





























































































































































Текст
А.К.ПИКАЕВ
ДОЗИМЕТРИЯ
В РАДИАЦИОННОЙ ХИМИИ
АКАДЕМИЯ НАУК СССР
П32
ОРДЕНА ТРУДОВОГО КРАСНОГО ЗНАМЕНИ ИНСТИТУТ ФИЗИЧЕСКОЙ ХИМИИ
А. К. ПИКАЕВ
ДОЗИМЕТРИЯ
В РАДИАЦИОННОЙ ХИМИИ
О
ИЗДАТЕЛЬСТВО «НАУК А»
МОСКВА 1975
У НИ,
УДК 541.15+539.12.08
Дозиметрия в радиационной химии. П и к а е в А. К. М., «Наука», 1975, стр. 312.
Монография посвящспа различным методам дозиметрии ионизирующего излучения, используемым в радиационной химии. Рассматриваются основные понятия и единицы дозиметрических измерений, особенности взаимодействия ионизирующего излучения с веществом, описываются источники ионизирующих излучений и дается краткая характеристика применения физических методов дозиметрии в радиационной химии. В книге обобщен и систематизирован обширный экспериментальный и теоретический материал, касающийся химических методов дозиметрии гамма- и рентгеновского излучений, потоков электронов и тяжелых заряженных частиц, импульсного и реакторного излучений.
Книга рассчитана па широкий круг научных работников, инженеров, студентов и аспирантов, специализирующихся в радиационной химии и смежных областях науки (радиобиологии, радиационной физике и др.).
Таблиц 65. Иллюстраций 87. Библ. 1634 назв.
20503-215 „„ „
П 055(02)-75 106-75
© Издательство «Наука», 1975 г.
ПРЕДИСЛОВИЕ
Успешное проведение любого радиационно-химического исследования зависит в первую очередь от правильного определения поглощенной дозы. В этой книге сделана попытка рассказать заинтересованному читателю о методах дозиметрии ионизирующего излучения, используемых в радиационной химии. Она написана на основе обширного экспериментального и теоретического материала, опубликованного примерно до начала 1972 г. В ряде случаев были учтены и более поздние результаты.
Хотя книга в целом посвящена дозиметрии, в ней нашли отражение и некоторые смежные вопросы. Это относится главным образом к видам и источникам ионизирующего излучения. Цель такого рассмотрения состоит в том, чтобы показать читателю, с какими излучениями и радиационными полями имеют дело в этой области химии.
В книге в главе I излагаются основные понятия и единицы измерения в дозиметрии ионизирующего излучения. В ней рассматриваются различные виды излучения (фотонное, электронное, потоки тяжелых заряженных частиц, нейтронное и др.), а также особенности взаимодействия его с веществом. В главе II описываются источники ионизирующих излучений (изотопные источники, ускорители, рентгеновские трубки и т. п.), применяемые в радиационной химии, В главе III дается сжатая характеристика использования физических методов дозиметрии (калориметрических, ионизационных и т. д.) химиками-радиационниками.
В радиационной химии доза, как правило, измеряется химическими методами. Поэтому им посвящена большая часть книги. В главе IV приводятся основные положения химической дозиметрии и методики различных дозиметрических расчетов. Глава V представляет собой обзор дозиметрических систем, являющихся водными растворами. Значительное место отводится здесь ферро-сульфатной дозиметрической системе (дозиметру Фрикке), широко применяемой в радиационной химии, радиобиологии и некоторых других областях науки. Подробно обсуждаются цериевый и глюкозный дозиметры, дозиметры на основе водных растворов бихромата калия, щавелевой кислоты, бензола и многие другие.
В главах VI, VII и VIII обобщаются сведения о применении соответственно органических жидкостей, газов и твердых тел в качестве дозиметрических систем. В них детально описываются дози
метрические свойства циклогексана, закиси азота, полимеров, стекол, щелочпогалоидных соединений и т. и.
В главе IX рассматриваются особенности дозиметрии импульсного излучения. Наконец, в главах X и XI излагается химическая дозиметрия потоков тяжелых заряженных частиц и реакторного излучения.
В книге, наряду с подробным обсуждением дозиметрических характеристик различных систем, в некоторых случаях приводится механизм их радиолиза и даются практические рекомендации для правильного применения этих систем при определении дозы. Кроме того, в книгу включено несколько таблиц справочного характера, необходимых для проведения дозиметрических расчетов.
Таким образом, книга охватывает широкий круг вопросов, связанных с ионизирующими излучениями и радиационной химией. Естественно, это могло привести к недостаткам в построении книги и изложении фактического материала. Автор будет благодарен всем лицам за любые критические замечания и пожелания.
Автор выражает глубокую признательность В. И. Спицыну, Н. Е. Брежневой, Л. Т. Бугаенко и П. Я Глазунову за ценные замечания.
А. К- Пикаев
ВВЕДЕНИЕ
Радиационная химия изучает химические превращения веществ под действием излучений высокой энергии. Зародилась она в конце XIX в. в результате открытия рентгеновских лучей и явления радиоактивности. Развитие ее сначала шло медленными темпами. Однако во время второй мировой войны (в 1942 г. радиационная химия получила свое название) и после нее развитие этой области физической химии достигло существенного прогресса в связи с возникшими потребностями атомной энергетики и технологии. В данный период радиационная химия выполняла преимущественно вспомогательные функции. Ей необходимо было дать ответ на многочисленные вопросы, которые ставились перед ней атомной промышленностью. В число зтих вопросов входили радиационная стойкость различных материалов, используемых в ре актор остр оенип, радиолитические превращения в системах, применяемых в технологии выделения ядерного горючего, и т. п. Радиационная химия успешно отвечала на зти вопросы.
К настоящему времени радиационная' химия стала самостоятельной областью науки. Здесь сделаны многие крупные открытия, имеющие общехимическое значение. Существенным достижением явились также разработка и внедрение в промышленность ряда радиационно-химических процессов. Тем самым мы стали свидетелями зарождения радиационно-химической технологии.
Сейчас радиационно-химические исследования проводятся во многих странах. При этом используются разнообразнейшие источники излучений высокой энергии, способные создавать самые различные диапазоны доз и мощностей доз. Естественно, успех любого исследования в области радиационной химии зависит в первую очередь от того, насколько правильно и точно определены доза и мощность дозы. Дозиметрия — это количественный аспект радиационной химии, и каждый радиационно-химический эксперимент начинается с измерения дозы.
В табл. 1 приведены диапазоны доз, с которыми сталкиваются в некоторых областях науки и техники [11. Эти диапазоны могут быть измерены различными современными методами (табл. 2). Однако в радиационной химии и радиационно-химической технологии, для которых характерно использование доз до 108—1012 рад, чаще всего применяются химические методы дозиметрии. Обусловлено это главным образом тем, что они позволяют определять дозу непосредственно в реакционном сосуде, не прибегая к слож-
Таблица 1
Диапазоны доз, используемые в различных областях науки и техники
Область применения Диапазон доз, рад Область применения Диапазон доз, рад
Радиобиологическая защита Радиотерапия Радиобиология 10-3—103 102—104 102—103 Радиационная химия Радиационно-химическая и ядерная технология 103—108 1О-10’2
ным расчетам. Кроме того, в данном случае величину дозы возможно найти с помощью обычных химических операций.
Историю развития дозиметрии (в первую очередь химической дозиметрии) можно разделить на три периода [4]: ранний период (1900—1927 гг.), второй период (1927—1945 гг.) и современный период (начиная с 1945 г.).
Таблица 2
Диапазоны доз, измеряемые различными методами [1—3]
Методы Диапазон доз, рад
Ионизационные Калориметрические Химические в том числе фотографические Люминесцентные Сцинтилляционные от 10-в до ~108 от 102 до ~ 103 от 0,1—1,0 до ~1010 от 10-2 до 104 от 10-з—10-2 до 101 от 10-0 до IO-2
Конец XIX в., как уже говорилось, ознаменовался открытием рентгеновских лучей и явления радиоактивности. Очень быстро эти открытия нашли широкое применение в медицинской практике (в диагностике и рентгено-и радиотерапии). Было установлено, что рентгеновское и радиоактивные излучения оказывают воздействие на процессы в человеческом организме. Поэтому возникла проблема измерения доз, поглощаемых живыми тканями. С этой целью стали предлагаться разнообразные химические дозиметры.
В 1902 г. Г. Хольцкнехт [5] описал «хроморадиометр», который представлял собой небольшие диски сплавленной смеси хлористого калия и соды. Эта смесь при действии рентгеновских лучей изменяла свой цвет. Несколько позже в качестве дозиметра потоков
этих лучей было рекомендовано использовать таблетки и пасты из платиносинеродистого бария [6, 7]. При облучении они изменяли свой цвет от светло-зеленого до темно-оранжевого. Степень изменения окраски зависела от количества поглощенной энергии излучения. Дозиметры на основе этого соединения находили применение до второй мировой войны [8]. Однако описанные дозиметрические системы обладали рядом существенных недостатков. В их состав входили элементы с высоким атомным номером. Поэтому их эффективный атомный номер существенно отличался от эффективного атомного номера мягких тканей человеческого тела, что при изменении длины волны излучения приводило к значительному различию в величинах энергий, поглощенных этими тканями и дозиметрами. Кроме того, облученные таблетки или диски на свету довольно быстро цринимали свою первоначальную окраску.
Некоторое применение находили в то время также фотографические методы дозиметрии [9]. Величина поглощенной энергии определялась по почернению фотобумаги или фотопластинок.
В 1907 г. Г. Шварц [10] предложил «каломельный радиометр», представлявший собой водный раствор оксалата аммония и двухлористой ртути. При действии рентгеновских лучей на этот раствор наблюдалось образование каломели. Количество осадка или степень помутнения служили мерой поглощенной энергии.
Л. Фройнд [11] рекомендовал растворы йодоформа в хлороформе в качестве дозиметра. Однако эти растворы чувствительны к действию света и тепла. Использование их усложнялось самопроизвольным окислением, отсутствием линейной зависимости количества превращения от величины дозы и зависимостью показаний от энергии излучения.
В 1910 г. У. Брзгг [12] высказал предположение о возможности измерения дозы и мощности дозы по ионизирующей способности излучений высокой энергии. Полная теория этого вопроса была дана в работах Л. Грея [13, 14] в 1929—1936 гг. Тем самым был сформулирован общеизвестный принцип Брэгга—Грея (см. стр. 87), ставший основой ионизационных методов дозиметрии излучений, высокой энергии.
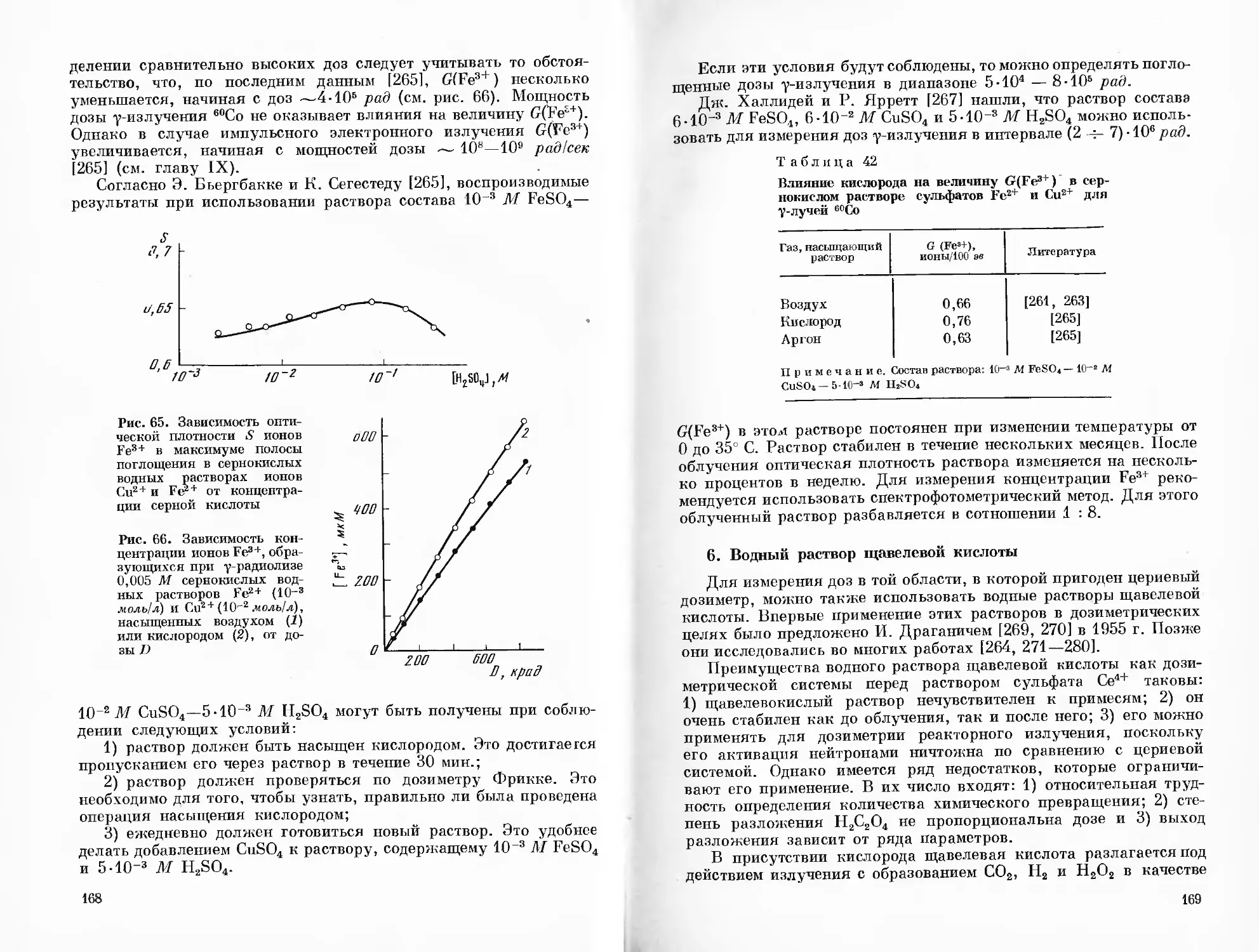
Выделение второго периода в самостоятельный этап развития дозиметрии вызвано главным образом тем, что в 1927—1929 гг. Г. Фрикке и С. Морзе [15—19] предложили разбавленный (4-10Б М) водный раствор сернокислого закисного железа в 0,4 М H2SO4, насыщенный воздухом, для дозиметрии рентгеновских лучей, а также прогрессом в области ионизационных методов. При действии излучения на указанный раствор Fe2+ окисляется в Fe8+. G(Fe3+), как можно рассчитать на основании экспериментальных результатов работ [15, 16], составляет примерно 18 ионов /100 эв. Данные, приведенные в более, поздней работе Г. Фрикке и Э. Браунскомбе [20], показывают, что G(Fe3+) = 16,4 иона/ /100 эв. Указанная концентрация была выбрана по той причине,
что массовый коэффициент поглощения рентгеновских лучей для этого раствора такой же, как и для воздуха 121]. Было найдено 116, 22], что начальный выход Fe3+ не зависит от концентрации ферросульфата в пределах от 10-Б до 10'2 М и что в процессе окисления Fe2+ существенную роль играет кислород, присутствующий в растворе, а также величина pH. В 1932 г. Н. А. Шишаков [23] подтвердил возможность применения сернокислых растворов ферросульфата для дозиметрии рентгеновского излучения. Величина G (Fe3+), вычисленная из его экспериментальных данных, составляет около 16 ионов/100 эв. Позднее ферросульфатная дозиметрическая система, часто называемая «дозиметром Фрикке», исследовалась многими авторами. Сейчас она находит широкое применение в радиационной химии, радиобиологии и некоторых других смежных областях науки.
В 1928 г. П. Гюнтнер и др. 124] нашли, что при действии рентгеновских лучей и у-лучей радия на хлороформ пли его насыщенный водный раствор происходит образование соляной кислоты. Концентрация кислоты пропорциональна энергии, поглощенной системой.
В. Стенстрём п А. Ломан [25—27] наблюдали обесцвечивание метиленового голубого в водном растворе под действием рентгеновского излучения и рекомендовали использовать это явление для дозиметрии. Позже этот процесс исследовался В. Зейтцем [28] и К. Циммером [29]. Возможность применения смеси двухлористой ртути и щавелевокислого калия для целей дозиметрии ионизирующих излучений изучалась в 1932 г. Э. В. Шпольским и С. В. Платоновым [30].
В рассматриваемый период, благодаря большим успехам, достигнутым в области радиоэлектроники, широкое распространение получили ионизационные методы дозиметрии, основанные, как отмечалось, на определении ионизации различных газов (главным образом воздуха) под действием излучения. Внедрению этих методов в практику способствовало то обстоятельство, что эффективный атомный номер воздуха близок к эффективному атомному номеру мягких человеческих тканей. Вследствие распространения ионизационных методов внимание ученых к разработке простых и надежных химических дозиметров значительно ослабло. Даже ферросульфатная система, которая, как известно сейчас, во многих отношениях удовлетворяет требованиям идеальной дозиметрической системы, почти не использовалась в то время для определения величины дозы. Другая причина этого состоит в медленном развитии самой радиационной химии в данный период.
Более подробно методы дозиметрии в первые два периода рассмотрены в книгах [31—34].
Современный период, начавшийся после второй мировой войны, характеризуется интенсивной разработкой химических методов дозиметрии. Появление мощных источников ионизирующего излучения, потребности ядерной энергетики и технологии, а также
необходимость разработки надежных способов защиты от вредного действия проникающей радиации стимулировали бурное развитие таких отраслей науки, как радиационная химия, радиобиология и т. п. Успешное развитие этих отраслей науки немыслимо без наличия простых и точных методов определения величины поглощенной дозы для различных видов излучения, уровней доз и мощностей дозы и других условий проведения эксперимента. Особые требования к дозиметрии предъявила радиационно-химическая технология. Для нее необходимы дозиметры, позволяющие измерять большие дозы, а также дозиметры-индикаторы, фиксирующие облучение системы до конкретной дозы.
К настоящему времени предложены многочисленные дозиметрические системы, с помощью которых возможно определять дозы при самых различных условиях облучения. Сейчас специалист по радиационной химии может в принципе подобрать дозиметрическую систему, пригодную для измерения больших или малых доз, высоких или низких мощностей дозы, для определения доз рентгеновского и у-излучения, электронов, тяжелых заряженных частиц, реакторного излучения или осколков деления, для нахождения доз при работе с реакционными сосудами разной конфигурации и т. п.
Для современного периода характерно также дальнейшее усовершенствование ионизационных методов и существенный прогресс калориметрических методов дозиметрии. Бурное развитие в этот период получила дозиметрия с помощью твердых тел (стекол, пластмасс, щелочногалоидных кристаллов и т. д.). Здесь достигнуты существенные успехи, и в последнее время такую дозиметрию стали рассматривать как особую область дозиметрии, называемую твердофазной дозиметрией — англ, solid state dosimetry (см., например, [1. 21). Вызвано это главным образом тем, что изменения, происходящие в таких системах при облученип.часто фиксируются сугубо физическими методами (люминесцентными, сцинтилляционными и т. д.)'
Развиваются также фотографические методы. Однако они практически не находят применения в радиационной химии и поэтому не будут рассматриваться в настоящей книге.
ЛИТЕРАТУРА
1. В. К. Broszkiewicz. Solid State and Chemical Radiation Dosimetry in Medicine and Biology. Vienna, IAEA, 1967, p. 213.
2. J. W. Boag. Ibid., p'. 349.
3. H. Fricke, E. J. Hart. Radiation Dosimetry (Ed. F. H. Attix and W. C. Roesch), vol. 2. N. Y., 1966, p. 167.
4. II. В. Верещинский, А. К. Пикаев. Введение в радиационную химию. М., Изд-во АН СССР, 1963, гл. IX.
5. G. Holzknecht. Compt. rend. cong. internat. d’electrol. et de radiol. med., 2, 377 (1902).
6. B. Sabouraud, N. Noire. Presse med., 2, 825 (1904).
7. W. Hampson. Arch. Roentgen. Ray, 16, 235 (1911).
8. 0. Glasser. Radiology, 37, 211 (1941).
9. R. Kienbock. Fortschr. a. d. Geb. d. Rontgenstrablen, 9 , 276 (1906).
10. G. Schwarz. Fortschr. a. d. Geb. d. Rontgenstrablen, 11, 114 (1907).
11. L. Freund. Wien. Klin. Wochschr., 17, 412 (1904).
12. W. H. Bragg. Phil. Mag., Ser. 6, 20, 385 (1910).
13. L. H. Gray. Proc. Roy. Soc., A122, 647 (1929).
14. L.H.Gray. Proc. Roy. Soc., A156, 578 (1936).
15. H. Fricke, S. Morse. Am. ,1. Roent. Rad. Ther., 18, 430 (1927).
16. H. Fricke, S. Morse. Phil. Mag., 7, 129 (1929).
17. H. Fricke, S. Morse. Strablentherapie, 26, 749 (1927).
18. H. Fricke, S. Morse. Ibid., p. 757.
19. H. Fricke. Phys. Rev., 31, 1117 (1928).
20. H. Fricke, E. R. Brownscombe. J. Am. Cliem. Soc., 55, 2358 (1933).
21. H. Fricke, B. W. Petersen. Am. J. Roent. Rad. Ther., 17, 611 (1927).
22. H. Fricke, E. J. Hart. J. Chem. Phys., 3,’60 (1935).
23. TV. A. Schischakov. Phil. Mag., 14, 138 (1932).
24. P. Gtintner, H. D. von der Horst, G. Cronheim. Z. Elektrochem., 34, 616 (1928).
25. W. StenstrSm, A. Lohman. Radiology, 16, 322 (1931).
26. W. Stenstrom, A. Lohman. Radiology, 21, 29 (1933).
27. W. Stenstrom. A. Lohman. Radiology, 22, 304 (1934).
28. W. Seitz. Strablentherapie, 61, 148 (1938).
29. K. G. Zimmer. Naturwiss., 32, 375 (1944).
30. Э. В. Шполъский, С. В. Платонов. Ж. физ. химии. 3, 121 (1932).
31. М. Неменов. Рентгенотерапия. Пг., ГИЗ, 1920.
32. Г. Ней. Рентгеновские лучи. М., ОГИЗ, 1928.
33. А. Н. Кронгауз. Дозиметрия рентгеновских лучей. Москва — Харьков, Госиздат черной и цветной металлургии, 1941.
34. Я. Л. Шехтман. Рентгеновская дозиметрия. М., Изд-во Народного комиссариата здравоохранения СССР, 1941.
Глава I
ИОНИЗИРУЮЩИЕ ИЗЛУЧЕНИЯ
И ОСОБЕННОСТИ ИХ ВЗАИМОДЕЙСТВИЯ С ВЕЩЕСТВОМ.
ОСНОВНЫЕ ПОНЯТИЯ II ЕДИНИЦЫ
Для осуществления радиационно-химических реакций используют излучения высокой энергии. Эти излучения при прохождении через среду производят ее ионизацию. Поэтому излучения, с которыми имеют дело в радиационной химии, называют ионизирующими излучениями. Конечно, из этого определения не следует, что они производят только ионизацию. Помимо данного процесса, при взаимодействии их с веществом наблюдается возбуждение молекул среды. Основываясь на этом, А. Чарлзби [1] предложил термин «атомные излучения». Однако этот термин не получил широкого распространения в радиационной химии.
Ионизирующие излучения делятся на непосредственно ионизирующие и косвенно ионизирующие излучения [2, 3J. Непосредственно ионизирующим излучением называется ионизирующее излучение, состоящее из заряженных частиц, которые имеют кинетическую энергию, достаточную для ионизации при столкновении. Косвенно ионизирующим излучением называется ионизирующее излучение, состоящее из фотонов или незаряженных частиц, взаимодействие которых со срепой приводит к возникновению непосредственно ионизирующего излучения.
К ионизирующим излучениям относят фотонное излучение (электромагнитное излучение с малой длиной волны) и корпускулярное излучение.
Фотонное излучение включает у- и рентгеновское излучения. у-Излучением' называется фотонное излучение атомных ядер. Рентгеновское излучение представляет собой совокупность тормозного и характеристического излучений. Тормозное излучение — это фотонное излучение с непрерывным спектром энергий, возникающее при изменении кинетической энергии заряженной частицы при ее движении в кулоновском поле. Характеристическим излучением называется фотонное излучение, возникающее при изменении энергетического состояния атома.
К корпускулярному излучению относят потоки быстрых электронов, протонов, гелионов, нейтронов, тяжелых многозарядных ионов, (3-частицы, а-частицы, атомы отдачи, возникающие в результате ядерных реакций, продукты ядерных реакций деления.
В радиационной химии применяется также термин — смешанное излучение. Под смешанным излучением понимают ионизирующее излучение, состоящее из частиц различного вида или из
частиц и фотонов. Например, со смешанным нейтронным и у-излу-чением часто имеют дело при работах на атомных реакторах.
Взаимодействие ионизирующих излучений с веществом обычно подразделяют на три стадии [4—6]: 1) физическую; 2) физико-химическую и 3) химическую. На первой стадии происходит образование вторичных электронов, их замедление до уровня электронных состояний среды и т. п. На физико химической стадии имеет место возникновение промежуточных частиц (свободных радикалов, ионов, возбужденных молекул). Последние во время химической стадии взаимодействуют друг с другом или с молекулами среды, в результате чего образуются конечные стабильные продукты радиолиза.
В настоящей главе наряду с характеристикой различных видов ионизирующего излучения излагаются главные особенности физической стадии взаимодействия их с веществом, поскольку знание этих особенностей необходимо для правильного использования различных методов дозиметрии. Детально физическая стадия рассмотрена во многих работах (см., например, [7—141). Физико-химическая и химическая стадии кратко описываются при обсуждении химической дозиметрии.
1. Основные понятия и единицы
Важнейшей характеристикой ионизирующего излучения является его энергия. Единица энергии используемая обычно в радиационной химии,— это электронвольт (эв). Он равен энергии, прпобретаемой одним электроном (заряд 1,602-10 19 кулона) при прохождении разности потенциалов в один вольт. Ниже приводятся соотношения между электронвольтом и другими единицами энергии:
L ав = 1,602-10-12 эрг = 1,602- 10~1е дж\
1 ав/молек. = 6,02-1023 эв/.мо.1ь = 9,6-104 дж!моль — 23,05-10:! кал!молъ = = 23,05 ккал)молъ.
Электронвольт — сравнительно небольшая единица энергии. • Поэтому для характеристики энергии ионизирующих излучений, как правило, применяют единицы: килоэлектронвольт (кэв) и мегаэлектронвольт (Aloe), равные соответственно 103 и 106 ав. Излучения, используемые в радиационной химии, имеют энергию от '—0,1 нов до 20—30 Мэв.
Если ионизирующее излучение состоит из фотонов одинаковой энергии или частиц одного вида с одинаковой кинетической энергией, то такое излучение называется моноэнергетическим. Синоним этого термина — однородное излучение. В случае фотонного излучения часто используется также термин — монохроматическое излучение. Ионизирующее излучение, состоящее из фотонов или частиц различной энергии, называется немоноэнергетическим (или неоднородным).
Если фотоны или частицы в пучке имеют распределение по анергиям, то говорят об энергетическом спектре ионизирующего излучения. Различают «дискретный спектр излучения» (спектр состоит из отдельных линий) и «непрерывный спектр излучения». Вместо последнего термина применяется иногда термин — сплошной спектр излучения.
С 1 июля 1964 г. в Советском Союзе введен в действие ГОСТ 8848—63 «Единицы радиоактивности и ионизирующих излучений» fl5], основанный на системе СИ 116]. Применительно к ионизирующим излучениям этот стандарт установил единицы для следующих величин: плотности потока ионизирующих частиц или фотонов, интенсивности излучения, поглощенной дозы излучения, мощности поглощенной дозы излучения, экспозиционной дозы рентгеновского и у-излученпй и мощности экспозиционной дозы рентгеновского и у-излучений. ГОСТ 8848—63 допускает также применение единиц, рекомендованных Международной комиссией по радиологическим единицам (МЕРЕ) [17]. Эти единицы обычно называют внесистемными.
Рассмотрим отдельно каждую из названных величин и единицы их измерения.
Плотность Ф потока частиц или фотонов (квантов) — это отношение числа частиц или фотонов (квантов) ДУГ, проникающих за некоторое время Д/ в объем элементарной сферы, к площади поперечного сечения Д« этой сферы, т. е.
Единицей измерения этой величины стандарт установил число частиц или фотонов в 1 сек. на 1 м2. Однако в радиационных исследованиях чаще пспользуется единица — число частиц или фотонов в 1 сек. на 1 см2. Единицы измерения потока в этом случае записываются так: альфа-частиц/ (сек-см2), бета-частиц/ (сек-см2), нейтрон/ (сек-см2), гамма-фотон/ (сек-см2). Обычно слова «альфа-частиц», «бета-частиц», «нейтрон», «гамма-фотон» ради сокращения заменяют на соответствующие буквы греческого и латинского алфавитов: а, Р, п, у.
Интенсивность излучения I — это отношение энергии ДЕ ионизирующих частиц или фотонов, проникающих за некоторое время Д/ в объем элементарной сферы, к площади поперечного сечения As этой сферы, т. е.
ДЕ
/-ДюДГ (2)
В качестве единицы измерения интенсивности излучения установлен ватт на квадратный метр (вт/м2). На практике обычно используются единицы: Мэе! (см2-сек) или эрг! (см2-сек).
Поглощенная доза излучения D — это отношение энергии ионизирующих частиц или фотонов ДЕ, отданной ионизирующим из-
ного излучения, а Ат должно быть настолько мало, чтобы оно заметно не влияло на радиационное поле. Единицей кермы в системе СИ является джоуль на килограмм (дж/кг).
2. Общие закономерности радиоактивного распада
Один из источников ионизирующих излучений — распад радиоактивных изотопов. Поэтому представляется целесообразным рассмотреть кратко общие закономерности этого процесса.
Скорость радиоактивного распада атомов одинакового типа в каждый момент времени t пропорциональна количеству атомов N, т. е.
где X — постоянная распада (характеристическая константа), т. е. число атомов, распадающихся в 1 сек. Уравнение (6) представляет скорость процесса первого порядка.
Интегрируя уравнение (6) по времени от 0 до t, получаем
JV = Л'о<ГХг, (7)
где No и N — количество атомов в моменты времени 0 и t.
Уравнение (7) можно записать в виде
1п(ЛуМ|) = — It. (8)
Если In (N/Nn) отложить по ординате, a t — по абсциссе, то графически уравнение (8) изображается прямой линией с отрицательным углом наклона. Наклон тем болыпе, чем быстрее идет распад.
Важнейшей характеристикой радиоактивного изотопа является период полураспада, представляющий собой промежуток времени, за который распадается половина ядер, существовавших сначала. Его обозначения — t, 2, Ti2, Т или т. В настоящей книге принято обозначение ii2. Очевидно, по истечении 6/2 распадается половина имевшихся радиоактивных ядер, по истечении 2 62— снова половина, так что остается У4 часть от начального количества ядер. Можно дать следующее соотношение между уменьшением No и кратностью периода полураспада:
Кратность 012345 6 7 89 10
Число нераспавшпхся ядер, % 100 50 25 12,5 0,25 3,12 1,56 0,78 0,39 0,2 0,1
В зависимости от того, каким (коротко- или долгоживущим) является радиоактивный изотоп, Л/2 измеряется в секундах, минутах, днях пли годах. Изотопы, известные в настоящее время, имеют величины t,,. от 10'7 сек. до 10В * * 11 лет.
Из определения tv2 следует, что прп t = б-2 N =NJ2. Отсюда
In 2 0,693
Ч=~ = -г-- (0>
Другая важная характеристика радиоактивного изотопа — активность (обозначим ее буквой а). Она равна числу ядер изотопа, распадающихся в единицу времени, т. е.
Из уравнений (10) и (6) находим
а = -ХЛ. (И)
т. е. активность равна произведению постоянной распада на количество имеющихся радиоактивных ядер.
ГОСТ 8848 —63 в качестве единицы активности в радиоактивном источнике установил распад в секунду (расп/сек).
Внесистемной единицей активности является кюри, определяемая как активность препарата данного изотопа, в котором происходит 3,700-1010 актов распада в одну секунду. Применяются также единицы, дробные или кратные кюри", пикокюрп (пкюри) = = 3,7-10“2 расп/сек-, нанокюрп (нкюри) =37 расп1сек\ микрокюри {мккюри) =3.7• расп/сек-, милликюри (мкюри) = 3,7 107расп/сек', килокюри (ккюри) =3,7-1013 расп!сек и мегакюри (Мкюри) — = 3,7-1016 расп/сек.
Понятие «активность» часто применяют для обозначения количества радиоизотопа. Например, говорят: «1 кюри 60Со», что строго означает: «количество ®°Со с активностью 1 кюри». Отметим также, что число ядерных превращений не всегда соответствует числу испущенных частиц п еще реже — числу испущенных у-кваптов. Активность характеризует лпшь скорость ядерных превращений.
у-Излучателп часто характеризуют по понизацпп, производимой у-лучами в воздухе и измеренной в одинаковых условиях. С этой целью используется величина — гамма-эквивалент. Единицей его является миллиграмм-эквивалент радия (мг-экв радия). Один мг-экв радия — это гамма-эквпвалент радиоактивного препарата, у-пзлучепие которого при данной фильтрации и тождественных условиях измерения создает такую же мощность экспозиционной дозы, как п у-пзлученпе 1 мг Государственного эталона радия СССР в равновесии с основными дочерними продуктами распада при платиновом фильтре толщиной 0,5 мм. Гамма-эквивалент М изотопа (мг-экв радия) связан с его активностью а (мкюри) соотношением
Л/=а7<у/8,4, (12)
где К — ионизационная гамма-постоянная [ее размерность — р • см2/(час мкюр и) ].
Цифра 8,4 в формуле (12) представляет собой мощность экспозиционной дозы (p/час), измеренную на расстоянии 1 см от источника радия активностью 1 мкюри, находящегося в равновесии с основными дочерними продуктами распада и окруженного платиновым фильтром толщиной 0,5 мм. Для 6РСо к 137Cs, на ходя
щих широкое использование в радиационной химии в качестве источников у-из л учения, К-( равны соответственно 12,93 и 3,10 р см2 / (час • мкюри) [12].
Легко найти число ядер N изотопа, соответствующее 1 кюри.
В этом случае из уравнения (11) имеем
X2V = 3,7-1010 расп/сек.
Тогда
3 7-1010 3,7-Ю10/,,
-v= = -ода— = 5,34-10^, (13)
где измерено в сек.
Масса М изотопов, соответствующая 1 кюри, равна (в скобках указаны единицы измерения й2)
NA , .
Л/ = g 02-1023 = 8186- 10-!МЧ (сек.) = 5,3• 1СНМt,(мпн.) =
= 3,2-10~10Л^2(час.) = 7,7-10-вЛ(1д(дней) = 2,8-10-Mt, 2(лет), (14)
где А — атомный вес изотопа.
Нетрудно показать, что активность аг (в кюри) 1 г любого радиоактивного изотопа равна
1,13-1013 1,88-1011 3,110е 1,310» 3,57-103 * 5 * * * *
Я1 — ^4(t/s (сек.) — At,,f (мин.) — . 1f,,2 (час.) “ Л/„2 (дней) ~ At, 2 (лет) ’
(15)
Следующее понятие — удельная активность. Это — активность на единицу массы или объема (например, кюри/г, кюри/л и т. п.).
Существует также понятие — средняя продолжительность жизни радиоактивного изотопа. Обозначим ее символом т. Очевидно,
т = 1/Х = 1,443А,2. (16)
Величина т равна времени, по истечении которого начальная активность уменьшится в 1/е раз (т. е. до 36,8%).
3. Фотонное излучение
Фотонное излучение, применяемое в радиационной химии,
имеет энергии от ~ 0,1 кэв до ~ 20—30 Мэв. Его часто характе-
ризуют также длиной волны. Соотношение между длиной волны
X и энергией Е таково:
Я = йс/Х, (17)
где h —постоянная Планка (6.63-10-27 эрг-сек)-, с —скорость
света (2,998* 1010 см/сек).
Таблица 3
Характеристики различных видов излучении
Излучение Длина о волны, А Энергия, эв Излучение Длина о волны, А Энергия, эв
Инфракрасное Видимое Ультрафиолетовое >8000 8000—4000 4000—1000 <1,6 1,6—3,1 3,1—12,4 Рентгеновское Т-Излученме <1000 <0,3 >100 >4-10‘
После подстановки численных значений формула (17) преобразуется к виду
£=12400/7., (18)
где Е выражена в эв, а X — в А (1 А = 10-8 см).
Длину волны фотонного излучения в ряде случаев измеряют в Х-единицах. Одна Х-единнца равна 10-11 см.
В радиационной химии используют два вида фотонного излучения: у-излучение и рентгеновское излучение. В табл. 3 с целью сравнения приведены характеристики обычного светового излучения, рентгеновского излучения и у-излучения.
Отметим, что, основываясь на энергетических характеристиках, резкой границы между ультрафиолетовым светом и рентгеновским излучением провести нельзя. Так, свет способен производить ионизацию некоторых веществ (процесс фотоионизации). Различие здесь состоит в том, что электрон, образующийся в этом процессе, не обладает энергией, достаточной для дальнейшей ионизации. Рентгеновские и у-лучи генерируют электроны, имеющие такую энергию. В этом случае в результате одного акта поглощения энергии средой возникает несколько пар ионов.
у-Из.гу чение
у-Излучение возникает при распаде радиоактивных изотопов. Оно возможно только при переходе ядра из возбужденного энергетического состояния в более низкое состояние (в частности, при переходе из возбужденного состояния в основное). у-Лучи моно-хроматичны или же имеют небольшое число дискретных энергий.
Поскольку у-лучи образуются при возвращении возбужденного ядра в более нпзкое энергетическое состояние, то энергия у-л у чей не может быть выше ~ 3 Мэв. Если энергия возбуждения ядра выше этого значения, то ядро распадается с испусканием нуклона или а-частицы.
Наиболее часто в радиационной химии используется у-излу-чение 60Со и 137Cs. Первый изотоп получают в ядерном реакторе
по реакции
69Со + п -> G0Co -)- г. (19)
137Cs выделяют из продуктов реакций деления, осуществляемых в ядерном реакторе.
С0Со имеет период полураспада 5,27 года *. Схема его распада такова:
«'Со—(20) Сначала при распаде атома G0Co выделяется [3-частица (максимальная энергия 0,308 Мэе) и образуется атом 6UNi. [3-Частицы полностью поглощаются в материале контейнера, в который помещен препарат ®°Со. Атомы fi0Ni выделяют примерно равное число у-кв антов с энергиями 1,332н 1,173Мэв, так что средняя энергия у-кв антов, образующихся при распаде С0Со, равна 1,25 Мэв.
Период полураспада 137Cs составляет 30 лет. Схема его распада следующая:
137Cs— ->13713а. (21)
Максимальная энергия [3-частпц, образующихся в этом процессе, равна 1,18 Мэв (8%) и 0,52 Мэв (92%), а энергия у-квантов — 0,6616 Мэв (82%).
В качестве источников у-излучення иногда применяются также отработанные тепловыделяющие элементы (твэлы) из ядерных реакторов и радиационные контуры при ядерных реакторах.
На раннем этапе развития радиационной химии в качестве источника -р-изл учения использовался радий, помещенный в контейнер с толщиной стенок, достаточной для полного поглощения [3- и а-частиц. В настоящее время радий как источник у-из л учения не применяется.
Рентгеновское излучение
Заряженные частицы, испытывающие ускорение прп взаимодействии с электрическим полем электронов атома или полем ядер, теряют свою энергию, испуская электромагнитное излучение. Это излучение называется тормозным. Потерн энергии на тормозное излучение существенны только для быстрых электронов. Если же тормозятся тяжелые заряженные частицы, то потери их энергии на тормозное излучение малы. Эти потерн в (т/М)2 раз меньше по сравнению с потерями для электронов (т и М — массы электрона и тяжелой заряженной частицы).
Для торможения электронов обычно используют мишени — материалы с высокими атомными номерами (чаще всего —вольфрам).
* Здесь и далее характеристики радиоактивного распада изотопов приводятся по книгам [18, 19].
Тормозное излучение испускается в виде непрерывного спектра с энергией квантов почти от нуля до максимальной энергии тормозящихся частиц. На рис. 1 в качестве примера приведен энергетический спектр тормозного излучения, полученного торможением электронов с энергией 0,25 Мэв вольфрамовой мишенью [20]. На этом рисунке два пика при 58 и 70 кэв соответствуют характеристическому излучению вольфрама. Это излучение обусловлено электронами, переходящими с внешних оболочек атомов материала мишени на вакантные внутренние оболочки. В случае
Рис. 1. Энергетический спектр рентгеновского излучения, полученного торможением электронов с энергией 0,25 Мэв в вольфрамовой мишени
I — интенсивность,
Ь’ — энергия излучения
вольфрама линии при указанных энергиях соответствуют переходам между внешними оболочками и /f-оболочкой. Однако интенсивность характеристического излучения составляет лишь малую часть общей интенсивности.
Как уже говорилось, совокупность тормозного и характеристического излучения называют рентгеновским излучением *.
Из рис. 1 видно, что значительная часть рентгеновских лучей имеет низкую энергию. Очевидно, эта часть поглощается в поверхностном слое облучаемого вещества. Вследствие этого распределение поглощенных доз в образце будет неравномерным. Для того, чтобы распределение стало более равномерным, используют металлические фильтры, помещаемые между выходным окном машины, генерирующей излучение, и образцом.
Рентгеновское излучение обычно подразделяют па длинноволновое (эффективная длина волны более 0,25 А или энергия менее 50 кэв) п коротковолновое (эффективная длина волны менее 0,25 А или энергия более 50 кэв). Часто вместо этих терминов используют соответственно термины мягкое и жесткое излучение. В радиационной химии длинноволновое рентгеновское излучение применяется крайне редко.
Для тормозного излучения существует понятие — граничная длина волны. Это — наименьшая длина волны в его спектре. Она, очевидно, соответствует максимальной энергии в спектре.
* В английской литературе это излучение называется Х-пзлучением.
Взаимодействие фотонного излучения с веществом
Для фотонов с энергией от 20 кэв до 10 Мэе возможны следующие процессы взаимодействия с веществом: фотоэлектрический эффект, комптоновское (некогерентное) рассеяние, образование электронно-позитронных пар, томсон-рэлеевское (когерентное) рассеяние, флуоресценция, тормозное излучение, аннигиляционное излучение, когерентное рассеяние на молекулах, потенциальное (дельбруковское) рассеяние, томпсоновское рассеяние на ядрах, ядерное резонансное рассеяние, ядерный фотоэффект *. Основными из них являются первые три процесса.
Фотоэлектрический эффект. Фотоэлектрическим эффектом (фотоэффектом) называется процесс взаимодействия фотонного излучения со связанным электроном, при котором вся энергия первичного фотона hv поглощается атомом и из последнего выбивается электрон с энергией
E=hv — Ec, (22)
где Ес — энергия связи выбитого электрона в атоме. Схематически этот процесс показан на рис. 2.
Фотоэлектрон е~
hv
Задающий ротон
Рис. 2. Схема фотоэлектрического эффекта
В результате выбивания связанного электрона в атоме появляется свободный уровень. Он заполняется одним из наружных электронов, и акт поглощения фотона заканчивается испусканием вторичного мягкого характеристического излучения, называемого флуоресценцией. Это излучение не имеет места в том случае, когда энергия возбуждения атома передается одному из электронов. Это — явление Оже, и испускаемые при зтом электроны называют электронами Оже. Флуоресцентное излучение наблюдается в материалах с большим атомным номером. В материалах с низким атомным номером преобладает испускание электронов Оже.
Главную роль в фотоэффекте играют электроны Л'-оболочки, так как они имеют наибольшую энергию связи. Если энергия фотона меньше энергии связи электрона 7х-оболочки, то может
* Когерентное (или упругое) взаимодействие — это процесс, при котором сумма кинетических энергий взаимодействующих частиц до взаимодействия и после него остается неизменной. Некогерептное (неупругое) взаимодействие — это процесс, при котором часть кинетической энергии взаимодейст-вующей системы передается образовавшимися свободными частицами или квантами.
быть выбит электрон с L- и более высоких оболочек. Поэтому сечение* фотоэффекта имеет скачки при энергиях фотонов, равных энергиям связи электронов. На рис. 3 в качестве примера приведена зависимость сечения фотоэффекта для свинца от энергии фотона [8].
Вероятность фотоэффекта возрастает с увеличением атомного номера материала и уменьшается с ростом энергии фотона. В ча
стности, вероятность этого процесса уменьшается при увеличении энергии Е фотона приблизительно по закону Е~э (при Е < 0.5 Мэв) и по закону Е~г (при Е 0,5
Мэв).
Подчеркнем, что в ослаблении фотонного излучения при прохождении его через среду фотоэффект играет преобладающую роль для излучений с энергией ниже 0,1 Мэв и для сред с атомным номером более 20.
Комптоновское рассеяние. Комптоновским рассеянием называется процесс взаимодействия фотонного излучения с веществом, в котором фотон в результате упругого столкновения с электроном теряет часть своей энергии и изменяет направление своего первоначального движения, а из атома выбивается электрон'отдачи. В ли-
тературе встречаются и другие Рис. 3. Зависимость сечения фотоэф-названияэтого процесса: эффект фекта о для свинца от энергии фото-Комптона, некогерентное рас- па ЕФ
сеяние. Электрон отдачи ча-
сто называют также комптоновским электроном. Схематически процесс комптоновского рассеяния показан на рис. 4.
Энергия Е электрона отдачи равна
Е = hvo — hv,
(23)
где hvn — энергия первичного фотона, a hv — энергия рассеянного фотона.
* Вероятность протекания того или иного процесса характеризуется сечением, которое измеряется в см2. 10~24 см2 составляет барн. Сечение означает отношение числа элементарных актов какого-либо процесса в 1 сек. на атом, ядро или частицу к числу падающих частиц на 1 см2 в 1 сек. Оно обычно обозначается букьой а.
фотонов связанными атомными электронами, при котором атом ни возбуждается, ни ионизируется. Этот процесс протекает, в основном, в случае фотонов низкой энергии и материалов с большими атомными номерами. Рассеянные фотоны имеют ту же длину волны, что и первичные, т. е. материалу не передается никакой энергии. Имеет место только ослабление первичного пучка.
Фотораспад ядер (или ядерный фотоэффект) — это процесс взаимодействия фотонного излучения с ядрами, приводящий к испусканию нейтрона, протона или а-частицы. Он имеет место в том случае, когда энергия фотона превышает энергию связи соответствующей частицы в ядре. Фотораспад характерен для фотонного излучения высокой энергии (выше 10—15 Мэв для материалов с низкими атомными номерами и выше ~ 7 Мэв для материалов с большими атомными номерами). Исключение составляют реакции (у, п) для дейтерия и бериллия, которые начинают протекать с энергией фотонов 2,23 и 1,6(55 Мэв соответственно (см. также стр. 37).
Ослабление интенсивности
и поглощение энергии фотонного излучения при прохождении через вещество
В результате протекания различных процессов взаимодействия с веществом интенсивность потока фотонного излучения при прохождении через вещество уменьшается. Возьмем узкий монохроматический пучок фотонов, падающий на прибор-детектор, измеряющий интенсивность (рис. 8). Если между этим прибором и источником излучения находится какой-либо поглотитель толщиной AZ, помещенный в пучок, то интенсивность I пучка при прохождении через этот поглотитель уменьшится на величину А/,
А/-_- pJAZ, (28)
где ц — коэффициент пропорциональности, называемый линейным коэффициентом ослабления.
После перегруппировки уравнения (28) и интегрирования его от 0 до Z получаем
I = 7oe-l^, (29)
где 10 — начальная интенсивность излучения.
Уравнение (29) выражает общеизвестный экспоненциальный закон ослабления.
Для пучка фотонов с различными энергиями закон ослабления выражается формулой
I = Zie-W 4. /2e-W -р ... + (30)
где 7Ъ 12, . . ., I; — начальные интенсивности фотонов с энерги
ями Et, Ег, . . ., Et, а щ, ц2, . . ., рг — соответствующие линейные коэффициенты ослабления.
Уравнение (29) было выведено для узкого параллельного пучка фотонов. В этом случае детектор регистрирует только первичпое нерассеянное излучение. Если же пучок широкий, то детектор, помимо нерассеянных фотонов, регистрирует также фотоны, многократно рассеянные в поглотителе (рис. 9). Говоря по-иному,
Рис. 8. Ослабление узкого монохроматического пучка фотонов при прохождении через вещество
1 — источник излучения; 2 — коллиматор; з — поглощающая среда; 4 — прибор-детектор
Рис. 9. Ослабление широкого пучка фотонов при прохождении через вещество Обозначения те же, что на рис. 8
детектор будет измерять большее количество фотонов. Вклад многократно рассеянного излучения по сравнению с первичным излучением учитывается с помощью так называемого фактора накопления (англ.— buildup factor). Его обозначают буквой В. Тогда для случая широкого пучка уравнение (29) трансформируется к виду
f = BIoe-{xl. (31)
Если I измеряется в см, то ц имеет размерность см'1. На практике чаще используют массовые коэффициенты ослабления ц/р, где р — плотность поглощающего вещества в г!см?. Очевидно, р/р не зависит от плотности и имеет размерность см?!г.
По определению МКРЕ [21], массовый коэффициент ослабления для косвенно ионизирующих частиц — это частное от деления dN па произведение р, N и dl, где N —число частиц, падающих перпендикулярно на слой материала с толщиной dl и плотностью р, a dN — число частиц, которые осуществляют взаимодействие в этом слое. Следовательно,
р 1 dN
Р ~~ PN dl ' <32>
При этом термин «взаимодействие» относится к любым процессам, в которых происходит изменение энергии или направления косвенно ионизирующих частиц.
При использовании р/р уравнение (29) имеет вид
/ = /oe-(W₽),n, (33)
где т — масса столбика поглощающего вещества с сечением 1 см2 и толщиной I.
При выводе рассмотренных соотношений предполагалось, что пучок у-лучей проходит через вещество, состоящее из одного сорта атомов (например, через металл). Если вещество имеет сложный химический состав, то р/р для него равно
Р Pi , Р* , ,
Т==/’1'рГ + ^'рГ + -" + Л^-’ С34)
где pj/pi, р^/Рг» • • •» Р;/р; —массовые коэффициенты ослабления различных составных частей, а р2, . . ., рг — их весовьте доли.
Для характеристики поглощающей способности среды относительно фотонного излучения часто применяют термин — толщина (или слой) половинного ослабления. Это — толщина поглощающего вещества, которая уменьшает интенсивность пучка в 2 раза. Она обычно обозначается символом di2. В табл. 4 приведены значения du, для некоторых веществ в случае у-лучей 60Со и 137Cs (20, 221.
Таблица 4
Толщины половинного ослабления (в слг) для узкого пучка у-лучей
Изотоп Вода Алюминий Бетон * Свинец
«"Со и 4,6 5,2 1,06
lr<7Cs 8,1 3,4 3,8 0,57
* Уд. вес 2,35 g/CJH3.
Из уравнения (29) легко вывести соотношение между р и При I = d,/2 I равно /0/2. Тогда
7о/2 = /ое-И<11/г (35)
и elidl/l=2, (36)
т. е. ц = 0,693/d1/2. (37)
Фотоны не имеют определенных величин пробегов. Поэтому
используется термин — средний пробег В фотонов в веществе.
Величины /?, ц и сА/2 связаны соотношениями
Л = 1/р, (38)
Я = 1,443а,/г. (39)
В случае рентгеновского излучения dv, —это толщина поглотителя, ослабляющего интенсивность нефильтрованного излучения в 2 раза. При прохождении этого вида излучения через поглотитель длипноволновая часть спектра ослабляется сильнее коротковолновой. Поэтому для фильтрованного излучения dv2 будет больше, чем для нефильтрованного. Возрастание dve после фильтрации — относительная особенность рентгеновских лучей по сравнению с у-лучами. Для последних di/2 не изменяется при ослаблении пучка.
В приложении к рентгеновскому излучению существует понятие — степень (коэффициент) однородности (или неоднородности). Степень неоднородности — это отношение dv2 для фильтрованного излучения к d./,, для нефильтрованного излучения. Обратное отношение называется степенью однородности. Очевидно, для моноэнергетического излучения эти отношения равпы единице; для немоноэнергетического излучения степень однородности всегда < 1. а степень неоднородности — > 1.
Для рентгеновского излучения применяется термин — эффективный линейный коэффициент ослабления р. Если толщина половинного ослабления некоторого немопохроматического излучения такая же. как у монохроматического с коэффициентом ослабления ц. то помонохроматическому излучению приписывают эффективную длину волны Х.1фф, равную длине волны монохроматического излучения [231. Существуют и другие определения понятия Х.,фф. Например, Хяфф равна такой длине волны, которая делпт спектр излучения на две части так, что суммарная интенсивность излучения с ZZ Хафф равна интенсивности излучения с X ^эфф [231. Понятие' часто вводится в связи ^ослаблением мощности дозы в пучке при его фильтрации. В этом случае Х1фф равна длине волны' такого однородного пучка рентгеновских лучей, мощность дозы в котором ослабляется при прохождении некоторого слоя облучаемой среды во столько же раз, во сколько ослабляется интенсивность данного неоднородного пучка.
В зависпмостп от энергии фотонного излучения и атомного номера материала в ослаблении интенсивности пучка фотонов играет главную роль одни из трех рассмотренных выше процессов взаимодействия излучения с веществом. В табл. 5 приведены диапазоны энергии фотонов, в которых доминирует один из этих трех процессов И21.
Каждый из трех основных процессов взаимодействия фотонного излучения с веществом характеризуется соответствующими Линейным и массовым коэффициентами ослабления.
Таблица 5
Диапазоны энергий Е фотонов, в которых преобладает один из трех основных процессов
Вещество Фотоэффект Комптоновское рассеяние Образование пар
Воздух Е < 20 кэв 20 кэв Е <^23 Мэв Е > 23 Мэв
Алюминий £ < 50 кэв 50 кэв < Е < 15 Мэв Е > 15 Мэв
Железо Е 120 кэв 120 кэв < Е <9,5 Мэв £>9,5 Мэв
Свинец Е < 0,5 Мэв 0,5 Мэв <Е <i,7 Мэв £>4,7 Мэв
Линейные коэффициенты ослабления обозначаются символами т, и и х соответственно для фотоэффекта, комптоновского рассеяния и образования пар. Обозначения массовых коэффициентов ослабления таковы: т/р, о/p и х/р (р — плотность материала в г/см3). Очевидно,
P=t + s + x, (40)
Линейный коэффициент ослабления о состоит из двух частей. Одна часть os характеризует передачу энергии начального фотона рассеянному фотону, а другая часть оа — передачу энергии начального фотона электрону отдачи. Тогда
с = с6 + са- 02)
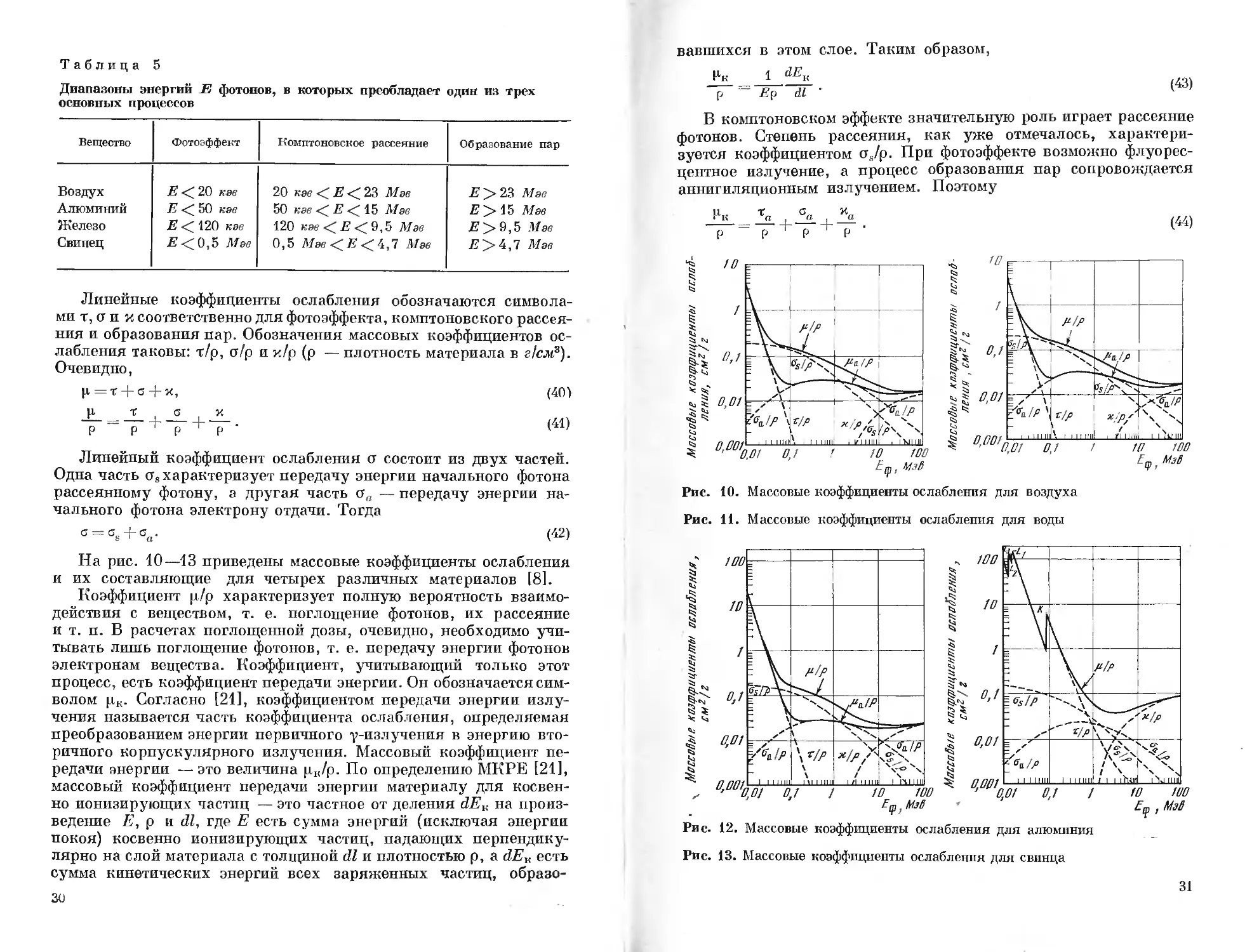
На рис. 10—13 приведены массовые коэффициенты ослабления и их составляющие для четырех различных материалов [8].
Коэффициент ц/р характеризует полную вероятность взаимодействия с веществом, т. е. поглощение фотонов, их рассеяние и т. п. В расчетах поглощенной дозы, очевидно, необходимо учитывать лишь поглощение фотонов, т. е. передачу энергии фотонов электронам вещества. Коэффициент, учитывающий только этот процесс, есть коэффициент передачи энергии. Он обозначается символом р,к. Согласно [21], коэффициентом передачи энергии излучения называется часть коэффициента ослабления, определяемая преобразованием энергии первичного у-излучения в энергию вторичного корпускулярного излучения. Массовый коэффициент передачи энергии —это величина цк/р. По определению МКРЕ [21], массовый коэффициент передачи энергии материалу для косвенно ионизирующих частиц — это частное от деления dEK на произведение Е, р и dl, где Е есть сумма энергий (исключая энергии покоя) косвенно ионизирующих частиц, падающих перпендикулярно на слой материала с толщиной dl и плотностью р, a dEv есть сумма кинетических энергий всех заряженных частиц, образо
вавшихся в этом слое. Таким образом,
Нк___1 dEu
р Ер dl
(43)
В комптоновском эффекте значительную роль играет рассеяние фотонов. Степень рассеяния, как уже отмечалось, характеризуется коэффициентом сц/р. При фотоэффекте возможно флуоресцентное излучение, а процесс образования пар сопровождается аннигиляционным излучением. Поэтому
Рис. 11. Массовые коэффициенты ослабления для воды
Рис. 13. Массовые коэффициенты ослабления для свинца
Рис. 12. Массовые коэффициенты ослабления для алюминия
При этом
Ха t [ б \
Р Р \ /'V / ’
ха х‘ / 2тс2„
---- — ----I ___J_____
i р р \ “ /гт
(45)
(46)
где 6 — средняя энергия, выделившаяся с флуоресцентным излучением, в расчете на фотон, поглощенный при фотоэффекте; hv — энергия падающего фотона; 2mc~/kv — поправка на излучение при аннигиляции позитрона.
В литературе часто встречается термин «массовый коэффициент поглощения» (обозначение рц/р). Он равен
Ид____т ад V.
Р — Р 1 р р
(47)
Уравнения (44) и (47) не учитывают образования тормозного излучения. Этот процесс в случае фотонов очень высокой энергии и материалов с большим атомным номером может привести к существенному уменьшению количества энергии, поглощенной материалом. В большинстве других случаев роль этого процесса ничтожна.
С целью учета образования тормозного излучения используется массовый коэффициент поглощения энергии (его обозначение — Р-еп/р)- Он равен
Hen
Ик
—с1-4 * 6)-
(48)
где G — доля энергии вторичных заряженных частиц, которая расходуется на образование тормозного излучения в материале.
Вопрос о коэффициентах [х„/р, |лк/р н Реп^Р обсуждается также в главе IV. Там же приведены соответствующие таблицы.
Для химических соединений и их смесей значения p,G/p, р.к/р и реп/р находятся из величин этих коэффициентов для элементов их составляющих. Расчет производится тем же способом, что и в случае р/р.
Таким образом, при прохождении фотонного излучения через вещество главным процессом является образование вторичных электронов. Последние осуществляют ионизацию и возбуждение атомов и молекул среды, что в конечном итоге и приводит к радиационно-химическим превращениям.
4. Корпускулярное излучение
Корпускулярные излучения образуются в результате внутри
ядерпых процессов или же генерируются специальными машинами.
Электроны
Электрон — отрицательно заряженная частица (заряд —1; он равен 1,602-10-19 кулона)-, его масса покоя равна 1/1837 массы покоя протона. В зависимости от типа процесса, в котором образуются электроны, в литературе используются названия: Р~лучи (или P-частицы) и быстрые электроны.
Р-Луч — электрон, возникающий в результате распада радиоактивного изотопа. Однако в настоящее время этот термин почти не используется; вместо него широко распространен термин «р~ча-стица». В табл. 6 приведены характеристики некоторых изотопов, применяемых в радиационной химии в качестве источников р-частиц.
Таблица 6
Характеристики изотопов — источников Р-частиц
Изотоп Максимальная энергия р-частиц, Мэв Средняя энергия р-частип, Мэв Период полураспада
Тритий Сера-35 Фосфор-32 Равновесная смесь строиций-90 4-+ иттрий-90 0,0186 0,167 1,710 B0Sr 0,546; 8(IY2,27 0,0056 0,0488 0,70 90Sr 0,205; 80 * * * * * * * * * 90Y 0,93 12,262 года 87,9 дня 14,28 дня 9(lSr 28 лет; 90Y 64 часа
80Sr выделяют из продуктов реакций деления, осуществляемых в ядерном реакторе. Он распадается по следующей схеме:
"Sr —-"Y —-* "Zr. (49)
Изотопы 3Н, 35S и 3аР получают в ядерном реакторе в результате процессов
6Li + п —>3Н + а, (50)
35Cl + n->35S-f-p, (51)
MS + n—зз8 + г, (52)
3]Р + п— 32Р + т, (53)
sas + n —88Р+р. (54)
Схемы распада этих изотопов таковы:
3Н — ->3Не, (55)
35S—-«Cl, (56)
32pJU:ffiS. (57)
P-Частицы, испускаемые радиоактивными изотопами, имеют непрерывный спектр энергий, простирающийся от нуля до макси-
2 А. К. Пикаев
33
мальной энергии. Это явление вызвано тем, что при Рраспаде энергия распределяется между электронами и возникающими одновременно с ними антинейтрино. Последние не имеют ни массы, ни заряда; их воздействие на вещество, через которое они проходят, ничтожно.
Максимальную энергию -Ешах имеет очень малое число |3-ча-стиц, образующихся при радиоактивном распаде. Поэтому гораздо более важной характеристикой является средняя энергия Ё р-частицы. Она определяется из выражения
•Emax
У EN (Е) dE
---------- max J N(E)dE О
(58)
где N (Е) — число р-частпц с энергией между Е и Е + dE. Средняя энергия Р-частицы равна примерно одной трети максимальной энергии. С точностью до ~10% Ё можно рассчитать из Етях с помощью следующих формул:
Е = 0,ЗЗЕтах при
Е =0,43 (Ятах — 0,14) при
Ё = 0,59 (£шах — 0,5) при
^тах<0,6 Мае, 0,6<Дтат<1,2 Мае, ^'тах^'^’З Мае.
(59)
(60)
(61)
На рис. 14 и 15 приведены в качестве примера спектры энергии Р-частиц, образующихся при распаде 32Р и 90Sr + 90Y [20]. Для
Рис. 14. Энергетический спектр
N — число, Е — энергия частиц
Рис. 15. Энергетический спектр (3-частиц 80Sr + 80Y N — число, Е — энергия частиц
пары изотопов 90Sr -| 90Y Е равно 1,13 J/эв. Это значение представляет собой сумму Е для 90Sr (0,205 Мэв) и 90Y (0,93 Мэв).
Быстрые электроны — это электроны, генерируемые специальными машинами (ускорителями). В этих машинах (более подробно они рассматриваются в главе II) электроны испускаются нака
ленной металлической нитью, лентой или спиралью (катодом) и затем ускоряются до требуемой энергии. Иногда быстрые электроны, имеющие сравнительно низкую энергию, называют катодными лучами. Быстрые электроны, как правило, характеризуются мо-ноэнергетичностыо. В радиационной химии они используются гораздо чаще, чем [3-частицы.
Античастицей электрона является позитрон (или р+-частица), обладающий той же массой и тем же (по противоположным по знаку) зарядом. Позитроны образуются при распаде некоторых изотопов (например, 64Сн и 22Na). В радиационной химии эти частицы не используются.
Тяжелые заряженные частицы
К тяжелым заряженным частицам относят быстрые протоны, дейтроны, гелионы, а-частицы. Их свойства приведены в табл. 7.
Таблица 7
Свойства тяжелых заряженных частиц
Частица Символ Заряд Масса покоя
а.е.м. * г-10“»
Протон р пли Н+ +1 1,00728 1,672
Дейтрон d или D+ +1 2,01410 3,344
а-Частица а +2 4,00273 6,644
Гелион Не2* +2 4,00273 6,644
* Атомная единица массы (масса ядра ,гС равна 12 а.е.м.).
Протоны (ядра атомов водорода), дейтроны (ядра атомов дейтерия) и гелионы (ядра атомов гелия) получают с помощью специальных машин (ускорителей). а-Частицы (ядра атомов гелия) образуются в процессах радиоактивного распада некоторых изотопов *. Опн моноэнергетичны. В табл. 8 приведены характеристики изотопов, применяемых в радиационной химии в качестве источников а-частпц.
Из числа изотопов, приведенных в табл. 8, наибольшее применение в радиационной химии находит 210Ро. Это — естественный радиоактивный изотоп. Однако с целью приготовления на его основе источника а-пзлучения его обычно получают в ядерном реакторе по реакциям:
2n9Bi + л -»210Bi + т, (62)
210Bi-^210po + p. (63)
* Иногда этот вид ионизирующего излучения называют а-лучами.
Таблица 8
Характеристики изотопов — источников «-частиц
Изотоп Период полураспада Энергия а-частиц, Мэв
Полонпй-210 138,4 дней 5,305
Радий-226 1,62-103 лет 4,777 (94,3%); 4,589(5,7%)
Радон-222 3,83 дней 5,49
Плутоний-239 2,44-Ю4 лет 5,1
Америций-241 458 лет 5,5
Период полураспада 210Bi равен 5 дням. Распад 210Ро происходит по схеме:
21ор0 20бр]; _
(64)
Нейтроны
Нейтрон (обозначается буквой п) — электрически нейтральная частица с массой 1,675*10~24 г или 1,00867 а.е.м. Он несколько тяжелее протона (см. табл. 7).
Нейтрон — нестабильная частица. Он распадается на протон, электрон и антинейтрино с й, ~ 12 мин. В одном акте распада выделяется энергия 0,78 Мэе. В настоящее время различными методами могут быть получены нейтроны с энергиями от 10-2 до ~ 4-10е эе.
Одним из важнейших источников нейтронов являются ядер-ные реакции, осуществляемые с помощью тяжелых заряженных частиц. Под действием а-частиц радиоактивных изотопов почти во всех легких элементах (с порядковым номером Z 20) протекает реакция (а, п). В общем виде эту реакцию можно записать следующим образом:
л/Ч-4Не-^л+3(2 + 2) + п + <2, (65)
где А — массовое число элемента и Q — энергия реакции.
Реакция (65) является экзотермической для бериллия, бора, углерода-13, кислорода-17, магния-25 и магнпя-26 [13]. Для приготовления источников нейтронов чаще всего используются бериллий и бор (реже). Порошки этих элементов смешиваются с радиоактивными а-из луча телями (радием, полонием, плутонием и др.). Иногда применяются химические соединения радиоактивного элемента с легким изотопом (например, РпВе13). Некоторым преимуществом характеризуется источник полоний + бериллий. Для него весьма низок фон у-излучения.
Энергетический спектр нейтронов, генерируемых бериллиевыми источниками, простирается от очень низких энергий до максимальных энергий, которые несколько меньше суммы энергий а-частиц и реакции. На рис. 16 приведены спектры нейтронов, испускаемых некоторыми бериллиевыми источниками 113].
Описанные радиоизотопные источники дают пучки нейтронов низкой интенсивности (— 10Б—107 п!се.к). Поэтому они практически не применяются в радиационной химии.
Рис. 16. Энергетические спектры нейтронов, генерируемых некоторыми источниками
1 — Ро 4- ве;
2 — Ra 4- ве;
8 — Ри 4- ве.
N — число.
Е — энергия нейтронов
Для получения нейтронов используются также нейтронные генераторы, в которых нейтроны образуются в результате действия ускоренных тяжелых частиц па некоторые элементы. Чаще всего с этой целью применяются реакции: 3H(d, и)4Не; 2H(d, п) sHe; 7Li(d, п) 8Ве; ®Be(fl!, zi)wB; 12C(d, ?i)13N. Из их числа наибольшее распространение получили первые две реакции. В них возникают почти моноэнергетические нейтроны: реакция 3H(d, п) 4Не дает нейтроны с энергией 14 Мэв, а реакция 2H(d, м)3Не — нейтроны с энергией 2,5 Мэв.
Нейтроны образуются и в -у,и-реакциях, когда энергия у-кванта превышает энергию связи нейтрона в ядре. Минимальная энергия падающего па мишень излучения, которая необходима для инициирования ядерной реакции, называется пороговой. Например, для 2Н и ®Ве пороговые энергии у,п-реакпий равны соответственно 2,23 и 1,67 Мэв. Источники, в которых для генерации нейтронов используются у,лг-реакцип, называются фотонейтрон-ными. Из их числа наибольшее употребление получили сурьмяно-бериллиевые источники. В них источником у-излученпя является 124Sb (энергия у-лучей 1,7 Мэв, = 60 дней). Образующиеся нейтроны имеют среднюю энергию, равную 0,024 Мэв [13].
Для генерации нейтронов вместо у-излучения может быть использовано и жесткое рентгеновское излучение, возникающее при торможении электронов в мишенях. Этот метод используется для получения нейтронов высокой энергии.
Наиболее интенсивные потоки нейтронов получают при делении тяжелых ядер в атомном реакторе. Нейтроны, сопровождающие деление, подразделяют на мгновенные и запаздывающие. Более 99% нейтронов испускается в пределах длительности самого процесса деления. Это — мгновенные нейтроны. Они имеют
широкий спектр энергий (от нескольких килоэлектронвольт до 18 Мэе), причем максимальное число нейтронов приходится на энергию — 1 Мэв. Запаздывающие нейтроны связаны с [3-распадом некоторых осколков деления (изотопов брома и иода), энергия возбуждения дочерних продуктов которых превышает энергию связи нейтронов. Выход этих нейтронов по сравнению с мгновенными нейтронами очень мал.
Одной из важнейших характеристик нейтронов является их энергия. Еще не существует общепринятой классификации нейтронов по энергиям. В табл. 9 в качестве примера приведена классификация нейтронов, данная в книге [12].
Таблица 9
Классификация нейтронов по энергиям
Нейтроны Диапазон энергий Нейтроны Диапазон энергий
Холодные Тепловые Надтепловые Медленные ЕС 0,026 эе 0,01 <£<0,1 66 0,1 <£< 100 эе 0,1 < £ < 103 эе Промежуточные Быстрые Очень быстрые Сверхбыстрые 1,0 < £ < 500 кэв 0,5<£< 10 Мэе 10<£<50 Мэе £>50 Мэв
Другие виды корпускулярного излучения
В радиационной химии находят применение также продукты некоторых ядерных реакций деления. Среди них важнейшими являются продукты реакций 6Li(n. а)Т и 10В(7г. a)7Li, т. е. а-частицы и атомы отдачи трития в первой реакции и а-частпцы и атомы отдачи 7Li во второй реакции. Эти реакции происходят под действием тепловых нейтронов. Они имеют весьма большие сечения.
Энергии а-частиц, образующихся в указанных реакциях, равны соответственно 2,05 и 1,50 Мэв. Атомы отдачи имеют энергии 2,73 Мэв (для Т) и 0,85 Мэв (для 7Li).
Применяются, кроме того, осколки деления тяжелых ядер (например, 235U) в атомном реакторе. Пробег этих осколков крайне мал, и их кинетическая энергия обычно рассеивается в виде тепла внутри топливных элементов реактора. Однако предложены некоторые методики (см. главу II), позволяющие использовать их для осуществления радиационно-химических реакций.
В радиационной химии иногда находят применение тяжелые многозарядные ионы, получаемые в ионных ускорителях. В частности, в работе [24] исследовался радиолиз некоторых жидкостей под действием ядер С6+,
Взаимодействие заряженных частиц с веществом
Заряженные частицы при прохождении через среду теряют свою энергию постепенно, в результате многократного столкновения с электронами среды.
Основным механизмом взаимодействия заряженных частиц с веществом является электромагнитное взаимодействие с электронами вещества (ионизация) и кулоновским полем ядра и электронов (тормозное излучение). Первый вид взаимодействия, кроме непосредственно ионизации, включает также возбуждение атомов и молекул. Потери энергии заряженной частицей в результате этого вида взаимодействия называют ионизационными потерями, а потери энергии за счет второго вида взаимодействия — радиационными.
Ионизация, возбуждение и тормозное излучение — это неупругие процессы взаимодействия. Средние ионизационные потери энергии на единице пути (или тормозная способность) определяются формулами Бете [7].
Для тяжелых заряженных частиц формула Бете имеет следующий вид:
dE 4ле4г2 Z Г mv2E 6 1
~ = mv2 Nop [1п 1(1 — З2) “ Р2 — Т] ’ (66)
где —dEldx — средняя потеря энергии на единице пути (Е — кинетическая энергия частицы, х — координата в направлении движения частицы); ze — заряд частицы; е — заряд электрона; т — масса покоя электрона; v — скорость частицы; No — число Авогадро; р — плотность среды; Z — атомный номер; А — атомный вес; р = v/c (с — скорость света); I — средний потенциал возбуждения атомов среды; 6/2 — поправка па поляризационный эффект (см. стр. 127).
Для нерелятпвистской области энергий, когда Е <^_ Л/с2 (Л/ — масса покоя частицы) и 2nZ/137 <</ р, формула (66) трансформируется к виду
dE 4ne4z2 Z 2mv2
~~~dx~ ~ mv2 ДТ ln ~7 • (G7)
В случае электронов для релятивистских энергий
dE 2ле4 Z Г mv2E г____
- —Iй 2 Z2 (1 — р2) - (2 -3*- 1 + Р2) 1п2 +
+ 1 —р2V1-32)2 —б] • (68)
Наконец, для нерелятивистских электронов потери энергии на единицу длины пути можно рассчитать по формуле
dE 2л/’4 Z 1,16mv2
~ dx = mv2 N<$ .11п 27 ' <69)
Средний потенциал возбуждения является мерой энергии связи электронов с ядром. Его значения увеличиваются с ростом Z. Обычно I определяется экспериментально. Значения I для некоторых сред приведены в табл. 10.
Таблица 10
Значения средних энергий возбуждения [25]
Среда I, эв Среда Г, эв Среда I, 9в
Н2 18,7 Kr 381 NaJ 433
Не 42,0 Ag 487 LiJ 473
Li 38,0 Sn 516 Метан 44,6
Be 60,0 Xe 555 Этилен 54,6
С 78,0 W 748 Полиэтцлен 54,6
N 85,0 Au 797 Ксилол 61,0
О 89,0 Pb 826 Толуол 62,1
Ne 131 U 923 Ацетилен 63,6
Mg 156 H20 65,1 Полистирол 63,6
Al 163 CO2 85,9 Стильбен 65,2
Ar 210 AgCl 384 Люцит * 65,6
Fe 273 AgBr 434 Антрацен 67,0
Cu 314
* Один из продажных заграничных сортов полиметилметакрилата (англ. Lucite).
Величина — dEldx зависит от скорости частицы и ее заряда. При одной и той же энергии ионизационные потери на единицу пути для электрона будут во много раз меньше, чем для а-частицы. Например, при энергиях порядка нескольких мегаэлектронвольт потери для электрона примерно в 1000 раз меньше, чем для а-частицы. Вследствие этого при равных энергиях путь электрона в веществе (глубина проникновения) гораздо больше, чем путь а-частицы.
Помимо ионизационных потерь, для заряженных частиц имеют место радиационные потери, т. е. потери энергии на тормозное излучение. Для тяжелых заряженных частиц радиационные потери малы по сравнению с ионизационными. В случае электронов отношение потерь энергии на излучение к потерям на ионизацию описывается выражением
(-^ЛМрзд EZ
800 •
Для электронов радиационные потери пропорциональны Z2 (в тонких мишенях) и почти пропорциональны Е. При высоких энергиях преобладают радиационные потери. С уменьшением
энергии играют большую роль ионизационные потери. При некоторой энергии, называемой критической (Л’кр), ионизационные и радиационные потери на единице пути станут сравнимы. Екр зависит от природы вещества. Для водорода Екр = 500 Мэв, тогда как для свинца Екр — 11 Мэв [26].
Таким образом, полные потери энергии на единице пути заряженной частицы равны сумме ионизационных и радиационных потерь:
dE \ _ / _dE_\ / dE \
dx /поли \ dx /ион dx /рад
(71)
На практике часто пользуются массовой тормозной способностью, обозначаемой символом S/p или mS. По определению МКРЕ [21], массовая тормозная способность материала для заряженных частиц есть частное от деления dEs на din р, где dEs — средняя энергия, потерянная заряженной частицей данной энергии при прохождении ею длины пути dl, а р — плотность среды. Таким образом, —
При этом dEg включает потери энергии на ионизацию, возбуждение и излучение.
Массовая тормозная способность, очевидно, не зависит от плотности. В качестве единицы измерения S/p служит, например, кэв‘См2/мг. На рис. 17 показана зависимость S/p для некоторых материалов в случае быстрых электронов от их энергии.
Для электронов при прохождении их через вещество характерен процесс упругого рассеяния па ядрах. В этом процессе часть кинетической энергии электрона передается ядру, а сам электрон при этом изменяет направление своего движения. Для тяжелых заряженных частиц (особенно для а-частиц) процесс упругого рассеяния мал по сравнению с ионизацией.
Истинный путь частиц в веществе, или истинный пробег, определяется, исходя из полных потерь энергии
dE
) (-й^/^полн
'О
(73)
где Ео — начальная энергия частицы.
Величину пробега часто измеряют в единицах мг/см2 или г/см2. Использование этих единиц вызвано тем, что при таком выражении толщина материала не зависит от его плотности.
Быстрые электроны и ^-частицы вследствие их многократного рассеяния при прохождении через вещество не имеют определенных значений пробегов. Действительно, даже электроны с одинаковой начальной энергией вследствие, например, рассеяния могут
полностью потерять свою энергию в различных точках пространства, Поэтому часто для их характеристики используют термины: практический (или экстраполированный) пробег и максимальный пробег (или максимальная глубина проникновения). Поясним эти термины с помощью рис. 18. На нем по оси ординат отложено число N моиоэнергетических электронов, прошедших через слой d поглотителя; значения d (например, в г/см2) отложены по осп абсцисс. При измерении N таким путем, чтобы регистрировались
Рис. 17. Массовая тормозная способность S/p воды (7), графита (2) п воздуха (3) как функция энергии Е электронов (а — ионизационные потери; б — радиационные потери)
Рис. 18. Зависимость числа N моноэпергетических электронов, прошедших через поглотитель, от толщины d последнего
и электроны, рассеянные под большими углами, кривая, характеризующая поглощение, будет иметь наклон, возрастающий с увеличением d. После более или менее длинного прямолинейного участка АВ кривая совпадает с осью абсцисс или становится почти параллельной ей (участок CD). То обстоятельство, что участок CD кривой может не совпадать с осью абсцисс, вызвано тем, что детектор регистрирует и вторичное рентгеновское излучение, возникающее при прохождении электронов через вещество. Практический пробег Rпр определяется экстраполяцией прямой А В до пересечения с продолжением CD. Максимальный пробег 7?1Пах характеризуется величиной d от 0 до точки С.
Поглощение р-частпц вследствие их немоноэпергетичности начинается с малых глубин проникновения. Поэтому N с ростом d убывает почти экспоненциально [в соответствии с уравнением (29)]. Отличие соответствующей кривой от экспоненты состоит в том, что экспонента не пересекается с осью абсцисс, тогда как число Р-частиц на определенной глубине поглотителя равно нулю (за вычетом фона).
В случае электронов применяют также термин — истинный (или полный) пробег /?ист электронов. Это — фактическая длина пути электрона вдоль искривленной или изломанной траектории. Иногда истинный пробег называют просто длиной пути электронов. Проекция истинного пробега на первоначальное направление движения электрона представляет собой /?юах, т. е. 7?И(;т _> Rmax- В газах истинный пробег находят с помощью снимков в камере Вильсона; в твердых и жидких веществах он рассчитывается теоретически.
Для p-излучения иногда применяют термин — толщина половинного ослабления. Он аналогичен такому же термину в случае фотонного излучения.
Таблица 11
Пробеги [1-частиц
Изотоп -^тах’ сле НИСт в воздухе, см Изотоп Rmax» см R ИСТ в воздухе, f'M
Алюминий Вода Алюминий Вода
зн 0,0002 0,0 н)55 0,65 90Sr 0,066 0,18 185
35S 32р 0,012 0,29 0,032 0,79 31 770 90у 0,40 1,1 1020
В табл. 11 приведены максимальные пробеги р-частпц ряда изотопов в алюминии п воде, а также истинный пробег их в воздухе [20]. На рис. 19 показаны истинные пробеги быстрых электронов различной энергии в воде [23]. Отметим, что приведенные в табл. 11 значения 7?тах относятся только к р-частицам с Ешах, которых в энергетическом спектре очень мало. Средние значения /?гаах с учетом всего спектра Р-частпц примерно в 5 раз меньше.
Очевидно, глубина проникновения электронов в вещество прямо пропорциональна энергии электронов и обратно пропорциональна плотности вещества. Имеется ряд эмпирических соотношений, связывающих /?ших и энергию электронов. Например, по формуле Фэзера
/?тах = 0,543Е -0,160 (74)
можно пайтп Лтлх (в г/см2) при энергиях электронов и максимальных энергиях р-частпц (в Мэв) в диапазоне от 0,7 до 15 Мэв. При энергиях электронов, больших 1 Мэв, приближенная формула для нахождения Rmax такова:
Ятах~0,5Е. (75)
Энергия, передаваемая электронами среде, распределяется в ней неравномерно. На рис. 20—22 показано распределение доз
по глубине для случая воды [27]. Из этих рисунков видно, что на малых глубинах имеет место возрастание дозы. Это обусловлено, в частности, попаданием сюда электронов, рассеянных на больших глубинах. Затем наблюдается спад, вызванный поглощением и рассеянием электронов. Очевидно, рассеянные электроны не вносят какого-либо вклада в дозу на данной глубине.
В отличие от электронов путь тяжелых заряженных частиц в веществе прямолинеен. Для них 7?тах ~ 7?ист. В случае «-частиц это иллюстрируется рис. 23, на котором по оси ординат отложено число N а-частиц, прошедших определенное расстояние d. На нем пробег обозначен буквой R.
Рис. 20. Распределение глубинных доз электронов с энергиями 0,5 (?), 1 (2), 2 (3) и 3 (4) Мэв в воде
Рис. 19. Зависимость истинного пробега 2?ист электронов в воде от их энергии Е
Рис. 21. Распределение глубинных доз электронов с энергиями 3 (2), 4 (2), 5 (3) п 6 {4) Мэв в воде
Рис. 22. Распределение глубинных доз электронов с энергиями 6 (1), 8 (2), 10 (3), 12 (4), 14 (5) и 16 (6) Мае в воде
В табл. 12 приведены средние пробеги а-частиц ряда изотопов в воздухе и воде [20, 28], а в табл. 13 — средние пробеги протонов и гелионов различной энергии в воде [28]. На рис. 24 показаны пробеги некоторых тяжелых многозарядных ионов различной энергии в воде [29]. Средние пробеги осколков деления 235U в различных материалах даны в табл. 14 [30].
Рис. 23. Зависимость числа 7V а-частиц, прошедших расстояние d от источника
Из-за малой роли процесса рассеяния тяжелых заряженных частиц при их прохождении через вещество кривые распределения доз по глубине облучаемого образца имеют отчетливо вы-
Таблица 12
Средние пробеги R а-частиц некоторых изотопов
Изотоп R, мк
Воздух* Вода
210ро 3,8-10* 38,9
226Ra 3,3-10* 33,0
222Rn 4,0-10* 41,1
* При 15° С и давлении 760 мм рт. ст.
Таблица 13
Средние пробеги R протонов и гелионов в воде
Энергия частицы. Мэв R, мк Энергия частицы, Мэв R, мк
Протон Гелион Протон Гелион
1 23 5,3 6 486 47,0
2 73 10,1 7 642 60,3
3 147 16,8 8 ; 813 75,5
4 241 25,1 9 1004 91,6
5 355 35,2 10 1211 108,4
Рис. 24. Пробеги R тяжелых многозарядных ионов в воде
1 — С; 2 — N; 3 — О; 4 — F; S — Ne
Рис. 25. Распределение доз в воде для дейтронов с энергией 190 Мэв (1), электронов с энергией 16,4 Мэв (2) и рентгеновских лучей с максимальной энергией 200 кэв (3)
раженный максимум вблизи конца пробега. На рис. 25 с целью сравнения приведены кривые распределения глубинных доз в воде для электронов с энергией 16,4 Мэв, рентгеновских луней с максимальной энергией 200 кэв и дейтронов с знергпей 190 Мэв [23]. На нем кривые отнесены к одинаковым значениям максимальной дозы.
Таблица 14
Средние пробеги осколков деления 235U в различных материалах
Материал Пло гность Пробег Материал Плотность Пробег
мк JAZi'CM2 мк мгц-м?
ио2 10,9 9,9 10,8 Си 8,9 5,8 5,2
U3O« 7,3 13,7 10,0 Вода 1,0 2,1 2,1
и А1 18,9 2,7 6,7 13,7 12,7 3,7 Воздух 1,23-Ю-3 2,2-10* 2,7
Взаимодействие нейтронов с веществом
Основными процессами взаимодействия нейтронов с веществом являтся неупругое рассеяние, упругое рассеяние и захват ядрами. При этом решающую роль при прохождении нейтронов через ве
щество играет взаимодействие их с ядрами. Взаимодействие нейтронов с электронами имеет гораздо меньшее значение, поскольку из-за отсутствия у нейтронов электрических зарядов силы, действующие между нейтроном и электроном, очень малы.
Неупругое рассеяние. В этом процессе образуется возбужденное ядро. Процесс происходит лишь в том случае, если энергия бомбардирующего нейтрона превышает минимальную энергию возбужденного ядра. На тяжелых ядрах неупругое рассеяние наблюдается при энергиях в несколько сотен килоэлектронвольт, а на легких —выше 1 Мэв.
При неупругом рассеянии нейтрон п', испущенный возбужденным ядром, имеет энергию, которая меньше энергии бомбардирующего нейтрона п. Переход возбужденного ядра в основное состояние сопровождается у-излучением. Таким образом, неупругое рассеяние — это реакция (и, п', у). Иногда оно представляет собой реакции (п, р, у) и (п, а, у).
Упругое рассеяние. В этом процессе нейтрон теряет тем большую энергию, чем меньше масса рассеивающего ядра и чем больше угол рассения. Если рассеяянпе происходит на тяжелых ядрах, то изменение энергии нейтрона невелико. Оно иногда обозначается ядерной реакцией вида (п, п).
Упругое рассеяние бывает двух видов: потенциальное рассеяние и резонансное рассеяние. Потенциальным рассеянием обычно называется рассеяние, являющееся простым отклонением нейтрона под действием поля ядерных сил. Это — простейший процесс взаимодействия нейтрона с веществом. Резонансным рассеянием называется рассеяние, связанное с промежуточным состоянием составного ядра. В случае быстрых нейтронов и легких ядер наблюдается главным образом резонансное рассеяние.
При упругом рассеянии энергия Е, передаваемая ядру нейтроном при одном столкновении, равна
4/7 '
= (Л7 -f- 1)*^° cns2 (7^)
где М — отношение массы ядра отдачи к массе нейтрона; Ео — энергия нейтрона и 0 — угол между направлением движения нейтрона и ядра отдачи.
При усреднении по всем углам рассеяния средняя потеря энергии на одно упругое рассеяние равна
/ Д£\ ЧА
\ Ес, /Ср - (А +1)2 ’ <77>
где А — атомный вес.
Максимальная энергия, которая может быть передана ядру с атомным весом А, составляет
/ А£ \ 4Л
\ Ео /max (А +1)2 ‘ (7^
В радиационной химии большое значение имеет процесс упругого рассеяния быстрых нейтронов на ядрах атомов водорода. Это обусловлено следующими обстоятельствами: 1) во многих системах (вода, водные растворы, полимеры, углеводороды и др.) значительную часть ядер составляют протоны (в воде их, например, около 2/3); 2) передача энергии протону максимальна по сравнению с другими ядрами [из формул (77) и (78) видно, что для протона (А£7Еп)ср = 0,5 и (А£7Е0)тах = 11 и 3) сечение процесса рассеяния больше, чем на других ядрах. На рис. 26 показана
Рис. 26. Зависимость эффективного сечения с процесса упругого рассеяния нейтронов на ядрах атомов водорода от энергии нейтронов
зависимость эффективного сечения процесса упругого рассеяния нейтронов на протонах от энергии нейтронов.
В результате рассеяния нейтронов на ядрах атомов водорода образуются протоны отдачи, которые и производят ионизацию и возбуждение молекул среды.
Захват ядрами. В этом процессе возникает возбужденное составное ядро, переходящее в основное состояние путем испускания в большинстве случаев одного или нескольких у-квантов. Это — радиационный захват, или реакция (п, у). Вероятность этого процесса уменьшается при увеличении энергии нейтрона, и для быстрых нейтронов она мала по сравнению с вероятностью процесса рассеяния.
Захват нейтрона ядром может приводить к испусканию протона или ос-частицы. В качестве примеров можно привести реакции 14N(«, р)14С, 10В(л, oc)7Li, eLi(zz, ос) Г. Последние две реакции часто используются в радиационно-химических исследованиях (см. также стр. 38). Энергия протонов, возникающих в реакции 14N(«, р)14С, составляет 0,66 Мэв.
Для тяжелых элементов характерны реакции деления. Некоторые из них идут под действием быстрых нейтронов (например, в случае 238U), другие — при воздействии тепловых нейтронов (например, в случае 235U и 239Ри). Продукты деления обладают высокой энергией и способны вызвать ионизацию и возбуждение среды, в которой содержится делящийся материал (см. также стр. 82).
Прохождение нейтронов через вещество. Потеря энергии быстрыми нейтронами до ~ 1 эв в результате рассеяния на ядрах ве
щества называется обычно замедлением, тогда как уменьшение энергии нейтронов от ~ 1 эв до тепловых энергий — термализацией. Тепловые нейтроны диффундируют в веществе до тех пор, пока не произойдет процесс захвата ядрами.
Как уже отмечалось, вероятность протекания того или иного процесса характеризуется сечением. В случае нейтронов в зависимости от вида процесса применяются следующие обозначения поперечных сечений: = рзе1 пли Ss = p3s —для упругого
рассеяния; Sc = рзс — для захвата нейтрона ядром; Sa = рз„ — для поглощения нейтрона ядром; Sjn = рзгп —для испускания нейтрона с энергией, меньшей, чем у исходного (т. е. для неупругого рассеяния); = рзу —для радиационного захвата и S/ = рз? — для деления ядра. Сумма поперечных сечений, представляющая собой вероятность столкновения нейтрона с ядром и называемая полным сечением, обозначается символом S(. Полное сечение характеризует эффективность экспоненциального ослабления нейтронного потока веществом. Величина з( = Б(/р называется эффективным поперечным сечением.
Основываясь на определении сечения взаимодействия, можно написать следующее уравнение для ослабления потока нейтронов:
Ф = Фо ехр (- 3 - (79)
' i '
где Фо — начальный поток нейтронов; Ф — поток нейтронов после прохождения слоя вещества I (в см)’, N —число ядер сорта i в 1 см3 и щ — эффективное сечение взаимодействия нейтронов с ядрами сорта i (в ел12).
Если сравнить уравнения (29) и (79), то видно, что для нейтронов
(80) г
Средний свободный пробег нейтронов в веществе вычисляется по формуле
7? = ^—. (81)
? Ni<5i
Значения Н для воды и полиэтилена приведены в табл. 15 [23].
Таблица 15
Средние свободные пробеги В нейтронов (в сл«)
Энергия нейтронов Вода Полиэтилен Энергия нейтронов Вода Полиэтилен
0,025 эв 0,28 0,24 1 Мэв 2,45 2,30
1—50 кэв 0,68 0,45 10 Мэв 11 7,7
5. Линейная передача энергии. «Шпоры»
Ионизирующие излучения часто характеризуют величиной линейной передачи энергии (англ. — linear energy transfer); при этом обычно используют сокращение ЛПЭ (англ.— LET). Согласно [21], ЛПЭ заряженных частиц в среде есть частное от деления й^лпэ на dZ, гДе ^лпэ — средняя энергия, локально переданная среде заряженной частицей данной энергии при прохождении ею расстояния dl, т. е.
Й/?ЛПЭ
лпэ - dl • (82)
Термин «локально переданная» может относиться или к максимальному расстоянию от трека, пли к максимальному значению
0,10
0,0 в
0,00
0,00
0,02
о,'-,—>--------*
'V 1,0 !0
О, М.чО
Рпс. 27. Зависимость ЛПЭ для воды от начальной энергии электронов (7, 2) п фотонов (3)
Z — величины начальной ЛПЭ; 2— величины средней ЛПЭ для полного пути электрона; 3 — величины средней ЛПЭ для электронов, возникших из фотонов указанной энергии в комптоновском процессе и процессе образования пар
дискретной потери энергии частицей, вне которого потери уже не рассматриваются как локальные. При этом соответствующие пределы должны быть указаны. В качестве единиц измерения ЛПЭ обычно используют кэв/мк или эв/А.
Линейная передача энергии отличается от тормозной способности. Первое понятие относится к энергии, переданной ограниченному объему; второе — к потере энергии независимо от того, где эта энергия поглощена. ЛПЭ характеризует потерю энергии ионизирующей частицей дискретными порциями, в результате чего в объеме материала создаются микрообластп с высокой локальной мощностью дозы. Эти микрообласти называются «шпорами» (апгл. — spurs). В ппх создаются первичные продукты радиолиза: свободные радикалы, ионы, возбужденные молекулы, имеющие высокую локальную концентрацию. Для у-излу-чения и быстрых электронов «шпоры» расположены далеко друг от друга. В случае тяжелых заряженных частиц расстояние между ними весьма небольшое, и они сразу же после своего образования сливаются в сплошную цилиндрическую колонку.
Обычно считается, что тормозная способность и ЛПЭ различаются па величину энергии Z?8, уносимой из трека ионизирующей частицы вторичными, пли 6-электронами:
dE
ЛПЭ=^Г-Е&-
Таблица 16
Начальные тормозные способности Ь'11ач излучения *
воды для различных видов
Вид излучения ®нач> Эв/А
Электроны (2 Мае) «Средние» комптоновские электроны из 7-лучей 60Со 0-Частицы Т (средняя энергия 5,6 кае) Протоны (100 Мае) Типичные «средние» электроны из рентгеновских лучей (£тах = 0,22 Мае) Типичные «средние» электроны из рентгеновских лучей (£тах = <М Мае) Дейтроны (10 Мае) Гелпопы (30 Мае) а-Частицы 21(Ро (5,3 Мае) Продукты ядерной реакции 6Li (n, a) Т Продукты ядерной реакции 10В (л, a) 7Li Продукты деления 236П 0,018 0,026 0,065 0,065 0,07 0,1 0,82 2,4 9,0 -10 -17 -180
Теоретическое минимальное значение 0,018 эв А
Для тяжелых частиц различие в величинах dEldx и ЛПЭ незначительно; однако для электронов оно существенно.
В радиационной химии часто отождествляют тормозную способность и ЛПЭ. Хотя такое отождествление и не вполне оправданно, тем не менее оно позволяет в известной степени характеризовать влияние вида излучения и его энергии на радиационно-химические выходы для той или иной системы. В табл. 16 приведены значения начальных тормозных способностей воды для различных видов излучения [31, 32]. На рис. 27 показана зависимость величии ЛПЭ для воды от начальной энергии электронов и фотонов (точнее: электронов, генерированных ими в процессах комптоновского рассеяния и образования пар) [33].
6. Радвацнонно-химическпй выход
Главной количественной характеристикой любой реакции, протекающей под действием ионизирующего излучения *, является радиационно-химический выход. Он равен числу молекул, ионов, свободных радикалов, атомов и т. п., возникающих (или разлагающихся) в системе при поглощении ею 100 эв энергии
* Любые химические процессы, происходящие в веществе или системе под действием ионизирующего излучения, называют радиолизом.
ионизирующего излучения. Он обозначается буквой G. Для обозначения выхода образования какого-либо вещества после буквы G в круглых скобках пишут формулу этого вещества [например, G (Се3+) есть число образовавшихся ионов Се3+ на 100 эв]. Для написания выхода разложения перед формулой соответствующего вещества ставится знак минус [например, G (—СН3ОН) обозначает число молекул СН3ОН, разлагающихся при поглощении 100 эе].
Для определения G необходимо знать дозу и концентрацию образовавшегося или разложившегося в результате облучения вещества. Тогда G можно рассчитать с помощью формулы
сЛМОО G=-^
(84)
где с —концентрация вещества, М; N —число Авогадро; D — доза, эв!л.
ЛИТЕРАТУРА
1. А. Чарлзби. Ядерпые излучения и полимеры. М., ИЛ, 1962.
2. Сборник рекомендуемых терминов, ч. I. М., Изд-во АН СССР, 1963.
3. Радиологические величины и единицы, серия технических докладов № 26. Вена, МАГАТЭ, 1964.
4. R. L. Platzman. Radiation Biology and Medicine (Ed. W. D. Claus). N. Y., 1956, p. 15.
5. A. Kuppermann. J. Chem. Educ., 36, 279 (1959.
6. A. Kuppermann. Nucleonics, 19, No. 10, 38 (1961).
7. Экспериментальная ядерная физика (под ред. Э. Сегрэ). М., ИЛ, 1955, т. 1, ч. 2.
8. Радиационная дозиметрия (под ред. Дж. Хайна и Г. Браунелла). М., ИЛ, 1962, гл. 2.
9. К. К. Аглинцев. Дозиметрия ионизирующих излучений. М., Гостехиз-дат, 1957.
10. Н. А. Власов. Нейтроны. М., «Наука», 1971.
11. М. Ф. Юдин. Дозиметрия фотонного излучения. М., Стандартгиз, 1970.
12. Н. Г. Гусев, Л. Р. Киммель, В. П. Машкович, Б. Г. Пологих, А. П. Суворов. Защита от ионизирующих излучений, т. 1. Физические основы защиты от излучений. М., Атомиздат, 1969, стр. 315.
13. Г. В. Горшков. Проникающие излучения радиоактивных источников. Л., «Наука», 1967.
14. Б. П. Голубев. Дозиметрия и защита от ионизирующих излучений. М., Атомиздат, 1971.
15. Государственный стандарт СССР. Единицы радиоактивности и ионизирующих излучений. ГОСТ 8848—63.
16. В. А. Б азаку ца. Международная система единиц. Харьков, Изд-во Харьковского ун-та, 1973.
17. ICRU Report 10b. Physical Aspects of Irradiation. National Bureau of Standards, Handbook No 85. Washington, 1964.
18. С. M. Lederer, J. M. Hollander, T. Perlman. Table of Isotopes, 6th Edition. N. Y., John Wiley and Sons, Inc., 1968.
19. И. П. Селинов. Изотопы. M., «Наука», 1970.
20. Дж. Спинкс, Р. Вудс. Введение в радиационную химию. М., Атомиздат, 1967.
21. ICRU Report 10a. Radiation Quantities and Units. National Bureau of Standards, Handbook No 84. Washington, 1962.
22. А. В. Бибергаль, У. Я. Маргулис, Е. И. Воробьев. Защита от рентгеновских и гамма-лучей. М., Мсдгиз, 1955.
23. Р. Егер. Дозиметрия и защита от излучений (физические и технические константы). М., Атомиздат, 1961.
24. В. Н. Schuler. J. Phys. Chem., 71, 3712 (1967).
25. М. J. Berger, S. M. Seltzer. Natl. Aeronautics and Space Administration Rep. NASA SP 3012; цит. по A. Ж. Boyd. Determination of Absorbed Dose in Reactors, Technical Reports Series No 127. Vienna, IAEA, 1971, p. 7.
26. А. А. Воробьев, В. А. Кононов. Прохождение электронов через вещество. Томск, Изд-во Томского ун-та, 1966.
27. A. Brynfolfsson. Proc. Intern. Conference on Radiation Research, U. S. Army Natick Lab., Natick, Mass. (USA), 1963, p. 116.
28. Д. E. Ли. Действие радиации на живые клетки. М., Атомиздат, 1963.
29. A. Mozumder, A. Chatterfee, J. L. Magee. Adv. Chem. Ser., 81, 27 (1968).
30. A. W. Boyd. Determination of Absorbed Dose in Reactors, Technical Reports Series No 127. Vienna, IAEA, 1971, p. 7.
31. C. J. Hochanadel. Comparative Effects of Radiation (Ed. M. Burton et al.). New York— London, 1960, p. 151.
32. A. J. Swallow. Radiation Chemistry. An Introduction. New York — London, John Wiley and Sons, Inc., 1973, p. 31.
33. H. Fricke, E. J. Hart. Radiation Dosimetry (Ed. F. H. Attix and W. C. Roesch), vol. 2. N. Y., 1966, p. 167,
Глава II
ИСТОЧНИКИ ИОНИЗИРУЮЩИХ ИЗЛУЧЕНИИ
В радиационной химии используются разнообразные источники ионизирующего излучения. Их можно разделить па два класса: аппаратурные и изотопные. К первому классу относятся рентгеновские трубки, ускорители заряженных частиц, атомные реакторы; ко второму — установки с радиоактивными изотопами.
В настоящей главе кратко рассматриваются принципы действия аппаратурных источников и конструктивного выполнения изотопных установок, а также излагаются рабочие характеристики источников ионизирующего излучения, наиболее часто использующихся в радиационной химии. Более подробно заинтересованный читатель может ознакомиться с этими вопросами в специальных книгах [1 —9].
1. Изотопные гамма-установки
Из числа изотопных установок наибольшее распространение в радиационной химии находят установки с радиоактивным изотопом С1)Со. Сейчас известны установки с этим изотопом, имеющие активность в несколько миллионов грамм-эквивалентов радия.
Промышленность пашей страны выпускает стандартные источники с изотопом виСо, активность которых составляет от 5 до 3000 г-экв радия. Они представляют собой чаще всего цилиндры, находящиеся в тонкой оболочке из алюминия или нержавеющей стали. |3-Частицы с энергией 0,306 Мэв, образующиеся при распаде этого изотопа, полностью поглощаются в самом кобальтовом препарате и оболочке, в которую он помещен. Поэтому такие источники можно рассматривать как практически чистые у-излуча-телп.
Стандартные источники собираются в облучатели различной конфигурации. Чаще всего применяются облучатели в виде полого цилиндра («беличье колесо») и плоскости. В радиационной химии обычно используются облучатели первого вида, так как в этом случае достигается равномерная и достаточно высокая мощность дозы во внутренней сравнительно небольшой полости облучателя. Облучатели в виде плоскости находят применение в радиационно-химической технологии, поскольку такая форма облучателя позволяет проводить конвейерную транспортировку материала через зону облучения.
Максимально достигнутая мощность дозы в случае источников с изотопом 60Со равна примерно 5000 —6000 рад/сек. Эта величина зависит от удельной радиоактивности препарата, которая имеет известные пределы. Однако, чем больше суммарная активность, тем больше величина облучаемого объема.
Конструкция изотопных кобальтовых установок весьма разнообразна (см., например, [1—3, 10—29]). Они различаются по типу защиты в нерабочем состоянии (вода, бетон и т. и.), по способам перевода облучателя из нерабочего состояния в рабочее (механический, пневматический, электромагнитный, гидравлический и ДР-)-
Согласно [3], изотопные установки можно разделить на следующие классы:
1. Исследовательские установки с неподвижным облучателем, не требующие специального помещения (так называемые «само-защпщенные» установки).
2. Исследовательские установки с подвижным облучателем постоянной конфигурации, пе требующие специального помещения.
3. Исследовательские установки с подвижным облучателем, требующие специального помещения.
4. Исследовательские универсальные установки с подвижным облучателем, требующие специального помещения. В них предусмотрена возможность изменения конфигурации облучателя, что позволяет моделировать различные радиационно-химические аппараты.
5. Опытно-промышленные и промышленные установки с неподвижным облучателем, не требующие специального помещения.
6. Опытно-промышленные и промышленные установки с неподвижным облучателем, требующие специального помещения.
7. Опытно-промышленные и промышленные установки с подвижным облучателем, требующие специального помещения.
К этим классам следует добавить еще один класс установок, а именно: исследовательские установки с неподвижным облучателем, требующие специального помещения. В соответствии с приведенной классификацией это — класс 2а.
Очевидно, в радиационно-химических исследованиях используются установки первых четырех классов. Установки последних трех классов находят применение в радиационно-химической технологии.
Установки 1 и 2 классов весьма удобны для проведения ради ациоппо-хпмпческпх экспериментов с небольшими образцами. В первое десятилетие после второй мировой войны было описано несколько небольших установок со сравнительно ппзкой активностью 60Со (см., например, [30—331). В качестве примера рассмотрим установку Дж. Гормлея и К. Хохападеля [30]. Общий вид ее показан на рис. 28. Она состоит из массивного свинцового блока
с двумя кубическими полостями, в которые могут быть помещены исследуемые образцы. Верхняя часть установки представляет собой свинцовый цилиндр, внутри которого находится препарат кобальта. С помощью специального приспособления цилиндр может передвигаться от одной полости к другой. Для облучения источник 60Со (его активность — 300 кюри) опускается из верхнего цилиндра. Мощность дозы для данного источника равна ~ 80 рад!сек.
В настоящее время в Советском Союзе выпускается несколько видов «самозащищепных» гамма-установок. В них облучатель находится внутри массивного свинцового блока, а исследуемый образец вводится в зону облучения. Характеристики этих устано-
Рис. 28. Общий вид источника у-из л учения в0Со 130]
вок приведены в табл. 17 [10, 27, 281. На рис. 29 в качестве примера показан внешний вид установки МРХ-у-100. Установка состоит из свинцового контейнера, играющего роль биологической защиты, облучателя, штока с рабочей камерой, механизма подъема и поворота штока. Облучатель является цилиндрическим. Он представляет собой кассету с источниками у-излучения 60Со. Кассета имеет 36 трубчатых ячеек, расположенных по окружности и скрепленных между собой. В каждую ячейку помещается по два кобальтовых источника. Облучатель находится внутри контейнера.
Обычно установки этого типа поставляются с облучателями с активностью до ~25000 г-экв радия. Однако по специальному
Рис. 29. Общий вид установки МХР-у-100
заказу активность может быть повышена до 40000 г-зкв радия. В частности, в Институте физической химии АН СССР уже в течение нескольких лет эксплуатируется установка \1РХ у 100, облучатель которой имел начальную активность ~ 40000 г-экв радия.
За границей широко распространены сходные установки типа «Gammacell», поставляемые Канадской комиссией по атомной энергии. Общий вид одной из таких установок («Gammacell-220») приведен на рис. 30. Установка «Gammacell-220» может иметь активность до 30000 кюри-, объем рабочей камеры составляет 2,5 — 3 л\ мощность дозы в рабочей камере — до ~550 рад/сек [34].
Только что рассмотренные установки относятся к первому классу. Ко второму классу принадлежат, например, установки К-300, К-600 и К-1400, сконструированные в Физико-химическом институте им. Л. Я. Карпова. Их характеристики приведены в табл. 18. В этих установках исследуемый объект, находящийся в специальном приспособлепип. помещается в рабочую камеру, закрывающуюся свинцовой пробкой. Источник излучения размещен в
Рис. 30. Общий вид установки «Gammacell- 220»
Таблица 17
Характеристики самозащищенных гамма-установок, выпускаемых промышленностью СССР
Тип установки Активность f0Co, кюри Мощность дозы, радIсек Габариты, см Вес, т Объем рабочей камеры, л
РХ-Т-ЗО 20 000 440 130X140X270 5,5—6 4,4
МРХ-уЮЭ 20 000 550 130X140X270 5 1,2
РХМ-т-20 12 500 250 150X150X320 5,5-6,5 4,4
ЛБМ-уЛМ 2 400* 55 50x50: 121,5 0,85 0,3
* В этой установке излучателем является изотоп 137Cs.
Таблица 18
Характеристики отечественных кобальтовых установок с подвижным облучателем
Установка Тип защиты Активность, г-же радия Максимальная мощность дозы, рад/сек Литература
К-300 «Сухая» защита 280 100 [13]
К-600 То же 500 200 [2]
К-1400 >> 1 440 300 [19]
К-20000а » 21000 1100 [20]
К-200006 » 18 000 1000 [20]
УК-70000 Водяная защита 70 000 1800 ПИ
К-60000 «Сухая» защита 45 000 1200 [22]
К-120000 То же 130 000 5200 [3]
ГУГ-120000 Водяная защита 120 000 1800
УГУ-20О000 То же 210 000 4&I [26]
рабочем контейнере, закрытом пробкой. Последующие операции: подъем пробки пз рабочего контейнера, перевод контейнера в рабочее положение, подъем облучателя в рабочую камеру па время проведения эксперимента — осуществляются дистанционно.
Представителем установок класса 2а является установка УК11-ЗиииО (установка киевская подводная с активностью 30UO0 г-экв радия) [18]. Схема * ее приведена на рис. 31. Облучатель 1 находится па дне бассейна под слоем воды толщиной 4 м, являющейся биологической защитой и термостатирующей средой. Поплавко
*7 — бассейн установки УК-70 ООО, описание которой дапо ниже.
вая блокировка 2, состоящая из свинцового П-образного (в плане) экрана и качающейся свинцовой крышки с противовесом, «срабатывает» в случае аварийной утечки воды из бассейна. Крышка удерживается в приподнятом состоянии поплавком 3. При снижении уровня воды в бассейне примерно на 30 см равновесие нарушается, и крышка опускается на экран, закрывая облучатель. Исследуемые образцы помещаются в герметическую кассету 4, которая при помощи ручного привода 5 доставляется к облучателю в каретке, двигающейся по плоской направляющей 6. Мощность дозы для этой установки при суммарной активности 31 500 г-экв радия составляет 3000 рад/сек.
Рис. 31. Схема установки УКП-30000
Впоследствии вместо описанной установки в том же бассейне были размещены две гамма-облучательные установки с активностью 25 000 и 100 000 г-экв радия [35]. В центральной секции последней из них мощность дозы превышает 5000 рад, сек.
Другой пример установок этого класса — это установка МГУП-30000 (малогабаритная гамма-установка подводная с активностью 30000 г-экв радия), смонтированная в Институте физической химии АН СССР. Она предназначена для облучения образцов объемом до -~40 смя. Цилиндрический источник постоянно находится подводой на глубине 4,5 м. Герметизированный реакционный сосуд с исследуемым объектом опускается по трубе в источник на тросе электропривода. Мощность дозы в реакционном сосуде составляет ~6000 рад /сек.
Сейчас в радиационной химии стали все более широко использоваться установки третьего класса. Обусловлено это прежде всего необходимостью проведения экспериментов с большими образцами. В этих установках облучатель в нерабочем положении находится за защитой (в бассейне с водой, бетонной камере и т. п.). Для облучения каких-либо объектов он подводится в «горячую» камеру, в которой размещены эти объекты. После окончания облучения источник вновь перемещается за защиту.
Существует два типа защиты в нерабочем положении источника: водяная защита и «сухая» защита (бетой, свинец, чугунная дробь и т. п.). Ниже в качестве примера дается описание двух установок третьего класса: одной — с водяной защитой и второй — с «сухой» защитой.
На рпс. 32 показана схема установки УК-70000 (установка киевская с активностью 70000 г-экв радия) [17]. Эта установка состоит из «горячей» камеры, бассейнов с водой, облучателя, электромеханической системы перемещения облучателя с приспособлением для аварийного сброса, системы блокировки, сигнализации и контроля, пульта управления, насосного и вентиляционного хозяйства. «Горячая» камера 1 —бетонное помещение размером 4 X 4 X 2,9 м (толщина бетонных стен — 2 м; перекрытие изготовлено из железобетона толщиной 1,5 ж). Камера имеет лабиринтный вход 2, закрывающийся металлической дверью 3. В камере находится рабочий бассейн 4 (зеркало воды 1,5 X 1,5 ж, глубина 4,3 м). В нижней части бассейн «горячей» камеры соединен шлюзовым устройством 5 с приемным бассейном 6, который аналогичен по конструкции и находится вне камеры. Над этим бассейном проходит монорельс, несущий электроталь 7 грузоподъемностью 2 т. Облучатель поднимается со дна бассейна на уровень рабочего стола 10 в охранный сосуд 9 с помощью тельфера 8. Управление установкой осуществляется с главного пульта 77.
Облучатель рассматриваемой установки (90 стандартных препаратов 60Со) состоит из 30 алюминиевых трубок, которые расположены в виде цилиндра. Начальная активность кобальтовых
препаратов была в пределах 650 —900 г-же радия. Внутри охранного сосуда мощность дозы по окончании зарядки достигала 1800 padjcex. Мощность дозы на рабочем столе была 15—600 рад/сек.
Позже активность рассмотренной установки была повышена до 120000 г-зкв радия [35]. При этом максимальная мощность дозы в центральной части установки достигла 2820 padiceK', на рабочем столе мощность дозы в зависимости от расстояния до облучателя колебалась от 56 до 620 рад/сек.
Схема установки К-20000, созданной в Физико-химическом институте им. Л. Я. Карпова [3, 20], дана на рис. 33. В этой установке принят вариант «сухой» защиты. В рабочей камере установки находятся контейнер 1 с кассетой 2, содержащей кобальтовые препараты, и свинцовой пробкой 5, рабочий стол 4, механизмы узлов подъема пробки, перемещения контейнера и подъема псточ ника 6, 13, 14, прппособления для аварийного опускания источника 7, 8 и аварийного закрывания контейнера запасной пробкой 3 и экраном 10. Камера имеет бетонные стены (толщина 1.5 м), бетонный потолок (2 м) и лабиринтный вход. Пульт управления 12 и световая сигнализация 11 находятся в отдельном зале. В помещении на втором этаже расположен физпко химический
Рис. 32. Схема установки УК-70000 а — вертикальный разрез; б — план
пульт У, который связан с рабочей камерой различными коммуникациями. Порядок проведения экспериментов на этой установке следующий: исследуемые объекты помещаются на рабочий стол, затем подсоединяются необходимые коммуникации, после чего облучатель дистанционно переводится из положения хранения на рабочий стол (для этого производят подъем пробки, передвижение контейнера в рабочее положение, подъем облучателя из контейнера в центр рабочего стола), по окончании облучения эти операции осуществляются в обратном порядке. Облучатель установки представляет собой полый цилиндр из нержавеющей стали высотой 190 см с наружным диаметром 14 см и содержит 56 стандартных препаратов 60Со с активностью ~ 400 г-экв радия каждый.
Примерами отечественных установок четвертого класса могут служить установки К-60000, К-120000, ГУГ-120000 и УГУ-200000. В первых двух установках применяется «сухая» защита, в третьей и четвертой — водяная защита. Отличительной особенностью первых трех установок является то, что кассеты с кобальтовыми препаратами находятся в изогнутых трубах-каналах из нержавеющей стали. Конфигурация облучателя может меняться
й
//
/2
Рис. 33. Схема установки К-20000
путем изменения расположения этих труб в рабочей камере. Можно также варьировать суммарную активность облучателя, поднимая различное число кассет. Установка УГУ-200000 (универсальная многокамерная гамма-установка активностью 210 000 г-экв радия) состоит из двух рабочих камер. При проведении облучения в одной из камер в другой можно осуществлять подготовку следующего опыта. Облучение образцов можно вести также под водой в баке установки. Характеристики указанных установок приведены в табл. 18. Там же даны характеристики других отечественных кобальтовых установок с подвижным облучателем.
В настоящем разделе рассмотрены преимущественно советские кобальтовые установки. Читатель, интересующийся устройством зарубежных установок, может найти их описание, например, в работах [36—43].
Другой изотоп, который перспективен для использования в у-установках,— это 137 Cs. Имеются сообщения о способах изготовления у-источников 137Cs из порошкообразного хлорида цезия, помещаемого в ампулы из нержавеющей стали с двойными стенками [44—46]. По данным [44], удельная активность при этом может достигать 25 кюри!г CsCl. Описаны также источники на основе цезиевых стекол (в частности, на основе цезийбороспликат-ного стекла) [36]. Однако в настоящее время изотопные установки с 137Cs в радиационной химии применяются редко.
Особую группу источников у-излучения составляют радиационные контуры. Согласно [3], радиационным контуром называется система, состоящая из генератора активности, находящегося в активной зоне реактора, облучателя, расположенного вне нейтронного поля реактора, и связывающих их коммуникаций, по которым циркулирует активируемое вещество, так что количество радиоактивных ядер в системе непрерывно пополняется, а энергия, выделяемая при распаде этих ядер, используется в облучателе в виде «чистого» у-излучения. Чаще всего в качестве активируемого вещества применяются жидкометаллические сплавы из элементов с большим сечением захвата нейтронов, причем образующиеся при этом радиоактивные изотопы должны иметь сравнительно малые периоды полураспада. К ним относятся сплавы In—Ga— Sn, In—Ga и др.
Радиационные контуры построены за рубежом и в Советском Союзе (Институт физики АН Латвийской ССР, Институт физики АН Грузинской ССР, Томский политехнический институт им. С. М. Кирова, филиал Физико-химического института им. Л. Я. Карпова [47]). Активности их достигают нескольких десятков тысяч г-экв радия. Радиационные контуры находят некоторое применение в научных исследованиях, а также для моделирования радиационно-технологических процессов. Более подробное описание этих установок можно найти, например, в книге [47] и статьях [48-521.
2. Источники р- и а-пзлучеппй
Выполнено весьма большое число радиационно-химических исследований с |3- и «-излучениями радиоактивных изотопов. Источники этих излучений можно подразделить на два типа: внутренние и внешние. В первом случае радиоактивный изотоп вводится непосредственно в исследуемую систему, а во втором — источник находится вне этой системы. Внутренние источники в свою очередь делятся на гомогенные и гетерогенные. Примерами гомогенных источников могут быть соль 36S или 32Р, растворенная в водном растворе, тритий в виде газа либо в воде, введенной в газовую смесь или водный раствор. В качестве таких источников, кроме упомянутых, применялись изотопы 14С (в случае некоторых органических жидкостей), 210Ро и 239Рп (в случае водных растворов), 232Вп (в случае газовых систем). В настоящее время наиболее часто используются тритий и 210Ро. О гетерогенном источнике можно говорить, если радиоактивный препарат вводится в изучаемую систему в виде слоя на пластинке носителя или же если радиоактивность находится в осадке. Последний случай очень часто встречается в технологии переработки и выделения ядерного горючего.
Внешние источники p-излучения применяются в радиационной химии крайне редко. Например, p-излучение 9°Sr—"Y использовалось в исследованиях радиационной газофазной полимеризации тетрафторэтилена [53]. Полимеризация проводилась в стеклянном сосуде емкостью 5 л, который был снабжен манометром и тонкостенной стеклянной гильзой. В последнюю и вводился источник P-излучения 90Sr. К настоящему времени описаны весьма интенсивные внешние источники p-излучения. В частности, в работе [54] приведены данные об источнике р-пзлучения 9°Sr на основе силиката стронция. Источники находятся в тонкостенных ампулах из нержавеющей стали. Мощность дозы на поверхности составляет — 6000 рад!сек. Имеется сообщение [55] о методе приготовления Р-излучателей с изотопом 98Кг, находящимся в виде клатратного соединения с гидрохиноном. При использовании криптона, содержащего 5% изотопа 98Кг, можно получать препараты с удельной активностью — 3 кюри/г.
Известно лишь несколько случаев применения внешних источников «-излучения в радиационной химии. В качестве примера на рис. 34 приведена схема устройства для облучения жидкостей «-частицами 219Ро [56]. Источник «-излучения (1) представляет собой металлический 210Ро, нанесенный на поверхность тантала. а-Частицы проходят через слой гелия внутри держателя (5 — ввод гелия и 4 — выход гелия), два окна из очень тонких пластинок слюды (5 — окно держателя и 7 — окно стеклянной ячейки с исследуемой жидкостью). Ячейка 7 спабжена магнитной мешалкой 9 и 10. Начало и окончание облучения системы осуществляют-
3 А. К. Пикаев
65
Рнс. 34. Схема установки для облучения жидкостей а-частицами 21сРо
ся с помощью затвора 2, который при включении размещается между источником и ячейкой. Для фиксации положения ячейки относительно источника используется латунная шайба 6.
3. Ускорители заряженных частиц
В последнее десятилетие в радиационной химии широкое распространение получили электронные ускорители. Это обусловлено, во-первых, успехами в области ускорительной техники и, во-вторых, исключительно большими возможностями этих машин для создания самых разнообразных условий облучения. Действительно, с их помощью можно проводить радиационно-химические эксперименты в стационарном режиме (непрерывное облучение) и в условиях импульсного облучения (при длительности импульсов до 10~10—10~и сек.), при низких мощностях дозы (— 103 рад/сек) и при сверхвысоких мощностях дозы (до 1014—1016 рад/сек). Некоторые из них находят применение в импульсном радиолизе —
методе радиационной химии, позволяющем регистрировать короткоживущие продукты радиолитических превращений и прямым путем изучать их свойства (см. стр. 74). Наконец, ускорители электронов используются и в радиационно-химической технологии (см., например, книгу [57]). Ускорители тяжелых заряженных частиц применяются исключительно в экспериментально-теоретических исследованиях (преимущественно для выяснения влияния ЛПЭ на радиолиз различных систем).
Ускорители могут служить не только как источники заряженных частиц. С их помощью возможно получать тормозное рентгеновское излучение и нейтроны.
Ускорители заряженных частиц делятся на две группы: ускорители прямого и непрямого действия. К первой группе относятся ускорители, в которых ускоряемая частица приобретает полную энергию при однократном прохождении высоковольтного промежутка. При этом энергия электрона (в Мэв} численно равна разности потенциалов ускоряющей трубки (в Мв). Такими ускорителями являются ускоритель Кокрофта—Уолтона, электростатический генератор (ускоритель Ван-дер-Граафа), резонансный трансформатор и т. д. Ко второй группе принадлежат ускорители, в которых ускоряемая частица многократно проходит ускоряющие промежутки, постепенно накапливая энергию. Ускорители непрямого действия бывают циклические (спиральная или круговая траектория ускоряемой частицы) и линейные (прямолинейная траектория). К этой группе ускорителей относятся линейный ускоритель, бетатрон, циклотрон, синхротрон и т. д.
Ниже рассматриваются принципы действия и рабочие характеристики ускорителей, наиболее часто применяемых в радиационной химии.
Ускоритель Кокрофта — Уолтона. Этот ускоритель состоит из двух частей (см. рис. 35): вертикальной секционированной трубки прямого ускорения (на рис. 35 она находится на переднем плане) и каскадного вентильно-конденсаторного умножителя напряжения.
Умножитель напряжения создает высокую разность потенциалов (до 1,5—1,8 Мв}, и электроны, генерированные вольфрамовым катодом в трубке прямого ускорения, приобретают в этой трубке энергию, соответствующую указанной разности потенциалов. Техника использования этого ускорителя в радиационно-химических экспериментах описана в статье [58].
Электронный пучок, создаваемый рассматриваемым ускорителем, характеризуется моноэнергетпчностыо. Обычно ускоритель Кокрофта — Уолтона работает в стационарном режиме, т. е. генерирует непрерывный пучок электронов. Одпако с помощью специальных схем можно получать как одиночные импульсы, так и импульсы, следующие с определенной скоростью. Подробная схема для генерации импульсов длительностью 5-10-6 сек. прп-
ведена в работе [59]. Пучок из вакуумной ускорительной трубки выводится в атмосферу через выпускное окно, изготовленное обычно из тонкой бериллиевой фольги.
Сила тока в непрерывном пучке — до 1,5—2 ма, в одиночном импульсе продолжительностью 5-10-7 сек.— до 0,8 а [60]. С помощью этого ускорителя в Советском Союзе в конце 50-х — нача ле 60-х годов выполнены разнообразные радиационно-химические работы, часть из которых освещена в книге [61].
Недостатками ускорителя Кокрофта — Уолтона являются его громоздкость и сравнительно низкая энергия электронного пучка, а достоинствами — простота эксплуатации и надежность в работе.
Рис. 35. Общий вид ускорителя Кокрофта — Уолтона на 1,2—1,5 Мэв 68
Электростатический генератор (ускоритель Ван-дер-Граафа). Этот ускоритель, предложенный в 1927—1931 гг. Ван-дер-Граафом, является самым распространенным среди ускорителей прямого действия. По данным [62], в 1962 г. в радиационных исследованиях использовалось 75 электростатических генераторов. Внешний вид одного из таких ускорителей показан на рис. 36.
Ускорители данного типа обычно генерируют непрерывные пучки электронов с энергией от 0,5 до 5 Мэв. Принцип работы их сравнительно прост (см. рис. 37). Электрический заряд переносится изолированным от земли ленточным транспортером от зарядного устройства к высоковольтному электроду. Этот заряд нано-
Рис. 36. Внешний вид электростатического генератора на 4 Мэв (бак поднят)
сится на поверхность транспортера вблизи нижнего шкива; величина заряда удваивается с помощью перезарядного устройства. Верхний шкив, расположенный под высоковольтным колпаком — кондуктором, находится под полным напряжением, создаваемым генератором. Вакуумная ускорительная трубка располагается рядом с транспортером. Она состоит из катода (источника электронов) и электронно-оптической системы для фокусировки пучка. Вся система помещена в бак, заполненный газом (смесью N2 и
Рис. 37. Схема электростатического генератора
1 — зарядный гребень;
2 — блок питания;
3 — нижний шкив;
4 — лента из изоляционного материала;
5 — верхний шкив;
6 — металлическая полусфера;
7 — металлический кожух;
8 — блок питания нити накала;
9 — нить накала;
ю — делитель напряжения;
11 — вакуумированная ускорительная трубка;
12 — катушка развертки;
13 — выпускное окно
О2 или SF6) под большим давлением, что необходимо для исключения коронного разряда. Выпуск пучка из трубки осуществляется через окно, изготовленное из тонкой алюминиевой или титановой фольги.
В табл. 19 приведены характеристики непрерывных пучков электронов, создаваемых некоторыми ускорителями Ван-дер-Граафа. Импульсное излучение получают с помощью специальных электронных схем (см., например, [63]). Характеристики импульсов электронов, генерируемых рядом ускорителей Ван-дер-Граафа, даны в табл. 20.
Резонансный трансфюрматор. Главными частями этого ускорителя являются низкочастотный (50—180 гг?) трансформатор и высоковольтная ускорительная трубка, помещаемые в металлический бак со сжатым газом (фреон, элегаз и др.). Трансформатор генерирует высокое напряжение; коаксиально в него вмонтирована ускорительная трубка. Сам трансформатор состоит из первичной и вторичной обмотки; между ними находится магнито-провод. Электроны испускаются инжектором с управляющим электродом. Питание инжектора, управляющего электрода и накала катода производится напряжением с части вторичной обмотки. Схема резонансного трансформатора показана па рис. 38 [78]. Подробно устройство и принцип работы этих машин рассмотрены в обзоре [79].
Таблица 19
Характеристики непрерывных пучков электронов, генерируемых ускорителями Ван-дер-Граафа
Ускоритель Энергия, Мэв Ток в пучке, ма Литература
ЭГ-2,5 (СССР) 1—2,5 -0,25 [7]
«High Voltage Eng. Corp.» 1,5 1,67 [62]
3,0 1,0 [62]
4,0 1,0 [62]
Таблица 20
Характеристики импульсов электродов, генерируемых различными ускорителями
Тип ускорителя Энергия электронов, Мэв Длительность импульсов, мксек. Максимальный ток в импульсе, а Литература
Ускоритель типа Кокрофта — Уолтона Ускоритель типа Ван-дер Граафа 0,8—1,2 0,5 п 5 0,8 [59, 60]
«High Voltage Eng., Ltd.» 2,5 0,5-2 2,5 [64]
3 0,001—0,1 5 [65]
3 5 1 [65]
«Mitsubishi» 3 0,3—3,5 0,2 [66]
Линейный ускоритель «Metropolitan Vickers Ltd.» 1,8 2 0,5 [67]
Линейный ускоритель «Associated Electric Industries Ltd.» 4 0,2 или 2 0,1 [68]
Линейный ускоритель «Applied Radiation Corp » 13 0,4-5 0,15 [69]
Линейный ускоритель «Varian 7 1,5—5 0,18 [70]
Associated» 4 0,006—0,05 0,5 [61]
4 1,6 0,32 [71]
Линейный ускоритель «Vickers 48 0,035 1,0 [72]
Eng.» 48 -2-Ю-5 15 [72]
Линейный ускоритель «Applied 11 0,5—10 2,5 [73]
Radiation Corp.» 19—21 0,01 20 [73]
Линейный ускоритель «У-12» 4—5 2—3 0,1—0,2 [74]
Линейный ускоритель «Электро-ппка» 5 4 1,0 [75]
Ускоритель «Febetron 703» («Field Emi'^ion Corp.») 2 0,05 5-103 [76]
Ускоритель «Feoetron-706» («Field Emission Corp.») 0,5—0,6 0,003 до ~10* [77]
Рис. 38. Схема резонансного трансформатора
1 — газоохладитель; 2 — днище котла; з — упругий прижим: 4 — первичная обмотка;
5 — вторичная обмотка; в — медный разрезной цилиндр; z — инжектор; л — кольцевой магнитопровод; 9 — крышка керна; 10 — вентилятор; 11 — крышка мапштопровода;
12 — конусный магнитопровод; 13 — головка; 14 — система управления инжектором;
15 — изоляционная лента; 16 — секция центрального керна; 17 — эквипотенциальные зкраны; 1з — ускорительная трубка; 10 — котел; 20 — донный магнитопровод
Резонансные трансформаторы появились в конце 30-х годов. В то время они применялись для генерации рентгеновских лучей в терапевтических целях. С 1948 г. они стали использоваться как источники быстрых электронов. Сейчас резонансные трансформаторы выпускаются в СССР, США, ФРГ, Японии. Институтом ядер-ной физики Сибирского отделения АН СССР разработан резонансный трансформатор, имеющий следующие характеристики [80]:
Напряжение питания первичной обмотки 220/380 в
Частота 50 ец
Максимальное напряжение на вторичной обмот- 1,7 Мв ке в режиме холостого хода
Диапазон изменения ускоряющего напряжения 0,4—1,5 Мв
Средняя мощность пучка (при напряжении на 25 кет вторичной обмотке 1,5 Ми)
Импульсная мощность пучка 150 кет
Среднее значение силы тока в основном режиме 17 ма
Резонансные трансформаторы находят широкое применение в радиационно-химической технологии. В частности, на базе этого ускорителя производится радиационное модифицирование кабельной полиэтиленовой изоляции [57].
Линейный ускоритель. С помощью линейных ускорителей, которые появились в начале 50-х годов, электроны могут быть ускорены до очень больших энергий. Принцип действия линейного ускорителя состоит в использовании высокочастотного электрического поля, приложенного к линейной периодической системе
Рис. 39. Внешний вид линейного ускорителя «У-12»
электродов, причем частота этого поля постоянна и находится в резонансе с движением частиц. Одна и та же частица проходит все ускоряющие промежутки при одной и той же фазе электрического поля и приобретает энергию в каждом промежутке. На рве. 39 показан внешний вид линейного ускорителя «У-12», часто используемого в радиационно-химических исследованиях в СССР.
Схема типичного линейного ускорителя приведена на рис. 40 [81 ]. Бегущие волны высокочастотной мощности генерируются магнетроном или клистроном. Эти волны направляются в волновод. В последний вводятся импульсы электронов, которые ускоряются волной до высокой энергии. Подробно устройство и принципы действия различных электронных ускорителей рассмотрены, в частности, в работах [9, 81].
Характерной особенностью линейных ускорителей является то, что они генерируют не непрерывный пучок электронов, а импульсы электронов, следующие с определенной частотой. С их помощью можно получать и одиночные импульсы электронов. При этом облучаемая система за каждый импульс получает значительную дозу. Вследствие этого линейные ускорители широко применяются в импульсном радиолизе.
Рис. 40. Схема типичного линейного ускорителя
1 — ввод высокочастотной мощности; 2 — питание волновода; з — электронная пушка' 4 — нагрузка; 5 — волновод; 6 — обратная связь; 7 — область формирования пучка; 8 — блок сдвига фазы; 9 — ускоряющая часть; ю — фокусирующие катушки; ц — выпускное окно
Обычная длительность импульсов — от микросекунд до наносекунд (см. табл. 20). Однако импульс имеет тонкую структуру. На рис. 41 в качестве примера показана тонкая структура электронного импульса, генерируемого линейным электронным ускорителем фирмы «Applied Radiation Corp.» в Аргоннской национальной лаборатории США [73]. Такая тонкая структура также используется в настоящее время в импульсном радиолизе. Характеристики импульсов, создаваемых различными линейными ускорителями, приведены в табл. 20.
В СССР большая серия линейных ускорителей, предназначенных для использования в радиационной химии и радиационно-химической технологии, разработана Московским инженерно-физическим институтом (серия У) и Научно-исследовательским институтом электрофизической аппаратуры им. Д. В. Ефремова в Ленинграде (серия ЛУЗ). Параметры этих ускорителей приведены в табл. 21 [9, 74, 82—86].
Рис. 41. Тонкая структура электронного импульса, генерируемого линейным ускорителем фирмы «Applied Radiation Corp.» (США)
1 — макроимпульс;
2 — отдельный микроимпульс.
I — ток в импульсе
Ускоритель «Febetron». В 60-х годах фирмой «Field Emission Corp.» (США) были сконструированы очень мощные и сравнительно дешевые импульсные электронные ускорители: «Febetron-705» и «Febetron-706», нашедшие широкое применение в радиационной химии (преимущественно для импульсного радиолиза). На рис. 42 показан внешний вид первого из этих ускорителей. Пх рабочие характеристики приведены в табл. 20.
Таблица 21
Параметры отечественных линейных ускорителей серий У и ЛУЭ
Тип ускорителя Энергия, Мэв Мощность пучка, кет Длительность импульса, мксек. Число импульсов, в сек.
У-10 3 1,0 2,5 400
У-12 5 0,6 2,2—3,0 400
У-13 10 0,6 3,0 400
У-16 1,4—2 1,2 2,5 400
У-27 10 5 3-5 430
У-33 3 И 3-5 430
ЛУЭ-8-5 8 5—7 2,8 31—500
ЛУЭ-8-5В 8 5 2,8 31—500
ЛУЭ-15-10 13 До 10 5,5; 2,5; 0,5 150; 300; 600
.1УЭ-15-10В 13 До 10 5,5 150
Рис. 42. Внешний вид ускорителя «Febetгоп-705»
Рис. 43. Принципиальная схема ускорителя «Febetron-705»
1 — выходное окно; 2 — магнит; 3 — эмиссионная трубка; 4 — внешняя изолирующая камера; 5 — генератор импульсов; 6 — модульная камера; [7 — передняя коническая камера
На рис. 43 дана принципиальная схема ускорителя «Febetron-705» [641. Он состоит из трех камер. Внешняя камера с целью общей изоляции заполнена фреоном под давлением. В центральной камере, содержащей модульную трубу, находится воздух под высоким давлением. В передней конической камере, залитой трансформаторным маслом, находится эмиссионная трубка и фокусирующее устройство. Модульная труба содержит 80 модулей, причем каждый из них включает два высоковольтных керамических конденсатора. Модули соединены параллельно; они заряжаются триггерным импульсом на 10 кв до напряжения 20—35 кв. Разряд модулей происходит серийно; высоковольтный импульс (1—2 Мв)
Рис. 44. Форма электронного импульса, создаваемого ускорителем «Febetron-705»
налагается на катод эмиссионной трубки, которая и генерирует электронный импульс. Главной частью эмиссионной трубки является катод. Он изготовлен из очень остроконечных вольфрамовых игл. Когда к этим иглам приложено высокое напряжение, они вследствие эффекта эмиссии поля испускают большое количество электронов.
Ускорители типа «Febetron» — мощные источники электронных импульсов. В частности, с помощью ускорителя «Febe-tron-706» возможно получать мощности дозы до — 2-1016 рад!сек. На рис. 44 в качестве примера приведена форма электронного импульса, создаваемого ускорителем «Febetron-705» [64].
Бетатрон. Действие этого ускорителя заряженных частиц основано на том, что изменяющееся во времени магнитное поле индуцирует в окружающем пространстве вихревое электрическое поле. Электрон, двигаясь в таком поле по круговой траектории и находясь под непрерывным воздействием электрической силы, приобретает определенную энергию. Когда энергия ускоряемых частиц достигнет требуемой величины, они направляются на мишень с целью генерации тормозного рентгеновского излучения или же выводятся из машины в виде электронного пучка. Поскольку электроны «впрыскиваются» в магнитное поле импульсами, то генерируемое бетатроном излучение является импульсным. Бетатронное излучение может иметь весьма высокую энергию (до 300 ЛГэв). Детально с принципами работы рассматриваемого ускорителя можно ознакомиться, например, в книгах [87—89].
В настоящее время бетатроны в радиационной химии используются сравнительно редко. Большее применение они находят в радиотерапии и металловедении.
Электронные ускорители можно использовать и для генерации тормозного рентгеновского излучения. С этой целью на пути электронного пушка ставят мишени из материала с высоким атомным номером (вольфрама, золота и т. п.). Получаемое излучение имеет непрерывный спектр энергий, причем максимальная эпергпя равна энергии электронов. Максимальные средние мощности * дозы такого излучения — порядка нескольких тысяч рад/сек.
С помощью многих описанных ускорителей можно также генерировать пучки тяжелых заряженных частиц. В этом случае вместо вольфрамового или танталового катода применяются соответствующие «инжекторы» ионов. В частности, в ускорителе Ван-дер-Граафа в качестве «инжектора» используется плазма, созданная газовым разрядом. Однако в радиационной химии наиболее распространенным источником положительных ионов является циклотрон.
Циклотрон. Эта машина используется для ускорения попов до больших скоростей без применения высокого напряжения. Поны образуются в газовом разряде низкого давления. Они ускоряются электрическим полем высокой частоты, приложенным между двумя полукруглыми полыми электродами, называемыми дуантами. Последние находятся внутри вакуумной камеры симметрично между полюсами электромагнита. Ионы, пройдя полукруг внутри одного из дуаптов, возвращаются к диаметральной между-электродной щели. При этом время, необходимое иону для прохождения полуокружности, равно времени изменения полярности электрического поля высокой частоты. Пон претерпевает новое ускорение, и вследствие более высокой скорости путь его внутри второго дуанта лежит вдоль полуокружности большего радиуса (см. рис. 45). Угловая скорость в однородном магнитном поле постоянна; поэтому время, необходимое для прохождения полуокружности, не зависит от скорости иона. Тем самым ноны ускоряются каждый раз, когда пересекают щель. Двигаются они по полуокружности все большего и большего радиуса. Отклонение ионов от спирального пути производится с помощью отрицательно заряженной отклоняющей пластппы. Пучок попов выходит наружу через тонкое окно в вакуумной камере. Подробно принципы действия и устройство циклотронов изложены, например, в книгах [б, 87, 90].
Пучок ионов, генерированный циклотроном, является, по существу, непрерывным. Дейтроны (ионы дейтерия) и протоны
* При работе с сериями импульсов различают мгновенную и среднюю мощности дозы. Мгновенная мощность дозы — это мощность дозы в расчете на время действия импульсов. Средней называется мощность дозы в расчете на все время облучения, включая промежутки между импульсами.
ускоряются с помощью этой машины обычно до энергий -— 20 Мае, а гелионы (ядра гелия) — до энергий ~ 40 Мэв. Такие циклотроны дают потоки попов порядка 1013 — 1014 и он/сек (ток в пучке — до нескольких десятков микроампер). Для получения пучков ионов более высокой энергии используются особые машины: протонный синхротрон и циклотрон с модулированной частотой.
Ускорители заряженных частиц применяются и для генерации нейтронов. С этой целью используются преимущественно ядерпые реакции типа (р, п) и (d,n). К ним относятся реакции: D (d, п) 3Не, 7Li (р, п) 7Ве, Т (р, п) 3Не, Т (d, п) 4Не п др. Для получения нейтронов пучок заряженных частиц направляется на мишень (например,
Рис. 45. Схема многократного ускорения иона между дуапта-ми и отклонения пучка в циклотроне
па тонкий слой дейтерия или трития, отделенный фольгой от вакуумной системы). Реакции типа (р, п) используются как источники нейтронов сравнительно малой энергии; реакции типа (d, п) позволяют получать нейтроны более высоких энергий (до 20 Мэв). Нейтроны, генерируемые этим способом, характеризуются моно-энергетичпостью. Интенсивность нейтронных пучков, получаемых с помощью рассмотренных генераторов, достигает — 2-Ю11 п!сек.
Рентгеновские трубки. Эти аппараты, как следует из их названия, служат для генерации рентгеновского излучения. Обычно они рассматриваются отдельно. Однако, поскольку в них также происходит ускорение заряженных частиц (электронов), они включены в этот раздел.
Рентгеновская трубка состоит из вольфрамового катода и массивного анода, помещенных в вакуумированный стеклянный баллон. Для создания потока электронов катод нагревается; электроны ускоряются под действием потенциала, приложенного между катодом и анодом. Попадая на апод, изготовляемый, как правило, из вольфрама, ускоренные электроны генерируют рентгеновское излучение. Величина указанного выше потенциала определяет максимальную энергию рентгеновских лучей. В пределе эта энергия составляет несколько сотен килоэлектронвольт.
Рентгеновские трубки широко используются для диагностических и терапевтических целей. Применяются они п в радиационной химии. Если до второй мировой войны и в первые годы после нее эти аппараты служили довольно часто химикам-радиационни-
кам в качестве источников ионизирующего излучения, то сейчас применение их в радиационной химии ограничивается главным образом студенческими практикумами. Из числа отечественных рентгеновских аппаратов для проведения радиационно-химических экспериментов наиболее подходящими являются установки РУП-300 и РУП-400. Методические особенности использования рентгеновских трубок в радиационной химии рассмотрены в статье [91].
Заслуживает специального рассмотрения вопрос о переделке рентгеновского дефектоскопического аппарата РУП-400-5 в ускоритель электронов. Эта переделка, произведенная авторами работ [92—94]. состоит в замене у трубки аппарата непроницаемого для электронов антикатода выпускным окном из алюминиевой фольги толщиной 20—30 мк, а также в создании системы постоянной откачки до вакуума 10~5—10~® мм рт. ст. Энергия электронов, генерируемых этим аппаратом, равна 0,3 Мэв, а мощность дозы составляет несколько мегарад в сек. Установка весьма подходит для облучения плепок и покрытий из полимерных материалов толщиной до 0,5 мм.
4. Ядерный реактор
как источник ионизирующего излучения
Первый ядерный реактор был сконструирован в 1942 г. в Чикаго. В настоящее время ядерные реакторы имеют многие важные применения; используются они и как источники ионизирующих излучений. Мы не будем сколь-лпбо подробно останавливаться на принципах действия ядерного реактора (они общеизвестны) и физических аспектах этой проблемы, которые детально изложены в специальной литературе (см., например, [95—97]). Здесь будет проведено лишь краткое рассмотрение особенностей излучений этого аппарата, представляющих особый интерес для химиков-радпационнпков. Обстоятельная характеристика реакторного излучения дана в работах [3, 98. 99].
Любой образец, помещенный в ядерный реактор, подвергается воздействию нейтронов и у-лучей. Кроме того, на него могут действовать р-частпцы, которые образуются прп распаде продуктов деления, а также других радиоактивных изотопов, возникших в результате захвата нейтронов различными педелящпмися материалами реактора. Взаимодействие образца с нейтронами и '(’-лучами может привести к появлению быстрых электронов, позитронов, протопов и а-частиц. Другой вид излучения реактора — это осколки деления. Однако они имеют малые пробеги и их энергия, как правило, поглощается в самом ядерпом горючем. Деление сопровождается образованием нейтрино. Эти частицы практически не взаимодействуют с веществом, и поэтому их можно не учитывать при рассмотрении радиационно-химических эффектов, производимых реакторным излучением. Природа возникновения разлпч-
пых видов излучения реактора иллюстрируется рис. 46, заимствованным из работы [1U0].
В большинстве исследовательских реакторов на тепловых нейтронах в качестве делящегося материала используется 235U. Распределение энергии процесса деления по различным компонентам излучения приведено в табл. 22 [89]. В случае плутония распределение энергии аналогично.
Некоторые сведения о нейтронном излучении реактора уже были приведены в главе I (см. стр. 37). у-Излучение реактора подразделяется на 4 вида: мгновенное у-излучение, сопровождающее деление; у-излучение распада продуктов деления; у-излучение, образующееся в результате захвата нейтронов ядрами различных конструкционных материалов (это так называемое захватное у-излучение), и у-излучение, возникающее при неупругом рассеянии быстрых нейтронов. Общая энергия осколков деления составляет ~ 80% от энергии деления. Вследствие этого осколки деления представляют значительный интерес с точки зрения использования их кинетической энергии для осуществления радиа-
1’ис. 46. Природа различных видов реакторного излучения
Таблица 22
Распределение энергии процесса деления 235U под действием тепловых нейтронов но различным компонентам реакторного излучения
Вид излучения Средняя энергия на одно деление, Мэв Поглощение энергии в реакторе
Осколки деления 167 Полностью поглощается в уране или оболочке
Мгновенные нейтроны 5 Энергия теряется главным образом в замедлителе
Мгновенное 7-излучение 6—7 50% энергии поглощается в уране •1; остальное — в замедлителе и конструкционных материалах
Излучение продуктов деления 6—7
(З-Излучение продуктов де- 8 Почти полностью поглощается в
леппя уране и оболочке
Захватное р-излучеппе 6*2 Поглошается в замедлителе и конструкционных материалах
Нейтрино 12 Не поглощается в реакторе
Суммарная поглощаемая энергия на одно деление 200
** Зависит от размеров и плотности урана.
*2 Зависит от степени обогащения, типа замедлителя, конструкционных и других материалов в реакторе.
ционно-химических процессов. Эти частицы, характеризующиеся высоким значением ЛПЭ, применяются в радиационной химии при изучении особенностей влияния линейной передачи энергии на радиолиз. Проблема использования указанной кинетической энергии включает ряд технических трудностей, обусловленных, в частности, малыми величинами их пробегов (см. табл. 14). Для этого разработаны различные методики. Например, для облучения смесей азота и кислорода с целью синтеза азотной кислоты предлагался реактор с топливом в виде тонких пленок с большой поверхностью [10Ц. Другие описанные источники осколков деления, использованные в радиационно-химических исследованиях, таковы: растворы солей уранила [102], тонкие взвеси окпслов урана в воде [103] или аммиаке [104], тонкие покрытия окисла урана на алюминии пли кварце [105], а также на стекловолокне [104].
Исследуемые образцы облучаются в активной зоне реактора или вне ее. В первом случае облучение осуществляется в специальных каналах, вводимых в активную вону обычно вместо какого-либо тепловыделяющего элемента (твэла) или группы твэлов.
Имеется два вида главных источников у-излучения, связанных с реактором. Это, во-первых, радиационные контуры. Описание их уже было дано на стр. 64. Во-вторых, к таким источникам относятся отработанные твэлы. Они могут использоваться после выгрузки их из реактора или после извлечения отдельных изотопов (2”®[J, 23!>Рп и т. п.). Однако сложность обращения с ними, а также сильное изменение интенсивности излучения во времени существенно ограничивают их возможное применение в радиационной химии и радиационно-химической технологии.
ЛИТЕРАТУРА
1. Radiation Sources (Ed. A. Charlesby). Oxford, Pergamon Press, 1964.
2. A. X. Брегер. Источники ядерных излучений и их применение в радиационно-химических процессах. М., Изд-во ВИНИТИ, 1960.
3. А. X. Брегер, Б. И. Вайнштейн, Н. II. Сыркус, В. А. Гольдин, Л. В. Чепель. Основы радиационно-химического аппаратостроения. М., Атом-пздат, 1967.
4. М. Ливингстон. Ускорители. М., ИЛ, 1956.
5. В. И. Векслер. Ускорители атомных частиц. М., Изд-во АН СССР, 1956.
6. А. А. Коломенский, А. Н. Лебедев. Теория циклических ускорителей. М., Физматгиз, 1962.
7. Е. Г. Комар. Ускорители заряженных частиц. М., Атомиздат, 1964.
8. В. А. Петухов, В. II. Котов. Современные ускорители частиц. М., Изд-во «Наука», 1965.
9. О. А. Вальднер. Линейные ускорители электронов. М., Атомиздат, 1966.
10. Д. А. Каушанский. Атомная энергия, 23, 475 (1967).
11. Б. Маноеиц. Труды II Международной конференции по мирному использованию атомной энергии (Женева, 1958). Избранные доклады иностранных ученых, т. 10. М., Атомиздат, 1959, стр. 230.
12. В. И. Синицын. Радиационные установки и их применение. М., ЦИТЭИН по атомной науке п технике, 1961.
13. А. X. Брегер, В. А. Белынский, С. Д. Прокудин. Атомная энергия, 1, № 4, 131 (1956).
14. А. X. Брегер, В. Б. Осипов, В. А. Гольдин. Радиоактивные изотопы и ядерпые излучения в народном хозяйстве СССР, т. 1. М., Гостоптехиз-дат, 1961, стр. 227.
15. А. С. Кузьминский, Г. С. Никитина, Е. В. Журавская, Л. А. Оксентъе-вич, Л. Л. Суница, 11. И. Витушкин. Труды II Международной конференции по мирному использованию атомной энергии. Доклады советских ученых, т. 4. М., Атомиздат, 1959, стр. 266.
16. А. В. Бибергаль, В. Л. Карпов, В. И. Синицын. Там же, т. 6, стр. 200.
17. Г. II. Пьянков, Н. В. Кулюпина. Атомная энергия, 19, 77 (1965).
18. Г. 11. Пьянков, М. А. Брашкин, II. В. Кулюпина. Атомная энергия, 19, 75 (1965).
19. А. X. Брегер, В. А. Белынский, В. Л. Карпов, С. Д. Прокудин. Действие ионизирующих излучений на органические и неорганические системы. М., Изд-во АН СССР, 1958, стр. 379.
20. А. X. Брегер, В. А. Белынский, В. Л. Карпов, С. Д. Прокудин, В. Б. Осипов. Труды Всесоюзной научно-технической конференции по применению радиоактивных и стабильных изотопов и излучений в народном хозяйстве и науке. Получение изотопов. Мощные гамма-установки. Радиометрия и дозиметрия. М., Пзд-во АН СССР, 1958, стр. 182.
21. А. В. Бабушкин, И. В. Вознесенская, II. Г. Жиров, В. И. Затуловский, Ю. Л. Хмельницкий. Там же, стр. 189.
22. A. X. Брегер, В. Б. Осипов, В. А. Гольдин. Атомная энергия, 8. 441 (1960).
23. С. М. Берлянт, В. Е. Дроздов, Э. Э. Финкель, П. А. Орленка, Л. М. Суроегин, А. X. Брегер, В Л. Карпов, В. А. Зорин. Атомная энергия, 21, 64 (I960).
24. Г. И. Волгин, Г. Н. Лисов, Д. Л/. У'орговицкий, В. К. Дроздов. Сб. «Радиационная химия», т. 2. М., Атомпздат, 1972, стр. 473.
25. В. Б. Осипов. Там же, стр. 475.
26. Е. А. Жихарев, В. М. Евдокимов, В. А. Борисевич, А. В. Полющиц. Там же, стр. 485.
27. Д. А. Каушанский, Г. М. Паиченков. Там же, стр. 491.
28. Д. А. Каушанский. Гамма-установка для микробиологических и радиационно-химических исследовании МРХ-у-100. М., Атомиздат, 1969.
29. Мощная радиационная техника (под ред. С. Джефферсона). М., Атомпздат. 1967.
30. J. А. Ghormley, С. J. Ilochanadel. Rev. Sci. Instruni., 22, 473 (1951).
31. H. A. Schwarz, A. O. Allen. Nucleonics, 12, No. 2, 58 (1954).
32. M. Burton. Nucleonics, 13, No. 10, 74 (1955).
33. В. Г. Фирсов. Атомная энергия, 2, 182 (1957).
34. Nucleonics, 16, No 7, 59 (1958).
35. Г. fl. Пьянков, .1/. .1. Брашкин, .1. M. Кабакчи, И. Н. Федчишин, В. П. Свиргун, Я. И. Лаврентович, А. Г. Старенький, Л, Л/. Коваленко, Л. Я. Гайче.нко. Изотопы в СССР, № 31, 3 (1973).
36. О. A. Kuhl, D. S. Ballantine. Radiation Sources (Ed. A. Charlesby). Oxford, Pergamon Press, 1964, p. 35.
37. Nucleonics, 16, No. 7, 108 (1958).
38. Nucleonics, 17, No. 7, 88 (1959).
39. Nucleonics, 19, No. 2, 96 (1961).
40. P. Leieque. Large Radiation Sources, vol. 1. Vienna, IAEA, 1960, p. 141.
41. 1). IF. George, J. JX. Gregory. Large Radiation Sources, vol. 1. Vienna, IAEA, 1960, p. 95.
42. J. Tunstall. Nucleonics, 18, No. 2, 100 (1960).
43. Nucleonics, 17, No. 5, 176 (1959).
44. A.F. Rupp. Proc. Intern. Conference on Peaceful Uses of Atomic Energy (Geneva, 1955), vol. 14. N. Y., 1956, p. 128.
45. Изотопы, источники излучений и радиоактивные материалы. Каталог, изд. 2. М., Атомиздат, 1962.
46. Nucleonics, 21, No. 5, 28 (1963).
47. А. С. Диндун, В. В. Гавар, Э. Я. Томсон. Радиационные контуры — источники гамма-излучения. Рига, «Зинатне», 1969.
48. Ю. С. Рябухин, С. Г. Тулькес, В. А. Гольдин, А. X. Брегер, А. С. Диндун, Б. С. Дорофеев, Ж. П. Упит. Industrial Uses of Large Radiation Sources, vol. 2. Vienna, IAEA, 1963, p. 189.
49. Г. И. Кикнадзе, В. Г. Гамбарян, Б. И. Литвинов, Р. Б. Людвигов, 3. Г. Размадзе, Л. И. Фельдман, В. М. Чантурия. Атомная энергия, 19, 176 (1965).
50. Г. И. Кикнадзе, А. II. Десипри, Д. М. Захаров, Л. В. Мельникова. Атомная энергия, 19, 178 (1965).
51. Е. Л. Ивантер, И. И. Рыбкин, С. П. Добровольский, А. X. Брегер, Ю. В. Давыдов, К. Ф. Козъменков, С. А. Требин, М. II. Беляев, В. А Сергеев, К. К. Кременецкий, В. И. Смирнов, Б. А. Савин. Сб. «Радиационная химия», т. 2. М., Атомпздат, 1972, стр. 516.
52. С. Г. Стоенко, В. Е. Дроздов. Там же, стр. 521.
53. Е. В. Волкова, А. В. Фокин, П. В. Зимаков. В. М. Беляков. Труды II Всесоюзного совещания по радиационной химии. М., Изд-во АН СССР, 1962, стр. 465.
54. F. N. Case, Т. S. Mackey. Large Radiation Sources for Industrial Processes, Vienna, IAEA, 1969, p. 633.
55. D. Chleck, C. Ziegler. Nucleonics, 17, No 9, 130 (1959).
56. E. J. Hart, J. Terandy. Rev. Sci. Instrum., 29, 962 (1958).
57. Э. Э. Финкель, С. С. Лещенко, P. JI. Брагинский. Радиационная химия и кабельная техника. М., Атомиздат, 1968.
58. Н. Я. Глазунов, Г. Б. Радзиевский. Сб. «Действие ионизирующих излучений на неорганические и органические системы». М., Изд-во АН СССР, 1958, стр. 395.
59. И. Я. Глазунов, А. К. Пикаев. Докл. АН СССР, 130, 1051 (1960).
60. А. К. Пикаев, П. Я. Глазунов. Докл. АН СССР, 154, 1167 (1964).
61. А. К. Пикаев. Импульсный радиолиз воды и водных растворов. М., «Наука», 1965.
62. Е. A. Burrill. Radiation Sources (Ed. A. Charlesby). Oxford. Pergamon Press, 1964, p. 85.
63. И. Hamler, K. Johnson, T. Klipperl. Nucl. lustrum. Methods, 47, 23 (1967).
64. IF. A. Seddon, C. Willis, M. J. Young, E. B. Selkirk. Report AECL — 3853, March 1971.
65. J. И'. Hunt, J. K. Thomas. Radiation Res., 32, 149 (1967).
66. 5. Arai, A. Kira, M. Imamura. J. Phys. Chem., 74, 2102 (1970).
67. E. J. Hart, J. IP. Boag. J. Am. Chem. Soc., 84, 4090 (1962).
68. J. P. Keene. J. Sci. lustrum., 41, 493 (1964).
69. L. M. Dorfman, 1. A. Taub, R. E. Buhler. J. Chem. Phys., 36, 3051 (1962J.
70. R. L. McCarthy, A. MacLachlan. Trans. Faraday Soc., 56, 1187 (I960).
71. IF. D. Felix, B. L. Gall, L. M. Dorfman. J. Phys. Chem., 71, 384 (1967).
72. M. J. Bronskill, J. IF. Hunt. J. Phys. Chem., 72, 3762 (1968).
73. II . J. Ramler, G. Macrogenes, E. Johnson. Report ANL— 7492, August 1968.
74. О. А. Вальднер, А. А. Глазков, A. II. Финогенов. Приборы и техника эксперимента, As 3, 29 (1963).
75. А. П. Быков, В. П. Марин, Л. В. Чепелъ, Ф. В. Шемаров. Всесоюзное научно-техническое совещание по использованию ускорителей в народном хозяйстве и медицине. Доклад № 4, 1971.
76. С. Willis, О. A. Miller, А. Е. Rothwell, A. W. Boyd. Adv. Chem. Ser., 81, 539 (1968).
77. G. A. Kenney, D. C. Walker. J. Chem. Phys., 50, 4074 (1969).
78. E. Абрамян, С. Вассерман, В. Вечеславов, В. Гапонов, В. Горбунов, А. Егоров, А. Алфимов. Large Radiation Sources for Industrial Processes. Vienna, IAEA, 1969, p. 661.
79. W. F. Westendorp. Radiation Sources (Ed. A. Charlesby). Oxford, Pergamon Press, 1964, p. 171.
80. E. А. Абрамян, В. А. Гапонов. Атомная энергия, 20, 385 (1966).
81. С. W. Miller. Radiation Sources (Ed. A. Charlesby). Oxford, Pergamon Press, 1964, p. 197.
82. О. А. Вальднер, E. Г. Пятнов, В. Д. Селезнев. Атомная энергия, 23, 467 (1967).
83. О. А. Вальднер, В. Ф. Викулов, О. С. Милованов, Е. Г. Пятнов, И. А. Смирнов. Ускорители, выл. 11. М., Атомиздат, 1969, стр. 16.
84. О. А. Вальднер, О. С. Милованов, В. А. Останин, В. Ф. Викулов, В. М. Светлов, И. С. Щедрин. Ускорители, вып. 12. М., Атомпздат, 1970, стр. 83.
85. В. М. Левин, В. Н. Мунтян, В. М. Николаев, В. В. Румянцев. Large Radiation Sources for Industrial Processes. Vienna, IAEA, 1969, p. 627.
86. В. M. Левин, В. M. Николаев, А. И. Семенов, Л. II. Фомин, В. И. Шахов. Сб. «Радиационная химия», т. 2. М., Атомпздат, 1972, стр. 509.
87. А. А. Воробьев. Ускорители заряженных частиц. М.— Л., Госэнерго-издат, 1949.
88. Л. М. Ананьев, А. А. Воробьев, В. И. Горбунов. Индукционный ускоритель электронов — бетатрон. М., Атомиздат, 1961.
89. А. А. Воробьев, В. А. Москалев. Сильноточный бетатрон и стереобетатрон. М., Атомиздат, 1969.
90. М. Ливингстон, М. Розе, М. Намиас. Циклотрон. М.— Л., Гостехтеор-издат, 1948.
91. В. И. Затуловский, Д. И. Нарядчиков. Сб. «Действие ионизирующих излучений на неорганические и органические системы». М., Изд-во АН СССР, 1958, стр. 406.
92. Д. М. Марголин, Л. А. Васильев. Сб. «Проблемы физической химии», выл. 3. М., Госхимиздат, 1963, стр. 134.
93. Г. Н. Пьянков, А. П. Мелешевич, Е. Г. Ярмилко, А. М. Кабакчи, С. И. Омельченко. Радиационная модификация полимерных материалов. Киев, «Техника», 1969, стр. 31.
94. Л. В. Чепелъ, Л. А. Васильев, Б. И. Витинг, Л. Н. Кузнецова. Сб. «Радиационная химия», т. 2. М., Атомиздат, 1972, стр. 501.
95. А. Вейнберг, Е. Вигнер. Физическая теория ядерных реакторов. М., ИЛ, 1961.
96. А. Д. Галанин. Теория ядерных реакторов на тепловых нейтронах. М., Атомиздат, 1959.
97. Е. М. Балабанов. Ядерные реакторы. М., Воениздат, 1957.
98. С. D. Bopp, W. W. Parkinson. Radiation Sources (Ed. A. Charlesby). Oxford, Pergamon Press, 1964, p. 1.
99. A. W. Boyd. Determination of Absorbed Dose in Reactors. Vienna, IAEA, 1971, p. 7.
100. D. C. Billington, J. H. Crawford. Radiation Damage in Solids. Princeton University Press, 1961.
101. M. Steinberg. Adv. Nucl. Sci. Technol., vol. 1. N. Y., Academic Press, 1962, Chapter 7.
102. J. W. Boyle, C. J. Hochanadel, T. J. Sworski, J. A. Ghormley, W. F. Kiff er. Proc. Intern. Conference on Peaceful Uses of Atomic Energy (Geneva, 1955), vol. 7. N. Y., 1956, p. 576.
103. B. L. Steele, S. Gordon, С. E. Dryden. Nucl. Sci. Eng., 15, 458 (1963).
104. B. Manowitz, M. Steinberg, J. W. Sutherland, P. Harteck, S. Dondes, J. H. Cusack. Third United Nations Intern. Conference on the Peaceful Uses of Atomic Energy. Geneva, 1964, paper 292.
105. A. W. Boyd, H. W. J. Connor. Can. J. Chem., 42, 1418 (1964).
Глава III
КРАТКАЯ ХАРАКТЕРИСТИКА
ПРИМЕНЕНИЯ ФИЗИЧЕСКИХ МЕТОДОВ ДОЗИМЕТРИИ В РАДИАЦИОННОЙ химии
В предисловии отмечалось, что поглощенная доза может быть определена физическими или химическими методами. Физические методы дозиметрии основаны на измерении величины какого-либо физического эффекта, обусловленного поглощением в веществе определенного количества энергии ионизирующего излучения. Таким эффектом могут быть ионизация, выделение тепла свечение и т. п. В зависимости от природы этого эффекта существуют различные физические методы дозиметрии: ионизационный, калориметрический, метод измерения заряда, индуцированного пучком заряженных частиц в веществе, сцинтилляционный и др. Многие из них являются абсолютными. Особенно это относится к калориметрическому методу, позволяющему измерять поглощенную дозу непосредственно в радах.
В радиационной химии физические методы используются преимущественно для калибровки химических дозиметров, а также в тех случаях, когда с помощью химических методов дозу определить невозможно (в частности, когда радиационно-химический выход в дозиметрической системе в условиях эксперимента неизвестен). Кроме того, физические методы применяются для поддержания одинаковых условий облучения в различных сериях опытов.
В настоящей главе кратко излагаются принципы наиболее распространенных физических методов дозиметрии и даются типичные примеры их применения в радиационной химии. Детально эти методы рассмотрены в специальной литературе (см., например, 11-9J). ‘
1. Ионизационный метод
В ионизационном метоле дозиметрии используется способность излучений высокой энергии производить ионизацию. В основе этого метода лежит принцип Брэгга — Грея [10—12J, гласящий, что количество ионизации в газовой полости есть мера энергии, поглощенной в окружающем материале. «Газовой полостью» может быть любой материал, отличающийся от окружающего материала. Однако обычно это — наполненная газом полость внутри твердого тела. Соответствующие приборы, называемые ионизационными камерами (твердое тело есть стенки камеры), чаще всего применяются для дозиметрии рентгеновского и у-излучений.
Принцип Брэгга — Грея математически выражается соотношением, носящим имя этих же ученых,
Е = ИУ7 s , (1)
т т т. ’ v '
где Ет — энергия, поглощенная единицей массы твердого тела;
— ионизация в единице массы газа; sm — отношение массовых тормозных способностей твердого тела и газа; W — средняя энергия, необходимая для образования в газе одной пары ионов.
Рассматриваемый принцип справедлив при следующих допущениях: 1) спектр электронов, образованных в твердом теле (т. е. в стенках камеры), не изменяется при наличии газовой полости; 2) генерация электронов в результате фотонных взаимодействий в полости ничтожна. Этп допущения выполняются в том случае, если размеры полости малы по сравнению с пробегом электронов, проходящих через нее. Однакс, если газ и твердое тело имеют одинаковый атомный состав (например, газ — это этилен, а твердое тело — полиэтилен), то это ограничение отпадает, поскольку, в соответствии с теоремой Фано [13J. «вереде данного состава, через которую проходит равномерный поток первичного излучения (типа у-лучей или нейтронов), поток вторичного излучения также равномерный; он не зависит от плотности и изменения последней от точки к точке».
Как видно из уравнения (1), доза Ет в материале стенок камеры может быть рассчитана из измеренной величины Ут1 если известны W и s,„.
Очень часто газом, наполняющим камеру, является воздух. Для него величина W в случае рентгеновского и у-излучений составляет 33,73 +0,15 эв [14]. МЕРЕ [15] рекомендует в случае воздуха использовать в расчетах значевие W = 33,7 эв (5,39-10 11 эрг) для рентгеновского и у-излучениЁ! с энергией квантов выше 30 кэв Отметим, что величина W зависит от природы газа и вида излучения. Например, в воздухе для o'-частиц W = 31,98 + 0,05 эв [14]. в гелии для рентгеновского и у-излучений W = 41,5 эв [14].
Массовые тормозные способности ряда элементов и воды по отношению к воздуху приведены в табл. 23 [16].
Принцип действия ионизационных камер общеизвестен. Если напряжение, приложенное к камере, увеличивать, то измеряемый ионизационный ток возрастает до тех пер, пока не достигнет величины насыщения (см. рис. 47). Это происходит тогда, когда все ионы, образованные излучением в полости, достигнут электродов. При меньших напряжениях часть образовавшихся ионов исчезает в результате рекомбинации. При сравнительно высоких напряжениях ионы приобретают энергию, достаточную для вторичной ионизации пии столкновениях. Вследствие этого ионизационный ток вновь возрастает.
Для данной камеры величина тока насыщения пропорциональна интенсивности излучения. Чаще всего методом измерения иони-
Таблица 23
Массовая тормозная способность по отношению к воздуху
Энергия электронов, Мэв S7n (без Учета эффекта плотности) sm (° Учетом °Ф“ фекта плотности
Н G N о р Ga Вода G Вода
0,001 3,564 1,079 1,018 0,965 0,962 0,598 1,254 1,079 1,254
0,002 3,223 1,062 1,014 0,973 0,752 0,685 1,223 1,062 1,223
0,003 3,084 1,055 1,013 0,976 0,776 0,720 1.211 1,055 1,211
0,004 3,004 1,051 1,012 0,978 0,791 0,741 1,203 1,051 1,203
0,005 2,949 1,049 1,011 0,979 0,800 0,755 1,198 1,049 1,198
0,006 2,904 1,045 1,0Ю 0,979 0,806 0,764 1,193 1,045 1,193
0,007 2,877 1,045 1,011 0,981 0,813 0,773 1,192 1,045 1,192
0,008 2,851 1,044 1,010 0,982 0,818 0,780 1,189 1,044 1,189
0,009 2,830 1,043 1,010 0,982 0,821 0,785 1,188 1,043 1,188
0,01 2,811 1,042 1,010 0,983 0,825 0,790 1,186 1,042 1,186
0,02 2,709 1,037 1,009 0,985 0,843 0,816 1,177 1,037 1,177
0,03 2,661 1,034 1,008 0,986 0,851 0,828 1,172 1,034 1,172
0,04 2,630 1,033 1,008 0,987 0,857 0,836 1,169 1,033 1,169
0,05 2,609 1,032 1,008 0,987 0,860 0,841 1,168 1,032 1,168
0,06 2,592 1,031 1,008 0,988 0,863 0,846 1,166 1,031 1,166
0,07 2,579 1,030 1,008 0,988 0,866 0,849 1,165 1,030 1,165
0,08 2,568 1,030 1,007 0,988 0,868 0,852 1,164 1,030 1,164
0,09 2,559 1,029 1.007 0,988 0,869 0,854 1,163 1,029 1,163
0,1 2,550 1,029 1,007 0,989 0,871 0,856 1,162 1,029 1,162
0,2 2,502 1,027 1,006 0,990 0,879 0,869 1,158 1,023 1,158
0,3 2,476 1,025 1,006 0,990 0,884 0,875 1,155 1,019 1,155
0,4 2,458 1,024 1,006 0,991 0,887 0.880 1.154 1,014 1,154
0,5 2,444 1,024 1,006 0,991 0,889 0,883 1,153 1,010 1,152
0,6 2,433 1,023 1,006 0,991 0,891 0,886 1,152 1,007 1,148
0,7 2,424 1,023 1,006 0,992 0,893 0,888 1,151 1,003 1,144
0,8 2,417 1,022 1,006 0,992 0,894 0,890 1,150 1,000 1,140
0,9 2,410 1,022 1,006 0,992 0,896 0,892 1,150 0,997 1,134
1,0 2,404 1,022 1,006 0,992 0,897 0,894 1,149 0,994 1,133
2 2,366 1,020 1,005 0,993 0,903 0,903 1,146 0,971 1,105
3 2,347 1,019 1,005 0,993 0,907 0,908 1,144 0,954 1,086
4 2,333 1,018 1,005 0,994 0,909 0,912 1,143 0,942 1,071
5 2,324 1,018 1,005 0,994 0,911 0,914 1,142 0,932 1,059
6 2,316 1,017 1,005 0,994 0,912 0,916 1,141 0,923 1,049
8 2,305 1,017 1,005 0,994 0,914 0,919 1,140 0,909 1,032
10 2,297 1,016 1,004 0,995 0,916 0,921 1,139 0,898 1,019
• Эффект плотности рассматривается на стр. 127.
зации в полости служит регистрация тока. Типичная электрическая схема, используемая в этом случае, показана на рис. 48. Величина ионизационного тока в условиях насыщения зависит не только от интенсивности излучения, но также от конструкции камеры. Обычно токи имеют значения от 0,1 до 100 мка [17].
Рис. 47. Типичная зависимость ионизационного тока от напряжения, приложенного к камере
Уже указывалось, что доза может быть вычислена по уравнению (1). Камеры, предназначенные для абсолютного измерения дозы в рентгенах, называются нормальными. В СССР такие измерения производятся эталонными установками, используемыми
Рис. 48. Типичная схема регистрации ионизационного тока
1 — камера;
2 — источник электрического поля;
з — электрометр
в следующих диапазонах энергий у-излучепия: 5—20, 20—60, 60—250 и 250—3000 кэв. Эти установки воспроизводят рентген с погрешностью, не превышающей 1,5—2%. Имеющиеся в лаборатории ионизационные камеры градуируют с помощью таких нормальных камер. Часто калибровку осуществляют в известных полях рентгеновского или у-излучений.
Ионизационные камеры обычно дают экспозиционную дозу в рентгенах. Поглощенная доза в воздухе, соответствующая 1 р, равна 0,869 рад (см. стр. 15). Тогда поглощенная доза в воздухе £)Возд, равная экспозиционной дозе R рентген, составляет
Двозд = 0,869/7 рад. (2)
При переходе от РВозД к дозе в исследуемой системе Dcne[ необходимо использовать формулу (см. также стр. 120).
(И(.,т/р)сисг сист
Деист = Двоздщ /Н) = 0.8697? - ,п, = /7? рад, (3)
“Иет/Ювозд Ювозд
где (|1с„/р)сист и (цег,/р)возд — массовые коэффициенты поглощения энергии для системы и воздуха.
В табл. 24 и 25 даны величины коэффициента / для ряда элементов и систем. Значения иеп/р приведены в главе IV. Там же обсуждается более подробно вопрос о пересчете поглощенной дозы при переходе от одной системы к другой.
Таблица 24
Значения множителя / для воды [16]*
Энергия фотонов, Мэв f •Энергия фотонов, Мэв f •Энергия фотонов, Мэв f Энергия фотонов, Мэв f
0,010 0,912 0,050 0,892 0,20 0,973 0,80 0,965
0,015 0,889 0,060 0,905 0,30 0,968 1,0 0,965
0,020 0,879 0,080 0,931 0,40 0,966 1,5 0,964
0,030 0,869 0,10 0,948 0,50 0,965 2,0 0,965
0,040 0,879 0,15 0,962 0,60 0,966 3,0 0,962
• Значения, приведенные в работе [16], пересчитаны с использованием Ш = 33,7эв.
Таблица 25
Значения множителя / для ряда элементов и полимеров [18]
Элемент или полимер Энергия фотонов, Мэв
0,1 0 5 1,0 2,0 5,0 10,0
н 1,54 1,74 1,74 1,35 1,74 1,60
с 0,811 0,877 0,877 0,834 0,877 0,864
N 0,844 0,877 0,877 0,877 0,877 0,876
О 0,877 0,877 0,877 0,894 0,77 0,880
F 0,905 0,832 0,830 0,880 0,835 0,850
Na 1,072 0,837 0,840 0,932 0,843 0,877
Mg 1,255 0,864 0,852 0,99 0,870 0,910
А1 1,395 0,845 0,842 0,991 0,852 0,900
Si 1,70 0,875 0,867 1,06 0,876 0,945
S 2,28 0,884 0,870 1,100 0,877 0,968
Cl 2,55 0,850 0,832 1,11 0,840 0,940
Fe 8,34 0,864 0,819 0,820 1,0 1,261
Полиэтилен 0,91 1,0 1,0 1,0 0,97 0,91
Натуральный каучук 0,90 0,99 0,98 0,99 0,96 0,90
Политетрафторэтилен 0,88 0,84 0,84 0,84 0,85 0,86
Ионизационный метод многократно (соответствующие ссыпки приведены на стр. 137) использовался для калибровки ферросуль-фатного дозиметпа (дозиметра Фрикке) *, широко применяемого в радиационной химии. При этой калибровке обычно измеряют ионизационный ток в полости. Исходя из этого, рассчитывают по соотношению Брэгга — Грея поглощенную дозу в материале стенки камеры, а затем и в дозиметрическом растворе. После этого определяют скорость образования ионов Fe3+, используя для этой цели сосуд, идентичный ионизационной камере и помещенный в то же положение относительно источника ионизирующего излучения. В этих экспериментах, помимо величин W и smr необходимо знать точный объем камеры. Этот сбъем сравнительно легко найти, заполнив его ртутью и затем взвесив ее. Определение объема может быть исключено, если примененная в калибровке камера проградуирована по нормальной камере. Эта методика использовалась, в частности, А. Фрегеном [19], который измерил G (Fe3+) для рентгеновских и у-лучей различной энергии.
Ионизационные камеры могут использоваться не только для дозиметрии фотонного излучения. Они применяются также для определения доз электронов высокой энергии и нейтронов. Например, Дж. Лафлин и сотр. f20| измерили G (Fe3+) длч дозиметра Фрикке ионизационным методом в случае электронов с энергией 10 и 20 Мэв. В этих опытах ионизация в воздушной полости опрепе-лялась в тех же условиях, в каких проводилось облучение электронами раствора ферросульфата. Полость находилась внутри полистиролового блока.
При дозиметрии потоков быстрых нейтронов используют способность их образовывать в водородсодержащих средах протоны отдачи. Поэтому стенки камер изготавливаются из материалов, богатых водородом (например, из полиэтилена). Для протонов величина W в случае воздуха составляет 35 эл(5,6-'|0 11 эрг) [16]. Ионизационные камеры пригодны, кроме того, для дозиметрии смешанного нейтронного и у-пзлучения реактора. В этом случае для оценки вклада каждого вида излучения в суммарную дозу измеряют ионизацию с помощью двух камер: 1) со стенками пз водородсодержащего вещества и 2) со стенками пз вещества, не содержащего водорода (папример, пз графита, алюминия плп тефлона). Часто камерой второго типа бывает камера со стенками из графита и полостью, заполненной СО,. Более подробно с ионизационными методами дозиметрии реакторного излучения можно ознакомиться. прочитав обзор [17]. В случае нейтронных потоков поглощенная доза в материале стенки камеры Dcr (в рад) равна
^ = 0,90^,
где .7т — заряд ионов в воздушной полости (единицы СГСЭ/с.мя)
* Дозиметр Фрикке подробно описывается на стр. 135.
(5)
и sm — отношение тормозных способностей материала стенки и воздуха для протонов, генерированных нейтронами.
Отметим, что в уравнении (4) стоит коэффициент 0,90, а не 0,869. Это обусловлено тем, что для протонов в воздухе, как уже отмечалось, W = 35 эв.
Для перехода от дозы в материале стенки к дозе в исследуемой системе НСист применяют формулу
сист ст 2 W^ct
где <т# — сечение рассеяния нейтронов для атома сорта i; ki — средняя доля потери энергии нейтронов, связанная с атомом сорта i и Nf — число атомов сорта i на единицу массы.
Значения о/с для Н, С, N, О, S и Р и энергий нейтронов от 0,1 до 10 Мэв приведены в табл. 26 [16].
Ионизационные камеры находят довольно широкое применение в радиационной химии для мониторирования пучков электронов высокой энергии и тормозного рентгеновского излучения, генерируемых различными ускорителями. В качестве примера рассмотрим кратко камеры, использованные с этой целью в Институте физической химии АН СССР в случае ускорителя-^Кокроф-
Таблица 26
Величины произведения о7с (в барн) для нейтронов
Энергия. М.>в н (к 0,5‘JJ) с (It = 0,1431) X (К = 0,1254) О (к = 0,1116) р (fc = 0,0611) S (4- = 0,0592)
о,1 6,3 0,66 0,6 0,30 0,21 0,42
0,15 5,4 0,63 0,51 0,39 0,15 0,24
0,2 4,8 0,60 0,46 0,40 0,13 0,41
0,3 3,95 0,56 0,40 0,44 0,15 0,13
0,4 3,45 0,52 0,36 0,67 0,13 0,18
0,5 3,1 0,49 0,30 0,65 0,17 0,12
0,6 2,8 0,46 0,20 0,36 0,14 0,11
0,8 2,4] 0,41 0.23 0,33 0,15 0,14
1,0 2,12 0,39 0,25 0,89 ~18 0,17
1,5 1,70 0,30 0,23 0,25 ~18 0,14
2 1,44 0,24 0,20 0,18 0,23 0,17
3 1,14 0,17 0,21 0,13 0,23 0,19
4 0,95 0,27 0,23 0,21 0,16 0,17
5 0,83 0,19 0,18 0,13 0,15 0,15
6 0,72 0,14 0,18 0,16 0,14 0,15
8 0,59 0,21 9,18 0,13 0,12 0,12
10 0,47 0,16 0,16 0,13 0,12 0,11
та — Уолтона [21, 22] (описание его было приведено на стр. 67). Для контроля указанных видов излучения камера помещается непосредственно у выходного окна ускорителя (электроны) или рентгеновской мишени насадки (тормозное излучение). При работе с электронным пучком применялась камера, ионизационный объем которой ограничен двумя алюминиевыми фольгами толщиной 12 мк. Собирающим электродом является третья (средняя) фольга. Ионизационный объем этой камеры имеет высоту 0,5 см. Напряжение, необходимое для создания условий насыщения, равно — 2 кв.
На рис. 49 приведена схема рентгеновской мишени-насадки и ионизационной камеры для контроля интенсивности тормозного излучения [22]. Стенки (толщина 0,5 с.и) камеры изготовлены из плексигласа. Сверху камера покрыта аквадагом (водным коллоидом графита). Собирающий электрод представляет собой металлическое кольцо, находящееся внутри камеры.
Рис. 49. Схема рентгеновской мшпепи-насадкп и ионизационной камеры
1 — мишень-насадка; 2 — ионизационная камера; 3 — выпускное окно ускорителя; 4 — схема регистрации ионизационного тока; 5 — ячейка с исследуемой системой. ИЛ — неоновая лампочка; mA — миллиамперметр
На основании изложенного можно заключить, что ионизационная камера может выполнять функции абсолютного дозиметра, если известна величина И' для газа, наполняющего полость. Кроме того, прокалиброванная ионизационная камера может служить вторичным стандартом в дозиметрии различных видов излучения. Легкость изготовления камер и простота аппаратурного оформления также являются достоинствами этого метода. К недостаткам следует отнести наличие для некоторых камер «хода с жесткостью», т. е. зависимости чувствительности от энергии излучения. Этот эффект наблюдается в тех случаях, когда газ и материал стенок камеры имеют различные эффективные атомные номера. При этом «ход с жесткостью» наиболее характерен в области низких энергий излучения. Особые сложности возникают также при использовании ионизационного метода для дозиметрии импульсного излучения. В этом случае существенно уменьшается эффективность собирания ионов по сравнению с непрерывным излучением.
Подробно ионизационный метод дозиметрии ионизирующих излучений изложен в работах [17, 23—26]. Сведения о конструкции ионизационных камер, предназначенных для различных целей, можно найти в статьях [17, 25—271.
2. Калориметрический метод
Калориметрический метод дозиметрии ионизирующих излучений основан на измерении повышения температуры облучаемой среды. Увеличение температуры в результате поглощения энергии излучения сравнительно небольшое. Например, в воде оно составляет приблизительно 2 град/Мрад. Поэтому для определения дозы рассматриваемым методом необходимо применять чувствительные способы измерения температуры. Особенно это характерно для низких мощностей дозы.
Калориметрия как метод дозиметрии начала применяться в начале этого столетия [28—30]. Она была использовала для нахождения энергии в случае небольших источников радиоактивного излучения. В 20-е годы дапный метод находил некоторое применение в дозиметрии рентгеновского излучения [31, 32]. Такое ограниченное использование калориметрии было обусловлено в первую очередь успешным развитием ионизационных методов. Однако в 40—50-е годы калориметрический метод претерпел своеобразное второе рождение. Вызвано это было достижениями в области микрокалорпметрии, появлением высокоинтепсив-пых источников ионизирующих излучений и введением рада в качестве единицы поглощенной дозы.
В настоящее время калориметрический метод широко используется в дозиметрической практике. По сравнению с ионизационным методом он имеет преимущества. При работе с калориметрами не нужно знать величину W, а часто и значение sm, что су
щественно облегчает проведение дозиметрических измерений. Это — абсолютный метод дозиметрии в полном смысле слова. Он, правда, менее чувствителен, чем ионизационный метод. Однако сравнительно низкие дозы, которые не могут быть измерены калориметрическим методом, почти не применяются в радиационной химии.
Принцип калориметрического метода ясен из соотношения
<1Т 1
~dlT = ~Мс (гРад!кал), (О
где dT — изменение температуры теплоемкостью с при поглощении
тела с массой М и удельной энергии излучения dE.
Рис. 50. Графитовый адиабатический калориметр
1 — калориметр;
2 — градуировочный нагреватель;
3 — прокладки из полистирола;
4 — термопара;
5 — термопары адиабатического контроля;
6 — нагреватель;
7 — оболочка;
8 — вакуумированное пространство
Известны различные варианты рассматриваемого метода дозиметрии. Наиболее широко применяются адиабатические и изотермические калориметры. В калориметрах первого типа теплообмен с наружной средой сведен к минимуму. В них скорость увеличения температуры образца пропорциональна скорости выделения тепла в образце. Калориметры второго типа работают в равновесных условиях. В них тепловой поток от образца равен скорости нагрева образца.
Приведем несколько примеров конструкций калориметров для дозиметрии рентгеновского и у-излучений. На рис. 50 показано устройство адиабатического калориметра, использованного для калибровки химических дозиметров [33]. Поглотителем в нем является графит. Этот материал удобен тем, что расход энергии ионизирующего излучения на химические реакции в нем ничтожен. Кроме того, он характеризуется сравнительно высокой теплопроводностью. Нагреватель служит для введеппя известного количества энергии при градуировке калориметра, т. е. при нахождении коэффициента пропорциональности между введенной энергией и повышением температуры. Последняя в рассматриваемом калориметре измеряется термопарой. Очень часто для этой
цели применяются также термисторы. Они изготовлены Из полупроводников, чье сопротивление уменьшается с ростом температуры.
Другими материалами поглотителей в калориметрах служат алюминий, вода и полиэтилен. Использование последнпх двух материалов осложняется расходом энергии излучения на химические реакции в них. К. Хоханадель и Дж. Гормли [34] применили адиабатический калориметр с водой в качестве поглотителя для калибровки дозиметра Фрпкке. Схема этого калориметра представлена на рис. 5'1. Вода заливалась в герметичную тонкостен-
Рис. 51. Калориметр для измерения поглощенной дозы у-излученпя в воде ] — тонкостенная колба из пирекса; 2 — вода; .? - апьсзон; J — медная оболочка; 5 — термопары; 6 — источник у-излучения С0Со; 7 — кожух источника; 8 —теплоизоляция из пенопласта
4 А. К. Пикаев
97
ную стеклянную колбу с посеребренными стенками и облучалась до тех пор, пока не устанавливалось равновесие в радиолизе воды и не прекращалось тем самым поглощение энергии в химических реакциях. В этом калориметре температура измерялась медь-константановыми термопарами. Температура медной оболочки с помощью нагревателя, вмонтированного в защитный блок источника, непрерывно поддерживалась равной температуре воды в колбе. Тем самым исключалась потеря тепла в окружающую среду. В расчетах дозы была сделана поправка на поглощение
Рис. 52. Водно-ледяной калориметр
1 — источник у-излучения "“Со; 2 - облучателышй канал; г — полихлорвпниловый ко-жух; 4 — капилляр; 5 — корковая пробка; 6 — колба от термоса; 7 — вода со льдом
излучения стенками колбы. Было принято, что величина энергии, поглощенной стеклом, пропорциональна его весу. Кроме того, было сделано допущение, что отношение доз, поглощенных в дозиметре Фрпкке и воде, равно отношению их удельных весов. В результате было найдено, что для у-лучей 60Со G (Fe3+) равен 15,6 + 0,3 пона/100 эв.
Калориметрическая калибровка дозиметра Фрмкке осуществлялась также в других работах. Ссылки на них приведены на стр. 137.
Согласно [35], для калибровки химических дозиметров весьма удобен водно-ледяной калориметр. Устройство его ясно из рис. 52. Принцип действия этого калориметра основан на уменьшении объема льда и воды вследствие плавления льда при поглощении системой энергии ионизирующего излучения. Это уменьшение объема измеряется с помощью капилляра, прокалиброванного взвешиванием количества ртути, помещенного между двумя метками. Точность измерения дозы этим калориметром составляет — 0,4%.
Адиабатические калориметры специальной конструкции позволяют определять сравнительно низкие мощности дозы у-из-
лучения. Например, в работе [36] описан калориметр для измерения мощностей дозы — 1 рад!сек с погрешностью 1,5—2%.
Изотермические калориметры чаще всего применяют для дозиметрии реакторного излучения. Это обусловлено тем, что в каналах реактора, охлаждаемых циркулирующей водой с постоянной температурой, пет необходимости создавать особые изотермические условия. В таких калориметрах температура оболочки поддерживается постоянной. Для них справедлива формула [37
dT ! dT\
dx \ dx /изм
Ь (Дюгл Д)бол) ’
(7)
где dT/dx — скорость изменения температуры поглощающего вещества; (dT/dx)il3M — измеряемое изменение температуры поглотителя; 7’погл и Т„еол — температуры поглощающего вещества и оболочки; к — коэффициент, характеризующий теплообмен между поглощающим веществом и оболочкой.
При использовании рассматриваемого метода для дозиметрии электронных пучков необходимость в сложной мпкрокалоримет-рической аппаратуре отпадает, поскольку увеличение темпера
туры поглотителя в этом случае сравнительно большое вследствие высоких мощностей дозы. Ниже в качестве примера излагается конструкция нескольких калориметров, примененных в дозиметрии этого вида излучения.
В работе [38] описан неравновесный изотермический калориметр для измерения энергии электронного пучка, генерируемого ускорителем Кокрофта — Уолтона (энергия электронов — до 1,2 Мэв). Схема этого калориметра приведена на рис. 53. Поглотитель 7 состоит пз плоской алюминиевой пластинки, покрытой со стороны входа пучка тонкими слоями бериллия и графита для уменьшения обратного рассеяния электронов. Внутри поглотителя вмонтирован электрический калибровочный нагреватель. Снизу к поглотителю плотно прижат термистор. Поглотитель подве
Рис. 53. Схема калориметра-коллектора
шен на капроновых питях внутри кожуха 2. Последний имеет
двойные стенки, между которыми циркулирует веда из термостата. К кожуху присоединен вакуумный насос ЦВЛ-100. позволяющий создавать в системе давление — (О-4 гпорр. Входным окном калориметра служит тонкая алюминиевая фольга 3. Кроме того, в верхней части кожуха располагалась алюминиевая диафрагма 4. Цифрой 5 обозначены стеклянные изоляторы для включения в схему термистора, нагревателя и корпуса поглотителя. Изменение температуры поглотителя записывалось на диаграммной ленте потенциометра ЭПП-09. При градуировке прибора тепловая
Рис. 54. Схема проточного калориметра
1 — внешний цилиндр;
2 — внутренний цилиндр:
3 — тонкая пленка.
Стрелки указывают направление движения жидкого поглотителя
мощность, выделяемая калибровочным нагревателем, сравнивалась со скоростью роста температуры поглотителя. Эти величины были пропорциональны. В расчеты дозы вносились поправки на теплообмен между поглотителем и кожухом, а также на выделение тепла в термисторе.
Рассмотренный калориметр был использован для дозиметрии не только непрерывного, но п импульсного пучка ускорителя Кокрофта — Уолтона. В последнем случае градуировка прибора осуществлялась при импульсном пагреве. Для этого конденсатор известной емкости, заряженный до определенного напряжения, разряжался через сопротивление калибровочного нагревателя.
При нахождении энергии электронов параллельно с измерением нагрева поглотителя за счет пучка производилась регистрация электронного тока в поглотителе (непрерывный пучок) пли заряда электронов в нем (одиночные импульсы). Зная дозу и количество электронов, легко рассчитать их энергию (см. стр. 103).
Согласно [39], для дозиметрии электронных пучков очень удобны калориметры проточного типа. В таких калориметрах движущийся поглотитель непрерывно выводит тепло из зоны облучения. Схема подобного калориметра приведена на рис. 54 [39]. Он состоит пз двух стеклянных цилиндров, вставленных один в другой. Со стороны входа пучка калориметр закрыт тонкой пленкой. Жпдкпй поглотитель (обычно вода) движется между цилиндрами, омывает пленку и поступает во внутренний цилиндр, где нагревается под действием электронного пучка. Температура поглотителя измеряется па выходе пз прибора. Калориметр, кроме того, снабжен термостатом со стабилизированным расходом жид
кости и прибором для контроля за ее расходом. Градуировка осуществляется градуировочным электронагревателем. При постоянных величинах расхода жидкости и разности температур окружающей среды п жидкости на входе в калориметр скорость увеличения температуры жидкости на выходе служит мерой мощности дозы электронного излучения. В расчеты вносилась поправка на отражение электронов и образование тормозного рентгеновского излучения. С помощью данного калориметра авторы работы [391 провели измерения мощности пучка электронов (энергия 1 Мэв), генерируемого ускорителем У-16.
В работах [40, 411 описан калориметрический метод дозиметрии мощных электронных импульсов ускорителя типа «Febet-гои-705» (энергия электронов — 2 Мэв). При исследованиях радиолиза жидкостей доза определялась с помощью калориметра, поглотитель которого представлял собой тонкий графитовый, алюминиевый или никелевый диск [401. Толщина последнего была меньше пробега электронов данной энергии. Поглотитель крепился в ячейке нейлоновым кольцом, которое служило также тепловой изоляцией. Ячейка была изготовлена из нержавеющей стали. Входным окном ячейки служила мембрана из этого же материала. Температура измерялась медь-коиоантановой термопарой. Схема этого устройства приведена на рис. 55. а. Во время измерений поглотитель находился в ячейке в том же положении, в котором затем облучался исследуемый образец. На рис. 55, б приведена также найденная зависимость показаний термопары от времени после, воздействия на алюминиевый диск^пмпульсом электронов. Отклонения 1 и 2 обусловлены изменениямиТинтенспв-ности^потока электронов в импульсе и~'наводкамп.” Величина
Рис. 55. Калориметрический метод дозиметрии импульсного излучения электронного ускорителя «Febetron-705»
а — калориметр (1 — мембрана из нержавеющей стали; 2 — поглотитель; з — термопара;
— ограничительное кольцо; 5 — нейлоновый держатель); б — изменение показаний термопары во времени после воздействия электронным импульсом
\v, полученная экстраполяцией кривой 3 до времени t = О, пересчитывалась в температуру, а затем в дозу за импульс. Для перехода от дозы в поглотителе к дозе в изучаемой жидкости учитывалось различие в тормозных способностях этих сред. В зависимости от материала диска и его толщины (чем тоньше диск, тем больше доза) доза изменялась от 1,65 (алюминиевый диск толщиной 0,439 см) до 5,7 Мрад (графитовый диск толщиной 0,081 см). Соответствующее этим дозам увеличение температуры равно 17,9 и 65,5° С.
Рис. 56. Зависимость средней дозы D в ячейке с газом от толщины d алюминиевого поглотителя
При изучении радиолиза газообразных систем под действием электронных импульсов ускорителя «Febetron-705» для измерения дозы применялись диски различной толщины, и доза находилась путем экстраполяции показаний калориметра до нулевой толщины поглотителя [41]. Кроме того, диски помещались в ячейке в различные положения, и находилась средняя доза для всего объема ячейки. Эти операции вызваны тем, что ячейки были сравнительно большого объема (— 50мл). Однако даже для такого объема газа при давлении 1 апгм эквивалентная толщина поглотителя весьма небольшая. На рис. 56 как пример показана зависимость средней дозы в ячейке от толщины алюминиевого поглотителя.
Из приведенного рассмотрения ясно, что в большинстве случаев аппаратурное оформление калориметрического метода довольно сложное. Поэтому чаще всего он применяется для калибровки более простых дозиметров.
В настоящем разделе была дана лишь краткая характеристика калориметрического метода. Детально он изложен в книге [42] и обзорах [37, 43, 44].
3. Метод измерения тока или заряда, индуцированного пучком заряженных частиц в облучаемой системе
При работе с пучками заряженных частиц очень часто дозу определяют путем регистрации тока или заряда, индуцированного пучком в облучаемой системе. Очевидно, такое определение
йозможно в том случае, если известна средняя энергия частиц в системе. При этом для электронных пучков доза D (в эв/сл<3) равна
6,25-102V£t
D =------v---, (8)
где I — ток в системе (в а); Е — средняя энергия частиц в системе (в Мэв); t — время облучения (в сек.); v — объем системы (в см3).
Для тяжелых частиц, имеющих заряд более единицы, необходимо в знаменателе дроби в правой части уравнения (8) поставить величину заряда. Например, для гелионов, имеющих заряд +2, в знаменателе должна быть цифра 2.
Данный метод пригоден для нахождения дозы в случае токопроводящих систем. Чаще всего он используется в исследованиях радиолиза водных растворов электролитов.
G целью иллюстрации рассмотрим, как метод измерения тока электронного пучка применялся Р. Шулером и А. Алленом [45] для калибровки ферросульфатного дозиметра. В их опытах источником электронов с энергией 2 Мэв служил ускоритель Ван-дер-Граафа. Определение энергии электронов, входящих в ячейку, осуществлялось путем нахождения пороговых энергий образования фотонейтронов в бериллии и дейтерии под действием тормозного рентгеновского излучения, генерированного электронным пучком в золотой мишени. Нейтроны, возникающие в бериллии или D2O, направлялись на родиевую фольгу, которая в результате этого активировалась. Количество активных атомов родия возрастает линейно при увеличении энергии электронов выше пороговой. Пересечение прямых, выражающих зависимость количества активных атомов от энергии электронов, с осью абсцисс дает значение пороговой энергии. Если найденная таким путем величина энергии совпадает с имеющейся в литературе, то это означает, что напряжение ускорителя прокалибровано правильно. Энергия электронов частично теряется при прохождении через мембрану ячейки, в которой находится ферросульфатная дозиметрическая система. В цитированной работе соответствующая поправка определялась измерением количества образованных ионов Fe3+ в зависимости от толщины мембраны (при прочих равных условиях) и экстраполяцией полученной прямой до нулевой толщины мембраны. Ток в растворе (его величина варьировалась в диапазоне от 10-10 до 10-7 а) регистрировался с помощью интегратора. Раствор во время облучения перемешивался магнитной мешалкой. В расчетах дозы учитывались поправки на возникновение в дозиметре Фрикке тормозного излучения и обратное рассеяние электронов. Таким путем было найдено, что G(Fel+) — 15,45 + 0,11 иона/100 эв.
Кроме метода определения пороговых энергий образования фотонейтронов, применяются также методы нахождения энергии электронного пучка по отклонению электронов в магнитном поле и их поглощению в некоторых материалах (обычно в алюминии).
Путем измерения тока электронного пучка, приходящегося на единицу поверхности облучаемого объекта, можно в случае объектов, состоящих из элементов с низким атомным номером (например, в случае воды) и сравнительно большими энергиями Е электронов (например, при Е 5 Мэе), когда рассеяние электронов весьма мало, рассчитать поглощенную дозу £)вх на входе в объект. Формула для расчета Z)BX (в Мрад) имеет следующий вид:
рвх = 10'’Шт, (10)
где I — сила тока (в а/с.м2); t — время облучения (в сек.) и sm — тормозная способность (в Мэв-см2[г).
Рис. 60. Схема контроля пучка электронов ускорителя
1 — выходное окно ускорителя;
2 — металлическая сетка;
3 — микроамперметр;
4 — электронный потенциометр ЭПП-09
Цилиндр Фарадея обычно используется для контроля пучков заряженных частиц. В случае токопроводящих жидкостей (например, водных растворов электролитов) электронный ток измеряют платиновым зондом, впаянным в радиолитическую ячейку. При этом зонд подсоединен к микроамперметру или интегратору. Возможны и другие способы постоянного контроля за мощностью дозы. Например, в работе [49] с этой целью применялась схема, показанная на рис. 60. Пучок электронов (энергия — 0,4 Л1эв) проходил через редкую сетку из вольфрамовой проволоки диаметром 0,5 мм. Величина тока на сетке записывалась на диаграммной ленте самопишущего электронного потенциометра ЭПП-09.
4. Определение поглощенных доз
в случае внутренних источников излучения
В главе II отмечалось, что в радиационной химии находят некоторое применение внутренние источники излучения, которые представляют собой радиоактивные изотопы, равномерно распределенные внутри исследуемой системы. Поскольку пробеги |3- и а-частиц обычно гораздо меньше размеров сосуда, в который помещена эта система, то вся энергия, переданная при распаде ионизирующим частицам полностью поглощается системой. Естественно, при сравнительно малых размерах сосуда энергия,
Переданная у-лучам и антинейтрино, практически не вносит вклада в поглощенную дозу; поэтому данную энергию можно не рассматривать.
Пусть удельная активность [3- или а-излучателя в системе равна с кюри/г, а средняя энергия частицы составляет Е Мэв. Тогда мощность поглощенной дозы
Р - 3,7 10В * * 11 * * * *с£ [as/ (г-сек)] - 593с£ рад/сек. (И)
Вследствие распада радиоактивных изотопов уравнение (11) характеризует мощность дозы в системе на момент измерения активности. Поэтому при определении дозы необходимо учитывать это изменение Р во времени. Из законов радиоактивности следует, что
ct = сие~7л, (12)
где с0 и с ( — удельные активности (в кюри/г) изотопа в начале облучения и по истечении времени t (в сек.); X — постоянная распада (в сек-1).
Тогда доза D, поглощенная системой за время t, равна
i i
D = 3,7- 1О1ПсоЕ е~*л<И (эв/г) = 593со£ e~y'ldt (рад). и о
Интегрируя, получаем
3,7-101Ссо£ 593со£ >,
D = ----(1 - е'7') (ю/г) = (1 - е~и) (рад).
Очевидно, наибольшая доза Da может быть получена при полном распаде радиоактивного изотопа, т. е. при t—► оо. В этом случае
3,7-1О16со£ 593во£
=-------х------(вб/в) = (рад^
(15)
В проведенных выше расчетах не было принято во внимание
то обстоятельство, что некоторая часть р- или tz-частиц может покидать систему, не передавая ей свою энергию. Рассмотрим,
каким образом учитывался этот эффект в работе [50] при определении G (Fe3+) для ферросульфатной дозиметрической системы при действии р-частиц 32Р. Дозиметрический раствор находился в
сферической колбе. Авторы цитируемой работы нашли, что по-
теря энергии p-излучения через стенки колбы уменьшается при бвеличении радиуса г колбы. В соответствии с этим величина
y(Fe3+) была получена экстраполяцией экспериментальной кри-
вой зависимости измеренного выхода Fe3+, т. е. выхода без вне-
сения поправки на потери энергии через стенки, от 1/г до нулевого
значения 1/г.
Для измерения активности обычно берут небольшую часть исследуемой системы. В принципе активность пробы можно определить любым способом. В частности, в работе [51] при калибровке дозиметра Фрикке активность измерялась с помощью 4л-пропорционапьного и 4л-жидкостного сцинтилляционного счетчиков.
5. Другие физические методы дозиметрии
Кроме физических методов дозиметрии ионизирующих излучений, рассмотренных выше, известны также активационный, сцинтилляционный и другие методы.
Активационный метод применяется преимущественно в дозиметрии нейтронных потоков. В этом методе доза находится по количеству радиоактивных атомов, образовавшихся в веществе-детекторе под действием нейтронов.
Сцинтилляционный метод дозиметрии основан на измерении интенсивности вспышек света в люминесцирующих веществах при прохождении через них ионизирующих излучений. Люминесци-рующими веществами могут быть неорганические и органические твердые сцинтилляторы (например, сульфид цинка, активированный серебром; антрацен), органические пластмассовые сцинтилляторы (например, полистирол с добавкой и-терфенила), жидкостные органические сцинтилляторы (например, раствор п-терфенила в ароматическом соединении), газовые сцинтилляторы (например, ксенон). Световые вспышки регистрируются фотоэлектронным умножителем со специальной электронной схемой.
Для измерения дозы широко применяются различные радиационные эффекты, имеющие место в твердых телах. Этот раздел дозиметрии, как уже отмечалось, называют твердофазной дозиметрией. Сюда, очевидно, относится и упомянутый выше сцинтилляционный метод с использованием твердых люмпнесцирую-щих веществ. В настоящей книге твердофазной дозиметрии посвящена специальная глава, поскольку многие дозиметры на основе твердых тел используются в радиационной химии.
ЛИТЕРАТУРА
1. К. К. Лглинцев. Основы дозиметрии ионизирующих излучений. М., Мед-гиз, 1955.
2. К. К. Аглинцев. Дозиметрия ионизирующих излучений. М., Гостехиздат, 1957.
3. Радиационная дозиметрия (под ред. Дж. Хайна и Г. Браунелла). М., ИЛ, 1958.
4. G. М. Whyte. Principles of Radiation Dosimetry. N. Y., 1959.
5. В. И. Иванов. Дозиметрия ионизирующих излучений. М., Атомиздат, 1964.
6. Измерение поглощенной дозы в нейтронных полях и в смешанных полях «гамма-излучение и нейтроны». М., Атомиздат, 1964.
7. Determination of Absorbed Dose in Reactors, Technical Reports Series No. 127. Vienna, IAEA, 1971.
8. Б. II. Голубев. Дозиметрия и защита от ионизирующих излучений. М., Атомиздат, 1971.
9. Radiation Dosimetry (Ed. F. H. Attixand VV. C. Roesch). N. Y., Academic Press 1966.
10. W. H. Bragg. Phil. Mag., Ser. 6, 20, 385 (1910).
11. L.H.Gray. Proc. Roy. Soc., A122, 647 (1929).
12. L.H.Gray. Proc. Roy. Soc., A156, 578 (1936).
13. U.Fano. Radiation Res., 1, 237 (1954).
14. Neutron Fluence Measurements, Technical Reports Series No 107. Vienna, IAEA, 1970.
15. ICRU Report 10b. Physical Aspects of Irradiation, National Bureau of Standards, Handbook No 85. Washington, 1964.
16. ICRU Report (1956). National Bureau of Standards, Handbook No 62. Washington, 1957.
17. К. K. Mehta. Determination of Absorbed Dose in Reactors, Technical Reports Series No 127, Vienna, IAEA, 1971, p. 167.
18. Ф. А. Махлис. Радиационная физика it химия полимеров. М., Атомиздат, 1972.
19. А. О. Fregene. Radiation Res., 31, 256 (1967).
20. J. Zsula, A. Liuzzi, J. S. Laughlin. Radiation Res., 6, 661 (1957).
21. П. Я. Глазунов, Г. Б. Радзиевский. Сб. «Действие ионизирующих излучений на неорганические и органические системы». М., Изд-во АН СССР. 1958, стр. 395.
22. А. Б. Ликосе, П. Я. Глазунов. Изв. АН СССР, ОХН, 1959, 1137.
23. В. Векслер, Л. Грошев, Б. Исаев. Ионизационные методы исследования излучений. М.— Л., Гостехиздат, 1950.
24. Ф. Спирс. Радиационная дозиметрия (под ред. Дж. Хайна и Г. Браунелла). М., ИЛ, 1958, стр. 9.
25. Дж. Боуг. Там же, стр. 134.
26. J. И'. Boag. Radiation Dosimetry (Ed. F. H. Attix and W. C. Roesch), vol. 2. N. Y., Academic Press, 1966, p. 2.
27. А. П. Комар, С. II. Круглов, II. В. Лопатин. Дозиметрия больших доз. Ташкент, «Фан», 1966, стр. 206.
28. Р. Curie, A. Laborde. Compt. rend., 136, 673 (1903).
29. Е. Rutherford, И. Barnes. Nature, 68, 622 (1903).
30. H. A. Callendar. Proc. Phys. Soc., 23, 1 (1911).
31. W. Rump. Z. Phys., 44, 396 (1927).
32. E. Stahel. Strablentherapie, 33, 296 (1929).
33. Мощная радиационная техника (под ред. С. Джефферсона). М., Атомиздат, 1967, стр. 270.
34. С. J. Hochanadel, J. A.Ghormley. J. Chem. Phys., 21, 880 (1953).
35. A. Brynjoljsson. Radiation Preservation of Food. Vienna, IAEA, 1973, p. 549.
36. Ю. И. Брегадзе, Б. M. Исаев, В. Б. Маркович, А. II. Ромашкевич, А. В. Тултаев. Радиационная дозиметрия и спектрометрия ионизирующих излучений. Ташкент, «Фан», 1970, стр. 78.
37. Дж. Лафлин, С. Дженна. Радиационная дозиметрия (под ред. Дж. Хайна и Г. Браунелла). М., ИЛ, 1958, гл. 9.
38. Г. Б. Радзиевский, II. Я. Глазунов, А. И. Варин. Приборы и техника эксперимента, № 2, 105 (1967).
39. Э. Д. Глушкова, К. И. Никулин, 10. Н. Тимко, А. II. Шанъгина. Радиационная дозиметрия и спектрометрия ионизирующих излучений. Ташкент, «Фан», 1970, стр. 58.
40. С. Willis, О. A. Miller, А. Е. Rothwell, A. W. Boyd. Radiation Res., 35, 428 (1968).
41. С. Willis, О. A. Miller, А. Ё. Rothwell, А. И'. Boyd. Adv. Chem. Ser., 81, 539 (1968).
42. H. С. Шиманская. Калориметрия ионизирующих излучений. М., Атомиздат, 1973.
43. J. S. Laughlin, S.Genna. Radiation Dosimetry (Ed. F. II. Attix and W. C. Roesch), vol. 2. N. Y., Academic Press, 1966, p. 389.
44. B. Sautiez. Determination of Absorbed Dose in Reactors, Technical Reports Series, No 127. Vienna, IAEA, 1971, p. 121.
45. R. H. Schuler, A. O. Allen. J. Chem. Phys., 24, 56 (1956).
46. А. А. Воробьев, Б. А. Кононов. Прохождение электронов через вещество. Томск, Изд-во Томского ун-та, 1966.
47. П. Я. Глазунов, А. К. Пикаев. Докл. АН СССР, 150, 1051 (1960).
48. Э. Харт, М. Анбар. Гидратированный электрон. М., Атомиздат, 1973, стр. 226.
49. О. П. Верхградский, 10. Ф. Голодный, Г. Н. Пьянков, И. П. Червецова, А. М. Кабакчи. Дозиметрия больших доз. Ташкент, «Фан», 1966, стр. 223.
50. D. М. Donaldson, N. Miller, J. Chim. phys., 52, 578 (1952).
51. M. Peysach, J. Steyn. J. S. Afric. Chem. Inst., 13, 34 (1960).
Глава IV
ОСНОВНЫЕ ПОЛОЖЕНИЯ ХИМИЧЕСКОЙ ДОЗИМЕТРИИ ИОНИЗИРУЮЩИХ ИЗЛУЧЕНИЙ
Химики-радиационники в своих исследованиях, как уже отмечалось, измеряют дозу чаще всего с помощью химических методов. Под химическими методами дозиметрии ионизирующих излучений обычно понимают такие методы дозиметрии, которые основаны на определении химических изменений, происходящих в некоторых системах при их облучении. Системы, используемые с этой целью, называются дозиметрическими. В принципе любая система, для которой точно известен радиационно-химический выход и более или менее хорошо установлен механизм радиолиза, может быть употреблена для дозиметрии. Этими системами в зависимости от условия проведения эксперимента могут быть жидкости, твердые тела или газы. Прибор с дозиметрической системой называется химическим дозиметром. Чаще всего он состоит из контейнера (или ячейки) и вещества или его раствора.
К настоящему времени опубликовано большое количество оригинальных работ (включая статьи в сборниках [1 —10]), касающихся химических методов дозиметрии ионизирующих излучений. Этим методам посвящены монографии [11, 12], отдельные главы книг [13—16] и многочисленные обзоры [17—591.
1. Требования, предъявляемые к химическим дозиметрам
Любая дозиметрическая система, согласно [17, 26], должна удовлетворять определенным требованиям. Выход радиационнохимического превращения (величина G) должен быть достаточно высоким и не должен зависеть в широких пределах от следующих факторов: 1) вида излучения; 2) энергии излучения; 3) дозы и мощности дозы; 4) концентрации реагентов; 5) температуры; 6) любых других условий, которые могут изменяться во время облучения (pH раствора, содержания растворенных газов и т. п.). Кроме того, 7) дозиметрическая система должна быть стабильной как до облучения, так и после него; 8) аналитические методы для определения химического превращения должны быть простыми и быстрыми; 9) необходимо, чтобы для приготовления дозиметра возможно было использовать реактивы обычной степени чистоты.
Дозиметрическую систему, отвечающую всем этим требованиям можно назвать идеальной. До настоящего времени еще неизвест
но ни одной такой системы. Из числа описанных в литературе дозиметрических систем, как будет показано ниже, наиболее полно удовлетворяет обсужденным требованиям ферросульфатная система.
2. Классификация химических дозиметров
Возможны два типа классификации химических дозиметров. Один из них основывается на видах и уровнях радиации, определяемых тем или иным дозиметром, а другой — на агрегатном состоянии дозиметрической системы.
Согласно [201, по первому типу классификации химические дозиметры можно разделить на три группы. К первой группе относятся химические дозиметры, с помощью которых возможно определять в случае рентгеновского, у- и электронного излучений дозы выше 10s рад. Эти дозиметры находят наиболее широкое • применение в радиационной химии. Вторая группа включает химические дозиметры, которые позволяют определять дозы этих видов излучения ниже 10s рад. В радиационной химии системы с такой высокой чувствительностью почти не требуются. Они используются в качестве средств индивидуального дозиметрического контроля и в радиобиологии. В третью группу входят химические дозиметры, предназначенные для измерения величины энергии, передаваемой среде нейтронами. Сюда же, возможно, следует отнести и дозиметры, позволяющие определять дозы в случае тяжелых заряженных частиц. Не видимому, в особую группу целесообразно вследствие их специфики выделить дозиметры, применяемые для определения доз импульсного излучения.
Резкой границы между описанными группами не существует. Очевидно, изменяя чувствительность дозиметрической системы, определяемую величиной выхода химического превращения и чувствительностью метода регистрации этого превращения, можно измерять различные дозы. С целью иллюстрации этого положения приведем следующий пример. Дозиметр Фрикке (детально этот дозиметр рассматривается в следующей главе) используется, как правило, для нахождения доз в диапазоне 4-103—4-Ю4 рад. В этом случае для анализа образующихся при облучении ионов Fe3+ применяется спектрофотометрический метод по поглощению света этими ионами при длине волны 304 нм при использовании оптических кювет с длиной поглощающего слоя, равной 1 см. Однако если для анализа ионов Fe3+ брать кюветы с длиной поглощающего слоя 10 см, то с помощью этого дозиметра можно регистрировать дозы порядка 400 рад. При добавлении бензола или бензойной кислоты к раствору ферросульфата G(Fe3+) возрастает примерно в 4 раза. Следовательно, в этом случае можно измерять дозы около 100 рад. Кроме того, небольшая модификация делает возможным применение дозиметра Фрикке для дозиметрии импульсного излучения. Наконец, если в раствор ферросульфата ввести
борную кислоту или сернокислый литий, то эта система становится пригодной для измерения потоков тепловых нейтронов.
По второму типу классификации химические дозиметры могут быть разбиты на группы, включающие: 1) газообразные; 2) жидкие и 3) твердые дозиметрические системы. К первой группе относятся, например, дозиметры на основе газообразной закиси азота и некоторых углеводородов, ко второй —водные растворы, гели, индивидуальные жидкости и смеси жидкостей и, наконец, к третьей — пластмассы, разнообразные стекла, щелочногалоид-ные кристаллы и др. В последнее время дозиметры на основе стекол и кристаллов стали относить к особой области дозиметрии, называемой твердофазной дозиметрией (см. стр. 9 и 108).
К химическим методам дозиметрии можно отнести также фотографический метод. В нем о величине дозы судят по степени почернения фотопленки или фотопластинки. В радиационной химии он практически не используется. Поэтому в настоящей книге он не обсуждается. Химики-радиацпонники сталкиваются с этим методом лишь при индивидуальном дозиметрическом фотоконтроле, т. е. при контроле доз излучения, получаемых отдельными лицами. Сотруднику, имеющему дело с радиоактивными изотопами или ионизирующими излучениями, выдается кассета со специальными сортами пленок. Через определенное время пленки обрабатываются, и доза находится по степени почернения. Подробно фотографический метод дозиметрии изложен, например, в обзоре [60] и книгах [61, 62].
Обсудим кратко, в какой степени выполняются изложенные выше требования той или иной группой химических дозиметров. Вид излучения и его энергия в общем случае оказывают влияние на выходы продуктов радиолиза жидких и твердых систем. Особенно это характерно для дозиметров на основе водных растворов. Объясняется данное явление тем, что радиационные превращения происходят за счет химически активных частиц (свободных радикалов, ионов, возбужденных молекул), возникающих в системе при действии излучения. Эти частицы образуются в небольших областях (треках или «шпорах») и поэтому имеют неравномерное распределение по объему облучаемой системы. Расстояние между такими областями зависит от величины ЛПЭ, т. е. от вида излучения и его энергии. Чем выше значение ЛПЭ, тем ближе друг к другу расположены области ионизации и возбуждения, а значит, тем вероятнее протекают реакции химически активных частиц между собой и тем меньшая доля их расходуется в реакциях с растворенным веществом. Величина зтого эффекта (его часто называют трековым эффектом) определяется механизмом реакций химически активных частиц с растворенным веществом. В отдельных случаях роль трекового эффекта может быть существенно уменьшена путем использования сравнительно концентрированных растворов, поскольку при больших концентрациях растворенные вещества могут подавлять внутритрековые реакции хи
мически активных частиц друг с другом. Для индивидуальных жидкостей трековый эффект, как правило, выражен гораздо менее резко, и здесь в принципе можно подобрать такие реакции, выходы которых практически не зависят от величины ЛПЭ. Например, по данным [63, 64], С(Н2) в облученном циклогексане практически не зависит от ЛПЭ. Однако для выходов других продуктов радиолиза этой системы такая зависимость имеет место [64, 65]. В газообразных системах вследствие их низкой плотности химически активные частицы распределены равномерно, и поэтому роль трекового эффекта при их облучении ничтожна.
Влияние мощности дозы наиболее резко проявляется, если в дозиметрической системе при радиолизе протекают цепные реакции. Если обрыв цепей происходит в результате рекомбинации радикалов, то выход обратно пропорционален мощности дозы в степени 0,5. Очевидно, такие системы малопригодны для дозиметрии. Однако в том случае, когда обрыв цепей обусловлен реакциями радикалов с растворенными веществами, выход не зависит от мощности дозы. Примером подобных дозиметрических систем могут служить водные растворы ферросульфата, содержащие кислород и бензол или бензойную кислоту (см. следующую главу).
F Большинство дозиметрических систем характеризуется отсутствием цепных процессов при облучении. Поэтому выходы радиационно-химических превращений в них не зависят от мощности дозы в весьма широких пределах. Однако при очень высоких мощностях дозы, создаваемых, например, импульсным электронным излучением, проявляется так называемый эффект «перекрывания треков». В результате заметно возрастает значение реакций химически активных частиц друг с другом, что может привести к изменению выхода измеряемого превращения. В случае водных растворов этот эффект можно исключить, используя достаточно высокие концентрации растворенных веществ.
При облучении концентрация реагентов в дозиметрической системе изменяется, причем происходит уменьшение количества исходных реагентов и накопление продуктов радиолиза. Их соотношение и реакционная способность относительно первичных химически активных частиц, возникающих при облучении, определяют верхнюю границу диапазона доз, в котором выход не зависит от дозы. В общем случае линейная зависимость количества химического превращения от дозы выполняется, если все продукты радиолиза стабильны в радиационно-химическом отношении и если выход не зависит от концентрации исходных компонентов. Независимость выходов от концентрации имеет место, например, в случае разбавленных водных растворов (~ 10~3 М). Однако при повышении концентрации раствора выходы, как правило, изменяются вследствие захвата растворенным веществом части первичных химически активных продуктов в «шпорах» или треках и вовлечения возбужденных молекул в реакции. При
этом в зависимости от механизма радиационно-химических реакций выходы могут или увеличиваться, или уменьшаться. В водных растворах на выходы может оказывать влияние и величина pH. Объясняется это, во-первых, тем, что выходы первичных химически активных частиц радиолиза воды несколько зависят от этого фактора, и, во-вторых, изменением природы и реакционной способности этих частиц в зависимости от pH.
Часто на радиолиз жидких дозиметрических систем существенное влияние оказывает природа насыщающего газа. Степень этого влияния зависит, естественно, от механизма радиационно-химических реакций в конкретных системах. Например, <?(Fe3+) в дозиметре Фрикке уменьшается почти в 2 раза при дозах, когда происходит полное израсходование кислорода, присутствовавшего в растворе. Однако показания цериевого дозиметра не зависят от того, имеется ли в растворе кислород или нет.
Многие дозиметры па основе твердых тел характеризуются чувствительностью к температуре. Например, радиационные дефекты в стеклах и щелочногалоидных кристаллах эффективно «отжигаются» при высоких температурах. Для жидких дозиметрических систем, в которых при облучении не протекают цепные процессы, температурные эффекты выражены гораздо менее резко, поскольку выходы первичных химически активных частиц в них обычно мало зависят от температуры.
Часто для приготовления дозиметров необходимо использовать тщательно очищенные реактивы, так как даже ничтожные примеси оказывают во многих случаях существенное влияние на выходы. Требуемая степень чистоты реактивов зависит от типа дозиметра. Однако можно дать общее правило [33]: неорганические дозиметры чувствительны к органическим примесям и, наоборот, органические дозиметры чувствительны к неорганическим веществам, способным претерпевать окислительно-восстановительные превращения. Специальной очистке должны быть подвергнуты и контейнеры (или ячейки), в которые помещаются дозиметрические системы.
3. Калибровка дозиметрических систем
Для определения поглощенной дозы с помощью химического дозиметра необходимо знать величину выхода превращения, которое претерпевает система при облучении. Измерение значения G обычно осуществляется сравнением поглощенной дозы, найденной каким-либо прямым методом (калориметрическим, ионизационным и др.), с количеством химического превращения в этой системе при данной дозе. В главе III было приведено несколько примеров калибровки дозиметра Фрикке.
Если для какой-либо дозиметрической системы величина G установлена весьма точно и эта величина сохраняет постоянное значение при изменении условий облучения в широких пределах,
то эту систему можно использовать в качестве вторичного стандарта, т. е. в тех случаях, когда необходимо определить G для других систем. Наиболее часто с этой целью применяется фер-росульфатная система.
4. Некоторые особенности дозиметрических измерений с помощью химических методов
Дозиметрические системы, являющиеся жидкостями пли газами, облучаются в ячейках. Эти ячейки должны удовлетворять следующим основным требованиям.
1. Материал, из которого изготовлена ячейка, должен быть химически инертным относительно дозиметрической системы. Например, некоторые металлы нельзя использовать для изготовления ячеек в случае систем, состоящих из 0,4 М раствора H2SO4.
2. Стенки ячейки не должны оказывать каталитического эффекта на химические реакции, протекающие в системе при облучении. Например, по данным Ф. Лямпе и др. [661, нержавеющая сталь в определенных условиях катализирует радиационно-химическое разложение закиси азота.
3. В случае рентгеновского и у-пзлучений толщина стенок ячейки должна быть достаточно большой по сравнению с длиной пробега электронов, генерированных в материале стенки. В табл. 27 приведены максимальные пробеги электронов, генерированных фотонами различных энергий, в некоторых материалах [33, 671. Из этой таблицы видно, что, например, для у-лучей 60Со толщина стенки стеклянной ячейки должна быть не менее 1,5 мм. Если это требование не выполняется, то могут быть получены завышенные результаты, поскольку часть электронов, возникших в материале стенок, попадает в дозиметрическую систему и вызовет дополнительное химическое превращение.
4. Для жидких дозиметрических систем размеры ячейки должны быть большими по сравнению с длиной пробега в жидкости
Таблица 27
Максимальные пробеги электронов, генерированных фотонами различной энергии
Энергия фотонов, Мэе Максимальный пробег, мм 1 Энергия фотонов, Мэв Максимальный пробег, мм
Вода Стекло *1 Полиэтилен *2, Вода Стекло ** Полиэтилен *а
0,1 0,14 0,07 0,14 4,0 20 10 20
0,5 1,0 0,5 1,0 8,0 40 20 40
1,0 3 1,5 3
♦* Уд. вес 2,7 г/сл13. *2 Уд. вес 0,92 г/сии3.
вторичных электронов, генерированных фотонами в дозиметрической системе и стенках ячейки. В общем случае это требование исходит из условия соблюдения электронного равновесия между дозиметрической системой и окружающей ее средой, выражающегося уравнением Брэгга — Грея для электронных энергий во всем энергетическом спектре вторичных электронов:
где [(реп/р)Лп£1ст и [(реп/р)/т^1ж есть отношения массовых коэффициентов поглощения энергии (цеп/р) к массовой тормозной способности (,„5) для электронов в жидкости (ж) и материале стенки (ст). Очевидно, для / ^> 1 большая часть энергии будет передаваться электронами от стенок раствору, чем в противоположном направлении. Обратное явление имеет место при / < 1. В первом случае выход будет возрастать при уменьшении размеров ячейки, а во втором — падать. Г. Фрикке и Э. Харт [33] отмечают, что для ячеек пз стекла и полиэтилена, наполненных водой, величина / равна соответственно 1,11 и 0,98 при воздействии у-лучей 60Со. Следовательно, для стеклянных ячеек относительные выходы при уменьшении их размеров будут возрастать, достигая в пределе 1,11. В полиэтиленовых ячейках максимальное падение выходов с уменьшением их размеров составляет всего 2%.
Очевидно, условие электронного равновесия соблюдается в том случае, когда дозиметрическая система и материал стенки характеризуются радиационным подобием, т. е. в них при облучении протекают одни и те же физические процессы. Важными характеристиками в этом отношении являются эффективный атомный номер и электронная плотность среды. В табл. 28 приведены эти характеристики для ряда систем и материалов. Эта таблица составлена по данным А. М. Кабакчи и сотр. [111. Если эффективные атомные номера двух сред равны, то в них количество энергии, переданной вторичным электронам, и распределение вторичных электронов по энергиям будут одинаковыми. Говоря другими словами, в этом случае / = 1. Из табл. 28 видно, что для пластмасс и водных систем величины эффективных атомных номеров отличаются незначительно друг от друга. Поэтому для них значение / близко к 1.
Влияние размеров ячейки на величины выходов отмечалось рядом авторов [68—73]. Так, по данным [69], при -у-радиолпзе ферросульфатпой системы в стеклянных ячейках с внутренним диаметром 4 мм G(Fes+) на 6% выше, чем в ячейках большего диаметра. В полистироловых ячейках этот эффект не наблюдается.
Т. Берлин и Ф. Чен [72, 73] подробно исследовали влияние размеров кварцевых ячеек на величину G(Fe®+) в ферросульфат-ной дозиметрической системе. На рис. 61 показана зависимость отношения дозы, поглощенной этой системой в ячейке с радиусом
то эту систему можно использовать в качестве вторичного стандарта, т. е. в тех случаях, когда необходимо определить G для других систем. Наиболее часто с этой целью применяется ферросульфатная система.
4. Некоторые особенности дозиметрических измерений с помощью химических методов
Дозиметрические системы, являющиеся жидкостями или га-зами, облучаются в ячейках. Эти ячейки должны удовлетворять следующим основным требованиям.
1. Материал, из которого изготовлена ячейка, должен быть химически инертным относительно дозиметрической системы. Например, некоторые металлы нельзя использовать для изготовления ячеек в случае систем, состоящих из 0,4 М раствора H2SO4.
2. Стенки ячейки не должны оказывать каталитического эффекта на химические реакции, протекающие в системе при облучении. Например, по данным Ф. Лямпе и др. [661, нержавеющая сталь в определенных условиях катализирует радиационно-химическое разложение закиси азота. и »
3. В случае рентгеновского и у-излучепий толщина стенок ячейки должна быть достаточно большой по сравнению с длиной пробега электронов, генерированных в материале стенки. В табл. 27 приведены максимальные пробеги электронов, генерированных фотонами различных энергий, в некоторых материалах [33, 67]. Из этой таблицы видно, что, например, для у-лучей 60Со толщина стенки стеклянной ячейки должна быть не менее 1,5 мм. Если это требование не выполняется, то могут быть получены завышенные результаты, поскольку часть электронов, возникших в материале стенок, попадает в дозиметрическую систему и вызовет дополнительное химическое превращение.
4. Для жидких дозиметрических систем размеры ячейки должны быть большими по сравнению с длиной пробега в жидкости Таблица 27
Максимальные пробеги электронов, генерированных фотонами различной энергии
Энергия фотонов, Мэв Максимальный пробег, мм | Энергия фотонов, Мэв Максимальный пробег, мм
Вода Стекло Полиэтилен *2 Вода Стекло ♦* Полиэтилен *2
0,1 0,14 0,07 0,14 4,0 20 10 20
0,5 1,0 0,5 1,0 8,0 40 20 40
1,0 3 1,5 3
** Уд. вес 2,7 г/см3. Уд. вес 0,92 г/см?.
вторичных электронов, генерированных фотонами в дозиметрической системе и стенках ячейки. В общем случае это требование исходит из условия соблюдения электронного равновесия между дозиметрической системой и окружающей ее средой, выражающегося уравнением Брэгга — Грея для электронных энергий во всем энергетическом спектре вторичных электронов:
где [(p-en/pJ/m'S'lcT и КНеп/рУт^ж есть отношения массовых коэффициентов поглощения энергии (цеп/р) к массовой тормозной способности (mS) для электронов в жидкости (ж) и материале стенки (ст). Очевидно, для / > 1 большая часть энергии будет передаваться электронами от стенок раствору, чем в противоположном направлении. Обратное явление имеет место при / < 1. В первом случае выход будет возрастать при уменьшении размеров ячейки, а во втором —падать. Г. Фрикке и 3. Харт [33] отмечают, что для ячеек из стекла и полиэтилена, наполненных водой, величина / равна соответственно 1,11 и 0,98 при воздействии у-лучей 6®Со. Следовательно, для стеклянных ячеек относительные выходы при уменьшении их размеров будут возрастать, достигая в пределе 1,11. В полиэтиленовых ячейках максимальное падение выходов с уменьшением их размеров составляет всего 2%.
Очевидно, условие электронного равновесия соблюдается в том случае, когда дозиметрическая система и материал стенки характеризуются радиационным подобием, т. е. в них при облучении протекают одни и те же физические процессы. Важными характеристиками в этом отношении являются эффективный атомный номер и электронная плотность среды. В табл. 28 приведены эти характеристики для ряда систем и материалов. Эта таблица составлена по данным А. М. Кабакчи и сотр. [И]. Если эффективные атомные номера двух сред равпы, то в них количество энергии, переданной вторичным электронам, и распределение вторичных электронов по энергиям будут одинаковыми. Говоря другими словами, в этом случае / = 1. Из табл. 28 видно, что для пластмасс и водных систем величины эффективных атомных номеров отличаются незначительно друг от друга. Поэтому для них значение / близко к 1.
Влияние размеров ячейки на величины выходов отмечалось рядом авторов [68—73]. Так, по данным [69], при у-радиолизе ферросульфатной системы в стеклянных ячейках с внутренним диаметром 4 мм G(Fes+) на 6% выше, чем в ячейках большего диаметра. В полистироловых ячейках этот эффект не наблюдается.
Т. Берлин и Ф. Чен [72, 73] подробно исследовали влияние размеров кварцевых ячеек на величину G(Fe3+) в ферросульфат-пой дозиметрической системе. На рис. 61 показана зависимость отношения дозы, поглощенной зтой системой в ячейке с радиусом
г, к дозе для бесконечно большой ячейки от радиуса сферической кварцевой ячейки. Как видно из этого рисунка, в случае кварцевых ячеек G(Fe3+) начинает заметно возрастать при г <С 4 мм.
Однако в работах [74—76] отмечалось, что стенки оказывают заметное влияние на выходы и в сравнительно больших ячейках. Например, согласно [74], при восстановлении у-лучами 60Со водных растворов Се4+ в стеклянных ячейках воспроизводимые результаты можно получить только при работе с ячейками объемом
Рис. 61. Зависимость отношения дозы, поглощенной в фер-росульфатной дозиметрической системе, находящейся в сферической кварцевой ячейке с радиусом г, к дозе для бесконечно большой ячейки от величи- * ны г
100 мл. Причины этого явления неясны. Быть может, оно обусловлено особенностями очистки стенок ячейки.
Для дозиметров на основе газов химические реакции происходят преимущественно за счет электронов, генерированных фотонами в стенках ячейки. В случае у-л у чей 60Со количество
Таблица 28
Характеристики некоторых сред
Среда Плотность, г/гльз Эффективный атомный номер для фотоэффекта Эффективный атомный номер для процесса образования пар . Электронная плотность, 1023 электроны г
Воздух 0,001293 7,64 7,36 3,03
Вода 1,00 7,42 6,60 3,34
Дозиметр Фрикке (0,1 М H2SO4) 1,003 7,44 6,50 3,34
Дозиметр Фрикке (0,4 М H2SO4) 1,024 7,68 6,84 3,33
Полиэтилен 0,92 5,46 4,78 3,43
Полистирол 1,1 5,79 5,28 3,24
Полпметилметакрнлат 1,18 6,46 5,83 3,26
Политетрафторэтилен 2,20 8,42 8,24 2,88
Стекло № 23 2,51 12,86 11,61 2,98
Стекло нейтральное 2,46 12,32 11,25 3,00
Стекло термостойкое 2,35 11,43 10,23 3,00
Кварц 2,65 11,60 10,81 3,01
химического превращения здесь практически пропорционально объему ячейки.
При работе с заряженными частицами следует учитывать, что они имеют сравнительно малые пробеги. Этим в принципе и должен определяться здесь выбор толщины стенок используемой ячейки. Возможность значительного поглощения энергии стенками ячейки необходимо иметь в виду и в случае фотонов малой энергии. В табл. 29 приведены данные о поглощении таких фотонов нейтральным стеклом [11].
Таблица 29 .
Поглощение фотонов различной энергии (в %) в нейтральном стекле
Энергия фотонов, Мае [Поглощение в слое толщиной Энергия фотонов, Мае Поглощение в слое толщиной
0,25 мм 0,5 лш 1,0 мм 0,25 мм 0,5 мм 1,0 мм
0,012 46,0 68,8 90,3 0,030 4,5 8,9 16,0
0,014 29,7 50,8 75,8 0,040 2,4 5,1 9,7
0,016 16,8 31,0 52,3 0,05') 1,0 2,2 4,0
0,020 14,2 26,5 46,2 0,070 <1,0 1,0 1,5
5. Как правило, ячейки перед проведением дозиметрии должны быть тщательно очищены. Специфические особенности очистки ячеек применительно к конкретным дозиметрическим системам обсуждаются в соответствующих главах книги.
6. Рекомендуется дозиметрические опыты проводить в тех же ячейках или сосудах, что и дальнейшие эксперименты по изучению радиолиза той или иной системы. При этом необходимо строго соблюдать одинаковую геометрию опытов, т. е. одинаковое расположение дозиметра и исследуемой системы относительно источника излучения.
При дозиметрических измерениях необходимо стремиться к тому, чтобы дозиметрическая система и исследуемый объект характеризовались радиационным подобием. Если же по каким-либо причинам нельзя подобрать радиационно-подобную дозиметрическую систему, то после измерения количества химического превращения в использованной системе следует рассчитать величину дозы, поглощенной ею, а затем величину дозы, поглощенной исследуемым объектом.
5. Расчет величины дозы, поглощенной исследуемым объектом, исходя из показаний дозиметра
Величину дозы, поглощенной дозиметрической системой, вычисляют, исходя из количества химического превращения и значения G этого превращения. Если доза D выражается в эв/мл, то общая формула для этого расчета такова:
£> = «100/0, (2)
где п —число молекул вещества, образующегося или разлагающегося при облучении 1 мл системы. При переходе к молярной концентрации этого вещества (обозначим ее буквой с) формула (2) преобразуется к виду
£> = 6,024-1022-с/0 эе/мл (3)
пли
£•=6,024-1025 с/G эв/л. (4)
Для нахождения дозы в эв/г и рад используются следующие выражения:
D = 6,024- Ю22-с/(Ор)эз/г, (5)
где р — плотность дозиметрической системы в г/см\
9,64-10“-с
11 D = —Ор— рад
(6)
Наиболее часто дозиметрическая и исследуемая системы имеют различные характеристики поглощения энергии ионизирующего излучения (радиационное подобие отсутствует). В этом случае необходимо осуществить пересчет показаний дозиметра применительно к данному объекту. Рассмотрим, каким путем осуществляется такой пересчет в случае рентгеновского и ^-излучений.
В общем виде формула для описываемого пересчета имеет следующий вид:
(Реп/Р)сист
Цшст' ^дозиметр 7р) СВ Г ’
М еп!1 'дозиметр V дозиметр'
где D и Dдозиметр поглощенные дозы для исследуемой и дозиметрической систем соответственно (в одинаковых единицах — электронвольтах на грамм или радах); (р,СТ1/р)Сист и (рЛп/ /р)дозиметр — массовые коэффициенты поглощения энергии для тех же систем; (Всист/Вд03иметр) — отношение факторов накопления для тех же систем.
Согласно [77], фактор накопления при определении поглощенной дозы данным химическим дозиметром есть отношение истинной поглощенной дозы в дозиметре к поглощенной дозе, которая была бы измерена в дозиметре, если бы отсутствовало рассеянное излучение.
Отношения (-BchctZ-Bhio) для точечного изотропного источника
у-излучения с0Со
„ Расстояние от н______р.. п
Система источника, см СИС1
Углеводород (СНг)п 15,8 31,6 63,2 0,977 0,959 0,949
0,1 М водный раствор щавелевой кислоты 15,8 1,000
31,6 1,000
63,2 1,000
Дозиметр Фринке (10-3 М FeSOj, 0,4 М H2SO4) 15,8 1,006
31,6 1,011
63,2 1,014
10~3 М FeSOi, 10-« М C11SO4, 5-10-3 М H2SO4 15,8 1,002
в Н2О 31,6 1,004
63,2 1,005
6-10-3 Ы FeSCh, 6-10-2 М C11SO4, 15,8 1,014
5-10-3 AfHaSOa в Н2О 31,6 1,024
63,2 1,031
IO’2 М Ce(SO4)2, 0,4 М H2SO4 в Н-2О 15,8 1,028
31,6 1,047
63,2 1,060
0,1 М Ce(S04)2, 0,4 М H2SO4 в Н2О 15,8 1,217
31,6 1,365
63,2 1,463
0,4 М Ce(SO4)2, 0,4 М H2SO4 в Н2О 15,8 1,720
31,6 2,216
63,2 2,545
Воздух (75,56% Ьт2, 23,15% Oj, 1,29% Аг) 15,8 1,006
31,6 1,013
63,2 1,017
LiF 15,8 1,019
31,6 1,037
63,2 1,051
Кварц SiO> 15,8 1,150
31,6 1,263
63,2 1,333
Система Расстояние от источника, см ^СИСТ^Н20
Поливинилхлорид (С2НзС1)п 15,8 1,347
31,6 1,597
63,2 1,756
Люцит (CsHsO)n 15,8 0,987
31,6 0,977
63,2 0,971
Влияние отношения (5сист/Ддозиметр) на расчеты дозы было подробно рассмотрено А. Брньелфсоном [77]. Он рассчитал эти коэффициенты для различных дозиметрических систем в случае точечного изотропного источника у-из лучения 60Со. Результаты некоторых его расчетов показаны в табл. 30. В этой таблице Т?н2о обозначает соответствующий коэффициент для воды.
Рассмотрение данных, приведенных в табл. 30, показывает, что для у-лучей 60Со отношение Всист/БдозимеТр равно ~ 1 в тех случаях, когда используются небольшие объемы, исследуемая система состоит из элементов с низким атомным номером и дозиметром является какой-либо разбавленный водный раствор. В этих условиях ослабление потока у-лучей 60Со мало и практически одинаково для обеих систем.
В радиационно-химической практике поглощенные дозы часто относят не к единице массы, а к единице объема. В этом случае при Всист/ВДозиметр = 1 формула (7) переходит в следующее выражение:
(Рст/р \:ист Рсист
Чист = Чозиметр /ц, /р) в > (°)
Vr'cn'1'дозиметр ‘ дозиметр
где Рдозимртр и Рсист — плотность дозиметрической^ и исследуемой систем (в г/см9). Если £>дозиметр выражена в электронвольтах на грамм, a Рсист необходимо получить в радах, то формула для соответствующего пересчета такова:
Чист
1,602-Ю-и
(Нета/Р)С ИСТ (Рет/Р^дозиметр
Чозиметр-
(9)
Когда Dдозиметр измерена в эв/мл, а Рсист требуется выразить в радах, то следует использовать формулу
Чист = 1,602.10-4
(Мр)сист (Рет/Р) дозиметр
1
р ^дозиметр1
‘дозиметр
(10)
0’^04с0о0'т4'^о0с0 0с000^04х^сгн0^ од со со од од о оо сг ог ОО I— Г- nF 04 о О СО г- г- с-О 1~— ОЪ оО Ю СО 04 04 ОД О1 04 04 04 04 04 04
со сс а с: л oq т-t СС' с с сс со с сс со о сссс Ci-nsf т-ССОСо'оС ссосс ссссссссо
OO0404vfQ0c01000t-C0vfOcC>t—041^-10 одгн^ою'нссссосог'юсо--< сх> оо оо г-’^NFr^NFNFI>-NFcOO4O4o4O4O4O4O4O4O4O4'«H’^i^^^ c0OO^nF04^OOOOOOOOOOOOOOOOOO £0 NF о о о о о о о о о о о о о о о"о~о"о"о"сГсГ СО 'гЧ
НО О0 НО
аблица 31 (окончание}
Значения реи/р (в i'.u-'i] для некоторых материалов
Энергия фотонов, Мэв Полистирол (С8Н8)П Полиметилметакрилат (С5НЬО2)П Полиэтилен (СН2)П Вода Воздух 0,4 М водный раствор H2SO4
0,010 1,82 2,91 1,69 4,79 4,61 5,36
0,015 0,495 0,783 0,461 1,28 1,27 1,45
0,020 0,193 0,310 0,180 0,512 0,511 0,585
0,030 0,0562 0,0899 0,0535 0,149 0,148 0,169
0,040 0,0300 0,0437 0,0295 0,0677 0,0668 0,0761
0,050 0,0236 0,0301 0,0238 0,0418 . 0,0406 0,0460
0,060 0,0218 0,0254 0,0225 0,0320 0,0305 0,0344
0,080 0,0217 0,0232 0,0228 0,0262 0,0243 0,0271
0,10 0,0231 0,0238 0,0243 0,0256 0,0234 0,0260
0,15 0,0263 0,0266 0,0279 0,0277 0,0250 0,0277
0,20 0,0286 0,0287 0,0303 0,0297 0,0268 0,0296
0,30 0,0309 0,0310 0,0328 0,0319 0,0287 0,0319
0,40 0,0318 0,0318 0,0337 0,0328 0,0295 0,0327
0,50 0,0321 0,0322 0,0340 0,0330 0,0296 0,0330
0,60 0,0318 0,0319 0,0337 0,0329 0,0295 0,0328
0,80 0,0310 0,0311 0,0329 0,0321 0,0289 0,0320
1,0 0,0300 0,0301 0,0319 0,0309 0,0278 0,0308
1,5 0,0275 0,0275 0,0291 0,0282 0,0254 0,0281
2,0 0,0252 0,0253 0,0267 0,0260 0,0234 0,0259
3,0 0,0219 0,0220 0,0232 0,0227 0,0205 0,0227
4,0 0,0198 0,0199 0,0209 0,0206 0,0186 0,0206
5,0 0,0182 0,0184 0,0192 0,0191 0,0174 0,0191
6,0 0,0171 0,0173 0,0180 0,0180 0,0164 0,0180
8,0 0,0155 0,0158 0,0162 0,0166 0,0152 0,0166
10,0 0,0145 0,0148 0,0151 0,0157 0,0145 0,0157
Формулы (9) и (10) так же, как и формула (8), справедливы при Всист/^дозиметр = 1 •
В главе I были даны определения коэффициентов ро/р, рк/р и реп/р. Величины этих коэффициентов имеются во многих работах (см., например, [78—801). Поскольку в расчетах дозы при переходе от одной системы к другой используется обычно реп/р, в табл. 31 и 32 приведены величины этого коэффициента для ряда элементов и некоторых материалов. Отметим, что в случае систем, состоящих из легких элементов, коэффициенты р,а/р, рк/р и р.еп/р отличаются друг от друга незначительно.
В радиационной химии очень часто используются системы, включающие элементы с малым порядковым номером, и энергии излучения от 0,1 до 3 Мэв. В этих случаях преобладающим при
взаимодействии фотонов с веществом является комптоновский эффект. Здесь справедливо соотношение
(йуп^Р)сист_____(^М{)]Сист
(^еп^дозиметр (^;^Р1дозимстр
где Ai и Z; есть атомный вес и порядковый номер элемента г в системе, а р; — его весовая доля.
Атомный номер, определяемый из суммы (Zg/Ag), обычно называется эффективным атомным номером, т. е. принимается, что сложное вещество или смесь веществ состоят из атомов одного вида с атомным номером Z. Для указанных выше условий
____ „ Z{
= (12>
Поэтому в данных условиях формула (7) при Всист/Вд03ИМеТр = = 1 преобразуется к виду
(Z/Л) __ тч * спи -
^сист = -^дозиметр c/tд\ ‘ I1*5)
Для систем, состоящих из атомов легких элементов, Z/A близко к 0,5. Например, для воды Z/A составляет 0,555, а для воздуха — 0,499. Поэтому значения р.рп/р в рассматриваемой области энергий почти равны для таких систем, и в этом случае при приближенных расчетах можно положить, что (ZM)ci1Ct ~(ZIA )ЦОзиметр> т. е. DCI1CT Лдозйметр, если обе последние величины выражены в эв1г или рад.
|В случае систем, состоящих из атомов легких элементов, и энергий фотонов 0,1—3,0 Мэв при пересчетах можно также применять формулу
''F п сист
^сист F -^дозиметр»
дозиметр
где ^дозиметр и ^сист — электронные плотности дозиметрической и исследуемой систем. Электронная плотность определяется выражением
F = No (pi -£ +> +------Ь рп , (15)
где No —число Авогадро.
При энергиях фотонов в несколько мегаэлектронвольт пересчет существенно усложняется тем обстоятельством, что химические превращения в системе могут быть вызваны в значительной степени вторичными электронами, генерированными в окружающей среде и попадающими в раствор. Поэтому системы необходимо
окружать тонкими слоями соответствующих материалов с тем, чтобы создать электронное равновесие [33]. Для этой области энергий весьма желательно, чтобы дозиметрическая и исследуемая системы были в высокой степени радиационно-подобными.
При работе с электронными пучками наиболее часто встречаются два случая.
1. Размеры ячеек с дозиметрической и исследуемой системами таковы, что они обеспечивают полное поглощение пучка. В этом случае пересчет показаний дозиметрической системы не требуется. Необходимо вносить лишь небольшие поправки, связанные с различиями в обратном рассеянии электронов и образованием тормозного рентгеновского излучения. Однако для водных и органических систем при энергиях до 4 Мэв эти поправки весьма малы [81—831. Следует отметить, что если дозиметрическая и исследуемая системы отличаются по своим характеристикам, то для них истинные облучаемые объемы будут различны, а значит, будут различными и истинные мощности поглощенной дозы, создаваемые электронами в них. В жидкостях равномерность облучения в известной мере может быть достигнута перемешиванием.
2. Размеры ячеек с системами достаточно малы, и в системах поглощается лишь небольшая доля энергии электронов. Очевидно, этот случай наиболее характерен для электронов с энергиями порядка 5 Мэв и выше, создаваемых линейными ускорителями. Здесь для пересчета показаний дозиметрической системы следует использовать формулу
D — D (таксист
^сист ^дозиметр ( S'! ’
чп 'дозиметр
где (mS)дозиметр к (тп^)сист — массовые тормозные способности дозиметрической и изучаемой систем соответственно. В этой формуле Расист и ^дозиметр выражены в эв!г или рад.
Относительные величины массовых тормозных способностей были приведены в табл. 23. В табл. 33 даны значения mS для воздуха и воды [84].
При высоких энергиях электронов заметную роль играет так называемый поляризационный эффект, или эффект плотности. Он обусловлен поляризацией атомов среды в электрическом поле движущейся частицы. Вследствие этого происходит своеобразная защита отдаленных атомов среды и уменьшение поля на некотором расстоянии от частицы. Это вызывает уменьшение величины потери энергии. Рассматриваемый эффект, очевидно, зависит от плотности среды, т. е. от числа поляризуемых атомов в 1 см3. Он наиболее характерен для твердых и жидких веществ. В газах этот эффект ничтожен до энергий 100 Мэв. Для воды он проявляется уже при энергиях электронов выше 1 Мэв. Поэтому при пересчете показаний дозиметрической системы, особенно в случае систем
Таблица 53
Массовые тормозные способности (в ЛГэв-сл12/г) воздуха и воды для электронов различной энергии Е
Е, Мэв Воздух Вода Е, Мэв Воздух Вода
0,010 19,71 23,21 0,90 1,683 1,906
0,015 14,42 16,91 0,95 1,679 1,899
0,020 11,55 13,51 1,0 1,676 1,893
0,025 9,737 11,37 1,1 1,673 1,885
0,030 8,479 9,884 1,2 1,675 1,880
0,035 7,552 8,794 1,3 1,675 1,887
0,040 6,840 7,956 1,4 1,679 1,876
0,045 6,273 7,292 1,5 1,683 1,877
0,050 5,812 6,751 1,6 1,689 1,878
0,055 5,429 6,303 1,7 1,695 1,880
0,060 5,106 5,924 1,8 1,701 1,883
0,065 4,829 5,600 1,9 1,708 1,886
0,070 4,590 5,320 2,0 1,714 1,889
0,075 4,380 5,075 2,2 1,729 1,897
0,080 4,195 4,859 2,4 1,743 1,905
0,085 4,031 4,667 2,6 1,757 1,914
0,090 3,884 4,496 2,8 1,771 1,922
0,095 3,752 4,341 з,о 1,786 1,931
0,10 3,632 4,202 3,5 1,820 1,953
0,15 2,862 3,304 4,0 1,852 1,974
0,20 2,472 2,850 4,5 1,884 1,994
0,25 2,240 2,580 5,0 1,913 2,014
0,30 2,088 2,401 5,5 1,942 2,032
0,35 1,984 2,280 6,0 1,969 2,051
0,40 1,908 2,190 6,5 1,995 2,068
0,45 1,852 2,123 7,0 2,020 2,085
0,50 1,810 2,071 7,5 2,045 2,102
0,55 1,777 2,032 8,0 2,068 2,119
0,60 1,752 2,000 8,5 2,091 2,135
0,65 1,732 1,975 9,0 2,115 2,152
0,70 1,717 1,955 9,5 2,137 2,167
0,75 1,705 1,939 10,0 2,159 2,183
0,80 1,696 1,926 20,0 2,534 2,470
0,85 1,688 1,915
с весьма различной плотностью, следует вносить поправки на поляризацию. Например, для воды при энергии электронов 16 Мэв поправка составляет 14% [851. Если дозиметрическая система и исследуемый объект имеют одинаковое агрегатное состояние и их плотность различается не очень сильно, то с достаточной
для многих практических целей точностью в расчеты можно не включать поправку на поляризацию.
Для тяжелых заряженных частиц (протоны, дейтроны, гелионы), генерируемых аппаратурными источниками, на практике имеет место исключительно первый случай из числа описанных для электронов.
ЛИТЕРАТУРА
1. Selected Topics in Radiation Dosimetry. Vienna, IAEA, 1961.
2. In-Pile Dosimetry. Vienna, IAEA, 1965.
3. Дозиметрия больших доз. Ташкент, «Фан», 1966.
4. Solid State and Chemical Dosimetry in Medicine and Biology. Vienna, IAEA, 1967.
5. Дозиметрия интенсивных потоков ионизирующих излучений. Ташкент, «Фан», 1969.
6. Радиационная дозиметрия и спектрометрия ионизирующих излучений. Ташкент, «Фан», 1970.
7. Manual on Radiation Dosimetry (Ed. N. W. Holm and R. J. Berry). N. Y., 1970.
8. Determination of Absorbed Dose in Reactors. Vienna, IAEA, 1971.
9. Дозиметрия и радиационные процессы в дозиметрических системах. Ташкент, «Фан», 1972.
10. Dosimetry in Agriculture, Industry, Biology and Medicine. Vienna, IAEA, 1973.
11. A. M. Еабакчи, Я. И. Лаврентоеич, В. В. Пеньковский. Химическая дозиметрия ионизирующих излучений. Киев, Изд-во АН УССР, 1963.
12. В. Broszkiewi.cz. Chemiczne Metody Dosimetrii Promieniowania Jonizu-jacego. Warszawa, 1971.
13. И. В. Верещинский, А. К. Пикаев. Введение в радпапионную химию. М., Изд-во АН СССР, 1963, гл. IX.
14. Дж. У. Т. Спинкс, Р. Дж. Вудс. Введение в радиационную химию (перевод с англ.). М., Атомиздат, 1967, гл. 4.
15. Г. Н. Пьянков, А. П. Мелешевич, Е. Г. Ярмилко, А. М. Еабакчи, С. И. Омельченко. Радиационная модификация полимерных материалов. Киев, «Техника», 1969, гл. 3.
16. М. Франк, В. Штольц. Твердотельная дозиметрия ионизирующего излучения. М., Атомиздат, 1973, гл. 2.
17. N. Miller, J. Wilkinson. Disc. Faraday Soc., 12, 50 (1962).
18. N. Miller. Actions Chimiques et Biologiques des Radiations, 2 Serie (Ed. M. Haissinsky). Paris, 1956, p. 145.
19. Д. Топлин. Радиационная дозиметрия (под ред. Дж. Хайна и Г. Браунелла). М., FIJI, 1958, стр. 298.
20. А. М. Еабакчи, В. А. Грамолин. Усп. химии, 27, 459 (1958).
21. W. Minder. Chimia (Switz.), 12, 17 (1958).
22. D. Bertram. Kernenergie, 1, 697 (1958).
23. H. Bildstein. Atompraxis, 5, 163 (1958).
24. B. D. Dove. Nucl. Power, 4, 124 (1959).
25. И. E. Соколова. Мед. радиол., 4, 4 (1959).
26. С. Таймути, Р. Гласс, Б. Дивер. Труды II Международной конференции по мирному использованию атомной энергии. Избранные доклады иностранных ученых, т. 10. М., Атомиздат, 1959, стр. 409.
27. J. Weiss. Intern. J. Appl. Rad. Isotopes, 4, 89 (1960).
28. F. Behounek. Atom. Energy Rev., 1, 37 (1963).
29. В. Миндер. Радиационная химия (под ред. Г. Молера). М., Атомиздат, 1963, стр. 107.
5 А. К. Пикаев
129
30. Л1. Tsuda. Encyclopedia of X Rays and Gamma Rays (Ed. G. L. Clark). N. Y., Reinhold Publishing Co., 1963, p. 294.
31. F. S. Dainton. Radiation Dosimetry (Ed. G. AV. Reed). New York — London, 1964, p. 167.
32. Z. Spumy. Atom. Energy Rev., 3, 61 (1965).
33. II. Fricke, E. J. Hart. Radiation Dosimetry (Ed. F. H. Attix and W. C. Roesch), vol. 2. N. Y., 1966, p. 167.
34. R. K. Broszkiewicz. Solid State and Chemical Dosimetry in Medicine and Biologv- Vienna, IAEA, 1967, p. 213.
35. R. J. Shalek, С. E. Smith. Ann. N. Y. Acad. Sci., 161, 44 (1969).
36. N. W. Holm. Large Radiation Sources for Industrial Uses. Vienna, IAEA, 1969, p. 555.
37. N. W. Holm, Z. P. Zagorski. Manual on Radiation Dosimetry (Ed. N. W. Holm and R. J. Berry). N. Y., 1970, p. 83.
38. W. L. McLaughlin. Ibid., p. 129.
39. J. lUeiss, F. X. Rizzo. Ibid., p. 231.
40. E. M.Fielden, TV. W. Holm. Ibid., p. 288.
41. K. Sehested. Ibid., p. 313.
42. E. Bjergbakke. Ibid., p. 319.
43. E. J. Hart. Ibid., p. 327.
44. E. Bjergbakke. Ihid., p. 323.
45. E. J. Hart, E. M. Fielden. Ihid., p. 331.
46. TV. W. Holm. Ibid., p. 337.
47. T. R. Johnson. Ibid., p. 341.
48. I. Dvomik. Ihid., p. 345.
49. C. Artandi. Ibid., p. 353.
50. C. G. Orton. Ihid., p. 357.
51. B. Whittaker. Ibid., p. 363.
52. TV. Goldstein. Ibid., p. 371.
53. W. L. McLaughlin. Ibid., p. 377.
54. R. Sheldon. Ibid., p. 411.
55. I. Draganic. Determination of Absorbed Dose in Reactors. Vienna, IAEA, 1971, p. 197.
56. S. Hayakawa. Ibid., p. 223.
57. II. К. Соколова. Химические методы дозиметрии в радиобиологии. М., Атомиздат, 1972.
58. А. К. Пикаев. Усп. химии, 41, 1696 (1972).
59. I. G. Draganic, В. L. Gupta. Dosimetry in Agriculture, Industry, Biology and Medicine. Vienna, IAEA, 1973, p. 351.
60. R. A. Dudley. Radiation Dosimetry (Ed. F. H. Attix and W. C. Roesch.), vol. 2. N. Y., Academic Press, 1966, p. 326.
61. K. Berker. Filmdosimetrie. Berlin, Springer-Verlag, 1962.
62. В. Ф. Koe.we. Фотографическая дозиметрия ионизирующих излучений. М., Атомиздат, 1964.
63. R. Н. Schuler, А. О. Allen. J. Am. Chem. Soc., 77, 507 (1956).
64. W.G. Burns, J. R. Parry. Nature, 201, 814 (1964).
65. J. W. Falconer, M. Burton. J. Phys. Chem., 67, 1743 (1963).
66. F. W. Lampe, L. Kevan, E. R. Weiner, W. H. Johnston. J. Phys. Chem., 71, 1528 (1967).
67. A. T. Nelms. Natl. Bur. Stand. (USA), Circular 577, Suppl., 1958.
68. J. Weiss. Nucleonics, 10, No 7, 28 (1952).
69. Дж. Вейсс, А. Аллен, Г. Шварц. Материалы Международной конференции по мирному использованию атомной энергии (Женева, 1955), т. 14. М., Физматиздат, 1958, стр. 216.
70. Дж. Гормлей. Цит. по [69].
71. J. Puig, J. Sutton. J. Chim. phys., 56, 699 (1959).
72. T. E. Burlin, F. K. Chan. Intern. J. Appl. Rad. Isotopes, 20, 767 (1967).
73. T. E. Burlin, F. K. Chan. Solid State and Chemical Dosimetry in Medicine and Biology. Vienna, IAEA, 1967, p. 393.
74. TV. F. Barr, R. H. Schuler. I. Phys. Chem., 63, 808 (1959).
75. R. Jarrett, J. Halliday. Activities Rept. 15, U. S. Natick Labs., 1963; цит. no [33].
76. K. Sehested, A. Brynfolfsson, N. W. Holm Rise Rept. 62 (1963).
77. A. Brynfolfsson. Adv. Chem. Ser., 81, 550 (1968).
78. R. T. Berger. Radiation Res., 15, 1 (1961).
79. R. D. Evans. Radiation Dosimetry (Ed. F. H. Attix and W. C. Roesch), vol. 1. N. Y., Academic Press. 1968, p. 93.
80. Руководство по радиационной защите для инженеров, т. 1. М., Атомиздат, 1972.
81. J. Saldick, А. О. Allen. J. Chem. Phys., 22, 438 (1954).
82. J. Saldick, A. O. Allen. Ibid., p. 1777.
83. R. H. Schuler. Proc. II United Nations Intern. Conference on Peacefu Uses of Atomic Energy, vol. 21. Geneva, 1958, p. 213.
84. H. Bischel. Radiation Dosimetry (Ed. F. H. Attix and W. C. Roesch), vol. 1. N. Y., Academic Press, 1968, p. 157.
85. J. Zsula, A. Liuzzi, J. S. Laughlin. Radiation Res., 6, 661 (1957).
Глава V
ВОДНЫЕ РАСТВОРЫ
КАК ДОЗИМЕТРИЧЕСКИЕ СИСТЕМЫ
В радиационной химии для дозиметрии рентгеновского и у-из-лучений и потоков быстрых электронов используются различные системы. Наиболее часто с этой целью применяются водные растворы. Поэтому рассмотрение химических методов определения дозы будет начато с дозиметрических систем этого типа.
1. Общая характеристика
Как правило, рассматриваемые дозиметрические системы представляют собой разбавленные водные растворы. Поэтому химические превращения в них осуществляются преимущественно в результате косвенного действия ионизирующего излучения, т. е. за счет продуктов радиационно-химического разложения воды. Такими продуктами являются гидратированный электрон (^aq), Н, ОН. Н2 и Н2О2 *.
Радикалы eaq, Н и ОН образуются в «шпорах». Концентрация радикалов в них довольно высока, и в начальные моменты времени эти частицы главным образом реагируют друг с другом. Соответствующие реакции таковы:
e-q + ОН —> ОН“, (1)
% + % — -Нз + 2ОН-, (2)
e-q + НзО+ -> НзО Н + НгО, (3)
е~+Н _Л±-»Н2+ОН-, (4)
ОН+ОН — Н2О2, (5)
II + II - Н2, (6)
II + ОН — Н2О. (7)
По истечении некоторого промежутка времени (~ 10~8 — 10-10 сек.) «шпоры» за счет диффузии настолько расширяются, что концентрация радикалов в них становится соизмеримой с
* При действии ионизирующего излучения на воду образуется также в качестве первичного продукта радиолиза атомарный кислород [1, 2]. По последним данным [2], для у-лучей GI’Co минимальное значение G(O) составляет 0,008 атомаДООэв.
концентрацией растворенного вещества. С этого момента становятся вероятными реакции типа радикал—растворенное вещество, которые и вызывают в значительной степени химические превращения в дозиметрической системе.
В результате реакций в «шпорах» образуются молекулярные продукты радиолиза воды — ГД и Н2О2. Их начальные выходы, а также выходы радикалов, избежавших гибели в «шпорах» *, приведены в табл. 34, составленной по последним литературным данным в этой области (см., например. [3—8]).
Таблица 34
Выходы продуктов радиолиза воды под действием у-лучей ®°Со и электронов с, энергией 1 Мэв и выше
pH Ge-е aq GH GOH GH2O2 gh2
0—2 3,05 0,6 2,95 0,8 0,45
4—9 2,8—2.9 0,6 2,8—2,9 0,75 0,45
12—13 3,05 0,55 2,9 0,75 0,4
Как уже упоминалось, для рентгеновского и у-излучений и быстрых электронов «шпоры» находятся сравнительно далеко друг от друга. Для излучений с высокими величинами ЛПЭ они расположены настолько близко друг к другу, что сразу же после своего возникновения сливаются, образуя сплошную цилиндрическую колонку. Это приводит к возрастанию начальной концентрации радикалов, что в свою очередь обусловливает увеличение выходов Н, и Н202 и уменьшение выходов радикалов. Поэтому для рассматриваемых дозиметрических систем характерна зависимость показаний от ЛПЭ.
Кроме того, в случае разбавленных водных растворов при высоких мощностях позы имеет место конкуренция реакций радикал—радикал и радикал—растворенное вещество, прстекаю-щая в объеме раствора. Этот эффект, очевидно, также может оказать влияние на результаты измерения дозы. Таким образом, недостатком дозиметров на основе разбавленных водных растворов является зависимость их показаний от ЛПЭ излучения и мощности дозы при высоких ее значениях. Для устранения этого не
* В радиационной химии воды и водных растворов выходы этих продуктов обозначают символампОх nGXj (X = e~q’ И пли ОН: Х2 = Н2 или Н2О2). Таким обозначением подчеркивается, что эти выходы являются начальными. Рассматриваемые выходы могут отличаться от наблюдаемых экспериментально. Например, Н2О2 может возникать не только в «шпорах», но и в некоторых условиях за счет рекомбинации радикалов ОН в объеме раствора. Очевидно, в этом случае G(H2O2) > GH 0 .
достатка необходимо повышать концентрацию растворенного ве щества.
В растворах средней и высокой концентраций, помимо эффекта косвенного действия излучения, возможны и другие эффекты, которые могут вызвать изменение выхода. Во-первых, с ростом концентрации растворенного вещества могут в некоторой степени подавляться реакции радикалов в «шпорах», что приведет к увеличению вклада этих частиц в радиолитические превращения растворенного вещества. Во-вторых, не исключено, что в таких растворах в реакции может вовлекаться «возбужденная вода». В-третьих, в концентрированных растворах может быть заметным эффект прямого действия излучения на растворенное вещество.
Несколько слов о свойствах радикалов — продуктов радиолиза воды. Гидратированный электрон в кислой среде в результате очень быстрой реакции (3) трансформируется в атом Н. Наоборот, в щелочной среде происходит превращение Н в ейд *
Н + ОН-—> eaq + НгО. (8)
Реакция (8) обратимая; рК равновесия составляет 9,6.
Гидроксильный радикал в щелочной среде претерпевает электролитическую диссоциацию
ОН + ОН" д2 О- 4- НгО. (9)
Величина рК этой диссоциации составляет 11,9.
В растворах, содержащих кислород, весьма эффективно протекают реакции
^q+Os-O;, (10)
Н -f- Ог —’ НОг, (И)
в результате которых образуется гидроперекиспый радикал НО2 или его диссоциированная форма — О2. Поэтому данные частицы играют существенную роль в химических превращениях в дозиметрических растворах, в которых присутствует кислород. Для радикала НО2 характерны следующие равновесия:
НО2 zt Н+ + О~, (12)
Н+ -р НОг 72 Н20+. (13)
Для первого равновесия значение рК равво 4,4, а для второго — j 0 **
* Свойства eaq подробно изложены в книгах [8, 9].
** Ссылки на соответствующие оригинальные работы приведены, например, в книге [9].
Подробно с механизмом радиолиза воды и радиолитических превращений растворенных веществ заинтересованный читатель может ознакомиться, прочитав книги [10, 111.
2. Дозиметр Фрикке
Из числа дозиметрических систем наибольшее распространение в радиационной химии получила ферросульфатная система. Она представляет собой (1—5)-10 3 М водный раствор сернокислого закисного железа в 0,4 М H2SO4, содержащий 10 3 молъ!л NaCl и насыщенный воздухом. Раствор такого состава часто называют дозиметром Фрикке. При действии ионизирующего излучения на этот раствор двухвалентное железо окисляется в трехвален-тпое.
Afescamte.w радиолитических превращений
Общепринятый в настоящее время механизм радиолитических превращений в дозиметре Фрикке был первоначально предложен Ф. Кренцем и Г. Дьюхерстом [12] в 1949 г. Он включает реакции
(И) и (14)—(17):
Fe2+ + ОН Fe3+ + ОН-, (14)
Fe2+ + НОг -» Fe3+ + НО~, (15)
Н+ + НО" — НгОг, (16)
Fe2+ + НгОг — Fe3+ + ОН + ОН-. (17)
Учитывая то обстоятельство, что к настоящему времени установлено образование сйч в качестве первичного продукта радиолиза воды, к этой совокупности реакций необходимо добавить еще реакцию (3).
Новые сведения о механизме окисления ионов Fe21’ в ферро-сульфатной системе получены с помощью метода импульсного радиолиза [13—15]. Найдено, что константы скорости реакций (14) и (15) в хлорнокислых растворах равны соответственно 2,3-•108 и 1,4-10е л/(молъ-сек). Согласно [13—15], образование ас социата FeSO4 в сернокислых растворах оказывает малое влияние на скорость этих реакций. В реакции (15) в качестве промежуточного соединения возникает комплекс Fe8+HO2.
Еще в 1934 г. Ф. Габер и Дж. Вейс [16] постулировали, что реакция (17) является источником радикалов ОН. В настоящее время эта гипотеза является общепринятой. Согласно [17], в 0,5 М H2SO4 fc17 = 61,9 л/(молъ-сек).
Из совокупности реакций (3), (11) и (14)—(17) легко найти, что для дозиметра Фрикке
G (Fe3+) = 3G - -f- ЗСц - ^он “Ь ,оН^)
/}ы.г;од трехвалентного железа для рентгеновского м у-излучений и быстрых электронов
До 1955 г. результаты измерений G(Fe3+) были противоречивы. Лишь после ряда весьма точных определений этой величины, проведенных в 1953—1956 гг., было окончательно установлено, что G(Fe3+) для данных видов излучения в очень широком диапазоне энергий составляет 15,5—15,6 иона/100 эв. Рассмотрим методы определения G(Fe3+), использованные различными авторами.
Методы определения абсолютной величины G(Fe3+)
Известны четыре метода определения абсолютной величины G(Fe3+) или калибровки ферросульфатного дозиметра: калориметрический, ионизационный, измерения тока в растворе в случае заряженных частик и измерения абсолютной активности радиоактивного изотопа, введенного в раствор. Подробно эти физические методы дозиметрии рассматривались в главе III. Там же даны соответствующие примеры.
В табл. 35 приведены величины G(Fe3+) для у-излучения 60Со и других видов излучения с примерно теми же значениями ЛПЭ, полученные различными исследователями с помощью указанных методов.
В некоторые из значений 6’(Fe3+), приводимые в литературе, Г. Фрикке и Э. Харт [51J внесли небольшие попиэвки, обусловленные следующими соображениями. Во-первых, для определения G(Fe3+) ионизационными методами, как уже отмечалось, необходимо знать величину энергии W, требующуюся для образования одной пары ионов в воздухе. В настоящее время принято [52J. что И — 33,7 эв. До 1964 г. расчеты G(Fe3+) осуществлялись на основе значения W, отличающегося от приведенного. Поэтому соответствующие величины G(Fe3+) были исправлены с учетом нового значения W. Во-вторых, ряд измерений G(Fe3+) проводился для 0,05 М растворов H2SO4. При переходе к 0,4 М H2SO4 результаты этих измерений должны быть увеличены на 2 %. В соответствующих значениях 6’(Fe3+), включенных в табл. 35, эти поправки учтены.
На основании данных табл. 35 можно заключить, что G(Fes+) в случае у-излучения 60Со равен 15,6. Эту величину G(F°3+) необходимо использовать во всех экспериментах с указанным видом излучения, проводимых при комнатной температуре.
Влияние энергии на G(Fe3+)
Как видно из табл. 35, G(Fe3+) практически не зависит от энергии рентгеновского и у-излучений и быстрых электронов в диапазоне от 0,66 до 16 Мэв. Объясняется это тем, что данные виды излучения в этом диапазоне энергий имеют близкие значения ЛПЭ. При достаточно низких энергиях рассматриваемых видов
Таблица 35
Величины G(Fe3+) для f-излучения 60Со и других видов излучения с близкими значениями ЛП )
Вид излучения Начальная или эффективная энергия, Мэв Метод измерения G(Fe3+) G(Fc’+), ионы; 100 эв Литература
у-Лучи всСо 1,25 Калориметричес- 15.6 [18]
кий То же 15,8 [19]
» 15,7 [20]
» 15,42 [21]
» 15,45 [22]
» 15,1 [23]
» 15,8м [24]
» 15,57 [25]
Ионизационный 15,9*2 [26, 27]
» 15,55 [28]
» 15,6 [29]
у-Л учи 137Cs » 15,7 [24]
0,66 Но низ аци о инып 15,7м [26]
у-Лучп 1921г » 15,6м [24]
0,382 1 Тонизацпопный 15,4м [24]
Рентгеновские 24,5*3 Ионизационный 15,2*2 [30]
луч п 22*з » 15,6 [29]
2о*з » 16,0*2 [28]
10 » 15,9* [26]
7,6 » 16,8М>*2 [27]
7,6 » 16,1м [24]
7,6 Калориметричес- 16,0м [24]
КИЙ 15,7*2
4, О*3 Ионизационный [28]
1,3 >> 16,4 [31]
Электроны 33 К ало р иметрп ноский 15,56 [32]
30 Ион из ацио нный 16,3 [33]
25 » 15,9 [34]
20 Калориметрический 15,56 [25]
20 Ионизационный 16,0 [34]
20 Калориметричес- 15,17 [35]
кий
18 То же 15,35 [36]
17 Ионизационный 15,8 [34]
16 » 15,9*2 [37]
16 Калорпметрнчес- 15,2 [35]
КИЙ
15,9 То же 15,4 [36]
Вид излучения Начальная или эффективная энергия, Мэв Метод измерения G(Fe3+) G(Fe3+), ионы/100 эв Литература
Электроны 15 Ионизационный 16,1 [38]
13,25 Калориметрический 15,49 |36]
6,3 Ионизационный 15,7 *2 [37]
6,3 » 15,7 133]
6,3 Калориметрический 15,32 [35]
2,0 Измерение тока в растворе 15,45 [39]
2,0 То же 15,6 [40]
1—1,2 » 15,1 141]
1,0 » 15,6 |40|
1,0 » 15,3 [39]
0,8—0,9 » 15,55 [42]
Р-Лучи 9 Y 0,93 Ионизационный 15,7*' [24]
р-Лучи 32Р 0,69 Измерение активности в растворе 15,4 |43-45]
0,69 То же 15,2 [45]
0,69 Калориметрический 15,6 *1 [24]
0,69 Ионизационный 15,65 *i 124]
0,69 Измерение активности в растворе 15,6 [46]
Протоны 660 Измерение тока в растворе 16,9 [47]
592 То же 16,2 [48]
16.) » 16,5 [49]
157 » 16,1 [50]
** V,05 М H2SOj, для 0,4 М H2SO4 величина G(Fe3+) увеличена на 2% Пересчитано для IV = 33,7 эв.
*• Максимальная анергия.
Таблица 36
Величины G(Fes+) для электронов низкой энергии
Виц излучения Средняя энергия, Мэв G(Fe3+), ионы/100 эв Литература
Р-Лучи трития 0,0057 13,05*1; 12,95*1 [24]
0,0057 12,7 [53]
0,0057 12,9 [54]
Р-Лучи MS 0,0495 14,7*1 [24]
Таблица 36а
Величины C?(Fe3+) для рентгеновских лучей низкой энергии
Эффективная энергия, Мае G(Fe3+), ионы/100 эв Литература Эффективная энергия, Мэв G(Fe3+), ионы 100 эв Литература
0,008 13,8*0 *г (55] 0,048 14,3 [60]
0,01 14,5*i> *2 [55] О,05*з 14,0 [61]
0,012 14,6 [56] 0,056 15,4*1, *2 [27]
0,014 14,8*1, *а [57] 0,062 13,8*’> *2 [62]
0,015*3 14,2*2 [58] 0,075 14,7*2 [26]
0,02*з 12,7 [58] 0,1 14,6*! [24]
0,021 13,5*1, *2 [27] 0,1 14,9*1 [24]
0,023 15,2*2 [59] 0,1 14,9 [61]
0,025 13,7 [60] 0,175 14,8 [63]
0,03*3 15,2*2 [38] 0,2*3 14,4 [29]
0,033 15,1*1,*2 [27] 0 2*3 15,8*2 [38]
0,035 15,2*1, *2 [59] 0,3*з 14,3*2 [64]
0,035 14,4*> [24] 0,3*з 14,6*2 [28]
0,04*з 15,8*2 [38]
0,045 14,5*1 [24]
*1 0,05 М H2SO4; для 0,4 М H2SO4 величина G(Fe’+) увеличена на 2%.
*2 Пересчитано для И‘ =33,7 эв.
*3 Максимальная энергия.
излучения вследствие заметного возрастания ЛПЭ значения G(Fe3+) уменьшаются. В табл. 36, 36а приведены величины G(Fe3+) для электронов и фотонов низкой энергии.
Сравнение данных табл. 35, 36 и 36а показывает, что для фотонов уменьшение G(Fe3+) становится заметным начиная с энергий порядка 0,1—0,3 Мэв. В этих случаях применение дозиметра Фрикке для точных измерений становится возможным только тогда, когда известна эффективная энергия используемого излучения.
Влияние мощности поглощенной дозы на G (Fe3*)
Нижний предел независимости G(Fe3+) от мощности поглощенной дозы точно не установлен. Согласно Н. Миллеру [65J, G(Fe3+) постоянен в диапазоне мощностей дозы от 0,1 до 4-103 рад/сек. Очевидно, этот нижний предел лимитируется скоростью самопроизвольного окисления ионов Fe2+ (см. стр. 142). Р. Шулер и А. Аллен [39] показали, что в случае непрерывного электронного потока с энергией 2Мэв G(Fe3+) не зависит от мощности дозы приблизительно до 10® рад/сек при условии эффективного перемешивания раствора во время радиолиза. При более высоких мощностях дозы наблюдалось уменьшение G(Fe3+), вызванное локальным расходом кислорода в облучаемом объеме раствора.
Использование импульсного электронного излучения позволило определить величину G(Fe3+) при значительно более высоких мощностях дозы. Оказалось, что, начиная примерно с Ю8 рад/сек G(Fe3+) уменьшается (см. главу IX).
Влияние концентраций кислорода и ионов Fe2 на G (Fe:!+)
Кислород, присутствующий в ферросульфатном дозиметрическом растворе, оказывает существенное влияние на радиолиз этой системы. В его отсутствие G(Fe3+) в 1,9 раза меньше.
В дезаэрированных растворах величина G(Fe3+) определяется протеканием реакций (3), (14), (17), (19) и (20):
Fe2+ 4- Н FeH2+, (19)
FeH2+ -|1К Fe3+ + Hz. (20)
Отсюда для дезаэрированных растворов
G (Fe>4 = G _ + GH + G0H + 2GHiO2. (21)
eaq
Согласно [13], среднее значение /с)9 из различных измерений составляет 1.8-107 л/(моль-сек). Гидрид FeH2+ имеет полосу оптического поглощения с Хтах = 330 нм. По данным [13], в хлорнокислом растворе kin = 1.06-104 л/(молъ-сек).
По многочисленным измерениям [66—73], G(Fe3+) г дезаэри-рованном растворе Fe2+ в 0,4 М H2SO4 при радиолизе под дейст вием 7-лучей и быстрых электронов равен 7,90—8,22 иона^ОО эв. Точное значение этого выхода составляет 8,2 иона/100 эв.
Рис. 62. Зависимость концентрации образовавшегося Fe3 + при -у-радиолизе 10~2 М раствора Ге2+ в 0,4 М H2SO4 от дозы D
Очевидно, по мере израсходования кислорода, присутствующего в растворе, скорость окисления Fe2+ пни дальнейшем облучении будет постепенно уменьшаться и, в конце концов, выход достигнет значения 8,2 иона/ЮО эв. На рис. 62 показана зависимость концентрации образующегося Fe3+ от поглощенной дозы для раствора ферросульфата, насыщенного воздухом (по данным 174]). Подробное исследование влияния концентрации кислорода на величину G(Fe‘!+) проведено в работах [73. 75].
В 10~я М растворе Fe2+ в 0,4 М H2SO4, насыщенном воздухом, G(Fe3+) сохраняет постоянное значение до того момента, пока
не будет израсходовано ~ 70 - 80% ионов Fe2+, что соответствует дозе ~ (4 —5)-10‘ рад.
При повышении концентраций ионов Fe2+ и кислорода G(Fe3+) несколько возрастает. По последним измерениям [70], при у-радио-лизе 10 2 М раствора Fe2+ в 0,4 М H,SO4, насыщенного кислородом, G(Fe3+) = 16,07 пона/100 эв.
По данным Ф. Дейнтона и Г. Саттона [77], G(Fe3+) снижается при концентрациях Fe2+ менее 10 1 М. Объясняется это сравнительно низким значением Л17. В 10 3 М растворе период полупревращения Fe2+ за счет этой реакции равен ~ 14 сек. Поэтому в обычных условиях пост-эффекты в дозиметре Фрикке не наблюдаются. Однако при малых концентрациях Fe2+ (< 10 4 М) они становятся заметными, и можно в принципе достичь такого положения, когда реакция (17) не будет протекать за время облучения. Вследствие этого измерение концентрации Fe3+ в таких растворах сразу после облучения даст заниженное значение дозы. Если же облученный раствор нагреть или дать ему постоять определенное время после радиолиза, то G(Fe3+) достигнет величины, соответствующей более концентрированным растворам. Уменьшение G(Fe3+) при концентрации Fe2+ ниже 10 4 М наблюдал также Г. Дьюхерст [78J. Дт. Гормли и К. Хоханадель [79] отметили небольшое увеличение G(Fe3+) в дозиметре Фрикке при концентрациях Fe2+ выше 10 2 М.
Таким образом, G(Fe3+) в ферросульфатной дозиметрической системе практически не зависит от концентрации Fe2+ в диапазоне Ю-4—ю-2 М
Влияние концентрации серной кислоты па G(Fe3+)
Влияние концентрации серной кислоты на величину G(Fe3+) в дозиметрическом растворе изучалось в ряде работ [24, 32, 70, 78, 80—93]. Наиболее детально окисление Fe2+ при высоких pH было исследовано А. Алленом и сотр. [82, 92]. Эти авторы нашли, что с ростом pH G(Fe3+) несколько уменьшается. Соответствующие данные [92] приведены ниже:
pH
0,4 Л/ H2SO4
1,30
1,88
2,75
G (Fe«+), ионы, 100 эв
15,5
15,52
15,19
14,60
pH G (Fes+), ионы,'100 эз
3,40
4,40
5,80
14,07
14,03
13,95
Эти данные были получены при использовании доз ниже 150 рад. При pH 1 концентрация образующегося Fe3+ не пропорциональна дозе [82, 92J. Очевидно, это обстоятельство усложняет использование таких растворов для измерения доз. Следовательно, 0,05 М H2SO4— это наинизшая концентрация серной кислоты, при которой дозиметр Фрикке пригоден для определения доз.
Таблица 37
Влияние температуры на G(Fe3+)
Интервал температур, °C Возрастание G(Fe»+), %-'°С Литература Интервал температур, °C Возрастание G(Fe3+), %/°С Литература
4—54 0 [94, 95] 31—60 0,4 [99]
5-50 0 [96] 15—30 0,11« [25]
20—45 0 [26] 30—45 0,38*1 [25]
25-65 0 [18] 15—30 0,14*2 [25]
0—70 0,04 [97] 30—45 -0,11*2 [25]
2—65 0.,09 [98]
** Облучение в полистироловых ампулах (объем 4 мл).
*з Облучение в полиэтиленовых ампулах (объем Я,5 мл).
Проводились измерения величины G(Fe3+) при концентрациях H2SO4, превышающих 0,4 М. Г. Дьюхерст [78] нашел, что G’(Fe3+) в 1 М П 2SO4 имеет ту же величину, как и в 0,4 М H2SO4. М. А. Про-скурнкн и сотр. [84] сообщили, что при концентрации H2SO4 выше 4 М G(Fe3+) составляет 60 ионов/100 эв. Позже их результаты не были подтверждены [85—91]. Было показано, что G(Fe3+) практически не зависит от концентрации H2SO4, по крайней мере, до 2,5 М. Таким образом, G(Fe3+) мало зависит от концентрации серной кислоты в диапазоне 0,05 — 2,5 М.
Влияние температуры на G(Fe3+)
По-видимому, G’(Fe3+) несколько увеличивается при возрастании температуры облучения. Однако литературные данные об этом эффекте противоречивы (табл. 37).
Стабильность раствора при хранении
При стоянии ферросульфатный раствор, насыщенный воздухом, медленно окисляется. По данным Р. Хуффмана и Н. Дэвидсона [100], скорость этого процесса пропорциональна концентрации ионов Fe2+ во второй степени и концентрации кислорода в первой степени. В 0,01 М растворе FeSO4 скорость самопроизвольного окисления ионов Fe2+ при 25° С составляет ~ 8-10~3 молъ/(л-час). Ионы СБ катализируют рассматриваемый процесс (см. стр. 144). При проведении дозиметрических измерении следует вносить поправку на самопроизвольное окисление Fe2+.
Г. Кубота [101] исследовал стабильность дозиметра Фрикке при 150—250° С. Оказалось, что при этих температурах происходит увеличение его оптической плотности при 305 нм на 0,2—0,4. Это вызвано окислением ионов Fe2+ кислородом воздуха, причем степень окисления зависит от объема воздуха над раствором.
Влияние примесей на G(V&*)
Влияние органических примесей
Еще в 1934 г. Г. Харкер [102] нашел, что присутствие небольших количеств некоторых органических соединений оказывает существенное влияние на скорость радиолитического окисления Fe2+. В 1951—1952 гг. Г. Дьюхерст [103, 104] исследовал влияние добавок алифатических спиртов на величину G(Fe3+) в дозиметрическом растворе. Им было обнаружено, что введение спирта в дозиметр Фрикке приводит к увеличению G(Fe3+). Аналогичный эффект оказывают и многие другие органические вещества [43, 105—125J. Из имеющихся литературных данных об этом эффекте следует, что он зависит от природы и строения органического вещества, концентрации последнего и концентрации ионов Fe2+.
Наиболее вероятно, объяснение рассматриваемого эффекта состоит в том, что органические примеси конкурируют с ионами Fe2+ за радикалы ОН; возникающие при этом органические радикалы взаимодействуют с кислородом, образуя перекисные радикалы; последние окисляют Fe2+ по цепному механизму. Например, в дозиметре Фрикке в присутствии этилового спирта возможны реакции (3), (И), (14) — (17) и (22) - (27) [103, 104, 126-129J:
СНзСНгОН + ОН СНзСНОН 4 Н2О, (22)
СНзСНОН 4 Ог -> СНзСН(О2)ОН, (23)
Н+ + рег+ СНзСЩСМОН Fe3+ 4- СН3СН(ООН)ОН, (24)
Н+ 4- Fe2+ 4- CHsCH(OOH)OH -» Fe3+ 4- СНзСН(О)ОН 4 Н2О, (25)
Н+ 4- Fe2+ 4 СН3СН(О)ОН -> Fe3-*- 4- СНзСНО 4- Н2О, (26)
СНзСНгОН 4 СН3СН(О)ОП -> СНзСНО 4- СНзСНОН -f- Н2О. (27)
Ингибирующий эффект хлорид-понов
В 1951—1952 гг. Г. Дьюхерст [103,104] показал, что возрастание G(Fe3+) в присутствии органических примесей подавляется добавлением к раствору хлорид-ионов. Например, влияние 10~4 молъ!л этилового спирта на G(Fe3+) исключается при введении в раствор 10~2 молъ/л NaCl. М. Бэртон и сотр. [112] наблюдали аналогичное действие добавки NaCl в случае бензола и фенола.
Объяснение ингибирующего действия хлорид-ионов состоит в том, что ионы С1_ конкурируют с органической примесью (обозначим ее RH) за радикалы ОН:
С1~ 4- Н+ 4 ОН С1 4 Н2О, (28)
С1 4- Cl- -» С1“, (29)
RH 4- ОН —» R 4- Н20. (30)
Образующиеся С1 и С12, подобно радикалу ОН, окисляют один ион Fe2+. Константа /с28 равна в 0,4 М H2SO4 4 -10s л/(молъ- сек) [130]. В обычной лабораторной дистиллированной воде концентрация
органических примесей низка, и поэтому добавка 10-3 моль!'л NaCl бывает, как правило, достаточной, чтобы выполнялось условие: fc,8 [СН| > *30 [RHJ, т. е. чтобы реакция (30) была подавлена. Дж. Вейс и др. [74J нашли, что добавление указанного количества NaCl к воде из различных источников устраняет в пределах ошибок опыта влияние содержащихся в ней органических примесей.
Отметим, что для некоторых органических веществ с двойной связью ингибирующий эффект хлорид-ионов не проявляется [43]. Наоборот, в ряде случаев (например, в присутствии аллилтиомочевины) G(Fe3+) после добавления NaCl еще больше увеличивается. Это явление обусловлено, по-видимому, конкуренцией ненасыщенных соединений и ионов Fe2+ за атомы С1 или С12:
Fe2+ + Cl Fe3+ + CH, (31)
Cl 4- СН=СН----> — CHCI - СИ -. (32)
Вследствие этого в присутствии кислорода сохраняется возможность образования перекисного органического радикала и последующего цепного окисления Fe2+.
Хлорид-ионы оказывают некоторое каталитическое действие на термическое окисление Fe2+ на воздухе. Этот эффект становится заметным при концентрации NaCl выше 10-2 М. Поэтому использование растворов с таким содержанием ионов С1~ включает ряд трудностей. С. А. Брусенцева и П. И. Долин [131] исследовали влияние концентрации СП на величину GfFe3+) в насыщенном воздухом растворе. Было обнаружено, чю G(Fe3+) не изменяется при концентрациях КС1 до 0,1 М. Однако в растворе, где содержание СП равно 1 М, G(Fe3+) возрастает до 16,5 иона/ЮОэв. Это увеличение, вероятно, объясняется окислением Fe2+ свободным хлором, образующимся в концентрированных растворах СП в результате прямого действия излучения на растворенное вещество [132, 133]. Поданным ]134], G(Fe3+) несколько возрастает даже в том случае, если концентрация ионов СП равна 10 2 М. Таким образом, можно заключить, что добавка СП не должна превышать 10-2 моль!л.
Недавно А. Своллоу и сотр. [135] исследовали методом импульсного радиолиза кинетику реакций в ферросульфатной системе в присутствии ионов СП. Ими было найдено, что константы скорости реакций С1 и С1й с ионами Fe2+ равны соответственно 5,9-10я и 1,4-107 л/(молъ- сек). На основании этих величин, а также значений ки, к№, к33 и k3i (по данным [135], к<м = 2,1 • 1010 л7(молъ-сек), /с33 — 4,3• 10я л/(молъ-сек} пk3i = 2,1 -1010 л/(молъ-сек) при ионной силе 1)* легко найти, что в растворе, содержащем 10-3 молъ/л Fe2+ и СП, все радикалы ОН реагируют с ионами СП, давая С1:
СП 4 ОН —» СЮН- (33)
СЮН- + Н+ С1 + Н2О. (34)
* Реакция (33) обратимая. Согласно [135J, константа скорости распада СЮН-на ОН и СП равна 6,1-109 сек-1.
При этом ~80% атомов С1 непосредственно взаимодействует с Fes+, а 20% атомов С1 — с ионами С1“, давая С12. Таким образом, в растворе указанного состава вместо реакции (14) протекают реакции (31) и (35):
Fe?+ + С1~ -> Fe3+ + 2С1~. (35)
Влияние неорганических примесей
Н. Миллер и Дж. Уилкинсон [136] показали, что использование соли Мора вместо сернокислого закисного железа, т. е. введение в раствор ионов аммония, не оказывает влияние на величину G(Fe3+). Позднее это было подтверждено в работе [74]. Дж. Вейс и сотр. [137] нашли, что в нейтральных и щелочных растворах (NH4)2SO4 в результате облучения наблюдается образование нитрита. В сильнокислых растворах этот процесс не происходит. Таким образом, для приготовления дозиметрического раствора можно применять как FeSO4-7H„O, так и (NH4)2SO4-FeSO4-6H2O.
В ряде случаев неорганические примеси оказывают влияние на величину G(Fe3+). Согласно [131], G(Fe3+) заметно изменяется в присутствии ионов Вг“. По данным [138], начальный выход Fe2+ в соляной кислоте такой же, как и в сернокислой среде. Однако наблюдается уменьшение G(Fe3+) при сравнительно больших дозах. Дж. Вейс и сотр. [801 наблюдали влияние фторид-и фосфат-ионов на процесс окисления Fe2+ при высоких значениях pH. Существенно влияет на величину G(Fe3+) наличие в растворе ионов Сп2+ [139]. В присутствии ионов Се3+ и NOg, по данным М. А. Проскурнина и сотр. [140], происходит возрастание G(Fe3+).
Однако маловероятно, чтобы в соли Мора или сернокислом закисном железе и серной кислоте марок «ч.д.а.» или «х.ч.» рассмотренные выше примеси присутствовали в значительных количествах и вызывали существенное изменение G(Fe3+).
И. Миллер и Дж. Уилкинсон [136], детально исследовав радиолитическое окисление Fe2+ в растворах, приготовленных из препаратов аналитической чистоты, поставляемых различными фирмами, показали, что расхождения в величинах G(Fe3+) для проверенных реактивов не превышают 2%. Правда. Дж. Дэвис и Дж. Лоу [141] обнаружили, что отдельные сорта серной кислоты содержат некоторые примеси (вероятно, главным образом SO2), которые могут привести к ошибке в несколько процентов при измерении дозы ферросульфатным раствором.
Методы определения концентрацией ионов Fe3+, используем ые в дозиметрических измерениях
Для определения концентрации ионов Fe3+, описаны следующие основные методы анализа: потенциометрическое титрование облученного раствора растворами перманганата калия, бихиомата калия или сернокислого окисного церия, колориметрическое или спектрофотометрическое определение Fe2+ с о-фенантроли-
ном, колориметрическое или спектрофотометрическое определение Fe3+ с тиоцианатом и прямое спектрофотометрическое определение Fe3+ в ультрафиолетовой области спектра.
Потенциометрический метод анализа состоит в определении концентрации Fe3+ по убыли Fe2+ в растворе, подвергнутом облучению, по сравнению с необлученным раствором. Г. Фрикке и С. Морзе [142| использовали с этой целью бихромат калия и сернокислый окисный церий, а М. А. Проскурнин и сотр. [114] — перманганат калия. По рекомендации Г. Фрикке и Э. Харта 151], для нахождения конечной точки титрования удобно применять о-фе-нантролиновый комплекс двухвалентного железа. Потенциометрический метод дает удовлетворительные результаты при измерении доз в диапазоне 4-103 — 5-104 рад [51].
Определение Fe3+ с о-фенантролином также производится нахождением разницы концентраций Fe2+ в необлученпом и облученном растворах. Окраска о-фепантролинового комплекса Fe2+ измеряется при pH 4—5 и длине волны 510 нм. Методика проведения анализа подробно излагается в разделе 3 этой главы. Согласно [143], при использовании о-фенантролипового метода определения Fe3+ возможно измерять дозы от 103 до 3-104 рад.
Тиоцианатный метод заключается в измерении оптической плотности тиоцианатного комплекса Fe3+ при длине волны 480 нм [144[. Подробное исследование этого метода в применении к ферро-сульфатной дозиметрии было проведено Н. Фригерио и В. Генри [145|. Эти авторы для определения Fe3+рекомендуют добавлять один объем насыщенного водного раствора NH4CNS к 3 объемам облученного раствора; интенсивность появляющейся красной окраски измеряется при 475 нм, причем в качестве раствора сравнения используется необлученпый раствор, с которым проделаны те же операции. Молярный коэффициент экстинкции тиоцианатного комплекса в этих условиях равен 1,56-104 л/(молъ-см). Он не зависит от температуры в интервале 19—31° С. Интенсивность окраски не меняется в течение 5—30 мин. после добавления NH4CNS. По данным работы [145], с помощью этого метода можно определять дозы от 50 до 5-103 рад.
Б. Гупта [146] ввел в сернокислый раствор ферросульфата краситель ксиленоловый оранжевый. Этот краситель образует комплекс с Fe3+. Максимум полосы оптического поглощения комплекса находится при 540 нм, причем в 0,05 A7 H2SO4 молярный коэффициент экстинкции при этой длине волны равен 1,34-104 л/(молъ-см). Найдено, что, измеряя интенсивность этого поглощения после облучения раствора, можно определять дозы от ~ 300 до 3500 рад.
Наиболее широкое применение в настоящее время находит прямой спектрофотометрический метод определения Fe3+ по поглощению света в ультрафиолетовой части спектра, введенный в радиационную химию Т. Хардвиком [147] в 1952 г. В нем оптическую плотность облученного раствора сравнивают с оптической плотностью необлученного раствора при 304—305 или 224 нм 146
(гораздо реже) и по разнице находят концентрацию Fe3+. Опти ческий спектр ионов Fe3+ в 0,4 М H2SO4 (относительно 0,4 М H2SO4) показан на рис. 63. Согласно [136, 137], подчинение закону Ламберта — Бэра в пределах 0,5% наблюдается вплоть до 10“2 молъ/л FeSO4. Молярный коэффициент экстинкции Врез+ ионов Fe3+ в 0,4 М H3SO4npn 304 нм возрастает при увеличении температуры (0,69%/граЗ [148]).
Молярный коэффициент экстинкции ионов Fe3+ в 0,4 М HoS04 в максимуме полосы оптического поглощения (при длинах волн
Рис. 63. Оптические спектры поглощения ионов Fe3+ (7) и Fe2+ (2) в 0,4 М растворе H2SO4 (относительно 0,4 М H2SO4)
S — оптическая плотность
302—305 нм) измерялся во многих работах (см., папример, [18, 20, 25, 29, 34, 37, 39, 43,44, 58, 59, 62-64, 67, 68, 78, 94, 134, 138, 141, 147—189[). Измерения проводились при различных температурах. Если найденные в этих работах величины в привести к температуре 25е С с использованием температурного коэффициента 0,69% /град, то эти величины колеблются в интервале от 2100 до 2340 л1(молъ-см), т. е. в пределах ~10%. В обзоре [51[ принято значение 2197 л/(моль-см) при 304 нм и 25° С.
В последние годы в литературе уделяется большое внимание анализу ошибок, обусловленных применением спектрофотометрического метода определения ионов Fe3+ [134, 189—192]. Согласно [189J, главный источник неопределенности в величине е, измеренной различными авторами, состоит в использовании неодинаковых спектрофотометров. Найдено 9 %-ное расхождение в значениях егез! . полученных с помощью 10 различных спектрофотометров.
Учитывая сравнительно большой разброс литературных величин е, для точных дозиметрических измерений коэффициент экстипкции необходимо определять с помощью того спектрофотометра, который используется в данной лаборатории для анализа Fe3+. С этой целью необходимо приготовить стандартные растворы Fe3+/ Последние можно получить, например, растворением спектрально чистой железной проволоки в серной кислоте и последующим окислением Fe2+ перекисью водорода. Определить еге»+ можно также добавлением к раствору Fe2+ известного объема стандартного раствора Се4+ в количестве, недостаточном для полного окисления Fe2+. При использовании этого раствора для нахождения tFe3+ следует вносить поправки на поглощение света ионами Се3+.
По данным работы [153], Рсе-<+ в 0,4 М HaSO4 равен 24,4 ± 2,4 при 305 нм и 26,4 ± 2,5 при 304 нм.
Двухвалентное железо не мешает определению концентрации Fe3+ измерением оптической плотности раствора при 304—305 нм. Значение еие^ при 304 нм равно ~ 1 л/(молъ-см) 151, 147J.
Согласно {1491, чувствительность метода может быть увеличена в 2 раза, если для определения Fe3+ использовать максимум при 224 нм. При этой длине волны Сее<+ в 0,4 М H2SO4 равен 4565 л/(молъ- см) при 25° С, причем температурный коэффициент евез+ в этом случае составляет всего 0,13%/град. Однако еве*+ при 224 нм выше, чем при 304 нм [при 224 нм он равен 20 л/(молъ-см)\.
Молярный коэффициент экстинкции Fe3+ зависит от концентрации серной кислоты. По данным работы [153J, °.Fe3+ при 304 нм в 0,05 М Н2ЬО4на 0,6% выше, а в 0,005 М H2SO4 на 2,4% ниже, . чем в 0,4 М H2SO4. Согласно Ф. Дейнтону и Ф. Джонсу [158], в 2 М H2SO4 cfcs+ на 2,7% ниже, а в 5 М H2SO4 на 26% выше, чем в 0,4 М H2SO4. При высоких концентрациях серной кислоты максимум оптического поглощения ионов Fe3+ несколько сдвигается в сторону более коротких длин волн. Как следует из работы [158], Хтах для ионов Fe3+ в 5М растворе H2SO4 находится при 295 нм.
Зависит от концентрации кислоты и температурный коэффициент Ргез+. К. Хендерсон и Н. Миллер [153] нашли, что в 0,05 и 0,005 М H2SO4 он равен соответственно 0,6 и 1,1%/град.
Предлагались и другие методы анализа Fe3+ в облученном ферросульфатном растворе: полярографический [193, 194], электрохимический (по изменению окислительно-восстановительного потенциала) [195, 196], магнетохимический [197J и радиометрический 1198—201J. Г. Рудстам и Т. Сведберг [198J добавляли к дозиметрической системе радиоактивный изотоп S8Fe. После облучения Fe3+ отделялся от Fe2+ экстракцией тиоцианатного комплекса изоамиловым спиртом. Величина дозы находилась путем измерения распределения 59Fe между органической и водной фазой. Цитированные авторы заявили, что этим методом можно определить дозы порядка 1 рад. Однако позже описанный метод был проверен С. Прибичевичем [199], который нашел, что низший предел измеряемой дозы составляет ~40 рад. О. Гал [200] предложила видоизменение радиометрического метода. В нем ионы Fe3+ отделя ются от ионов Ге2+на ионообменной колонке из фосфата циркония. Низший предел в этом случае равен 10 рад. К. Хасегава 1201] применил для разделения колонку с фосфатом титана.
По данным [202], чувствительность дозиметра Фрикке можно повысить применяя для анализа ионов Fe3+ метод прямого счета отдельных фотонов ультрафиолетового света. Этот метод примерно в 30—35 раз чувствительнее спектрофотометрического, и низший предел измеряемых доз при использовании оптической кюветы с толщиной слоя поглощающей жидкости 1 см равен 120 рад.
Недавно У. Маклафлин и Э. Бьергбакке [203J предложили
использовать метод электрохимической потенциометрии для анализа ионов Fe3+ в облученных сернокислых (pH 2,7) растворах Fe2+ (10-3 М), содержащих ионы F~ (10 4 М). В этом методе с помощью фторидного электрода измеряется комплексообразование Fe3h с F-. Применение его дает возможность работать с малыми объемами (~ 10 5 мл) раствора. При этом диапазон определяемых доз такой же, как в случае спектрофотометрического метода (от ~ 5 до 40 крад).
Границы применимости дозиметра Фрикке
Исходя из изложенного выше, можно сделать определенные выводы о границах применимости растворов ферросульфата для дозиметрии рентгеновскою и у-излучений и быстрых электронов. Нижний предел измерения дозы, очевидно, зависит оз чувствительности метода анализа трехвалентного железа. Например, при прямом спектрофотометрическом метопе определения Fe3h при использовании Zrnax = 304 -=- 305 нм с достаточной сзепенью точности (в пределах 1—2%) можно измерить изменение оптической плотности, равное 0.1. Эго соответствует дозе ~ 3- 10s рад [74]. В табл. 38 охарактеризована дозиметрическая чувствительность различных методов анализа понов Fe3+.
Таблица 38
Низший предел измерения дозы ферросульфатным раствором при использовании различных методов анализа ионов Fe3+
Метоп анализа Низший предел измерения дозы, рад Литература
Потенциомет рический 4-103 [51]
Спектрофотометрический Xmax = Зи4 -г- 305 нм, 1 = 1 см 3-103 [74]
Хтах = 304 305 нм, 1 = 10 см (3 -4)-102 [И7]
\пах = t = 1 (•'< (1,5- -2,0) -10s [149]
о-Фенантролпповый 1-Ю3 [143]
Измерение оптического поглощения комплекса роз+ с ксиленоловым оранжевым (Х]Пах 540 им, 1 = 1 см) 300 [146]
Прямой счет фотонов ультрафиолетового света (Хтах = 304 = 305 им ,1 = 1 см) 120 [202]
Тиоцианатный 50 [145]
Радиометрический (с 59Ре) 10 [200]
Примечание. I — толщина поглощающего слоя.
Верхний предел измерения дозы с Помощью ферросульфатного дозиметра ограничивается израсходованием кислорода, присутствующего в растворе. Это соответствует, как видно из рис. 62, примерно 4-101 рад (для раствора, насыщенного воздухом). По данным работы [741, этот предел можно повысить приблизительно в 5 раз, если использовать раствор, насыщенный кислородом и содержащий 5 -10 2 моль/л ферросульфата. Ф. Хаасброэк [204J сообщил, что увеличение концентрации Fe2+ до 5-10 2 М и непрерывное пропускание воздуха или кислорода через раствор позволяют расширить верхний предел до 106 рад. В этих условиях G(Fe3+) не зависит от дозы вплоть до израсходования 40% начального количества ионов Fe2+. При больших степенях превращения G(Fe3+) уменьшается, но если учесть этот эффект, то можно измерять дозы и выше 10е рад. Однако в этом случае G(Fe3') несколько выше [76[. Кроме того, при работе с такими сравнительно концентрированными растворами следует обращать особое внимание на самопроизвольное окисление ионов Fe2+. Для 5-10"2 М раствора скорость этого процесса равна (2—4)-10 6 мольЦп• час), а при пропускании кислорода она возрастает до 1,25 • 10'5 молъ/(л• час) 1100].
П рактичеекие. рекомендации для измерения дозы с помощью дозиметра Фрикке
При проведении дозиметрии раствор ферросульфата заливают в сосуд, который помещают в строго определенное положение относительно источника ионизирующего излучения. Размеры и форма дозиметрических сосудов практически не оказывают влияния та процесс окисления ионов Fe2+ в том случае, если их внутренний диаметр больше 8 мм |74] (см. также стр. 153).
Иногда при дозиметрии необходимо, чтобы материал сосуда и дозиметр Фрикке имели одинаковый атомный состав. В этих случаях используют сосуды из пластмасс. Многие исследователи [26, 27, 31, 63, 136, 141 205, 206| считают, что наряду со стеклянными сосудами возможно применять также сосуды из политетрафторэтилена или полиметилметакрилата; данные о возможности использования полиэтиленовых сосудов противоречивы. При хранении ферросульфатпого раствора в полистироловых сосудах появляется заметное оптическое поглощение в ультрафиолетовой области спектра без облучения и, кроме того, дозиметр дает при этом неправильные показания [207, 208]. По данным Г. Свенссона и др. [209|, на воспроизводимость показаний ферросульфатного дозиметра, помещенного в полистироловый сосуд, оказывает влияние отношение поверхности сосуда к его объему и время хранения раствора до облучения. Если указанное отношение равно 5 см~3 (объем сосуда 3 см3), то дозиметр дает правильные и воспроизводимые результаты, несмотря на то что до облучения раствор стоял в этом сосуде в течение 2—3 час. Когда это отношение составляет 27 см~' (объем сосуда 0,4 см3), то хранение раствора до
облучения даже в течение 15 мин. приводит к неправильным показаниям.
Г. Фрикке и Э. Харт [511 отмечают, что даже два образца полимерного материала одного и того же состава могут содержать различные количества примесей. Поэтому в каждом отдельном случае при работе с пластмассовыми сосудами требуется специальная проверка. Необходимо хранить сосуды заполненными дозиметрическим раствором и заливать свежий раствор непосредственно перед измерениями [511. Применять металлические сосуды не рекомендуется, так как многие металлы реагируют с 0,4 М H2SO4. Правда, по дачным [210|, можно пользоваться сосудами из нержавеющей стали, обработанными в течение нескольких минут горячей 96%-ной азотной кислотой.
Для приготовления дозиметрического раствора рекомендуется использовать следующую пропецуру: 0,4 г FeSO4-7H2O или 0,5—0,6 г соли Мора, 50—60 мг NaCl и 22 лм концентрированной серной кислоты (уд. вес 1,84) растворяют в 1 л дистиллированной воды. Применяемые реактивы должны быть марки «ч.д.а.» или «х.ч.». При проведении особо точных измерений необходимо использовать дважды перегнанную воду и серную кислоту, обработанную сначала 5%-ным раствором перекиси водорода (1 —2 капли на 22 л/л), а затем 0,1 .V раствором КМцО4 до появления слабой розовой окраски [111].
Для рентгеновских лучей со средней эффективной энергией менее 0,25 Мэв становится важным эффект фотоэлектрического поглощения излучения средой, и поэтому 0,4 М HgSO4 поглощает энергию таких лучей в большей степени, чем вода. Например, для рентгеновских лучей с энергией 0,025 Мэв это возрастание поглощения составляет 14%. В таких случаях целесообразно использовать 0,05 М раствор серной кислоты. При этом следует учитывать, что в такой среде G(Fe3+) несколько ниже, чем в 0,4 М H2SO4 (см. стр. 141).
При проведении дозиметрии ферросульфатным методом необходимо хотя бы приблизительно оценить величину экспозиции, выше которой нельзя проводить облучение. В противном случае вследствие падения величины G(Fe3+), связанного с израсходованием кислорода, присутствовавшего в растворе, результаты измерения дозы будут неправильными. Если используется спектрофотометрический метод определения Fe3+ (при Атах = 304-н 305 нм), то экспозиции следует выбирать такими, чтобы разность оптической плотности облученного и необлученного растворов не превышала 1,5. В случае других методов определения Fe,+ время экспозиции всегда можно найти, зная, что уменьшение G(Fe3+) вследствие расхода кислорода становится заметным начиная с концентрации Fe3+, равной —(6,5—7)-10-4 М.
Когда толщина слоя облучаемой системы больше величины слоя полного поглощения данного вида излучения (особенно часто с этим приходится сталкиваться в случае электронных пучков),
дозиметрический раствор во время облучения необходимо перемешивать. Для этой цели особенно удобны магнитные мешалки Без перемешивания из-за локального расхода кислорода в облучаемой зоне раствора излом на кривой зависимости концентрации Fe3+ от дозы произойдет раньше, чем израсходуется весь кислород, присутствовавший в растворе. Если по какой-либо причине перемешивание раствора невозможно осуществить, то необходимо построить кривую зависимости количества возникшего Fe3+ от времени облучения для данной ячейки и измерение дозы производить, используя линейный участок этой кривой.
Как уже отмечалось выше, eFe=+ в 0,4 М H„SO4 при 304—305 нм зависит от температуры. Поэтому для получения точных данных кюветное отделение спектрофотометра или кювету необходимо термостатировать либо во всяком случае отмечать температуру образца при измерении оптической плотности (с последующим внесением соответствующей поправки в расчеты дозы).
Если определение концентрации ионов Fe3+ образовавшихся при облучении, проводится спектрофотометрическим метолом, то величину дозы D в рад можно рассчитать по формуле
7V (Е — 7?о) • 100
D = (W - 8гем) G(Fe3+) /PZ - (36>
где N — число Авогадро (6,02-1023 молек./лголь); Е — оптическая плотность облученного раствора; Ео— оптическая плотность необлученного раствора; ерез+ — молярный коэффициент экстинкции [в л!(моль • см) I ионов Fe3+ при выбранной длине волны; вре!+ — то же для ионов Fe2+; р — плотность раствора (р = 1,024 г!см? для 0,4 М H2SO4); I — длина поглощающего слоя кюветы (в сж); / — коэффициент перехода от электронвольт к радам (/ = 6,24-1013 эв!рад).
Когда измерения проводятся при Хтах = 304 нм и 25° С [cie+ = 2197 лЦмолъ-см) и t'Fe2+- = 1 л!(молъ-см)}, то при I = 1 см после подстановки численных значений в формулу (36) для тех видов излучения, для которых G(Fe3+) = 15,6 иона'100 эв, получаем:
D= 2,75-10*(Е — Еп) рад. (37)
Когда определение концентрации Fe3+ проводится при Хгаах = 224 нм и 25° С, соответствующая формула такова:
D = 1,33-1O1(E —£о) рад. (38)
Формула для расчета дозы в эв/мл в том случае, когда концентрация Fe3+ измеряется при 304 нм и 25° С, имеет следующий вид:
D = 1,76 • 10« (Е — Ее) эв/м I. (39)
Если же ионы Fe3t определяются по поглощению света при 224 нм, то
D = 0,85• 1018 (Е — Ее) эв/мл. (40)
Таблица 39
Коэффициенты перехода от измеренной при 304 нм разности Е — Ёо для ионов Fe3+ в 0,4 М JEhSOi к дозе в случае дозиметра Фрикке*
Температура, ° С eFe3+ ’ л/(люль сл0 (при Зи4 нм) [Fc3+], ЛГ10-* Доза
эв[мл- 10“ рад 104
20 2121 4,715 1,820 2,846
21 2136 4,682 1,808 2,826
22 2152 4,647 1,794 2,804
23 2167 4,615 1,781 2,785
24 2182 4,583 1,769 2,766
25 2197 4,552 1,757 2,747
26 2212 4,521 1,746 2,728
27 2227 4,490 1,733 2,710
28 2242 4,460 1,722 2,692
29 2258 4,429 1,710 2,673
30 2273 4,399 1,698 2,655
31 2288 4,371 1,687 2,638
32 2303 4,342 1,667 2,620
33 2318 4,314 1,665 2,603
34 2333 4,286 1,654 2,587
35 2349 4,257 1,643 2,569
* Чтобы получить значения [Fe3+] или дозы, необходимо приводимые в трех последних колонках цифры умножить на экспериментальную величину разности Е — Ео.
В случае других температур для перехода от величины разности Е—Ео при 304 нм к дозе удобно пользоваться данными табл. 39, заимствованной из обзора [511*.
Недавно [211] было найдено, что G(Fe3+) в ферросульфатной дозиметрической системе зависит от отношения внутренней поверхности сосуда к его объему. С учетом этого эффекта формула (37) приобретает следующий вид:
2,75.10»
D~~ Iк (A/v) (Е—^' (41>
где А — внутренняя поверхность сосуда (в см2)-, v — объем раствора (в см?) и к — констапта (для стеклянных сосудов она равна 1,78-Ю-2 см1 [211]).
Приведенные выше формулы правильны для расчета величины энергии, поглощенной в воде, разбавленных водных растворах и
* Вновь подчеркнем, что для получения наиболее точных результатов необходимо измерить е с помощью спектрофотометра, имеющегося в лаборатории. В расчетах дозы при этом следует пользоваться, очевидно, найденной величиной е.
других близких по атомному составу системах. Если необходимо знать величину дозы для систем, отличающихся от ферросульфат-ной по атомному составу, то при расчетах дозы, исходя из показаний дозиметра Фрикке, необходимо принимать во внимание соображения, высказанные в главе IV.
3. Модифицированные дозиметры Фрикке
Предлагались различные модификации дозиметра Фрикке. Ф. Хаасброэк [204] исследовал возможность использования дез-аэрированных растворов ферросульфата в дозиметрии. Уже отмечалось, что G(Fe3+) в таких растворах равен 8,2 иона/100 эв. Оказалось, что G(Fe3+) в 5-10-2 М растворе ферросульфата в 0,4 71/ H2SO4 не зависит от дозы до 10® рад (при этой дозе расходуется 25% ионов Fe2+). Если же использовать калибровочные кривые, то верхний предел измерения дозы этим раствором можно повысить до 4-10е рад. Согласно [212], с помощью дезаэрированного 0,5 1/ водного раствора Fe2+ в 0,4 .1/ HaSO4 можно определять дозы до 8-10® рад.
Г. Кубота [101] показал, что дезаэрированный раствор, содержащий 0,4 молъ/л H2SO4, 2-10-3 молъ/л FeSO4 и 10-3 молъ/л NaCl, можно использовать для дозиметрии у-излучения при высоких температурах (вплоть до 250° С). Растворы облучались в запаянных ампулах. Было найдено, что при 150—250 С G(Fe3+) = = 8,1 иопаИОО эв, т. е. в пределах ошибки опыта величина выхода соответствует значению его для комнатной температуры. По данным этой работы, описанный дозиметр пригоден в этих условиях для измерения доз в диапазоне от 104 до 6-104 рад.
В предыдущем разделе говорилось о том, что в присутствии органических веществ G(Fe3+) в дозиметре Фрикке существенно возрастает.
Например, по данным Э. Харта [105], при введении муравьиной кислоты в этот дозиметр G(Fe3+) достигает 225 ионовЙОО эв. Однако эта система непригодна для дозиметрических целей, поскольку G(Fe3+) сильно зависит от дозы. К тому же показания трудно воспроизводимы.
Гораздо лучшие результаты получаются, если к дозиметру Фрикке добавлять бензол или бензойную кислоту [115—120, 125, 213—215]. Л. И. Карташева и А. К. Пикаев [125] обнаружили, что с помощью серпокислых водных растворов Fe2+, насыщенных воздухом и бензолом, могут быть измерены дозы в диапазоне от ~ 100 до 2,5'103 рад. Выход Fe3+ в таких растворах зависит от концентрации бензола и ионов Fe2+, причем наибольшее значение выхода (66 ионов/100 эв) было получено для 2,5-10 4 71/ раствора Fe2+ в 0,4 М H2SO4, насыщенного бензолом (~ 1,5-10~2 молъ/л) и воздухом. В растворе этого состава G(Fe3+) сохраняется постоянным даже при израсходовании ~ 75% начального количества ионов Fe2+.
Концентрацию ионов Fe3+, образовавшихся при облучении, рекомендуется определять о-фенантролиновым методом (по убыли Fe2+), поскольку прямое спектрофотометрическое определение Fe3+ по поглощению света при 304 нм дает завышенные примерно на 10°6 результаты (в этой области спектра, кроме Fe3+, поглощает свет другое вещество, возникающее в результате радиолиза). В работе 1125] применялась методика, предложенная А. Алленом и У. Ротшильдом [75]. Обычно для анализа употреблялось 4 мл облученного раствора, к нему добавлялось 4 мл ацетатного буфера, 2 мл раствора фторида натрия и 2 мл о-фенантро линов ого реактива. Полученный раствор разбавлялся водой до 25 мл. Величина pH для конечного раствора составляла 4,2—4,4. Оптическая плотность раствора измерялась спектрофотометрически при длине волны 510 нм с использованием кювет с длиной поглощающего слоя от 1 до 5 см в зависимости от величины дозы.
По литературным данным [78 , 81, 136, 143, 146, 150, 169, 216—221], молярный коэффициент экстинкции о-фенантролино-вого комплекса Fe2+ при 510 нм колеблется в пределах от 10660 до 11800 л/(моль • см). Молярный коэффициент экстинкции комплекса о-фенантролина с Fe3+ составляет 3,3 [136] или 2,5% [216] от величины коэффициента для комплекса двухвалентного железа.
В работе [125] было найдено, что G(Fe3+) в рассматриваемой системе практически не зависит от мощности дозы у-излучения (исследовался диапазон от 0,64 до 192 рад/сек). величины pH от 0,4 до 1, температуры от 7 до 42" С, времени стояния раствора после облучения до 26 час. (после завершения пост-эффектов), введения ионов Fe3+ (до 10-3 /17), С1~ (до 10-3 М) и NO3 (до 10-3 М). G(Fe3+) одинаков как для аэрированных растворов, так и для растворов, насыщенных кислородом. Выход Fe3+ не зависит также от того, на какой воде, обычной дистиллированной или бидистил-лированной, очищенной радиолизом и фотолизом (см. стр. 162), приготовлен раствор.
Недостатком описываемой системы является наличие пост-эффектов. Поэтому измерение G(Fe3+) необходимо проводить через 1,0—1,5 часа после облучения. Выход Fe3+ зависит от концентрации Fe2+ и бензола. Увеличение концентрации Fe2+ от 2,5-10-4 до 5-10-4 М приводит к снижению G(Fe3+) на ~ 10%, а уменьшение концентрации С6Н6 от ~ 1,5-10-2 до 6-10-3Л7 — к падению G(Fe3+) на 5% [121, 122, 213]. Очевидно, при использовании данной системы для определения доз необходимо контролировать концентрацию раствора. При этом постоянство концентрации С6Н6 может быть достигнуто добавлением капли бензола к предварительно насыщенному им раствору. Эта операция не оказывает влияния па величину G(Fe3+). Выход Fe3+ уменьшается при высоких мощностях поглощенной дозы (~ 107—108 рад/сен), создаваемых импульсным электронным излучением [124, 2131. Следовательно, рассматриваемая система непригодна для дозиметрии этого вида излучения.
Г. Адамс и У. Болквелл [115, 1161 в 1957 г. обнаружили, что 0,5 М раствор Н^Од, содержащий примерно по 10-3 молъ/л ферросульфата и бензойной кислоты, очень устойчив при хранении. В оптическом спектре этого раствора имеются две полосы поглощения с максимумами при 232 и 271,5 нм. При облучении оптическая плотность раствора уменьшается. Снижение ее при длинах волн 260,5 нм, т. е. в минимуме, и 271,5 нм пропорционально поглощенной дозе. Согласно Г. Тиленсу [117], минимальная доза, которая может быть измерена с помощью этой системы, равна 40 рад.
Подробное исследование характеристик дозиметра Фрикке с добавками бензойной кислоты было проведено А. Фаучитано и др. [215]. Эти авторы для анализа Fe3+ использовали тиоцианатный метод. Они добавляли к 10 мл облученного раствора 1,2 мл 6 М раствора KCNS. Окраска смеси измерялась при длине волны 465 нм в кювете с толщиной поглощающего слоя 4 см. В качестве раствора сравнения применялся необлученный раствор, с которыми были проделаны те же операции. Было найдено, что G(Fe3+) зависит от концентраций Fe2+, бензойной кислоты и кислорода. Для этих растворов характерны пост-эффекты (в зависимости от концентрации они длятся до ~2 час.). Некоторые из полученных цитированными авторами результатов приведены в табл. 40. Из нее видно, что с помощью описанной системы можно определять очень малые дозы (до 5 рад}.
Таблица 40
Характеристики дозиметра Фрикке с добавками бензойной кислоты [215]
Состав раствора * G(Fe3+), ионы/100 эв Диапазон измеряемых доз, рад
10-4 М FeSO.i- 3-Ю-3 М С6НБСООН—0,4 М HaSO* 59 8—100
5-10-4 м FeS04—1,5 • 10-3 Л/ С6Н5СООН-0,1 М H2SO4 66,6 7—100
5• 10-4 М FeSO4 — 1,5 - IO-3 М СвНбСООН — — 0,4 Л/НгЭСЦ 57 8—100
IO-3 М FeSOi — 3-Ю-3 MCeHsCOOH—0,4 Л/ H2SO4 83,3 5—100
* В первых трех случаях раствор насыщен воздухом, в четвертом — кислородом.
Сернокислые водные растворы ферросульфата с добавками бензойной кислоты исследовались также в работах [146, 222]. Б. Гупта [1461 предложил использовать эти растворы в присутствии красителя ксиленолового оранжевого для измерения малых доз. Ионы Fe3+, как уже упоминалось, образуют с этим красителем комплекс, имеющий полосу оптического поглощения с Хтах = = 540 нм. Было найдено, что с помощью насыщенного воздухом раствора состава: 2-10-4 молъ/л Fe2+, 5-10—Б молъ/л Fe3+,
2*10~4молъ/л красителя, 5-10-3 молъ/л С6Н-,СООНи 2,5*10-г молъ/л H2SO4 — можно определять дозы ~ 1 рад. Для нахождения таких малых доз необходимо применять кюветы с толщиной поглощающего слоя 10 см. Выход Fe3+ для раствора указанного состава равен 65 ионам/100 эв.
Э. Бенн и др. [222] нашли, что низший предел определения дозы с раствором ферросульфата, бензоата натрия и о-фенантроли-на в водно-этанольной смеси (состава 1:1) равен 20 рад.
4. Цериевая дозиметрическая система
Цериевая дозиметрическая система представляет собой раствор Се4* в 0,4 М H2SO4, насыщенный воздухом. Уже давно известно [223—227]. что под действием ионизирующих излучений Се4* в сернокислом водном растворе восстанавливается до Се3+. Использование этой реакции в дозиметрических целях было предложено Т. Хардвиком [2261 и Дж. Вейсом [227] в 1952 г. К настоящему времени установлено, что с помощью данной системы можно измерять дозы до ~ 108 рад.
Механизм радиолитических превращений
В цериевой дозиметрической системе восстановление Се4+ происходит за счет атомов Н, радикалов НО2 и перекиси водорода [реакции (3), (11), (42) — (44)].
Се4* + Н -> Се3* -ИН+, (42)
Се4* + НО2 Се3* + Н+ + О2, (43)
Се4* -ь Н2О2 — Се3* 4- Н+ h НО2. (44)
Радикалы ОН обратно окисляют ионы Се3+ по реакции
Се3* + ОН Се4* + ОН-. (45)
Отметим, что как атом Н, так и радикал НО2 восстанавливают по одному" иону Се4*. Говоря по-иному, присутствие кислорода в растворе пе оказывает влияния па величину G(Ce3+). Исходя из указанной совокупности реакций, легко найти, что
С (Се3*) = G - 4- GH -|- 20 н о — Оон, (46)
Величина выхода трехвалентного церия
Величина б?(Се3+) измерялась многими авторами. В табл. 41 приведены значения С(Се3*) для разбавленных растворов Се4* (10-4—10~2 М) в 0,4 М H»SO4, определенные в различных работах.
Как видно из табл. 41, величина G(Ce3+) для разбавленных (10~4—10-2 М) растворов сульфата Се4* в 0,4 М H2SO4 для рентгеновских и у-лучей и быстрых электронов равна 2,3—2,5 иона/100 эв.
Таблица 41
Величины G(Ce3+) для разбавленных водных растворов сульфата Се4+ в 0,4 М H2SO4 в случае рентгеновского и у-излучений и быстрых электронов *!
G(Ce’+), ионы/100 эв Литература G(Ce3+), ионы/100 эв Литература G(Ce»+), ионы/100 эв Литература
2,33 *2 [226] 2,32 [66] 2,44 [237]
2,65 [227] 2,50 [231, 232] 2,47 [89]
2,52 [18] 2,40 [233] 2,49 [238]
2,39 [178] 2,36 [234] 2,48 [145]
2,45 [228, 229] 2,35 [235] 2,48 [239]
2,34 [230] 2,3—2,5 [236] 2,54 [240]
“ Здесь не указаны результаты ранних работ, в которых приведены завышенные значения С(Сез+).
** Значение G(Ce3+), приведенное в работе [226], пересчитано, исходя HgG(Fe’+) = = 15,6 иона 100 ав.
Отметим, что теоретическое значение G(Ce3+), рассчитанное на основе величин GH, Ge- , Goh и Gh,o,, составляет 2,34 иона/100 эв. aq
Имеются указания 1229], что при низких дозах начальные выходы Се3+ заметно превышают 2,3—2,5 иона/100 эв. По данным работы [229], это обусловлено тем, что в начальный момент облучения в растворе практически не имеется ионов Се3+. Указанное явление, если не приняты соответствующие меры предосторожности, приведет к неправильному измерению сравнительно низких доз.
Определение G(Ce3+) проводилось, как правило, сравнением химического превращения в цериевой системе с показаниями фер-росульфатного дозиметра. В работах [18, 232] G(Ce8+) измерялся калориметрическим методом.
Зависимость GfCe3+> от энергии излученггя и мощности дозы
Величина G(Ce3+) не зависит от энергии излучения в широком диапазоне. По данным работы [66], для у-излучепия * 60Со G(Ce3+) равен 2,33, а для электронов с энергией 2 Мэв — 2,32 иона/100 эв. С. Таймути и др. [232] определили выход Се8+ для у-излучения
60Со и быстрых электронов с энергиями 1, 8, 10, 18 и 24 Мэв. Было показано, что для этих энергий в пределах экспериментальной ошибки G(Ce3+) равен 2,50 иона/100 эв. Согласно Г. Джонсону и Дж. Вейсу [229], G(Ce3+) для рентгеновских лучей с максимальной энергией 200 кэв равен 3,15 иона/100 эв. Однако, по более поздним данным [145], С(Се3+) для рентгеновских лучей с примерно той же максимальной энергией (250 кэв) составляет 2,55 иона/
/100 эв. Как следует из работы [233], G(Ce3+) для рентгеновских лучей с энергией 10 и 8 кэв равен 2,7 и 2,9 нона /100 эв соответственно. В случае |3-излучения трития G(Ce3+) = 2,84 иона/100 эв [2411.
Таким образом, можно заключить, что G(Ce3+) не зависит от энергии рентгеновских и у-лучей и быстрых электронов, по крайней мере, в диапазоне от 0,25 до 24 Мэв.
Величина G(Ce3+) не изменяется при увеличении мощности дозы примерно до 108 рад/сек [235, 236, 242]. При более высоких мощностях дозы выход Се3+ возрастает. Соответствующие данные обсуждаются в главе IX.
Влияние концентрации кислорода, серной кислоты и ионов Се4+ на величину Gf'Ce3*/
По данным Т. Хардвика [226], G(Ce3+) одинаков как для деза-эрированпых растворов, так и для растворов, насыщенных воздухом.
Т. Хардвик [226] и Дж. Бойль [89] нашли, что G(Ce3+) не зависит от концентрации серной кислоты в диапазоне от 0,4 до 1 М. В более концентрированных растворах выход Се3+ уменьшается [89, 236]. Например, в 4 М H2SO4 G(Ce3+) равен 1,7 [891 или 1,55 иона/100 эв [236]. Наоборот, при понижении концентрации кислоты G(Ce3+) несколько увеличивается. По данным [89, 236], в 0,05 М II2SO4 G(Ce3+) = 2,55 ч- 2,6 иона/100 эв.
G(Ce3+) не зависит от концентрации ионов Се4+ в диапазоне ~ 10~Б — 5-10-2 М [89, 177, 226—228]. При концентрациях выше 5-10-2 М G(Ce3+) увеличивается. Дж. Харлан и Э. Харт [231] исследовали действие у-лучей 60Со на 0,39 М раствор Ce(HSO4)4 в 0,4 М H2SO4. Для этого раствора концентрация образующегося Се3+ не пропорциональна дозе. По мере роста дозы G(Ce3+) в этом растворе уменьшается от 3,1 до 1,7 иона/100 эв. Повышенное значение G(Ce3+) при сравнительно небольших дозах объясняется здесь тем, что вследствие высокой концентрации попов Се4+ некоторая часть гидратированных электронов или атомов Н, обычно рекомбинирующих в треках, вовлекается в реакцию восстановления четырехвалентного церия. Уменьшение G(Ce3+) при дальнейшем облучении обусловлено постепенным израсходованием ионов Се4+, а также накоплением в растворе ионов Се3+, которые акцептируют все большее количество радикалов ОН в треках и тем самым понижают Gh2o2-
Таким образом, G(Ce3+) не зависит от концентрации ионов Се4+ в весьма широких пределах. Это обстоятельство благоприятствует возможности измерения очень высоких доз.
Влияние темпериту ры на величину G(C&3+)
Согласно Т. Хардвику [226], G(Ce3+) постепенно уменьшается с ростом температуры (примерно на 10% при увеличении температуры от 6 до 73° С). X. Хотта и К. Шимада [243] исследовали зависимость G(Ce3+) от температуры в интервале 20—90° С. Подобно Т. Хардвику, они пашли, что G(Ce3+) уменьшается при возрастании температуры. С. Таймути и др. [232] также изучали температурную зависимость G(Ce3+). Они обнаружили, что G(Ge3+) практически не зависит от температуры ниже 35° С. Однако при более высоких температурах G(Ce3+) заметно уменьшается, достигая 2,23 иона/100 эв при 61° С.
Стабильность раствора при хранении
С. Никитич и Дж. Райт [244] обнаружили, что разбавленные водные растворы сульфата окиси церия весьма неустойчивы к действию света. Поэтому рекомендуется приготовлять 0,1 1/ раствор сульфата Се4+ в 0,4 М H2SO4. Этот раствор при хранении в темноте устойчив в течение многих месяцев. Непосредственно перед проведением дозиметрических опытов зтот раствор разбавляется 0,4 М серной кислотой до необходимой концентрации. Разбавленные растворы можно хранить в темноте лишь в течение нескольких часов. Позже наблюдения, сделанные С. Никшичем и Дж. Райтом, были подтверждены в работе [232].
Стабильность цериевой дозиметрической системы при повышенных температурах была исследована Г. Кубота [101]. Этот автор обнаружил, что при нагревании раствора в запаянной ампуле до 150° С оптическая плотпость его несколько уменьшается. Увеличение температуры до 200“ С и выше приводит к появлению желтого аморфного осадка [по-видимому, гидроокиси Се(III)]. При повышении концентрации серной кислоты от 0,4 до 1,5 М осадок не выпадает; однако оптическая плотность раствора все же заметно уменьшается.
Влияние примесей и чистоты воды и реактивов на величину G(Cc3*)
Еще в 1937 г. Г. Кларк и У. Коэ [223] наблюдали существенное возрастание скорости восстановления Се4+ под действием рентгеновских лучей в присутствии небольших количеств органических соединений. Согласно М. Лефору [233], добавка 10-5 молъ/л бензола к раствору сульфата Се4+ приводит к увеличению G(Ce3+) от 2,4 до 8,2 иона/100 эв. По данным Т. Сворского [245, 246], такое же возрастание G(Ce3+) происходит в присутствии 10-2 молъ/л муравьиной кислоты. С. Таймути и др. [232] отметили увеличение G(Ce3+) от 2,5 до 3,55 иона/100 эв при добавлении 2-10“5% муравьиной кислоты.
Возрастание G(Ce3+) до 7,92 иона/100 эв наблюдается в присутствии ионов Т1+ [178]. Введение в раствор сернокислого окисного церия ионов NO3 приводит к значительному увеличению 6(Се3+) [247—251], причем этот эффект тем больше, чем выше концентрация нитрат-ионов. По данным М. Лефора [233], в присутствии бромид-ионов 6(Се3+) уменьшается. Для раствора, содержащего 0,1 молъ/л Вт- G(Ce3+) составляет 1,5 иона 100 эв. Имеются указания [229], что G(Ce3+) в хлорнокислых растворах несколько выше, чем в сернокислых.
На показания цериевой дозиметрической системы существенное влияние оказывает чистота воды и используемых реактивов. Растворы, приготовленные на обычной дистиллированной воде, дают неправильные результаты. Поэтому для приготовления раствора Се4+ обычно рекомендуется использовать трижды перегнанную воду с применением специальных методов ее очистки. Эти методы описаны в следующем разделе.
Чистота используемых реактивов также влияет на показания цериевого дозиметра. С. Таймути и др. [232] исследовали пять препаратов, поставляемых различными фирмами. Оказалось, что лишь два из них дают удовлетворительную воспроизводимость результатов и устойчивость растворов при хранении. Очистку продажных препаратов можно осуществить перекристаллизацией из трижды перегнанной воды, к которой добавлена серная кислота (примерно 5%).
Согласно С. Таймути и др. [232], сосуды для цериевого дозиметрического раствора должны быть изготовлены из стекла (желательно из стекла «пирекс»). При хранении раствор должен соприкасаться лишь со стеклянными поверхностями. Перед использованием сосуды для облучения тщательно очищаются. Для этого они промываются дистиллированной водой, горячей концентрированной азотной кислотой, а затем несколько раз трижды перегнанной водой. Часто сосуды обрабатывают паром. В некоторых случаях (особенно при использовании новых сосудов) перед проведением опытов их наполняют тщательно очищенной водой и облучают, чтобы разрушить органические примеси, адсорбированные стеклом. Обычно сосуды до облучения хранят заполненными цериевым дозиметрическим раствором.
Н. Берр и Р. Шулер [66] отметили, что в случае у-излучения 60Со трудно получить воспроизводимые результаты при работе с растворами Се4 + даже при соблюдении всех мер предосторожности, описанных выше. Они нашли, что данные хорошо воспроизводятся, если облучение раствора осуществляется в больших ячейках (объем около 100 мл) и к раствору перед облучением добавлено небольшое количество ионов Се3+ (~ 10-4 молъ/л).
Следует подчеркнуть, что разбавленные растворы Се4 + более чувствительны к органическим примесям, чем концентрированные.
Ф. Мунош и М. Миро [2381 с целью подавления влияния органических примесей на показания цериевого дозиметра предложили использовать добавки сульфата окиси меди. По их данным, эти добавки в случае 10~3 М и более концентрированных растворов оказывают такое же действие, как и добавки ионов С1_ для дозиметра Фрикке. Недавно это наблюдение подтвердили Д. Бонгир-вар и Дж. Вейс [252]. Опи нашли, что введение ионов Сп2+ в цериевую дозиметрическую систему существенно повышает ее стабильность. Например, растворы, содержащие 2,5-IO-2 моль/л Се4+ и 5-10-2 молъ/л Си2+ или 5-Ю-2 молъ/л Се4+ и 0,1 молъ/л Си2+, дают одинаковые результаты как сразу же после приготовления, так и через 3 месяца хранения. Воспроизводимость показаний при этом почти такая же, как в случае дозиметра Фрикке. Раствор первого состава пригоден для измерения доз в диапазоне 1—8 Мрад, а раствор второго состава — в интервале 1—15 Мрад. Выход Се3+ в присутствии иопов Сп2+ такой же, как и в их отсутствие.
В сернокислых растворах Се4+, содержащих Сн2+, могут про-
текать реакции (3), (И), (42) — (45) и (47) — (51).
Си2+ + ОН —» Cu3+ + ОН-, (47)
Сезт + Си3+ -» Сс4+ + Си2+, (48
Cu2++ Н -» Си+ + Н+Д (49)
Си2++ НОа -» Си+ + Н+ + О2, (50)
Се4+ + Си1 -» Се3+ + Си2+. (51)
Отметим, что образование Сп3+ в результате реакции между Си2+ и ОН показано методом импульсного радиолиза [253,2541. Из совокупности приведенных реакций следует, что G(Ce3+) в присутствии ионов Сп2+ выражается уравнением (46), т. е. тем же уравнением, как и в отсутствие Си2+.
Р. Метьюс [255] обнаружил, что добавка ионов Се3+ в цериевую систему существенно улучшает ее дозиметрические свойства: она становится менее восприимчивой к примесям и более стабильной к действию света, уменьшается зависимость G(Ce3+) от температуры. По его данным, для приготовления растворов можно использовать обычную воду, если концентрация введенных ионов Се3 + превышает 3-10-3 М. Однако при этом необходимо иметь в виду, что G(Ce3+) при высоких концентрациях Се3+ уменьшается. Например, для у-радиолиза G(Ce3+) составляет 2,36 иона/100 эв, если концентрация Се3+ равна 3,14-103 М, и 2,11 иона/100 эв при содержании Сс3+ в растворе 7,83-Ю-2 молъ/л.
Способы очистки воды
В радиационной химии водных систем обычно используется тщательно очищенная вода. Ниже описываются наиболее распространенные способы, с помощью которых получают воду, достаточно чистую для приготовления растворов (в том числе и цериевого дозиметра).
В Аргоннской национальной лаборатории США применяется следующий способ [51]. Дистиллированная вода сначала перегоняется из раствора, содержащего 1 г NaOH и 1 г КМпО4 в 1 л. Водяной пар при этом пропускается через кварцевую трубку, нагретую до 700° С. Пар конденсируется, и вода вновь перегоняется (на этот раз из раствора, в 1 л которого содержится 1 г К2Сг2О7 и 1 г H2SO4). Пар опять пропускается через кварцевую трубку при температуре 700° С, затем он конденсируется, и вода хранится в кварцевой колбе. Во время перегонки через указанные растворы непрерывно барботируется очищенный кислород (для предохранения воды от «переброса» и окисления летучих органических веществ в водяном паре при прохождении его через нагретые кварцевые трубки). Па всех стадиях очистки не допускается применение каких-либо резиновых или пластмассовых пробок, соединений и т. п.
Дальнейшая очистка воды осуществляется облучением ее. В работе [256] с этой целью радиолизу подвергалась дезаэрирован-пая вода; после облучения она вновь перегонялась. В Брукхей-венской национальной лаборатории США [257] применяется следующая методика. Трижды перегнанная вода, насыщенная воздухом, облучается в течение нескольких часов на источнике у-из-лучения 60Со до тех пор, пока концентрация Н2О2 не достигнет ~ 10“4 М. Образовавшаяся перекись водорода затем разлагается воздействием ультрафиолетового света. Согласно [258], у-облуче-ние воды в присутствии водорода снижает концентрацию О2 до величины менее 10-7 М.
Опыт лаборатории, руководимой автором этой книги, показывает, что для многих целей можно ограничиться трехкратной перегонкой воды с последующим ее радиолизом и фотолизом. Стадию пропускания пара через нагретые кварцевые трубки можно исключить.
Как правило, степень чистоты воды проверяют, приготовив на ее основе ферросульфатный дозиметрический раствор. Если концентрация образовавшихся ионов Fe3+ пропорциональна дозе и скорость окисления идентична в присутствии и в отсутствие 10~3 молъ1л NaCl, то эта вода является достаточно чистой. Низкая электропроводность воды пе может служить критерием ее чистоты. Это лишь свидетельствует о том, что в ней сравнительно мало ионов. Вода может иметь низкую электропроводность и при большом содержании органических веществ (например, углеводородов), пе образующих ионов при растворении.
Методы определения
концентрации трехвалентного церия
При дозах ~ 10° рад облученные растворы можно анализировать титрованием раствором ферросульфата с использованием о-фенантролинового комплекса Fe2+ в качестве индикатора [231].
Г. Фрикке и Э. Харт [51] отмечают, что в этом методе ионы Fe2+ окисляются не только нонами Се4+, по также надсерными кислотами. Поэтому титрование не дает истинное значение концентрации Се3+. Однако выход надкислот низок, и такое небольшое различие в величине найденной концентрации Се3+ не оказывает влияния на полезность рассматриваемого дозиметра, поскольку его можно прокалибровать с учетом суммарного химического превращения. Б. Уайттекер [234] концентрацию Се3+ находил потенциометрическим титрованием раствора сульфата Се4+ (4-10-3 М) до и после облучения раствором соли Мора в 0,4 М HaSO4.
Рис. 64. Оптические спектры поглощения ионов Се4+ (1) и Се3+ (2) в 0,4 М растворе H2SO4 (относительно 0,4 М H2SO4)
S — оптическая плотность
Наиболее удобно концентрацию Се3+, образовавшегося при радиолизе, определять прямым спектрофотометрическим измерением количества ионов Се4+ в исходном и облученном растворе по поглощению света в ультрафиолетовой области спектра. Этот метод был предложен в 1952 г. Т. Хардвиком [226]. Оптическую плотность растворов измеряют при длине волны 320 нм, что соответствует максимуму поглощения для ионов Се4+ (см. рис. 64). Молярный коэффициент экстинкции ионов Се4+ в 0,4 М HaS04 при 320 нм определялся в работах [18, 68, 89, 153, 154, 167, 226, 229, 232, 259]. Значения коэффициента, приводимые в этих работах, колеблются в пределах от 5550 до 6100 л/(моль-см). Г. Фрикке и Э. Харт [51] рекомендуют использовать значение 5610 л/(молъ• см) (при 25° С).
Величина есен- не зависит от температуры в области 15—25° С [229]. В 0,05 М HaSO4 еСе1- ниже, а в 4 М HaSO4 выше, чем в 0,4 М HaSO4 [153, 229, 246]. По данным Т. Сворского [246], значения все«+ при 320 нм в 0,05 и 4 Л/ H2SO4 равны соответственно 5245 и 6650 л/(молъ-см). Трехвалентный церий не мешает определениям при этой длине волны. Согласно К. Хендерсону и Н. Миллеру [153], все»+ при 320 нм в 0,4 М HaSO4 составляет 2,7 л/(молъ-• см).
Оптическая плотность ионов Се4+ в 0,4 М H2SO4 при 320 нм составляет 1,12 (кювета с толщиной поглощающего слоя1сл<), когда концентрация сульфата окиси церия равна 2-10-4 М. Очевидно, если для дозиметрии используются более концентрпрован-
ные растворы, то их перед анализами следует разбавлять 0,4 М серной кислотой. С. Таймути и др. [232] показали, что в этом случае удовлетворительные результаты могут быть получены при 200-кратном разбавлении, если эта операция проводится очень тщательно и если колбы и пипетки предварительно прокалиброваны. Таким образом, спектрофотометрический метод определения концентрации Се3+ возможно использовать для растворов с начальной концентрацией ионов Се4+ до (3 н- 4) -10-2 М.
Н. Фригерио и В. Генри [145] предложили использовать для определения Се3+ флуоресцентный метод. По их наблюдениям, ионы Се4+ количественно тушат флуоресценцию флуоресцеина в водном растворе. Применяя этот метод, можно измерять дозы в диапазоне 50—5-103 рад.
Границы применимости цериевого дозиметра
Если применяется спектрофотометрический метод определения концентрации Се3+ (при использовании кюветы с толщиной поглощающего слоя 1 см), то нижний предел измерений с помощью цериевого дозиметра составляет ~ 104 рад. Флуоресцентный метод анализа Се3+, как отмечалось выше, позволяет расширить этот предел до 50 рад.
Верхний предел измерений дозы зависит от концентрации ионов Се4+. Соответствующие данные приводятся ниже:
[Се‘+], М Диапазон измеряемых доз, Мрад [Се*+], м Диапазон измеряемых доз, Мрад
2-10-* 0,01— 0,1 2-10-2 1,2—6,0
ю-3 0,1-0,3 5-Ю-2 6,0—15,0
4-Ю-3 0,3—1,2 0,39 До ~102
Выше уже говорилось, что G(Ce3+) равен 2,3—2,5 независимо от концентрации Се4+ в 0,4 М H2SO4 вплоть до ~ 5-10~2 М. При больших концентрациях G(Ce3+) возрастает (если дозы не слишком высокие). Дж. Харт и Э. Харлан [231] использовали 0,39 М раствор Ce(HSO4)4 для определения больших доз. Для этого раствора почти линейная зависимость концентрации Се3+ от дозы сохраняется до 5-107 рад. При более высоких дозах G(Ce3+) уменьшается. Было обнаружено следующее эмпирическое соотношение между G(Ce3+) и концентрацией образовавшихся ионов Се3+ (в молъ/л):
С (Се3+) = 3,1 (1 — 1,16 [Сез+]). (52)
Чтобы найти дозу (в рад), можно использовать формулу [231]
D = — 2.32 • 108 lg (1 —1,16 [Сез+]). (53)
Максимальная доза, которую можно измерить с помощью 0,39 М раствора Ce(HSO4)4 в 0,4 М H2SO4, равна ~ 108 рад [231].
Подчеркнем, что концентрация насыщенного при комнатной температуре раствора Ce(HSO4)4 в 0,4 М HaSO4 равна 0,65 М. Однако, как отмечают Г. Фрикке и Э. Харт 151], если этот раствор облучать, то при высоких дозах из него выпадает осадок. Поэтому предельная концентрация Ce(HSO4)4, пригодная для дозиметрических целей, составляет 0,39 М.
Практические рекомендации
для проведения дозиметрии
с помощью цериевой системы
Для приготовления дозиметрического раствора обычно применяют сернокислый окисный церий или аммонийцерий (IV) сульфат *>. Если необходимо приготовить концентрированный раствор, то используют Ce(HSO4)4.
Применение растворов Се4+ различной концентрации позволяет постепенно изменять в широком диапазоне значение эффективного атомного номера дозиметрической системы. Соответствующие данные имеются, например, в книге [260]. Эта особенность рассматриваемого дозиметра весьма важна с точки зрения подбора условий радиационного подобия дозиметрической и исследуемой систем.
Формула для расчета величины дозы D (в эв!мл) из определенной разности оптической плотности раствора до и после облучения при длине волны 320 нм имеет следующий вид (для толщины поглощающего слоя 1 см) **:
D = 4,47 1018 (Е — Ео). (54)
Для расчета дозы (в рад) формула (54) трансформируется в уравнение
D = 7,16 • 101 (Е — Е0)/р, (55)
где р — плотность раствора.
Для разбавленных растворов Се4+ в 0,4 М H2SO4 р = = 1,024 г!см?. В случае концентрированных растворов Се4+ следует учитывать изменение их плотности, обусловленное присутствием значительных количеств соли Се4+.
5. Ферросульфатиая дозиметрическая система
с добавками ионов Си2+
В работах [261, 262] было предложено использовать ферросуль-фатный раствор с добавками ионов Сп2+ для измерения доз в диапазоне от 4-104 до 107 рад. Расширение диапазона определяемых
* Ионы аммония, как и в случае ферросульфатной системы, не оказывают влияния на показания цериевого uoniMetpa.
** Формула (54) приведена для vCe4-, = 5610 лЦмоль- см) иС(Се3+) = 2,4 иона/ /100 эв.
доз по сравнению со стандартным дозиметром Фрикке обусловлено тем, что здесь G(Fe3+) существенно ниже. Он равен 0,66 иона/100 эв, когда концентрация серной кислоты составляет 0,005 М [261, 262].
Величина G(Fe3-1) в данной дозиметрической системе определяется совокупностью реакций (3), (И), (14), (17), (49), (50), (56) и (57) [262].
Cu^+e^Cu-*-, (56)
Fe3* + Cu+ Fe2+ -J- Cu2+. (57)
Очевидно,
G (Fe3+) = Gon-J 2G -G - GH. (58)
Следует отметить, что в этой системе кислород в отличие от дозиметра Фрикке не расходуется при радиолизе.
Описываемая дозиметрическая система исследовалась рядом авторов [261—267]. Ниже кратко излагаются выводы цитированных работ.
Стандартный дозиметрический раствор имеет следующий состав: 10-3 М FeSO4, 10-2 М CuS04 и 5-10-3 М H2SO4. Показания его сравнительно малочувствительны к органическим примесям [51, 261]. Поэтому для его приготовления можно применять биди-стиллированную воду и реактивы обычной степени чистоты. Ионы Си2+ понижают термическую устойчивость ферросульфата. По данным В. П. Трусовой и др. [264], раствор стабилен только в течение двух часов после приготовления. Поэтому в опытах необходимо использовать свежеприготовленные растворы. Из работы [268] следует, что термическую стабильность можно существенно повысить, если раствор предварительно облучить до такой дозы, чтобы оптическая плотность ионов Fe3+ при 304 нм была равна 0,6 (при толщине поглощающего слоя 1 см).
Измерение концентрации ионов Fe3+, возникающих при облучении, рекомендуется производить спектрофотометрпчески по поглощению света при длине волны 304 нм. По последним данным [265], ерез+ при этой длине волны для раствора указанного выше состава равен 2167 л! (молъ-см). Зависимость экстинкции ионов Fe3+ от концентрации серной кислоты показана на рис. 65 [265].
Концентрация кислорода оказывает некоторое вл ие а величину G(Fe3+) (см. табл. 42). Варьирование концентрации кислоты па 50% приводит к изменению G(Fe3+) всего на 5% [263]. Однако, если концентрацию H2SO4 увеличить до 0,4 711, то, по данным Э. Харта [262], G (Fe3+) возрастает до 4,0 ионов,'100 эв.
Использование раствора указанного выше состава позволяет измерять дозы до — 106 рад. Путем увеличения концентрации ионов Fe,+ и Си2+, а также введения ионов F~ или SO4~ в раствор этот предел можно повысить до — 107 рад [262]. Однако при опре
делении сравнительно высоких доз следует учитывать то обстоятельство, что, по последним данным [265], G(Fe3+) несколько уменьшается, начиная с доз —4-105 рад (см. рис. 66). Мощность дозы у-излучения в0Со не оказывает влияния на величину G(Fe£+). Однако в случае импульсного электронного излучения G(Fe3+) увеличивается, начиная с мощностей дозы <— 108—109 рад/сек [265] (см. главу IX).
Согласно Э. Бьергбакке и К. Сегестеду [265], воспроизводимые результаты при использовании раствора состава 10~3 М FeSO4—
Рис. 65. Зависимость оптической плотности S ионов Fe3+ в максимуме полосы поглощения в сернокислых водных растворах ионов Сн2+ и Fe2 + от концентрации серной кислоты
Рис. 66. Зависимость концентрации ионов Fe3+, образующихся при у-радиолизе 0,005 М сернокислых водных растворов Fe2+ (10~3 моль!л) и Си2+ (10"2 моль/л), насыщенных воздухом (1) или кислородом (2), от дозы D
10-2 М CuSO4—5-10-3 М H2SO4 могут быть получены при соблюдении следующих условий:
1) раствор должен быть насыщен кислородом. Это достигается пропусканием его через раствор в течение 30 мин.;
2) раствор должен проверяться по дозиметру Фрикке. Это необходимо для того, чтобы узнать, правильно ли была проведена операция насыщения кислородом;
3) ежедневно должен готовиться новый раствор. Это удобнее делать добавлением CuS04 к раствору, содержащему 10 ~3 М FeSO4 и 5-10-3 М H2SO4.
Если эти условия будут соблюдены, то можно определять поглощенные дозы у-излучения в диапазоне 5-104 — 8-105 рад.
Дж. Халлидей и Р. Ярретт [267] нашли, что раствор состава 6-10-3 М FeSO4, 6-10-2 М CuSO4 и 5-10~3 М H2SO4 можно использовать для измерения доз у-излучения в интервале (2 -4- 7) • 10е рад.
Таблица 42
Влияние кислорода на величину G(Fe3+)’ в сернокислом растворе сульфатов Fe2+ и Си2+ для 7-лучей 60Со
Газ, насыщающий раствор G (Fe3+), ионы/100 эв Литература
Воздух 0,66 [261, 263]
Кислород 0,76 [265]
Аргон 0,63 [265]
Примечание. Состав раствора: 10~3 М FeSO4 — 10~® М CuSO4 —5-10-® М H2SO4
G(Fe3+) в этом растворе постоянен при изменении температуры от 0 до 35° С. Раствор стабилен в течение нескольких месяцев. После облучения оптическая плотность раствора изменяется на несколько процентов в неделю. Для измерения концентрации Fe3+ рекомендуется использовать спектрофотометрический метод. Для этого облученный раствор разбавляется в сотношении 1 : 8.
6. Водный раствор щавелевой кислоты
Для измерения доз в той области, в которой пригоден цериевый дозиметр, можно также использовать водные растворы щавелевой кислоты. Впервые применение этих растворов в дозиметрических целях было предложено И. Драганичем [269, 270] в 1955 г. Позже они исследовались во многих работах [264, 271—280].
Преимущества водного раствора щавелевой кислоты как дозиметрической системы перед раствором сульфата Се4+ таковы: 1) щавелевокислый раствор нечувствителен к примесям; 2) он очень стабилен как до облучения, так и после него; 3) его можно применять для дозиметрии реакторного излучения, поскольку его активация нейтронами ничтожна по сравнению с цериевой системой. Однако имеется ряд недостатков, которые ограничивают его применение. В их число входят: 1) относительная трудность определения количества химического превращения; 2) степень разложения Н2С2О4 не пропорциональна дозе и 3) выход разложения зависит от ряда параметров.
В присутствии кислорода щавелевая кислота разлагается под действием излучения с образованием СО2, Н2 и Н2О2 в качестве
Рис. 67. Зависимость Дс для 0,1 М раствора щавелевой кислоты от дозы у-из-лучепия в0Со
главных продуктов. Радиолиз ее в этих условиях подробно исследован И. Драганичем и сотр. [281]. В отсутствие воздуха механизм радиолитических превращений в нейтральных водных растворах Н2С2О4 весьма сложен. Кроме СО2, возникают формальдегид, глиоксаль и глиоксалевая кислота [280, 282]. Как отмечает И. Дра-ганич [283], при измерении высоких доз с использованием закрытых сосудов кислород быстро расходуется, и в этом случае превалируют реакции, протекающие в отсутствие кислорода. При облучении щавелевая кислота в водном растворе разлагается, и по степени ее разложения можно судить о величине дозы.
По данным И. Драганича [272], выход разложения щавелевой кислоты равен — 4,9, причем он не зависит от дозы вплоть до 30% разложения кислоты. В этих условиях выход постоянен в области начальных концентраций от 2,5-10-2до 0,6 М. При больших степенях разложения доза и концентрация распавшейся щавелевой кислоты связаны логарифмическим соотношением
lg D — a 1g (со — с) 4- Ъ, (59)
где D — доза (ъэв/мл); с0 и с— начальная концентрация кислоты и ее концентрация после облучения (в моле к./ /мл)’, а и Ъ — константы, зависящие от концентрации кислоты до и после облучения.
получены несколько иные результаты, что и при малых степенях разложения
кислоты G(—Н2С2О4) зависит от дозы. На рис. 67 в качестве примера показана зависимость Ас = с0 — с от поглощенной дозы у-лучей для 0,1 М раствора Н2С,О4, найденная в работе [279].
По данным И. Драганича [275], концентрация щавелевой кислоты в зависимости от времени облучения изменяется в соответствии с уравнением первого порядка
Однако позднее были Было найдено [264, 2791,
D - act) 1g — ,
(60)
где а — коэффициент пропорциональности (его размерность эв/молек.); с0 и с выражены в молек./лм, a D — в эе!мл.
Согласно И. Драганичу [275], для у-излучения 60Со коэффициент а в уравнении (60) равен 41,5 эв/молек., если начальная концентрация раствора варьируется в интервале от 5-10-2 до 0,6 М, а степень разложения изменяется в диапазоне между 15
и 60%, т. е. когда 0,07 1g (с0/с) < 0,40. Правильность уравнения (60) была подтверждена тщательными исследованиями Н. Хольма и К. Сегестеда [279]. Однако, по данным этих авторов, коэффициент пропорциональности получается неодинаковым при работе на различных установках у-излучения 60Со. По их мнению, это вызвано так называемым «эффектом продолжительности» облучения. В качестве одних из первичных продуктов радиолиза возникают глиоксаль и глиоксалевая кислота [280], которые вступают в некоторые медленные реакции конденсации. Если время «жизни» этих продуктов равно или больше времени облучения, то они в большей степени будут реагировать с первичными радикалами из воды, что приведет к более низкой величине выхода разложения Н2С2О4. По этой причине коэффициент а должен измеряться на конкретной установке у-излучения 60Со.
Предлагались следующие методы определения степени разложения щавелевой кислоты: 1) перманганатометрическое титрование [270]; 2) спектрофотометрический анализ с использованием бензидинового комплекса Сн2+ [271, 272, 284] и 3) титрование раствором NaOH [272, 279, 285]. Перманганатометрический метод, как выяснилось [264, 279], дает неправильные и плохо воспроизводимые результаты, поскольку, кроме Н2С2О4, перманганатом оттитровываются и некоторые продукты радиолиза. Второй метод гораздо лучше, так как продукты радиолиза не оказывают влияния на результаты анализа. В этом методе используется способность щавелевой кислоты образовывать комплексное соединение с бензидиновым комплексом Сн2+. Это комплексное соединение имеет резкий максимум при 246 нм. Молярный коэффициент экстинкции при Хгаах равен 2490 + 250 л/(моль-см) [272, 284]. Температурный коэффициент е составляет—0,7% на градус Цельсия. Закон Ламберта — Бера выполняется для концентраций комплексного соединения до ~2-10-4 М. Поэтому если в дозиметрии используются концентрированные растворы, то они перед анализом должны быть разбавлены в соответствующее число раз. Значение е несколько зависит от чистоты бензидина. По этой причине для каждой партии бензидина должны проводиться соответствующие калибровочные измерения. По данным Н. Хольма и К. Сегестеда [279], наиболее хорошие результаты получаются, если для анализов применять следующую методику:
1) Раствор А: растворяют 51,4 мг хлорида бензидина в 10 мл 30%-ной (по объему) уксусной кислоты и разбавляют дистиллированной водой до 250 мл.
2) Раствор Б: растворяют 159,7 мг ацетата окиси меди в 250 мл дистиллированной воды.
3) Перед анализом растворы АиБ смешиваются в соотношении 1 : 4 (по объему), и затем приливается исследуемый раствор.
Как следует из работы [279], хранение растворов АиБ отдельно в течение месяца не оказывает влияния на результаты анализов. Однако, если их смешать и затем хранить в течение этого
периода, то такая смесь становится неустойчивой к действию ультрафиолетового света и разлагается в спектрофотометре.
Титрование раствором NaOH дает воспроизводимые результаты, особенно в случае больших доз. Перед титрованием из облученного раствора должна быть удалена углекислота. М. Матсуи [285] достигал этого нагреванием раствора на водяной бане в течение 30 мин.
Водные растворы щавелевой кислоты можно использовать для измерения доз в области от 10е до—2 -108 рад. Верхний предел соответствует почти полному израсходованию щавелевой кислоты в насыщенном водном растворе ее. Ниже приводятся концентрации растворов щавелевой кислоты, которые следует применять для определения различных диапазонов доз [51]:
[ЩС.О*], м Диапазон измеряемых доз, Мрад [II2C2O4J, м Диапазон измеряемых доз, Мрад •
0,025 0,3—1,6 0,100 3,2—16
0,050 1,6—8 0,600 16—160
Для приготовления растворов можно брать щавелевую кислоту обычной степени чистоты и лабораторную дистиллированную воду [271]. Сосуды должны быть изготовлены из стекла или кварца. Их следует запаивать пли закрывать пришлифованными пробками так, чтобы объем газовой фазы над раствором был минимальным. Они должны быть достаточно прочными, чтобы выдержать давление СО2 и Н2, образующихся в процессе облучения. Согласно Р. Хаммелу [286], при применении сосудов из нержавеющей стали получаются невоспроизводимые результаты. Для получения воспроизводимых данных необходимо растворы помещать в такие сосуды залитыми в полиэтиленовые мешочки. Растворы в стеклянных или кварцевых сосудах устойчивы при обычном хранении в течение месяцев. Изменение температуры облучения в диапазоне 20 —80° С не оказывает заметного влияния на показания дозиметра [271]. Вопрос о возможности применения растворов щавелевой кислоты для дозиметрии импульсного излучения обсуждается в главе IX.
Дозиметрические характеристики водных растворов щавелевой кислоты изучались также А. В. Васильевым и сотр. [287]. По их данным, G(—Н2С2О4) зависит от степени а разложения щавелевой кислоты. Эта зависимость характеризуется формулой
G (— Н2С2О4) = Go (—{Н2С2О4) + кос, (61)
где G0(H2C2O4) — выход разложения при а = 0 (для у-излучения 6GCo он равен 4,45 молек./100 эв) и к — константа (в случае у-радиолиза она составляет —1,69).
7. Водный раствор бихромата калия
В работах [288—291] было рекомендовано использовать для целей дозиметрии сернокислые водные растворы К.аСг2О7. Согласно [291], эти растворы пригодны для измерения доз от 5-104 до 5-10й рад. Г. Де Баэр и др. [290] нашли, что G(—СгаО7”) для 1,5-Ю-3 и 2,5-10 4 М растворов бихромата в 0,5 М HaSO4 равен соответственно 0,390 и 0,408 иона 400 зв (у-излучение ЙОСо). Сходные данные были получены А. М. Кабакчн и сотр. [2911. Они определили, что G(—СгаО7~) в 0,4 М H2SO4 составляет 0,395 ±0,01 иона/100 эв.
Очевидно, восстановление ионов Cr(Vl) до Сг(111) происходит за счет атомов Н и перекиси водорода. Часть образовавшихся ионов Сг(Ш) окисляется радикалами ОН обратно в шестивалентный хром. Отсюда
G(-Cr2O?-) 4/е (G _ + GH ± 2СНЛ - С(Ш). (6?Л
eaq
Уменьшение концентрации Сг2О7” определяется спектрофотометрическим методом. В оптическом спектре поглощения этих ионов в 0,5 М HaSO4 имеются три максимума: при 260, 350 и 440 нм (рис. 68). Г. Де Баэр и др. [290] рекомендуют измерять оптическую плотность необлученного и облученного растворов Сг2О7~ при 350 или 440 нм (в зависимости от концентрации раствора). Молярные коэффициенты экстинкции при этих длинах волн, когда оптическая плотность раствора определяется против 0,5 М раствора HaSO4, равны соответственно 2442 и 385 л/(моль-см). Согласно А. М. Кабакчн и сотр. [291], в 0,4 М HaS04 коэффициенты экстинкции попов СгаО?“ равны 2640 (при Хтах = = 350 нм) и 410 л! (моль• см) (при ХЦ1ах _. 435 нм).
А. М. Кабакчн и сотр. [291] рекомендуют использовать 3,33-10-4 и 2,5-10”4 N растворы К2Сг2О7 в 0.4 М H2SO4. Для приготовления растворов необходимо применять бидистпллирован-ную воду; с растворами на обычной дистиллированной воде результаты трудно воспроизводятся. G(—Сг,О7 ) не зависит от мощности дозы в исследованном диапазоне (38 4- 1300 рад!сек), Одна-
ко он уменьшается при увеличении температуры раствора. При 80° С G(—Сг2О?‘) равен 0,23 иона/100 эв.
Выход разложения зависит от концентрации кислоты в растворе. По данным [290], он на 1,2% выше в 0,45 М HaSO4 и на 0,2% ниже в 0,55 М H2SO4, чем в 0,5 М H2SO4.
8. Водные растворы углеводов
С. В. Стародубцев и сотр. [292—295] предложили измерять дозы в диапазоне от 106 до 5-108 рад с помощью водных растворов глюкозы и сахарозы. При облучении эти углеводы разлагаются, в результате чего снижается оптическая активность растворов. Поэтому уменьшение угла вращения плоскости поляризации, что определяется с помощью поляриметра, может служить мерой поглощенной дозы. По данным цитированных авторов, водные растворы глюкозы очень стабильны при хранении. Растворы сахарозы менее устойчивы; к тому же для них характерен эффект последействия. Показания глюкозной дозиметрической системы не зависят от температуры облучения вплоть до 80° С.
Позднее пригодность водных растворов углеводов для дозиметрии ионизирующего излучения исследовалась во многих работах [287, 296—305]. Согласно [296, 297], величину дозы в случае растворов глюкозы можно также определять по увеличению вязкости или электропроводности и выделению газообразных продуктов при радиолизе. На этом основании были сконструированы циркуляционные устройства, позволяющие непрерывно контролировать дозу в интенсивных полях ионизирующего излучения (в диапазоне от 10® до 8-108 рад).
По данным А. Гласса [298], 10%-ный водный раствор мальтозы, насыщенный воздухом и содержащий 0,02% NH4OH, пригоден для измерения доз в диапазоне 2,5-107—108 рад.
Подробное исследование возможности использования растворов оптически активных веществ в дозиметрии выполнено О. Су-шным и др. [299]. В табл. 43 приведено определенное ими уменьшение угла вращения аБ78 плоскости поляризации при длине волны 578 нм в результате у-облучения для 2%-ных растворов ряда веществ.
Наиболее детально в работе [299] были исследованы водные растворы сахарозы. Было найдено, что использование их усложняется образованием кислоты, которая оказывает каталитическое действие на гидролиз сахарозы до глюкозы и фруктозы. Это, очевидно, в свою очередь влияет на изменение угла вращения плоскости поляризации. Применение фосфатного или бикарбопат-ного буферов (pH 7,5) позволяет поддерживать значение pH постоянным, и в этом случае наблюдается линейная зависимость изменения а от дозы.
Р. Рассел [300] изучил водные растворы глюкозы. По его данным, эти растворы могут использоваться для измерения доз, 174
Изменение угла вращения плоскости поляризации 2%-ных водных растворов ряда веществ в результате у-облучеиия
Растворенное вещество «87 Я
Для необлу-ченных растворов Для облученных растворов до доз
0,5 Мрад 4 Мрад 50 Мрад
Сахароза 4-1,400 4-1,380 4-1,230 4-0,170
Лактоза 4-1,245 4-1,320 4-1,090 4-0,330
Фруктоза —1,790 —1,810 -1,580 —0,495
tZ-Маннптол 4-0,010 4-0,010 4-0,010 —1,150
l-Аскорбиповая кислота 4-0,475 4-0,475 4-0,500 4-0,330
Z-Винная кислота 4-0,335 -0,330 4-0,225 —0,110
d-Камфарсульфоновая кислота * 4-4,955 4-4,950 4-4,880 4-3,700
♦ В Опытах использовался 22%-ный раствор.
по крайней мере, до 2,5-108 рад. При этом растворы могут облучаться в стеклянных или металлических ячейках.
Р. Венкатарамани [301] исследовал изменение угла вращения плоскости поляризации для водных растворов сахарозы, глюкозы, фруктозы и арабинозы в результате облучения. Было найдено, что из их числа наиболее хорошими дозиметрическими характеристиками обладают водные растворы арабинозы, насыщенные воздухом. Для 3 и 5%-ных растворов этого углевода логарифм угла вращения линейно зависит от дозы у-пзлучения 60Со до 8,3-107 и 9,8-107 рад соответственно. На показания не влияет изменение мощности дозы в исследованном диапазоне (от 50 до 200 рад/сек).
Согласно [287], выход G разложения глюкозы в водном растворе под действием у-излучения 60Со зависит от начальной концентрации с0 глюкозы и не зависит от концентрации ее после облучения и поглощенной дозы. Зависимость G от с0 выражается формулой
G = Go -|- ксо, (63)
где Go — выход разложения в разбавленном растворе (при у-радиолизе он равен 1,897 молек./100 эв); к — константа (для у-излучения опа составляет 1,08), а концентрация с0 выражена в моль/л.
В. В. Генералова и сотр. [302] подробно исследовали возможность применения водного раствора глюкозы как стандартной дозиметрической системы. Ими было найдено, что оптимальная
концентрация глюкозы в растворе при определении доз в диапазоне от 1 до 400 Мрад является 20?6-ной. Растворы готовятся на дистиллированной воде из глюкозы марки «для инъекции». Начальная концентрация устанавливалась по углу вращения плоскости поляризации через сутки после приготовления раствора. Раствор разливался в стеклянные ампулы, последние запаивались в атмосфере воздуха, затем приготовленные образцы подвергались стерилизации при 80° С в течение 1 часа.
Как следует из работы (3021, зависимость изменения оптической активности растворов глюкозы не является линейной функцией дозы D. Эта зависимость описывается эмпирической формулой
1 <Ро
D = yln^, (64)
где <р0 и <р — углы вращения плоскости поляризации соответственно необлученного и облученного растворов глюкозы; к — параметр, зависящий от концентрации глюкозы. В случае 20 %-кого раствора к для у-лучей 60Со равно 3,9-10-3 Мрад-1. Согласно 1304], относительная погрешность измерения дозы у-излучения этим раствором составляет 7°ь с доверительной вероятностью 0,95.
Е. П. Ковалева п др. [3051 предложили использовать количество водорода, выделяющегося при облучении 0,3, 1,2 и 2,5 М растворов глюкозы, в качестве меры дозы. Начальные значения С(Н2) для растворов этих концентраций равны соответственно 0,3, 1,5 и 2,0 молек./ЮО эв. Выходы Н2 постоянны при изменении дозы до ~500 Мрад, мощности дозы от 50 до 104 рад'сен, температуры от 20 до 70° С и ЛПЭ излучения от 0,02 до 1 эо/А. Водород определяется методом газовой хроматографии.
9. Водные растворы бензола или бензойной кислоты
Многими авторами [306—335] установлено, что при облучении водных растворов бензола, насыщенных воздухом, образуются фенол, водород, перекись водорода, муконовый или (1-оксимуко-новый альдегид. Ралпацпонпо-хпмпческую реакцию образования фенола в этой системе различные исследователи [306, 307, 309, 313, 314, 329—334, 336] предлагали применять для дозиметрии ионизирующих излучений. С этой целью рекомендовано использовать нейтральный, аэрированный и насыщенный (~2-10~2 М) водный раствор бензола.
Для раствора указанного выше состава выход фенола, по данным [319], равен 1,90—2,05 молек./100 эв (у-излучеппе 60Со). Выход практически пе зависит от pH раствора. В 0,4 М H2SO4 он составляет 2,0—2,2 молек./ЮО эв [319], а при pH 13 2,1 — 2,3 молек./ЮО эв. G(C6HbOII) не изменяется при уменьшении концентрации бензола до ~10~4 М.
В настоящее время общепринято, что фенол при радиолизе рассматриваемой системы образуется в результате следующих реакций [325, 3281:
(65)
(66)
(67)
G(CeHsOH) в отсутствие кислорода существенно ниже. Поэтому кривая зависимости концентрации образующегося фенола от дозы является прямой линией до -~6-104 рад. В этой точке на кривой наблюдается излом, вызванный израсходованием кислорода в растворе.
На показания дозиметра не оказывает влияния изменение температуры от 15 до 60° С. Растворы бензола характеризуются высокой стабильностью до облучения и после него. Органические примеси также не влияют сколь-либо заметно на ход радиолиза этих растворов. Для изготовления сосудов можно использовать различные материалы: стекло, кварц, полиэтилен, нержавеющую сталь, алюминий и др.
По данным работ [319, 325], G(CeH5OH) снижается при высоких мощностях дозы, начиная примерно с 108 рад!сек. Подробнее вопрос о величинах выхода фенола при таких мощностях дозы обсуждается в главе IX.
Для определения фенолав облученных растворах бензола предлагались разнообразные аналитические методики. Его можно определять с помощью реактива Фолина, представляющего собой раствор смеси вольфрамата натрия, ортофосфорной кислоты и МоО3 [306], который в присутствии фенольных соединений окрашивается. М. Керр [329] предложил спектрофотометрический метод. Фенольные соединения определяются по поглощению света при 273 нм. Интенсивность поглощения повышается, если к раствору добавить щелочь до pH 12 [314, 3291. Однако при этой длине волны свет поглощается и самим бензолом. Поэтому перед анализом его необходимо удалить (например, продуванием щелочного раствора азотом). По данным М. А. Проскурнипа и сотр. [332], фенол надежнее всего определять по окраске красителя, возникающего при сочетании фенола с диазотированным п-нптроанили-ном. Очень чувствительный способ определения фенола был описан Н. Кляйном [3311. Этот способ основан на измерении окраски 2,6-диброминдофенола, образующегося в результате реакции фе
нола с 2,6-дибром-К-хлорбензохинонимином. Если использовать этот метод, то можно находить очень низкие дозы (~ 10 рад).
М. Дей и Г. Штейн [306, 307] показали, что с дозиметрической точки зрения аналогично водным растворам бензола ведут себя и водные растворы бензойной кислоты и их солей. При радиолизе этих растворов возникают о-, м- и n-оксибензойные кислоты [337 — 345]. Кроме того, происходит декарбоксилирование бензойной кислоты.
У. Армстронг и др. [337, 340] применили весьма чувствительный спектрофотофлуорометрический метод определения салициловой кислоты, образующейся при радиолизе водных растворов бензоата кальция. В работе [3401 применялся аэрированный 10-3 М раствор бензоата кальция (pH ~ 6). Определение проводилось сравнением флуоресценции облученного раствора при 400 нм (возбуждение флуоресценции ультрафиолетовым светом с длиной волны 290 нм) с флуоресценцией стандартного раствора сали- • циловой кислоты. Интенсивность флуоресценции не зависит от величины pH при pH 6. Описанным методом можно измерять дозы в диапазоне от 16 до 5-103 рад.
Другой чувствительный метод дозиметрии с помощью бензоат-ного раствора был разработан А. Даунсом [339]. Он предложил измерять 14СО2, выделяющуюся при радиационно-химическом разложении С6Н;,14СООН. Этот метод позволяет определять дозы в диапазоне от 10 до 100 рад.
10. Водные растворы хлорированных углеводородов и смеси хлорированный углеводород — вода
При облучении хлорированных углеводородов (хлороформ, трихлорэтилен и др.) в индивидуальном состоянии или их водных растворов выделяется соляная кислота в количествах, пропорциональных дозе. Это интересное свойство указанных систем привлекло внимание многих ученых [346—365] с точки зрения использования его для дозиметрии.
Такой интерес объясняется тем, что НС1 возникает с большим выходом по цепному механизму, а индикаторы, применяющиеся для измерения изменений кислотности вблизи pH 7, характеризуются высокой чувствительностью. Поэтому рассматриваемые системы можно использовать для определения очень низких доз.
В чистых веществах могут быть достигнуты высокие значения G(HC1) (до 104 молек./100 эв). Поскольку с помощью индикаторов можно обнаружить очень малые концентрации кислоты (~10-87И), то, как отмечается в обзоре [51], с их помощью в принципе можно определять дозы менее 1 рад. Однако показания в этом случае трудновоспроизводпмы. Они, кроме того, зависят от мощности дозы и температуры облучения. Поэтому чистые хлорированные углеводороды не находят применения в дозиметрической практике. Гораздо лучшую воспроизводимость можно получить путем вве-
Характеристики некоторых дозиметрических систем на основе хлорированных углеводородов [51, 260]
Состав системы Диапазон измеряемых доз, рад
Насыщенный водный раствор трихлорэтилена (0,1%), содержащий 2,1-10-3% фенолового красного 20—4-102
Тот же раствор, содержащий 1,8- 10'3% хлорфенолового красного 10г—103
Тот же раствор, содержащий 0,15% цитратно-фосфат-ного буфера (pH 6) и 1,8-10-®% хлорфенолового красного 3-102—3-Ю3
Тот же раствор, содержащий 0,25% цптратно-фосфат-ного буфера (pH 6), 0,1 % тиомочевины и 1,8-10-3% хлорфенолового красного 2-Ю3—2-Ю4
Тот же раствор, содержащий 0,6% цптратно-фосфатно-го буфера (pH 6), 0,4% тиомочевппыи 1,8-10~3% хлорфенолового красного 1,2-10’—1,5-Ю5
Тот же раствор, содержащий 5% цптратно-фосфатного буфера (pH 6), 0,4% тиомочевппы и 1,8-10-3% хлорфенолового красного 5-10’—2-10е
0,8% водный раствор СНС13, содержащий 0,025% бромкрезолового пурпурного 102-5-103
Двухфазная смесь тетрахлорэтилена с водой (4:1 по объему), содержащая 0,1% этилового спирта, 0,2% тио-мочевины и 1,8-10-3% хлорфенолового красного 2—12
Та же смесь, содержащая 0,2% ионола *1, 0,2% тпо-мочевипы п 1,8-10_3% хлорфенолового красного 10-2-102
Та же смесь, содержащая 0,2% тиомочевины, 0,1% цптратно-фосфатного буфера (pH 6) и 1,8-10~3% хлорфеполрвого красного *- 50-5-102
Та же смесь, содержащая 0,2% ионола, 0,2% тиомочевины, 0,25% цитратно-фосфатного буфера (pH 6) и 1,8-10-3% хлорфенолового красного Ю2—103
Та же смесь, содержащая 0,2% ионола, 0,2% тпомоче-вины, 1,5% цитратно-фосфатного буфера (pH 6) и 1,8-10~3% хлорфенолового красного 5-102—6-103
Та же смесь, содержащая 0,2% ионола, 0,2% тиомо-чевппы, 4% цптратно-фосфатного буфера (pH 6) и 1,8-10—3% хлорфеполового красного 1,5-103—2,5-10’
Та же смесь, содержащая 0,5% ионола, 0,2% тпомо-чевипы, 10% цптратно-фосфатного буфера (pH 6) и 1,8-10-3% хлорфенолового красного 5-103—1,5.103
Та же смесь, содержащая 0,5% ионола, 0,2% tiiomi-чевины, 50% цптратно-фосфатного буфера (pH 6) и 1,8-10-3% хлорфенолового красного 5-10’—8-105
Та же смесь, содержащая 0,5% ионола, 0,2% тиомочевины, 100% цитратно-фосфатного буфера (pH 6) и 1,8-10-з% хлорфенолового красного *’ Ионол —это 2,6-ди-трет-бутил-тг-крезол. 8-10’—2-106
*2 Буфер находится в верхней водной фазе, а ионол — практически полностью в нижней тетрахлорэтиленовой фазе.
депия в раствор некоторых ингибиторов цепной реакции (резорцина, тиомочевины и т. и.). Дозиметрическая система при этом становится менее чувствительной к действию излучения, и удовлетворительная воспроизводимость достигается при определении доз ~Ю—20 рад. Такие системы находят применение как персональные дозиметры. В обзоре [511 отмечается, что две американские фирмы «Edgerton, Germenhausen and Grier, Inc.» и «General Dynamics» серийно выпускают персональные дозиметры на основе хлорированных углеводородов.
Рассматриваемые дозиметрические системы вследствие многочисленных трудностей, возникающих при работе с ними, не используются в радиационной химии. Поэтому здесь дается лишь краткая характеристика их.
В табл. 44 приведены наиболее характерные примеры, показывающие зависимость диапазона измеряемых доз от состава системы [51, 260]. Обычно применяются два типа систем: однофазные (растворы хлорированных углеводородов в воде или органическом растворителе) и двухфазные (хлорсодержащий углеводород, покрытый слоем воды). Чаще всего количество химического превращения определяется по изменению окраски кислотно-основного индикатора, введенного в систему. Можно использовать и метод электропроводности. В работе [358] было показано, что если к раствору хлороформа в воде добавить смесь KJ и KJO3, то в данной системе вследствие образования соляной кислоты происходит реакция:
10“ + 5J- + 6Н- -» ЗН2О + 3J2. (68)
Выделившийся иод легко измеряется обычными аналитическими методами (например, титрованием с раствором тиосульфата).
11. Водные растворы красителей и их лепкооснований
Многие красители в водных растворах имеют высокие значения коэффициентов экстинкции. Поэтому небольшое изменение в концентрации красителя приводит к заметному изменению оптической плотности таких растворов, что, очевидно, способствует их возможному использованию в целях дозиметрии. Это обстоятельство привлекло внимание многих исследователей.
Наиболее подробно изучались аэрированные водные растворы метиленового голубого [154, 307, 366—375]. При облучении такие растворы обесцвечиваются. Процесс происходит, в основном, за счет радикалов ОН, поскольку восстановленные формы красителя окисляются кислородом обратно в краситель. По данным М. А. Проскурнина и сотр. [372], начальный выход обесцвечивания равен 1,3 молек./ЮО эв. При сравнительно больших дозах выход составляет ~1 молек./ЮО эв.
Согласно Б. Лафуэнте и др. [373], на величину выхода не влияет изменение температуры облучения от 7 до 66° С. Зависимость количества превращения от дозы имеет экспоненциальный характер [374]. Степень разложения сильно зависит от pH. При высоких мощностях дозы выход снижается. Так, из работы [154] следует, что при мощности дозы ~1010 рад!сек выход разложения красителя в 5-Ю~6 М растворе примерно в 2—4 раза меньше, чем в аналогичном растворе при мощности дозы ~ 100 рад/сек. Варьируя концентрацию метиленового голубого от 5- Ю-® до 2- Ю~8ЛГ, можно с помощью этой системы измерять дозы в диапазоне от 103 до 5-106 рад. Изменение окраски определяется колориметрическим или спектрофотометрическим методами. При хранении в темноте растворы устойчивы в течение нескольких месяцев. Г. Фрикке и Э. Харт [51] считают, что аэрированные водные растворы метиленового голубого можно использовать в тех случаях, когда требуются не очень точные измерения.
Для дозиметрических целей предлагались водные растворы и других красителей: метилового оранжевого [376, 377], фенолового красного [376], индигокармина [378—381], хлорфенолового красного [382], эозина [383], резазурина [375], уранина S [384], кристаллического фиолетового [170], малахитового зеленого [385], метилового зеленого [385], бромкрезолового зеленого [386], тионина [387] и т. д. По данным А. М. Кабакчи и сотр. [376], с помощью растворов метилового оранжевого (выход обесцвечивания равен 1,27 молек./ЮО эв) можно находить дозы от 10—20 до 2-Ю4 рад-, применяя растворы фенолового красного (выход обесцвечивания составляет 0,12 молек./ЮО эв), можно определять дозы в диапазоне 4-Ю2—Юв рад.
У. Армстронг и Г. Грант [388, 389] исследовали возможность использования водных растворов лейкооснований триарилметановых красителей в дозиметрии. В этом случае о величине дозы судят по появлению окраски красителя. Проведенные исследования показали, что наиболее удобным для целей дозиметрии является раствор, содержащий Ю-4 молъ/л 4,4'-(5-хлор-2-тенилиден)-бггс-(П,П-диметиланилина), Ю-4 молъ!л соли Мора, 0,1 молъ/л NaCI и 7-Ю-3 молъ/л соляной кислоты. Концентрация образующегося красителя (выход равен 1,1 молек./ЮО эв) пропорциональна дозе до 2-Ю3 рад. Выход красителя не зависит от энергии излучения в пределах 0,16—2,0 Мэв и мощности дозы в диапазоне 0,067 —4 рад/сек. При более высоких мощностях дозы наблюдается послерадиационное обесцвечивание образовавшегося красителя. Ниже 30° С выход мало зависит от температуры. С ростом температуры выход увеличивается.
С помощью рассмотренной системы визуальным наблюдением окраски можно определить дозы порядка 250 рад. Меньшие дозы можно измерить спектрофотометрически по поглощению света при длине волны 633 нм. Нижний предел измерений составляет примерно 20 рад. Крупным недостатком системы является то, что
ее необходимо тщательно оберегать от контакта с воздухом. Даже при стоянии раствора при комнатной температуре в нем через две недели появляется окраска.
Недавно Б. Гупта [390] предложил применять щелочные водные растворы ксиленолового оранжевого для измерения больших доз. При облучении краситель обесцвечивается. Изменение окраски наблюдают при X = 580 нм и pH 10 в присутствии буферного раствора состава 0,025 молъ/л Na2CO3 и 0,025 молъ/л NaHCO3. В этих условиях е = 3,12-104 лЦмолъ-см). Диапазон определяемых доз зависит от концентрации красителя. Если концентрация равна 2-10—4 М, то этот диапазон составляет 1—50 крад, а при концентрации красителя выше 10-3 М он расширяется до 0,5 Мрад. Выходы обесцвечивания красителя для растворов этих концентраций равны соответственно 0,80 и 1,22 молек./100 эв. При введении тиомочевины в раствор выход обесцвечивания уменьшается. Вследствие этого можно измерять гораздо большие дозы. Например, щелочной Ю-3 М раствор красителя в присутствии 10-2 молъ/л тиомочевины пригоден для нахождения доз до 10 Мрад. Согласно [391], обесцвечивание ксиленового оранжевого происходит преимущественно в результате действия радикалов ОН.
У. Маклафлин и сотр. [392—395] описали дозиметры на основе растворов нитрилов триарилметановых красителей в различных растворителях. При облучении этих растворов возникает окраска, по интенсивности которой и судят о величине дозы. В качестве растворителей были использованы некоторые органические жидкости, а также водный раствор уксусной кислоты. Дозиметры на основе растворов в органических растворителях будут рассмотрены в главе VI. В этом разделе приводятся результаты исследования дозиметрических характеристик уксуснокислых растворов этих цианидов. Наиболее успешными были опыты с нитрилом парарозанилина (4,4',4"-триаминотрифенилацетонитрилом). Использовались 5-10-3 М растворы этого нитрила в 20—50%-ной уксусной кислоте. Для такого раствора, насыщенного воздухом, выход образования красителя равен 0,206 молек./100 эв и не зависит от дозы до 80 крад. Введение в раствор нитробензола уменьшает выход и тем самым расширяет диапазон измеряемых доз. Если концентрация этого вещества равна 5-10—2 М, то выход составляет 0,029 молек./100 эв, причем он не зависит от дозы до 400 крад. Концентрация образовавшегося красителя находится спектро-фотометрически (Хтах == 547 нм).
Красители, введенные в водные гели желатина или агар-агара, можно использовать для визуального наблюдения дозного поля и ориентировочного определения глубинной дозы [307, 370, 375, 396—3991. Гель готовится добавлением желатина или агар-агара (обычно 1%) к горячей воде.
12. Водные растворы нитратов
При радиолизе водных растворов нитратов образуется нитрит. Этот процесс нашел применение в дозиметрических целях.
В работах [400 , 401] рекомендовано использовать 1—3 М водные растворы KN03 пли NH4NO3 для определения доз в диапазоне 103—105 рад. Возникающий при облучении нитрит измеряется по окраске красителя (Хтах = 520 нм), появляющегося в кислой среде в присутствии NO2 в результате диазосочетания сульфаниловой кислоты и а-нафтиламина.
Ррс. 69. Зависимость количества разложившихся ионов NH* от дозы D (температура ~ 50 °C) при у-радиолизе 6 М раствора NH4NO3, содержащего 2,5 моль!л метилового спирта
А. К. Пикаев и Т. П. Жесткова [402, 403] предложили применять нагретые концентрированные водные растворы, содержащие метиловый или этиловый спирты, для измерения больших доз у-излучения. Нитрит аммония, образующийся при радиолизе, разлагается в водных растворах при температурах выше 33“ С:
К H4N Ог -> N2 + 2НгО. (69)
Тем самым при облучении нагретых растворов NH4NO3 исключается накопление ионов NO2, что приводит к линейной зависимости количества химического превращения от дозы в широком диапазоне доз. Введение спирта препятствует реакции радикалов ОН с питрит-ионами и обусловливает возрастание выхода NO2. Было найдено, что при 45—55° С G(—NH^) в 6 М растворе NH4NO3, содержащем 2,5 молъ/л СН3ОН, не зависит от дозы до ~6-108 рад (рис. 69). В этих условиях G(—NH4) равен 3,4 иона/100 эв. Убыль концентрации ионов NH4 легко определяется формальдегидным способом [404], оспованным на реакции
4NH+ + 6СН2О -» (СН2Ж + 4Н+ + 6Н3О (70)
Образовавшаяся кислота затем оттитровывается раствором КОН с фенолфталеином в качестве индикатора. Дозу с помощью этой системы можно находить и по выделению азота. Нижнее предельное значение дозы, которое может быть измерено с раствором указанного состава при использовании формальдегидного способа
определения расхода NH4, равно ~6-107 рад. При проведении дозиметрии необходимо учитывать то обстоятельство, что удельный вес раствора несколько уменьшается при облучении. Рассматриваемая система наиболее пригодна для дозиметрии в случае мощных источников у-излучения 60Со (с активностью в десятки тысяч грамм-зквивалентов радия и выше). Нагревание растворов здесь может быть осуществлено за счет тепла, выделяющегося при радиоактивном распаде 60Со. С этой целью временно выключают водяное охлаждение источника.
Для дозиметрии высокоинтенсивных потоков у-лучей 60Со рекомендован также •водный раствор AgNO3 [4=051.
13. Другие дозиметрические системы
на основе водных растворов
Предлагались многие другие дозиметрические системы на основе водных растворов. Однако сколь-либо широкого применения в радиационной химии эти системы не нашли. Поэтому мы ограничимся табл. 45, в которой приведены дозиметрические характеристики некоторых из них, и кратко рассмотрим свойства двух систем из их числа.
10-3 М раствор KJ в воде, насыщенный воздухом, был предложен в качестве дозиметрической системы Э. Хартом и П. Уолш [421]. Если в большинстве систем, описанных в настоящем разделе, доза определяется по количеству химического превращения растворенного вещества, то этот дозиметр основан на измерении газовыделения в результате радиационно-химического разложения воды. По данным работы [421], в стационарных условиях у-радиолиза О'(П2 + О2) = 0,575 молек./ЮО эв. Выход не зависит от температуры в диапазоне от 8 до 90r С. Для приготовления растворов необходимы тщательно очищенные вода и иодид калия. Очевидно, измерение скорости газовыделения позволяет определять мощность дозы. Описание установки, используемой с этой целью, дано в главе XI.
Мощность дозы можно определять также электрохимическим методом. Впервые он был использован с этой целью в работе [437]. В цитированной работе измерялась скорость радиолитического образования водорода в системе Pt/0,4 М раствор HaSO4-|-+ 3-10-®—2-10-2 N раствор щавелевой кислоты. Эта скорость находилась с помощью платинового электрода. Ток, проходящий через раствор, пропорционален этой скорости в том случае, если поверхность электрода велика по сравнению с объемом раствора. С помощью данной системы можно находить мощности дозы до 750 рад/сек (доза при этом не должна превышать ~5-107 рад).
Опыты по электрохимическому методу определения мощности дозы были продолжены А. Г. Васильевым и сотр. [440—444]. Эти авторы исследовали электрохимическое окисление продуктов радиолиза трех систем: растворов H2SO4, сернокислых растворов
Характеристики некоторых дозиметрических систем на основе водных растворов
Система X имическое превращение Диапазон оп-ределяеьшх доз, рад Литература
Водный раствор HCOONa Разложение формиата IO3 —8-Ю7 [406 — 4081
Раствор полиакриламида Изменение вязкости 102—10‘ [409 — 413]
Водно-спиртовый раствор молибдата аммония, содержащий соляную кислоту Образование синей окраски BP— 106 [414]
Водно-тетрагицрофурано-вый раствор молибдата аммония, содержащий соляную кислоту То же 105 — Ю7 [414]
Водный раствор хинина Разложение хинина 0,5—103 [415, 416]
Водный раствор хлораль гидрата Образование НС1 103 — 10* [356, 365, 417 — 419]
Водный гель состава 0,5—0,75% агар-агара и 0,05 молъ/л хлоральгид-рата Образование НС1 2-юз—2-105 [420]
10~3 М раствор KJ, насыщенный воздухом Газовыделение (На и О2) До нескольких М рад [421, 422]
0,1 М раствор KJ (в буферном растворе с pH-—5) Образование J2 Ю« _ 10б [423]
Вода, насыщенная воздухом Образование Н2О2 103 —5-103 [424]
Вода, насыщенная воздухом или азотом Образование NOa иХО<Г От 10е —107 до 1,2-10» [425]
Водный щелочной раствор В г О- Разложение ВгО~ До 108 [426]
Водный раствор HgCla и щавелевой кислоты (pH 5) Образование Hg2C12 240-3-103 [427]
Раствор Ti4+ в 0,4 М H2SO1 Восстановление Ti4+ 106 _ юн [428, 429]
Водный раствор терефталевой кислоты Образование окситере-фталевой кислоты 1 —103 [430[
1—2 М водный раствор аланина Образование NHs 106 _ 108 [431]
0,05 Л/ раствор какодилата натрия (pH 5,5) Разложение какодилата натрия 5-105 —5-106 [432]
Водно-этапольный раствор Fe3+, НС1, формиата натрия и фенантролина Восстановление Fe3+ 20-1,5-Ю3 [222]
Водный раствор 4-амино-1, 2, 4-триазола Изменение электропроводности 5-103—2-106 [433]
Т а б л и ц а 45 (окончание).
Система Химическое превращение Диапазон определяемых доз, рад Литература
0,015 М водный раствор пероксиламиндисульфоната калия Разложение растворенного вещества — [434)
Раствор Sn2+ в 6Л/ НС1 Образование Sn4+ Ют—7.1 о5 [435]
3 М водный раствор метилового спирта Образование этиленгликоля 3-105 — 10’ [436]
Pt/0,4 М H2SO4 4-3 • IO-3— 2-10-2 N щавелевой кислоты в воде Образование водорода (измеряется электрохимически) До 5-10’ [437]
Pt/0,4 М H2SO4 + IO-3 М FeSO4 в воде Образование Fe3+ (измеряется электрохимически) См. * [438]
Hg2Cl2/0,4 М ffcSO4 р + lO-ч — Ю-з м Fe2+ в воде То же До ~6-10‘ [196, 439]
Pt/0,25 М H2SO4 + 2,5-.10-2 м UO2SO4 в воде Окисление продуктов радиолиза (измеряется электрохимически) До 2-10’ [440 — 442]
Pt/0,1 м k2so4 + io-j— 5-10-2 М NaNOsBBOfle То же До ' ~108 [442 — 444]
Pt/водный раствор H2SO4, содержащий кислород * Измерение мощностей дозы » в диапазоне 10< — 10’ рад'час. До -108 [442]
UO2SO4 и растворов NaNO3 в присутствии K2SO4. Блок-схема соответствующей установки показана на рис. 70. Было найдено, что линейная зависимость между силой тока окисления продуктов
Рис. 70. Блок-схема электрохимического дозиметра
1 — вспомогательный электрод;
2 — измерительный электрод;
з — электрод сравнения;
4 — ячейка-датчик с раствором;
5 — источник постоянного тока;
6 — регулятор напряжения;
7 — самопишущий прибор силы тока;
8 — потенциометр
радиолиза и мощностью дозы у-излучения 60Со сохраняется в диапазоне 50—103 рад/сек (для первых двух растворов) и до 3-103 рад/сек (для 5• 10_3 71/ раствора NaNO3). Сила тока не зависит от дозы вплоть до десятков мегарад.
К химическим методам дозиметрии, основанным на использовании водных растворов, примыкает метод, состоящий в предварительном облучении твердых щелочногалоидных соединений (NaCl, КВг и т. п.) и последующем измерении люминесценции при растворении этих соединений в воде [445, 446]. Общеизвестно, что при радиолизе многих щелочногалоидных соединений образуются стабильные /’-центры. Электроны этих F-центров и являются источниками свечения при растворении облученных кристаллов. Люминесценция есть результат реакции между электронами и Х2 (X — атом галогена) [447] или деактивации возбужденных молекул воды, возникших при передаче молекулам воды энергии электронов [448, 449]. Согласно [446], в случае NaCl рассматриваемый метод позволяет определять дозы от 1 до 107 рад.
ЛИТЕРАТУРА
1. А. О. Allen. Radiation Res., Suppl., 4, 54 (1964).
2. W. G. Brown, E. J. Hart. Radiation Res., 51, 248 (1972).
3. E. J. Hart. Actions Chimiques et Biologiques des Radiations (Ed. M. Hais-sinsky), 10 Serie. Paris, 1966, p. 1.
4. M. Haissinsky. Actions Chimiques et Biologiques des Radiations. (Ed. M. Haissinsky), 11 Serie. Paris, 1967, p. 133.
5. А. А. Зансохова, T. П. Жесткова, А. К. Пикаев. Химия высоких энергий, 4, 454 (1970).
6. К. Sehested, Н. Corfitzen, Н. Fricke. J. Phys. Chem., 74, 211 (1970).
7. В. H. Bielski, A. О. Allen. Intern. J. Rad. Phys. Chem., 1, 153 (1968).
8. Э. Харт, M. Анбар. Гидратированный электрон. M., Атомиздат, 1973.
9. А. К. Пикаев. Сольватированный электрон в радиационной химии. М., «Наука», 1969.
10. А. О. Аллен. Радиационная химия воды и водных растворов. М., Атомиздат, 1963.
11. I. G. Draganic, Z. D. Draganic. Radiation Chemistry of Water and Aqueous Solutions. Academic Press, New York— London, 1971.
12. F. H. Krenz, H. A. Dewhurst. J. Chem. Phys., 17, 1337 (1949).
13. G. G. Jayson, B. J. Parsons, A. J. Swallow. Intern. J. Rad. Phys. Chem., 3, 345 (1971).
14. G.G. Jayson, B. J. Parsons, A. J. Swallow. J. C. S. Faraday I, 68, 2053 (1972).
15. G.G. Jayson, B. J. Parsons, A. J. Swallow. J. C. S. Faraday I, 69, 1079 (1973).
16. F. Haber, J. J. Weiss. Proc. Roy. Soc., A147, 332 (1934).
17. J. P. Beene. Radiation Res., 22, 14 (1964).
18. C. J. Hochanadel, J. A. Ghormley. J. Chem. Phys., 21, 880 (1953).
19. R. M. Lazo, II A. Dewhurst, M. Burton. J. Chem. Phys., 22, 1370 (1954).
20. N. W. Holm, A Brynfolffsson, J. E. Maul. Selected Topics in Radiation Dosimetry. Vienna, IAEA, 1961, p. 371.
21. J. P. Keene, J. Law. Phys. Med. Biol., 8, 83 (1963).
22. P. Milvy, S. Genna, N. Barr, J. S. Laughlin. Proc. II United Nations Intern. Conference on Peaceful Uses of Atomic Energy, vol. 21. Geneva, 1958, p. 142.
23. B. Radak. Nucleonics, 22, No 11, 52 (1964).
24. A. O. Fregene. Radiation Res., 31, 256 (1967).
25. C. Petterson, G. Hettinger. Acta Radiologica, 6, 160 (1967).
26. R. J. Shalek, W. К. Sinclair, J. C. Calkins. Radiation Res., 16, 344 (1962).
27. J. L. Haybittie, R. D. Saunders, A. J. Swallow. J. Chem. Phvs., 25, 1213 (1956).
28. J. V. Davies, D. Greene, J. P. Keene, J. Law, J. B. Massey. Phvs. Med. Biol., 8, 97 (1963).
29. W. K. Sinclair, R. J. Shalek. Radiology, 70, 92 (1958).
30. 7?. W. Hummel, J. W. T. Spinks. J. Chem. Phys., 20, 1056 (1952).
31. J. TVeiss, W. Bernstein, J. В. H. Cuper . J. Chem. Phys., 22 1593 (1954).
32. C. Pettersson. Ark. Phys., 34, 385 (1967).
33. W. Minder. Selected Topics in Radiation Dosimetry. Vienna, IAEA, 1961, p. 315.
34. H. Liesem, W. Pohlit. Z. phys. Chem. (Neue Folge), 35, 352 (1962).
35. J. Geisselsoder, K. Koepke, J. S. Laughlin. Radiation Res., 20, 423 (1963).
36. P. R. Almond. Phys. Med. Biol., 12, 13 (1967).
37. J. Zsula, A. Liuzzi, J. S. Laughlin. Radiation Res., 6, 661 (1957).
38. S. Rosinger. Proc. II United Nations Intern. Conference on Peaceful Uses of Atomic Energy, vol. 21. Geneva, 1958, p. 209.
39. R. H. Schuler, A. O. Allen. J. Chem. Phys., 24, 56 (1956).
40. J. Saldick, A. O. Allen. J. Chem. Phys., 22, 438 (1954).
41. H. Я. Глазунов, А. К. Пикаев. Докл. АН СССР, 130, 1051 (1960).
42. А. К. Пикаев, П. Я. Глазунов. Изв. АН СССР, ОХН, 1959, 2244.
43. D. М. Donaldson, N. Miller. J. Chim. phys., 52, 578 (1955).
44. M. Peisach, J. Steyn. Nature, 187 , 58 (I960).
45. M. Peisach, J. Steyn. J. S. Aftic. Chem. Inst., 13, 34 (1960).
46. A. M. Baptista, J. G. daSilva. Nucl. Sci. Abstr., 17, 4597 (1963).
47. С. А. Брусенцева, П. И. Долин. Докл. АН СССР, 131, 117 (1960).
48. Р. Bonet-Maury, О. Bagno, A. Weill-Cohen. Personnel Dosimetry for Radiation Accidents. Vienna, IAEA, 1965, p. 109.
49. L. Ehrenberg, E. Saeland. JENER Puhi. No 8 (1954).
50. P. Bonet-Maury. Compt. rend., 251, 3087 (1960).
51. H. Fricke, E. J. Hart. Radiation Dosimetry, 2nd Edition (Ed. F. H. Attix and W. C. Roesch), vol. 2. N. Y., 1966, p. 167.
52. ICRU Report 10b, Natl. Std. Handbook 85 (1964).
53. E. Collinson, F. S. Dainton, J. Kroh. Proc. Roy. Soc., A265, 422 (1962).
54. W. R. McDonell, E. J. Hart. J. Am. Chem. Soc,, 76, 2121 (1954).
55. M. Cottin, M. Lefort. J. Chim. phys., 53, 267 (1956).
56. J. Geisselsoder, C. J. Karzmark. Phys. Med. Biol., 14, 61 (1969).
57. II. Gevantman, J. T. Pestaner. J. Chem. Phys., 31, 140 (1959).
58. A. T. Red path, J. Law, J. R. Greening, K. J. Randle. Solid State and Chemical Radiation Dosimetry in Medicine and Biology. Vienna, IAEA, 1967, p. 241.
59. M.Friley, M. Lefort. J. Phys. Rad., 20, 516 (1959).
60. J. Law. Phys. Med. Biol., 14, 607 (1969).
61. M.H.Back, N. Miller. Nature, 179, 321 (1957).
62. J. Novotny, Z.Spumy. Intern. J. Appl. Radiation Isotopes, 19, 403 (1968).
63. J. Baarli, P. Borge. Acta Radiologica, 47, 203 (1957).
64. W. M. Dale. Brit. J. Radiol., 30, 340 (1957).
65. N. Miller. Nature, 171, 688 (1953).
66. N. F. Barr, R. H. Schuler. .1. Phys. Chem., 63, 808 (1959).
67. T. Sawai. Proc. Japan Conference on Radioisotopes, 5, No 3, 112 (1963).
68. H. Hotta. Bull. Chem. Soc. Japan, 33, 442 (1960).
69. N. F. Barr, C. G. King. J. Am. Chem. Soc., 76, 5565 (1954).
70. II. A. Dewhurst. Disc. Faraday Soc., 12, 255 (1952).
71. Б. В. Эршлер, В. Г. Фирсов. Изв. АН СССР, ОХН, 1958, 633.
72. N.F.Barr, С. G. King. J. Am. Chem. Soc., 78, 303 (1956).
73. W. G. Rothschild, A. O. Allen. Radiation Res., 8, 101 (1958).
74. Дж. Вейсс, А. Аллен, Г. Шварц. Материалы Международной конференции по мирному использованию атомной энергии (Женева, 1955), т. 14. М., Фпзматиздат, 1958, стр. 216.
75. A. О. Allen, W. G. Rothschild. Radiation Res., 7, 591 (1957).
76. K. Sehested, E. Bfergbakke, O. L. Rasmussen, H. Fricke. J. Chem. Phys., 51, 3159 (1969).
77. F. S. Dainton, H. C. Sutton. Trans. Faraday Soc., 49, 1011 (1953).
78. H. A. Dewhurst. Ibid., p. 1174.
79. J. A. Ghormley, C. J. Hochanadel. Цпт. no [74].
80. T. Rigg, G. Stein, J. Weiss. Proc. Roy. Soc., Л211, 375 (1952).
81. С. B. Amphlett. Disc. Faraday Soc., 12, 144 (1952).
82. A. O. Allen, V. D. Hogan, W. G. Rothschild. Radiation Res., 7, 603 (1957).
83 G. Gilat, G. Stein. Proc. Il I’nited Nations Intern Conference on Peaceful Uses of Atomic Energy, vol. 29. Geneva, 1958, p. 43.
84. M. А. Проскурин, В. Д. Орехов, А. И. Чернова. Сборник работ по радиационной химии. М., Изд-во АН СССР, 1955, стр. 79.
85. N. Miller, М. Н. Back. J. Chim. phys., 52, 644 (1955).
86. G. R. A. Johnson. Ibid., p. 645.
87. H. LeBail, J. Sutton. .1. Chim. phys., 53, 430 (1956).
88. Б. В. Эршлер, В. Г. Фирсов. Изв. АН СССР, ОХН, 1958, 644.
89. J. W. Boyle. Radiation Res., 17, 427 (1962).
90. J. W. Boyle. Ibid., p. 450.
91. Л. T. Бугаенко, Хуан Гуан-линь, H. А. Бах. Докл. АН СССР, 149, 1099 (1963).
92. В. Н. J. Bielski, А. О. Allen. Intern. J. Rad. Phys. Chem., 1, 153 (1968).
93. T. J. Hardwick. .1. Chem. Phys., 31, 226 (1959).
94. W. Minder, A. Liechte. Experientia, 2. 410 (1946).
95. W. Minder. Brit. J. Radiol., 24, 435 (1949).
96. H. Fricke. Army Chem. Corp. Symp. No 4, part 1, 1950, p. 24.
97. H. A. Schwarz. J. Am. Chem. Soc., 76, 1587 (1954).
98. C. J. Hochanadel, J. A. Ghormley. Radiation Res., 16, 653 (1962).
99. T. J. Hardwick. Can. J. Chem., 31, 881 (1953).
100. R. E. Huffman, N. Davidson. J. Am. Chem. Soc., 78, 4836 (1956).
101. H. Kubota. .1. Inorg. Nucl. Chem., 28 , 3053 (1966).
102. G. Harker. Nature, 133, 378 (1934).
103. H. A. Dewhurst. J. Chem. Phys., 19, 1329 (1951).
104. H. A. Dewhurst. Trans. Faraday Soc., 48, 905 (1952).
105. E. J. Hart. J. Am. Chem. Soc., 74, 4174 (1952).
106. M. Cottin, M. Haissinsky, C. Vermeil. Compt. rend., 235, 542 (1953).
107. C. Vermeil, M. Cottin, M. Haissinsky. J. Chim. phys., 49, 437 (1952).
108. C. Vermeil. ,1. Chim. phys., 52, 587 (1955).
109. C. Vermeil. Ann. chim., 1, 641 (1956).
110. G. R. A. Johnson. J. Chim. phys., 52, 594 (1955).
111. P.G. Glay, G. R. A. Johnson., J. Weiss. J. Phys. Chem., 63, 862 (1959).
112. К. C. Kurien, P. V. Phung, M. Burton. Radiation Res., 11, 283 (1959).
113. J. Teply, J. Bednar. Proc. II United Nations Intern. Conference on Peaceful Uses of Atomic Energy, vol. 29. Geneva, 1958, p. 71.
114. M. А. Проскурнин, Я. M. Колотыркин. Труды II Международной конференции по мирному использованию атомной энергии. Доклады советских ученых, т. 4. М., Атомиздат, 1959, стр. 211.
115. G. D. Adams, IF. R. Balkwell. Radiation Res., 7, 298 (1957).
116. W. R. Balkwell, G. D. Adams. Ibid., p. 591.
117 G. Thielens. Selected Topics in Radiation Dosimetry. Vienna. IAEA, 1961, p. 321.
118. A. Rosinger, R.Glocker, J. Goubeau. Z. phys. Chem. (Neue Folge), 13, 1 (1957).
119. C. R. Maxwell. Radiation Res., 9, 150 (1958).
120. С. С. Кузнецова, И. К. Соколова. Радиобиология, 2, 626 (1962).
121. Л. И. Карташева, А. К. Пикаев. Докл. АН СССР, 163, 1155 (1965).
122. Л. И. Карташева, А. К. Пикаев. Химия высоких энергий, 1, 22 (1967).
123. L. I. Kartasheva, А. К. Pikaer. Proc. II Tihany Symposium on Radiation Chemistry. Budapest, 1967, p. 399.
124. Л. II. Карташева, А. К. Пикаев. Химия высоких энергий, 3, 10 (1969).
125. Л. И. Карташева, А. К. Пикаев. Радиационная дозиметрия и спектрометрия ионизирующих излучений. Ташкент, «Фаи», 1970, стр. 84.
126. Л. Э. Столярчик, А. К. Пикаев. Докл. АН СССР, 141, 1147 (1961).
127. L. Stolarczyk. Nukleonika, 7 , 245 (1962).
128. L. Stolarczyk. Nukleonika, 8, 313 (1963).
129. L. Stolarczyk. Ibid., p. 391.
130. M. Anbar, J. K. Thomas. J. Phys. Chem., 68, 3829 (1964).
131. С. А. Брусенцева, II. И. Долин. Ж. физ. химии, 34, 2513 (1960).
132. А. М. Кабакчи. Ж. физ. химии, 30, 1906 (1956).
133. С. А. Брусенцева, II. И. Долин. Труды I Всесоюзного совещания по радиационной химии. М., Изд во АН СССР, 1958, стр. 39.
134. R. К. Broszkiewicz, Z. Bulhak. Phys. Med. Biol., 15, 549 (1970).
135. G.G. Jayson, B. J. Parsons, A. J. Swallow. J. C. S. Faraday I, 69, 1597 (1973).
136. N. Miller, J. Wilkinson. Disc. Faraday Soc., 12, 50 (1952).
137. T. Rigg, G. Scholes, J. Weiss. J. Chem. Soc., 1952, 3034.
138. H. A. Schwarz. J. Am. Chem. Soc., 79, 534 (1957).
139. E. J. Hart. Radiation Res., 2, 33 (1955).
140. А. И. Чернова, В. Д. Орехов, М. А. Проскурнин. Сб. «Действиеионизирующих излучений на неорганические и органические системы». М. Изд-во АН СССР, 1958, стр. 28.:
141. J. V. Davies, J. Law. Phys. Med. Biol., 8, 91 (1963).
142. H. Fricke, S. Morse. Am. J. Roent. Rad. Ther., 18, 430 (1927).
143. H. Bouziques, J. Schneidhauer, R. Brian. Selected Topics in Radiation Dosimetry. Vienna, I/'EA, 1961, p. 331.
144. E. R. Johnson, A. O. Allen. J. Am. Chem. Soc., 74, 4147 (1952).
145. N. A. Frigerio, V. D. Henry. Solid State and Chemical Radiation Dosimetry in Medicine and Biology. Vienna, IAEA, 1967, p. 257.
146. B. L. Gupta. Dosimetry in Agriculture, Industry, Biology and Medicine.
Vienna, IAEA, 1973, p. 421.
147. T. Hardwick. Can. J. Chem., 30, 17 (1952).
148. K. Scharf, R. M. Lee. Radiation Res., 16, 115 (1962).
149. I. G. Draganic, N. W. Holm, J. E. Maul. Rise Report 22 (1961); цит. no
150. H. Hotta, S. Ohno. Bull. Chem. Soc. Japan, 34, 1640 (1961).
151. R. Broszkiewicz, S. Mine. Nukleonika, 8, 157 (1963).
152. A. Bryn/olffsson, N. W. Holm. Metrology of Radionuclides. Vienna, IAEA, 1960, p. 419.
153. С. M. Henderson, N. Miller. Radiation Res., 13, 641 (1960).
154. F. Hutchinson. Radiation Res., 9, 13 (1958).
155. J. Rotblat, H. C. Sutton. Proc. Roy. Soc., A255, 490 (1960).
156. R. M. Lazo. J. Chem. Phys., 22, 1370 (1954).
157. M. Lefort, P. Douzou. J. Chim. phys., 53, 536 (1956).
158. F. S. Dainton,F. T. Jones. Trans. Faraday Soc., 61, 1681 (1965).
159. H. L. Atkins, W. Bennett-Corniea, W. M. Garrison. J. Phys. Chem., 71, 772 (1967).
160. G. Czapski, G. Stein. J. Phys. Chem., 63, 850 (1959).
161. C. Senvar. Comm. Fac. Sci. Univ. Ankara, 8B, 12 (1960).
162. M. C. Anta, M. H. Mariano. ,T. Chim. phys., 57, 59 (1960).
163. M. Lefort. Compt. rend., 245, 1623 (1957).
164. N. F. Barr, J. Geisselsoder, J. S. Laughlin. Radiation Res., 14, 291 (1961).
165. J. Geisselsoder. Radiation Res., 20, 423 (1963).
166. J. Weiss, N. Miller. J. Phys. Chem., 63, 888 (1959).
167. M. Lefort, X. Tarrago. J. Phys. Chem., 63, 833 (1959).
168. J. Weiss. J. Chem. Phys., 22, 1593 (1954).
169. J. W. Boyle, S. Weiner, C. J. Hochanadel. J. Phys. Chem., 63, 892 (1959).
170. Г. А. Михальченко, И. Клейнман. Радиохимия, 4, 479 (1962).
171. С. N. Trumbore, Е. J. Hart. J. Phys. Chem., 63, 867 (1959).
172. J. W. Boyle, H. A. Mahlmart. Radiation Res., 16, 416 (1962).
173. И. Драганич, M. Симич, H. Милашин, Б. Радак. Труды I Всесоюзного совещания по радиационной химии. М., Изд-во АН СССР, 1958, стр. 64.
174. N. Miller. Radiation Res., 9, 633 (1958).
175. Е. J. Hart, W. J. Ramler, S. R. Rocklin. Radiation Res., 4, 378 (1956).
176. R. W. Hummel. Can. J. Chem. , 31, 1203 (1953).
177. J. Rabani, G. Stein. Radiation Res., 17, 327 (1962).
178. T. J. Sworski. Radiation Res., 4, 483 (1956).
179. T. Rigg, W. Taylor, J. РКей.?. J. Chem. Phys., 22, 575 (1954).
180. A. J. Swallow. J. Chim. phys., 52, 585 (1955).
181. R. G. McIntosh, R. L. Eager', J. W. T. Spinks. Can. J. Chem., 42, 2033 (1964).
182. R. L. S. Willix, W. M. Garrison. Radiation Res., 32, 452 (1967).
183. G. R. Freeman. J. Chem. Phys., 33, 71 (1960).
184. M. H. Mariano, M. L. Santos. Radiation Res., 32, 905 (1967).
185. Ю. E. Тиликс, К. К. Шварц, Д. Тоне. Радиационная физика. П. Рига, «Зинатне», 1964, стр. 35.
186. Н. A. Schwarz, А. О. Allen. J. Am. Chem. Soc., 77, 1324 (1955).
187. J. Nagi, A. Sanielevici. Strahlenther., 133, 561 (1967).
188. S. S. Kumar, R. E. Faw. Nucl. Instr. Meth., 57, 175 (1967).
189. T. H. E. Bryant. T. P. Ridler. Health Phys., 15, 263 (1968).
190. K. Scharf. Phys. Med. Biol., 16, 77 (1971).
191. J. J. McFarlane, R. F. Reeve, W. T. Spragg. Intern. J. Appl. Radiation Isotopes, 19, 883 (1968).
192. J. Law. Phys. Med. Biol., 15, 301 (1970).
193. Z. P. Zagorski. Nukleonika, 5, 253 (1960).
194. N. Matsuura. Radioisotopes, 11, 173 (1962).
195. В. И. Медведевский, H. А. Бах, E. В. Журавская. Сборник работ по радиационной химии. М., Изд-во АН СССР, 1955, стр. 71.
196. К. Sugimoto. Bull. Univ. Osaka Pref., All, 75 (1963).
197. S'. Y amaguchi. Naturwiss., 47, 176 (1960).
198. G. Rudstam, T. Svedberg. Nature, 171, 648 (1953).
199. S. M. Pribicevic. NR — 001 — 0022 (1961).
200. O. S.Gal. Intern. J. Appl. Radiation Isotopes, 13, 304 (1962).
201. K. Hasegawa. Radioisotopes, 15, 69 (1966).
202. R. G. Waggener, C. W. Hiatt, L. F. Rogers, P. Zanca. Radiation Res., 45, 244 (1971).
203. W. L. McLaughlin, E. Bjergbakke. Dosimetry in Agriculture, Industry, Biology and Medicine. Vienna, IAEA, 1973, p. 383.
204. F. G. Haasbroek. Цит. no [51].
205. H. C. Sutton. Phys. Med. Biol., 1, 153 (1956).
206. E. J. Hall, R. Oliver. Brit. J. Radiol., 34, 397 (1961).
207. D. V. Cormack, R. W. Hummel, H. E. Johns, J. W. T. Spinks. J. Chem.
Phys., 22, 6 (1954).
208. R. Loevinger, C. J. Karzmark, M. Weissbluth. Radiology, 77, 906 (1961).
209. H. Svensson, C. Pettersson, G. Hettinger. Solid State and Chemical Radia-
tion Dosimetry in Medicine and Biology. Vienna, UAEA, 1967, p. 251.
210. В. II. Трусова, Ю. С. Рябухин. Атомная энергия, 15, 526 (1963).
211. A. Brynjolfsson. Radiation Preservation of Food. Vienna, IAEA, 1973, p. 549.
212. G. Meshitsuka, T. Sawai. Tokyo Metropolitan Isotopes Centre, Ann. Rept., 1, 71 (1962).
213. Л. И. Карташева, А. К. Пикаев. Intern. J. Rad. Phys. Chem., 1, 243 (1969).
214. J. Geisselsoder, M. J. Kingkade, J. S. Laughlin. Radiation Res., 20, 363 (1963).
215. A. Faucitano, F. F. Martinotti, V. Maxia. Ricerca Scient., 38, 214 (1968).
216. K. Coathsworth, E. Collinson, F. S. Dainton. Trans. Faraday Soc., 56, 1008 (1960).
217. W. W. Brandt, G.F. Smith. Analyt. Chem., 21, 1313 (1949).
218. D. M. Donaldson, N. Miller. Proc. II United Nations Intern. Conference on Peaceful .Uses of Atomic Energy, vol. 29. Geneva, 1958, p. 88.
219. R. W. Hummel, J. W. T. Spinks. Can. J. Chem., 31, 250 (1963).
220. G. Dobson, G. Hughes. Trans. Faraday Soc., 57, 1117 (1961).
221. В. H. Шубин, П. И. Долин. Докл. АН СССР, 134, 891 (1960).
222. Е. Benn, R. G. Post, М. Е. Wacks. Trans. Am. Nucl. Soc., 10, 53 (1967).
223. G. L. Clark, W. S. Coe. J. Chem. Phys., 5, 97 (1937).
224. M. Haissinsky, M. Lefort, H. LeBail. J. Chim. phys., 48, 208 (1951).
225. M. Haissinsky, M. Lefort. J. Chim. phys., 48, 209 (1951).
226. T. J. Hardwick. Can. J. Chem., 30, 23 (1952).
227. J. Weiss. Nucleonics, 10, No 7, 28 (1952).
228. G. R. A. Johnson, J. Weiss. J. Chem. Phys., 22, 752 (1954).
229. G. R. A . Johnson, J. Weiss. Proc. Roy. Soc., A240, 189 (1957).
230. A'.F. Barr, R. H. Schuler. Radiation Res., 5, 468 (1956).
231. J. T. Harlan, E. J. Hart. Nucleonics, 17, No 8, 102 (1957).
232. S. T. Taimuty, L. H. Towle, D. L. Peterson. Nucleonics, 17, No 8, 103 (1957).
233. M. Lefort. J. Chim. phys., 54, 782 (1957).
234. B. Whitetaker. Nature, 160, 1302 (1957).
235. А. К. Пикаев, П. Я. Глазунов. Изв. АН СССР, ОХН, 1960, 940.
236. А. К. Пикаев, П. Я. Глазунов. Изв. АН СССР, ОХН, 1964, 1944.
237. Э. Харт, Г. Кох, Б. Петри, Дж. Шульман, С. Таймути. Г. Викоф. Труды II Международной конференции по мирному использованию атомной энергии. Избранные доклады иностранных ученых, т. 10. М., Атомиздат, 1959, стр. 419.
238. F. J. Munoz, М. Miro. Intern. J. Appl. Radiation Isotopes, 15, 159 (1964).
239. G. L. Kochanny, A. Timnick, C. J. Hochanadel, C. D. Goodman. Radiation Res., 19, 462 (1963).
240. R. R. Hentz, Farhataziz, D. J. Milner, M. Burton. J. Chem. Phys., 46, 4154 (1967).
241. E. Collinson, F. S. Dainton, J. Kroh. Nature, 187, 475 (1960).
242. А. К. Пикаев, П. Я. Глазунов. Докл. АН СССР, 135, 902 (1960).
243. Н. Hotta, К. Shimada. Bull. Chem. Soc. Japan, 33, 114 (I960).
244. <S. W. JVicksic, J. R. Wright. Nucleonics, 13, N 11, 104 (1955).
245. T. J. Sworski. J. Am. Chem. Soc., 78, 1768 (1956).
246. T. J. Sworski. Radiation Res., 6, 645 (1957).
247. T. J. Sworski. J. Am. Chem. Soc., 77, 4689 (1955).
248. H. A. Mahlman, G. K. Schweitzer. J. Inorg. Nucl. Chem., 5, 213 (1958).
249. H. A. Mahlman. J. Phys. Chem., 64, 1598 (1960).
250. А. К. Пикаев П. Я. Глазунов, А. А. Якубович. Кинетика и катализ, 4, 835 (1963).
251. Т. J. Sworski, R. W. Mathews, Н. A. Mahlman. Adv. Chem. Ser., 81, 164 (1968).
252. D. R. Bongiwar, J. Weiss. Radiation Preservation of Food. Vienna, IAEA, 1973, p. 525.
253. J. H. Baxendale, E. M. Fielden, J. P. Keene. Pulse Radiolysis (Ed. M. Ebert et al.). London — New York, 1965, p. 217.
254. J. H. Baxendale, E. M. Fielden, J. P. Keene. Proc. Roy. Soc., A286, 320 (1965).
255. R. W. Mathews. Intern. J. Appl. Radiation Isotopes, 22, 199 (1971).
256. H. Fricke, E. J. Hart, H. P. Smith. J. Chem. Phys., 6, 229 (1938); Radiation Res., 17, 262 (1962).
257. A. O. Allen. R. A. Holroyd. J. Am. Chem. Soc., 77, 5852 (1955).
258. E. J. Hart, E. M. Fielden. Adv. Chem. Ser., 50, 253 (1965).
259. A. J. Medalia, B. J. Byrne. Analyt. Chem., 23, 453 (1951).
260. A. M. Кабакчи, Я. И. Лаврентович, В. В. Пеньковский. Химическая дозиметрия ионизирующих излучений. Киев, Изд-во АН УССР, 1963.
261. Е. J. Hart, Р. D. Walsch. Radiation Res., 1, 342 (1954).
262. Е. J. Hart. Radiation Res., 2, 33 (1955).
263. Ft. D. Jarrett, J. W. Halliday. Activities Rept. 15 (V. S. Natick Labs.); цит. no [51].
264. В. П. Трусова. Г. С. Бологова, Р. А. Николаева, А. Г. Васильев. Дозиметрия больших доз. Ташкент, «Фаи», 1966, стр. 75.
265. Е. B/ergbakke, К. Sehested. Adv. Chem. Ser., 81, 579 (1968).
266. H.G. Swope. Argonne Natl. Lab. Kept. ANL-5819; цит. no [51].
267. J. W. Halliday, R. D. Jarrett. Adv. Chem. Ser., 81, 604 (1968).
268. N. W. Holm, R. D. Jarrett. Неопубл, работа; цит. по [51].
269. I. G. Draganic. J. Chim phys., 52, 511 (1955).
270. I. G. Draganic. Ibid., p. 595.
271. I. G. Draganic. J. Chim. phys., 56, 9 (1959).
272. I. G. Draganic. Nucleonics, 21, No 2, 33 (1963).
273. I. G. Draganic, В. B. Radak, E. M. Markovic. Intern. J. Appl. Radiation Isotopes, 16, 145 (1965).
274. В. В. Генералова. Дозиметрия больших доз. Ташкент, «Фан», 1966, стр. 29.
275. I. G. Draganic. Proc. II Tihany Symposium on Radiation Chemistry. Budapest, 1967, p. 139.
276. I. G. Draganic, I. Markovic. Radiation Res., 35, 587 (1968).
277. В. B. Radak, I\I. Karapandzic. O.Gal. Nucleonics, 22, No 11, 52 (1964).
278. P. Claes, P. Huyskens, J.-L. Galliez, S. Rzad. J. Chim. phys., 61, 271 (1964).
279. IV. W. Holm, K. Sehested. Adv. Chem. Ser., 81, 568 (1968).
280. A. Sehested, E. Bjergbakke, N. И7. Holm. Proc. II Tihany Symposium on Radiation Chemistry. Budapest. 1967. rp. 149.
281. Z. D. Draganic, I. G. Draganic, M. M Kosanic. J. Phys. Chem., 68, 2085 (1964).
282. I. G. Draganic. J. Chim. phys., 56, 18 (1959).
283. I. G. Draganic. Ibid., p. 16.
284. Z. D- Draganic. Analyt. Chim. Acta, 28, 394 (1963).
285. M. Matsui. Sci. Paper I. P. C. R. Japan, 53, No 1528 (1954): цит. no [279].
286. R. W. Hummel. Proc. II Tihany Symposium on Radiation Chemistry. Budapest, 1957, p. 147.
287. В. П. Трусова, Г. С. Бологова, А. В. Васильев. Атомная энергия, 26, 457 (1969).
288. О. Bagno. Compt. rend., 256, 3516 (1963).
289. Т. Asai, S. Ikeguchi. Radioisotopes, 14, 73 (1965).
290. J. C. DeBaere, M. J. DeProost, T. K. Van Eisen. Solid State and Chemical Radiation Dosimetry in Medicine and Biology. Vienna, IAEA, 1967, p. 281.
291. H. В. Кулюпина, О. И. Вакуленко, С. К. Мегедь, А. М. Кабакчи. Дозиметрия интенсивных потоков ионизирующих излучений. Ташкент, «Фан», 1969, стр. 9.
292. С. В. Стародубцев, HI. А. Абляев, В. В. Генералова. Атомная энергия, 8, 264 (1960).
293. С. В. Стародубцев, Ш. А. Абляев, В. В. Генералова. Труды Ташкентской конференции по мирному использованию атомной энергии, т. I. Ташкент, Изд-во АН УзбССР, 1961, стр. 159.
294. С. В. Стародубцев, В. В. Генералова. Изв. АН УзбССР, серия физ.-мат. наук, 1963, 46.
295. С. В. Стародубцев, В. В. Генералова, II. X. Абдукадырова. Изв. АН УзбССР, серия физ.-мат. наук, 1965, 19.
296. В. В. Генералова, Р. Гуламова. Дозиметрия интенсивных потоков ионизирующих излучений. Ташкент, «Фан», 1969, стр. 142.
297. В. В. Генералова, Р. Р. Гуламова. Радиационная дозиметрия и спектрометрия ионизирующих излучений. Ташкент, «Фан», 1970, стр. 133.
298. A. L. Glass. Nucleonics, 20. No 12, 66 (1962).
299. О. Suschny, Н. Frittum, Н. Eisenlohr. Solid State and Chemical Radiation Dosimetry in Medicine and Biology, Vienna, IAEA, 1967, p. 317.
300. R. D. Russell. Intern. J. Appl. Radiation Isotopes, 21, 143 (1970).
301. R. Venkataramani. Radiation Effects, fl, 15 (1971).
302. В. В. Генералова, M. Б. Кишиневская, Б. А. Вайншток. Атомная энергия, 26, 31 (1969.)
303. В. М. Ленченко, М. Г. Анцилевич, Л. А. Софиенко, М. Б. Кишиневская, А. Н. Цой. Дозиметрия и радиационные процессы в дозиметрических системах. Ташкент, «Фан», 1972, стр. 95.
304. В. В. Братаноеский, В. В. Генералова, М. Н. Гурский, А. В. Тултаее. Там же, стр. 100.
305. Е. Л. Ковалева, Е. П. Петряев, Е. П. Калязин. Там же, стр. 167.
306. М. Day, G. Stein. Nature, 164, 671 (1949).
307. М. Day, G. Stein. Nucleonics, 8, No. 2, 34 (1951).
308. M. А. Проскурнин, E. В. Барелко. Сборник работ по радиационной химии. М., Изд-во АН СССР, 1955, стр. 99.
309. Р. Phung, М. Burton Radiation Res., 2, 1 (1955).
310. М. Daniels, G. Scholes, J. Weiss. J. Chem. Soc., 1956, 832.
311. P. Phung, M. Burton. Radiation Res., 7, 199 (1957).
312. E. В. Барелко, Л. И. Карташева, П. Д. Новиков, М. А. Проскурнин. Труды I Всесоюзного совещания по радиационной химии. М., Изд-во АН СССР, 1958, стр. 89.
313. J. Goodman, J. Steigman. J. Phys. Chem., 62, 1020 (1958).
314. T. Johnson, J. Martin. Nucleonics, 20, No. 1, 83 (1962).
315. E. В. Барелко, Л. И. Карташева, M. А. Проскурнин. Труды II Всесоюзного совещания по радиационной химии. М., Изд-во АН СССР, 1962, стр. 221.
316. Н. С. Christensen. Nukleonik, 7, 1 (1965).
317. К. С. Kurien, Р. V. Phung, М. Burton. Radiation Res., И, 283 (1959).
318. Е. В. Барелко, Л. И. Карташева, М. А. Проскурнин. Докл. АН СССР, 116, 74 (1957).
319. Л. И. Карташева, А. К. Пикаев. Ж. физ. химии, 41, 2855 (1967).
320. I. Loeff, G. Stein. Nature, 184, 901 (1959).
321. I. Loeff, G. Stein. J. Chem. Soc., 1963 , 2623.
322. В. С. Жихарев, H. А. Высоцкая. Укр. хим. ж., 33, 551 (1967).
323. В. С. Жихарев, Н. А. Высоцкая. Ж. физ. химии, 42, 360 (1968).
324. В. С. Жихарев, Н. А. Высоцкая. Химия высоких энергий, 2, 249 (1968).
325. L. М. Dorfman, I. A. Taub, В. Е. Biihler. J. Chem. Phys., 36, 3051 (1962).
326. Л. И. Карташева, А. К. Пикаев. Химия высоких энергий, 6, 90 (1972).
327. G. Stein, J. Weiss. J. Chem. Soc , 1951, 3265.
328. Л. И. Карташева, 3. С. Булановская, Е. В. Барелко, Я. М. Варшавский, М. А. Проскурнин. Докл. АН СССР, 136, 143 (1961).
329. М. Е. J. Carr. Nature, 167, 363 (1951).
330. Т. J. Sworski. J. Chem. Phys., 20, 1817 (1952).
331. TV. Klein Health Phys., 6, 212 (1961).
332. M. А. Проскурнин, E. В. Барелко, Л. И. Карташева. Докл. АН СССР, 121, 671 (1958).
333. F. W. Bloch, Е. J. Haasbroek, D. R. McKenzie. Nucleonics, 26, No. 3, 3 (1964).
334. В. Caire, L. Ordinaire. Solid State and Chemical Radiation Dosimetry in Medicine and Biology IAEA, Vienna, 1967, p. 287.
335. I. Balakrishnan, M. P. Reddy. J. Phys. Chem., 74, 850 (1970).
336. G. Ahnstroem, C.G. Rosen. Acta Chem. Scand., 19, 263 (1965).
337. W- A. Armstrong, G. W. Grant. Can. J. Chem., 38, 845 (i960).
338. W- A. Armstrong, B. A. Black, G. W. Grant. J. Phys. Chem., 64, 1415
(1960).
339. A. M. Downes. Australian J. Chem., 11, 154 (1958).
340. W- A. Armstrong, R. A. Facey, D. W. Grant, W. G. Humphreys. Radiation Res., 19, 120 (1963).
341. H. Loebl, G. Stein, J. Weiss. J. Chem. Soc., 1951, 405.
342. A. Sakumoto, G. Tsuchikasi. Bull. Chem. Soc. Japan, 34, 660 (1961).
343. A. Sakumoto, G. Tsuchikasi. Ibid., p. 663.
344. W. A. Armstrong, D. W. Grant. Nature, 182, 747 (1958).
345. W. A. Armstrong, W. G. Humpreys. Can. J. Chem., 42, 1092 (1964).
346 P.Giintner, H. D. ton der Horst, G. Cronheim. Z. Electrochem., 34, 616 ’ (1928).
347. W- Minder, H. Knuchel, P. GUntner. Experientia, 4, 219 (1948).
348. W. Minder, II. Heydrich. Disc. Faraday Soc., 12, 305 (1952).
349 J. W- Schulte, J.F. Suttle, R. Wilheim. J. Am. Chem. Soc., 75, 2222 (1953).
350. J- W. Schulte. J. Am. Chem. Soc., 79, 4643 (1957).
351. R- W. Hummel, J. W. T. Spinks. J. Chem. Phys., 20, 1056 (1952).
352. R. W. Hummel, A. R. van Cleave, J. W. T. Spinks. Can. J. Chem., 32, 522 (1954).
353. Д. Тэплин. Радиационная дозиметрия (под ред. Дж. Хайна и Г. Браунелла). М., ИЛ, 1958, стр. 298.
354. S. G. Sigoloff. Selected Topics in Radiation Dosimetry. Vienna, IAEA, 1961, p. 337.
355. D.G. Olt, W. II. Schweitzer, J. A. Sayeg, P. S. Harris. Health Phys., 7, 20 (1961).
356. R. F. Plat ford, J. W. T. Spinks. Can. J. Chem., 37, 1022 (1959).
357. R. A. Glass, W. E. Wilson, A. H. Samuel. Stanford Res. Inst. Rep. AD 270014 (1961); цит. no [51].
358. А. К. Пикаев, П. Я. Глазунов. Изв. АН СССР, ОХН, 1959, 1137.
359. S’. G. Sigolojf. Nucleonics, 14, No 10, 54 (1956).
360. I. Dvornik, U. Zee, A. Ante, F. Ranogajec. Solid State and Chemical Radiation Dosimetry in Medicine and Biology. Vienna, IAEA, 1967, p. 273.
361. G. Taplin, C. Douglas. Nucleonics, 13, No 1, 88 (1956).
362. G. Taplin. Intern. J. Appl. Radiation Isotopes, 2, 180 (1957).
363. Дж. Тэплин. Доклады иностранных ученых на Международной конференции по мирному использованию атомной энергии (Женева, 1955). Дозиметрия ионизирующих излучений. М., Гостехтеориздат, 1956, стр. 248.
364. Z. Spumy. Jaderna Energ., 10, 326 (1963).
365. L. Reyes, L. S. A. Reyes. Rev. Mex. Fis., 17, 263 (1968).
366. W. Stenstrom, A. Lohman. Radiology, 21, 29 (1933).
367. W. Stenstrom, A. Lohman. Radiology, 22, 304 (1934).
368. Я. Л. Шехтман, А. А. Красновский, И. В. Верещинский. Докл. АН СССР, 74, 767 (1950).
369. S. Davisson. Nucleonics, 11, No. 7, 22 (1953).
370. М. J. Day, G. Stein. Radiation Res., 6, 666 (1957).
371. E. Hayon, G. Scholes, J. Weiss. J. Chem. Soc., 1957, 301.
372. В. Д. Орехов, А. И. Чернова, M. А. Проскурнин. Сборник работ по радиационной химии. М., Изд-во АН СССР, 1955, стр. 85.
373. В. Lafuente, S.Goldblith, В. Proctor. Intern. J. Appl. Radiation Isotopes, 3, 119 (1958).
374. Я. Л. Шехтман, T. Г. Ратнер. Труды I Всесоюзного совещания по радиационной химии. М., Изд-во АН СССР, 1958, стр. 106.
375. В. Е. Proctor, S. A. Goldblith. Nucleonics, 7. N 2, 83 (1950).
376. Н. В. Малахов, О. Ф. Чебурков, А. М. Набакчи. Труды II Всесоюзного совещания по радиационной химии. М., Изд-во АН СССР, 1962, стр. 243.
377. К. S. Kargoankar, М. S. Тarankanath. Intern. J. Appl. Radiation Isotopes, 14, 405 (1963).
378. L. Mongini, E. Zimmer. J. Chim. phys., 50, 491 (1953).
379. M. C. Anta. Proc. II United Nations Intern. Conference on Peaceful Uses of Atomic Energy, vol. 29. Geneva, 1958, p. 99.
380. M. C. Anta, M. L. R. Santos. Intern. J. Appl. Radiation Isotopes, 4, 261 (1959).
381. M. C. Anta. J. Chim. phys., 57, 728 (1960).
382. E. Weber, R. Schuler. J. Am. Chem. Soc., 74, 4415 (1952).
383. F. Patti. J. Chim. phys., 52, 77 (1955).
384. F. Patti. Ibid., p. 38.
385. .7. Patko. Z. Szentirmay, Z. Dezsi, M. Gorgey. Acta Univ. Debrecen., Ser. phys. chem., 12, 29 (1966).
386. K. Sugimoto. Ann. Rep. Rad. Center Osaka Pref., 3, 114 (1962).
387. C. Cabot, W. B. Mason, J. T. Bickmore. Radiation Res., 7, 307 (1957).
388. W. A. Armstrong, G. A. Grant. Radiation Res., 8, 375 (1958).
389. W. A. Armstrong, G. A. Grant. Can. J. Chem., 36, 1398 (1958).
390. B. L. Gupta. Intern. J. Appl. Radiation Isotopes (в печати); цит. по I. G. Draganic, В. L. Gupta. Dosimetry in Agriculture, Industry, Biology and Medicine. Vienna, IAEA, 1973, p. 351.
391. B.L. Gupta, E.J. Hart. Radiation Res., 48, 8 (1971).
392. W. L. McLaughlin, L. Chalkley. Trans. Am. Nucl. Soc.. 10, 52 (1967).
393. W. L. McLaughlin. Manual on Radiation Dosimetry (Ed. by N. W. Holm and R. J. Berry). N. Y., Marcel Decker, Inc., 1970, p. 377.
394. W. L. McLaughlin. E. K. Hussmann. H. H. Eisenlohr, L. Chalkley. Intern. J. Appl. Radiation Isotopes, 22. 135 (1971).
395. W. L. McLaughlin. Dosimetry in Agriculture, Industry, Biology and Medicine. Vienna, IAEA, 1973, p. 362.
396. J. Weiss. Intern. J. Appl. Radiation Isotopes, 4, 89 (1958).
397. В. E. Proctor, S. A. Goldblith. Nucleonics, 7, No. 2, 83 (1950).
398. S. A. Goldblith, В. E. Proctor, O. A. Hammerli. Ind. Eng. Chem., 44, 310 (1952).
399. J. Pestaner, F. Gevantman. Radiation Res., 9, 166 (1958).
400. G. Geissler. Strahlenther., 123, 451 (1964).
401. G. Orban. J. Patko, I. Berta. Acta Univ Debrecen., Ser. Phys. Chem., 11, 13 (1965).
402. А. H. Пикаев. T. П. Жесткова. Авт. свпд. СССР 369526 (1973); Бюлл. изобр., № 10, 135 (1973).
403. Т. П. Жесткова, А. К. Пикаев. Химия высоких энергии, 6, 92 (1972 .
404. Г. Шарле. Методы аналитической химии, ч. 2. М., «Химия», 1969. стр. 680.
405. J. J. Law. Health Phys., 17, 338 (1969).
406. Т. J. Hardwick, W. S.Guentner. J. Phys. Chem., 63, 896 (1959).
407. T. J. Hardwick. Radiation Res., 12, 5 (I960).
408. J. Janovsky, B. Bartonicek, J. Bednar. Coll. Czech. Chem. Commun., 28, 2245 (1963).
409. A. L. Boni. Radiation Res., 14 . 374 (1961).
410. С. С. Кузнецова. Радиобиология, 5, 775 (1961).
411. M. В. Raju, Е. W. Merrill. .1. Polymer Sci., B2, 13 (1964).
412. M. B. Baju. Phys. Med. Biol., 10. 515 (1965).
413. R. F. ПеПоп, R. J. Schultz. Radiology, 82, 120 (1964).
414. P. M. Oza, P. S. Bao. Bull. Chem. Soc. Japan, 40, 239 (1967).
415. N. F. Barr, M. B. Stark. Radiation Res., 9, 89 (1958).
416. N.F.Barr, M. B. Stark. Radiation Res., 12, 1 (I960).
417. H. L. Andrews, P. A. Shore. J. Chem. Phys., 18, 1165 (1950).
418. G. R. Freeman, A. B. van Cleave, J. W. T. Spinks. Can. J. Chem., 31,
1164 (1953).
419. W. С. Moos. Intern. J. Appl. Radiation Isotopes, 23, No. 11, 538 (1972).
420. H. Andrews, R. Murphy, E. LeBrun. Rev. Sci. Instrum., 28, 329 (1957).
421. E. J. Hart, P. D. Walsch. Radiation Res., 1, 342 (1954).
422. A. Gordon, E. J. Hart. Proc. II United Nations Intern. Conference on Peaceful Uses of Atomic Energy, vol. 29. Geneva, 1958, p. 13.
423. M.Fiti. L. Tudor. Rev. Roum. Phys., 12, 583 (1967).
424. D. J. Savage. Analyst, 76, 224 (1956).
425. M. T. Дмитриев. Атомная энергия, 15, 52 (1963).
426. С. Н. Cheek, V. J. Linnenbom. Rept. Naval Research Lah., N 10 (1965).
427. P. M. Oza. P. S. Bao. Z. phys. Chem. (Neue Folge), 57, 265 (1968).
428. K. Hasegawa. Radioisotopes, 16, 423 (1963); C. A., 68, 26024 (1968).
429. T. Shiokawa, K. Hasegawa. Nature, 195, 592 (1962).
430. W. A. Armstrong, R. A. Facey, D.W. Grant, W. G. Humphreys. Can. J. Chem., 41, 1575 (1963).
441 А. П. Ибрагимов, А. В. Туйчиев. Атомная энергия, 18, 185 (1965).
432 И- 8. Levinson, Е. В. Garber. Nature, 207, 751 (1965).
433. Ф- И. Билетская. Мед. радиол., 12, № 3. 63 (1967).
434. V. A. Sharpaty, D. М. Margolin, N. V. Zakatova, К. G. Tanova. Proc. II Tihany Symposium on Radiation Chemistry. Budapest, 1967, p. 187.
435. K. Sugimoto, S. Oae. Bull. Chem. Soc. Japan, 35, 1159 (1962).
436. E. П. Ковалева. Л. В. Жильцова, E. П. Калязин, E. П. Петряев. Изв. АН БССР, серия физ. энерг. наук, № 3, 24 (1971).
437. Г. 3. Гочалиев, Ц. И. Залкинд. Труды II Всесоюзного совещания по радиационной химии. М., Изд во АН СССР, 1962, стр. 741.
438. С. Z. Lewin, В. Н. Kozlow. Пат. США 3179581 (1963); С. А., 63, 12637 (1965).
439. К. Sugimoto. Nucl. Sci. Abstr., 17, 30097 (1963).
440 А. Г. Васильев, E И. Вайль, Л. 3. Калмыков. Химия высоких энергий, 3, 175 (1969).
441. А. Г. Васильев, Е. И. Вайль, Л. 3. Калмыков. Радиационная дозиметрия и спектрометрия ионизирующих излучений. Ташкент, «Фан», 1970, стр. 71.
442. Л. 3. Калмыков, А Г. Васильев, Е. И. Вайль. Атомная энергия, 26, 386 (1969).
443. Л. 3. Калмыков, А. Г. Васильев, Е. И. Вайль. Химия высоких энергий, 3, 259 (1969).
444. Е. И. Вайль, А. Г. Васильев, Л. 3. Калмыков. Радиационная дозиметрия и спектрометрия ионизирующих излучений Ташкент, «Фан», 1970, стр. 64.
445. К. К. Шварц, Ю. Е. Тиликс, Д. К. Тоне. Дозиметрия интенсивных потоков ионизирующих излучений. Ташкент, «Фан», 1969, стр. 35.
446. N. A. Atari. К. V. Ettinger, J.H.Fremlin. Radiation Effects, 17, 45 (1973).
447. G. Ahnstrom, III Intern. Congress of Radiation Research. Book of Abstracts, Cortina d’Ampezzo, 1966, p. 18.
448. J. Mittal. Nature. Phys. Sci., 230, 160 (1971).
449. Ю. E. Тиликс, Л. T. Бугаенко, Я. A. Тетерис, К. К. Шварц, В M. Бяков, Р. А. Кан. Изв. АН ЛатвССР, серия физ., № 5, 19 (1970).
Глава VI
ДОЗИМЕТРЫ
НА ОСНОВЕ ОРГАНИЧЕСКИХ ЖИДКОСТЕЙ
Предлагалось сравнительно большое число дозиметрических систем, представляющих собой органические жидкости или растворы различных веществ (чаще всего красителей или их лейко-оснований) в них. В табл. 46 приведены общие характеристики этих систем.
Важной особенностью некоторых дозиметров на основе органических жидкостей (например, циклогексана) является то, что их показания практически не зависят от величины ЛПЭ. Подробно этот вопрос рассматривается в главе X.
Многие из дозиметрических систем, приведенных в табл. 46, не находят какого-либо применения в радиационной химии. Поэтому в настоящей главе подробно рассматриваются только важнейшие из этих систем.
1. Циклогексан
Главными продуктами радиолиза циклогексана являются водород, циклогексен и дициклогексил. Их выходы при различных дозах у-излучения 60Со приведены в табл. 47. Данные этой таблицы получены в одной из последних работ [6], посвященных рассматриваемому вопросу.
Можно представить следующую совокупность основных реакций, объясняющих возникновение указанных выше продуктов радиолиза:
CeHia — СсНи -р Н, СеН12 —СвНю -р Hz, CeHiz -pH—» СеНи -р Н-2, СеНи -р СеНи —» CizHz», СбНи -р СеНи —» СеНю -р CeHi-2.
(1)
(2)
(3)
(4)
(5)
Подробно механизм радиолиза циклогексана рассмотрен в обзоре [591.
Для дозиметрических целей предложено использовать процесс образования Н2. По данным различных авторов [1, 4, 6—11, 60—75], для у- и рентгеновских лучей и быстрых электронов П(Н2) колеблется от 5,25 до 5,85 молек./100 эв. С?(Н3) несколько уменьшается при увеличении дозы На рис 71 приведена изме-
Таблица 4Й
Характеристики дозиметров на основе органических жидкостей
Система Измеряемое превращение Диапазон измеряемых доз, рад Литература
Циклогексан Образование Н-2 До нескольких мегарад [1-111
Бензол То же До 108 [12—15]
Ацетонитрил Образование CN- 6.10s—1,5-Ю7 и выше [16]
Метиловый спирт Образование этиленгликоля 5-105—108 [17]
Этиловый спирт Образование 2,3-бутанди-ола 5-105 — 108 [17]
Хлороформ, кислотно основной индикатор Образование НС1 До 103 [18, 19]
Четыреххлористый углерод Образование СЬ 10» —9-10» [20]
Перфтор-и-гексан или полностью фторированный керосин Образование CFi и C-2Fe 3-106 —4-10’ [21]
Перфторметилциклогек-сан Образование CFa; разложение перфторметилцик-логексана 3-10’ —4,2-10’ [22, 23]
Раствор лейкооснования кристаллического фиолетового в метилэтилкетоне Образование красителя 15-1,5-10» [24, 25]
Раствор нитрила гекса-(оксиэтил)параризани лина [4, 4', 4"-трис-(ди-Р-оксиэтиламино)трифе-нилацетонитрила] в 2-мет-оксиэтаноле Образование красителя 10» — 10» [26, 27]
Раствор нитрила парарозанилина (4, 4' 4"-три-аминотрифенилацето нитрила) в 3-метоксиэтаноле Образование красителя 10» —105 [26—28]
То же с добавкой нитробензола То же До—IO8 [28]
Раствор дифенилтпокарб-азола (дитизона) в СНС13 или ССЦ Изменение окраски 2-3-10» [29 — 35]
Раствор метилового желтого, 2-окси-З-нитрофенил-азо-3-нафтола или реза-зурина в смеси ССЦ и этанола То же 2-102 —9-Ю3 [29, 36]
Раствор резазурина в смеси 80% ССЦи20% этанола » До 2-10» [37]
Таблица 46 (окончание)
Система Измеряемое превращение Диапазон определяемых доз, рад Литература
Раствор бромкрезолового зеленого в смеси 90% СС1г и 10% этанола Изменение окраски До 5-Ю2 [38]
Раствор а, а'-дифенил-З-пикрилгидразила в СНС1з, бензоле или октане То же —• [39, 40]
Раствор ферроцена в ССЦ Образование Fn+—FeCl*-(Fn — ферроцен) До 107 [41]
Раствор лейкооснования малахитового зеленого или лейкооснования кристаллического фиолетового в СНС1з или СНВгз Образование красителя ~5 [42]
Раствор метилового желтого в кумоле Изменение окраски 41O5 — 2-10’ [43]
Растворы красителей в бензоле То же До 10» [44]
Раствор а, х'-дифепилД-пикрилгпдразипа в мета ноле » — [45]
Раствор бепзплацетата в циклогексане Деэтерификация бензил-ацетата 1,5-10» — 3-107 [46]
Раствор ацетилацето на тов Сг3+ пли Со3+ в толуоле или этилбензоле Изменение окраски До 5-Ю7 [47]
Раствор цис-полибутадп-ена в органических растворителях Изомеризация в транс-изомер До 5-Ю8 ]48]
Раствор хлорбензола, ,и-дихлорбензола или тетрахлорэтилена в смеси этанола с толуолом пли 2,2,4-триметилпептаном, содержащий тимолсульфофталеин и до 0,1% воды Образование НС1, приводящее к изменению окраски До 8-Ю3 [49,50]
Раствор хлорбензола в этаноле, содержащем 4 объема. % воды Образование НС1 о 2-Ю7 [51-56]
Раствор полистирола в CCI4 Изменение вязкости раствора 102 — 108 [57]
Раствор полиизобутилена в гептане и ССЦ То же 103 __ мы [58]
ренная в работе [10] зависимость G(H2) от дозы для у-излучения 60Со. Согласно Г. Фримэну [67], зависимость G(H2) от дозы удовлетворительно описывается уравнением
G (Нг) = 5,37 - 8,2 • 10~3Г>, (6)
где D — доза (в единицах 1019 эв/мл).
Как видно из табл. 47 и рис. 71, для доз до нескольких мегарад изменение G(H2) лежит в пределах ±5%. Очевидно, указанная величина является верхним предельным значением дозы, которое можно измерять с помощью этого дозиметра.
Таблица 47
Выходы главных продуктов у-радиолиза циклогексана [6]
Доза, Afptu) G (Н2), молек.,100 эв G (СвН10), молен ./100 эв G (CjgHzg), молек./100 эв
0,5 5,44 3,36 1,61
1,0 5,27 3,32 1,4.2
2,0 — 3,18 1,70
4,0 4,86 2,87 1,41
8,0 4,81 2,38 1,27
Наиболее вероятно, падение G(H2) с ростом дозы обусловлено захватом атомов Н циклогексеном, образующимся при радиолизе:
Н + СбНю СбНп. (7)
Поэтому с целью уменьшения зависимости G(Ha) от дозы рекомендовано [76, 77] использовать циклогексан с добавками циклогексена. Согласно [76], G(H2) для циклогексана, содержащего 8,5% циклогексена, равен 2,93 молек./100 эв.
Рис. 71. Зависимость G (Н2) от дозы D при у-радиолизе циклогексана (мощность дозы ~ 100 рад!сек}
D-КГ™ эВ/г
G(H2) при радиолизе жидкого циклогексана не зависит от температуры [11, 78]. На выход Н2 оказывает малое влияние ЛПЭ излучения. По этой причине циклогексан находит применение в дозиметрии реакторного излучения и потоков тяжелых заряженных частиц (см. главы X и XI). По-видимому, в указанном выше диапазоне доз G(H2) мало зависит от мощности дозы при
обычных ее значениях. По данным работы [79], зависимость выходов продуктов радиолиза от мощности дозы имеет место только в присутствии акцептора радикалов. Таким акцептором, в частности, является один из продуктов радиолиза, а именно: циклогексен. Поэтому влияние мощности дозы может наблюдаться при сравнительно больших дозах. При очень высоких мощностях дозы (1011—Ю14 рад!сек) G(H2) существенно ниже [6], чем в случае у-излучения 60Со (см. главу IX).
При проведении дозиметрии циклогексан с целью удаления растворенных газов подвергают нескольким циклам замораживания, вакуумирования и плавления до остаточного давления менее 10-5 мм рт. ст. Количество образовавшегося водорода определяется обычной вакуумной техникой или методом газовой хроматографии.
2. Бензол
Ароматические углеводороды, в том числе и бензол, характеризуются сравнительно высокой радиационной стойкостью*. Главными продуктами радиолиза бензола являются водород, ацетилен и полимер. Однако выходы этих продуктов малы. Согласно [80], среднее значение выхода водорода составляет 0,039 молек./ЮО эв. Эта величина не зависит от дозы до 108 рад [15], мощности дозы вплоть до очень высоких ее значений [12] (см. главу IX) и температуры в диапазоне от 8 до 160° С [81]. Очевидно, это обстоятельство делает бензол весьма перспективным с точки зрения использования его в химической дозиметрии.
3. Раствор лейкооснования
кристаллического фиолетового в метилэтилкетоне
Н. А. Бах и сотр. [24, 25] предложили использовать образование кристаллического фиолетового при облучении раствора его лейкооснования в метилэтилкетоне для измерения доз в диапазоне 15—1500 рад. Выход красителя (2,0 молек./ЮО эе) не зависит от мощности дозы в исследованном интервале (0,05—25 рад!сек) и температуры облучения раствора в диапазоне от —85 до 50° С. Выход одинаков для у-лучей е0Со, рентгеновских лучей с энергией 0,08 Мэв и а-частиц 210Ро.
В опытах цитированных авторов концентрация раствора лейкооснования составляла 5-Ю-3 М. Лейкооснование и растворитель должны быть высокой степени чистоты; они не должны содержать ни восстановительных, ни окислительных примесей. Растворы облучались в цилиндрических ампулах, запаянных в атмосфере азота. Определение концентрации образовавшегося красителя удобнее всего осуществлять спектрофотометрически, применяя
* Детально механизм радиолиза ароматических углеводородов обсужден в обзоре [80].
эти ампулы в качестве кювет. Молярный коэффициент экстинкции кристаллического фиолетового в максимуме полосы оптического поглощения равен Ю5 л'(молъ-см).
4. Растворы тетрахлорэтилена, хлорбензола и дихлорбензола в органических растворителях
И. Дворник и сотр. [49—55] рекомендуют применять для дозиметрии ионизирующего излучения растворы тетрахлорэтилена, хлорбензола или да-дихлорбензола в органических растворителях. Сначала ими были описаны дозиметры на основе растворов С2С14, С6Нг,С1 или CfiH4Cl2 в смеси этанола (10%) с толуолом или 2,2,4-триметилпентаном, содержащей до 0,1 % воды [49, 50], а затем — дозиметры на основе растворов хлорбензола в этаноле, содержащем 4 объемн. % воды [51—55]. При облучении указанных систем образуется HCI; количество этого вещества и служит мерой дозы.
К системам первого типа добавлялась калиевая соль тимолсульфофталеина. С ее помощью определялась концентрация I1C1. Максимум полосы поглощения кислой формы индикатора (она имеет красную окраску) находится при 533 нм; его калиевая соль (она желтого цвета) в указанной области спектра не поглощает свет. Хлористый водород взаимодействует с этой солью, трансформируя тимолсульфофталеин в кислую форму, и по увеличению оптической плотности раствора при 533 нм находят количество HCI. Концентрация тимолсульфофталеина в зависимости от состава системы варьировалась в диапазоне 2-10“®—4-10-5 If.
Было найдено, что в этанольно-толуольных растворах G(HC1) зависит от концентрации хлорбензола или дихлорбензола (см. табл. 48). Влияние условий облучения было подробно проверено в случае растворов, содержащих 5 объемн. % хлорбензола. G(HC1), как было обнаружено, не зависит от содержания воды (от 0,01 до 0,3%), дозы (до 8000 рад) и мощности дозы (от 165 Таблица 48
Зависимость (7(НС1) от концентрации хлорбензола или дпхлорбензола при у-радиолизе их растворов в смесях толуол — этанол (10%) [49]
Концентрация хлорбензола или дихлорбензола, объемн. % G (НС1), молек. 100 эв Концентрация хлорбензола или ди хлорбензола, объемн.% G (НС1)„ молек., 100 эз
Хлорбензол Ди хлорбензол Хлорбензол Дихлорбензол
1 1,57 3,33 5 3,29 4,57
2 2,36 3,97 7 3,56 4,74
3 2,83 4,25 10 3,87 4,86
4 3,09 4,40 20 4,30 —
до 1050 рад!мин). Недостатком является наличие пост-эффекта (его продолжительность — около 1 часа).
Для 2,5%-ного раствора тетрахлорэтилена в смеси 2,2,4-триметилпентан—этанол (10%) G(HC1) = 8,4 молек./100 эв [49]. Выход постоянен при изменении температуры от —10 до 35е С и мощности дозы от 1,3 до 133 рад!мин. Существенный недостаток системы — наличие довольно медленного пост-эффекта. Согласно [501, в случае 10%-ного раствора хлорбензола в смеси 2,2,4-трпметилпентана и этанола (10%) (?(НС1) = 5,05 молек./100 эв.
Растворы хлорбензола в смеси этанол—вода (4%) предназначены для измерения больших доз (до 20 Мрад) [51—55]. Как и в случае систем первого типа, (?(НС1) зависит от концентрации хлорбензола (см. табл. 49). В то же время выход не зависит от мощности дозы вплоть до весьма больших ее значений (см. главу IX). Присутствие кислорода в системе оказывает малое влияние на величину G(HC1). Дозиметр нечувствителен к примесям. Найдено, в частности, что С(НС1) одинаков как в присутствии 0,04% ацетона или бензола, так и в отсутствие этих добавок. Не было обнаружено какого-либо температурного эффекта в диапазоне 20-90° С.
Тайл и ц а 49
Зависимость G(HC1) от концентрации хлорбензола и дозы при у-радиолизе растворов этого вещества в смеси этанол -вода (4%) в отсутствие кислорода [55]
Концентрация хлорбензола, объемн. % Доза, Мрад G(Cl-), ионы/100 эв Концентрация хлорбензола, объемн. % Доза, Мрад G(C1~), ионы/ЮО эв
4 4-10 3,61 20 2—20 5,67
10 3—15 4,96 40 0—2 6,22
20 0—2 "5 1 40 2—20 6,10
В случае хлорбензольного дозиметра доза находится путем измерения концентрации ионов Н+ или СК, возникших в результате облучения. Рекомендовано [53] концентрацию ионов Н+ находить титрованием со спиртовым раствором щелочи при использовании бромфенолового синего в качестве индикатора, а концентрацию ионов С1~ — титрованием со спиртовым раствором Hg(NO3)2 (индикатор — дифенилкарбазон). Недавно [56, 82, 83] применительно к этому дозиметру описан еще один способ определения ионов С1~ — осциллометрическое титрование со спиртовым раствором Hg(NO3)2.
5. Растворы нитрилов трнфенилметановых красителей в органических растворителях
У. Маклафлин и сотр. 126—281 описали новый метод дозиметрии, основанный на применении растворов нитрилов триарилметановых красителей в органических растворителях. Принцип этого метода уже был описан в предыдущей главе. В этом разделе излагаются результаты, полученные при исследовании растворов нитрила парарозанилина в 2-метоксиэтаноле. Максимум полосы оптического поглощения красителя, возникающего при облучении, находится в этом растворителе при 549,5 нм. 5• 10-3 М раствор указанного нитрила в 2-метоксиэтаноле, как было найдено, пригоден для определения доз до 150 крад, при этом выход образования красителя равен 0,234 молек./100 эв. Введение нитробензола в раствор приводит к уменьшению выхода и расширению диапазона измеряемых доз. Для 5 • 10_3 Л/ раствора, содержащего 3-10-2 молъ/л нитробензола, выход равен 0,057 молек./100 эв, а максимальная доза, которая может быть найдена с помощью этого раствора, составляет 900 крад. Показания дозиметра не зависят от энергии фотонов и электронов в диапазоне от 24 кэв до 1,25 Мэв. Однако выход возрастает при увеличении температуры. Кроме того, он сильно зависит от величины pH раствора. Другой недостаток дозиметра — его чувствительность к действию света.
6. Растворы полимеров в органических растворителях.
Мономеры
Облучение мономера или полимера в растворе или в чистом виде обусловливает полимеризацию мономера и деструкцию или сшивание полимера. Эти изменения можно легко фиксировать путем измерения вязкости при постоянной температуре. Данная особенность находит применение в дозиметрии ионизирующих излучений.
Впервые использование явления деструкции полимеров для целей дозиметрии было предложено П. Александером и др. [84] в 1954 г. Они облучали полиметилметакрилат и затем измеряли вязкость его растворов в хлороформе. Было найдено, что эта методика пригодна для определения доз в диапазоне 5-105—5-107 рад. Позднее возможность применения полимеров в дозиметрии изучалась многими авторами. Результаты, полученные при исследовании твердых полимеров, подробно обсуждаются в главе VIII. В этом разделе рассматриваются дозиметры на основе растворов полимеров в органических растворителях, а также дозиметры на основе мономеров.
При облучении растворов полимеров происходит их деструкция под действием свободных радикалов, возникающих при радиолизе растворителя. Это приводит к уменьшению вязкости раствора. Этот эффект может применяться для определения доз
в весьма широких диапазонах. Для нахождения малых доз используются разбавленные растворы полимеров с высоким молекулярным весом, а для нахождения больших доз — концентрированные растворы полимеров с низким молекулярным весом.
П. Фенг [57] нашел, что насыщенные воздухом растворы полистирола в четыреххлористом углероде могут использоваться для измерения поглощенных доз от 102 до 108 рад. В этой методике раствор облучается непосредственно в приборе для определения вязкости — вискозиметре проточного типа.
Подробное исследование дозиметрических характеристик растворов полиизобутилена в гептане и четыреххлористом углероде, а также жидкого полиизобутилена было выполнено Л. Висне-ром [58]. В своих опытах он использовал полимер марки «Oppanol», выпускаемый западногерманской фирмой «Badische Anilin und Soda Fabrik». Было найдено, что в зависимости от молекулярного веса полимера растворы в гексане пригодны для нахождения доз в следующих диапазонах (в скобках указан молекулярный вес): до 3-10® рад (2-1U5—3-105), до 109 рад (~5000) и до ~1010 рад .(500—1500), а раствор полимера с молекулярным весом 2-Ю5— 3-105в четыреххлористом углероде — в диапазоне 103—ЗЛО1 pad. Вязкость растворов определялась с помощью вискозиметров Уббелоде или Оствальда. Сходные результаты были получены при использовании полиизобутилена марки «Vistanex» [85].
Рассмотренные системы характеризуются стабильностью до облучения и после него. Их показания не зависят от температуры облучения, мощности дозы и наличия кислорода в растворе.
В 1950 г. А. Прево [86] предложил использовать для целей дозиметрии явление радиационной полимеризации. Однако, по мнению А. Чарлзби [87], измерения здесь сильно затруднены зависимостью скорости процесса от мощности дозы и наличия примесей в мономерах. Согласно М. Талат-Эрбену и др. [88], для дозиметрии у-излучения можно применять процесс полимеризации метакрилонитрила с последующей регистрацией оптического поглощения кетиминных связей /С—C=N— полимера при длине волны 4950 нм. Этим методом можно измерять дозы до 2-107 рад. В цитированной работе мономер тщательно очищался, но даже в этом случае точность определений дозы составляет всего +10%. К тому же не было изучено влияние мощности дозы в достаточно широком интервале.
При облучении мономеров или полимеров, в которых преобладают соответственно процессы полимеризации или сшивания, при достаточно высоких дозах имеет место гелеобразование. Поскольку это наблюдается, как правило, при вполне определенных дозах, то было предложено [89—92] .использовать такие системы в дозиметрах с «конечной точкой» или дозиметрах-индикаторах, т. е. для установления того момента, когда исследуемый объект получит данную дозу. В этом методе в систему вво
дится металлический шарик или в ней создается пузырек воздуха. Когда происходит гелеобразование, то шарик или пузырек остаются неизменными, если ампулу перевернуть. В качестве примера рассмотрим дозиметр, описанный в работе [89]. Он представляет собой небольшой прозрачный желатиновый сосуд, заполненный жидкой стирол-полиэфирной смесью. Внутри нее помещен металлический шарик. В результате облучения жидкость частично затвердевает. При перевертывании сосуда шарик опускается до границы раздела между жидкостью и твердым телом. После соответствующей калибровки дозу можно определить путем измерения положения шарика. «Конечная точка», т. е. полное затвердение жидкости, зависит от состава смеси. С помощью этого дозиметра можно находить дозы в диапазоне 1,5-104—2,5-10® рад. По-видимому, такие дозиметры могут найти применение в радиационнохимической технологии.
ЛИТЕРАТУРА
1. R. Н. Schuler, А. О. Allen. J. Am. Chem. Soc., 77, 507 (1955).
2. H. A. Dewhurst. J. Phys. Chem., 61, 1466 (1957).
3. J. Preve, G. Gaudemaris. Compt. rend., 250, 3470 (1960).
4. W.G. Burns, J. R. Parry. Nature, 201, 814 (1964).
5. A. W. Boyd, H. W. J. Connor. Can. J. Chem., 42, 1418 (1964).
6. A. W. Boyd, C. Willis, O. A. Miller, A. E. Rothwell. Adv. Chem. Ser., 82, 456 (1968).
7. E. Proksch, H. Bildstein. Intern. J. Appl. Radiation Isotopes, 16, 56 (1965).
8. R. A Holroyd, К. G. Golliher, A. W. Thiele. Trans. Am. Nucl. Soc., 10, 54 (1967).
9. P. J. Dyne, J. A. Stone. Can. J. Chem., 39, 2381 (1961).
10. A. W. Boyd, H. IP. J. Connor, J. J. Pieroni. CRNL Rep. AECL — 2203 (1965).
11. В. И. Макаров, Л. С. Полак. Нефтехимия, 7, 219 (1967).
12. С. Willis, О. A. Miller, А. Е. Rothwell, A. W. Boyd. Radiation Res., 35, 428 (1968).
13. W.G. Burns, J. D. Jones. Trans. Faraday Soc., 60, 2022 (1964).
14. R. H. Schuler. Trans. Faraday Soc., 61, 100 (1965).
15. T. Gdumann, R. H. Schuler. J. Phys. Chem., 65, 703 (1961).
16. G. H. Miller, J. J. Martin. Ann. N. Y. Acad. Sci., 105, 629 (1963).
17. E. П. Ковалева. Автореферат канд. дисс. Минск, Ин-т ядерной энергетики АН БССР, 1972.
18. Д Тэплин. Радиационная дозиметрия (под ред. Дж. Хайна и Г. Браунелла). М., ИЛ, 1958, стр. 298.
19. Дж. Тэплин. Доклады иностранных ученых на Международной конференции по мирному использованию атомной энергии (Женева, 1955). Дозиметрия ионизирующих излучений. М., Гостехтеориздат, 1956, стр. 248.
20. В. В. Генералова. Дозиметрия больших доз. Ташкент, «Фан», 1966, стр. 29.
21. Е. Proksch. Solid State and Chemical Dosimetry in Medicine and Biology. Vienna, IAEA, 1967, p. 307.
22. E. Proksch. Intern. J. Appl. Radiation Isotopes, 22, 241 (1971.)
23. P,Gehringer, P. Krenmayr, E. Proksch. Proc. Ill Tihany Symposium» on Radiation Chemistry. Budapest, 1972, p. 371.
f 24. H. А. Бах, Г. Г. Бабичева, В. А. Ларин. Докл. АН СССР, 134, 1079 (1960).
25. II. А. Бах, Г. Г. Бабичееа, В. А. Ларин. Труды II Всесоюзного совещания по радиационной химии. М.. Пзд-во АН СССР, 1962, стр. 738.
26. W. L. McLaughlin. Manual on Radiation Dosimetry (Ed. by N. W. Holm and R. J. Berry). N. Y., Marcel Decker, Inc., 1970, p 377.
27. W. L. McLaughlin, E. Hussmann, H. II. Eisenlohr, J. Chalkley. Intern. J. Appl. Radiation Isotopes, 22, 135 (1971).
28. W. L. McLaughlin. Dosimetry in Agriculture, Industry, Biology and Medicine. Vienna, IAEA, 1973, p. 362.
29. G. L. Clark. P. E. Bierstedt. Radiation Res., 2, 199 (1955).
30. J. Wilkinson, II. J. Fitches. Nature, 179, 863 (1957).
31. D. Hoffmann, C.F. Michel. Strahlenther., 112, 54 (1960).
32. J. Patko, Z. Szentirmay, Z. Dezsi. Acta Univ. Debrecen., Ser. Phys. Cbim., 11, 27 (1965).
33. S. Kawanishi. Nippon Igaku Hoshasen, 20, 1035 (1965); цит. no Nucl. Sci Abstr., 20, 25417 (1966).
34. D. Bertram. Kernenergie, 2, 329 (1959).
35. D. Bertram. Kernenergie, 3, 556 (I960).
36. G. L. Clark, P. E. Bierstedt. Radiation Res., 2, 295 (1955).
37. Z. Spumy. Jaderna Energ., 4, 294 (1958).
38. G. Orban, J. Patko, I. Berta. Magyar Radiol., 14, 162 (1962); цит. no Nucl. Sci. Abstr., 18, 12475 (1964).
39. A. Chapiro, J. W. Boag, M. Ebert, L. H.Gray. J. Chim. phys., 50, 468 (1953).
40. L. Bouby, A. Chapiro. J. Chim. phys., 52, 645 (1955).
41. E.K.Gustorf. Z. Naturforsch., 19, 170 (1964).
42. J. F. Suttle. Rep. LA-1615 (1953).
43. J.F. Kircher, B. W. King, M. J. Oestman. P. Schall, G. D. Calkins. Proc. II United Nations Intern. Conference on Peaceful Uses of Atomic Energy, vol. 21. Geneva, 1958, p. 199.
44. W. H. Cropper, A. W. Snyder. Rep. AECL — 802 (1959).
45. M. Corval, A. Chapiro, C. Cousin. Compt. rend., 235, 799 (1952).
46. A. McLachlan. J. Org. Chem., 29, 1598 (1964).
47. Atlantic Research Nucl. Corp., TID — 17356 (1962); цит. по R. K. Brosz-kiewicz. Chemiczne Metody Dosimetrii Promieniowania Jonizujacego. Warszawa, 1971.
48. J. Morton, A. Golub. J. Am. Chem. Soc., 80, 1794 (1958).
49. I. Dvornik, U. Zee, A. Anic, F. Ranogafec. Solid State and Chemical Dosimetry in Medicine and Biology. Vienna, IAEA, 1967, p. 273.
50. I. Dvornik, U. Zee, M. Baric, D. Razem. Handling of Radiation Accidents. Vinna, IAEA, 1969, p. 225.
51. I. Dvornik. Manual on Radiation Dosimetry (Ed. N. W. Holm and R. J. Berry). N. Y., Marcel Decker, Inc., 1969, p. 345.
52. I. Dvornik, U. Zee, F. Ranogafec. Food Irradiation. Vienna, IAEA, 1966, p. 81.
53. I. Dvornik, D. Razem, M. Baric, Large Radiation Sources for Industrial Processes. Vienna, IAEA, 1969, p. 613.
54. D. Razem, I. Drornik. Radiation Preservation of Food. Vienna, IAEA, 1973, p. 537.
55. D. Razem, I. Dvornik. Dosimetry in Agriculture, Industry, Biology and Medicine. Vienna, IAEA, 1973, p. 405.
56. G. Foldiak, Z. Horvath, V. Stenger. Ibid., p. 367.
57. F. J. Feng. Nucleonics, 16, No. 10, 114 (1958).
58. L. Wiesner. Selected Topics in Radiation Dosimetry. Vienna, IAEA, 1961, p. 361.
59. В. Крамер. Углеводороды. Аспекты радиолиза (под ред. Ю. Уанье и Т. Гейманна). М., «Мир», 1972, стр. 163.
60. L. J. Forrestall, W. Н. Hamill. J. Am. Chem. Soc., 83, 1535 (1961).
61. M. Burton, J. Y. Chang, S. Lipsky, M. P. Reddy. Radiation Res., 8, 203 (1958).
62. J. S. Mehlin, S. Lipsky, J. Phys. Chem., 68, 3297 (1964).
63. S.K.Ho. G. R. Freeman. Ibid., p. 2189.
64. M. Cher, C. S. Hollingsworth. B. Browning. J. Chem. Phys., 41, 2270 (1964).
65. R. H. Schuler. J. Phys. Chem., 61, 1472 (1957).
66. J. Kroh, S. Karolczak. Bull. Acad. Polon. Sci., Ser. Chim., 12, 157 (1964).
67. G. R. Freeman. J. Chem. Phys., 33, 71 (1960).
68. E. S. Weight, P. Walker. J. Chem. Soc., 1960, 2225.
69. P. J. Dyne, J. Denhartog. Can. J. Chem., 40, 1616 (1962).
70. G. E. Adams, J. H. Baxendale, R. D. Sedgwick. J. Phys. Chem., 63, 854 (1959).
71. С. E. Klotz, Y. Raef, R. H. Johnsen J. Phys. Chem., 68, 2040 (1964).
72. W. S.Guentner, T. J. Hardwick, P. P. Nejak. J. Chem. Phys., 30, 601 (1959).
73. R. H. Schuler. Proc. II Intern. Conference on Peaceful Uses of Atomic Energy, vol. 21. Geneva, 1958, p. 213.
74. J. P. Manion, M. Burton. J. Phys. Chem., 56, 560 (1952).
75. H. A. Dewhurst. .1. Phys. Chem., 63, 813 (1959).
76. P. Lutgen. P. Claes. Bull. Soc. Chim. Beiges, 74, 581 (1965).
77. E. Proksch. Solid State and Chemical Dosimetry in Medicine and Biology, Vienna, IAEA, 1967, p. 316.
78. M. Hamashima, M. P. Reddy, M. Burton. J. Phys. Chem., 62, 246 (1958).
79. P. J. Dyne, J. W. Fletcher. Can. J. Chem., 38, 851 (1960)
80. Ю. Уанъе. Углеводороды. Аспекты радиолиза (под ред. Ю. Уанье и Т. Геймапна). М., «Мир», 1972, стр. 68.
81. Т. Gaumann. Helv. Chim. Acta, 46, 2873 (1963).
82. G. Foldiak. Large Badiation Sources for Industrial Processes. Vienna, IAEA, 1969, p. 621.
83. Zs. Horvath, E. Banyai, G. Foldiak. Radiochim. Acta, 13, 150 (1970).
84. P. Alexander, A. Charlesby, M. Ross Proc. Roy. Soc., A223, 392 (1954).
85. A. W. Carviker. Rev. Sci. Instrum., 37, 1045 (1966).
86. A. Prevot. Compt. rend., 230. 288 (1950).
87. А. Чарлзби. Ядерпые излучения и полимеры. М.. ИЛ, 1962, стр. 108.
88. М. Talat-Erben, Е. Vnseren, N. Seber. Solid State and Chemical Dosimetry in Medicine and Biology. Vienna, IAEA, 1967, p. 301.
89. F. E. Hoecker, I. W. Atkins. Intern. J. Appl. Radiation Isotopes, 3, 31 (1958).
90. F. E. Hoecker, H. L. Hiebert. Radiology, 72, 254 (1959).
91. F. E. Hoecker. Radiology, 76, 116 (1961).
92. F. E. Hoecker. Health Phys., 8, 381 (1962).
Глава VII
ГАЗООБРАЗНЫЕ ВЕЩЕСТВА
КАК ДОЗИМЕТРИЧЕСКИЕ СИСТЕМЫ
В радиационной химии (преимущественно при исследовании радиолиза газов) находят применение в дозиметрических целях некоторые газообразные вещества. К их числу относятся углеводороды (главным образом метан [1, 2], /t-пентан [3], этилен [4—10] и ацетилен [5, 11—18]), закись азота [4, 7, 19—45], другие окислы азота [29, 46], кислород [47—491, воздух [29], газообразный сероуглерод [50]. Характеристики газообразных дозиметрических систем представлены в табл. 50.
1. Углеводороды
В случае метана, этана, н-пентана, этилена и пропилена величина дозы определяется по количеству водорода, образующегося в результате облучения. Наиболее удобно измерять возникающий водород методом газовой хроматографии.
При радиолизе метана [53] выход водорода не зависит от дозы до 5-107 рад, мощности дозы в исследованном интервале (от 10 до 8-104 рад!сек), температуры от 20 до 100° С и давления от 13 мм рт. ст. до 25 атм.
Из числа углеводородов в дозиметрическом отношении наиболее подробно исследован этилен. Для у-излучепия 60Со и электронов с энергией 1—2 Мзв измеренные значения G(H2) колеблются в пределах 1,2—1,33 молек./ЮО эв [5, 6, 8—10, 60—63]. По данным работ [5, 6], G(H2) не зависит от давления этилена в диапазоне от 0,2 до 15 атм. Изменение температуры также не оказывает влияния на величину G(H2). Выход водорода практически постоянен при обычных мощностях дозы (~102—105 рад!сек). Однако при высоких мощностях дозы (~1013—1014 рад/сек) G(H2) сильно зависит от давления [7](см. главу IX). В лаборатории Г. Фримэна [64] была проведена проверка ряда газообразных дозиметрических систем. Было найдено, что наиболее удобным из их числа является этилен (давление — несколько десятых атмосферы, температура 25° С).
При облучении ацетилена образуются преимущественно полимер высокого молекулярного веса и бензол; выход же водорода весьма мал (см., например, [65]). В результате этого давление газа в ячейке уменьшается. Эта особенность и используется в целях дозиметрии [5, 11—18]. С помощью ацетилена можно измерять
Таблица 50
Характеристики газообразных дозиметрических систем
Система Измеряемое превращение Выход превращения, молек.ДОО эе Диапазон доз, рад Диапазон мощностей дозы, рад1сек
Метай Образование Па 5,5—6,9 (12, 40, 51 -56] До 5-Ю7 10-8-101
Этан То же 3,6—3,7 [57, 58] —’ —
н-Пентап » 6 [59] — —
Этилен » 1,2—1,33 [5, 6, 8—10, 60—63] До 8-Ю7 102—10®
Пропилен » 1,1 [63] — —
Ацетилен Образование полимера и бензола, приводящее к уменьшению давления газа в ячейке G(-CaHa) = =70—110 [17] До ~ 107 Выход зависит от мощности дозы
Закись азота Используется в основном процесс образования Na G(Na) = 10 [26] 105—3-10® До 2-108
Кислород Образование Оз 6,2 [47, 49] До ~ 3-10“ При мощностях дозы Ю1*—1013 рад/сек выход существенно выше
Воздух Образование NOa 1,4—-1,45 (атмосферное давление, 20— 30е С) [29] До 1,5-10® —
Сероуглерод Разложение CSa 15,4 (23° С) J50] — Выход постоянен при обычных мощностях дозы
дозы до ~107 рад. Однако радиационно-химический выход процесса зависит от температуры, давления и мощности дозы. Так, по данным работы [17], при варьировании этих параметров G(—С2Н2) изменяется в интервале 70—110 молек./100 эв. Очевидно, это является существенным недостатком ацетиленовой дозиметрической системы.
2. Закись азота
N20 при радиолизе разлагается с образованием азота, кислорода и окиси азота. Общая начальная реакция разложения выражается уравнением
4NzO —» 3Na "Ь О2 -р 2NO. (1)
Окись азота затем окисляется в двуокись:
2NO + 0-2-> 2NO2. (2)
При сравнительно высоких давлениях реакция (2) в газовой фазе протекает быстро. Однако при давлениях ниже 10 мм рт. ст. эта реакция медленная, и она идет на стенках ячейки или в низкотемпературной ловушке, используемой в анализе. В этих условиях может иметь место и реакция
4NO + Ог —» 2N2O3. (3)
Впервые использование закиси азота в качестве дозиметрической системы было предложено П. Хартеком и С. Дондесом [19, 201. Позже возможность применения NO2 для дозиметрии ионизирующих излучений исследовалась многими авторами [4, 7, 21-45].
Имеющиеся в литературе данные о выходах продуктов радиолиза N2O противоречивы. Приводились следующие величины этих выходов для у-лучей 60Со и электронов (в молек./100 эв) при обычных мощностях дозы: G(—N2O) — Ю,9 4- 12,0119,28 , 29 , 39, 66]; G(N2) = 9,2 -г-12,4 [4, 19, 22-29, 33, 41, 42, 44, 66]; G(O2) = = 1,3 н-4,0 119, 24—26, 28, 33, 66]; G(NO) = 3,9 ~ 5,4 [24— 26, 331; G(NOa);= 2,3 4,4 [19, 25, 281; G(Na + Оа) = 11,4
-: 14,0 [19, 22, 26, 30]. Очевидно, эти расхождения вызваны тем, что на результаты измерения выходов существенное влияние оказывают условия проведения экспериментов. В частности, одной из причин может быть то обстоятельство, что на стенках ячеек могли присутствовать следы воды, которая ускоряет реакцию NO и О2 [231. Недавняя калибровка [26] этой системы в случае электронов с энергией 1 Мэв дала следующие значения начальных выходов: G(N2) = 10,0 + 0,2 молек./100 эв; G(O2) = 4,0 ± ± 0,4 молек./100 эв; G(NO) = 3,9 ± 0,3 молек./100 эв и G(--N2O) = = 12,0 + 0,4 молек./100 эв.
Выходы продуктов радиолиза N2O не зависят от температуры в диапазоне от —81 до 120ц С [22]. При более высоких температурах выход неконденсирующихся газов (N2 и О2) резко возрастает [22]. Согласно [26], G(N2) равен 10 молек./100 эв при 24° С и 20 молек./ /100 эв при 200° С. По данным работы [35], G(N2) при 376° С примерно в 8 раз больше, чем при 40° С. Низший температурный предел использования этой системы определяется точкой кипения N2O(—88,5° С). На выход неконденсирующихся газов не оказы
вает влияния изменение давления от 50 мм рт. ст. до почти критического его значения [19, 23].
Выходы не зависят от дозы до ~3-108 рад. При дозе ~3-109 рад устанавливается равновесное состояние, поскольку азот, кислород и окислы азота вступают в обратные реакции [67, 681. Мощность дозы вплоть, по крайней мере, до ~2-108 рад!сек не оказывает влияния на величину G(N2) [7, 36, 45] (см. главу IX). Выход характеризуется практически одинаковыми значениями для различных видов излучения. Благодаря этому, N2O может применяться для измерения доз тяжелых заряженных частиц и нейтронов (см. главы X и XI). Наконец, закись азота весьма устойчива при хранении.
Для анализа продуктов радиолиза N.2O можно использовать различные методы. Можно применять обычную технику измерения давления [19, 20]. После облучения ячейка присоединяется к вакуумной системе. Неразложившаяся закись азота и появившаяся при радиолизе двуокись азота конденсируются в ловушках, охлаждаемых жидким азотом. Давление азота и кислорода находится при помощи манометра. Затем в систему вводится водород, и после его сжигания определяется соотношение между азотом и кислородом. Согласно 1211, для переноса несжижаю-щихся газов из облученной ячейки в вакуумную систему можно применять насос Тепплера. С. В. Стародубцев и Е. В. Яковлева [30, 31] для регистрации доз использовали термопарные лампы ЛТ-2, наполненные закисью азота. Через час после облучения окислы азота вымораживались без вскрытия ампул, а количество азота и кислорода измерялось после установления температурного равновесия с помощью вакууметра. Если вымораживание производить сразу же после облучения, то получаются невоспроизводимые результаты (по-видимому, из-за реакции NO с О2). Выход N2 + О2, измеренный в этих работах, равен 11,7 молек./ /100 эв (точность ±Ю%). Варьирование давления Х2О от 0,1 до 760 мм рт. ст. позволяет определять дозы 104—109 рад. При больших дозах измерения можно производить колориметрически без вскрытия ампулы (по окраске образовавшейся двуокиси азота) [19, 20]. Наконец, анализ можно осуществить хроматографическим (например, применением колонок из молекулярных сит [23]) и масс-спектрометрическим методами.
При проведении дозиметрии необходимо применять медицинскую закись азота. Перед употреблением она должна быть очищена путем нескольких циклов замораживания и размораживания. Ячейка должна быть «промыта» очищенной закисью азота.
Облучение закиси азота проводят обычно в ячейках из стекла или кварца. При работе с ячейками из нержавеющей стали необходимо помнить о том, что этот материал оказывает каталитическое действие на процесс разложения закиси азота [4]. По данным Ф. Лампе и др. [4], прирост выхода азота AG(N2) за счет
atofo эффекта выражается следующей эмпирической формулой:
„ 4,2
AG(.V2)=-^- (4)
где р—давление N2O (в атм) и I — мощность дозы (в Мрад/час).
Из формулы (4) следует, что при р == 1 атм и I 10 Мрад/час значение AG(Na) ничтожно по сравнению с обычной величиной ^(Ng), равной ~Ю молек./ЮО эв. Максимальная мощность дозы, создаваемая источниками у-из л учения 60Со, которые используются в радиационной химии, как раз и составляет ~10 Мрад/час. Поэтому при работе с у-лучами 60Со применять ячейки из нержавеющей стали для проведения дозиметрии с закисью азота не рекомендуется.
Одним из недостатков рассматриваемой дозиметрической системы является то, что на результаты измерений может оказывать влияние так называемый стеночный эффект. Этот эффект может сильно сказаться, если выход разложения NaO рассчитывается, исходя, например, из показаний дозиметра Фрикке или другого дозиметра на основе водного раствора. По данным Дж. Хирна и Р. Хаммела [23], при расчете дозы с учетом различий в электронных плотностях NaO и дозиметра Фрикке G(N2) Для у-лучей 60Со составляет 15,9 молек./ЮО эв, если применяется цилиндрическая ячейка объемом 42,7 мл, и 11,8 молек./ЮО эв при использовании ячейки объемом 353,5 мл.
Как отмечается в обзоре [691, для ячеек объемом 100 мл или менее разложение N2O под действием у-лучей 60Со происходит преимущественно за счет вторичных электронов, возникших в стенках ячейки. Слоя стекла или кварца в 1— 2 .м.ч (обычная толщина ячеек) достаточно, чтобы поглотить электроны, генерированные вне ячейки. В этих условиях концентрация продуктов радиолиза должна быть почти пропорциональна объему ячейки. Говоря по-иному, ячейка с N2O должна быть прокалибрована в известном поле у-излучения. Очевидно, подобным недостатком страдают и другие дозиметры на основе газообразных систем.
3. Другие окислы азота
М. Т. Дмитриев [291 допускает возможность применения других окислов азота (NO и NO2) для дозиметрии ионизирующего излучения. При у-радиолизе NO (давление 1 атм, температура 20° С) выходы равны [291: G(-NO) = 14,8; G(N.>) = 3,8; G(N2O) = = 0,4 и G(NOa) = 7,2 молек./ЮО эв. Выходы мало зависят от температуры в диапазоне от 0 до 450° С. Для выходов радиолиза NO2 характерны существенное увеличение с ростом температуры и заметное уменьшение при увеличении давления. Однако в работе [39] не исследовалось влияние мощности дозы па ход радиолиза этих окислов. Отсутствие этих сведений препятствует использованию NO и NO2 в дозиметрических целях.
4. Кислород и азотно-кислородные смеси
Возможность применения газообразного кислорода в качестве дозиметрической системы обсуждалась в работах [47—49]. При его облучении возникает озон. Последний легко определяется иодидным способом или прямым спектрофотометрическим методом, причем первый метод более чувствителен, чем второй. Недавно А. Бойд и др. [70] подробно исследовали стехиометрию реакции между и О3. Они нашли, что при pH 7 одна молекула О3 выделяет 1,53 молекулы J2. Эту стехиометрию необходимо учитывать при проведении анализов озона. Для определения озона А. Бойд и др. [70] использовали следующую методику. Ячейка с облученным газом охлаждается жидким азотом, и к полученной жидкости через кран добавляется несколько миллилитров иодидного реактива. Смесь встряхивается до тех пор, пока пе достигнет комнатной температуры. Затем содержимое ячейки переносится в мерную колбу и разбавляется до метки водой. Ионы J3» возникающие в результате реакции между О3 и J-, определяются спектрофотометрически по поглощению света при длине волны 358 нм. Во втором методе используется то обстоятельство, что газообразный озон имеет оптическую полосу поглощения в ультрафиолетовой части спектра с максимумом при 254 нм. Коэффициент экстинкции при этой длине волны и нормальных температуре и давлении равен 132 [17] или 135 см^1 [72].
Выход О3 при обычных мощностях дозы измерялся многими авторами [47, 49. 73—76]. По последним данным [47, 49], G(O3) при этих условиях равен 6,2 ± 0,6 молек./ЮО эв. Он пе зависит от температуры в исследованном диапазоне (77 ч~ 298° К). При высоких мощностях дозы (~Ю1а ч- 10,я рад! сек) G(OS) гораздо больше: он равен 12,8—13,8 молек./ЮО эв [47—49, 77] (см. главу IX).
Дозиметрические характеристики азотно-кислородных смесей, включая воздух, были исследованы М. Т. Дмитриевым [29]. При их радиолизе образуются NO, NO2, N2O и О3. По данным [29], дозу наиболее удобно измерять по концентрации образующейся двуокиси азота. Она может быть определена различными методами: манометрическим (вакуумная разгонка), спектрофотометрическим (поглощение света с длинами волн 390—450 нм) и химическим (растворение NO„ в воде и последующий анализ ионов NO2). Выход NO2 постоянен до дозы ~l,5*109 рад (воздух, у-излучение 60Со, давление 1 атм). Однако на величину G(NO?) оказывает влияние температура облучения. Как следует из работы [29],’для воздуха при давлении 1 атм G(NO2) в случае у-излучения 60Со увеличивается от 1,35 до 1,46 молек./ЮО эв при пеоеходе от 0 к 30° С; для 200° С выход равен 3,3 молек./ЮО эв, а для 750° С — 0,7 молек./ЮО эв. Кроме того, G(NO2) зависит от состава смесей и их давления. Зависимость G(NO2) от мощности дозы в работе [29] не исследоваласд.
Согласно [78J, при мощности дозы электронного излучения ~3-1012 рад/сек выход NO, в азотно-кислородных смесях весьма мал. Например, при дозе 1,6-10® рад G(NO2) < 0,15 молек./ /100 эв. В этих условиях главным продуктом радиолиза является озон. При указанной мощности дозы G(O3) для воздуха при комнатной температуре и давлениях выше ~ 0,4 атм равен 10,3 молек./100 эв (для у-радиолиза этой системы в тех же условиях G(O3) составляет ~3,5 молек./100 эв [291).
5. Сероуглерод
Согласно [50J, сероуглерод в газовой фазе может использоваться в качестве дозиметрической системы. При радиолизе CS? разлагается с образованием серы и полимерного продукта, в результате чего происходит уменьшение давления в ячейке. Это уменьшение и служит мерой дозы.
Было найдено, что для у-лучей ®°Со, электронов и протонов с энергией ~3 Мэв G(—CS2) = 15,4 молек./100 эв при 23° С. Выход не зависит от мощности дозы при обычных ее значениях. Поглощенная энергия Е (в эв) рассчитывается, исходя из величины уменьшения давления Др при комнатной температуре, с помощью формулы
£ = 2,12 - 1017гДд, (5)
где v — объем газа (в ел3); Др измерено в торрах.
ЛИТЕРАТУРА
1. В. L. Tarmy. Ind. Eng. Chem., Int. Ed., 53, 147 (1961).
2. R. W. Hummel, J. A. Hearne. Chem. Ind., 1961, 1827.
3. R. A. Back, E. A. Cherniak, E. Collinson, W. Cooper, F. S. Dainton, G. M. Meaburn, IN. Miller, W. H. Stafford, G. Л. Swen, P. S. Timmons, D. C. Walker, D. Wright. Proc. II United Nations Intern. Conference on Peaceful Uses of Atomic Energy, vol. 29. Geneva. 1958, p. 125.
4. F. W. Lampe, L. Ketan, E. R. Weiner, W. H. Johnston. .1. Phys. Chem., 71, 1528 (1967).
5. M. C. Sauer, L. M. Dorfman. J. Phys. Chem., 66, 322 (1962).
6. 5. Srinivasan, W. E. Smith. Intern. J. Appl. Radiation Isotopes, 17, 643 (1966).
7. C. Willis, O. A. Miller, A. E. Rothwell, A. W. Boyd. Adv. Chem. Ser., 81, 539 (1968).
8. R. A. Back, T. W. Woodward, K. A. McLaughlan. Can. J. Chem., 40, 1380 (1962).
9. W. J. Holtslander, G. R. Freeman. Can. J. Chem., 45, 1649 (1967).
10. A. R. Anderson, J. U. T. Best. Nature, 216, 576 (1967).
11. L. M. Dorfman, F. J. Shipko. J. Am. Chem. Soc., 77, 4723 (1955).
12. F. W. Lampe. J. Am. Chem. Soc.. 79, 1055 (1957).
13. F. W. Lampe. Nucleonics, 18, No. 4, 60 (1960).
14. F. W. Lampe. Radiation Res., 10, 691 (1959).
15. A. Delle Site, A. Mele. J. Phys. Chem., 69, 4033 (1965).
16. G. J. Mains, H. Niki, M. H. Wijnen. J. Phys. Chem., 67, 11 (1963).
17. F. H. Field. J. Phys. Chem., 68, 1039 (1964).
18. Сб. «Радиолиз углеводородов» (под ред. А. В. Топчиева и Л. С. Полака). М., Изд-во АН СССР, 1962, стр. 189.
19. Р. Harteck, S. Dondes. Nucleonics, 14, No. 3, 66 (1956).
20. С. Дондс. Материалы Международной конференции по мирному использованию атомной энергии (Женева, 1955), т 14. М., Физматиздат, 1958, стр. 213.
21. R. Е. Simpson. Health Phys., 8, 143 (1962).
22. D. A. Flory. Nucleonics, 21, No. 12, 50 (1963).
23. R. W. Hummel, J. A. Hearne. Radiation Res., 15, 254 (1961).
24. G. P. Burtt, J. P. Kircher. Radiation Res., 9, 1 (1957).
25. G. R. A. Johnson. J. Inorg. Nucl. Chem., 24, 461 (1962).
26. F. T. Jones, T. J Sworski. J. Phys. Chem., 70, 1546 (1966).
27. D A. Flory. Radiation Res., 14, 466 (1961).
28. M. T. Дмитриев, Л. В. Сараджев. Ж. физ. химии, 35, 727 (1961).
29. М. Т. Дмитриев. Атомная энергия, 15, 52 (1963).
30. С. В. Стародубцев, Е. В. Яковлева. Дозиметрия больших доз. Ташкент, «Фан», 1966, стр. 91.
31. С. В. Стародубцев, Е. В. Яковлева. Дозиметрия интенсивных потоков ионизирующих излучений. Ташкент. «Фан», 1969, стр. 29
32. И7. J. Holtslander, G. R. Freeman. Can. J. Chem., 45, 1661 (1967).
33. N. Takao, S. Shida, Y. Hatano, H. Yamazaki. Bull. Chem. Soc. Japan, 41, 2221 (1968).
34. A. W. Boyd. Nucleonics, 22, No. 7, 6 (1964).
35. R. Garden, P. Ausloos, J. Res. Natn. Bur. Stand.. 69A, 79 (1965).
36. C. Willis, A. W. Boyd, D. A. Armstrong. Can. J. Chem., 47, 3783 (1969).
37. E. П. Петряев, E. П. Ковалева. Пзв. АН БССР, серия физ.-энерг. н., № 1, 49 (1970).
38. Е. В. Сазонова. Радиационная дозиметрия и спектрометрия ионизирующих излучений. Ташкент, «Фан», 1970, стр. 183.
39. М. Т. Дмитриев. Ж. прикл. химии, 33, 808 (1960).
40. L. Dolle. Proc. Il United Nations Intern. Conference on Peaceful Uses of Atomic Energy, vol. 29. Geneva, 1958, p. 367.
41. R. W. Hummel, J A. Hearne Nature, 188, 734 (1960).
42. F Moseley, A. E. Truswell. AERE Rep.— 3078 (1960).
43. I. Takatani. Genshiryoki Kogyo, 9, 60 (1963): цит. no Nucl. Sci. Abstr., 17, 20364 (1963).
44. K. Furukawa, S. Shida. J. Nucl. Sci. Tech. (Tokyo), 3, 41 (1966).
45. A. W. Boyd, C. Willis, O. A. Miller. Can. J. Chem., 47, 351 (1969).
46. T. Takaga, J. SekimitiJ Genshiryoki Kogyo, 9,60 (1963); цит. no Nucl. Sci. Abstr., 19, 13433 (1965).
47. A. W. Boyd, C Willis, R. Cyr, D. A. Armstrong Can. J. Chem., 47, 4715 (1969).
48. J. A. Ghormley, C. J. Hochanadel, J. W. Boyle. J. Chem. Phys., 50, 419 (1969).
49. C. Willis, A. W. Boyd, M. J. Young, D. A. Armstrong. Can. J. Chem., 48, 1505 (1970).
50. O. Janssen, A. Henglein, D. Perner. Z. Naturforsch., 19b, 1005 (1964).
51. K. Yang, P. J. Manno. .1. Am. Chem. Soc., 81, 3507 (1959).
52. P. Ausloos, R. Gorden, S. G. Lias. J. Chem. Phys., 40, 1854 (1964).
53. J. Maurin. J. Chim. phys., 59, 15 (1962).
54. R.W. Hummel. Disc. Faraday Soc., 36, 75 (1963).
55. P. S. Rudolph. Adv. Chem. Ser., 82, 101 (1968).
56. R. W. Hummel. Trans. Faraday Soc., 62, 59 (1966).
57. H. Gillis. J. Phys. Chem., 67, 1399 (1963).
58. R. A. Holroyd, R. W. Fessenden. Ibid., p. 2743.
59. J. H. Futrell. J. Phys. Cheiu , 64, 1634 (1960).
60. G. G. Meisels. J. Am. Chem. Soc., 87, 950 (1965).
61. G.G. Meisels. J. Chem. Phys., 41, 51 (1964).
62. L. M. Dorfman, M. C. Sauer. J. Chem. Phys., 32, 1886 (1960).
бЗ. К. Yang, Р. L. Gant. J. Phys. Chem., 65, 1861 (1961).
64. G. R. Freeman. Actions Chimiques et Biologiques des Radiations (Ed M. Haissinsky), 14 Serie. Paris, 1970, p. 73.
65. G. R. Freeman. Radiation Res. Rev., 1, 1 (1968).
66. P. Harteck, S. Dondes. J. Chem. Phys., 27, 546 (1957).
67. P. Harteck, S. Dondes. J. Chem. Phys., 29, 234 (1958).
68. P. Harteck, S. Dondes. J, Phys. Chem., 63, 956 (1959).
69. H. Fricke, E. J. Hart. Radiation Dosimetry (Ed. F. H. Attix and W. C. Roesch), vol. 2. N. Y., 1966, p. 167.
70. A. W. Boyd, C. Willis, R. Cyr. Analyt. Chem., 42, 670 (1970).
71. W. B. DeMore, O. Paper. J. Phys. Chem., 68, 412 (1964).
72. M. Griggs. J. Chem. Phys., 49, 857 (1968).
73. J. F. Kircher, J. S. McNulty, J. L. McFarling, A. Levy. Radiation Res., 13, 452 (1960).
74. G. R. A. Johnson, J. M. Warman. Disc. Faraday Soc., 37, 87 (1964).
75. J. 7'. Sears, J. W. Sutherland. J. Phys. Chem., 72, 1168 (1968).
76. H. А. Бунеев, С. Я. Пшежецкий, И. А. Мясников. Сб. «Действие ионизирующих излучений на неорганические и органические системы». М., Изд-во АН СССР, 1958, стр. 129, 133.
77. G. М. Meaburn, D. Perrier, J. LeCalve, М. Bourene. J. Phys. Chem., 72, 3920 (1968).
78. C. Willis, A. W. Boyd, M. J. Young. Can. J. Chem., 48, 1515 (1970).
Глава VIII
ДОЗИМЕТРЫ НА ОСНОВЕ ТВЕРДЫХ ТЕЛ
При действии ионизирующего излучения на твердые тела в них в зависимости от природы происходят различные превращения: изменяется окраска, возникают парамагнитные частицы, появляется способность светиться при нагревании и т. д. Эти превращения находят широкое применение в дозиметрии.
Из числа дозиметров на основе твердых тел наибольшее распространение получили полимеры, галогениды щелочных и щелочноземельных металлов, стекла. Чаще всего в целях дозиметрии используются следующие явления, происходящие с ними в результате облучения: окрашивание, обесцвечивание, радиофотолюминесценция (свечение при действии света), радиотермолюми-
Таблица 51
Характеристики наиболее распространенных твердых дозиметрических систем
Система Измеряемое явление Диапазон доз, рад Диапазон мощностей дозы, рад/сек
Фосфатное стекло с добавкой Ag Боросиликатное стекло с до- Окрашивание 2-103—10® До 105
То же 103—5-Ю5 До 10®
бавкой Со
Полиметилметакрилат » 5-Ю3—3-Ю’ До Ю8
Полистирол » 10s—Ю8 —
Целлофан с добавкой краси- Изменение ок- 5-10’—3-Ю7 До ~ 10”
теля раски
LiF Радиотермолюминесценция 0,01—5-10s До lO’i—1012
CaFa: Ми То же 5-10"»—103 До 102
Фосфатное стекло с добавкой Ag (с высоким атомным номером) Радиофотолюминесценция 10-2-10’ До 105
Фосфатное стекло с добавкой Ag (с низким атомным номером) То же 0,1—103 До 105
Антрацен Деградация люминесценции Ю5—5-107 До 103
несценция (свечение при нагревании), деградация люминесценции (уменьшение интенсивности свечения некоторых люминофоров). В табл. 51, составленной по данным Г1J, приведены общие характеристики некоторых распространенных дозиметров на основе твердых тел.
Отметим, что в настоящее время дозиметры на основе стекол и кристаллов относят к так называемым твердофазным дозиметрам, и метод измерения дозы с их помощью есть один из физических методов дозиметрии. Полимерные дозиметры рассматриваются обычно как химические дозиметры.
1. Полимеры
Применению полимеров в целях дозиметрии ионизирующих излучений посвящена обширная литература [2—158J. Из нее следует, что практически все обычные полимеры — полиметилметакрилат, поливинилхлорид, полиэтилен, полистирол и т. п.— могут служить в качестве дозиметрических систем.
Известны полимерные дозиметры различных типов: твердые полимеры в виде пластинок, цилиндров и т. п. (как с добавками, так и без них), полимерные пленки, окрашенные полимерные пленки, жидкие полимеры и растворы полимеров. Особенно часто для дозиметрии используется свойство ряда полимеров изменять оптические спектры поглощения в ультрафиолетовой и видимой областях под действием ионизирующего излучения. При этом, как правило, имеет место линейная зависимость между измене нием оптической плотности и величиной дозы. Доза может быть найдена измерением и других превращений, происходящих в некоторых полимерах при облучении: путем определения вязкости раствора облученного полимера (полиметилметакрилат, полиизобутилен), увеличения поверхности полимерных пластинок (полиэтилен), концентрации свободных радикалов, возникших при радиолизе (политетрафторэтилен), возрастания веса полимера при облучении в атмосфере кислорода (полиэтилен) и т. п.
В табл. 52 приведены характеристики некоторых дозиметрических систем рассматриваемого типа. Как видно из таблицы, эти системы могут служить для нахождения больших доз.
Дозиметры на основе полимеров, несмотря на многие достоинства, имеют ряд недостатков. Во-первых, их показания зависят от сорта пластмассы. Промышленные сорта многих полимеров содержат пластификаторы, примеси мономеров и т. п. Естественно, это оказывает влияние на результаты определения дозы. Поэтому для точных измерений необходимо осуществлять калибровку каждой партии используемого промышленного полимера или же синтезировать полимер в лаборатории. Во-вторых, для некоторых облученных полимеров характерны пост-эффекты. В частности, это наблюдается в случае полиметилметакрилата, полистирола и поли* этилентерефталата.
Характеристики полимерных дозиметрических систем
Система Измеряемое превращение Диапазон доз, рад Литература
Полпметилметакрп-лат *1 или полиизобутилен Изменение вязкости раствора облученного поли мера До 108 [2, 28]
Полиакриламид Деструкция полимера в водном растворе 50 -7,5-10» [31]
ГГолпметил метакрилат или полистирол Изменение оптической плотности в области 390—450 нм 5-10!—2-107 [3]
Полистирол Изменение оптической плотности при 330 нм 6-10»— 4,2-107 [144]
Полиметилметакрп-лат Изменение оптической плотности при 280, 292, 300 или 314—316 нм 10s—3-101 [4, 5, 27, 50, 53, 127,132 140,142,144]
Полиэтилентерефта лат *2 Изменение оптической плотности при 325—330 нм 10»—10» [4, 5, 132]
То же » 5-10>—10» 10»—4-10’ [17] [152]
Поливинилхлорид Изменение оптической плотности при 396 нм 10 s—107 [8,18, 132 140]
Сополимер поливинилхлорида с полиметилметакрилатом, содержащий 7% пластификатора (вини-проз) Изменение оптической плотности при 390 нм 5.10 s—2-Ю7 [147]
Поливинилхлорид, содержащий 15% пластификатора (винипласт) То же 5-10’—2-107 [147]
Полихлорвинилвини-лиден (C4HsCl3)n Изменение оптической плотности при 260 нм 10‘—107 [57]
Поливинилфторид Изменение оптической плотности при 315 нм До З-Ю7 [56]
Полиизобутилен, полиметилметакрилат, стирол (-изобутилен Изменение вязкости раствора облученного полимера 10s—5-108 [6]
Поливинилпирроли-дон Изменение оптической плотности при 350—380 нм Ю»—108 [138]
Полиамид (CsHnNO)n Изменение оптической плот ности при 315 нм 10s—1010 [42]
Система Измеряемое превращение Диапазон доз, рад Литература
Диацетат и триацетат целлюлозы Изменение оптической плотности при 280 нм 107—108 [13]
Диацетат целлюлозы Изменение оптической плотности при 320 нм 2.10’—8-Ю7 [24, 143, 150,151]
Триацетат целлюлозы Изменение оптической плотности при 280, 300, 320 или 330 нм 5-105—108 [23, 24, 50, 67, 132, 144, 148]
Нитрат целлюлозы Изменение оптической плотности при 320—340 нм 10’—Ю7 [126]
Ацетат-бутират целлюлозы Изменение оптической плотности при 320 нм 10’—10’ [134]
Целлюлоза Изменение интенсивности желтой окраски после обработки облученного полимера раствором NaOH 10’—108 [16]
Полиэтилен Изменение транс-ненасы-щенности (измерения в инфракрасной области спектра) 5-107—10’ [9, 37, 38, 51, 151]
Изменение гель-фракции (сшивание) Д~(3-М). • 107 [150]
Увеличение веса в атмосфере кислорода До 5-Ю7 [33]
Увеличение поверхности пластинки толщиной 1 мм при 170°С 5-loss’107 [Ю]
Изменение оптической плотности при 270 или 240 нм 107—2-Ю9 [51, 52, 141, 144, 145]
Полипропилен Изменение оптической плотности при 245 нм 5.10s— 2-Ю7 [152]
Меламин (CsHeNe)n Изменение оптической плотности в области 340—420 нм 5-102—107 [12]
Пол итетрафторэтилен (тефлон) Образование свободных радикалов (измерение методом ЭПР) 10’-108 [85, 85а]
Политрихлорфторэти- Изменение оптической плот- 5-10’— [150]
лен ности при 230 нм 7-Ю7
Поликарбонат Изменение скорости разло -(женин в 6,25 М растворе ]NaOH при 70,4° С 107—10» [154]
* 1 В продажу этот полимер поступает под названиями: плексиглас (англ. — Plexiglass), люцит (англ.— Lucite), перспекс (англ. — Perspex) и метаплекс (англ. — Metaplex).
•’Заграничные коммерческие нгзыняч этого полимера: мелинекс (англ. — Melinex), милар (англ. — Mylar), гозгдфш (знг.ч. — Hostap’un) и эстрофол (англ.—Estrofol).
В настоящее время в дозиметрии ионизирующих излучений широкое применение находят пленки различных полимеров (так называемые пленочные дозиметры). Чаще всего они используются для определения доз излучений с малой проникающей способностью и для нахождения пространственного распределения поглощенной энергии. Отметим, что показания некоторых дозиметров на основе полимеров мало зависят от величины ЛПЭ. Другая область применения пленочных дозиметров состоит в использовании их в радиационных процессах в качестве дозимстров-индй-
Рис. 72. Зависимость разности оптической плотности А А триацетатной пленки от дозы D при различных длинах волн 1 — 320 нм\ 2 — 300 н.и;
3 — 280 нм
каторов дозы. Для контроля таких процессов необходимы простые и надежные средства определения степени облучения изделия. С зтой целью полоски полимерных пленок, которые предварительно прокалиброваны, помещаются в каждую упаковку или наклеиваются на каждое облучаемое изделие. Обычно здесь используются пленки, содержащие красители, и о величине дозы судят по изменению окраски.
Приведем два примера. За границей при радиационной стерилизации лекарственных препаратов и медицинского оборудования используются индикаторы «Мегараи» (Венгрия). Эти индикаторы при определенной дозе меняют цвет с зеленого на красный. Г. Н. Пьянков и др. [143] для нахождения оптимальных режимов радиационного модифицирования полиэтиленовых изделий на Броварском заводе пластмасс применяли в качестве дозиметров пленки окрашенного целлофана и диацетата целлюлозы.
А. М. Кабакчи и сотр. [23] подробно исследовали дозиметрические характеристики пленок триацетата целлюлозы. В своих опытах они использовали промышленную непластифицированную пленку, а также пленки, изготовленные в лаборатории. Было найдено, что показания не зависят от мощности дозы у-из л учения в изученном диапазоне (от 10 до 2,5-103 рад!сек). Пленки пригодны для измерения доз электронов при мощностях дозы до 2-107 рад/сек. Для одной и той же дозы изменение оптической плотности тем больше, чем короче длина волны, которая выбрана для оптических измерений (см. рис. 72). Варьирование интервала определяемых доз производится также применением пленок различной толщины.
Для нахождения пространственного распределения поглощенной энергии у-из л учения 60Со А. М. Кабакчи и сотр. [23] предлагают помещать пленку триацетата целлюлозы между пластинками полиметилметакрилата. Пластинки необходимы для соблюдения условий электронного равновесия. Эта система затем ориентируется в любом выбранном направлении дозного поля. После облучения оптическая плотность измеряется по всей длине пленки. Пленки можно использовать и для определения пространственного распределения поглощенной энергии в случае электронов.
Согласно [8, 18, 19], пленки из поливинилхлорида пригодны для определения доз в диапазоне 0,5—6 Мрад. Метод измерения пространственного распределения дозы с помощью пленок из этого полимера предложен в работе [19]. Как следует из работы [132], максимальное значение дозы, которое можно определить с помощью таких пленок, колеблется от 2,5 до 6 Мрад в зависимости от сорта полимера.
Многие авторы [32, 39, 47, 48, 53, 54, 86, 87, 102, 110—122, 131, 133, 135, 137,143,144,146,149,153,153а] рекомендуют использовать для дозиметрии полимеры с различными добавками. Чаще всего такими добавками являются красители или их лейкоосно-вания. В этом случае о величине дозы судят по изменению окраски. Полимеры обычно применяют в виде пленок. В табл. 53 приведены дозиметрические характеристики некоторых полимерных плецок с добавками.
Из числа подобных систем хорошо зарекомендовали себя целлофановые пленки, содержащие красители [32, 113]. Согласно [ИЗ], диметоксидифенил-диазо-бпс-8-амино-1-нафтол-5,7-дисульфоновая кислота, введенная в целлофан, отличается высокой стабильностью при хранении и почти не подвергается воздействию света, тепла и изменения pH. Пленки не изменяют своих оптических свойств при хранении в темноте в течение двух лет. При облучении краситель необратимо обесцвечивается. Степень обесцвечивания, которая пропорциональна дозе, измеряется на спектрофотометре при длине волны 655 нм.
Рассматриваемый дозиметр можно использовать для определения доз в диапазоне 105—IQFpad. К. Уиллис и др. [118] нашли, что эти пленки пригодны и для дозиметрии импульсного электронного излучения (см. главу IX).
По данным работы [53], отечественный промышленный зеленый целлофан характеризуется неравномерным распределением красящего вещества. Поэтому для проведения дозиметрии необходимо отбирать участки пленки с примерно одинаковой оптической плотностью.
Пленки из поливинилхлорида, содержащие метиловый фиолетовый, были рекомендованы для дозиметрии Э. Хенлеем и А. Миллером [111]. Р. Вейл и Дж. Фаррелл [119] взяли патент на дозиметр-указатель дозы, представляющий собой пленку га-
Таблица 53
Дозиметрические характеристики полимерных пленок с добавками
8 А. К, Пикаев
225
Полимер Добавка Измеряемое превращение Диапазон доз, рад Диапазон мощностей дозы, рад/сек Литература
Полиметилметакрилат Полистирол Поли-4-хлорсти-рол Диацетат целлю- лозы 1,4-Диаминоаптрахиноп Бензолазо-а- нафтиламип 1,4-Диаминоантрахипоп 1,8-Нафтолле и-12' -бензимидазол /мраис-Стпльбеи Лейкооспование кристаллического фиолетового, тетрабромэтан Лейкооспование малахитового зеленого 1,4-Диамппоантрахипон Обесцвечивание красителя (изменение оптической плотности при 530 нм) Обесцвечивание красителя (изменение оптической плотности ирп 490—500 нм) Обесцвечивание красителя (изменение оптической плотности при 540 нм) Обесцвечивание красителя (изменение оптической плотности при 280—420 нл() Изомеризация транс-стильбе-иа в цис-изомор (изменение оптической плотности при 324 нм) Появление окраски Появление окраски (изменение оптической плотности при 430 пли 630 нм) Обесцвечивание красителя (изменение оптической плотности при 550 нм) 5-Ю3—5-10е До 2-10’ 5-10s—9-10’ До ~ 5- 10s 2- 10s—10s 103—10е 5-10*—1,5-10’ 10R—10’ 10’—105 103—10s 1,6-102— 6-10’ 3-102— 7,5-Ю13 0,1—Ю2 3-Ю2—юн 10s—10s [149] [47] [149] [102] [86] [74] [87] [149]
Таблица 53 {окончание)
Полимер Добавка Измеряемое превращение Диапазон поз, рад Диапазон мощностей дозы, рад/сек Литература
Поливинилхлорид Эпоксидная смола Полиамид (нейлон), полпвпнпл- *5 пирролидон, целлюлоза и др. * Дозиметрические х ной промышленность м до 100—300 Мрад. Метиловый фиолетовый Кристаллический фиолетовый 1,4-Диампноантрахинон Метиленовый голубой Кислотный зеленый Конго красный Метиловый оранжевый Лейкооспование кристаллического фиолетового, СНВгз Нитрилы триарплмета-новых красителей арактеристики окрашенных л ю, приведены в работах [140, Обесцвечпвацде красителя (изменение оптической плотности при 600 нм) Обесцвечивание красителя (изменение оптической плотности при 570 нм) Обесцвечивание красителя (изменение оптической плотности при 550 нм) Обесцвечивание красителя Обесцвечивание красителя Изменение оптической плотности при 520 и 580 нм Изменение интенсивности отраженного света при 536 пли 584 нм Появление окраскд (изменение оптической плотности при 440, 490, 532, 570 или 610 нм) Появление окраски (изменение оптической плотности при 430, 560, 580, 600 и 625 нм) денок поли эти лен терефталата и регев 52]. В частности, с помощью полиэтр 5-Ю3—108 103-1,5-10"’ 10"’—1,2-10’ До 4-Ю5 До 2,5-103 До 1,5.10s До ~(1,5—2)-10s До ~4,4-105 101—108 ерированной целлюло лентерефталатных пле 103-103 IO3—105 До - 1018 зы, выпускаешь нок можно из [111] [149] [149] [146] [146] [146] [146] [73] [131] IX отечествен-мерять дозы
логенсодержащего полимера или сополимера винилхлорида и ви-нилиденхлорида с добавкой чувствительного к кислотам краси-сителя (диметиловый желтый, тимоловый синий, конго красный и т. п.). При облучении из полимера выделяется кислота, которая вызывает изменение цвета красителя. Сходны дозиметры, запатентованные в Японии и Дании [121, 122].
С. Мацумото и Т. Цукада [121] разработали дозиметр на основе сополимера винилхлорида и винилацетата или поливинили-денхлорида с добавкой 3,3,5,5-тетрабромкрезолсульфофталеина и n-диметиламиноазобензола. Он изготовляется в виде таблеток. В зависимости от дозы эти таблетки имеют следующий цвет (дозы в радах указаны в скобках): голубовато-зеленый (0); желто-зеленый (104); темно-зеленый (5-104); желто-краспый (105); оранжево-красный (5-10®) и красный (10е).
В дозиметре, предложенном Н. Хольмом и Дж. Маулем [122], красителем является метилоранж. При облучении его окраска переходит из желтой в красную. Несколько отличается принцип действия полимерного дозиметра, запатентованного К. Пфёртне-ром [120]. Этот дозиметр представляет собой прозрачный полимер (например, виниловый полимер или полиэфирную смолу), содержащий галогенированный углеводород (CHBr3, CHJ3 и т. п.) или неорганическое вещество (JBr, JC1, HgCl2 и т. п.) и лейкооснова-ние красителя. При облучении галогенсодержащее вещество разлагается, лейкооснование окисляется, и дозиметр приобретает окраску.
Как было обнаружено Г. Тэплиным и К. Молином [117], с помощью твердого поливинилового спирта, окрашенного тетразолом синим, можно определять дозы в диапазоне от 10® до 10® рад. Дозиметрические характеристики пленок из поливинилового спирта, содержащих метиленовый голубой,, были детально изучены А. М. Кабакчи и сотр. [39, 54]. При облучении этих пленок краситель обесцвечивается, и доза измеряется по уменьшению оптической плотности при 660 нм. При действии у-лучей ®°Со радиационно-химический выход обесцвечивания метиленового голубого равен 1,8 молек./100 эв. Пленки пригодны для определения доз в диапазоне 5-104 — 10® рад. Показания дозиметра не зависят от мощности дозы в изученном интервале (от 10 до 104 рад!сек), энергии излучения и температуры во время облучения (исследовался диапазон от —196 до 50° С). Цитированные авторы рекомендуют использовать эти пленки и для нахождения пространственного распределения дозы.
Пленки готовятся следующим образом [39]. Смесь 200 см? 10%-ного водного раствора поливинилового спирта и 40 см? 10_® М водного раствора метиленового голубого выливается на горизонтально установленную стеклянную пластинку размером 60 X 31 см?, защищенную от попадания пыли. Через 5—7 суток при комнатной температуре большая часть воды испаряется, и пленка легко отделяется от стекла. Пленка затем выдерживается
до постоянного веса в эксикаторе над прокаленным хлористым кальцием. По этой методике получаются плотные, равномерно окрашенные пленки толщиной ~ 90 мк. Для изготовления пленок использовался технический поливиниловый спирт ВТУ—М— 582—55, очищенный трехкратным переосаждением из водного раствора метиловым спиртом. Метиленовый голубой был марки «ч.д.а.». Стеклянная пластинка перед употреблением тщательно промывалась водой с содой, высушивалась и перед нанесением смеси растворов протиралась ватой, смоченной в этиловом спирте.
И. М. Блаупштейн и др. [47, 481 исследовали возможность использования пленок из полиметилметакрилата, окрашенных бензолазо-а-нафтиламином, для дозиметрии ионизирующего излучения. Оказалось, что эти пленки можно применять для определения доз до 3-106 рад при мощностях дозы 102 — 4-103 рад/сек. При облучении краситель разлагается, что сопровождается уменьшением оптической плотности пленки при 490—500 нм. По величине уменьшения судят о дозе (выход разложения красителя составляет 0,62 молек./100 эв; молярный коэффициент экстинкции при 490 нм равен 3,86-10* л/моль-см). По данным [471, пленки из полистирола, содержащие этот краситель, могут быть использованы для измерения доз до 2-107 рад. Правда, точность определения поглощенной дозы с помощью полистироловых пленок хуже.
За границей выпускаются дозиметры на основе красного по-лиметипметакрилата (марки «Perspex Red 400» и «Red Perspex»). Использование этих дозиметров основано на изменении окраски красителя, введенного в полимер [34, 35, 64, 1141. Дозу определяют путем измерения оптической плотности при 635 нм. При комнатной температуре эта оптическая плотность практически не изменяется по крайней мере в течение 2 суток после облучения.
Однако при более коротких длинах волн оптическая плотность за это время несколько возрастает, а при более длинных волнах — уменьшается. По этой причине рекомендовано регистрацию оптической плотности проводить при 635 + 1 нм. Краситель обесцвечивается также под действием кислорода. R связи с этим необходимо применять пластинки толщиной ire менее 3 мм. При такой толщине диффузия кислорода воздуха в полимер практически не оказывает влияния на оптическую плотность. Показания дозиметров колеблются от одной партии к другой. Поэтому каждую партию калибруют по ферросульфатному дозиметру. С помощью красного полиметилметакрилата можно определять дозы в диапазоне 105 — 5-106 рад.
Представляют интерес дозиметры, предложенные У. Маклафлином и др. [69—72, 131, 1351. Действие этих дозиметров основано на использовании предшественников ди- или триамипотрп-фенилметановых красителей (см. также стр. 182 и 205). Общая
формула этих соединений такова:
сх
где Rj, R2 и R3 могут быть NH2, N(CH3)2 и т. п.; R4, R5 и Re-обычно атом Н, но может быть СН3 и т. п., а X есть Н, ОН, ОСН3, CN и т. д. (чаще всего CN).
Рассматриваемые вещества в виде 10~3 — Ю1 М растворов входят в состав различных гелей и смол. Они могут применяться также в форме покрытий, тонких пленок на полимерной основе и т. п. В результате облучения дозиметр приобретает интенсивную окраску. При этом оптическая плотность пропорциональна дозе в широких пределах. Например, пленки из полиэтилеп-терефталата, покрытые «раствором» нитрила фуксина в нейлоне, и пленки из ацетата целлюлозы, покрытые гелем нитрила парарозанилина в желатине, позволяют определять дозы в диапазоне 2—30 Мрад. Первая система приобретает при облучении розовую окраску разных оттенков, а вторая — красную.
На показания таких систем малое влияние оказывают изменения температуры от —80 до 4-100° С и мощности дозы до 1015 рад/сек', показания одинаковы для у-лучей, электронов и протонов. Кроме того, они стабильны при хранении. В настоящее время дозиметры этого типа выпускаются серийно американской фирмой «EG and G».
Пленки указанного выше состава удобны для нахождения пространственного распределения дозы в случае электронов. С этой целью полоски пленок помещаются внутри исследуемого материала перпендикулярно направлению электронного пучка или под углом к мему. Подробно методика измерений изложена в работах (133, 1391.
Очень чувствительный дозиметр, предназначенный для измерения малых доз (порядка 0,5 рад), был разработан Г. Остером и Б. Бройде [115]. Он представляет собой пленку, приготовленную выпариванием водного раствора, содержащего 3,2% поливинилового спирта, 0,04% метиленового голубого, 7% нитрата свинца, 17% этилендиаминтетрауксусной кислоты и 8,5% глицерина. Полученная пленка с целью обесцвечивания облучалась ультрафиолетовым светом. Под действием ионизирующего излучения лейкоформа красителя окисляется, в результате чего происходит окрашивание пленки. Выход превращения красителя 104 молек./100 эв, что указывает на цепной характер реакции.
Л. Харрах [86, 87] подробно исследовал возможность использования полистироловых и полигалостироловых пленок с неко-230
горыми добавками в дозиметрии ионизирующих излучений. По его данным [86], полистироловые пленки, содержащие тпранс-стильбен, пригодны для определения доз в диапазоне 2-105 — Ю« рад. В них транс-стильбен при облучении изомеризуется в ^ас-изомер. Это превращение и рекомендуется применять для дозиметрии. В следующей работе [87] этого автора изучались по-лигалостироловые пленки, содержащие лейкоформу малахитового зеленого. При действии ионизирующего излучения пленка окрашивается. Интенсивность появляющейся окраски может служить мерой дозы в интервале 5-104— 1,5-107 рад. Показания полистироловых и полигалостироловых пленок с указанными добавками не зависят от мощности дозы до Ю14 рад/сек, т. е. они могут быть использованы в дозиметрии импульсного электронного излучения. Поэтому они рассматриваются подробно в главе IX.
К дозиметрам па основе полимеров в некоторой степени примыкают системы, состоящие из мономеров, которые под действием излучения полимеризуются [26,123, 1241- Однако, поскольку эти системы до облучения представляют собой жидкости, они были рассмотрены в главе VI. В той же главе были указаны дозиметрические системы, являющиеся жидкими полимерами [7, 14, 21, 22]. Дозиметры па основе растворов полимеров в воде [31] и органических растворителях [79—81] описаны в главах V и VI.
Другой способ измерения дозы основан на изменении интенсивности люминесценции некоторых веществ, введенных в полимеры, в результате облучения (см., например, [48, 96—105, 125, 155—158]. В частности, по данным [98, 102], интенсивность фото- и радиолюминесценции полистироловых плепок, содержащих люминесцирующие добавки — 1,8-пафтоилен-1',2'-бепз-имидазол или его хлор- и метоксипроизводные, падает с ростом дозы у-излучепия, что может быть использовано для определения доз в диапазоне 107 — Ю10 рад. При этом показания не зависят от мощности дозы, температуры облучения и времени хранения в широких пределах. Фотолюминесценция облученных плепок возбуждается светом с длиной волны 405 нм, а радиолюминесценция — а-частицами 239 Рп.
2. Стекла
Исследования свойств стекол с точки зрения использования их в дозиметрии ионизирующих излучений проводились многими авторами [20, 40, 46, 53, 126, 159—296]. В результате этих исследований установлено, что с помощью стекол могут быть определены как малые дозы (до 102 3 рад), так и большие дозы (до 1010 рад). Стеклянные дозиметры находят широкое применение в радиобиологии, радиотерапии, биофизике и т. п. Стекла предлагались и в качестве персональных дозиметров. Взято большое число патентов (см., например, 1297—315]) на использование различных
Дозиметричеркие характеристики стекол, изменяющих окраску при облучвнии
Точный состав в оригинальных работах ие указан.
стекол в дозиметрии. Однако в радиационной химии они применяются сравнительно редко. Поэтому в настоящем разделе дается лишь краткая характеристика этих систем. Более подробно заинтересованный читатель может ознакомиться с ними в обзорах [1, 238, 316—318] и книгах [319—322].
При измерении дозы с помощью стекол используются следующие явления: 1) изменение окраски под действием излучения; 2) радиофотолюминесценция; 3) радиотермолюминесценция; 4) используются также сцинтилляционные свойства специальных стекол.
Способность стекол окрашиваться под действием излучений обычно применяется для измерения доз 108 — 104 рад. Наиболее вероятно, окрашивание часто обусловлено возникновением F-центров (F-центр — это электрон, захваченный анионной вакансией). Свойство окрашиваться при облучении характерно для фосфатных стекол с добавками серебра или кобальта, силикатных и боросиликатных стекол с добавками кобальта, силикатных стекол с добавками ванадия, марганца и железа, висмутборатных стекол с добавками трехокиси мышьяка и т. п. В дозиметрии используется и способность некоторых окрашенных стекол обесцвечиваться при облучении. В табл. 54 приведены дозиметрические характеристики некоторых стекол, которые характеризуются изменением окраски при облучении.
С помощью фосфатных стекол, активированных серебром (см. табл. 55), можно по появлению при облучении дополнительной полосы поглощения с максимумом при ~ 320—350 нм определять дозы от 103 до 2-10s рад [161]. Показания их не зависят от мощности дозы до ~ 10s рад/сек. Однако на измерения оказывает влияние энергия ионизирующего излучения. Кроме того, возникшее оптическое поглощение нестабильно после облучения. Эти стекла находят гораздо большее применение как радиофотолюминесцентные дозиметры.
Таблица 55
Составы люминесцирующих стекол (в вес. %)
Ag К Li Ва Be Mg в р А1 О Марка и тип стекла Литература
4,3 7,7 — 10,8 .— 28,4 4,7 44,1 Стекло с высоким Z [164]
4,3 — 1,9 — — 3,1 — 33,7 4,7 52,3 Bausch and Lomb (стекло с низким Z) [163]
4,2 — 3,6 — — — 0,8 33,3 4,6 53,5 Toshiba (стекло с низким Z) [237]
2,4 —- 2,5 —. 0,5 — — 33,8 3,5 52,5 С. Е. С. [256]
4,6 — 4,7 .—- —- 0,9 — 34,1 3,1 52,8 Schott I [242]
0,6 — 7,3 — — — 1,° 34,7 0,5 55,9 Schott [250]
Для измерения низких доз (до 103 рад) используется способность некоторых облученных стекол светиться при освещении или нагревании. Наибольшее применение здесь получили фосфатные стекла, активированные серебром. В табл. 55 приведены составы таких стекол.
На рис. 73 показаны оптические свойства фосфатных стекол, активированных серебром [238]. Как видно из рисунка, при облучении в стекле возникает новая полоса оптического поглощения. Она обусловлена образованием атомарного серебра. Возбуждение светом в этой полосе приводит к появлению оранжевой
Рис. 73. Оптические свойства фосфатных стекол, активированных серебром 1 — поглощение необлученного стекла;
2 — полоса поглощения, возникающая при облучении;
з — люминесценция как результат^освещения в полосе 2
люминесценции, что и используется как мера дозы. С ростом дозы интенсивность полосы поглощения увеличивается, и эта полоса перекрывается с полосой испускания. Поэтому интенсивность свечения при повышении дозы проходит через максимум (см. рис. 74 [2381). С помощью этих стекол можно измерять дозы в диапазоне от 1 до ^lO3 рад.
И. А. Бочвар и др. [262] разработали термолюминесцентные дозиметры (марки ИКС) из алюмофосфатных стекол, позволяющие измерять дозы в диапазоне от 2-10—2 до (1-е-2)-10® рад. Эти стекл'а имеют следующий состав: MgO-P2O5 — А12О3-ЗР2ОБ (марка 5" 26); SrO-P2O5 — А12О3-ЗР2ОБ (марка 340 3); Ы2О-Р2ОБ— — А12О3-ЗР2О5 (марка 2° 3) и SrO • Р2ОБ — SiO2-P2O5— 2(А12О3--ЗР2ОБ) (марка 580-1). Кроме того, они содержат 0,1% Мп. Пик свечения наблюдается при температуре 350—360° С.
Дж. Шульман и сотр. [239] использовали для дозиметрии способность облученного бората лития, активированного марганцем (обычно 0,1%), светиться при нагревании. Этим методом можно измерять очень низкие дозы (порядка 1 рад и менее). Детальное исследование этой дозиметрической системы выполнено недавно Ч. Архундпа и др. [323]. По их данным, борат лития, содержащий 0,34% Ма, может применяться для определения доз до ~ 103 рад.
Для определения низких доз можно использовать сцинтилляционные свойства некоторых стекол. С этой целью применяются преимущественно литиевые и боратные стекла, содержащие, кроме SiO2, окислы натрия, магния и алюминия. В качестве активатора в такие стекла вводится Се2О3.
Стекла как дозиметрические системы обладают рядом преимуществ по сравнению с другими системами. Они характеризуются простотой изготовления в промышленных масштабах, устойчивостью при хранении, компактностью, возможностью многократного использования (стекла, которые приобрели окраску в результате облучения, можно обесцветить путем нагревания до 300—500° С). Вместе с тем они страдают рядом существенных недостатков. Прежде всего их показания вследствие сравнительно высоких эффективных номеров зависят от энергии излучения. На результаты измерений дозы с их помощью оказывает влияние и
Рис. 74. Зависимость интенсивности I радиофотолюминесценции фосфатного стекла, активированного серебром, от экспозиционной дозы у-пзлученмя
температура облучения. Окраска облученных стекол изменяется во времени («выцветание» или «фэдипг»), Особенно быстро этот процесс протекает при повышенных температурах. Например, в случае фосфатных стекол, активированных серебром, ь «выцветание» составляет 8—20% в течение первых 24 час. после облучения (161]. Для боросиликатных стекол, содержащих кобальт, оно равно ~ 2% за 24 часа [165]. Наиболее стабильны в этом отношении висмутсвинцовоборатные стекла с добавками As2Oa, для них «выцветание» ничтожно 1165, 166]. Отмеченные недостатки обусловливают необходимость периодической калибровки стекол (например, по ферросульфатному раствору) при использовании их в качестве дозиметров.
По данным [324], природный бразильский кварц имеет три пика (при 110, 220 и 310° С) на кривой радиотермолюминесцен-ции. Интенсивность пика при 220° С характеризуется хорошей стабильностью и линейной зависимостью от дозы до 104 рад. Это свойство облученного кварца рекомендовано использовать для дозиметрии у-излученпя 60Со.
3. Галогениды щелочных
и щелочноземельных металлов
Применению галогенидов щелочных и щелочноземельных металлов в ^дозиметрии посвящено большое число работ [232, 238, 241, 284, 325—399]. Такой интерес к этим системам обусловлен следующими их свойствами. Во-первых, они окрашиваются при облучении. Во-вторых, облученные кристаллы (как чистые, так
и содержащие некоторые добавки) характеризуются наличием свечения при нагревании. В-третьих, используется явление радиофотолюминесценции.
Окрашивание рассматриваемых кристаллов обусловлено образованием /'-центров. Однако F-цептры в чистых кристаллах быстро разрушаются. Поэтому способность таких кристаллов к окрашиванию при облучении не находит применения в дозиметрии.
Стабильность /'-центров повышается введением в кристалл щелочного металла и насыщеция его водородом. Например, по данным [325], кристаллы КВг, обработанные таким способом, пригодны для измерения доз в диапазоне от 10 до 103 рад. В них при радиолизе возникает полоса поглощения с максимумом при 630 нм. Интенсивность окраски в течение 3 суток после облучения уменьшается на 16,7%, а затем остается постоянной.
Метод дозиметрии ионизирующих излучений, основанный на способности щелочногалоидных кристаллов окрашиваться при облучении, не получил широкого распространения. Объясняется это, прежде всего, тем, что интенсивность окраски зависит от температуры радиолиза и энергии излучения.
В настоящее время в различных областях науки для измерения доз широко применяется способность облученных галогенидов щелочных и щелочноземельных металлов светиться при нагревании. В результате радиолиза в кристалле возникают захваченные электроны (/’-центры) и захваченные «дырки» (Н- или F-центры). Под действием света или тепла электрон освобождается из /-центра и диффундирует в полосу проводимости. Он может прорекомбинировать с «дыркой». При этом энергия рекомбинации может выделиться в виде света. Если кристалл содержит добавки активатора, то «дырки» захватываются молекулами или ионами активатора. Электрон в этом случае рекомбинирует с молекулой или ионом активатора. В результате этого активатор возбуждается и люминесцирует.
В качестве люминесцентных дозиметров предлагались LiF, природный CaF2, синтетический CaF2 с добавками Мн, NaCl или КС1 с добавками Ag. Наибольшее распространение получили первые три дозиметра.
Фтористый литий в виде монокристаллов может быть использован для измерения доз от 0,01—0,1 до 105 рад [238, 344, 346, 352, 355]. Показания его не зависят от мощности дозы до 1011 — 1012 рад1сек [353]. Кроме того, поскольку LiF имеет низкий эффективный атомный номер, то на результаты измерений дозы с его помощью не оказывает влияния энергия излучения вплоть до сравнительно малых ее значений (до — 0,1 Мэв). Он может быть также использован для определения доз нейтронов (см. главу XI).
В настоящее время дозиметры на основе LiF выпускаются промышленностью США, ГДР, Бельгии и некоторыми научными
учреждениями, в том числе Институтом физики АН Латвийской ССР. Широкое распространение за границей получил дозиметр марки TLD-100 (США). .Зависимость интенсивности радиотермолюминесценции этого дозиметра от температуры и спектр свечения показаны па рис. 75 и 76 [1J. Интенсивность пика при 210° С практически не зависит от времени хранения облученного образца (за год храпения при комнатной температуре опа уменьшается менее чем на 5%). Зависимость интенсивности пика при 210° С от дозы у-изл учения приведена на рис. 77. Как видно, линейность показаний сохраняется до доз ~ 10Б рад. Тепловая обработка в течение 1.5 часа при 400° С и затем в течение 24 час. при 80° С приводит к восстановлению исходных свойств дозиметра. Эта операция обусловливает также уменьшение интенсиг
Рис. 75. Зависимость интенсивности I радиотермолюминесценции облученного LiF (TLD-100) от температуры (доза 1()4р; скорость нагревания 20°С/мип.)
g L_i-------1--------L_
350 050 550
Л, нм
Рис. 76. Спектр радиотермолюминесценции облученного LiF (TLD-100)
Г — интенсивность радиотермолюминесценции
Рис. 77. Зависимость интенсивности I радиотермолюминесценции LiF (TLD-100) при 210° С от экспозиционной дозы у-излучения с0Со
Рис. 78. Зависимость интенсивности I радиотермолюминесценции облученного дозиметра CaF2 : Ми от температуры (доза 104 />; скорость нагревания 20 еС,мпп.)
ности низкотемпературных пиков свечения (при 100 и 150° С) [346]. Дозиметр, предложенный Институтом физики АН Латвийской ССР, содержит Mg (0,3 вес. %) и Са (0,01 вес. %) [389, 390]. По своим характеристикам он сходен с дозиметром TLD-100.
Несколько большей чувствительностью по сравнению с LiF обладает фторид кальция, активированный марганцем. С его помощью могут быть определены дозы порядка 1О-3 рад [238]. На рис. 78 показана зависимость интенсивности свечения облученного CaF2 : Мп от температуры [1|. Спектр радиотермолюминесценции этого дозиметра приведен на рис. 79 [1], а зависимость
Рис. 79. Спектр радиотермолюминесценцпи облученного дозиметра CaF„ : Мп Г — интенсивность радиотермолюминесценции
Рис. 80. Зависимость интенсивности I радиотермолюминесценцпи CaF2 : Мп при 260 °C от экспозиционной дозы у-излучения 60Со
интенсивности свечения от дозы иллюстрируется рис. 80 [346] Этот дозиметр имеет «ход с жесткостью».
LiF и CaF2 : Мп могут быть использованы в виде порошка. Однако необходимо, чтобы порошок находился в вакууме или инертной атмосфере. В противном случае фоновая люминесценция образца из-за трения между гранулами или взаимодействия с воздухом существенно понижает чувствительность дозиметра. Например, для порошка LiF по этой причине низший предел измерения дозы составляет 10 рад [344]. Монокристаллы же можно применять для определения гораздо меньших доз [352].
«Выцветание» CaF2 : Мп при комнатной температуре составляет ~10% в течение первых 16 час. после облучения, затем 1% в течение нескольких дней, так что через 80 дней интенсивность свечения равна ~60% от исходной [1].
Природный фторид кальция (минерал флюорит) характеризуется высокой интенсивностью радиотермолюминесценции. Это свойство используется в дозиметрических измерениях [357, 358]. Флюорит имеет три пика на кривой радиотермолюминесценции при 70—190, 150—190 и 250—300° С. Интенсивность последнего пика характеризуется стабильностью при хранении при комнат
ной температуре и линейной зависимостью от дозы в диапазоне от —10 3 - 10’2 до 5-103 — 104 рад.
Рассмотренные дозиметры дают относительные показания. Поэтому перед употреблением их необходимо калибровать. Более подробные сведения о них можно найти в обзорах [1, 316, 318, 354, 355, 392] и книгах [320, 321, 389|.
4. Сульфат кальция
Кроме LiF и CaF2, широкое распространение в качестве термолюминесцентного дозиметра находит сульфат кальция, активированный некоторыми добавками [330, 399—417]. Согласно [403. 406], кривая радиотермолюминесценцпи CaSO4: Мп имеет один пик при 80—100° С; спектр свечения характеризуется максимумом при —500 нм. С помощью CaS04: Мп возможно определять как низкие дозы (10-4 — 10 я и даже 5-10-6 рад), так и высокие (до 104 рад). Недостатком его является довольно болошая скорость «выцветания»: 40—85% заЗ дня после облучения. CaSO4: : Sm обладает большей чувствительностью и меньшей скоростью «выцветания», чем CaSO4 : Мп [405, 406]. По данным [400], дозиметр на основе CaSO4 с добавками Мп и Sm в отдельных случаях можно применять для измерения доз до 10е рад. Согласно [399], сульфат кальция, активированный диспрозием или тулием, пригоден для определения доз в диапазоне от 10~2 до 10s рад.
В табл. 56 сравнены характеристики термолюминесцентных дозиметров на основе различных твердых тел. Из нее видно, что наибольшей чувствительностью из числа дозиметров рассматриваемого типа обладает дозиметр из CaSO4 : Мп.
Ш. Алимов и др. [400] обнаружили, что в CaS04 с различными добавками при облучении возникают парамагнитные центры,
Табл и’ц а 56
Характеристики термолюминесцентных дозиметров на основе твердых тел
‘Дозиметр Диапазон измеряемых доз, рад Температура главного пика радиотермолюминесценции, °C Максимум спектра свечения, нм
Алюмофосфатиос стекло с добавкой Мп (ИКС, СССР) 0,02-2- 10s 350—355 560
Борат лития с добавкой Ми (США) 0,01—106 200 605
LiF (TLD-100, США) 0,01—105 210 400
LiF с добавкой Mg (200 s, ГДР) 1—105 170 400
LiF -ф 0,3 вес. % Mg +0,01 вес. % Са (Институт физпки, СССР) 0,01—10® 230 400
CaF2: Мп (США) 5-Ю"8—105 260 500
CaF-2 (Бельгия) 2-Ю-3—10* 260 380
CaSO4: Мп (Чехословакия) Ю-з_1оз 100—110 -500
концентрация которых пропорциональна дозе. Регистрация этих центров осуществляется методом ЭПР. Наиболее чувствителен сульфат кальция с добавками Zn. В этом случае низший предел измерения дозы составляет ~ 100 рад. Отметим, что методу ЭПР присущи большие абсолютные ошибки определения концентрации парамагнитных центров (примерно + 30%). Позтому при проведении дозиметрии необходимо проводить калибровку рассматриваемой системы применительно к парамагнитным эталонам, имеющимся в лаборатории.
5. Другие дозиметры на основе твердых тел
Предлагались многие другие дозиметры на основе твердых тел, принцип действия которых заключается в измерении химических или физических превращений, происходящих в них при облучении. В. В. Генералова и др. [4181 исследовали возможность использования твердых углеводов в дозиметрии у-излучения. Эти авторы обнаружили, что изменение угла вращения плоскости поляризации растворов, приготовленных из облученных порошков глюкозы, мальтозы и сахарозы, пропорционально дозе в довольно широком диапазоне. Для 20%-ных растворов облученной глюкозы эта пропорциональность сохраняется в пределах от 107 до 7-10® рад. Повышение температуры радиолиза глюкозы до 70° С не оказывает существенного влияния на показания. Однако в случае сахарозы повышение температуры от 40 до 80° С приводит к ускорению разложения углевода.
Для определения дозы можно применять и измерение показателя преломления 20%-ных растворов облученных глюкозы и сахарозы. С этой целью использовался рефрактометр (измерения проводились при длинах волн 589,3 и 546,8 нм) [4181. Было обнаружено, что коэффициент преломления указанных растворов линейно уменьшается при возрастании дозы в интервале 5-107 — 8-10® рад, а показания не зависят от температуры (до 70° С) и присутствия воздуха.
Регистрировать дозу можно измерением оптической плотности водных растворов некоторых облученных углеводов. Так, растворы сахарозы и мальтозы характеризуются устойчивыми максимумами поглощения при 263—265 нм. В случае 0,05%-ного раствора облученной сахарозы оптическая плотность при 265 нм пропорциональна дозе до 7-10® рад. Однако при измерениях необходимо точно фиксировать температуру, поскольку выход продукта, ответственного за это поглощение (по-видимому, им является диокси ацет он), возрастает на 2—3% при увеличении тем пературы на 1° С.
Г. С. Бологова и др. [419] применили весовой метод. Навески глюкозы в 3—6 г облучались в стеклянных пробирках с притертыми пробками, в которых имелись капиллярные отверстия. В результате облучения вес уменьшался за счет выделения газо
образных продуктов. Точность этого метода составляет + 13,5% при 25—30° С. Если облучение проводить при больших температурах, то разложение глюкозы значительно ускоряется.
В. В. Генералова и М. Б. Кишиневская [420, 421] предложили использовать метод диффузного отражения. В этом случае измеряется коэффициент диффузного отражения в ультрафиолетовой области спектра для углеводов, облученных в виде таблеток (с ростом дозы он уменьшается). Были исследованы таблетки глюкозы и глюконата кальция. Оказалось, что глюконат кальция обладает более хорошими дозиметрическими характеристиками, чем глюкоза. Для него величина коэффициента отражения при длине волны 260 нм не зависит от температуры облучения до 70° С. Для регистрации отражения применялся спектрофотометр СФ-4, снабженный приставкой ПДО-1. Метод позволяет измерять дозы у-излучения в диапазоне 104 — 10® рад.
Наконец, Е. П. Ковалева и др. [422] как меру дозы использовали количество водорода, выделяющегося из твердой глюкозы при ее облучении. По их данным, G(H2) = 2,14 молек./100 эв. Метод можно применять при дозах до 10® рад.
И. Драганич и др. [423: 424] рекомендуют применять твердую щавелевую кислоту Н2С2О4-2Н2О для дозиметрии ионизирующего излучения. При облучении она разлагается, причем начальный выход разложения для у-лучей 60Со равен 6 молек./100 эв. Было найдено, что с помощью зтой системы можно определять дозы от 108 до 10® рад. Доза находится по уравнению
где g0 и g — вес кислоты до и после облучения; а — коэффициент пропорциональности (для у-излучения 60Со он равен 1,81-• 10® рад [424]).Степень разложения измеряется весовым методом или после растворения спектрофотометрически либо титрованием (см. стр. 171). Отметим, что при у-радиолизе выход разложения безводной щавелевой кислоты примерно на 15% больше, чем выход разложения Н2С2О4-2Н2О [425].
У. Брэдшоу и др. [426] предложили использовать порошкообразный аланин для измерения доз в диапазоне 102 — 105 рад. Метод основан на измерении образующихся при облучении свободных радикалов, концентрация которых при комнатной температуре изменяется всего примерно на 5% в течение 90 дней. Определение концентрации радикалов проводится методом ЭПР.
И. Такашима и др. [427] исследовали возможность применения метода мессбауэровской спектроскопии для определения больших доз у-излучения. В качестве дозиметрических систем были использованы FeSO4-7H2O, смеси антрацен—FeCl2 и перилен—FeCl2. Было найдено, что ферросульфат и указанные смеси пригодны для нахождения доз соответственно в диапазонах 108 — 1010 и 107 — 108 рад. При облучении в этих системах воз-
пикает трехвалентное железо, и интенсивность его сигнала в мессбауэровском спектре пропорциональна дозе.
Согласно [331, 428J, с помощью сульфидов стронция, кальция и магния, активированных Sm или Ей, можно измерять низкие дозы (например, 10 2 рад с помощью MgS: Ей или MgS: Sm). Для этих облученных кристаллов характерно свечение при действии инфракрасного света. Оптимальные длины волн для возбуждения свечения следующие: ~1000 нм для MgS и SrS; 1250 нм для CaS.
Как следует из работ [429—431J, измерением термолюминесценции облученной окиси бериллия при 160° С можно определять дозы то ~105 рад. Чувствительность этого дозиметра близка к чувствительности LiF.
В ряде работ (см. например, [432—434J) предлагалось использовать для дозиметрии ионизирующих излучений явление эк-зоэлектроппой эмиссии (испускание электронов низкой энергии), происходящее при нагревании некоторых твердых тел (ВеО, смесь ВеО с ZnO, Li2B4O7: Мп. BaS04 и др.). Как выяснилось, этим методом можно измерять дозы от ~ 10 4 до 2-104 рад.
По данным работ [156, 159, 160, 435—4421, для измерения больших доз (до 5-10я рад) могут быть использованы некоторые органические люминесцирующие вещества (нафталин, антрацен, n-терфенил, и-кватерфенпл и др.). В результате облучения происходит уменьшение интенсивности фотолюминесценции этих веществ. Хотя степень уменьшения интенсивности свечения непропорциональна дозе, этим методом можно определять дозы в диапазоне от 5-105 до 5-10я рад. Сходные результаты можно получить при использовании растворов n-терфенила в бензоле [440J. С помощью этой системы можно измерять дозы у-излучения в интервале 10Б — 107 рад.
О. П. Верхградский и др. [441] наносили антрацен в виде тонких пленок на некоторые металлы и щелочногалоидные кристаллы и затем по степени деградации фотолюминесценции этого вещества судили о величине поглощенной дозы в пленках. Было найдено, что в пленках толщиной от 6,7-10-4 до 3,2-10-3 мг!смг, нанесенных на алюминий, золото и КС], эта степень почти не отличается от степени деградации фотолюминесценции антрацена в гомогенных условиях. При толщинах менее 6,7-10’4 мг!смг в случае алюминия и КС1 тушение люминесценции антрацена резко возрастает. Это свидетельствует о перераспределении энергии ионизирующего излучения в тонких слоях.
М. С. Юнусов и др. [443J исследовали дозиметрические характеристики рубина и цинкогерманата натрия, содержащего двухвалентный марганец. Было обнаружено, что первый люминофор можно применять для определения доз до 10е рад путем измерения термолюминесценции, а второй — для нахождения доз в интервале 10—10я рад путем измерения выхода хемилюминесценции.
ЛИТЕРАТУР^
1. «S’. Hayakawa. Determination of Absorbed Dose in Reactors. V ienua,IAEA, 1971, p. 223.
2. P. Alexander, A. Charlesby, M. Ross. Proc. Roy. Soc., A223, 392 (1954).
3. I. Fowler, M. Day. Nucleonics, 13, No. 12 52 (1955).
4. J. W. Boag, G. W. Dolphin, J. Rotblat. Radiation Res., 9, 589 (1958).
5. J. W. Boag, G. W. Dolphin, J. Rotblat. Pbys. Med. Biol., 2, 383 (1958).
6. J. F. Kircher, B. W. King, M. J. Oestman, P. Schall, G. D. Galkins. Proc. II United Nations Intern. Conference on Peaceful Uses of Atomic Energy, vol. 21, Geneva, 1958, p. 199.
7. F. E. Hoecker, 1. W. Watkins. Intern. J. Appl. Radiation Isotopes, 3, 31 (1958).
8. C. Artandi, A. Stonehill. Nucleonics, 16, No. 5, 118 (1958).
9. A. Charlesby, W. H. Davison. Chem. and Ind., 1957, 232.
10. H. Wilski. Atomkernenergie, 4, 402 (1959).
11. E. H. Cornish, D. M. Citting. Nature, 183, 1804 (1959).
12. M. Birnbaum, J. Schulman,G. Seren. Rev. Sci. Instrum., 26, 457 (1957).
13. W. Goebel. Nukleonik, 1, 227 (1959).
14. E. P. Hoecker, II. L. Hiebert. Radiology, 72, 254 (1959).
15. M. Lefort, X. Tarrago. Intern. J. Appl. Radiation Isotopes, 7, 323 (1960).
16. R. Bosworth, J. Ernst, J. Garnett. Intern. J. Appl. Radiation Isotopes, 11, 152 (1961).
17. V. H. Ritz. Radiation Res., 15, 460 (1961).
18. C. Artandi, A. Stonehill. Nucl. Instrum. Methods, 6, 279 (1960).
19. C. Artandi. Nucleonics, 17, No. 10, 62 (1959).
20. A. Scharmann. Selected Topics in Radiation Dosimetry. Vienna, IAEA, 1961, p. 511.
21. F. E. Hoecker. Radiology, 76, 116 (1961).
22. F. E. Hoecker. Health Phys., 8, 381 (1962).
23. О. П. Верхградский, И. H. Череецоеа, А. М. Кабакчи. Сб. «Дозиметрия больших доз». Ташкент, «Фан», 1966, стр. 79.
24. О. Н. Верхградский, IO. Ф. Голодный, Г. Н. Пъянкое, И. II. Череецоеа, А. М. Кабакчи. Там же, стр. 223.
25. R. D. Jarrett, J.W. Halliday. Adv. Chem. Ser., 81, 605 (1968).
26. M. Talat-Erben, E. Vnseren, N. Seber. Solid State and Chemical Ra--diation Dosimetry in Medicine and Biology. Vienna, IAEA, 1967, p. 301.
27. G. Davidson, H. C. Sutton. Intern. J. Appl. Radiation Isotopes, 15, 741 (1964).
28. P. Alexander, R. M. Black, A. Charlesby. Proc. Roy. Soc., A232, 31 (1955).
29. M. Frank, W. Stolz. Kernenergie, 8, 553 (1965).
30. W. Stolz. Isotopenpraxis, 3, 83 (1967).
31. A. L. Boni. Radiation Res., 14, 374 (1961).
32. A. Charlesby, R. J. Woods. Intern. J. Appl. Radiation Isotopes, 14, 413 (1963).
33. A. L. Bradshaw, A. M. Maysent. Brit. J. Radiol., 37, 219 (1963).
34. R. Whitetaker. Brit. J. Radiol., 36, 779 (1963).
35. C. G. Orton. Phys. Med. Biol., 11, 551 (1966).
36. T. Grunewald, G. Rumph. Atompraxis, 11, 95 (1965).
37. B. J. Lyons. J. Polymer. Sci., A3, 777 (1965).
38. Г. H. Пьянков, А. П. Мелешевич, E. Г. Ярмилко, A. M. Кабакчи, С. И. Омельченко. Радиационная модификация полимерных материалов. Киев, «Техника», 1969, стр. 65.
39. Я. И. Лаерентоеич, А. И. Левон, Г. Н. Мельникова, .4. М. Кабакчи. Атомная энергия, 19, 273 (1965).
40. Е. G. Hoffmann. Kerntechnik, 5, 439 (1963).
41. N. Kishi. Kobunshi, 9, 95 (1960).
42. I. Kugler, A. Scharmann. Atomkernenergie, 4, 23 (1959).
43. W. Masaru, L. Yogo. J. Radiation Res. Japan, 4, 68 (1963).
44. W. S. Moos. Atompraxis, 6, 477 (1960).
45. M. Mozisek. Jaderna Energ., 9, 81 (1963).
46. T. Tuchscheerer. Atomkernenergie, 9, 371 (1964).
47. 11. M. Блаунштейн, H. У. Рахимбаева. Радиационная дозиметрия и спектрометрия ионизирующих излучений. Ташкент, «Фан», 1970, стр. 111.
48. И. М. Блаунштейн, Б. А. Вайншток, В. В. Генералова, Н. У. Рахим-баева. Там же, стр. 114.
49. А. Г. Васильев, В. П. Трусова. Там же, стр. 24.
50. Г. С. Бологова, А. Г. Васильев, Р. А. Николаева, В. П. Трусова. Там же, стр. 102.
51. И. М. Блаунштейн, Ф. Т. Золотаревская, В. И. Кубатина. Там же, стр. 139.
52. И. М. Блаунштейн, В. И. Кубатина, Н. У. Рахимбаева. Дозиметрия интенсивных потоков ионизирующих излучений. Ташкент, «Фан», 1969, стр. 21.
53. В. П. Трусова, Г. С. Бологова, Р. А. Николаева, А. Г. Васильев. Дозиметрия больших доз. Ташкент, «Фан», 1966, стр. 75.
54. Я. И. Лаврентович, А. М. Кабакчи. Радиационная химия полимеров. М., «Наука», 1966, стр. 253.
55. К. Heuss. Strahlentlierapie, 110, 628 (1959).
56. W. С. Windley, Т. Elleman. .1. Nucl. Energy, 21, 803 (1967).
57. К. К. Harris, IF. E. Price. Intern. J. Appl. Radiation Isotopes, 11, 114 (1961).
58. L. A. Wall, D. M. Brown. J. Res. Nat. Bur. Stand., 57, 131 (1959).
59. C. G. Orton. Phys. Med. Biol., 11, 377 (1966).
60. C. G. Orton. Intern. J. Appl. Radiation Isotopes, 19, 297 (1968).
61. F. X. Bizzo, G. Cunningham, L. Galanter. Trans. Am. Nucl. Soc., 10,
53 (1967).
62. R. J. Berry, C. G. Orton. Phys. Med., Biol., 11, 475 (1966).
63. B. Whitetaker, C. A. Lowe. Intern. J. Appl. Radiation Isotopes, 18, 89 (1967).
64. С. H. Marshall, C.G. Orton. Phys. Med. Biol., 11, 563 (1966).
65. H. Hinrichs. Atomkernenergie, 2, 41 (1957).
66. Y. Oshima, R. Tanaka. Oyo Butsuri, 36 , 515 (1967); C. A., 67, 96047 (1967).
67. О. П. Верхградский, Г. H. Пьянков, И. II. Червецоса, А. М. Кабакчи. Укр. физ. ж., 12, 1902 (1967).
68. О. П. Верхградский, И. Н. Червецова, А. М. Кабакчи. Атомная энергия, 20, 512 (1966).
69. W. L. McLaughlin, L. Chalkley. Phot. Sci. Eng., 9, 159 (1965).
70. W. L. McLaughlin. Intern. J. Appl. Radiation Isotopes, 17, 85 (1966).
71. W. L. McLaughlin, L. Chalkley. Radiology, 84, 124 (1965).
72. W. L. McLaughlin, L. Chalkley. Trans. Am. Nucl. Soc., 10, 52 (1967).
73. £. Ishikawa. Rikagaku Ken. Hokoku, 35, 389 (1959); C. A., 54 , 20445 (1961).
74. W. Stolz, K. Prokert. Kernenergie, 8, 425 (1965).
75. >S’. A. Goldblith, R. J. Mateles. Nucleonics, 16, No. 6, 102 (1958).
76. Ф. А. Махлис. Приборы и техника эксперимента, 1965 , 204.
77. F. Е. Hoecker, J. W. Watkins. Intern. J. Appl. Radiation Isotopes, 3, 31 (1958).
78. H. A. Kostalas. Intern. J. Appl. Radiation Isotopes, 18, 783 (1967).
79. P. Y. Feng. Nucleonics, 16, No 10, 114 (1958).
80. L. Wiesner. Selected Topics in Radiation Dosimetry. Vienna, IAEA, 1961, p. 361.
81. A. W. Carviker. Rev. Sci. Instrum., 37, 1045 (1966).
82. J. W. Lentsch, R. A. Finston. Phys. Med. Biol., 12, 543 (1967).
83. P. J. Ebert, K. A. Hardy, D.G. Cadena. Radiation Res., 26, 178 (1965).
84. B. Whitelaker. Intern. J. Appl. Radiation Isotopes, 19, 427 (1968).
85. H. S. Judeikis, H. Hedgpeth, S. Siegel. Radiation Res., 35, 247 (1968).
85а. В. К. Посольский, Б. А. Вонсяцкий, Я. И Лаврентович, А. №. Кабакчи. Атомная энергия, 35, 427 (1973).
86. L. A. Harrah. Radiation Res., 39 , 223 (1969).
87. L. A. Harrah. Radiation Res., 41, 229 (1970).
88. L. A Harrah. Trans. Am. Nucl. Soc , 17, 278 (1970).
89. K. 11. Chadwick. Radiation Res., 44, 282 (1970).
90. A'. Tamura, J. Oshirna, K. Yotsumoto, H. Sunaga. Japan J. Appl. Phys., 9, 1148 (1970).
91. J. A. Simmons. Phys. Med. Biol., 15, 748 (1970).
92. A’. Prokert, W. Stolz. Isotopenpraxis, 6, 325 (1970).
93. W Mehringer. Phys. Med. Biol., 16, 311 (1971).
94. 11. J. Berry, С. H. Marshall. Phys. Med. Biol., 14, 585 (1969).
95. F. Gazea, M. JNicolae. Rev. Romaine Phys., 16, 385 (1971).
96. И. M. Розман, К. Г. Килин. Атомная энергия, 2, 70 (1957).
97. В. В. Генералова, М. Н. Гурский, О. А. Гундер, Г. В. Желин-ская, А. Г. Сизых. Монокристаллы, сцинтилляторы и органические люминофоры, вып. 4. Харьков, 1969, стр. 201.
98. В. В. Генералова, Б. А. Вайншток, Н. Ф. Левченко. Там же, стр. 194.
99. F. II. Attix. Nucleonics, 17, No 4, 142 (1959).
100. Е. Brannen, G. L. Old. Phys. Med. Biol., 5, 37 (1960).
101. L. Keszthelyi, I. Berkes, I. Demeter, l.Fodor. Nucl. Instrum. Meth., 10, 193 (1961).
102. В. В. Генералова, Б. А. Вайншток, H. Ф. Левченко. Химия высоких энергий, 3, 90 (1969).
103. Е. А. Либерман. Биофизика, 1, 575 (1956).
104. К. Mallet, II. Constans. Compt. rend., 249, 953 (1959).
105. Б. А. Вайншток, В. В. Генералова, II. Ф. Левченко. Радиационная дозиметрия и спектрометрия ионизирующих излучений. Ташкент, «Фан», 1970, стр. 178.
106. Я. И. Лаврентович, А. Б. Зверев, А. А. Беликовский, А. М. Кабакчи. Химия высоких энергий, 3, 146 (1969).
107. А. Б. Зверев, Я. И. Лаврентович, А. М. Кабакчи. Там же, стр. 453.
108. Я. И. Лаврентович, Г. С. Якименко, А. Г. Старенький, А. М. Кабакчи. Там же, стр. 464.
109. G. М. Murray. Nucleonics, 21, No. 12, 50 (1962).
110. М. Day, G. Stain. Nucleonics, 8, No. 2, 34 (1951).
111. E. Henley, A. Miller. Nucleonics, 9, No. 6, 62 (1951).
112. E. J. Henley. Nucleonics, 12, No. 9, 62 (1954).
113. E. Henley, D. Richman. Anal. Chem., 28, 1580 (1956).
114. M. Day,G. Stein. Nature, 168, 644 (1951).
115. G. Oster, B. Broyde. Nature, 184, 545 (1959).
116. J. J. Furrell, R. L. Vale. Nucleonics, 21, No. 11, 78 (1963).
117. G. Taplin, K. Molin. Radiation Res., 14, 510 (1961).
118. C. Willis, O. A. Miller, A. E. Rothwell, A. W. Boyd. Radiation Res., 35, 1580 (1968).
119. R. L. Vale, J. J. Farrell. Брит. пат. 920689 (1963).
120. К. Pjoertner. Пат. ФРГ 1142038 (1963).
121. A. Matsumoto, T. Tsukada. Японск. пат. 13539 (1967).
122. N. W. Holm, J. E. Maul. Датск. пат. 98773 (1961).
123. A. Prevot. Compt. rend., 230, 288 (1950).
124. А. Чарлзби. Ядерные излучения и полимеры. М., ИЛ, 1962, стр. 108.
125. Л. Я. Малкее, Ф. Ш. Муртазин, Л. В. Шубина, М. 11. Гурский. Сб. «Дозиметрия больших доз». Ташкент, «Фан», 1966, стр. 83.
126. М. JNicolae. Dosimetry in Agriculture, Industry, Biology and Medicine. Vienna, IAEA, 1973, p. 555.
127. К. H. Chadwick. Ibid., p. 563.
128. К. H. Chadwick. Atompraxis, 15, No. 3, 181 (1969).
129. R. J. Berry, J. F. В. Dealler. Phys. Med. Biol., 15, 748 (1970).
130. К. H. Chadwick, J. J. Ten Bosch, W. F. Verheist. Dosimetry in Agriculture, Industry, Biology and Medicine. Vienna, IAEA, 1973, p. 569.
131. W. L. McLaughlin, P. E. lljortenberg, В. B. Badak. Ibid., p. 577.
132. R. K. Broszkiewicz, Z. Bulhak. Ibid., p. 599.
133. H. Eisen, M. Rosenstein, J. Silverman. Ibid., p. 615.
134. F. X. Rizzo, К. Krishnamurthy. Trans. Am. Nucl. Soc., 12, No 1,61 (1969).
135. W. L. McLaughlin. Manual on Radiation Dosimetry (Ed. N. W. Holm and R. ,T. Berry). N. Y., Marcel Decker, Inc., 1970, p. 377.
136. C. G. Orton. Ibid., p. 357.
137. G. N. Flanagan. Pbotogr. Sci. Eng., 12, 117 (1968).
138. W. L. McLaughlin. Цит. no [131].
139. W. L. McLaughlin, E. K. Hussmann. Large Radiation Sources for Industrial Processes. Vienna, IAEA, 1969, p. 579.
140. А. К. Дмитриев, H. Г. Коньков, C. JO. Крылов. Сб. «Дозиметрия и радиационные процессы в дозиметрических системах». Ташкент, «Фан», 1972, стр. 57.
141. В. А. Муминов, И. В. Шубин, Ф. Т. Золотаревская. Там же, стр. 62.
142. К. И. Никулин, Л. М. Кухтина, Л. 3. Зиганшин. Там же, стр. 66.
143. Г. Н. Пьянков, В. С. Жихарев, А. Н. Кордикова, А. М. Кабакчи. Там же, стр. 103.
144. И. М. Блаунштейн, М. Б. Кишиневская, Н. У. Рахимбаева. Там же, стр. 107.
145. И. М. Блаунштейн, Ф. Т. Золотаревская, А. Г. Икрамов. Там же, стр. 110.
146. В. С. Грамматикати, М. П. Гринев, 3. Ф. Ершова, Л. Л. Козлов, Т. Г. Литвинова, Л. М. Михайлов, А. А. Молин, Г. М. Панченков. Там же, стр. 113.
147. Т. Г. Литвинова, М. П. Гринев, В. С. Грамматикати, Л. М. Михайлов, Г. Ф. Марголина. Там же, стр. 118.
148. Л. С. Актабаева, В. А. Зюбко, Б. М. Терентьев. Там же, стр. 130.
149. Л. М. Коваленко, Л. II. Гайченко, Я. И. Лаврентович. Там же, стр. 174.
150. Я. И. Лаврентович, Л. М. Коваленко, А. Г. Старенький, А. А. Беликовский, А. М. Кабакчи. Там же, стр. 178.
151. Я. И. Лаврентович, Г. С. Якимченко, А. Б. Зверев, А. М. Кабакчи. Атомная энергия, 27, 296 (1969).
152. Г. М. Панченков, Л. Л. Козлов, Л. А. Молин, 3. Ф. Ершова. Тезисы докладов Второй Всесоюзной конференции по прикладной радиационной химии (Обнинск, 1973). Черкассы, 1973, стр. 119.
153. Я. И. Лаврентович, Л. Я. Хорликова, А. Г. Старенький, В. В. Генералова, А. М. Кабакчи. Там же, стр. 120.
153а. Л. М. Коваленко, Л.Н. Гайченко, Я. И. Лаврентович, А. М. Кабакчи. Атомная энергия, 31, 510 (1971).
154. A.L. Frank, Е. V. Benton. Radiation Effects, 2, 269 (1970).
155. M. H. Гурский, Г. В. Желинская. Дозиметрия и радиационные процессы в дозиметрических системах. Ташкент, «Фан», 1972, стр. 122.
156. А. Г. Сизых, В. П. Мирошникова. Там же, стр. 126.
157. В. А. Вайншток, Я. Ф. Левченко. Там же, стр. 135.
158. Г. В. Желинская, А. Г. Сизых. Там же, стр. 147.
159. С. Таймути, Р. Гласс, Б. Дивер. Труды II Международной конференции по мирному использованию атомной энергии. Избранные доклады иностранных ученых, т. 10. М., Атомиздат, 1959, стр. 409.
160. Э. Харт, Г. Кох, Б. Петри, Дж. Шульман, С. Таймути, Г. Викоф. Там же, стр. 419.
161. J. Н. Schulman, С. С. Klick, Я. Rabin. Nucleonics, 13, No 2, 30 (1955).
162. И. Rabin, W. E. Price. Nucleonics, 13, No 3, 33 (1955).
163. R. G. Ginther, J. H. Schulman. Nucleonics, 18, No 4, 92 (1960).
164. J. H. Schulman, W. Schurcliff, B. Ginther, F. H. Attix. Nucleonics, 11, No 10, 52 (1953).
165. TV. J. Kreidl, G. E. Blair. Nucleonics, 14, No. 1, 82 (1956).
166. iS. Davison, S. Goldblith, В. E. Proctor. Ibid., p. 34.
167. G. E. Blair. J. Am. Ceramic Soc., 43, 426 (1960).
168. N. J. Kreidl, G. E. Blair. Nucleonics, 14, No 1, 56 (1956).
169. W. A. Hedden, J.F. Kircher, B. W. King. J. Am. Ceramic. Soc., 43, 413 (1960).
170. J. Paymal, M. Bonnaud, P. LeClerc. J. Am. Ceramic. Soc., 43, 430 (1960).
171. W. A. Weyl, J. H. Schulman, R. J. Ginther, L. W. Evans. J. Electrocbem. Soc., 95, No. 2, 70 (1949).
172. J. H. Schulman, R. J. Ginther, С. C. Click, R. S. Alger, R. A. Levy. J. Appl. Phys., 22, 1479 (1951).
173. L. Kiigler. Atomkernenergie, 4, 67 (1959).
174. J. Paymal, M. Bonnaud, P. LeClerc. Glass Ind., 40, 771 (1959).
175. A. M. Bishay. Phys. Chem. Glasses, 2, No 2, 33 (1961).
176. A. M. Bishay. Nucleonics, 19, No. 9, 88 (1961).
177. K. Otley, W. Weyl. J. Appl. Phys. 23, 499 (1952).
178. K. Przibram, E. Kara-Michailova. Ber., 131, 511 (1962).
179. K. Przibram, E. Kara-Michailova. Ber., 134, 233 (1965).
180. W. Weyl. Ind Eng. Chem., 34, 1035 (1942).
181. R. Alger, R. Levy. Phys. Rev., 81, 657 (1951).
182. J. H. Schulman, H. Etzel. Science, 118, 184 (1953).
183. В. В. Ткаченко, Б. А. Брискман, Л. M. Коваленко, Я. И. Лаврентович, II. Ф. Орлов. Атомная энергия, 35, 210 (1973).
184. I. Kugler. Atomkemenergie. 4, 105 (1959).
185. R. Yokota, S. Naka/ima. Health Phys., 5, 219 (1961).
186. R. Yokota J. Phys. Soc Japan, 20, 1537 (1965).
187. R. J. Ginther, j. H. Schulman. Trans. Nucl. Sci., 5, No 3, 92 (1958).
188. L. Bollinger, G. Thomas, R. Ginther. Rev. Sci. Instrum., 30, 1135 (1959).
189. D.G. Anderson, J. Dracass, T. P. Flanagan. J. Electronics and Control!, 7, 463 (1959).
190. R. E. Ginther. Trans. Nucl. Sci., 7, No. 2/3, 28 (1960).
191. D. G. Anderson, J. Dracass, T. P. Flanagan. Proc. 5th Intern. Conference on Instruments and Methods, vol. 2. Stockholm, 1960, p. 616.
192. G. Thomas, L. Bollinger, R. Ginther. WAX. Am. Phys. Soc., 5, 226 (1960).
193. A. M. Bishay. J. Am. Ceramic Soc., 44, 231 (1961).
194. F. Kirk, G. Slaughter, R. Ginther. Nucl. Instrum, and Methods, 13, 313 (1961).
195. L. Bollinger. G. Thomas. Rev. Sci. Instrnm., 32, 1044 (1961).
196. L. Bollinger, G. Thomas, R. Ginther. Nucl. Instrum, and Methods, 17, 97 (1962).
197. D. G. Anderson. Advances in Glass Technology. N. Y., 1963, p. 429.
198. C. Coceva. Nucl. Instrum, and Methods, 21, 93 (1963).
199. A7. F. Barr, M D. Stark, J. S Laughlin. Radiology, 76, 113 (1961).
200. M. Hodara, M. Friedman, G. J. Hine. Radiology, 73, 693 (1959).
201. A. Rigert, J. W. T. Spinks. Intern. J. Appl. Radiation Isotopes, 3, 125 (1958).
202. TV. F. Barr, M. D. Stark, J. Hands, J. C. Laughlin. Health Phys., 7, 48 (1961).
203. G. J. Hine, M. Hodara, M. Freidman. Radiology, 78, 44 (1962).
204. A. Rigert, H. Johns, J. W. T. Spinks. Nucleonics, 14, No. 11, 134 (1956).
205. 5. Kondo. Health Phys., 4, 21 (1960).
206. S. Kondo. Health Phys., 7, 25 (1961).
207. D. N. Tapper, M. M. Hold, C. L. Kowar. Health Phys., 8, 207 (1962).
208. M. M. Nold, D. N. Tapper, C. L. Kowar. Ibid., p. 217.
209. 5. Kondo, T. Kato. Ann. Rep. Natl. Inst. Genetics of Japan, 10, 155 (1960).
210. C.G. Amato, S. J. Maisky. Radiology, 76, 290 (1961).
211. S. J. Maisky, C. G. Amato, B. Roswit, С. B. Reild, S. M. Unger, С. C. Spreckels, E. Villason. Trans. N. Y. Acad. Sci., 22, 104 (1959).
212. S. J. Maisky, C.G. Amato, C.B. Reid, С. C. Spreckels, L. Maddalone. Am. J. Roentg. Rad. Ther., 85, 568 (1961).
213. B. Roswit, S. J. Maisky, C. G. Amato, С. B. Reid, L. Maddalone. Radiology, 76, 295 (1961).
214. B. Roswit. Am. J. Roentg. Rad. Ther., 85, 572 (1961).
215. R. Yokota, S. Nakajima, J. Uesugi. Radioisotopes, 10, 387 (1961).
216. P. Levy. J. Am. Ceramic Soc., 43, 389 (1960).
217. С. M. Бреховских. Атомная энергия, 8, 37 (1960).
218. С. М. Бреховских. Стекло и керамика, № 1, 10 (1958).
219. С. М. Бреховских, И. II. Борисова. Стекло и керамика, № 1, i (1962).
220. 3. Спурны. Атомная энергия, 10, 172 (1961).
221. А. М. Bishay. J. Am. Ceramic Soc., 44, 545 (1961).
222. TV. Kreidl, J. Hensler. J. Am. Ceramic Soc., 38, 423 (1955).
223. Г. Я. Васильев, А. Ф. Усатый, Ю. С. Лазуркин, А. М. Марков. Докл. АН СССР, 125, 1219 (1959).
224. Р. Aters. J. Opt. Soc. Am., 51, 1251 (1961).
225. A. M. Bishay. J. Am. Ceramic Soc., 45, 389 (1962).’
226. R. S. Barker, D. A. Richardson, E. A. McConvey, R. Rimmer. Nature, 187, 133 (1960).
227. R. Bouman. Nucleonics, 14, No. 6, 90 (1956).
228. Z. Spumy. Jaderna Energ., 6, 163 (1960).
229. M. Tashira, S. Sakka, N. Soga. Jogyo Kyokai Sci., 68, 191 (1960).
230. 5. Kondo. J. Phys. Soc. Japan, 13, 224 (1958).
231. T. Brustad, T. Henriksen. Am. J. Roentg. Rad. Ther., 8U, 117 (1958).
232. J. H. Schulman, F. H. Attix, E. West, R. Ginther. Rev. Sci. Instrum., 31, 1263 (1960).
233. E. Piesch. Atompraxis, 10, 268 (1964).
234. C. G. Amato, S. J. Maisky, V. P. Bond. Radiology, 76, 292 (1961).
235. S. J. Maisky, C.G. Amato, Г. P. Bond, J. S. Robertson, B. Roswit. Radiation Res., 21, 462 (1964).
236. J.F. Fowler. Nucleonics, 21, No 10, 60 (1963).
237. R. Yokota, S. Nakajima. Health Phys.. 11, 241 (1965).
238. J. H. Schulman. Solid State and Chemical Dosimetry in Medicine and Biology. Vienna, IAEA, 1967, p. 3.
239. R.D. Kirk, J. H. Schulman, E. J. TFest, A. E. Nash. Ibid., p. 91.
240. J. Kastner, D. Eggenherger, A. Longnecker, D. King, D. Schutt. Ibid., p. 115.
241. R. Regulla, H. Pychlau, F. Washmann. Ibid., p. 121.
242. K. Berger. Ibid., p. 131.
243. R. Maushart, E. Piesch. Ibid., p. 157.
244. В. В. Ткаченко. Радиационная дозиметрия и спектрометрия ионизирующих излучений. Ташкент, «Фан», 1970, стр. 169.
245. Н. 3. Андреева, И. Н. Добрецоеа, Н. Ф. Орлов, В. М. Трофимов. Там же, стр. 96.
246. Z. Spumy. Personnel Dosimetry for Radiation Accidents. Vienna, IAEA, 1965; p. 137.
247. R. S. Barker, D. A. Richardson. .1. Am. Ceramic Soc., 44, 552 (1961).
248. K. Becker. Kerntechnik. 6, 199 (1964).
249. K. Becker. Health Phys.. 6. 523 (1965).
250. K. Becker. Nucl. Instrum, and Methods, 36, 323 (1965).
251. K. Becker. Nukleonik, 5, 154 (1963).
252. C.G. Amato, S. J. Maisky, B. Roswit. Trans. N. Y. Acad. Sci., 23, 357 (1961).
253. J. A. Auxier. С. H. Bernard, W. T. Thornton. Selected Topics in Radiation Dosimetry. Vienna, IAEA, 1961, p. 503.
254. K. Becker. Health Phys., 8, 812 (1965).
255. K. Becker. Personnel Dosimetry for Radiation Accidents. Vienna, IAEA, 1965, p. 169.
256. II. Francois, Y. Bourbigot, A. M. Grand-Clement, G. Portal, G. Soudain. Ibid., p. 319.
257. K. Becker. T. N at urf orach., 19A, 1223 (1964).
258. С. H. Bernard, W. T. Thornton, J. A. Auxier. Health Phys., 4, 236 (1961).
259. A. M. Bishay. J. Chem. UAR, 6, 135 (1963).
260. B.G. Blaylock J. P. Witherspoon. Health Phys., 11. 549 (1965).
261. И. А. Бочвар. II. Б. Кериим-Маркус, А. А. Моисеев, T. II. Прошина,
В. В. Якубик. Атомпая энергия, 19, 311 (1965).
262. И. А. Бочеар, Т. Б. Кериим-Маркус, А. А. Васильева, Т. И. Прошина, 3. М. Сырицкая, В. В. Якубик. Атомная энергия, 15, 48 (1963).
263. Т. Brustad, Т. Henriksen. Am. J. Roentg. Rad. Ther., 81, 509 (1959).
264. T. Brustad, T. Henriksen. Rrit. J. Radiol., 31, 163 (1958).
265. J. S. Cheka. Health Phys., 8, 551 (1962).
266. J. S. Cheka. Health Phys., 10, 303 (1964).
267. J. Gegelman, A. B. Callahan, G. P. Fulton. Radiation Res., 6, 548 (1957).
268. E. Eklund, R. Timmerman. Trans. Am. Nucl. Soc., 4, 329 (1961).
269. M. Frank, IF. Stolz. Kernenergie, 8, 541 (1965).
270. K. D. Friddell. Trans. Nucl. Sci., 11, No. 5, 155 (1964).
271. H. J. Hardt. Personnel Dosimetry for Radiation Accidents. Vienna, IAEA, 1965, p. 127.
272. H. J. Hardt, A. Rudloff. Atompraxis, 9, 45 (1963).
273. R. W. Hawley. Ind. Atom., 9, No 1, 87 (1965).
274. K. Hayakawa, K. Nunome, K. Kawase, S. Ikura. Nagoya Kogyo Gijutsu Shikensho Hokoku, 14, 338 (1965).
275. И. Б. Нериим-Маркус, В. В. Якубик, И. А. Бочеар, 3. М. Сырицкая, А. А. Васильева, Т. И. Прошина. Стекло и керамика, 16, 75 (1963).
276. Н. Kiejer, R. Maushart, F. Piesch.'Atompraxis, 11, 88 (1965).
277. N. J. Kreidl, G. E. Blair. Nucleonics, 17, No. 10, 58 (1959).
278. С. K. Menkes. Health Phys., 12, 429 (1966).
279. G. A. Monk. Nucleonics, 10, No 11, 52 (1952).
280. U. Proesch. Radiation Chemistry. Budapest, 1964, p. 453.
281. F. H. Ritz, С. H. Cheek. Radiation Res., 25, 537 (1965).
282. J. H. Schulman. Proc. II United Nations Intern. Conference on Peaceful Uses of Atomic Energy, vol. 21. Geneva, 1958, p. 220.
283. E. Tochilin, J. T. Lyman, F. H. Attix, E. J. West. Radiation Res., 19, 200 (1963).
284. G. A. M. Webb. Personnel Dosimetry for Radiation Accidents. Vienna, IAEA, 1965, p. 149.
285. R. Yokoto, S. Sakai. Selected Topics in Radiation Dosimetry. Vienna, IAEA, 1961, p. 497.
286. В. M. Трофимов, Д. Г. Галимов, И. Н. Добрецова, Н. Ф. Орлов, Д. М. Юдин. Атомная энергия, 30, 74 (1971).
287. В. М. Трофимов, Н. Ф. Орлов, Н. 3. Андреева. Атомпая энергия, 27, 155 (1959).
288. J. Barthe, D. Blanc, L. Commanay, J. L. Teyssier, H. Francois. Health Phys., 18. 573 (1970).
289. Г. В. Бюргановская, E. Г. Гвоздева, А. И. Хованович. Атомная энергия, 21, 38 (1966).
290. V. Labau, M. Nicolae. M. Matache, V. Spiridon. Dosimetry in Agriculture, Industry, Biology and Medicine. Vienna, IAEA, 1973, p. 51.
291. J. Barthe,D. Blanc, J.M.Wauquier, L. Commanay, H. Francois. Ibid., p. 67.
292. D. F. Regulla. Ibid., p. 215.
293. Г. С. Бологова, Б. А. Брискман, В. В. Ткаченко, В. П. Трусова. Дозиметрия и радиационные процессы в дозиметрических системах. Ташкент, «Фан», 1972, стр. 162.
294. И. А. Бочеар, Д. В. Викторов, В. В. Ткаченко, В. М. Трофимов. Там же, стр. 182.
295. Ю. С. Рябухин, В. В. Ткаченко, Г. С. Бологова, Т. В. Вахлакова, Г. М. Обатуров, А. Г. Васильев. Мед. радиол., 14, № 8, 66 (1959).
296. В. П. Трусова, Г. С. Бологова, А. Г. Васильев. Атомная энергия, 26, 457 (1969).
297. М. Adie. Пат. США 3089957 (1963).
298. R. Bedier, S. Carpentier, Н. Francois, А. М. Grand-Clement, J. 'Menerel. Франц, пат. 1327099 (1963); брит. пат. 974157 (1964).
299. А. М. Bishay. Пат США 3052637 (1962).
300. S. Carpentier, R. Delarue, Н. Francois, А. М. Grand-Clement, С. Portal, S. Soudain. Франц, пат. PV—8203 (1965).
301. W. В. Clarke, R. J. Meltzer. Hat. США 3136689 (1964).
302. Compagnie de Saint-Gobain. Брит. пат. 912192 (1963); 989078 (1965).
303. D. Hale. Пат. США 3073955 (1965).
304. H. P. Hood. Пат. США 3173850 (1965).
305. C. R. Hornier, W. E. Gray. Пат. США 3169188 (1965).
306. R. Just. Пат. США 3093734 (1963).
307. R. Just, A. Waly. Пат. США 3042082 (1962).
308. P. G. LeClerc. Канад, пат. 655091 (1963).
309. J. A. Paymal. Hat. США 3046400 (1962).
310. J. H. Schulman. Брит. пат. 981888 (1965).
311. В. Shenker, В. P.Fabricand. Пат. США 3100262 (1963).
312. R. Yokota, J. Muto, K. Hukuda. Пат. ФРГ 2135656 (1972).
313. Matsushita Electric Ind. Co. Франц, пат. 2114801 (В) (1972).
314. В. М. Трофимов, Н. Ф. Орлов, Н. 3. Андреева. Авт. свид. СССР 281666 (1972); Бюлл. изобр., № 22, 248 (1972).
315. В. М. Трофимов, Н. Ф. Орлов, Н. 3. Андреева. Авт. свид. СССР 314352 (1972); Бюлл. изобр., № 33, 176 (1972).
316. J. Н. Schulmann. Solid State Dosimetry (Ed. by S. Amelinckx et., al.). New York—London—Paris, 1969, p. 107.
317. K. Becker. Ibid., p. 153.
318. J. R. Cameron. Manual on Radiation Dosimetry (Ed. N. W. Holm and R. J. Berry). N. Y., Marcel Dekker, 1970, chap. 5.
319. Г. В. Бюргановская, В. В. Варгин, Н. А. Леко, Н. Ф. Орлов. Действие излучений на неорганические стекла. М., Атомиздат, 1968.
320. J. R. Cameron, Й. Suntharalingam, G. Й. Kenney. Thermoluminescent Dosimetry. L., 1968.
321. M. Франк, В. Штольц. Твердотельная дозиметрия ионизирующего излучения. М., Атомиздат, 1973.
322. А. М. Кабакчи, Я. И. Лаврентович, В. В. Пеньковский. Химическая дозиметрия ионизирующих излучений. Киев, Изд-во АП УССР, 1963, стр. 104.
323. С. Archundia, A. Moreno, М. Navarrete, S. Alva. Dosimetry in Agriculture, Industry, Biology and Medicine. Vienna, IAEA, 1973, p. 187.
324. Y. Ishikawa, T. Morii. Bull. Inst. Chem. Res. Kyoto Univ., 44, 84 (1966).
325. В. С. Бубнов, И. Б. Кериим-Маркус, Т. Н. Смирнова. Мед. радиол., 5, № 3, 61 (1961).
326. D. Kahn. Acta Radiol., 46, 563 (1965).
327. Н. Friedmann, С. Glover. Nucleonics, 10, No 6, 24 (1958).
328. W. Burns, B. Lockjer, J. Sci. Instrum., 32, 316 (1955).
329. F. Daniels, C. A. Boyd, D. F. Saunders. Science, 117, 343 (1953),
330. W. Kossel, U. Mayer, H. C. Wolf. Naturwiss., 41, 209 (1954).
331. В. А. Антонов-Романовский, И. Б. Кериим-Маркус, М. С. Прошина, 3. А. Трапезникова. Конференция АН СССР по мирному использованию атомной энергии (Москва, июль 1955 г.). Сессия Отделения физ,-мат. наук. М., Изд-во АН СССР, 1956, стр. 239.
332. R. J. Ginther, R. D. Kirk. J. Electrochem. Soc., 104, 365 (1957).
333. H. W. Etzel, J. H. Schulman. J. Chem. Phys., 22, 1549 (1954).
334. С. E. Mandeville, H. O. Albrecht. Phys. Rev., 91, 566 (1953).
335. J. R. Cameron, F. Daniels, N. Johnson, G. Kenney. Science, 134, 333 (1961).
336. J. H. Schulman, H. J. Ginther, R. D. Kirk, H. S. Goulart. Nucleonics, 18, No. 3, 92 (I960).
337. J. H. Schulman, E. J. West. Rev. Sci. Instrum., 34, 863 (1963).
338. C. Brooke, R. Schayes. Solid State and Chemical Dosimetry in Medicine and Biology. Vienna, IAEA, 1967, p. 31.
339. P. R. Almood, A. Wright, J. E. Lontz. Ibid., p. 53.
340. S. Benner, J. M. Johansson, L. Lindskoug, P. T. .Nyman. Ibid., p. 65.
341. M. Lorthioir. Ind. Atom., 8, No. 11, 113 (1963).
342. G. N. Kenney, J. R. Cameron, D. W. Zimmerman. Rev. Sci. Instrum., 34, 769 (1963).
343. J. R. Cameron, D. W. Zimmerman, G. N. Kenney, R. Buch, R. Bland, R. Grand. Health Phys., 10, 25 (1964).
344. C. J. Karzmark, J. White, J. F. Fowler. Phys. Med. Biol., 9, 273 (1964).
345. D. W. Zimmerman, C. R. Rhyner, J. R. Cameron. Health Phys., 12, 525 (1966).
346. M. J. Marrone, F H. Attix. Health Phys., 10, 431 (1964).
347. J. R. Cameron, L. DeWerd, J. Wagner, C. Wilson, K. Doppke, D. W. Zimmerman. Solid State and Chemical Dosimetry in Medicine and Biology. Vienna, IAEA, 1967, p. 77.
348. В. E. Bfarngand, U. Jones. Ibid., p. 99.
349. M. Michailovit. Ibid., p. 107.
350. P. R. Almond, R. J. Shalek. Ibid., p. 149.
351. J. S. Laughlin, R. J. Shalek, J.Ovadia, J.G. Holt. Phys. Med. Biol., 10, 429 (1965).
352. E. M. Fielden, E. J. Hart. Adv. Cbem. Ser., 81, 585 (1968).
353. N. Goldstein, E. Tochilin Radiation Res., 32,- 280 (1967).
354. Z. Spumy. At. Energy Rev., 3, No. 2, 61 (1965).
355. J. G. Fowler, F. H. Attix, Radiation Dosimetry (Ed. F. H. Attix and W. C. Roesch), vol. 2. N. Y., 1966, chap. 13.
356. A. R. Reedy, K. Ayyanger,G. L. Brownell. Radiation Res., 40, 552 (1969).
357. N. Grogler, F. G. Houtermans, H. Staufer. Proc. II United Nations Intern. Conference on Peaceful Uses of Atomic Energy, vol. 21. Geneva, 1958, p. 226.
358. L. Luchner. Z. Phys., 149, 435 (1957).
359. J. E. Anderson, S. E. Williams. Aust. J. Pbys., 14, 368 (1961).
360. L. L. Anderson, T. L Duffy, J. Sedlet, D. P. O'Neil. Personnel Dosimetry for Radiation Accidents. Vienna, IAEA, 1965, p. 645.
361. P. Braunlich, A. Scharmann. Nucleonik, 4, 65 (1962).
362. W. D. DeVore, E. A. Fonts. Radiology, 84, 128 (1965).
363. R. C. Fix, R. C. McCall. Trans. Am. Nucl. Soc., 6, 413 (1963).
364. J. G. Fowler, E. Shuttleworth, V. Scarcer, J. T. Whitte, C. J. Karzmark. Nature, 207, 997 (1965).
365. M. Frank. Kernenergie, 6, 76 (1963).
366. M. Frank, H.J. Dubrau, H. Lingertat, L. Herforth. Kernenergie, 7, 570 (1964).
367. M. Frank, L. Herforth Kernenergie, 5, 173 (1962).
368. P. Goerlich, H. Karras, W. Luedke, H. Mothes, R. Reimann. Phys. Status Solidi, 3, 478 (1963). »
369. N. Haring. M. Schon. Selected Topics in Radiation Dosimetry. Vienna, IAEA, 1961, p. 541.
370. J. S. Handloser. Personnel Dosimetry for Radiation Accidents. Vienna, IAEA, 1965, p. 115.
371. L. Herforth, M. Frank. Czech. J. Phys., 13B, 219 (1963).
372. N. Itoh, T. Suita. Technology Rep. Osaka Univ., 12, 291 (1962).
373. C. J. Karzmark, J. F. Fowler, J. White. Intern. J. Appl. Radiation Isotopes, 17, 161 (1966)
374. «9. J. Maisky, C.G. Amato, B. Roswit, С. B. Reid, C. Spreckels. Radiology, 78, 277 (1962)
375. R. C. McCall, L. E. Babcock, R. C. Fix. Radiation Res., 19, 200 (1963).
376. R. C. McCall, R. C. Fix. Health Phys., 10, 602 (1964).
377. G. P. Naylor. Phys. Med. Biol., 10, 564 (1965).
378. R. C. Palmer, E. F. Blase, V. Poirier. Intern. J. Appl. Radiation Isotopes, 16, 737 (1965).
379. J. Patko, M. Berta. Magy. Radiol., 16, 303 (1964).
380. D A Patterson, H. Friedman. J. Opt. Soc. Am., 47, 1136 (1957).
381 W. F Schmid, R.W. Mooney. J. Electrochem. Soc., 104, 340 (1963).
382. J. H. Schulman, F. H. Attix, E. J. West, R. J. Ginther-’Selected Topics cr“' in Radiation Dosimetry. Vienna, IAEA, 1961, p. 531.
383. G. Busuoli, A. Cavallini, A Tasso, O. Rimondi Phys. Med. Biol., 15, 673 (1970).
384. A. S. Harasawa, A. Kitahara, M. Saitoh. Jap. J. Appl. Phys., 9, 1552 (1970).
385. M. H. Дьяченко, M. И. Сеульская. Вопросы эксп. клпнич. радиол., № 6, 197 (1970).
386. F. S. W. Hwang. Nature, 227, 269 (1970).
387. W. J. Vaughan, L. O. Miller. Health Phys., 18, 578 (1970).
388. M. R. Mayhugh, R. V. Christy, A. M. Johnson. J. Appl. Phys., 41, 2968 (1970).
389. К. К. Шварц, 3. А Грант, T. К. Меже, M. M. Грубе. Термолюминесцентная дозиметрия. Рига, «Зинатне», 1968.
390. М. М. Грубе, К. К. Шварц. Изв. АН ЛатвССР, серия физ. и техн, наук, 4, 37 (1970).
391. К. К. Шварц, IO. Е. Тиликс, Д. К. Тоне. Дозиметрия интенсивных потоков ионизирующих излучений. Ташкент, «Фан», 1969, стр. 35.
392. J. R. Cameron. Solid State Dosimetry (Ed. By S. Amelinckx et al.). New York—London—Paris, 1969, p. 695.
393. С. E. /Vollmann, E. E. Smolko. Dosimetry in Agriculture, Industry, Biology and Medicine. Vienna, IAEA, 1973, p. 143.
394. W. Pohlit. Ibid., p. 155.
395. M. A. Gomaa, A. M. El-Naggar. Ibid., p. 165.
396. P. Majborn, L. Botter-Jensen, P. Christensen. Ibid., p. 169.
397. T. Niewiadomski. Ibid., p. 199.
398. M. R. Sachdev, R. K. Nukkoo, S. Somasundaram, C.N. Wadhwani. Ibid., p. 229.
399. E. Blum, K. D. Bewley, J.D. Heather. Ibid., p. 277.
400. Ш. А. Алимов, A. P. Красная, Б. M. Носенко, Л. С. Ревзин, В. Я. Яс-колко. Дозиметрия больших доз. Ташкент, «Фан», 1966, стр. 71.
401. Б. М. Носенко, Л. С. Ревзин, В. Я. Ясколко. Ж. техн, физики, 26, 2046 (1956).
402. Б. М. Носенко, Л. С. Ревзин, В. Я. Ясколко. Докл. АН УзбССР, 4, 3 (1956).
403. Z. Spumy. Kernenergie, 5, 611 (1962).
404. Z. Spumy. Jaderna Energ., 8, 166 (1962).
405. A. P. Красная, Б. M. Носенко, Л. С. Ревзин, В. Я. Ясколко. Атомная энергия, 10, 630 (1961).
406. В. А. Архангельская, Б. И. Вайнберг, В. М. Кодюков, Т. К. Разумова. Атомная анергия, 8, 559 (1960).
407. Н. Peter. Atomkernenergie, 5, 453 (1960).
408. В. А. Архангельская, Б. И. Вайнберг, Т. К. Разумова. Оптика и спектроскопия, 4, 681 (1958).
409. В. Bjarngard. Rev. Sci. Instrum., 33, 1129 (1962).
410. В. Bjarngard. Proc. Ill United Nations Intern. Conference on Peaceful Uses of Atomic Energy, vol. 14. N. Y., 1965, p. 5.
411. J. Felszerfalvi, J. Patko. Magy. tudom. Akad. atom. kut. Intez. kosl., 4, 169 (1962).
412. J. Felszerfalvi, J. Patko. Acta Univ. Debrecen., Ser. Phys., Chem., 9, 5 (1963).
413. J. Patko. Fiz. Szle, 12 1 (1962).
414. J. Patko. Acta Univ. Debrecen., Ser. Phys. Chem., 8, 85 (1962).
415. K. Watanade. Phys. Rev., 83, 785 (1951).
416. G. Rauh, F. Struss. Radiobiol. Radiother., 4, 499 (1963).
417. N. Fiel, H. Keppel. Naturwiss., 42, 634 (1955).
418. В. В. Генералова, M. Б. Кишиневская, Г. В. Поляк. Дозиметрия больших доз. Ташкент, «Фан», 1966, стр. 87.
419. Г. С. Бологова, А. Г. Васильев, Р. А. Николаева, В. П. Трусова. Радиационная дозиметрия и спектрометрия ионизирующих излучений. Ташкент, «Фан», 1970, стр. 102.
420. В. В. Генералова, М. Б. Кишиневская. Там же, стр. 128.
421. В. В. Генералова, М. Б. Кишиневская. Атомная энергия, 27, 564 (1969).
422. Е. П. Ковалева, Е. П. Петряев, Е. П. Калязин. Дозиметрия и радиационные процессы в дозиметрических системах. Ташкент, «Фан», 1972, стр. 167.
423. О. Gal, I. Draganic. Intern. J. Appl. Radiation Isotopes, 22, 753 (1971).
424. O. Gal, L. Petkovic, L. Josimovic, I. Draganic. Intern. J. Appl. Radiation Isotopes, 19, 645 (1968).
425. O. Gal, P. J. Baugh, G. O. Phillips. Intern. J. Appl. Radiation Isotopes, 22, 321 (1971).
426. W. W. Bradshaw, D. G. Cadena, G. W. Crawford, H. A. W. Spetzler. Radiation Res., 17, 11 (1962).
427. Y. Takashima, Y. Nakayama, L. Chandler. Dosimetry in Agriculture, Industry, Biology and Medicine. Vienna, IAEA, 1973, p. 303.
428. R. E. Sanborn, E. L. Beard. Rep. UCRL— 12371 (1965); цит. по [1].
429. E. Tochilin, N.Goldstein, W.G. Miller. Health Phys., 16, 1 (1969).
430. G. Scarpa. Phys. Med. Biol., 15, 667 (1970).
431. G. Scarpa, G. Benincasa, L. Ceravolo. Dosimetry in Agriculture, Industry, Biology, and Medicine. Vienna, IAEA, 1973, p. 207.
432. K. Becker, J. S. Cheka, M. Oherhofer. Health Phys., 19, 391 (1970).
433. K. Becker At. Energy Rev., 8, No 1, 173 (1970).
434. U. Brunsmann, M. Euler, W. Kriegseis, A. Sharmann. Dosimetry in Agriculture, Industry, Biology and Medicine. Vienna, IAEA, 1973, p. 255.
435. J. B. Birks. Proc. Phys. Soc., A63, 1044 (1950).
436. J. B. Birks. Ibid., p. 1294.
437. F. H. Attix. Nucleonics, 17, No. 10, 60 (1959).
438. J. H. Schulman, П. W. Etzel, J. G. Allard. J. Appl. Phys., 28, 792 (1957).
439. В. В. Генералова, M. II. Гурский, Л. И. Спектор, Л. Я. Малкее. Дозиметрия интенсивных потоков ионизирующих излучений. Ташкент, «Фан», 1969, стр. 25.
440. В. В. Генералова, М. Н. Гурский, Г. М. Рудакова, П. С. Самойлов, А. Н. Цой. Дозиметрия больших доз. Ташкент, «Фан», 1966, стр. 95.
441. О. П. Верхградский, В. Н. Дорошенко, И. Н. Червецова. Дозиметрия и радиационные процессы в дозиметрических системах. Ташкент, «Фан», 1972, стр. 132.
442. А. Г. Сизых, Л. II. Спектор, Л. Я. Малкее. Там же, стр. 143.
443. М. С. Юнусов, Г. П. Гринченко, А. Султанов, А. Н. Цой. Там же, стр. 138.
Глава IX
ДОЗИМЕТРИЯ ИМПУЛЬСНОГО ИОНИЗИРУЮЩЕГО
ИЗЛУЧЕНИЯ
В последнее десятилетие в радиационной химии стали часто применяться источники импульсного ионизирующего излучения. Особенностью этих источников является генерация за малые промежутки времени (микросекунды и менее) сравнительно больших доз (до -10е рад). Говоря по-иному, часто приходится иметь дело с весьма высокими мощностями дозы (до ~1014 — 101В рад/сек). При таких мощностях дозы в случае конденсированных сред существенную роль играют реакции рекомбинации первичных продуктов радиолиза в объеме системы. Например, этими реакциями в водных растворах могут быть процессы с участием еДр Н и ОН.
В газообразных системах в рассматриваемых условиях могут протекать реакции рекомбинации электронов и первичных положительных ионов, не имеющие места при низких мощностях дозы. В частности, в случае закиси азота при сверхвысоких мощностях дозы происходит замена реакции присоединения электрона к молекуле N2O
е~ -|- N2O —» продукты (1)
процессом нейтрализации ионов Г1]
е~ -|- N30+ -» продукты. (2)
Очевидно, описанные процессы оказывают влияние на выходы стабильных продуктов радиолиза. Для случая водных растворов особенности радиолитических превращений при высоких мощностях дозы подробно рассмотрены в книге f2j.
Естественно, протекание указанных процессов следует учитывать при использовании той или иной системы для дозиметрии импульсного излучения. Соответствующие исследования, выполненные в различных лабораториях, показывают, например, что практически для всех дозиметров на основе водных растворов при таких мощностях дозы характерно изменение выхода измеряемого химического превращения. Ранние работы по дозиметрии импульсного излучения рассмотрены в книгах 12, 3J и обзорах [4, 5J.
В радиационно-химических экспериментах с импульсным излучением требуется знать следующие основные параметры: дозу за импульс, поглощенную системой, продолжительность им
пульса и форму импульса. Последние два параметра легко измеряются с помощью осциллографа. На рис. 81 в качестве примера приведены осциллограммы импульсов электронов, генерируемых тремя типами ускорителей [6—8]. Импульсы электронов, получаемые с помощью линейных ускорителей, имеют топкую структуру. Один из примеров такой структуры уже был показан на рис. 41 (см. главу II).
Если продолжительность импульса —10'6?сек., то возникающие короткоживущие продукты радиолиза имеют, как правило,
Рис. 81. Осциллограммы электронных импульсов, создаваемых линейным ускорителем [6] (я), ускорителем типа Ван-дер-Граафа (7] (б) и ускорителем типа Кокрофта — Уолтона [8] (е)
гораздо больше длительно-
периоды полупревращения, которые
сти импульса. Реакции с участием этих продуктов, в основном,
протекают после прохождения импульса. Поэтому в таких реакциях большую роль играет доза за импульс (ей пропорциональна концентрация короткоживущих продуктов), чем мощность дозы (она характеризует скорость образования этих продуктов).
В импульсном радиолизе имеют дело с двумя видами дози-
метрических измерений, зависящими от целей эксперимента. Если необходимо определить величину радиационно-химического выхода какого-либо продукта, то требуется знание средней дозы во всем объеме образца. В опытах с оптической регистрацией короткоживущих частиц необходимо знать оптическую плотность короткоживущей частицы и соответствующую ей дозу. В этом случае из-за неравномерности дозного поля внутри ячейки из
меряют поглощенную дозу вдоль пути регистрирующего светового пучка, а не во всем облучаемом объеме.
1. Дозиметр Фрикке
По данным различных авторов [8—22J, (?(Fe3+) в дозиметре Фрикке уменьшается, начиная с мощностей дозы ~108 рад/сек. По мнению авторов работ [8, 9, 12, 14, 17—19], это уменьшение
вызвано конкуренцией реакций
Fe2+ + ОН Fe3+ + ОН-, (3)
Н -|- Ог —» НОг, (4)
Н ф ОН — НгО. (5)
Г. Шварц [3J считает, что рассматриваемый эффект связан с протеканием реакции
НОг + ОН — НгО + Ог. (6)
К. Сегестед и др. [22J отмечают, что главной причиной снижения G(Fe3+) при импульсном облучении растворов ферросульфата является конкуренция реакций (5), (7) и (8) с реакцией (4) за атомы Н.
Н + Н - Н2, (7)
Н -|- НО‘2 —* Н2О2. (8)
В работах [12, 16, 19J найдено, что уменьшение G(Fe3+) при высоких мощностях дозы еще больше, если раствор содержит NaCl. Возможно, это обусловлено конкуренцией за атомы С1, образовавшиеся в реакции [2J
С1- + ОН + Н+ — С1 + НгО, (9)
между атомами Н
С1 + Н -> Cl- + Н+ (IO-
ii ионами Fe2+
Fe2+ 4- Cl Fe3+ + Cl~. (11)
Увеличение концентрации ионов Fe2+ до 10~2 М и насыщение раствора кислородом приводят к тому, что G(Fe3+) становится практически независимым от мощности дозы вплоть до ~6-•109 рад/сек [9, 14, 16, 19, 20, 22J *. В табл. 57 приведены резуль-
Таблица 57
Значении G(Fe3+) для ферросульфатной дозиметрической системы при высоких мощностях дозы (длительность импульса электронов— 1,4 мксек)
Мощность дозы, [раЭ/сек G(Fe3+), ионы/100 эв Мощность дозы, ра0/сек G(Fe’+), ионы/100 эв
10-3 Af раствор Fe2<- в 0,4 М H2SO4 (насыщение воздухом) 10~2 М раствор Fe2+B0,4Af H2SO4 (насыщение кислородом) 10-3 М раствор Fe2+ в 0,4 М H2S04 (насыщение воздухом) 10~2 М раствор Fe2+ в 0,4М HsSO, (насыщение кислородом)
<108 15,60 16,07 5,72-109 13,76 15,88
7,14-108 15,33 16,07 1,14-101п 12,68 15,65
1,43-10’ 15,04 16,07 2,29-1010 11,33 15,20
2,86-Ю9 14,55 16,02 4,57-1010 9,64 14,61
* При у-радиолизе раствора указанного состава G(Fe3+) = 16,07 иона/100 эв
таты измерений G(Fe3+) для растворов ферросульфата указанных выше составов, выполненных недавно К. Сегестедом и др. [20, 22J. Согласно [16J, G(Fe3+) в насыщенном кислородом 10~2 М растворе Fe2+ в 0,4 М H2SO4 составляет 11,2 иона/100 эв при мощности дозы ~ 1011 рад/сек.
Таким образом, насыщенный кислородом 10’2 М раствор Fe2+ в 0,4 М H2SO4 (его иногда называют «супердозиметром» Фрикке) пригоден для определения доз импульсного излучения при мощ ностях дозы до~ 6*109 рад/сек. При проведении дозиметрии не рекомендуется добавлять NaCl к этому раствору. Для приготовления раствора в этом случае необходимо использовать очищенную воду (по крайней мере, трижды перегнанную) и реактивы марки «х. ч.»
2. Другие дозиметрические водные системы
В работах [13, 14, 17, 18, 24J было обнаружено, что G(Ce3+) в цериевой дозиметрической системе (10~4 — 10~3 М раствор Се4+ в 0,4 М H2SO4) возрастает, начиная с мощности дозы ~108 рад/сек. Например, для 2-10‘4 М раствора Се4+ в 0,4 М H2SO4 G(Ce3+) при мощности дозы ~-8-109 рад/сек равен ~ 3,3 пона/100 эв [23J. Этот эффект удовлетворительно объясняется конкуренцией реакций
ОН + ОН —* Н2О2, (12)
Се3+ + ОН Се4+ + ОН-. (13)
Следовательно, цериевая система малопригодна для дозиметрии импульсного излучения при мощностях дозы выше —108 рад/сек.
Согласно I25J. выход обесцвечивания метиленового голубого в водных растворах существенно уменьшается в условиях импульсного электронного облучения. Так, было найдено, что в случае 5*10 6 М раствора этого красителя, насыщенного воздухом, доза, необходимая для уменьшения оптической плотпостп этого раствора до 37% от исходной, при мощности дозы ~ 1010 рад/сек примерно в 2—Л раза больше, чем при низких мощностях дозы.
Э. Бьергбакке и К. Сегестед [26] исследовали зависимость G(Fe3+) в 5-10 3 М растворе H2SO4, содержащем 10“3 молъ/л Fe2+ и 10 2 молъ/л Сп2+, от мощности дозы при импульсном электронном облучении. Они нашли, что G(Fe3+) в дезаэрированпом растворе практически не зависит от мощности дозы до 5 -108 рад/сек, а в растворе, насыщенном кислородом.— до 108 рад/сек. Для первого раствора G(Fe3+) равен 0,65, для второго — 0,75 иона/100 эв. Выход не зависит от дозы в диапазоне 5-104 -8-10е рад. При больших мощностях дозы G(Fe3+) возрастает.
Более подробно радиолиз водных растворов FeSO4—CuSO4— H2SO4 при высоких мощностях дозы исследовался Р. Фенгом
9 А. К. Пикаев 257
и др. [271. Эти авторы нашли, что G(Fe3+) с ростом мощности дозы, кислотности раствора и концентрации кислорода может достигнуть 4 ионов/100 эв. Однако в том случае, когда раствор содержит большие количества ионов Си2+, зависимость G(Fe3+) от мощности дозы выражена в гораздо меньшей степени. Для раствора, содержащего Fe2+, Н+, О2 и Сн2+ в количествах 103, Ю'2, 2,4-Ю4 и 5-10'2 моль!л соответственно, G(Fe3+) не зависит от мощности дозы до ~ (1,0 -г- 1,4)-1010 рад/сек (выход Fe3+ равен 0,58 иона/100 эе). При несколько большей концентрации Сн2+ (0,1 Л/) в этом растворе G(Fe3+) постоянен до мощности дозы 3* • 1&°рад/сек.
При высоких мощностях дозы увеличиваются выходы Fe2+ в 5-Ю-3 М растворе H2SO4, насыщенном воздухом и содержащем Fe3+, Cu2+ и муравьиную кислоту [171. Даже при мощности дозы ~108 рад/сек G(Fe2+) в растворах, содержащих сравнительно большие количества Си2+ или НСООН, меньше величины выхода для у-радиолиза.
Поведение дозиметра на основе водного раствора щавелевой кислоты при высоких мощностях дозы исследовалось в работах [18, 29]. В них найдено, что выход разложения щавелевой кислоты (при дозе, соответствующей 25% ее разложения) не зависит от мощности дозы до 2-Ю8 рад/сек, если концентрация кислоты равна 5-Ю-2 М, до 2-109 рад/сек при концентрации 0.1 М и до 2- Ю10 рад/сек при концентрации 0,2 М. Выход разложения Н2С2О4 равен 4,9 молек./ЮО эв.
А, Андерсон и Э. Харт [30J измерили выходы продуктов радиолиза насыщенного кислородом раствора, содержащего 10-2 молъ/л НСООН и 5-Ю-4 или 5-Ю-3 молъ/л H2SO4, при мощности дозы ~ 3-Ю® рад/сек. Оказалось, что они равны следующим величинам (в молек./ЮО эв): G(H2O2) = 3,25; G(CO2) = 2,75; G(—О2) = 2,82 и G(H2) = 0,43. т. е. практически равны выходам в случае у-излучения 6()Со.
А. К. Пикаев и П. Я. Глазунов [17] нашли, что G(Fe3+) в дезаэрированных сернокислых растворах Fe2+ при импульсном электронном облучении существенно меньше, чем при у-радио-лизе. Так, даже при мощности дозы 5-Ю7 рад/сек G(Fe3+) в 3-•10-3 М растворе Fe2+ в 0,4 М H2SO4 равен ~ 6,7 иона/100 эв.
Выходы фенола в растворах бензола, насыщенных воздухом или кислородом, сильно зависят от мощности дозы при импульсном радиолизе [6, 31, 32J. Например, согласно [31, 32] при мощности дозы 3,6-10® рад/сек G(C6HBOH) = 0,8 молек./100 эв (при у-радиолизе выход фенола составляет 1,9—2,1 молек./ЮО эе).
А. К. Пикаев и др. [32—34J обнаружили, что G(Fe3+) в сернокислых растворах Fe2+, содержащих кислород и бензол или этиловый спирт, существенно уменьшается при высоких мощностях дозы. Например, по данным Г32, 33J. при мощности дозы ~ 5-108 рад/сек G(Fe3+) в 2,5-Ю-4 М растворе Fe2+ в 0,4 М H2SO4, насыщенном бензолом и воздухом, равен ~ 15 ионам/ЮО эв
(для у-излучения 60Со G(Fe3+) в этом растворе составляет 64 иона/100 эв).
Рядом авторов [16, 29, 35—37J исследовался импульсный радиолиз воды при различных значениях pH. Г. Фрикке и Э. Харт [4J рекомендуют использовать дезаэрированный раствор H2SO4 с pH 3—4 для дозиметрии импульсного излучения. Для этого раствора G(H2O2) = G(H2) = 1,16 молек./ЮО эв при мощностях дозы до ~ 2- 16я рад/сек [36J. Согласно К. Уиллису, и др. [37J, G(H2) в дезаэрированной нейтральной воде уменьшается от ~1 до ~0,8 молек./ЮО эв при увеличении мощности дозы от ~ 1,6-•Ю12 до ~1,6-1014 рад/сек. При мощности дозы ~ 1,6-Ю14 рад/сек G(H2) увеличивается от 0,8 до 1,1 молек./ЮО эв при уменьшении pH от 7 до 1.
3. Использование процесса образования гидратированного электрона для дозиметрии
Э. Харт и сотр. [4, 38—42J предложили применять для дозиметрии импульсного излучения процесс образования гидратированного электрона в Ю~2 М растворе NaOH, насыщенном водородом (7-Ю~4 молъ/л Н2). В этом растворе eaq, возникшие в результате действия импульса, при дозах за импульс от 1 до 100— 150 рад исчезают преимущественно за счет рекомбинации *:
Н о
+ —— Ю + 0Н-. (14)
Гидратированный электрон характеризуется интенсивным оптическим поглощением в видимой области спектра (максимум полосы находится при 720 н.м). Согласно последним измерениям [43[, молярный коэффициент экстинкции eaq при 715 нм равен 1,85-Ю4 л/(молъ-см). Очевидно, это способствует измерениям его концентрации спектрофотометрическим методом. Однако <?aq — короткоживущая частица. Период полупревращения ее в растворе указанного выше состава равен ~25 мксек [40J. Поэтому для регистрации оптического поглощения eaq необходимо применять быстрые спектрофотометрические методы. Эти методы подробно описаны в книгах [2, 3, 38J.
В рассматриваемом способе дозиметрии измеряется кинетика спада оптического поглощения caq. Для нахождения дозы из этих измерений необходимо знать начальную оптическую плотность Ео гидратированного электрона и произведение G х в
* Радикалы ОН реагируют с Н2; ионы Н + нейтрализуются гидроксильными ионами, а атомы Н трансформируются в e“q (реакция: Н -р ОН~ —» + Н2О). Поэтому могут участвовать только в реакции (14) и в реакции с водой. Однако при использованном pH равновесие реакции + Н20
Н + ОН~ сдвинуто влево.
(е коэффициент экстинкции saq) при использованной длине волны. Величина Ео находится путем экстраполяции кинетической кривой до t = 0 (t — время). Значение G х ₽ при 700 нм для раствора, насыщенного водородом (pH 12), равно 121-Ю5 [л/(молЪ'См)1 -молек./100 ав. Формула для расчета дозы D (в рай/импульс) в этом случае имеет следующий вид:
n O,994-lO97?o
l,21-105Z —7.8-103 —,
(15)
Где I — длина пути регистрирующего света в ячейке (в см).
Описываемый метод дозиметрии, по данным [40J, позволяет измерять дозы в диапазоне от 1 до 100—150 pad/импульс. При дозах, меныпих 1 раб/импульс, точные измерения затруднительны. При дозах за импульс, превышающих 100—150 рад, из-за неполной трансформации радикалов ОН в eaq произведение G х X 8 уменьшается. Для повышения верхнего предела измеряемых доз необходимо снизить pH раствора до 9 и вместо Н2 использовать, например, метиловый спирт (его концентрация должна быть 10~3 М). В таком растворе Н и ОН не превращаются в eaq, и поэтому 6X8 при 700 нм становится равным 5,0-104 [л/(молъ--с.м)]-молек./100 эв. С помощью этого раствора можно определять дозы за импульс до 104 рад [4J.
Данный метод дозиметрии нельзя применять для импульсов, продолжительность которых превышает несколько микросекунд [40J. Объясняется это тем, что в этом случае длительность импульса становится сопоставимой с периодом полупревращения eaq, т. е. значительная часть eaq расходуется уже во время действия импульса. Однако при низких дозах за импульс (~ 1 рад) период полупревращения eaq достаточно большой, так что можно применять импульсы продолжительностью до 10 3 сек. Э. Фпль-ден и Э. Харт [40J нашли, что ограничения могут быть определены пз следующего соотношения: для доз за импульс ~100 рад минимальная мощность дозы должна быть 107 рад/сек, а в случае доз ~1 рад эта мощность дозы равна 103 рад/сек. Дозиметр дает одинаковые показания при использовании как электронных импульсов, так и импульсов рентгеновских лучей.
Дозиметрический раствор можно применять неоднократно. В работе [40J было обнаружено, что этот раствор, запаянный в кварцевую ячейку, давал воспроизводимые результаты при хранении в течение двух лет и при поглощении общей дозы в несколько мегарад. Следы кислорода, которые могут быть в исходном растворе, удаляются предварительным облучением. Если раствор по какой-либо причине окажется облученным при большой мощности дозы, то возникшая в результате этого перекись водорода может быть удалена воздействием излучения с низкой мощностью дозы.
Э. Харт и М. Анбар [381 для измерения доз при больших мощностях дозы (до 1010 рад/сек для микросекундных импульсов или до Ю1а рад/сек для наносекундных импульсов) рекомендуют следующую методику. В растворе, не содержащем акцептора eaq, максимальная концентрация этих частиц достигается к концу импульса. Однако, если в растворе в сравнительно большом количестве присутствует какой-либо электронный акцептор, то во время действия импульса устанавливается стационарная концентрация eaq. Путем измерения этой концентрации находится мощность дозы. Для расчетов используется формула:
, MS] [e-q]CT.6.02-10*
1 =--------77^---------’ (16)
G (сач>
где I — мощность дозы, эв'(л-сен); к — константа скорости реакции акцептора S с eaq, л/(моли-сек)-, ISI и [еа(1]ст — соответст венно молярная концентрация S и стационарная концентрация eaq во время действия импульса; G(eaq) — выход eaq (2,7 электрона/100 эв).
В качестве акцептора eaq может быть взята хлорная кислота при концентрациях 10-3 — 10-2 М. Для ионов Н+ величина к составляет 2,2-1010 л/(молъ-сек) [44]. Некоторая неопределенность рассматриваемой методики состоит в том, что G (eaq), возможно, зависит от концентрации акцептора [45].
Недостатком рассмотренных методов дозиметрии является сложность аппаратуры для измерения оптического поглощения eaq. Поэтому они доступны только лабораториям, в которых проводятся исследования в области импульсного радиолиза с оптической регистрацией короткоживущих частиц.
4. Дозиметры на основе полимерных систем
Для дозиметрии импульсного излучения рекомендованы различные полимеры. Дж. Боуг [46] предлагает использовать с этой целью пластинки из полиметилметакрилата толщиной от одного до нескольких миллиметров. При облучении в этом полимере возникает оптическое поглощение с максимумом при 292 нм. При этой длине волны оптическая плотность линейно зависит от дозы в диапазоне от 5-104 до — 10е рад.
Л. Харрах [47. 48] исследовал полистироловые и полита л о-стпроловые пленки, содержащие некоторые добавки. В работе [47] им было найдено, что полистироловые пленки, содержащие транс-стильбен, пригодны для измерения доз в диапазоне 2-105 * * — 108 рад
при мощностях дозы от 300 до 7,5-1013 рад/сек', при этом показания не зависят от энергии излучения в интервале 1,25-Ю-3 — 4,7 Мэв. Пленки готовятся из бензольных растворов полисти-
рола (10 вес.%) и транс-стильбена (0,025 вес.%). При облучении
пленок происходит изомеризация траяс-стильбепа в z/uc-изомер. За этим превращением удобно следить спектрофотометрически по поглощению света при 324 нм (при этой длине волны коэффициент экстинкции транс-стильбена более чем на порядок выше коэффициента экстинкции г/ос-стильбена). При этом находится отношение Н = (Do — Di)ID0, где Do — исходная оптическая плотность пленки при 324 нм и Dt — ее оптическая плотность при данной дозе. Радиационно-химический выход расхода транс-стильбена равен 0,73 молек./100 эв. Точность измерения этим методом при дозах более 1,5-10® рад равна +5%; при меньших дозах она составляет 4- 10%.
В следующей работе Л. Харрах [48] показал, что полигало-стироловые пленки, содержащие метоксид малахитового зеленого, могут использоваться для измерения доз в диапазоне 5-104 — 1,5-107 рад при мощностях дозы до 10й рад/сек. Пленки получались из бензольных или хлороформенных растворов, содержащих 10 вес.% полимера и 0,1 вес.% красителя. При этом использовалась лейкоформа красителя, приготовленная медленным добавлением метанольного раствора малахитового зеленого к 1 М раствору метоксида натрия при быстром перемешивании и последующей перекристаллизации из бензола с обработкой активированным углем. Облучение приводит к окрашиванию пленок. Интенсивность окраски измерялась спектрофотометрически по поглощению света при 630 нм (для доз до — 3-10® рад) или 430 нм (для доз 10® — 1,5-10’ рад). Коэффициент экстинкции красителя при 430 нм равен — 20% от коэффициента экстинкции при 630 нм. Влияние мощности дозы проверялось в случае поли-4-хлорстиро-ловых пленок. Показания для мощностей дозы до — 1014 рад'сек, создаваемых импульсами электронов длительностью 2-10-8 сек. (энергия — 2 Мэв), отличаются всего на 2% от показаний для •у-излучения ®°Со при мощности дозы 300 рад'сек. Для мощности дозы 1015 рад/сек зто различие составляет 8%. Рассматриваемые измерения были проведены в вакууме. В присутствии воздуха чувствительность пленок для у-излучения на 25% выше чувствительности их при облучении в вакууме. В случае поли-4-бром-стироловых пленок кислородный эффект выражен мепее резко (он составляет — 4%). Наличие кислородного эффекта является одним из недостатков описываемого метода дозиметрии. Другой его недостаток состоит в том, что показания сильно зависят от присутствия примесей в пленках.
К. Уиллис и др. [49] изучили возможность применения синпх целлофановых пленок [фирма «Dupont Со.» (США)| для дозиметрии импульсного электронного излучения ускорителей типа «Febetrou». В этих пленках, как уже упоминалось (см. стр. 224), содержится краситель — диметоксилифенил-диазо-бнс-8-амино-1-наф-тол-5,7-дисульфоновая кислота. Краситель в результате облучения обесцвечивается. Степень обесцвечивания измеряется на спектрофотометре при длине волны 655 нм. К. Уиллис и др. об-
нарушили, что эти пленки могут быть использованы для определения доз в диапазоне 5-105 — 1,4-107 рад.
В обзоре [50] отмечается, что показания дозиметров на основе пленок, содержащих нитрилы триарилметановых красителей, не зависят от мощности дозы до — 1015 рад/сек. Принцип действия этих дозиметров рассмотрен в предыдущей главе.
В пленках, как правило, происходит неполное поглощение энергии излучения. Поэтому они могут быть применены как относительные дозиметры, нуждающиеся в предварительной калибровке.
5. Органические жидкости как дозиметрические системы
К. Уиллис и др. [49] нашли, что С(Н2) при радиолизе жидкого бензола под действием импульсов электронов, генерируемых ускорителем «Febetron-705» (мощность дозы 1014 рад/сек), равен 0-04 молек./100 эв. Выход водорода такой же, как и в случае низких мощностей дозы.
Указанные авторы обнаружили следующую особенность дозиметрических измерений при сверхвысоких мощностях дозы с помощью бензола (и, возможно, других жидкостей, плохо проводящих ток). За время импульса в жидкость вводится заряд, равный нескольким сотням микрокулонов. Время релаксации проводимости в этих жидкостях такое же или даже больше длительности импульса (— 3-10“8 сек.). Поэтому высокое электрическое поле, наведенное в жидкости, в начальный период действия импульса будет отражать электроны, поступающие в нее в последующие периоды действия импульса. В результате измеренная доза в этих условиях будет гораздо меньше. Очевидно, данный эффект будет тем значительнее, чем толще облучаемый образен. Согласно [49], этот эффект ничтожен для образцов толщиной 0,2—1,0 см (для них G(H2) = 0.04 молек. 100 эв). Однако он существен для образцов бензола толщиной 1,5 см [в этом случае G(H,) = 0,02—0,03 молек./ /100 эв].
А. Бойд и др. [51] исследовали радиолиз циклогексана под действием импульсов электронов при мощностях дозы до — 1014 рад/сек. Было найдено, что в этих условиях С(Н2), хотя и меньше, чем при у-радиолизе, но имеет постоянную величину (4 молек./ '100 эв) в диапазоне мощностей дозы от 6-101'2 до 1,3-Ю14 рад/сек. С(Н3) не зависит и от дозы в изученном интервале (4• 105 — 2,4--107 рад).
II. Дворник и др. [52] рекомендуют использовать для дозиметрии импульсного электронного излучения этанольные растворы хлорбензола, содержащие 4 объемн. % воды. С помощью этой системы доза измеряется по образованию ионов Н* или СП. Было найдено, что G(HC1) при содержании С6Н5С1 в растворе более 10 объемн. % практически одинаков как для у-лучей 60Со, так и для импульсного электропиого излучения с мощностью дозы
10s рад/сек (длительность импульсов была равна 7-10"6 сек., а энёргия электронов — 10 Мэе). Величины G(HC1) в молек./ЮО эв составляют (в скобках указаны концентрации С6Н5С1 в объемных процентах): 4,53 (10); 4,95 (15); 5,19 (20); 5,34 (30); 5,47 (40). Способы анализа ионов Н+ и С1~ в облученных растворах рассмотрены на стр. 204.
Согласно [53], дезаэрированный раствор нитрила парарозанилина в 2-метоксиэтаноле может быть применен для измерения доз импульсного электронного излучения (мощность дозы — 3 • 10е рад/сек) до 15—20 крад. Методика дозиметрии с помощью этой системы описана в главе V.
6. Газообразные дозиметрические системы
В работах [1, 54—58] изучена возможность использования этилена, кислорода и закиси азота для дозиметрии импульсного электронного излучения. По данным [54], 6'(И2) при радиолизе этилена при мощности дозы — Ю14 рад/сек зависит от давления-Выход водорода уменьшается от 1,9 до 1,04 молек./ЮО эе при увеличении давления от 0,5 до 10 атм. При у-радиолизе этилена (давление 1 атм) G(H2) = 1,2—1,3 молек./ЮО эе (см. главу A ll).
Выход озона при радиолизе газообразного кислорода под действием импульсов электронов (мощность дозы Ю12 — 3-Ю13 рад/ /сек) существенно выше, чем при -у-радиолизе [56—59]. Согласно [57], при указанных мощностях дозы С(О3) = 12,8 + 0,6 молек./ /100 эв (при у-радиолизе выход О3 составляет всего 6,24^ 0,6 молек./ /100 эв). Точное значение мощности дозы, при которой G(O3) изменяется, не установлено. Поэтому газообразный кислород можно использовать как дозиметрическую систему при мощностях дозы Ю12 — 3-Ю18 рад/сек, для которых G(O3) независимо от мощности дозы равен 12,8 молек./ЮО эе [57]. В этих условиях С(О3) постоянен при изменении давления в диапазоне 0,04—1 атм и дозы в интервале 0,05—0,35 Мрад. Озон в образцах, облученных импульсами электронов, удобнее всего определять спектрофотометрическим методом по поглощению света при 254 нм- (см. стр. 215).
При переходе от мощностей дозы — Ю2 — Ю3 рад/сек к мощностям дозы — Ю12 рад/сек G(N2) при радиолизе газообразной закиси азота возрастает примерно на 20%. Для у-радиолиза выход равен ~ 10 молек./ЮО эе (см. главу VII), а для радиолиза под действием мощных импульсов электронов он составляет 12,4 молек./ЮО эв [1, 54, 55]. По данным [1], при давлении N2O, равном 1 атм, это изменение выхода происходит в диапазоне мощностей дозы 1,6-• Юв — 1,6-Ю11 рад/сек. При импульсном радиолизе G(N2) не зависит от давления N2O в интервале 0,5—5,0 атм [54]. При давлении 1 атм- он не изменяется при увеличении температуры до 200° С [55]. Выход N2 постоянен до дозы 6,4-10е рад. Согласно [60], G(N2) равен 12,4 молек./ЮО эв даже при мощности дозы—3-Ю14 рад/сек.
А. Бойд ii др. [61] обнаружили следующую особенность дозиметрии импульсного электронного излучения с помощью газообразных систем. Исследуя влияние давления на выходы радиолиза газообразных НО, НВг и N2O при мощности дозы 1,6-1013 рад/сек, они нашли, что при давлениях менее 0,5 атм выходы Н2 в НС1 и НВг, а также выходы N2 в К2О существенно выше, чем при давлениях более 1 атм. На рис. 82 в качестве примера показана зависимость 6'(N2) от давления N2O при радиолизе в этих условиях. Данный эффект объяснен ускорением вторичных электронов в
Рис. 82. Зависимость G(N2) при радиолизе N2O от давления р (импульсное электронное излучение, мощность дозы 1,6-1013 рад/сек)
1 — облучение в кварцевых ячейках;
2 — облучение в ячейках из нержавеющей стали
кратковременном электрическом-поле, создаваемом мощным электронным импульсом. Аналогичное явление имеет место и при радиолизе N2O и H2S под действием электронов с энергией 0,6 Мэв и мощностью дозы — 3-1014 рад/сек [60]. В этом случае выходы продуктов радиолиза постоянны только при давлениях выше 1.2 (для N2O) и 1 атм (для H2S). При радиолизе СО2 выход СО равен 7,7 молек./100 эв при давлениях выше 1 атм.
’ 7. Другие дозиметрические системы
По данным [62—64], показания термолюминесцентного дозиметра LiF не зависят от мощности дозы до 2-1011 рад/сек. Подробное изучение пригодности монокристаллов LiF для дозпметрпи импульсного излучения было проведено Э. Фильденом и Э. Хартом [40]. Указанные авторы нашли, что показания этого дозиметра не зависят от мощности дозы в интервале от 0,9 до 1,5-16е рад/ /сек. В своих опыт ах они использовали у-излучение 6ОСо и импульсы рентгеновских лучей длительностью 10_6, 9,5-10~8 и 1,1 -10—8 сек. Было обнаружено, что с помошью LiF можно измерять очень низкие дозы (вплоть до 10-2 /шб/гмпульс).
Фтористый литий как термолюминесцентный дозиметр импульсов электронов при мощностях дозы до 1012 рад/сек исследовался также в работе [65]. В ней. кроме того, рассмотрена возможность использования термолюминесцентных свойств CaF2 : Мп для дозиметрии этого вида излучения.
К. Шмидт и У. Бэкк [66] предложили калориметрический метод
дозиметрии импульсного электронного излучения с регистрацией тепловых изменений кондуктометрическим способом. Метод состоит в следующем. Кварцевая ячейка с двумя плоскопараллельными платиновыми электродами в виде круглых фолег, наполненная электролитом, помещается за коллиматором, вырезающем пучок электронов диаметром 2,5 см. Когда в ячейку попадает электронный импульс, поглощенная доза вызывает повышение температуры электролита, что в свою очередь приводит к изменению его электропроводности (—- 2% на градус). Изменение электропроводности измеряется чувствительным мостом. Этим методом могут быть определены дозы > 103 рад. Наиболее подходящим электролитом является фосфатный буфер (pH — 7), который показывает ничтожное изменение электропроводности вследствие радиационно-химических реакций. В расчеты дозы необходимо вносить поправку на поглощение энергии платин эвыми фольгами.
8. Методы измерения дозы вдоль пути светового пучка в импульсном радиолизе с оптической регистрацией короткоживущих частиц
Как уже отмечалось, в опытах по импульсному радиолизу с оптической регистрацией короткоживущих частиц необходимо знать дозу вдоль пути регистрирующего светового пучка. Вызвано это тем, что в этих опытах распределение дозы в объеме раствора бывает, как правило, неравномерным (например, облучению подвергается только часть раствора, поперечное сечение светового пучка меньше поперечного сечения пучка электронов, входящих в ячейку, и т. п.). В таких случаях дозу находят путем измерения оптического поглощения какого-либо продукта, чьи оптические характеристики известны. В работах [67, 68] с этой целью рекомендуется использовать оптическое поглощение ионов Fe3+, возникающих при облучении сернокислого раствора ферросульфата. Для этой системы кинетическая кривая достигает плато через несколько секунд после подачи импульса. Г. Адамс и др. 169] применили водный раствор ферроцианида, в котором плато достигается в течение миллисекунд. Другая система, рекомендованная с этой целью,— водный раствор KCNS [70]. У. Флетчер и др. [71] использовали 2-10'3 М раствор KCNS, насыщенный кислородом. В этом растворе в результате реакции радикалов ОН с рода-нид-понами возникают ион-радикалы (CNS)3. Молярный коэффициент экстинкции этой частицы при 475 нм равен 7350 лf (моль-• см), a G[(GNS)2] = G (ОН) =2,9 [72, 73]. Раствор CNS~ применялся также в других работах по импульсному радиолизу (см., например, [74—761). Недавно К. Иона и др. [77] использовали 10-2 М раствор KCNS, насыщенный воздухом, для дозиметрии одиночных импульсов тонкой структуры в случае линейного ускорителя (энергия электронов — 19 Мэв). Длительность этого импульса составляет 50 псек, а доза за импульс равна — 2 крад.
Очевидно, рассматриваемый вопрос можно решить, используя процесс образования гидратированных электронов (см. стр. 259). Эта методика применялась, в частности, в работах [77, 78].
9. Некоторые практические рекоменцацпи
Дозиметрию импульсного излучения при мощностях дозы менее — 1012 рад/сек можно проводить в стеклянных ячейках. При больших мощностях дозы использование этих ячеек не рекомендуется. По данным А. Бойда и др. [51], при мощностях дозы выше 101а рад/сек стеклянные ячейки быстро растрескиваются. Эти авторы опыты по дозиметрии при таких высоких мощностях дозы осуществляли в ячейках из нержавеющей стали. Отметим, что для высоких мощностей дозы каталитический эффект нержавеющей стали на процесс радиационно-химического разложения N2O (см. стр. 213) ничтожен.
При работе с импульсным электронным излучением вследствие неравномерного распределения поглощенной дозы по глубине облучаемого объема и в поперечном сечении пучка средняя мощность поглощенной дозы в облученном объеме отличается от средней мощности дозы во всем объеме. Это особенно проявляется в тех случаях, когда размеры пучка в поперечном сечении меньше диаметра используемой ячейки и когда толщина слоя вещества в ячейке превышает^ величину пробега электронов.
Средняя доза D в облученном объеме равна [14]:
опти
(18)
(19)
где dv — элемент объема раствора; D — общая доза (элемент объема dr получает дозу Ddv). Интегрирование производится по всему облучаемому раствору, исходя из распределения интенсивности в поперечном сечении пучка и вдоль пути электронов в растворе.
А. Андерсон [15] находил контуры облученного объема по окрашиванию, возникающему под действием излучения в блоке из по-лиметплметакрилата. Блок после облучения разрезался па тонкие полоски. Спектрофотометрически измерялось радиальное распределение оптической плотности и рассчитывалась средняя ческая плотность Ё для облученного объема по формуле
Ё = E*dv I Edv.
Затем находились эффективный облученный объем г.,фф »эфф =^Edv}2 / ^E4v
и средняя мощность дозы Р в облученном объеме
D р =----z ,
^ФФТ
где D — общая доза за импульс, эв', т — длительность импульса,. сек. Если г?,Цфф выражено в мл, то Р, очевидно,— в эвЦмл-сек).
При проведении дозиметрии импульсного электронного излучения необходимо строго соблюдать геометрию опытов. Очень важно при этом, чтобы диаметр пучка при входе его в ячейку был оди-наковым во всех опытах. В противном случае эффективный облученный объем, а значит, и средняя мощность дозы в нем при равном токе пучка будет изменяться от опыта к опыту, что, естественно, исказит результаты эксперимента. За выполнением этого условия необходимо наиболее строго следить, когда диаметр пучка меньше диаметра ячейки.
Контроль положения и размеров электронного пучка можно осуществлять различными методами. Дж. Ротблат и Г. Сэттон [14] использовали с этой целью фотопленку. Они помещали ее сразу же за ячейкой и по почернению ее судили о диаметре пучка. Очевидно, этот метод мало удобен, так как он требует больших затрат времени на обработку фотопленки. Дж. Томас и Э. Харт [16] следили за размерами пучка по образованию при облучении коричневого окрашивания в тонких ппрексовых пластинках пли по обесцвечиванию синих целлофановых пленок, находящихся непосредственно перед ячейкой. Можно, по-видимому, применять для этого и пленки из других полимеров.
А. К. Пикаев и др. [12, 19] перед каждой серией опытов юстировали электронный пучок по свечению кварцевой пластинки, вмонтированной в держатель, который навинчивался на выпускное окно ускорителя. За положением светящегося пятна следили с пульта управления ускорителем с помощью зеркала и телевизионной установки.
При определении дозы импульсного излучения «супердозпмет-ром» Фрикке следует принимать во внимание также то обстоятельство, что локальные дозы в некоторых точках раствора могут превышать среднюю дозу в облученном объеме. Особенно это характерно для электронов с энергией от 1 до 2—3 Мэв. Поэтому даже в тех случаях, когда средняя доза значительно меньше дозы, необходимой для полного израсходования кислорода в растворе, может происходить локальный расход этого вещества. При низких мощностях дозы этот эффект устраняется перемешиванием раствора во время облучения. При импульсном же облучении эта операция ничего не дает. Поэтому значение дозы, при которой наблюдается локальный расход кислорода, определяется экспериментально. Ее можно_найти, построив кривую, выражающую зависимость G (Fe^+) от I) при какой-либо одной высокой мощности дозы. Величину D можно изменять, давая в раствор различное число импульсов, причем промежутки между импульсами должны быть настолько малыми, насколько это возможно. Это необходимо для того, чтобы кислород в промежутках между импульсами не успевал диффундировать в реакционную зону из объема раствора.
При нахождении дозы вдоль пути регистрирующего света в
О ’НЮ СО CM CM СМ Ю ' ТЛ’-'
V V VV V V V VVV^VvvJ
СО Ю
О ci
Таблица 58
Дозиметрические системы для импульсного излучения
импульсном радиолизе с оптической регистрацией короткоживущих частиц необходимо учитывать следующее обстоятельство. При высоких мощностях дозы в случае водных растворов, как уже отмечалось, может иметь место конкуренция реакций: радикал — радикал и радикал — растворенное вщцество. Поэтому концентрация введенного в раствор вещества, которое в реакции с радикалами дает продукт с известными оптическими характеристиками, должна быть достаточной для полного захвата зтих радикалов. Например, по данным [74], при дозе — 3,6 -104 рад за импульс длительностью — 3-10-8 сек. ионы CNS- полностью акцептируют радикалы ОН лишь тогда, когда концентрация этих ионов равна 0,1 М.
Проведенное рассмотрение различных дозиметрических систем показывает, что химические методы дозиметрии импульсного излучения характеризуются рядом особенностей по сравнению с аналогичными методами для у-радиолиза. В первую очередь это относится к изменению выходов продуктов радиолиза. Поэтому при определении дозы необходимо прежде всего правильно выбрать дозиметрическую систему, показания которой не зависят от мощности дозы в исследуемом диапазоне мощностей. Помощь читателю при выборе систем может оказать табл. 58.
На основании данных, приведенных в табл. 58, можно сделать следующие общие выводы. Наиболее универсальной дозиметрической системой является жидкий бензол. Выход водорода при радиолизе его постоянен во всем диапазоне мощностей дозы, с которыми могут иметь дело исследователи в настоящее время. В случае водных растворов область мощностей дозы, в которой выход не зависит от мощности дозы, обычно тем шире, чем выше концентрация растворенного вещества. Из газообразных систем для дозиметрии импульсного излучения наиболее подходит закись азота. Выход N2 при радиолизе ее изменяется всего на 20% при переходе от у-излучения к электронному излучению ускорителей типа «Febetron». Наконец, можно отметить и полимерные системы, количество химического превращения в которых часто не зависит от мощности дозы в широких пределах.
ЛИТЕРАТУРА
1. С. С. Willis, A. W. Boyd, D. A. Armstrong. Can. J. Chem., 47, 3783 (1969).
2. А. К. Пикаев. Импульсный радиолиз воды и водных растворов. М., «Наука», 1965.
3. М. S. Matheson, L. М. Dorfman. Pulse Radiolysis. Cambridge (U.S.A.) — London, The M.I.T. Press, 1969.
4. H. Fricke, E. J. Hart. Radiation Dosimetry, 2nd Edition (Ed. F. H. Attix and W. C. Roesch), vol. 2. N. Y., 1966, p. 167.
5. А. К. Пикаев. Усп. химии, 41, 1696 (1972).
6. L. M. Dorfman, I. A. Taub, R. E. Biihler. J. Chem. Phys., 36, 3051 (1962)..
7. J. W. Hunt, J. K. Thomas. Radiation Res., 32, 149 (1967).
8. П. Я. Глазунов, А. К. Пикаев. Докл. АН СССР, 130, 1051 (1960).
9. А. Н. Пикаев, П. Я. Глазунов, jfpvii. АН СССР, 154, 1167 (1964).
10. J. P. Keene. Radiation Res., 6, 424 (1957).
11. А. К. Пикаев, II. Я. Глазунов. Изв. АН СССР, ОХН, 1959, 224.
12. А. К. Пикаев, П. Я. Глазунов, В. И. Спицын. Докл. АН СССР, 150. 1077 (1963).
13. Н. С. Sutton, J. Rotblat. Nature, 180, 1332 (1957).
14. J. Rotblat, H. C. Sutton, Proc. Roy. Soc., A255, 490 (1960).
15. A. R. Anderson. J. Phys. Chem., 66, 180 (1962).
16. J. K. Thomas, E. J. Hart. Radiation Res., 17, 408 (1962).
17. А. К. Пикаев, П. Я. Глазунов. Труды II Рсесоюзного совещания по радиационной химии. М., Изд-во АН СССР, 1962, стр. 109.
18. А. К. Пикаев, П. Я. Глазунов. Труды Ташкентской конференции по мирному использованию атомной энергии, т. 1. Ташкент, Изд-во АН УзбССР, 1961, стр. 354.
19. А. К. Пикаев, П. Я. Глазунов, В. И. Спицын. Изв. АН СССР, серия хим., 1965, 401.
20. К. Sehested, Е. Bjergbakke, О. L. Rasmussen, Н. Fricke. J. Chem. Phys., 51, 3159 (1969).
21 В. М. Моралев, Л. Т. Бугаенко. Химия высоких энергий, 2, 461 (1968).
22. К. Sehested, Е. Bjergbakke, N. W. Helm, Н. Fricke. Dosimetry in Agriculture, Industry, Riology and Medicine. Vienna, IAEA, 1973, p. 397.
23. H A. Schwarz. Radiation Res., Suppl. 4, 89 (1964).
24. А. К. Пикаев, П. Я. Глазунов. Изв. АН СССР, серия хим., 1964, 1944.
25. F. Hutchinson. Radiation Res., 9, 13 (1958).
26. Е. Bjergbakke, К. Sehested. Adv. Chem. Ser., 81, 579 (1968).
27. P. Y. Feng, A. Brynjolfson, J. M. Halliday, R. D. Jarrett. J. Phys. Chem., 74, 1221 (1970).
28. K. Sehested, E. Bjergabakke, N. W. Holm. Proc. II Tihany Symposium on Radiation Chemistry. Rudapest, 1967, p. 149.
29. N. W. Holm, S. Sehested. Adv. Chem. Ser., 81, 568 (1968).
30. A. R. Anderson, E. J. Hart. J. Phys. Chem., 66, 70 (1962).
31. Л. И. Карташева, А. К. Пикаев. Ж. физ. химии, 41, 2855 (1967).
32. Л. И. Карташева, А. К. Пикаев. Intern. J. Radiat. Phys. Chem., 1, 243 • (1969).
33. Л. И. Карташева, А. К. Пикаев. Химпя высоких энергий, 3, 10 (1969).
34. Л. Э. Столярчик, А. К. Пикаев. Докл. АН СССР, 141, 1147 (1961).
35. А. К. Пикаев, Г. К. Сибирская, Г. Г. Рябчикова, П. Я. Глазунов. Кинетика и катализ. 6, 41 (1965).
36. Н. Fricke, J К. Thomas. Radiation Res., Suppl., 4, 35 (1964).
37. C. Willis, A.W. Boyd, A. E. Rothwell, O. A. Miller. Intern. J. Radiat. Phys. Chem., 1, 373 (1969).
38. Э. Харт, M. Анбар. Гидратированный электрон. M., Атомиздат, 1973.
39. Е. J. Hart. Actions Chimiques et Biologiques des Radiations (Ed. M. Hais-sinsky), 11 Serie. Paris, 1966, p. 1.
40. E. M. Fielden, E. J. Hart. Adv. Chem. Ser., 81, 585 (1968).
41. E. J. Hart. Health Phys., 12, 641 (1966)
42. E. M. Fielden, E. J. Har . Radiation Res., 27, 525 (1966).
43. E. M. Fielden, E. J. Hart. Radiation Res., 32, 564 (1967).
44. M. Anbar, P. Neta. Intern. J. Radiat. Phys. Chem., 18, 493 (1967).
45. A. M. Koulkes-Pujо, B. D. Michael, E. J. Hart. Intern. J. Radiat Phys. Chem., 3, 333 (1971).
46. J. W. Boag. Actions Chimiques et Biologiques des Radiations (Ed. M. Hais-sinsky), 6 Serie. Paris, 1963, p. 4.
47. L. A. Harrah. Radiation Res., 39, 223 (1969).
48. L. A. Harrah. Radiation Res., 41, 229 (1970).
49. C. Willis, O. A. Miller, A. E. Rothwell, A. W. Boyd. Radiation Res., 35, 1580 (1968).
50. W. L. McLaughlin, P. E. Hjortenberg, В. B. Radak. Dosimetry in Agriculture, Industry, Biology and Medicine. Vienna, IAEA, 1973, p. 577.
51. A. W. Boyd, C. Willis, O. A. Miller, A. E. Rothwell. Adv. Chem. Ser., 82, 456 (1968).
52. I. Dvornik, D. Razem,, M. Baric. Large Radiation Sources for Industrial Processes. Vienna, IAEA, 1969, p. 613.
53. W. L. McLaughlin. Dosimetry in Agriculture, Industry, Biology and Medicine. Vienna, IAEA, 1973, p. 362.
54. C. Willis, O. A. Miller, A. E. Rothwell, A. W. Boyd. Adv. Chem. Ser., 81, 539 (1968).
55. A. W. Boyd, C. Willis, 0. A. Miller. Can. J. Chem., 47, 351 (1969).
.56. J. A. Ghormley, C. J. Hochanadel, J. W. Boyle. J. Chem. Phys., 50, 419 (1969).
57. C. Willis, A. TV. Boyd, M. J. Young, D. A. Armstrong. Can. J. Chem., 48, 1505 (1970).
58. A. W. Boyd, C. Willis, R. Cyr, D. A. Armstrong. Can. J. Chem., 47, 4715 (1969).
59. G. M. Meabum, D. Perner, J. LeCalve, M. Bourene. J. Phys. Chem., 72, 3920 (1969).
60. C. Willis, A. W. Boyd, O. A. Miller. Radiation Res., 46, 428 (1971).
61. A. W. Boyd, D. A. Armstrong, C. Willis, O. A. Miller. Radiation Res., 40, 255 (1969).
62. N. Goldstein, E. Tochilin. Radiation Res., 32, 280 (1967).
63. N. Goldstein, E. R. Schleiger, E. Tochilin. Health Phys., 13, 806 (1967).
64. M. J. Marrone,F. H. Attix. Health Phys., 10, 431 (1964).
65. B. Alsop, H. Weiss, E. R. Epp. Radiation Res., 35 , 483 (1968).
66. К. H. Schmidt, W. L. Buck. Radiation Res., 39, 471 (1969).
67. L. M. Dorfman, I. A. Taub. J. Am. Chem., Soc., 85, 2370 (1963).
68. G. Czapski, L. M. Dorfman. J. Phys. Chem., 68, 1169 (1964).
69. G. E. Adams, J. W. Boag, B. D. Michael. Trans. Faraday Soc., 61, 492 (1965).
70. E. M. Fielden, N. W. Holm. Manual on Radiation Dosimetry (Ed. H. W. Holm and R. J. Berry). N. Y., Marcel Decker, Inc., 1970, p. 288.
71. J.W.Fletcher, P. J.Richards, W. A. Seddon. Can. J. Chem., 48, 1645 (1970).
72. G. E. Adams, J. W. Boag, J. Currant, B. D. Michael. Pulse Radiolysis (Ed. M. Ebert et al.). London—New York, 1965, p. 117.
73. J. H. Baxendale, P. L. T. Bevan, D. A. Stott. Trans. Faraday Soc., 64, 2389 (1968).
74. M. Daniels. J. Phys. Chem., 73, 3710 (1969).
75. M. Simic, P. Neta, E. Hayon. Ibid., p. 3794.
76. А. К. Пикаев, Г. К. Сибирская, Е. М. Ширшов, П. Я. Глазунов, В. И. Спицын. Докл. АН СССР, 200, 393 (1971).
77. С. D. Jonah, Е. J. Hart, М. S. Matheson. J. Phys. Chem., 77, 1838 (1973).
78. С. А. Кабакчи, А. А. Зансохова, А. К. Пикаев. Химия высоких энергий, 8 , 255 (1974).
Глава X
ДОЗИМЕТРИЯ ПУЧКОВ
ТЯЖЕЛЫХ ЗАРЯЖЕННЫХ ЧАСТИЦ
В радиационной химии широко используются тяжелые заряженные частицы (а-частицы, гелионы, протоны, дейтроны, многозарядные ионы). Они характеризуются более высокими значениями ЛПЭ, чем у-лучи 60Со и быстрые электроны. Это обстоятельство обусловливает изменение выходов радиолитических превращений во многих системах. Поэтому при проведении дозиметрии пучков тяжелых заряженных частиц с помощью таких систем необходимо a priori знать величину ЛПЭ. Естественно, это является определенным недостатком таких систем. Очевидно, более перспективны в этом направлении дозиметрические системы, для которых выход превращения не зависит от ЛПЭ.
1. Дозиметр Фрикке
G (Fe3+) в дозиметре Фрикке уменьшается с ростом величины ЛПЭ использованного излучения. Соответствующие данные приведены в табл. 59. Как следует из нее, для данного вида тяжелой заряженной частицы G (Fe3+) существенно зависит от ее энергии. Поэтому при проведении дозиметрии этих видов излучения с помощью дозиметра Фрикке необходимо предварительно знать начальную энергию частицы.
Оригинальный метод дозиметрии излучений с высокими значениями ЛПЭ предложен в работах [21, 22]. Айторы этих работ, проанализировав литературные данные о влиянии ЛПЭ на выходы продуктов радиолиза водных растворов и выполнив некоторые эксперименты с а-частицами 210Ро и р-частицами трития, обнаружили, что G(H2) в 0,4 М растворе H2SO4 связан с величинами ЛПЭ зависимостями, показанными на^ рис. , 83. В свою очередь зависимость G(H2) от отношения концентраций Н2 и Fe3+ в облученных одинаковое время 0,4 М растворе H2SO4 и дозиметре Фрикке выражается кривой, представленной на рис. 84. Поэтому, измерив экспериментально это отношение, можно по кривой рис. 84 найти G(H2), а значит, и поглощенную дозу. Достоинством этого метода является то, что при измерении дозы с его помощью нет необходимости a priori знать энергию использованного излучения. Эту энергию в принципе можно определить из зависимости между G(H2) и ЛПЭ (см. рис. 83). В работе [22] описанным методом была проведена дозиметрия реакторного излучения (см. следующую главу).
Таблица 59
Значения G(Fes|-) в дозиметре Фрикке для тяжелых заряженных частиц
Энергия излучения, Мэв G (Fc3+), ионы/100 эв Литература Энергия излучения, Мэв G (Fe3+), ионы/100 эв Литература
Протоны Дейтроны
22,9 12,8 [1] 9,38 9,08 [3]
22,6 12 6 [1] 7,30 8,06 [3]
22,5 12 8 1] 6,20 8,09 [3]
18,8 12,4 11] 5,09 7,22 13]
18,5 12,3 [1] 3,91 7,21 [3]
18,3 12,5 [1] 3,47 6,90 [3]
15,6 11,8 |1] 18 11,2 [2]
15,3 И 8 fl] 16 10,9 12]
15,0 11,8 |1] 14 10,7 12]
11,7 И,4 [1] 12 10,4 12]
11,2 11,3 [1] 10 10,0 [2]
10,9 11,3 11] 8 9,7 12]
8,4 11,28 [2] 6 9,1 [2]
1,99 8,0 [3]
1,79 7,92 [3] Гелионы
1 ,59 7,80 13]
1,27 7 45 [3] 30 7,9 (2|
0,98 7,19 |3] 25 7,5 [2]
0,63 6,89 [3] 20 7,1 [2]
0,46 6,29 [3] 15 6,5 [2]
0,29 7,16 [3]
0,65 6,12 [4] 7-Частицы 210Ро
0,58 0,39 0,38 0,30 0,26 0,25 0,20 5,60 5,24 6,з 6,5 6,2 7,0 7,7 [4] (4) 15] 15] 14) 15) ]5] 5,3 5,3 5,3 5,3 3,4 3,4 5,1 5,5 5,2 5,6 4,8 4,7 [10] [И] 112] (13) ]14] [15, 16]
0,15 0,15 9,5 6,44 (5] 14] Продукты деления 235U
0,10 12,3 [5] 152 3,0 [17]
0,085 15,8 15] Продукты ядерной реакции
Дейтроны 14N(zt, p)uC
21,16 10,86 13] 0,626 7 117]
18,70 17,34 10,79 10,50 13] [3] Быстрые нейтроны *3
14,36 10,14 [3] 14,6 11,5 (18]
12,0 9,81 [3] 14,1 12,4 [19]
14,0 10,5 [20]
♦* Значения G(Fe®+) для продуктов ядерных реакций 10В (n, a) 7Li й eLi(n, а) Т приведены в табл. 65.
*в Здесь не приводятся величины G(Fe®+) для а-частиц 210Ро, равные 5,9—6,2 [6—9]. Эти значения, очевидно, завышены.
Радиационно-химические превращения производятся заряженными частицами, возникшими при упругих и неупругих взаимодействиях быстрых нейтронов с ядрами Н и О.
По данным [22], ферросульфатный дозиметр, в котором концентрация ионов Fe2+ равна М, пригоден для измерения доз до 5-105 рад в случае а-частиц 210Ро и до — 1,3-105 рад в случае смешанного излучения 210Ро и трития с ЛПЭ 0,5 эв/А.
Позднее методика, предложенная в работах [21, 22], использовалась Е. П. Ковалевой и др. [231 для нахождения величины
ЛПЗ.зв/Л
Рис. 83. Зависимость <7(Н2) в 0,4 М растворе H2SO4 от величины ЛПЭ
Рис. 84. Зависимость G(H2) от отношения концентраций Н2 и Fe3+ в облученных одинаковое время 0,4 М растворе Н28О4 и дозиметре Фрикке
ЛПЭ излучения реактора (см. также главу XI). Указанными авторами применялись дезаэрированный 5• 10 2 М раствор Fe2+ в 0,4 М 112SO4 с добавкой 10-3 моль!л NaCl и насыщенный воздухом 0,4 М раствор H2S04 с добавкой 2 • 10~5 молъ/л КВг.
2. Цериевая дозиметрическая система
В случае цериевой дозиметрической системы G(Ce3+) сравнительно мало зависит от энергии тяжелых заряженных частиц. Соответствующие данные приведены в табл. 60. Как видно из нее, изменение величины G(Ce3') в зависимости от вида частицы и ее энергии в исследованном диапазоне незначительно. Эта особенность данной системы делает перспективным ее использование при дозиметрии потоков тяжелых заряженных частиц.
3. Циклогексан
Р. Шулер и 4. Аллен [27] нашли, что выход молекулярного водорода при радиолизе циклогексана не зависит от ЛПЭ. Позже этот вывод был подтвержден другими авторами [28—311. В табл. 61 приведены величины G(H2) при радиолизе циклогексана под действием излучений с различными значениями ЛПЭ.
Величины G (Се31) при радиолизе цериевой системы под действием излучений с высокими зпачепиями ЛПЭ
Вид излучения Энергия, Мэв G(Ce* * 3 4 * *+), ионы/100 эв Литература
Протоны 0.05-0,3 2,5 [5]
10,9—22,5 2,9 [1]
Дейтроны 18 2,84 [24]
Гелионы 33 2,92 [24]
а-Частицы 210Ро 3,4 2,88 [25]
5,3 2,94 [26]
5,3 3,2 [И]
Продукты ядерной реакции 10В (п, a)’Li 2,33 2,94 [24]
Таблица 61
Величины G(H2) при радиолизе циклогексана под действием излучений с различными значениями ЛПЭ *
Вид излучения ЛПЭ, лз,А С(Н2), молек. 100 эз Литература
Т-Лучи, быстрые электроны 0,02 5,66 [28]
Быстрые электроны 0,02 5,25 [27]
Дейтроны, гелионы 0,5< ЛПЭ<2,0 5,2—5,4 [27]
Гелионы 22 5,33 [28]
Излучение ядерпого реактора Макс. 10 5,2 [29]
Продукты деления 236U 200 7,7 [30]
* Перечень работ, в которых измерялся ЫН,) для т- и рентгеновского излучений и быстрых электронов, приведен в главе VI.
Из табл. (51 видно, что изменение 6’(Н2) имеет место только
для продуктов деления 235U. G(112) несколько уменьшается при вы-
соких дозах [321. Однако для доз до 1,7-107 * * рад изменение G(H2) лежит в пределах + 5% [321.
4. Полимеры
А. М. Кабакчи и сотр. [33—371 подробно исследовали влияние
величины ЛПЭ на радиационно-.химические процессы в полиме-
рах. Было найдено, что некоторые из этих систем пригодны для
дозиметрии потоков тяжелых заряженных частиц. В случае пленок
диацетата целлюлозы варьирование значения ЛПЭ от 0,02 до
—4,4 зв/А, температуры от —196 до 120° С и мощности дозы в ши-
роких пределах не влияет на изменение оптической плотности пленок при 320 нм. Измеряемый диапазон доз составляет 2-10® — 7-107 рад [33]. Величина ЛПЭ не оказывает влияния и на выход деструкции диацетата целлюлозы. При этом линейная зависимость изменения молекулярного веса полимера от дозы наблюдается в диапазоне 5-10® — 4-Ю7 рад [34]. По данным работы [35], для полиэтилена низкой плотности отсутствует влияние величины ЛПЭ (использовалось у-излучение ®°Со, дейтроны с энергией 14 Мэв и гелионы с энергией 28 Мэв) на скорости образования гель-фрак-ции и mpaiic-впниленовых связей (вплоть до дозы 108 рад).
Согласно А. М. Кабакчи и сотр. [38, 39], выход обесцвечивания метиленового голубого, введенного в пленку из поливинилового спирта, мало зависит от величины ЛПЭ. Соответствующие данные приведены в табл. 62.
Таблица 62
Выход обесцвечивания метиленового голубого в пленках из поливинилового спирта [38, 39]
Вид излучения Энергия, Л1э? ЛПЭ, эв/А Мощность дозы, рад/сек G, молек./ /109 st
Т-Пзлучение 6 Со 1,25 0,02 10—103 1,8
Т-л Нейтронное излучение ядерного реактора — 0,8 6-Юз 1,7
Дейтроны 14 0,75 10’ 1,7
Гелионы 28 3,0 10’ 1,6
Продукты ядерноп реакции ’"В («, a)7Li * 2,33 20 — 1,06
* В «том случае пленка содержит борную кислоту; для т-лучей 60Со выход обесцвечивания метиленового голубого в поливиниловой пленке в присутствии борной кислоты равен 1,42 молек., 100 эв.
Выход обесцвечивания не зависит от дозы в диапазоне 104 — 10® рад. Методики приготовления пленок и измерения степени обесцвечивания метиленового голубого приведены на стр. 228.
Показания пленок из окрашенного целлофана (см. стр. 224) также не зависят от ЛПЭ [40]. Однако точность измерений здесь невысока, особенно при использовании сс-частиц. Согласно [41], окрашенные целлофановые пленки можно применять^для_ нахождения доз до 300 Мрад.
Согласно [421, дозиметры, представляющие собой полимерные пленки с добавкой нитрила трифенилметанового красителя (например, нитрила парарозанилина), дают одинаковые показания в случае у-лучей, электронов и протонов.
Недавно подробное исследование влияния ЛПЭ на свойства окрашенных полимеров было выполнено в работе [431. В ней найдено, что ЛПЭ мало влияет на характер обесцвечивания красителей в поливинилхлориде и диацетате целлюлозы. Например, выходы обесцвечивания 1,4-диаминоантрахинона в диацетате целлюлозы (измерения проводились при длине волны 550 нм) составляют 0,172, 0,176 и 0,171 молек./100 эв соответственно для Y-излучения 60Со, дейтронов с энергией 13 Мэв и гелионов с энергией 23 Мэв.
5. Другие дозиметрические системы
С ростом ЛПЭ G(Fe3+) в 0,05 М растворе серной кислоты, содержащем 10~3 молъ/л FeSO4 и 10~2 молъ/л CuSO4 и насыщенном воздухом, увеличивается. Данные о влиянии ЛПЭ на G(Fe3+) в этой системе приведены в табл. 63.
Таблица 63
Величины G(Fe3+) для сернокислого водного раствора FeSO4 и CuSO4 в случае излучений с высокими значениями ЯП )
Энергия излу чения, M.ie G (Fe3+), ионы/100 эв Литература Энергия излучения, 'Мэз G (Fe3+), ионы, 130 эв Литература
1,99 1,79 1,59 1,27 0,98 0,63 0,46 0,29 21,24 18,80 17,42 14,54 12,20 9,62 7,59 6,55 5,45 4,14 3,35 Протоны 1,58 1,62 1,64 1,67 1,79 2,03 2,18 2,60 Дейтроны 1,02 1,06 1,07 1,11 1,15 1,24 1,34 1,42 1,57 1,67 1,81 [3] [31 [3] [3] [3] [3] [3] [3] [31 [3] [3] [3] [3] [3] [3] [3] [3] [3] [3] 5,3 3,5 3,4 2,0 1,5 1,0 0,6 0,2 Продукт! 2,33 2,33 д-Частицы 2,44 2,1 2,3 1,04 2,0 2,2 2,55 3,4 ,1 ЯД?рп.Н1 J 10B( n, a)7 Li 2,0 2,16 [44] [44] [44] [44] [44] [44] [44] [44] еакции [45] [46J
Согласно [23J, G(—Cr2O?~) для сернокислого водного раствора бихромата (2,5-10-3 молъ/л К2Сг2О7 в 0,4 М H2SO4) при ЛПЭ = =2 эв/А равен 0,43 иона/100 эв. Это значение близко к величине для у-излучения 60Со. По данным работ [23, 47], ЛПЭ можно найти путем измерения отношения G(Fe3+)/G:(—Сг2О7 ) (выход Fe3+ определяется в случае дезаэрированного раствора ферросульфата).
А. М. Кабакчи и сотр. [48] нашли, что выходы нитрита в концентрированных водных растворах нитрата натрия, содержащих реактив Грисса *, мало зависят от величины ЛПЭ. Так, для 6 М раствора NaNO3 6?(NO2) составляет 4,8 и 4,0 иона/100 эв соответственно для у-лучей 60Со и а-частиц 238Ри.
В работе [49] проводилось измерение G(NO2) для водных растворов нитрата аммония при действии нейтронов с энергией 14,1 Мэв **, генерированных в ядерной реакции 3Н (d, и)4Не. В растворы до облучения вводился реактив Грисса для связывания образующихся нитрит-ионов. В этих условиях значения G(NO2) для 4 и 8 М растворов составляют соответственно 5,0 и 5,7 иона/100 эв. Параллельные эксперименты с у-лучами ®°Со дали более низкие величины G(NO2) для растворов указанных концентраций (4,4 и 4,6 иона/100 эв).
Согласно [50], выход НС1 для 10%-ного раствора хлорбензола в смеси 2,2,4-триметилпентан — этанол (10 объемн.%) (раствор насыщен воздухом и содержит 2-10“6 молъ/л калиевой соли тимолсульфофталеина и до 0,1% воды) практически одинаков для у-лучей 60Со и нейтронов с энергией 13,55 Мэв. Для у-лучей G(HC1) равен 5,05, а для нейтронов — 4,76 молек./100 эв.
В работах [23, 51, 52] обнаружено, что выходы этиленгликоля и 2,3-бутандиола в 3 М водных растворах соответственно метанола и этанола не зависят от величины ЛПЭ излучения в диапазоне 0,02—0,7 эв/А. По образованию этих гликолей можно измерять дозы в интервале 5-105 — 1,5-107рад (раствор CHSOH) и 5-105 — 4-107 рад (раствор С2Н60Н) (см. также стр. 295).
По данным Н. А. Бах и сотр. [53, 54], выход красителя в растворе лейкооснования кристаллического фиолетового в метилэтил-кетоне не зависит от величины ЛПЭ, составляя 2 молек./100 эв для рентгеновских лучей с энергией 0,08 Мэв, у-лучей в0Со и а-частиц 210Ро.
Для газообразных систем при сравнительно низких давлениях роль трековых эффектов весьма мала. Поэтому выходы химических превращений в таких системах практически не зависят от величин ЛПЭ в широких пределах. В литературе имеются данные [55—57] об отсутствии влияния ЛПЭ на выходы продуктов радио
* Реактив Грисса предназначен для определения нитрит-ионов. Он представляет собой уксуснокислый раствор а-нафтиламина и сульфаниловой кислоты. В присутствии NO” возникает красная окраска.
** См. примечание к табл. 59.
лиза газообразной закиси азота. Согласно [58J, при очень высоких мощностях дозы электронного излучения эти выходы несколько возрастают. Отмеченное явление, как указывалось в главе IX, обусловлено тем, что в этих условиях происходит замена реакции (1) реакцией (2). Возможно, что такое же явление имеет место и в случае излучений с высокими величинами ЛПЭ [59J. Как следует из работы [60J, С(Н2) для этилена практически одинаков как для ^-излучения 60Со, так и для протонов и гелионов различной энергии. Данные о выходах продуктов радиолиза закиси азота и этилена были приведены в главе VII.
По данным [61J, алюмофосфатные стекла, активированные марганцем, можно применять для нахождения доз пучков тяжелых заряженных частиц. Одинаковые показания, как было найдено, дают протоны (энергия 7 Мэв), дейтроны (энергия 14 Мэв) и гелионы (энергия 28 Мэв). Доза измеряется по появлению окраски (длина волны 560 нм), обусловленной образованием Мп8+. В случае у-излучения 60Со интенсивность возникающей окраски примерно в 1,5 раза больше по сравнению с тяжелыми заряженными частицами.
6. Некоторые практические рекомендации
Тяжелые заряженные частицы при прохождении через конденсированную среду теряют свою энергию на весьма малом пути. Это обстоятельство следует учитывать при определении доз рассматриваемых видов излучения. Например, G(Fes+) в дозиметре
ffffff /ООО и, off./мин.
Рис. 85. Зависимость G(Fe3+)Ha61 от скорости перемешивания г растворов ферросульфата в 0,4 М H2SOi, насыщенных воздухом
1 — облучение дейтронами с энергией 19,4 Мэв, 10—1 М Fe"+, 1=8-10-" а;
— облучение гелионами с энергией 34 Мэе, 10~" М Fe"+, 1 = 10-" а;
— облучение гелионами с энергией 34 Мэе, 10-" М Fe"+, 1 = 5-10-" а
2
з
Фрикке зависит от того, присутствует ли кислород в растворе или же нет (см. главу V). Как правило, при работе с внешними пучками тяжелых заряженных частиц толщина слоя жидкости значительно превышает величину пробега частицы. Поэтому в отсутствие перемешивания могут создаться такие условия, когда скорость расхода кислорода в этом тонком слое жидкости будет превышать скорость его диффузии из необлучаемого объема. Однако при сравнительно больших мощностях дозы даже перемешивание раствора с высокой скоростью не может обеспечить кислородом облучаемую зону в местах высокой плотности ионизации. На рис.
85 показана зависимость наблюденного выхода Fe3+ от силы тока пучка гелионов или дейтронов для насыщенных воздухом растворов ферросульфата [21. Как видно из этого рисунка, G(Fe3+) заметно уменьшается при токах, превышающих 10~8 а. Увеличение скорости перемешивания в этом случае не приводит к возрастанию G(Fe®+). Таким образом, при проведении дозиметрии пучков тяжелых заряженных частиц с помощью систем, для которых выход превращения зависит от наличия кислорода, необходимо использовать перемешивание и небольшие мощности дозы.
ЛИТЕРАТУРА
1. G. L. Kochanny, A. Timnick, С. J. Hochanadel, С. D. Goodman. Radiation Res., 19, 462 (1963).
2. R. H. Schuler, A. О. Allen. J. Am. Chem. Soc.. 79. 1565 (1957).
3. E. J. Hart, W. J. Ramler, S. R. Rocklin. Radiation Res., 4, 378 (1956).
4. И. Драгаиич, M. Симич, H. Милашин, Б. Радак. Труды I Всесоюзного совещания по радиационной химии. М., Пзд-во АН СССР, 1958, стр. 64.
5. J. Pucheault, R. Julien. Proc. Ill Tihany Symposium on Radiation Chemistry. Budapest, 1973, p. 1191.
6. M. Haissinsky, M. C. Anta. Compt. rend., 236, 1161 (1953).
7. N. Miller. Trans. Faraday Soc., 50, 690 (1964).
8. R. C. McDonnell, E. J. Hart. J. Am. Chem. Soc., 76, 2121 (1954).
9. E. Collinson, F. S. Dainton, J. Kroh. Proc. Roy. Soc., A265, 407 (1962)..
10. C. J\. Trumbore, E. J. Hart. J. Phys. Chem., 63, 867 (1959).
11. M. Lefort, X. Tarrago. J. Phys. Chem., 63, 833 (1959).
12. C. N. Trumbore, J. Am. Chem. Soc., 80, 1772 (1958).
13. M. Lefort. Compt. rend., 245, 1623 (1957).
14. " N. Miller. Radiation Res., 9, 633 (1958).
15. E. J. Hart, J. Terandy. Rev. Sci. Instrum, 29, 962 (1958).
16. С. B. Senvar, E. J. Hart. Proc. II United Nations Intern. Conference on Peaceful Uses of Atomic Energy, vol. 29. Geneva, 1958, p. 19.
17. L. Ehrenberg, E. Saeland. JENER Publ. No. 8 (1954).
18. R. C. Axtmann, J. A. Licari. Radiation Res., 12, 511 (1964).
19. А. Г. Васильев, Д. В. Викторов, И. М. Розман, В. В. Ткаченко. Химия высоких энергий, 3, 269 (1969).
20. W. A. Armstrong, W. G. Humpreys. Can. J. Chem., 42, 1092 (1964).
21. M. В. Владимирова. Атомная энергия, 17, 222 (1964).
22. М.В. Владимирова, А. А. Баталов, И. А. Куликов, Л. Г. Шулятикова. Дозиметрия больших доз. Ташкент, «Фан», 1966, стр. 163.
23. Е. П. Ковалева, А. М. Афанасьев, Л. Т. Бугаенко, В. М. Бяков, Е. Н. Калязин, Е. П. Петряев. Вести. АН БССР, серия физ.-энерг. наук, № 2, 29 (1970).
24. N. F. Barr, R. Н. Schuler. .1. Phys. Chem., 63, 808 (1969).
25. J. Weiss, N. Miller. Ibid., p. 888.
26. M. C. Anta, M. H. Mariano, M. L. Santos. J. Chim. phys., 61, 577 (1964).
27. R. H. Schuler, A. O. Allen. J. Am. Chem. Soc., 77, 507 (1955).
28. W. G. Burns, J. R. Parry. Nature, 201, 814 (1964).
29. F. Mooseley, A. E. Truswell. Rep. AERE—R 3078 (1960); цит. no I. Draganic. Determination of Absorbed Dose in Reactors. Vienna, IAEA, 1971, p. 197.
30. A. W. Boyd, H. W. J. Connor. Can. J. Chem., 42, 1418 (1964).
31. W. G. Burns. J. Phys. Chem., 65, 2261 (1961).
32. P. J. Dyne, J. A. Stone. Can. J. Chem., 39, 2381 (1961).
33. Я. И. Лаврентович, А. Б. Зверев, А. А. Беликовский, A. M. Кабакчи. Химия высоких энергий, 3, 147 (1969).
34. А. Б. Зверев, Я. И. Лаврентович, А. М. Еабакчи. Там же, стр. 453.
35. Я. И. Лаврентович, Г. С. Якименко, А. Г. Старенький, А. М. Кабакчи. Там же, стр. 44.
36. Я. И. Лаврентович, Г. С. Якименко, А. Б. Зверев, А. М. Кабакчи. Атомная энергия, 27, 296 (1969).
37. Я. И. Лаврентович, Л. Е. Коваленко, А. Г. Старенький, А. А. Беликовский, А. М. Кабакчи. Дозиметрия и радиационные процессы в дозиметрических системах. Ташкент, «Фан», 1972, стр. 178.
38. Я. И. Лаврентович, А. И. Левон, Г. Н. Мельникова, А. М. Кабакчи. Атомная энергия, 19, 273 (1965).
39. Я. И. Лаврентович, А. М. Кабакчи. Радиационная химия полимеров. М., «Наука», 1966, стр. 253.
40. A. Charlesby, R. J. Woods. Intern. J. Appl. Radiation Isotopes, 14, 413 (1963).
41. Л. M. Коваленко, Л. H. Гайченко, Я. И. Лаврентович, А. М. Кабакчи. Атомная энергия, 31, 510 (1971).
42. W. L. McLaughlin, Р. Е. Hjortenberg, В. В. Radak. Dosimetry in Agriculture, Industry, Biology and Medicine. Vienna, IAEA, 1973, p. 577.
43. Л. M. Коваленко, Л. H. Гайченко, Я. И. Лаврентович. Дозиметрия и радиационные процессы в дозиметрических системах. Ташкент, «Фап», 1972, стр. 174.
44. A. Gordon, Е. J. Hart. Radiation Res., 15, 440 (1961).
45. Е. J. Hart, P. D. Walsch. Radiation Res., 1, 342 (1954).
46. P. Sigli, J. Pucheault. J. Chim. phys., 70, 717 (1973).
47. E. П. Петряев, E. П. Калязин, E. П. Ковалева. Дозиметрия и радиационные процессы в дозиметрических системах.„Ташкент, «Фан», 1972, стр. 170.
48. О. Ф. Чебурков, К. В. Малахов, В. А. Грамолин, А. М. Кабакчи. Труды II Всесоюзного совещания по радиационной химии. М., Изд-во АН СССР, 1963, стр. 159.
49. А. Г. Васильев, Д. В. Викторов, И. М. Розман, В. В. Ткаченко. Химия высоких энергий, 3, 373 (1969).
50. I. Dvornik, U. Zee, М. Baric, D. Razem. Handling of Radiation Accidents. Vienna, IAEA, 1969, p. 225.
51. A. M. Афанасьев, Л. T. Бугаенко, В. M. Бяков, Е. П. Калязин, Е. П. Ковалева, Ф. Т. Ничипоров, Е. П. Петряев. Вестн. АН БССР, серия физ.-энерг. наук, № 4, 23 (1970).
52. Е. П. Ковалева. Автореферат канд. дисс. Минск, Ин-т ядерной энергетики АН БССР, 1972.
53. Н. А. Бах, Г. Г. Бабичева, В. А. Ларин. Докл. АН СССР, 134, 1079 (1960).
54. Н. А. Бах, Г. Г. Бабичева, В. А. Ларин, Труды II Всесоюзного совещания по радиационной химии. М., Изд-во АН СССР, 1962, стр. 738.
55. М. Т. Дмитриев. Атомная энергия, 15, 52 (1963)
56. R. W. Hummel, J. A. Hearne. Radiation Res., 15, 254 (1961).
57. В. W. Hummel, J. A. Hearne. Nature, 188, 734 (1960).
58. C. Willis, A. W. Boyd, D. A. Armstrong. Can. J. Chem., 47, 3783 (1969).
59. I. Draganic. Determination of Absorbed Dose in Reactors. Vienna, IAEA, 1971, p. 197.
60. A. R. Anderson, J. V.F. Best. Nature, 216, 576 (1967).
61. В. В. Ткаченко, Б. А. Брискман, Л. M. Коваленко, Я. И. Лаврентович. Атомная энергия, 35, 210 (1973).
Глава XI
ХИМИЧЕСКИЕ МЕТОДЫ ДОЗИМЕТРИИ
РЕАКТОРНОГО ИЗЛУЧЕНИЯ
На практике часто имеют дело с реакторным излучением, состоящим из потоков нейтронов и у-лучей. Дозиметрия этого вида излучения химическими методами основана на правиле аддитивности [1J: суммарная реакция, протекающая под действием смешанного излучения, равна сумме реакций, индуцированных отдельно каждым компонентом этого излучения. Оно выражается формулой
G=XG„+/vGV _ W
где G — выход превращения в дозиметрической системе; Gn и Gy — выходы этого превращения за счет нейтронов и у-лучей соответственно; /п и /т — доли вклада нейтронов и у-лучей в суммарную дозу.
При работе на реакторах энергию излучения, поглощаемую системой (точнее — мощность дозы), обычно выражают в милливаттах на грамм или в ваттах на грамм. Соотношения между этими единицами и радами на секунду или радами на час таковы: 1 рад/ /сек = 10 5 ет/г; 1 мет/г = 3,6-10s рад/час.
При проведении радиационно-химических экспериментов с реакторным излучением приходится обычно решать два вопроса: 1) определение суммарной поглощенной дозы смешанного гамма-нейтронного излучения и 2) определение дозы каждого отдельного компонента смешанного излучения. Для дифференциации доз различных компонентов реакторного излучения используются следующие особенности взаимодействия нейтронов с веществом. В главе I отмечалось, что при действии быстрых нейтронов на различные среды, состоящие из водорода, углерода, азота, кислорода и небольших количеств других легких элементов, энергия поглощается в основном в результате процесса упругого рассеяния нейтронов ядрами атомов водорода. В этом процессе возникают протоны отдачи. Поэтому применением двух дозиметрических систем — водородсодержащей и не содержащей атомов водорода — можно в принципе разграничить дозы быстрых нейтронов и у-лучей. Для измерения потоков тепловых нейтронов в систему обычно вводят соединения элементов (чаще всего — 6Li, ,0В и 23®U), имеющих большие поперечные сечения захвата этих нейтронов.
Предлагались различные способы решения указанных выше вопросов с помощью химических дозиметрических систем. Сводка таких систем дана в табл. 64. Из их числа наиболее подробно исследованы водный раствор щавелевой кислоты, закись азота и циклогексан.
1. Водный раствор щавелевой кислоты
По мнению И. Драганпча [1J, который провел обширные исследования радиолиза растворов щавелевой кислоты в Н2О и D2O [26, 30, 65J, с помощью этой системы возможно определять дозы для многих объектов с низкими значениями эффективного атомного номера. Как уже отмечалось (см. стр. 169), при облучении водных растворов щавелевой кислоты происходит ее разложение, причем главным продуктом радиолиза является СО2. Скорость разложения вплоть до 60% подчиняется уравнению первого порядка, которое можно представить в следующей форме, удобной для использования:
„ , с<>
D = ас,, 1g — , (2)
где D — доза; с0 и с — концентрации щавелевой кислоты до и после облучения; а — коэффициент пропорциональности. Этот коэффициент не зависит от с0 в диапазоне от 0,05 до 0,6 М, мощности дозы до 10в рад/сек [66J и температуры до — 80° С. Было найдено [28J, что он увеличивается с ростом ЛПЭ излучения.
Для реакторного излучения коэффициент а зависит от вклада нейтронов и у-лучей в суммарную дозу (т. е. от величин fn и /Y) и соответствующих значений а для каждого компонента (т. е. от ап и aY). Уравнение, связывающее эти величины, имеет следую-щии вид Z
= /Y/«Y + /„/«„. (3)
Очевидно, уравнение (2) можно применять для расчета дозы в воде в том случае, когда, кроме с0 и с, известно также а для данных условий облучения в реакторе. Этот коэффициент может быть определен экспериментально (например, путем использования метода калориметрии), вычислен с помощью уравнения (3), когда известны члены, входящие в его правую часть, или взят из измерений в аналогичных условиях на другом реакторе. Отметим, что он равен aY для отражателя нейтронов или тепловой колонны. В этом случае необходимо для приготовления растворов использовать О2О вследствие меньшего поперечного сечения захвата тепловых нейтронов. Согласно [281, при отношениях доз у-лучей и нейтронов, равных от 3 : 2 до 2 : 3, коэффициент а равен 49 эв/молъ (+ 4%); ошибка больше (+10%), если это отношение лежит в пределах от 4 : 1 до 1 : 4. По мнению И. Драганича [1], ап не должно сильно зависеть от спектра нейтронов реактора.
Сводка систем, рекомендованных для дозиметрии реакторного излучения
Литература [2-Н] [12, 13] [4, 5, 10] *-1 [15-24] [19] LQ (М [3, 24, 26- 32] [14] [33] [34-36]
Диапазон измеряемых доз, рад 10s—107 5-10’—108 1 00 О СО i 1 о» О J о »г-< LQ СМ 1 6 т—’ Ю С X 2 —1 1 Г-Н мощностей дозы более 10s рад/час 1 5-Ю5— 1,5-107
Метод измерения пренращения к © о г H e- о Я о о >©< о Си к ф к о л Обычная техника измерения На и Н2О2 Поляриметрический э D S =1 э □ 5 2 » S ф □и -о S к ф в* и Ри о & о ср сб Спектрофотометрический; титрование NaOH Измерение скорости газовыде-ления (На + Ог) Спектрофотометрический Спектрофотометрический и хроматографический
Химическое превращение Образование Fe3+ То же Восстановление Се4+ с г с: с Ё С С £ а JS t с Й с г О' с вание На и НаОа Изменение оптического вращения иоразование кислотных продуктов, приводящее к увеличению электропроводности Образование газообразных про- а э 2 Разложение Н2С2О4 1 1 1 С С 5 а D Й D $ 2 •о U Образование NOa~ Образование этиленгликоля
Система Водный раствор Fe2+ в 0,4 М HeSOi Водный раствор Fe2+ и Си2+ в 0,05 М H2SO4 Водный раствор Се4+ в 0,4 М H2SO4 Водный раствор НСООН в присутствии кислорода Водный раствор глюкозы Водный раствор щавелевой кис- лоты Водный раствор К J в присутствии кислорода Водный раствор N FUN Оз 3 М водный раствор метанола
Таблица 64 (продолжение)
Система Химическое превращение Метод измерения превращения Диапазон измеряемых доз, рад Литература
3 М водный раствор этанола Водный раствор Сг2О;2- в 0,4 М H2SO4 Образование 2,3-бутанднола Восстановление Cr (VI) Хроматографический Спектрофотометрический 5- Юз-4-107 [37] [34, 36]
Метиловый спирт Образование этиленгликоля Спектрофотометрический и хро- 5.10s—108 [37]
Этиловый спирт Циклогексан Образование 2,3-бутапдиола Образование Н2 матографический Хроматографический Стандартная вакуумная техни- 5.105—108 До 1 6-107 [37] [38-42]
Бензол Дифенил То же Г а зовыдепе ние ка То же — [41]
Тетрахпорметан Тетрахлорэтилен или хлороформ Образование С1з Образование НС1 Спектрофотометрический Титрование NaOH До 10° 2-103—2-106 До 2 • 105 [43] [44] [45]
Полностью фторированные углеводороды Образование CF4 и C2Fe Хроматографический 2-10е—5-Ю7 [46—49]
Декан Газовыделение Стандартная вакуумная техни- — [50]
Твердая щавелевая кислота Разложение Н2С2О4 ка Спектрофотометрический, тит- Ю8—10° [51]
Твердая глюкоза фондированные полимеры (CFCl=CF2)n Образование Из Деструкция полимера ровапие, взвешивание Хроматографический Вискозиметричссклй До 10° До 1010 [25, 37] [52]
Таблица 64 (окончание)
Диапазон Литература
Система Химическое превращение Метод измерения превращения измеряемых доз, рад
Диацетат целлюлозы Деструкция полимера Спектрофотометрический 2-10е—7-107 [53—55]
Пленки из поливинилового спирта, содержащие метиленовый голубой Обесцвечивание красителя » 104—10s 108-Ю9 [56]
Полиметилметакрилат Деструкция полимера » [57]
Полиэтилен Образование транс-винилено- Спектроскопический (измере- 107—108 [54]
вых связей пия в инфракрасной области при 966 сл1-1)
Сшивание Измерение величины гель-фрак-ции До ~3-107 [55]
Политрихлорфторэтилен Деструкция Спектрофотометрический (измерения при 230 нм) 5.10S—6-107 [55]
Закись азота Разложение N3O; образование Стандартная вакуумная тех- 10s—5-10° [52, 58—63]
Na и Оз ника; хроматографический
Воздух Образование NOa Стандартная вакуумная техника, спектрофотометрический, растворение в воде с последующим определением NOa~ До ~10° [61]
Смесь окиси и двуокиси угле- Обмен 14С между СОа и СО Разделение методом хромато- — [64]
рода графин и счет сцинтилляций
Метан Образование На Хроматографический До 5-Ю7 [63]
Выбор начальной концентрации раствора зависит от измеряемого диапазона доз (см. стр. 172). Для получения наибольшей точности необходимо подбирать условия облучения таким образом, чтобы степень разложения 112С2О4 была в интервале от 40 до 60%. Методики анализа щавелевой кислоты были приведены в главе V.
Водные растворы Н2С2О4 стабильны при хранении, в них не наблюдаются пост-эффекты. Они характеризуются отсутствием наведенной радиоактивности. В этом состоят преимущества данной дозиметрической системы. Однако она имеет существенный недостаток, а именно: кривая зависимости количества разложившейся кислоты от дозы не является линейной. Поэтому для точных измерений (особенно в случае больших доз) ее необходимо прокалибровать для данных рабочих условий [1J.
Совместное использование растворов Н?С2О4 в Н2О и D2O позволяет оценивать вклад у-лучей и нейтронов в суммарную поглощенную дозу. Однако, поскольку различия в поглощающей способности между Н2О и D2O сравнительно небольшие, такая оценка является лишь весьма приближенной.
2. Водные растворы углеводов
При облучении водных растворов глюкозы или мальтозы происходит разложение этих веществ, что проявляется в уменьшении оптической активности растворов. Согласно [15, 16J, для смешанного реакторного излучения это изменение связано с дозой D (в Мрад) уравнением»
n_ALi
D~ а lg 1
(4)
где а — коэффициент, зависящий от природы и концентрации растворенного вещества; £0 — оптическое вращение раствора до облучения и Л£ — абсолютное изменение угла вращения после облучения. Для растворов глюкозы с концентрацией от 5 до 35% коэффициент а изменяется от 8-10~3 до 2-10‘3.
Растворы глюкозы могут быть использованы для определения доз в диапазоне от 0,1 до 800 Мрад. Выход не зависит от мощности дозы в интервале от 5 до 100 Мрад/час. По данным [15, 16J, выход изменяется не более чем на 10—15% при переходе от у-лучей к быстрым нейтронам. Растворы стабильны до и после облучения; они не становятся радиоактивными в результате воздействия излучения реактора^
В. В. Генералова и сотр. [18] сконструировали петлевую дозиметрическую установку с циркуляцией водного раствора глюкозы. Раствор непрерывно облучается в активной зоне реактора; при этом производится постоянный и дистанционный контроль за химическими превращениями дозиметрической системы. В цитированной работе измерялось изменение удельного вращения плоскости поляризации облучаемых растворов. Верхний продел опре
деляемых в этом случае доз составляет — 10п рад. По данным [19], при использовании этой установки для непрерывного измерения дозы можно также регистрировать увеличение электропроводности растворов глюкозы, обусловленное образованием кислотных продуктов.
Согласно [67], водные растворы глюкозы, содержащие Н3ВО3, пригодны для измерения потоков тепловых нейтронов.
Е. П. Ковалева и др. [25] предложили использовать выделение водорода из водных растворов глюкозы как меру суммарной дозы реакторного излучения. Выходы водорода равны 0,64, 1,5 и 2,0 молек./ЮО эв соответственно для 0,3, 1,2 и 2,5 М растворов. Они не зависят от мощности дозы в интервале 50—Ю4 рад/сек, температуры облучения в диапазоне 20—70° С и величины ЛПЭ в пределах 0,02-1 эв/к.
3. Определение дозы реакторного излучения параллельным использованием двух водных дозиметрических систем
М. В. Владимирова и сотр. [6, 8] предложили проводить параллельное измерение концентрации иопов Fe3+ в дозиметре Фрпк-ке и молекулярного водорода в 0,4 М растворе H2SO4, образующихся при облучении, для нахождения дозы реакторного излучения. Эксперименты проводились на реакторе ВВР. Величина ЛПЭ для смешанного излучения (у-лучи и быстрые нейтроны) указанного реактора составляет 0,28 эе/А. Значения 6?(П2) и G(Fe3+) для этого излучения равны соответственно 0,58 и 12,6 молек./ /100 эв [6, 8|. По данным цитируемых работ, максимальная доза, которую можно измерить с помощью дозиметра Фрикке, равна —- 10® рад. Насыщение раствора кислородом расширяет этот предел до — 5-10® рад. Для раздельного определения доз у- и нейтронного излучения этим методом необходимо знать величины G(H2) и 6?(Fe3+) для быстрых нейтронов. В работах [6, 8] значение (?(Н2) для быстрых нейтронов рассчитывалось, исходя из энергетического спектра быстрых нейтронов в реакторе ВВР и величин 6?(П2) для протонов отдачи каждой энергии. Было найдено, что это значение равно 1,0 молек./ЮО эв.
Рассматриваемый способ дозиметрии позволяет найти величину ЛПЭ излучения путем измерения отношения С(П2) / G(Fe3+). Соответствующая методика была приведена в главе X (см. стр. 273).
Позднее сходный метод был описан в работе [34]. В ней для дозиметрии реакторного излучения использованы следующие пары водных систем: дезаэрированный 5-Ю’2 М раствор Fe2+ в присутствии 10’3 молъ/л NaCl и насыщенный воздухом 0,4 М раствор H2SO4 с добавкой 2-10'® молъ/л КВт; первый из этих растворов и насыщенный воздухом 2,5-Ю-3 М раствор К2Сг2О7 в 0,4 М H2SO4. Согласно [37], с помощью этих пар можно определять эф-
фективнуто величину ЛПЭ и мощность дозы реакторного излучения соответственно до 10 эв/А и 105 рад1сек. По данным [36J, применение водных растворов Fe2+ и Сг2О?~ позволяет измерять дозы в диапазоне 5-10® — 5-10® рад.
4. Закись азота
В главе VII уже говорилось о том, что N2O при радиолизе разлагается и количество образующихся азота и кислорода может служить мерой дозы в диапазоне от 10® до 3-10® рад и даже выше. Поскольку N,0 в обычных условиях является газом, то ЛПЭ излучения при радиолизе не оказывает влияния на выход разложения. Эта особенность находит свое применение в дозиметрии смешанного реакторного излучения [52, 58—63, 681. Данные о выходах продуктов радиолиза N.,0 были приведены в главе VII (см. стр. 212).
Поглощенная доза в дозиметре на основе N2O при облучении в реакторе главным образом обусловлена у-лучами и продуктами ядерной реакции 14N (и, р)14С. Рассчитаем дозу за счет этой реакции [69J. Сечение захвата тепловых нейтронов атомами 14N равно 1,75- 1(Г24 ел.2. Энергия протона и атома отдачи 14С составляет 0,626 Мэв. Тогда при потоке тепловых нейтронов, равном, например, 1014 нейтронов/сж2-сск энергия, полностью поглощаемая одним граммом N2O за счет рассматриваемой реакции, составляет
£1 • (28/44) (6,02 IO*3) (1,75 - 10-2*) • 1 10’* • (0,626 - 10»)
- . (6,24 • 1015) = мет!г
Протон, образующийся в этой реакции, имеет энергию 0,59 Мэв. Пробег такого протона в воздухе при 15° С и давлении 1 атм равен — 1 см. Пробег 14С очень мал, и эти атомы отдачи полностью поглощаются в N2O. Таким образом, для того, чтобы получить только дозу у-лучей, необходимо в показания этого дозиметра вносить поправку на дозу за счет реакции 14N (п, р)14С.
Согласно [58], N2O может быть использована для измерения потоков тепловых нейтронов. С этой целью рекомендуется вводить в дозиметрический сосуд несколько миллиграммов 23®UO2. Изотоп 23®U под действием тепловых нейтронов претерпевает деление. Продукты деления вызывают разложение закиси азота, причем степень разложения гораздо больше, чем в отсутствие 23®UO2 (т. е. за счет у-излучения). Таким образом, облучая N2O в отсутствие и в присутствии 23®U можно найти дозу у-лучей и определить поток теп* ловых нейтронов.
Особенности дозиметра на основе N2O и способы измерения химических превращений в закиси азота при облучении подробно рассмотрены в главе VII (см. стр. 212).
5. Циклогексан
При радиолизе циклогексана, наряду с другими продуктами, образуется молекулярный водород. Предлагалось [38—42, 70J по количеству водорода измерять поглощенную дозу реакторного излучения. 6'(Н2) мало зависит от ЛПЭ излучения (см. табл. 61) и постоянен до доз 1 7-107 рад. Зависимость 6?(Н2) от дозы для смешанного реакторного излучения приведена на рис. 86. Циклогексан стабилен при хранении; он не становится радиоактивным в результате облучения в реакторе.
Рис. 86. Зависимость G(H2) от дозы при радиолизе циклогексана под действием смешанного п, у-излучения ядерного реактора (70% нейтронов и 30% у-лучей)
1 — мощность дозы 2-104 рад/сек-, и— мощность дозы 2-105 рад/сек
о /
• 2
---1_______।
В /2
и-1О~гЬ,зв/г
По данным А. Бойда и др. [70, 711, по выделению D2 из полностью дейтерированного циклогексана CeD12 можно оценить дозу у-излучения, а по выделению Н2 из циклогексана CeHJ2 — суммарную дозу за счет быстрых нейтронов и у-лучей.
Методики приготовления образцов циклогексана для дозиметрии и анализа Н2 описаны в главе VI (см. стр. 202).
Циклогексановый дозиметр рекомендован [1, 70, 71] для измерения поглощенных доз в материалах состава (СН2)„.
6. Другие дозиметрические системы
Вода Н2О и D2O. Еще в 1952 г. предлагалось [72] с целью дозиметрии реакторного излучения использовать Н2О и D2O. В Н2О водород и перекись водорода возникают главным образом за счет быстрых нейтронов и у-лучей, а в D2O — преимущественно за счет у-излучения.
Водный раствор буры. В 1951 г. П. Боне-Мари [73] использовал для определения больших потоков тепловых нейтронов (до 1017 нейтронов/сж2) водные растворы буры. В этих растворах под действием продуктов реакции 10В (и, a)7Li выделялась перекись водорода, концентрация которой служила мерой дозы. Определение Н2О2 проводилось с помощью титанового реактива.
Водный раствор феррина. Ж. Пюшю [4, 74] исследовал возможность применения водных растворов феррина (комплексное соединение трехвалентного железа с о-фенантролином) для раздельного измерения доз у-лучей и быстрых нейтронов. При облучении этих растворов происходит восстановление Fe(III) в Fe(II), причем радиационно-химический выход данного превраще
ний при действии у-лучей примерно в 15 раз больше, чем прИ действии быстрых нейтронов. Однако эта система имеет недостаток. связанный с послерадиационным восстановлением Fe3+.
Водный раствор К.Т. 9. Харт и сотр. Г14, 75] разработали метод дозиметрии смешанного гамма-нейтронного излучения ядер ного реактора, состоящий в измерении количества газа (Н2 и О2), выделяющегося при радиолизе водного раствора иоцида калия. Уравнение, выражающее зависимость скорости газовыделения при облучении раствора KJ от мощности дозы, имеет следующий вид [14|:
(dn\ G, /ЛЕ\ Gn fdE\
\ dt /Y+„ — 100 \ dt 100 \ dt ! ' (5)
где n — суммарное количество образующихся молекул Н2 и О2; t — время в минутах; G.( и Gn — выходы Н2 О2 за счет у-излуче-ния и быстрых нейтронов (протонов отдачи) соответственно; Еу и Еп — поглощенная энергия у-излучения и быстрых нейтронов (протонов отдачи) соответственно.
Как видно из уравнения (5), чтобы рассчитать E.t и Еп, необхо
димо знать Gy, Gn и отношение Е.(/Еп (скорость газовыделения
Рис. 87. Схема прибора для измерения газовыделения при ра дполизе водного раствора KJ
определяется экспериментально). GY и Gn находятся в специально поставленных для этого опытах. Отношение Еу/Еп возможно найти с помощью какой-либо другой системы. Э. Харт и П. Уолш использовали для этой цели водный раствор муравьиной кислоты (в присутствии кислорода).
Схема прибора для измерения газовыделения показана на рис. 87. Из бюретки А периодически удаляется дибутилфталат со скоростью, равной скорости газовыделения из ячейки В с раствором, помещенной в реактор. Манометр С наполнен дибутилфталатом; он служит в качестве индикатора для уравнивания давления. Краны D, Е, F и G открыты в начальной стадии газовыделения; краны D и F закрываются во время измерений. Колба /7, частично заполненная водой, предназначена для уравнивания давления и температуры. Ячейка соединена с измерительной системой по
средством алюминиевых трубок; для
сочленений используется каучук.
По данным тех же авторов [14, 75J, если в раствор KJ ввести бор
ную кислоту, то раствор становится пригодным для определения потоков тепловых нейтронов. Добавка Н3ВО3 приводит к возрастанию скорости газовыделения вследствие протекания ядер-ной реакции 10В (п, a)7Li. Доза рассчитывается по уравнению
(dn \ / dn \ GHSBO3 /££нзВОя_\
dt /н3В03 ~ \ dt /у+п 100 \ dt /
где п — количество молекул Н2 и О2, образующихся при действии различных видов реакторного излучения (индекс «у п» обозначает газовыделение в растворе без добавки Н3ВО3, а индекс «Н3ВО3» — газовыделение в растворе с добавкой Н3ВО3); t — время в минутах; Ь’нщо., — поглощенная энергия за счет ядер-ной реакции 10В (п, cx)7Li; 6?н3во3 — выход Н2 и О2 в случае продуктов ядерной реакции 10В (и, a)7Li.
Перегруппировывая уравнение (6), получаем
^НзВОз dt
100 Г/ dn \ / dn \
GH3B03 /Н3ВО3 \ dt М"
(7)
Таким образом, для определения дозы, обусловленной реакцией тепловых нейтронов с 10В, необходимо знать лишь величину ^НзВОз-
Дозиметр Фрикке с добавкой борной кислоты или сульфата лития. Для измерения потоков тепловых нейтронов часто применяется дозиметр Фрикке с добавкой Н3ВО3 или Li2SO4-H2O. Впервые эта система в качестве дозиметра для смешанного потока тепловых нейтронов и у-лучей была предложена Э. Силандом и Л. Эренбергом [76J. Позже она исследовалась многими авторами [3, 6, 8, 77—80J. В табл. 65 приведены измеренные этими авторами значения G(Fe3+).
Таблица 65
Величины G(Fe3+) для продуктов ядерных реакций 10В(»г, a)7Li и 6Li(п, a)Т
Ядерная реакция G(Fe3+), ЙОНЫ/100 96 Литература Ядерная реакция G(Fe3+), ионы/100 эз Литература
10В (п, a)’Li 4,1 4,2 4,22 [77] [68] [79] 6Li(n, a) Т 5,4 5,2 5,0 5,69 5,65 [77] [78] [80] [79] [6, 8]
Борная кислота не мешает определению Fe3+ спектрофотометрическим методом (при длине волны 304—305 нм). Однако в присутствии Li2SO4 молярный коэффициент экстинкции Fe34
в 0,4 М H2SO4 при этой длине волны несколько возрастает (791. Это следует учитывать при проведении дозиметрических измерений.
Дозиметр Фрикке с добавкой Н3ВО3 или Li2SO4 дает величину дозы в радах или в электронвольтах на миллилитр. Потоки тепловых нейтронов обычно измеряют числом нейтронов на 1 см2 в 1 сек. \п/(см2-сек)\. Пересчет производится следующим образом. Пусть Р — мощность дозы [в эвЦмл-сек)\ при данной концентрации Н3ВО3 или Li2SO4; / — поток тепловых нейтронов [в п/(см2-•сек)!; М — молярная концентрация борат-ионов или Li+; о — сечение захвата тепловых нейтронов атомами В или Li (в см2); Ео — энергия, выделяемая в результате одного распада В или Li (в эв/п), и 7V — число Авогадро. Тогда
1000
f = ~E9MN<s n/^CMi' С(’к^ &
Для реакции 10В (п, a)7Li значение Ео равно 2,33-10® эв/п *. Сечение захвата тепловых нейтронов атомами 10В составляет 3,84-•10~21 см2. Отсюда для естественной смеси изотопов бора сечение реакции 10В (n, a)7Li равно 7,4-10~22 см2. Тогда
Р
/ = 9,63 • 10- -у- пЦсм* сек). (9)
В случае реакции 6Li (п, а)Т Ео = 4,66-10® эв/п. Величина о для атомов ®Li 9,5-10~22 см2. Тогда для естественной смеси изотопов находим, что о = 7,1 -10~23 см2. Следовательно,
Р
f = 5,02-10 ° -ду- п/(см2 - сек). (10)
Водный сернокислый раствор Fe2+ и Сиа+. Величина G(Fe3a') в дозиметре Фрикке при действии атомов отдачи ядерных реакций в 3—4 раза меньше, чем в случае у-излучения. Поэтому при определении доли тепловых нейтронов в смешанном потоке приходится иметь дело со сравнительно высоким фоном, что, естественно, приводит к не очень точным результатам. Этот недостаток в не которой степени был устранен Э. Хартом и П. Уолш [82]. Указан ные авторы предложили применять для дозиметрии реакторного излучения водный раствор, содержащий 5-10 3 молъ/л H2SO4, 10-2 молъ/л CuSO4, 5-10“3 молъ/л FeSO4 и 1,8-10~2 молъ/л Н3ВО3. В этой системе G(Fe3+) равен 2.0 и 0,66 иона/100 эв соответственно для продуктов реакции 10В (n, cc)7Li и у-излучения 60Со. Поэтому фон, создаваемый у-компонентом смешанного излучения, является сравнительно низким. Однако данная система, как выяснилось позже [83, 84J, характеризуется некоторой неустойчивостью
* Величины Ёо и а для реакций 1(|В (и, a)’Li и “Li (re, а)Т приводятся по данным [81].
после облучения, что весьма неудобно, поскольку образцы не могут быть сразу же проанализированы из-за наведенной активности в материале ячейки.
Метиловый и этиловый спирты и их водные растворы. Подробное исследование возможности использования метилового и этилового спиртов и их водных растворов было проведено в работах [34—37J. Было найдено, что по образованию этиленгликоля и 2,3-бутандиола в 3 М водных растворах соответственно метанола и этанола можно измерять дозы смешанного излучения реактора в диапазоне 5-Ю5 — 1,5-107 рад (раствор метанола) и 1,5-Ю5 — 4-107 рад (раствор этанола). Выходы гликолей не зависят от мощности дозы в интервале 102 — 105 рад/сек и величины ЛПЭ в диапазоне 0,02—0,7 эв/А. При больших значениях ЛПЭ выходы этих продуктов уменьшаются. Выходы сравнительно мало зависят от температуры. Например, выход этиленгликоля возрастает всего примерно на 30% при увеличении температуры от комнатной до — 300° С. В сухих спиртах выходы указанных гликолей не зависят от дозы до 10а рад. Однако в этом случае наблюдается сильная зависимость выходов от температуры.
Хлорированные углеводороды. Согласно [44, 45, 85, 861, для дифференциации доз у-излучения и быстрых нейтронов можно использовать дозиметры на основе хлорированных углеводородов. Принцип метода заключается в следующем. Например, дозиметр, состоящий из хлорсодержащего углеводорода, покрытого водным раствором pH-индикатора, или из водного раствора хлорированного углеводорода, чувствителен как к у-лучам, так и к быстрым нейтронам. Измерить же одну у-составляющую возможно с помощью тетрахлорэтилена, который не содержит водорода и потому практически не претерпевает химических изменений при действии быстрых нейтронов. Однако недостатком подобных дозиметрических систем является сравнительно высокое поперечное сечение захвата тепловых нейтронов хлором [47J.
Фторированные углеводороды. Е Прёкш [46—481 предложил использовать перфторалканы как дозиметры, не чувствительные к быстрым нейтронам. Наиболее подробно были изучены перфтор-w-гексан и два полностью фторированных сорта керосина (низко-кипящий продукт состава — C12F2e и высококипящий продукт состава — C14F30). Было найдено, что эти вещества пригодны для дозиметрии у-излучения в смешанном потоке. Оказалось, в частности, что с помощью перфтор-н-гексана можно измерять дозы у-излучения от 3 до 40 Мрад. Величина дозы определяется по выходу газообразных продуктов радиолиза. Анализ их осуществляется методом газовой хроматографии. При этом измеряются пики, обусловленные CF4 или C2Fe. Для перфтор-н-гексана G(CF4) и G(C2F6) равны соответственно 0,18 и 0,10 молок./100 эв. Выходы практически не зависят от мощности дозы в исследованном диапазоне (от 14 до 3 -104 рад/сек). Преимуществом перфторалканов перед длоралканами является то, что в них не появляется наведенная
радиоактивность и что они менее чувствительны к быстрым нейтронам.
Подробное изучение радиолиза перфторметилциклогсксана под действием у-излучения 60Со, смешанного излучения реактора и быстрых нейтронов было проведено в работе [49J. Было найдено, что выходы различных продуктов зависят в некоторой степени от многих параметров (дозы, мощности дозы, температуры облучения и т. п.). Наименьшей зависимостью от этих параметров характеризуется выход разложения перфторметилциклогек-сана.
Отметим, что степень вклада быстрых нейтронов в общую поглощенную дозу зависит от их доли в смешанном излучении реактора. В частности, выход разложения дезаэрированного перфтор-метилциклогексана под действием быстрых нейтронов (т. е. за счет чаряжениых частиц, возникших при упругих и неупругих взаимодействиях быстрых нейтронов с ядрами С и F) равен примерно половине выхода разложения при у-радиолизе [49].
Дозиметры на основе твердых тел. В литературе имеется довольно большое число работ (см., например, [24, 25, 33, 37, 50— 56, 87—106]) по исследованию возможности применения различных твердых веществ для дозиметрии реакторного излучения. Рассмотрим результаты некоторых из этих работ. Согласно [51J. твердая щавелевая кислота П2С2О4-2П2О пригодна для измерения доз в диапазоне 108 — 109ра<3. При облучении она разлагается. Выход разложения G(—Н2С2О4 • 2Н2О) для реакторного излучения, для которого вклад гамма- и нейтронной компонент составляет 82 и 18% соответственно, равен 8,3 молек./100 эв: в этих условиях G(CO2) = 12,7 молек./100 эв. Аналитические методы определения степени разложения такие же, как и в случае водных растворов этой кислоты. Когда разложение составляет более 15%, можно применять весовой метод. В этом случае облученный образец перед взвешиванием нагревается в течение 2 час. при 95° С для того, чтобы удалить газообразные продукты. Для расчета дозы D используется формула
По
D=alg—, (11)
где п0 и п — количество вещества до и после облучения.
Как отмечается в работах [25, 37], для определения суммарных поглощенных доз смешанного излучения реактора может быть использована твердая глюкоза. С ее помощью по образованию водорода можно измерять дозы до 10у рад.
По данным [53—55], для измерения суммарных поглощенных доз гамма-нейтронного излучения ядерного реактора с успехом могут применяться пленки диацетата целлюлозы. О величине дозы удобно судить по изменению оптической плотности пленок при 320 нм (см. также главы VIII и X). Согласно [55|, с помощью пленок диацетата целлюлозы, содержащих 4-1010 атомов бора ца 1 г
образца, можно измерять потоки тепловых нейтронов в интервале 1014 — Ю1в н/с.И2.
Л. М. Кабакчи и сотр. [561 нашли, что с помощью пленок из поливинилового спирта, окрашенных метиленовым голубым, можно измерять суммарные поглощенные дозы гамма-нейтронного излучения ядерного реактора в диапазоне от Ю4 до 106 рад. Для этого вида излучения выход обесцвечивания составляет 1,7 молек./ /100 эв [56, 991. Если в такие пленки ввести борную кислоту, то они становятся пригодными для обнаружения потоков тепловых нейтронов от 1012 до 1014 п/см2. По данным цитируемых авторов, поливиниловый спирт хорошо «совмещается» с борной кислотой вплоть до концентрации 3-Ю20 атомов бора на 1 г продукта без изменения прозрачности. Однако борная кислота влияет на химические процессы в пленках при облучении. Например, для пленок, содержащих 2,5-1020 атомов бора на 1 а поливинилового спирта, выход обесцвечивания красителя под действием у-лучей 60Со равен 1,42 молек./ЮО эв (для пленок, не содержащих Н3ВО3, выход составляет 1,8 молек./ЮО эв). Выход обесцвечивания для продуктов реакции 10В (и, a)7Li равен 1,06 молек./ЮО эв [56, 991-Методики приготовления пленок и измерения степени обесцвечивания красителя были приведены в главе VIII.
Как следует из работы [54], по образованию лгранс-вииилёно-вых связей при облучении полиэтилена можно определять суммарные дозы реакторного излучения в диапазоне Ю7 — Ю8 рад. При этом измеряется увеличение оптической плотности полиэтилена в инфракрасной области (при длине волны 966 еж-1). Для дозиметрии реакторного излучения можно использовать и явление сшивания полиэтилена [55]. В этом случае определяется величина гель-фракции облученного полиэтилена. По данным [55], для того чтобы найти дозу, обусловленную преимущественно гамма-составляющей реакторного излучения, необходимо применять по-литрихлорфторэтилен. Этот полимер не содержит водорода, и поэтому быстрые нейтроны оказывают на него малое влияние. Дозу находят здесь путем измерения оптической плотности облученного полимера при 230 нм. Однако эта система имеет существенный недостаток, вызванный сравнительно высоким поперечным сечением захвата тепловых нейтронов хлором.
Термолюминесцентный дозиметр CaF2 '• Ми нечувствителен к нейтронам. Однако, если его покрыть eLi, то в этом случае можно зарегистрировать поток тепловых нейтронов 5-Ю3 п!см2 [92]. Если же CaF2 : Мп покрыть полиэтиленом, то он становится чувствительным к быстрым нейтронам [1031. Обычный LiF содержит — 7,4% ®Li. Этот изотоп, как уже отмечалось, имеет большое поперечное сечение захвата тепловых нейтронов. Кроме дозиметра TLD-100 (см. стр. 237), разработаны также другие дозиметры на основе LiF: TLD-700, содержащий 99,993% 6Li, и TLD-600, обогащенный 6Li [103, 104]. Использование TLD-700 в паре с TLD-ЮО или TLD-600 позволяет разграничивать дозы тепловых нейт-
ропов и у-лучей. С этой целью можно применять также пару Li2B4O, : Мп и TLD-700 [1021.
; Люминесцентные стекла, как правило, характеризуются низкой чувствительностью относительно быстрых нейтронов. Например, чувствительность фосфатных стекол, активированных серебром, по отношению к быстрым нейтронам составляет 1—3,5% чувствительности их относительно у-лучей 60Со [97, 981.
Согласно [105], отечественные термолюминесцентные детекторы ИКС па основе алюмофосфатных стекол типа 50-26 можно применять для регистрации дозы, обусловленной гамма-составляющей реакторного излучения. По данным [106], с этой целью можно применять алюмофосфатные стекла, активированные марганцем. Вклад быстрых нейтронов очень мал (несколько процентов и даже долей процента). Доза находится по появлению окраски, вызванной образованием Мп3+. Измерения оптической плотности проводятся при 560 нм.
7. Некоторые практические рекомендации
Одной из трудностей, возникающих при проведении дозиметрии реакторного излучения, является возможная активация сосудов и дозиметрических систем. Как правило, стекло сильно активируется под действием тепловых нейтронов. Кроме того, наиболее распространенные сорта стекла обычно содержат бор. Поэтому тепловые нейтроны могут частично поглощаться стенками стеклянного сосуда. Это вызвано протеканием реакции 10B(n, a)’Li. По оценке А. М. Кабакчи и сотр. [107], сосуд из термостойкого стекла со степкой толщиной 1 мм ослабляет поток тепловых нейтронов па 7,2%.
Сосуды для дозиметрических систем при работе со смешанным излучением реактора должны изготавливаться из кварца или пластмасс. Очевидно, пластмассовые сосуды можно применять в тех случаях, когда пластмасса не оказывает влияния на показания дозиметра. Возможно также использование сосудов из мягких сортов стекла, не содержащих бора.
При определении потоков тепловых нейтронов необходимо учитывать образование у-излучения, сопровождающего распад возбужденных ядер 7Li или Т. Например, в реакции 10В (n, a)7Li возникают у-лучи с энергией 0,48 Мэв. Естественно, это излучение может вызывать химические превращения в дозиметрической системе, что исказит результаты измерений. Очевидно, чем меньше диаметр дозиметрического сосуда, тем меньше вклад этого вида излучения в измеряемую дозу. По данным [671, в случае водных растворов при диаметре сосуда, равном 1, 2, 3, 4, 10 и 20 см, этот вклад составляет соответственно 1, 2, 3, 4, 8 и 13%. Следовательно, при измерении потоков тепловых нейтронов на основе реакции 10В (п, a)’Li диаметр дозиметрического сосуда не должен превышать 3—4 см.
Необходимо отметить еще одну особенность дозиметрии реакторного излучения. Как известно, ЛПЭ этого вида излучения зависит от энергетического спектра быстрых нейтронов и соотношения потоков нейтронов и у-лучей. В случае одного и того же реактора па эти параметры сильное влияние оказывает расстояние сосуда с дозиметрической системой от активной зоны. Поэтому для успешного проведения радиационно-химического эксперимента следует не только точпо соблюдать идентичность положения дозиметра и облучаемого образца относительно активной зоны реактора, но и знать для этого положения величину выхода превращения в дозиметрической системе (естественно, если выход зависит от ЛПЭ).
ЛИТЕРАТУРА
1. 1. Draganic. Determination of Absorbed Dose in Reactors. Vienna, IAEA, 1971, p. 197.
2. J. Wright. Disc. Faraday Soc., 12, 60 (1952).
3. J. Sutton, 1. Draganic, H. Hering. Proc. Intern. Conference on Peaceful Uses of Atomic Energy, vol. 14. Geneva, 1956, p. 160, 301.
4. J. Pucheault. J. Cbini. phys., 53, 705 (1956).
5. A. A. Ghosh-Mazumdar, H. N. Singh. J. Scient. Ind. Res., 20B, 509 (1966).
6. M. В. Владимирова, А. А. Баталов, И. А. Куликов, Л. Г. Шулятикова. Атомная энергия, 20, 509 (1966).
7. М. В. Владимирова. Атомная энергия, 17, 222 (1964).
8. М. В. Владимирова, А. А. Баталов, И. А. Куликов, Л. Г. Шулятикова. Дозиметрия больших доз. Ташкент, «Фан», 1966, стр. 163.
9. Ю. Е. Тиликс, Д. К. Тоне, В. Я. Калъкис. Радиационная физика. II. Рига, Изд-во АН ЛатвССР, 1964, стр. 39.
10. Ю. С. Рябухин, В. В. Ткаченко, Г. С. Бологова, Т. В. Вахлакова, Г. М. Обатуров, А. Г. Васильев. Мед. радиол., 14, № 8, 66 (1969).
11. Ю. С. Рябухин. Дозиметрия и радиационные процессы в дозиметрических системах. Ташкент, «Фан», 1972, стр. 185.
12. G. Ahnstrom, G. Guerrieri, М.Giacometti, L. DiPallo. Rep. RT/B10, No. 64, 1964, p. 26; цит. no [1J.
13. J. Jaspert. Rep. CEA-CEN, Int./Pi—171127 (1963); цит. no [1J.
14. E. J. Hart, P. Walsch. Proc. II United Nations Intern. Conference on Peaceful Uses of Atomic Energy, vol. 29. Geneva, 1958, p. 38.
15. С. В. Стародубцев, Ш. А. Абляев, В. В. Генералова. Атомная энергия, 8, 264 (1960).
16. С. В. Стародубцев, В. В. Генералова, Г. В. Поляк. Радиационная физика. II. Рига, Изд-во АН ЛатвССР, 1964, стр. 27.
17. В. В. Генералова. Дозиметрия больших доз. Ташкент, «Фан», 1966, стр. 29.
18. И. X. Абду Кадырова, В. В. Генералова, Н. А. Ремискевич. Там же, стр. 168.
19. И. X. Абдукадырова, В. В. Генералова. Дозиметрия интенсивных потоков ионизирующих излучений. Ташкент, «Фан», 1969, стр. 157.
20. С. В. Стародубцев, В. В. Генералова. Изв. АН УзбССР, серия физ-мат. наук, 1963, стр. 46.
21. Л. Г. Гуревич, И. X. Абдукадырова. Дозиметрия интенсивных потоков ионизирующих излучений. Ташкент, «Фан», 1969, стр. 61.
22. С. В. Стародубцев, И. X. Абдукадырова, В. В. Генералова. Изв. АН УзбССР, серия физ.-мат. наук, 1966, 62.
23. Б. Г. Дзантиев, Е. II. Ковалева. Вести. АН БССР, серия физ.-техн, наук, № 1, 47 (1967).
24. Г. С. Бологова, Б. А. Брискман, В. В. Ткаченко, В. П. Тусова. Дозиметрия и радиационные процессы в дозиметрических системах. Ташкент, «Фан», 1972, стр. 162.
25. Е. П. Ковалева, Е. П. Петряев, Е. П. Калязин. Там же, стр. 167.
26. I. Draganic. J. Chim. phys., 52, 595 (1955).
27. I. Draganic. J. Chim. phys., 56, 9 (1959).
28. /. Draganic, В. Radak, V. Markovic. Intern. J. Appl. Radiation Isotopes, 16, 145 (1965).
29. O. Gal, S. Pribicevic, S. Constantinovic, I. Draganic. Bull. Inst. Nucl. Sci. Boris Kidrich, 13, 54 (1962).
30. V. Marcovic, I. Draganic. Radiation Res., 36, 588 (1968).
31. J.Fenger. Riso Rep., No. 67 (1963); цит. по [1].
32. K. Gopakumar. Int. Rep. Tromhay, India (1968); цит. по [1].
33. В. В. Ткаченко. Радиационная дозиметрия и спектрометрия ионизирующих излучений. Ташкент, «Фан», 1970, стр. 169.
34. Е. П. Ковалева, А. М. Афанасьев, Л. Т. Бугаенко, В. М. Бяков, Е. П. Калязин, Е. П. Петряев. Вести. АН БССР, серия физ.-энерг. паук, № 2, 29 (1970).
35. А. М. Афанасьев, Л. Т. Бугаенко, В. М. Бяков, Е. П. Калязин, Е. П. Ковалева, Ф. Т. Ничипоров, Е. П. Петряев. Вести. АН БССР, серия физ.-энерг. паук, № 4, 23 (1970).
36. Е. П. Петряев, Е. П. Калязин, Е. П. Ковалева. Дозиметрия и радиационные процессы в дозиметрических системах. Ташкент, «Фан», 1972, стр. 170.
37. Е. П. Ковалева. Автореферат канд. дисс. Минск, Ин-т ядерной энергетики АН БССР, 1972.
38. A. W. Boyd, Н. W. J. Connor. Can. J. Chem., 42, 1418 (1964).
39. I. Kules, М. Gees. Proc. II Tihany Symposium on Radiation Chemistry. Budapest, 1967, p. 319.
40. I. Kiss, K. Pinter, M. Roder. Ibid., p. 391.
41. R. A. Holroyd, K.G. Golliher, A. W. Thiele. Trans. Am. Nucl. Soc., 10, No. 1, 54 (1967).
42. E. Proksch, H. Bildstein. Intern. J. Appl. Radiation Isotopes, 16, 56 (1956).
43. In-Pile Dosimetry. Vienna, IAEA, 1965.
44. Z. Spumy, J. Sulgova, J. Hruska. Jaderna Energ., 9, 329 (1963).
45. <У. C. Sigoloff. Nucleonics, 14, No. 10, 54 (1956).
46. E. Proksch. Atompraxis, 13, 190 (1967).
47. E. Proksch. Solid State and Chemical Radiation Dosimetry in Medicine and Biology. Vienna, IAEA, 1967, p. 307.
48. E. Proksch. Intern. J. Appl. Radiation Isotopes, 22, 441 (1971).
49. P. Gehringer, P. Krenmayr, E. Proksch. Proc. Ill Tihany Symposium on Radiation Chemistry, vol. 1. Budapest, 1973, p. 371.
50. C. D. Bopp, W. W. Parkinson, R. L. Towns, W. K. Kirkland. Rep. ORNL — 3213, 1961, p. 95; цит. по [1].
51. O. Gal, I. Draganic. Intern. J. Appl. Radiation Isotopes, 22, 753 (1971).
52. E. Jeltsch. Atomkernenergie, 14, 369 (1969).
53. Я. И. Лаврентович, А. Б. Зверев, А. А. Беликовский, A. M. Кабакчи. Химия высоких энергий, 3, 147 (1969).
54. Я. И. Лаврентович, Г. С. Якименко, А. Б. Зверев, А. М. Кабакчи. Атомная энергия, 27, 296 (1969).
55. Я. И. Лаврентович, Л. Е. Коваленко, А. Г. Старенький, А. А. Беликовский, А. М. Кабакчи. Дозиметрия и радиационные процессы в дозиметрических системах. Ташкент, «Фан», 1972, стр. 178.
56. Я. И. Лаврентович, А. И. Левон, Г. И. Мельникова, А. М. Кабакчи. Атомная энергия, 19, 273 (1965).
57. G. Ahnstrom, L. Ehrenberg. Selected Topics in Radiation Dosimetry. Vienna, IAEA, 1961, p. 603.
58. P. Harteck, S. Dondes. Nucleonics, 14, No. 3, 66 (1956).
59. R. E. Simpson. Health Phys., 8, 143 (1962).
60. D. A. Flory. Nucleonics, 21, No. 12, 50 (1963).
61. M. T. Дмитриев. Атомная энергия, 15, 52 (1963).
62. E. В. Сазонова. Радиационная дозиметрия и спектрометрия ионизирующих излучений. Ташкент, «Фан», 1970, стр. 183.
63. Е. И. Петряев, Е.Л. Ковалева. Вестн. АН БССР, серия физ.-энерг. наук, № 1, 49 (1970).
64. R. В. Thomas, J. К. Linacre. In-Pile Irradiation Equipment and Techniques (Proc. Symp. Harwell, 1966). AERE, 1966; цит. по [1].
65. 1. Draganic. Nucleonics, 21, No. 2, 33 (1963).
66. K. Sehested, E. Bjergbekke, N. W. Holm. Proc. II Tihany Symposium on Radiation Chemistry. Budapest, 1967, p. 149.
67. Г. С. Бологова, С. П. Добровольский, И. В. Захарова, В. П. Трусова. Радиационная дозиметрия и спектрометрия ионизирующих излучений. Ташкент, «Фан», 1970, стр. 165.
68. A. W. Boyd. Nucleonics, 22, No 7, 6 (1964).
69. A. W. Boyd. Determination of Absorbed Dose in Reactors. Vienna, IAEA, 1971, p. 85.
70. A. W. Boyd, H. W. Connor, J. J. Pieroni. CRNL — Rep. AECL — 2203 (1965); цит. по [1].
71. A. W. Boyd. In-Pile Dosimetry. Vienna, IAEA, 1966; цит. по [1].
72. M. Corbal, A. Chapiro, C. Cousin. Compt. rend., 235, 799 (1952).
73. P. Bonet-Maury. Brit. J. Radiology, 24, 284 (1951).
74. J. Pucheault. Comp, rend., 240, 772 (1952).
75. E. J. Hart, S. Gordon. Nucleonics, 12, No. 4, 40 (1954).
76. E. Saeland, L. Ehrenberg. Acta Chem. Scand., 6, 1113 (1952).
77. L. Ehrenberg, E. Saeland. Nucl. Sci. Abstr., 9, N 3, 899 (1955).
78. R. McDonnell, E. J. Hart. J. Am. Chem., Soc., 76, 2121 (1954).
79. R. H. Schuler, JS.F. Barr. J. Am. Chem. Soc., 78, 5766 (1956).
80. I. Dargenic, J. Sutton. J. Chim. phys., 52, 327 (1955).
81. A. W. Boyd. Determination of Absorbed Dose in Reactors. Vienna, IAEA, 1971, p. 7.
82. E. J. Hart, P. D. Walsch. Radiation Res., 1, 342 (1954).
83. E. J. Hart, H. W. Koch, B. Petree, J. H. Schulman, S. 1. Taimuty, H. O. Wyckoff. Proc. II Intern. Conference on Peaseful Uses of Atomic Energy, vol. 10. Geneva, 1958, p. 523.
84. А. I. Taimuty, R. A. Glass, B. S. Deaver. Proc. II Intern. Conference on Peaceful Uses of Atomic Energy, vol. 21. Geneva, 1958, p. 204.
85. Д. Тэплин. Радиационная дозиметрия (под ред. Дж. Хайна и Г. Браунелла). М., ИЛ, 1958, стр. 298.
86. А1. С. Sigoloff. Selected Topics in Radiation Dosimetry. Vienna, IAEA, 1961, p. 337.
87. С. В. Стародубцев, M. H. Гурский, A. H. Цой. Дозиметрия больших доз. Ташкент, «Фан», 1966, стр. 156.
88. И. Г. Берзина. Там же, стр. 173.
89. М. Н. Гурский, А. Н. Цой. Радиационная дозиметрия и спектрометрия ионизирующих излучений. Ташкент, «Фан», 1970, стр. 174.
90. Б. А. Вайншток, В. В. Генералова, Н. Ф. Левченко. Там же, стр. 178.
91. И. А. Бочвар, Т. Б. Кериим-Маркус, А. А. Васильева, Т. И. Прошина, 3. М. Сырицкая, В. В. Якубик. Атомная энергия, 15, 48 (1963).
92. JV. Haring, М. Schon. Proc. IX Intern. Congress (Munich, 1979); цит. по A. Hayakawa. Determination of Absorbed Dose in Reactors. Vienna, IAEA, 1971, p. 223.
93. A. R. Reedy, K. Ayyangar, G. L. Brownell. Radiation Res., 40, 552 (1962).
94. G. W. Endres, L. F. Kocher. Rep. BN WL—1080 (1969); цит. no A. Hayakawa, Determination of Absorbed Dose in Reactors. Vienna, IAEA, 1971, p. 223.
6S. C. J. Karzmark, J. White, J.F. Fowler. Phys. Med. feiol., 9, 273 (1964).
96. J. Kastner, B. G. Oltman. Health Phys., 12, 1125 (1966).
97. S. Kondo. Health Phys., 4, 21 (1960).
98. С. H. Bernard, W. T. Thornton, J. A. Auxier. Health Phvs., 4 236 (1961).
99. Я. И. Лаврентович, A. M. Кабакчи Радиационная химия полимеров. М., «Наука», 1966, стр. 253.
100. G. Busuoli, A. Caiallini, A. Fasso, О. Rimondi. Phys. Med. Biol., 15, 673 (1970).
101. M. A. Gomaa, A. M. El-JNaggar. Dosimetry in Agriculture, Industry, Biology and Medicine. Vienna, IAEA, 1973, p. 165.
102. B. Majborn, L. Botter-Jensen, P. Christensen. Ibid.,p. 169.
103. E. Blum, K. D. Bewley, J. D. Heather. Ibid., p. 277.
104. M. Nicolae. Ibid., p. 555.
105. И. А. Бочвар, Д. В. Викторов, В. В. Ткаченко, В. М. Трофимов. Дозиметрия и радиационные процессы в дозиметрических системах. Ташкент, «Фан», 1972, стр. 182.
106. В. В. Ткаченко, Б. А. Брискман, Л. М. Коваленко, Я. И. Лаврентович, Н. Ф. Орлов. Атомная энергия, 35, 210 (1973).
107. А. М. Кабакчи, Я. И. Лаврентович, В. В. Пеньковский. Химическая дозиметрия ионизирующих излучений. Киев, Изд-во АН УССР, 1963.
ЗАКЛЮЧЕНИЕ
Изложенный в книге материал показывает, что в настоящее время радиационная химия располагает многими методами дозиметрии. Читатель мог убедиться, что с их помощью возможно в лабораторных условиях определять с большой точностью дозы различных видов излучения: рентгеновских и у-лучей, быстрых электронов, тяжелых заряженных частиц, нейтронов, осколков деления. Вместе с тем в этой области есть и нерешенные проблемы. Остановимся на некоторых из них.
Во введении отмечалось, что современный этап развития радиационной химии характеризуется разработкой и внедрением в промышленность нескольких процессов, в которых используется энергия ионизирующего излучения. Однако проблема технологической дозиметрии еще далека от своего решения.
Существующие технологические радиационные процессы можно разделить на две большие группы. В первую группу следует включить радиационно-химические процессы (радиационно-химический синтез, полимеризация, радиационная модификация материалов и т. д.), во вторую — радиационную стерилизацию лекарственных препаратов и медицинского оборудования, радиационное консервирование пищевых продуктов и т. п.
В радиационно-химической технологии нередко нет необходимости измерять поглощенные дозы, поскольку степень облучения определяется здесь путем контроля количества или качества целевого продукта [1—4]. Такие процессы обычно характеризуются параметрами источника ионизирующего излучения, геометрией аппаратуры, скоростью подачи сырья и т. п. И если целевой продукт получается в количестве, соответствующем заданным технологическим условиям, или же он в результате радиационной обработки приобретает необходимые качества, это означает, что процесс протекает нормально. В противном случае должны быть изменены, например, условия облучения. Н. Хольм [2] предлагает этот метод контроля радиационно-химического процесса называть «дозиметрией эффекта».
Дозиметрические измерения проводятся на стадиях лабораторной и полупромышленной отработки радиационно-химического процесса. Кроме того, эти измерения необходимы в случае радиационных процессов второй группы. Здесь объект, как правило, облучается до строго определенной дозы (например, при стерили-
зации лекарственных препаратов или медицинского оборудования эта доза равна 2,5 Мрад), и поэтому требуется знать, что каждый образец «получил» такую дозу. Дозиметры, фиксирующие конкретные дозы, называются дозиметрами-индикаторами (см. также стр. 223). К настоящему времени, как это уже было отмечено в главе VIII, описано всего несколько таких дозиметров. Они являются преимущественно полимерными пленками, содержащими красители или их лейкооспования. Очевидно, дальнейшие исследования с целью разработки дозиметров-индикаторов для различных диапазонов доз представляют существенный интерес.
Согласно [2], другое перспективное направление в технологической дозиметрии — это поиски систем, с помощью которых можно осуществлять автоматическую регистрацию дозы. В связи с этим важное значение имеют сконструированные В. В. Генераловой и сотр. [5—81 специальные петлевые устройства. В этих устройствах через поле радиации постоянно циркулирует дозиметрическая жидкость. В качестве такой жидкости применялся водный раствор глюкозы. В результате облучения в нем происходит увеличение вязкости и электропроводности, а также наблюдается газовыделение. Путем измерения этих изменений можно непрерывно контролировать поглощенные дозы в диапазоне 106 — 8-108 рад. Контроль осуществляется с помощью релейных устройств. Рассмотрим в качестве примера принцип действия релейного устройства, работающего на основе изменений электропроводности раствора [8].
Устройство состоит из электролитической ячейки с раствором глюкозы, усилителя низкой частоты, электронного реле и блоков питания. К электродам ячейки подается напряжение порядка 1,5—2 в. Через раствор протекает ток, который при облучении возрастает вследствие уменьшения сопротивления. Выходной сигнал из ячейки после усиления и выпрямления подается на вход электронного реле. Последнее собрано по мостовой схеме. Оно «срабатывает» (загорается красная сигпальная лампочка), когда в схему из ячейки поступает ток, равный некоторой заранее заданной величине. Изменение дозы (т. е. тока), при которой загорается лампочка, осуществляется варьированием напряжения, подаваемого в схему. Описанное релейное устройство удовлетворительно функционирует в диапазонах температур 35 —60° С и мощностей дозы 100—3000 рад/сек.
Недавно сконструированы и другие подобные устройства. В работе [91 описан метод непрерывной регистрации поглощенных доз у-излучепия в интервале 0,06—1 Мрад с помощью 18%-ного водного раствора глюкозы. Доза измеряется по увеличению оптической плотности облученного раствора при 265 нм. Раствор непрерывно протекает через радиационно-химический аппарат и затем через измерительную кварцевую кювету. Оптическая плотность раствора в этом методе регистрируется с помощью фотоумножителя ФЭУ-39. Е. П. Петряев и др. [10] предложили ис
пользовать 3 М водный раствор метанола для технологической дозиметрии смешанного реакторного излучения. Ими найдено, что выход этиленгликоля не зависит от скорости протока раствора в диапазоне от 0 до 620 мл/час. Способ пригоден для определения доз в интервале 0,5 — 100 Л/pad (при температурах от 20 до 300° С). В статье [11] представлена проточная схема для осуществления дозиметрии реакционного объема с использованием закиси азота в случае пучков быстрых электронов. В этой схеме доза измеряется по количеству выделившегося водорода, определяемому методом газовой хроматографии.
Рассмотренные устройства являются первыми в области автоматизации процессов измерения поглощенной дозы в радиационно-химической технологии. Желательны дальнейшие работы в этом направлении с целью создания более простых и надежных устройств.
В радиационных процессах облучению часто подвергаются объекты, состоящие из самых разнообразных материалов, имеющие различные размеры, конфигурацию и т. п. Это обусловливает существенные трудности при проведении дозиметрических измерений. Отметим некоторые из таких трудностей.
При действии у-излучения на сравнительно большие объекты доза вследствие рассеяния квантов значительно зависит от природы материала объекта (см. также стр. 120). Поэтому для определения дозы необходимо использовать дозиметрическую систему, которая по своим характеристикам близка к материалу облучаемого объекта. В дозиметрии электронных потоков это усложняющее обстоятельство не имеет места. В этом случае доза в дозиметре и объекте пропорциональна их тормозным способностям.
При осуществлении тех нологических процессов во многих случаях (особенно при работе с электронными пучками) необходимо знать пространственное распределение дозы в облучаемом объекте. В последнее время это выполняется преимущественно с помощью пленочных дозиметров. Особенности соответствующих измерений подробно изложены в недавнем обзоре У. Мак-Лафлина и др. 112]. Там же сделан анализ возможных ошибок при использовании этих дозиметров.
Свою специфику имеет дозиметрия в случае гетерогенных систем. Особенно это относится к измерениям дозы для полимеров, находящихся в контакте с металлами. Как следует из работы А. X. Брегера и сотр. [13], доза в поверхностных слоях полимера, граничащих с металлом, существенно отличается от дозы в толще полимера. Согласно [12—14], для определения доз в таких гетерогенных системах удобны пленочные дозиметры.
Другая нерешенная проблема — это проблема единства измерений поглощенной дозы при проведении радиационно-химических исследований 12, 15]. По ГОСТ 16263—70 [16] единство измерений — состояние измерений, при котором результаты выражены в узаконенных единицах и погрешности измерений известны с заданной
вероятностью. В настоящее время в радиационной химии широкб применяется дозиметр Фрикке. Этот дозиметр изучен весьма обстоятельно учеными разных стран. В США он принят в качестве стандартного для определения поглощенной дозы в случае рентгеновского и у-излучений [17]. Рекомендуется использовать дозиметр Фрикке как стандартный и в нашей стране [15].
Отметим, что в том случае, если молярный коэффициент экстинкции ионов Fe3+ в 0,4 М растворе H2SO4 находится с помощью спектрофотометра, имеющегося в лаборатории (см. стр. 147), погрешность измерения дозы рассматриваемым методом не превышает 3—5%. Предлагается [15] в качестве стандартного употреблять также глюкозный дозиметр, действие которого, как уже отмечалось (см. стр. 174), основано на изменении угла вращения плоскости поляризации 20%-ного раствора глюкозы при облучении. Рабочий диапазон этого дозиметра — от 5 до 100 Мрад. Как показала недавняя метрологическая проверка данного дозиметра [1-8], погрешность измерения дозы у-излучения составляет здесь 7% с доверительной вероятностью 0,95. Однако широкому распространению глюкозного дозиметра препятствует отсутствие линейной зависимости количества превращения от дозы и недостаточная изученность механизма радиолитических превращений в концентрированных водных растворах глюкозы.
Единству измерений должен способствовать выпуск отградуированных дозиметров в централизованном порядке. Согласно [15], в нашей стране таким путем изготавливаются стеклянные дозиметры СГД-8 для измерения экспозиционной дозы в интервале 103—106 р. Кроме того, выпущена опытная партия цветовых индикаторов дозы, представляющих собой полимерные пленки с добавками люминесцирующих красителей. Они предназначены для визуальной оценки дозы (с погрешностью 25—30%) в диапазоне 0,2—5 Мрад. Несомненно, ассортимент отградуированных дозиметров, выпускаемых в централизованном порядке, должен быть расширен. Необходим также выпуск эталонной аппаратуры (в первую очередь калориметров), пригодной для калибровки различных дозиметров.
ЛИТЕРАТУРА
1. A. W. Holm. Radiation Dosimetry (Ed. F. H. Attix and E. Tochilin), 2nd Edition, vol. 3. N. Y., Academic Press, 1969, chap. 33.
2. A. TV. Holm. Large Radiation Sources for Industrial Processes. Vienna, IAEA, 1969, p. 555.
3. К. H. Chadwick. Dosimetry in Agriculture, Industry, Riology and Medicine. Vienna, IAEA, 1973, p. 3.
4. В. Б. Осипов. Дозиметрия и радиационные процессы в дозиметрических системах. Ташкент, «Фан», 1972, стр. 12.
5. В. В. Генералова. Дозиметрия больших доз. Ташкент, «Фан», 1966, стр. 29.
6. И. X. Абдукадырова, В. В. Генералова, Н. А. Ремискевич. Там же, стр. 168.
7. В. В. Генералова, Р. Гуламова. Дозиметрия интенсивных потоков ионизирующих излучений. Ташкент, «Фан», 1969, стр. 142.
8. В. В. Генералова, Р. Р. Гуламова. Радиационная дозиметрия и спектрометрия ионизирующих излучений. Ташкент, «Фан», 1970, стр. 133.
9. В. А. Гольдин, Е. Д. Проценко, Э. Л. Мендельсон, В. П. Базов, Ю. М. Курзин, Т. М. Гаврилова. Тезисы докладов Второй Всесоюзной конференции по прикладной радиационной химии (Обнинск, 1973). Черкассы, 1973, стр. 118.
10. Е. П. Петряев, Е. П. Калязин, Е. П. Ковалева. Дозиметрия и радиационные процессы в дозиметрических системах. Ташкент, «Фан», 1972, стр. 170.
11. О. Н. Крутецкая, Н. Е. Орлова, Ю. А. Панин, Л. Б. Упадышев. Там же, стр. 68.
12. W. L. McLaughlin, Р. Е. Hfortenberg, В. В. Radak. Dosimetry in Agriculture, Industry, Biology and Medicine. Vienna, IAEA, 1973, p. 577.
13. Ф. А. Махлис, A. X. Врегер, E. H. Смагин. Дозиметрия больших доз. Ташкент, «Фан», стр. 62.
14. Е. Eisen, М. Rogenstein, J. Silverman. Dosimetry in Agriculture, Industry, Biology and Medicine. Vienna, IAEA, 1973, p. 615.
15. Ю. И. Брегадзе, В. В. Генералова. Дозиметрия и радиационные процессы в дозиметрических системах. Ташкент, «Фан», 1972, стр. 3
16. ГОСТ 16263—70 «Государственная система обеспечения единства измерений. Метрология. Термины и определения».
17. ASTM Bull. No. 239 (1959).
18. В. В. Братановский, В. В. Генералова, М. Н. Гурский, А. В. Тултаев. Дозиметрия и радиационные процессы в дозиметрических системах. Ташкент, «Фан», 1972, стр. 100.
ОГЛАВЛЕНИЕ
Предисловие................................................. 3
Введение.................................................... 5
Литература........................................... 9
Глава I.Ионизирующие излучения и особенности их взаимодействия с веществом. Основные понятия и единицы ... 11
1. Основные понятия и единицы......................... 12
2. Общие закономерности радиоактивного распада .... 16
3. Фотонное излучение................................. 18
4. Корпускулярное излучение........................... 32
5. Линейная передача энергии. «Шпоры»................. 50
6. Радиационно-химический выход....................... 51
Литература.......................................... 52
Глава II. Источники ионизирующих излучений .................. 54
1. Изотопные гамма-установки.......................... 54
2. Источники ₽- и а-излучений......................... 65
3. Ускорители заряженных частиц....................... 66
4. Ядерный реактор как источник ионизирующего излучения 80
Литература.......................................... 83
Глава III. Краткая характеристика применения физических методов дозиметрии в радиационной химии.............................. 87
1. Ионизационный метод................................ 87
2. Калориметрический метод............................ 95
3. Метод измерения тока или заряда, индуцированного пучком заряженных частиц в облучаемой системе............ 102
4. Определение поглощенных доз в случае внутренних источников излучения.................................. 106
5. Другие физические методы дозиметрии............... 108
Литература......................................... 108
Глава IV. Основные положения химической дозиметрии ионизирующих излучений................................................. Ш
1. Требования, предъявляемые к химическим дозиметрам 111
2. Классификация химических дозиметров............... 112
3. Калибровка дозиметрических систем................. 115
4. Некоторые особенности дозиметрических измерений с помощью химических методов.............................. 116
5. Расчет величины дозы, поглощенной исследуемым объектом, исходя из показаний дозиметра.................... 120
Литература....................................... 129
Глава V. Водные растворы как дозиметрические системы........ 132
1. Обедая характеристика............................... 132
2. Дозиметр Фрикке..................................... 135
3. Модифицированные дозиметры Фрикке................... 154
4. Цериевая дозиметрическая система.................... 157
5. Ферросульфатная дозиметрическая система с добавками ионов Си2+............................................ 166
6. Водный раствор щавелевой кислоты................... 169
7. Водный раствор бихромата калия...................... 173
8. Водные растворы углеводов........................... 174
9. Водные растворы бензола или бензойной кислоты .... 176
10. Водные растворы хлорированных углеводородов и смеси:-хлорированный углеводород — вода....................... 178
11. Водные растворы красителей и их лейкоосновапий ... 180
12. Водные растворы нитратов........................... 183
13. Другие дозиметрические системы па основе водных растворов ............................................... 184
Литература.......................................... 187
Глава VI. Дозиметры на осневе органических жидкостей......... 198
1. Циклогексан........................................ 198
2. Бензол............................................. 202
3. Раствор лейкооснования кристаллического фиолетового в метилэтилкетот................................... 202
4. Растворы тетрахлорэтилена, хлорбензола и дихлорбензола в органических растворителях.......................... 203
5. Растворы нитрилов трифенилметановых красителей в органических растворителях.......................... 205
6. Растворы полимеров в органических растворителях. Мономеры ............................................. 205
Литература.......................................... 207
Глава VII. Газообразные вещества как дозиметрические системы 210
1. Углеводороды....................................... 210
2. Закись азота .................................. 212
3. Другие окислы азота................................ 214
4. Кислород и азотно-кислородные смеси................ 215
5. Сероуглерод........................................ 216
Литература.......................................... 216
Глава VIII. Дозиметры на основе твердых тел......... 219
1. Полимеры........................................... 220
2. Стекла............................................. 231
3. Галогениды щелочных и щелочноземельных металлов 235
4. Сульфат кальция.................................... 239
5. Другие дозиметры па основе твердых тел.... 240
Литература.......................................... 243
Глава IX. Дозиметрия импульсного ионизирующего излучения . . . 254
1. Дозиметр Фрикке.................................... 255
2. Другие дозиметрические водные системы...........е 257
3. Использование процесса образования гидратированного электрона для дозиметрии............................... 259
4. Дозиметры на основе полимерных систем.............. 261
5. Органические жидкости как дозиметрические системы 263
6. Газообразные дозиметрические системы............... 264
7. Другие дозиметрические системы..................... 265
8. Методы измерения дозы вдоль пути светового пучка в импульсном радиолизе с оптической регистрацией короткоживущих частиц......................................... 266
9. Некоторые практические рекомендации............... 267
Литература ........................................ 270
Глава X. Дозиметрия пучков тяжелых заряженных частиц .... 273
1. Дозиметр Фрикке.................................... 273
2. Цериевая дозиметрическая система................... 275
3. Циклогексан........................................ 275
4. Полимеры........................................... 276
5. Другие дозиметрические системы..................... 278
6. Некоторые практические рекомендации................ 280
Литература.......................................... 281
Глава XI. Химические методы дозиметрии реакторного излучения 283
1. Водный раствор щавелевой кислоты................... 284
2. Водные растворы углеводов.......................... 288
3. Определение дозы реакторного излучения параллельным использованием двух водных дозиметрических систем 289
4. Закись азота ...................................... 290
5. Циклогексан ................................ . ... 291
6. Другие дозиметрические системы..................... 291
7. Некоторые практические рекомендации................ 298
Литература.......................................... 299
Заключение..................................................... 303
Литература........................................... 306
Алексей Коистангггинсеич Ликаее
Дозиметрия в радиационной химии
Утверждено к печати
ордена Трудового Красного Знамени
Институтом физической химии
Академии наук СССР
Редакторы И. Д. Казаринова^ Н. Г. Явкина
Художник Э. Л. Эрмаи
Художественный редактор Н. Н. Власик
Технический редактор П. С. Кашина
Сдано в набор 24/VH-1974 г. Подписано в печать 29/Х-1974 г. Формат 60X90V1,в. Бумага № 2. Усл. печ. л. 19,5. Уч.-изд. л. 21,3. Тираж 1900. Т-13446. Тип. зак. 930. Цена 1р. 63 к.
Издательство «Наука»
103717 ГСП, Москва, К-62, Подсосенский пер., 21
2-я типография издательства «Наука»
121099, Москва, Г-99, Шубинский пер., 10