомянуто, способен адсорби-
ровать вообще все газы, кроме гелия, создавая при этом вакуум.
Известно также, что даже при комнатной температуре уголь хорошо
поглощает пары жидких углеводородов. На этом основан один из
методов промышленного извлечения газолина из нефтяных газов.
Многочисленные исследования показали, что чем больше моле-
кулярный вес углеводородов, тем полнее и быстрее они поглощаются
углем из газовой смеси. При пропускании газа, содержащего раз-
личные углеводороды, через уголь метан при комнатной температуре
почти не задерживается, тогда как этан поглощается. Момент про-
скока зависит от скорости пропускания газа, от концентрации того
или иного углеводорода и длины адсорбционного слоя. На способ-
ности угля поглощать углеводороды основаны некоторые методы
раздельного определения метана и более тяжелых углеводородов.
При малых концентрациях углеводородов в воздухе или в каком-
169
либо другом газе удается довольно точно и полно отделить метан
от более тяжелых углеводородов., пропуская его через трубку с ак-
тивированным углем [18]. Эта методика нашла применение при ана-
лизе малых концентраций углеводородов. Схема определения такова.
Газ, освобожденный от углекислоты и водяных паров, пропускают
через трубку с активированным углем при определенной температуре.
В прошедшем газе определяют содержание метана путем сожжения.
Предварительный анализ исходного газа путем сожжения дает сумму
всех углеводородов. По разнице можно вычислить содержание тяже-
лых углеводородов. Применяя различные температуры, начиная
от температуры жидкого воздуха, можно произвести выделение
отдельных углеводородных компонентов путем создания вакуума и
постепенного повышения этой температуры.
Однако более удобным и точным является так называемый «хрома-
тографический» метод, который может быть применен для разделе-
ния углеводородов. При одном из вариантов этого метода анали-
зируемый газ пропускается через сорбент, а затем через этот сорбент,
в частности уголь, пропускается воздух [19]. При пропускании газа
наиболее хорошо сорбируемые компоненты поглощаются в первых
же слоях угля. Трудно поглощаемые компоненты проходят дальше
и задерживаются уже в следующих слоях. Совсем не поглощающиеся
компоненты проходят через уголь, не задерживаясь. По мере пропу-
скания воздуха первоначально из угля выделяются те компоненты,
которые наиболее легко передвигаются по углю; затем из него вы-
деляется следующий компонент, лучше сорбируемый, чем первый.
Дальнейшее пропускание воздуха ведет к выделению третьего ком-
понента, сорбируемого лучше второго, и т. д.
Этим путем, подбирая соответствующие адсорбент, длину трубки
и температуру, можно разделить углеводородные газы на отдельные
компоненты. Каждый компонент является, таким образом, смешан-
ным с тем воздухом, который пропускается через адсорбент. Опре-
деление горючих индивидуальных компонентов в воздухе может
производиться путем сожжения или конденсации при низких тем-
770
пературах. На фиг. 73 представлен один из графиков подобного
анализа, показывающий разделение метана, этана и пропана.
Кроме указанного так называемого «элюентного» метода хрома-
тографического анализа, может применяться метод вытеснительной
хроматографии. Вытеснительный метод заключается в том, что погло-
щенное адсорбентом вещество, в частности тот или иной углеводород,
вытесняется из адсорбента другим веществом, адсорбируемым в еще
большей степени. Известны и другие варианты хроматографического
анализа.
В качестве адсорбента при анализе углеводородных газов реко-
мендуется применять в зависимости от условий анализа также сили-
кагель и некоторые другие вещества.
Используя хроматографический метод, автор, А. М. Туркельтауб
и А. А. Жуховицкий разработали прибор для определения индиви-
дуальных углеводородов в газовых смесях, не требующий примене-
ния глубокого охлаждения.
§ 5. Методы определения предельных и непредельных углеводородов
с применением абсорбции и некоторых реакций
Алканы и цикланы сравнительно трудно вступают в такие реак-
ции, которые позволили бы простым путем проводить разделение
на индивидуальные соединения. Значительно легче вступают в реак-
ции непредельные углеводороды. Однако и в этом случае способность
к реакции распространяется в той или иной степени на весь гомоло-
гический ряд, что опять-таки затрудняет определение индивидуаль-
ных углеводородов. Для простейших углеводородов и при наличии
в смеси лишь небольшого числа компонентов в некоторых случаях
аналитические задачи могут решаться путем применения специаль-
ных растворителей и реагентов. Подобные методы определения угле-
водородов могут быть, в частности, использованы для исследования
отдельных фракций, полученных при разгонке углеводородных смесей
на описанных в настоящей главе приборах. Растворители и реагенты,
применяемые для определения углеводородов, можно помещать в
обычные газовые пипетки на каком-либо из приборов для общего
анализа.
Различные реакции, применяемые для определения углеводородов
Алканы, как уже упомянуто выше, не способны вступать в какие-
либо характерные реакции, которые могли бы служить для качествен-
ного или количественного определения индивидуальных компо-
нентов.
Сожжение в присутствии кислорода или с помощью окиси меди
позволяет производить анализ лишь на суммарное содержание угле-
водородных газов.
В литературе имеются указания, что для качественного опреде-
ления метана в воздухе можно воспользоваться реакцией окисления
метана озоном, которая хорошо идет даже при комнатной темпера-
туре. При этой реакции образуется формальдегид.
СН4-Ь 2О3 = НСНО 4- Н2О + 2О2
17!
Образовавшийся формальдегид можно определить с помощью
какой-либо характерной реакции, в частности с помощью морфин-
серной кислоты, дающей в присутствии формальдегида красно-фиоле-
товое или сине-фиолетовое окрашивание, постепенно переходящее
в синее.
Этан в присутствии озона окисляется до уксусного альдегида
и уксусной кислоты.
Следует, однако, учесть, что этен и пропен в присутствии озона
также образуют форьмальдегид.
Непредельные углеводороды значительно легче вступают в реак-
ции; некоторые реакции являются весьма характерными.
Этен поглощается ацетатом окиси ртути Hg(CH3COO)2. При по-
следующем подкислении этен выделяется.
Аммиачный раствор Си2С12, поглощающий СО, хорошо погло-
щает также этен и ацетилен.
Раствор марганцевокислого калия окисляет этен с образованием
углекислоты, муравьиной, уксусной и щавелевой кислот.
Пропен поглощается упомянутыми поглотителями подобно этену.
Еще более активным в химическом отношении является ацетилен.
Помимо хорошо идущих реакций с серной кислотой и бромной во-
дой ацетилен быстро окисляется раствором КМпО4.
Йодноватый ангидрид J2O5 окисляет С2Н2 с выделением иода
C2H2 + J2O5=J2+2CO2 + H2O К
Следует учесть, что аналогичную реакцию дает и окись углерода.
Ацетилен легко вступает в реакции с солями ртути и серебра.
Аммиачный раствор солей серебра количественно поглощает аце-
тилен.
Особенно характерную реакцию дает ацетилен с раствором ни-
трата меди. В присутствии ацетилена появляется розовое, а затем
красное окрашивание и образуется красный осадок ацетиленистой
меди (Си2С2). Эта реакция может быть использована для определения
ацетилена колориметрическим путем, сравнивая получаемую окраску
со стандартной шкалой.
Для получения наилучших результатов рекомендуется раствор,
в котором на 100 см3 воды взято 0,5 г нитрата меди, растворенной
в 20 см3 воды с добавкой аммиака в количестве 0,53 г.
В раствор добавляют 2,25 г солянокислого гидроксиламина,
растворенного в 33 см3 воды, 4,5 см3 2%-ного раствора желатина
в 33 см3 этилового спирта. Реактив следует приготовлять в день от-
бора пробы, но составляющие его растворы вполне устойчивы и могут
приготовляться заранее.
Сероводород, [если он есть в газе, предварительно поглощают
ацетатом свинца.
На основании данных, приведенных в табл. 18, можно в некоторых
случаях с помощью различных растворителей отделить одни угле-
водородные газы от других. Так, например, с помощью этилового
спирта можно определить содержание пропана и бутана (вместе с
изобутаном и более тяжелыми углеводородами) в газе, содержащем
772
Таблица 18
Растворимость углеводородов в различных растворителях
Газ Количество газа в см3, растворимое в 100 см3 растворителя при 20° и 760 мм рт. столба
вода этиловый спирт (абсолют- ный) бензол гексан ацетон
Метан . . 33 46 49 59 ——
Этан . . . 47 220 — 340 —
Пропан . . 37 800 1450 —. —
Бутан . . . 36 1800 Более 3000 — —
Этен . . . 13 270 290 300 2400
Пропен . . 22 1200 — —
Ацетилен 103 680 400 3100
еще метан, этан и другие компоненты, встречающиеся в природных
газах (О2, n2, редкие газы). Присутствие непредельных углеводо-
родов мешает проведению подобного анализа.
Для определения пропана и бутана в присутствии метана и этана
необходимо принять некоторые меры предосторожности. Абсолютный
спирт сначала насыщают метаном и этаном. Чтобы привести в равно-
весие раствор метана и этана с анализируемым газом, пропускают
через раствор две или три порции газа по 100 смг и затем выбрасы-
вают их. После этой операции спирт готов для поглощения всех угле-
водородов, исключая метан и этан.
Еще большую разницу в растворимости легких и тяжелых угле-
водородов имеют такие растворители, как бензол и гексан (а также
гептан, хлороформ, этиловый эфир). В этих случаях, однако, сле-
дует учитывать значительную упругость паров самих растворителей.
При разделении углеводородов пары растворителей должны быть
удалены. В частности, для этой цели могут быть использованы сор-
бенты (уголь, силикагель).
При использовании растворителей для разделения углеводород-
ных газов следует учесть очень хорошую растворимость ацетилена
в ацетоне. Растворимость ацетилена в ацетоне резко возрастает при
понижении температуры или при повышении давления. При — 80°
один объем ацетона может растворить около 2000 объемов ацети-
лена, что сопровождается увеличением объема жидкости. При дав-
лении 12 ат один объем ацетона может растворить около 300 объемов
ацетилена.
Проводя разгонку углеводородных газов, как это описано в на-
стоящей главе, можно разделить анализируемую смесь на ряд фрак-
ций, в которых с помощью вышеупомянутых поглотителей (серная
кислота, бромная вода) и растворителей можно довольно точно опре-
делить многие индивидуальные предельные и непредельные угле-
водороды даже при весьма сложном составе смеси.
173
Концентрированная серная кислота, особенно дымящая, погло
щает не только непредельные углеводороды с одной двойной связью,
как этен, пропен и более тяжелые, но также и другие, относящиеся
к ряду ацетилена и более сложного строения.
Серная кислота поглощает, кроме того, ароматические углеводо-
роды и тяжелые углеводороды метанового ряда. Чистый метан прак-
тически не поглощается даже дымящей серной кислотой. Этан при
длительном контакте с дымящей серной кислотой слегка погло-
щается. Однако это поглощение происходит настолько медленно,
что не может служить препятствием для отделения с помощью
серной кислоты метана и этана от непредельных углеводородов.
Пропан уже заметно поглощается дымящей серной кислотой,
а бутан поглощается хорошо и быстро. Это обстоятельство следует
учитывать при анализе смесей предельных и непредельных угле-
водородов. Использовать серную кислоту для точного определения
непредельных можно лишь в том случае, если в газе отсутствуют
или предварительно удалены из него пропан, бутан и более тяжелые
углеводороды метанового ряда.
Для определения индивидуальных углеводородов из смеси, состоя-
щей только из газообразных алкенов, серная кислота различной
крепости использовалась многими исследователями.
По Добрянскому, для раздельного определения этена, пропена и
изобутена следует первоначально впустить газ в пипетку с 63—
64%-ной кислотой, поглощающей только изобутен [20]. После этого
газ впускают в пипетку с 83—84%-ной кислотой для поглощения
пропена. Для поглощения этена используется 100%-ная серная
кислота, содержащая SO3.
Другие исследователи для отделения этена от более тяжелых
алкенов рекомендуют 87%-ную серную кислоту. Эта кислота погло-
щает пропен и более тяжелые алкены, а этен почти не затрагивает.
Для лучшего разделения некоторые авторы рекомендуют добавлять
в серную кислоту сульфаты серебра и никеля, являющиеся в этом
процессе катализаторами.
Насыщенная бромная вода быстро и полностью поглощает этен
с образованием дибромэтана:
СН2 = СН2 + Вг2 = СН2Вг — СН2Вг
Бромная вода поглощает пропен несколько медленнее, чем этен,
но с жидким бромом реакция проходит очень быстро. Поэтому для
полного поглощения пропена требуется применение насыщенной
бромной воды, содержащей избыток жидкого брома.
Насыщенная бромная вода поглощает также ацетилен. В то же
время слабые растворы бромной воды почти не поглощают ацетилена,
так как реакция идет крайне медленно.
Определение предельных и непредельных углеводородных газов
с применением алюминия
Простая однократная разгонка смеси тяжелых углеводородных
газов не дает полного разделения компонентов. Если взять смесь
только алканов, то вместе с этаном будет отгоняться частично прс-
/74
пан, а вместе с пропанол! — бутан и т. д. Только путем многократ-
ных перегонок можно разделить смесь на отдельные компоненты.
Присутствие непредельных углеводородов еще более затруд-
няет разделение смеси на отдельные компоненты. Если мы имеел!
некоторый объем жидкой смеси, например 1—2 см3, то испарение
вышекипящего компонента происходит преимущественно с поверх-
ности. Испарение же из более глубоких слоев жидкости происходит
уже с некоторым затруднением. По этой причине полное удаление
вышекипящего компонента из смеси происходит с большим трудо.м
и требует значительного времени. Если же смесь подогревать и за-
ставлять ее интенсивно кипеть, то вместе с вышекипящил! компонен-
том уходит и некоторое ,ко-
личество нижекипящего ком-
понента.
Исследования, производив-
шиеся автором по методике
микроанализа углеводород-
ных газов, показали, что ма-
лые их количества разделя-
ются значительно лучше. Это
явление станет понятным,
если мы учтем, что очень
малые количества газа, Гсгу-
щаясь в жидкость, образуют
весьма тонкую пленку на
внутренней поверхности той
стеклянной трубки, в которой
Фиг. 74. Отношение упругостей паров газо-
образных углеводородов.
1 — этен — пропен; 2 — этан — пропан; з —
этен — этан.
производится разгонка смеси.
При этих условиях испарению молекул вышекипящего угле-
водорода не препятствуют другие углеводороды, вследствие чего разде-
ление смесей на отдельные компоненты происходит более эффек-
тивно.
Вопрос о выборе температур разгонки имеет первостепенное зна-
чение. Необходимо выбрать такие температуры, при которых раз-
ница в упругостях паров разделяемых углеводородов была бы Л1а-
ксимальной. На фиг. 74 приведены кривые отношений упругостей
паров некоторых углеводородов в зависимости от температуры.
Мы видим, что с понижением температуры отношения упругостей
паров углеводородов увеличиваются, откуда можно сделать вывод,
что чем ниже температура фракционного испарения, тем легче раз-
деляются компоненты. Однако использовать для разделения такие
весьма низкие температуры, как температура жидкого кислорода
и т. д., невозможно, так как упругость паров тяжелых углеводоро-
дов при этом так низка, что их откачка будет происходить настолько
медленно, что анализ с замером на ртутных манометрах станет прак-
тически невозможным. Поэтому для разрешения поставленной за-
дачи необходимо выбрать наиболее низкие температуры, при которых
процесс разделения и откачка углеводородов не отнимали бы слиш-
ком много времени.
175
Как видно из фиг. 74, отношения упругостей паров отдельно
предельных и непредельных углеводородов достаточно велики. Так,
например, для пары этан—пропан в интервале —120—до —140°
это отношение равно от 30 до 50; для пары этен—пропен при —120°—
около 45 и т. д.
Таким образом, для каждого гомологического ряда отношения
упругостей паров соседних членов ряда являются при низких тем-
пературах значительными величинами. В то же время углеводороды
различных гомологических рядов, но имеющие в молекуле одина-
ковое число углеродных атомов, имеют весьма близкие упругости
паров, поэтому отношения этих упругостей представляют собой срав-
нительно небольшие величины. Так, например, отношения упруго-
стей паров этена и этана при температурах от —140 до —150° нахо-
дятся в пределах от 3 до 4; для пропена и пропана эти величины
еще меньше, вследствие чего разделение этих компонентов весьма
затруднено.
При разделении углеводородов мы должны, следовательно, ожи-
дать, что этан будет испаряться вместе с этеном, пропан вместе с
пропеном и т. д. Таким образом, при успешном разрешении задачи
фракционного испарения углеводородных смесей мы получим в ре-
зультате три фракции. В каждой фракции будут находиться угле-
водороды с одинаковым числом углеродных атомов.
Вопрос о дальнейшем анализе этих фракций первоначально пред-
полагалось разрешить путем замера кривых испарения с помощью
микроманометра. Однако, учитывая необходимость применения при-
бора в производственных условиях и желательность максимального
упрощения конструкции прибора и техники анализа, было решено
использовать другой способ. Этот способ заключается в разложении
полученной фракции в присутствии алюминия (без доступа воздуха)
в кварцевой трубке. Как это было установлено еще в 1907 г. проф. Ли-
довым и Кузнецовым [10], алюминий при нагревании до температуры
красного каления разлагает углеводороды. Углерод связывается
алюминием, а водород остается свободным. Отношение объема обра-
зовавшегося водорода к объему взятого газа зависит от количества
водородных атомов в молекуле испытуемого углеводорода.
При разложении этена выделяется двукратный объем водорода,
при разложении этана—трехкратный объем и т. д. Следовательно,
если мы имеем два углеводорода с разным числом водородных ато-
мов, то по количеству выделившегося водорода можно установить
процентное содержание каждого из углеводородов, поскольку здесь
имеет место простая линейная зависимость.
Таким образом, можно дать следующую схему анализа смеси угле-
водородных газов. Очень небольшое количество газа подвергается
фракционному испарению, в результате которого получаются три
фракции: 1) этан + этен, 2) пропан + пропен и 3) бутан + бу-
тены.
В третьей фракции находятся также и все более тяжелые угле-
водороды (пентан и т. д.), что же касается ацетилена, то о нем речь
будет итти ниже.
176
Каждая из полученных фракций разлагается с помощью нагре-
того до красного каления алюминия.
Устройство прибора схематически изображено на фиг. 75. Бю-
ретка 2 имеет уравнительный сосуд 7. К бюретке присоединены про-
мывалка 3 с раствором едкого кали и трубка 4 с фосфорным ангидри-
дом. 5 и 6 — баллончики для конденсации газов. К баллончикам
Фиг. 75. Схема прибора для определе-
ния углеводородов с применением алю-
миния.
присоединены ртутные манометры. Для создания высокого вакуума
в приборе служит баллончик 7 с активированным углем, присоеди-
ненный к прибору через трехходовый кран. Трубка 5 служит для
конденсации газов. К одному из ходов крана 8 присоединена квар-
цевая трубка 9, в которой нахо-
дится алюминий. Другой ход
крана 8 идет к крану 10 и слу-
жит для откачки из прибора
газов, что производится масля-
ным насосом и углем. Краны 11
и 12 служат для разъединения
баллончиков 5 и 6 и трубки 4
с фосфорным ангидридом.
Ход анализа следующий. В
приборе прежде всего создается
вакуум сначала масляным насо-
сом через кран 10, а затем с
помощью баллончика с углем 7,
погруженного в жидкий воздух.
Вакуум создается также в труб-
ках 3 и 4. Анализируемый газ
забирается в бюретку. Запор-
ной жидкостью в бюретке служит раствор поваренной соли или
сернокислого натрия. Баллончик 5 погружается в жидкий воз-
дух. Кран 11 закрывается, а из бюретки в трубки 5, 6 и 4 мед-
ленно впускается анализируемый газ. Кран 12 открывается, и газ
попадает в баллончик 5, где и конденсируются тяжелые углеводороды.
Так как некоторое количество тяжелых углеводородов остается в
трубках 3 и 4, то их промывают воздухом, впуская его через кран
бюретки в баллончик 5. Все тяжелые углеводороды попадают таким
образом в баллончик 5. Трубки 3 и 4 служат для удаления из газа
и воздуха углекислоты и паров воды. После промывки кран 12 за-
крывают, а в баллончике 5 через краны 11,8 и 10 создают вакуум
сначала масляным насосом, а затем углем. После того как манометр
баллончика 5 покажет нуль, необходимо продолжать откачку углем
еще несколько минут, чтобы удалить последние остатки метана и
других газов. Закрыв кран 77, удаляют жидкий воздух, в который
был погружен баллончик 5, дают ему нагреться до комнатной тем-
пературы и по манометру замеряют давление тяжелых газообразных
углеводородов. Зная объем баллончика 5 (что определяют заранее),
вычисляют суммарное процентное содержание тяжелых углеводо-
родов в анализируемом газе. Объем газа для анализа определяют
по бюретке.
12 в, А. Соколов.
777
В баллончике 5 мы имеем, таким образом, следующие компоненты:
этан, этен, пропан, пропен, бутан и бутены, а также и некоторые
другие, более тяжелые углеводородные газы, если таковые присут-
ствовали в анализируемой смеси.
Далее тяжелые углеводороды, находящиеся в баллончике 5, разго-
няются на три фракции: 1) этан + этен, 2) пропан пропен и 3) бу-
тан, бутены и более тяжелые углеводороды. Чтобы выяснить усло-
вия, при которых возможна подобная разгонка, был проведен ряд
опытов с индивидуальными углеводородами и их искусственными сме-
сями. Испытания индивидуальных углеводородов и разгонка их
смесей производились следующим образом. Сначала углеводороды
конденсировались в баллончике 5, как это описано выше, затем бал-
лончик 5 погружался в дьюаровский сосуд, где поддерживалась та
или иная низкая температура. Для поддержания постоянной тем-
пературы в дьюаровский сосуд помещали широкую медную трубку
с баллончиком 5 и термометром и термопарой.
После того как в баллончике 5 устанавливалась определенная
температура, кран 11 открывали и баллончик 6 погружали в жидкий
воздух при закрытом кране 8. Углеводороды, которые при темпера-
туре баллончика 5 имеют некоторую упругость паров, хотя бы исчи-
сляемую десятыми или сотыми долями миллиметра, переходят при
этом в баллончик 6. Через некоторое время кран 11 закрывают, уда-
ляют жидкий воздух от баллончика 6 и измеряют количество скон-
денсировавшегося газа. Эту операцию проводят до тех пор, пока
количество газов, конденсирующихся в баллончике 6, не перестанет
увеличиваться.
Проведенные опыты показали, что отделение этана и этена от
остальных компонентов можно произвести подобным образом при
температуре минус 144—145°. В табл. 19 приведены данные окон-
чательных опытов по отделению этана от пропана и этена от пропена;
Таблица 19
Данные по отделению этана от пропана и этена от пропена
Этан + пропан, мм
Этен 4-пропен, мм
Газ
4J
3"
S
Ч
О
X
о
U.
о
ь
ск
И И
X
о
н
rt
ь
о
о
Г аз
с2н6
с3н8
36
28,5
Таким образом, при температуре —145° этан и этен должны от-
гоняться от остальных углеводородов. Точность производившихся
замеров по манометру лежит в пределах 1 мм.
178
Следующая серия опытов касалась подыскания такой температуры»
при которой отгонялись бы пропан и пропен. Подобную разгонку,
как выяснилось, можно произвести при температурах от —120° до
—122°. В табл. 20 приведены данные окончательных опытов по от-
делению пропана и пропена от остальных углеводородов.
Таблица 20
Данные по отделению пропана и пропена
Газ
С3н8
с4н10
Пропан 4-бутан, мм
к
со
о о
38
37
Газ
Пропен + бутен, мм
38
51
Таким образом, при температурах от —121 ° до —122° пропан и про-
пен будут отделяться от остальных углеводородов. При температу-
рах от—121° до —122° в остатке будут находиться бутан, бутены и
более тяжелые углеводороды.
Для окончательной проверки разработанной методики был про-
веден анализ смеси, искусственно составленной из шести колшонен-
тов—этана, этена, пропана, пропена, бутана и бутена. Результаты
анализа представлены в табл. 21.
Таблица 21
Данные анализа смеси из шести компонентов
Газ Количество взятого газа, мм Количество взятого газа по фрак- циям, мм Отогнано при —145°, мм Отогнано при температурах от — 121° до —122°, мм Остаток, мм
п о to to ® * 88 148 236 238 — —
С3Нв с3н8 18 39 57 — 56 —
С4Н8 31 24 55 — — 54 (по разнице)
12*
179
Полученные результаты показывают, что в пределах ошибки
измерений при указанных выше температурах разделение фракций
происходит удовлетворительно.
Дальнейший ход анализа заключается в разложении каждой из
фракций с помощью алюминия. После разгонки фракция этана и
этена направляется в кварцевую трубку, которая для этой цели по-
гружается в жидкий воздух, а краны, которые ведут к баллончику
с этой фракцией, открываются. После того как фракция соберется
в трубке 9, кран 8 закрывают. Трубку 9 нагревают до красного ка-
ления.
Предварительный замер фракции производят по манометру 13
(до нагрева). Нагрев ведут до тех пор, пока объем газа не перестанет
увеличиваться. Потребное для этого время устанавливают опытным
путем. При надлежащей температуре разложение углеводородов
происходит полностью за 20—30 мин. После окончания реакции дают
кварцевой трубке остыть до комнатной температуры, а затем замеряют
давление по манометру 13. Отношение этого давления к первоначаль-
ному (до нагрева) дает водородный коэфициент, по которому с по-
мощью графика определяют содержание каждого из компонентов
анализируемой фракции. Приводим пример подобного расчета. Со-
гласно табл. 21 во фракции этана и этена содержалось 63% этана
(148 мм из 236) и 37% этена (88 мм из 236). После отгонки из первой
фракции для анализа с алюминием было изъято 12 мм (по маномет-
ру 13). После 30-минутного нагревания давление газа поднялось
до 33 мм. Дальнейшее нагревание не вызывало повышения давле-
ния. Водородный коэфициент для этой фракции составляет, таким
образом, , т. е. 2,66, что соответствует 66% содержания этана
во фракции и 34% этена. Расхождение с составом взятого газа,
как видно, находится в пределах погрешностей измерений.
Следует отметить, что проверка точности реакции предельных
углеводородов с алюминием показала, что реакция идет достаточно
полно для большинства практических целей. Для этана, пропана
и бутана количество углеводорода, не вошедшего в реакцию, было
в пределах 1% от взятого количества. Опыты, проведенные автором
с пропеном, показали, что и здесь реакция идет достаточно полно
и количество неразложившегося углеводорода лежит в пределах 1%.
Таким образом, разработанная методика позволяет проводить
анализы тяжелых углеводородов, состоящих из упомянутых шести
компонентов.
Из непредельных углеводородов мы не касались еще ацетилена.
Первые же опыты с ацетиленом показали, что он задерживается в
трубках 3 и 4. Повидимому, здесь имеет место поглощение ацетилена
фосфорным ангидридом.
§ 6. Определение газолина в газе
Горючие природные газы в большинстве случаев содержат в боль-
шем или меньшем количестве пары газолина. Извлечение газолина
является крупной отраслью промышленности, занимающейся ути-
180
лизацией природного газа, поэтому измерение содержания газолина
в газе является одним из важных определений при анализе угле-
водородных газов. В зависимости от содержания газолина в газе
решается вопрос, следует ли строить газолиновый завод для извле-
чения газолина из данного газа и, если следует, каким способом
извлекать газолин из газа.
Для измерения содержания газолина в газе путем определения
индивидуальных углеводородов описанными выше методами суще-
ствует ряд других методов, которые можно разделить на две группы:
косвенные и прямые.
Косвенные методы определения газолина
К косвенным методам измерения содержания газолина в газе
относится определение удельного веса газа и его растворимости в со-
ляровом масле.
Способ определения газолина по удельному весу весьма груб
и ориентировочен; при некоторых условиях он может повести к
совершенно ошибочным заключениям.
Удельный вес остальных компонентов, кроме газолина, входящих
в состав горючего природного газа, колеблется от 0,55 для метана
до 2,0 для бутана; удельный вес газолина еще больше. Следовательно,
чем выше удельный вес газа, тем большего содержания газолина в
нем можно ожидать. Но это утверждение было бы совершенно спра-
ведливо, если бы в горючем газе не встречались углекислота и азот
и если бы метан, этан, пропан и бутан входили в состав газа всегда
в одной и той же пропорции. Ни того, ни другого на самом деле нет.
В горючем газе могут встречаться СО2 и N2, а также и СН4, С2Н6
и т. д. в разных количествах.
Так как горючий природный газ в большинстве случаев представ-
ляет собой смесь всех перечисленных углеводородов, то его удель-
ный вес колеблется от 0,6 до 1,5. В очень редких случаях удельный
вес газа бывает еще выше. Газы с большим удельным весом обычно
содержат больше газолина. На основании испытаний различных образ-
цов газа составлена таблица содержания газолина в газах различ-
ного удельного веса (табл. 22). Эта таблица основана на результатах
сотен определений, произведенных в различных местностях, и в
ней даны средние цифры содержания газолина в газах (удельный
вес воздуха принят равным единице).
Следует заметить, что практические данные очень часто и резко
расходятся с величинами, приведенными в табл. 22.
Газ может иметь самый большой удельный вес, упомянутый в
таблице, именно 1,5, и в то же время совершенно не содержать газо-
лина. Например, газ может состоять из чистой СО2 и, следовательно,
иметь удельный вес 1,5. Чтобы не впасть в ошибку, связанную с
присутствием СО2, газ рекомендуется сначала очистить от СО2 при
помощи КОН, а затем уже определять его удельный вес. Но и при
таком определении мы никогда не гарантированы от ошибки. Газ
может состоять из пропана с небольшой примесью бутана и этана
181
Таблица 22
Удельный вес газа и содержание в нем газолина
Уд. вес Содержание газолина, см3 на 1 м3 газа Уд. вес Содержание газолина, см3 на 1 м3 газа Уд. вес Содержание газолина, см3 на 1 м3 газа
0,60 8,0 0,77 143 0,90 254
0,65 42,8 0,78 152 0,95 295
0,66 52,2 0,79 160 1,00 338
0,67 60,2 0,80 170 1,05 381
0,68 66,8 0,81 179 1,10 424
0,69 77,6 0,82 187 1,15 466
0,70 85,6 0,83 195 1,20 509
0,71 93,6 0,84 203 1,25 552
0,72 103 0,85 213 1,30 595
0,73 111 0,86 221 1,35 638
0,74 120 0,87 229 1,40 680
0,75 128 0,88 235 1,45 723
0,76 135 0,89 243 1,50 766
и не содержать совершенно газолина, имея в то же время удельный
вес больший 1,5.
Таким образом, способ определения газолина по удельному весу
газа ненадежен и им можно пользоваться только, когда нет возмож-
ности произвести определение другим путем. Перед определением
удельного веса всегда следует уда-
лять СО2 или, определив ее содержа-
ние, вводить соответствующие по-
правки.
Природный горючий газ раство-
рим в таких жидкостях, как соляро-
вое масло, лигроин и т. п.
Степень растворимости зависит
от содержания в газе высших угле-
водородов. Чем больше содержание
газолина в газе, тем больше раство-
римость его в масле.
Для определения растворимости
газа можно употреблять прибор, изо-
браженный на фиг. 76.
Пипетка для поглощения имеет
Фиг. 76. Прибор для определе-
ния газолина путем его раство-
рения.
два шара 7 и 2; емкость первого
135—140 сл/3, емкость второго 35 см3. В уравнительный сосуд нали-
вается ртуть; поднятием уравнительного сосуда заполняют шар 7
ртутью; в шар 2 наливают 35 см3 свежего солярового масла. Опуская
уравнительный сосуд, масло переводят в шар 7, затем поворотом
крана сообщают шар 7 с бюреткой 3, в которую введено 100 см3 испы-
туемого газа из баллона для взятия проб газа 4. Способ перевода
182
газа из баллона 4 в бюретку ясен из фигуры. Так как результаты
испытания получаются приблизительные, то большой точности в
измерении объема газа не требуется, и поэтому бюретка вместо крана
имеет зажим.
Таблица 23
Растворимость газа И содержание
в нем газолина
Растворимость газа, % Содержание газо- лина, смя на 1 mz газа
10 Не содержится
20 67
25 200
30 330
40 670
70 1060
Опусканием уравнительного сосуда 100 см'л газа переводят в
шар 7. После встряхивания пипетки в течение 3 мин. газ переводят
обратно в бюретку и измеряют оставшийся объем. Затем газ снова
переводят в пипетку и после встряхивания — опять в бюретку. Эту
операцию повторяют до тех пор, пока объем газа не перестанет умень-
шаться, что будет означать окончание растворения. Растворимость
чистого метана в масле равна 11—12%, растворимость чистого эта-
на 68%. Приблизительное содержание газолина в газе может
быть вычислено при помощи табл. 23.
Растворимость газа является недостаточно надежным признаком
для определения газолина по тем же причинам, какие были указаны
и для удельного веса.
Прямые методы определения газолина
Прямые способы определения газолина состоят в извлечении его
из газа тем или иным способом. Извлечь газолин из газа можно тремя
способами: компрессией, поглощением при помощи солярового масла
и поглощением при помощи активированного угля. Всеми тремя
методами пользуются для определения газолина в газе.
Для определения содержания газолина при помощи компрессии
употребляют специальные компрессоры небольшого размера, снаб-
женные мотором, газовым счетчиком, холодильником и прочими
приспособлениями для извлечения газолина из газа; подобная уста-
новка делается передвижной, для чего ее помещают на автомашину*
Для извлечения газолина из газа можно пользоваться также
ручным компрессором, изображенным на фиг. 77. Этот компрессор
состоит из цилиндра, содержащего определенное количество газа
при атмосферном давлении, ручного гидравлического насоса, при
183
Фиг. 77. Ручной компрес-
сор для определения газо-
лина в газе.
помощи которого достигается необходимое давление для сжижения
паров газолина, стеклянной измерительной трубки, в которой соби-
рается ц измеряется газолин, и манометра, показывающего, при
каком давлении произошло сжижение.
Определение газолина при помощи ручного компрессора про-
изводится следующим образом. Холодную воду впускают через вен-
тиль 1 и действуют ручным насосом до тех пор, пока вода не польется
через трубку 2, что означает полное вытеснение воздуха из прибора.
Испытуемый газ присоединяют к трубке 2,
после чего выпускают из прибора воду
через вентиль 5; манометр показывает да-
вление газа. Через вентиль 7 опять пуска-
ют воду и действуют насосом до тех пор,
пока в измерительной трубке 4 не появит-
ся газолин. Из измерительной трубки газо-
лин можно выпустить через кран, изме-
рить количество его и подвергнуть раз-
личным испытаниям.
Масляные абсорберы. Известно
несколько систем абсорберов, применяю-
щихся для определения газолина при по-
мощи поглощения маслом.
На фиг. 78а изображена одна из подоб-
ных систем.
Аппарат (фиг. 78а) состоит из отрезка
6" трубы, разделенной на пять отделений,
которые могут соединяться последователь-
но. Каждое отделение имеет 3/4" трубку
для ввода газа и 2" трубку для отвода,
доходящую почти до дна камеры. Из отвер-
стия в верхней части камеры выходит 3/g'?
трубка, свернутая в спираль диаметром
3" с семью оборотами, через которую
проходит испытуемый газ и где происхо-
дит наибольшее поглощение. Для произ-
водства определения в каждое отделение
абсорбера наливают 2700 см3 очищенного
солярового масла или вообще достаточное количество, чтобы уровень
масла был на 2" выше верхней части б” трубки и значительно выше
входного отверстия змеевика. Наиболее существенное требование,
которое предъявляется к маслу, состоит в том, чтобы его начальная
температура кипения была как можно выше, чтобы при последующей
перегонке можно было достигнуть количественного отделения погло-
щенного газолина. Применяемое масло имеет удельный вес 36° Вё
и начальную температуру кипения 250°. В большинстве случаев
пользуются только первыми тремя отделениями, четвертым отделе-
нием пользуются только, если имеют богатый газолином газ, пятое
же отделение не наполняется маслом, а служит для удержания частиц
масла, увлекаемых газом из предыдущих отделений. К входному
184
концу трубки присоединяют газовый счетчик. Испытуемый газ мед-
ленно впускают при закрытом вентиле на выпускной линии. Когда
в абсорбере установится постоянное давление, открывают выпуск-
Фиг. 78а. Масляный абсорбер для определения газолина в газе
Фиг. 786. Отгонка газолина из масла.
ной вентиль, допускающий прохождение газа через счетчик с желае-
мой скоростью. Входящий в абсорбер газ проходит пузырьками через
масло, поглощающее газолин. Назначение змеевика — обеспечить
продолжительный и тесный контакт между маслом и газом. После
185
прохождения нужного объема газа входной кран закрывают, а дав-
ление в абсорбере спускают через счетчик. Из каждого отделения
масло выпускают и объем его тщательно измеряют, из каждого от-
деления берут 1000 см3 его для разгонки.
В колбу емкостью 500 см3 помещают 400 см3 масла; колбу соеди-
няют с холодильником, представляющим собой медную трубку, про-
ходящую через металлический ящик с ледяной водой (фиг. 786).
Колбу нагревают сначала слабо, а затем сильнее, отгоняемый же
газолин собирают в градуированный цилиндр. Колбу нагревают
так, чтобы температура паров была не выше 190—195°. Определив
количество газолина, отогнанное из пробы масла, легко вычислить
содержание его в газе.
Поглощение газолина зависит от скорости прохождения газа
через масло, от содержания газолина в газе, от объема газа, прошед-
шего через абсорбер, от давления газа и от температуры масла.
В табл. 24 приведены оптимальные условия для абсорбции газа
маслом.
Таблица 24
Оптимальные условия для работы масляного абсорбера
Максимальная скорость газа, мг1час Давление, ат Объем газа, прошедшего через абсор- бер, JW3 Содержание газолина, л на 100 м3
11,2 21 22,4 1,66
5,6 10,5 11,2 3,32
2,8 5,2 5,6 6,64
1,4 2,6 4,2 9,96
0,6 Атмосферное 2,8 13,28
— — 1,4 26,56
— — 0,7 53,12
Температура масла в абсорбере имеет большое влияние на эф-
фективность извлечения газолина. В ряде испытаний, проведенных
с газом, содержащим незначительное количество газолина, установ-
лено, что при изменении температуры масла в пределах 10—15° из-
менение объема поглощенного газолина достигало 30—40%.
Прибор, изображенный на фиг. 79, состоит из трех металличе-
ских цилиндров 7, 2, 3 диаметром 12,5 см, двух манометров и газо-
вого счетчика 4, соединенных между собой последовательно. Газ
входит в каждый абсорбер через трубку, доходящую до самого дна
и имеющую ряд мелких отверстий. В верхней части каждого абсор-
бера имеется трубка для выхода газа и трубка, через которую в аб-
сорбер наливается масло; последняя трубка снабжена крышкой на
нарезке.
В каждый абсорбер наливают около 7 л солярового масла. Обычно
пропускают газ через абсорбер под давлением б—8 ат.. Вентили
186
открывают так, чтобы через счетчик проходило около 30 л газа в
одну минуту. Когда желаемое количество газа пройдет через при-
бор, закрывают вентиль на впускной линии и спускают давление в
приборе через счетчик до атмосферного.
Из каждого абсорбера берут по 1 л масла для разгонки на при-
боре, изображенном на фиг. 79. Масло наливают в медный котел,
снабженный холодильником. Для нагревания масла через него про-
пускают водяной пар, получаемый из отдельного котла. Перегонку
продолжают в течение 20 мин. после того, как температура паров
в котле с маслом достигнет 98°. Газолин собирают в охлаждаемом
льдом градуированном цилиндре, находящемся под холодильником.
Рекомендуется сделать разгонку двух образцов масла из каждого
абсорбера. Зная количество отогнанного газолина, количество про-
шедшего через прибор
газа и измерив тщатель-
но количество масла,
имеющегося в каждом
абсорбере, легко вычи-
слить содержание газо-
лина в газе.
Масляные абсорберы
дают удовлетворитель-
ные результаты при
Фиг. 79. Масляный абсорбер.
определении содержания
газолина в газе, но они
слишком громоздки, что
затрудняет пользование ими, особенно в полевых условиях.
Для определения газолина в газе весьма удобным и портативным
является прибор, в котором газолин поглощается активированным
углем. Этот прибор состоит из газового счетчика, набора трубок
с активированным углем и реометра; кроме того, к прибору прила-
гается диафрагменный измеритель, трубка Пито, U-образный мано-
метр и различные принадлежности, которые требуются при работе.
Счетчик трубки с углем, реометр и все принадлежности помещаются
в металлическом ящике. Общий вид прибора изображен на фиг. 80, а.
На откидной крышке ящика помещается реометр, к которому
присоединен вентиль; через этот вентиль впускается испытуемый
газ. Реометр представляет собой металлическую трубку, в середине
которой имеется пластинка с маленьким отверстием; перед пластин-
кой и после нее металлическая трубка имеет отводы с кранами, при-
соединяющимися к U-образному манометру, наполненному водой.
Наличие пластинки с малым отверстием ведет к образованию разницы
давления газа до пластинки и после нее. Другой конец реометра
присоединен при помощи каучука к газовому счетчику. При испы-
тании газа на содержание газолина к выходной трубке газового счет-
чика присоединяют при помощи каучука металлический цилиндр
с активированным углем, который и абсорбирует газолин. Когда
достаточное количество газа пройдет через прибор, уголь пересы-
пают в колбу и отгоняют газолин, измерив количество которого, легко
187
вычислить содержание газолина в газе, так как объем прошедшего
через прибор газа известен. Устройство угольного поглотителя изоб-
ражено на фиг. 80, б. Металлическая трубка емкостью около 250 ем3
имеет на обоих концах нарезки, на которые навинчиваются крышки 2
и 4, снабженные для герметичности резиновыми прокладками. Внутри
трубки около одного из концов имеется металлическая сетка 3, под-
держивающая уголь. Для пропускания газа через абсорберу с него
снимают крышки и на тот конец, где нет сетки, надевают специаль-
ную крышку, в середину которой
впаяна изогнутая медная трубка 7;
эту трубку и npi соединяют к вы-
ходному концу счетчика.
Когда прибор не работает, все
трубки с углем должны плотно
закрываться.
Наличие в приборе реометра
и газового счетчика дает возмож-
Фигг. 80. Прибор для определения газолина с помощью активи-
рованного угля.
ность определить также удельный вес испытуемого газа, а прила-
гаемые к прибору диафрагменный измеритель и трубка Пито позво-
ляют определить дебит газа.
Приступая к испытанию газа, прежде всего определяют дебит
газа; предварительно следует удостовериться, что давление газа
при открытой скважине достигло постоянной величины. Если сква-
жина была закрыта, газ следует выпускать в атмосферу в течение
времени, пока давление не станет постоянным.
Для дебита до 10 тыс. м3]сутки можно употреблять диафрагмен-
ный измеритель, который ввинчивают в головную насадку сква-
жины; все остальные отверстия в насадке при этом должны быть
закрыты. Манометр наполняют водой до нулевой метки и присоеди-
няют к ниппелю диафрагменного измерителя при помощи каучуко-
вой трубки. В измеритель вставляют такую диафрагму, чтобы ди-
188
ференциальное давление (разница уровней воды) на манометре было
от 7 до 18 см. Когда диференциальное давление достигнет постоян-
ной величины, его замечают. Определив удельный вес газа, как опи-
сано ниже, вычисляют дебит газа, пользуясь специальными табли-
цами. Если дебит газа превышает Ютыс. м^сутки, применяют трубку
Пито.
Для определения удельного веса газа диафрагменный измеритель
и манометр соединяют так, как описано выше, чтобы диференциаль-
ное давление было несколько более 20 см.
В U-образную трубку реометра, находящегося на крышке при-
бора, наливают воду до нулевой метки; шкала реометра сделана
подвижной, что облегчает ее установку; краны U-образного мано-
метра должны быть открыты. Когда прибор не работает, краны мано-
метра должны быть закрыты, иначе при опускании крышки прибора
вода из манометра выльется.
Закрыв вентиль, находящийся с правой стороны реометра, диа-
фрагменный измеритель соединяют с реометром, для чего каучук,
идущий из измерителя, надевают на трубку с вентилем. Левую сто-
рону реометра присоединяют к входной трубке счетчика. Открыв
краны манометра, открывают вентиль так, чтобы диференциальное
давление было равно 20 см; это давление должно быть постоянным
в течение всего определения.
При постоянном диференциальном давлении, равном 20 см, заме-
чают по секундомеру время, необходимое для пропуска через прибор
определенного количества газа, например 30 л. Время, потребное
на прохождение равного объема воздуха, обычно дается при при-
боре как постоянная величина; эту постоянную величину необходимо
время от времени проверять, а при точных измерениях определять
при каждом испытании газа.
Время, потребное для прохождения определенного объема воз-
духа, удобнее всего определить, присоединив реометр к линии, подаю-
щей воздух; если такой линии нет, продувают через прибор 2—3 л
воздуха ртом и умножают получаемое время на соответствующий
коэфициент, показывающий, во сколько раз объем продутого воз-
духа меньше объема прошедшего через прибор газа. Желательно
воздух, идущий из легких, освобождать от СО2 и паров воды, про-
пуская его через раствор соды или едкого кали. При некоторой прак-
тике этот способ продувания может дать удовлетворительные ре-
зультаты, если опыт проделать несколько раз и взять среднее из
полученных величин.
Более удовлетворительный метод измерения времени, потреб-
ного на прохождение воздуха, состоит в продувании воздуха через
прибор при помощи двойной резиновой груши или какого-либо на-
гнетающего насоса.
Удельный вес газа вычисляется по формуле
где d —удельный вес газа;
189
7'—время прохождения газа;
/ — время прохождения такого же объема воздуха.
Приступая к определению содержания газолина в газе, диафраг-
менный измеритель, реометр и счетчик соединяют так же, как и при
определении удельного веса, оставляя ту же самую диафрагму1.
Пропустив через прибор 15—20л для вытеснения находящихся в при-
боре воздуха и газа от предыдущего испытания, закрывают вентиль
реометра. Берут адсорбер, наполненный свежим активированным
углем, снимают с него обе крышки и на тот конец, где нет сетки, на-
винчивают крышку с отводной трубкой, которую и соединяют каучу-
ком с выходным концом счетчика. Адсорбер можно затем поместить
на его прежнее место в приборе, но необходимо смотреть, чтобы газ
имел свободный выход со дна адсорбера.
Перед определением необходимо убедиться, что прибор не дает
течи. Записав показания стрелок счетчика, открывают вентиль,
чтобы скорость прохождения газа через прибор была в пределах
7—9 л1мин. Эта скорость будет достигнута, когда диференциальное
давление реометра будет в пределах 12—20 см.
Через угольный поглотитель должно пропускаться такое коли-
чество газа, чтобы после отгонки получить из угля около 25 см3 газо-
лина. Потребное количество газа можно приблизительно определить,
исходя из его удельного веса, по табл. 25.
Содержание газолина в табл. 25 выведено на основании удель-
ного веса газа. Как было указано выше, подобное определение яв-
ляется весьма приблизительным, а во многих случаях дает совер-
шенно ложные результаты, поэтому, определяя содержание газолина
при помощи угля, мы можем получить после отгонки такое количество
газолина, которое будет значительно уклоняться от указанной выше
нормы в 25 см3.
Если количество отогнанного из угля газолина будет больше
40 см3 или меньше 12 см3, то полученную величину содержания газо-
лина в газе мы не должны считать совершенно правильной, В пер-
вом случае, т. е. когда газолина получено более 40 см3, уголь был
почти насыщен газолином, поэтому можно опасаться, что из послед-
них порций газа уголь не поглотил всего газолина; содержание газо-
лина в газе в этом случае получится уменьшенным. Кроме того, уголь
обладает свойством в большей степени поглощать тяжелые углеводо-
роды, чем легкие; по мере насыщения угля более тяжелые углеводо-
роды вытесняют легкие, поэтому газолин, полученный из насыщен-
ного угля, будет более тяжелым, чем полученный из угля, насыщен-
ного углеводородами. Во втором случае, т. е. когда газолина полу-
чено менее 12 см3, можно опасаться, что результат является преуве-
личенным вследствие поглощения углем более легких углеводородов;
в этом случае отогнанный из угля газолин будет легкокипящим.
1 Если испытание производится в лаборатории, то диафрагменный изме-
ритель, разумеется, не требуется и испытуемый газ непосредственно впускают в
прибор через вентиль.
190
Таблица 25
Количество газа, потребное для определения газолина с помощью угля
Уд. вес Содержание газолина, см3 на 1 м3 газа Потребное количество газа, л/3 Уд. вес Содержание газолина, см3 на 1м3 газа Потребное количество газа, м3
0,60 8,0 3,54 0,84 203 0,122
0,65 42,8 0,558 0,85 213 0,116
0,66 52,2 0,493 0,86 221 0,113
0,67 60,2 0,416 0,87 229 0,107
0,68 66,8 0,374 0,88 235 0,105
0.69 77,6 0,323 0,89 243 0,102
0,70 85,6 0,291 0,90 254 0,099
0,71 93,6 0,263 0,95 295 0.085
0,72 103 0,243 1,00 338 0.074
0,73 111 0,226 1,05 381 0,065
0,74 120 0,207 1,10 424 0,059
0,75 128 0,195 1,15 466 0,054
0,76 135 0,184 1,20 509 0,048
0,77 143 0,175 1,25 552 0,045
0,78 152 0,164 1,30 595 0,0425
0,79 160 0,156 1,35 638 0,0396
0,80 170 0,147 1,40 680 0,0368
0,81 179 0,139 1,45 723 0,0340
0,82 187 0,133 1,50 766 0,0340
0,83 195 0,127 — — —
Результаты сравнительных испытаний, производящихся с раз-
личными количествами одного и того же газа и с применением одина-
ковых поглотителей, содержавших 250 см3 угля, показали, что при
количестве отогнанного из угля газолина от 12 до 40 см3 результаты
вычисляемого содержания газолина в газе совпадают в пределах
2—3%. Если при отгонке получится более 40 см3 или менее 12 см3
газолина, то испытание рекомендуется повторить, соответственно
уменьшив или увеличив количество пропускаемого через прибор
газа.
После окончания пропускания газа через прибор поглотитель
следует плотно завинтить крышками, наклеить ярлычок с обозна-.
чением места взятия газа, его дебита, температуры воздуха и прочих
данных и отправить в лабораторию для отгонки газолина.
Как было указано выше, рекомендуется пропускать газ через
поглотитель со скоростью 7—9 л]мин.
При испытании очень бедного газолином газа скорость пропу-
скания можно увеличить до 23 aJmuh без заметного влияния на ре-
зультаты. При испытании же очень богатого газа скорость пропу-
скания следует уменьшить до 1,5—2 л!мин. Рекомендуется брать
такую скорость прохождения газа, чтобы испытание продолжалось
не менее 30 мин.; вследствие этого теплота, получающаяся при по-
191
голщении газолина, успевает рессеяться, что способствует полу-
чению правильных результатов.
Количество газолина, поглощенное углем, зависит от темпера-
туры поглотителя. Чем ниже температура, тем больше количество
поглощаемого углем газолина и тем меньше удельный вес газолина.
Температура не имеет особого влияния на результаты испытания,
если отогнанный газолин выветривать до получения продукта желае-
мой упругости паров и выход вычислять по объему выветренного
продукта.
Процентное содержание воды в угле не оказывает значительного
влияния на поглотительную способность паров газолина. Уголь,
применяемый для определения газолина в газе, может иметь по край-
ней мере до 4% влаги. При высоком содержании влаги в угле его
способность поглощать наиболее летучие пары несколько умень-
шается.
При определении содержания влаги в угле можно пользоваться
также прибором, применяющимся для отгонки газолина, описан-
ным ниже; к 250 см? угля прибавляют 100 смъ нефти, керосина или
еще какого-либо тяжелого дестиллата, после чего производят от-
гонку.
Поглотительная способность угля, естественно, зависит от его
качества. Уголь, применяемый для определения газолина в газе, дол-
жен отвечать следующей спецификации: выдерживать 50-минутную
Фиг. 81. Испытание поглотительной способности угля.
пробу с четыреххлористым углеродом; проходить через сито с 8—14
отверстиями в квадратном дюйме; содержание влаги не должно пре-
вышать 4% по весу. Рекомендуется применять специальный акти-
вированный уголь. Для испытания поглотительной способности угля
можно пользоваться прибором, изображенным на фиг. 81.
Уголь помещают в стеклянную трубку 7, на дне которой имеется
фарфоровый диск с отверстиями: этот диск не позволяет углю про-
валиваться вниз. Трубка 7 присоединена к газовой горелке 2, в ко-
торую газ поступает в направлении, указанном стрелкой. Трубка 7
должна иметь диаметр около 15 мм\ высота слоя угля должна быть
около 10 см.
192
Фиг. 82. Прибор для отгонки газолина
из
Чтобы получать сравнимые результаты, количество испытуемого
угля во всех случаях должно быть одинаково и равно 16 см3. Рео-
метр 3 градуируют так, чтобы через него проходило 750 см3 воздуха
в минуту; эта скорость должна оставаться постоянной в продолже-
ние всего испытания.
Воздух, пропускаемый через уголь, проходит первоначально
через три промывалки с серной кислотой для удаления влаги, а за-
тем через две промывалки с четыреххлористым углеродом, поме-
щенные в сосуд с тающим льдом. Воздух с парами четыреххлори-
стого углерода проходит затем через реометр, через уголь и попа-
дает в газовое пламя, в котором подвешена медная спираль. Высота
пламени должна быть око-
ло 30 мм. Уголь сначала
будет поглощать пары че-
тыреххлористого углерода,
и в горелку будет посту-
пать чистый воздух. Как
только уголь насытится и
пары четыреххлористого
углерода попадут в горел-
ку, пламя сейчас же окра-
сится в зеленый цвет. Вре-
мя, протекшее с момента
пуска воздуха до появле-
ния зеленой окраски пла-
мени, замечают; оно харак-
теризует поглотительную
способность угля. Чем
больше это время, тем вы-
ше поглотительная способность угля; следовательно, 60-минутный
уголь обладает большей поглотительной способностью, чем 50- или
40-минутный.
Отгонка газолина из угля. Прибор, применяемый
для отгонки газолина из угля, изображен на фиг. 82. Он состоит из
колбы 3, боковая трубка которой 8 соединена при помощи пробки
с медным змеевиком холодильника 7, наполненного кусками льда
с водой. Отгоняемый газолин попадает в градуированный цилиндр
10 емкостью 50 см3, помещенный в стакан 9 со льдом и водой. К труб-
ке, через которую попадает в цилиндр 10 газолин, присоединен на
каучуке угольный поглотитель 11. Колба нагревается горелкой 6;
7, 4 и 5 — держатели для колбы и горелки. Отгонку газолина из
угля можно производить или с глицерином, или с ртутью, или с по-
мощью водяного пара. При отгонке газолина с глицерином в колбу
наливают приблизительно 150 см3 чистого глицерина1. Берут пу-
стой угольный поглотитель и пересыпают в него испытуемый уголь;
для этого открытые концы обоих поглотителей соединяют и пере-
ворачивают их так, чтобы уголь пересыпался в пустой поглотитель.
1 Чистота применяемого глицерина не должна быть ниже 90%.
13 в. А. С околов, 193
Цель этой операции — перевести наверх уголь, наименее насыщен-
ный газолином (газ пропускают через уголь сверху вниз, поэтому
нижняя часть угля наименее насыщена газолином). Около 40 см3
наименее насыщенного газолином угля насыпают в отдельный погло-
титель 77 и присоединяют его к прибору для отгонки. Остальной
уголь всыпают в колбу.
Через пробку в колбу входит термометр 2, градуированный до
360°, шарик которого должен находиться немного ниже боковой
трубки колбы.
Первое время колбу подогревают очень осторожно, чтобы пре-
пятствовать образованию пены в глицерине. Когда глицерин подо-
греется, горелку устанавливают против центра колбы и регулируют
пламя так, чтобы вся отгонка продолжалась не более 45 мин. Когда
температура достигнет 100—110°, горелку отнимают и дают колбе
остыть в течение 5 мин. Затем 40 см3 угля, находившегося в погло-
тителе 77 для задержки паров газолина, отходящих вместе с некон-
денсирующимися парами во время отгонки, пересыпают насколько
возможно быстро в колбу. Отгонку продолжают, пока не будет до-
стигнута температура 250°.
Отогнанный газолин собирают в градуированный цилиндр 10',
зная объем полученного газолина, легко вычислить его содержание
в газе.
Холодильник 7 рекомендуется снабжать приспособлением для
перемешивания смеси льда и воды. Это перемешивание производится
путем пропускания воздуха через смесь.
Полученный газолин можно выветривать до получения желае-
мого удельного веса.
Кроме глицерина, отгонку газолина из угля можно производить
при помощи ртути; преимущество этого метода состоит в том, что
уголь после каждой отгонки остается сухим и после отделения ртути
может вновь применяться для испытания; кроме того, при употреб-
лении ртути можно не бояться таких явлений, как вспенивание гли-
церина и его обугливание. При пользовании ртутью уголь остается
на ее поверхности; пары ртути проникают в уголь и вытесняют газо-
лин. Прежде чем разобщить колбу и холодильник, необходимо как
следует охладить весь прибор, чтобы защитить работающего от ядо-
витых паров ртути. Змеевик холодильника следует делать стеклян-
ным, так как ртуть растворяет многие металлы. Большая часть ртути
отделяется от угля фильтрованием, но небольшое количество ее всегда
будет оставаться в угле, несмотря на все предосторожности при раз-
делении. Ртуть, остающаяся в угле, будет амальгамировать метал-
лический адсорбер, вследствие чего во время испытания газа в ад-
сорбере может образоваться течь. Для предотвращения этого при
пользовании ртутью вместо глицерина рекомендуется употреблять
адсорберы из цельнотянутых железных трубок.
Применение глицерина и ртути имеет ряд неудобств. Гораздо
удобнее пользоваться методом, применяемым в наших лабораториях
и заключающимся в отгонке газолина с помощью перегретого водя-
ного пара.
194
Метод отгонки в этом случае является очень простым. Через
герметичный патрон с углем пропускают перегретый водяной пар,
поступающий затем в холодильник. Водяной пар вытесняет газолин,
который вместе с водой после холодильника попадает в приемник.
Газолин легко отделяется от воды, и его граница с водой в стеклян-
ном градуированном цилиндре хорошо заметна. После окончания
отгонки и пропускания некоторого количества воздуха патрон с
углем готов для последующего определения.
Анализом газа при помощи всех описанных выше методов можно
определить количество газолина, которое может быть извлечено
из газа тем или иным способом. Но ни одно из этих испытаний не
может дать указаний на то, что представляет собой полученный газо-
лин: состоит ли он из пентана, гексана и гептана или является смесью
их, растворены ли в газолине бутан и пропан, и в каких количествах.
Все эти вопросы могут быть разрешены только путем разгонки газа
при низких температурах, описанной в начале этой главы.
13*
ГЛАВА ПЯТАЯ
МЕТОДЫ ОПРЕДЕЛЕНИЯ НЕОРГАНИЧЕСКИХ
ГАЗОВ, В ЭЛЕМЕНТАРНЫЙ СОСТАВ КОТОРЫХ
ВХОДЯТ АЗОТ, СЕРА, ГАЛОИДЫ И НЕКОТОРЫЕ
ДРУГИЕ ЭЛЕМЕНТЫ
§ 1. Определение окислов азота и аммиака
Определение о кислое азота
разообразные окислы азота NO и NO2 являются объектом ана-
• литических исследований главным образом при производстве
серной и азотной кислот. В природных газах окислы азота могут
встречаться лишь в незначительных концентрациях. Небольшие
количества окислов азота могут присутствовать в атмосферном воз-
духе. Закись азота в небольших концентрациях встречается в поч-
венном воздухе. Что касается глубинных природных газов, то ка-
ких-либо данных о наличии в них окислов азота не имеется; при-
сутствие в них NO и NO2 можно считать исключенным. Присутствие
N2O в глубинных природных газах сомнительно.
В табл. 26 приведены некоторые основные свойства газообраз-
ны:: окислов азота.
Таблица 26
Свойства газообразных окислов азота
Свойства N2O NO no2
Удельный вес (уд. вес воздуха принят равным единице) 1,5208 1,0367 1,5906
Вес 1 л газа при 0э и 760 мм рт. столба, г . . • . 1,977 1,3402 2,0568
Температура плавления. °C -102,4 -160,9 -9,3
» кипения, °C . -88,7 —150,2 4-21.3
Критическая температура, °C .... 36,5 -96 —
Критическое давление, ат 71,7 64 —
Растворимость в воде при 0°, объем газа в одном объеме воды 1,305 0,0738
Растворимость в воде при 20°, объем газа в одном объеме воды ... . 0,670 0,0470 ——
196
Закись азота — довольно устойчивое химическое соединение. До
сих пор не удалось найти таких веществ, которые при комнатной
температуре легко вступали бы в соединение с закисью азота и могли
бы служить для определения закиси азота методом поглощения.
Кислоты практически не действуют на закись азота. Растворы ще-
лочей оказывают лишь слабое действие на закись азота, главным
образом путем физического растворения. Точно так же на закись
азота слабо действуют и растворы обычно применяемых окислителей.
Для определения закиси азота некоторые исследователи реко-
мендовали воспользоваться повышенной растворимостью закиси азота
в абсолютном этиловом спирте. Действительно, один объем спирта
растворяет при 20° около трех объемов закиси азота. Однако повы-
шенной растворимостью в спирте обладают также углеводородные
газы, поэтому в присутствии углеводородов указанный метод вообще
неприменим. Кроме того, следует учесть, что и многие другие газы
обладают хотя и небольшой, но вполне заметной растворимостью
в спирте, вследствие чего полное отделение от этих газов закиси азота
невозможно.
Повышенной растворимостью закиси азота в спирте можно вос-
пользоваться для ее концентрирования при отсутствии в газовой
смеси других газов, хорошо растворимых в спирте. Во многих слу-
чаях для аналогичных целей можно воспользоваться, и притом с боль-
шим успехом, повышенной растворимостью закиси азота в воде.
Коэфициент растворимости закиси азота в воде при 0° составляет 1,3.
Этот коэфициент значительно меньше упомянутого коэфициента
растворимости закиси азота в спирте. Однако вода имеет то преиму-
щество, что в ней плохо растворяются углеводородные газы; кроме
того, водяные пары являются обычным компонентом газовой смеси,
а применение спирта вызывает появление в газовой смеси нового
компонента (пары спирта), что представляет ряд неудобств при даль-
нейшем анализе.
Применяя последовательно несколько пипеток с водой, автору
этих строк и А. М. Туркельтаубу удавалось поглощать более 90%
закиси азота из образцов подпочвенного воздуха, содержащего,
кроме того, и примесь углеводородных газов. Концентрация угле-
водородных газов при этом значительно не уменьшалась. Однако
эта методика довольно сложна, поэтому она может иметь лишь огра-
ниченное применение.
Большинство методов, предложенных для определения и иденти-
фикации закиси азота, основано на реакциях, происходящих при
высокой температуре. Один из наиболее употребительных методов
заключается в сожжении закиси азота в присутствии водорода. Смесь
закиси азота с водородом пропускают через нагретый пламенем газо-
вой горелки платиновый капилляр или через трубку с накаленной
платиновой проволокой. Происходит следующая реакция:
n2o+h2 = n2+h2o
Уменьшение объема смеси соответствует (за вычетом Н2О) объему
имевшейся закиси азота. Для полноты реакции рекомендуется иметь
197
избыток водорода, который затем может быть удален сожжением
над окисью меди, а остающийся азот — измерен. Измерение умень-
шения объема газовой смеси после сожжения и измерение оставше-
гося азота позволяют определить как содержание закиси азота, так
и свободного азота, имевшегося первоначально в закиси азота.
Указанная методика может дать удовлетворительные резуль-
таты лишь при отсутствии в газовой смеси таких компонентов, кото-
рые сами легко сгорают или разлагаются. Присутствие углеводоро-
дов осложняет упомянутую реакцию закиси азота и водорода. В от-
сутствии кислорода в той или иной степени будет происходить тер-
мический распад углеводородов, что не позволит правильно изме-
рить величину контракции. При сгорании углеводородов образу-
ющийся углекислый газ будет частично вступать в реакцию с водо-
родом, восстанавливаясь до СО согласно уравнению
СО2 + Н2 = Н2О + СО
А 1 А А *
Так как закись азота в свою очередь вступает в реакцию с окисью
углерода, та и в этом случае точное измерение контракции невоз-
можно. Если продукты упомянутых реакций вместе с избыточным
водородом подвергнуть дальнейшему сожжению с окисью меди,
то водород и СО окисляются и после удаления СО2 щелочью можно
измерить суммарный объем всего азота, как содержавшегося в за-
киси азота, так и имевшегося в свободном виде в газовой смеси. Таким
образом, задача определения закиси азота в этом случае может бьпъ
решена лишь при отсутствии свободного азота в газовой смеси.
Для определения окиси азота некоторые исследователи реко-
мендуют применять раствор сернокислого железа. Этот реагент при-
готовляют, растворяя одну часть сернокислой соли железа в двух
частях воды и слегка подкисляя раствор серной кислотой. Приго-
товленный раствор помещают в обычную газовую пипетку. Этот
метод определения окиси азота не может дать точных результатов,
если в газе присутствует закись азота, главным образом вследствие
большой растворимости закиси азота в воде и разнообразных водных
растворах.
Для определения окиси азота предложено сожжение с добавкой
окиси углерода
2NO + 2СО = 2СО2 4-N2
Для количественного сожжения рекомендуется немедленно уда-
лять образующийся углекислый газ, помещая в пипетку несколько
кубических сантиметров раствора едкого натра.
Титрометрический метод определения окиси азота основан йа
применении подкисленного раствора марганцевокислого калия.
В этом случае имеет место следующая реакция:
1 ONO + 6KMnO4+9H2SO4 = 3K2SO4 + 6MnSO4 + 10HNO3 4- 4 H2O
Реакция происходит медленно, поэтому рекомендуется применять
приспособления, обеспечивающие большую поверхность контакта
газа с реагентом.
В присутствии перекиси водорода окись азота окисляется до
азотной кислоты согласно реакции
2NO 4- ЗН2О2 = 2HNO3 + 2Н2О
В этом случае применяется 3%-ный раствор перекиси водорода.
Образующаяся кислота титруется раствором едкого кали с приме-
нением в качестве индикатора фенолфталеина.
Одним из методов определения окиси азота является поглощение
ее раствором едкого кали или едкого натра при пропускании воз-
духа. В результате реакции образуется азотистокислая соль, коли-
чество которой определяется колориметрически с помощью специаль-
ного реактива путем сравнения со стандартными растворами азотисто-
кислой соли. Этот реактив приготовляют следующим образом. 0,5 г
сульфоанилиновой кислоты растворяют в 150 см3 разбавленной
уксусной кислоты; 0,1 г а-нафтиламина кипятят в 20 см3 воды, бес-
цветный раствор сливают с фиолетового остатка и смешивают со
150 см3 разбавленной уксусной кислоты. Затем оба раствора смеши-
вают и держат в хорошо закупоренной склянке. Анализируемый
раствор едкой щелочи должен нейтрализоваться уксусной кисло-
той и иметь слабо кислую реакцию. В присутствии окиси азота ука-
занный реактив дает оранжевое окрашивание.
Для поглощения окиси азота рекомендовалась также концентри-
рованная серная кислота (95%-ная), содержащая 2% концентриро-
ванной азотной кислоты. Окись азота поглощается с образованием
нитрозилеерной кислоты согласно следующей реакции:
HNO3 + 2NO 4- 3H2SO4 = 3NOHSO4 -f- 2Н2О
Содержание окиси азота вычисляют по азоту нитрозилсерной
кислоты, определяемой титрованием марганцевокислым калием.
Двуокись азота NO2 при невысоких температурах частично поли-
меризуется, переходя в N2O4. При температуре ниже —1Г двуокись
азота состоит только из молекул N2O4; по мере повышения темпе-
ратуры растет и количество молекул NOa.
При контакте с водой двуокись азота образует азотную и азо-
тистую кислоты, а также и окись азота согласно уравнениям
N2O4 -Ь Н2О=Н N Э3 4г н NO2
3N2O4 4- 2 Н2О=4 Н NO3 4- 2NO
При пропускании двуокиси азота через разбавленный раствор
едкого натра в отсутствии воздуха, т. е. в отсутствии кислорода,
образуется смесь нитрата и нитрита натрия. В присутствии кисло-
рода вследствие окисления NO возрастает содержание нитрата.
Двуокись азота быстро поглощается концентрированной серной
кислотой с образованием нитрозилсерной кислоты и азотной кис-
лоты согласно уравнению
N2O44- H2SO4=NOHSO4+ HNO3
799
Двуокись азота может быть обнаружена путем поглощения
в разбавленном растворе едкого натра с последующим подкислением
раствора уксусной кислотой и прибавлением упомянутого выше
реактива, дающего в присутствии двуокиси азота оранжевое окра-
шивание.
Задача анализа сильно осложняется, когда в газе присутствуют
различные окислы азота вместе с другими газами кислотного харак-
тера, как, например, СО2, SO2, НС1 и др. Один из путей анализа в
этом случае заключается в поглощении газов раствором щелочи.
Образующуюся смесь солей исследуют обычными методами хими-
ческого анализа. Закись азота при пропускании газа через щелочь
не задерживается, следует только учесть значительную раствори-
мость закиси азота. При точном анализе необходимо откачать эту
растворенную закись азота.
Сложнее обстоит дело с точным определением в исходном газе
окиси азота, поскольку в присутствии воздуха она переходит в дву-
окись азота.
В газах, образующихся в процессе производства серной кислоты,
присутствует, кроме окислов азота, также и SO2.
В настоящее время известен ряд химических методов, позволя-
ющих делать анализ подобной смеси газов. Для этого обычно SO2
и окислы азота окисляют с помощью перекиси водорода в H2SO4 и
HNO3, после чего титрованием щелочью определяют общую кислот-
ность газа. Для определения H2SO4 в этой смеси кислот существуют
объемные методы анализа: стеаратный, хроматный, бензидиновый.
Определив H2SO4, находят по разности количество HNO3 и пере-
счетом получают содержание окислов азота в газе.
Однако для практических целей требуется раздельное опреде-
ление в газе NO2 и NO. Применяемые для этой цели химические
методы состоят в длительном просасывании газа через ряд реакти-
вов, поглощающих NO2 и NO, с последующим титрованием этих
растворов в лаборатории. Для цеховых условий эти анализы не-
удобны по своей громоздкости, трудоемкости и длительности. Этим
путем невозможно своевременно обнаруживать и предотвращать
нарушения режима промышленных установок, так как пока длится
анализ, процесс может претерпеть новые изменения. Кроме того,
анализы, требующие длительного просасывания газа через погло-
тители, могут дать лишь средние цифры за большой отрезок времени,
а средние цифры способны иногда исказить истинную картину.
Колориметрический контроль нитрозных газов. В 1936 г.
И. Н. Кузьминых и Э. Я. Турхан [21] предложили и разработали
метод контроля нитрозных газов, основанный на колориметриче-
ском принципе.
Как показала практика, этим путем, не прибегая к каким-либо
химике-аналитическим операциям (взвешивание, титрование, изме-
рения объема газа и т. п.), можно легко и быстро определять содер-
жание NO2 и NO в газах.
Колориметрический метод контроля основан на том, что NO2 имеет
200
желто-бурую окраску, в то время как NO бесцветна. Как показали
наблюдения, интенсивность окраски газа при одной и той же толщине
слоя пропорциональна концентрации NO2. Это дает возможность
по окраске газа судить о содержании в нем двуокиси азота. Если
же газу дать еще некоторое время для окисления содержащейся в
нем NO в NO2, то можно определить и сумму NO2 -f- NO.
Применительно к потребностям сернокислотных заводов было
разработано несколько конструкций приборов для колориметри-
ческого газового контроля.
Устройство колориметра и пользование им.
Для количественных отсчетов авторы предложили пользоваться
заранее приготовленными эта-
лонами в виде стеклянных
запаянных трубок с опреде-
ленным содержанием NO2.
Колориметры этого типа (с
набором из 6—7 эталонов)
под названием колориметров
Унихима нашли широкое при-
менение на сернокислотных
заводах.
Практикой установлено,
что для анализа выхлопных
газов, где суммарное содер-
жание окислов азота обычно
не превышает 0,3%, наибо-
лее подходящей является дли-
на трубки в 1 м. Для такого
колориметра обычно берут
эталоны со следующими кон- фиг. 83. Схема колориметра для опреде-
центрациями NO2: 0,00; 0,05; ления NO и no2.
0,10; 0,15; 0,20; 0,25; 0,30%.
Набор эталонов помещают в деревянный футляр в виде ящика
сечением 15 х 15 см и длиной, равной длине трубки. Как показано
на фиг. 83, в центре прибора помещается трубка для анализируемого
газа. Одним своим концом она присоединяется к башне или газо-
ходу, откуда требуется отобрать пробу газа. Другой конец трубки
присоединяется к такой точке системы, где имеется меньшее давле-
ние, чем в точке отбора пробы. Эталоны располагаются вокруг
этой трубки, что позволяет сопоставлять окраску анализируемого
газа с любым из эталонов и таким образом отсчитывать концент-
рации NO2, производя в случае надобности интерполяцию.
Для газов, содержащих 0,3—1,5% NO2 4- NO, длину прибора
уменьшают до 50 см, для газов с 3%NO2 -f- NO —до 20 см. Первый
отсчет, который показывает процент NO2 в газе, производят в мо-
мент просасывания газа. Чем выше содержание NO2, тем энергичнее
идет в газе реакция окисления NO в NO2 и, следовательно, тем бы-
стрее нужно просасывать газ через прибор, чтобы получить правиль-
ные результаты. После первого отсчета движение газа в трубке пре-
201
кращают, закрывая краник на газовой линии, и газ в трубке оста-
вляют на некоторое время для окисления NO в NO2. При анализе
выхлопных газов сернокислотных систем достаточно точные данные
для второго отсчета можно получать, выдерживая газ в трубке 10—
15 мин.
Однако при этом значительная часть NO еще не успевает окис-
литься в NO2 и, если не принять этого во внимание, получатся зани-
женные результаты для NO. Даже если между первым и вторым
отсчетами проходит 1 час, при содержании в газе 0,05 — 0,2% NO
не успевает окислиться 0,01—0,02% NO, Чем ниже содержание NO
в газе, тем больше по своей относительной величине та доля, которая
не успевает за определенное время окислиться в NO2. Наоборот,
при высокой нитрозности газа относительная величина этой поправки
становится меньше, а следовательно, сокращается и потребный про-
межуток времени между первым и вторым отсчетами. Как найти
величину поправки, подробно описано в следующем разделе.
Чтобы колориметры давали правильные показания, нужно при
пользовании ими соблюдать следующие правила.
В колориметры ни в коем случае не следует забирать газ без
фильтрации его от брызг и тумана серной кислоты, иначе мутность
газа будет маскировать его окраску и затруднять отсчеты. Без филь-
трации стенки трубки быстро покрываются налетом кислоты, что
также затрудняет пользование колориметром. Как правило, при
вводе в колориметр анализируемый газ надо просасывать через
брызгоуловитель в виде стеклянной трубочки длиной 50—70 мм,
диаметром 7—10 мм с вложенным в нее тампоном из асбестовой или
бумажной ваты. Чем больше содержится в газе тумана серной кис-
лоты, тем чаще следует менять тампон. В хвосте башенных и камер-
ных систем, где содержание кислотного тумана в газе обычно не-
велико, достаточно менять тампоны один раз в сутки.
Если газы содержат мало паров воды, то сушить их перед вводом
в колориметр нет надобности. При значительной влажности газов
требуется их осушка. Большая влажность газа может привести к
неверным результатам анализа, так как влага, конденсируясь на
стенках трубки, поглощает NO2, превращая окислы азота в азот-
ную кислоту:
3NO2 + Н2О=2HNO3 4- NO
в результате чего отсчеты для окислов азота получаются ниже истин-
ных. Здесь не только следует отфильтровывать газы от капелек кис-
лоты, но и пропускать их перед входом в колориметр через осуша-
ющее вещество, например через Р2О5, СаС12, а лучше всего через
прокаленный сульфат меди.
Среднюю трубку колориметра, куда вводится анализируемый
газ, необходимо периодически промывать и сушить. Во избежание
перебоев в работе колориметра следует иметь в запасе несколько
готовых сухих трубок. Частота смены трубок определяется опытом;
как упомянуто выше, на разных заводах она различна и зависит от
содержания в газе тумана влаги.
Линия, подводящая газ к колориметр}', не должна иметь неплот-
ностей, иначе вследствие разрежения здесь может происходить под-
сос постороннего воздуха и показания прибора получатся ниже
истинных. Колориметры следует располагать возможно ближе к точке
отбора пробы газа; при этом надо свести до минимума применение
резиновых трубок, используя их лишь для соединения встык стек-
лянных трубок, посредством которых и следует подводить газ в коло-
риметр. Резиновые трубки значительно разъедаются окислами
азота, что приводит к подсосам воздуха; кроме того, резина (осо-
бенно свежая) способна абсорбировать окислы азота, а это также
может привести к искажению результатов анализа.
При колориметрическом анализе газа с высоким содержанием
окислов азота следует обращать особое внимание, чтобы засасыва-
ние анализируемого газа в прибор производилось с возможно боль-
шей скоростью, иначе NO может заметно окислиться в NO2 еще на
пути к прибору.
Кроме того, необходимо, чтобы при поступлении в колориметр
газ, двигаясь по подводящей линии, успевал принять температуру,
близкую к температуре эталонов, иначе более высокая температура
газа обусловит в нем уменьшение степени полимеризации NO2 в
N2O4 (по сравнению с этим явлением в эталонах). Поскольку окраску
газу придает только NO2, a N2O4 бесцветна, отсчет по колориметру
даст при этом цифру для NO2 несколько выше истинной. Разница
в температуре на несколько градусов здесь практического значения
не имеет; надо только, чтобы не было слишком большой разницы
температур. Для второго отсчета по колориметру, что делается обыч-
но не менее чем через 10—15 мин. после взятия пробы газа, эти со-
ображения, конечно, не имеют практического значения, поскольку
температура газа за это время становится одинаковой с температурой
эталонов.
Для правильности отсчетов по колориметрам очень важное зна-
чение имеет надлежащее освещение прибора. При слабом накале
лампы и желтизне света трудно различить окраску эталонов. Свет
должен быть сильным, обязательно белым и притом рассеянным, что
достигается установкой между колориметром и источником света
матового стекла или хотя бы листа белой фильтровальной бумаги.
Поправки к показаниям колориметра на
недоокисление окиси азота. Как указывалось выше,
при пользовании колориметром в показания прибора следует всегда
вводить поправку на недоокисление NO. Величины поправок, вы-
численные авторами метода для наиболее распространенных условий
(нормальное давление, температура 15°, содержание кислорода
6—7%), приведены в табл. 27.
Чтобы найти по таблице поправку на недоокисление, берут раз-
ность между вторым и первым отсчетами и точно фиксируют проме-
жуток времени между отсчетами. Для больше^ надежности ре-
зультата эту операцию можно с одной и той же пробой газа про-
делать дважды: сделать отсчет с поправкой на недоокисление сначала
через 10 мин., а затем еще раз через 5 мин. Если условия, при
Поправки на недоокисление NO
Таблица 27
Найденные по колори- метру концентрации NO, О/ /0 Поправки на недоокисление NO в NO2 при 15е (в %) при интервале времени между отсчетами
10 мин. 15 мин. 30 мин. 60 мин.
До 0,05 0,05 0,04 0,02 0,01 '
0,06—0,15 . 0,06 0,04 0,02 0,01
0,16—0,25 0,07 0 05 0,03 0,02
которых идет окисление NO в NO2 в трубке, значительно отличаются от
указанных выше (иное содержание кислорода, иная температура
или давление значительно меньше 760 мм рт. столба), нужно рас-
считать другую таблицу поправок, пользуясь для этого уравнением
скорости реакции окисления NO в NO2. В изданном НИУИФ сбор-
нике «Контроль производства серной кислоты» дана таблица попра-
вок на недоокисление NO для температур 5° и 15° (табл. 28).
Таблица 28
Поправки на недоокисление NO при 5° и 15°
Найденные по колори- Поправки на недоокисление NO в NO2
при 5° при 15°
метру концентрации NO, при интервале времени между отсчетами
о/ /0 10 мин. 15 мин. 10 мин. 15 мин.
0,04- 0,08 0,04 0,03 0,05 0,04
0,08—0,10 0,05 0,03 0,05 0,04
0,10—0,15 0,05 0,04 0,05 0,04
0,15-0,20 0,06 0,04 0,06 0,05
0,20-0,25 0,06 0,04 0,07 0,05
Замена газовых эталонов окрашенными
стеклами. Неоднократно возникал вопрос о замене эталонов
с газообразной NO2 светофильтрами в виде окрашенных стеклянных
пластинок. На этом принципе И. Н. Кузьминых и Э. Я. Турхан
сконструировали лабораторный точный колориметр.
Замена эталонов, заполненных газом, окрашенными стеклами
может упростить устройство колориметра. Однако при этом необхо-
димо учитывать следующее:
1) органические красители с течением времени меняют свои от-
тенки (в частности, от действия света), поэтому нельзя поручиться
за долговечность покрытых ими эталонных пластинок, в то время
как эталоны в виде запаянных трубок с NO2 имеют неограниченный
срок службы;
204
2) в связи с полимеризацией в газе NO2 в N2O4 интенсивность
окраски газа при одной и той же его нитрозности может меняться
в зависимости от температуры.
Если, например, газ содержит 0,3% NO2 (видимая концентрация),
то в нем находятся в форме бесцветного полимера N2O4 следующие
количества NO2:
при 30° 0,01%
» 15°......................0,03%
» 5° 0,07%
» 0° 0,10%
Поскольку колориметр должен показывать величину полной ни-
трозности газа, включая и полимер N2O4, необходимо окрашенную
пластинку отградуировать на определенную температуру. При вся-
ком отклонении от этой температуры отсчет по светофильтру будет
неточен и придется иметь дело с поправками на полимеризацию,
что, конечно, не упростит, а усложнит пользование колориметром.
При более высоких концентрациях NO2 в газе и более низких темпе-
ратурах поправка на полимеризацию будет больше. Между тем обыч-
ные эталоны (запаянные трубки с газом) дают правильные показа-
ния при любых температурах, так как газ в эталоне и анализируе-
мый газ имеют одну и ту же температуру и потому в одинаковой мере
меняют свою окраску вследствие полимеризации NO2 в N2O4. По-
этому для газов с значительным содержанием окислов азота замена
эталонов с газом окрашенными стеклами едва ли оправдает себя.
Для колориметрического контроля выхлопных газов башен-
ных систем окрашенные стекла с успехом могут применяться. Прибор,
сконструированный для этих целей Михальчуком и Муратовым,
состоит из двух трубок длиной 1 м, одна из которых заполнена ана-
лизируемым газом, другая наглухо закрыта и содержит чистый воз-
дух. При анализе газа ко второй трубке путем поворота диска при-
кладывают пластинку с набором отградуированных светофильтров
п подбирают тот светофильтр, окраска которого совпадает с окра-
ской исследуемого газа. В остальном прибором пользуются так же,
как обычным колориметром с газовыми эталонами.
Для изготовления светофильтров берут целлулоид (старая фото-
или кинопленка), который хорошо окрашивается органическими
красками и обладает прозрачностью, а также хорошей механиче-
ской прочностью. Прокрашивание целлулоида производят в спир-
товых растворах красителей — ауранции и нейтральрота. Комби-
нация таких отдельно окрашенных пленок позволяет получить свето-
фильтры, хорошо имитирующие окраску нитрозных газов с малыми
концентрациями NO2.
Лабораторный колориметрический прибор. Как видно из схемы
фиг. 84, колориметр состоит из неподвижно закрепленной трубки 1
(закрытой с торцов плоскими стеклами), в которой находится ис-
следуемый газ, стеклянной кюветы 2, которая передвигается в верти-
кальном направлении, экрана 6 для получения рассеянного света
и окуляра 4, состоящего из двух призм Френеля и наблюдательной
205
трубки. Положение кюветы 2 отмечают на шкале 5, помещенной с
задней стороны колориметра, и определяют толщину d слоя рас-
твора красителя,
Фиг. 84. Схема ла-
бораторного коло-
риметра для опре-
деления NO и NO2.
налитого в кювету, через которую проходит свет.
В кювету погружают стеклянную трубку 3, закры-
тую снизу плоским стеклом, укрепленную непо-
движно. От экрана 6, освещенного ярким белым
светом, отраженный свет проходит через трубку
7 с анализируемым газом, а также через слой
эталонной жидкости, который заключен между
плоским дном кюветы 2 и дном стеклянной труб-
ки 3.
Эталонную жидкость, окраска которой имити-
рует окраску нитрозных газов с небольшой (до
0,5%) концентрацией NO2, подбирают эмпирически.
При высоте трубки 7 в 80 см применима смесь
водных растворов метилоранжа и а-нафтолоранжа
в отношении 85 : 15. Тот и другой растворы берут
концентрации 1,25 мг]л. Эти растворы имеют зеле-
новатый и красноватый оттенок. Изменяя соот-
ношение этих веществ в растворе, можно полно-
стью устранить влияние цветности различных сор-
тов стекла.
Контрольные опыты, проведенные с этим стан-
дартным раствором, показали, что между толщиной
слоя раствора и концентрацией NO2 в газе наблю-
дается удовлетворительное соотношение. Именно, если взять слой
газа толщиной 50 см, то в зависимости от его нитрозности нужна
следующая толщина слоя раствора для получения одинаковой
окраски (табл. 29):
Таблица 29
Зависимость плотности газа от толщины
слоя раствора
Содержание в газе NO2 % Толщина слоя эта- лонной ЖИДКОСТИ d, см Отношение с d
0,52 14,0 0,037
0,36 9,4 0,038
0,18 4,6 0,039
0,09 2,3 0,039
Такая пропорциональность подтверждает для данного случая
возможность колориметрического отсчета в газе концентрации NO2.
Опыты показали также, что для газа с содержанием NO2 менее 0,1%
получается слишком малая толщина слоя стандартного раствора,
что не позволяет достаточно точно отсчитывать его концентрацию.
206
Попытка обойти это затруднение дальнейшим уменьшением концен-
трации красителей в растворе не дала хорошего результата: окраска
получилась слишком бледной и трудно сличаемой с окраской газа.
Поэтому при анализах малонитрозного газа для достижения не-
обходимой точности пришлось увеличить толщину слоя газа (и со-
ответственно высоту прибора) до 80 см.
Оба световых пучка (фиг. 84) проходят через призму Френеля
и затем попадают в окуляр, где глаз видит разделенный пополам
круг. Кювета 2 передвигается вверх и вниз, вследствие чего изме-
няется толщина слоя эталонной жидкости и тем самым изменяется
окраска одного из полей, видимых в окуляр. Двигая кювету, находят
положение, при котором окраска полей в окуляре становится оди-
наковой. Шкалу кюветы 5 заранее градуируют опытным путем в
лаборатории, причем получается график, в котором каждому поло-
жению кюветы отвечает определенная объемная концентрация в
газе NO2.
При появлении в трубке налета кислоты можно разобрать эту
трубку, протереть ее и снова ввести прибор в действие. Чтобы кис-
лые газы, попадая из атмосферы в эталонную жидкость, не могли
изменить ее окраску, эта жидкость должна быть слабощелочной,
для чего в ней растворяют около 10 г едкой щелочи в I л.
Полезно иметь контрольный эталон (в виде запаянной трубки
с 0,2—0,3%N02), поместив который на место трубки с газом,
можно проверить, не изменилась ли со временем окраска эталонной
жидкости.
Как и для любого другого колориметра, газ перед поступлением
в прибор должен просасываться через фильтр в виде стеклянной
трубки (длиной 5—7 см, диаметром 1 см) с ватным тампоном. Если,
как это бывает, например, в азотнокислотном производстве, газ со-
держит много паров воды, необходима еще осушка газа (путел! про-
пускания его через прокаленный сульфат меди). Первый отсчет по
колориметру делают сразу же после заполнения трубки 7 анализи-
руемым газом или во время просасывания газа через эту трубку.
Затем краники, расположенные перед трубкой 7 и после нее, запи-
рают и взятый на анализ газ оставляют в трубке 7 на 10—20 мин.
При этом большая часть NO окисляется в NO2. После этого произ-
водят второй колориметрический отсчет, который дает сумму NO2 +
4- NO. Отрезок времени между первым и вторым отсчетами должен
быть точно известен, чтобы по таблице или графику взять поправку
на недоокисление NO в NO2. Чтобы избежать ошибок вследствие
окисления NO на пути газа в колориметр, следует прибор располагать
возможно ближе к точке отбора пробы газа и, кроме того, пропускать
газ с значительной скоростью — порядка 200 мл}мин.
Бесцветный полимер N2O4 прибором не определяется. Поэтому
к каждому отсчету по колориметру для получения истинного содер-
жания окислов азота в газе необходимо вводить поправку «на поли-
меризацию», которая тем меньше, чем выше температура газа и ниже
концентрация NO2.
207
Величина поправок вследствие реакции
2NO2 = NaO,+13,6 ккал
может быть вычислена по уравнению
rz _ Йо2 _ аг
Р“ Pn804 “ >
где pNo2 и Pn2o4 (или а и ft) — равновесные парциальные давления
NO2 и N2O4, ат',
КР — константа равновесия, которая зависит от температуры
следующим образом:
t°, °C........... О 9,5 20 29,5 40,4 50,9
Кр, ат........... 0,0179 0,0404 0,0951 0,178 0,451 0,906
Скорость реакции полимеризации весьма велика; равновесие
наступает почти мгновенно.
Если, например, содержание NO2 в газе (без учета полимеризации)
равно 1% и температура 40°, а общее давление равно 1 ат, то можно
написать систему из следующих двух уравнений:
а-Ь 2ft = 0,01,
g2: ft = O,451.
Отсюда
а* 4- 0,225 а -0,00225 = О,
а = 0,0095 ат,
т. е. почти вся имеющаяся в газе двуокись азота находится при 40°
в форме NO2 и лишь 5% ее полимеризовано в N2O4. С понижением
температуры и повышением нитрозности газа степень полимери-
зации увеличивается. При расчетах употребляются обычно графики
поправок на полимеризацию NO2.
Применяя колориметрический метод для анализа газов, сле-
дует иметь в виду, что объем газов зависит от температуры и давле-
ния гораздо больше, чем объем жидкостей. Поэтому при градуи-
ровке прибора, как и при изготовлении эталонов для обычных коло-
риметров, следует оговаривать те условия (температуру и давле-
ние), для которых дана шкала.
Определение аммиака
В некоторых промышленных, а также и природных газах может
встречаться аммиак.
Удельный вес аммиака по отношению к воздуху равен 0,5971,
что соответствует плотности аммиака при 0° и 760 мм рт. столба
0,7708 г/л.
При определении аммиака следует учитывать, что он очень хорошо
растворяется в воде. Один объем воды растворяет следующие коли-
чества аммиака при его давлении в 760 мм рт. столба:
208
при 0° ... 1176 объемов аммиака
8° ... 947 » »
» 13° ... 837 » »
» 16° ... 775 » »
при 20°
» 24°
» 28°
702 объема аммиака
639 объемов »
586 » »
Аммиак хорошо растворяется также в спирте и эфире. Наиболее
чувствительным реагентом для обнаружения малейших следов ам-
миака является K2HgJ4 в растворе едкого кали (реактив Несслера).
Когда на этот раствор действует аммиак, получается желтое окра-
шивание.
Для количественного определения аммиака пропускают анализи-
руемый газ через слабый (0,02 N) раствор серной кислоты. После
окончания пропускания газа раствор титруют для определения коли-
чества оставшейся кислоты, откуда и вычисляют концентрацию ам-
миака в анализируемом газе.
§ 2. Определение сернистых газообразных соединений
(H2S, SO2, COS)
Определение сероводорода (H2S)
Удельный вес сероводорода по отношению к воздуху равен 1,190.
При 0° и 760 мм рт. столба вес 1 л равен 1,539. Температура кипения
сероводорода —60°, температура плавления —83°.
Сероводород в значительной степени растворяется в воде. 1 л
воды при 760 мм рт. столба растворяет в себе следующее количество
сероводорода:
при 0°........... 4,670 л при 20°........... 2,582 л
» 5°. 3,977 » » 25°........... 2,282 »
» 10° ....... 3,399 » » 30°........... 2,037 »
» 15°. 2,945 »
Присутствие сероводорода в газе легче всего обнаруживается
по запаху или при помощи бумажки, смоченной раствором уксусно-
кислого свинца; в присутствии H2S бумажка чернеет.
Сероводород можно определять путем поглощения растворами
едких щелочей, нитратом серебра и сульфатом меди.
Весовым путем сероводород можно определять при помощи фос-
форномедной соли или пемзы, пропитанной крепким раствором
серномедной соли.
Мелкие кусочки пемзы обливают горячим концентрированным
раствором серномедной соли, а затем хорошо высушивают. На 100 г
пемзы берут 40—60 г серномедной соли. Полученный препарат поме-
щают в V-образную трубку, через которую пропускают анализируе-
мый газ. Перед трубкой с серномедной солью рекомендуется ставить
трубку с хлористым кальциел! для осушки газа.
При пропускании через раствор иода в иодистом калии серо-
водород количественно поглощается по уравнению
H2S + 2J = 2HJ+S
Реакция идет точно по приведенному уравнению, если раствор
достаточно разведен и защищен от прямого действия солнечного
14 В. А. Соколов. 209
света. Ненасыщенные углеводороды и цианистый водород также
вступают в реакцию с иодом, поэтому их присутствие в газе мешает
определению H2S.
Сероводород определяется пропусканием известного количества
газа через 0,1 N или 0,01 N раствор иода и титрованием не вошед-
шего в реакцию иода.
1 см3 0,1 N раствора иода соответствует 1,104 см3 H2S при 0° и
760 мм рт. столба.
Точное количественное определение сероводорода не может про-
изводиться обычным путем с помощью поглощения в газоаналити-
ческой аппаратуре. Следующий метод дает точные результаты, если
он проведен достаточно тщательно. Он заключается в пропускании
измеренных порций анализируемого газа через раствор едкого натра
или едкого кали или через аммиачный раствор хлористого кадмия
или сульфата цинка, подкислении раствора соляной кислотой и
титровании иодом освобожденного сероводорода.
Аппаратура. 20 см3 поглощающего раствора помещают в колбу
емкостью 500 см3 и разбавляют водой до 200 см3. Колбу закрывают
резиновой пробкой с двумя отверстиями, куда вставлены две стек-
лянные трубки для впуска и выпуска газа. Впускная трубка должна
быть достаточно длинной и с одной стороны доходить до дна колбы,
а с другой — на 12—15 см выступать над пробкой, закрывающей
колбу. Выпускная трубка должна иметь внутри колбы короткий
конец. Для удобства соединений обе трубки отгибаются под пря-
мым углом сейчас же по выходе их из пробки. Впускную трубку со-
единяют небольшой резиновой трубкой с краном магистрали для
отбора проб, по которой газ протекает через нее более или менее
равномерно. Если резиновой трубки нет, то трубка, из которой от-
бирается проба газа, должна хорошо промываться сильной струей
газа перед присоединением колбы и газ выпускается через одну или
две горелки в течение всего времени производства анализа. Если
для анализа применяется раствор едкого натра или едкого кали, то
выпускная трубка колбы может присоединяться непосредственно
к измерительной аппаратуре; если же в качестве реагента применяется
аммиачный раствор, то газ пропускают через небольшую промыв-
ную склянку с 5%-ным раствором серной кислоты для удаления
паров аммиака, включаемую между колбой с реагентом и измери-
тельной аппаратурой.
Установка для измерения газа. Наиболее удоб-
ным для измерения является тщательно проградуированный мок-
рый счетчик. Вместо счетчика могут применяться 4-л аспиратор и
1-л градуированный сосуд. Аспиратор наполняют водой, отверстие
закрывают резиновой пробкой с одним отверстием, через которое
аспиратор соединяется с поглотительным сосудом. Поступающий
в аспиратор газ вытесняет из него воду через нижнее отверстие. Об
объеме поступившего газа судят по объему вытесненной воды. Для
производства анализа достаточно 3 л газа. Измеренный объем газа
приводят к нормальным условиям путем пересчета температуры и
210
давления (см. таблицы приведения к нормальному объему). Темпе-
ратуру измеряют термометром, вставленным в трубку, соединяющую
аспиратор и колбу с раствором. Давление измеряют манометром,
присоединенным в любом месте трубки, соединяющей измеритель-
ную установку и колбу с раствором.
Показания манометра складывают с показанием барометра; в
полученное таким образом общее давление газа вводят поправку на
давление паров воды, которые в данном случае могут считаться на-
сыщающими газ.
Титрование сероводорода. После того как описанный выше объем
газа пропущен через поглощающий раствор, колбу отключают от
газовой линии, подающую трубку промывают приблизительно 100 ем3
воды и разбавленной соляной кислотой (1 : 1); затем в колбу при-
ливают около 5 см3 раствора крахмала, раствор подкисляют той же
соляной кислотой и немедленно оттитровывают освободившийся серо-
водород до появления устойчивой синей окраски раствора. Титро-
вание производят децинормальным (0,1 N) раствором иода или иодно-
ватокислой соли, который быстро приливают с легким взбалтыва-
нием. Количество сероводорода рассчитывают, исходя из количества
кубических сантиметров раствора, употребленного для титрова-
ния.
Приводим расчет. 1 см3 0,1 N титрованного раствора иода или
иодноватокислой соли соответствует 1,17 см3 объема сероводорода.
Умножая количество кубических сантиметров титрованного рас-
твора на 1,17, получаем объем сероводорода, содержащегося во взя-
той пробе газа. Затем определяем полный объем газа. Так как угле-
кислота была поглощена вместе с сероводородом, то полный объем
газа определяется приведением измеренного объема к нормальным
условиям, как это указывалось выше, путем прибавления объема
сероводорода, найденного титрованием, и деления этой суммы на
100% минус процент углекислоты в газе:
измеренный объем газа х фак-
тор приведения к нормальным
условиям + 1,17 х число см3
Общий объем первоначального газа =-----гл7Го^аСТ~В°Ра И°Дгл-*
г 100%—процент СО2
Процент углекислоты получаем из анализа. Содержание серо-
водорода может быть подсчитано по следующим формулам:
т, тт п , о см3 раствора иода х 0,0017
Количество граммов H2S на 1 л? = --------1---- •
г л газа взятого для анализа ’
п ? г г- г H2S на 100 см3
Процент n2S по объему =-^“ибоТг- '
Необходимые реактивы. Крахмальный индикатор.
1. Эмульсию из 6 г крахмала в 100 см3 холодной воды осторожно
приливают к 1 л кипящей воды. После кипячения в течение 5 мин.
14* 211
сосуд и его содержимое охлаждают и для сохранения раствора к нему
приливают раствор из б г хлористого цинка в 50 см3 холодной воды.
После тщательного смешения раствор оставляют на 24 часа, взбал-
тывая его время от времени. После осаждения более тяжелых частиц
оставшуюся жидкость декантируют и к ней прибавляют 3 г йоди-
стого калия.
2. К эмульсии, состоящей из б г растворенного крахмала в 25 см3
холодной воды, осторожно добавляют раствор 1 г едкого натра при-
близительно в 10 см3 воды, все это размешивают до желатинизации.
Образовавшуюся массу переносят в больший сосуд и разбавляют
до 1000 см3. После добавления и растворения 3 г йодистого калия
раствор готов к употреблению.
Аммиачный раствор хлористого кадмия
Хлористый кадмий........................... 5 г
Вода .................................. 375 см3
Аммиак................................... 625 »
Аммиачный раствор сульфата цинка
Сульфат цинка........................... 10 г
Аммиак ................................... 50 см3
Вода в количестве, необходимом для доведе-
ния раствора до....................... 1000 »
Раствор едкого кали или едкого натра
Едкий кали или едкий натр ............... 20 г
Вода . . .............................. 1000 см3
0,1 N раствор иодноватокислого калия
Йодноватокислый калий .....................3,7 г
Йодистый калий...........................12 »
Едкий кали................................1 ь
Вода в количестве, необходимом для доведе-
ния раствора до ................... ..... 1000 см3
Иод и иодистый калий помещают в градуированную однолитро-
вую склянку, к ним приливают 50 см3 холодной воды и встряхивают
склянку до растворения иода. Затем раствор разбавляют водой до
Гл и оставляют стоя’ть не менее 24 час., после чего устанавливают
титр раствора.
Титрование растворов. Растворы иода или иодно-
ватокислого -калия титруют 0,1 N раствором тиосульфата натрия,
который в свою очередь титруют 0,17V раствором марганцевокислого
калия. Марганцевокислый калий титруют 0,1 N раствором щавелево-
кислого натрия. Раствор приготовляют и титруют следующим обра-
зом. 3,17 г марганцевокислого калия растворяют приблизительно
в 950 см3 свежей кипящей воды, оставляют до охлаждения, а затем
разбавляют до требуемой концентрации свежей дестиллироваиной
водой. Точно так же растворяют 6,7 г щавелевокислого натрия в
300 см3 воды и разбавляют затем до 1000 см3. Для титрования пер-
манганата 50 см3 раствора щавелевокислого натрия помещают в
212
стакан емкостью 400 см3, содержащий 200 ои3 воды и 20 см3 раз-
бавленной серной кислоты (1 : 1). Содержимое стакана нагревают
до 80° и раствор перманганата вливают в него из бюретки сначала
медленно, а затем быстрее до тех пор, пока одна капля не сообщит
раствору устойчивую розовую окраску. Перманганат может быть
и большей концентрации. В этом случае количество приливаемой
воды находят умножением на 19 разницы между 50 и фактическим
количеством кубических сантиметров использованного перманга-
ната. Раствор тиосульфата натрия приготовляют растворением
24,82 г этой соли в 400—500 см3 воды и разбавлением до 1000 см3.
Раствор должен быть децинормальным, однако он нуждается в про-
верке перманганатом. Точно так же должны быть проверены рас-
творы иода и йодата.
Проверка производится следующим образом. К 300 см3 воды, на-
ходящейся в стакане емкостью 600 см3, приливают 10 см3 соляной
кислоты и 10 см3 раствора йодистого калия (10%-ного). К ним при-
ливают 25 см3 титрованного раствора перманганата и оттитровы-
вают освободившийся иод раствором тиосульфата. Раствор взбалты-
вают. Когда окраска раствора станет бледнеть, приливают крах-
мал, а затем медленно приливают тиосульфат до окончательного
обесцвечивания. Таким же образом титруют 25 см3 1 7V раствора
иода и йодистого калия. Полученные результаты дают возможность
подсчитать нормальность раствора. В случае необходимости уста-
навливают растворы иода и йодистого калия точно децинормальные.
Один из способов определения сероводорода в газе заключается
в том, что в бюретку с трехходовым краном набирают 100 см3 газа.
В бюретке и в уравнительном сосуде применяют в этом случае
воду, содержащую немного крахмального раствора. После запол-
нения бюретки газом в нее впускают через один из ходов крана рас-
твор иода известной концентрации. Впуская по каплям раствор
иода в бюретку и встряхивая ее, наблюдают за оставшимся в ниж-
ней части бюретки раствором крахмала. Посинение раствора, не
пропадающее при встряхивании бюретки, означает, что окисление
сероводорода окончилось. По количеству израсходованного на реак-
цию иода вычисляют содержание H2S в газе.
Колориметрическим путем сероводород можно определять с по-
мощью бумажек, пропитанных азотнокислым серебром или уксусно-
кислым свинцом. В обоих случаях бумажку помещают в соответ-
ствующий шприц или микроаспиратор, в который забирают опре-
деленную порцию анализируемого газа. Окраску бумажки сравни-
вают со стандартной шкалой.
Указанные колориметрические методы применяются главным обра-
зом для определения малых концентраций сероводорода в воздухе.
Определение двуокиси серы (SO2)
Двуокись серы или сернистый ангидрид имеет удельный вес по
отношению к воздуху 2,2639. При 0° и 760 мм рт. столба вес 1 л ра-
вен 2,9267 г. Температура кипения SO9 равна —10°, температура
плавления —72,7°.
213 '
Сернистый ангидрид легко растворим в воде. 1 л воды при 760 мм
рт. столба растворяет двуокиси серы:
при 8°.................58,7 л при 20°....................36,4 л
•> 10°................53,9 » » 24° . . . ... 32,3 »
> 14°................45,6 » » 30°............ . 27,3 »
Двуокись серы количественно поглощается растворами едких
щелочей. В отсутствии углекислоты SO2 можно определять погло-
щением едким кали. Если есть оба газа., то количество SO2 опре-
деляют титрованием, путем пропускания определенного объема газа
через раствор брома в воде и осаждением образующейся серной кис-
лоты хлористым барием или же измерением количества раствора
иода известной концентрации, который обесцвечивается определен-
ным количеством газа.
В последнем случае газ просасывают через 0,1 N раствор иода,
окрашенный крахмалом, пока раствор не обесцветится. Зная коли-
чество газа и количество раствора, можно вычислить и содержание
SO2. Поглощение SO2 раствором иода происходит по уравнению:
SO2 4- 2Н2О + J2 = 2Н J 4- H2SO4
На каждый кубический сантиметр 0,1 N раствора иода
идет 1,094 см3 SO2 в сухом виде при 0° и 760 мм рт. столба.
Можно также, пропустив известное количество газа через рас-
твор иода известной концентрации, оттитровать серноватистонатрие-
вой солью избыток иода.
При одновременном присутствии в газе сернистого ангидрида и
сероводорода анализ рекомендуется проводить путем отбора двух
параллельных проб. В одной из них определяют сероводород путем
пропускания газа через раствор азотнокислого серебра с упомяну-
тым выше колориметрическим определением, а в другом определяют
суммарно сероводород и сернистый газ путем пропускания через
раствор иода.
Рекомендуется также следующий метод. Сернистый газ окисляют
в 5%-ном растворе КС1О3. Сероводород при этом окисляется незначи-
тельно •— на 1—2%. Газ, пропускаемый через раствор КС1О3, посту-
пает затем в раствор иода, где поглощается сероводород.
Для определения SO2 в присутствии окислов азота в качестве
экспресс-метода рекомендуется пропускание анализируемого газа
через титрованный раствор хлористого бария, к которому доба-
влена перекись водорода. При этом образуется сернокислый барий
и раствор мутнеет. Фиксируя с помощью фотонефелометра момент
прекращения нарастания мути и объем пропущенного газа, соот-
ветствующий точке эквивалентности, можно вычислить процентное
содержание SO2 в газе.
Сероводород реагирует с азотнокислым серебром согласно реак-
ции
H2S + 2AgNO3 = Ag2S 4- 2HNO3
При незначительных концентрациях сероводорода образуется
темнокоричневый коллоидальный раствор сульфида серебра. Содер-
214
жание сероводорода можно определить с помощью этой реакции
колориметрически, сравнивая окраску раствора со стандартной
шкалой.
Определение сероокиси углерода (COS)
Удельный вес по отношению к воздуху равен 2,105; 1 л при 0°
и 760 мм рт. столба весит 2,721 г. При 760 мм рт. столба 1 л воды
растворяет сероокиси углерода:
при 0°..........; 1,348 л при 20° 0,568 л
» 10°........... 0,844 » » 30° . 0,407 »
Температура плавления ее —138°, температура кипения —48°.
Раствор едкого кали поглощает COS настолько медленно, что
можно освободить COS от примесей СО2 и H2S, пропуская газ через
разбавленный раствор КОН.
Спиртовый раствор КОН быстро поглощает COS. Раствор гото-
вят растворением 1 части КОН в 2 частях воды, куда добавляют
равный объем спирта.
В отличие от сероводорода COS почти не поглощается кислым
растворол! сернокислой меди.
Сероводород в присутствии COS титруется 0,1 N раствором иода.
Один объем COS, смешанный с 1V2 объемами кислорода, сгорает
с сильным взрывом, образуя СО2 и SO2. При соприкосновении с на-
каленной платиной COS распадается на СО и серу.
§ 3. Определение цианистоводородной кислоты и циана в газах
Цианистоводородная кислота и циан содержатся во многих газах,
получающихся в промышленности, и их определение приобретает
все большее и большее значение. В газах коксовых печей также со-
держится цианистая кислота. Приводим здесь два метода определе-
ния цианистой кислоты в газе коксовых печей [22].
МЕТОД I
Цианистая кислота в коксовом газе
Ход анализа. В каждую из двух промывных склянок
емкостью 250 см3 помещают 100 см3 поглощающего раствора (см.
ниже) и присоединяют их к газовой линии или к линии для взятия
проб газа. Склянки включают в линию последовательно с мокрым
счетчиком. Через промывные склянки пропускают 140 л газа со ско-
ростью около 5 л[час. После этого содержимое промывных склянок
сливают в мерную колбу емкостью 500 см3 и хорошо промывают
склянки водой. Мерную колбу доливают водой до метки и ее содер-
жимое тщательно смешивают. Осажденный сернистый свинец отфиль-
тровывают на сухом фильтре и с помощью пипетки переводят 200 см3
очищенного фильтрата в стакан для титрования емкостью 400 см3.
Стакан ставят на лист черной бумаги и вливают в него 5 см3 10%-ного
раствора йодистого калия; туда же приливают из бюретки, постоянно
215
помешивая, 0,1 N раствор азотнокислого серебра до появления
устойчивой желтой мути. Появление мути считается концом реакции.
Так как раствор поглощает сероводород и углекислоту, заме-
ренное количество газа делят на 100% минус процент поглощенных
сероводорода и углекислоты; это делают для того, чтобы найти дей-
ствительный объем газа, взятого для анализа. Этот объем затем при-
водят к нормальным условиям. Результат анализа подсчитывают
затем по следующей формуле:
иг-хт 1 э см 0,1 N AgNO3 • 0,0054
количество граммов HCN на 1 л/3 = —--’-5----------.
м3 газа, взятого для анализа
Необходимые растворы
Децинормальный раствор азотнокислого серебра
Азотнокислое серебро................................. 16,989 г
Вода в количестве, необходимом для доведения раствора
ДО ; .... ;........................................ 1000 СМ3
Для проверки этого раствора разбавляют 25 см3 в стакане ем-
костью 400 см3. Разбавленную соляную кислоту (1 : 5) по каплям
приливают в стакан до тех пор, пока не окончится выпадение осадка.
После этого стакан ставят в место, защищенное от света, до осажде-
ния осадка на дно стакана. Затем отфильтровывают хлористое серебро
на взвешенное стекло или в алундовый тигель и промывают его
1%-ной соляной кислотой и дважды холодной водой. Осадок вы-
сушивают при 110°, очень осторожно прокаливают, охлаждают и
взвешивают.
Вес хлористого серебра, разделенный на 0,3584, дает количество
кубических сантиметров 0,1 N раствора, эквивалентного 25 см3 упо-
требленного раствора.
Поглощающий раствор
Едкий натр . ........... . . ; 150 г
Азотнокислый свинец.......... 50 »
Вода . . , . : ............ 1000 см3
Едкий натр растворяют приблизительно в половине воды, пред-
назначенной для приготовления раствора, а в остающейся части
воды растворяют азотнокислый свинец. Затем два раствора сливают
и перемешивают до тех пор, пока не растворится выпавший вначале
осадок.
МЕТОД II
Весовой метод. В этом анализе применяется поглоти-
тельная система, состоящая из шести промывных склянок и счет-
чика. Первые три склянки содержат по 100 см3 раствора уксусно-
кислого свинца, полученного путем растворения 100 г уксуснокис-
лого свинца в 1000 см3 воды, содержащих 30 см3 ледяной уксусной
кислоты. Раствор поглощает сероводород. Последние три склянки
содержат по 60 см3 0,1 N раствора едкого кали. Газ пропускают
216
через склянку в течение 4 час. со скоростью 50—80 л!час. Растворы
трех последних склянок переводят в мерную колбу емкостью 500 см3,
куда добавляют воду до метки, и полученный раствор хорошо пере-
мешивают. Порции по 50 см3 обрабатывают нагретым 10%-ным рас-
твором сернокислой окиси железа. Добавляют избыток хлорного
железа; при подкислении разбавленным раствором серной кислоты
выпадает берлинская лазурь. Осадок фильтруют и промывают горя-
чей водой до удаления серной кислоты, после чего осадок (окись
железа) прокаливают и взвешивают. Вес окиси железа умножают
на 0,8376 и получают эквивалентный вес циана (CN), который затем
переводят в граммы на кубический метр газа.
Объемный метод. Берлинскую лазурь можно оттитро-
вать сейчас же 0,02 N раствором едкого натра. Раствор едкого натра
приливают понемногу до тех пор, пока не разложится берлинская
лазурь. Во время приливания раствора едкого натра содержимое
сосуда держат горячим для ускорения реакции. Избыток едкого
натра титруют 0,02 N раствором кислоты. Жидкость необходимо
нагревать и быстро мешать, так как в присутствии избытка едкого
натра берлинская лазурь сообщает жидкости зеленую окраску. Когда
окраска раствора станет светложелто-зеленой, реакция окончена.
1 сл/3 0,02N NaOH = 0,00078024 г C2N2 или 0,0008105 г HCN
§ 4. Определение галоидов и галоидоводородных кислот
Определение хлора и хлористого водорода
Одним из методов определения хлора является восстановление-
его до соляной кислоты с помощью мышьяковистой кислоты.
Реакция в этом случае идет таким путем:
As2O3 + 2 С12 + 2Н,0 = As2O5 + 4НС1
Ион хлора определяют с помощью нитрата серебра по помутне-
нию раствора азотнокислого серебра. Следует указать, что анало-
гично действуют и другие галоиды, а также синильная кислота,
которая с азотнокислым серебром тоже дает нерастворимое соеди-
нение. Для проведения указанных определений приготовляют рас-
твор мышьяковистой кислоты и 0,002 N концентрации.
Для проведения измерений отбирают пробу воздуха и пропускают
через два последовательно соединенных поглотителя, содержащих
по 5 см3 раствора мышьяковистой кислоты. Воздух пропускают со
скоростью 20—25 л]час. Необходимо пропустить не менее 20 л воз-
духа. '
После окончания пропускания воздуха жидкость из каждого
поглотителя сливают в мерный цилиндр, причем каждый поглоти-
тель споласкивают небольшим количеством воды. При анализе объем
жидкости в мерном цилиндре доводят до одной и той же величины,
например 15 см3', определенное количество этого раствора, напри-
мер 5 см3, наливают в колориметрическую пробирку и помещают
ее в колориметр. Одновременно готовят стандартную шкалу в десяти
217
колориметрических пробирках с разным количеством раствора хло-
ристого калия. В пробирки стандартной шкалы и в пробу прибавляют
по 1 с.и3 1%-ного раствора азотной кислоты и 1 см310%-ного раствора
азотнокислого серебра.
Раствор тщательно перемешивают и через несколько минут сра-
внивают муть пробирок со стандартной шкалой на чернол! фоне.
Указанный метод применяется главным образом для определе-
ния малых количеств хлора, а именно, меньших 0,005 мг'[Л.
При более высокой концентрации хлора применяют титрометри-
ческий метод. Воздух пропускают через титрованный раствор
мышьяковистой кислоты, окисляя последнюю до мышьяковой кис-
лоты. Избыток мышьяковистой кислоты определяют иодометриче-
ски :
As2O3 + 2 J2 + 4NaHCO3 = As2O5 4- 4NaJ + 4CO2 + 2H2O
По разности вычисляют искомую концентрацию хлора.
Бром, фтор и цианистый водород мешают этому определению.
Хлористый водород с нитратом серебра дает муть; по интенсив-
ности этой мути можно определить концентрацию хлористого водо-
рода в газе. Для этой цели можно пользоваться вышеупомянутым
колориметрическим методом, применяя стандартную шкалу, но в
этом случае метод не специфичен, так как другие галоиды и синиль-
ная кислота также дают помутнение раствора азотнокислого серебра.
При одновременном присутствии хлора и хлористого водорода
пользуются реакцией окисления мышьяковистой кислоты, которая
при действии хлора переходит в мышьяковую. Хлористый водо-
род в этих условиях растворяется в поглотительной жидкости, не
изменяя ее титра. В одной пробе определяют общее количество ионов
хлора при помощи нитрата серебра, а в другой титрометрически
определяют количество хлора. По разности определяется концентра-
ция хлористого водорода.
Определение брома, бромистого водорода и фтористого водорода
Бром и бромистый водород можно определять, как хлор и хло-
ристый водород, колориметрическим путем с применением нитрата
серебра и мышьяковистой кислоты. Подобные определения не ха-
рактерны в присутствии других галоидов или цианистого водорода.
Определение фтористого водорода основано на том, что фтори-
стый водород и соли фтористоводородной кислоты дают бесцветные
соединения с солями окисного железа
FeC 13 + 6NaF = Na3FeF6 + 3NaCl
Избыток хлорного железа определяют колориметрически с рода-
нидом калия. При этом употребляют обратную стандартную шкалу,
так как при больших концентрациях фтористого водорода полу-
чается менее интенсивная окраска.
218
Определение галоидов и галоидоводородных кислот при совместном их
присутствии
Описанные выше методы применяются главным образом как экс-
пресс-методы при анализе воздуха. Для точного количественного
определения галоидов и галоидоводородных кислот при совместном
их присутствии эти газы следует поглотить, пользуясь слабой ще-
лочью и мышьяковистой кислотой. Получившиеся растворы в даль-
нейшем исследуют обычными методами аналитической химии.
ГЛАВА ШЕСТАЯ
МИКРОАНАЛИЗ ГАЗОВ
§ 1. Общий газовый микроанализ
Под термином «микроанализ газа» подразумевают весьма разно-
образные методы газового анализа, позволяющие определять
небольшие количества или небольшие концентрации тех или иных
компонентов.
Многочисленные методы лшкроанализа имеют весьма различную
чувствительность и точность определения.
При общем анализе, описанном в главе III. обычно берут 100 см3
газа и приводят определения с точностью до 0,1—0,2 см3.
На том же принципе, что и в обычном общем анализе, можно по-
строить приборы, позволяющие, например, анализировать 5—10 см3
газа. По сравнению с обычным общим анализом такой метод по суще-
ству тоже можно назвать микроанализом. Известны специальные
приборы и методы, позволяющие анализировать газ в количестве до
1 см3 и даже до нескольких кубических миллиметров.
Во всех этих случаях речь идет о микроанализе газа, хотя эти
методы совершенно различны по своим принципам и чувствительности
определений.
Указанные методы позволяют проводить тот или иной вид ана-
лиза с различным и в то же время с очень небольшим количеством
газа.
С другой стороны, существуют методы, которые позволяют опре-
делять в анализируемом газе малые концентрации тех или иных
компонентов—порядка десятых и сотых долей процента. Известны
также методы, позволяющие определять содержание отдельных ком-
понентов в различных газовых смесях при концентрациях порядка
тысячных и десятитысячных долей процента. В этих случаях коли-
чество газа, которое берется для анализа, может быть очень велико.
Здесь не имеют существенного значения количество газа и объем
отбираемой пробы, а представляет интерес только концентрация
тех или иных интересующих нас компонентов.
Все упомянутые методы, обладающие разной чувствительностью
и разной точностью и основанные на самых разнообразных прин-
ципах, объединяют общим понятием «микроанализ». Те методы,
которые позволяют проводить анализ сравнительно небольших коли-
220
честв газа, но не столь малых, как, например, 1 см3 или еще меньше,
называют методами полумикроанализа.
Под термином «полумикроанализ» подразумевают также методы,
позволяющие определять такие концентрации интересующих нас
компонентов, как десятые и сотые доли процента.
В настоящей главе дается описание методов микроанализа и ме-
тодов полумикроанализа, позволяющих анализировать малые коли-
чества газа и определять малые концентрации различных компонен-
тов. Определение малых количеств или малых концентраций угле-
кислоты, водорода, кислорода, окиси углерода, азота можно назвать
общим микроанализом. В некоторых случаях при подобном микро-
анализе определяют и отдельные непредельные углеводороды, кото-
рые могут поглощаться соответствующими реагентами.
Описываемые в данном параграфе методы основаны на тех же
принципах, что и общий анализ. Для этой же цели применяют в
некоторых случаях вакуумные установки с использованием вымора-
живания отдельных компонентов газовой смеси (§ 2).
Микроанализ на углеводородные газы основан на применении
специальной аппаратуры, в связи с чем его описание дается в от-
дельном параграфе. Описание методов микроанализа редких газов
дано в главе VII.
Помимо методов, в которых используются поглощение, сожже-
ние и вымораживание отдельных компонентов газовой смеси, для
микроаналитических определений используются и различные физи-
ческие и, в частности, оптические методы, которые описаны
в главе VIII.
Прибор Института химической физики для общего газового
микроанализа позволяет проводить этот анализ с количеством газа
порядка 1 см3 и даже менее [23].
Общий вид прибора представлен на фиг. 85.
На станине 6, покоящейся на лапках 7 с установочными винтами,
укреплена главная часть прибора — измерительный капилляр 7,
внутренний диаметр которого равен приблизительно I мм. Деления,
нанесенные непосредственно на капилляре, позволяют отсчитывать
1 мм3 газа. Капилляр окружен стеклянной муфтой 4, заполненной
водой, температура которой измеряется термометром 5.
Конец капилляра входит в камеру 2, наполненную ртутью. Объем
этой камеры можно плавно изменять при помощи микрометриче-
ского винта 3. Другой конец капилляра изогнут, как показано на
чертеже, и погружен в чашку 0, наполненную ртутью до краев. Эта
чашка при помощи ряда деталей закреплена на станине 6. Микро-
метрический винт, входящий в тройник 5, позволяет плавно изменять
высоту чашки.
В центре чашки находится держатель 10 баллончиков 11. Этот
держатель при помощи барашка можно поднимать и поворачивать.
Поворот возможен только на такой высоте, когда баллончики не
могут задеть кончика капилляра. Посадка баллончиков на место
устроена таким образом, что исключается всякая возможность по-
227
ломки. Держатель несет на себе четыре баллончика, так что возможно
производить четыре параллельных газовых анализа.
На чертеже видно, как кончик капилляра входит в баллончик,
касаясь его дна. Для введения в баллончик пробы газа, анализ кото-
рого желательно произвести, служит капилляр 12. Этот капилляр
соединяется короткой каучуковой трубкой или, лучше, припаивается
к капилляру, ведущему к сосуду с исследуемым газом. Капилляр 12
предварительно заполняется ртутью. Сосуд, содержащий газ, кау-
чуковой трубкой соединяется с воронкой с ртутью. Газ в сосуде дол-
жен храниться также над ртутью. Подняв воронку, можно вытолк-
нуть необходимый для анализа объем газа в баллончик. Введя в
баллончик небольшой пузырек газа, например 0,2—0,7 ел/3, и дей-
Фиг. 85. Микроанализатор Института химической физики.
ствуя держателем 10, приподнимают баллончик, поворачивают его
на 180° и опускают так, чтобы он сел на кончик капилляра 7.
Вращая винт 3 и всасывая ртуть в сосуд 2, легко ввести в капил-
ляр весь находящийся в баллончике объем газа. Этот газ заключен
между двумя столбикалш ртути. Доведя мениск левого столбика
ртути до нуля шкалы, легко отсчитать объем газа при атмосферном
давлении. Капилляр расположен горизонтально и находится на
одном уровне с поверхностью ртути в чашке 9. Температуру газа
отсчитывают по термометру. При изменении температуры или давле-
ния во время анализа легко ввести соответствующие поправки.
После определения объема газа его снова переводят в баллончик,
вращая винт 3 в противоположную сторону. Действуя барашком,
держатель 10 и баллончик 77 с газом приподнимают и поворачивают
на 60°. Далее его опускают на поглотитель одного из газов, име-
ющихся в смеси.
222
Вследствие небольших количеств газа в качестве поглотителей
употребляют почти исключительно твердые адсорбенты, крупинки
которых помещают в небольшой платиновой петле, впаянной в стек-
лянную палочку. После того как газ поглотится (на это требуется,
как показали опыты, от 1 до 5 мин.), баллончик снова передвигают
на кончик капилляра и оставшийся газ всасывают.
Новое измерение объема газа позволяет вычислить процент по-
глощенного газа в смеси. Пуская в ход различные адсорбенты, можно
произвести полный газовый анализ.
Примерный ход анализа для смеси, состоящей из ацетилена,
окиси углерода, углекислого газа, воды и кислорода, следующий:
1) вода поглощается хлористым кальцием или фосфорным анги-
дридом;
2) углекислый газ поглощается едким кали;
3) кислород поглощается желтым фосфором;
4) окись углерода окисляется и поглощается свежеосажденной и
высушенной при 10—20° окисью серебра;
5) ацетилен поглощается сплавом Си2С12 с 5% едким кали.
Возможно поглощать сухими адсорбентами и некоторые другие
газы.
Для нормальной работы микрогазоанализатора необходимо, чтобы
в камеру 2 не проникал воздух. Эта камера наполняется ртутью
через имеющееся в ее верхней части отверстие, закрывающееся
винтом. Этот винт должен быть хорошо затянут, чтобы обеспечить
газонепроницаемость. В камеру 2 входит еще винт 3, служащий
для засасывания и выталкивания ртути. Здесь уплотнение дости-
гается при помощи сальника, показанного на чертеже. В сальник
помещена асбестовая нить, слегка смазанная вазелином. Вазелин
следует брать очищенный, не содержащий сернистых соединений,
во избежание загрязнения ртути.
Если сальник начинает пропускать воздух, необходимо слегка
подтянуть гайку сальника и вытеснить воздух из камеры 2, доли-
вая ртуть через верхнее отверстие в камере, после чего надо закрыть
это отверстие винтом.
В сальники помещена асбестовая нить, смазанная очищенным
вазелином, и они держат давление на 3—5 см ниже атмосферного.
Ниже даются некоторые указания по изготовлению реактивов-
поглотителей и методика пользования ими.
Поглотитель для углекислого газа. Кусочек едкого кали, служа-
щий для поглощения углекислого газа, сплавляют в платиновой
петельке. Шарик оставляют на некоторое время на воздухе, чтобы
он покрылся слоем влаги, так как совершенно сухой шарик погло-
щает СО2 очень медленно.
Поглотитель для кислорода. Фосфор для поглощения кислорода
плавится в специальной ложечке, имеющей вид глубокой чашечки,
которую погружают в воду с температурой 50°. Затем в чашечку
с расплавленным фосфором вводят платиновую петельку держалки,
223
чашечку вынимают из теплой воды на воздух и немедленно обливают
холодной водой из водопровода, чтобы фосфор застыл и его можно
было вынуть на платиновой проволочке из чашечки. Шарик фос-
фора должен сохраняться под ртутью и не подвергаться действию
солнечных лучей, так как при освещении он переходит в неактив-
ную форму, не поглощающую кислорода.
В наборе принадлежностей, необходимых для микрогазового ана-
лиза, имеется пустая склянка для фосфора, в которую следует по-
местить шарики фосфора, приготовленные, как указано выше. К на-
бору приложена для этой цели железная ложечка. Фосфор не прИт
лагается вследствие опасности его самовоспламенения при перевозке.
Поглотитель для окиси углерода. Специально приготовленная
сухая окись серебра является вполне удовлетворительным адсор-
бентом для окиси углерода. Окись серебра осаждают из раствора
AgNO3 посредством крепкого раствора КОН или NaOH и тщательно
промывают декантацией. Затем осадок отделяют от жидкости на
фильтре и отжимают от влаги посредством листа фильтровальной
бумаги. Слегка влажный осадок прессуют в цилиндрики с помощью
тисков в специальной форме.
Эти цилиндры сушат при комнатной температуре в эксикаторе
над серной кислотой.
Вместо платиновой петельки для укрепления кусочка адсорбента
употребляют прямую платиновую проволочку, кончик которой сма-
чивают небольшим количеством концентрированного раствора жид-
кого стекла, которым и приклеивают AgaO. Кусочек AgaO должен
иметь размер около 1,5—2 мм. Должны быть приняты меры предо-
сторожности, чтобы не покрыть большей части поверхности Ag2O
жидким стеклом. После затвердевания жидкого стекла, на что
требуется около 15 мин., нет опасности утери поглотителя, когда он
вводится в баллончик. Сушка уменьшает давление пара цемента до
величины, не играющей роли в анализе. Адсорбция обычно за-
канчивается в 10 мин. При реакции не образуется никаких летучих
продуктов.
Реакция поглощения окиси углерода может быть представлена
следующим образом:
2Ag2O ~г СО = 2Ag + Ag2CO3
Поглотитель для этена. Для поглощения этена применяют ша-
рики пористого стекла диаметром 2 мм. Шарик прикрепляют к пря-
мой платиновой проволочке, которую нагревают и конец которой
вводят в шарик. Для анализа на этен шарик погружают медленно
в дымящую серную кислоту (в серной кислоте должно быть рас-
творено 20% SO3). Поры шарика заполняются жидкостью. Шарик
вытирают куском фильтровальной бумаги, чтобы удалить избыток
кислоты с внешней поверхности, и затем вводят обычным путем в газ.
Происходит почти мгновенное поглощение. Через 2 мин. кислоту
удаляют и в баллончик вводят слегка влажный шарик едкого кали,
чтобы поглотить пары трехокиси серы. Это поглощение протекает
221
также очень быстро. После следующих 2 мин. твердый поглотитель
удаляют и измеряют уменьшение объема, связанное с удалением
ненасыщенных углеводородов.
Поглотитель для ацетилена. Для поглощения ацетилена при-
готовляют густую пасту из влажной хлористой меди и разбавлен-
ного раствора едкого кали. Эту пасту помещают в платиновую петлю
держалки и нагревают слегка досуха. Нагревают осторожно, чтобы
не перегреть твердый поглотитель до появления черного цвета.
Шарик поглотителя полностью поглощает ацетилен менее чем в
5 мин. Ацетиленистая медь, образующаяся при реакции, удерживается
реагентом и не загрязняет ртути, когда удаляется из реакционной
системы.
Описанная методика может быть использована во всех случаях,
когда в распоряжении исследователя имеется лишь очень небольшой
объем газа.
Прибор позволяет определять только те компоненты, для которых
имеются избирательно действующие поглотители. На нем нельзя
провести анализ газа методом сожжения. К недостатку прибора
относится также необходимость использования открытой части
с ртутью, что неизбежно приводит к разбрызгиванию ртути со всеми
вытекающими отсюда последствиями.
Общий газовый микроанализ может проводиться и на приборах,
конструкции которых аналогичны описанным в главе III, но с та-
кими изменениями, которые вызываются малыми объемами анализи-
руемого газа. Бюретка в подобном приборе представляет собой трубку
того или иного диаметра в зависимости от употребляемых для ана-
лиза количеств газа. Можно брать для этого толстостенные капил-
ляры диаметром 1—3 мм, при которых объем бюретки при ее длине
50 см будет от 0,4 до 3,6 см3. При таком приборе желательно иметь
набор из трех бюреток объемом 0,4—0,5; 2—3 и 5—7 см3. Бюретка
в верхней части имеет кран, присоединенный к гребенке с набо-
ром поглотителей и трубкой для сжигания газа. Уравнительный
сосуд бюретки представляет собой небольшой баллончик. Бюретку
и уравнительный сосуд заполняют водой или для более точного ана-
лиза лучше ртутью, которая не оказывает вредного действия, так
как здесь нет разбрызгивания ртути и баллончик может быть гер-
метично закрыт все время, за исключением тех моментов, когда про-
изводится замер газа или перевод его из одной части прибора в дру-
гую. Для большей точности отсчета параллельно бюретке идет трубка
такого же диаметра, сообщающаяся с атмосферой и закрывающаяся
с помощью каучуковой трубки с заглушкой.
Поглотители (раствор КОН, пирогаллол и т. п.) помещают в
U-образные трубки небольшого диаметра. Верхняя часть трубок имеет
капилляры диаметром 1—2 мм. Растворы наливают таким обра-
зом, чтобы при открытом конце поглотительной трубки, сообща-
ющемся с атмосферой, уровень жидкости стоял на определенной
метке, до которой этот уровень доводился бы и после поглощения
того или иного компонента. Этим путем обеспечивается атмосферное
15 в. а. Соколов. 225
давление в капиллярах при замере. В гребенке прибора и в капил-
лярах перед анализом должен быть азот.
Для сожжения применяется трубочка диаметром около 1 см со
впаянной платиновой проволокой. Эта трубка с уравнительным со-
судом заполнена ртутью. При впуске кислорода уровень ртути пони-
жается, обнажая платиновую спираль. После этого включают на-
кал и производят впуск и сожжение анализируемого газа. Можно
проводить и дробное сожжение, если включить в прибор маленькую
узкую трубку с окисью меди.
На этом приборе можно провести анализ малых количеств^ газа
таких компонентов, как СО2, О2, СО, Н2, СН4, N2, а в некоторых
случаях (при отсутствии тяжелых парафинов) и С2Н4 и С2Н2.
Вышеописанные приборы позволяют проводить общий микро-
анализ малых образцов газа, но они не пригодны для определения
очень малых концентраций тех или иных компонентов, а также и для
анализа очень малых количеств газа, исчисляемых кубическими мил-
лиметрами. Кроме того, на этих приборах нельзя производить раз-
дельное определение углеводородов.
§ 2. Вакуумные установки для определения малых количеств СО2,
н2, со, n2, н2о
Ниже описываются некоторые вакуумные установки для анализа
очень малых количеств газа. В одной установке источник газа соеди-
няется при помощи U-образной трубки, погруженной в жидкий воз-
дух, с ртутным насосом и ртутным микроманометром [24]. Аппарат
устроен таким образом, чтобы газы, откачанные насосом, можно было
собирать над ртутью и смешивать с кислородом или водородом, а
затем привести в контакт с накаленной платиновой нитью и возвра-
тить в соединительные трубки, причем они не касались бы никаких
кранов и никаких других веществ, кроме стекла, ртути и платины.
При помощи этого аппарата можно проводить количественный ана-
лиз газа при его объеме лишь в несколько кубических миллиметров,
определяя при этом Н2О, СО2, СО, Н2, О2 и N2 (последний по раз-
ности). Метод анализа заключается в следующем. Газ собирается
при помощи насоса в маленькую колбочку емкостью около 1 см3,
содержащую тонкую и короткую (2 мм) платиновую проволоку.
Эта проволока накаливается до темнокрасного каления примерно
в течение 2 мин., потом газ возвращается в канализацию, где и из-
меряется давление ртутным микроманометром, чтобы определить
уменьшение объема газа. Это сокращение соответствует присут-
ствию кислорода и окиси углерода или водорода. Получающиеся
углекислота или водяной пар вымораживаются жидким воздухом.
После этого вводится известное количество кислорода в избытке
и снова измеряется давление. При нормальном анализе количество
газа, вычисленное по приращению давления, показанному мано-
метром, должно точно согласоваться с измерением объема введен-
ного кислорода. Далее смесь кислорода и исследуемого газа снова
при помощи насоса собирается в маленькую колбочку для сжига-
22б
ния, где нить накаливается второй раз. После откачки газа hJkoh-
денсирования образовавшейся углекислоты и воды снова замечается
показание манометра. После замещения жидкого воздуха твер-
дой углекислотой в ацетоне углекислота испаряется, в то время как
водяной пар остается в ловушке. По увеличению давления можно
вычислить количество углекислоты, а по нему и количество присут-
ствовавшей вначале окиси углерода. Так как сумма количеств водо-
рода и окиси углерода известна, то можно вычислить и количество
водорода.
Этот метод анализа дает удовлетворительные результаты’ для
газов, выделяющихся при нагревании стекла ламп накаливания,
за исключением случая, когда нить нагревается продолжительное
время в присутствии газа, содержащего водород. Это исключение
Фиг. 86. Аппарат для микроанализа.
представляет собой результат «активности», получаемой при таких
условиях водородом, который, как показали дальнейшие работы,
переходит в атомное состояние.
Водяной пар в этой установке определяется по разности давле-
ний, когда U-юбразная трубка находится при обыкновенной темпе-
ратуре и когда она охлаждается твердой углекислотой с ацетоном.
Эти давления измеряются при помощи ртутного микроманометра.
Однако известно, что подобные измерения не дают хороших ре-
зультатов, когда газ содержит пары воды.
На фиг. 86 схематически представлен другой аналогичный вакуум-
ный прибор. Для устранения неточностей замеров вследствие присут-
ствия водяного пара здесь применяют ртутный манометр с опти-
ческим отсчетом [25].
На схеме установки (фиг. 86), необходимой для производства
анализа, не показаны ни вторичный насос, который соединен с при-
бором при помощи трубки 1, ни та емкость, в которой получается
15*
227
бравшийся для анализа газ и которая присоединяется к трубке 2,
ни аппарат для получения кислорода, водорода или окиси углерода,
который позволяет вводить в прибор определенные количества газов;
этот последний аппарат присоединяется к трубке 3. Кислород полу-
чается электролитическим путем, а окись углерода — из серной и
муравьиной кислот.
Все ртутные барометрические затворы отпускаются, т. е. откры-
ваются, затем производится откачка до самого совершенного ва-
куума аппарата и приемника, в котором собирались подлежащие
исследованию газы, и аппарата для получения газов. После этого
закрывают затворы 4 и 5, которые изолируют аппарат для анализа
от насоса и генератора известных газов. Анализируемый газ (напри-
мер, газ, выделяемый металлической нитью при накаливании) на-
правляется в прибор 6 через трубку 2. Углекислота и водяной пар
конденсируются в ловушке с жидким воздухом 12. Далее измеряется
манометром 11 давление газов, оставшихся несконденсированными.
Зная общий объем системы, можно определить объем газа при
нормальных условиях (760 мм рт. столба и 0°). При помощи ртут-
ного насоса 8, нижний резервуар которого ставится попеременно
на сообщение с атмосферой и с насосом предварительного вакуума,
газы собираются в трубку 9, в которой имеется маленькая плати-
новая нить накаливания. Во время этой операции затвор 10, есте-
ственно, должен оставаться закрытым. Путем замены жидкого
воздуха баней с ацетоном и твердой углекислотой можно испарить
сконденсированную углекислоту и на манометре 11 измерить ее давле-
ние. Далее углекислота откачивается при помощи вторичного насоса
через трубку 7, для чего опускается затвор 7, который затем снова
закрывается. После этого закрывается затвор 4 и ловушка 12 снова
нагревается до комнатной температуры. Ртутный манометр с опти-
ческим отсчетом 13 позволяет измерить давление, которое полу-
чается в объеме (предполагается, известном) правой ветви манометра
трубки 13 и левой ветви затвора 4. Таким образом можно опреде-
лить присутствовавшие вначале количества СО2 и Н2О. После этого
ловушка снова охлаждается жидким воздухом.
Анализ неконденсирующихся газов, собранных в трубке 9, про-
изводится следующим путем. Предполагается, что газы не содер-
жат одновременно кислорода и какого-либо углеводорода. Через
затвор 5 вводится измеренное количество водорода. Для контроля
измеряется его давление в системе, затем его собирают в трубке 9
при помощи ртутного насоса. Далее производят сжигание, накаливая
платиновую нить, и открывают затвор 10, чтобы получающиеся угле-
кислота и вода могли распространиться по соединительным труб-
кам вместе с кислородом в избытке и негорючими газами. Угле-
кислота и водяной пар конденсируются в трубке 13. Измеряется да-
вление кислорода и негорючих газов, после чего их снова собирают в
трубке 9. Далее, как и прежде, измеряют получившиеся количества
СО2 и Н2О (количество Н2О по разности между новой и старой вели-
чиной количеств сконденсированных газов). Полученные величины
непосредственно определяют содержание окиси углерода и водорода
228
в данной смеси. Если эта смесь содержала кдкой-либо определенный
углеводород, например СН4, то количества углекислоты и воды по-
зволяют вычислить содержание СО, СН4 и Н2 при условии, что изве-
стен общий объем газов, т. е. если сделано определение, как об этом
говорится далее, объема газов, не конденсирующихся при темпера-
туре жидкого воздуха. Во всех случаях найденные количества угле-
кислоты и водяного пара позволяют определить количество кисло-
рода, пошедшего на сжигание. Его вычитают из количества при-
бавленного кислорода. Если начальный газ не содержит ни кисло-
рода, ни азота, ни редких газов, то эта разница должна представлять
собой общее количество газа, действительно содержавшегося в труб-
ке 9, т. е. газа, последнее измерение которого манометром дает его
объем. Таким образом, можно непосредственно узнать, содержала
ли данная смесь по крайней мере один из газов — кислород, азот
или редкие газы.
Если начальная смесь не содержала ни кислорода, ни азота, то
анализ на этом заканчивается. В противном случае разность между
введенным кислородом и использованным будет меньше объема со-
бранных в трубке 9 газов, которые могут содержать только кислород,
азот и редкие газы. Тогда вводят в трубку 9 измеренный избыток
окиси углерода, производят сжигание и открывают затвор 10. Изме-
ряют, как выше было указано, количество образовавшейся углекис-
лоты и по нему вычисляют соответствующее количество кислорода.
Если начальная смесь не содержала кислорода, то это количество
должно равняться разности между введенным кислородом и исполь-
зованным. Если вычисленное количество больше разности между
кислородом введенным и использованным, то отклонение в точности
должно давать количество кислорода, присутствовавшего вначале.
В том и другом случае разница между общим количеством анализи-
руемого газа и количествами, найденными для его составных ча-
стей — Н2О, СО2, горючих газов и О2 — и будет представлять собой
содержание азота (или редких газов).
Если данная смесь содержит одновременно кислород и углеводо-
роды, то метод не может быть применен.
Приведем пример такого анализа газа, выделенного проволокой
из ферросилиция, нагретой до 750—800°. Давления указаны в де-
сятых долях миллиметра:
1. Н2О сконденсированная и отделенная ... О
2. СО2 сконденсированная и отделенная . . . 33,5 или 3,86 мм2
3. Газы «сухие •............................898
4. О2 добавленный ..........................535
5. Сумма........................... ... 1433
6. Количество продуктов, оставшихся после
сжигания и конденсации Н2О и СО2 . . . 605
7. Сжатие вследствие образования Н2О и СО2 828
8. 7з приходится на долю О2................276
9. 2/з приходится на долю СО + На .........552
10. СО2, образовавшаяся, сконденсированная и
затем испаренная, откуда равное количество
СО ........................................ 437 или 50,3 мм?
220
11. Н2 по разности...........................115 или 13,22 мк?
«сухие» газы по п. 3 . •................898
СОЧ-Н2 по п. 9 .........................552
346 или 39,8 мм3
12. Разность (N2? + O2?)..................... 346 или 39,8 л/л/3
О2 прибавленный 6,4......................535
О2 потребленный 6,8......................276
13. Разность 259 (<346, т. е. прибавленная СО) 259
14. «Сухие» газы по п. 6 (N2 ]-О2)...........605
СО прибавленная ..................... . 1583
15. Сумма...................................2188
16. Количество, оставшееся после сжигания и
конденсации образовавшейся СО2..............1415
17. Количество, исчезнувшее вследствие образо-
вания СО2 ..................................773
18. его, приходящаяся на долю кислорода.
Это количество то же, что в п. 13 .... 258
Таким образом, смесь не содержала свободного кислорода, и ее
состав был следующий:
Н2О ................. 0 0
СО2 ....................................... 3,86 мм3 3,6%
СО ........................................ 50,30 » 46,85%
Н2..........................................13,22 » 12,35%
Na........................................ 39,80 » 37,20%
По мнению автора прибора, при общих количествах газа 5 лш3
ошибки в определении главных составных частей могут достигать 6%.
На фиг. 87 представлен еще один прибор для микроанализа газа
[32]. Этот аппарат имеет ртутный насос 8 с трубкой для сжатия газа 9}
который через кран сообщается с распределительной трубкой 6.
К распределительной трубке также через кран присоединена кол-
бочка 7, содержащая спиральку из железа, предназначенную для
поглощения кислорода, камера 2 с электродами из платины для того,
чтобы при помощи искры производить сожжение горючих газов с
кислородом, маленькая колбочка 3, содержащая кусочки едкого
кали для поглощения углекислоты, и разрядная трубка 5 для наблю-
дения за цветом и спектром разряда. Перед трубкой 5 в баллончике 4
находится листочек золота, назначение которого — поглощение
паров ртути, которые мешают спектральным наблюдениям. Источник
газа присоединяется к трубке 7.
Так как весь объем аппарата предварительно измерен путем со-
общения с баллоном известного объема, то нетрудно узнать объем
впущенного в прибор газа при атмосферном давлении. После пере-
ведения всего газа в сосуды 7—2 и при подъеме ртути до колбочки 7
железная спиралька накаливается. Давление газа, собираемого в
трубке 9} измеряют снова. Таким образом определяют содержание
кислорода. Аналогичным образом после впуска газа в колбочку 3,
содержащую едкий кали, определяют остаток газа и узнают содер-
жание СО2. Если имеются еще и другие газы, то вводят либо кисло-
род, либо водород и производят сожжение в баллоне 2.
Описанные выше вакуумные установки для микроопределения
таких газов, как СОа, Н2, СО, N2, и паров воды и разработанные
230
зарубежными исследователями имеют существенные недостатки мето-
дического и конструктивного характера. Следует прежде всего отме-
тить, что все определения паров воды, служащие для определения
водорода как свободного, так и входящего в элементарный состав
других газов, методически неудовлетворительны и дают явно оши-
бочные результаты независимо от системы применяемого микромано-
метра.
В описанных приборах совершенно не учтена возможность нали-
чия в анализируемых газах углеводородов и'некоторых других газов,
как, например, закиси азота, которые,
вания, очень часто встречаются в
самых разнообразных образцах газа,
не говоря уже о природных, где ука-
занные компоненты почти всегда встре-
чаются в тех или иных количест-
вах.
Прибор, изображенный на фиг. 86,
неудобен и в конструктивном отноше-
нии. Так, например, ртутный мано-
метр с закрытым капилляром неудо-
бен для газоаналитических опреде-
лений, так как требуется дополни-
тельный ртутный насос для перекачки
газа. В приборе, изображенном на
фиг. 87, применение кранов и ртути
не является удачным решением во-
проса анализа малых количеств газа
из-за подтекания кранов и загрязне-
ния всей системы вследствие контакта
ртути со смазкой.
Правильный микроанализ выше-
как показали наши исследо-
Фиг. 87. Аппарат для микро-
анализа.
указанных компонентов представляет
большие трудности, и этот вопрос не
мог быть решен без разработки микро-
анализа на углеводороды и некоторые другие газы. Методы микро-
анализа углеводородных газов, разработанные в Советском Союзе,
позволили более правильно решить вопрос и об общем микроанализе.
Методика полного микроанализа с определением как СО2, Н2, О2,
так и углеводородов и некоторых других компонентов описывается
ниже после изложения методики углеводородного микроанализа.
Определение малых концентраций углекислого газа, окиси углерода и водорода
Одним из наиболее простых методов определения углекислого
газа является метод контракции [33, 15]. Пробу газа, содержащую
малые концентрации углекислоты, вводят в соответствующий бал-
лон, после чего она приводится в контакт со щелочью. По уменьше-
нию давления вследствие поглощения углекислоты можно определить
и ее содержание в газе. Для измерения этого уменьшения давления
применяют различные приемы. К баллону присоединяют какой-либо
231
манометр или микроманометр, показания которого и служат мери-
лом концентрации углекислоты. В наиболее простых приборах мано-
метром служит присоединенная к баллону трубка, содержащая рас-
твор щелочи.
Чувствительность подобной установки зависит от диаметра трубки,
служащей манометром. Чем уже трубка, тем до известного предела
выше чувствительность прибора. Наивысшим пределом чувстви-
тельности подобного прибора являются величины порядка одной-
двух сотых долей процента.
В то же время необходимо учесть, что быстрое поглощение угле-
кислоты щелочью является причиной неточности подобных опреде-
лений. Пока производится впуск газа в прибор, некоторое количе-
ство углекислоты уже будет поглощено щелочью и, таким образом,
результаты анализа будут преуменьшены. Поэтому для точного опре-
деления малых концентраций углекислоты рекомендуется такое
устройство, чтобы газ первоначально забирался в соответствующий
баллон, а уже потом в этот баллон вводилась щелочь, служащая
для поглощения углекислоты.
Можно воспользоваться и такими твердыми поглотителями, ко-
торые действуют не столь быстро и поэтому дают возможность про-
извести замер количества газа до начала его поглощения.
К баллону, содержащему твердую щелочь, присоединяют резер-
вуар, заполненный водой или ртутью и служащий для впуска не-
которого количества газа. К баллону со щелочью присоединяют
наклонный манометр, содержащий керосин или какое-либо легкое
масло. После впуска газ путем диффузии и конвекции начинает по-
ступать через сетку в верхнюю часть камеры, где помещается твер-
дая щелочь. По показанию наклонного манометра, предварительно
прокалиброванного, определяют содержание углекислоты.
Так как в данном случае измеряется падение давления за счет
поглощения, то количество газа не имеет значения. Поэтому не тре-
буется определения объема взятого газа. Чувствительность подобного
прибора может быть достаточно большой, соответствующей чувстви-
тельности наклонного манометра. Во всяком случае при небольшом
наклоне манометра можно определять концентрации углекислоты
порядка сотых и тысячных долей процента [27]. Для получения наи-
более точных результатов необходимо вводить поправку на измене-
ние объема газа за счет смещения жидкости в манометре. Для про-
ведения измерений можно воспользоваться и иными микроманомет-
рами, упомянутыми в главе I.
Описанный прибор предварительно калибруется искусственными
смесями с известным содержанием углекислоты.
Как уже было упомянуто выше, получение таких искусственных
смесей с малой концентрацией углекислоты связано с различными
затруднениями. Необходимо, чтобы бюретка, в которой готовится
искусственная смесь газа или воздуха с углекислотой, была предва-
рительно прогрета и с помощью вакуума из стенок бюретки удалена
углекислота. Иначе при приготовлении смесей эта углекислота вый-
дет из стеклянных стенок и изменит ее концентрацию.
232
Калибровка прибора заключается в анализе нескольких искус-
ственно составленных смесей газа. Для этого берут, например, 100 ои3
атмосферного воздуха, очищенного от углекислоты и разбавленного
тем или иным количеством атмосферного воздуха, содержащего,
как известно, 0,3—0,4% углекислого газа. При различных соотно-
шениях этих газов можно получить смесь с различным и притом с
очень небольшим содержанием углекислоты. Для точной калибровки
содержание углекислоты в воздухе должно быть определено каким-
либо иным способом, например титрометрическим, который описы-
вается ниже. Для калибровки можно приготовлять искусственные
смеси путем добавок малых, точно измеренных количеств углекис-
лоты. Если удаление углекислого газа из стенок бюретки затру-
днительно, то всякая искусственная смесь должна быть проверена
каким-либо иным методом.
Одним из наиболее надежных методов определения малого коли-
чества или малой концентрации углекислого газа является метод
титрования.
Общая схема определения углекислоты путем титрования заклю-
чается в том, что анализируемый газ собирают в какую-либо бюретку
или иную емкость, но с соблюдением упомянутых выше предосторож-
ностей, а затем направляют в барботер, где происходит поглощение
углекислоты баритовой водой.
После окончания пропускания газа через баритовую воду ее тит-
руют соответствующим раствором соляной кислоты. Зная титр до и
после анализа, можно путем соответствующих расчетов определить
концентрацию и количество углекислоты во взятом газе аналогично
тому, как это производится на вышеупомянутых приборах для опре-
деления углеводородов.
Для ориентировочного определения углекислого газа можно поль-
зоваться также методом, основанным на появлении кристаллов угле-
кислого бария в баритовой воде [37] при приведении ее в контакт с
анализируемым газом, как это описано в предыдущем параграфе.
Следующий метод, который может применяться для определения
углекислого газа, основан на измерении электропроводности рас-
твора баритовой воды или какой-либо другой щелочи, поглощающей
углекислый газ. Этот метод называется кондуктометрическим.
Анализируемый газ попадает в реакционный сосуд, в котором
имеются два электрода, присоединенные к потенциометру. Пропуская
определенное количество газа и замечая изменение электропровод-
ности раствора, можно определить концентрацию и количество угле-
кислоты. Прибор должен быть предварительно прокалиброван искус-
ственными смесями, содержащими определенный процент углекислого
газа.
Методика определения малых количеств СО имеет много общего с
методикой определения СО2. Разница заключается только в том, что
при определении СО этот газ следует сначала сжечь и перевести та-
ким образом в углекислый газ. Дальнейшие определения будут про-
водиться уже с СО2, как это упомянуто выше.
Микросожжение СО проводится так же, как и при обычном общем
233
анализе. Для этого сожжения можно пользоваться окисью меди, а
также пятиокисью иода, как это описано в главе III.
Определение водорода в малых концентрациях представляет зна-
чительные трудности вследствие наличия метана. Даже при темпе-
ратуре жидкого воздуха упругость паров метана достаточно велика
(76 мм рт. столба), и путем охлаждения при атмосферном давлении
нельзя понизить его концентрацию в газовой фазе меньше чем
до 10%. Путем охлаждения можно удалить из газовой фазы толь-
ко более тяжелые углеводороды.
Водород при этом охлаждении не конденсируется, но он смешан
с метаном. Если дальнейший анализ проводить обычным путем, ме-
тодом дробного сожжения, то трудно достигнуть чувствительности,
большей 0,2—0,3%.
Известны методы, при которых содержание водорода опреде-
ляется путем сожжения по количеству образовавшихся паров воды.
Для этой цели Б. М. Накашидзе предложен способ измерения элек-
тропроводности хлористого кальция при поглощении водой. При-
бор состоит из баллона с стеклянной трубкой, покрытой тонким слоем
хлористого кальция, к которому присоединены электроды, позво-
ляющие измерять электропроводность этой трубки.
Для сожжения следует применять окись меди при температуре
300°, когда сгорает только водород. В присутствии метана, прово-
дя полное сожжение с применением накаленной платины, не-
обходимо определять углекислый газ, образовавшийся от сожжения
метана, и воду, образовавшуюся от сожжения метана и водорода.
Введя поправку на воду, образовавшуюся в результате сожжения
метана, по разнице можно вычислить и содержание водорода в газе.
В этом случае также трудно определить малое количество водорода
в присутствии большого количества метана.
Значительно большей чувствительностью при анализе на водород
отличаются физические методы газового микроанализа, описывае-
мые в главе VIII.
§ 3. Микроанализ на углеводородные газы с применением
вакуумных установок и глубокого охлаждения
Микроанализ на углеводородные газы получил широкое практи-
ческое применение в геохимических методах поисков нефти. Методы
микроанализа на углеводородные газы могут быть использованы
также для определения многих других органических газообразных
и парообразных соединений, в частности для обнаружения присут-
ствия углеводородов, а также других органических веществ в воз-
духе.
Методы микроанализа на углеводородные газы представляют так-
же интерес для разрешения многих научных и научно-технических
вопросов, например при изучении проблем происхождения нефти,
проблемы миграции нефтяных газов и т. д.
В настоящее время для количественного микроанализа на угле-
водородные газы используются различные методы. Некоторые из
234
этих методов основаны на применении глубокого охлаждения и вы-
мораживании отдельных индивидуальных углеводородов. Другие
методы основаны на применении сожжения углеводородов с опре-
делением продуктов этого сожжения в виде углекислого газа. Эти
методы сожжения позволяют определить суммарное микросодержа-
ние углеводородов, а при разделении углеводородов на индивидуаль-
ные компоненты каким-либо иным путем сожжение позволяет иден-
тифицировать эти углеводороды.
Для микроанализа на углеводородные газы применяются также
сорбенты, позволяющие в известных пределах проводить разделение
углеводородной смеси на отдельные индивидуальные компоненты.
Для определения индивидуальных компонентов могут быть исполь-
зованы некоторые физические, в частности оптические, методы ана-
лиза, подробно описанные в главе VIII.
Ниже описываются методы микроанализа на углеводородные
газы с применением глубокого охлаждения и фракционировки этих
газов.
Стационарные ртутные приборы
Стационарные ртутные приборы, применяемые при микроанализе
на углеводородные газы, представляют собой высоковакуумные стек-
лянные установки, снабженные специальными ртутными затворами
для впуска газа в прибор и для изолирования по ходу анализа одних
частей прибора от других [27]. В обычных газоанализаторах эту
роль выполняют краны, однако при той чувствительности, которая
требуется от приборов, применение кранов в вакуумной части, вообще
говоря, нежелательно. Малейшая негерметичность кранов может
значительно затруднить проведение анализа и исказить его резуль-
таты. Ниже приводятся описания схем нескольких применяющихся
стационарных ртутных приборов.
Задачей этих приборов является прежде всего определение малых
концентраций (до 10~5%) углеводородных газов, содержащихся в
воздухе или ином негорючем газе.
Одна из первоначальных схем стационарного ртутного прибора
представлена на фиг. 88. Прибор состоит из следующих частей. Впу-
скной ртутный затвор 7 представляет собой стеклянную трубку с
резервуаром на конце, по всей длине которой проходит тонкий капил-
ляр, по которому поступает анализируемый газ. В систему прибора
включены барботеры с раствором едкого кали, трубка с твердым ед-
ким кали и трубки с фосфорным ангидридом, ртутный насос 2, труб-
ка для сожжения с платиновой проволокой 3, накаливаемой элек-
трическим током. Для получения глубокого вакуума применяется
баллончик с углем 4, погружаемый в жидкий воздух, змеевики 5 и 6
служат для конденсации углеводородов и продуктов их сожжения.
В ртутных затворах ртуть поднимается или опускается с помощью
трехходовых кранов, присоединяемых или к вакууму, или к атмо-
сфере.
К ртутному насосу 2 присоединен капилляр, изогнутый так, как
показано отдельно на фиг. 88, а. Другой конец этого капилляра при-
235
соединен к ртутному затвору. Объем капилляра между несколькими
нанесенными на нем метками точно измерен. Подобное соединение
ртутного насоса и затвора очень удобно, так как позволяет пользо-
ваться насосом 2 и для откачки, и для замера малых количеств газа
в капилляре 8. Для данного случая подобная конструкция более
рациональна, чем обычный манометр Мак Леода, в котором вакуум
создается значительно медленнее и который не может быть исполь-
зован одновременно и как насос. Для замера газ загоняют в капил-
ляр 8 и поднимают ртуть до тех или иных меток. По давлению
в другой трубке затвора 9 можно вычислить объем
газа при норлтальном давлении.
Фиг. 88. Стационарный ртутный прибор.
Методика анализа такова. В приборе предварительно создают
полный вакуум, после чего, опуская и поднимая впускной затвор 7,
забирают некоторое количество газа в прибор. Впуск газа продолжа-
ют до тех пор, пока в насосе не наберется достаточное количество
газа, т. е. когда ртуть в насосе будет стоять у метки 0, а замер пока-
жет давление, соответствующее тому, которое необходимо, чтобы
объем газа составлял при нормальных условиях, например, 100 см3.
Объем газа можно замерить и перед его впуском в прибор. После
того как необходимое количество газа набрано в насос, манипулируя
затворами, несколько раз прогоняют газ по системе насос—трубка
3 — змеевики 5 и 6 — насос. Эта циркуляция проводится для окон-
чательного осушения газа с помощью включенной в систему труб-
ки 7, содержащей Р2О5. После этого змеевик 5 погружают в дьюаров-
ский сосуд с жидким воздухом и продолжают циркуляцию газа; в
змеевике 5 задерживаются все тяжелые углеводороды. После окон-
чания циркуляции газ загоняют в трубку для сожжения 3. Затем
233
производят циркуляцию газа при накаленной платиновой прово-
локе в трубке 3. Метан в газе при этом сгорает, а пары воды, обра-
зовавшиеся при сожжении, задерживаются в трубке 7. Змеевик 6
погружен в дьюаровский сосуд с жидким воздухом. После оконча-
ния сожжения продолжают циркуляцию через змеевик 6. В змеевике
конденсируется углекислый газ, образовавшийся при сожжении
метана.
Затем в приборе создается высокий вакуум. Газ, находившийся
в приборе, откачивают предварительно масляным насосом через ка-
кой-либо из ртутных затворов. Когда давления в приборе будет уже
недостаточно, чтобы преодолеть столб ртути в затворе, откачку про-
должают насосом 2, направляя газ в баллончик с углем. Откачку
змеевиков 5 и 6 прекращают только тогда, когда при сжатии газа
в капилляре насоса практически не будет заметно смещения ртути.
После этого, отняв дьюаровский сосуд, измеряют количества газов,
сконденсировавшихся в змеевиках 5 и 6.
Объем верхней части капилляра между метками обычно около
20—40 мм3. Если разница уровней ртути в капилляре и в манометре
составит 1 мм, то это означает, что в капилляре имеется (при объеме
40 мм) = 0,053 мм3 газа.
Для анализа обычно берут 100 см3 (при нормальных условиях),
поэтому откачанное количество газа (0,053 мм3) составит 53 милли-
онных долей процента.
Другая схема стационарного ртутного прибора представлена на
фиг. 89. Прибор состоит из ртутного насоса 5, простого ртутного за-
твора 6, сложного затвора 7 с баллонами 7, 2, 3 и 4, впускного за-
твора 8, бюретки с уравнительным сосудом 9, барботеров 10 с рас-
твором КОН, трубки 77 с твердым КОН, трубок 12 и 13 с фосфор-
ным ангидридом, двух змеевиков 14 и 15, трубки для сожжения с
платиновой спиралью 16 и баллона с углем 77, присоединенного к
прибору при помощи трехходового крана.
К уравнительным сосудам насоса и затворов присоединены краны.
Вторые ходы кранов присоединены к вакууму, а третьи — к атмо-
сфере. В насосах и затворах залита ртуть, в бюретку 9 — вода.
В баллонах 7, 2, 3 и 4 капилляры (диаметр 0,5 мм) имеют разную
длину: капилляр 4 имеет длину 11 см, 3 — 9 см, 2 — 1 см yl 1 —5 см.
Для измерения объема углеводородов служит градуированный капил-
ляр 18, вторая трубка (левая) затвора 6 служит при измерениях
манометром.
Перед анализом в приборе создается полный вакуум при помощи
насоса и угля, погружаемого в дьюаровский сосуд с жидким воз-
духом.
В затворе 7 уровень ртути устанавливается так, чтобы капилляр
в баллоне 7 был погружен в ртуть на глубину 4 см, в баллоне 2 —
на 6 см и т. д. Ртуть во всех баллонах 7,2,3 и 4 стоять на одной
высоте.
Перед впуском газа поднимают воду в сосуд до верха и вытесняют
бывший там воздух или газ через кран. Капилляр, идущий вправо
237
от сосуда 9, погружен в ванну с водой ;'на капилляр^надеваетсяАбу-
тылка с анализируемым газом. Сначала газом промывают баллоны 10,
выпуская газ через кран. Опуская уровень воды в сосуде 9, забирают
газ до метки. Объем сосуда 9 вместе с капилляром, идущим к впуск-
ному затвору 8, измеряют заранее (100 гл/3)/поэтому объем взятого
газа при нормальных условиях можно вычислить, зная барометри-
ческое давление.
Фиг. 89. Стационарный’ртутный прибор.
Опуская и поднимая ртуть во впускном затворе 8, впускают;газ
небольшими порциями в прибор. При этом змеевики 14 и 15 погружа-
ются в дьюаровские сосуды с жидким воздухом, а спираль в трубке
16 накаливается электрическим током до красного каления. Газ из
трубки 13 поступает через узкий капилляр (диаметр 0,5 мм), идущий
по всей длине затвора. Проходя через трубки 11 и 12, газ окончатель-
но освобождается от следов СО и паров воды. В змеевике 15 задержи-
ваются тяжелые углеводороды. При прохождении через трубку для
сожжения 16 в газе сгорает метан. Пары воды, образовавшиеся при
238
сожжении., задерживаются фосфорным ангидридом в трубке 13,
а углекислый газ, образовавшийся от сгорания, задерживается в
змеевике 14. Газ попадает затем в насос 5 и через затвор 6 откачи-
вается масляным насосом. Когда прохождение газа через баллоны
7, 2, 3 и 4 установится, ртуть во всех баллонах будет стоять на раз-
ной высоте. Разница давления газа в соседних баллонах будет равна
2 см. Ниже всего ртуть будет стоять в баллоне 4, а выше ^всего в бал-
лоне 7.
Вследствие присутствия ртути и капилляров в баллонах 7У, 2, 3
и 4 газ в змеевиках и трубках 72, 13 и 16 будет несколько задержи-
ваться, и, таким образом, будет обеспечиваться полнота сожжения
и конденсации в змеевиках 14 и 15.
Когда весь газ из сосуда 9 будет переведен в прибор и давления
в трубке 72 будет недостаточно, чтобы пройти через слой ртути в
баллоне 4, опускают ртуть во всем затворе 7. Насосом и углем со-
здается вакуум во всем приборе. Таким образом, весь газ, попавший
в прибор, пройдет по линии 75, 16, 13 и 14. При создании оконча-
тельного вакуума в змеевиках ртуть в затворе 7 опускают до уровня,
лежащего несколько ниже трубки, идущей к углю. Когда оконча-
тельный вакуум в змеевиках будет достигнут (проверяется на капил-
ляре 18}, уровень ртути в насосе 5 устанавливается на 2—3 см ниже
трубки, идущей к баллону 7, отнимают от змеевика 14 дьюаровский
сосуд, дают змеевику нагреться и откачивают насосом углекислый
газ, сконденсировавшийся в змеевике 14, в капилляр 18, где и изме-
ряют его объем (затвор б при этом должен быть поднят).
При откачке СО2 нельзя опускать уровень ртути в насосе до труб-
ки, идущей к змеевику 15.
Откачанное количество СО2 удаляют углем в сосуде 77. Откачку
повторяют несколько раз до тех пор, пока не будет откачан весь СО2
из змеевика 14.
Следует заметить, что уже первая откачка дает обычно 70—80%
всего углекислого газа, имевшегося в змеевике 14. Объем откачан-
ного углекислого газа согласно уравнению сожжения равен объему
сгоревшего метана. После этого поднимают ртуть в затворе на такую
высоту, чтобы закрыть капилляр в баллоне 7. Опуская ртуть ниже
трубки, идущей к змеевику 15, и отняв дьюаровский сосуд от змее-
вика 15, откачивают и измеряют тяжелые углеводороды.
Следует учесть, что в змеевике 15 помимо таких тяжелых угле-
водородов, как этан, пропан и т. д., будут конденсироваться вообще
всякие газы и пары, которые при температуре жидкого воздуха имеют
очень малую упругость паров и которые не поглощаются упомяну-
тыми реактивами — КОН и Р2О5, помещенными в трубках 10, 11
и 72. Из газов неорганического характера в змеевике 14, в частности,
будет конденсироваться закись азота. В связи с этим конденсиру-
ющиеся и не откачивающиеся из змеевика 14 газы принято называть
тяжелой фракцией. Поскольку в тяжелой фракции могут находиться
различные газы, то были разработаны приборы, позволяющие про-
водить анализ самой тяжелой фракции, а также продуктов сожжения,,
конденсирующихся в змеевике 15 и называемых легкой фракцией.
239
Один из подобных приборов представлен на фиг. 90. Как и на
стационарном приборе, изображенном на фиг. 88, имеется возмож-
ность несколько раз прогонять анализируемый газ через змеевики
и сжигательную колонку. Подобное устройство позволяет подвергать
дальнейшему анализу то количество тяжелой фракции, которое было
замерено в капилляре 18.
Фиг. 90. Стационарный циркуляционный прибор.
В приборе, изображенном на фиг. 90, анализируемый газ из бю-
ретки направляется через поглотители 1 с раствором КОН и впуск-
ной ртутный затвор 2 в трубки 3 и 4 с твердым КОН и Р2О5, откуда
он идет далее в змеевик 5, баллон с платиновой проволокой для сож-
жения газа 6, проходит через трубку 7 с Р2Об, змеевик 8 и через на-
сос 9 и ртутный затвор откачивается из прибора. В змеевике 8 кон-
денсируются продукты сожжения, а в змеевике 5 конденсируется
тяжелая фракция. После создания вакуума в приборе и удаления
240
жидкого воздуха конденсат из первого или из второго змеевика мо-
жет быть откачан и измерен в капилляре 10. После этого измерения
конденсат может быть переведен в баллончик 11, содержащий КОН.
Тяжелая фракция после измерения может быть переведена в бал-
лон 6 и сожжена. Кислород или воздух для этого сожжения заби-
рается в баллон 6 обычным способом через бюретку и очищается,
проходя через змеевик 5, охлаждаемый жидким воздухом. Продукты
сожжения тяжелой фракции (кроме воды, задерживаемой Р2О5 в
трубке 7) конденсируются в змеевике 8,
духом, затем замеряются в капилляре
и подвергаются дальнейшему анализу
в баллончиках 11 и 12, содержащих
КОН и Р2О5.
Перед вакуумной частью прибора,
но до поглотителей с КОН рекомендует-
ся поставить дополнительную сжигатель-
ную колонку (фиг. 91). В этом случае
анализ можно провести тремя способами.
Первый — это обычный анализ, ход ко-
торого описан выше и при котором
определяют отдельно тяжелую и легкую
фракции; он называется раздельным.
Второй—проводится таким образом,
что охлалаждается только второй змее-
вик. Тяжелая и легкая фракции под-
вергаются суммарному сожжению и за-
меряются продукты этого сожжения,
сконденсированные во втором змеевике;
он называется суммарным. Третий —
анализ проводится с применением ко-
лонки предварительного сожжения. Угле-
водороды, содержащиеся в газе, сго-
рают, и в первом змеевике конденсируются только негорючие ком-
поненты. Этот способ называется анализом с предварительным сож-
жением и служит для определения в газе негорючих компонентов,
попадающих в тяжелую фракцию.
Конденсирующаяся в первом змеевике тяжелая фракция может
содержать, помимо углеводородов, также и некоторые другие орга-
нические и неорганические соединения. Для исследования тяжелой
фракции и выяснения ее состава применяют ее сожжение с опреде-
лением продуктов сожжения. Кроме того, тяжелая фракция может
подвергаться разгонке, поскольку отдельные ее компоненты при
определенных температурных условиях можно отделить друг от
друга, хотя эта операция представляет некоторые трудности. При тем-
пературе около —145° откачивается этан, при —130° пропан.
Испытания стационарных ртутных приборов и проверка их пока-
заний. Проведение анализа на каком-либо из вышеописанных при-
боров заключается прежде всего в пропускании через прибор
16 В. А. Соколов. 241
охлаждаемом жидким воз-
Фиг. 91. Колонка предвари-
тельного сожжения.
7 — замерная пипетка; 2 — барбо-
теры для раствора КОН; 3 — ко-
лонка предварительного сожжения;
4 — трехходовый вакуумный кран;
5 — вакуумметр; 6 — колонка для
твердого КОН; 7 — впускной за-
твор.
образца газа с последующим определением объема задержавшихся
в приборе углеводородов (а также некоторых других компонентов,
если они присутствуют) и углекислого газа, образовавшегося в ре-
зультате сожжения. Однако, если мы пропустим через только что
собранный прибор совершенно чистый воздух, лишенный каких-
либо углеводородов, то получим некоторые показания по тяжелой
и по легкой фракциям. Это объясняется тем, что с внутренней по-
верхности стекла выделяются пары воды и углекислый газ, а кроме
того, из фосфорного ангидрида и едкого кали при прохождении через
них чистого воздуха выделяются газообразные и парообразные при-
меси. Все эти выделения будут задерживаться в змеевиках и при
откачке ртутным насосом будут собираться в капилляр, создавая
впечатление о наличии в образце углеводородных газов. Поэтому-
только что собранные приборы перед вступлением в нормальную
эксплоатацию для проведения анализов должны пройти предвари-
тельно очистку и промывку.
После того как прибор собран, высушен и в него налита чистая
ртуть, необходимо создать в нем хороший вакуум. Предварительный
вакуум создается масляным насосом, вакуум же более совершенный
создается углем, погруженным в жидкий воздух. Проверка вакуума
производится насосом, которым в капилляре определяют количество
откачанных газов и паров. Вакуум в приборе проверяется, чтобы
убедиться в отсутствии каких-либо трещин в приборе, т. е. в его пол-
ной герметичности. Кроме того, при откачке удаляется значитель-
ная часть вредных примесей, могущих находиться в реактивах, а
также частично пары воды и углекислый газ, адсорбированные стек-
лом. Для наибольшей эффективности удаления из стекла адсорби-
рованных в нем газов и паров рекомендуется основные части прибора
прогреть до 200—300°. Прогревать необходимо змеевики, соедини-
тельные трубки, идущие от змеевиков к ртутному насосу, сам ртут-
ный насос и ртутные затворы. Однако этого нагревания с откачкой
все-таки недостаточно для подготовки прибора к проведению ана-
лизов. Наибольшее затруднение представляет удаление паров воды,
адсорбированных стеклом. Удаление паров воды до такой степени,
чтобы остающимся их количеством можно было пренебречь,— про-
цесс медленный и требующий многократных откачек и многократных
пропусканий через прибор очищенного воздуха.
В конце концов, после надлежащей отработки прибор начинает
давать по обеим фракциям небольшие, но более или менее постоянные
показания. Эти показания называют собственными показаниями
прибора, и для хороших приборов они составляют величины порядка
10~5% при анализе 100 см3 чистого атмосферного воздуха. Эти вели-
чины обычно сбрасывают с показаний, полученных при анализах
исследуемых газов. Собственные показания приборов должны время
от времени проверяться и не выходить из установленной нормы.
Собственные показания приборов при раздельном анализе и при
анализе с предварительным сожжением меньше, чем при анализе с
суммарным сожжением. Если анализируемые пробы имеют невысо-
кую концентрацию тяжелой фракции, то необходимо путем более тща-
212
тельной обработки приборов добиваться снижения собственных пока-
заний до желаемого уровня. В производственных условиях для ана-
лиза проб с невысокими концентрациями рекомендуется выделять
отдельные приборы с наиболее низкими собственными показаниями.
На этих приборах не следует делать анализов проб с высокими кон-
центрациями.
При весьма тщательной обработке собственные показания хоро-
шего прибора на тяжелую фракцию могут быть доведены до 5—10 х
х 10~5%. При проведении сожжения собственные показания прибо-
ров обычно выше, составляя величины от 20 • 10~5% до 40 • 10-5%.
Проверка показаний приборов, их чувствительности и точности
проводится обычно с помощью искусственных смесей воздуха с угле-
водородными и иными газами. По анализу смесей воздуха с метаном
проверяют полноту сожжения метана и точность его определений.
Анализ смесей воздуха с пропаном или бутаном позволяет проверить
точность определений тяжелых углеводородов, а суммарное сож-
жение и получающееся в результате углеродное число позволяют
судить о полноте сожжения и качественной характеристике тяжелой
фракции.
Для проверки приборов рекомендуется применять также смеси
воздуха с закисью азота и смеси воздуха с закисью азота и угле-
водородами. Закись азота N2O является вполне устойчивым соеди-
нением и попадает в тяжелую фракцию, если находится в анализи-
руемом газе. В табл. 30 приведены результаты анализов подобных
искусственных смесей с применением колонки предварительного
сожжения.
Данные, приведенные в табл. 30, а также другие исследования
показали, что в колонке предварительного сожжения имеет место
частичное разложение закиси азота. В то же время в основной сжи-
гательной колонке при тех условиях, когда углеводороды сгорают,
закись азота остается почти нетронутой.
Поэтому вследствие указанного частичного разложения закиси
азота углеродные числа для пропана и бутана бывают занижены.
Этан в условиях стационарного ртутного микроанализа конден-
сируется в первом змеевике любого из описанных выше приборов.
Однако полнота этой конденсации зависит от конструкции змеевика
и от его температуры.
При употреблении жидкого кислорода для охлаждения змееви-
ков значительная часть этана может пройти через змеевик и попасть
в легкую фракцию. При употреблении жидкого азота проскок этана
будет уже ничтожным. Ряд опытов также показал, что применение
змеевиков в отношении проскока газов не имеет преимуществ по
сравнению с простыми цилиндрическими трубками, в которые газ
поступает, проходит по более широкой части и уходит через внутрен-
нюю более узкую трубку.
Проведенные исследования показали, что закись азота при темпе-
ратуре жидкого воздуха конденсируется в первом змеевике не пол-
ностью, а часть ее попадает в основную сжигательную колонку,
а затем во второй змеевик. Это обстоятельство осложняет анализ,
16* 243
Таблица 30
Результаты анализа
Наименование № смеси Раздельный анализ Анализ с предвари- тельным сожжением Анализ с суммарным сожжением Углеродное число Примечание
тяжелая фракция легкая фракция тяжелая фракция легкая фракция
смеси
Искусственн ые смеси, 10“5°/ э
Смесь тана воздуха и ме- 69 80 312 424 1,08
То же Л 69 48 360 — — 432 1,05
69 92 313 — —— 460 1J
» 70 37 552 — — 607 1,03
» 70 37 147 — — 239 1,2
» 70 55 258 — — 313 1,00
» 70 48 264 — 376 1,2
» 71 74 147 — — 368 1,6
» 72 193 340 18 46 607 1,1
» 72 31 193 18 37 276 1,2
» 72 74 166 — 352 1,4
» 83 156 1150 — — 1316 1,1
Смесь тана Среднее 1,12
воздуха, ме- и пропана То же 2 2 392 313 248 350 . — 1459 1453 3,1 3,5
» 2 368 276 — — 1398 3,0
» 3 400 320 — — 1566 3,1 Для
» 3 349 276 — — 1625 3,9 тяжелой
» 3 368 •331 — — 1263 2,5
» 4 344 176 — ' 1298 3,0 фракции
)> 5 92 202 — — 515 3,4
» 6 169 96 — — 664 3,5
» 4 384 176 — 1351 3,0 J
Среднее — 3,2
Смесь Смеси закиси аз о т а и углеводородов
метана и за- азота 7 388 1185 465 75 1544 —
КИСИ
То же 7 432 1303 456 144 1557
» 8 828 1187 160 16 1542 —
» 9 512 1157 64 112 1680 —
» 3 752 937 204 106 1677 .—
» 3 739 766 119 119 1462
3 693 1109 454 504 1848 —
» 3 857 794 113 50 1805 —
>> 5 1008 2160 850 68
Пропан и закись 166 1724 2,0
азота 13 865 197 28
Пропан 13 1389 202 175 18 2443 1,7 Для
» 13 1342 212 193 37 2776 1,9
» 13 1454 239 230 9 1 2488 1,65 тяжелой
» » • 16 16 394 464 136 144 95 96 0 8 । 1172 1204 3,1 2,6 фракции
» 16 707 258 16 88 1890 2,5
2U
особенно в тех случаях, когда концентрация закиси азота очень ве-
лика по сравнению с концентрацией метана. Небольшой проскок
закиси азота может исказить определяемый объем СО2, по которому
судят о содержании метана. Для устранения этих погрешностей реко-
мендуется для охлаждения змеевиков применять жидкий азот и для
ускорения необходимо очень медленно пропускать газ последователь-
но через два змеевика. Если не соблюдать указанных предосторож-
ностей, то в легкой фракции (конденсат второго змеевика) можно
ожидать помимо СО2, образовавшейся при сожжении метана, неко-
торые количества N2O, а также СО2, образовавшейся при сожжении
того этана, который проскочил через первый змеевик.
Как легкая, так и тяжелая фракции могут подвергаться на упо-
мянутых приборах и дальнейшему анализу. Легкая фракция может
быть направлена в баллончик с КОН для поглощения СО2, тяжелая
фракция сжигается, а затем СО2 поглощается КОН. Проведенные
исследования показали, что определение N2O таким путем не дает
точных результатов, так как частично сорбируется твердым едким
кали. Большая часть закиси азота остается непоглощенной и может
быть идентифицирована различными способами. В частности, для
этих целей применялось разложение закиси азота накаленной медью
с освобождением азота и также разложение раскаленной до 1000°
платиной с разложением закиси азота на кислород и азот и с при-
менением спектральных наблюдений.
Следует учесть, что раздельное определение N2O и углеводородов
с помощью одного сожжения представляет большие методические
трудности. Закись азота начинает разлагаться частично при тех
температурах, которые необходимы для полного сожжения всех
углеводородов.
Микроразгонка углеводородов
Смесь микроколичеств углеводородных газов и паров для опре-
деления индивидуальных компонентов может разгоняться при низ-
ких температурах.
Первоначальные опыты по разгонке малых количеств газов про-
водились автором настоящей книги путем наблюдения за показаниями
чувствительного микроманометра, присоединенного к содержащему
сконденсированные газы змеевику, температура которого постепенно
поднималась, начиная с температуры жидкого воздуха.
На фиг. 92, а представлен образец графика, получающегося при
подобной микро разгонке, когда в газе имеются углекислота и вода,
т. е. компоненты, температуры кипения которых резко отличаются
друг от друга. По удалении дьюаровского сосуда с жидким возду-
хом через 15—20 сек. температура поднимается и углекислый газ
испаряется, что и замечается по показаниям микроманометра. Затем
в течение некоторого времени (1,5—2 мин.) зайчик микроманометра
стоит неподвижно на одном и том же делении, после чего начинается
испарение паров воды и наблюдается новая подвижка зайчика ми-
кроманометра.
245
Подобный упрощенный метод микроразгонки может давать удо-
влетворительные результаты при наличии в смеси газов., сильно
отличающихся друг от друга по упругостям паров. Разделить угле-
водороды таким путем очень трудно.
Для решения этой задачи Б. М. Накашидзе разработана уста-
новка., в которой имеется специальная колонка для микроразгонки .,
соединенная с микроманометром, а также ряд приспособлений, позво-
лявших изолировать отдельные фракции и производить повторные
Фиг. 92. Графики микроразгонки.
а: 1 — начало испарения СОа; 2 — начало десорбции и испарения НвО;
б'. 1 — этан; 2 — закись азота; 3 — пропан; 4 — бутан; 5 — н-пентан;
б — н-гексан.
их разгонки для более тщательного разделения индивидуальных
компонентов. Разгоночная колонка, помещенная в дьюаровский
сосуд, нагревается очень медленно, температура ее контролируется
помещенной в колонке термопарой, соединенной с потенциометром.
При этих условиях уже по первоначальной кривой разгонки можно
судить о составе смеси углеводородов, даже если общий ее объем
составляет несколько десятков кубических миллиметров.
На фиг. 92, б дан график подобной микроразгонки смеси угле-
водородов, в которой была еще закись азота. На горизонтальной
оси графика нанесена температура, а на вертикальной — величины
давлений, характеризующие количества испаряющегося при данной
температуре газа. Путем повторной перегонки отдельных фракций
246
можно выделить индивидуальные компоненты углеводородных га-
зов (этан, пропан, изобутан, бутан) и легких жидких углеводородов.
Если в газе присутствуют, кроме того, цикланы или алкены,
то можно выделить фракции, содержащие 2—3 компонента, которые
подвергаются уже отдельному анализу, как это описывается ниже.
Анализ микроколичеств углеводородных газов. Несколько изменяя
и дополняя схемы вышеописанных вакуумных установок, можно
построить прибор, который позволит провести детальное определение
индивидуальных углеводородных газов, а также наиболее часто встре-
чающихся неорганических газообразных веществ.
Подобный прибор разработан Б. М. Накашидзе. Он отличается
рядом методических и конструктивных особенностей, из которых
следует отметить: 1) включение в замерную дугу прибора микро-
колонки для сожжения; 2) проведение замеров газа при температуре
100° во всех тех случаях, когда определяется Н2О; 3) введение в
схему прибора микроэффузиометра для определения плотности газа.
При детальном микроанализе на углеводородные газы прежде
всего вымораживаются тяжелые углеводороды при температуре жид-
кого воздуха или, еще лучше, жидкого азота. Метан вместе с другими
неконденсирующимися примесями при этом откачивается, и эти
газы могут подвергаться дальнейшему анализу.
В сконденсировавшейся части газа, вообще говоря, могут нахо-
диться как различные углеводороды (предельные и непредельные),
так и некоторые неорганические соединения, как N2O и другие, ко-
торые не задерживаются в реактивах (КОН и Р2О5) и конденсиру-
ются при температуре жидкого азота или жидкого кислорода.
Первый этап микроанализа сконденсированной фракции заклю-
чается в проведении микро разгонки, позволяющей выделить фрак-
ции закиси азота, этана, пропана и т. д.
Если в газе нет других компонентов, кроме метановых углеводо-
родов и закиси азота, то указанные фракции будут представлять собой
эти компоненты в более или менее чистом виде.
Если же в конденсате содержатся непредельные углеводороды
и цикланы, некоторые их производные и примесь неорганических
веществ, то в выделенных фракциях могут содержаться, помимо
основного компонента, и еще одно-два вещества. Так, например,
во фракции этана можно предполагать примесь этена, а также за-
киси азота, во фракции пропана может присутствовать пропен
и т. д.
После выделения той или иной фракции проводится ее микроана-
лиз. Содержащиеся во фракции индивидуальные компоненты опре-
деляются путем сожжения фракции в упомянутой микроколонке
с добавкой некоторого количества чистого кислорода и с последу-
ющим точным замером образовавшихся СО2 и Н2О.
Избыток кислорода удаляется путем откачки до высокого вакуума
при вымораживании СО2 и Н2О в микроколонке и замерной дуге.
В результате определяются углеродные и водородные числа ана-
лизируемых углеводородов.
Углекислый газ отделяют от воды при температуре твердой СО2.
Пары воды измеряются при 100°. СО2 и Н2О удаляются поглощением
КОН и Р2О5, помещенных в ртутных затворах, снабженных специ-
альными клапанами.
Микроэффузиометр служит для определения плотности исследуе-
мой фракции. Как показали проведенные опыты, микроэффузиометр
может давать точные определения плотности газа даже при его коли-
чествах в несколько десятков кубических миллиметров.
Зная плотность газа и его углеродное и водородное числа, можно
выяснить состав фракции, даже если в ней присутствуют углеводо-
роды различных рядов.
Значительное затруднение при анализе легких фракций создает
закись азота. Наиболее рационально закись азота выделять путем
микро разгонки. Если же закись азота содержится в какой-либо из
фракций, то при сожжении в условиях яркожелтого накала плати-
новой проволоки закись азота разлагается.
Ряд опытов показал, что при соприкосновении с ртутью кислород
разложившейся закиси азота окисляет ртуть и остается только азот.
Таким образом, при разложении закиси азота и поглощении кисло-
рода оставшийся объем азота равен исходному объему закиси азота.
Присутствие азота можно проверить спектральными наблюдениями.
При сожжении смеси закиси азота с углеводородами образую-
щиеся азот и кислород удаляются при вымораживании продуктов
сожжения углеводородов, т. е. СО2 и Н2О, и откачке микроколонки
до высокого вакуума.
Для удаления сернистых соединений Б. М. Накашидзе введена
в систему прибора трубка с окисью свинца.
В результате анализа отдельных фракций можно получить де-
тальный состав первоначального конденсата. Для углеводородов
тяжелее С4 указанная методика еще недостаточно разработана.
Газы, откачанные при первоначальной конденсации и состоящие
из метана, азота, водорода, кислорода и окиси углерода, также мо-
гут подвергаться дальнейшему анализу. Если количества этих газов
достаточно велики, их можно проанализировать на обычных при-
борах для общего анализа (типа ТИ и др.) или на аналогичных при-
борах для полумикроанализа.
Если количество упомянутых газов мало, то на той же микроана-
литической установке эти газы можно подвергнуть сожжению и дру-
гим упомянутым операциям. Зная плотность газовой смеси, коли-
чество взятого для сожжения кислорода и определив углеродные и
водородные числа, можно определить состав упомянутой смеси. После
поглощения продуктов сожжения (СО2 и Н2О) в остатке будут нахо-
диться азот и не вошедший в реакцию кислород. По плотности этой
смеси можно определить содержание обоих газов.
Для удаления кислорода можно использовать и другой метод —
окисление кислородом красной меди. Для этой цели медная спи-
раль, накаливаемая электрическим током, помещается в отдельной
колонке.
Остающийся азот может содержать примесь редких газов. Ана-
248
лиз на редкие газы производится особыми методами, описываемыми
в главе VIII.
По вышеописанной схеме можно проводить полный микроанализ
газовой смеси, состоящей из углеводородов и ряда неорганических
газов (СО2, H2S, О2, N2, N2O и др.) в количествах, начиная
с нескольких кубических миллиметров.
§ 4. Микроанализ на углеводородные газы без применения
низких температур
Одним из методов, применяемых для суммарного определения
микроколичеств углеводородов, является сожжение этих углеводо-
родов с последующим наблюдением уменьшения объема. Необходимо
отметить, что при всяком сожжении углеводородов в воздухе или
кислороде в результате реакции происходит некоторая контракция
(уменьшение объема), поскольку объем образующейся углекислоты
меньше объема вошедшего в реакцию кислорода. При использовании
этого метода анализируемый газ прежде всего освобождается от угле-
кислоты. Можно увеличить чувствительность метода, если наблюдать
не только контракцию от сожжения, но и провести поглощение обра-
зовавшейся углекислоты. Анализируемый газ в этих случаях вво-
дится в камеру, которая имеет платиновую спираль, накаливаемую
электрическим током. К этой камере присоединена трубка с жидкой
щелочью. После ввода газа и установления уровня щелочи включают
ток и производят сожжение газа. Анализируемый газ, разумеется,
должен содержать достаточное количество кислорода, чтобы сож-
жение произошло полностью. Трехминутного накала обычно бывает
достаточно для проведения полного сожжения. После окончания
сожжения наблюдают за положением уровня щелочи. Первоначально
вследствие повышения температуры имеется даже повышенное давле-
ние в камере. После охлаждения, когда камера примет первоначаль-
ную температуру, смещение уровня щелочи вследствие падения да-
вления в камере служит показателем концентрации сгоревших угле-
водородов.
Подобная методика, разумеется, не может быть точной вслед-
ствие колебаний температуры. Иначе говоря, трудно ожидать, что
при помощи такого метода можно определить концентрацию меньше
десятых долей процента.
Для повышения чувствительности подобного прибора можно
применить компенсационную камеру, в которой имеется атмосфер-
ный воздух и где также имеется спираль, нагреваемая электриче-
ским током. Соответствующим подбором сопротивления можно до-
биться того, что при наличии атмосферного воздуха в обеих камерах
уровень щелочи не будет смещаться при накале обеих спиралей.
Если в одну из камер ввести анализируемый газ, то смещение
уровня щелочи будет показывать содержание углеводородов или
вообще любых горючих веществ.
В приборе (фиг. 93) показания манометра (узкой трубки со ще-
лочью) не точно пропорциональны концентрации углеводородов,
249
поскольку понижение уровня щелочи тормозится постепенным сни-
жением давления в компенсационном баллоне. Зная объем баллона
и диаметр трубки, нетрудно сделать расчеты для калибровки при-
бора на действительное содержание углеводородов в смеси.
При аккуратной работе на приборе можно достигнуть чувстви-
тельности порядка 0,01%.
Для микроанализа на углеводороды используются различные
методы, основанные
Один из методов
Впуск
анализируе-'"*-
мого газа
на поглощении углекислоты баритовой водой,
основан на определении появления углекислого
бария в виде мути или в виде от-
дельных кристаллов. Схема подобного
прибора представлена на фиг. 94а.
Анализируемый газ очищается от
углекислоты и поступает в колонку
сожжения. После этой колонки газ
попадает в камеру, где имеется пла-
U типовая петля с пленкой баритовой
воды.
Кон
Фиг. 93.
Наблюдая в микроскоп за момен-
том появления первых кристаллов
углекислого бария, можно судить q
содержании в газе углекислоты и по
углекислоте о содержании углеводо-
родов. Подобный прибор должен быть
предварительно прокалиброван ис-
кусственными смесями, содержащими
углеводороды. Примером калибровки
является диаграмма, изображенная
на фиг. 946.
В другом приборе (фиг. 95) ана-
лизируемый газ попадает в камеру
с платиновой спиралью, где и про-
изводится его сожжение. Как в этом, так и в предыдущих слу-
чаях предполагается, что микроанализу на содержание углеводо-
родов подвергается газ, содержащий достаточное для сожжения ко-
личество кислорода. Если кислорода мало, то его нужно добавить.
После сожжения поворачивают кран с широким ходом и через него
в камеру опускают петлю с пленкой баритовой воды (фиг. 95). Для
набора баритовой воды служит стеклянная трубка с отверстием 7.
Такая же трубка, содержащая соляную кислоту 2, служит для ней-
трализации баритовой воды. Первоначально платиновую петлю оку-
нают в раствор баритовой воды, а затем производят определение;
далее пленку с кристаллами барита окунают в трубку с соляной
кислотой. Широкие трубки прибора, в которых имеется твердая
щелочь, позволяют манипулировать указанными трубками. Выдви-
гая их из притертых охранных трубок, можно проводить указанные
операции. Твердая щелочь в широких трубках препятствует поступ-
лению углекислоты из атмосферного воздуха внутрь прибора. Анализ
на подобном приборе проводится достаточно быстро. Однако точность
250
Фиг. 94а. Баритовый прибор.
7 —показания прибора № 1; 2—показания прибора №* 2; J — показания
прибора № 3.
таких определений невелика и лежит при аккуратной работе в пре-
делах ±15—20%.
Более точный метод разработан в результате дальнейшей моди-
фикации указанных приборов, в которых определяется титр бари-
товой воды до и после поглощения углекислоты. Подобные приборы
применяются при газовой съемке [28]. Схема прибора приведена
на фиг. 96.
251
Анализируемый газ, очищенный от щелочи в барботерах, посту-
пает в камеру сожжения, откуда идет в специальные поглотители с
баритовой водой. После окончания сожжения баритовую воду ти-
труют соляной кислотой с применением в качестве индикатора фенол-
фталеина.
Употребляя разбавленные растворы соляной кислоты, можно до-
биться весьма высокой чувствительности определений СО2 — порядка
В другой модификации этого прибора производится параллель-
ное определение как метана, так и суммы более тяжелых углеводоро-
Фиг. 95.
дов с применением угля в каче-
стве сорбента.
Анализируемый газ разделя-
ют на две части, из которых одна
идет непосредственно на суммар-
ное сожжение, а другая прохо-
дит предварительно через уголь
и затем уже через колонку сож-
жения. После колонок сожжения
каждая из двух частей газа по-
ступает в поглотитель с бари-
товой водой, где и производится
определение СО2 описанным вы-
ше способом.
Для более детального ана-
лиза углеводородных газов на
основе вышеупомянутых прибо-
ров Н. М. Туркельтаубом [29]
разработан так называемый «хро-
матографический» газоанализа-
тор, основанный на применении
метода М. С. Цвета.
Как и в других приборах
для микроанализа на углеводо-
роды, анализируемый газ пред-
варительно проходит через по-
глотители 2 и 3 с раствором
КОН и трубку 4 с твердый! КОН (фиг. 97). В тех случаях, когда
через прибор требуется пропускать совершенно чистый и лишенный
горючих газов воздух, он впускается через колонку предварительного
сожжения 7, присоединенную к первому поглотителю.
Проведение анализа начинают с промывки очистительной системы
исследуемым газом в количестве 200—400 мм.
Затвор 5 содержит на дне немного ртути, а поверх нее воду.
После этого анализируемый газ через краны 6 и 7 разделяют на
два параллельных потока. Одна часть газа проходит через сжига-
тельную колонку 73, где сгорают все углеводороды, после чего посту-
пает в поглотительный сосуд 75, содержащий титрованный раствор
едкого бария (0,005 N), а затем в аспиратор 9, заполненный водой и
252
снабженный уравнительной склянкой 11. Аспиратор проградуиро-
ван по объему, что и позволяет замерять количество взятого для
анализа газа.
Вторая часть газа проходит через «хроматографическую» трубку
25, содержащую 10 г угля КАД (с равновесной влажностью бб%).
В угле задерживаются этан и более тяжелые углеводороды. Через
барботер 28, содержащий раствор КОН, газ, содержащий метан,
поступает затем в сжигательную колонку 12, поглотительный сосуд
14 с титрованным раствором едкого бария и замеряется в аспираторе
8. В сосуды 14 и 15 вводят раствор едкого бария предварительно
из микро бюреток 20 и 21.
Фиг. 96. Титрометрический газоанализатор.
t
К прибору приключены две склянки (не показанные на фиг. 97)/
Одна из них содержит раствор Ва(ОН)2, другая — раствор НС1.
Из этих склянок производится наполнение микро бюреток 20 и 21
указанными растворами. По количеству пошедшей на титрование
кислоты (0,005 N раствор) определяют в поглотителях 14 и 15 коли-
чество двуокиси углерода, образовавшейся при сожжении углеводо-
родов. Индикатором служит фенолфталеин.
Краны 16, 17 служат для регулировки впуска газа, краны 18
и 19 — для впуска растворов в поглотительные сосуды. Отработан-
ные растворы из поглотителей 14 и 15 спускаются в сливную склян-
253
ку 22. Шарики бюреток 20 и 21 могут сообщаться с атмосферой через
защитный барботер 24 с раствором КОН и через кран 23. Краны
26 и 27 закрывают трубку с углем 25.
После того как анализируемый газ пройдет указанным образом
через прибор и соберется в аспираторах 8 и 9, включают колонку
предварительного сожжения и промывают всю очистительную си-
стему, просасывая через нее 200—400 мл воздуха. Предварительное
сожжение производится для того, чтобы освободить атмосферный
воздух от случайных загрязнений горючими газами. После этого
воздух впускается равномерно в обе секции прибора. Одна часть
воздуха проходит через ту секцию газоанализатора, где проводилось
Фиг. 97. Хроматографический газоанализатор.
сожжение всех углеводородов и поглощение образовавшейся угле-
кислоты. Проходящий воздух, промывая все соединения, способ-
ствует также перемешиванию раствора едкого бария при титровании
в поглотительном сосуде 15.
Вторая часть воздуха в количестве 400 мл, проходя через трубку
с углем 25, вымывает метан, который сжигается в колонке 12, а обра-
зовавшаяся углекислота поглощается в сосуде 14 и титруется. После
пропускания 500 мл через трубку 25 воздух направляют в сосуд 14,
предварительно заполненный свежим раствором едкого бария, а в
поглотительном сосуде 15 производят контрольный анализ на отсут-
ствие горючих в пропускаемом воздухе. После того как весь метан
вымыт из трубки 25 (пропускаемый воздух первоначально не содер-
жит никаких углеводородов) при дальнейшем пропускании воздух
начинает вымывать этан.
Выделение этана заканчивается, когда через трубку 25 пройдет
еще 2000 мл. Весь этот этан сгорает в колонке 12, и образующаяся
254
С02 поглощается в сосуде 14} где титрованием и определяется ее
количество.
В результате подобного анализа в газе определяется содержание
метана, этана и по разности СО2 от сожжения более тяжелых угле-
водородов, поскольку суммарное сожжение всех углеводородов про-
водится отдельно.
Прибор предназначен для определения очень малых концентраций
углеводородов в воздухе, в азоте или каком-либо другом газе, кото-
рый при сжигании не дает кислых продуктов.
Прибор может быть использован для определения не только угле-
водородов, но и других горючих газов и паров.
Прибор позволяет определять концентрации углеводородов, начи-
ная с 1 • 10-5%.
Перед проведением анализа необходимо проверить прибор путем
определения титра едкого бария в токе воздуха и контрольного ана-
лиза 500 мл очищенного от углеводородов воздуха.
Описанная методика хроматографического газового микроанализа
может применяться и для определения более тяжелых индивидуаль-
ных углеводородов. Для этой цели следует применять специальные
сорбенты, так как уголь слишком хорошо поглощает тяжелые угле-
водороды, начиная с пропана.
Для многих практических и научных целей представляет большой
интерес определение малых концентраций углекислого газа в той
или иной газовой смеси. Совершенно очевидно, что необходимо в
этом случае принять меры, чтобы в анализируемый газ не могла
попасть углекислота из какого-либо внешнего источника. В связи
с этим подобные определения, когда требуется проанализировать
газ на СО2 с чувствительностью порядка сотых или тысячных долей
процента, необходимо принимать различные меры предосторожности
и проводить анализ непосредственно на месте получения этого
газа.
Искажение результатов анализов может быть прежде всего вслед-
ствие попадания в пробу атмосферного воздуха.
Как известно, в атмосферном воздухе всегда содержится неболь
шое количество углекислоты — около 0,03%. Попадание этого воз-
духа в анализируемую пробу может значительно исказить результат
микроанализа на углекислый газ.
Другая причина, вызывающая искажение результатов анализа,
заключается в том, что всякая проба газа, помещенная в стеклянную
или иную какую-либо тару и соприкасающаяся с каучуковыми или
другими пробками, неизбежно вступает в газообмен с теми газами,
которые сорбированы стенками бутылей или каучуковыми соеди-
нениями. В частности, углекислый газ довольно хорошо сорбируется
стеклом, и если в какую-либо бутыль, бюретку, пипетку мы наберем
чистый, лишенный углекислоты газ, то после весьма короткого пре-
бывания в этой посуде газ будет уже содержать заметное количество
углекислоты, выделившейся из стенок бутыли или бюретки. Во всех
тех случаях, когда требуется определять малые концентрации угле-
кислоты и приходится производить отбор пробы, необходимо тща-
255
тельно подготовить тару для этой пробы путем предварительной
ее дегазации длительным нагревом и созданием высокого вакуума.
Только в этих случаях можно предполагать, что количество газов,
выделившихся из стеклянных стенок, будет невелико.
Иметь такую чистую пробу и устранить влияние выделения газов
из стенок очень трудно. Поэтому рекомендуется эти определения про-
водить так, чтобы анализируемый газ непосредственно на месте его
получения поступал в аналитический прибор.
В известной степени все сказанное относится и к другим газам,
но наибольшее значение это имеет для углекислого газа.
§ 5. Бактериальный метод определения углеводородных газов
Для определения качественного состава углеводородных газов
предложено воспользоваться способностью некоторых видов бакте-
рий окислять определенные углеводородные газы.Эти бактерии могут
развиваться в атмосфере только определенных углеводородов. Есть
бактерии, способные окислять метан, эти же бактерии могут окис-
лять также пентан, гептан и более тяжелые углеводороды, но не
окисляют такие углеводороды, как этан и пропан. Некоторые бак-
терии окисляют этан и более тяжелые углеводороды, но не окисляют
метан. В табл. 31 приведены данные о способности этих бактерий
к окислению различных углеводородов [30].
Таблица 31
Использование газообразных и жидких углеводородов бактериями
Культура бактерий Углеводород
ме- тан ЭТЙН X Н "Ан -И
Метанокисляющие
Этанокисляющие - -f- ! -т- ? — -г- -р*
Пропанокисляющие — — -г j У У У ! У
Окисляющие пентан и высшие ! I
углеводороды — - I -- ! У ! i J_ । i I i
Указанные обстоятельства позволили предложить способы опре-
деления углеводородов в газах с помощью бактерий (исследования
Могилевского, Боковой, Кузнецовой и Кузнецова).
Газовую смесь предварительно очищают от жидких углеводородов,
чтобы иметь возможность установить присутствие в газе их низших
гомологов. Для этой цели газ пропускают через специальные бак-
териальные фильтры, которые представляют собой стеклянную ко-
лонку, заполненную песком с соответствующей культурой бакте-
рий. Развитие бактерий, окисляющих метан в газе, очищеннол! от
этана и тяжелых углеводородов, служит доказательством присутствия
метана. При определении газообразных гомологов метана необходима
256
дальнейшая очистка газа. Так, для определения наличия этана газ
должен очищаться от пропана и более тяжелых углеводородов.
Развитие бактерий, окисляющих этан, в газе, предварительно
очищенном от паров жидких углеводородов и пропана, служит дока-
зательством присутствия этана. Аналогичным образом можно опре-
делить присутствие пропана.
Эта методика может служить для качественного и полуколиче-
ственного анализа углеводородных газов. Интересно применение
водородокисляющих бактерий, что позволяет качественно определить
присутствие водорода в природных газах. Под каждый колпак, куда
вводится анализируемый газ, вводят помимо культуры бактерий
определенное количество воздуха, поскольку действие бактерий за-
ключается именно в окислении углеводородов с помощью кислорода
воздуха.
Бактериальный метод обладает довольно высокой чувствитель-
ностью. Концентрации углеводородов в десятые и сотые доли про-
цента и даже ниже могут быть уже замечены.
§ 6. Определение вредных примесей в воздухе
Определение вредных примесей в воздухе представляет одну из
задач санитарно-химических исследований в целях охраны труда и
техники безопасности [31].
Следует отметить, что в воздухе промышленных предприятий
и в атмосфере могут присутствовать не только примеси ядовитых
газообразных веществ, но и вредные твердые вещества, находящиеся
в воздухе в виде пыли; в воздухе может присутствовать также в виде
мельчайших капелек «туман» вредных жидких веществ.
В связи с этим полный анализ воздуха на вредные примеси за-
ключается не только в проведении специального газового анализа
или микроанализа, но и обычного анализа тех твердых и жидких
веществ, которые тем или иным путем улавливаются из анализируе-
мого воздуха.
Применяемые методы в большинстве случаев заключаются в том,
что исследуемый воздух предварительно пропускается через погло-
титель, улавливающий определяемую вредную примесь, а затем
поглотитель исследуется обычными химическими методами. Очень
часто для определения вредных примесей в воздухе пользуются
экспресс-методами, дающими приближенные количественные данные.
Так как по определению всех вредных примесей в воздухе
имеются специальные руководства [31], то в настоящем параграфе
кратко даются основные принципы лишь тех методов, которые при-
меняются для определения газообразных вредных веществ. Эти
газообразные вредные вещества могут присутствовать в воздухе про-
мышленных предприятий обычно в очень небольших концентрациях.
Галоиды и галоидоводородные кислоты. Для
определения галоидов и галоидово до родных кислот пользуются реак-
циями с мышьяковистой кислотой и нитратом серебра с применением
нефелометрических или титрометрических измерений, аналогично
17 в. А. Соколов.
257
тому, как это описано в главе V. Для определения фтористоводород-
ной кислоты и ее солей используют ее реакцию с треххлористым
железом, избыток которого определяют колориметрически.
Сернистые соединения. Сернистый газ окисляют хло-
ратом калия КС1О3 до серной кислоты. В присутствии хлорида бария
образуется муть BaSO4, которая сравнивается со стандартной шка-
лой.
Сероводород определяют с помощью нитрата серебра. Окрашен-
ный в желто-коричневый цвет коллоидный раствор сульфида серебра
сравнивается со стандартной шкалой.
Окислы азота. Определение окислов азота производится
обычно колориметрическим путем с применением сульфофеноловой
кислоты (для окислов, окисленных до HNO3) и реактива Грисс-Илло-
свайя.
Цианистый водород. Для определения цианистого водо-
рода применяют колориметрический метод, используя реакцию с
Na2S4O6. Образующийся роданид дает с FeCl3 красное окрашивание.
Применяется также реакция с пикриновой кислотой, при которой
раствор приобретает оранжево-красную окраску.
Мышьяковистый и фосфористый водород. Эти
соединения окисляют до мышьяковистой и фосфорной кислот. При-
меняя раствор молибдата аммония в серной кислоте, получают голу-
бое окрашивание и проводят колориметрическое измерение.
Пары ртути. Анализируемый воздух пропускают через
раствор иода в иодистом калии. Образующийся иодид ртути дает
с солями меди в присутствии восстановителя комплексное соедине-
ние CuJ • HgJ2 розового цвета. По интенсивности окраски получаю-
щегося осадка путем сравнения со стандартной шкалой определяют
содержание ртути в воздухе.
Двуокись и окись углерода. Для определения С02
следует рекомендовать методы, описанные в главе VI, в частности
титрометрический прибор (фиг. 96—97), удалив из него очиститель-
ную систему, содержащую КОН.
Для определения СО пользуются методом с применением J2O5.
Образующуюся СО2 определяют титрометрическим или иным путем.
Углеводороды и другие горючие вещества.
В воздухе могут присутствовать помимо углеводородных газов (ме-
тан, этан и др.) также пары жидких углеводородов (бензин, керосин,
бензол и др.) и других горючих веществ (спирты, эфиры, органиче-
ские кислоты, альдегиды и т. п.). Для суммарного определения этих
горючих веществ применяют сожжение и определение образующейся
СО2. Для этой цели следует рекомендовать описанный в главе VI
титрометрический газоанализатор.
Индивидуальные газообразные углеводороды определяют мето-
дами микроанализа, описанными выше (глава VI).
Ацетилен можно определять колориметрически, применяя реак-
цию с солями закиси меди. При этом образуется красно-фиолетовое
окрашивание, обусловленное коллоидным раствором ацетиленистой
меди.
258
Бензол и толуол. Толуол нитруют до тринитротолуола;
бензол в этих условиях переходит в динитробензол. Образовавшиеся
нитросоединения извлекают метилэтилкетоном или эфиром. В первом
случае бензол с метилэтилкетоном колориметрируют с едкой щелочью,
а толуол с аммиаком и этанолом. Во втором случае, т. е. при извле-
чении эфиром, бензол колориметрируют в ацетоне, а толуол в спир-
товой среде.
Галоидные производные углеводородов.
Для определения галоидных производных производят сожжение
и продукты сожжения пропускают через мышьяковистую кислоту.
Нефелометрическим путем и с помощью AgNO3 определяют
в мышьяковистой кислоте ионы галоидов и пересчитывают на опре-
деляемое вещество.
Не содержащие галоидов углеводороды определяют при этом
по количеству СО2.
Метанол и этанол. Метанол окисляют перманганатом
калия до формальдегида, который определяют колориметрически
с фуксинсерной кислотой.
Этанол окисляют титрованным раствором бихромата калия до
уксусной кислоты. По количеству затраченного окислителя судят
о количестве этанола.
ГЛАВА СЕДЬМАЯ
ОПРЕДЕЛЕНИЕ РЕДКИХ ГАЗОВ
§ 1. Основные физические и химические свойства редких газов
Основные физические свойства редких газов приведены в табл. 32.
Из всех редких газов наименьшим удельным весом обладает
гелий. Именно это и дало возможность применять гелий для напол-
нения дирижаблей. Помимо небольшого удельного веса гелий об-
ладает и другими исключительными свойствами: температуры сжи-
жения и плавления гелия являются наиболее низкими из достигну-
тых в настоящее время; температура плавления гелия отстоит от
температуры абсолютного нуля всего на 0,8°. В табл. 32 для срав-
нения указаны также свойства водорода, кислорода и азота.
Таблица 32
Основные физические свойства редких газов
Газ Уд. вес (уд. вес воз- духа принят равным единице) при нор- мальных условиях Вес 1 л при 0° и 760лш рт. столба, г Температура кипе- ния, °C Температура плав- ления, °C 1 Критическая тем- пература, °C Критическое давле- ние, ат
Г елий 0,138 0,1784 —268,9 —272,2 —267,9 2,2
Неон 0,695 0,8985 —245,9 —248,5 —228,7 26,8
Аргон 1,378 1,7833 -185,85 —189,3 — 122,4 48,0
Криптон 2,886 3,733 — 152,9 - 156,6 —62,5 54,3
Ксенон 4,553 5,887 —107,1 — 111,5 + 16,6 58,2
Кислород ..... 1,1054 1,4290 -183 —218 —118,8 49,7
Азот 0,96736 1,2506 —195,8 —209,8 —147,1 33,5
Водород 0,06951 0,08987 -253 -259 —239,9 12,8
Все многочисленные попытки получить устойчивые химические
соединения редких газов с другими элементами оканчивались до
сего времени неудачно. Однако известно, что при тихом разряде
в гелии в присутствии ртутных паров образуется небольшое коли-
260
чество газообразного гелида ртути. Имеются указания на получение
соединений редких газов с галоидами и некоторыми другими эле-
ментами. Все подобные соединения являются очень нестойкими.
Аргон, криптон и ксенон обладают способностью образовывать
гидраты, в которых одна молекула газа связана с несколькими моле-
кулами воды. При 0° аргон дает гидрат при давлении около 100 ат,
криптон уже при 0° и 14,5 ат, а ксенон соответственно при 1,4° и
1,45 ат.
Диффузия редких газов через твердые тела
Накаленные докрасна железо и платина непроницаемы для гелия
и других редких газов. Кварц проницаем для гелия даже при ком-
натной температуре, а при высоких температурах диффузия идет
очень быстро. Различные исследователи наблюдали, что при тем-
пературе 1100° давление гелия в кварцевом баллоне в течение не-
скольких часов резко падает. При 510 и 220° диффузия также весьма
заметна. Скорость йадения давления приблизительно пропорцио-
нальна величине давления гелия. Следует заметить, что для таких
газов, как N2, О2 и СО2, кварц мало проницаем до 1000°. Дальней-
шие исследования в этой области показали, что при комнатной тем-
пературе наполненный гелием кварцевый шар поверхностью 50 см2
и с толщиной стенок 0,8 мм пропускал сквозь стенки в 1 час 1 см3
гелия. Другие исследователи дают несколько иные величины для
скорости диффузии. Были проведены измерения скорости диффузии
газов через кварцевое стекло, через стекло пайрекс и через йенское
тугоплавкое. Для азота и воздуха диффузии не наблюдалось; для
водорода не наблюдалось диффузии через стекло пайрекс и йенское
стекло до температуры 640°. Водород диффундировал заметно лишь
через кварц, а гелий диффундировал через все названные материалы.
Для стекла пайрекс скорость диффузии гелия под давлением в 1 ат
при 610° была равна 5,2 • 10-4 см^/час через 1 см2 поверхности при
толщине стенок в 1 мм. При 1200° диффузия гелия через 1 см2 кварца
при толщине в 1 мм и давлении в 1 ат составляет 0,007 см3/час. Дру-
гие измерения показали, что скорость диффузии гелия через кварц
при 480° равна 2 • 10~5 см3/час через 1 см2. Подобные разногласия,
вероятно, объясняются различием применяемых материалов.
Через обыкновенное стекло гелий диффундирует значительно
медленнее. При 400° диффузия через стекло в 100 раз медленнее,
чем через кварц.
При помощи очень чувствительного метода определения гелия
была обнаружена диффузия гелия из атмосферы сквозь стекло в ва-
куум при комнатной температуре.
Методы определения редких газов
Обычные методы газового анализа, основанные на поглощении
отдельных компонентов газовой смеси различными химическими
реагентами, при определении редких газов совершенно неприменимы,
поскольку основным отличительным свойством всех редких газов
261
является их химическая инертность. Невозможность определения
редких газов химическим путем привела к тому, что все современные
методы анализа на редкие газы базируются главным образом на их
физических свойствах.
Смесь редких газов может состоять максимум из пяти компонен-
тов: Не, Ar, Ne, Кг, Хе. Радиоактивную эманацию мы сюда не
включаем, так как ее количество, могущее встретиться в газовой смеси,
настолько ничтожно, что не повлияет на результаты анализа.
Радиоактивные газы определяются с помощью специальных методов.
Чтобы провести полный анализ на редкие газы с определением
каждого из упомянутых пяти компонентов, необходимо предвари-
тельно удалить из испытуемого газа все остальные составные части
газовой смеси, кроме редких. Определение каждого из пяти компо-
нентов при помощи физических методов очень трудно, особенно при-
нимая во внимание, что некоторые из редких газов (Кг, Хе) встре-
чаются лишь в крайне незначительных количествах. Вследствие
этого методика полного анализа на редкие газы до настоящего вре-
мени является еще весьма сложной и несовершенной. Для большин-
ства целей нет надобности определять все пять редких газов, а до-
статочно определить лишь гелий и аргон. Поэтому на практике
часто пользуются методами и аппаратами, позволяющими определять
гелий и аргон, пренебрегая неоном, криптоном и ксеноном, тем более
что содержание их очень мало по сравнению с гелием и аргоном.
Для определения редких газов существует ряд методов и довольно
много различных конструкций приборов.
Разработаны методы, в которых предварительно из смеси газа
удаляют химическим путем все составные части, кроме редких. После
этого при помощи угля, охлаждаемого жидким воздухом, сумму
редких газов разделяют на две фракции. Откачанная из угля фрак-
ция представляет собой сумму Не + Ne, а поглощенная Аг + Кг -р
+ Хе. Содержание Ne, Кг, Хе обычно очень мало, поэтому сумму
Не -р Ne принимают практически за гелий, а сумму Аг + Кг + Хе—
практически за аргон.
Для тех случаев, когда представляет интерес только содержание
гелия, существуют методы, при которых испытуемый газ непосред-
ственно впускают в ампулку с углем, охлаждаемым жидким воз-
духом. Из угля откачивают только гелий и неон.
Известны также методы, в которых после удаления из газа всех
компонентов, кроме редких, определяют и гелий и аргон путем
определения физических свойств смеси (удельный вес и др.), пренебре-
гая содержанием Ne, Кг и Хе.
Помимо упомянутых существуют еще специальные методы для
определения неона в смеси с гелием и методы полного анализа при-
родного газа с определением всех редких газов.
Очистка редких газов от прочих газообразных элементов и соединений
Если в газе необходимо определить не только Не и Ne, но и Аг,
Кг и Хе, то его обязательно приходится подвергать очистке для уда-
ления всех компонентов смеси, кроме редких. Такие газы, как СО2,
262
H2S, можно легко удалить путем поглощения едким кали., кисло-
род — при помощи пирогаллола или гидросернистого натрия, а угле-
водороды, водород и окись углерода — путем сожжения. Удобнее
всего эту очистку производить, анализируя газ. При определении
редких газов состав остальной части представляет также интерес,
а потому общий анализ газа все равно приходится делать. Поэтому
для экономии времени и испытуемого газа определение редких
газов лучше всего производить в остатке после общего анализа,
состоящем из азота и редких газов.
Удаление СО2, О2, Н2, СО не представляет больших затрудне-
ний. Сожжение газа можно проводить с окисью меди или в чистом
кислороде с накаленной платиновой спиралью. Применялся и такой
способ удаления горючих составных частей: углеводороды погло-
щались металлическим кальцием при 600—700°, а водород сжигался
в чистом кислороде в присутствии палладия. Сожжение углеводо-
родов в чистом кислороде с накаленной платиновой спиралью обеспе-
чивает полноту сожжения. Однако сожжение с окисью меди имеет
то преимущество, что не требуется вводить кислород, который дол-
жен быть очень чистым при точном анализе на редкие газы.
Для удаления азота из смеси редких газов пользуются щелочно-
земельными и щелочными металлами, главным образом кальцием,
магнием и литием. Применяют также и смеси металлического маг-
ния с соединениями кальция, который при нагревании восстанав-
ливается и быстро и полностью поглощает азот. В табл. 33 приведены
данные, характеризующие поглощение азота различными металлами
при назревании до светлокрасного каления.
Кальций является наиболее употребительным металлом для по-
глощения азота. Реакция кальция с азотом становится заметной
Таблица 33
Скорость поглощения азота различными металлами
Металл Количество поглощен- ного N2, с.и3 Металл Количество поглощен- ного N2, гл«3
после 15 мин. после 1 часа после 15 мин. после 1 часа
1) 1 г Mg 2) 1 г Li 3) 1 г Са ; 4) 1 г Mg, 5 г СаО . . 5) 1 г Mg, 3 г СаО . . 6) 1 г Mg, 8 г СаО . . 80 94,5 14,5 73,5 112 50 31,4 7) 1 г Mg, 5 г СаО (свежепрокаленной) 8) 1 г Mg, 5 г СаО (свежепрокаленной), 0,1 г Na 9) 1 г Mg, 5 г СаО (свежепрокаленной), 0,25 г Na 10) 1 г Mg, 5 г СаО (свежепрокаленной), 0,11 г Li 86,4 201 196 169 1 1 122,5 287 , 326,2 228 i
263
уже при 300°. О поглощении азота при более высоких температурах
мнения различных исследователей расходятся. Некоторые считают,
что кальций начинает поглощать азот лишь при температуре около
800°, другие полагают, что оптимальная температура поглощения
лежит в пределах 650—700°. Высказывалось мнение, что различные
препараты кальция ведут себя по-разному. «Активные» поглощают
азот при меньшей температуре; «неактивные» реагируют лишь
выше 800°.
Металлический литий начинает поглощать азот уже при комнатной
температуре [32]. При 440° реакция проходит очень бурно с выде-
лением большого количества тепла. Смесь Li, Na и CaF2 поглощает
азот уже при температуре около 200°.
§ 2. Определение гелия и неона
Прибор Лукашука и Хлопина для определения гелия [33], как
и другие аналогичные приборы, представляет собой в основном мано-
метр Мак Леода, к которому присоединена трубка с углем. Уравни-
тельный резервуар 7 (фиг. 98) через кран присоединяется к насосу,
и путем откачивания воздуха или его впускания уровень ртути в са-
мом баллоне микроманометра 2 можно поднять или опустить. К микро-
манометру 2 присоединена U-образная трубка 5, в расширении кото-
рой помещается активированный уголь. U-образная трубка оканчи-
вается краном 4, присоединяемым к источнику газа; через другой
ход этого крана можно производить откачку соединительной
трубки.
Предварительно прибор эвакуируют через кран 5 при помощи
хорошего масляного или ртутного насоса. При откачке ртуть из бал-
264
лона 2, разумеется, должна быть переведена в баллон 1. Закрыв
кран 5, погружают U-образную трубку с углем в жидкий воздух.
Уголь поглотит остатки воздуха. Объем остающегося после этого
воздуха измеряется микроманометром в трубке 6 точно так же, как
в дальнейшем будет измеряться объем гелия. Если остающийся объем
воздуха невелик (0,1—0,5 л/л/3), то приступают к анализу, для чего
впускают в прибор известное количество испытуемого газа (напри-
мер, 100 гл/3). Впустив газ, закрывают кран 4. Уголь, находящийся
в расширении U-образной трубки, поглотит все газы, кроме гелия
и неона. При впуске и поглощении газа ртуть в трубке 7 поднимают
высоко, чтобы газ никоим образом не мог попасть в баллон 2. Для
полного поглощения всех газов, кроме Не + Ne, обычно вполне
достаточно 10—15 мин. Для наблюдения за степенью очистки гелия
применяется разрядная трубка 8. Когда разрядная трубка покажет
чистый спектр гелия, опускают ртуть в баллоне 2. Гелий из угля
переходит в баллон 2, а затем, подняв ртуть, загоняют гелий в ка-
пилляр б, объем которого невелик (обычно 1 гл/3, разделенный на
100 частей). По разнице в уровнях ртути в капилляре бив трубке 7
можно вычислить объем гелия. При большом количестве гелия одной
откачки может оказаться недостаточно. Поэтому рекомендуется произ-
водить две или три откачки, пользуясь краном 9.
После нескольких анализов уголь в U-образной трубке следует
прогреть до 300—400° при одновременной откачке масляным или
ртутным насосом.
Другой вариант прибора для определения гелия представляет
собой бюретку 7 (фиг. 99, а), имеющую вверху трехходовый кран,
к которому присоединен баллон с углем [42]. Бюретка имеет в ниж-
ней части расширение J, которое соединяется с уравнительным сосу-
дом 4 с ртутью; в этот же уравнительный сосуд входит и конец мано-
метра. При помощи крана 6 и какого-либо насоса (ручного, водоструй-
ного или масляного) можно поднимать и опускать уровень ртути
в бюретке на желаемую высоту. Верхняя часть бюретки представ-
ляет собой узкую трубку емкостью около 1 см3, разделенную до
0,01 см3, средняя часть бюретки имеет объем от 10 до 50 см3, разде-
ленный до 0,2 см3, полная же емкость бюретки равна 300—400 см3.
Манометр 5 имеет шкалу, проградуированную на миллиметры.
Предварительно через краны 7 и 8 откачивают адсорбированные
газы из угля, нагревая его до 300—400°. Откачку лучше всего произ-
водить масляным насосом, а затем можно откачивать и бюреткой 7,
пользуясь ею как ртутным насосом. Когда подготовка прибора закон-
чена, в бюретку через кран 7 набирают пробу газа. Объем бюретки
до метки 9 точно измеряется заранее. Установив ртуть на этой метке,
отмечают показания манометра, откуда и вычисляется объем газа,
взятый для анализа.
Повернув кран 8, сообщают бюретку с баллоном 2, охлаждаемым
жидким воздухом. Уровень ртути доводят до крана бюретки 8. Газ
из трубки между кранами 7 и 8 откачивают через кран 7, затем,
закрыв кран 7, сообщают трубку с баллоном 2 для поглощения
остатков газа.
265
Как и во всех других приборах для определения гелия, перед
откачиванием гелия необходимо несколько раз опустить и поднять
ртуть в бюретке, чтобы собрать приставшие к стенкам бюретки пу-
зырькилгаза. Собранные остатки газа также направляют в баллон 2.
После "того как газ пробудет здесь 10—15 мин., можно отка-
чать гелий, для чего следует опустить уровень ртути в бюретке и
открыть кран 8. Затем, закрыв кран 7 так, чтобы между его тремя
Фиг* 99.
ходами совершенно не было сообщения, поднимают ртуть в бюретке.
При заметном количестве откачанного гелия его направляют в шарик
между кранами 7 и 8. Откачку продолжают, пока не будет откачан
весь гелий. Откачанный газ собирают таким образом в шарик, а затем
переводят в бюретку и тщательно измеряют. Поскольку объем трубки
с шариком, предварительно измеренный, известен, так же как и
объем бюретки, легко ввести соответствующую (очень неболь-
шую) поправку на гелий, находящийся в трубке между кра-
нами 7 и 8.
Спектральные наблюдения можно производить в верхней части
трубки, идущей от баллона 2 к манометру 5, пользуясь впаянными
электродами или безэлектродным разрядом.
Определение чистоты гелия. Вышеописанные приборы предна-
значены главным образом для определения малых концентраций
редких газов. На практике могут встретиться и такие случаи, когда
266
требуется определить в каком-либо редком газе примесь иных газо-
образных веществ, иначе говоря, требуется определить чистоту об-
разца того или иного редкого газа.
Для качественного решения этого вопроса можно воспользоваться
спектральными наблюдениями; для количественного же определе-
ния примесей необходимо проводить специальный анализ. Часто
о чистоте можно судить на основании определения удельного веса
газа или иных физических свойств (см. главы VIII и IX).
Для анализа гелия в целях определения суммарного содержания
Примесей можно использовать прибор, изображенный на фиг. 99, б.
Анализируемый гелий забирается в бюретку 1, замеряется и направ-
ляется в сосуд с углем 2, охлаждаемый жидким воздухом. Спустя
некоторое время (15—20 мин.), гелий откачивают с помощью ртутного
насоса 3 в бюретку, где и замеряют. Разница в измерениях пока-
зывает содержание примесей.
Чистоту того или иного редкого газа можно определять и дру-
гими описанными в настоящей главе приборами (например, изобра-
женным на фиг. 99, а). Анализируемый гелий или неон пропускается
через уголь, предварительно откачанный и охлаждаемый жидким
воздухом. Гелий или неон пол-
ностью откачивается. После это-
го жидкий воздух удаляют и
из угля откачивают задержав-
шиеся в нем газы, представляю-
щие собой примеси, имевшиеся
в гелии или в неоне.
Прибор с применением уг-
ля, охлаждаемого сухим льдом.
Этот прибор, сконструирован-
ный А. А. Черепенниковым,
отличается от других подобных
приборов с угольным поглоще-
нием тем, что в нем вместо жид-
кого воздуха применяют для
охлаждения угля твердую угле-
кислоту [35]. При температуре
—78° из угля можно откачать
аргон и азот, так что непосред-
ственное определение гелия этим
способом невозможно. По этой
причине указанный прибор скон- фиг> юо.
струирован таким образом, что
позволяет произвести особую
фракционировку газа, подвергая гелий постепенной очистке. Прибор
изображен на фиг. 100. 7 и 2 — большие резервуары емкостью до
500 см3 каждый, 3— колокол, в который во время анализа собирается
газ, содержащий гелий, 4 и 5 — баллончики с углем, 6 — разрядная
трубка, 7 — малый колокол, 8—манометр. Предварительно в при-
267
боре создают вакуум} затем вводят анализируемый газ в количестве
500 см3, который поглощается частично углем в баллонах 4 и 5,
охлаждаемых твердой углекислотой. Спустя некоторое время после
перевода газа в прибор приступают к откачке газового остатка, в
котором находится весь гелий, заключавшийся в первоначальной
порции газа.
Откачиваемый не поглощенный углем газ собирают в колоколе 3.
Откачку повторяют несколько раз, причем из угля частично выде-
ляется поглощенный газ. Весь гелий находится в откачанном газе.
После четырех-пяти откачек в колоколе собирается около половины
первоначального объема пробы. Остальной газ, находящийся в угле
и не содержащий гелия, удаляют из прибора откачкой с прогревом
баллонов с углем. Операции поглощения и откачки газа продолжают
много раз, пока объем газа не перестанет уменьшаться. Этот остаю-
щийся объем представляет собой гелий 4- неон. Чистоту отдельных
фракций определяют спектроскопически в разрядной трубке. Остав-
шийся объем гелия измеряют в малом колоколе 7.
Вследствие большого числа фракционированных перегонок опре-
деление гелия на этом приборе отнимает в несколько раз больше
времени, чем определение на аналогичных приборах с применением
жидкого воздуха.
§ 3. Раздельное определение аргона и гелия
В приборах для раздельного определения гелия и аргона [36]
производится разделение всех редких газов на две группы: Не-|- Ne
и Аг Кг + Хе. Так как содержание Ne, Кг и Хе обычно мало
по сравнению с Не и Аг, то первую фракцию практически принимают
за Не, а вторую за Аг. Первоначальный прибор, разработанный для
раздельного определения Не и Аг, иМеет сходство с прибором, ко-
торым пользовались для очистки редких газов при их открытии в
атмосферном воздухе. Разница заключалась в том, что в приборе
для определения Не и Аг имеется трубка с углем, охлаждаемая жид-
ким воздухом, что и позволяет очищенную смесь редких газов раз-
делить на две фракции: непоглощенную, состоящую из Не 4- Ne,
и поглощенную углем, состоящую из Аг 4- Кг 4- Хе. Для погло-
щения азота применяется металлический кальций или смесь каль-
ция и магния. Прибор Мурё (для определения Не и Аг) состоит из
бюретки, простого или автоматического ртутного насоса и системы
трубок с реагентами для очистки редких газов от всех прочих. Кроме
того, в прибор включена трубка с кокосовым углем. Для сожжения
горючих составных частей газа в приборе употребляют окись меди,
помещенную в тугоплавкую трубку и нагреваемую до светлокрасного
каления. Для удаления продуктов горения служат КОН и Р2О6.
Для поглощения азота употребляют металлический кальций, поме-
щенный, как и окись меди, в тугоплавкую трубку и нагреваемый
до светлокрасного каления.
В приборе предварительно создают полный вакуум, прогревая
при этом уголь, окись меди и кальций до температуры 300° для уда -
268
ления абсорбированных газов. Испытуемый газ вводят в бюретку,
а затем при помощи ртутного насоса заставляют газ циркулировать
по системе трубок с КОН, Р2О5 и СпО и Са, нагревая при этом трубки
с СиО и Са до светлокрасного каления. Время от времени, закрыв
кран бюретки, весь несгоревший и непоглощенный газ откачивают
в бюретку и измеряют его объем. Когда этот объем перестанет умень-
шаться, можно считать, что он состоит только из редких газов. За
степенью очистки можно следить по спектру, наблюдаемому при по-
мощи разрядной трубки.
Сожжение углеводородных газов при помощи окиси меди пред-
ставляет значительные трудности. Процесс идет очень медленно,
и сгорание нацело последних остатков углеводородов происходит
Фиг. 101.
с большим трудом. Для улучшения процесса рекомендуется в при-
боре проводить два цикла сожжения. В первом сгорает основная
масса углеводородов, а во втором сгорают остатки. На фиг. 101 изо-
бражен подобный прибор с двумя циклами, с усовершенствованиями
А. А. Черепенникова [35]. В этом приборе имеются два ртутных ав-
томатических насоса и две системы трубок с КОН, Р2О5, СиО и Са.
Для большей точности измерения объемов рядом с бюреткой 1 поме-
щен барометр 2. Главная масса газов сжигается и поглощается в ле-
вом цикле прибора, остаток собирается в маленькую бюретку 3, и
процесс заканчивается в правом цикле прибора.
Остаток, состоящий только из редких газов, разделяется на две
фракции, для чего его заставляют циркулировать через трубку с
углем 4, охлаждаемую жидким воздухом. Вследствие того, что оста-
ток редких газов обычно невелик, угля в трубку помещают немного —
обычно 0,5 г. Когда по спектру убеждаются, что непоглощенными
269
остались лишь гелий и неон, их откачивают в бюретку и измеряют.
Сумма Аг + Кг 4- Хе определяется по разности или непосредственно
путем откачки в бюретку и измерения. Для тех случаев, когда тре-
буется определить в образце газа только гелий (+неон), А. А. Чере-
пенников ввел к прибору добавление в виде особого баллона
с б—8 г активированного кокосового угля. Это добавление пред-
ставляет собой особый угольный цикл.
На указанном приборе можно со значительной точностью опре-
делить как Не (4-Ne), так и Аг(+Кг4~Хе), однако сложность
этого прибора, особенно с дополнительными циклами, является его
недостатком.
На фиг. 102 изображен другой прибор для раздельного определе-
ния редких газов [37]. В этом приборе в качестве основного поглоти-
теля применяют металлический кальций, поскольку опытными ра-
Фиг. 102.
ботами установлено, что металлический кальций, нагретый прибли-
зительно до 700°, поглощает все газы, кроме редких. Из всех газов,
кроме редких, только водород поглощается металлическим кальцием
несколько хуже, чем остальные газы. Прибор состоит из следующих
частей: 7—трубка с Р2Об, 2 — колба, объем которой точно изве-
стен, 3 и 4 — микроманометры, 5 и 6 — трубки с активированным
углем, 7 — тугоплавкая трубка с металлическим кальцием, нагре-
ваемая через обмотку электрическим током, 8 — трубка с палладие-
вым катализатором, 9 — ртутный манометр.
Предварительно весь прибор эвакуируют при прогреве трубок 5,
6 и 8. Производят полную откачку прибора, что проверяется под-
нятием ртути в микроманометре 3 и капилляре 10.
Затем в прибор впускают газ. О количестве впущенного газа
судят по показанию манометра 9} поскольку объем отдельных частей
прибора точно известен согласно предварительной калибровке. На-
гревая трубку 7, добиваются полного поглощения всех газов, кроме
редких. Остаток, состоящий из редких газов, измеряют обычным
270
о
Фиг. 103.
способом в капилляре 10. Если в газе присутствует водород, то его
сжигают, добавив к газу кислород, при помощи палладиевого ката-
лизатора, нагреваемого в трубке 8 небольшой электрической печью.
Кислород подводят к прибору из от-
дельной нагреваемой трубки с мар-
ганцевокислым калием. Спектроско-
пические наблюдения производят в
капилляре 10, концы которого обмо-
таны снаружи проводами вторичной
обмотки индукционной катушки.
Остаток, состоящий только из редких
газов, разделяют на две фракции:
Не + Ne и Аг + Кг + Хе, обычным
способом — при помощи угля в труб-
ке 5, охлаждаемой жидким воздухом.
Обе фракции измеряют в капил-
ляре 10.
На фиг. 103, а представлена еще
одна схема прибора для определения
Не и Аг: 1 — бюретка, 2 — уравни-
тельный сосуд, 3 — трубка с углем,
4 — тугоплавкая трубка с металли-
ческим кальцием, 5 — манометр.
Предварительно прибор подготовляют
так же, как и другие приборы для
определения Не и Аг, описанные вы-
ше. Анализируемый газ через осуши-
тельную трубку (не показанную на
фиг. ЮЗ, а) забирается в бюретку,
измеряется и направляется в труб-
ку 4, нагреваемую до 700—800°. По-
сле окончания поглощения всех газов,
кроме редких, объем последних изме-
ряется. Трубка 3 охлаждается жид-
ким воздухом, и в нее впускается
остаток, состоящий только из редких
газов; аргон, криптон и ксенон погло-
щаются углем, а гелий с неоном отка-
чиваются и измеряются.
При исследовании какого-либо газа помимо определения редких
газов всегда приходится делать общий анализ газа с определением Н2,
N2, СО2, О2, H2S, СО и суммы углеводородов. Поэтому рекомендуется
в прибор для определения гелия и аргона впускать тот остаток, ко-
торый получается после общего анализа и который состоит из азота
и редких газов с небольшой примесью остальных газов.
Определение гелия и аргона без применения низких температур
В описанных выше приборах, где применяется удаление азота
кальцием, не поглощенный углем газ представляет собой сумму
271
Не -г Ne, а поглощенный сумму Ar + Кг 4- Хе. Многочисленные
наблюдения показали, что содержание Ne, Кг и Хе в природных
газах очень мало по сравнению с содержанием Не и Аг, поэтому
не поглощенный углем газ принимают за гелий, а поглощенный —
за аргон. Во всяком случае для большинства практических целей
подобное предположение вполне допустимо. Таким образом, при
всех анализах, когда интересующим объектом является гелий или
аргон, можно считать, что смесь редких газов состоит из двух ком-
понентов. Следовательно, анализ этой бинарной смеси может произ-
водиться путем определения какого-либо физического свойства этой
смеси. Подобный метод анализа на редкие газы и был предложен
автором настоящей монографии. Анализ бинарной смеси можно про-
изводить путем измерения удельного веса или коэфициента прелом-
ления или путем сравнения теплопроводности анализируемой смеси
и «стандартного» газа. Схема прибора, основанного на подобных
измерениях, представлена на фиг. ЮЗ, б. Этот прибор состоит из бю-
ретки 7, трубки с металлическим кальцием 7, манометра 2 и газовых
микровесов или камеры для сравнения теплопроводности газа 3 [34].
После поглощения азота в трубке с кальцием 4 оставшиеся редкие
газы забираются в бюретку 7 и направляются в газовые микровесы
или камеру для сравнения теплопроводности газа 4 (более подробное
описание физических методов газового анализа дано в главе VIII).
По удельному весу газа можно рассчитать содержание двух его
компонентов — гелия и аргона (пренебрегая количеством Ne,
Кг и Хе). Газовые микровесы предварительно должны быть отка-
чаны и тщательно выверены.
В тех случаях, когда пользуются методом сравнения теплопро-
водности, применяют две камеры. В одну из них поступает анали-
зируемая смесь, а в другой находится эталонный газ, представляю-
щий собой в данном случае чистый гелий или чистый аргон. В каме-
рах имеются тонкие платиновые проволоки, нагреваемые электри-
ческим током. Проволоки соединены по схеме мостика Уитстона.
Концентрацию гелия в аргоне непосредственно показывает стрелка
гальванометра.
Теплопроводность газа в значительном диапазоне концентраций
почти не зависит от давления газа. Это позволяет при измерениях не
добиваться точного совпадения давления анализируемой смеси и
давления эталонного газа, находящегося во второй камере.
Чтобы можно было менять давление в камере с эталонным газом
во время анализа, следует к этой камере присоединить отдельную
бюретку с манометром.
Для проведения наиболее точных измерений следует вводить
поправку на изменение показаний прибора в зависимости от давле-
ния газа в камере.
§ 4. Полный анализ на редкие газы
Проведение полного анализа на редкие газы — очень сложная
задача, так как следует определить все пять редких газов по отдель-
ности, из которых три — Ne, Кг, Хе — встречаются лишь в крайне
272
незначительных количествах. Для полного анализа на редкие газы
можно предложить следующий метод.
Если мы имеем бинарную смесь газов, то содержание каждого
из компонентов может быть определено, как это описано выше, путем
измерения физических свойств смеси, например удельного веса,
теплопроводности и т. п. Так как полная смесь редких газов состоит
из пяти компонентов, то прежде всего ее необходимо разделить на
несколько фракций, чтобы в каждой фракции было не больше двух
компонентов. Дальнейшая задача заключается в испытании физи-
ческих свойств этих фракций, состоящих из бинарных смесей, откуда
и можно вычислить содержание каждого из компонентов.
Для полного анализа на редкие газы можно пользоваться при-
бором [34], схематически изображенном на фиг. 104, at 1—бюретка,
2 — трубка с металлическим кальцием, 3 и 4 — трубки с углем,
5 — гальванометр, 6 — аппарат для сравнения теплопроводности
газов, состоящий из четырех камер — одна для испытуемого газа
и три для «стандартов». При помощи переключателя можно провода
из камеры с испытуемым газом присоединять к любой камере со
стандартным газом. Прибор предварительно эвакуируют и из угля
полностью удаляют абсорбированные газы путем продолжительной
откачки при нагревании угля до 300—400°. Откачку необходимо
производить при помощи хорошего масляного или ртутного насоса.
Для работы прибора необходима лишь одна трубка с углем 3, дру-
гая же 4 может употребляться для получения вакуума в приборе
и откачки газов из угля трубки 3. Погружая трубку 4 в жид-
кий воздух и нагревая трубку 3 до 300—400°, полностью удаляем
из трубки 3 абсорбированные газы, соединив трубки 3 и 4 через
краны.
В прибор следует впускать газ, состоящий только из азота и ред-
ких газов и освобожденный от водяных паров и других газов. На-
гревая металлический кальций, удаляя из смеси азот, будем иметь
в остатке только редкие газы. Погрузив трубку 3 в жидкий воздух
и сообщив ее с остальными частями прибора через кран, разделим
редкие газы на две фракции Не + Ne и Аг + Кг + Хе. Гелий и
неон следует нацело откачать и затем направить в камеру аппарата 6,
провода которой присоединяют к камере со стандартным газом,
представляющим собой чистый неон или гелий.
Определив Не и Ne и удалив их из камеры аппарата 6, из угля
откачивают аргон, что можно сделать, подняв температуру угля до
минус 120°. Чистоту аргона можно определять также в аппарате 6,
причем провода камеры с испытуемым газом присоединяют к стан-
дартной камере с аргоном. Наконец, подняв температуру угля до
комнатной (или выше), откачивают криптон и ксенон, которые также
направляются в камеру аппарата 6, из которой предварительно дол-
жен удаляться аргон. Для определения криптона и ксенона эталоном
служит или чистый криптон, или чистый ксенон. Градуировку галь-
ванометра можно производить, очищая в этом же приборе гелий
или неон, аргон и т. п., получаемые из отдельных присоединенных
к прибору баллонов.
18 в. А. Соколов.
273
На фиг. 104, б представлена упрощенная схема прибора Хлопина,
Герлинга и Барановской £38]. Прибор состоит из ртутного микро-
манометра .7, к которому присоединена трубка из тугоплавкого
а
К насосам
Фиг. 104.
стекла 2; в трубке 2 находится металлический кальций, нагреваемый
до 650°. Помимо этого к прибору присоединена трубка с углем
служащая для поглощения тяжелых редких газов, а также и другие
части, назначение которых понятно из схемы.
274
на сравнении
спектральных
анализируемого
эталона. Для
Аргон откачивается из угля при —120°. Прибор предназначен для
определения ксенона, который и откачивался из угля при 200°.
Для определения
криптона и ксенона
предложен метод, осно-
ванный
яркости
линий
газа и
этого фракцию Аг -|-
+ Кг + Хе обогащают
криптоном и ксеноном,
для чего подвергают ее
циркуляции над углем,
охлаждаемым до темпе-
ратуры от—30 до —50°.
Криптон и ксенон по-
глощается углем, а
главная Aiacca аргона
с небольшой примесью
криптона и ксенона
3
вакуу"
впуск газа
a
остается непоглощен-
ной. Удалив непогло-
щенную часть и вы-
делив из угля нагрева-
нием фракцию Кг + Хе,
производят спектрофо-
тометрические измере-
ния интенсивности ли-
ний криптона (5870,9 А)
и ксенона (4671,2 А),
сравнивая их с интен-
сивностью этих же ли-
ний в эталонах, пред-
ставляющих собой смеси
криптона и ксенона с
чистым аргоном, не со-
держащим криптона и
ксенона.
Дальнейшее усовер-
шенствование этого ме-
б
Фиг. 105. Прибор для полного анализа на ред-
кие газы.
тода связано с изуче-
нием зависимости ин-
тенсивности линий крип-
тона, ксенона, и аргона от давления газа. В результате этого
исследования составлена табл. 34, при помощи которой можно
определить содержание Кг и Хе в смеси с аргоном по давлению,
при котором одинаковы интенсивности соседних линий криптона
18*
275
(5870,9 А) и аргона (5860,5 А) и ксенона (4671,2 А) и аргона
(4702,5 А).
Т а б л.и ц а 34
Давления, при которых интенсивности характеристических линий Кг и Хе
одинаковы с интенсивностью характеристических линий Аг
Содержание Кг в Кг 4-Аг, % 5871 (Кг)- Содержание Хе в Хе + Аг, % 4671 (Хе) = = 4702,5 А
5860,5 (А) 5912,3 (А)
ММ мм ми
0,046 12.5 0,020 12,7
0,098 4,8 11,5 0,041 5,6
0,118 4,3 10,0 0,059 2,7
0,15 2,9 7,4 0,078 1,9
0,20 2,3 6,0 — —
0,32 1,5 3,7 — —
0,43 1,4 3,0 — ,—
Эта таблица справедлива лишь в том случае, если разрядная трубка
имеет совершенно определенные размеры, которые в этих опытах
были следующими: длина трубки 6 см, диаметр 1,2 мм, емкость 5 смг.
Приборы для микроанализа на редкие газы с ртутными затворами
Содержание редких газов обычно очень незначительно, и поэтому
описанные выше приборы представляют собой вакуумные установки.
Как уже упоминалось, применение обычных вакуумных кранов в
таких установках имеет значительные неудобства и ведет, особенно
при малых концентрациях, к большим аналитическим погрешностям.
Для устранения этих неудобств автором были сконструированы для
углеводородного микроанализа описанные в предыдущей главе при-
боры, где путем использования ртутных затворов анализируемый
газ не соприкасается в вакуумной части с кранами. Схемы этих при-
боров использованы при конструировании аналогичных установок
для микроанализа на редкие газы.
Одна из установок (фиг. 105, а) предназначена для микроанализа
на гелий, а другая (105, б)—для полного микроанализа на редкие
газы.
Установка для микроанализа на гелий (Ц-Ne) состоит из ртут-
ного насоса 1 с замерной дугой 2, трубки с углем 3, барботера с
ртутью 4, трубки с фосфорным ангидридом 5 и ртутного затвора 7..
Замеренный объем анализируемого газа впускается через кран
б, освобождается от паров воды и попадает в трубку с углем 3, охлаж-
денную жидким воздухом. После впуска газа ртуть в барботере 4
отъединяет трубку 3 от впускной части. За полнотой поглощения
наблюдают по спектру, даваемому разрядной трубкой 8.
По окончании поглощения всех газов, кроме Не (Ч-Ne), с помощью
насоса 7 несколько раз откачивают Не -ф- Ne, собирают их в шари-
276
ках затвора 7, а затем замеряют в капилляре 2. После окончания
измерения удаляют дьюаровский сосуд от трубки 3, откачивают
выделяющий газ, пользуясь насосом 1 и краном 9.
Установка для полного микроанализа на редкие газы (фиг. 105, б)
состоит из ртутного насоса 1, тугоплавкой трубки с кальцием 2, трубки
с углем 3, малого ртутного насоса 4, ртутного затвора с двумя отво-
дами 5, трубки с фосфорным ангидридом 6 и ртутного затвора 7.
Откачка прибора производится через боковой отвод затвора 7
и через кран 8. В прибор впускается замеренное количество газа
и направляется в трубку 2, нагретую до 700°. Рекомендуется
впускать в прибор газ, уже прошедший общий анализ, т. е. освобож-
денный от углеводородов. Для дожигания небольших количеств
водорода в приборе имеется сжигательная колонка 9 и трубка с
Р2О5 10. За поглощением газа в трубке 2 наблюдают по положению
ртути в затворе 5. Количество оставшегося газа замеряется с помощью
ртутного насоса 7.
Для дожигания в прибор впускается несколько кубических сан-
тиметров чистого кислорода через трубку 6, предварительно тща-
тельно откачанную. Газ собирают в 9 и 10 и производят его сож-
жение. Пары воды поглощаются Р2О5 в трубке 10, а оставшийся
кислород — кальцием в трубке 2.
По окончании удаления азота и других газов, кроме редких, что
определяется с помощью разрядной трубк и 77, производится тща-
тельный замер объема редких газов в капилляре 72. Откачанные
редкие газы направляются в трубку с углем 3, охлаждаемую жидким
воздухом. Перед этила выключают трубку 2, поднимая затвор 5.
Непоглощенные в трубке 3 газы, т. е. Не + Ne, откачивают и
замеряют. После этого с помощью малого ртутного насоса 4 смесь
Не 4- Ne направляют в камеру 13 для сравнительных измерений
теплопроводности, что позволяет определить раздельно содержание
Не и Ne. Смесь Не + Ne удаляется затем из прибора через затвор
и кран 8. Вместо прибора для измерений теплопроводности можно
применить газовые микровесы или микроэффузиометр.
Из трубки 3 при температуре —120° откачивают затем аргон,
измеряют его объем и проверяют чистоту в камере 13. Аналогичным
образом уже при 100—200° откачивают смесь Кг + Хе, измеряют их
объем и определяют их раздельно с помощью камеры 13.
Определение редких газов в минералах и водах
Методы определения редких газов в минералах и водах отлича-
ются от методов определения редких газов в природных газах лишь
тем, что первые требуют предварительной обработки исследуемого
материала для извлечения из него газа. Самое определение редких
газов в минералах производится на тех же приборах, которые были
описаны выше.
Для извлечения газов из минералов существует несколько
способов, а именно: измельчение, нагревание и разложение.
Минералы, содержащие значительные количества гелия, как
монацит, торианит, сами отдают гелий. Например, куски монацита
277
в сутки отдают около 0,002 см3 гелия из 1 кг минерала. Торианит
в вакууме отдает в сутки из 1 кг около 0,06 см3 гелия. По-
теря гелия монацитом и торианитом идет значительно быстрее,
чем образование гелия в минерале в результате радиоактивного рас-
пада. Поэтому при обычных лабораторных условиях минерал через
1,5—2 года теряет почти весь свой гелий. При измельчении минерала
отдача газа увеличивается с уменьшением размеров частиц. Путем
измельчения можно извлечь из минерала лишь часть содержащихся
в нем газов. Для более полного извлечения рекомендуется применять
вакуумную шаровую мельницу (см. главу II).
Вторым способом извлечения редких газов из минералов является
их нагревание. Для полного извлечения гелия необходимо нагрева-
ние до 1000—1200°. Нагревать минерал до такой температуры удоб-
нее всего в вакууме в стальной трубе; такими материалами, как кварц
и фарфор, пользоваться нельзя, так как через них при 1000—1200°
гелий быстро диффундирует. Нагревать следует до тех пор, пока из
минерала не перестанет откачиваться газ.
Третий способ извлечения редких газов представляет собой хими-
ческое разложение минералов при помощи азотной или серной кис-
лоты. Смесь измельченного минерала и кислоты нагревают в туго-
плавкой стеклянной или фарфоровой трубке в атмосфере СО2. Раз-
ложение может быть проведено с помощью прибора, изображенного
на фиг. 51 (глава II).
Xлопин, Герлинг и Барановская для растворения уранинита и
выделения из него газов применяли кипячение в соляной кислоте [38].
В выделенных из минералов редких газах могут оказаться и дру-
гие газы, как N2, СО2 и т. п., которые также необходимо определить,
когда требуется полный анализ газа.
Для извлечения редких газов из воды применяют способы, опи-
санные в главе II. К прибору для анализа на редкие газы присоеди-
няют колбу и между прибором и колбой вставляют трубку с Р2О;-
для осушения газа. Прибор, колба и все соединения предварительно
эвакуируют, а затем в колбу через кран впускают исследуемую воду;
последняя, попадая в безвоздушное пространство, вскипает и вы-
деляет все растворенные газы, которые и забираются в прибор для
анализа.
§ 5. Определение радиоактивных эманаций
Во всех природных газах всегда имеется некоторое количество
радиоактивной эманации. Содержание эманации в газах настолько
мало, что его часто выражают числом атомов, а не в каких-либо объ-
емных или весовых единицах.
В 1 см3 атмосферного воздуха содержится в среднем примерно
1 атом эманации; содержание эманации в некоторых почвенных газах
превышает содержание эманации в воздухе в несколько сот раз, но и
в этом случае объем эманации в 1 л газа чрезвычайно мал.
Например, 1000 атомов эманации при нормальных условиях имеют
объем, равный приблизительно 4 • 10-14 мм3.
Несмотря на такую ничтожную концентрацию, содержание эма-
нации в природных газах можно очень точно измерить вслед-
ствие радиоактивного распада эманации и необычайной чувстви-
тельности специально применяемых для этой цели методов изме-
рения.
Сама радиоактивная эманация природных газов не имеет прак-
тического применения, но измерение ее содержания представляет
интерес, поскольку наличие эманации свидетель-
ствует о присутствии радиоактивных элементов.
Помимо радиоактивных эманаций (эманация
радия, эманация тория, эманация актиния), су-
ществующих в природных условиях, известны
различные радиоактивные газы, получаемые ис-
кусственным путем. В состав этих газов входят
искусственные радиоактивные элементы, предста-
вляющие собой изотопы обычных нерадиоактив-
ных элементов.
Для измерения количества радиоактивных
эманаций и других радиоактивных газов при-
меняют специальные электроскопы и электро- фиг- ,06- Электро-
метры, а также счетчики частиц, выделяющихся скоп-
из ядер радиоактивных атомов при их распаде.
Устройство электроскопа с камерой для измерения радиоактив-
ности газа изображено схематически на фиг. 106. Этот электро-
скоп приспособлен для измерений по а-лучам. Стержень 1 проходит
через янтарный изолятор. К верхней части стержня, которая сделана
плоской, приклеен очень тонкий листочек алюминиевой фольги,
в вырезе которого наклеена кварцевая нить. Если сообщить стержню
(электроду) некоторый заряд, то листочек отойдет от стерженька,
как это изображено на фиг. 106. Утечка электрического заряда через
янтарь и нейтрализация его ионами воздуха вызывают приближе-
ние листочка к стержню или, как говорят, его падение. Это падение
листочка наблюдают в микроскоп, в окуляре которого имеется шкала,
что дает возможность, пользуясь секундомером, определять скорость
движения листочка, выражая ее числом делений, проходимых в ми-
нуту. Когда внутри прибора нет радиоактивных веществ, листочек
движется очень медленно, именно 0,2—0,3 деления в минуту. Эту
величину скорости движения листочка, когда в приборе нет радиоак-
тивных веществ, называют «натуральным рассеянием», которое опре-
деляют всегда перед измерением радиоактивности.
Ниже электроскопа, где помещается листочек, расположена иони-
зационная камера 2, герметически закрытая и имеющая два крана —
один вверху, а другой внизу, расположенные на противоположных
стенках. После измерения натурального рассеяния в ионизацион-
ную камеру вводят исследуемый газ. Закрыв один из кранов, можно
через другой откачать воздух из камеры, а затем впустить в камеру
газ. Эту операцию следует повторить 2—3 раза, чтобы удалить из
камеры остатки воздуха. Наполнив окончательно камеру исследуемым
газом, оба крана закрывают. Вместо откачки можно наполнить ка-
меру путем пропускания через нее газа. После прохождения 10—
27.9
20-кратного объема можно считать, что в камере воздуха нет, а имеется
только газ. . •
Объем камеры, а следовательно, и объем впускаемого в камеру
газа должен быть известен.
Перед камерой следует поставить трубку с Р2О5 для осушения
газа. Иначе при влажном газе может иметь место большая утечка
заряда по поверхности янтарного изолятора.
После введения испытуемого газа в камеру краны закрывают;
измерения же производят спустя 3,5 часа. В течение 3,5 часа число
«-частиц, испускаемых радиевой эманацией, растет вследствие обра-
зования и распада RaA — RaB—RaC, а по истечении этого времени
эманация приходит в равновесие с RaA—RaB—RaC и активность
ее остается постоянной, зависящей только от распада самой эманации.
Через 3,5 часа после впуска эманации в камере производят измере-
ние точно таким же способом, как и измерение натурального рас-
ссянич
Вычитая натуральное рассеяние прибора, получаем активность
эманации, выраженную в делениях в минуту или в вольтах в минуту.
Чтобы определить абсолютное количество эманации, необходимо
на том же приборе произвести измерения известного количества эма-
нации. В качестве такого эталона эманации применяют обычно рас-
твор, содержащий известное количество радия. Эманация все время
образуется из радия, и на основании законов распада и образования
радиоактивных веществ легко вычислить количество эманации, на-
ходящееся в данный момент в растворе известного количества радия.
Выделив из раствора эту эманацию и переведя ее в эманационный
электроскоп, измерим активность выделенной эманации. Допустим,
что выделенная из раствора радия эманация в количестве 1 • 10~8
кюри дала активность 60 в)мин. Если испытуемый газ дал активность
120 в)мин, то совершенно очевидно, что в объеме газа, находящемся
в камере, имеется 2 • 10~8 кюри эманации. Распад всякого радиоак-
тивного вещества, в том числе и эманации, происходит по формуле
где —количество эманации во время /;
Nq—количество эманации в начальный момент;
Я—постоянная распада эманации;
/—время, протекшее от начального момента;
е — основание натуральных логарифмов.
Величину e~lt для эманации обычно находят из таблиц.
Образование эманации из радия происходит по следующему за-
кону:
где Nt — количество эманации в момент /;
Noo—количество эманации, которое находится в равновесии с
данным количеством радия (иначе говоря, максимальное ко-
личество эманации, могущее быть в данном количестве радия);
/ — время, протекшее с момента начала образования эманации.
280
Раствор радия, служащий эталоном, хранится или в промывание,
или в стеклянном барботере. Концы обеих трубок должны запаиваться
и лишь в крайнем случае закрываться каучуками со стеклянными
палочками. Если имеется свежий эталон, с которым не сделано еще
ни одного измерения, то из него предварительно следует удалить эма-
нацию, что производится пропусканием воздуха через раствор в
течение 15—20 мин. Воздух должен проходить через раствор мел-
кими пузырьками. После такой промывки можно считать, что вся
эманация из раствора удалена. Концы трубок после эманации сле-
дует запаять, а время окончания продувания заметить: это будет
начальный момент для накопления эманации. Через 1—2 суток эма-
нацию из раствора переводят в камеру электроскопа, присоединив
промывалку или барботер к крану камеры (фиг. Юб) и заставив воз-
дух, прежде чем попасть в камеру, проходить через раствор. Это
производят путем предварительной откачки камеры. 15-минутного
прохождения воздуха через раствор достаточно, чтобы перевести
эманацию из раствора радия в камеру. После окончания этой опера-
ции трубки промывалки или барботера следует запаять. Момент
окончания продувания раствора замечают, — это и будет конечный
момент накопления эманации. Количество образовавшейся эмана-
ции вычисляют по формуле
Nt представляет собой то количество эманации, которое находится
в равновесии с радием, находящимся в растворе. Так как количество
радия нам известно, то известно и Nx.- Количество эманации, нахо-
дящееся в равновесии с 1 г радия, называется «кюри», а соответствую-
щее количество для I мг радия называется «милликюри». Следова-
тельно, если мы имеем в растворе 1 • 10-8 г радия, то равно
1 • 10“8 кюри. В приведенной формуле t — время накопления эма-
нации—известно. Величина для различных величин t находится
по табл. 35. На основании этих данных можно вычислить и Nt> т. е.
количество эманации, находящейся в камере. Измерение эманации
в этом случае производят так же, как описано выше, а именно, через
3,5 часа после ее введения в камеру. После измерения вычисляют,
какая скорость движения листочка (или скорость падения потен-
циала) соответствует, например, 1 • 10-8 или 1 . 10~9 кюри. Поль-
зуясь этой константой прибора, вычисляют содержание эманации в
исследуемых газах.
Содержание эманации дают обычно в единицах кюри на 1 л или
на 1 см3 газа.
Величина, равная 1 • 1О'10 кюри, называется «эман».
При измерениях необходимо следить, чтобы во время стояния
эманации в течение 3,5 часа камера была герметически закрыта;
Янтарный изолятор электроскопа необходимо держать в чистоте;
для предохранения от влаги к. электроскопу присоединяется осу-
шитель.
Этим способом измерения активности эманации пользуются также
281
для измерения количества радия. Если мы имеем упомянутый выше
эталонный раствор с известным количеством радия, то за определен-
ный промежуток времени в этом растворе может накопиться только
определенное количество эманации, которое вычисляется по выше-
приведенной формуле. Зная, какой активности (в вольтах в минуту)
соответствует, например, 1 • 10~8 кюри, можно определить коли-
чество радия в любом растворе. Измерив активность эманации этого
исследуемого раствора и зная время накопления ее, можно вычислить
и то количество эманации, которое будет находиться в равновесии
с радием, имеющимся в исследуемом растворе, по формуле
N - N‘
У‘ i_e-« ’
Nt — наблюденная активность;
е~11 ~находимая из таблиц величина, соответствующая времени
накопления.
Определив Nm в единицах кюри, мы определяем и количество
радия; например, 1 . 10~8 кюри, очевидно, соответствует 1 • 10~8 г
радия в растворе.
Вместо электроскопа, изображенного на фиг. 106, к ионизацион-
ной камере можно присоединить тот или иной электрометр, пред-
ставляющий собой более совершенный тип измерительного
прибора.
Наиболее чувствительными приборами для радиоактивных изме-
рений являются счетчики частиц (а, 0, у). Имеется несколько систем
счетчиков, описываемых в специальной литературе [40].
Вышеописанными способами может быть измерено не только содер-
жание эманации радия, но и содержание эманации тория и актиния,
а также искусственных радиоактивных газов. Следует только учесть,
что эманации тория и актиния распадаются очень быстро и для точ-
ного их измерения пользуются специальными методами.
Для измерения малых концентраций эманации ее предварительно
концентрируют из больших объемов исследуемого газа путем погло-
щения или путем конденсации эманации.
Такие жидкости, как керосин, масло, сероуглерод, толуол и дру-
гие, обладают свойством сильно поглощать эманацию при охлажде-
нии. При пропускании газа через охлажденный сосуд с абсорбирую-
щей жидкостью вся эманация будет задержана абсорбентом. Выклю-
чив сосуд и нагрев его до комнатной температуры, можно, пропуская
через жидкость воздух, перевести эманацию в ионизационную камеру
электроскопа для измерения. Объем прошедшего газа известен,
поэтому легко вычислить и содержание эманации в нем.
Вместо упомянутых жидкостей для поглощения эманации можно
употреблять активированный уголь. Свежепрокаленный уголь уже
при комнатной температуре адсорбирует эманацию, если скорость
прохождения через него газа невелика. При охлаждении уголь пол-
ностью адсорбирует эманацию.
282
Распад эманации радия
Таблица 35
Время е-’л Время е~4 Время Время е-*1
дни часы дни часы ДНИ часы ДНИ часы
0 1,0000 -— 35 0,7676 3 22 0,4914 11 6 0,1302
—> 0,5 0,9962 — 36 0,7620 4 — 0,4844 11 12 0,1244
— 1 0,9925 37 0,7560 4 4 0,4696 11 18 0,1189
— 2 0,9850 — 38 0,7504 4 8 0,4557 12 — 0,1136
— 3 0,9786 — 39 0,7448 4 12 0,4424 12,5 — 0,1038
4 0,9703 — 40 0,7391 4 16 0,4293 13 -— 9,48-10~2
— 5 0,9629 — 41 0,7336 4 20 0,4165 13,5 — 8,66-10~ 2
— 6 0,9557 — 42 0,7282 5 — 0,4041 14 — 7,91-102
— 7 0,9485 — 43 0,7225 5 4 0,3921 14,5 — 7,22-10-2
5 8 0,9413 44 0,7171 5 8 0,3804 15 — 6,6- io—2
— 9 0,9343 —— 45 0,7117 5 12 0,3691 15,5 6,03.10“2
— 10 0,9272 — 46 0,7064 5 16 0,3581 16 — 5,5-10-2
— 11 0,9203 — 47 0,7010 5 20 0,3475 16,5 — 5,03-10-2
— 12 0,9134 2 48 0,6960 6 — 0,3371 17 4,59-10~2
— 13 0,9064 2 2 0,6865 6 4 0,3271 17,5 — 4,19-Ю-2
— 14 0,8996 2 4 0,6750 6 8 0,3174 18 — 3,83-10-2
— 15 0,8929 2 6 0,6651 6 12 0,3079 18,5 — 3,5-10“2
— 16 0,8861 2 3 0,6561 6 16 0,2988 19 — 3,2 10—2
— 17 0,8795 2 10 0,6451 6 20 0,2899 19,5 2,92-10—2
—— 18 0,8729 2 12 0,6357 7 — 0,2812 20 2,67-10-2
— 19 0,8662 2 14 0,6272 7 6 0,2689 21 — 2,22-10~2
— 20 0,8597 2 16 0,6165 7 12 0,2569 22 — 1,86-10~2
— 21 0,8533 2 18 0,6075 7 18 0,2454 23 1,55 10-2
— 22 0,8468 2 20 0,5994 8 — 0,2346 24 — 1.29-10—2
— 23 0,8405 2 22 0,5892 8 6 0,2242 25 1.08.10—2
—— 24 0,8343 3 — 0,5806 8 12 0,2143 26 8,09-10—‘{
25 0,8278 3 2 0,5729 8 18 0,2048 27 —- 7,5 10—3
— 26 0,8216 3 4 0,5631 9 --- 0,1957 28 — 6,26.10~3
— 27 0,8155 3 6 0,5549 9 6 0,1871 29 — 5.22-10—3
—. ; 28 0,8093 3 8 0,5476 9 12 0,1788 30 — 4,35-10~3
— ; 29 0,8032 3 10 0,5381 9 18 0,1709 35 — 1,76-10-3
—' ! 30 0,7973 3 12 0,5303 10 — 0,1633 40 —• 7,11 -10—4
— 31 0,7911 3 14 0,5234 10 6 0,1561 45 2,87-10 ~4
— I 32 0,7852 3 16 0,5142 10 12 0,1491 50 1,16 10“4
--- ! зз 0,7793 3 18 0,5068 10 18 0,1425 60 1 1,9-10
— | 34 0,7734 3 20 0,5002 11 — 0,1362 80 — 5,06-10“7
283
Чтобы собрать эманацию из большого объема, можно пользо-
ваться также низкими температурами. В этом случае газ предвари-
тельно пропускают через систему трубок с КОН и Р2О5 для удаления
паров воды и углекислоты. Высушенный и освобожденный от угле-
кислоты газ поступает в охладитель, представляющий собой систему
трубок, погруженных в сосуд с жидким воздухом. После оконча-
ния пропускания газа охладитель закрывают, вынимают из жид-
кого воздуха и присоединяют к эвакуированной ионизационной
камере. При постепенном подогревании до комнатной температуры
эманация из охладителя переходит в ионизационную камеру. Послед-
ние остатки эманации в охладителе переводят в камеру путем впуска
в камеру воздуха через охладитель.
В течение 3,82 суток эманация радия распадается наполовину;
практически активность эманации в течение месяца падает до нуля,
поэтому определить эманацию можно лишь в пробах газа, взятых
недавно. Удобнее всего, разумеется, измерять содержание эманации
на месте нахождения источника газа. Если содержание эманации
измеряется в пробе газа, взятой недавно, то необходимо вводить
поправку на распад эманации с момента взятия пробы.
ГЛАВА ВОСЬМАЯ
ФИЗИЧЕСКИЕ МЕТОДЫ ГАЗОВОГО АНАЛИЗА
Описанные в предыдущих главах методы газового анализа осно-
ваны в той или иной степени на использовании химических реак-
ций. В некоторых из этих методов химические реакции между опре-
деляемыми компонентами и применяемыми реагентами играли ре-
шающую роль в процессе анализа. Тем не менее и в этих случаях
помимо химических реакций необходимы определения физического
характера, как, например, измерения объема и давления газа.
В других описанных выше методах физические и физико-химиче-
ские процессы и определения играли главную роль при анализе,
а химические реакции—второстепенную. Так, например, методы опре-
деления углеводородных газов с применением низкотемпературной
разгонки основаны главным образом на физических и физико-химиче-
ских методах. Химические реакции применяют здесь для очистки
газа, для контроля за разгонкой, а также для более полного анализа
(на О2, СО2, N2 и др.), используя при этом уже другие методы и при-
боры.
|При анализе на редкие газы физические и физико-химические
процессы и определения точно так же играют главную роль. Хими-
ческие реакции служат лишь для предварительной очистки газа.
В то же время существуют методы газового анализа, основанные
исключительно на физических явлениях и процессах. В этих мето^
дах не применяются никакие реагенты или применение их имеет чисто
вспомогательный характер.
В настоящей главе описываются подобные методы, которые можно
назвать физическими методами газового анализа. Сюда относятся,
в частности, различные оптические методы, а также анализ газа с
помощью масс-спектрографа. Следует, однако, заметить, что выделе-
ние этих физических методов в отдельную группу имеет, конечно,
условный характер, поскольку и здесь приходится в ряде случаев
сочетать эти физические методы с применением тех или иных химиче-
ских реагентов.
§ 1. Спектральный газовый анализ
В главе о методах определения редких газов уже было упомянуто
о качественном спектральном газовом анализе. Присутствие характер-
ных линий того или иного редкого газа в спектре газовой смеси решает
285
вопрос о присутствии этого газа. Для проведения подобных определе-
ний анализируемый газ под очень небольшим давлением впускают
в разрядную трубку, электроды которой соединены с полюсами ин-
дукционной катушки. Свечение в газе при разряде наблюдают с по-
мощью спектроскопа или спектрографа. Эти наблюдения ведутся,
следовательно, по спектрам испускания. Интенсивность свечения
характерных спектральных линий зависит от концентрации соот-
ветствующих компонентов газовой смеси, однако указанные на-
блюдения носят лишь качественный характер.
Количественный спектральный анализ многих твердых материалов
и растворов разработан с достаточной степенью точности. Много-
численные преимущества спектрального метода вызвали быстрое его
внедрение в различные отрасли промышленности. Возможность точно
измерить мельчайшие следы примесей в тех или иных материалах
и возможность проведения анализа малых проб, а также высокая
скорость определений без потери точности представляют особенные
преимущества спектральных методов.
Количественный анализ смесей газов при помощи спектральных
методов до последнего времени не разработан в удовлетворитель-
ной степени. Существуют некоторые затруднения, до сих пор задер-
живавшие применение спектрографа в газовом анализе. Относи-
тельная интенсивность спектральных линий газов-компонентов может
изменяться в зависимости от общего давления газа в разрядной труб-
ке, даже если относительные количества компонентов остаются по-
стоянными.
Этих трудностей можно легко избежать, если всегда вводить в
разрядную трубку одно и то же количество газа. Однако почти все
газы исчезают в разрядной трубке во время прохождения разряда
вследствие их поглощения и разложения. Скорость исчезновения
различна для различных газов, а потому соотношение нескольких
компонентов смеси газов не остается постоянным во время разряда.
Вследствие этого относительная интенсивность спектральных линий
компонентов не остается постоянной. Скорость исчезновения газов
также зависит от силы тока разряда и от применяемого вольтажа.
Помимо поглощения и разложения в разрядной трубке компонен-
тов газовой смеси имеется еще одно затруднение, заключающееся
в том, что интенсивность спектральных линий одного элемента на-
ходится в некоторой зависимости от количества других элементов.
Указанные обстоятельства ограничивают возможность количе-
ственного газового анализа по спектрам испускания. Компоненты
анализируемой газовой смеси, представляющие собой химические
соединения, как, например, углеводороды или окислы элементов,
кроме того, разлагаются под действием разряда. Большие затруд-
нения представляет также присутствие азота, который нельзя уда-
лить из газовой смеси до удаления других компонентов и который
имеет очень сложный полосатый спектр, делающий крайне затрудни-
тельным рассмотрение линий других элементов. По этим причинам
газовый анализ по спектрам испускания не получил применения,
кроме отдельных случаев, когда требуется лишь качественный анализ
286
простых газовых смесей и, в частности, качественный анализ смесей
редких газов.
Другим видом спектрального газового анализа является изуче-
ние спектров поглощения при прохождении инфракрасных лучей
через исследуемое вещество. Каждое жидкое и газообразное вещество
поглощает лучи только определенной длины волны, благодаря чему
по спектрам поглощения можно судить о составе исследуемого ве-
пропускают инфракрасные лучи,
Фиг. 107. Схема инфракрасного спектрографа.
щества.
Преимущество этого метода заключается в том, что само анализи-
руемое вещество, через которое
не подвергается каким-ли-
бо изменениям, как при
получении спектров испу-
скания.
Общая схема прибора
для получения спектров
поглощения газами инфра-
красных лучей представле-
на на фиг. 107 [41]. Источ-
ник инфракрасного излу-
чения 7 представляет со-
бой обычно металлический
стержень или ленту, через
которые пропускают элек-
трический ток, доводящий
их до красного накала.
Излучение фокусируется
вогнутым зеркалом 2 на щель инфракрасного спектрографа 3. Как
зеркало 2, так и другие зеркала прибора алюминированы с передней
стороны.
Между щелью спектрографа и источником излучения помещают
кювету 4 длиной в несколько сантиметров, имеющую окна из камен-
ной соли. В эту кювету вводят анализируемый газ.
Инфракрасные лучи, пройдя анализируемый газ и щель спектро-
графа, попадают после отражения на зеркало 5, затем проходят
через призму из каменной соли 6, которая пропускает через себя
инфракрасные лучи и разлагает их в спектр. После прохождения
лучей через 6 они попадают на зеркало 7, затем снова через призму 6
попадают на зеркало 5 и через щель 8 и зеркало 9 на термостолбик 10.
Попадание инфракрасных лучей на термостолбик вызывает возникно-
вение электрического тока, который и замеряют зеркальным галь-
ванометром.
Вращая зеркало 7, можно целиком просмотреть спектр, получив
соответствующую кривую зависимости показаний гальванометра от
длины волны. Имеются инфракрасные спектрографы, позволяющие
автоматически записывать спектр поглощения с помощью соответ-
ствующих усилителей или фотографического устройства.
Описанный прибор позволяет определять примесь одного газа
к другому, когда их спектры поглощения различны. Для проведения
287
измерений выбирают такие характерные полосы поглощения опре-
деляемого газа, на которые не налагаются полосы поглощения дру-
гих компонентов газовой смеси. Выбрав такие полосы поглощения,
производят калибровку прибора, применяя различные концентрации
интересующих нас компонентов.
Наиболее простой задачей, которую может разрешать инфракрас-
ный спектрограф, является определение чистоты индивидуального
газообразного соединения или элемента. Спектр поглощения может
показать наличие или отсутствие примесей в исследуемом газообраз-
ном веществе.
Инфракрасный спектрограф позволяет со значительной точностью
анализировать и многие двойные газовые смеси даже в тех случаях,
когда имеем дело с изомерами углеводородов и их производных,
мало отличающихся друг от друга по химическим и физическим
свойствам. Так, например, известно, что с помощью инфракрасного
спектрографа можно анализировать смесь нормального бутана и
изобутана. Анализ отнимает 15 мин., а точность его около 0,5%.
Поскольку ни азот, ни кислород не поглощают инфракрасную
часть спектра, легко определять примеси разнообразных газов в
воздухе, как, например, углекислоту, этан, пары органических ве-
ществ. Согласно некоторым исследованиям чувствительность этих
определений достигает нескольких тысячных долей процента.
Анализ многокомпонентных смесей с помощью инфракрасного
спектрографа представляет уже значительные затруднения, так как
каждое индивидуальное соединение обычно дает ряд полос погло-
щения, которые налагаются друг на друга. Выбор таких полос,
которые вызываются только одним компонентом, представляет боль-
шие затруднения, если этих компонентов много. Какие компоненты
могут присутствовать в газовой смеси, должно быть предварительно
известно.
При проведении подобного анализа можно пользоваться и комби-
нированным методом, применяя те или иные поглотители или дру-
гие способы разделения газовых смесей. При применении инфракрас-
ного спектрографа анализируемый газ рекомендуется очистить от
водяных паров, пропуская его через осушитель (Р2О5). Эта очистка
необходима, так как водяные пары имеют полосы поглощения в
инфракрасной части спектра.
Следует отметить, что ширина полос поглощения зависит от тем-
пературы; чем ниже температура, тем меньше ширина полос. Чем
резче полосы поглощения, тем больше число компонентов, которые
можно определить в газовой смеси. Поэтому при анализе многоком-
понентных смесей выгодно насколько возможно понижать темпера-
туру исследуемого газа вплоть до температуры жидкого воздуха.
§ 2. Газовый интерферометр
Под преломляющей способностью газа подразумевают величину,
равную показателю преломления минус единица, или
/? = л-1,
288
где /?—преломляющая способность газа;
п — показатель преломления.
Преломляющая способность газа пропорциональна его плот-
ности или
R X
= const.
а
Так как плотность газа пропорциональна давлению, под которым
он находится, то преломляющая способность газа следует газовым
законам постольку, поскольку сам газ им подчиняется. Если /?0 —
преломляющая способность при нормальных условиях, т. е. при 0°
и 760 мм рт. столба, то преломляющая способность газа R при дру-
гой температуре t и давлении р будет
р__р 273 ' р
* <0 273 4-1 760 *
Преломляющую способность смеси газов довольно точно можно
вычислить по правилу смешения, а именно:
где /?2 и т. д. —преломляющие способности отдельных компо-
нентов;
р —давление смеси;
р3 и т. д.—парциальные давления компонентов.
смеси из двух газов, содержащей х % одного компонента
р__р iOQ х । р х
к Л1 100 ' ^2 юо •
Pi,
Для
Для сравнения преломляющей способности газов применяют
специальные газовые интерферометры. Устройство газового интер-
ферометра лабораторного типа схематически изображено на фиг. 108.
Лучи света от лампы 7, проходя через узкую вертикальную щель,
при помощи линзы 2 делаются параллельными, после чего проходят
через две вертикальные щели 3 шириной 4 мм, длиной 27 мм, нахо-
дящиеся на расстоянии 12 мм одна от другой. Верхние половины
лучей света проходят через воздух до пластинки 6, которая откло-
няет свет вниз для того, чтобы верхние пучки сделать смежными
с нижними пучками, проходящими через трубки для газа 4 и 5. Верх-
ние и нижние пучки лучей остаются разделенными узкой темной
линией. В противном случае без пластинки 6 в поле зрения будет
широкая темная полоса, являющаяся тенью стенок металлических
трубок.
Верхние пучки света после пластинки 6 попадают в телескоп 7
и рассматриваются через окуляр 8. Вследствие интерференции обоих
верхних пучков лучей мы увидим через 8 ряд темных и светлых по-
лос. Поскольку верхние пучки лучей имеют разные оптические пути,
эти полосы имеют совершенно определенное, постоянное положение
и по отношению к ним производится установка полос в нижней части
19 в. А. Сокол он.
289
поля зрения. Нижние пучки лучей, пройдя через трубки 4 и 5, содер-
жащие сравниваемые газы, попадают каждый на стеклянную пла-
стинку, наклоненную под углом 45° к оптической оси. Одна из пла-
стинок неподвижна, а другая может вращаться вокруг горизонталь-
ной оси, перпендикулярной к пучкам света. На фиг. 108 схемати-
чески указано положение этих двух пластинок 9 и 10, причем пла-
стинка 9 укреплена неподвижно, а 10 может вращаться. Нижние
пучки лучей рассматриваются через телескоп 7. Вследствие интер-
ференции видны темные и светлые полосы: одни в верхней половине
поля зрения, от лучей, прошедших через воздух, другие в нижней,
от лучей, прошедших через камеры 4 и 5.
Появление интерференционных полос объясняется следующим
образом. Свет из источника 1 (фиг. 108, б) проходит через две щели"2
и 3, и получившиеся два пучка света сходятся в плоскости 4; в точку 5,
находящуюся на равном расстоянии от щелей 2 и 3, оба луча при-
ходят с одинаковой фазой, и в результате получается светлая по-
лоса. Длина путей лучей до точки 6 различна. Если длина 2—6 отли-
чается от длины 3—6 на половину или на нечетное число полуволн,
то пучки света будут приходить к точке 6 с противоположными
фазами и в точке 6 получится темная полоса. Если же она будет от-
личаться на целое число волн, то в точке 6 будет светлая полоса.
Таким образом, в плоскости 4 мы будем иметь попеременно светлые
и темные полосы. При монохроматическом свете будем иметь попе-
ременно полосы темные и полосы того цвета, каким обладает источ-
ник, а при белом свете средняя полоса будет белая, а остальные —
окрашенные, причем внутренний край будет фиолетовым, а внешний
красным.
Нижние половины лучей света проходят через газовые камеры 4
и 5 (фиг. 108, а), каждая длиной 100 см; концы камер закрыты плоско-
параллельными стеклянными пластинками. Каждая камера имеет
две трубки для впуска и выпуска газа. Если состав газа в обеих
трубках различен, то оптические пути лучей будут неодинаковы
290
и интерференционные полосы в точке 8 будут смещены со своего
нормального положения. Вращая компенсационную пластинку 70
микрометрическим винтом, можно изменить длину оптического пути
луча, идущего через одну из камер, и сделать пути обоих лучей оди-
наковыми; это будет достигнуто, когда интерференционные полосы
верхней и нижней половин поля зрения совпадут. Тот угол, на кото-
рый пришлось повернуть пластинку 70, чтобы сделать одинаковыми
пути лучей, является мерой разницы между преломляющими спо-
собностями газов в камерах.
Точность показаний интерферометра при прочих равных усло-
виях зависит от длины камер; чем длиннее камера, тем точнее ре-
зультат. Газы в обеих камерах должны быть сухими и иметь оди-
наковые температуру и давление.
Интерферометр калибруют перед употреблением, для чего в
одну из камер впускают стандартный газ, а в другую тот же стан-
дартный газ с известным содержанием того компонента, который
в дальнейшем требуется определять.
При работе с интерферометром прежде всего находят «нулевую
точку» прибора, наполняя обе камеры сухим стандартным газом и
замечая положение микрометрического винта. Пластинка 70 должна
быть так повернута, чтобы интерференционные полосы обеих половин
поля зрения совпадали. После этого в одну из камер впускают испы-
туемый газ и, передвигая микрометрический винт, опять доводят
до совпадения интерференционные полосы. По калибровке находят
содержание определяемого компонента.
Этот метод применяется главным образом при анализе смесей,
состоящих из двух известных газов.
Помимо описанного лабораторного прибора изготовляют также
переносный газовый интерферометр, устроенный так же, как и лабо-
раторный, но с камерами меньшей длины (фиг. 108, в).
В табл. 36 приведены величины преломляющей способности раз-
личных газов. Так как некоторые из них заметно отклоняются от
газовых законов, то преломляющая способность при низких пар-
циальных давлениях может отличаться от величины, вычисленной
из отношения давлений. В этих случаях следует пользоваться так
называемой «идеальной преломляющей способностью», вычисленной
из преломляющей способности при очень низких давлениях.
Газовый интерферометр применяется в различных областях газо-
вого анализа [42].
Дымовые газы. В дымовых газах нужно в первую очередь опре-
делить содержание двуокиси углерода и кислорода. Для анализа
дымовых газов вполне применим газовый интерферометр. Два отсчета
до и после удаления СО2 дают ее содержание. Остаток сравнивают
с воздухом и по калибровочной кривой для О2 ф- Н2 находят содер-
жание кислорода. При неполном сгорании или при топливе с боль-
шим содержанием летучих или серы нужно считаться с заметными
примесями окиси углерода, водорода, метана и сернистого газа. Их
также можно определить интерферометрическим путем. Сравнение
19* 297
Таблица 36
Величины преломляющей способности газов при 0° и 760 мм
рт. столба линий натрия
Газ Преломляющая Идеальная преломляющая
способность способность
Воздух . 2917-10~7 2916-10-7
Метан 4415-10~ 7 4408-10-7
Этан 7660-10~7 7578-10-7
Пропен 11200-10~7 —
Этен 7266-10“7 7209-10“7
Ацетилен 6020-10-7 597110“7
Окись углерода 3347-10“7 3346-10“7
Углекислый газ 4498-10“7 4467-10“7
Кислород 2706-10“7 2704-10“7
Азот 2972-10“7 2971-10“7
Водород 1387-10~7 1388-10“7
Сероводород 6440-10“ 7 —
Сернистый газ 6760-10“ 7 6628-10“7
Фосфористый водород . . . 7890-10“7 —
Водяной пар 2570-10“ 7 —
Г елий 342-10“ 7 342-10“7
Неон 671 • 10~7 671 10“7
Аргон 2809-10“7 2807 -10“7
Криптон ......... 4270-10~7 4259-10“7
Ксенон 7020-10~7 6969-10“7
найденного количества О2 с вычисленным из содержания СО2 ука-
зывает, в каких случаях нужно считаться с этими примесями.
Рудничный газ. Наиболее широкое применение газовый интер-
ферометр получил для анализа рудничного газа на метан и СО2. Для
этой цели сконструирован специальный переносный прибор, в кото-
ром интерферометр и поглотители смонтированы в общем кожухе
и шкала компенсатора непосредственно дает содержание СН4. Сравне-
ние газа, освобожденного от влаги (с помощью СаС12) и от СО2
(с помощью натронной извести), с воздухом, очищенным аналогич-
ным образом (взятым вне шахты), дает содержание метана. Второй
отсчет газа, не освобожденного от СО2, дает содержание последнего.
Достижение точности до 0,1% СН4 и СО2 не представляет затруд-
нений. В приборе обычного типа с большой кюветкой точность
может быть доведена до 0,01%.
292
Коксохимическая промышленность. Применению газового интер-
ферометра в коксохимической! производстве посвящен ряд работ.
Наиболее актуально, вероятно, интерферометрическое определение
содержания бензола в коксовом газе. Обычные методы (выморажива-
ние или поглощение активированным углем с последующей отгон-
кой из него) требует много времени. Интерферометрическое сравнение
коксового газа до и после поглощения из него бензола углем дает
более надежные и быстрые результаты.
Аналогичным путем можно определять содержание аммиака в
коксовом газе, поглощая его серной кислотой. Сероводород также
может быть найден интерферометрическим путем.
Следует также отметить применение интерферометра для проверки
взрывчатости воздуха, наполняющего пустые цистерны от бензола,
нефтеналивные суда и т. д. Комбинируя инте]фб рометр с определе-
нием плотности и пользуясь графиком или практическими форму-
лами, можно довести быстроту анализа Н2, СН4, С6С6 и H2S в коксо-
вом газе до 10—12 мин.
Анализ воздуха. Интерферометр позволяет легко контролировать
содержание СО2 в воздухе, например в закрытых помещениях, с
точностью до 0,1—0,05%, а также других токсических и посторонних
примесей.
В производстве жидкого кислорода и в азотной промышленности
он очень удобен для определения отношения О2 : N2. Интерферометр
также нашел применение в анализе благородных газов, при произ-
водстве газонаполненных лампочек и т. д.
Интересно применение интерферометра в воздухоплавании при
проверке проницаемости оболочки дирижаблей путем исследования
разных мест оболочки. Точность определения составляет 0,1% Н2
в воздухе. Изучение непроницаемости тканей для аэростатов в лабо*
раторных условиях также сильно выигрывает в простоте и точности
при пользовании интерферометром.
§ 3. Оптико-акустический метод газового анализа
Разработанный проф. М. Л. Вейнгеровьш [43] оптико-акустический
метод газового анализа основан на явлении звучания газов и паров
при действии на них прерываемого со звуковой частотой потока ин-
фракрасных лучей.
Сущность этого явления заключается в том, что при поглощении
инфракрасных лучей газ нагревается, что вызывает повышение его
давления. Если поток инфракрасных лучей прерывистый, то в газе воз-
никает пульсация давления, т. е. появляется звук. Высота тона за-
висит от частоты прерывания, а сила звука от способности данного
газа поглощать инфракрасные лучи и концентрации этого газа. Ука-
занный звуковой эффект не будут давать только газы, совсем не по-
глощающие инфракрасных лучей, как азот, кислород и водород.
Это обстоятельство создает, в частности, благоприятные предпосылки
293
для определения оптико-акустическим методом примесей различных
газообразных соединений в воздухе.
Первоначальная схема аналитического прибора, основанного на
вышеуказанном принципе, изображена на фиг. 109. Источник ин-
фракрасных лучей представляет собой спираль или стержень из
специального сплава, нагреваемый электрическим током. Неболь-
шой моторчик вращает диск, имеющий ряд прорезей, благодаря
чему прерывистый пучок инфракрасных лучей попадает в камеру,
содержащую анализируемый газ. Возникающие при этом в газе коле-
бания улавливаются микрофоном. Получающийся в микрофоне элек-
трический ток передается гальванометру.
Имеется несколько вариантов проведения анализа на основе
описанной схемы.
Наиболее простой вариант заключается в том, что гальванометром
замеряется сила тока, возникающая при попадании инфракрасных
лучей в камеру с анализи-
руемым газом. Чем выше кон-
центрация того или иного
соединения в инертном к дей-
ствию инфракрасных лучей
газе (например в воздухе),
тем больше и сила тока.
В этом случае прибор предва-
рительно калибруется с по-
,ЛЛ л „ мощью эталонов, содержащих
Фиг. 109. Оптико-акустическии газоанали- , г “*
затор. различные известные концен-
трации определяемого компо-
нента.
Преимуществом прибора является возможность мгновенного полу-
чения результата анализа для отдельных образцов исследуемой газо-
вой смеси. Прибор особенно удобен для непрерывного анализа струи
газа, которая в этом случае должна с постоянной скоростью прохо-
дить через камеру. Постоянство скорости должно автоматически регу-
лироваться для постоянства давления газа в камере. Необходимо также
поддерживать постоянство интенсивности инфракрасного излучения.
Для большого удобства и повышения точности анализа применяют
диференциальную схему. Пучок инфракрасных лучей разделяют
на две равные части, которые идут в две газовые камеры. В одной
камере находится анализируемый газ, а в другой тот же газ, но ли-
шенный определяемого компонента. Микрофоны и гальванометр
соединяют таким образом, чтобы можно было заметить разницу тока
от обеих камер.
Описанное устройство позволяет анализировать газовые смеси,
в которых помимо инертных к действию инфракрасных лучей газов
имеется только один компонент, содержание которого и требуется
определить.
Для анализа многокомпонентных газовых смесей предложены
иные схемы использования этого явления звучания газа под дей-
ствием инфракрасных лучей.
294
В одной из этих схем исследуемая газовая смесь помещается на
пути лучей, направляемых на индикатор.
Ослабление силы звука, происходящее вследствие поглощения
радиации исследуемым газом, является мерой концентрации смеси.
Применяя подобные -газовые инфракрасные фильтры, можно в не-
которых случаях анализировать смеси газов, состоящие из несколь-
ких компонентов.
Оптико-акустический газоанализатор, приспособленный для ра-
боты с радиацией, разложенной в спектр с помощью инфракрасного
монохроматора, называется спектрофоном. С помощью спектрофона
можно исследовать многокомпонентные газовые смеси, а также изу-
чать инфракрасные спектры поглощения. Кривые, показывающие
зависимость силы звука от длины волны, характеризуют спектр
поглощения газа._______________________________________________
Оптико-акустический метод обладает большой чувствительностью
и может применяться, например, для определения в воздухе при-
месей углеводородных газов и паров, а также для других аналитиче-
ских исследований. Чувствительность некоторых приборов сейчас
достигает 10-3%.
§ 4. Анализ газа с помощью масс-спектрографа
Идея применения масс-спектрографа для целей газового анализа
(в частности, для анализа углеводородных газов) была выдвинута [б]
после изобретения этого прибора,
примененного первоначально для раз-
деления и определения изотопов.
Общая схема масс-спектрографа
представлена на фиг. НО [44].
Движение частиц в масс-спектро-
графе определяется следующим ура-
внением:
Н*г*
п К Е ’
где fi — масса иона В атомных еди- Фиг. 110. Схема масс-спектрографа,
ницах;
п —число элементарных зарядов иона;
Н —магнитное поле;
г—радиус траектории иона в магнитном поле, см;
к — коэфициент пропорциональности;
Е — энергия иона, в.
Масс-спектральный анализ многокомпонентных газовых смесей
представляет большие трудности, так как отдельные газы по-разному
ионизируются, а сложные молекулы при ионизации дают серию иони-
зированных осколков (оскольчатый спектр). По этим причинам при
масс-спектральном газовом анализе необходимо применение эталон-
ных газовых смесей для калибровки прибора. Необходимо также
магнитное управление ионным лучом. Для устранения перезарядки
295
ионов степень вакуума при масс-спектральном газовом анализе
должна быть еще выше, чем при определении изотопов. Для ускорения
записи ионных пик требуется применение быстропишущих электрон-
ных потенциометров [45].
Масс-спектральный газовый анализ обладает большой чувстви-
тельностью. Лучшие приборы этого типа позволяют определять при-
меси к основному газу до 1 : 100000. Точность определений дости-
гает 1%. Для анализа требуется очень небольшое количество газа,
исчисляемое десятыми долями кубического сантиметра. Имеются
установки, позволяющие определять в газе до 16—20 компонентов.
Однако сложность и дороговизна масс-спектрографа ограничи-
вают его применение в газовом анализе. Следует также учесть, что
многие разработанные в последнее время у нас в Союзе приборы для
.микроанализа газа по своей относительной чувствительности, а тем
более по простоте превосходят масс-спектрограф. Эти приборы тре-
буют при анализе большего количества газа, однако для большинства
практических задач получение образцов газа объемом 0,2—1,0 л
и даже больше обычно не представляет затруднений.
§ 5. Определение удельного веса газа
Под удельным весом газа понимают отношение веса газа к весу
такого же объема воздуха. Так как вес определенного объема газа
зависит от температуры и давления, то все определения удельного
веса газа делаются по отношению к сухому воздуху при 0° и 760 мм
рт. столба. Вес 1 л сухого воздуха, освобожденного от углекислоты,
равен 1,2928 г при 0° и 760 мм рт. столба.
Удельный вес газа зависит от его состава. Так как согласно физи-
ческим законам всякий газ в смеси занимает объем, равный объему
всей смеси, то удельный вес смеси газов равен среднему арифмети-
ческому из произведений удельного веса каждой составной части на
ее долю в смеси. Отсюда следует, что, зная удельный вес газа, можно
сделать некоторые заключения о его химическом составе; в частности,
если мы имеем смесь только двух газов, то по удельному весу можно
легко вычислить процентное содержание каждого из компонентов,
разумеется, если известно, какие именно два газа входят в состав
смеси.
При наличии смеси из двух газов для вычисления процентного
содержания каждого из них проще всего пользоваться графическим
способом. На двух ординатах откладывают удельный вес: на одной
ординате отмечают точку, соответствующую удельному весу одного
из газов смеси, а на другой — точку другого газа. Соединяют эти
точки прямой линией. Точку пересечения одной ординаты с абсцис-
сой обозначают как 0, точку пересечения другой ординаты обозна-
чают как 100%. Абсциссу между 0 и 100% делят на 100 или другое
число частей. Если 0 поставлен у ординаты, на которой нанесен мень-
ший удельный вес, то обозначения 0 и 100% относятся к содержанию
газа с большим удельным весом. Для газа с меньшим удельным весом
обозначения должны быть обратные. Определив удельный вес газа,
296
по графику находят процентное содержание каждого из компонен-
тов. Совершенно ясно, что, зная процентное содержание одного из
газов, легко найти и процентное содержание другого, вычтя про-
центное содержание первого из 100%. Если смесь газов состоит более
чем из двух компонентов, то по удельному весу нельзя узнать состав
газа, но удельный вес газа при одновременном химическом анализе
может дать ценные сведения о тех составных частях газа, которые
химическим способом не определяются. Например, при исследова-
нии углеводородных газов можно определить содержание в газе угле-
кислоты и кислорода, а затем по удельному весу судить о содержа-
нии тяжелых углеводородов.
Точным способом определения удельного веса газа является его
взвешивание, для чего употребляют стеклянные баллоны с простым
или трехходовым краном. Если установка, при помощи которой бал-
лон наполняется исследуемым газом, имеет кран для откачки, то
баллон должен иметь простой кран, в противном случае необходим
трехходовый кран или простой, но с притертой пробкой на конце,
имеющей отдельный ход.
После тщательной откачки баллона его наполняют исследуемым
газом, который хорошо высушивают, пропуская его через трубку
с фосфорным ангидридом.
При наполнении замечают то давление, под которым находится
газ в баллоне, для чего употребляется ртутный манометр. Соеди-
нительные трубки должны быть также заполнены исследуемым
газом.
При наполнении газом баллона следует защищать его каким-
либо колпаком от температурных колебаний и течений воздуха. Не-
обходимо также измерять температуру баллона во время наполнения
его газом. Закрыв кран, баллон отъединяют от источника газа, пере-
носят к весам, дают принять температуру окружающего воздуха и
взвешивают с максимальной точностью.
После взвешивания баллона с газом определяют вес пустого бал-
лона, откачав из него воздух масляным или ртутным насосом.
Вычтя из веса баллона с газом вес пустого баллона, получим чистый
вес газа в объеме баллона. Чтобы привести вес газа к нормаль-
760. т
ному давлению и температуре, его умножают на величину ’
где р — давление; Т — абсолютная температура в баллоне в момент
закрытия его крана при наполнении газом.
Для получения удельного веса газа необходимо знать объем бал-
лона или повторить всю операцию взвешивания с воздухом или дру-
гим газом, вес которого известен.
Для определения объема баллон наполняют дестиллированной
водой, имеющей ту же температуру, как и газ в момент наполнения
баллона. Вода должна заполнить весь баллон и канал крана, но в
трубке, идущей вверх от крана, воды быть не должно. Если канал
крана достаточно широк, то наполнять баллон водой удобнее всего
через капиллярную трубку, пропущенную через канал крана. Если
в трубку, идущую вверх от крана, попадет вода, ее необходимо уда-
207
лить. Баллон с водой взвешивают и, вычтя из полученного веса вес
пустого баллона, находят вес воды в объеме баллона. Разделив полу-
ченный вес воды на удельный вес воды при температуре наполнения,
получают объем баллона. Зная объем баллона и вес газа в его объеме,
находят вес 1 л газа, а отсюда получают удельный вес, разделив вес
1 л газа на вес 1 л воздуха.
Объем баллона хотя и незначительно, но будет меняться в зави-
симости от температуры, а так как комнатная температура изменяется
не в очень широких пределах, то, измерив однажды объем баллона
при различных температурах и построив соответствующий график,
определяют обычно только вес газа в объеме баллона, измеряя объем
баллона лишь изредка и в случаях, требующих большой точности.
При определении объема баллона необходимо следить за тем,
чтобы при наполнении его водой на стенках не осталось пузырьков
воздуха. Кран баллона для взвешивания воздуха должен пришли-
фовываться самым тщательным образом. Смазка крана должна быть
высококачественной. Ни в канале крана, ни на наружных частях
его около шлифа смазки не должно быть. Не следует допускать какого-
либо соприкосновения баллона для взвешивания с ртутью; капельки
ртути застревают в кране, пристают к смазке, и от них очень трудно
освободиться, присутствие же их может сильно влиять на точность
взвешивания.
Удельный вес газа можно определять, не прибегая к определению
объема баллона. Определив вес газа в объеме баллона, проделывают
ту же операцию с воздухом или другим газом, удельный вес кото-
рого точно известен. Разделив вес газа на вес воздуха, получают
удельный вес газа. Никаких приведений к нормальным условиям не
требуется, если газ и воздух взвешены при одинаковой температуре
и давлении; в противном случае следует вводить поправку, умножая
вес газа и вес воздуха на приведенную выше величину.
Емкость баллонов для взвешивания газа обычно равна 200—
300 ел/3. Применяют и баллоны большего объема. Вес большинства
природных газов в подобных объемах выражается десятыми долями
грамма. При очень малом количестве исследуемого газа чувствитель-
ность аналитических весов недостаточна. Например, вес воздуха
в баллоне емкостью 10 ел/3 равен 12,9 мг, а вес водорода —0,89 мг.
Подобные веса могут определяться с достаточной точностью с по-
мощью микровесов, но при еще меньших количествах исследуемого
газа рекомендуется пользоваться для определения удельного веса
специальными газовыми весами, описываемыми ниже.
Путем взвешивания удельный вес газа определяется с большой
точностью, но это определение отнимает много времени и может быть
сделано только в хорошо оборудованной лаборатории. Когда же
большой точности не требуется, применяют более простые способы
определения удельного веса газа, основанные на истечении газов
из малых отверстий. Приборы для определения удельного веса газов
методом истечения носят общее название «эффузиометр». На подоб-
ном приборе удельный вес газа можно определить в течение несколь-
ких минут с точностью до 1—2%.
298
Теория истечения газов под давлением из малого отверстия при-
водит к простому соотношению между скоростью истечения газа и
его плотностью, которое можно вывести на основании следующих
соображений.
Пусть газ, находясь под давлением р, вытекает из узкого отвер-
стия, имеющего площадь s, в пространство, где имеется давление pv
Обозначим линейную скорость истечения газа через о, а плотность
газа через D. Отсюда следует, что в единицу времени через отверстие
протекает масса газа Л1, равная:
М а= svD.
Работа /?, которая при этом затрачивается, будет равна:
R^utp-pjs.
По теореме живых сил имеем
откуда
т. е. скорость истечения газа обратно пропорциональна квадратному
корню из его плотности.
Если через одно и то же отверстие заставить последовательно вы-
текать одинаковые объемы двух различных газов при условии, что
давления р и рх в обоих случаях остаются неизменными, то из по-
следней формулы мы найдем, что
D _ vl
Di ~ V9' ’
где D и и— плотность и скорость истечения первого газа;
Di и те же величины для второго газов.
Скорость истечения газа обратно пропорциональна времени, по-
требному на истечение определенного объема газа. Так как объем
газа в обоих случаях одинаков, то, очевидно, отношение скоростей
можно заменить обратным отношением времен истечения обоих газов;
следовательно,
D _ Т2 „
оГ-~т^’
где Т и 7\—время истечения того и другого газов.
Из последней формулы видно, что, измеряя время истечения раз-
личных газов при одинаковых внешних условиях, можно определить
их плотности по отношению к одному из них, например к воздуху,
принимая его плотность за единицу.
299
Следовательно, в окончательном виде формула для определения
удельного веса газа методом истечения будет иметь вид:
где D— удельный вес исследуемого газа;
Т—время его истечения;
Dx—удельный вес газа, с которым производят сравнение;
7\—время его истечения.
Если газ сравнивается с воздухом, удельный вес которого при-
нимается за единицу, то формула получает еще более простой вид:
Фиг. 111. Приборы для опреде-
ления удельного веса газа.
щие видеть метки на узких
Для вычисления удельного веса
исследуемого газа требуется, следо=
вательно, возвести в квадрат время
истечения его в секундах и разде-
лить на квадрат времени истечения
воздуха.
Прибор для определения удель-
ного веса, основанный на истечении
газа, изображен на фиг. 111, а. Стек-
лянный цилиндр диаметром 7—8 см
закрыт металлической крышкой,
сквозь которую проходит стеклянная
трубка 7, открытая с нижнего конца,
в верхней части она присоединена к
металлическому трехходовому крану
и окружена защитной металлической
трубой, имеющей прорезы, позволяю-
частях стеклянной трубки. Защитная
труба составляет одно целое с трехходовым краном. Вся эта часть
вынимается из стеклянного цилиндра, если отвинтить гайку 2. Вверх
от крана идет металлическая трубка с крышкой, имеющая внутри пла-
тиновую пластинку с маленьким отверстием. Трехходовый кран на
рукоятке имеет метки, показывающие, с чем соединена трубка 7;
в том положении, какое имеется на чертеже, трубка 7 соединяется
с боковым ходом крана. Прибор наполнен водой почти до верха. Тре-
буется определить скорость истечения газа из трубки 7, для чего
по секундомеру измеряют время, необходимое для поднятия уровня
воды от нижней до верхней метки трубки. В металлической крышке
стеклянного цилиндра имеется отверстие, которое закрывается завин-
чивающейся металлической пробкой. Если эту пробку завинтить и
закрыть кран, то вода из прибора не выльется; в таком виде прибор
годен для переноски. При работе прибора должно быть сообщение
между воздухом, имеющимся над водой в цилиндре, и атмосферой,
для чего металлическую пробку следует открыть, вывинтив ее на
несколько оборотов. Целиком вывинчивать пробку не рекомендуется,
300
чтобы не потерять ее; внутри же пробки имеется специальный канал,
через который после нескольких оборотов цилиндр будет сообщаться
с внешним воздухом. Чтобы набрать в трубку 1 газ, поступают сле-
дующим образом: источник газа при помощи каучука соединяют с
боковым отростком крана, который должен быть повернут, как ука-
зано на чертеже. Если давление газа достаточно велико, то газ будет
входить в трубку 7, вытесняя воздух (трубку 7 предварительно на-
полняют водой, для чего ее соединяют с атмосферой). Если давление
газа мало, то для наполнения газом трубки 7 ее постепенно подни-
мают, развинтив гайку 2, но, разумеется, не вынимая ее открытого
конца из воды. Когда вся трубка наполнится газом, т. е. когда уро-
вень воды опустится за нижнюю метку, поворачивают кран на 90°
по часовой стрелке (чтобы длинная черточка на рукоятке была вер-
тикальна, а короткая шла от нее влево) и опускают трубку 7 в ци-
линдр. Газ при этом будет выходить через отверстие в платиновой
пластинке. Когда газ выйдет и уровень воды в трубке 7 перестанет
повышаться, кран ставят в первоначальное положение и опять на-
полняют трубку газом. Повторяя эту операцию несколько раз, про-
мывают таким образом газом все соединительные трубки, кран и
верхнюю часть трубки 7. Когда набирают газ уже для определения
удельного веса, необходимо, чтобы уровень воды был ниже метки
на 1—2 см. Закрыв кран, трубку 7 следует совершенно отъединить
от внешнего воздуха и от источника газа. Длинная черта на рукоятке
при этом должна быть горизонтальной, а короткая итти вверх. Взяв
секундомер и приготовившись к наблюдениям, поворачивают кран
на 90° против часовой стрелки. Газ начинает выходить через узкое
отверстие, и в момент прохождения уровня воды через нижнюю
метку пускают в ход секундомер. При выпуске газа трубка 7 должна
сообщаться с внешним воздухом только через отверстие в платиновой
пластинке; ни в коем случае не допустимо такое положение, чтобы
газ выходил одновременно и через боковой ход крана. Длинная черта
на рукоятке обязательно должна итти вертикально, а короткая —
влево от нее. Когда уровень воды дойдет до верхней метки, секундо-
мер останавливают. Если газ наполняет трубку 7 собственным напо-
ром, можно и не закрывать совершенно кран перед выпуском газа.
При некотором навыке можно успеть приготовиться к наблюдениям,
если после наполнения трубки 7 газом сейчас же повернуть кран для
выпуска газа через узкое отверстие. Если же для наполнения газом
трубку 7 приходится поднимать, то обязательно полностью закрывать
кран после набора газа.
Первоначально опыт истечения проделывают несколько раз с
воздухом, а затем несколько раз с исследуемым газом, причем в обоих
случаях перед измерением несколько раз промывают трубку 7 от остат-
ков газа или воздуха соответственно. При наполнении прибора воз-
духом все операции выполняют точно так же, как и при наполнении
газом, сняв, разумеется, с крана каучук.
Удельный вес газа находят по приведенной выше формуле
. т2
301
где у — искомый удельный вес;
Т — среднее арифметическое из измерений времени истечения газа;
7\ —та же величина для воздуха.
Другой аналогичный прибор представлен на фиг. 111, б. Стек-
лянный баллон 7 сообщается одним концом с уравнительным сосу-
дом 2, а другим с трехходовым краном. Баллон и частично уравни-
тельный сосуд наполняют водой. Через левую трубку трехходового
крана наполняют прибор газом или воздухом; через правую, имею-
щую внутри платиновую пластинку с узким отверстием, газ выпу-
скают. Баллон 7 имеет одну метку внизу, другую вверху и поме-
щается в широкой стеклянной трубке, в которую наливается вода,
служащая для поддержания постоянной температуры газа и воз-
духа. Источник исследуемого газа присоединяется каучуком к левой
трубке трехходового крана. При достаточном давлении газ сам на-
полняет баллон 7, вытесняя воду; если же давление газа мало или
если в баллон 7 набирают воздух, то опускают уравнительный сосуд
и закрывают кран в тот момент, когда уровень воды будет на не-
сколько сантиметров ниже метки. Наполнив баллон 7 газом, закры-
вают кран, поднимают уравнительный сосуд и выпускают газ через
узкое отверстие в правой трубке, повернув соответствующим обра-
зом кран. Наполнив и выпустив газ из баллона 7 два или три раза
для промывки соединительных трубок и прибора от остатков воз-
духа, набирают газ окончательно так, чтобы, когда уравнительный
сосуд будет поднят и установлен на соответствующей подставке,
уровень воды был на 2—3 см ниже метки. Соединив баллон 7 с пра-
вым ходом, где имеется отверстие, пускают в ход секундомер в мо-
мент прохождения уровня воды через нижнюю метку. При прохож-
дении уровня через верхнюю метку секундомер останавливают. Опыт
повторяют несколько раз с исследуемым газом и берут среднее из
измерений. Измерение скорости истечения воздуха можно произво-
дить до или после измерений с газом. В обоих случаях перед изме-
рением следует два-три раза промыть прибор, выпуская воздух через
узкое отверстие. Если сначала производят измерения с воздухом,
то предварительная промывка прибора все равно обязательна, так
как в большинстве случаев нельзя быть уверенным, что в приборе
не осталось газа от предыдущего опыта. Определив среднее из вре-
мени истечения воздуха и среднее из времени истечения газа, нахо-
дят удельный вес газа по указанной выше формуле.
В описанных приборах в качестве вытесняющей жидкости употреб-
ляется вода. Если исследуемый газ сильно растворим в воде, то из-
мерения этими приборами уже будут неточными. Для определения
удельного веса легко растворимых в воде газов можно заменить воду
ртутью (в приборе фиг. 111, б). Углеводородные природные газы в
воде плохо растворимы, поэтому их удельный вес можно определять
с точностью до 1—2% на всех приборах, конструкция которых осно-
вана на методе истечения и в качестве вытесняющей жидкости при-
меняется вода.
Помимо взвешивания и метода истечения удельный вес газа можно
определить при помощи так называемых газовых весов, основанных
302
на законе, по которому всякое тело, помещенное в газ, теряет в своем,
весе столько, сколько весит вытесненный им газ. Ниже описывается
одна из конструкций газовых весов.
Металлическая камера весов имеет двойные стенки, между кото-
рыми наливается вода, обеспечивающая постоянную температуру
газа и воздуха. В камере помещается коромысло 1 (фиг. 112), имеющее
на одном конце полый металлический запаянный цилиндр, а на дру-
гом массивный противовес. Коромысло опирается двумя стальными
иголками на агатовую пластинку и имеет две шайбочки, передви-
гающиеся на нарезке и служащие для установления коромысла в
равновесии и для регулировки его чувствительности. В боковую
стенку, которая находится около противовеса, вделано толстое стекло,
имеющее горизонтальную черту. Противовес на стороне, обращенной
7ZZZZZ2ZZZZZZZZZ2ZZZZZZZZZZZZZZZZZZ22Z
gZZZZZZZZZZZZSZZSStegZZZZZ^^^^^K
Фиг. 112. Газовые весы.
к стеклу, также имеет горизонтальную черту. При помощи лупы 2
легко можно заметить момент совпадения черты на противовесе
с чертой на стекле. Камера весов может сообщаться с внешним воз-
духом, исследуемым газом и вакуумом при помощи двух вентилей 3
и 4. Боковая крышка, прилегающая к противовесу, имеет нарезку
и прокладку. Отвинтив крышку, можно выдвинуть подставку вместе
с коромыслом.
Через боковой отвод 5 весы соединяются с ртутным манометром
(не показанным на фигуре).
Как известно, плотность газа прямо пропорциональна его да-
влению; сила, с которой газ действует снизу вверх на находящееся
в нем тело, пропорциональна плотности газа, а следовательно, и его
давлению. Предположим, что в двух различных газах коромысло
весов находится в равновесии (совпадение черты противовеса с чер-
той на стекле). Это означает, что при тех давлениях, при которых
коромысло находится в равновесии, оба газа имеют одинаковый
удельный вес. Отсюда следует, что чем меньше удельный вес данного
газа, тем большее давление необходимо, чтобы привести коромысло
весов в равновесие. Если р — давление воздуха, при котором коро-
303
мысло будет в равновесии, а /к — та же величина для испытуемого
газа, то искомый удельный вес D будет равен:
Агатовая подставка, на которую опирается коромысло, должна
быть совершенно горизонтальна, что достигается подвинчиванием
уравнительных винтов и проверяется постукиванием пальцем по
весам; если при этом коромысло сдвигается в одну какую-либо сто-
рону, это означает, что агатовая подставка имеет в эту сторону уклон.
Весы перед работой проверяют на плотность всех соединений; для
этой цели откачивают из весов воздух, а затем, закрыв вентиль,
через который производилась откачка, наблюдают за манометром.
Если в течение 5 мин. не будет никакого понижения уровня ртути
в манометре, то можно считать плотность всех соединений удовле-
творительной. После этой проверки наполняют весы сухим воздухом,
впуская его через трубку с хлористым кальцием или фосфорным
ангидридом. В конце осушительной трубки имеется вата, чтобы вос-
препятствовать попаданию пыли в камеру весов. Воздух откачивают
через вентили 3 и 4 при помощи хорошего масляного или ртутного
насоса. Если в весах находился газ от предыдущего определения,
то его удаляют путем нескольких повторных откачек и наполнений
прибора воздухом. Впускать воздух через вентиль следует очень
осторожно и закрывать его нужно в тот момент, когда черта на коро-
мысле совпадет с чертой на стекле. Рекомендуется сначала набрать
в весы воздуха больше, чем следует, а затем, закрыв вентиль 3, вы-
пустить излишек воздуха через вентиль 4; который допускает более
тонкую регулировку и позволяет изменять давление очень медленно
и постепенно. Когда будет достигнуто равновесие, отсчитывают дав-
ление манометра. Также отсчитывают и атмосферное давление по
барометру. После этого весы наполняют газом, удельный вес кото-
рого требуется определить. Для наполнения весов газом поступают
следующим образом. Вентиль 3 при помощи толстостенного каучука
соединяют с трехходовым краном ртутной бюретки или насоса. Затем,
оставляя вентиль 3 открытым, откачивают воздух из весов и из кау-
чука. Свободную трубку трехходового крана присоединяют к источ-
нику газа и из нее также откачивают воздух. После откачки опуска-
нием уравнительного сосуда газ набирают в бюретку или насос.
Закрыв вентиль 4} через который производилась откачка, соеди-
няют бюретку с весами и поднятием уравнительного сосуда напол-
няют весы газом. Наполнять весы газом следует осторожно, медленно
поворачивая трехходовый кран насоса, чтобы газ не входил в камеру
весов резкой и сильной струей. Первую порцию впущенного газа
следует опять откачать для промывки весов от остатков воздуха.
После этого, набрав в насос новую порцию газа, впускают его опять
в камеру весов до равновесия коромысла. Рекомендуется впустить газа
больше, чем следует, а затем, как уже упоминалось выше, закрыв вен-
тиль 3, выпустить осторожно излишек через вентиль 4. Давление,
которое будет показывать манометр в момент совпадения черты ко-
304
ромысла с чертой на стекле, необходимо заметить. Если равновесие
коромысла наступило при некотором вакууме, то разницу уровней
ртути в манометре следует вычесть из атмосферного давления. Если
равновесие наступило при давлении больше атмосферного, то эту
разницу следует прибавить к атмосферному давлению, так как для
определения удельного веса необходимо знать полное давление; это
относится как к воздуху, так и к испытуемому газу. В качестве при-
мера определения удельного веса газа можно привести следующий
расчет:
Барометрическое давление............................... 750 мм
Показание манометра с воздухом в приборе:
левое колено......................................... 390 »
правое » 396 »
Разница уровней ртути в манометре.......................—6 мм
Так как показания манометра означают некоторый вакуум в при-
боре (по сравнению с атмосферой), то для получения абсолютной
величины давления воздуха в весах следует из атмосферного давле-
ния вычесть разницу показаний манометра. Следовательно, давление
воздуха, при котором коромысло весов находится в равновесии равно
750 — 6 = 744 мм.
Показания манометра с газом в приборе:
левое колено...................................... 600 мм
правое ’ . .................................. 186 »
Разница уровней ртути в манометре . ;.............4-414 мм
Следовательно, давление газа, при котором коромысло находится
в равновесии, равно
750 + 414— 1164 мм.
Удельный вес газа равен
744
•11+=0,639.
При опытах с воздухом иногда равновесие еще не наступает и
при атмосферном давлении. В этом случае следует давление поднять
выше атмосферного, для чего воспользоваться бюреткой или ртут-
ным насосом.
Чувствительность коромысла можно изменять, поднимая или
опуская шайбочку, находящуюся на вертикальном стерженьке коро-
мысла. Положение равновесия коромысла можно изменять передви-
жением шайбочки на горизонтальном стержне. При определении
плотности очень легких газов (содержащих главным образом водо-
род или гелий) необходимо в качестве газа, с которым производится
сравнение, вместо воздуха употреблять чистый водород, так как
при той большой разнице давлений, которая необходима для равно-
весия коромысла в легком газе и в воздухе, манометр, имеющий длину
80 см, окажется слишком малым. Пользуясь водородом как стандарт-
ным газом, необходимо изменить положение равновесия коромысла,
чтобы оно наступало приблизительно при атмосферном давлении.
20 В. А. Соколов.
305
При определении удельного веса газа методом истечения и при
помощи газовых весов температура воздуха, взятого в прибор, бу-
дет иногда отличаться от температуры испытуемого газа. В этом слу-
чае в величину удельного веса газа следует ввести поправку по сле-
дующей формуле:
г. _ D
Uq~ Л + at •
где Do — истинный удельный вес;
D— то же, измеренный;
t — разница в температуре воздуха и газа;
а —коэфициент расширения газа, равный 0,00367.
Если газ холоднее воздуха, то в знаменателе следует взять знак
плюс, если же теплее, то знак минус.
В большинстве случаев температура воздуха и газа в течение
опыта меняется очень незначительно, и эту поправку следует вводить
только при желании получить максимально точные результаты.
Преимущество газовых весов перед другими методами определе-
ния удельного веса газов в том, что объем исследуемого газа не играет
роли. Поэтому при помощи специально устроенных газовых микро-
весов можно определить удельный вес очень незначительных количеств
газа. Так, например, автором были построены газовые микровесы,
в которых для определения удельного веса требуется только 1 см3
газа при нормальном давлении. Устройство газовых микровесов
в принципе такое же, как и описанных выше газовых весов, только
размер баллона и внутренний свободный объем сделаны минималь-
ными.
Как уже было упомянуто выше (глава VI), удельный вес очень
малых количеств газа можно определять с помощью «микроэффузио-
метра». Б. М. Накашидзе построил такой микроэффузиометр, с по-
мощью которого можно со значительной точностью определить удель-
ный вес газа при его количестве 0,1 см3 и даже менее.
§ 6. Определение теплотворной способности газов
Когда газ используется как топливо, то химический состав его
не играет существенной роли, важна лишь величина его теплотворной
способности. Поэтому определение теплотворной способности необ-
ходимо для многих практических целей. Зная точно состав газа,
легко вычислить его теплотворную способность, так как теплотвор-
ная способность каждого из компонентов хорошо известна, но точ-
ный и полный анализ углеводородного газа не всегда бывает из-
вестен, поэтому теплотворную способность газа часто определяют
экспериментальным путем, что сделать значительно легче, чем про-
извести полный анализ газа.
Определение величины теплотворной способности газа дает воз-
можность сделать некоторые выводы о составе газа. В некоторых
случаях, в частности, когда в газе присутствует лишь один горючий
компонент, измерение теплового эффекта при сожжении газа дает
вполне определенный ответ о содержании этого горючего компонента.
306
Теплотворная способность газа определяется при помощи спе-
циальных калориметров, из которых наибольшим распространением
пользуется калориметр, представленный на фиг. 113. При проведении
определений к калориметру А присоединяют газовый счетчик В,
регулятор давления С и весы для определения количества воды.
Определенное количество газа, указываемое счетчиком, при постоянном
давлении воздуха поступает в горелку, где и сжигается полностью
при достаточном доступе воздуха. Теплота, выделяющаяся при сго-
рании газа, поглощается водой, проходящей по калориметру, поэтому
температура выходящей из калориметра воды выше температуры
входящей. Зная разницу температур входящей и выходящей воды
Фиг. 113. Газовый калориметр.
и количество протекшей воды при сгорании известного количества
газа, легко вычислить теплотворную способность газа. Исследуемый
газ входит в газовый счетчик через патрубок 34, а через каучук 28
направляется в регулятор давления С. Регулятор давления пред-
ставляет собой открытый вверху металлический цилиндр, внутрь
которого входит металлический колпак; в цилиндр наливается вода,
а газ впускается под колпак. Металлические пластинки 41 наклады-
ваются на колпак и служат для регулировки давления, которое
определяется манометрол! 40. Вывинтив крышку 42, можно выпустить
воду из регулятора давления 43 через нижний конец .манометра.
Из регулятора давления через трубку 44 газ направляется в горелку
23, где и сжигается; за пламенем горелки наблюдают при помощи
зеркала 24, а за температурой поступающего в счетчик газа по термо-
метру 12.
20* 307
Калориметр представляет сооой металлический цилиндр, внутри
которого имеется еще несколько цилиндров; между стенками одних
протекает вода, между стенками других — воздух с продуктами
горения. Все эти цилиндры, по которым протекают вода и газы,
изолированы воздушной прослойкой от внешнего цилиндра. Весь
калориметр помещаемся на трех ножках 21 с установочными вин-
тами 22; калориметр при работе должен стоять вертикально, для
чего имеется отвес О. Полость калориметра между внешним цилин-
дром и всеми внутренними частями сообщается с атмосферным возду-
хом через трубку 15. Воздух, поступающий через промежуток между
горелкой 23 и калориметром, и продукты горения проходят вверх
по цилиндру 45, а затем вниз, по промежутку между двумя метал-
лическими цилиндрическими стенками, за которыми протекает вода;
при этом теплота, выделившаяся при горении, поглощается водой,
и воздух с продуктами горения в выходную трубку 16 поступает
уже холодным; для замера температуры выходящих газов имеется
термометр 13, тяга регулируется задвижкой 17. При сгорании угле-
водородных газов всегда образуется вода, которая конденсируется
в упомянутом выше промежутке между стенками и стекает вниз через
трубку 18 в измерительный цилиндр 20. Вода в калориметр поступает
по каучуку 2; предварительно она поступает в воронку 4 с постоянным
уровнем. После трубки 2 вода поступает в более широкую трубу;
излишняя вода, перелившись через край этой широкой трубы, ухо-
дит по трубке 3 в раковину. Между трубками 2 и 3 имеется еще трубка,
по которой вода поступает в калориметр. Благодаря подобному
устройству воронки вода поступает в калориметр всегда при постоян-
ном уровне. Количество поступающей воды регулируется краном 7.
Пройдя весь путь внутри калориметра, вода выходит через воронку 5
и трехходовый кран 6 или в раковину через трубку 8, или в ведро 19
через трубку 7, смотря по тому, как повернут кран 6. Благодаря
устройству воронок 4 и 5 разница в уровнях входящей и выходящей
воды все время остается постоянной, равной Н, чем обусловливается
равномерная скорость протекания воды по калориметру. Темпера-
тура входящей в калориметр воды измеряется термометром 10, а тем-
пература выходящей—термометром 77, укрепленным втулками 51.
Выходящая из калориметра вода перед термометром проходит через
специальное приспособление для перемешивания 47. Если прекра-
тить доступ воды в калориметр, то оставшуюся в нем воду можно
выпустить через кран 26. Воду взвешивают на весах 25; вместо взве-
шивания количество воды можно определить также по объему мер-
ным цилиндром; 46, 48, 49, 50, 52 — крепления различных частей
калориметра, 37 — установочные плиты счетчика, 34 — кран для
сообщения с воздухом, 39 — манометр, 29 — кран с регулировкой
скорости протекания газа, 38 — уровень, 32 — воронка, через кото-
рую счетчик наполняется водой, 30—трубка, через которую изли-
шек воды вытекает из счетчика, 31 — кран, 33 — камера со стек-
лом, через которое виден уровень воды в счетчике, 35 — кран для
выпуска лишней воды, 36 — втулка на нарезке, закрывающая от-
верстие, через которое можно выпустить всю воду из счетчика.
308
Перед тем как пускать в ход весь аппарат, газовый счетчик сле-
дует наполнить водой через крышку в воронке 32; уровень воды в
счетчике должен быть доведен до черты на стекле камеры 33; счет-
чик должен стоять горизонтально по уровню 38. По наполнении счет-
чика водой краны 34 и 31, а также крышку воронки 32 следует за-
крыть. Перед определением следует проверить плотность всех соеди-
нений. Для этой цели, соединив счетчик с регулятором давления и
горелкой, закрывают кран горелки и впускают в счетчик газ. Если
нигде утечки нет, стрелка счетчика двигаться не будет.
Первые порции газа, попавшего в горелку, смешиваются с
остатками воздуха в счетчике и регуляторе давления, поэтому перед
тем как зажечь горелку некоторое время выпускают эту смесь на
воздух. Когда горелку зажгут, то, впуская воздух в нижнюю часть
ее, делают пламя несветящимся, добиваясь таким образом полного
сгорания газа. Высота пламени должна быть небольшая—4—5 см
Перед тем как ввести горелку, калориметр наполняют водой, кото-
рая впускается по трубке 2. Кран 6 должен быть повернут таким
образом, чтобы вода из калориметра стекала в раковину. Скорость
прохождения воды по калориметру регулируется вентилем 7, а тем-
пература входящей и выходящей воды измеряется термометрами
10 и 11 с делениями до 0,1°. Для большей точности отсчеты по тер-
мометрам делаются при помощи луп 14. Воду, поступающую в кало-
риметр, можно брать из водопровода, но при желании получить наи-
более точные результаты следует брать воду из отдельного резервуара
емкостью около 100 л, расположенного на некоторой высоте над
прибором; в этом резервуаре температура воды комнатная, температу-
ра же водопроводной воды может быть значительно ниже комнатной
и подвергаться значительным колебаниям во время опыта, что пони-
жает точность определения. Впускать воду в калориметр следует
осторожно, чтобы вода не переливалась через край воронок 4 и 5.
Убедившись в том, что из калориметра вода вытекает, вводят го-
релку в калориметр, а под трубку 18 ставят какой-либо стаканчик.
За пламенем наблюдают при помощи зеркала 24; главная цель этих
наблюдений — смотреть, чтобы пламя не погасло и чтобы оно было
несветящимся, т. е. чтобы происходило полное сгорание. Теплота,
выделяющаяся при горении, поглощается водой, проходящей по
калориметру, поэтому температура выходящей воды начнет повы-
шаться и между температурой входящей и температурой выходящей
воды образуется некоторая разница. Температура входящей воды
может также несколько подняться, но разница между температурами
входящей и выходящей воды будет увеличиваться до тех пор, пока
не наступит равновесие, т. е. когда эта разница будет постоянной.
Равновесие означает, что вся теплота, выделяющаяся при горении,
поглощается водой, протекающей по калориметру. При одном и том
же количестве сжигаемого газа разница в температурах входящей
и выходящей воды зависит от количества воды, проходящей через
калориметр: чем больше это количество, тем меньше разница. Наи-
выгоднейшие условия опыта—когда разница равна 10—12°. При
«сухом» природном газе подобная разница получается, когда на 1 л
309
воды, проходящей через калориметр, приходится около 1,5 л сжигае-
мого газа. Когда установится разница в 10—12°, что достигается
регулировкой вентилем 1 количества поступающей в калориметр
воды, приступают к определению теплотворной способности газа.
В тот момент, когда стрелка счетчика проходит через нулевое
давление, поворачивают кран 6 таким образом, чтобы вода из кало-
риметра попала в ведро 19, и одновременно под трубку 18 подста-
вляют сухой мерный цилиндр 20, отставив стаканчик, который ста-
вился лишь для того, чтобы конденсирующаяся вода не капала на
стол. С момента поворота крана 6 вся вода, проходящая калориметр,
попадает в ведро 19. Обычно вполне достаточно сжечь 20—30 л газа;
в момент, когда необходимое количество газа будет сожжено, кран 6
ставят в прежнее положение, чтобы вода из калориметра стекала в
раковину, и отставляют цилиндр 20. Во время сжигания газа не-
обходимо часто отмечать температуру, показываемую обоими термо-
метрами; это даст возможность проверять постоянство разницы тем-
ператур; среднее из показаний первого термометра вычитается из
средней величины показаний второго термометра, и полученная раз-
ница принимается за окончательный результат.
Скорость сжигания газа не должна быть очень большой; вода в
калориметре в течение часа должна поглощать не больше 1000 ккал,
следовательно, если теплотворная способность газа равна 8000 ккал
на 1 м8, то скорость сжигания подобного газа не должна превышать
125 л}час. Зная количество сожженного газа, количество воды, про-
шедшей за время сожжения через калориметр, и разницу между
температурами выходящей и входящей воды, можно вычислить тепло-
творную способность газа.
Объем сожженного газа, отмеченный счетчиком, следует привести
к нормальным условиям. Скорость выходящих из калориметра газов
при помощи задвижки 17 необходимо поддерживать такой, чтобы
температура их была равна температуре газа, входящего в счетчик.
Если же между этими температурами имеется разница, то в вычис-
ление вводят поправку, учитывая, что на один объем сгоревшего газа
через отверстие в нижней части калориметра засасывается 7—8 объ-
емов воздуха и что теплоемкость 1 м8 воздуха равна около 0,3 ккал.
Все то количество теплоты, которое образовалось при горении газа,
поглощено водой; поэтому произведение из веса воды в килограммах
на разницу между показаниями термометров 10 и 11 будет равно
тому количеству теплоты в килокалориях, которое выделилось при
сожжении взятого объема газа. Разделив полученное количество
теплоты на объем газа, получим теплотворную способность газа:
где Н —теплотворная способность газа, ккал 1м8;
W — количество воды, г;
/—разница температур входящей и выходящей воды,
О —объем газа, л.
310
Продуктами горения углеводородного газа являются углекислота
и вода; при переходе паров воды в жидкое состояние выделяется
теплота парообразования, поэтому теплотворная способность газа,
продукты горения которого охлаждены ниже 100°, отличается от
теплотворной способности того же газа, продукты горения которого
имеют температуру выше 100°, даже если ввести поправку на разность
температур отходящих газов. Различают «верхнюю теплотворную
способность», в которую входит вся теплота, выделяющаяся при
горении и охлаждении отходящих газов до комнатной температуры,
и «нижнюю теплотворную способность», которая равна верхней тепло-
творной способности за вычетом теплоты, выделившейся при конден-
сации водяного пара. Верхнюю теплотворную способность Н вычи-
сляют по приведенной выше формуле, а нижнюю теплотворную спо-
собность по формуле
wt—600 к
°" G
где К—количество воды, набранное во время опыта в мерный ци-
линдр 20, см3.
Произведя вычисления по приведенным формулам, получаем
величину теплотворной способности газа в условиях опыта. Чтобы
получить абсолютную величину теплотворной способности, необхо-
димо объем газа привести к нормальным условиям и ввести поправку
на присутствие водяных паров, так как газ, проходя через счетчик
с водой, насыщается ими.
Поправочный фактор вычисляется по формуле
273.(В + Р1 — S)
J60- (273+ tg)
где В —барометрическое давление в момент опыта, приведенное
к 0°, мм рт. столба;
Pi—давление в счетчике, определяемое манометром 39, мм рт.
столба;
S —упругость водяных паров при температуре газа, мм рт.
столба;
tg—температура газа в счетчике, °C;
/—поправочный фактор счетчика, так как его показания могут
быть не совсем точными.
Для перевода показаний водяного манометра в миллиметры ртут-
ного столба следует величину водяного столба в миллиметрах раз-
делить на 13,6. Чтобы ввести поправки, объем газа G следует умно-
жить на F; следовательно, мы будем иметь окончательно:
wt -600 к
о- GF
Правильность определения теплотворной способности газа в силь-
ной степени зависит от правильности показаний счетчика; для про-
311
верки показаний счетчика употребляется точно выверенная литро-
вая колба.
Автоматические калориметры. Если сделать постоянным отно-
шение между количеством сжигаемого газа и количеством воды,
проходящей через калориметр, то разница температур входящей и
выходящей воды будет прямо пропорциональна теплотворной спо-
собности газа. Разницу температур удобно измерять при помощи тер-
мопары, один спай которой погружен во входящую, а другой в—выхо-
дящую воду. Если температуру входящей воды поддерживать по-
стоянной, то температура выходящей воды будет прямо пропорцио-
нальна теплотворной способности газа; в этом случае температуру
выходящей воды можно измерять самопишущим прибором, который
можно прокалибровать так, чтобы он непосредственно показывал
теплотворную способность.
На этих принципах основан автоматический самопишущий газо-
вый калориметр.
Микрокалориметры. Для определения теплотворной способности
газа вышеупомянутым калориметром требуется значительное коли-
чество газа, а именно, несколько десятков литров, что делает не-
возможным применение этого прибора для испытания небольших
образцов газа. Если требуется определить теплотворную способность
небольшого образца газа, можно применять микрокалориметры.
Для определения теплотворной способности на микрокалориметре,
представленном на фиг. 114, а, требуется всего 30 сл/3 газа. Принцип
действия этого калориметра заключается в том, что смесь газа с воз-
духом взрывается в специальной пипетке, а количество образовав-
шегося тепла определяется по увеличению объема воздуха, находя-
щегося в резервуаре, окружающем пипетку.
Исследуемый газ вводят в бюретку 7 через кран 6, опустив урав-
нительный сосуд с ртутью 3 и открыв краны 4 и 5. В баллоне 2 на-
ходится нормальный объем воздуха, равный в сухом состоянии при
0° и 760 мм рт. столба 30 см3. В бюретке и в баллоне поверх ртути
имеется немного воды, так что газ всегда насыщен водяными парами.
Когда уровни ртути в нижних частях бюретки и баллона 2 сравня-
ются, это означает, что в бюретке имеется такое количество газа,
которое при 0° и 760 мм рт. столба равно 30 см3. Бюретка и баллон
помещены в охранную трубку, наполненную водой. После взятия
газа пипетку 7 емкостью около 200 см3 наполняют водой из бутыли 8.
а затем в нее переводят газ из бюретки 7. Затем поворачивают кранР
так, чтобы в пипетку 7 входил воздух. Вода из пипетки 7 будет сте-
кать в бутыль 8, наполнит ее и поднимется по трубке, вставленной
в пробку; когда вода установится здесь на том же уровне, как и г
трубке, под пипеткой 7, краны 9 и 10 закрывают. Резервуар, окру-
жающий пипетку 7, сообщается с резервуарол! 72, содержащим ке-
росин, от которого идет наклонный манометр со шкалой. Конец мано-
метра, присоединенный к резервуару 72, заполнен также керосином;
другой конец манометра сообщается с резервуаром 13 и резиновой
312
грушей 14. Количество керосина в резервуаре 12 таково, что нить
керосина в наклонном манометре при открытом кране 15 стоит на
нулевом давлении. Количество воздуха в резервуаре 77 должно быть
таким, какое было взято при градуировании шкалы манометра. Не-
обходимо, чтобы весь прибор был установлен точно горизонтально
по уровню.
Закрыв кран 15, наблюдают, стоит ли керосиновый манометр
на нуле. Если этого нет, выжидают, пока манометр установится.
Затем, пустив ток в индукционную катушку 16, получают в ре-
зервуаре 7 искровый разряд, который и взрывает смесь газа с воз-
духом. Как только произойдет вспышка, керосиновый манометр нач-
нет подниматься, но прежде чем манометр достигнет максимума,
следует открыть кран 9, чтобы впуском воздуха компенсировать раз-
режение, происшедшее в результате сокращения объема газовой
смеси при сгорании. Если воздуха не впускать в пипетку, то мы
определим теплотворную способность газа при постоянном объеме,
тогда как обычно требуется определять теплотворную способность
при постоянном давлении. После взрыва газа столбик керосина в
манометре в течение 20—30 сек. поднимется до максимума, где и
остановится на некоторое время; положение максимума замечают
на шкале. На этом заканчивается опыт. Пипетку 7 освобождают от
газов, подняв бутыль 8, и прибор вновь готов для следующего опре-
деления; необходимо только подождать, пока температура воздуха
в резервуаре 77 совершенно сравняется с комнатной и жидкость в.
манометре упадет до первоначального положения.
313
Шкалу манометра градуируют так, что на ней непосредственно
отсчитывается теплотворная способность исследуемого газа.
Шкалу градуируют при помощи какого-либо чистого горючего
газа, теплотворная способность которого хорошо известна. Берут
определенное количество газа и получают одну точку; затем берут
двойное количество и получают вторую; между этими двумя точками
наносят все остальные.
Описанный микрокалориметр приспособлен главным образом для
определения теплотворной способности светильного газа, водяного
газа и т. п. При определении же теплотворной способности природ-
ных горючих газов отношение между объемом пипетки для взрыва
и объемом бюретки должно быть увеличено, в противном случае
кислорода, имеющегося в воздухе в объеме пипетки, окажется не-
достаточно для полного сожжения газа; можно, разумеется, при
испытании брать меньше 30 см3 газа, но тогда конструкцию бюретки
следует несколько изменить, чтобы эти небольшие объемы измерялись
с достаточной точностью. Вместо уменьшения объема испытуемого
газа можно оставить объем прежним, но производить сожжение не
с воздухом, а с кислородом.
На фиг. 114, б представлена другая схема микрокалориметра.
Пипетка 1 емкостью около 100 см3 служит одновременно для
взрыва газа и для измерения его объема. Пипетка 7 имеет двойные
стенки, между которыми налит керосин; к наружной стенке припаяна
капиллярная трубка 2. Для установки уровня керосина в трубке 2
служит винт 3. Пипетка сообщается каучуком с уравнительным со-
судом 4} наполненным разбавленной серной кислотой (1 : 10). В ниж-
ней части пипетки впаяны платиновые электроды, соединенные с
индукционной катушкой 5. Аккумулятор 6 может давать ток или
в катушку, или непосредственно в пипетку.
Исследуемый газ вводят через верхний кран пипетки и измеряют;
так как верхняя часть пипетки узкая, то отсчет объема следует про-
извести достаточно точно. Для испытания природного горючего газа
берут 5—8 см3. После замера газа через третий ход верхнего крана
вводят в пипетку воздух; как только кислота стечет до нижнего кра-
на пипетки, закрывают верхний и нижний краны. Перед сжиганием
газа отмечают положение столбика керосина в капилляре. После
сжигания газа керосин нагреется и начнет расширяться, поднимаясь
по капилляру; наблюдая за подъемом керосина, отмечают наивысшее
положение столбика. Для сравнения сжигают гремучий газ, который
получают, заполнив пипетку кислотой из уравнительной бутылки и
пропустив через платиновые электроды ток непосредственно из ак-
кумулятора; гремучего газа следует брать около 20 см3. Наблюдае-
мое расширение керосина считают пропорциональным количеству
тепла, выделяющегося при горении. Теплотворная способность испы-
туемого газа будет равна:
где G —объем испытуемого газа, см3;
314
Л: —подъем керосина, мм;
С?!—объем гремучего газа, см3;
/rj—подъем керосина при сжигании гремучего газа, мм;
//—теплотворная способность гремучего газа, кал.
Для получения большей точности следует сжигать такое коли-
чество гремучего газа, чтобы вызываемый им подъем керосина был
одинаков с подъемом керосина при сжигании испытуемого газа.
На описанных микрокалориметрах можно определять только
аысшую теплотворную способность.
ГЛДВА ДЕВЯТАЯ
АВТОМАТИЧЕСКИЕ ГАЗОАНАЛИЗАТОРЫ
При промышленном производстве газов, а также в различных
промышленных установках, где состав газов может служить
средством контроля за протекающими в установке процессами, тре-
буется во многих случаях проведение непрерывного газового ана-
лиза с автоматической записью результатов анализа.
Иногда при исследовании природных газов требуется непрерыв-
ный анализ с автоматической записью. Так, например, подобный
анализ требуется при газовом кароттаже.
Следует учесть, что автоматический анализ, как правило, отнюдь
не точнее простого анализа отдельной пробы газа на каком-либо из
приборов, описанных в предыдущих главах. Часто непрерывный газо-
вый анализ с автоматической записью уступает по точности анализу
отдельных образцов газа. Кроме того, автоматический анализ приспо-
соблен главным образом к исследованию бинарных смесей, а именно,
к определению примеси одного газа (или группы схожих по свой-
ствам газов) к другому, основном)/ газу. Непрерывный анализ много-
компонентных газовых смесей в настоящее время еще слабо разра
ботан. Непрерывный автоматический или полуавтоматический га-
зовый анализ применяется, когда требуется непрерывно следить
за составом получающихся или выделяющихся газов и когда обыч-
ный, так сказать, «ручной анализ» не в состоянии обеспечить нужную
быстроту определений или обходится значительно дороже автомати-
ческого анализа. В связи с этим приборы для непрерывного анализа
в большинстве случаев основаны на физических методах определения,
обеспечивающих немедленное получение результатов анализа, обыч-
но в виде показаний стрелки гальванометра и соединенного с ним
самописца. Быстрое получение результата анализа особенно важно,
когда это влечет за собой необходимость немедленного вмешатель-
ства в ход контролируемой установки, поэтому при непре-
рывном анализе часто применяют автоматические реле, регулирую-
щие ход контролируемого процесса, или соответствующие сигнали-
заторы.
Ниже описываются наиболее употребительные приборы для не-
прерывного газового анализа.
316
§ 1. Автоматические химические газоанализаторы
В основу химических газоанализаторов для определения СО2
положено поглощение этого газа раствором едкого кали (фиг. 115, а)
[13, 45]. Для засасывания газов в сосуд с раствором едкого кали
используется напор струи воды из водоподающей сети. Вода, не-
прерывной струей стекающая по трубе вниз, образует в ней разре-
жение и засасывает газы по трубке 7. Засосанные газы проходят через
измерительный сосуд 6 в трубку 2 и выходят наружу.
Вода через воронку попадает в нижний сосуд 3, плавающий в
закрытом резервуаре 4, наполовину наполненном дестиллированной
водой. Вода, поступающая в сосуд 5, заполняет сифонную трубку 5
слева и вытесняет дестиллированную воду из резервуара 4 в изме-
рительный сосуд 6.
317
Когда поднимающаяся в сифоне вода дойдет до уровня, сифон
начинает действовать и опоражнивает сосуд 3. Вода из измерителя
возвращается в резервуар 4. Таким образом, вода в измерителе пе-
риодически поднимается и спадает.
Когда поднимающаяся в измерителе вода дойдет до уровня, про-
хождение через него газов прекращается: газы выходят наружу через
верхний гидравлический затвор 12. Дальнейший подъем воды про-
талкивает газы в верхний резиновый мешок 9 до тех пор, пока вода
не дойдет до метки Ь.
С этого момента в резиновый мешок 9 могут попасть лишь те газы,
которые заключаются в ведущей к нему трубке; содержимое же соб-
ственно измерителя проталкивается по капиллярной трубке в со-
суд с едким кали 8, в котором происходит поглощение СО2.
Измеритель рассчитан таким образом, что объем его от линии с
до черточки d на капиллярной трубке равен 100 см3.
Сифон установлен таким образом, что начинает забор воды из
сосуда 3 как раз в тот момент, когда уровень воды в измерителе до-
ходит до линии d. Таким образом, в сосуд с едким кали попадает ровно
100 см3 газов, которые в свою очередь вытесняют часть раствора ед-
кого кали в колокол 10. Воздух из колокола выходит наружу через
открытые боковые трубочки. Когда уровень едкого кали доходит
до метки /, сообщение трубочек с наружным воздухом прерывается
и колокол начинает подниматься, приводя в движение записываю-
щее перо 11. Внутренний объем колокола таков, что первые 80 см3
газов, поступающих в сосуд с едким кали, поднимают уровень жид-
кости в колоколе от е до /. Цилиндрическая часть колокола над /
имеет такие размеры, что остальные 20 см3 газов (предполагается,
что они не будут поглощены, даже частично, едким кали) заставят
пишущее перо провести черту по всей ширине диаграммы.
Так как СО2 поглощается едким кали, то пишущее перо проводит
черту не по всей ширине, а соответственно тому количеству газов,
которое поступает в цилиндрическую часть колокола после погло-
щения СО2.
Перо прекращает запись в тот момент, когда сифон начинает опо-
ражнивать сосуд 3; давление во всех частях прибора падает, перо
опускается и вновь повторяются все описанные выше явления.
Другой газоанализатор, основанный на том же принципе погло-
щения СО2 раствором едкого кали, представлен по схеме фиг. 115, б.
Цилиндрический резервуар 1 наполняют водой посредством труб-
ки 2. Когда уровень воды доходит до начинает действовать си-
фон 3, который опоражнивает резервуар 1. Измеритель 4, служа-
щий для засасывания определенного объема газов, остается напол-
ненным водой до тех пор, пока уходящая из резервуара 1 вода не
опустится до уровня Л2; в этот момент газам открывается доступ в
прибор через трубку 5. Так как столб воды в измерителе 4 тяжелее
столба воды в трубке 6, вода из него вытекает в резервуар 1 и засасы-
вает газы через трубки 5 и 6.
Вода, продолжая уходить, достигает уровня Д2, сифон прекра-
щает свое действие и сосуд 1 опять начинает наполняться.
318
Поднимающаяся вода доходит до уровня А2, прерывает сообще-
ние с газами, идущими через трубку 5, и вытесняет их из измерителя 4
через трубку 7 в сосуд 8, наполненный раствором едкого кали (КОН),
который поглощает СО2. Остатки газов по трубке 9 уходят в коло-
кол 10, к которому прикреплено записывающее перо, проводящее
на диаграмме 12 черту, длина которой обратно пропорциональна
количеству поглощенных газов.
Фиг. 116.
В тот момент, когда черта на диаграмме проведена, вода начинает
опять опускаться и, дойдя до уровня Л2, открывает выхлопную труб-
ку 77, через которую выходят наружу газы из колокола 10, и прибор
опять готов для нового анализа.
Частота анализов зависит от скорости, с какой наполняется водой
резервуар 7; ее можно регулировать при помощи водоподающего
крана.
В приборе, изображенном на фиг. 116, измерение содержания
СО2 в газах сводится к измерению разрежения, создаваемого вслед-
ствие поглощения СО2 сухим NaOH.
319
Газ через трубку 11 проходит в пористый сосуд 7 и по выходе из
него разделяется на две струи, как указано на чертеже стрелками.
Газы, подвергающиеся анализу, следуют по пути, указанному
жирными стрелками, проходят через патрон 2, наполненный сухим
NaOH, который поглощает СО2, и, наконец, выходят через отверстие 3
и кран 4 в трубку 5, ведущую к аспиратору.
Газы, проходя через отверстие 76, находятся под влиянием раз-
режения, производимого всасывающим действием особого аспира-
тора. Так как едкий натр поглощает СО2 в коробке 2, в которой по-
мещен патрон с едким натром, разрежение увеличивается. Если вса-
сывающее действие аспиратора постоянно, величина вакуума в короб-
ке 2 зависит только от содержания СО2 в дымовых газах. Коробка 2
посредством трубки 6 и трехходового крана 7 сообщается с указа-
телем, состоящим из обыкновенной манометрической ртутной трубки,
градуированной непосредственно на процентное содержание СО2;
8 — сосуд с ртутью, сообщающийся с наружным воздухом. Для пра-
вильности работы аппарата необходимо, чтобы дымовые газы имели
постоянную скорость, т. е. необходимо, чтобы всякие изменения
силы тяги в трубу или силы засасывания аспиратора автоматически
компенсировались и сводились на-нет.
Для автоматической компенсации изменений силы тяги часть
газов, как уже упомянуто, по выходе из пористого сосуда 7 идет в
сосуд 9 с керосином, проходит поверх его и особой боковой трубкой,
запирающейся краном 10, соединяется с аспиратором. Таким обра-
зом, поверх керосина в сосуде 9 создается такое же разрежение, ка-
кое существует в трубке 77. В сосуд 9 погружена почти до дна осо-
бая, открытая сверху трубка, через которую проходит в сосуд 9 воз-
дух и пузырьками поднимается к поверхности, откуда удаляется
по упомянутой трубке.
Если тяга в трубу увеличивается, разрежение над поверхностью
керосина усиливается и воздух начинает быстрее поступать в сосуд 9,
что уменьшает в нем разрежение; наоборот, если тяга уменьшается,
разрежение уменьшается и воздух поступает через трубку слабее,
что увеличивает разрежение. Благодаря этому регулятору разре-
жение над поверхностью керосина остается приблизительно постоян-
ным, чем и достигается постоянство скорости дымовых газов. Вели-
чина разрежения измеряется U-образной трубкой 13. Чтобы достичь
заранее установленной величины разрежения, нужно только отрегу-
лировать положение трубки 72 (поднимая или опуская ее) так, чтобы
уровни жидкости в U-образной трубке совпадали с указателями
шкалы 14.
Постоянство засасывающего действия аспиратора создается спе-
циальным регулятором.
Патрон с едким натром вставляют в резиновое кольцо. Когда
весь запас NaOH в патроне перешел в Na2CO3, нужно отвинтить
нижнюю половину коробки, вынуть отработавший патрон 2 из коль-
ца 75 и вставить новый.
На фиг. 117 представлена еще одна схема автоматического газо-
анализатора.
320
Для периодического засасывания газов и продвижения определен-
ного объема их в сосуд с раствором едкого кали используется струя
воды с давлением 0,5 ат. Вода через трубку 7 входит в расширение 2,
на дне которого находится немного ртути, и через дырочки в верх-
ней части его проникает в сосуд 3, из которого вытесняет ртуть в
трубки 4, 5, 6. Трубка 4 расширяется наверху в сосуд грушевидной
формы, соединяющийся посредством трубки 7 с наружным возду-
хом.
Фиг. 117.
Трубка 5 имеет два расширения 8 и 9, соединенные друг с другом
узкой трубочкой. Ртуть из сосуда 3 поднимается в трубках 4} 5, 6
до тех пор, пока давлением той же воды вся ртуть из расширения 2
не будет вытеснена по трубке 10 в грушевидное расширение трубки 4.
Так как ртуть в расширении 2 не находится в связи с ртутью в со-
суде 3, а расположена несколько выше, ртуть в трубке 10 выли-
вается вся, в то время как уровень ртути в трубке 5 и расширениях
8 и 9 только достигает нижнего края цилиндрического сосуда, нахо-
дящегося над обоими расширениями 72 и 77.
Когда вся ртуть из трубки 2 вытеснится в грушевидную полость
трубки 4, воздух получает свободный доступ к воде; давление в со-
21 В, А, Соколов. 321
суде 3 падает, ртуть из трубок 4, 5 и 6 поступает обратно в сосуд 3
и попадает в расширение 3, переливаясь через отверстия. Сообщение
с воздухом прекращается, и давление воды опять обусловливает
подъем ртути в трубках. Отступающая из шаровидного расшире-
ния 9 ртуть создает в нем пустоту и засасывает дымовые газы через
трубку 77.
Подъем ртути в трубках вытесняет газ из шара 9 через 72, 13,
14, 15, 16, 77 и 18 в сосуд 19 с раствором едкого кали, где происходит
поглощение СО2. Остаток газов выходит по трубкам 20 и 6 на воз-
дух до тех пор, пока поднимающаяся в трубке 6 ртуть не прервет
сообщения с воздухом. Дальнейшее движение ртути вверх вытес-
няет остатки дымовых газов в измерительный колокол. Колокол
поднимается, поворачивает колесико 21, захватывающее шкивок 22.
К последнему прикреплен на металлической цепочке штифт, кото-
рый проводит черту на бумаге, натянутой на барабан 23, вращаю-
щийся вокруг своей оси при помощи помещенного внутри часового
механизма. Бумага сворачивается с барабана 23, и готовые диаграм-
мы наворачиваются на барабан 24.
Ширина диаграммы выбирается такая, чтобы полная амплитуда
колокола (когда его наполняют полным объемом дымовых газов до
поглощения СО2) соответствовала всей ширине диаграммы. При та-
ких условиях черта, проводимая штифтом, пропорциональна остат-
кам дымовых газов и, следовательно, чистая часть диаграммы про-
порциональна количеству СО2.
Вода, поступающая из крана 25, поднимается одновременно по
трубке 1, змеевику 31 и трубке 26 в цилиндр 27, в котором ходит пор-
шень. Шток поршня соединен с трехходовым краном, имеющим ртут-
ный запор.
Под давлением воды поршень поднимается и остается в таком
положении до тех пор, пока сосуд не имеет сообщения с наружным
воздухом. Как только это сообщение устанавливается, давление под
поршнем падает и поршень под влиянием пружины опускается. Каж-
дое поднятие поршня перемещает ртутный вентиль таким образом,
что попеременно трубка 16 сообщается то с трубкой 77, то с трубкой 28.
После того как первая порция газов прошла по трубке 77, трех-
ходовый кран передвигается поднятием поршня в такое положение,
что газы проходят по трубке 28 и попадают в фарфоровую трубку 29,
наполненную окисью меди. Трубка 29 обогревается электрической
печью 30. СО сгорает в СО2, и вся масса газов по трубке 18 проходит
в сосуд с едким кали 19 и т. д.
Черта на диаграмме, соответствующая новой величине остатков
газов, будет короче, и разность между первой и второй чертой дает
количество СО.
Для получения сравнимых результатов необходимо, чтобы ана-
лиз газов происходил при постоянных температурах и давлении и
одинаковой степени их влажности.
Постоянное давление газов достигается благодаря системе ртут-
ных запоров-вентилей. Для получения постоянной температуры
вода из крана, поднимающаяся к поршню по трубке 7 змеевиков 31..
322
проходит вдоль колокола, в котором собираются измеряемые газы,
охлаждаемые таким образом протекающей водой.
Для насыщения газов парами воды в цилиндрической части над
обоими шаровидными расширениями наливается поверх ртути не-
много воды, сквозь которую должен проходить засасываемый по
трубке 11 газ. Таким образом, он одновременно и охлаждается, и
насыщается парами воды.
§ 2. Анализ газа методом сравнения теплопроводности
Абсолютную величину теплопроводности газа определить до-
вольно трудно, но сравнить теплопроводность некоторого «стандарт-
ного газа» с исследуемым гораздо легче.
Если мы имеем некоторую камеру, стенки которой поддержива-
ются при постоянной температуре, и в этой камере имеется тонкая
проволока, накаливаемая электрическим током, то температура про-
волоки зависит от теплопроводности того газа, который наполняет
камеру. Для газа с большой теплопроводностью температура про-
волоки ниже, чем для газа с малой теплопроводностью.
При соответствующих размерах камеры потеря тепла проволокой
через излучение, через конвекцию, а также вследствие теплопровод-
ности проводов, подводящих ток к проволоке, может быть сведена
к минимуму и температура проволоки будет зависеть главным обра-
зом от теплопроводности газа. От температуры проволоки зависит
сопротивление; поэтому, измеряя сопротивление проволоки в ка-
мере, мы получил! величины, определяющие температуру проволоки,
а следовательно, и теплопроводность газа, наполняющего камеру.
Сравнивая сопротивления двух проволок, окруженных двумя га-
зами, мы найдем отношение теплопроводностей этих газов. Если
состав одного газа известен и постоянен, а другой газ имеет такой
же состав, но содержание одного из компонентов может меняться, то,
сравнив теплопроводности газов, можем определить и содержание
этого компонента, если предварительно проградуируем прибор.
Схема прибора для анализа газа путем сравнения теплопровод-
ностей изображена на фиг. 118, п. Здесь 1 и 2 — две металлические
камеры с проходящими через них платиновыми проволоками, 3 и 4 —
постоянные сопротивления, 5 — гальванометр, 6 — амперметр, 7 —
батарея, 8 — реостат. Величина электрического тока, идущего от
батареи 7, при измерениях должна оставаться постоянной; это до-
стигается регулировкой при помощи реостата 8 и амперметра 6. Если
две одинаковые камеры 1 и 2 наполнены одним и тем же газом, то
температура проволок будет одинакова и при равных сопротивле-
ниях 3 и 4 гальванометр покажет отсутствие тока. Если в камере
находятся газы с различной теплопроводностью, сопротивления
плечей мостика будут различны и через гальванометр пойдет ток.
Чем больше разница в теплопроводности газов, тем больший ток
пойдет через гальванометр. При измерениях в одну из камер впу-
скают стандартный газ, а в другую исследуемый. Перед измерениями
прибор необходимо калибровать, для чего в одну из камер впу-
21* 323 -----------
скают стандартный газ, а в другую — тот же стандартный газ, сме-
шанный с известным количеством определяемого компонента. Заме-
чая положения стрелки гальванометра при различном процентном
содержании определяемого компонента, получают калибровку при-
бора, при помощи которой уже определяют содержание этого компо-
нента в исследуемом газе. Величина переменного сопротивления 9
подбирается при калибровке.
Большая скорость протекания газа может повести к ошибкам.
Поэтому в описанном приборе применяют такое устройство камер,
чтобы газовый поток непосредственно не проходил мимо проволок
Фиг. 118. Газоанализатор, основанный на сравнении
теплопроводностей газов.
(фиг. 118, в). Изменение давления газа в камере существенного зна-
чения не имеет, так как теплопроводность газа в некотором диапа-
зоне давлений от него не зависит. Для повышения чувствительности
применяют также сдвоенные камеры, как это показано на фиг. 118, в.
Параллельно с гальванометром 1 здесь включен самописец 2. Пла-
тиновая проволока в камерах не должна при измерениях нагреваться
выше 400°.
Вместо схемы, изображенной на фиг. 118, а, можно применять
несколько иную схему, изображенную на фиг. 118, б. Эта схема отли-
чается от предыдущей тем, что гальванометр 5 нулевой. Компенса-
ция производится при помощи движка 6, передвигаемого по про-
волоке, на линейке которой и наносится калибровка прибора. Сопро-
321
тивления 3 и 4 подбирают таким образом, чтобы на проволоку 7 при-
ходился требуемый интервал процентного содержания определяе-
мого газа.
При необходимости получить более точные результаты пользу-
ются потенциометром.
Стандартный газ во всех схемах приборов должен быть или су-
хим, или насыщенным водяными парами, смотря по тому, является
ли исследуемый газ сухим или насыщенным парами.
Главное применение описанный метод находит при построении
самопишущих приборов для анализов газа, непрерывно доставляе-
мого каким-либо источником. Газ проходит через камеры, и галь-
ванометр или самописец отмечает содержание определяемого компо-
нента.
Сравнением теплопроводностей можно определить содержание
только одной составной части, если же в смеси газов меняется содер-
жание двух или больше компонентов с различными теплопроводно-
стями, то этот метод не дает правильных результатов. В последнем
случае этот метод можно комбинировать с сожжением газа или с
адсорбцией отдельных компонентов, определяя, например, тепло-
проводность до и после сожжения или до и после адсорбции и т. д.
Описанный прибор применяется чаще всего для определения СО2
в топочных и иных газах, но может применяться и для других
определений. Чем больше разница в теплопроводности определяемого
компонента и остальной части газа, тем выгоднее условия для при-
менения данного метода.
§ 3. Автоматический газовый анализ, основанный на измерении
теплоты сгорания
Для ряда технических целей нефтяной и газовой промышленности
требуется непрерывный автоматический или полуавтоматический ана-
лиз на содержание углеводородных газов. Широкое применение по-
лучил подобный анализ при газовом кароттаже буровых скважин,
при контроле за содержанием горючих газов в воздухе и для многих
других целей. Подобный анализ еще ранее получил применение для
контроля за содержанием СО в топочных газах.
Общая схема анализа сходна со схемой, основанной на сравне-
нии теплопроводностей газов и описанной в предыдущем параграфе.
Схема основана на применении мостика Уитстона, в котором в ка-
честве одного из плечей включена платиновая проволока, накали-
ваемая электрическим током до температуры, обеспечивающей сго-
рание углеводородных газов. Платиновая проволока помещена
в камеру 7, через которую просасывается анализируемый газ (фиг.
119). Камера устроена таким образом, чтобы пропускаемый газ не-
посредственно не омывал накаленной проволоки, а доходил до нее
благодаря диффузии через отверстия в охранной трубке. Другое
плечо мостика представляет собой такую же платиновую проволоку,
накаливаемую ‘ электрическим током и помещенную в такую же ка-
меру 2, но содержащую воздух, лишенный углеводородов. Третье
325
и четвертое плечи 3 и 4 представляют собой постоянные сопротивле-
ния. Амперметр 5 контролирует силу тока, идущего через платино-
вые проволоки 6 и регулируемую реостатом 7.
При пропускании через камеру 1 чистого воздуха гальванометр 8
должен оставаться в покое, что обеспечивается соответствующим
подбором сопротивлений 3 и 4.
Если в анализируемом газе имеются углеводороды, то они будут
сгорать на проволоке, благодаря чему ее температура будет выше,
чем проволоки, находящейся в чистом воздухе. Нарушение ба-
ланса в мостике обнаруживается по отклонению стрелки гальвано-
метра. Шкала гальванометра
Фиг. 119. Газоанализатор для го-
рючих газов с применением сож-
жения.
проградуирована на процентное содер-
жание углеводородов. Присоединен-
ный к прибору самописец регистри-
рует на ленте концентрации углево-
дородов через определенные проме-
жутки времени (например, через каж-
дые 15 сек.).
Для правильных показаний при-
бора требуется, чтобы скорость про-
хождения анализируемого газа через
камеру 7 была постоянной. Поэтому
прибор снабжен реометром или иным
приспособлением, показывающим ско-
рость течения газа. С помощью соот-
ветствующего регулирующего крана
скорость пропускания анализируе-
мого газа через камеру 7 поддержи-
вают постоянной, следя за показа-
ниями реометра (ротаметра). Калиб-
ровка прибора точно так же должна
проводиться при той же скорости
пропускания газа.
Чувствительность описанного прибора при определении метана
достигает 0,1—0,2%. Вся шкала гальванометра обычно соответствует
5%. Если стрелка гальванометра выходит за пределы, то, включая
дополнительное сопротивление, изменяют значения шкалы, что по-
зволяет измерять и более высокие концентрации углеводородов.
Необходимо следить, чтобы при всех указанных определениях в
газе присутствовало достаточное для сожжения количество кислорода.
Известно, что различные индивидуальные газы начинают гореть
при различной телшературе. Так, например, водород начинает сго-
рать раньше углеводородов. Из предельных углеводородов позднее
всех начинает сгорать метан. Это обстоятельство позволяет при раз-
личном накале платиновой проволоки раздельно определять водород
и углеводороды, а в углеводородном газе раздельно метан и тяже-
лые газообразные углеводороды. Следует учесть, что подобное раз-
дельное определение, особенно для углеводородных газов, не яв-
ляется точным. Однако для некоторых практических целей и прибли-
женное раздельное определение метана и более тяжелых угле-
326
водородов представляет интерес. В частности, подобное раздельное
определение применяется при газовом кароттаже для того, чтобы раз-
личать газоносные и нефтенос-
ные горизонты.
На вышеописанно/м принци-
пе строятся и переносные полу-
автоматические газоанализаторы
на горючие газы. Образец газа
забирается в камеру 7, вклю-
чается ток и наблюдается отброс
стрелки гальванометра.
На фиг. 120 представлен
схематически прибор, разрабо-
танный М. М. Файнбергом [47].
Анализируемый газ засасывает-
ся поршнем и поступает в одну
из камер. При включении на-
кала наблюдается максима ль-
Фиг. 120. Переносный прибор.
ный отброс стрелки гальванометра, который характеризует содер-
жание горючего газа.
§ 4. Прочие методы автоматического газового анализа
Определение кислорода
Современные автоматические газоанализаторы на кислород осно-
ваны или на измерении теплового эффекта от сжигания избытка
горючих газов в присутствии газа, содержащего кислород, или на
измерении магнитной восприимчивости этого газа [47].
На фиг. 121, а представлена схема прибора, в котором сжигание
происходит на платиновой нити, нагретой электрическим током до
светлокрасного каления. Анализируемый газ проходит через легко
кипящее жидкое горючее (петролейный эфир и т. п.) и, имея избы-
ток горючих паров по отношению к кислороду, поступает в камеру
с накаленной платиновой спиралью. Тепловой эффект от сгорания
газа, пропорциональный содержанию кислорода, измеряется с по-
мощью схемы мостика Уитстона, как описано выше (§ 2—3). Ско-
рость поступления жидкого горючего в испаритель поддерживается
постоянной с помощью соответствующего регулятора. Прибор позво-
ляет определять концентрации кислорода, начиная с 0,1—0,2%.
Известны автоматические газоанализаторы на кислород, осно-
ванные на применении двух термометров сопротивления, на одном
из которых нанесен катализатор. Камеры с термометрами сопроти-
вления помещены в печь, нагретую до температуры сгорания газа.
Если в газе нет водорода или других горючих компонентов, то доба-
вляют водород, постоянный ток которого поступает из электролизера.
Разница температур термометров сопротивления измеряется обыч-
ным путем по схеме мостика Уитстона и характеризует процентное
содержание кислорода.
327
Аналогичный прибор известен и в другом варианте, в котором
один из термометров сопротивления (или одна группа слоев термо-
батареи) помещается в порошкообразный или зерненный катализа-
тор (палладиевый или платино-палладиевый). Если катализатор
действует без подогрева, то вся эта система, т. е. термометр и ката-
Фиг. 121. Газоанализаторы на кислород.
лизатор, располагается в сосуде с вакуумной рубашкой (типа со-
суда Дьюара), как это представлено на фиг. 121,6.
Следует указать, что для автоматического определения кислорода
наибольший интерес представляет магнитный метод, основанный
на высокой магнитной восприимчивости кислорода, резко отлича-
ющей его от других газов, как это видно из следующего.
328
Газ (при 20° С) Объемная магнитная восприимчивость
Кислород Азот Водород Углекислый газ . . Метан Этен Окись азота .... —1-0,151 -0,00055 -0,0002 —0,00093 —0,00054 —0,00054 +0,05
В этих приборах используются различные явления, связанные
с высокой магнитной восприимчивостью кислорода. Так, например,
известны приборы, основанные на изменении теплопроводности газа
в магнитном поле, что зависит от величины магнитной восприимчи-
вости. Уменьшение теплопроводности газа в магнитном поле пропор-
ционально содержанию кислорода. Однако чувствительность подоб-
ных приборов невелика.
В других приборах используется так называемый магнитный
ветер, возникающий в негомогенном магнитном поле при наличии
температурного градиента в парамагнитном газе (т. е. в газе с поло-
жительной магнитной восприимчивостью).
С повышением температуры магнитная восприимчивость кисло-
рода уменьшается, поэтому более холодный кислород будет как бы
втягиваться в поле магнита, а более горячий — выталкиваться.
В автоматических магнитных газоанализаторах используется охлаж-
дающее действие этого конвекционного газового потока на нагре-
вательный элемент, повышающий температуру газа и помещенный в
магнитном поле (фиг. 121, в). В одном из приборов этого типа газ
проходит через кольцевую камеру с горизонтальной соединительной
трубкой, имеющей нагревательную обмотку. У края обмотки рас-
положены полюсные башмаки постоянного магнита. Нагреватель-
ная обмотка, разделенная на две секции, служит термоманометром,
измеряющим скорость течения газа. Первая по ходу газа секция
охлаждается, а вторая нагревается потоком газа. Секции обмотки
являются плечами мостика, и разность температур секций, характе-
ризующая концентрацию кислорода в газе, фиксируется самопишу-
щим гальванометром.
Определение углекислого газа
Действие газоанализатора основано на том факте, что вязкость
СО2 меньше вязкости воздуха, а плотность СО2 больше. Вследствие
меньшей вязкости СО2 легче проходит через капилляры, а благо-
даря большей плотности медленнее проходит сквозь отверстие в диа-
фрагме.
В приборе газы и воздух заставляют с одинаковой скоростью
проходить сначала через капиллярную трубку, а затем через отверстие
в диафрагме и измеряют разность давлений в трубках (фиг. 122).
329
Фиг. 122. Ав-
томатический
определитель
удельного веса.
При анализе топочных газов эта разность зависит исключительно
от присутствия СО2, потому что в отношении вязкости и плотности
СО почти не отличается от воздуха.
Через капилляр 7 и отверстие в диафрагме 2 засасываются ана-
лизируемые газы, а через 3 и 4 — воздух.
Так как газы, содержащие СО2, проходят через капилляры легче,
а через отверстие в диафрагме труднее воздуха, то вакуум в маноме-
трической трубке, соединенной с воздушной трубкой, больше ва-
куума в манометрической трубке, соединенной с газо-
вой трубкой.
Разность этих вакуумов является мерой содер-
жания СО2. Подобный прибор, сравнительно про-
стой по устройству, может применяться не только
для определения СО2, но и других газов при упо-
мянутых условиях в отношении вязкости и плот-
ности газов.
Автоматический замер удельного веса газа
На фиг. 123 представлена схема одного из прибо-
ров, употребляемых для автоматического определе-
ния удельного веса газа.
Прибор представляет собой как бы газовые весы,
на одно плечо которых давит воздух, а на другое
анализируемый газ (фиг. 123, а). Небольшой электро-
мотор приводит во вращение два одинаковых вен-
тилятора, которые вращаются с одинаковой ско-
ростью, но в противоположных направлениях. Один
вентилятор засасывает воздух, а другой — анализи-
руемый газ. В верхней камере 7 прибора создается
вихрь из анализируемых газов 2; в нижней 3 такой же вихрь
из воздуха 4 в противоположном направлении. Каждый из этих вих-
рей действует на крыльчатые диски, стремясь придать им вращатель-
ное движение в соответствующем направлении. Диски соединены
между собой системой шарнирно связанных рычагов так, чтобы вра-
щательные моменты, создаваемые вихрями, могли передаваться на
сопряженные оси (верхний вращательный момент на нижнюю ось,
а нижний момент на верхнюю ось).
Если бы вихри состояли из газов одинаковой плотности, диски
заняли бы положение, указанное на фиг. 123, б. Вихрь от верхнего
вентилятора стремится повернуть диск в направлении, указанном
стрелкой. Результирующая сила Fr направлена вверх по соедини-
тельному рычагу р. Эта сила стремится повернуть ось О2> к которой
прикреплена стрелка указателя, слева направо. Вращение нижнего
вентилятора создает силу F2, направленную вниз по рычагу р и стре-
мящуюся повернуть ось Ot справа налево.
Если вихри состоят из газов одинаковой плотности, то система
рычагов, шарнирно соединяющая оси дисков, располагается сим-
метрично относительно горизонтальной плоскости, проходящей по-
330
средине между обоими дисками. Рычаг р стоит вертикально; стрелка
указывает на нуль.
Если верхний вентилятор будет создавать вихрь из газов, обла-
дающих вследствие содержания СО2 большей плотностью, а нижний
будет вдувать воздух, то сила Fr будет больше F2. Эта сила Fr создает
вокруг оси О2 момент вращения больший, чем момент вращения,
созданный силой F2 вокруг оси Ог; нижний диск повернется слева
направо на больший угол, чем верхний; система рычагов займет дру-
гое положение, и стрелка передвинется направо. Равновесие насту-
пит тогда, когда вращательные моменты вокруг обеих осей будут
равны.
Как уже упоминалось выше, измерение удельного веса газа во
многих случаях помогает проводить достаточно точный анализ га-
зовой смеси, особенно когда интересующий нас компонент обладает
удельным весом, резко отличающим его от других газов.
Так, например, с помощью описанных приборов можно опреде-
лять содержание метана в природных газах. Для этой цели перед
Фиг.
123.
Автоматический определитель удельного веса.
прибором для определения удельного веса газа следует поставить
химические поглотители для СО2 и О2 и трубку с активированным
углем для поглощения тяжелых углеводородов. Попадающий в при-
бор газ будет состоять практически из азота и метана, поэтому шкалу
прибора можно проградуировать по содержанию метана. Пропуская
тот же газ через другой прибор, но без применения трубки с углем,
можно судить о содержании тяжелых углеводородов в газе (по раз-
331
нице показаний второго и первого приборов). Подобная схема анализа
может применяться при газовом кароттаже. Аналогичным образом
определения удельного веса можно использовать и для решения
других аналитических задач.
0.1
0.2
J' / / / /
0°С160ммНд
Фиг. 124. Автоматический определитель удельного веса.
Вышеописанный прибор не обладает большой чувствительностью
и точностью и используется главным образом в тех случаях, когда
требуется измерять присутствие в газе СО2, имеющей значительно
больший удельный вес, чем другие компоненты газовой смеси.
Фиг. 125. Автоматический определитель удельного веса.
В последнее время А. Якимовым [48] разработана новая схема
прибора для автоматического определения удельного веса газа.
Газовые весы, непрерывно показывающие удельный вес проходя-
щего через весы газа, представлены на фиг. 124. Коромысло весов
связано через магнитную муфту с пишущим механизмом. На фиг. 125
332
представлена схема разработанных автором газовых весов с непре-
рывными показаниями. Большая чувствительность достигается пу-
тем применения металлических баллонов 1 и 2} имеющих большой
объем (по 3—5 л) и подвешенных к коромыслу весов 7. В камеру,
где помещается один из баллонов, впускается газ, затем свободно
выходящий из камеры через трубки 3, 4 и 5, а также через отвер-
стие 6. Вторая камера сообщается с атмосферой.
М. М. Файнбергом разработан прибор для автоматического опре-
деления удельного веса газов, основанный на применении термо-
анемометра. [49].
ЛИТЕРАТУРА
1. X во ль сон О. Д., Курс физики. Гос. изд-во, 1923.
2. Смирнов А. С., Технология углеводородных газов. Гостоптехиздат,
1946.
3. Оствальд, Лютер, Друкер, Физико-химические измерения.
ОНТИ, 1935.
4. С о к о л о в В. А., Газовая съемка. ОНТИ, 1936.
5. И о ф ф е А. Ф., Техника физического эксперимента. Гос. изд-во, 1929*
6. Соколов В. А., Методы исследования природных газов. ОНТИ, 1932.
7. Г е л л е р Е. М., Оператор по газовой съемке (инструкция). Гостоп-
техиздат, 1944.
8. Савченко В. П., Сборник «Природные газы», № 9, 1935; № 11,
1936 и др.
9. Элинсон М. М., «Зав. лабор., № 6, 1949.
10. Лидов А. П., Анализ газов. Гос. изд-во, 1923.
11. Деннис и Н и к о л ь с, Газовый анализ. Гос. изд-во, 1929.
12. Кнорре, Тепловые расчеты при анализе газов. Химиздат, 1948.
13. Мовшович, Газоанализаторы. Гос. изд-во, 1930.
14. Вейнберг, Жидкий поглотитель водорода. «Зав. лабор.» № 6, 1939.
15. С о к о л о в В. А., «Нефт. хоз.» № 5, 1930.
16. Наметкин С. С., Соколов В. А. и др., Анализ естественных
газов. «Журн. прикл. хим.» № 23, 1931.
17. П о д б и л ь н я к. «Ind. Eng. Chem., Ап. Ed.», 3, 177, 1931 и др.
18. Соколов В. А., Прямые геохимические методы поисков нефти. Гос-
топтехиздат, 1947.
19. Туркельтауб А. М., «Зав. лабор.» № 6, 1949.
20. Добрянски й, «Нефт. хоз.» № 9, 1925.
21. Кузьминых И. Н., Контроль нитрозных газов. Госхимиздат,
1945.
22. Справочник по газовому делу, т. II, перевод. Гостоптехиздат, 1940.
23. Прибор для микрогазового анализа. Издание Ин-та химической физики,
Ленинград, 1934.
24. Лангмюр. «J. Am. Chem. Soc.», 34, 1310, 1912.
25. Райдер. «J. Am. Chem. Soc.», 40, 1656, 1918.
26. Д ю н у а й е, Техника высокого вакуума. Гос. науч. тех. изд-во, 1931.
27. Гуревич М. Г., «Зав. лабор.» № 6, 1949.
28. Файнберг М.М. и Туркельтауб Н.М., «Зав. лабор.» № 11
1945.
29. Т у р к е л ь т а у б Н. М., «Зав. лабор.» № 6, 1949.
30. В о к о в а Е. Н., Кузнецова В. А., Кузнецов В. И., «Докл.
Акад, наук» VI, № 7, 1947.
31. А л е к с е е в а М. А., Андронов Б. Е., Гурвиц С. С., Жит-
кова А. С., Определение вредных веществ в воздухе производственных поме-
щений. Госхимиздат, 1949.
333
32. Захарьевский В. А., Сборник «Анализ газов». Издание Моск,
нефт. ин-та, 1933.
33. Л у к а ш у к А. И., Хлоп ин В. Г., «Доклады Р. А. Н.», 1924.
34. Соколов В. А., Гелий и другие редкие газы. ОНТИ, 1933.
35. Черепенников А. А., Сборник «Природные газы» № 2, изд.
Союзгеолразведки, 1931.
36. X лопин В. Г., Сборник «Природные газы», №2, 1931.
37. Панет и П е т е р с. «Ztschr. Phys. Chem.», 134, 1928.
38. X л о п и н В. Г., Г е р л и н г А. Г. и Б а р а н о в с к а я Н. В.,
«Изв. Акад, наук» № 6, 1947.
39. Каминер Б. Б. и Потоловский П. А.., «Зав. лабор.»
№ 6, 1949.
40. К о р ф С., Счетчики электронов и ядерных частиц. Перевод. Изд-во.
иностр, литер., 1947.
41. Райт Н. «Ind. Eng. Chem., An. Ed.», № 1, 1941.
42. Б p о д с к и й А. И., Труды Всесоюзн. конферен. по аналитич. химии,
1939.
43. В ейнгеров М. Л., Успехи физических наук, т. XXI, вып. 4., 1939.
Тезисы докладов совещания по газовому анализу. Изд. Акад, наук, 1949.
44. Астон, Изотопы и масс-спектры. Перевод И. Л. 1942.
45. Трофимов А. В., Тезисы докладов совещания по газовому ана-
лизу. Изд. Акад, наук, 1949.
46. Файнберг М. М., «Зав. лабор.» № 3, 1939.
47. ФайнбергМ. М., «Зав. лабор.» № 6, 1949.
48. Якимов, «Докл. Акад, наук», LVI, № 4, 1947.
49. Файиберг М. М., Докл. Акад, наук, т. LXIX, № 2, 1949.
СОДЕРЖАНИЕ
Стр
Глава 1. Общие сведения и техника газоаналитических определений . . . 3
§ 1. Понятие о газовом анализе и его подразделениях. Законы газо-
вого состояния ................................................. 3
§ 2. Приборы и приспособления общего назначения, применяемые
в газовом анализе ................................ &
§ 3. Измерения объема и давления газа .......................... 34
§ 4. Стеклодувные вспомогательные работы ....................... 4б
Глава II. Отбор газовых проб. Извлечение растворенных и сорбированных
газов ................................................................ 56
§ 1. Отбор проб природных и промышленных газов ................. 56
§ 2. Извлечение растворенных газов ............................. 66
§ 3. Извлечение газов, сорбированных твердыми телами ........... 68
Глава III. Общий газовый анализ ...................................... 72
§ 1. Назначение общего газового анализа. Основные физико-хими-
ческие свойства газов, определяемых при общем анализе (СО2,
О2, Н2, СО, N2, простейшие углеводороды) ...................... 72
§ 2. Методика общего газового анализа .......................... 82
§ 3. Приборы, применяемые при общем газовом анализе .... 95
§ 4. Реагенты, применяемые при общем газовом анализе, и их дей-
ствие на различные газы ......................................... 12 1
§ 5. О результатах и точности общего газового анализа.......... 129
§ 6. Некоторые методы определения СО2, СО и Н2 ................ 131
Глава IV. Анализ углеводородных газов ............................... 139
§ 1. Углеводородные газы и их свойства......................... 139
§ 2. Анализ углеводородных газов с применением разгонки при низ-
ких температурах ............................................. 142
§ 3. Приборы низкотемпературной разгонки с применением ректи-
фикационной колонки .......................................... 152
§ 4. Анализ углеводородных газов с помощью адсорбентов......... 169
§ 5. Методы определения предельных и непредельных углеводородов
с применением абсорбции и некоторых реакций................... 171
§ 6. Определение газолина в газе .............................. 180
Глава V. Методы определения неорганических газов, в элементарный состав
которых входят азот, сера, галоиды и некоторые другие элементы 196
§ 1. Определение окислов азота и аммиака....................... 196
§ 2. Определение сернистых газообразных соединений (H2S, SO,,
COS) .......................................................' e 209
§ 3. Определение цианистоводородной кислоты и циана в газах . . 215
§ 4. Определение галоидов и галоидоводородных кислот........... 217
335
Стр.
Глава VI. Микроанализ газов ........................................... 220
§ I. Общий газовый микроанализ .................................. 220
§ 2, Вакуумные установки для определения малых количеств СО,,
Н2 ,СО, N2, Н2О.................................................226
§ 3. Микроанализ на углеводородные газы с применением вакуум-
ных установок и глубокого охлаждения .......................... 234
§ 4. Микроанализ на углеводородные газы без применения низких
тейиператур.................................................... 249
§ 5. Бактериальный метод определения углеводородных газов , . 256
§ 6. Определение вредных примесей в воздухе ..................... 257
Глава VII. Определение редких газов................................. 260
§ 1. Основные физические и химические свойства редких газов . . . 260
S 2. Определение гелия и неона .................................. 264
§ 3. Раздельное определение аргона и гелия ...................... 268
§ 4. Полный анализ на редкие газы ............................... 272
§ 5. Определение радиоактивных эманаций ........................ 278
Глава VIII. Физические методы газового анализа......................... 285
§ 1. Спектральный газовый анализ ................................ 285
§ 2. Газовый интерферометр ...................................... 288
§ 3. Оптико-акустический метод газового анализа.................. 293
§ 4. Анализ газа с помощью масс-спектографа ..................... 295
§ 5. Определение удельного веса газа ............................ 296
§ 6. Определение теплотворной способности газов.................. 306
Глава IX. Автоматические газоанализаторы .............................. 316
§ 1. Автоматические химические газоанализаторы................... 317
§ 2. Анализ газа методом сравнения теплопроводности.............. 323
§ 3. Автоматический газовый анализ, основанный на измерении теп-
лоты сгорания ............................................. . 325
§ 4. Прочие методы автоматического газового анализа.............. 327
Ведущий редактор Л. А. Львова
Техн. ред. А. С. Полосина
Корректор В. А. Богословский
Т-04028. Подписано К печати 10/V 1950 г. Печ. л. 21.
Тираж 2000 экз. Формат 60X921/'ig- 10,5 б. Л.
Уч. изд. л. 24,18. Ц. 17 руб. Зак. № 2930/119А
Типогр. «Кр- Печатник».
Ленинград, Международный пр., 91.