








































































Автор: Ластовский Р.П. Поспелов А.М.
Теги: органическая химия химия химическая промышленность химические реакции
Год: 1966




Текст
Редакционная коллегия
Доктор хим. наук Р. П. Ластовскнй (гл. редактор),
инженер-технолог А. М. Поспелов (зам. гл. редактора),
канд. хим. наук Е. А. Божевольнов,
доктор хим. наук А. В. Бромберг, канд. техн, наук
В. Г. Брудзь,
доктор хим. наук В. М. Дзиомко,
капд. хим. наук Г. А. Певцов
СОДЕРЖАНИЕ
З-Амипокарбазол. В. В. Кояин, Г. Н. Сутягина ............... 5
З-Амино-1, 2, 4-триазолкарбоновая-5 кислота. Г. И. Чипен .... 9
Аценафтенон-1-спиро-(2,4')-2', З'(СО), 6', 5'(СО)-дибензоилен-1', 4'-
дигидропиридин. Г. Я. Дубур, |Г. Я- Ванаг |............. 12
2-М-Ацетиламинопиридин. Г. Ф. Дрегвйль, А. П. Мартынюк ... 16
8-Ацетоксикарбостирил. В. М. Дзиомко, И. А. Красавин, Ю. П.
Радин, Н. И. Мирошкина................................. 18
Бензиловый эфир М-[]1-(2-пиридил) этил]-N-фенилдитиокарбамино-
вой кислоты. Н. Ф. Казаринова, Т. И. Тумбина, К- А. Соломко 20
N, Ы'-Бис-(карбоксиметил)-4,4'-дипиридилий дихлорид. Н. Ф. Каза-
ринова, К. А. Соломко, М. И. Котеленец.................. 22
Боратран. М. Г. Воронков, Г. И. Зелчан......................24
тргт-Бутил-2-тиенилсульфид. Б. П. Федоров, Ф. М. Стоянович 26
9-Винил-1,2-беизакридин. Б. С. Танасейчук, И. Я- Постовский 28
Дибепин. Г. Я. Дубур, |Г. Я. Ванаг |, Я- Р- Улдрикис........ 30
2-\'-Диизопропилфосфиниламинопириди1'. Г. Ф. Дрегваль, Н. В. Ко-
валенко, А. П. Мартынюк .................................... 36
2-[{1-(М,М-Диметиламино)этил]пиридии. Н. Ф. Казаринова, Н. В. Джи-
гирей, Н. А. Шабаева........................................ 38
Диметиловый эфир имидазол-4,5-дикарбоновой кислоты. Н. Б. Ви-
ноградова, Н. В. Хромов-Борисов............................. 40
2,6-Дистирилпирон-4. Н. С- Вульфсон, Е. В. Савинкова............ 45
2,2-'-Дитиенилсульфид. Б. П. Федоров, Ф. М Стоянович........... 48
4,5-Дифенилимидазол. А. Д. Гарновский........................... 51
1,,5-Дифенил-пиразолидон-З. Н. А. Захарова, Н. В. Хромов-Борисов,
С. /7. Гафт................................................. 54
2,3-Дихлор-1,4-диоксаи. М. Г. Воронков, Э. П. Попова............ 57
2-[₽-(Ы,Ы-Диэтиламино)этил] пиридин. Н. Ф. Казаринова, II. В. Джи-
гирей, Н. А. Шабаева........................................ 59
Диэтилацеталь 2-этилмеркапто-5-этил-3-тиофеиальдегида.
Я- Л. Гольдфарб, М. А. Калик, М. Л. Кирмалова............... 61
Имидазол-4,5-дикарбоновая кислота. Н. Б. Виноградова, Н. В. Хро-
мов-Борисов . . . . <...................................... 63
2-Меркапто-5-этил-3-теиилиденимин Я. Л- Гольдфарб, М. А. Ка-
лик, М. Л. Кирмалова........................................ 66
2-Метил-5-изоиропил-5-(а-метокси) этил-1.3-диоксан. А. В. Богат-
ский, Н. Л. Гарковик....................................... 68
3-Метил-5-пропилпиррол-(2-азо-2')-фенол.В. М. Дзиомко, К. А. Ду-
наевская ................................................... 70
4-Метил-1-феиилпиразолидон-3. Р. Б. Журин, О. Е. Шульгина . . 72
2-[р-(И-Морфолил) этил] пиридин. Н. Ф. Казаринова, Н. В. Джи-
гирей ...................................................... 75
3
З-Нитрамино-1, 2, 4-триазол. Г. И. Чипен................... 77
р-(5-Нитрофурил-2)-акриловая кислота, К. К. Бентер, С. А. Гил-
лер, В. В. Цируле...................................... 80
4-Окси-3-ацетил-6-метил-пиридон-2. Н. С. Вульфсон, Г. М. Сухо-
тина ................................................. 83
8-Окси-2-гидразииохинолин. И. А. Красавин, В. М. Дзиомко, Н. И.
Мирошкина............................................. 86
8-Окси-2-метоксихииолии. В. М. Дзиомко, И. А. Красавин, Ю. П.
Радин.................................................. 88
4-Окси б-стирнлпирон-2. Н. С. Вульфсон, Е. В. Савинкова .... 90
4-Окси-3-циииамоил-6-метилпирои-2. Н. С. Вульфсон, Е. В. Савин-
кова ................................................. 92
1,2,3,4,5,6,7,8-Октагидродибеизофуран (I) и 2-метил-2,3-циклопеи-
тано-4,5-циклогексено-(Д5)-2, 3, 4,5- тетрагидрофураи (II). С. В.
Светозарский, К. Л. Феллер, Ю. Ю Саматов, Е. Н. Зильбер-
ман, Г. А. Разуваев ..................... 95
2-Пиридилметанол. Н. Ф. Казаринова, Н. А. Рыбак, В. В. Балы-
кина ..................................................... 98
2-Стирил-6-метилпироны-4. Н. С. Вульфсон, Е. В. Савинкова . . . 100
Тетрафурфурилоксисилан. Э. Лукевиц, Ю. П. Рамадан, С. А.
Киллер .......................... 103
2-Тиотиазанон-4. Е. В. Владзимирская, И. М. Туркевич.......1С6
1-(,и-Толил)-пиразолидон-3. Р. Б. Журин, О. Е. Шульгина .... 10.)
8-(л-Толуолсульфоииламино) хинальдин. Б. В. Парусников, В. М.
Дзиомко, И. А. Красавин.................................ИЗ
1, 2, 4-Триазолинтион-5. И. Л. Шегал, И. Я. Постовский .... 116
1, 2, 4-Триазолои-3-карбоновая-5 кислота. Г. И. Чипен......119
1-Фенил-3,6-диметил-пиразоло-(4,5с)-пиридон-4. Н. С. Вульфсон,
Г. М. Сухотина ...................... 122
4-Фенил-л/-диоксаи. А. В. Богатский, Г. Л. Камалов.........125
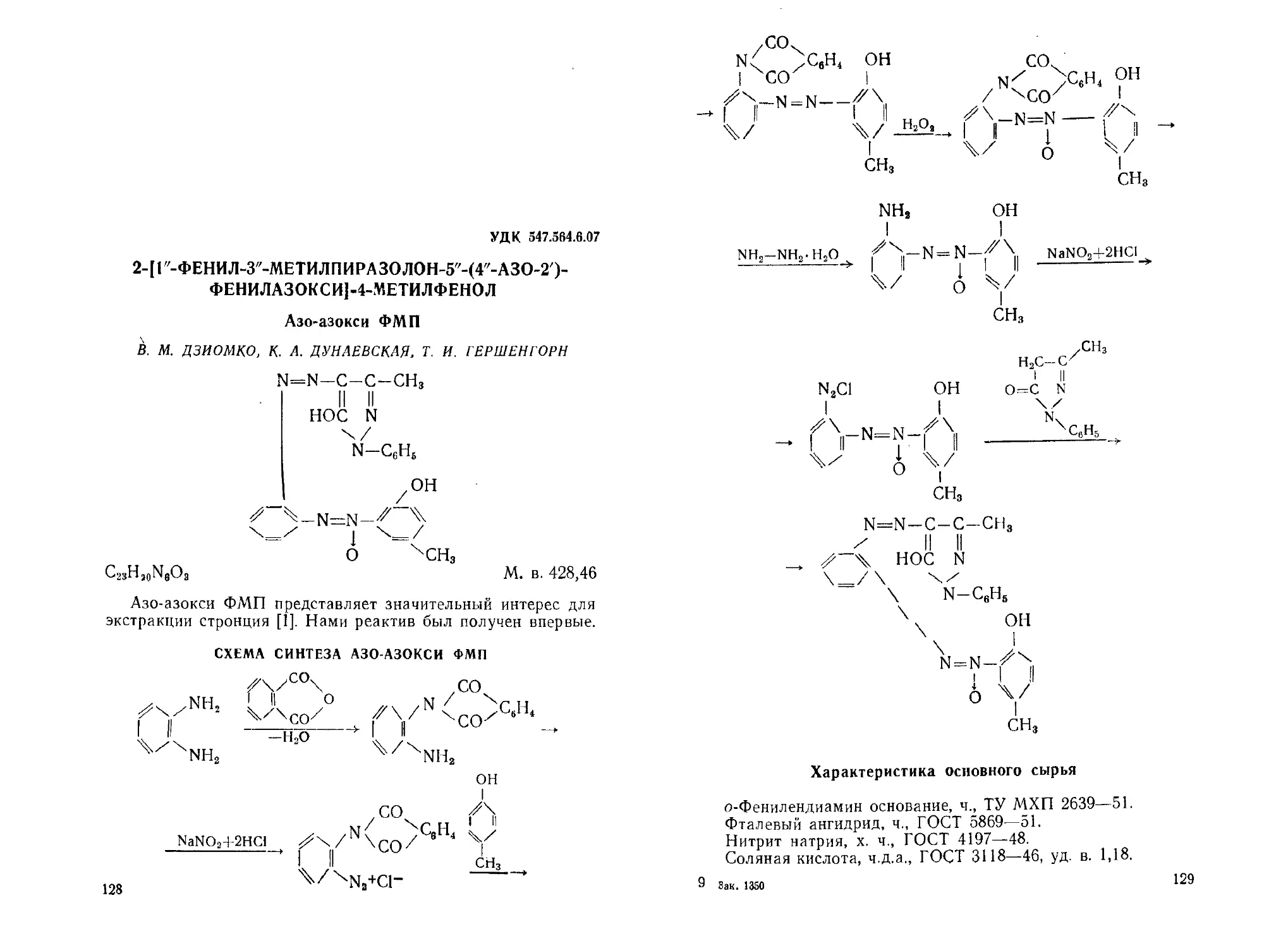
2- [Г'-Феиил-З" -метилпиразолои-5"- (4"-азо-2') -феиилазоксн] -4-ме-
тилфенол. В. М. Дзиомко, К. А. Дунаевская, Т. И. Гершенгорн 128
1-Фенил-пнразолидои-З. Н. И. Симонова, В. В. Пигулевский, Н. А.
Захарова, Н. В. Хромов-Борисов, С. И. Гафт.............133
1-Фенилснлатран. М. Г. Воронков, Г. И. Зелчан..............138
Р-Фенилтиофеи. М. Г. Воронков, А. Н. Переферкович..........141
2-Фуроил- и 2-тиеиоилбромметилеифосфораиы. А. А. Григоренко,
М. И. Шевчук, А. В. Домбровский........................144
2-Фуроил- и 2-тиеноилметилеитрифеиилфосфораны. А. В. Домбров-
ский, М. И. Шевчук, А. А. Григоренко...................147
2-Хлор-8-оксихинолин. И. А. Красавин, В. М. Дзиомко, Н. И. Ми-
рошкина, Ю. П. Радин...................................150
2-Этилмеркапто-5-этил-3-тиофеиальдегид. Я. Л. Гольдфарб, М. А.
Калик, М. Л. Кирмалова .................. 153
Этил-(5-этил-2-тиенил) сульфид. Я. Л. Гольдфарб, М. А. Калик,
М. Л. Кирмалова ..................... 156
1-Этоксисилатраи. М. Г. Воронков, Г. И. Зелчон.............159
Алфавитный перечень соединений, описанных в настоящем сбор-
ниме......................................................161
УДК 547.759.32.07
З-АМИНОКАРБАЗОЛ
В. В. КОРИН, Г. Н. СУТЯГИНА
—/\-nh2
В I
NH
Cl2HI0N2 М. в. 182,22
При синтезе 3-аминокарбазола пользуются многостадий-
ным методом [1]. Вначале получают 9-нитрозокарбазол дей-
ствием нитрита натрия на карбазол, смешанный с ледяной
уксусной кислотой. Затем проводят нитрование 9-нитрозосое-
динения смесью азотной и уксусной кислот. Полученный
9-нитрозо-З-нитрокарбазол подвергают омылению в спирто-
вом растворе едкого кали. Наконец, полученный таким обра-
зом 3-нитрокарбазол подвергают восстановлению. Различные
авторы предлагают применять следующие способы восстанов-
ления 3-нитрокарбазола: оловом в среде соляной кислоты [2],
Пинковой пылью в щелочной среде [3J, гидросульфитом нат-
рия в спиртовом растворе едкого кали [4], спиртовым раство-
ром сернистого аммония [5], сернистым натрием в водной
среде [6] и сернистым натрием в спиртовой среде (7].
Мы остановились на последнем из перечисленных способов,
так как он дает хорошие результаты при несложном оформ-
лении.
Нами найдено, что процесс омыления нитро-нитрозосоеди-
нения может быть совмещён с восстановлением нитрозосое-
Динения, а это приводит к некоторому повышению выхода
3-аминокарбазола.
Характеристика основного сырья
Карбазол, ч., ВТУ РУ 463—51.
Уксусная кислота, ч., ледяная, ГОСТ 61—51.
5
Нитрит натрия, ч., ГОСТ 4197—48.
Азотная кислота, ч., ГОСТ 4461—48.
Ацетон, ч., ГОСТ 2603—51.
Натрий сернистый, ч., ГОСТ 2053—43.
Спирт этиловый, ректифицированный, ГОСТ 8314—57.
Соляная кислота, ч., ГОСТ 3118—46.
Условия получения
I. Синтез 9-нитрозокарбазола
______________/\
| || II | + NaNO2 + СН3СООН ->
+ CH3COONa + Н2О
В стеклянном стакане, помещенном в водяную баню, раст-
воряют 20 г (0,12 М) карбазола в 160 мл (2,5 М) ледяной
уксусной кислоты при нагревании. Раствор охлаждают до 10°
и в него в течение 1 часа небольшими порциями вносят 9 г
(0,13 Л1) измельченного нитрита натрия. Смесь перемешива-
ют еще 2 часа, после чего температуру ее поднимают до 40°
и вносят еще 1,3 г (0,02 М) нитрита натрия. После охлажде-
ния до 10° выпавшее нитрозосоединение отфильтровывают,
промывают на фильтре водой и сушат. Получается вещество
с т. пл. 79—81,5°. Из фильтрата после разбавления его водой
выделяется еще некоторое количество загрязненного продукта
с т. пл. 77—79°.
Перекристаллизованный из спирта 9-нитрозокарбазол
представляет собой игольчатые кристаллы желтого цвета с
т. пл. 81—82° [8, 9, 1].
Выход перекристаллизованного продукта равен 19,9 г, что
составляет 85% от теоретического.
II. Получение 9-нитрозо-З-нитрокарбазола
<?\_____/\ ____/V-NO,
| || || | + HNO3 -> | || || | + H2O
N—О N=O
В стеклянном стакане, помещенном в водяную баню, раст-
воряют 20 г (0,1 М) нитрозокарбазола в 160 мл (2,5 Л4) ле-
дяной уксусной кислоты при 40° и после охлаждения до 10°
6
нитруют смесью 12,5 г (0,2 Л1) азотной кислоты и равного
по весу количества ледяной уксусной кислоты. Нитрующую
смесь вводят в реакционную массу равномерными порциями
при постоянном перемешивании в течение 1 часа. После вне-
сения всей смеси массу выдерживают еще 2 часа при темпе-
ратуре 10° и постоянном перемешивании. Затем осадок от-
фильтровывают, промывают на фильтре водой и сушат. По-
лученный 9-нитрозо-З-нитрокарбазол имеет т. пл. 156—159°.
Для очистки его кипятят в течение 5 минут с 5-кратным по
весу количеством ацетона. В результате такой обработки тем-
пература плавления продукта повышается до 162—163° [8, 1].
Выход очищенного 9-нитрозо-З-нитрокарбазола равен
10,6 г, что составляет 43% от теоретического (см. примеча-
ние 1).
Ш. Получение 3-аминокарбазола
S'"'-___/Z_ no,
4 | || II I + 6Na2S + 7H2O ->
Z\Z
N=O
Z \_____/ 4 _ NH
->4| II || | ’ 4 3Na2S2O3 + 4NaNO2 + 2NaOH
Z/\/\Z
NH
Загружают 10 г нитрозо-3-нитрокарбазола в круглодон-
ную колбу, снабженную обратным холодильником, добавля-
ют 10-кратное по весу количество 96%-ного этилового спирта
и вносят 10-кратное по весу количество сернистого натрия.
Смесь кипятят в течение 5 часов на водяной бане. После
окончания восстановления реакционную массу фильтруют и
из фильтрата посредством разбавления водой выделяют
3-аминокарбазол. Выделенный продукт отфильтровывают,
промывают на фильтре большим объемом воды и высушива-
ют при комнатной температуре в темноте. Сырой амин пла-
вится при 230°.
Для очистки 3-аминокарбазола его переводят в солянокис-
лую соль действием соляной кислоты. Полученный раствор
фильтруют и действием раствора едкого кали вновь выделя-
ют амин в виде мелких кристаллов телесного цвета. После
однократной очистки температура плавления 3-аминокарба-
зола повышается до 245° [8, 1] (см. примечание).
Выход очищенного продукта равен 6,3 г, что составляет
83% от теоретического.
Примечания:
1. Очистка 9-нитрозо-З-нитрокарбазола ацетоном производится в круг-
лодоииой колбе, помещенной на водяную баню и снабженной обратным
холодильником. Прежде чем выделять продукт после кипячения, реакци-
онную массу необходимо охладить. Очистка 9-нитрозо-З-нитрокарбазола
приводит и уменьшению выхода продукта (примерно в 2 раза).
7
2. Очистку сырого 3-аминокарбазола можно также осуществлять путем
однократной перекристаллизации его из толуола. Температура плавления
продукта прн этом также повышается до 245°. Но при дальнейшей очист-
ке от остатков толуола выход очищенного 3-амннокарбазола несколько
ниже, чем по описанному способу.
ЛИТЕРАТУРА
1. Р. К. Эйхман, В. О. Лукашевич, Е. А. Силаева, ПОХ,
6 93 (1939)
2. О. JRuff, Y. Stein. Вег., 34,1668 (1931).
3. Герм, пат., 46 438; Frdl., 2, 447.
4. Р. Ziersch. Вег., 42, 3797 (1909).
5. Герм. пат. 134938; Frdl., 6, 61.
6. Герм. пат. 139568; Frdl., 7, 71.
7. G. Carl, Schwalbe, S. Wolff. Вег., 44, 234 (1911).
8. Н. Lindemann, Вег., 57, 555 1924.
9. Герм. пат. 128853; Frdl., 6, 53.
Поступила в ноябре 1964 г.
Томский политехнический институт
УДК 547.852.9.07
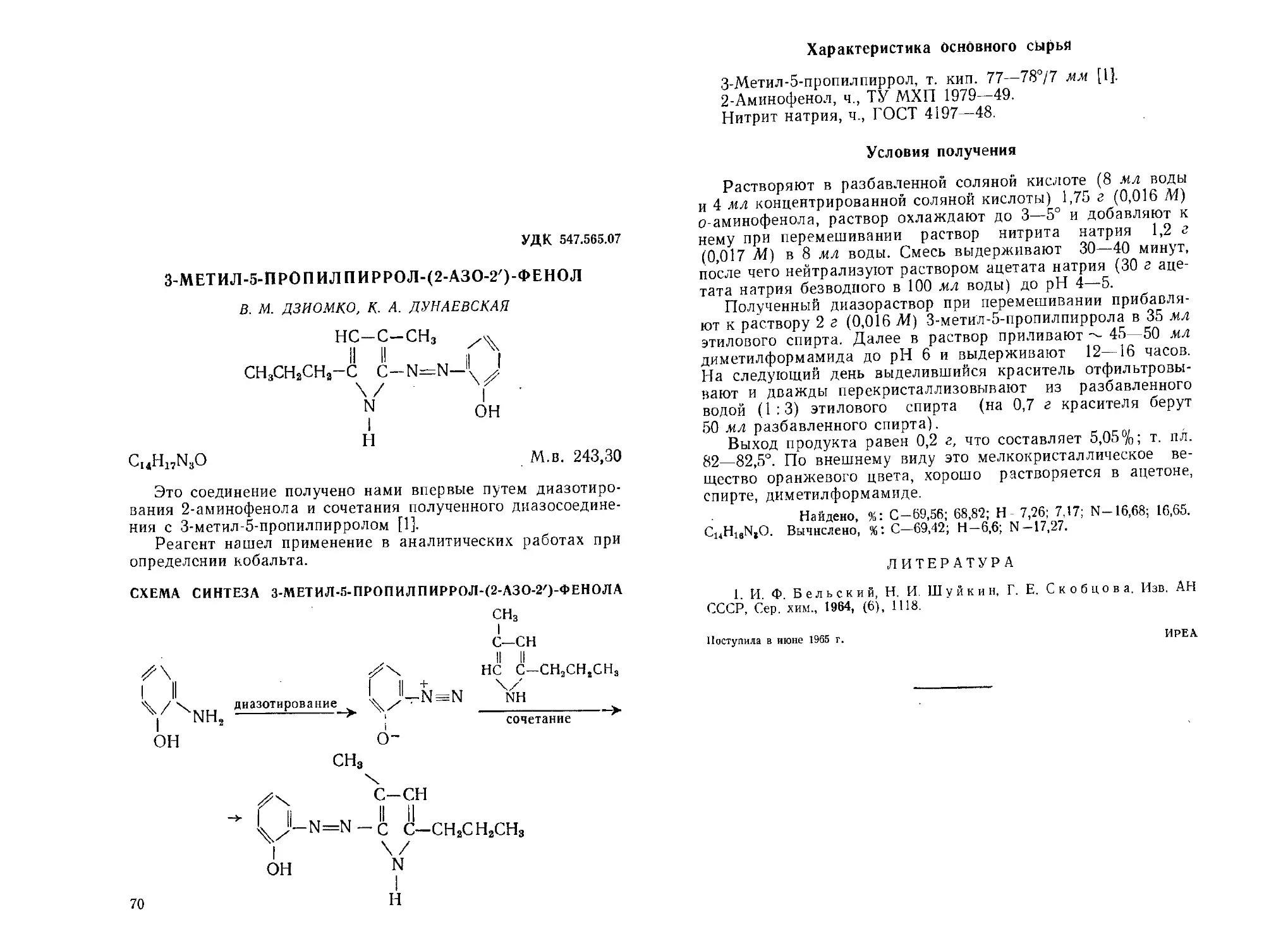
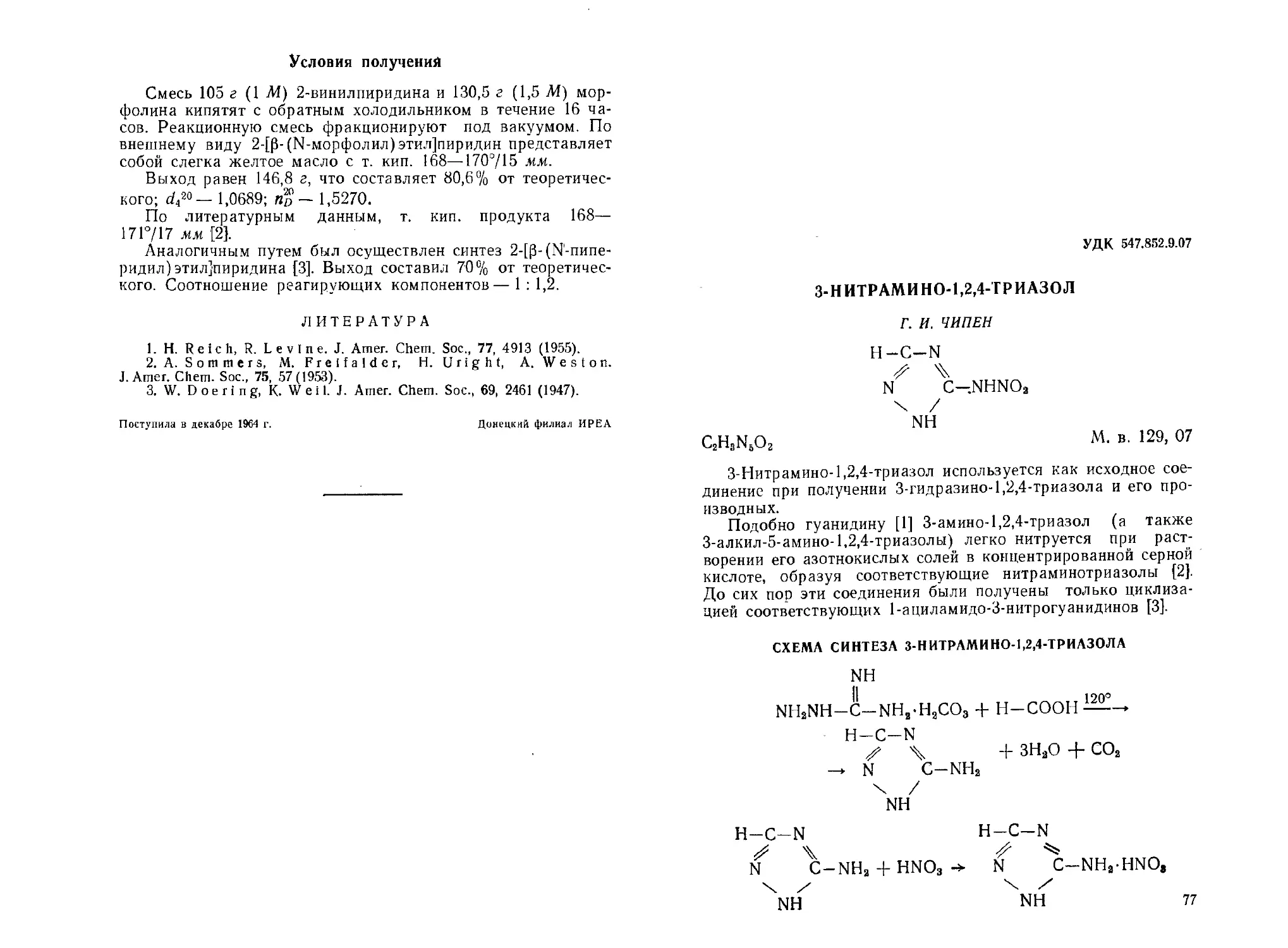
З-АМИНО-1,2,4-ТРИАЗОЛКАРБОНОВАЯ-5 КИСЛОТА
Г. И. ЧИНЕН
h2n-c-n
N С-СООН-1/2НаО
\н
C3H4N4Oa-l/2 Н2О
М.в. 137,14
3-Амино-1,2,4-триазолкарбоновая-5 кислота является ис-
ходным продуктом при получении 3-диазо-1,2,4-триазолкар-
боновой-5 кислоты, из которой в свою очередь можно полу-
чить целый ряд производных 1,2,4-триазола (см. статью 1,2,4-
триазолон-З-карбоновая-5 кислота в настоящем выпуске).
Согласно данным [1, 2], 3-амино-1,2,4-триазолкарбоновую-5
кислоту получают циклизацией оксалиламиногуанидина, ко-
торый легко образуется при кипячении водных растворов ок-
салата аминогуанидина. Оксалат аминогуанидина в свою оче-
редь получают из щавелевой кислоты и бикарбоната амино-
гуанидина. Однако, как показали опыты, при проведении ре-
акции получения 3-а.мино-1,2,4-триазолкарбоновой-5 кислоты,
согласно методике [2], с увеличенными загрузками как по-
бочный продукт в значительных количествах образуется ди-
гуанилгидразид щавелевой кислоты, который в дальнейшем
при щелочной циклизации образует соответствующий диамино-
битриазол [3] (см. также [4]). Образование днгуанилгидрази-
да щавелевой кислоты не наблюдается при «обратном» по-
рядке прибавления реагентов, т. е. при добавлении бикарбо-
ната аминогуанидина к раствору щавелевой кислоты.
Проводя реакцию циклизации оксалиламиногуанидина в
сильнощелочной среде, можно значительно сократить время
реакции (от восьми до одного часа).
9
Аминотриазолкарбоновую кислоту получают также из ди-
азоуксусного эфира, нагревая его с концентрированным рас-
твором едкого кали [5—7].
СХЕМА СИНТЕЗА З-АМИНО-1,2,4-ТРИАЗОЛ КАРБОНОВОЙ-5
КИСЛОТЫ
NH
|| СООН
NHaNH—С—NH,- НаСО3+ | --->
СООН
NH
II
CONHNH-C-NH,
—* | +СОа+2НаО
СООН
NH
II
CONHNH—С—NHa
| 4-NaOH—>
СООН
HaN—С—N
/ Ч
N С-COONa+2H2O
NH
HaN—С—N
N C —COONa-f-HCI--->
\h
HaN —C—N
—> N C-COOH +NaCl
NH
Характеристика основного сырья
Аминогуанидинбикарбонат, РУ-901—53.
Щавелевая кислота, ч., ГОСТ 5873—51.
Натр едкий, ч„ ГОСТ 4328—48.
Кислота соляная, ч., ГОСТ 3118—46,
10
Условия получения
В трехлитровой колбе приготовляют раствор 252 г (2 Af)
щавелевой кислоты в 2000 мл воды. К полученному раствору
при температуре 50—60° небольшими порциями прибавляют
238 г (1,75 /И) бикарбоната аминогуанидина (см. примеча-
ние 1). Для завершения реакции ацилирования колбу вы-
держивают в кипящей водяной бане 6 часов. После охлажде-
ния раствор декантируют, а осадок оксалиламиногуанидина
растворяют прибавлением раствора 100 г (2,5 М) едкого
натра в 1500 мл воды, Щелочной раствор кипятят один час
с обратным холодильником, охлаждают, фильтруют через
складчатый фильтр и подкисляют соляной кислотой до сла-
бокислой реакции. Осаждается аминотриазолкарбоновая кис-
лота, которую отфильтровывают, промывают холодной водой
и сушат на воздухе.
Выход равен 190—195 г, что составляет 79—81%, считая
па бикарбонат аминогуанидина. Продукт содержит 0,5 М во-
ды и плавится при 182—183° с разложением (см. примеча-
йте 2).
Примечания:
1. Скорость прибавления бикарбоната амииогуанидина лимитируется
выделением углекислого газа. После прибавления бикарбоната аминогуа-
нидинл наблюдается выделение в осадок оксалиламииогуанидияа.
2. Температура плавления аминотриазо.пкарбоновой кислоты, по лите-
ратурным данным, 182° (с разл.) [1], 182—183° (с разл.) [2].
ЛИТЕРАТУРА
1. J. Thiele, W. М а n с h о t. Liebigs Ann. Chem., 303, 33 (1898).
2. Препаративная органическая химия, ГХИ, М„ 1959, стр. 764.
3. Г. И. Ч и п е н, В. Я. Г р и н ш т е й н. Изв. АН Латв. ССР, Сеп.
хим., 1965, № 2, 204.
4. Пат. США 2744116, Ch. А., 51, 489 (1957).
5. Т. Curtins, A. Da rapsky, Е. М u 11 е г. Вег., 40, 1194 (1907).
6. A. Hantzsch, О. Silberrard. Вег., 33, 76 (1900).
7. Lehrman п. Вег., 33, 3679 (1900).
Поступила в декабре 1964 г.
Институт
органического синтеза
АН Латв- ССР
УДК 547.822.1.07
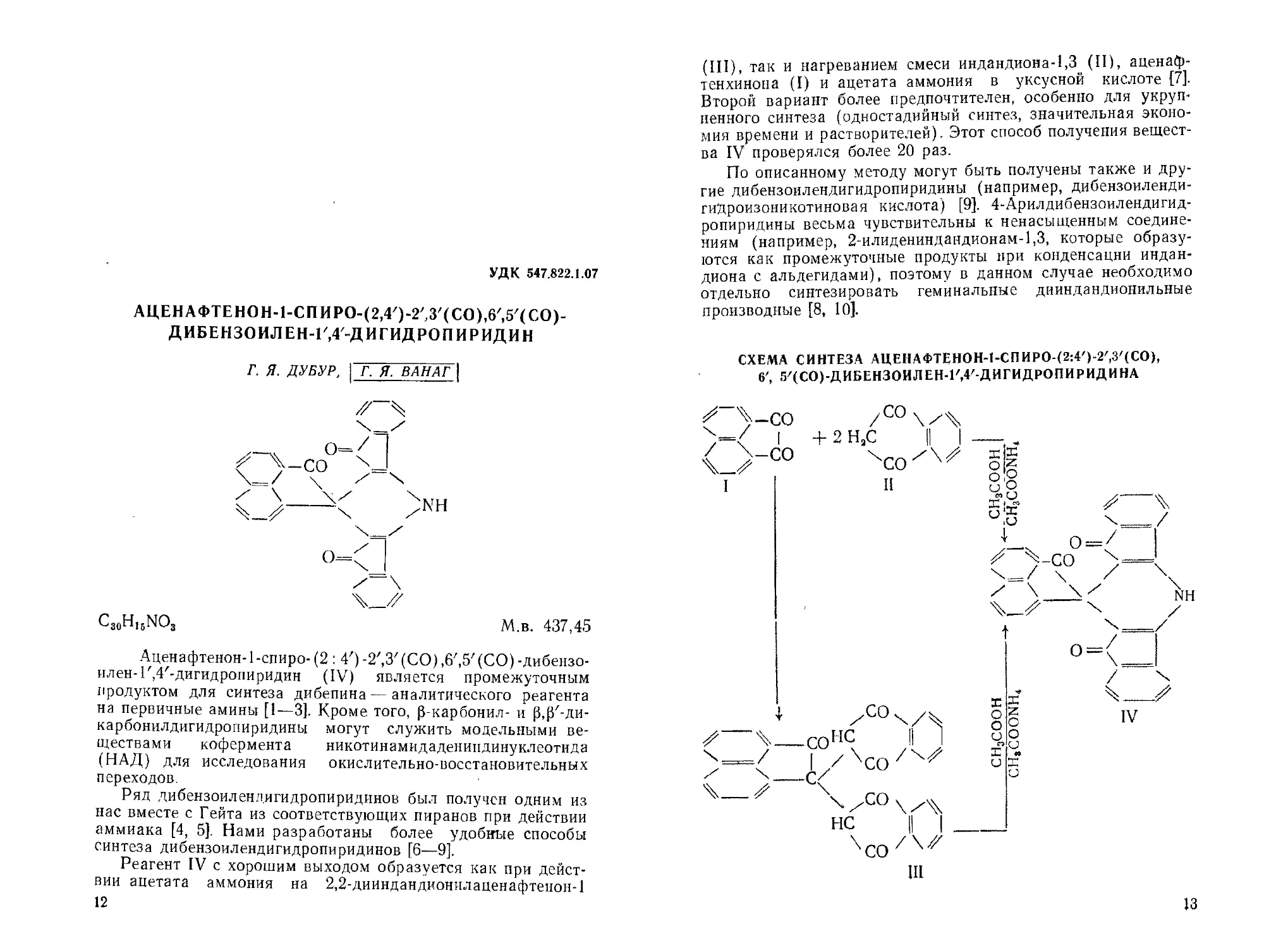
АЦЕНАФТЕНОН-1-СПИРО-(2,4/)-2/3/(СО),6',5/(СО)-
ДИБЕНЗОИЛ ЕН-Г,4-ДИГИДРОПИРИДИН
Г. Я. ДУ БУР, | Г. Я. ВАНАГ |
М.в. 437,45
Аценафтенон-1-спиро-(2 : 4')-2',3'(СО),6',5'(СО)-дибензо-
илен-Г, 4'-дигидропиридин (IV) является промежуточным
продуктом для синтеза дибепина — аналитического реагента
на первичные амины [1—3]. Кроме того, p-карбонил- и р,р'-ди-
карбонилдигидропиридины могут служить модельными ве-
ществами кофермента никотинамидаденипдинуклеотида
(НАД) для исследования окислительно-восстановительных
переходов.
Ряд дибензоилендигидропиридинов был получен одним из
нас вместе с Гейта из соответствующих пиранов при действии
аммиака [4, 5]. Нами разработаны более удобные способы
синтеза дибензоилендигидропиридинов [6—9].
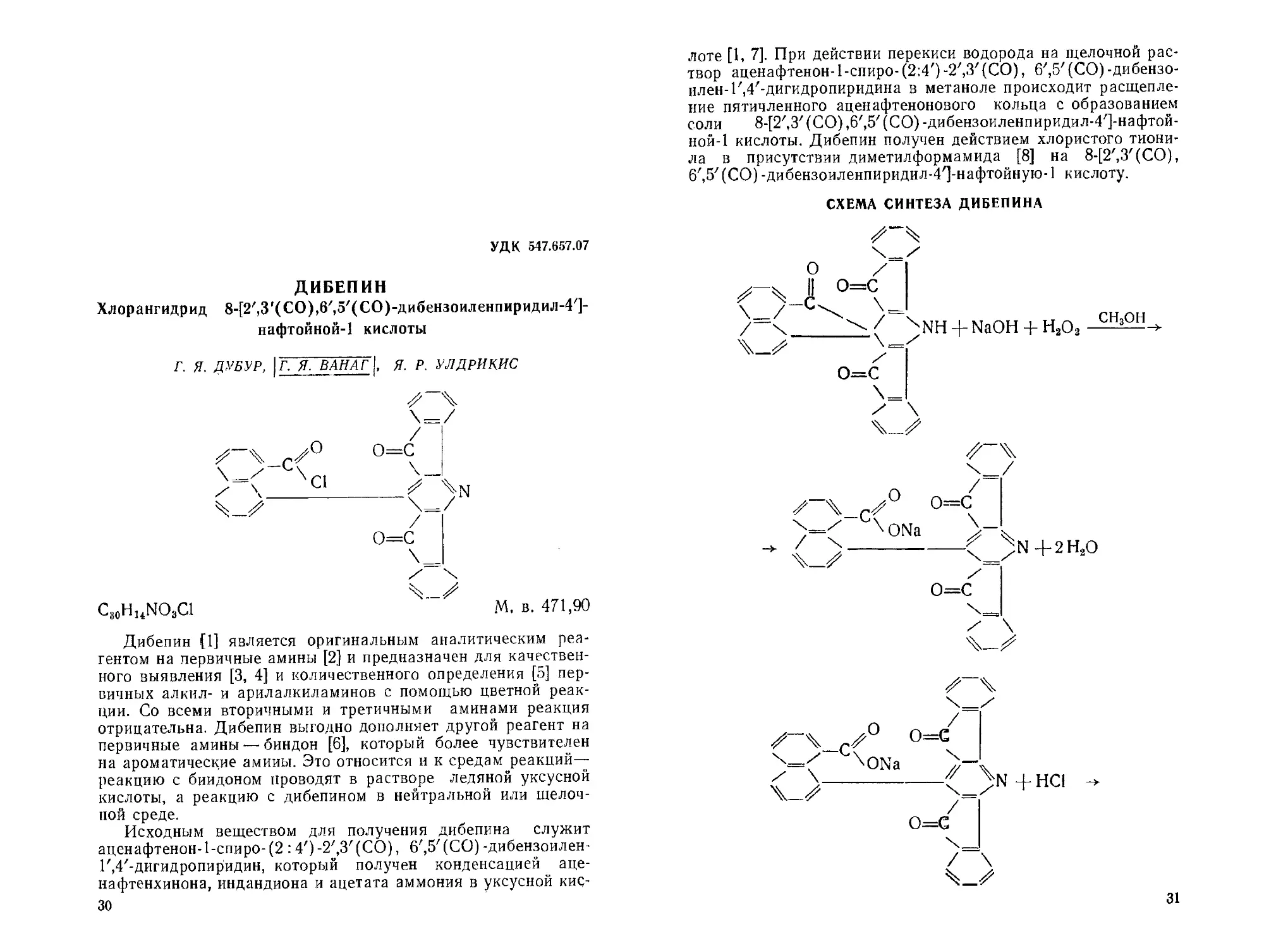
Реагент IV с хорошим выходом образуется как при дейст-
вии ацетата аммония на 2,2-дииндандионилаценафтенон-1
12
(Ill), так и нагреванием смеси индандиона-1,3 (II), аценаф-
тенхинона (I) и ацетата аммония в уксусной кислоте [7].
Второй вариант более предпочтителен, особенно для укруп-
ленного синтеза (одностадийный синтез, значительная эконо-
мия времени и растворителей). Этот способ получения вещест-
ва IV проверялся более 20 раз.
По описанному .методу могут быть получены также и дру-
гие дибензоилендигидропиридины (например, дибензоиленди-
ги'дроизоникотиновая кислота) [9]. 4-Арилдибензоилендигид-
ропиридины весьма чувствительны к ненасыщенным соедине-
ниям (например, 2-илидениндандионам-1,3, которые образу-
ются как промежуточные продукты при конденсации индан-
диона с альдегидами), поэтому в данном случае необходимо
отдельно синтезировать геминальные дииндандионильные
производные [8, 10].
СХЕМА СИНТЕЗА АЦЕИАФТЕНОН-ЬСПИРО-(2:4')-2',3'(СО),
6', 5/(СО)-ДИБЕНЗОИЛЕН-1',4/-ДИГИДРОПИРИДИНА
13
Характеристика основного сырья
Аценафтенхинон, ч., СТУ 79-5-27—62.
1,3- Индандион, ч„ ВТУ МГ УХП 127—58.
Уксусная кислота, 98%-ная, ГОСТ 61—51.
Аммоний уксуснокислый, ГОСТ 3117—51:
Условия получения
В круглодонную колбу емкостью 2 л с нормальным шли
фом, соединенную с обратным холодильником Либиха, поме
щают 18 г (0,0989 А1) аценафтенхинона и 860 мл (907 г)
98 %-ной уксусной кислоты и кипятят в течение 30 минут (см.
примечание 1). Прибавляют 28,8 г (0,197 АН индандиона и
6 мл (7,1 г) концентрированной соляной кислоты и кипятят
10 минут. Затем к реакционной смеси добавляют 66 г
(0,84 М) ацетата аммония и несколько кусочков пористого
вещества (см. примечание 2) и кипятят в течение 2 часов.
Выпавший кристаллический осадок отделяют (в горячем ви-
де), промывают 40 мл этилового спирта, кипятят в 200 мл
98%-ной уксусной кислоты в течение 30 минут, отделяют в
горячем виде и промывают 40 мл этилового спирта (см. при-
мечание 3).
Выход продукта равен 27—30 г, что составляет 62,5—
69,5% от теоретического. По внешнему виду это красно-ко-
ричневое кристаллическое вещество с т. пл. 260—264° (см.
примечание 4).
Примечания:
I. Ацеиафгенхинон медленно растворяется в уксусной кислоте, необ-
ходимо добиваться почти полного его растворения.
2. В ходе реакции образуется осадок, который мешает равномерному
кипению
3. Фильтрат уксусной кислоты собирают отдельно и перегоняют.
4. Реагент IV малорастворим в обычных органических растворите-
лях (кроме пиридина и спиртовой щелочи). После описанной обработки
вещество достаточно чисто.
ЛИТЕРАТУРА
I. Г. Я. В а и а г, Г. Я. Ду бур. Авт. свид. 143030; Бюлл. изобр.,
№ 23 (1961).
2. Г. Я. Ду бур, Г. Я. В а н а г. Изв. АН Латв. ССР, сер. хим.,.
3. Л. Я. Лейт и с, Г. Я. Д у б у р, ("А. В. Шиманская, Г. Я. Ва-
на г. Изв. АН Латв. ССР, сер. хим., 1963, 41.
4. Г. Я. В а н а г, Л. С. Гейта. Ж. общ. химии, 26, 511 (1956).
5. Л. С. Гейта, Г. Я. В а н а г. Ж. общ. химии, 27, 3109 (1957).
6. Г. Я. В а н а г, Г. Я. Дубур. Ж. общ. химии, 27, 2729 (1957).
7. Г. Я. В а н а г, Г. Я. Дубур. Ж. общ. химии, 30 1898 (1960).
8. Г. Я. Дубур. Сб. «Циклические ₽-днкетоны», Рига, АН Латв.
ССР, 1961, стр. 259.
14
9. Г. Я. Дубур, Г. Я. В а и а г. Изв. АН Латв. ССР, сер. хим.,
1962, 119.
10. Г. Я. Дубур, Г. Я- В а и а г. Изв. АН Латв. ССР, сер. хим.,
1962, 287.
Поступила в декабре 1964 г.
Институт
органического синтеза
АН Латв. ССР
УДК 547.821.2.07
2- N-АЦЕТИЛАМИНОПИРИДИН
Г. Ф. ДРЕГВАЛЬ, А. П. МАРТЫНЮК
C7H8N2O
NHCOCH3
М. в. 136,15
2-М-Ацетиламинопиридин представляет интерес в качестве
исходного соединения для синтетических работ.
Его получают взаимодействием 2-аминопиридина и уксус-
ного ангидрида [1, 2]. Нами уточнены условия получения и
выделения продукта реакции и наши результаты соответст-
вуют литературным данным.
СХЕМА СИНТЕЗА 2-1Ч-АЦЕТИЛАМИНОПИРИДИНА
/Ч /X
II | + (СН3СО)2О -> || | + СНзСООН
XN/4*NH2 Xn^XNHCOCH3
Характеристика основного сырья
2-Аминопиридин, ч., т. пл. 56—58°.
Уксусный ангидрид, ч. д. а., ГОСТ 5815—52.
Условия получения
В двугорлую колбу помещают 30 г (0,32 2И) 2-аминопи-
ридина и медленно прибавляют 32,7 г (0,32 2И) уксусного ан-
гидрида. Реакция идет с выделением тепла, при этом 2-ами-
нопиридин переходит в раствор, который окрашивается в
16
красный цвет. Затем реакционную смесь нагревают до ки-
пения и кипятят 4 часа. Уксусную кислоту отгоняют в ваку-
уме водоструйного насоса при 60—70 мм. В остатке получа-
ют светло-коричневое масло, которое вскоре закристаллизо-
вывается.
Выход 2-Ы-ацетиламинопиридина равен 34,7 г, что состав-
ляет 82% от теоретического; т. пл. 68—69°.
Для очистки продукт можно перегнать в вакууме при
127—12877 мм или перекристаллизовать из смеси бензол —
петролейный эфир (2:1); т. пл. 71°.
По литературным данным, т. пл. продукта 71° [1, 2].
ЛИТЕРАТУРА
1. Нем, пат. 406 206; Chem. Zbl., 1925, 1, 1534.
2. Camps. Arch. Pharm., 240, 349 (1902); Chem. Zbl, 1902, II, 647.
Поступила в декабре 1964 г.
Донецкий филиал ИРЕЛ
2 Зак. 1350
УДК 547.831.7.07
8-АЦЕТОКСИКАРБОСТИРИЛ
2-Окси-8-ацетоксихинолин
В. М. ДЗИОМКО, И. А. КРАСАВИН, Ю. П. РАДИН,
Н. И. МИРОШКИНЛ
//\/Х
I II I
W4
I I 0
СНэСОО н
//\/ч
I II J
VW\
I он
СН3СОО
CnH9NO3
М.в. 203,20
Известны два способа получения 8-ацетоксикарбостирила.
Один из них состоит в ацетилировании 2,8-диоксихииолина
кипящим уксусным ангидридом [1]. Метод, которым автор ра-
боты [1} получил 2,8-диоксихинолин (нагревание 8-оксихино-
лина с расплавленной щелочью в серебряном тигле при 380°),
неудобен, так как требует применения жестких эксперимен-
тальных условий и дает загрязненный продукт.
Другой путь заключается во взаимодействии 8-оксихино-
лин-1-оксида с уксусным ангидридом при комнатной темпе-
ратуре в течение нескольких дней (выход 88%) (2].
Второй способ был нами проверен и уточнен.
Получение 8-ацетоксикарбостирила
I л | 4-(Сн8со)2о -4 । । । +сн3соон
Ч/Xn^
I I I I О
ОН о СНаСОО Н
В коническую колбу емкостью 1 л помещают 265 мл
(286 а; 2,8 М) свежеперегнанного уксусного ангидрида и ра-
створяют в нем 45,1 г (0,28 М) 8-оксихинолин-1-оксида (см.
18
примечание 1) при кратковременном подогревании колбы теп-
лой водой. Раствор защищают от действия влаги воздуха и
оставляют на 12—15 дней при комнатной температуре (см.
примечание 2). Перед выделением продукта колбу помеща-
ют на 2—3 часа в ледяную воду, затем осадок отсасывают,
тщательно отжимают, промывают на фильтре ацетоном (3
раза по 10 мл) и высушивают в вакууме.
Выход 8-ацетоксикарбостирила равен 45,5—47,8 г (80—
84% от теоретического); т. пл. 249—249,5° (см. примечания
3 и 4).
Если разбавить фильтрат двойным объемом воды, высу-
шить осадок и перекристаллизовать его из 15—20 мл уксус-
ного ангидрида, можно получить еще 2—3 г вещества с т. пл.
244—245° (см. примечание 5).
Общий выход равен 47,5—50,7 г (83,5—89,2%)
Примечания:
I. Применялся 8-оксихинолии-1-оксвд с т. пл. 138,5—139°, полученный
известными методами [2, 3] и дважды перекристаллизованный из бензола.
2. После остывания раствора вещество опять выпадает в осадок.
При стоянии количество осадка постепенно уменьшается и через 2—3 су-
ток образуется прозрачный раствор; еще через 1—2 суток начинает выде-
ляться продукт реакции.
3. По литературным данным, т. пл. 8-ацетоксикарбостирила 244—
247° (из ледяной уксусной кислоты) [1, 4J; 247,5—248,5° (из этанола) [5|;
250° (из этанола) [2].
4. Продукт пригоден для синтетических работ. Отрицательная реак-
ция с хлорным железом указывает иа отсутствие примеси 2,8-диоксихи-
нолина. Для дальнейшей очистки Можно применить перекристаллизацию из
диметилформамида.
5. Вещество, выделенное посредством разбавления фильтрата водой,
обычно содержит примесь диоксипроизводного; под действием уксусного
ангидрида последнее превращается в 8-ацстоксикарбостирил.
ЛИТЕРАТУРА
1. J. D i a m а п t. Monatsh., 16, 760 (1895).
2. J. Р. Phillips, Е. М. Barra 11, R. Breese. Trans. Kentucky
Acad. Set., 17, 135 (1956); Chetn. Abstrs, 51,11349b (1957).
3. I. Mutase, Y. Demtira. Mem. Fac. Set., Kynshu Univ., Ser.
C4, № 3, 175 (1961); Chem. Abstrs, 58, 3390b (1963).
4. S. Koshimura, A. Hamada, T. Otaki, K. Deguchi. Ann.
Rept. Research Inst. Tuberc., Kanazawa Univ., 12, 2, 9 (1954); Chem.
5Ь’Т. Ohta, Y. Morl, S. Taka gi, Jr. Yakugaku Zasshi, 78,697 (1958).
Поступила в сентябре 1965 г.
ИРГ.А
УДК 547.551.43.07
БЕНЗИЛОВЫЙ ЭФИР К-[Н2-ПИРИДИЛ)ЭТИЛ]-1^
ФЕНИЛДИТИОКАРБАМИНОВОИ КИСЛОТЫ
И. Ф. КАЗАРИНОВА, Т. И. ТУМБИНА, К. А. С0Л0МК0
/ \ СЛ
( J-CH2-CH2-N-C-S-CH2CeH6
s
C31Hj0N2Sj M.b. 365,54
Эфиры N-(2-пирндилэтил)-N'-фенилтиокарбаминовой кис-
лоты в литературе не описаны. Для синтеза этих эфиров ис-
пользована методика, описанная Кеннардом [1] для эфиров
N- (2-пиридилалкил) -N-метилднтнокарбамнновой кислоты.
Подобные соединения могут представить интерес в каче-
стве пестицидных препаратов,
СХЕМА СИНТЕЗА БЕНЗИЛОВОГО ЭФИРА К-[₽-(2-ПИРИДИЛ)ЭТИЛ]-
N-ФЕНИЛДИТИОКАРБАМИНОВОЙ КИСЛОТЫ
\ фн'
I J!—СН2—СН2—NH+NaOH+CSs ->
Чг
СвНб
I ILcH2-CH2-N-C-S-Na+H,O
||
S
I J-CHj-CH.-N-C-SNa+CICHAHg
||
20 S
S \ свн«
f J—CH2CHaN —С—SCH,CeHB-f-NaCl
X’/ ||
S
Характеристика основного сырья
2-[(1-(М-Фениламино)этил]пиридин, т. кип. 190—204°/12лш.
Сероуглерод, технический.
Натр едкий, технический.
Бензил хлористый, ч., ТУ МХП 50—49.
Условия получения
К смеси, состоящей из 9 г (0,045 М) 2-[р-(М-фениламино)
этил]пиридина и 1,8 г (0,045 М) едкого натра в 16 мл воды,
постепенно, прн непрерывном перемешивании, добавляют 4 г
(0,05 М) сероуглерода. Полученный раствор перемешивают
в течение 1,5 часа, после чего к нему добавляют 5,5 г
(0,045 М) хлористого бензила. Реакционную смесь нагревают
на водяной бане в течение часа, а затем выдерживают при
комнатной температуре в течение ночи. Выпавший осадок от-
фильтровывают, промывают водой и перекристаллизовывают
из метанола (см. примечание).
Выход реактива равен 8,7 г, что составляет 53% от тео-
ретического; т. пл. 111—112°.
Найдено, %: N-7,42; 7,61; S—17,14; 17,52.
C21H3oS2N2. Вычислено, %: N -7,6; S—17,5.
Примечание. ,
По аналогичной методике могут быть получены другие S-алкилзаме-
шейные эфиры ЬЦМ2-пиридил)этил]-М-фенилдитиокарбамииовой кислоты.
ЛИТЕРАТУРА
1. К. Kennard, D. В u г n е s s. J. Organ. Chem., 24, 464 (1954).
Поступила в декабре 1964 г.
Донецкий филиал ИРЕА
УДК 547.828.07
N,N'- Б И С- (К А Р БО КС И М ЕТ И Л) -4,4'-Д И П И Р И Д И Л И Й
ДИХЛОРИД
И. Ф. КАЗАРИНОВА, К. А. СОЛОМКО, М. И. КОТЕЛЕНЕЦ
2С1-
HOOCHjC
СН2СООН
c14hun8cia
М.в. 345,18
Четвертичные соли 4,4'-дипиридил а широко известны как
соединения с высокой гербицидной активностью [1, 2]. О син-
тезах 4,4'-дипиридилиевых солей имеется лишь ряд патентных
сообщений [1]. Для синтеза N,N'-6hc-(карбоксиметил)-4,4'-
дипиридилий дихлорида нами был использован обычный ме-
тод получения четвертичных аммониевых солей, заключаю-
щийся во взаимодействии основания с соответствующим га-
лоидным алкилом.
СХЕМА СИНТЕЗА N.N' -БИС-(КАРБОКСИМЕТИЛ)-4,4'-ДИПИРИДИ-
ЛИЙ ДИХЛОРИДА
Л II II i + 2С1СН,СООН - 1,+ || || -I
Ч/ /Ч/
НООСН2С СН2СООН
Характеристика основного сырья
4,4'-Дипиридил, техн., т. пл. 69—71°, ВТУ РУ 661—52.
Монохлоруксусная кислота, ч., ГОСТ 5836—51.
Изопропиловый спирт, о. ч„ ТУ ОРУ 50—57.
22
Условия получения
К раствору 15,6 г (0,1 Л!) 4,4'-дипиридила в 10 мл изопро-
пилового спирта добавляют раствор 18,9 г (0,2 М) монохлор-
уксусной кислоты в 20 мл изопропилового спирта. Получен-
ную смесь кипятят в течение 6 часов. Выпавший после охлаж-
дения Н.М'-бис-(карбоксиметил)-4,4/-дипиридилий хлорид от-
фильтровывают и промывают этиловым спиртом и эфиром.
Выход равен 24,2 г, что составляет 70% от теоретического,
т. разл. выше 300°.
Примечание.
По аналогичной методике из 4,4/-дипиридила и этиленхлоргидрина
может быть получен N.N-бис-(Р-оксиэтил)-4,4-'-дипирндилий. дихлорид с
выходом 48% от теоретического и т. разл. выше 300°.
ЛИТЕРАТУРА
[.Брайан, Др ей в ер, Хомер, Джонс. Англ. пат. 813531:
РЖхим., № 5, 19213, 1960.
2. J. Crons hey. The new bipyridyl_herbicides. Imp. Cheat. Ind., 1960.
Поступила в декабре 1964 г.
Донецкий филиал ИРЕА.
УДК 553.637.07
БОРАТРАН
(2,2',2"-Ам инотр иэтил) бор ат
М. Г. ВОРОНКОВ, Г. И. ЗЕЛЧАН
CeH12O3NB
М.в. 156,97
Известно несколько способов получения боратрана из три-
этаноламина и борного ангидрида [1], борной кислоты [2, 3]
или низших триалкилборатов [1, 3].
Наиболее удобный метод синтеза боратрана основан на
реакции триэтаноламина с борной кислотой. Однако и он
имеет несколько модификаций. Взаимодействие компонентов
в отсутствие растворителя с отгонкой в вакууме образующей-
ся воды продолжается около 10 часов [2]. Процесс можно
проводить и в присутствии растворителя (диметилформамид)
с удалением воды также путем отгонки [3].
Нами разработан простой и удобный метод получения
боратрана из триэтаноламина и борной кислоты с использо-
ванием в качестве растворителя изоамилового спирта. Он от-
личается меньшей продолжительностью (~2,5 часа) и высо-
ким выходом продукта (около 90%). Выделяющаяся в про-
цессе реакции вода удаляется путем непрерывной азеотроп-
ной отгонки с возвратом растворителя в реакционную смесь.
Образующийся боратран выкристаллизовывается при охлаж-
дении непосредственно из реакционной смеси, выделяется
фильтрацией и обладает высокой степенью чистоты.
24
СХЕМА СИНТЕЗА БОРАТРАНА
N(CHaCH2OH)3 + (НО)3В N(CHaCH,O)3 В %- ЗН2О.
I__________________________________-t
Характеристика основного сырья
Триэтаноламин, ч., СТУ 12 10113—61.
Борная кислота, х. ч., СТ ГОХП 27—1830.
Изоамиловый спирт, ч. д. а., ГОСТ 5830—51.
Условия получения
В круглодонную колбу емкостью 0,5 л, соединенную че-
рез водоотборную ловушку с обратным холодильником, поме-
щают 89,4 г (0,6 М) триэтаноламина (см. примечание 1),
37,1 г (0,6 Л4) борной кислоты и 300 мл изоамилового спир-
та. Смесь кипятят до прекращения выделения воды в ловуш-
ке. Теоретическое количество воды (32,4 мл) выделяется за
1,5—2 часа. При медленном охлаждении реакционной смеси
(прозрачный светло-желтый раствор) из нее выпадают белые
кристаллы боратрана, которые отсасывают, промывают аце-
тоном и сушат в вакууме (см. примечание 2).
Выход боратрана с т. пл. 237—238° равен 78,0 г, что со-
ставляет 83% от теоретического. Отгонкой растворителя из
маточного раствора дополнительно можно выделить еще 6,6 г
чистого вещества. Общий выход боратрана составляет 90%
от теоретического.
Примечания:
1. Для синтеза использовался продажный триэтаноламин, предвари-
тельно перегнанный в вакууме.
2. Боратран хранят в запаянных ампулах или плотно закрытых бан-
ках для предохранения от влаги воздуха, под действием которой он мед-
ленно гидролизуется.
ЛИТЕРАТУРА
1. F. Н е i п, R. В и г k h а г d t. Z. anorgan. und allgem. Chem., 268,
159 (1952).
2. H. C. Brown, E. A. Fletcher. J. Amer. Chem. Soc., 73, 2808
(1951).
3. A. A. S c h 1 e p p n i к, C. D. G u t s c h e. J. Organ. Chem., 25, 1378
(1960).
Поступила в декабре 1964 г. Институт органического синтеза
АН Латв. ССР
УДК 547.569.2.07
/П/шп-БУТИЛ-2-ТИЕНИЛСУЛЬФИД
Б. П. ФЕДОРОВ, Ф. М. СТОЯНОВИЧ
\ V SC(CH3)g
\ Qz
C8H12S2
М. в. 172,31
трет-Бутил-2-тиенилсульфид в литературе не описан. Для
его синтеза мы использовали методику, разработанную для
получения третичных алкиларилсульфидов [1] (см. примеча-
ние 1). Полученный в этих условиях трет-бутил-2-тиенилсуль-
фид не содержит примеси изобутил-2-тиенилсульфида. Этим
путем, а именно, присоединением соответствующих замещен-
ных тиофентиолов к изобутилену в кислой среде, могут быть
получены трет-бутил-2-тиенилсульфиды, замещенные в тиофе-
новом кольце [4].
СХЕМА СИНТЕЗА трет- БУТИЛ-2-ТИЕНИЛСУЛЬФИДА
сн
/-SH +
sz сн.
, 75%H2SO,
хснз
^S-C<CH3
\сн3
Характеристика основного сырья
2-Тиофентиол, свежеприготовленный, т. кип. 76—77°/40 мм
[5].
Изобутилен, ректифицированный (96,3%), без дополни-
тельной очистки.
26
Условия получения
В трехгорлую круглодонную колбу емкостью 0,5 л с хо-
рошо работающей мешалкой, трубкой для ввода газа, опу-
щенной под слой жидкости, и холодильником, охлаждаемым
сухим льдом и ацетоном или рассолом (температура от ми-
нус 10 до минус 20°), помещают 88 г 75 %-ной серной кисло-
ты, охлаждают ее до минус 10" и начинают пропускать изо-
бутилен до тех пор, пока не поглотится 25,2 г газа (см. при-
мечание 2). Заменяют трубку для ввода газа на более ко-
роткую, не доходящую до жидкости, и, пропуская инертный
газ (азот или аргон), прибавляют в течение 10 минут 16,4 г
2-тиофентиола при хорошем размешивании, поддерживая тем-
пературу около 0°. Температуру доводят до комнатной и раз-
мешивают 1,5 часа. Затем реакционную массу выливают в ле-
дяную воду, продукт экстрагируют эфиром, эфирный слой
промывают водой, холодным 5%-ным раствором едкого нат-
ра, вновь водой и высушивают прокаленным сернокислым
магнием. Остаток после удаления растворителя перегоняют.
Выход трет-бутил-2-тиенилсульфида равен 16,2 г, что со-
ставляет 67% от теоретического; т. кип. 74,5—75,574 мм;
83—8479 мм; 88—90712 мм; tig — 1,5460; 20 — 0,9809.
Найдено, %: С-55, 81; 55, 63; Н-7, 01;6, 97; S—37, 10; 37, 10.
C8H12S2. Вычислено, %: С—55, 76; Н—7,02; S—37,21.
Примечания:
1. Синтез трег-бутил-2-тиенилсульфида мы пытались осуществить пос.
ледовательным действием серы и третичного бромистого бутила на 2-ти-
еииллитин [2]. Сульфид был получен с очень низким выходом (около
10%), и в чистом виде его выделить не удалось. Хиггинс и Гэррет в сво-
ей недавно опубликованной работе [3] также не смогли получить этот
сульфид взаимодействием натриевой соли 2-тиофентиола с трет-бутил-
галогенидом Причина, вероятно, заключается в легкоидущем в щелочной
среде отщеплении галоидоводорода от молекулы третичного галоидного
алкила. Сульфид нельзя также получить взаимодействием 2-тиениллития
с дитретичнобутилдисульфидом, который, по-видимому, в силу пространст-
венных затруднений не вступает в реакцию и почти целиком был возвра-
щен обратно.
2. Привес определяют, время от времени взвешивая колбу с раство-
ром под тягой. Первое взвешивание делают не раньше, чем через 1,5 ча-
са после начала пропускания. Время пропускания обычно колеблется от
2 до 3 часов. Необходимо следить, чтобы при взвешивании реакционная
масса не состояла нз двух слоев, так как верхним слоем может быть скон-
денсированный непрореагпровавший изобутилен.
ЛИТЕРАТУРА
1. V. N. Ipatieff, Н. Pines, В. S. Friedmann. J. Amer. Chem.
Soc., 60, 273 (1938).
2. Я. Л. Гольдфарб, М. А. Калик, М. Л. Кирмалова. Ж.
общ. химии, 29, 2034 (1959).
3. R. W. Higgins, R. Garrett. J. Organ. Chem, 27, 2168 (1962).
4. Б. П. Федоров, Ф. M. Стоянов и ч. Ж. общ. химии (в печати).
5. Я. Л. Гольдфарб, М. А. Калик, М. Л. Кирмалова. Ж.
общ. химии, 32, 292 (1962).
Поступила в декабре 1961 г.
ИОХ АН СССР
27
УДК 547.835.9.
9-ВИНИЛ-1,2-БЕНЗАКРИДИН
Б. С. ТАНАСЕЙЧУК, И. Я. ПОСТОВСКИЙ
Ci9H13N
М. в. 255,32
9-Винил-1,2-бензакридин в литературе не описан. Он нами
получен посредством нагревания 9-метил-1,2-бензакридина с
формальдегидом в среде спирта в присутствии гидрохлорида
триэтиламина или в присутствии незначительных количеств
соляной кислоты.
СХЕМА СИНТЕЗА 9-ВИНИЛ-1,2-БЕНЗАКРИДИНА
сн8 п /ч СН=СН,
II । НС^ NRtHCI I' । ।
\//\/ Ч./'Ч \н ’ 3 \//\/
I и I I---------------------> I II II
Характеристика основного сырья
9-Метил-1,2-бензакридин, т. пл, 143—145° (1].
Формалин, ГОСТ 1625—45.
Условия получения
В круглодонную колбу емкостью 50 мл, снабженную об-
ратным холодильником, помещают 1,2 г 9-метил-1,2-бензакри-
28
дина (см. примечание 1), 0,65 мл формалина (см. примеча-
ние 2), 0,69 г гидрохлорида триэтиламина (см. примеча-
ние 3) и 5 мл этилового спирта. Содержимое колбы нагрева-
ют на воздушной бане до выпадения осадка (2—4 часа). Оса-
док, представляющий собой 9-винил-1,2-бензакридин, от-
фильтровывают и промывают спиртом.
Выход продукта равен 0,4—0,5 г, что составляет 30—40%
от теоретического; т. пл. 230—232°. При перекристаллизации
из бензола по внешнему виду это длинные бесцветные иглы с
т. пл. 232—233°. В концентрированной серной кислоте 9-ви-
нил- 1,2-бензакридин растворяется с образованием желтого
раствора.
При продолжении нагревания маточного раствора через
1—2 часа выпадет дополнительное количество продукта.
Примечания:
1. Особое внимание должно быть обращено на чистоту 9-метил-1,2-
бензакридина, так как выход 9-вииил-1,2-беизакридина снижается, если
исходный препарат имеет температуру плавления ниже, чем указано в
методике.
2. Формалин берут продажный.
3. Вместо гидрохлорида триэтиламина можно взять гидрохлорид
циклоалкиламииа (пиперидина, морфолина или пирролидина) или же ве-
сти реакцию в присутствии одной капли концентрированной соляной кис-
лоты, но в этом случае время иагрева увеличивается до 10—15 часов.
ЛИТЕРАТУРА
1. А. Е. Порай-Кошиц, Г. С. Тер-Саркисян. Изв. АН СССР,
ОХН, 771 (1951).
Поступила в декабре 1964 г.
Уральский политехнический институт
УДК 547.657.07
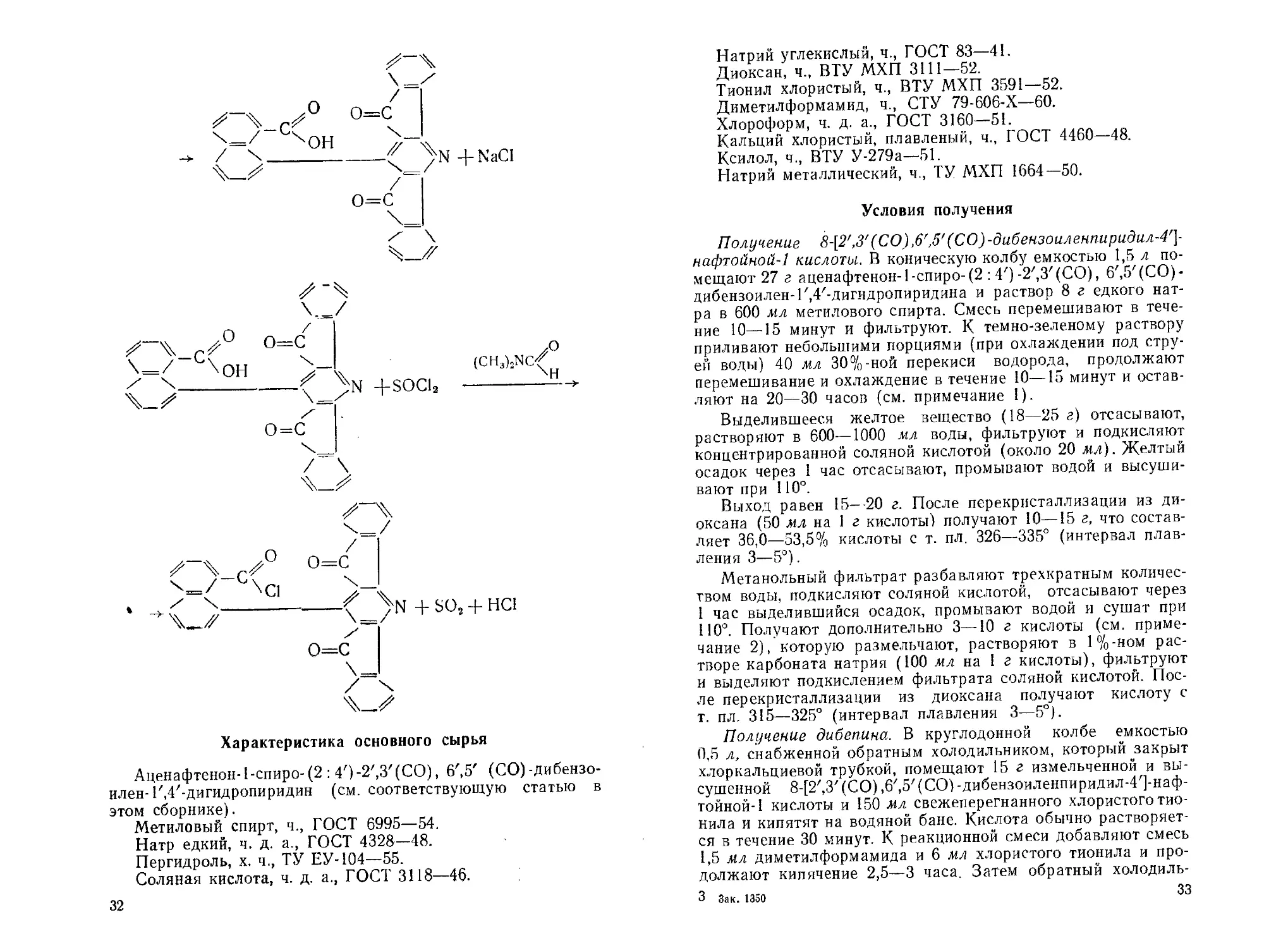
ДИБЕПИН
Хлорангидрид 8-[2/,3'(СО),6,,5/(СО)-дибензоиленпиридил-4/]-
нафтойной-1 кислоты
Г. я. ДУБУР, [г. Я. В АН А Г \, Я. Р. УЛДРИКИС
C30HuNO3Cl
М. в. 471,90
Дибепин [1] является оригинальным аналитическим реа-
гентом на первичные амины [2] и предназначен для качествен-
ного выявления [3, 4] и количественного определения [5] пер-
вичных алкил- и арилалкиламинов с помощью цветной реак-
ции. Со всеми вторичными и третичными аминами реакция
отрицательна. Дибепин выгодно дополняет другой реагент на
первичные амины — биндон [6], который более чувствителен
на ароматические амииы. Это относится и к средам реакций—
реакцию с биидоном проводят в растворе ледяной уксусной
кислоты, а реакцию с дибепином в нейтральной или щелоч-
ной среде.
Исходным веществом для получения дибепина служит
аценафтенон-1-спиро-(2 :4') -2',У (СО), 6',5'(СО) -дибензоилен-
1',4'-дигидро пиридин, который получен конденсацией аце-
нафтенхинона, индандиона и ацетата аммония в уксусной кис-
30
лоте [1, 7]. При действии перекиси водорода на щелочной рас-
твор аценафтенон-1-спиро-(2:4')-2/,3,(СО), 6',5'(СО)-дибензо-
илен-1',4'-дигидропиридина в метаноле происходит расщепле-
ние пятичленного аценафтенонового кольца с образованием
соли 8-[2/,3/(СО),6/,5/(СО)-дибензоиленпиридил-4л]-нафтой-
ной-1 кислоты. Дибепин получен действием хлористого тиони-
ла в присутствии диметилформамида [8] на 8-[2',3'(СО),
6',5' (СО) -дибензоиленпиридил-4']-нафтойную-1 кислоту.
СХЕМА СИНТЕЗА ДИБЕПИНА
\ONa
---+ НС|
О=С
31
°^с/
Cxci
---------\_/N + so, + НС1
о—сГ
c__^
Характеристика основного сырья
Аценафтенон-1-спиро-(2 : 4л)-2/,3/(СО), 6',5' (СО)-дибензо-
илен- 1',4'-дигидропиридин (см. соответствующую статью в
этом сборнике).
Метиловый спирт, ч., ГОСТ 6995—54.
Натр едкий, ч. д. а., ГОСТ 4328—48.
Пергидроль, х. ч., ТУ ЕУ-104—55.
Соляная кислота, ч. д. а., ГОСТ 3118—46.
32
Натрий углекислый, ч., ГОСТ 83—41.
Диоксан, ч., ВТУ МХП 3111—52.
Тионил хлористый, ч., ВТУ МХП 3591—52.
Диметилформамид, ч., СТУ 79-606-Х—60.
Хлороформ, ч. д. а., ГОСТ 3160—51.
Кальций хлористый, плавленый, ч., ГОСТ 4460—48.
Ксилол, ч„ ВТУ У-279а—51.
Натрий металлический, ч., ТУ МХП 1664—50.
Условия получения
Получение 8-[2',3'(СО),6',5'(СО)-дибензоиленпиридил-4']-
нафтойной-1 кислоты. В коническую колбу емкостью 1,5 л по-
мещают 27 г аценафтенон-1-спиро-(2 : 4') -2/,3/(СО), 6',5'(СО)-
дибензоилен-1',4'-дигидропиридина и раствор 8 г едкого нат-
ра в 600 мл метилового спирта. Смесь перемешивают в тече-
ние 10—15 минут и фильтруют. К темно-зеленому раствору
приливают небольшими порциями (при охлаждении под стру-
ей воды) 40 мл 30%-ной перекиси водорода, продолжают
перемешивание и охлаждение в течение 10—15 минут и остав-
ляют на 20—30 часов (см. примечание 1).
Выделившееся желтое вещество (18—25 г) отсасывают,
растворяют в 600—1000 мл воды, фильтруют и подкисляют
концентрированной соляной кислотой (около 20 мл). Желтый
осадок через 1 час отсасывают, промывают водой и высуши-
вают при 110°.
Выход равен 15-20 г. После перекристаллизации из ди-
оксана (50 мл на 1 г кислоты) получают 10—15 г, что состав-
ляет 36,0—53,5% кислоты с т. пл. 326—335° (интервал плав-
ления 3—5°).
Метанольный фильтрат разбавляют трехкратным количес-
твом воды, подкисляют соляной кислотой, отсасывают через
1 час выделившийся осадок, промывают водой и сушат при
110°. Получают дополнительно 3—10 г кислоты (см. приме-
чание 2), которую размельчают, растворяют в 1%-ном рас-
творе карбоната натрия (100 мл на 1 г кислоты), фильтруют
и выделяют подкислением фильтрата соляной кислотой. Пос-
ле перекристаллизации из диоксана получают кислоту с
т. пл. 315—325° (интервал плавления 3—5°).
Получение дибепина. В круглодонной колбе емкостью
0,5 л, снабженной обратным холодильником, который закрыт
хлоркальциевой трубкой, помещают 15 г измельченной и вы-
сушенной 8-[2',3' (СО) ,6',5' (СО) -дибензоиленпиридил-4']-наф-
тойной-1 кислоты и 150 мл свежеперегнанного хлористого тис-
нила и кипятят на водяной бане. Кислота обычно растворяет-
ся в течение 30 минут. К реакционной смеси добавляют смесь
1,5 мл диметилформамида и 6 мл хлористого тионила и про-
должают кипячение 2,5—3 часа. Затем обратный холодиль-
3 Зак. 1350 33
нпк заменяют нисходящим и отгоняют из реакционной массы
в вакууме (с водоструйным насосом) на водяной бане хло-
ристый тионил. После прекращения перегонки остаток охлаж-
дают, прибавляют 15 мл хлороформа (высушенного над хло-
ристым кальцием), перемешивают, через 15 минут отсасыва-
ют, промывают на фильтре 25 мл хлороформа и сушат при
60—80° в течение 2 часов. Получают 11 —13 г желтого веще-
ства, которое перекристаллизовывают из безводного ксилола
(120 мл на 1 г; см. примечания 3 и 4).
Выход дибепина после перекристаллизации равен 8—11 г,
что соответствует 51—70% от теоретического, считая на кис-
лоту.
Дибепин представляет собой желтое кристаллическое ве-
щество, которое плавится с разложением в интервале 280—
300°. При длительном хранении гидролизуется влагой воздуха
(см. примечание 5).
Примечания:
1. Если в реакционной смеси после 20—30 часов видно зеленое или
красное вещество или проба раствора после прибавления щелочи приоб-
ретает зеленый цвет, то к реакционной смеси прибавляют раствор 4 г ед-
кого натра в 6 мл воды и 20 мл 30%-ной перекиси водорода и оставляют
еще на 20—30 часов.
2. Кислота, полученная из метанольного раствора, более загрязнена,
поэтому необходима дополнительная очистка.
3. Следует тщательно избегать присутствия воды в ксилоле. Исполь-
зуют высушенный, перегнанный над натрием ксилол.
4. Размельченный дибепин прибавляют к горячему ксилолу, кипятят
не более 15 минут. Следует учесть, что при перекристаллизации дибепина,
содержащего кислоту, возможно образование ангидрида 8-[2',3'(СО),
6',5'(СО)-дибензоиленпиридил-4']-нафтойной-1 кислоты, весьма труднорас-
творимого вещества. В этом случае полученный дибепин дает мутный рас-
твор в диоксане. Следует избегать длительного воздействия влаги воздуха
на дибепин.
5. При этом образуется кислота. Качество дибепина можно проверить
хроматографически. Тонкослойная хроматография [9] (на пластинке 9Х
X12 см, силикагель КСК, система н-бутанол:концентрированный водный
раствор аммиака = 4:1, для кислоты /?/ = 0,42, для дибепина Rf - 1)
позволяет обнаружить наличие кислоты в дибепине.
Хроматография на бумаге «Ленинградская быстрая» в системе изопро-
панол : концентрированный водный раствор аммиака : вода = 14:1:5 дает
Rf = 0,61 (дибепин) и Rf = 0,76 (кислота).
ЛИТЕРАТУРА
1. Г. Я. В а н а г, Г. Я. Дубур. Ж. общ. химии, 30, 1898 (1960).
2. Г. Я. В а н а г, Г. Я. Дубур. Авт. свид. 143030; Бюлл. изобр.,
№ 23 (1961).
3. Г. Я. Дубур, Г. Я. В а н а г. Тр. Комис, по аналпт. химии АН
СССР, Органический анализ, 13, 429 (1963).
4. Г. Я. Дубур, Г. Я. В а н а г. Изв. АН Латв. ССР, сер. хим, 1962,
№ 1, 25.
5. Л. Я. Лейт ис, Г. Я. Дубур, М. В. Шиманская, Г. Я. В а-
II а г. Изв. АН Латв. ССР, сер. хим., 1963, 1, 41.
6. Губен-Вейль. Методы органической химии, т. II (Методы ана-
лиза), 1963, стр. 656.
34
7. Г. Я. Дубур, Г. Я. В а и а г. Методы получения химических ре-
активов и препаратов, вып. 14, М„ ИРЕА, 1966, стр. 12.
8. Н. Н. Bosshard, R. Могу, М. Schmid, Н. Zollinger.
Helv. chim. acta, 42, 1653(1959).
9. A. A. A x p e м, А. И. Кузнецова. Тонкослойная хроматогра-
фия, M., Наука, 1964.
Поступила в июне 1965 г.
Институт органического синтеза
АН Латв. ССР
3*
УДК 547.822.7.07
2-№Д И ИЗОПРОПИЛ ФОСФИН ИЛАМИНОП ИРИД ИН
Г. Ф. ДРЕГВАЛЬ, Н. В. КОВАЛЕНКО, А. П. МАРТЫНЮК
!^LNHP-(OC8H,-Z)2
CnH19NaO3P М.в. 258,28
2-Ы-Диизопропилфосфиниламинопиридин представляет ин-
терес в качестве физиологического активного препарата и ре-
актива для синтеза других интересных фосфорорганических
соединений. Взаимодействие хлорангидридов диалкилфосфор-
ных кислот с 2-аминопиридином изучали несколько ученых
[1, 2].
Нами проверена методика получения этого продукта и
разработаны общие условия получения 2-№-диалкилфосфи-
ниламинопиридинов.
СХЕМА СИНТЕЗА 2-М-ДИИЗОПРОПИЛФОСФИНИЛАМИНО-
ПИРИДИНА
/Ч х°
у Lnh + рС + (с’н»ьN
2 ХС1
/Ч ,о
-II 1-nhp€ +(C3H6)3N.HC1
\N/ НЧ-(ОС3Н;-Д)2
Характеристика основного сырья
Диизопропилхлорфосфат, свежеперегн энный, т. кип. 97—
100715 мм; п2° — 1,4185.
36
2-Аминопиридин, ч., т. пл. 56—58д.
Триэтиламин, ч., ТУ РУ 796—53.
Бензол, х.. ч., ГОСТ 5955—51.
Условия получения
В четырехгорлую колбу, снабженную обратным холодиль-
ником, мешалкой, капельной воронкой, загружают 9,4 г
(0,1 2И) 2-аминопиридина, 20,2 г (0,2 М) триэтиламина и
20 мл сухого бензола.
При перемешивании и охлаждении ледяной водой прибав-
ляют 20 г (0,1 М) диизопропилхлорфосфата. Затем реакцион-
ную смесь нагревают на водяной бане в течение часа, дают
ей охладиться до комнатной температуры и отфильтровывают
осадок солянокислого триэтиламина и основного продукта.
Осадок дважды промывают водой по 15 мл для удаления со-
лянокислого триэтиламина и сушат.
Выход 2-М'-диизопропилфосфиниламинопиридина равен
9,65 г (37,4%). Для выделения основного количества продук-
та упаривают бензольный фильтрат в вакууме водоструйного
насоса при 60—70 мм рт. ст. В остатке получают 2-диизопро-
пилфосфиниламинопиридин. Выход 15,45 г (60%).
Общий выход неочищенного продукта равен 25,1 г, что со-
ставляет 97,4% от теоретического; т. пл. 132—133,5°.
После кристаллизации из метанола получают 20 г (77,5%)
вещества с т. пл. 135—136°.
По этой методике могут быть получены:
2-М-Диэтилфосфиниламинопиридин с выходом 90%; т. пл.
86—87°.
2-М-Дипропилфосфиниламинопиридин с выходом 87%;
т. пл. 96—97°.
ЛИТЕРАТУРА
1. Б. П. Луговкии, Б. А. Арбузов. Ж. общ. химии, 22, 2041
(1952).>
2. Б. А. Арбузов, В. М. Зороастров а, М. П. Осипова. Изв.
АН СССР, ОХН, 12, 2163 (1961).
Поступила в декабре 1964 г.
Донецкий филиал ИРЕА
УДК 547.821.42.07
2-[|3-( М,1Ч-ДИМЕТИЛАМИНО)ЭТИЛ]ПИРИДИН
Н. Ф. КАЗАРИНОВА, Н. В. ДЖИГИРЕИ, Н. А. ШАВАЕВА
II I /СНЗ
1 J-ch2ch2-n<
W « \снз
c9hun2
М.в. 150,22
2-[|3-(М,Ы-Диметиламино)этил] пиридин представляет ин-
терес для синтеза биологически активных соединений.
По литературным данным, его получают конденсацией
2-винилпиридина с хлоргидратом диметиламина в воде [1].
При проверке этой методики оказалось, что выход продукта не
превышает 20%. С целью повышения выхода нами была из-
менена методика выделения вещества: вместо раствора едко-
го натра для нейтрализации кислой реакционной смесн был
использован поташ и изменено соотношение реагирующих
компонентов — диметиламин вводился в реакцию не в дву-
кратном, а в полуторакратном избытке.
СХЕМА СИНТЕЗА 2-[₽-(Ы,М-ДИМЕТИЛАМИНО)ЭТИЛ]ПИРИДИНА
/ Н2о
U-ch=ch, + NH>HC1
хСНа
I J-CH2CH2-Nx +НС1
N' хсн3
Характеристика основного сырья
2-Винилпиридин, ч., свежеперегнанный, т. кип. 60°/17 мм,
ВТУ TCP 2—60.
Диметиламин солянокислый, ч., ВТУ МХП 2885—51.
38
Условия получения
К 21 г (0,2 Af) 2-винилпиридина прибавляют 24,6 г
(0,3 А1) диметиламина солянокислого в 30 мл воды. Смесь
кипятят 5 часов, затем охлаждают, обрабатывают поташом
до образования кашицы и экстрагируют эфиром. Последний
отгоняют, а полученный продукт перегоняют под вакуумом.
Выход 2-[р-(М,М-диметиламино)этил]пиридина равен 15 г,
что составляет 50% от теоретического; т. кип. 102—
104715 мм- d420 — 0,9474;4°- 1,5040.
По литературным данным, т. кип. продукта 101 —
103717 мм [1].
ЛИТЕРАТУРА
1. F. F. Blicke, S. L. Hughes. J. Organ. Chew., 26,3257(1961).
Поступила в декабре 1964 г.
Донецкий филиал ИРЕА
УДК 547.784.1.07
ДИМЕТИЛОВЫЙ ЭФИР ИМИДАЗОЛ-4,5-ДИКАРБОНО-
ВОЙ кислоты
Н. Б. ВИНОГРАДОВА, Н. В. ХРОМОВ-БОРИСОВ
СН3ООС—С—N
II II
СН3ООС-С СН
NH
C,HSNA М.в. 184,15
Диметиловый эфир имидазол-4,5-дикарбоновой кислоты
используется для синтеза алкилированных амидов, являю-
щихся лекарственными препаратами [1, 2]. По литературным
данным, диметиловый эфир имидазол-4,5-дикарбоновой кис-
лоты можно получить или при пропускании хлористого водо-
рода через кипящую суспензию этой кислоты в метаноле, или
при ее этерификации метанолом в присутствии 100%-ной сер-
ной кислоты с последующей нейтрализацией минеральных
кислот карбонатами [3]. Проверка показала, что в обоих слу-
чаях выходы эфира непостоянны, могут сильно колебаться и
наряду с эфиром получаются соли карбметоксиимидазолмо-
нокарбоновой кислоты [1].
Однако более высокие выходы эфира достигаются по пер-
вому способу, поэтому данный метод был нами детально раз-
работан с внесением следующих изменений.
1) Для того, чтобы иметь возможность осуществить про-
цесс этерификации не только в лаборатории, но и в заводских
условиях, целесообразно проводить, предварительное насы-
щение метанола хлористым водородом на холоду до 35—40 %-
ного; содержания соляной кислоты и кипятить полученный
раствор с имидазол-4,5-дикарбоновой кислотой.
2) Нейтрализацию хлористого водорода содовым раство-
ром необходимо проводить при пониженной температуре (не
40
выше 15°), так как при более высоких температурах увеличи-
вается выход побочного продукта — натриевой соли 4-карб-
метоксиимидазол-5-карбоновой кислоты.
3) Перекристаллизацию полученного эфира следует осу-
ществлять не из воды, а из спиртов для отделения небольшой
примеси побочного продукта.
СХЕМА СИНТЕЗА ДИМЕТИЛОВОГО ЭФИРА ИМИДАЗОЛ-4,5-
ДИКАРБОНОВОЙ КИСЛОТЫ
HOOC-C-N II || +2СН3ОН — ноос-с сн СНзООС-С-N > II II +2Н2О СН3ООС-С сн
\н NH to
побочная реакция:
HOOC-C-N
II II +СН8ОН—>
ноос-с сн
NH
СН3ООС—С—N
II II
ноос-с сн
8 NH
сч
то
Z
СН3ООС-С-N
II II
NaOOC-C СН
\н
использование побочного продукта:
CH3OOC-C-N
II II +НС1
NaOOC-C СН
СН3ООС-С-N
* II II +NaCl
НООС-С СН
NH
CH3OOC-C-N
II || +СН3ОН
НООС-С сн
(НС1)
CH3OOC-C-N
II II +НаО
СНзООС-С сн
NH
41
Характеристика основного сырья
Имидазол-4,5-дикарбоновая кислота, техническая. Белый
порошок с желтоватым оттенком, т. пл. в интервале 274—
280° (см. примечание 1).
Метанол, ч., ГОСТ 6995—54.
Условия получения *
Предварительно производится насыщение 430 мл метано-
ла прн 0э сухим хлористым водородом, генерируемым из
900 мл концентрированной соляной кислоты при постепенном
прибавлении к ней по каплям 1 л концентрированной серной
кислоты (колбу с метанолом необходимо охлаждать льдом с
солью).
Хлористый водород сушится пропусканием через 2 склян-
ки с концентрированной серной кислотой, после которых ста-
вится пустая буферная склянка. В результате насыщения
получают метиловый спирт удельного веса 1,0 с концентра-
цией хлористого водорода 35—40%. Объем полученного рас-
твора 600 мл (см. примечание 3). В колбу для этерификации
загружают 40 г имидазол-4,5-дикарбоновой кислоты и 200 мл
метанола, насыщенного хлористым водородом (см. примеча-
ние 4). Нагревают 1 час при механическом перемешивании на
водяной бане с температурой 75—80°, затем прекращают ки-
пение, добавляют 200 мл насыщенного хлористым водородом
метанола и нагревают еще 1 час в тех же условиях (см. при-
мечание 5). Затем проводят дополнительную добавку 200 мл
насыщенного хлористым водородом метанола. Через 15—20
минут после его добавления осадок имидазол-4,5-дикарбоно-
вой кислоты полностью растворяется. Смесь снова кипятят
при перемешивании 30 минут и .охлаждают (см. примеча-
ние 5). Затем раствор переносят в двухлитровый стакан, ох-
лаждаемый льдом с солью до температуры 5° (см. примеча-
ние 6) и медленно, при работающей мешалке, вливают посте-
пенно холодный 12—14%-ный раствор углекислого натрия,
причем наблюдается сильное вспенивание. Расход безводной
соды на нейтрализацию составляет около 70—100 г. Нейтра-
лизацию раствором соды проводят до pH 4 по универсаль-
ной индикаторной бумажке при температуре не выше 15°. При
этом выпадает эфир имидазол-4,5-дикарбоновой кислоты в
виде густой массы и лишь иногда в виде мелкого осадка.
Продукт отфильтровывают и высушивают при температуре
80°. Вес сухого неочищенного эфира составляет 35 г или
74,5% от теоретического выхода, т. пл. 195—198° (см. приме-
чания 7 и 8). Фильтрат сохраняют. Эфир содержит около 1 —
1,5% побочного продукта — натриевой соли 4-карбметокси-
* См. примечание 2.
42
имидазол-5-карбоновой кислоты. Указанная примесь не вре-
дит синтезу амидов [2]. В случае необходимости проводят
очистку эфира путем перекристаллизации из метилового, эти-
лового или изопропилового спиртов (см. примечание 9). Пос-
ле одной перекристаллизации эфир получается в аналитиче-
ски чистом виде с т. пл. 201—203°, выход составляет 60% от
теоретического.
Найдено, %: N—15,6; 15,84.
C7H8N2O4. Вычислено, %: N—15,2.
Для получения дополнительного количества (10—15% от
теоретического) эфира используется следующая методика:
ьодно-метанольный фильтрат, оставшийся после нейтрализа-
ции, упаривают на 2/3 и охлаждают. Через 2 часа отфильтро-
вывают натриевую соль 4-карбметоксиимидазол-5-карбоно-
вой кислоты (см. примечание 10) и высушивают при 80—
100°. Вес продукта 10—36 г. Вещество содержит примесь не-
органических солей. Для перевода в 4-карбметоксиимидазол-
5-карбоновую кислоту 36 г вещества помещают в 400 мл
воды, смесь нагревают с мешалкой до кипения (~10 минут) и
подкисляют соляной кислотой по бумажке конго, затем ох-
лаждают, через 2 часа фильтруют и высушивают при 80—100°,
выход 9 г. Полученный таким образом монометиловый эфир
используют для синтеза диметилового эфира имидазол-4,5-
дикарбоновой кислоты, прибавляя его к этерифицируемой
кислоте или этерифицируя отдельно.
Выход эфира из эфирокислоты составляет 7,6 г с т. пл.
195—198°. Суммарный выход диметилового эфира имида-
зол-4,5-дикарбоновой кислоты с использованием побочного
продукта составляет 85% от теоретического.
Примечания:
1. Имидазол-4,5-дцкарбоновая кислота получается окислением бенз-
имидазола хромовым ангидридом (см. стр. 63 настоящего сборника),
содержит следы солей хрома, придающие кристаллам кислоты желтоватый
оттенок. Удалять эту примесь не следует, так как соли хрома являются
катализаторами процесса этерификации.
2. Необходимым условием успешного проведения процесса является
сухость и чистота всех составных частей прибора.
3. Раствор необходимо хранить закупоренным при температуре <5°.
4. Реакцию можно проводить в колбе с обратным холодильником без
мешалки, при периодическом взбалтывании от руки. В случае применения
механической мешалки необходимы затвор с вазелиновым маслом и вто-
рой холодильник, пропущенный через боковой тубус колбы, соединенный,
как и при ручном взбалтывании, со склянкой Тищенко, содержащей воду
для поглощения хлористого водорода.
5. Более длительное кипячение понижает выход эфира.
6. В условиях производства охлаждение осуществляют сильным то-
ком воды через рубашку куба, ио процесс при этом затягивается на 25—
35 часов.
7. Выход эфира на заводе «Фармакон» по этой методике составлял
“8% от теоретического, т. пл. 197—200°, и содержание побочного продук-
та равнялось 1%.
43
8. По литературным данным, неочищенный эфир плавится при 198—
200°, после перекристаллизации температура плавления 200—203°, 200—
202° [3].
9. Перекристаллизацию эфира производят с применением активирован-
ного угля из расчета: 1 г эфирр^ра 32 мл этилового или 20 мл изопро-
пилового спирта. Если требуется’выделить побочный продукт, содержа-
щийся в неочищенном эфире и нерастворимый в спирте, уголь применять
не следует.
10. Натриевая соль 4-карбметокси-имидазол-5-карбоновой кислоты
может быть выделена в аналитически чистом виде после кипячения с ме-
танолом н нескольких перекристаллизаций из воды с углем [1].
Вещество не плавится до 300°.
Найдено, %: N—11,68; 11,65.
C6H6N2O4Na. Вычислено, %: N—11,9.
ЛИТЕРАТУРА
1. Н. Б. Виноградова, Н. В. Хромов-Борисов. Ж. общ. хи-
мии, 31, 1466 ,(1961).
2. Н. В. X р о м о в-Б орисов, С. В. Аничков, Ю. С. Бородкин,
Н Б. Виноградова. Авт. свид. 160712; Бюлл. изобр., № 5 (1963).
3. R. А. В а х t е г, F. S. Spring. J. Chem. 5ос„ 232 (1945).
Поступила в декабре 1964 г.
ИЭМ АМН СССР
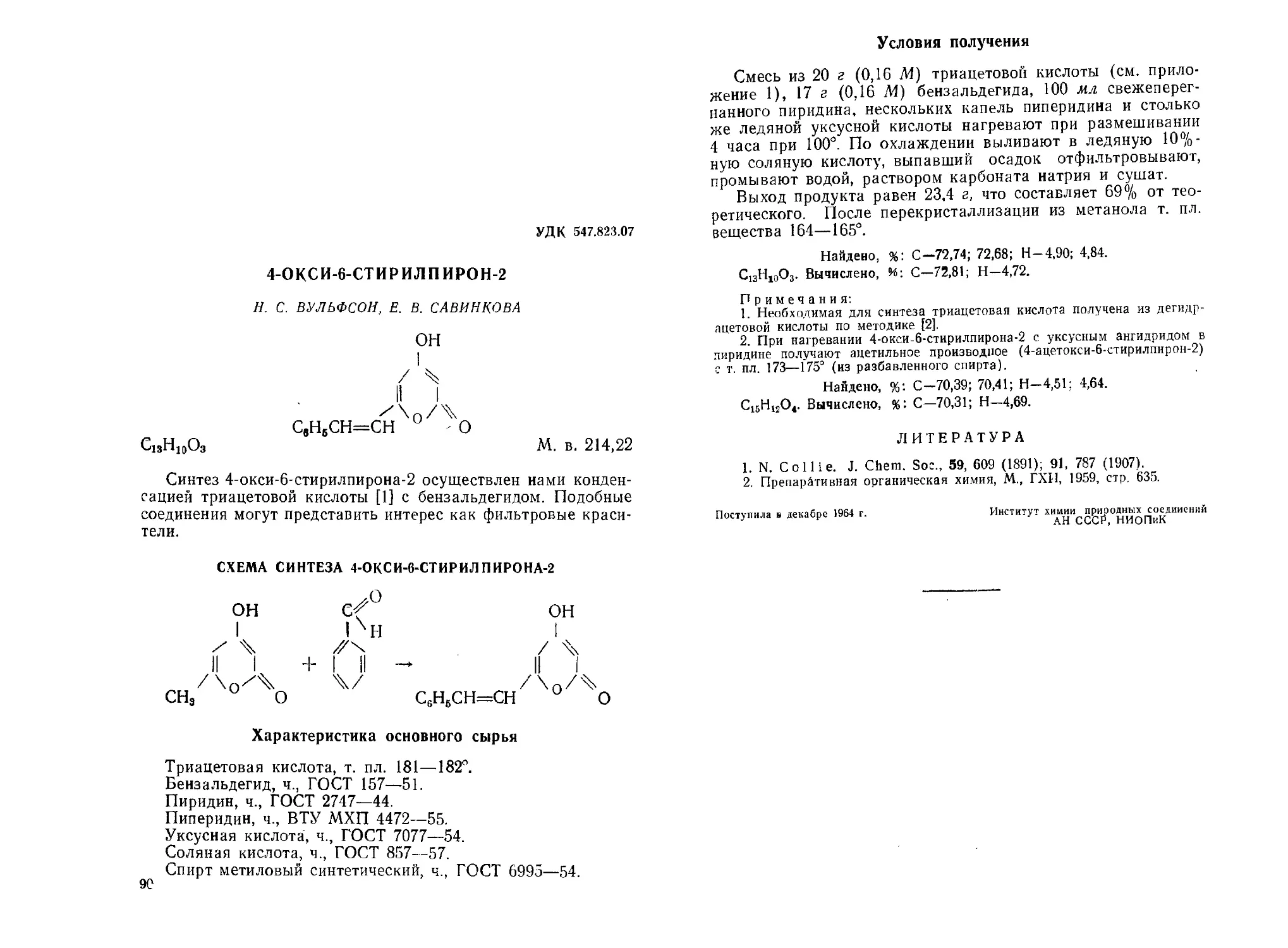
УДК 547.823.07
2.6-ДИСТИРИЛПИРОН-4
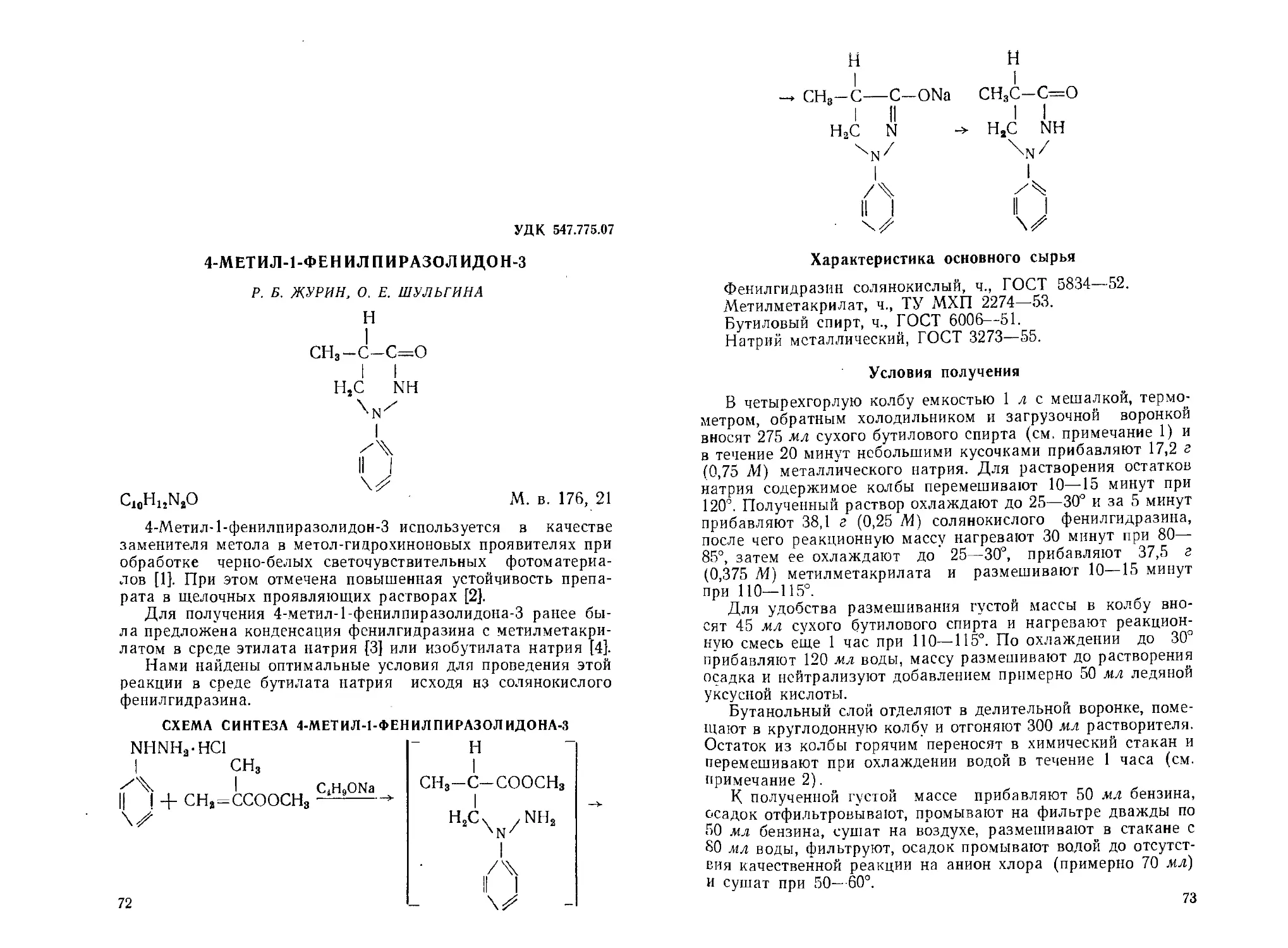
Н. С. ВУЛЬФСОН, Е. В. САВИНКОВА
О
||\
J 11
свн8сн=сн Ч°/Хчсн=снс6н5
С31Н16О3 М. в. 300,35
2,6-Дистирилпирон-4 был ранее получен конденсацией
2,6-диметилпирона-4 с бензальдегидом [1].
Нами синтез этого вещества осуществлен перегруппиров-
кой 4-окси-З- циннамоил-6-стирилпирона-2 в сернокислой сре-
де, последний мы получили С-ацилированием 4-окси-6-стирил-
пирона-2 с последующей конденсацией образовавшегося про-
дукта по Кляйзену-Шмидту с бензальдегидом.
Подобные соединения могут представить интерес как
фильтровые красители.
СХЕМА СИНТЕЗА 2,6-Д И СТИР ИЛ ПИРОНА-4
ОН
I
/ Ч сн.соон
II I------------*
едсн^сн7 ХО/1%
он
А >сосн3
/ V/ С6Н5СНО
II L c5h5n **
45
он
А ,СОСН=СНС6Н5
/ \/ 68 H2SO4
II I С3Н5ОН
CeHjCH^H^o/^O
о
Г 1
С6Н5СН = СН/Хо/ХСН=СНС6Н6
Характеристика основного сырья
Бензальдегид, ч., ГОСТ 157—51.
Пиридин, ч„ ГОСТ 2747—44.
Пиперидин, ч., ВТУ МХП 4472—55.
Серная кислота, ч., ГОСТ 2184—59.
Соляная кислота, ч., ГОСТ 857—57.
Уксусная кислота, ч., ГОСТ 7077—54.
Метиловый спирт, синтетический, ч., ГОСТ 2222—54.
Этиловый спирт, ГОСТ 5963—51.
Условия получения
Синтез 4-окси-3-ацетил-6-стирилпирони-2. Смесь из 13.г
(0,06 Л4) 4-окси-6-стирилпирона-2 (см. стр. 90), 50 мл ледя-
ной уксусной кислоты и 2 г полифосфорной кислоты (см. при-
мечание 1) нагревают на водяной бане 2 часа при темпера-
туре 85—90°. Затем реакционную массу выливают на лед, вы-
павший темный осадок отфильтровывают, промывают водой
и сушат.
Выход продукта равен 11,8 г, что составляет 76% от тео-
ретического. После перекристаллизации из разбавленного ме-
танола т. пл. вещества 159—160°.
Найдено, %: С-70,28; 70,22; Н -4,71; 4,74.
С„Н12О4. Вычислено, %: С—f 0,30; Н—4,60.
Синтез 4-окси-3-циннамоил-6-стирилпирони-2. Смесь из
5 г (0,019 44) 4-окси-3-ацетил-6-стирилпирона-2, 3 г (0,019 44)
бензальдегида, 50 мл свежеперегнанного пиридина, несколь-
ко капель пиперидина и столько же ледяной уксусной кисло-
ты нагревают при размешивании 5 часов при 90—100°. По
охлаждении реакционную массу выливают в ледяную 10%-
ную соляную кислоту, выпавший осадок отфильтровывают,
промывают водой, раствором карбоната натрия и сушат.
46
Выход продукта равен 4 г, что составляет 60% от теорети-
ческого; т. пл. 180—182° (из метанола).
Найдено, %: С-75,87; 75,91; Н-4,82; 4,77.
C22HJ6O4. Вычислено, %: С—76,00; Н—4,65.
Синтез 2,6-дистирилпирона-4. Смесь из 3 а (0,009 М) 4-
окси-3-циннамоил-6-стирилпирона-2 растворяют в 100 мл эти-
лового спирта, добавляют 200 мл 10%-ной серной кислоты и
еще 50 мл этилового спирта до полного растворения и кипя-
тят 50 часов. Затем спирт отгоняют, выпавший осадок от-
фильтровывают и перекристаллизовывают из этилового
спирта.
Выход продукта равен 1,9 г, что составляет 72% от теоре-
тического; т. пл. 167—168°. По литературным данным, т. пл.
вещества 168° [1] (см. примечание 2).
Примечания:
1. Полифосфорная кислота получена из ортофосфорной кислоты и
пятиокиси фосфора [2].
2. Для идентификации 2,6-дистирилпирона-4 он был получен нами
также из 2-стирил-6-метилпирона-4 путем конденсации его с бензальдеги-
дом в условиях реакции Кляйзена-Шмидта [3].
ЛИТЕРАТУРА
1. Bonn, Trotter, Me Kenzie. Proc. Chem. Soc., 30, 206 (1914).
2. H. С. Вульфсон, P. Б. Ж у p и и. Ж. общ. химии, 29, 3677 (1959).
3. Н. С. Вульфсон, Е. В. Савинкова. Ж. общ. химии, 34
(1964).
Поступила в декабре 19б4'Т. Институт химии природных соединений
АН СССР, НИОПиК
УДК 547.569.2.07
2,2'-ДИТИЕНИЛ СУЛЬФИД
Б, П. ФЕДОРОВ, Ф. М. СТОЯНОВИЧ
\ s / s /
C8HeS3 М. в. 198,32
2,2'-Дитиенилсульфид был впервые выделен Челлендже-
ром и Гаррисоном [1] при попытке получения 2-тиофен-
тиола. Незначительное количество дитиенилсульфида обра-
зуется при пиролизе 2,2'-дитиенилдисульфида [2].
Нами разработан препаративный метод синтеза 2,2'-ци-
тиенилсульфида из 2-гиениллития и 2,2'-дитиенилдисульфи-
да [3]. Этот метод использовался ранее для синтеза других
арил- и алкил-2-тиенилсульфидов [4—8]. В основу получения
необходимого промежуточного продукта 2,2'-дитиенилдисуль-
фида положен видоизмененный способ Гольдфарба с сотруд-
никами [9].
СХЕМА СИНТЕЗА 2,2'-ДИТИ ЕН ИЛ СУЛЬФИДА
H-C4H9Li . S , . K3Fe(CN)6
48
Характеристика основного сырья
Тиофен марки «чистейший».
Титрованный раствор н-бутиллития в эфире [10].
Сера черенковая, ч„ ТУ ОРУ 83—57.
Калий железистосинеродистый, х. ч., ГОСТ 4207—48.
Условия получения
Синтез 2,2'-дитиенилдисульфида. В четырехгорлую кругло-
донную колбу емкостью 2,5 л, снабженную двурогим фор-
штоссом, мешалкой, термометром, капельной воронкой, труб-
кой для пропускания инертного газа (см. примечание 1) и
холодильником, защищенным хлоркальциевой трубкой, поме-
щают 168 г (2 А1) тиофена и 200 мл абсолютного эфира (см.
примечание 2) и при комнатной температуре, охлаждая колбу
водой, прибавляют 1385 мл эфирного раствора н-бутиллития
(1,48 н.) в течение 40 минут. Полученную смесь выдерживают
при комнатной температуре полчаса, затем охлаждают до 0—•
5° и при этой температуре цорциями в течение 20—25 минут
прибавляют 64 г (2 М) серы (см. примечание 3). Реакцион-
ную массу выдерживают 30—40 минут при комнатной темпе-
ратуре до исчезновения серы и образования желтого раство-
ра, затем вновь охлаждают до минус 10° и осторожно пор-
циями при взбалтывании выливают в делительную воронку,
куда помещено 2 л охлажденного до 0° 2н. раствора едкого
натра. Слои разделяют, эфирный слой экстрагируют 1 л 4н.
раствора едкого натра. Щелочные растворы, промытые эфи-
ром, присоединяют к основному щелочному слою и весь ще-
лочной раствор вливают при размешивании в охлажденный
до 0° раствор 800 г железистосинеродистого калия в 4 л во-
ды (см. примечание 4). Желтый осадок дисульфида отфиль-
тровывают с отсасыванием, сырым растворяют в бензоле, бен-
зольный раствор отделяют от воды и нерастворившихся неор-
ганических веществ, бензол отгоняют и остаток перегоняют
под давлением не выше 0,1 мм.
Выход перегнанного дисульфида равен 150—160 г, что со-
ставляет65—70% от теоретического; т. кип. 112—115°/0,01 мм;
115—145°/0,02 мм (см. примечание 5); т. пл. 55—56°.
Синтез 2,2'-дитиенилсульфида. Раствор 2-тиениллития из
42 мл (0,53 М) тиофена и 350 мл (0,5 Л4) эфирного раствора
н-бутиллития готовят так, как описано выше при получении
дитиенилдисульфида. К полученному раствору приливают в
течение 10 минут раствор 115 г (0,5 М) дитиенилдисульфида в
250 мл эфира. Смесь нагревают при размешивании с обрат-
ным холодильником в течение 5 часов, после чего вылива-
ют в воду, продукт экстрагируют эфиром. Эфирный слой про-
мывают разбавленной щелочью, водой и сушат прокаленным
4 Зак. 1350 49
сернокислым магнием. Эфир удаляют и остаток перегоняют
под вакуумом. . .
Выход 2,2'-дитиенилсульфида равен 70—75 а, что состав-
ляет 71—76% от теоретического (см. примечание 6); т. кип.
129—130,572,2 мм; п^—1,6603, — 1,3161.
Примечания:
1. Все операции с бутиллитием проводят под током свободного от
влаги и кислорода инертного газа (азота или лучше аргона), который
предварительно пропускают для очистки через систему, включающую
щелочной раствор пирогаллола, концентрированную серную кислоту, хло-
ристый кальций, пятиокись фосфора и твердое едкое кали.
2. Абсолютирование эфира проводилось следующим образом: медицин-
ский эфир для наркоза выдерживают над твердым КОН в течение 1—2
дней, затем сливают и ставят над .металлическим натрием. После I —
2 дней стояния эфир используют без дополнительной очистки и перегонки.
3. Черенковую серу размалывают, просеивают через сито 14 меш и
удаляют остатки влаги отгонкой с бензолом на роторном испарителе.
4. Необходимо как можно быстрее проводить обработку реакционной
массы щелочью и не оставлять щелочной раствор стоять, а при окисле-
нии следить, чтобы окислитель был все время в избытке, в противном
случае дисульфид выпадает в виде масла, увлекая за собой неокислив-
шнйся тиофентиол, и выход значительно снизится.
5. Дисульфид легко перегревается при кипении, и пределы кипения
иногда довольно широки, но это не отражается на его температуре плав-
ления.
6. В одном из опытов был достигнут выход 90%.
ЛИТЕРАТУРА
1. F. Challenger, J. В. Harrison. J. Inst. Petrol Technolog., 21,
135 (1935).
2. E. Ko ft. Амер. пат. 2571370; С. А., 48, 2117 (1954).
3. Б. П. Федоров, Ф, М. С т о я н о в и ч. Ж. общ. химии, 33, 2251
(1963).
4. Т. S. М п г t h у, L. J. Ра nd у а, В. D. Til a k. J. Set. Ind. Res,
20В, 169 (1961),
о
5. S. G г о п о w i t z, Р. М о s е s, R. Н а к а п s s о п. Ark. Kemi, 16, 267
(1960).
6. V. V. О h a is a s, В. D. Т 11 а к. Proc. Indian Acad. Set., 39A, 14
(1954).
7. S. Gronowitz, P. Moses, А, В. H 6 r n f e 1 d t, R. Hakans-
s о n. Ark. Keml, 17, 165 (1960),
8. Б. П. Федоров, Ф. M. Сто янович, Ж. общ. химии, 31, 238
(1961).
9. Я. Л. Гольдфарб, М. А. Калик, М. Л. Кирмалова. ЖОХ,
32, 222 (1962).
10. Г. Гилман. Сб. «Органические реакции», вып. 8, М., ИЛ, 1956,
стр. 359.
Поступила в декабре 1964 г. ИОХ АН СССР
УДК 547.781.1.07
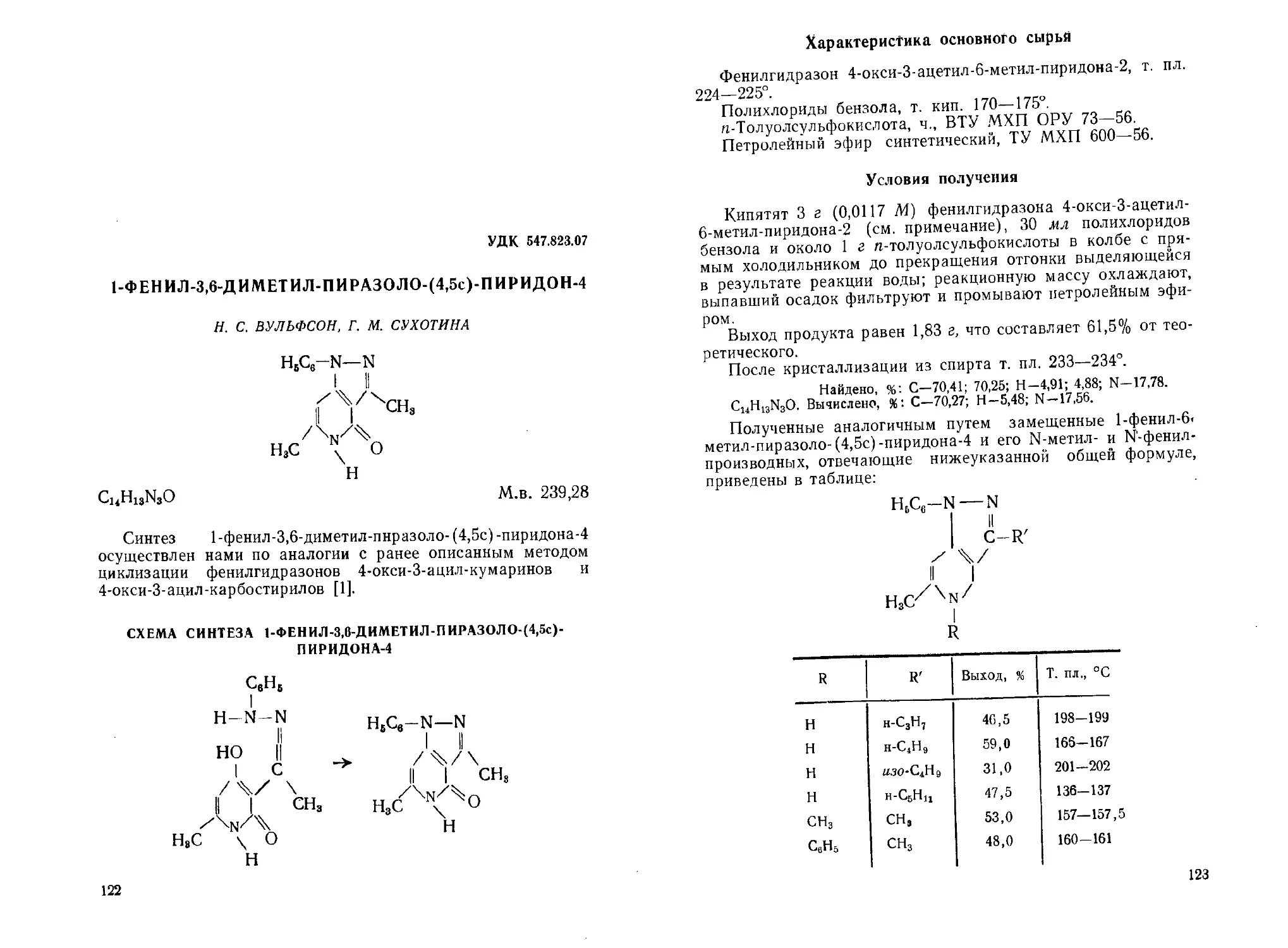
4,5-ДИФЕНИЛ ИМИДАЗОЛ
А. Д. Г АРНО ВС КИИ
ir ZCH
снА/
t-eH5 ]
H
cI6h12n2
M. B. 210,2
4,5-Дифенилимидазол может быть получен при конденса-
ции бензила, аммиака и формальдегида [1, 2]; при взаимодей-
ствии бензоина, аммиака и формальдегида в присутствии аце-
тата меди [3, 4]; из бензила, уротропина и уксуснокислого
аммония в уксуснокислой среде [5]; из бензоина, муравьиной
кислоты и карбоната аммония [6, 7]. Довольно широко приме-
няются также формамидные синтезы, разработанные в пос-
леднем десятилетии [8—11].
Среди указанных способов получения 4,5-дифенилимида-
зола наибольший интерес представляет синтез Радзишевско-
го [1, 2] по простоте выполнения операций и сравнительной
доступности исходных веществ. Предложенные ранее вариан-
ты этого метода многостадийны, приводят к образованию по-
бочных продуктов, требуют длительного времени ц дают низ-
кие выходы основного продукта.
Нами было найдено [12, 13], что при взаимодействии бен-
зила, аммиака и формалина в среде метанола при низкой
температуре (5—8°) 4,5-дифеиилимидазол получается с выхо-
дом 75—80%.
Преимуществом этого способа является одиостадийность
синтеза, короткий срок течения процесса (6—7 часов), полу-
чение лишь одного побочного продукта, легко отделяющегося
4* 51
от основного. Способ применим также для получения 2-ме-
тил-4,5-дифенилимида.зола (из уксусного альдегида, бензила
и аммиака). При введении в реакцию изомасляного, бензой-
ного, и-нитробензойного альдегидов соответствующие 4,5-ди-
фенилимидазольные производные получаются лишь с незна-
чительным выходом; при применении фурфурола, 5-галоид-
Фурфуролов, о-хинонов (нафтохинона-1,2; фснантренхинона-
9, 10 и других) происходит осмоление реакционной смесн.
Положительных результатов в последнем случае удается до-
биться при проведении реакции в уксуснокислой среде в при-
сутствии ацетата аммония [13, 14].
СХЕМА СИНТЕЗА 4,5-ДИФЕН ИЛ ИМИДАЗОЛА
СвН6
С-0 + 2NH3 + СН2О -
,С=О
с6н/
С6Н6Х ,N4
II сн + ЗН2О
/с\ /
С6н/ V
н
Характеристика основного сырья
Бензил (дибензоил), ч., ТУ МХП 2619—51, т. пл. 94°.
Формалин, 40%-ный, уд. в. 1,08.
Условия получения
В трехгорлой колбе, снабженной эффективной механичес-
кой мешалкой, термометром и трубками для ввода и вывода
аммиака, растворяют 21 г (0,1 М) бензила и 40 мл формали-
на в 500 мл метанола (см. примечание 1). Охладив реакцион-
ную смесь до 3—5°, пропускают в течение 2 часов сильный
ток аммиака (см. примечание 2). При этом раствор становит-
ся прозрачным, а затем начинается выделение белых кристал-
лов побочного продукта. Осадок отфильтровывают, подогре-
вают фильтрат (тяга!) до 40—50° и разбавляют 400 мл теп-
лой воды. Полученный раствор оставляют стоять на 4—5 ча-
сов, а затем отсасывают бесцветные (иногда с розоватым от-
тенком) кристаллы 4,5-дифенилимидазола, многократно про-
мывают их водой до исчезновения запаха аммиака и высуши-
вают в вакуум-эксикаторе над хлористым кальцием. Выход
52
Неочищенного 4,5-дифенилимидазола с т. пл. 224—227° равен
17,2 г, что составляет 82% от теоретического. При перекри-
сталлизации из смеси спирта и бензола или (лучше) неболь-
шого количества диоксана получаются бесцветные игольча-
тые кристаллы с т. пл. 231—232°, хорошо растворимые в спир-
те, дноксане, трудно в бензоле, эфире, не растворимые в воде.
Выход равен 15,7 г (~75%).
Изучены дипольные моменты [13, 15, 16], ИК-спектры [17]
и другие свойства 4,5-дифенилимидазола и его производных
[12, 15—18].
Примечания:
1. Не следует добиваться полного растворения бензила, так как он
растворяется полностью при пропускании аммиака через реакционную
смесь.
2. Работу необходимо вести в вытяжном шкафу.
ЛИТЕРАТУРА
1. А. Р a d z i s z e w s k i. Ber., 15, 1493, 2701 (1882).
2. A. Pinner. Ber., 38, 1537 (1905).
3. R. Weidenhagen, R. Herrmann, H. Wegner. Ber., 70,
570 (1937).
4. R. W e i d e n h a ge n, H. Wegner, Z. Wirtschaftsgruppe Zucke
rind, 87, Tech. Tl. 755 (1937); Chem. Abstrs, 32, 8416 (1938).
5. D. D a vl d s о n, M. Weiss, M. Jelling. J. Organ. Chem., 2,
319 (1937).
6. A. Novell!. Chem. Abstrs, 34, 1659 (1940).
7. Q. G. Novas, M. d e la M о r e n a C la vet, F. M. Archil-
1 а. РЖхим, 1957, реф. 15400.
8. H. В r e d e r e c k, G. T h e i 1 i g. Ber., 86, 88 (1953).
9. H. Bredereck, R. G о m p p e r. Ber., 87, 726 (1954).
10. H. В r e d e r e c k, R. G о m p p e r, D. Hayer. Ber., 92, 338 (1959).
11, H. Bredereck, F. Effenberger, F. Marguez, K. Ocke-
witz. Ber., 93, 2083 (1960).
12. A. M. Симонов, А. Д. Г a p н о в с к и й. Ж. общ. химии, 31, 114
(1961).
13. А. Д. Г а р и о в с к и й. Строение и реакционная способность систем,
содержащих имидазольное кольцо, Автореферат диссертации, РГУ, Ро-
стов-на-Доиу, 1961.
14. Е. S t е с k, A. Day. J. Amer. Chem. Soc., 65, 452, (1943).
15. О. А. Осипов, A. M. Симонов, В. И. Минкин, А. Д. Гра-
новский. Докл. АН СССР, 137, 1374 (1961).
16. В. И. Минкин, О. А. Осипов, А. Д. Гарновский, А. М.
Симонов. Ж. физ. химии, 36, 469 (1962).
17. А. М. Симонов, А. Д. Г а р н о в с к и й, Ю. Н. Ш е й н к е р, Б. И.
X р и с т и ч, С. С. Трофимова. Ж. общ. химии, 33, 571 (1963).
18. А. Д. Г а р н о в с к и й, А. М. Симонов, В. И. Минкин, В. Д.
Дионисьев. Ж. общ. химии, 34, 272 (1964).
Поступила в декабре 1964 г.
Ростовский-на-Дону университет
УДК 547.775.07
1,5-ДИФЕНИЛ-ПИРАЗОЛИДОН-З
Н. А. ЗАХАРОВА, Н. В. ХРОМОВ-БОРИСОВ. С. И. ГАФТ
сн —с=о
-CH NH
\N/
CI6H14N2O
М. в. 238,29
1,5-Дифенил-пиразолидон-З подобно 1-фенил-пиразоли-
доиу-3 (фенидону) может применяться в качестве добавки к
проявляющим растворам, содержащим гидрохинон, несколь-
ко увеличивая его активность как проявляющего вещества.
В литературе имеются указания на синтез 1,5-дифенил-пи-
разолидона-3 из этилового эфира коричной кислоты и фенил-
гидразина [1] и из амида коричной кислоты и фенилгидрази-
на [2].
В основу предлагаемого нами синтеза положен метод,
описанный в американском патенте [1]. Одиако в отличие от
описанной методики мы проводили синтез продукта в атмос-
фере азота, несколько уменьшили время реакции и изменили
последовательность прибавления реагентов, что позволило
увеличить выход 1,5-дифенил-пиразолидона-З до 40%.
СХЕМА СИНТЕЗА 1,5-ДИФЕНИЛ-ПИРАЗОЛИДОНА-З
СН—С—О
I II I.....
|-СН ! осан6
; HN-H
сна-с=о
^~СН NH
H-N
+ СаН6ОН
к/1
Характеристика основного сырья
Этиловый эфир коричной кислоты, ч., —1,5617, ВТУ
РУ 697 - 52.
Фенилгидразин основание, ч., ГОСТ 8750—58.
Натрий металлический, ч., ТУ МХП 1664—50.
Спирт этиловый сухой, ГОСТ 8314—57.
Условия получения
В трехгорлой литровой колбе, снабженной обратным хо-
лодильником, механической мешалкой, капельной воронкой и
трубкой для подвода азота, растворяют в 150 мл абсолютного
спирта 6,9 г (0,3 М) металлического натрия. После растворе-
ния натрия при перемешивании в один прием приливают
38,6 мл (40,5 г; 0,23 М) этилового эфира коричной кислоты
(см. примечание 1), при этом наблюдается выпадение белого
осадка, После этого под током азота в течение 20 минут при
непрерывном перемешивании по каплям прибавляют 19,7 мл
(21,6 г; 0,2 М) фенилгидразина основания. Реакционную
смесь при непрерывном перемешивании под током азота ки-
пятят 10,5 часов (см. примечание 2). По окончании нагрева
к реакционной массе прибавляют 60 мл воды, смесь переме-
шивают и приливают 80 мл 50 %-ной уксусной кислоты. Сра-
зу начинает выпадать осадок. Смесь оставляют стоять иа ночь
при температуре 3—7°. На другой день осадок 1,5-дифенил-
пиразолидона-З отфильтровывают, тщательно отжимают и на
фильтре промывают 300 мл воды и 100 мл эфира. После вы-
сушивания на воздухе вес продукта равен 27—33 г. Его очи-
щают перекристаллизацией из 100 мл смеси ацетона с бензо-
лом (1 :2).
Получают 22,15—22,77 г вещества с т. пл. 161 —163°, что
составляет 40,7— -41,4% от теоретического выхода. По литера-
турным данным, т. пл. 1,5-дифенил-ппразолидона-З 157—
159° [1, 2].
Найдено, %-. N—11,52; 11,52.
Ci6HuN2O. Вычислено, %: N—11,76 (см. примечание 3).
Примечания:
1. Этиловый эфир коричной кислоты получен при кипячении корич-
ной кислоты с абсолютным этиловым спиртом в присутствии небольшого
количества концентрированной серной кислоты [3].
2. При нагреве реакционной массы можно делать перерывы, оставляя
колбу с реакционной смесью под азотом.
3. Из маточника, после отделения основного осадка, при стоянии вы-
падает еще небольшой белый осадок. Это, вероятно, побочнообразующий-
ся 1,3-дифенил-пнразолидон-5 за счет обратного порядка присоединения
фенилгидразина к этиловому эфиру коричной кислоты. Осадок отфильтро-
вывают, промывают небольшим количеством эфира и сушат. Его вес 3 г;
1 пл. нечеткая — 166—179°. После двух перекристаллизаций из ацетона
(70 мл) и спирта (50 мл) получают 1 —1,3 г 1,3-дифенил-пиразолидона-5
с т. пл. 183—185° (см. примечание 4).
55
Найдено, %: N—11,63; 11,51.
CjbHhNjO. Вычислено, %: N—11,76.
4. 1,3-Дифенил-пиразолидон-5 в литературе не описан. Соединение
было получено также встречным синтезом — сплавлением коричной кисло-
ты с фенил!ндразином с выходом, не превышающим 17—19% от теорети-
ческого. После двух—трех перекристаллизаций 1,3-дифепил-пиразолидон-5
получен в аналитически чистом состоянии с т. пл. 184—186°. Депрессии с
веществом, выделенным в вышеописанном синтезе, не наблюдалось.
ЛИТЕРАТУРА
1. J. D. Kendall, G. F. Duffin, A. J. A xf о rd. Пат. США,
2688024; РЖхим., 1956, 23 543.
2. J. D. К е П d а 11, G. F. D u f f 1 п. Пат. США, 2704762; С. А., 50,2680
(1956).
3. Е. Fischer, A. Speler. Вег., 28, 3254 (1895).
Поступила и декабре 1964 г.
ПЭМ АМН СССР
УДК 547.841.07
C^HgOjCIs
2,3-ДИХЛОР-1,4-ДИОКСАН
М. Г. ВОРОНКОВ. Э. П. ПОПОВА
НгС/°\СНС1
н.с' Jchci
\0/
М, в. 156,99
Распространенный способ получения 2,3-дихлор-1,4-ди-
оксана основан на хлорировании 1,4-диоксана газообразным
хлором в жидкой или паровой фазе, а также в растворе. При
хлорировании диоксана в среде четыреххлористого углерода
пои 90° выход 2,3-дихлор-1,4-диоксана составляет всего 25%
[Ц.
Более высокий выход (69%) 2,3-дихлор-1,4-диоксана по-
лучается при хлорировании диоксана в отсутствие раствори-
теля при 90° [2, 3]. Применение хлористого йода или хлори-
стого олова в качестве катализатора повышает выход продук-
та еще больше [4].
Нами разработан удобный метод синтеза 2,3-дихлор-1,4-
диоксана путем хлорирования диоксана хлористым сульфу-
рилом.
СХЕМА СИНТЕЗА 2,3-ДИХЛОР-1,4-ДИОКСАНА
н2с/°\сн2
Н2С* JcHa + 2S°2C12
НаС/°\СНС1
H2cl Jchci + 2НС1 + 2S°2
57
Характеристика исходного сырья
1,4- Диоксан, ч„ ТУ ГКХ 1558—61.
Хлористый сульфурил, ч., ВТУ МХП 3591—52.
Условия получения
Реакцию проводят в трехгорлой колбе емкостью 1 л, снаб-
женной мешалкой с герметизирующим затвором, обратным
холодильником и капельной воронкой. В колбу помещают
88,1 г (1 М) 1,4-Диоксана (см. примечание 1) и из капельной
воронки медленно прибавляют 270 г (2 Л1) хлористого суль-
фурила (см примечание 2). Реакция начинается самопроиз-
вольно и протекает с разогреванием, сопровождаясь выделе-
нием газов (SO2 и НС1) (см. примечание 3). После того как
весь хлористый сульфурил введен в реакцию, и выделение га-
зов почти прекратилось, колбу подогревают на водяной бане
до полного прекращения выделения НС1 и SO2. Избыток не-
прореагировавшего диоксана отгоняют, а остаток перегоняют
в вакууме.
Выход 2,3-дихлор-1,4-диоксана равен 107,5 г, что состав-
ляет 79,3% от теоретического, считая на прореагировавший
диоксан (68,4% на исходный); т. кип. продукта 82—83°/14 мм;
По —1,4970; №—1,4498, По литературным данным, т. кип.
82,4714 мм; т. пл. 30°.
Примечания:
1. Диоксан предварительно кипятят с обратным холодильником над
твердым едким кали в течение 8 часов и затем перегоняют над металли-
ческим натрием (проверка на присутствие перекисей).
2. Если в диоксане содержатся следы перекисей, реакция начинается
сразу. В противном случае взаимодействие реагентов происходит после
некоторого индукционного периода (5—10 минут).
3. В реакцию вводится предварительно перегнанный хлористый суль-
фурил.
ЛИТЕРАТУРА
1. R. К. Summer bell, Н. Е. Lun k. J. Amer. Chem. Soc., 79,
4802 (1957).
2. J. Boeseken. F. Tellegen, P. C. Henriques. Proc. Roy. Soc.
Amsterdam, 34, 631 (1931).
3.1. Boeseken, F. Tellegen, P. C. Henriques. Rec. trav.
chim., 50, 90'J (1931).
4. J. J. К u с e r a, P. C. Carpenter. J. Amer. Chem. Soc., 57, 2346
(1935).
Поступила в декабре 1964 г. ' Институт органического синтеза
АН Латв. ССР
УДК 547.821.42.07
2-[3-(К,КДИЭТИЛАМИ НО)ЭТИЛ)П ИРИДИИ
Н. Ф. КАЗАРИНОВА, Н. В. ДЖИГИ РЕИ. И. А. ШАБАЕВА
CjiHjgNj
С2Н6
ch3ch2n^
чс2нб
М. в. 178, 27
2-[|3-(М-Диэтиламино)этил1пиридин представляет интерес
для синтеза биологически активных соединений.
По литературным данным, его получают с выходом 12%
при взаимодействии 2-винилпиридина с диэтиламином без ка-
тализатора [1]. С целью повышения выходов конечного про-
дукта конденсация 2-винилпиридина с диэтиламином нами
проведена в присутствии уксусной кислоты.
СХЕМА СИНТЕЗА 2-[₽-(И,М-ДИЭТИЛАМИНО)ЭТИЛ|ПИРИДИНА
/
\n^xCH=CH2 + (C2H6)2NH
/ X-
" \ А /СгН5
XN^xCH2CH2nC
U
Характеристика основного сырья
2-Винилпиридин, свежеперегнанный, т. кип. 60°/17 мм.
Диэтиламин, ч., СТУ 12-10-105—61.
Уксусная кислота, х. ч., 98%-ная, ГОСТ 65—51.
59
Условия получения
Смешивают 157,5 г (1,5 М) 2-винилпиридина с 146 г (2 М)
диэтиламина и добавляют 50 мл уксусной кислоты. Смесь
кипятят в течение 16 часов. Охлаждают, подщелачивают 10—
15%-ным раствором едкого натра до pH 8—9. Органический
слой отделяют и перегоняют под вакуумом.
Выход 2-[р-(М,М-диэтиламино)этил]пиридина в виде бес-
цветного масла равен 198 г, что составляет70% оттеоретичес-
кого; т. кип. 115—120°/8 мм; d420 — 0,9345; Пр—1,4990.
По литературным данным, т. кип. продукта 82—83°/2 мм
[11-
ЛИТЕРАТУРА
1. W. Doering, R. Weil. J. Amer. Chem. Soc., 69, 24G1 (1947).
Поступила в декабре 1964 г. Донецкий филиал ИРЕА
УДК 547.73.07
ДИЭТИЛАЦЕТАЛЬ 2-ЭТИЛМЕРКАПТО-5-ЭТИЛ-3-
ТИОФЕНАЛЬДЕГИДА
Я. Л. ГОЛЬДФАРБ, М. А. КАЛИК. М. Л. КИРМАЛОВА
СН(ОС2Н{)2
CjsHjjOaSa
М. в. 274, 44
Диэтил ацеталь 2-этилмеркапто-5-этил-3-тиофенальдегида
получен по методу, описанному для диэтилацеталя 2-тио-
фенальдегида [1].
СХЕМА СИНТЕЗА ДИЭТИЛАЦЕТАЛЯ 2-ЭТИЛМЕРКАПТО-5-ЭТИЛ-
3-ТИОФЕНАЛЬДЕГИДА
сно
+ СН(ОС2Н6)з
SC2Hs
СН(ОС2Н6)2
' \ с / х
н5с2 ь SC2H6
Характеристика основного сырья
2-Этилмеркапто-5-этил-3-тиофенальдегид [2], см. стр. 153.
о-Муравьиный эфир, ч., СТУ 79-570-Х—60.
Спирт этиловый, абсолютный, ГОСТ 5962—51.
61
Условия получения
Смесь 100,2 г (0,5 М) 2-этилмеркапто-5-этил-3-тиофеналь-
дегида, 81,5 г (0,55 А4) о-муравьиного эфира, 300 мл абсо-
лютного этилового спирта и трех капель концентрированной
соляной кислоты кипятят в литровой круглодонной колбе с
обратным холодильником, снабженным трубкой, заполненной
сульфатом магния, в течение 4 часов. Охлажденный раствор
нейтрализуют 0,5 н. спиртовым раствором едкого кали. Раст-
воритель отгоняют, остаток разбавляют равным объемом
воды.
Органический слой отделяют, водный экстрагируют эфи-
ром и высушивают объединенный эфирный раствор сульфа-
том магния. После отгонки эфира ацеталь перегоняют в ва-
кууме с елочным дефлегматором, отбирая фракцию с т. кип.
134—138° при 2 мм.
Получают 123 г ацеталя, что составляет 90% от теорети-
ческого выхода. После повторной перегонки т. кип. продукта
137—138°/2 мм; п™ —1,5193; d420 — ! ,0537.
ЛИТЕРАТУРА
1. Я. Л. Гольдфарб, П. А. Константинов. Изв. АН СССР,
ОХН, 1957, 217.
2. Я. Л. Гольдфарб, М. А. Калик. М. Л. Кирмалова. Изв.
АН СССР, ОХН, 1962, 701.
Поступила в декабре К64 г.
ИОХ АН СССР
УДК 547.784.1.07
ИМИДАЗОЛ-4,5-ДИКАРБОНОВАЯ КИСЛОТА
Н. Б. ВИНОГРАДОВА. И. В. ХРОМОВ-БОРИСОВ
НООС-С— N
II II
НООС-С сн
NH
C5H4N2O4 М.в. 156,09
Имидазол-4,5-дикарбоновая кислота и ее гомологи явля-
ются узловыми соединениями при синтезе целого ряда лекар-
ственных препаратов: антифеинов [1], флагила [2], а также
некоторых красителей (3} и имидазола [4], употребляемого в
органическом синтезе. Основным из описанных в литературе
методов получения этой кислоты является синтез с промежу-
точным выделением динитрата винной кислоты, легко разла-
гающегося на воздухе [4]. Однако применение этого метода
нежелательно из-за взрывоопасности динитрата винной кис-
лоты.
Нами предлагается метод, представляющий собой вари-
ант и более подробную детализацию ранее описанного в ли-
тературе метода Эфроса и Хромова-Борисова, заключающе-
гося в окислении бензимидазола хромовой смесью в кислой
среде [5J. Этот одностадийный синтез практически более удо-
бен, чем метод с применением динитрата винной кислоты. В
ходе изучения процесса окисления нами были внесены неко-
торые технологические изменения, позволившие повысить вы-
ход и уточнить оптимальные условия синтеза имидазол-4,5-
Дикарбоновой кислоты этим методом.
СХЕМА СИНТЕЗА ИМИДАЗОЛ-4,5-ДИКАРБОНОВОЙ КИСЛОТЫ
___Ы HOOC-C-N
| || f +9О—>2СО2+ || || +НаО
НООС-С СН
NH \/
NH
63
Характеристика основного сырья
Бензимидазол, технический. Температура плавления в пре-
делах 167—170°. Допускается темная окраска кристаллов.
Бензимидазол получается из технического ортофенилендиами-
на по известному методу [6].
Серная кислота, ч., ГОСТ 4204—48, концентрированная.
Двухромовокислый калий, мелкорастертый, технический,
ГОСТ 4220—48.
Условия получения
Помещают 40 г (0,34 Л1) сухого бензимидазола в двух-
литровый фарфоровый стакан или двухлитровую колбу с
широким боковым тубусом, где находится 650 мл заранее
приготовленного раствора 80%-ной серной кислоты уд. в. 1,727
при 20° (готовится постепенным прибавлением 435 мл кон-
центрированной серной кислоты к 160 мл воды). Реакцион-
ную смесь нагревают при перемешивании до температуры 85°,
при этом весь бензимидазол растворяется (см. примечание 1).
Затем прекращают нагрев, пускают в ход энергично дейст-
вующую мешалку и начинают периодически добавлять к ре-
акционной смеси 225 а (2,25 М) кристаллического хромового
ангидрида порциями по 8—10 г. В результате окисления
происходит некоторое разогревание реакционной смеси и ее
вспенивание. Введение СгО3 проводят при температуре реак-
ционной смеси 79°, не давая ей остывать или подниматься вы-
ше 90° (см. примечание 2). По окончании прибавления би-
хромата калия смесь выдерживают при перемешивании для
естественного охлаждения до 35—40°, затем постепенно
при перемешивании приливают 1500 мл холодной воды и
оставляют для кристаллизации при охлаждении водой на 12—
15 часов (см. примечание 3). Продукт отфильтровывают, про-
мывают 3 раза водой по 200 мл, отжимают на фильтре и су-
шат при 80—100°. Его темпера’тура плавления в интервале
274—280° в пределах двух градусов (см. примечание 4). При
плавлении кислота разлагается со вспениванием [4, 5], по ли-
тературным данным, она плавится при 280° [4].
Выход кислоты равен 37—38,7 г, что составляет 70—73%
от теоретического. Имидазол-4,5-дикарбоновая кислота пред-
ставляет собой белый порошок с желтоватым или фиолето-
вым (после переосаждения) оттенком (см. примечание 5). В
случае необходимости производят очистку кислоты путем пе-
реосаждения из щелочного раствора (см. примечание 6). Пос-
ле проведения двух повторных переосаждений получается
кислота в виде белого порошка, в аналитически чистом виде
64
с температурой плавления в интервале 274—280° в пределах
двух градусов.
Найдено, %: N—17,95; 18,20.
C5H4N3O4. Вычислено, %: N—17,90.
Примечания:
1. Практически удобнее растворить бензимидазол в отдельном сосуде
и затем вылить раствор в приготовленный стакан или колбу с мешалкой
и термометром, которые должны быть погружены в глубокую пустую во-
дяную баню для того, чтобы иметь возможность при сильном росте тем-
пературы смеси (92—95°) периодически понемногу подливать туда холод-
ную воду.
2. Допускать остывания реакционной смеси ниже 79° равно как и
искусственный подогрев во время окисления не следует, потому что в та-
ких условиях снижается выход кислоты.
3. Уменьшение времени отстаивания до 2—3 часов снижает выход
кислоты на 5—6%. Уменьшение количества воды с 1500 до 600 мл мало
снижает выход, но сильно затрудняет фильтрацию (разрывы фильтров).
4. Величина температуры плавления имидазол-4,5-дикарбоновой кисло-
ты очень сильно зависит от характера нагревания капилляра.
5. Окраска объясняется присутствием следов солей хрома. Продукт
вполне пригоден для дальнейших синтезов без очистки.
6. Переосаждение кислоты осуществляется следующим образом. Рас-
творяют 15,2 г сухого едкого натра в 320 мл воды, добавляют 20 г сухой
нмидазол-4,5-дикарбоиовой кислоты и кипятят с обратным холодильни-
ком 10—15 минут до полного растворения кислоты. Затем прекращают
кипячение, добавляют 1 г активированного угля, кипятят еще 15 минут и
фильтруют. Фильтрат подкисляют соляной кислотой до кислой реакции
(по бумажке конго), через 3 часа отфильтровывают выпавшую кислоту,
промывают ее тремя порциями воды по 20 мл и сушат при 70—100°. Вы-
ход очищенной кислоты около 16 г. Температура плавления в интервале
274—280° в пределах двух градусов [4].
ЛИТЕРАТУРА
1. Н. Б. Виноградова, Н. В. Хромов-Борисов. Ж. общ. хи-
мии, 31, 1466 (1961).
2. М. Negwer. Organisch—chetnische Arznelmittel, 44 (1961).
3. Jam una Datt Tewari. Quarterlu Journ. Indian Chem. Soc., 4,
201, Allahabad.
4. Сб. «Синтезы органических препаратов», вып. 3, 1952, стр. 254, z
5. Л. С. Эфрос, Н. В. Хромов-Борисов и др. Ж. общ. химии,
26, 455 (1956).
6. Сб. «Синтезы органических препаратов», вып. 2, 1949, стр. 85.
Поступила в декабре 1964 г. ИЭМ АМН СССР
5 Зак. 1350
УДК 547.415.3.07
2-МЕРКАПТО-5-ЭТИЛ-3-ТЕНИЛ ИДЕНИМИН
Я. Л. ГОЛЬДФАРБ, М. А. КАЛИК, М. Л. КИРМАЛОВА
XCH=NH
или
h6c^4s /Xs/
/CH=NH8
H6C2 s s-
c7h9ns2
M. в. 171,28
2-Мсркапто-5-этил-3-тенилиденимин является первым
представителем соединений ароматического типа, несущих од-
новременно альдиминную и меркаптогруппы у соседних ато-
мов углерода. Будучи комплексообразующим агентом, он вме-
сте с тем служит исходным соединением для получения ряда
других комплексообразователей. Так, при взаимодействии с
первичными аминами и диаминами образуются основания
Шиффа, содержащие меркаптогруппы [1].
СХЕМА СИНТЕЗА 2-МЕРКАПТО-5-ЭТИЛ-3-ТЕНИЛ ИДЕНИМИНА
ZCH(OC2H6)2
+ 2Na
H5Ca/XS /xsc2h6
CH=NH
66
Характеристика основного сырья
Диэтилацеталь 2-этилмеркапто-5-этил-3-тиофенальдегида
[1], см. стр. 61.
Аммиак газообразный.
Натрий металлический, ч., ГОСТ 3273—55.
Условия получения
В чстырехгорлую колбу емкостью 750 мл, снабженную
мешалкой, термометром, трубкой для ввода аргона и трубкой,
заполненной едким кали, конденсируют 400 мл аммиака (см.
примечание 1) и добавляют раствор 40 г (0,146 Л1) диэтил-
ап.еталя 2-этилмеркапто-5-этил-3-тиофенальдегида в 40 мл су-
хого эфира. Затем при минус 70° и перемешивании в токе ар-
гона вводят небольшими порциями 6,8 г (0,296 Л4) металли-
ческого натрия. Синяя окраска раствора вначале быстро исче-
зает, но сохраняется после добавления последней порции нат-
рия. Через 15—20 минут избыток натрия разлагают неболь-
шим количеством сухого хлористого аммония, при этом синяя
окраска исчезает. Аммиак испаряют в токе аргона и к остат-
ку добавляют 100 мл бензола, 50 мл метанола и 150 мл воды.
Водный слой отделяют, экстрагируют бензолом, приливают
10 мл 26%-ного водного аммиака, охлаждают, добавляют по
каплям разбавленную соляную кислоту (1:1) до pH 8 и
оставляют на ночь в холодильном шкафу.
Выпавший желтый кристаллический осадок отфильтровы-
вают, промывают водой и высушивают. Получают 22,4 г 2-
меркапто-5-этил-З-тенилиденимина с т. пл. 127° с разложени-
ем (см. примечание 2), что соответствует 88,5% от теоретиче-
ского выхода. После перекристаллизации из бензола и хло-
роформа т. пл. продукта 135° с разложением (см. примеча-
ние 3).
2-Меркапто-5-этил-3-тенилиденимин труднорастворим в
спирте, эфире, бензоле, гептане, хлороформе, хорошо раство-
рим в этилацетате, тетрагидрофуране, диоксане.
Примечания:
1. Газообразный аммиак из баллона для высушивания пропускают че-
рез 3 колонки с твердым едким кали.
2. Добавляя к фильтрату еще немного соляной кислоты (1:1) до
pH 7—8, получают дополнительное количество 2-меркапто-5-этнл-3-тенили-
денимина.
3. Аналогичным образом получают 2-меркапто-5-метил-3-тенилиден-
iiMHH, т. пл. 155—156,5° с разложением [2].
ЛИТЕРАТУРА
1. Я. Л. Гольдфарб, М. А. Калик, М. Л. Кирмалова. Изв.
АН СССР, ОХН, 1962, 701.
2. Я. Л. Гольдфарб, М. А. Калик, М. Л. Кирмалова. Изв.
АН СССР, сер. хим., 1966, 897.
Поступила в декабре 1964 г. ИОХ АН СССР
5* 67
УДК 547.841.07
2-МЕТИЛ-5-ИЗОПРОП ИЛ-5-(а-МЕТОКСИ)ЭТИЛ-1,3-
ДИОКСАН
А. В. БОГАТСКИИ, Я. Л. ГАРКОВИК
г-С3Н7х ,СН2-0 ч /СН3
/СС /С\
CHg-CtT CH2-OZ
осн3
СПН22О3 М. в. 202,29
Способы получения алкоксиалкилзамещенных 1,3-диокса-
нов в литературе не описаны. Нами в 1963—1964 гг. получен
ряд алкоксиалкилзамещенных 1,3-диоксанов [1], оказавшихся
интересными с теоретической точки зрения ввиду их стерео-
химических особенностей и физиологической активности [2].
СХЕМА СИНТЕЗА 2-МЕТИЛ-5-ИЗОПРОПИЛ-5-( а-МЕТОКСИ)ЭТИЛ-
1,3-ДИОКСАНА
г-С3Н, СН2ОН
n-CH3C6H4SOsH
с +О=с-СН3 ------------------------>-
/ । ° абс. бензол
сн3-сн СН2ОН н
I
OCHS
z-CjHj СН2-0 сн3
С С +НаО
СН3-СН "сНа-с/ \
ОСНз
Характеристика основного сырья
2-Изопроиил-2-а-метоксиэтилпропандиол-1,3, получаемый
по методике [3].
Уксусный альдегид, ч., ТУ МХП 2633—51.
Ионит КУ-1 в Н-форме или я-толуолсульфокислота, х. ч.,
ТУ ОРУ 73—56.
Бензол, перегнанный, т. кип. 80,5—81°, ГОСТ 5955—51.
68
Условия получения
Вариант 1. В колбу емкостью 100 мл, снабженную обрат-
ным холодильником с насадкой Дина-Старка, загружают
50 мл абсолютного бензола, 10 г (0,056 Л!) 2-изопропил-2-а-
метоксиэтилпропандиола-1,3 (см. примечание), 5 г (0,04 М)
паральдегида и каталитическое количество (0,05 е) п-толуол-
сульфокислоты. Смесь нагревают до 80—90° в течение двух
часов до выделения теоретического количества воды в насад-
ке Дина-Старка. Реакционную массу после охлаждения про-
мывают 5%-ным раствором соды и водой до нейтральной
реакции среды, затем сушат над безводным порошкообраз-
ным поташом. После отгонки бензола продукт реакции очи-
щают перегонкой в вакууме.
Выход 2-метил-5-изопропил-5- (а-метокси) этил-1,3-диокса-
на равен 9,5 г, что составляет 85% от теоретического; т. кип.
83°/5 мм; «д—1,4478; d^' — 0,9850.
Найдено, %: С—65,19; Н—11,19.
СПНИО3. Вычислено, %: С—65,30; Н —10,96.
Нами предлагается также и другой способ получения
алкоксиалкилзамещенных 1,3-диоксанов с применением ка-
тионита (вариант 2). Этот способ имеет преимущество перед
вышеприведенным, поскольку в нем облегчается обработка
реакционной массы после синтеза и выход продукта увеличи-
вается.
Вариант 2. В колбу емкостью 100 мл, снабженную обрат-
ным холодильником с насадкой Дина-Старка, загружают
50 мл абсолютного бензола, 10 г (0,056 М) 2-изопропил-2-(а-
метокси)этилпропандиола-1,3, 5 г (0,04 М) паральдегида и
0,8 г смолы КУ-1 (вН+-форме). Смесь нагревают до 80—90°
в течение двух часов до выделения теоретического количест-
ва воды в насадке Дина-Старка. После отгонки бензола про-
дукт перегоняют в вакууме, причем выход продукта увеличи-
вается до 89% от теоретического.
Примечание.
2-Изопропил-2-(а-метокси)-этилпропандиол-1,3 получен но методике
[3] с т. кип. 12475 мм; nW — 1,4650; — 1,0228.
ЛИТЕРАТУРА
1. А. В. Богатский, Н. Л. Г аркови к. Ж. общ. химии, 34
5 (1964).
2. А. В. Богатский, И. Л. Г аркови к. Тезисы конференции ге-
тероциклов в органической химии, Киев, Об-во «Знание», 1964.
3. Г. В. Пьянкова, А. В. Богатский, А. А. Колесник. Ж.
общ. химии, 34, 1199 (1964).
Поступила в декабре 1964 г.
Одесский государственный
университет
УДК 547.565.07
3-МЕТИЛ-5-ПРОПИЛПИРРОЛ-(2-АЗО-2')-ФЕНОЛ
В. М. ДЗИОМКО, к. А. ДУНАЕВСКАЯ
НС—С—СН3
II И II I
СН3СН2СН2-С C-N^N-Л^1
ОН
cuh17n80
М.в. 243,30
Это соединение получено нами впервые путем диазотиро-
вания 2-аминофенола и сочетания полученного диазосоедине-
ния с З-метил-5-пропилпирролом [1].
Реагент нашел применение в аналитических работах при
определении кобальта.
СХЕМА СИНТЕЗА 3-МЕТИЛ-5-ПРОПИЛ ПИРРОЛ-(2-АЗО-2')-ФЕНОЛА
С—СН
I II
ОН
I
о-
II II
НС С-СН2СНгСН3
\н
сочетание "*"
СН3
С-СН
I II м м II II
\>-N=N-c С-СН2СН2СН3
ОН
Характеристика основного сырья
З-Метил-5-пропилпиррол, т. кип. 77—78°/7 мм [1}.
2-Аминофенол, ч., ТУ МХП 1979—49.
Нитрит натрия, ч., ГОСТ 4197—48.
Условия получения
Растворяют в разбавленной соляной кислоте (8 мл воды
и 4 мл концентрированной соляной кислоты) 1,75 г (0,016 М)
о-аминофенола, раствор охлаждают до 3—5° и добавляют к
нему при перемешивании раствор нитрита натрия 1,2 г
(0,017 Л!) в 8 мл воды. Смесь выдерживают 30—40 минут,
после чего нейтрализуют раствором ацетата натрия (30 г аце-
тата натрия безводного в 100 мл воды) до pH 4—5.
Полученный диазораствор при перемешивании прибавля-
ют к раствору 2 г (0,016 М) З-метил-5-пропилпиррола в 35 мл
этилового спирта. Далее в раствор приливают 45—50 мл
диметилформамида до pH 6 и выдерживают 12—16 часов.
На следующий день выделившийся краситель отфильтровы-
вают и дважды перекристаллизовывают из разбавленного
водой (1:3) этилового спирта (на 0,7 г красителя берут
50 мл разбавленного спирта).
Выход продукта равен 0,2 г, что составляет 5,05%; т. пл.
82—82,5°. По внешнему виду это мелкокристаллическое ве-
щество оранжевого цвета, хорошо растворяется в ацетоне,
спирте, диметилформамиде.
Найдено, %: С-69,56; 68,82; Н- 7,26; 7,17; N—16,68; 16,65.
Ci4HleNiO. Вычислено, %: С—69,42; Н—6,6; N—17,27.
ЛИТЕРАТУРА
1. И. Ф. Бельский, Н. И. Шуйкин, Г. Е. Скобцова, Изв. АН
СССР, Сер. хим., 1964, (6), 1118.
Поступила в июне 1965 г.
ИРЕА
УДК 547.775.07
4-МЕТИЛ-1-ФЕН ИЛ ПИРАЗОЛ ИДОН-3
Р. Б. ЖУРИН, О. Е. ШУЛЬГИНА
н
СНз-С-С=О
I I
Н,С NH
Ч7
с10ним,о
М. в. 176, 21
4-Метил-1-фенилпиразолидон-3 используется в качестве
заменителя метола в метол-гидрохиноновых проявителях при
обработке черно-белых светочувствительных фотоматериа-
лов [1]. При этом отмечена повышенная устойчивость препа-
рата в щелочных проявляющих растворах [2].
Для получения 4-метил-1-фенилпиразолидона-3 ранее бы-
ла предложена конденсация фенилгидразина с метилметакри-
латом в среде этилата натрия [3] или изобутилата натрия [4}.
Нами найдены оптимальные условия для проведения этой
реакции в среде бутилата натрия исходя нз солянокислого
фенилгидразина.
Н
СХЕМА СИНТЕЗА 4-МЕТИЛ-1-ФЕНИЛ ПИРАЗОЛИДОНА-3
NHNHa-HCl
I СНз
II I + СН2-ССООСН3 c^-0Na
СН3-С-СООСН3
Н2С\ znh2
\N/
72
Характеристика основного сырья
Фенилгидразин солянокислый, ч., ГОСТ 5834—52.
Метилметакрилат, ч., ТУ МХП 2274—53.
Бутиловый спирт, ч., ГОСТ 6006—51.
Натрий металлический, ГОСТ 3273—55.
Условия получения
В четырехгорлую колбу емкостью 1 л с мешалкой, термо-
метром, обратным холодильником и загрузочной воронкой
вносят 275 мл сухого бутилового спирта (см. примечание 1) и
в течение 20 минут небольшими кусочками прибавляют 17,2 г
(0,75 М) металлического натрия. Для растворения остатков
натрия содержимое колбы перемешивают 10—15 минут при
120°. Полученный раствор охлаждают до 25—30° и за 5 минут
прибавляют 38,1 г (0,25 М) солянокислого фенилгидразина,
после чего реакционную массу нагревают 30 минут при 80—
85°, затем ее охлаждают до 25—30°, прибавляют 37,5 г
(0,375 М) метилметакрилата и размешивают 10—15 минут
при 110—115°.
Для удобства размешивания густой массы в колбу вно-
сят 45 мл сухого бутилового спирта и нагревают реакцион-
ную смесь еще 1 час при 110—115°. По охлаждении до 30°
прибавляют 120 мл воды, массу размешивают до растворения
осадка и нейтрализуют добавлением примерно 50 мл ледяной
уксусной кислоты.
Бутанольный слой отделяют в делительной воронке, поме-
щают в круглодонную колбу и отгоняют 300 мл растворителя.
Остаток из колбы горячим переносят в химический стакан и
перемешивают при охлаждении водой в течение 1 часа (см.
примечание 2).
К полученной густой массе прибавляют 50 мл бензина,
осадок отфильтровывают, промывают на фильтре дважды по
50 мл бензина, сушат на воздухе, размешивают в стакане с
80 мл воды, фильтруют, осадок промывают водой до отсутст-
вия качественной реакции на анион хлора (примерно 70 мл)
и сушат при 50—60°.
Выход 4-метил-1-фенилпиразолидона-3 равен 31,9 г, что
составляет 72,5% от теоретического, считая на солянокислый
фенилгидразин; т. пл. 132—134°. После кристаллизации из
разбавленного спирта (1:1) т. пл. продукта 135—136°. По
литературным данным, вещество имеет т. пл. 135° [3].
Примечания:
1. Сухой бутилозый спирт получен с помощью отгонки примерно 1/4
части растворителя (по объему).
2. Без перемешивания получается менее чистый продукт.
ЛИТЕРАТУРА
1. Яп. пат. 11036; РЖхим, 1964, 5Н455П.
2. G. С. A He tag. Phot. Set. Engng., 2, 213 (1958); С. A., 53, 1967
(1959).
3. Англ. пат. 703669; Zbl., 10180 (1955).
4. Н. И. С и м о и о в а, Ю. Е. Усаиов. Ж. Всесоюзн. хим. об-ва
им. Д. И. Менделеева, 7, 239 (1962).
Поступила в декабре 1964 г. НИОПиК
УДК 547.821.42.07
2-[₽-( N-МОРФОЛ ИЛ)ЭТИЛ]ПИРИДИН
Я. Ф. КАЗАРИНОВА, Н. В. ДЖИГИРЕИ
О
CnHleN2O
М. в. 192, 26
Нами проверена и подтверждена методика получения
2-[|3-(Ы-морфолил)этил]пиридина из 2-винилпиридина и мор-
фолина [1, 2]. В условия получения внесены лишь небольшие
изменения, касающиеся количеств реагирующих компонентов,
а именно: морфолин вводился в реакцию не в двукратном, а
в полуторакратном избытке по отношению к 2-винилпириди-
ну [1].
СХЕМА СИНТЕЗА 2-[₽-(N- МОРФОЛ ИЛ)ЭТИЛ]ПИРИДИНА
/ Ч
11 1 /°\ / ч
Ч^хсн=сна+| । -> ц । _
\N/ XN^\CH2CH2-N/
н
Характеристика основного сырья
2-Винилпиридин, ч., свежеперегнанный, т. кип. 60°/17 мм,
ВТУ TCP 2—60.
Морфолин, ч„ МРТУ 6-09-443—63.
75
Условия получения
Смесь 105 г (1 Л4) 2-винилпиридина и 130,5 г (1,5 Л4) мор-
фолина кипятят с обратным холодильником в течение 16 ча-
сов. Реакционную смесь фракционируют под вакуумом. По
внешнему виду 2-[р- (М-морфолил)этил]пиридин представляет
собой слегка желтое масло с т. кип. 168—170°/15 мм.
Выход равен 146,8 г, что составляет 80,6% от теоретичес-
кого; <Л20— 1,0689; йр - 1,5270.
По литературным данным, т. кип. продукта 168—
171°/17 мм [2}.
Аналогичным путем был осуществлен синтез 2-[(3-(N-пипе-
ридил) этил]пиридина [3]. Выход составил 70% от теоретичес-
кого. Соотношение реагирующих компонентов—1:1,2.
ЛИТЕРАТУРА
1. Н. Reich, R. Levine. J. Amer. Chem. Soc., 77, 4913 (1955).
2. A. Sommers, M. Freifalder, H. Uright, A. Weston.
J. Amer. Chem. Soc., 75, 57(1953).
3. W. Doering, K. Weil. J. Amer. Chem. Soc., 69, 2461 (1947).
Поступила в декабре 1964 г.
Донецкий филиал ИРЕА
УДК 547.852.9.07
3-НИТРАМИН0-1,2,4-ТРИА30Л
Г. И. ЧИПЕН
H-C-N
C2H3N6O2
N C-NHNOa
NH
М. в. 129, 07
3-Нитрамино-1,2,4-триазол используется как исходное сое-
динение при получении 3-гидразино-1,2,4-триазола и его про-
изводных.
Подобно гуанидину [1] 3-амино-1,2,4-триазол (а также
3-алкил-5-амино-1,2,4-триазолы) легко нитруется при раст-
ворении его азотнокислых солей в концентрированной серной
кислоте, образуя соответствующие нитраминотриазолы (2].
До сих пор эти соединения были получены только циклиза-
цией соответствующих 1-ациламидо-З-нитрогуанидинов [3].
СХЕМА СИНТЕЗА 3-НИТРАМИНО-1,2,4-ТРИАЗОЛА
NH
NH2NH-C-NHa-H2CO3 + Н-СООН —-
H-C-N
X + ЗН2О -J- со2
— N C-NH2
NH
H-C-N H-C-N
N C-NHa + HNO3 -> N C-NH2-HNO,
NH NH
77
H-C-N H-C-N
H2SO4
N C-NH2-HNO3 -5—-* N C-NHNO2 + H2o
XNH \h
Характеристика основного сырья
Аминогуанидинби карбонат, РУ-901—53.
Муравьиная кислота, ч., ГОСТ 1706—42.
Азотная кислота, ч„ ТУ АУ 112—56.
Серная кислота, ч., ГОСТ 4204—48.
Условия получения
Получение азотнокислой соли З-амино-1,2,4-триазола. В
круглодонную колбу емкостью 500 мл помещают 136 г (1 М)
бикарбоната аминогуанидина и прибавляют 50 мл муравьи-
ной кислоты. Смесь осторожно нагревают на горячей водяной
бане до полного растворения бикарбоната. После этого со-
держимое колбы нагревают при 120° в течение 5 часов (см.
примечание 1). После охлаждения полученную массу раство-
ряют при кипячении в 100 мл этилового спирта и фильтруют.
К фильтрату приливают 60 мл азотной кислоты и оставляют
кристаллизоваться. Выделившийся нитрат аминотриазола
фильтруют, промывают небольшим количеством спирта и су-
шат на воздухе.
Выход равен 90—93 г, что составляет 61—63% от теоре-
тического; т. пл. продукта 173—175° (см. примечание 2).
Получение З-нитрамино-1,2,4-триазола. Прибавляют 73,5 г
(0,05 Л1) нитрата аминотриазола небольшими порциями при
медленном перемешивании к 125 мл концентрированной сер-
ной кислоты (см. примечание 3). Температуру реакционной
среды путем наружного охлаждения водой поддерживают ни-
же 35°. После прибавления нитрата аминотриазола смесь вы-
держивают при комнатной температуре 15 минут, затем ее
выливают в смесь 300 г тонкоизмельченного льда и 200 мл
воды. Выпавший нитраминотриазол фильтруют и, не высуши-
вая, перекристаллизовывают из 1000 мл воды.
Выход 3-нитрамино-1,2,4-триазола равен 106—110 г, что
составляет 82—85% от теоретического; т. пл. продукта 216°
(с разл.) (см. примечания 4,5).
Примечания:
1. Смесь нагревали инфракрасной лампой. О получении 5-амино-1,2,4-
триазола см. [4].
2. Согласно литературным данным, нитрат З-амино-1,2,4-триазола пла-
вится при 174° [5]. Для следующей стадии синтеза нитрат аминотриазола
использовался без очистки.
3. Нитрат аминотриазола прибавляли к серной кислоте в течение
15 минут.
78
4. По литературным данным, 3-ннтрамино-1,2,4-триазол плавится
при 217° с разложением [3].
5. По такой же методике можно получить 3-алкил-5-нитрамино-1,2,4-
гриазолы.
ЛИТЕРАТУРА
1. A. F. McKay. Chem. Revs., 51, 329 (1952).
2. Г. И. Чипен, В. Я- Гринштенн, Р. П. Пр ей май. Ж. общ.
химии, 32, 454 (1962).
3. R. A. Henry. J. Amer. Chem. Soc., 72, 5343 (1950).
4. Сб. «Синтезы органических препаратов», вып. 6, 1956, стр. 9.
5. J- Thiele, W. Mane hot. Liebigs. Ann. Chem, 303, 33 (1898).
Поступила в декабре 1964 г.
Институт органического синтеза
АН Латв. ССР
79
УДК 547.391.1.07
|3-(5-НИТРОФУРИЛ-2)-АКРИЛОВАЯ КИСЛОТА
К. к. ВЕНГЕР. С. А. ГИЛЛ ЕР. В. В. ЦИРУЛЕ
O2N-^ 5-СН=СН-СООН
C,H6NO6 М. в. 183, 13
Для синтеза р-(5-нитрофурил-2)-акриловой кислоты пред-
ложены разные методы: нитрование р-(фурил-2)-акриловой
кислоты в среде уксусного ангидрида [1—5], конденсация
5-нитрофурфурола с уксусным ангидридом в присутствии ук-
суснокислого натрия по Перкину [1, 6} и кислотный гидролиз
этилового эфира |3-(5-нитрофурил-2)-акриловой кислоты, по-
лучаемого или нитрованием этилового эфира (3-(фурил-2)-ак-
риловой кислоты в среде уксусного ангидрида [1], или взаи-
модействием 5-нитрофурфурола с карбэтоксиметилентрифе-
нилфосфораном по Виттигу [7]. Попытка нитрования р- (фу-
рил-2) -акриловой кислоты смесью серной и азотной кислот не
увенчалась успехом [2].
Наиболее простым путем получения р-(5-нитрофурил-2) -
акриловой кислоты является нитрование р-(фурил-2)-акрило-
вой кислоты в уксусном ангидриде. Выход при этом без при-
менения катализаторов составляет 40—58% от теории.
Нами предложен способ нитрования р- (фурил-2)-акрило-
вой кислоты в уксусном ангидриде в присутствии катализато-
ра (концентрированной серной кислоты), что увеличивает вы-
ход р-(5-нитрофурил-2)-акриловой кислоты до 70—75% [5].
Стадию нитрования р-(фурил-2)-акриловой кислоты про-
водят в безводных условиях при низкой температуре (—25°),
а после завершения реакции реакционную смесь выливают
на смесь льда с водой.
Продуктом реакции является транс-р-(5-нитрофурил-2)-
акриловая кислота, т. пл. 234—6° (с разл.).
80
цис-fl- (5-Ннтрофурил-2) -акриловая кислота с т. пл. 176°
(с разл.) получена с небольшим выходом нитрованием цис-
p. (фурил-2)-акриловой кислоты или декарбоксилированием
5-нитрофурфурилиденмалоновой кислоты [6].
СХЕМА СИНТЕЗА р- (5-НИТРОФУРИЛ-2)-АКРИЛ ОВОЙ КИСЛОТЫ
СН=СН—СООН + HNO3 + (СН3СО)8О _LH^SO<J—
хо'
-> O2N-Y ^-СН=СН—СООН + 2СН3СООН
Характеристика основного сырья
транс-fl- (Фурил-2)-акриловая кислота, ТУ TCP 461р—61.
Уксусный ангидрид, ч., ГОСТ 5815 -52.
Серная кислота, ч., ГОСТ 4204—48.
Азотная кислота концентрированная, ГОСТ 701—58.
Условия получения
В трехгорлую колбу емкостью 500 мл, снабженную меха-
нической мешалкой, термометром, капельной воронкой и
хлоркальциевой трубкой, помещают 97,4 г (0,95 М) уксусно-
го ангидрида и медленно при перемешивании и охлаждении
(температура в колбе от — 10 до 0°) из капельной воронки
прибавляют 37,8 г (0,6 М) азотной кислоты (уд. вес 1,5) и
1 г (0,01 Л4) концентрированной серной кислоты (уд. вес
1,84) (см. примечание 1). Реакцию проводят в безводных ус-
ловиях. Нитрующую смесь охлаждают до минус 25° и при
этой температуре добавляют по каплям, в течение 1 часа, из
сухой капельной воронки раствор 27,6 г (0,2 Л4) fj-(фурил-
о-акриловой кислоты в 108 г (1,05 Л4) уксусного ангидрида.
К концу прибавления выделяется кристаллический продукт
нитрования. Смесь перемешивают при той же температуре
еще в течение 1 часа, выливают при перемешивании в 0,3 кг
измельченного льда с 0,3 л воды и перемешивают в течение
4 часов. Выпавшую fl-(5-нитрофурил-2)-акриловую кислоту
отсасывают, промывают 300 мл холодной воды и оставляют
на 24 часа под 350 мл воды. Затем отсасывают, сушат сперва
на воздухе, а потом в вакуум-эксикаторе над пятиокисыо
фосфора.
Выход р-(5-нитрофурил-2'1-акриловой кислоты равен 25,6—
27,4 г, что составляет 70—75% от теоретического; т. пл. 230—
235° (с разл.) (см. примечание 2)'.
Примечания:
1. При смешивании уксусного ангидрида с азотной и серной кислота-
ми выделяется значительное количество тепла. Экзотермической является
также реакция нитрования ₽-(фурил-2)-акриловой кислоты.
6 Зак. 1350 81
В качестве охлаждающего агента для проведения данного синтеза
рекомендуется ацетон, к которому добавляют соответствующее количество
твердой углекислоты для поддержания в реакционной колбе заданной
температуры.
2. Полученная ₽- (5-нитрофурил-2)-акриловая кислота для большинст-
ва целей вполне чиста. В случае необходимости ее можно очистить пере-
кристаллизацией из уксусной кислоты. Очищенный препарат представляет
собой светло-желтый кристаллический порошок, т. пл. 234—6° (с разл.).
ЛИТЕРАТУРА
1. Н. Gilman, G. F. Wright. J. Amer. Chem. Soc., 52, 2550
(1930).
2. T. Sasaki. Bull. Chem. Soc. Japan, 27, 398 (1954).
3. H. Saikachi, R. Kimura, H. Hoshida. J. Pliarm. Soc. Ja-
pan, 73, 1132(1953); C. A., 48, 12072(1954).
4. Y. Arata, T. О has hl, K. Aoki, M. Koseki, K. Sakai.
J. Pharm. Soc. Japan, 76, 211 (1956); C. A., 50, 14698 (1956).
5. К. К. Вентер, С. А. Гил л ер, В. В. Ц и руле. Изв. АН Латв.
ССР, сер. хим., 1962, 131.
6. Н. Saikachi, A. Tanaka. Yakngakn Zasshi, 83, 240(1963).
7. К. К. Вентер, С. А. Г и л л е р, В. Ф. Кучеров, В. В. Ц и р у л е,
А. М. Карклыня. Докл. АН СССР, 140, 1073 (1961).
Поступила в декабре 1964 г. Институт органического синтеза
АН Латв. ССР
УДК 547.823.07
4-ОКСИ-3-АЦЕТИЛ-6-МЕТИЛПИРИДОН-2
Н. С. ВУЛЬФСОН, Г. М. СУХОТИНА
он
| со-сн8
c8h9no3
м. в. 167,17
Для синтеза 4-окси-3-ацетил-6-метилпиридона-2 нами при-
менена реакция С-ацилирования кетоенолов алифатическими
кислотами в присутствии полифосфорной кислоты по анало-
гии с ранее описанным методом [1].
В качестве примера типового синтеза приводим синтез
4-окси-3-ацетил-6-метилпиридона-2.
СХЕМА СИНТЕЗА 4-ОКСИ-3-АЦЕТИЛ-6-МЕТИЛПИРИДОНА-2
но
ОН
СНзСООН
| СО-СНз
Характеристика основного сырья
4-Окси-6-метилпиридон-2, т. пл. 330° (с разложением).
Уксусная кислота, х. ч„ ГОСТ 7077—54.
6*
83
о-Фосфорная кислота, ч., ГОСТ 6552—58.
Фосфорный ангидрид, ч., ВТУ 521—41.
Условия получения
Смесь из 2 г (0,016 Л4) 4-окси-6-метилпиридона-2 (см.
примечание 1), 10 мл уксусной кислоты и 50 мл полифосфор-
ной кислоты (см. примечание 2) нагревают при размешива-
нии до возникновения интенсивно-красного окрашивания
(около 120°), затем размешивание продолжают еще 6 часов
при 90—100° и по охлаждении выливают в воду со льдом.
Выпавший осадок 4-окси-3-ацетил-6-метилпиридона-2 от-
фильтровывают, промывают водой, сушат.
Выход продукта равен 1,82 г, что составляет 67,9% от тео-
ретического. После кристаллизации из спирта т. пл. 259,5—
260,5°.
Найдено, %: С -57,45; 57,19; Н-5,55; 5,82; N- 8,53; 8,51.
CsH0NO3. Вычислено, %: С—57,48; Н—5,43; N— 8,38.
Фенилгидразон, т. пл. 224—225° (см. примечание 3).
Найдено, %: С-65,05; 65,05; Н-6,07; 6,11; N—16,12; 15,89.
CuH16NaO2. Вычислено, %: С—65,35; Н-5,87; N- 16,35.
Полученные аналогичным путем замещенные 4-окси-6-ме-
тилпиридона-2 и его N-метил- и N-фенилпроизводные, отве-
чающие нижеуказанной общей формуле, приведены в таблице.
ОН
I CO-R'
/^/
R
R R' Выход, % Температу- ра плавле- ния, °C Фенил- гидразоны, температура плавления, °C
Н Н н н н сн3 П р и м е ч а 1. Необходи н N-фенилпроизЕ с2н6 н-С3Н7 н-С4Н9 U3O-C4Hg H-C5Htl СН3 СН3 и я: иый для СИН юдные получ 34,0 28,0 • 29,9 22,4 28,0 26,5 60,0 геза 4-окси-б ают методам 225,5-226 183,5-184 188—188,5 157,5-158,5 175 175,5 133,5—134,5 223-224 метилпиридо И [2], [3], [4] 196-197 218-220 224-225 227-228 194—195 234—235 239-240 т-2 и его N-метил соответственно.
84
2. Полифосфорную кислоту получают растворением 100 г фосфорного
ангидрида в 100 мл фосфорной кислоты.
3. Фенилгидразон получают кипячением в спирте эквимолекулярных
количеств кетона и фенилгидразина.
ЛИТЕРАТУРА
I. Н. С. Вульфсон, Р. Б. Ж у р и н. Ж. общ. химии, 29, 3677 (1959);
Ж. Всес. хим. об-ва им. Д. И. Менделеева, 1960, № 3, 352; Ж. общ. хи-
лии, 31, 281 (1961).
2. N. Collie, W. Myers. J. Chem. Soc., 61, 723(1898).
3. F. Arndt, В. E i s t e r t, H. Scholz, E. Aron. Ber„ 69, 2373
(1936).
4. П. П e т p e н к o-K P и т ч e н к о, H. 3 о пев а. ЖРФХО, 45, 1098
(1913)
Поступила в декабре 1964 г.
Институт химии
природных соединений
АН СССР, НИОПиК
УДК 547.831.6.07
8-ОКСИ-2-ГИДРАЗИНОХИНОЛ ИН
(8-Окси-2-хинолил)гидразин
И. А. КРАСАВИН, В. М. ДЗИОМКО, Н. И. МИРОШКИНА
c,h9n,o
V/'W\nHNH2
ОН
М. в. 175,20
В литературе 8-окси-2-гидразинохинолин не описан. Он
был получен нами с выходом 92—96% при кратковременном
нагревании 2-хлор-8-оксихинолина с избытком гидразин-гид-
рата.
8-Окси-2-гидразинохинолин малорастворим в воде, но рас-
творяется в разбавленных кислотах и во многих органических
растворителях. Из бензола кристаллизуется в бледно-желто-
ватых иголках с т. пл. 180,5—181,5° (с предварительным по-
краснением при 170°).
Различные производные 8-окси-2-гидразинохинолина явля-
ются хелатообразующими соединениями.
Получение 8-окси-2-гидразинохииолина
I || | +2(N8HrH2O)_| || | +
4/W\ci ^/xn^\nhNH2
он он
+ N2H4-HC1 + 2HSO
В круглодонную колбу емкостью 250 мл со шлифом поме-
щают 5,39 г (0,03 М) 2-хлор-8-оксихиполина (см. примеча-
86
ние 1) и 30 мл (0,615 М) 99,5%-ного гидразин-гидрата
(ч. д. а.). Присоединяют обратный холодильник и погружают
колбу на 30 минут в глицериновую баню, заранее нагретую
до 128°. Образующийся вскоре прозрачный раствор периоди-
чески помешивают вручную. Затем его охлаждают до 5—8°,
причем реакционная масса закристаллизовывается, добавля-
ют 60 мл дистиллированной воды и комки измельчают стек-
лянным стержнем до исчезновения частиц оранжевого цве-
та. Когда образуется однородная светло-желтая суспензия,
осадок отсасывают, промывают водой и высушивают в ваку-
ум-эксикаторе.
Выход сырого 8-окси-2-гидразинохинолнна равен 4,85—
5,05 а (92,3—96,1% от теоретического); т. пл. 173—174° (с
покраснением).
После перекристаллизации из 550—600 мл бензола полу-
чают 4,1—4,2 г (78—80%) светло-желтых игл с т. пл. 178,5—
179,5° (краснеет при 165°) (см. примечание 2).
Примечания:
1. Применялся 2-хлор-8-оксихинолин с т. пл. 81—81,5°, полученный по
методу, описанному в настоящем сборнике.
2. Образец с т. пл. 180,5—181,5° (покраснение при 170°) был приго-
товлен кристаллизацией из бензола и проанализирован.
Найдено, %: С -61,87; 61,84; Н—5,14; 4,92; N-24,31; 24,25.
C9H9N3O. Вычислено, %: С—61,70; Н—5,18; N—23,99.
Поступила в сентябре 1965 г.
ИРЕА
УДК 547.831.8.05
8-ОКСИ-2-МЕТОКСИХИНОЛИН
В. М. ДЗИОМК.О, И. А. КРАСАВИН. Ю. П. РАДИН
^\/ Ч
I II I
^/XN^\oCH8
он
c10h9no2
М. в. 175, 19
В литературе 8-окси-2-метоксихинолин не описан, но ра-
нее нами было отмечено [!}, что он образуется (с выходом
20—23%) как побочный продукт реакции метосульфата 8-ок-
си-1-метоксихинолиния с водным раствором едкого кали.
Теперь нами установлено, что 8-окси-2-метоксихинолин
может быть получен с выходом 70—73% при взаимодействии
метосульфата 8-окси-1-метоксихинолиния с метилатом натрия
в метаноле.
8-Окси-2-метоксихинолин представляет собой бесцветное
кристаллическое вещество, летучее с водяным паром, сравни-
тельно малорастворимое в холодной воде, но прекрасно раст-
воряющееся во всех органических растворителях, а также в
разбавленных растворах щелочей и кислот. Он кристаллизу-
ется из гептана в белоснежных иглах с т. пл. 50,5—51°.
Получение 8-окси-2-метоксихинолина
+ 2CH3ONa
CH3SOr
ОН ОСН3
+ NaCH3SO4 + 2СН3ОН
88
ONa
N осн
4-СНзСООН -
+ CHgCOONa
В трехгорлой колбе емкостью 500 мл, снабженной мешал-
кой, капельной воронкой и обратным холодильником, защи-
щенным хлоркальциевой трубкой, готовят раствор 6,9 г
(0,3 г-ат) металлического натрия в 120 мл абсолютного мета-
нола и охлаждают его в бане со льдом.
При нагревании с обратным холодильником растворяют
28,7 г (0,1 М} метосульфата 8-окси-1-метоксихинолиния (см.
примечание 1) в 80 мл абсолютного метанола и охлаждают
до 3—5°. Этот раствор прибавляют по каплям за 1,5 часа к
раствору метилата натрия, который размешивают и охлажда-
ют ледяной водой.
Образовавшуюся желтую, постепенно загустевающую мас-
су перемешивают еще 1,5—2 часа при 0—2°, а затем остав-
ляют на ночь при комнатной температуре.
На следующий день разбавляют водой (150—200 мл), пе-
реливают в одногорлую колбу и полностью отгоняют метанол
в вакууме водоструйного насоса. В случае необходимости до-
бавляют к остатку еще 100—150 мл воды, чтобы получить
прозрачный раствор, затем нейтрализуют его 20%-ной уксус-
ной кислотой до pH 7 и подвергают перегонке с водяным па-
ром. Кристаллы, выделившиеся из дистиллята при охлажде-
нии и стоянии, отсасывают, промывают водой и высушивают
в вакуум-эксикаторе.
Выход 8-окси-2-метоксихинолина равен 12,3—12,8 г (70,3—
73,1% от теоретического); т. пл. 49,5—50° (см. примечания
2 и 3).
Примечания:
1. Применялся сырой метосульфат 8-окси-1-метоксихинолииия, полу-
ченный по описанному нами ранее методу [2].
2. Вещество можно перекристаллизовать из гептана (2—3 мл на 1 г),
но обычно в этом нет необходимости.
3. Образец с т. пл. 50,5—51° был приготовлен повторной перегонкой
е паром и перекристаллизацией из гептана и проанализирован.
Найдено, %: С-68,41; 68,71; Н—5,22; 5,13; N—7,73; 8,05.
C10H8NO2. Вычислено, %: С—68,56; Н—5,18; N—8,00.
ЛИТЕРАТУРА
1. В. М. Дзиомко, И. А. Красавин, Ю. П. Радин. Методы
получения химических реактивов и препаратов, вып. 13, М„ ИРЕА, 1965,
стр. 44.
2. И. А. Красавин, В. М. Дзиомко, Ю. П. Радин. Там же,
ст р. 68.
Поступила в сентябре 1965 г. ИРЕА
89
УДК 547.823.07
4-ОКСИ-6-СТИРИЛ ПИРОН-2
Н. С. ВУЛЬФСОН, Е. В. САВИНКОВА
ОН
С,Н6СН=СН
СазН^Оз
М. б. 214,22
Синтез 4-окси-6-стирилпирона-2 осуществлен нами конден-
сацией триацетовой кислоты [1] с бензальдегидом. Подобные
соединения могут представить интерес как фильтровые краси-
тели.
СХЕМА СИНТЕЗА 4-ОКСИ-6-СТИРИЛПИРОНА-2
Характеристика основного сырья
Триацетовая кислота, т. пл. 181—182°.
Бензальдегид, ч., ГОСТ 157—51.
Пиридин, ч., ГОСТ 2747—44.
Пиперидин, ч., ВТУ МХП 4472—55.
Уксусная кислота, ч., ГОСТ 7077—54.
Соляная кислота, ч., ГОСТ 857—57.
Спирт метиловый синтетический, ч., ГОСТ 6995—54.
Условия получения
Смесь из 20 г (0,16 М) триацетовой кислоты (см. прило-
жение 1), 17 г (0,16 М) бензальдегида, 100 мл свежеперег-
нанного пиридина, нескольких капель пиперидина и столько
же ледяной уксусной кислоты нагревают при размешивании
4 часа при 100°. По охлаждении выливают в ледяную 10%-
ную соляную кислоту, выпавший осадок отфильтровывают,
промывают водой, раствором карбоната натрия и сушат.
Выход продукта равен 23,4 г, что составляет 69% от тео-
ретического. После перекристаллизации из метанола т. пл.
вещества 164—165°.
Найдено, %: С—72,74; 72,68; Н-4,90; 4,84.
С13Н1()О3. Вычислено, И: С—72,81; Н—4,72.
Примечания:
1. Необходимая для синтеза триацетовая кислота получена из дегидр-
ацетовой кислоты по методике [2].
2. При нагревании 4-окси-6-стирилпирона-2 с уксусным ангидридом в
пиридине получают ацетильное производное (4-ацетокси-6-стирилпирон-2)
с т. пл. 173—175° (из разбавленного спирта).
Найдено, %: С-70,39; 70,41; Н-4,51; 4,64.
С16Н12О4. Вычислено, %: С—70,31; Н—4,69.
ЛИТЕРАТУРА
1. N. Collie. J. Chem. Soe., 59, 609 (1891); 91, 787 (1907).
2. Препаративная органическая химия, М., ГХИ, 1959, стр. 635.
Поступила в декабре 1964 г. Институт химии природных соединений
АН СССР, НИОПиК
УДК 547.823.07
4-ОКСИ-3-ЦИННАМОИЛ-6-МЕТИЛ ПИРОН-2
Н. С. ВУЛЬФСОН, Е. В. САВИНКОВА
ОН
СОСН=СН-А Э
С16н12о4
СНз/Ч 0/Ч-0
М. в. 256,25 при R=H
Синтез замещенных 4-окси-3-циннамонл-6-метилпиронов-2
осуществляется конденсацией дегидрацетовой кислоты [1] с
бензальдегидами в условиях реакции Кляйзена-Шмидта. Кон-
денсация дегидрацетовой кислоты с бензальдегидами изуче-
на мало. Впервые она была проведена в спиртовом растворе
щелочи [2], позднее в хлороформном растворе в присутствии
пиперидина [3] и совсем недавно в пиридине в присутствии
пиперидина [4]. Нами осуществлен синтез и разделение ве-
щества на геометрические изомеры.
Соединения данного класса могут представить интерес
как фильтровые красители.
В качестве примера типового синтеза приводим синтезы
и разделение на цис-транс-изомеры 4-окси-3-циннамоил-6-
метилпирона-2 и 4-окси-3-(4/-нитроциннамоил)-6-метилпиро-
на-2.
СХЕМА СИНТЕЗА 4-ОКСИ-3-ЦИННАМОИЛ-6-МЕТИЛ ПИРОНА-2
ОН
/Ч/СОСНз 0
А ) + у
СНв/ХО/х\О - Hz 4=XR
92
он
где R: Н; о-, м-, n-окси; о-, н-хлор; о-метокси; и-диметиламино
Характеристика основного сырья
Бензальдегид, ч., ГОСТ 157—51.
Пиридин, ч., ГОСТ 2747—44.
Пиперидин, ч., ВТУ МХП 4472—55.
Уксусная кислота, ч., ГОСТ 7077—54.
Соляная .кислота, ч., ГОСТ 857—57.
Бензол, ч., ГОСТ 8448—61.
Петролейный эфир, синтетический, ТУ МХП 600—58.
Спирт метиловый синтетический, ч„ ГОСТ 6995—54.
Условия получения
Получение 4-окси-3-циннамоил-6-метилпирона-2. Нагрева-
ют при размешивании 25,2 г (0,15 М) дегидрацетовой кисло-
ты, 15,9 г (0,15 М) бензальдегида, 120 мл свежеперегнанно-
го пиридина, несколько капель пиперидина и столько же ле-
дяной уксусной кислоты в течение 45 минут при 45—50° и 15
минут при 100°. По охлаждении выливают в ледяную 10%-
ную соляную кислоту, осадок отфильтровывают, промывают
водой, раствором карбоната натрия, сушат, кристаллизуют из
-тилового спирта и получают 23,1 г вещества, что составляет
% от теоретического, с т. пл. 98—125° (см. примечание 1).
зобной кристаллизацией продукта из разбавленного мета-
ла выделяют сначала цис-4-окси-З-циннамоил-б-метилпи-
iH-2 с выходом 2,5 г, что составляет 6,5% от теоретического.
По внешнему виду это желтые иглы с т. пл. 105—106°.
Найдено, %: С - 70,31; 70,21; Н- 4,76; 4,60.
С16Н12О4. Вычислено, %: С-70,31; Н—4,69.
Выход транс-изомера равен 15,5 г, что составляет 40,4%
от теоретического. По внешнему виду это также желтые иглы
ст. пл. 129—130°.
Найдено, %: С-70,29; 70,33; Н- 4,64; 4,72.
Г15Н12О4. Вычислено, %: С—70,31; Н—4,69.
При проведении конденсации в кварцевой колбе и облу-
чении УФ-светом выход цис-изомера повышается до 9,4 г,
что составляет 24,5% от теоретического (см. примечание 2).
Получение 4-окси-З-(4'-нитроциннамоил)-6-метилпирона-2.
Конденсируют 25,2 г (0,15 М) дегидрацетовой кислоты и
93
22,6 г (0,15 М) n-нитробензальдегида, как описано выше. По
окончании конденсации реакционную массу охлаждают до 0°,
размешивают при этой температуре 3—4 часа и отфильтровы-
вают выпавший в осадок транс-изомер.
Выход вещества равен 21,0 г, что составляет 47% от тео-
ретического; т. пл. после перекристаллизации из смеси диок-
сана и этилового спирта (1 : 1) 246—247°.
Найдено, %: N—4,64; 4,69.
C16HUO6N. Вычислено, Н: N—4,65.
Оставшийся пиридиновый фильтрат выливают в избыток
охлажденной 10%-ной соляной кислоты и после многократ-
ных перекристаллизаций из смеси спирта и этилацетата вы-
деляют ^ас-изомер с выходом 3,9 г, что составляет 22% от
теоретического; т. пл. 165—166°.
Найдено, %: N—4,68; 4,62.
C16HnO6N. Вычислено, %: N—4,65.
Аналогичным путем нами получены нижеследующие заме-
щенные 4-окси-3-циннамоил-6-метил пироны-2.
R Изомер Температу- ра плавле- ния, 1С Раствори- тель кри- сталлизации Выход, %
о-Хлор транс 116-117 Этанол 36,0
п-Хлор транс 155-156 Этанол 53,5
.м-Нитро транс 171-172 Этанол 60,0
о-Окси транс 186 - 188 Метанол 67,0
лг-Окси транс 181-183 Этанол 61,0
п-Окси транс 260-262 Диоксан 68,5
о-Метокси транс 153-154 Этанол 73,0
п-Диметил- амино транс 198—200 Этанол 71,0
Примечания:
1. Хроматограмма продукта на бумаге марки «быстрая», пропитан-
ной фосфатным буфером (pH 3), свидетельствует о его неоднородности.
2. При кипячении /(ис-изомера в бензоле в присутствии пиперидина
илн следов соляной кислоты он превращается в транс-изомер.
ЛИТЕРАТУРА
1. Препаративная орг. химия, М., Госхимиздат, 1959, стр. 635.
2. W. J. Hale. J. Amer. Chem. Soc., 33, 1131(1911).
3. R. H. Wiley. J. Amer. Chem. Soc., 77, 5102 (1955).
4. A. J. Birch, D. W. Cameron. J. Chem. Soc., 1960, 4395.
П оступила в декабре 1964 г. Институт химии природных
соединений АН СССР, НИОПиК
УДК 547.722.07
1,2,3,4,5,6,7,8-ОКТАГИДРОДИБЕНЗОФУРАН (I) И
2-МЕТИЛ-2,3-ЦИКЛОПЕНТАНО-4,5-ЦИКЛОГЕКСЕНО-
(Д5)-2,3,4,5-ТЕТРАГИДРОФУРАН (II)
С. В. СВЕТ03АРСКИЙ, К. Л. ФЕЛЛЕР, Ю. Ю. САМИТОВ,
Е. И. ЗИЛЬБЕРМАН, Г. А. РАЗУВАЕВ
С12Н1вО М. в. 176,25
О сн3
(II) -
CltH18O М. в. 178,27
1,2,3,4,5,6,7,8-Октагидродибензофуран (I) получают разло-
жением соответствующего d-сультона в присутствии медного
порошка. Нами предложен другой способ его получения —
взаимодействием циклогексанона с концентрированной сер-
ной кислотой при комнатной температуре [1]. Наряду с окта-
гидродибензофураном получено не описанное в литературе
соединение (II). К преимуществам предложенного способа
относятся доступность исходных продуктов, относительная
простота, удобство методики синтеза и выделения продуктов
реакции.
2-, 3- и 4-Метилциклогексаноны взаимодействуют с кон-
центрированной серной кислотой аналогично циклогексанону
с образованием метилзамещенных фурановых соединений [2].
СХЕМЫ СИНТЕЗА 1,2,3,4,5,6,7,8-ОКТАГИДРОДИБЕНЗОФУРАНА И
2-МЕТИЛ-2,3-ЦИКЛОПЕНТАНО-4,5-ЦИКЛОГЕКСЕНО-( Л6)-2,3,4,5-
ТЕТРАГИДРОФУРАНА
2
+_Н38О£
(О)
I II » I
\/Х/\/ + 2НаО
О
(I)
95
2 Г Г° |/Ч| |/Х| +н2о
\/ \^\/\
О СНз
(И)
Характеристика основного сырья
Циклогексаион, ч., СТУ 10244—63.
Серная кислота, концентрированная, ГОСТ 4204—48.
Бензол, х. ч., ГОСТ 5955—54.
Условия получения
1. В термостойком стакане емкостью 3 л смешивают
250 мл циклогексанона и 250 мл бензола. Затем, охлаждая
стакан смесью льда и соли, при постоянном перемешивании
из капельной воронки прикапывают 250 мл концентрирован-
ной серной кислоты в течение 10 минут так, чтобы температу-
ра реакционной массы держалась в интервале 20—30° (см.
примечание 1). Полученную смесь перемешивают еще в тече-
ние одного часа при комнатной температуре, после чего к ней
прибавляют 1000 мл дистиллированной воды при перемеши-
вании стеклянной палочкой (см. примечание 2). К всплывшему
органическому слою добавляют 150 мл бензола и содержи-
мое стакана переносят в делительную воронку. Бензольный
экстракт отделяют, промывают водой до нейтральной реак-
ции (см. примечание 3). Затем бензол отгоняют на водяной
бане, а остаток разгоняют в вакууме на установке с елочным
дефлегматором. Отбирают фракцию с т. кип. 117—13572 мм,
которую затем подвергают ректификации на лабораторной
вакуумной колонке эффективностью в 35 т.т. (флегмовое чис-
ло 10—12).
Получают: а) 100 г 2-метил-2,3-циклопентано-4,5-циклогек-
сено-(А5)-2,3,4,5-тетрагидрофурана (II), что составляет 47%
от теоретического; т. кип. 8571,5 мм; — 1,5060; б/420 —
1,067; 2,4-динитрофенилгидразон с т. пл. 167° (из метилового
спирта);
б) 5 г промежуточной фракции с т. кип. 86—9671,5 мм;
в) 15 г октагидродибензофурана (I), что составляет 7%
от теоретического; т. кип. 96°/1,5 мм; п‘%—1,5198; d420—
1,0379; аддукт с малеиновым ангидридом, т. пл. 167° (из
этилацетата).
II. К смеси 200 мл 2-циклогексенил-циклогексанона [3] и
200 мл бензола добавляют 200 мл концентрированной серной
кислоты (20—30°, 10 минут). После перемешивания при ком-
натной температуре в течение 2 часов реакционную массу
перерабатывают, как описано выше.
96
Получают 10 г окта гидродибензофура на (I), что состав-
ляет 5% от теоретического, и 60 а 2-метил-2,3-циклопентано-4,
5-циклогексено-(Д5)-2,3,4,5-тетрагидрофурана (И), что состав-
ляет 30% от теоретического (см. примечание 4).
Примечания:
1. Сначала кислоту добавляют осторожно (возможен резкий скачок
температуры), а к концу реакции прибавление можно ускорить. Реакци-
онная масса имеет интенсивный красный цвет.
2. При добавлении воды реакционная масса разогревается, поэтому
стакан охлаждают водой и только после этого добавляют бензол.
3. При промывании бензольного экстракта не рекомендуется сильное
встряхивание, так как при этом может образоваться устойчивая суспензия,
которая долго не расслаивается. При работе с бензолом необходимо от-
сутствие нагревательных приборов.
4. Соединения (I) и (II) неустойчивы на воздухе и имеют резкий за-
пах. Оба вещества рекомендуется хранить в ампулах, запаянных под ва-
куумом,
ЛИТЕРАТУРА
1. С. В. С в е т о з а р с к и й, К- Л. Феллер, Ю. Ю. Самитов,
Е. Н. Зильберман, Г. А. Разуваев. Изв. АН СССР, ОХН, 1964,
121.
2. К. Л. Феллер, С. В. С в е т о з а р с к и й, Ю. Ю. Самитов,
Е. Н. 3 и л ь б е р м а н, Г. А. Разуваев. Изв. АН СССР (в печати).
3. Н. Gault, L. Daltroff, J. Eck-Tridon. Bull. Soc. chim.,
12, 952 (1945).
Поступила в ноябре 1964 г.
НИХП, г. Дзержинск
УДК 547.822.8.07
2-ПИРИДИЛМЕТАНОЛ
Н. Ф. КАЗАРИНОВА, Н. А. РЫБАК. В. В. БАЛЫКИНА
L ^-CHjOH
C6H8NO М. в. 110,13
2-Пиридилметанол является исходным продуктом для по-
лучения разнообразных производных пиридина. Его получа-
ют при взаимодействии N-окиси 2-пиколина с ангидридами
кислот [1—4]. Обычно в реакции используется чистая перег-
нанная N-окись. Нами установлено, что для получения 2-пи-
ридилметанола с успехом может быть использован неочищен-
ный ацетат N-окиси, получаемый при окислении 2-пиколина
перекисью водорода в уксусной кислоте, после отгонки из ре-
акционной массы воды и уксусной кислоты.
Предлагаемый метод позволяет значительно упростить по-
лучение 2-пиридилметанола, сократив весьма трудоемкую
стадию выделения и очистки N-окиси.
СХЕМА СИНТЕЗА 2-ПИРИДИЛМЕТАНОЛА
Н, Оз
СН3СООН
•СНзСООН
HCI
(СН3СО)3О
\
L J-ch2oc
//°
хсн3
-СН2ОН
98
Характеристика основного сырья
2-Пиколин, технический.
Перекись водорода, х. ч., ТУ ЕУ 104—55, 30%-ная, меди-
цинская.
Уксусный ангидрид, ч., ГОСТ 5815—52.
Условия получения
Смесь 93 а (1 М) 2-пиколина, 300 г (4,8 М) уксусной кис-
лоты и 150 мл перекиси водорода нагревают 3 часа на ки-
пящей водяной бане. Затем снова прибавляют 80 мл переки-
си водорода и нагревание продолжают еще в течение 4 часов.
По окончании нагревания реакционную смесь упаривают в
вакууме водоструйного насоса для удаления воды и уксусной
кислоты. К остатку, представляющему собой ацетат N-окиси
2-пиколина, прибавляют 50 мл хлороформа, нагревают реак-
ционную смесь на водяной бане до кипения и медленно по
каплям прибавляют 204 г (2 М) уксусного ангидрида. Затем
реакционную смесь кипятят еще 1 час и на водоструйном на-
сосе отгоняют хлороформ и уксусный ангидрид. В остатке по-
лучают темно-коричневую вязкую массу, представляющую
собой «сырой» ацетат 2-пиридилметанола. К этому ацетату
2-пиридилметанола прибавляют 45 мл разбавленной соляной
кислоты (1:1) и смесь кипятят на водяной бане 4—5 часов
(см. примечание). Затем отгоняют воду, разбавленную соля-
ную и уксусную кислоты. Остаток после отгонки нейтрализу-
ют содой до нейтральной реакции (по конго), отфильтровы-
вают и промывают на фильтре хлороформом. Фильтрат су-
шат безводным сульфатом натрия, отгоняют хлороформ, а
продукт очищают перегонкой под вакуумом.
Выход 2-пиридилметанола равен 78,5 г, что составляет
72% от теоретического; т. кип. 98—100°/7 мм; пд— 1,5248.
По литературным данным, т. кип. продукта 102°/8
1,5444.
Примечание.
Для омыления пиридилметанолацетата может быть использован
10%-ный раствор едкого натра. В этом случае 2-пиридилметанол сразу
же из щелочной реакционной массы легко извлекается хлороформом.
ЛИТЕРАТУРА
1. О. В u 1111, J. М а у n а г d. J. Amer. Chem. Soc., 76, 1370 (1954).
2. A. Ber son, T. Cohen. J. Amer. Chem. Soc., 77, 1281, 3(1955).
3. G. К о b a j a s h 1, S. Furukawa. C. A., 46, 10948 (1952).
4. J. Vozza. J. Organ. Chem., 27, 3856 (1962).
Поступила в декабре 1964 г. Донецкий филиал ИРЕА
7*
УДК 547.823.07
2-СТИРИЛ-6-МЕТИЛ ПИРОНЫ-4
Н. С. ВУЛЬФСОН, Е. В. САВИНКОВА
С14Н18О2
СН=СНС6Н6
М. в. 212, 24
Синтез 2-стирил-6-метилпирона-4 был осуществлен нагре-
ванием 4-окси-3-циннамоил-6-метилпирона-2 в концентриро-
ванном растворе соляной кислоты [1] или в смеси ледяной ук-
сусной и концентрированной соляной кислот [2].
Нами уточнены условия синтеза, и реакция распростране-
на на ряд замещенных 4-окси-3-циннамоил-6-метилпиронов-2.
Показано, что нуклеофильные заместители облегчают, а
электрофильные тормозят эту реакцию.
В качестве примера типовых синтезов приводим синтезы
2-стирил-6-метилпирона-4 и его З'-оксипроизводного.
СХЕМА СИНТЕЗА 2-СТИРИЛ-6-МЕТИЛ ПИРОНА-4
ОН
। СОСН=СНСвНБ
II I [Н+]
/\ 0 А С2НбОН
CHS U О
100
б
II
II II
сн/ 0 сн=сн-/ J
= R
где R: Н;о-, м-, и-ОН; о-, п-CI; м-, n-NO2; о-ОСН3
Характеристика основного сырья
Спирт этиловый, ГОСТ 5963—51.
Спирт изобутиловый, ч„ ГОСТ 6016—51.
Соляная кислота, ч„ ГОСТ 857—57.
Серная кислота, ч., ГОСТ 2184—59.
Бензол, ч., ГОСТ 8448—61.
Петролейный эфир синтетический, ТУ МХП 600—58.
Условия получения
Получение 2-стирил-6-метилпирони-4. Смесь из 3 г
(0,01 Л4) 4-окси-3-циннамоил-6-метилпирона-2 (см. стр. 92)
растворяют в 100 мл этилового спирта, добавляют .200 мл
10%-ной серной кислоты и еще 50 мл этилового спирта до
полного растворения и кипятят 50 часов (см. примечание 1).
Затем спирт отгоняют, выпавший осадок отфильтровывают,
промывают ледяной водой и сушат. Полученный осадок
(2,2 г, что составляет 89% от теоретического) кипятят в бен-
золе и фильтруют горячим. Фильтрат разбавляют петролей-
ным эфиром и выпавший осадок мономера кристаллизуют из
воды или разбавленного спирта.
Выход равен 1,2 г. что составляет 48,5% от теоретическо-
го; т. пл. 121 —122°.
Найдено, %: С 78,86; 78,84; Н-5,61; 5,70.
СцН12О2. Вычислено, %: С—79,10; Н—5,65.
Нерастворившееся в бензоле вещество перекристаллизо-
вывают из разбавленного спирта и получают димер 2-сти-
рил-б-метилш/рона-4 (см. примечание 2).
Выход равен 0,5 г, что составляет 20% от теоретического;
т. пл. 230—232°.
Найдено, %: С-79,00; 78,80; Н—5,49; 5,58.
(Ci4H12O2)2. Вычислено, %: С—79,10; Н-5,65.
Получение 2- (3'-оксистирил) -6-метилпирона-4. Смесь из
0,5 г (0,002 М) 4-окси-3-(3''-оксициннамоил)-6-метилпирона-2
растворяют в 20 мл изобутилового спирта, добавляют 20 мл
10%-ной серной кислоты и кипятят 10 часов. По окончании
реакции массу охлаждают, нейтрализуют раствором карбона-
101
та натрия, изобутиловый слой отделяют и растворитель отго-
няют. Выпадает осадок 2-(3/-оксистирил) -6-метилпирона-4.
Выход равен 0,34 г, что составляет 81% от теоретического.
После перекристаллизации из метилового спирта т. пл. веще-
ства 209—210°.
Найдено, %: С-73,47; 73,26; Н-5,38; 5,27.
С14Н12О3. Вычислено, %: С—73,67; Н—5,30.
Аналогичным путем могут быть получены следующие за-
мещенные 2-стирил-6-метилпироны-4.
R Раствори- тель Кислота Выход, % Температу- ра плавле- ния, °C
о-Хлор Этанол Концентрированная соляная 9,0 108-109
п-Хлор Изобутанол Концентрированная соляная 8,0 128-130
м-Нитро — Концентрированная соляная 6,0 163-164
о-Окси Этанол 10% -ная серная 78,0 195-197
п-Окси Этанол 10% -ная серная 82,0 223-224
о-Метокси Изобутанол 3%-ная соляная 68,0 134—136
Примечания:
1. Реакция проходит аналогично и с 90%-ной и с концентрированной
серной кислотами, а также с 3%-ной соляной кислотой.
2. Мономер 2-стирил-6-метилпирона-4 при кипячении в бензоле в при-
сутствии пиперидина или следов соляной кислоты превращается в димер.
ЛИТЕРАТУРА
1. R. Н. Wiley. J. Amer. Chem. Soc., 77, 5102 (1955).
2. A. J. Birch, D. W. Cameron. J. Chem. Soc., 1960, 4395.
Поступила в декабре 1964 г.
Институт химии
природных соединений
АН СССР, нИОПиК
УДК 547.724.1.07
ТЕТРАФУРФУРИЛОКСИСИЛАН
Фурфурилсиликат
Э. ЛУК.ЕВИЦ, 10. II. РОМАДАН, С. А. ГИЛЛЕР
CaoHgoOgSi
М. в. 416,46
Тетрафурфурилоксисилан получают при взаимодействии
фурфурилового спирта с этиловым эфиром ортокремневой
кислоты [1—7], дисульфидом кремния [8] и четыреххлористым
кремнием в присутствии пиридина {4—6].
Наиболее удобным методом, позволяющим быстро, просто
и с хорошим выходом синтезировать тетрафурфурилоксиси-
лан, является реакция фурфурилового спирта с четыреххло-
ристым кремнием в присутствии пиридина.
Нами разработана предлагаемая ниже методика получе-
ния тетрафурфурилоксисилана по этому способу.
103
Схема синтеза тетрафурфурилоксисиланА
4 И. СН2ОН + S1C14 + 4C6H6N
Si + 4C6H5N-HC1
Характеристика основного сырья
Фурфуриловый спирт, ТУ МХП 2623—51 (см. примеча-
ние 1).
Четыреххлористый кремний, ч., ВТУ МХП 3490—52.
Пиридин, ч., ГОСТ 1625—61 (см. примечание 2).
Условия получения
В литровую трехгорлую круглодонную колбу, снабженную
мешалкой с герметизирующим затвором, капельной воронкой
и обратным холодильником, закрытым хлоркальциевой труб-
кой, помещают 98,1 г (1 М) фурфурилового спирта, 79,1 г
(1 М) пиридина и 250 мл абсолютного эфира. Колбу охлаж-
дают ледяной водой и при интенсивном перемешивании по
каплям прибавляют раствор 42,4 г (0,25 М) четыреххлори-
стого кремния в 50 мл абсолютного эфира. После прибавле-
ния четыреххлористого кремния реакционную смесь нагрева-
ют на водяной бане до 40° в течение 3 часов. После охлажде-
ния осадок солянокислого пиридина отсасывают и промыва-
ют эфиром. Растворитель отгоняют в вакууме водоструйного
насоса, при этом остаток в колбе закристаллизовывается (см.
примечание 3). После перекристаллизации его из смеси пет-
ролейного эфира с толуолом получают 72—78 г тетрафурфу-
рилоксисилана, что составляет 70—75% от теоретического вы-
хода; т. ил. 38—39° (см. примечание 4).
Примечания:
1. Фурфуриловый спирт сушат сульфатом магния и перед употребле-
нием перегоняют в вакууме (10—15 .юи).
2. Пиридин сушат над едким кали и быстро перегоняют над натрием.
3. Иногда необходимо более сильное охлаждение, чтобы вызвать кри-
сталлизацию тетрафурфурилоксисилана. Продукт можно также вымора-
живать из растворителя.
Перегонка в вакууме обычно ведет к понижению выхода тетрафур -
фурилоксиснлана вследствие частичного осмоления; т. кип. фурфурилокси-
силана 203—205°/3 мм.
4. По этой методике можно синтезировать и другие фурфурилоксиси-
ланы с общей формулой
R„Si ОСН2II II
\ 'СИ
104
4—д
ЛИтёрАтурА
1. Н. Rothrock. Пат. США 2276094 (1942); С. А„ 36, 4630 (1942),
2. J. Rust, Пат. США 2300812 (1943); С. А., 37, 2103 (1943).
3. Р. Garner. Пат. США 2396692 (1946); С. А., 40, 3766 (1946).
4. Э. Лукевиц, Ю. Р о м а д а и, С. Г ил л ер. Изв. АН Латв. ССР,
7, 59 (1961).
5. И. В. Каменский, И. К. Санин, В. В. К о р ш а к. Пласт, мас-
сы, 1962, № 3, 8.
6. И. К. Санин, И. В. Каменский, В. И. Итииский. Авт.
спид. 140060; Бюлл. изобр., № 15, 19 (1961).
7. И. К. Санин, И. В. Каменский, В И. И т и н с к и й. Авт.
свид. 140059; Бюлл. изобр., № 15, 19 (1961).
8. J. Culbertson, Н. Erasmus, R. Fowler. Пат. США
2569455 (1951); С. А., 46, 3084 (1952).
Поступила в декабре J964 г.
Институт органического синтеза
АН Латв. ССР
УДК 547.86.07
2-ТИОТИАЗАНОН-4
Е. В. ВЛАДЗИМИРСКАЯ, И. М. ТУРДЕВИЧ
О
HaCf 'nH
f| I
н2с c=s
CtH6NOS2 М.в. 147,21
2-Тиотиазанон-4 рассматривается как сырье для получе-
ния ускорителей вулканизации каучука, а также фунгицид-
ных и пестицидных средств {1}. При нагревании с ароматичес-
кими аминами образует ариламиды S-тиокарбаминилтио-
гидракриловой кислоты (ArNH.CO.CH2.CH2.CS.NH2), которые
легко гидролизуются с образованием ариламидов тиогидр-
акриловой кислоты (ArNH.CO.CH2.CH2.SH), причем послед-
ние нашли применение для горячей, стабилизации хлорсодер-
жащих полимеров [2].
Для синтеза 2-тиотиазанона-4 вводят дитиокарбаминат,
аммония в реакцию с 0-йодпропионовой [3] или 0-хлорпропио-
новой кислотой [4], либо с 0-пропиолактоном [5] и образовав-
шуюся S-тиокарбаминилтиогидракриловую кислоту ангидри-
зуют нагреванием с треххлористым фосфором, а также с ук-
сусным ангидридом или его смесью с серной кислотой. Наи-
лучшие выходы в процессе ангидризации (42,4%) были по-
лучены [4] при быстрой отгонке уксусного ангидрида. Пред-
лагаемый нами метод более удобен и повышает выход про-
дукта ангидризации до 73%.
106
СХЕМА СИНТЕЗА 2-ТИОТИАЗАНОНА-4
2 С1СН2СН2СООН + Na2CO3 2 ClCH2CH2COONa +Н,О+СО2
H2N-CS-SNHt+ClCHaCH2COONa
-* H2N-CS.S.CH2CH2COONa-J~NHtCl
H2N-CS-S-CH2CH2COONa -|-НС1 ->
-> HaN-CS-SCH2CH2COOH f-NaCI
соон
CHa nh2
I I -H2O ->•
CH2 c=s
xsz
о
II
/СХ
H2C NH
I I
H2C c=s
xsz
Характеристика Основного сырья
Дитиокарбаминат аммония (см. примечание 1).
Р-Хлорпропионовая кислота (см. примечание 2).
Натрий углекислый кристаллический, ч., ГОСТ 84—41.
Соляная кислота, ч., ГОСТ 3118—46.
Натрий углекислый кислый, ч., ГОСТ 4201—48.
Условия получения
Синтез S-тиокарбаминилтиогидракриловой кислоты. В
плоскодонную колбу емкостью 2 л помещают 217 г (2 Л4)
Р-хлорпропионовой кислоты, растворяют ее в 300 мл воды и
нейтрализуют раствор прибавлением к нему 286,2 г (1 М)
кристаллического карбоната натрия. Полученный раствор
Р-хлорпропионата натрия охлаждают льдом и при переме-
шивании прибавляют к нему кристаллический дитиокарбами-
нат аммония, приготовленный из 148 мл (2,46 М) сероугле-
рода. Смесь выдерживают в течение суток в холодильном
шкафу. Затем ее охлаждают до минус 5° и подкисляют мед-
ленным прибавлением 500 мл концентрированной соляной
кислоты при перемешивании. При этом выделяется 5-тиокар-
баминилтиогидракриловая кислота в виде желтоватых кри-
сталлов, которые отфильтровывают, тщательно промывают
водой (см. примечание 3) и сушат на воздухе.
Выход вещества равен 153 г, что составляет 46,3% от тео-
ретического, считая на 0-хлорпропионовую кислоту. Получен-
ное таким путем вещество достаточно чисто (см. примеча-
ние 4) для дальнейшего превращения в 2-тиотиазанон-4.
Получение 2-тиотиазанона-4. Помещают 33,0 г (0,2 Л4)
107
S-тиокарбаминилтйогидракриловой кислоты в круглодонную
колбу емкостью 100 мл и нагревают в масляной бане при
120°/7—10 мм в течение 8 часов. Полученный продукт расти-
рают в ступке с 50 мл насыщенного раствора двууглекислого
натрия (см. примечание 5), фильтруют, промывают тщатель-
но водой и высушивают на воздухе.
Получают 21,6 г 2-тиотиазанона-4, что составляет 73,4%
от теоретического, в виде желтых кристаллов; т. пл. 116—
119°. Для большинства дальнейших превращений препарат
достаточно чист; перекристаллизация из спирта приводит к
повышению температуры плавления до 120—12Г.
Примечания:
1. Дитиокарбаминат аммония приготовляют из сероуглерода и аммиа-
ка по методике [6].
2. ₽ -Хлорпропионовую кислоту получают из акрилонитрила и соля-
ной кислоты по методу [7].
3. Рекомендуется кристаллы растереть с 100 мл воды в ступке и за-
тем еще раз отфильтровать.
4. Продукт реакции содержит некоторое количество 2-тиотиазанона-4
и имеет поэтому заниженную температуру плавления (около 100°). Это
загрязнение не имеет значения, так как в следующем этапе синтеза
S-тиокарбаминплтиогидракриловую кислоту превращают именно в 2-тио-
тиазанон-4. Т. пл. чистой S-тиокарбаминнлтиогидракриловой кислоты со-
ставляет 125—126° (из воды).
5. При растирания наблюдается выделение СОа в результате взаимо-
действия двууглекислого натрия с неизмененной S-тиокарбаминилтиогидр-
акриловой кислотой; образовавшаяся натриевая соль указанной кислоты
переходит в раствор.
ЛИТЕРАТУРА
1. В. F. Goodrich. Брит. пат. 626486 (1949).
2. Т. Н. S h е 1|1 е у, N. J. S р о 11 s w о о d. Пат. США 2614095
(1951).
3. В. Holmberg. Вег., 47, 159 (1914).
4. А. П. Г р н щ у к, И. Р. Б а р и л я к. Ж. общ. химии, 33, 3972 (1963).
5. Т. L. Gresham, J. Е. J a n s е п, Е. W. Shaver. J. Amer.
Chem. Soc., 70, 1001 (1948).
6. Сб. «Синтез органических препаратов», вып. 4, М., ИЛ, 1953,
стр. 436.
7. А. П. Гр ищу к, С. Н. Баранов. Ж. прикл. химии, 33, 487
(1960).
Поступила в мае 1964 г. Львовский
медицинский институт
УДК 547.775.07
1-(.«-Т0ЛИЛ)-ПИРА30ЛИД0Н-3
Р. Б. ЖУРИН, О. Е. ШУЛЬГИНА
н2с---с=о
нА /NH
4N
Ci0HijN2O
М. в. 176,21
1-(м-Толил)-пиразолидон-3 может применяться в качест-
ве активатора гидрохинона в проявителях, предназначенных
для обработки черно-белых светочувствительных фотомате-
риалов [1].
Для получения 1-(ai-толил) -пиразолидона-3 были предло-
жены: взаимодействие л<-толилгидразина с амидом акрило-
вой кислоты [2], циклизация м-толилгидразида 0-оксипропио-
новой кислоты [3] и способ, ПОЗВОЛЯЮЩИЙ исходить ИЗ Л1-ТО-
луидина и метилакрилата [4].
Нами был использован последний метод, преимуществом
которого являются легкодоступные исходные вещества.
СХЕМА СИНТЕЗА 1-(л -ТОЛ ИЛ )-ПИРАЗОЛ ИДОНА-3
nh2 HNCH3CH3COOCH3
I I
/Ч
II I + ен2=снсоосн3и |
\^хсн8 х^хсн8
109
HNCH2CHaCOOCH3
ONNCH2CH2COOCH3
ifl + HNO2—►ff
\^чснв
ONNCH2CH2COOCH3 ~h2nnch2ch2cooch3
I I
H /4
II I —— II I
WXCH3 X//XCH3
H2C----c=o
I
H2C\ /NH
4 N7
Характеристика основного сырья
л-Толуидин, ч., ГОСТ 2119—25.
Метилакрилат, ТУ МХП 2471—53.
Нитрит натрия, ч., ГОСТ 6194—52.
Уксусная кислота ледяная, ГОСТ 61—51.
Цинковая пыль, 95%-ная.
Хлороформ, ч., ГОСТ 3160—51.
Сульфат натрия безводный, ч., ГОСТ 4166—48.
Условия получения
Получение метилового эфира ^-(м-метилфениламино)-
пропионовой кислоты. В двугорлую колбу емкостью 0,5 л,
снабженную мешалкой и обратным холодильником, вносят
107,1 г (1 М) л-толуидина, 120,8 г (1,4 М) метилакрилата и
50 мл ледяной уксусной кислоты. Реакционную смесь при
размешивании нагревают на кипящей водяной бане в тече-
ние 2 часов, затем охлаждают до комнатной температуры,
выливают в 200 мл воды и экстрагируют 100 мл хлороформа.
Экстракт промывают 50 мл воды, 2 раза 60 мл 5%-ного раст-
вора бикарбоната натрия и снова 50 мл воды и выдержи-
вают в течение ночи над 50 г безводного сульфата натрия.
Затем экстракт фильтруют для удаления сульфата, отго-
няют хлороформ на водяной бане и при разгонке остатка в
вакууме получают 130 г метилового эфира р-(л-метилфенил-
110
амино)-пропионовой кислоты, что составляет 67% от теоре-
тического. По внешнему виду это бесцветная подвижная жид-
кость с т. кип. 154—16072 .иж; л* — 1,5382 (см. примеча-
ние 1). По данным литературы, вещество имеет т. кип. 167—
16878 жж —1,5382 [4].
Получение метилового эфира П-нитрозо-$-(м-метилфенил-
амино)-пропионовой кислоты. В четырехгорлую колбу с ме-
шалкой, термометром, капельной воронкой и трубкой для
отвода выделяющихся газов вносят 139,3 г (0,72 Л4) метило-
вого эфира р-(ж-метилфениламино)-пропионовой кислоты и
75 мл ледяной уксусной кислоты. К образовавшемуся раство-
ру при охлаждении льдом и перемешивании прибавляют по
каплям охлажденный раствор 49,8 г (0,72 Л4) нитрита натрия
в 72 мл воды. При этом температура реакционной смеси не
должна превышать 5°.
Массу выдерживают 1 час при комнатной температуре.
Выделившееся масло извлекают 100 мл хлороформа, вытяж-
ку промывают 50 мл воды, 2 раза 60 мл 5%-ного раствора
бикарбоната натрия и снова 100 мл воды, выдерживают над
50 г безводного сульфата натрия в течение ночи, фильтруют,
из раствора отгоняют в вакууме хлороформ (при этом темпе-
ратура в бане не должна превышать 55°) (см. примечание
2) и получают в остатке 151,2 г технического нитрозоэфира,
что составляет 94% от теоретического.
Получение 1 -(м-толил)-пиразолидона-З. В четырехгорлую
колбу емкостью 1,5 л с мешалкой, термометром, обратным
холодильником и загрузочной воронкой вносят 158,5 г
(0,7 М) технического метилового эфира М-нитрозо-0-(ж-ме-
тилфениламино) -пропионовой кислоты и 165 мл ледяной ук-
сусной кислоты. К полученному раствору при температуре
0—5° и энергичном размешивании небольшими порциями
прибавляют 97 г цинковой пыли, после чего смесь выдержи-
вают 16 часов при комнатной температуре. Затем прибавля-
ют 920 мл воды и кипятят 3 часа на солевой бане.
Избыток цинка удаляют фильтрованием горячей реакци-
онной массы. Фильтрат охлаждают, выпавший осадок от-
фильтровывают, промывают 200 мл воды, сушат на воздухе
и кристаллизуют из 240 мл четыреххлористого углерода.
Выход 1-(ж-толил)-пиразолидона-3 равен 54,1 г, что со-
ставляет 31% от теоретического, считая на исходный ж-толу-
Вдин; т. пл. 113—114°.
По литературным данным, вещество имеет т. пл. 114—
Н6° [4].
Примечания:
1. При перегонке кубового остатка можно получить 18—20 г вещест-
ва C15II21NO.1 с т. кип. 20076 льи; я»-1,5289.
Ill
Образование этого вещества является, по-видимому, результатом присое-
динения двух молекул метилакрилата к л-толуидину.
2. При температуре выше 55° может произойти бурное разложение
ннтрозоэфира,
ЛИТЕРАТУРА
1. Фр. пат. 1106813; Zbl., 37, 10546 (1958).
2. Австрал. пат. 162025; Zbl., 44, 12580 (1958).
3. Амер. пат. 2743279; Zbl., Б, 1475 (1958).
4. Р. Б. Ж у р и н, О. Е. Лишенок, В. Л. А б р и т а л и н, Н. И. Си-
монова. Ж. общ. химии, 31, 8, 2758 (1961).
Поступила п декабре 1984 г. НИОПиК
УДК 547.831.2.07
8-( я-ТОЛУОЛСУЛ ЬФОН ИЛАМИ НО)ХИНАЛЬДИН
Б. В. ПАРУСНИКОВ, В. М. ДЗИОМКО, И. А. КРАСАВИН
CivHigNjOjS
М.в. 312,40
8- (n-Толуолсульфониламино) хинальдин получен нами с
90%-ным выходом при взаимодействии 8-аминохинальдина с
н-толуолсульфохлоридом в условиях, подобных применяв-
шимся для синтеза аналогичных производных 8-аминохино-
лина [1, 2]. Он кристаллизуется из этанола в бесцветных пла-
стинках с т. пл. 152,5—153° и дает с ионами цинка и кадмия
люминесцентную реакцию, отличающуюся высокой чувстви-
тельностью.
8-(Бензолсульфониламино) хинальдин (т. пл. 161 —161,5°)
и 8-(мезитиленсульфониламино) хинальдин (т. пл. 165,5—166°)
могут быть получены с 75%-ным выходом при взаимодейст-
вии 8-аминохинальдина с соответствующими сульфохлорида-
ми в таких же условиях.
8 Зак. 1350 113
СХЕМА СИНТЕЗА 8-(л-Т0ЛУ0ЛСУЛЬФ0Н ИЛАМИНО)ХИНАЛЬДИНА
I II I
nh2
SO2C1 / ч
I I II I
/\ x/w\
II I + C6HBN -> I СН3 +
NH
I I
CH3 so2
+ CBH6N-HC1
CH3
Условия получения
В коническую колбу емкостью 25—50 мл помещают 1,19 г
(0,0075 М) 8-аминохинальдина (см. примечание 1) и 5 мл су-
хого перегнанного пиридина (см. примечание 2). К образо-
вавшемуся раствору при охлаждении льдом и размешивании
добавляют за 5—10 минут раствор 1,57 г (0,0082 М) п-толу-
олсульфохлорида (см. примечание 3) в 5 мл пиридина. Вы-
держивают при комнатной температуре 1 час и столько же
времени на кипящей водяной бане, затем охлаждают до 5—
10° и выливают в 150 мл дистиллированной воды. Осадок
отсасывают, тщательно промывают на фильтре водой до ис-
чезновения запаха пиридина и высушивают в вакуум-экси-
каторе над едким натром.
Выход сырого продукта равен 2—2,1 г, что составляет
86—90% от теоретического. После перекристаллизации из
70—80 мл этанола получают 1,6—1,7 г бесцветных пластинок
с т. пл. 152—152,5° (см. примечания 4 и 5).
Примечания:
1. Применялся 8-аминохинальдин с т. пл. 54—55°, очищенный путем
перегонки с водяным паром. Он был получен с выходом 73% при восста-
новлении 8-нитрохинальдииа железным порошком в 50%-иой уксусной
кислоте (методика восстановления аналогична применявшейся для синтеза
5-хлор-8-аминохинолина [3]).
2. Чтобы получить реагент, пригодный для люминесцентного анализа,
следует принять меры против загрязнения вещества следами ионов тя-
желых металлов. Необходимо пользоваться перегнанными растворителями
и дистиллированной водой.
3. Применялся п-толуолсульфохлорид с т. пл. 68—69°, перед употреб-
лением перегнанный в вакууме.
4. Чистый образец с т. пл. 152,5—153° был приготовлен повторной
перекристаллизацией из этанола и проанализирован.
Найдено, %-. С—65,04; 65,43; Н—5,12; 4,94; N—8,69;
9,00; S—10,17; 10,27.
C17H16N2OaS, Вычислено, %: С—65,36; Н—5,16; N—8,97; S—10,27.
114
5. При взаимодействии 8-аминохинальдина с бензолсульфохлоридом
пли мезитиленсульфохлоридом в аналогичных условиях были получены
соответствующие производные:
8-(Бензолсульфониламиио) хинальдин (выход 75%)—бесцветные иглы,
г. пл. 161—161,5° (из этанола).
Найдено, %: С-64,66; 64,39; Н-4,90; 4,88; N-9,11;
9,29; S—10,52; 10,60.
Ci6H14N2O2S. Вычислено, %: С-64,41; Н—4,73; N-9,39; S—10,74.
8- (Мезитиленсульфониламино) хинальдин (выход 75%) —бесцветные
иглы, т. пл. 165,5—166° (из этанола).
Найдено, %: С-67,19; 67,05; Н—6,04; 6,08; N—8,13;
7,86; S-9,01; 9,28-
С1эН2оМ,0,8. Вычислено, %: С-67,03; Н—5,92; N—8,23; S-9,42.
ЛИТЕРАТУРА
1. В. М. Дзиомко, И. А. Красавин. Методы получения хими-
ческих реактивов н препаратов, вып. 4—5, М., ИРЕА, 1962, стр. 67.
2. И. А. Красавин, В. М. Дзиомко. Там же, стр. 69.
3. В. М. Дзиомко, И. А. Красавин. Методы получения хими-
ческих реактивов и препаратов, вып. 2, М., 14РЕА, 1961, стр. 98.
Поступила в январе 1966 г. ИРЕА
8*
УДК 547.792.7.07
1,2,4-ТРИАЗОЛ ИНТИОН-5
Меркаптотрназол
И. Л. ШЕГАЛ, И. Я. ПОСТОВСКИЙ
НС—NH
c2h3n3s
М.в. 101,13
1,2,4-Триазолинтион-5 был впервые получен в две стадии
исходя из тиосемикарбазида [1]. Недостатком этого способа
является малый выход на второй стадии; кроме того, реак-
ция плохо поддается контролю и конечный продукт реакции,
как правило, загрязнен побочными продуктами.
Этот метод является единственным препаративным мето-
дом получения незамещенного 1,2,4-триазолинтиона-5, в то
время как производные этого соединения могут быть получе-
ны многими путями. Один из них заключается во взаимодей-
ствии 4-замещенного тиосемикарбазида с этиловым эфиром
карбоновой кислоты (в частности, муравьиной) в присутствии
метилата натрия [2].
Нами найдено, что этот метод с небольшими изменениями
может быть распространен на незамещенный тиосемикарба-
зид. 1,2,4-Триазолинтион-5 получается при многочасовом ки-
пячении тиосемикарбазида с этиловым эфиром муравьиной
кислоты в присутствии эквимолекулярного количества мети-
лата натрия в среде безводного метанола. Преимуществом
этого способа является возможность работы с большими за-
грузками и чистота получаемого продукта.
116
Схема синтеза 1,2,4-триазолинт йона-5
HaN—С-NH-NH3+C2H5OCH+CH3ONa
НС—N HC-NH
II II II I
-> N C-SNa 4-НС1 -> N C=S
NH NH
Характеристика основного сырья
Тиосемикарбазид, ч., ТУ МГ УХП 279—59.
Этиловый эфир муравьиной кислоты, ч., ВТУ РУ 1285—56.
Натрий металлический.
Метанол, ГОСТ 6995—54.
Условия получения
К 200 мл метанола (см. примечание 1), помещенного в
трехгорлую колбу емкостью 500 мл, снабженную обратным
холодильником с хлоркальциевой трубкой и мешалкой с за-
твором, при перемешивании порциями добавляют 13,8 г ме-
таллического натрия (см. примечание 2). После того как весь
натрий прореагирует, в колбу загружают 54,6 г тиосемикар-
базида и 48 мл предварительно высушенного над хлористым
кальцием этилового эфира муравьиной кислоты. Образовав-
шийся прозрачный раствор кипятят 25 часов (см. примеча-
ние 3), причем по истечении первых 8 часов в реакционную
массу добавляют 24 мл этилового эфира муравьиной кисло-
ты и еще через 8 часов— 12 мл этого эфира. Реакционную
массу в виде прозрачного раствора переносят в колбу Кляй-
зена и отгоняют под 'вакуумом метанол до образования твер-
дой массы, которую растворяют в 100 мл воды и переносят
в стакан емкостью 250 мл; колбу Кляйзена промывают 20 мл
воды и промывные воды присоединяют к предыдущей порции
(см. примечание 4). Раствор кипятят с углем, фильтруют, ох-
лаждают и нейтрализуют концентрированной соляной кисло-
той до кислой реакции на лакмус. Если осадок'после охлаж-
дения нейтрализованного раствора не выпадает, то раствор
следует упарить до половинного объема и выпавший после
охлаждения осадок отфильтровать.
Выход 1,2,4-триазолинтиона-5 равен 33,2 г, что составляет
65% от теоретического; т. пл. 215—216°.
1,2,4-Триазолинтион-5 хорошо растворим в воде и спирте,
нерастворим в эфире, бензоле и хлороформе.
117
Примечания:
1. Для реакции метанол следует предварительно обезводить обработ-
кой магнием.
2. Металлический натрий следует добавлять небольшими порциями:
слишком быстрое добавление может привести к энергичному кипению и
даже выбросу через холодильник.
3. Реакцию ке обязательно вести непрерывно. При остановке реак-
ции на ночь не следует убирать хлоркальциевую трубку.
4. При уменьшении продолжительности реакции выходы несколько
уменьшаются и остается часть иепрореагировавшего тиосемикарбазида,
который не растворяется в воде и может быть отфильтрован.
ЛИТЕРАТУРА
1. М. Freund. Вег., 29, 2483 (1896).
2. М. Р ess on, G. Polmanss, S. Dupin. C. r., 248, 1677 (1959).
Поступила в ноябре 1964 г.
Уральский
политехнический институт
УДК 547.874.07
1,2,4 ТРИАЗОЛОН-З-КАРБОНОВАЯ-5 КИСЛОТА
Г. И. ЧИПЕН
О=С—NH
Hl/ 'с-соон
C3H3N,O, М.в. 129,07
1,2,4-Триазолон-3-карбоновую-5 кислоту используют для
синтеза 1,2,4-триазолона-З путем термического декарбоксили-
рования.
Наиболее удобным методом получения триазолонкарбоно-
вой кислоты является разложение 3-диазо-1,2,4-триазолкарбо-
новой-5 кислоты в кислой среде [1].
В настоящей работе дана новая методика получения 3-ди-
азо-1,2,4-триазолкарбоновой-5 кислоты. Для разложения ди-
азотриазолкарбоновой кислоты использована 25%-ная сер-
ная кислота.
1,2,4-Триазолон-3-карбоновую-5 кислоту можно также по-
лучить из диэтилового эфира ди-(а,р-дикарбэтоксигидрази-
но)-малоновой кислоты [2] или при щелочном гидролизе ди-
семикарбазида щавелевой кислоты [3].
СХЕМА СИНТЕЗА 1,2,4-ТРИАЗОЛОН-З-КАРБОНОВОЙ КИСЛОТЫ
HaN—С—N
// X
N С-СООН+-NaNO2+-HCl
\н
119
HO—N=N—C—N
/ Ч
N C-COOH+H2O+NaCl
^NH
HO-N=N-C-N
4
N C-COOH
25%H2SO4
--------->
О—C—NH
HN C-COOH + Na
Характеристика основного сырья
3-Амино-1,2,4-триазолкарбоновая-5 кислота (см. статью
на стр. 9).
Натрий азотистокислый, ч., ГОСТ 4197—48.
Серная кислота, ч., ГОСТ 4204—48.
Условия получения
Получение 3-диазо-1,2,4-триазолкарбоновой-5 кислоты. В
двухлитровой колбе приготавливают раствор из 180 мл
(^2,1 М) концентрированной соляной кислоты в 300 мл во-
ды, прибавляют 68,5 г (0,5 М) З-амино-1,2,4-триазолкарбо-
новой-5 кислоты и нагревают до растворения. После охлаж-
дения до комнатной температуры к раствору прибавляют
500 г льда (см. примечание 1) и в течение 2—3 минут при
механическом перемешивании приливают раствор 40 г азо-
тистокислого натрия (0,58 Л4) в 100 мл воды. Раствор про-
должают перемешивать еще полчаса, поддерживая темпера-
туру в пределах 0—4° (наружное охлаждение ледяной во-
дой). Выпавшую диазотриазолкарбоиовую кислоту отфиль-
тровывают на воронке Бюхнера и, не высушивая (см. приме-
чание 2), используют для следующего синтеза.
Получение 1,2,4-триазолон-3-карбоновой-5 кислоты. Диазо-
триазолкарбоновую кислоту переносят в двухлитровую кол-
бу с 640 мл 25%-ной серной кислоты и медленно нагревают.
При температуре 40—50° начинается разложение диазосоеди-
нения с выделением азота. Реакция дальше протекает экзо-
термически (см. примечание 3). Для полного завершения ре-
акции раствор нагревают до 95—100° и немедленно охлажда-
ют (см. примечание 4). Выпавшую 1,2,4-триазолон-З-карбоно-
120
вую-§ кислоту фильтруют, промывают водой и сушат на воз-
духе. Выход равен 41,0 г, что составляет 56%, считая на 3-
амино-1,2,4-триазолкарбонрвую-5 кислоту; т. пл. 196° с разло-
жением. После перекристаллизации из воды (1 г кислоты из
40 мл воды) т. пл. продукта 205° с разложением.
Выход вещества при перекристаллизации составляет 68%
(см. примечание 5).
Примечания:
1. После прибавления, льда часть аминотриазолкарбоновой кислоты
выделяется в виде мелкозернистого осадка.
2. Сухая диазотриазолкарбоновая кислота взрывоопасна [1].
3. В некоторых случаях разложение диазотриазолкарбоновой кислоты
протекает бурно. Ход реакции можно контролировать, погружая колбу в
сосуд с ледяной водой.
4. При температуре около 100° тпиазолонкарбоиовая кислота разла-
1ается с выделением углекислого газа.
3. Продукт представляет собой кристаллическое соединение, содержа-
щее молекулу кристаллизационной воды, которую ои теряет при 120°.
По литературным данным, 1,2,4-триазолон-3-карбоновая-5
кислота плавится с разложением при 205° [1], при 218° [2] и
при 210° [3].
ЛИТЕРАТУРА
1. W. М a n с h о t, R. Noll. Liebigs Ann. Chem., 343, 1 (1905).
2. R. Stolle. Ber., 57, 1558 (1924).
3. H. Q e h 1 e n. Liebigs Ann. Chem., 577, 237 (1952).
Поступила в декабре 1964 r.
Институт
органического синтеза
АН Латв. ССР
УДК 547.823.07
1-ФЕНИЛ-3,6-ДИМЕТИЛ-ПИРАЗОЛО-(4,5с)-ПИРИДОН-4
Н. С. ВУЛЬФСОН, Г. М. СУХОТИНА
Н8Се—N— N
I I!
сн3
C14H13N3O
М.в. 239,28
Синтез 1-фенил-3,6-диметил-пнразоло-(4,5с)-пиридона-4
осуществлен нами по аналогии с ранее описанным методом
циклизации фенилгидразонов 4-окси-З-ацил-кумаринов и
4-окси-З-ацил-карбостирилов [1].
СХЕМА СИНТЕЗА 1-ФЕНИЛ-3,6-ДИМЕТИЛ-ПИРАЗОЛО-(4,5с)-
ПИРИДОНА-4
свн8
H-N-N
HsCe—N—N
НО II
/w\
II I СН
СНз
122
Характеристика основного сырья
Фенилгидразон 4-окси-3-ацетил-6-метил-пиридона-2, т. пл.
224—225°.
Полихлориды бензола, т. кип. 170—175°
«-Толуолсульфокислота, ч., ВТУ МХП ОРУ 73—56.
Петролейный эфир синтетический, ТУ МХП 600—56.
Условия получения
Кипятят 3 г (0,0117 М) фенилгидразона 4-окси-З-ацетил-
6-метил-пиридона-2 (см. примечание), 30 мл полихлоридов
бензола и около 1 г n-толуолсульфокислоты в колбе с пря-
мым холодильником до прекращения отгонки выделяющейся
в результате реакции воды; реакционную массу охлаждают,
выпавший осадок фильтруют и промывают иетролейным эфи-
ром.
Выход продукта равен 1,83 г, что составляет 61,5% °т тео-
ретического.
После кристаллизации из спирта т. пл. 233—234°.
Найдено, %: С-70,41; 70,25; Н—4,91; 4,88; N-17.78.
CMH13N3O. Вычислено, %: С-70,27; Н—5,48; N-17,56.
Полученные аналогичным путем замещенные 1-фенил-6<
метил-пиразоло-(4,5с)-пиридона-4 и его N-метил- и N'-фенил-
производных, отвечающие нижеуказанной общей формуле,
приведены в таблице:
Н6Се—N — N
II
C-R'
R R' Выход, % Т. пл., °C
Н н-С3Н7 40,5 198-199
Н н-С4Н9 59,0 165-167
н и.зо-С4Н9 31,0 201-202
н 4-СбНц 47,5 136-137
СН3 СН, 53,0 157—157,5
С6н5 сн3 48,0 160-161
123
Примечание.
Феннлгидразон 4;окси-3-ацетнл-6-метнл-пирндона-2 получен путем кй;
пячения в спирте эквимолекулярных количеств кетона и фенилгидразнна.
ЛИТЕРАТУРА
1. Н. С. Вульфсон, Р. Б. Ж у р и н. Ж. общ. химии, 31, 3381 (1961);
32, 991 (1962).
Поступила в декабре 1964 г. Институт химии природных соединений АН СССР, НИОПиК
УДК 547.841.07
4-ФЕН ИЛ-лг-ДИОКСЛН
А. В. ВО Г АТСКИ К, Г. Л. КАМАЛОВ
/СНз-О
сн2 сна
хсн -о х
I
СвН6
CjoHiaOg
М. в. 164,20
Одним из наиболее распространенных способов получения
-И-диоксанов является реакция Принса, заключающаяся во
взаимодействии олефинов с формальдегидом (либо другими
альдегидами) в присутствии кислотного катализатора [1, 2].
Нами исследован синтез 4-фенил-.и-диоксана из стирола и
формальдегида с применением в качестве катализатора ио-
нообменной смолы КУ-1- За основу была взята методика
Шрайнера и Рюби, где в качестве катализатора используется
серная кислота [3, 4].
Варьированием температуры синтеза, а также количества
катализатора на 1 моль исходного стирола нами усовершен-
ствована эта методика, что позволило проводить синтез 4-фе-
нил-л-диоксана в более мягких температурных условиях с не-
меньшими выходами целевого продукта. Кроме того, обработ-
ка реакционной смеси после проведения синтеза значительно
упростилась.
СХЕМА СИНТЕЗА 4-ФЕН ИЛ-л-ДИОКСАНА
C6Hs-GH=CHa+CH,0
КУ-1
125
CH2-0
—> сн2 сн2
ХСН - с/
С6Н5
Характеристика основного сырья
Формалин, 37%-ный.
Стирол, перегнанный под вакуумом (см. примечание 1).
Ионит КУ-1 в Н-форме (см. примечание 2).
Бензол перегнанный, т. кип. 80,5—81°.
Условия получения
В колбу емкостью 1 л, снабженную механической мешал-
кой и обратным холодильником, помещают 337,5 г (4,15 Л1)
37%-ного формалина, 156 г (1,5 Л1) стирола и 45 а ионита
КУ-1.
Смесь нагревают на масляной бане или на колбонагрева-
теле при температуре 70—75° в течение 7 часов, затем охлаж-
дают и катализатор отделяют фильтрованием. Оставшиеся на
фильтре зерна смолы промывают 50 мл бензола, который
соединяют с полученным ранее фильтратом (см. примеча-
ние 3). К раствору прибавляют еще 200 мл бензола, при этом
раствор расслаивается. Водный слой еще два раза экстраги-
руется по 200 мл бензола, бензольные вытяжки соединяют
вместе, бензол отгоняют и остаток фракционируют в вакууме.
Сначала при давлении 2 мм и при температуре до 94° от-
деляют головной погон, затем собирают главную фракцию
при 96—9872 мм (см. примечание 4).
Выход 4-фенил-л1-диоксана равен 192—195 г, что состав-
ляет 78—79% от теоретического.
Примечания:
1. Стирол отмывается от гидрохинона в делительной воронке 10—
15%-ным раствором едкого кали или едкого натра до тех пор, пока про-
мывные воды не будут прозрачными. После этого его промывают водой
от щелочи (по фенолфталеину) и сушат над прокаленным хлористым
кальцием в холодильном шкафу в течение 1—2 суток. Высушенный сти-
рол перегоняется в вакууме; т. кип. 38—4071 мм.
2. Продажная смола переводится в Н-форму по обычной методике [5].
3. После промывки бензолом смола 2—3 раза промывается 50 мл аце-
тона, высушивается на воздухе и может быть использована в повторных
циклах синтеза без заметной потери каталитической активности.
4. Более подробные примечания можно найти у Шрайнера и Рюби [4].
126
ЛИТЕРАТУРА
1. Гетероциклические соединения, т. 6, стр. 30—46.
2. А. С е р р е й. Справочник по органическим реакциям, М., Госхнм-
издат, 1962.
3. Н. Г. Полянский. Успехи химии, т. 31, 1962, стр. 1046.
4. Сб. «Синтез органических препаратов», вып. 5, 1954, стр. 76.
5. К. И. С а л д а д з е, А. Б. Пешков, В. С. Титов. Ионообменные
высокомолекулярные смолы, М., Госхимиздат, 1960.
Поступила в декабре 1964 г.
Одесский государственный
университет
УДК 547.564.6.07
2- [ 1 "-ФЕНИ Л-3"-МЕТИ Л П И РА 30 ЛОН-5"-(4"- АЗО-2 )-
ФЕНИЛАЗОКСИ]-4-МЕТИЛФЕНОЛ
Азо-азокси ФМП
В. М. ДЗИОМКО, к. А. ДУНАЕВСКАЯ, Т. И. ГЕРШЕНГОРН
N=N—С—С—СН3
II II
HOC N
N—С6Н6
CssHjoNgOg
/он
N—N—
Х=Х 1
О 'СН3
М. в. 428,46
Азо-азокси ФМП представляет значительный интерес для
экстракции стронция [1]. Нами реактив был получен впервые.
СХЕМА СИНТЕЗА АЗО-АЗОКСИ ФМП
128
NaNO3+2HCl
^/xn2+ci-
ОН
co
N=N—С—С—CH3
/ II II
z^~ 4 HOC N
X=/ \ Xn-c6h5
Характеристика основного сырья
о-Фенилендиамин основание, ч., ТУ МХП 2639—51.
Фталевый ангидрид, ч., ГОСТ 5869—51.
Нитрит натрия, х. ч., ГОСТ 4197—48.
Соляная кислота, ч.д.а., ГОСТ 3118—46, уд. в. 1,18.
9 Зак. 1350 1 29
Метанол, ч., ГОСТ 6995—54.
n-Крезол, ч., ВТУ МГ УХП 402—59.
Углекислый натрий, безводный, ч., ГОСТ 4530—48.
Уксусная кислота, х. ч., ГОСТ 61—51.
Этанол, ч., ГОСТ 5962—51.
Перекись водорода, медиц., ГОСТ 5815—-52.
Уксусный ангидрид, ч.д.а., ГОСТ 5815—52.
Гидразингидрат, ч., ГОСТ 5832—51.
Мочевина, ч., ГОСТ 6691—53.
1-Фенил-3-метил-5-пиразолон, ч., ВТУ РУ 466—51.
Едкий натр, ч.д.а., ГОСТ 4328—48.
Изоамиловый спирт, ч., ГОСТ 5830—51.
Четыреххлористый углерод, ч., ГОСТ 5827—51.
Условия получения
Синтез солянокислой соли 2-амино-1 -фталоиламинобен-
зола [2].
В трехгорлую круглодонную колбу емкостью 3 л, снаб-
женную мешалкой, обратным холодильником и термометром,
загружают 108,0 г (1 44) о-фенилендиамина, 148 г (1 44) фта-
левого ангидрида и добавляют туда 1000 мл метилового
спирта (см. примечание 1). Содержимое колбы при переме-
шивании медленно нагревают на водяной бане до 37° в тече-
ние^-30 минут. Затем добавляют 180 мл концентрированной
соляной кислоты, при этом образуется суспензия и темпера-
тура поднимается до 42—50° (см. примечание 2). Реакцион-
ную массу далее нагревают 15 минут до кипения и кипятят
30 минут. Затем охлаждают до комнатной температуры и
фильтруют. На фильтре осадок промывают 200 мл метилово-
го спирта в 2 приема и сушат па воздухе в темном месте до
постоянного веса.
Выход равен 150—156 г, что составляет 53,9—56,2% от
теоретического; т. пл. 227—230°.
Получение 2'-фталоиламино-6-окси-3-метилазобензола [3].
Смешивают в стакане 48 г (0,174 44) солянокислой соли
2-амиио-1-фталоиламинобензола с 140 мл концентрированной
соляной кислоты. Смесь перемешивают 15—20 минут без ох-
лаждения, затем в баню загружают ледяную воду, а в стакан
прибавляют 500 г льда и раствор 15,2 г (0,22 44) нитрита нат-
рия в 200 мл воды (см. примечание 3). Выдержка при диазо-
тировании 1,5 часа.
Для сочетания диазораствора с n-крезолом в трехгорлую
круглодонную колбу емкостью на 3 л, снабженную мешал-
кой, термометром, обратным холодильником и капельной во-
ронкой, загружают 23,6 г (0,22 М) «-крезола, 1400 мл мети-
лового спирта (см. примечание 1) и затем профильтрованный
диазораствор и раствор безводного углекислого натрия
(270 г в 1 л дистиллированной воды) одновременно посте-
130
пенно прибавляют к спиртовому раствору n-крезола (см. при-
мечание 4). После этого реакционную массу выдерживают
: —1,5 часа при 0°, подкисляют соляной кислотой (•'-350 мл)
Ю кислой реакции по конго красному. Выделившийся краси-
тель отфильтровывают, сушат на воздухе до постоянного йе-
са и перекристаллизовывают из уксусной кислоты (118—
125 жл).
Выход равен 38—39,7 г, что составляет 81,0—82,6% на
продукт, взятый на перекристаллизацию и 60,8% на соляно-
кислую соль 2-амино-1-фталоиламинобензола. По внешнему
виду, это кристаллы красного цвета; т. пл. 160—162°.
Получение 2'-фталоиламино-6-окси-3-метилазоксибензола.
В круглодонную колбу емкостью Зле обратным холодильни-
ком (на корковой пробке или шлифе) загружают 30 г 2'-фта-
лоиламино-6-окси-З-метилазобензола, 800 мл ледяной уксус-
ной кислоты и 400 мл уксусного ангидрида. Смесь нагревают
до температуры 90° в бане до полного растворения краси-
теля. Затем раствор охлаждают до комнатной температуры и
прибавляют 135 мл 29—30%-ной перекиси водорода (см.
примечание 5). Смесь нагревают 6 часов при температуре в
бане 70—80°, затем раствор охлаждают до комнатной темпе-
ратуры, прибавляют вторую порцию перекиси водорода
(135 мл) и продолжают нагревание еще 10—15 часов. Окис-
ление считается законченным, когда раствор из темно-крас-
ного становится оранжево-желтым. В противном случае на-
гревание продолжают. Окисленный раствор фильтруют и вы-
ливают в ледяную воду (4 л) (см. примечание 6). Выделив-
шийся осадок отфильтровывают, промывают дистиллирован-
ной водой (•'-1 л) до исчезновения кислой реакции по конго
красному в фильтрате и сушат на воздухе в темном месте до
постоянного веса. По внешнему виду это порошок желтого
цвета; т. пл. 135—140°.
Выход равен 10,8—13,0 г, что составляет 35,0—41,9% от
теоретического.
Получение 2-[1"-фенил-3"-метилпиразолон-5"-(4"-азо-2')-
фенилазокси]-4-метилфенола. В круглодонную колбу ем-
костью 0,5 л с обратным холодильником загружают 3 г
2'-фталоиламино-6-окси-3-метилазоксибензола, 180 мл этило-
вого спирта и 1 мл гидразингидрата. Смесь кипятят 6 часов
при температуре в бане 85—90°, затем охлаждают до ком-
натной температуры, добавляют 180 мл концентрированной
соляной кислоты и кипячение продолжают еще 6 часов при
температуре в бане 100°. Затем раствор охлаждают до ком-
натной температуры, фильтруют, фильтрат разбавляют
180 мл воды. К полученному спирто-водному фильтрату при-
бавляют при перемешивании избыток нитрита натрия 3,5—4 г
(см. примечание 7). Температуру поддерживают в интервале
0—5°. Дают выдержку 30 минут и затем прибавляют в два
9*
131
приема ~3 г мочевины в течение 30—40 минут. Далее прово-
пят сочетание с 1-фенил-3-метил-5-пиразолоном. Для этого в
стеклянный стакан емкостью 2 л с мешалкой и ледяной ба-
ней загружают 1,44 г 1-фенил-3-метил-5-пиразолона, прибав-
ляют 10 мл этилового спирта, раствор углекислого натрия
(50 г углекислого натрия в 150 мл воды) и 180 мл 20%-ного
раствора едкого натра. В раствор постепенно при перемеши-
вании приливают диазораствор. Температуру поддерживают
в интервале 0—3° Выдержку дают 3—4 часа. Затем раствор
подкисляют соляной кислотой до кислой реакции по конго.
Выделившийся осадок отфильтровывают, промывают водой
(-~ 50 мл) и последовательно перекристаллизовывают из изо-
амилового спирта (200 мл) и четыреххлористого углерода
(150 мл}.
Выход продукта равен 0,26—0,40 г, что составляет 7,6—
11,7% от теоретического, считая на 2'-фталонламино-6-окси-3-
метилазоксибснзол; т. пл. 218—221° (в пределах Г).
Примечания:
1. Вместо метилового спирта можно работать с этиловым спиртом.
2. Соляную кислоту необходимо добавлять постепенно в четыре прие-
ма (в течение 15 минут), иначе температура поднимается выше 42—50°.
3. Нитрит натрия прибавляют медленно при перемешивании в тече-
ние ~ 1—1.5 часа, следя за ходом реакции по йодкрахмальной бумажке
и бумажке конго, температуру поддерживают в интервале 0—3°.
4. Прибавление продолжается 1,5 часа.
5. Содержание перекиси водорода определяется прямым титрованием
раствором перманганата по ГОСТ 177—55.
6. Выдержка 30 минут.
7. За ходом реакции следят по йодкрахмальной бумажке.
ЛИТЕРАТУРА
1. Ф. П. Горбенко. В. М. Дзиомко, А. А. Надежда, К. А. Ду-
наевская. Авт. свид. 171658, Бюлл. изобр., № 11 (1965).
2. J. Arie nt, J. М а г h a n. Coll. Czech, chem. Comm., 26,98 (1961).
3. В. M. Дзиомко, К. А. Дунаевская. Ж. общ. химии, 30, 628
(1960).
Поступила в августе 1965 г.
ПРЕД
УДК 547.772.2.07
1-ФЕНИЛ-ПИРАЗОЛИДОН-З
Фенидон
Н. И. СИМОНОВА, В. В. ПИГУЛЕВСКИИ, И. А. ЗАХАРОВА,
Н. В. ХРОМОВ-БОРИСОВ, С. И. ГАФТ
СН2—С=О
СН2 NH
C9H10N2O М.в. 162,19
1-Фенил-пиразолидон-З находит широкое применение в
процессе фотографического проявления. При введении фени-
дона в проявляющий раствор, содержащий гидрохинон, рез-
ко увеличивается активность последнего как проявляющего
вещества.
Из числа опубликованных в литературе методов синтеза
фенидона наиболее интересными являются: конденсация
фенилгидразина с p-галогенопропионовыми кислотами или
их эфирами [1]; взаимодействие фенилгидразина с эфиром
акриловой кислоты [2] или ее амидом [3]; циклизация эфира
Р-(а-фенилгидразино) -пропионовой кислоты [4] или фенилгид-
разида p-оксипропионовой кислоты [5, 6]; кислотный гидро-
лиз 3-амино-пирозолинов [7]; взаимодействие фенилгидразина
с пропиолактоном [8] или акрилонитрилом [9]; конденсация
Р-фенилгидразинкарбоновой кислоты с метиловым эфиром
акриловой кислоты [10, 11].
В нашей прописи приведен синтез 1-феннл-пиразолидона-З
из анилина и метилового эфира акриловой кислоты. Метод
предложен Ленинградским институтом киноинженеров
133
(ЛИКИ) [12, 13] и проверен в Институте экспериментальной
медицины АМН СССР.
СХЕМА СИНТЕЗА 1-ФЕНИЛ-ПИРАЗОЛИДОНА-З
,0
свнБ-nh2+ch2=ch-с<
хосн3
CeH6-NH-СН2-СНг-cf
ХОСН3
//°
CeH6-NH-CH2CH2-Cf
OCH3+NaNO2H-CH3COOH ->
-> C6H6-N-CH2CH2Cf +CH3COONa+H2O
I хосн3
NO
CeH6-N— CH2CHS-C^° 4-2H2 Zn4-CH,COOH
I OCH3
NO
-> C6H6-N—CH2CHa-cf +H2o
I XOCH3
nh2
CeH6-N-CH2CH2-cf° нагрев._>
I XOCH3
nh2
ch2-c=o
-> I I +CH3OH
CH2 NH
XNZ
zS
\//
Характеристика основного сырья
Анилин, ч., ГОСТ 5819—51, свежеперегнанный.
Метиловый эфир акриловой кислоты (метилакрилат), тех-
нический, свежеперегнанный,«д1,6 — 1,4035.
134
Уксусная кислота, х. ч., 98%-ная (либо ледяная),
ГОСТ 61—51.
Нитрит натрия, ч., ГОСТ 4197—48.
Цинковая пыль, ч.
Условия получения
Синтез метилового эфира ft-анилинопропионовой кислоты.
В литровой колбе, снабженной обратным холодильником, сме-
шивают 45,5 мл (46,5 г; 0,5 Л4) свежеперегнанного анилина,
46 мл (44,8 г; 5%-ный избыток против рассчитанного) перег-
нанного и стабилизированного гидрохиноном метилового эфи-
ра акриловой кислоты и 25 мл 98 %-ной уксусной кислоты
(см. примечание 1). Колбу погружают в водяную баню, ба-
ню доводят до кипения и реакционную массу выдерживают
в кипящей воде 3 часа. Смесь охлаждают и переносят в де-
лительную воронку, в которую заранее наливают 100 мл во-
ды; остатки реакционной массы смывают небольшими пор-
циями эфира (всего 65 мл) и присоединяют к основной мас-
се. Содержимое делительной воронки взбалтывают и отделя-
ют водный слой; эфирный слой, окрашенный в красный цвет,
дважды промывают водой по 50 мл за прием (см. примеча-
ние 2). Красный эфирный слой высушивают прокаленным
сульфатом натрия. После отгонки эфира получают 78,8—
82,63 г метилового эфира (З-анилинопропионовой кислоты в
виде красного масла, которое прн встряхивании иногда за-
кристаллизовывается.
Нитрозирование метилового эфира $-анилинопропионовой
кислоты. Растворяют в 50 мл 98%-ной уксусной кислоты
78,8—-82,63 г указанного эфира, смесь охлаждают льдом до
0° и при энергичном перемешивании прикапывают раствор
44,5—46 г нитрита натрия в 70—75 мл воды (см. примеча-
ние 3). Скорость прибавления нитрита натрия регулируют так,
чтобы температура реакционной массы поддерживалсь в пре-
делах 3—7°. По прибавлении раствора нитрита натрия ох-
лаждение снимают и реакционную смесь перемешивают в
течение часа при комнатной температуре (см. примечание 4).
Продукт реакции переносят в делительную воронку и экстра-
гируют 50 мл эфира, экстракт промывают 50 мл воды и вы-
сушивают над прокаленным сульфатом натрия. После отгон-
ки эфира получают 86—88,2 г нитрозоэфнра в виде темно-
красного масла.
Получение 1 -фенил-пиразолидона-3. В двухлитровой трех-
горлой колбе, снабженной энергичной мешалкой, термомет-
ром, холодильником, растворяют 86—88,2 г метилового эфи-
ра N-нитрозо-р-анилинопропионовой кислоты в 140 мл 98 % -
ной уксусной кислоты и при энергичном перемешивании в
течение 2—2,5 часа небольшими порциями прибавляют 100 г
135
(в пересчете на сухую) активированной цинковой пыли (см.
примечание 5). Сильного разогревания, как правило, не наб-
людается, и реакционную массу лишь время от времениохлд^
ждают водопроводной водой. После прибавления цинковой пы-
ли реакционную смесь в течение 3—4 часов энергично пере-
мешивают при комнатной температуре и оставляют на ночь.
На другой день добавляют 800 мл воды и при энергичном пе-
ремешивании кипятят в течение трех часов, после чего избы-
ток цинка удаляют фильтрованием горячей реакционной сме-
си. Фенидон выделяется в виде капель темноокрашенного
масла. При охлаждении и трении стеклянной палочкой быст-
ро наступает кристаллизация. Смесь оставляют стоять до сле-
дующего дня (лучше при температуре 3—5°), отфильтровыва-
ют выпавший осадок, который на фильтре промывают 50—
70 мл холодной воды. Маточник и промывную воду сохраня-
ют. Осадок высушивают на воздухе. Вес полученного про-
дукта колеблется в пределах 40,0—30,6 г, т. пл. нечеткая:
108—114°. Осадок кристаллизуют из спирта с расчетом 5 мл
ректификата на 1 г фенидона. Кристаллизация наступает при
трении о стенки сосуда. Осадок отфильтровывают, спиртовой
фильтрат отделяют и используют в дальнейшей работе. Оса-
док фенидона промывают 2—3 раза небольшими порциями
бензола (по 20—30 мл за прием). Вес полученного продукта
составляет 15,2—17,3 г с т. пл. 121 —122°.
По литературным данным, т. пл. продукта 121° [12, 13].
Маточник и промывную воду, оставшиеся после отделения
осадка фенидона, обрабатывают 5 раз хлороформом по
100 мл за прием. Хлороформные вытяжки высушивают про-
каленным сульфатом натрия; после отгонки хлороформа
остается 12,75—20,8 г желтого масла, которое при даче за-
травки фенидона и трении становится медоподобным, полной
кристаллизации не наступает (см. примечание 6). Этот про-
дукт растворяют в спиртовом маточнике, оставшемся после
кристаллизации основного осадка, добавляют уголь, кипятят
и фильтруют. Спиртовой фильтрат упаривают в небольшом
вакууме до объема в 50—60 мл, вызывают кристаллизацию
трением, прибавляют 40 мл сухого бензола, перемешивают и
отфильтровывают осадок, который на фильтре промывают
2—3 раза сухим бензолом (по 20—30 мл за прием). Таким
образом получают еще 10,61—10,90 г осадка фенидона с
т. пл. 121—122°.
Общий выход фенидона равен 26,1—27,91 г, что составля-
ет 32,42—34,4% от теоретического.
П р имела пия:
1. При применении ледяной уксусной кислоты в каждой операции па
каждые 25 мл ледяной уксусной кислоты добавляют 1 мл воды.
2. Иногда после второй промывки вода плохо отделяется, тогда до-
бавляют 15—20 г сульфата натрия и взбалтывают, после этого водный
слой отделяется хорошо.
136
3. Нитрита натрия берут 50%-ный избыток из расчета 0,565 г нитри-
та натрия на каждый грамм технического метилового эфира (3-анилино-
пропионовой кислоты.
4. Если применять охлаждение льдом с солью, то продукт реакции
может отделиться в виде слоя тяжелого темно-красного масла, которое
собирается на дне колбы. Обычно смесь бывает однородной.
5. 100 г цинковой пыли заливают 175 мл примерно 5%-ной соляной
кислоты (берут 150 мл воды и 25 мл концентрированной соляной кислоты
уд. в. 1,18). Как только начнут выделяться пузырьки водорода, цинковую
пыль переносят па воронку Бюхнера и отмывают соляную, кислоту водой.
6. Суммарный вес сырого фенидона—осадка и медоподобной массы —
в сумме составляет 51,4—53,15 г.
ЛИТЕРАТУРА
1. С. F. Behringer u. J б h п е. Герман, пат. 53 834; ZbL, 1891, 1,
112; Вег., 34, 234 (1891).
2. J. D. Kendall, G. F. Duffin, A. J. Axford. Амер. пат.
2688024; РЖхим., 1956, 23543.
3. J. D. Kendall, G. F. Duffin. Амер. пат. 2 704 762; С. А., 50,
2680(1956).
4. С. Harris, О. Loth. Вег., 29,513(1896).
5. Т. S. Donovan. Амер, пат, 2843598; С. А., 53,2256 (1959).
6. G. A. Reynolds, J. F. Tinker. Амер. пат. 2743279; С. А.,
51,488(1957).
7. G. F. Duffin, A. J. Axford. J. Chem. Soc., 1954,414; Брит,
пат. 679677; С. А., 48,736 (1954).
8. J. D. Kendall. Брит. пат. 650911; С. А., 46,144 (1952).
9. J. D. Kendall, G. F. Duffin. Брит. пат. 679678; С. А., 48,
736 (1954).
10. В. В. Пи гуле вс к ий, Н. И. Симонова. Тр. Ленинградск.
ни-та киноинженеров, вып. 6, 1961, стр. 13.
II. Н. И. Симонова, В. В. Пи гул ев с кий. Авт. свид. 113570,
1957; РЖхим , 1959, 58350.
12. Н. И. Симонова. Тр. Ленинградск. ин-та кинбинжеперов,
вып. 6, 1961, стр. 19.
13. Н. И. Симонова. Авт. свид. 129659, 1959; РЖхим., 1961, 22Л351.
Поступила в декабре 1964 г. ЛИКИ и ИЭМ АН СССР
УДК 546.281'13:
1-ФЕНИЛСИЛАТРАН
Фенил(2,2',2"-аминотриэтокси) силан
М. Г. ВОРОНКОВ, Г. И. ЗЕЛЧАН
CiaH17O3NSi М.в. 251,35
Запатентован Способ получения 1-фенилсилатрана с выхо-
дом до 71% посредством переэтерификации фенилтриэтокси-
силана триэтаноламином в среде бензола [1]. Выделяющийся
при этом этиловый спирт удаляется из реакционной смеси
азеотропной отгонкой.
Нами разработан новый простой и удобный метод [2] по-
лучения 1-фенилсилатрана с использованием более доступ-
ного и дешевого, чем фенилтриэтоксисилан исходного сырья.
Он основан на реакции расщепления триэтаноламином про-
дукта гидролиза фенилтрихлорсилана в присутствии щелоч-
ного катализатора. Процесс осуществляется путем азеотроп-
ной отгонки образующейся воды с инертным растворителем
(ксилол) и завершается очень быстро (за 1—2 часа). Выход
1-фенилсилатрана составляет 83% от теоретического.
Метод применим для синтеза различных 1-алкил-, арил-,
циклоалкил-, аралкил- и алкенилсилатранов.
138
СХЕМА СИНТЕЗА 1-ФЕНИЛСИЛАТРАНА
CgHjSiClg -f~ (1,5 + х)НаО х/п [C6H6SiO1>s_x(OH)aJ„ -И
+ ЗНС1
(х = 0 — 1,5)
кон
Чп [CeH6SiOi.5-x (ОН)!Х]„ + (НОСНаСН3)3 N---------’
->CeH5Si (ОСНаСНг)3 N 4- (1,5 + х) Н2О
Характеристика основного сырья
Фенилтрихлорсилан, ТУ 60755.
Триэтаноламин, ч., СТУ 12-10-113—61.
Ксилол, смесь трех изомеров, ч., ВТУ У 279а—51.
Условия получения
Исходный фенилполисилоксанол [CsHsSiO 1,5-х (ОН)П]„ го-
товится прибавлением фенилтрихлорсилана небольшими
порциями к большому избытку дистиллированной воды при
интенсивном перемешивании. Выпавший белый осадок от-
фильтровывается, промывается большим количеством дистил-
лированной воды (до отрицательной реакции на С1~) и су-
шится на воздухе до постоянного веса (см. примечание 1).
В круглодонную колбу емкостью 0,5 л, снабженную меха-
нической мешалкой и обратным холодильником, соединенным
с водоотборной ловушкой, помещают 14,2 г (0,11 М) (см.
примечание 2) фенилполисилоксанола, 14,9 г (0,10 А1) три-
этаноламина (см. примечание 3), 0,1 г порошкообразного ед-
кого кали и 300 мл ксилола.
Реакционную смесь нагревают при перемешивании до ин-
тенсивного кипения и прекращения отделения воды в ловуш-
ке. Теоретическое количество воды (2,7 мл) выделяется за
1—2 часа. Полученный'раствор фильтруют горячим и медлен-
но охлаждают. При этом из него выпадают игольчатые бе-
лые кристаллы, которые отфильтровывают, промывают пет-
ролейным эфиром и сушат в вакууме (см. примечание 4).
Выход 1-фенилсилатрана с т. пл. 207—208° 20,9 г или 83%
от теоретического. После повторной перекристаллизации из
ксилола получают 17,8 г (70% от теоретического) чистого ве-
щества с т. пл. 210,3—211,3°.
Примечания:
1. Можно пользоваться и влажным продуктом гидролиза фенилтри-
хлорсилана.
2. Количество фенилполисилоксанола приведено для случая х = 0.
139
3. Для синтеза использовался продажный триэтаноламин, предвари-
тельно перегнанный в вакууме.
4.1-Фенилсилатраи является очень сильным ядом. Он в два раза ток-
сичнее стрихнина или синильной кислоты и поэтому требует очень осто-
рожного обращения.
ЛИТЕРАТУРА
1. Westinghouse Electric Corp. (А. В. Flneston), пат. ФРГ 1131681
(1962); Chem. Abstrs, 58, 4598 (1963).
2. Al. Г. Воронков, Г. И. Зел чан. Авт. свид. 162139; Бюлл.
изобр., № 9 (1964).
Поступила в ноябре 1964 г.
Институт
органического синтеза
АН Латвийской ССР
УДК 547.732.07
р-ФЕНИЛТИОФЕН
М. Г. ВОРОНКОВ, А. И. ПЕРЕФЕРКОВИЧ
свнБ—с—сн
II II
НС сн
М. в. 160,23
До сих пор р-фенилтиофен получали несколькими метода-
ми: действием трехсернистого фосфора на ' фенилсукцинат
натрия [1, 2], реакцией нитрозоацетанилида с тиофеном [3, 4]
или дегидрогенизацией р-фенилдигидротиофена [5], являюще-
гося в свою очередь продуктом многостадийного синтеза.
Броун и Воронков [6] синтезировали р-фенилтиофен путем
осернения а, p-диметилстирола, а также наблюдали образова-
ние его при действии серы на вторично-бутилбензол [7].
Из перечисленных способов препаративное значение имеет
лишь реакция осернения а,р-диметилстирола, отличающаяся
своей простотой, доступностью исходных реагентов и не усту-
пающая по выходам другим способам.
Предлагаемый видоизмененный метод синтеза состоит из
двух стадий, первой из которых является получение аф-ди-
метилстирола из метилэтилфенилкарбинола (ранее описанное
в литературе [8, 9]) без выделения последнего..
СХЕМА СИНТЕЗА ₽-ФЕНИЛТИОФЕНА
1. С2Н6Вг + Mg -> C2H5MgBr
OMgBr
C?H6MgBr + C5H6-C-CH3 -> CeHs-C-CH2
II I I
О H3C CH8 141
OMgBr он
CSH-“C-CH3 10% HCU C6H8-C-CHa
II II
H3C CH3 H3C CH3
OH
CeH5-C - CH3 CeH5-C=CH + H3O
II II
H3C CH3 H3C CH3
2.
CeH5—C=CH + 3S -> CeII6—C-CH
II II II
H3C CH3 HC CH
Характеристика основного сырья
Этил бромистый, ч., ТУ МХП 80—47,
Магний металлический (стружка), ГОСТ 804—56,
Ацетофенон, ч., ТУ ОРУ 28—55.
Сера черенкован, ч., ТУ ОРУ 83—57.
Условия получения
1. Синтез а.,$-диметилстирола. В литровую трехгорлую
колбу, снабженную капельной воронкой, мешалкой со ртут-
ным затвором и обратным холодильником, помещают 12,16 г
(0,5 М) магниевой стружки и 50 мл абсолютного эфира и до-
бавляют по каплям при перемешивании 2,7 г (0,025 М) бро-
мистого этила. Прн этом наблюдается помутнение и неболь-
шое повышение температуры. Далее прикапывают 51,8 г
(0,415 Л1) бромистого этила, растворенного в 125 мл абсо-
лютного эфира, с такой скоростью, чтобы эфир слабо кипел.
Под конец смесь осторожно нагревают на водяной бане, по-
ка не растворится весь магний.
К полученному реактиву Гриньяра прикапывают при пе-
ремешивании раствор 48,06 г (0,43 /И) ацетофенона, раство-
ренного в 47 мл эфира. Смесь нагревают еше-3 часа на водя-
ной бане, охлаждают и медленно прибавляют при достаточ
ном охлаждении 100/о-ную соляную кислоту до полного раст-
ворения осадка. Эфирный слой отделяют, а водный слой два-
жды экстрагируют эфиром. От соединенных эфирных вытя-
жек отгоняют эфир, добавляют к остатку кристаллик йода и
отгоняют образующуюся при дегидратации воду (см. приме-
чание). Остаток подвергают фракционированной перегонке в
вакууме.
142
Общий выход а,р-диметилстирола с т. кип. 63—6479 мм
равен 34,4 г, что составляет 65% от теоретического; т. кип.
188- 190/°760 ял; d?* — 0,9102; п?°- 1,5345.
2. Осернение а$-диметилстирола. В трехгорлую кругло-
донную колбу, снабженную пришлифованным обратным холо-
дильником, термометром и мешалкой, помещают НО г
(0,83 М) а,р-диметилстирола и 80 г (2,5 М) серы. Верхний
конец холодильника соединяют с ловушкой, наполненной
35%-ным раствором едкого натра, для поглощения выделяю-
щегося сероводорода. Массу при перемешивании нагревают
до температуры кипения (190°), при которой начинается
обильное выделение сероводорода. Реакционную смесь нагре-
вают до 205° и выдерживают при этой температуре приблизи-
тельно 7 часов до полного прекращения выделения, серово-
дорода.
Продукт реакции (темно-коричневую смолистую массу)
перегоняют с перегретым водяным паром. Дистиллят экстра-
гируют эфиром, экстракт высушивают над хлористым каль-
цием и эфир отгоняют. Из остатка осаждают 75%-ным эти-
ловым спиртом кристаллы fj-фенилтиофена с т. пл. 91—92°.
Выход продукта равен 67—68 г, что составляет 50—51 %
от теоретического.
Для получения чистого вещества его дважды возгоняют
при нормальном давлении. р-Фенилтиофен представляет со-
бой блестящие серебристо-белые пластинчатые кристаллы со
своеобразным запахом; т. пл. 91,5—92,0°; т. кип. 254—
2567758 мм.
Примечание.
Вместе с водой отгоняется некоторое количество “«Р-диметилстирола,
который отделяется, сушится над хлористым кальцием и возвращается в
перегонную колбу.
ЛИТЕРАТУРА
1. A. Chrzaszc. zewska. Rocz. Chem., 5, 32 (1025).
2. I. L. Me 11 e r, H. J. Backer. Rec. trav. chim., 72, 491 (1953).
3. E. Bamberger. Ber., 30, 360 (1897).
4. J. D e g a n i, M. Pallotti, A. T u n d o. Ann. chim., 51, 434
(1961).
5. H. ff у n be rg, A. L e g о t b e 11 s, V. D e r p 1 о e g. J. Amer, Chem.
Soc., 79, 1972 (1957).
6. А. С. Броун, M. Г. Воронков. Ж. общ. химии, 17, 1162 (1947).
7. М. Г. Воронков, А. С. Броун. Ж. общ. химии, 18, 70 (1948).
8. А. К lag с s. Вег., 35, 2641, 3507 (1902).
9. К. Au wers, F. Е i s е n 1 о h г.. J. Pract. Chem., 82, 90 (1910).
Поступила в ноябре 1964 г. Институт органического синтеза
АН Латв. ССР
УДК 661.718.1.07
2-ФУРОИЛ- И 2-ТИЕНОИЛБРОММЕТИЛЕНФОСФО-
РАНЫ
А. А. ГРИГОРЕНКО, М. И. ШЕВЧУК, А. В. ДОМБРОВСКИЙ
/ — С—СВг = Р(С6Н6)3
О
Ca4H18BrO2P М. в. 449,29
I о J-C-CBr=P(CeHs)3
S II
О
C24H18BrOPS М. в. 465,35
Известно [1], что фосфораны склонны к реакциям электро-
фильного замещения. Реагируя с кислотами, хлорангидрида-
ми кислот, галоидными алкилами и т. щ, они образуют фос-
фониевые соли различной степени устойчивости, которые при
дегидрогалогенировании легко превращаются вгалоидпроиз-
водные фосфоранов [2, 3]. Последние представляют интерес,
так как н<: их основе по реакции Виттига могут быть получе-
ны непредельные соединения, содержащие при углероде с
двойной связью атом галоида.
Нами разработана методика получения 2-фуроилбромме-
тилентрифенилфосфорана и 2-тиеноилбромметилентрифенил-
фосфорана из 2-фуроилметилентрифенилфосфорана и 2-тие-
ноилметилентрифенилфосфорана. Последние при взаимодейст-
вии с бромом в растворе ССЦ образуют с количественными
выходами а-бромзамещенные бромиды (II), при дегидробро-
мировании которых водным раствором едкого кали получа-
ются соответственно 2-фуроилбромметилентрифенилфосфоран
(Ша) и 2-тиеноилбромметилентрифенилфосфоран (III6).
Бромфосфораны Ша, Шб представляют собой вполне
устойчивые кристаллические вещества, умеренно раствори-
мые в обычных органических растворителях.
144
СХЕМА СИНТЕЗА 2-ФУРОИЛ- И 2-ТИЕНОИЛБРОММЕТИЛ ЕН-
ФОСФОРАНОВ
LCO_CH=P(C.H,),
А 4
I
Lco-CHBr-P (С6Н6)3
X'
в;
- llXxJ-CO-CBr=P(C6H5)3
III
а: .Х —О
б: X—S
Характеристика основного сырья
Четыреххлористый углерод, ч., т. кип. 76,8°.
2-Фуроилметилентрифенилфосфоран, т. пл. 237—239° (см
стр. 147 в этом сборнике).
2-Тиеноилметилентрифенилфосфоран, т. пл. 225—226°
(там же).
Условия получения
I. 2-Фуроилбромметилентрифенилфосфоран (Ша). В
трехгорлую колбу, снабженную капельной воронкой, обрат-
ным холодильником и мешалкой, помещают раствор
18,5 г (0,05 А4) 2-фуроилметилентрифенилфосфорана (1а) в
300 мл четыреххлористого углерода и при перемешивании в
течение 40 минут приливают из капельной воронки раствор
8 г (2,6 мл\ 0,05 Л4) брома в 50 мл четыреххлористого угле-
рода. От нескольких капель раствора брома содержимое кол-
бы мутнеет, а под конец введения на стенках колбы начина-
ют выделяться кристаллы. После прибавления всего брома
мешалку останавливают и через 2 часа выделившуюся свет-
ло-желтого цвета кристаллическую соль бромистого 2-фу-
роилбромметилтрифенилфосфония (Па) отделяют, промы7
вают сухим эфиром и сушат на воздухе.
Выход продукта количественный, равен 26,2 г; т. пл. 245—
247°; после перекристаллизации из смеси этилацетат—мета-
нол (1:1) т. пл. 247—248°.
Для получения 2-фуроилбромметилентрифенилфосфорана к
раствору 15,9 г (0,03 М) фосфониевой соли в 80 мл метило-
10 Зак. 1350 145
оого спирта при перемешивании в течение 20 минут прибав-
ляют водный раствор едкого кали (2 г КОН в 40 мл воды).
Затем содержимое колбы перемешивают еще 2 часа. Выде-
лившийся осадок 2-фуроилбромметилентрифенилфосфорана
(Ша) отделяют, промывают и сушат при 50—60°.
Выход продукта равен 12,7 г, что составляет 94% от тео-
ретического; т. пл. 179—180°
II. 2-Тиеноилброммети.лентрифенилфосфоран (III6). Ана-
логичным образом из 2-тиеноилметилентрифенилфосфорана
(16) получают бромистый 2-тиеноилбромметилтрифенилфос-
фоний (116) с выходом 90% и т. пл. 246—247° (из смеси этил-
ацетат— метанол, 1:1), из которого дегидробромированием
получают 2-тиеноилбромметилентрифенилфосфоран с т. пл.
183—184° (из метанола); выход 65%,
ЛИТЕРАТУРА
1. М. И. Шевчук, А. В. Домбровский. Ж. общ. химии, 34, 916
(1964).
2. Н. А. Несмеянов, С. Т. Жужликова, О. А. Реутов До-
клады АН СССР, 151, 856 (1963).
3. G. Markl. Вег., 95, 3003 (1962).
Поступила в ноябре 1964 г.
Черновицкий
государственный
университет
УДК 661.718.1.07
2-ФУРОИЛ- И 2-ТИЕНОИЛМЕТИЛЕНТРИФЕНИЛ-
ФОСФОРАНЫ
А. В. ДОМБРОВСКИЙ, М. И. ШЕВЧУК, А. А. ГРИГОРЕНКО
II 11-С-СН=Р(С5Н6)3
О II
о
С24Н19ОаР М. в. 370,38
J—С—СН=Р (СвН6)3
SZ II
о
C24HI9OPS М. в. 386,55
Открытая в 1953 году реакция Виттита [1] явилась отлич-
ным методом синтеза непредельных соединений на основе
трифенилфосфоранов и карбонильных соединений [2]. Однако
получение непредельных соединений, содержащих ядра фу-
рана и тиофена, по этой реакции до сих пор не было осуще-
ствлено.
В настоящей работе описывается разработанный нами до-
ступный и удобный метод получения 2-фуроил- и 2-тиеноил-
метилентрифенилфосфоранов. Путем бромирования в эфир-
но-диоксановом растворе [3] 2-ацетилфурана (1а) и 2-аце-
тилтиофена (16) получают 2-(ы-бромацетил)-фуран (Па) и
2-(w-бромацетил)-тиофен (Пб), которые гладко вступают в
реакцию с трифенилфосфином, образуя практически с коли-
чественными выходами четвертичные фосфониевые соли
(III). При дегидробромировании фосфониевых солей спирто-
вым раствором этилата натрия получаются с высокими выхо-
дами соответствующие фосфораны (IV).
СХЕМА СИНТЕЗА 2-ФУРОИЛ- И 2-ТИЕНОИЛМЕТИЛ ЕНТРИФЕНИЛ-
ФОСФОРАНОВ
;-СО-СНз + Вга -* \ /- СО - СН2Вг
х z Хх z
10* I II
+(С6Н6)3Р Э
147
CO — СН2—Р (С6Н6)3
X
р)Г CaH5ONa ,
в спирте
III
IV
а :Х = О
б : X = S
Характеристика основного сырья
2-Ацетилфуран, т. кип. 64—66713 мм; т. пл. 27—28°.
2-Ацетилтиофен, т. кип. 214—214,57760 мм.
2- (со-Бромацетил)-фуран, т. кии. 97—9873 мм ; «д —
1,5848; <7420— 1,774.
2-(ю-Бромацетил)-тиофен, т. кип. 143—14574 мм; п2£ —
1,6308; d^ — 1,753.
Трифенилфосфин, т. пл. 79°.
Условия получения
2-Фуроилметилентрифенилфосфоран (IVa). Раствор
26,3 г (0,1 Af) трифенилфосфина в 60 ли сухого бензола по-
мещают в трехгорлую колбу емкостью 250 мл, снабженную
мешалкой, капельной воронкой и хлоркальциевой трубкой.
При перемешивании прибавляют из капельной воронки в те-
чение 30 минут раствор 18,9 г (0,1 А1) 2-(ш-бромацетил)-фу-
рана (Па) в 50 мл бензола. Сразу же начинается выделение
кристаллического осадка. Перемешивание продолжают еще
30 минут, а затем образовавшийся мелкокристаллический
осадок бромистого 2-фуроилметилтрифенолфосфония (П1а)
отфильтровывают, промывают бензолом и сушат на воздухе.
Выход фосфониевой соли (Ша) равен 41,2 г, что состав-
ляет 91% от теоретического; т. пл. 275—276° (из смеси 2:1
этилацстат — метанол).
Для получения фосфорана (IVa) помещают в трехгорлую
колбу, снабженную мешалкой, капельной воронкой и хлор-
кальциевой трубкой, взвесь 33,9 г (0,075 М) фосфониевой со-
ли (Ша) в 75 мл безводного этилового спирта и при переме-
шивании прибавляют из капельной воронки в течение 45 ми-
нут раствор этилата натрия, приготовленный из 1,84 г
(0,08 >Л4) натрия и 50 мл безводного спирта (см. пр^меча*
148
ние 1). После прибавления примерно двух третей раствора
этилата натрия осадок полностью растворяется. Через 45 ми-
нут после прибавления всего раствора этилата содержимое
ко^бы выливают в стакан с водой (около 200 мл). Выделив-
шийся осадок фосфорана (IVa) через 3 часа отделяют, пос-
ледовательно промывают водой и 25%-ным водным спиртом
до исчезновения реакции на ион брома и сушат при 60—80°.
Выход продукта равен 23,3 г, что составляет 84% от тео-
ретического.
2-Фуроилметилентрифенилфосфоран представляет собой
бесцветные кристаллы с температурой плавления 237—239°
и может быть использован в таком виде для реакции с кар-
бонильными и другими соединениями. После перекристалли-
зации из спирта он плавится при 239—239,5°.
2-Тиеноилметилентрифенилфосфоран (JV6). Аналогичным
образом из 2- (о-бром а цетил) -тиофена (Пб) и трифенилфос-
фина получается (см. примечание 2) бромистый 2-тиеноил-
метилтрифосфоний (Шб) с т. пл. 262—263° (из смеси этил-
ацетат— метанол, 4:1) и выходом 94%, из которого путем
дегидробромирования получают 2-тиеноилметилентрифенил-
фосфоран (IV6).
Выход продукта составляет 86%; т. пл. 225—226° (из эти-
лового спирта).
Примечания:
1. От большого избытка этилата натрия реакционная смесь темнеет
и значительно уменьшается выход фосфорана.
2. При получении бромистого 2-тиеноилметнлтрифени.пфосфония реак-
цию необходимо проводить в растворе сухого толуола, нагревая реакци-
онную смесь в колбе с обратным холодильником при 70—80° в течение
30 минут.
ЛИТЕРАТУРА
1. Г. Витт и г. Успехи химии, 26, 1141 (1957).
2. Л. А. Яновская. Успехи химии, 30, 813 П96Г).
3. А. В. Домбровский, М. И. Шевчук, В. П. Кравец. Ж.
общ. химии, 32, 2278 (1962); 33, 1135 (1963).
Постуяила в ноябре 1964 г. Черновицкий государственный
университет
УДК 547.831.8.05
2-ХЛ О Р-8-0 КС И X И Н О Л И Н
И. А. КРАСАВИН, В. М. ДЗИОМКО, Н. И. МИРОШКИНА,
Ю. И. РАДИН
ОН
C9H6C1NO М. в. 179,61
Первое упоминание о 2-хлор-8-оксихинолине содержится
в патенте (1950 г.), однако способ его получения и характе-
ристики не указываются [1].
В 1961 г. 2-хлор-8-оксихинолин (т. пл. 82,5—83,5°) был
синтезирован нами из 8-ацетоксикарбостирила, но эти дан-
ные не были тогда опубликованы.
В 1964 г. появилось сообщение {2] о получении 2-хлор-8-
оксихинолина посредством нагревания 8-(п-толуоЛсульфо-
нилокси)карбостирила с пятихлористым фосфором при 140—
160° в течение 10 часов и кипячения образовавшегося
(выход 90,7%) 2-хлор-8-(п-толуолсульфонилокси) хинолина
со спиртовым раствором едкого кали.
Выход 2-хлор-8-оксихинолина 87,2%; т. пл. 63—64°, т. е.
почти на 20° ниже, чем у полученного нами образца; причины
этого не ясны.
Для синтеза 2-хлор-8-оксихинолина нами была использо-
вана общая методика, предложенная Михайловым для прев-
ращения хинолонов в хлорхинолины [3]. Кратковременное
нагревание 8-ацетоксикарбостирила с избытком хлорокиси
фосфора с последующим кислотным гидролизом ацетоксипро-
изводного дало 2-хлор-8-оксихинолин с выходом 88—92%
(считая на 8-ацетоксикарбостирил).
150
2-Хлор-8-оксихинолин представляет собой бесцветное кри-
сталлическое вещество, летучее с водяным паром, малораст-
воримое в холодной воде, но хорошо растворяющееся во
в^ех органических растворителях и в разбавленных растворах
щелочей и кислот; из циклогексана или из водного этанола
кристаллизуется в бесцветных иглах с т. пл. 82,5—83,5°.
Приготовленный из него образец 2-хлор-8-(н-толуолсуль-
фонилокси) хинолина имел т. пл. 132,5—133° (по данным [2],
т. пл. 132—134°).
Получение 2-хлор-8-оксихинолина
рос'3 ? р/
CH8COO и СН8СОО
НС1 "Х
—- I li I
Ч/ Xn^cI
он
В круглодонную колбу емкостью 200 мл с пришлифован-
ным обратным холодильником, защищенным хлоркальциевой
трубкой, помещают 20,3 г (0,1 М) 8-ацетоксикарбостирила
(см. примечание. 1) и 18 мл (30,7 г; 0,2 Л1) свежеперсгнанной
хлорокиси фосфора. Колбу погружают в глицериновую баню,
заранее нагретую до 120°, и выдерживают при этой темпера-
туре в течение 30 минут, периодически помешивая вручную
(см. примечание 2).
Затем охлаждают содержимое колбы до 5—7° и осторож-
но добавляют к нему 15 г дробленого льда. Когда разложе-
ние избыточной хлорокиси фосфора закончится, приливают
60 мл 35%-ной соляной кислоты и колбу нагревают на кипя-
щей водяной бане в течение 30 минут.
Потом реакционную массу охлаждают, выливают в 250 мл
воды и подщелачивают раствором 100 г едкого кали в 400 мл
воды. Размешивают до полного растворения осадка, взбал-
тывают с активированным углем и фильтруют. Фильтрат ней-
трализуют 10%-ной уксусной кислотой до pH 7. Образовав-
шуюся суспензию экстрагируют хлороформом (3—4 раза по
100 мл; см. примечание 3) и экстракт сушат безводным суль-
фатом натрия в течение ночи. Хлороформ отгоняют, остатки
его удаляют под вакуумом и продукт (17—18 г) перекристал-
лизовывают из 35—40 мл циклогексана (см. примечание 4).
Выход 2-хлор-8-оксихинолина равен 14,6—15,4 г (81,4—
85,8% от теоретического в расчете на 8-ацетоксикарбости-
рил); т. пл. 81—81,5° (см. примечание 5).
151
Упаривайием маточника можно выделить еще 1—2 г ме/
нее чистого вещества.
Общий выход 16,1 —16,6 г (89,7—92,5%).
Примечания:
1. Применялся 8-ацетоксикарбостирил с т. пл. 249—249,5°, получен-
ный по методу, описанному в настоящем сборнике. /
2. Через 7—10 минут после начала нагревания твердое вещество раст-
воряется, а еще через 8—10 минут содержимое колбы начинает закри-
C1 аллизовываться.
3. Экстракцию применять не обязательно; в одном опыте выпавший
осадок был просто отфильтрован и было получено 15,85 г (88,3%) про-
дукта с т. пл. 81,5—82°. Можно также отогнать 2-хлор-8-оксихинолин с
водяным паром.
4. Вместе циклогексана для перекристаллизации можно применить
60%-ный водный этанол.
5. Чистый образец 2-хлор-8-оксихинолина с т. пл. 82,5—83,5° был при;
готовлен перегонкой с водяным паром и перекристаллизацией из 60%-ного
этанола и проанализирован.
Найдено, %: С-60,47; 60,23; Н—3,58; 3,63; С1-19,39; 19,52;
N —8,00; 8,09.
C9H6C1NO. Вычислено, %: С-60,18; Н—3,37; Cl-19,74; N-7,80.
ЛИТЕРАТУРА
1. Пат. США 2524725 (1950); Chem. Abstrs, 45, 3272е (1951).
2. М, Натала, К. Funakoshi. Yakugaku Zasshi, 84, 28 (1964).
3. Г. И. Михайло в. Химические реактивы и препараты, вып. 24,
М., ИРЕА, 1961, стр. ИЗ.
Поступила в сентябре 1965 г, ИРЕА
УДК 547.732.07
2-ЭТИЛМЕРКАПТО-5-ЭТИЛ-3-ТИОФЕНАЛЬДЕГИД
Я. Л. ГОЛЬДФАРБ, М. А. КАЛИК., М. Л. КИРМАЛОВА
----СНО
Н6Са-< SC2H6
S х
CsH12OS2 М.в. 200,32
2-Этилмеркапто-5-этил-3-тиофенальдегид является проме-
жуточным продуктом в синтезе комплексообразующих соеди-
нений ряда тиофена, несущих свободную меркаптогруппу [1].
До настоящего времени в литературе не было описано пря-
мое форматирование алкил-(5-алкил-2-тиенил) сульфидов, но
были известны случаи введения альдегидной группы в 5-по-
ложение тиофенового кольца алкил-2-тиенилсульфидов с по-
мощью М’,М-диметилформамида [2] или N-метилформанили-
да [3] в присутствии хлорокиси фосфора. Мы применили обе
эти методики для формилировання алкил-(5-алкил-2-тиенил)-
сульфидов, несколько видоизменив их.
СХЕМА СИНТЕЗА 2-ЭТИЛМЕРКАПТО-5-ЭТИЛ-3-ТИОФЕНАЛБДЕГИДА
/СН3
сж
чсно
НБС2-Г>Х + или POCI^
s sc2H6
(СН3)2 NCHO
---СНО
Характеристика основного сырья
Этил-(5-этил-2-тиенил) сульфид [4], см. стр. 156.
Хлорокись фосфора, ч., МРТУ 6-09-337—63.
N-Метилформанилид [5].
М',М-Диметилформамид, ч., СТУ 79-606-Х—60 (см. приме-
чание 1).
Условия получения
А. В четырехгорлую круглодонную колбу емкостью
500 мл, снабженную мешалкой, термометром, капельной во-
ронкой и хлоркальциевой трубкой, помещают 87 г (0,64 М)
N-метилформанилида и 98 г (0,64 М) хлорокиси фосфора и
оставляют стоять в течение 40 минут. Затем при 25—35°, ох-
лаждая колбу водой, постепенно прибавляют 100 г (0,58 М)
этил-(5-этил-2-тиенил) сульфида, перемешивают ири 50—60° в
течение двух часов и оставляют на ночь. На следующий день
темную вязкую массу переносят в стакан со смесью 800 г
льда и 250 мл воды и перемешивают 20—30 минут. Выделив-
шееся масло отделяют, а водный слой экстрагируют тремя
порциями эфира (по 150 мл). Органический слой и эфирные
вытяжки объединяют и промывают разбавленной соляной
кислотой (30 г концентрированной НС1 на 250 мл воды) для
удаления следов N'-метиланилина. Водные вытяжки вновь
экстрагируют эфиром. Объединенный эфирный раствор про-
мывают насыщенным раствором бикарбоната натрия, водой и
сушат сульфатом магния. Эфир отгоняют, остаток перегоня-
ют в вакууме с елочным дефлегматором и получают 94,4 г
2-этилмеркапто-5-этил-3-тиофенальдегида, что составляет
81,3% от теоретического выхода; т. кип. 132—134° при 4 мм.
Эту фракцию можно использовать для дальнейших превраще-
ний без дополнительной очистки. После повторной перегонки
т. кип. продукта 118—119° при 2 мм; п2£ — 1,5889; сЦ20—
1.1536 (см. примечание 2).
2,4-Динитрофенилгидразон, перекристаллизованный из
смеси спирта с ксилолом, плавится при 150—150,5°.
Б. К перемешиваемому раствору 100 г (0,58 М) этил-(5-
этил-2-тиенил) сульфида в 60 мл (0,82 М) М,Г*1-диметилформ-
амида при 0—5° постепенно прибавляют 98 г (0,64 М) хлор-
окиси фосфора. Затем реакционную массу перемешивают в
течение часа при 20°, нагревают один час при 50—60° и остав-
ляют стоять на двое суток. Темный раствор выливают в смесь
800 г льда и 500 мл раствора ацетата натрия (взято 200 г
кристаллического ацетата натрия), экстрагируют четыре ра-
за эфиром (по 150 мл). Эфирный раствор обрабатывают, как
указано выше, и получают 90,4 г 2-этилмеркапто-5-этил-3-тио-
фенальдегида, что составляет 77,6% от теоретического вы-
хода; т. кип. 132—140° при 4 мм; —1,5842.
154
П р I, м е ч а н й я:
1. И,М-Диметнлформамид смешивают с сУхйм бензолом (10%
по объему), отгоняют последний, затем остаток сушат окисью бария и пе-
регоняют.
2. Этим методом были получены также 2-мег илмеркапто-5-метнл-З-
тиофенальдегид [6] и 2-бепзилмеркапто-5-этил-3-тпофенальдегид [I].
ЛИТЕ Р.А ТУРА
1. Я. Л. Гольдфарб, М. А. Калик, М. Л. Кирмалова. Изв.
АН СССР, ОХН, 1962, 701.
2. J. Cymerman-Craig, J. W. Loder. J. Chem. Soc., 1954,237.
3. E. Profit. Monatsber. Dtsch. Akad. Wiss., Berlin, 1,180(1959).
4. Я. Л. Гольдфарб, M. А. Калик, 1Л. Л. Кирмалова. Ж-
общ. химии, 29, 2034 (1959).
5. Синтезы органических препаратов, сб. 3, ИЛ, 1952, стр. 320.
6. Я. Л. Гольдфарб, М. А, Калик, М. Л. Кирмалова. Ж.
общ. химии, 30, 1012 (1960).
Поступила в декабре 1964 г.
ИОХ АН СССР
УДК 547.569.2.07
ЭТИЛ-(5-ЭТИЛ-2-ТИЕНИЛ )СУЛЬФИД
Я. л. ГОЛЬДФАРБ, М. А. КАЛИК. М. Л. КИРМАЛОВА
H6c/4S /XSC2H6
CSH1S52 М.в. 172,31
Алкил-(5-алкил-2-тиенил) сульфиды являются исходными
веществами в синтезе целого ряда производных тиофена, в
том числе 5-алкил-2-тиенилмеркаптанов, замещенных различ-
ными функциональными группировками.
Этил-(5-этил-2-тиенил) сульфид впервые получен нами [1].
До настоящего времени в литературе не описан синтез этого
сульфида, но имеется ряд работ по получению алкил-(2-тие-
нил) сульфидов исходя из 2-йодтиофена, магния и серы и га-
лоидного алкила [2, 3] или из 2-гаЛоидтиофена и меркаптида
меди [4] и др. Авторы настоящей работы получили с выходом
50,5% [1] этил-(о-этил-2-тиенил) сульфид из 5-бром-2-этилтио-
фена, магния, серы и йодистого этила [2]. 5-Бром-2-этилтио-
фен синтезирован с выходом 75% из 2-этилтиофена.
Использование литийзамещенных 2-алкилтиофенов более
удобно, так как дает возможность повысить выход сульфидов
до 75—80%, считая на 2-алкилтиофен; при этом отпадает
необходимость синтеза 2-галоидтиофенов.
СХЕМА СИНТЕЗА ЭТИЛ-(5-ЭТИЛ-2-ТИЕНИЛ)СУЛЬФИДА
Н6Са-\ / - Н5С2-^ — ->
S ' S
-> нЕса-^ „/-SL1 н6с2-€ >-sc2H5
О ' о
156
Характеристика основного сырья
2-Этилтиофен [5].
н-Бутй’ллитий (см. примечание 1).
Сера черенковая, ч., ТУ МХП ОРУ 83—57 или серный
цвет, ГОСТ 702—41.
Этил йодистый, ч., ТУ МХП 98—51.
Условия получения
Синтез проводят в токе инертного газа (см. примеча-
ние 2). В четырехгорлую круглодонную колбу емкостью 2 л,
снабженную мешалкой, термометром, обратным холодильни-
ком с хлоркальциевой трубкой и трубкой для ввода инертно-
го газа, помещают 112,2 г (1 Л4) 2-этилтиофена в 100 мл су-
хого эфира, добавляют при перемешивании при 0° эфирный
раствор 70,4 г (1,1 Л!) н-бутиллития и кипятят смесь в тече-
ние часа. В охлажденный раствор при 0° прибавляют неболь-
шими порциями 32 г (1 М) сухой мелкорастертой серы и
смесь нагревают в течение нескольких минут до полного рас-
творения серы. Затем при 0—5° к раствору приливают 171,6г
(1,1 М) сухого перегнанного йодистого этила и кипятят смесь
в течение 7 часов; при этом выпадает осадок йодистого лития
(см. примечание 3).
Затем реакционную смесь при охлаждении льдом обраба-
тывают 400 мл 15%-ного водного раствора хлористого аммо-
ния до полного растворения осадка. Эфирный раствор отде-
ляют, а водный экстрагируют эфиром. Объединенный эфир-
ный раствор промывают три раза 5%-ным водным раствором
щелочи, водой и сушат хлористым кальцием.
После удаления эфира остаток перегоняют в вакууме с
елочным дефлегматором высотой 20—25 см и отбирают фрак-
цию с т. кип. 118—122°/22 мм.
Выход продукта 134 г, что составляет 77,9% от теорети-
ческого. Эта фракция может быть использована для дальней-
ших синтезов. Но для окончательной очистки сульфид пере-
гоняют над натрием; т. кип. 103—105°/10 мм; — 1,0577;
га® — 1,5539 (см. примечание 4).
Примечания:
I. Эфирный раствор и-бутиллнтия получают по методу [7], концентра-
ция раствора около 0,1 г/мл.
2. Лучше всего применять аргон или азот, не содержащий кислорода.
Газ высушивают пропусканием через две склянки с концентрированной
серной кислотой.
3. Первые 2—3 часа смесь необходимо кипятить в тот же день, до-
бавляя сухой эфир для компенсации потерь, с током газа. Во время ноч-
ной выдержки реакционную смесь следует хранить под азотом.
4. Аналогичным образом нами получены метил-2-тиеиилсульфид, ме-
тил- (5-метил-2-тиеиил)сульфид, этил-2-тиенилсульфид [1] и бензил-(5-
этил-2-тиеиил) сульфид [6].
157
ЛИТЕРАТУРА
1. Я. Л. Гольдфарб, М. А. Калик, М. Л. Кирмалова. Ж.
общ. химии, 29, 2034 (1959).
2. Синтезы органических препаратов, 7, ИЛ, 195G, стр. 41.
3. R. A d a m s, A. F е г г е 111. Amer. chem. Soc., 51, 4927 (1959).
4. E. Prof ft. Chemiker Zig., 82, 295 (1958).
5. N. P. В u u - H о i, N. Hoan, N. H. К h о i. J. Organ. Chem., 15,
959(1950).
6. Я. Л. Г о л ь д ф а р б, м. А. Калик, М. Л. Кирмалова. Изв.
АН СССР, ОХН, 1962, 701.
7. Н. О 11 m а п, J. Beet, С. Brannen и др. J. Amer. Chem. Soc
71,1499 (1949).
Поступила в декабре 1964 г.
ИОХ АН СССР
УДК 661.718.5.07
1-ЭТОКСИСИЛАТРАН
Этокси(2,2',2"-аминотриэтокси)силан
М. Г. ВОРОНКОВ, Г. И. ЗЕЛЧАН
C8H17O4NSi
М. в. 219,31
Имеющиеся в литературе весьма скудные сведения о пу-
тях синтеза 1-этоксисилатрана являются противоречивыми.
По одним данным [1], при взаимодействии триэтаноламина с
тетраэтоксисиланом образуется 1-этоксисилатран с выходом
85%, имеющий т. пл. 35—37°. По другим данным [2], 1-этокси-
силатран имеет т. пл. 100—102°, однако условия его получе-
ния не приводятся.
Нами найдено, что 1-этоксисилатран гладко образуется
при переэтерификации тетраэтоксисилана триэтаноламином в
присутствии каталитических количеств гидроокиси щелочно-
го металла в среде инертного растворителя (ксилол). Его
выход составляет 91 % от теоретического. Перекристаллизо-
ванное вещество имеет т. пл. 102—103°.
СХЕМА СИНТЕЗА 1-ЭТОКСИСИЛАТРАНА
Si (ОС,Н5)4 + (HOCH2CH,)sN
— C2H5OSi(OCH2CH2)s N + 3CSH5OH
|| 159
Характеристика основного сырья
Тетраэтоксисилан, ч., ЕУ-64—54.
Триэтаноламин, ч., СТУ 12-10-113—61.
Ксилол, смесь трех изомеров, ч., ВТУ У279—51.
н-Гептан, ч., ГОСТ 4375—48.
Условия получения
В перегонную колбу с елочным дефлегматором помещают
20,8 г (0,1 Л4) тетраэтоксисилана (см. примечание 1), 14,9 г
(0,1 М) триэтаноламина (см. примечание 2), 0,1 г порошко-
образного едкого кали и 75 мл ксилола. Реакционную смесь
медленно нагревают —- так, чтобы температура переходящих
паров не превышала 80°. Нагревание ведут до прекращения
отгонки этилового спирта. За два часа выделяется теорети-
ческое количество спирта (13,8 г). К охлажденной ниже 100°
реакционной смеси прибавляют равный объем н-гептана и
охлаждают ее до комнатной температуры. Выпавшие белые
кристаллы отсасывают, промывают петролейным эфиром и
сушат в вакууме.
Выход 1-этоксисилатрана с т. пл. 99—101° составляет
19,9 г (91% от теоретического). После перекристаллизации
из н-гептана получают 14,7 г (67%) совершенно чистого ве-
щества с т. пл. 102—103°,
Примечания:
1. Тетраэтоксисилан очищается перегонкой над натрием и имеет
т. кип. 166—168°.
2. Для синтеза использовался продажный триэтаноламин, предвари-
тельно перегнанный в вакууме.
ЛИТЕРАТУРА
1. А. В. F i n е s t о п е. Пат. США 2953545 (1960); С. А„ 85, 4045
(1961); Пат. ФРГ 1131681 (1962); С. А., 58, 4598 (1963).
2. С. L. Frye, G.E. Vogel, J. A. H a 11. J. Amer. Chem. Soc.,
83, 996 (1961).
Поступила в декабре 1964 г.
Институт органического синтеза
АН Латвийской ССР
АЛФАВИТНЫЙ ПЕРЕЧЕНЬ СОЕДИНЕНИЙ,
ОПИСАННЫХ В НАСТОЯЩЕМ СБОРНИКЕ
Азотнокислая соль 3-амино-1,2,4-триазола .......................... 78
З-Аминокарбазол..................................................... 5
3-Амино-1,2,4-триазолкарбоновая-5 кислота .......................... 9
Аценафтенов-1-спиро-(2,4')-2',3'(СО),6',5'(СО)-дибепзоилен-1',4' -ди-
гидропиридин .................................................. 12
2-14-Ацетиламинопиридин............................................ 16
8-Ацстоксикарбостирил...........................................• 18
2-Бевзилмеркапто-5-метил-3-тиофевальдегид........................ 155
Бензиловый эфир М-[р-(2-пиридил)этил]-14-фенилдитиокарбамиво-
вой кислоты......................................•............. 20
Бензил-(5-этил-2-тиенил)сульфид ................................. 157
8-(Бензолсульфоннламино)хинальдин................................ 113
М,М'-Бис-(карбоксиметил)-4,4'-дипвридилий дихлорид................. 22
N,N'-Bhc-(B оксвэтил)-4,4'-дипиридилиЙ дихлорид .................. 23
Боратран..................•........................................ 24
лгрет-Бутил-2-тиеиилсульфид •...................................... 26
9-Винил-],2-бензакридин.............................•............. 28
3-Лиазо‘1,2,4-тряазолкарбоновая-5-кислота......................
8-[2',3'(СО), 6', 5'(СО)-Дибензоиленпиридил-4']-вафтойпая-1 кислота
Дибепин........................................................
2-Г4-Диизопропилфосфиниламинопиридин..........•................
2-[₽-(М,М-Диметиламино)этил]пиридин ...........................
Диметиловый эфир имидазол-4,5-дикарбоновой кислоты ............
аф-Диметилстирол...............................................
2-5!-Дипропилфосфиннламинопиридии..............................
2,6-Дистирилпирон-4............................................
2,2'-Дитиеннлдисульфнд.........................................
2,2'-Цитиенилсульфид...........................................
1,5-Д ифеии л-3,6-диметил-пиразол о-(4,5)-пиридон-4............
4,5-Дифенилимидазол ...........................................
1,5-Дифенилпиразолидон-З.......................................
1,3-Дифенилпиразолидон-5.......................................
2,3-Дихлор-1,4-диоксаи . . . . ........................• . . .
2-[|3-(М,М-Диэтиламино)этил] пиридин...........................
Диэтилацеталь 2-этилмеркапто-5-этил-3-тиофенальдегида..........
2-М-Диэтилфосфиниламинопиридин.................................
11 Зак. 1350
120
33
30
36
38
40
142
37
45
49
48
123
51
54
56
57
59
161
Имидазол-4,5-дикарбоновая кислота................................. 63
8-(Мезитиленсульфониламино) хинальдин............................ 113
2-Меркапто-5-метил-3-тенилиденимип................................ 67
2-Меркапто-5-этил-3-тенилиденимин................................. 66
2-Метил-5-изопропил-5-(а-метокси)этил-1,3-диоксан................. 68
2-Метилмеркапто-5-метил-3-тнофенальдегид.......................... 155
3 Метил-5-пропилпиррол-(2-азо-2')-фенол....................• . . 70
2-Метил-2,3-циклопентано-4,5-циклогексено- (Дь) -2,3,4,5 -тетрагидро-
фуран . . •................................................... 95
Метиловый эфир ₽-анилинопропионовой кислоты................... 135
Метиловый эфир р-(_и-метилфениламино)пропионовой кислоты . . 110
Метиловый эфир N-нитрозо-р (_и-метилфениламино)-пропионовой
кислоты .....................•.............................. 111
Метил-2-тиенилсульфид........................................... 157
4-Метил-1-фенилпиразолидон-3..................................... 72
Метил-(5-метил-2-тиенил)сульфид.................................. 157
2-[8-(Г\'-Морфолил)этил]пиридин................................... 75
Натриевая соль 4-карбметокси-имидазол-5-карбоновой кислоты . . 44
3-Нитрамино-1,2,4-триазол......................................... 77
9-Нитрозо-З-нитрокарбазол.......................................... 6
9-Нитрозокарбазол.................................................. 6
₽-(5-Нитрофурил-2)-акриловая кислота.............................. 80
4-Окси-3-ацетил-1,6-диметилпиридон-2.............................. 84
4-Окси-3-ацетил-6-метцлпиридон-2.................................. 83
4-Окси-3-ацетил-6-стирилпирон-2................................... 46
4-Окси-3-ацетил-1-фенил-6-метил-пиридон-2......................... 84
4-Оксн-3-бутироил-6-метил-пиридои-2............................... 84
4-Окси-3-н-валероил-6-метил-пиридон-2.............•............... 84
8-Окси-2-гидразинохинолин......................................... 86
4-Окси-3-изовалероил-6-метил-пиридин-2............................ 84
4-Окси-3-н-капроноил-6-метил-пиридин-2............................ 84
8-Окси-2-метоксихннолин........................................... 88
т/7Я«с-4-Окси-3(4'-нитроциннамоил)-6-метилпирон-2................. 92
4-Окси-3-(4'-нитроциинамоил)-6-метилиирон-2....................... 93
тра«с-4-Окси-3-(2'-оксициннамоил)-6-метилпирои-2...........• • . 94
т/>а«с-4-Окси-3-(3'-оксициннамоил)-6-метилпирон-2..........• . 94
яцга«с-2-Окси-3-(4'-оксициниамоил)-6-метилпирон-2................. 94
4-Окси-3-пропионил-6-метилпиридон-3............................... 84
2-{3'-Оксистирил)-6-метилпирон-4................................. 101
4-Окси-6-стирилпирон-2............................................ 90
тра«с-4Окси-3-(2'-хлорциинамоил)-6-метилпирон-2 ....... 94
трл«с-4-Окси-3 (4'-хлорциниамоил)-6-метилпирон-2.................. 94
4-Окси-3-циннамоил-6-метилпирон-2................................. 92
4-Окси-3-циннамоил-6-стирилпирон-2................................ 46
1,2,3,4,5,6,7,8-Октагидродибензофураи......................• . . 95
2-[р-(И-Пиперидил) этил] пиридин.................................. 76
2-11иридилметанол . . . •......................................... 98
Солянокислая соль 2-амино-1-фталоиламинобензол.................... 130
2-Стирил-6-метилпирон-4 ................•........................ 100
Тетрафурфурилоксисилан........................................... 103
2-Тиеноилбромметиленфосфоран...................................... 144
2-Тиеноилметилентрнфенилфосфоран.................................. 147
S-Тиокарбаминилтиогидракриловая кислота........................... 107
162
2-Тиотиазанон-4.................................................
1-(л-Толил)пиразолидон-3............•...........................
8-(я-Толуолсульфониламино)хииальдин.............................
1,2,4-Триазолинтион.............................................
1,2,4-Триазолон-3-карбоновая-5 кислота..........................
106
109
113
116
119
1-Фенил-3-н-амил-6-метил-пиразоло-(4,5с)-пиридон-4............... 123
1-Фенил-3-н-бутил-Ь-метилпиразоло-(4,5с)-пиридон-4.........• . 123
1-Фснил-3,6-диметил-пиразоло-(4,5с)-пиридон-4...........• • . . 123
4-Фенил-лг-диоксан............................................... 125
]-Фенил-3-изобутил-6-метил-пиразоло-(4,5с)-пиридои-4............. 123
2-[1"-Фенил-3’-метилпиразолон-5"-(4"-азо-2')-фенилазокси]-4-метил-
фенол......................................................... 128
1-Фенилпиразолидон-З............................................. 133
1-Фенил-и-пропил-6-метил-пиразоло-(4,5с) пиридои-4............... 123
1-Фенилсилатран.................................................. 138
р-Фенилтиофен................................................... 141
1-Фенил-3,5,6-триметил-пиразоло-(4,5с)-пиридон-4 . . •........... 123
2"-Фталоиламино-6-окси-3-метилазобензол.......................... 130
2"-Фталоиламино-6-окси-3-метилазоксибензол....................... 131
2-Фуроилбромметилентрифенилфосфоран............................ 147
2-Фуроилметилентрифенилфосфоран.................................. 147
2-Хлор-8-оксихинолин.............................•.............. 150
2-Этилмеркапто-5-этил-3-тиофенальдегид ......................... 153
Этил-(5-этил-2-тиенил)сульфид . . , •........................... 156
Этил-2-тиенилсульфид.........................................• 157
1-Этоксисилатран.............................................• 159
11*
Редактор R. Г. Козлов
Техн, редактор А. И, Пирожкова
Корректор Л. Я. Кузнецова
Сдано в набор 20.11.65 г. Подписано к печати 13. 6. 1966 г.
Формат бумаги 60X90% Печ. л. 10,25 Уч. изд. 7,6
Т-05385 Заказ 1350 Тираж 1000 экэ. Цена 53 коп.
Типография ВАХЗ
Замеченные опечатки
Страница Строка Напечатано Следует читать
24 5 сверху (формула) N 7 N 4
S3 9 снизу А. Д. Гра- А. Д. Гар-