









































































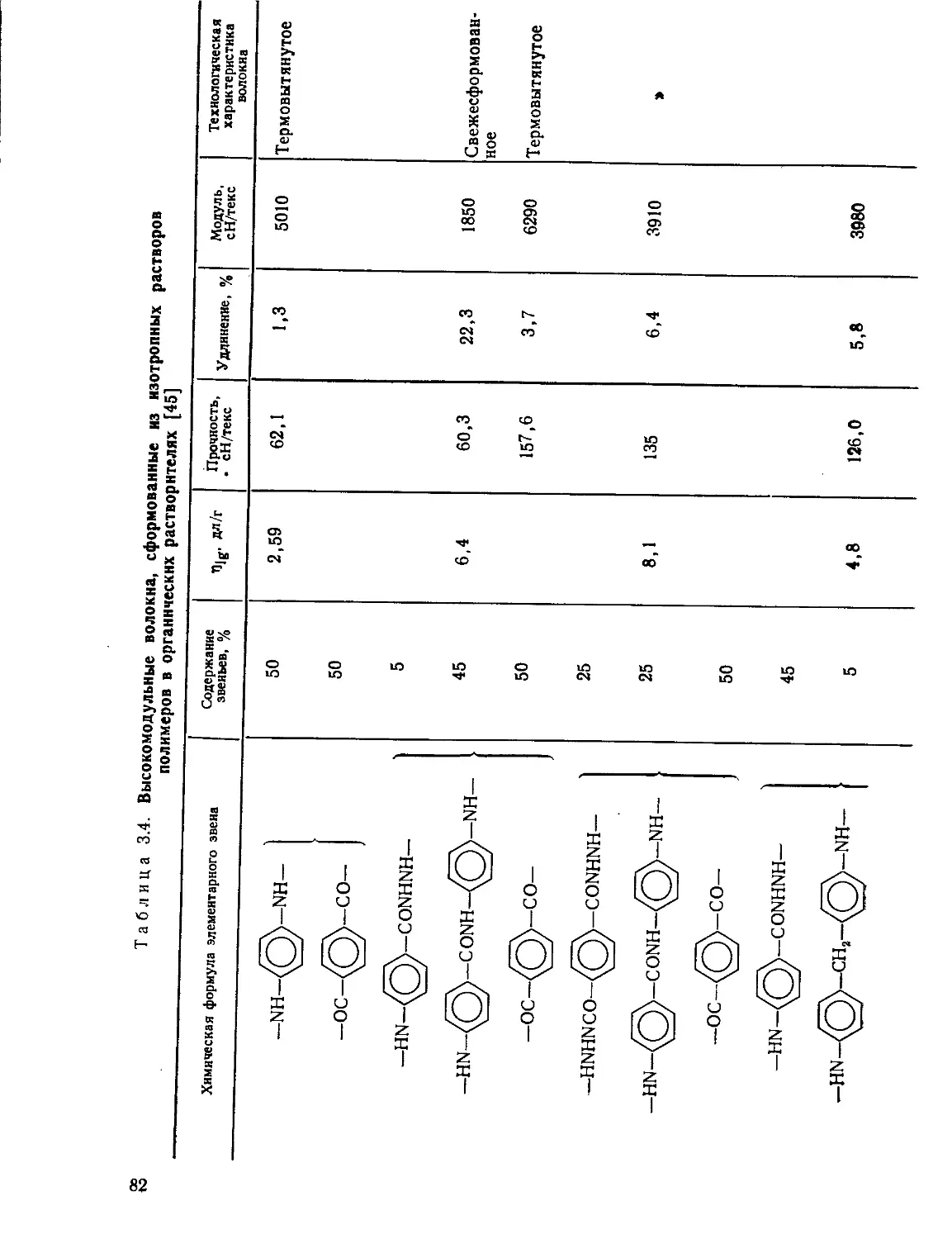



























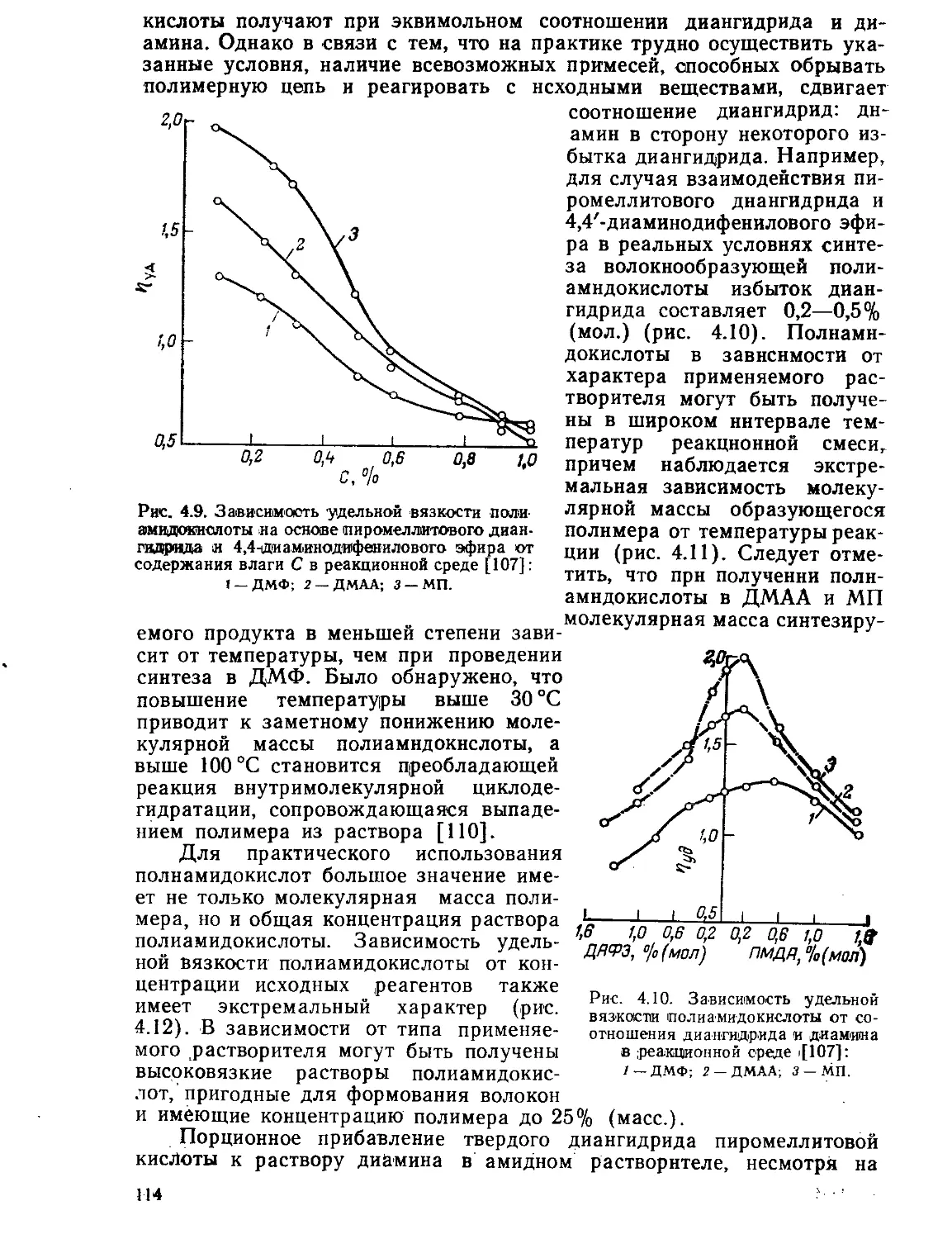



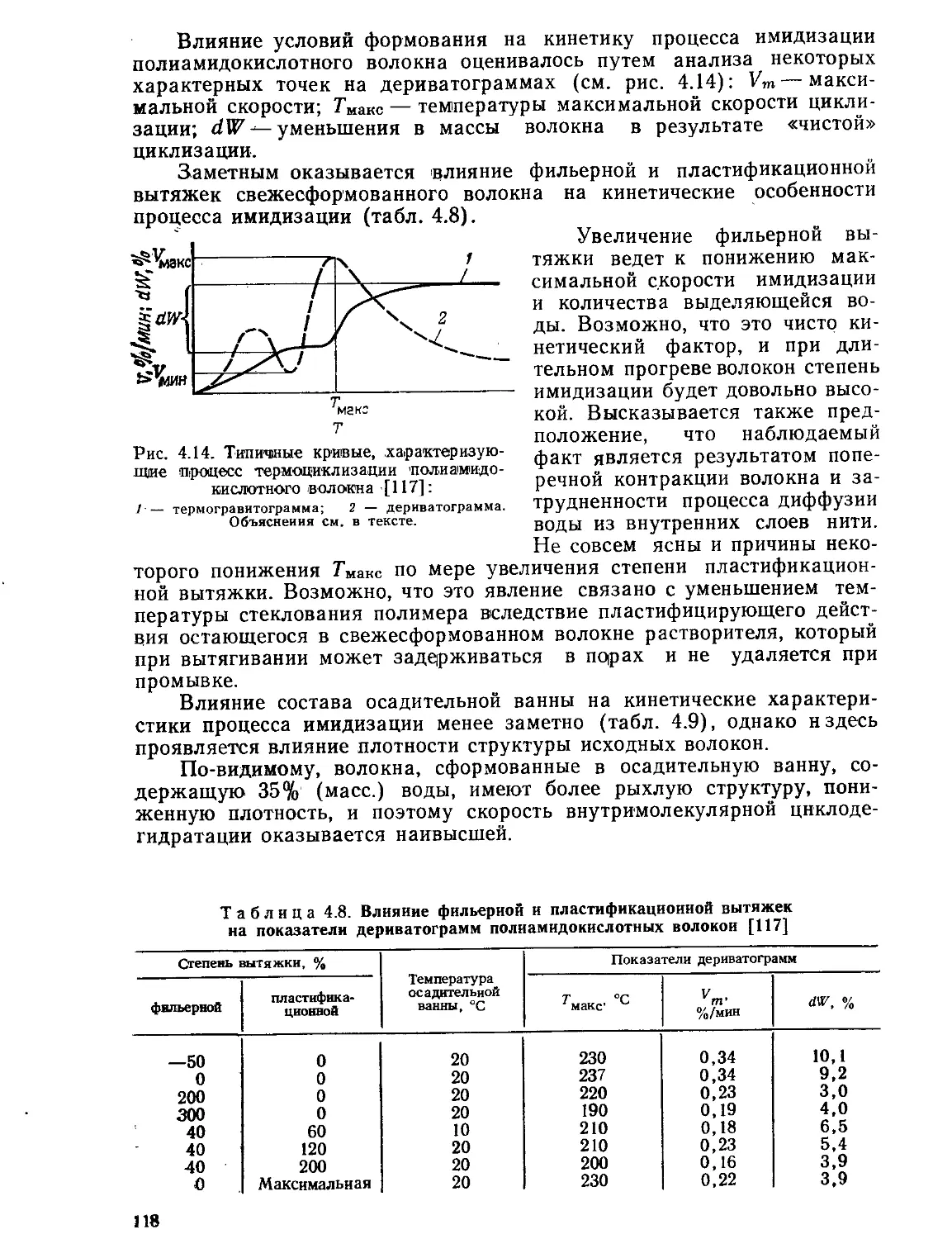


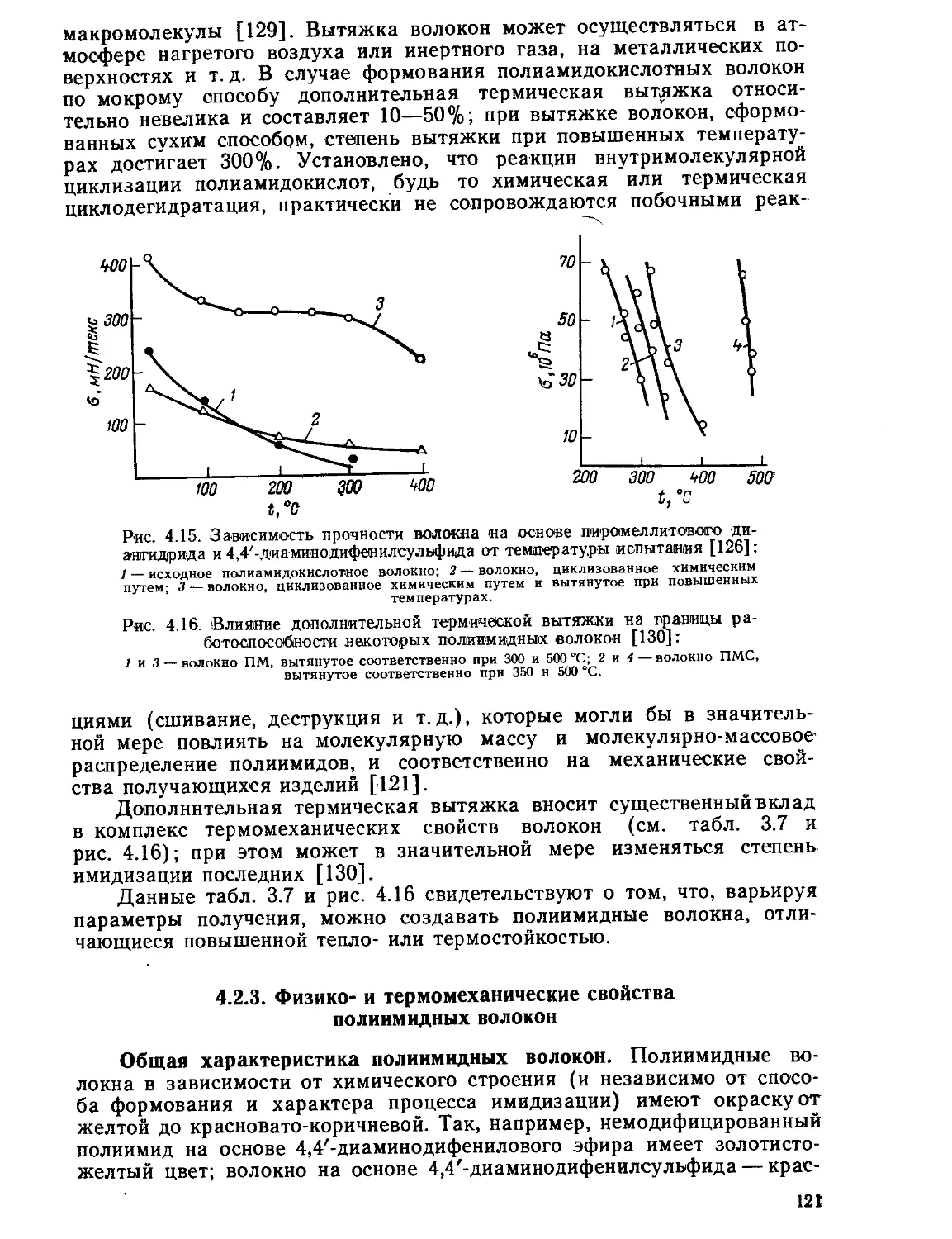


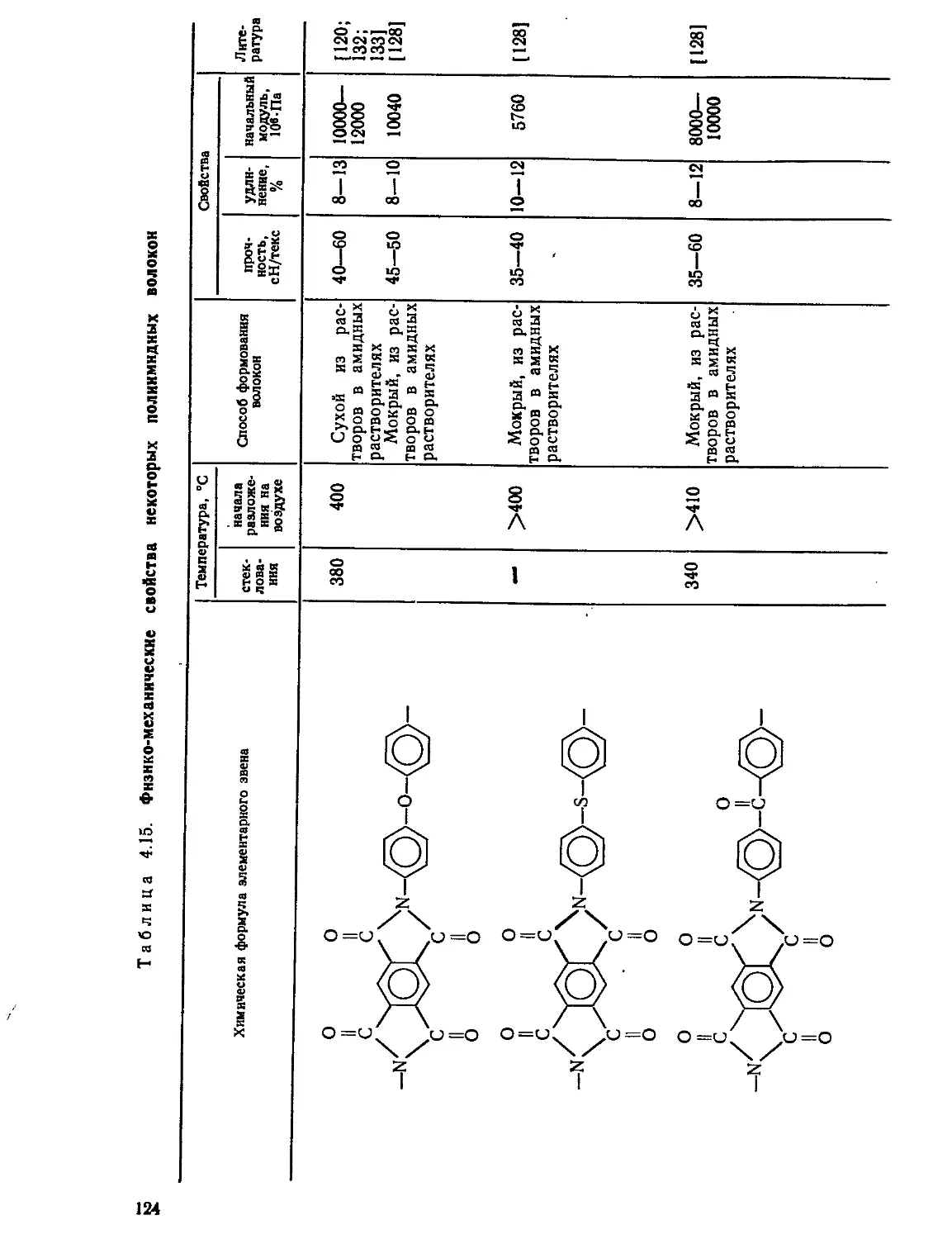





















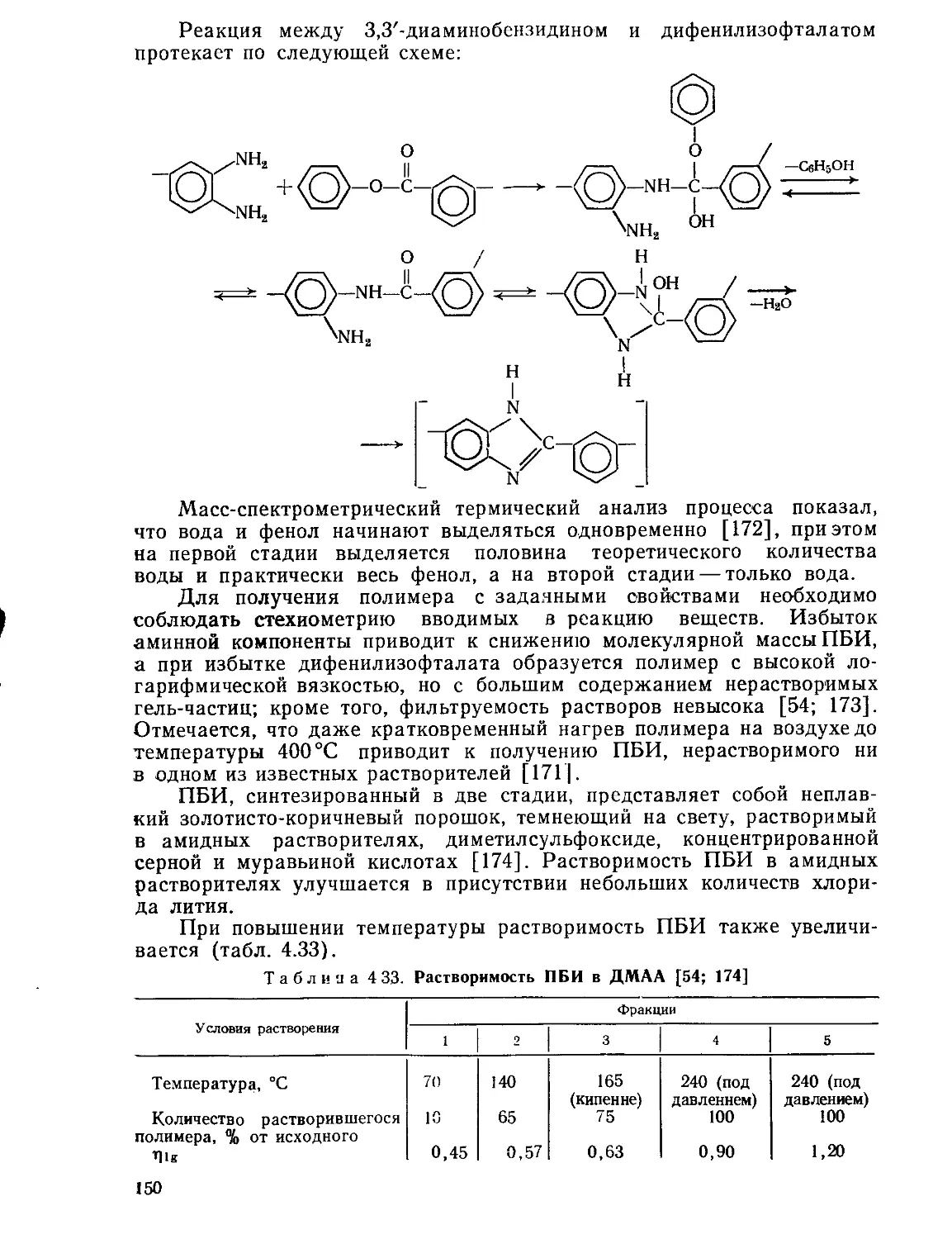



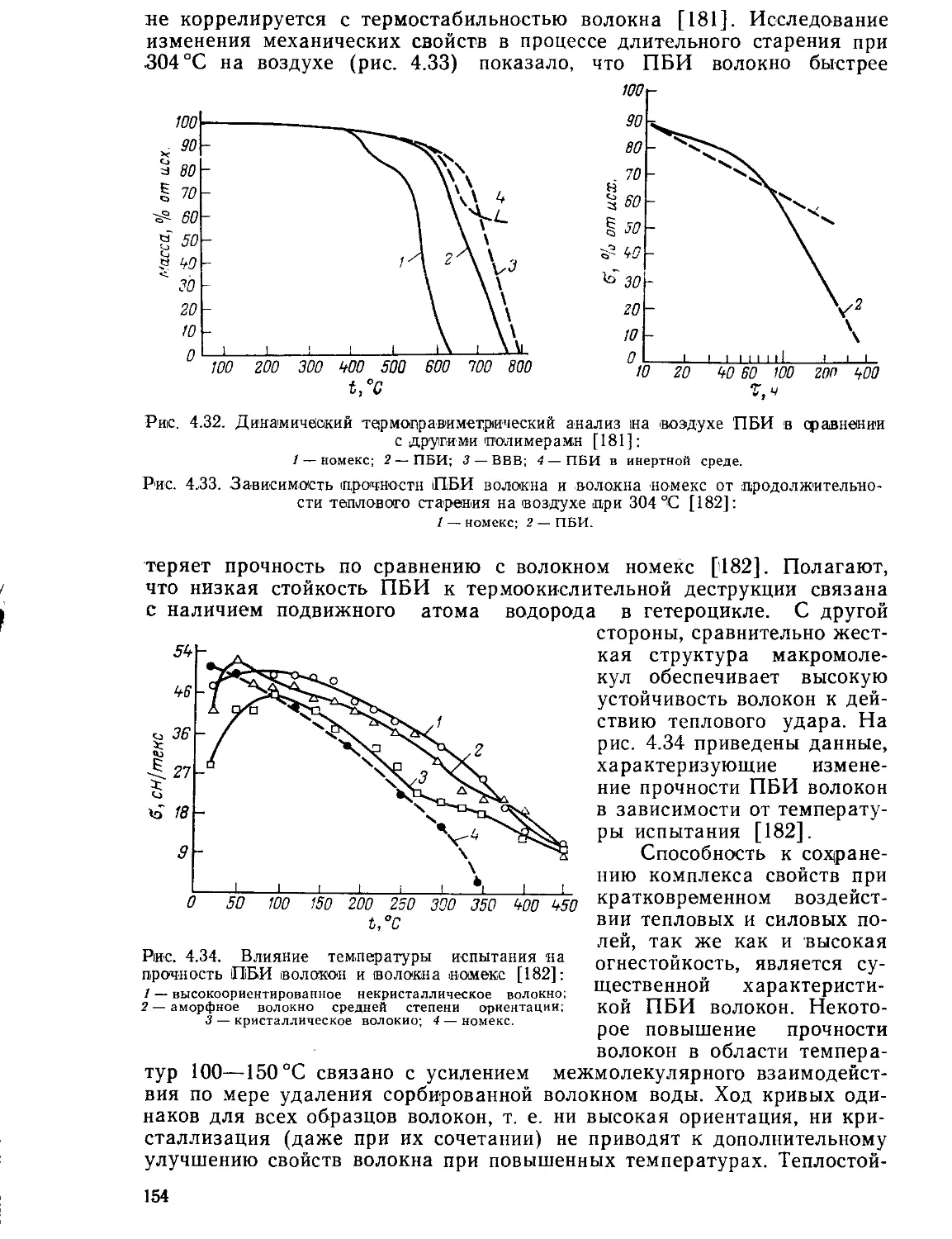




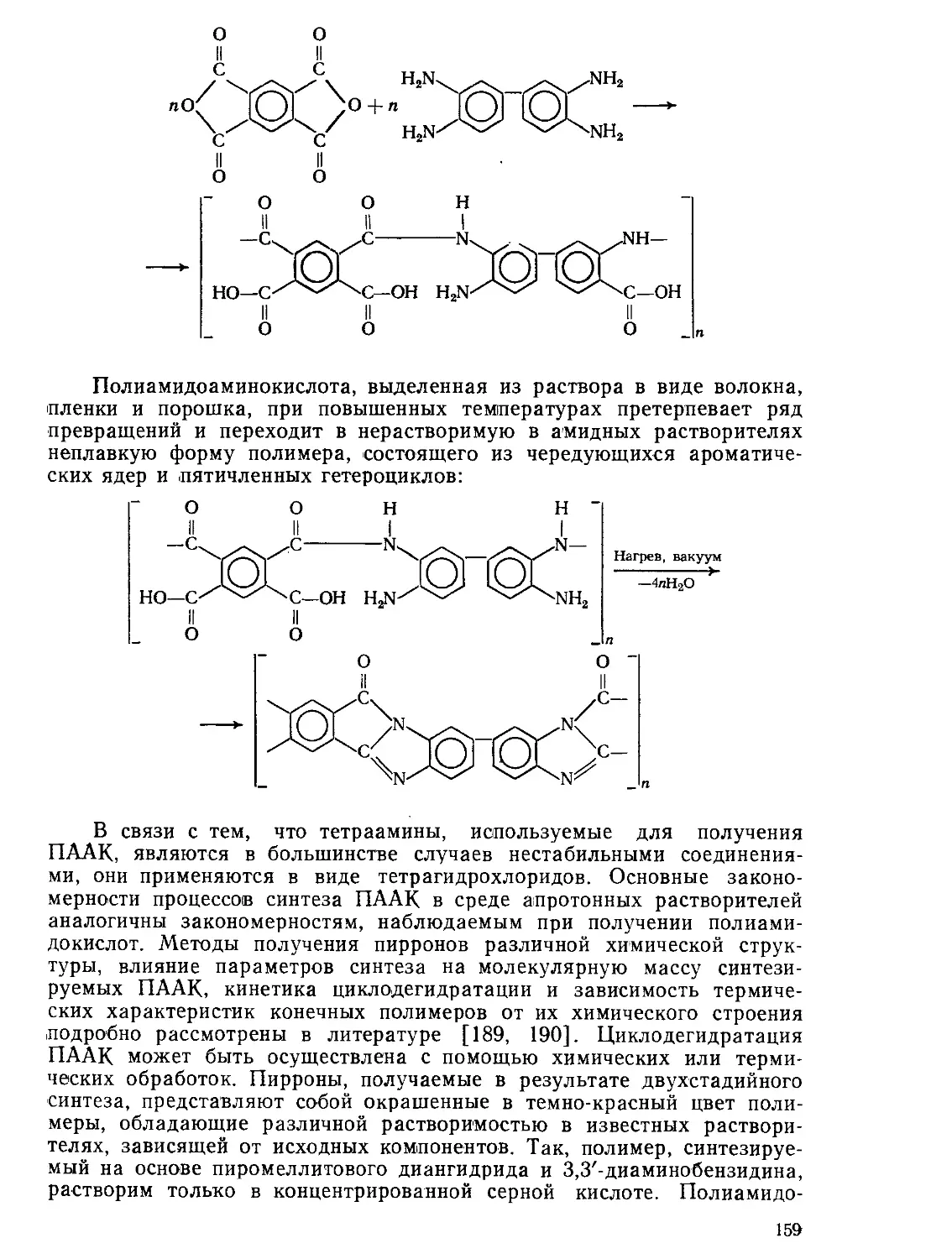



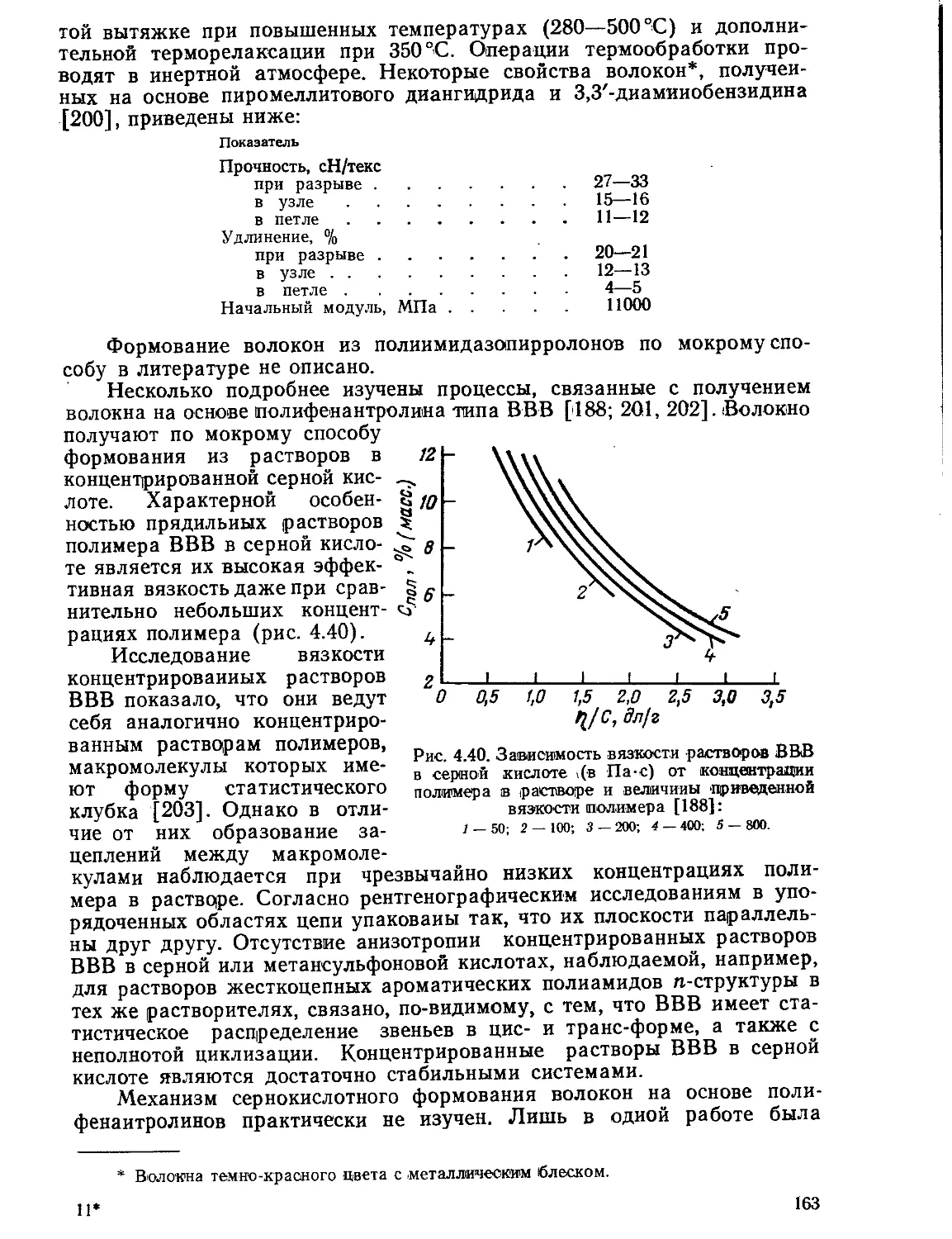





























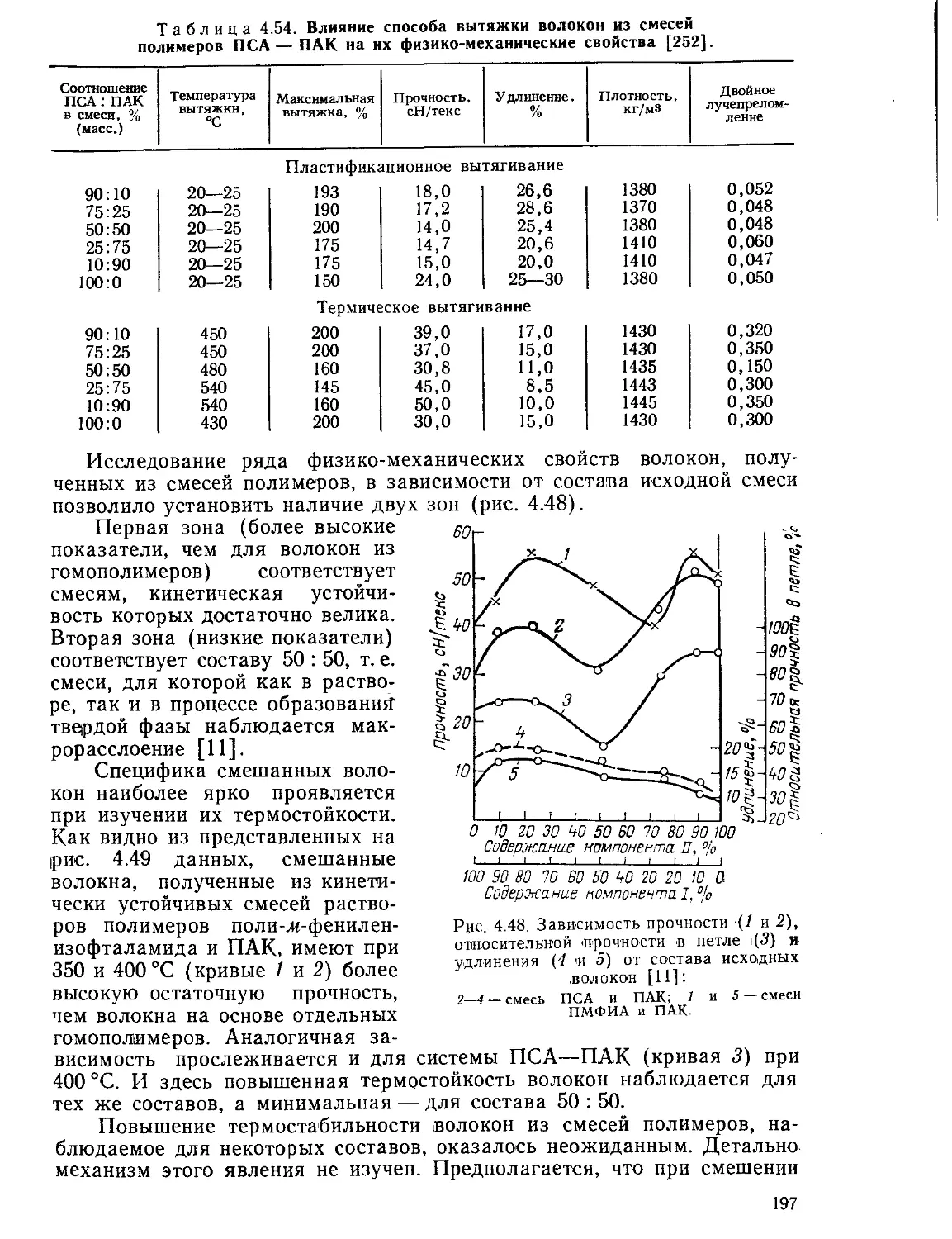
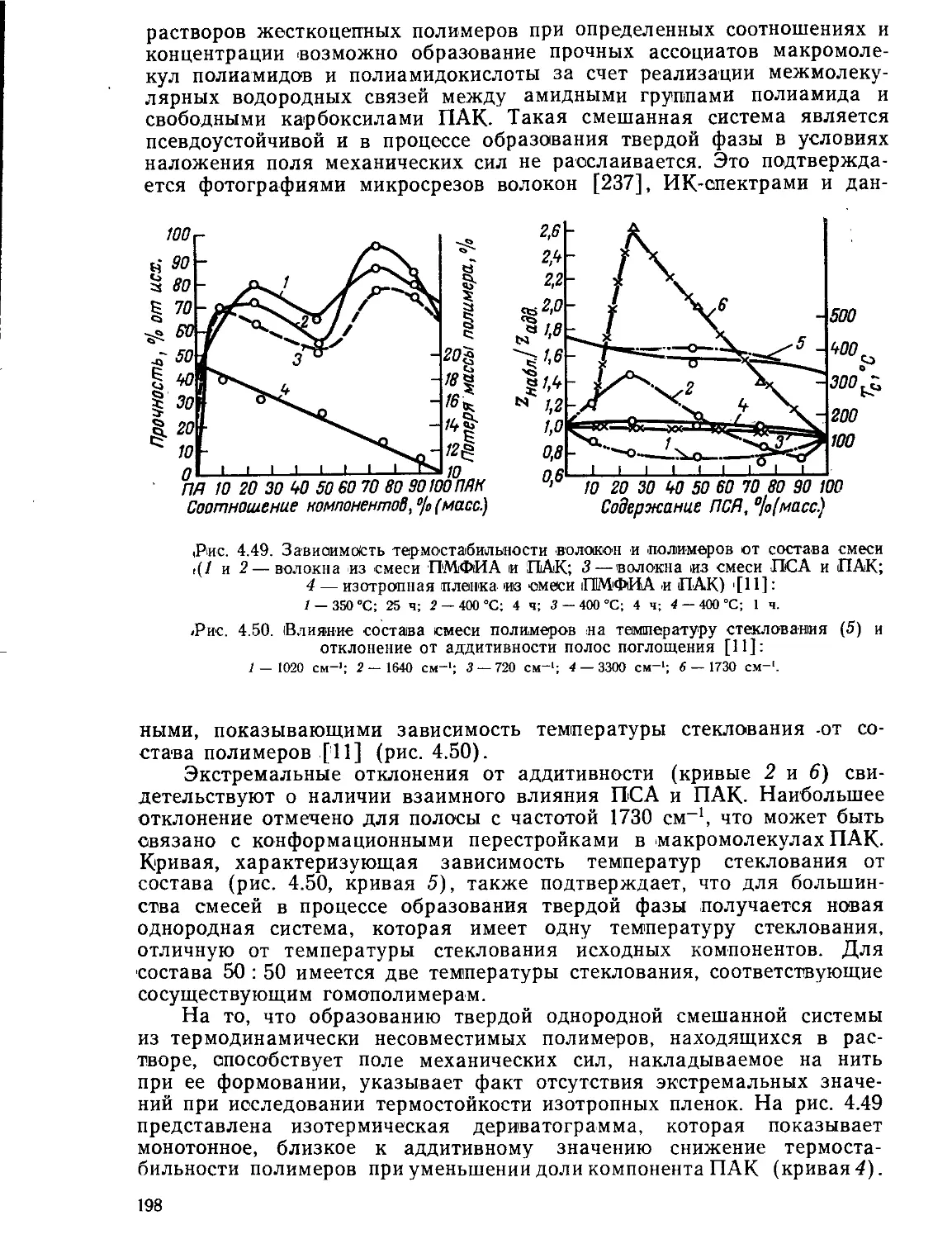










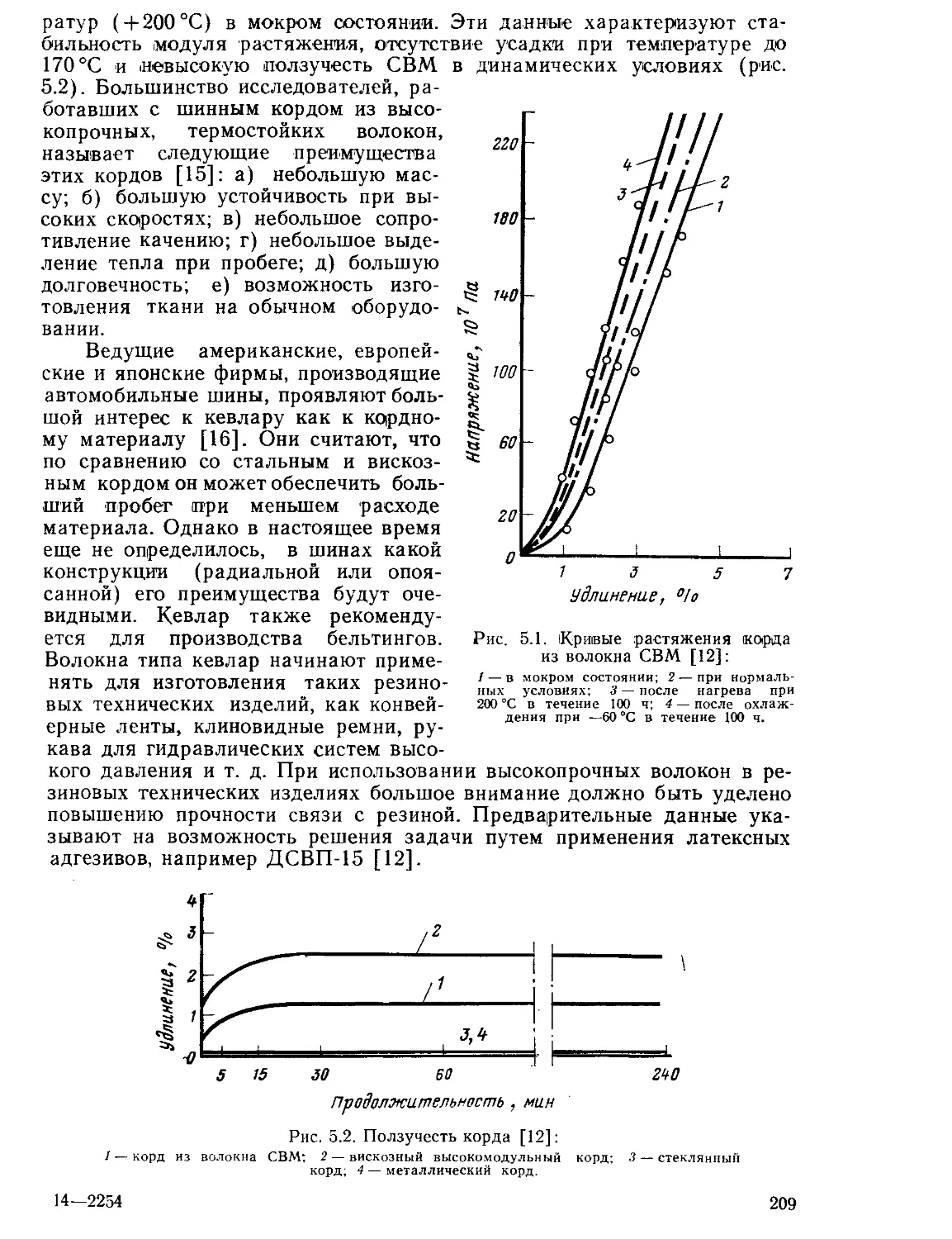




















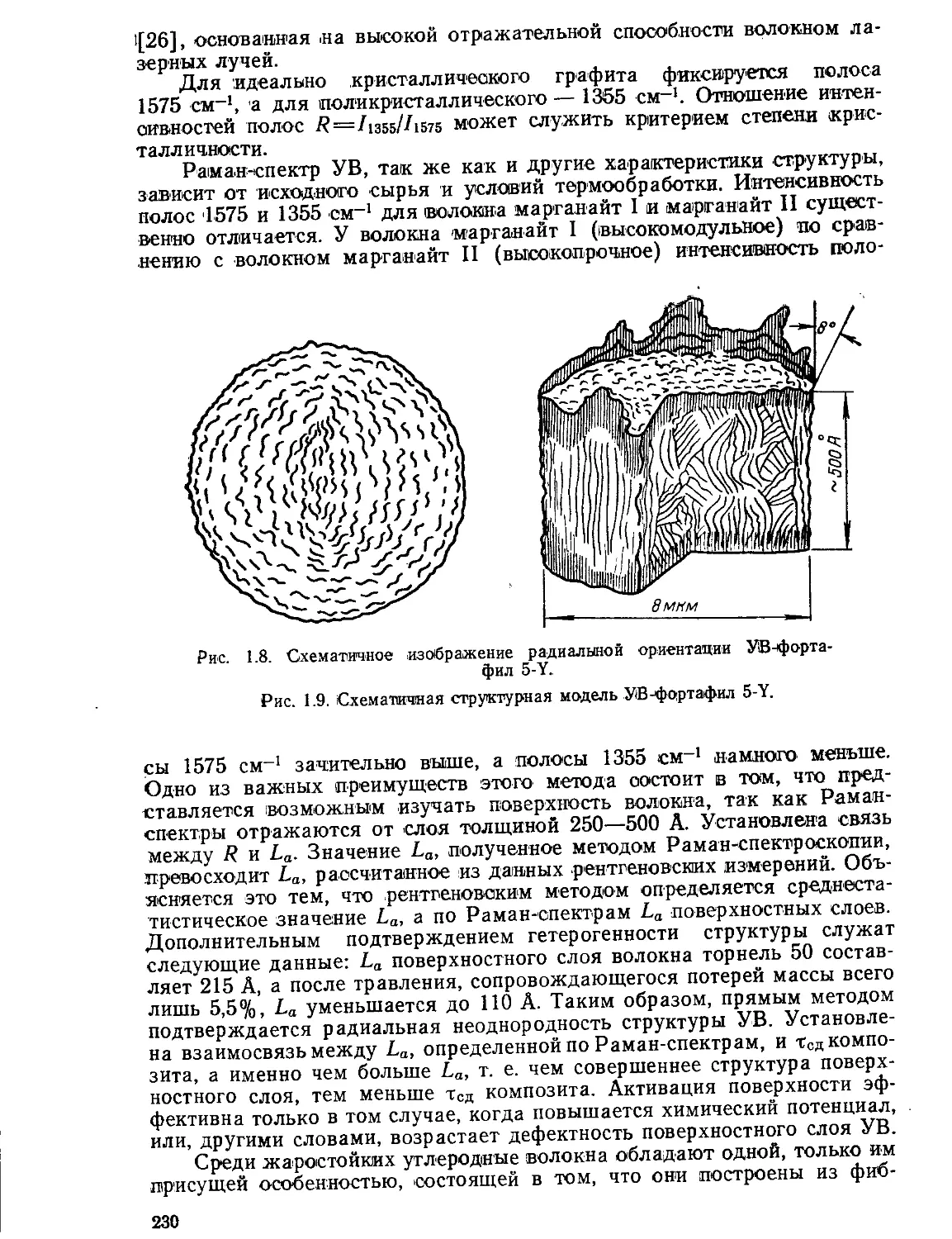














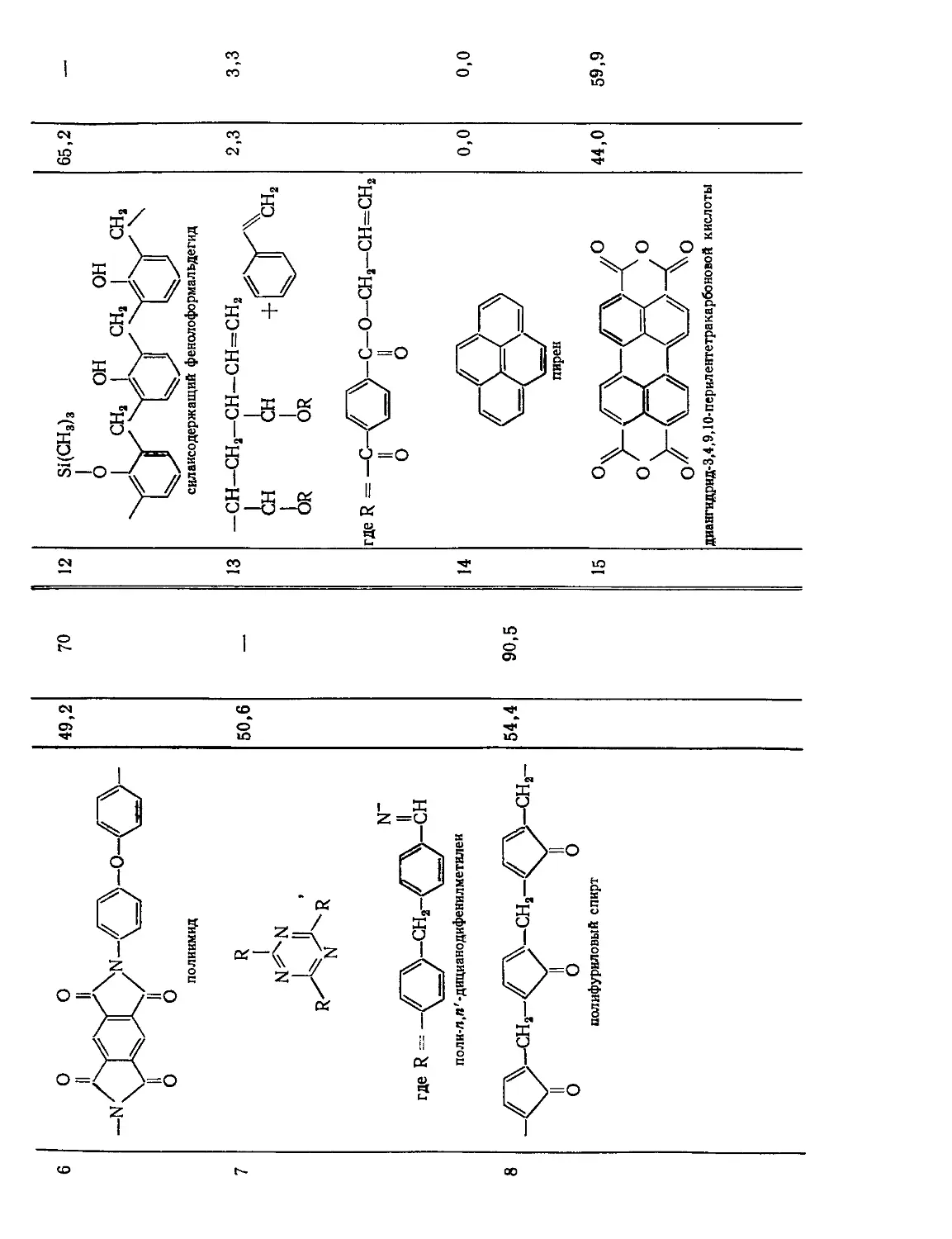






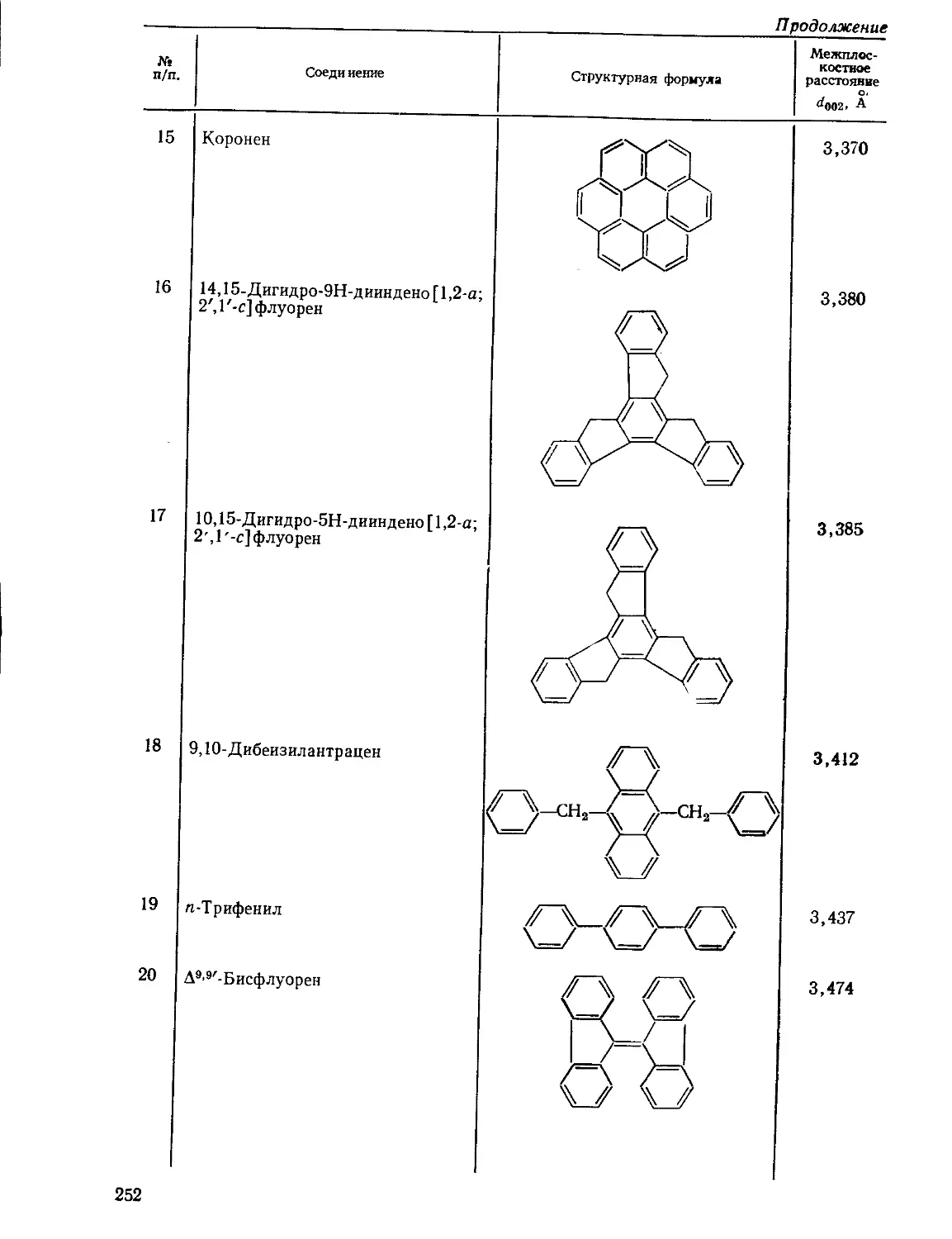
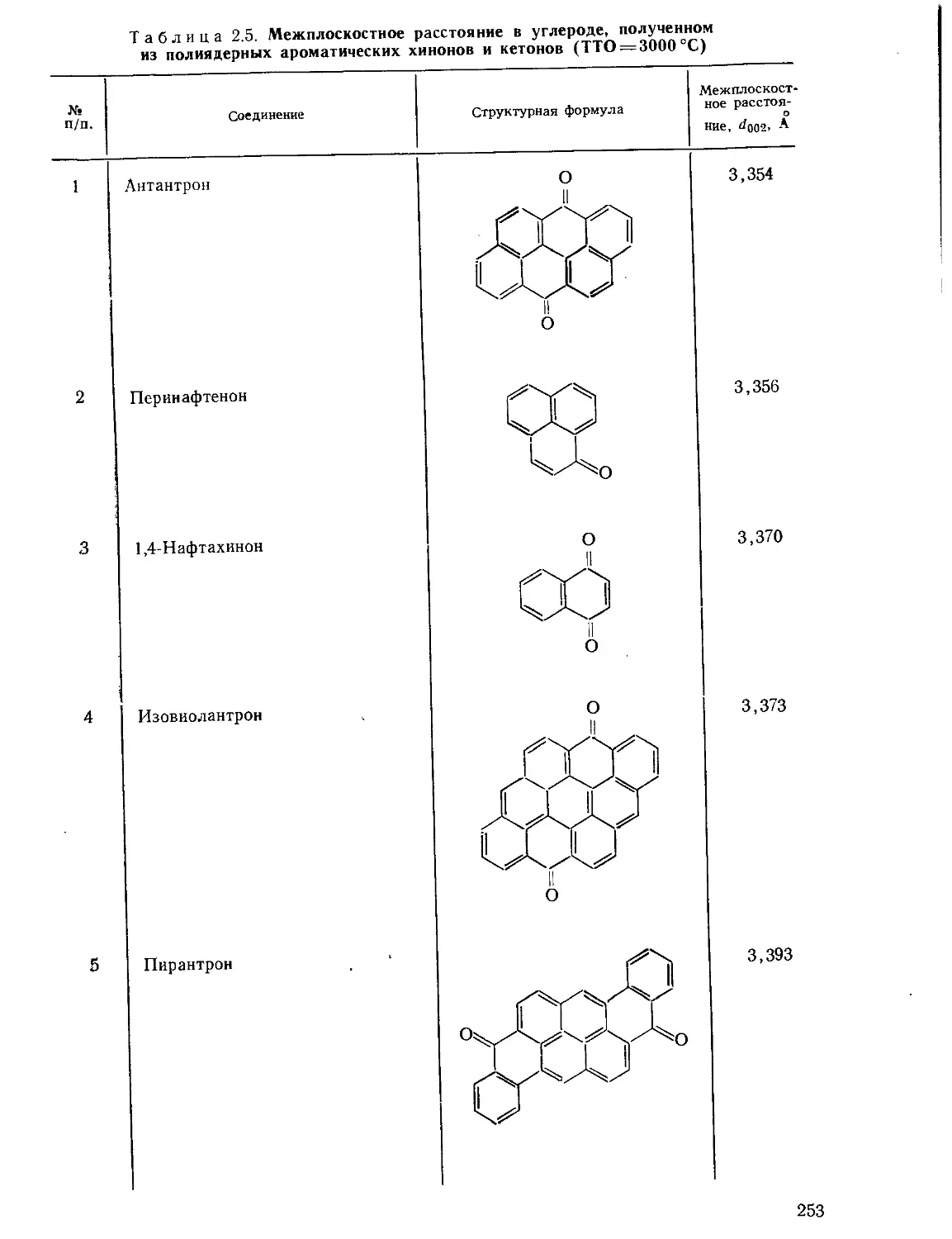

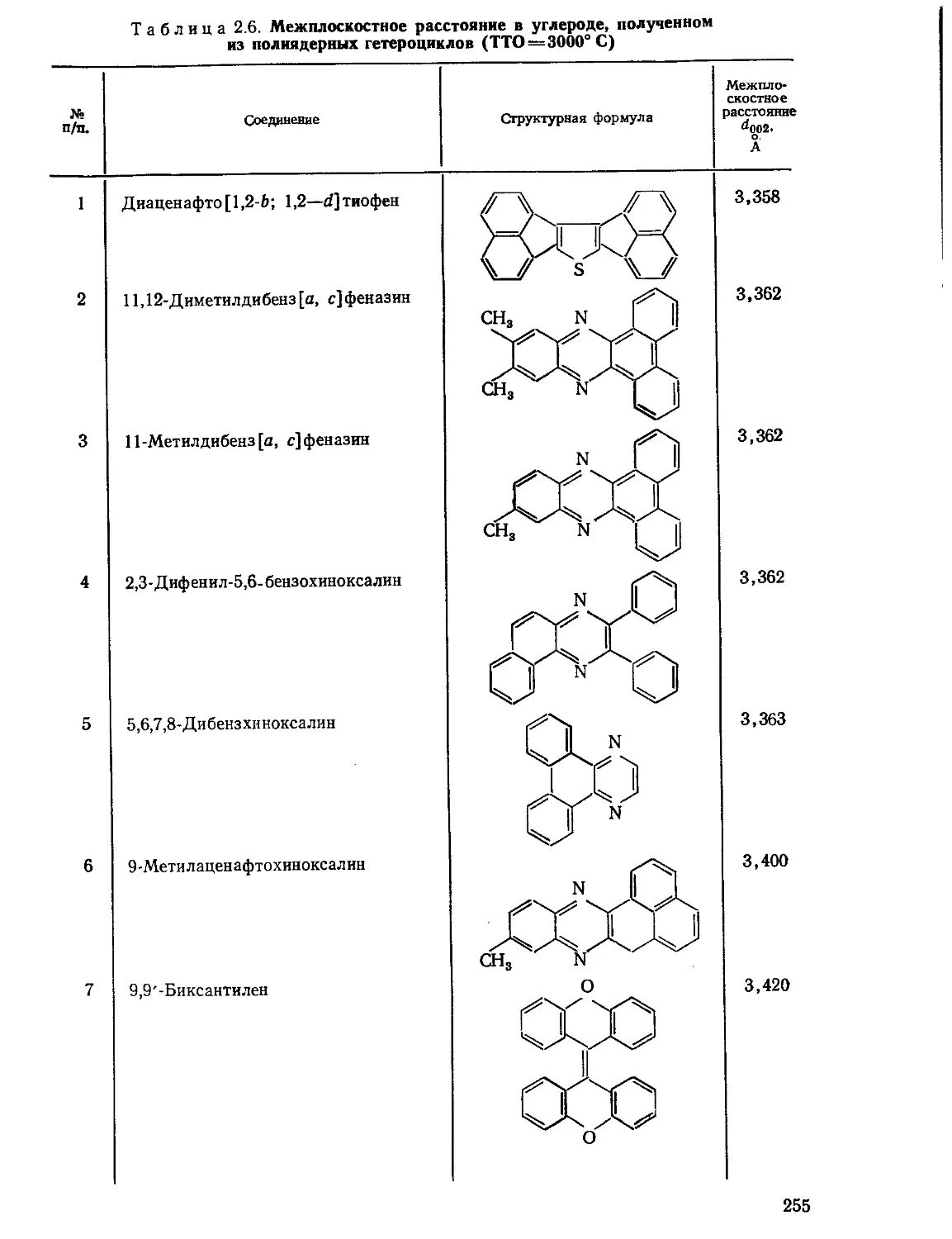










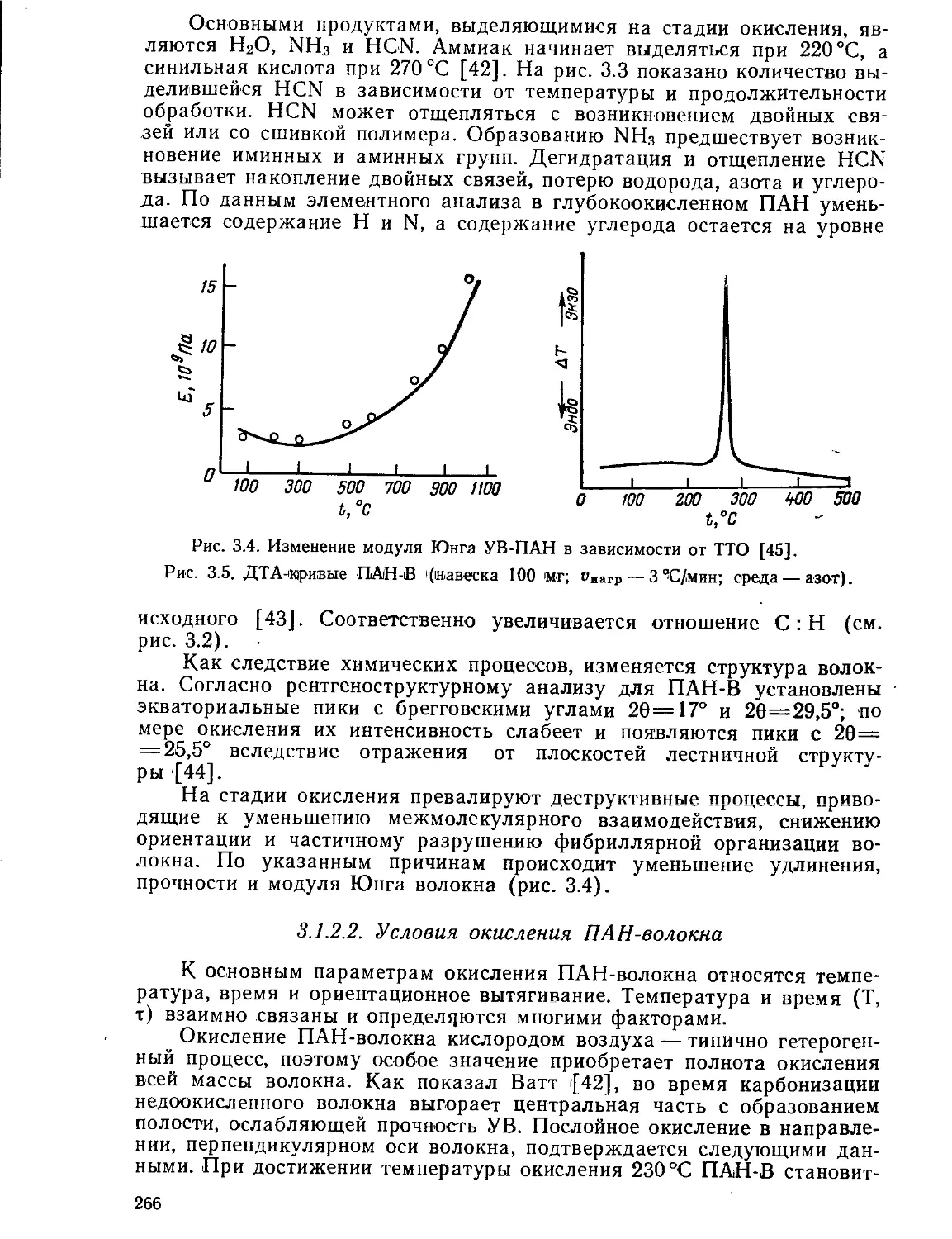


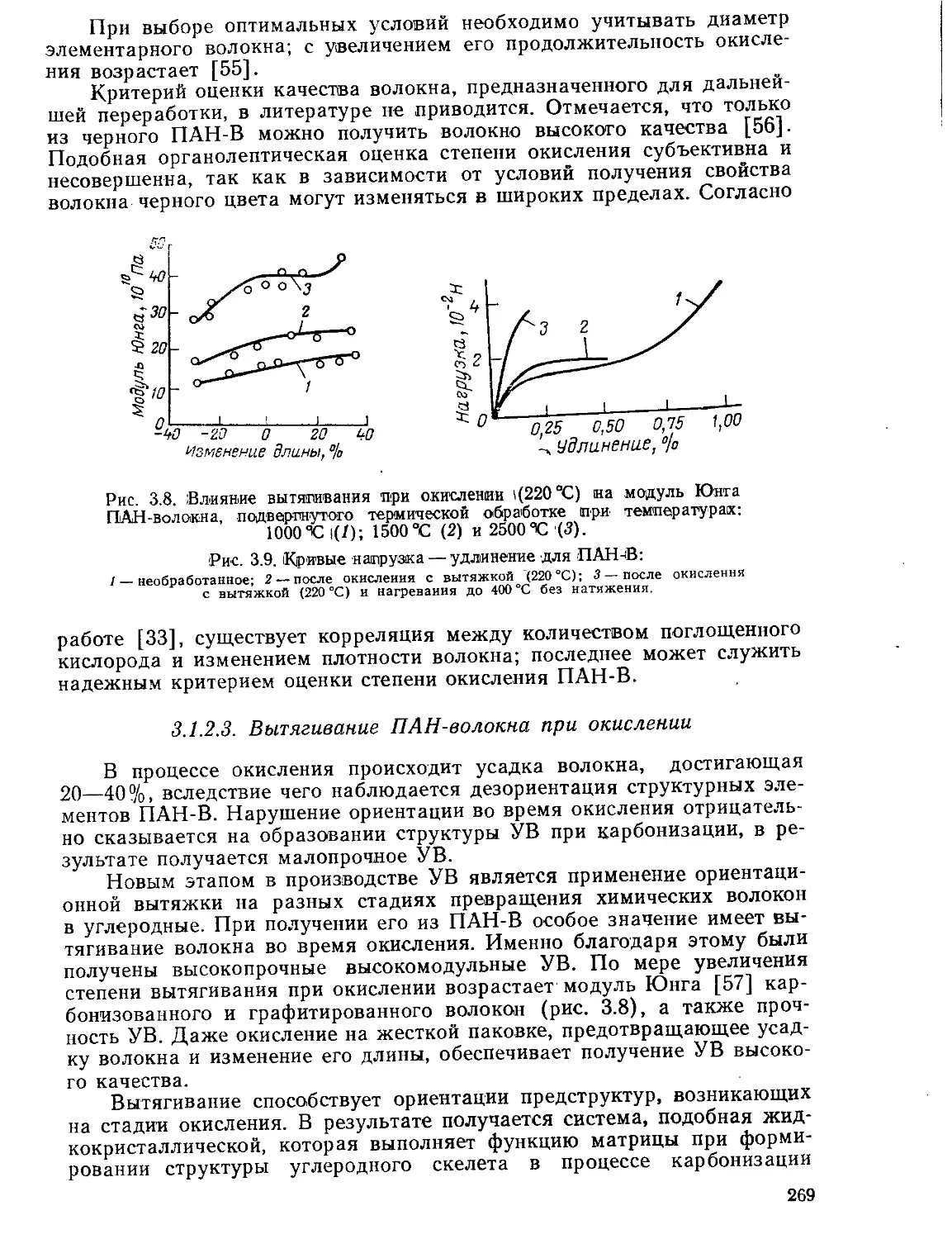


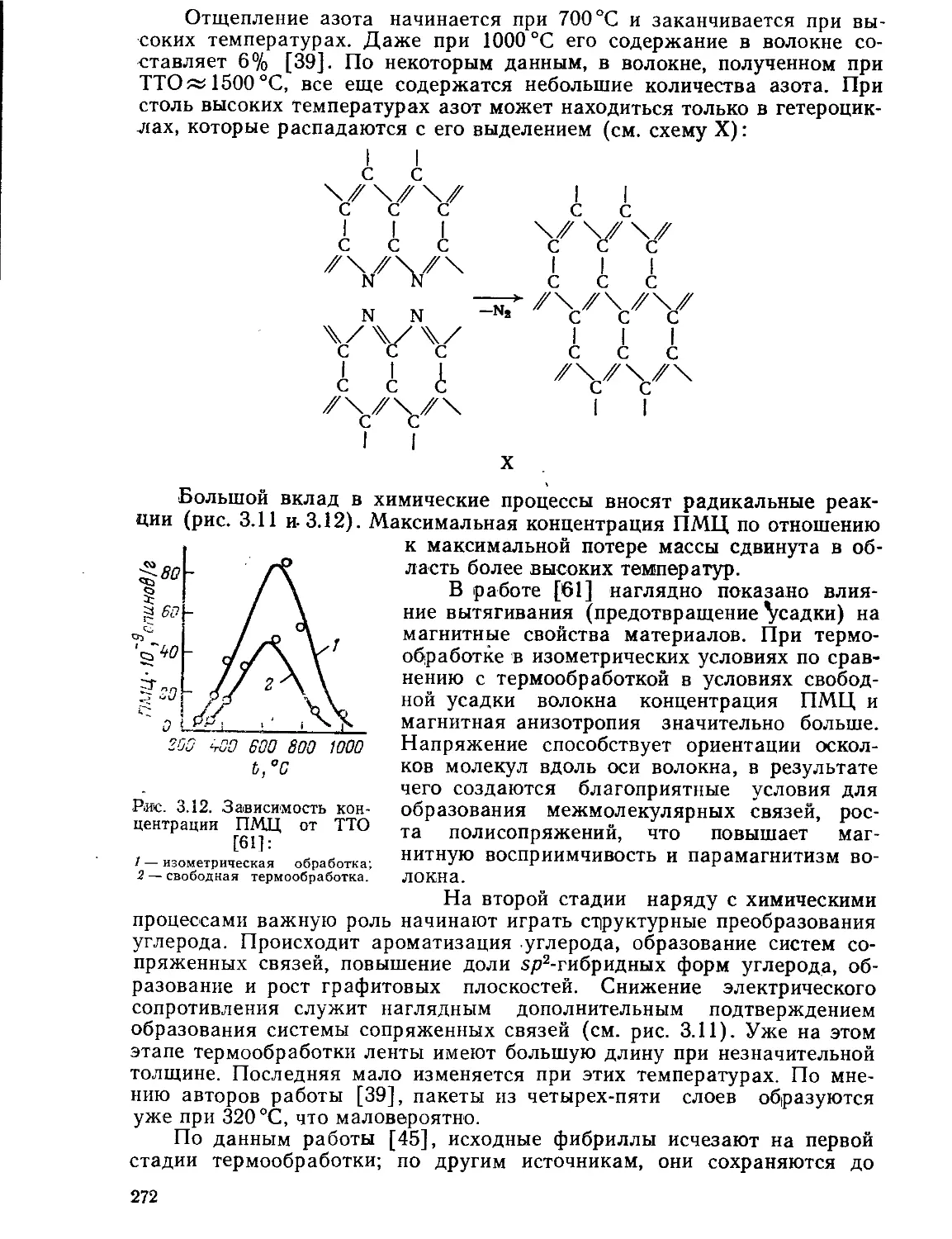










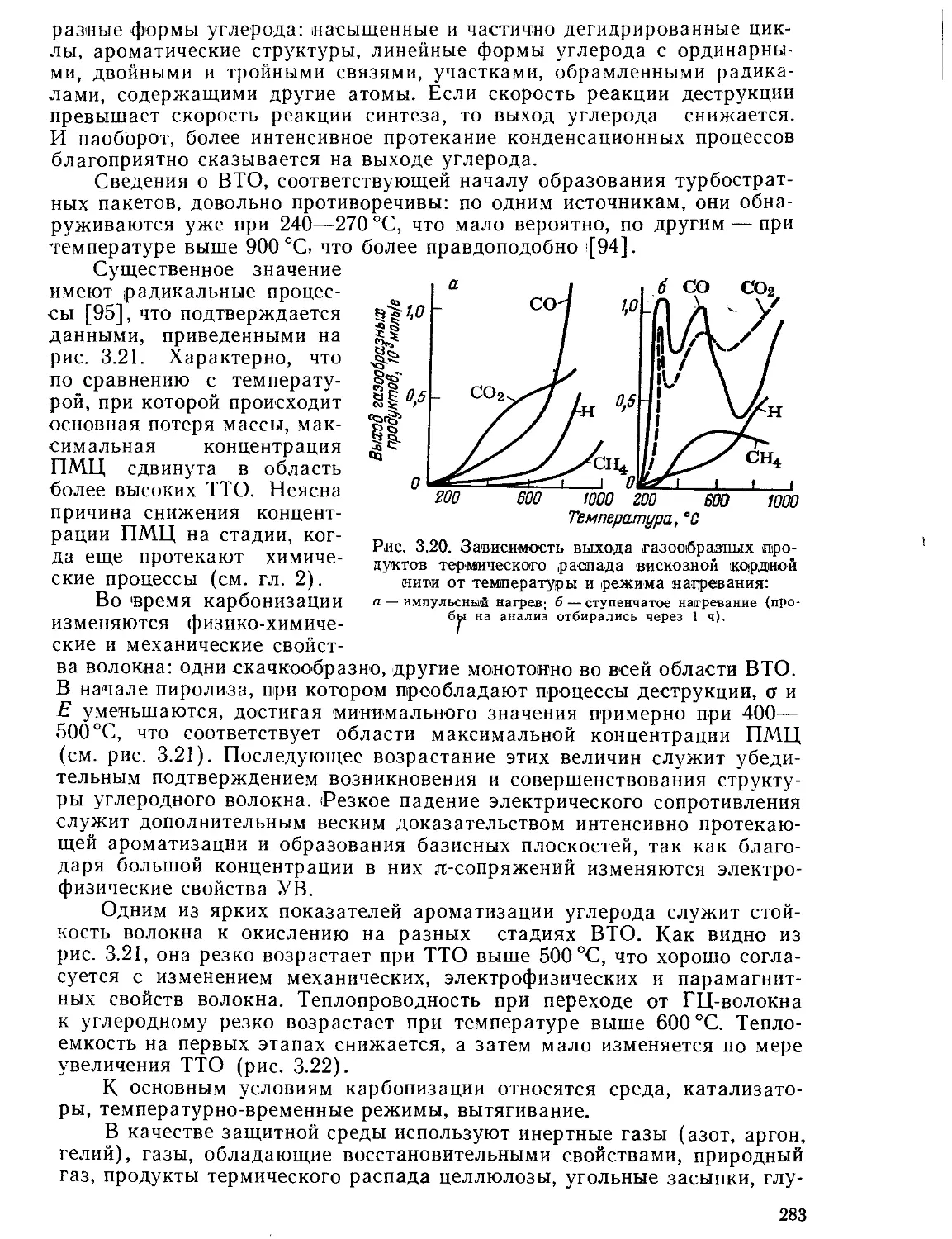
























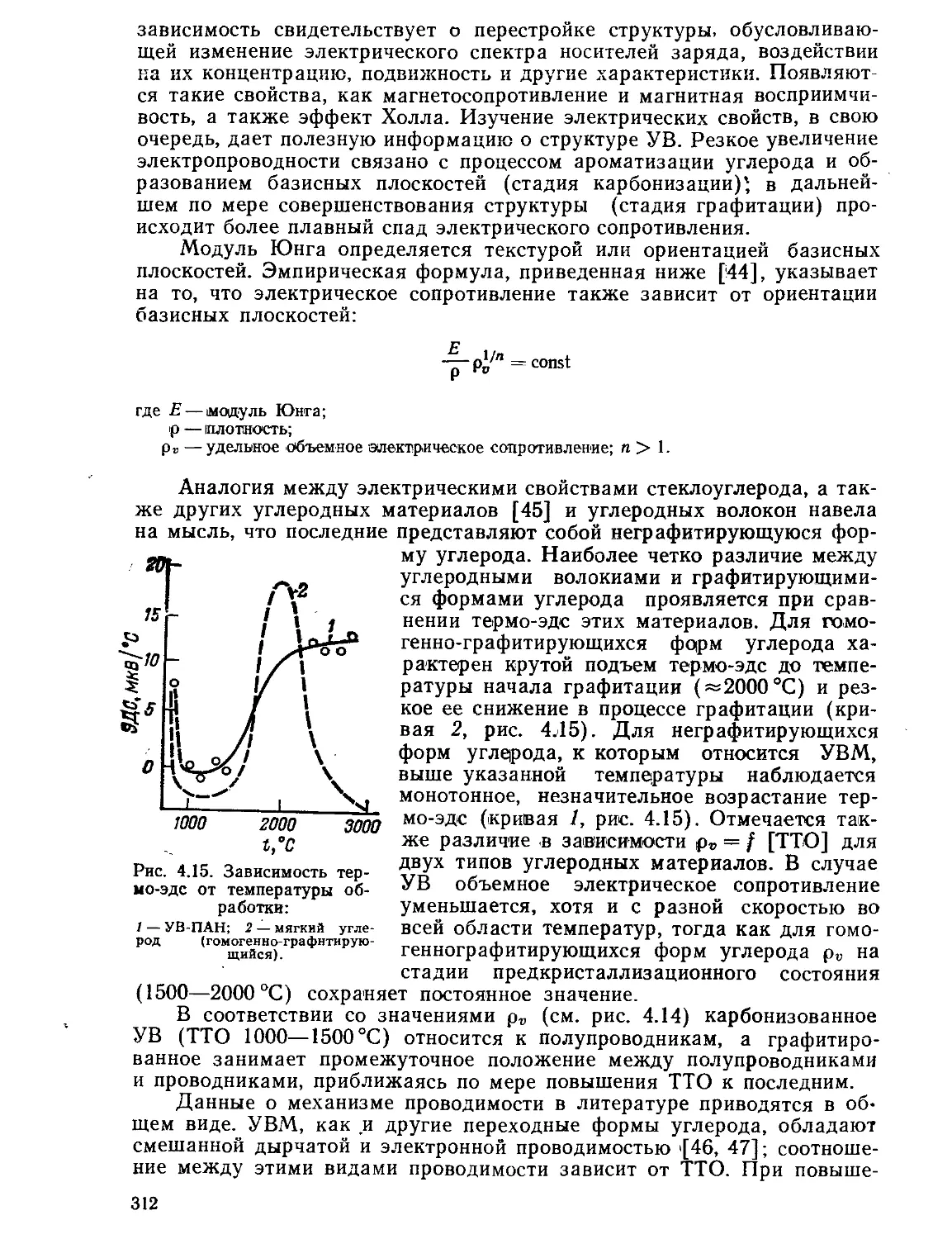




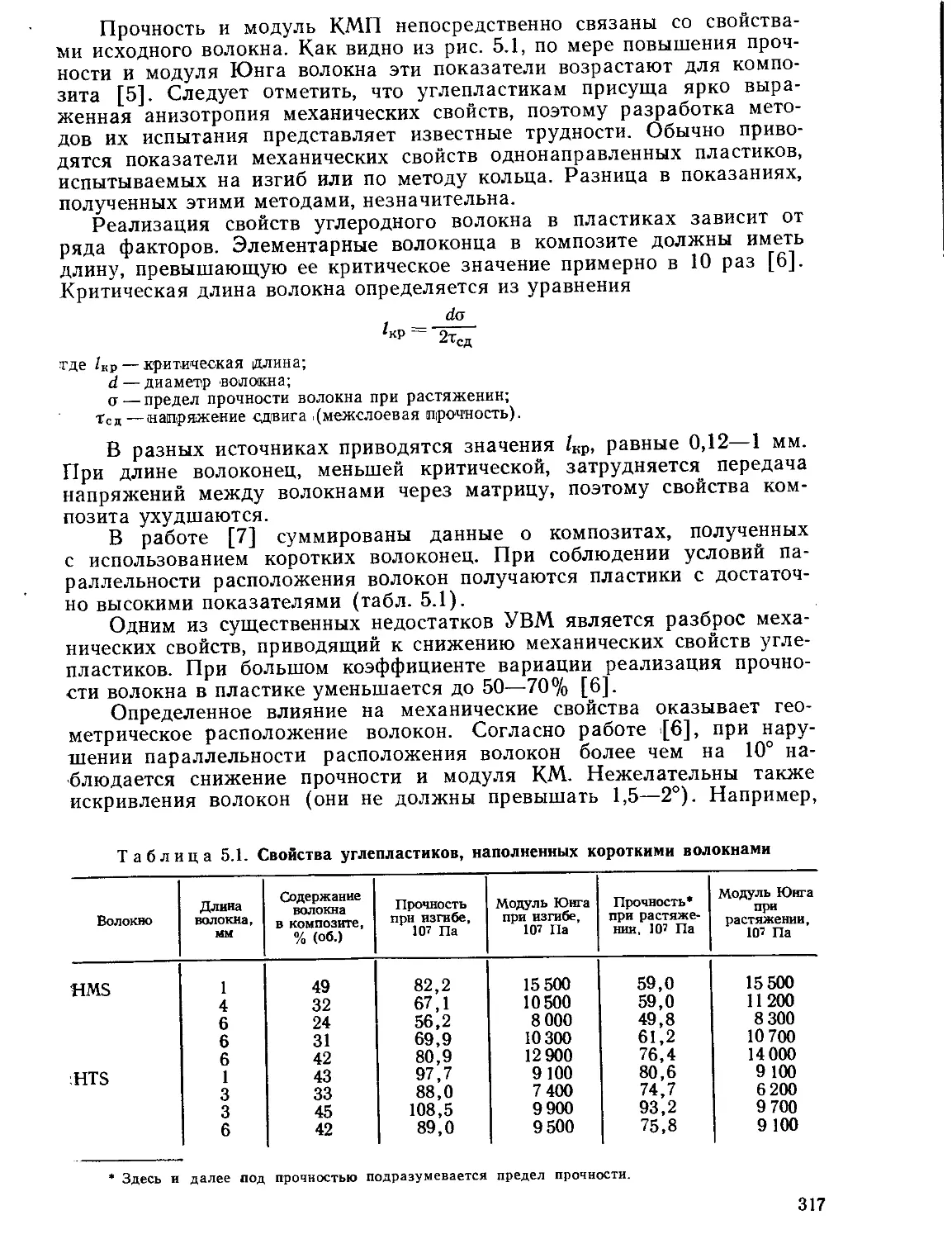




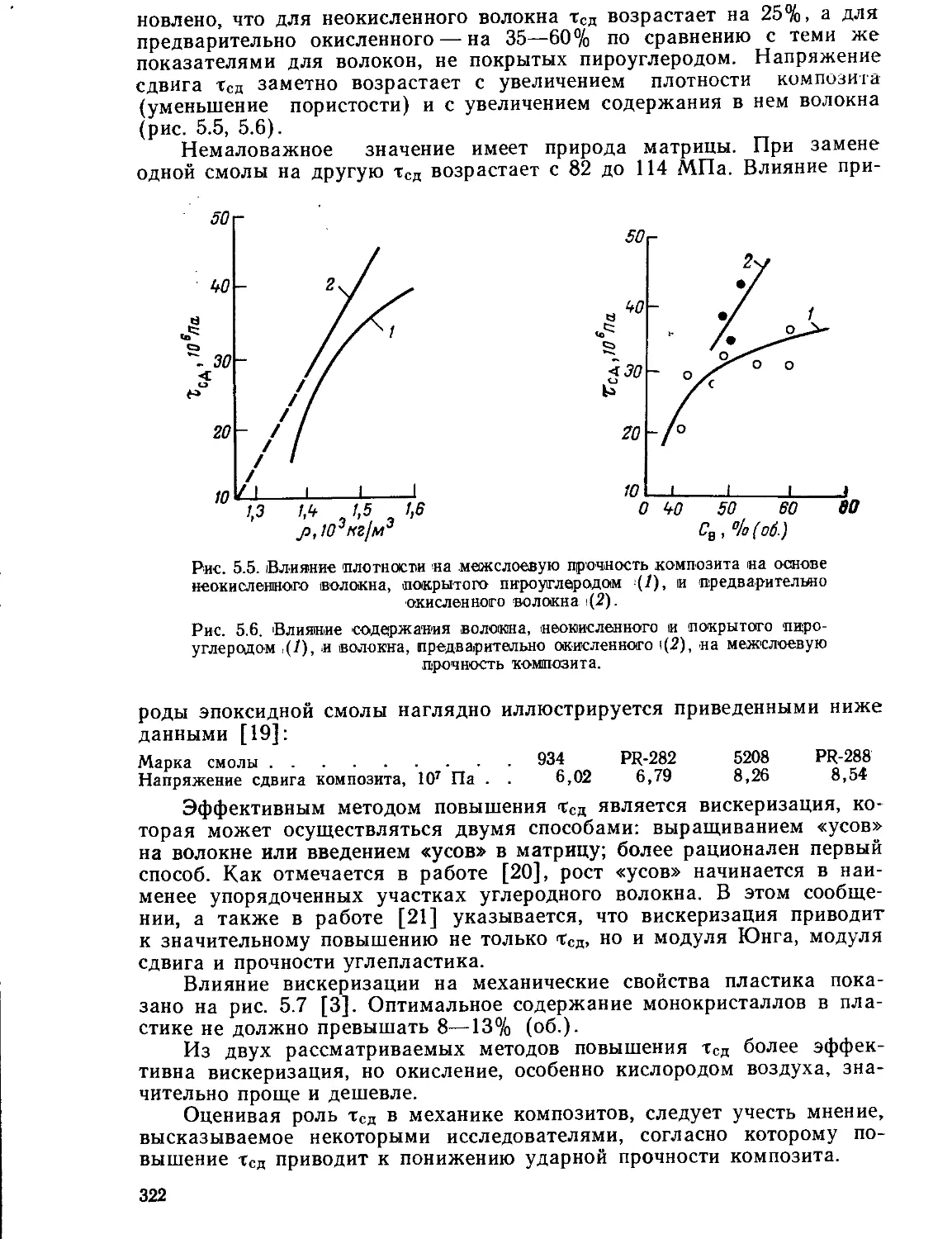

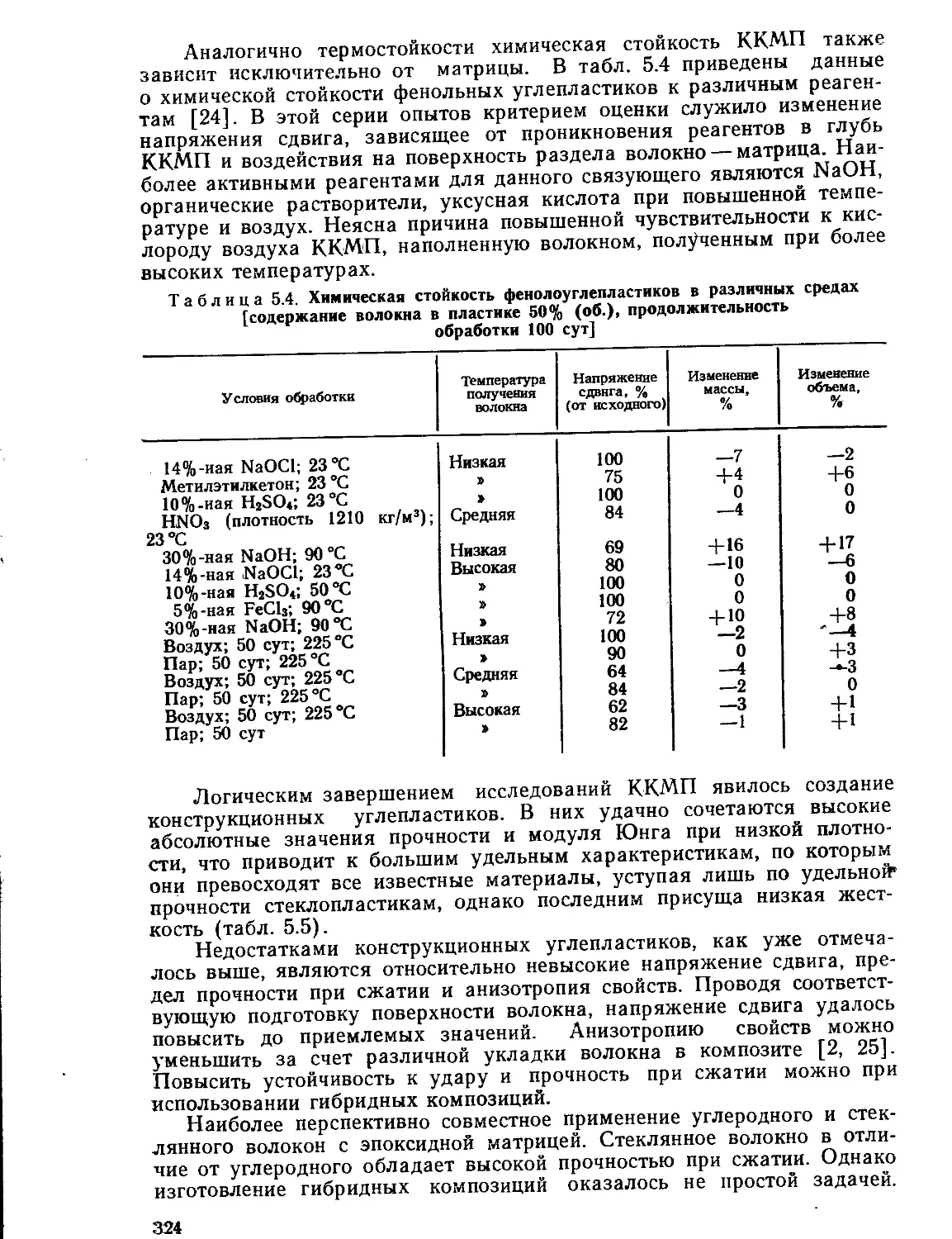


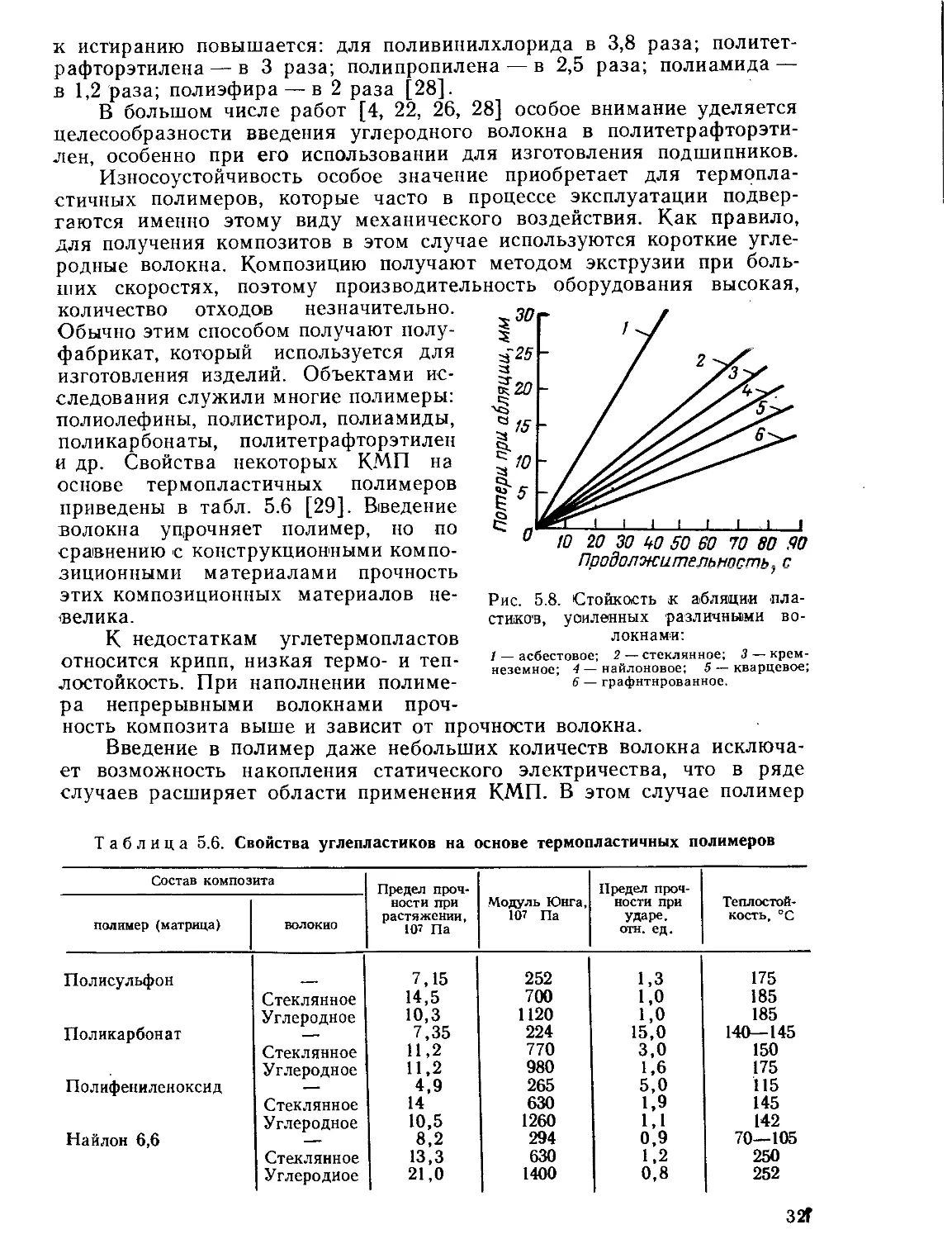




























































































Текст
ХИМИЧЕСКИЕ
ВОЛОКНА
РЕДАКЦИОННАЯ КОЛЛЕГИЯ
АСЛАНОВА М. С., КОНКИН А. А., КУДРЯВЦЕВ Г. И„
ПАКШВЕР А. Б., ПАНКОВ С. П„
ПЕРЕПЕЛКИН К- Е. (зам. председателя),
РОГОВИН 3. А. (председатель), СЕРКОВ А. Т.
МОСКВА • 1978
ТЕРМО-ЖАРОСТОЙКИЕ
И НЕГОРЮЧИЕ ВОЛОКНА
Под ред. А. А. КОНКИНА
'«НГ-
ИЗДАТЕЛЬСТВО «ХИМИЯ»
УДК 677.494:536.49
Термо-, жаростойкие и негорючие волокиа/Под ред.
А. А. Конкина. — М.: Химия, 1978. 424 с., ил.
Монография является шестой книгой из серии «Химиче-
ские волокна». (В ней описаны обладающие особыми свойства-
ми волокна: термостойкие, жаростойкие и негорючие; подробно
рассмотрены физико-химические основы получения этих новых
материалов; (большое внимание уделено областям применения
и перспективам использования таких волокон в народном хо-
зяйстве.
Книга предназначена для научных и инженерно-техниче-
ских работников промышленности химических волокон, а так-
же для специалистов тех отраслей промышленности, где ис-
пользуются Эти волокна. Она (мажет быть полезна аспирантам
и студентам химико-техиологических вузов.
424 с., 117 табл., 155 рис., список литературы 1063 ссылки.
Авторы: Конкин. А. А., Кудрявцев Г. И., Щетинин А. М.,
Дружинина Т. В., Мухин Б. А.
31412-049
Т 050(01)-78 J49’78
© Издательство «Химия», 1978 г.
ОГЛАВЛЕНИЕ
Предисловие............................. 6
Часть I. Термостойкие волокна. Г. И. Кудрявцев,
А. М. Щетинин.......................... "
Часть II. Жаростойкие Чуглеродные) волокна.
А. А. Конкин..........................217
Часть III. Негорючие волокна. Т. В. Дружинина,
Б. А. Мухин...........................342
Предметный указатель ..................417
ПРЕДИСЛОВИЕ
Шестая книга монографии «Химические волокна» посвящена во-
локнам, обладающим специфическими свойствами и предназначенным
для применения в самых различных областях. Эти новые материалы
удачно названы «волокнами третьего поколения». К важнейшим из них
относятся термостойкие, жаростойкие, негорючие и некоторые другие
волокна. Каждому из этих волокон присущи ценные, а по ряду показа-
телей уникальные механические и физико-химические свойства.
Интерес к новым волокнам появился 10—15 лет тому назад, и в
наши дни продолжаются интенсивные исследования, включающие
поиски исходных материалов, способы получения, изучение свойств и
областей применения этих волокон.
Наиболее «старыми» среди новых являются негорючие целлюлоз-
ные волокна. Свойство негорючести этим волокнам придается путем
специальных дополнительных обработок существующих химических во-
локов.
Термостойкие волокна получаются, как правило, формованием из
растворов полимеро®. Из-за большой жесткости макромолекул, зача-
стую наличия сопряженных связей, интенсивного межмолекулярного
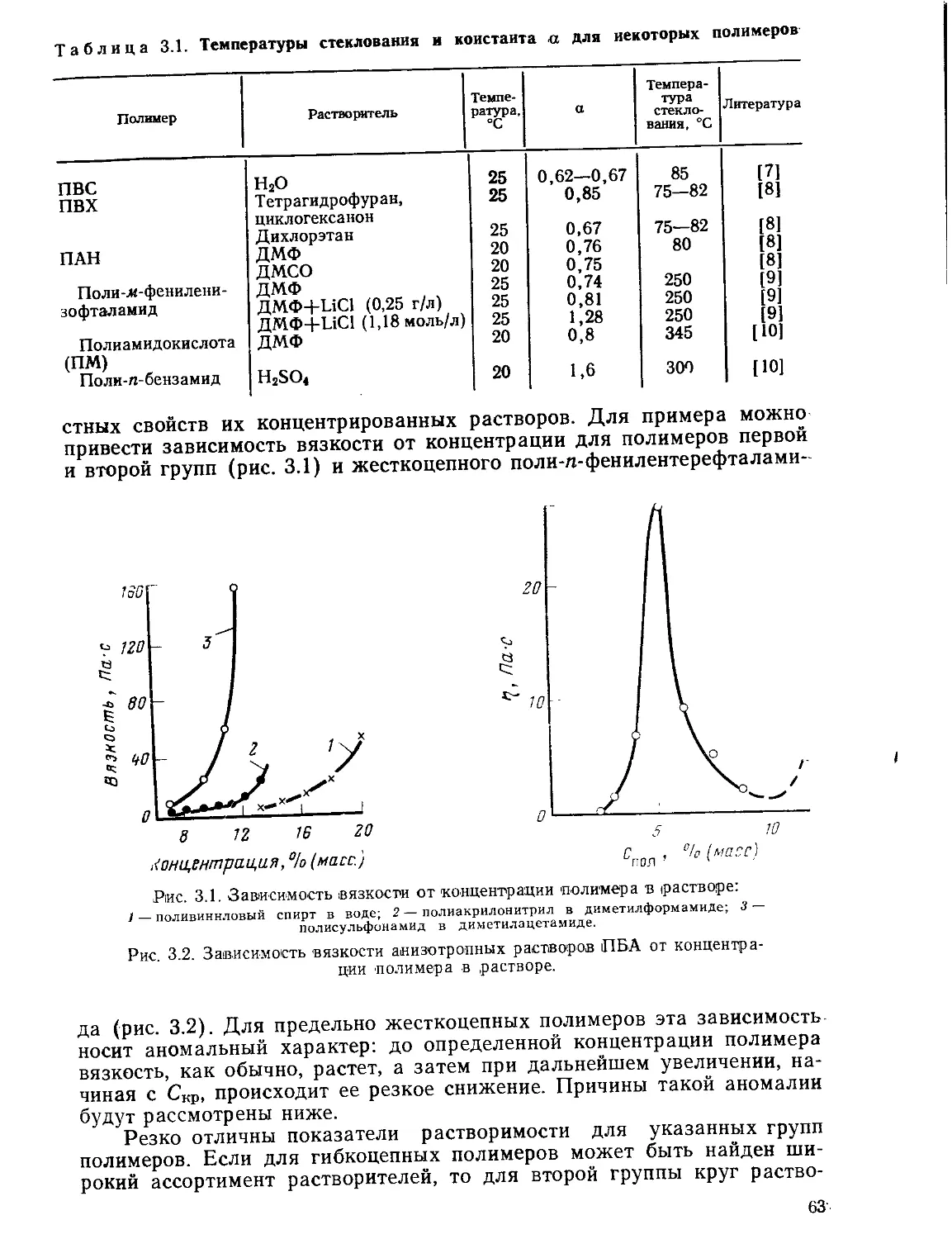
взаимодействия, специфичного жидкокристаллического состояния рас-
творов полимеров, особых методов синтеза волокнообразующих поли-
меров, при получении этих волокон возникали сложные технические
проблемы, которые успешно были преодолены.
Для жаростойких, в том числе углеродных волокон, появилась не-
обходимость в разработке новых методов, так как традиционные спо-
собы получения химических волокон оказались непригодными. Метод
получения жаростойких волокон основан на термодеструкции некото-
рых типов химических волокон, проводимой в инертной среде при стро-
го регулируемых температурно-временных режимах.
В последние годы опубликовано большое число патентов, ориги-
нальных и обзорных статей, посвященных различным аспектам получе-
ния и применения волокон третьего поколения, поэтому назрела необ-
ходимость в обобщении материала и целесообразности издания спе-
циальной книги, входящей в монографию «Химические волокна».
В связи с научно-технической новизной, особыми областями при-
менения некоторых типов волокон (термостойких, жаростойких) в ли-
тературе преимущественно рассматриваются физико-химические осно-
вы процессов получения волокон; технология и особенности аппаратур-
ного оформления процессов пока еще не освещаются. Это отразилось на
содержании книги, в которой излагаются принципы получения, свойст-
ва и области применения новых волокон. Потенциальные возможности
перечисленных волокон далеко еще не использованы, поэтому они пред-
ставляют собой материалы не только настоящего, но и будущего.
Книга, вероятно, не лишена недостатков, поэтому авторы заранее
благодарят читателей, которые выскажут свои замечания по содержа-
нию книги.
АВТОРЫ
Часть I
ТЕРМОСТОЙКИЕ
ВОЛОКНА
ПРИНЯТЫЕ СОКРАЩЕНИЯ
Г1МФА — гексаметилфосфортриамид
ДМАА —N -дам етил ацет амид
ДМСО — диметилсульфоксид
ДМФ — диметилформамид
ИФХ —дихлорангидрид изофталевой кислоты
МП — N-метилп'ирролидон
МФДА — л-феиилендиамин
ПА —полиамиды
ПБИ — полибензимидазолы
ПБО — полибензооксазолы
ПИ — полиимиды
ПОД — полиоксадиазолы
ПФДА — я-фенилендиамин
ПХО — полихиноксалины
ТММ —М,М,(М'М'-тетраметилмочевина
ТФК —терефталевая кислота
ТФХ —дихлорангидрид терефталевой кислоты
СОДЕРЖАНИЕ
Введение..............................
Литература ...........................
Глава 1. Исходные мономеры и растворители, применяемые в производстве тер-
мостойких ВОЛОКОН.......................................................14
1.1. Основные классы волокноюбравующих термостойких полимеров .... 14
1.2. Исходные вещества, применяемые для получения термостойких волокон . . 14
1.3. Растворители........................................................
Литература..............................................................39
Глава 2. Общие принципы синтеза термостойких волокиообразующих полимеров 40
2.1. Реакции, протекающие по типу полиацилирования......................42
2.1.1. Механизм реакций ацилирования полифункциональных аминов и их
производных..........................................................43
2.1.2. Роль реакционной среды в реакциях полиацилирования...........50
2.1.3. Побочные процессы при реакциях полиацилирования..............51
2.2. Реакции полигетероциклизации.......................................54
Литература............................................................. 59
Глава 3. Основные принципы формования термостойких волокон .... 61
3.1. Введение.............................................................61
3.2. Получение волокон по мокрому методу..................................64
3.2.1. Растворимость термостойких полимеров...........................64
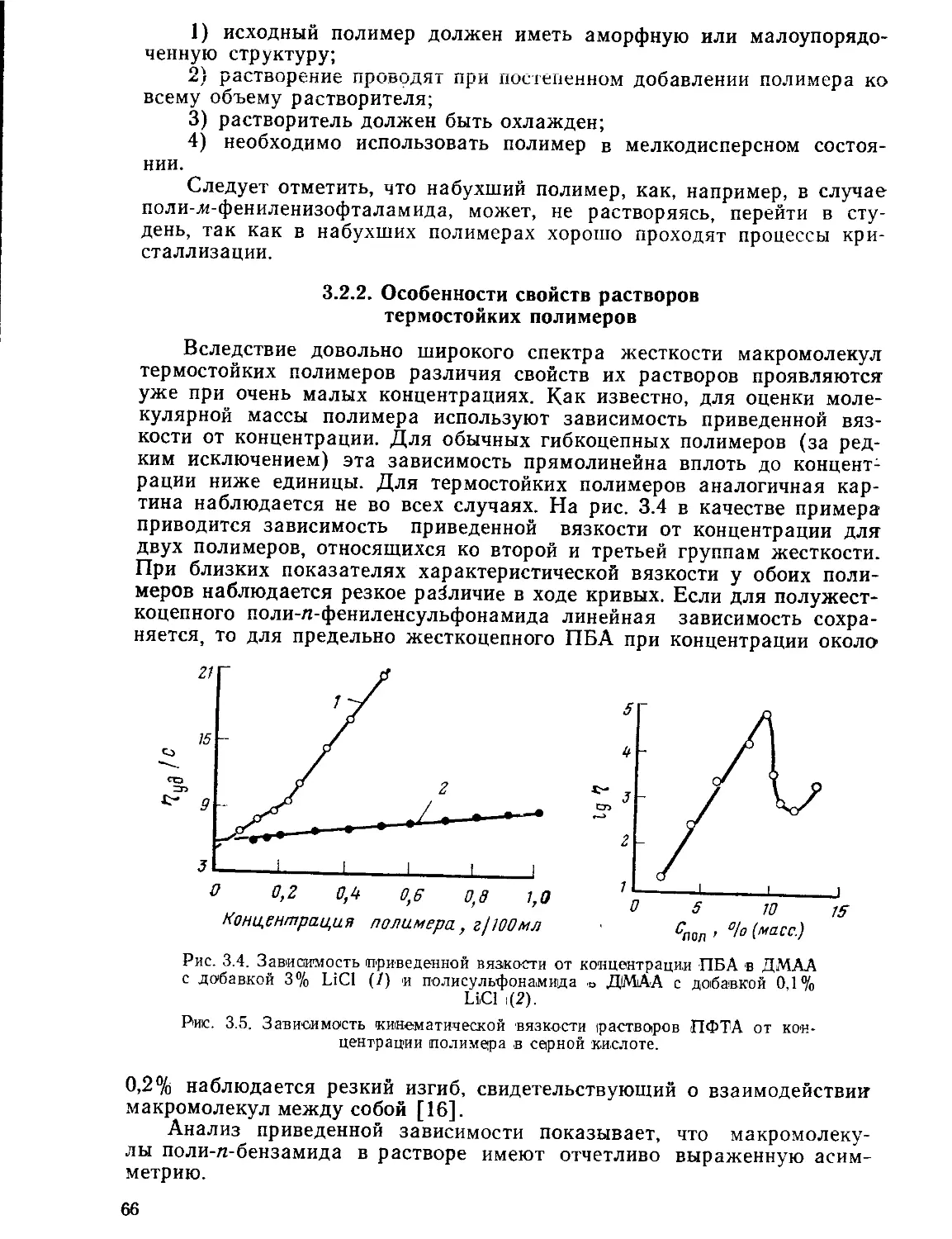
3.2.2. Особенности свойств растворов термостойких полимеров .... 66
3.2.3. Приготовление прядильных растворов.............................69
3.2.4. Закономерности формования термостойких высокопрочных высокомо-
дульных волокон........................................................71
3.2.5. Ориентационное вытягивание свежесформованных волокон .... 75
3.3. Специфические процессы получения термостойких волокон................78
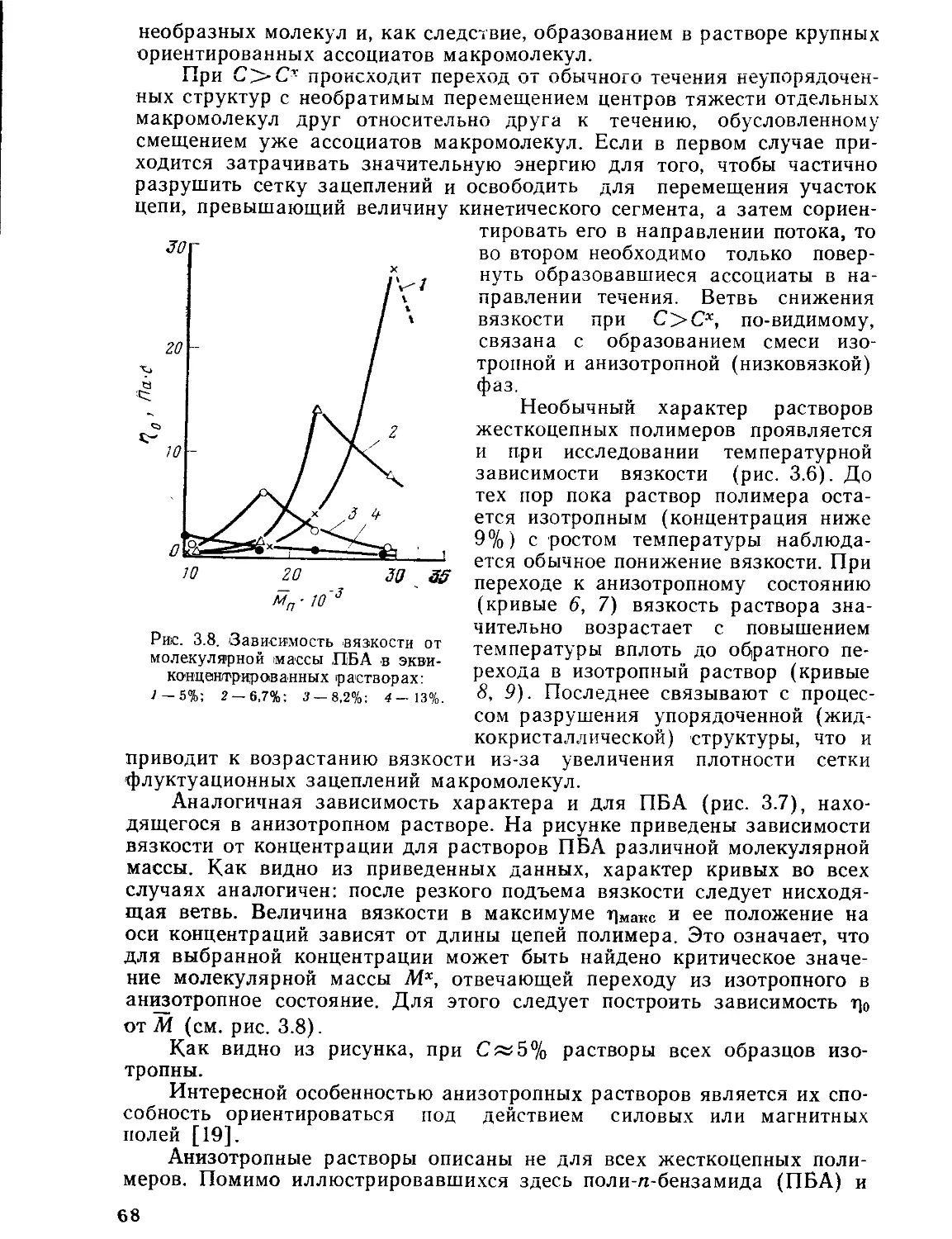
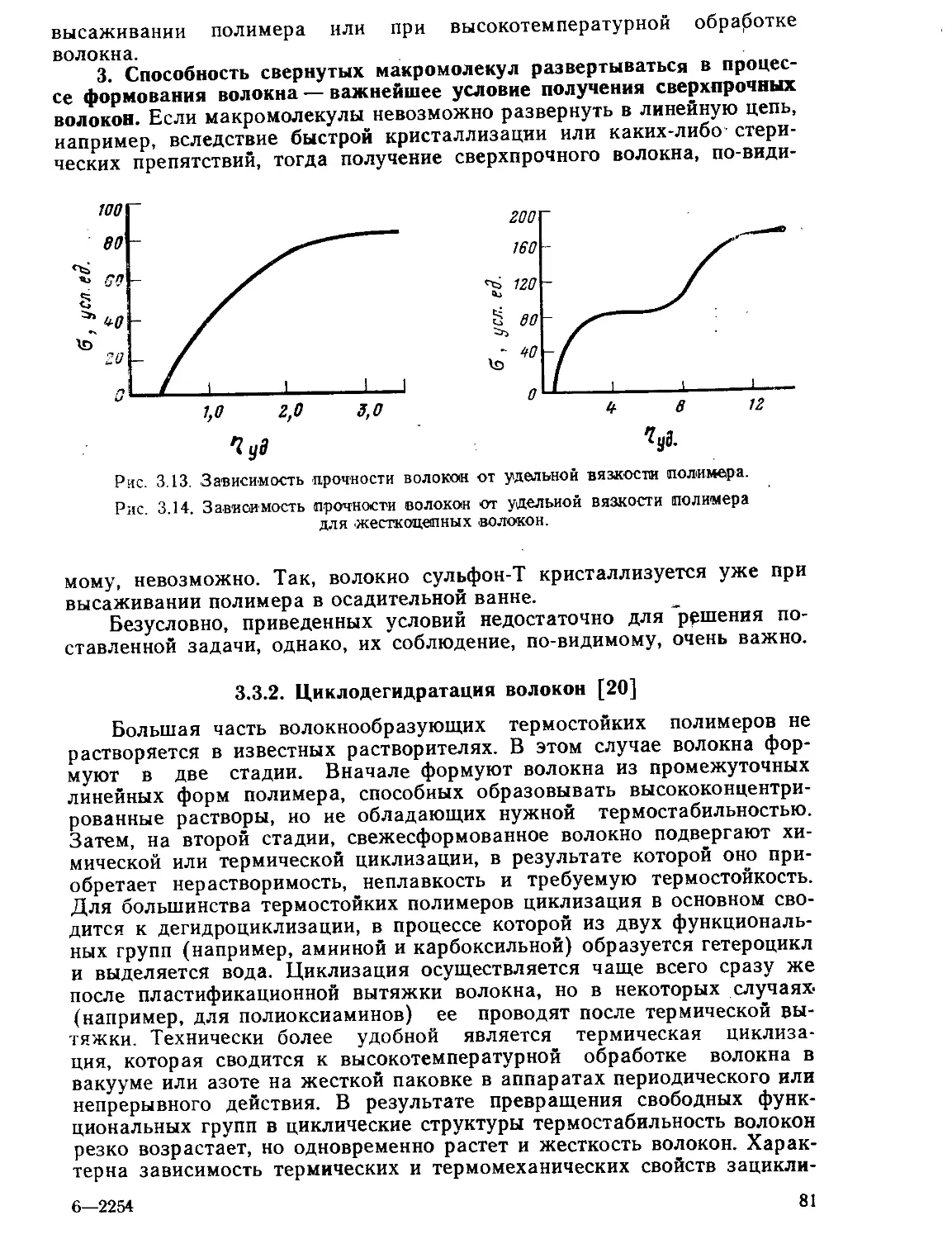
3.3.1. Получение высокомодульных сверхпрочных волокон................78
3.3.2. Циклодегидратация волокон......................................81
3.3.3. Термостабилизация волокон......................................86
3.4. Сухой метод формования термостойких волокон..........................87
3.5. Сухо-мокрый метод формования термостойких волокон....................89
Литература................................................................91
Глава 4. Получение и свойства термостойких волокон..........................92
4.1. Волокна на основе полностью ароматических полиамидов...................92
4.1.1. Синтез полиамидов................................................92
4.1.2. Некоторые особенности изотропных растворов ароматических поли-
амидов ................................................................95
4.1.3. Формование полиамидных волокон...................................97
4.1.4. Ориентационное упрочнение волокон................................99
4.1.5. Физико-механические свойства полиамидных волокон................101
4.1.6. Термомеханические свойства полиамидных волокон ...... 106
4.1.7. Термо- и светостабилизация полиамидных волокон..................107
4.2. Полиимидные волокна.................................................112'
4.2.1. Получение растворов полимеров...................................112
4.2.2. Получение волокон...............................................116
4.2.3. Физико- и термо механические свойства полиимидиых волокон . . 121
4.2.4. Структурные особенности полиамидных волокон.....................129
4.3. Полиоксадиавольные волокна............................................130
4.3.1. Получение волокон по двухстадийиому способу.....................133
4.3.2. Получение полиоксадиазольных волокон по одностадийному способу 135-
4.3.3. Свойства растворов полиоксадиазолов в концентрированной серной
кислоте...............................................................137
9»
4.3.4. Формование полиоксадиаэольных волокон из растворов в серной кис-
лоте .................................................................138
4.3.5. Упрочнение полиоксадиазояьных волокон.....................138
4.3.6. Физико- и термомеханические свойства полиоксадиазольных волокон 140
4.4. Полибеизимидазольиые волокна....................................148
4.4.1. Получение и свойства полимеров............................148
4.4.2. Получение и физико-механические свойства полибензимидаэольных
волоком........................................................151
4.4.3. Термомех аиичесние свойства полибензимидаэольных волокон ... 153
4.4.4. Химическая и радиационная стойкость полибензимидаэольных волокон 155
4.5. Волокна лестничного строения....................................157
4.5.1. Методы синтеза и свойства лестничных полимеров............158
4.5.2. Получение волокон на основе лестничных и полулестничных полимеров 162
4.5.3. Физико-механические свойства волокон........................... 165
4.5.4. Термомеханические свойства лестничных волокон.............165
4.5.5. Другие физико-химические свойства волокон.................172
4.5.6. Волокнообразующие лестничные полимеры других типов .... 173
4.6. Полиамидоимидные волокна........................................176
4.6.1. Получение волокон................................................176
4.6.2. Физико- и термомеханические свойства волокон.....................180
4.7. Термостойкие волокна других типов...............................181
4.7.1. Политиади азольные волокна.......................................181
4.7.2. Волокна из полифенилентриазолов..................................183
4.7.3. Полйбензоксазольные волокна......................................184
4.7.4. Термостабильность волокон........................................185
4.8. Химическая и физическая модификация термостойких волокон .... 185
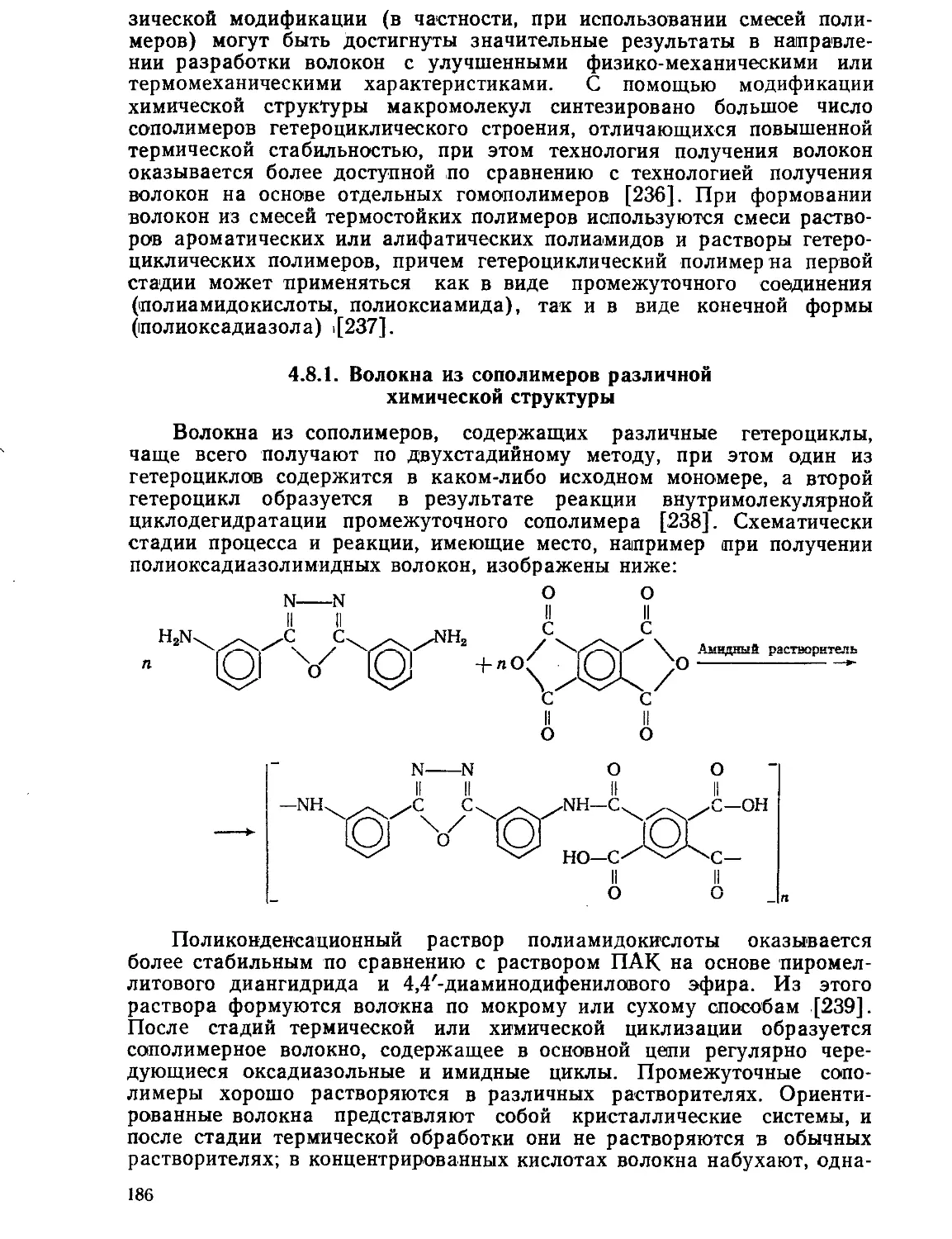
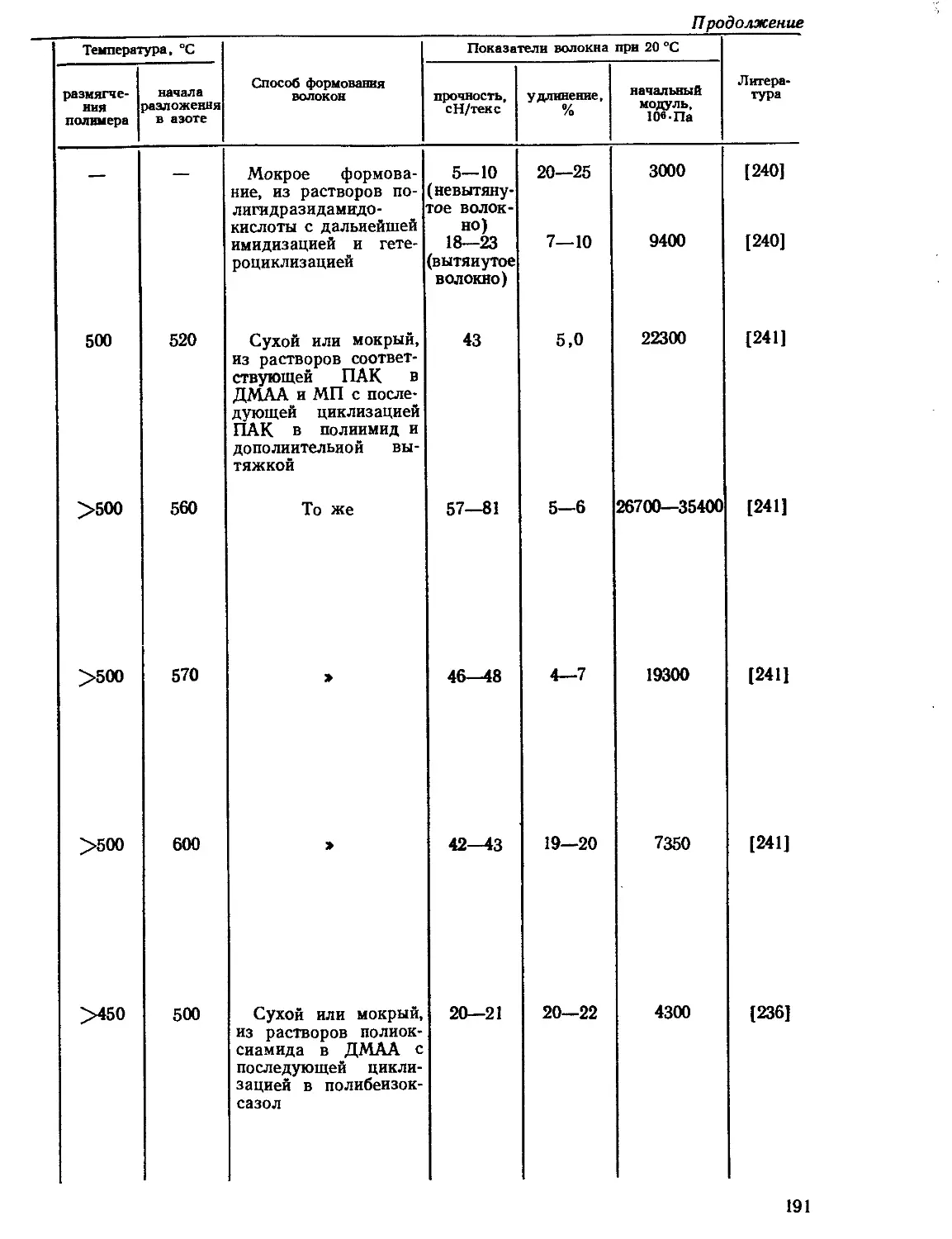
4.8.1. Волокна из сополимеров различной химической структуры .... 186
4.8.2. Волокна на основе смесей полимеров...............................195
Литература .................................................................200
Глава 5. Области применения термостойких волокон......................205
5.1. Фильтровальные и защитные материалы............................ 206
5.2. Антифрикционные полимерные покрытия............................ 207
5.3. Производство шинного корда.......................................207
5.4. Конструкционные материалы........................................210
5.5. Прочие области применения........................................214
Литература ...........................................................215
ВВЕДЕНИЕ
Термостойкими называют такие волокна, которые длительное время
сохраняют необходимые эксплуатационные свойства при температурах
выше области разложения химических волокон массового применения
(например, гидратцеллюлозных, полиамидных, полиэфирных, полиакри-
лонитрильных и др.).
Условно температурной границей считают 200°C, так как все
известные волокна выше этой температуры для эксплуатации не при-
годны.
В волокнах, подвергающихся действию высоких температур, про-
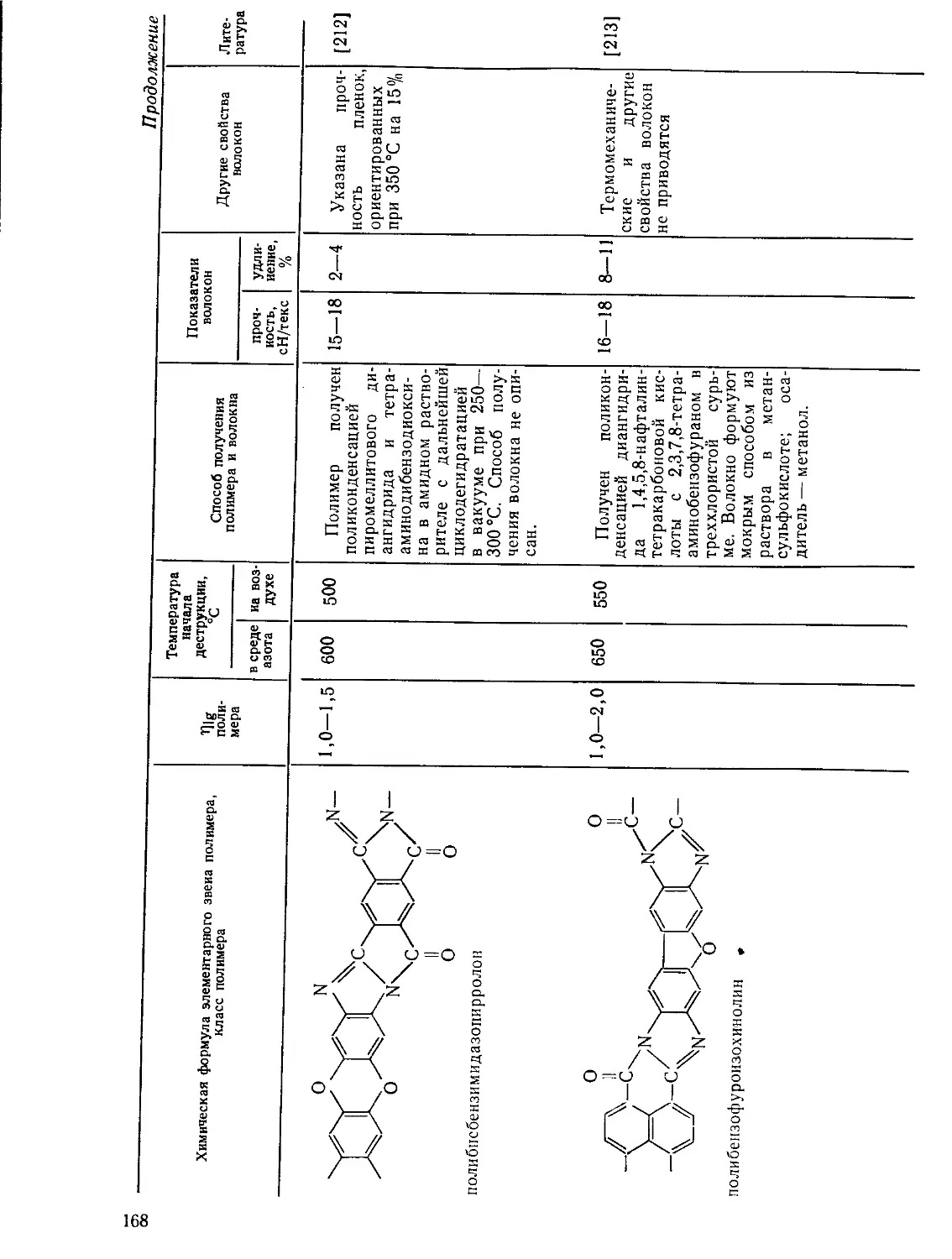
текают два существенно различающихся процесса. Первый из них —
обратимый процесс, который связан с физическими явлениями в поли-
мерах при их нагревании, приводящими к потере заданной формы во-
локна (усадка, размягчение, плавление). Протекание этого процесса в
основном определяется только температурой, хотя некоторое влияние
на количественные показатели этого процесса может оказывать и вре-
менной фактор вследствие релаксационной природы полимеров. Обра-
тимый процесс условно оценивается критерием теплостойкости.
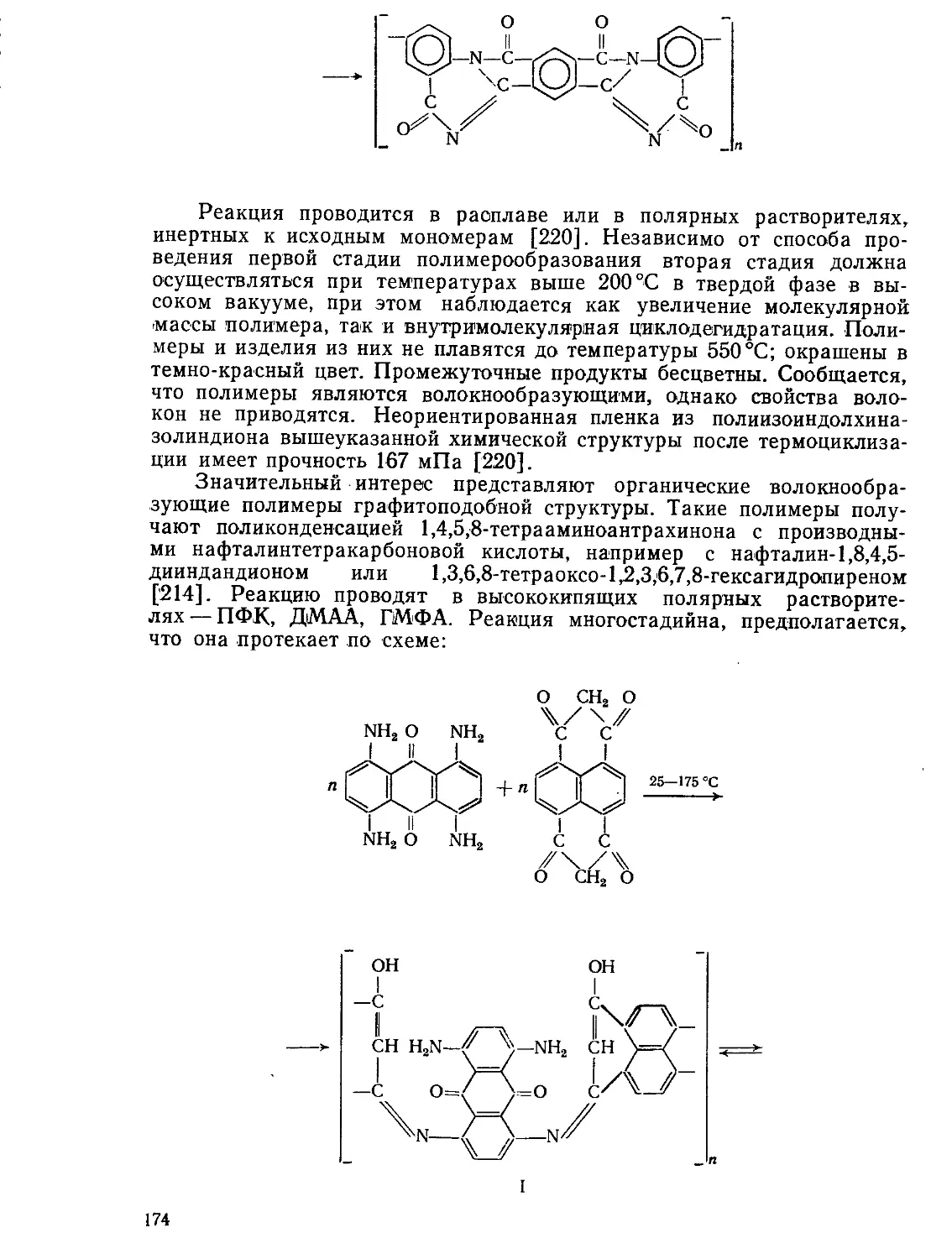
Второй процесс представляет собой необратимое разложение (де-
струкцию) волокна при воздействии тепла и окружающей среды (кис-
лород, вода и др.). Последний зависит от температуры и времени. Кри-
терием его является термостойкость. Методы определения этих харак-
теристик приводятся во многих монографиях [1, с. 27; 2].
Критерии тепло- и термостойкости оценивают различные процессы,
но для характеристики эксплуатационных свойств волокон, они должны
рассматриваться совместно, так как изделия, за редким исключением,
подвергаются деформациям и нагрузкам непосредственно в зоне высо-
ких температур.
Истинной характеристикой термостойкости волокна является пре-
дельная температура, при которой не наблюдается изменений механи-
ческих и других свойств как необратимого, так и обратимого харак-
тера.
Имеются два пути получения волокон с повышенной термостой-
костью. Первый связан с модификацией известных синтетических воло-
кон (капрон, нитрон и др.). Второй основывается на получении волокон
из специально синтезируемых высокотермостойких полимеров.
Модификация синтетических волокон сводится к применению реак-
ций в цепях полимеров (полимераналогичные превращения), радиаци-
онно-химической модификации, применению смесей полимеров, термо-
стабилизирующих добавок и других методов структурно-химической
модификации.
11
Среди изученных реакций в цепях полимеров с целью повышения
термостойкости волокон наибольшее число относится к структурирова-
нию (сшивкам) полиамидных [3, с. 220] и полиакрилонитрильных во-
локон [4]. Исследования показали, что этот метод позволяет заметно
повышать теплостойкость волокон, что же касается термостабильности,
то она изменяется незначительно, так как химическая структура основ-
ной макроцепи не изменяется. Поэтому, основываясь на ранее дан-
ной характеристике термостойкости волокон, можно сделать вы-
вод, что метод химической модификации волокон является неперспек-
тивным.
Радиационно-химическая модификация также практически не уве-
личивает термостабильности волокон, по-видимому, по тем же самым
причинам, что и реакции полимераналогичных превращений.
Кроме того, при радиационно-химической модификации так же,
как и при реакциях в цепях полимеров значительно ухудшаются меха-
нические свойства нитей, так как реакции проводятся выше температу-
ры стеклования волокон. Все остальные виды модификации — примене-
ние смесей полимеров, термостабилизация, структурно-химическая мо-
дификация и др. приводят к положительным результатам только в при-
ложении к некоторым видам высокотермостойких волокон. Поэтому они
будут описаны в соответствующих разделах.
Второй путь получения термостойких волокон, базирующийся на
волокнообразующих полимерах, специально предназначенных для ис-
пользования при высоких температурах, оказался более перспективным,
хотя и он пока не привел к решению проблемы получения волокон с
термостойкостью выше 300 °C.
Оказалось, что несмотря на наличие нескольких тысяч синтезиро-
ванных термостойких полимеров, только очень немногие из них нашли
применение для формования волокон. Это объясняется очень жесткими
требованиями к волокнообразующим полимерам.
1. Полимеры должны обладать способностью переходить в вязко-
текучее состояние, не разлагаясь, или растворяться в известных рас-
творителях.
2. Химическая структура полимера должна быть линейной и удов-
летворять определенным критериям [1].
3. Молекулярная масса полимера должна быть выше нижнего кри-
тического предела, значения которого зависят от структуры полимера
и метода формования.
Наряду с изложенными требованиями важнейшим фактором яв-
ляется также технико-экономическая целесообразность получения дан-
ного типа волокна и, особенно, доступность исходного сырья.
Создание новых волокнообразующих термостойких полимеров яв-
ляется задачей чрезвычайно сложной, так как основывается исключи-
тельно на эмпирическом поиске. До сих пор остается в силе утвержде-
ние Мелвила [5] о том, что практически нет достоверных данных
о взаимосвязи механических свойств с химическим составом, и об ос-
новных физических причинах проявления этих свойств. Не будет боль-
шой ошибкой указать в первую очередь на отсутствие четких корреля-
ций между теплостойкостью и термостойкостью полимеров и механиче-
скими (в том числе термомеханическими) свойствами получаемых на их
основе волокон.
При выборе полимеров для получения термостойких волбкон с за-
данными свойствами некоторыми теоретическими предпосылками мож-
12
но пользоваться только при прогнозировании теплостойкости создавае-
мого волокна, причем полученные данные являются чисто качествен-
ными [6].
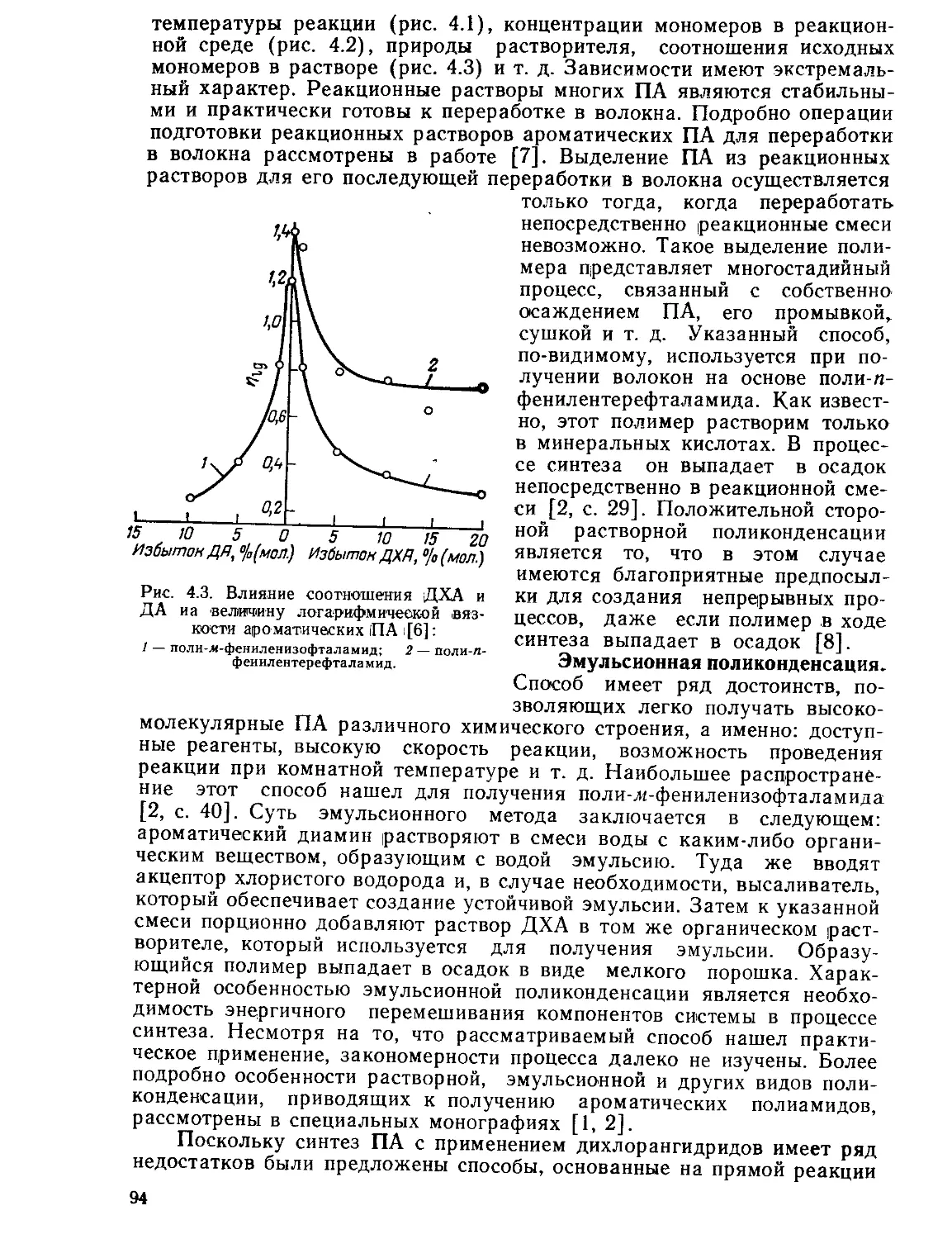
В основу такого прогноза может быть положена известная теория
прочности Журкова. Если экспериментальным путем определить
энергию активации разрыва наиболее слабых связей макромолекулы
полимера то, применяя уравнение о=—(<70—и преобразуя
* Y т0
его применительно к процессу разрыва нити на обычном динамомет-
ре, как рекомендует С. П. Папков [7], в формулу По = 0,064 —1 •
можно вычислить температуру, при которой прочность исходного во-
локна может уменьшаться на заданную величину, например вдвое.
Исследования показали, что расчетные данные дают хорошее качест-
венное совпадение с экспериментальными [6].
При прогнозировании термостабильности полимеров и волокон от-
сутствует возможность даже такого расчета. Поэтому обычно руковод-
ствуются рядом установленных формальных связей, полученных на ос-
новании анализа обширного экспериментального материала [1; 2; 8].
Кратко их можно свести к следующему.
1. Более жесткие макромолекулы оказываются термостабильнее
гибкоцепных одного и того же полимерного класса.
2. Ароматические и гетероциклические полимеры стабильней али-
фатических и циклоалифатических полимеров.
3. Отсутствие водородных атомов в макромолекулах должно при-
водить к снижению термоокислительной деструкции полимера.
4. Жесткоцепные лестничные полимеры должны иметь повышенную
термостойкость, так как полимерные цепи не разрушаются при разрыве
одиночной связи.
Перечисленные общие соображения согласуются с тем эксперимен-
тальным материалом, который накоплен к сегодняшнему дню. В самом
деле, в литературе приводится очень много примеров получения воло-
кон из относительно доступных жирноароматических полимеров [9] или
алициклических полиамидов [10]. Однако эксплуатационные свойства
таких волокон оказались низкими по механическим показателям, а по
термостабильности не выше обычных полиамидных и полиэфирных во-
локон.
И, наоборот, волокна из полностью ароматических полиамидов мо-
гут успешно эксплуатироваться при температурах до 250 °C.
Для волокон на основе гетероциклических и, особенно, лестничных
полимеров температурная граница эксплуатации выше еще на 50—
100 °C. Возможно, что дальнейшие исследования в области элементоор-
ганических полимеров или смешанных органо-минеральных систем по-
зволят достигнуть границ 400—500 °C.
В I части монографии будут рассмотрены процессы получения и
свойства волокон на основе полностью ароматических полимеров, поли-
гетероариленов и подобных им полимерных систем.
По требованиям, предъявляемым такими отраслями промышленно-
сти, как авиационная, электротехническая и др., волокна не только
должны быть термостойкими, но и одновременно должны иметь доста-
точно высокие, а в ряде случаев и сверхвысокие механические свойст-
ва; обладать высокой стойкостью к жесткому и ультрафиолетовому об-
лучению, быть химически стойкими, негорючими и, наконец, иметь
13
удовлетворительные эксплуатационные свойства в условиях глубокого
холода и вакуума.
В настоящее время термостойкие волокна все чаще рассматрива-
ют как «носители» особых свойств, делающих пригодными их для экс-
плуатации в экстремальных условиях. Последняя глава посвящена
применению термостойких волокон в различных отраслях техники.
ЛИТЕРАТУРА
1. Фрейзер А. Г. Высокотермостойкие полимеры. М., «Химия», 1974. 296 с.
2. Коршак В. В. Химическое строение и температурные характеристики полимеров.
М., «Наука», 1970. 417 с.
3. Кудрявцев Г. И. и др. Полиамидные волокна. М., «Химия», 1976. 264 с.
4. Кудрявцев Г. И., Ж.ВХО им. Д. И. Менделеева, 1966, т. 11, с. 665—678.
5. Мелвил Г., «Успехи химии», 1966, т. 35, с. 1030—1046.
6. Кудрявцев Г. И. Международный симпозиум по химическим волокнам. Пре-
принты. Калинин, 1974, секция 4, с. 5—10.
7. ГТапков С. П., Хим. волокна, 1971, № 2, с. 10—14.
8. Прокопчук Н. Р. и др. Хим. волокна, 1976, № 6, с. 44—49.
9. Кудрявцев Г. И., Волохина А. В., ЖВХО им. Д. И. Менделеева, 1966, т. 11,
с. 665—672.
10. Вольф Л. А., Меос А. И. Волокна специального назначения. М., «Химия»,
1971. 224 с.
ГЛАВА 1
ИСХОДНЫЕ МОНОМЕРЫ И РАСТВОРИТЕЛИ, ПРИМЕНЯЕМЫЕ
В ПРОИЗВОДСТВЕ термостойких волокон
1.1. ОСНОВНЫЕ КЛАССЫ ВОЛОКНООБРАЗУЮЩИХ
ТЕРМОСТОЙКИХ ПОЛИМЕРОВ
В последние 10—15 лет был синтезирован ряд новых термостойких
органических полимеров, которые по устойчивости к действию повы-
шенных температур значительно превосходят ранее известные полиме-
ры. Характерной особенностью строения таких полимеров является то,
что их макромолекулы состоят из ароматических колец (типа бензола,
дифенила, нафталина и т. д.), соединенных между собой различными
гетероциклами или амидной связью. Многие из синтезированных поли-
меров оказались волокнообразующими и из них по различным способам
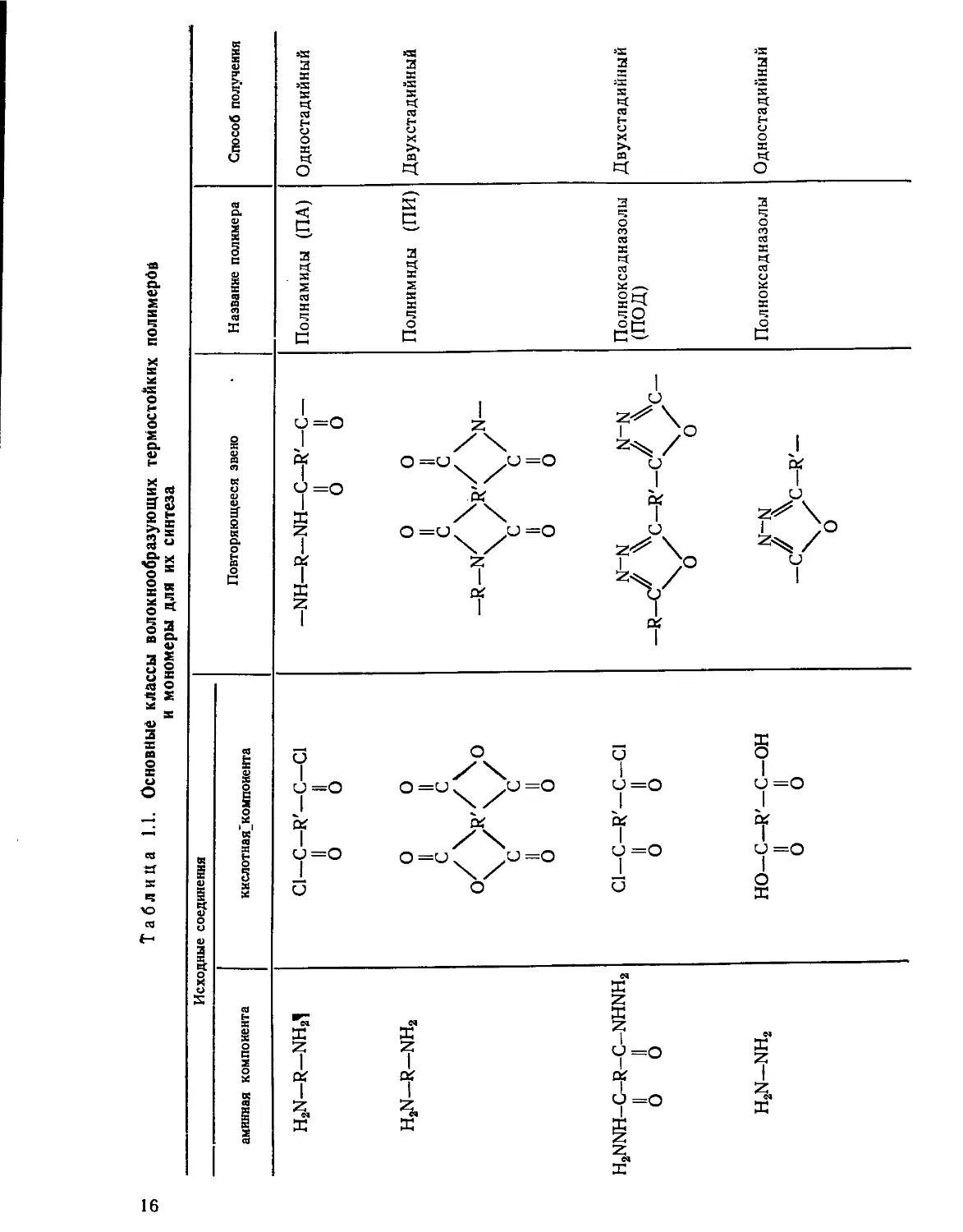
формования были получены волокна. В таблице 1.1 приведены основные
классы волокнообразующих термостойких полимеров и мономеры, ис-
пользуемые для их синтеза.
Как видно из приведенной таблицы, тип связи между ароматиче-
скими ядрами определяет принадлежность полимеров к тому или иному
классу. Характер этой связи в значительной степени влияет как на тех-
нологию получения полимеров и волокон на их основе, так и на физи-
ко-механические и термические свойства промежуточных и конечных
изделий.
1.2. ИСХОДНЫЕ ВЕЩЕСТВА, ПРИМЕНЯЕМЫЕ
ДЛЯ ПОЛУЧЕНИЯ ТЕРМОСТОЙКИХ ВОЛОКОН
Основными исходными мономерами для получения практически
всех термостойких волокнообразующих полимеров являются много-
функциональные ароматические соединения, содержащие две, три или
14
четыре реакционноспособные группы, а также гидразин, который при-
меняется для получения полиамидогидразидов или полиоксадиазолов.
В последнем случае амидогидразидная группировка в результате реак-
ции внутримолекулярной циклизации превращается в устойчивый
1,3,4-оксадиазольный цикл. В табл. 1.2 представлены некоторые моно-
меры, нашедшие широкое распространение в качестве исходных продук-
тов для получения волокнообразующих термостойких полимеров, а так-
же мономеры, являющиеся перспективными, однако производство
последних ограничено по экономическим или технологическим сообра-
жениям. Подробные сведения о методах синтеза, очистке и свойствах
многих из указанных в табл. 1.2 мономеров содержатся в монографии
[1].
1.3. РАСТВОРИТЕЛИ
Ароматические полимеры, пригодные для получения термостойких
волокон, практически не растворяются в известных органических рас-
творителях. Это обстоятельство в сочетании с неплавкостью указанных
полимеров длительное время оказывалось препятствием для синтеза
исходных полимеров, так как для большинства термостойких полимеров
поликонденсация в растворе является практически единственным спо-
собом их получения. И в настоящее время, несмотря на то, что имеется
ряд технологически пригодных растворителей и разработаны основы
теории растворов жесткоцепных высокомолекулярных соединений, под-
бор новых растворителей осуществляется эмпирически. Характерно при
этом, что термостойкие полимеры растворяются лишь в системах, обла-
дающих высокой полярностью. К такого рода веществам относятся ор-
ганические апротонные растворители, такие, как М,М-диметилацетамид,
N-метилпирролидон, гексаметилфосфортриамид, М,М-диметилформ-
амид, диметилсульфоксид и т. д. Некоторые полимеры, например аро-
матические полиамиды, растворимы в N-метилкапролактаме, адипонит-
риле, сульфолане. Практически универсальным растворителем для
большинства термостойких волокнообразующих полимеров являются
концентрированные кислоты, такие, как серная, олеум, полифосфорная,
хлор- или метансульфоновая. Ниже приведены характеристики некото-
рых органических и неорганических растворителей, применяемых в про-
изводстве термостойких волокнообразующих полимеров и волокон на
их основе.
М,М-Диметилацетамид (ДМАА). ДМАА относится к продуктам,
выпускаемым в большинстве промышленно развитых стран. Основной
способ синтеза ДМАА — взаимодействие уксусной кислоты или уксус-
ного ангидрида с диметиламином по реакции
н3сч Н3С, “
Н3ССООН+ /NH -------► ,N—С—СН3+Н2О
н3с/ н3с/
с дальнейшей очисткой путем ректификации [18; 20]. ДМАА — про-
зрачная, довольно подвижная жидкость, обладающая специфическим
запахом аминов. Смешивается с большинством жидкостей, в том числе
с водой, спиртами и т. д. во всех отношениях. Ограниченно растворим в
метиленхлориде. Гигроскопичен. В водных растворах, в особенности в
присутствии кислот и оснований, медленно гидролизуется. Гидролиз
становится особенно заметным при температуре выше 100 °C, однако
15
о
Таблица 1.1. Основные классы волокнообразующих термостойких полимеров
и мономеры для их синтеза
Исходные соединения Повторяющееся звено Название полимера Способ получения
аминная компонента кислотная^компоиента
H2N-R-NH21 С1—С—R'—С—С1 II II О О —NH—R—NH—С—R'—С— II II О О Полиамиды (ПА) Одностадийный
h2n—r—nh2 О О II II с с °\ /R \ /° с с II II О О О О II II с с —R— n/ yN~ с с7 II II О О Полнимнды (ПИ) Двухстадийный
h2nnh-c-r-c-nhnh2 II II О О Cl-C-R'-C-Cl II II О О N-N N-N —R—С" Хз—R'—С^^С— О О Полноксадназолы (ПОД) Двухстадийный
h2n-nh2 НО—С—R'-C-OH II II О О N-N V R, I\ О Полноксадназолы Одностадийный
ьэ
s
Примечание. R. R', R" — ароматические радикалы соответствующей валентности.
Полибензимидазо-
лы (ПБИ)
Полибензоксазо-
лы (ПБО)
Одностадийный
Двухстадийный
Полиимидазопир- Одно- и двухста-
ролоны (пирроиы) дийные
Полихиноксалины
(ПХО)
Одно- и двухста-
дийиые
Таблица 1.2. Соединения, применяемые для синтеза термостойких полимеров
Название, химическая формула Молеку- лярная масса гпл- °с Растворимость . Токсичность, пожаро- н взрывоопасность Стабильность соединения, основное применение Литература
I. Бифункциональные соединения
Терефталевая кислота (ТФК)
СООН
Изофталевая кислота (ИФК)
ноос—
-СООН
Ароматические дикарбоновые кислоты, их хлорангидриды, эфиры
166,14 Не плавит- Плохо рас- Токсичное, Стабильно. [1, с. 4131
166,14 ся (субли- мируется) творима в во- де, незначи- тельно раство- ряется в горя- чем метаноле (15 г в 100 мл при 200 °C), в диметилформ- амиде (6,7 г в 100 мл), в ди- метилсульфок- сиде (20 г в 100 мл) пожаро- и взрывоопасное вещество. ПДК в рабочей зоне составляет 0,1 мг/м3; ПДК в открытом во- доеме — 0,01 мг/л. В ви- де пыли взры- воопасна в сме- сях с воздухом (нижний пре- дел взрыво- опасности 52 г/м3). Тем- пература само- воспламенения облака пыли — 591 °C Выпускается во многих стра- нах в промыш- ленном мас- штабе. Приме- няется в чи- стом виде для получения по- лиоксадиазолов при односта- дийном способе синтеза; может быть примене- на для получе- ния ароматиче- ских полиами- дов при ката- литической по- ликонденсации с диаминами 12]; ]3]
Не плавит- Нераствори- Образует по- Стабильно. (1, с. 413]
ся (сублими- руется) ма в воде, ча- стично раство- ряется в мета- ноле (при обычных темпе- ратурах) . При повышенных температурах жароопасные смеси пыли с воздухом; взве- шенная в воз- духе пыль взрывоопасна, нижиий предел взрывоопасно- сти 35 г/м3. Широко приме- няется для син- теза дихлоран- гидридов, на- шедших приме- нение в химии и технологии ароматических полиамидов, [3]
>5
216,20
315—320
^разложе-
нием)
2,6-Нафталиндикарбоновая кислота
НООС-(0©_сооН
4,4'-Дифенилдикарбоновая кислота
ноос
соон
216,20
Выше 300
(с разложе-
нием)
242,24 Выше 300
(190 °C) рас- творима в воде Температура самовоспламе- полибензимнда- золов, полибен-
(16,5 г/л) и в ледяной уксус- ной кислоте Нераствори- нения 605 °C. Нет данных зоксазолов и других полиме- ров Стабильно. [1. с. 418—
ма в обычных органических растворителях В обычных То же Выпускается в промышленном масштабе. Ос- новное приме- нение—для по- лучения поли- оксадиазолов ароматических полиамидов Стабильно. 419] [1, с. 418-
условиях не- растворима в органических растворителях; при нагревании растворима в бензоле, ледя- ной уксусной кислоте и эти- ловом спирте В обычных Токсические За рубежом ос- воен промыш- ленный синтез. Применяется для получения ароматических полиамидов Стабильно. 419] [1, с. 420]
условиях не- растворима в воде и органи- ческих раство- рителях. При нагревании ча- стично раство- рима в спиртах н уксусной кис- лоте характеристики ие изучены. Горючий про- дукт Выпуск ограни- чен. Применя- ется для полу- чения аромати- ческих поли- амидов
8
Название, химическая формула Молеку- лярная масса Гпл- °с
Дихлорангидрид терефталевой кислоты (ТФХ) 203,02 78—81
С1ОС—СОС1
Дихлорангидрид изофталевой кислоты (ИФХ)
203,02 42—43
С1ОС—I
I—СОС1
Продолжение
Растворимость Токсичность, пожаро- и взрывоопасность Стабильность соединения, основное применение Литература
Растворим в бензоле, ксило- ле, четыреххло- ристом углеро- де и других уг- леводородах; в ДМАА, ДМФ, ДМСО и дру- гих амидиых растворителях Хорошо рас- творим в угле- водородах и амидных рас- творителях 1 Токсичен. ПДК в воздухе рабочих поме- щений 0,1 мг/м3. Горюч. Темпе- ратура воспла- менения 650 °C. Температура вспышки 210 °C (в закрытом тигле) Токсичен. ПДК в воздухе рабочих поме- щений состав- лиет 0,1 мг/м3. Температура воспламенения 260 °C. Нижний предел воспла- менения при концентрации 83 г/м3 Нестабильное соединение. В присутствии влаги гидроли- зуется с выде- лением хлори- стого водорода и терефталевой кислоты. Выпускается в крупном мас- штабе в боль- шинстве про- мышленно раз- витых стран. Применяется для синтеза ароматических полиамидов, ПВО и других термостойких полимеров Нестабиль- ный продукт, гидролизуется во влажной ат- мосфере. Про- мышленный продукт. Ис- пользуется при получении тер- мостойких аро- матических по- лиамидов [в частности, но- [1, с. 393; 4; 5, с. 129] [1,’с. 393; 4; 5, с. 129;!6, с. 5Ц
Дихлорангидрид
кислоты
4,4'-дифенилдикарбоновой
279,02
сюс
СОС1
Дихлорангидрид 2,6-нафталиндикарбоновой
253,08
кислоты
^ТОО-сое.
184—188
(способен к
сублимации
при понижен-
ном давле-
нии)
Хорошо рас- творим в обыч- Токсикология мало изучена. мекс (США), фенилон (СССР)] Стабильность несколько вы- [1, с. 396; 7]
ных и хлориро- ванных углево- дородах, тетра- гидрофуране, в амидных рас- творителях Предполагают, что токсиче- ское действие аналогично хлорангидри- дам тере- и изофталевой кислот ше, чем у ди- хлорангидри- дов тере- и изофталевой кислот. Произ- водится в опытном мас- штабе. Исполь- зуется для син- теза аромати- ческих поли- амидов
Растворим в хлорированных углеводородах, в тетрагндро- фуране, в амидных рас- творителях Токсические характеристики мало изучены Во влажной атмосфере гид- ролизуется до дикарбоновой кислоты с вы- делением хло- ристого водо- рода. Выпус- кается в опыт- ном масштабе. Основное при- менение—сырье для получения ароматических полиамидов [1. с. 3931
Продолжение
Нмваиие, химическая формула
Диметиловый эфир терефталевой кислоты
Дифениловый эфир изофталевой кислоты
Молеку- лярная масса гпл> °с Растворимость
194,18 140,6 При нагре- вании хорошо растворим во многих органи- ческих раство- рителях. На холоду раство- рим в дихлор- этане, диоксане Н хлороформе. Плохо раство- рим в воде
328,14 134—138 Нерастворим в воде. При на- гревании рас- творим во мно- гих органиче- ских соедине- ниях
Токсичность, пожаро- н взрывоопасность Стабильность соединения, основное применение лтература
Кристалличе- ский продукт. Мо.ч-.гт образо- вывать пыль. Токсичен. ПДК в рабочей зоне 0,1 мг/м8. С воз- духом пыль об- разует взрыво- опасные смеси. Горюч Стабильный продукт, во всех промыш- ленно развитых странах выпус- кается в боль- ших объемах. Представляет значительный интерес с точки зрения замен . дихлоранг идри да терефтале- вой кислоты при получении ароматических полиамидов и других полиме- [1, с. 390]
Кристалличе- ский продукт. Горюч. Токси- кологические характеристики не изучены Стабильный продукт. Вы- пускается в опытном мас- штабе. Основ- ное примене- ние — синтез полибензимида- золов [1, с. 484]
Ароматические дйамииы
n-Фёнилендиамин (ПФДА)
108,14
139—141; Слабо р не-
способен к творим в воде,
сублимации хорошо в амид-
ных раствори-
Бесцветный
кристалличе-
ский продукт,
быстро темнею-
Дихлоргид-
рат п-фенилен-
диамина явля-
ется более
стойким про-
[1, с. 435;
6, с. 75,
237; 8; 9]
ж-Фенилендиамин (МФДА)
108,14 6
телях, в этило- щий на возду- дуктом. Произ-
вом спирте и диэтиловом эфире хе. Малостаби- лен. Токсичный продукт. Вызы- вает сильное раздражение кожи, астмати- ческие явления, поражает пе- чень. ПДК в воздухе рабо- чих помещений составляет 0,1 мг/м3, в от- крытых водое- мах—0,01 мг/л. Горюч. Теплота сгорания 3500 кДж/моль. Нижний предел воспламенения аэровзвеси 26 г/м3. водится в про- мышленном масштабе. На- ходит примене- ние для полу- чения термо- стойких арома- тических поли- амидов и поли- имидов. Пред- полагают, что ПФДА являет- ся одним из компонентов высокопрочного термостойкого полиамидного волокна кевлар
Хорошо рас- Токсичен. Нестабильно, [1, с. 433
творим в воде, этиловом спир- те и амидных растворителях Воздействует иа печень. Так же, как и в слу- чае п-феиилен- диамина, сле- дует избегать контакта с пылью. ПДК в рабочей зоне 0,1 мг/м3 быстро темнеет на воздухе. Производится в промышлен- ном масштабе. Применяется для синтеза ароматических полиамидов, является со- ставляющей во- локон иомекс (США), фени- лен (СССР)- Ю]
ьэ
Название, химическая формула Молеку- лярная масса
Бензидин HaN—/ 184,14
4,4'-Диаминодифенилоксид HaN— 200,26
4,4'-Диаминодифенилсульфон 248,32
Продолжение
гпл-°с Растворимость Токсичность, ^пожаро- и вз рыво оп асность Стабильность соединения, основное применение ! Литература
127—129 193—195 (склонен к сублимации) 176—178 Слабо рас- творим в воде, органических растворителях. Хорошо раство- рим в амидных растворителях Нерастворим в спирте и во- де, хорошо растворим в ацетоне и амидных рас- творителях Плохо рас- творим в воде, хорошо — в спирте и амид- ных раствори- телях Горюч. Ниж- ний предел вос- пламенения 41 г/м3. Токси- чен. Обладает канцерогенны- ми свойствами. Рекомендуетсн избегать кон- такта с пылью И парами бен- зидина Токсичен. Вы- зывает раздра- жение кожи, может воздей- ствовать на пе- чень. Горюч Продукт уме- ренно токсичен. ПДК пыли в воздухе рабо- чих помещений 2 мг/м3. Горюч. Нестабильно. В промышлен- ности выпуска- ется в виде сульфатов или хлоргидратов. Применяется для синтеза по- либензнмидазо- лов, может быть использо- ван при получе- нии ароматиче- ских полиами- дов Стабилен. Выпускается в промышленном масштабе. При- меняется для получения аро- матических по- лиимндов и по- лиамидов. На- шел примене- ние для полу- чения волокон аримид ПМ (СССР). Стабилен. В рнде стран выпускается в большом мас- штабе. Исполь- зуется в меди- [1, с. 429] [1, с. 440] [И]
198,29
4,4'-Диаминодифенилметан
346,42
М,М'-л-Фенилен-бис- (л-аминобеизамид)
346,42
К1Ч'-Бис(3-аминофенилизофталамид)
8
92—93 Плохо рас- В смеси с воз- духом образует горючие смеси. Нижний предел воспламенения 37 г/м3. Темпе- ратура само- воспламенения 555 °C Токсичен. цине; является перспективным при получении ароматических полиамидов Стабилен. п, с. 439]
творим в воде, хорошо в спир- тах, эфирах, ацетоне, бензо- ле и амидных растворителях Раздражает кожу. Воздей- ствует на пе- чень. Наблюда- лись случаи цианоза рабо- чих, соприка- сающихся с большими ко- личествами пы- ли диаминоди- фенилметана. Горюч Выпускается в промышленном масштабе. При- меняется при получении по- лиимидов и по- лиамидоимидов 442]
213—214 Плохо рас- творим в воде, хорошо в спир- тах, амидных растворителях Предполага- ют, что соеди- нение малоток- сично. Нормы ПДК не уста- новлены. Го- рюч Стабилен. Выпускается в крупном мас- штабе. Нашел применение для получения аро- матических «упорядочен- ных» полиами- [1. с.
227—228 Растворим в горячей воде, мети лен хлори- де, диметилаце- тамиде и в сме- сях воды с аце- тоном Токсикологи- ческие характе- ристики мало изучены дов Стабилен. Выпускается в опытном мас- штабе. Нашел применение при синтезе арома- тических «упо- рядоченных» полиамидов 11. с. 443]
КЗ
Название, химическая формула Молеку- лярная масса
2,5-Бис (л-амннофеиил) -1,3,4- окса диазол 252,30
4,4'-Диамииоазобеизол
212,28
n-Аминобеизойная кислота
137,15
Продолжение
Twi- °с Растворимость Токсичность, пожаро- и в з ры воопасиост ь Стабильность соединения, основное применение Литература
260—262 Растворим в горячей воде, спирте, ацетоне и амидных рас- творителях Токсикологи- ческие характе- ристики мало изучены. Горюч Стабилен. Представляет большой инте- рес для получе- ния ароматиче- ских волокно- образующих полиамидов и полиимидов. Выпускается в опытном мас- штабе (1, С. 447]
238—241 Нерастворим в воде, раство- рим в спирте, амидных рас- творителях и уксусной кис- лоте Токсикологи- ческие характе- ристики мало изучены Стабилен. Применяется для получения азокрасителей. Представляет значительный интерес для по- лучения термо- стойких поли- амидов, поли- имидов и дру- гих полимеров, содержащих азосвязи [1, с. 441]
186—187 Растворима в воде, спирте, эфире, амид- ных раствори- телях Кристалличе- ский продукт. Токсичен. Ток- сикологические характеристики изучены недо- статочно. Го- рюча. Аэро- взвесь взрыво- При хране- нии в темноте без доступа кислорода ста- бильна. Про- мышленный продукт. Ис- пользуется для получения раз- [12; 13]
ж-Амииобеизойиая кислота
137,15
HjN—
-С ООН
Хлорангидрид n-амииобеизойной кислоты
155,59
Ь9
174—176 Хорошо рас- опасна; нижний предел воспла- менения 39,5 г/м3; тем- пература само- воспламенения 576 °C Токсические личных моно- меров, приме- няемых для по- лучения термо- стойких волок- нообразующих полимеров раз- личных классов и для получе- ния высоко- прочных высо- комодульных полиамидных волокон Используется [13]
(склонен к творима в го- свойства не для синтеза ис-
сублимации) рячей воде, спирте, ацетоне и эфире изучены ходных моно- меров, приме- няемых для по- лучения термо- стойких поли- меров
Не плавится Нерастворим в большинстве обычных орга- нических рас- творителей. Растворим в амидных рас- творителях (при этом идет реакция поли- мерообразова- ния) Токсичен. ПДК в возду- хе рабочих по- мещений не установлены. Горюч Нестабилен. Хлорангидрид п-аминобензой- ной кислоты су- ществует толь- ко в виде хлор- гидрата. Про- изводится в больших мас- штабах. Ис- пользуется для получения вы- сокопрочных полиамидных волокон [12]
П родолжение
___Название, химическая формула
Гидразинсульфат
H2N-NH2.H2SO4
4,4'-Дифенилметанднизоцианат
OCN
NCO
Молеку- лярная масса гпл- °с Растворимость Токсичность, пожаро- н взрывоопасность Стабильность соединения, основное применение Литература
130,14 245 Плохо рас- творим в хо- лодной воде. Слабо раство- рим в кислотах Токсичен Стабилен. Промышленный продукт. При- меняется для получения по- лигидразидов и полиоксадиазо- лов (в том чис- ле для волокна оксалон) [U, с,, 1631
250,27 40 Разлагается в большинстве растворителей. Очищается пе- регонкой Обладает вы- сокой токсич- ностью. Воздей- ствует на сли- зистую оболоч- ку глаз и на открытые уча- стки кожи. ПДК рабочей зоны 0,02 мг/м3. Горюч Нестабилен. Требует осо- бых условий при хранении. Склонен к са- мополимериза- ции. Произво- дится в про- мышленном масштабе. Ис- пользуется для получения вы- сокомолекуляр- ных полимоче- вин, полурета- нов и огнестой- ких полиамидо- имидов П. с. 291]
Диангидрид пиромеллитовой кислоты
11. Тетрафункциональные соединения
Кислородсодержащие мономеры
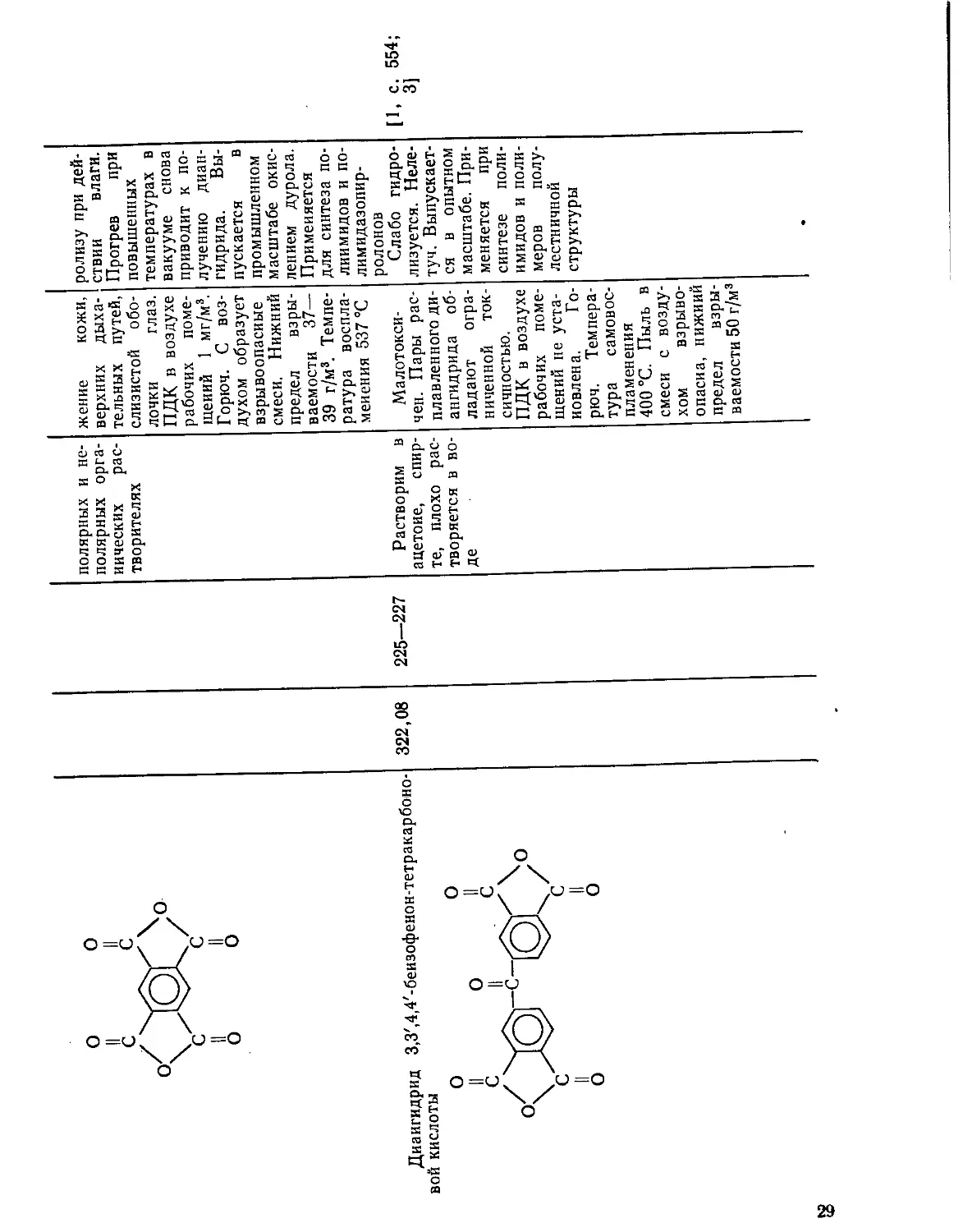
218,06 285—287 Растворяется Токсичен, вы- Стабилен. [1, с. 553J
в различных зывает раздра- Подвержен гид-
Диаигидрид
вой кислоты
3,3',4,4'-беизофенон-тетракарбоно-
322,08
8
полярных и не- жение кожи, релизу при дей-
225—227 полярных орга- нических рас- творителях Растворим в верхних дыха- тельных путей, слизистой обо- лочки глаз. ПДК в воздухе рабочих поме- щений 1 мг/м3. Горюч. С воз- духом образует взрывоопасные смеси. Нижний предел взры- ваемости 37— 39 г/м3. Темпе- ратура воспла- менения 537 °C Малотокси- ствии влаги. Прогрев при повышенных температурах в вакууме снова приводит к по- лучению диан- гидрида. Вы- пускается в промышленном масштабе окис- лением дурола. Применяется для синтеза по- лиимидов и по- лимидазопир- ролонов Слабо гидро- [1, с. 554;
ацетоне, спир- те, плохо рас- творяется в во- де чен. Пары рас- плавленного ди- ангидрида об- ладают огра- ниченной ток- сичностью. ПДК в воздухе рабочих поме- щений не уста- новлена. Го- рюч. Темпера- тура самовос- пламенения 400 °C. Пыль в смеси с возду- хом взрыво- опасна, нижиий предел взры- ваемости 50 г/м3 лизуется. Неле- туч. Выпускает- ся в опытном масштабе. При- меняется при синтезе поли- имидов и поли- меров полу- лестничной структуры 3]
Co
о
Название, химическая формула Молеку- лярная масса Т Of 1 пл’ G
440 (в азоте)
(кислота при
сушке при
температуре
выше ПО °C
превращается
в ангидрид)
Диангидрид 3,4,9, 10-перилентетракарбоновой
кислоты
392,3 Выше 360
190,16
140—150
(с разложе-
нием)
Продолжение
Растворимость Токсичность, пожаро- и взрывоопасность Стабильность соединения, основное применение Литература
Растворим в уксусной кисло- те. Нераство- рим в воде Не растворя- ется в обычных органических растворителях, растворим в серной, уксус- ной кислотах и щелочах Существует в виде дигидра- та. Растворим в горячей воде и спиртах Токсические характеристики мало изучены. Горюч. Невзры- воопасен в аэ- ровзвеси с воз- духом. Ниж- ний предел вос- пламеняемости в смеси с воз- духом— 113,4 г/м3. Тем- пература само- воспламенения 485 °C. Сведения о токсичности от- сутствуют. Го- рюч. Невзрыво- опасен в сме- си с воздухом (нижний пре- дел воспламе- няемости выше 260 г/м3). Тем- пература само- воспламенения 610 °C Сведений о токсичности и горючести нет Стабилен при хранении в без- водных услови- ях. В при- сутствии вла- ги гидроли- зуется. Выпус- кается в опыт- но-промышлен- ном масштабе. Применяется для получе- ния полимеров лестничной . и полулестничной структуры Стабилен. Вы- пускается в опытно-про- мышленном масштабе. При- меним для по- лучения поли- меров лестнич- ной структуры Малостаби- лен. Выпуска- ется в неболь- ших количест- вах. Исполь- зуется для по- лучения высо- [1, с. 553; 3] [1, с. 557; Ш [1, с. 556]
Тетрааминобеизол
13
З.З'-Диамннобеизидин
21
со
8,20
4,30
Тетраминь 274—276 Растворим в Обладает комолекуляр- ных термостой- ких полихинО- ксалииов Свободный [1. С. 571;
(основание) и выше 360 (тетрахлор- гидрат) амидных рас- творителях. Плохо раство- рим в воде канцерогенны- ми свойствами. Горюч тетрамин окис- ляется на свету и в присутст- вии влаги. Хлоргндрат — стабильное со- единение. Про- изводится в промышленном масштабе. При- меняется для получения поли- меров лестнич- ной структуры 15] 571]
178—179 (основание); выше 300 (тетрахлор- гидрат) Нерастворим в воде, раство- рим в метано- ле, амидных растворителях Свободный амин обладает умеренной ток- сичностью; тет- рахлоргидрат высокотоксичен и обладает канцерогенны- ми свойствами (по другим ис- точникам, осно- вание также является кан- церогенным). Горюч Основание быстро окис- ляется на све- ту, на воздухе, медленнее — в среде азота. Хлоргидрат также неустой- чив. За рубе- жом оба про- дукта выпуска- ются в про- мышленном масштабе. При- меняется для синтеза поли- бензимидазолов и полимеров полулестничной структуры [1, с.
Название, киническая формула Молеку- лярная масса гпл.°с
З.З'ЛЛ'-Тетраминодифенилоксид [QL <nh2 230,26 150—152
H2N^^ nh2
2,3,5,6-Тетраминодибензофуран H2N^ zNH, 228,28 —
4NH2
3,3'-Дикарбоксибензидин НООС. /СООН хнх 272,28 —
Продолжение
Растворимость Токсичность, пожаро- и взрывоопасность Стабильность соединения, основ- ное применение Литература!;
Растворим в амидных рас- творителях и ДМСО, в воде, в спирте, соля- ной и уксусной кислотах Токсические характеристики мало изучены. Горюч. Темпе- ратура само- воспламенения 500 °C. Нижний предел взры- ваемости аэро- взвеси 20 г/м3 Производит- ся в опытном масштабе. Про- дукт малоста- билен. Более стабилен тетра- хлоргидрат. Является пер- спективным для получения полимеров по- лулестиичной структуры [1, с. 574J
Растворим в амидиых рас- творителях Сведения о токсических ха- рактеристиках и горючести от- сутствуют Неустойчив. Производится в небольших количествах. Представляет интерес для по- лучения поли- меров лестнич- ной или полу- лестиичной структуры [16]
Растворим в амидиых рас- творителях, слабо раство- рим в горячей воде Токсические характеристики ие изучены. Го- рюч Производит- ся в опытном масштабе. Ис- пользуется для получения по- либензоксази- нонов. Амиды этого соедине- ния применяют для получения полихиназоло- нов [17]
даже при комнатной температуре, несмотря на малую скорость гидро-
лиза, длительное хранение ДМАА (в присутствии даже незначительных
количеств воды) приводит к накоплению заметных количеств уксусной
кислоты и диметиламина. Например, при содержании воды 0,44% в
системе за 5 сут накапливается около 0,6% уксусной кислоты, которая,
действуя в свою очередь как катализатор, еще более ускоряет процесс
гидролиза. С уксусной кислотой ДМАА образует азеотропную смесь
состава 78:22 % (масс.). Азеотропная смесь кипит при температуре
170 °C (при 0,1 МПа) и при ректификации остается в кубовом остатке.
Растворяющее действие ДМАА резко усиливается в присутствии хлори-
дов лития, кальция, магния и некоторых других солей. Обладает об-
щетоксичным и местным раздражающим действием. Действует
на центральную нервную систему, печень, почки. Способен проникать
через неповрежденные кожные покровы и кумулировать в организме.
Предельно допустимая концентрация: в воздухе рабочих помещений —
1 мг/м3, в атмосферном воздухе — 0,05 мг/м3, в открытых водоемах —
20 мг/л. Наличие ДМАА в водах, идущих на биологическую очистку, в
количестве до 400 мг/л не препятствует жизнедеятельности бактерий.
ДМАА нашел широкое практическое применение как растворитель,
причем непосредственно при синтезе, для ароматических полиамидов,
полиамидокислот, полиоксиамидов, полиаминоамидокислот, полигидра-
зидов и ряда других полимеров. Применяется для растворения поли-
бензимидазолов при получении волокна ПБИ. В связи с тем, что ДМАА
используется в качестве реакционной среды при синтезе полимеров,
требования к его чистоте оказываются очень жесткими. Самыми неже-
лательными примесями в этом случае являются вода и диметиламин,
так как оба этих продукта легко вступают в реакцию с дихлорангидри-
дами или ангидридами соответствующих ароматических кислот, что при-
водит к получению низкомолекулярных полимеров. Физические свой-
ства ДМАА представлены далее (см. табл. 1.4).
Диметилформамид (ДМФ). Из многих способов получения ДМФ
наиболее экономичными являются синтез его из метанола, аммиака и
углекислого газа, а также из диметиламина и муравьиной кислоты [21].
ДМФ представляет собой прозрачную жидкость, обладающую специфи-
ческим запахом. Хорошо смешивается с водой и с другими полярными
органическими растворителями. Легко летуч. Как и ДМАА, ДМФ, явля-
ясь амидом, склонен к гидролизу, причем скорость гидролиза увеличи-
вается при температурах выше 100 °C. Конечным продуктом гидролиза
являются диметиламин и хмуравьиная кислота. Присутствие солей, кис-
лот и оснований катализирует гидролиз. По характеру токсичес-
кого воздействия на организм аналогичен ДМАА. ПДК в воздухе
рабочих помещений составляет 10 мг/м3 [6, с. 39 и 266]. Для открытых
водоемов ПДК составляет 10 мг/л. Интересно отметить, что в сточных
водах, направляемых на биологическую очистку, может содержаться
до 1000 мг/л ДМФ [22]. Горюч. Образует с воздухом взрывоопасные
смеси. Растворяющая способность ДМФ по отношению к некоторым
термостойким полимерам или промежуточным продуктам близка к
растворяющей способности ДМАА. ДМФ используется как раствори-
тель поли-м-фениленизофталамида при получении волокон номекс и
фенилон [23, с. 149; 24] в особенности, в сочетании с лиофильными
солями типа хлорида лития или кальция.
Диметилсульфоксид (ДМСО). Промышленный метод получения
ДМСО базируется на прямом синтезе из метанола и сероводорода
3—2254
33
[25] по следующей схеме
СН3ОН + H2S--->- (CH3)2S=O 4- Н2О
ДМСО — слегка желтоватая, маслянистая жидкость, отличается гиг-
роскопичностью, хорошо смешивается с водой, спиртами, циклическими
углеводородами, кетонами, эфирами и т. д.
Предполагают, что ДМСО образует с водой гидраты. ДМСО, как
и ДМФ, не пригоден для использования в качестве реакционной среды
в тех случаях, когда одним из реагентов являются дихлорангидриды
ароматических дикислот, так как сам легко вступает в реакцию с ди-
хлорангидридами [23, с. 15], однако он хорошо растворяет ароматиче-
ские полигидразиды и полиамиды, а также некоторые типы полигетеро-
ариленов. Зависимость физических свойств ДМСО от температуры при-
ведена в табл. 1.3.
Таблица 1.3. Влияние температуры на физические свойства ДМСО [26]
Свойство Температура. °C
20 40 60 80 100 130
Плотность, кг/м3 1100 1082 1063 1047 1022
Динамическая вязкость, 10~3 Па-с 2,47 1,64 1,12 0,84 0,68 0,57
Мольный объем, см3/моль 71,0 72,1 73,4 74,6 —
Показатель преломления и“ 1,4783 1,4695 1,4600 1,4517 — —
Удельная электропроводность, 10-6 См-м-1 3,00 4,3 5,8 7,1 — —
Давление паров, Па 49 220 1000 1870 4550 —
ДМСО — сильный донорный растворитель, смешивается с различ-
ными акцепторами с выделением тепла, склонен к образованию соль-
ватов. Может содержать в виде примесей меркаптаны. Горюч. Токсиче-
ские свойства, по мнению ряда исследователей, выражены менее ярко
по сравнению с ДМАА и ДМФ. Некоррозионноактивен по отношению к
металлам.
N-Метилпирролидон (МП). В настоящее время выпускается в про-
мышленном масштабе. Синтезируется по следующей схеме [27, 28]:
Н2С---СН2
Н2С С=О
'чо/
+
ch3nh2
Н2С—сн2
+ NH3 > | | +NaOH
Н2С с=о
I
н
Н2С---сн2
Н2С с=о
Н2С—сн2
С1СН3 + I I
Н2С с=о
сн3
Na
МП хорошо смешивается с водой, спиртами, эфирами, кетонами,
углеводородами, некоторыми галоидуглеводородами и растительными
маслами. С хлористым водородом дает хлоргидрат, плавящийся при
температуре 85°C. Образует комплексные соединения с галоидами ни-
келя, кобальта и железа.
34
В нейтральном растворе МП очень стабилен. Под действием горя-
чих щелочей и кислот гидролизуется. МП характеризуется меньшей
токсичностью по сравнению с ДМАА и ДМФ. ПДК в воздухе рабочих
помещений составляет 100 мг/м3 [6, с. 18]. Горюч. В последнее время
нашел широкое применение в качестве реакционной среды при получе-
нии полностью ароматических ПА и некоторых типов гетероцикличе-
ских термостойких полимеров.
Гексаметилфосфортриамид (ГМФА). Промышленный продукт, по-
лучаемый по схеме [29]:
РОС13 + 6HN(CH3)2 -> OP[N(CH3)2]3 4- 3H2N(CH3)2C1
Бесцветная жидкость, смешивающаяся в любых соотношениях с водой,
полярными и неполярными органическими соединениями. Избиратель-
но сольватирует хлорированные углеводороды и служит экстрагирую-
щим агентом для извлечения последних из водных смесей. Образует
комплексы с бромидами кобальта и кадмия. Устойчив к гидролизу в
инертных средах. Загорается с трудом. Токсикологические характери-
стики практически не изучены, однако предполагается, что пары ГМФА
канцерогенны. ГМФА широко используется в химии и технологии коор-
динационных соединений, а также в электрохимии. В последнее время
нашел применение для получения термостойких полиамидов [30; 31],
которые плохо растворяются в других амидных растворителях. При
этом используются смеси ГМФА с другими амидными растворителя-
ми— такими, как ДМАА и МП. Оказалось, что смеси ГМФА с указан-
ными соединениями обладают повышенной растворяющей способностью
по сравнению с растворяющей способностью отдельных компонентов
смеси. Некоторые физические свойства ГМФА приведены в табл. 1.4.
Другие растворители. Из других органических растворителей, кото-
рые в настоящее время считаются промышленно перспективными, нуж-
Н2С—СН2
I I
но отметить сульфолан Н2С СН2 и Н,М,М',Ат/-тетраметилмочевину
SO2
О
Н3Сх II /СН3
\N—С—Nz
н3с/ \сн3
Сульфолан — торговое название двуокиси тетрагидротиофена
[32—33]. Он представляет собой при обычной температуре бесцветное
твердое вещество с приятным запахом, растворимое в воде, этиловом
спирте, бензоле, ацетоне и других веществах, нетоксичен. Обладает вы-
сокой гигроскопичностью. Кроме реакционной среды при получении
термостойких полимеров, сульфолан находит применение в качестве
экстрагента ароматических углеводородов из нефтяных фракций, пла-
стифицирующей и стабилизирующей добавки, а в производстве химиче-
ских волокон как растворитель полиакрилонитрила или его сополи-
меров.
Г4,Ы,М',М'-Тетраметилмочевина (ТММ) как растворитель описана
в меньшей мере, хотя за рубежом производится в больших объемах.
Является легкоподвижной жидкостью, смешивающейся в любых соот-
ношениях с водой, спиртами и углеводородами. Так же, как и сульфо-
лан, гигроскопична; токсические свойства не изучены. Получил широ-
кое распространение в качестве реакционной среды при получении аро-
3* 35
магических полиамидов «-структуры [23, с. 18]. Ниже приведены неко-
торые физические характеристики сульфолана и ТММ:
Сульфолан тмм
Температура плавления, °C 28 —1,2
Температура кипения, °C Плотность кг/м3 283 176,5
при 30 °C 1260 960
при 100 °C 1200
Динамическая вязкость, Па-с
при 30 °C 9,8-10-3 1,28-10-з
при 100 °C 2,5-Ю-з
Дипольный момент, D Удельная теплоемкость при 100 °C (373 К), —, 3,37
кДж/(кг-К) Диэлектрическая проницаемость 502 —
при 30 °C 44 23,1
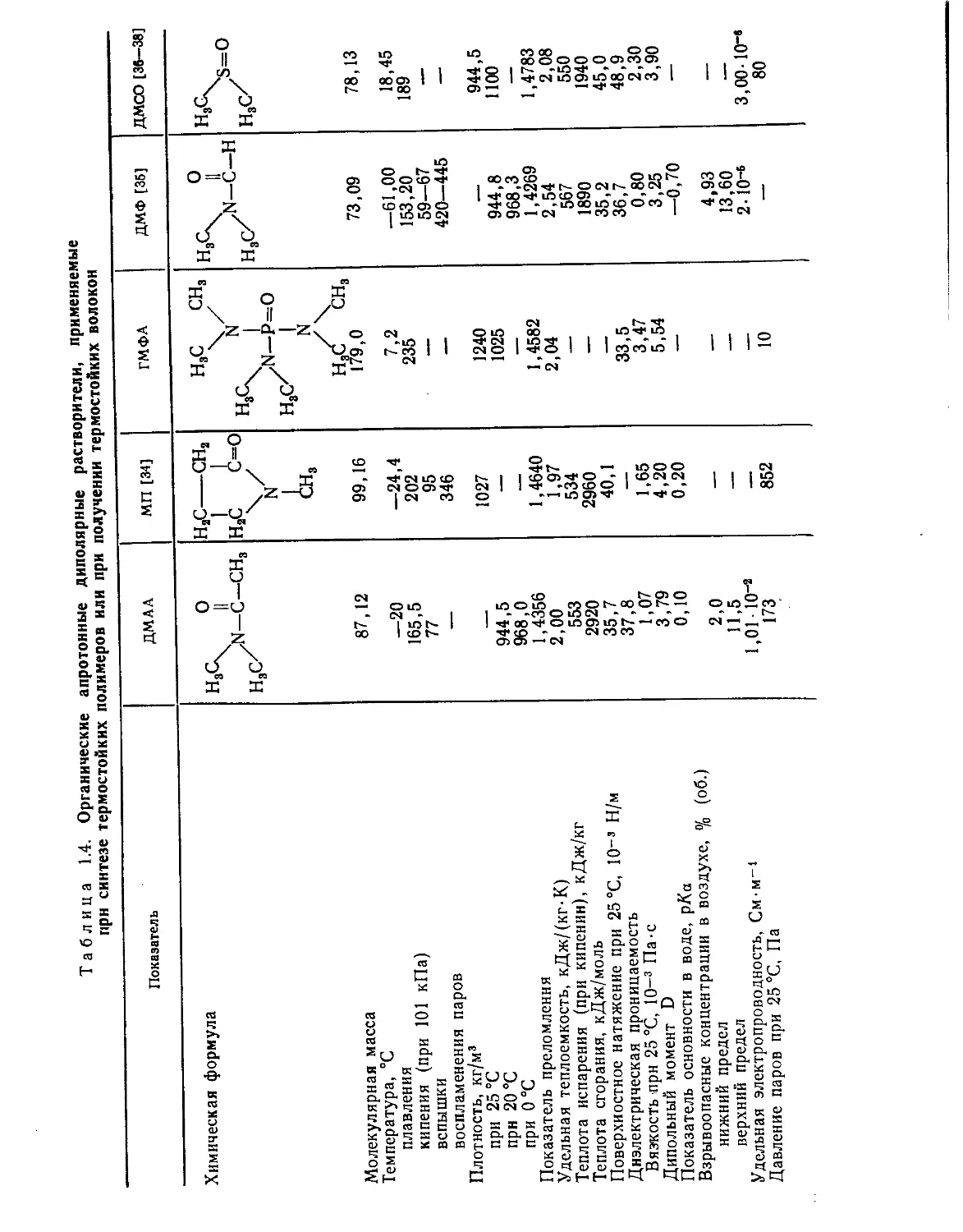
Физико-химические свойства органических апротонных диполярных
растворителей, нашедших широкое распространение при получении
термостойких полимеров и волокон на их основе, представлены в
табл. 1.4.
В последнее время появилось сообщение о том, что некоторые фос-
форорганические растворители в смеси с хлористым литием растворяют
высокомолекулярный поли-п-фенилентерефталамид [39]. К таким рас-
творителям относятся: М,М,Ь1',М'-тетраметилметилфосфондиамид
СН3Р(О) [Ь1(СНз)2]2; М,М-диметилдиметилфосфинамид (CH3)2P(O)N
(СН3)2; И,М,М',№-тетраметилэтилфосфондиамид С2Н5Р(О) [N(CH3)2]2;
триэтилфосфиноксид (С2Н5)3РО и ряд других. Наиболее доступным из
них является первый растворитель. Это бесцветная стабильная жид-
кость, 7’кип=120°С (при 4 кПа), является практически единственным из
органических растворителей, пригодных для растворения указанного
полимера.
Полифосфорная кислота. Полифосфорная кислота, применяемая в
качестве реакционной среды при синтезе некоторых гетероциклических
волокнообразующих термостойких полимеров, представляет собой си-
ропообразную, гигроскопичную жидкость, состоящую из равновесных
смесей орто-, пиро-, тетра- и триполифосфорных кислот, содержащих
от 72,4 до 88,7% фосфорного ангидрида [40, с. 36]. В промышленности
производство полифосфорной кислоты осуществляется двумя способами:
1) упариванием экстракционной фосфорной кислоты (мокрый
способ);
2) сжиганием желтого фосфора до фосфорного ангидрида с после-
дующим насыщением фосфорной кислоты получаемыми парами (терми-
ческий способ).
Второй способ дает возможность получать полифосфорную кислоту
высокой степени чистоты.
В связи с тем, что полифосфорная кислота является продуктом на-
сыщения ортофосфорной кислоты фосфорным ангидридом, ее физиче-
ские свойства резко зависят от многих факторов (концентрации ангид-
рида, температуры и т. д.).
В табл. 1,5 представлены данные, характеризующие влияние кон-
центрации фосфорного ангидрида на свойства полифосфорной кислоты.
Одной из главных характеристик полифосфорной кислоты, влияющей
на процесс синтеза термостойких полимеров, является ее вязкость, в
большой степени зависящая от концентрации и температуры (табл. 1.6).
36
Таблица 1.4. Органические апротонные диполярные растворители, применяемые
прн синтезе термостойких полимеров или при получении термостойких волокон
Показатель ДМАА МП [34] ГМФА ДМФ [35] ДМСО [36-38]
Химическая формула НаС\ ? Н2С—сн2 1 1 Н3С СН3 ис О Н3(к
/N-C-CH, нас с=о иг /N-C-H н3с/ н сА0 нас
>N—Р=О
I н3с/ |
СН, N
Молекулярная масса 87,12 99,16 НаС/ \на 179,0 73,09 78,13
Температура, °C
плавления —20 —24,4 7,2 —61,00 18,45
кипения (при 101 кПа) 165,5 202 235 153,20 189
вспышки 77 95 —— 59—67
воспламенения паров — 346 420—445 —1
Плотность, кг/м3
при 25 °C — 1027 1240 —. 944,5
прн 20 °C 944,5 — 1025 944,8 1100
при 0°С 968,0 — — 968,3 ——
Показатель преломления 1,4356 1,4640 1,4582 1,4269 1,4783
Удельная теплоемкость, кДж/(кг-К) 2,00 1,97 2,04 2,54 2,08
Теплота испарения (при кипении), кДж/кг 553 534 — 567 550
Теплота сгорания, кДж/моль 2920 2960 —— 1890 1940
Поверхностное натяжение при 25 °C, 10~3 Н/м 35,7 40,1 — 35,2 45,0
Диэлектрическая проницаемость 37,8 —— 33,5 36,7 48,9
Вязкость прн 25 °C, 10-3 Па-с 1,07 1,65 3,47 0,80 2,30
Дипольный момент D 3,79 4,20 5,54 3,25 3,90
Показатель основности в воде, рХа 0,10 0,20 ч — —0,70 —
Взрывоопасные концентрации в воздухе, % (об.)
нижний предел 2,0 — — 4,93 —
верхний предел 11,5 — —— 13,60 *—
Удельная электропроводность, См-м-1 1,01-io-2 — 2-10-6 3,00-10-«
Давление паров при 25 °C, Па 173 852 10 — 80
Таблица 1.5. Влияние концентрации фосфорного ангидрида
на физические свойства полифосфорной кислоты [40, с 75]
Концентрация фосфорного ангидрида, % (масс.) Концентрация кислоты, % (масс.) ГКИП' С Плотность, кг/мз (25 °C) ) Удельная теплоемкость ср, кДжДкг-К)
72,65 100,70 250,3 0,1674
74,92 103,10 290,5 1928 0,1632
76,13 104,71 322,2 1913 0,1630
78,38 108,16 368,7 1950 0,1593 .
81,74 112,80 501,1 2042 0,1540
83,49 — 536,3 2064 —
87,68 120,99 — 2101 0,1258
88,79 122,53 — 2126 0,1280
ПФК с содержанием Р2О5 до 77,6% способна выдерживать сильное
переохлаждение без заметной кристаллизации. При более высоких
концентрациях фосфорного ангидрида и в присутствии механических
примесей кристаллизация становится заметной, однако кислота не рас-
слаивается.
ПФК электропроводна, причем электропроводность увеличивается
с повышением температуры и уменьшается с ростом концентрации.
При 20 °C удельная электропроводность ПФК, содержащей 75,4% Р2О5,
равна 9,28-10~1 С мм-1, а при концентрации Р2О5 82,6% —0,639-1О-1
Смм~‘. При 200°С электропроводность этих кислот равна 398-10-1 и
155-10-1 Смм-1 [40, с. 80].
Растворение ПФК в воде сопровождается выделением значитель-
ного количества тепла. ПФК негорюча и токсикологически менее опасна
по сравнению с другими минеральными кислотами, не обладает запа-
хом, пары не раздражают дыхательных путей. При прямом контакте с
кожей человека вызывает ожоги. ПДК в рабочей зоне составляет
1 мг/м3, в воздухе населенных мест — 0,05 мг/м3 [6, с. 57]. Коррозион-
ная активность ПФК по сравнению с ортофосфорной кислотой и с дру-
гими минеральными кислотами выражена менее ярко.
Серная кислота и олеум. Серная кислота является практически
универсальным растворителем для большинства термостойких полиме-
ров; ее свойства как растворителя хорошо известны. Следует отметить,
что как технологически пригодный растворитель для получения волокон
из различных термостойких полимеров серная кислота используется
сравнительно недавно. Серная кислота — вязкая жидкость, молекулы
которой связаны между собой сильными водородными связями. Благо-
даря своим координирующим свойствам и высокой диэлектрической
проницаемости она способствует ионизации и, следовательно, диссо-
циации многих соединений, в том числе и полимерных. Физические
Таблица 1.6. Зависимость динамической вязкости (в Па с) ПФК
от температуры и концентрации [40, с. 78]
Концентрация, % (масс.) Температура, °C
Р2О5 НзРОа 20 40 60 80 100 120 140
72,84 100,56 0,312 0,116 0,055 0,031
75,41 104,14 0,645 0,218 0,112 0,058 0,031 0,019 0,013
76,44 105,56 0,974 0,296 0,121 0,059 —
78,30 107,93 6,88 1,29 0,410 0,166 0,076 0,044 0,027
79,82 110,05 14,00 2,300 0,691 0,266 0,123 0,068 0,042
38
Таблица 1.7. Физико-химические свойства метай- и хлорсульфоиовых
кислот [42; 43]
Свойство Метансульфоновая кислота Хлорсульфоновая кислота Примечание
Химическая форму- ла CHsSO3H CISOjH Хлорсульфоновая кислота — токсичный
Молекулярная мас- са 96,10 116,50 продукт (при хране- нии и нагревании
Физическое состоя- Бесцветная Бесцветная, сильно возможно выделение
ние ЖИДКОСТЬ дымящая на воздухе жидкость SO3 и С12). Пары вызывают тяжелые
Температура плав- ления, °C Температура кипе- — —80 поражения дыхатель- ных путей
167 (при 1,3 кПа 155—156 (частично Токсические свой-
ния, °C может разлагать- ся) разлагается) ства метансульфоно- вой кислоты не изу-
Плотность (при 20 °C), кг/м’ 1481 1770 чены. Применяются для растворения по-
Теплота испарения, кДж/моль — 53,5 (при кипении) лиоксадиазолов, аро- матических полиами-
Удельная теплоем- кость, кДж/(кг-К) 1,2 дов л-структуры и лестничных полиме- ров
свойства безводной серной кислоты приведены в литературе [38, с. 93].
Практическое применение для синтеза волокнообразующих поли-
гетероциклов, в частности полиоксадиазолов, нашел олеум — дымящая
маслянистая опаЯесцирующая, чрезвычайно гигроскопичная жидкость
[41].
Свойства олеума в сильной мере определяются концентрацией сво-
бодного SO3 в серной кислоте [41, с. 50].
Пары SO3 токсичны. Предельно допустимая концентрация в возду-
хе рабочих помещений составляет 1 .мг/м3 [6, с. 16].
Олеум используется в качестве реакционной среды непосредствен-
но для одностадийного синтеза ПОД из ароматических дикислот и про-
изводных гидразина, при этом ПОД остается в растворе. После разбав-
ления серной кислотой смесь практически готова к формованию.
Кроме этого, серная кислота или олеум нашли применение как
растворители при получении высокопрочных волокон из растворов пол-
ностью ароматических ПА пара-структуры, при получении волокон
лестничной или полулестничной структуры. Технико-экономические рас-
четы показали, что этот путь перспективен, хотя имеются затруднения
из-за высокой коррозионной активности серной кислоты и ее растворов.
В литературе часто указывается на возможность применения в ка-
честве растворителей для некоторых типов высокотермостойких поли-
меров метан- и хлорсульфоновой кислот. Некоторые физико-химические
свойства этих кислот приведены в табл. 1.7.
ЛИТЕРАТУРА
1. Мономеры для поликонденсации. Под ред. В. В. Коршака. М., «Мир», 1976.632 с.
2. Термостойкие пластики. Экспресс-информация. М., ВИНИТИ, 1976, Xs 19,
реф. 137.
3. Взрывоопасность химических веществ. Экспресс-информация. Северодонецк,
ВНИИТБХП, 1970, сер. 1, вып. 6.
4. Соколов Л. Б. Поликонденсационный метод синтеза полимеров. М., «Химия»,
1966. 336 с.
5. Пожарная опасность веществ и материалов. Под ред. И. В. Рябова. М., «Строй-
издат», 1970. 336 с.
39
6. Беспамятное Г. П. и др. Предельно допустимые концентрации вредных веществ
в воздухе. Изд. 2-е. Л., «Химия», 1975. 456 с.
7. Work Т. S., J. Chem. Soc., 1940, № 9, р. 1315—1321.
8. Термостойкие пластики. Экспресс-информация. М., ВИНИТИ, 1976, № 12, реф. 82.
9. Взрывоопасность химических веществ. Экспресс-информация. М., НИИТЭХИМ,
1974, вып. 6.
10. Пожаро- н взрывоопасность химических веществ. Экспресс-информация. М.,
НИИТЭХИМ, 1976, вып. 10 (20).
11. Пожаро- и взрывоопасность химических веществ. Экспресс-информация. М.,
НИИТЭХИМ, 1975, вып. 7 (17).
12. Craaf R., Langer W., J. prakt. Chem., 1937, Bd. 148, S. 161—172.
13. Взрыво- и пожароопасность химических веществ. Экспресс-информация. М.,
НИИТЭХИМ, 1975, вып. 1 (10).
14. Одрит Л., Огг Б. Химия гидразина. Под ред. Я- М. Варшавского. М., Издатин-
лит, 1954. 237 с.
15. Stille J. К., Mainen Е. L., J. Polymer. Sci., 1966, A-l, v. 4, р. 665—670.
16. Пат. США 3743624 (1973).
17. Joda N., Ikeda К.., J. Polymer Sci., 1967, A-l, v. 5, p. 2354—2364.
18. Пат. США 2667511 (1954).
19. Leader G. R., Gormely I. F., J. Am. Chem. Soc., 1951, v. 73, p. 5731—5738.
20. Паркер Д., «Успехи химии», 1963, т. 32, с. 1270—1295.
21. Wolfgang I., Alexander L., Chem.-Reifung. Chem. Apparatur, 1960, Bd. 84, S. 239.
22. Пакшвер Э. А. В кн.: Карбоцепные синтетические волокна. Под ред. К. Е. Пе-
репелкина. М., «Химия», 1973. 596 с.
23. Соколов Л. Б. и др. Термостойкие ароматические полиамиды. М., «Химия»,
1975. 253 с.
24. Информация ВНИИСВ. Хим. волокна, 1968, № 6, с. 72—73.
25. Abadie-Moumert F. A., «Papeterie», 1959, № 3, р. 187—196.
26. Schlafer М. L., Schaffernicht W„ Angew. Chem., 1960, Bd. 8, Ns 17, S. 618—625.
27. Промышленный органический синтез. Экспресс-информация. М., ВИНИТИ, 1963,
№ 3, реф. 29.
28. Remand I., Rev. prod, chim., 1962, v. 65, № 1301, p. 28—36.
29. Normant H., Angew. Chem., 1967, Bd. 79, Ns 23, S. 1029—1050.
30. Престон Д. Международный симпозиум по химическим волокнам. Препринты.
Калинин, 1974, секция 1, с. 15—>18.
31. Федоров А. А'., Соколов Л. Б., Высокомол. соед., 1973, сер. Б, т. 15, № 1,
с. 74—77.
32. Rev. prod, chim., 1964, v. 67, № 1320, p. 173—179.
33. Максимова И. В. и др., Хим. пром, за рубежом, 1975, № 9, с. 43—60.
- 34. Геллер Б. Э., и др., «Труды ВНИИВ». М., Гизлегпром, 1959, вып. 5. 191 с.
35. Васенко Е. И., Дубровский С. М„ ЖФХ, 1953, т. 27, № 2, а 281—289.
36. Chem. Rundschau, 1958, № 2, S. 533—535.
37. Douglas T. S.. J. Am. Chem. Soc., 1948, v. 70, № 60, p. 2001—2008.
38. Гутман В. Химия координационных соединений в неводных средах. М., «Мир»,
1971 220 с
39. Пат.’США’3748299 (1973).
40. Михайлин А. Д., Постников И. Н„ Кленицкий А. И., Фосфор и фосфорная кис-
лота. Труды НИУИФ, 1969, вып. 211, с. 36—44.
41. Справочник сернокнслотчика. М., «Химия», 1971. 744 с.
42. Краткая химическая энциклопедия. Т. 5. М., «Советская энциклопедия», 1967,
с. 726.
43. Handbook of Chemistry and Physics. Cleveland. Chemical Rubber Publishing Co.,
1956.
ГЛАВА 2
ОБЩИЕ ПРИНЦИПЫ СИНТЕЗА ТЕРМОСТОЙКИХ
ВОЛОКНООБРАЗУЮЩИХ ПОЛИМЕРОВ*
Подавляющее большинство термостойких полимеров получают ме-
тодами поликонденсации. Гораздо реже применяют для этой цели по-
лимеризацию.
Теоретические основы поликонденсационных методов синтеза по-
лимеров освещены в ряде работ [I—5]. Процессы поликонденсации
* Глава напясана совместно с Ю. Л. Панкратовым.
40
можно классифицировать по типу протекания реакции (равновесная и
неравновесная), способам проведения и аппаратурного оформления и
по механизму процесса.
Для неравновесной поликонденсации характерны необратимость и
отсутствие обменных реакций в условиях синтеза. Необратимость может
быть вызвана либо очень высокими скоростями процесса, либо стабиль-
ностью образующихся химических структур, например, пяти- или шести-
членных циклов в основной цепи полимера при полигетероциклизации.
На примере синтеза ПА можно проследить, как повышение скорости по-
ликонденсации при переходе от менее активных к более активным моно-
мерам приводит к необратимости процесса. По реакционной способности
производные дикарбоновых кислот располагаются в ряд: карбоновые
кислоты <эфиры< ангидриды Сгалогенангидриды.
Взаимодействие карбоксильных групп с аминогруппами при отсут-
ствии катализаторов начинается при температурах выше 150 °C. Не-
смотря на высокие температуры, процесс протекает сравнительно мед-
ленно (константы скоростей 10~s—10~3 моль-л-1-с-1) и носит равновес-
ный характер. Более реакционноспособные ангидриды кислот могут
взаимодействовать с диаминами при невысоких температурах; при этом
равновесный характер процесса сохраняется [6, с. 17; 7]. При исполь-
зовании дихлорангидридов температуру реакции можно снизить вплоть
до температуры замерзания растворителя. Несмотря на это, реакция
протекает с высокой скоростью (10~3—106 моль • л-1 • с-1) и практически
нацело сдвинута в сторону образования полимера; процесс становится
практически необратимым.
Для всех поликонденсационных процессов характерны общие за-
кономерности, в частности зависимость молекулярной массы от глуби-
ны превращения и от избытка одной из реагирующих функциональных
групп. Однако практическая необратимость быстро протекающих реак-
ций поликонденсации вносит свою специфику. Так как скорости реак-
ции намного превышают скорости диффузии мономеров (Ур^Уд), то
поликонденсация протекает прежде всего в тех зонах, где успел уста-
новиться контакт взаимодействующих веществ. Общие закономерности
поликонденсации проявляются именно в этих реакционных зонах.
При отнесении же ко всему объему они предстают в искаженном
виде. Это касается, например, зависимости молекулярной массы поли-
мера от избытка одной из функциональных групп. В определенных ус-
ловиях становится возможным получение высокомолекулярного поли-
мера при неэквимольном соотношении мономеров. Так, при медленном
дозировании дихлорангидрида, взятого с 50%-ным избытком, удалось
получить полиамиды той же молекулярной массы, что и при эквива-
лентном соотношении мономеров [8, с. 56]. Подобное «несоблюдение»
принципа эквивалентности является кажущимся. Попадая в реакцион-
ную зону, мономер, взятый в избытке, сразу реагирует с эквимольным
количеством второго мономера. А поскольку образующийся полимер
не подвержен деструктивным реакциям под действием имеющегося из-
бытка второго мономера, то при поступлении следующей порции веще-
ства реакция развивается лишь в одном направлении — в направлении
роста цепи. Фактически в реакционной зоне автоматически поддержи-
вается эквивалентное соотношение мономеров. При больших скоростях
дозирования мономеров возникает опасность появления местных пере-
гревов, что приводит к повышению роли побочных процессов и сниже-
нию молекулярной массы полимера. Таким образом, в принципе суще-
4 Г
ствует возможность регулирования молекулярной массы за счет изме-
нения скорости дозирования одного из мономеров.
Как и для необратимых полимеризационных процессов, молеку-
лярная масса продуктов неравновесной поликонденсации определяется
не термодинамикой, а кинетикой, точнее, соотношением скоростей основ-
ной и побочной реакций.
Быстро протекающие процессы неравновесной поликонденсации ха-
рактеризуются низкими значениями энергии активации (4—
40 кДж/моль) и положительным тепловым эффектом.
Способы проведения поликонденсации разнообразны. Процесс мож-
но провести в расплаве, твердой фазе, растворе, эмульсии и на границе
раздела фаз. Применимость поликонденсации в расплаве и твердой фазе
ограничена низкой термостойкостью исходных мономеров и неплав-
костью полимеров. Из всех способов наибольшее распространение по-
лучила поликонденсация в растворе — низкотемпературная в амидных
растворителях и высокотемпературная в растворителях типа ПФК,
олеума и других дегидратирующих агентов. Меньшее практическое
применение нашли межфазная и эмульсионная поликонденсация.
Несмотря на большое разнообразие функциональных групп, при-
нимающих участие в синтезе волокнообразующих термостойких поли-
меров, можно выделить две большие группы процессов, различающихся
по механизму.
1) процессы, в основе элементарных актов которых лежат реакции
ацилирования. Назовем их «процессами полиацилирования»;
2) реакции полигетероциклизации.
Приведенная классификация является упрощенной. Ряд термостой-
ких полимеров получают методами окислительной поликонденсации,
поликоординации и другими методами [3, с. 248; 9]. Однако эти спосо-
бы не получили пока широкого распространения для синтеза волокно-
образующих термостойких полимеров.
2.1. РЕАКЦИИ, ПРОТЕКАЮЩИЕ ПО ТИПУ ПОЛИАЦИЛИРОВАНИЯ
В качестве ацилирующих агентов применяют производные дикар-
боновых, реже — сульфокислот, фосфиновых и фосфоновых кислот.
Ацилируемыми мономерами являются либо ароматические диамины и
их производные, либо ароматические диолы. Реакции протекают по типу
нуклеофильного замещения с атакой неподеленной пары электронов
атома азота (в аминах) или кислорода (в диолах) на карбонильный
атом углерода (в кислотной компоненте). Общая схема ацилирования
имеет следующий вид:
О.
—R-NH2+ /C-Rj
у/
НО- о
I. I II
—R—N — С—Rj —R—NH—С—Rj+HX (1)
Н X
или
О.
—R—ОН-f- V—Rj
х/
О
II
—R—О—С—Rj+HX
(2)
(R и Ri — ароматические радикалы, X—ОН, Cl, Br, F, ОСНз, ОС6Н5 и
т. д.). На заключительной стадии ацилирования происходит разрыв свя-
42
зей азот (кислород) —водород и углерод — X с образованием низкомо-
лекулярного продукта НХ. При использовании диангидридов реакция
не сопровождается выделением низкомолекулярного продукта.
О
с но но
/ \/ |, II I II
—R—NH2 + O R, —R—N+---С. ?=+ — R—N—/ (3)
''''''Н /К1\ /Rl\
\ н О-—с/ Х НО—CZ Х
о И 11
° о о
В зависимости от природы ацилирующих агентов завершающая
стадия реакции может быть равновесной или неравновесной, что и опре-
деляет тип протекания процесса в целом. При использовании дикарбо-
новых кислот, их диэфиров и диангидридов реакция носит равновесный,
а при использовании дигалогенангидридов—’неравновесный характер.
При полиацилировании диаминов и их производных (схемы 1 и 3)
образуются полиамиды и их производные (реакции полиамидирования),
а при полиацилировании ароматических диолов (схема 2) — ароматиче-
ские сложные полиэфиры (полиарилаты).
2.1.1. Механизм реакций ацилирования
полифункциональных аминов и их производных
Эти реакции являются основой получения не только ароматических
полиамидов, но и начальной стадией синтеза ПИ, ПБИ, ПВО, ПОД,
пирронов и ряда других полигетероариленов.
Известно [10, с. 118], что для реакций алифатических диаминов с
алифатическими дикарбоновыми кислотами величины констант равно-
весия лежат в пределах 4—10; реакции протекают со скоростью
10~5—10-3 л-мол-1-с-1 и имеют высокие энергии активации. Аромати-
ческие диамины имеют более низкую основность по сравнению с али-
фатическими [И]; они, например, не способны давать устойчивые соли
с ароматическими кислотами. Поэтому для этих систем можно ожидать
уменьшения констант скоростей и равновесия и увеличения энергии
активации процесса по сравнению с системами из алифатических моно-
меров. Высокие энергии активации и низкая реакционная способность
мономеров заставляют проводить поликонденсацию ароматических
диаминов с ароматическими дикарбоновыми кислотами при высоких
температурах (до 350°C). Температуры плавления термостойких поли-
меров лежат вблизи или выше температур разложения, поэтому, не-
смотря на высокие температуры поликонденсации, полимер быстро за-
твердевает. Поликонденсация при этом практически прекращается и
молекулярная масса полимера остается низкой. Чтобы сдвинуть равно-
весие в сторону образования высокомолекулярного полимера, поликон-
денсацию продолжают в твердой фазе, прибегая при этом к дальней-
шему повышению температуры и глубокому вакууму. Продолжитель-
ность процесса резко возрастает. К тому же следует учитывать неста-
бильность мономеров (особенно, ароматических диаминов), тетраминов
и др. Из термостойких полимеров поликонденсация в расплаве и в
твердой фазе получила некоторое распространение при синтезе волок-
нообразующих полибензимидазолов '[12]. Попытки прибегнуть к этому
43
способу для синтеза ароматических полиамидов [13], полиимидов
[6, с. 8], пирронов [14] и др. приводят к получению низкомолекулярных
полимеров.
Применение катализаторов в реакциях ароматических диаминов с
ароматическими дикарбоновыми кислотами позволяет ускорить дости-
жение равновесия и проводить процесс при более низких температурах
(100—250°C) в высококипящих растворителях [15].
Переход к более реакционноспособным диэфирам и, особенно, к
диангидридам ароматических дикарбоновых кислот приводит к воз-
можности снижения температуры полиамидирования. Так, например,,
для взаимодействия диангидридов с ароматическими диаминами до-
статочны низкие температуры (0—50°C). Более того, повышение тем-
пературы нежелательно из-за ускорения побочных реакций, снижающих
ММ полимера. Высокие значения констант скоростей и равновесия ре-
акций ацилирования ангидридами кислот сближают их по ряду деталей
механизма с типично необратимыми процессами ацилирования поли-
аминов галогенангидридами.
Для необратимых процессов полиамидирования схема ацилирова-
ния выглядит следующим образом [7]:
а-комплекс
Переходному состоянию (о-комплексу) предшествует образование
комплекса с переносом заряда (КПЗ) в результате донорно-акцептор-
ного взаимодействия между молекулами диаминов и их производных
(доноры электронов) и диангидридов или галогенангидридов (акцепто-
ры электронов). Аналогичная схема, возможно, имеет место и при
взаимодействии диаминов с дикарбоновыми кислотами, хотя это не под-
тверждено литературными данными. Отмечают [7], что донорно-акцеп-
торное взаимодействие способствует созданию такой ориентации моно-
меров в системе, которая благоприятствует протеканию реакции, сни-
жая величину предэкспоненциального множителя и энергию активации.
Образование КПЗ зафиксировано для ряда систем: галогенангидри-
ды — третичные и первичные амины [7; 16—18], пиромеллитовый диан-
гидрид— диамины [19] и др. В ряде случаев удалось выделить КПЗ
как индивидуальные химические соединения. КПЗ—энергетически не-
устойчивая система. Под влиянием внешних факторов происходит пол-
ный перенос электрона от донора к акцептору и образуется о-комплекс.
Обе стадии (образование КПЗ и a-комплекса) являются равновесными»
имеют высокую скорость и низкую энергию активации. Константа рав-
новесия реакции образования ст-комплекса зависит от полярности рас-
творителя [19; 20] и электронной характеристики мономеров [20—22].
Например, в реакции пиромеллитового диангидрида с производными
ПФДА замена тетрагидрофурана на более полярный ДМАА приводит
44
к полному переносу заряда с образованием ион-радикала, способствуя
тем самым значительному увеличению константы равновесия. Анало-
гичный результат достигается при введении электрондонорного мости-
ка в молекулу диамина или электроноакцепторного — в молекулу дига-
логенангидрида.
Лимитирующей стадией ацилирования дигалогенангидридами яв-
ляется необратимый распад о-комплекса с разрывом связей азот — во-
дород и углерод — галоген и образование новой, амидной связи и гало-
геноводорода. Скорость этой стадии зависит от активности ацилирую-
щего агента (см. ниже).
Молекулярная масса полимеров при необратимой поликонденсации
определяется соотношением скоростей основной реакции и побочных
(химических и физических) процессов. Скорость собственно поликон-
денсации зависит от реакционной способности мономеров, природы ре-
акционной среды, концентрации, температуры, чистоты исходных мо-
номеров.
Реакционная способность мономеров играет первостепенное значе-
ние при необратимой поликонденсации. Выше указывалось, что ацили-
рование диаминов и их производных протекает через стадию донорно-
акцепторного взаимодействия мономеров. Естественно ожидать, что
реакционная способность мономеров будет связана с электроноакцеп-
торной или электронодонорной способностью соответственно производ-
ных дикарбоновых кислот и производных диаминов.
Электроноакцепторную способность производных дикарбоновых
кислот можно оценить по величине их сродства к электрону, а также
по энергии активации образования КПЗ с аминами. В табл. 2.1 приве-
дены значения этих величин для производных фталевых кислот [23].
Таким образом, сродство к электрону возрастает в ряду: альдеги-
ды < фторангидриды < хлорангидриды < бромангидриды < циан-
хлорангидриды. В том же ряду уменьшается энергия активации обра-
зования КПЗ, что облегчает протекание реакции ацилирования. Счи-
тают [20; 23], что для галогенангидридов увеличение константы скоро-
сти ацилирования в приведенном ряду связано не только с повышением
их сродства к электрону, но и со снижением прочности связи углерод —
галоген. Известно [20], что энергия связи углерод — галоген составляет:
448 кДж/моль для С—F; 280 кДж/моль — для С—С1; 226 кДж/моль
для С—Вг и 190 кДж/моль для С—I.
Таблица 2.1. Сродство к электрону н способность к комплексообразованию
производных фталевых кислот в реакциях с n-фенилендиамином (ПФДА)
и М,М,М,М-тетраметил-п-фенилеиднамином (ТМ-ПФДА)
Кислотный компонент Сродство к электрону, эВ Энергия полос переноса заряда, эВ
для ПФДА для ТМ-ПФДА для ПФДА для ТМ-ПФДА
Терефталальдегид 0,06 0,08 2,92 2,76
Терефталоилфторид 0,12 0,19 2,86 2,65
Терефталоилхлорид 0,30 0,59 3,68 2,25
Терефталонлбромид 0,32 0,61 2,66 2,23
Терефталоилциаиид 0,78 — 2,20 —
2,5-дициан-терефталоил-хлорид 1,17 — 1,81 —
45
Таблица 2.2. Теплоты взаимодействия изо- и терефталоилхлорида
с ароматическими диаминами в диметилацетамиде при 25 °C [24]
Диамины Теплота реакции АН, кДж/моль
с изофталоилхлоридом с терефталоилхлоридом
ж-Фенилендиамии 235 236
4,4'-Диамннодифенилсульфнд 233 227
4,4'-Диаминодифенилсульфон 205 200
Бензидин 240
ч-Фенилендиамин 255
3,3'-Диамино дифенилизофталамид 232
Реакционную способность производных дикарбоновых кислот мож-
но полуколичественно оценить по теплотам их взаимодействия с ами-
нами (табл. 2.2).
Приведенные в таблице теплоты взаимодействия являются алгеб-
раической суммой собственно теплот поликонденсации и теплот раство-
рения твердого дихлорангидрида. Теплоты взаимодействия ТФХ с бен-
зидином и ПФДА определить не удалось, так как полиамиды выпадали
в осадок. Близость величин теплот взаимодействия обоих дихлорангид-
ридов с каждым из диаминов свидетельствует о близкой реакционной
способности изо- и терефталоилхлорида. По-видимому, реакционная
способность дихлорангидридов мало зависит от изомерии.
Реакционная способность полиаминов в реакциях ацилирования
связана с их основностью [25], которую оценивают по величинам по-
тенциала ионизации и констант диссоциации в воде или органических
растворителях [26, с. 462]. В свою очередь основность определяется
плотностью электронного облака неподеленных р-электронов у атома
азота [27]. Установлен следующий ряд по основности диаминов [25,
27]: «-фенилендиамин>4,4'-диаминодифенилсульфид>4,4'-диаминоди-
фенилоксид>л*-фенилендиамин>бензидин. В работе [25] показана
симбатная связь между основностью и реакционной способностью ди-
аминов. Основность диаминов зависит от их строения.
Для ароматических диаминов с двумя (или более) фениленовыми
ядрами большое значение имеет природа «шарнирной» связи. Мостико-
вые связи электроноакцепторного характера (—SO2—;—СО—и др.) уси-
ливают «стягивающее» действие ароматических фрагментов, приводя к
дальнейшему снижению основности аминогрупп; электронодонорные
мостики (—О—, —S—, —СН2— и др.) вызывают противоположный эф-
фект. Введение в ядро атомов галогена, гидроксильных и карбоксиль-
ных групп (особенно в орто-положении к аминогруппе) понижает реак-
ционную способность аминогрупп. При наличии объемных заместите-
лей пониженная реакционная способность может быть результатом сте-
рических препятствий. В ряде случаев низкую реакционную способ-
ность связывают с таутомерными превращениями диаминов [28].
Низкая основность некоторых диаминов особенно неблагоприятно
сказывается при попытках их поликонденсации с производными дикар-
боновых кислот, обладающих более слабой ацилирующей способностью.
Так например, если поликонденсация 4,4'-диаминодифенилсульфона с
ТФХ приводит к получению высокомолекулярного полиамида, то попыт-
46
ка синтеза полиамидокислоты на основе того же диамина и пиромел-
литового диангидрида оказывается безуспешной [7; 29].
В ряде случаев снижение реакционной способности аминогрупп яв-
ляется благоприятным фактором. Так, например, пониженная основ-
ность, а следовательно, и реакционная способность аминогрупп в
орто-положении к амидной связи препятствует гелеобразованию на
первых стадиях синтеза полиамидоаминокислот (промежуточных про-
дуктов для получения пирронов).
Как и в случае дихлорангидридов, реакционную способность ди-
аминов можно полуколичественно охарактеризовать по теплотам их
взаимодействия с различными ацилирующими агентами, например по
теплотам поликонденсации или теплотам образования хлоргидратов
[24]. Поскольку теплота поликонденсации зависит от строения как ди-
амина, так и ацилирующего агента, то с помощью этой величины мож-
но дать лишь сравнительную оценку активности диаминов.
Особенность бифункциональных ароматических соединений (и али-
фатических с числом метиленовых групп меньше 6) — изменение реак-
ционной способности функциональных групп, после того как прореаги-
рует первая из них [30, 31], т. е. (Ki и К.2— константы аци-
лирования). Различие в активностях возникает вследствие изменения
основности аминогрупп или электрофильности ацилирующих групп.
Большое отклонение величины Ki/Лг от единицы может привести к
«особому виду поликонденсационных процессов» [31]. Участие в реак-
ции мономера с -Ki/ZG^l равносильно введению почти монофункцио-
нального соединения. Наибольший интерес могли бы представить про-
цессы, в которых Ki/Ka-Cl. Такие процессы по ряду закономерностей
приближаются к свободнорадикальной полимеризации. Нарушается ха-
рактерная для поликонденсации зависимость между молекулярной мас-
сой полимера и глубиной превращения. Согласно расчетам [31], при
/С1/К2~0,01 высокая молекулярная масса может быть достигнута уже
при 70%-ном превращении. К настоящему времени известны лишь не-
многие примеры поликонденсации бифункциональных соединений, для
КОТОРЫХ Х1//<2<1 [32].
Для большинства ароматических диаминов отношения основностей
первой и второй аминогрупп (рКа, /рКа2) лежат в пределах 4,0—6,0.
Минимальное значение рКа, /рКа2 = 1,09 отмечено у 4,4'-диаминодифе-
нилметана. Обе группы почти равноценны. Максимальная величина
Кл/К.2 наблюдается у n-фенилендиамина ПФДА при его ацилировании
бензоилхлоридом. В порядке убывания К1/К2 диамины можно располо-
жить в ряд [33]: 2,6-диаминопиридин>п-фенилендиамин>бензидин>
> .м-фенилендиамин > 4,4'-диаминодифенилоксид > 4,4'-диаминоди-
фенилметан.
Для дихлорангидридов различие в активностях первой и второй
функциональных групп может достигать больших величин. Так, при
ацилировании п- и .м-фенилендиамина ТФХ в бензоле при 25 °C значе-
ние K.JK2 для ТФХ составляет соответственно 15,0 и 5,6.
Выявлены некоторые общие закономерности:
1) различие в активностях первой и второй функциональных групп
проявляется в большей степени для пара-, чем для .мета-изомеров;
2) величины К.1/К.2 практически не зависят от природы реакцион-
ной среды [25], если она не меняет механизма реакции;
3) активности функциональных групп уравниваются {KJK2—>-1)
по мере их пространственного удаления в молекуле мономера или об-
47
разующегося полимера, что показано на примере синтеза поли-ж-фе-
ниленизофталамида [24].
Реакционная способность различных функциональных групп имеет
особо важное значение при поликонденсации тетрафункциональных
соединений. Если все четыре функциональные группы имеют близкую
реакционную способность, то появляется возможность получения раз-
ветвленного или сшитого полимера, переработка которого в волокно
практически невозможна. При синтезе полибензоксазолов и полибенз-
оксазоламидоимидов и ряда других гетероциклических полимеров эта
опасность исключена, поскольку гидроксильные и карбоксильные груп-
пы нереакционноспособны (при отсутствии катализаторов) в условиях
низкотемпературной поликонденсации. Они вступают в реакцию (с об-
разовавшейся амидной связью) в более жестких условиях. Осложнения
возникают при поликонденсации тетрафункциональных соединений с
однотипными функциональными группами. Близкая реакционная спо-
собность всех аминогрупп в тетраминах показана на примере 3,3'-ди-
аминобензидина, молекулярная диаграмма которого свидетельствует о
почти равных величинах электронной плотности у всех четырех атомов
азота [34].
Для предотвращения гелеобразования прибегают либо к специаль-
ным методам синтеза, либо к подавлению активности свободных ами-
ногрупп путем их замены в исходном тетрамине на менее реакционно-
способные группы [35], либо к блокированию ангидридных групп (на
завершающей стадии реакции) различными аминами [36].
Реакционную способность регулируют подбором пар мономеров,
растворителей или применением катализаторов. Для получения высоко-
молекулярных полимеров нужны мономеры не с максимально высокой,
а с оптимальной реакционной способностью [30]. Использование слиш-
ком реакционноспособных соединений повышает вероятность протека-
ния побочных реакций; резко возрастают требования к чистоте исход-
ных мономеров. Если при ацилировании диаминов дифторангидридами
высокая молекулярная масса полимеров достигается при содержании
воды в системе до 10%, то при использовании более реакционноспособ-
ных дихлорангидридов допустимое количество воды не должно превы-
шать 0,05—0,1% [37].
Вопросы катализа реакций необратимого полиацилирования разра-
ботаны недостаточно. Для низкомолекулярных соединений в качестве
катализаторов ацилирования применяют карбоновые кислоты [38—40]
н третичные амины, в том числе соединения пиридинового ряда [41, 42].
Катализ более эффективен при ацилировании ангидридами, чем гало-
генангидридами [40]. В общем случае, чем слабее ацилирующий агент,
тем сильнее каталитическая активность карбоновых кислот. Это прави-
ло справедливо и при синтезе полимеров. Показана [37] каталитиче-
ская роль уксусной кислоты при поликонденсации изофталоилфторида
с рядом ароматических диаминов. Скорость поликонденсации и ММ
возрастают с повышением концентрации катализатора и мономеров.
Отмечают [37], что катализ эффективен лишь при использовании мало-
основных мономеров. Попытка применения уксусной кислоты в качестве
катализатора при поликонденсации изофталоилхлорида с теми же
диаминами оказалась безуспешной. Более того, присутствие уксусной
кислоты привело к некоторому снижению ММ полиамида.
В ряде случаев прослеживается корреляция между основностью
третичных аминов и молекулярной массой полиамидов [43]. В других
48
случаях эта корреляция отсутствует. Действие третичных аминов зави-
сит от реакционной среды. В реакциях полиацилирования, сопровож-
дающихся выделением хлористого водорода, третичные амины выпол-
няют роль его акцептора. Их присутствие является желательным при
синтезе полиамидов в растворителях (точнее нерастворителях) типа
нитробензола, ацетонитрила, циклогексанона, бензонитрила и т. д.
Здесь они являются и акцепторами хлористого водорода и катализато-
рами. При этом образующийся хлоргидрат третичного амина также
оказывает ускоряющее действие. При синтезе же полиамидов в амид-
ных растворителях, которые сами играют роль акцептора хлористого
водорода, попытки использования третичных аминов в качестве катали-
заторов оказались безуспешными [44]. Возможно, в этих средах доми-
нирующую роль приобретают побочные реакции третичных аминов с
дихлорангидридами.
В отличие от галогенангидридов реакции ангидридов кислот с ами-
нами замедляются в присутствии третичных аминов [41].
В литературе отмечают [9, 45] каталитическую роль амидных рас-
творителей в реакциях ацилирования диаминов и диолов дигалогенгид-
ридами. В связи с этим реакции этого типа называют акцепторно-ката-
литической поликонденсацией. По-видимому, взаимодействие дихлоран-
гидрида с амидным растворителем приводит сначала к образованию
реакционноспособного орто-ацилированного комплекса с повышенной
ацилирующей активностью [45]:
О О
° ХСН3 ° ° И^о—с—R-с—С1
Н3С—С—+ Cl—С—R—С—Cl > Н3С—cf /СН3
ХСН3
\сн3
Наряду с гаммой всевозможных побочных превращений орто-
комплекс реагирует с аминогруппой с образованием амидной связи:
О О
Н I' 'I
I/O—С—R—С—С1 Н/ /Н
Н3С—С\ /СН3 + )n—R'—Nx ►
Vх н/ \н
ХСН3
О О 0 гм
1! II II Xе 3
--► H2N—R'—NH—С—R—С—С1 4-Н3С—С—N4 . НС1
\сн.
Ряд авторов [46; 47, с. 44; 48; 49] сообщает, что в определенных
условиях наличие в реакционной системе воды не мешает получению
высокомолекулярных полимеров. Эти данные носят противоречивый
характер. Общеизвестно, что гидролиз ангидридных и, особенно хлор-
ангидридных групп, является причиной снижения ММ полиамидокислот
и полиамидов. Тем не менее положительная активность воды отмечена
для реакций пиромеллитового диангидрида с рядом ароматических ди-
аминов [46]. При этом оптимальное количество воды зависит от приро-
ды диамина [46]. Для системы пиромеллитовый диангидрид — 4,4'-ди-
аминодифенилкарбонат оно составляет 0,12 моль на 1 моль пиромелли-
тового диангидрида [46]. При большем количестве воды преобладает
4—2254 49
гидролиз ангидридных групп. Любопытно, что в той же системе эффект
каталитического действия воды проявляется лишь при содержании кар-
бонатных (—ОСО—) групп >40°/о.
Вода играет положительную роль при синтезе полиамидов по
эмульсионному и межфазному способам. Она способствует диффузии
диаминов из водной в органическую фазу (зону реакции), отводу хло-
ристого водорода из этой зоны, повышает поверхностное натяжение на
границе раздела фаз и т. д. Показано [48], что в водноорганических
системах, содержащих хорошо смешивающиеся с водой органические
жидкости, резкое уменьшение содержания воды в органической фазе
эмульсионной системы приводит к снижению молекулярной массы
полиамида. Этот факт приходится связывать с каталитическим действи-
ем воды. Предполагаемый механизм катализа [49] сводится к образо-
ванию комплекса, в котором облегчено протекание заключительной
стадии ацилирования (распад о-комплекса и отщепление галогенводо-
рода):
O=C--N—Н
R Аг
-О—С—N+—Н
НОН
О—Н
Н—ОН--О—Н
Во всех вышеприведенных примерах каталитическое действие тех
или иных соединений связано с усилением ацилирующей способности
производных дикарбоновых кислот. Хуже обстоит дело с ацилируемыми
мономерами. Для диаминов вообще неизвестны вещества, способные
увеличить их активность в реакциях ацилирования.
Особую группу катализаторов составляют каталитические системы,
используемые при поликонденсации ди (тетра) карбоновых кислот с
ди (тетра) аминами, а также в реакциях полигетероциклизации. Описа-
ние этих катализаторов будет приведено ниже.
2.1.2. Роль реакционной среды
в реакциях полиацилирования
В качестве реакционной среды при растворных способах полиаци-
лировання применяют главным образом органические растворители.
Важно, чтобы хорошей растворимостью обладал хотя бы один из
мономеров поликонденсационной системы. При межфазной и эмульси-
онной поликонденсации используют двухфазные системы, каждая из
которых растворяет лишь один из мономеров. В качестве органической
среды при этих способах применяют тетрагидрофуран, диоксан, нитро-
бензол, ацетофенон, циклогексан, циклогексанон, четыреххлористый уг-
лерод, тетрахлорэтан и множество других. Их роль в данном случае
ограничивается переводом в раствор дигалогенангидридов. Эти же ве-
щества хотя и с меньшим успехом могут быть использованы и при низ-
котемпературной растворной поликонденсации. Но гораздо перспектив-
нее использовать при этом способе высокополярные апротонные рас-
творители типа амидных растворителей (ДМАА, ДМФ, МП, гексаме-
тилтриамида фосфорной кислоты, ТММ, ДМСО и ряда других). Эти
соединения растворяют в большинстве случаев как мономеры, так и
полимер.
Прекрасными растворителями для большинства термостойких по-
лимеров являются соединения типа сильных неорганических (концент-
рированная серная кислота и олеум, ПФК, метансульфокислота, рас-
плавленные кислоты Льюиса и др.) или органических (типа сульфоно-
вых) кислот.
Эти соединения будут рассмотрены при описании реакции полиге-
тероциклизации в растворе.
Основные характеристики растворителей, влияющие на скорость
поликонденсации и молекулярную массу, — полярность, растворяющая
способность и способность к взаимодействию с мономерами.
Показана [43] корреляция между ММ полимера и полярностью
растворителя. Неоднократно отмечалось преимущество использования
гексаметилфосфорамида и МП по сравнению с менее полярным ДМАА
при низкотемпературной поликонденсации, особенно с участием менее
реакционноспособных мономеров [50; 51].
Существует прямая связь между полярностью растворителя и его
растворяющей способностью по отношению к полимеру. Тем не менее,
хотя амидные растворители являются высокополярными, их примени-
мость для синтеза ряда термостойких полимеров оказывается ограни-
ченной. Термостойкие полимеры характеризуются сильным межмолеку-
лярным взаимодействием; многие из них имеют жесткую «стержнеоб-
разную» конфигурацию макромолекул или их агрегатов. При растворе-
нии таких полимеров большое значение приобретает энтропийный фак-
тор; кроме того, требуется применение растворителей, обладающих по-
вышенным сродством к полимеру. В этих случаях для повышения рас-
творимости прибегают либо к добавкам неорганических лиофильных
солей типа хлоридов лития, кальция и др., либо к применению смесей
растворителей. Механизм растворяющего действия обеих систем рас-
сматривается в специальном разделе.
Влияние растворяющей способности реакционной среды на ММ по-
лимера обнаруживается при синтезе плохо растворимых полимеров.
В этих случаях продолжительность пребывания полимера в растворе
при его синтезе становится фактором, определяющим его ММ. С выпа-
данием полимера в осадок поликонденсация либо резко замедляется
(если осадок имеет аморфную структуру), либо практически прекра-
щается (при кристаллическом характере осажденного полимера). При-
менение бинарных систем (амидный растворитель — соль) или смесей
органических растворителей позволяет увеличить продолжительность
пребывания полимера в растворе при синтезе и повысить его молеку-
лярную массу [52]. Так, например, благодаря применению систем с по-
вышенной растворяющей способностью (а также катализаторов) до-
стигнуто получение поли-п-фенилентерефталамида с высокой молеку-
лярной массой.
2.1.3. Побочные процессы при реакциях полиацилирования
Поликонденсация прекращается при израсходовании функциональ-
ных групп или замедляется при нарушении контакта между ними.
В идеальном (чисто гипотетическом) случае все молекулы мономеров
должны объединиться в одну гигантскую сверхмолекулу с практиче-
ски бесконечно большой молекулярной массой. На практике этот вари-
ант невозможен вследствие физических и химических ограничений.
4*
51
Вследствие первых рост цепи не прекращается, но столь резко за-
медляется, что практически процесс оказывается приостановленным.
Растворы термостойких полимеров имеют очень высокую динамическую
вязкость, величина которой уже на ранних стадиях синтеза полимеров
даже при умеренной концентрации исходных мономеров может превы-
шать 100 Па-с. В столь вязкой среде контакт функциональных групп
затруднен; скорость поликонденсации резко снижается. В связи с этим
получение концентрированных растворов высокомолекулярных полиме-
ров, особенно необходимых для сухого и сухо-мокрого формования,
представляет собой сложную проблему.
Химические процессы, конкурирующие с основной реакцией, в от-
личие от физических приводят к прекращению роста цепи. Их роль
сводится к дезактивации конечных функциональных групп.
Среди побочных химических реакций наибольшую опасность пред-
ставляет гидролиз ангидридных и, особенно, галогенангидридных групп.
При поликонденсации диаминов с дихлорангидридами по низкотемпе-
ратурному и эмульсионному способам этот вид побочных реакций имеет
превалирующее значение. При межфазном способе роль гидролиза ме-
нее значительна. Чем выше активность ацилирующего агента, тем в
большей степени он подвержен гидролизу. Это не удивительно, посколь-
ку гидролиз можно рассматривать как частный случай реакции ацили-
рования (ацилирование воды). Выше уже отмечалось, что чем сильнее
ацилирующий агент, тем жестче должно быть требование к содержанию
воды в поликонденсационной системе. Для получения высокомолекуляр-
ных ПА на основе диаминов и дихлорангидридов допустимое количест-
во воды составляет 0,05—0,1%. Менее жесткие требования предъявля-
ются при поликонденсации диаминов с дифторангидридами и с диан-
Гидридами.
Весьма распространенной группой побочных реакций производных
дикарбоновых кислот являются их реакции с растворителями, особен-
но, растворителями амидного типа.
Основность амидных растворителей гораздо ниже основности аро-
матических диаминов. Показатель основности рКа диметилацетамида
в воде составляет 0,19, в то время как для большинства ароматических
диаминов эта величина превышает 1,0. Поэтому для большинства амид-
ных растворителей скорости их ацилирования намного ниже скоростей
поликонденсации, что и делает возможным проведение синтеза в этих
растворителях.
Исключение составляют некоторые амидные растворители типа
ДМФ, а-пирролидона, а также ДМСО и некоторые другие.
Побочные реакции дихлорангидридов с амидными растворителями
протекают через стадию образования комплекса, который в дальней-
шем претерпевает изменения, превращаясь в соединения типа амиди-
нов, производных кетонов и т. д.
Интенсивность побочных реакций производных дикарбоновых кис-
лот с растворителем зависит от природы растворителя и химической
структуры ацилирующего агента (табл. 2.3.).
Из таблицы видно, что наибольшую активность по отношению к
хлорангидридам имеют ДМФ, ДМСО и а-пирролидон. Теплоты поли-
конденсации изофталоилхлорида с МФДА (см. табл. 2.2) и его взаи-
модействия с ДМФ близки, соответственно 251 и 222 кДж/моль; ско-
рости обоих процессов соизмеримы. Это делает практически невозмож-
ным синтез полиамидов (или их производных) с участием дихлорангид-
52
Таблица 2.3. Теплоты взаимодействия и энергии активации реакции
хлорангидридов с растворителями [45, 53]
Растворитель Хлорангидрид Теплота реакции, кДж /моль Энергия активации, кДж/моль
Диметнлформамид Бензонлхлорид — 7,3
Изофталоилхлорид 222 ——
Диметилацетамид Беизоилхлорид — 23,2
Изофталоилхлорид 57,2 ——
Беизоилхлорид — 83,6
Диметилпропионамид — 83,6
Диметилсульфоксид Изофталоилхлорид 226 —
АГ-Метилпирролидон » 44 —
Тетраметилмочевнна » 18 —
ц-Пирролидон 176 —
ридов в ДМФ, ДМСО и а-пирролидоне. Наиболее благоприятными для
синтеза растворителями (с точки зрения малой интенсивности побочных
процессов) являются гексаметилфосфорамид, АЛметилпирролидон, тет-
раметилмочевина, тетраметиленсульфон, диметилпропионамид и неко-
торые другие. Отечественной промышленностью пока не освоено произ-
водство этих растворителей. Что касается гексаметилфосфорамида, то
он вряд ли найдет широкое применение в промышленности ввиду его
канцерогенности [54]. В настоящее время наибольшее распространение
в лабораторной и полупромышленной практике получил диметилацета-
мид. Хотя побочные реакции с хлорангидридами протекают в этом рас-
творителе с более низкой скоростью, чем поликонденсация, тем не менее
их приходится учитывать.
Кроме реакций с растворителем хлорангидриды способны взаимо-
действовать с амидной связью с образованием амидоилхлорида.
Эта реакция также приводит к обрыву цепи.
В среде амидных растворителей возможна побочная реакция, свя-
занная с образованием хлорангидрида соответствующей кислоты (му-
равьиной, уксусной и т. д.). Считают [45], что образование, например,
ацетилхлорида происходит в результате реакции ДМАА с двумя моле-
кулами хлористого водорода
? /снз
н3сс—г/ + нс!
ХД13
сн,
HC1 \
СН3СОС1 + HN-HCI
сн3
Наряду с ацетилхлоридом выделяется диметиламин. Оба соединения
обрывают растущую полимерную цепь.
53
Дезактивация аминогрупп может наступить в результате вышеупо-
мянутой реакции с ацетилхлоридом, либо за счет образования соли
ионного типа с хлористым водородом. Хлористый водород, выделяю-
щийся при поликонденсации дихлорангидридов с диаминами, вступает
во 1взаимодейств!ие с аминогруппами, давая продукты двоякого рода:
молекулярное соединение Р—NH2-HC1 (хлоргидрат диамина) и соеди-
нение ионного типа Р—NH3CI- [38; 55; 56]. Литературные сведения по
реакционной способности хлоргидратов носят противоречивый харак-
тер. С одной стороны утверждают [38], что они столь же реакционно-
способны, что и свободные амины; по сведениям других авторов [55],
образование хлоргидратов приводит к снижению скорости поликон-
денсации; при этом меняются оптимальные условия проведения реак-
ции. Практика показывает, что процессы низкотемпературной раствор-
ной поликонденсации носят более длительный характер, чем это долж-
но быть судя по константам ацилирования. Не исключено, что участие в
реакции хлоргидратов диаминов является одной из причин снижения
суммарной скорости процесса.
В отличие от хлоргидратов соединения ионного типа нереакцион-
носпособны; что и делает необходимым удаление хлористого водорода
из сферы реакции, несмотря на необратимый характер взаимодействия
диаминов с дихлорангидридами. Роль акцепторов хлористого водорода
могут выполнять неорганические основания (при межфазном способе),
третичные амины или амидные растворители.
2.2. РЕАКЦИИ ПОЛИГЕТЕРОЦИКЛИЗАЦИИ
1 Процессы полигетероциклизации лежат в основе получения таких
полигетероариленов, как ПИ, ПВО, ПБИ, ПХО, ПОД, ПБОАИ и дру-
гих полимеров полулестничной и лестничной структуры. Пяти- и шести-
членные гетероциклические структуры, образующиеся в цепи полимера,
имеют ароматический характер; они являются электронными аналогами
бензола или конденсированных ароматических систем [57, с. 97, 166,
241]. Поэтому их присутствие способствует повышению термостойкости
волокнообразующих полигетероариленов. При многостадийном способе
синтеза реакциям полигетероциклизации предшествуют процессы обра-
зования промежуточных полимеров, главным образом, полиамидов со
свободными функциональными группами в орто-положении к амидной
связи. При одностадийном способе обе реакции протекают параллель-
но; суммарный процесс бывает трудно расчленить на отдельные стадии.
Иногда под полигетероциклизацией (поликонденсацией) или полицик-
локонденсацией подразумевают совокупность процессов, ведущих к
образованию полимеров с гетероциклами в цепи (если только эти цик-
лы заранее не «предусмотрены» в составе мономеров).
Обычно для синтеза полимеров с гетероциклами в цепи прибегают
к три- или тетрафункциональным соединениям. В случае полиоксадиа-
золов оба мономера являются бифункциональными. Образование окса-
диазольного цикла происходит за счет перестройки внутри гидразидной
связи. Замыкание циклов может быть результатом полимераналогич-
ных превращений. Наконец, циклические структуры могут возникать в
результате реакций хелатирования. Например, обработка полигидрази-
Дов Солями Двухвалентных металлов приводит к образованию хелатных
полимеров с циклическими структурами типа:
54
N—N N—NH
/ X \
—C C— —C C—
I | И I II
о о о о
Реакции полигетероциклйзации могут сопровождаться отщеплени-
ем низкомолекулярных продуктов (вода, фенолы, спирты, аммиак и др.)
или протекают по изомеризационному механизму. Если побочным низ-
комолекулярным соединением является вода, то подобные реакции на-
зывают поликлодегидратацией. Эти реакции часто встречаются на
практике.
Механизмы реакций полигетероциклизации детально не изучены.
Отмечают [7] избирательный характер этих процессов. Свободные
функциональные группы реагируют предпочтительно со «своей», а не
с «чужой» связью. Селективному протеканию процесса способствует
предварительное образование циклического переходного состояния.
Например, в полиамидокислотах подобный комплекс возникает за счет
смещения электронной плотности от амидной связи через бензольное
кольцо к карбоксильной группе [7]:
Величина этого смещения может зависеть от природы заместителей.
Например, (СНз)2-группы в орто-положении к аминогруппам значитель-
но усиливают смещение электронного облака и меняют энергетику про-
цесса. Если энергия активации имидизации полиамидокислоты на осно-
ве пиромеллитового диангидрида и бензидина составляет 105 кДж/моль,
то замена последнего на З.З'-диамино-М.М.М'.Ы'-тетраметилбензидин
приводит к снижению энергии активации до 40 кДж/моль.
Вместе с тем не исключена возможность и межмолекулярного про-
текания реакций свободных функциональных групп. Селективное про-
текание реакции затруднено при поликонденсации тетрафункциональ-
ных соединений с близкой реакционной способностью функциональных
групп. При этом процессы разветвления и сшивки развиваются уже на
стадии полиацилирования. Выше были указаны принципиальные пути
предотвращения гелеобразования при синтезе полиаминоамидокислот.
Реакции разветвления описаны также для завершающей стадии синте-
за полихиноксалинов [58]:
о:
Число разветвлений на одну макромолекулу линейно возрастает с по-
вышением молекулярной массы. Реакции разветвления возможны так-
же при имидизации, циклизации полиоксиамидов, полиаминоимидов
и т. д.
Дать общую схему механизма процессов полигетероциклизации
трудно. Каждый конкретный случай требует отдельного рассмотрения.
Полагают [59], например, что циклизация полигидразидов проте-
кает через стадию образования имидольной формы гидразидной группы:
Н Н
—R—С—N—N—С—Rj— —R-C=N—N=C—Rj
II II II
О О он он
N---N
II II
—R-С С—Ri-
\)/
Не исключено, что амидоимидольная таутомерия играет роль и в
других реакциях полигетероциклизации с участием амидных групп.
Имеются сведения о некоторых деталях механизма имидизации. Счи-
тают [6, с. 1], что в процессе имидизации принимают участие как
«обычная», так и солевая формы поли амидокислот:
.СО—NH—R'
4 С—О- H3N—R"—
II
О
В первом случае имидизация протекает через стадию образования про-
О
II
межуточного соединения типа —R —> хотя доказательств.
но' \)Н
существования этих структур не приводится. Солевые формы полиами-
докислот также способны к имидизации. Циклизация в этом случае
протекает по следующей схеме:
/СО—NH—R'—
ХС—О" H3N—R"—
II
С
-н2о _ /CO-NH-R'-
\со—NH—R"—
СО
При поликонденсации мономеров с нитрильными группами полигетеро-
циклизация протекает по изомеризационному механизму — без отщеп-
ления низкомолекулярного продукта. Так, при циклизации полициан-
амидов имеет место нуклеофильная атака атома углерода CN-группы
неподеленной парой электронов атома азота с последующим переходом
протона к нитрильному атому азота [60; 61]:
56
Предложен и другой механизм (непосредственная атака атома азо-
та протоном), однако, по предварительным данным А. Н. Праведнико-
ва [61], первый механизм является предпочтительным.
Некаталитическая полигетероциклизация требует, как правило,,
жестких условий (высокие температуры, глубокий вакуум, большая
продолжительность). Применение катализаторов позволяет проводить
процесс в гораздо более мягких условиях. В реакциях полициклодегид-
ратации используют различные дегидратирующие агенты типа ангидри-
дов органических кислот, комплекса ДМФ-серный ангидрид, серной
кислоты, олеума, ПФК, расплавленных кислот Льюиса, сульфоновых
кислот и др. При этом сильные дегидратирующие агенты (Нг5О4,
олеум, ПФК, кислоты Льюиса) могут одновременно служить раствори-
телями промежуточного и конечного полимеров.
Общей особенностью процессов полигетероциклизации является
зависимость кинетики и энергетических параметров от жесткости поли-
мерной цепи [7]. Она проявляется тем отчетливее, чем более высокую
жесткость имеют макромолекулы конечного циклизованного полимера.
При полициклизации ряда полимеров зафиксировано наличие по край-
ней мере двух стадий с различными величинами энергии активации.
При этом завершающие стадии характеризуются более высокими зна-
чениями этого параметра. Так, например, энергия активации имидиза-
ции полиамидокислоты на основе пиромеллитового диангидрида и
4,4'-диаминодифенилового эфира возрастает по мере протекания реак-
ции от 95 до 125 кДж/моль [6, с. 45]. Одновременно возрастает пред-
экспоненциальный множитель. Непосредственным результатом описан-
ной выше зависимости кинетики процесса от гибкости молекулярной
цепи является трудность достижения полной завершенности реакции
полициклизации. В большинстве случаев степень циклизации не пре-
вышает 95—98%. Практически это приводит к тому, что конечные по-
лимеры имеют дефектную химическую структуру, являясь, по выраже-
нию В. В. Коршака «разнозвенными», что неблагоприятно отражается
на их термических характеристиках.
В зависимости от условий реакции и реакционной среды различают
твердофазную полициклизацию и полициклизацию в растворе. Первую
проводят на волокне; во втором случае используют растворители, об-
ладающие дегидратирующей активностью. Каждый из способов имеет
свои отличительные особенности.
Особенностью твердофазного способа является зависимость скоро-
сти и степени циклизации от фазового состояния полимера, что нагляд-
но показано на примере циклизации полигидразидных волокон [7].
Процесс чаще проводят в присутствии катализаторов (органические
57
кислоты, их ангидриды, третичные амины и т. д.). Некаталитическую
полициклизацию осуществляют в области температур 100—450 °C в со-
четании с глубоким вакуумом или атмосферой инертного газа. Приме-
нение вакуума или инертной атмосферы связано с опасностью термо-
окислительной деструкции и необходимостью удаления выделяющихся
низкомолекулярных продуктов, могущих вызвать деструкцию полимера
или чисто механическое разрушение физической структуры волокна.
«Термическая» циклизация характеризуется сравнительно высокими
значениями энергии активации (обычно выше 85 кДж/моль). Процесс
протекает с невысокой скоростью, носит продолжительный характер и
часто приводит к снижению физико-механических показателей готового
волокна. Для предотвращения этого часто прибегают к программиро-
ванному повышению температуры при проведении процесса.
Использование катализаторов (обычно в сочетании с дегидрати-
рующими агентами) позволяет снизить энергию активации и повысить
скорость полициклизации. Для полигидразидов, например, отмечено
15-кратное увеличение константы скорости циклизации в присутствии
катализаторов. Имидизацию проводят при комнатной температуре за
гораздо более короткий срок, чем при термической циклизации. Мягкие
условия каталитической (химической) циклизации сводят к минимуму
роль деструктивных процессов и в итоге обеспечивают получение воло-
кон с более высокими физико-механическими характеристиками.
Полициклизация в растворе лишена многих недостатков, присущих
твердофазным способам. Из общих соображений для гомогенной поли-
циклизации можно ожидать повышения скоростей и степени завершен-
ности реакции; отпадает необходимость учета фазового состояния поли-
мера; нет опасности разрушения волокна и т. д. Препятствием к при-
менению этого способа служит низкая растворимость полигетероариле-
нов с циклами в цепи. Для повышения растворимости полимеров с ге-
тероциклами можно прибегнуть либо к их химической модификации,
либо к подбору сильнодействующих растворителей. Первый путь пока
представляется менее перспективным из-за значительного снижения
термических характеристик полимера. Практическое применение полу-
чила полициклизация в серной кислоте, олеуме и полифосфорной кис-
лоте. Физическая характеристика этих растворителей приведена выше.
В последние годы в лабораторной практике стали применять кислоты
Льюиса и растворители сульфонового типа. Растворяющая способность
серной и полифосфорной кислот связана с протонированием гетероато-
мов и ароматических ядер; кислот Льюиса — с возникновением коорди-
национных связей между этими кислотами и гетероатомами и аромати-
ческими ядрами полимера [62]. Наряду с высоким растворяющим дей-
ствием эти соединения являются сильными дегидратирующими агента-
ми, что собственно и определяет их применимость в качестве реакцион-
ной среды для полициклодегидратации. Помимо этого ПФК обладает
и каталитическим действием [63]. Считают, что она образует соль с
диаминами, способствует повышению реакционной способности электро-
фильного углеродного атома карбонильной группы. Показано присутст-
вие фосфора в цепи полимера. Комплекс ПФК с амином находится в
равновесии со свободным амином
R+NH3O"— "О О - II II —Р—О—Р— —ОН R—NH2+HO— ~ О О “ II II _р—о—Р— —он
1 1 он он_ п 1 1 он он_ п
58
С ростом температуры равновесие сдвигается в сторону увеличения
концентрации свободных аминогрупп, что приводит к повышению моле-
кулярной массы полимера. Большую роль играет состав ПФК- Отме-
чают [64], что повышение содержания Р2О5 в ПФК до 85% способст-
вует увеличению ММ полимеров за счет усиления ее дегидратирующего
действия. Дальнейшее повышение концентрации Р2О5 может неблаго-
приятно отразиться на ММ из-за резкого нарастания вязкости самой
ПФК. Достаточно сказать, что при увеличении концентрации ортофос-
форной кислоты от 105 до 117% вязкость повышается в 50 раз [65].
Наибольшее практическое применение ПФК нашла при синтезе лест-
ничных полимеров и, особенно, полибензимидазобензофенантролинов.
Концентрированная серная кислота является уникальным раство-
рителем для полимеров и эффективным дегидратирующим средством.
В качестве реакционной среды реакций полигетероциклизации она ус-
пешно используется при одностадийном способе синтеза волокнообра-
зующих полн-1,3-4-оксадиазолов [66]. Чаще используют не саму серную
кислоту, а олеум. Поглощая воду, серная кислота постепенно снижает
свою активность как дегидратирующий агент; кроме того, с понижением
концентрации кислоты в водных растворах повышается ее способность
вызывать деструкцию полимера и возрастает коррозионная активность.
Описан синтез лестничных полимеров на основе тетракарбоновых
кислот и тетраминов в расплавленных хлоридах сурьмы, цинка, олова,
алюминия, висмута и их эвтектических смесях [62]. Процесс проводят
при 170—250 °C. Получают полимеры с тце до 3,5. Этот способ, однако,
не вышел за рамки лабораторных исследований.
По литературным данным, существуют различные точки зрения на
равновесный или неравновесный характер полициклизации. Возможно,
характер протекания процесса зависит от способа его проведения или
других факторов. Обратимый процесс зафиксирован, например, при вы-
сокотемпературной имидизации полиамидокислот, если не обеспечено
эффективное удаление воды из сферы реакции [6, с. 46]. Этот факт,
однако, еще не может служить аргументом в пользу обратимости поли-
циклизации, поскольку противоположный процесс осложняется рядом
дополнительных реакций, влияющих на его кинетику. Но так или иначе
на практике приходится сталкиваться с процессами «расциклизации».
Это явление наблюдается, в частности, при контакте растворов полиок-
садиазолов с большими массами воды, например, при их переосажде-
нии или формовании из растворов в концентрированной серной кислоте.
ЛИТЕРАТУРА
1. Морган П. У. Поликонденсационные процессы синтеза полимеров. Пер. с англ.
Под ред. Я- С. Выгодского, Б. Р. Лившица. М.—Л., «Химия», 1970. 448 с.
2. Соколов Л. Б. Поликонденсационный метод синтеза полимеров. М., «Химия»,
1966. 336 с.
3. Кормак В. В., Виноградова С. В. Равновесная поликонденсация. М., «Наука»,
1968. 444 с.
4. Кормак В. В., Виноградова С. В. Неравновесная поликонденсация. М., «Наука»,
1972. 696 с.
5. Соколов Л. Б. Докт. дис. М., НИФХИ им. Л. Я- Карпова, 1965.
6. Адрова Н. А. и др. Полиимиды — новый класс термостойких полимеров. Л.,
«Наука», 1968. 211 с.
7. Праведников А. Н. и др., Высокомол. соед., 1973, сер. А, т. 15, № 2, с. 349—353.
5 9
8. Савинов В. М. Канд. дне. М., МХТИ, им. Д. И. Менделеева, 1967.
9. Коршак В. В. Высокомол. соед., 1976, сер. А, т. 18, № 7, с. 1443—1459.
10. Химия и технология синтетических высокомолекулярных соединений. Вып. I. Под
ред. В. В. Коршака. хМ„ изд-во АН СССР, 1959, с. 593.
П. Краткая химическая энциклопедия. Под ред. И. Л. Киуияица. Т. 1. М., «Совет-
ская энциклопедия», 1961, с. 195—198.
12. Заявка США 281971 (1975).
13. Федотова О. Я. и др., Высокомол. соед., 1960, т. 2, № 6, с. 899—903.
14. Пат. США 3549594 (1971); Davans F., Marvel С. S., J. Polymer Sci., 1965, A, v. 3,
№ 10, р. 3549—3572.
15. Пат. США 3639358 (1972); 3870685 (1975).
16. Кардаш И. Е. и др., ДАН СССР, 1966, т. 169, № 4, с. 876—879.
17. Кальниньш К. К. и др., ДАН СССР, 1971, т. 200, № 4, с. 850—852.
18. Кальниньш К. К. и др., ДАН СССР, 1972, т. 204, № 4, с. 876—879.
19. Ардашников А. Я. и др., ДАН СССР, 1965, т. 164, № 6, с. 1293—1295.
20. Кардаш И. Е., Праведников А. Н., ЖФХО, им. Д. И. Менделеева, 1975, т. 20,.
№ 1, с. 14—19.
21. Литвиненко Л. М. и др. Изв. АН СССР, ОХН, 1962, № 10, с. 1737—1744.
22. Бендер М. Механизмы катализа нуклеофильных реакций производных карбоно-
вых кислот. Пер. с аигл. Под ред. В. К. Антонова. М., Издатинлит, 1964. 192 с.
23. Пыхтина Е. В., Кардаш И. Е., Праведников А. Н., ДАН СССР, 1974, т. 215»
№ 2, с. 380—383.
24. Соколова Д. Ф., Соколов Л. Б., Высокомол. соед., 1974, сер. А, т. 16, № 6, с. 1369.
25. Курицин Л. В. и др., Высокомол. соед., 1974, сер. Б, т. 16, № 7, с. 532—535.
26. Стилл Д., Кэмпбелл Т. Мономеры для поликонденсации. Пер. с англ. Под ред..
В. В. Коршака. М., «Мир», 1976. 632 с.
27. Коршак В. В. и др., ДАН СССР, 1968, т. 181, № 4, с. 885—887.
28. Vanghun G. В., Rose F., Brown G. R., J. Polymer Sci., 1971, A-l, v. 9, № 4, p. 1117—•
1138; «Термостойкие пластики», 1972, № 24, с. 5—16.
29. Ардашников А. Я-, Кардаш И. Е., Праведников А. И., Высокомол. соед., 1971»
сер. А, т. 13, № 8, с. 1863—1869.
30. Курицин Л. В., Соколов Л. Б., Высокомол. соед., 1972, сер. А, т. 14, № 10, с. 2028—
2032.
31. Соколов Л. Б., Никонов В. 3., Федотова М. И., Высокомол. соед., 1971, сер. Б,,
т. 13, № 6, с. 459—462.
32. loda N., High Polymer Japan, 1966, v. 15, № 2, p.' 166—172.
33. Hodgkin F., J. Polymer Sci., Polymer Chem. Ed., 1976, v. 14, № 2, p. 409—431;
«Термостойкие пластики», 1976, № 26, с. 1—17.
34. Коршак В. В., Русанов А. Л., Кацарава Р. Д., Высокомол. соед., 1969, сер. А,
т. 11, № 9, с. 2090—2101.
35. Коршак В. В. и др., Высокомол. соед., 1973, сер. А, т. 15, № 5, с. 969—976.
36. Пат. США 3640960 (1972).
37. Наумов В. С., Соколов Л. Б., Горелик Л. М., Высокомол. соед., 1971, сер. А,
т. 13, с. 1522—1530.
38. Литвиненко Л. М., Александрова Д. М., Укр. хим. жури., 1960, т. 26, № 5, с. 621.
39. Литвиненко Л. М., Александрова Д. М. Укр. хим. журн., 1961, т. 27, № 4, с. 487.
40. Литвиненко Л. М., Семенюк Г. В., Олейник Н. М., ДАН СССР, 1967, т. 176, № 5,
с. 1107—1110.
41. Литвиненко Л. М., Кириченко А. И., ДАН СССР, 1967, т. 176, № 1, с. 97—100.
42. Литвиненко Л. М., Кириченко А. И., Рудаков Е. С., «Кинетика и катализ», 1962,
т. 3, с. 651—660.
43. Медведь С. С., Соколов Л. Б., Соколова Д. Ф., Высокомол. соед., 1972, сер. А,
т. 14, № 6, с. 1313—1318.
44. Семенова А. С., Васильева-Соколова Е. А., Хим. волокна, 1969, № 5, с. 2—4.
45. Herlinger Н. е. a., Appl. Polymer Symp., 1973, № 21, р. 201—209.
46. Колесников Г. С. и др., Высокомол. соед., 1970, сер. А, т. 12, № 9, с. 1968—1973.
47, Соколов Л. Б. и др., Термостойкие ароматические полиамиды. М., «Химия», 1975.
253 с.
48. Соколов Л. Б., Кудим Т. В., Высокомол. соед., 1965, т. 7, № 11, с. 1899—1904.
49. Соколов Л. Б. и др., Высокомол. соед., 1971, сер. Б, т. 13, с. 359—362.
50. Одноралова В. Н. и др., Хим. волокна, 1973, № 1, с. 48—50.
51. Одноралова В. Н. и др., Хим. волокна, 1973, № 5, с. 9—12.
52. Федоров А. А. и др., Высокомол. соед., 1973, сер. Б, т. 15, № 1, с. 74—77.
53. Швецова Л. Н. и др., ЖВХО им. Д. И. Менделеева, 1972, т. 17, № 4, с. 476.
54. Chem. Eng. News, 1975, v. 53, № 39, p. 17.
60
55. Савинов В. М„ Соколов Л. Б., Высокомол. соед., 1973, сер. Б, т. 15, № 4,
56 Соколов3 Л. Б., Загуменнова Т И.., Серова Л. Я., Высокомол. соед., 1972, сер. Б,
т. 14, Xs 5, с. 386—389. „
57. Пакетт Л. Основы современной химии гетероциклических соединений. Пер. с
англ. Под ред. В. Г. Яшунского. М., «Мир», 1971. 352 с.
58. Hagnauer G. L., Mulligan G. D., «Macromolecules», 1973, v. 6, Ns 4, p. 477—482;
«Термостойкие пластики», 1974, № 5, с. 24—29.
59. Кронгауз Е. С. и др., Высокомол. соед., 1967, сер. А, т. 9, Ns 6, с. 1369—1372.
60. Телешов Э. Н. и др. Сборник рефератов докладов XI Менделеевского съезда по
общей и прикладной химии. М., «Наука», 1974, с. 148—149.
61 Васильева И. В. и др., ДАН СССР, 1971, т. 201, № 4, с. 850—853.
62 Lowell S., «Nuova chim.», 1974, v. 50, Ns 7, p. 63—66; «Термостойкие пластики»,
1974, № 48, с. 15—16.
63. Колесников Г. С. и др., Высокомол. соед., 1969, сер. А, т. 11, Ns 7, с. 1465—1471;
РЖХим, 1963, реф. 187124.
64. Могнонов В. М., Изынеев А. А., Дорошенко Ю. Е. Изв. Сиб. отд. АН СССР.
Сер. хим. наук, 1974, вып. 3, № 7, с. 100—105.
65. Корецкая А И., Савинова В. М. Способы получения полимеров лестничной и по-
лулестничной структуры. Ч. I. М., НИИТЭХИМ, 1974. 45 с.
66. Раскина А. Б., Волохина А. В., Окромчедлидзе Н. П. В кн.: Получение и свой-
ства полиоксадиазольных волокон. М., НИИТЭХИМ, 1974, с. 12—20.
ГЛАВА 3
ОСНОВНЫЕ ПРИНЦИПЫ ФОРМОВАНИЯ
термостойких волокон
3.1. ВВЕДЕНИЕ
Термостойкие волокна получают из полимеров, температура плав-
ления которых лежит в области их термического распада. Поэтому ме-
тоды формования из расплава или размягченного состояния для них
неприемлемы. Практически все термостойкие волокна формуют из рас-
творов. Принципиально не исключена возможность получения волокон
по межфазному способу непосредственно в процессе образования поли-
мера из мономеров [1]. Однако получить волокна с удовлетворитель-
ными механическими свойствами этим способом пока не удается. Во-
локно в процессе образования имеет очень рыхлую структуру, непод-
дающуюся уплотнению известными технологическими приемами (вы-
тяжкой, термообработкой и т. д.). Перспективными могут быть способы
получения волокон из дисперсий и из набухшего состояния под сверх-
высоким давлением. Первый из них разработан пока применительно к
получению политетрафторэтилена, а второй еще находится в стадии ла-
бораторных исследований.
В производственных и экспериментальных условиях волокна форму-
ют из растворов по мокрому, сухому и сухо-мокрому способам. В лите-
ратуре также описаны только эти три способа. Наибольшее распростра-
нение получил мокрый способ формования, хотя технико-экономические
показатели его из-за низких скоростей формования значительно уступа-
ют двум другим способам. Единственной положительной характеристи-
кой мокрого способа является возможность широкого регулирования
структуры формуемого волокна и, следовательно, большая возможность
получения заданных физико-механических свойств в готовом волокне.
61
Прежде чем рассматривать конкретные условия формования тер-
мостойких волокон, следует напомнить, что практически все они полу-
чаются из жесткоцеппых полимеров, жесткость которых, однако, сильно
различается. Эти различия сказываются не только в процессе при-
готовления прядильных растворов, но и при формовании воло-
кон особенно из «предельно» жесткоцепных полимеров. Поэтому в
литературе делаются попытки классифицировать термостойкие поли-
меры и волокна по степени жесткости молекулярной цепи. Отнесе-
ние полимеров к жестко- и гибкоцепным может основываться на осо-
бенностях термодинамики их разбавленных и концентрированных рас-
творов. Количественной мерой равновесной жесткости макромолекул
полимера является величина статистического сегмента Куна А или пер-
систентная длина цепи. Как правило, величина статистического сегмен-
та Куна равна удвоенной персистентной длине цепи [2]. Иногда для
оценки жесткости макромолекул используют значение показателя а в
уравнении Марка — Хувинка [rj] =kMa.
Однако последняя зависимость эмпирическая, поскольку а зависит
от молекулярной массы полимера и термодинамической характеристики
растворителя. С. Я. Френкель оценивает гибкость макроцепей условно
с помощью параметра Ди Марцио [3, с. 94]:
г______ЯГ.С
«ж.с + «г.с
где пг. с и пж. с —соответственно число гибких и жестких связей.
К сожалению, общепризнанной количественной классификации по-
лимеров по жесткости до сих пор нет.
Поэтому мы вынуждены здесь пользоваться чисто условной качест-
венной классификацией полимеров и формуемых из них волокон:
1) гибкоцепные полимеры (ПВХ, ПВС, ХПВХ, ПАК и др.);
2) полужесткие полимеры, имеющие структуру ароматических ко-
лец или гетероциклов разреженных гибкими шарнирными связями типа
—S—, —О—, —SO2—, —СО—, а также содержащие метаизомерные
звенья;
3) предельно жесткоцепные полимеры, содержащие в структуре
ароматические или полигетероариленовые связи с высокой степенью
делокализации электронов.
Для гибкоцепных полимеров сегмент Куна А не превышает 2,0 нм.
Для полужестких полимеров он может быть равен нескольким десяткам
нанометров. Так, например, для поли-льфениленизофталамида А =50 А
[4]. Предельно жесткоцепные полимеры имеют А выше 1000 А. Так, по
данным Цветкова [5], для поли-п-бензамида А=2100 А, что соответст-
вует 320 мономерным звеньям в цепи. По другим данным [6, с. 28],
персистентная длина поли-п-бензамида и поли-п-терефталамида равны
соответственно ~ 400 и 200 А.
По значениям эмпирической константы а в уравнении Марка —
Хоувинка полимеры первой и второй группы очень близки между собой,
тогда как для жесткоцепных полимеров а>1,5 (табл. 3.1).
Для полимеров второй группы, так же как и для гибкоцепных по-
лимеров, в разбавленных растворах характерна конформация стати-
стического клубка [11], в то время как для третьей группы присуща
стержневая, вытянутая форма макроцепи. Принципиальные различия
указанных трех классов полимеров проявляются при сравнении вязко-
62
Таблица 3.1. Температуры стеклования и константа а для некоторых полимеров
Полимер Растворитель Темпе- ратура, °C а Темпера- тура стекло- вания, °C Литература
пвс Н2О 25 0,62—0,67 85 7]
пвх Тетрагидрофуран, 25 0,85 75—82 8]
ПАН циклогексанон Дихлорэтан ДМФ 25 20 0,67 0,76 75—82 80 00 00
Поли-ж-фенилени- ДМСО ДМФ 20 25 0,75 0,74 250 [8] [91
зофталамид ДМФ-j-LiCl (0,25 г/л) 25 0,81 250 9]
Полиамидокислота ДМФ+LiCl (1,18моль/л) ДМФ 25 20 1,28 0,8 250 345 9] 10]
(ПМ) Поли-п-бензамид H2SO, 20 1,6 300 [10]
стных свойств их концентрированных растворов. Для примера можно
привести зависимость вязкости от концентрации для полимеров первой
и второй групп (рис. 3.1) и жесткоцепного поли-л-фенилентерефталами-
,<они,ентраи,ия, % (масс.}
Рис. 3.1. Зависимость вязкости от концентрации полимера в растворе:
1 — поливиниловый спирт в воде; 2 — полиакрилонитрил в диметилформамиде; 3 —
полисульфонамид в диметилацетамиде.
Рис. 3.2. Зависимость вязкости анизотропных растворов ПБА от концентра-
ции полимера в растворе.
да (рис. 3.2). Для предельно жесткоцепных полимеров эта зависимость
носит аномальный характер: до определенной концентрации полимера
вязкость, как обычно, растет, а затем при дальнейшем увеличении, на-
чиная с СКР, происходит ее резкое снижение. Причины такой аномалии
будут рассмотрены ниже.
Резко отличны показатели растворимости для указанных групп
полимеров. Если для гибкоцепных полимеров может быть найден ши-
рокий ассортимент растворителей, то для второй группы круг раство-
63
рителей ограничен так называемыми апротонными растворителями-—
ДМФ, ДМАА, N-метилпирролидоном, тетраметилмочевиной и некото-
рыми другими редкими растворителями типа фторуксусной кислоты.
Полимеры третьей группы, как правило, растворимы только в силь-
ных минеральных кислотах (концентрированная H2SO4, Н3РО4 и др.)
или амидных растворителях с добавками LiCl, СаС1г и др.
Имеется ряд других отличий между тремя группами полимеров,
которые будут изложены в процессе рассмотрения мокрого способа
формования волокон.
3.2. ПОЛУЧЕНИЕ ВОЛОКОН ПО МОКРОМУ МЕТОДУ
Технология формования термостойких волокон по мокрому способу
аналогична технологии получения многотоннажных синтетических во-
локон (нитрон, ПВХ, ПВС и др.) и осуществляется на аналогичном
оборудовании. Однако большая жесткость макроцепей термостойких
полимеров обусловливает некоторые особенности технологических ус-
ловий формования волокон. Прежде всего это относится к третьей
группе — предельно жестким полимерам.
3.2.1. Растворимость термостойких полимеров
Как и для обычных синтетических волокон, технологический про-
цесс производства начинается с получения концентрированных прядиль-
ных растворов и, в частности, с растворения полимеров.
Самопроизвольное растворение, протекающее с понижением энер-
гии Гиббса (изобарно-изотермического потенциала), возможно при
резком возрастании энтропии или при значительном изменении энталь-
пии системы за счет энергии взаимодействия между полимером и рас-
творителем. Из-за жесткости цепей число конформаций макромолекул
мало изменяется при растворении. Поэтому определяющим фактором
при растворении может быть только энтальпия системы. Очевидно,
перевод в раствор жесткоцепных полимеров возможен при условии
реализации энергии взаимодействия между полярными группами
растворяемого полимера и молекулами растворителя. Большинство
полимеров второй группы хорошо растворяются в апротонных раство-
рителях, высокая полярность которых способствует увеличению взаи-
модействия полимер — растворитель [1]. Для полиамидов и аналогич-
ных им полимеров (полиамидокислот, полиоксамидов) этот процесс
может быть схематически изображен следующим образом:
О
II I
---R—С—N—R----- О
I II
н С—сн,
: + I
о N—СН3
II I сн
---R—С—N—R-----
Н
<64
о
I! I
----R—C—N—R-------
। °
H II I
; + — R—C—N—R-------
б X
I! H
H3C-C-N(CH3)3
В ряде случаев стабильность растворов полимеров (особенно по-
лиамидов) оказывается недостаточной, и растворы желатенизируют.
Тогда в раствор добавляются лиофильные соли типа хлорида лития,
кальция и др. Предполагается [13], что при растворении указанных со-
лей в амидных растворителях происходит диссоциация соли и сольва-
тация катиона растворителя через кислород, на котором локализована
большая электронная плотность:
СН3—N----С=О- -Li+
I I
сн3 сн3
Анионы хлора при этом взаимодействуют с водородом амидной группы
полимера с образованием устойчивого комплекса.
Большое значение для растворимости имеет исходное фазовое со-
стояние полимера. Если полимер находится в кристаллическом состоя-
нии, то для его перевода в раствор
необходимо затратить энергию, превы-
шающую энергию кристаллической
решетки. Для жесткоцепных полиме-
ров, особенно относящихся к третьей
группе, в этом случае необходимо при-
менять очень сильные растворители.
Такими растворителями могут быть,
по-видимому, только концентрирован-
ные кислоты. Даже в таком универ-
сальном растворителе, как концентри-
рованная серная кислота, растворение
наблюдается в узком интервале кон-
центрации кислоты [14]. На рис. 3.3
Рис. 3.3. Зависимость растворимости
ИФТА от концентрации серной -кис-
лоты:
[Удельные вязкости ПФТА: 20 (/, 4, 5)',
8 (2, 6); 4 (3, 7); температуры; 40 °C (/—-3);
80 °C (4); 90 °C (5—7)1.
представлена зависимость раствори-
мости (концентрации насыщенного
раствора) поли-/г-фенилентерефтала-
мида от концентрации серной кислоты
для полимеров разной вязкости. Как
следует из приведенных данных, мак-
симальная растворяющая способность серной кислоты лежит в преде-
лах 99—100%. В амидных апротонных растворителях такие полимеры
не растворяются. Например, поли-«-бензамид растворяется в ДМАА
только в тех случаях, когда полимер не закристаллизован или находит-
ся в несовершенной паракристаллической форме.
В связи с этим приводится ряд практических рекомендаций [15,
с. 76] по получению стабильных прядильных растворов жесткоцепных
полимеров:
5—2254
65
1) исходный полимер должен иметь аморфную или малоупорядо-
ченную структуру;
2) растворение проводят при постепенном добавлении полимера ко
всему объему растворителя;
3) растворитель должен быть охлажден;
4) необходимо использовать полимер в мелкодисперсном состоя-
нии.
Следует отметить, что набухший полимер, как, например, в случае
поли-тг-фениленизофталамида, может, не растворяясь, перейти в сту-
день, так как в набухших полимерах хорошо проходят процессы кри-
сталлизации.
3.2.2. Особенности свойств растворов
термостойких полимеров
Вследствие довольно широкого спектра жесткости макромолекул
термостойких полимеров различия свойств их растворов проявляются
уже при очень малых концентрациях. Как известно, для оценки моле-
кулярной массы полимера используют зависимость приведенной вяз-
кости от концентрации. Для обычных гибкоцепных полимеров (за ред-
ким исключением) эта зависимость прямолинейна вплоть до концент-
рации ниже единицы. Для термостойких полимеров аналогичная кар-
тина наблюдается не во всех случаях. На рис. 3.4 в качестве примера
приводится зависимость приведенной вязкости от концентрации для
двух полимеров, относящихся ко второй и третьей группам жесткости.
При близких показателях характеристической вязкости у обоих поли-
меров наблюдается резкое различие в ходе кривых. Если для полужест-
коцепного поли-л-фениленсульфонамида линейная зависимость сохра-
няется, то для предельно жесткоцепного ПБА при концентрации около
Концентрации полимера, г) 100 мл
Рис. 3.4. Зависимость приведенной вязкости от концентрации ПБА в ДМАА
с добавкой 3% LiCl (/) и полисульфонамида ДМАА с добавкой 0,1%
LiCl (2).
Рис. 3.5. Зависимо1сть кинематической вязкости растворов ПФТА от кон-
центрации полимера в серной кислоте.
0,2% наблюдается резкий изгиб, свидетельствующий о взаимодействии
макромолекул между собой [16].
Анализ приведенной зависимости показывает, что макромолеку-
лы поли-л-бензамида в растворе имеют отчетливо выраженную асим-
метрию.
66
При переходе от разбавленных к концентрированным (прядиль-
ным) растворам термостойких полимеров различия между гибкоцепны-
ми и жесткоцепными системами становятся еще более заметными. Если
полимеры первой и второй групп сохраняют свойства обычных изотроп-
ных растворов, то жесткоцепные полимеры образуют в определенных
условиях анизотропные растворы.
Рис. 3.6. Зависимость 'кинематической вязкости растворов ПФТА в серной
кислоте от температуры при различных концентрациях полимера в растворе
H2SO4:
/ — 2,0%; 2-6,0%; 3-8,0%; 4-9,0%; 5-9,5%; 6-10%; 7-10,5%; 3-11,0%;,
9 — 11,6%.
Рис. 3.7. Зависимость динамической вязкости от концентрации растворов
поли-п-бензамида различной молекулярной массы при 20 °C:
7 — 41=9890, [ц]=0,47; 2 —М=17500, [п] = 1,12; 3 — Л! = 22200, [711 = 1,68; 4 — 41 = 29200.
[41=2,96.
Поскольку свойства изотропных растворов полимеров исчерпываю-
ще описаны в многочисленных монографиях, останавливаться на них
вряд ли целесообразно. Свойства анизотропных растворов полимеров
рассматриваются пока лишь в нескольких работах [17; 18], поэтому не-
которые принципиальные особенности их следует подчеркнуть.
Наиболее характерной особенностью анизотропных растворов яв-
ляется аномальная зависимость вязкости от концентрации и температу-
ры. На рис. 3.5 показано аномальное поведение растворов ПБА и ПФТА
от концентрации их в растворе. До определенной концентрации наблю-
дается обычный рост вязкости. При концентрации, которую называют
критической, кинематическая вязкость резко падает, а затем снова воз-
растает. При концентрации, отвечающей критической Сх и максимуму
вязкости, происходит переход от прозрачной к мутной опалесцирующей
жидкости, двулучепреломляющей в недеформированном состоянии. Это
объясняется самопроизвольным упорядочением системы жестких стерж-
5* 67
необразных молекул и, как следствие, образованием в растворе крупных
ориентированных ассоциатов макромолекул.
При С^>СХ происходит переход от обычного течения неупорядочен-
ных структур с необратимым перемещением центров тяжести отдельных
макромолекул друг относительно друга к течению, обусловленному
смещением уже ассоциатов макромолекул. Если в первом случае при-
ходится затрачивать значительную энергию для того, чтобы частично
разрушить сетку зацеплений и освободить для перемещения участок
цепи, превышающий величину кинетического сегмента, а затем сориен-
Рис. 3.8. Зависимость вязкости от
молекулярной массы ,ПБА в экви-
концентрированных растворах:
7-5%; 2 — 6,7%; 3 — 8,2%; 4—13%.
тировать его в направлении потока, то
во втором необходимо только повер-
нуть образовавшиеся ассоциаты в на-
правлении течения. Ветвь снижения
вязкости при С>СХ, по-видимому,
связана с образованием смеси изо-
тропной и анизотропной (низковязкой)
фаз.
Необычный характер растворов
жесткоцепных полимеров проявляется
и при исследовании температурной
зависимости вязкости (рис. 3.6). До
тех пор пока раствор полимера оста-
ется изотропным (концентрация ниже
9%) с ростом температуры наблюда-
ется обычное понижение вязкости. При
переходе к анизотропному состоянию
(кривые 6, 7) вязкость раствора зна-
чительно возрастает с повышением
температуры вплоть до обратного пе-
рехода в изотропный раствор (кривые
8, 9). Последнее связывают с процес-
сом разрушения упорядоченной (жид-
кокристаллической) структуры, что и
приводит к возрастанию вязкости из-за увеличения плотности сетки
флуктуационных зацеплений макромолекул.
Аналогичная зависимость характера и для ПБА (рис. 3.7), нахо-
дящегося в анизотропном растворе. На рисунке приведены зависимости
вязкости от концентрации для растворов ПБА различной молекулярной
массы. Как видно из приведенных данных, характер кривых во всех
случаях аналогичен: после резкого подъема вязкости следует нисходя-
щая ветвь. Величина вязкости в максимуме т]Макс и ее положение на
оси концентраций зависят от длины цепей полимера. Это означает, что
для выбранной концентрации может быть найдено критическое значе-
ние молекулярной массы Мх, отвечающей переходу из изотропного в
анизотропное состояние. Для этого следует построить зависимость т]о
от М (см. рис. 3.8).
Как видно из рисунка, при С~5% растворы всех образцов изо-
тропны.
Интересной особенностью анизотропных растворов является их спо-
собность ориентироваться под действием силовых или магнитных
полей [19].
Анизотропные растворы описаны не для всех жесткоцепных поли-
меров. Помимо иллюстрировавшихся здесь поли-п-бензамида (ПБА) и
68
ПФТА в литературе приводится несколько примеров для сополиамидов
в концентрированных серной, метилсерной, хлорсульфоновой и фтори-
стоводородной кислотах [18]. В амидно-солевых растворах анизотроп-
ные растворы получены только для ПБА и поли-2-хлор-п-фенилентере-
фталамида. Имеются неподтвержденные данные о возможности получе-
ния анизотропных растворов полифенилен-1,3,4-оксадиазолов в кон-
центрированной серной кислоте. Пока не объяснено отсутствие анизо-
тропии растворов для многих известных жесткоцепных полимеров, на-
пример лестничных полимеров, полибензимидазолов и др.
3.2.3. Приготовление прядильных растворов
Технологический процесс приготовления прядильных растворов
включает три основных этапа:
1) получение прядильного раствора полимера;
2) фильтрацию прядильного раствора;
3) обезвоздушивание прядильного раствора.
Прядильные растворы термостойких полимеров в большинстве слу-
чаев получают непосредственно в процессе поликонденсации исходных
мономеров, а поэтому единственной операцией на этой технологической
стадии является доведение концентрации полимера до требуемых зна-
чений, иногда совмещенная с нейтрализацией выделившейся свободной
соляной кислоты (в случае применения в качестве одного из мономе-
ров хлорангидрида дикарбоновой кислоты).
Сочетание в едином процессе синтеза полимера с получением пря-
дильного раствора несомненно является наиболее прогрессивным мето-
дом, позволяющим сократить такие технологические стадии, как выде-
ление полимера из реакционной среды, отмывку полимера от раство-
рителя и других загрязнений, сушку, измельчение, транспортировку
сухого полимера к месту приготовления прядильного раствора и, нако-
нец, такой ответственной операции, как растворение полимера в соот-
ветствующем растворителе. Однако в некоторых случаях такое сочета-
ние не удается реализовать. В основном это относится к тем случаям,
когда необходимо иметь высококонцентрированные прядильные раство-
ры, например, при сухо-мокром методе формования и др. Тогда прихо-
дится создавать всю названную технологическую цепочку. Наиболее от-
ветственной, оказывающей большое влияние на последующий процесс
формования, является стадия растворения полимера. Основные факто-
ры, определяющие условия приготовления прядильных растворов,—про-
должительность, температура, концентрация полимера и аппаратурное
оформление процесса — достаточно полно описаны в литературе [20,
с. 10].
Скорость растворения полимера зависит от скорости диффузии
молекул растворителя в частицы полимера. Как правило, продолжи-
тельность растворения обратно пропорциональна кубу площади гранул
полимера. Поэтому казалось бы, наиболее тонкое измельчение полиме-
ра до пылевидного состояния должно быть целесообразным. Однако
пылевидный полимер трудно замешивается с растворителем и легко
образует комочки набухшей массы, диффузия в которые сильно затруд-
нена. Поэтому степень дисперсности полимера должна иметь оптималь-
ную величину в пределах 0,05—0,3 мм и достаточно узкий фракционный
состав. Высококонцентрированные растворы полимеров, таких, напри-
мер, как ПБА, ПФТА, при низких температурах представляют собой
твердый гель, температура плавления которого тем выше, чем больше
69
молекулярная масса полимера и его концентрация в прядильном рас-
творе. Так, например, температура плавления геля с концентрацией
ПФТА, равной 18—19%, и удельной вязкостью >15 составляет
70—90 °C.
Вместе с тем при температуре выше 80 °C начинается интенсивная
Деструкция полимера. Поэтому важной проблемой при приготовлении
высококонцентрированных прядильных растворов является снижение
температуры плавления геля. Проще всего температуру плавления сни-
зить путем введения в основную цепь макромолекул шарнирных групп
или гибких звеньев [21]. К сожалению, этот путь ограничен, так как
при введении большого числа гибких звеньев понижается термостой-
кость полимера. Содержание таких звеньев не должно быть >10%.
В патенте [22] указывается на возможность снижения температуры и
вязкости геля ПФТА путем добавления в концентрированную H2SO4
небольших количеств других минеральных кислот или некоторых орга-
нических соединений. Облегчить растворимость геля при умеренных
температурах можно путем применения аппаратов «реологического дей-
ствия» [23, с. 543]. Быстрота растворения полимера в этих аппаратах
достигается благодаря большим градиентам скорости и напряжениям
сдвига, создаваемым быстро вращающимися шнеками или растирате-
лями с узкими щелями между вращающимися и неподвижными частя-
ми аппарата.
Важным обстоятельством, которое необходимо учитывать при при-
готовлении высококонцентрированных растворов, является небольшая
разница между температурой плавления геля и температурой интенсив-
ной деструкции полимера. Поэтому при конструировании аппаратов-
растворителей необходимо предусмотреть небольшой перепад темпера-
туры на стенках реактора и в его центре.
Второй важной технологической стадией процесса приготовления
прядильного раствора является фильтрация. Фильтрующий материал
должен обладать следующими свойствами:
1) способностью хорошо задерживать твердые частички;
2) небольшим гидравлическим сопротивлением потоку фильтрата;
3) химической стойкостью;
4) способностью к набуханию при соприкосновении с раствором;
5) обладать достаточной механической прочностью.
Применение в качестве растворителей термостойких полимеров
таких сильно действующих агентов, как амидно-солевые системы или
концентрированные минеральные кислоты при повышенных температу-
рах, резко ограничивает возможность выбора текстильных материалов
в качестве фильтрующих перегородок. Вследствие этого проблема на-
дежной фильтрации растворов термостойких полимеров до сих пор
остается открытой. По-видимому, перспективными материалами могут
быть минеральные пористые перегородки, керамические фильтры, а для
первой фильтрации — фильтры с намывным слоем.
Высокая вязкость прядильных растворов затрудняет процесс их
обезвоздушивания. Простое выстаивание раствора в стандартных емко-
стях при обычном или пониженном давлениях, так же как и центрифу-
гирование, требует большого времени (несколько суток) [18]. Недавнб
был предложен новый способ дегазации высоковязких прядильных рас-
творов, основанный на последовательном повышении избыточного дав-
ления (до 0,3 МПа) и создании вакуума в аппарате обезвоздушивания
70
При этом из раствора удаляются только крупные пузырьки возду-
ха, диаметр которых выше критического. Мелкие же пузырьки, сум-
марный объем которых не достигает пределов насыщения, растворяют-
ся в прядильном растворе при включении давления воздуха или азота.
Если давление во всех трубопроводах, включая фильерный комплект,
не снижается, формование проходит хорошо. Этот способ, по-видимому,
найдет широкое применение в практике производства термостойких во-
локон. Для интенсификации процесса авторы предложили дальнейшее
усовершенствование [24]. Учитывая тот факт, что скорость дегазации
определяется разностью равновесных концентраций газа в жидкости и
над нею, то, если прядильный раствор дополнительно насытить ка-
ким-либо инертным газом, процесс выделения пузырьков газа и воздуха
под вакуумом значительно ускоряется.
Процесс подготовки прядильных растворов к формованию может
быть реализован и без стадии их обезвоздушивания в специальных
эвакуаторах. Для этого необходимо проводить растворение полимера
под вакуумом [30] и в дальнейшем исключить попадание воздуха на
всех переходах вплоть до выхода раствора из фильеры прядильной ма-
шины. До сих пор нет сведений о внедрении этого способа в промыш-
ленности.
В технической литературе приводится очень мало материалов по
аппаратурному и технологическому оформлению процессов подготовки
термостойких полимеров к формованию.
3.2.4. Закономерности формования термостойких
высокопрочных высокомодульных волокон
Рассматривая вопросы формования волокон, необходимо подчерк-
нуть, что основные принципы и закономерности образования нитей яв-
ляются общими как для волокон с обычными механическими свойства-
ми, так и для высокопрочных высокомодульных волокон. Те и другие
волокна получаются из жесткоцепных полимеров, и хотя высокопрочные
высокомодульные волокна пока получены только из предельно жестко-
цепных полимеров, закономерности формования во многих случаях яв-
ляются аналогичными. Некоторые наблюдаемые существенные разли-
чия в большей степени связаны с состоянием прядильного раствора
(анизотропное или изотропное), чем со степенью жесткости полимера.
Как будет показано далее, в принципе можно получить любое термо-
стойкое волокно с высокими физико-механическими характеристиками,
за исключением особых случаев, связанных с невозможностью получе-
ния высокомолекулярного продукта или быстрой его кристаллизуе-
мостью при высаживании.
Общие принципы формования термостойких волокон. Первой ста-
дией получения любого синтетического волокна является осаждение
полимера, т. е. превращение вытекающих из фильеры струек раствора
в гелеобразные нити. Этот процесс фазового превращения в системе по-
лимер — растворитель детально рассмотрен в работах С. П. Папкова
и его сотрудников [25]. В литературе преимущественно описываются
два возможных механизма фазового распада: нуклеационный и спино-
дальный [26].
Независимо от механизма распада конечная морфологическая
картина образующихся студней, по-видимому, аналогична. Структура
фазовых образований зависит от природы полимера и условий его
71
осаждения. В литературе описывается большое разнообразие структур-
но-морфологических картин фазового распада полимерных растворов
[27]. Для гибкоцепных полимеров, как правило, характерны глобу-
лярные образования и свернутые пачки молекул, тогда как при за-
студневании растворов жесткоцепных полимеров в тех же условиях
наблюдаются сплошные сетки фибриллярных образований [27]. При-
чина таких различий будет описана ниже. Здесь только следует под-
черкнуть, что принципиальные отличия между гибкоцепными и жестко-
цепными полимерами на данной стадии образования волокна сводятся
к тому, что в первом случае при образовании полимерного геля макро-
молекулы успевают складываться и образовывать ламелярные структу-
ры; во втором случае они фиксируются в распрямленной форме.
Генезис структуры волокон, формуемых мокрым способом, на-
чавшись в растворе, продолжается на стадии отверждения жидкой нити
под действием осадительной ванны. Состав осадительной ванны до не-
которой степени может направленно изменять структуру свежесформо-
ванного волокна. Некоторые исследователи [28, с. 112] считают, что
характер осадительной ванны является одним из важнейших парамет-
ров, определяющих структуру и свойства волокна. Однако в этом воп-
росе остается еще много неясного. Последние работы по формованию
волокон, особенно из предельно жесткоцепных полимеров, показывают,
что надмолекулярная структура гелеобразной нити и особенно ее мор-
фология не всегда определяют конечные механические свойства ни-
ти. Можно лишь утверждать, что для гибкоцепных волокон кинетика
структурообразования зависит от процессов массо- и теплообмена.
Ряд авторов [29] разделяет осадители на жесткие и мягкие.
Для первых скорость диффузии осадителя внутрь прядильной струйки
достаточно велика и соизмерима со скоростью диффузии растворителя.
Для осадителей второй группы скорость диффузии растворителя из ни-
ти значительно выше скорости диффузии осадителя. В зависимости от
природы осадителя форма поперечного среза и пористость структуры
нити различны. Так, в жестких осадителях высаживание полимера в
формуемой струйке происходит во всем объеме при достаточно высокой
концентрации осадителя внутри струйки, вследствие чего свежесфор-
мованное волокно имеет правильную форму, близкую к кругу, однород-
ный срез и рыхлую пористую структуру. Воздействие жестких осадите-
лей на процесс формования различных волокон являлось предметом
многих исследований [30]. Несмотря на это, единого мнения о меха-
низме структурообразования гель-волокон до сих пор нет. Так, напри-
мер, в работе [31] структура свежевысаженного гель-волокна из поли-
л/-фениленизофталамида (ПФИА) объясняется механизмом капельного
переноса осадителя. В момент контакта осадителя с раствором полиме-
ра на поверхности струйки образуется тонкая поверхностная оболочка.
Предполагается, что при действии сильных (жестких) осадителей такая
оболочка сразу переходит в стеклообразное состояние. Образующаяся
система, внутри жесткой оболочки которой заключен истинный раствор,
не является устойчивой. При действии на нить напряжений («выход-
ные» напряжения, фильерная вытяжка, гидродинамическое сопротивле-
ние ванны и т. д.) оболочка разрывается и раствор вступает в прямой
контакт с осадителем. Образуются направленные к центру волокна
вакуоли.
Следует отметить, что хотя морфологическая картина гелеобразной
нити, получающейся в жестких осадительных ваннах, практически оди-
72
какова для всех термостойких волокон, описываемый механизм фор-
мования наблюдается только для ПФИА. Общим для всех волокон,
высаженных в жесткие осадительные ванны, является низкая дефор-
мируемость на стадии пластификационной вытяжки и низкие физико-
механические показатели свежесформованной нити.
В мягких осадительных ваннах внешне как бы создаются более не-
однородные условия для осаждения полимера по сечению волокна.
В результате быстрой десольватации внешнего слоя образуется плот-
ная рубашка, которая становится основным регулятором диффузион-
ных процессов, препятствуя равномерной усадке волокна под действи-
ем возникающих напряжений, что приводит к сжатой, бобовидной фор-
ме сечения волокна.
При анализе данных о влиянии характера осадительной ванны на
свойства волокна было установлено [32], что при применении мягких
осадительных ванн, характеризуемых медленным структурообразовани-
ем, удается получить волокна с мелкими равномерными порами. Такие
волокна обладают большой способностью к пластической деформации
и эффективной ориентации. Несмотря на неровный срез и наличие не-
однородности (ярко выраженная рубашка и ядро) волокна, сформован-
ные в мягкие осадительные ванны, почти всегда имеют лучшие физико-
механические показатели, чем волокна, сформованные в жесткие оса-
дители. Исключением являются предельно жесткоцепные волокна (при-
чины будут рассмотрены ниже). Несмотря на явные преимущества
мягких осадительных ванн, в производственных условиях, они не
всегда могут применяться, так как в этом случае требуется очень
большой путь нити в осадительной и пластификационной ванне.
Важным фактором, влияющим на формование волокна, является кон-
центрация прядильного раствора. В ряде работ [33] показано, что для
гибкоцепных полимеров с увеличением концентрации полимера в пря-
дильном растворе снижается стойкость его к действию осадителей и
замедляются диффузионные процессы. Для растворов с большой кон-
центрацией вследствие повышения осаждающей способности осадителя
наблюдается быстрое образование поверхностного слоя струйки. Обра-
зовавшаяся оболочка замедляет массобмен. Вследствие этого образу-
ются неоднородные в поперечном сечении волокна с ухудшенной спо-
собностью к пластификационному вытягиванию. Аналогичная картина
характерна и для термостойких волокон, хотя для каждого волокна
существует своя оптимальная концентрация полимера в прядильном
растворе. Последняя также зависит от состава осадительной ванны.
Для полимеров полужесткой структуры (сульфон Т, полиимиды и др.)
оптимальная концентрация, при которой получаются волокна с лучши-
ми физико-механическими характеристиками, как правило, в 1,5—2,5
раза выше, чем для волокон предельно жесткой структуры, если не при-
нимать во внимание специальные методы формования последних (из
размягченных гелей) [20].
Влияние фильерных вытяжек на процесс формования волокна,
по-видимому, носит общий характер. Как правило, за исключением так
называемого сухо-мокрого метода формования, фильерное вытягивание
не используется для эффективной ориентации структурных элементов.
Наоборот, при формовании волокон в условиях нулевой и даже неболь-
шой отрицательной вытяжки лучше всего реализуются возможности
релаксации нормальных напряжений, вследствие чего, отводимая струя
находится в ненапряженном гидродинамическом равновесном состоя-
73
нии. Это облегчает процессы последующего пластификационного вытя-
гивания. Как показано в работе [34], при формовании волокон из гиб-
коцепных полимеров фильерная вытяжка мало влияет на прочность
получаемых волокон. Применение больших фильерных вытяжек почти
всегда приводит к отрицательным результатам, по-видимому, из-за воз-
никновения локальных напряжений и разрывов сплошности. Только в
случае формования термостойких волокон из сернокислотных раство-
ров применяют небольшие положительные фильерные вытяжки, так как
вытекающие из фильеры струйки раствора имеют более высокую плот-
ность, чем осадительная ванна и поэтому при свободном истечении они
стремятся опуститься на дно прядильной ванны.
Особенности формования волокон из анизотропных растворов. Ра-
нее уже отмечалось, что характерным свойством предельно жесткоцеп-
ных полимеров является их способность к самоупорядочению. Равно-
весному состоянию жесткоцепных полимеров отвечает максимально
упорядоченная система, что проявляется в возникновении жидкокри-
сталлического состояния в умеренно концентрированных растворах.
Для гибкоцепных и полужестких полимеров типична тенденция к раз-
ориентации, поскольку равновесному термодинамическому состоянию
отвечает статистическое взаимное расположение макромолекул. Эти
принципиальные различия в структуре растворов накладывают отпеча-
ток и на закономерности формования волокон из полимеров третьей
группы. Прежде всего следует остановиться на различии явлений ори-
ентации макромолекул. Первый этап ориентации макромолекул осуще-
ствляется, как известно, при входе раствора в отверстия фильеры
[3] и затем в осадительной и пластификационной ваннах за счет гра-
диента скорости в направлении оси волокна. При этом, если ориенти-
руются полимеры первой и второй групп, то после снятия растягиваю-
щего усилия (т. е. после уменьшения градиента продольной скорости до
нуля) за какие-то доли секунды наступает тепловая разориентация по-
лимера, которая протекает тем быстрее, чем ниже вязкость системы.
Иное положение имеет место для анизотропных растворов, образуемых
предельно жесткоцепными полимерами. Во-первых, такие растворы лег-
че ориентируются, так как требуется всего лишь развернуть уже упо-
рядоченные агрегаты молекул вдоль оси волокна. Ориентированная
система оказывается более устойчивой, ее не может существенно нару-
шить тепловое движение макромолекул, так как равновесное состояние
является упорядоченным. Было показано [35] на примере ПБА, что
даже при обычном (без наложения механического поля) высаживания
этого полимера из анизотропного раствора в ДМАА возникают над-
молекулярные структуры в виде резко асимметричных образований.
Такая структурная организация полимера позволяет получать даже при
небольших кратностях вытяжки высокоориентированные волокна. Изло-
женные принципиальные различия предопределяют и некоторые другие
отличия в формовании анизотропных растворов. Изменение состава
осадительной ванны оказывает гораздо меньшее влияние на свойства
жесткоцепных волокон. Если для гибкоцепных полимеров более пред-
почтительными являются мягкие ванны, то для таких волокон, как
ПФТА и ПБА, одинаково применимы как мягкие, так и жесткие ванны.
Коагуляция нити из растворов жесткоцепных полимеров происхо-
дит почти сразу же по выходе из фильеры, причем прочность гелеоб-
разной набухшей нити достаточно высокая (от 5 до 15 сН/текс). Сине-
ретические процессы в формуемой нити проходят в незначительной
74
степени, концентрация полимера в гелеобразной струйке очень низкая
(не выше 10—15%), причем она почти не изменяется вплоть до стадии
сушки волокна.
Волокна из изотропных растворов способны к большим пластифи-
кационным вытяжкам, тогда как при формовании анизотропных раство-
ров вследствие того, что ориентация наблюдается уже в осадительной
ванне, можно ограничиться минимальной пластификационной вытяжкой.
Специфическая структура жесткоцепного полимера, осажденного из
анизотропного раствора, приводит к очень низким величинам внутрен-
них напряжений в полимере после сушки. Если для гибкоцепных поли-
меров процесс осаждения связан с образованием пространственной сет-
ки, состоящей из отдельных анизотропных элементов, связанных между
собой узлами, в которых возникают остаточные внутренние напряже-
ния из-за быстрого стеклования, то специфика структуры полимеров в
анизотропном состоянии иная. Анизометрические элементы, образую-
щиеся при осаждении полимера из раствора, не связаны между собой
монолитными узлами, а контакты между ними являются флуктуирую-
щими. Поэтому при удалении низкомолекулярных компонентов (остат-
ки растворителя и осадителя) путем сушки коллапс структуры не при-
водит к существенным внутренним напряжениям. В результате после
сушки может возникать система с невысокими внутренними напряже-
ниями и незначительными дефектами [36]. Подтверждением этому ме-
ханизму, по мнению авторов, является низкая пористость после сушки
жесткоцепных волокон в сопоставлении с волокнами из гибкоцепных
полимеров (табл. 3.2).
Таблица 3.2. Пористость и удельная свободная поверхность
волокон из гибко- и жесткоцепных полимеров
Полимер Суммарный объем пор, 10* смз/г Удельная поверхность, м2/г
Полн-^-капроамид 86 11,6
Полигексаметилентерефталамнд 85 14,2
ПБА 1,1 0,11
ПФТА 1,1 0,10
Не останавливаясь на некоторых других менее важных отличиях
в формовании волокон из анизотропных растворов, следует указать,
что многие вопросы еще мало изучены и истинный механизм формова-
ния неясен. Подтверждением этому является факт наличия ряда термо-
стойких полимеров, по жесткости структуры относящихся ко второй
группе и не образующих анизотропных растворов, но вместе с тем фор-
мующихся подобно предельно жесткоцепным полимерам из анизотроп-
ных растворов. И наоборот, такой предельно жесткоцепной полимер,
как ПБА при формовании волокна из изотропного раствора (при кон-
центрациях полимера в растворе ниже критической) ведет себя как
полужесткий полимер.
3.2.5. Ориентационное вытягивание
свежесформованных волокон
В большинстве случаев свежесформованные волокна подвергаются
ориентационному вытягиванию непосредственно по выходе из осади-
тельной ванны. Вытяжка осуществляется в присутствии пластификато-
75
pa [37], которым является растворитель, остающийся в волокне. Вы-
тяжка волокон может проводиться в специальных пластификационных
ваннах при 30—90 °C или в среде острого пара при 100—НО °C. Иссле-
довано [39] влияние пластификационной вытяжки на свойства волокон.
Как видно из рис. 3.9, степень вытяжки, прочность волокна и двойное
лучепреломление связаны симбатно. Для целого ряда термостойких
полимеров дополнительная ориентация макромолекул может быть до-
стигнута путем вытягивания при повышенных температурах [39].
Известно, что переход к ориентированному состоянию гибкоцепных
полимеров связан с разрушением исходной изотропной структуры. Для
Рис. 3.10. Зависимость прочности ,(/), максимальной кратности вытяжки >(2)
и удлинения (3) от температуры для ПМФИА.
(I—IV— см. объяснения в тексте).
Рис. 3.9. Зависимость коэффициента двойного лучепреломления поли-л-фе-
ниленизофталамида (/) и прочности ПМФИА '(2) и поли-3,3'-дифенилсуль-
фонтерефталамида (3) от кратности вытяжки.
большинства волокон, содержащих ароматические и гетероциклические
структуры с гибкими шарнирными связями, ориентационный переход
может быть реализован без труда. Они легко подвергаются одноосной
деформации на 200% и более. По своей природе эта деформация высо-
коэластическая и связана с распрямлением цепей. Однако из-за наличия
стерических затруднений вследствие больших размеров циклов процесс
распрямления цепей часто бывает затруднен, что приводит к особенно-
сти их ориентации. Так, например, в ряде работ [40; 47] была установ-
лена бимодальная зависимость прочности ориентированных волокон от
температуры вытяжки. Эта бимодальная зависимость связана с сущест-
вованием четырех температурных областей вытяжки, в каждой из кото-
рых наблюдаются релаксационные переходы. На рис. 3.10 представлен
типичный пример зависимости прочности Р и максимальной кратности
вытяжки ТСмакс от температуры для волокон из поли-лг-фениленизофтал-
амида. Хорошо видно наличие четырех температурных (релаксацион-
ных) областей, в каждой из которых наблюдается своя специфика
структурообразования. В первой температурной области (200—260°C)
эффективное упрочнение осуществляется в режиме вынужденной эла-
стичности, а во второй (260—300 °C)—в режиме истинной высокоэла-
стичности, когда интенсивные релаксационные процессы снижают проч-
ность Р, а кратность вытягивания Хмакс растет. В третьей области
76
указывалось, самопроизволь-
1200
400\-------L______I_______I
250 350 450 550
t°C
Рис. 3.11. График изменения на-
пряжения полиоксадиазольных во-
локон при изометрическо'м нагреве.
(300—350 °C) доминирующими становятся кристаллизация и образова-
ние ориентированной структуры волокна. При этом растет прочность,
а кратность вытяжки снижается. В четвертой области (360°C) проис-
ходит деструкция и вязкое течение полимера; прочность при этом сни-
жается. Аналогичная картина наблюдается, по-видимому, для всех не
предельно жестких термостойких волокон. Это подтверждается данны-
ми изометрического нагрева для полиоксадиазольных волокон
(рис. 3.11) [41].
Принципиально иная картина наблюдается для жесткоцепных во-
локнообразующих полимеров. Как ранее
ное упорядочение их макромолекул при-
водит к тому, что уже в исходном раство-
ре обеспечивается относительно строгое
взаимное расположение ассоциатов мак-
ромолекул. При формовании и пласти-
фикационной вытяжке ориентация ста-
новится еще более совершенной — упоря-
доченными становятся не только макро-
молекулы внутри ассоциатов, но и сами
ассоциаты. Так как термодинамически
равновесным состоянием жесткоцепных
макромолекул является максимально
упорядоченное состояние, то достигнутая
ориентация волокон устойчива и тепло-
вая разориентация практически сводится
к нулю.
Естественно, что дополнительная го-
рячая вытяжка для жесткоцепных волокон не требуется. Вместо этого
их подвергают кратковременной термообработке при 500—600°C под
небольшим натяжением. Это оказывается достаточным, чтобы ориен-
тация и структурная организация макромолекул волокна достигла
равновесной. По данным [42], в таких волокнах углы разрриентации
проходных и держащих нагрузку сегментов 0 практически равны и не
Таблица 3.3. Характеристика строения аморфных участков
высокоориентированных волокон (0] и 02 — углы разориентации
соответственно макромолекул, «держащих нагрузку» и всех
проходных сегментов; n/W — доля «держащих нагрузку» цепей
от числа цепей в кристаллите)
Полимеры 91 02 0к n/N
Гибкоцепные полиэтилен низкого давления 16,5 4,1 4,0 0,06
поливиниловый спирт 26 18 5,1 0,14
полиоксиметилен 39 14 4 0,05
поликапроамид 31,5 21,1 — 0,09
Жесткоцепные поли-1,3-фениленизофталамид 32 32 — 0,11
поли-1,4-фенилентерефталамид 15,3 15,3 — 0,66
полипирромелитимид-4,4'-диаминодифе- 16,0 16,0 8 0,5
нилоксид полипирромеллитимид-4,4'-диаминотрифе- 33,0 33,0 'II 0,13
нилоксид
77
превышают 15°, причем доля «держащих нагрузку» цепей в аморфных
участках достигает 0,66 против максимально возможных 0,14 для гибко-
цепных полимеров (табл. 3.3).
Волокна из предельно жесткоцепных полимеров подвергают тер-
мическим вытяжкам только с целью повышения модуля упругости. Если
модуль упругости нетермовытянутого волокна не превышает 80—
90 ГПа, то после термической вытяжки он может быть повышен до
150 ГПа (для поли-га-фенилентерефталамида).
Необходимо отметить, что скорость вытягивания термостойких во-
локон при повышенных температурах относительно невелика (не более
20 м/мин). В научно-технической литературе отсутствуют сведения о
влиянии скорости вытягивания на механические свойства получаемых
волокон.
3.3. СПЕЦИФИЧЕСКИЕ ПРОЦЕССЫ ПОЛУЧЕНИЯ
ТЕРМОСТОЙКИХ ВОЛОКОН
В отличие от обычных синтетических волокон технического назна-
чения к термостойким волокнам предъявляется ряд специфических тре-
бований:
1) длительное сохранение высоких механических характеристик при
высоких температурах;
2) способность выдерживать высокие статические и динамические
нагрузки;
3) морозостойкость до —260 °C;
4) стойкость к радиации и т. д.
В общем виде эти требования можно считать критерием пригод-
ности термостойких волокон для экстремальных условий эксплуатации.
Вполне понятно, что обычными традиционными методами формо-
вания этих особых свойств достичь невозможно. Поэтому в технологии
производства термостойких волокон имеется ряд специфических про-
цессов, связанных с особыми условиями при получении или последую-
щими обработками готовых волокон.
3.3.1. Получение высокомодульных сверхпрочных волокон
При разработке проблемы получения термостойких волокон впер-
вые были получены химические волокна со сверхвысокой прочностью
при разрыве и высоким модулем упругости. Характерно ли это только
для этих типов волокон? Прежде чем попытаться ответить на постав-
ленный вопрос, рассмотрим некоторые общие условия получения воло-
кон с высокой прочностью и с этой целью обобщим ранее изложенное.
Проблема получения сверхпрочных волокон для технических целей
связана в первую очередь с решением двух задач:
1) достижением максимальной и устойчивой ориентации макромо-
лекул;
2) получение особой надмолекулярной структуры с минимумом
макро- и микродефектности волокна.
Между ориентацией макромолекул и прочностью волокон сущест-
вует определенная корреляция, которая обычно характеризуется зави-
симостью прочности одноосного растяжения от среднего угла разори-
ентации макромолекул, которая может быть представлена графиком
78
(рис. 3.12). Как видно из рисунка, только при достижении очень малых
углов разориентации начинает резко возрастать прочность волокна.
Поэтому для получения высокопрочных волокон необходимо стремиться
к максимальным величинам ориентации. При существующей техноло-
гии производства для обычных химических волокон эта цель недости-
жима. Для большинства волокнообразующих полимеров термодинами-
ческому равновесию отвечает разупорядоченное, изотропное состояние.
Ориентация достигается путем приложения однооснонаправленного
механического поля (ориентационная вытяжка волокна). Однако одно-
временно с ориентирующим действием
усилия действует энтропийный фактор
(тепловая разориентация). В результа-
те достигается какой-то компромисс
между ориентирующим влиянием ме-
ханического поля и р азориентирующим
действием тепла. Увеличение напря-
жения при одноосной ориентации с
целью повышения степени ориентации
приводит к появлению микро- и макро-
дефектов в структуре волокна. Роль
дефектов и неполной ориентации вид-
на из того, что реальная прочность
снижается почти на целый десятичный
порядок по сравнению с теоретически
рассчитанной. Вместо предполагаемой,
приложенного механического
Рис. 3.12. Корреляция прочности и
среднего угла разориентации макро-
молекул в волокнах:
а _ угол разориентации; Оо — максималь-
ная прочность при а = 0°; а а — прочность
при угле а.
согласно ряду теоретических расчетов
[43], прочности 6,0—8,0 ГПа реаль-
ные многотоннажные волокна имеют
прочность не выше 1,0 ГПа. Наиболее
простым путем для приближения
прочности к требуемой является ис-
пользование в качестве волокнообра-
зующего такого полимера, термодинамическому равновесию которого
отвечает упорядоченное состояние. Реализация этого пути оказалась
возможной при использовании предельно жесткоцепных полимеров типа
поли-п-бензамида, поли-п-фенилентерефталамида и аналогичных им
полимеров третьей группы жесткости.
Эти полимеры при определенных критических концентрациях об-
разуют анизотропную жидкокристаллическую систему. Порядок в такой
системе еще не достигает характерного для истинного кристалла, а яв-
ляется промежуточным между твердым и жидким состоянием. Но это
как раз и позволяет достичь высокой упорядоченности полимера с од-
ной стороны и сохранить высокую текучесть растворов, что позволяет
формовать из них волокна обычным способом. Сформованное волокно
обладает термодинамически устойчивой упорядоченностью. В опреде-
ленных условиях повышенные температуры (процесс термообработки)
даже способствует дальнейшему упорядочению.
Как было ранее указано, данный путь реализован в производст-
венных условиях и на его основе создана технология получения высо-
копрочных и высокомодульных волокон типа кевлар, кевлар 49
и др. Однако высокомодульные и сверхпрочные волокна могут быть
получены не только из жесткоцепных полимеров, но и из гибкоцепных
полимеров при условии применения новых методов упрочнения.
79
Как было отмечено [44], превращение струи в волокно при формо-
вании волокон из расплава или раствора может рассматриваться с по-
зиции принципа эквивалентности физической кинетики полимеров, ко-
торый сводится к тому, что термодинамическое поведение системы рас-
тянутых гибкоцепных макромолекул эквивалентно поведению системы
жесткоцепных макромолекул в отсутствие внешних полей. Не останав-
ливаясь здесь на теоретических доказательствах этого принципа [44],
остановимся на практических выводах из этой теории. Принцип «тер-
модинамической эквивалентности» обосновывает второй путь решения
проблемы упрочнения. Он сводится к созданию условий высокой ори-
ентации макромолекул любой жесткости и обеспечению быстрой фикса-
ции распрямленных и ориентированных макромолекул, например путем
переохлаждения или кристаллизации, для предотвращения процессов
разориентации и образования складчатых структур. Для реализации
этого пути применяют особые условия формования. Процесс «эффек-
тивной» ориентации переносится со стадии вытяжки сформованного
волокна на стадию выхода раствора или расплава из отверстий филье-
ры. С этой целью формование ведут при больших скоростях сдвига.
Судя по литературным данным, таким способом удается получать по-
лиолефиновые и полиамидные волокна с прочностью до 2,5—3,5 ГН/м2.
Правда, сведений о промышленном внедрении этого метода пока нет.
Особые усилия формования, по-видимому, необходимо применять
для гибкоцепных полимеров. Для полужестко- и жесткоцепных поли-
меров можно ограничиться некоторой модификацией существующей
технологии. Из известных в настоящее время условий получения
высокопрочных и высокомодульных волокон можно выделить сле-
дующие.
1. Получение полимера высокой молекулярной массы. До недавне-
го времени предполагалось, что прочность волокна при некотором зна-
чении молекулярной массы достигает предела, что отражается на фор-
ме кривой о—г]уд (рис. 3.13). При этом насыщение начиналось при от-
носительно невысоких молекулярных массах. Поэтому считалось неце-
лесообразным работать с высокомолекулярными полимерами, техноло-
гия переработки которых в то время была не разработана. После освое-
ния методов переработки высоковязких полимеров было установлено,
что зависимость прочности от молекулярной массы имеет иной харак-
тер (рис. 3.14). После довольно значительного плато при каком-то
критическом значении М прочность снова резко возрастает. По данным
С. П. Папкова, для ПФТА критической молекулярной массой является
30 000—35 000.
2. Формование полимера в волокно через стадию анизотропного со-
стояния. Опыт показывает, что оптимальным вариантом является про-
явление анизотропного состояния в прядильных растворах. Тогда тех-
нологический процесс формования не требует особых перестроек, вы-
сокопрочные волокна получаются из тех же самых полимеров.
В табл. 3.4—3.6 показано, что волокна, сформованные из анизотропных
растворов, как правило, имеют более высокие механические характери-
стики, чем волокна, сформованные из изотропных растворов, однако
при этом удлинения ниже [45].
Анизотропия растворов облегчает процесс получения волокон, од-
нако, имеется ряд примеров получения сверхпрочных волокон и из
изотропных растворов (см. табл. 3.4). [46]. Предполагается, что и в
этих случаях анизотропное состояние реализуется, но происходит оно при
80
высаживании полимера или при высокотемпературной обработке
волокна.
3. Способность свернутых макромолекул развертываться в процес-
се формования волокна — важнейшее условие получения сверхпрочных
волокон. Если макромолекулы невозможно развернуть в линейную цепь,
например, вследствие быстрой кристаллизации или каких-либо стери-
ческих препятствий, тогда получение сверхпрочного волокна, по-види-
Рис. 3.13. Зависимость прочности волокон от удельной вязкости полимера.
Рис. 3.14. Зависимость прочности волокон от удельной вязкости полимера
для жесткоцепных волокон.
мому, невозможно. Так, волокно сульфон-Т кристаллизуется уже при
высаживании полимера в осадительной ванне.
Безусловно, приведенных условий недостаточно для решения по-
ставленной задачи, однако, их соблюдение, по-видимому, очень важно.
3.3.2. Циклодегидратация волокон [20]
Большая часть волокнообразующих термостойких полимеров не
растворяется в известных растворителях. В этом случае волокна фор-
муют в две стадии. Вначале формуют волокна из промежуточных
линейных форм полимера, способных образовывать высококонцентри-
рованные растворы, ио не обладающих нужной термостабильностью.
Затем, на второй стадии, свежесформованное волокно подвергают хи-
мической или термической циклизации, в результате которой оно при-
обретает нерастворимость, неплавкость и требуемую термостойкость.
Для большинства термостойких полимеров циклизация в основном сво-
дится к дегидроциклизации, в процессе которой из двух функциональ-
ных групп (например, аминной и карбоксильной) образуется гетероцикл
и выделяется вода. Циклизация осуществляется чаще всего сразу же
после пластификационной вытяжки волокна, но в некоторых случаях-
(например, для полиоксиаминов) ее проводят после термической вы-
тяжки. Технически более удобной является термическая циклиза-
ция, которая сводится к высокотемпературной обработке волокна в
вакууме или азоте на жесткой паковке в аппаратах периодического или
непрерывного действия. В результате превращения свободных функ-
циональных групп в циклические структуры термостабильность волокон
резко возрастает, но одновременно растет и жесткость волокон. Харак-
терна зависимость термических и термомеханических свойств зацикли-
6—2254 81
Таблица 3.4. Высокомодульные волокна, сформованные из изотропных растворов
полимеров в органических растворителях [45]
Химическая формула элементарного эвеиа Содержание звеньев, % Djg. дл/г Прочность, • сН/текс Удлинение, % Модуль, сН/текс Технологическая характеристика волокна
—NH— О-™- 50 2,59 62,1 1.3 5010 Термовытянутое
-ОС- ©~С0~ 50
—HN—(Q ©—CONHNH- 5
~hn~O - CONH- (^2^ >—NH- 45 6,4 60,3 22,3 1850 Свежесформован- ное
-ОС- ©-со- 50 157,6 3,7 6290 Термовытянутое
—HNHNCO- CONHNH— 25
CONH— -NH— 25 8,1 135 6,4 3910
-ОС- ©-со- 50
-HN— ^-CONHNH- 45
"HN-© NH- 5 4,8 126,0 5,8 3980
50
2
35
13
50
50
25
25
50
20
30
Формование из 20<>/п-ных (масс.) растворов в 100% HjSO*.
7,0 217,8 4,6 5580
3,4 162,0 4,9 4410 Свежесформован
3,3 162,0 5,7 5080 ное*
Таблица 3.5. Высокомодульные ароматические волокна, сформованные
из анизотропных растворов полимеров в органических растворителях [45]
Химическая формула элементарного звена Содержание звеньев, % (мол.) Растворитель < дл/г Прочность, сН/текс Удлинение, % Модуль, сН/текс Технологическая характеристика волокна
—NH- 50 ГМФТА+ДМАЦ+ 4-L1C1 3,64 103,5 1,3 10000 Термовытянутое
-°с-О —СО— 50
С1\
-™~© -NH— 50 ДМАЦ+LiCl 3,73 129,6 1,4 9230
-ос-© -СО- 50
-HN-© -со- 100 ДМАЦ+LiCl 2,4 165,0 8,1 2550 Свежесформован- ное
-ос-©- -со—
HN-(g }-NH 50 ДМАЦ+LiCl 3,2 119,7 1,9 6870 Термовытянутое
с/ ~HN~^c^~-NHC0 —— -со- 50
-™-© —NH— 10 TMM+LiCl 1,4 117,0 1,4 8230
-ос-© -СО— 10
-HN-© -со- . 80
Определялась прн 30 °C в HjSO* (0,5 г/100 мл).
Таблица 3.6. Высокомодульные волокна, сформованные
из анизотропных растворов в минеральных кислотах [46]
Структура звена Содержание звеньев, % (мол.) nlg. дл/г Концентрация полиме- ра в растворе, % (мол.) Растворитель Прочность, сН/текс Удлинение, % Модуль, сН/текс Те хнологиче ска я характеристика волокна
NH 50 4,8 20 100,2%-ная H2SO4 234,0 3,7 6750 Свежесформован- ное
-°с-О~с°~ 50 189,0 2,2 8550 Термовытянутое
—NH— 43 6,2 10 100,8%-ная 81,0 10,1 2410 Свежесформован- ное
—ОС—<^>-СО- 43 180,9 2,0 9580 Термовытянутое
—HN—СО— 14
С\
NH 50 3,9 20 100%-ная H2SO4 162,0 6,5 3330 Свежесформован- ное
—ос—(Q)-co— 50
NH— 50 3,7 20 HFH2SO4 (1:1) - 189,0 4,8 58|0 >
-°с-О-с°- 50
hn-^Q^-nh 50 5,3 20 100%-ная H2SO4 162,0 5,8 4230 »
г^1-со- 50
-OC-lyJ N
зованного волокна от последующей термической вытяжки. Эта зависи-
мость хорошо иллюстрируется данными для полиимидного волокна
«аримид» [47]. Как видно из таблицы 3.7, существует две зоны. В «низ-
котемпературной» зоне (300—350 °C) термовытянутые волокна имеют
максимальную термостойкость, тогда как в высокотемпературной облас-
ти вытяжек (450—500 °C) термостойкость волокон заметно снижается,
зато повышается теплостойкость и модуль упругости [47].
Таблица 3.7. Физико-механические характеристики аримидных волокон
Температура вытяжки, °C Физико-механические характеристики при 20эС Прочность при 300 СС, % от исходной Удлинение при 300 °C, % от исходного Термостойкость после прогрева в течение 100 ч при 300 °C Степень имнднзацин, % Акустический ! модуль, МПа Число двойных изгибов при о=50 МПа
прочность, сН/текс удлинение, % начальный модуль, МПа прочность, % от исходной удлинение, % от исходного
Невытя- нутое во- локно 27,1 19,6 5770 43,3 119 92,0 55 100 7090 9556
320 37,8 9,5 6190 35,5 69,5 71,5 95 95,4 8660 2304
350 45,5 9,7 7150 36,6 ИЗ 81,8 83 86,7 8910 2415
380 46,2 5,5 6830 45,6 ПО 66,6 82 96,7 8320 —
400 43,2 8,0 6990 45,9 84 51,6 58 96,7 8740 2302
450 45,4 11,0 6300 40,9 105 47,7 61,5 95,0 7930 1761
500 53,1 7,2 7100 51,5 96 70,1 66,6 94,0 9740 1562
550 49,6 6,2 5880 48,0 75 71,5 50,0 76,7 9710 1053
Наблюдаемая зависимость характерна почти для всех термостой-
ких волокон, содержащих в цепи циклические структуры. Объясняется
она частичной расциклизацией при температурах, оптимальных для
ориентационного упрочнения волокон. Поэтому параметры технологи-
ческого процесса должны всегда корректироваться в зависимости от
заданных свойств. Для высокопрочных волокон они должны быть ины-
ми, чем для высокотермостойких или высокотеплостойких волокон.
3.3.3. Термостабилизация волокон
Термостойкие полимеры по своей природе казалось бы не должны
нуждаться в дополнительной термостабилизации. Однако оказалось, что
термостойкость полимеров и полученных из них волокон не одна и та
же. Волокна всегда имеют более низкую термостабильность, что объяс-
няется следующим.
1. Термостойкость полимеров оценивается по температуре начала
интенсивного выделения газообразных продуктов термического распа-
да, тогда как у волокон начинается снижение физико-механических ха-
рактеристик задолго до заметного выделения продуктов распада.
2. В процессе переработки полимера в волокно в последнем накап-
ливаются различные примеси: соли металлов, продукты разложения
растворителей и т. д., которые могут являться инициаторами термо-
окислительной деструкции полимерной цепи.
3. Высокотемпературная технологическая обработка волокон (го-
рячая вытяжка, термофиксация и т. д.) приводит к частичной расцикли-
86
зации структурных единиц макромолекулы и накоплению стабильных
радикалов и перекисей.
Все это приводит к тому, что известные термостойкие волокна, не-
смотря на различия в химической структуре, отличаются незначительно
по термостабильности. Так, например, термостабильность волокна из
полибензимидазобензофенантролина (волокно ВВВ) не выше термо-
стабильности полиимидного волокна, хотя оно и имеет лестничную
структуру. Считают, что это объясняется большой дефектностью и не-
совершенством лестничной структуры пирронов.
Для исправления дефектов химической структуры волокон в техно-
логической практике часто прибегают к помощи добавок — антиокси-
дантов, стабилизаторов, циклизующих агентов и т. д. В ряде случаев
это приводит к заметным результатам. Так, например, при применении
некоторых фосфорорганических соединений термостабильность полиок-
садиазольного волокна значительно возрастает [48]. Для полиимидных
волокон эффективным оказалось применение фосфорной кислоты.
В отличие от формования синтетических волокон из расплава все
добавки вводят в волокно не перед формованием или на стадии приго-
товления полимера, а путем пропитки свежесформованного волокна.
Только отдельные стабилизаторы, нерастворимые в осадительной ван-
не, добавляются при подготовке прядильного раствора к формованию.
3.4. СУХОЙ МЕТОД ФОРМОВАНИЯ ТЕРМОСТОЙКИХ
ВОЛОКОН
Метод сухого формования применяется только для тех полимеров,
которые растворяются в достаточно летучих растворителях, таких как
ДМАА, ДМСО, МП и другие. Применительно к предельно жестким
полимерам способ сухого формования не описан. Сообщается, что но-
мекс, конекс, а также волокна из полигетероциклических и отдельных
полулестничных полимеров хорошо формуются на машинах сухого фор-
мования. Указывается, что сухое формование является основным спо-
собом переработки высоковязких высококонцентрированных поликон-
денсационных сиропов, нейтрализованных гидроокисями щелочных
металлов [49]. Свежесформованные волокна, как правило, аморфны,
легко подвергаются ориентационному упорядочению и после дополни-
тельных обработок имеют хорошие физико-механические характеристи-
ки. Некоторые исследователи утверждают [50], что основные законо-
мерности процесса сухого формования являются общими для всех во-
локон и практически не зависят от природы полимера и растворителя.
Не отрицая правомерность таких утверждений, все же следует учиты-
вать, что от формования ацетатных волокон сухое формование термо-
стойких волокон отличается не только необходимостью применения
более высококипящих растворителей, чем ацетон; прядильные машины
отличаются устройством прядильных шахт, распределением газовых
потоков, способами отвода и последующей обработки нити и т. д. [50].
При формовании из растворов высококипящих растворителей необходи-
мо применять инертный газ, предохраняющий от возможных хлопков и
загорания. Можно использовать в качестве инертного газа отработан-
ные топочные газы, смесь двуокиси углерода и азота, двуокись азота
или перегретый пар повышенного давления. Параметры формования по
сухому способу обычных и термостойких волокон приведены в табл. 3.8.
87
Таблица 3.8. Технологические характеристики формования некоторых волокон по сухому способу
Волокно Характеристика растворов Условия формования Свойства волокон Литература
H]g. цп/г растворитель концен- трация , % (масс.) динами- ческая вяз- кость, Па-с температура, °C скорость формова- ния, м/мнн прочность, Н/текс удлинение, %
шахты газа
Полиакрилонитрильное 60000* ДМФ 15—40 75 160—180 200 180—220 0,44—0,68 10—16 [50]
Полиуретановое ДМФ, ДМСО или ДМАА 15-18 40—70 250 50—100 0,88 520—650 [50]
Полибензимидазольное 0,7—0,8 ДМАА+5% LiCl 25 300 240 50—350 0,47—0,53 23 [50]
На основе ароматиче- ских полиамидов (поли- .и-фениленизофталамид) 1.1—1.7 МП (ДМАА)+5% L1C1 17—24 200—270 230—270 200 0,795 6—8 [49]
Пол нами доимидное 0,9—1,6 МП (ДМАА) 18—30 300 200 280 150—200 0,44—0,61 13—20 [50]
Полиимидное 1,5—3,5 ДМФ 18—20 200 200 280 300 0,44—0,66 17 [50]
Полиамидгидразидное 4—7 ДМАА 7—10 250—270 200 1,57—1,77 2—4 [45]
На основе полимеров лестничной структуры 0,9 ДМСО 25 215—225 255 80 0,33 21 [50]
Молекулярная масса.
3.5. СУХО-МОКРЫЙ МЕТОД ФОРМОВАНИЯ ТЕРМОСТОЙКИХ ВОЛОКОН
В последние годы получил распространение сухо-мокрый метод
формования [51].
Этот способ (рис. 3.15) основан на том, что образующиеся при
истечении из фильеры струйки раствора прежде, чем попасть в осади-
тельную ванну, проходят через воздуш-
ную (газовую) прослойку, в которой
происходит частичное испарение раст-
ворителя с поверхности струйки и
образование тонкой наружной оболоч-
ки, замедляющей диффузию осадителя
внутрь формирующегося волокна.
В патентной литературе [49] сравни-
ваются мокрый и сухо-мокрый методы
формования некоторых «-ароматиче-
ских полиамидов и полиамидогидра-
зидов. Указывается, что по сухо-мок-
рому способу были получены волокна
с прочностью в два раза выше, чем
прочность волокон, полученных по
мокрому и сухому способам. По-види-
мому, эти утверждения недостаточно
обоснованы, так как если не прини-
мать во внимание преимуществ сухо-
мокрого способа в высоких скоростях
формования, то с точки зрения физико-
механических свойств волокон оба
способа являются равноценными, осо-
бенно при применении органических
растворителей. Так, например, сооб-
щается [51], что поли-п-бензамидное
волокно, сформованное сухо-мокрым
способом, имело прочность 153 сН/текс,
мокрым способом—189 сН/текс, а
полиамидгидразидное волокно, сфор-
Рмс. 3.15. Принципиальная схема по-
лучения полиамидных волокон по су-
хо-моирому способу:
/ — фильерный комплект; 2 — фильера;
3 — газовая или воздушная камера; 4 —
нить; 5 — осадительная ванна; 6 — напор-
ная барка; 7— диск; 8 — приемное устрой-
ство; 9 — приемная барка; 10— насос; 11 —
прядильная трубка.
мованное сухо-мокрым способом —
171 сН/текс. Возможно, что явные преимущества сухо-мокрый способ
имеет при формовании волокон из высококонцентрированных растворов.
в серной кислоте.
Формованием из растворов в концентрированной серной кислоте
получают волокна на основе полигексаметилентерефталамида [52]к
полиарилен-1,3,4-оксадиазолов [53], полибензимидазолов [54], арома-
тических полиамидов [51] и других волокнообразующих полимеров
[55]. Для получения высокопрочных волокон необходимо применять
полимеры с высокой молекулярной массой и концентрацией. Такие
прядильные растворы являются высоковязкими и нетекучими. Чтобы
перевести их в текучее состояние, необходим нагрев до 80—90 °C. Оса-
дительную ванну при этом необходимо охлаждать, поэтому единствен-
но приемлемым способом для таких растворов является формование па
сухо-мокрому способу. По данным [56], высокомолекулярный полигид-
разидоксадиазол формуют из сернокислотного раствора, нагретого да
120 °C, через фильеру, расположенную на расстоянии 1,27 см от поверх-
89.
ности водной осадительной ванны со скоростью 239,8 м/мин. Волокна
после вытяжки при 409 °C имели прочность 81 сН/текс при удлинении
4,3%. При формовании из сернокислотных анизотропных растворов
ароматических полиамидов пара-структуры получают волокна с проч-
ностью выше 200 сН/текс. Так, например, из 20%-ного раствора поли-
п-фенилентерефталамида с T]ig=5,4 в 99,7 %-ной серной кислоте при
95 °C через фильеру, расположенную на расстоянии 1 см от поверхно-
сти осадительной ванны, были получены нити с прочностью 205 сН/текс,
удлинением 2,8% и начальным модулем 106,0 ГПа. Прочность и удлине-
ние элементарных волокон несколько выше—соответственно 234 сН/текс
и 3,7%. Фюрмование волокон из гелеобразного состояния в серной кис-
лоте по сухочмокрому способу позволяет получать высокопрочные волок-
на без термической обработки. Дополнительная термическая вытяжка
при 525 °C приводит к снижению прочности до 184 сН/текс и удлинения
до 1,7%. Модуль при этом повышается до 143,9 ГПа.
Таким образом, сухо-мокрый способ в случае формования из твер-
дых набухших полимерных гелей позволяет не только повысить ско-
рость формования до 400 м/мин и более, но также регулировать про-
цесс структурообразования и обеспечивать высокие физико-механиче-
ские характеристики волокон. В ряде случаев он позволяет по-новому
решать проблему регенерации растворителей и обезвреживания произ-
водства.
В заключение настоящей главы можно сделать следующие выводы.
1. Методы формования и аппаратурное оформление технологиче-
ского процесса для большинства термостойких волокон аналогичны
применяемым в многотоннажном производстве обычных синтетических
волокон, формуемых из растворов.
2. Основные закономерности процесса формования термостойких
волокон с гибкими и полужесткими макромолекулами такие же, как
для волокон из полиакрилонитрила, поливинилхлорида, поливинилово-
го спирта и других полимеров.
3. Принципиальные отличия в механизме формования наблюда-
ются для волокон, получаемых из предельно жесткоцепных полимеров.
Они обусловлены, главным образом, легкостью образования последни-
ми жидкокристаллического состояния и их свойством самопроизволь-
ного упорядочения как в жидкой, так и в твердой фазе.
4. Способность предельно жесткоцепных полимеров находиться при
термодинамическом равновесии в упорядоченном состоянии облегчает
получение сверхпрочных и высокомодульных термостойких волокон,
типичными представителями которых являются волокна кевлар, вниив-
лон и др.
5. Закономерности, выявленные при изучении процессов формова-
ния сверхпрочных термостойких волокон, позволяют сформулировать
фундаментальные основы образования высокопрочных волокон из боль-
шинства волокнообразующих полимеров: а) получение высокомолеку-
лярного полимера, не образующего складчатых цепей или способного
легко разворачиваться; б) создание в процессе нитеобразования струк-
туры предельно ориентированных распрямленных макроцепей; в) воз-
можно быстрое и полное затормаживание процессов тепловой дезориен-
тации при коагуляции полимера.
6. Термостабильность созданных в настоящее время термостойких
волокон находится примерно на одном уровне, однако, для некоторых
волокон при дополнительных обработках она может быть повышена.
90
ЛИТЕРАТУРА
1. Михайлов И. В. и др., Высокомол. соед., 1960, № 4, с. 991—995.
2. Цветков В. Н. и др., ДАН СССР, 1975, т. 224, № 5, с. 1126—1129.
3. Френкель С. Я., Ельяшевич Г. К. В кн.: Теория формования химических волокон.
М., «Химия», 1975. 280 с.
4 Битовская М. Г. и др., Высокомол. соед., 1976, сер. А, т. 18, № 3, с. 691—695.
5. Цветков В. Н. и др., ДАН СССР, 1975, т. 224, № 5, с. 1128—1129.
6. Иовлева М. М., Ефимова С. Г., Прозорова Г. Е. В кн.: Теория формования хи-
мических волокон. М„ НИИТЭХИМ, 1975. 185 с.
7. Перепелкин К. Е. Докт. дис. Л., ЛИТЛП им. С. М. Кирова, 1967.
8. Справочник по химии полимеров. Киев, «Наукова думка», 1971. 533 с.
9 Некрасов И. К., Хабарова К. Г., Высокомол. соед., 1971, сер. А, т. 13,
с. 1707—1709.
10. Wallach М. L., J. Polymer Sci., 1967, А-2, v. 5, р. 653—659.
11. Некрасов И. К, и др., Высокомол. соед., 1972, сер. А, т. 14, с. 866—867.
12. Паркер Д., «Успехи химии», 1963, т. 32, с. 1270—1275.
13. Савинов В. М. Канд. дис. М., МХТИ, им. Д. И. Менделеева, 1967.
14. Соколова Т. С. и др., Хим. волокна, 1974, № 1, с. 26—29.
15. Соколов Л. Б. и др., Термостойкие ароматические полиамиды. М., «Химия»,
1975. 168 с.
16. Голубев В. М. и др., Высокомол. соед., 1974, сер. Б, т. 16, с. 699—701.
17. Папков С. П. Теория формования химических волокон. Труды ВНИИВ, Мытищи,
1975. 185 с.
18. Пат. США 3671542 (1972), 3673143 (1972); фр. пат. 1526745 (1967); англ. пат.
1198081 (1970).
19. Платонов В. А. и др., Хим. волокна, 1975, № 5, с. 70.
20. Роговин 3. А. Основы химии и технологии химических волокон. Т. 2. М.,
«Химия», 1974. 360 с.
21. Пат. США 3671542 (1972), 3767756 (1973); заявка ФРГ 2346227 (1974).
22. Фр. заявка 2134581 (1972).
23. Химия (справочное руководство). Под ред. Ф. Г. Гавриченкова, Л., «Химия»,
1975. 576 с.
24. Матвеев В. С. и др., Хим. волокна, 1971, № 4, с. 6.
25. Папков С. П. Международный симпозиум по химическим волокнам. Препринты.
Калинин, 1974, секция 1, с. 73.
26. Smolders Н., Koll.-Z., 1971, Bd. 243, S. 14.
27. Иовлева М. М. Международный симпозиум по химическим волокнам. Препринты.
Калинин, 1974, секция 1, с. 179; Краснов Е. П. Докт. дис. М., МГУ, 1976.
28. Папков С. П. В кн.: Теория формования химических волокон. М., «Химия»,
1976. 280 с.
29. Фихман А. Д. Канд. дис. Л., ЛИТЛП им. С. М. Кирова, 1965.
30. Grdbe V., Meyer К, Faserf. u. Textilt., 1959, Bd. 10, S. 211—215; Gensrich N. J.,
Grope A., Faserf. u. Textilt., 1970, Bd. 21, S. 163—164; Epstein M. E„ Rosenthal A. J.,
Text. Res. J., 1966, v. 36, p. 813—819.
31. Руднева Л. Д. и др., Хим. волокна, 1975, № 2, с. 13—15.
32. Межиров М. А., Пакшвер Э. А., Хим. волокна, 1968, № 3, с. 13—14; Начин-
кин О. И., Перепелкин К. Е., Хим. волокна, 1963, № 1, с. 13—47; Савицкий А. В.,
Горшкова И. Я-, Хим. волокна, 1973, № 3, с. 4—5.
33. Начинкин О. И., Хнм. волокна, 1973, № 2, с. 12—13; Ефимова С. Г. и др., Высоко-
мол. соед., 1972, сер. Б, т. 14, с. 1527—1531; Hiretny Tauda, J. Chem. Soc. Japan,
Ind. Chem. Sect., 1963, v. 66, № 5, p. 72—74; Grdbe V., Hoger N., Faserf. u. Textilt.,
1968, Bd. 19, S. 398—399.
34. Перепелкин К. E. Докт. дис. Л., ЛИТЛП им. С. М. Кирова, 1967; Захаров В. С.
Канд, дис., М., ВНИИВ, 1972.
35. Папков С. П. и др., Высокомол. соед., 1973, сер. Б, т. 15, с. 370—375.
36. Папков С. П., Хим. волокна, 1977, № 3, с. 5—13.
37. Муравьева Н. И., Конкин А. А., Хим. волокна, 1972, № 2, с. 8—10; Глухое-
дов Н. П. и др., ДАН СССР, 1969, т. 187, с. 597—599.
38. Краснов Е. П. Международный симпозиум по химическим волокнам. Препринты.
Калинин, 1974, секция 1, с. 124—128; Пакшвер Э. А. и др.. Хим. волокна, 1976,
№ 1, с. 27—29.
39. Толкачев Ю. А., Краснов Е. П. Международный симпозиум по химическим волок-
нам. Препринты. Калинин, 1974, секция 3, с. 25—27.
40. Краснов Е. П. и др., Хнм. волокна, 1971, № 1, с. 48—51.
41. Волохина А. В., и др.. Хим. волокна, 1975, № 5, с. 39—40.
91
42. Утевский Л. Е. Международный симпозиум по химическим волокнам. Пре-
принты. Калинин, 1974, секция 1, с. 114—117.
43. Перепелкин К. Е. Международный симпозиум по химическим волокнам. Пре-
принты. Калинин, 1974, секция 7, с. 18—39.
44. Френкель С. Я- Международный симпозиум по химическим волокнам. Препринты.
Калинин, 1974, секция 1, с. 18—19; Френкель С. Я., Ельяшевич Г. К. В кн.: Тео-
рия формования химических волокон. М., «Химия», 1975. 280 с.
45. Black W. В., Preston A. High-Modulus Wholly Aromatie Fibers. N. — Y., Marcel Dek-
ker, 1973. 348 p.
46. Кудрявцев Г. И. и др.. Хим. волокна, 1973, № 3, с. 78—80.
47. Кудрявцев Г. И. Международный симпозиум по химическим волокнам. Пре-
принты. Калинин, 1974, секция 4, с. 1—9.
48. Одноралова В. Н. и др. Международный симпозиум по химическим волокнам.
Препринты. Калинин, 1974, секция 4, с. 13—15.
49. Кудрявцев Г. И., Панкратов Ю. Л., Щетинин А. М. Основы технологии произ-
водства ароматических полиамидов и волокон на их основе. М., НИИТЭХИМ,
1976. 81 с.
50. Шейн Т. И. и др.. Хим. волокна, 1976, № 1, с. 11—28.
51. Волохина А. В. и др.. Хим. волокна, 1975, № 1, с. 23—30.
52. Quyan R. G., Text. Res. J., 1970, v. 40, № 8, p. 677—679.
53. Imai I., J. Appl. Polymer Sci., 1970, v. 14, № 1, p. 225.
54. Пат. США 3441640 (1969).
55. Gloor W. N., Appl. Polymer Symp., 1967, Ns 6, p. 151.
56. Заявка ФРГ 2257707 (1973).
ГЛАВА 4
ПОЛУЧЕНИЕ И СВОЙСТВА ТЕРМОСТОЙКИХ ВОЛОКОН
В данной главе рассмотрены способы получения и свойства не-
скольких классов термостойких волокон, работы в области которых
вышли за рамки лабораторных исследований. Это — волокна на основе
полностью ароматических полиамидов, полиимидов, полиоксадиазолов,
лестничных полимеров и другие. Производство некоторых волокон, та-
ких, как полибензоксазольные, полихиноксалиновые и политиадиазоль-
ные, несмотря на их высокие термические свойства, не получило пока
развития. Причиной этого является отсутствие сырьевой базы, либа
сложность технологии, а комплекс физико-механических характеристик
получаемых волокон лишь не намного выше комплекса свойств уже из-
вестных волокон. В главе также кратко рассмотрены возможные пути:
модификации термостойких волокнообразующих полимеров и волокон
на их основе.
4.1. ВОЛОКНА НА ОСНОВЕ ПОЛНОСТЬЮ
АРОМАТИЧЕСКИХ ПОЛИАМИДОВ
4.1.1. Синтез полиамидов
Исходными соединениями для синтеза полиамидов являются аро-
матические диамины, с одной стороны, и ароматические дикислоты или
их производные — с другой стороны. Принципиально для получения по-
лиамидов могут быть использованы все известные способы поликон-
денсации: в расплаве, в растворе, межфазная или эмульсионная поли-
конденсация и даже поликонденсация в твердой фазе. Однако для по-
лучения волокнообразующих полиамидов применяют в основном два
92
метода — поликонденсацию в растворе и в эмульсии [1]. Расплав-
ный способ не нашел применения из-за того, что практически все поли-
амиды разлагаются ниже температуры плавления. Гетерофазные мето-
ды синтеза, такие, как газофазная или межфазная поликонденсация,
хотя в принципе и приводят к образованию высокомолекулярных поли-
амидов, также не нашли практического выхода и применяются в основ-
ном для получения небольших количеств полиамидов [2, с. 11]. Поли-
конденсация полиамидов в твердой фазе известна давно, однако только
в последнее время появились сообщения о синтезе волокнообразующих
полностью ароматических полиамидов [3].
Рис. 4.1. Зависимость логарифмической вязкости ароматических ПА от
температуры синтеза [2, 4]:
1 — поли-Ж'феннлентерефталамнд; 2 — поли-л-фениленизофталамид.
Рис. 4.2. Зависимость логарифмической .вязкости ароматических ПА от кон-
центрации мономеров в реакционной среде [5]:
1 — поли-З.З'-днфеииленсульфонизофталамид; 2 — полн-З.З'-дифеннленсульфонтере-
фталамид; 3 — полн-4,4'дифеннленсульфонизофталамид; 4 — полн-4,4'-дифениленсуль-
фонтерефталамнд.
0,4 Ofi 0,8 1,0
С, моль / ЮООмл
Поликонденсация в растворе. Реакцию проводят в амидных раство-
рителях, из которых чаще всего применяются ДМАА, МП или ГМФА.
С целью усиления растворяющей способности к указанным веществам
иногда добавляют хлориды лития, кальция, магния. Реакция идет по
схеме
О О
II II
nH2N—R—NH2 4-nCl—С— R'— С—Cl —
--->• [—HN—R—NH—CO—R'— CO—]„ + (2n - 1) HC1
Диамин или хлоргидрат диамина растворяется в ДМАА или другом
растворителе; смесь, как правило, охлаждается и затем в нее вводится
дихлорангидрид (ДХА). Наиболее приемлемым с точки зрения получе-
ния высокомолекулярных полиамидов считается способ, когда ДХА
вводится в твердом виде. Особенностью поликонденсации в растворе
является необходимость перемешивания высоковязких сред. Экзотер-
мический характер реакции ДХА с диаминами требует отвода выделяю-
щегося тепла. Хлористый водород, образующийся в результате реакции
и обладающий высокой коррозионной активностью по отношению к ме-
таллам, должен быть нейтрализован. Молекулярная масса ПА при
растворных способах поликонденсации зависит от многих факторов:
93
температуры реакции (рис. 4.1), концентрации мономеров в реакцион-
ной среде (рис. 4.2), природы растворителя, соотношения исходных
мономеров в растворе (рис. 4.3) и т. д. Зависимости имеют экстремаль-
ный характер. Реакционные растворы многих ПА являются стабильны-
ми и практически готовы к переработке в волокна. Подробно операции
подготовки реакционных растворов ароматических ПА для переработки
в волокна рассмотрены в работе [7]. Выделение ПА из реакционных
растворов для его последующей переработки в волокна осуществляется
Рис. 4.3. Влияние соотношения ДХА и
ДА иа величину логарифмической вяз-
кости ароматических 1ПА i [6]:
1 — поли-л-фениленизофталамид; 2 — поли-л-
феиилентерефта ламид.
только тогда, когда переработать
непосредственно реакционные смеси
невозможно. Такое выделение поли-
мера представляет многостадийный
процесс, связанный с собственно
осаждением ПА, его промывкой,
сушкой и т. д. Указанный способ,
по-видимому, используется при по-
лучении волокон на основе поли-п-
фенилентерефталамида. Как извест-
но, этот полимер растворим только
в минеральных кислотах. В процес-
се синтеза он выпадает в осадок
непосредственно в реакционной сме-
си [2, с. 29]. Положительной сторо-
ной растворной поликонденсации
является то, что в этом случае
имеются благоприятные предпосыл-
ки для создания непрерывных про-
цессов, даже если полимер в ходе
синтеза выпадает в осадок [8].
Эмульсионная поликонденсация.
Способ имеет ряд достоинств, по-
зволяющих легко получать высоко-
молекулярные ПА различного химического строения, а именно: доступ-
ные реагенты, высокую скорость реакции, возможность проведения
реакции при комнатной температуре и т. д. Наибольшее распростране-
ние этот способ нашел для получения поли-л/-фениленизофталамида
[2, с. 40]. Суть эмульсионного метода заключается в следующем:
ароматический диамин растворяют в смеси воды с каким-либо органи-
ческим веществом, образующим с водой эмульсию. Туда же вводят
акцептор хлористого водорода и, в случае необходимости, высаливатель,
который обеспечивает создание устойчивой эмульсии. Затем к указанной
смеси порционно добавляют раствор ДХА в том же органическом раст-
ворителе, который используется для получения эмульсии. Образу-
ющийся полимер выпадает в осадок в виде мелкого порошка. Харак-
терной особенностью эмульсионной поликонденсации является необхо-
димость энергичного перемешивания компонентов системы в процессе
синтеза. Несмотря на то, что рассматриваемый способ нашел практи-
ческое применение, закономерности процесса далеко не изучены. Более
подробно особенности растворной, эмульсионной и других видов поли-
конденсации, приводящих к получению ароматических полиамидов,
рассмотрены в специальных монографиях [1, 2].
Поскольку синтез ПА с применением дихлорангидридов имеет ряд
недостатков были предложены способы, основанные на прямой реакции
94
диаминов с дикарбоновыми кислотами [9], или реакции с участием
сероуглерода [10]. Получены высокомолекулярные полиамиды, обла-
дающие хорошими волокнообразующими свойствами, однако эти спо-
собы пока не вышли из стадии поисковых работ.
Рис. 4.4. Влияние добавок на стабиль-
ность прядильных растворов ПМФИА
в ДМ00 [11]:
1 — вода; 2— солянокислый диэтиламин; 3 —
солянокислый диметиламин; 4 — хлорид; 5 —
хлорид лития.
4.1.2. Некоторые особенности изотропных растворов
ароматических полиамидов
Как уже указывалось выше, синтез полиамидов проводится путем
низкотемпературной поликонденсации в растворе ДМАА, содержащем
хлориды кальция или лития [2, с. 30]. После завершения реакции обра-
зования полимера проводится
нейтрализация выделившегося
хлористого водорода окисями или
гидроокисями металлов, при этом
в реакционной среде достигается
определенная концентрация хло-
рида металла, и получаемая ком-
позиция оказывается стабильной
и пригодной для формования. В
том случае, когда полиамиды по-
лучают другими методами, на-
пример путем эмульсионной поли-
конденсации диаминов и дихлор-
ангидридов, и когда во время
синтеза полимер выпадает в виде
порошка, для растворения сухого
полимера используют смеси раст-
ворителя с хлоридами лития или
кальция. Первый путь оказывает-
ся более предпочтительным и, по-видимому, он является основным при
получении растворов таких волокнообразующих полимеров и волокон,
как номекс [7], фенилон [11, с. 12], сульфон-Т [12] и волокон из упо-
рядоченных сополиамидов [13].
На примере волокнообразующих ПМФИА и поли-4,4'-дифенил-
сульфонтерефталамида, т. е. полиамидов, не образующих анизотроп-
ных систем, изучена стабильность растворов и влияние на нее таких
факторов, как концентрация полимера, температура, добавки различ-
ных солей, воды, продолжительности выдерживания в определенных
условиях (рис. 4.4). Установлено [11, с. 12, 34], что причиной низкой
стабильности растворов полиамидов в чистом растворителе является
локальная кристаллизация полимера в растворе; исследование кинетики
формирования надмолекулярных структур в растворах показало, что
процесс гелеобразования, медленно идущий в начальных стадиях, рез-
ко ускоряется в конце; вязкость растворов нарастает вплоть до застуд-
невания. Опыты по формованию волокон из растворов ПМФИА, вы-
держанных различное время в одинаковых условиях, показали [11,
с. 17], что прядомость раствора (определяемая по фильерной вытяж-
ке) достигает максимального значения при длительности термостатиро-
вания, отвечающей окончанию первой стадии формирования надмоле-
кулярной структуры, т. е. до резкого изменения характеристики систе-
мы. Зависимость прочности волокон от выдерживания прядильного
95
раствора при определенной температуре также имеет экстремальный
характер (рис. 4.5). Средний размер надмолекулярных частиц умень-
шается со временем термостатирования до определенных размеров, а
дальше начинается их агрегация и увеличение размеров. Интересно
отметить, что фазовая диаграмма состояния системы ПМФИА—ДМАА
характеризует растворы как системы с аморфным разделением, имею-
щими нижнюю критическую температуру смешения в области 30—40°C
[11, с. 12]. Изучение реологических свойств системы ПМФИА — раство-
ритель позволило строго определить концентрационный интервал обра-
зования термодинамически стабильных растворов и критическую тем-
пературу растворения поли-
Рис. 4.5. Зависимость прочности волокон на осно-
ве ПМФИА (/) и размеров надмолекулярных
образований в прядильном растворе (2) от про-
должительности термостатирования (/=90 °C)
ПН-
мера, что важно для разра-
ботки технологического ре-
жима переработки раство-
ров ПМФИА в волокна [14].
При изучении влияния
добавок солей к метаста-
бильному раствору поли-4,
4'-дифенилсульфонтерефта-
ламида в ДМАА, таких
как хлориды лития, каль-
ция, кадмия, было обнару-
жено, что зависимость вяз-
кости получаемых систем от
концентрации соли в раст-
воре носит экстремальный
характер, причем максимум
вязкости растворов прихо-
дится на концентрацию со-
ли 1—2% (масс.) {11, с. 34].
Аналогичным образом характеристическая вязкость полимера зависит
от концентрации соли в бинарном растворителе. Авторы полагают, что
термодинамические свойства бинарных растворителей оказывают влия-
ние на конформацию макромолекул в растворе и что это должно ска-
заться на технологических особенностях переработки растворов в
волокна.
Предлагаются различные способы повышения стабильности
растворов полимеров типа ПМФИА. Одним из интересных предложе-
ний является добавка поверхностно-активных веществ к прядильным
растворам, что одновременно улучшает перерабатываемость последних.
Стабильность прядильных растворов полидифенилсульфонтерефталами-
да в ДМАА, не содержащем добавок солей, можно повысить термообра-
боткой растворов в течение 30 мин [11, с. 34]. Отмечается усиление
растворяющего эффекта смеси амидных растворителей по отношению
к полимеру, причем каждый из отдельных растворителей практически
не растворяет волокнообразующий полиамид. Правда, формование во-
локон из смесей амидных растворителей, за редким исключением, не
практикуется. Универсальным растворителем для ароматических поли-
амидов является концентрированная серная кислота. Отмечается, что
растворяющая способность серной кислоты в отношении ароматических
полиамидов зависит от ее концентрации [16]. Немаловажную роль
играет и химическая природа растворяемого полиамида, а также его
молекулярная масса.
9G
Особенности получения и поведения концентрированных растворов
ароматических полиамидов, способных образовывать анизотропные си-
стемы, рассмотрены в предыдущей главе.
4.1.3. Формование полиамидных волокон
Практически все волокна из ароматических полиамидов получают-
ся формованием из растворов. Следует, однако, отметить, что принципи-
ально возможными способами получения термостойких полиамидных
волокон, кроме указанных, могут быть также формование волокон не-
посредственно в процессе синтеза (при межфазной поликонденсации) и
формование волокон из дисперсий полимеров, аналогично получению
волокон на основе политетрафторэтилена [18, 19]. Эти способы, прав-
да, не нашли широкого применения, по-видимому, потому, что в первом
случае получаются низкопрочные волокна, а во втором — принципиаль-
ной трудностью является получение устойчивой коллоидной системы с
высокой степенью дисперсии полиамида. На сегодняшний день, как уже
указывалось в предыдущей главе, наибольшее внимание уделяется
трем способам формования волокон из ароматических полиамидов, а
именно: сухому, мокрому и сухо-мокрому.
Способ сухого формования волокон из ароматических полиамидов
имеет ряд преимуществ. В первую очередь необходимо отметить высо-
кие скорости формования и сравнительно высокие концентрации поли-
мера в прядильном растворе; кроме того, большая часть растворителя,
испаряющегося в прядильной шахте, легко регенерируется. В связи с
тем, что при формовании волокон из ароматических полиамидов при-
меняются высококипящие растворители, аппаратурное оформление про-
цесса имеет ряд особенностей, касающихся устройства прядильной
шахты, распределения газового потока, способов отвода нити и т. д.
Более подробно специфические особенности процессов сухого формова-
ния рассмотрены в обзоре [20]. Одним из недостатков сухого способа
формования полиамидных ароматических волокон является необходи-
мость и трудность промывки сравнительно тонких волокон от остатков
неорганических солей. В присутствии следов хлоридов лития или каль-
ция в готовых волокнах значительно снижается термостойкость послед-
них, и поэтому указанное обстоятельство необходимо иметь в виду, в
особенности в случае использования волокон при повышенных темпера-
турах [21]. С технологической точки зрения формование волокон по
сухому способу из изотропных и анизотропных растворов соответствую-
щих полиамидов практически должно проходить одинаково. Различа-
ются они лишь тем, что для волокон, формуемых из растворов полу-
жестких ароматических полиамидов, характерны значительные степени
дополнительной вытяжки, в то время как для предельно жесткоцепных
полиамидных волокон дополнительная вытяжка невелика [22].
Мокрое формование волокон из ароматических полиамидов осу-
ществляется как из органических растворителей, так и из растворов
концентрированных кислот, причем последний способ в настоящее вре-
мя получает все более широкое признание. Технология формования
полиамидных волокон мокрым способом аналогична технологии формо-
вания обычных синтетических волокон, например поливинилспиртовых,
полиакрилонитрильных.
В случае мокрого формования волокон из полиамидов типа
ПМФИА из растворов в амидных растворителях рядом авторов отме-
7—2254
97
чается возможность получения различной структуры свежесформован-
ного волокна путем варьирования условий осаждения полимера из рас-
творов. При исследовании механизма образования первичной структуры
волокна на основе ПМФИА и поли-4,4'-дифенилсульфонизофталамида
при мокром способе формования указывается на решающее влияние
диффузионных процессов осадителя и растворителя в системе струйка
прядильного раствора — осадительная ванна [23; 24]. Механизм этого
явления рассмотрен в предыдущей главе. В патентной литературе в ка-
честве осадительных ванн для ПМФИА и аналогичных ему полимеров
предлагаются различные композиции; концентрированные растворы со-
лей типа хлоридов кальция или лития, различных роданидов, широкий
температурный интервал осадительных ванн, содержащих тройные сме-
си, и т. д. [25].
Для предельно жесткоцепных полиамидов типа поли-п-фениленте-
рефталамида, полибензамида, их сополимеров при формовании из рас-
творов по мокрому способу влияние составов осадительной ванны на
свойства волокон сказывается в меньшей степени, чем для более «гиб-
коцепных» полиамидов, хотя некоторое различие между изотропными и
анизотропными растворами полимера одного и того же типа имеется
[И, с. 24].
Интересной особенностью некоторых предельно жесткоцепных по-
лиамидов, не образующих анизотропных растворов, является возмож-
ность перехода в анизотропное состояние струйки прядильного раство-
ра при действии на нее осадителя [26, с. 93].
Формование волокон по мокрому способу из растворов в различных
растворителях, в особенности это относится к полиамидам с промежу-
точной жесткостью цепи, предпочтительно тем, что, меняя состав оса-
дительной ванны, можно получать волокна с различной макро- и
микроструктурой, способных хорошо окрашиваться; при этом спо-
собе формования легко регулировать фильерные вытяжки, тепло- и
массообмен и т. д. Однако существенным недостатком способа мокрого
формования являются низкие скорости (8—20 м/мин), открытые по-
верхности испарения летучих компонентов. Считают, что мокрый спо-
соб наиболее пригоден для получения штапельных полиамидных воло-
кон. Комплексные нити получают в основном по методу сухого формо-
вания [27, с. 35].
При сухо-мокром способе формования ароматических полиамидных
волокон из струйки прядильного раствора полимера в летучем раство-
рителе, еще не попавшего в осадительную ванну, испаряется часть рас-
творителя, на поверхности струйки возникает тонкая полимерная плен-
ка, которая является регулятором диффузионных процессов при движе-
нии струи прядильного раствора в осадительной ванне [28]. При необ-
ходимости воздушная прослойка между фильерой и поверхностью осади-
тельной ванны заполняется нагретым газом. Полагают, что при формо-
вании ПА волокон по этому способу из растворов в концентрированной
серной кислоте на поверхности струйки также может образовываться
тонкая пленка полимера вследствие высокой гигроскопичности кислоты
и частичного осаждения полимера в результате взаимодействия раство-
ра с парами воды [29], что также приводит к регулированию диффузи-
онных процессов. В связи с тем, что любая вязкая жидкость при прину-
дительном входе в узкие капилляры накапливает значительную энергию
(так называемую «энергию входа»), при истечении в свободную зону в
струе возникают благоприятные условия для релаксации напряжений.
98
Это особенно важно для анизотропных растворов, когда при прохожде-
нии прядильного раствора через фильеру происходит значительная ори-
ентация жидкокристаллических агрегатов, реализуются большие филь-
ерные вытяжки и при попадании в осадительную ванну только фикси-
руется ненапряженная и уже ориентированная «волоконная заготовка»
[26, с. 93]. По-видимому, всем вышесказанным можно объяснить чрез-
вычайно высокие механические показатели свежесформованных воло-
кон типа ПФТА, которые не подвергались дополнительному вытягива-
нию или термообработке (глава 3). К числу недостатков сухо-мокрого
способа получения термостойких волокон из высококонцентрированных
анизотропных растворов полиамидов следует отнести сложность техно-
логического процесса и его аппаратурного оформления. Ряд технологи-
ческих особенностей сухо-мокрого формования, принципиальных схем,
влияние гидродинамики, воздушной прослойки и других факторов на
стабильность процесса рассмотрен в обзоре [30].
4.1.4. Ориентационное упрочнение волокон
Волокна, формуемые по мокрому методу, вытягиваются непосред-
ственно сразу же после осадительной ванны в присутствии пластифи-
катора [31]. Полиамидные волокна, получаемые по сухому способу,
предварительно отмываются от солей и остатков растворителя, высуши-
ваются и затем вытягиваются при повышенных температурах в одну
или две стадии [32]. Для волокон, формуемых из растворов гибкоцеп-
ных ароматических полиамидов, фильерная вытяжка мало влияет на
механические свойства [26, с. 124]. Вытягивание в среде пластификато-
ра, приводя при правильно подобранном режиме к переходу от изотроп-
ного состояния к ориентированному, решающим образом сказывается
как на прочности волокон, так и на их структурных особенностях
(рис. 3.9). Метод подбора условий пластификационного вытягивания
свежесформованных волокон ПМФИА, основанный на оценке подвиж-
ности структурных элементов гелеобразного волокна, рассмотрен в ра-
боте [33]. Следует отметить, что при пластификационной вытяжке ПА
волокон степень кристалличности практически не изменяется.
Для целого ряда ароматических полиамидов ориентация и кристал-
лизация макромолекул достигаются путем ориентационного вытягива-
ния при повышенных температурах [34]. На рис. 4.6 представлена за-
висимость прочностных свойств некоторых волокон (на основе ПМФИА,
ПФТА, поли-4,4-дифенилоксидтерефталамида и поли-4,4'-дифенилокси-
дизофталамида) от температуры вытяжки [35; 36]. Сложный характер
кривых объясняется, по-видимому, структурными превращениями в во-
локнах при одновременном воздействии температурных и силовых по-
лей. Анализируемые зависимости интересны тем, что они обнаружены
для волокон, полученных на основе полиамидов с различной жест-
костью цепи и различной способностью образовывать анизотропные си-
стемы в растворах. Для всех приведенных образцов характерны более
или менее выраженные бимодальные зависимости прочности волокон от
температуры вытяжки. Полагают, что бимодальный характер зависимо-
сти связан с существованием по крайней мере четырех релаксационных
областей ориентации.
Природа и механизм переходов, наблюдаемых при исследовании
зависимости прочности жесткоцепных волокон от температуры вытяж-
ки, обсуждались ранее (см. гл. 3). Несмотря на одинаковый харак-
7* 99
тер, кривые несколько различаются. Во-первых, экстремальные точки
для различных образцов лежат в различных температурных областях,
что связывают с различиями в кинетической гибкости полимерных це-
пей, которая уменьшается в исследованном ряду: поли-м-фениленизо-
фталамид > поли-4,4-дифенилоксидизофталамид > поли-4,4-дифенил-
оксидтерефталамид>поли-л-фенилентерефталамид (температуры стек-
лования для исследованных полимерных систем составляют соответст-
венно 270—275, 280—310, 320—340 и 340—360°C). Кроме того, для бо-
лее жесткоцепного ПФТА первый максимум выражен менее ярко. Нали-
чие нескольких релаксационных областей процесса высокотемператур-
ной ориентации доказывается с помощью кривых изометрического на-
' t,°C
Рис. 4.6. Зависимость прочности ПА волокон от температуры вытягивания
[35; 36]:
J — поли-4,4-дифенилоксидтерефталамид; 2 — поли-4,4х-дифеиилоксидизофталамид; 3 —
ПМФИА; 4 — ПФТА.
грева и рентгенограммами термообработанных волокон. Рентгенострук-
турный анализ свидетельствует о том, что до высокотемпературной вы-
тяжки волокна имеют аморфную или аморфно-кристаллическую структу-
ру. Ориентация при повышенных температурах приводит к резкой кри-
сталлизации волокон, причем для макромолекул с меньшей кинетиче-
ской подвижностью характерна более сильная дефектность в попереч-
ном направлении. Наблюдается так называемая «одномерная кристал-
личность» [37; 38]. Предполагают, что сильная дефектность в попереч-
ном направлении у жесткоцепных полиамидов обусловлена релаксаци-
онным механизмом структурообразования при ориентации в высоко-
температурном поле. Макромолекулы ароматических полиамидов с вы-
сокой жесткостью и сильным межмолекулярным взаимодействием, как
известно, требуют больших времен релаксации, что несопоставимо с
реальными временами термообработки (максимальная продолжитель-
ность термообработки для различных волокон из ароматических поли-
амидов составляет несколько секунд [32]). При столь кратковременном
контакте с нагретой поверхностью в обрабатываемых волокнах про-
цессы ориентации и структурообразования не могут пройти полностью и
в большинстве случаев возникает напряженная структура, что отра-
жается на кривых изометрического нагрева [35]. Кристаллическая
структура с трехмерным порядком была получена для некоторых воло-
кон из ароматических полиамидов с высокой жесткостью цепи лишь в
100
Таблица 4.1. Влияние условий термообработки сополиамидиых волокон,
содержащих оксанилидные звенья, на их механические свойства [39]
Состав сополнамида, % (мол.) Свойства волокон
исходное вытянутое при 200—250 СС термообра ботанное при 500—550 °C
4.4-диамино- оксаиилид диамин ХФ1 прочность, сН/текс разрывное . удлинение, % i начальный мо- дуль, 103 Па прочность, сН/текс разрывное удлинение, % начальный мо- дуль, МПа прочность, сН/текс . разрывное удлинение, % начальный мо- дуль, 10е Па
0,09 0,09* 0,18 6,6 23,4 50,0 7250 70,0 2,4 49200 103,5 1,8 77500
0,60 0,40* 1,00 4,3 24,3 45,0 10400 — — —- 118 1,4 110000
0,03 0,03** 0,06 3,9 23,4 60,0 — 77,2 4,1 34600 100 6,0 28000
0,65 0,35** 1,00 2,0 15,0 31,0 5950 26,4 3,5 16900 — — •—
* п-Фенилендиамин; *• — 4,4-диаминоднфенилоксид.
условиях длительного высокотемпературного отжига [37]. Резкое влия-
ние термообработки на механическую прочность волокон заметно на
примере получения сополиамидиых волокон, содержащих звенья
4,4-диаминооксанилида [39]. Волокна получали по мокрому способу
из реакционных растворов. Промытые и высушенные волокна подвер-
гали сначала термовытяжке на 60—70% при 200—250°C*, а затем
термообработке в натянутом состоянии при 500—550°C в течение 1,2 с.
В табл. 4.1 представлены данные, показывающие влияние состава со-
полиамида и условий термообработки на свойства волокон. Исходные
волокна имели аморфно-кристаллическую структуру, а термообработан-
ные волокна характеризовались высокой степенью кристалличности.
Волокна из полиамидов типа ПМФИА вытягивают при температу-
рах, лежащих в интервале от 220 °C, вплоть до температур плавления
под определенным натяжением, достаточным для того, чтобы предот-
вратить усадку нитей более чем на 10%. Наиболее эффективные резуль-
таты получаются при непрерывной термообработке. При этом в качест-
ве нагревательных элементов используются металлические или керами-
ческие поверхности, горячий газ, кипящий слой. Полученные волокна
имеют не только повышенные механические характеристики, но и вы-
сокую стабильность размеров [32]. Продолжительность контакта с на-
гревателями— 1—3 с.
При формовании волокон из анизотропных растворов жесткоцеп-
ных полиамидов через газовую прослойку в охлаждаемую осадитель-
ную ванну создаются настолько благоприятные условия для ориента-
ции волокна непосредственно в процессе формования, что дополнитель-
ная вытяжка при повышенных температурах оказывается излишней.
Она может быть применена, как уже указывалось, только для повыше-
ния начального модуля [40, 41].
4.1.5. Физико-механические свойства полиамидных волокон
Анализ технической и патентной литературы показал, что имеется
большое число волокнообразующих полиамидов, из которых различны-
ми способами формования могут быть получены достаточно прочные
* Здесь и далее указывается температура нагревательного элемента, а не волокна.
101
о
Ь5
Таблица 4.2. Свойства высокопрочных высокомодульных волокон из жесткоцепных гомополиамидов [22, 40, 77; 78]*
Химическая формула элементарного эвена Tllg. ДЛ/Г Прочность, сН/тенс Удлинение, % Начальный модуль, 108.Па
-NH— (^>-NH-CQ-(^>-C0- 4,8 206 5,0 62000
-NH-4^-NH-CO-<^S-^\ 3,6 153 4,9 60500
-NH-^Q^-NH-CO—^N\-CO— 5,3 162 5,8 60500
—NH—(O)-NH-CO-<fQ\—co- 3,9 162 6,5 47500
' X21
—NH—NH—CO—CO— 3,1 144 2,5 100000
X2H3
-NH-<^^-NH-CO-<^^—CO— 3,7 189 4,8 82300
\ci -nh-^Q^-co-** 5,o 108—188 4,0—7,0 57600
-NH—CO— 3,9 207 4,8 87500
-NH—(^^-CO-NH-^^—NH-CO-(^)-CO— 4,3 163 3,9 79500
* Растворитель — H2SO4; способ формования — сухо-мокрый.
** Сухое формование из органического растворителя.
Таблица 4.3. Свойства высокопрочных сополиамидных волокон
на основе га-фенилендиамина и дихлорангидрида терефталевой кислоты,
содержащих гибкоцепные звенья третьего компонента [40; 79; 80]
Химическая формула элементарного звена третьего компонента Добавка, % (мол.) 41g. дл/г Проч- ность, сН/текс Удлинение, % Начальный модуль, 10» Па
h2n—(О)-СНг—О-Н2 5,0 3,8 135 4,1 75000
h2N~^O>~cH2^H2~C^~nH2 7,5 3,4 189 3,8 94000
5,0 4,3 216 6,2 67000
H2N—nh2 5,0 4,2 153 5,4 59200
ci— CO—N=N—COCI 5,0 3,4 162 4,6 64300
CI-CO—(^j-COCl 5,0 3,9 180 4,6 74700
CICO—у—COCI (транс-) 25,0— 50,0 3,4— 4,3 162— 198 4,5 70000
C1CO—CH=CH—COCI 40,0 3,3 162 3,7 70200
CICO—(CH2)8—COCI 5,0 2,7 144 4,1 59000
, , z /NH2 50 6,3 275 2,5 143000
NH2-(O/-c\ tot
bl
1 H
NH2—COCI 25 5,0 288 5,4 130000
волокна. Основные физико-механические свойства некоторых поли-
амидных волокон приведены в литературе [16—76].
Приведенные литературные данные [16—76] охватывают круг аро-
матических полиамидных волокон, механическая прочность которых по
современным представлениям является «средней». Достижением послед-
них 5—8 лет можно считать разработку способов получения высоко-
прочных высокомодульных волокон, прочность которых превышает
130—140 сН/текс (и может достигать 288 сН/текс). Волокна с такой
прочностью были получены преимущественно на основе предельно жест-
коцепных полиамидов. В табл. 4.2 представлены данные, характеризую-
щие механические свойства высокопрочных высокомодульных волокон,
полученных на основе некоторых гомополиамидов.
1ЛО
Специфика жесткоцепных полиамидов, из которых могут быть по-
лучены высокопрочные высокомодульные волокна, заключается также
в том, что при введении в макромолекулы указанных ПА других звень-
ев с более или менее высокой гибкостью прочность получаемых воло-
кон не снижается. При этом образуются статистические сополиамиды,
обладающие хорошей волокнообразующей способностью (табл. 4.3).
В связи с недостатками способа формования волокон из растворов
в H2SO4 (высокая вязкость прядильных растворов ПА, агрессивность
H2SO4) заслуживают внимания предложения по получению высокопроч-
ных высокомодульных волокон из полимеров «-структуры, содержащих
наряду с амидными гидразидные связи [64; 81]. Полимеры или сополи-
меры такого типа оказываются хорошо растворимыми в ДМСО. Фир-
мой «Монсанто» разработана технология получения высокопрочного
высокомодульного волокна типа «Х-500» [82]. Интересны высокопроч-
ные сополимерные полиамидные волокна, содержащие хиназолиндионо-
вые, хиназолоновые или гидантоиновые циклы. Волокна формуют по
сухому или мокрому способам из органических растворителей. Проч-
ность волокон достигает 180 сН/текс [84].
Из большого числа различных полиамидных волокон практическое
применение нашли лишь некоторые. К ним относятся: номекс, конэкс и
фенилон, получаемые на основе ПМФИА; сульфон-Т, формуемый из
растворов ароматического полисульфонамида «-структуры и волокно
типа ЗМП, разработанное фирмой «Монсанто» [85]. Имеются также
сообщения о создании фирмой «Дюпон» опытно-промышленного произ-
водства полиамидного волокна под названием НТ-4. Предполагается,
что это волокно является модифицированным ПФТА [86]. Из высоко-
прочных необходимо назвать волокно кевлар. Свойства указанных во-
локон приведены в табл. 4.4.
Таблица 4.4. Свойства некоторых волокон из ароматических ПА
Свойство Номекс [7] фенилон [87] Сульфон-Т [12] НТ-4 [88] Кевлар [89]
Ассортимент Волокно, нить Волокно, нить Волокно, нить Волокно, нить Нить
Прочность, сН/текс Удлинение, % Начальный модуль, МПа 50 15—20 12300 45—50 15—20 13000 35—40 16—18 6000 30—40 6—8 16000 225 3—5 68500—128000
Прочность (% от исходной) 90 90 65 80 49
в узле в петле в мокром состоянии Плотность, кг/м3 95 75 1380 95 80 1380 75 80 1450 80 75 1480 78 1450 1,5—2,0
Равновесное влагопоглоще- 4—5 4—5 5—6 4,0
ние, % Устойчивость одиночного во- 6-Ю5 5 105 1,2-10® — 6 10*
локна к двойным изгибам (чис- ло циклов до разрыва) 1200
Устойчивость к нстнранню (число циклов до разрыва) —
Усадка, %: в воде на воздухе при 300 °C Кислородный индекс, % 0,5 28—29 1,5 5—6 28—29 1,0 0,5 28—32 1,о 0 35—40 0,5 0,5 28—30
Окрашиваемость Хорошая Хорошая Хорошая Хорошая
iru
105
Таблица 4.5. Теплостойкость некоторых волокон из ароматических полиамидов
Химическая формула элементарного звена Прочность (% от исходной) при температурах Литература
200 °C 250 °C 300 °C 350 °C 400 °C 450 °C 500 °C
—NH—NH—со—со— —NH—СО-(^>-СО- -NH-^-SO.-^-NH-CO-^-CO- 90 80 83 65 65 70 40 54—60 50 15 38 7 40 33 25 16 16 [91, С. 270] [66;'92[ [12]
-nh-©>_o_0_nh~co-©“co_ о 85 70 54 35 — —< — [37]
У ©-NH-CO_©-CO- N N 0 64 47 34 26 23 22 7-8 [67]
—NH—С7" NH—CO—CO— и ii KJJ N N N N 57 40 37 20 8 3 — [67]
-NH-<^>-C/ 'c-^CS.-C// XC-(^-NH-CO-r^-CO- HC S s CH 4/ 62 '— 47—50 29 19 17 — [67; 68]
Волокна из ароматических ПА обладают хорошими электроизоля-
ционными свойствами, стабильностью размеров, укрывистостью, высо-
кой стойкостью к действию химических реагентов кислого характера и
органических растворителей. Радиационная стойкость волокон из аро-
матических ПА превышает стойкость обычных синтетических волокон
[87]. Неожиданным оказалась сравнительно низкая стойкость ПА во-
локон к действию УФ-облучения (рис. 4.7). Более подробно физико-хи-
мические свойства ароматических ПА рассмотрены в монографии [2].
4.1.6. Термомеханические свойства полиамидных волокон
Ароматические полиамиды явились первым классом полимеров, на
основе которых в промышленном масштабе были получены термостойкие
волокна, способные выдерживать действие повышенных температур (до
300—350°C). Важнейшими характеристиками, определяющими возмож-
ность применения указанных волокон при повышенных температурах,
являются их тепло- и термостабильность.
В табл. 4.5 приведены данные, характеризующие теплостойкость не-
которых волокон из ароматических полиамидов. Прочность волокон по
мере повышения температуры испытания уменьшается. При 300°C прак-
тически все волокна, за исключением волокон из ПФТА, теряют полови-
ну (или более) исходной прочности. Правда, ряд волокон сохраняет
прочность до температур порядка 500 °C, но при столь высоких темпера-
турах их термическая стабильность оказывается недостаточной. Указы-
вается, что для повышения теплостойкости и стабильности размеров ПА
волокна необходимо в течение 0,5—-5,0 с обрабатывать при температуре
выше их температуры стеклования [32]. Сделана попытка повысить
теплостойкость волокон на основе 4,4/-диаминобензофенона и терефта-
левой кислоты путем дополнительного сшивания диаминами в присут-
ствии кислот Льюиса {59]. Теплостойкость при 300 °C удалось повысить
почти в два раза. Для других волокон аналогичные работы не прово-
дились.
Термостабильность—более важный показатель волокон, предна-
значенных для эксплуатации при повышенных температурах. В ряде
работ приведены сведения, характеризующие основной показатель,
предъявленный к термостойким волокнам, — их способность сохранять
комплекс физико-механических свойств и в первую очередь прочность,
Рис. 4.7. Зависимость прочности
ПА волокна от среды и продолжи-
тельности УФ-облучения [90; 104]:
1 — облучение на воздухе; 2 — облуче-
ние в среде азота.
при длительном воздействии повышенных
температур. Некоторые данные суммиро-
ваны в табл. 4.6.
В таблице приведена термостойкость
как чисто ароматических полиамидных
волокон, так и полиамидных волокон,
содержащих в основной цепи макромоле-
кул шарнирные мостики типа —SO2— и
—О—, а также гетероциклические груп-
пировки. Как видно из представленных
в таблице данных, в области температур
до 250 °C практически все ПА волокна
оказываются довольно устойчивыми к
термоокислительной деструкции. При пе-
реходе к температурам 300—350 °C тер-
106
мостабильность волокон резко падает. Наиболее термостабильными
оказываются полиамидные волокна с гетероциклическими группиров-
ками, не содержащими атомов водорода. Полагают, что при замене
части амидных группировок на различные гетероциклы, не содержа-
щие атомов водорода или других, легко отщепляющихся групп атомов,
термостабильность ПА волокон будет увеличиваться. В качестве при-
мера можно привести волокно на основе полиоксадиазоламида [67].
В области 400 °C практически ни одно полиамидное волокно нерабо-
тоспособно длительное время. Для сравнения в таблице приведена
термостабильность волокон на основе ароматического полиоксадиа-
зола [91]. Видно что ПОД волокно в области температур до 350°C
является более термостабильным, чем волокна на основе ароматиче-
ских полиамидов. Кроме того, следует отметить, что падение механи-
ческой прочности волокон в результате теплового воздействия начи-
нается при температурах, далеких от температур начала интенсивной
термоокислительной деструкции. Существует мнение [2, с. 183], что
предельные рабочие температуры изделий из ароматических полиами-
дов, в том числе и волокон, определяются их тепло- и термостой-
костью. При этом для полиамидов с относительно низкими температура-
ми стеклования предельная температура эксплуатации ограничивается
температурой стеклования (физический фактор), а для поли-
амидов с высокими температурами стеклования — устойчивостью хими-
ческих связей. Поэтому высказывается мысль, что нетермостабилизи-
рованные волокна на основе ароматических полиамидов .и-структуры
могут применяться в изделиях, длительно работающих при температу-
рах, не превышающих 250 °C [94]. Волокна из полиамидов «-структу-
ры, а также из полиамидов с устойчивыми гетероциклическими
группировками способны к эксплуатации в течение нескольких сотен
часов при 250—300 °C. Для условий длительной эксплуатации при тем-
пературах выше 300 °C волокна из ароматических полиамидов, по-ви-
димому, не могут быть рекомендованы.
4.1.7. Термо- и светостабилизация полиамидных волокон
Термостабилизация. Волокна из ароматических полиамидов, на-
ходясь в условиях воздействия температурных полей, постепенно изме-
няют свои механические характеристики, причем процессы деструкции,
снижающие механические свойства волокон, протекают в температур-
ной области, весьма далекой от температурных областей плавления
или разложения исходных полимеров. Для ароматических полиамидов
наиболее вероятными процессами, протекающими под действием тепла
и кислорода воздуха, могут быть реакции гидролиза, окисления, струк-
турирования и гомолитического распада. Замечено [26, с. 155], что
тип реакции влияет на изменение свойств изделия. Так, например,
разрыв макромолекулярной цепи приводит к потере прочности и элас-
тических свойств волокна, тогда как сшивка макромолекулярных
цепей в меньшей мере сказывается на изменении прочности. Обнару-
жено также, что чем выше температура начала термического разложе-
ния ароматических ПА, тем выше термостойкость волокна на их осно-
ве, однако прямая корреляция между этими величинами отсутст-
вует [95].
Ряд полиамидов хорошо выдерживает термообработку в вакууме,
а при нагревании на воздухе их термостабильность оказывается крайне
107
Таблица 4.6. Термостабильность волокон из ароматических полиамидов
Химическая формула элементарного звена
Прочность волокна после экспозиции на
воздухе, % от исходной
250 °C (100 ч) 300 °C (100 ч) 350 °C (50 ч) 400 °C (50 ч)
Литература
78 50—60 Разрушается — [91]
— 70 45 — [92]
90 80 55 15 [12]
80 10—15 Разрушается — [37]
— 65 20 — [37]
96 90 50 15 [67]
00
CJ3
о
СО
О
ю
О
со
109
низкой, что, например, хорошо иллюстрируется на примере волокон на
основе поли-4,4'-дифенилоксидтерефталамида [37]. Полагают, что в
гетероцепных ПА волокнах кроме реакций гомолитического распада
возможны и гетеролитические реакции, поскольку связь углерод — гете-
роатом является полярной и подвергается действию агрессивных сред
в первую очередь [96]. Таким образом, стабилизация должна сво-
диться не только к образованию более прочных связей (химических,
меж молекулярных и т. д.) в структуре волокна, но и связей, устойчи-
вых в гидролитических средах. Термостабилизации препятствуют при-
меси в ПА волокнах, подвижный атом водорода в амидной группе, кон-
цевые карбоксильные или аминогруппы [96]. Можно констатировать,
что, несмотря на значительные успехи в области получения волокон из
неплавких термостойких полиамидов, ПА волокон, способных дли-
тельно работать в области температур 300 °C и выше, практически нет.
В связи с этим большое значение приобретает проблема их термоста-
билизации. На первый план выдвигается стабилизация волокон против
термоокислительной деструкции. Однако и здесь не удалось добиться
желаемых результатов. В первую очередь это связано с тем, чтоболь-
шинство известных термостабилизаторов сами оказываются неработо-
способны при температурах выше 250—300 °C, что только ускоряет
процессы деструкции волокон. Механизм деструкционных процессов в
таких ориентированных напряженных системах, какими являются
волокна на основе жесткоцепных полиамидов, неясен. Все это вызвало
необходимость решать задачу пока чисто эмпирически. Показано, что
в ряде случаев введение галогена в цепь полиамидов несколько повы-
шает термостабильность последних [97]. Предполагают, что системы,
содержащие группировки диоксибензофенона, будут обладать способ-
ностью «самозалечивания» [98]. Обнаружено, что добавки в качестве
антиоксиданта 0,5—10% (от массы полимера) карбоната никеля [99]
к волокну номекс повышают его термостабильность. К такому же ре-
зультату приводит обработка волокон типа ПМФИА соединениями мо-
либдена в присутствии галогенов, окисями висмута, марганца или алю-
миния [100]. Для тканей на основе ПМФИА некоторый эффект дает
обработка их растворами или дисперсиями акриловой кислоты с
последующей термообработкой при температурах до 180 °C в присут-
ствии катализаторов типа щавелевой кислоты. Имеются рекомендации
по кратковременной высокотемпературной обработке волокон номекс
при 360—400 °C с целью повышения их термостабильности и гидроли-
тической устойчивости [101]. Для повышения термостойкости волокон
на основе поли-4,4'-дифенил-сульфонтерефталамида или ПМФИА
предлагается их структурно-физическая модификация путем формова-
ния из растворов смесей термодинамически несовместимых полимеров.
Это направление является технологически приемлемым и, по-видимому,
найдет распространение [102]. Считается, что термостабильность суще-
ствующих волокон можно повысить путем более тщательной очистки от
низкомолекулярных примесей [26, с. 155], однако экспериментальных
данных пока нет.
Светостабилизация. Исследование механизма реакций, протекаю-
щих в ароматических полиамидах под действием света, позволило уста-
новить, что фотолиз соответствующих полимеров сопровождается рас-
щеплением амидной связи и образованием бензоильного радикала и
радикала анилина [103]. Фотодеструкция полиамидных волокон, прохо-
дящая на воздухе, в инертной атмосфере, а также в присутствии паров
ПО
воды [90, 104] свидетельствует об ускорении процесса распада макро-
молекулярных цепей в активной (по отношению к полимеру) среде
(см. рис. 4.7). Увеличение плотности упаковки структуры в результате
ориентационной вытяжки полиамидных волокон несколько замедляет
процесс фотодеструкции, однако применение светостабилизаторов ока-
зывается более эффективным [103]. При выборе светостабилизато-
ров обычно учитывается, что они должны обладать максимально
высоким коэффициентом поглощения света в области длин волн по-
рядка 290—400 нм. Обязательным условием являются также химиче-
ская и термическая стабильность и малая летучесть светопоглотите-
лей. Установлено, что хими-
ческая связь стабилизатора
с полимером может значи-
тельно повысить устойчи-
вость волокон из ароматиче-
ских ПА к действию света.
При этом часть энергии, по-
глощаемой полимером, лег-
ко передается на светостаби-
лизатор [ЮЗ]. К такому же
эффекту приводит наличие
водородных связей между
полиамидом и стабилизато-
ром. Менее действенными
оказываются вещества, за-
щитный механизм которых
основан на экранировании
амидных связей полимера.
Наиболее эффективными
светостабилизаторами ПА
Рис. 4.8. Влияние добавок стабилизатора на изме-
нение относительной вязкости ПМФИА при УФ-
облучении [103; 104]:
/ — исходный полимер: 2 и 3 — полимер, содержащий со-
ответственно 1 % (масс.) 2(2'-окси-5-трет-бутилфенил)-
бензтриазол-карбанилида и 3% (масс.).
волокон являются некото-
рые аминофенолы, замещенные дибензофеноны, триазины и бензтриа-
золы. Например, добавки 2-(2/-окси-5/-т’рет’-бутилфенил) бензтриазол-
карбанилида к ПМФИА замедляет изменение молекулярной массы
полимера при УФ-облучении (рис. 4.8). При введении в цепь того же
полимера некоторых аминофенолов светостойкость волокна номекс
увеличивается вдвое [105]. Обработка водными дисперсиями производ-
ных бензтриазолов волокон номекс и кевлар позволяет использовать
их длительное время в условиях прямого солнечного облучения [106].
Технологически удобнее вводить добавки светостабилизаторов в пря-
дильные растворы полиамидов, при этом процесс получения волокон
практически не изменяется [86]. Несмотря на то, что вопросам светоста-
билизации волокон на основе полностью ароматических полиамидов
уделяется сравнительно мало внимания, считают, что перспективы в этом
направлении весьма благоприятны. По-видимому, уже в ближайшее
время можно ожидать появления светостабилизаторов, эффективных по
отношению к волокнам из ароматических полиамидов [103].
Заканчивая рассмотрение способов получения и свойств во-
локон на основе ароматических ПА, необходимо подчеркнуть следую-
щее.
1. Этот класс полимеров и волокон является наиболее изученным
по сравнению с другими классами термостойких волокнообразующих
полимеров.
ill
2. Из ароматических полиамидов были впервые в промышленном
масштабе получены волокна, отличающиеся повышенными термически-
ми свойствами по сравнению с известными синтетическими волокнами,
а также высокопрочные высокомодульные волокна.
3. В теоретическом отношении жесткоцепные ароматические по-
лиамиды являются чрезвычайно интересными объектами, и их исследо-
вание продолжает интенсивно развиваться.
4.2. ПОЛИИМИДНЫЕ ВОЛОКНА
Полиимидные волокна относятся к волокнам, получаемым по двух-
стадийному методу: на первой стадии получают форполимер в виде
полиамидокислоты, которая затем в готовом изделии в результате
реакции внутримолекулярной полициклизации превращается в поли-
R = О, S, СН2, СО
Процесс получения полиимидных волокон включает несколько
стадий:
1) подготовку исходных мономеров и растворителя;
2) синтез полиамидокислоты в среде амидного растворителя;
3) фильтрацию и обезвоздушивание раствора полиамидокислоты;
4) формование волокна;
5) имидизацию;
6) дополнительную вытяжку полиимидного волокна при повышен-
ных температурах.
4. 2.1.Получение растворов полимеров
Подготовка исходных мономеров и растворителей. Основными
исходными мономерами, применяемыми в производстве полиимидных
волокон, являются, как указывалось выше, диангидриды ароматиче-
ских тетракислот и ароматические диамины.
112
Основное требование к исходным продуктам и растворителю —
высокая степень чистоты. Наибольшее распространение из исходных
продуктов получили диангидрид пиромеллитовой кислоты и 4,4'-диа-
минодифениловый эфир. Подготовка исходных продуктов заключается
в их сушке при 80—230 °C в вакууме с целью удаления воды (из
диамина) и дополнительной циклизации пиромеллитовой кислотЬг
Амидные растворители очищаются путем вакуумной перегонки или
ректификации.
Синтез полиамидокислот. Синтез полиамидокислот проводится в
среде полярных растворителей. Несмотря на большое число работ,
посвященных синтезу полиамидокислот, вопрос о влиянии условий про-
ведения процесса синтеза на молекулярную массу конечного продукта
изучен недостаточно. Как и при синтезе полностью ароматических
полиамидов, технологические приемы проведения процесса, а именно:
способ введения компонентов в реакцию, соотношение диангидрид —
диамин, температура реакции, тип применяемого растворителя, кон-
центрация реагентов в реакционной среде и т. д., оказывают решаю-
щее влияние на молекулярную массу образующейся полиамидокисло-
ты, а также на ее стабильность [107]. Отмечается, что одним из воз-
можных способов регулирования молекулярной массы полиамидокис-
лоты в процессе ее синтеза является способ проведения реакции
ацилирования (табл. 4.7).
Из данных таблицы видно, что независимо от типа применяемого
растворителя полиамидокислота с достаточно высокой удельной вяз-
костью получается только при порционном дозировании твердого
диангидрида пиромеллитовой кислоты в раствор диамина. Ароматиче-
ские диамины, как правило, хорошо растворимы в амидных раствори-
телях, и этот способ получения высокомолекулярных волокнообра-
зующих полиамидокислот различного химического строения является
наиболее распространенным. Наличие влаги в исходном растворителе
или в смеси растворителя с диамином, согласно одним данным [107],
приводит к понижению молекулярной массы полиамидокислоты
(рис. 4.9); согласно другим данным [108], влага оказывает каталити-
ческое влияние на процесс образования полиамидокислоты.
При использовании высокочистых исходных продуктов и раствори-
телей, не содержащих влаги и реакционноспособных примесей типа
диметиламина, высокомолекулярные волокнообразующие полиамидо-
Таблица 4.7. Влияние способа введения пиромеллитового диангидрида
и 4,4 -диаминодифенилоксида в реакцию на удельную вязкость полиамидокислоты
в различных растворителях [107]
Способ проведения реакции т4уд полнамндокислоты в различных растворителях
ДМФ ДМАА мп
Сливание растворов диангидрнда и диамина 0,08 0,17 0,12
Введение смесн твердых диамина и диангндрида 0,19 0,38 0,34
в растворитель Введение растворителя в смесь твердых днамнна и 0,28 0,37 0,40
диангидрида Введение твердого днангидрида в раствор диамина 1,30 1,62 1,84
8—2254
113
кислоты получают при эквимольном соотношении диангидрида и ди-
амина. Однако в связи с тем, что на практике трудно осуществить ука-
занные условия, наличие всевозможных примесей, способных обрывать
полимерную цепь и реагировать с исходными веществами, сдвигает
Рис. 4.9. Зависимость удельной вязкости поли-
амидокислоты на основе пиромеллитового диан-
гидрида и 4,4-диаминодифенилового эфира от
содержания влаги С в реакционной среде [107]:
1 —ДМФ; 2 —ДМАА; 3 —МП.
соотношение диангидрид: ди-
амин в сторону некоторого из-
бытка диангидрида. Например,
для случая взаимодействия пи-
ромеллитового днангидрнда и
4,4'-диаминодифенилового эфи-
ра в реальных условиях синте-
за волокнообразующей поли-
амндокислоты избыток диан-
гидрида составляет 0,2—0,5%
(мол.) (рис. 4.10). Полнамн-
докислоты в зависимости от
характера применяемого рас-
творителя могут быть получе-
ны в широком интервале тем-
ператур реакционной смеси,,
причем наблюдается экстре-
мальная зависимость молеку-
лярной массы образующегося
полимера от температуры реак-
ции (рис. 4.11). Следует отме-
тить, что прн получении полн-
амндокислоты в ДМАА и МП
молекулярная масса синтезиру-
емого продукта в меньшей степени зави-
сит от температуры, чем при проведении
синтеза в ДМФ. Было обнаружено, что
повышение температуры выше 30 °C
приводит к заметному понижению моле-
кулярной массы полиамндокнслоты, а
выше 100 °C становится преобладающей
реакция внутримолекулярной циклоде-
гидратации, сопровождающаяся выпаде-
нием полимера из раствора [ПО].
Для практического использования
полнамидокислот большое значение име-
ет не только молекулярная масса поли-
мера, но и общая концентрация раствора
полиамидокислоты. Зависимость удель-
ной вязкости полиамидокислоты от кон-
I- । I g,51 । । , ।
г, в 1,0 0,6 0,2 0,2 0,6 1,0 19
ДЙ<РЭ, % (мол) пмдд, % (мол)
центрации исходных реагентов также
имеет экстремальный характер (рис.
4.12). В зависимости от типа применяе-
Рис. 4.10. Зависимость удельной
вязкости полиамидокислоты от со-
отношения диангидрида и диамина
мого растворителя могут быть получены в реакционной среде [107]:
высоковязкие растворы полиамидокис- г —дмф; 2-дмаа; з-мп.
лот, пригодные для формования волокон
и имеющие концентрацию полимера до 25% (масс.).
Порционное прибавление твердого диангидрида пиромеллитовой
кислоты к раствору диамина в амидном растворителе, несмотря на
114
то, что при этом образуются высокомолекулярные продукты реакции,
имеет ряд недостатков. Часть из них связана с тем, что при таком
способе синтеза не всегда удается хорошо гомогенизировать реакцион-
ную смесь и трудно выдержать температурные условия реакции из-за
локальных перегревов в результате экзотермичности процесса и т. д.
Поэтому для улучшения условий проведения процесса предлагается
предварительно диспергировать исходные диамины и диангидриды
Рис. 4.11. Зависимость удельной вязкости полиамидокисдоты в различных
реакционных средах от температуры реакции [107]:
1 — ДМФ; 2 —ДМАА; 3 — МП.
Рис. 4.12 Зависимость удельной вязкости полиамидокислоты от концентра-
ции исходных веществ в реакционной среде [Ю7]:
/ — ДМФ; 2 —ДМАА; 3 — МП.
кислот в инертном органическом растворителе (бензине, бензоле, то7
луоле и др.); затем при контролируемой температуре в дисперсию мо*
номеров медленно вводить полярный растворитель [111]. Скорость
реакции полимерообразования увеличивается в 3—5 раз по сравнению
с обычным способом синтеза. Получаемые сиропы пригодны для формо-
вания волокон по сухому методу.
Некоторые закономерности синтеза высокомолекулярной полиами-
докислоты на основе пиромеллитового диаигидрида и 4,4'-диаминоди-
фенилового эфира, описанные выше, характерны также для процесса
получения полиамидокислот на основе других диангидридов тетракар-
боновых кислот и диаминов; более подробно эти процессы рассмот-
рены в монографии [112].
Стабильность растворов полиамидокислот. Характерной особен^
ностью растворов полиамидокислот является их нестабильность: удель-
ная вязкость растворов уменьшается во времени (рис. 4.13) незави>
симо от типа применяемого растворителя. При этом > изменяются
8*
116
эффективная вязкость растворов и молекулярная масса полиамидо-
кислоты.
Повышение температуры растворов приводит к более заметному
падению удельной вязкости полиамидокислоты. Причины, вызываю-
щие изменёйие вязкости растворов, окончательно не установлены. Ряд
авторов считает, что основной причиной снижения молекулярной массы
полиаМидокйслоты является гидролиз амидной связи полимера [113].
Другие авторы полагают, что большая роль в этом процессе принадле-
жит структурным превращениям [114]. Тем не менее всеми авторами
подчеркивается необходимость хранения прядильных растворов поли-
амидокислот при температурах,
ЗД* близких к О °C, в условиях, ис-
Рис. 4.13. |Кинетика изменения удельной
вязкости 1лолиамидожислоты иа основе пиро-
меллитового диангидрида и 4,4'-диаминоди-
фенилового эфира в различных растворите-
лях [107]:
1 и 2 —ДМФ; 3 и 4 — ДМАА; 5 и б — МП
(—при 4 °C;-------------при 20 °C).
ключающих контакт раствора с
влагой воздуха.
Заметное уменьшение моле-
кулярной массы полиамидокислот
наблюдается и при переосажде-
нии их как в воду, так и в инерт-
ные растворители [115].
Фильтрация и обезвоздуши-
вание прядильных растворов по-
лиамидокислот. Для формования
волокон используются реакцион-
ные растворы полиамидокислот.
Подготовка прядильных раство-
ров к формованию затруднений
не вызывает. Желательным яв-
ляется проведение стадий фильт-
рации и обезвоздушивания при температурах не выше 10 °C. Для
проведения этих стадий используется обычное оборудование, применя-
емое в производстве химических волокон, формуемых из растворов.
4. 2.2. Получение волокон
Как уже указывалось, полиимидные волокна получаются по двух-
стадийному способу: сначала из раствора полиамидокислоты формуют
волокйа, которые затем путем имидизации превращаются в полиимид-
ные, отличающиеся высокой термической стабильностью. Волокна на
основе полиамидокислот различного химического строения могут быть
получены по мокрому или сухому способам формования. В качестве
растворителей применяют амидные растворители или ДМСО.
Мокрое формование полиамидокислотных волокон. Процесс полу-
чения полиамидокислотных волокон по способу мокрого формования
включает стадию волокнообразования, предварительную пластифика-
ционную вытяжку свежесформованной нити на воздухе или в пласти-
фикацйонных средах, отмывку волокна от растворителя и компонентов
осадительной ванны и сушку. В качестве осадительных ванн приме-
няют смесь растворителя с водой либо инертные осадители типа спир-
тов или ароматических углеводородов. Применяют и многокомпо-
нентные осадительные ванны, состоящие из воды, растворителя и не-
которых солей типа роданидов щелочных металлов [116]. Свежесфор-
мОваиные волокна имеют невысокие физико-механические характери-
116
стики, пластификационная вытяжка приводит к повышению прочности
до 20—22 сН/текс [117—119]. Высушенное волокно подвергается ими-
дизации.
Сухое формование полиамидокислотных волокон. При формова-
нии полиамидокислотных волокон по сухому методу концентрация
раствора форполимера в амидных растворителях составляет 15—
25% (масс). Важной характеристикой при этом способе формования
является эффективная вязкость раствора. Оптимальной считают вяз-
кость 180—200 Па-с, измеренную при.30°C [120]. Характеристическая
вязкость используемых полиамидокислот составляет 1,4—3,4. Отмеча-
ется, что волокна, получаемые по сухому способу, имеют лучшие
эластические характеристики и повышенную термическую стабильность
по сравнению с волокнами, полученными по мокрому способу. Темпе-
ратура прядильного раствора, подаваемого на формование, не должна
превышать 80 °C. Температура шахты —200—210О|С; температура
инертного газа, подаваемого прямотоком, составляет 265—280 °C [120].
Так, например, при формовании волокна из 25%-ного раствора
полиамидокислоты на основе пиромеллитовой кислоты и 4,4'-диамино-
дифенилового эфира в ДМАА, имеющей характеристическую вязкость
1,3, выдерживаются следующие параметры процесса: температура
прядильного раствора — 65 °C; температура азота по длине шахты —
200—-280 °C; скорость формования—150 м/мин. По-видимому, при
прохождении струйки прядильного раствора и свежесформованного
волокна в шахте происходит частичная имидизация полиамидокис-
лоты.
Свежесформованные волокна содержат от 20 до 35% растворите-
ля, что обусловливает необходимость дополнительной отмывки
волокна, так как оставшийся в волокне растворитель оказывает су-
щественное влияние на последующие превращения полиамидокислоты
в полиимид, а также на механические характеристики получаемых
волокон.
Имидизация и дополнительная термическая вытяжка волокон.
Необходимой стадией процесса получения полиимидных волокон яв-
ляется перевод полиамидокислоты в полиимид. Этот, процесс может
быть осуществлен воздействием на полиамидокислотное волокно либо
дегидратирующими агентами в присутствии катализаторов (химический
способ), либо термообработкой высушенных полиамидокислотных воло-
кон в инертной среде, на воздухе или в вакууме (термический способ}
[109; 121—123]. Химический способ имидизации в технологическом от-
ношении оказывается менее удобным по сравнению с термическим, так
как при этом возникает необходимость применения больших количеств
смесей циклодегидратирующих агентов и катализаторов, волокно после
обработки смесью реагентов должно быть отмыто от указанных ком-
понентов и высушено перед стадией дополнительной термической вы-
тяжки [121].
Влияние некоторых технологических параметров формования по-
лиамидокислотных волокон на кинетику термической циклизации по-
лиимидного волокна изучено [117] методом динамической термограви-
метрии. Была использована полиамидокислота на основе пиромелли-
тового диангидрида и 4,4'-диаминодифенилсульфида, полученная Я
среде ДМФ. Волокна формовали по мокрому способу в ванну, содер*
жащую ДМФ, воду и роданид кальция. Типичные кривые, характери-
зующие процесс термоциклизации волокна, приведены на рис. 4.14.
117
Рис. 4.14. Типичные кривые, характеризую-
щие процесс термоциклизации полиамидо-
кислотного волокна [117]:
1 — термогравитограмма; 2 — дериватограмма.
Объяснения см. в тексте.
Влияние условий формования на кинетику процесса имидизации
полиамидокислотного волокна оценивалось путем анализа некоторых
характерных точек на дериватограммах (см. рис. 4.14): Vm — макси-
мальной скорости; Тмакс — температуры максимальной скорости цикли-
зации; dW уменьшения в массы волокна в результате «чистой»
циклизации.
Заметным оказывается влияние фильерной и пластификационной
вытяжек свежесформованного волокна на кинетические особенности
процесса имидизации (табл. 4.8).
Увеличение фильерной вы-
тяжки ведет к понижению мак-
симальной скорости имидизации
и количества выделяющейся во-
ды. Возможно, что это чисто ки-
нетический фактор, и при дли-
тельном прогреве волокон степень
имидизации будет довольно высо-
кой. Высказывается также пред-
положение, что наблюдаемый
факт является результатом попе-
речной контракции волокна и за-
трудненности процесса диффузии
воды из внутренних слоев нити.
Не совсем ясны и причины неко-
торого понижения Тмакс по мере увеличения степени пластификацион-
ной вытяжки. Возможно, что это явление связано с уменьшением тем-
пературы стеклования полимера вследствие пластифицирующего дейст-
вия остающегося в свежесформованном волокне растворителя, который
при вытягивании может задерживаться в порах и не удаляется при
промывке.
Влияние состава осадительной ванны на кинетические характери-
стики процесса имидизации менее заметно (табл. 4.9), однако н здесь
проявляется влияние плотности структуры исходных волокон.
По-видимому, волокна, сформованные в осадительную ванну, со-
держащую 35% (масс.) воды, имеют более рыхлую структуру, пони-
женную плотность, и поэтому скорость внутримолекулярной цнклоде-
гидратации оказывается наивысшей.
Таблица 4.8. Влияние фильерной и пластификационной вытяжек
на показатели дериватограмм полиамидокислотных волокон [117]
Степень вытяжки, % Температура осадительной ванны, °C Показатели дериватограмм
фильерной пластифика- ционной Т °C 1 макс’ и m %/мин dW, %
—50 0 20 230 0,34 10,1
0 0 20 237 0,34 9,2
200 0 20 220 0,23 3,0
300 0 20 190 0,19 4,0
40 60 10 210 0,18 6,5
40 120 20 210 0,23 5,4
40 200 20 200 0,16 3,9
О Максимальная 20 230 0,22 3,9
118
Таблица 4.9. Влияние состава осадительной ванны на показатели
дериватограмм имидизации волокна
Состав осадительной ванны, % (масс.) Плотность волокна, кг/мЗ Показатели дериватограмм
Ca(CNS)« ДМФ н2о Т °C макс’ vm, %/мин <№. %
50 25 25 1441 210 0,18 3,4
50 10 40 1439 210 0,18 3,8
30 35 35 1436 190 0,19 4,0
25 50 25 1438 210 0,17 2,3
Промывка свежесформованного волокна также влияет на процесс
имидизации (табл. 4.10).
Т а б л и ц а 4.10. Влияние условий промывки свежесформованного полиамидокислотного
волокна на показатели дериватограмм термической имидизации
Условия промывки Показатели дериватограмм
среда температура, °C 7 °г 1 макс’ ь Vm. %/мин dW, %
Спирт 60 230 0,32 7,5
Ацетон 65 220 0,30 4,7
Вода 80 215 0,16 2,3
60 150 0,11 3,2
45 155 0,21 3,0
Предполагают [117], что промывка волокна водой создает более
«мягкие» условия для термоимидизации.
Наибольшее влияние на термическую циклизацию полиамидокис-
лотных волокон оказывает предварительная химическая циклизация.
Замечено, что если обработать полиамидокислотные волокна смесью
уксусного ангидрида и пиридина, то скорость термоциклизации и коли-
чество выделившейся при этом воды с увеличением продолжительно-
сти химической обработки волокна резко уменьшаются (табл. 4.11).
Полиамидокислотные волокна, полученные по способу сухого фор-
мования, подвергают термо- или химической циклизации, а затем
после отмывки и сушки вытягивают при повышенных температурах.
Указывается [117] на положительную роль остающегося в волокне
растворителя, создающего пластифицирующий эффект. При добавле-
Таблица 4.11. Влияние предварительной химической циклизации
полиамидокислотного волокна на показатели дериватограмм
термической имидизации [117]
Продолжительность химической обработки, ч Содержание имидных циклов, % Показатели дериватограмм
7 °Г 2 макс’ ь %/мин dW. %
1 52 200—210 0,09 2,0
3 62 160—180 0,08 1,7
. 6 64 270 0,03 1,3
12 74 290 0,03 1,2
24 81 330 0,01 0,2
119
нии некоторых пятичленных гетероциклов, таких, как N-формилимид,
N-бензоилбензимидазол, к прядильному раствору раствор полиами-
докислоты стабилизируется, облегчается формование волокон по
сухому способу и ускоряется процесс имидизации волокон. Для окон-
чательной циклизации волокна прогревают при 300°C в течение
30 мин [124]. Быстрая циклизация полиамидокислоты (в течение не-
скольких минут) достигается при добавлении в прядильный раствор
мономерных гетероциклических кислот, содержащих третичный атом
азота, таких, как никотиновая, изоникотиновая и т. д. Прядильные
растворы стабильны в течение нескольких часов при повышенной тем-
пературе [125].
Химическая циклодегидратация, хотя и приводит к созданию в
макромолекуле имидной структуры, практически не отражается на
физико-механических характеристиках волокон; при термической цикло-
дегидратации в значительной степени изменяются как физико-, так и
термомеханические характеристики нитей (табл. 4.12).
Таблица 4.12. Физико-механические свойства некоторых полиимидных волокон,
циклизованных химическим и термическим способами [128]
Показатель ПМ ПМС ПФГ
I и I 11 » 1 и
Линейная инти, текс плотность 35,6 29,0 35,6 29,0 35,6 29,0
Прочность, сН/текс 27—30 45—50 18—20 35—40 28—35 50—55
Удлинение, % - 21—25 8—10 25—26 10—12 25—30 8—10
Начальный 10’ Па . модуль, 10400 5760 12000—15000 25000—32000
Плотность, кг/м3 . . 1340 1410 1440 1460 1380 1430
* ПМ — волокно на основе пиромеллитового днангидрнда и 4,4'-диамииодифенилоксида;
ПМС —пиромеллитовый диаигндрнд н 4,4'-диаминодифенилсульфид; ПФГ — пиромеллитовый
диангидрид и бис(4-аминофеннловый эфир) гидрохинона; I — полиамидокислотиое волокно; II —
волокно после термической цнклодегндратацнн.
Сочетание химической циклизации и дополнительной термической
вытяжки дает возможность получать полиимидные волокна с высокой
термостойкостью [128] (рис. 4.15, табл. 4.13).
Термическая вытяжка полиимидных волокон, прошедших стадии
химической или термической имидизации, проводится при температурах
300—600 °C в зависимости от химической структуры основной цепи
Таблица 4.13. Влияние условий циклизации полиамидокислоты
на свойства волокон [126]
Волокно Прочность, 10® -Па Параметры долговечности*
прн 20 °C при 300 °C UQ, кДж/моль V
Исходное (нециклнзованное) Химически циклизованное и до- полнвтельно вытянутое прн повы- шенной температуре 200 420 20—30 300 120—135 230—280 0,55—0,60 1,12—1,20
* Параметры долговечности Uo и V определены из формулы температурно-временной зависи-
мости прочности химических волокон [127].
120
макромолекулы [129]. Вытяжка волокон может осуществляться в ат-
мосфере нагретого воздуха или инертного газа, на металлических по-
верхностях и т. д. В случае формования полиамидокислотных волокон
по мокрому способу дополнительная термическая вытяжка относи-
тельно невелика и составляет 10—50%; при вытяжке волокон, сформо-
ванных сухим способом, степень вытяжки при повышенных температу-
рах достигает 300%. Установлено, что реакции внутримолекулярной
циклизации полиамидокислот, будь то химическая или термическая
циклодегидратация, практически не сопровождаются побочными реак-
Рис. 4.15. Зависимость прочности волокна на основе пиромеллитово1го ди-
ангидрида и 4,4'-днаминодифенилсульфида от температуры испытания [126]:
1 — исходное полиамидокислотное волокно; 2 — волокно, циклизованное химическим
путем; 3— волокно, циклизованное химическим путем и вытянутое при повышенных
температурах.
Рис. 4.16. Влияние дополнительной термической вытяжки на границы ра-
ботоспособности некоторых полиамидных волокон [130]:
1 и 3 — волокно ПМ, вытянутое соответственно при 300 и 500 °C; 2 и 4— волокно ПМС,
вытянутое соответственно прн 350 н 500 °C.
циями (сшивание, деструкция и т. д.), которые могли бы в значитель-
ной мере повлиять на молекулярную массу и молекулярно-массовое
распределение полиимидов, и соответственно на механические свой-
ства получающихся изделий [121].
Дополнительная термическая вытяжка вносит существенный вклад
в комплекс термомеханических свойств волокон (см. табл. 3.7 и
рис. 4.16); при этом может в значительной мере изменяться степень
имидизации последних [130].
Данные табл. 3.7 и рис. 4.16 свидетельствуют о том, что, варьируя
параметры получения, можно создавать полиимидные волокна, отли-
чающиеся повышенной тепло- или термостойкостью.
4. 2.3. Физико- и термомеханические свойства
полиимидных волокон
Общая характеристика полиимидных волокон. Полиимидные во-
локна в зависимости от химического строения (и независимо от спосо-
ба формования и характера процесса имидизации) имеют окраску от
желтой до красновато-коричневой. Так, например, немодифицированный
полиимид на основе 4,4'-диаминодифенилового эфира имеет золотисто-
желтый цвет; волокно на основе 4,4'-диаминодифенилсульфида — крас-
121
новато-коричневый. Замечено, что окраска сохраняется и при раство-
рении соответствующих полимеров или волокон в таких растворителях,
как азотная кислота или расплав треххлористой сурьмы [118].
Органические растворители, включая амидные, не растворяют ли-
нейные немодифицированные полиимидные волокна. Это затрудняет
определение молекулярной массы и молекулярно-массового распреде-
ления волокнообразующих полиимидов. Установлено, что полиимиды
могут быть растворены в кипящих азотной или серной кислотах, а
также в расплаве треххлористой сурьмы. Логарифмическая вязкость
волокнообразующих полимеров, определенная в указанных раствори-
телях, составляет 1,0—1,3 [118]. Зависимость прочности полиамидо-
кислотных волокон- от молекулярной массы соответствующих полиме-
ров носит S-образный характер; предел прочности при растяжении
достигает максимального значения при молекулярной массе, равной
~ 140 000, и при дальнейшем росте молекулярной массы прочность
волокон практически не изменяется [121].
В настоящее время волокна выпускаются, по-видимому, только в
виде комплексной нити [118; 131].
Физико-механические свойства полиимидных волокон. Несмотря
на то, что ароматические полиимиды являются одним из интересных
и исследованных классов полимеров, сведений о волокнах на их осно-
ве, по сравнению с ароматическими полиамидами имеется немного.
Наиболее изученными можно считать волокна на основе полипнромел-
литимидов. По физико-механическим свойствам полиимидные волокна
близки к свойствам синтетических волокон, выпускаемых в промыш-
ленных масштабах (полиэфирным и волокнам из алифатических по-
лиамидов), однако, полиимидные волокна значительно превосходят по-
следние по стойкости к действию повышенных температур.
В СССР разработана технология получения термостойкого поли-
имидного волокна аримид ПМ и его модифицированного аналога —
аримид-Т [131]. Физико-механические свойства этих волокон в срав-
нении с полиимидным волокном фирмы «Дюпон» [118] представлены
в табл. 4.14.
Кроме указанных волокон, свойства которых изучены достаточно
подробно и для которых уже определены области применения, име-
ется еще целый ряд волокон из полиимидов различного химического
строения. В табл. 4.15 приведены термические характеристики полиме-
ров, способы формования и основные физико-химические показатели
волокон.
Полиимидные волокна характеризуются также высокой устойчи-
востью к сдвиговым напряжениям, обладают повышенной прочностью
в петле, в мокром состоянии, к знакопеременным нагрузкам, истира-
нию. Отмечается сравнительно высокая устойчивость полиимидных
волокон к ударным нагрузкам [118].
Термомеханические свойства полиимидных волокон. Термомеха-
нические свойства полиимидных волокон превосходят не только термо-
механические свойства обычных промышленных волокон, но и оказы-
ваются выше по сравнению с аналогичными показателями волокон из
ароматических полиамидов. Исходные полимеры являются неплавки-
ми. Температура стеклования большинства полиимидов превышает
300 °C. Более или менее заметная термоокислительная деструкция по-
лиимидных волокон наблюдается при температуре выше 450 °C; в ва-
кууме деструкция начинается при более высоких температурах. О вы-
122
Таблица 4.14. Физико-механические свойства волокон аримид ПМ и аримид-Т
в сравнении с полиимидным волокном фирмы «Дюпон» [118; 131]
Свойства волокна Величина показателей
аримид ПМ аримид-Т полиимидное волокно фирмы «Дюпои»
Линейная плотность нити, текс Прочность прн разрыве, сН/текс в кондиционных условиях 13,3—33,3
45—50 45—60 62
в петле 42—50 28—48 52,2
в мокром состоянии 42—50 — —
Удлинение, % в кондиционных условиях 6—8 — 13
в петле — — 10
в мокром состоянии 8—10 — —
Начальный модуль, 10е Па 10400 15000 10000
Плотность, кг/м3 1410—1430 1450 1410
Равновесное влагопоглощеиие, % (при 65%-ной относительной влажности) Степень эластичности при растяжении 1,0—1,5 1,0—1,5
100
на 4%, % Усадка, % в воде при 95% 0 0 0,5
на воздухе при 300 °C 1—2 1—2 0,5
Устойчивость к действию многократ- 1200—3500 12000 2000000
ных изгибов (число циклов до разрыва) (о=120 МПа) (о=50 МПа) (а=12 МПа)
Устойчивость к истиранию (число 4000—33000 —
циклов) Кислородный индекс, % 38 50 34—35
сокой теплостойкости полиимидных волокон свидетельствуют как
термомеханические кривые (рис. 4.17), так и кривые, характеризую-
щие зависимость прочности от температуры испытания (рис. 4.18).
Из приведенных данных ясно, что полиимидные волокна при тем-
пературе порядка 400°C сохраняют свыше 30% исходной прочности.
Заметная усадка волокон наблюдается только при температурах по-
рядка 500 °C, т. е. полиимидные волокна обладают высокой стабиль-
ностью размеров при повышенных температурах, даже при действии
нагрузок (рис. 4.19). При кратковременных воздействиях полиимид-
ные волокна способны выдерживать температуру до 500—550 °C.
При изучении температурной зависимости модуля упругости по-
лиимидных волокон было отмечено чрезвычайно малое уменьшение
последнего с повышением температуры (рис. 4.20). Предполагают, что
это связано с высокой жесткостью макромолекул полиимидов, а также
с тем, что температуры плавления или размягчения лежат вблизи
температур разложения соответствующих полимеров. Из-за высокой
жесткости основной макромолекулы эластические свойства полиимид-
ных волокон сохраняются при температуре жидкого азота [128].
Полиимидные волокна отличаются высокой стойкостью к длитель-
ному действию повышенных температур. При длительном нагревании
на воздухе волокна окисляются с образованием летучих продуктов.
Если нагревание ведется в инертной атмосфере, то наблюдаются поте-
ри массы примерно до 40% при 650°C, а затем масса остатка не из-
меняется даже при повышении температуры до 850 °C {136].
Отмечено, что стабильность к действию повышенных температур
для полиимидных волокон значительно ниже стабильности соответст-
вующих полимеров. При нагревании волокон типа ПМ при 200 °C свой-
123
Таблица 4.15. Фнзнко-механические свойства некоторых полиимидных волокон
Химическая формула элементарного звена Температура, °C Способ формования волокон Свойства Лите- ратура
стек- лова- ния начала разложе- ния на воздухе проч- ность, сН/текс удли- нение, % начальный модуль, 10«-Па
“N\ О II с 'с О II с с 380 400 Сухой из рас- творов в амидных растворителях Мокрый, из рас- творов в амидных растворителях 40—60 45-50 8—13 8—10 10000— 12000 10040 (120; 132; 133] [128]
II О II О
О II с О II с >400 Мокрый, из рас- творов в амидных растворителях 35—40 10—12 5760 [128]
—n' II О с7 II О )n—
О II с О II с О —х II / < 340 >410 Мокрый, из рас- творов в амидных растворителях 35—60 8—12 8000— 10000 [128]
—n( II о (2 II О )N—/Г Л\—с—/ГЛ\_
в
390 >400 Мокрый, из рас- творов в ДМФ 22—35 11—16 — [129]
390 >450 Мокрый, из рас- творов в ДМФ 22—40 8—11 —’ [129]
— 400 Сухой, из рас- творов в ДМФ Мокрый, из рас- творов в ДМФ 55—75 50—55 6—10 8-10 25000— 32000 [119] [128]
Химическая формула элементарного звена
Продолжение
Температура, °C Способ формования волокон Свойства Лите- ратура
стек- лова- ния начала разложе- ния на воздухе прочность, сН/текс удли- нение, % началь- ный модуль, 10е »Па
>- 400 400 Сухой, нз раство- ров в ДМФ 40—80 6—10 20000 [134J
320 400 Мокрый, из рас- творов в ДМФ 35—40 10—12 — [134]
210 400 Сухой, нз раство- ров в амидных растворителях Мокрый, нз рас- творов в амидных растворителях 45—50 20—36 10 16 12500 8200 [135] [135]
ства их практически не изменяются [128]. Однако при нагревании на
воздухе при 280—300 °C, т. е. в области, далекой от температур тер-
мической деструкции полимеров, механические свойства волокон
Рис. 4.17. Термомеханические кривые полиимидных волокон в сравнении
с волокном капрон [128]:
1 — ПФГ; 2 — ПМ; 3 — ПМС; 4 — капрон.
Рис. 4.18. Зависимость прочности полиимидных и других химических воло-
кон от температуры испытания [87; 118; 128]:
Рис. 4.19. Зависимость удлинения при разрыве некоторых полиимидных
волокон от температуры [128]:
1 — капрон; 2—ПФГ; 3 — ПМ и ПМС.
Рис. 4.20. Зависимость модуля упругости некоторых полиимидных волокон
от температуры испытания [118; 128]:
1 — ПФГ; 2 — ПМ; 3 — ПМС; 4 — полиимидное волокно фирмы «Дюпон».
начинают ухудшаться (рис. 4.21). Предполагают, что ухудшение ме-
ханических характеристик волокон является результатом повышения
хрупкости полимеров, обусловленного образованием поперечных свя-
зей, а не следствием резкого уменьшения молекулярной массы. При
исследовании процесса термоокислительной деструкции полимеров при
350 °C было найдено [118], что логарифмическая вязкость полимеров,
определенная в расплаве смесей треххлористой сурьмы и мышьяка,
127
Рис. 4.21. Термостабильность полиимидных
волокон на воздухе прн различных темпера-
турах [87; 118; 128]:
/ — ПМС (300 °C); 2 —волокно фирмы «Дюпон»
(283 °C) и ПМ (300 °C); 3 — ПФГ (300 °C); 4 — фе-
нидон (300 °C); 5 — ПМ. ПФГ и ПМС (100 и 200 °C).
возрастает по мере увеличения продолжительности прогрева до тех
пор, пока волокно не утрачивает способность растворяться. При на-
гревании полиимидных волокон при 350 °C в вакууме падение проч-
ности волоком сопровождается незначительным выделением двуокиси
углерода.
Для полиимидных, как и для полиамидных волокон, характерным
оказывается резкое падение прочности в результате прогрева на воз-
духе при температурах выше 350 °C. Термоокислительная стабильность
полиимидных волокон может быть значительно повышена без сущест-
венного изменения механических
характеристик заменой в диамин-
ной компоненте атома кислорода
на серу или карбонильную груп-
пу [137]. Так, прочность поли-
пиромеллитимидного волокна на
основе 4,4-диаминодифенилового
эфира после прогрева при 350 °C
на воздухе в течение 20 ч состав-
ляет около 50% от исходной, в то
время как волокно на основе
4,4-диаминодифенилсулыфида,
прогретое в тех же условиях, те-
ряет лишь 8—10% исходной" проч-
ности. Этот факт объясняют тем,
что сульфидная группа играет
защитную роль, превращаясь под
действием кислорода воздуха при
повышенных температурах в
-сульфоксидную или сульфоновую без разрушения связи основной ва-
лентности [138]. Повышенная термическая стабильность волокон на
основе 4,4'-диаминобензофенона объясняется отсутствием в цепи свя-
зей, энергетически менее стойких, чем связь азот — углерод.
Химическая и гидролитическая стабильность полиимидных во-
локон; Химическая и гидролитическая стабильность полиимидов и не-
которых полиимидных волокон изучена довольно хорошо. Полинмид-
ные волокна не растворяются и не набухают в кипящих органических
растворителях; волокна устойчивы к действию кипящей воды [128].
Перегретый водяной пар разрушает полиимидное волокно на основе
4,4'-диаминодифенилового эфира и пиромеллитового диангидрида
£118]. В табл. 4.16 приведены данные, характеризующие химическую
и гидролитическую стойкость полиимидных волокон.
Щелочи и концентрированные кислоты при нагревании разрушают
волокно. Кипячение в воде, вызывая понижение прочности волокон,
по-видимому, оказывает пластифицирующий эффект, так как обработка
волокон и пленок в вакууме после кипячения приводит к восстановле-
нию механических свойств [139].
Радиационная стойкость и стойкость к УФ-излучению полиимид-
мых волокон. Полиимиды обладают исключительно высокой стойкостью
к воздействию излучений высоких энергий [ПО]. Прочность волокна
ПФГ, облученного на воздухе при 25 °C электронами с энергией около
одного МэВ, интенсивностью 0,2 Мрад/с при суммарной дозе около
1000 Мрад уменьшается всего иа 20% [128].
128
Таблица 4.16. Действие различных химических реагентов
на полиимидные волокна типа ПМ (температура 95—100 °C) [118, 128]
Реагент Продолжительность воздействия, ч Снижение разрывной прочности, %
Вода 100 15*
1400 4*
Соляная кислота, 10%-ная 100 74
260 71
Серная кислота, 10%-иая 100 37
260 52
Фтористоводородная кислота, 10%-ная 150 36
Едкий натр, 0,4%-ный 4 57
Гипохлорит натрия, 0,5%-ный 150 37
ДМАА (при 166 °C) — 0**
* Усадка 0,25%.
** Усадка 2,0%.
Волокна и пленки на основе пиромеллитового диангидрида и
4,4'-диаминодифенилового эфира существенно не изменяют своих ха-
рактеристик после облучения электронами с энергией 2 МэВ дозой
10 000 Мрад [140]. Стойкость к УФ-излучению изделий из полиимидов
(волокон, пленок) по-разному оценивается различными авторами. По
данным [128], волокно аримид ПМ сохраняет механические характе-
ристики после УФ-облучения на воздухе в течение 260 ч. Механиче-
ские характеристики полиимидного волокна типа ПФ Г после облуче-
ния лампой ПРК-2 в течение 200 ч не изменяются. Промышленные
волокна типа капрона теряют почти половину исходной прочности в
результате облучения лампой ПРК-2 в течение 30 ч. Данных по более
длительному облучению полиимидных волокон не имеется; длительным
испытаниям подвергались только пленки [141]. Экспозиция полиимид-
ных пленок (3000—6000 ч) на воздухе приводит к потере эластических
свойств; во влажной атмосфере скорость падения механических
свойств в результате УФ-облучения возрастает. Основной причиной
изменения механических характеристик полиимидных материалов яв-
ляется фотохимическая деструкция, сопровождающаяся разрывом
молекулярной цепи полимера с образованием свободных карбоксиль-
ных групп (при облучении в сухой атмосфере), и гидролиз макромо-
лекул с возникновением свободных гидроксильных групп и амино-
групп. По-видимому, полиимидные волокна, предназначаемые для
изделий, длительно работающих в условиях воздействия УФ-облуче-
ния, необходимо обрабатывать светостабилизаторами.
4.2 .4. Структурные особенности полиимидных волокон
Полиамидокислотные волокна, независимо от способа формования
и степени пластификационной вытяжки, обладают аморфной структу-
рой со слабо развитым одномерным порядком вдоль оси ориентации.
Химическая имидизация не приводит к существенным изменениям в
надмолекулярной структуре волокон. Термическая имидизация сопро-
вождается кристаллизацией волокон. В работе '[142] приводятся ре-
зультаты рентгенографического изучения некоторых полиимидных во-
локон. Отмечается, что, несмотря на хороший порядок вдоль оси
волокна, поперечная упаковка несовершенна. Кроме того, интенсив-
9—2254
129
ность рефлексов малоугловых рентгенограмм уменьшается с ростом
термической вытяжки полиимидных волокон (рис. 4.22).
Исходя из данных рентгеноструктурного анализа, авторы пред-
ставляют модель надмолекулярной структуры ПИ волокон в виде
широких, но разрыхленных фибрилл; доля проходных макромолекул,
по-видимому, близка к 100%. Предполагают, что достигнутая проч-
ность полиимидных волокон (70—
Рис. 4.22. Кривые малоуглового рас-
сеяния рентгеновских лучей в мери-
диональном направлении для поли-
нмндных волокон ПФГ с различной
степенью вытяжки [142]:
/ — невытянутое; 2—100%; 5 — 160%.
100 сН/текс) может быть повышена за
счет совершенствования технологии их
получения [142].
Продолжается поиск полимеров,
пригодных для получения новых поли-
нмидных волокон (табл. 4.17). Как
видно, полимеры имеют высокие тем-
пературы стеклования. Механические
свойства неориентированных пленок
оказались довольно высокими [143].
Отмечается возможность химической
модификации полиимидов за счет меж-
молекулярной сшивки по группам
С=О и СН2, что должно привести к
улучшению термических характери-
стик изделий и их огнестойкости.
Одной нз наиболее трудных проблем,
стоящих перед создателями полиимид-
ных волокон, является достижение
100%-ной степени циклизации имидных
звеньев и подбор оптимальных усло-
вий имидизации. Не выяснены пути
стабилизации полиамидокислотных
растворов и термической стабилизации
полимеров, имеющих имидные гетеро-
циклы.
4.3. ПОЛИОКСАДИАЗОЛИНЫЕ ВОЛОКНА
Способность гидразидной группировки в определенных условиях
превращаться в цикл была использована для получения нового клас-
са термостойких волокнообразующих полимеров — полиоксадиазолов
(ПОД). ПОД волокна могут быть получены по двухстадийному и од-
ностадийному способам.
Первый способ включает две основные стадии:
а) получение высокомолекулярных, хорошо растворимых в поляр-
ных растворителях полигидразидов (ПГ) по схеме:
nH2NNHCO—R—CONHNHa + лС1—СО—R'—СО—Cl ->
---> [—NHNHCO—R-CONHNH—СО—R'—СО—]„
Полигидразиды являются волокнообразующими полимерами, из рас-
творов которых методами сухого или мокрого формования получают
волокна;
130
Таблица 4.17. Новые волокнообразующие полиимиды
Химическая формула элементарного звена
а
Характеристика полимера Свойства ПИ-пленок Лите- ратура
”1в ПАк тс, °с т разл на воз* Духе, °C проч- ность, 106-ПА мо- дуль, 106.ПА удли- нение, %
0,54 315—350 450 186 9300 2,8 [143|
0,54 340 450 153 7500 2,6 [143]
0,54 325—364 430—450 163 7100 2,9 [143J
kL
Химическая формула элементарного эвена
П родолжение
Характеристика полимера Свойства ПИ-пленок Лите- ратура
ПАК гс, °с т разл иа воз- проч- ность. Юв-Па МО- ДУЛЬ, 100-Па удли- нение, %
0,54 347 450 140 6000 2,9 1143]
> 2,0 — 450 242 — 14,0 [144]
2,22 — 450 242 — 14,0 [144]
б) циклодегидратацию полигидразидов, приводящую к созданию
системы чередующихся ароматических ядер и 1,3,4-оксади азольных
циклов:
[—NHNHCO—R—CONHNHCO—R'—СО—]„
Нагрев
—2лН2о"
Как уже указывалось, оксадиазольный цикл по своей электронной
структуре близок к ароматическому бензольному ядру [54, с. 113],
поэтому он имеет высокую термическую стабильность и, являясь по-
лярным в отличие от бензольного цикла, придает образующимся по-
лимерам способность растворяться в сильнополярных растворителях,
таких, как концентрированная серная, хлорсульфоновая и полифосфор-
ная кислоты. Это дало возможность получать волокнообразующие по-
лиоксадиазолы по одностадийному способу.
Одностадийный способ синтеза волокнообразующих ПОД заклю-
чается в проведении реакции поликонденсации дигидразидов аромати-
ческих кислот в жидких конденсирующих средах (олеум, полифосфор-
ная кислота, хлорсульфоновая кислота и др.) или поликонденсации
ароматических дикислот с гидразином или его производными, в част-
ности с гидразинсульфатом, гидразинхлоридом, в тех же реакционных
средах:
nH2NHNHCO—R—CONHNH2 ----—-------
—R—G С—
nHOOC—R—СООН + nH2NNH2 -H2SO4
В данном способе в одну стадию совмещены процессы полимеро-
образования, роста полимерной цепи и внутримолекулярной циклоде-
гидратации. Достоинством этого способа является доступность исход-
ного сырья и реакционной среды и возможность получения высокомо-
лекулярного полиоксадиазола. Последний находится в растворе, из
которого непосредственно могут быть сформованы волокна.
4.3.1. Получение волокон по двухстадийному способу
Из нескольких возможных способов синтеза полигцдразидов:
в расплаве, на границе раздела фаз, низкотемпературная растворная
поликонденсация — наиболее приемлемым является последний, так как
в этом случае молекулярная масса образующегося полимера оказыва-
ется достаточной для получения прочных волокон [54, с. 113]. Реакция
проводится в амидных растворителях. Основные закономерности син-
теза полигидразидов аналогичны закономерностям, наблюдаемым при
получении волокнообразующих ароматических полиамидов, и описаны
выше.
Растворы ПГ в амидных растворителях оказываются довольно
стабильными и могут быть переработаны в волокна по сухому, мокро-
му или сухо-мокрому способам формования. При получении ПОД
волокон по двухстадийному способу применяют, как правило, сухой
133
Таблица 4.20. Свойства ориентированных полигидразидных волокон [145J
Показатели волокон Полиизофтал- гидразид Сополигидразид тере- и изофталевой кислот
Прочность, сН/текс 46,8 54,0
Разрывное удлинение, % 14,0 8,0
Начальный модуль, 10е Па 15000 19700
Прочность в узле, сН/текс 19,0 10,0
Удлинение при испытании в узле, % 7,0 4»2
Температура плавления, °C 320 335
Плотность, кг/м3 1443 1452
Влагопоглощение, % 4,5 4,3
Кристалличность Умеренная Высокая
способ формования, при этом синтез проводится в амидном растворите-
ле; затем полимер переосаждается, отмывается от растворителя, хло-
ридов и других примесей, сушится и снова растворяется в ДМСО.
Сложный процесс подготовки прядильных растворов связан с тем, что
концентрация полимера в реакционных растворах невелика, раство-
ры могут быть переработаны только по мокрому способу (прочность
волокон при этом оказывается невысокой). Растворение переосажден-
ного высокомолекулярного полигидразида в ДМСО дает возможность
получить стабильные растворы с концентрацией 15—20% (масс.); су-
хое формование с последующей ориентацией приводит к получению
комплексных нитей с хорошими механическими характеристиками
(табл. 4.20).
Полигидразидные волокна — стабильные полимерные системы, ко-
торые непосредственно без термической обработки могут быть приме-
нены для получения изделий с хорошим комплексом свойств, однако
они не могут быть использованы для эксплуатации при повышенных
температурах. Внутримолекулярная циклизация значительно улучшает
термическую стабильность получающихся изделий. Для превращения
полигидразидных волокон в полиоксадиазольные предварительная
ориентация исходных волокон нежелательна. Перевод полигидразидов
в полиоксадиазолы может быть осуществлен как химическим, так и
термическим путем; на практике полигидразидные волокна превра-
щают в полиоксадиазольные путем длительного нагревания в вакууме
при повышенных температурах. Применение кислых катализаторов
позволяет резко ускорить процесс циклодегидратации [146]. Предва-
рительная ориентация полигидразидных волокон оказывает сущест-
венное влияние на ход процесса циклизации (табл. 4.21).
Вытянутые полигидразидные волокна обладают кристаллической
структурой; это обстоятельство считают одной из причин медленного и
Таблица 4.21. Влияние вытяжки на степень циклодегидратации
полигидразидных волокон
Степень вытяжки, % Равновесная степень превращения при различных температурах, %
290 °C 300 °C 310 °C
0 81,5 90,7 92,0
54 63,2 63,2 87,3
134
Таблица 4.22. Физико-механические свойства ПОД волокна [146]
Условия циклизации Элементарная нить Комплексная нить
проч- ность, сН/текс удли- нение, % начальный модуль, 10«Па прочность в петле, сН/текс проч- ность, сН/текс удли- нение, % начальный модуль, 10«Па прочность в петле, сН/текс
Без катализато- 36,0 10,0 21000 12,6 29,7 3,о 22000 27,7
ра С катализато- ром 72,0 10,0 29200 47,7 60,3 6,0 30700 50,0
неполного превращения полигидразидных волокон в полиоксадиазоль-
ные. Проведение процесса циклодегидратации в присутствии катализа-
торов кислого характера позволяет достигать больших глубин превра-
щения. Типичные физико-механические характеристики ПОД волокон,
получаемых по двухстадийной схеме, приведены в табл. 4.22.
4.3.2. Получение полиоксадиазольных волокон
по одностадийному способу
Способ образования ПОД волокон через стадию полигидразидов,
хотя и дает возможность получать изделия с высокими физико-меха-
ническими характеристиками, из-за многостадийности оказывается тех-
нологически неудобным. Одностадийный синтез ПОД с высоким выхо-
дом осуществляется в результате реакции дикарбоновых ароматиче-
ских кислот с гидразином или его производными в сильнополярных
конденсирующих жидкостях (олеуме, полифосфорной кислоте, хлор-
сульфоновой кислоте) [147]. Реакция протекает по схеме:
Нагрев, олеум
пНООС—R—COOH+nH2NNH2H2SO4--------->•
N---N "
+ ЗпН2О + nH2SO4
п
Влияние некоторых условий поликонденсации в олеуме аромати-
ческих дикислот с гидразинсульфатом при получении волокнообразую-
щих полиоксадиазов рассмотрено в работе [148]. Синтез осуществляли
с использованием смеси тере- и изофталевых кислот (соотноше-
ние 7:3) в среде 30%-ного олеума. Зависимость логарифмической вяз-
кости образующегося ПОД от температуры реакции и концентрации
полимера в реакционном растворе носит экстремальный характер
(рис. 4.23). Максимум значений логарифмической вязкости достигается
при температурах от 120 до 160 °C и при концентрации образующего-
ся ПОД порядка 8—13% (масс.). С увеличением концентрации обра-
зующегося ПОД выше 14% (масс.), т. е. с ростом концентрации реаги-
рующих веществ, увеличивается количество воды, выделившейся в
результате реакции полимерообразования и внутримолекулярной цикли-
зации, что снижает концентрацию олеума и обрывает реакцию поли-
гетероциклизации. При температуре реакционной среды выше 160 °C
усиливаются побочные реакции, такие, как окисление гидразинсульфа-
та. Это также приводит к снижению логарифмической вязкости полиок-
135
садиазола. Кинетика поликонденсации смеси изо- и терефталевых
кислот с гидразинсульфатом свидетельствует о непрерывном нараста-
нии молекулярной массы полиоксадиазола (рис. 4.24). Введение моно-
функциональных соединений, например, n-толуиловой или бензойной
кислот, позволяет получать полиоксадиазолы определенной молекуляр-
ной массы [148].
Соотношение дикарбоновых кислот и гидразинсульфата также
оказывает существенное влияние на конечную молекулярную массу
полиоксадиазола. Максимальную логарифмическую вязкость полимера
Рис. 4.23. Зависимость равновесных значений логарифмической вязкости
ПОД от температуры реакции \1) и 'концентрации полимера в реакционной
среде i(2) ‘[148].
Рис. 4.24. Зависимость логарифмической вязкости ПОД от продолжитель-
ности реакции поликонденсации (/) и концентрации n-толуиловой кислоты
в реакционной среде (2) [148].
при проведении реакции поликонденсации в среде олеума удавалось
получить при 10——20%-ном избытке гидразинсульфата [149].
Исследования реакции полигетероциклизации непосредственно в
процессе синтеза с помощью ИК-спектроскопии показали, что обра-
зование гидразидных и оксадиазольных звеньев в полимере проходит
одновременно [149]. Эти реакции ускоряются и проходят более глубо-
ко при повышении концентрации олеума в реакционной среде. В ряде
случаев в качестве реакционной среды предлагается хлорсульфоновая
кислота [150]. Преимущества процесса проведения синтеза в хлор-
сульфоновой кислоте:
возможность синтеза ПОД с высокой концентрацией образующе-
гося полимера и с пониженной по сравнению с олеумом динамической
вязкостью растворов;
возможность получения ПОД с высоким значением логарифмиче-
ской вязкости; при проведении синтеза в растворе хлор сульфоновой
кислоты значение логарифмической вязкости полимеров достигает 30;
волокна, полученные из раствора полимера в хлорсульфоновой кис-
лоте, обладают более высокой хемостойкостью, термостойкостью и име-
ют повышенную степень белизны.
По-видимому, полифосфорная кислота менее пригодна для получе-
ния волокнообразующих ПОД по сравнению с олеумом и хлорсуль-
фоновой кислотой. При применении двух последних реагентов имеется
возможность сразу использовать реакционные системы в качестве пря-
дильных растворов, в то время как после проведения синтеза в ПФК
образующийся ПОД необходимо выделять из реакционной среды, от-
мывать, сушить и снова растворять в концентрированной серной
кислоте.
Рис. 4.25. Зависимость эффектив-
ной вязкости растворов ПОД раз-
личной концентрации при 20 °C от
концентрации серной кислоты
[151]:
7—2%; 2-3%; 3-4%; 4—5,5%;
5—6%. (ПОД содержит ЗО°/о изо- и 70%
терефталевых звеньев).
4.3.3. Свойства растворов полиоксадиазолов
в концентрированной серной кислоте
Реологические характеристики концентрированных растворов во-
локнообразующих ПОД зависят от концентрации серной кислоты [151]
(рис. 4.25). Зависимость эффективной вязкости растворов полиокса-
диазола от концентрации H2SO4 носит
экстремальный характер; перегиб на
кривых начинается в области концентра-
ции серной кислоты 99% (масс.), при
этом наблюдается минимум вязкости
раствора. С увеличением концентрации
полимера особенно быстро повышается
вязкость растворов, содержащих свобод-
ный SO3. Авторы полагают, что в этой
области изменяется не только форма
макромолекул полиоксадиазола, но и ха-
рактер структурообразования и проч-
ность структуры раствора (табл. 4.23).
Значения характеристической вяз-
кости полиоксадиазола проходят через
максимум, а энергии вязкого течения —
через минимум при концентрации кисло-
ты, равной 99,3%. Изменение величины
энергии вязкого течения в зависимости
от качества растворителя свидетельству-
ет о заметных изменениях в структуре
концентрированного раствора. Причиной
такого поведения ПОД в концентриро-
ванной серной кислоте является протони-
зация кислотой полимера, атомы которо-
го имеют неподеленную пару электронов
и способны к акцептированию протона
серной кислоты [151; 152, с. 112]. При
растворении ПОД в 98—99 %-ной серной
кислоте происходит наиболее полная протонизация полимера; при по-
нижении концентрации кислоты начинается процесс протонизации
Таблица 4.23. Влияние концентрации серной кислоты на характеристическую
вязкость полимера и кажущуюся энергию вязкого течения (АН) концентрированного
раствора полиоксадиазола [концентрация раствора 5,5% (масс.), т=9,8 кПа] [151]
Концентрация серной кислоты, % И ДН, кДж/моль
93,0 2,30 3,20
96,8 2,40 3,18
98,6 3,30 3,13
99,3 7,25 2,71
100,0 4,60 3,73
102,1 4,10 3,78
137
воды, а при повышении содержания SO3 происходит самопротонизация
H2SO4. Эти факты свидетельствуют о том, что, меняя концентрацию
серной кислоты, можно влиять на эффективную вязкость прядильного
раствора полиоксадиазола, что, в свою очередь, оказывается удобным
при осуществлении технологического процесса получения ПОД-волокон
из растворов в концентрированной серной кислоте.
4.3.4. Формование полиоксадиазольных волокон
из растворов в серной кислоте
Растворы полиоксадиазолов, полученные одностадийным способом,
имеют высокую вязкость [151]. Поэтому их разбавляют концентриро-
ванной серной кислотой и после фильтрации и обезвоздушивания пере-
рабатывают в волокна по мокрому способу формования. При исследо-
вании процесса формования ПОД волокон из сернокислотных растворов
было установлено {153—155], что одним из важных факторов, опреде-
ляющих механические свойства волокон и их структурные особенности,
является состав осадительной ванны, а также химическое строение по-
лимера. Применение воды в качестве осадителя приводит к получению
хрупкого волокна с пористой структурой. Одной из причин появления
пористости является высокий тепловой эффект взаимодействия кислоты
с водой. Прочность волокон, сформованных в воду, оказывается ниже,
чем у волокон, сформованных в водно-сернокислотную осадительную
ванну (табл. 4.24).
Таблица 4.24. Влияние состава исходного полимера и условий формования
на свойства полиоксадиазольных волокон [154] (концентрация прядильного
раствора 10% (масс.); г] 1g =2,11; температура 20 °C)
Состав ПОД Осадительная ванна Свойства волокон до термической вытяжки Свойства волокон после термической вытяжки
ТФК ИФК содержание содержание плотность, прочность, удлинение, прочность, удлинение,
% воды, % КГ/м3 сН/текс % сН/текс %
50 50 100 1390 12,2 35,0
40 60 1420 33,8 37,5 36,0 4—6
50 50 1420 35,0 36,0 38,0 4—6
70 30 — 100 — 18—25 20 25—40 4—8
50 50 — 35—40 8 40—60 4—8
75 25 — 100 1396 16,2 35,0 25—30 6—10
40 60 1410 23,6 46,0 54,9 12
Оптимальной осадительной ванной при формовании ПОД волокон
по мокрому способу является водно-кислотная, с содержанием кислоты
50—55% (масс.) [154]. В более мягких условиях, при применении
70%-ной сернокислотной осадительной ванны, коагуляция полимера
проходит очень медленно. При использовании в качестве осадительных
ванн сульфатов натрия или аммония, хлоридов цинка и растворов
других солей прочность полученных волокон не превышала
29,7 сН/текс [153—154].
4.3.5. Упрочнение полиоксадиазольных волокон
Свежесформованные волокна, получаемые по мокрому способу из
растворов в серной кислоте, имеют невысокие физико-механические ха-
рактеристики; прочность при разрыве составляет 5—15 сН/текс, раз-
138
рывное удлинение — 50—80%. Упрочнение таких волокон проводится в
две стадии: мокрое неотмытое волокно вытягивается в пластифицирую-
щей жидкости или на воздухе на 200—400%, затем отмывается от рас-
творителя и компонентов осадительной ванны, высушивается и допол-
нительно вытягивается при 270—500 °C [156]. Ориентационное вытяги-
вание в обоих случаях приводит к улучшению физико-механических
показателей и изменению структуры волокон (рис. 4.26).
Исходное волокно аморфно; данные рентгеноструктурных исследо-
ваний образцов, максимально ориентированных в пластифицированном
Рис. 4.26. Влияние 'пластификационной вытяжки на свойства ПОД воло-
кон [1561:
1 — максимальное напряжение изометрического нагрева; 2 — начальный модуль; 3 —
прочность; 4 — двойное лучепреломление; 5 — удлинение.
состоянии, говорят о том, что волокно избранного химического состава
практически не кристаллизуется в условиях высокоэластической
деформации [156]. Ориентационное упрочнение при степенях вытяжки
выше 300% оказывается неэффективным. Пластификационная вытяж-
ка, приводя к улучшению физико-механических свойств волокон,
не дает возможности получить высокотеплостойкое волокно. Тепло-
стойкость при 300°C волокон, имеющих оптимальную (или максималь-
ную) степень вытяжки, составляет 20—28%. Дополнительная высоко-
температурная вытяжка, степень которой невелика по сравнению со
степенью пластификационной вытяжки, оказывает решающее влияние
на повышение теплостойкости волокон, а также на дальнейшее улуч-
шение физико-механических характеристик последних (табл. 4.25).
Обращает на себя внимание бимодальный характер зависимости
прочности волокна от температуры вытяжки, а также монотонное
возрастание теплостойкости волокона по мере увеличения температу-
ры дополнительной вытяжки (рис. 4.27). Природа указанной бимо-
дальной зависимости прочности жесткоцепных волокон от температу-
ры вытягивания обсуждалась в гл. 3.
При исследовании процесса термического упрочнения полиокса-
диазольных волокон было замечено, что при простой термообработке
волокон, вытянутых в пластифицированном состоянии, их механиче-
ская прочность также увеличивается, однако эффект ориентационного
139
Таблица 4.25. Влияние температуры дополнительной вытяжки
иа свойства ПОД волокон [156]
Условия вытягивания Механические показатели Плот- ность, КГ/мЗ Дп Теплостой- кость при 300 °C, %
Т, °C X» прочность, сН/текс удлинение, % начальный модуль, 10«Па
Исходное 36,8 15,4 9790 0,229 25,0
волокно** 270 1,26 51,0 11,2 14600 1420 0,300 30,9
300 1,27 50,5 9,0 14600 — 31,7
320 1,27 65,0 7,8 26200 1424 0,451 34,0
350 1,30 53,2 8,4 15600 1420 0,444 32,9
380 1,30 48,8 4,8 20500 1422 0,461 40,0
400 1,30 66,2 4,0 34000 1425 0,482 46,2
420 1,37 63,7 4,0 33000 1430 0,701 49,0
450 1,46 74,5 4,0 32500 1448 0,692 52,0
480 1,36 45,0 2,2 32000 1450 0,672 59,0
500 1,36 36,9 1,9 32800 1450 0,385 61,0
* Максимальная кратность вытяжки прн данной температуре. Вытягивание проводилось в
воздушной атмосфере в электрообогреваемой трубке.
** Исходное волокно имело предварительную вытяжку, равную 4.
вытягивания при тех
[148] (рис. 4.28).
же температурах
оказывается более заметным
Прочность ,10 Па
Рис. 4.27. Зависимость прочности (/) и теплостойкости (2) ПОД волокон от темпера-
туры дополнительной вытяжки [156].
Рис. 4.28. Зависимость механических свойств ПОД волокон от характера термической
обработки:
!~4 — прочность и начальный модуль волокон; 1 н 3 — вытянутых при повышенных температурах;
2 и 4 — прогретых в статических условиях [1481-
4.3.6. Физико- и термомеханические свойства
полиоксадиазольных волокон
Несмотря на большое внимание со стороны исследователей к во-
локнообразующим полиоксадиазолам, имеется лишь несколько типов
волокон, свойства которых описаны подробно. Наиболее изученными
являются следующие ПОД волокна.
140
1. Волокно из полимера химической структуры (тип I):
которое было получено двухстадийным способом [146].
2. Волокно из полимера формулы (тип II):
Это волокно было одним из первых, полученных по одностадийному
способу [153; 154].
3. Волокно, имеющее химическую структуру (тип III):
Это волокно выпускается отечественной промышленностью под назва-
нием «оксалон» [157].
4. Имеются сведения о ПОД волокне, полученном из терефталевой
кислоты и гидразинсульфата (тип. IV). Полимер синтезируют по одно-
стадийному способу в растворе олеума при повышенных температурах;
формование волокна осуществляют из реакционных растворов в водно-
сернокислотные ванны [158]. Основные физико-механические свойства
волокон указанных типов приведены в табл. 4.26.
Сопоставляя основные механические свойства приведенных воло-
кон, можно отметить, что они близки между собой и что их прочность
находится на уровне прочности обычных химических волокон. Жест-
кость ПОД волокон, оцениваемая величиной начального модуля, ока-
зывается гораздо выше жесткости распространенных химических во-
локон.
Основным достоинством ПОД волокон по сравнению с другими
химическими волокнами и по сравнению с некоторыми типами термо-
стойких волокон является высокая стойкость к действию повышенных
температур. Исходные полимеры имеют высокие температуры стекло-
вания (выше 300°C). Деструкция наблюдается при температурах выше
450 °C. Температурная зависимость прочности полиоксадиазольных
волокон представлена на рис. 4.29. Стойкость к длительному тепло-
вому воздействию у полиоксадиазольных волокон является, по-видимо-
му, более высокой по сравнению с полиамидными термостойкими волок-
нами. Так, продолжительность нагревания при 300 °C на воздухе, при
которой прочность ПОД волокна снижается на 50%, составляет 700 ч
(рис. 4.30). Для волокон типа номекс это время составляет 150—200 ч
[146]. Термостабильность ПОД волокон при более высоких темпера-
турах характеризуется данными рис. 4.31.
Гидролитическая стабильность ПОД волокон также выше стабиль-
ности полиимидных и полиамидных волокон. Волокно оксалон сохра-
няет 50% исходной прочности при кипячении его в 10%-ном растворе
щелочи в течение 24 ч; номекс и полиимидное волокно типа ПМ в этих
141
Таблица 4.26.Физико-механические свойства некоторых полиоксадиазольных волокон
( Показатель Тип волокна
I П III IV
Способ получения полимера Двухстадийный Одностадийный Одностадийный Одностадийный
Способ получения волокна Сухое формова- ние полигидразида с последующей циклизацией Мокрый, из рас- творов в концент- рированной серной кислоте Мокрый, из рас- творов в концент- рированной серной кислоте Мокрый, из кон- денсационных рас- творов в концент- рированной серной кислоте
Прочность в кондиционированном состоянии, сН/текс Удлинение прн разрыве в кондиционированных уело- 60,3 54,9 50—70 40—10
6 12,0 4-8 5—10
ВИЯХ, % Удлинение в мокром состоянии, %
— — 4—8 4—8
Начальный модуль, 10е Па 30700 19050 30000—54000 29000
Относительная прочность в мокром состоянии, % —— 73-94 85—89
(от ИСХ.)
Плотность, кг/м3 1443 1410 1430 —
Равновесное влагосодержание (/=20 °C, относитель- ная влажность 65%), % •— 4,5 2,3 6—8 (нетермо- обработанное во- локно)
Степень эластичности при растяжении, % 1 76—83 76,8
Усадка
в воде при 95 °C, % 0,5 —— 0,5 —•
на воздухе прн 300 °C, % 0,5 — 0,7
Устойчивость к действию многократных изгибов — —• 3000—7000 3000—5000
(<т= 120 МПа) —число циклов до разрыва
Устойчивость к истиранию (число циклов до разру- шения) — 1— 30000—40000 —
Температура
плавления, °C Не плавится
стеклования, °C 330—350 — 320—330 350
Кислородный индекс (для тканей), % 30—35 (модифицирован- ное)
условиях разрушаются [2; 128; 157]. Стойкость полиоксадиазольных
волокон к действию кислых сред близка к стойкости полиамидных и
полиимидных волокон.
Неожиданной оказалась
низкая стойкость ПОД воло-
кон к действию открытого пла-
мени; немодифицированные
полиоксадиазольные волокна,
так же как и полигидразидные,
воспламеняются, поддержива-
ют горение и не затухают даже
после вынесения их из пламе-
ни [145].
ПОД волокна оказывают-
ся также стойкими к действию
УФ-облучения. Так, волокно
оксалон теряет 13% исходной
прочности после УФ-облучения
в течение 10 ч [157]. Свето-
стойкость таких волокон близ-
ка к светостойкости полиамид-
Рис. 4.29. Зависимость прочности ПОД воло-
кон от температуры испытания [146; 157; 158]:
1—волокно, полученное по двухстадийному методу;
2 — оксалон; 3 — волокно на основе терефталевой кис-
лоты и гидразина.
ного волокна типа номекс.
Предполагают, что введение
светостабилизаторов должно
повысить этот показатель во-
локон [159]. Хороший эффект
дает сшивка ПОД волокон трехфункциональными ароматическими кис-
лотами [160].
Рис. 4.30. Зависимость прочности ПОД волокон от продолжительности нагревания иа-
воздухе при 300 [7; 146; 157; 158]:
/ — волокно, полученное по двухстадийному методу; 2 — оксалон; 3 — номекс; 4 — волокно на ос-
нове терефталевой кислоты и гидразина.
Рис. 4.31. Тер1моста<бильность ПОД волокон при 350—400'’С [146; 157; 162]:
1 — модифицированное волокно оксалон (350 °C); 2— оксалон (350 °C); 3 — ПОД волокно, получен-
ное двухстадийным методом (400 °C).
Причиной относительно невысокой термостабильности волокон на
основе полиоксадиазолов, получаемых формованием сернокислотных
растворов в водно-сернокислотные осадительные ванны, является не-
14S
полная степень циклизации. Показано, что степень циклизации поли-
мера, содержащегося в поликонденсационном сиропе, и волокна, сфор-
мованного из указанного сиропа, неодинакова; в волокне степень
циклизации на 20—15% меньше [161]. Процесс раскрытия циклов про-
исходит при формовании, последующая ориентационная вытяжка,
проходящая при повышенных температурах, практически не изменяет
степени циклизации волокон [148]:
Характер обработки полимера или волокна
Степень
циклизации
Полимер в растворе.......................................... 97,0
Свежесформованное волокно............................... 76,1
Волокно, дополнительно вытянутое при температуре:
350 °C................................................... 75,6
400 °C................................................... 75,6
450 °C................................................... 79,0
Волокно, прогретое при 350°C в течение 25 ч на воздухе . . 84,6
Была сделана попытка увеличить степень циклизации ПОД воло-
кон путем дополнительной термообработки в присутствии некоторых
кислот: бензолсульфокислоты, ортофосфорной и борной кислот
(табл. 4.27) [162].
Таблица 4.27. Влияние природы кислоты и условий термообработки
на термомеханические свойства модифицированных ПОД волокон [162]
Модифицирующий агент и оптимальная концентрация его на волокне Прочность волокна. сН/текс Степень циклизации, % Прочность волокна после прогрева при 350 °C, % от исходной
ДО обра- ботки после обра- ботки До обра- ботки после обработки 30 ч 50 ч 100 ч
Исходное 46,4 76,1 40,0 25,0 0
Бензолсульфокнслота (1,18%) Ортофосфорная кислота (1,2%) Смесь ортофосфорной ибен- золсульфокнслот (0,73 % + +0,83%) Борная кислота (5%) 46,4 38,8 76,1 94,7 59,0 44,6 23,5
46,4 46,9 76,1 78,1 67,7 62,4 42,5
46,4 39,0 76,1 84,6 74,9 62,3 40,6
46,4 50,8 76,1 76,1—78,1 61,6 45,9 32,3
Полагают [162], что термостабильность полифениленгидразид-
оксадиазольных волокон можно повысить путем обработки кислотами,
при этом в ряде случаев повышается степень циклизации волокна.
Однако, по-видимому, добавки кислот, ингибирующие процесс термо-
окислительной деструкции ПОД волокон, могут обусловливать термо-
стабилизирующий эффект не только за счет повышения степени цикли-
зации. Одним из возможных механизмов ингибирования процесса
термоокислительной деструкции является блокирование концевых гид-
разидных групп модифицирующими кислотами.
Гидролиз полиоксадиазолов при формовании волокон из серно-
кислотных растворов в водно-кислотных осадительных ваннах может
быть уменьшен путем применения нескольких осадительных ванн с
последовательно уменьшающейся концентрацией серной кислоты
144
Таблица 4.28. Влияние условий формования
на свойства полиоксадиазольных волокон [163]
Условия формования Свойства волокон до прогревания Свойства волокон после прогревания
прочность, сН/текс удлинение, % гигро- скопич- ность, % потеря массы после прогрева при 375 °C в течение 48 ч, % прочность по- сле прогрева прн 200 °C в течение 1000 ч, % от исходной гигроско- пичность после прогрева 1000 ч прн 200 °C, %
Двухванное формова- ние: 1-я осадительная ванна — 56% (масс.) серной кислоты; 12% (масс.) сульфата аммо- ния; температура 35 °C; 2-я ванна — 18% (масс.) серной кислоты, темпера- тура 40 °C 49,5 41 4,9 9,4 49,5 4,8
Однованное формова- ние [56% (масс.) HjSO< н 10% (масс.) сульфата аммония] 42,3 30 23,5 36,9 5,2
[163]. Волокна, сформованные предлагаемым способом, имеют более
высокую стойкость к термоокислительной деструкции (табл. 4.28).
Термостабильность полиоксадиазольных волокон может быть
также повышена путем введения в основную цепь макромолекул аро-
матических фрагментов, содержащих атомы галогена, например бро-
мированную терефталевую кислоту. Исходные полимеры синтезируют
поликонденсацией терефталевой (или изофталевой) и бромированной
терефталевой кислот и гидразинсульфата в олеуме. Волокна получают
из сиропа по обычной технологии: свежесформованное волокно под-
вергают пластификационной вытяжке на 200—260% на воздухе, про-
мывают водой, сушат и дополнительно вытягивают при 400—420 °C
[164]. Прочность модифицированных таким образом ПОД волокон
составляет 35—40 сН/текс при удлинении 3—4%. Влияние концентра-
ции брома на стойкость полученных волокон к термоокислительной
деструкции можно проследить из данных табл. 4.29.
Галогенирование ПОД волокон можно проводить непосредственно
при синтезе полимеров в олеуме путем добавления к поликонденсаци-
онному раствору волокнообразующего полиоксадиазола различных
Таблица 4.29. Зависимость термостойкости ПОД волокон
от содержании в них брома [164]
Содержание брома в волокне, % (масс.) Термостойкость бромсодержащего полиоксадназольного волокна (% прочности от исходной) прн 350 °C после выдерживания на воздухе
10 я 25 ч 50 ч 100 ч
0 45—55 40—45 25—30 Разрушается
1,28 69—70 63—65 30—33 >
3,35 80—83 70—71 59—60 30—31
5,02 90—100 — 85—88 32—35
8,64 80—81 72—74 70—73 31—32
10—2254
145
галогенов, лучше всего брома [165]. В среде олеума происходит ча-
стичное замещение водорода ариленовых групп на галоген:
Нагрев, олеум
------------->
Волокна формуют по мокрому способу в сернокислотные осадительные
ванны с последующей пластификационной и дополнительной термиче-
ской вытяжкой. Полученные волокна имеют повышенную термоста-
бильность, приобретают самогасимость и обнаруживают более высокую
устойчивость к действию знакопеременных нагрузок [165]. Замечено
также, что обработка прядильных растворов полиоксадиазолов или
свежесформованных гелеобразных волокон, содержащих гидразидные
связи, аминами или аммиаком также приводит к некоторому повыше-
нию термостабильности ПОД волокон [166]. Некоторые способы мо-
дификации ПОД волокон, способствующие улучшению их термических
характеристик, рассмотрены в обзоре [167]. Термостабильность воло-
кон может быть улучшена при введении в макромолекулярную цепь
различных циклов: бензоксазолыных, имидных бензтиазольных и др.
(см. раздел 4.8).
Прочность полиоксадиазольных волокон, получаемых по одно- или
двухстадийному способам, не превышает 60—70 сН/текс. Выяснено, что
она может быть несколько повышена путем введения в цепь полимера
алкилгидразидных связей. Последнее достигается в результате замены
части исходной дикарбоновой кислоты ее диметиловым эфиром при
получении полиоксадиазолов по одностадийному методу поликонденса-
цией в олеуме [168]. Применение сухо-мокрого способа способствует
увеличению скорости формования до 240 м/мин и улучшению физико-
механических характеристик волокон (табл. 4.30).
Сухо-мокрый способ получения синтетических волокон, позволяю-
щий резко улучшить свойства нитей из ароматических полиамидов,
может быть использован и для формования ПОД волокон. При этом,
изменяя условия формования (молекулярную массу исходного полиме-
рн, температуру и концентрацию прядильного раствора, состав и тем-
пературу осадительной ванны), удается в значительной степени изме-
нять свойства волокон [168]. Так же как и в случае ароматических
полиамидов, оказывается возможным использовать высококонцентри-
рованные растворы полимера в серной кислоте [концентрация поли-
мера— 15—20% ('масс.)]. Эти растворы при обычных условиях пред-
ставляют собой твердые тела. Поэтому прядильный раствор перед
стадией волокнообразования нагревают до 130—140 °C. Полиоксадиа-
золы, растворенные в концентрированой серной кислоте, при повышен-
ной температуре оказываются нестабильными из-за гидролиза групп
—CONHNHCO—, которые содержатся практически во всех полимерах
независимо от способа синтеза. Предложен способ формования ПОД
волокон с повышенными механическими характеристиками [169]. По
этому способу исходный полиоксадиазол синтезируют конденсацией
ароматических дикислот с гидразинсульфатом в хлорсульфоновой кис-
лоте или смеси хлорсульфоновой и серной кислот. Молекулярная масса
полимера выше, чем при использовании олеума в качестве реакционной
среды. Полимер выделяют из реакционной среды, а затем растворяют
146
*0 I
Таблица 4.30.Свойства модифицированных полиоксадиазольных волокон,
полученных по мокрому и сухо-мокрому способам формования [168]
Мономеры Соотно- шение компо- нентов ”18 полимера Условия формования Скорость формо- вания, м/мин Свойства получаемых волокон
способ темпе- ратура фильеры, °C расстояние до ванны, см осадительная ваниа прочность, сН/текс удлинение, % начальный модуль, 10» Па
Терефталевая кислота 100 3,3 Сухо-мокрый 195 1,27 52-%-ная кислота серная 48,7 46,8 5,1 16830
Мокрый 40 — То же 10,0 45,0 5,6 15000
Терефталевая кислота и димети- ловый эфир тере- фталевой кислоты 50:50 50:50 3,3 3,2 » Сухо-мокрый 40 195 1,27 14-%-мая кислота Вода серя а я 10,0 240,0 85,5 81 4,2 4,3 27090 23220
75:25 3,6 (Мокрый 40 40%-ный рас- твор сульфата ам- мония 10,0 76,5 4,9 —
85:15 3,3 » 195 1,27 13 %-кая кислота серная 148,3 71,2 3,7 20790
90:10 3,3 » 160 1,27 То же 30,4 39,6 3,4 —
в серной кислоте, причем концентрация прядильного раствора, точнее
геля, составляет 15—25% (масс.). Зависимость прочности ПОД во-
локон, формуемых по сухо-мокрому способу, от молекулярной массы
исходного полимера имеет экстремальный характер, что видно из дан-
ных табл. 4.31.
Таблица 4.31. Влияние тце исходного полимера на физико-механические
свойства полиоксадиазольных ПОД волокон, формуемых
по сухо-мокрому способу [169]*
T]]g полимера Температура прядильного раствора, °C Свойства волокна
”lg прочность, сН/текс удлинение, %
2,65 75 2,48 71,1 6,4
3,08 85 2,85 100,0 5,5
3,76 90 3,12 110,0 5,5
4,50 ПО 3,98 113,8 7,2
6,40 130 5,26 128,0 4,8
8,12 145 6,00 135,0 4,6
12,00 172 5,03 100,8 3,4
* Состав полиоксадиазола: 80% (мол.) терефталевой кислоты и 20% (мол.) изофталевой
кислоты. Концентрация прядильного раствора — 17,5% (масс.).
По мере увеличения логарифмической вязкости исходного полиме-
ра температура прядильного раствора, поступающего на формование,
должна быть также увеличена. При этом усиливается гидролиз исход-
ного полимера и становится заметным при температуре выше 130°С,
однако экстремальная зависимость прочности получаемых волокон от
логарифмической вязкости сохраняется.
Рассмотренные способы получения и модификации ПОД волокон
свидетельствуют о том, что на основе доступного исходного сырья
получены новые синтетические термостойкие волокна, свойства которых
могут быть изменены в широких пределах. Ценные свойства в сочета-
нии с дешевой сырьевой базой делают полиоксадиазольные волокна,
несомненно, одними из наиболее перспективных термостойких мате-
риалов.
4.4. ПОЛИБЕНЗИМИДАЗОЛЬНЫЕ ВОЛОКНА
4.4.1. Получение и свойства полимеров
Полностью ароматические полибензимидазолы были синтезиро-
ваны Марвелом [170] в 1961 г. конденсацией в расплаве ароматиче-
ских бис(2,4-диаминофенилов) с ароматическими дикислотами по
схеме:
148
В качестве кислотной компоненты могут быть использованы как
ароматические дикислоты, так и их производные (метиловые или фе-
ниловые эфиры, дихлорангидриды, иминоэфиры и т. д.), однако лучшие
результаты получаются при применении дифениловых эфиров. Исполь-
зование дифениловых эфиров кислот для синтеза ПБИ дает возмож-
ность получать высокомолекулярные продукты (молекулярная масса
достигает 100 000). При реакции свободных ароматических дикислот с
бис-о-диаминами образуются низкомолекулярные полимеры вследствие
декарбоксилирования кислотной компоненты при повышенных темпе-
ратурах. С метиловыми эфирами дикислот принципиально возможно
образование достаточно высокомолекулярных продуктов, однако реак-
ция осложняется побочными процессами метилирования аминогрупп, на-
рушающими соотношение между концевыми группами. Высокая реак-
ционная способность дихлорангидридов приводит к замещению всех
аминогрупп в аминной компоненте, что сопровождается гелеобразова-
нием [171]. Наиболее широкое распространение получил полимер, син-
тезируемый на основе 3,3'-диаминобензидина и дифенилового эфира
изофталевой кислоты. ПБИ волокно указанного состава выпускается
в опытно-промышленном масштабе [54, с. 65], однако, по последним
сведениям, развитие волокна приостановилось и перспективы его рас-
ширения неясны из-за появления негорючего волокна кайнол [86].
Процесс производства ПБИ волокна включает обычные стадии,
характерные для процессов получения термостойких волокон
[54, с. 65]:
1) синтез полимера;
2) растворение полимера в амидных растворителях (чаще всего
используется ДМАА);
3) формование, промывка и сушка волокна;
4) дополнительная вытяжка волокна при повышенных темпера-
турах;
5) структурная модификация волокна, приводящая к повышению
степени его кристалличности.
Реакция образования ПБИ проводится в две стадии: сначала по-
лучают форполимер, осуществляя эту стадию в расплаве, который
быстро затвердевает. Форполимер, находящийся в виде вспененной
массы, измельчают до порошкообразного состояния и затем загружают
во второй реактор, где реакция проводится при температуре выше
300°C в вакууме или в токе азота, причем предпочтение отдается
второму способу, так как при этом получают полимер, концентриро-
ванные растворы которого обладают лучшей фильтруемостью
(табл. 4.32).
Таблица 4.32. Свойства ПБИ, полученного в вакууме
и в среде азота
Условия синтеза* Tjjg полимера Показатель фильтруемости, г/(см2-мин)
Форполимер 0,24
Вакуум 1,00 0,15
Азот при атмосферном давлении 0,89 0,35
* Продолжительность синтеза 3 ч, температура 375 °C.
149<
Реакция между 3,3'-диаминобензидином и дифенилизофталатом
протекает по следующей схеме:
Масс-спектрометрический термический анализ процесса показал,
что вода и фенол начинают выделяться одновременно [172], при этом
на первой стадии выделяется половина теоретического количества
воды и практически весь фенол, а на второй стадии — только вода.
Для получения полимера с заданными свойствами необходимо
соблюдать стехиометрию вводимых в реакцию веществ. Избыток
аминной компоненты приводит к снижению молекулярной массы ПБИ,
а при избытке дифенилизофталата образуется полимер с высокой ло-
гарифмической вязкостью, но с большим содержанием нерастворимых
гель-частиц; кроме того, фильтруемость растворов невысока [54; 173].
Отмечается, что даже кратковременный нагрев полимера на воздухе до
температуры 400 °C приводит к получению ПБИ, нерастворимого ни
в одном из известных растворителей [1711.
ПБИ, синтезированный в две стадии, представляет собой неплав-
кий золотисто-коричневый порошок, темнеющий на свету, растворимый
в амидных растворителях, диметилсульфоксиде, концентрированной
серной и муравьиной кислотах [174]. Растворимость ПБИ в амидных
растворителях улучшается в присутствии небольших количеств хлори-
да лития.
При повышении температуры растворимость ПБИ также увеличи-
вается (табл. 4.33).
Таблица 433. Растворимость ПБИ в ДМАА [54; 174]
Условия растворения Фракции
1 о 3 4 5
Температура, °C 70 140 165 (кипение) 240 (под давлением) 240 (под давлением)
Количество растворившегося полимера, % от исходного 13 65 75 100 100
Шк 0,45 0,57 0,63 0,90 1,20
150
При растворении ПБИ в ДМАА, не содержащем добавок хлорида
лития, растворы нестабильны (наблюдается процесс обратного выде-
ления полимера из раствора). Это может быть обусловлено химиче-
скими факторами: процессами сшивки макромолекул ПБИ в щелочной
среде (ДМАА имеет основную реакцию) в присутствии кислорода;
поэтому все операции по растворению, обезвоздушиванию и фильтра-
ции необходимо проводить в инертной атмосфере. Кроме того, неста-
бильность концентрированных растворов ПБИ в ДМАА связана с
протеканием процессов локальной кристаллизации. Хотя процесс и об-
ратим, и нагревание расслоившейся системы вновь приводит к ее го-
могенизации, необходимы добавки, увеличивающие стабильность рас-
творов. Кроме хлоридов лития в качестве добавок используют N-ме-
тилморфолин, триэтиламин и другие третичные амины [54].
4.4.2. Получение и физико-механические свойства
полибензимидазольных волокон
Основным методом получения ПБИ волокон является сухое фор-
мование из концентрированных растворов ДМАА [54]. В принципе
волокна могут быть получены и по способу мокрого формования; при
этом применяют как амидные растворители, так и концентрированную
серную кислоту [175]. При формовании по сухому способу используют
прядильные растворы с концентрацией ПБИ 20—30% (масс.), содер-
жащие 1—2% (масс.) хлорида лития. Процесс формования осущест-
вляется в инертной атмосфере (азот или двуокись углерода), или в
атмосфере перегретого пара [54; 176]. Отмечается, что даже при
незначительном содержании кислорода в прядильной шахте свойства
готового волокна ухудшаются; концентрация кислорода не должна пре-
вышать 2,5% (масс.). Нить, выходящую из шахты, вытягивают на
5—10%, принимают на паковку и подвергают промывке.
Промывка волокна может быть осуществлена периодическим спо-
собом путем циркуляции воды через перфорированные бобины с во-
локном или при прохождении одиночной нити через систему орошае-
мых роликов [177]. Свежесформованные волокна из ПБИ имеют
сравнительно невысокую механическую прочность (8—15 сН/текс) и
высокое удлинение (70—120%). Эти показатели могут быть улучшены
путем дополнительной вытяжки при температуре выше 450—500 °C.
Волокно, поступающее на термическую вытяжку, должно быть тща-
тельно высушено. Ориентационное вытягивание волокна из ПБИ при
повышенных температурах помимо тщательной сушки имеет ряд осо-
бенностей: во-первых, для получения высоких механических характери-
стик волокно перед вытяжкой необходимо длительное время выдержи-
вать при той же температуре, при которой осуществляется процесс
ориентации; во-вторых, предварительный прогрев должен проводиться
в инертной атмосфере. Нить, поступающая на вытяжку, должна иметь
определенное натяжение [54]. Повышение степени ориентации волокна
практически не сказывается на кристалличности и плотности образ-
цов. Для получения ПБИ волокна, имеющего прочность в пределах
30—40 сН/текс, применяют одноступенчатое вытягивание. Двухступен-
чатое вытягивание дает возможность получать волокна с прочностью
до 55 сН/текс [54, с. 86].
Интересны результаты исследования [54; 178], показывающие
влияние фракционного состава исходного ПБИ на механические харак-
151
теристики волокон. Исследовались волокна, полученные из нефракцио-
нированного полимера и из полимера, имеющего максимальное и сред-
нее значение молекулярной массы с узким молекулярно-массовым
распределением (табл. 4.34).
Таблица 4.34. Свойства ПБИ волокон, полученных
из нефракционированного и фракционированных полимеров
Полимер Невытянутое волокно Вытянутое волокно
прочность, сН/текс удлинение, % прочность, сН/текс удлинение, % начальный модуль, 10®-Па
Нефракционированный (щ8=0,72) 14,6 100 39,9 13,5 14400
Фракционированный высокомолекулярная фракция с T]ig = l,2 20,5 138 52,3 19,2 18000
фракция с ц ig =0,72 6,0 76 24 ;з 23 87С0
Волокно, полученное из высокомолекулярного ПБИ, по всем
показателям превосходит как волокно, сформованное из нефракциони-
рованного, полидисперсного полимера, так и волокно, полученное из
фракционированного полимера, имеющего сравнительно невысокую мо-
лекулярную массу.
Характерной особенностью ПБИ волокна, формуемого на основе
3,3'-диаминобензидина и дифенилизофталата, является низкая кри-
сталличность. Кристалличность полимера и волокна может быть повы-
шена путем дополнительной обработки образцов фенолом или муравьи-
ной кислотой [54, с. 86]. Показано, что обработка волокна фенолом
при повышенных давлениях и температурах дает возможность значи-
тельно увеличить степень кристалличности волокон, но при этом не-
ожиданно изменяются их механические характеристики: уменьшается
прочность и увеличивается удлинение (табл. 4.35). Релаксация наблю-
дается даже при обработке волокон на жестких паковках.
Таблица 4.35. Влияние обработки фенолом на свойства ПБИ волокна
Условия обработки в автоклаве Свойства волокон
концентрация фенола, % (масс.) температура, продолжи- тельность, ч кристалличность прочность, удлинение, о/ /о
Исходный об- — — Аморфный 40,1 18,4
разец
14 250 2 Умеренная 23,6 39,5
14 250 4 > 24,8 43,6
20 200 2 Слабая 13,8 79,5
20 250 2 Сильная 22,6 48,0
Вытягивание при повышенных температурах ПБИ волокна, обра-
ботанного предварительно растворами муравьиной кислоты, также при-
водит к получению высококристаллических образцов, однако прочность
их не достигает прочности волокон, полученных по обычному способу
[54, с. 86].
Сравнение механических показателей ПБИ волокон, сформован-
ных различными методами из органических растворителей и серной
кислоты, показывает, что наиболее прочные волокна получают, при-
меняя сухой способ формования (табл. 4.36).
152
Таблица 4.36. Влияние способа получения на физнко-механическне
характеристики ПБИ волокон
Способ получения и параметры формования волокон Свойства волокон Литература
прочность, сН/текс удлинение, % начальный модуль, 106-Па
Сухое формование, дополни- тельная вытяжка на 200— 350% при 480—520 °C 44—54 8—24 12 600—16800 [54]
Мокрое формование из рас- творов в ДМАА; осадительная ваниа — смесь этиленгликоля с водой. Вытяжка иа 150— 250% при 450—600 °C с допол- нительной релаксацией на 30— 50% при 300—600 °C 32—33 15—25 [175]
Мокрое формование из рас- твора в коицентрироваиной серной кислоте; осадительная н пластификационная ванны — смесь кислоты и воды; допол- нительная вытяжка при темпе- ратуре выше 450 °C 40—41 15—20 10 800—12 000 [179]
Ниже представлены данные, характеризующие некоторые физико-
механические и текстильные свойства ПБИ волокон, полученных на
основе З.З'-диаминобензидина и дифенилизофталата [180]:
Вид волокна................................... Комплексная
нить '
Линейная плотность, текс.................................. 20
Прочность, сН/текс...................................... 50,0
Удлинение, %..................................... 10—12
Начальный модуль, МПа................................. 16750
Плотность, кг/м3........................................ 1320
Равновесное влагопоглощение, % (при 65%-ной
относительной влажности)......................... 13—15
Прочность в мокром состоянии, % от исходной . . 80—90
Кислородный индекс, %............................ 30—43
Температура плавления, °C......................... 560 (с разложением)
Усадка при 450 °C, %............................. 8
Отмечается, что ПБИ волокна могут выпускаться и в резаном
виде, в основном для переработки в смеси с хлопком. Волокно легко
окрашивается в темные тона красителями различных классов. Усадка
волокна при повышенных температурах может быть снижена путем
дополнительной обработки хлорсульфоновой кислотой, а также бро-
мом, пиридином и хлорокисью фосфора [86]. При этом кислородный
индекс повышается до 50—60%.
4.4.3. Термомеханические свойства
полибензимидаэольных волокон
Термогравиметрические и масс-спектроскопические исследования
ПБИ на основе 3,3'-диаминобензидина и дифенилизофталата, прове-
денные в вакууме и в кислородсодержащей атмосфере, показали до-
вольно высокую стойкость полимера к действию повышенных темпера-
тур (рис. 4.32), однако высокая термическая стабильность полимера
153
не коррелируется с термостабильностью волокна [181]. Исследование
изменения механических свойств в процессе длительного старения при
304 °C на воздухе (рис. 4.33) показало, что ПБИ волокно быстрее
Рас. 4.32. Динамический тфмоправиметрический анализ на 'воздухе ПБИ в орав нении
с другими полимерами [181]:
1 — номекс; 2 — ПБИ; 3— ВВВ; 4 — ПБИ в инертной среде.
Рис. 4.33. Зависимость прочности ПБИ волокна и волокна номекс от продолжительно-
сти теплового старения на воздухе при 304 °C [182]:
1 — номекс; 2 — ПБИ.
теряет прочность по сравнению с волокном номекс [482]. Полагают,
что низкая стойкость ПБИ к термоокислительной деструкции связана
с наличием подвижного атома водорода в гетероцикле. С другой
Рис. 4.34. Влияние температуры испытания на
прочность ПБИ волокон и волокна номекс [182]:
1 — высокоориентированное некристаллическое волокно;
2 — аморфное волокно средней степени ориентации;
3 — кристаллическое волокно; 4 — номекс.
стороны, сравнительно жест-
кая структура макромоле-
кул обеспечивает высокую
устойчивость волокон к дей-
ствию теплового удара. На
рис. 4.34 приведены данные,
характеризующие измене-
ние прочности ПБИ волокон
в зависимости от температу-
ры испытания [182].
Способность к сохране-
нию комплекса свойств при
кратковременном воздейст-
вии тепловых и силовых по-
лей, так же как и высокая
огнестойкость, является су-
щественной характеристи-
кой ПБИ волокон. Некото-
рое повышение прочности
волокон в области темпера-
тур 100—150 °C связано с усилением межмолекулярного взаимодейст-
вия по мере удаления сорбированной волокном воды. Ход кривых оди-
наков для всех образцов волокон, т. е. ни высокая ориентация, ни кри-
сталлизация (даже при их сочетании) не приводят к дополнительному
улучшению свойств волокна при повышенных температурах. Теплостой-
154
кость ПБИ волокон значительно превосходит теплостойкост!) волокна
номекс; при температуре испытания 450 °C они сохраняют 20—30%
исходной прочности. Волокно номекс в этих условиях разрушается.
4.4.4. Химическая и радиационная стойкость
полибензимидазольных волокон
Изучение гидролитической стойкости ПБИ волокон в сравнении с
гидролитической стойкостью номекса показало, что ПБИ уступает по
этому показателю волокну на основе ароматического полиамида
(табл. 4.37).
Таблица 4.37. Химическая стойкость ПБИ волокна и волокна номекс
при комнатной температуре [182]
Номекс
ПБИ волокно
Среда, концентрация Усадка, % Остаточная прочность, %
16 ч 96 ч 192 ч 16 ч 96 с 192 ч
Муравьиная кис- лота, 10%-ная 57,0 — 58,0 48,1 — 48 3
Серная кислота, 10%-ная 37,7 — 40,1 86,2 — 79,1
Серная кислота, 70%-ная — 43,2 — — 82,7 —
Азотная кисло- та, 10%-ная Соляная кисло- та, 35%-ная 32,5 — 35,0 56,1 — 69,9
30,6 — 30,2 76,5 — 78,1
Фтористоводо- родная кислота, 40%-ная Едкий натр 31,6 79,6
10%-ный — 11,8 — — 87,8 —
40%-ный — 34,8 — — 53,1 —
3,0
8,8
6,6
11,0
Усадка, %
16 ч 96 ч
8,9
10,0
3,8
6,6
192 ч
9,3
10,8
1,3
0,8
Остаточная
прочность, %
16 ч 96 ч 192 ч
104,0
119,0
91,2
96,2
95,2
94,1
82,1
83,1
98,4
80,7
98,3
51,5
Обработка химическими реагентами ПБИ волокна в свободном
состоянии даже при комнатной температуре приводит к сильной усад-
ке волокна. Полагают [182], что это связано только с релаксацией
волокна и что химических превращений ПБИ не испытывает, однако
экспериментальный материал не приводится. Косвенным подтвержде-
нием сказанному является более высокая гидролитическая стойкость
ПБИ волокон по сравнению с волокном номекс, определенная при повы-
шенных температурах (табл. 4.38).
Для ПБИ волокна, как и для волокон из ароматических поли-
амидов, характерна низкая светостойкость.
Высокая скорость фотодеструкции ПБИ волокна связана, как по-
лагают, с интенсивным поглощением света полимером [183]. В про-
цессе светостарения резко падает молекулярная масса полимера и
значительно ухудшаются его механические свойства (рис. 4.35). Так,
например, после старения в течение месяца летом на открытом воздухе
прочность ПБИ волокон падает вдвое, а износостойкость составляет
лишь 11% от начальной величины [183]. Введение в волокно свето-
стабилизаторов, эффективных для волокон из ароматических полиами-
155
Таблица 4.38. Химическая стойкость волокон ПБИ и номекс
при повышенных температурах [182]
ПБИ волокно
(8 ч, 95 °C)
Номекс
(8 ч, 95 °C)
ПБИ волокно
(102 ч, 60 °C)
Номекс
(102 ч, 60 °C)
Показатель
Усадка, %
Прочность,
от исходной
14,2
91,4
50—5550—5521—22
28,8
63,4
18,8
31,2
76—90 0,20 56,7
дов, не приводит к успеху [182]. Незначительный эффект повышения
светостойкости ПБИ волокон наблюдается лишь при обработке послед-
них тетраоксидом осмия, уксусным ангидридом, при применении спе-
Рис. 4.36. Зависимость прочности ПБИ волокон от температуры и продол-
жительности у-облучения [185]:
/ — исходный ПБИ; 2—1ч; 3—18,2 ч
Рис. 4.35. Влияние УФ-облучения на изменение прочности i(7 и 2) и лога-
рифмической вязкости (3) [183] ПБИ волокон. (О—ПБИ; ф — но-
циальных покрытий или прививке к макромолекуле ПБИ мономеров,
стойких к УФ-облучению [86; 184].
Рядом исследователей отмечается сравнительно высокая радиа-
ционная стойкость ПБИ волокон, превышающая радиационную стой-
кость волокон из ароматических полиамидов. При этом радиационная
стойкость ПБИ волокон мало изменяется в результате суммарного
воздействия излучения и тепла (рис. 4.36) [185].
Данные, приведенные выше, свидетельствуют о том, что на основе
полибензимидазолов возможно получение волокон, обладающих комп-
лексом высоких физико-механических свойств и имеющих при этом
высокую гигроскопичность, хорошую устойчивость к тепловым ударам
156
и исключительную огнестойкость. Широкое развитие работ в этой
области тормозится, по-видимому, из-за сложной технологии получения
волокон, и возможно, ограниченной сырьевой базы одного из исходных
мономеров — дифенилового эфира ароматической кислоты, сведения о
промышленном производстве которых отсутствуют.
4.5. ВОЛОКНА ЛЕСТНИЧНОГО СТРОЕНИЯ
Волокнообразующие полимеры лестничного строения привлекают
особое внимание по целому ряду обстоятельств. Впервые на э/ом
классе была показана возможность получения волокон из полимерных
систем, макромолекулы которых состоят как бы из двух линейных
цепей, соединенных между собой большим числом химических связей.
Рис. 4.37. Снижение молекулярной массы моноцепного (/) и двухцепного
(2) полиэфиров цри гидролизе [186].
Рис. 4.38. Скорость снижения молекулярной массы V моноцепного (1) и
двухцепного (2) полимеров при термодеструкции [187].
Исходя из основного положения о том, что механические свойства
полимеров определяются их молекулярной массой, можно полагать, что
разрыв одной химической связи в макромолекуле не должен привести
к падению молекулярной массы и соответственно к изменению меха-
нических характеристик волокон из таких полимеров. Заметное умень-
шение молекулярной массы, происходящее под действием тепла, гид-
ролиза и других факторов наблюдается при одновременном разрыве
противоположных химических связей в одном цикле, что менее веро-
ятно, чем разрыв одной связи. Кроме этого, вероятность рекомбинации
радикалов благодаря эффекту клетки в случае лестничных полимеров
оказывается выше, чем в случае одноцепных макромолекул. Эти пред-
положения подтверждаются сравнением стабильности лестничного по-
лимера при гидролизе со стабильностью полимера с одинарной цепью,
проведенным на примере двухцепного и одноцепного полиэфиров
(рис. 4.37). Теоретическим расчетом с использованием модели Монте-
Карло было также показано, что молекулярная масса линейного поли-
мера при деструкции должна уменьшаться значительно быстрее, чем
157
молекулярная масса лестничного полимера; начальная скорость дест-
рукции лестничного полимера при этом ниже (рис. 4.38). Подтвержде-
нием этому является некоторое повышение-температур начала терми-
ческой и термоокислительной деструкции полифенантролинов по срав-
нению с полиимидами (рис. 4.39). Однако, вопреки предположениям и
расчетам, это повышение оказалось не столь значительным. Разница
в температурах начала термоокислительной деструкции полиимидов и
полифенантролинов составляет 50°C; при термической деструкции
(в среде азота) она составляет примерно 50—80°C.
О 200 МО 600 800
Температура, °C
Рис. 4.39. Динамический термограви-
метрический анализ полимера ВВВ и
полиимида на основе пиромеллитово-
го диангидрида и бензидина [109-
188]:
1 и 2 —ВВВ соответственно в азоте и на
воздухе; 3 и 4 — полннмнд соответственно
в азоте н на воздухе.
Исходными веществами для
синтеза волокнообразующих поли-
меров лестничного строения являют-
ся тетрафункциональные соедине-
ния — диангидриды тетракарбоно-
вых кислот и тетр амины. В качест-
ве диангидридов наибольшее рас-
пространение нашли ангидриды
пиромеллитовой и нафталинтетра-
карбоновой кислот; в качестве тет-
раминов — 3,3'-диаминобензидин.
Лестничные полимеры, получаемые
с использованием диангидридов
кислот, построенных из одного или
нескольких бензольных колец, на-
пример диангидрида пиромеллито-
вой кислоты, называют полиимида-
зопирролонами, а полимеры, синте-
зируемые на основе кислот с кон-
денсированными ядрами, в частно-
сти нафталинтетракарбоновой ки-
слоты — полибисбензимидазобензо-
содержащие между циклами простых
фенантролинами*. Полимеры, не
связей, называют лестничными; полимеры, включающие одинарные свя-
зи между ароматическими ядрами, — блок-лестничными [189].
4.5.1. Методы синтеза и свойства лестничных полимеров
Полимеры лестничной или блок-лестничной структуры могут быть
получены различными методами: в расплаве, растворе, поликонденса-
цией на границе раздела фаз или в твердой фазе [190, с. 58]. Подобно
другим полигетероциклическим полимерам они могут быть синтезиро-
ваны одно- или двухстадийным способами. Для пирронов наиболее ши-
роко распространенным методом синтеза является двухстадийный, ос-
нованный на поликонденсации ароматических бис (2,4-диаминофенилов)
с пирромеллитовым диангидридом или диангидридами других кислот в
среде апротонных растворителей (например, ДМСО, ДМАА и др.) при
низких или повышенных температурах [191]. На этой стадии получает-
ся промежуточное высокомолекулярное волокнообразующее соедине-
ние — полиамидоаминокислота (ПААК):
“'Первые в дальнейшем будут сокращенно называться -пирронами, вторые —
полифенантролинам1и.
158
Полиамидоаминокислота, выделенная из раствора в виде волокна,
пленки и порошка, при повышенных температурах претерпевает ряд
превращений и переходит в нерастворимую в амидных растворителях
неплавкую форму полимера, состоящего из чередующихся ароматиче-
ских ядер и пятичленных гетероциклов:
В связи с тем, что тетраамины, используемые для получения
ПААК, являются в большинстве случаев нестабильными соединения-
ми, они применяются в виде тетрагидрохлоридов. Основные законо-
мерности процессов синтеза ПААК в среде апротонных растворителей
аналогичны закономерностям, наблюдаемым при получении полиами-
докислот. Методы получения пирронов различной химической струк-
туры, влияние параметров синтеза на молекулярную массу синтези-
руемых ПААК, кинетика циклодегидратации и зависимость термиче-
ских характеристик конечных полимеров от их химического строения
подробно рассмотрены в литературе [189, 190]. Циклодегидратация
ПААК может быть осуществлена с помощью химических или терми-
ческих обработок. Пирроны, получаемые в результате двухстадийного
синтеза, представляют собой окрашенные в темно-красный цвет поли-
меры, обладающие различной растворимостью в известных раствори-
телях, зависящей от исходных компонентов. Так, полимер, синтезируе-
мый на основе пиромеллитового диангидрида и 3,3'-диаминобензидина,
растворим только в концентрированной серной кислоте. Полиамидо-
15»
аминокислоты, находящиеся в реакционном растворе, могут быть непо-
средственно использованы для получения волокон по сухому илн
мокрому методам формования, но эти вещества в отличие от полиами-
докислот используются крайне редко. Основным недостатком, ограничи-
вающим практическое применение конденсационных растворов ПААК
для получения волокон, является их быстрая желатинизация [189].
Предполагают, что процесс желатинизации является следствием поли-
электролитной формы полимера, а также связан с тем, что свободные
амино- или карбоксильные группы одной молекулы могут вступать в
реакцию со свободными карбоксильными группами или аминогруппа-
ми другой молекулы, что приводит к появлению сшитых структур.
Возможны также реакции сшивки с участием свободных аминогрупп
соседних макромолекул и молекул исходного диангидрида кислоты:
Несмотря на то, что в последнее время был предложен ряд спо-
собов предотвращения желатинизации поликонденсационных раство-
ров ПААК (добавка к раствору третичных аминов [192], обработка
диазометаном с целью перевода свободных карбоксильных групп в
сложноэфирные [193]), в литературе нет сведений об использовании
реакционных растворов ПААК для формования волокон соответствую-
щей химической структуры.
Большое практическое значение для получения лестничных или
блок-лестничных волокнообразующих полимеров (в особенности для
получения полифенантролинов) имеет одностадийный способ синтеза
указанных полимерных систем в полифосфорной кислоте. Проведение
реакции поликонденсации диангидридов кислот с ароматическими тет-
рааминами в среде ПФК позволяет синтезировать более высокомоле-
кулярные полимеры с высокой степенью циклизации, обладающие
хорошей растворимостью в таких растворителях, как метансульфоно-
вая и серная кислоты и в концентрированных растворах гидроокисей
натрия и калия [194, 195].
160
Процесс образования волокнообразующих полифенантролинов в
одну стадию в среде ПФК при повышенных температурах осущест-
вляется по схеме:
Блок-лестничный полимер приведенного химического строения и
волокно ВВВ на его основе исследованы довольно подробно.
Повышенная растворимость полифенантролинов, получаемых ука-
занным методом, объясняется следующим:
1) большей легкостью образования конденсированной системы,
состоящей из комбинации пяти- и шестичленных гетероциклов; это
уменьшает вероятность побочных реакций и, следовательно, возмож-
ность возникновения межмолекулярных сшивок;
2) применением ПФК, являющейся не только катализатором кон-
денсации, но и эффективным сравнительно мягким дегидратирующим
агентом;
3) гетерогенностью реакционной смеси, связанной с незначитель-
ной растворимостью исходных диангидридов в ПФК, в результате чего
каждая новая порция диангидрида вступает в реакцию по мере израс-
ходования предыдущих порций.
Молекулярная масса полифенантролинов, синтезируемых по одно-
стадийному способу в среде ПФК, зависит от целого ряда факторов:
температуры реакции и ее продолжительности, концентрации ПФК,
соотношения исходных мономеров, концентрации реагентов и других
[196]. Важным фактором является концентрация ПФК, так как вяз-
кость реакционной среды зависит от содержания Р2О5 в фосфорной
кислоте [197]. При проведении реакции в ПФК тщательно удаляют
кислород из реакционной среды с 'целью предотвращения окисления
тетрааминов, а также хлористый водород перед началом дозирования
диангидридов (в случае использования хлоргидратов тетрааминов).
Необходимо отметить, что получение высокомолекулярных полифенан-
тролинов двухстадийным синтезом затруднено из-за низкой реакцион-
ной способности соответствующих диангидридов [194, 197].
Полиамидоаминокислоты, выделенные из реакционных растворов,
представляют собой аморфные продукты, хорошо растворимые в апро-
тонных растворителях, однако стабильность «вторичных» растворов
оказывается кратковременной, и системы быстро застудневают.
Полиимидазопирролоны и полифенантролины также имеют аморфную
структуру и интенсивную окраску: темно-красную (пирроны) и темно-
коричневую или черную с металлическим блеском (полифенантро-
лины) .
11—2254
161
Высокомолекулярные волокнообразующие лестничные или блок-
лестничные полимеры растворимы в ограниченном круге растворите-
лей, при этом характерной особенностью полифенантролинов является
растворимость их в концентрированных кислотах и щелочах. Обработ-
ка полифенантролинов разбавленными растворами щелочей приводит
к улучшению растворимости конечного продукта в концентрированной
серной кислоте [198]. Полимер типа ВВВ, выделенный из реакцион-
ного раствора в ПФК, во влажном состоянии обрабатывают водными
растворами щелочей с концентрацией 0,1—1,0 н. при 80—100°С в те-
чение 0,5—2,0 ч. Процессы, происходящие при обработке щелочью, не
описаны, и их механизм неясен. Замечено, что величина логарифмиче-
ской вязкости полимера на начальных стадиях обработки несколько
падает, а затем остается постоянной. Предполагают, что при таких
обработках разрушаются амидные связи в тех местах макромолекулы,
где не прошли процессы циклодегидратации. В результате получается
полимер с повышенной степенью циклизации, более высокими терми-
ческими характеристиками, а волокно, сформованное из обработанного
щелочью полифенантролина, обладает лучшими механическими харак-
теристиками.
Определение молекулярной массы ПААК на основе пиромеллитовой
кислоты и 3,3'-диаминобензидина показало, что для полимера с лога-
рифмической вязкостью, равной 1,45, молекулярная масса составляет
15000, а циклизация практически не сказывается на величине послед-
ней [197]. Для полифенантролина ВВВ, имеющего значения характе-
ристической вязкости в пределах 1,65—1,70 дл/г (характеристическая
вязкость определялась в 96%-ной серной кислоте), среднемассовая
молекулярная масса составляет 65000 [199].
Величина характеристической вязкости BiBB, определенная веер-
ной кислоте различной концентрации, зависит от степени диссоциации
серной кислоты, что видно из приводимых ниже данных:
Концентрация H2SO4, % [р], дл/г
104,0 2,78
99,7 2,03
97,4 1,94
97,0* 1,42
* Насыщена K2SO4.
Эти данные свидетельствуют о том, что, несмотря на большую ка-
жущуюся жесткость, лестничный полимер ведет себя в растворах
так же, как полиоксадиазолы, т. е. протонизация полимера в сильной
степени зависит от степени диссоциации серной кислоты.
4.5.2. Получение волокон на основе лестничных
и полулестничных полимеров
Волокна из полиимидазопирролонов принципиально могут быть
получены как способом сухого, так и мокрого формования [200]. При
сухом формовании используют реакционные растворы ПААК, которые
концентрируют до 20—25% (масс.) и формуют в шахту, продуваемую
азотом с температурой 225—250 °C. Отмечено, что продолжительность
пребывания нагретого раствора ПААК перед фильерой должна быть
минимальна. Свежесформованное волокно подвергают двухступенча-
162
той вытяжке при повышенных температурах (280—500 °C) и дополни-
тельной терморелаксации при 350 °C. Операции термообработки про-
водят в инертной атмосфере. Некоторые свойства волокон*, получен-
ных на основе пиромеллитового диангидрида и 3,3'-диамииобензидина
[200], приведены ниже:
Показатель
Прочность, сН/текс
при разрыве.........................27—33
в узле.........................15—16
в петле.........................11—12
Удлинение, %
при разрыве.........................20—21
в узле.........................12—13
в петле.........................4—5
Начальный модуль, МПа................. 11000
Рис. 4.40. Зависимость вязкости растворов ВВВ
в серной кислоте >(« Па-с) от концентрации
полимера в растворе и величины ‘Приведенной
вязкости полимера [188]:
1 — 50; 2 — 100; 3 — 200; 4 — 400; 5 — 800.
Формование волокон из полиимидазопирролонов по мокрому спо-
собу в литературе не описано.
Несколько подробнее изучены процессы, связанные с получением
волокна на основе полифенантролина типа ВВВ [188; 201, 202]. Волокно
получают по мокрому способу
формования из растворов в
концентрированной серной кис-
лоте. Характерной особен-
ностью прядильных растворов
полимера ВВВ в серной кисло-
те является их высокая эффек-
тивная вязкость даже при срав-
нительно небольших концент-
рациях полимера (рис. 4.40).
Исследование вязкости
концентрированных растворов
ВВВ показало, что они ведут
себя аналогично концентриро-
ванным растворам полимеров,
макромолекулы которых име-
ют форму статистического
клубка [203]. Однако в отли-
чие от них образование за-
цеплений между макромоле-
кулами наблюдается при чрезвычайно низких концентрациях поли-
мера в растворе. Согласно рентгенографическим исследованиям в упо-
рядоченных областях цепи упакованы так, что их плоскости параллель-
ны друг другу. Отсутствие анизотропии концентрированных растворов
ВВВ в серной или метансульфоновой кислотах, наблюдаемой, например,
для растворов жесткоцепных ароматических полиамидов n-структуры в
тех же растворителях, связано, по-видимому, с тем, что ВВВ имеет ста-
тистическое распределение звеньев в цис- и транс-форме, а также с
неполнотой циклизации. Концентрированные растворы ВВВ в серной
кислоте являются достаточно стабильными системами.
Механизм сернокислотного формования волокон на основе поли-
фенаитролинов практически не изучен. Лишь в одной работе была
* Волокна темно-красного цвета с металлическим блеском.
11*
163
сделана попытка исследовать влияние молекулярной массы полимера,
температуры и состава осадительной ванны на свойства волокна ВВВ
[204], причем в качестве критерия был использован так называемый
«фактор растяжения»* [205]. Осадительными ваннами являлись вод-
ные растворы серной кислоты. Найдено, что волокно с оптимальными
свойствами получается при формовании раствора полимера с
T]ig=2,6—3,1 в 70%-ную серную кислоту при температуре около 60°C.
Свежесформованное волокно отмывается от растворителя, высуши-
вается и затем вытягивается при повышенных температурах. Влияние
температуры вытягивания на механические показатели волокна ВВВ
иллюстрируется данными табл. 4.39.
Таблица 4.39. Влияние температуры вытягивания на степень вытяжки
и механические свойства волокна ВВВ [201]
(rjig полимера=2,0; вытяжка проводилась в среде аргона)
Температура вытяжки, СС Максимальная вытяжка, % Прочность, сН/текс Удлинение, % Начальный модуль, Юв-Па
170 31 13,4 18,1 7160
230 31 13,5 14,2 7300
290 31 13,6 12,3 9320
340 31 15,1 10,0 6 000
400 40 18,2 5,1 10650
425 53 20,9 4,5 16 700
500 60 25,8 4,2 16 700
550 60 28,1 4,0 17000
590 65 32,1 3,9 17 970
620 55 31,5 3,9 17 000
650 76 34,4 2,9 23 900
Отмечается [201], что вытяжка проводится с низкой скоростью,
продолжительность пребывания волокна в зоне вытягивания состав-
ляет примерно 20 с. За это время волокно успевает прогреться до
необходимой температуры. Было отмечено, что при осуществлении
термической вытяжки с одинаковой кратностью, но при различных
температурах прочность волокна повышается по мере увеличения тем-
пературы вытяжки (табл. 4.40).
Таблица 4.40. Зависимость механических свойств волокна ВВВ
от температуры вытяжки (вытягивание проводилось в среде аргона;
степень вытяжки постоянна и равна 55%; rpg = 2,0)
Температура вытяжки, °C Прочность, сН/текс Удлинение, % Начальный модуль, 106-Па
425 21,6 4,5 16500
450 22,8 4,5 16700
500 24,6 4,6 16700
550 25,9 4,7 17000
590 30,8 4,8 19000
620 31,5 3,9 17000
650 32,4 2,9 23000
* Фактор .растяжения (Ф. ,р.) равен Р-/1/2,где Р — прочность волокна, а I — удли-
нение.
164
Как уже указывалось выше, свойства волокна ВВВ зависят
не только от молекулярной массы полимера, но и в значительной
мере от его предыстории. Согласно [198], волокно ВВВ, сформованное
из полимера, который был обработан раствором щелочи, имеет более
высокую прочность по сравнению с волокном, сформованным из необра-
ботанного полимера (табл. 4.41).
Таблица 4.41. Влияние обработки исходного полимера щелочью
на свойства волокон ВВВ
Температура, °C Степень вытяжки Обработанный полимер Необработанный полимер
прочность, сН/текс удлинение, % прочность, сН/текс удлинение, %
525 50 29,7 14,4 22,4 14,8
80 37,3 6,8 34,6 5,1
НО .— — 29,6 6,0
550 50 29,7 9,3 26,6 11,8
80 37,2 8,5 40,9 4,5
НО —. — 31,3 3,6
575 50 37,0 10,9 24,7 11,6
80 44,2 5,0 36,0 5,6
ПО — — 39,6 4,6
Применение органических осадительных ванн, состоящих из амид-
ных растворителей, таких, как ДМАА, МП, ДМФ или их смесей, дает
возможность получать волокна ВВВ, обладающие плотной непористой
структурой [206].
4.5.3. Физико-механические свойства волокон
Несмотря на то, что в последние годы получено большое число
полимеров лестничной или блок-лестничной структуры, волокон лест-
ничного строения известно немного. Механические характеристики
практически всех известных волокон лестничной или блок-лестничной
структуры, а также способы их получения приведены в табл. 4.42.
Прочность волокон, химическая структура которых указана в
таблице, оказывается невысокой по сравнению с прочностью волокон
из других гетероциклических полимеров, например полиимидов, по-
либензимидазолов. Необходимо отметить, что, такие физико-механиче-
ские свойства лестничных волокон, как влагопоглощение, усталостные
характеристики и другие, в технической литературе не указаны. Более
подробно описаны свойства волокна лола, полученного советскими ис-
следователями путем мокрого формования сернокислотных растворов
полимера, имеющего предположительно блок-лестничную структуру
(табл. 4.43) [216].
4.5.4. Термомеханические свойства лестничных волокон
Волокнообразующие полимеры лестничной и полулестничнон
структуры являются неплавкими, а температура начала термиче-
ской или термоокислительной деструкции превышает 500 °C.
Температуру стеклования не удалось определить ни для
одного лестничного или полулестничного волокнообразующего
165
$
Таблица 4.41 Волокна лестничного или блок-лестничиого строении
Химическая формула элекёйТйрНого эвена полимера, класс полимера П1й поли- мера ‘ Температура начала деструкции, °C Способ получения полимера и волокна Показатели волокон Другие свойства волокон Лите- ратура
в среде азота на воз- духе проч- ность, сН/текс Удли- нение, %
N N О О полибисбензимидазобензофенантролин (ВВВ) 1,0—4,0 650— 700 550— 600 Полимер получен поликонденсацией 3,3'-диаминобензиди- на и диаигидрида 1,4,5,8-нафталиитет- ракарбоновой кисло- ты в среде ПФК. Волокно формовали из раствора в кон- центрированной H2SO4; осадительная ванна — водный рас- твор H2SO4 40—45 3,0— 3,5 Термомеханиче- ские свойства см. раздел 4.54 (188; 196; 201]
N N II 11 О О полиимидазопирролон 0,92 600 450— 480 Двухстадийный.' ПААК синтезирована в среде ДМСО. Реак- ционный раствор с концентрацией 25% (масс.) формовали по сухому способу в сре- де азота. Волокно подверга- лось ступенчатой вы- тяжке (на 20% при 280 °C и иа 20% при 500 °C) и дополни- тельной термообра- ботке в течение 15 мин, при 350°C 27—33 20—21 Механические свойства волокон практически не из- меняются при дей- ствии горячих ще- лочей (нагревание в течение 10—12 ч в 10%-ном раство- ре NaOH и при выдерживании на воздухе в течение 5 ч при 400 °C) [200; 207]
N N
2,0—3,0
поли&исбензимидазобензофенантролин .(BBL)
2,2
полибисбензимидазофенантролин, содержащий
антрахиноновые циклы
650— 550— Полимер получают
700 600 поликонденсацией тетрааминобензола н диангидрида нафта- лин- 1,4,5,8-тетракар- боновой кислоты в ПФК. Волокно фор- муют так же, как и ВВВ; отмечается очень высокая вяз- кость прядильных растворов BBL прн низких концентраци- ях полимера (3—4%)
600 550 Полимер получен поликонденсацией диангидрида нафта- лин-1,4,5,8-тетракар- боновой кислоты и 1,2,5,6-тетраминоан- трахинона в ПФК при 200—240 °C. Волокно формуют из 10%-кого раство- ра полимера в ДМСО, содержащем небольшие добавки щелочи, воды и дн- тионата натрия. Спо- соб формования — мокрый. Осадитель — вод- ный раствор соляной кислоты
Конкретные ме-
ханические свой-
ства волбкон не
приводятся; отме-
чается, что они
близки к свойст-
вам волокон типа
ввв
Механические
свойства волокон
не приводятся.
Отмечается, что
на воздухе волок-
но от желтого или
красноватого цве-
та переходит в
темный цвет
[208;
209]
[210;
211]
Химическая формула элементарного звена полимера,
класс полимера
nig
поли-
мера
1,0—1,5
полибпсбензимидазопирролон
1,0—2,0
полибензофуроизохинолин *
Продолжение
Температура начала деструкции, °C Способ получения полимера и волокна Показатели волокон Другие свойства волокон Лите- ратура
в среде азота на воз- духе проч- ность, сН/текс удли- нение, %
600 500 Полимер получен поликонденсацией пиромеллитового ди- ангидрида и тетра- аминодибензодиокси- на в амидном раство- рителе с дальнейшей циклодегидратацией в вакууме при 250— 300 °C. Способ полу- чения волокна не опи- сан. 15—18 2—4 Указана проч- ность пленок, ориентированных при 350 °C на 15% [212]
650 550 Получен поликон- денсацией диангидри- да 1,4,5,8-нафталин- тетракарбоновой кис- лоты с 2,3,7,8-тетра- аминобензофураном в треххлористой сурь- ме. Волокно формуют мокрым способом из раствора в метан- сульфокислоте; оса- дитель — метанол. 16—18 8—11 Термомеханиче- ские и другие свойства волокон не приводятся [213]
0,47
лестничный полимер графитоподобной структуры
1,0
1полихи1ноксали1н
>550 >550 Поликонденсацией 10-16 8—10 Другие свойства
1,4,5,8-тетрааминоан- трахннона с 1,3,6,8- тетроксо-1,2,3,6,7,8- гексагидропиреном в ПФК. Волокно фор- мовали из 14%-ных растворов в метан- сульфокислоте, осади- тель — водный рас- твор метансульфокис- лоты (хруп- кое во- локно) волокон не приво- дятся
530 510 Поликонденсацией диаминобензидина и 1,4-бис (фенилглиок- салил) бензола в хло- роформе и в твердой фазе. Волокно фор- муют по мокрому способу из 20%-ного раствора полимера в л-крезоле; осади- тель — метанол. 18,0 10—12 Термомеханиче- ские свойства не описаны
1214]
[215]
полимера или волокна. Исследования показали, что волокна
лестничного строения, в частности ВВВ, отличаются высокой
теплостойкостью. На рис. 4.41 представлены данные, характеризующие
Таблица 4.43. Физико-механические показатели волокна лола [207]
Показатель Исходное Упрочненное
ВОЛОКНО волокно
Линейная плотность, текс 25—50 17—33
Прочность, сН/текс 15—20 35—40
Удлинение, % 15—25 2—6
Начальный модуль (акустический), 10е Па 9000 28000
Плотность, кг/м3 1450 1350
Равновесное влагосодержаиие (при 20 °C и 65%-ной отно- сительной влажности), % 8,0 3,3
Относительная прочность, % 100,0 98,0
в мокром состоянии
в узле 82,0 50,0
в петле 98,5 45,0
Усадка, %
в кипящей воде 1,50 1,30
при 300 °C на воздухе 0,45 0,14
при 350 °C иа воздухе 0,60 0,23
при 400 °C на воздухе 0,70 0,30
зависимость прочности волокон ВВВ и лола от температуры испыта-
ния (для сравнения здесь же приведена теплостойкость ПБИ во-
локна).
Приведенные данные свидетельствуют о том, что теплостойкость
лестничного волокна ВВВ, даже не прошедшего стадию дополнитель-
но 200 300 W0 500 SOO 700 800
t,°C
Рис. 4.41. Зависимость прочности
лестничных волокон от температуры
испытания [188; 216; 217]:
1 — ПБИ волокно; 2— волокно лола, тер-
мообработанное; 3— ВВВ; 4 — ВВВ, об-
работанное в пламени пропановой горелки.
ной термообработки, превышает теп-
лостойкость всех известных термостой-
ких волокон. Теплостойкость волок-
на лола близка к теплостойкости ВВВ.
Термостабильность лестничных или
блок-лестничных волокон, вопреки
ожиданиям и, несмотря на очень вы-
сокие температуры начала термоокис-
лительной деструкции, оказалась не-
намного выше термостабильности во-
локон из полиоксадиазолов (рис. 4.42
и 4.43), в особенности при температу-
рах, превышающих 300 °C.
Термические свойства волокна
ВВВ могут быть улучшены путем до-
полнительной обработки при темпера-
турах выше 800 °C [217]. Волокно
подвергают кратковременному предва-
рительному нагреву при 500—700 °C (в течение 0,2 с) и затем термооб-
работке при 1200 °C в пламени пропановой горелки в атмосфере аргона
в течение 0,001—0,05 с. Предполагают, что в результате таких обрабо-
ток изменяется химическая структура полимера, происходит декарбони-
170
лирование полифенантролина и образуется полимер новой структуры:
После обработки ВВВ в пламени пропановой горелки прочность
волокна увеличивается на 3—10% по сравнению с исходным, волокно
Рис. 4.42.. Влияние теплового
старения на прочность волокон
лола и 'ПОД .волокна [157,
216]:
1 и 2 — лола, соответственно исход-
ное и термоупрочнеяное; 3 — ПОД
волокно.
(температура 300 °C).
становится более эластичным, и, главное, повышаются его теплостой-
кость и термостабильность [217]. Так, если теплостойкость исходного
волокна при 600 °C составляет 45—50%, то после термообработки она
Рис. 4.43. Зависимость прочности во-
локон ПОД, ВВВ и лола от продол-
жительности старения при повышен-
ных температурах на воздухе [146;
162; 188; 216]:
1 и 2 — волокно лола соответственно исход-
ное и термоупрочненное (350 °C); 3 и 5 —
ПОД соответственно при 350 и 400 °C;
4 — ВВВ (360 °C).
повышается до 60% (см. рис. 4.41). Волокно разрушается при 900 °C.
После 30-часового нагревания при 360 °C на воздухе потеря прочности
волокна незначительна, в то время как исходное волокно теряет почти
171
Таблица 4.44. Огнестойкость изделий из волокна лола [216]
Изделие из волокна лола Температура воспла- менения в среде кислорода Кислородный индекс, %
Исходная ткань 595 54
Ткань, обработанная фосфор- н галогенсодер- жащнми антипиренами 600 63
Трикотаж, обработанный фосфорсодержащими реагентами 600 70
половину исходной прочности. Отмечается, что волокна на основе
лестничных полимеров являются наиболее огнестойкими из термостой-
ких волокон [218]. Волокно лола в пламени пропановой горелки
(1200 °C) в среде воздуха лишь раскаляется докрасна, но не воспла-
меняется [216]. Стойкость волокна лола к действию открытого пла-
мени, охарактеризованная по величине кислородного индекса, иллю-
стрируется данными табл. 4.44.
4.5.5. Другие физико-химические свойства волокон
Лестничные волокна являются одними из немногих термостойких
волокон, которые обладают высокой хемостойкостью (табл. 4.45).
Таблица 4.45. Химическая стойкость волокна лола [216]
Среда Условия испытания Сохранение прочности волокон, % от исходной
температура, °C продолжи- тельность, ч
H2SO4 (10%-ная) 20 150 98,5
100 5 100,0
H2SO4 (50%-ная) 20 100 84,7
124 5 83,3
КОН (10%-ный) 20 150 98,5
100 5 100,0
М,М-Диметилформамид 20 150 90,0
150 5 99,4
Волокно лола устойчиво к действию разбавленных и концентриро-
ванных кислот, щелочей и органических апротонных растворителей.
В 96%-ной серной кислоте волокно, прошедшее стадию термовытяжки,
лишь незначительно набухает. Рядом авторов отмечается исключитель-
но высокая радиационная стойкость пирронов: изделия из пирроно»
практически не теряют прочность и эластичность после облучения
дозой до 10 000 Мрад при бомбардировке электронами с энергией
2 Мэв в вакууме, а пленки на основе пиромеллитовой кислоты и
Таблица 4.46. Светостойкость волокна ВВВ [188]
Экспозиция, ч Изменение свойств
прочность, сН/текс удлинение, % начальный модуль, 108-Па
Исходное 35,1 3,4 21000
100 34,2 3,1 21500
200 38,7 3,8 19000
400 36,0 3,4 22000
172
З^'-диаминобензидина устойчивы к дозам вплоть до 22 000 Мраддаже
на воздухе при воздействии пучка электронов с энергией 3 Мэв [197].
К сожалению, данные, характеризующие радиационную устойчивость
волокон лестничного строения, отсутствуют.
Практически все химические волокна, в том числе и термостойкие,
изменяют свои механические характеристики под действием УФ-облу-
чения, и для них требуется защита светостабилизаторами. Полифенан-
тролиновое волокно типа ВВВ в этом смысле обладает исключитель-
ными показателями (табл. 4.46).
4.5.6. Волокнообразующие лестничные полимеры
других типов
К лестничным полимерам, близким по строению к пирронам, от-
носятся полихиноксалины и полиизоиндолхиназолиндионы. Первые
сведения о них появились в 1966—1967 г.г. [219], при этом указыва-
лось, что полимеры обладают высокой термостабильностью, высокой
молекулярной массой; некоторые из них хорошо растворяются в до-
ступных растворителях с образованием концентрированных растворов.
Высокомолекулярные полихиноксалины получают путем поликонденса-
ции ароматических тетраминов с тетракарбонильными соединениями
типа диглиоксалей:
Полимер синтезируют в две стадии. На первой стадии в среде
амидных растворителей, хлороформе, диоксане получают полимеры,
которые самопроизвольно выпадают из раствора в виде низкомолеку-
лярных порошков. Порошок выделяется, и вторая стадия проводится
при высоких температурах в вакууме, причем дальнейшее увеличение
молекулярной массы осуществляется в твердой фазе. Некоторые
полимеры растворимы в крезоле, муравьиной, серной или метансуль-
фоновой кислотах; на основе одного из них сформовано волокно (см.
табл. 4.42).
Полиизоиндолхиназолиндионы также получают двухстадийным
способом. Исходными реагентами для них являются диангидриды
ароматических тетракарбоновых кислот и ароматические бис(2-амино-
карб-4-аминофенилы):
173
Реакция проводится в расплаве или в полярных растворителях,
инертных к исходным мономерам [220]. Независимо от способа про-
ведения первой стадии полимерообразования вторая стадия должна
осуществляться при температурах выше 200 °C в твердой фазе в вы-
соком вакууме, при этом наблюдается как увеличение молекулярной
массы полимера, так и внутримолекулярная циклодегидратация. Поли-
меры и изделия из них не плавятся до температуры 550 °C; окрашены в
темно-красный цвет. Промежуточные продукты бесцветны. Сообщается,
что полимеры являются волокнообразующими, однако свойства воло-
кон не приводятся. Неориентированная пленка из полиизоиндолхина-
золиндиона вышеуказанной химической структуры после термоциклиза-
ции имеет прочность 167 мПа [220].
Значительный интерес представляют органические волокнообра-
зующие полимеры графитоподобной структуры. Такие полимеры полу-
чают поликонденсацией 1,4,5,8-тетрааминоантрахинона с производны-
ми нафталинтетракарбоновой кислоты, например с нафталин-1,8,4,5-
дииндандионом или 1,3,6,8-тетраоксо-1 Д,3,6,7,8-гексагидропиреном
[214]. Реакцию проводят в высококипящих полярных растворите-
лях— ПФК, ДМАА, ГМФА. Реакция многостадийна, предполагается,
что она протекает по схеме:
NH2 О nh2
nh2 о nh2
174
При нагревании эквимольных количеств исходных реагентов в
среде полярного растворителя до температуры порядка 175 °C в ре-
зультате взаимодействия аминогрупп тетрамина с карбоксильными
группами диангидрида образуется незациклизованный промежуточный
продукт I. Равновероятность образования двух типов структур I не
доказана. Выделенный из реакционной среды промежуточный продукт
структуры 1 подвергают дальнейшей термообработке при 200—250 °C
в вакууме или в инертной среде. В результате реакции соседних
амино- и карбонильных групп проходит частичная внутримолекулярная
циклизация с образованием полимера структуры II, содержащего
внутри макроциклов реакционноспособные группы. Продукты, имею-
щие структуру I и II, растворимы в концентрированной серной кисло-
те, а также в смешанном растворителе, состоящем из ДМСО, КОН,
Na2SO4 и содержащего 1—2% (масс.) воды. Указывается, что из
8%-ных растворов в любом из этих растворителей по мокрому
способу формования могут быть получены волокна, которые после до-
полнительной термообработки при 350—400 °C в среде азота приобре-
тают высокую термическую стабильность и имеют химическую струк-
туру типа III. Полимер структуры III может быть получен при обра-
ботке в инертной атмосфере или в вакууме полимера структуры II; он
растворим в метан-сульфокислоте. Механические свойства волокна из
такого полимера и способ получения волокна приведены в табл. 4.42.
Более подробно способы синтеза различных полимеров лестнич-
ной или блок-лестничной структуры рассмотрены в литературе [189].
Здесь же необходимо отметить, что, несмотря на труднодоступность
исходного сырья, многостадийность процессов получения как лестнич-
175
ных полимеров, так и соответствующих волокон, последние представ-
ляют значительный практический и научный интерес и являются са-
мыми тепло- и термостойкими. Дальнейшие исследования в области
получения лестничных волокон направлены как на поиск новых хими-
ческих структур, обеспечивающих высокую термостабильность изде-
лиям на их основе, так и на усовершенствование способов синтеза
исходных полимеров. В этом смысле определенный интерес представ-
ляет способ получения блок-лестничного полимера типа ВВВ путем
обработки полибензимидазолов фосгеном в присутствии треххлористо-
го алюминия [221]. Процесс идет по следующей схеме:
А1С13: СОС12
Обнаружено [208], что при проведении синтеза полимера типа BBL
в ПФК в присутствии небольшого количества кислот Льюиса может
значительно повыситься молекулярная масса полимера, при этом по-
лимер обладает хорошей растворимостью в концентрированной серной
кислоте и образует растворы, пригодные для формования волокон.
Полагают, что применение в качестве реакционной среды расплавлен-
ных кислот Льюиса даст возможность синтезировать высокомолекуляр-
ные волокнообразующие полимеры типа ВВВ, полихиназолиндионы,
полихиноксалины, в том числе и фенилированные полихиноксалины
[222], интерес к которым в последнее время значительно возрос
[189; 223].
4.6. ПОЛИАМИДОИМИДНЫЕ ВОЛОКНА
4.6.1. Получение волокон
Полностью ароматические полиамидоимидные волокна привле-
кают внимание исследователей тем, что сочетают в себе свойства во-
локон из полностью ароматических полиамидов и полиимидов. Извест-
ны три способа получения полимеров и волокон на их основе [224].
Первый способ основан на взаимодействии хлорангидридан-
гидрида тримеллитовой кислоты с ароматическими диаминами, наи-
большее распространение из которых получили двухъядериые
(4,4'-диаминодифенилметан и 4,4'-диаминодифенилоксид). Реакция
идет по схеме:
176
Процесс образования полимера на стадии получения полиамидо1
кислоты осуществляется в амидных растворителях. Хлористый водо-
род, выделяющийся при этом, необходимо нейтрализовать до стадии
имидизации. Волокна формуют из растворов полиамидокислоты сухим
способом. Отмытое от растворителя и хлоридов волокно подвергают
имидизации в вакууме при 240—250 °C. Имидизованное волокно под-
вергают термовытяжке при температуре до 350 °C.
Второй способ. В том случае, когда хотят избежать стадии
имидизации, в качестве исходных продуктов применяют мономеры, в
частности дихлорангидриды, содержащие имидные циклы. Однако при
этом способе синтеза также необходимо удалять хлористый водород,
что в ряде случаев оказывается технологически неудобно. Таким путем
получают упорядоченные полиамидоимиды и волокна на их основе,
причем волокна формуют преимущественно по сухому способу. Свой-
ства волокна на основе упорядоченного полидифенилметантримеллит-
имидоамида приведены в табл. 4.47.
Третий способ. Был найден прямой синтез полиамидоимидов
путем взаимодействия триметиллитового ангидрида с ароматическими
диизоцианатами по реакции:
12—2254
177
Таблица 4.47. Физико-механические свойства
Химическая формула элементарного звена
(сополимер гриме ллитовой кислоты, 4,4-дифеиилметандиизоцианата и изофталевой кислоты)
(сополимер тримеллитовой кислоты, 4,4-дифенилметандиизоциоиата и терефталевой кислоты)
178
полнамидоимидиых волокон
Температу- ра начала разложения на воздухе, °C Способ получения волокон Механические свойства Литература
прочность, сН/текс удлинение, % модуль растяже- ния, 10бПа
380—400 Сухой, из растворов в МП Мокрый, из амидных растворите- лей 36—45 25—50 15—20 10—25 810 700—800 [86; 224]
— Сухой, из растворов в амидных растворителях 40—41 12—13 945 [224]
380—400 Мокрый, из растворов в амидных растворителях 40—60 10—20 800—1300 1224]
360—400 Сухой, из растворов в МП с по- следующей вытяжкой 25—40 10—18 400—600 [227]
360—400 Сухой, из растворов в МП, с по- следующей вытяжкой 47—67 10—15 — [227]?
12*
Реакция проводится в полярных амидных растворителях. В связи
с высокой реакционной способностью диизоцианатов растворители и
мономеры должны быть тщательно очищены. Основным достоинством
указанного способа является отсутствие побочных продуктов реакции,
так ,как двуокись углерода полностью удаляется. Согласно данным
ИК-спектроскопии, имидизация, идущая непосредственно в реакцион-
ной смеси, протекает достаточно глубоко. Раствор полиамидоимида
после фильтрации и обезвоздушивания направляется на формование.
Формование волокон осуществляется по сухому или мокрому спосо-
бам. В прядильный раствор перед формованием могут вводиться раз-
личные добавки, красители, матирующие присадки и т. д. При формо-
вании волокон по сухому способу используются полимеры с логариф-
мической вязкостью 0,9—1,6. Растворы имеют концентрацию 18—30%
(масс.). В качестве растворителей применяют МП и ДМАА, иногда
содержащие добавки легкокипящих инертных растворителей (толуол
и т.д.) [225]. Скорость формования 150—200 м/мин; фильера должна
иметь температуру 60—1ЙЬ°С [226]. Свёжесформованные волокна,,
содержащие 10—20% (масс.) растворителей, промывают водой, сушат
в вакууме или в инертной атмосфере.
Для получения волокон по мокрому способу формования приме-
няют полимеры с логарифмической вязкостью 1,0—2,5 [225]. Концент-
рация прядильных растворов 8^15% (масс.).
Механические свойства свежесформованных волокон относитель-
но невысоки (примерно 10—15 сН/текс) при удлинении 50—80% •
Ориентационное вытягивание при температурах 200—350 °C приводит
к повышению прочностных характеристик волокон. Физико-механиче-
ские свойства волокон на основе полидифенилметантримеллитамидо-
имида и полидифенилоксидтримеллитимидоамида приведены в
табл. 4.47. Из представленных данных видно, что анализируемые во-
локна обладают средней механической прочностью.
Были сделаны попытки получить сополимеры и волокна на основе
-тримеллитовой кислоты, 4,4,-дифенилметандиизоцианата и третьего
компонента. В-.качестве последнего применялись изофталевая и.терег
<фталевая кислоты, 4,4/-дифенилдикарбоновая кислота, а также пиро-
меллитовый диангидрид [227]. Во всех случаях процесс получения
полимеров и волокон протекал без осложнений, а основные характе-
ристики сополимерных волокон оказывались близки к характеристикам
волокон, полученных из гомополимеров.
4.6.2. Физике- и термомеханические свойства волокон
Из полиамидоимидных волокон наиболее известно волокно на
основе тримеллитовой кислоты и 4,4'-дифенилметандиизоцианата, вы-
пускаемое в промышленном масштабе фирмой «Рон-Пулен-Текстиль»
(Франция) под торговой маркой «кермель» [86, 228]. Свойства волок-
на кермель приведены ниже:
Линейная плотность, текс.......................0,2—0,3
Прочность при растяжении, сН/текс .... 24,5—44,1
Удлинение, %...................................10—20
Начальный модуль, 10® Па....................... 7800
Плотность, кг/м3.................................. 1390
Равновесное влагопоглощение, % (при 65%-ной
180
относительной влажности)........................2,22—3,4
Усадка, %
в воде, при 95 °C........................... 0,6
на воздухе, при 190°C........................... 0,6
Удельная теплоемкость, кДж/(кг-К) .... 1,29
Кислородный индекс, %...........................30—32
। । 1 । । -
200 400 600 800 1000
Ъ,ч
Рис. 4.44. Влияние теплового старения
при 260 X на прочность полиамидо-
имадных волокон [128; 224]:
1 _ на основе 4,4'-диаминодифенилметана;
2 —на основе 4,4'-диаминодифенилового
эфира; 3 — полиимидное ПМ (при 300 °C).
Несмотря на то, что эти волокна получены на основе полностью
ароматического полиамидоимида, их термостабильность оказывается
относительно невысокой по сравнению с полностью ароматическими
полиимидами и полиамидами (рис. 4.44).
Термостойкость полиамидоимид-
ных волокон в значительной мере
зависит от их химического «троения.
Волокна с оксидной группировкой
оказываются более стойкими к термо-
окислительной деструкции, чем волок-
на с дифенилметановой группой. После
1000 ч прогрева при 260 °C на воздухе
волокна первого типа сохраняют около
80% первоначальной прочности, в то
время как волокна второго типа со-
храняют только 30%. При 380—400 °C
эти волокна начинают разлагаться с
обугливанием как в среде азота, так
и в воздушной атмосфере [224].
Теплостойкость полиамидоимид-
ных волокон достаточно высока. Так,
при 260 °C прочность обоих типов во-
локон составляет выше 50% от исХ'
полиамидоимидных волокон является их огнестойкость. Волокна него-
рючи, при действии открытого пламени только обугливаются. По вели-
чине кислородного индекса полиамидоимидные волокна сравнимы с
волокнами на основе ароматических полиамидов, практически не уса-
живаются в пламени. Положительным свойством волокон является ма-
лое выделение дыма и отсутствие ярко выраженных токсичных продук-
тов сгорания и термоокисления {86; 224].
Основным ДОСТОИНСТВОМ
4.7. ТЕРМОСТОЙКИЕ ВОЛОКНА ДРУГИХ ТИПОВ
Из других термостойких волокон, которые не нашли практическо-
го применения, можно отметить политиадиазольные, полифенилен-
триазольные и полибензоксазольные. Указывается также, что в прин-
ципе можно получить волокна на основе полибензтиазолов [229],
однако их термические свойства не описаны.
4.7.1. Политиадиазольные волокна
Политиадиазольные волокна по своей химической структуре близ-
ки к оксадиазольным. Основной стадией процесса получения политиа-
диазолов является синтез полиоксатиогидразидов или политиогидрази-
дов высокой молекулярной массы [230]. Из многих способов синтеза
указанных продуктов наиболее эффективным оказалось взаимодейст-
181
Таблица 4.48. Фнзико-хнмнческие н механические свойства некоторых полибензоксазольных, полнтиадиазольных
н полифенилентрназольных волокон
Химическая формула элементарного звена
Температура, °C Способ получения волокон Проч- ность, сН/текс Удли- нение , % Модуль растяже- ния, 10вПа Лите- ратура
стекло- вания полимера начала разложения на воздухе
— >450 Мокрый, из растворов в МП, с вытяжкой в 5 раз при 280 °C и цик- лизацией в вакууме при 325—375 °C 20—23 6-8 — [233]
>450 Мокрый, из 30%-ных растворов полиоксиами- да в ДМАА, с вытяжкой при 270 °C в 3,7 раза и циклизацией при 280 °C в азоте в течение 4 ч 17—19 29—30 2800 [234]
>360 >550 Мокрый, из 28%-ных растворов в ДМАА. Оса- дитель — смесь воды и ДММА. Дополнительная вытяжка при 220 °C 18—25 16—22 — [235]
320—340 >450 Сухой, из растворов в ДМАА промежуточных политногидразидов 30—35 14—15 12000 [54]
265 400 Сухой или мокрый, нз растворов в муравьиной кислоте, вытяжка в две стадии при 300—360 °C 18—28 8—18 3500— —10000 [54]
вие ароматических полигидразидов высокой молекулярной массы с
пятисернистым фосфором в среде кипящего пиридина [54, с. 121]:
о
О
II II
—R—С—NHNH—С—
48 ч
+ P2S3
30 мин
Промежуточные продукты реакции хорошо растворимы в ДМАА
с образованием стабильных растворов. Волокна из растворов проме-
жуточных продуктов формуют по сухому способу. После отмывки от
растворителя и сушки волокно вытягивают при 250—275 °C на нагре-
той поверхности, и в процессе термической вытяжки происходит непо-
средственно образование тиадиазольного цикла. В отличие от образо-
вания оксадиазольного цикла тиадиазольный цикл замыкается очень
быстро и степень циклизации оказывается высокой [230; 54, с. 121].
Физико-механические свойства поли-1,3,4-тиадиазольного волокна при-
ведены в табл. 4.48.
4.7.2. Волокна из полифенилентриазолов
Ароматические полигидразиды являются хорошим исходным мате-
риалом для получения других полимеров. Обработка ароматических
полигидразидов анилином в присутствии ПФК приводит к полифени-
лентриазолам [54, с. 99]
Макромолекулы полифенилентриазола содержат термостабильные
структурные фрагменты: звенья бензола и гетероциклические группи-
ровки N-замещенного триазола. Полимеры обладают хорошей раство-
римостью в амидных растворителях и муравьиной кислоте. Волокна
могут быть получены по способам сухого или мокрого формования.
Свежесформованные волокна обладают высокой пластичностью, хоро-
шо вытягиваются при повышенных температурах, однако, несмотря на
это, степень ориентации, кристалличность и механические показатели
нитей оказываются довольно низкими (табл. 4.48) [54].
183
4.7.3. Полибензоксазольные волокна
Волокнообразующие ароматические полибензоксазолы (ПВО),
макромолекулы которых состоят из бензоксазольных циклов и аро-
матических фрагментов, получают методом двухстадийного синтеза из
бис (2-ОКСИ-4-аминофенилов) и дихлорангидридов ароматических ди-
карбоновых кислот [231]:
На первой стадии различными методами поликонденсации [190,
с. 70], из которых наиболее эффективной является низкотемператур-
ная поликонденсация в амидных растворителях, получают промежу-
точные полиоксиамиды; полиоксиамиды затем термической циклоде-
гидратацией превращают в ПВО. Промежуточные продукты растворя-
ются в амидных растворителях, обладают хорошей волокнообразую-
щей способностью. Циклодегидратацию проводят на готовых волокнах
при 200—500 °C в вакууме или инертной атмосфере [190]. Аромати-
ческие ПВО характеризуются высокой термостабильностью. Термогра-
виметрический анализ полимеров показывает, что разложение их на
воздухе начинается при 450—500 °C. В настоящее время имеется боль-
шое число ПВО, отличающихся высокой термической стабильностью.
Ароматические ПВО не растворяются в органических растворителях,
поэтому единственным методом получения волокон на их основе явля-
ется сухое или мокрое формование промежуточного полиоксиамида и
последующая циклодегидратация ориентированных полиоксиамидных
волокон. Отмечается трудность достижения 100%-ной циклизации во-
локон [190; 232], связанная с одновременно протекающими процессами
термодеструкции и разрушения физической структуры волокон.
Физико-механические свойства волокон из поли-2,2'-лг-фенилен-5,5'-
дибензоксазола приведены ниже:
Линейная плотность, текс............................. 10—30
Прочность, сН/текс.................................. 35—65
Удлинение, %............................................... 1—8
Начальный модуль, 10е Па.............................. 15000—17000
Прочность в петле, % от исходной............................ 82
Плотность, кг/м3........................................... 1300
Усадка при 300—400 °C, %...................................... 0
Ползучесть, % (при нагрузке 10% от разрывной, 300 °C и
продолжительности действия 25 ч)....................... 3
Потеря прочности после УФ-облучения в течение 85 ч, % 12
184
Отмечаются негорючесть ПБО волокон, их высокие диэлектриче-
ские характеристики и высокая стойкость к действию кислот и щело-
чей [190].
Механические свойства известных ПБО волокон указаны в
табл. 4.48.
4.7.4. Термостабильность волокон
Включение бензоксазольных, тиадиазольных и других циклов в
макромолекулы полимеров приводит к получению высокотермостойких
продуктов (см. табл. 4.48). Термостабильность некоторых волокон при-
ведена на рис. 4.45.
Рис. 4.45. Влияние продолжительно-
сти прогрева при 300 ‘С на воздухе
на прочность волокон [54; 230; 231]:
1 — политиадиазольиое; 2 — ПБО; <3 — по-
лифеиилеитриазольное; 4 — политиадиа-
зольное (при 400 °C).
(иа оси абсцисс приведена продолжитель-
ность прогрева в я).
Полифенилентриазольное волокно вопреки предположениям ока-
залось нетермостабильным. По-видимому, это связано с неполнотой
превращения гидразидной группировки в фенилентриазольную. Поли-
Рис. 4.46. Зависимость прочности во-
локон от температуры испытания [54;
230; 231];
/ — ПБО; 2— политиадиазольиое; 3 —по-
лифеиилентриазольное.
бензоксазольное и политиадиазольиое волокна имеют высокую термо-
стабильность. Теплостойкость политиадиазольных и ПБО волокон на-
ходится также на высоком уровне (рис. 4.46). Но, как уже указыва-
лось ранее, труднодоступность исходного сырья, многостадийность
технологии их получения не позволили развить технологию получения
этих волокон в промышленном масштабе.
4.8. ХИМИЧЕСКАЯ И ФИЗИЧЕСКАЯ МОДИФИКАЦИЯ
ТЕРМОСТОЙКИХ ВОЛОКОН
Исследования в области синтеза термостойких полимеров и полу-
чения волокон на их основе показали, что путем химической модифи-
кации структуры полимеров (например, сополиконденсацией), или фи-
185
зической модификации (в частности, при использовании смесей поли-
меров) могут быть достигнуты значительные результаты в направле-
нии разработки волокон с улучшенными физико-механическими или
термомеханическими характеристиками. С помощью модификации
химической структуры макромолекул синтезировано большое число
сополимеров гетероциклического строения, отличающихся повышенной
термической стабильностью, при этом технология получения волокон
оказывается более доступной по сравнению с технологией получения
волокон на основе отдельных гомополимеров [236]. При формовании
волокон из смесей термостойких полимеров используются смеси раство-
ров ароматических или алифатических полиамидов и растворы гетеро-
циклических полимеров, причем гетероциклический полимер на первой
стадии может применяться как в виде промежуточного соединения
(полиамидокислоты, полиоксиамида), так и в виде конечной формы
(полиоксадиазола) >[237].
4.8.1. Волокна из сополимеров различной
химической структуры
Волокна из сополимеров, содержащих различные гетероциклы,
чаще всего получают по двухстадийному методу, при этом один из
гетероциклов содержится в каком-либо исходном мономере, а второй
гетероцикл образуется в результате реакции внутримолекулярной
циклодегидратации промежуточного сополимера [238]. Схематически
стадии процесса и реакции, имеющие место, например при получении
полиоксадиазолимидных волокон, изображены ниже:
Поликонденсационный раствор поли амидо кислоты оказывается
более стабильным по сравнению с раствором ПАК на основе пиромел-
литового диангидрида и 4,4'-диаминодифенилового эфира. Из этого
раствора формуются волокна по мокрому или сухому способам [239].
После стадий термической или химической циклизации образуется
сополимерное волокно, содержащее в основной цепи регулярно чере-
дующиеся оксадиазольные и имидные циклы. Промежуточные сопо-
лимеры хорошо растворяются в различных растворителях. Ориенти-
рованные волокна представляют собой кристаллические системы, и
после стадии термической обработки они не растворяются в обычных
растворителях; в концентрированных кислотах волокна набухают, одна-
186
ко полного растворения не наблюдается [236]. Примерно по такому же
принципу получают волокна, включающие тиадиазольные, бензимида-
зольные, бензоксазольные и другие гетероциклические группировки.
Физико-механические характеристики некоторых сополимерных волокон,
содержащих различные гетероциклы, приведены в табл. 4.49.
Анализ данных, приведенных в табл. 4.49, свидетельствует о том,
что на основе различных сополигетероциклических систем могут быть
получены волокна с хорошими физико-механическими характеристика-
ми. Отмечается, что в случае, когда конечной стадией процесса полу-
чения полигетероциклической сополимерной системы является имиди-
зация, механические свойства волокон, в частности прочность при
разрыве, оказываются выше по сравнению с теми случаями, при ко-
торых на последней стадии осуществляется замыкание оксадиазоль-
ного или бензоксазольного циклов. Это связывают с характером про-
цессов, приводящих к созданию циклов [236]. В отличие от имидных
циклов, которые образуются в ПАК при сравнительно невысоких тем-
пературах (150—170 °C) и при этом полимер не деструктирует, бенз-
оксазольные циклы образуются при 300—375 °C; последнее обстоя-
тельство обусловливает частичное разрушение как химической, так и
физической структуры исходных волокон.
Термомеханические характеристики волокон смешанной полигете-
роциклической структуры оказываются довольно высокими. Полимеры
и волокна на их основе не плавятся, разложение при нагревании в
азоте наблюдается при 500—550 °C; отмечают, что на воздухе термо-
стабильность полимеров оказывается более низкой, однако экспери-
ментальный материал не приводится. Данные, характеризующие теп-
лостойкость некоторых смешанных полигетероциклических волокон,
представлены в табл. 4.50. Для сравнения приведена также теплостой-
кость волокон на основе гомополимеров ПОД, ПИ, ПБИ.
Из данных таблицы видно, что теплостойкость некоторых сополи-
мерных полиоксадиазольных и полибензоксазольных волокон в области
температур 300—350 °C близка к теплостойкости гомополиоксадиазоль-
ных и полиимидных волокон.
Теплостойкость волокон на основе немодифицированного ПБИ,
макромолекулы которого связаны между собой большим числом водо-
родных связей, превышает теплостойкость сополимерных гетероцикли-
ческих волокон. Сополимерные гетероциклические волокна в ряде
случаев показывают и высокую термостабильность (табл. 4.51).
Как видно из данных, приведенных в таблице, стойкость некото-
рых типов полибензоксазолимидных и полиоксадиазолимидных воло-
кон к термоокислительной деструкции при 300 °C оказывается более
высокой, чем тот же показатель для волокон из гомополимеров.
Предполагают, что это обусловлено более полной имидизацией и от-
сутствием термически нестабильных связей в макромолекулах сополи-
меров, однако нужно отметить, что в работах, посвященных изучению
термомеханических свойств сополигетероциклических волокон, степень
циклизации конечных изделий не оценивается. По-видимому, все же
циклизация не достигает 100%; свидетельством этому является срав-
нительно низкая термостабильность практически всех сополигетеро-
циклических волокон при 350 °C (табл. 4.52).
Прогрев при 350 °C на воздухе приводит к потемнению волокон.
Волокна становятся хрупкими, теряют способность растворяться в
нолярных растворителях. Отличительной особенностью некоторых со-
187
Таблица 4.49. Физико-механические свойства некоторых сополимерных
№№ п/п
Химическая формула элементарного звена сополимера
188
гетероциклических волокон
Температура, °C Способ формования волокон Показатели волокна при 20 °C Литера- тура
размягче- ния полимера начала разложения в азоте прочность, сН/текс удлинение, % начальный модуль, 106.Па
>500 520 Сухой, из раство- ров ПАК с дальней- шей циклизацией и термовытяжкой 46—49 5—6 14700—17400 [236; 239]
485 500 То же 56—63 5—9 15900—24200 [236; 239]
500 520 Сухой, из раство- ров ПАК с дальней- шей циклизацией без термовытяжки 35 19 12600 [236; 239]
— — Мокрое формова- ние, из растворов по- лигидразид амидокис- лоты с дальнейшей имидизацией и гете- роциклизацией 10—13 3-5 7000 [240]
— — Мокрое формова- ние, из растворов полигидразидамидо- кислоты с дальней- шей имидизацией и гетероциклиз ацией 30—33 3—4 18000 [240]
— — То же 20—25 5—6 — [240]
189
Химическая формула элементарного звена сополимера
ММ п/п
190
Продолжение
Температура, °C Способ формования волокон Показатели волокна при 20 °C Литера- тура
размягче- ния полимера начала разложения в азоте прочность, сН/текс удлинение, % начальный модуль, 10«Па
— — Мокрое формова- ние, из растворов по- лигидразидамидо- кислоты с дальнейшей имидизацией и гете- роциклизацией 5—10 (невытяну- тое волок- но) 18—23 (вытянутое волокно) 20—25 7—10 3000 9400 [240] [240]
500 520 Сухой или мокрый, из растворов соответ- ствующей ПАК в ДМАА и МП с после- дующей циклизацией ПАК в полиимид и дополнительной вы- тяжкой 43 5,0 22300 [241]
>500 560 То же 57—81 5—6 26700—35400 [241]
>500 570 » 46—48 4—7 19300 [241]
>500 600 » 42—43 19—20 7350 [241]
>450 500 Сухой или мокрый, из растворов полиок- сиамида в ДМАА с последующей цикли- зацией в полибеизок- сазол 20—21 20—22 4300 [236]
191
Химическая формула элементарного звена сополимера
14
15
16
полигетероциклических волокон является высокая стойкость к дейст-
вию открытого пламени. Величина кислородного индекса для волокон
8 и 10 достигает 42% [245].
Сополигетероциклические волокна, за редким исключением, ока-
зываются довольно неустойчивыми к ультрафиолетовому облучению.
Таблица 4.50. Влияние температуры испытания на прочность
некоторых сополигетероциклических волокон
Волокно* Прочность при различных температурах испытания, % от исходной Лите- ратура
20 °C 100 °C 200 °C 300 °C 350 °C 400 °C 450 °C 500 °C
1 100 77 60 41 35 27 22 16 [236]
2 100 72 51 22 15 11 8 — [236]
3 100 75 46 26 И — — — [236]
8 100 73 73 48 44 33 31 35 |241|
9 100 —— 31 25 23 15 — |241|
10 100 — — 40 — 21 20 — |241[
Полиоксадиазольное 100 83 61 38 21 20 — — [146]
Полиимидное 100 77 57 50 31 27 13 —— [ 118]
Полибензимидазольное 100 105 97 80 68 37 30 — [182]
* Номера волокон см. в табл. 4.49.
192
П родолжение
Температура, °C Способ формования волокон Показатели волокна при 20 °C Литера- тура
размягче- ния полимера начала разложения в азоте прочность, сН/текс удлинение, % начальный модуль, 106-Па
>450 500 Сухой или мокрый, из растворов полиок- сиамида в ДМАА с последующей цикли- зацией в полибенз- оксазол 12—13 10—11 8100 [236]
>450 460 Сухой или мокрый, из растворов поли- гидразида, содержа- щего тиадиазольный цикл, с дальнейшим превращением в окса- диазол 11—13 3—5 6000 [242]
>450 >500 Мокрый, из раство- ров сополимера в концентрированной серной кислоте, с до- полнительной вытяж- кой в воде на 500% 36—40 12—20 [243]
>400 >500 Мокрый, из раство- ра в концентрирован- ной H2SO4 43—45 30—36 [244]
Указывается, что их светостойкость может быть повышена путем вве-
дения в волокна светопоглотителей, однако конкретные сведения в ли-
тературе не приводятся [238].
Сравнительно высокую светостойкость полибензоксазолимидного
волокна (№ 10, табл. 4.53) авторы работы [241] просто констатируют,
не приводя никаких объяснений.
Изучение смешанных полигетероциклических систем к действию
щелочей показало [246], что некоторые полибензоксазолимиды явля-
ются гидролитически стойкими; кипячение в течение 30 мин ориентиро-
ванных пленок из полибензоксазолимидов в 10%-ном растворе щелочи
не изменяет механических и эластических свойств пленок. Полиимид-
ная пленка, обработанная в тех же условиях, полностью разруша-
ется. Полибензоксазолимиды стойки не только к действию щелочей,
но и к действию серной, соляной и азотной кислот; в дымящей азотной
кислоте полибензоксазолимиды разлагаются [246]. Гидролитическая
стабильность полиоксадиазолимидных волокон практически не иссле-
дована.
Химическая модификация цепей жесткоцепных полимеров, т. е.
получение сополимеров, представляется довольно перспективным на-
правлением. Используя этот метод, можно получать жесткоцепные
полимеры, содержащие различные гетероциклические группировки в
13—2254 193
Таблица 4.51. Влияние продолжительности теплового старения
на воздухе прн 300 °C на прочность сополигетероцнклнческнх
волокон
Волокно* Прочность (в % от исходной) после экспозиции Лите- ратура
3 ДНЯ 7 дней 14 дней 21 день 35 дней 49 дней 56 дней 70 дней
1 97 90 79 71 70 59 46 . [236]
2 80 62 60 59 40 38 30 — [236]
3 120 105 — 99 100 105 — — [236]
8 88 79 79 64 70 15 — |2411
9 100 99 81 80 73 83 40 30 [241|
10 94 87 70 65 — 1 1— — — [241|
11 118 115 105 105 73 68 — 37 [241|
Полноксадназольное 98 87 70 62 44 22 — — Ц46|
Полнимидное (при 283 °C) 70 60 54 48 45 40 — — [118]
* Номера волокон см. в табл. 4.49.
Таблица 4.52. Влияние продолжительности теплового старения
на воздухе при 350 °C на прочность сополигетероцнклнческнх волокон
Волокно* Прочность (в % от исходной) после экспозиции Литература
24 ч 48 ч 72 ч 120 ч
1 52 52 35 Разрушается [236]
2 67 29 14 » [236]
Ч 86 63 42 23 [241]
* Номера волокон см. в табл. 4.49.
основной макромолекулярной цепи. Значительный интерес представ-
ляют сополимеры, не содержащие атомов водорода, или сополимеры,
имеющие в составе макромолекул лестничные или хиноксалиновые
фрагменты. Отмечается, что полиимиды, не содержащие водорода
характеризуются высокими показателями термостойкости как в инерт-
ной среде, так и на воздухе [247]. Сополиимиды, включающие фени-
лированные хиноксалиновые группировки
СО СО
194
Таблица 4.53. Влияние УФ-облучения на прочность
сополигетероциклических волокон
Волокно* Прочность (в % от исходной) после экспозиции Литература
20 ч 40 ч 80 ч 140 ч
1 92,0 82,7 73,1 63,5 [236]
2 71,4 70,0 68,2 61,9 [236]
3 93,5 73,1 34,0 [236]
9 95,0 — 83,0 48,0 [241]
10 98,9 98,6 96,7 94,4 [241]
11 76,2 76,0 65,0 59,0 [241]
12 86,5 81,1 73,4 69,4 [236]
* Номера волокон см. в табл. 4.49.
имеют такую же термостабильность, как и немодифицированные по-
лиимиды, и при этом способны растворяться в органических раствори-
телях [248]. И хотя волокна на основе сополимеров последних хими-
ческих структур не описаны, указывается, что сополимеры являются
высокомолекулярными, пленко- и волокнообразующими.
4.8.2. Волокна на основе смесей полимеров
Использование при получении термостойких волокон физических
смесей полимеров практикуется сравнительно недавно [237]. Говоря о
смеси полимеров как об исходном материале для получения термо-
стойких волокон, следует прежде всего иметь в виду их совмести-
мость, и в первую очередь совместимость в растворе, так как термо-
стойкие полимеры не плавятся. Известно [249, с. 51], что явление
молекулярной совместимости двух полимеров, даже при использовании
полимеров близкой химической структуры, встречается крайне редко.
Смесь полимеров, находящихся в растворе одного и того же раствори-
теля, как правило, расслаивается, однако этот процесс является кине-
тическим и в ряде случаев удается получать хотя термодинамически и
несовместимые смеси, но со стабильностью вполне достаточной для
того, чтобы можно было провести все операции, характерные для про-
цессов получения волокон (смешение, фильтрацию, обезвоздушива-
ние и т. д.).
Некоторые особенности процессов формования и упрочнения и спе-
цифика свойств смешанных волокон были изучены на системах: аро-
матический полиамид — полиамидокислота. В качестве полиамида в
одних случаях был использован полимер на основе 4,4'-диаминодифе-
нилсульфона и терефталевой кислоты; в других — полимер на основе
.и-фенилендиамина и изофталевой кислоты; в качестве полиамидокис-
лоты применяли полимеры на основе пиромеллитового диангидрида
или диангидрида 3,3', 4,4'-дифенилтетракарбоновой кислоты и 4,4'-
диаминодифенилового эфира. Растворителями являлись ДМАА или
ДМФ [11, с. 39; 237; 250]. Процесс получения смешанных волокон со-
стоит из нескольких стадий:
1) получения поликонденсационных растворов ароматических по-
лиамидов или полиамидокислот по обычным режимам;
13* 195
/,2 1,6 1,8 2,0 2,2 2,Ь 2,6
К
Рис. 4.47. Зависимость физико-
мехаиических свойств волокон из
смесей полимеров от кратности
термической вытяжки и способа
имидизации [251]:
1 и 2 — прочность; 3 и 4 — удлинение
(— химическая циклизация ПАК;
------термическая циклизация).
2) смешения растворов ПА и ПАК;
3) формования волокон из смесей растворов полимеров по мокро-
му способу с проведением пластификационной вытяжки свежесформо-
ваниых волокон;
4) имидизации ПАК (термическим или химическим методом);
5) дополнительной термической вытяжки полученных смешанных
волокон.
Показано, что при выделении твердой фазы из смесей полимеров,
находящихся в растворе, можно получать волокна ,со структурой, близ-
кой к однородной. Подобное явление наблюдается только ® случае об-
разования анизотропной твердофазной
системы. При этом во всех случаях, когда
смесь полимеров является кинетически
устойчивой, структура образующихся во-
локон близка к гомогенной. В тех слу-
чаях, когда формование ведется из рас-
творов, кинетическая стабильность кото-
рых низка, в структуре волокон наблю-
даются разрывы сплошности в результа-
те приложения даже таких незначитель-
ных напряжений, которые вызываются
фильерной вытяжкой. Другой особен-
ностью, наблюдаемой при формовании
волокон из смесей полимеров, является
действие осадительных ванн со слабой
коагулирующей способностью. Обнаруже-
но, что осаждение смешанной твердой
фазы в таких ваннах протекает быстрее
по сравнению с осаждением гомополи-
меров и в момент коагуляции происходит частичное расслоение си-
стемы [1,1].
В связи с тем, что в исследованных смесях одной из составляю-
щих является полиамидокислота, характер ее циклизации обусловли-
вает поведение волокон при дополнительной термической вытяжке.
Обнаружено [251], что дополнительную термическую вытяжку смешан-
ных волокон невозможно осуществить, если амидокислотные группы
ПАК не зациклизованы, хотя волокно на основе гомополимера ПАК
может быть ориентировано при повышенных температурах и до стадии
имидизации. Для смешанных волокон, циклизация ПАК в которых
осуществлялась химическим методом, степень предельно достижимой
дополнительной термической вытяжки оказывается меньше, чем для
волокон, подвергнутых термической имидизации. Удлинение также
различно и зависит от степени вытяжки и характера имидизации
(рис. 4.47). Характерной особенностью ориентационного упрочнения
волокон из исследованных смесей является большая эффективность
термического вытягивания по сравнению с вытяжкой свежесформован-
ных волокон в пластифицированном состоянии [252]. Если после пла-
стификационного вытягивания двойное лучепреломление и плотность
волокон из смесей ПСА—ПАК практически такие же, как и у волокон
на основе гомополимера ПСА, а прочность их даже несколько ниже,
то в результате термического вытягивания картина меняется
(табл. 4.54).
196
Таблица 4.54. Влияние способа вытяжки волокон из смесей
полимеров ПСА— ПАК на их физико-механические свойства [252].
Соотношение ПСА: ПАК в смеси, % (масс.) Температура вытяжки, °C Максимальная вытяжка, % Прочность, сН/текс Удлинение, % Плотность, КГ/мЗ Двойное лучепрелом- ление
Пластификационное вытягивание
90:10 20—25 193 18,0 26,6 1380 0,052
75:25 20—25 190 17,2 28,6 1370 0,048
50:50 20—25 200 14,0 25,4 1380 0,048
25:75 20—25 175 14,7 20,6 1410 0,060
10:90 20—25 175 15,0 20,0 1410 0,047
100:0 20—25 150 24,0 25—30 1380 0,050
Термическое вытягивание
90:10 450 200 39,0 17,0 1430 0,320
75:25 450 200 37,0 15,0 1430 0,350
50:50 480 160 30,8 11,0 1435 0,150
25:75 540 145 45,0 8,5 1443 0,300
10:90 540 160 50,0 10,0 1445 0,350
100:0 430 200 30,0 15,0 1430 0,300
Исследование ряда физико-механических свойств волокон, полу-
ченных из смесей полимеров, в зависимости от состава исходной смеси
позволило установить наличие двух зон (рис. 4.48).
Первая зона (более высокие
показатели, чем для волокон из
гомополимеров) соответствует
смесям, кинетическая устойчи-
вость которых достаточно велика.
Вторая зона (низкие показатели)
соответствует составу 50 : 50, т. е.
смеси, для которой как в раство-
ре, так и в процессе образований'
твердой фазы наблюдается мак-
рорасслоение [11].
Специфика смешанных воло-
кон наиболее ярко проявляется
при изучении их термостойкости.
Как видно из представленных на
рис. 4.49 данных, смешанные
волокна, полученные из кинети-
чески устойчивых смесей раство-
ров полимеров поли-л-фенилен-
изофталамида и ПАК, имеют при
350 и 400 °C (кривые 1 и 2) более
высокую остаточную прочность,
чем волокна на основе отдельных
и 1U ZU JU НО 50 ВО 70 80 90 100
Содержание компонента П,
।__।__।__।__1 । । । । 1 ।
100 90 90 70 ВО 50 90 20 20 Ю 0
Содержание компонента I, °1о
Рцс. 4.48. Зависимость прочности (1 и 2),
относительной 'Прочности в петле i(3) и
удлинения (4 и 5) от состава исходных
.волокон [11]:
2—4 _ смесь ПСА и ПАК; 1 и 5 —смеси
ПМФИА и ПАК.
гомополимеров. Аналогичная за-
висимость прослеживается и для системы ПСА—ПАК (кривая 3) при
400 °C. И здесь повышенная термостойкость волокон наблюдается для
тех же составов, а минимальная — для состава 50 : 50.
Повышение термостабильности волокон из смесей полимеров, на-
блюдаемое для некоторых составов, оказалось неожиданным. Детально
механизм этого явления не изучен. Предполагается, что при смешении
197
растворов жесткоцепных полимеров при определенных соотношениях и
концентрации возможно образование прочных ассоциатов макромоле-
кул полиамидов и полиамидокислоты за счет реализации межмолеку-
лярных водородных связей между амидными группами полиамида и
свободными карбоксилами ПАК. Такая смешанная система является
псевдоустойчивой и в процессе образования твердой фазы в условиях
наложения поля механических сил не расслаивается. Это подтвержда-
ется фотографиями микросрезов волокон [237], ИК-спектрами и дан-
Соотношение компонентов,°/о (масс.) Содержание ПСЯ, °1а(масс}
,Рис. 4.49. Завиоимо1сть термостабилыюсти волокон и полимеров от состава смеси
•(/ и 2 — волокна из смеси ПМФИА и ПАК; 3 — волокна из смеси ПСА и ПАК;
4 — изотропная пленка ив смеси ПМФИА и ПАК) '[11]:
/ — 350 °C; 25 ч; 2 — 400 °C; 4 ч; 3 — 400 °C; 4 ч; 4 — 400 °C; 1 ч.
«Рис. 4.50. Влияние состава смеси полимеров на температуру стеклования (5) и
отклонение от аддитивности полос поглощения [И]:
/ — 1020 см-'; 2 — 1640 см-1; 3 — 720 см-'; 4 — 3300 см-'; « — 1730 см-'.
ными, показывающими зависимость температуры стеклования -от со-
става полимеров [И] (рис. 4.50).
Экстремальные отклонения от аддитивности (кривые 2 и 6) сви-
детельствуют о наличии взаимного влияния ПСА и ПАК. Наибольшее
отклонение отмечено для полосы с частотой 1730 см-1, что может быть
связано с конформационными перестройками в макромолекулах ПАК.
Кривая, характеризующая зависимость температур стеклования от
состава (рис. 4.50, кривая 5), также подтверждает, что для большин-
ства смесей в процессе образования твердой фазы получается новая
однородная система, которая имеет одну температуру стеклования,
отличную от температуры стеклования исходных компонентов. Для
состава 50 : 50 имеется две температуры стеклования, соответствующие
сосуществующим гомополимерам.
На то, что образованию твердой однородной смешанной системы
из термодинамически несовместимых полимеров, находящихся в рас-
творе, способствует поле механических сил, накладываемое на нить
при ее формовании, указывает факт отсутствия экстремальных значе-
ний при исследовании термостойкости изотропных пленок. На рис. 4.49
представлена изотермическая дериватограмма, которая показывает
монотонное, близкое к аддитивному значению снижение термоста-
бильности полимеров при уменьшении доли компонента ПАК (кривая 4).
198
Формование из механических смесей жирноароматического по-
лиамида и полиоксадиазолов также приводит к повышению их прочно-
сти и термостойкости [253]. Так, например, у волокон, сформованных
из смеси полиоксадиазола и полипиперазинтерефталамида (в соотно-
шении 80:20) прочность после прогрева в течение 1000 ч при
250 °C на воздухе не только падает, но и увеличивается с 40,0 до
43,2 сН/текс; волокна из гомополимеров, прогретые в тех же условиях,
теряют от 15 до 25% прочности. Отмечен факт повышения прочности,
термостойкости и эластичности для смесей волокон из полибензоксазо-
лов и полиакрилонитрила [254]. Волокна получали из смеси ПАН
(10%) и полиоксиамида на основе 3,3'-диоксибензидина и изофталевой
кислоты по способу мокрого формования. Свежесформованное волок-
но вытягивали на воздухе, а затем дополнительно при 500 °C. Цикли-
зованное волокно имеет прочность около 60 сН/текс, эластичность
67%, выдерживает около 600 двойных изгибов и сохраняет 93% проч-
ности от исходной после прогрева при 300 °C в течение 24 ч. Прочность
полибензоксазольного волокна, не содержащего добавки ПАН, состав-
ляет около 40 сН/текс, эластичность 45%, волокно разрушается после
300 двойных изгибов и теряет половину прочности после прогрева
при 300 °C.
Смеси полиамидов и полиоксадиазолов применяют также для
повышения стойкости волокон к действию УФ-лучей [255]. Полимеры
растворяют в концентрированной серной кислоте, формование осу-
ществляется по мокрому способу. В качестве полиоксадиазольного
компонента использован полимер на основе терефталевой кислоты и
гидразинсульфата, в качестве полиамидного компонента — полигекса-
метилентерефталамид. Влияние состава смеси на светостойкость во-
локон можно проследить по табл. 4.55.
Таблица 4.55. Влияние соотношения полиамида и полиоксадиазола
на стойкость волокон к УФ-облучению [255]
Соотношение ПА : ПОД (масс.) Прочность до облучения, сН/текс Прочность после облучения, сН/текс
при разрыве в узле прн разрыве в узле
100:0 75,6 63,9 37,8 23,4
99:1 72,9 63,9 43,2 22,5
98:2 69,4 64,8 67,5 63,0
80:20 73,8 61,2 73,8 58,5
70:30 67,5 63,0 72,0 61,2
55:45 63,9 57,6 63,9 56,7
40:60 58,5 51,3 61,3 61,3
Физическая модификация ПБИ волокон, сформованных из смеси
сернокислотных растворов ароматических ПБИ и полихиноксалина по
мокрому способу, приводит к повышению термостабильности поли-
бензимидазольного волокна, при этом волокна имеют высокие механи-
ческие характеристики и хорошую теплостойкость [237].
Рассмотренный выше материал позволяет сделать вывод о том,
что с помощью метода структурной модификации волокон из жестко-
цепных полимеров могут быть получены интересные результаты.
И хотя экстремальные эффекты проявляются не во всех случаях,
199
этому направлению в дальнейшем, по-видимому, будет уделяться зна-
чительное внимание.
В заключение, давая оценку приведенным материалам, необходимо
еще раз подчеркнуть следующее. Проблему создания волокнистых ма-
териалов для эксплуатации при 180—250 °C, по-видимому, можно
считать решенной. С другой стороны, несмотря на то что в настоящее
время синтезировано большое число термостойких полимеров, лишь
некоторые из них могут быть использованы для получения волокон,
способных длительно выдерживать температуру 250—300 °C. Ряд гете-
роциклических полимерных волокон может быть применен в изделиях,
кратковременно соприкасающихся с температурами порядка 350—
450 °C. К сожалению, пока нет не только органических волокон, спо-
собных длительно выдерживать воздействие температур выше 350 °C,
но не ясны и принципиальные пути их получения.
Большим достижением химии и технологии синтетических волокни-
стых материалов было получение высокопрочных высокомодульных
волокон на основе жесткоцепных термостойких ароматических по-
лиамидов.
ЛИТЕРА ТУРА
1. Соколов Л. Б. Поликонденсационный метод синтеза полимеров. М., «Химия»,
1966. 336 с.
2. Соколов Л. Б. и др. Термостойкие ароматические полиамиды. М., «Химия»,
1975. 256 с.
3. Заявка ФРГ 2258567 (1973).
4. Федоров А. А. Канд. дис. М., МХТИ им. Д. И. Менделеева, 1969.
5. Савинов В. М. и др., Пласт, массы, 1967, № 6, с. 25—27.
6. Савинов В. М., Соколов Л. Б., Хим. волокна, 1965, № 4, с. 22—25.
7. Англ. пат. 871578 (1962).
8. Пат. США 3884881 (1975).
9. Пат. США 3870685 (1975).
10. Metneger IF., Polymer Prepr., 1976, v. 17, № 1, p. 163—168.
11. Жиздюк Б. И. и др. Международный симпозиум по химическим волокнам.
Препринты. Калинин, 1974, секция 4, с. 12—18.
12. Информация ВНИИВ. Хим. волокна, 1969, № 4, с. 78.
13. Black IF. В., Preston J. In: Man-Made Fibers. V. 2. New York, Interscience Publishers,
1968, p. 297.
14. Циперман P. Ф. и др.. Хим. волокна, 1974, № 4, с. 16—18.
15. Иовлева М. М. и др., Высокомол. соед., 1974, сер. Б, т. 16, № 3, с. 219—221.
16. Фр. заявка 2010753 (1970), пат. США 3671542 (1972).
17. Пат. США 3673143 (1972).
18. Михайлов И. В. и др., Высокомол. соед., 1960, т. 2, № 7, с. 991—996; Хим.
волокна, 1960, № 6, с. 10—14.
19. Сигал М. Б., Козиорова Т. И., Хим. волокна, 1966, № 6, с. 1—6.
20. Шейн Т. И. и др., Хим. волокна, 1976, № 1, с. 11—18.
21. Пат. США 3300450 (1967).
22. Англ. пат. 1198082 (1970), 1262002 (1972); пат. США 3637606 (1972).
23. Руднева Л. Д. и др., Хим. волокна, 1975, № 2, с. 13—17; № 3, с. 1—7.
24. Щетинин А. М., Кудрявцев Г. И., Хим. волокна, 4968, № 1, с. 22—26.
25. Фр. пат. 2066108 (1971), яп. пат. 17751 (1973); фр. заявка 2125346 (1972); авт.
свид. СССР 322435; Открытия. Изобр. Пром, образцы. Товарн. знаки, 1971, №35.
26. Иовлева М. М., Бандурян С. И., Папков С. П. Теория формования химических
волокон. Труды ВНИИВ, Мытищи, 1975, с. 93—103.
27. Основы технологии получения ароматических полиамидов и волокон на их
основе. Обзор. Сер.: Промышленность химических волокон. М., НИИТЭХИМ,
1976. 81 с.
28. Юницкий В. П. и др., Хим. волокна, 1970, № 5, с. 11—15.
29. Пат. США 3767756 (1973).
30. Серков А. Т. и др., Хим. волокна, 1975, № 1, с. 30—36.
200
31. Яп. пат. 52-143 (1972).
32. Англ. пат. 871581 (1961); 877885 (1961); пат. США 3287324 (1966).
33. Пакшвер Э. А. и др., Хим. волокна, 1976, № 1, с. 27—29.
34. Пат. США 3133138 (1964).
35. Соколова Т. С. и др., Хим. волокна, 1974, № 3, с. 25—28.
36. Толкачев Ю. А., Краснов Е. П. Международный симпозиум по химическим во-
локнам. Препринты. Калинин, 1974, секция I, с. 148—151.
37. Г речушникова Л. Д. Канд. дис. Калинин, ВНИИСВ, 1974.
38. Preston J., Black W. В., J. Polymer Sci., 1965, В, v. 3, p. 845—851.
39. Пат. США 3738964 (1973).
40. Пат. США 3767756 (1973).
41. Фр. заявка 2134582 (1972).
42. Пат. США 3324086 (1967).
43. Семенова А. С., Соколова-Васильева Е. А., Хим. волокна, 1969, № 5, с. 2—4.
44. Англ. пат. 901159 (1962).
45. Пат. США 3049518 (1962).
46. Пат. США 3240758 (1966).
47. Пат. США 3006899 (1961).
48. Пат. США 3296201 (1966).
49. Пат. США 3801528 (1974).
50. Пат. США 3827998 (1974).
51. Пат. США 3836498 (1974).
52. Англ. пат. 876491 (1961).
53. Бельг, пат. 569760 (1958).
54. Новое в производстве химических волокон. Под ред. 3. А. Роговина. М., «Мир»,
1968. 226 с.
55. Яп. пат. 15555 (1972).
56. Пат. США 3598768 (1971).
57. Пат. США 3804791 (1974).
58. Щетинин А. М., Кудрявцев Г. И., Kmi. волокна, 1968, № 1, с. 22—25.
59. Колот В. Н. и др., Хим. волокна, 1974, № 4, с. 72; пат. США 3353123 (1967).
60. Пат. США 3931119 (1976).
61. Пат. США 2696482 (1954).
62. Муравьева Н. И., Конкин А. А., Хим. волокна, 1972, № 2, с. 8—11.
63. Preston J., Smith R. W., J. Polymer Sci., 1970, A-l, v. 8, № 7, p. 1841—1849.
64. Фр. заявка 2151108 (1973).
65. Preston J., J. Appl. Polymer Sci., 1972, v. 16, № 12, p. 3237—3244.
66. Holsten G. R., Preston I., Appl. Polymer. Symp., 1969, № 9, p. 67—76.
67. Preston J., Black W. B., J. Polymer Sci., 1967, C, № 19, p. 17—26.
68. Англ. пат. 1104411 (1968).
69. Фр. пат. 1574409 (1969).
70. Англ. пат. 1227509 (1971).
71. Англ. пат. 1222050 (1971).
72. Preston J., Appl. Polymer Symp., 1969, № 9, p. 75—81.
73. Kunzel H. E. e. a., Makromol. Chem., 1970, Bd. 138, S. 23—44.
74. Kunzel H. E. e. a., Makromol. Chem., 1969, Bd. 130, S. 103—136.
75. Фр. пат. 1566254 (1970).
76. McDonald R. N., Sharkey W. H., J. Polymer. Sci., Polymer Chem. Ed. 1973, v. 11,
№ 10, p. 2519—2536.
77. Пат. США 3671542 (1972).
78. Hannell J. W., «Polymer News», 1970, № 1, p. 8—9.
79. Заявка ФРГ 2277869 (1975).
80. Шейн T. И. и др. Новое в области термостойких полимеров и волокон. Обзор.
М„ НИИТЭХИМ, 1974. 57 с.
81. Заявка ФРГ 2144126 (1973).
82. Black W. В., Brucke W., J. Macromol. Sci., 1973, A, v. 7 (I), р. 3—41.
83. Заявка ФРГ 2232504 (1973).
84. Заявка ФРГ 2248662 (1974); 2248663 (1974); 2259123 (1974).
85. Sakamoto S„ Kasen Geppo, Japan Chem. Fibres Mon., 1974, v. 27, № 9, p. 54—63.
86. Mohajer A. A., Ferguson W. J., Text. Progr., 1976, v. 8, № 1, p. 97—128.
87. Информация ВНИИСВ. Хим. волокна, 1968, № 6, с. 72—73.
88. Pamm G„ Hentschel R. A., Lenzinger Ber., 1974, № 36, S. 57—64.
89. Dappen W„ «Kunststoffe», 1973, Bd. 63, № 12, S. 919—928.
90. Bach H. C„ Plack W., Appl. Polymer Symp., 1969, № 9, p. 167—176.
91. Фрейзер А. Г. Высокотермостойкие полимеры. Под ред. А. Н. Праведникова.
М., «Химия», 1971. 296 с.
201
92. Информация ВНИИВ. Хим. волокна, 1972, № 6, с. 20—21.
93. Пат. США 3527732 (1970).
94. Роговин 3. А., Хим. волокна, 1976, № 1, с. 36—39.
95. Кудрявцев Г. И., Хим. волокна, 1969, Ns 4, с. 23—30.
96. Беляков В. К. и др., Высокомол. соед., 1973, сер. А, т. 15, Ns 7, с. 1483—4490.
97. Авт. свид. СССР 186191; Изобр. Пром, образцы. Товарн. знаки, 1968, № 18; авт.
свид. СССР 285233; Открытия. Изобр. Пром, образцы. Товарн. знаки, 1970, №3.
98. Коршак В. В. и др., Высокомол. соед., 1966, т. 8, № 8, с. 1608—.1611.
99. Фр. заявка 2235171 (1975).
100. Англ, заявка 1307601 (1973).
101. Пат. США 3560137 (1971).
102. Кудрявцев Г. И., Федорова Р. Г., Хим. волокна, 1975, № 5, с. 28—31.
103. Khster В., Herlinger Н., Angew. makromoL Chem., 1974, Bd. 40—41, S. 265—
276.
104. Day M„ Wiles D. M., Text. Res. J., 1974, v. 44, p. 888—892.
105. Яп. пат. 21439 (1974).
106. Заявка ФРГ 2364756 (1975).
107. Чернова А. Г. и др., Пласт, массы, 1972, № 4, с. 26—29; Johnson Е. L., J. Appl.
Polymer Sci., 1971, v. 15, № 11, р. 2825—2839.
108. Закощиков С. А., Руженцева Г. А., Высокомол. соед., 1966, т. 8, с. 1231—1233.
109. Bower G. М., Frost L. М., J. Polymer Sci., 1963, A-l, v. 1, р. 3135—3138.
ПО. Sroog С. Е. е. a., J. Polymer Sci., 1966, A-l, v. 4, p. 1373—1376.
111. Фр. заявка 2077433 (1971).
112. Адрова M. А. и др. Полиимиды — новый класс термостойких полимеров. Л.,
«Наука», 1968. 210 с.
113. Frost L. М., Kesse J., J. Appl. Polymer. Sci., 1964, v. 9, p. 1039—1044.
114. Корчмарчик О. С. и др. Конф. «Термостойкие волокна». Тезисы докл. Мытищи,
ВНИИВ, 1972, с. 21—22.
115. Павлов А. В., Чернова А. Г., Пинаева Н. К, Высокомол. соед., 1972, сер. Б,
т. 14, с. 415—417.
116. Пат. США 3179614 (1965).
117. Оприц 3. Г., Лазуткина Т. П., Кудрявцев Г. И., Хим. волокна, 1970, Ns 4,
с. 10—13.
118. Irwin R. S., Sweeny W., J. Polymer Sci., 1967, C, Ns 19, p. 52—65.
119. Коржавин Л. H. Канд. due. Л., ЛИТЛП им. С. M. Кирова, 1970.
120. Фр. пат. 1432038 (1965), англ. пат. 1038736 (1965), пат. США 3415782 (1968).
121. Wallach М. L., J. Polymer Sci., 1968, А-2, v. 6, Ns 5, р. 953—960; 1969, А-2, v. 7,
Ns 8, р. 1435—1438.
122. Wallach М. L., Polymer Prepr., 1967, v. 8, Ns 1, p. 656—660.
123. Kreuz J. A., e. a., J. Polymer Sci., 1966, A-l, v. 4, Ns 10, p. 2607—2617.
124. Пат. США 3575936 (1971).
125. Пат. США 3541057 (1971).
126. Худошев И. Ф., Оприц 3. Г., Кудрявцев Г. И., Хим. волокна, 1972, Ns 2,
с. 39-41.
127. Аскадский А. А., Слонимский Г. Л., «Физика твердого тела», 1964, вып. 5, Ns 6,
с. 1430—1436.
128. Оприц 3. Г. и др., Хим. волокна, 1970, Ns 3, с. 61—64.
129. Пат. США 3179634 (1965).
130. Кудрявцев Г. И. Международный симпозиум по химическим волокнам. Пре-
принты. Калинин, 1974, секция 4, с. 5—13.
131. Информация ВНИИВ. Хим. волокна, 1969, Ns 5, с. 66.
132. Термостойкие пластики. Экспресс-информация. М., ВИНИТИ, 1971, Ns 33,
реф. 187.
133. Англ. пат. 1038738 (1965).
134. Прохоров О. С. Канд. дис. Л., ЛИТЛП им. С. М. Кирова. 1971.
135. Термостойкие пластики. Экспресс-информация. М., ВИНИТИ, 1972, Ns 47,
реф. 202
136. Berr С. Е., SPE Trans., 1965, v. 5, р. 105—ПО.
137. Токарев А. В., Любова Т. А., Кудрявцев Г. И., Высокомол. соед., 1967, сер. Б,
т. 9, с. 634—637.
138. Кудрявцев Г. И., Хим. волокна, 1969, Ns 4, с. 23—30.
139. Термостойкие пластики. Экспресс-информация. М., ВИНИТИ, 1972, № 28,
реф. 83.
140. Коршак В. В. Термостойкие полимеры. М., «Химия», 1969. 381 с.
141. Термостойкие пластики. Экспресс-информация. М., ВИНИТИ, 1972, № 12,
реф. 51.
202
142. Туйчиев Ш. Р. и др., Высокомол. соед., 1971, сер. А, т. 13, № 7, с. 1463—1468.
143 Bell V. L., J. Plymer Sci., Polymer Chem. Ed., 1976, v. 14, № 1, p. 225—235.
144. Пат. США 3835120 (1974). n
145 Frazer A H., Wallenberger F. T„ J. Polymer Sci., 1964, A-2, v. 2, p. 1137—1157;
пат. США 3130182 (1964).
146. Frazer A. H., Wilson D. R., Appl. Polymer Symp., 1969, № 9, p. 89—101.
147. Iwakura I., Uno K., Hara S., J. Polymer Sci., 1965, A-2, v. 1, p. 45—51.
148. Семенова А. С. и др. Международный симпозиум по химическим волокнам.
Препринты. Калинин, 1974, секция 4, с. 25—27.
149. Кронгауз Е. С., Коршак В. В., Вириша 3. О., Высокомол. соед., 1970, сер. А,
т. 12, с. 135—139; яп. пат. 43-635 (1968).
150. Фр. заявка 2119913 (1973).
151. Волохина А. В. и др.. Хим. волокна, 1974, № 4, с. 13—17.
152 Неводные растворители. Под ред. Р. Баддингтона. М., «Химия», 1971. 315 с.
153. Imai Е, J. Appl. Polymer Sci., 1970, v. 14, p. 225—230.
154. Varma I. K-, Sambandam R. M., Varma D. S., Angew. makromol. Chem., 1973,
Bd. 32, S. 53—60.
155. Англ. пат. 1252508 (1971).
156. Волохина А. В., Худошев И. Ф., Батикьян Б. А., Хим. волокна, 1975, № 5,
с. 14—17.
157. Информация ВНИИВ. Хим. волокна, 1971, № 2, с. 64.
158. Заявки на авт. свид. СССР 1491109 (1970), 2022045 (1974).
159. Яп. пат. 71040 (1972).
160. Яп. пат. 85690 (1972).
161. Одноралова В. Н. и др., Высокомол. соед., 1974, сер. Б, т. 16, с. 842—844.
162. Одноралова В. Н. и др., Хим. волокна, 1975, № 2, с. 10—12.
163. Яп. пат. 29298 (1974).
164. Кравченко Т. В., Богданов М. Н., Волохина А. В., Хим. волокна, 1975, № 3, с. 17.
165. Англ. пат. 1343077 (1974).
166. Яп. пат. 14197 (1973), 17657 (1974).
167. Получение и свойства полиоксадиазольных волокон. Обзорная информация. Сер.
хим. М„ НИИТЭХИМ, 1974. 66 с.
168. Фр. заявка 2161090 (1973).
169. Швейц, заявка 2056188 (1973).
170. Vogel Н„ Marvel С. S„ J. Polymer Sci., 1961, v. 50, р. 511—516.
171. Marvel С. S., J. Polymer Sci., 1967, A-l, v. 5, p. 7—15.
172. Gray D., Scnulmann S., J. Macromol. Sci., 1967, A, v. 1, p. 395—401.
173. Пат. США 3549603 (1968), 3509108 (1969), 3551389 (1969).
174. Термостойкие пластики. Экспресс-информация. М., ВИНИТИ, 1975, № 24, реф. 163.
175. Пат. США 3441640 (1969), 3619453 (1971).
176. Пат. США 3502756 (1969).
177. Пат. США 3747479 (1973).
178. Wiff G. R., Wereta A., J. Polymer Sci., Polymer Phys. Ed., 1975, v. 13, № 2,
p. 275—284.
179. Пат. США 3441660 (1969).
180. Gorlach H„ «Chemiefasern», 1968, Bd. 18, № 8, S. 575—579.
181. Beloglav L. R., Angew. makromol. Chem., 1974, Bd. 40—41, S. 465—483.
182. Singleton R. W., Appl. Polymer Symp., 1969, № 9, p. 133—144.
183. Термостойкие пластики. Экспресс-информация. M., ВИНИТИ, 1975, № 28,
реф. 195.
184. Пат. США 3856549 (1974).
185. Ross I. Н., Opt Р. С., Polymer Ргерг., 1968, v. 9, р. 1078—1087.
186. Bailey В., Polymer Ргерг., 1967, v. 8, р. 165—168, 292—296.
187. Tessier М. М., J. Polymer Sci., 1966, v. 4, р. 2521—2537; Polymer Ргерг., 1967, v. 8,
р. 152—157.
188. Gloor W. H., Appl. Polymer Symp., 1969, № 9, p. 159—166.
189. Способы получения полимеров лестничной и полулестничной структуры. Обзор.
Сер.: Важнейшие проблемы промышленности химических волокон. Ч. I и II. М.,
НИИТЭХИМ, 1974, ч. I, 46 с.; ч. II, 77 с.
190. Химия и технология высокомолекулярных соединений. Итоги науки. Сер. «Химия».
Под ред. А. Н. Праведникова. М., ВИНИТИ, 1971. 166 с.
191. Bell V. L., Pesdirtz W. В., J. Polymer Sci., 1965, В, v. 3, p. 977—981.
192. Пат. США 3640960 (1972).
193. Авт. свид. СССР 360688 (1972); Открытия. Изобр. Пром, образцы. Товарн. знаки,
1972, № 36.
194. Davans F., Marvel С. S„ J. Polymer Sci., 1965, A-l, v. 3, p. 3549—3556.
203
195. Gibbs W. E„ J. Macromol. Sci., 1968, A, v. 2, p. 1968—1973.
196. Пат. США 3505277 (1970), 3574170 (1971).
197. Bell V. L., Jewell R. A., J. Polymer Sci., 1967, A-l, v. 5, p. 3043—3060.
198. Пат. США 3574171 (1971).
199. Термостойкие пластики. Экспресс-информация. М., ВИНИТИ, 1971, № 26,
реф. 115.
200. Пат. США 3414453 (1968).
201. Gloor IF. Н., Polymer Prepr., 1966, v. 7, р. 819—825.
202. Gloor IF. H., Appl. Polymer Symp., 1967, № 6, p. 151—163.
203. Berry С. C„ Disc. Faraday Soc., 1970, № 49, p. 121—162.
204. Пат. США 3539677 (1970).
205. Rosenthal A., Text. Res. J„ 1966, v. 36, p. 596—599.
206. Пат. США 3562380 (1971).
207. Англ. пат. 1101641 (1965).
208. Пат. США 3792204 (1974).
209. Deusen R. L., van, J. Macromol. Sci., 1968, A, v. 2, № 7, p. 1291—1298.
210. Пат. США 3624249 (1971).
211. Marvel C. S„ J. Polymer Sci., 1971, A-l, v. 9, p. 691—698.
212. Авт. свид. СССР 308031 (1971); Открытия. Изобр. Пром, образцы. Товарн. знаки,
1971, № 21; англ. пат. 1323606 (1973).
213. Пат. США 3743624 (1973).
214. Пат. США 3635896 (1972); Hurley S. A., Dutt R. К-, Marvel С. S„ J. Polymer Sci.,
1972, A-l, v. 10, № 4, р. 1243—1261.
215. Пат. США 3661850 (1972).
216. Информация ВНИИВ. Хим. волокна, 1975, № 3, с. 36.
217. Пат. США 3575941 (1971).
218. Термостойкие пластики. Экспресс-информация. М., ВИНИТИ, 1974, № 12,
реф. 70.
219. Stille J. К-, Williamson I. R., J. Polymer Sci., 1967, A-l, v. 2, p. 3867—3872; 1966,
A-l, v. 4, p. 551—562; 1967, B, v. 5, p. 989—992.
220. Англ. пат. 1123424 (1971), пат. США 3639342 (1972).
221. Пат. США 3767616 (1973).
222. Пат. США 3826783 (1974).
223 Новое в области получения термостойких полимеров и волокон. Обзорная ин-
формация. Сер. «Промышленность химических волокон». М., НИИТЭХИМ,
1975. 43 с.
224. Pigeon R., Allard Р., Angew. makromol. Chem., 1974, Bd. 40—41, S. 139—158.
225. Фр. пат. 1603108 (1971); англ. пат. 1268267 (1972).
226. Пат. США 3727696 (1973).
227. Фр. заявка 2045566 (1971).
228. Erbers G., Ehrler Р., Textil praxis intern., 1974, № 3, S. 16—24.
229. Russo M., Mater, plast. ed elast., 1968, v. 34, № 7, p. 739—743.
230. Frazer A. H., Fitzgerald W. P., J. Polymer Sci., 1968, C, № 24, p. 25—38.
231. Kubota T., Nakanishi R., J. Polymer Sci., 1964, B, v. 2, p. 655—659.
232. Андриченко Ю. Д. Канд. дис. M., МТИ, 1971.
233. Новое в области термостойких полимеров и волокон. Обзор. М., НИИТЭХИМ,.
1972. 44 с.
234. Англ. пат. 1392935 (1975).
235. Пат. США 3896104 (1976).
236. Preston J., Dewinter W., Black W. В., J. Polymer Sci., 1972, A-l, v. 10, p. 1377—
1389.
237. Пат. США 3441640 (1969); Федорова P. Г., Кудрявцев Г. И., Крутикова Н. С.,.
Хим. волокна, 1970, № 6, с. 71—73.
238. Preston J., Black W. В., Appl. Polymer Symp., 1969, № 9, p. 107—117.
239. Пат. ФРГ 2247567 (1969).
240. Термостойкие пластики. Экспресс-информация. М., ВИНИТИ, 1972, № 42, реф. 184.
241. Preston J., Black W. В., Dewinter W., Appl. Polymer Symp., 1969, № 9, p. 145—
158.
242. Memeger W., Frazer A. H., Appl. Polymer Symp., 1969, № 9, p. 119—131.
243. Пат. США 3803100 (1974).
244. Фр. заявка 2124362 (1972).
245. Термостойкие пластики. Экспресс-информация. М., ВИНИТИ, 1974, № 39, реф. 260.
246. Термостойкие пластики. Экспресс-информация. М„ ВИНИТИ, 1972, № 22, реф. 87.
247. Hirsch S. S., J. Polymer Sci., 1969, A-l, v. 7, p. 15—21; Vangahn G. R., J. Polymer
Sci., 1971, A-l, v. 9, p. 1117—1124.
248. Augl S. M„ Duffy S. V., J. Polymer Sci., 1971, A-l, № 9, p. 1343—1349.
204
249. Папков С. П. Физико-химические основы производства искусственных и синте-
тических волокон. М„ «Химия», 1972. 312 с.
250. Федорова Р. Г., Кудрявцев Г. И., Хим. волокна, 1973, № 5, с. 18—20.
251. Федорова Р. Г., Кудрявцев Г. И., Хим. волокна, 1976, № 2, с. 66.
252. Федорова Р. Г., Кудрявцев Г. И., Хим. волокна, 1975, № 6, с. 28—29.
253. Яп. пат. 4316 (1974).
254. Авт. свид. СССР 324318 (1972); Открытия. Изобр. Пром, образцы. Товарн. знаки,
1972, Ns 2.
255. Яп. пат. 44178 (1973).
ГЛАВА 5
ОБЛАСТИ ПРИМЕНЕНИЯ ТЕРМОСТОЙКИХ ВОЛОКОН
Получение высокотермостойких синтетических волокон явилось
важнейшим достижением полимерной науки и промышленности за
последние двадцать лет. Высокотермостойкие волокна позволили резко
Таблица 5.1. Сравнительная характеристика волокон [1]
Обычные волокна* Термостойкие волокна
Показатель тип волокна значение показа- теля тип волокна значение показа- теля
Прочность, сН/текс Модуль упругости, МПа Устойчивость к многократным изги- бам (число циклов до разрыва прн на- грузке 120 МПа) Горючесть (кисло- родный индекс) Термостойкость, % Морозостойкость (предельная темпера- тура) Устойчивость к жесткому излучению (% к исходному) Г игроскопичность при относительной влажности 65% (95%), % Теплостойкость (предельная темпера- тура эксплуатации), °Г Найлон 6,6 Капрой Лавсан Капрон Капрон Лавсан Капрон Лавсан (стаби- лизированный) Анид 80—90 2500—4000 4000—6000 11000—12000 29,0 22—25 30,0 (при 150 °C в тече- ние 100 ч) 72,0 (при 200 °C в тече- ние 24 ч) —40 °C свм Кевлар 49 СВМ Терлон СВМ Аримид Т Полифен Аримид Т Оксалон Аримид 400 350 120000 150000 8000—9000 60,0 70,0 40,0 (при 400 °C в течение 25 ч) 85,0 (при 300 °C в течение 100 ч) —270 °C
Капрон Нитрон Вискозное Капрон Лавсан Вискозное 0 (300 Мрад) 30 (500 Мрад) 13 (30) 3,8 (6,0) 230 150—180 Аримид Оксалон С СВМ ВВВ, лола 90 (10000 Мрад) 12,0 (25) 6,0 (18) 500—550
Стойкость к свето- погоде (уменьшение прочности после 100 недель), % Нитрон 20,0 СВМ 5,0—9,0
* Выпускаются в промышленном масштабе.
205
расширить границы применения химических волокон и повысить их
характеристики (табл. 5.1).
Большинство термостойких волокон имеет высокие электроизоля-
ционные показатели, особенно при повышенных температурах. Низкие
плотности органических волокон делают их экономически более вы-
годными, чем металлические или стеклянные нити.
Новый высокий уровень технических показателей термостойких
волокон открыл широкие возможности для использования текстиль-
ных материалов в технике.
5.1. ФИЛЬТРОВАЛЬНЫЕ И ЗАЩИТНЫЕ МАТЕРИАЛЫ
Наиболее исследованной и, по-видимому, одной из самых широ-
ких является область использования термостойких волокон для изго-
товления фильтровальных материалов. В таких отраслях народного
хозяйства, как цветная металлургия, производство сажи и цемента,,
выбрасываемые технологические газы уносят с собой ценные пылевид-
ные компоненты, причем температура их превышает 250—300 °C. Для
извлечения пылевидных компонентов газы подвергают фильтрации,
предварительно охлаждая их до более низких температур. Применяе-
мые в настоящее время фильтры из полиакрилонитрильных, полиэфир-
ных нитей и стеклянные ткани имеют очень малый срок эксплуатации
из-за низких характеристик исходных волокон. Сообщается [1], что при
применении вместо стеклянных тканей материалов из термостойких
волокон срок эксплуатации фильтров увеличивается в несколько раз.
В связи с особой остротой проблемы по охране окружающей среды
возникла также задача очистки отходящих газов от доменных и мар-
теновских производств с одновременной утилизацией тепла. По-види-
мому, решение этой задачи будет связано с применением высокотер-
мостойких волокон как фильтровального материала.
Термостойкие волокна, которые, как правило, получают из арома-
тических и полигетероариленовых полимеров, оказались не только
термостойкими, но и малогорючими и даже негорючими [2]. Это от-
крывает большие перспективы их использования для изготовления
негорючих текстильных изделий.
В настоящее время одним из главных направлений применения
волокна номекс является защитная одежда для персонала, работаю-
щего в условиях воздействия тепла или огня [3]. Используемые для
этой цели материалы представляют собой обычные ткани или трико-
таж, а также нетканые материалы [4]. Сообщается, что защитные
ткани полотняного переплетения из номекса, хотя и дороже хлопка в
три раза, служат при эксплуатации в 6—12 раз дольше. Такая защит-
ная одежда применяется в химической, нефтехимической, нефтяной,
металлургической промышленности [5], пожарными и автогонщика-
ми [6].
Легким (136 гс/м2) тканям, используемым для защитной одежды,
придают «фитильные» свойства с помощью специальной авиважной
обработки [7]. Нижнее белье из номекса, так же как и алюминизиро-
ванная верхняя одежда, используются персоналом космодромов [8].
Защитные перчатки из волокна сульфон-Т и номекса во многих слу-
чаях с успехом заменяют асбестовые в электроламповой промышлен-
ности, при проведении газо- и электросварки, в металлургических про-
изводствах и других горячих цехах.
206
5.2. АНТИФРИКЦИОННЫЕ ПОЛИМЕРНЫЕ ПОКРЫТИЯ
Новая область применения термостойких волокон появилась в
связи с открытием уникальных антифрикционных свойств этих воло-
кон. Особый интерес представляет использование волокон в виде поли-
мерных покрытий для подшипников скольжения. Покрытия представ-
ляют собой композиционные полимерные материалы, армированные тка-
нями из одного или двух типов волокон. В последнем случае одно из
них является высокопрочным, другое антифрикционным. Материал
может наноситься на поверхность любой из сопряженных деталей узла
трения (вал, втулка, сфера и т. д.). Толщина покрытия в зависимости
от назначения и предполагаемого ресурса работы может колебаться
от 0,2 до 1,0 м/м. Покрытия обладают хорошими упругими свойствами.
Интервал рабочих скоростей без смазки составляет 1 м/с, со смаз-
кой— выше 12—15 м/с. Давление при работе двигателей в отсутствие
ударного нагружения ~5,6-108 Па, в условиях ударного нагруже-
ния— 2,8-108 Па. Коэффициент трения колеблется от 0,03 до 0,08 в
зависимости от нагрузки и скорости. Продолжительность работы таких
покрытий в зависимости от скоростей и давлений составляет до
50 тыс. ч. Сообщается [9], что полимерные антифрикционные покры-
тия успешно применяются в автомобилестроении, авиа- и ракетострое-
нии с 1962 г.
В настоящее время эти материалы внедряются в автомобильную,
станкостроительную и судостроительную промышленности, в дорожное,
сельскохозяйственное, химическое машиностроение и другие отрасли
хозяйства. Первой автомобилестроительной фирмой, применившей тка-
невые покрытия из волокна тефлон, была фирма «Фиат». В настоящее
время шаровые шарниры подвески передних колес и шарниры тяг ру-
левого управления имеют тканевые покрытия и устанавливаются во
всех моделях легковых и грузовых автомобилей. Все узлы работают
без смазки и рассчитаны на весь срок службы машин [9]. Автомо-
бильные заводы США, кроме указанных узлов, применяют эти покры-
тия в крестовинах карданных валов (вместо игольчатых подшипников),
в подшипниках шкворней поворотных цапф, в стойках амортизаторов,
в выжимных подшипниках муфт сцепления, шарнирах управления
топливной аппаратуры и других деталях [10].
Наряду с волокном тефлон начинают применять и другие термо-
стойкие и высокопрочные волокна, причем износостойкость таких по-
крытий оказывается гораздо выше, особенно при нагрузках да
2,5-108 Па.
5.3. ПРОИЗВОДСТВО ШИННОГО КОРДА
Появление высокопрочных и высокомодульных волокон типа кев-
лар, Х-500, вниивлон, СВМ открыло новые перспективы в производст-
ве шинного корда. По рекламным данным, перспективными волокнами
для армировании шин являются кевлар [11] и вниивлон [12].
Обладая исключительно высокой прочностью, эти волокна обеспе-
чивают также прекрасную стабильность размеров и стойкость к высо-
ким температурам. Волокна могут перерабатываться в шинный корд
на существующем оборудовании для синтетических волокон. В табл. 5.2
приведены свойства корда кевлар в сравнении с другими кордными
материалами [13].
207
Таблица 5.2. Свойства кевлара и других кордиых волокон
Свойства Кевлар Стальное Стеклянное Вискозное Полиэфир- ное (дакрон) Найлон 6,6
Прочность, сН/текс 185 32 84 55 80 85
Модуль, 109 Па 62 156 51,7 16,5 13,4 5,5
Удлинение, % 4,0 2,0 4,0 11,0 14,5 18,0
Прочность в петле, сН/текс 100 14 31 34 55 60
Удлинение в петле, % 2,0 0,9 0,9 7,0 9,0 12,0
Усадка при 160 °C, % 0,2 0 0 0 6,0 6,8
Удлинение (при 24 °C в те- чение 24 ч при нагрузке 9 сН/текс) 0,5 0,7 0,5 4,9 2,1 4,8
Ползучесть (30 мин), % 0,03 0,03 0,03 1,4 0,3 0,4
Температура стеклования, О/^ 300 — — — 75 50
Чл Усталостная прочность* (число циклов) 60000 55 3 22000 — —
Плотность, кг/м3 1440 7850 2560 1520 1380 1140
Температура плавления, 500 — — — 250 250
Диаметр элементарного во- локна, мм 0,0121 0,2—0,25 — — — 0,0272
* При нагрузке 5,3 сН/текс.
Преимущества кевлара в прочности и изгибоустойчивости по срав-
нению с другими кордными волокнами хорошо видны из приведенных
данных. Термостойкость кевлара так высока, что даже при 200 °C его
модуль и прочность в 2—3 раза выше, чем у обычных химических во-
локон при комнатной температуре. Волокно отличается низкой растя-
жимостью, но выдерживает большое число изгибов. Другим волокнам
кевлар уступает по устойчивости к осевому сжатию. Высокие показате-
ли имеет корд из волокна СВМ (табл. 5.3) {14].
На рис. 5.1 приведены кривые растяжения корда СВМ, снятые после
воздействия на него в течение 100 ч низких (—60 °C) и высоких темпе-
Таблица 5.3. Свойства высокомодульных кордов
Показатели Корд из СВМ Металли- ческий корд (22Л15) Стеклян- ный корд* Вискозный высокомо- дульный корд
Толщина нити, мм 0,61 0,88 0,45 0,67
Разрывное напряжение, 10е Па 2560 2500 1900 680
Удлинение при разрыве, % 4,5 2,5—3,0 4,3 12,0
Прочность (после воздействия при 200 “С в течение 2 ч), % от исходной 90 100 100 60
Прочность в мокром состоянии, % от ис- ходной 90 100 100 70
Плотность, кг/м3 1430 7800 1700 1520
Изгибоустойчивость при нагрузке 4,4мПа и а = 60° (число циклов, тыс.) 1000 0,16 20 20
Устойчивость к растяжению при Р = 7,5 см 5 — Не выдерживает
(число циклов, ТЫС.)
* Прн 20 °C.
208
ратур ( + 200 °C) в мокром состоянии. Эти данные характеризуют ста-
бильность модуля растяжения, отсутствие усадки при температуре до
в динамических условиях (рис.
170 °C и .невысокую ползучесть СВМ
5.2). Большинство исследователей, ра-
ботавших с шинным кордом из высо-
копрочных, термостойких волокон,
называет следующие преимущества
этих кордов [15]: а) небольшую мас-
су; б) большую устойчивость при вы-
соких скоростях; в) небольшое сопро-
тивление качению; г) небольшое выде-
ление тепла при пробеге; д) большую
долговечность; е) возможность изго-
товления ткани на обычном оборудо-
вании.
Ведущие американские, европей-
ские и японские фирмы, производящие
автомобильные шины, проявляют боль-
шой интерес к кевлару как к кордно-
му материалу [16]. Они считают, что
по сравнению со стальным и вискоз-
ным кордом он может обеспечить боль-
ший пробег при меньшем расходе
материала. Однако в настоящее время
еще не определилось, в шинах какой
конструкции (радиальной или опоя-
санной) его преимущества будут оче-
видными. Кевлар также рекоменду-
ется для производства бельтингов.
Волокна типа кевлар начинают приме-
нять для изготовления таких резино-
вых технических изделий, как конвей-
ерные ленты, клиновидные ремни, ру-
Рис. 5.1. (Кривые растяжения корда
из волокна CBM [12]:
1 — в мокром состоянии; 2—при нормаль-
ных условиях; 3— после нагрева при
200 °C в течение 100 ч; 4 — после охлаж-
дения при —60 °C в течение 100 ч.
J
7
кава для гидравлических систем высо-
кого давления и т. д. При использовании высокопрочных волокон в ре-
зиновых технических изделиях большое внимание должно быть уделено
повышению прочности связи с резиной. Предварительные данные ука-
зывают на возможность решения задачи путем применения латексных
адгезивов, например ДСВП-15 [12].
Рис. 5.2. Ползучесть корда [12]:
/ — корд из волокна СВМ; 2— вискозный высокомодульный корд; 3— стеклянный
корд; 4 — металлический корд.
14—2254
209
5.4. КОНСТРУКЦИОННЫЕ МАТЕРИАЛЫ
Химические волокна уже более тридцати лет применяются для ар-
мирования пластических масс. Создание высокопрочных и высокомо-
дульных химических волокон стимулировало дальнейшее развитие этой
области применения. По имеющимся данным термостойкие волокна мо-
гут перерабатываться в конструкционные материалы различными спо-
собами (табл. 5.4).
Таблица 5.4. Способы использования термостойких волокон
в пластиках
Волокно Пластик Связующее Способ переработки
Из химически сшитого ПАН Полиимидные Из ароматиче- ских полиамидов Текстолит термо- стойкий Текстолит термо- стойкий Однонаправленный, намоточный Текстолит с повы- шенной жесткостью Фенольное, поли- имидное, кремнийор- ганическое Полиимидное, крем- иийоргаиическое, фе- нольное Эпоксидное, изо- циануратиое, поли- имидное Эпоксидное, фе- нольное, полиимидное Прессование » Протяжка, намотка Прессование
Для создания новых конструкционных материалов, по-видимому, бу-
дут использованы волокна типа кевлар (PRD-49). По опубликованным
данным [17], кевлар выпускается в различных модификациях, но ос-
новными являются две модификации, одна из которых предназначает-
ся для армирования пластических масс, а другая — для изготовления
резиновых технических изделий. Характеристики пряжи из кевлара-49
представлены ниже:
Прочность, МПа.............................
Удлинение, %.............................
Модуль, 106-Па.............................
Работа обрыва..............................
Плотность, кг/м3...........................
Влагоемкость, %............................
Кривая напряжение — деформация .
Применение.................................
Модификация
тип III
2800
2,0
153000
0,215
1450
1,5
Г уковская
Армирован-
ные пластики
Модификация
тип IV
3100
ч ч
84000
0,379
1450
4,0
Не Гуковская
Резиновые
технические
изделия
Высокие прочность и модуль наряду с низкой плотностью обуслов-
ливают бесспорные преимущества кевлара перед другими высокопроч-
ными материалами, такими, например, как стеклянное, стальное и дру-
гие волокна (рис. 5.3).
Кроме того, несмотря на высокий модуль, кевлар обладает значи-
тельно более высокими текстильными свойствами по сравнению с угле-
родными, стеклянными, борными и другими волокнами. Прочность в
петле составляет 40—50%, а в узле — 30% от нормальной прочности:
Прочность в узле, 106-Па.............................784
Прочность в петле, 106-Па...........................1340
Остаточная прочность после ткачества, % . 90
210
Кевлар достаточно термостабиле». После эксплуатации в течение
1 мин при 200 °C модуль уменьшается всего на 18%, а прочность— на
25%. При длительном воздействии в течение 450 ч при 235 °C прочность
уменьшается на 30%. Волокно не поддерживает горения, не плавится,
стойко к воздействию ряда растворителей, жидкому топливу, смазоч-
ным материалам. Сильные кислоты и основания при высоких темпера-
турах действуют разрушающе. Основ-
ным недостатком кевлара как компо-
зиционного материала является низ-
кая прочность при сжатии. В направ-
лении оси волокон она составляет все-
го 15—20% от прочности при растя-
жении. Предполагают, что низкая
прочность при сжатии однонаправлен-
ных органоволокнитов обусловлена от-
ступлением от закона Гука и микро-
фибриллярностью структуры. Дефор-
мация при сжатии волокна сопровож-
дается отделением этих фибрилл.
Несмотря на некоторые недостат-
ки, композиционные материалы из ке-
влара нашли широкое применение в
самолетостроении. Впервые они были
применены в самолете Z.-1010 в виде
проемов, обтекателях крыльев и других узлов (рис. 5.4) [18].
В табл. 5.5 приводятся основные конструкционные узлы самолетов,
которые могут изготавливаться из органических композиционных ма-
териалов [18] (рис. 5.5).
Модуль, 109Па
Рис. 5.3. Показатели удельных меха-
ничесюих свойств различных волокон:
1 — стальное; 2 — вискозное; 3 — стеклян-
ное; 4 — полиэфирное; 5 — найлон 6,6; 6 —
кевлар 29; 7 — кевлар-49.
потолочных панелей и оконных
Рис. 5.4. Высокопрочная паяешь из PRD-4S.
Рис. 5.5. Обтекатель Боинга-737.
Сообщается [19], что волокно кевлар используется в автомобильной
промышленности для изготовления матов и кабин. Основным пре-
имуществом этих материалов является чрезвычайно высокая ударная
прочность. Кроме того, малая плотность органических волокон обуслов-
ливает уменьшение массы армированных материалов до 30% по срав-
нению со стеклянным волокном и до 50% по сравнению с алюминие-
выми сплавами. Необходимо особо подчеркнуть, что технология полу-
чения пластических масс, армированных кевларом, такая же, как тех-
нология получения стеклопластиков.
14* 21 г
Таблица 5.5. Перечень деталей летательных аппаратов, изготовленных
из органоволокнитов на основе полимерного волокна кевлар-49
Объект Фирма Детали
Вертолет «Боинг Вертол» Створки батареи, грузовые двери, горизои-
ВО-105 (США) совместно с «Мессершмидт» (ФРГ) тальный стабилизатор, щиток управления, хво- стовое оперение, двери кабин, ограждение ве- дущего вала, панели корпуса, обтекатели дви- гателей и трансмиссий, хвостовой лонжерон, обшивка нижней части фюзеляжа, панели по- ла, боковые панели фюзеляжа н крыша ка- бины
СН-46 «Боинг Вертол» (США) Антенный обтекатель л
Аэробус «Три- «Локхид» Потолочные панели, боковые панели ин-
стар L-1011» (США) терьера, паиелн илюмииаторов, заливы меж- ду крылом и фюзеляжем, перегородки грузо- вого отсека, задние и передние кромки руля высоты, панели горизонтального и вертикаль- ного стабилизаторов, элероны, панели крыла, задние и передние кромки крыла, облицовка грузового отсека, вентиляционные короба
«Боинг-737» «Боинг» (США) Закрылки, лопатки ротора, баки для горю- чего, камеры сгорания двигателя, элементы крыла, подставки, двери грузового люка
Фирмой «Дюпон» разработаны тканые и нетканые материалы на ос-
нове |кевл'ара-49, предназначенные для замены стеклонолокнистых на-
полнителей в пластиках авиационного и космического назначения [20].
Рекомендуется также применение кевлара ib термоизоляции пони-
женной плотности, для обшивок сотовых панелей и др. Помимо эконо-
мии массы, эффективность замены других волокнистых наполнителей
органотекстолитами обусловлена высокой ударной вязкостью послед-
них. В табл. 5.6 приведены результаты испытания на ударную вязкость
методом падающего шарика сотовой панели с обшивками из эпоксид-
ных органо- и стеклотекстолитов.
Нетканые материалы на основе кевлара изготавливают из штапель-
ных волокон, прошитых в направлении, перпендикулярном к плоскости
Таблица 5.6. Характеристика эпоксидных органо- и стеклотекстолитов
Марка ткаин (волокно) Переплетение Толщина слоя, мм Работа, затраченная на разруше- ние, Дж
однослой- ная обшивка двухслой- ная обшивка трехслой- иая обшивка
181 (кевлар-49) Сатиновое 0,254 2,88 5,6 4,6
181 (е-стекловолокно) » 0,254 2,88 5,75 6,9
281 (кевлар-49) Полотняное 0,254 5,2 5,75 6,91
2542 (е-стекловолокно) 0,254 2,54 — —
328 (кевлар-49) » 0,330 5,75 6,35 —
Экспериментальная ткань из кев- » 0,508 7,5 — —
лара-49
212
Таблица 5.7. Свойства органоволокнитов
Связующее Волокно Степень наполне- ния, % Пло тность, кг/м3 Прочность, 10« Па Модуль упругости при растяже- нии, 10в’Па
Эпоксидная смола Кевлар-49 48 1230 210,9 14760
ВР-907
Эпоксидная смола Стеклянное 50 2000 84,0 14760
ЕМ-7302
Фенольная смола Кевлар-49 40 1240 168,0 9843
61113 номекс/стеклянное 40 1300 8,40 5624
Полиэфирное 411-45 Кевлар-49 40 1310 182,0 11250
Стеклянное 40 1530 147,0 9843
Полиэфирное Кевлар-49 30 1400 154,0 12650
Стеклянное 35 1700 126,0 9843
Найлон 612 Кевлар-49 26 1320 147,0 9139
Стеклянное 31 1700 105,0 8436
материала. В табл. 5.7 приведены свойства органоволокнитов на осно-
ве нетканых материалов в сочетании с термопластичными и термореак-
тивными связующими. Для сравнения приведены свойства стеклопла-
стиков, наполненных рубленым стеклянным волокном.
В настоящее время исследуется возможность применения нетканых
материалов в сочетании с углеродными
из кевлара с большим успехом приме-
няются в комбинации с графитовыми,
борными, стеклянными и стальными
волокнами в производстве канатов,
кабелей, парашютов, напорных емко-
стей, спортинвентаря, высокоскорост-
ных маховиков, пуленепробиваемых
жилетов, обтекателей антенн, конст-
рукционных материалов для авто- и
судостроения. Американский институт
авиации и космонавтики разработал
метод намотки оболочек ракетных дви-
гателей диаметром 754 мм из волокна
кевлар-49, которые при испытаниях
разрушались при давлении 750 МПа
(рис. 5.6) [17]. Оболочки из стеклян-
ной ровницы и углеродного волокна
волокнами. Комплексные нити
Рис. 5.6. Топливный сосуд ракетного
двигателя.
выдерживали давление соответственно в пределах 660 и 630 МПа. При-
менение волокна кевлар позволило снизить массу оболочки на 35% [21].
Успешное применение высокомодульных высокопрочных органиче-
ских волокон во многом зависит от их адгезии к различным связую-
щим.
В работе [22] приводятся обобщенные данные по адгезии волокна
к иономеру (ИМ), полиэтилену (ПЭ), найлону 12 (тип Surlyn-1558,
фирма «Дюпон»), поликарбонату (ПК), полиметилметакрилату
(ПММ). В качестве наполнителя использовалось волокно кевлар-49 с
прочностью 2800 МПа, модулем растяжения 133000 МПа, относитель-
ным удлинением 2%, диаметром волокна 10 мкм трех видов:
1) комплексная нить, не обработанная аппретом, с низкой круткой;
2) волокно, пропитанное клеем марки ДР 5259-18;
213
3) не обработанная пряжа после кипячения в воде в течение
6,5 сут.
Были определены следующие показатели адгезионной прочности
и сдвиговых напряжений при трении: для ИМ соответственно 197,8 н
1,078 МПа; для ПЭ — 12,7 и 1,54 МПа; для ПММ — 54,6 и 3,07 МПа;
для ПК — 39,2 и 2,66 МПа; для Н-12 — 46,4 и 2,04 МПа. Адгезионная
прочность стеклянного волокна с эпоксидными и полиэфирными связу-
ющими находится в пределах 4,23—24,5 МПа, т. е. лежит в той же об-
ласти значений. Было сделано предположение, что экспериментальные
данные для кевлара занижены из-за дефектов на поверхности раздела
фаз, неполного смачивания, наличия микропустот и других факторов,
приводящих к концентрации напряжений на концах волокна и обу-
словливающих образование трещин.
Эксплуатационные свойства органопластиков определяются не толь-
ко прочностью, но и ползучестью при комнатной и повышенных темпе-
ратурах. В работе [23] приводятся результаты изучения ползучести
композита кевлар-49 — EKIA-4617 в сравнении с ползучестью волокна,
не пропитанного эпоксидной смолой. Композиты проявляют нестацио-
нарную ползучесть вплоть до 1000 ч, причем форма кривой зависит от
напряжения. При низких напряжениях матрица вначале ползет с мень-
шей скоростью, чем волокно кевлар, а затем картина меняется. При
нагрузках 50 и 80% от предела прочности материала при 24 °C наблю-
дается линейная зависимость деформации от напряжения. При нагру-
жении в течение 1000 ч с напряжением 2100 МПа и 24 °C конечная де-
формация составляла 0,15%; при 149 °C образцы выдерживали нагруз-
ку до 988 МПа и имели удлинение 0,2%. Деформация ползучести на
первом этапе развивается более интенсивно, чем в конце испытания,
где она практически достигла предельных значений.
Обобщая приведенные данные, ^следует отметить, что органические,
и особенно высокопрочные, волокна, отвечают требованиям, предъявляе-
мым к армирующим 'волокнам. Они имеют очень высокие прочности,
модуль эластичности, ударную вязкость и в сочетании с низкой плот-
ностью обеспечивают хорошие механические свойства, электрические
характеристики и достаточную стабильность свойств.
Обладая, кроме того, удовлетворительными текстильными свойства-
ми, эти волокна составляют реальную конкуренцию другим армирую-
щим материалам, таким, как стеклянные, углеродные, борные и другие
минеральные нити.
5.5. ПРОЧИЕ ОБЛАСТИ ПРИМЕНЕНИЯ
Большое разнообразие уникальных свойств термостойких волокон
превратило их в потенциально перспективный материал в самых раз-
личных областях техники и быта. Так, например, в США номекс все
шире начинает применяться для отделки интерьеров общественных зда-
ний (госпитали, школы, отели, залы приемов, концертные залы и др.) и
средств транспорта вследствие его негорючести и относительной без-
вредности продуктов разложения. Окрашенные ковры из номекса, со-
держащие до 1 % металлических нитей для снятия электростатических
зарядов, используются в самолетах «Боинг-707» фирмой «Сабена» [24].
Имеются сообщения о применении номекса и подобных волокон (поли-
имидных) для изготовления парашютов, электроизоляции, армирующих
материалов, панелей судовых и других транспортных средств [25]. Са-
214
мым крупным потребителем номекса является бумажная промышлен-
ность, использующая его для получения фибридов и специальной высо-
копрочной синтетической бумаги. Такая бумага незаменима в качестве
электро- и теплоизоляции в двигателях электровозов, конденсаторах
и изделиях электротехнической промышленности [26]. Хорошие резуль-
таты получены при использовании полых волокон из номекса в процес-
сах обратного осмоса для обессоливания воды. Эти волокна выдержи-
вают давление в 6 (раз большее, чем полые ацетатные волокна.
Наряду с ароматическими полиамидными волокнами широкое при-
менение начинают приобретать и полиимидные волокна. Имеются со-
общения [27] о применении кермеля для защитной одежды в горячих
цехах промышленных предприятий, для одежды гонщиков, военного
персонала и даже спального белья в госпиталях. В резинотехнической
промышленности эти волокна применяются для изготовления рукавов,
шлангов и т. д. В качестве декоративной пожаробезопасной отделки
общественных зданий полиимидные волокна имеют преимущества пе-
ред номексом [28].
Волокно кинол, получаемое из фенолоформальдегидной смолы, мо-
жет использоваться в виде штапельных тканей для изготовления за-
щитной одежды, обивки самолетов, мебели в больницах и госпиталях,
а также пламяшреграждающих занавесей [29].
Смеси кинола и номекса применяются для повышения огнестойко-
сти номекса и облегчения окраски изделий [30]. Полое волокно кинол,
превращенное путем термообработки в углеродное, имеет очень хоро-
шие сорбционные свойства и является ионообменным [31].
Ткани из термостойких волокон, наполненные, окислами тяжелых
металлов, используются для защиты от радиации.
ЛИТЕРАТУРА
1. Фрейзер А. Г. Высокотермостойкие полимеры. М., «Химия», 1971. 270 с. ; Хим. во-
волокна, 1975, № 3, с. 73—75.
2. Жевлакод А. Ф. и др., Хим. волокна, 1976, № 5, с. 28—29.
3. Kuhner G., Spinner, Weber, Textil v. 1971, Bd. 89, S. 405—410.
4. Sheales O. L., Text. Inst. Ind., 1971, v. 9, p. 10—13.
5. Chem. Week, 1971, v. 108, № 6, p. 47—48.
6. Text. World, 1971, v. 121, № 3, p. 31—32; Nenworen Rep., 1972, № 10, p. 2—8.
7. Daily News Rec., 1973, v. 11, № 1, p. 22—25.
8. N.A.S.A. Spec. Rut, 1971, № 5096, p. 155—165.
9. Techn. Rundschau, 1971, Bd. 63, № 26, S. 5—7.
10. Ing. Digest, 1973, v. 12, p. 94—99.
11. Wilfong P. E., Zimmerman G., Rubb. World, 1972, v. 166, № 4, p. 40—44, 46—50.
12. Нагдаседа И. П. и др. Международный симпозиум по химическим волокнам. Пре-
принты. Калинин, 1974, секция 6, с. 114—118.
13. Text. World, 1974, v. 124, р. 21—25.
14. Wolf R. F., Rubb. Age, 1971, v. 105, № 8, p. 25—30; Нагдаседа И. П. и др. Между-
народный симпозиум по химическим волокнам. Препринты. Калинин, 1974, секция 6,
с. 115—118.
15. Chem. Week, 1974, v. 114, № 3, р. 9—11; 1975, v. 116, № 6, р. 15—19.
16. Chem. Eng. News, 1974, v. 52, № 2, p. 7—11; Chem. Fibres Monthly (Japan), 1973,
v. 26, № 8, p. 70—75.
17. Bruins P. F. Un saturated Polyester Technology. N. Y., 1976. 437 p.
18. New Ind. a. Appl. Mater., 1974, № 2, p. 609—627; РЖХим, 1975, реф. 10T706.
19. «Kunststoff-Rundschau», 1973, Bd. 21, Ns 12, S. 592; РЖХим, 1975, реф. 11T154.
20. Symp. a. Exhib. Calif, 1974, v. 19; РЖХим, 1975, реф. 10T193.
21. Martel I. B„ «Paper», 1975, Ns 1335, p. 5; РЖТ, 1975, № 7, c. 10—11.
22. Eagles T. B. e. a., J. Appl. Polymer Sci., 1976, v. 20, Ns 2, p. 435—448.
215
23. Erichsen P. H., «Komposites», 1976, Bd. 7, № 3, S. 189—193.
24. Tufting a. Needling, 1973, № 7, p. 9—10.
25. Plast. a. Rubber Wukly, 1971, № 10, p. 10—12.
26. Chem. Ind., 1973, Bd. 25, S. 67—68.
27. Mod. Text. Magaz., 1974, v. 55, № 8, p. 37—40.
28. Tufting a. Needling, 1973, № 40, p. 4—5.
29. Daily News Rec., 1974, v. 12, № 2, p. 2—3.
30. Daily News Rec., 1973, v. 11, № 1, p. 12—13; 1974, v. 12, № 8, p. 16—18.
31. Chem. Eng., 1972, v. 79, № 16, p. 52—55.
Часть II
ЖАРОСТОЙКИЕ
(УГЛЕРОДНЫЕ)
ВОЛОКНА
ПРИНЯТЫЕ СОКРАЩЕНИЯ
ВК— вискозная кордная нить
ВТО—високотемпературная обработка
ГЦ-В — гидратцеллюлозное волокно
КМ—композиционные материалы, композиты
КМК— композиционные материалы иа основе углеродного волокна
и керамической матрицы
КММ—композиционные материалы на основе углеродного волокна
и металлической матрицы
ККМП — конструкционные композиционные материалы на основе
углеродного волокна и полимерных связующих
КМП— композиционные материалы на основе полимерных связую-
щих
КМУ композиционные материалы на основе углеродной матрицы
МКЦ — монокарбоксицеллюлоза
МУР—малоугловое 'рассеяние, дифференциальное рассеяние под
малыми углами
ПАН-В — полиакрилонитрильное волокно
ПВ — пековое волокно
ПВС—поливиниловый спирт
ПВХ—поливинилхлорид
ПМЦ — парамагнитные центры
СП — ciCII'cnb |П0ЛИ'МСрИЗаЦИИ
ТГА — термогравиметрический анализ
ТТО—-температура термообработки
УВ — углеродное волокно
УВ-ГЦ— углеродное волокно на основе гидратцеллюлозного волокна
УВМ — углеродные волокнистые материалы
УВ-ПАН — углеродное волокно на основе полиакрилонитрильного во-
локна
УВ-П — углеродное волокно на основе пекового волокна
УВ-ФФС—углеродное волокно на основе фенолоформальдегндного во-
локна
УМ— углеродные .материалы
УУТ —• углеродная упрочненная ткань
ФФС — фенолоформальдегидная смола
ФФС-В — волокно из фенолоформальдегидных смол
СОДЕРЖАНИЕ
Введение.................................................................
Г лава 1. Структура углеродных волокон...................................
1.1. Особенности структуры переходных форм углерода......................
1.2. Особенности структуры углеродных волокнистых материалои.............
Литература...............................................................
Глава 2. Исходные материалы и некоторые особенности получения углерод-
ных волокон .............................................................
2.1. Линейные полимеры...................................................
2.2. Пространственные полимеры...........................................
2.3. Пеки................................................................
2.4. 'Некоторые особенности процесса перехода полимер —► углерод ....
Литература...............................................................
Г лава 3. Получение углеродных волокнистых материалов....................
3.1. Получение углеродных волокнистых материалов на основе ПАН-волокна
3.1.1. Требования к исходному ПАН-волокну............................
3.1.2. Термическое окисление ПАН-волокна.............................
3.1.2.1. Физико-химические процессы, протекающие при окислении ПАН-
волокна .......................................................
3.1.2.2. Условия окисления ПАН-волокна..........................
3.1.2.3. Вытягивание ПАН-волокна при окислении..................
3.1.3. Высокотемпературная обработка ((карбонизация и графнтация) окис-
ленного ПАН-волокна..................................................
3.1.3.1. Физико-химические процессы, протекающие при высокотемпе-
ратурной обработке ПАН-волокна.................................
3.1.3.2. Условия проведения карбонизации ((графитами) волокна
3.2. Получение углеродных волокнистых материалов на основе ГЦ-®олокна .
3.2.1. Основные закономерности термической деструкции целлюлозы .
3.2.2. Основные закономерности и условия проведения карбонизации
3.2.3. Основные закономерности процесса графнтании углеродного волокна
3.3. Получение углеродных волокнистых материалов на основе пеков и феноль-
ных смол..............................................................
3.3.1. Пеки..........................................................
3.3.2. Фенольные смолы ...........................................
Литература...............................................................
Глава 4. Свойства углеродных волокнистых материалов......................
4.1. Макроморфологня.....................................................
4.2. Механические свойства...............................................
4.3. Термические свойства................................................
4.4. Химические .свойства................................................
4.5. Физические свойства.................................................
Литература...............................................................
Глава 5. Применение углеродных волокон...................................
5.1. Композиционные материалы на основе полимерных связующих (КМП) .
5.1.1. Общие замечания...............................................
5.1.2. (Конструкционные композиционные материалы на основе полимерных
связующих (ККМП).....................................................
5.1.3. Другие типы композиционных материалов на основе полимерных свя-
зующих ((КМП)........................................................
5.2. Композиционные материалы на основе углеродного волокна и углеродной
матрицы >(КМУ)........................................................
5.3. Композиционные материалы на основе углеродного волокна и керамической
матрицы (КМК)............................................................
5.4. Композиционные материалы на основе углеродного волокна н металлической
матрицы (КММ).........................................................
5.5. Применение композиционных материалов................................
5.6. Применение собственно углеродных волокнистых материалов . . . .
Литература...............................................................
220
222
222
225
235
236
238
243
248
256
259
259
260
260
262
262
266
269
270
270
273
275
276
281
285
287
287
291
295
298
298
299
305
307
309
313
314
315
315
316
326
328
331
332
334
336
338
219
ВВЕДЕНИЕ
В связи с развитием ракето- и самолетостроения, высокотемпера-
турной техники, освоением космоса возникла острая необходимость в-
жаростойких волокнистых материалах, предназначенных для эксплуа-
тации при высоких температурах. Наряду с жаростойкостью в зависи-
мости от назначения и области применения к этой группе материалов
предъявляется ряд других жестких требований. Традиционные природ-
ные и химические, включая термостойкие, волокна не удовлетворяли
этим требованиям.
Создание волокон новых типов сводилось к изысканию исходных
соединений и в большинстве случаев к разработке новых методов полу-
чения волокна. И эта сложная научно-техническая проблема в корот-
кие сроки успешно была решена.
В настоящее время известно большое число жаростойких волокон,
которые находятся на различных стадиях технического развития: одни
из них не вышли за рамки лабораторных исследований, другие выпус-
каются в опытном или промышленном масштабе. К важнейшим жаро-
стойким волокнам относятся: углеродное, борное, на основе окислов;
различных элементов (А12О3, ZrO2 и др.), карбидов (SiC-волокно), нит-
рида бора, металлов и сплавов и др.
Благодаря разнообразным ценным, а по ряду показателей уни-
кальным механическим и физико-химическим свойствам среди жаро-
стойких особое положение занимают углеродные волокна. В углерод-
ных волокнах удачно сочетаются высокие прочность и модуль Юнга с-
низкой плотностью, поэтому по удельным показателям (отношение
прочности и модуля к плотности) они превосходят все жаростойкие во-
локна. Высокопрочные высокомодульные волокна вырабатываются с
прочностью 2500—3500 МПа (250—350 кгс/мм2) и модулем Юнга 200—
600 ГПа (20 000—60 000 кгс/мм2) при плотности'1,6-103—1,8-103 кг/м3.
Им присуща необычайно высокая теплостойкость; в инертной среде или
при глубоком вакууме механические свойства волокна до 1800—2200 °C
практически не изменяются. Волокна обладают исключительной хи-
мической стойкостью. Они стойки ко всем агрессивным средам, за
исключением окислителей при повышенной температуре. Эти волок-
на разнообразны по электрофизическим свойствам (от полупроводни-
ков до проводников с проводимостью, характерной для металлов) и
могут иметь необычайно развитую поверхность (до 1000—2000 м2/г).
Ценные свойства предопределяют возможность применения волокна или
пластиков на его основе в самолета- и ракетостроении, при создании
космических аппаратов, в химическом и общем машиностроении, судо-
строении, текстильном машиностроении, для изготовления спортинвен-
таря, в качестве теплозащитных и абляционностойких материалов, ан-
220
тистатических и антифрикционных добавок к полимерам, при изготов-
лении нагревательных элементов разнообразного назначения, получении
сорбционно-активных материалов, носителей катализаторов и др. Уг-
леродные волокна и ткани можно выпускать в различной текстильной
форме, что также является одним из существенных их достоинств. По-
тенциальные возможности, заложенные в волокнах, далеко еще не ис-
черпаны, и их с полным основанием можно считать не только материа-
лом настоящего, но и будущего.
Углеродные волокнистые материалы в ощутимых количествах нача-
ли вырабатываться примерно с 1958 г. В настоящее время они изго-
товляются в США, Англии, Японии, Франции, ФРГ. Ниже при-
веден перечень основных фирм, выпускающих углеродные волокна:
Celanese Advanced Engineering Composites (Селаниз эдванст инжении-
ринг композите); Great Lakes Carbon Corp. (Грейт лейке карбон кор-
порейшен); Hercules Inc. (Геркулес инкорпорейшен); Hitco, Morganite,
.Modmor Inc. (Хитко, Марганайт Модмор инкорпорейшен); Union
Carbide Inc. (Юнион карбайд инкорпорейшен); Stackpole Fibres Со.
1пс. (Стакпол фойбз компани инкорпорейшен); Corborundum, Wittakers
Со. (США) (Карборундум, Витакере компани); Courtaulds Ltd.
(Куртолдс лимитед); Morganite Modmor Ltd. (Марганайт Модмор ли-
митед); Bristol Composite Materials (Бристол композит материалз);
[Rolls — Royce Ltd. (Роле — Ройс лимитед) [Англия]; Nippon Carbon Со.
(Ниппон карбон компани); Toray Industries Inc. (Торей индастриес
инкорпорейшен); Toho Beslon (Тохо Беслон); Kureha Chemical Industry
Со., Ltd. (Куреха кемикл индастри компани лимитед); Mitsubischi
Rayon Со. (Митсубиси рейон компани) [Япония]; Le Carbone Lerraine
(Ле Карбон Лорен) [Франция]; Sigri Elektrographit GmbH (Зигри
электрографит) |[ФРГ].
На первом этапе волокна использовались преимущественно в воен-
ной промышленности, поэтому о масштабах производства публикуются
противоречивые и неполные сведения. Все же объемы их производства
невелики, и пока еще стоимость этих волокон довольно высока. В за-
висимости от свойств волокне и природы используемого для их полу-
чения пр едм а тер и ала на международном рынке цена углеродного во-
локна составляет 50—250 долларов/кг. Этот важный показатель тесно
связан с масштабом производства; по прогнозам в ближайшие годы
стоимость волокна снизится до 10—25 дол ле ров/кг.
Вначале сведения об этих волокнах появлялись преимущественно
в патентах; за последние 5—7 лет опубликовано большое число ориги-
нальных и обзорных статей, начинают появляться книги, в которых
преимущественно описываются свойства и области их применения; о
принципах получения волокна приводятся краткие сведения. Наиболее
полно обобщена литература по этой проблеме в книге автора «Углерод-
ные и другие жаростойкие волокнистые материалы», выпущенной изда-
тельством «Химия» в 1974 г.
В предлагаемой читателям части монографии рассматриваются
принципы подбора исходного сырья, современные представления о
структуре углеродных волокон и ее формировании на разных стадиях
термообработки, физико-химические основы получения, свойства и об-
ласти применения собственно углеродного волокна и композиционных
материалов с различными связующими. Большой интерес представля-
ет аппаратурно-технологическое оформление процесса получения угле-
родного волокна. К сожалению, сведения об этом в литературе не при-
221
водятся. Поэтому мы лишены возможности рассмотреть эту крайне
важную составную часть процесса получения углеродных волокнистых
материалов.
Автор выражает глубокую благодарность коллегам по работе и
особенно кандидату технических наук Н. Ф. Конковой за помощь, ока*
занную при подборе материала и в работе над рукописью.
ГЛАВА 1
СТРУКТУРА УГЛЕРОДНЫХ волокон
1.1. ОСОБЕННОСТИ СТРУКТУРЫ ПЕРЕХОДНЫХ ФОРМ УГЛЕРОДА
Для четырехвалентного углерода известны три валентных состоя-
ния, соответствующие sp3-, sp2-, sp-гибридизации электронов атомов уг-
лерода. При зр3-гибридизации углерод образует четыре энергетически
равноценные о^связи, при зр2-пибридизации три о-связи и одну л-связь,
а sp-гнбридное состояние соответствует двум ст-связям и двум л-связям.
Алмаз представляет собой пространственный полимер, состоящий
из атомов углерода зр3-гибридизации с тетраэдрическим расположени-
ем валентных связей. Графит является ‘слоистым полимером, постро-
енным из атомов углерода зр2-гибридизации, расположенных в одной
плоскости. В карбине [1], имеющем линейную полимерную цепочку
полиенового (—С = С—С = С—)п или кумуленового (=С=С=С=
= С=С = )П типа, реализуется sp- гибридное состояние атомов углерода.
Таким образом, алмаз, графит и карбин относятся к простейшим
кристаллическим формам трех гибридных разновидностей углерода. На-
ряду с кристаллическими известно большое число аморфных и частич-
но-кристаллических переходных углеродных веществ. К ним относятся:
угли различной степени метаморфизма, сажи, кокс, волокна, пленки,,
мембраны, стеклоуглерод, пеноуглерод, .пироуглерод, пирографит и др.
Как отмечает В. И. Касаточкин [2]: «Здесь мы встречаемся с редким
случаем непрерывных изменений физических и физико-химических
свойств однокомпонентной системы, зависящей только от структуры, а
не от состава, как это обычно наблюдается для многокомпонентных
систем».
Углерод переходных форм обладает разнообразными технически
ценными свойствами и имеет большое практическое значение. Широ-
кий диапазон изменения свойств связан с возможностью набора и со-
четания различных гибридных форм и особенностями структуры угле-
рода. Эти формы углерода сложны и мало изучены. К основным эле-
ментам его структуры относятся: базисные ленты, турбостратные па-
кеты, аморфный углерод, состоящий из набора различных гибридных
форм, и надатомные образования высшего порядка. Структурная орга-
низация и свойства существенно зависят от природы исходного сырья,
используемого для получения углерода, и условий термообработки, осо-
бенно высокотемпературной (ВТО).
Переходные формы углерода подразделяются на гомогенно-трафи-
тирующиеся и неграфитирующиеся. Углеродные волокнистые материа-
222
Рис. 1.1. Структура графита.
лы (УВМ) относятся к гомогенно-неграфитируюшимся формам угле-
рода.
Базисные ленты переходных форм углерода по строению аналогич-
ны графитовым плоскостям. Они состоят из атомов углерода, образую-
щих гексагональную сетку, при этом атомы располагаются в вершинах
шестигранника, а ординарные и двойные связи последовательно череду-
ются. Особенностью плоскостей является большая насыщенность сопря-
женными связями. Расстояние между атомами углерода в цикле и ве-
личина угла между ними составляют 1,417 А и 60° соответственно [3,
с. 22—27]. В зависимости от условий получения углеродных материалов
(УМ) размеры лент могут ко-
лебаться в широких пределах,
достигая тысячи ангстрем.
В идеальной структуре
графита (рис. 1.1) плоскости
расположены параллельно
друг другу; межплоскостное
расстояние doo2 составляет
3,356 А[4, с. 10—12]. Строгая
повторяемость слоев наблюда-
ется через один слой, т. е. трех-
мерная структура графита мо-
жет быть представлена после-
довательно чередующимися
слоями ававав...; расстояние
между идентично расположен-
ными слоями составляет 6,69 А.
Атомы углерода одной плос-
кости находятся над и под центрами правильных шестиугольников.
Подобная система соответствует гексагональной структуре с четырьмя
атомами углерода в ячейке.
Турбостратная структура углерода и сходна, и отличается
от структуры графита. Ее основой являются базисные плоскости,
строение которых аналогично графитовым плоскостям. Определенное
число соединенных между собой плоскостей образует турбостратные
кристаллиты (пакеты). В отличие от графита в пакетах отсутствует ка-
кая-либо взаимная ориентация плоскостей в направлении, перпендику-
лярном базисным плоскостям, т. е. они расположены под различными
углами друг к другу. В строго’ кристаллографическом понимании паке-
ты по структуре нельзя отнести к кристаллам, так как в них отсутству-
ет трехмерная упорядоченность. В каждой плоскости пакета атомы уг-
лерода расположены в строгом порядке, поэтому пакеты являются как
бы двухмерными кристаллами. Однако условно в литературе принято
турбостратную структуру углерода называть кристаллической, что с
практической точки зрения вполне оправдано.
Межплоскостное расстояние в пакетах колеблется в пределах
3,36—3,95 А; оно значительно больше, чем в графите. Форма и разме-
ры турбостратных пакетов характеризуются параметрами La, Lc, d002;
La> Lc — размеры кристаллитов в направлениях, параллельном и пер-
пендикулярном базисным плоскостям (La, Lc — соответственно ширина
и толщина пакета); doo2 по аналогии с графитом—межплоскостное рас-
стояние. Значение dOoz рассчитывается из рентгеновских данных по фор-
223
муле Вульфа—Брегга, a Lc и La — по формулам Селякова—Шеррера
[5]:
пк = 2d sin 0
а Р100 COs 0100
0.89А,
С РоО2 COs 0002
где А — длина волны рентгеновского излучения;
0 — угол отражения данной плоскости;
Роог и Pioo — исправленная ,(с учетом экспериментального уширения) толщина линий,
отраженных от плоскостей 002 и 100.
Параметры пакетов зависят от
рис. 1.2 для иллюстрации приведено
Рис. 1.2. Влияние высокотемпературной
обработки |(ВТО) на межплоскостное
расстояние dQ02 (/), ширину La (2) и
толщину Lc (3) пакета шунгита (пара-
метры исходного шунгита:
Д»2=3,497 А; £а=67 А; 7^=18 А).
происхождения УМ и ВТО. На
значение параметров для шунгита,
относящегося к неграфитирую-
щимся формам ископаемых уг-
лей, в зависимости от ВТО. На
рис. 1.2 можно выделить три об-
ласти изменения параметров па-
кета: до температуры 1000—
1400 °C они мало ^меняются; в
пределах 1400—1800 °C происхо-
дит резкое уменьшение г/оог и
возрастание La и Lc, выше ука-
занных температур наблюдается
более монотонное изменение па-
метров пакета. По абсолютным
значениям La>Lc, a J002 даже
при ТТО = 2800°С остается боль-
ше межплоскостного расстояния
в кристаллах ррафита. С учетом
межплоскостного расстояния,
равного 3,436 А, при ТТО =
=2800 °C пакет состоит из 25
плоскостей. Размеры пакетов
стеклоуглерода, полученного из
фенолоформальдегидной смолы
(ФФС), значительно меньше
размеров пакетов термообрабо-
танного шунгита. В литературе
приводятся и другие данные о размерах пакетов.
В углероде переходных форм содержится набор пакетов различных
размеров, поэтому рентгенографически определенные значения La, Lc,
d002 являются среднестатистическими.
В рассматриваемых материалах сосуществуют кристаллические
турбостратные структуры и аморфный углерод. Наименее изученной яв-
ляется аморфная часть УМ, так как практически отсутствуют методы
ее исследования. В аморфной части, вероятно, находятся атомы углеро-
да sp3-, sp2- и sp-гибридизации. Число возможных вариантов углерод-
углеродных связей огромно. Они могут отличаться межатомными рас-
224
стояниями, распределением электронной плотности и энергией [6].
Различный среднестатистический набор углерод-углеродных связей вно-
сит существенный вклад в свойства и тем самым определяет многообра-
зие углеродных веществ.
Из-за отсутствия надежных методов трудно определить количест-
венное соотношение аморфной и кристаллической фракций в УМ.
В коксе, полученном в результате термодеструкции ПВХ, содержание
аморфной фракции составляет 35% [?]•
Кристаллические и аморфные фрикции углерода химически связа-
ны между собой и образуют пространственное полимерное тело.
1.2. ОСОБЕННОСТИ СТРУКТУРЫ УГЛЕРОДНЫХ
ВОЛОКНИСТЫХ МАТЕРИАЛОВ
Структурная организация УВМ наследуется от исходного сырья.
Химические волокна представляют собой высокоориентированные, по-
строенные из фибрилл системы, из которых образуются анизотропные
УВ; при применении в .качестве исходных материалов олигомеров по-
лучается изотропное волокно. В последние годы разработаны способы
получения УВ, позволившие значительно сгладить это различие.
Рассмотренные в разделе 1.1 структурные элементы присущи УВМ.
Вместе с тем им свойственна более богатая и сложная, особенно для
анизотропных волокон, структурная организация, включающая в себя
фибриллы, специфические поры и характеризующаяся высокой ориен-
тацией элементов структуры вдоль оси волокна. Помимо природы сырья
структура УВМ зависит от условий термообработки, особенно ВТО.
На ранней стадии карбонизации (600—900 °C) происходит арома-
тизация углерода с образованием базисных плоскостей, которые имеют
ограниченные размеры и беспорядочно расположены в массе мате-
риала. Видимо, большая часть углерода представлена линейными фор-
мами различной гибридизации. При ТТОД>1000°С происходит дальней-
шая ароматизация с превращением линейных форм углерода в полисо-
пряженные гексагональные системы.
Относительно температуры, соответствующей началу образования
турбостратных структур, приводятся противоречивые сведения. По од-
ним источникам [8], согласно данным, полученным с помощью элект-
ронного микроскопа высокой разрешающей способности, уже при 320 °C
образуются пакеты, состоящие из четырех 'слоев Lc, а при 800°C число
слоев в пакете возрастает до шести (Lc=17 А). В обзоре [9] приводят-
ся данные об образовании графитоподобных структур при ТТО до
1100 °C, но их количество невелико, и они обнаруживаются лишь после
удаления (травления) менее упорядоченных областей (при ТТО=
= 1000°C пакет состоит из четырех-пяти слоев). В то же время, по
Фиалкову <[10], хорошо воспроизводимые значения La, Lc, dm2 полу-
чаются лишь при ТТО>1800°С. При применении в качестве исходного
материала ПАН-В образование пакетов происходит на более ранней
стадии термообработки по сравнению с другими видами сырья. Бес-
спорно, и это показано многими исследователями l[il 1—13], что размеры
кристаллов (La, Lc) возрастают при ВТО, причем наиболее интенсивно
при ТТО>1800—2000 °C (рис. 1.3, 1.4). Как правило, по размеру и
скорости прироста La превосходит £с.
По данным Ока до и сотр. [14], размеры кристаллитов не зависят
от скорости нагрева и определяются лишь ТТО. Иного, более достовер-
15—2254
225
него мнения придерживается В. Я. Варшавский и сотр. [13], согласно
которому скорость (нагревания вносит вклад в процесс структурообра-
зования. При кратковременной обработке (в течение 1 мин) система
Рис. 1.3. Влияние температуры обработки УВ-ПАН на La при разной продолжи-
тельности процесса:
1 — 1 мин; 2— 90 мин; 3— 1 мин (в присутствии бора).
Рис. 1.4. Влияние температуры обработки УВ-ПАН на £с '(кривые 1—3) и <р/2
(кривая 4) при продолжительности процесса:
1, 4—1 мии; 2 — 90 мин; 3— 1 мин (в присутствии бора).
остается неравновесной, увеличение продолжительности до 90 мин дает
возможность получить волокно с более равновесной структурой. Усло-
вия деформации также влияют на параметры кристаллитов. Снижение
усадки УВ-ПАН с 12% (полная усадка) до 7% приводит к росту раз-
меров кристаллитов, особенно пара-
метра La [13]. Влияние вытягивания
на разных стадиях ВТО наглядно так-
же подтверждено данными работы
[15]. Например, в условиях свободной
усадки размеры La составляют 170 А,
а при вытягивании на 23% возрастают
Рис. 1.5. Влияние скорости вытягива-
ния УВ-ПАН (ТТО=2900 °C) на раз-
меры Lc (перпендикулярно оси во-
локна).
до 250А (ТТО = 2970°С, УВ-ПАН).
Как показано Перретом и Руландом
[16], размеры пакетов зависят от ско-
рости деформации. Значение Lc начи-
нает интенсивно увеличиваться прн
скоростях деформации выше 0,1 и
0,05 с-1 (рис. 1.5.). Влияние фактора
времени (скорости деформации) сви-
детельствует о значительной сопротив-
ляемости системы перестройке струк-
туры, проявляющейся даже при очень
высоких температурах.
На кристаллизацию влияют не
только кинетика и термодинамика этих процессов, но и предструктура
углеродного материала, полученного на ранних стадиях термообработ-
ки. Сопоставление рис. 1.2 с рис. 1.3 и 1.4 показывает, что шунгит по
сравнению с УВ-ПАН при ТТО до 2000—2400 °C способен к образова-
нию более крупных кристаллов. В свою очередь, для УВ-ПАН харак-
226
терна более легкая кристаллизуемость по сравнению с УВ-ГЦ. В стек-
лоуглероде установлено наличие турбостратных структур, но в нем
размеры кристаллитов гораздо меньше, чем в УВМ и других углерод-
ных материалах.
На рис. 1.3 и 1.4 приведены пакеты сравнительно небольших
размеров. По другим источникам {10, 17], La для УВ-ПАН и УВ-ГЦ
достигает 200—300 A, a Lc составляет 100—150 А (ТТО = 3000 °C). В об-
зоре [8] для УВМ с ТТО=2500°С даны следующие размеры кристал-
литов: La = 70—120 A, Lc—60 А; по другим источникам эти данные за-
вышены; указывается, что при повышении ТТО от 1100 до 2300 °C Lc
возрастает с 10 А до 40 A, a La от 20 А до 71 А. При температурах,
близких к реальным условиям получения графитированных У В (1500—
2400°C), размеры Lc составляют 30—50 А (8—14 слоев в пакете). Та-
ким образом, УВМ можно отнести к поликристаллическим системам,
состоящим из набора мелких кристаллов.
Другим важным параметром кристаллической ячейки является
межплоскостное расстояние d002- Для гомогенно-графитирующихся ма-
териалов по мере повышения ТТО происходит распад боковых ради-
калов и линейных гибридных форм углерода, сопровождающийся рос-
том размеров кристаллов, сближением базисных плоскостей с одновре-
менным их азимутальным поворотом и превращением в трехмерно-упо-
рядоченную структуру графита. Для графитирующихся материалов, по
мнению В. И. Касаточкина и сотр. [18, 19], изменение йоог может слу-
жить критерием оценки степени графитании, определяемой по уравне-
нию
_ 3,43 — d002
3,43 — 3,356
где у — относительное сближение слоев;
3,43 — исходное межплоскостное расстояния;
4оо2 — текущее значение межллоскостного расстояния;
3,356 — межплоскостное расстояние для предельно графитированного искусственного
графита. Значение у изменяется в пределах от 0 до 1.
Значение d002 зависит от многих факторов, поэтому приводимые
сведения противоречивы. Для гомогенно-графитирующихся материалов
(нефтяной кокс) максимальная скорость уменьшения йоог наблюдается
при 2000—2500 °C и совпадает со скоростью прироста La и Lc [20,
с. 30]. В случае некристаллизующихся УМ <Фюг является одним из па-
раметров, характеризующих степень совершенствования структуры. По
мере повышения ТТО наблюдается уменьшение Д>о2- Например, у шун-
гита резкое уменьшение d002 отмечается при ТТО«1500 °C и совпадает
с началом интенсивного роста La и Lc (см. рис. 1.2). Согласно [16],
для УВМ наблюдается сравнительно монотонный ход кривой doo2=f
(ВТО) (рис. 1.6). Аналогичная закономерность отмечается в работе
[10]. До температур 2400—2500 °C Д>ог уменьшается без заметного
укрупнения кристаллитов, величина которых начинает резко возрастать
лишь в пределах 2500—2900°C (УВ-ГЦ). Для УВ-ПАН doo2 мало из-
меняется при ТТО 1000—2400 °C и только выше 2400 °C заметно умень-
шается [11, 13]. Для УВМ минимальное значение </002 составляет около
3,4 А (для графита — 3,35 А).
Ориентация относится к одному из важнейших элементов структу-
ры и является характерной особенностью УВМ.
15»
227
До недавнего времени предметом исследования являлась ориен-
тация базисных плоскостей вдоль оси волокна (аксиальная). В послед-
ние годы установлено, что радиальная наряду с аксиальной ориентаци-
ей влияет на механические свойства волокна. Продольная (осевая, ак-
сиальная) ориентация определяется рентгенографическим или электрон-
но-микроскопическим методом, радиальная — с помощью поляризаци-
онного метода или сканирующей электронной микроскопии.
УВМ, полученные из химических волокон, могут обладать необы-
чайно высокой осевой ориентацией, и именно благодаря этому им при-
Рис. 1.7. Зависимость предельной ориентации волокна Z от температуры
обработки:
1 — УВ-ПАН; 2 — УВ-ГЦ.
Рис. 1.6. Зависимость межплоскостного расстояния от температуры обра-
ботки УВ-ГЦ.
суща анизотропия физико-механических свойств. Осевая ориентация
зависит от природы исходного сырья [22], ВТО >[21], условий термооб-
работки, особенно вытягивания в процессе получения УВМ.
Различие в степени ориентации УВ из ПАН и ГЦ |[16] на-
глядно иллюстрируется рис. 1.7. Ориентация исходного ПАН-В выше
ГЦ-В. На начальных стадиях термообработки в обоих случаях наблю-
дается снижение ориентации, но для ГЦ-В оно гораздо больше, чем
для ПАН-В. При ТТО>400—500°C ориентация УВ-ПАН возрастает
значительно интенсивнее, поэтому при одинаковых ТТО она для
УВ-ПАН гораздо выше, чем для УВ-ГЦ. Лишь при очень высоких ТТО
и вытягивании УВ-ГЦ значения Z для обоих УВ становятся соизмери-
мыми. Образование из ПАН-В лестничного полимера на ранних стади-
ях термообработки способствует сохранению и росту ориентации. С по-
вышением ТТО ориентация УВ возрастает, особенно при ТТО >2000 °C
(см. рис. 1.4). Подобная закономерность отмечается во многих работах.
Однако совершенствование структуры УВ-ГЦ запаздывает по сравне-
нию с УВ-ПАН по ТТО (рис. 1.7).
Согласно [23], ориентация лент вдоль оси волокна является средне-
статистической величиной. Отсюда становится понятным разное значе-
ние модуля Юнга при одинаковой средней ориентации УВ-ПАН и
УВ-ГЦ. Для улучшения механических свойств УВ необходимо до бивать -
228
ся снижения неравномерности ориентации вдоль оси волокна и макси-
мальной однородности структуры (радиальной ориентации) в попереч-
ном сечении волокна. Плоскости, расположенные на границе раздела
поры — углерод, искривляются, и из-за этого несколько снижается ори-
ентация [24].
Как и следовало ожидать, вытягивание волокна в процессе термо-
обработки способствует ориентации плоскостей. Для углеродных воло-
кон, полученных при высоких ТТО и вытягивании, характерна необы-
чайно высокая аксиальная ориентация базисных плоскостей (угол по-
луширины <р/2 достигает 6—8°). Для УВМ, полученных из ФФС или
пеков, по мере роста ТТО наблюдается совершенствование структуры
[25], но волокно остается изотропным вплоть до ТТО=2700°С. Только
вытягивание в сочетании с высокой ТТО приводит к ориентации плос-
костей и приближению волокна по структуре и свойствам к УВ-ГЦ
[8]-
Элементарные нити неоднородны по поперечному сечению. Ради-
альная (поперечная) ориентация зависит от тех же факторов, что и
аксиальная, поэтому она изменяется симбатно с последней. Эта зако-
номерность особенно четко проявляется для слоев, близлежащих к по-
верхности волокна. Радиальная ориентация рассмотрена в работах (И,
12, 23]. Объектами исследования являлись УВ, полученные из ПАН-В
куртель (круглый срез) и орлон (гантелевидный срез). Показано, что
присущая ПАН-В неоднородность поперечного сечения наследуется в
УВ. На рис. 1.8 схематично изображена радиальная ориентация УВ.
Наиболее типичным показателем радиальной неоднородности является
наличие ядра и оболочки, сохраняемых в УВ. Особое внимание следу-
ет уделять полноте окисления ПАН-В. При неполном окислении наруж-
ные слои достаточно хорошо ориентированы, далее ориентация осла-
бевает, а при очень быстром нагревании недоокисленного ПАН-В в
центре УВ образуется полость. Согласно [11], УВ, полученное из ор-
лона, имеет более совершенную радиальную ориентацию по сравнению
с УВ, полученным из волокна куртель. В первом случае в процессе
окисления происходит более равномерное образование лестничного по-
лимера по всему объему волокна. С увеличением ТТО и уменьшением
скорости нагревания совершенствуется структура и уменьшается гра-
диент радиальной ориентации волокна. Обработка под натяжением так-
же приводит к пониженному градиенту радиальной ориентации УВ-ГЦ.
На рис. 1.9 приведена трехмерная модель высокомодульного
УВ-ПАН, на которой видна слоистая структура, напоминающая го-
дичные кольца роста древесины, переплетенные базисные ленты, рас-
положенные под разными углами к оси волокна, и пустоты (поры).
Радиальная ориентация заметно влияет на свойства УВ. При быст-
ром охлаждении (закалке) волокна появляются остаточные напряже-
ния, приводящие к образованию дефектов и понижению прочности во-
локна. Влияние радиальной ориентации становится особенно заметно
при получении высокомодульного УВ, т. е. при высоких ТТО. Экстре-
мальная зависимость o=f (ТТО) связана с появлением остаточных на-
пряжений '[12].
Следует отметить, что совершенствование структуры (осевой и ра-
диальной ориентации) влечет за собой снижение активности поверхно-
сти (химического потенциала), уменьшение смачиваемости волокна
связующими и снижение сдвиговой прочности (тсд) композита [12].
Одним из методов исследования УВМ является Раман-спектроскоп<ия
229.
if26], основанная на высокой отражательной способности волокном ла-
зерных лучей.
Для идеально кристаллического графита фиксируется полоса
1575 см-1, а для поликристаллического — 1355 см-1. Отношение интен-
сивностей полос Т?=/1355//1575 может служить критерием степени крис-
талличности.
Раман-спектр УВ, так же как и другие характеристики структуры,
зависит от исходного сырья и условий термообработки. Интенсивность
полос 1575 и 1355 см-1 для волокна марганайт I и марганайт II сущест-
венно отличается. У волокна марганайт I (высокомодульное) по срав-
нению с волокном марганайт II (высокопрочное) интенсивность поло-
Рис. 1.8. Схематичное изображение радиальной ориентации УВ-форта-
фил 5-Y.
Рис. 1.9. Схематичная структурная модель УВ-фортафил 5-Y.
сы 1575 см-1 зачительно выше, а полосы 1355 см-1 намного меньше.
Одно из важных преимуществ этого метода состоит в том, что пред-
ставляется [возможным изучать поверхность волокна, так как Раман-
спектры отражаются от слоя толщиной 250—500 А. Установлена связь
между R и La. Значение La, полученное методом Раман-спектроскопии,
превосходит La, рассчитанное из данных рентгеновских измерений. Объ-
ясняется это тем, что рентгеновским методом определяется среднеста-
тистическое значение La, а по Раман-спектрам La поверхностных слоев.
Дополнительным подтверждением гетерогенности структуры служат
следующие данные: La поверхностного слоя волокна торнель 50 состав-
ляет 215 А, а после травления, сопровождающегося потерей массы всего
лишь 5,5%, La уменьшается до ПО А. Таким образом, прямым методом
подтверждается радиальная неоднородность структуры УВ. Установле-
на взаимосвязь между La, определенной по Раман-спектрам, и тСд компо-
зита, а именно чем больше La, т. е. чем совершеннее структура поверх-
ностного слоя, тем меньше тсд композита. Активация поверхности эф-
фективна только в том случае, когда повышается химический потенциал,
или, другими словами, возрастает дефектность поверхностного слоя УВ.
Среди жаростойких углеродные волокна обладают одной, только им
присущей особенностью, состоящей в том, что они построены из фиб-
230
рилл, наличие которых доказано экспериментально с помощью элект-
ронного микроскопа. Фибриллы в измененном виде наследуются от ис-
ходного волокна, оставаясь ориентированными вдоль оси волокна.
Наличие фибрилл в УВ впервые было обнаружено Бэкапом и Тан-
гом '[27], а также подтверждено в более поздних работах многих ис-
следователей. Четко выраженная фибриллярность УВ видна на
рис. 1.10. Строение фибрилл мало изучено. В этой связи следует отме-
тить, что структура фибрилл химических и природных волокон изуча-
ется более 50 лет и за это время было предложено большое число
схем, каждую из которых трудно доказать и трудно опровергнуть. УВ
просты по химическому составу, Но из-за большого многообразия со-
четания углерод-углеродных связей н кристаллических структур созда-
ются сложные, трудно поддающиеся
изучению надмолекулярные образова-
ния высшего порядка — фибриллы. По
Руланду [28—31], фибриллы состоят
из переплетенных лент (рис. 1.11). На
темнопольных электронно-микроскопи-
ческих снимках видны контуры боль-
шого числа фибрилл. В направлении
оси волокна чередуются темные и яр-
кие области, вызванные муаровым
эффектом, свидетельствующим о не-
однородности структуры микрофиб-
рилл. Последние состоят из прямых
участков, в которых плоскости хорошо
ориентированы в направлении оси
волокна, и участков, в которых мицро- Рис- 110. Фибриллярная структура
фибриллы сморщены вследствие нару- углеродного волокна,
шения ориентации плоскостей вдоль
оси волокна. Муаровый эффект является следствием наложения двух
микрофибрилл со слоями, расположенными под малыми углами друг
к другу. Фибриллы разделены порами, ориентированными вдоль оси
волокна. УВ диаметром 7—10 мкм состоит из десятка тысяч фибрилл.
В результате вытягивания и повышения ТТО происходит выпрям-
ление изогнутых участков базисных лент, улучшение их ориентации,
рост размеров кристаллов и, как следствие этого, возрастание модуля
Юнга волокна [23].
По Джонсону с сотр. [32—36], УВ представляет собой гетероген-
ную систему, состоящую из прямых базисных лент и пор; Агрегаты па-
кетов разделены гранями и порами диаметром 10 А. Цепи агрегатов,
расположенные вдоль оси волокна, группируются в фибриллы длиной
более 1 мкм. Грани раздела между агрегатами кристаллов могут быть
расположены под разными углами к оси волокна. Модель структуры
УВ, предложенная Джонсоном и сотр; и дополненная Купером и Майе-
ром [37], приведена на рис. 1.12. Наличие муарового эффекта, по
Джонсону, обусловлено отражением от разориентированных граней.
Толщина пакетов, участвующих в построении фибрилл, согласно
различным источникам составляет 20—ПО А, что соответствует 7—
30 слоям.
Из двух приведенных моделей следует отдать предпочтение модели
Руланда, которая в первом приближении описывает надмолекулярную
организацию УВМ. Однако этим моделям присущ ряд недостатков.
231
1. Согласно предложенным моделям, УВМ состоят только из
эр2-гибридной формы атомов углерода, поэтому неясным остается ха-
рактер связей между пакетами в фибриллах. Учитывая только ван-дер-
ваальсовые силы взаимодействия между плоскостями, трудно объяс-
нить, исходя из этих схем, высокие механические свойства УВМ.
2. Модели описывают наиболее совершенную структуру УВМ (гра-
фитированные волокна). Из них выпадают карбонизованные волокна,
полученные при относительно низких температурах (900—1500°C), ко-
торые имеют достаточно высокие механические свойства и построены
из фибрилл.
Рис. 1.11. Модель фибриллярной структуры УВ по Руланду.
Рис. 1.12. Модель структуры УВ по Джонсону.
Иное представление о структуре переходных форм углерода выдви-
нуто В. И. Касаточкиным [38, 39]. По его мнению, в подобных мате-
риалах содержатся атомы углерода трех гибридных состояний (sp-,
sp2-, sp3-). Основу организованной структуры УВ составляют турбо-
стратные пакеты, соединенные между собой линейными боковыми це-
почками (sp-, sp2-, зр3-гибридизации), которые представляют собой
аморфный углерод. В неграфитирующихся формах углерода пакеты
сшиты термически устойчивыми полииновыми (—С^С—)п или куму-
леновыми ( = С = С = С=С = )П цепочками, а в гомогенно-графитиру-
ющихся — менее термостабильными цепочками полиенового типа
(—СН = СН—)п- Видимо, эту модель первичных элементов структуры
следует признать наиболее рациональной, но автором не рассматрива-
ются структуры высшего порядка (фибриллы).
По нашему мнению, наиболее приемлемой является следующая
надмолекулярная организация УВ. Основой структуры являются базис-
ные ленты, ориентированные вдоль оси волокна. Турбостратные крис-
таллы (пакеты), построенные из базисных плоскостей (лент), представ-
ляют наиболее совершенную структурную организацию УВ. Размеры
лент значительно превышают размеры пакетов. Поэтому по аналогии с
химическими волокнами ленты являются проходными, участвуя в пост-
роении большого числа пакетов, и образуют пространственный полимер.
Ленты состоят из прямых участков, входящих в пакеты, и искривлен-
ных участков, заполняющих межпачечное пространство. Концы лент
могут находиться внутри пакетов и в межпачечном пространстве, со-
здавая дефектность структуры.
232
Ленты и пакеты имеют большое число дефектов, через которые
обеспечивается связь между пакетами с помощью линейных цепочек
углерода различных гибридных состояний. Между лентами, находящи-
мися вне пакетов, а также между пакетами имеются поры. Размеры
фибрилл в поперечном направлении (их ширина) определяются наи-
меньшими энергиями суммарных связей между первичными элементами
структуры за счет пор и дефектов.
В зависимости от ВТО меняются размеры пакетов, число и раз-
меры переходных лент и лент, входящих в пакеты, соотношение между
аморфным (линейными формами) и организованным углеродом, число
и характер дефектов. Формирование структуры по мере повышения
ТТО определяет физико-механические свойства УВМ.
Одним из характерных элементов структуры являются закрытые
поры, которые могут занимать до 30% объема волокна. Поры имеют
иглоподобную форму; они расположены между базисными плоскостями
и ориентированы вдоль оси волокна. Средний размер1 пор — длина
200—300 А, диаметр 10—20 А. Гетерогенность УВМ обусловливается
диаметром пор (<^пор), расстоянием между ними (/пор), площадью по-
верхности пор (Sr) и общей пористостью, определяемой по МУР. Зна-
чение dnop определяется ВТО и мало зависит от природы исходного
сырья. С ростом ТТО происходит уменьшение числа пор и укрупнение
их размеров, влекущее за ‘собой увеличение /пор и уменьшение Sv. Пе-
рераспределение числа и размеров пор происходит симбатно с увели-
чением размеров пакетов. Пористость описывается также параметром
Порада (Тр), представляющим собой среднюю длину сечения пор в
направлении, перпендикулярном оси волокна. Между параметром Lp
и механическими свойствами волокна [32] существует зависимость, по-
казанная на рис. 1.13. Имеются и другие представления о роли пори-
стости. Так, по Бекану и Шалимову [40], поры в ориентированных во-
локнах не влияют на их Механические свойства (подробнее см. гл. 4).
Еще менее изучена ‘структура и ее связь с механическими свойст-
вами изотропных УВМ. До недавнего времени исследовались изотроп-
ные волокна, полученные из пеков и ФФС без ориентационного вытяги-
вания. Эти волокна имеют те же первичные элементы структуры, но
размеры кристаллов гораздо меньше (Lc—10 A, La=V7 А) и базисные
плоскости слабо ориентированы вдоль оси волокна. По данным [25],
с ростом ТТО происходит совершенствование структуры УВ-ФФС, но
даже при ТТО=2700°С они остаются изотропными. Прямые доказа-
тельства наличия в них фибрилл отсутствуют.
При получении изотропных волокон с ориентационным вытягива-
нием происходит сближение их структуры со структурой анизотропных
волокон. Графитация под натяжением способствует ориентации базис-
ных плоскостей вдоль оси волокна и росту пакетов до размеров (La=
= 175 A; Lc=136 А), ‘соизмеримых с размерами пакетов анизотропных
волокон [41]. При просмотре под электронным микроскопом обнаруже-
ны фибриллоподобные элементы, расположенные вдоль оси волокна.
Авторы работы [42] предлагают для стеклоуглерода (изотропный УМ)
модель перепутанных (рис. 1.14) лент, соединенных межмолекулярны-
ми связями с широким набором энергий. Прямые участки имеют разме-
ры около 100 А, толщина пакетов 40 А, что согласуется со значени-
ем Lc.
И все же остается невыясненной специфика структуры, обусловли-
вающая столь разнообразные свойства УВМ.
233
Как указывалось выше, УВМ относятся к неграфитирующимся
формам углерода. В этом состоит особенность УВМ, н это является
достоинством их, так как в случае гомогенной графитации они, приоб-
ретая графнтоподобную структуру, потеряли бы свои ценные свойства.
Причины неграфитируемости УВМ и других УМ, даже при нагревании
до 3000 °C, не ясны. Ряд исследователей это явление связывает с тем,
что в неграфитирующемся углероде содержатся более мелкие турбо-
стратные кристаллы, базисные плоскости искривлены в большей степе-
ни, пакеты соединены более термостойкими линейными формами угле-
рода.
Рис. 1.13. Зависимость параметра £р \(1) и предела прочности при растя-
жении о (2) от температуры графитации.
'Рис. 1.14. Структурная модель перепутанных лент стеклоуглерода.
При высоких ТТО (2500—2600 °C и выше) происходит частичная
гетерогенная графитации (испарение—^конденсация) с отложением
вторичных продуктов разной формы на поверхности УВМ [43].
Для гомогенно-графитирующихся материалов при ТТО, лежащей
в пределах 1500—1900 °C, а для неграфитирующихся в пределах 1500 и
2200°C наблюдается своеобразное изменение их свойств, соответству-
ющее предкристаллизационному состоянию. Характерным для этой ста-
дии является возрастание энтальпии (уменьшение теплоты сгорания)
н увеличение энтропии [3, с. 138—178].
Возрастание энтропии свидетельствует о преобладании процессов
разупорядочения структуры над ее упорядочением, а возрастание эн-
тальпии — о преимущественном протекании процессов, приводящих к
увеличению внутренней энергии. На этой стадии превалируют реакции
рекомбинации и синтеза над реакциями деструкции [44]. На ста-
дии предкристаллизации происходит переход материала в псевдопла-
стичное состояние и создаются благоприятные условия для совершенст-
вования структуры при более высоких ТТО. Действительно, выше ука-
занных пределов температур для гомогенно-графитирующихся УМ на-
чинается графитация, а для неграфитирующихся материалов — более
интенсивное изменение параметров структуры (возрастание La, Lc,
уменьшение dona)-
На структуру и свойства УВМ влияет ряд химических элементов,
вводимых в исходное волокно или на промежуточных стадиях обработ-
ки. К таким элементам относятся бор, хром, ванадий и др. [45, 46].
Бор участвует в структурообразовании и способствует улучшению ме-
234
панических свойств [13], в частности повышается модуль Юнга волок-
на. Например, введение 1% (масс.) бора вызывает увеличение модуля
Юнга с 424 до 537 ГПа.
Неожиданным оказался богатый
спектр переходных состояний в УВМ.
Информация о переходных состояних
получена в результате изучения внутрен-
него трения методом крутильных коле-
баний [47]. В работе [48] исследовалось
внутреннее трение УВ-ПАН, полученных
при разных ВТО. Установлено большое
число максимумов и минимумов на кри-
вой зависимости внутреннего трения от
температуры испытания. Предваритель-
ная обработка волокна при 1000 °C обед-
няет набор спектров и снижает общую их
интенсивность (рис. 1.15). Интерпретация
этих данных довольно трудная. Обилие
30 к
20 -
701 1___L—1___1—J I____1 L
О 100 300 500 700
200 400 600 800
t, °с
экстремальных точек свидетельствует о
наличии большого числа переходов, про-
являющихся для столь жесткой системы,
как УВМ, при относительно невысоких
температурах испытания. Это лишний раз
подтверждает предположение о многооб-
Рис. 1.15. Температурная зависи-
мость внутреннего трения (Q-1)
УВ-ПАН, обработанного при
1500 °C, до '(/) и после (2) про-
грева при 1000 °C.
разии элементов структуры различной
подвижности. Авторы работы [48] изменение внутреннего трения свя-
зывают с релаксацией напряжения в менее упорядоченных областях
волокна.
ЛИТЕРАТУРА
1. Коршак В. В. и др., «Вестник АН СССР», 1968, № 9, с. 89; авт. свид. на откры-
тие 107 (1971); Открытия. Изобр. Пром, образцы. Товарн. знаки, 1972, № 6, с. 3.
•2. Касаточкин В. И., «Химия твердого полимера», 1969, № 4, с. 33.
3. Шулепов С. В. Физика углеграфитовых материалов. М., «Металлургия», 1972.
250 с.
4. Уокер Ф. Химические и физические свойства углерода. М., «Мир», 1969. 366 с.
5. Жумалиева К-, Усенбаев К., Касаточкин В. И., «Химия твердого топлива», 1974,
№ 4, с. 104—110.
6. Мортимер К. Теплоты реакций и прочность связей. М., «Мир», 1964. 287 с.
7. Franklin R. Е„ Acta Cryst., 1950, v. 3, р. 107; 1951, v. 4, р. 253; Proc. Roy. Soc., 1951,
A, v. 209, p. 196.
8. Goodhew P. J., Clarke A. G., Bailey G. E„ Mater. Sci. a. Eng., 1975, v. 17, p. 3—30.
9. Варшавский В. Я., «Химия и технология высокомолекулярных соединений», 1976,
т. 8, с. 67—120.
10. Фиалков А. С. и др., «Химия твердого топлива», 1968, № 6, с. 101.
11. Le Maistre С. М., Diefendorf R. J. New Horizans Mater, a. Process., Azussa, Colif.,
1973, p. 158—164.
12. Le Maistre C. W., Diefendorf R. J., Polymer Prepr., 1973, v. 14, p. 105.
13. Варшавский В. Я. и др. В ки.: Структура, свойства и применение углеродных во-
локнистых материалов. Мытищи, ВНИИВ, 1975, с. 4—12.
14. Okado Т., Sekinguchi А., Тshit Т., 5th Carbon conference, 1961, р. 497.
15. Jahnson J. M., Marjoram J. B„ Rose P. «Nature», 1969, v. 221, № 5178, p. 387.
16. Perret R., Ruland W., Kallond Z., «Polymer», 1973, v. 14, p. 34—40.
17. Chinda A., Bull. Osaka Ind. Inst., 1961, v. 12, p. 110.
18. Касаточкин В. И., Финкельштейн Г. Б., ДАН СССР, 1963, т. 149, № 3, с. 629.
.19. Скрипченко Г. Б-, Касаточкин В. И. В кн.: Структурная химия углерода и углей.
М., «Наука», 1969, с. 67.
235
20. Конкин А. А. Углеродные и другие жаростойкие волокнистые материалы. М.г
«Химия», 1974. 374 с.
21. Касаточкин В. И. и др., ДАН СССР, 1972, т. 205, с. 1090—1092.
22. Волков Ю. В. и др. В кн.: Структура, свойства и применение углеродных волок-
нистых материалов. Мытищи, ВНИИВ, 1975, с. 24—29.
23. Diefendorf К. G., Tokarsky Т„ Polymer Eng. a. Sci., 1975, v. 15, № 3, р. 150—
159.
24. Sharp J. V. e. a. International Conference on Carbon Fibres, their Place in Modern
Technology, London, 1974, paper № 5.
25. Волков Ю. В. и др. В кн.: Структура, свойства и применение углеродных волок-
нистых материалов. Мытищи, ВНИИВ, 1975, с. 43—47.
26. Tuinstra F., Koenig G. L., J. Composite Mater., 1970, v. 4, p. 492.
27. Bacon R., Tang M., «Carbon», 1964, v. 2, p. 221.
28. Ruland W., J. Appl. Phys., 1967, v. 38, p. 3585.
29. Fourdeux A., Perret R., Ruland W. International Conference on Carbon Fibres, their
Composites and Applications, London, 1971, paper № 9.
30. Fourdeux A., Perret R., Ruland W., Compt. rend., 1970, Ser. C, v. 271, p. 1495.
31. Fourdeux A. e. a., Compt. rend., 1969, Ser. C, v. 269, p. 1597.
32. Johnson J., Tyson C., J. Phys., Ser. D, Brif. J. Appl. Phys., 1970, v. 3, p. 526.
33. Johnson J., W., Polymer Prepr., 1968, v. 9, № 2, p. 1316—1323.
34. Johnson J., Marjoram G. R., Rose P. J., «Nature», 1969, v. 221, № 5178, p. 357.
35. Johnson J., International Conference on Carbon Fibres, their Composites and Appli-
cations, London, 1971, paper № 39.
36. Johnson J., «Nature», 1970, v. 226, p. 750—781.
37. Cooper G. A., Mayer N. J., J. Mater. Sci., 1971, v. 6, № 1, p. 60.
38. Касаточкин В. И. В кн.: Структурная химия углерода и углей. М., «Наука»,
с. 7—17.
39. Касаточкин В. И., «Химия твердого топлива», 1969, № 4, с. 33.
40. Bacon R., Schalamon W., Polymer Prepr., 1968, v. 9, p. 2.
41. Hawthorne H. e. a., «Nature», 1970, v. 227, p. 946; Hawthorne H. International Con-
ference on Carbon Fibres, their Composites and Applications, London, 1971, paper
№ 13.
42. Jenkin G. M., Kawarumo K-, «Nature», 1971, v. 231, № 5299, p. 175.
43. Фиалков А. С. и др., ДАН СССР, 1972, т. 205, № 5, с. 1168—1169.
44. Касаточкин В. И. и др. В кн.: Структурная химия углерода и углей. М., «Наука»,
1969, с. 27—34.
45. Ермоленко И. Н., Выговский И. И., Люблинер И. П., Изв. АН БССР. Сер. хим.
наук, 1973, № 6, с. 43; 1974, № 4, с. 78.
46. Шашкова Г. Н. и др., ЖФХ, 1973, т. 47, № 10, с. 2495.
47. Иванов Н. В. и др., «Механика полимеров», 1974, № 3, с. 520—521.
48. Варшавский В. Я. и др. В кн.: Структура, свойства и применение углеродных во-
локнистых материалов. Мытищи, ВНИИВ, 1975, с. 12—19.
ГЛАВА 2
ИСХОДНЫЕ МАТЕРИАЛЫ И НЕКОТОРЫЕ ОСОБЕННОСТИ
ПОЛУЧЕНИЯ УГЛЕРОДНЫХ ВОЛОКОН
В результате термического распада органических соединений, про-
водимого в инертной среде, образуются летучие продукты (газы и смо-
лы) и твердый остаток — углерод, называемый коксовым остатком или
коксовым числом. Для получения углеродных волокнистых материалов
(УВМ) используются исключительно полимеры или олигомеры (по-
следние на промежуточных стадиях обработки превращаются в поли-
меры). Между исходным материалом и УВМ существует генетическая
связь, так как оба они представляют собой полимеры.
Полимеры, предназначенные для получения УВМ, должны удовлет-
ворять следующим требованиям: иметь волокнистую форму, не пла-
236
виться при термической обработке, обеспечивать высокий выход угле-
рода и высокие механические и физико-химические свойства углерод-
ного волокна.
Предшественниками УВМ являются органические волокна, ибо, ис-
ходя только из полимеров волокнистой формы, можно получить угле-
родные волокна. УВМ имеют разнообразную текстильную форму, кото-
рая определяется геометрией исходного материала. При использовании
нитей получается углеродная нить, из жгута вырабатывается углерод-
ный жгут, из тканых материалов — углеродные ткани различной струк-
туры, при резке углеродного жгута получается штапельное волокно
(войлок) или кноп. Разработаны способы изготовления тканей (полот-
но, трикотаж) из углеродных нитей, а также гибридных тканей на ос-
нове углеродных и органических или стеклянных волокон.
Одним из необходимых условий получения УВМ является примене-
ние неплавких полимеров, так как в противном случае вместо волокна
в результате термической обработки получится коксовый остаток в ви-
де 'спекшейся массы. Впрочем, не исключена возможность использова-
ния плавящихся органических волокон, которые, однако, путем предва-
рительной обработки переводятся в неплавкое состояние.
Стоимость УВМ в значительной мере зависит от стоимости исход-
ного волокна. Это особенно важно в связи с резким повышением цен на
химическое сырье, поэтому увеличение выхода углерода оказывает
большое влияние на технико-экономические показатели производства
УВМ.
К основным видам сырья, пригодного для получения УВМ, относят-
ся химические волокна, а также пеки, фенолоформальдегидные и дру-
гие смолы, обладающие волокнообразующими свойствами.
Проблема термостойкости и термодеструкции полимеров давно
привлекала внимание исследователей. Это объясняется прежде всего
тем, что во время эксплуатации изделия из полимеров подвергаются
термо- и термоокислительной деструкции, следствием чего является
снижение их физико-механических свойств и сокращение срока службы.
С развитием техники появилась потребность в термостойких полимерах
и волокнах, предназначенных для эксплуатации при температурах
300—500 °C. Для решения этой задачи необходимо было более углуб-
ленное изучение взаимосвязи между строением и свойствами полимеров
[1]. Наряду с термостойкими создано большое число жаростойких во-
локон, среди которых одно из первых мест занимают углеродные во-
локна.
При разработке процессов получения УВМ по-новому начали изу-
чать проблему термической деструкции полимеров. Если раньше задача
сводилась к изысканию путей повышения термостойкости или синтеза
термостойких полимеров, то в данном случае оказалось необходимым
изыскать способы целенаправленной термической деструкции полиме-
ров, обеспечивающие высокий выход углерода и получение УВМ с за-
данными свойствами.
Изучение термической деструкции полимеров имеет помимо при-
кладного немаловажное теоретическое значение.
В настоящее время не представляется возможным сформулировать
научно обоснованные требования к полимерам, предназначенным для
получения УВМ. Тем не менее огромный опыт, накопленный при изуче-
нии термического распада полимеров, установленные закономерности,
а также исследование процессов получения УВМ позволяют обсудить
237
некоторые общие требования, которым должны удовлетворять исходные
материалы (предматериалы).
Получение УВМ основано на термической деструкции органических
волоко», проводимой в строго заданных и контролируемых условиях.
Процесс связан с тремя состояниями и двумя переходами: исходный
органический полимер (волокно) в результате термической обработки
превращается в ароматический полимер-; ароматический — во время
карбонизации — в углеродный полимер. Этот сложный переход сопро-
вождается изменением структуры при сохранении элементов первона-
чального полимерного скелета.
В результате термической деструкции отщепляются иные кроме
углерода атомы или группы атомов с частичной потерей углерода, и в
конечном итоге образуется углеродное волокно. Эти сложные процессы
сопровождаются одновременным протеканием большого числа после-
довательных и параллельных гетеролитических и гомолитических реак-
ций, в результате которых образуются разнообразные продукты распа-
да. Выразить эти реакции в виде конкретных химических уравнений не
представляется возможным. Однако можно выделить две наиболее ти-
пичные группы реакций. К первой группе относятся деполимеризация,
протекающая по гетеролитическому или радикальному механизму, об-
разование межмолекулярных химических связей и переход линейного
полимера в пространственный, внутри- и межмолекулярная циклиза-
ция, более глубокая деструкция полимеров с образованием газообраз-
ных и жидких летучих продуктов и фрагментов деструкции, видимо,,
плоскостной формы, являющихся предшественниками формирования
углерода. Реакции этой группы протекают при температурах до 300—
350 °C, причем на ранних стадиях преобладают гетеролитические реак-
ции; с повышением температуры все большее значение приобретают го-
молитические реакции. В этом интервале температур наблюдается наи-
большая потеря массы полимера.
В пределах 300—1000 °C существенную роль играют реакции вто-
рой группы: рекомбинация и поликонденсация за счет ранее образовав-
шихся функциональных групп, возникновение предструктур и аромати-
ческих планарных фрагментов, участвующих в построении переходных
волокнистых форм углерода, и постепенное обогащение коксового ос-
татка углеродом. При этом роль химических реакций постепенно снижа-
ется, начинают преобладать физические процессы формирования струк-
туры углерода. Эта стадия превращения органических волокон в угле-
родные наименее изучена.
Ниже рассматриваются основные, наиболее типичные закономерно-
сти термической деструкции материалов, являющихся источниками по-
лучения УВМ.
2.1. ЛИНЕЙНЫЕ ПОЛИМЕРЫ
Химические волокна являются одним из основных материалов, ис-
пользуемых для производства УВМ. Волокна построены из линейных
полимеров и относятся к высокоориентированным системам. Это накла-
дывает некоторые особенности на процесс их превращения в углеродное
волокно.
Как уже указывалось, к сырью, из которого получают УВМ, предъ-
является основное требование —• высокое содержание углерода в исход-
ном материале. Однако не всегда существует прямая связь между со-
238
держанием углерода в исходном волокне и выходом углеродного волок-
на. Последний определяется многими факторами, среди которых нема-
ловажную роль играет энергия диссоциации химических связей. В ли-
тературе приводятся противоречивые сведения о численном значении
этих величин. Ниже представлены данные, заимствованные из моногра-
фии Т. И. Темниковой [2]:
Тип связи Энергия связи, кДж/моль Тип связи Энергия связи, кДж/moj
с—н 358,5—413,76 с=с 536,9—808,2
с—с 263—347,2 с=о 603,4—760,9
С—О 314,2—358,2 С—С1 293,3—331,8
О—н 460,9—464,2 С—N 224,17—289,3
N—Н 347,7—391,3 C=N 351,9
с=с 423,8—612,6 C=N 624,3—856,01
Из сопоставления значений энергии связей С—С с энергией связи
атомов углерода с другими атомами видно, что термическим путем не-
возможно, не нарушая связь С—С, осуществить отщепление от поли-
мера других атомов без потери углерода.
Насыщенные полиуглеводороды (полиэтилен, полипропилен и др.)
характеризуются наиболее высоким содержанием углерода. Тем не ме-
нее, как показал Мадорский [3], линейный полиэтилен (содержание уг-
лерода 85,7%) при нагревании в инертной среде (гелий) до 500—800 °C
полностью распадается до низкомолекулярных ненасыщенных и насы-
щенных углеводородов.
В полиэтилене имеются два типа связей —С—С— и —С—Н, при-
чем энергия связей —С—Н больше энергии связей —С—С—, поэтому
под влиянием теплового воздействия прежде всего разрываются связи
С—С основной цепи, что приводит к низкому выходу коксового остат-
ка. Аналогичная картина наблюдается для полипропилена.
Необычайно большие значения энергии связей С = С и С=С обу-
словливают высокую теплостойкость переходных форм углерода.
Кроме энергии связи между атомами на термостойкость полимеров
влияют другие факторы. К ним относятся межмолекулярные физические
связи (дипольный эффект 36,45 кДж/моль); дисперсионные связи 8,38—
25,14 кДж/моль; водородные связи 25,14—41,9 кДж/моль, которые за-
висят от химического состава полимера, характера и ориентации надмо-
лекулярных образований и, как правило, повышают теплостойкость по-
лимера.
Существенное влияние на термические характеристики оказывает
резонансная стабилизация (167,6—293 кДж/моль), присущая некото-
рым циклическим структурам и полимерам с системой сопряженных
связей [4]. Большая концентрация л-сопряжений обусловливает высо-
кую теплостойкость УВМ.
По-иному реагируют на тепловое воздействие ненасыщенные поли-
углеводороды. При определенных условиях термообработки из ненасы-
щенных алифатических соединений с достаточным выходом получается
коксовый остаток, что обусловлено наличием связей С=С и их склон-
ность к образованию сетчатых структур. Вследствие циклизации и по-
239
следующего дегидрирования из 1,2-полибутадиена образуется лестнич-
ный полимер по следующей схеме:
СН2 СН2 СН2 СН2
„„ „ Cr(CO)6/AiR3 CH CH CH CH BF3/AICI3
пСН2=СН—СН=СН2------------->- 1111 — ----►
сн сн сн сн
II II II II
сн2 сн2 сн2 сн2
сн2 сн2 сн2 сн2
Используя этот принцип, авторы работ [5, 6] получили УВМ типа
«плутон». Аналогичным способом происходит циклизация 1,2-, 3,4- или
1,4-полиизопрена '.[7, 8].
Своеобразно ведут себя некоторые карбоцепные полимеры. Реак-
ция цепной полимеризации является равновесной, поэтому при опре-
деленных условиях возможна деполимеризация полимеров. Так как
энергия активации прямой реакции .меньше энергии активации обрат-
ной реакции, то последняя протекает при высоких температурах. Тем-
пература, при которой скорости прямой и обратной реакций становят-
ся одинаковыми, называется предельной температурой Гпр. Эта темпе-
ратура определяется по изменению термодинамических функций. По-
лимеризация возможна, когда
ДГ<ДЯ —ТД$ (1)
Состояние равновесия процессов полимеризацияч^деполимеризация оп-
ределяется уравнением
ДГ = ДЯ—ТД5 = 0 (2)
Из уравнения (2) находят предельную температуру
Тпр = ДЯ/Д5 (3)
где ДЯ — разность энергии активации обратной и црямой реакций.
При температуре выше Тщ, начинает превалировать реакция деполиме-
ризации.
Для большинства карбоцепных полимеров значение Тщ, составляет
200—350 °C. Деполимеризация возможна, когда теплостойкость полиме-
ра выше Тпр. Под влиянием теплового воздействия деполимеризация
становится основной реакцией термического распада карбоцепных по-
лимеров, содержащих четвертичный атом углерода в основной цепи
'[9]. Например, при термодеструкции поли-а-метилстирола, полистиро-
ла, полиметилметакрилата выход мономеря составляет 65—90%.
Некоторые гетероцепные полимеры также деполимеризуются под
влиянием теплового воздействия; например., при деполимеризации поли-
формальдегида выход коксового остатка близок к нулю. При нагрева-
нии в вакууме хлопковая целлюлоза деполимеризуется с высоким вы-
240
ходом (до 50%) левоглюкозана, который является видоизмененным
элементарным звеном целлюлозы.
Приведенными примерами не исчерпывается число полимеров, рас-
падающихся до мономеров по цепному механизму.
Деполимеризация гетероцепных полимеров происходит при отно-
сительно невысоких темпер ату рах под влиянием реагентов, избира-
тельно воздействующих на гетеросвязь. Следы воды при повышенной
температуре вызывают гидролиз целлюлозы, полиамидов, полиэфиров
(сложных) и других соединений. При небольшом содержании кисло-
ты, которая может являться первичным продуктом распада, резко уско-
ряется гидролиз полимеров. Именно по этой причине при назревании
до 120—150 °C резко падает степень полимеризации целлюлозы без по-
тери массы.
Наличие слабых связей в полимере также вызывает распад макро-
молекул.
Реакции деполимеризации при получении УВМ играют отрица-
тельную роль по трем причинам: во-первых, снижается выход углерода;
во-вторых, с уменьшением СП ниже определенного предела уменьшает-
ся прочность исходного и зависящая от нее прочность углеродного во-
локна; в-третьих, снижение СП может привести к нарушению структур-
ной организации волокна, которая влияет на формирование промежу-
точных продуктов распада полимера, определяющих структуру УВМ.
Большое влияние оказывают реакционноспособные функциональ-
ные группы полимера, склонные к внутри- и межмолекулярной цикли-
зации и образованию межмолекулярных химических связей. Как пра-
вило, реакции протекают в обоих направлениях. Циклизация происхо-
дит с образованием наиболее устойчивых шестизвенных циклов. Иногда
в результате циклизации получается лестничный полимер. Образова-
ние межмолекулярных связей влечет за собой превращение линейного
полимера в сетчатый. Типичными полимерами, претерпевающими по-
добные превращения, могут служить ПВХ, ПВС, ПАН, ГЦ и др.
Не часто, но все же бывают случаи, когда из-за отщепления ато-
мов или групп атомов в полимере возникают диеновые структуры. По-
добные реакции имеют место при отщеплении НС1 от ПВХ, НаО от
ПВС. При последующем взаимодействии диеновых структур1 по реакции
Дильса—Альдера [10] образуется сетчатый полимер, сшитый с по-
мощью циклов:
II I II , II I
НС НС СН I СН I
I 11 \ /
СС1 СС1 конденсация С1С СС1 отщепление НС! С Cz
II + I ------------------> I II------------------------- I II +ЗНС1
СН СН НС СН С\ Сч
I II / '"Vci
cic cic cci । cci y
II I II 1 II 1
Термическая деструкция полимеров в большинстве случаев проис-
ходит по цепному механизму. Рекомбинация макрорадикалов может
приводить к образованию сшитого полимера:
СН—снх-
•СН—СНХ'
•СН—снх^
•СН—снх^
16—2254
241
Способность линейных полимеров к образованию межмолекуляр-
ных связей и лестничных структур на ранних стадиях термообработки
играет важную роль в процессах получения УВМ, так как повышается
теплостойкость полимера и соответственно выход углерода; образуются
планарные промежуточные структуры, способствующие переходу угле-
рода в графитоподобные ленты; повышается температура стеклования,
что благоприятно сказывается на сохранении надмолекулярных образо-
ваний (фибрилл) исходного волокна и волокнистой формы материала.
Повышение ТСт при увеличении ТТО отмечается также в работе [11].
Достигнутый эффект зависит от природы полимера, характера и
густоты сетки и типа внутримолекулярных структур. Даже полиэтилен,
при термическом распаде которого крайне низок выход углерода, после
предварительной сшивки проявляет склонность к образованию коксово-
го остатка {12]. В работах ,[13, 14] было показано, что структурирова-
ние полистирола способствует образованию кокса. Из сополимера сти-
рола с дивинилбензолом или дивинилбензола с этилвинилбензолом вы-
ход кокса составляет всего 6%, а из тривинилбензола [3], имеющего
густую сетку связей, выход кокса повышается до 60% (при ТТО 800 °C
кокс на 99% состоит из углерода). Высокий выход кокса из ПВХ (до
92% от теоретического) связан с межмолекулярной циклизацией.
Кислород в органических соединениях влияет на выход углерода
и его структуру. Кислород облегчает дегидрирование, и его содержа-
ние до определенного предела повышает выход коксового остатка.
Большое содержание кислорода нежелательно, так как, являясь окис-
лителем, он за счет отщепления СО и СО2 уменьшает выход углерода.
Для многих полимеров и олигомеров, особенно способных перехо-
дить в вязкотекучее состояние, окисление является необходимым пред-
варительным условием получения УВМ.
Наряду с упомянутым облегчением дегидрирования кислород спо-
собствует образованию межмолекулярных связей и тем самым придает
неплавкость полимеру, ускоряет химические процессы, приводящие к
получению промежуточных структур, участвующих в образовании угле-
рода. Подробнее роль кислорода рассматривается в гл. 3; здесь же
приводится только несколько примеров.
Окисление полиэтилена вследствие образования межмолекулярных
связей повышает выход углерода. Согласно [13], предварительное
окисление сополимера дивинилбензола и этилвйнил бензол а повышает
выход коксового остатка с 6 до 50% (ТТО 600—700°C). Предваритель-
ное окисление ПВХ, ПВС, ПАН (см. гл. 3) благоприятно сказывается
на выходе углерода.
Присутствие кислорода в составе полимеров играет немаловажную
роль в формировании структуры графита. Соединения с большим со-
держанием кислорода дают жесткие, пористые коксы, размеры крис-
таллических образований которых не изменяются в широком диапазоне
ТТО. Из веществ с содержанием кислорода ниже критического выход
кокса уменьшается, но в этом случае с повышением ТТО наблюдается
рост и упорядочение кристаллитов [15, 16].
Выше рассмотрена роль наиболее типичных реакций на примере
относительно простых полимеров. В табл. 2.1 приведены данные о вы-
ходе коксового остатка из линейных полимеров различных состава и
строения [17]. Для этих систем выход коксового остатка также зави-
сит от факторов, предвидеть которые заранее невозможно. Лишь по
выходу углерода можно судить о направлении течения реакций. Высо-
242
кий выход углерода (выше 50%) отмечается из полифурилового спирта
и модифицированных фенольных смол. Кислород и сера между бен-
зольными циклами незначительно снижают (№ 1, 2), а метиленовые
группы (№ 4) резко уменьшают выход углерода. Видимо, в последнем
случае прежде всего претерпевают разрыв алифатические связи С—С
с образованием летучих продуктов. Сложноэфирные смолы дают низ-
кий выход углерода (№ 13). Высокий выход коксового остатка из азот-
содержащих гетероциклов и силиконовых полимеров объясняется, ве-
роятно, тем, что в остатках кроме углерода содержатся азот и крем-
ний соответственно.
На основании анализа термической деструкции линейных полиме-
ров можно сделать следующий вывод. Во многих случаях вследствие
подвижности исходных макромолекул и промежуточных продуктов при
термическом распаде образуются достаточно стабильные фрагменты,
способные к построению планарных ароматических графито подобных
плоскостей; вместе с тем они достаточно термостойки, чтобы обеспечить
высокий выход углерода. Природа протекающих реакций и конечный
результат зависят от химического состава и структуры полимеров. Ис-
пользование полимеров для изготовления УВМ определяется также
возможностью получения исходного органического волокна на их ос-
нове.
2.2. ПРОСТРАНСТВЕННЫЕ ПОЛИМЕРЫ
По сравнению с линейными пространственные полимеры обладают
двумя наиболее типичными особенностями: беспорядочным пространст-
венным расположением атомов и высокой термостойкостью как исход-
ных полимеров, так и промежуточных продуктов их распада. Термо-
стойкость полимеров обеспечивает высокий выход углерода. Жесткость
системы и беспорядочное расположение атомов наследуются в полу-
ченном из них коксе. Кокс имеет менее организованную структуру; раз-
меры кристаллитов меньше, а межплоскостное расстояние больше; ха-
рактерным для него является изотропность свойств, поэтому он отно-
сится к особой разновидности углерода — стеклоуглероду [18].
Различная термическая стойкость линейных и пространственных
полимеров обнаруживается при исследовании потери массы в процессе
нагревания. Для линейных полимеров характерна потеря массы в уз-
ком интервале температур; для пространственных полимеров — в более
широком температурном интервале, что свидетельствует о повышенной
термостойкости исходного полимера и промежуточных продуктов его
распада. Характерно, что при разрыве одной связи линейного полиме-
ра среднечисловая молекулярная масса уменьшается вдвое; для прост-
ранственного полимера разрыв не только одной, но и большого числа
связей мало сказывается на молекулярной организации полимера.
В последнем случае создаются благоприятные условия для рекомбина-
ции радикалов с восстановлением структурного каркаса полимера.
Изучению термического распада пространственных полимеров по-
священо большое число работ, однако установить научно обоснован-
ные критерии пригодности полимеров для получения УВМ труднее, чем
для линейных полимеров.
Обстоятельные исследования связи строения и выхода коксового
остатка из фенольных и эпоксидных смол были проведены Маккеем
[17]. Полученные им данные суммированы в табл. 2.2 и 2.3.
16*
243
Таблица 2.1. Выход кокса и эффективность превращении углерода (С/Со) из различных линейных полимеров (ТТО = 840°С)
Полимеры
Выход
кокса,
%
Эффективность
превращения
углерода
(С/Со), %
Полимеры
Выход
кокса,
%
Эффективность
превращения
углерода,
(С/Со), %
полисульфон
48,0
61, 46
82,7
3 —сн2—сн-сн2—сн—сн2—сн— 23,9 96,2
1 1 С1 С1 С1
поливинилхлорид
поли-л-ксилен
11
I
сн3
полиметилфенилсилнкон
88,9
ОН
58,7
72,8
дифенилоксидфенолоформальдегид
полибензимидазол
7
50,6
N“
где R =
поли-л,л'-дицианодифенилметилеи
8
полифуриловый спирт
54,4
90,5
12
Si(CH3)3
65,2
О ОН ОН
I СНа I СНа I сна
хУтгтг
силаисодержащий фенолоформальдегид
13
—сн—сна—сн—сн=сн2
OR
‘2
2,3
3,3
OR
где R =
—С—О—СНа—СН=СН2
0,0
44,0
0,0
59,9
Таблица 2.2. Выход кокса и эффективность превращения
углерода С/Со из различных фенольных смол (ТТО=840 °C)
№ п/п. Основа Выход кокса, % Эффективность превращения углерода, С/Со, %
1 он + нсно фенол формальдегид 60,4 76,4
2 ОН + нос—сон фенол глиоксаль 48,2 60,3
3 ОН + СНО О фенол фурфурол 61,6 80,3
4 ОН + СНО фенол бензальдегид 42,3 48,7
5 но ОН + нсно резорцин формальдегид 56,9 78,1
6 НО. ОН + СНО О резорцин фурфурол 59,6
7 но—он +НСНО л,л-дигидродифенил формальдегид 64,5 80,5
8 но—сн2—он + нсно бис(н-гндроксифенил)метан формальдегид 60,0 74,8
9 О но—V- S—он + нсно \=/ II \=/ О бис(л-гидроксифенил)сульфон формальдегид 56,5 73,0
10 ОН + нсно л-фенилфенол формальдегид 8,8 10,2
11 НО ,== +НСНО \)Н 1,5-нафталннднол формальдегид 62,2 80,0
Таблица 2.3. Выход кокса из эпоксидных смол
| № п/п. 1 Эпоксисистема О (R= - СН2-СнХн2) Отверждающие реагенты
f3b.nh2c2h5 nh2 nh2 \CHv Х-ССНз оАоУо
выход кокса, %
1 2 3 4 5 6 7 сн2—сн—сн2—сн^ 1 1 сн2 сн2 OR OR полиаллилглициловый эфир СН3 ко_/гА—с—or f сн3 2,2' - ди(п*глицидилоксифенил )пропаи СН3 СН3 СН3 R0 R° R0/C/L сн2 сн2 сн2^ эпокси-о-крезолформальдегид OR OR OR I СН2 1 СН2 1 сн2— ХУ ХУ ХУ эпоксифенолоформальдегцд R°x^/ 0R резорцнндиглицидиловый эфир R-N—R О OR эпокси-п-аминофеиол RO—/ OR — сн—сн — Ro—XAZ \/~\'_qr 1,1' ,2,2/-тетра-(л-глицидилоксидифенил)этаи 5,8 24,3 37,6 49,8 16,4 5,4 54,2 2,3 6,8 40,3 44,5 4,9 21,9 29,4
Фенолоформальдегидная смола дает высокий выход коксового ос-
татка; резорцин—несколько меньший; видимо, избыток кислорода от-
щепляется в виде окислов углерода (см. табл. 2.2). Из дигидродифенила
отмечается наиболее высокий выход углерода. Наличие группы СН2
в бис (л-гидроксифенил) метане мало сказывается, a SO2 несколько сни-
жает выход кокса (см. табл. 2.2). Блокирование одной гидроксильной
группы в дигидродифениле резко снижает выход кокса. При замене
формальдегида на глиоксаль вследствие возрастания подвижности си-
стемы выход кокса уменьшается. Причины различного эффекта, вызы-
ваемого фурфуролом и бензальдегидом, неясны.
Выход кокса из эпоксидных смол зависит от вида отвердителя и
эпоксидного компонента. Для алифатических эпоксисоединений харак-
терен низкий выход кокса (табл. 2.3). Из доступных соединений наи-
более высокий выход отмечается для модифицированной новолачной
смолы (см. табл. 2.3, № 4). В качестве отвердителей наиболее эффек-
тивен РзВ-МН2С2Н5, наименее пригодны ангидриды кислот.
В литературе приводятся и другие данные о выходе кокса из про-
странственных полимеров, но, поскольку скорость нагревания влияет на
конечный результат, сравнивать эти данные с данными табл. 2.2 и 2.3
некорректно (неизвестны условия проведения эксперимента). В табл.
2.2 и 2.3 приведены значения, полученные в одинаковых условиях. Они
не являются оптимальными для конкретных соединений, но метод
оценки выхода кокса при одинаковых условиях термообработки вполне
оправдан.
2.3. ПЕКИ
Пеки относятся к перспективным видам сырья, из которого могут
быть получены УВМ. Это объясняется высоким содержанием углерода
(85—96%), а также высоким выходом коксового остатка. Интерес к
пекам значительно возрос после получения их в мезофазном (жидко-
кристаллическом) состоянии.
Пеки представляют собой смесь разнообразных ароматических и
гетероароматических соединений. В связи со сложным составом пеков
термические превращения их изучают на модельных соединениях —
ароматических и гетероароматических многочленных циклических со-
единениях известных состава и строения.
Можно выделить следующие состояния и стадии перехода от орга-
нических продуктов к углероду и графиту: исходное органическое веще-
ство и его пиролитическое превращение в плавкий кокс; пиролиз по-
следнего и его переход в неплавкий пек (кокс); карбонизация кокса с
получением углерода и, наконец, графитация (высокая температура
термообработки), приводящая к получению искусственного графита.
По сравнению с сетчатыми, а тем более линейными полимерами
пеки и их модели обладают рядом специфических свойств. Как прави-
ло, пеки представляют собой смесь ароматических многоядерных си-
стем с большой .концентрацией л-сопряжений. Для таких соединений ха-
рактерна высокая термо- и теплостойкость, способность давать сигна-
лы ЭПР, свидетельствующая о сильной делокализации л-электронов и
наличии стабильных свободных радикалов [4, 19]. На их термические
свойства влияет также резонансная стабилизация '[4], достигающая
167,6—293,3 кДж/моль.
Другая особенность состоит в том, что в пеках отсутствуют реак-
ционноспособные функциональные группы. Наиболее типичными для
248
них являются связи С—С разных гибридных форм углерода и связи
С—Н; возможно также включение в циклы гетероатомов, боковых али-
фатических радикалов и атомов кислорода.
Наконец, в зависимости от структуры модельные соединения и пе-
ки подразделяются на планарные (плоскостные) и иепланарные. Пла-
нарные соединения превращаются в искусственный графит, непланар-
ные дают неграфитирующийся углерод.
Из-за большой (концентрации л-сопряжений значительно ослабля-
ется связь С—Н, которая распадается при тепловом воздействии с обра-
зованием свободных радикалов. К одной из основных реакций относит-
ся полирекомбинация, приводящая к возрастанию размеров молекул,
увеличению отношения С : Н и изменению физико-химических свойств
пека. Источником пополнения свободных радикалов для поддержания
этой реакции служит распад более (слабых связей С—Н. Помимо ради-
кальных возможны чисто термические превращения молекул, связан-
ные, например', с перестройкой циклов.
Согласно работе ([20], газофазный пиролиз антрацена (I) (ТТО=
=700°C) приводит к присоединению по месту 9 с образованием биант-
рила (И), дибензоперилена (III) и бензантреиа (IV). В результате ря-
да последующих превращений образуется полимер (V), переходящий
в графит с </оо2=3,354 А.
При газофазном пиролизе флуорена (VI) образуется А9’9'-бифлуо-
рен (VII). В результате дальнейшего превращения получаются продук-
ты термической перестройки: тетрабензанафталин (VIII) или руби-
цен (IX).
249
Таблица 2.4. Межплоскостное расстояние в углероде, полученном
вз ароматических углеводородов (ТТО=3000 °C)
№ п/п. Соединение Структурная формула Межплос- костное расстояние СН ^002» А
1 Натуральный графит — 3,354
2 9,9'-Биантрил Сф 3,354
3 9,10-Диметил антрацен (XX) СН3 I 3,354
[1 't J. \ СН3
4 Дибенз[6, к] хризен 3,354
5 Аценафтилен 3,356
6 5,6,11,12-Тетрафенилнафтацен и > tTO' 3,356
7 Биаценафтилиден d d 99 3,357
dx5
250
Продолжение
№ п/п. Соединение Структурная формула Межплос- костное расстояние О) ^002» A
8 Декациклен 1 Г ТО /==/ух ЦЧ 3,358
9 Овален 3,358
10 Гексафенилэгаи об О4-Ю 00 3,360
11 Пентацеи 3,363
12 Рубицен 3,363
13 Пирантреи ,qco c6r 3,363
14 Квантарилен Z^V_ZX_ZX-/~^ M )=K M M 3,368
251
Продолжение
Xf п/п. Сое ди некие Структурная формула Межплос- костное расстояние о. 4оо2, а
15 Коронен
3,370
16
14,15-Дигидро-9Н-диин дено [ 1,2-а;
2', l'-с] флуорен
17
18
10,15-Дигидро-5Н-дииндено[1,2-а;
2', 1'-с] флуорен
9,10-Дибеизилантрацен
19 п-Трифенил
20 Д9.9'-Бисфлуорен
3,380
3,385
3,412
3,437
3,474
252
Таблица 2.5. Межплоскостное расстояние в углероде, полученном
из полиядерных ароматических хинонов и кетонов (ТТО = 3000°С)
№ п/п. Соединение Структурная формула Межплоскост- ное расстоя- о ние, ^оо2> А
1 Антантрон О ХХГ) иХХ II о 3,354
2 Перинафтенон ut 3,356
3 1,4-Нафтахинон о оэ II о 3,370
4 Изовиолантрон гссс II О 3,373
5 Пирантрон / /° О 3,393
253
Продолжение
Оба продукта из-за дефектности ароматических структур- дают плохо
графитирующийся углерод (d002=3,375 А). В цитируемой статье приво-
дятся и другие примеры превращения модельных соединений в углерод
разной формы.
В обстоятельной работе Эдстрома и Левиса [21] изучена связь
между строением ароматических углеводородов, ароматических хино-
нов и кетонов и полиядерных гетероциклов и их способностью под
влиянием теплового воздействия превращаться в углерод разной
структуры (турбостратная, графитоподобная). Как видно из табл. 2.4,
ароматические планарные углеводороды (см. табл. 2.4, № 2—11) пре-
вращаются в графитирующийся углерод с doo2=3,354—3,360 А, непла-
254
Таблица 2.6. Межплоскостное расстояние в углероде, полученном
из полиядерных гетероциклов (ТТО=3000° С)
Ns п/п. Соединение Структурная формула Межпло- скостное расстояние ^002. о, А
1 Диаценафто [1,2-6; 1,2—d] тиофен СО ОТ 3,358
2 11,12-Диметилдибенз [а, с] феназин СН3 N XX XX сн3 /П 3,362
3 11 -Метилдибенз [а, с] феназин 3,362
4 2,3-Дифенил-5,6-бензохиноксалин n О 3,362
5 5,6,7,8-Дибензхиноксалин C'jl n дП СУ 3,363
6 9-Метилаценафтохиноксалин N f ] ХХХСО СН3 N 3,400
7 9,9'-Биксантилен О °Г° ООО О 3,420
255
парные ароматические циклы (ем. № 18—20) и соединения, занимаю-
щие промежуточное положение (№ 12—17), превращаются в уг-
лерод с doo2 = 3,363—3,474 А. Отмеченная взаимосвязь между структу-
рой предшественника и образующейся формой углерода не всегда
наблюдается. Так, соединения № 5 и № 8 (табл. 2.4), видимо, претер-
певают перестройку на промежуточных стадиях термообработки. Со-
единения № 14 и № 15 — планарные, но планарность, видимо, наруша-
ется, и в итоге образуется углерод с большим d002- Не ясна причина
возникновения неграфитирующегося углерода из соединения № 19,
имеющего регулярное строение. Видимо, в линейных ароматических уг-
леводородах недостаточно поляризована связь С—Н и равновероятен
разрыв связей С—С и С—Н. Поэтому образуется коксовый остаток не-
совершенной структуры (с большим й!оог)-
Почти из всех ароматических хинонов и кетонов образуется угле-
род с большим doo2 (см. табл. 2.5). Видимо, отщепление кислорода вле-
чет за собой нарушение планарности (исключением являются соедине-
ния № 1 и № 2).
Полиядерные гетероциклы можно подразделить на три группы: от-
носительно хорошо графитирующиеся (табл. 2.6, № 1); плохо графи-
тирующиеся (№ 6 и № 7); соединения, занимающие среднее положе-
ние (№ 2—5).
В цитируемых работах, к сожалению, отсутствуют данные о выхо-
де углерода из различных полициклических ароматических соединений.
По другим опубликованным источникам, некоторые из этих соединений
дают низкий выход углерода [17, 23].
Интерес к Многоциклическим ароматическим углеводородам осо-
бенно возрос в связи с возможностью их нахождения в мезофазном
(жидкокристаллическом) состоянии; из них, согласно литературным
данным [24], получаются УВМ высокого качества.
Мезофазные предшественники позволят решить одну важную науч-
ную проблему. Из (способных к графитации многоядерных систем воз-
можно получить УВМ графитовой структуры. Сопоставление такого во-
локна с обычным волокном, состоящим из переходных форм углерода,
позволит четко выявить взаимосвязь между структурой и свойствами
УВМ. Вероятно, УВМ графитовой структуры будут обладать куда
большей анизотропией и невысокими механическими свойствами. Эти
исследования могли бы наглядно подтвердить, что высокая степень со-
вершенства структуры (кристалличность) не всегда желательна для
обеспечения комплекса высоких механических свойств волокон.
2.4. НЕКОТОРЫЕ ОСОБЕННОСТИ ПРОЦЕССА ПЕРЕХОДА
ПОЛИМЕР УГЛЕРОД
Переход полимер1—►углерод по некоторым схемам термодинамиче-
ски выгоден, так как он сопровождается уменьшением свободной энер-
гии. В табл. 2.7 приведен расчет изменения термодинамических пара-
метров (энтальпии АТ/, энтропии AS, свободной энергии AF) при 25 °C
для гипотетических реакций термической деструкции полиэтилена и по-
литетрафторэтилена [22], из которого видно, что даже при 25 °C в не-
которых случаях из-за значительного уменьшения AF должен осу-
ществляться самопроизвольный переход указанных полимеров в гра-
фит. Аналогичная закономерность, по-видимому, распространяется и на
другие полимеры. В действительности полимеры стойки при более вы-
256
Таблица 2.7. Термодинамические параметры термического распада
полиэтилена и политетрафторэтилена (расчетные данные для 25 °C)
№ реакции Направление деструкции ДЯ, кДж/моль ДЗ, кДж/(моль-К) ДР, кДж/моль
1 сн2—сн2 — —> сн2=сн2 93,65 143,6 51,08
2 > VaCeHe + H, 68,98 144,09 26,1
3 >- 2С (графит) + 2Н2 40,27 190,69 —17,14
4 >- Н-алкены А 4,57 9,05 2,01
5 I СН3(СН2)33СН=СН2 ► С (графит) + СН4 —33,69 115,6 —68
6 cf2-cf2 > CFa=CF2 192,74 188,56 138,27
7 ► 2С (графит) + 2F2 812,86 297,49 720,68
8 > С (графит) + CF4 —113,13 146,65 —149,22
соких температурах. Это лишний раз подтверждает, что изменение AF
указывает лишь на возможность течения процесса, но отнюдь не опре-
деляет реальность его протекания. Не менее важное, а иногда решаю-
щее значение имеют кинетические факторы. Известно большое число
термодинамически неустойчивых систем, существующих бесконечно дли-
тельное время. Примером могут служить янтарь, копал н другие иско-
паемые, аморфное состояние которых термодинамически неустойчиво,
но в данном фазовом состоянии из-за больших кинетических барьеров
они существуют миллионы лет. В то же время в геологические эпохи
создавались благоприятные условия для превращения полимеров (рас-
тений) в кристаллические аллотропные формы углерода и ископаемые
угли различной стадии метаморфизма.
Процессу превращения полимеров в углерод присущ ряд особенно-
стей. Из литературы известно, что карбонизация (графитация) может
совершаться за очень короткое время, исчисляемое минутами или до-
лями минуты. Уже сам по себе факт перехода от органического к угле-
родному волокну за столь короткое время — фактически при тепловом
ударе — заслуживает особого внимания. Сочетание малых времен с вы-
сокой температурой вызывает необходимость новых решений проблем
кинетики термического распада полимеров.
Переход органического волокна в углеродное происходит при не-
обычайно высоких скоростях. Так, если принять температурный коэф-
фициент реакции 2—2,5, то при ТТО, равной 2500°C (нагрев от темпе-
ратуры 100 до 2500°C), скорость реакций возрастает в 2240—2,5240 раза.
Огромные скорости химических реакций были известны и раньше. При-
мером могут служить взрывные процессы. Между этими процессами су-
ществует общность и различие. К общим чертам относятся огромные
скорости реакций, выделение газообразных и других летучих соедине-
ний. При взрыве высвобождается огромная энергия; надо полагать, по-
добное явление имеет место при карбонизации полимеров.
Для взрывчатых веществ подбирается такой состав соединений, ко-
торые полностью превращаются в газообразные продукты. Результатом
17—2254
257
карбонизации полимеров является образование (с большим выходом)
коксового остатка—углерода. Поскольку карбонизация (графитания),
как и взрыв, сопровождается выделением огромного количества энер-
гии, то продукт реакции (кокс) сильно обедняется запасом внутренней
энергии. Во время карбонизации уменьшается свободная энергия и
остаток — кокс — оказывается в глубокой «термодинамической яме»,
что, как уже упоминалось выше, придает УВМ, как, впрочем, и другим
родственным материалам, особые свойства, в частности необычайно вы-
сокую теплостойкость, по которой они превосходят все органические и
большинство 1неор1ганических соединений нашей планеты.
Как было показано в свое время Н. Н. Семеновым, в основе взры-
ва лежат радикальные цепные процессы, протекающие с вырожденным
развитием кинетических цепей. Можно предположить, что по анало-
гичному механизму происходит карбонизация полимеров. Помимо ана-
логии прямые эксперименты подтверждают это положение. Однако
максимальная концентрация ПМЦ и ее убывание почти до нуля (в пре-
делах чувствительности приборов) происходит раньше завершения хи-
мических процессов. {Сведения о ПМЦ приводятся для гаиролизованно-
го остатка, охлажденного до нормальной температуры, т. е. фиксиру-
ются долгоживущие радикалы (пост-эффект).] Объяснить своеобраз-
ный ход кривой ПМЦ-f (ТТО) большим вкладом, вносимым в цепной
процесс маложивущими (активными) радикалами, трудно, так как аро-
матизация (накопление л-сопряжений), происходящая с повышением
температуры, приводит к стабилизации и снижению активности радика-
лов. Для понимания механизма реакций, протекающих при ТТО выше
400—500 °C, необходимо определение концентрации ПМЦ непосредст-
венно в условиях пиролиза.
Своеобразна кинетика пиролитических процессов. Карбонизации
одновременно подвергаются десятки и сотни тысяч плотно упакованных
волоконец диаметром 12—15 мкм и, несмотря на большую потерю мас-
сы и усадку, материал сохраняет волокнистую форму. Этот простой
факт дает исключительно ценную информацию о макромеханизме пиро-
литических процессов. В массе каждого элементарного волоконца про-
текают сложные физико-химические превращения органического волок-
на в углеродное. Поверхность волокон (служит границей раздела реак-
ционных пространств. Сохранение формы свойственно не только волок-
нам, но и более массивным образцам (спички и стопка листов бумаги,
подвергнутые карбонизации, сохраняют свою форму). Единственным
необходимым условием замкнутости реакционного пространства явля-
ется неплавкость полимера. Одна из возможных причин столь необыч-
ного явления связана с огромными скоростями реакций, при которых
они не выходят за рамки, ограниченные поверхностью волокна. Даже
для относительно невысоких температур (250—450°C), соответствую-
щих наиболее бурному течению реакций и максимальной потере массы,
скорость реакций (отсчет от нормальной температуры) возрастает в
(2—2,5)25—(2—2,5)45 раз. Видимо, огромное значение имеют матричные
эффекты, лежащие в основе пиролиза полимеров. Макромолекулы игра-
ют роль микроматриц, а фибриллы — макроматриц. Фибриллы явля-
ются самостоятельной микромассой, в которой совершаются физико-хи-
мические превращения.
Сохранение фибрилл, хотя и в видоизмененной форме, в УВМ и
роль структуры исходного волокна подтверждают значение макрома-
тричного эффекта, создаваемого фибриллами.
258
Исследования кинетики и механизма пиролитических превращений
органических волокон в углеродные, по существу, еще не начаты, но
они могут стать новым этапом в развитии науки о полимерах.
ЛИТЕРАТУРА
1. Коршак В. В. Термостойкие полимеры. М., «Наука», 1969. 410 с.
2. Темникова Т. И. Курс теоретических основ органической химии. М., «Химия»,
1962. 1006 с.
3. Мадорский, «Химия и технология полимеров», 1961, № 3, с. 77.
4. Фрайзер А. Т. Высокотермостойкие полимеры. М., «Химия», 1971. 294 с.
5. Фольмерт, «Химия и технология полимеров», 1967, № 9, с. 104.
6. Проспект фирмы «Minnesoto Minning and Manufacturing Co.», 1961.
7. Laszkiewiez B„ «Polymery», 1967, v. 12, № 8, p. 359.
8. Gaylord N. e. a., J. Am. Chem. Soc., 1963, v. 85, p. 641.
9. Стрепихеев А. А., Деревицкая В. А., Слонимский Г. Л. Основы химии высокомоле-
кулярных соединений. М., «Химия», 1967. 513 с.
10. Вассерман А. Реакция Дильса — Альдера. М., «Мир», 1968. 131 с.
11. Литвинов И. А., Каргин В. А., Высокомол. соед., 1973, сер. А, т. 15, с. 1615—1620.
12. Winslow F. W. е. a., J. Am. Chem. Soc., 1955, v. 77, р. 4751.
13. Winslow F. W. e. a., J. Polymer Sci., 1955, v. 16, p. 101.
14. Jellinek H. H., J. Polymer Sci., 1949, v. 4, p. 13.
15. Асеева P. M. и др. В кн.: Структурная химия углерода и углей. М., «Наука»,
1969, с. 161—200.
16. Gibson J., Halohen N., Riley H., J. Chem. Soc., 1946, № 6, p. 456; Smith T. D., J. Chem.
Soc., 1952, № 3, p. 923; Hofman K, Sinkel S., Z. anorg. Chem., 1940, Bd. 85, S. 245;
Blalden H. E., Gibson J., Rilley H., «Fuel», 1940, v. 19, p. 24.
17. Mackay H. A., «Carbon», 1970, v. 8, p. 517.
18. Goodnew R. J., Clark A. J., Bailey J. E., Mater. Sci. Eng., 1975, v. 17, p. 3—30.
19. Берлин А. А. и др. Высокомол. соед., 1959, т. 1, № 12, с. 1817—1820.
20. Lewis J. C., Polymer Prepr., 1973, v. 14, № 1, p. 380.
21. Edstrom T., Lewis J. C„ «Carbon», 1969, v. 7, p. 85—91.
22. Wall L. A., SPE Journal, 1960, v. 16, № 8, p. 810.
23. Madison J. J., Roberts R. N„ Ind. Eng. chem., 1958, v. 50, № 2, p. 237.
24. Chambers W. E., Meehan. Eng., 1975, v. 97, № 12, p. 37—41.
ГЛАВА 3
ПОЛУЧЕНИЕ УГЛЕРОДНЫХ ВОЛОКНИСТЫХ МАТЕРИАЛОВ
Объектами исследований, посвященных получению УВМ, служило
подавляющее большинство природных и химических волокон. В резуль-
тате проведенных работ было установлено, что наиболее приемлемыми
для этих целей оказались полиакрилонитрильные (ПАН-В) и гидрат-
целлюлозные (вискозные ГЦ-В) волокна. ПАН-В используется преиму-
щественно для производства высокопрочного высокомодульного УВ, а
для УВМ другого ассортимента и назначения применяется ГЦ-В. Пре-
имуществом ПАН-В является больший выход углерода (около 40% от
массы полимера) и менее сложная технология получения УВМ благода-
ря особенностям структуры исходного полимера и промежуточных про-
дуктов. Однако в процессе переработки ПАН-В выделяется синильная
кислота, и это является их существенным недостатком. ГЦ-В гораздо
дешевле и доступнее ПАН-В. Эти критерии зависят от конъюнктурных
технико-экономических факторов, и поэтому могут существенно повли-
ять на выбор исходного сырья.
К перспективным видам сырья относятся разнообразные пеки, бо-
гатые содержанием углерода, и фенольные смолы. В связи с разра-
17* 25»
боткой способов получения мезофазных (жидкокристаллических) пе-
ков к ним проявляется повышенный интерес.
В данной главе рассматриваются принципы получения УВМ из
трех перечисленных видов сырья. Попытки применения других волок-
нистых материалов не увенчались успехом 1[ 1, с. 205—208].
3.1. ПОЛУЧЕНИЕ УГЛЕРОДНЫХ ВОЛОКНИСТЫХ МАТЕРИАЛОВ
НА ОСНОВЕ ПАН-ВОЛОКНА
Процесс получения УВМ из ПАН-В включает текстильную подго-
товку материала, окисление, высокотемпературную обработку (карбо-
низация и графитация), подготов.ку поверхности УВМ и получение пре-
прегов. В этом разделе главы рассматриваются требования к исход-
ному сырью, процессы окисления ПАН-В и его высокотемпературная
обработка (ВТО).
3.1.1. Требования к исходному ПАН-волокну
Исследования, связанные с использованием в качестве предмате-
риала ПАН-В, впервые были начаты в СССР {2]. В то время в науч-
ной и патентной литературе отсутствовали сведения о применении
ПАН-В для этих целей. Затем интенсивные исследования начали про-
водиться в Японии [3] и несколько позже в ряде научных учреждений
Англии с использованием волокна куртель [4]. В 1966 г. появилась
первая научная публикация Ватта i[5] на эту тему. В США на первом
этапе создания производства УВМ сырьем служило ГЦ-В. Лишь в кон-
це 60-х годов и начале 70-х годов ряд фирм США начал производство
УВМ на основе ПАН-В по лицензиям японских и английских фирм.
В настоящее время высокопрочное высокомодульное волокно выраба-
тывается в Англии, Японии, США и Франции.
Свойства исходного ПАН-волокна оказывают большое влияние на
качество УВ. К числу важнейших его показателей относятся: химиче-
ский состав, структура, механические свойства и дефекты.
Объектами исследования служили ПАН-гомо-вол окно и волокна из
сополимеров акрилонитрила. Изучались промышленные волокна, содер-
жащие в качестве сомономера метилакрилат, метилметакрилат, винил-
ацетат и др. Введение сомономеров ускоряет циклизацию и снижает
тепловой эффект в процессе термоокисления, что облегчает проведение
этой стадии получения УВ. Продолжаются поиски новых сомономерных
волокон. Предложено [6] использовать новый тип сополимера акрило-
нитрила с гидроксиалкилированными производными общей формулы:
СН2=С—X
R—С—ОН
I
н
где X —CN, СООН, фенильная или нафтильная группа;
R — атом водорода, алкил, арил, или алкиларильный радикал с числом атомов угле-
рода не более 12.
Из волокон этого типа получены УВ, в которых высокая прочность
сочетается с высоким модулем Юнга ’[1, с. 140]. По патенту [7] реко-
260
мендуется применять тройной сополимер акрилонитрила, N-винилпир-
роллидона и метилакрилата (мольное соотношение 95:3:2), из кото-
рого получено УВ с прочностью 7 ГПа (700 кгс/мм2). Волокно, содер-
жащее в качестве второго компонента амидоонсим, видимо, отличается
тем, что амидооксимные группы при нагреве распадаются, ускоряя тем
самым циклизацию и образование межмолекулярных связей, благодаря
чему продолжительность окисления сокращается до 30 мин [8]. Не-
смотря на большое число работ, в литературе не приводится сведений
о том, какой тип волокна находит практическое применение. В Англии
основным сырьем служит волокно куртель, в других странах — видимо,
промышленные волокна, но их состав и показате-
ли неизвестны.
Существует несколько способов получения
ПАН-волокон, отличающихся типом применяемо-
го растворителя и методом формования (сухой
и мокрый), от которых зависят структура и мор-
фология волокна. Эти факторы влияют на тер-
мохимические превращения полимера, образова-
ние структуры УВ и его свойства. Роль условий
получения и, следовательно, структуры наглядно
показана в работах [9—11] на примере волокон,
сформованных в органическую и водно-диметил-
формамидную ванны. Условия формования
(осаждения полимера) влияют на надмолекуляр-
ную организацию, величину поверхности, темпе-
ратурный интервал экзотермических эффектов,
Рис. 3.1. Влияние прочно-
сти ПАН-В «а прочность
(/) и модуль Юнга (2)
УВ.
максимальную скорость потери массы и количе-
ство поглощенного при термоокислении кислорода. Установленно, что
условия формования имеют большее значение, чем химический состав
ПАН-волокна.
Высокие степени вытягивания в различных средах повышают ориен-
тацию и прочность ПАН-В, что благоприятно сказывается на механиче-
ских свойствах У В. Действительно, как видно из рис. 3.1, по мере уве-
личения прочности ПАН-В заметно возрастают прочность и модуль
Юнга УВ [12]. Аналогичная закономерность установлена в ряде других
работ [13, 14]. Особенно существенное значение имеет ориентация
ПАН-В, так как организованные надмолекулярные образования служат
матрицей при формировании структуры углерода и обусловливают ме-
ханические свойства УВ. Однако вытягивание ПАН-В не должно пре-
вышать оптимальных значений, выше которых начинают возрастать де-
фекты ПАН-В; дефекты обычно переходят на УВ и снижают его проч-
ность. Необходимость применения высокопрочных ПАН-В не является
бесспорной. В работе Байлея и Кларка [15] приводятся данные о во-
локне куртель, имеющем низкую прочность, из которого получается УВ
с высокими механическими показателями.
Поскольку во время окисления волокно подвергается вытягиванию,
следует учитывать термомеханические свойства ПАН-В. В работе [16]
указывается, что при нагревании в изотермических условиях (до
320°C) выявлено шесть областей различного поведения ПАН-В.
На свойства УВ большое влияние оказывают загрязнения ПАН-В.
В результате выгорания инородных включений во время карбонизации
на поверхности УВ возникают дефекты, снижающие его прочность.
В этом плане заслуживает внимание работа [17], в которой показано,
261
что при получении ПАН-волокна в воздушной среде, очищенной от пы-
ли, повышается не только прочность полученного из него УВ, но исче-
зает экстремальное значение на кривой (ТТО). Поэтому следует
отдать предпочтение прямому методу получения ПАН-В из растворов,
так как в этом случае оно менее загрязнено инородными частицами.
При использовании УВ для изготовления конструкционных компо-
зитов особое внимание уделяется коэффициенту вариации механиче-
ских свойств УВ, особенно по прочности. Весомый вклад в этот пока-
затель вносит исходное волокно. Неоднородность ПАН-В возникает
главным образом на стадии формования. При формовании сухим мето-
дом получается более однородное волокно, чем при формовании мокрым
методом '[18]. Основным дефектом ПАН-В является неравномерность
сечения, или площади поперечного среза, волокна. Поэтому наблюдает-
ся большая разница в коэффициентах вариации, определяемых для ком-
плексных или элементарных нитей. В последнем случае он гораздо
выше.
На технико-экономические показатели влияет линейная плотность
нити ПАН-В; с ее увеличением снижается стоимость УВ.
3.1.2. Термическое окисление ПАН-волокна
3.1.2.1. Физико-химические процессы, протекающие
при окислении ПАН-волокна
Окисление — важнейшая стадия технологического процесса получе-
ния УВ. Предварительное окисление облегчает дегидрирование полиме-
ра и, что особенно важно, создает условия для образования предструк-
туры, обеспечивающей создание оптимальной структуры углерода и
приобретение УВ ценных механических свойств. Превратить ПАН-В в
УВ можно, не прибегая к окислению, но практически этот способ не-
приемлем, так как при этом увеличивается длительность технологиче-
ского цикла, происходит более глубокая деструкция полимера, сопро-
вождающаяся снижением выхода углерода.
На стадии окисления протекают сложные химические процессы и
структурные превращения. Несмотря на большое число работ, в лите-
ратуре приводятся различные и подчас противоположные мнения об ос-
новных химических процессах, протекающих на этой стадии.
При термообработке на воздухе потемнение ПАН не сопровождает-
ся потерей массы и азота, но методом ИК-спектроскопии установлено,
что число групп CN уменьшается. Хоутц [19] предложил структуру чер-
ного ПАН, состоящую из гетероциклов, содержащих двойные сопряжен-
ные связи —C=N— (см. схему I). Систематическое изучение терми-
ческого окисления ПАН выполнено Грасси и сотр. [20—24]. По их дан-
ным, первой стадией является миграция третичного водорода к азоту с
образованием иминной группы (см. схему II). Последующая миграция
иминного водорода к группе CN приводит к образованию тетрагидро-
пиридиновых (нафтиридиновых) циклов. Переход водорода может про-
исходить внутри макромолекул и между макромолекулами. При внутри-
молекулярной циклизации сохраняется линейная форма макромолеку-
лы (см. схему III), а в случае межмолекулярной миграции водорода
наряду с циклизацией имеет место сшивка с образованием сетчатого
полимера (см. схему IV).
262
СН сн сн
•СН2—СН—СН2—СН'
I I
CN CN
—СН + —CN--> —С—C=NH
II
CN CN
/CHj CHa СН^/СНа!
СН СН СН СН СН
III NN
I I I
c c c c c c
N м N I I I
CH CH CH
^CHa^CH^^CHa^
IV
Иное предложение о направлении течения реакции при термообра-
ботке ПАН выдвинуто А. А. Берлином с сотр. [25] и другими исследо-
вателями [26, 27]. По их мнению, окислительное дегидрирование про-
исходит с образованием полиеновой структуры с системой сопряженной
связи в основной цепи (см. схему V).
СН СН СН
I I I
CN CN CN
V
В последнее время о предпочтительном течении реакций, приводя-
щих к продукту V, указывается также в работе [28].
Наконец, по Шурцу '[29], основным направлением является образо-
вание межмолекулярных связей азометинового типа (см. схему VI).
—СН2—СН— —СН2—СН—
CN C=NH
»- I
CN-----------— СН2—С—
I I
—СН,—СН— CN
VI
263
Следует отметить, что, используя один и тот же метод (ИК-спект-
роскопию) для исследования кинетики реакции, авторы многих работ
приходят к различным, подчас взаимоисключающим выводам. Такое
положение объясняется отчасти различными условиями проведения ре-
акции, но главным образом субъективностью трактовки данных, полу-
ченных этим методом.
Из рассматриваемых схем наиболее достоверными следует при-
знать схемы Хоутца и Грасси. Ими, а также многими другими исследо-
вателями [30—33] показано, что с ужесточением условий термообра-
ботки уменьшается интенсивность полосы CN (2240 см-1)> появляется
и постепенно усиливается полоса 1620 (группы —C=N — в цикле),
уменьшается полоса 1445 см-1 (группы СН2). Трактовка полосы
1620 см-1 затруднена, так как ее трудно отличить от близлежащей по-
лосы 1640 см-1, относящейся к валентным колебаниям групп С=С.
Термоокислительная деструкция — сложный процесс, который со-
провождается многими реакциями, в том числе не исключается окисли-
тельное дегидрирование с образованием двойных связей С=С в основ-
ной цепи. Однако главной и основной реакцией является циклизация.
Дополнительным подтверждением этой схемы служат данные по изу-
чению УФ-спектров окисленного ПАН [34].
Циклизация вследствие образования стойких шестизвенных циклов,
протекающая с уменьшением свободной энергии, выгодна термодинами-
чески.
На интенсивность химических процессов при термообработке ПАН
влияют многие факторы. В присутствии кислорода заметно ускоряются
циклизация и потемнение ПАН. Продолжительность термообработки
также влияет на глубину химических превращений. Действие фактора
времени заметно в первые 2—4 ч.
Новые связи во время циклизации образуются после миграции од-
ной пары электронов группы CN по схеме
СН, СН,
\н \н —сн НС—
+бс сб+ +С с
III III
N N N N-
VII
Эффективная отрицательная атака на атом азота способствует цикли-
зации. Водород при p-углероде под влиянием группы CN приобретает
кислый характер, поэтому наличие оснований вызывает более сильную
атаку на атом азота. Из рассмотренного становится очевидным, что
щелочи, амины [24], амиды щелочных металлов [35], нуклеофильные
реагенты [36] типа органических кислот, фенолов и другие соединения
катализируют циклизацию. Даже незначительное содержание некото-
рых примесей может оказать заметное влияние на циклизацию и на-
правление течения реакции [37]. На процесс образования сопряженных
связей влияют регулярность строения макромолекул, молекулярная мас-
са и надмолекулярная структура полимеров. Хироси [38] показал, что
для ПАН синдиотактической структуры циклизация протекает при бо-
лее низкой температуре по сравнению с ПАН атактической структуры.
Одно из существенных преимуществ ПАН-В как исходного сырья
по сравнению с другими волокнами состоит в том, что образование на
264
стадии окисления лестничного полимера обусловливает повышение ТСт
и термостойкости промежуточных продуктов, сохранение ориентации и
структуры волокна. Промежуточные соединения играют роль матрицы и
способствуют формированию структуры углеродного волокна [39, 40].
По указанным причинам повышается выход углерода, достигается по-
лучение УВ совершенной структуры с повышенными механическими
свойствами и упрощается технологический процесс. Размеры зациклизо-
ванных участков, по разным источникам, составляют 5—15 элементар-
ных звеньев {41] и зависят от длины стереорегулярных участков ПАН.
Высказывается мнение, что они определяют размеры турбостратных па-
Рис. 3.2. Изменение химического состава ПАН-В в зависимости от продол-
жительности термообработки [33] |(температура 200°C):
1 — отношение С : Н; 2 — содержание кислорода.
Рис. 3.3. Выделение HCN при термической обработке ПАН-В на воздухе
при различной температуре и разной продолжительности (содержание HCN
дано в % от теоретически возможного при отщеплении всех групп CN
® виде HCN),
кетов [39]. Циклизация происходит по цепному механизму: для гомо-
полимеров, вероятно, по свободнорадикальному механизму, а для сопо-
лимеров— по ионному [39].
Накопление кислорода в полимере при мягких условиях окисле-
ния интенсивно протекает, согласно одним источникам, по истечении 6 ч
(рис. 3.2), а по другим [41] —в течение 1 ч. Содержание кислорода в
волокне достигает 10—16%. Химизм взаимодействия кислорода возду-
ха с полимером неясен. Методом ИК-спектроскопии в окисленном во-
локне установлено наличие групп СО, СООН и ОН. Авторы [41] счи-
тают, что образование межмолекулярных связей за счет гидроксильных
групп играет большую роль в сохранении структуры и ориентации во-
локна, так как циклизация влечет за собой уменьшение интенсивности
межмолекулярных водородных связей. Помимо перечисленных функ-
циональных групп приводятся иные формы связи кислорода с полиме-
ром [1, с. 144—145]. По мнению авторов [33], окисляется не исходный
полимер, а продукт его превращения, в частности связи С=С, явля-
ющиеся акцепторами кислорода, а группы =NH играют роль катали-
заторов циклизации. Отмечается повышенная реакционная способность
зациклизованных участков по отношению к кислороду воздуха.
К недостаткам подавляющего числа работ относится отсутствие
прямых химических методов идентификации функциональных групп и
их количественного определения в окисленном полимере. Наличие толь-
ко одних данных ИК-спектроскопии явно недостаточно для выяснения
процесса взаимодействия кислорода воздуха с ПАН-В.
265
Основными продуктами, выделяющимися на стадии окисления, яв-
ляются Н2О, NH3 и HCN. Аммиак начинает выделяться при 220°C, а
синильная кислота при 270 °C [42]. На рис. 3.3 показано количество вы-
делившейся HCN в зависимости от температуры и продолжительности
обработки. HCN может отщепляться с возникновением двойных свя-
зей или со сшивкой полимера. Образованию NH3 предшествует возник-
новение иминных и аминных групп. Дегидратация и отщепление HCN
вызывает накопление двойных связей, потерю водорода, азота и углеро-
да. По данным элементного анализа в глубокоокисленном ПАН умень-
шается содержание Н и N, а содержание углерода остается на уровне
Рис. 3.4. Изменение модуля Юнга УВ-ПАН в зависимости от ТТО [45].
Рис. 3.5. ДТА-кривые ПАН-В |(навеска 100 мг; Онагр — 3°С/мин; среда — азот).
исходного [43]. Соответственно увеличивается отношение С:Н (см.
рис. 3.2).
Как следствие химических процессов, изменяется структура волок-
на. Согласно рентгеноструктурному анализу для ПАН-В установлены
экваториальные пики с брегговскими углами 26=17° и 26=29,5°; по
мере окисления их интенсивность слабеет и появляются пики с 26=
= 25,5° вследствие отражения от плоскостей лестничной структу-
ры [44].
На стадии окисления превалируют деструктивные процессы, приво-
дящие к уменьшению межмолекулярного взаимодействия, снижению
ориентации и частичному разрушению фибриллярной организации во-
локна. По указанным причинам происходит уменьшение удлинения,
прочности и модуля Юнга волокна (рис. 3.4).
3.1.2.2. Условия окисления ПАН-волокна
К основным параметрам окисления ПАН-волокна относятся темпе-
ратура, время и ориентационное вытягивание. Температура и время (Т,
т) взаимно связаны и определяются многими факторами.
Окисление ПАН-волокна кислородом воздуха — типично гетероген-
ный процесс, поэтому особое значение приобретает полнота окисления
всей массы волокна. Как показал Ватт {42], во время карбонизации
недоокисленного волокна выгорает центральная часть с образованием
полости, ослабляющей прочность УВ. Послойное окисление в направле-
нии, перпендикулярном оси волокна, подтверждается следующими дан-
ными. При достижении температуры окисления 230°C ПАН-В становит-
266
ся нерастворимым в ДМФ; при температуре ниже 230 °C—частично рас-
творимым, при этом растворяется сердцевина волокна [45]. Неполнота
окисления проявляется даже в УВ, при травлении которого разруше-
нию подвергается в первую очередь сердцевина волокна. По мнению
В. М. Бондаренко и сотр. [46], окисление протекает в кинетической
области; более рыхлая структура сердцевины УВ связана с неоднород-
ностью ПАН-В. Доказательством служило неодинаковое количество по-
глощенного кислорода ПАН-В различного диаметра. Однако опыты
проводились при общем малом поглощении кислорода, поэтому они не-
достаточно убедительны.
Рис. 3.6. Изменение температуры начала экзотермического эффекта в зави-
симости от продолжительности предварительной термообработки при
200 ЧС:
1 — волокно-О; 2 — волокно-М.
Рис. 3.7. Зависимость прочности УВ от продолжительности окисления
ПАН-В [42] ;(температура 225 ЧС).
Циклизация и окисление сопровождаются выделением большого
количества тепла (рис. 3.5). При большой массе волокна тепло не ус-
певает рассеиваться, происходит перегрев волокна, интенсивная дест-
рукция полимера, сопровождающаяся большой потерей углерода, за-
трудняется формирование структуры УВ. Вследствие протекания ука-
занных процессов получается УВ низкого качества. Выделение тепла в
результате циклизации подтверждается тем, что после предварительно-
го прогрева волокна при температуре ниже температуры окисления эк-
зотермический эффект в процессе окисления резко уменьшается. Замет-
но снижается также температура начала экзотермического эффекта
(рис. 3.6).
Снижение экзотермического эффекта позволяет создать более ра-
циональный технологический процесс окисления ПАН-В. Экзотермиче-
ский эффект зависит от многих факторов. В работе [11] отмечается
влияние способов формования волокна. Введение в состав волокна не-
больших количеств сомономеров благоприятно сказывается на тепло-
вом режиме окисления. Как правило, начало экзотермических реакций
сдвигается в область более низких температур, расширяется темпера-
турный диапазон экзотермического эффекта и снижается его величина
[47]. Существенным является введение в волокно добавок, например
кислот Льюиса [1, с. 167], или обработка волокна фосфорной кислотой
[48], гидроксиламином ([49] и другими соединениями.
Приводимые в литературе температурные режимы можно подраз-
делить на три группы: мягкие (до 220°C), средние (220—250°C), жест-
267
кие (выше 250°C). Естественно, что продолжительность процесса
снижается с повышением температуры. Приводится значение этого пара-
метра, находящееся в пределах 0,5—24 ч. По первому варианту, раз-
работанному в Англии, окисление проводилось при 220 °C в течение
24 ч, т. е. в мягких условиях, но продолжительное время.
Продолжительность окисления является одним из важнейших пара-
метров, определяющих качество УВ. Как видно из рис. 3.7, существует
оптимальное время окисления, соответствующее максимальным прочно-
стям УВ. Наличие экстремальных значений свидетельствует по меньшей
мере о двух противоположно действующих факторах: образование лест-
ничного полимера на первой стадии обработки благоприятно сказыва-
ется на прочности УВ; на второй — начинают преобладать деструктив-
ные процессы, приводящие к снижению прочности УВ. Необходимо так-
же учитывать продолжительность разогрева волокна до заданной
температуры окисления. Уменьшение скорости нагрева (увеличение про-
должительности) приводит к повышению прочности волокна, но на мо-
дуль этот фактор не влияет (табл. 3.1).
Окисление является наиболее продолжительной стадией технологи-
ческого процесса получения УВ, поэтому изыскиваются пути ее сокра-
щения.
Рассмотренные выше способы снижения экзотермического эффекта
дают возможность сократить продолжительность окисления. Введение
в волокно солей металлов IV группы также позволяет снизить продол-
жительность операции до 1—2 ч (температура 230 °C) (50]. При ис-
пользовании в качестве катализаторов алкилсульфонатов щелочных
металлов [51] продолжительность окисления составляет 12 мин (темпе-
ратура 300°C). Волокно из сополимера акрилонитрила и гидроксилсо-
держащих соединений [6] окисляется в течение 3—12 ч (температура
230—260°C). Заслуживают внимания серосодержащие катализаторы
(SO2, COS, меркаптаны и др.) или элементарная сера [52]. Термиче-
скую обработку волокна в присутствии подобных соединений предпоч-
тительно проводить в интервале температур 200—300 °C в течение 1—
20 ч. Представляет интерес применение двухстадийного сокращенного
метода окисления с использованием смеси Ог+НС! или O2+CI2 '[53].
Наряду с сокращением продолжительности увеличивается выход угле-
рода. Двухстадийный метод рекомендуется также проводить при при-
менении других катализаторов '[50]. В случае введения в прядильной
раствор кислоты Льюиса, в частности комплекса SnCl4 с ДМФ, и его
прогрева перед формованием волокна до 140—160 °C происходит цик-
лизация полимера, что дает возможность сократить продолжительность
окисления до 30 мин [54]. Однако этот на первый взгляд заманчивый
метод из-за нестабильности прядильного раствора неперспективен.
Таблица 3.1. Влияние продолжительности подъема температуры
до 205 °C иа свойства УВ [44]
Продолжительность разогрева до тем- пературы окисления*, мии Прочность, 10? Па Модуль Юнга, 1010 Па
0 91 11,9
30 140 12,6
180 154 13,3
* Продолжительность окисления 10 ч при 205 *С.
268
При выборе оптимальных условий необходимо учитывать диаметр
элементарного волокна; с увеличением его продолжительность окисле-
ния возрастает [55].
Критерий оценки качества волокна, предназначенного для дальней-
шей переработки, в литературе не приводится. Отмечается, что только
из черного ПАН-В можно получить волокно высокого качества [56].
Подобная органолептическая оценка степени окисления субъективна и
несовершенна, так как в зависимости от условий получения свойства
волокна черного цвета могут изменяться в широких пределах. Согласно
Рис. 3.8. Влияние вытягивания при окислении i (220 °C) на модуль Юнга
ПАН-волокна, подвергнутого термической обработке при температурах:
1000ЧС |(/); 1500°C (2) и 2500 ЧС (3).
Рис. 3.9. Кривые нагрузка — удлинение для ПАН-В:
7— необработанное; 2 — после окисления с вытяжкой (220 °C); 3— после окисления
с вытяжкой (220 °C) и нагревания до 400 °C без натяжения.
работе [33], существует корреляция между количеством поглощенного
кислорода и изменением плотности волокна; последнее может служить
надежным критерием оценки степени окисления ПАН-В.
3.1.2.3. Вытягивание ПАН-волокна при окислении
В процессе окисления происходит усадка волокна, достигающая
20—40%, вследствие чего наблюдается дезориентация структурных эле-
ментов ПАН-В. Нарушение ориентации во время окисления отрицатель-
но сказывается на образовании структуры УВ при карбонизации, в ре-
зультате получается малопрочное УВ.
Новым этапом в производстве УВ является применение ориентаци-
онной вытяжки на разных стадиях превращения химических волокон
в углеродные. При получении его из ПАН-В особое значение имеет вы-
тягивание волокна во время окисления. Именно благодаря этому были
получены высокопрочные высокомодульные УВ. По мере увеличения
степени вытягивания при окислении возрастает модуль Юнга [57] кар-
бонизованного и графитированного волокон (рис. 3.8), а также проч-
ность УВ. Даже окисление на жесткой паковке, предотвращающее усад-
ку волокна и изменение его длины, обеспечивает получение УВ высоко-
го качества.
Вытягивание способствует ориентации предструктур, возникающих
на стадии окисления. В результате получается система, подобная жид-
кокристаллической, которая выполняет функцию матрицы при форми-
ровании структуры углеродного скелета в процессе карбонизации
269
волокна. В этом заключается одно из преимуществ применения в качест-
ве исходного сырья ПАН-В. При вытягивании на стадии окисления по-
следующие операции можно проводить без вытяжки.
Высоких деформаций (вытягивания) можно достичь, видимо, до
момента образования густой сетки межмолекулярных связей, препятст-
вующих развитию деформации. Вытягивание сильноструктурированных
волокон приводит к появлению дефектов и к их обрыву. Характер кри-
вых нагрузка — удлинение исходного и термообработанного волокон
дает информацию об оптимальных условиях проведения процесса вы-
тягивания волокна (рис. 3.9) [58].
3.1.3. Высокотемпературная обработка
(карбонизация и графитация) окисленного ПАН-волокна
3. 1.3.1. Физико-химические процессы, протекающие
при высокотемпературной обработке ПАН-волокна
В процессе высокотемпературной обработки осуществляется пере-
ход от органического к углеродному волокну, сопровождающийся слож-
ными химическими и структурными преобразованиями полимера, аро-
матизацией углерода и формированием структуры углеродного волок-
на. Одновременно происходит изменение физико-химических и механи-
ческих свойств материала. Отобразить эти процессы в виде конкретных
химических уравнений не представляется возможным. Этот сложный
Рис. 3.10. Выделение летучих продуктов при пиролизе окисленного ПАН-В:
1 — НаО; 2 —СОа; 3 — СО; 4— HCN; 5 — NH3; 6 — СН„; 7 — Na.
переход мбжно разделить на три основных стадии: при температурах
200—600°C протекают наиболее важные химические процессы; в интер-
вале 400—il200°C формируются основные элементы структуры УВ; при
температурахвыше 1200°C происходят преимущественно физические из-
менения, связанные с совершенствованием структуры УВ.
Основная потеря массы на первой стадии наблюдается в довольно
узкой области температур (200—400°C), хотя химические процессы
на этом не заканчиваются. Летучими продуктами термического
270
распада являются: NH3, HCN, N2, СО2, СО, НгО, СН4. Кинетика [59]
их выделения в зависимости от температуры показана на рис. 3.10. По-
добные сведения приводятся также в работе [42]. Отщепление воды в
результате дегидратации происходит при температурах 250—600 °C с
максимумом около 400 °C. Источником СО является реакция декарбок-
силирования, протекающая примерно в том же интервале температур.
Максимум образования СО сдвинут в область более высоких темпера-
тур; СН4 и Нг выделяются в относительно небольших количествах. Для
NH3 установлены два незначительно различающихся максимума (пер-
Рис. 3.11. Зависимость
концентрации ПМЦ ((/)
и удельного объемного
электрического сопротив-
ления (2) ПАН-<В от
ТТО.
вый.примерно при 370°C, второй — около 570°C). Ниже приведена одна
из возможных схем (VIII) выделения NH3 [60]:
^СН2 J2H2
СН СН СН
С С
N N NH
СН сн
VIII
Окисление волокна подавляет выход аммиака. HCN образуется в ши-
роком диапазоне температур (300—900 °C) с двумя максимумами (при
350 и 750°C), причем второй максимум гораздо больше первого. Пер-
вый максимум соответствует выделению HCN из незациклизованных
участков цепи с образованием двойных связей; второй — является след-
ствием межмолекулярного взаимодействия глубоких продуктов распа-
да и сопровождается возникновением графитоподобных структур (см.
схему IX).
СН СН
сн сн .
—HCN
I I I
с с с
271
Отщепление азота начинается при 700 °C и заканчивается при вы-
соких температурах. Даже при 1000 °C его содержание в волокне со-
ставляет 6% [39]. По некоторым данным, в волокне, полученном при
ТТО» 1500°C, все еще содержатся небольшие количества азота. При
столь высоких температурах азот может находиться только в гетероцик-
лах, которые распадаются с его выделением (см. схему X):
Большой вклад в химические процессы вносят радикальные реак-
ции (рис. 3.11 и-3.12). Максимальная концентрация ПМЦ по отношению
200 ‘тОО 600 800 1000
6, °C
Рис. 3.12. Зависимость кон-
центрации ПМЦ от ТТО
[61]:
/ — изометрическая обработка;
2 — свободная термообработка.
к максимальной потере массы сдвинута в об-
ласть более высоких температур.
В работе [61] наглядно показано влия-
ние вытягивания (предотвращение \садки) на
магнитные свойства материалов. При термо-
обработке в изометрических условиях по срав-
нению с термообработкой в условиях свобод-
ной усадки волокна концентрация ПМЦ и
магнитная анизотропия значительно больше.
Напряжение способствует ориентации оскол-
ков молекул вдоль оси волокна, в результате
чего создаются благоприятные условия для
образования межмолекулярных связей, рос-
та полисопряжений, что повышает маг-
нитную восприимчивость и парамагнитизм во-
локна.
На второй стадии наряду с химическими
процессами важную роль начинают играть структурные преобразования
углерода. Происходит ароматизация углерода, образование систем со-
пряженных связей, повышение доли зр2-гибридных форм углерода, об-
разование и рост графитовых плоскостей. Снижение электрического
сопротивления служит наглядным дополнительным подтверждением
образования системы сопряженных связей (см. рис. 3.11). Уже на этом
этапе термообработки ленты имеют большую длину при незначительной
толщине. Последняя мало изменяется при этих температурах. По мне-
нию авторов работы [39], пакеты из четырех-пяти слоев образуются
уже при 320 °C, что маловероятно.
По данным работы [45], исходные фибриллы исчезают на первой
стадии термообработки; по другим источникам, они сохраняются до
272
800 °C, а при более высоких ТТО начинают вновь появляться фибриллы,
характерные для УВ.
Вполне закономерно изменение на этой стадии механических
свойств. Прочность (рис. 3.13) и модуль Юнга (см. рис. 3.4) вначале
из-за преобладания деструктивных процессов уменьшаются, а с момента
начала формирования структуры УВ эти показатели возрастают. Мак-
симальная скорость роста модуля Юнга наблюдается выше 700 °C.
Плотность волокна, несмотря на потерю массы, возрастает.
На третьей стадии термообработки продолжается увеличение доли
зр2-гибридных форм углерода и уменьшение других гибридных форм,
переход линейных форм углерода в графитоподобные. Перераспределе-
ние связей углерод—углерод можно отнести к своеобразным химиче-
Рис. 3.13. Изменение прочности ПАВ-В в зависимости от ТТО на ранней
стадии термообработки.
Рис. 3.14. Влияние ТТО иа модуль Юига (4) [64] и прочность УВ по
данным различных авторов:
.’—[66’; 2—U, с. 1961; 3— [641.
ским процессам. Результатом этих превращений является увеличение
размеров базисных лент, турбостратных кристаллов, совершенствование
фибрилл (см. гл. 1) и изменение физико-механических свойств волокна.
3. 1.3.2. Условия проведения карбонизации
(графитации) волокна
К основным параметрам процесса относится среда, температура,
продолжительность процесса и вытягивание.
Защитной средой при высокотемпературной обработке служит азот,
который наиболее доступен среди инертных газов. В лабораторной
практике кроме азота применяется гелий и аргон; иногда обработку
осуществляют в глубоком вакууме.
В условиях высоких температур резко возрастают скорости реак-
ций, поэтому к чистоте азота предъявляются высокие требования;
содержание кислорода в азоте должно быть минимальным.
В процессе карбонизации в результате глубоких химических прев-
ращений промежуточных продуктов распада на поверхности УВ осаж-
дается аморфный углерод, снижающий качество волокна, особенно тСд
композита. Для удаления [63] этого углерода предложено к инертному
газу добавлять кислород (5-10“6 — 25-10-5%), окисляющий аморфный
углерод. Необходимо соблюдать точную дозировку кислорода, так как
при избытке его происходит окисление волокна и ухудшение его свойств,
а также уменьшение срока службы нагревателей.
Важнейшим параметром является ТТО. С увеличением ТТО изме-
няются структура и механические свойства волокна, поэтому, закаичи-
18—2254
273
вая процесс при разных ТТО, можно получать углеродные волокна
с различными свойствами и предназначенные для разных целей.
С ростом температуры обработки происходит спонтанное совершен-
ствование структуры; в частности, улучшается ориентация волокна, спо-
собствующая росту модуля Юнга (см. рис. 3.14). Размеры турбострат-
ных кристаллов возрастают: La до 250 A, Lc до 100 А. Наблюдается бо-
лее сложная зависимость прочности от ТТО. В ранних работах [1,
с. 195—196] указывалось, что вначале с повышением температуры проч-
ность возрастает, а затем уменьшается. По одним данным, критическая
температура составляет 1200°С, по другим (см. рис. 3.14)—достигает
1500 °C. Поэтому появились волокна двух типов: высокопрочное, тип II
(2,5—3,2 ГПа, модуль Юнга 200—250 ГПа) и высокомодульное, тип I
(прочность 2—2,5 ГПа, модуль Юнга более 300 ГПа). Эти волокна име-
ют различные (разрывные деформации: для волокна типа I — около 1%,
для волокна типа II—около 0,5%. В более поздних работах описан
процесс, при котором прочность волокна по достижении максимального
значения с ростом температуры не изменяется, а так как модуль Юнга
продолжает возрастать, то было получено волокно типа III, сочетающее
в себе высокую прочность и высокий модуль Юнга (прочность около
3 ГПа, модуль Юнга 300 ГПа) (см. рис. 3.14). Различная зависимость
прочности а и модуля Юнга Е от ТТО объясняется тем, что на о влияет
большое число факторов; в частности, большое значение имеют дефек-
ты волокна (см. гл. 4). По мнению Гибсона [64], переход от периодиче-
ского к непрерывному способу позволил получить волокна типа III.
Своеобразно, но вполне закономерно изменяется плотность УВ.
Вначале (примерно до 1000 °C) с увеличением ТТО она круто возрастает,
затем начинает убывать, достигая минимума примерно при 1500 °C, т. е.
на стадии предкристаллизационного состояния, что дополнительно под-
тверждает разупорядочение структуры волокна в этой области темпера-
тур, и затем снова возрастает. Электрическое сопротивление резко сни-
жается при возрастании температуры обработки до 1400—1500 °C; при
более высокой температуре оно уменьшается незначительно (рис. 3.15).
В лабораторных условиях графитацию проводят при температуре
до 3000 °C. На практике максимальная температура, видимо, не превы-
шает 2400—2600 °C, так как эксплуатация оборудования при более
высоких температурах с практической точки зрения мало приемлема.
В зависимости от назначения волокна процесс может заканчиваться при
более низких температурах (1000—2000 °C) с получением карбонизован-
ного волокна. Содержание углерода в графитированном волокне выше
99%, в карбонизованном — до 95%.
Опубликованные сведения о продолжительности карбонизации и
графитации противоречивы. В патентах приводятся большие интерва-
лы времен, чем в научных статьях; применяемые на практике режимы
неизвестны. Согласно различным литературным источникам [1, с. 189—
191; 198], продолжительность высокотемпературной обработки состав-
ляет от нескольких минут до 2,5 ч. В этой связи не лишне напомнить,
что, по данным Гибсона (см. ниже), при графитации материала, по-
лученного из целлюлозы, физические процессы структурообразования
заканчиваются в течение 1 мин. УВ-ПАН имеют более совершенную
структуру, поэтому вряд ли разумна в этом случае длительная графи-
тация.
Переход от органического к углеродному волокну целесообразно
подразделить на две стадии:
274
1) низкотемпературная; на этой стадии происходят основные хими-
ческие процессы и наблюдается максимальная потеря массы; подъем
температуры должен быть медленный;
2) высокотемпературная (структурные преобразования); эта ста-
дия должна протекать при быстром подъеме температуры. Вероятно,
чем медленнее протекают процессы на первой стадии получения УВ,
тем более благоприятные условия создаются для образования совер-
шенной структры, определяющей свойства УВ. Однако слишком медлен-
ные процессы невыгодны по экономическим соображениям из-за сниже-
ния производительности оборудования. В подобных случаях выбирают
разумные временные режимы, обес-
печивающие получение продукции
высокого качества без снижения
производительности оборудования.
ПАН-волокна обладают уни-
кальными, пожалуй, только им при-
сущими свойствами, облегчающими
получение из них углеродного волок-
на. Вытягивание во время окисления
позволяет проводить последующую
карбонизацию без вытягивания или
с незначительной вытяжкой или, на-
конец, даже с небольшой усадкой.
Высокотемпературная обработка со-
Рис. 3.15. Влияние ТТО на плотность
(/) н удельное электрическое сопро-
тивление (2) УВ [64].
провождается самопроизвольным со-
вершенствованием структуры [65], что обеспечивает получение волокна
с высокими показателями и упрощает технологию. Все же, по данным
работы [66], вытягивание способствует росту La, улучшению ориентации
и соответственно повышению модуля Юнга и прочности. В этом случае
легче осуществить процесс, при котором одновременно возрастают проч-
ность и модуль Юнга волокна.
Физико-химическим основам получения УВМ на основе ПАН-В по-
священа обширная литература, однако сведения о реальных параметрах
технологического процесса и оборудовании в ней отсутствуют.
3.2. ПОЛУЧЕНИЕ УГЛЕРОДНЫХ ВОЛОКНИСТЫХ МАТЕРИАЛОВ
НА ОСНОВЕ ГЦ-ВОЛОКНА
Целлюлозные волокна наряду с ПАН-волокном являются одним
из основных видов сырья, применяемого для получения УВМ. На их
основе изготовляются УВМ различных текстильных форм (нити, жгуты,
ткани, нетканые материалы, войлок и др.), разнообразных свойств, на-
значения и областей применения (теплозащитные, высокопрочные,
с заданными электрофизическими, сорбционными свойствами и другими
характеристиками).
Для получения волокна хорошего качества большое значение имеет
свойство исходного сырья. Наибольшее внимание уделялось искусствен-
ным волокнам. Исследовалась вискозная текстильная и кордная нить,
медно-аммиачная нить, волокна типа «фортизан», полинозное, ацетат-
ное [67, 68]. Результаты исследования показали, что наиболее приемле-
мыми являются гидратцеллюлозные нити. Судя по литературным дан-
ным, вероятно, для этих целей используется кордная нить (ВК). Вискоз-
ная кордная нить по сравнению с текстильной более однородна по
18*
275
поперечному сечению [69, с. 219—239]. Радиальная неоднородность не-
желательна из-за различной усадки при карбонизации внутренних и
внешних слоев, вызывающей напряжение и снижение прочности УВ.
Вискозный корд имеет специфическую структуру, характеризующуюся
мелкокристаллическими надмолекулярными образованиями и высокой
ориентацией вдоль оси волокна..
Взаимосвязь между свойствами ГЦ-В и УВМ имеет большое значе-
ние, но в литературе приводятся очень скудные сведения по этому во-
просу. По Бекану и Тангу [70], ориентация вискозного волокна опреде-
ляет ориентацию элементов структуры и, следовательно, механические
свойства УВМ. Руланд [71] на основании рентгеноструктурного анализа
приходит к другому выводу. По его данным, связь между ориентацией
исходного и углеродного волокна отсутствует. В работе [72] установле-
но влияние природы ГЦ-волокна на Sv УВМ, но других данных не при-
водится. Бош и Левин [73] исследовали влияние кристалличности и ори-
ентации ГЦ-1волокна на пиролиз в вакууме и в воздушной среде. Как
видно из приведенных ниже данных, увеличение степени вытягивания
повышает скорость реакции, протекающей на первой стадии пиролиза:
Степень вытяги-
вания, % ; ... 20 30 40 50 60
Константа скоро-
сти К-Ю2, мин-‘ 2,30 2,34 3,23 3,18 5,56
Начальная скорость реакции определяет выход остатка. Показана
зависимость выхода остатка от потери массы на начальной стадии, про-
порциональной скорости реакции. Авторы считают, что при пиролизе
вначале происходит дегидратация с образованием межмолекулярных
связей, а затем более глубокая деструкция промежуточных продуктов
распада.
Большое внимание уделяется подготовке волокна. Авиважные пре-
параты относятся к нежелательным примесям, так как ухудшают свой-
ства УВМ. Это, по-видимому, справедливо, но объяснение этого факта
не приводится. Для удаления органических примесей исходное волокно
(ткани) обрабатывается органическими растворителями или поверх-
ностно-активными веществами [74]. Влага также относится к нежела-
тельным примесям, поэтому’ рекомендуется перед карбонизацией исход-
ный материал подвергать сушке в течение 0,5—15 ч при температуре не
ниже 100—230 °C [75—77].
3.2.1. Основные закономерности термической
деструкции целлюлозы
Процесс карбонизации целлюлозы целесообразно подразделить на
две стадии: пиролиз целлюлозы, сопровождающийся наиболее быстрым
течением химических процессов и потерей массы материала, и собствен-
но карбонизация, завершающаяся получением УВМ с содержанием
углерода до 95%.
Термическую деструкцию целлюлозы начали изучать более 70 лет
тому назад. Успешные работы по приданию негорючести целлюлозным
материалам и особенно получение УВМ послужили дополнительным
стимулом для проведения более широких исследований в этой области.
Однако, несмотря на огромное число работ, механизм термической дест-
рукции целлюлозы остается невыясненным. Ниже рассматриваются ос-
новные закономерности этого сложного процесса.
276
Потеря основной массы полимера происходит в узкой области
температур, в пределах 200—350°C (рис. 3.16). К основным продуктам
термического распада целлюлозы относятся низкомолекулярные летучие
соединения, смолы и коксовый остаток — углеродное волокно. В лету-
чих продуктах обнаружено большое число соединений, среди которых
идентифицированы: НгО, СО, СОг, СН4, С2Н4, ацетон, глиоксаль, гли-
кольальдегид, 5-(гидроксиметил)-фурфурол, формальдегид, акролеин,
метилэтилкетон и др. [78, 79]. Дифференциальная кривая общего выде-
Рис. 3.16. Дифференциальные и интегральные кривые исходного ГЦ-В (/) и волокна,
содержащего NH4C1 (2).
Рис. 3.17. Дифференциальная кривая выделения газов при пиролизе целлюлозы.
ления газов [80] приведена на рис. 3.17. Максимальная скорость про-
цесса соответствует ТТО около 350 °C.
К нежелательным продуктам распада относятся смолы, в результа-
те разложения которых образуется аморфный углерод, осаждающийся
на поверхности УВ и ухудшающий его свойства.
Уже давно [81] в продуктах термического распада целлюлозы об-
наружен левоглюкозан, представляющий собой видоизмененное элемен-
тарное звено целлюлозы. О. П. Голова и сотр. [82] устайовили, что вы-
ход левоглюкозана из хлопковой целлюлозы составляет 47%, а из гид-
ратцеллюлозы— не превышает 9,3%. Наиболее достоверное объясне-
ние этого явления дано в работе [83]: широкий набор комформеров
группы СНгОН (по данным ИК-спектроскопии) обусловливает высо-
кий выход левоглюкозана из хлопковой целлюлозы.
Существенное влияние на термический распад целлюлозы оказыва-
ют антипирены, специфика действия которых сводится к следующему.
Антипирены увеличивают [84] выход кокса и газообразных продуктов
(рис. 3.16) и значительно уменьшают выход левоглюкозана и смол
(табл. 3.2). В присутствии антипиренов разложение целлюлозы начи-
нается при более низкой температуре и распространяется на более ши-
рокую область температур. Максимальная скорость потери массы цел-
люлозы, обработанной антипиренами, уменьшается и сдвигается в об-
ласть более низких температур, но суммарная скорость деструкции воз-
растает. Эффективное действие проявляется при небольшом содержании
(1—2%) антипирена в волокне. Механизм действия антипирена заклю-
чается в том, что в его присутствии на первых стадиях термообработки
более интенсивно протекает дегидратация целлюлозы, вследствие чего
277
Таблица 3.2. Влияние содержания смеси В+ВА* на выход (%) продуктов
пиролиза хлопковой целлюлозы [79] (температура 420 °C, продолжительность
пиролиза 6 мин)
Содержание смеси В+ВА, % от массы целлюлозы Смолы Углеродный остаток Левоглюкозан 5-Гидроксиме- тил фурфурол 1,6-Ангидро- глюкофура- ноза
метод ИК-спек- троскопии хроматографи- ческий метод
0 68 8,1 36 36 2,1 1,5
1,05 36 26 11 13 2,0 1,2
2,1 25 — 5,3 5,1 1,5 0,5
4,2 12 40 1,0 0,6 0,5 0
8,4 , 5,1 — 0 0,05 0,005 0
12,4 3,8 47 0 0 — —•
* В+ВА— смесь буры и борной кислоты в соотношении 7:3.
подавляются реакции, приводящие к образованию смолообразных про-
дуктов, и прежде всего отщепление левоглюкозана. Напримерч дегидра-
тация чистой целлюлозы происходит при температурах 240—260 9С, а
целлюлозы, содержащей NH4C1, — при 155—190 °C, т. е. дегидратация
сдвинута в область более низких температур [84]. Аналогично антипире-
нам действуют кислоты и основания Льюиса.
Для изучения физико-химических процессов, сопутствующих пиро-
лизу, широко используется метод ДТА и ТГА. На кривых ДТА отме-
чаются наиболее типичные области: эндотермический эффект при темпе-
ратуре примерно 120 °C, соответствующий десорбции физически связан-
ной воды, и при 150—200 °C — обусловленный дегидратацией, деполи-
меризацией и распадом связей. Глубокий эндотермический эффект при
280 °C связан с интенсивной термической деструкцией и потерей массы
полимера. При температурах 350—370 °C появляется четко выраженный
экзотермический эффект, связанный с вовлечением в реакцию продук-
тов распада (выделение СО2, СО, образование новых связей и др.).
В присутствии антипирена тепловые эффекты смещаются в область
более низких температур.
По кривым ТГА представляется возможным определить основные
константы (порядок реакции, энергию активации, предэкспоненциаль-
ный коэффициент) реакции пиролиза целлюлозы. Установлено, что пи-
ролиз целлюлозы протекает по двум механизмам: на начальных ста-
диях — по реакции псевдонулевого порядка, на более поздних — по
реакции псевдопервого порядка. Первая стадия характеризуется мень-
шим значением энергии активации (83,8—146,6 кДж/моль), чем вторая
(175,98—224,6 кДж/моль), и более глубоким термическим распадом.
Несмотря на одинаковый состав, целлюлоза в зависимости от пре-
дыстории имеет различные структуру, макро- и микроморфологию,
влияющие на ее поведение при пиролизе. Согласно Мадорскому [85,
с. 254], по скорости деструкции целлюлозные препараты располагаются
в следующий ряд: вискозное волокно > волокно фортизан > хлопок >
>гидратцеллюлоза. Филлипом и сотр. [86] найдена следующая после-
довательность изменения скоростей реакции: вискозный корд > тек-
стильная нить > хлопок. Монокарбоксилцеллюлоза (МКП) менее тер-
мостабильна [87] по сравнению с целлюлозой: зона термического рас-
пада целлюлозы находится в пределах 250—370 °C, а зона МКП, содер-
жащей 15,2% групп СООН, сдвинута в область 170—350 °C. Для МКП
богаче набор пиков эндо- и экзотермических эффектов. Замена СН2ОН
278
3.18). Появление
280 320 360
t,9C
Рис. 3.18. Зависимость
интенсивности им-
пульсов 1т от темпе-
ратуры пиролиза для
волокон, находивших-
ся в разном исходном
состоянии:
1 — сухое свободное; 2 —
влажное свободное; 3 —
сухое фиксированное;
4 — влажное фиксирован-
ное.
целлюлозы на группу СООН исключает возможность ее деполимериза-
ции с образованием лекоглюкозана.
Из-за гетерогенности целлюлозы пиролиз может протекать на раз-
личных структурных уровнях, что наглядно показано в работах [88, 89].
Для наблюдения за пиролизом авторы применяли методы МУР и обыч-
ного рентгеноструктурного анализа. Установлено, что при достижении
определенной температуры появляются рефлексы МУР, интенсивность
которых вначале возрастает, а затем падает (рис.
рефлексов МУР свидетельствует о наличии в поли-
мере фракции с различной электронной плотностью.
По мнению авторов, на начальной стадии происхо-
дит пиролиз в аморфных областях, а кристалли-
ческие фракции служат сдерживающим каркасом,
или матрицей. Дополнительным подтверждением
подобной схемы служит упорядочение структуры,
сохранение больших периодов и доля кристалличе-
ской фракции на начальных стадиях пиролиза. На
второй стадии (выше 320°C) разрушается кристал-
лическая фракция, и функции каркаса начинает вы-
полнять частично пиролизованная аморфная фрак-
ция. Снижение интенсивности рефлексов МУР
является следствием разрушения кристаллической
фракции. Действительно, согласно данным многих
авторов, кристаллическая структура целлюлозы
исчезает при 280—300 °C.
Интересные данные приведены в работах '[80,
83]. По информации, полученной с помощью метода
спектроскопии для природной целлюлозы (хлопко-
вая, древесная) и вискозной текстильной нити,
начало структурной разупорядоченности предшест-
вует химическим превращениям, оно значительно
удалено от активной стадии термического распада;
в случае кордной нити химические превращения
начинаются раньше, чем структурные изменения.
Это дает основание полагать, что более стабильная
структура кордной нити служит матрицей и предопределяет более со-
вершенную структурную организацию полученного из нее УВМ.
Пиролиз целлюлозы сопровождается сложными физико-химиче-
скими превращениями, протекающими по различным механизмам. На
ранних стадиях термообработки главную роль играют гетеролитические
реакции. С повышением температуры все больший вклад начинают вно-
сить гомолитические реакции. Целлюлоза представляет собой гетеро-
циклический полимер, обогащенный относительно лабильными к тепло-
вому воздействию гидроксильными группами; это дополнительно услож-
няет изучение ее распада. В результате многочисленных исследований
накоплен обширный экспериментальный материал, установлен ряд бес-
спорных фактов, однако их трактовка в большинстве случаев затрудне-
на, а в связи со сложностью и многообразием протекающих процессов
особенно большие трудности возникают при попытках представить тер-
мическую деструкцию целлюлозы в виде конкретных уравнений.
Пиролизу обычно подвергается целлюлоза, содержащая сорбиро-
ванную влагу. Уже при температурах около 120 °C в незначительных
количествах образуются кислые продукты распада, являющиеся ката-
279
лизаторами гидролиза целлюлозы. Поэтому на начальной стадии термо-
обработки при относительно низких температурах происходит гидролиз
целлюлозы, ведущий к снижению СП и изменению свойств целлюлозы,
зависящих от СП. Процесс гидролиза не сопровождается потерей мас-
сы. По указанным причинам собственно термодеструкции подвергается
не исходная, а гидролизованная целлюлоза.
Другой важнейшей реакцией является дегидратация, которая на-
чинается при температурах около 200—220 °C. Именно эта реакция во
многом предопределяет возможность получения УВ из целлюлозы. Де-
гидратация может протекать по меньшей мере по трем реакциям: внут-
римолекулярная реакция (см. схему XI); внутримолекулярная реакция
нуклеофильного замещения (см. схему XII); межмолекулярная реакция
(см. схему XIII). По схеме XI отщепляется вода и появляется в звене
двойная связь; по схеме XII образуется внутрициклическая эфирная
связь, а по схеме XIII — межмолекулярная эфирная связь. Благодаря
дегидратации, с одной стороны, повышается термостойкость промежу-
точных продуктов распада и тем самым сохраняется форма волокна при
более высоких температурах термообработки и, с другой, — что не ме-
нее важно, подавляется реакция деполимеризации. В результате дегид-
ратации образуется промежуточный продукт — дегидроцеллюлоза.
СНоОН
н он
СН2ОН
XIII
>
280
230 270 ЗЮ 350
t, °C
Рис. 3.19. Изменение интенсив-
ности полос поглощения (часто-
ты указаны в скобках), отно-
сящихся к валентным колеба-
ниям:
С=О (1720 см-') (Ц; С=С
(1620 СМ-’) (2); С—Н (2920 см-')
(3); О—Н (3410 см-1) (4); высоко-
упорядоченные молекулярные труп-
пировки целлюлозы (1375 см-1) (5)-
С помощью метода ИК-спектроскопии многими авторами подтверж-
дено течение реакции дегидратации i[l, с. 74—93]. На рис. 3.19 показа-
но изменение интенсивности полосы поглощения групп ОН (3410 см-1)
и других групп в зависимости от ТТО; видно резкое уменьшение содер-
жания гидроксильных групп в пределах 200—300 °C [90, 91]. Одновре-
менно в полимере происходит накопление групп СО, связей С=С и др.
Деполимеризация с выделением левоглюкозана начинается вы-
ше 250—280 °C и распространяется в область более высоких температур.
В реакции участвует часть целлюлозы, не
подвергнутая дегидратации.
Деполимеризация и дегидратация явля-
ются конкурирующими реакциями. Строго
их разделить невозможно, и в определенной
области температур протекают обе реакции,
но дегидратация начинается при более
низких температурах, и тем самым созда-
ются благоприятные условия для получе-
ния УВМ. Если начальную стадию пиролиза
проводить при низких температурах, соот-
ветствующих дегидратации (для этого по-
требуется длительное время), то значитель-
но повышается выход углерода [92]. Пред-
ложено большое число схем перехода от
целлюлозного к углеродному волокну, ра-
зобранных нами ранее [1, с. 74—93]. Ниже
(см. с. 282) приводится, на наш взгляд, бо-
лее правдоподобная «фурфурольная» схе-
ма распада целлюлозы, предложенная Шин-
до [93].
Приведенными примерами не исчерпывается большое многообразие
химических процессов, протекающих при пиролизе целлюлозы.
3.2.2 Основные закономерности
и условия проведения карбонизации
Первая стадия термических превращений (пиролиз) заканчивается
при ТТО 300—350 °C, при этом получается волокно, содержащее всего
лишь 60—70% углерода. На второй стадии (карбонизации) протекают
дальнейшие химические процессы и структурные преобразования, при-
водящие в итоге к получению УВМ. Как видно из приведенных ниже
данных, с повышением ТТО волокно обогащается углеродом и
происходит уменьшение числа других, унаследованных от целлюлозы
атомов:
Температура, °C . . . 370 420 550 650 750 850
Состав, °/0 углерод . . . 84,8 88,9 91,8 93,8 95,4 95,8
водород . . . 4,55 3,69 3,01 2,27 1,40 0,84
кислород ности) . (по раз- . . 10,65 7,41 5,19 3,93 3,20 3,36
О составе летучих продуктов, выделяющихся на стадии карбониза-
ции, в литературе приводится мало сведений. Содержание смолообраз-
ных веществ в продуктах термического распада невелико. Преимущест-
венно отщепляются низкомолекулярные соединения: Н2О, СО, СОг,
карбонилсодержащие соединения, насыщенные и ненасыщенные углево-
дороды. Выход различных летучих продуктов в зависимости от ВТО по-
281
казан на рис. 3.20. В результате реакций диспропорционирования воз-
никают карбоксильные и карбонильные группы, которые являются ис-
точником образования СО2 и СО.
При этом условия нагревания влияют на кинетику выделения га-
зов, а наличие экстремальных точек свидетельствует о разном механиз-
ме реакций, ведущих к отщеплению СО и СО2. Водород и углеводороды
интенсивно начинают выделяться при ТТО > 400 °C.
По мере повышения температуры, несмотря на упрощение элемент-
ного состава, строение остатка усложняется. Схему перехода от целлю-
лозы к углероду представить трудно, так как целлюлоза является гете-
роциклическим полимером, и поэтому переходу предшествует полный
распад макромолекулы и элементарных звеньев (отщепление кислоро-
да). Видимо, в этом процессе важную роль играют матричные эффекты.
Некоторые схемы превращения целлюлозного волокна в углеродное рас-
смотрены в монографии [1, с. 96—102].
В общем виде термическая деструкция целлюлозы сопровождается
двумя группами реакций, а именно: деструкцией полимера и промежу-
точных продуктов и синтезом (конденсационные процессы), приводящим
к образованию новых типов связей С—С. В отличие от целлюлозы,
имеющей лишь один тип связей С—С ($р3-гибридизация), в промежу-
точных продуктах и карбонизованном УВ присутствуют самые разнооб-
282
разные формы углерода: насыщенные и частично дегидрированные цик-
лы, ароматические структуры, линейные формы углерода с ординарны-
ми, двойными и тройными связями, участками, обрамленными радика-
лами, содержащими другие атомы. Если скорость реакции деструкции
превышает скорость реакции синтеза, то выход углерода снижается.
И наоборот, более интенсивное протекание конденсационных процессов
благоприятно сказывается на выходе углерода.
Сведения о ВТО, соответствующей началу образования турбострат-
ных пакетов, довольно противоречивы: по одним источникам, они обна-
руживаются уже при 240—270 °C, что мало вероятно, по другим — при
температуре выше 900 °C. что более правдоподобно {94].
Существенное значение
имеют радикальные процес-
сы [95], что подтверждается
данными, приведенными на
рис. 3.21. Характерно, что
по сравнению с температу-
рой, при которой происходит
основная потеря массы, мак-
симальная концентрация
ПМЦ сдвинута в область
более высоких ТТО. Неясна
причина снижения концент-
рации ПМЦ на стадии, ког-
да еще протекают химиче-
ские процессы (см. гл. 2).
Во 'время карбонизации
изменяются физико-химиче-
ские и механические свойст-
Рис.
цу<КТОВ
нити от температуры и режима нагревания:
а — импульсный нагрев; б — ступенчатое нагревание (про-
бы на анализ отбирались через 1 ч).
3.20. Зависимость выхода газообразных про-
*ro<TYMiwtJ£w>vn>T‘n па/чпатто глиттили
* д VV11W1 V» ip WU
ва волокна: одни скачкообразно, другие монотонно во всей области ВТО.
В начале пиролиза, при котором преобладают процессы деструкции, о и
Е уменьшаются, достигая минимального значения примерно при 400—
500°C, что соответствует области максимальной концентрации ПМЦ
(см. рис. 3.21). Последующее возрастание этих величин служит убеди-
тельным подтверждением возникновения и совершенствования структу-
ры углеродного волокна. Резкое падение электрического сопротивления
служит дополнительным веским доказательством интенсивно протекаю-
щей ароматизации и образования базисных плоскостей, так как благо-
даря большой концентрации в них л-сопряжений изменяются электро-
физические свойства УВ.
Одним из ярких показателей ароматизации углерода служит стой-
кость волокна к окислению на разных стадиях ВТО. Как видно из
рис. 3.21, она резко возрастает при ТТО выше 500 °C, что хорошо согла-
суется с изменением механических, электрофизических и парамагнит-
ных свойств волокна. Теплопроводность при переходе от ГЦ-волокна
к углеродному резко возрастает при температуре выше 600 °C. Тепло-
емкость на первых этапах снижается, а затем мало изменяется по мере
увеличения ТТО (рис. 3.22).
К основным условиям карбонизации относятся среда, катализато-
ры, температурно-временные режимы, вытягивание.
В качестве защитной среды используют инертные газы (азот, аргон,
гелий), газы, обладающие восстановительными свойствами, природный
газ, продукты термического распада целлюлозы, угольные засыпки, глу-
283
бокий вакуум. По данным работ [90, 91] и др., проведение процесса
в вакууме нецелесообразно из-за технических сложностей; к тому же
в этих условиях уменьшается выход УВ. В среде аргона выход увели-
чивается вследствие более интенсивного протекания дегидратации. При-
чины влияния аргона (нейтральная среда) на реакцию дегидратации
неясны. Видимо, практическое применение находят экономически оправ-
данные защитные среды, такие, как азот и природный газ.
В процессе карбонизации в реакционную систему вводят добавки,
которые подразделяются на две группы: катализаторы и вещества, мо-
дифицирующие УВ. При производстве УВМ в качестве катализаторов
Рис. 3.21. Влияние ТТО на изменение прочности (/), концентрации ПМЦ (2), стойкости
к окислению [кислород воздуха, температура 400 °C, продоЛ|Ж1ителыю1сть 30 мин] (3)
и электрического сопротивления (4) ГЦ-В.
Рис. 3.22. Влияние ТТО иа коэффициент тегмопрсхводиости i(/) и теплоемкость 1(2)
УВ-ГЦ.
используются антипирены (бура, борная кислота, диаммонийфосфат,
хлорид аммония и др.) [96—98]. Рекомендуются соли сильных кислот и
аммониевого основания — сульфат аммония, хлорид аммония и др.
[77, 99]; галогениды переходных металлов периодической системы и
парообразные соединения железа и др. Перечисленные соединения спо-
собствуют повышению выхода УВ и сокращению длительности процесса.
Для изменения теплофизических, электрофизических и других
свойств УВ в систему вводят тугоплавкие и термостойкие соединения
(соли и оксиды гафния, бора, свинца, магния, кадмия, ванадия, цирко-
ния, кремния и др.) [100].
Из рассмотренных закономерностей пиролиза целлюлозы со всей
очевидностью вытекает, что температурно-временные режимы оказыва-
ют влияние на выход, структуру и свойства УВМ. Нижний предел тем-
пературы карбонизации составляет 900—1000 °C, границу между верх-
ним пределом и графитацией установить трудно. Чаще всего приводятся
температуры 1300—1500 °C.
Одна из характерных особенностей получения УВ-ГЦ состоит
в длительности процесса, что является недостатком применения ГЦ-во-
локон как исходного сырья. Особенно медленно протекает первая ста-
дия — пиролиз. По различным, преимущественно патентным источни-
кам [75, 101], продолжительность карбонизации составляет 10—250 ч.
Предпринимаются попытки сократить продолжительность процесса. По
данным Стронга [102] предварительное окисление ГЦ-волокон позволя-
284
ет сократить продолжительность процесса и получить УВ с высокими
показателями (0=1,79 ГПа, £==374 ГПа). В работе В. О. Горбачевой
с corp. [403] установлено влияние скорости нагревания на последова-
тельность химических реакций. С повышением скорости нагревания воз-
растают эндотермические пики, интенсивность потери массы; эндотерми-
ческие пики смещаются в область повышенных температур, замедляется
выделение газа, т. е. полимер становится как бы более термостойким и
лучше сохраняется его структура. По данным работы [104], при мед-
ленном (ступенчатом) повышении температуры по сравнению с быстрым
(импульсным) больше выход кокса и газообразных продуктов (см.
рис. 3.20) и меньше выход смол. Более благоприятные условия при мед-
ленном подъеме температуры обусловлены большим влиянием конден-
сационных процессов по сравнению с деструкцией полимера.
При карбонизации в свободном состоянии происходит усадка во-
локна в продольном направлении на 20—40%, совпадающая по темпе-
ратуре с максимальной потерей массы полимера. Из-за усадки происхо-
дит разориентация структуры, поэтому получаются УВ с невысокими
механическими свойствами (о=300—500 МПа). Только благодаря при-
менению вытягивания удалось из ГЦ-волокна получить УВ высокого ка-
чества.
Как показали М. В. Шаблыгин и сотр. [105; 106], вытягивание
(напряжение) влияет на кинетику термического распада, а именно ста-
дия глубоких химических преобразований протекает в более узкой
области, максимальная скорость термического распада сдвинута в об-
ласть более низких температур, ИК-спектры подтверждают более ин-
тенсивное протекание дегидратации полимера.
3.2.3. Основные закономерности процесса графитации
углеродного волокна
Графитация — завершающая стадия технологического процесса, на
которой углеродное (карбонизованное) волокно подвергается высоко-
те^мпературной обработке при 1800—2500 °C. Графитация является энер-
гоемким и сложным процессом, удорожающим волокно, поэтому в зави-
симости от требований к материалам и областей его применения
конечным продуктом могут быть углеродное и графитовое волокна.
При графитации главным образом протекают структурные превра-
щения и соответственно изменяются свойства материала. На этой ста-
дии происходит обогащение волокна углеродом до содержания его не
менее 99%; потеря массы волокна составляет 5—15%, и если волокно
находится в свободном состоянии, то оно усаживается на 3—6%. Види-
мо, основными продуктами распада являются углеводороды и СО.
В процессе графитации происходят дальнейшая ароматизация углерода
и совершенствование структуры (см. гл. 1). Глубина протекания этих
процессов и изменение свойств волокна зависят от параметров графита-
ции. К важнейшим из них относятся: среда, температура, продолжи-
тельность и вытягивание.
В условиях высоких температур защитной средой может служить
азот или аргон. По экономическим соображениям, видимо, используется
азот с минимальным содержанием в нем кислорода.
Несмотря на большую жесткость системы, при столь высоких тем-
пературах (1800—2500 °C) физико-химические и структурные превра-
щения во время графитации завершаются за очень короткое время. По
285
данным Гибсона и Ланглойса [107J, графитация заканчивается за не-
сколько секунд, поэтому ее продолжительность определяется техниче-
скими возможностями оборудования.
На первом этапе развития производства карбонизация и графита-
ция проводились в свободном состоянии, и волокно значительно усажи-
валось. В этих условиях получается волокно несовершенной структуры,
приближающееся к стеклоуглероду, с низкими механическими свойст-
вами (прочность 700—800 МПа, модуль Юнга 50—60 ГПа). Следую-
щим этапом явилось применение вытягивания при получении УВ-ГЦ,
что привело к совершенствованию структуры и значительному улучше-
ние. 3.23. Зависимость модуля Юига от полуширины угла отражения от плоскости 002.
Рис. 3.24. Влияние степени вытягивания при графитации на .модуль Юнга графитиро-
ваиного ГЦ-В (температура графитации 2780—2870 X, продолжительность 0,2—0,27 с).
Рис. 3.25. Влияние температуры графитации на модуль Юнга графитированного ГЦ-В
(степень вытягивания 40%, продолжительность графитации 0,25 с).
нию механических свойств волокна. Анализ литературных данных по-
казывает, что для получения волокна высокого качества вытягивание
необходимо проводить на стадии карбонизации и графитации.
Бэкон и Шаламон [108] изучали рентгеновским методом ориента-
цию волокна, которая оценивалась по интенсивности отражения от пло-
скости 002. В результате вытягивания этот показатель уменьшается, что
свидетельствует об улучшении ориентации волокна. Связь между сте-
пенью ориентации и модулем Юнга наглядно иллюстрируется рис. 3.23.
Прямая связь между степенью вытягивания и модулем Юнга установ-
лена в цитируемой выше работе [107] (рис. 3.24). Изекиль и Спейн
[109] также исследовали влияние вытягивания на свойства графитиро-
ванного волокна. Установлено, что по мере возрастания степени вытяги-
вания увеличивается модуль Юнга и уменьшается диаметр волокна. За-
висимость между степенью вытягивания и прочностью менее четкая:
как правило, по мере вытягивания прочность возрастает, но в отдель-
ных случаях эта закономерность не соблюдается. Последнее связано
с большой неравномерностью волокна.
При графитации с одновременным вытягиванием четко проявляется
роль температуры: с ее повышением модуль Юнга возрастает
(рис. 3.25). В отличие от данных, приведенных в работе [109], в других
источниках указывается на одновременное возрастание модуля Юнга
и прочности волокна.
Осуществление вытягивания на обеих стадиях термообработки поз-
волило получить УВ-ГЦ (волокно торнель 50) с прочностью около 2 ГПа
286
н модулем Юнга 350 ГПа, а в лабораторных условиях (торнель 100)
с прочностью 3,5 ГПа и модулем Юнга 703 ГПа [ПО].
В заключение следует отметить, что УВ-ГЦ с высокими механиче-
скими показателями получают при проведении графитации с вытяжкой
в условиях очень высоких температур (не менее 2800 °C). Практически
это осуществить довольно трудно. Тот же эффект, но при более низких
температурах получается, если в качестве предматериала использовать
ПАН-В. Поэтому целесообразнее при производстве высокопрочного
высокомодульного волокна применять ПАН-В.
3.3. ПОЛУЧЕНИЕ УГЛЕРОДНЫХ ВОЛОКНИСТЫХ МАТЕРИАЛОВ
НА ОСНОВЕ ПЕКОВ И ФЕНОЛЬНЫХ СМОЛ
3.3.1. Пеки
Работы японских исследователей [62] по получению углеродного
MP-волокна из ПВХ-пека послужили стимулом дальнейших исследова-
ний, особенно по изысканию более доступных источников сырья. Преж-
де всего привлекли внимание нефтяные и каменноугольные пеки. Пре-
имуществом пеков по сравнению с другими источниками сырья являет-
ся их доступность, низкая стоимость, высокое содержание углерода и
соответственно высокий выход УВМ. Перечисленные факторы предопре-
деляют благоприятные технико-экономические предпосылки производ-
ства и применения УВМ. Согласно [111], стоимость пека составляет
30 центов/фунт, а ГЦ-В и ПАН-В — 10—30 долларов/фунт. В зависимо-
сти от свойств стоимость УВЛ\ из псков колеблется в ппе^елах 10—
125 долларов/кг [112, 113], а из ГЦ-В и ПАНчВ— 50—250 долларов/кг.
Вероятно, большинство пеков содержит канцерогенные вещества,
что является их недостатком, поэтому в процессе получения УВМ из пе-
ков необходимо применять меры безопасности для защиты обслуживаю-
щего персонала.
Пеки в зависимости от происхождения подразделяются на природ-
ные (нефтяные, каменноугольные и др.) и синтетические, и по структуре
на обычные и мезофазные (жидкокристаллические).
Получение волокна из пеков состоит из следующих основных
стадий: подготовка пека, формование пекового волокна (ПВ), отверж-
дение ПВ и высокотемпературная обработка (карбонизация, графита-
ция).
Обыкновенные пеки. Пеки представляют собой сложную смесь орга-
нических соединений различного состава и разного строения. Подготов-
ка пеков для придания им требуемых свойств зависит от их происхож-
дения и сводится к отгонке низкомолекулярных фракций и дополнитель-
ной термообработке. Последняя проводится для повышения молекуляр-
ной массы пека; для интенсификации этого процесса рекомендуется
в пек добавлять соединения, легко распадающиеся с образованием сво-
бодных радикалов. По данным [62], пеки
дующим требованиям:
Исходный материал
Нефтяной пек .......................
Смесь асфальта с каменноугольной смолой
Каменноугольная смола...............
В литературе приводятся и другие требования к составу пеков. Важ-
ными, характеристиками пеков являются температура размягчения и
287
должны удовлетворять еле-
Содержание Отношение
углерода Н:С
86,1—86,7 1,06—1,09
88,7—89,0 0,75—0,81
92,4—92,7 0,47—0,49
средняя молекулярная масса (400—2000). Эти показатели определяют
условия формования и свойства ПВ [1, с. 235—243].
Формование волокна из пеков (смеси олигомерных продуктов)
является сложной стадией технологического процесса. Обычно эти во-
локна формуются при температуре расплава 100—350 °C со скоростями
300—800 м/мин. Особенность процесса состоит в том, что применяются
фильеры с большим диаметром отверстия (около 0,3 мм), поэтому для
достижения нужного диаметра ПВ и соответственно УВ формование
необходимо осуществлять при очень больших фильерных вытяжках
[1114]. При вытяжках (100 000—500 000%) достигается высокая
ориентация ПВ, сохраняющаяся в УВ-П, модуль которого составляет
140 ГПа [115].
Для придания неплавкости ПВ подвергается окислению. Опробова-
но большое число жидких и газообразных окислителей [116]. Наиболее
приемлемой оказалась двухстадийная обработка волокна: вначале озо-
нированным воздухом (70 °C, 3 ч), затем воздухом (260 °C, 1—3 ч).
Карбонизация заканчивается при температуре 800—1200 °C и про-
водится со скоростью подъема температуры 5—10°С/мин. Графитация
осуществляется в условиях, описанных в предыдущих разделах этой
главы.
Фирма «Kureha Carbon Fibre» («Куреха карбон файбер») [Япония]
организовала производство УВМ на основе нефтяного пека, полученно-
го при высокотемпературном крекинге нефти. В результате слияния
фирм «Kureha Chemical Industry» («Куреха кемикал индастри») и
«Toyabo Spenning Со.» («Тойобо спиннинг компани») образовалась
фирма «Taiyo Какеп» («Тайё какен»), которая производит УВМ двух
типов: KCF-100 и KCF-200 [117]. Свойства этих УВМ приведены ниже:
Диаметр, мкм....................................
Прочность, 107 Па...............................
Модуль Юнга, 1010 Па............................
Относительное удлинение, %......................
Содержание углерода, %..........................
Плотность, 103 кг/м3............................
Влагопоглощение, %..............................
Порог окисления, °C.............................
KCF-100 KCF-200
10—12 10—12
70—100 70—100
4—5 4—5
1,5—2,5 1,5—2,5
95—98 99
1.6—1,7 1,5
10 2
310 390
УВ-П представляет собой типичный стеклоуглерод.
Способы упрочнения, применяемые для УВ-ПАН и УВ-ГЦ, были
использованы для улучшения механических свойств УВ-П. Применение
вытягивания на стадии карбонизации, и особенно графитации способст-
вует совершенствованию структуры (рост размеров кристаллитов, улуч-
шение ориентации, уменьшение dQQ2) и значительному повышению проч-
ности и модуля Юнга волокна [118]. По данным [119], из нефтяного
пека получено волокно с прочностью 2,6 ГПа и модулем Юнга до
630 ГПа. Однако столь высокие показатели достигаются при высоких
температурах обработки, что практически мало приемлемо.
Мезофазные пеки. Получение мезофазных пеков знаменует собой
новый этап в развитии производства УВМ на основе доступного сырья.
Этот способ впервые разработан совместными усилиями фирм «Union
Carbide Corp.» («Юнион карбайд ко*рпорейшен») [США] и «Taiyo
Kaken Со.» («Тайё какен компани») [Япония].
В США созданы производства по выпуску 40 т/г волокна под назва-
нием «торнель-Р» [ИЗ]. Особенность метода состоит в том, что благо-
даря склонности мезофазных пеков к упорядочению представляется воз-
288
можность получать УВМ с высокой ориентацией и высокими механиче-
скими свойствами без применения вытягивания при карбонизации и гра-
фитации. Мезофазные пеки являются серьезными конкурентами УВМ,
получаемых с использованием в качестве предматериала ГЦ-В и
ПАН-В. К 1980 г. предполагается выпускать волокно торнель-Р стои-
мостью 12,5—25 долларов/кг (в 1975 г. стоимость волокна составляла
30—50 долларов/фунт, или 75—125 долларов/кг). К этому времени стои-
мость УВМ-ПАН будет составлять 56 долларов/кг [113, 120].
Мезофазные пеки получаются из натуральных и синтетических пе-
ков. Указывается на возможность использования нефтяных, каменно-
угольных [68] и нафтеновых пеков, способных давать мезофазные
структуры [121]. Следовательно, в качестве исходного сырья могут при-
меняться только пеки определенных типов [123]. Согласно [122], пеки
должны содержать органические молекулы планарного строения, вклю-
чающие не менее семи ароматических циклов. По химическому составу
к пекам предъявляются следующие требования (%) [121]:
Углерод..............................92—96
Водород..............................4—8
Кислород, сера, азот, не более ... 4
Фракция, нерастворимая в пиридине . 2
Для нефтяного пека допускается более низкое содержание углерода
(около 86%) [124]. Видимо, наиболее приемлемыми являются нефтя-
ные пеки.
Для получения мезофазного пека обычный пек подвергается термо-
обработке. Содержание мезофазы зависит от температуры и продолжи-
тельности этого процесса. Во время термообработки образуются мезо-
сферы, которые наблюдаются в поляризованном свете [125]. В литера-
туре, преимущественно в патентах, приводятся различные режимы тер-
мообработки. Согласно [126], термообработка нефтяного пека прово-
дится при 400 °C в течение 24 ч в атмосфере азота, при этом содержание
мезофазы достигает 73%; при сокращении продолжительности термо-
обработки до 14 ч содержание мезофазы снижается до 52%. Примерно
такие же условия приводятся в патенте [127]. Возрастание вязкости
и наличие неплавких и нерастворимых фракций затрудняет образование
мезофазы [128—130].
Мезофазные пеки являются двухфазными системами, состоящими
из жидкокристаллической и аморфной фракций. Такие системы обла-
дают своеобразными реологическими свойствами, зависящими от соот-
ношения фаз, молекулярной массы и ММР фракций и других факторов.
Согласно [123], они могут обладать пластичными и тиксотропными
свойствами. В последнем случае затрудняется или исключается возмож-
ность формования ПВ. Высокое содержание мезофазы затрудняет фор-
мование, но дальнейшая переработка ПВ облегчается. По одним источ-
никам, содержание мезофазы в пеке должно составлять 40—90%, по
другим — 55—65%; вязкость расплава пека 2—20 Па-с при температуре
250—450 °C.
Для получения синтетических пеков рекомендуется применять аро-
матические углеводороды, склонные к образованию планарных продук-
тов. Добавка кислородсодержащих гетероциклов, в частности ангидри-
дов, способствует образованию мезофаз [131—134]. Влияние планарно-
сти промежуточных радикалов [131] на склонность пеков к графитации
наглядно иллюстрируется приведенными ниже данными:
19—2254
289
Исходное соединение Промежуточный радикал Конформация радикала Межплоскост- ное расстояние». о ^002- А
Ацетонафтилеи . . Фсналенил Планарная 3,356
Поливинилхлорид — — 3,357
9,9'-Биантрил . . Антрил Планарная 3,357
9,9'-Бифлуореи . . Флуоренил Непланариая 3,374
9-Хлор-9-фенилфлуорен . . . . . . 9-Фенилфлуо- ренил > 3,401
9-Хлор-9-метнлфлуорен .... . . 1,3-Бисдифенил- еналлнл > 3,438
Связь между строением исходных продуктов и их способностью да-
вать графитирующийся кокс рассмотрена в гл. 2. Описано получение
синтетических пеков из хризена, фенантрена, перилена, бензопирена,
изовиолантрена и других соединений в присутствии 1—10% кислот
Льюиса в качестве катализатора [122, 135].
Формование волокна проводится при 340—380 °C (вязкость 3—
6 Па- с) [135]. Скорость формования, по разным источникам, 30—
900 м/мин. Уже во время формования под влиянием возникающих на-
пряжений осуществляется высокая ориентация пекового волокна, в ко-
тором просматриваются фибриллярные образования [129, 136]. При
карбонизации до 1000 °C наблюдается дезориентация, но выше этой
температуры, и особенно выше 2500 °C, резко возрастает ориентация и
увеличиваются размеры надмолекулярных структур [136, 137].
Подготовка волокна зависит от содержания в нем мезофазы. При
содержании мезофазы менее 85% волокно подвергается отверждению
(обработка воздухом при 250—400 °C в течение 4—50 мин) [121, 125]
или предварительно экстрагируется аморфная фракция с последующим
отверждением волокна. При содержании мезофазы больше 85% отверж-
дение исключается из технологического цикла и ПВ непосредственно
подвергается карбонизации.
ПВ имеет достаточно высокое относительное удлинение (около
5%), поэтому его можно подвергнуть текстильной переработке с после-
дующей карбонизацией, что позволяет получать высококачественные
ткани [138].
Карбонизация проводится при 1000—2000 °C в течение 5—60 мин,
графитация — при 2800—3000 °C. Основная потеря массы ПВ в отличие
от большинства полимеров происходит в довольно широкой области
температур (400—700°C). Выход углеродного волокна при ТТО 1000°C
составляет 87%. Предварительная окислительная обработка ПВ благо-
приятно сказывается на выходе УВ. Графитированное волокно, получен-
ное без вытягивания, имеет высокую ориентацию (<р/2«5°), большие
размеры кристаллитов, малое значение ^оог, высокую плотность; отме-
чается наличие трехмерных элементов структуры в волокне [121,
125, 136].
Свойства ПВ в зависимости от ТТО приведены ниже [128]:
1000 °C 1500 °C 2000 °C 2800 °C
Прочность, 107 Па 70 100—115 — 175-256-
Модуль Юнга, 10ю Па 14 17—24 — 53—85
Плотность, 103 кг/м3 Удельное электрическое сопротив- 1,25—1,40 2,1—2,2 — 2,1—2,2
ление, 10-в Ом м —- 8—12 — 1,5—2
La, А 25—60 — 80—100 >1000
doo2, А 3,45—3,55 3,40—3,43 —- 3,36—3,37
<р/2, град 30—40 — 10-20 5—10
290
По данным [125], высокопрочные ПВ имеют прочность 2,18—
2,8 ГПа и модуль Юнга 200—250 ГПа, а высокомодульные — прочность
1,75—2,5 ГПа и модуль Юнга 350—490 ГПа. Согласно [138], при гра-
фитации без натяжения получается ПВ с прочностью 1,4 ГПа и моду-
лем Юнга 350 ГПа при диаметре волокна 8 мкм.
В структурном отношении углеродные волокна из мезофазных пе-
ков представляют большой интерес, однако волокна с высокими механи-
ческими свойствами получаются только при очень высоких ТТО, что
практически трудно выполнимо. Приводимый многими авторами довод
о существенных преимуществах этого волокна перед другими в связи
с проведением графитации без вытягивания не состоятелен, так как
в инженерном плане операция вытяжки не вызывает затруднений. При
практически приемлемых ВТО получаются ПВ по свойствам, уступаю-
щие УВ-ПАН и находящиеся на одном уровне с УВ-ГЦ.
3.3.2. Фенольные смолы
Исходные смолы и требования к ним. Фенолоформальдегидные смо-
лы (ФФС) относятся к перспективным видам сырья для получения
УВМ. По этому вопросу, однако, опубликовано ограниченное число ра-
бот, но, видимо, проводятся интенсивные исследования. Получение УВМ
из фенольных смол включает три основных стадии: формование ФФС-В,
отверждение волокна и высокотемпературная обработка. Наиболее
сложными являются стадии формования и особенно отверждения.
Изучены новолачные, резольные, фуриловые смолы, смеси смол,
а также смеси смол с пеками. Наиболее приемлемыми оказались ново-
лачные смолы, получаемые из фенола и формальдегида в присутствии
кислых катализаторов [139]. Эти смолы обладают способностью пла-
виться, имеют линейную форму и, что наиболее важно, в них отсутству-
ют реакционноспособные группы, поэтому в расплавленном состоянии
не протекают процессы структурирования, благодаря чему не изменяет-
ся вязкость и облегчается формование волокна. Резольные смолы тер-
мореактивны, поэтому в процессе формования возможно увеличение
вязкости расплава, а следовательно, нарушение стабильности формова-
ния и изменение свойств волокна [140]. Кроме того, выход углерода из
резольных смол ниже (45 вместо 60%). Для синтеза ФФС рекомендует-
ся применять смесь фенола и гексаметилентетрамина (CH2)6N4. При
нагревании этой смеси до 100—150 °C гексаметилентетрамин разлагает-
ся с образованием формальдегида; при взаимодействии последнего
с фенолом образуется ФФС [141, 142]. Молекулярная масса ФФС (но-
волачных) составляет 500—1200 {140, 143]. При молекулярной массе
выше 1500 смола имеет высокую вязкость, поэтому в процессе формо-
вания ФФС-В необходимо повышать температуру, что нежелательно,
так как возможно структурирование смолы и нарушение стабильности
формования. При молекулярной массе ниже 500 возможно слипание
элементарных волоконец.
Формование и отверждение ФФС-волокна. ФФС, используемые для
получения ФФС-В, представляют собой олигомеры, и формование во-
локна из них представляет известные трудности. Обычно применяются
фильеры с большим диаметром отверстий (0,08—1,9 мм), и для по-
лучения ФФС-В и соответственно УВМ требуемого диаметра формова-
ние необходимо проводить с высокими фильерными вытяжками. По-
следние определяются прядомостью расплава, которая, в свою очередь,
19*
291
зависит от свойств смолы; рекомендуется применять фильеры с боль-
шим отношением l/d (где I — длина, d — диаметр отверстия фильеры).
Температура формования волокна из новолачных смол находится
в пределах 100—150°С (вязкость при этой температуре 20—50 па-с),
а для резольных смол, имеющих более низкую молекулярную массу,
она составляет 40—80 °C [140, 143—145]. Т. Н. Козиоровой и сотр.
[146] определена оптимальная температура формования, которая близ-
ка к температуре каплеобразования смолы. Авторами проведены си-
стематические исследования и рекомендуются следующие условия фор-
мования ФФС-В из смолы марки 121:
Температура, °C.............................136—138
Скорость, м/мин............................. 200—1 000
Диаметр отверстия фильеры, мм .... 0,05
Фильерная вытяжка, %......................... 13 000—17 000
Диаметр волокна, мкм........................ 15—40
Сформованное ФФС-В имеет низкие механические показатели, за-
трудняющие его дальнейшую переработку; к тому же оно плавко. Для
придания волокну требуемых свойств необходимо его отвердить; эту
операцию рекомендуется проводить в две стадии: обработка формаль-
Рис. 3.26. Изменение длины
нити (1) и потери массы (2)
при карбонизации ФФС-В,
дегидом в присутствии кислот в газообразной
или жидкой среде; термообработка вначале
в среде воздуха, насыщенного озоном, затем
в воздушной среде при повышенной темпера-
туре [146, 147]. В результате отверждения
прочность волокна возрастает с 35—80 МПа
до 140—180 МПа. Отверждение является наи-
более сложной стадией получения УВМ из
ФФС, определяющей возможность практичес-
кого использования ФФС в качестве предмате-
риала. В этом случае возникают трудности
при инженерном оформлении процесса от-
верждения.
Отверждение волокна формальдегидом
происходит постепенно от периферии к цент-
ру по мере диффузии реагента. При неполном
завершении процесса отверждения путем растворения сердцевины
можно получить полое ФФС-В и соответственно изготовить полое
УВМ [147].
Указывается, что при применении резольных смол или использо-
вании при синтезе ФФС гексаметилентетрамина вместо формальдеги-
да можно ограничиться отверждением путем термообработки волокна.
Представляет интерес введение в смолу циклических соединений
формальдегида, в частности триоксана [148]. Триоксан устойчив
в условиях формования волокна, но при нагревании выше температуры
формования разлагается с выделением формальдегида; в результате
взаимодействия последнего с новолачным олигомером в присутствии
кислых катализаторов происходит структурирование волокна и значи-
тельно упрощается стадия отверждения волокна. Помимо этого, триок-
сан служит пластификатором и облегчает формование волокна. Опти-
мальное содержание триоксана в полимере составляет 10% (масс.).
Высокотемпературная обработка ФФС-волокна. Высокотемператур-
ная обработка ФФС-В может проводиться в две стадии: карбонизация
(получение карбонизованного волокна) и графитация (получение гра-
292
фитированного волокна). Конечная температура карбонизации 800—
1100 °C. Подъем температуры осуществляется со скоростью 20—
100°С/ч. До 300 °C нагревание рекомендуется проводить медленно (со
скоростью 1 °С/мин), а затем быстро [142]. В процессе карбонизации
наблюдаются усадка и потеря массы волокна (рис. 3.26). Эти процессы
заканчиваются при температуре около 600 °C [149]. Выход карбонизо-
ванного волокна из новолачной смолы составляет 50—60%, а из ре-
зольной— около 45%. В отличие от многих полимеров потеря массы
ФФС происходит в широкой области температур (300—600°C), что
обусловлено структурированием полимера и повышением его термоста-
бильности. Термическая деструкция ФФС представляет сложный про-
Рис. 3.27. Влияние температуры карбонизации на прочность УВ. Условия
карбонизации: среда — азот; скорость подъема температуры —0,7 °С/мин;
выдержка при конечных температурах — 30 мин.
Рис. 3.28. Изменение размеров кристаллитов в зависимости от ТТО:
‘~La- 2~Lc-
цесс, сопровождающийся одновременно протеканием большого числа
реакций, выделением газообразных продуктов, смол и формированием
углеродного скелета (кокса). Тероном [150] среди продуктов распада
идентифицировано 30 соединений с температурой кипения, лежащей
в пределах 50—250 °C, в том числе фенол, о-, м- и га-крезол; 2,4- и
2,6-ксилол; 2,4,6,-триметилфенол, бензол; толуол, о- и л-ксилол и др.
Авторы работы [151] на основании собственных исследований и лите-
ратурных данных предложили следующую схему распада фенольных
смол (см. с. 294).
Ароматизация углерода начинается при ТТО выше 350 °C.
Характер изменения прочности волокна при карбонизации иллюст- •
рирует рис. 3.27, из которого видно, что до 900 °C прочность мало изме-
няется, в пределах 900—1000 °C возрастает, а затем уменьшается
[144]. В обычных условиях высокотемпературной обработки из ФФС
получаётся низкомодульное низкопрочное УВ (прочность не выше
500 МПа, модуль Юнга 20—30 ГПа). В результате вытягивания удалось
получить УВ с прочностью до 2,1 ГПа и модулем Юнга 170 ГПа. Вытя-
гивание необходимо проводить как на стадии карбонизации, так и на
стадии графитации, так как осуществление этой операции только на
одной из этих стадий малоэффективно [139, 141]. Сообщается [152]
о перспективности использования ФФС для получения УВМ с развитой
поверхностью. При ТТО 800—900 °C можно получить волокно с по-
верхностью 500—1000 м2/г; при дополнительной активации поверхность
волокна увеличивается до 2000—3000 м2/г. Представляет интерес полу-
чение полых УВМ.
293
Н20, со, сн4, н2
УВ-ФФС относятся к типичному стеклоуглероду. По мере повы-
шения ТТО уменьшается d002 и возрастают размеры кристаллитов
(рис. 3.28), особенно заметно выше 1500 °C, но по абсолютному значе-
нию они невелики и характерны для стеклоуглерода.
В заключение следует отметить, что по сравнению с обычными пе-
ками ФФС имеют следующие преимущества: синтез фенольных смол
проще предварительной подготовки пеков; ФФС стандартны по составу
и свойствам. Особого внимания заслуживает тот факт, что ФФС не об-
ладает канцерогенными свойствами. К недостаткам ФФС относится
сложность процесса отверждения ФФС-В, а также трудность получе-
ния из ФФС высокопрочных высокомодульных углеродных волокон.
ЛИТЕР АТ У РА
1. Конкин А. А. Углеродные и другие жаростойкие волокнистые материалы. М.,
«Химия», 1974. 375 с.
2. Авт. свид. СССР 138324 (1959); Изобр. Пром, образцы. Товарн. знаки, 1966,
№ 23, с. 173.
3. Яп. пат. 304892 (1962).
4. Фр. пат. 1430803 (1965); англ. пат. 1110791 (1968); Standage А. Е., Prescott R.,
«Nature», 1966, v. 211, р. 169.
5. Watt W., Phillips I. N„ Johnson W., «Engineer» (London), 1966, v. 221, p. 815.
6. Фр. заявки 2001496 (1971), 2059388 (1971), яп. пат. 22658 (1972).
7. Яп. пат. 28986 (1973).
8. Англ. пат. 1365165 (1974).
9. Волков Ю. В. и др., Хим. волокна, 1976, № 2, с. 47—49.
10. Волков Ю. В. и др. В кн.: Структура, свойства и применение углеродных во-
локнистых материалов. Мытищи, ВНИИВ, 1975, с. 24—29.
11. Горбачева В. О. и др., Хим. волокна, 1973, № 5, с. 16.
12. Яп. пат. 32146 (1972).
13. Moreton R. International Conference on Carbon Fibres, their Composites and Appli-
cations, London, 1971, paper № 12.
14. Фр. пат. 1502253 (1965).
15. Bailey J., Klarke A., Chem. Brit., 1970, v. 6, № 11, p. 484.
16. Толке A. M. и др., «Механика полимеров», 1974, № 4, с. 628—633.
17. Moreton R„ Watt W„ «Carbon», 1974, v. 12, № 5, p. 543—554.
18. Силиванова Л. Ф., Пакшвер Э. А., Полатовская P. А., Хим. волокна, 1974, № 5,
с. 47—50.
19. Houtz R. C., Text. Res. J., 1950, v. 20, p. 786.
20. Grassie N„ Neill T., J. Chem. Soc., 1956, p. 3929.
21. Grassie N., Novel N., J. Polymer. Sci., 1960, v. 48, № 150, p. 79.
22. Grassie N., Neill T., J. Polymer. Sci., 1959, v. 39, p. 211.
23. Grassie N., Hay N., Neill T„ J. Polymer. Sci., 1958, v. 31, p. 205.
24. Grassie N., Hay N„ J. Polymer. Sci., 1962, v. 56, p. 189.
25. Берлин А. А., Дубинская A. M., Московский Ю. С., Высокомол. соед., 1964,
т 6 1938
26. Fester W.', Mell. Textilber., 1966, Bd. 47, S. 673.
27. Conley R„ Bioron J., J. Appl. Polymer. Sci., 1963, v. 7, p. 1757.
28. Литовченко Г. Д. и др., Ж- прикл. спектроск., 1975, т. 23, № 2, с. 251—253.
29. Schurz I., J. Polymer. Sci., 1958, v. 28, p. 438.
30. Токата T., Хирон Д., Танияна М., «Химия и технология полимеров», 1965,
№ 1, с. 26.
31. Hoh J., 1и Н„ J. Polymer Sci., 1966, В, v. 4, № 10, р. 721.
32. Комлякова Л. И. и др., Хим. волокна, 1976, № 2, с. 49—51.
33. Азарова М. Т. и др. Международный симпозиум по химическим волокнам. Пре-
принты, Калинин, 1974, секция 5, с. 56—60.
34. Драбкин И. А. и др., ДАН СССР, 1964, т. 159, с. 197.
35. Overberger С. G., Juki Н., Irakava N., J. Polymer. Sci., 1960, v. 45, p. 127.
36. Takata T„ Bull. Chem. Soc. Japan, 1962, v. 35, p. 1438.
37. Grassie N., Me Neill L„ J. Polymer. Sci., 1958, v. 33, p. 171.
38. Хироси С., «Химия и технология полимеров», 1965, № 1, с. 88.
39. Goodhew Р. J., Clarke A. J., Bailey J. Е„ Mater. Sci. a. Eng., 1975, v. 17, p. 3—30.
295
40. Каргин В. А., Литвинов И. А., Высокомол. соед., 1965, т. 7, № 2, с. 226.
41. Clarke A. !., Bailey J. Е. International Conference on Carbon Fibres, their Place in
Modern Technology, London, 1974, paper № 2.
42. Watt W„ Johnson D. J., Parker M. E. International Conference on Carbon Fibres,
their Place in Modern Technology, London, 1974, paper № 1.
43. Burlant M. G., Parsons I. L., J. Polymer Sci., 1956, v. 22, p. 249.
44. Bane О. P., Manacha L. M., «Carbon», 1974, v. 12, p. 417—423.
45. Волков Ю. В. и др. В кн.: Структура, свойства и применение углеродных волок-
нистых материалов. Мытищи, ВНИИВ, 1975, с. 29—36.
46. Бондаренко В. М. и др. В кн.: Структура, свойства и применение углеродных
волокнистых материалов. Мытищи, ВНИИВ, 1975, с. 20—24.
47. Яп. пат. 43579 (1973).
48. Яп. пат. 42813 (1973).
49. Авт. свид. СССР 389012 (1973); Открытия. Изобр. Пром, образцы. Товарн. знаки,
1973, № 29, с. 78.
50. Фр. пат. 1602487.
51. Пат. США 3850876 (1974).
52. Фр. заявки 2023746, 2026637 (1971).
53. Фр. заявка 2207088 (1974).
54. Пат. США 3729549 (1973).
55. Фр. пат. 1430803.
56. Пат. США 3285696.
57. Watt W., Johnson W„ Polymer Prepr., 1968, v. 9, № 2, p. 1245.
58. Watt W., «Carbon», 1972, v. 10, № 2, p. 121—143.
59. Filder A. K., Fitzer E., Pozploch F., «Carbon», 1973, v. 11, № 4, p. 226; Ganlin C. A.,
Me Donald W. R., Gilmartin D. E., New Horisons Mater, a. Process., Azussa, Calif.,
1973, p. 142.
60. Hay J., J. Polymer Sci., 1968, A-l, v. 6, p. 2127.
61. Пшеничкин П. А. и др., Хим. волокна, 1976, № 3, с. 16—17.
62. Otani S., «Carbon», 1965, v. 3, p. 31; Otani S., J. Soc. Chem. Ind. Japan, 1958, v. 61,
p. 447; Otani S., ibid., p. 1324; Otani S., Ichikawa I., J. Soc. Chem. Japan, 1962, v. 65,
p. 1617.
63. Фр. заявка 2057903 (1971).
64. Gibson D. W. New Horisons Mater, a. Process., Azussa, Calif., 1973, p. 165—174.
65. Chinda A., Bull. Osaka Ind. Inst., 1961, v. 12, № 2, p. 110.
66. Johnson J. M„ Marjoram J. R., Rose P. G., «Nature», 1969, v. 221, № 5178, p. 357.
67. Фр. пат. 1274825 (1961).
68. Англ. пат. 906479 (1962).
69. Конкин А. А. и др. Производство шинного корда. М., «Химия», 1964. 480 с.
70. Bacon R„ Tang М. М., «Carbon», 1964, v. 2, р. 221.
71. Ruland W.. J. Appl. Phys., 1967, v. 38, p. 3585.
72. Тюлина M. P. и др. В кн.: Новые химические волокна технического назначения.
Л., «Химия», 1973, с. 60—65.
73. Basch A., Lewin М„ J. Polymer. Sci., Polymer Chem. Ed., 1974, v. 12, p. 2053—2063;
1973 v. 11 ". 3071 3095.
74. Пат.’США 3179605 71965), 3290489 (1966).
75. Фр. пат. 1406529 (1965).
76. Пат. США 3116975 (1964).
77. Фр. пат. 146564 (1966).
78. Klason Р., Heidenstam G., Norlin Е., Z. angew. Chem., 1909, Bd. 22, S. 1205; 1910,
Bd. 23, S. 1252.
79. Byrne G. A., Gardiner D„ Holmes F. H., J. Appl. Chem., 1966, v. 16, № 13, p. 81.
80. Волкова И. С. и др. Международный симпозиум no химическим волокнам. Пре-
принты. Калинин, 1974, секция 2, с. 197—200.
81. Pictet A., Sarasin J., Helw. chim. acta, 1918, v. 1, p. 87.
82. Голова О. П., Крылова Р. Г., Николаева И. И., Высокомол. соед., 1959, т. 1,
с. 1295; Голова О. П., Крылова Р. Г., ДАН СССР, 1957, т. 116, с. 419; Голо-
ва О. П., Пахомов А. М., Андриевская Е. А., ДАН СССР, 1957, т. 115, с. 1122;
Голова О. П., Пахомов А. М., Андриевская Е. А., Изв. АН СССР, ОХН, 1957,
№ 12, с. 1499; Голова О. П., Пахомов А. М., Андриевская Е. А., ДАН СССР,
1957, т. 112, с. 430.
83. Жбанков Р. Г., Бычкова Г. С., Конкин А. А., Хим. волокна, 1976, № 1, с. 31—32;
№ 4, с. 51—53.
84. Гаврилов М. 3., Ермоленко И. Н., «Вестник АН БССР». Сер. хим. наук, 1975,
№ 3, с. 32—36.
296
85. Мадорский С. Термическое разложение органических полимеров. М., «Мир»,
1967. 328 с.
86. Phillip В., Baudiseh J., Klugmann Н., Faserf u. Textilt., 1967, Bd. 18, № 10,
S. 461.
87. Соловьева Л. В. и др., Изв. АН БССР. Сер. хим. наук, 1973, № 3, с. 32—35.
88 Домбровская И. П. и др., Высокомол. соед., 1972, сер. Б, т. 14, № 10,
с. 723—724.
89 Домбровская И. П. и др., Высокомол. соед., 1975, сер. А, т. 17, № 7,
с. 1555—1559.
90. Бирюкова Г. П. и др., Высокомол. соед., 1973, сер. А, т. 15, № 7, с. 1573—1577.
91. Шаблыгин М. В., Бирюкова Г. П., Михайлов Н. В. В кн.: Новые химические во-
локна технического назначения. М., «Химия», 1973, с. 50—55.
92. Kilzer F. J., Broido A., «Pyrodynamics», 1965, v. 2, р. 151.
93. Shindo A., Nakanishi J., Somo J., Polymer Prepr., 1968, v. 9, № 2, p. 1333.
94. Фиалков А. С. и др., «Химия твердого топлива», 1968, № 3, с. 116.
95. Буянова В. К., Донкин А. А., Хим. волокна, 1977, № 1, с. 53, 54;
Losly N. N., Blaclack М. D. Second Conference Industry Carbon and Graphite, Lon-
don, April, 1965.
96. Schmidt D., Jones W., Chem. Eng. Progr., 1962, v. 58, № 10, p. 42.
97. Kohler O., Sperk E., Ber. Deut. Keram. Ges., 1966, Bd. 43, № 3, S. 199.
98. Гаврилов H. 3., Ермоленко И. H., «Вестник АН БССР». Сер. хим. наук, 1976,
№ 1, с. 23—28.
99. Пат. США 3235329 (1966).
100. Пат. США 3242000 (1966), 3281261 (1966), 3374102 (1968); фр. пат. 1508760
(1968).
101. Англ. пат. 894458 (1962); пат. США 3107152 (1963), 3116975 (1965).
102. Strong S. L., J. Mater. Sci., 1974, v. 9, № 6, p. 993.
103. Горбачева В. О. и др., Хим. волокна, 1975, № 4, с. 39—40.
104. Эвентова И. Л. и др., Хим. волокна, 1974, № 4, с. 29.
105. Шаблыгин М. В., Никитина О. А., Хуснулин Г. Н. Международный симпозиум
по химическим волокнам. Препринты. Калинин, 1974, секция 1, с. 226—230.
106. Шаблыгин М. В. и др., Хим. волокна, 1974, № 3, с. 72.
107. Gibson D. W„ Langlois G. В., Polymer Prepr., 1968, v. 9, № 2, p. 1376.
108. Bacon R., Schalamon W. A., Appl. Polymer Sympos., 1969, № 9, p. 285.
109. Изекил H., Спайн M. В. В кн.: Новое в производстве химических волокон. Под
ред. 3. А. Роговина и С. П. Папкова. М. «Мир», 1968, с. 169.
110. Proc. Eng., 1970, v. 41, № 14, р. 10.
111. Proc. Eng., 1975, v. 46, № 11, р. 27.
112. Volk Н. F. 4th International Carbon and Graphite Conference, Extended Abstracts
of Papers, London, 1974, sept., p. 23—27.
113. Chem. Eng., 1975, v. 82, № 26, p. 85.
114. Яп. пат. 21509 (1968), фр. пат. 2067619 (1971).
115. Яп. пат. 10254 (1972), 10255 (1972).
116. Англ. пат. 1091890 (1967); фр. пат. 2078849 (1971); яп. пат. 28013 (1970), 46009
(1972); фр. заявка 2010075 (1970).
117. Okuda Kensuke, Chem. Econ. a. Eng. Rev., 1974, v. 63, № 2, p. 34.
118. Otani S., Kokudo G„ Koitabashi T., Bull. Chem. Soc. Japan, 1970, v. 43, № 10,
p. 3291; Hawthorne H. e. a., «Nature»», 1970, v. 227, p. 946; Chem. Age, 1970, v. 100,
№ 2662, p. 8.
119. Hawthorne H., International Conference on Carbon Fibres, their Composites and
Applications, London, 1971, paper № 13.
120. Fudzivara M., Eng. Mater., 1974, v. 22, № 9, p. 37.
121. Пат. США 3919376 (1976).
122. Англ. пат. 1310769 (1973).
123. Didchenko R., Barr J., Chwostiak S. 12th Biennial Conference on Carbon, 1975,
28 July.
124. Яп. пат. 10252 (1972).
125. Chambers W. E„ Meehan. Eng., 1975, v. 97, № 12, p. 37—41.
126. Пат. США 3919387 (1976).
127. Пат. США 3787541 (1975).
128. Marsh Н., «Fuel», 1973, v. 52, № 3, p. 205.
129. Фр. заявка 2178193 (1973).
130. Cornford C., Marsh H. 4th International Carbon and Graphite Conference, Extended
Abstracts of Papers, London, 1974, sept., paper 205.
131. Lewis J. L„ Singer L. E„ «Carbon», 1969, v. 7, p. 93—99.
132. Evans S„ Marsh H„ «Carbon», 1971, v. 9, p. 733—746.
297
133. Evans S., Marsh H„ ibid., p. 747—753.
134. Marsh H. e. a., «Carbon», 1973, v. 11, p. 424—426.
135. Фр. заявка 2178193 (1973).
136. Fujmaki Hiroto e. a., «Tanso», 1975, v. 27, № 80, p. 3—10.
137. Dieffendorf R., Takarsky E., Polymer. Eng. a. Sci., 1975, v. 15, № 3, p. 150.
138. Proc. Eng., 1975, v. 46, № 11, p. 27.
139. Economy G„ J. Mater. Sci., 1971, v. 6, p. 1151.
140. Фр. заявка 2019821 (1970), англ. пат. 1258277 (1971).
141. Фр. пат. 2017543 (1970).
142. Kawamura К., Jonkins М„ J. Mater. Sci., 1970, v. 5, р. 262.
143. Пат. США 365199 (1972).
144. Tamado S., lamamoto М„ Appl. Polymer Symp., 1969, № 9, p. 339.
145. Пат. США 3650102 (1972).
146. Казиорова Т. И. и др., Хим. волокна, 1975, № 2, с. 18.
147. Казиорова Т. Н. и др. Хим. волокна, 1975, № 1, с. 73.
148. Яп. пат. 17392 (1973).
149. Фр. заявка 2216227 (1974).
150. Херон Дж., Синтетические высокомолекулярные материалы. Экспресс-информа-
ция, ВИНИТИ, 1961, № 3, с. 35.
151. Eitzer Е., Schafer W„ «Carbon», 1970, v. 8, р. 353.
152. Фр. заявка 2173198 (1973).
ГЛАВА 4
свойства углеродных волокнистых материалов
Упругопрочностные и физико-химические свойства УВМ определя-
ются видом исходного сырья, условиями получения, дополнительными
специальными обработками и другими факторами. Требования к свой-
ствам волокон зависят также от их назначения, и поэтому подбираются
соответствующие оптимальные условия получения волокна.
4.1. МАКРОМОРФОЛОГИЯ
К основным элементам макроморфологии относятся характер по-
перечного среза и поверхность волокна. Эти показатели, особенно вид
поперечного среза, наследуются от исходного волокна. При формовании
Рис. 4.1. Поперечные срезы углеродных волокон:
а — на основе вискозного корда; б — из нефтяного пека.
из раствора по сухому или мокрому способу получаются волокна, по-
перечный срез которых имеет изрезанную форму (бобовидная, фасо-
леподобная, гантелевидная и др.). Исключением является ПАН-В кур-
298
тель: форма его среза круглая. В процессе формования из расплава,
как правило, получаются волокна, имеющие круглую форму среза. На
рис. 4.1 приведены срезы волокон двух типов. Из-за неправильной фор-
мы среза трудно определить площадь поперечного сечения и как след-
ствие этого произвести расчет прочности нити. Неправильная форма
среза влияет на распределение напряжений, возникающих при нагру-
жении волокна. Изрезанная, и в частности бобовидная, форма, свойст-
венная большинству УВ, полученных из ПАН-В и ГЦ-В, затрудняет
контакт матрицы с волокном, что нарушает монолитность композита.
Рис. 4.2. Поверхность углеродного волокна, полученного из пека.
Рис. 4.3. Поверхность углеродного волокна, полученного из ГЦ-В в условиях,
способствующих образованию вторичного углерода.
Поверхность волокна определяется теми же факторами. Волокна
из пеков имеют гладкую (рис. 4.2), а из ГЦ-В — шероховатую поверх-
ность. Существенно влияют условия получения волокна. При значи-
тельном отложении вторичного углерода в результате разложения про-
дуктов распада иногда поверхность приобретает неровную чешуйчатую
форму (рис. 4.3). С повышением ТТО вторичный углерод выгорает, и
поверхность УВМ становится более гладкой.
4.2. МЕХАНИЧЕСКИЕ СВОЙСТВА
В зависимости от назначения УВМ изготовляются с различными,
а в ряде случаев уникальными, упругопрочностными показателями.
Среди жаростойких материалов УВМ занимают особое положение бла-
годаря сочетанию высоких механических свойств с низкой плотностью.
К важнейшим механическим показателям УВМ относятся прочность а,
модуль Юнга Е и разрывная деформация I. Для УВМ характерна ярко
выраженная анизотропия механических свойств. Ниже рассматривают-
ся преимущественно показатели волокна при растяжении (приложение
механического поля вдоль оси волокна).
Существует три способа определения прочности УВМ: определение
прочности одиночной нити, комплексной нити и полосок ткани извест-
ной ширины (обычно 5 см). При использовании УВ для изготовле-
299
ния композиционных материалов можно разрывать одиночные нити„
жгутик, пропитанный связующим, или пластик. Предпочтение следует
отдать первому способу, так как при определении прочности по двум
последним получаются завышенные результаты [1]. В случае некругло-
го сечения поперечного среза из большого числа определений диаметра
или площади находят поправочный коэффициент, который учитывают
при расчете а. Прочность определяется на разрывных машинах типа
«Инстрон» или на других приборах. В связи с большой хрупкостью, ко-
торая, как показали Л. Е. Утевский и сотр. [2], возрастает с ТТО и
диаметром волокна, особые требования предъявляются к зажимам.
Предварительно нити приклеиваются к рамкам и затем подвергаются
испытанию.
Модуль Юнга УВ, так же как прочность, определяют несколькими
способами. При испытаниях волокна на разрывных машинах по углу
наклона прямой напряжение — деформация находят модуль Юнга (ста-
тический). При этом одновременно определяется разрывная деформа-
ция волокна I.
Простой и доступный метод основан на измерении скорости ультра-
звука. Зависимость модуля Юнга (динамического) от скорости ультра-
звука выражается уравнением
Е~ А
где Е — модуль Юнга;
v — скорость ультразвука;
р — плотность;
А — коэффициент пропорциональности, зависящий от прибора.
Описаны [3] способы определения модуля Юнга рентгеновским, оп-
тическим, резонансным и другими методами. Динамический модуль не-
сколько превышает статический модуль Юнга.
Модуль сдвига находят с помощью крутящего маятника (торсион-
ный метод).
Механические свойства УВМ определяются структурной организа-
цией углерода, при этом выявляется существенная аналогия между
структурой и свойствами химических и углеродных волокон.
Для УВ характерен разброс механических свойств, что объясняет-
ся неоднородностью исходного волокна, усугубляющейся термообра-
боткой. Типичные гистограммы УВ приведены на рис. 4.4.
Таблица 4.1. Свойства волокна «модмор»
Высокомодульное волокно* (тип I) Высокопрочное волокно** (тип II)
Значения проч- ность, 107 Па модуль Юнга, 1010 Па диа- метр, мкм проч- ность при сдвиге, 107 па проч- ность, 107 Па модуль Юнга, 10Ю Па диа- метр, мкм проч- ность при сдвиге, 107 Па.
Среднее Минимальное Максимальное Коэффициент вариации, % 172 137 207 9,5 42,6 30,9 47,8 6,7 7,8 7,0 9,7 4,3 6,22 4,9 7,0 280 242 324 7,7 26,3 27,1 28,4 4,9 8,1 7,6 8,6 2,9 8,26 5,95 10,5
* Длина волокна при испытании 5 см; разрывное удлинение 0,41%.
** Длина волокна при испытании 5 см; разрывное удлинение 1,07%.
300
Высокий коэффициент вариации является одним из недостатков
УВМ, так как снижается надежность эксплуатации изделий. В табл. 4.1
приведены показатели волокна «модмор», для которого коэффициент
вариации относительно небольшой [4]. По другим источникам, значение
его достигает 20—25%. Следует отметить, что способ измерения и
расчета о влияет на значение коэффициента вариации.
С увеличением длины волокна возрастает суммарное число дефек-
тов, приводящих к снижению о волокна ;[5]. В логарифмических коор-
динатах зависимость 1g (/) (где I—длина образца) выражается пря-
мой линией (рис. 4.5). По данным Ватта {6], при увеличении длины от
Рис. 4.4. Гистограмма карбониаованного УВ, полученного на основе ПАН-В
куртель [3].
Рис. 4.5. Зависимость средней прочности УВ при растяжении от базы испы-
тания:
1 — модмор-1; 2 — модмор-П; 3 — ВМН; 4 — ВМН-С; 5 — ВМН-4; 6 — ВЭН-210.
0,5 до 5 см прочность УВ уменьшается на 33%. В другой работе того
же автора [7] приводятся данные, согласно которым при изменении
длины от 0,5 до 10 см разница в прочности достигает 50%. При экстра-
поляции I—>-0 прочность волокна составляет 7 ГПа. Заметно изменяют-
ся гистограммы распределения прочности волокна; с увеличением дли-
ны волокна максимум о сдвигается в область более низких значений
(рис. 4.6).
На прочность УВ по тем же причинам влияет масштабный эффект.
В ряде работ показано, что с уменьшением диаметра (или площади
поперечного сечения) о волокна возрастает.
Наряду со структурой существенное влияние на прочность оказы-
вают дефекты УВ. Они наследуются от исходного волокна и возникают
в процессе высокотемпературной обработки. Общепринято, что дефекты
ограничивают прочность УВ. Они подразделяются на внутренние и
внешние. По Шарпу и сотр. [8], наиболее типичными являются пустоты
в виде двойного конуса, беспорядочно расположенные в массе волокна.
Возможны также включения в виде пузырьков [9].
На границе раздела фаз под влиянием возникающих напряжений
происходит дополнительная кристаллизация углерода. В этих участках
плоскости, обрамляющие поверхности раздела, хаотически расположе-
ны вдоль оси волокна. Дезориентация слоев и возникновение дополни-
тельных напряжений на границе раздела фаз приводят к снижению
прочности волокна. Влияние этого фактора становится особенно замет-
ным для волокон, полученных при высоких ТТО. С этим связывается
экстремальная зависимость c—f (ТТО).
301
С ростом ТТО происходит слияние мелких пор, вследствие чего
число их уменьшается, а размеры увеличиваются.
По Джонсону [10], одной из причин разрыва волокна является на-
личие раковин и трещин. Трещины могут возникать под влиянием на-
пряжений, появляющихся при охлаждении волокна [11], или вследст-
вие выгорания примесей. Роль примесей наглядно подтверждена в ра-
боте [12]. Авторы получали ПАН-В в воздухе, очищенном от пыли.
В этом случае a=f (ТТО) непрерывно возрастает, а для контрольного
образца, приготовленного в обычных условиях, omax достигается при
1400 °C, а затем уменьшается. Более 95% инородных частиц (в среднем
восемь частиц на 1 мм длины волокна) находится на поверхности
Рис. 4.6. Гистограммы зависимости прочности углеродного волокна от базы
испытании:
а — длина 10 см; средняя прочность 1,86 ГПа; б —длина 5 см; средняя прочность 2,1
ГПа; в — длина 0,5 см; средняя прочность 2,8 ГПа.
ПАН-В [13]. Из такого ПАН-В получается низкопрочное (1,16 ГПа^
УВ.
Высказывается мнение о том, что экстремальная зависимость o=f
(ТТО) обусловлена вкладом сдвига в разрыв волокна по мере роста
ТТО. Возрастанию сдвига дополнительно способствуют дефекты волок-
на и разориентации плоскостей на границе раздела фаз.
Картину, характеризующую роль дефектов, можно получить травле-
нием поверхности волокна. В результате травления залечиваются дефек-
ты, поэтому возрастает о, что наглядно подтверждается гистограммами
прочности до и после травления волокна. Для высокомодульного волок-
на, полученного при высокой ТТО, наибольший вклад вносится внутрен-
ними дефектами; для высокопрочного волокна, полученного при более
низких ТТО, — внешними.
На основании исследований, проведенных с помощью электронного
микроскопа высокой разрешающей способности, поверхности и характе-
ра разрыва были предприняты попытки установить зависимость между
числом дефектов и о. По мнению авторов работы [9], 90% разрывов
связано с наличием пузырьков в волокне. Найдена зависимость прочно-
сти от диаметра пузырьков: волокно с о=1,5 ГПа разрушается при на-
личии пузырьков диаметром 2 мкм, а с прочностью 2,2 ГПа — при диа-
метре пузырьков менее 1 мкм. Рейнольдсом {14] в УВ найдены дефек-
ты размером более 1 мкм. Теоретические расчеты показывают, что для
высокомодульного волокна гарантируется прочность 2,14 ГПа и модуль
Юнга 400 ГПа при устранении всех дефектов размером более 0,2 мкм,
расположенных перпендикулярно оси волокна, а для высокопрочного
УВ — прочность 3,15 ГПа и модуль Юнга 240 ГПа при устранении де-
фектов диаметром более 0,05 мкм. Приводятся данные, согласно кото-
302
рым с уменьшением размеров дефектов от 0,5 до 0,3 мкм прочность во-
локна возрастает с 4,2 до 5,5 ГПа.
Залечивание внешних дефектов приводит к повышению прочно-
сти УВ. Выше отмечалось положительное влияние травления волокна.
Аналогичный эффект достигается при ионной бомбардировке УВ плаз-
мой аргона [15]. Введение бора в волокно способствует возрастанию
его модуля Юнга [16].
По мнению Ватта [6], наличие фибрилл препятствует распростра-
нению и росту трещин и тем самым приводит к самоупрочнению во-
локна. Среди жаростойких волокон это свойство присуще только УВ.
Типичным макроморфологическим дефектом является центральная
полость, появляющаяся в результате нарушения технологического про-
цесса получения УВ [8]. В этом случае <т снижается из-за уменьшения
эффективной площади поперечного сечения волокна.
Особого внимания заслуживает рассмотрение микропор, имеющих
форму призм. Эти микропоры расположены между базисными плоско-
стями и ориентированы вдоль оси волокна. Они свойственны высокомо-
дульным высокопрочным волокнам, изготовленным при ориентацион-
ном вытягивании. По мнению авторов работы [17], микропоры не влия-
ют на механические свойства волокна.
По прочности УВМ можно подразделить на три группы: низкопроч-
ные 500 МПа, средней прочности 500—1500 МПа и высокопрочные —
более 2 ГПа. Модуль Юнга УВМ может изменяться в широких преде-
лах: 30—700 ГПа. В табл. 4.2 приведены механические свойства высо-
копрочных высокомодульных волокон [18, 19].
Таблица 4.2. Свойства высокопрочных высокомодульных УВ,
выпускаемых в СССР н ведущими зарубежными фирмами
Марка волокна Страна Прочность, 107 Па Модуль Юнга, ЮЮ Па Диаметр, мкм Плотность, 103 кг/мЗ Удлинение при разрыве, %
Торнел-40 США 175 28,0 6,9 1,56 0,8
Торнел-50 200 35,0 6,6 1,63 0,8
Торнел-300 210 26,0 1 W — 0,8
Торнел-400 280 20,0 — — 1,0
Ригилор АС Франция 200 20,0 12,4 1,75 1,5
Ригилор AG 190 42,0 11,0 2,0 0,5
Модмор I Англия 170—250 40—45 7,8 2,0 0,5
Модмор II 280 27 8,1 1,8 0,8—1,0
Торейка Т300А Япония 250—310 22—25 8,4 1,78 0,9—1,2
Торейка М40А 200—250 37—43 7,9 1,8 0,5—0,7
HMS США 200 36,5—40 1,88 0,5—0,7
HT-S 250—290 24 8 1,73 1,0
УВМ с высокими механическими свойствами подразделяются на
две группы: высокомодульные (модуль Юнга 300—700 ГПа, прочность
2—2,5 ГПа) и высокопрочные (прочность 2,5—3,2 ГПа, модуль Юнга
200—250 ГПа). В последнее время получены волокна, в которых соче-
таются высокая прочность и высокий модуль Юнга.
К замечательным особенностям УВ относится низкая их плотность,
поэтому по удельным значениям механических свойств (отношение
прочности и модуля Юнга к плотности) они превосходят все известные
волокна и конструкционные материалы, что наглядно иллюстрируется
диаграммами (рис. 7, а и б) [8].
303.
Особого внимания заслуживает
Юнга УВ, который увеличивается с
очень высокий удельный модуль
ориентацией базисных плоскостей
Рис. 4.7. Удельные значения прочности и модуля Юнга углеродных и других волокон
и конструкционных материалов:
а —удельная прочность волокон; б — удельный модуль Юнга конструкционных материалов;
1 — Е-стекло; 2 — карбонизованное волокно; 3 — графитированное волокно; 4 — борное волокно;
5 — волокно из карбида бора; 6 — вольфрамовое волокно; 7— бериллиевое волокно; 8 — сапфировое
волокно (АЬОз); 9— волокно из карбида кремния; 10 — графитовые «усы»; // — сплав А1; 12 —
сплав Ti; 13 — сплав Mg; 14 — сталь.
(рис. 4.8), ТТО и степенью вытягивания. Согласно работе [20], модуль
Юнга возрастает с увеличением длины фибрилл: при длине фибрилл
200—400 А и 400—1500 А модуль Юнга равен 224 ГПа и 420 ГПа соот-
4.8. Зависимость модуля Юнга УВ
ориентации базисных плоскостей
(ЧР/2).
ветственно. Высокопрочные высокомодульные УВ, как правило, изготов-
ляются в виде нитей, жгутиков и лент.
УВМ средней прочности (ст = 500—1500 МПа, £ = 50—150 ГПа)
вырабатываются в виде тканей различных структур, пряжи, нитей, жгу-
тов, войлока, кнопа и других текстильных форм. Разница в свойствах
УВМ этой группы, вырабатываемых ведущими фирмами, незначитель-
ная. В табл. 4.3 для иллюстрации приведены свойства углеродных тка-
ней [3, с. 273].
-304
Таблица 4.3. Некоторые характеристики углеродных тканых материалов
Материал Прочность по- лоски шири- ной 1 м, кН Удлинение при разрыве, % Масса 1 м2, г Толщина, мм Плотность, 103 кг/мЗ Диаметр элементар- ной инти, мкм Содержание углерода, %
основа уток основа уток
WCB 8,2 7,9 8,6 8,4 257,7 0,6 1,50 8,38 99,6
G-1550 8,9 4,2 2,0 4,9 210,2 0,3 1,62 10,16 99,9
WCK 6,8 6,1 — — 274,7 0,6 1,50 7,62 99,6
ССА-1 3,1 1,6 — — 254,3 0,4 1,49 9,91 93,8
«Плутон «В>: 3,6 3,6 —- — 197,0 0,6 1,53 — 84,8
VCK 2,3 2,4 — — 288,2 0,7 1,49 10,16 99,1
VCA 1,1 6,8 — — 254,3 0,7 1,45 8,38 94,4
CSCC-2 — — — 254,3 0,4 1,50 7,62 99,3
GSGS-2 — — — — 254,3 0,4 1,50 7,62 —
Выше рассмотрены механические свойства УВМ, полученных на
основе ГЦ-В и ПАН-В. Исследовались почти все известные химические
и природные волокна, однако УВМ на их основе имеют более низкие
механические свойства*. Исключением могут служить волокна из пеков
и ФФС.
4.3. ТЕРМИЧЕСКИЕ СВОЙСТВА
Переход от органического к углеродному волокну сопровождается
резким уменьшением свободной энергии и образованием нового поли-
Ъ,°С
Рис. 4.9. Зависимость прочности различных материалов от температуры
|(инертная среда):
/ — высокомодульное УВ; 2 — стеклянное волокно; 3 — борное волокно; 4 — сталь;
5 —титан; 6 — алюминий.
Рис. 4.10. Зависимость модуля Юнга различных материалов от температуры
((инертная среда):
I — углеродные «усы»; 2 — высокомодульное углеродное волокно; 3 — графит марки
ЕСА.
л-сопряжений. Именно благодаря этому УВМ обладают исключитель-
но высокой теплостойкостью, превосходя почти все известные материалы
[21] (рис. 4.9 и 4.10). В инертной среде а и Е до температуры около
1500 °C практически не изменяются.
* Дополнительные сведения о механических свойствах УВМ приведены в моно-
графии [3].
20—2254
305
Термостойкость УВМ зависит от структуры, характера поверхно-
сти, ТТО и других факторов. Влияние ТТО на стойкость УВМ к кисло-
роду воздуха при повышенных температурах иллюстрируется приведен-
ными ниже данными [22]:
Степень
ТТО Условия обработки сохранения а,
%
Низкая...................................100 сут, 250 °C 85
Средняя..................................100 сут, 250 °C 125
Высокая..................................100 сут, 250 °C 100
Низкая...................................100 сут, 300 °C Разрушается
Средняя..................................100 сут, 300 °C 145
Высокая..................................100 сут, 300 °C 95
средних ТТО
обусловлена
Повышенная стойкость волокон при
устранением дефектов в результате воздействия окислителя.
Р. М. Левит и сотр. [23] для оценки реакционной способности
предложили реакцию "
Рис. 4.11. Интегральные ки-
нетические кривые выхода
CS2 при 800 °C для УВ, по-
лученного при ТТО:
1 — 800 “С; 2 — 1000 °С^ 3 —
1600 °C; 4 — 2000 °C.
взаимодействия УВМ с газообразной серой при
высоких температурах. Сера является мягким
окислителем, поэтому по кинетике процесса
легко оценить особенности структуры и термо-
стойкость волокна. К преимуществам метода
относится простота улавливания и возможность
количественного определения основного про-
дукта реакции — сероуглерода и образующего-
ся в небольших количествах HjS. Из рис. 4.11
видно значительное влияние ТТО на реакцион-
ную способность УВМ.
Предельная температура длительной экс-
плуатации УВМ в воздушной среде составля-
ет 300—400 °C (300 °C для карбонизованного
волокна, 400 °C для графитированного) [1].
Для повышения стойкости волокна к кис-
лороду воздуха предложен ряд методов. Сре-
ди них следует отметить введение добавок в.
прядильный раствор, пропитка исходного, час-
тично карбонизованного или карбонизованно-
го волокна (тканей) соединениями, которые
в результате термообработки образуют покрытия (оксиды, карбиды),,
обладающие высокой стойкостью к окислению, или методы, основанные
на нанесении из газовой фазы веществ (пироуглерод, карбиды), повы-
шающих стойкость УВМ к окислителям.
Введение в прядильный раствор добавок неперспективно, так как
резко ограничивается число соединений, совмещающихся с прядильным
раствором. Исключение составляет силикат натрия, растворяющийся
в вискозе. При карбонизации вискозного волокна, содержащего сили-
кат натрия, образуется оксид кремния или карбид кремния [24].
Несложно получить покрытие на исходном или карбонизованном
волокне. Например, при нанесении на УВМ буры в сочетании с моче-
виной [25] и последующей термообработке на поверхности нити обра-
зуется слой нитрида бора >[1—4% (масс.)]. В случае предварительной
обработки ацетатом циркония УВМ покрывается слоем карбида цирко-
ния [26].
306
Таблица 4.4. Свойства углеродных волокон в зависимости от ТТО
и степени модификации пироуглеродом и тугоплавкими соединениями кремния
ТТО, °C Привес, % (масс.) Элементный состав, % Температурный интервал экзо- термического эффекта, °C Температура начала интен- сивной потери массы, °C Потеря массы при 900 °C, %
с Н зола
900 95,8 0,2 4,0 380—550 400 72
1450 —. 98 0,35 1,65 300—640 402 63
1450 18* —- — — 540—800 595 45
2200 — 99,4 Следы Следы 500—688 535 47
2200 18* — —— — 535—710 600 42,5
2400 — 100 Отсут. Отсут. 490—710 630 32,5
2400 18* — — — 565—860 650 30,5
2000 3** 93,3 Следы 6,6 500—740 600 40
2000 15** 86,8 > 13,0 560—740 610 38
2000 20** 78,8 в 21,2 590—785 640 32
2000 — 98,6 в 1,4 440—680 540 47
* Пироуглерод.
•• SiC, SiOj, SiO, Si.
Наибольший эффект достигается при применении газофазового ме-
тода нанесения пироуглерода, карбида кремния, нитрида бора. В каче-
стве источника пироуглерода используются углеводороды (метан, этан,
пропан, бензол и др.); для образования карбида кремния применяется
смесь хлорсиланов с углеводородами. Источником углерода может слу-
жить также углеродное волокно. Газом-носителем служит аргон, азот,
водород, гелий. Изменение термических характеристик [27] УВМ за-
висит от условий термообработки, количества нанесенного пироуглерода
или карбида кремния (табл. 4.4). Для покрытия УВМ нитридом бора
применяется смесь BCI3+NH3, в которой аммиак является источником
азота [28], а при нанесении алюминия — пары А1(С4Нд)з [29]. Покры-
тие пироуглеродом производится в газовой среде при 900—1200 °C,
а карбидами и боридом — при 1500 — 1800 °C.
По мере увеличения защитного слоя возрастает эффективность, од-
нако при этом повышается хрупкость УВ, что осложняет их переработ-
ку и применение. Оптимальной считается толщина слоя не более 8 мкм,
привес 1—4% (масс.).
При нанесении защитных покрытий из газовой фазы залечиваются
трещины, и поэтому возрастает прочность УВ.
4.4. ХИМИЧЕСКИЕ СВОЙСТВА
Углеродные волокна характеризуются высокой химической стой-
костью к большинству агрессивных сред; она зависит от ТТО, структу-
ры и поверхности волокна, типа исходного сырья и других факторов.
Из данных табл. 4.5 следует, что модуль Юнга практически не изменяет-
ся при воздействии на высокомодульное волокно кислот [30], за ис-
ключением HNO3. Длительное воздействие (около года) светопогоды,
воды, минеральных кислот (H2SO4, НС1, HNO3), раствора КОН (40%)
при комнатной температуре не оказывает влияния на механические
свойства УВ [31]. Чувствительность УВ к действию окислителей
(HNO3, гипохлорит натрия и др.) наблюдается при повышенных темпе-
ратурах. В мягких условиях воздействия окислителей происходит трав-
ление поверхности, способствующее устранению дефектов и возраста-
20*
307
Таблица 4.5. Химическая стойкость в различных агрессивных средах
высокомодульного УВ, полученного на основе ПАН-В
(продолжительность воздействия 257 сут)
Реагенты Температура, °C Диаметр, мкм Прочность, 107 Па Модуль Юнга, 1010 Па
Контрольный образец — 6,2 214,4 40,4
НС1, 50%-иая 50 5,9 188,2 41,4
H2SO4, 50%-ная 50 6,3 153,8 40,0
HNO3, 50%-иая 50 6,8 146,9 33,8
NaOH, 50%-ная 50 6,5 177,2 36,5
НзР2О6, 50%-ная 50 6,5 158,6 42,1
Н3РО4 50 6,5 171,0 42,1
СН3СООН, ледяная — 6,1 196,5 43,4
НСООН, 90%-ная 20 6,1 184,8 42,1
Сульфобензойная кислота, 32%-ная — 6,5 205,5 44,1
нию прочности УВМ. Этот прием используется для повышения тСд угле-
пластиков, Химическая стойкость УВМ обеспечивает стабильность
свойств композиционных материалов. По сравнению с углеродными
стеклянные волокна обладают значительно меньшей химической стой-
костью. Последние нестойки к 10%-ным растворам НС1 и H2SO4 (при
температуре 65 °C потеря массы волокна составляет 35%), а также
к соде и щелочам. Даже длительное воздействие воды вызывает сни-
жение механических свойств стеклянных волокон.
Влияние ТТО на химическую стойкость наглядно иллюстрируется
следующими данными: волокно, полученное при ТТО 900—1100 °C,
обработанное концентрированной HNO3 (температура 120 °C, продол-
жительность 5 ч), полностью разрушается; при ТТО 1300— 1-800 °C во-
локно сохраняет 40—80% начальной прочности; при ТТО>2000°С и
указанных выше условиях обработки свойства образцов не изменяются
[31]. Аналогичные результаты были получены Бирне и Джефрисом
[32] при окислении карбонизованного и графитированного УВ смесью
Н3РО4+К2СГ2О7+Н2О.
Установлено, что в первую очередь разрушается аморфный углерод,
о чем свидетельствует уменьшение интенсивности фона рассеивания
рентгеновских лучей после травления. Представляют интерес приведен-
ные ниже данные, полученные при воздействии на УВМ КгСг2О7 в при-
сутствии H2SO4 или Н3РО4:
Тип углеродного волокна Кислота Продолжи- тельность обработки, ч Темпера- тура, °C Потеря массы, %
Карбонизованное .... . н3РО4 5 133 71,2
Графитированное .... . Н3РО4 5 133 63,2
Карбонизованное .... . H2SO4 4,25 но 51,5
Графитированное .... . H2SO4 4,2 по 100
Повышенная чувствительность графитированного волокна к рас-
твору H2SO4 + K2Cr2O7+H2O объясняется химическим взаимодействием
графитированного волокна с H2SO4, способствующей разрыхлению
структуры, облегчающей проникновение окислителя внутрь волокна.
Неожиданной оказалась более высокая химическая стойкость
УВ-ГЦ по сравнению с УВ-ПАН, полученных при одинаковой ТТО
[31]. УВ-ПАН имеет более совершенную структуру по сравнению
308
с УВ-ГЦ, особенно на ранних стадиях термообработки, и казалось бы
его химическая стойкость должна быть выше. В действительности на-
блюдается иная закономерность. Видимо, специфические (невыяснен-
ные) структурно-морфологические особенности существенно влияют на
химическую стойкость волокна. Методом сканирующей электронной
микроскопии для УВ-ПАН установлено при травлении наличие более
рыхлого ядра, унаследованного от исходного ПАН-В. В том случае,
когда ПАН-В по структуре однородно, сердцевина полученного из него
УВ-ПАН не вытравливается.
В работе [33] показано, что волокно торнель при 125—300 °C
взаимодействует с калием (образуется СвК), в результате чего электро-
проводность его повышается в 20—28 раз.
При обработке соединениями бора происходит его внедрение в УВ,
приводящее к преобразованию структуры и развитию ориентации ба-
зисных плоскостей параллельно оси волокна, вследствие чего возра-
стают модуль Юнга и электропроводность [34]. Обработка УВ хлори-
дами брома или иода приводит к его пластификации, облегчая текстиль-
ную переработку УВ. При нагревании до 125°C бром удаляется [35].
Механизм пластифицирующего действия брома неясен. Практическая
значимость этого метода для облегчения текстильной переработки во-
локна сомнительна. При контакте графита с металлами ускоряется
коррозия металла; это обстоятельство необходимо учитывать при экс-
плуатации УВМ и углепластиков [36].
4.5. ФИЗИЧЕСКИЕ СВОЙСТВА
Физические свойства зависят от предыстории УВМ, поэтому све-
дения о них, приводимые в литературе, противоречивы. Ниже представ-
лены наиболее характерные физические константы УВМ:
Волокно
Плотность, 103 кг/м3 карбонизованиое 1,30—1,65 графитированное 1,3—1,9
Удельная поверхность, м2/г 0,3—1000 0,15—3
Термический коэффициент линейного расширения
а-106, 1/°С 4 2
Удельная теплоемкость, кДж/(кг-К) 0,66 0,66
Коэффициент теплопроводности Л, Вт/(м-К) . 0,837—20,934 83,74—125,6
Удельное объемное электрическое сопротивление
р, 10—6 Ом-м 0,4—70 0,003—0,6
Тангенс угла диэлектрических потерь (прн 1010 Гц) . . 0,17—0,42 0,25—0,33
рн 6,2 6,5
Температура сублимации, °C — 3600
Гигроскопичность, % 0,1—10 1
Плотность УВМ, как правило, возрастает с ТТО. Лишь на стадии
предкристаллизационного состояния (при ТТО 1800—2000 °C) она не-
сколько уменьшается. Плотность графита составляет 2260 кг/м3; она
значительно превосходит плотность УВМ, что обусловлено менее совер-
шенной структурой и большей пористостью УВМ. Наблюдаемая иногда
низкая плотность графитированного волокна объясняется тем, что
в процессе графитации карбонизованного волокна, имеющего развитую
поверхность, происходит закупорка пор, которая приводит к снижению
кажущейся плотности УВМ. При любом методе определяется не истин-
ная, а кажущаяся плотность УВМ, зависящая от объема закрытых пор.
309
Между плотностью и модулем Юнга существует прямая взаимосвязь;
плотность возрастает пропорционально модулю Юнга [17]. Среди жа-
ростойких и высокопрочных волокон УВ обладают наименьшей плот-
ностью, что благоприятно сказывается на их удельных показателях ме-
ханических свойств.
Одним из характерных свойств УВ, особенно подвергнутых специ-
альной обработке, является развитая пористость и огромная удельная
поверхность, достигающая 2000 м2/г. К основным параметрам сорбцион-
но-активных УВ относятся средний радиус пор, распределение пор по
радиусам, объем пор и предельный сорбционный объем. Для опреде-
ления пористости используются ме-
тоды микроскопии, малоуглового
рентгеновского рассеяния (внутрен-
няя пористость), сорбция, ртутная
порометрия и др. Чаще применяется
метод сорбции. При этом следует
учитывать возможность протекания
чисто физической сорбции, а также
капиллярную конденсацию, хими-
ческую адсорбцию или абсорбцию.
Достоверные результаты получают-
ся при наличии физической сорб-
ции. Для УВ характерны поры раз-
личного диаметра, поэтому важное
значение приобретает размер моле-
кул сорбата, что наглядно показа-
но в работе [37]. Согласно кривым
сорбции-десорбции, приведенным в
одних источниках, в УВМ содержат-
Рис. 4.12. Кривые распределения пор по
эффективным радиусам:
1 — УВ на основе ГЦ-В; 2 — УВ на основе
ПАН-В.
ся микро- и переходные поры; по
другим источникам — микро- и ультрапоры. Размер пор колеблется в
пределах 3—50 А |[38]. Типичные кривые распределения пор по радиу-
сам приведены на рис. 4.12.
Удельная поверхность карбонизованных волокон составляет 0,5—
120 м2/г. При графитации происходит закупорка пор и уменьшение по-
верхности, приближающейся по значению к геометрической поверхности
волокна. Активацией волокна водяным паром или двуокисью углерода
при повышенной температуре удается значительно повысить пористость
и удельную поверхность волокна. В процессе активации наблюдается
частичная потеря массы волокна. Наиболее эффективно активации под-
дается волокно, полученное при относительно низкой ТТО. С повыше-
нием последней пористость и поверхность волокна уменьшаются. Удель-
ную поверхность и пористость можно регулировать ТТО, условиями ак-
тивации, типом исходного волокна, из которого получено УВМ [39—42].
Например, для активированного УВМ, полученного при относительно
высоких ТТО, характерна однородная пористая структура, а для УВМ,
полученного при низкой ТТО (600—800 °C), — развитая система пор
разных диаметров [40]. В работе [40] указывается на возможность по-
лучения ультрапористых УВМ. По данным ртутной порометрии [38],
суммарная пористость в УВМ достигает 1,5 см3/г, при этом 0,5 см3/г
приходится на микропоры радиусом 2,9—6,8 А.
Необычайно развитые пористость и удельная поверхность предо-
пределяют области применения УВМ.
310
УВМ представляют собой типичное гидрофобное вещество, однако
из-за капиллярной конденсации паров воды карбонизованные волокна
могут иметь высокую гигроскопичность.
Для УВМ характерна низкая поверхностная энергия, значительно
меньшая, чем для стеклянного волокна, поэтому УВМ отличаются
слабым сродством к связующим и связанным с этим низким напряже-
нием сдвига композитов.
УВМ имеют отрицательный термический коэффициент линейного
расширения вдоль оси волокна, приводящий к накоплению остаточных
напряжений при тепловом воздействии на волокно. Максимальное от-
Рис. 4.13. Зависимость теплопроводности углеродной ткани от температуры
осаждения пироуглерода и ТТО:
1 — ткань, графитированная при 3000 °C н уплотненная пнроуглеродом; 2 — ткань, кар-
боиизоваииая при 800 °C и уплотненная пироуглеродом; 3 — ткань, карбонизованиая
при 800 °C, не уплотненная пнроуглеродом.
Рис. 4.14. Изменение удельного объемного электрического сопротивления
УВ-ПАН в зависимости от ТТО.
рицательное значение термического коэффициента линейного расшире-
ния соответствует 0 °C.
Коэффициент теплопроводности УВМ, особенно графитированных,
приближается к металлам: для стали он составляет 20,93 —
54,43 Вт/ (м • К), для алюминия он равен 25,02 Вт/(м • К).
Эффективная теплопроводность зависит от текстильной формы ма-
териала. Как показано в работе [43], для тканей она возрастает мень-
ше с увеличением ТТО и продолжительности термообработки, чем для
волокон. Уплотнение пироуглеродом увеличивает число контактов меж-
ду нитями, что . приводит к повышению теплопроводности ткани
(рис. 4.13). В зависимости от области применения УВМ высокая тепло-
проводность может являться достоинством или недостатком. Теплоем-
кость УВМ находится в пределах, близких к твердым телам.
Ни одно физическое свойство твердых тел не изменяется в таких
широких пределах, как электропроводность. Удельное объемное элек-
трическое сопротивление (Ом-м) составляет для изоляторов 105—1015,
полупроводников 105—10~6, проводников меньше 10~6. В процессе пре-
вращения органических волокон в углеродные осуществляется переход
через все зоны проводимости. Исходные волокна являются диэлектри-
ками, в процессе термообработки до 1200—1500°С происходит резкое
уменьшение электрического сопротивления, при более высоких ТТО оно
продолжает уменьшаться, но менее интенсивно (рис. 4.14). Подобная
311
зависимость свидетельствует о перестройке структуры, обусловливаю-
щей изменение электрического спектра носителей заряда, воздействии
на их концентрацию, подвижность и другие характеристики. Появляют-
ся такие свойства, как магнетосопротивление и магнитная восприимчи-
вость, а также эффект Холла. Изучение электрических свойств, в свою
очередь, дает полезную информацию о структуре УВ. Резкое увеличение
электропроводности связано с процессом ароматизации углерода и об-
разованием базисных плоскостей (стадия карбонизации)', в дальней-
шем по мере совершенствования структуры (стадия графитации) про-
исходит более плавный спад электрического сопротивления.
Модуль Юнга определяется текстурой или ориентацией базисных
плоскостей. Эмпирическая формула, приведенная ниже [44], указывает
на то, что электрическое сопротивление также зависит от ориентации
базисных плоскостей:
-|-p’/n = const
где Е — модуль Юнга;
р — плотность;
р„ — удельное объемное электрическое сопротивление; п > 1.
Аналогия между электрическими свойствами стеклоуглерода, а так-
же других углеродных материалов [45] и углеродных волокон навела
на мысль, что последние представляют собой неграфитирующуюся фор-
Рис. 4.15. Зависимость тер-
мо-эдс от температуры об-
работки:
/ — УВ-ПАН; 2 — мягкий угле-
род (гомогенно-графнтирую-
щийся).
му углерода. Наиболее четко различие между
углеродными волокнами и графитирующими-
ся формами углерода проявляется при срав-
нении термо-эдс этих материалов. Для гомо-
генно-графитирующихся форм углерода ха-
рактерен крутой подъем термо-эдс до темпе-
ратуры начала графитации («2000 °C) и рез-
кое ее снижение в процессе графитации (кри-
вая 2, рис. 4.15). Для неграфитирующихся
форм углерода, к которым относится УВМ,
выше указанной температуры наблюдается
монотонное, незначительное возрастание тер-
мо-эдс (кривая 1, рис. 4.15). Отмечается так-
же различие в зависимости р„ = / [ТТО] для
двух типов углеродных материалов. В случае
УВ объемное электрическое сопротивление
уменьшается, хотя и с разной скоростью во
всей области температур, тогда как для гомо-
геннографитирующихся форм углерода р„ на
стадии предкристаллизационного состояния
(1500—2000 °C) сохраняет постоянное значение.
В соответствии со значениями р„ (см. рис. 4.14) карбонизованное
УВ (ТТО 1000—1500°C) относится к полупроводникам, а графитиро-
ванное занимает промежуточное положение между полупроводниками
и проводниками, приближаясь по мере повышения ТТО к последним.
Данные о механизме проводимости в литературе приводятся в об-
щем виде. УВМ, как и другие переходные формы углерода, обладают
смешанной дырчатой и электронной проводимостью {46, 47]; соотноше-
ние между этими видами проводимости зависит от ТТО. При повыше-
312
НИИ ТТО, сопровождающемся совершенствованием структуры и увеличе-
нием числа л-электронов, запретная зона проводимости УВМ умень-
шается, поэтому возрастает электропроводность, которая для волокон,
полученных при высоких конечных температурах обработки, по абсо-
лютному значению приближается к электропроводности проводников.
В результате металлизации (покрытие палладием, никелем, медью)
карбонизованных УВМ (при ТТО 800—1000 °C) электропроводность их
значительно повышается [48].
Разнообразные электрофизические свойства открывают широкие
возможности практического использования УВМ для различных целей.
Однако следует иметь в виду, что из-за резкого снижения электриче-
ского сопротивления при ТТО до 1000 °C трудно получить УВМ с задан-
ными электрофизическими свойствами.
ЛИТЕРА ТУРА
1. Gibson D. W. New Horizons Mater, a. Process., Azussa, Calif., 1973, p. 165—
174.
2. Утевский Л. А. и др., «Механика полимеров», 1973, № 5, с. 950—952.
3. Конкин А. А. Углеродные и другие жаростойкие волокнистые материалы. М., «Хи-
мия», 1974. 375 с.
4. Gill Р. М. Carbon Fibres in Composite Materials Published for the Plastics Institute,
London, 1972, p. 67.
5. Туманов A. T. и др., «Механика полимеров», 1975, № 2, с. 248—257.
6. Watt W., Kunststoffe-Plast., 1970, Bd. 17, № 11, S. 444.
7. Watt W., «Carbon», 1972, v. 10, № 2, p. 121—143.
8. Sharp J. V. e. a. Conference on Carbon Fibres, their Place in Modern Technology, Lon-
don, 1974, paper № 5.
9. Sharp J. V., Burnay S. G. International Conference on Carbon Fibres, their Compo-
site and Applications, London, 1971, paper № 10.
10. Johnson J. W., Appl. Polymer Sym., 1969, № 9, p. 229.
11. Goodhew C. J., Clarke A. I., Boiley J. E., Mater. Sci. a. Eng., 1975, v. 17, p. 3—30.
12. Moreton R„ Watt W., «Nature», 1974, v. 247, p. 360.
13. Moreton R. International Conference on Carbon Fibres, their Composite and Applica-
tions, London, 1971, paper № 2.
14. Reynolds W. International Conference on Carbon Fibres, their Composite and Appli-
cations, London, 1971, paper № 52.
15. Johnson J. W„ «Carbon», 1969, v. 7, № 6, p. 659.
16. Allen S., Coofer T., Mayer R., «Nature», 1969, v. 224, p. 684.
17. Bacon R., Schalamon W., Polymer Prepr., 1968, v. 9, p. 2.
18. Конкин A. A. и dp., Хим. волокна, 1977, № 3, с. 65.
19. Diefendorf, Tokarsky, Polymer Eng. a. Sci., 1975, v. 15, № 3, p. 150—159.
20. Watt IF., Johnson W., «Polymer», 1968, v. 9, p. 1245.
21. Кобец Л. П„ Гуняев Г. M. В кн.: Пластики конструкционного назначения. Под
ред. Е. П. Тростянской. М., «Химия», 1974, с. 204—246.
22. Hart G. L., Pritenard G. International Conference on Carbon Fibres, their Place in
Modern Technology, 1974, London, paper № 34.
23. Левит P. И., Райкин В. Г., Ивин В. Д. В ки.: Новые химические волокна техниче-
ского назначения. Л., «Химия», 1973, с. 198.
24. Визон И., Робертсон Дж. В кн.: Новое в производстве химических волокон. Под
ред. 3. А. Роговина и С. П. Папкова. М., «Мир», 1968, с. 188.
25. Пат. США 3551484 (1967).
26. Пат. США 3356525 (1967).
27. Робимов И. П. и dp. Структура, свойства и применение углеродистых волокнистых
материалов. Труды ВНИИВ, Мытищи, 1975, с. 36—42.
28. Фр. пат. 2358140.
29. Англ. пат. 1255658 (1971).
30. Judd N. S. W. International Conference on Carbon Fibres, their Composites and Appli-
cations, London, 1971, paper № 32.
31. Мелешко А. И., Горбачева В. О., Федюков Е. М. Структура, свойства и примене-
ние углеродных волокнистых материалов. Труды ВНИИВ, Мытищи, 1975,
с. 47—50.
32. Byrne G„ Jeffries R., Chem. a. Ind., 1971, № 29, p. 809.
313.
33. Herink С., Perret R., Ruland W., «Nature», 1968, v. 220, № 5162, p. 63.
34. Фиалков А. С. и др. Тезисы III Всесоюзной конференции по композиционным ма-
териалам. М., Ин-т металлургии им. Байкова, 1974, с. 128.
35. Warner D. R. International Conference on Carbon Fibres, their Place in Modern
Technology, 1974, London, paper № 3.
36. Brawn A. B. International Conference on Carbon Fibres, their Place in Modern
Technology, London, 1974, paper № 35.
37. Бавер А. И. и др., «Химия твердого топлива», 1971, № 2, с. 149.
38. Фридман Л. И. и др., Хим. волокна, 1977, № 1, с. 11; Левит Р. М., Райкин В. Г.
Международный симпозиум по химическим волокнам. Препринты. Калинин, 1974,
секция 1, с. 186—188.
39. Фридман Л. И. и др., Изв. АН БССР, Сер. хим. наук, 1974, № 2, с. 37.
40. Ермоленко И. Н., Морозова А. А., Гаврилов М. 3., ДАН БССР, 1974, т. 18,
№ 3, с. 234.
41. Фридман Л. И., Морозова А. А., Ермоленко И. Н., ДАН БССР, 1974, т. 18, № 12,
с. 1087—1090.
42. Ермоленко И. Н. и др., Изв. АН БССР, Сер. хим. наук, 1975, № 5, с. 20.
43. Лутков А. И., Захаров Ю. Н., Михайлов В. Н., «Химия твердого топлива», 1974,
№ 4, с. 125—128.
44. Тростянская Е. Б., Кобец Л. П., Гуняев Г. М., Пласт, массы, 1971, № 5,
с. 846—850.
45. Усенбаев К. Исследование структуры и -свойств переходных форм углерода.
Фрунзе, изд-во «Мектен», 1970. 153 с.
46. Касаточкин В. И., Каверов А. Т„ ДАН АН СССР, 1958, т. 120, № 5, с. 1007—1010.
47. Фиалков А. С. и др., «Химия твердого топлива», 1968, № 6, с. 191.
48. Абрамчук В. Е. и др. Тезисы III Всесоюзной конференции по композиционным ма-
териалам. М., Ин-т металлургии им. Байкова. 1974, с. 130—-131.
ГЛАВА 5
ПРИМЕНЕНИЕ УГЛЕРОДНЫХ ВОЛОКОН
Композиционные материалы КМ (композиты) представляют собой
системы, состоящие из волокон различной текстильной формы и свя-
зующих (матрица). В данной главе рассматривается применение компо-
зиционных материалов на основе углеродных волокнистых материалов
(УВМ) и собственно УВМ. Эти материалы относятся к новым, обладаю-
щим ценными механическими свойствами материалам с благоприятной
перспективой развития производства и применения их в народном хо-
зяйстве. В качестве связующих используются полимеры, углерод, кера-
мика, металлы (сплавы). Композиционные материалы подразделяют на
четыре типа в соответствии с используемой матрицей: углеродные во-
локна — полимерная матрица (КМП), углеродное волокно — углерод-
ная матрица (КМУ), углеродное волокно — керамика (КМК) и угле-
родное волокно — металлы (сплавы металлов) (КММ). В композитах,
как правило, сочетаются вещества с контрастными свойствами (волок-
но—матрица), и именно благодаря этому они приобретают новые техни-
чески ценные характеристики.
По сложившейся традиции под КМ обычно подразумеваются кон-
струкционные материалы с высокими механическими показателями.
€ появлением углеродных волокон значительно расширилось понятие
о КМ; к ним теперь относят наряду с конструкционными другие ком-
позиты, обладающие разнообразными свойствами и имеющие многооб-
разные области применения. Эти особенности КМ обусловлены изме-
няющимися в широком пределе свойствами углеродных волокон и свя-
зующих.
За короткое время разработаны процессы получения УВМ и КМ на
их основе, однако области применения этих новых материалов пока еще
314
ограниченны и ценные свойства их полностью не использованы. Такое
положение объясняется рядом причин, среди которых (особенно это
касается конструкционных КМ) немаловажное значение имеют трудно-
сти, связанные с преодолением традиций и психологических барьеров.
КМ являются наиболее материалоемкой сферой применения углеродных
волокон, и поэтому их использование во многом определяет перспекти-
ву и объемы производства, а следовательно, и стоимость УВМ.
5.1. КОМПОЗИЦИОННЫЕ МАТЕРИАЛЫ
НА ОСНОВЕ ПОЛИМЕРНЫХ СВЯЗУЮЩИХ (КМП)
5.1.1. Общие замечания
Для получения КМП применяются УВМ различной текстильной
формы: нити, жгуты, ленты, ткани, кноп, войлок и др. Помимо внешней
формы к волокнам в зависимости от назначения КМ предъявляются
различные требования. Волокна, предназначенные для получения кон-
струкционных пластиков, должы обладать высокими механическими
свойствами; волокна, используемые для получения теплозащитных
КМП, — высокой абляционной стойкостью; волокна, применяемые в ка-
честве нагревателей или антистатических компонентов, — заданными
электрофизическими свойствами и т. д. В ряде случаев оправдано при-
менение двух типов волокон в сочетании с одной матрицей. При этом
недостатки одного волокна компенсируются преимуществами другого;
в результате получается КМП лучшего качества. Не исключена возмож-
ность использования слоистых пластиков, в которых сочетается одно
волокно и связующие двух типов.
В качестве связующих (матрицы) служат термореактивные и тер-
мопластичные полимеры. Выбор связующего определяется назначением
КМП. Наиболее широко применяются термореактивные смолы. В тер-
мопластичные полимеры углеродные волокна вводят для придания ма-
териалам тех или иных специфических свойств.
Одна из особенностей КМП состоит в том, что оба компонента (во-
локно и связующее) являются высокомолекулярными соединениями.
К полимерным связующим предъявляются следующие требования:
высокая адгезия к волокну, определяющая напряжение сдвига; хорошая
смачиваемость волокна и связанная с ней пористость композита; тех-
нологичность при переработке; достаточно высокие механические свой-
ства, обеспечивающие наиболее полную реализацию свойств волокна
в композите.
Технология изготовления КМП во многом заимствована из техно-
логии получения стеклопластиков и описана в ряде монографий [1, 2].
Поэтому отметим лишь, что наиболее распространенными являются ме-
тоды прессования, осуществляемые в различных вариантах, и метод
намотки, реализуемый в двух основных вариантах (сухая и мокрая на-
мотка).
Для углеродных волокон характерна высокая хрупкость, малая
устойчивость к истиранию, малая изгибоустойчивость. В связи с этим
при их переработке в пластики для предохранения волокна от повреж-
дений необходимо соблюдать ряд условий: поддерживать низкое давле-
ние прессования, избегать малых углов перегибов, сводить по возмож-
ности к минимуму углы трения и др.
315
5.1.2. Конструкционные композиционные материалы
на основе полимерных связующих (ККМП)
Классическим примером природного конструкционного материала
является древесина, в которой силовым каркасом служат целлюлоз-
ные волокна, а связующим — лигнин, полиозы и пектиновые вещества.
Благодаря низкой плотности (кажущаяся плотность древесины 300—
1040 кг/м3) древесина характеризуется высокими удельными значения-
ми прочности и жесткости; она широко применяется в технике. До не-
давнего времени древесина служила одним из основных конструкцион-
ных материалов в самолетостроении, где предъявляются наиболее жест-
Рис. 5.1. Зависимость механических свойств композитов от механических
свойств УВ I(содержание УВ ,в композите 60%):
а — прочность композита при изгибе; б — модуль изгиба композита.
кие требования к конструкционным материалам. В 40-х годах древесина
была заменена металлами и сплавами. Однако возможности, заложен-
ные в металлах и сплавах, практически исчерпаны. За последние 25 лет
прочность алюминиевых сплавов повысилась на 100—150 МПа (10—
15 кгс/мм2), конструкционных сталей на 300 МПа, титана — примерно
на такую же величину; средний прирост теплостойкости составлял 8—
10°С/год [3]. Специальным способом удалось получить стали с проч-
ностью 3 ГПа, но повышение прочности повлекло за собой необычай-
ную чувствительность к концентрации напряжения в местах дефектов,,
избежать которых невозможно, к хрупкому излому стали, а также сни-
жению стойкости к коррозии в местах концентрации напряжений.
Новым этапом в развитии конструкционных материалов явилось
создание стеклопластиков, которые находят широкое применение. Вы-
сокопрочные высокомодульные волокна стимулировали появление угле-
пластиков с высокими легко регулируемыми механическими свойствами.
Одно из замечательных свойств углепластиков состоит в том, что рост
трещин в этих материалах под влиянием внешнего механического поля
задерживается вследствие поглощения энергии разрыва волокна [4].
Свойства конструкционных углепластиков зависят от свойств во-
локна и связующего. Важное значение имеет также технология изго-
товления композита. Для конструкционных КМП наиболее приемлемы-
ми оказались эпоксидные смолы, которые обладают достаточной адгези-
ей к волокну, не требуют применения высоких давлений при прессова-
нии и в процессе отверждения не выделяют низкомолекулярных летучих
соединений, повышающих пористость композита.
316
Прочность и модуль КМП непосредственно связаны со свойства-
ми исходного волокна. Как видно из рис. 5.1, по мере повышения проч-
ности и модуля Юнга волокна эти показатели возрастают для компо-
зита [5]. Следует отметить, что углепластикам присуща ярко выра-
женная анизотропия механических свойств, поэтому разработка мето-
дов их испытания представляет известные трудности. Обычно приво-
дятся показатели механических свойств однонаправленных пластиков,
испытываемых на изгиб или по методу кольца. Разница в показаниях,
полученных этими методами, незначительна.
Реализация свойств углеродного волокна в пластиках зависит от
ряда факторов. Элементарные волоконца в композите должны иметь
длину, превышающую ее критическое значение примерно в 10 раз [6].
Критическая длина волокна определяется из уравнения
do
1кр=^
где /кр — критическая длина;
d — диаметр волокна;
о — предел прочности волокна при растяжении;
Тед —напряжение сдвига (межслоевая прочность).
В разных источниках приводятся значения 1кр, равные 0,12—1 мм.
При длине волоконец, меньшей критической, затрудняется передача
напряжений между волокнами через матрицу, поэтому свойства ком-
позита ухудшаются.
В работе [7] суммированы данные о композитах, полученных
с использованием коротких волоконец. При соблюдении условий па-
раллельности расположения волокон получаются пластики с достаточ-
но высокими показателями (табл. 5.1).
Одним из существенных недостатков УВМ является разброс меха-
нических свойств, приводящий к снижению механических свойств угле-
пластиков. При большом коэффициенте вариации реализация прочно-
сти волокна в пластике уменьшается до 50—-70% [6].
Определенное влияние на механические свойства оказывает гео-
метрическое расположение волокон. Согласно работе [6], при нару-
шении параллельности расположения волокон более чем на 10° на-
блюдается снижение прочности и модуля КМ. Нежелательны также
искривления волокон (они не должны превышать 1,5—2°). Например,
Таблица 5.1. Свойства углепластиков, наполненных короткими волокнами
Волокно Длина волокна, мм Содержание волокна в композите, % (Об.) Прочность прн изгибе, 107 па Модуль Юнга при изгибе, 107 Па Прочность* при растяже- нии, 10? Па Модуль Юнга при растяжении, 10? Па
HMS 1 49 82,2 15 500 59,0 15 500
4 32 67,1 10 500 59,0 11200
6 24 56,2 8 000 49,8 8 300
6 31 69,9 10300 61,2 10 700
6 42 80,9 12 900 76,4 14 000
:HTS 1 43 97,7 9100 80,6 9 100
3 33 88,0 7 400 74,7 6 200
3 45 108,5 9900 93,2 9 700
6 42 89,0 9500 75,8 9100
* Здесь и далее под
прочностью подразумевается предел прочности.
317
при использовании некрученого волокна реализация его свойств в КМ
составляет 100%, тогда как крученого волокна (120 витков/м) —всего
29—45% (прочность) и 31—37% (модуль Юнга).
Большое значение имеет сродство матрицы к волокну, в том числе
характер поверхности и связанная с этим смачиваемость волокна. Пло-
хая смачиваемость способствует появлению пористости. Согласно ра-
Рис. 5.2. Влияние пористости на механические свойства углепластика:
1 — модуль Юнга; 2 — межслоевая прочность; 3 — прочность при изгибе.
боте [8], пористость мало сказывается на модуле сдвига и межслое-
вой прочности, но заметно снижает оИзг (рис. 5.2). Однако известны №
другие данные. В обстоятельной работе Митчелла [9] показано, что-
плотность, которая определяется пористостью композита, заметно ска-
зывается на модуле композита (рис. 5.3). По данным Д. М. Карпино-
са и сотр. [10], увеличение пористости с 2 до 10% понижает вдвое не
только а, но и тсд.
Рис. 5.3. Изменение модуля Юнга в зависимости от плотности композита.
Рис. 5.4. Зависимость прочности (/, 2) и модуля Юнга (3, 4) при изгибе
от степени наполнения пластика волокнам [2]:
1, 3 — кремнийорганическая матрица; 2, 4 — эпоксидная матрица.
Степень наполнения КМ волокном является одним из важных фак-
торов, определяющих механические свойства композита. С увеличе-
нием содержания волокна до определенного предела наблюдается по-
вышение прочности и модуля Юнга, затем показатели КМ начинают
ухудшаться, что обусловлено недостаточным количеством полимера;
318
для получения монолитного композита (рис. 5.4). Оптимальные свой-
ства пластика соответствуют 60—70 %-ному содержанию волокна.
К особенностям ККМП относится высокая выносливость к статиче-
ским и динамическим нагрузкам по сравнению со стеклопластиками
и ее сохранение в большом интервале низких температур вплоть до
—196 °C. Это положительное свойство ККМП обусловлено малой дефор-
мируемостью волокна и, следовательно, связующего, что препятствует
развитию трещин. Приведенные ниже данные наглядно иллюстрируют
преимущества ККМП по сравнению с другими материалами {11]:
Материал
Стеклопластик
Титан
Алюминий
Эпоксиугле-
пластик
Сохранение прочности после
10’ циклов, %
30
52
55
80
По данным работы [12], прочность КМП в зависимости от числа
циклов испытания выражается эмпирическим уравнением
акмп=4 — BlgN
где А и В — постоянные;
N — число циклов.
Характеристика выносливости пластика для разных волокон при-
ведена в табл. 5.2.
Беван и Старгеон [13] исследовали усталостные свойства эпокси-
углепластиков, усиленных высокопрочным и высокомодульным волок-
ном при температурах от —40 до +120 °C. Критерием оценки служили
изменения прочности при изгибе Стизг и сдвиге тсд; установлены высокие
усталостные свойства углепластиков.
Наряду с бесспорно ценными механическими свойствами ККМП не
лишены недостатков. Для них характерна низкая межслоевая проч-
ность (напряжение сдвига тСд), 20—30 МПа. В результате многочис-
ленных исследований были изысканы пути повышения тСд. Современ-
ные УВ обеспечивают получение эпоксиуглепластиков с тсд 60—
100 МПа, а по некоторым данным — даже 120 МПа.
Повышение межслоевой прочности пластика достигается двумя
путями: подготовкой поверхности волокна и вискеризацией. На тсд,
по-видимому, влияют следующие факторы: поверхность (ее характер
и величина), структура волокна и наличие реакционноспособных функ-
циональных групп. Базисные плоскости волокна имеют малую поверх-
ностную энергию и низкую реакционную способность. Наличие дефек-
та б л и ц а 5.2. Усталостные свойства эпоксиуглепластиков
Волокно А* В* Прочность после 10’ цик- лов, 10’ Па Сохранение прочности после 107 цик- лов, %
Z-2-1 (фирма «Ниппон карбон») 123 5,8 80 60
Z-2-2 (фирма «Ниппои карбон») 143 10,4 79 67
Т-300 А (фирма «Торей») 154 60,0 68 49
* Постоянная в приведенном выше эмпирическом уравнении.
31»
тов в плоскостях, включая появление неспаренных электронов, благо-
приятно сказывается на свойствах композита. Отрицательную роль,
видимо, играет отложение на поверхности волокна вторичного угле-
рода.
Значение структуры наглядно иллюстрируется приведенными ниже
данными [14] о зависимости тсд композита от модуля Юнга углерод-
ного волокна:
Модуль Юнга, ГПа...................... 35 182 462
Напряжение сдвига, МПа................. 94,5 28,0 14,0
Подготовка поверхности сводится к окислению углеродного волок-
на газообразными или жидкими окислителями. Условия окисления не-
обходимо подбирать с расчетом, чтобы возрастание тсд композита не
сопровождалось заметным падением прочности волокна. Основными
параметрами процесса служат температура и продолжительность об-
работки, полезно также следить за потерей массы волокна. Условия
обработки определяются также ТТО при получении волокна. Высоко-
модульное волокно, полученное при более высоких ТТО, обладает по-
вышенной стойкостью к окислению. Поэтому для его обработки требу-
ются более жесткие условия по сравнению с волокном, полученным
при более низкой конечной ТТО. Независимо от условий окисления
для волокна с низким <тсд во всех случаях достигается больший эффект.
Среди газообразных окислителей наибольшее внимание привле-
кает кислород воздуха. В работе [14] исследовалась область темпера-
тур 400—600 °C. Ниже приводятся данные об эффективности предва-
рительного окисления воздухом волокна с начальным модулем
420 ГПа. Условия обработки следующие: окислитель — кислород воз-
духа; температура 550 °C, продолжительность 15 ч, потеря массы во-
локна 1%. Для получения композита использовалась полиэфирная
смола марки SR17449 холодного отверждения и эпоксидная смола
«шелл 828» горячего отверждения.
Образец т , 10’ Па
Неокисленное волокно, смола SR17449 . . . 2,03
Окисленное волокно, смола SR17449 . . . . 4,06
Неокисленное волокно, смола «шелл 828» . 2,10
Окисленное волокно, смола «шелл 828» . . . 7,00
В патенте [15] описано применение кислорода в смеси с газом-
носителем; обработку волокна проводили при 475—600 °C в течение
2,5—7,5 мин.
Эффективна также сухая смесь кислорода и хлора [16] (табл.5.3).
В результате обработки волокна указанной смесью (температура
1250—1450 °C, продолжительность 0,3—0,8 с, потеря массы волокна
не более 2%, потеря прочности не более 15%) тсд композита возра-
стает в 2—3 раза.
Обработка жидкими окислителями — гипохлоритом натрия, азот-
ной кислотой, реагентом Хаммерса (смесь H2SO4 + NaNO3+KMnO4) —
проводится при невысоких температурах. Например, в работе [14]
указывается, что окисление УВ типа II гипохлоритом натрия проводи-
лось при обычной температуре в течение 2—63 ч (прочность УВ почти
не изменялась).
Результаты, полученные при обработке, зависят от свойств исход-
ного волокна: при тсд исходного композита 35 МПа тСд после обработки
320
Таблица 5.3. Влияние условий обработки на свойства углеродного волокна
и напряжение сдвига композита
Условия обработки Потеря массы, % Прочность волокна а, 107 Па Напряжение сдвига г t0? Па
температура, °C скорость дви- жения нити, м/ч продолжитель- ность обработки, с состав газовой смеси CI2: О2, %
1350+20 1800 0,4 93:7 0,8 207 8,7
1350+ 20 1800 0,4 97:3 0,1 193 5,9
1350+ 20 1350 0,5 97:3 0,1 197 9,4
1350 ±20 1800 0,4 77:23 0,3 102 9,0
1350 1800 0,7 75:25 0,5 86 8,9
Необработан- — — — 215 3,2
ное
Примечание. Для исследований использовалось УВ не основе ГЦ. смола эпоксидная
марки ERZ 2256.
возрастает до 84 МПа, а при тСд исходного 70 МПа—до 80 МПа. Такая же
закономерность наблюдается для УВ, окисленного кислородом воздуха.
В работе [117] применялось волокно АС (высокопрочное) и AG (высоко-
модульное); первое окислялось азотной кислотой, второе — наиболее
стойкое — реактивом Хаммерса. Авторы работы <[17] установили связь
между Тсд и содержанием кислотных групп в УВ. Полученные ими дан-
ные приведены ниже:
Исходное волокно Поверх- ность (по БЭТ). м2/Г Содержание кислотных групп, экв/г (нейтрализация NaOH) Напряже- ние сдвига тсД’ 107 Па
Волокно AG 0,37 3 2,2
Волокно окисленное AG-H 0,23 400 6
Волокно окисленное AG-H
быстро нагретое в азоте . . . 0,45 38 3,05
медленно нагретое в азоте . . 0,18 0 2,3
Волокно AG-Н метилированное (ката-
лизатор — кислота Льюиса) . . — 180 3,45
Из приведенных данных видно, что после нагревания окисленного
УВ в азоте (температура 300 °C) тсд падает; метилирование кислотных
групп вызывает аналогичный эффект.
Авторы работы [17] считают, что механизм действия жидких окис-
лителей, приводящих к повышению тсд композита, различный: при об-
работке HNO3 волокна AG возникающая шероховатость поверхности
способствует улучшению адгезии и тсд; на свойства других волокон
существенно влияют кислотосодержащие группы, химически взаимо-
действующие с матрицей.
Положительная роль функциональных групп отмечается также ав-
торами работы [14].
Рисе и сотр. [18] считают, что предварительная обработка эла-
стомерами, химически взаимодействующими с УВ, благоприятно ска-
зывается на тСд композита (тсд повышается с 56 до 89 МПа). Помимо
этого, эластомер выполняет функцию демпфера, в результате чего повы-
шается ударная прочность композита.
Митчелл [9] исследовал влияние исходного и предварительно
окисленного волокна, покрытых пироуглеродом, на тсд; при этом уста-
21—2254
321
новлено, что для неокисленного волокна тсд возрастает на 25%, а для
предварительно окисленного — на 35—60% по сравнению с теми же
показателями для волокон, не покрытых пироуглеродом. Напряжение
сдвига тСд заметно возрастает с увеличением плотности композита
(уменьшение пористости) и с увеличением содержания в нем волокна
(рис. 5.5, 5.6).
Немаловажное значение имеет природа матрицы. При замене
одной смолы на другую тсд возрастает с 82 до 114 МПа. Влияние при-
Рис. 5.5. Влияние плотности па межсловную прочность композита на основе
неокисленного волокна, покрытого пироуглеродом (1), и предварительно
окисленного волокна i(2).
Рис. 5.6. Влияние содержания волокна, неокисленного и покрытого пиро-
углеродом (/), и волокна, предварительно окисленного 1(2), на межслоевую
прочность композита.
роды эпоксидной смолы наглядно иллюстрируется приведенными ниже
данными [19]:
Марка смолы....................... 934 PR-282 5208 PR-288
Напряжение сдвига композита, 107 Па . . 6,02 6,79 8,26 8,54
Эффективным методом повышения тСд является вискеризация, ко-
торая может осуществляться двумя способами: выращиванием «усов»
на волокне или введением «усов» в матрицу; более рационален первый
способ. Как отмечается в работе [20], рост «усов» начинается в наи-
менее упорядоченных участках углеродного волокна. В этом сообще-
нии, а также в работе [21] указывается, что вискеризация приводит
к значительному повышению не только тСд, но и модуля Юнга, модуля
сдвига и прочности углепластика.
Влияние вискеризации на механические свойства пластика пока-
зано на рис. 5.7 [3]. Оптимальное содержание монокристаллов в пла-
стике не должно превышать 8—13% (об.).
Из двух рассматриваемых методов повышения тсд более эффек-
тивна вискеризация, но окисление, особенно кислородом воздуха, зна-
чительно проще и дешевле.
Оценивая роль тсд в механике композитов, следует учесть мнение,
высказываемое некоторыми исследователями, согласно которому по-
вышение тсд приводит к понижению ударной прочности композита.
322
к недостаткам композиционных материалов на основе пластиков
относится невысокая прочность на сжатие <тСж- Этот показатель замет-
но улучшается при вискеризации и поверхностной обработке волокна.
Например, в результате вискеризации прочность КМП на сжатие воз-
растает с 400 до 1300 МПа. Увеличение диаметра волокна приводит
к росту Ос®, однако для углеродных
Для повышения ударной прочности
предложено покрывать поверхность
волокна эластомерами, применять гиб-
ридные композиции из углеродного и
стеклянного волокна; при этом одно-
временно повышается сопротивление
сжатию.
Как уже отмечалось, для углепла-
стиков характерна анизотропия меха-
нических свойств, более ярко выра-
женная, чем для боро-, и тем более
для стеклопластиков. Для текстильных
форм пространственного переплетения
анизотропия выражена значительно
меньше, чем для пластиков, армиро-
ванных нитями [2]. Некоторое умень-
шение анизотропии достигается при
перекрестной укладке волокна, в част-
волокон этот путь неприемлем.
Рис. 5.7. Влияние содержания ните-
видных кристаллов нитрида кремния
на предел прочности композита при
сжатии,(/), сдвиге (2) и модуль сдви-
га |(3)-
иости ортогональной со взаимно перпендикулярным расположением
волокон.
Термостойкость ККМП полностью определяется матрицей. Соглас-
но работе [22], температурный интервал эксплуатации ККМП в зависи-
мости от матрицы лежит в следующих пределах:
Матрица
Политетра- Поли-
фторэтилен амид-6
Фенол о-
формаль- Эпок сид над
дегидная смола
смола
Температурный интервал экс-
плуатации, °C................ 108—120 185—200 250
150—200
В связи с синтезом большого числа термостойких полимеров ве-
дутся исследования по их использованию в качестве матрицы. В рабо-
те [23] отмечается, что в США получены углепластики с температурой
эксплуатации 350 °C. Г. М. Гуняевым и сотр. [2] приводятся несколько
отличные результаты длительного (около 1000 ч) ресурса работы КМ.
с различными связующими:
Температура
Смола (матрица) эксплуатации
°C
Эпоксидные............................. 200
Фенолоформальдегидные .... 250
Кремнийорганические.................... 300
Полиимидные............................ 330
Полихнноксолиновые..................... 360
Отмечается, что в ряде случаев прочность ККМП снижается при
применении термостойких смол. Последнее обусловлено их склон-
ностью к образованию трещин из-за плохой адгезии к волокну, высо-
кими остаточными напряжениями из-за усадки волокна и смолы при
отверждении и другими факторами.
21*
324
Аналогично термостойкости химическая стойкость ККМП также
зависит исключительно от матрицы. В табл. 5.4 приведены данные
о химической стойкости фенольных углепластиков к различным реаген-
там [24]. В этой серии опытов критерием оценки служило изменение
напряжения сдвига, зависящее от проникновения реагентов в глубь
ККМП и воздействия на поверхность раздела волокно — матрица. Наи-
более активными реагентами для данного связующего являются NaOH,
органические растворители, уксусная кислота при повышенной темпе-
ратуре и воздух. Неясна причина повышенной чувствительности к кис-
лороду воздуха ККМП, наполненную волокном, полученным при более
высоких температурах.
Таблица 5.4. Химическая стойкость фенолоуглепластиков в различных средах
[содержание волокна в пластике 50% (об.), продолжительность
обработки 100 сут]
Условия обработки Температура получения волокна Напряжение сдвига, % (от исходного] Изменение массы, % Изменение объема, %
14%-иая NaOCl; 23 °C Низкая 100 —7 —2
Метилэтилкетон; 23 °C » 75 +4 +6
10%-иая H2SO4; 23 °C » 100 0 0
HNO3 (плотность 1210 кг/м3); 23 °C Средняя 84 —4 0
30%-ная NaOH; 90 °C Низкая 69 +16 + 17
14%-ная NaOCl; 23°С Высокая 80 —10 -
10%-ная H2SO4; 50°C » 100 0 0
5%-ная FeCl3; 90°C » 100 0 0
30%-ная NaOH; 90°C » 72 + 10 +8
Воздух; 50 сут; 225 °C Низкая 100 —2 '—4
Пар; 50 сут; 225 °C » 90 0 +3
Воздух; 50 сут; 225 °C Средняя 64 --4 —3
Пар; 50 сут; 225 °C » 84 —2 0
Воздух; 50 сут; 225 °C Высокая 62 —3 +1
Пар; 50 сут » 82 —1 +1
Логическим завершением исследований ККМП явилось создание
конструкционных углепластиков. В них удачно сочетаются высокие
абсолютные значения прочности и модуля Юнга при низкой плотно-
сти, что приводит к большим удельным характеристикам, по которым
они превосходят все известные материалы, уступая лишь по удельной'
прочности стеклопластикам, однако последним присуща низкая жест-
кость (табл. 5.5).
Недостатками конструкционных углепластиков, как уже отмеча-
лось выше, являются относительно невысокие напряжение сдвига, пре-
дел прочности при сжатии и анизотропия свойств. Проводя соответст-
вующую подготовку поверхности волокна, напряжение сдвига удалось
повысить до приемлемых значений. Анизотропию свойств можно
уменьшить за счет различной укладки волокна в композите [2, 25].
Повысить устойчивость к удару и прочность при сжатии можно при
использовании гибридных композиций.
Наиболее перспективно совместное применение углеродного и стек-
лянного волокон с эпоксидной матрицей. Стеклянное волокно в отли-
чие от углеродного обладает высокой прочностью при сжатии. Однако
изготовление гибридных композиций оказалось не простой задачей.
324
Таблица 5.5. Свойства углепластиков и других конструкционных материалов
Материал Плотность, 103 кг/мЗ Прочность, 107 Па Модуль Юнга. 10Ю Па Удельная прочность, 10* Па мз/кг Удельный модуль упругости, 10? Па-мЗ/кг
Эпоксиуглепластик 17,2 82,2 10,8
с высокопрочным УВ 1,6 130
с высокомодульным УВ 1,7 56 20,4 33,0 12,0
Эпокеистеклопластик (S-во- локно) 2,12 192 6,9 91 3,25
Сталь Сплав 7,8 100 20,7 12,8 2,66
алюминия 2,7 34,5 6,9 12,8 2,55
магния 1,8 27,6 4,4 15,4 2,45
титана . 4,5 93,1 11 26 2,45
Наиболее целесообразно получение смешанных тканых материалов из
этих волокон. Такие работы были начаты в то время, когда высокопроч-
ное и высокомодульное волокна «модмор» получали в виде коротких
отрезков (1—1,5 м), потому эти работы не дали тогда положительных
результатов. Ткачество даже непрерывных углеродных нитей из-за их
хрупкости, низкой устойчивости к трению представляет большие труд-
ности, и тем не менее они были преодолены. При переработке углерод-
ных волокон требуется: подбор аппретирующих средств, желательно
без применения органических растворителей, снижение скорости тка-
чества, а следовательно, уменьшение производительности ткацких стан-
ков, кондиционирование воздуха в цехе, строгий контроль за нитепрово-
дящими узлами и др.
Успехи, достигнутые при переработке углеродных волокон в тканые
материалы, позволили получить гибридные ткани. Филлипс и Ловелл
[26] сообщили о получении лент из углеродных жгутиков, скрепленных
стеклянным волокном, а также тканей из углеродных и стеклянных во-
локон. Содержание углеродных волокон в ткани составляло 10, 25, 35,
45% (масс.). Для пластика, полученного из ленты, в которой основой
служило углеродное, а утком — стеклянное волокно, а также для конт-
рольного образца стеклопластика приводятся следующие характери-
стики:
Контрольный
Гибридный образец из
композит стеклопластика
Предел прочности при изгибе, МПа .... 766 880
Модуль Юнга, ГПа........................... 52,8 37
Напряжение сдвига, МПа..................... 43 39,6
* Содержание волокна в композите 60% (15% углеродного, 45% стеклянного).
Как видно из приведенных данных, для гибридных композитов
жесткость и напряжение сдвига выше, но прочность несколько ниже,
чем для стеклопластика. К сожалению, в работе не приводятся сведе-
ния о прочности композитов при сжатии.
Высокомодульные углеродные волокна (наиболее жесткие) трудно
подвергнуть текстильной переработке и трудно получить из них смешан-
ные ткани.
Из-за жесткости и хрупкости УВ при транспортировке возможно его
повреждение. Переработка, особенно методом намотки, также, затруд-
325
йена. По указанным причинам все большее применение находят пре-
преги [2, 23, 26], представляющие собой углеродный волокнистый мате-
риал, пропитанный неотвержденным связующим. Наиболее удобной тек-
стильной формой является волокно в виде лент, полученных ткачест-
вом или раскладкой жгутиков. Оправдал себя метод протяжки пропи-
танных жгутиков большой длины через узкие каналы. Препреги содер-
жат требуемое количество связующего и подвергаются переработке
прессованием или намоткой для получения изделий с их последующим
холодным или горячим отверждением. Наиболее важной характеристи-
кой препрегов является жизнеспособность, так как в случае отвержде-
ния связующего переработка препрегов в изделия исключается. Приво-
дятся противоречивые сведения, по которым стабильность свойств пре-
прегов сохраняется 3—12 месяцев. Жизнеспособность препрегоП опре-
деляется свойствами смолы и условиями хранения (при низких темпе-
ратурах «продолжительность жизни» значительно увеличивается).
5.1.3. Другие типы композиционных материалов
на основе полимерных связующих (КМП)
Конструкционные углепластики являются одним из основных, но
не единственным видом композиционных углепластиков. Сочетание
углеродных волокон с различными полимерами позволяет получить
технически ценные материалы. Да и сами конструкционные пластики
можно применять для самых разнообразных целей.
КМП обладают исключительно ценными термическими характери-
стиками. Они могут использоваться как теплозащитные материалы, но
максимально возможная температура эксплуатации определяется мат-
рицей, термостойкость которой относительно невысокая.
В связи с развитием ракетостроения и освоением космоса возник-
ла потребность в новых материалах, способных кратковременно про-
тивостоять высоким температурам. При прохождении ракетами или
космическими кораблями нижних слоев атмосферы на поверхности
аппаратов, особенно носовых частей, развивается температура 5000—
6500 °C. В двигателях ракет также создаются большие тепловые на-
грузки [27]: температура 2516 — 3066 °C, скорость газового потока
1589,5—1840 м/с. Ни один из известных в природе материалов не мо-
жет длительное время противостоять столь высоким температурам. На
границе движущейся ракеты и воздуха возникают сложные физико-
химические процессы — абляция. Оказалось, что наибольшей абляци-
онной стойкостью обладают КМ с фенолоформальдегидной смолой. На
рис. 5.8 показана абляционная стойкость пластиков, наполненных раз-
личными волокнами.
В отличие от полимеров КМП обладают повышенной теплопро-
водностью. В тех случаях, когда ее необходимо уменьшить, в состав
углепластиков вводят волокна с малой теплопроводностью. Высокая мо-
розостойкость позволяет использовать КМП в криогенной технике. Бла-
годаря низкому термическому коэффициенту расширения КМП прису-
ща стабильность формы при различных температурах.
Введение в полимеры углеродных волокон значительно снижает
коэффициент трения, и как следствие этого резко возрастает износо-
устойчивость углепластиков. Этому способствует также повышенная
теплопроводность КМП по сравнению с полимерами. Устойчивость
326
к истиранию повышается: для поливинилхлорида в 3,8 раза; политет-
рафторэтилена — в 3 раза; полипропилена — в 2,5 раза; полиамида —
в 1,2 раза; полиэфира — в 2 раза [28].
В большом числе работ [4, 22, 26, 28] особое внимание уделяется
целесообразности введения углеродного волокна в политетрафторэти-
лен, особенно при его использовании для изготовления подшипников.
Износоустойчивость особое значение приобретает для термопла-
стичных полимеров, которые часто в процессе эксплуатации подвер-
гаются именно этому виду механического воздействия. Как правило,
для получения композитов в этом случае используются короткие угле-
родные волокна. Композицию получают методом экструзии при боль-
ших скоростях, поэтому производительность оборудования высокая,
количество отходов незначительно.
Обычно этим способом получают полу-
фабрикат, который используется для
изготовления изделий. Объектами ис-
следования служили многие полимеры:
полиолефины, полистирол, полиамиды,
поликарбонаты, политетрафторэтилен
и др. Свойства некоторых КМП на
основе термопластичных полимеров
приведены в табл. 5.6 [29]. Введение
волокна упрочняет полимер, но по
сравнению с конструкционными компо-
зиционными материалами прочность
этих композиционных материалов не-
велика.
К недостаткам углетермопластов
относится крипп, низкая термо- и теп-
лостойкость. При наполнении полиме-
ра непрерывными волокнами проч-
Рис. 5.8. Стойкость к абляции пла-
стиков, усиленных различными во-
локнами:
I — асбестовое; 2 — стеклянное; 3 — крем-
неземное; 4 — найлоновое; 5 — кварцевое;
6 — графитированное.
ность композита выше и зависит от прочности волокна.
Введение в полимер даже небольших количеств волокна исключа-
ет возможность накопления статического электричества, что в ряде
случаев расширяет области применения КМП. В этом случае полимер
Таблица 5.6. Свойства углепластиков на основе термопластичных полимеров
Состав композита Предел проч- ности при растяжении, 107 Па Модуль Юнга, 107 Па Предел проч- ности при УДаре. отн. ед. Теплостой- кость, °C
полимер (матрица) ВОЛОКНО
Полисульфон 7,15 252 1,3 175
Стеклянное 14,5 700 1,0 185
Углеродное 10,3 1120 1,0 185
Поликарбонат — 7,35 224 15,0 140—145
Стеклянное 11,2 770 3,0 150
Углеродное 11,2 980 1,6 175
Полифениленоксид — 4,9 265 5,0 115
Стеклянное 14 630 1,9 145
Углеродное 10,5 1260 1,1 142
Найлон 6,6 — 8,2 294 0,9 70—105
Стеклянное 13,3 630 1,2 250
Углеродное 21,0 1400 0,8 252
32Г
наполняется короткими волокнами, к которым не предъявляют вы-
соких требований по механическим свойствам.
Из углеродного кнопа в смеси с целлюлозными волокнами на
обычных бумагоделательных машинах получают электропроводящие
бумаги с различными номиналами сопротивления. На основе бумаги
готовят слоистые пластики, которые в ближайшее время найдут широ-
кое применение в качестве нагревателей различного назначения.
В зависимости от связующего КМП обладают различной химиче-
ской стойкостью. На основе термореактивных смол, и в частности фе-
нолоформальдегидных, как наиболее доступных и дешевых, получают
достаточно стойкие композиты (см. табл. 5.4). Такие химически стой-
кие КМП, безусловно, представляют большой интерес прежде всего
для химического машиностроения.
5.2. КОМПОЗИЦИОННЫЕ МАТЕРИАЛЫ НА ОСНОВЕ
УГЛЕРОДНОГО ВОЛОКНА И УГЛЕРОДНОЙ МАТРИЦЫ (КМУ)
Необычные свойства углерода послужили стимулом для проведе-
ния исследований по получению композитов с углеродной матрицей.
Композиты, представляющие собой систему углеродное волокно —
углеродная матрица (КМУ), являются еще одним подтверждением
многообразных свойств углеродных материалов.
Известны два основных способа получения КМУ. Первый основан
на карбонизации углепластиков. В процессе карбонизации происходит
термодеструкция матрицы, сопровождающаяся усадкой и образова-
нием большого числа пор. Для устранения этого недостатка операцию
пропитка — карбонизация повторяют несколько раз. Повторную про-
питку рекомендуется проводить пеками [30], одним из преимуществ
которых является повышенное содержание углерода и высокий выход
коксового остатка. По этим же причинам предлагается применять в ка-
честве исходного материала углеродное волокно, пропитанное пеком
[31]. Как правило, завершающей операцией является уплотнение ком-
позита пироуглеродом. Второй способ получения КМУ заключается
в том, что на углеродное волокно наносят пироуглерод, причем опера-
цию повторяют также несколько раз.
Свойства КМУ зависят от свойств исходного волокна, матрицы
и ее связи с волокном, выхода коксового остатка, свойств кокса, чис-
ла циклов пропитка — карбонизация и условий проведения процесса.
В качестве исходной матрицы используются преимущественно фено-
лоформальдегидные смолы, которые в наибольшей степени удовлетво-
ряют необходимым требованиям. Пироуглерод наносят из газовой
фазы при разложении метана или других углеводородов.
Большое значение имеет текстильная форма волокна. Возможно
применение тканей, лент, войлока, жгутов (нитей), объемных струк-
тур. Главное требование к исходной форме волокна — обеспечить
изотропность композита, добиться высокой межслоевой прочности,
определяющей механические свойства композита и его устойчивость
к тепловому удару. Наиболее полно требованиям к исходной форме
волокна удовлетворяют трехмерные структуры или войлок.
При использовании нитей (лент) получают однонаправленный
КМУ с максимальной прочностью. Улучшение сдвиговых характеристик
достигается соответствующей укладкой волокон. Оптимальные углы
составляют от ± 10 до ± 20°. При укладке волокна под углом 20° проч-
328
ность в поперечном направлении возрастает вдвое с одновременным
повышением прочности в продольном направлении.
Композиты на основе углеродной матрицы получают в виде ли-
стов [9]. Резаное волокно замешивают с раствором ПВС, обрабатыва-
ют раствором ФФС, подсушивают и карбонизуют до ТТО=ЮОО°С.
Затем полуфабрикат уплотняют пироуглеродом при ТТО 2000—
2400 °C, используя в качестве источника углерода метан, пропан или
природный газ. Прочность листа толщиной 5 мм составляет 294 Н
(30 кгс) на полоску шириной 5 см.
По мнению авторов [32—34], процесс начинается на границе раз-
дела фаз с образованием прочной связи углеродное волокно — смола,
при этом волокно служит своеобразной матрицей. Ориентационный
эффект волокна, возникающий вследствие напряжений на границе раз-
дела фаз из-за их различного термического коэффициента расширения
(18-10—6—23-10-6 1/°С для волокна; 3,5-10-6 1/°С для смолы), приво-
дит к появлению анизотропии композита.
В работе [35] исследовано влияние свойств исходного волокна и
условий его получения на свойства КМУ.
Существенное влияние на свойства КМУ оказывают свойства
углеродных волокон, и в частности температура получения волокна и
композита. Если ТТО при получении волокна выше ТТО при карбониза-
ции композита, свойства волокна практически не меняются, а в случае
более высокой ТТО при карбонизации они изменяются так же, как при
получении углеродного волокна.
С увеличением содержания волокна в композите его прочность
возрастает (рис. 5.9).
Уплотнение КМУ углеродом происходит в результате разложения
метана (как источника углерода) при температуре 1100 °C. Скорость
газового потока и повышение давления благоприятно сказываются на
свойствах КМУ [35] (табл. 5.7). Оптимальное давление составляет
26,6—39,9 кПа. По другим источникам, целесообразно поддерживать
давление на уровне 2,66 кПа. КМУ обладают целым рядом уникаль-
ных свойств. Им прежде всего присуща необычайно высокая тепло-
стойкость. Так, в инертной среде прочность остается неизменной вплоть
Таблица 5.7. Влияние условий получения на свойства КМУ
(в поперечном направлении)
Условия процесса Прочность КМУ Модуль КМУ
давление, кПа (мм рт. ст.) скорость пода- чи газа, мл/мин графитирован- ного, 10? Па иеграфитирован- ного, 10? Па графитирован- ного, ЮЮ Па н еграфитированно- го, 1010 Па
6,65 (50) 18,3 (100) 33,25 (250) 66,5 (500) 100 250 500 100 250 500 100 250 500 100 250 500 27,5 27,5 41,3 34,4 43,4 49,6 44,5 40,0 52,2 44,8 27,5 44,1 26,8 30,3 32,4 34,4 39,2 38,6 48,2 43,4 61,4 44,8 43,4 4,82 5,51 8,26 5,51 8,96 7,57 8,26 6,89 6,20 9,65 4,14 7,57 4,14 5,51 6,2 1,38 4,82 6,20 2,76 8,96 8,96 7,57 9,65
329-
до температуры 1000 °C, модуль Юнга практически не зависит от тем-
пературы испытания. Необычайно высокую теплостойкость КМУ среди
жаростойких материалов иллюстрирует рис. 5.10. В этом композите
сочетается химическая инертность наполнителя и матрицы. Такой ма-
териал стоек ко всем, кроме сильнодействующих окислителей, химиче-
ским реагентам. Он обладает также необычайно высокими фрикцион-
ными свойствами.
Механические свойства в зависимости от исходных материалов и
условий получения, а также назначение композита меняются в широ-
Рис. 5.10. Зависимость удельной прочности от температуры испытания жаро-
стойких материалов:
I — ориентированный КМУ на высокомодульиом волокне н пироуглеродной матрице;
2— эпоксиборопластик; 3 — алюминий, армированный борным волокном; 4— никель,
армированный высокомодульиым углеродным волокном (60°/в-ное наполнение); 5 —
бериллий; 6 — нержавеющая сталь; 7 — сплав RENE-41; 8 — углепластик «carbitex»;
9 — сплав ТД — никель; 10 — КМУ на основе углеродного войлока и пироуглеродной
матрицы; 11 — графит АТУ.
ких пределах. Предел прочности при растяжении составляет 300—
1200 МПа, модуль Юнга 155—211 ГПа, прочность при истирании
300 МПа. В табл. 5.8 приведены данные о свойствах углерод-угле-
родных композитов, полученных на основе войлока; эти композиты
обладают достаточно хорошими механическими свойствами [19] и мо-
гут служить конструкционным материалом. Плотность КМУ составля-
ет 1300—1500 кг/м3; коэффициент теплопроводности изменяется от
Таблица 5.8. Свойства углерод-углеродных пластиков, полученных
на основе войлока и других материалов
Материал Плотность волокна. Юз кг/м’ Содержание волокна. % (об.) Прочность прн изгибе, 107 Па Прочность при сдвиге, 107 Па
Модмор II G-1550 на основе гидратцеллю- лозы Войлок на основе гидратцеллю- лозы 1,69 1,84 1,81 1,87 1,71 1,80 3 10 18,4 21,7 34 46 5 15 22,4 16,5 14,4 7,7 3,56 4,2 3,56
330
9.88 Вт/м при 425 °C до 11,598 Вт/м при 1025 °C; коэффициент терми-
ческого расширения в пределах 20—60°C составляет 0,5-10—6 1/°С,
а при 60—1000 °C—1,10~6 1/°С. Ползучесть композита очень низкая.
5.3. КОМПОЗИЦИОННЫЕ МАТЕРИАЛЫ НА ОСНОВЕ
УГЛЕРОДНОГО ВОЛОКНА И КЕРАМИЧЕСКОЙ МАТРИЦЫ (КМК)
При сочетании углеродного волокна с керамикой можно получить
материал с уникальными свойствами. Для керамики характерны хи-
мическая инертность и особенно высокая стойкость к окислителям, тер-
мостойкость, достаточно высокие прочность и жесткость. К недостат-
кам керамики относятся низ-
кая усталостная прочность,
хрупкость, низкое сопротивле-
ние тепловому удару и удар-
ным нагрузкам. Введение в
керамику (стекло) углеродных
волокон позволит в значитель-
ной мере устранить перечис-
ленные недостатки керамики.
В свою очередь, керамика мо-
жет защитить углеродное во-
локно от действия кислорода
воздуха; это позволяет полу-
чать композиты, которые мож-
но эксплуатировать в воздуш-
ной среде при высоких темпе-
ратурах. На рис. 5.11 приведе-
йлюминиевая Никелевая керамическая
Матрица,
Рис. 5.11. Интервалы рабочих температур ком-
позиционных материалов с различной матри-
цей.
ны максимально возможные предельные температуры эксплуатации
композитов с различными матрицами.
Получение КМК оказалось достаточно сложной технической проб-
лемой, и в этом направлении проводятся интенсивные поиски, которые
пока еще не вышли за рамки лабораторных исследований.
Химическая инертность керамики затрудняет получение компози-
тов. Попытки сочетать волокна с расплавленным стеклом приводят
к окислению волокна и потере им механических свойств. В работе
[36] для нанесения керамики на волокно предложено применять смесь
следующего состава [% (об.)]: керамика — 5, поливинилацетат (склеи-
вающее вещество) — 10, метилэтилкетон — 85. Скорость пропускания
Таблица 5.9. Свойства композитов с матрицей из стекла и стеклокерамики [38J
Состав композита Характер структуры Прочность, 10? Па Работа разруше- ния, КДж/м2
матрица волокно содержание волокна, % (Об.) пористость, % (об.) при изгибе при меж- слоевом сдвиге
открытая закрытая
Стекло пи- Высокомодуль- 45,9 0,6 1,1 45,9 5,9 3,1
реке ное Высокопрочное 48,9 0,3 4,9 57,5 7,6 3,6
Стеклокера- Высокомодуль- 49,5 7,3 1,6 55,8 3,2 4,5
мика ное Высокопрочное 45,7 4,7 9.0 57,4 2,6 10,3
331
шнура через ванну 2 м/мин. Свойства композита зависят от температу-
ры и давления прессования. Повышение температуры (приводятся пре-
делы 1000—1400 °C) и давления (6,3 МПа и 10,5 МПа) способствует
увеличению прочности композита и уменьшению его пористости. Опти-
мальное наполнение композита волокном составляет 40% (об.). Полу-
чены композиты с пределом прочности при изгибе 1000 МПа, при сдви-
ге — 70 МПа.
локном, равна
toooy
900 -
& 800 - о
«а о
L 700-
ь* 800-
200 -
X
ЮО - *
Ударная вязкость стекла, армированного углеродным во-
104 Дж/м2, а обычного стекла — 10 Дж/м2, т. е. в 1000 раз
меньше. В работе [37] и в табл. 5.9
° [38] приводятся свойства КМК на
° о о основе стеклянной матрицы.
° ° ° Наиболее интересны термические
характеристики КМК. В инертной сре-
де (азот) прочность композита при
температурах до 800 °C мало меняется
и значительно превосходит прочность
| | матрицы — стекла (рис. 5.12). Однако
। ।____, на воздухе преимущества композита
000 ООО 800 не столь существенны. Максимальная
температура длительной эксплуатации
составляет 400 °C. Видимо, кислород
проникает внутрь материала и окис-
ляет углеродное волокно. Авторы ра-
боты [37] сочетали углеродное волок-
но с цементом, в результате механи-
ческие свойства композита резко воз-
росли. Однако значительная разница
г, °C
Рис. 5.12. Влияние температуры на
изменение прочности стеклокерамики
и композита стеклокерамика — угле-
родное волокно в среде азота:
X — исходная стеклокерамика; О — компо-
зиты стеклокерамика — углеродное во-
локно.
в цене цемента и углеродного волокна пока исключает возможность
применения последнего для получения подобных композитов.
5.4. КОМПОЗИЦИОННЫЕ МАТЕРИАЛЫ НА ОСНОВЕ
УГЛЕРОДНОГО ВОЛОКНА И МЕТАЛЛИЧЕСКОЙ МАТРИЦЫ (КММ)
В КММ удачно сочетаются и взаимно дополняются свойства метал-
лов и УВМ. Введение углеродного волокна в металлы позволяет зна-
чительно повысить теплостойкость, стойкость к тепловым и ударным на-
грузкам, усталостные характеристики и уменьшить ползучесть металлов
[39]. Многие металлы и сплавы являются конструкционными материа-
лами, но они зачастую не удовлетворяют требованиям, предъявляемым
к ним по механическим показателям. Наполнение таких металлов УВ
позволит резко улучшить их механические свойства, особенно такие,
как прочность и модуль Юнга. В свою очередь, металлы защищают во-
локно от действия кислорода воздуха, что наряду с увеличением тепло-
стойкости приводит к повышению предельных температур эксплуатации
КММ. Однако если кислород диффундирует между зернами металла
к границе раздела фаз, то некоторые металлы катализируют окисление
углерода.
Получение КММ связано с техническими трудностями, но в этом
направлении ведутся интенсивные исследования, что объясняется пер-
спективностью рассматриваемых материалов.
При получении КММ следует учитывать возможность взаимодей-
ствия волокна с рядом металлов с образованием карбидов и раствори-
мость углерода в расплаве металла. В результате этих процессов про-
332
исходит разупрочнение углеродного волокна. Например, карбиды алю-
миния, титана, хрома образуются при температуре 500 °C, а кобальта —
при 600—800 °C [40]. Никель не образует карбидов, но растворяется
в волокне и разрушает его. Температура и продолжительность контак-
та определяют глубину протекания этих процессов и вызываемое ими
нежелательное явление. В. И. Касаточкин и А. С. Фиалков [41] выска-
зали оригинальную мысль, которая сводится к тому, что за счет л-элек-
тронов происходит химическое взаимодействие углеродного волокна
с металлом. Этому способствует склонность базисных плоскостей волок-
на к образованию слоистых соединений. К физико-механическим аспек-
там проблемы относятся внутренние напряжения термического и меха-
нического происхождения. Необходимо также учитывать факторы, ко-
торые имеют место при получении любых композитов (смачиваемость,
текстильная форма волокна, равномерность распределения волокна, ад-
гезия и др.). Способность взаимодействия волокна с металлами умень-
шается с повышением модуля Юнга волокна. Вискеризация УВМ приво-
дит к упрочнению связи на границе раздела фаз волокно — металл.
С учетом перечисленных моментов разрабатываются различные
способы получения КММ: пропитка углеродного волокна расплавами
металлов или диффузионное спекание, предварительное покрытие УВ
металлом при низких температурах (электрохимический метод нанесе-
ния металла, разложение газообразных соединений металлов), предва-
рительное получение полуфабрикатов плазменным напылением метал-
ла, пропускание волокна через расплав металла, горячее прессование
в режиме контактного плавления и др.
Основными параметрами получения КММ являются температура
и давление прессования, а также продолжительность контакта УВ с ме-
таллом при высокой температуре.
Наибольший практический интерес представляют композиты алю-
миний — УВ, никель — УВ. Алюминий и его сплавы являются одним из
основных конструкционных материалов, но алюминий характеризуется
относительно низкими механическими свойствами. Введение в алюми-
ний УВ благоприятно сказывается на этих показателях. Для компози-
ции никелевая матрица — УВ характерна высокая температура эксплуа-
тации (см. рис. 5.10).
С целью предотвращения образования карбидов и разрушения во-
локна перспективным оказалось предварительное нанесение на волокно
барьерного слоя TiC [42], SiC [43, 44]. В случае никелевой матрицы
эффективно предварительное нанесение на волокно никеля [45], пред-
варительное легирование волокна никель-титановым сплавом [46]. По
данным Олда [47], композиции сплав (Ni+Al)—УВ неперспективны.
Обнадеживающие результаты были получены для композиции сплав
(Pb + Sn) — УВ. В этом случае удается повысить прочность сплава.
В работе [48] рекомендуется композит на основе медной матрицы.
Свойства КММ, как и других композитов, зависят от свойств и со-
держания волокна в нем, условий получения композита, включающих
большое число параметров, и других факторов.
Из приведенных в табл. 5.10 данных видно, что разработаны мето-
ды, позволяющие получить КММ с высоким наполнением волокном.
При оптимальных условиях — пропитка волокна расплавом алюминия,
содержащим 11 % кремния, — удалось получить композит с высокой
прочностью 1190 МПа. Все алюминиевые композиты имеют достаточно
высокую прочность; исключение составляет композит алюминий — вы-
333
Таблица 5.10. Свойства композитов с металлической матрицей
Композит Способ получения Объем- ное напол- нение, % Предел прочности при рас- тяжении, 107 Па Коэффициент использования» %
способ нанесения металла последующая обработка
Л* В*
Никель—торнель 50 Химический Горячее прессование 50 83 80 100
Никель—торнель 50 Электрохимический То же 40 79 80 по
Никель—высокомо- 50 77 80 100
дульное УВ Алюминий—низко- модульное УВ » 35 48 80 80
Алюминий—высо- Пропитка распла- — 45 119 85 —
копрочное высокомо- дульное УВ вом алюминия, содер- жащим 11% (об.) Si
Алюминий—высо- Пропитка под дав- — 35 35 61 58
комодульиое УВ пением
Алюминий—тор- Пропитка распла- — 28 90 130 —
иель 50 вом алюминия, содер- жащим 13% (об.) Si
То же То же — 35 67 100 —
Алюминий—высо- Из газовой фазы Горячее 35 54 80 —
копрочное высокомо- дульиое УВ прессование
Медь—высокопроч- Электрохимический То же 35 55 100 —
иое УВ Медь—низкомо- дульиое УВ > 40 91 100 100
* А — коэффициент использования, рассчитанный по о исходного волокна.
В — коэффициент использования, рассчитанный по о волокна, извлеченного из композита.
сокомодульное УВ (см. табл. 5.10). Аналогичные результаты получены
для композитов на основе никелевой и медной матрицы [40]. Композит
никель — УВ имеет высокую теплостойкость (см. рис. 5.10). Согласно
данным работы [2], максимальная температура эксплуатации компози-
та никель — УВ составляет 950 °C, что значительно выше температуры
эксплуатации никеля. По данным В. С. Ивановой [39], предельная тем-
пература эксплуатации КММ повышается до 0,8—0,9 Тил металла, тогда
как для металлов она не превышает 0,6—0,7 Тпл.
В большинстве случаев (см. табл. 5.10) коэффициент реализации
прочности волокна в композите довольно высок.
При использовании металлической матрицы лучшие результаты по-
лучаются с монокристаллами («усами») [1] и борным волокном, но
они, особенно «усы», гораздо дороже УВМ, поэтому придается столь
большое внимание использованию для этих целей УВ.
5.5. ПРИМЕНЕНИЕ КОМПОЗИЦИОННЫХ МАТЕРИАЛОВ
В последнее время довольно четко определилось использование КМ
на основе УВМ для замены традиционных материалов и в областях,
где они являются незаменимыми.
Благодаря удачному сочетанию высоких механических свойств
с низкой плотностью конструкционные КМП являются перспективными
материалами для использования прежде всего в авиа-, вертолетострое-
нии и космической технике. В этом направлении ведутся широкие иссле-
дования и испытания в США, ФРГ, Англии, Швеции. В литературе
334
[49—54] приводятся данные о целесообразности применения КММ
в следующих элементах конструкции самолетов: крыльях, фюзеляже,
элементах управления и стабилизации, балках полов, секциях пола и
лонжерона, носовой и несущей кромках винтов, дверях, оболочках об-
текателей крыла, шасси, гасителях скорости, секциях крыла и др.
Применение КМП значительно улучшает тактико-технические дан-
ные самолетов. Роль КМП наглядно иллюстрируется данными на при-
мере самолета ИЛ-62, приведенными в работе А. Т. Туманова [3]. Их
использование может обеспечить снижение взлетной массы на 17%,
увеличение дальности полета при сохранении взлетной массы на 15%,
увеличение полезной нагрузки на 20%, уменьшение линейных размеров
самолета на 12%. В общем случае отношение массы конструкции само-
лета к его взлетной массе может быть снижено на 7—10%, а отношение
массы конструкции двигателя к его мощности — на 8—10%, что недо-
стижимо при использовании традиционных материалов. В ряде источ-
ников [55, 56] указывается, что масса самолета за счет применения
КМП может быть уменьшена на 25—35%. Особенно огромный выигрыш
получается для аппаратов с вертикальным взлетом (экономия массы
30%, увеличение подъемной силы на 100%) [54].
Высокая химическая стойкость предопределяет возможность ис-
пользования конструкционных КМП в судо- и химическом машино-
строении [24]. Огромное число бакового оборудования в химической
промышленности эксплуатируется в агрессивных средах при давле-
ниях 0,03—0,05 МПа. Для изготовления такого оборудования можно
применять волокна со средней прочностью, стоимость которых значи-
тельно ниже стоимости высокопрочных волокон. Для изготовления хи-
мического оборудования целесообразно использовать композиционные
материалы на основе ФФС; они наиболее доступны и химически стойки.
Для снижения стоимости оборудования его можно изготовлять из стек-
ло- и углепластиков, при этом два-три слоя углепластика служат в ка-
честве антикоррозионного материала. Внедрение КМП в химическую
промышленность дает возможность заменить дорогостоящие химически
стойкие специальные стали. Рекомендуется применение КМП в тек-
стильном машиностроении [27].
Длительные сроки испытания новых материалов в авиастроении
привели к диспропорции между созданными мощностями производства
этих материалов и их потреблением авиационной промышленностью.
В сложившейся ситуации одним нз основных потребителей конструк-
ционных КМП оказались производители спортивного инвентаря [23].
Из КМП изготовляют клюшки для гольфа, обода ракеток для тенниса
и бадмингтона, корпуса гоночных автомобилей, лыжи, корпуса лодок,
удилища, лыжные палки, клюшки для хоккея и др.
В связи с высокой стоимостью КМП перспективным оказалось при-
менение пластин (реек) для придания жесткости отдельным элементам
конструкции самолетов, спортивного инвентаря, лодок и других изделий.
Заслуживает внимания применение комбинированных материалов,
например КМП в сочетании со стеклопластиком, алюминием, древеси-
ной и другими материалами. Применение КМП облегчает изделие и
придает ему жесткость. Из рис. 5.13 четко видно возрастание жесткости
алюминия прн использовании углепластика [57].
Композиционные материалы с металлической матрицей (КММ)
применяются в тех же областях, где н сами металлы (сплавы), но они
обладают преимуществами по прочности, жесткости и теплостойкости.
335
Наибольшей теплостойкостью, стойкостью к тепловым ударам,
абляционной устойчивостью и химической стойкостью обладают КМУ.
Эти материалы используются в ракетостроении, в узлах, испытываю-
щих наибольшие тепловые нагрузки. Среди композиционных материа-
лов [58] КМУ обладают рядом существенных преимуществ (табл. 5.11).
Рис. 5.13. Диаграмма на-
грузка (изгиб)—деформация
алюминиевой пластины тол-
щиной 1,5 мм до (/) и пос-
ле 1(2) армирования двумя
пластинами углепластика
толщиной 0,25 мм.
В них сочетаются прекрасные тепловые ха-
рактеристики и высокие механические показа-
тели, особенно их удельные значения. Наибо-
лее удачны трехмерные структуры, заготовки
которых изготовляются на специально сконст-
руированных для этих целей ткацких стан-
ках. Такие материалы применяются для защи-
ты носовых кромок космических аппаратов
многократного использования [59]. О приме-
нении КМУ в ракетной технике США сообща-
лось также в докладе [35]. КМУ является
незаменимым теплозащитным материалом при
больших тепловых нагрузках. Превосходно
зарекомендовали себя КМУ в качестве тор-
мозных устройств самолетов [35]. Сообща-
лось об их использовании при создании топ-
ливных элементов.
КМУ являются также прекрасным мате-
риалом для химического машиностроения.
Решение проблемы использования КМК
находится пока еще на стадии, не позволяю-
щей оценить их практическую значимость.
Электрофизические свойства УВМ позволяют на их основе изготовлять
гибкие и жесткие нагреватели, предназначенные для обогрева жилых
помещений, животноводческих ферм, киосков и других сооружений, а
также для отверждения бетона в зимних условиях. Решение этой проб-
лемы приобретает особенно важное значение для районов Крайнего Се-
вера. Разработан процесс получения слоистых декоративных пластиков
для обогрева помещений. Эти материалы успешно прошли испытание.
Полимеры находят все большее применение в быту и технике. Во
многих случаях электризация изделий из них создает немалые трудно-
сти при эксплуатации, а иногда по этой причине исключается возмож-
ность их использования. Введение в полимеры небольшого количества
УВ устраняет их электризуемость. Добавка УВМ значительно повышает
антифрикционные свойства полимеров. Применение УВМ для этих це-
лей может иметь огромное практическое значение в связи с использова-
нием полимеров для изготовления подшипников и в других узлах тре-
ния машин. В США политетрафторэтиленовые подшипники выпускают-
ся исключительно с добавками углеродного волокна.
5.6. ПРИМЕНЕНИЕ СОБСТВЕННО УГЛЕРОДНЫХ
ВОЛОКНИСТЫХ МАТЕРИАЛОВ
Высокая термостойкость позволяет применить УВМ в качестве теп-
лозащитных средств. Для этих целей пригодны различные волокнистые*
формы, но, пожалуй, наиболее целесообразно использовать войлок. При
плотности ПО кг/м3 коэффициент теплопроводности войлока составляет
336
Таблица 5.11. Свойства материалов,
применяющихся в конструкциях иеохлаждающегося сопла ракет
Материал Температура плавления (сублимации), °C Плотность, 103 КГ/мЗ Прочность при изгибе или растяжении, 10? Па Модуль при изгибе или растя- жении, 10Ю Па Теплопро- водность. ВТ/(м К) Применение
Обычный си- ликатный гра- фит 3700 1,65 2—3,4 3,4—6,9 40—120 Для защиты внутренней по- верхности, кон- тактирующей с горячими газа- ми
Высококаче- ственный синте- тический гра- фит Композиция ФФС — асбе- стовое волокно 3700 1,88 3,4—4,8 10—17,2 40—86 То же
Обугливание при Т>500 °C 1,67 6,2—7,6 0,4—0,69 Тепловой барь- ер для защиты внешней по- верхности, кон- тактирующей с горячими газа- ми
Композиция ФФС — крем- ниевое волокно То же 1,7 16,5—18,6 —- 0,86—1,4 То же
Сталь (4*/о углерода) 1550 7,84 62,0 20,7 49 Конструкцион- ный материал
Сплав алю- миния 570 2,83 о/ 6.89 146 То же
Композиция углеродная матрица — уг- леродное во- локно типа модмор I 3700 1,68 34—52 13—17 1—2 Конструкцион- ный материал; также для за- щиты материа- ла, контакти- рующего с го- рячими газами
0,381 Вт/(м-К), удельная теплоемкость при 20 °C равна 0,63 кДж/(кг-К).
Углеродные ткани являются прекрасными тепловыми экранами.
Благодаря высокой и легко изменяемой в широких пределах элек-
тропроводности УВМ находят разнообразное применение. Сообщается
об изготовлении из углеродных тканей обогреваемых одеял, грелок и
других приспособлений. Для районов Крайнего Севера особое значение
приобретает обогреваемая одежда. На основе углеродной ткани, кото-
рая в виде полосок монтируется в одежду, выпускаются обогреваемые
костюмы. Ткань обеспечивает равномерный нагрев, а небольшая масса
нагревателей позволяет осуществлять почти мгновенный нагрев и мгно-
венное охлаждение ткани, что.облегчает автоматическое регулирование
температуры. Описаны также специальные костюмы для персонала, ко-
торый обслуживает линии высокого напряжения; благодаря электропро-
водности ткани обеспечивается безопасность работы в такой одежде.
Углеродные ткани можно применять для самолетов с целью предотвра-
щения обледенения. Исследуется возможность применения углеродных
волокон для изготовления сеток, радиоламп, термопар, антенн, устано-
вок для улавливания пыли и др. На основе углеродных волокон изготов-
22—2254
337
ляются электропроводящие бумаги с различным сопротивлением, имею-
щие разнообразные области применения.
Высокая химическая стойкость предопределяет применение УВМ
для фильтрации агрессивных сред, очистки дымовых газов и других
целей.
Для углеродных волокон вследствие их малого диаметра (2—
12 мкм) характерна необычайно развитая поверхность; благодаря та-
кой поверхности сводятся к минимуму внутренние и внешние диффузи-
онные сопротивления массообмена и значительно облегчается скорость
его протекания.
Следует учесть также, что УВ имеют чрезвычайно развитую по-
ристость и активную поверхность, достигающую 2000 м2/г. УВМ различ-
ных текстильных форм можно применять в виде тонких слоев с малым
гидродинамическим сопротивлением [60]. Исходя из этого, были наме-
чены пути использования УВМ как сорбентов. Их рекомендуется при-
менять для очистки воздуха [61—65], в противогазах [63, 66, 67], филь-
трах автомобильных кондиционеров [61], в респираторах [61], для
очистки и осветления жидкостей [61, 63], в фильтрах сигарет [68], при
изготовлении защитной одежды [61, 62, 64, 66, 69, 70], применяемой
в химической промышленности, и др.
Развитая поверхность позволяет использовать УВМ в качестве но-
сителей катализаторов [63, 71]. Получены катализаторы, содержащие
металлы Pd, Pu, Ро, Pt или их соли, обеспечивающие 100%-ную конвер-
сию СО в СОг.
На основе У В созданы иониты [72—74], в которых удачно соче-
таются высокие емкость (1—6 мг-экв/г) и химическая стойкость.
Указывается, что УВ используются в качестве химических источни-
ков тока.
Выше перечислены основные направления использования собствен-
но УВМ, которыми отнюдь не исчерпывается многообразие возможных
областей применения их в различных отраслях промышленности.
ЛИТЕРАТУРА
1. Карол-Порчинский П. Материалы будущего. М., «Химия», 1966, 237 с.; Современ-
ные композиционные материалы. Под ред. Л. Браутмана, Р. Корка. М., «Мир»,
1970. 350 с.; Композиционные материалы волокнистого строения. Под ред.
И. П. Францевич и Д. М. Карпиноса. Киев, «Наукова думка», 1970. 380 е.
2. Пластики конструкционного назначения. Под ред. Е. Б. Тростянской. М., «Химия»,
1974. 303 с.
3. Туманов А. Т„ «Вестник АН СССР», 1975, № 3, с. 37—44.
4. Diefendorf R., Tokarsky Е., Polymer Eng. a. Sci., 1975, v. 15, № 3, p. 150—159.
5. Tsukada S. e. a. International Conference on Carbon Fibres, their Composites and
Applications, London, 1971, paper № 5.
6. Туманов А. И. и др., «Механика полимеров», 1975, № 2, с. 248—257.
7. Dingle L. E. International Conference on Carbon Fibres, their Place in Modern
Technology, 1974, paper № 11.
8. Hancox N. L„ J. Mater. Sci., 1975, v. 10, № 2, p. 234.
9. Mitchell S. J. International Conference on Carbon Fibres, their Place in Modern
Technology, London, 1974, paper № 20.
10. Карпинос Д. M. и др. Тезисы III Всесоюзной конференции по композиционным
материалам. М., Ин-т металлургии им. Байкова, 1974, с. 122.
11. Фydзuвapa М., Eng. Mater., 1974, v. 22, № 9, р. 37.
12. Фу рута Г., Начучи Д., Матушима М„ Techn. Rep. Mat. Aerospace, 1974, v. 1, № 366,
> 1
338
13. Bevan L. I., Sturgeon J. B. International Conference on Carbon Fibres, their Place
in Modern Technology, London, 1974, paper № 32.
14. Clark D., Wodswor N. J., Watt W. International Conference on Carbon Fibres, thei,
Place in Modern Technology, London, 1974, paper № 7.
15. Англ, заявка 1366244 (1974).
16. Фр. заявка 2123366 (1972).
17. Donnet В., Papirer Е. International Conference on Carbon Fibres, their Place in Mo-
dern Technology, London, 1974, paper Na 9.
18. Riess J., Bourdeaux №., Brie M. International Conference n Carbon Fibres, their
Place in Modern Technology, London, 1974, paper № 8.
19. Gibson D. W. New Horizonz Mater, a. Process., Azussa, Cai it., 1973, p 165
20. Работное Ю. H. и др. Тезисы III Всесоюзной конференции по композиционным
материалам. М., Ин-т металлургии им. Байкова, 1974. с. I.
21. Сорокина Т. И. и др. Тезисы III Всесоюзной конференции по композиционным ма-
териалам. М., Ин-т металлургии нм. Байкова, 1974, с. 55-—56.
22. Okida К., Japan Plast. Age, 1974, v. 12, № 1, p. 35.
23. Robbins D., Hayes C. International Conference on Carbon Fibres, tbeir Place tn
Modern Technology, London, 1974, paper № 42.
24. Hart G. L., Pritchard I. International Conference on Carbon Fibres, tbeir Place in
Modern Technology, London, 1974, paper №34.
25. Гуняев Г. M. и др. Тезисы III Всесоюзной конференции по композиционным ма'.-
риалам. М., Ин-т металлургии им. Байкова, 1974, с. 54—55.
26. Phillips L. N„ Lavell D. R. International Conference on Carbon Fibres, their Place in
Modern Technology, London, 1974, paper № 10.
27. Конкин А. А. Углеродные и другие жаростойкие волокнистые: материалы М;
«Химия», 1974. 375 с.
28. Отани С., «Тидзюцу сире Мицубиси сэкино кабусики кайся», 1973, № 51, р. 45..
29. Segol С. L., 19th Nat. SAMPE. Symp. a. Exhib. Buena Park, Calif., 1974. 19- New
Ind. Appl. Mater. Technol. Azussa. Calif., 1974, p. 277.
30. Фр. заявка 2171414 (1973),
31. Англ, заявка 1339291 (1973).
32. Tavarna A. R., McAllister L. E., Adv. Compos, a. Carbon S. np., Am. Ceram. Soc.
Chicago, Columbus, Ohio, 1973, p. 149.
33. Cooper G. A. e. a., J. Mater. Sci., 1974, v. 9, № 5, p. 835.
34. Hiskigama J. e. a., «Carbon», 1974, v. 12, № 3, p. 249.
35. Hill Thomas C. R., Walker E. J. International Conference on Carbon Fibres, their
Place in Modern Technology, London, 1974, paper № U.
36. Pambell A. J., Phillips D. C., Bomen D. H. International Conference on Carbon Fibres,
Their Place in Modern Technology, London, 1974. paper № 16.
37. Briggs A., Bomen D. H. International Conference on Carbon Fibres, their Place in
Modern Technology, London, 1974, paper № 1 /.
38. Phillips L. N., J. Mater. Sci., 1974, v. 9, p. 1547.
39. Иванова В. С. Тезисы III Всесоюзной конференции по композиционным материа-
лам. М., Ин-т металлургии им. Байкой, 1974, с. 11—13.
40. Jackson Т. W., Metals Eng. Quart., 1969, v. 9, p. 22; hacker A. 4., Shipman C..
Cripmell P. A., Rieber. Sci. a. Tecnn., 1972, v. 5, p. 255; Backet A. A., Mater. Sci.
a. Eng., 1975, v. 17, p. 177; Lumley E. L. Gillin M. Preprints of the 4th Australien
Ceramic Conf., August, 1970.
41. Касаточкин В. И. и др. Тезисы III Всесоюзной конференции по композиционным
х териалам. М., Ин-т металлургии им. Байкова, 1974, с. 125—126.
42. B' UtmKoe А. Н. и др. Тезисы III Всесоюзной конференции по композиционным и
териалам. М., Ин-т металлургии им. Байкова, 1974, с. 73.
43. Салибеков С. Е. и др. Тезисы III Всесоюзной конференции по композиционным
материалам. М., Ин-т металлургии им. Бийкова, 1974, с. 74—75
44. Багров Г. Н. и др. Тезисы III Всесоюзной конференции по композиционным мате-
риалам. М., Ин-т металлургии им. Байконя, 1974, с. 131—132
45. Салибеков С. Е. и др. Тезисы III Всесоюзаой конференции но композиционным ма-
териалам. М., Ин-т металлургии им. Байкова, 1974, с. 75.
46. Гроза Н. И. и др. Тезисы III Всесоюзной конференции по композиционным мате-
риалам. М„ Ин-т металлургии им. Байкова, 1974, с. 78.
47. Old С. Е. International Conference on Carbon Fibres, thei» Place in Modern Technolo-
gy, London, 1974, paper № 13.
48. Mortimer D. A., Mickolas M„ Cripin R. M. International Conference on Carbon Fibres,
their Place in Modern Technology, London, 1974, paper № 15.
49. Gill R. M. Carbon Fibres in Composite Materials. Published for the Plastics Institute,
London, 1972.
22*
339
50. Кутников В. Ф. Тезисы III Всесоюзной конференции по композиционным материа-
лам. М., Ин-т металлургии нм. Байкова, 1974, с. 25.
51, Pinckney R. L. International Conference on Carbon Fibres, their Place in Modern
Technology, London, 1974, paper № 38.
52. Mayer N. J. International Conference on Carbon Fibres, their Place in Modern
Technology, London, 1974, paper № 39.
53. Вольгин А. С., «Механика полимеров», 1972, № 1, с, 105.
54. Синицын Е. Н. и др. Тезисы III Всесоюзной конференции по композиционным ма-
териалам. М., Ин-т металлургии им. Байкова, 1974, с. 26.
55. Гуняев Г. М. и др. Тезисы III Всесоюзной конференции по композиционным ма-
териалам. М., Ин-т металлургии им. Байкова, 1974, с. 14—16.
56. Синицын Е. Н. и др. Тезисы III Всесоюзной конференции по композиционным
материалам. М., Ин-т металлургии им. Байкова, 1974, с. 26—27.
57. Motoro F., Japan Plast. Age, 1973, v. 11, p. 32.
58. Parmec L. C. International Conference on Carbon Fibres, their Composites and
Applications, London, 1971, paper № 38.
59. Carnachan K. R., Kleger R. V., Demsey P. K. Adv. Compos, a. Carbon Symp., Am. Ce- •
ram. Soc., Chicago, Columbus, Ohio, 1973, p. 171.
60. Фридман П. И. и др., Хим. волокна, 1971, № 1, с. 11.
61. Arong I. N„ Macnair R. N., Text. Res. J., 1974, v. 44, № 11, p. 876.
62. Arong J. N., Macnair R. N„ Erikson R. S., Text. Res. J., 1973, v. 43, № 9, p. 539.
63. Швейц, пат. 514500 (1970).
64. Пат. США 3744534 (1973).
65. Weber S., «Textilveredlung», 1971, Bd. 6, № 5, S. 412.
66. Пат. ФРГ 2246572 (1973).
67. Пат. США 3849332 (1974).
68. Англ. пат. 1256048 (1970).
69. Arong J. N., Macnair R. N., Erikson R. L., Text. Res. J., 1972, v. 42, № 1, p. 60—63.
70. Пат. США 3769144 (1973).
71. Фр. пат. 2198893 (1974).
72. Авт. свид. СССР 286218 (1968); Открытия. Изобр. Пром, образцы. Товарн. знаки,
1970, № 34.
73. Яп. пат. 7523 (1972).,
74. Otam S. е. a.. Bull. Chem. Soc. Japan, 1972, v. 45, № 6, p. 1908.
340
Часть III
НЕГОРЮЧИЕ
ВОЛОКНА
содержание
Введение................................................................... 343
Глава 1. Процесс горения и механизм огнезащиты...............................345
1.1. Процесс горения.........................................................345
1J2. Механизм огнезащитного действия антипиренов.............................350
1.3. Методы определения огнестойкости текстильных материалов.................354
Глава 2. Целлюлозные волокна..................................................356
2.1. Разложение целлюлозы и механизм ог/.езащй.'.’ ...........................356
2.2. Методы придания огнестойкости целлюлозным воло«. '• к тканям . . 358
2.2.1. Введение добавок в прядильный раствор..............................358
2.2.2. Поверхностная обработка тканей.....................................359
2.2.3. Химическая модификация.............................................365
Глава 3. Полиамидные волокна...............................................372
3.1. Термическое и термоокислительное-разложение полиамидов................373
3.2. Методы придания огнестойкости полиамидным волокнам и тканям . . . 378
3.2.1. Поверхностная отделка тканей.................................. 378
3.2.2. Добавки антипиренов в полимер...................................382
3 2.3. Химическая модификация полимера и волокна....................383
Г лава 4. Полиакрилонитрильные волокна................................. 398
4.1. Ме’.сды придания огнестойкости полиакрилонитрильным волокнам . . 399
4.1.1. Химическая модификация........................................399
4.1.2. Формование волокон из смесей полимеров.................... . 402
4.1.3. Введение добавок в прядильный раствор при формовании волокна . 403
4.1.4. Поверхностная отделка волокон и тканей........................405
Глава 5. Полиэфирные волокна..........................................407
5.1. Термическое и термоокислительное разложение полиэтилентерефталата . . 4<:
5.2. Методы придания огнестойкости полиэфирным волокнам..................40
Литература............................................................411
ВВЕДЕНИЕ
Полимерные материалы с новыми ценными свойствами получают
не только путем синтеза новых высокомолекулярных соединений, но и
химической модификацией известных полимеров или изделий из них.
Чрезвычайно актуальной задачей, в частности, является получение
огнестойких полимерных материалов и текстильных изделий.
В настоящее время значительно возрос спрос как на огнестойкие
текстильные товары широкого потребления, так и на материалы спец-
назначения. Если раньше требование в отношении огнестойкости предъ-
являлось в основном к декоративным тканям, рабочей одежде и мате-
риалам для военных целей, то в последние годы в ряде стран приняты
законы, запрещающие применение воспламеняющихся материалов для
детской одежды и предметов домашнего обихода. Повысились требова-
ния к огнестойкости текстильных материалов, используемых в авиации,
автомобильной промышленности и других отраслях.
Возникают новые области использования химических волокон
в более жестких условиях эксплуатации (воздействие открытого пламе-
ни, высоких температур, повышенное содержание кислорода и т. д.).
Проблема создания негорючих текстильных материалов может ре-
шаться в двух направлениях — получение химических волокон на осно-
ве огнестойких полимерных материалов и придание огнезащитных
свойств существующим типам волокон.
По вопросам синтеза волокнообразующих полимеров, обладающих
повышенной стойкостью к действию высоких температур, и исследова-
нию в области получения волокон с огнезащитными свойствами на их
основе имеется обширная литература.
В данной части монографии обобщен и систематизирован материал,
относящийся к области разработки методов придания огнестойкости
химическим волокнам, выпускаемым в промышленном масштабе. Осве-
щено общее состояние проблемы огнестойких волокон, кратко изложены
принципы огнезащиты полимерных материалов й получения материалов
с огнезащитными свойствами на основе целлюлозных, полиамидных,
полиакрилонитрильных и полиэфирных волокон.
Традиционные природные и синтетические волокна, за исключением
галогенсодержащих, широко применяемые для изготовления товаров
народного потребления и в технике, являются горючими материалами.
Появившиеся в последнее время новые типы негорючих синтетических
волокон — термостойкие на основе ароматических полиамидов и поли-
эфиров, из гетероциклических и лестничных полимеров не могут удов-
летворить спрос на огнестойкие волокнистые материалы. Области их
применения ограничены только техническим сектором и то в небольшом
масштабе. Поэтому проблема уменьшения горючести известных типов
343
многотоннажных химических волокон приобретает особо важное
значение.
Проблема огнезащиты текстильных материалов возникла давно.
Первый патент на огнезащитную обработку бумаги и льняного волокна
смесью, состоящей из квасцов, сульфата железа и буры, был выдан
в 1735 г. В 1820 г. Гей-Люссак предложил использовать в качестве огне-
защитных средств хлорид и сульфат аммония.
Работы в этом направлении проводились постоянно. Однако до не-
давнего времени основным направлением научных исследований была
разработка методов придания негорючести целлюлозным волокнам и
тканям. С развитием промышленности синтетических волокон, увеличе-
нием объема их производства и применения возникла необходимость
разработки методов придания огнестойкости материалам из этих воло-
кон.
Исследование процесса горения различных типов химических волб-
кон, а также разработка методов получения огнестойких материалов
является предметом многочисленных исследований как в Советском
Союзе, так и за рубежом. Однако теория процессов горения и огнеза-
щиты синтетических волокон еще мало освещена в литературе. Способ-
ность большинства синтетических волокон плавиться затрудняет разра-
ботку методов придания им огнестойкости. Поэтому все чаще к термо-
пластичным волокнам предъявляются новые требования — огнестой-
кость, неплавкость.
Для придания полимерным материалам повышенной стойкости
к действию огня используются различные методы:
1) поверхностная обработка тканей;
2) введение добавок в полимер перед формованием;
3) химическая модификация волокон или изделий из них.
При поверхностной отделке тканей, простой в технологическом от-
ношении, часто ткани не придается стабильная огнестойкость, так как
отделочные материалы могут вымываться из ткани. Стабильная огне-
стойкость достигается путем введения добавок в полимер перед формо-
ванием; в этом случае добавки не вымываются, но этим путем для тер-
мопластичных волокон не достигается неплавкость. Химической моди-
фикацией удается получать волокна с высокими и стабильными огнеза-
щитными свойствами, однако относительная сложность проведения про-
цесса снижает в ряде случаев практическую ценность данного метода.
Выбор того или иного метода в каждом конкретном случае должен
определяться требуемой эффективностью огнезащиты, физико-механи-
ческими свойствами получаемых волокон и тканей, технологическим
оформлением процесса и технико-экономическими показателями.
Универсальных методов огнезащиты текстильных материалов не
существует вследствие того, что процесс горения различных полимеров
протекает по разным механизмам и зависит от химического состава и
строения полимера, а также характера продуктов, выделяющихся при
разложении. Так, многие известные антипирены, хорошо действующие
при введении в целлюлозный материал, оказываются неэффективными
для ряда синтетических волокон из-за иного механизма разложения
этих полимеров.
Несмотря на большое количество средств, предложенных для огне-
защиты полимерных материалов, можно считать, что удовлетворительно
решена только проблема получения огнестойких целлюлозных материа-
лов. Для синтетических волокон еще не разработано достаточно прием-
344
лемого в технологическом и аппаратурном оформлении эффективного
метода придания термопластичным материалам одновременно негорю-
чести и неплавкости.
ГЛАВА 1
ПРОЦЕСС ГОРЕНИЯ И МЕХАНИЗМ ОГНЕЗАЩИТЫ
1.1. ПРОЦЕСС ГОРЕНИЯ
Причиной возгорания текстильных материалов может быть откры-
тое пламя, нагретая поверхность с температурой выше 300 °C и искра
электрического тока.
Процессам пиролиза, горения и распространения пламени посвяще-
но большое число работ [1—5]. Горение рассматривается как процесс
окисления, возникающий в условиях самоускорения и сопровождающий-
ся выделением большого количества тепла и излучением света. Горение
полимерных материалов — сложный процесс, зависящий от многих фак-
торов. До настоящего времени не существует достаточно четкой теории
горения синтетических материалов. Известно, однако, что причиной их
воспламенения являются низкомолекулярные соединения, образующиеся
в процессе термического разложения полимера. Поэтому состав продук-
тов пиролиза и скорость их выделения оказывают значительное влия-
ние на горючесть материалов.
Схему горения упрощенно можно представить следующим обра-
зом. Тепло, выделяемое источником огня, поднимает температуру поли-
мера выше температуры разложения, что приводит к выделению лету-
чих горю*чих продуктов, которые смешиваются с воздухом и попадают
к фронту пламени, в результате чего происходит возгорание. Тепло от
воспламенения продолжает генерировать горючие газы, и горение про-
должается.
Условно процесс горения можно разделить на следующие стадии:
Г) нагревание вещества;
2) пиролиз;
3) воспламенение;
4) горение;
5) распространение пламени.
Горение может сопровождаться также побочными явлениями: вы-
делением дыма, токсичных газов и плавлением материала.
Нагревание. При нагревании поверхности материала скорость воз-
растания температуры зависит от большого числа факторов: характера
окружающей среды, температуры и скорости нагрева, теплопроводности
вещества, удельной теплоемкости, теплот плавления и испарения.
Причиной повышения температуры вещества могут служить также
экзотермические радикально-цепные реакции. Нагретый материал мо-
жет образовывать газовую или жидкую фазы, способные реагировать
с кислородом воздуха. Однако накопление в материале энергии за счет
химических процессов на этой стадии еще недостаточно и не может при-
вести к воспламенению.
Пиролиз. При достижении материалом температуры разложения
начинается образование продуктов пиролиза. Механизм пиролиза и ха-
рактер образующихся газов во многом определяют легкость возгорания
345
материала. В процессе разложения образуются горючие и негорючие
газы, жидкие продукты, обугленный остаток. Состав продуктов пироли-
за в основном и обусловливает воспламеняемость материала. Если об-
разуются негорючие газы (НС1, НВг, СОг, пары Н2О), например при
пиролизе ароматических, термостойких полимеров, то воспламенения (и
соответственно горения) не происходит.
Однако большинство органических материалов разлагается с обра-
зованием легколетучих горючих веществ, таких, как Н2, СН4, С2Н4,
С2Н6, СН2О, СО и др.
Текстильные материалы в пожарном отношении особенно опасны
в том случае, когда при пиролизе выделяется много горючих соедине-
ний, особенно если пиролиз протекает с большой скоростью.
Выделяющиеся в процессе горения наряду с горючими негорючие
газы имеют тенденцию в большей или меньшей степени замедлять го-
рение материала по следующим причинам:
1) тяжелые газы затрудняют диффузию окислителя к горящей
массе;
2) негорючие газы, поглощающие тепло, способствуют охлаждению
поверхности материала;
3) испарение воды и других негорючих жидких продуктов пиролиза
способствует понижению температуры материала.
При определенных условиях пиролиз происходит по всему объему
материала. Если при пиролизе одновременно с низкомолекулярными
органическими образуются низкокипящие полифункциональные соедине-
ния, на поверхности материала возможно образование пространствен-
ного полимера. Поскольку горение начинается с поверхности, слой об-
разующегося полимера затрудняет диффузию органических соединений,
выделяющихся при пиролизе, из массы полимерного материала к зоне
горения и тем самым затрудняет горение.
Снижение горючести материала во многом зависит от образования
твердых обугленных продуктов. Наличие таких продуктов замедляет
диффузию газов из материала и уменьшает дальнейшее термическое
разложение благодаря тепловой изоляции.
Степень разложения полимерных материалов определяется темпе-
ратурой разложения и суммарной теплотой процесса. При экзотермиче-
ском процессе разложения высвобождаемая энергия способствует повы-
шению температуры, в то время как при эндотермическом процессе ос-
новная часть тепла расходуется на разложение полимера.
На основании термогравиметрического анализа (ТГА) установлено,
что введение в полимерный материал замедлителей горения (так назы-
ваемых «антипиренов») приводит к снижению температуры начала раз-
ложения и повышению выхода карбонизованного остатка по сравнению
с исходным материалом. Процессу карбонизации полимеров особенно
способствуют фосфорсодержащие соединения [6].
Воспламенение. Горючие газы, выделяющиеся при пиролизе в при-
сутствии кислорода воздуха, воспламеняются, что приводит к началу
горения. Воспламенение зависит от следующих факторов:
1) температуры вспышки, при которой газы, образующиеся при
разложении, могут быть зажжены пламенем или искрой;
2) температуры самовоспламенения;
3) минимального количества кислорода, необходимого для поддер-
жания горения;
4) состава продуктов пиролиза.
346
Для термопластичных полимеров воспламеняемость зависит также
и от плавления, так как температура плавления, как правило, ниже тем-
пературы воспламенения и самовоспламенения и воспламенение мате-
риала в этом случае затруднено. Айнзеле [2] для различных синтетиче-
ских волокон приводит следующие данные по температуре их воспламе-
нения (табл. 1.1).
Таблица 1.1. Термические характеристики волокон
Волокно Температура плавления, °C Температура вос- пламенения от постороннего источника, °C Температура само- воспламенения, °C
Хлопок 350 400
Триацетатное ' - 340 480
Полиамидные
поликапроамид (найлон 6) 215 390 510
анид (найлон 6,6) 255 — 532
Полиакрилонитрильное 215—255 250 515—560
Полиэфирное 250 390 508
Полипропиленовое 165 375 495
Номекс 375—400 490 675
Автор считает, что чем больше разница между температурой плав-
ления и температурой воспламенения от постороннего источника, тем
труднее воспламенить термопластичное волокно. Однако высказанное
положение оправдывается только при испытании на горючесть по вер-
тикальному методу тонких легких тканей из термопластичных волокон.
В случае тяжелой плотной ткани пламя успевает распространиться
раньше, чем расплавится большая часть материала.
Горение. Горение так же, как и процессы воспламенения, зависит
от многих факторов. Суммарный тепловой эффект при горении, учиты-
вающий энергию на разрушение различных связей в материале, и теп-
ло, выделяемое при окислении продуктов, может быть как положитель-
ным, так и отрицательным. В первом случае избыток тепла приводит
к новышению температуры вещества, во втором — горение будет под-
держиваться при дополнительном подводе энергии.
Основными энергетическими факторами, влияющими на процесс
горения, являются теплота сгорания и энергия химических связей.
Исходя из общих представлений, наличие в макромолекуле поли-
мера химических связей с высокой энергией [7, с. 654] должно способ-
ствовать повышению стойкости материала к действию высоких темпера-
тур. Необходимо, однако, отметить, что наличие групп С—Br, С—С1,
С—1 с низкой энергией связи замедляет горение, так как разложение
таких соединений приводит к выделению атомов галогена, которые яв-
ляются эффективными антипиренами. В литературе [2; 8—10] приво-
дятся следующие величины теплот сгорания наиболее распространенных
1ипов волокон (табл. 1.2).
Из приведенных данных видно, что горение синтетических волокон
сопровождается значительным выделением тепла. Для замедления горе-
ния желательны более низкие значения теплот сгорания, поскольку,
если энергии, выделяемой при горении, недостаточно для поддержания
горения, пламя затухает, а чем больше тепла выделяется при горе-
нии, тем больше опасность распространения пламени. При этом на
347
Таблица 1.2. Теплота сгорания некоторых типон волокон
Волокно Температура пламени, °C Теплота сгорания. кДж/г
Хлопок 880 18,8
Вискозное 15,5
Шерсть 580 20,7
Полиэфирное 697 23,8
Найлон 6,6 875 33,0
Полиакрилонитрильное 855 36,0
Полипропиленовое 839 43,9
Полиэтиленовое — 46,4
распространение пламени основное влияние оказывает не общее коли-
чество тепла, выделяемое при сгорании 1 г волокна, а количество тепла,
выделяющееся в единицу времени.
Существенное влияние на общий энергетический баланс полимер-
ных материалов оказывают также межмолекулярные связи, в частности
водородные (энергия водородной связи 29,3—41,8,кДж/моль).
Суммируя вышесказанное, можно сделать вывод, что высокая
энергия химических и межмолекулярных связей, а также низкая тепло-
та сгорания будут повышать стойкость полимерных материалов к дей-
ствию высоких температур.
Для воспламенения и поддержания горения необходима определен-
ная концентрация кислорода в газовой среде. Фенимор и Мартин [11]
разработали метод испытания для определения минимальной концент-
рации кислорода в смеси с азотом, при которой поддерживается непре-
рывное горение материала.
Ниже приводятся минимальные концентрации кислорода в газовоз-
душной среде, необходимые для горения различных типов волокон (так
называемый «показатель кислородного индекса»):
Волокно
11олнэтиленовое
Полиакрилонитрильное
Триацетатное
Полипропиленовое
Вискозное
Хлопок
Найлон 6
Найлон 6,6
Показатель
кислородного
индекса, %
17,5
18,0
18,4
18,6
18,9
19,0
20,0
20,1
Волокно
Полиэфирное
Шерсть
Номекс
Ацетохлорин
Кайнол
Поливинилхлоридное
Поливиннлиденхлорндное
Фторлон
Политетрафторэтнленовое
Показатель
кислородного
индекса, %
22,0
25,2
28,2
34,0
35,0
37,1
60,0
69,0
95,0
Как видно из приведенных данных, наиболее низкий кислородный
индекс имеют целлюлозные, полиакрилонитрильные и полиолефиновые
ткани.
Распространение пламени. Процесс горения зависит от эффективно-
сти нагревания поверхности материала, т. е. от притока тепла. Если
этой энергии достаточно для пиролиза и воспламенения, то пламя рас-
пространяется. Скорость распространения пламени зависит от химиче-
ского строения вещества, а для ткани — и от ее технических характери-
стик: толщины и типа переплетения.
Термопластичные волокна могут затухать после воспламенения, так
348
как фронт пламени уносится с каплями расплава. Если эти волокна
используются в смеси с неплавкими волокнами, то последние препятст-
вуют стеканию капель, и расплавленный полимер сгорает, распростра-
няя пламя.
Побочные процессы, сопровождающие горение. Для некоторых во-
локон, в частности целлюлозных, характерно длительное тление обго-
ревшего материала, которое может приводить к повторному воспламе-
нению и поэтому представляет не меньшую огнеопасность, чем выделе-
ние горючих продуктов разложения.
В процессе тления твердый обугленный остаток, образовавшийся
при горении, окисляется кислородом воздуха до двуокиси углерода.
Окисление углерода до СО2 происходит при высоких температурах
(«700 °C) и сопровождается выделением большого количества теплоты
(393,5 кДж/моль), которое может обеспечить поддержание высоких
температур, а следовательно, и процесса горения. Поэтому тление мо-
жет явиться дополнительным источником пожара.
Синтетические волокна в этом отношении менее опасны, так как
процесс тления после сгорания возможен в значительно меньшей сте-
пени.
Процесс горения сопровождается также выделением дыма и ток-
сичных газов, которые представляют не меньшую опасность для чело-
века, чем огонь. Токсичность газообразных продуктов разложения по-
лимеров изучена крайне недостаточно, и поэтому оценка токсичности
газообразных продуктов разложения текстильных материалов сугубо
ориентировочна. *
Поскольку горение происходит при температурах выше 700 °C, то
практически все химические и природные волокна выделяют в доста-
точно большом количестве различные газообразные продукты, создаю-
щие токсичную концентрацию газов.
Основными токсичными газообразными компонентами, выделяю-
щимися прн разложении полимеров, являются окись углерода, циани-
стый водород, окись азота, фосген, фосфины, хлористый водород. Их на-
личие и количество в газообразной фазе определяется химической при-
родой полимера, а также объемом воздуха, в котором происходит горе-
ние. Окись углерода образуется при разложении почти всех углеродсо-
держащих волокон; цианистый водород выделяется при пиролизе поли-
акрилонитрила, полиамида, полиуретанов; фосфины — при разложении
фосфорсодержащих материалов, галогенсодержащие полимеры выделя-
ют галогенводород.
Выделение дыма в процессе горения зависит от количества посту-
пающего воздуха и влажности горящей ткани. При горении синтетиче-
ских волокон (за исключением полипропиленового) дыма выделяется
гораздо больше, чем при горении целлюлозных материалов. Дымообра-
зование для волокон уменьшается в ряду: полиэфирное > полиамид-
ное > целлюлозное > шерсть. Внимание к этому вопросу возрастает,
так как при пожаре выделение дыма затрудняет эвакуацию людей из
общественных мест, особенно при большой насыщенности помещений
полимерными материалами. В США в последнее время ввели дополни-
тельный критерий для оценки текстильных материалов, используемых
в авиации, —степень задымленности.
Ряд синтетических волокон, таких, как полиамидные, полиэфирное,
полиолефиновые, при повышенных температурах плавятся. Перед плав-
лением синтетические ткани сильно усаживаются. При загорании одеж-
34 9
ды из синтетических материалов сильная усадка способствует тесному
контакту выделяющегося тепла с кожей и прилипанию расплава, что
приводит к тяжелым ожогам.
1.2. МЕХАНИЗМ ОГНЕЗАЩИТНОГО ДЕЙСТВИЯ АНТИПИРЕНОВ
Несмотря на большое число работ, посвященных механизму огне-
защиты полимерных материалов, при введении даже наиболее распрост-
раненных антипиренов, таких, как галоген- и фосфорсодержащих со-
елинс’ий, механизм этого процесса остается невыясненным. Предло-
жер ;ые схемы действия антипиренов при горении недостаточно обосно-
ван и являются в большинстве случаев гипотетическими, что объяс-
няется сложностью процессов и многообразием реакций, протекающих
при гс рении чтих материалов. Тем не менее накопленный материал
предст <вл' .г большой интерес и облегчает выбор эффективных замед-
лителе. .орения.
Исследование процесса горения позволяет сделать вывод, что для
огнезащиты необходимо сократить, замедлить или исключить отдельные
стадии пиролиза и горения. Решить эту проблему можно различными
способами:
1) снижением температуры пиролиза с тем, чтобы этот процесс
протекал при более низких температурах, чем температура самовоспла-
менения, и был эндотермичен;
2) увеличением количества карбонизированного остатка и соответ-
ственно снижением количества выделяющихся горючих 1азов:
3) образованием негорючих газообразных соединений, которые за-
медляют процесс горения;
4) ингибированием реакции окисления за счет связывания реакцион-
носпособных гидроксильных радикалов, так как последние активно
участвуют в реакции экзотермического окисления окиси углерода. Эта
реакция является основным источником выделения тепла в процессе
горения.
Плейс [4] предлагает следующую схему основных реакций про-
цесса окисления газообразных продуктов пиролиза, способствующих
распространению горения с участием гидроксильного радикала:
СО + ОН---> СО2 + Н (1)
Н О2 ——> ОН О (2)
Все существующие методы придания огнестойкости текстильным
материалам основаны <на введении антипиренов.
В ряде случаев огнестойкие материалы получают и без введения
антипиренов путем изменения химического состава и строения полиме-
ра, например волокна на основе ароматических полиамидов, полимеров
с гетероциклами в цепи или галогенсодержащие.
Действие антипиренов проявляется как в жидкой, твердой, так и
в газовой фазе продуктов горения. В жидкой и твердой фазе антипире-
ны изменяют направление процесса пиролиза и снижают скорость диф-
фузии продуктов разложения к фронту пламени, а в газовой фазе — ин-
гибируют радикальные реакции окисления [3].
В настоящее время не существует универсальных методов огнеза-
350
щиты текстильных материалов. Это объясняется тем, что процесс го-
рения различных волокон протекает по разным механизмам и зависит
в основном от химической природы полимера и характера продуктов,
выделяющихся при термоокислительном разложении.
Наибольшее распространение в качестве замедлителей процесса го-
рения полимерных материалов получили галоген- и фосфорсодержащие
соединения.
В основе огнезащитного действия галогенсодержащих соединений,
как считает большинство исследователей [12], лежит образование
в процессе горения галогенводорода. Находясь в газообразном состоя-
нии, галогенводороды взаимодействуют с наиболее реакционноспособ-
ными гидроксильными радикалами, образующимися при радикальных
цепных реакциях разложения полимера, и тем самым способствуют об-
разованию менее реакционноспособных радикалов по схеме:
НХ + ОН---->Н2О + Х (3)
X + RH---► HX4-R (4)
R—углеводородный радикал; X—галоген
Снижение концентрации гидроксильных радикалов приводит к ин-
гибированию реакций (1) и (2) и как следствие к замедлению
горения.
Выдвинуто также предположение [13; 14], что негорючие галоген-
водороды разбавляют горючие газы пиролиза и образуемая газовая
смесь изолирует поверхность горящего материала от доступа кислорода
воздуха. По-видимому, газовоздушная смесь разбавляется также и га-
логенами, выделяющимися наряду с галогенводородами при нагревании
в присутствии галогенсодержащих антипиренов.
Что касается механизма действия галогенсодержащих антипиренов
в конденсированной фазе, то предполагают, что он основывается на
повышении степени карбонизации вещества [15; 16].
Эффективность галогенсодержащих антипиренов увеличивается
в ряду F<Cl<Br<I.
Это обусловлено большой легкостью образования галогенводорода
вследствие снижения энергии взаимодействия R— галоген в органиче-
ских соединениях. По данным Хагинса [17], энергии химической связи
углерод — галоген в этом ряду составляют соответственно 389,3; 280,4;
220; 180 кДж/моль. Кроме того, эти закономерности объясняются пони-
жением летучести галогенводородов с повышением молекулярной мас-
сы в указанном ряду, что увеличивает время нахождения галогенводоро-
дов в зоне огня. Очевидно, по этой причине алифатические галогенсодер-
жащие соединения являются более эффективными антипиренами, чем
ароматические [3].
Анализ литературы по огнезащите полимерных материалов гало-
генсодержащими соединениями показывает, что наиболее широко
используются хлор- и бромсодержащие соединения. Лайонс [3] на осно-
вании детального изучения большого экспериментального материала по
эффективности действия галогенсодержащих органических соединений
сделал вывод, что бромсодержащие вещества более эффективны, чем
аналогичные соединения хлора, в два раза по массе и в четыре раза
по мольному соотношению.
351
Применение иодсодержащих антипиренов, несмотря на высокую эф-
фективность, очень ограничено из-за высокой стоимости и низкой стой-
кости их к ультрафиолетовому излучению. Фторсодержащие соединения
практически не нашли применения, что обусловлено их низкой эффек-
тивностью вследствие высокой прочности связи С—F.
При использовании галогенсодержащих антипиренов, особенно
хлорпроизводных, для придания сравнительно высокой огнестойкости
необходимо вводить в материал большое количество этих соединений,
что приводит к значительному снижению физико-механических характе-
ристик, а при поверхностной обработке материала — к повышению
жесткости ткани.
Эффективность огнезащитного действия галогенсодержащих соеди-
нений значительно возрастает при добавлении синергически действую-
щих веществ (неорганических солей или оксидов металлов), что позво-
ляет уменьшить количество вводимого антипирена.
Наиболее эффективным синергическим веществом для соединений
хлора и брома оказалась трехокись сурьмы. Поэтому в многочисленных
патентах по огнезащите полимерных материалов рекомендуется при-
менение хлор- и бромсодержащих соединений в смеси с трехокисью
сурьмы. Оптимальным принимают эквимольное соотношение сурьмы и
хлора. При добавлении трехокиси сурьмы количество требуемого анти-
пирена снижается на 50%.
Ранее предполагалось, что синергическое действие SbjOs связано
с образованием летучих оксигалогенидов сурьмы, препятствующих до-
ступу горючих газов и кислорода воздуха к пламени '[18]. Позднее Пит-
су [13; 20] удалось показать, что огнезащитными свойствами обладает
тригалогенид сурьмы, образующийся в результате термических превра-
щений оксигалогенидов сурьмы.
Пите предполагает следующую схему огнезащитного действия этого
соединения:
RC1 ---► R’-f-HClf (5)
2HC14-SbaO3 ---»- 2[SbOCl] + НаО (6)
AQ
SbOCl ---->• SbCl3t+HaO (7)
SbCl3 + HaO ---► HClf + Sb4Oef (8)
На основании исследования термического разложения сурьмухлор-
содержащих антипиренов при температурах 245—655°C Пите предложил
выделить несколько стадий, сопровождающихся поглощением тепла:
AQ
245—280°С... .5SbOCl ---> SbCl3f + 2SbOClSbaO3 (9)
AQ
410—475 °C... .8SbOCl- 4SbaO3 ---> SbCl3f + 5SbOCl-5SbaO3 (10)
AQ
475—568 °C.... 3SbOCl -3SbaO3 ->• SbCl3f + 4Sd2O3 (11)
AQ
658 °C....2SbaO3 --->• Sb4Oet (12)
В интервале температур 245—565 °C образуется трихлорид сурьмы
в газообразном состоянии (/кип SbCl3=223°C), который замедляет про-
цесс горения материала. При пиролизе в конденсированной фазе SbCl3
352
может являться дегидратирующим агентом '[уравнение (8) J, интенсифи-
цирующим процесс карбонизации.
Таким образом, огнезащита материалов сурьмугалогенсодержащи-
ми антипиренами обусловлена протеканием эндотермических процессов
в сочетании с выделением продуктов, снижающих горючесть за счет
ингибирования радикальных реакций в газовой фазе, снижения кон-
центрации горючих газообразных продуктов и повышением степени кар-
бонизации в твердой фазе.
Фосфорсодержащие соединения как органические, так и неоргани-
ческие относятся к наиболее эффективным замедлителям горения поли-
мерных материалов. Введение фосфорсодержащих соединений в состав
полимерных материалов приводит к увеличению выхода карбонизиро-
ванного остатка и уменьшению количества выделяющихся горючих га-
зообразных продуктов. В отличие от галогенсодержащих антипиренов
соединения фосфора оказывают огнезащитное действие в основном
в жидкой и твердой фазах горения.
Фосфорорганические соединения в присутствии воды или под дей-
ствием окислителя распадаются в процессе горения на ангидриды и
фосфорсодержащие кислоты [21]. Известно, что кислоты наряду с осно-
ваниями Льюиса и антипиренами являются катализаторами процесса
дегидратации. Поэтому в присутствии выделившейся кислоты ускоряет-
ся отщепление воды и процесс дегидратации протекает более интенсив-
но, в результате чего повышается выход обугленного твердого остатка.
Окиси фосфора способствуют образованию стекловидного расплава, ко-
торым покрывается поверхность горящего материала, предотвращая
таким образом доступ кислорода [18; 22]. Это приводит к замедлению
окисления материала в твердой фазе при повышенных температурах.
Процесс окисления углерода ограничивается образованием окиси угле-
рода, что снижает теплоту горения, так как при окислении углерода до
СО выделяется тепла в 3,5 раза меньше, чем при окислении до СОг:
С 4- V2O2 -► СО 4- 110,4 кДж/моль (13)
С-|-О2 -->- СО24*394,7 кДж/моль (14)
Выдвинуто предположение о том, что пятивалентный фосфор пере-
ходит в процессе эндотермической реакции в трехвалентный, который в
процессе пиролиза проявляет себя как антипирен и замедляет горение.
Таким образом, замедление горения материалов в присутствии
соединений фосфора обуславливается каталитическим влиянием этих
соединений на процесс дегидратации, способствующим повышению
карбонизации вещества, ингибированием реакции экзотермического
окисления углерода и снижением общего количества тепла, выделяю-
щегося при пиролизе.
Эффективность действия фосфорсодержащих антипиренов можно
существенно повысить, используя их совместно с галоген- или азотсо-
держащими соединениями.
По мнению Хиладо [13], механизм синергического действия фосфо-
ра и галогена сводится к следующему: в присутствии фосфора вследст-
вие усиленной карбонизации вещества создаются условия для образо-
вания при разложении галогенсодержащих соединений галогенидов и
оксигалогенидов фосфора (например, трибромида, пентабромида, окси-
бромида фосфора). Эти соединения имеют сравнительно высокую моле-
кулярную массу (270—430 для бромидов), и поэтому летучесть их зна-
23—2254
353
чительно ниже соответствующих галогенводородов. Соответственно про-
должительность пребывания их в зоне горения увеличивается и пропор-
ционально возрастает эффективность их замедляющего действия на про-
цесс горения материала.
Таким образом, наблюдаемое значительное усиление огнезащитно-
го действия фосфорсодержащих антипиренов, используемых совместно
с галогенсодержащими соединениями, обусловлено тем, что соединения
фосфора способствуют дегидратации и повышению степени карбониза-
ции вещества, а галогениды фосфора замедляют процесс горения в га-
зовой фазе.
Синергическое действие фосфора и хлора проявляется в меньшей
степени, чем соединений фосфора и брома, так как хлориды фосфора
обладают большей упругостью пара, имеют более низкую температу-
ру испарения и поэтому находятся в зоне горения меньшее время.
Однако синергическое действие фосфора и хлора используется более
широко благодаря доступности хлорсодержащих соединений.
Механизм синергического действия фосфора и азота пока недоста-
точно изучен. Предполагают, что снижение содержания фосфора, необ-
ходимого для устранения горючести материала, при введении в мате-
риал аминного азота связано с выделением при горении негорючих га-
зов [23; 24]. Вилард [25] при изучении процесса горения целлюлозных
материалов показал, что огнезащитное действие соединений, содержа-
щих азот и фосфор, является не аддитивным, а синергическим.
Приведенные данные дают возможность сделать предположения:
о механизме действия различных типов антипиренов. Однако следует
отметить, что в большинстве случаев механизм действия этих реагентов-
требует дополнительной проверки и уточнения.
1.3. МЕТОДЫ ОПРЕДЕЛЕНИЯ ОГНЕСТОЙКОСТИ
ТЕКСТИЛЬНЫХ МАТЕРИАЛОВ
Для оценки огнезащитных свойств материалов рекомендовано мно-
го методов испытания. Большинство из них разработано применитель-
но к тканям.
Основными показателями огнестойкости текстильных материалов
являются:
1) легкость воспламенения материала;
2) скорость распространения пламени;
3) продолжительность горения после удаления источника огня:
4) продолжительность остаточного тления;
5) количество тепла, выделяющегося при горении;
6) сохранение формы при воздействии пламени.
При оценке огнезащитных свойств кроме продолжительности горе-
ния после удаления источника огня определяют также длину и пло-
щадь обугленного участка и потерю массы материала.
Предложенные методы отличаются по выбранному критерию оцен-
ки огнестойкости, продолжительности действия пламени на материал
(от 10 с до 2 мин) и положением образцов ткани по отношению к пла-
мени (вертикальное, под углом 45° и горизонтальное положение).
Вследствие этого встречаются трудности в объективной оценке данных,
опубликованных в литературе, по огнестойкости различных типов тек-
стильных материалов или эффективности действия разных антипиренов.
Кроме того, необходимо учитывать и тот факт, что в большинстве за-
354
рубежных методик к огнестойким материалам относятся текстильные
материалы, которые обладают свойствами самозатухания в течение 1—
20 с после устранения источника воспламенения.
Стандартные методы испытания огнестойкости текстильных мате-
риалов при различных положениях анализируемой ткани, широко при-
меняемые как за рубежом, так и в СССР, основаны на определении
длины обугленного участка и скорости распространения пламени [11;
26, с. 251—259].
При оценке огнезащитных свойств на результаты испытания оказы-
вает определенное влияние положение ткани. При вертикальном поло-
жении образца ткани к источнику огня создаются благоприятные усло-
вия для процесса тления, а при горизонтальном положении образца —
для остаточного горения. Метод испытания, при котором ткань поме-
щается по отношению к источнику огня под углом 45°, позволяет полу-
чать средние величины продолжительности тления и остаточного горения.
В Советском Союзе основные методики по определению пожаро-
опасности материалов разработаны Всесоюзным научно-исследователь-
ским институтом пожарной охраны. К ним относится метод «огневой
трубы» [27, с. 67], по которому огнестойкость материала оценивается
по потере массы образца. Материал относится к сгораемым, если поте-
ря массы составляет болеее 20%. Это очень жесткий метод испытания
и он не всегда может характеризовать огнестойкость текстильных ма-
териалов, особенно на основе термостойких полимеров, так как потеря
массы может происходить за счет выделения газообразных продуктов,
не приводящих к воспламенению материала.
Более точную характеристику дает калориметрический метод [28],
сущность которого заключается в определении «показателя возгораемо-
сти» в воздушном калориметре. «Показатель возгораемости» К харак-
теризуется отношением тепла, выделяющегося при горении материала
9с, к количеству тепла, выделенному источником зажигания qs.
Чем меньше количества тепла выделяется образцом при горении,
т. е. чем меньше значение К, тем выше огнестойкость материала. Для?
несгораемых материалов К=0, для сгораемых К=2,1. Между этими
значениями показателя К материалы характеризуются как трудносго-
раемые и трудновоспламеняемые.
Сложность этого метода определения огнестойкости материалов за-
трудняет его применение на практике.
Наиболее перспективным является метод кислородного индекса,
предложенный Фенимором и Мартинсом [11]. Основа метода состоит
в определении минимальной концентрации кислорода в смеси с азотом,
при которой поддерживается непрерывное горение материала. Сжига-
ние образца осуществляется в стеклянной камере, в нижнюю часть ко-
торой подается смесь кислорода и азота. Испытуемый образец закреп-
ляется в вертикальном положении к источнику огня и поджигается пла-
менем горелки сверху. Обычно материалы, не поддерживающие горе-
ние на воздухе, имеют кислородный индекс выше 21%.
Кроме описанных выше методов для определения огнестойкости
текстильных материалов предложено много других, которые, однако, не
получили широкого распространения.
Следует отметить, что для огнестойких текстильных материалов не-
обходимо учитывать не только стойкость их к действию огня, но и ме-
ханические свойства, внешний вид ткани, а также стойкость огнезащит-
ного эффекта к водным обработкам и химической чистке.
23*
ГЛАВА 2
ЦЕЛЛЮЛОЗНЫЕ ВОЛОКНА
Целлюлозные волокна (вискозное и хлопок) являются самыми мас-
совыми волокнами. Гигроскопичность, хорошая накрашиваемость, а так-
же высокие гигиенические свойства изделий из них определили широ-
кое использование этих волокон для изготовления товаров народного
потребления.
Целлюлозные текстильные материалы легко воспламеняются и име-
ют относительно низкий кислородный индекс (15—18%). Кроме того,
для целлюлозных волокон характерно наличие процесса тления, кото-
рый при определенных условиях может быть причиной воспламенения
материала.
Огнезащитой целлюлозных материалов занимаются давно. В этой
области накоплен обширный материал. Однако проблема снижения го-
рючести целлюлозных материалов полностью не решена и исследо-
вания в этой области продолжаются.
2.1. РАЗЛОЖЕНИЕ ЦЕЛЛЮЛОЗЫ И МЕХАНИЗМ ОГНЕЗАЩИТЫ
Термическая и термоокислительная деструкция целлюлозы изучены
довольно подробно. Вопросам механизма разложения целлюлозы, со-
ставу продуктов деструкции, изменению физико-химических и механи-
ческих свойств целлюлозы, влиянию различных факторов на процессы
деструкции посвящено большое число исследований.
Накопленный материал довольно полно обобщен и проанализиро-
ван в литературе [29, с. 327—344; 30, с. 46—93].
В данном разделе кратко рассмотрены причины легкой воспламе-
няемости целлюлозных материалов и влияние антипиренов на процесс
разложения целлюлозы.
Разложение целлюлозы — сложный процесс, сопровождающийся
протеканием многообразных реакций, приводящих к глубоким превра-
щениям целлюлозы. В общем виде можно представить, что при разло-
жении целлюлозы протекает два конкурирующих процесса:
1) дегидратация с последующим более глубоким распадом деги-
дратированной целлюлозы, приводящим к образованию карбонизован-
ного вещества, воды, двуокиси и окиси углерода и других продуктов;
2) деполимеризация с образованием преимущественно левоглюко-
зана и продуктов его превращения.
Энергии активации этих процессов соответственно равны 146,5 и
188,3—230,2 кДж/моль [31; 32].
При термодеструкции в области температур 200—280 °C превали-
рует реакция дегидратации целлюлозы; при дальнейшем повышении
температуры — процесс деполимеризации. Распад молекулы целлюлозы
может происходить по 1,4-р-глюкозидным связям с последующей изоме-
ризацией элементарного звена в молекулу левоглюкозана—1,6-ангид-
ро-р,а-Д-глюкопиранозу [33].
Наиболее интенсивно деструкция целлюлозы происходит в области
температур 280—350 °C. На этой стадии выделяется основная масса
летучих продуктов. В составе летучих продуктов разложения целлюло-
зы выделено и идентифицировано около 50 различных соединений [34].
Летучие продукты состоят из простейших газообразных соединений
(водород, метан, двуокись углерода), низкомолекулярных углеводоро-
356
дов, спиртов, карбонилсодержащих соединений, левоглюкозана и других
ангидридов сахаров.
Основным фактором, обуславливающим высокую воспламеняемость
целлюлозных материалов, является выделение большого количества ле-
воглюкозана. Как показали исследования [35], при термическом раз-
ложении обеззоленных образцов целлюлозы (содержание золы <
<0,001%) образуется около 70% левоглюкозана (от массы исходного
образца). В реальных условиях термоокислительного разложения тек-
стильных целлюлозных материалов выход левоглюкозана значитель-
но ниже теоретически возможного, так как наряду с деполимеризацией
протекает окислительная деструкция. Кроме того, на выход левоглюко-
зана большое влияние оказывают примеси и структура целлюлозы.
Помимо содержания летучих продуктов, образующихся при терми-
ческом разложении целлюлозы, большое влияние на поддержание про-
цесса горения оказывает тление угольного остатка. Благодаря тлению,
сопровождающемуся выделением большого количества тепла, повы-
шается теплота горения.
На основании представлений о механизме разложения целлюлозы
для огнезащиты целлюлозных материалов необходимо создание условий
за счет введения различных веществ, способствующих изменению на-
правления распада целлюлозы и приводящих к снижению образования
левоглюкозана, повышению дегидратации целлюлозы и ингибированию
процесса тления.
Предложено несколько теорий для объяснения огнезащитного дей-
ствия антипиренов на целлюлозу.
Газовая теория. Огнезащита материала осуществляется за
счет выделения негорючих газов НгО, NH3, СО2, галогенводородов и др.,
которые разбавляют летучие легковоспламеняемые продукты.
Теория покрывающего с л о я. Образование на поверхности
материала стеклообразного расплава, предотвращающего доступ кис-
лорода к целлюлозе.
Термическая теория, согласно которой антипирены отводят
тепло, поступающее от источника огня, с такой же скоростью, с какой
тепло поступает к целлюлозному материалу.
Следует, однако, отметить, что некоторые из предложенных теорий
не могут объяснить огнезащитного действия целого ряда используемых
в настоящее время антипиренов.
Наиболее распространенной теорией огнезащиты целлюлозы яв-
ляется теория каталитической дегидратации. Многочис-
ленные исследования по огнезащите целлюлозных материалов показали,
что введение в целлюлозу антипиренов приводит к дегидратации цел-
люлозы с образованием значительного количества карбонизованного
остатка. Повышенный выход кокса снижает количество тепла, выделяе-
мое целлюлозным материалом в процессе горения, тем самым подавляя
процесс воспламенения и распространения пламени. Процесс дегидра-
тации целлюлозы в основном катализируют кислоты, выделяющиеся
при разложении антипиренов, а также кислоты и основания Льюиса.
В этом отношении наиболее эффективны фосфорсодержащие антипире-
ны. Механизм их действия довольно сложный. Предполагают {36; 37],
что фосфорсодержащие соединения или продукты их разложения в про-
цессе пиролиза вначале взаимодействуют с целлюлозой с образованием
сложных эфиров, а в дальнейшем происходит пиролиз новых произ-
водных целлюлозы, сопровождающийся процессом деполимеризации и
357
образованием карбонизованного вещества. Протекание процесса раз-
ложения и присутствие антипирена зависит от природы используемого
антипирена. Высокая термическая стойкость фосфорсодержащих соеди-
нений или высокая их летучесть снижает возможность образования про-
межуточных продуктов, необходимых для фосфорилирования целлюло-
зы, и тем самым уменьшает эффективность этих соединений как анти-
пиренов [37]. Так как действие фосфора при разложении материала
проявляется в конденсированной фазе, содержание фосфора в коксовом
остатке может свидетельствовать об эффективности огнезащитного дей-
ствия антипирена на целлюлозу [38].
Добавление фосфорсодержащих антипиренов способствует умень-
шению потери массы целлюлозы при разложении и значительно сни-
жает выход горючих продуктов. Ингибирование экзотермической реак-
ции окисления угля до СОг проявляется в изменении соотношения СО
и СОг в газообразных продуктах распада целлюлозы. Вебстер показал
[39], что при разложении хлопка это соотношение составляет 0,11. Для
хлопка, обработанного диаммонийфосфатом, содержание СО в газооб-
разных продуктах значительно увеличивается, и отношение СО к СОг
становится равным 4,0.
Резюмируя вышеизложенное, можно заключить, что огнезащита
целлюлозных материалов может быть обеспечена соединениями, кото-
рые способствуют процессу дегидратации, повышению выхода карбо-
низованного остатка и уменьшению количества летучих воспламеняе-
мых веществ. Наиболее эффективны в этом отношении фосфорсодержа-
щие соединения. Однако в вопросах механизма огнезащиты целлюлозы
еще нет полной ясности ввиду многообразия и сложности процессов,
сопровождающих термоокислительное разложение целлюлозы.
2.2. МЕТОДЫ ПРИДАНИЯ ОГНЕСТОЙКОСТИ ЦЕЛЛЮЛОЗНЫМ
ВОЛОКНАМ И ТКАНЯМ
2.2.1. Введение добавок в прядильный раствор
Для гидратцеллюлозных волокон довольно перспективным может
быть введение антипирена в волокно на стадии формования. Достаточ-
ная простота этого способа позволяет быстро реализовать его на дей-
ствующих заводах, особенно если предусмотреть введение антипирена
в прядильный раствор непосредственно перед формованием. Основная
сложность заключается в получении тонкой дисперсии антипирена, так
как антипирены, добавляемые в прядильный раствор, нерастворимы
в водных растворах щелочи, и их вводят в раствор в виде водной сус-
пензии. Формование и отделка волокна проводится по обычному техно-
логическому режиму.
Для получения огнезащитных целлюлозных волокон в вискозу и
медноаммиачные растворы вводят дисперсии различных фосфорсодер-
щащих и хлорсодержащих полимеров, эфиры фосфорных кислот, а
также фосфонитрилхлорид (ФНХ) и его производные.
Эффективными с точки зрения огнезащиты являются добавки в вис-
козу фосфонитрилхлорида в виде смеси, содержащей 65% тримера
(РР4С1г)з, 15% тетрамера (PiNCb).», 15% высших циклических полиме-
ров (PNCl2)n и 5% линейных полимеров [40].
Вследствие высокой реакционной способности фосфонитрилхлорид
в свободном состоянии неустойчив и обычно существует в виде
358
гримера, напоминающего по структуре бензол. Температура плавления
(PNC12)3 — 114°С, (PNC12)4 — 123,5°С. Тример и тетрамер образуют
эвтектическую смесь, которая плавится при 88,5—89 °C. При 150—400 °C
фосфонитрилхлорид полимеризуется, образуя неорганический эласто-
мер. Смесь продуктов ФНХ добавляется в вискозу на машине для фор-
мования. Формование волокна осуществляется в осадительную ванну
состава: 9% H2SO4, 2% ZnSO4, 15% Na2SO4 H74% Н2О.
Высокое содержание фосфора (до 27%) в сочетании с большим
количеством азота и хлора делают фосфонитрилхлорид очень перспек-
тивным для огнезащиты целлюлозных материалов.
Во многих патентах [41; 42] рекомендуется получение огнестойких
вискозных волокон путем введения в прядильный раствор трис(2,3-ди-
бромпропил) фосфата в количестве до 30% или смеси его с продуктами
взаимодействия хлорида тетраоксиметилфосфония и мочевины (или ме-
ламина) с аммиаком. Указывается, что смеси более эффективны для
огнезащиты, чем один трис(2,3-дибромпропил) фосфат. Получил рас-
пространение метод [43] введения в вискозу водонерастворимых низко-
молекулярных полимеров фосфонитрила общей формулы:
R и R'—алкильный радикал
Количество добавляемых в вискозу алкоксипроизводных фосфо-
нитрила составляет около 30% от массы целлюлозы.
Огнестойкие гидратцеллюлозные волокна получены при добавлении
в прядильный раствор гексабромциклододекана i[44], гексабромбензола
[45] и других соединений.
В качестве хлорсодержащих полимеров как добавок в прядильный
раствор предлагают [46] поливинилхлорид и поливинилиденхлорид
с трехокисью сурьмы. Ввиду того, что 5ЬгО3 коагулирует вискозу,
указанные вещества рекомендуют вводить в медноаммиачные прядиль-
ные растворы.
Так как в процессе формования и при отделке свежесформованно-
го вискозного волокна часть антипирена удаляется из волокна, для по-
лучения хорошего эффекта по огнестойкости в прядильные растворы
вводят значительный избыток антипирена (не менее 20—30%). В гото-
вом волокне остается до 15—20% антипирена. Полученные таким обра-
зом гидратцеллюлозные волокна имеют устойчивый к различным обра-
боткам огнезащитный эффект и высокие физико-механические показа-
тели. Однако несмотря на это данный способ пока широкого промыш-
ленного применения не получил.
2.2.2. Поверхностная обработка тканей
Среди различных методов придания огнестойкости целлюлозным
материалам самое широкое распространение получила поверхностная
обработка тканей. Это обусловлено достаточно простой технологией от-
делки и возможностью осуществления ее на обычном красильно-от-
делочном оборудовании текстильных фабрик.
359
Если рассматривать исследования по огнезащитной обработке цел-
люлозных материалов в историческом аспекте, то следует отметить, что
этой проблемой начали заниматься очень давно и первые методы вклю-
чали обработку текстильных материалов неорганическими соединения-
ми [47—49]. В основном это были аммонийные соли различных кис-
лот, смеси буры и борной кислоты, TiCl4 и SbCh. Наиболее эффектив-
ные из них — фосфаты и бромиды аммония. При использовании этих
препаратов можно проводить быструю обработку (пропитка водными
растворами и сушка) больших количеств ткани. Однако огнезащитные
свойства материала неустойчивы к мокрым обработкам.
Применение препаратов, содержащих наряду с неорганическими
соединениями органические вещества, повышает устойчивость огнеза-
щитных пропиток к водным обработкам. В этом случае ткань обраба-
тывают водной эмульсией веществ либо, что менее целесообразно, рас-
творами в органических растворителях, а затем сушат при повышенной
температуре.
Одним из примеров такой обработки может служить пропитка цел-
люлозных тканей препаратами, основными компонентами которых яв-
ляются хлорпарафин, хлоркаучук и Sb2O3 [50]. Для придания огнестой-
кости на ткань следует нанести не менее 30—40% этих веществ.
Ряд методов включает применение препаратов на основе фосфор-
ной кислоты и азотсодержащих соединений (карбамида, дициандиами-
да, меламина, гуанидина) [51—54]. Ткань обрабатывают водным рас-
твором карбамида (или других азотсодержащих соединений) и фосфор-
ной кислоты (или диаммонийфосфата) и подвергают термообработке
при 150 °C и выше в течение нескольких минут. В процессе термообра-
ботки происходят сложные химические превращения, что в конечном
итоге приводит к образованию на поверхности ткани нерастворимого
продукта взаимодействия фосфорной кислоты и азотсодержащего осно-
вания и частично — к химическому присоединению фосфора к макромо-
лекуле целлюлозы.
При повышенных температурах нз карбамида образуется трициан-
мочевина:
Нагрев
с—nhconh2
h2nochn—с c-nhconh2
Ортофосфорная кислота переходит в метафосфорную кислоту:
t > 130 °с
пН3РО4 -----> [НРО3]„
При взаимодействии трицианмочевины с метафосфорной кислотой
образуется труднорастворимая соль следующего состава:
С—NHCONH2
/\
N N
I II -ННРО3]„
H2NOCHN—с с—nhconh2
V
360
C—NHCONH2 • [НРО3]„
// \
N N
---> I II
[HPO8]n-H2NOCHN—С С—NHCONH2.[HPO3]n
X /
N
Характер этих реакций изменяется в зависимости от состава ком-
понентов, входящих в реакционную смесь.
В условиях данной обработки при повышенной температуре в при-
сутствии органического основания возможно образование сложного
эфира целлюлозы с мета- или ортофосфорной кислотами.
Пропитка целлюлозных тканей водным раствором дициандиамида,
фосфорной кислоты и диаммонийфосфата с последующей термообра-
боткой при 170 °C (огнезащитная пропитка ОП) получила применение в
отечественной промышленности.
Существенными недостатками способа поверхностной обработки
ткани, основанного на использовании смесей указанного выше состава,
является нестойкость огнезащитного эффекта к щелочным обработкам
и обработкам жесткой водой, а также необходимость проведения термо-
обработки, что значительно снижает физико-механические характери-
стики материала. Прочность ткани при разрыве уменьшается на 20—
50% (в зависимости от состава используемого для отделки препарата),
а прочность на раздир — более чем на 50% (табл. 2.1).
С целью снижения потерь механических свойств текстильных ма-
териалов при огнезащитной.отделке предложена [51] последовательная
обработка ткани водными растворами меламина (при 90°C), гексаме-
тафосфата натрия и метафосфорной кислоты (при 20 °C) и водой, не
предусматривающая выдерживания ткани при высоких температурах.
Образующееся водонерастворимое соединение
/NH2(HPO3)„
^N~4
(HPO3)nH2N—СГ ,N
\N=CZ
\nh2(hpo3)„
II
осаждается в порах волокна и придает огнезащитные свойства целлю-
лозным материалам.
Однако большинство методов поверхностной огнезащитной отделки
целлюлозных материалов основано на применении препаратов, содер-
Таблица 2.1. Физико-механические свойства огнезащитной ткани,
полученной по способу огнезащитной пропитки
Показатели Исходная ткань Огнезащищенная ткань Изменение пока» зателей, %
Масса 1 м2, г 530 610 + 15
Прочность при разрыве, мН
по основе 1900 1600 —16
по утку 1700 1350 —21
Прочность иа раздир, мН 250 90
по основе 0^
по утку 250 90 —64
361
жащих соединения, способные в результате последующей термообработ-
ки образовывать на целлюлозном материале полимерные вещества,
в состав которых входит фосфор и азот. В ряде случаев возможно об-
разование химических связей между целлюлозой и антипиреном. Благо-
даря образованию на поверхности ткани сетчатого высокомолекулярно-
го соединения огнезащитный эффект более устойчив к различным вод-
ным обработкам и химической чистке.
С этой целью предложено использовать большое число различных
полифункциональных и ненасыщенных химических соединений, содер-
жащих фосфор, галоген и азот. Анализ имеющейся в этой области лите-
ратуры позволяет выделить несколько классов веществ, которые в раз-
личных вариантах используются для огнезащитной обработки целлю-
лозных материалов. Это композиции на основе тетраоксиметилфосфо-
нийхлорида (ТНРС) и полифункциональных азотсодержащих соеди-
нений: М,М',ЬГ'-триэтиленимидфосфорной кислоты (препарат АРО),
фосфонитрилхлорида и его производных.
Тетраоксиметилфосфонийхлорид (ТНРС) — реакционноспособное
тетрафункциональное соединение, получается при взаимодействии фос-
фина, формальдегида и соляной кислоты по схеме [55]:
РН3 + 4СН2О + НС1
НОН2С. /СН2ОН
Cl-
III
При 140—150 °C ТНРС вступает в реакцию поликонденсации с по-
лифункциональными соединениями, такими, как триметилоламин [56],
меламин [57], дициандиамид [58] и др., образуя сетчатый полимер,
обладающий свойствами антипиренов.
Наряду с основной реакцией ТНРС может взаимодействовать
с гидроксильными группами целлюлозы с образованием поперечных
связей:
О
2Целл—ОН + [(СН2ОН)4Р]+ СГ Целл—ОСН,—Р—СН2О—Целл
Активатор ।
сн2
Возможность протекания такой реакции была высказана Риверсом
[59], который синтезировал эфир целлюлозы с ТНРС.
Для связывания НС1, выделяющегося при поликонденсации ТНРС
с меламином и другими соединениями, процесс проводят в среде аммиа-
ка (ткань обрабатывают газообразными NH3 и дополнительно промы-
вают водным раствором аммиака) [60; 61]. Однако полностью устра-
нить деструктирующее действие НС1 на целлюлозу возможно только
при замене ТНРС на окись тетраоксиметилфосфония (ТНРОН), кото-
рая получается путем обработки соответствующего хлорида раствором
NaOH [62].
В настоящее время имеется много вариантов обработки целлюлоз-
ных материалов с применением ТНРС и ТНРОН. Изменяются темпера-
тура и продолжительность термообработки, состав и соотношение ком-
понентов в исходной композиции, добавляются активаторы ZnCl2,
NH4CI, способствующие протеканию реакции взаимодействия указанных
соединений с целлюлозой, и т. п.
362
Подробно составы препаратов для огнезащитной отделки целлю-
лозных материалов на основе ТНРС и его производных приведены в ли-
тературе [29, с. 451—461].
Метод придания огнезащитных свойств целлюлозным материалам
при использовании ТНРС получил название «пропитки Пробан» и на-
шел промышленное применение в США и западноевропейских странах.
Обычно при применении этого метода на ткань наносят около 20% по-
лимера, что способствует введению 2—3% фосфора в материал. Благо-
даря тому, что образующийся в результате такой обработки ткани по-
лимер имеет сетчатую структуру и не исключена возможность образо-
вания химических связей его с целлюлозой, он прочно удерживается на
волокне и обеспечивает относительно устойчивый эффект огнезащиты
к различным обработкам. Однако по токсикологическим соображениям
такие ткани не могут быть рекомендованы для изготовления одежды,
так как вызывают патологические изменения кожных покровов.
Достаточно высокие огнезащитные свойства целлюлозные материа-
лы приобретают в результате обработки тканей препаратами, содержа-
щими триэтиленимид фосфорной кислоты [63]. При термофиксации
в присутствии кислых активаторов Zn(BF4)2 при 140 °C триэтиленимид
фосфорной кислоты образует на поверхности ткани труднорастворимый
полимер с большим числом сшивок:
I
А=Н или —СН2—СН2—N—
Триэтиленимидофосфат может также реагировать и с целлюлозой:
О О
II Нагрев II
Целл—ОН + P(NCH2CH2)3 ---► Целл—OCH2CH2NP(NCH2CH2)2
Указанная поверхностная отделка целлюлозных тканей получила
название «огнезащитной пропитки АРО».
Наиболее эффективной с точки зрения огнезащиты является сов-
местная обработка текстильных материалов триэтиленимидом фосфор-
ной кислоты и ТНРС. Ткань пропитывают при комнатной температуре
водным раствором, содержащим 15,7% Р(О) (NCH2CH2)3, 17,3% ТНРС,
J__I
4,3% триэтаноламина, отжимают и подвергают термофиксации при
140 °C в течение 5 мин [64]. В этих условиях происходит поликонден-
сация РНРС и Р(О) (NCH2CH2)3 с образованием сшитого полимера:
O=P(NCH2CH2)3 + [(НОСН2)4Р] С1
Нагрев
363
II
„ ,CHaOCH2CH2HNP—
° / . I
II / 1
—ch2ch2hnp—nhch2ch2och2pch2o—
? x° J"
NHCH2CH2OCH2P—
4-HC1+HCHO
Такая обработка позволяет получать огнезащищенные целлюлозные
материалы при содержании на ткани до 10—15% продукта конденсации
вместо 20—25%, как это имеет место при обработке ткани каждым из
компонентов в отдельности, по-видимому, за счет фосфоразотного си-
нергизма (введение амминного азота).
Для поверхностной обработки целлюлозных тканей используют
также такие эффективные антипирены, как фосфонитрилхлорид и его
производные.
Вследствие высокой реакционной способности атома хлора относи-
тельно легко могут быть получены различные производные ФНХ. Син-
тезированы фосфонитриламиды (V), аллил (VI) и дибромпропил (VII),
производные фосфонитрилхлорида и другие соединения:
H2n/ И |XNH2; [NP(OCH2CH=CH2)2]„; [NP(OCH2CH-CH2)2]„
NN |J
\ f Br Br
P
\h2
V VI VII
Использование ФНХ или его аллил- и дибромпропилпроизводных тре-
бует применения органических растворителей или приготовления ста-
бильных водных эмульсий [65—68]. При частичном замещении хлора
в ФНХ на аминогруппу получаются водорастворимые соединения, ко-
торые можно наносить на ткань путем пропитки их водными раствора-
ми [68]. В процессе последующей термообработки при 140 °C ФНХ
и его производные образуют на поверхности материала нерастворимые
полимерные соединения или трехмерные продукты конденсации. Кроме
того, частично имеет место химическое присоединение этих веществ
к макромолекуле целлюлозы. В присутствии бифункциональных соеди-
нений, легко реагирующих с гидроксильными группами целлюлозы
(например, М,1>Г-диметилолмочевины), возможность химического при-
соединения к целлюлозе фосфонитриламида повышается. Полученные
по указанному методу огнестойкие целлюлозные материалы содержат
от 2,5 до 5% фосфора и 5—6% азота.
В работе [69] отмечается, что кислородный индекс для целлюлоз-
ных тканей, обработанных препаратами на основе фосфонитриламида,
можно повысить до 40%.
Известны также устойчивые к стиркам огнестойкие отделки целлю-
лозных тканей, которые основаны на применении соединений диалкил-
фосфонийамида карбоновой кислоты (препарат «Пироватекс») [70],
хлорида тетраметилфосфордиамида [(CH3)2N]2 Р(О)С1 [71] и других
фосфорсодержащих соединений. Эти соединения фиксируются на волок-
364
не вместе с метилоламином при 140—150 °C. Технология процесса обыч-
ная: пропитка — сушка — термообработка. Недостатками огнезащитной
поверхностной обработки ткани являются: ухудшение грифа ткани (по-
вышение жесткости), в ряде случаев — накрашиваемости, большая по-
теря прочности, особенно при раздире (в результате деструкции целлю-
лозы в процессе обработки антипиренами при 130—170°C), и снижение
огнезащитного эффекта за счет постепенного вымывания антипирена
при щелочных и водных обработках ткани. Все это в ряде случаев за-
трудняет промышленную реализацию рассмотренных методов orrfeaa-
щиты целлюлозных материалов.
2.2.3. Химическая модификация
Существенный интерес для получения негорючих целлюлозных ма-
териалов, обладающих устойчивым эффектом огнезащиты, является хи-
мическое присоединение антипирена к макромолекуле целлюлозы. Для
этой цели могут быть использованы методы полимераналогичных прев-
ращений и привитая сополимеризация.
Применяя реакции, известные для низкомолекулярных спиртов,
можно получить различные производные целлюлозй. Реакцию фосфори-
лирования целлюлозы соединениями трех- и пятивалентного фосфора
изучали многие исследователи [72—77], и в этой области накоплен
большой экспериментальный материал. Наиболее часто для синтеза
фосфорсодержащих эфиров целлюлозы используют хлорангидриды
кислот. Реакция этерификации целлюлозы хлорангидридами фосфор-
содержащих кислот проводится при повышенной температуре, в среде
органического растворителя, в присутствии основания для связывания
выделяющегося НС1. Степень замещения, а следовательно, и количество
введенного в целлюлозу фосфора изменяется в зависимости от условий
проведения реакции (температуры, концентрации этерифицирующего
реагента, продолжительности). С использованием хлорангидридов кис-
лот были синтезированы сложные эфиры целлюлозы с фосфорной [72],
метилфосфиновой, этилфосфиновой, метилфосфорной, фенилфосфиновой;
фенилфосфорной, дифенилфосфорной кислотами [73], содержащими
от 1 до 8% фосфора.
Возможно и фосфорилирование целлюлозы непосредственным воз-
действием кислот. Таким способом получены эфиры целлюлозы с фос-
фористой [74] и гипофосфористой [75] кислотами. Однако условия
реакции этерификации в этом случае более жесткие, чем при использо-
вании хлорангидридов, и полученные эфиры целлюлозы неустойчивы —
легко гидролизуются. Так, например, при кипячении в воде в течение
30 мин содержание фосфора снижается с 4,6 до 1,2%. Описано получе-
ние фосфорсодержащих эфиров целлюлозы путем переэтерификации
эфиров целлюлозы моноэтилфосфитом [76], ди-р-хлорэтилфосфитом,
ди-р-фторэтилфосфитом и дифенилфосфитом [77].
В результате фосфорилирования целлюлозные материалы приоб-
ретают огнезащитные свойства. На огнестойкость, как показано Рогови-
ным [73], существенно влияет строение фосфорсодержащего соедине-
ния (табл. 2.2).
При увеличении алкильного радикала в молекуле алкилфосфино-
вой кислоты снижается эффективность огнезащитного действия. Для
достижения одинакового эффекта по негорючести содержание фосфора
в эфире целлюлозы с этилфосфиновой кислотой должно быть в 2 раза
больше, чем в эфире с метилфосфиновой кислотой. Замена алкильного
365
Таблица 2,2. Влияние строения фосфорорганических кислот
на огнестойкость полученных эфиров целлюлозы
Эфир целлюлозы Строение элементар- ного звена Максимальное значение угла воспламенения*, град Минимальное со- держание фосфора, при котором до- стигается него- рючесть, %
Эфир целлюлозы и метнлфосфи- новой кислоты со О 5 ч/ Он J L 180 2,8
Эфир целлюлозы и этилфосфи- новой кислоты X о о ч/ CU с/" 180 4,09
Эфир целлюлозы и метилфос- фориой кислоты -°\р^° —о/ \эсн3 150 6,25
Эфир целлюлозы и фенилфосфи- иовой кислоты X <о о. J 1 160 7,4
Эфир" целлюлозы и фенилфос- форной кислоты ~v° -о/ \освн5 90 9,13
* Определение угла воспламенения как одного из методов оценки огнестойкости текстильных
материалов основано на поджигании полоски ткани размером 1X7 см при поднесении к образцу
источника огня под различными углами. Нанлучшей огнестойкостью обладают образцы, которые
не воспламеняются, если источник огня поднести снизу, т. е. под углом 180 °C.
радикала на арильный приводит к еще большему снижению эффектив-
ности фосфорсодержащего соединения как антипирена. Заметное пони-
жение огнестойкости эфиров целлюлозы с фосфорорганическими кисло-
тами наблюдается и при замене в этих кислотах связи С—Р на
С—О—Р. Это подтверждается на примере эфиров целлюлозы с аллил-
и арилпроизводными соответствующих кислот (табл. 2.2).
Однако, несмотря на дысокую эффективность огнезащиты целлюло-
зы в результате ее фосфорилирования, для практического применения
этот метод не может быть рекомендован ввиду сложности и длительно-
сти процесса и сравнительно высокой стоимости исходных продуктов.
Кроме того, в большинстве случаев реакция фосфорилирования прово-
дится в жестких условиях и сопровождается значительно^ деструкцией
целлюлозы, что приводит к резкому снижению прочности целлюлозных
материалов.
Одним из способов придания огнестойкости целлюлозным материа-
лам является синтез привитых сополимеров. Этот метод позволяет хими-
чески присоединять к целлюлозе полимеры, обладающие свойствами
антипиренов, или полимеры, способные придавать материалу огнеза-
щитные свойства при дальнейших химических превращениях. Привитые
сополимеры позволяют получать устойчивый к различным обработкам
огнестойкий эффект. Кроме того, существенным преимуществом приви-
тых сополимеров является то, что возможность осуществления прививки
непосредственно на волокно не приводит к утяжелению готовой ткани,
и ткань, полученная путем переработки модифицированного целлюлоз-
ного волокна, имеет высокие физико-механические показатели (в том
366
числе прочность на раздир), не повышается жесткость ткани, не ухуд-
шаются гигиенические свойства материала, в частности не уменьшается
воздухопроницаемость. Химическая модификация целлюлозных волокон
путем синтеза привитых сополимеров с целью придания огнезащитных
свойств получила широкое развитие в Советском Союзе благодаря ис-
следованиям, проведенным под руководством проф. 3. А. Роговина.
Для синтеза привитых сополимеров по реакции радикальной поли-
меризации могут быть использованы галоген- и фосфорсодержащие
виниловые мономеры.
Однако опубликованные работы по приданию огнезащитных
свойств целлюлозным волокнам путем привитой сополимеризации не-
многочисленны и большинство из них носит описательный характер,
без раскрытия механизма огнезащиты вводимого антипирена.
Описано [78] получение огнезащитных целлюлозных материалов
в результате прививки к целлюлозным волокнам поливинилиденхлори-
да (ПВДХ). Инициирование реакции привитой сополимеризации осу-
ществлялось при разложении предварительно введенных в макромоле-
кулу целлюлозы диазогрупп. В качестве исходного материала использо-
валась алкилированная целлюлоза, содержащая ароматические амино-
группы с у=20—22. Целлюлозный материал приобретает негорючесть
при содержании хлора, равном 30—32%. Для введения такого количест-
ва хлора необходимо присоединить к целлюлозе до 60% (от исходной
массы) привитого ПВДХ. При введении такого количества ПВДХ значи-
тельно ухудшаются механические показатели тканей. Следует отме-
тить, что уже в процессе алкилирования и диазотирования происходит
снижение прочности ткани на 20—30%. Сложность метода синтеза
привитых сополимеров целлюлозы с ПВДХ и резкое ухудшение механи-
ческих свойств полученных огнезащищенных целлюлозных материалов
делает этот способ неприемлемым для практического использования.
В работе [79] описан синтез привитых сополимеров с поли-а-фе-
нилвинилфосфиновой кислотой. Инициирование реакции привитой сопо-
лимеризации осуществляли путем диазотирования предварительно по-
лученных производных целлюлозы, содержащих ароматические амино-
группы. Синтез привитых сополимеров проводили из водных растворов
мономера при 80 °C в атмосфере инертного газа.
В принятых условиях протекает преимущественно реакция приви-
той сополимеризации:
ONaNO»+HCl
—осн3-------->-
NH2
/^\ Fe2+
---»- Целл—OCH2CH2SO2—<(_))—ОСН3 + СН2=С-Р—ОН -->
l7 \он
N+sN СГ
Целл—OCH2CH2SO
367
хотя частично может иметь место взаимодействие соли диазония с а-фе-
нилвинилфосфиновой кислотой с образованием простого эфира целлю-
лозы:
Целл—OCH2CH2SO2—ОСН3
I ? /С«Нб
N=N—О—Р—С=СН2
ОН
или
Целл—OCH2CH2SO2-/^j\—ОСН3
СвН5
О—Р—С=СН2
Содержание фосфора в привитом сополимере в сильной степени за-
висит от концентрации а-фенилвинилфосфиновой кислоты, что видно из
приведенных в табл. 2.3 данных, и от количества азота в исходном эфи-
ре целлюлозы, так как ароматические аминогруппы в этих условиях
являются центрами прививки.
Таблица 2.3. Влияние концентрации мономера
на состав фосфорсодержащих препаратов целлюлозы*
Концентрация моно* мера, % Содержание фосфора в привитом сополи- мере, % Привес образцов, % от массы исходной ткани Огнезащшцениость
5 0,71 Горит
10 3,06 17,7 Держит пламя 3 с
15 3,92 23,0 Не поддерживает горения
20 4,06 26,1 То же
* Температура реакции 80 °C, продолжительность — 60 мин, среда — аргон. Содержание азота
в исходном эфире целлюлозы— 1,3% (V —18).
При увеличении содержания азота в производном целлюлозы, со-
держащем ароматические аминогруппы, от 0,7% (у=Ю) до 1,7%
(у = 26) количество вводимого фосфора в волокно возрастает с 2,1 до
4,72%.
При содержании фосфора около 4% целлюлозные ткани не поддер-
живают горения и не тлеют после удаления источника огня.
Однако существенным недостатком этого способа является труд-
ность осуществления прививки а-фенилвинилфосфиновой кислоты, так
как необходима предварительная обработка целлюлозного материала
для получения промежуточного производного целлюлозы и проведение
синтеза в атмосфере инертного газа. Исследования по использованию
более простых методов инициирования привитой сополимеризации,
в частности окислительно-восстановительной системы Fe2+—Н2О2 (спо-
соб Бриджфорда) и метода предварительного введения в макромолеку-
лу целлюлозы перекисных групп при синтезе привитых сополимеров
целлюлозы с поли-а-фенилвинилфосфиновой кислотой не привели к по-
ложительным результатам.
368
Получены [80] огнестойкие материалы на основе привитых сополи-
меров целлюлозы с эфирами винилфосфиновой кислоты следующего
состава:
Целл—OCH2CH2SO2—/(^)\—[—СН2—СН—]п
I /ОС2Н3
О=р/
Целл-ОСН2СН25О2—(IQS—[—СН2—СН—]„
'-------------------' | /ОСН2СН2С1
° P\och2ch2ci
В качестве исходных мономеров для проведения реакции привитой
сополимеризации использовали ди-0,р'-хлорэтиловый эфир и диэтило-
вый эфир винилфосфиновой кислоты.
Реакцию проводили из 13%-ных водных эмульсий или растворов
в атмосфере азота при 65 °C. В присутствии кислорода воздуха реак-
ция привитой сополимеризации эфиров винилфосфиновой кислоты не
протекает. Полимеризация этих мономеров происходит с низкой скоро-
стью и в среде аргона, вследствие чего продолжительность реакции ве-
лика и количество фосфора в привитом сополимере не превышает 4%
(табл. 2.4).
Таблица 2.4. Состав и огнестойкость привитых сополимеров целлюлозы
с эфирами винилфосфиновой кислоты*
Эфир винилфосфиновой кислоты Продолжи- тельность реакции, ч Содержание в привитом со* полимере, % Огнестойкость. градусы
Р С1
Ди-£,р '-хлорэтиловый 33 4,33 8,24 160
2 2,7 5,1 150
Диэтиловый 33 2,8 — 90
* В качестве исходного продукта для прививка применяли эфир целлюлозы с 4-(0-оксиэтил-
сульфонил)анилином с у=30 (содержание азота 2%).
Огнезащитный эффект поли(ди^р,р'-хлорэтилового) эфира винил-
фосфиновой кислоты и полидиэтилового эфира винилфосфиновой кисло-
ты неодинаков. При одном и том же содержании фосфора в привитых
сополимерах целлюлозы модифицированная ткань с привитым поли-
мером на основе ди-|3,р'-хлорэтилового эфира винилфосфиновой кислоты
обладает более высокой огнестойкостью, чем с диэтиловым эфиром ви-
нилфосфиновой кислоты.
Можно предположить, что это обусловлено наличием в привитом
сополимере хлора наряду с фосфором, хотя не всегда одновременное
присутствие фосфора и галогена приводит к синергизму.
Исследования, проведенные на привитых сополимерах целлюлозы
с поли (ди-р,р'-хлорэтиловым) эфиром винилфосфиновой кислоты, пока-
зали, что существенное влияние на огнестойкость полученных материа-
лов оказывает длина привитой цепи фосфорсодержащего нолимера.
Наличие большого числа коротких цепей способствует повышению огне-
стойкости материала (табл. 2.5).
24—2254
369
Таблица 2.5. Влияние длины боковой цепи* в привитом
сополимере целлюлозы с поли(ди-Рф'-хлорэтиловым)
эфиром вииилфосфиновой кислоты на его огнестойкость
Состав исходного эфира целлюлозы Продолжитель- ность реакции, ч Содержание фосфора в привитом сополи- мере, % Огнестойкость, градусы
содержание азота, % степень замеще- ния
1,01 13,8 1 2,33 90
1,01 13,8 2 3,32 90
1,01 13,8 6 3,05 90
2,0 31,2 1 2,32 110
2,0 31,2 2 2,93 150
* О числе боковых цепей привитого полимера косвенно можно судить по степени замещения
целлюлозы при получении исходного эфира целлюлозы, используемого для реакции привитой со-
полимеризации, или содержанию ароматических аминогрупп, так как они являются центрами при-
вивки.
Присоединение фосфора к макромолекуле целлюлозы прочной кова-
лентной связью обеспечивает стабильность огнезащитных свойств
при различных обработках материала.
Однако вследствие недостаточной доступности фосфорсодержащих
мономеров, относительно высокой их стоимости и, как правило, слож-
ности синтеза привитых сополимеров целлюлозы с этими мономерами
по радикальному механизму приведенные огнезащитные целлюлозные
материалы, по-видимому, не найдут практического применения.
Разработка эффективного метода синтеза привитых сополимеров
с использованием окислительно-восстановительной системы на основе
ксантогената целлюлозы позволила осуществить прививку к целлюлозе
виниловых мономеров на основе эфиров фосфорных кислот и акрил-
амида [81] общей формулы:
О О OP'
11 II
СН2=СНС—NH— R— ОР
VI
Из привитых сополимеров целлюлозы, предложенных с целью при-
дания огнезащитных свойств материалам, практическое значение полу-
чили привитые сополимеры целлюлозы с фосфорнокислой солью
поли-2-метил-5-винилпиридина (ПМВП) [82] следующего состава:
Целл—[— СН2—СН—]„
(НРО3)„
СНз
Процесс получения таких огнезащищеиных целлюлозных материа-
лов состоит из двух стадий: синтеза привитых сополимеров целлюлозы
с ПМВП и последующей обработки привитого сополимера разбавлен-
ным раствором фосфорных кислот.
Для синтеза привитых сополимеров целлюлозы с ПМВП исполь-
зуется принцип инициирования, основанный на реакции передачи цепи
на полимер от радикала, образованного при каталитическом разложе-
нии Н2О2 ионами Fe2+, связанными с целлюлозой. Реакцию осущест-
вляют из водных растворов уксуснокислой соли метилвинилпиридина
(МВП) или эмульсией МВП. Если реакцию проводят в растворе, то
используют уксуснокислую соль МВП, если в эмульсии — МВП в основ-
ной форме.
При выборе условий реакции следует отдать предпочтение проведе-
нию привитой сополимеризации в эмульсии, так как в этом случае зна-
чительно повышается эффективность реакции привитой сополиме-
ризации.
Подробно реакция привитой сополимеризации целлюлозы с ПМВП
исследована в работах Р. М. Лившица, Б. П. Морина и 3. А. Роговина
[83—85]. Оптимизация условий синтеза привитых сополимеров целлю-
лозы с ПМВП с применением математических методов исследования
позволяет выбрать условия проведения реакции привитой сополимери-
зации, обеспечивающие получение привитых сополимеров целлюлозы
с ПМВП при максимальной конверсии мономера и наименьшем количе-
стве гомополимера.
При последующей обработке привитых сополимеров целлюлозы
с ПМВП разбавленными (2%-ными) водными растворами фосфорных
кислот (Н3РО4 или НРОз) можно ввести фосфор в модифицированное
целлюлозное волокно за счет образования в привитых цепях фосфорно-
кислой соли ПМВП. Количество фосфора, вводимого в волокно, в основ-
ном, определяется содержанием в нем привитого ПМВП.
При прививке фосфорнокислой соли ПМВП целлюлозный материал
приобретает огнезащитный эффект при введении около 4% фосфора.
Такие модифицированные волокна содержат до 30% (к массе исходного
волокна) фосфорнокислой соли ПМВП. Следовательно, даже несмотря
на благоприятные с точки зрения огнезащиты химическое строение и
свойства целлюлозы для получения негорючих материалов, необходимо
присоединить достаточно большое количество довольно эффективного
фосфоразотсодержащего антипирена. Вследствие наличия солевой свя-
зи между НРОз и третичным азотом ПМВП в процессе водно-содовых и
щелочных обработок происходит удаление метафосфорной кислоты и
материал теряет огнезащитные свойства. Однако их легко можно вос-
становить повторной обработкой раствором метафосфорной кислоты.
В результате прививки фосфорнокислой соли ПМВП механические
свойства вискозного волокна снижаются незначительно, что видно пз
приведенных ниже данных [86]:
Показатели
Линейная плотность, текс
Прочность, мН/текс
Удлинение, %
Исходное Огнезащищен-
вискозное ное
волокно волокно
0,166 0,208—0,192
180—210 150—190
19—20 17—19
При использовании привитой сополимеризации для получения огне-
защищенных целлюлозных материалов, как показывают проведенные
в этой области исследования, необходимо ввести (независимо от типа
антипирена и его эффективности) не менее 20—30% фосфорсодержа-
щего полимера [87]. Присоединение такого количества антипирена
к тканям вызывает значительное снижение механических свойств, осо-
бенно прочности на раздир, а также приводит к утяжелению веса ткани
и сильному ее уплотнению. Поэтому модификация готовых тканей и
изделий из них мало приемлема. При этом методе наиболее целесооб-
разно осуществлять прививку полимеров, обладающих свойствами анти-
24*
371
пирена, на волокно, а затем уже его текстильную переработку в пряжу
и ткань. Огнезащищенные вискозные волокна с фосфорнокислой солью
ПМВП хорошо перерабатываются в ткани и нетканые материалы как
в чистом виде, так и в смеси с другими волокнами.
ГЛАВА 3
ПОЛИАМИДНЫЕ ВОЛОКНА
Полиамидные волокна и ткани иногда классифицируют как мате-
риалы, с трудом поддерживающие горение или самозатухающие, вслед-
ствие того, что при поджигании ткани, находящейся в вертикальном по-
ложении, пламя не распространяется. Полиамид плавится и горит не-
большим пламенем только до тех пор, пока источник пламени находит-
ся в контакте с тканью. Через несколько секунд после удаления источ-
ника пламени горение прекращается, так как пламя уносится каплями
расплавленного полимера. Однако расплав, попадая на кожу, прилипает
к ней, вызывая тяжелые ожоги. Кроме того, ткани из смеси полиамида
с хлопком или вискозным волокном очень хорошо распространяют пла-
мя за счет так называемого «эффекта поддержки»; неплавкие целлю-
лозные волокна в процессе горения являются своеобразной сеткой, ко-
торая задерживает в зоне огня расплав полиамида, и он продолжает
гореть. Такие ткани представляют большую опасность в пожарном от-
ношении.
Огнезащитная обработка синтетических волокон, и в частности по-
лиамидных волокон, в настоящее время еще не получила должного
развития по сравнению с подобной обработкой целлюлозных волокон.
В литературе почти нет публикаций, касающихся систематических ис-
следований по получению огнестойких полиамидных волокон. В основ-
ном имеются патентные сообщения, где излагаются принципы придания
полиамидам огнестойкости, а теория процесса не рассматривается.
При этом большинство из предложенных способов не приводит к полу-
чению неплавкого волокна с высокой способностью к коксообразова-
нию, что снижает их практическую ценность. Кроме того, различные
методы оценки огнестойкости материалов приводят часто к тому, что
патентуемый способ при проверке в других условиях испытания часто
оказывается неэффективным.
Проблема создания негорючих полиамидных нитей может решать-
ся в двух направлениях: первое — получение негорючих волокон на
основе полностью ароматических полиамидов или полиамидов с гетеро-
циклами в цепи; второе — придание огнезащитных свойств многотон-
нажным волокнам из линейных алифатических полиамидов. В данном
разделе рассматриваются только методы получения огнестойких мате-
риалов на основе алифатических полиамидов.
Для повышения огнестойкости полиамидных нитей могут быть ис-
пользованы методы, применяемые для целлюлозных волокон. Это по-
верхностная отделка тканей, добавки антипиренов к полимеру перед
формованием волокна и химическая модификация готового волокна.
Поверхностная отделка тканей не придает материалу устойчивой огне-
стойкости. Добавки, вводимые в полимер, не вымываются при обра-
ботке, но полученные волокна плавятся. Сохраняет свою актуальность
проблема химической модификации готового волокна, которая наряду
с огнезащитой дает возможность получения неплавкого материала.
372
Прежде чем перейти к рассмотрению фактического материала по
приданию алифатическим полиамидам огнезащитных свойств, следует
кратко остановиться на процессе их пиролиза и термоокисления, с тем
чтобы понять причины, обусловливающие основную трудность в разра-
ботке методов получения полиамидных материалов с огнезащитными
свойствами.
Как известно, процессы пиролиза с образованием легколетучих го-
рючих продуктов предшествуют воспламенению большинства волокни-
стых материалов. Текстильные материалы легко воспламеняются при
условии, что температура и продолжительность пиролиза невелика, и если
выделяется много горючих соединений. Исходя из этого для повышения
устойчивости полимерных материалов к действию огня необходимо воз-
действовать на пиролиз в таком направлении, чтобы он протекал при
температурах более низких, чем температура самовоспламенения, был
эндотермичен, обусловливал образование трудновоспламеняемого остат-
ка, обогащенного углеродом, и чтобы при его протекании не выделялись
горючие летучие продукты.
В общей проблеме получения устойчивых к действию огня волокон
исследования деструкции таких материалов имеют важное значение,
так как знание механизмов окисления в различных температурных диа-
пазонах и характера превращений, происходящих в условиях действия
высоких температур, позволяет целенаправленно подбирать нужные ан-
типирены и регулировать свойства получаемых материалов.
3.1. ТЕРМИЧЕСКОЕ И ТЕРМООКИСЛИТЕЛЬНОЕ
РАЗЛОЖЕНИЕ ПОЛИАМИДОВ
В области термической и термоокислительной деструкции полиами-
дов накоплен обширный экспериментальный материал. Тем не менее
некоторые вопросы механизма реакции, характер и количество обра-
зующихся низкомолекулярных продуктов распада (особенно в области
высоких температур и при горении полиамидов) изучены недостаточно.
Исследования термической и термоокислительной деструкции полика-
проамидного волокна методом дифференциально-термического и термо-
гравиметрического анализов [88] показывают, что интенсивные потери
массы начинаются при температурах, близких к 300 °C, и проходят
в несколько стадий (рис. 3.1 и 3.2).
Однако и в низкотемпературной области (до 300°C) происходит
ряд превращений полимера, не связанных с собственно деструкцией
макромолекул (понимаемой как ряд актов, приводящих к разрушению
полимерной цепи), но оказывающих влияние на последующее разложе-
ние полимера.
Сопровождающееся эндотермическим эффектом небольшое (до 5%)
уменьшение массы поликапроамида (ПКА) в начальный период свя-
зано с удалением сорбированной влаги. Кроме того, в этой области про-
исходит плавление полимера (эндотермический пик при 225 °C) и его
частичное структурирование (экзотермический пик при 190—220 °C);
последнее согласуется с литературными данными [89] о потере раство-
римости полиамида после прогрева его в этой области.
Деструкция поликапроамидного волокна в высокотемпературной об-
ласти протекает в несколько этапов. Основная масса (88—90%) лету-
чих продуктов удаляется в интервале 300—500 °C. В среде инертного
газа этот этап характеризуется термограммой с двумя перекрывающи-
373.
мися эндотермическими пиками при 435 и 460 °C и перегибом при
480 °C. Эффективная энергия активации этих превращений составляет
144,4±12,5 кДж/моль (табл. 3.1), что хорошо согласуется с данными,
Рис. 3.1. Кривые ТГА (1 и 2) и скорое™ разложения поликапроамидного
волокна (3 и 4).
(--------в аргоне;--------на воздухе).
полученными другими методами [90—92]. Для поликапроамида терми-
ческое разложение сопровождается, в основном, отщеплением мономер-
ных звеньев капролактама [93].
374
Таблица 3.1. Параметры термической и термоокислительной
деструкции поликапроамидного волокна
Среда Интервал темпе- ратур, °C Потеря массы, % £эфф. КДж/моль Порядок реакции п
Аргон 300—500 88—90 144,4+12,5 1,1
500—600 6 (2 94—96) 167,4—209,3 —
Воздух 300—475 82 115,1± 12,5 0,7
475—560 12 (2 94) 150,7±20,9 0,6
В дальнейшем, однако, скорость газовыделения снижается и не-
большой остаток (около 6% от первоначальной массы) разлагается
в интервале 500—600 °C с £'эфф = 167,4—209.3 кДж/моль. В указанных
выше работах нет прямых данных, свидетельствующих об изменении
механизма процесса деструкции ПКА по ходу разложения. Однако
анализ экспериментальных результатов, приведенных в [90], подтверж-
дает отмеченное изменение направления деструкции в области высоких
температур.
Действительно, ЕЭфф, найденная Озава для термодеструкции найло-
на 6, равна 151,5+2,1 кДж/моль. Но на больших глубинах превраще-
ния (а^0,9) наблюдается отклонение экспериментальных данных от
линейной зависимости [90]. Расчет энергии активации для а>0,9 по
экспериментальным данным Озава его методом приводит к значениям
Дэфф 167,4 кДж/моль.
Хотя термогравиметрические кривые деструкции поликапроамида
на воздухе и в аргоне близки, тем не менее термограммы и рассчитан-
ные величины Е8фф различны. Это можно объяснить тем, что в присут-
ствии кислорода воздуха термические процессы деструкции, которые
дают основной вклад в потери массы, осложнены реакциями окисления.
С ростом температуры доля последних увеличивается Прежде всего
это сказывается на повышении степени структурирования: на послед-
нем этапе масса остатка достигает 12% (при нагревании в аргоне —
6%). Вследствие этого термограммы, характеризующие экзотермиче-
ские процессы в атмосфере воздуха, являются суммарными, так как
экзотермическое окисление частично компенсируется эндотермическими
процессами деструкции.
Анализируя имеющиеся данные, можно сказать, что для полика-
проамида при высоких температурах деструкция преобладает над тер-
мическим и термоокислительным структурированием.
Механизм термического разложения полиамидов довольно полно
рассмотрен в литературе [94—97]. Многочисленные факты свидетель-
ствуют о том, что разложение полиамидов протекает одновременно по
двум механизмам: радикально-цепной деструкции и гидролитического
расщепления амидных связей.
При относительно низких температурах преобладает гидролитиче-
ский разрыв амидных связей с последующим декарбоксилированием
и дезаминированием, а при более высоких температурах — процессы
гомолитического разрыва с выделением СОг и Н2 и сшивание полимера.
Полагают [94], что первичным актом термической деструкции полиами-
да является гомолитическое расщепление связи —НгС—NH—, находя-
щейся в 0-положении к карбонилу и являющейся наиболее слабой.
Свободнорадикальный механизм термического разложения поли-
амидов маскируется гидролитическими процессами за счет воды, прочно
375
сорбированной полимером, и воды, выделяющейся при разложении
полиамидов [98].
Штраус и Уолл [99] рассчитали кажущуюся энергию активации
термодеструкции поликапроамида, которая была равна 142,3 кДж/моль.
Тщательная промывка и высушивание привели к увеличению энергии
активации до 171,6—180 кДж/моль. Еще более тщательная очистка
полимера от следов катализатора и влаги, как считают авторы, может
привести к увеличению Е до 209,3—251,1 кДж/моль, т. е. до величины,
характеризующей радикальные процессы.
Если у исследователей нет сомнений относительно наличия гомоли-
тического разрыва полимерных цепей полиамидов под действием тепла,
то относительно места гомолитических разрывов и их механизма имеют-
ся различные мнения.
По данным Полинга [100], наиболее слабой является амидная
связь, энергия которой равна 203,4 кДж/моль. Штраус и Уолл отмеча-
ют, что наиболее слабой является связь—С—N— (Е=284,6кДж/моль),
в то время как энергия разрыва связи —С—С— составляет
334,9 кДж/моль. Однако из двух типов связи —С—N— : Н2С—NH— и
О
—С—NH— в полиамиде связь —НгС—NH— является более слабой, так
как амидная связь —С—NH—- усиливается за счет резонансной энер-
II
О
гии [101].
Закаревский В. А. и др. [102] оценивают энергию связи в молекуле
поликапроамида следующим образом:
1 2 3 4 5 6 7
—СО—СН2—СН2—СН2—СН2—СН2—NH—СО—
Энергии связи 3 и 4 приблизительно равны энергии разрыва связи
в простых углеводородах, т. е. «330,7 кДж/моль. Энергия разрыва свя-
зи —С—N— достаточно высока; для метиламина она равна
334,9 кДж/моль. Авторы полагают, что в поликапроамиде энергия раз-
рыва связи —Н2С—NH— должна быть такой же, поскольку длина этой
связи сохраняется 1,47 А. Амидная связь должна быть более прочной.
Прочной является связь 1 — 326,5 кДж/моль. По мнению авторов, ме-
нее прочными являются связи 2 и 5, так как в результате их разрыва
должны образовываться свободные радикалы с сильно делокализован-
ными неспаренными электронами, отчего энергия их понижается при-
мерно на 62,8 кДж/моль по сравнению с обычными —С—С-связями.
В образцах поликапроамида, подвергнутого механическому дроблению,
с помощью ЭПР были обнаружены радикалы —СН2—СН—;
—NH—СН—; —СО—СН—.
В процессе термического окисления полиамидов также осуществля-
ются одновременно реакции как радикальной, так и нерадикальной при-
роды. В настоящее время общепринято, что термоокисление полиами-
дов на воздухе протекает по тем же схемам, что и окисление их низко-
молекулярных аналогов.
Рафиков С. Р. [89] по аналогии с автоокислением низкомолекуляр-
ных амидов [103], а также учитывая факт ингибирования окисления
полиамидов антиоксидантами, предположил, что окисление протекает
по радикально-цепному механизму, включающему реакции распада и
376
структурирования. Позднее Нейман М. Б. с сотр. [105], изучая состав
летучих продуктов термоокислительной деструкции поликапроамида,,
привели подробную схему механизма термоокисления:
—CH2CONHCH2CH2— + О2-> —CH2CONHCHCH2 — + Н2О
0-0-
—ch2conhchch2— + о2 —> —ch2conh(!:hch2—
о—о. о—о—н
I I
—CH2CONHCHCH2— + RH ► -CH2CONHCHCH2- + R.
ROOH-f-RH -----------> RO- +R- +H20
0—0. 0—0
—CONH^HCH,------> —ch2conhc- c!h2ch2--->
.0 .Ck
---► —CONHC\ + CH2CH2—
I II
—ch2conhc^ —> —ch2conh2 + co
°\ch2ch2-----> CH2O + CH2CH2—
III
CH2CH2CH2----> CH3CHCH2
IV
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
Авторы считают, что инициирование цепного процесса окисления
полиамидов происходит путем отрыва водорода метиленовой группы,
находящейся в a-положении к азоту [уравнение (15)]. По месту отрыва
атома водорода происходит образование перекисного радикала и гид-
роперекиси [уравнения (16) и (17)]. При распаде гидроперекиси обра-
зуется вода [уравнение (18)]. При термоокислении полиамидов выде-
ление воды может приводить к гидролизу полимера и увеличению числа
концевых карбоксильных групп, при декарбоксилировании которых об-
разуется двуокись углерода [105]. Наряду с распадом перекисей может
происходить распад радикалов. Для углеводородов было установлено,
что перекисный радикал может изомеризоваться путем атаки свободной
валентностью соседней —С—С-связи. Дальнейший распад изомеризо-
ванного перекисного радикала приводит к разрыву цепи и образованию
молекулы с концевой альдегидной группой (I), а также алкоксильного
радикала (II) [уравнение (19)]. При распаде альдегида [уравне-
ние (20)] получается окись углерода. Радикал (II) распадается с раз-
рывом по связи —С—С— с образованием формальдегида и радикала
(III), который далее может изомеризоваться и присоединять кислород.
Следовательно, начальной ступенью окисления является образование
перекисных или гидроперекисных радикалов. Их распад и последующие
превращения продуктов окисления могут осуществляться по различным
механизмам. Первичная атака кислорода направлена преимущественно
на метиленовую группу, соседнюю к азоту. Однако, по-видимому, одно-
377
временно реализуется окисление и других метиленовых групп поли-
амида.
Термоокисление полиамидов сопровождается выделением СО2, СО,
Н2О и альдегидов с образованием карбонильных, карбоксильных и пе-
рекисных групп [105].
Кроме окислительных превращений при высоких температурах
имеют место процессы чисто термической деструкции и структурирова-
ния — основным деструктивным процессом является деполиризация по-
лиамидов, если в их цепи имеются структурные единицы, способные
выделять летучие циклические мономеры.
Таким образом, при термическом и термоокислительном разложе-
нии полиамидов при высоких температурах (400—500 °C) протекают
в основном деструктивные процессы с образованием большого количе-
ства низкомолекулярных летучих продуктов распада (до 85—90% от
первоначальной массы образца) [88; 106—108]. Выделяющиеся газы
образуют воспламеняющуюся смесь, которая загорается при 460 °C*
[108, с. 20].
Полиамиды характеризуются также очень высокими скоростью
(до 50 кг/ч) и теплотой горения (~30—33 МДж/кг); для вискозного
волокна теплота горения составляет ~15 МДж/кг [108; 113]. В этих
условиях затруднены процессы, приводящие к образованию сетчатой
структуры полимера, обусловливающей понижение горючести материа-
ла. В связи с этим проблема придания огнезащитных свойств мате-
риалам из алифатических полиамидов чрезвычайно сложна. Кроме того,
до настоящего времени не изучен механизм действия огнезащитных
средств на полиамиды, поэтому трудно подобрать хороший замедлитель
горения.
В настоящее время для полиамидных волокон известно относитель-
но небольшое число огнезащитных средств, но даже и они сравнитель-
но мало эффективны. Поэтому для получения огнестойких полиамидов
требуется введение большого количества антипиренов, что значительно
ухудшает физико-механические характеристики материала. Многие из-
вестные антипирены, хорошо действующие при обработке целлюлозно-
го волокна, плохо защищают полиамидные волокна вследствие иного
механизма разложения полиамидов.
Ниже рассматриваются существующие методы придания огнестой-
кости полиамидным волокнам и тканям.
3.2. МЕТОДЫ ПРИДАНИЯ ОГНЕСТОЙКОСТИ ПОЛИАМИДНЫМ ВОЛОКНАМ
И ТКАНЯМ
3.2.1. Поверхностная отделка тканей
Для обработки полиамидных нитей и тканей используют азот-, се-
ру- и галогенсодержащие продукты. Впервые для повышения устой-
чивости к действию огня изделий из полиамидов была применена по-
верхностная отделка ткани водными растворами карбамида с формаль-
дегидом. Количество наносимого аппрета при такой отделке составляло
в зависимости от назначения ткани от 20 до 100%, что обусловливало
* В литературе [109—112] приводятся и более высокие температуры самовоспла-
менения полиамида, например 510 и 530 °C, что обусловлено различием в методах испы-
тания.
378
большую жесткость ткани. Достаточно эффективной для полиамидов
оказалась тиомочевина. У полиамидных тканей, пропитанных 3%-ным
раствором тиомочевины [П4] , повышаются огнезащитные свойства,
однако эта обработка неустойчива к стирке. Полиамидные ткани при-
обретают огнестойкость при нанесении на них около 12% тиомочевины.
Для повышения устойчивости огнезащитной отделки тканей к мокрым
обработкам их покрывают тиомочевиноформальдегидной или мочевино-
формальдегидной смолой, в которой часть мочевины замещена на тио-
мочевину [115] или ее производные.
В патентной литературе предлагаются различные по составу рецеп-
ты для огнезащитной отделки полиамидной ткани, включающие форм-
альдегид и тиомочевину [116; 117], формальдегид и алкиленмочевину
[118], формальдегид и метилольные производные мочевины или тио-
мочевины, в частности метилолтиомочевины (HOCH2NHCSNHCH2OH)
[119], диметилолэтилентиомочевины (VIII) [120], формальдегид и ди-
метилолтиотриазин (IX) и др.
Н2С----СН2
HOH2CN nch2oh
II
S
VIII
R
N
\н2
hoh2cn nch2oh
II
s
IX
Обработка сводится к пропитке ткани или нити продуктом пред-
конденсации указанных выше веществ с последующей термообработкой
материала при 100 °C и выше в присутствии катализаторов (MgCh,
NH4CI). В результате конденсации альдегидов с мочевиной и ее про-
изводными образуется сетчатый полимер. Частично этот полимер хи-
мически связан с полиамидом, так как при обработке ткани формаль-
дегидом при повышенной температуре в присутствии катализатора не
исключена возможность образования метилольных производных поли-
амида, способных к реакции конденсации. Для повышения стабильно-
сти водных растворов и эмульсий огнезащитных композиций, содержа-
щих тиомочевиноформальдегидный конденсат, применяют этиленмоче-
вину
Н2С—NH
I /С==°
Н2С—NH
X
и бисульфитметилолмочевину (HOCH2NHCONHCH2SO3H) [121, 122].
До настоящего времени нет достаточно обоснованного объяснения,
почему отделка из тиомочевины, мочевины и формальдегида способст-
вует понижению горючести полиамидов. Полагают [123], что эффект
основан на снижении температуры плавления полиамида, а следова-
тельно, более быстром стекании капель расплавленного полимера и
удалении его из зоны огня. Существует также мнение, что это обуслов-
лено способностью мочевины при температуре пламени образовывать в
379
полиамиде поперечные связи, благодаря чему замедляется его разло-
жение. Образующийся при этом слой между пламенем и несгоревшим
полиамидом препятствует выделению горючих продуктов пиролиза и
приводит к затуханию пламени.
Кроме серуазотсодержащих соединений для огнезащитной обра-
ботки полиамидных изделий применяют мочевиноформальдегидную
смолу или аминопласт на основе формальдегида и аминотриазина с до-
бавками бромида аммония [124; 125]. Вместо бромида аммония могут
быть использованы органические бромсодержащие амины (бромиды
триэтаноламина, метиламина, диметиламина, этиленамина и других
низкомолекулярных водорастворимых алифатических аминов). В ли-
тературе указывается, что хорошие огнезащитные свойства полиамид-
ные ткани приобретают при введении около 10% брома.
Для огнезащитной отделки полиамидных тканей предлагают ис-
пользовать также мочевиноформальдегидные смолы в комбинации
с диамидом щавелевой кислоты [126], фосфорными соединениями, та-
кими как пирофосфат аммония, хлорэтиламинофосфат, трис (2,3-ди-
бромпропил) фосфат [127], трис(1,3-дихлорпропил) фосфат {128].
Огнестойкая отделка полиамидных тканей фосфорсодержащими
соединениями основана, главным образом, на использовании тетра (ок-
симетил) фосфонийхлорида (III) или окситрисоксиметилфосфина (XII)
[129].
уСН2ОН
О=Р—СН2ОН
\сн2он
хп
Эти соединения имеют примерно одинаковую реакционную способ-
ность и подобно формальдегиду реагируют с аминами. Обычно тка-
ни из полиамида пропитывают водным раствором, содержащим тет-
ра (оксиметил)фосфонийхлорид (18,5%), триметилоламин (8,5%), моче-
вину (8,8%), поверхностно-активное вещество (0,1%), и отжимают при
плюсовании (степень отжима 75%). После сушки при 110 °C тетраме-
тилфосфонийхлорид конденсируют с триметилолмеламином при 145 °C,
в результате чего на поверхности ткани образуется огнезащитный слой
из нерастворимого трехмерного продукта. Выделяющийся при этом хло-
ристый водород связывается мочевиной. Вместо триметилол аминов мож-
но использовать другие полифункциональные соединения азота, в част-
ности алифатические первичные амины, первичные, вторичные и третич-
ные ароматические амины и полиамины, а также амиды алифатических
и ароматических кислот [130—132]. Иногда применяют анилин, дифе-
ниламин и цианамид.
Существует много других огнезащитных отделок, содержащих фос-
форорганические соединения, такие как, например, триэтиленимид фос-
форной кислоты (IV) [433], хлорид трис(3-галогеи-2-оксипропил)оксиме-
тилфосфония (XIII) {134], галогенированные оксиды фосфина; оксид
трис (хлорметил) фосфина (XIV) или оксид феноксиметил бис(хлорме-
тил)фосфина (XV) [135], трис(1,3-дихлорпропил)фосфат (XVI) [128]
РСН2ОН1+ СГ
XIII
(где X—галоген)
380
>СН2С1
О=Р—СН2С1
\сн2с1
XIV
XV
,ОСНС1СН2СН2С1
О= Р—ОСНС1СН2СН2С1
\оСНС1СН2СН2С1
XVI
Однако фосфорсодержащие соединения не всегда эффективны для
полиамидных материалов.
Для полиамидных тканей технического назначения рекомендуют
обработку высокогалогенированными соединениями типа поливинил-
хлорида (ПВХ) [136; 137], поливинилиденхлорида (ПВДХ) [138], хло-
рированного полиэтилена [139], хлоркаучуков [140], хлорпарафинов,
хлбрамидов [141], галогенсодержащих N-метиламинов [142]. В боль-
шинстве случаев при поверхностной отделке полиамидных материалов
галогенсодержащие вещества используют в сочетании с трехокисью
сурьмы, обладающей синергическим действием. Композиции с ПВХ,
ПВДХ и хлорированным полиэтиленом содержат пластификаторы, чаще
эфиры фосфорной кислоты. Иногда в состав огнезащитной отделки
в качестве связующего добавляют полимерные латексы, например по-
лиакрилонитрильный [143; 144]. Полиамидные ткани, сетки или канаты
пропитывают водной эмульсией или суспензией огнезащитного состава
и сушат. Для получения определенного эффекта по огнезащите данный
состав наносится на ткань в больших количествах (30—50%), вслед-
ствие чего материал становится жестким.
Для огнезащитной отделки полиамидных тканей можно использо-
вать также хлорированные бензолы [145] и фосфонитрилхлорид [146],
ангидрид тетрахлорфталевой кислоты (XVII), хлорэндиковую кислоту,
полученную путем конденсации гексахлорциклопентадиена с малеино-
вым ангидридом (XVIII) [11], 1,2-бис (3,4-дибромциклогексил)-1,2-ди-
бромэтан (XIX) [147], димер гекса бром циклопента диена и другие соеди-
нения.
Вг Вг
XVII XVIII XIX
Несмотря на то, что имеется большое число патентов на огнестой-
кую отделку полиамидных тканей, в литературе не приводится сведений
о том, какие из них находят практическое применение. Критический
анализ опубликованных данных позволяет сделать вывод, что большая
часть предложенных композиций все-таки практического интереса не
представляет, так как во многих случаях поверхностная отделка ткани
неустойчива к многократным стиркам, химической чистке, ухудшает
физико-механические свойства материала, повышая в ряде случаев
жесткость тканей и снижая прочность на разрыв.
381
3.2.2. Добавки антипиренов в полимер
В большинстве патентов для получения огнестойких полимерных
композиций на основе полиамидов в качестве добавок предлагают ис-
пользовать галогенированные органические соединения. К ним отно-
сятся хлорированный дифенил, содержащий 20—70% хлора, гексабром-
дифенил, трис (2,3-дибромпропил) фосфат, хлорированный нафталин,
содержащий ^50% хлора [148], галогенированный дифенилсуль-
фид [149].
Для повышения огнезащитных свойств материала наряду с га-
логенорганическими соединениями предлагается вводить неорганиче-
ские соли или окислы металлов, оказывающие синергический эффект:
борат свинца, ZnO, SnOz, РЮ, СиО, РегОз, 8ЬгО3 в количестве 1—15%.
Кроме того, рекомендуют также использовать неорганические галоген-
содержащие вещества, например галогениды Zn, Cd, Pl или редкозе-
мельных металлов, и смеси 3—15% такого галогена с 1—5% SnOz
и СиО.
Различные фирмы США предлагают в качестве замедлителей горе-
ния для полимерных материалов бромсодержащие соединения под
общим названием «фламмекс», которые вводятся на стадии полиме-
ризации или переработки полимера. В частности, в качестве добавок
для полиамидов рекомендуют «фламмекс В10» — бромированное аро-
матическое соединение, содержащее 84,7% брома с taa, равной 378 °C,
и /кип 300°С. Фламмекс В10 — термостойкое вещество; летучие потери
при 300°С не превышают 2%.
Введение фламмекса В10 в расплав полиамида (найлона 6) в
количестве 20% или в комбинации с окисью сурьмы (соответственно
12% и 8%) придает полимеру и изделиям из него повышенную устойчи-
вость к действию огня. Однако при использовании фламмекса В10
необходимо учитывать тот факт, что он снижает вязкость расплава и
вызывает коррозию технологического оборудования. Поэтому стремятся
сократить продолжительность нахождения расплава при высокой темпе-
ратуре и предъявляют более жесткие требования к металлу, из кото-
рого изготавливают оборудование. Все поверхности, соприкасающиеся
с расплавом, должны быть хромированы или защищены каким-нибудь
способом.
В литературе указывается на возможность введения в расплав
полиамида фосфорсодержащих соединений, таких, как трифенилфосфат,
трифенилфосфит и их метил- и галогенпроизводные [150].
В патентах [151, 152] предлагается способ получения невоспла-
меняющегося полиамидного волокна из смеси найлона с фосфорсодер-
жащим соединением типа
Н2С-О °
I /Р—ОН
Н2С—СИ
XXI
(продуктом реакции этиленгликоля с РС13) и полимером ди-р.р'-хлор-
этилового эфира винилфосфиновой кислоты.
Обычно общее содержание добавок в огнестойких композициях
составляет до 30% от массы полиамидов. Их рекомендуют вводить до
382
и после реакции полимеризации. При добавлении к полимеру огнеза-
щитные вещества смешивают с гранулами или наносят на гранулы из
легколетучего растворителя, некоторые из них можно добавлять прямо
в расплав. Считается, что полученные системы термостойки при темпе-
ратурах переработки, но выделяют галогенводород или фосфорную
кислоту [153] при температуре пламени.
Однако необходимость введения в полиамид достаточно больших
количеств таких добавок приводит к резкому снижению физико-
механических характеристик материала. В основном такие смеси
рекомендуют использовать для электроизоляционных покрытий, хотя в
патентах указывается на возможность применения их для получе-
ния нитей. Кроме того, формование полиамидных нитей из расплава
при 260—280°C требует применения термически стабильных соеди-
нений.
В связи с этим многие эффективные огнезащитные средства на
основе галогенированных и фосфорсодержащих соединений не могут
быть добавлены к полиамидам, так как они при высоких температурах
выделяют кислые продукты пиролиза, которые способствуют расщеп-
лению связи —СО—NH— и тем самым — деструкции полимера. Все
это весьма ограничивает число веществ, используемых для этих целей.
3.2.3. Химическая модификация полимера и волокна
Химическая модификация полиамидов с целью придания огнестой-
кости довольно перспективна, так как позволяет наряду с устойчи-
выми огнезащитными свойствами получать неплавкие волокна. Иссле-
дования в этом направлении начаты сравнительно недавно, но уже
накоплен определенный экспериментальный материал как по химизму
процессов, так и по изучению свойств огнестойких полиамидных мате-
риалов.
Для получения огнестойких полиамидных волокон методом хими-
ческой модификации используют реакции полимераналогичных пре-
вращений и привитой сополимеризации.
Полимераналогичные превращения. Наличие подвижного водорода
у атома азота амидных групп полиамида позволяет осуществлять
различные химические превращения путем взаимодействия с хлоран-
гидридом и изоцианатами и др. Используя соединения, содержащие
фосфор или галогены, можно химически присоединить антипирены к
полиамидам и тем самым получить устойчивый к различным обработ-
кам эффект огнезащиты. В основном для химической модификации
полиамидов с целью повышения огнестойкости применяются фосфор-
содержащие соединения. Описано получение огнестойкого полиамида
путем фосфорилирования хлор ангидридам и метилфосфиновой кисло-
ты [146].
Реакция фосфорилирования поликапроамидного волокна дихлор-
ангидридом метилфосфиновой кислоты осуществлялась при мольном
соотношении компонентов (полиамид — хлоргангидрид) в присутствии
избыточного количества органического основания (2 моля пиридина на
1 моль хлорангидрида) в течение 1—8 ч при разных температурах.
Чтобы избежать гидролиза дихлорангидрида, реакцию проводили в
среде абсолютного толуола.
383
Взаимодействие дихлорангидрида метилфосфиновой кислоты
поликапроамидом в общем виде протекает по схеме:
[—NH(CH2)5CO—]„ + СН3РОС12 ->
О СН,
-----р------
—СО—N—(СН2)5—СО—N—(СН2)6—СО—N—(СН2)5—СО—NH(CH2)5—СО—
---> ' Н3С— Р=О
—СО—N—(СН2)5—СО—NH(CH2)5—CONH(CH2)5—СО—N(CH2)5—СО—
%|
н/Р-СНз
с
Изменяя температуру реакции в интервале 20—100°С, можно
ввести в полиамид до 5% фосфора (рис. 3.3), но при этом происходит
сильная деструкция полимера. Волокно становится хрупким и силь-
но темнеет. В оптимальных ус-
Рис. 3.3. Зависимость количества вводимого
фосфора (концентрация дихлорангидрида—
1 моль на элементарное звено поликапро-
амида) от условий реакции фосфорилиро-
вания (ф—-в среде толуол — пиридин
т= 1 ч; О — в диметилформамиде, т=8 ч).
ловиях, когда сохраняется волок-
нистая структура (100 °C, 8 ч),
степень замещения составляет
всего лишь 9,6% от теоретически
возможной, содержание фосфо-
ра— 2,3%. Такие модифициро-
ванные полиамидные волокна
становятся нерастворимы в кон-
центрированной серной кислоте,
но температура плавления поли-
амида снижается до 200—205 °C.
Следует отметить, что целлю-
лозные материалы, модифициро-
ванные аналогичным способом
(фосфорилированием дихлоран-
гидридом метилфосфиновой кис-
лоты), приобретают огнестой-
кость при содержании фосфора
2% [154], в то время как поли-
амидные волокна, содержащие
2,5% фосфора, полностью сгорают. Другие синтетические материалы,
такие, как полиуретаны -[153] и поливинилспиртовые волокна [155],
подобно целлюлозе также не поддерживают горения при незначитель-
ном содержании фосфора (1,5—2,5%). Это, по-видимому, обусловлено
различным механизмом разложения указанных полимеров. Одной из
причин, затрудняющих получение модифицированного полиамидного
волокна с высоким содержанием фосфора, является высокоориентиро-
ванная кристаллическая структура полиамидного волокна. При фосфо-
рилировании неориентированного полиамида (например, переосажденно-
го из раствора в муравьиной кислоте) количество вводимого фосфора
(в %) в одних и тех же условиях обработки может быть увеличено в 7 раз:
Полиамид (порошок) .... 8,5—8,6
Волокно...........................1,9—2,0
Техническая нить ................. 0,9—1,1
384
Таблица 3.2. Влияние условий реакции фосфорилирования
полиамидного волокна на содержание фосфора и свойства
модифицированного волокна (реакция проводилась в присутствии
пиридина при 40 °C в течение 1 ч)
Содержание дихлор- ангидрида метилфос- финовой кислоты в растворе абсолют- ного толуола, % Содержание фосфора, % Температура плавления, °C Удельная вяз- кость* Коксовое число** Отношение к дей- ствию огня
Исходное волокно 215 1,20 3,0 Горнт
1,5 2,2—2,5 193—199 0,86 7,0 »
50 5,6—6,8 133—137 0,42 13,8 »
100 (расплав) 8,2—8,7 115—122 0,03 31,8 Не поддержи- вает горения
* В 84%-ной серной кислоте (концентрация полимера — 1 г на 100 мл растворителя).
** Несгоревший остаток после выдерживания волокна при 450 °C в течение 30 мин на воздухе.
Существенное влияние на количество введенного фосфора оказы-
вает и концентрация дихлорангидрида (табл. 3.2).
Полученные авторами данные свидетельствуют о том, что поли-
амидное волокно приобретает огнестойкость только при содержании
фосфора 8—9%. Примерно такое же количество фосфора (7—9%)
содержат огнестойкие полиамиды на основе дикарбоновых кислот
с оксифосфиновой группой и различных диаминов [156]. Однако прове-
дение реакции фосфорилирования при высокой концентрации дихлор-
ангидрида, обеспечивающей введение в полиамидное волокно требуе-
мого количества фосфора, приводит к значительному снижению
молекулярной массы полимера. Причиной снижения молекулярной
массы поликапроамида является деструкция, вызываемая выделяющим-
ся при реакции НС1, и ацидолиз благодаря взаимодействию хлор-
ангидрида кислоты с амидными группами макромолекул.
При фосфорилировании полиамидных волокон монохлорангидри-
дом метилфосфиновой кислоты вследствие более низкой реакционной,
способности этого реагента ввести в полимер более 2,3% фосфора не
представляется возможным.
Химическое присоединение к полиамидному волокну такого актив-
ного антипирена, как фосфонитрилхлорид '[146], позволяет ввести в
макромолекулу полимера от 5 до 8% фосфора. Обработку волокна
проводили 20%-ным раствором фосфонитрилхлорида в абсолютном
толуоле при 100°С в течение 8 ч. Полученный модифицированный поли-,
амид также содержит 7—8% хлора. Однако, несмотря на введение в
полиамид наряду с фосфором хлора, полимер приобретает огнестой-.
кость при содержании фосфора не менее 7—8%.
В американских патентах [157; 158] предложена огнезащитная
отделка полиамидных тканей, основанная на присоединении к полиами-
ду фосфорсодержащих соединений с реакционноспособной изоцианат-
ной группой. Например, полиамидную ткань пропитывают смесью, со-
стоящей из продуктов реакции этиленгликоля, диэтилхлорфосфата и
димера 2,4-толуилендиизоцианата:
О О
II II
(Н5С2О) 2РОСН2СН2ОС OHN. С —N. ^NHOCOCH2CH2OP (ОС2Н5) 2
О <и ОС
Н.,(Х 1|
о
25—2254
385
Затем ткань нагревают в течение 3 мин до 180°iC. В этих условиях
димер распадается, и образовавшееся вещество с реакционноспособной
группой —NCO реагирует с подвижным водородом у атома азота поли-
амида. В противоположность мнению 3. А. Роговина с сотр. {146] авто-
ры этих патентов характеризуют полиамидную ткань, содержащую 2—
3% фосфора, как материал, обладающий пониженной горючестью. Что
касается галогенсодержащих соединений, взаимодействующих с поли-
амидом в процессе обработки тканн, то в патентной литератуое упоми-
наются 1,4,5,6,7,7'-гексахлор-5-карбонен-2-ацетилхлорид (XXII)
|ci- -С1
СН2СОСР
С1
XXII
и галогенсодержащие эпоксисоединения общей формулы:
X2YC(CH2)nCH—СН2, где п = 0—4; X=Y —Cl, Br, F.
Ткань рекомендуют пропитывать раствором 1,4,5,6,7,7'-гексахлор-
5-карбонен-2-ацетилхлоридом, содержащим акцептор кислоты, и после
испарения растворителя нагревать непродолжительное время при
200°С. При этой температуре хлорангидрид кислоты реагирует с поли-
амидом [159]. В случае галогенсодержащих эпоксисоединений, в част-
ности 1,1', Г'-трихлор-2,3-эпоксипропана (С1зССН—iCH2) [160], реакция
взаимодействия с водородом амидной группы полимера протекает при
более низкой температуре (НО°C). Количество присоединенного веще-
ства составляет около 20%. Полученные материалы называют само-
затухающими, так как они горят вблизи пламени и гаснут при удале-
нии источника огня.
Анализируя имеющиеся по этому вопросу литературные данные, *
можно сделать вывод, что использование данного метода для моди-
фикации полиамидных волокон более целесообразно с целью получения
не огнестойких волокон, а волокон с пониженной горючестью, так как
высокий эффект огнестойкости требует проведения реакции химических
превращений с высокой степенью замещения, что должно приводить к
ослаблению межмолекулярного взаимодействия и как следствие — к
снижению механических свойств материала.
Привитая сополимеризация. Данные об использовании метода
привитой сополимеризации для придания огнестойкости полиамидным
нитям немногочисленны. Бриджфорд [161] предложил с этой целью при-
вивать на полиамиды галогенсодержащие полимеры. Позднее появил-
ся ряд патентов на огнезащитную отделку полиамидных тканей путем
радиационной прививки поливинилхлорида [162] и поливинилиденхло-
рида {163] облучением частицами высокой и низкой энергии. В работе
[164] указывается на повышение огнестойкости полиамидных нитей в
результате прививки 70% поливинилфторида.
386
Из опубликованных данных видно, что наиболее широкие исследо-
вания проведены в области получения огнестойких полиамидных воло-
кон на основе сополимеров поликапроамида с полиакролеином. При-
вивка полиакролеина, с одной стороны, приводит к получению неплав-
ких волокон на основе алифатических полиамидов, а с другой сторо-
ны,— содержание реакционноспособных функциональных групп в при-
витых цепях позволяет осуществлять последующие превращения этого
производного полиамида введением различных антипиренов или цик-
лических звеньев путем термического нагревания модифицированных
Содержание привитого
полианролеина, °/о
Рис. 3.4. Влияние условий реакции привитой сополимеризации на содержа-
ние привитого полиакролеина:
/ — продолжительность реакции (Сц2О2=0,5% Смои=50%);
. 2 — концентрация перекиси водорода (^иои=50%, т=1 ч);
3 — концентрация акролеина <Сн2О2~°.3^1', т=1 ч).
Рис. 3.5. Зависимость содержания альдегидных групп от содержания при-
витого полиакролеина:
/ — расчетные данные; 2 — экспериментальные данные [О, ®, X — привес соответст-
венно в зависимости от ^н оа’ ^мои и т РеакУии)-
Проведенные исследования интересны в научном плане для выяс-
нения необходимого содержания фосфора в полиамиде для придания
ему огнестойкости.
Основная сложность заключается в синтезе привитых сополимеров^
так как известно [165, 166], что полимеризация акролеина может про-
текать как по ванильной и альдегидной группам, так и по механизму
1,4-присоединения, в результате чего в получаемом полимере содержат-
ся различные звенья (альдегидные группы, двойные связи и т. д.).
Наличие в макромолекуле полимера звеньев того или иного типа в боль-
шей степени зависит от условий проведения реакции полимеризации.
Для получения на основе привитых сополимеров огнестойких материа-
лов необходимо введение в боковые цепи альдегидных групп. В усло-
виях привитой сополимеризации, осуществляемой с использованием
окислительно-восстановительной системы Fe34- — Н2О2 [167], акролеин
полимеризуется в основном по винильным группам, что позволяет
ввести в полиамид значительное количество альдегидных групп. В рабо-
25*
387
30
-^600
-$500
-*400
-^200
100
^20
ООО
^10
20 40 60 80
Содержание привитого полиакролеина.,
&25
5
О'- О
Рис. 3.6. Изменение механических свойств
модифицированного 'поликапроамидного во-
локна в зависимости от содержания приви-
того полиакролеина:
/ — прочность; 2 — удлинение.
Получение сополимеров полиамида
те [168] синтез привитых сополимеров на основе поликапроамидного
волокна и полиакролеина по указанному выше методу проводили из
водных растворов акролеина при 70 °C и pH 7.
Количество привитого полиакролеина в значительной степени зави-
сит от условий проведения реакции (концентрации мономера и переки-
си водорода и продолжительности) (рис. 3. 4). Используя этот метод
прививки, можно получать модифицированные полиамидные волокна
с содержанием полиакролеина до 72% (от массы привитого сополиме-
ра). Однако выход привитого полиакролеина в исследуемых условиях
реакции не превышает 15% от
исходного мономера. В моди-
фицированных полиамидных
волокнах при содержании по-
лиакролеина до 64% методом
оксимирования [169] опреде-
ляется до 70—90% альдегид-
ных групп от расчетного коли-
чества. При дальнейшем повы-
шении содержания полиакро-
леина количество альдегидных
групп понижается, что обус-
ловлено возможностью проте-
кания реакции полимеризации
акролеина и по альдегидным
группам. Максимальное коли-
чество альдегидных групп, ко-
торое удалось ввести в поли-
амидное волокно в результате
прививки полиакролеина, со-
ставило 49% (или 24,5%‘ от
массы привитого сополимера)
(рис. 3.5).
с высоким содержанием при-
витого полиакролеина вызвано необходимостью введения в волокно при
последующем фосфорилировании сравнительно больших количеств
фосфорсодержащего антипирена. Ранее показано [146], что при при-
менении фосфорсодержащих замедлителей горения огнезащитный эф-
фект для поликапроамидного волокна достигается при содержании фос-
фора в волокне не ниже 8%. В идеальном случае для введения такого
количества фосфора может быть использован привитой сополимер поли-
капроамида, содержащий 33,5% полиакролеина. Однако в реальных
условиях, учитывая возможность протекания реакции полимеризации
акролеина по разным механизмам, вероятность побочных реакций
альдегидных групп и то, что степень замещения их на a-фосфорно-
кислые при высоком содержании полиакролеина, как будет показано
ниже, не превышает 60—70%, содержание привитого полиакролеина в
сополимере для введения необходимого количества фосфора должно
быть значительно выше (64%). Естественно, что прививка такого коли-
чества полиакролеина ухудшает механические характеристики волокна;
прочность снижается, удлинение в зависимости от содержания приви-
того полимера проходит через максимум (рис. 3.6). Путем рентгено-
графических исследований показано, что при содержании в волокне
полиакролеина 37—64% ухудшается ориентация кристаллитов по срав-
388
нению с исходным поликапроамидным волокном (угол разориентации в
модифицированном волокне, содержащем 64% полиакролеина, равен
14° (стл—200), а в исходном — 9°) и снижается степень кристаллич-
ности, о чем свидетельствует увеличение интенсивности аморфного гало
на рентгенограммах волокон.
Фосфорилирование привитого сополимера поликапроамида с поли-
акролеином (ПКА—ПА). Наличие в макромолекуле модифицированно-
ного полиамида реакционноспособных альдегидных групп позволяет
проводить полимераналогичные превращения с диметилфосфитом с
целью придания полиамидным волокнам огнестойкости. Фосфорилиро-
вание проводят в среде абсолютного толуола в присутствии 1 % диэтил-
амина при десятикратном мольном избытке диметилфосфита по отно-
шению к С=О группам. Реакция взаимодействия привитых сополиме-
ров ПКА—ПА с диметилфосфитом протекает, по-видимому, по следу-
ющей схеме:
Ч (CsHshNH
[—NHCH(CH2)4CO—]m + /Р(ОСН3)2 ----►
[-CH2-CH-]„
1^°
\н
[—NHCH(CH2)4CO—]m
[-CH2-CH-]„
(C2H5)2N-CH-P(OCH3)2
II
о
При использовании сополимеров ПКА—ПА с содержанием 26% аль-
дегидных групп степень замещения альдегидных групп, а следователь-
но, содержание фосфора в волокне сильно зависит от условий реакции
фосфорилирования (рис. 3.7). Полное замещение альдегидных групп
а-оксифосфорнокислыми наблюдается при проведении реакции в чистом
диметилфосфите при 140 °C в течение 6 ч. В результате фосфорилирова-
ния привитых сополимеров поликапроамида (содержание альдегидных
групп изменялось от 12 до 49%) количество фосфора закономерно
повышается с увеличением полиакролеина в сополимере, однако при
содержании альдегидных групп в привитом сополимере выше 26% сте-
пень превращения групп €=О снижается до 60—70% от теоретически
возможной. Максимальное количество фосфора, введенное в волокно,
составляет 11%.
Модифицированные поликапроамидные волокна с содержанием
фосфора выше 7% характеризуются повышенной стойкостью к действию
огня. Они не загораются при действии открытого пламени (под углом
45 0 к волокну) в течение 10 с. Однако эти волокна имеют низкую
прочность (30—50 мН/текс), что обусловлено большими структурными
изменениями поликапроамида в результате прививки полиакролеина и
фосфорилирования. На рентгенограммах такого волокна появляется
широкое распределение интенсивности, характерное скорее для рас-
плава, чем для аморфной части в твердом состоянии, что указывает на
более широкий набор по расстояниям между макромолекулами. Угол
разориентации увеличивается до 19°.
Превращения альдегидных групп в макромолекуле привитого сопо-
лимера поликапроамида при термоокислении. Наличие в привитой цепи
альдегидных групп, способных при тепловом воздействии к образова-
нию трехмерных структур, позволяет получать неплавкие модифициро-
ванные поликапроамидные волокна, обладающие хорошей формоустой-
чивостью при действии высоких температур, В зависимости от условий
26—2254 389
термоокисления процессы структурообразования в модифицированном
полиамидном волокне протекают по-разному. При медленном нагрева-
нии волокна (3 град/мин) химические превращения альдегидных групп
приводят к глубоким изменениям поликапроамида. Даже при неболь-
шом содержании привитого полиакролеина (до 10%) модифицирован-
ное волокно не плавится, и на его термограмме не обнаружено эндотер-
мического эффекта в области 200—220 °C,
обусловленного плавлением полимера. При
нагревании волокна до 200 °C с более высокой
скоростью (10—20 град/мин) структурирова-
ние сополимера протекает не по всему объему
волокна и, хотя волокно по-прежнему сохра-
няет волокнистую структуру (вплоть до
450 °C), термограмма фиксирует тепловой
эффект, ответственный за плавление поликап-
роамида. С увеличением содержания поли-
акролеина в волокне способность полимера к
структурообразованию закономерно возраста-
ет.
Следует отметить, что ни один из извест-
ных методов |[170—-172] структурирования по-
ликапроамидных волокон полифункциональ-
ными соединениями не дает такого значитель-
ного эффекта снижения плавкости полиамида,
как прививка полиакролеина. Для «сшитых»
волокон, описанных в указанных работах, на-
блюдается лишь смещение температуры пере-
хода в вязкотекучее состояние в область более
высоких температур по сравнению с исходным
поликапроамидом.
Используя способность поликапроамид-
ных волокон, модифицированных прививкой
полиакролеина, сохранять волокнистую струк-
туру при температурах выше 200 °C, получили
огнестойкие волокна путем длительного тер-
мического воздействия в присутствии кислоро-
да воздуха. Поликапроамидные волокна, со-
держащие 30—40% полиакролеина, вначале
нагревают на воздухе от комнатной темпера-
скоростью 7 град/мин, затем выдерживают при
гние 1—2 ч (под натяжением или в свободном
состоянии). Если в процессе термообработки удаляется не менее 50%
(от первоначальной массы), волокна становятся стойкими к действию
открытого пламени. После такой обработки волокно заметно обогаща-
ется углеродом (табл. 3.3).
Содержание кислорода в волокне уменьшается. Сравнительно не-
большое изменение содержания азота в волокне после термообработки
позволяет предположить, что он принимает участие в образовании
циклических звеньев.
Введение хлорсодержащих соединений в модифицированное поли-
капроамидное волокно. Определенный интерес представляет получение
огнестойких полиамидных волокон на основе поликапроамида, модифи-
цированного прививкой полиакролеина, путем введения хлорсодержа-
^ДМФ, °1°
I---1—1__I l I I
/ г 3 4 5 6
Т,ч
I--1--1--L—L__I I I
О 15 20 25 30 35 40 45
Рис. 3.7. Влияние условий
реакции фосфорилирования
на содержание фосфора в
модифицированном волок-
не:
1 — температура реакции (т-3 ч,
с дмф — 50%);
2 — концентрация диметилфос-
фита (t=140° С, т-3 ч);
3 — продолжительность реакции
(<=140°C, СдМФ=50%);
4 — содержание альдегидных
групп в привитом сополимере
(Т=6 ч, <=140 °C, СДМф =
= 100%).
туры до 350—400 °C со
этой темпепатупе в
390
Таблица 3.3. Влияние условий термоокисления иа элементарный состав
модифицированных ПКА волокон и огнестойкость
(содержание привитого полиакролеина 30%)
Волокно Потеря массы Ьолокном, % Элементарный состав, % Отношение к действию огня
с о N н
До термоокисления — 60,95 20,88 9,39 8,78 Горит
После термоокнсления (при 345— 355 °C) под натяжением: в течение 1 ч 41,0 68,98 14,44 9,26 7,32 >
в течение 2 ч 60,6 77,37 8,59 7,08 6,96 Не горит
Рис. 3.8. Зависимость содержания SbCh,
введенного в .модифицированное поликапро-
амидное волокно, от концентрации соли в
растворе в разных растворителях:
1 — бензол; 2 — метиловый спирт; 3 — ацетон.
щих антипиренов, в частности хлорида сурьмы, являющегося хорошим
замедлителем горения.
При применении этого антипирена для поликапроамидных волокон
даже при высоком содержании соли на волокне трудно обнаружить ог-
незащитный эффект вследствие
того, что при действии пламени
волокно быстро плавится и капли
расплава удаляются из зоны ог-
ня. Однако, если судить по коксо-
вому остатку, применение SbCl3
для поликапроамидного волокна
неэффективно.
Применение неплавкого по-
ликацроамидного волокна за счет
прививки полиакролеина позво-
ляет при обработке его SbCl3
получать модифицированные по-
лиамидные волокна с высокими
огнезащитными свойствами. Ис-
следования проводили на волок-
не, содержащем 16—21% полиакролеина. С целью равномерного рас-
пределения хлорида сурьмы волокна обрабатывали раствором соли в
различных низкокипящих растворителях — ацетоне, метиловом спирте,
бензоле, которые затем удаляли с волокна испарением при температу-
рах, соответствующих температуре кипения растворителя. В зависи-
мости от условий обработки на волокне можно фиксировать от 7 до 40%
хлорида сурьмы (рис. 3.8), который достаточно прочно удерживается
на волокне. Несмотря на то, что SbCl3 является инертным наполните-
лем, не исключена возможность частичного образования комплексов
SbCl3 с альдегидными группами привитого полиакролеина.
При введении около 15% хлорида сурьмы поликапроамидные во-
локна приобретают огнезащитный эффект — не поддерживают горения.
Физико-механические свойства этих волокон зависят от нагрузки на
волокно в процессе его обработки. При обработке волокна, находящего-
ся в свободном состоянии, прочность и удлинение резко снижаются
(соответственно до 40—60 мН/текс и 7—9%). При осуществлении обра-
ботки волокна под натяжением удается в значительной степени сохра-
нить механические показатели волокна (прочность 200—300 мН/текс,
удлинение 23—38%). Однако после водных обработок огнестойкость
этих волокон ухудшается.
26*
391
Исследование процесса термоокислительного разложения различ-
ных типов огнестойких полиамидных волокон. Для огнестойких ма-
териалов, полученных на основе органических соединений, большое зна-
чение имеют исследования термоокислительной деструкции (состав,
количество и токсичность продуктов разложения, образующихся в ре-
зультате действия высоких температур).
Имеющиеся в литературе сведения по этим вопросам для огнестой-
ких волокон, и для полиамидных в частности, очень ограничены.
Изучена [168] кинетика термоокислительной деструкции неплав-
ких огнестойких поликапроамидных волокон (полученных на основе
Рис. 3.9. Термограви1мет1риче1акие кривые ((скорость нагрева 3 прад/мин):
1 — поликапроамидное (ПКА) волокно; 2 — огнестойкое фосфорсодержащее (8.4%
фосфора) на основе привитого сополимера с полиакролеином (ПА) состава 36:64
(ПКА—ПА); 3— огнестойкое иа основё привитого сополимера с полиакролеииом со-
става 84: 16 (ПКА—ПА), содержащее 26% SbCla; 4— огнестойкое, полученное при тер-
моокислении привитого сополимера с полиакролеином состава 70 : 30 (ПКА—ПА);
5 — поликапроамидное волокно, модифицированное прививкой полиакролеииа состава
36 : 64 (ПКА—ПА).
привитых сополимеров с полиакролеином путем химического присое-
динения фосфорсодержащего соединения, введения хлорида сурьмы и
термообработки на воздухе), в условиях неизотермического нагревания
на воздухе в интервале температур 200—800 °C методом ДТА и ТГА
анализов; исследован также характер термических превращений, ка-
чественный и количественный состав продуктов разложения.
Природа антипирена, характер его связи с полимером, наконец,
структурные превращения полиакролеина, входящего в состав огне-
стойких полиамидных волокон, несомненно должны оказывать сущест-
венное влияние на процесс термоокисления поликапроамида.
Данные ТГА и ДТА свидетельствуют о сложности процессов, про-
текающих при нагревании исследуемых материалов. Температуры на-
чала разложения испытываемых волокон в зависимости от способа при-
дания им огнестойкости лежат в области 200—300 °C (рис. 3.9), и де-
струкция протекает в несколько этапов и, по-видимому, по различным
механизмам. Прежде всего обращает на себя внимание то, что привив-
ка полиакролеина снижает температуру начала разложения, и огнестой-
кие фосфор- и галогенсодержащие полиамидные волокна начинают раз-
лагаться на 50—100 °C раньше, чем исходное поликапроамидное волок-
но. При этом и эффективные энергии активации первого этапа разло-
392
Таблица 3.4. Параметры термоокислительной деструкции исследуемых волокон
Температурный интервал,
°C
Потеря массы, % от ис-
ходной
Еэфф, кДж/моль
Порядок реакции л
Исходное поликапроамидиое волокно
300—475 I 82 I 115,1 ±12,5 I 0,7
475—560 I 12 | 150,7+20,9 | 0,6
Огнестойкое фосфорсодержащее ПКА волокно
250—300 12 92,1+12,5 1
300—450 10 117,2—146,5 —
470—600 30 167,4—209,5 —
600—800 31 184,2+20,9 0,6
Огнестойкое хлорсодержащее ПКА волокно
190—310 33 69,0+8,4 0,9
ЗЮ—475 26 125,6—146,5 —
475—600 29 209,5 —
Огнестойкое волокно, полученное прн термоокислении волокна ПКА-ПА
300—600
85 | 159,0+12,5 | 1,3
жения несколько ниже: соответственно 92,1 ±12,5 и 69,0±8,4 кДж/моль
(для поликатроамидного волокна 115,1 ±12,5 кДж/моль) (табл. 3.4).
Рис. 3.10. Зависимость скорости разложения различных типов огнестойких
полиамидных волокон от температуры. Кривые 1—4 ом, рис. 3.9.
Дополнительная термообработка волокна, модифицированного привив-
кой полиакролеина, нивелирует это различие в температуре (см. рис.
3.9, кривая 4), но в этом случае скорости массовых потерь значительно
ниже, чем у исходного поликапроамидного волокна (рис. 3.10), а эффек-
тивные энергии активации выше (табл. 3.4). В то же время все иссле-
дуемые огнестойкие полиамидные волокна при нагревании на воздухе
393
в области температур, наиболее опасных для самовоспламенения поли-
капроамида (460—500 °C), разлагаются с гораздо меньшей скоростью,
чем исходное волокно. Так, максимум скорости потери массы галоген-
и фосфорсодержащих волокон примерно в 4 раза меньше, чем у поли-
капроамида, причем эти максимумы смещены соответственно до тем-
ператур 560 и 700 °C.
Если сравнить количество твердого остатка огнестойких волокон,
содержащих в качестве антипиренов фосфор и хлорид сурьмы, с исход-
Рис. 3.11. Зависимость средней
скорости газообразования от
температуры:
1 — поликапроамидное волокно; 2 —
огнестойкое фосфорсодержащее по-
лиамидное волокно, полученное иа
основе привитого сополимера с по-
лиакролеином.
медленном нагревании в
ным поликапроамидным волокном при кри-
тической температуре самовоспламенения
(450°C), то оказывается, что последнее в
выбранном режиме нагревания при данной
температуре уже практически полностью
разрушается (остаток массы 12%), тогда
как у огнестойких материалов при сохране-
нии волокнистой структуры остается от пер-
воначальной массы до 70% (фосфорсодер-
жащее) или до 30% (содержащее SbCl3).
Данные при термоокислении при мед-
ленном нагревании хорошо коррелируют с
результатами, полученными в условиях
теплового удара. В этом случае фосфорсо-
держащие модифицированные волокна так-
же разлагаются с меньшей скоростью по
сравнению с поликапроамидом (рис. 3.11),
а энергия активации газообразования при
тепловом ударе (500—800 °C), равная
153,2 кДж/моль, близка к Еэфф термоокисли-
тельной деструкции в условиях медленного
нагрева в той же области температур
(184,2 кДж/моль). В отличие от поликапро-
амида для исследуемых огнестойких воло-
кон процессы химических превращений при
атмосфере воздуха протекают со значитель-
ным выделением тепла, по-видимому, вследствие большой склонности
полиакролеина к реакциям окисления (рис. 3.12).
Данные ДТА полиамидных волокон, модифицированных прививкой
полиакролеина, показывают, что в интервале 170—300 °C с полиакро-
леином происходит сложный комплекс превращений, обусловленный,
по-видимому, протеканием различных конкурирующих процессов
(несколько экзотермических пиков и перегибов на кривой ДТА при
200, 210 и 265°C) (см. рис. 3.12, кривая 5). Вероятно, это процессы
окисления, структурирования и другие. Некоторые из них способствуют
повышению термостабильности, а другие препятствуют.
При предварительной термообработке волокна на воздухе превали-
руют, вероятно, процессы структурообразования, что и обеспечивает
повышение термостойкости волокон. У такого волокна отсутствуют экзо-
термические пики в области температур 200—350 °C, что свидетельству-
ет о более или менее полном завершении указанных процессов. Антипи-
рены действуют иначе. Так, при введении хлорида сурьмы лишь частич-
но подавляется нежелательный процесс окисления полиакролеина
(уменьшение интенсивности экзопика). Значительно эффективнее дейст-
вие фосфора, который в значительно большей степени подавляет
394
реакции окисления, но при этом не препятствует другим превращениям
полиакролеина, приводящим к повышению термостабильности волокна
при высоких температурах (выше 450°C).
Таким образом, имеющиеся данные позволяют предположить,
что огнезащитный эффект достигается за счет торможения процессов
окисления и разложения при температурах, близких к температуре
самовоспламенения, и образования трудновоспламеняемого карбонизо-
ванного продукта.
Рис. 3.12. Кривые ДТА различных типов огнестойких полиамидных волокон
(скорость нагревания 3 прад/мин). Обозначения см. подпись к рис. 3.9.
Этот вывод подтверждается анализом продуктов разложения во-
локон. Анализ продуктов термического разложения модифицированного
фосфорсодержащего и исходного поликапроамидного волокон показы-
вает, что введение фосфорсодержащего антипирена приводит к умень-
шению общего количества выделяющихся газообразных продуктов
(рис. 3.13), особенно в области высоких температур (700°C и выше), за
счет образования большого количества карбонизованного остатка.
Качественный состав газообразных продуктов почти не изменяется
(табл. 3.5). Основными газообразными продуктами термического раз-
ложения являются углеводороды, количество которых составляет
50—70% от объема выделяющихся газов.
Среди горючих газообразных продуктов, кроме углеводородов,
содержатся водород и окись углерода; выделяется также двуокись угле-
рода и аммиак, а при температурах выше 700°C — в небольшом коли-
честве цианистый водород. Однако введение фосфорсодержащего анти-
пирена приводит к значительному изменению количественного состава
395
газообразных продуктов. Существенным является то, что при термичес-
ком разложении огнестойкого волокна при 700 °C практически не обра-
зуется цианистый водород, а при 800 °C его выделяется в 7 раз меньше
по сравнению с разложением поликапроамидного волокна. Кроме этого,
гораздо меньше выделяется таких горючих газообразных веществ, как
аммиак, пропан, но в продуктах разложения повышается содержание
метана, этана и водорода.
Полученные кинетические кривые свидетельствуют о том, что коли-
чество выделяющихся газообразных продуктов устанавливается посто-
янным уже через 1—1,5 мин
Рис. 3.13. Зависимость объема га-
зообразных продуктов термическо-
го разложения волокон V от тем-
пературы:
1 — полиамидное волокно; 2 — огне-
стойкое поликапроамидное волокно.
(рис. 3.14).
Для термоокислительного разложе-
ния огнестойких фосфорсодержащих
поликапроамидных волокон характерно
большое выделение жидких смолообраз-
ных веществ при 200—560 °C. Поэтому
объем газообразных, а следовательно, и
токсичных веществ относительно неболь-
шой.
В составе выделяющихся газообраз-
ных веществ найдены двуокись углерода,
окись углерода, водяные пары, метдн и
цианистый водород. Как показывают
данные исследования продуктов терми-
ческого окисления фосфорсодержащего
поликапроамидного волокна другого ти-
па, практически весь фосфор остается в
карбонизованном остатке [173]. Следы
фосфинов обнаруживаются в летучих
продуктах только при дополнительном
сжигании смолистых веществ. Из газооб-
разных продуктов, выделяющихся при го-
рении поликапроамидного волокна, наиболее токсичным является ци-
анистый водород. Содержание его в составе газообразных продуктов
Таблица 3.5. Данные хроматографического анализа газообразных продуктов
термического разложения волокон
Температура, °C Твердый остаток, % (масс.) Смолистые вещества, % (масс.) Содержание газов, % (об)
С2Н4 СзЩ н2 сн4 со СС>2 NH3 HCN
Исходное поликапроамидное волокно
560 0 55,8 — 39,6 1,58 20,56 4,26 18 16 0
660 0 45,4 — 57,7 2,10 8,75 15,71 5,2 10,5 0
760 0 24,0 — 63,3 3,81 8,61 11,98 3,4 7,6 1,8
860 0 8,8 — 57,3 4,27 11,1 12,68 6,10 5,8 2,8
Огнестойкое фосфорсодержащее поликапроамидное волокно
500 37 27 0 0 0 86 0 13 7,6 0
600 30 22 6,5- 13 20,8 36,2 14,1 5,0 4,5 0
700 23 20 4,4 14,7 23,9 36,8 20,2 6,6 4,7 Следы
800 16 16 1,8 17,7 21,2 35,4 12,4 6,0 5,0 0,4
396
Таблица 3.6. Образование двуокиси углерода, аммиака и цианистого водорода
в процессе горения фосфорсодержащего ПКА волокна
при различных объемах подаваемого воздуха
V0/m, л/г со2 NH3 HCN
% (масс.) % (Об.) % (масс.) % (Об.) % (масс.) % (Об.)
1,59 45,0 14,9 1,08 0,92 0,26 0,14
2,06 41,4 10,2 0,64 0,36 0,70 0,30
2,73 50,2 9,3 0,81 0,40 0,56 0,17
3,40 66,6 9,7 0,81 0,30 0,35 0,08
4,59 69,6 7,53 0,54 0,15 — —
увеличением объема воз-
уменыпается. С учетом
HCN
Рис. 3.14. Кинетика выделения газообраз-
ных продуктов термического разложения
огнестойкого поликапроамидного волок-
на при различных температурах:
7 — 500° С- 2 — 600° С; 3 — 700° С; 4 — 800° С;
5 — 900 °C.
сильно зависит от соотношения между объемом подаваемого на горение
воздуха Уо и массой сжигаемого вещества т (табл. 3.6).
Максимальное количество HCN (0,7% от массы исходного волок-
на) образуется при соотношении Vo'
духа, подаваемого на горение, вых<
элементарного состава фосфорсо-
держащего поликапроамидного во-
локна объем воздуха, необходимый
для сгорания единицы массы во-
локна, составляет 5,7 л/г. В этом
случае соотношению Vo/m=2 соот-
ветствует коэффициент избытка
воздуха, равный 0,354. Для исход-
ного поликапроамидного волокна в
аналогичных условиях выделяется
7% HCN. Анализ полученных дан-
ных показывает, что содержание
цианистого водорода в газообразных
продуктах горения огнестойкого
поликапроамидного волокна снижа-
ется в 10 раз по сравнению с поли-
капроамидным волокном, что свя-
зано не только с меньшим содер-
жанием азота в волокне, но и с
уменьшением доли превращейия
азота в цианистый водород.
Так, если в поликапроамидном
волокне с содержанием азота
12,38% превращается при горении в HCN около 28,3%, то при горении
фосфорсодержащего модифицированного волокна (содержание азота
2,59%) превращение азота в цианистый водород составляет 13,5%.
Таким образом, токсичность газообразных продуктов, выделяемых
огнестойким фосфорсодержащим волокном, ниже, чем токсичность про-
дуктов, выделяемых исходным поликапроамидным волокном.
В литературе приводится ряд составов для придания полиамид-
ным нитям и тканям огнезащитных свойств, однако до настоящего вре-
мени не разработан метод получения негорючего неплавкого волокна
на основе алифатических полиамидов с высокими физико-механиче-
скими характеристиками.
397
ГЛАВА 4
ПОЛИАКРИЛОНИТРИЛЬНЫЕ ВОЛОКНА
Среди карбоцепных волокон наиболее широкое промышленное при-
менение нашли синтетические волокна из полимеров и сополимеров
акрилонитрила. К полиакрилнитрильным (ПАН) волокнам принято
относить волокна на основе сополимеров, в цепи которых содержится
не менее 85% звеньев полиакрилонитрила. Мягкий шерстеподобный
гриф, высокая объемная эластичность и низкая теплопроводность оп-
ределили целесообразность использования полиакрилонитрильных во-
локон для изготовления изделий народного потребления определенного
ассортимента (верхней одежды, драпировочных материалов, ковров
и др.).
ПАН волокна подобно целлюлозе относятся к легковоспламеня-
емым материалам. Температура воспламенения от постороннего источ-
ника сравнительно низкая — 250 °C. Показатель кислородного индекса
для этих волокон составляет 18,0%. На воздухе ПАН волокна горят с
высокой скоростью, причем при горении выделяются в значительном
количестве различные токсичные газы, в частности цианистый водород.
По термической и термоокислительной деструкции ПАН накоплен
большой экспериментальный материал. Термическая и термоокисли-
тельная деструкции ПАН в свете структурных превращений полимера
на стадии окисления подробно рассмотрены в литературе [30, с. 141].
В то же время наиболее сложные аспекты этих процессов (механизм
разложения и состав газообразных продуктов) остаются недостаточно
выясненными.
Характерной особенностью разложения полиакрилонитрила явля-
ется высокая способность его к образованию циклических структур.
Термогравиметрический анализ ПАН [174] показывает, что для этого
полимера нет области с резким падением массы до нуля, как это наблю-
дается при разложении полиамидов. При разложении ПАН образуется
большое количество карбонизованного вещества. Если в интервале
температур 400—500 °C полиамид разлагается полностью, ПАН сохра-
няет до 40% от исходной массы. Наибольшая скорость потери массы
полимера наблюдается при 280 °C.
К основным продуктам распада ПАН относятся нитрилы, циани-
стый водород, аммиак и вода, которые начинают выделяться еще на
низкотемпературной стадии разложения. Масс-спектрометрическим
анализом при распаде ПАН обнаружены [1, с. 328] акрилонитрил, ви-
нилацетонитрил, ацетонитрил, бутилонитрил и пропилонитрил.
По литературным данным [I], в среднем при пиролизе ПАН в глу-
боком вакууме акрилонитрила выделяется 5,2, а цианистого водорода
2,9% (от массы исходного полимера). На воздухе HCN выделяется
более интенсивно, чем в инертной среде. Роль кислорода, по-видимому,
сводится к ускорению циклизации, которая способствует выделению
HCN [175]. С увеличением температуры и продолжительности обработ-
ки количество образующегося цианистого водорода при термической и
термоокислительной обработке ПАН возрастает. Относительно состава
газообразных продуктов на разных стадиях разложения ПАН имеются
противоречивые данные, объясняемые и различными условиями прове-
дения термической деструкции и изменением скорости и направлений
реакций под влиянием большого числа факторов.
398
Высокая горючесть ПАН волокон, по-видимому, обусловлена тем,
что уже при низких температурах начинается пиролиз с выделением
легколетучих соединений, образующих с воздухом горючую газовую
смесь. Нижний предел воспламенения смеси с воздухом таких продук-
тов разложения ПАН, как акрилонитрила и ацетонитрила, составляет
около 3% (об.). Следовательно, даже при небольшой концентрации
этих веществ образуются горючие легковоспламеняемые газы. Темпе-
ратура самовоспламенения ПАН (515 °C) близка к температуре само-
воспламенения акрилонитрила (480 °C).
Анализируя данные, имеющиеся по термической и термоокислитель-
ной деструкции ПАН, можно сделать следующий вывод. Для снижения
горючести ПАН волокон необходимо предотвратить деполимеризацию,
приводящую к образованию нитрилов, и создать условия для реакции
циклизации.
4.1. МЕТОДЫ ПРИДАНИЯ ОГНЕСТОЙКОСТИ
ПОЛИАКРИЛОНИТРИЛЬНЫМ ВОЛОКНАМ
Из всех синтетических волокон ПАН волокнам могут быть легче
всего приданы огнезащитные свойства. Для этого применяют следующие
методы: химическую модификацию, формование из смесей полимеров,
введение добавок в прядильный раствор при формовании и поверх-
ностную отделку волокон или тканей.
Обстоятельный обзор патентов по повышению огнестойкости ПАН
волокон приведен в работах [10; 176, с. 23—35].
4.1.1. Химическая модификация
Метод химической модификации ПАН волокон с целью придания
мм огнестойкости включает сополимеризацию и полимераналогичные
превращения групп CN полимера.
Сополимеризация. Для повышения огнестойкости ПАН волокон
широко применяется метод радикальной сополимеризации акрилонит-
рила с различными мономерами винилового ряда, содержащими хлор,
бром или фосфор. Судя по литературным данным, для модификации
ПАН с целью повышения огнезащитных свойств предложено большое
число сомономеров к акрилонитрилу. Некоторые из используемых вини-
ловых мономеров приведены в табл. 4.1.
Обычно для улучшения эластичности и накрашиваемости ПАН
волокон при полимеризации вводят в небольшом количестве мономеры
с большими боковыми группами или с функциональными группами,
обладающими сродством к красителю. Вследствие этого при получении
волокнообразующих сополимеров акрилонитрила с повышенной огне-
стойкостью необходимо применять смеси мономеров из трех-четырех
компонентов, что в определенной степени усложняет процесс синтеза.
Содержание акрилонитрила в сополимерах может изменяться от 80 до
40%. Анализ приведенных литературных данных показывает, что при
модификации ПАН путем сополимеризации преимущественно исполь-
зовали в качестве мономера-антипирена соединения сложного строения.
Эти работы представляют в основном теоретический интерес и позво-
ляют выяснить пути повышения огнестойкости ПАН волокон. Однако
возможность практической реализации синтеза многих сополимеров
399
Таблица 4.1. Мономеры, используемые для сополимеризации с акрилонитрилом
с целью повышения огнестойкости ПАН
Мономер Формула Литература
Винилхлорид СН2=СНС1 [177, с. 345; 178}
Вииилиденхлорид СН2«=СС12 [179; 180, с. 222}
а-Хлоракрилоии- трил СН2=СС1 An [181]
Винилхлорид и ви- ни лиденхлорид СН2=СНС1, СН2=СС12 [182]
Вииилбромид СН2=СНВг [183; 184]
Вииилбромид и ви- нилхлорид (или ви- нилиденхлорид) СН2=СНВг, СН2=СНС1(СН2=СС12) [185—187J
Алкилэфиры аллил- фосфиновой кислоты CH2=CHCH2P(OR)2 и о [188]
Бис(2-хлорэтил)ви- иилфосфат (С1СН2СН2О)2РСН=СН2 11 о [189]
Эфиры а- и р-диал- килфосфоноакрило- вой кислоты О О II II СН2=С—С—OR R'O—Р—СН=СН—С—OR 1 1 II O=P(OR')2 or о [190]
Диалкил-2-галогеи- пропен-3-фосфоиаты СН2=С-СН2—P(OR)2 1 (1 X О (где X = Вг или С1) [191]
Дибутиловый эфир а-стирофосфиионой кислоты СН2=С—Р(ОС4Н7)2 АА [192]
Производные ви- иилфосфнновой кисло- ты R-P(OH)2 II О (где R = СН2=СН) [193]
Эфиры метилфосфи- новой кислоты /ОС2Н5 сн3-р( п \осн=сн2 О [194]
Тригалогенфенокси- алкилакрилаты (мет- акрилаты) R' О Х\ СН2=С-С—ORO—х х/ (где R =СН2—СН2 или СН,—СН; Ан3 R' =Н или СН3; X = Вг или С1) [195; 196}
400
маловероятна вследствие сложности синтеза мономеров, а также раз-
личия в константах сополимеризации акрилонитрила и предлагаемых
мономеров.
Наиболее просто в технологическом отношении получение сополи-
мера и волокон с огнезащитными свойствами на основе сополимеров
акрилонитрила с винилхлоридом или винилиденхлоридом. Волокно из
сополимера акрилонитрила (40%) и винилхлорида (60%), впервые
полученное фирмой «Карбид Карбон» (США), известно под названием
виньон N (комплексная нить) и дайнел (штапельное волокно) и выпус-
кается в промышленном масштабе. Волокно содержит 34% хлора и
считается огнестойким. Однако волокна на основе этих сополимеров
имеют низкую теплостойкость и настолько большую усадку, что их при-
менение в качестве волокна технического назначения нецелесообразно.
Волокно дайнел начинает размягчаться при температуре ниже 150 °C, а
при 100°C усаживается на 20% [197], в то время как усадка ПАН
волокна составляет 2%. Наблюдаемое ухудшение свойств волокна
обусловлено введением в макромолекулу полимера большого количества
винилхлорида, а небольшие добавки его малоэффективны. Волокна из
сополимеров акрилонитрила с винилиденхлоридом имеют лучшую
термо- и теплостойкость [179; 180]. Использование для сополимериза-
ции бромсодержащих соединений (в частности, винилбромида), явля-
ющихся более эффективными замедлителями горения, а также введе-
ние в галогенсодержащие сополимеры акрилонитрила синергически
действующих веществ (например, Sb2O3) позволяет получать огнестой-
кие ПАН волокна с меньшим содержанием второго компонента, что
положительно сказывается на комплексе физико-механических свойств
волокна. Поэтому важны выбор сомономера, повышающего огнестой-
кость, и его содержание в сополимере. Кроме того, на свойства волокон
оказывает влияние равномерность сополимера по составу.
Отмечая перспективность этого направления получения огнестой-
ких ПАН волокон, следует указать на его недостатки: снижение тепло-
стойкости, механических характеристик волокон и необходимость
использования в ряде случаев труднодоступных сомономеров.
Для придания огнестойкости ПАН волокнам можно использовать
химические реакции с функциональными группами полиакрилонитрила,
хотя исследования в этом направлении весьма ограничены. Однако
жесткие условия проведения этой реакции в гетерогенной среде без-
условно могут отрицательно сказаться на свойствах волокна.
Описано [198] получение огнестойкого волокна путем взаимодейст-
вия диметилфосфита с модифицированным полиакрилонитрилом, содер-
жащим альдегидные группы. Альдегидные трупы легко вступают в ре-
акцию с диметилфосфитом, но образовавшиеся эфиры оксифосфиновой
кислоты
—СН2—СН—
Н—С—ОН
О=Р(ОСН3)2
легко гидролизуются. Для устранения этого реакцию проводят в при-
сутствии диэтиламина:
401
II HN(C2H5)2
/—CH2—CH—CH2—CH—\ + nHP(OCH3)2 -----►
—СН2— СН— \— /—СН2—СН— X
I ] I \
CN }т Н-С—N(C2H5)2
\ I /
\ О=Р(ОСН3)2 /п
Обработка волокна проводилась 50%-ным раствором диметилфос-
фита в кислой среде при 120°C в течение 6ч в присутствии 1% диэтил-
амина. В зависимости от содержания альдегидных групп и условий
реакции фосфорилирования в волокно можно ввести различное коли-
чество фосфора. При содержании альдегидных групп 11% в волокно
вводится до 9% фосфора. ПАН волокна уже при содержании 3,5%
фосфора обладают высокой огнестойкостью и мало отличаются по меха-
ническим свойствам от стандартного волокна (табл. 4.2).
Таблица 4.2. Изменение физико-механических свойств ПАН
волокна при модификации
Показатели Исходное волокно Модифицированные волокна
содержание альдегидных групп 13% содержание фосфора 3,25%
Линейная плотность, текс 14,3 14,9 16,6
Прочность, мН/текс 355 320 300
Удлинение, % 11,2 12,4 19,6
Однако необходимость сравнительно сложных обработок при вве-
дении альдегидных групп в молекулу модифицированного ПАН и по-
следующее взаимодействие с диметилфосфитом значительно затрудняют
возможность практического использования этого метода для придания
огнестойкости ПАН волокном.
4.1.2. Формование волокон из смесей полимеров
Метод модификации химических волокон путем формования из
смесей полимеров в последние годы привлек внимание большого круга
исследователей и нашел применение в технологической практике.
Используя смеси различных полимеров, можно широко изменять свойст-
ва волокон. Переработка смесей полимеров, один из которых является
огнестойким, дает возможность получать модифицированные волокна с
огнезащитными свойствами.
Для повышения огнестойкости ПАН волокон предложены смеси
ПАН или его сополимеров, состоящие в основном из акрилонитрила, с
галогенсодержащими полимерами или сополимерами. В качестве
галогенсодержащих полимеров используют поливинилхлорид, поливи-
нилиденхлорид и сополимеры, содержащие более 50% винилхлорида
[199; 200]. Описано [201] получение модифицированного ПАН волокна
с повышенной огнестойкостью из смеси тройного сополимера (акри-
лонитрила с винилбромидом и акриламидом) и сополимера винилхло-
402
рида. Формование волокон осуществляется из растворов в диметилфор-
мамиде по мокрому способу. Оптимальные соотношения компонентов
смеси выбирают, исходя из требуемого эффекта огнестойкости и сохра-
нения физико-механических свойств ПАН волокна. Однако, как и в
случае получения волокон из сополимеров с галогенсодержащими моно-
мерами, для достижения высокой огнестойкости необходимо относитель-
но большое содержание второго компонента.
В заключение следует отметить, что повышение огнестойкости
ПАН волокон формованием из смесей с хлорсодержащими полимерами
имеет существенное преимущество перед получением волокон из анало-
гичных огнестойких сополимеров, которое заключается в том, что отпа-
дает необходимость синтезировать новые сополимеры акрилонитрила.
Изменение свойств в нужном направлении достигается в результате
использования полимеров, производство которых освоено промышлен-
ностью. Но, по-видимому, здесь имеются свои трудности, поэтому огне-
стойкие ПАН волокна из смесей полимеров не производятся в промыш-
ленном масштабе.
4.1.3. Введение добавок в прядильный раствор
при формовании волокна
Метод придания огнестойкости ПАН волокнам путем введения в-
прядильный раствор полимера добавок различных антипиренов заслу-
живает особого внимания, так как заманчиво, не изменяя существенно
технологического режима формования, получать модифицированные
ПАН волокна с огнезащитными свойствами. Судя по литературным дан-
ным, в этом направлении проводится много исследований.
Одним из условий успешного применения этого способа является
достаточно низкая себестоимость вводимых в прядильный раствор
веществ и нерастворимость их в осадительной ванне. Выполнение этих
условий обеспечивает сохранение антипирена в полимере в процессе
формования, а следовательно, высокую эффективность огнезащиты.
Добавки, используемые для придания огнестойкости полиакрило-
нитрильным волокнам, растворяют в прядильном растворе, дисперги-
руют или суспендируют. Затем осуществляют формование обычным
способом из растворов.в диметилфцрмамиде, роданидах или хлориде
цинка.
Большинство добавок представляют собой эфиры фосфорсодержа-
щих соединений, часто имеющие в алкильной боковой цепи галогены.
Перри [202] предложил для повышения огнестойкости ПАН волокна
вводить трис(дибромпропил) фосфат, который является очень активным
антипиреном, содержащим эффективно действующие на замедление
процесса горения атомы брома и фосфора. Кроме того, трис(дибромпро-
пил) фосфат хорошо совмещается с полиакрилонитрилом и поэтому при
формовании волокна не возникает осложнений. Для усиления эффекта
огнестойкости рекомендуют [203] добавлять в прядильный раствор
полиакрилонитрила в диметилформамиде смесь трис (дибромпро-
пил) фосфата с дифенилкрезилфосфатом.
Интересны композиции на основе бронированных теломеров, полу-
чаемых из ненасыщенных фосфатов [204]:
О
II Вг2
(СН2=СНСН2О)3Р=О + СС14 -► СН2—СНСН2ОР(ОСН2СН=СН2)2 --->
<ici3 ci
403
о
II
---> С13ССН2СНСН2ОР^ОСН2СН—СНД
i к i Вт Л
На бромсодержащие фосфаты синергическое действие оказывают
кальциевые соли фосфорных кислот. Согласно патенту [205], бромсо-
держащие фосфаты в сочетании с нерастворимыми кальциевыми солями
фосфорных кислот диспергируют в водные растворы ZnCl2 с 10%-ной
концентрацией полиакрилонитрила в присутствии 5—17% поверхностно-
активных веществ.
Достаточно активными замедлителями горения полиакрилонитрила
являются и негалогенированные фосфаты, такие, как триалкилфосфаты,
которые добавляются в прядильный раствор в количестве 5—30% от
массы полимера и сохраняются в волокне в процессе формования по
мокрому способу и при последующих обработках f206]. В то же время
трикрезилфосфат при водных обработках почти полностью экстраги-
руется.
ПАН волокно с повышенной огнестойкостью получают также путем
добавления в прядильный раствор 10—20% от массы полимера смеси
бромированного циклогексана (например, пентаброммонохлор-, три-
бромтрихлор-, тетрабромдихлор- или гексабромциклогексана) и карбо-
ната орто-, пиро- или метафосфорной кислоты {207].
Описано [208] применение гексабромбифенила или октабромбифе-
нила совместно с ЗЬгОз- Их добавляют в раствор соответственно в
количествах 15,7—26 и 3—5%.
Среди других фосфорсодержащих соединений в качестве добавок в
прядильный раствор полиакрилонитрила предложены третичный орга-
нический фосфин [209], диэтил (З-хлор-2-метиленпропил) фосфонат [210]
и 2-цианэтилтетраметилдиамидофосфат, который получается путем
конденсации 2-цианэтанола с тетраметилдиаминохлорфосфатом в при-
сутствии третичного амина:
(OW^O
/Р—Cl + HOCH2CH2CN ---► . ,Р—OCH2CHaCN
(CH3)2n/ (CH3)2rf/
В ряде патентов для повышения огнестойкости полиакрилонитриль-
ных волокон рекомендуют добавлять в прядильный раствор эфиры
галогенированных жирных кислот, содержащие бром [211; 212] или
амиды соответствующих кислот [213].
Фирма «Дюпон» предлагает [214] сурьмусодержащие композиции,
состоящие из соединений типа:
О=С—(X ,Х
I /Sb—Y
R—О7 \z
XXIII
X,Y,Z — алкоксисоединеиия с одним—четырьмя атомами С1 или Вт
К прядильному раствору полиакрилонитрила или сополимеров
акрилонитрила добавляют от 0,5 до 20% данного соединения (к массе
полимера) в зависимости от требуемого эффекта огнестойкости и со-
404
хранения физико-механических свойств волокна. Волокна формуют из
диметилформамидных растворов по сухому способу.
Хлорированные парафины, используемые для повышения огнестой-
кости и других синтетических полимеров, равномерно распределяются в
прядильном растворе ПАН и не вымываются из волокна при формова-
нии. Согласно литературным данным [215], парафины с содержанием
хлора более 50% растворяют в ССЦ и в виде 60—72%-ного раствора
вводят в прядильный раствор ПАН. Для получения волокна с огнеза-
щитными свойствами содержание хлорированного парафина должно
быть не ниже 20—25%.
Привлекает внимание исследователей способ получения полиакри-
лонитрильных волокон с повышенной огнестойкостью путем добавления
к раствору полиакрилонитрила полиэпигалогенгидринов с молекуляр-
ной массой 1200 [216; 217]. Полиэпигалогенгидрины обладают хоро-
шей совместимостью с акриловыми полимерами. Огнезащитный эффект
усиливается при введении фосфатов кальция или окиси сурьмы, оказы-
вающих синергическое действие в присутствии галогенсодержащих сое-
динений. Полученные волокна сохраняют огнезащитные свойства после
нескольких стирок.
4.1.4. Поверхностная отделка волокон и тканей
Огнезащитная отделка ПАН волокон и тканей, так же как и дру-
гих синтетических текстильных материалов, в настоящее время нахо-
дится на стадии разработки. В литературе по этим вопросам имеются
только патентные данные.
При большинстве способов огнезащитной отделки ухудшается
гриф тканей и их практически нельзя использовать для тканей бытово-
го назначения. При выборе соединений для поверхностной отделки
тканей, предназначенных для одежды, необходимо учитывать также их
токсичность и раздражающее действие на кожу. Все это значительно
ограничивает возможности при разработке метода огнезащитной отдел-
ки тканей из ПАН волокон, применяемых в основном для товаров
народного потребления.
Метод поверхностной отделки включает обработку волокна или
ткани органическими галоген-, фосфорсодержащими соединениями или
солями металлов.
Огнезащитную обработку ПАН волокон иногда рекомендуют сов-
мещать с технологическим процессом формования, вводя в осадитель-
ную ванну антипирены. В патенте [218] предлагается получение огне-
стойкого ПАН волокна путем добавления в осадительную ванну гекса-
бромбензола, гексахлорбензола или трихлоруксусной кислоты. Из дру-
гих галогенсодержащих соединений для огнезащитной обработки ПАН
волокна описано применение ароматических и алифатических углеводо-
родов с цепочкой, состоящей из 10—17 углеродных атомов [219, 220],
в частности хлорированных парафинов, содержащих 52% хлора. Обра-
ботанное волокно сушат при 100 °C на воздухе, а затем кипятят в тече-
ние 2 ч в буферном растворе с pH 4,9. Считают, что уже при содержа-
нии 3% хлора ПАН волокно обладает пониженной горючестью. Иногда
для усиления эффекта к галогенсодержащим соединениям добавляют
до 5% трехокиси сурьмы.
Фирма «Монсанто Кемикэл Компани» (США) предлагает для
огнезащитной обработки полиакрилонитрильных волокон водные 10—
405
75%-ные растворы фитина СбНб[ОРО(ОН)г]б с формальдегидом. Тка-
ни, пропитанные такими составами, сушат и подвергают термообработ-
ке при 125—165 °C в течение 5—20 мин. Несмотря на простоту об-
работки, этот метод нельзя считать перспективным из-за сложности
синтеза фитиновой кислоты, которая получается из пищевого сырья.
По данным американского патента [221], хорошие результаты по
огнестойкости достигаются при пропускании свежесформованного ПАН
волокна через ванну, содержащую 10%-ный раствор азиридинилфос-
финооксида или сульфида общей формулы (XXIV) с последующим
прогревом волокна при 100—140 °C в течение 1—6 мин
* /СН2
R—Р—N' |
и, \сн2
XXIV
Z = 0 или S; R,R'—азиридииил, аллокси, арокси или арил
В этих условиях на волокно наносится до 6% антипирена, количест-
во которого не снижается при крашении и стирках. Так же, как и для
полиамидов, для полиакрилонитрильных волокон рекомендуются также
композиции из мочевины, формальдегида и бромида аммония {222].
Однако во многих случаях для поверхностной отделки ПАН воло-
кон и тканей предлагают фосфор- и фосфоргалогенсодержащие органи-
ческие соединения довольно сложного строения, которые вряд ли
найдут в дальнейшем широкое применение.
Заслуживает внимания обработка полиакрилонитрильных материа-
лов солями металлов, являющихся катализаторами окислительного
сшивания макромолекул ПАН. Структурирование полимера замед-
ляет процесс деполимеризации ПАН с образованием горючих газооб-
разных продуктов и тем самым снижает горючесть материала. Наибо-
лее эффективны соли кобальта и германия [223]:
СоС12-6Н2О; Co(NO3)2H2O; CoSO4H2O; СоВг2-6Н2О;
[2СоСО3-ЗСо(ОН)2]; Со(СН3СОО)2; GeCl4-7H2O
Могут быть использованы также соли меди (например, CuSO4}
[224]. Волокна или ткань обрабатывают растворами солей из такого
расчета, чтобы на волокно было нанесено 0,1—3% металла, сушат и
подвергают кратковременной (1 мин) термообработке при 200—300 °C.
ПАН волокна, полученные из сополимеров, содержащих 4,5—4,7%
(мол.) гидрооксиметакрилата или оксиэтилметакрилата, с целью по-
вышения огнестойкости предлагают сшивать фосфорной кислотой [225].
Сшивку осуществляют при 50—150°C в среде 1 — 10%-ного водного
раствора Н3РО4.
Из различных методов, предложенных для огнезащитной отделки
полиакрилонитрильных текстильных материалов, по-видимому, наиболь-
ший интерес может представлять обработка антипиренами свежесфор-
мованного волокна в процессе формования.
Анализ литературных данных в области получения огнестойких
ПАН волокон показывает, что, несмотря на проводимые исследования,
эта проблема остается еще слабо разработанной.
ГЛАВА 5
ПОЛИЭФИРНЫЕ ВОЛОКНА
Полиэфирные волокна, получаемые преимущественно из полиэти-
лентерефталата, благодаря ценным потребительским свойствам и ши-
рокой сырьевой базе, нашли большое применение в технике и для
изготовления товаров народного потребления.
Полиэфирные волокна относятся к классу горючих материалов.
Это плавкие волокна, поэтому ткани из них в условиях пожара могут
представлять большую опасность для человека. Кислородный индекс
полиэфирного волокна 22%.
5.1. ТЕРМИЧЕСКОЕ И ТЕРМООКИСЛИТЕЛЬНОЕ
РАЗЛОЖЕНИЕ ПОЛИЭТИЛЕНТЕРЕФТАЛАТА
Нагревание полиэтилентерефталата (ПЭТФ) выше температуры
плавления приводит к изменению окраски полимера и увеличению
концентрации карбоксильных групп. При этом протекают два процес-
са — деструкция и структурирование, которые в равной степени при-
водят к ухудшению свойств материала.
При исследовании термической деструкции ПЭТФ при 307 °C
было отмечено [226], что скорость деструкции ПЭТФ выше, чем ско-
рость деструкции полиэтилена, и на основании этого был сделан вывод,
что при введении в углеводородную цепь сложноэфирных групп тер-
мическая стабильность полимера снижается. Полагают, что иницииро-
вание термической деструкции ПЭТФ происходит в результате разрыва
связи С—О, а не связи С—С, поскольку прочность первой меньше.
Близкие значения энергии активации деструкции ПЭТФ и модель-
ного соединения — гликольдибензоата (309,7 и 184,2 кДж/моль)
подтверждают предположения о разрыве в ПЭТФ эфирных связей по
закону случая [227]. Распад сложноэфирной связи в ПЭТФ, как указы-
вается в ряде работ {227—229], протекает через образование проме-
жуточного шестичленного комплекса, в котором происходит значитель-
ное перераспределение связей:
О - Н
II I
—С,Н4СООСН2СН2ООССвН4---> —с,н4с сноосс,н4------>
о—сн2
---> —С,Н4СООН + СН2=СНООСС,Н4—
Термическое разложение полиэтилентерефталата является сложным
процессом, при котором последовательно протекает ряд реакций, что
приводит к образованию довольно большого числа газообразных и
нелетучих продуктов. Изучение термического разложения ПЭТФ и низ-
комолекулярных алкильных эфиров ароматических кислот в области
температур 282—383 °C <[230] и 370—475 °C {231] показало, что про-
дукты пиролиза ПЭТФ аналогичны продуктам пиролиза модельных
соединений.
Гудинге {230] и Ритчи [232] при пиролизе гликольдибензоата
идентифицировали следующие продукты термического разложения: аце-
тальдегид, окись углерода, бензойную кислоту и винилбензоат
,(СбН5СООСН = СН2). При термическом разложении ПЭТФ в составе
407
газообразных продуктов обнаружены ацетальдегид, окись и двуокись
углерода и в небольшом количестве метан, а в конденсированной фа-
зе— терефталевая кислота, олигомеры ПЭТФ и ненасыщенные эфиры.
Из указанных выше соединений основными продуктами пиролиза
ПЭТФ являются ацетальдегид и терефталевая кислота. Предполагают
[227], что ацетальдегид образуется в результате взаимодействия карб-
оксильных и винилэфирных групп:
-СвН4С\
—СвН4СООН + СН2=СНООССвН4--► /О + СН3СНО
-свнХ
Продукты термической деструкции при определенных условиях
могут снова образовывать полиэфиры, вступая в реакцию с концевы-
ми гидроксильными группами исходного полимера [233]. Продукты
винилового типа способны к реакции полимеризации с образованием
производных поливинилбензоата, которые нестабильны, легко распада-
ются и могут вызывать окрашивание полимера [227].
На основании анализа продуктов деструкции гликольдибензоата
и ПЭТФ Гудинге [230] предложил следующий механизм термической
деструкции полиэтилентерефталата:
—СвН4СООСН2СН2ООССвН4-----> —С6Н4СООСН=СН2 + НООССвН4----►
---> —СвН4СООСНООСС6Н4-----> — СвН4СОООСвН4— + СН3СНО
сн3
—С6Н4СООСН=СН2 + ОНСН2СН2ООССвН4----->
---> —СвН4СООСН2СН2ООССвН4— + СН3СНО
' пСН3СНО --> СН3(СН=СН)„_1СНО + (п — 1) Н2О
—С6Н4СОООСвН4— +Н2о 2 —СвН4СООН
—СвН4СООСН2СН2ООССвН4—+ Н2О ---> —С6Н4СООН+НОСН2СН2ООССвН4—
Однако имеющихся данных еще недостаточно, чтобы сделать
окончательный вывод о механизме пиролиза ПЭТФ.
Данные о составе продуктов термического разложения ПЭТФ
дают возможность предположить, что горючесть полиэфирных волокон
обусловлена воспламенением таких продуктов пиролиза, как ацеталь-
дегид, терефталевая кислота и другие, которые выделяются в значи-
тельных количествах.
5.2. МЕТОДЫ ПРИДАНИЯ ОГНЕСТОЙКОСТИ
ПОЛИЭФИРНЫМ ВОЛОКНАМ
В настоящее время в литературе имеется очень мало данных о по-
лучении полиэфирных волокон, обладающих огнезащитными свойства-
ми, причем в основном это патентные данные.
Придание огнезащитных свойств полиэфирным волокнам может
быть осуществлено путем введения антипирена в расплав полимера
перед формованием или путем обработки готовых волокон и тканей.
Кроме того, возможно получение огнестойких волокон из сополиэфиров,
содержащих бромированные кислоты или дигликоли. По сообщениям
фирмы «Амоко», огнестойкие полиэфиры получены путем введения при
408
поликонденсации 2,5-дибромтерефталевой кислоты [234]. Огнестойкое
полиэфирное волокно дакрон 900Ф получено из сополиэфира, содержа-
щего продукт взаимодействия этиленоксида и тетрабромбисфенола
[235]. Температура плавления этого волокна составляет 235 °C; содер-
жание брома в нем — 6%. По физико-механическим свойствам это
волокно близко к волокну из полиэтилентерефталата, но уступает ему
по устойчивости к истиранию.
Это направление работ может представлять существенный инте-
рес, если будут удовлетворительно решены в технологическом отноше-
нии процессы этерификации и поликонденсации при синтезе сополи-
эфиров.
Анализ имеющихся литературных данных показывает, что в каче-
стве замедлителей горения для полиэфиров используются в основном
те же классы галоген- и фосфорсодержащих соединений, которые при-
меняются для огнезащиты полиамидов. Наибольшее распространение по-
лучили галогенсодержащие соединения, в частности содержащие бром.
Метод введения антипирена в расплав полиэфира, по сравнению
с поверхностной отделкой ткани, дает возможность получить волокно
с огнезащитным эффектом, устойчивым к различным обработкам.
Однако вследствие ограниченного числа термостабильных соединений,
используемых для этой цели, метод добавления огнезащитных средств
в процессе формования волокон из расплава должного применения
не получил. Что касается полиэфиров, то известно относительно не-
большое число патентов по приданию огнестойкости волокнам путем
введения различных ароматических и в меньшей степени алифатиче-
ских галогенсодержащих органических соединений в расплав полимера
перед формованием. Рекомендованы следующие соединения:
гекса- или пентабромбензол [236];
гексабромдифенил амин [237];
дека-, окта- или гексабромдифенил [238];
тетрахлор (или бром)бисфенол [239];
бромсодержащее ароматическое соединение
(С6Вг5—О—С6Н4—ОС6Вг5) ![240];
хлор- или бромзамещенные эфиры фталевой кислоты [241; 242];
бромфенилтлицидные эфиры [243];
дифенилхлорид и тетрафторфталевый ангидрид [244].
Обычно в расплав полиэфира добавляют 10—30% (масс.) антипи-
рена. В зависимости от содержания антипирена изменяются огнеза-
щитные и физико-механические свойства волокна. Чем больше анти-
пирена, тем выше огнестойкость волокна; однако при этом снижаются
механические показатели волокна.
В японском патенте [245] описано получение полиэфирного во-
локна с огнезащитными свойствами путем введения в расплав в про-
цессе формования арилполифосфата:
XXV
27—2254
409
Кислородный индекс волокна равен 28. Имеется сообщение, что
выпуск этого волокна под торговым названием «хайм» освоен в опыт-
но-промышленном масштабе. Волокно предназначается для изготовле-
ния декоративно-обивочного материала, занавесей [246].
Для поверхностной огнезащитной обработки полиэфирных тканей
используются соединения различных классов. В состав большинства
огнестойких аппретов входят галоген- и фосфорсодержащие соедине-
ния. Часто с целью закрепления на поверхности ткани галоген- и фос-
форсодержащих антипиренов рекомендуется в состав композиций вво-
дить полифункциональные азотсодержащие соединения или формаль-
дегид с мочевиной, тиомочевиной или производными мочевины, спо-
собными при определенных условиях (в частности, при термообра-
ботке) образовывать искусственную смолу.
Детальный перечень различных композиций для огнезащитной
отделки полиэфирной ткани, предлагаемых в патентной литературе,
приведен в обзоре [247]. Во многих случаях огнезащитная отделка
полиэфирной ткани аналогична отделке полиамидных текстильных ма-
териалов.
На обивочную мебельную ткань и ковровые изделия с целью огне-
защиты наносят тонкий слой хлорсодержащих полимеров, в частности
поливинилхлорида, полихлоропрена или поливинилиденхлорида. Фирма
«Мичиган кемикэл корпорейшен» (США) предлагает {248] для отделки
текстильных материалов из полиэфирного волокна препарат на основе
трис (2,3-дибромпропил) фосфата. Для обработки ткани обычно исполь-
зуется 20%-ная водная эмульсия трис (2,3-дибромпропил) фосфата.
Полиэфирную ткань, освобожденную от замасливателя, пропитывают
огнестойким аппретом, отжимают на плюсовке, оставляя на волокне до
60% эмульсии. Затем проводят термообработку при 125°C в течение
90 мин. После термообработки ткань промывают 0,1%-ным раствором
бикарбоната натрия при 75—85 °C и высушивают. При нанесении на
ткань 12% трис (2,3-дибромпропил)фосфата она приобретает огнеза-
щитные свойства. Огнезащитная отделка относительно устойчива к об-
работкам и выдерживает 50 стирок (при 60°C) и 50 химчисток пер-
хлорэтиленом. Огнезащитная отделка полиэфирной ткани этим препа-
ратом в настоящее время нашла наибольшее применение и рекомендо-
вана для тканей, предназначенных для изготовления одежды [249].
Довольно эффективным огнезащитным средством для полиэфирной
ткани является трибромфеноксиэтанол:
Вг—<р-°(сН2)2°Н
\вг
XXVI
Его наносят на ткань из растворов или эмульсий в количестве
10% [250].
В качестве огнестойких аппретов предлагают использовать также
следующие соединения:
тетраоксиметилфосфоний хлорид с мочевиной [251] (обработка
тканей осуществляется раствором или эмульсией с последующей тер-
мофиксацией) ;
410
продукт взаимодействия полигалогенных ароматических кислот
(хлорэндиковая) с тиомочевиной [252];
триметилолфосфинат, 2,4-толуилендиизоцианат, фенилизоцианат,
гексаметиленизоцианат, цианамид с формалином [253];
аммонийную соль бис (оксиметил) фосфиновой кислоты [254].
В патентной литературе для поверхностной отделки полиэфирных
тканей рекомендуется также много других композиций, которые не
нашли практического применения.
Предлагается [255] поверхностное хлорирование тканей из поли-
эфира газообразным хлором. Вместо хлора могут быть использованы
и другие соединения, способные в определенных условиях выделять
свободный хлор — хлористый сульфурил, пятихлористый фосфор, ги-
похлориты. Реакция хлорирования проводится в присутствии инициа-
тора (перекись бензоила, перекись водорода, персульфат аммония) или
под действием УФ-лучей. В зависимости от хлорирующего реагента и
типа инициатора температура реакции изменяется от 20 до 150 °C.
При хлорировании ткани с поверхности уже при содержании хлора 3—
5% значительно снижается горючесть полиэфирной ткани.
Следует отметить, что пока удовлетворительных результатов по
приданию огнестойкости полиэфирным волокнам и тканям в промыш-
ленном масштабе получить не удалось.
ЛИТЕРА ТУРА
1. Мадорский С. Термическое разложение органических полимеров. М., «Мир»,
1967. 328 с.
2. Einsele U., Lenzinger Вег., 1972, № 34, S. 108—118.
3. Loynes J. IF. The Chemistry and uses of Fire Retardants. New York — London, Wil-
ley Interscience, 1970. 324 p.
4. Place J., Chem. Proc. (Cr. Brit), 1972, v. 18, № 7, p. 9—10; пер.: Пожарная охрана,
Экспресс-информация. M„ ВИНИТИ, 1972, Ns 44, реф. 267, с. 15—16.
5. Lyssi Т., Lenzinger Вег., 1974, № 36, S. 197—212.
6. Hoke С. Е., SPE Journal, 1973, v. 29, Ns 5, р. 36—40.
7. Физер Л., Физер М. Органическая химия. Т. I. М., «Химия», 1970. 688 с.
8. Crawshaw G. Н., Duffield Р. A., Mehta Р. N., Appl. Polymer Сутр., 1971, № 18,
pt. 2, р. 1183—1197; пер.: Пожарная охрана. Экспресс-информация, М., ВИНИТИ,
1972, № 37, реф. 231, с. 14—27.
9. Таубкин С. И., Баратов А. Н., Никитина Н. С. Справочник пожароопасности твер-
дых веществ и материалов. М., изд-во Мин. коммун, хоз. РСФСР, 1961. 146 с.
10. Nametz R. С., Ind. Eng. Chem., 1970, v. 62, № 3, p. 41—53.
11. Fenimore C. P„ Martin F. G., Mod. Plast., 1966, v. 43, № 11, p. 141—148.
12. Piechota H., J. Cell. Plast., 1965, v. 1, p. 186—189.
13. Hilado C. J., J. Cell. Plast., 1968, v. 4, p. 339—343.
14. Nametz R. C., Ind. Eng. Chem., 1967, v. 59, Ns 5, p. 99—116.
15. Ingram A. M., J. Appl. Polymer Sci., 1964, v. 8, p. 2485—2495.
16. Fenimore С. P., Martin F. G„ «Combustion a. Flame», 1966, v. 10, p. 135 (взято из
обзора Lyssi T., Lenzinger Вег., 1974, Ns 36, S. 197—212).
17. Huggins M. L„ J. Am. Chem. Soc., 1953, v. 75, № 17, p. 4123—4133.
18. Dickert,E. A., Toone G. C„ Mod. Plast., 1965, v. 42, Ns 1, p. 197—199.
19. Pitts J. J., Scott P. H„ Powell D. G., J. Cell. Plast, 1970, v. 6, p. 35—37.
20. Pitts J. J., J. Fire a. Flammabil., 1972, v. 3, p. 51—84.
21. Bayer N. E., Plast. Technol., 1962, v. 8, № 11, p. 33—36.
22. Hillado C. J., Ind. Eng. Chem., 1968, v. 7, p. 81—93.
23. Tesoro G. C., Sello S. B., Willard I. I., Text. Res. J., 1969, v. 39, Ns 2, p. 180—
190.
24. Reewes W. A. e. a.. Text. Res. J., 1970, v. 40, № 3, p. 223—231.
25. Willard J. J., Wondra R. E„ Text. Res. J., 1970, v. 40, № 3, p. 203—210.
26. Симигин П. А., Зусман M. H., Райхлин Ф. И. Защитные пропитки текстильных
материалов. М., Гизлегпром, 1957. 299 с.
27*
411
27. Монахов В. Т. В кн.: Методы исследования пожарной опасности веществ. М.,
«Химия», 1972. 414 с.
28. Панченко И. П„ «Текстильная промышленность», 1973, № 1, с. 87—88.
29. Целлюлоза и ее производные. Под ред. Н. Ьайклза и л. Сегала. Т. 2. М., «Мир»,
1974. 510 с.
30. Конкин А. А. Углеродные и другие жаростойкие волокнистые материалы. М.,
«Химия», 1974. 375 с.
31. Williams I. Heat Transfer Fires. Washington, D. C. e. a. 1974, p. 197—237;
Пожарная охрана. Экспресс-информация. M., ВИНИТИ, 1976, № 13, реф. 61,
с. 16—23.
32. McCarter R. J., Text. Res. J., 1972, v. 42, № 12, p. 709—719.
33. Голова О. П„ Пахомов А. М„ Николаева И. И., Изв. АН СССР, ОХН, 1957,
№ 4, с. 519—521.
34. Schwenker R. F., Beck L. R„ J. Polymer Sci., 1963, C, № 2, p. 331—340.
35. Голова О. П., Крылова Р. Г., ДАН СССР, 1960, т. 135, с. 1391—1394.
36. Hendrix J. Е„ Drake G. L., J. Appl. Polymer Sci., 1972, v. 16, № 2, p. 257—274.
37. Drews M. J. e. a. Int. Symp. «Fire Safety Combustible Mater., Edinburgh», 1975,
p. 206—217; Пожарная охрана. Экспресс-информация. M., ВИНИТИ, 1976, № 27,
реф. 139, с. 16—23.
38. Drews М. J., Barker R. Н., J. Fire a. Flammabil., 1974, v. 5, № 2, р. 116—124;
Пожарная охрана. Экспресс-информация. М., ВИНИТИ, 1974, № 41, реф. 296,
с. 1—4. j
39. Webster С. Т., J. Text. Inst., 1967, v. 58, № 6, р. 471—473.
40. Harms J., Krassig H., Lenzinger Ber., 1974, № 39, S. 214—221.
41. Фр. пат. 2047566 (1971). 3
42. Англ, заявка 1299373 (1973).
43. Пат. ФРГ 2016153 (1970).
44. Фр. заявка 2137250 (1973).
45. Фр. заявка 2138400 (1973).
46. Пат. США 3556815 (1971).
47. Frieser Е., Mell. Textilber., 1958, Bd. 39, № 9, S. 1034—1037.
48. Карлов В. А. Придание негорючести текстильным материалам из целлюлозных
волокон. М., ЦНИИЛегпром, 1973. 48 с.
49. Заметта Б. В., Новиков П. В. Производство огнезащищенных текстильных мате-
риалов за рубежом. М., ЦНИИТЭИЛегпром, 1974. 32 с.
50. Quehl, Kurt, Mell. Textilber., 1953, Bd. 34, № 1, S. 69—71; № 2, S. 143—145.
51. Таубкин С. И. Основы огнезащиты целлюлозных материалов. М., изд-во Мин.
коммуи. хоз. РСФСР, 1960. 347 с.
52. Блехман Э. А. Канд. дис. М., МХТИ им. Д. И. Менделеева, 1950.
53. Bilger X., Mangeney G., Mell. Textilber., 1965, Bd. 46, № 3, S. 294—300.
54. Stubbs A. E., Whittaker L. J., Text. Manufact, 1959, v. 85, p. 1017.
55. Hoffmann A. F., J. Am. Chem. Soc., 1955, v. 77, № 14, p. 3923.
56. Jonis D. M., Noone T. M., J. Appl. Chem., 1962, v. 12, № 9, p. 397—405.
57. Reid J. D„ Text. Res. J., 1956, v. 26, № 2, p. 136—140.
58. Пат. США 3698854 (1972).
59. Reevers W. A. e. a., Text. Res. J., 1955, v. 25, № 1, p. 41—46.
60. Beninate L., Text. Res. J., 1969, v. 39, № 4, p. 368—374.
61. Фр. заявка 2172263 (1973).
62. Patat F., Derst P„ Angew. Chem., 1959, Bd. 71, № 3, S. 105—110.
63. Drake I. G., Guthrie I. D„ Text. Res. J., 1959, v. 29, № 2, p. 155—164.
64. Drake I. G„ Beninate I. V., Guthrie J. D„ Am. Dyestuff Reportes, 1961, v. 50, № 4,
p. 27—32. РЖХим 1961, реф. 19П351.
65. Hamalainen G., Guthrie L. D., Text. Res. J., 1956, v. 26, № 2, p. 141.
66. Англ. пат. 788785 (1958).
67. Яп. заявка 113 (1972), кл. ДО6т13/26.
68. Фр. пат. 1225073 (1959).
69. Евтеев А. М. и др., Хим. вол., 1975, № 5, с. 31.
70. Einsele U„ «Textil-Praxis», 1975, v. 30, № 12, S. 1672—1673, 1686—1687.
71. Leblanc R. B„ Banger I. H., Clark К. P„ Text. Chemist a. Colorist, 1975, v. 7, № 10,
ip. 172—174; пер.: Текстильная промышленность. Экспресс-информация. M„
ВИНИТИ, 1976, № 16, реф. 79, с. 17—21.
72. Muendel С. Н.. Selke W. A., Ind. Eng. Chem., 1955, v. 47, № 3, p. 374—379.
73. Роговин 3. А. и др., Высокомол. соед., 1963, т. 5, № 4, с. 506—511.
74. Петров К. А. и др., Высокомол. соед. Сб. «Целлюлоза и ее производные». М.,
изд-во АН СССР, 1963, с. 90—93.
412
75. П редводителев Д. А., Нифантьев Э. Е„ Роговин 3. А., Высокомол. соед., 1965,
т. 7, № 5, с. 791—794.
76. П редвадителев Д. А., Нифантьев Э. Е., Роговин 3. А., Высокомол. соед., 1965,
т. 7, Xs 6, с. 1005—1009.
77. Петров К. А. и др., Высокомол. соед. Сб. «Целлюлоза и ее производные». М.,
изд-во АН СССР, 1963, с. 86—89.
78. Исправникова А. Г., Слеткина Л. С., Роговин 3. А., Высокомол. соед., 1962, т. 4,
Xs 12, с. 1790—1795.
79. Жарова Т. Я. и др., Высокомол. соед., 1967, сер. А, т. 9, Хе 6, с. 698—703.
80. У Мей-янь, Роговин 3. А., Высокомол.. соед., 1963, т. 5, Х° 5, с. 706—711.
81. Harms J., Krassing Н., Lenzinger Вег., i974, Xs 36, S. 214—221.
82. Авт. свид. СССР 744109 (1972); Открытия. Изобр. Пром, образцы. Товарн. знаки,
1972, Xs 21.
83. Гулина А. А., Лившиц Р. М., Роговин 3. А., Высокомол. соед., 1965, т. 7, № 9,
с. 1529—1534.
84. Морин Б. П., Роговин 3. А., Высокомол. соед., 1976, сер. А, т. 18, Xs 10,
с. 2147—2160.
85. Лишевская М. О., Морин Б. П., Ровенькова Т. А. Международный симпозиум по
химическим волокнам. Препринты. Калинин, 1974, секция 3, с. 122—125.
86. Тюганова М. А., Кожанова Т. Я. Международный симпозиум по химическим во-
локнам. Препринты. Калинин, 1974, секции 3, с. 23—28.
87. Tjuganova М. A., Kopyev М. A., Lenzinger Вег., 1976, Bd. 40, S. 148—151.
88. Мухин Б. А., Дружинина Т. В., Межиковский С. М., Хим. волокна, 1973, X» 4,
с. 55—58.
89. Рафиков С. Р., Сорокина Р. А., Высокомол. соед., 1961, т. 3, Xs 1, с. 21—29.
90. Osava Т., Bull. Chem. Soc. Japan, 1965, v. 38, Xs 11, p. 1881—1884.
91. Inoue R., Sumoto M., Chem. High Polymers, 1954, v. 11, p. 169—173.
92. Straus S., Wall A., J. Res. Nat. Bur. Stand., 1958, v. 60, p. 39—42.
93. Рафиков С. А., Сорокина P. А., Высокомол. соед., 1959, т. I, Xs 4, c. 549—557.
94. Кемербик Б., Крое Г., Гролль В., «Химия и технология полимеров», 1961, № 4,
с. 53—80.
95. Goodman J., J. Polymer. Sci., 1954, v. 13, p. 175.
96. Левантовская И. И. В кн.: Старение и стабилизация полимеров. М., «Наука»,
1964, с. 197—236.
97. Achhammer G„ J. Res. Nat. Bur. Stand., 1951, v. 46, p. 391—395.
98. Рафиков С. P„ Архипова И. А., ЖОХ, 1964, т. 34, Xs 12, с. 4086—4090.
99. Straus S„ Wall A., J. Res. Nat. Bur. Stand., 1959, A, v. 63, Xs 3, c. 269—271.
100. Pauling L., Neimann K, J. Am. Chem. Soc., 1939, v. 61, Xs 7, p. 1862—1867.
101. Moore R. F„ «Polymer», 1963, v. 4, Xs 4, p. 493—496.
102. Закаревский В. А., Томашевский E. А., Бантизманский В. В., «Физика твердого
тела», 1967, т. 9, Х° 5, с. 1434—1438.
103. Sharkey W. Н„ Mochel W. Е„ J. Am. Chem. Soc., 1959, v. 81, Х° 12, р. 3000—
3005.
104. Коршак В. В., Рафиков С. Р., ДАН СССР, 1947, т. 56, с. 597—598.
105. Левантовская И. И. и др., Высокомол. соед., 1964, т. 6, Хе 10, с. 1885—1890.
106. Рафиков С. Р., Челнокова Г. Н„ Сорокина Р. А., Высокомол. соед., 1962, т. 4,
Хе 11, с. 1639—1645.
107. Рафиков С. Р., Архипова И. А. Синтез мономеров и полимеров. Труды инсти-
тута химических наук Казахской академии наук. Алма-Ата, 1970, т. 29, вып. 4,
с. 3—18.
108. Алексеев М. В., Исправникова А. Г. Пожарная профилактика при производстве
пластмасс и химических волокон. М., «Коммунальное хозяйство», 1966. 178 с.
109. Mark Н., Lenzinger Вег., 1972, Хе 33, S. 5—11.
ПО. Sayers L. W., Text. Inst. Ind., 1965, v. 3, Xe 7, p. 168—171.
111. Goldberg J. B„ Text. Ind., 1970, v. 134, Xe 6, p. 123—126.
112. Collins J. R., Plast. a. Polymers, 1972, v. 40, № 149, p. 289—293.
113. Craurhaw G. H., Duffield P. A., Mehta P. N., Appl. Polymer Symp., 1971, X> 18
p. 1183—1197.
114. Dorset B„ Text. Manufact., 1961, v. 87, p. 243.
115. Пат. США 2922726 (1960).
116. Robinette H. J., Mod. Text. Magaz., 1957, v. 38, Xe 6, p. 46—49.
117. Авт. свид. СССР 310960 (1969); Открытия. Изобр. Пром, образцы. Товарн. знаки,
1971, Хе 24.
118. Пат. США 2881152 (1959).
119. Пат. США 2999847 (1961).
120. Пат. США 2795513 (1957).
413
121. Пат. США 2854537 (1958).
122. Пат. США 2923644 (1960).
123. Соосе Т. F., Roth Р. В., Text. Res. J., 1956, v. 26, № 3, p. 223-^225.
i24. Пат. США 2953480 (I960).
125. Пат. США 3017292 (1962).
126. Frieser Е., Mell. Textilber., 1959, Bd. 40, № 4, S.-436—438.
127. Пат. США 3682692 (1972).
128. Пат. США 3607745 (1971).
129. Фр. пат. 1532006 (1968).
130. Англ. пат. 61985 (1956).
131. Пат. ФРГ 1102697 (1961).
132. Пат. ФРГ 1294340 (1969).
133. Пат. США 2886539 (1959).
134. Пат. США 3681126 (1972).
135. Пат. ФРГ 1282599 (1968).
136. Stepnitsky Н. Е., «Textilveredlung», 1973, Bd. 8, № 6, S. 293—310.
137. Пат. США 3637409 (1971).
138. Kubitsky К., Mell. Textilber., 1954, Bd. 35, № 1, S. 66—68.
139. Пат. США 2658880 (1953).
140. Пат. ФРГ 1150044 (1963).
141. Пат. США 2926097 (1960).
142. Пат. США 3620818 (1971).
143. Пат. США 3574164 (1971).
144. Пат. США 3574230 (1971).
145. Фр. пат. 1453415 (1966).
146. Мухин Б. А., Роговин 3. А. и др., Хим. волокна, 1973, № 1, с. 17—19.
147. Пат. США 3591507 (1971). .
148. Пат. США 3418267 (1968).
149. Пат. ПНР 55911 (1968).
150. Яп. пат. 2518 (1972).
151. Яп. пат. 1694 (1972).
152. Пат. США 2854434 (1958).
153. Кодолов В. И., Сапогова Л. А., Спасский С. С., Пласт, массы, 1969, № 10,
с. 40—43.
154. У Мей-янь. Канд. дне. М., МТИ, 1962.
155. Цейтлина Л. А., Меос А. И., Вольф Л. А., Хим. волокна, 1961, № 6, с. 22—24.
156. Фрунзе Т. М. и др., Изв. АН СССР, ОХН, 1958, № 6, с. 783—785.
157. Пат. США 2691566 (1954).
158. Пат. США 2691567 (1954).
159. Пат. США 3409386 (1968).
160. Пат. США 3498953 (1970).
161. Пат. США 3083118 (1963).
162. Голл. пат. 6601681 (1966).
163. Фр. пат. 2015100 (1970).
164. Усманов X. Ч., Юльчибаев А. А., Сирлибаев Т., Изв. вузов. Химия и химическая
технология, 1969, т. 12, № 3, с. 319—322.
165. Schulz R. С., Cherdron Н., Kern W., Makromol. Chem., 1957, Bd. 24, № 2, S. 141—
151.
166. Коральник H. Г. Сб научно-исследовательских работ Ташкентского текстильного
института: «Химия и химическая технология высокомолекулярных соединений»,
Ташкент, 1964, № 1(17), с. 46.
167. Авт. свид. СССР 361179 (1971); Открытия. Изобр. Пром, образцы. Товарн. знаки,
1973, № 1.
168. Muchin В. А. е. a., Faserf. u. Textilt., 1975, Bd. 26, № 9, S. 421—429.
169. Schulz R. C., Fauth H., Kern W., Makromol. Chem., 1956, Bd. 20, № 2, S. 161—
167.
170. Смольникова Л. Г., Конкин А. А., Хим. волокна, 1965, № 2, с. 28—30.
171. Смольникова Л. Г., Конкин А. А., Хим. волокна, 1965, № 3, с. 20—24.
172. Хакимова А. X., и др., Хим. волокна, 1965, № 6, с. 29—32.
173. Щеглов П. П. и др., Изв. вузов. Химия и химическая технология, 1974, т. 17,
№ 6, с. 890—894.
174. Смуткина 3. С. и др. В кн.: Структурная химия углерода и углей. Под ред.
В. И. Касаточкина. М, «Наука», 1969, с. 201—214.
175. Makschin W., Faserf. u. Textilt., 1969, Bd. 20, № 7, S. 321—329.
176. Жаркова M. А. и др., Способы получения негорючих синтетических материалов.
М., НИИТЭХИМ, 1974. 36 с.
414
Vl'l. Монкриф P. У. Химические волокна. М., «Легкая индустрия», 1964. 60/ с.
178. Rygeley Е. W„ Rayon Text. Monthly, 1948, v. 29, № 5, p. 69—73.
179. Mell. Textilber., 1962, Bd. 43, № 6, S. 582.
180. Роговин 3. А. Основы химии и технологии полимеров. Т. 2. М., «Химия»,
1974. 343 с.
181. Пат. США 3379699 (1968).
182. Яп. пат. 7204757 (1972).
183. Яп. заявка 4757 (1972).
184. Голл. пат. 6517131 (1966).
185. Голл пат. 65’7132 (1966).
186. Голл. пат. 6517074 (1966).
187. Голл. пат. 6517190 (1966).
188. Пат. США 2636027 (1953).
189. Пат. США 2888434 (1959).
190. Паг. США 2559854 (1951).
191. Пат. США 2827475 (1958).
192. Пат. США 2343261 (1950).
193. Колесников Г. С., Родионова Е. Ф„ Федорова Л. С., Высокомол. соед., 1959, т. 1,
№ 3, с. 367—377.
194. Кривошеева И. А. и др., Высокомол. соед., 1966, т. 8, № 11, с. 1960—1964.
195. Фр. пат. 1560562 (1969).
196. Фр. пат. 1560567 (1969).
197. Berger W. е. a., Faserf. u. Textilt., 1969, Bd. 20, № 10, S. 467—472.
198. Роговин 3. А. и др., Хим. волокна, 1966, № 3, с. 27—30.
199. Пат. США 3288888 (1966).
200. Пат. США 2949437 (1960).
201. Яп. паг. 25417 (1974).
202. Пат. ФРГ 1221761 (1966).
203. Авт. свид. СССР 245976; Открытия. Изобр. Пром, образны. Товарн. знаки,
1969, № 20.
204. Пат. США 3318978 (1967).
205. Пат. США 3242124 (1965).
206. Пат. США 2949432 (1960).
207. Пат. США 3213052 (1965).
208. Пат. ФРГ 2262779 (1973).
209. Англ. пат. 1147180 (1969).
210. Пат. ФРГ 2160021 (1973).
211. Англ. пат. 1004889 (1965).
212. Ял. пат. 7115127 (1971).
213. Пат. США 3313867 (1967).
214. Пат. США 3657179 (1972).
215. Яп. пат. 7203535 (1972).
216. Пат. США 3271343 (1966).
217. Пат. США 3271344 (1966).
218. Яп. пат. 7039057 (1970).
219. Фр. пат. 2056659 (1971).
220. Англ. пат. 1310341 (1973).
221. Пат. США 3582258 (1971).
222. Пат. США 3047425 (1962).
223. Пат. США 3656882 (1972).
224. Англ. пат. 1029867 (1966).
225. Пат. США 3544262 (1970).
226. Pohl Н. A., J. Am. Chem. Soc., 1951, v. 73, р. 5660—5661.
227. Кардаш И. Е., Праведников А. И., Медведев С. С., ДАН СССР, 1964, т. 156, № 3,
с. 658—661.
228. Sommers Е. Е„ Crowell J. I., J. Am. Chem. Soc., 1955, v. 77, № 20, p 5443—
5444.
229. O’Connor G. L., Nace H. R., J. Am. Chem. Soc., 1953, v. 75, № 9, p. 2118—
2123.
230. Гудинге E. П., «Химия и технология полимеров», 1961, № 3, с. 104—119,
231. Allan R. J., Lengar H. V., Ritchie P. D., J. Am. Chem. Soc., 1957, v. 79, № 5,
p. 2107—2113.
232. Allan R. J., Forman R. L., Ritchie P. D., J. Am. Chem. Soc., 1955, v. 77, № 4, p. 2717—
2721.
233. Zimmermann H., Faserf. u. Textilt., 1968, Bd. 19, № 8, S. 372—377.
234. Text. Ind., 1973, v. 137, № 7, p. 23.
415
235. Davis I. C., Chem. Eng., 1973, v. 80, № 9, p. 84—8b.
236. Яп. пат. 32430 (1972).
237. Яп. пат. 27137 (1972), кл. 25(1) D 32; РЖХим, 1973, реф. 13С1028П.
238. Англ, заявка 1340013 (1973).
239. яп. пат. 24171 (1972), кл. 42D12, Р/КХим. 1973, реф. 17С1017П.
240. Яп. пат. 14500 (1972), кл. 25(1) D31; РЖХим, 1973, реф. 12С391П.
241. Пат. США 3624024 (1971); РЖХим, 1972, реф. 16С648П.
242. Яп. пат. 19181 (1972); РЖХим, 1973, реф. 13С338П.
243. Яп. пат. 36248 (1972); С. А., 1973, v. 73, № 8, 44546 X.
244. Яп. пат. 24777 (1971); РЖХим, 1972, реф. 7С559П.
245. Яп. пат. 61725 (1973).
246. Le Blanc R. В., Text. Ind., 1974, v. 138, № 2, p. 115—120.
247. Stepniczka H. E., «Textilveredlung», 1975, Bd. 10, № 6, S. 234—243.
248. Text. Ind., 1972, v. 136, № 10, p. 29.
249. Suchecki S. M., Text. Ind., 1973, v. 137, № 1, p. 67—68.
250. Пат. США 3758335 (1973).
251. Пат. США 3642559 (1972); РЖХим, 1973, реф. 14С10099П.
252. Пат. США 3642525 (1972).
253. Пат. США 3697316 (1972); С. А., 1973, v. 78, № 8, 45006 h.
254. Пат. США 3600219 (1971).
255. Фр. пат. 1593829 (1970).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абляционная стойкость 326, 327
л-Аминобензойная кислота 27
n-Аминобензойная кислота 26
Аморфные участки волокон (строение) 77
Анизотропные растворы 74
Антипирены 277, 284, 350 сл.
механизм защитного действия 350 сл.,
357
Антифрикционные полимерные покры-
тия 207
Антрацен 249
Апротонные диполярные растворители 37
Аримидные волокна 86
аримид ПМ 123
— Т 123, 205
Ароматические высокомодульные волок-
на 84
Ароматические диамины 46
Ароматические полиамиды 95, 96
Ароматические углеводороды 250 сл.
Ацилирование (схема) 42, 44
Базисные ленты 231, 232, 312
параметры 273
Бензидин 24
ВВВ (волокно) 161 сл., 205
Виньон N (волокно) 401
Вискеризацня 322
Волокна
из ароматических полиамидов 104,
105
лестничного строения 157 сл., 166 сл.
-----методы синтеза и свойства
лестничных полимеров 158 сл.
----- получение 162 сл.
-----термомеханические свойства
165 сл.
-----физико-механические свойст-
ва 165
на основе полностью ароматических
полиамидов 92 сл.
-----смесей полимеров 195 сл.
Волокнообразующие полиимнды 131, 132
классы 14 сл.
требования 12, 13
Воспламенение 346, 347
Высокомодульные волокна 78 сл.
ароматические 84
из жесткоцепных гомополиамидов 102
Высокомодульные волокна
сформованные из анизотропных рас-
творов полимеров в минеральных
кислотах 85
----изотропных растворов полиме-
ров в органических растворителях
82, 83
Высокомодульные корды 208
Высокотемпературная обработка окис-
ленного ПАН-волокна 270 сл.
Вытягивание 285 сл.
кратность вытяжки 76
ориентационное 75 сл.
при окислении ПАН-волокна 269 сл.
Вязкость
зависимость от концентрации поли-
мера 63
Газовая теория огнезащитного действия
антипиренов 357
Газофазный пиролиз 249
Гексаметилфосфортриамид 35
Гибкоцепные полимеры 62, 77
Гидразинсульфат 28
Гидратцеллюлозные волокна (получение
углеродных волокон)
закономерности термической деструк-
ции целлюлозы 276 сл.
Гистограмма 301, 302
Глобулярные образования 72
Г омогенно-графитирующиеся материалы
227
Горение 345 сл.
побочные процессы 349
стадии 345
Графит
структура 223
формирование структуры 242, 243
Графитизация 233, 248, 257
окисленного ПАН-волокна 270 сл.
основные закономерности 285 сл.
температура 234
Графитоподобные структуры 271
Двойное лучепреломление 76
Дегидратирующие агенты 57
Дегидроцеллюлоза 280
Дериватограммы полиимидных волокон
118, 119
Джонсона модель фибриллярной струк-
туры 232
417
З.З'-Диамииобензидин 31
4,4'-Диаминоазобензол 26
4,4'-Диаминодифенилметан 25
4.4'-Диаминодифенилоксид 24
4,4'-Диаминоднфенилсульфон 24
Ди ангидрид
3,3',4,4'-бензофенонтетракарбоновой
кислоты 29
1,4,5.8-иафталинтетракарбоновой кис-
лоты 30
3,4,9,10-перилентетракарбоновой кис-
лоты 30, 245
пиромеллитовой кислоты 28
1,4-Диглиоксалилбензол 30
3,3'-Дикарбоксибензидин 32
Ди Марцио параметр 62
N.N-Диметилацетамид 15, 32
Диметиловый эфир терефталевой кисло-
ты 22
Диметилсульфоксид 33, 34
Диметилформамид 33
Динамическая вязкость, зависимость от
концентрации растворов 67
4,4'-Дифенилдикарбоновая кислота 19
4,4'-Дифенилметандиизоцианат 28
Дифениловый эфир изофталевой кисло-
ты 22
Дифенилоксид-фенолоформальдегид 244
Дихлорангидрид
4,4'-дифенилкарбоновой кислоты 21
изофталевой кислоты 20
2,6-нафталиндикарбоновой кислоты 21
терефталевой кислоты 20
Жесткоцепные полимеры 77
Застудневание 72
Изотропные растворы ароматических по-
лиамидов 95, 96
Изофталевая кислота 18
Инертные газы (защитная среда) 283,
285
Искусственный графит 249
Карбонизация
и графитизация окисленного ПАН-
волокна 270 сл.
кокса 248
основные закономерности и условия
281 сл.
пеков 290
полимеров 258
температура 293
Кевлар 205, 208, 212, 213
Кермель (волокно) 180
Кинематическая вязкость
зависимость от концентрации поли-
мера 66
-----температуры 67
Кинетика
пиролитических процессов 258
термического распада 258
Кислородный индекс 407, 410
Кислоты Льюиса 57, 58, 267
Кокс 241 с л.
Композиты
с матрицей из стекла и стеклокера-
мики 331
— металлической матрицей 324
Композиционные материалы (компо-
зиты)
на основе полимерных связующих
315 сл.
----- углеродного волокна и керами-
ческой матрицы 331 сл.
-----------металлической матрицы
332 сл.
--------— — углеродной матрицы
328 сл.
определение 314
применение 334 сл.
степень наполнения волокном 318
типы 314
Конструкционные материалы 210
на основе полимерных связующих
316 сл.
Коэффициент вариации 300, 301
Кратность вытяжки 76
Кривые
изометрического нагрева 100
малоуглового рассеяния рентгенов-
ских лучей полиимидных воло-
кон 130
ТГА и ТДА поликапроамидного во-
локна 374
Критическая молекулярная масса 80
Куртель 260, 261, 298, 301
Левоглюкозаи 277, 281
Лестничные полимеры 173 сл.
графитоподобной структуры 169
Линейные полимеры (термическая де-
струкция)
выход кокса 241 сл.
закономерности деструкции 238 сл.
эффективность превращения углерода
244 сл.
Лола (волокно) 170, 205
Льюиса кислоты 57, 58, 267
Макроматричный эффект 258
Макроморфология углеродных волокон
298 299
Матрица 314, 315. 322, 323, 328, 334
Межплоскостное расстояние в углероде
250 сл.
Межслоевая прочность пластика 319,
322
Метансульфоновая кислота 39
•V-Метилпирролидон 34, 35
Метод кислородного индекса 357
Методы придания огнестойкости ПАН-
волокнам
введение добавок в прядильный рас-
твор при формовании 403 сл.
поверхностная отделка волокон и
тканей 405, 406
химическая модификация 399 сл.
----- сополимеризация 399, 400
формование из смесей полимеров
402, 403
418
Механизм
огнезащитного действия антипиренов
350 сл.
фазового распада 71
Модмор (волокно) 300, 303, 325
Модуль Юига 274, 286, 291, 293, 300,
303 сл., 320, 330, 332
Мокрый способ формования 64 сл.
Мономеры 14 сл.
Насыщенные полиуглеводороды 239
Нафталинкарбоновая кислота 19
Неграфитирующиеся формы углерода
232, 249
Непланарные соединения 249
Низкотемпературнаи поликонденсация 95
Обезвоздушивание прядильных раство-
ров 70
Огнезащита
механизм 356 сл.
огнезащитная пропитка 361
Огнезащитное действие 357
Огнестойкость
текстильных материалов 354, 355
эфиров целлюлозы 367, 369
Окисление волокон 268
Оксалан 205
Органоволокниты 213
Ориентационное вытягивание волокон
75 сл.
Ориентационное упрочнение волокон
99 сл.
Ориентация
волокон 228, 229, 274, 286
структур 269
Отвердители 247, 248
.'Пачки 72
Пеки 248 сл., 287 сл.
мезофазные 289 сл.
обыкновенные 287
получение углеродных волокнистых
материалов 287
склонность к графитации 289, 290
стадии перехода от органических про-
дуктов к углероду и графиту 248
— получения волокна 287
Пилиимидазопирролоны (пирроны) 17
Пирен 245
Пиролиз 248, 345, 346
выделение летучих продуктов при пи-
ролизе окисленного ПАН-волокна
270
газофазный 249
кинетика 258
целлюлозы 276 сл.
Пироуглерод 307, 311, 321
Планарные (плоскостные) соединения
249
Пластификаторы 99
Плотность волокна 274, 275, 310
«Плутон» (УВМ) 240
Поверхностно-активные вещества 96
Показатель возгораемости 355
Полиакрилонитрильные волокна
высокотемпературная обработка окис-
ленного ПАН-волокна 270 сл.
Полиакрилонитрильные волокна
методы придании огнестойкости
399 сл.
Полиамидные волокна 372
методы придания огнестойкости
378 сл.
термическое и термоокислительное
разложение полиамидов 373 сл.
Полиамидоаминокислота 158 сл.
Полиамидоимидные волокна 176 сл.
получение 176 сл.
физико-механические свойства 179
Полиамидокислотные волокна см. Поли-
имидные волокна
Полиамидокислоты
синтез 113
стабильность растворов 115, 116
удельная вязкость 114, 115
Полиимидные волокна
волокнообразующие полимеры 131,
132
дериватограммы 118, 119
кривые малоуглового рассеяния рент-
геновских лучей 130
методы получения 116, 117
радиационная стойкость и стойкость
к УФ-излучению 128, 129
структурные особенности 129, 130
Полибензимидазол 17, 244
Полибензимидазольные волокна 148 сл.
получение и свойства полимеров
148 сл.
----- физико-механические свойства
151 сл.
термомеханические свойства 153 сл.
химическая и радиационная стой-
кость 155, 156
Полибензоксазольные волокна 182, 184
Полибензоксазолы 17
Полибензофуроизохинолии 168
Полибисбензимазобензофенантролин
ВВВ 166
BBL 167
Полибисбензимидазопирролон 168
Поливинилхлорид 244
Полигидразидные волокна 134
Полигидразиды 130
методы синтеза 133 сл.
Полиимидазопирролон 166
Полиимидные волокна 112 сл.
методы получения 116 сл.
общая характеристика 121, 122
получение растворов полимеров
112 сл.
структурные особенности 129, 130
физико-химические и термомеханиче-
ские свойства 121 сл.
Полиимиды 16
Поликонденсация в растворе 93, 94
Полимерные гетероциклы 255
Полимеры
графитоподобной структуры 174
классификация 62
лестничные 173
полужесткие 62
предельно жесткоцепные 62
419
Полимеры
пространственные 243 сл.
Полиметилфенилсиликон 244
Полиоксадиазольные волокна 130 сл.
влияние условий формования на свой-
ства 145
модифицированные 147
упрочнение 138 сл.
физико- и термомеханические свойст-
ва 140 сл.
формование 138
Полиоксадиазолы 16
свойства растворов 137, 138
Полисульфон 244
Политиадиазольные волокна 181 сл.
Полифен 205
Полифениленоксид 244
Полифенилсиликон 244
Полифенилентриазольные волокна 182 сл.
Полифосфорная кислота 36, 38
Полифуриловый спирт 245
Полихиноксалин 17, 169
Полициклогидратация 55
Полиэтилентерефталат (термическое и
термоокислительное разложение)
407, 408
Полиядерные ароматические хиноны и
кетоны (получение углерода) 253,
254
Полужесткие полимеры 62
Породе, параметр 233
Пористость
волокон 75, 310, 338
углеродных волокнистых материалов
233
Предельно жесткоцепные полимеры 62
Приведенная вязкость 66
Привитые сополимеры целлюлозы 370
Приготовление прядильных растворов
69 сл.
Придание огнестойкости
полиамидным волокнам и тканям
378 сл.
полиэфирным волокнам 408 сл.
целлюлозным волокнам и тканям
358 сл.
Применение
термостойких волокон 205 сл.
углеродных волокон 314 сл.
Принципы синтеза термостойких поли-
меров 40 сл.
Продукты
пиролиза 277, 281 сл.
термического распада 270 сл., 278
Пространственные полимеры 243 сл.
Прядильные растворы
прядомость 95
стабильность 65, 66
Радиационная стойкость и стойкость к
УФ-излучению полиимидных воло-
кон 128, 129
Радиационно-химическая модификация
12
Растворимость полимеров 64 сл.
420
Растворители 14 сл. *
апротонные диполярные 37
сильные 65
Растворы полимеров 66 сл.
Реакции
полигетероциклизации 54 сл.
— катализаторы 58
по типу полиацилирования 42 сл., 50,
51
разветвления 55
Резонансная стабилизация 239
Руланда модель фибриллярной структу-
ры 232
Светостабилизация полиамидных воло-
кон 110, 111
Светостойкость волокон 193
СВМ (волокно) 205, 209
Свойства углеродных волокнистых мате-
риалов
макроморфология 298, 299
механические 299 сл.
термические 305 сл.
физические 309 сл.
химические 309 сл.
Связующие 323
Серная кислота и олеум 38, 39
Стабильность прядильных растворов 65,
66, 95, 115, 116
Степень
дисперсности полимера 69
кристалличности 99
циклодегидратации полигидразидных
волокон 134
Стойкость к УФ-облучению
иолокон из смесей полимеров 199
сополигетероциклических волокон 195
Сополимерные гетероциклические волок-
на 188 сл.
е-Стекловолокно 212
Структура
графита 223
переходных форм углерода 222 сл.
совершенствование структуры 275
углеродных волокнистых материалов
225 сл.
Сульфолан (волокно) 35, 81
Сухо-мокрый метод формования 89, 96
Сухой метод формования
волокон 87, 88
технологические характеристики 88
Теория
каталитической дегидратации 357
покрывающего слоя 357
Тепловое старение волокон 171, 194
Теплостойкость 11, 105, 106
Теплота сгорания волокон 348
Температура
графитизации 234
стеклования 63
термической обработки 266, 272, 273,
284, 290, 293, 302, 306 сл.
Терефталевая кислота 18
Терлон 205
Термическая деструкция
линейных полимеров 238 сл.
Термическая деструкция
пространственных полимеров 243 сл.
целлюлозы 276 сл.
Термическая теория огнезащитного дей-
ствия антипиренов 357
Термическое и термоокислительное раз-
ложение
полиамидов 373 сл.
— механизм термоокнсления 377
— параметры деструкции 375
— скорости разложения 374
полиэтилентерефталата 407, 408
Термическое окисление ПАН-волокна
вытягивание при окислении 269 сл.
изменение химического состава во-
локна 265
продукты окисления 266
условия окисления 266 сл.
физико-химические процессы, проте-
кающие при окислении 262 сл.
Термодинамические параметры термиче-
ского распада полиэтилена и по-
литетрафторэтилена 257
Термомеханические кривые полиимидных
волокон 127
Термомеханические свойства волокон
из лестничных полимеров 165 сл.
полиамидных 106, 107, 127
полибензимидазольных волокон
153 сл.
полиимидных 122 сл.
Термоокислительное разложение огне-
стойких полиамидных волокон
392 сл.
кривые ДТА 395
параметры деструкции 393
скорость газообразования 394, 397
— разложения 393
термогравиметрические кривые 392
Термостабилизация волокон 86, 87
полиамидных 107 гл.
Термостабильность волокон 12, 185
из смесей полимеров 198
полиимидных 128
полиоксадиазольных 143 сл.
прогнозирование 13
Термостойкость 11, 106
полиоксадиазольных волокон 145
Термоциклизация полиимидных воло-
кон 118
Т\!,М,М',М'-Тетраметилмочевина 35, 36
Тетрамннобензол 31
2,3,5,6-Тетраминодибензофуран 32
З.З'ДД'-Тетраминодифенилоксид 32
Тетраоксиметилфосфоний 362
Тиомочевина 379
Торейка (волокно) 303
Торнел (волокно) 303
Трицианмочевина 360
Турбостратные пакеты 222 сл., 232, 283
параметры 224, 227, 265
— влияние температурной обработ-
ки 226
Турбостратные структуры 225, 254
Углепластики 323, 325, 336
на основе термопластичных полиме-
ров 327
наполненные короткими волокнами
317, 318
углерод-углеродные на основе войло-
ка 330
Углерод
гомогенно-графитирующиеся формы
232, 234
неграфитирующиеся формы 232, 249
структура переходных форм 222 сл.
Углеродные волокна
исходные материалы 236 сл.
макроморфология 298, 299
получение из гидратцеллюлозных во-
локон 276 сл.
структура 222 сл.
Углеродные волокнистые материалы
получение из гидратцеллюлозного во-
локна 275 сл.
----ПАН-волокна 260 сл.
-----пеков и фенольных смол 287
применение 336 сл.
свойства 298 сл., 305 сл.
структура 225 сл.
физические свойства 309 сл.
Углеродные тканые материалы 305
Угол разориентации волокон 77, 79
Удельная вязкость полимера 81
Удельная свободная поверхность воло-
кон 75
«Усы» 322
•м-Феннлендиамин 23
л-Фенилендиамин 22
Фенолоуглепластики 324 сл.
Фенольные смолы
высокотемпературная обработка
ФФС-волокна 292 сл.
выход кокса и эффективность пре-
вращения углерода 246
исходные смолы 291
схема распада 294
формование и отверждение ФФС-во-
локна 291, 292
Фибриллы 233, 258, 273, 303
Фибриллярная структура 231, 232
Физико- и термомеханические свойства
волокон
полиамидоимидных 180, 181
полиимидных 121 сл.
Физико-механические свойства волокон
из лестничных полимеров 165
полиамидных 101 сл.
полиамидоимидных 178, 179
полиимидных 122 сл.
Физические свойства углеродных волок-
нистых материалов 309 сл.
Фильтровальные и защитные материа-
лы 206
Фильтрующий материал 70
Флуорен 249
Формование
из анизотропных растворов 74, 75, 80
полиамидных волокон 97 сл.
термостойких высокопрочных высоко-
модульных волокон 71 сл.
421
Фосфонитрилхлорид 364
Фосфорилирование полиамидного волок-
на 385
Фталевые кислоты 45
ФФС (волокно) 291 сл.
Химическая и гидролитическая стабиль-
ность полиимидных волокон 128
Химическая и радиационная стойкость
полибензимидазольиых волокон
155, 156
Химическая и физическая модификация
термостойких волокон 185 сл.
Химическая модификация полиамидов с
целью придания огнестойкости
введение хлорсодержащих соединений
390 сл.
полимераналогичные превращения
383 сл.
превращение альдегидных групп при
термоокислении 389, 390
привитая сополимеризация 386 сл.
' фосфорилирование 389, 390
Химическая модификация целлюлозных
материалов 365 сл.
Химическая стойкость углеродных воло-
кон 307 сл.
Хлорангидрид n-аминобензойной кисло-
ты 27
Хлорсульфоиовая кислота 39
Хоутца и Грасси схемы окисления ПАН-
болокнм 26° 2&л
Целлюлозные волокна 356 сл.
Целлюлоза 277 сл.
разложение и механизм огнезащиты
356 сл.
термическая деструкция 276 сл.
«фурфурольная» схема распада 281
Циклогидратация волокон 81 сл.
Шинный корд (производство) 207 сл.
Шунгит 224, 226, 227
Эмульсионная поликонденсация 94
Энергия диссоциации химических свя-
зей 239
Эпоксидные смолы 247
Эпоксиуглепластики 319
Этиленмочевина 379
Юнга модуль см. Модуль Юнга
HMS (волокно) 303
HTS (волокно) 303
Александр Арсеньевич Конкин, Георгий Иванович Кудрявцев,
Александр Михайлович Щетинин, Тамара Викторовна Дружи-
нина, Борис Алексеевич Мухин
ТЕРМО-, ЖАРОСТОЙКИЕ
И НЕГОРЮЧИЕ ВОЛОКНА
Редакторы С. М. Беленькая, И. И. Машине кая
Технический редактор Г. И. Косачева
Художественный редактор И. В. Носов
Корректоры Т. С. Васина, И. А. Иванова
ИБ № 734
Сдано в наб. 12.10.77. Подп. в печ. 6.4.78. Т-07648. Формат бумаги 70Х100'/1в.
Бумага тип. №2. Гарн. литературная. Печать высокая. Усл. печ. л. 34,45.
Уч.-изд. л. 33,39. Тираж 3100 экз. Заказ 2254. Цена 5 р. 50 к. Изд. № 1093.
Издательство <Химия>. 107076. Москва, Стромынка, 13.
Московская типография № 11 Союзполиграфпрома при Государствен-
ном комитете Совета Министров СССР по делам издательств, поли-
графии и книжной торговли. Москва, 113105, Нагатинская ул., д. 1.