




















































































































































































































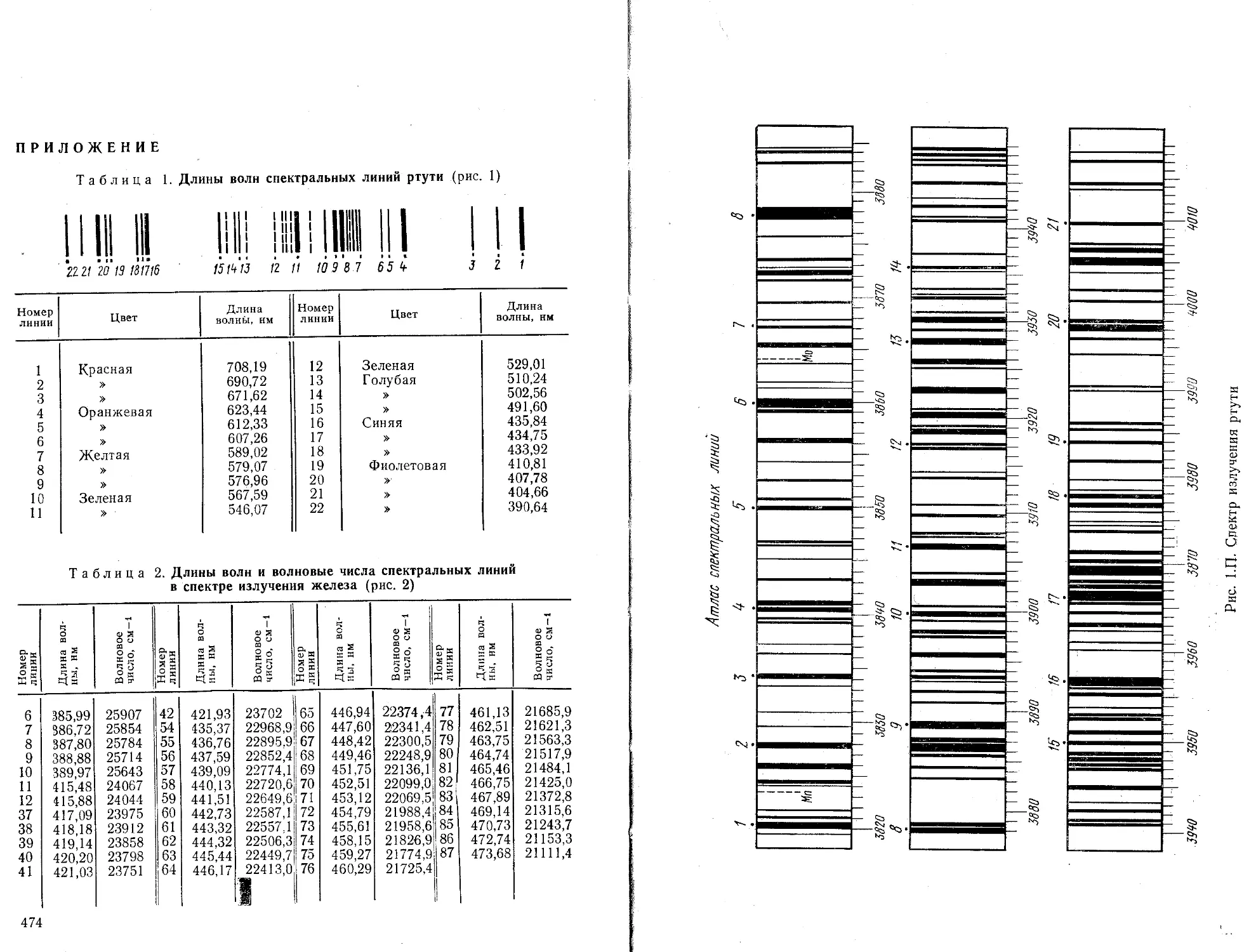









Текст
пределение параметров линейном зависимости
э?сп|риментальным данным (метод наименьших квадратов)
Ввод программы
№ № ном анд
Клавиши
А
Пояснения
№№ команд
Клавиши
00
10
20
ПРГ
П9
X.П8
П~Х4
X—П4
П-Х6
Х-П6
П-Х5
Fx"
Х--П5
П-Х6
4Ф
48
12
64
Ю
44
66
69
Ю
46
Ю
68
Перевод машины в режим „программирование11
X—П7
Запись значения у,- в ячейке 2
Обмен информацией регистра х и у
Запись значения х.- в ячейке 3
Вызов содержимого ячейки 4 Накопление и запись значения Ех/ У/
Вызов значения у, Накопление и запись значения Е у-
J I
Вызов значения X;
Накопление и запись значения Е х-’
Вызов значения Х;
30.
40
Х-П^у С/П
Fx2
n~xd
П-Х5
Х-П9
П-Х6
П-Х7
n-xd
•Ш
41
оЕ
10
50
67
13
66:
67
12
Й
яке
01
опление и запись чения Ех/
*
Пояснения
Вычисление общего числа точек
Вызов значения
(Sx,J2
Вызов значения
п
Вызов значения Lx Вычисление и запись значения
Вызов значения Еу,-
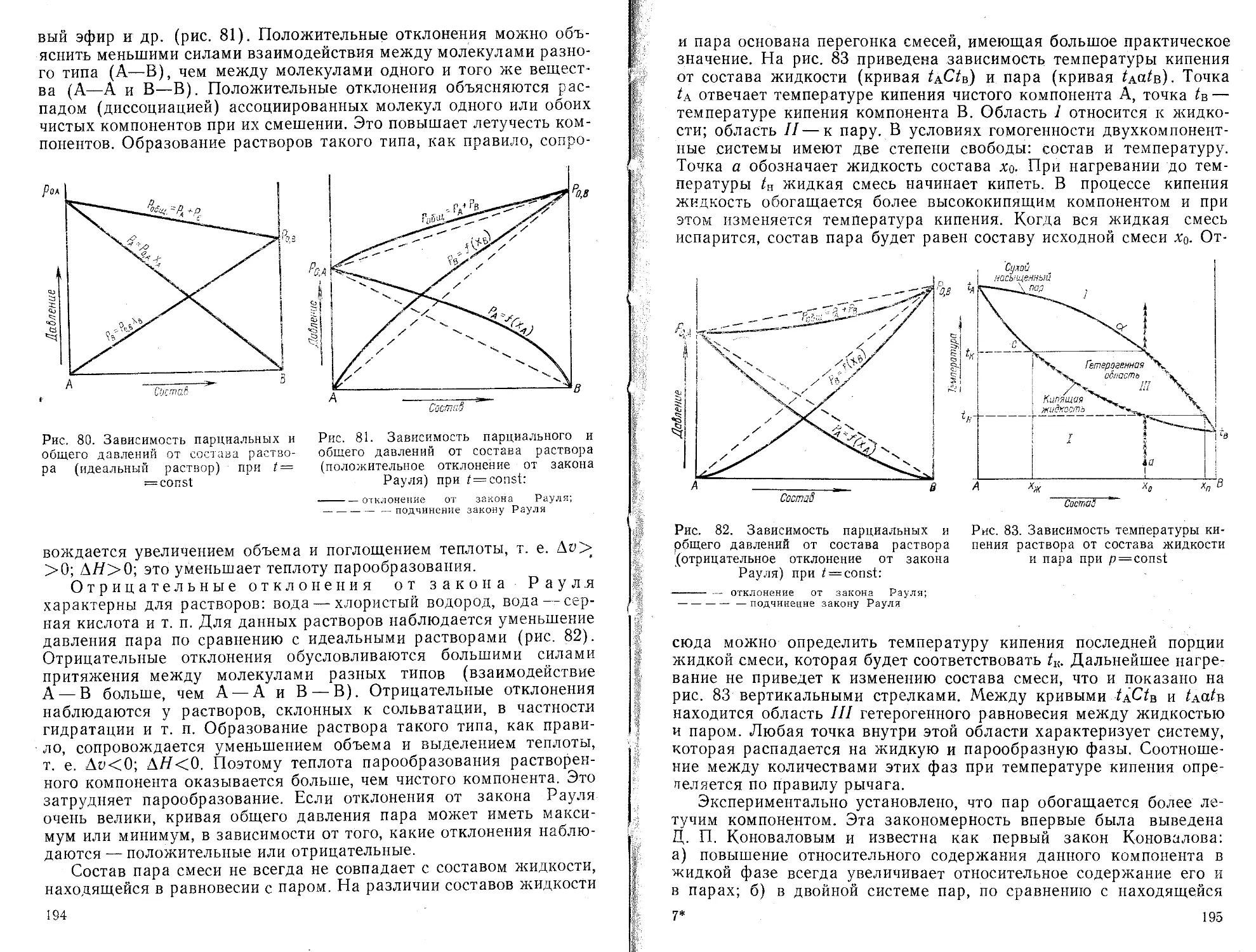
Вы^ов значения
Вызов значения
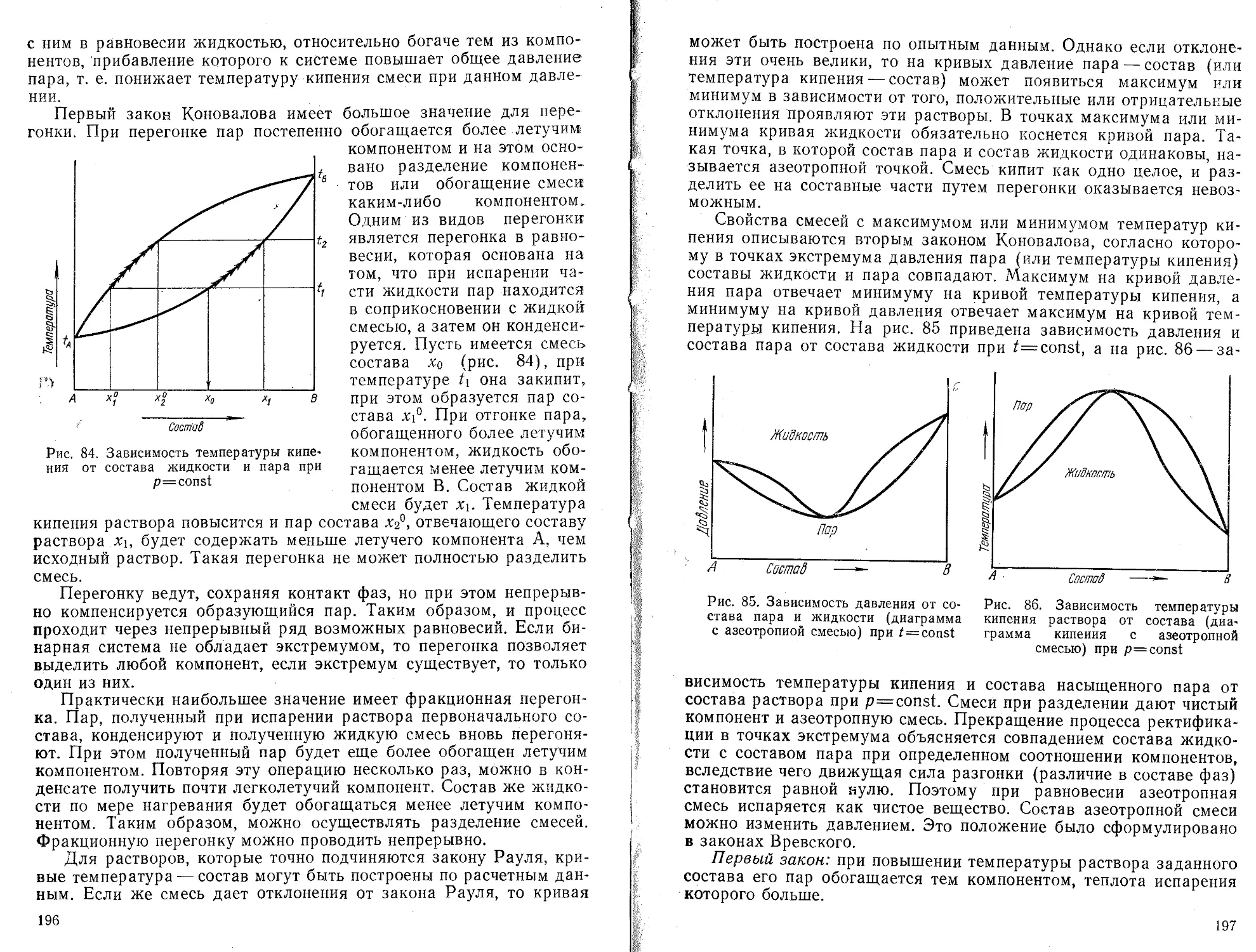
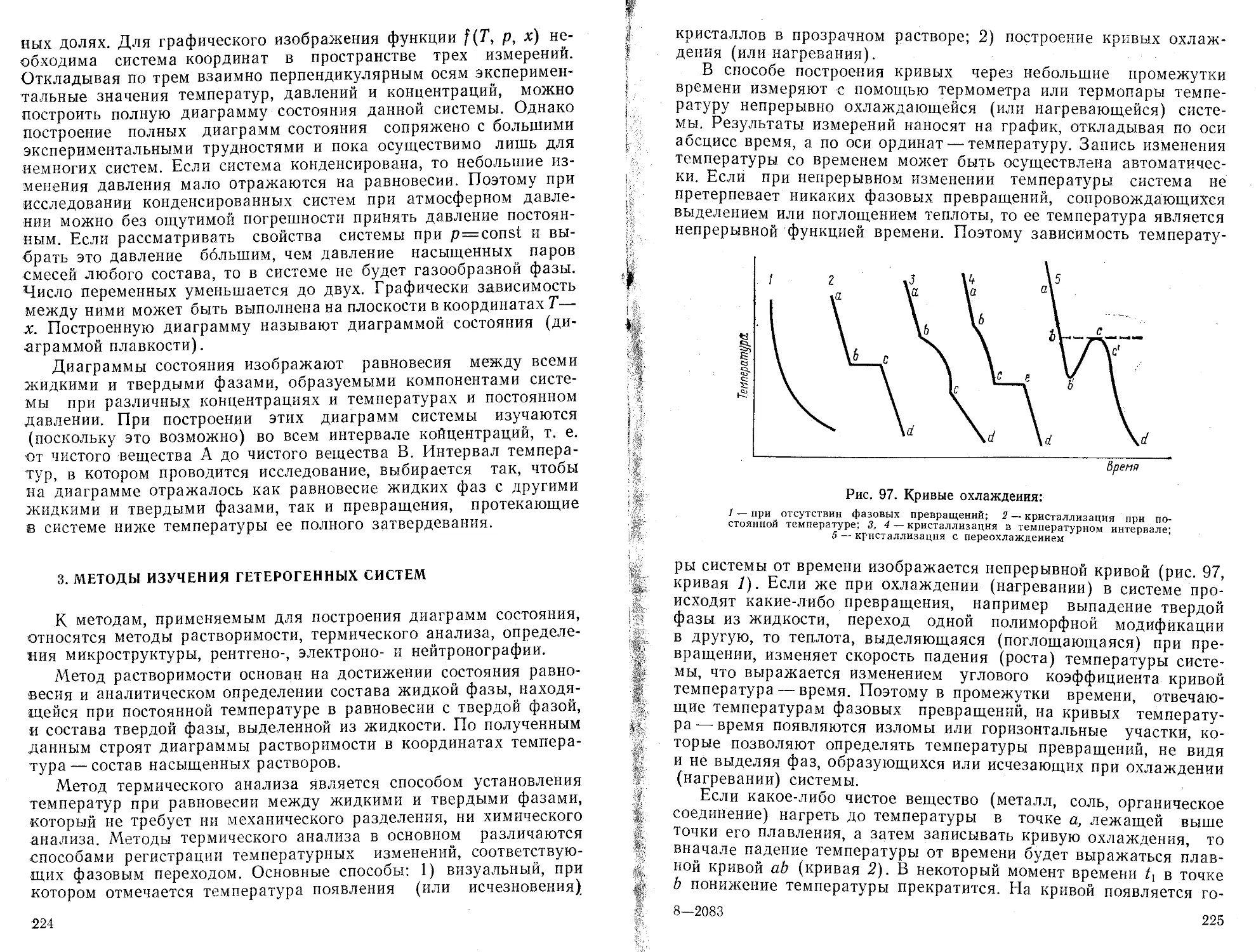
п
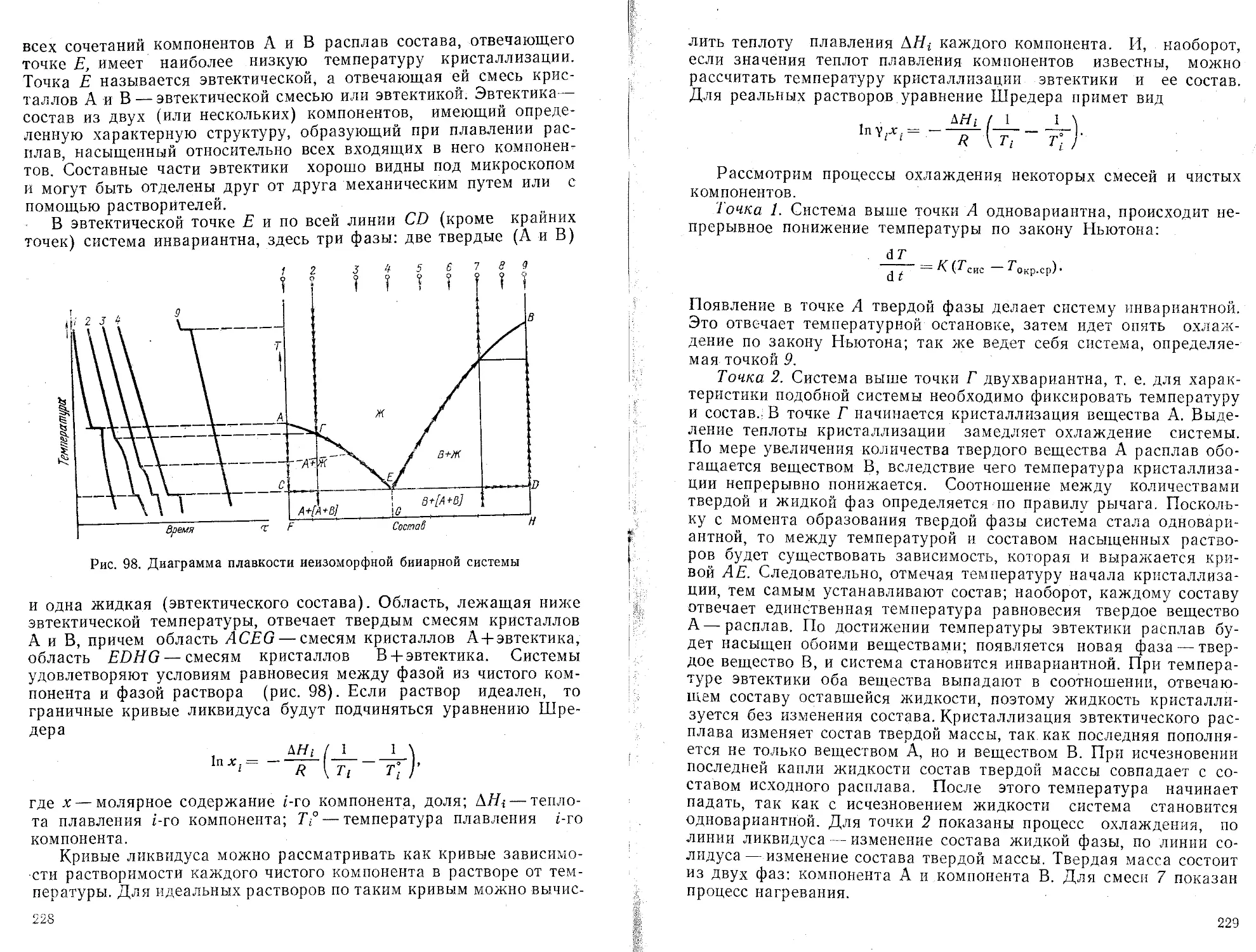
п
Г /
№ № команд
50
60
70
Клавиши Код
ШИ ММВЙЬМ
П-Х7
П-Xtf
Х--П2
П-Xd
«Ю
64
10
69
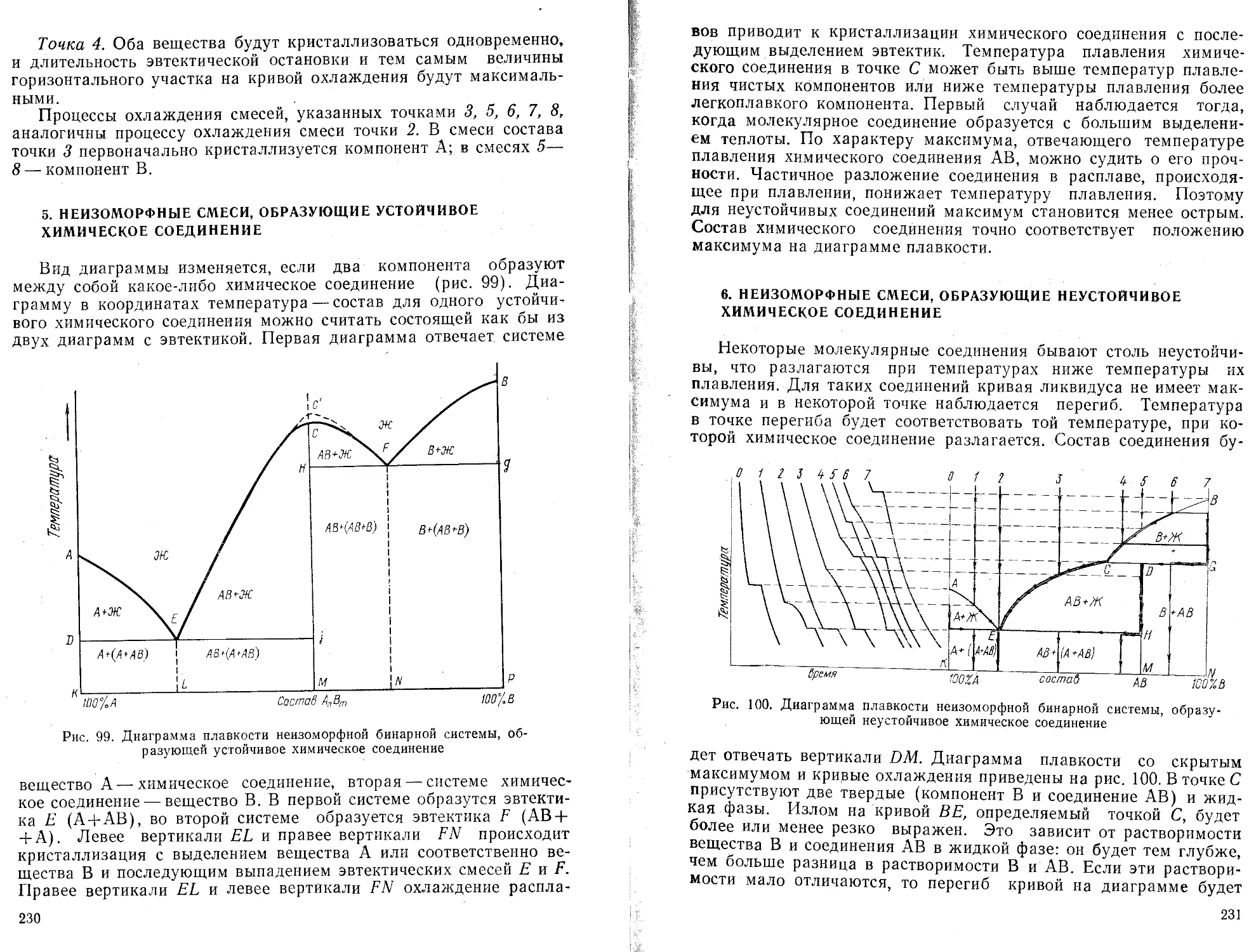
13
4Е
67
13
6 L
66
13
62
11
50
Пояснения
<
Вызов значения Ех/У/
Вызов значения Ех? —
Вычисление и запись значения b
Вызов значения Ех;-
Вызов значения П
Вычисление и запись
значения х =
Вызов значения b Вычисление „и запись значения Ьх
значения
значения
значения
п
Ьх
Вызов Е у, /п Вызов
Индикация значения а
величины: значение параметров а и b
, наилучшим образом описывающей полученные экспериментальные данные
Р ассч иты вае м ы е
в уравнении
прямой
Порядок вычислений
линии
Формулы для расчета
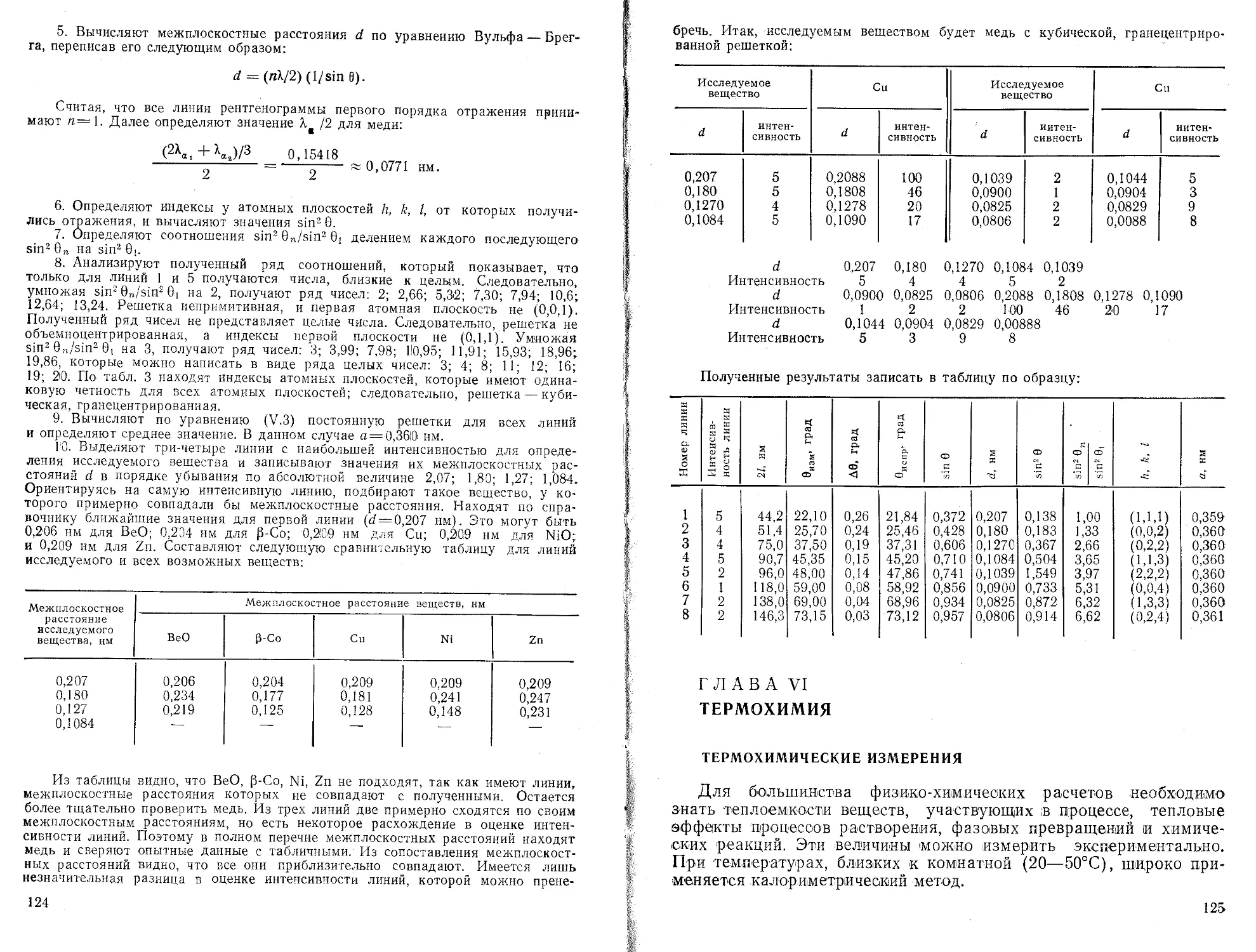
1. ПереЕзод машины в режим вычислений нажатием клавиши F АВТ В/О
2. Очистка памяти машины от результатов предыдущих вычислений нажатием клавиш Су Х~П a,b,c.d}
п
ь— п
Ь =
3. Введение в память машины попарно экспериментальных значений и расчет промежуточных результатов-сумм f'ExJ2 Еу-
п
fx/, Vj] “"Экспериментальные точки i л-общее число экспериментальных точек
4. Вычисление параметров а и b БП 30 С/П
5. Выписать значения b с индикатора, С/П—а
ББК 24.5
П69
УДК 541.1
Авторы:
Г. С. Каретников, | Н. А. Козырева, И. В. Кудряшов, Е. П. Старостенко, О. Б. Хачатурян
Рецензенты:
кафедра физической химии Казанского химико-технологического института им. С. М. Кирова (зав. кафедрой проф. Г. А. Добреньков) и проф. А. А. Равдель (Ленинградский технологический институт им. Ленсовета)
Практикум по физической химии: Учеб, пособие для сту-
П69
дентов химико-технол. спец, вузов//Каретников Г. С.|, Козырева Н. А., Кудряшов И. В. и др.; Под ред. И. В. Кудряшова.— 4-е изд., перераб. и доп. М.: Высш, шк., 1986.— 495 с., ил.
В пер.: 1 р. 40 к.
В практикуме описаны лабораторные работы, охватывающие весь курс физической химии. Каждой работе предпослано теоретическое введение. Особое внимание обращено на современные методы исследования; спектроскопию, рентгенографию, электронографию и др., а также рассмотрены классические методы исследо* вания; криоскопия, эбуллиоскопия, колориметрия, хроматография и т. п.
1805000000—216
001(01)—86
91—86
ББК 24.5
541
© Издательство «Высшая школа», 1974
© Издательство «Высшая школа», 1986, с изменениями
ПРЕДИСЛОВИЕ
Задачи, поставленные XXVII съездом КПСС перед наукой по интенсификации многих производственных процессов, могут быть решены благодаря знаниям законов физической химии.
Физическая химия органически сочетает в себе теорию и эксперимент. Путем экспериментальных исследований физическая химия обогащается новыми фактами, на базе которых делают обоб-* щения, разрабатывают теории. Выводы из теоретических положений, в свою очередь, контролируют опытом.
Практикум по физической химии по ряду причин не может выполняться синхронно с изложением курса лекций. Однако работа в физико-химической лаборатории только тогда имеет смысл и повышает квалификацию студента, когда она проводится с пониманием задач эксперимента. Поэтому описанию лабораторных работ предпосланы краткие теоретические введения, которые необходимы, чтобы с минимальной затратой времени понять смысл, теоретическое обоснование и следствия производимого эксперимента.
На лабораторных занятиях студент знакомится с устройством прибора или установки. При подготовке к выполнению лабораторной работы необходимо сначала по схеме, а затем непосредственно у прибора ознакомиться с его устройством и принципом работы. Во избежание порчи дорогостоящих приборов следует строго руководствоваться прилагаемыми инструкциями. При работе в физико-химической лаборатории следует строго соблюдать правила техники безопасности.
Совершенствование техники эксперимента ведет к ускорению получения экспериментальных данных и повышению степени точности и надежности их. За сравнительно короткое время стали получать такое количество экспериментальных данных, математическая обработка которых оказалась слишком трудоемкой. На помощь экспериментатору приходят ЭВМ. Они становятся неотъемлемой частью лабораторного оборудования.
Четвертое издание практикума отличается от предыдущих тем, что в большинстве лабораторных работ предложено математическую обработку полученных результатов проводить на ЭВМ. В при
3
ложении приведены программы некоторых расчетов на специализированном управляющем устройстве 15-ВСМ-5 (можно также воспользоваться калькуляторами и ЭВМ других марок). Введены новые лабораторные работы. Обращено более серьезное внимание на определение точности и надежности полученных экспериментальных данных.
Практикум написан следующими авторами: главы I—III |Г. С. Каретниковым |; главы IV—V Н. А. Козыревой; главы VI— XII и XVII—XIX, XXI И. В. Кудряшовым; главы XIII—XIV, XX О. Б. Хачатурян; главы XV—XVI Е. П. Старостенко. В создании практикума принимали участие все преподаватели кафедры физической химии МХТИ им. Д. И. Менделеева.
Авторы будут весьма признательны и благодарны читателям за жритические замечания и советы по содержанию практикума.
И. В. Кудряшов
Г Л А В A' I
МОЛЕКУЛЯРНАЯ СПЕКТРОСКОПИЯ
1. ВЗАИМОДЕЙСТВИЕ ЭЛЕКТРОМАГНИТНОГО ИЗЛУЧЕНИЯ
С МОЛЕКУЛАМИ ВЕЩЕСТВА
При взаимодействии электромагнитного излучения с веществом наблюдается уменьшение энергии излучения и увеличение энергии молекул вещества. Характер взаимодействия зависит от энергии действующего на вещество излучения. Молекулярная спектроскопия имеет дело с ограниченным диапазоном энергий излучения (табл. 1).
Поглощение электромагнитного излучения наблюдается строго избирательное, соответствующее строению молекул поглощающего вещества. Поглощаются веществом только те кванты излучения, энергия которых равна разности энергий энергетических уровней молекул, переходы между которыми разрешены правилами отбора. Поглощенная энергия «удерживается» молекулой вещества корот-
Т а б л и ц а 1. Энергии электромагнитного излучения в разных участках спектра
Область спектра Е, Дж/молекула К, см V, см-1
Микроволновая
Дальняя инфракрасная
Инфракрасная
Видимая
Ультрафиолетовая
Дальняя ультрафиолетовая (вакуумная ультрафиолетовая)
2-10~244-2-10~2i
2-10~214-8-10~21
8-10-214-2,5-10-19
2,5-10~194-5-10-19
5-10~194-1 • 10"18
1 • 10" 184-2-10“17
104-10“2
1 СМЦ-2,5 -10-3
2,5 • 10-3-=-8* 10-5
8-10-54-4-10~5
4 10~54-2-10-5
2- 10~5~ 10”6
0,14-100
100—400
4004-1,25-104
1,25- 1044-2,5-104
2,5-1044-5-104
5* 1044-106
кое время порядка 10“34-10~8 с. Таким образом, молекула «живет» в возбужденном состоянии столь короткое время, что вероятность поглощения второго кванта излучения или большего числа квантов практически равна нулю. Отсюда спектральными методами регистрируется поглощение только молекулами, находящимися в наиболее устойчивом энергетическом состоянии.
5
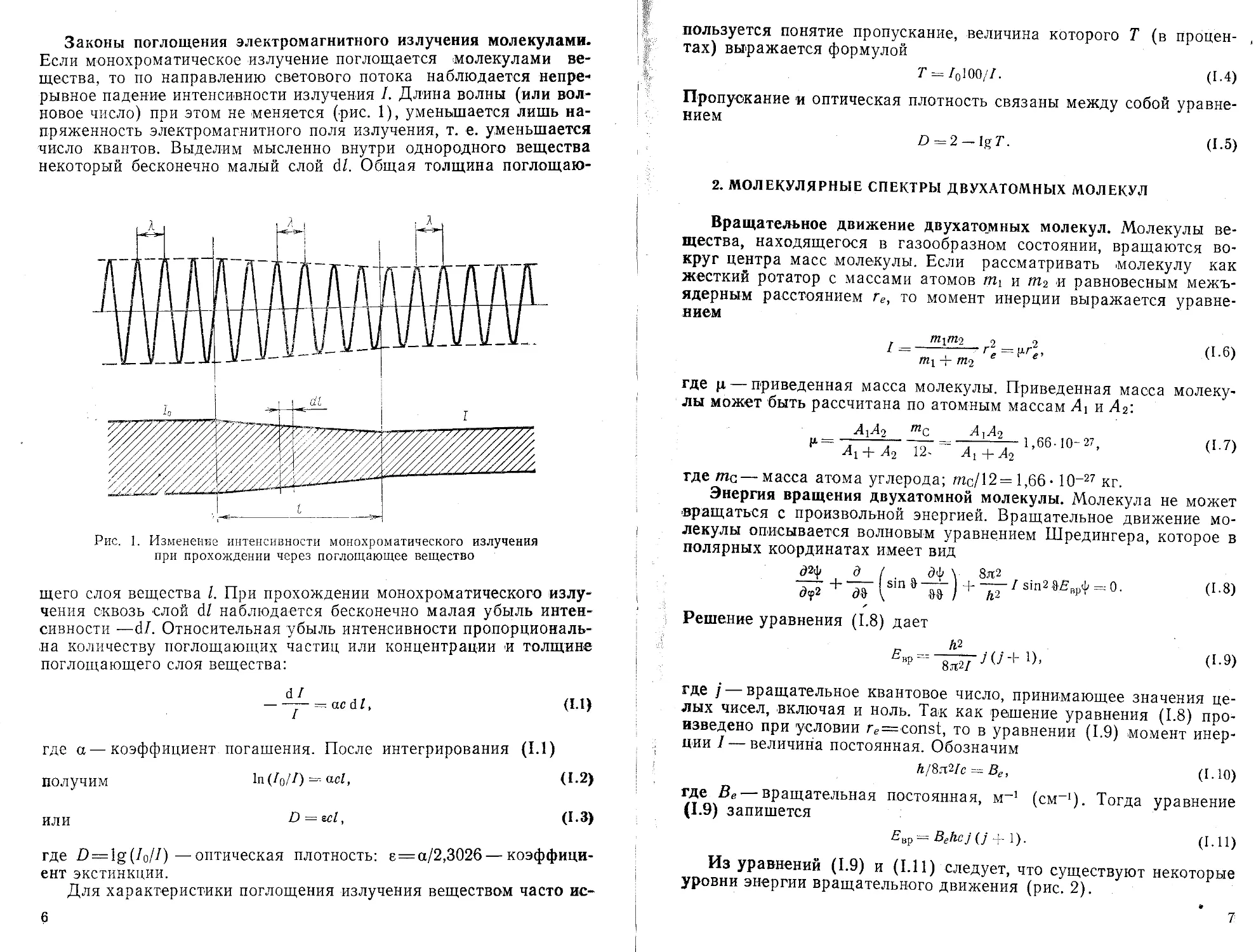
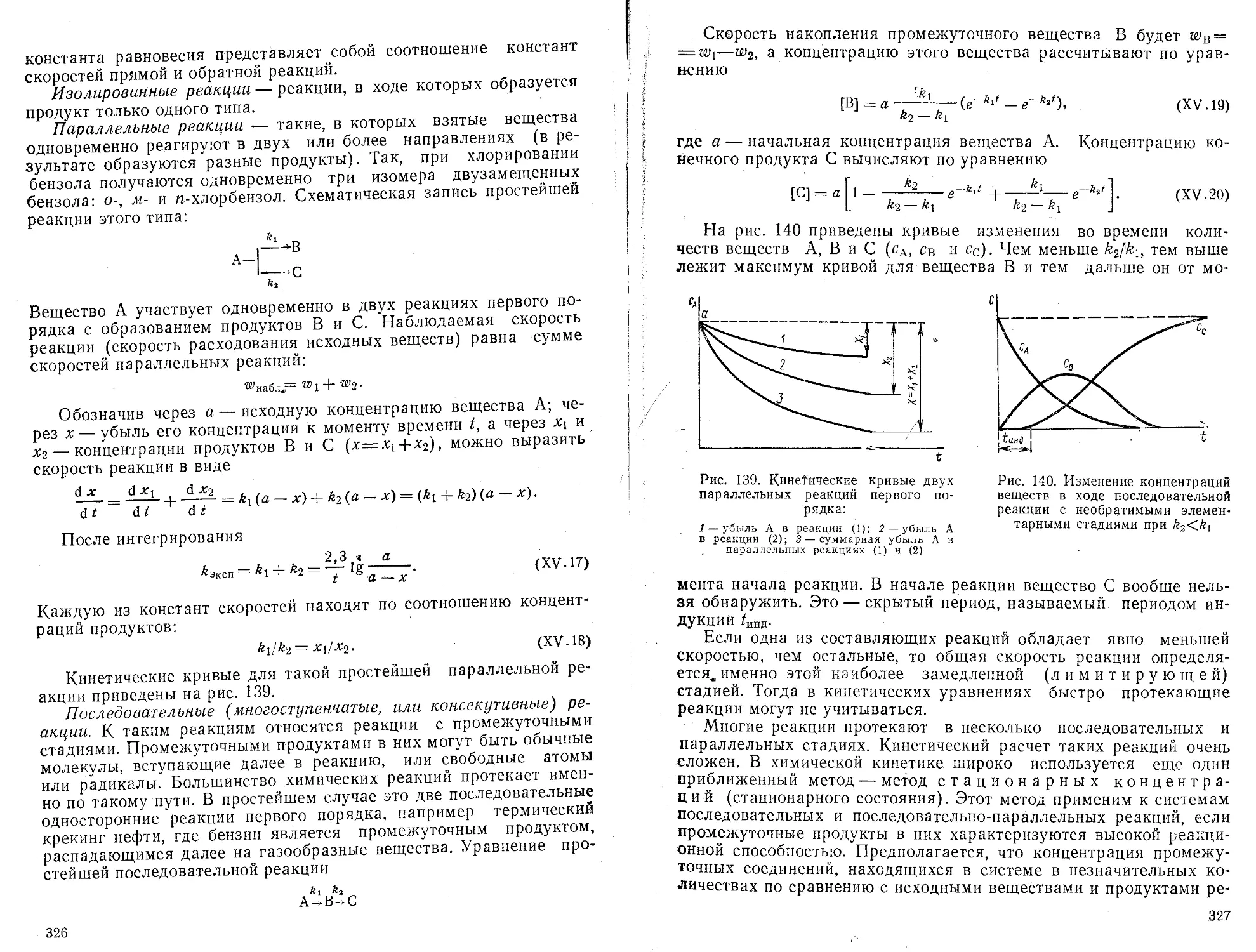
Законы поглощения электромагнитного излучения молекулами. Если монохроматическое излучение поглощается молекулами вещества, то по направлению светового потока наблюдается непрерывное падение интенсивности излучения /. Длина волны (или волновое число) при этом не меняется (-рис. 1), уменьшается лишь напряженность электромагнитного поля излучения, т. е. уменьшается число квантов. Выделим мысленно внутри однородного вещества некоторый бесконечно малый слой dl. Общая толщина поглощаю-
Рис. 1. Изменение интенсивности монохроматического излучения при прохождении через поглощающее вещество
щего слоя вещества I. При прохождении монохроматического излучения сквозь слой dl наблюдается бесконечно малая убыль интенсивности —dl. Относительная убыль интенсивности пропорциональна количеству поглощающих частиц или концентрации и толщине поглощающего слоя вещества:
= ас d Z,
<1Л)
где а—коэффициент погашения. После интегрирования (1.1)
получим ln(Z0/O =- сщ/, (1.2)
или = (1.3)
где £)“lg(/o//)—оптическая плотность: е=а/2,3026 — коэффициент экстинкции.
Для характеристики поглощения излучения веществом часто ис-
6
пользуется понятие пропускание, величина которого Т (в процен- , тах) выражается формулой
7^ = ZolOO/Z. (1.4)
Пропускание и оптическая плотность связаны между собой уравнением
£)—2 —IgZ.
(1.5)
2. МОЛЕКУЛЯРНЫЕ СПЕКТРЫ ДВУХАТОМНЫХ МОЛЕКУЛ
Вращательное движение двухатомных молекул. Молекулы вещества, находящегося в газообразном состоянии, вращаются вокруг центра масс молекулы. Если рассматривать молекулу как жесткий ротатор с массами атомов mi и т2 и равновесным межъядерным расстоянием ге, то момент инерции выражается уравнением
где ц — приведенная масса молекулы. Приведенная масса лы может быть рассчитана по атомным массам А и Ач\
«с
молеку-
1,66-10-27,
где та— масса атома углерода; тс/12= 1,66 • 10-27 кг.
Энергия вращения двухатомной молекулы. Молекула не может
•вращаться с произвольной энергией. Вращательное движение молекулы описывается волновым уравнением Шредингера, которое в полярных координатах имеет вид
02Ф д f дФ V 8я2
~— 1 sift I 1 _— ду>2 ‘ dfr [ W I №
Решение уравнения (1.8) дает
Д2
£вр = -8^7
где /— вращательное квантовое число, принимающее значения целых чисел, включая и ноль. Так как решение уравнения (1.8) произведено при условии re=const, то в уравнении (1.9) момент инерции 1 — величина постоянная. Обозначим
А/8л2/с==вг1 (1.Ю)
где Ве — вращательная постоянная, м-1 (см-1). Тогда уравнение (1.9) запишется
^вр — Behcj (j 4- 1) •
(I.11)
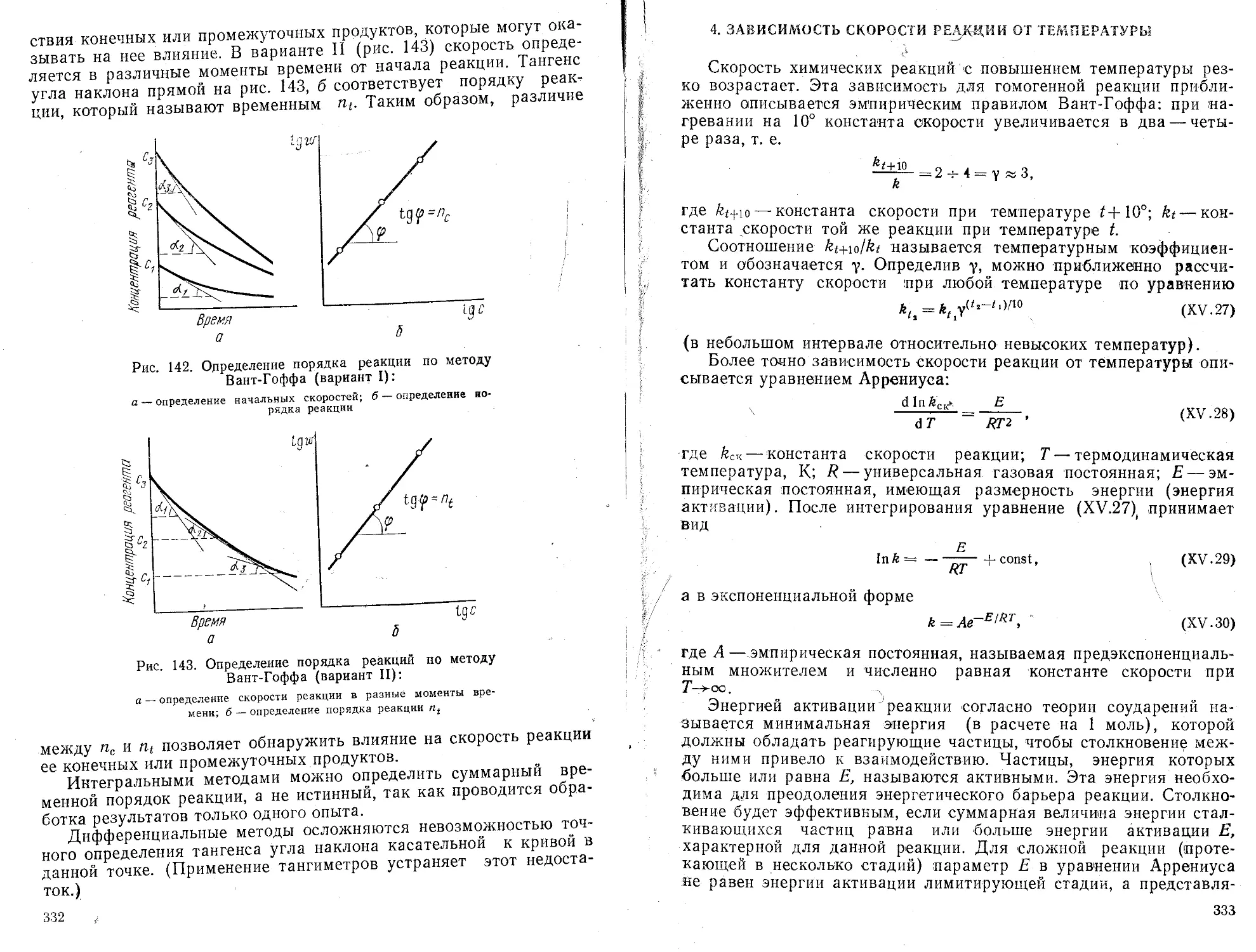
Из уравнений (1.9) и (1.11) следует, что существуют некоторые уровни энергии вращательного движения (рис. 2).
*
7
6----------j—
5 т-J—JOBs tic
4-------F------г0ВеЬс
3--------------12Behc
I
2 ——j—J--------OBgtic
1 -i’----------2BehC
0-Л-------------0
J ESp
Рис. 2. Вращательные энергетические уровни двухатомной молекулы одного и того же колебательного состояния
Вращательные спектры поглощения двухатомных молекул* Электромагнитное поле может взаимодействовать с электрическим диполем. Отсюда поглощение возможно только полярными молекулами. Переходы между уровнями энергии определяются правилом отбора
(1.12)
Энергия кванта при этом переходе описывается уравнением
£Кв - hcv - Е^ - £’р - Векс [Г (Г-Н) - J" (J" + 1)Ь
(1.13)
где ЕКв — энергия кванта поглощенного излучения, v — волновое число; £%, £Z/BP — энергии высокого и низкого соседних вращательных квантовых уровней соответственно; /'= = Из уравнения (1.13) вытекает выражение волнового числа вращательного спектра двухатомной молекулы как жесткого ротатора:
v - E^hc - 2Ве U + 1), (1.14)
где / — вращательное квантовое число уровня, с которого осуществлялся переход молекулы при поглощении ею кванта излучения.
Вращательный спектр поглощения наблюдается только у веществ, находящихся в газообразном состоянии. Это обусловлено тем, что энергия межмолекулярного взаимодействия между молекулами в жидком и твердом состоянии вещества превышает энергию вращения. Чисто вращательные спектры поглощения наблюдаются в микроволновой и дальней инфракрасной (ИК) области спектра.
Оптическая плотность линии поглощения во вращательном спектре при постоянной толщине поглощающего слоя определяется согласно уравнению (1.3) концентрацией молекул, находящихся на уровне, с которого происходит переход, и способностью молекулы взаимодействовать с квантом [е в уравнении (1.3)]. Можно принять, что коэффициент экстинкции не зависит от энергии вращения, следовательно, интенсивность линии в спектре поглощения определяется только количеством молекул, находящихся на уровне j. Количество же молекул на этом уровне рассчитывают по уравнению распределения Больцмана
хг xr -E./kT Nj = Nog}e ,
(1.15)
где Nj — число молекул на /-м вращательном квантовом уровне; g, — вырождение /-го вращательного уровня; Ej — энергия уровня; No— число молекул на нулевом уровне.
$
8
Решение уравнения (1.8) дает вырождение вращательных квантовых уровней. Под вырождением понимают число подуровней с одинаковой энергией Ej на одном вращательном уровне:
£у = 2/+1. (1.16)
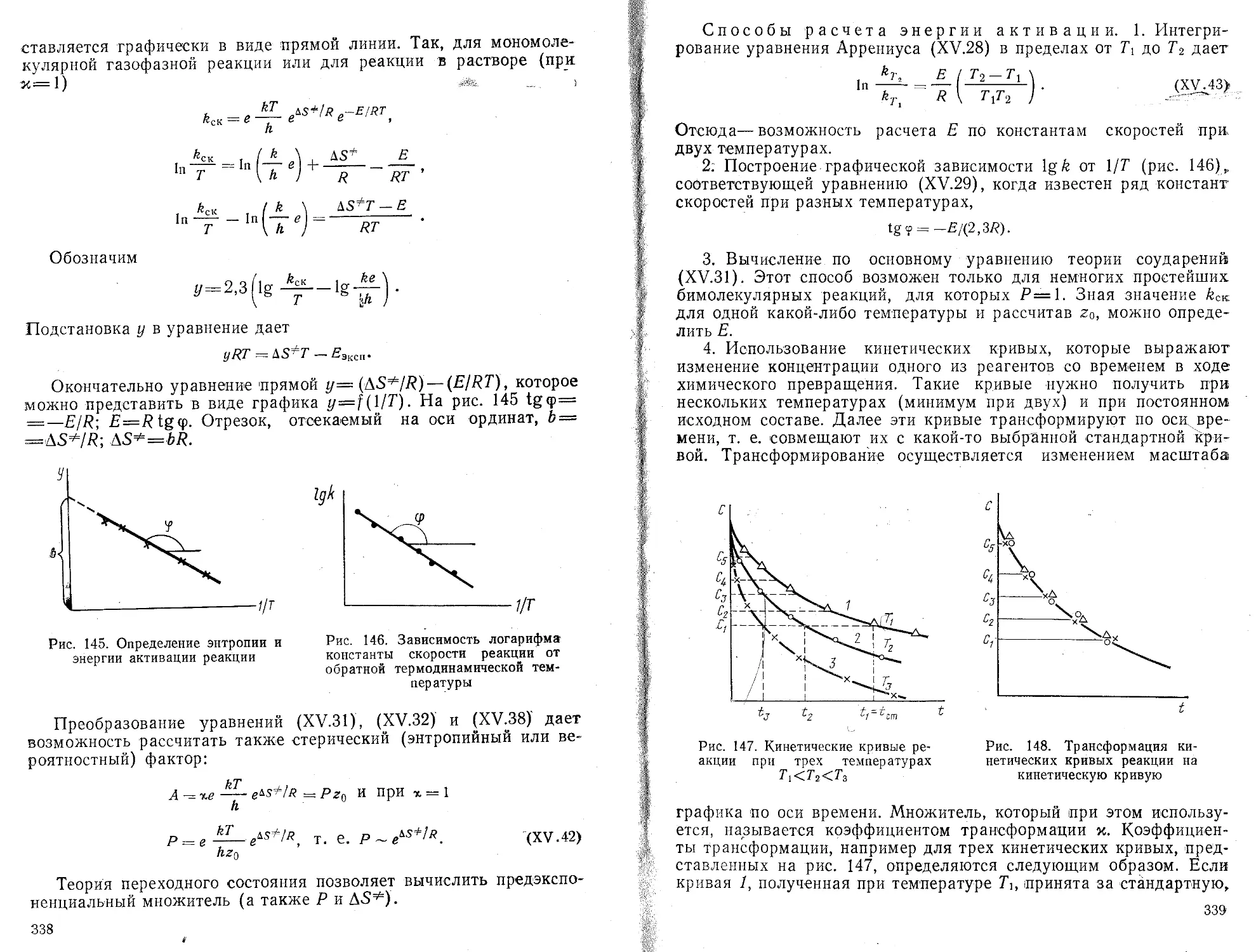
Таким образом, можно теоретически описать распределение оптических плотностей вращательных линий в спектре поглощения (рис. 3).
Определёние молекулярных констант Ве, 1 и ге. Разность волновых чисел соседних линий во вращательном спектре поглощения из (1.14) выражается уравнением
Av ™ 2Ве — hj&iZIc.
(117)
Таким образом, если ности волновых чисел
экспериментально определить значение раз-соседних линий во вращательном спектре по-
глощения, то можно рассчитать вращательную постоянную Ве, момент инерции I и по уравнениям (1.6) и (1.7) — равновесное межъядерное расстояние. Обычно определяют среднюю разность волновых чисел, измерив волновые числа двух ли-
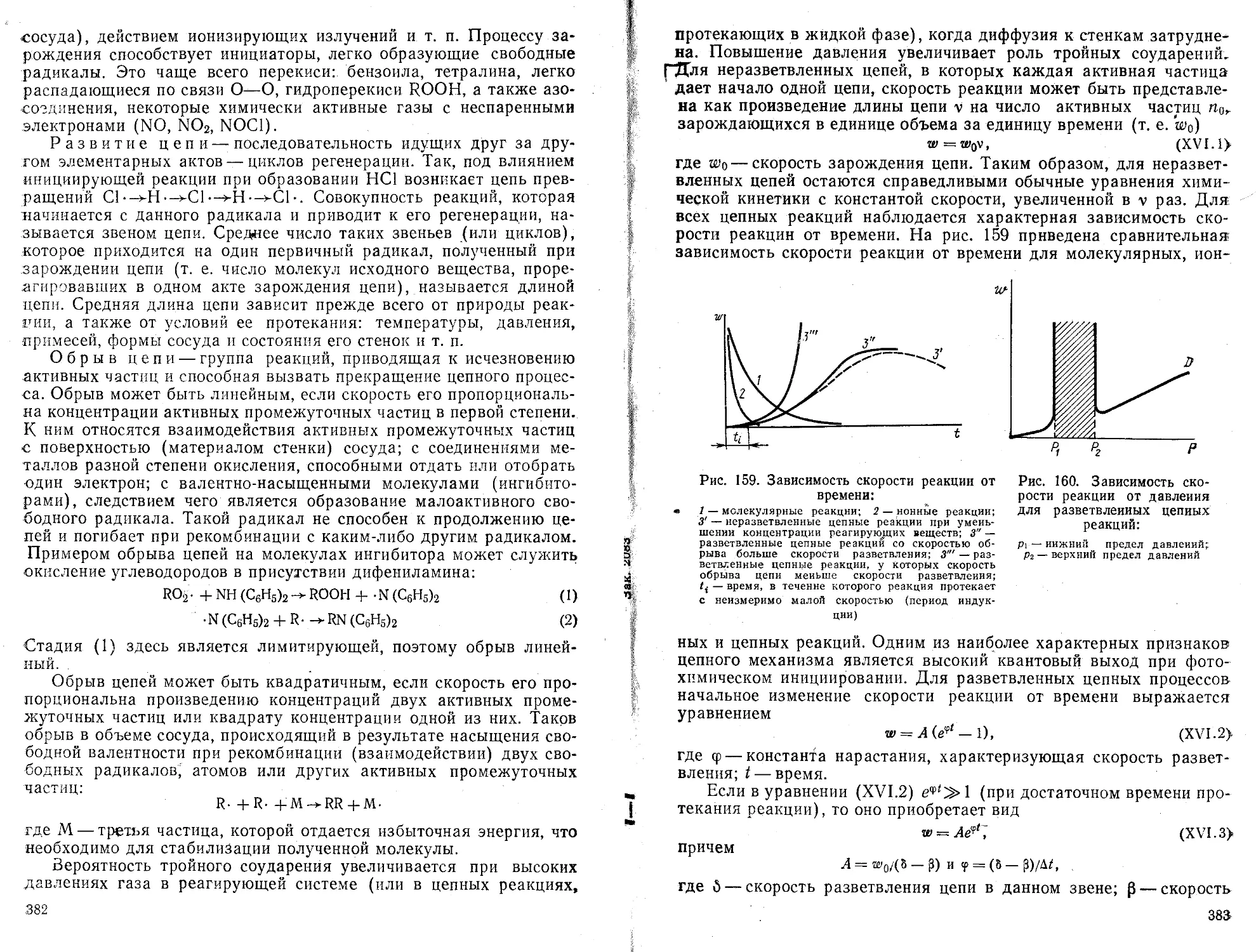
Рис. 4. Схема колебательного дви* жения в несимметричной двух* атомной молекуле
Рис. 3. Вращательные энергетические уровни двухатомной молекулы с учетом вырождения
ний, отстоящих друг от друга на несколько интервалов. Например, если измерены волновые числа v/ и v/+n, то
(1.18)
где Avcp —средняя разность волновых чисел; п — число линий. Так как на самом деле молекула не жесткий ротатор, то Av не остается постоянной. Увеличение энергии вращения ведет к росту центробежной силы, которая увеличивает межъядерное расстояние и, следовательно, момент инерции. Отсюда во вращательном спектре поглощения наблюдается постепенное уменьшение Av по мере увели
чения v. Поэтому и введено понятие среднего равновесного межъядерного расстояния ге.
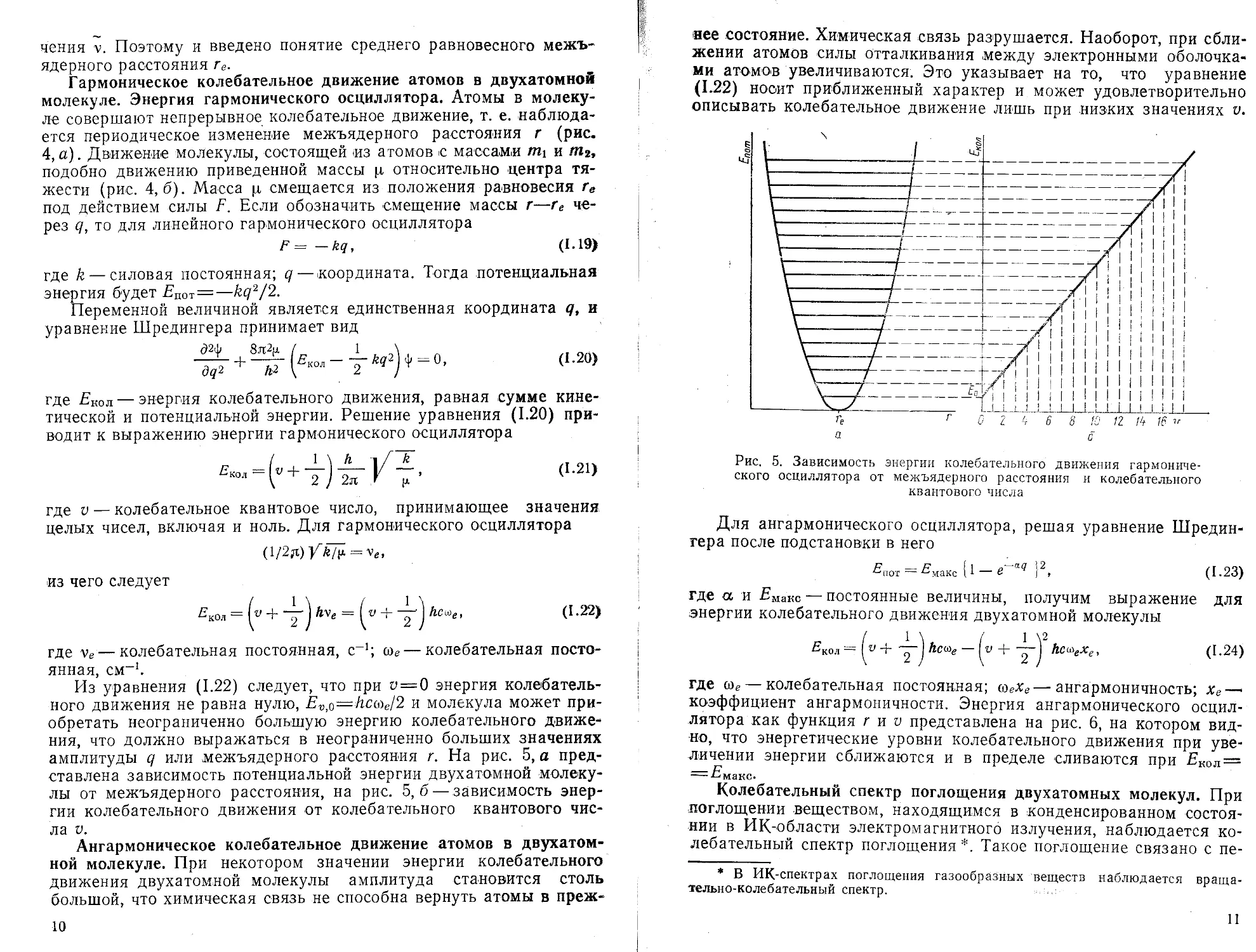
Гармоническое колебательное движение атомов в двухатомной молекуле. Энергия гармонического осциллятора. Атомы в молекуле совершают непрерывное колебательное движение, т. е. наблюдается периодическое изменение межъядерного расстояния г (рис, 4,а). Движение молекулы, состоящей из атомов с массами mi и подобно движению приведенной массы ц относительно центра тяжести (рис. 4,6). Масса ц смещается из положения равновесия ге под действием силы Е. Если обозначить смещение массы г—ге через q, то для линейного гармонического осциллятора
(1.19>
F = — kq,
где k — силовая постоянная; q — координата. Тогда потенциальная энергия будет Епот^—kq2/2.
Переменной величиной является единственная координата q, и уравнение Шредингера принимает вид
(Е20)
где Екол — энергия колебательного движения, равная сумме кинетической и потенциальной энергии. Решение уравнения (1.20) приводит к выражению энергии гармонического осциллятора
где v — колебательное квантовое число, принимающее значения целых чисел, включая и ноль. Для Гармонического осциллятора
из чего следует
(W) =
где уе — колебательная постоянная, с-1; — колебательная посто-
янная, СМ"1.
Из уравнения (1.22) следует, что при и —0 энергия колебательного движения не равна нулю, Ev$—hctoel2 и молекула может приобретать неограниченно большую энергию колебательного движения, что должно выражаться в неограниченно больших значениях амплитуды q или межъядерного расстояния г. На рис. 5, а представлена зависимость потенциальной энергии двухатомной молекулы от межъядерного расстояния, на рис. 5,6 — зависимость энергии колебательного движения от колебательного квантового числа V,
Ангармоническое колебательное движение атомов в двухатомной молекуле. При некотором значении энергии колебательного движения двухатомной молекулы амплитуда становится столь большой, что химическая связь не способна вернуть атомы в преж-10
нее состояние. Химическая связь разрушается. Наоборот, при сближении атомов силы отталкивания между электронными оболочками атомов увеличиваются. Это указывает на то, что уравнение (1.22) носит приближенный характер и может удовлетворительно описывать колебательное движение лишь при низких значениях V.
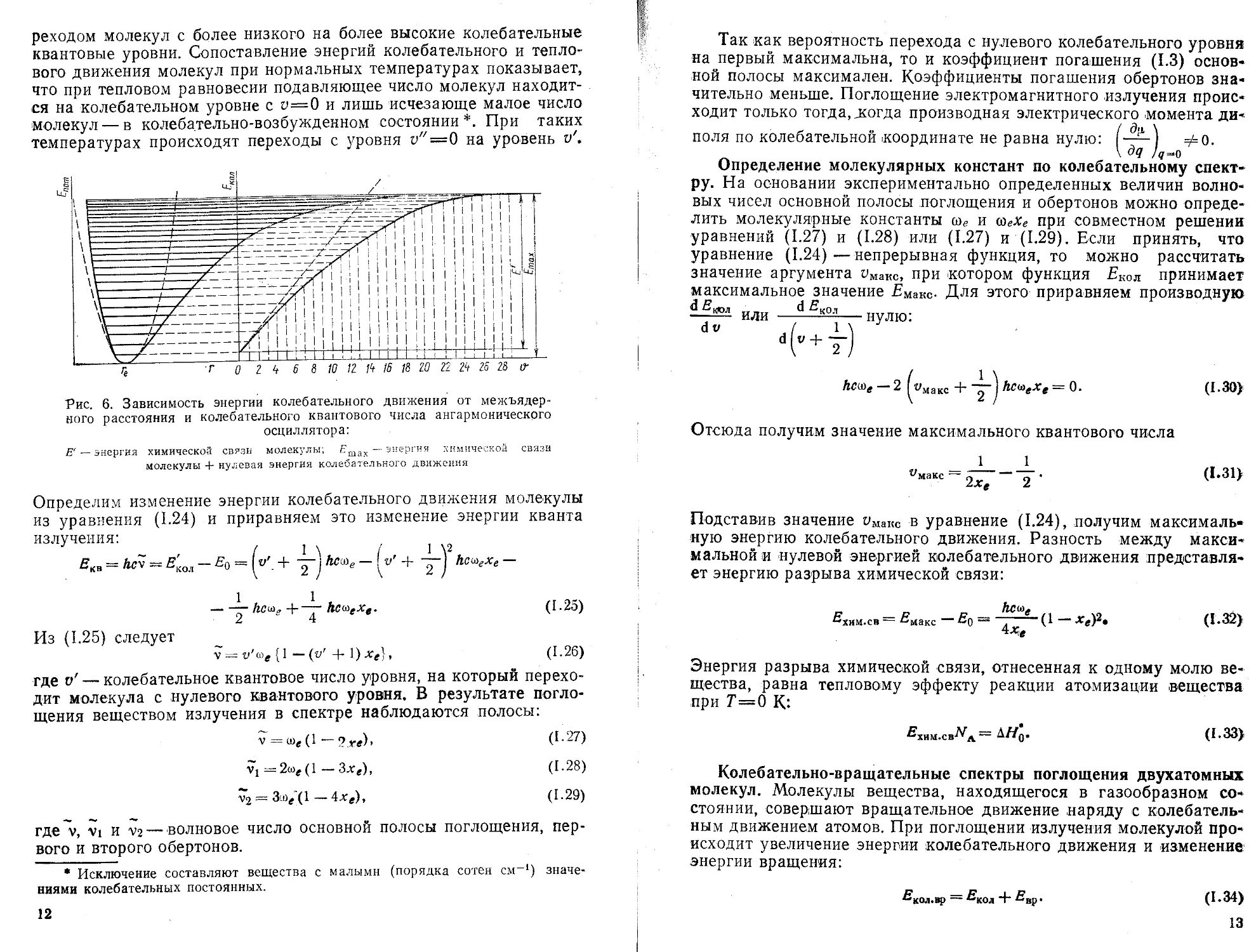
Рис. 5. Зависимость энергии колебательного движения гармонического осциллятора от межъядерного расстояния и колебательного квантового числа
Для ангармонического осциллятора, решая уравнение Шредингера после подстановки в него
р р 11 12
^пот — 11 & f f
где а и Вмакс — постоянные величины, получим выражение энергии колебательного движения двухатомной молекулы
(1.23)
для
(1.24)
где сое — колебательная постоянная; ыеХе—ангармоничность; коэффициент ангармоничности. Энергия ангармонического осциллятора как функция г и v представлена на рис. 6, на котором видно, что энергетические уровни колебательного движения при увеличении энергии сближаются и в пределе сливаются при ВКОл = ^^Вмакс.
Колебательный спектр поглощения двухатомных молекул. При поглощении веществом, находящимся в конденсированном состоянии в ИК-области электромагнитного излучения, наблюдается колебательный спектр поглощения *. Такое поглощение связано с пе-
* в ик-спектрах поглощения газообразных веществ наблюдается вращательно-колебательный спектр.
реходом молекул с более низкого на более высокие колебательные квантовые уровни. Сопоставление энергий колебательного и теплового движения молекул при нормальных температурах показывает, что при тепловом равновесии подавляющее число молекул находится на колебательном уровне с v=0 и лишь исчезающе малое число молекул — в колебательно-возбужденном состоянии*. При таких температурах происходят переходы с уровня у"=0 на уровень v'.
Рис. 6. Зависимость энергии колебательного движения от межъядерного расстояния и колебательного квантового числа ангармонического осциллятора:
Ег - -- энергия химической связи молекулы; £та — энергия химической связи молекулы 4- нулевая энергия колебательного движения
Определим изменение энергии колебательного движения молекулы из уравнения (1.24) и приравняем это изменение энергии кванта
излучения:
^кв “ “ ^кол — ^0 ~
Из (1.25) следует
(1.25)
(1.26)
где v' — колебательное квантовое число уровня, на который переходит молекула с нулевого квантового уровня, В результате поглощения веществом излучения в спектре наблюдаются полосы:
V =(ое(1 —
(L27)
Vj ~2wtf (1 — Зхе),
(1.28)
v2 = Зю/( 1 — 4xg), (1.29)
где v, vi и V2—волновое число основной полосы поглощения, первого и второго обертонов.
* Исключение составляют вещества с малыми (порядка сотен см-1) значениями колебательных постоянных.
12
Так как вероятность перехода с нулевого колебательного уровня на первый максимальна, то и коэффициент погашения (1.3) основ* ной полосы максимален. Коэффициенты погашения обертонов зна* чительно меньше. Поглощение электромагнитного излучения проис*
ходит только тогда, жогда производная электрического момента ди* поля по колебательной координате не равна нулю: =^о.
Определение молекулярных констант по колебательному спектру. На основании экспериментально определенных величин волновых чисел основной полосы поглощения и обертонов можно определить молекулярные константы и сол при совместном решении уравнений (1.27) и (1.28) или (1.27) и (1.29). Если принять, что уравнение (1.24) — непрерывная функция, то можно рассчитать значение аргумента оМакс, при котором функция Екол принимает максимальное значение Емакс- Для этого приравняем производную — или —d —нулю:
d v . / 1 \
(1.30)
Отсюда получим значение максимального квантового числа
11
^макс “ • (1.31)
Подставив значение аМакс в уравнение (1.24), получим максимальную энергию колебательного движения. Разность между макси* мальвой и нулевой энергией колебательного движения представляет энергию разрыва химической связи:
хим.св — *-*макс
(1.32)
Энергия разрыва химической связи, отнесенная к одному молю вещества, равна тепловому эффекту реакции атомизации вещества при Т—О К:
^ХИМ.СВ^д— ^//q.
(1.33)
Колебательно-вращательные спектры поглощения двухатомных молекул. Молекулы вещества, находящегося в газообразном состоянии, совершают вращательное движение наряду с колебательным движением атомов. При поглощении излучения молекулой происходит увеличение энергии колебательного движения и изменение энергии вращения:
Екол.вр — ^КОЛ 4“ Евр •
(1.34)
13
Волновое число линии поглощения в колебательно-вращательном спектре будет
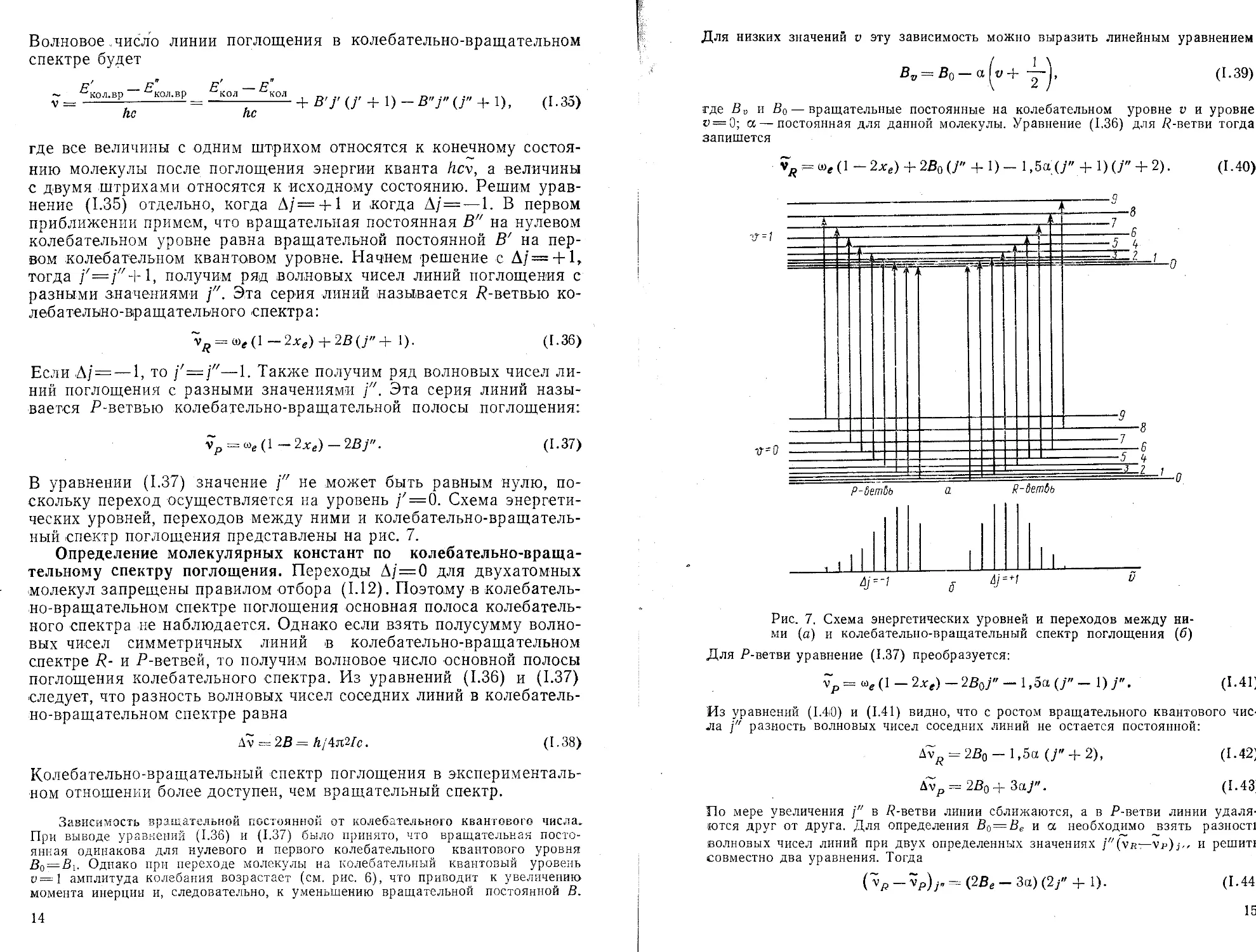
где все величины с одним штрихом относятся к конечному состоянию молекулы после поглощения энергии кванта hcv, а величины с двумя штрихами относятся к исходному состоянию. Решим уравнение (1.35) отдельно, когда Д/ = +1 и когда Д/= —1. В первом приближении примем, что вращательная постоянная Bz/ на нулевом колебательном уровне равна вращательной постоянной В7 на первом колебательном квантовом уровне. Начнем решение с Д/= + 1, тогда /z=/zz+l, получим ряд волновых чисел линий поглощения с разными значениями Эта серия линий называется 7?-ветвью колебательно-вращательного спектра:
\ (1 - 2хе) + 2В (у" + 1). (1.36)
Если Aj~ — 1, то j'=j"—1. Также получим ряд волновых чисел линий поглощения с разными значениями ///. Эта серия линий называется Р-ветвью колебательно-вращательной полосы поглощения:
(1.37)
В уравнении (1.37) значение jzz не может быть равным нулю, поскольку переход осуществляется на уровень /z=0. Схема энергетических уровней, переходов между ними и колебательно-вращательный спектр поглощения представлены на рис. 7.
Определение молекулярных констант по колебательно-вращательному спектру поглощения. Переходы Д/ = 0 для двухатомных молекул запрещены правилом отбора (1.12). Поэтому в колебательно-вращательном спектре поглощения основная полоса колебательного спектра не наблюдается. Однако если взять полусумму волновых чисел симметричных линий в колебательно-вращательном спектре 7?- и Р-ветвей, то получим волновое число основной полосы поглощения колебательного спектра. Из уравнений (1.36) и (1.37) следует, что разность волновых чисел соседних линий в колебательно-вращательном спектре равна
h[b&Ic. (1.38)
Колебательно-вращательный спектр поглощения в экспериментальном отношении более доступен, чем вращательный спектр.
Зависимость вращательной постоянной от колебательного квантового числа. При выводе уравнений (1.36) и (1.37) было принято, что вращательная постоянная одинакова для нулевого и первого колебательного квантового уровня Bq — Bi. Однако при переходе молекулы на колебательный квантовый уровень v=l амплитуда колебания возрастает (см. рис. 6), что приводит к увеличению момента инерции и, следовательно, к уменьшению вращательной постоянной В.
14
Для низких значении v эту зависимость можно выразить линейным уравнением
(1.39)
где Bv и Во — вращательные постоянные на колебательном уровне v и уровне 0; ос постоянная для данной молекулы. Уравнение (1.36) для Р-ветви тогда запишется
(1.40)
(1 - 2хе) 4- 2В0 (/" + 1) - 1,5а (Д' 4- 1) (/" 4- 2).
Рис. 7. Схема энергетических уровней и переходов между ними (а) и колебательно-вращательный спектр поглощения (б)
Для Р-ветви уравнение (1.37) преобразуется:
(1 — 2хг) — 2В0Г — 1,5а (Д' — 1) Д',
(1.41:
Из уравнений (1.40) и (1.41) видно, что с ростом вращательного квантового числа j'f разность волновых чисел соседних линий не остается постоянной:
AVp = 2В0 — l,5ct (J" + 2),
(1.42;
Avp - 2В0 + ЗаД'. (1.43.
По мере увеличения j" в Р-ветви линии сближаются, а в Р-ветви линии удаля* ются друг от друга. Для определения Во — Ве и ос необходимо взять разноси волновых чисел линий при двух определенных значениях /"(vj?—vp)p, и решит! совместно два уравнения. Тогда
( Vp — Vp)yff~(2Be — За) (2/'4-1). (1.44
15
Рис. 8. Относительное расположение МО по энергиям для двухатомных молекул Оз (а) и BN (б)
Электронное состояние двухатомных молекул. Энергия электронного состояния. При образовании химиче* ской связи происходит перекрывание атомных орбита-лей (АО) и образование молекулярных орбиталей (МО). Энергия МО определяется по уравнению Шре-
дингера
г» (1.45)
где ф — волновая функция;
Е — энергия электрона; Я— оператор Гамильтона;
Л2 -------(Vi
НОТ»
(1.46)
где Vi, V2 — операторы Лапласа; £Пот — потенци-
альная энергия.
В результате решения уравнения (1.45) определяют энергию связывающих и разрыхляющих МО и строят энергетическую диаграмму, т. е. относительное расположение МО по энергетическим уровням. В качестве
примера рассмотрим энергетическую диаграмму гомо-ядерной двухатомной молекулы О2 (рис. 8, а) и гете-роядерной молекулы BN (рис. 8, б). Электронная конфигурация молекулы О2
Ру*
*
молекулы BN
ру>
При поглощении энергии электромагнитного излучения в УФ-участке спектра электрон может переходить с занятой на свободную молекулярную орбиталь. Из рис. 8 видно, что молекула обладает некоторым набором квантовых энергетических уровней. Каждое электронное состояние молекулы характеризуется значением полного орбитального и спинового моментов количества движения.
Двухатомные молекулы имеют ось симметрии бесконечного порядка, совпадающую с осью молекулы. Величина проекции орби
16
тального момента L на ось молекулы является важной характеристикой электронного состояния молекулы. Она задается квантовым числом X. Состояния молекулы, соответствующие определенным квантовым числам, обозначают символами: ф,
X 0 1 2 3 ...
Символ S л Д' Ф ...
Кроме орбитального квантового числа % электронное состояние молекулы характеризуется спиновым квантовым числом S, которое определяет статистический вес или мультиплетность состояния
+ (1.47)
где g — статистический вес или мультиплетность; S — суммарный спин молекулярных электронов. Мультиплетность записывают в виде числа слева, вверху символа состояния. Например, означает Х=0 и£ = 0; Зл— и S—1. Электронные состояния двухатомных молекул могут различаться еще по свойствам симметрии. Если молекулярная волновая функция сохраняет свой знак при операции отражения в плоскости, проходящей через ось молекулы, то такое состояние положительное ( + ). Если же при операции отражения знак волновой функции меняется, то такое состояние отрицательное (—). Этот индекс записывается справа вверху символа. Например, !S+. Гомоядерные двухатомные молекулы обладают еще центром симметрии, расположенным в центре масс молекулы. Электронное состояние может быть четным (g) при наличии центра симметрии и нечетным (и) при отсутствии центра симметрии. Символ четности ставят справа внизу символа состояния.
Электронно-колебательные спектры поглощения двухатомных молекул. Разность энергий свободной и занятой молекулярной орбитали у большинства молекул достаточно велика (около 250 кДж/моль). Это означает, что термического возбуждения за счет соударений молекул при.их тепловом движении не наблюдается. При поглощении УФ-излучения наблюдается поглощение, связанное с переходом электрона на более высокую свободную МО. Электронный спектр молекулы представляет набор полос поглощения, и каждая полоса соответствует определенному электронному переходу.
Теория взаимодействия кванта электромагнитного излучения с молекулой вещества приводит к правилу отбора для двухатомных молекул. Поглощение энергии кванта электромагнитного излучения возможно, если удовлетворяются условия:
ДХ = О; zfcl; Д<5=0;
(-)->(-); ( + )-( + );
(«)-*(£); (£)->-(“)*
Если хотя бы одно из этих условий не удовлетворяется, то квантовый переход запрещен. Запрещенными являются также кванто- '
* Только для гомоядерных двухатомных молекул.
17
вне переходы, при которых происходит возбуждение более чем одного электрона в молекуле. При любых дозволенных электронных переходах происходит изменение электронного состояния химической связи, что влечет за собой изменение потенциальной энергии молекулы. При этом возможно, что при поглощении энергии не происходит изменения равновесного межъядерного расстояния (рис. 9, а) или происходит увеличение равновесного межъядерного
Рис. 9. Электронно-колебательный спектр поглощения двухатомной молекулы
расстояния (уменьшение энергии связи, рис. 9,6). Принцип Франка— Кондона показывает, что колебания атомов совершаются за время значительно большее, чем время перехода электрона. На рис. 9 переходы электрона показаны вертикальными линиями. Так как подавляющее большинство молекул при нормальной температуре находится на нулевом колебательном квантовом уровне, то наиболее вероятными будут переходы из этого наиболее вероятного состояния. Если равновесные межъядерные расстояния в невозбужденном и в возбужденном состояниях одинаковы, то наиболее интенсивной будет полоса поглощения 0 ->0 (рис. 9, а).
Если же равновесное межъядерное расстояние при возбуждении электрона увеличивается, то наиболее интенсивной будет полоса поглощения, соответствующая переходу электрона 0 Величина интенсивности зависит от относительного смещения потенциальных кривых (рис. 9,6). Если различие равновесных межъядерных расстояний значительное, то возможны такие переходы электрона, при
18
которых происходит диссоциация молекулы (рис. 9,6). В спектре это проявится в том, что 'Высокочастотный край полосы переходит из дискретного в сплошное поглощение.
Определение энергии химической связи по электронно-колебательному спектру. По волновому числу границы дискретного и сплошного поглощения можно определить энергию кванта излучения, необходимого для разрушения химической связи. Однако при фотодиссоциации один или иногда оба атома будут в электронновозбужденном состоянии. Электронное возбуждение атомов в продуктах диссоциации происходит за счет энергии того же кванта, который при действии на молекулу вызвал ее диссоциацию. Энергию разрыва химической связи рассчитывают по формуле
£р.с “ ^vmax “” ^возб > (1.48)
где £ВОзб — энергия возбуждения одного или обоих атомов, которую определяют по спектру излучения атома; vmax — волновое число границы сплошного поглощения.
Определение молекулярных констант по электронно-колебатель-но-вращательному спектру. Молекулы при электрическом разряде и достаточной разности потенциалов возбуждаются. При переходе молекулы или радикала из возбужденного электронно-колебательно-вращательного состояния на различные колебательно-вращательные уровни нулевого электронного уровня происходит излучение квантов светового потока с энергиями, равными разности энергий между высоким и низким энергетическими уровнями. Без учета энергии вращательного движения молекулы можно записать энергию электронно-колебательного уровня как сумму энергий электронного состояния и колебательного движения:
кол •
Энергия более высокого возбужденного энергетического будет
СОСТОЯНИЯ
hc^' —
2
ЭЛ.-КОЛ
ЭЛ
(1.50)
е*
а энергия более низкого энергетического уровня —
(1.51)
\ Колебательные постоянные сое и ангармоничности ($exe на более высоком и на более низком электронно-колебательных уровнях не одинаковы. Для отнесения линий в спектре обычно составляют таблицу, в которую записывают волновые числа линий спектра в виде прямоугольной схемы, столбец которой соответствует различным b/z, а строки — у7. Такая таблица называется таблицей Деландра. Если е и сильно различаются, т. е. кривые потенциальной энергии в невозбужденном и в возбужденном состояниях сильно смещены, то наблюдается сложная картина спектра. Спектральные
19
а
линии, соответствующие разным значениям Ду, накладываются друг на друга и отнесение линий к определенным переходам представляет значительную трудность. Если же разность и невелика, то линии в спектре группируют по величине разности квантовых чисел &v = v'—v". Получают ряд групп линий, соответствующих Ду = — 1, Ди=0 и Ду= + 1.
На основании экспериментальных данных хч линий в электронно-колебательном спектре могут быть определены со^, ®еХе для электронно-невозбужденного состояния. Тонкая структура полосы электронно - колебательно - вращательного спектра может быть получена только при съемке спектра на спектрографе с высокой дисперсией и достаточной разрешающей способностью.
На рис. 10 показаны возможные переходы и линии в спектре излучения двухатомной молекулы или радикала; вращательные линии не показаны. Из рис. 10 видно, что разность энергий Д£о-*о и AEo^i равна разности энергий колебательных квантовых уровней с колебательными квантовыми числами v= 1 и 0. Отсюда
(Л^О^-О “ Д^0->1)/^С ~ V0->0 v0 * (I • 52)
Разность волновых чисел линий в спектре излучения равна волновому числу, соответствующему переходу молекулы в электронно- невозбужденном состоянии с vf/—0 на v'=l, т. е. волновому числу основной полосы поглощения в колебательном спектре [см. (1.27)].
Разность энергий переходов
Д£1->1—Д£1^2 соответствует энергии перехода молекулы с колебательного квантового уровня v/7=l на колебательный квантовый уровень у/=2:
Рис. 10. Переходы (а) и спектральные линии (6) в электронно-колебательном спектре излучения (толщина линий, изображающих переходы, соответствует вероятности перехода; толщина линии в спектре — интенсивности)
(L53)
Сумма разностей (1.52) и (1.53) будет
( vl->2) + ( v0->0 ~~ VO >1) = V1»
(L54)
где vi — волновое число первого обертона (1.28).
По экспериментально полученным волновым числам линий в электронно-колебательном спектре можно определить сое и соеХ* при совместном решении уравнений (1.27) и (1.28), а по уравнению (1.32) вычислить энергию разрыва связи.
20
3. СПЕКТРЫ КОМБИНАЦИОННОГО РАССЕЯНИЯ
. Ц':- •• >. \
Колебание атомов в двухатомной молекуле описывается изменением колебательной координаты, В качестве колебательной координаты примем разность между межъядерным расстоянием и равновесным межъядерным расстоянием. Колебательная координата зависит от времени:
q = Q cos (2лсюе0> (1.55)
где 7, Q — колебательные координаты; со*— колебательная постоянная; t — время. Если вещество облучать монохроматическим световым потоком с частотой cv, то напряженность поля от времени будет изменяться по уравнению
Е = Ео c^s (2ncvt + <р),
(1.56)
где Е и Eq — напряженность поля при времени t и /=0; ф— сдвиг фазы Е относительно q.
Под действием поля световой волны происходит поляризация электронов в молекуле, что выражается в появлении наведенного электрического момента диполя р, величина которого пропорциональна напряженности поля:
Р = аЭлЕ, w (1.57)
где аэл — электронная поляризуемость молекулы, т. е. способность электронов смещаться под действием внешнего поля.
Так как поле переменное, то напряженность меняется во времени и наведенный дипольный момент также будет меняться во времени согласно урав
нению
Р = аэл^о cos (2jwvZ 4- <р).
(1.58)
Введем обозначение аЭлЕо=Цо, тогда
у = Ро cos (2ncv/ 4- ср). (1.59)
Зависимость электронной поляризуемости от колебательной координаты сложная, но вблизи положения равновесия колеблющихся ядер атомов (</—0) ее можно представить линейным уравнением
(д(1зл \ __ аэл — а0,Эл
dq q
или / да эл \
Дэл — 4~ “7 ) Я » (1.60)
\ vq /q~Q
где осо, эл — электронная поляризуемость в состоянии равновесия.
Подставив уравнение (1.60) и (1.58), получим
а0,эл 4-
q Eq cos (2jicW 4- ?).
Если теперь вместо q подставить ее значение из (1.55), то получим
.,Эл Q cos (2лсш^О 1 cos (2ncv/ 4~ ?). oq J
(1.61)
(1.62)
21
После некоторого преобразования уравнения (1.6.2) правую часть можно раз^ бить на три части:
pi — Р-о cos (2ncv£ +?) + “" (— Сеэл-) QEq cos [2jw (v 4~ ые) t + <р] +
(1.63)
III
Рис. 11. Схема энергетических переходов молекулы при поглощении монохроматического излучения и при рассеянии светового потока
Таким образом, наведенный электрический момент диполя молекулы меняется во времени. Вынужденное колебание молекулярного диполя есть не что иное как смещение электронов. Периодическое движение электронов вызывает излучение электромагнитного поля с частотой, равной частоте колебания электронов. Как видно ив уравнения (1.63), колебания диполя можно разложить на три слагаемых. Слагаемое I описывает колебания диполя -с частотой, равной частоте су монохроматического светового потока, которым Облучалось вещество. Слагаемые II и III описывают колебания диполя с измененными частотами c(v+<oe) и c(v—сое). Следовательно^ в рассеянном излучении будет наблюдаться три частоты: c(v-F<oe)y су и —(Ое). Рассеяние светового потока без изменения частоты
называется классическим или ре-леевским. Рассеяние светового потока с изменением частоты называется комбинационным, причем рассеяние с частотой с (у— называется стоксовым, а с частотой с(у-|-ол?) —антистоксовым. Интенсивность стоксовых линий значительно больше интенсивности антистоксовых линий. Как следует из уравнения (1.63), линии комбинационного рассеяния не наблюдаются, если при вынужденном колебании диполя электронная поляризуемость молекулы не ( дссэл \ меняется, т. е. -------- =0.
\ dq / ?=«0
Схема энергетических переходов, молекулы при поглощении монохроматического излучения и при рассеянии излучения молекулой представлена на рис. И. Так как интенсивность линий зависит от
количества молекул, поглощающих энергию монохроматического излучения, то антистоксовые линии в спектре комбинационного
рассеяния появляются в результате поглощения кванта монохроматического излучения молекулой, находящейся в. колебательновозбужденном состоянии, следовательно, интенсивность антистоксовых линий будет меньше интенсивности стоксовых и тем более линий классического рассеяния. Измерение частоты стоксовых линий в спектре комбинационного рассеяния дает возможность определять молекулярную константу сое:
а)е ~ V — V^K.p, (1.64)
где V —волновое число монохроматического излучения; ул.к.р— волновое число стоксовой линии комбинационного рассеяния.
4. МОЛЕКУЛЯРНЫЕ СПЕКТРЫ МНОГОАТОМНЫХ МОЛЕКУЛ
Вращательное движение многоатомных молекул. Вращательноколебательные спектры. Многоатомные линейные молекулы обладают двумя степенями свободы вращательного движения вокруг осей, проходящих через центр масс молекулы и перпендикулярно оси молекулы. Оба момента инерции одинаковы и, следовательно, одинаковы и вращательные постоянные, которые могут -быть определены из вращательного или вращательно-колебательного спектра по одному из уравнений (1.38), (1.42), (1.43). У молекул типа сферически симметричного волчка все три момента инерции одинаковы:
Ix=Iy^Iz. (1.65)
Равны и вращательные постоянные:
Bx = By = Bz. (1.66)
Из-за отсутствия электрического момента диполя у таких молекул вращательный спектр не наблюдается. Однако во вращательно-колебательном спектре наблюдаются /?-, Q-* < и Р-ветви. С помощью вращательно-колебательного спектра можно определить по Дур кли Дуд и уравнениям (1.38) или (1.42), (1.43) вращательную постоянную, момент инерции и равновесное межъядерное расстояние исходя из геометрии молекулы. У молекул типа симметричного волчка имеются два равных момента инерции. При этом возможны два варианта: a) Ix = Iy<h и Bx=By>Bz— вытянутый симметричный волчок; б) Ix<_Iy=Iz и Bx>By=B.z — сплющенный симметричный волчок. В чисто вращательном и во вращательно-колебательном спектрах наблюдается поглощение. Линии в спектрах описываются уравнениями (1.14), (1.36), (1.37). Отличительной особенностью вращательного и вращательно-колебательного спектра является распределение интенсивности линий в спектре. Это связано с иным
* Для многоатомных молекул разрешен переход А/ —О, в результате чего наблюдается Q-ветвь в колебательно-вращательном спектре.
23
вырождением вращательных квантовых уровней относительно вырождения уровней у двухатомных молекул:
^-(2/+ 1)2. . (1.67)
Колебательное движение. Колебательные спектры многоатомных молекул. Многоатомные молекулы обладают большим числом степеней свободы колебательного движения по сравнению с двухатомными молекулами. Для линейных многоатомных молекул число степеней свободы колебательного движения будет
Лол-ЗХ-5, (1.68)
для нелинейных
/KtM = 37V-6, (L69)
где N — число атомов в молекуле.
Из-за различия масс атомов и химических связей между ними каждое колебание осуществляется с вполне определенной частотой. Наиболее наглядной системой координат для описания колебательного движения ядер атомов является естественная система координат, которая задается значениями межъядерных расстояний и углов между направлениями связей:
02 = г2 — ге.^
4l = ri-re,i' <L70>
где reii — равновесное межъядерное расстояние; фез — равновесный угол.
Кинетическая и потенциальная энергии колебательного движения могут быть выражены уравнениями
Лсол
1 5L1 . .
^КИИ “ 2 ^iflflр П'71)
Лео л
Дют — 2 j 0*72)
где £кин, £пот — кинетическая и потенциальная энергии; — приведенная масса; qi, qj — координаты; qi=dqildt\ t — время, — квазиулругая постоянная связи между ядрами ь и /-атомов.
Если учесть, что ил -и ki^=k^ то уравнения (1.71) и (1.72)
можно записать , <
7кол 7КОЛ
^кии = 9 ^ifl fl Г (Е73)
Z t,J=l
24
(1.74)
Первые члены уравнения (1.73) и (1.74) характеризуют взаимодействия атомов, непосредственно связанных химической связью. У обоих соседних ядер атомов общая колебательная координата q,. Вторые члены выражают кинематическое взаимодействие и динамическое взаимодействие координат. В общем случае взаимодействие координат не равно нулю. Для решения задачи о форме колебаний и частотах колебаний удобнее перейти от естественных координат qi к их линейным комбинациям:
'кол
?z=£c<2z, (Ь75)
i~l
где Qi — нормальная координата; C/j— коэффициент. Тогда
(1.76)
где рг- и ki— коэффициенты, зависящие от p/j и
Энергия колебательного движения равна сумме кинетической и потенциальной энергии:
Л<ол ^Кол
В форме уравнения Лагранжа колебательное движение можно записать для z-й колебательной степени свободы
+ Ш = 0. (1.78)
Решение дифференциального уравнения (1.78) ищем в виде функции
Qi = Qz,ocos(2jiv^ 4- &.), (1.79)
где Vi — частота z-ro колебания; / — время;
<1
Q. = — sin (2jtv У -4- &z); Qi ~ — 4nv?Qz Qcos (2nv.Z + IL).
Подставим вторую производную Oi и функцию Oi в (1.78): z
f — 4ft2vzQz 0 cos (2jiv7 4- 6.) 4- k.Q. Q cos (2nvЛ 4- 6.) = 0. (1.79a)
После преобразования (1.79 а) получим
v,= 1/2л . (L80)
Колебательное движение в многоатомных молекулах исходя из решения (1.78) можно разделить на валентные и деформационные.
25
Валентные колебания — это те, при которых в основном изменяется межъядерное расстояние. При деформационных колебаниях преимущественно изменяется угол между направлениями химических связей. Если в молекуле несколько колебаний совершается с одинаковой частотой и, следовательно, с одинаковой энергией, то такие колебания называют вырожденными. Рассмотрим в качестве примера колебания в трехатомной линейной и нелинейной молекулах
в пятиатомной тетраэдрической молекуле:
Vi — валентное симметричное; v2 — деформационное симметричное; — валентное асимметричное^ V4 — деформационное асимметричное; v2 — дважды вырожденные; v3, V4 — трижды вырожденные
(рис. 12). Линейные трехатомные молекулы имеют четыре степени свободы колебательного движения. При колебаниях vi и V3 (рис. 12, а) меняются только межъядерные расстояния. Валентные колебания бывают симметричные (vi) и асимметричные (V3). При колебании V2 изменяется только угол между направлениями связей. Это деформационное колебание. Оба колебания можно представить в виде колебаний с одинаковой частотой во взаимно перпендикулярных плоскостях.
Тетраэдрические пятиатомные молекулы обладают девятью степенями свободы колебательного движения. Однако наблюдается только четыре типа колебательного движения в таких молекулах (,рис. 13). Интенсивность полосы поглощения прямо пропорциональна квадрату производной электрического момента диполя по коле* /Aj, \2
бательной координате — . Сопоставление интенсивностей по*
\dq = o
лос поглощения приведено в табл. 2.
Таблица 2. Интенсивности полос поглощения пятиатомиых тетраэдрических молекул и вырождение колебаний
Тип колебания
Обозначение колебания
Вырождение
Op. \2 0# /<7=0
Интенсивность
Валентное симметричное
Деформационное симметричное
Валентное асимметричное
Деформационное асим-
метричное
Vi
V3
v4
Не наблюдается Слабая
Очень сильная
Сильная
Сопоставление колебательных спектров многих органических соединений, обладающих одинаковыми группами атомов СН3, СН2, ОНГ NH2 и т. д., показало, что в спектрах присутствуют одни и те же или мало отличающиеся друг от друга частоты. Некоторые частоты можно привести в соответствие с колебаниями ядер атомов в отдельных атомных группах. Такие частоты принято называть характеристическими. Обнаружение характеристических частот в спектре какого-либо органического соединения позволяет сделать вывод о наличии в нем соответствующих групп атомов. Этот принцип лежит в основе спектрального группового анализа органических и некоторых координационных соединений. Однако этот принцип не может быть применен к неорганическим соединениям, поскольку массы колеблющихся атомов и характер химических связей между атомами сильно различаются.
Электронные спектры поглощения многоатомных молекул. Электронные спектры поглощения многоатомных молекул представляют собой набор полос поглощения. Каждая полоса соответствует переходу между электронными уровнями. В соответствии с энергиями электронных переходов электронные спектры делятся на спектры в вакуумной ультрафиолетовой, в ультрафиолетовой и в видимой частях спектра (см. табл. 1).
Молекулярные орбитали (МО) делятся на а-, л- и и-орбитали. о-МО — симметричная относительно оси, связывающей атомы в молекуле. л-МО — несимметричная относительно оси молекулы; п — несвязывающая. Несвязывающая молекулярная орбиталь обычно наблюдается у тех молекул, у которых имеется сильно электроотрицательная группа атомов или атом. Энергия таких электронов близка к энергии соответствующей атомной орбитали. При поглощении молекулой кванта электромагнитного излучения происходит электронный переход со связывающей на незанятую разрыхляющую (о*- или л*-МО) или с несвязывающей на незанятую разрыхляющую (о*- или л*-МО) (рис. 14). Правило отбора соблюдается и в этом случае.
Предельные углеводороды имеют только о-МО. При поглощении электромагнитного излучения предельными углеводородами возможны только переходы о->о*. Непредельные углеводороды имеют
27
а- и л-МО. В спектрах поглощения наблюдаются наряду с переходы л->л*. Переходы л->л* требуют меньше энергии и полосы наблюдаются в более длинноволновой области спектра, чем а-> ->сг*-переходы. Если в соединении имеется гетероатом, обладающий высокой электроотрицательностью, то в спектре наблюдаются переходы n->a* при отсутствии в молекуле л-связей или переходы п->л*, если в соединении имеются л-связи. На переходы и тем более п->л* требуется меньше энергии, чем на переходы и л->л*. Поглощение в ультрафиолетовой и даже в видимой части спектра связано с переходами с несвязывающей МО на разрыхляю-
Рис. 14. Электронные переходы молекул при поглощении квантов излучения в видимой и ультрафиолетовой области спектра
Рис. 15. Расщепление МО при сопряжении л-связей
щие. Интенсивность полос поглощения, соответствующих переходам и п->л*, меньше интенсивности полос переходов
и л->л*. Наличие полос поглощения в ультрафиолетовом или в видимом участке спектра с низкими значениями коэффициентов погашения означает наличие в поглощающих молекулах несвязывающей МО и, следовательно, сильно электроотрицательного атома или группы атомов. Если в молекуле органического соединения происходит сопряжение л-связей, то л-связывающая и л-разрыхляющая МО расщепляются на дзе МО (рис. 15). Расщепление МО наблюдается тем больше, чем большее число л-связей сопряжено. При поглощении углеводородами или другими соединениями с сопряженными связями наблюдаются переходы (л—л)->(л* + л*). Полоса такого перехода смещена относительно полосы перехода л->л* в сторону больших длин волн или меньших энергий.
Спектры комбинационного рассеяния многоатомных молекул. Отличительной особенностью спектров комбинационного рассеяния (СКР) многоатомных молекул от спектров двухатомных молекул является то, что многоатомные молекулы обладают большим числом степеней свободы колебательного движения и, следовательно, в спектрах наблюдается большее число линий комбинационного рассеяния. Число степеней свободы колебательного движения атомов в молекулах определяется уравнением (1.68) или (1.69).
СКР имеет преимущество перед ИК спектрами поглощения, которое заключается в простоте устройства приборов. В данных приборах используются стеклянная оптика, более дешевые приемники и источники излучения. В качестве приемника излучения широко применяются фотоэлементы и фотоумножители. В качестве источника монохроматического излучения применяются оптические квантовые генераторы, дающие монохроматическое излучение высокой интенсивности, что значительно облегчает исследования СКР газообразных и твердых кристаллических соединений. При исследовании СКР растворов в качестве растворителя можно применять воду. Это открывает широкие возможности исследования структуры неорганических, координационных соединений, ионов в растворах..
У веществ в газообразном состоянии наблюдаются вращательно-колебательные СКР. Вращательно-колебательные СКР позволяют определять моменты инерции молекулы и межъядерные расстояния. Вращательные и вращательно-колебательные СКР отличаются от ИК вращательно-колебательных или ИК вращательных спектров поглощения тем, что правило отбора для вращательных и вращательно-колебательных СКР Д/=±2. Колебательные СКР многоатомных молекул дают возможность определять частоты колебаний атомов в молекуле.
5. ПРИМЕНЕНИЕ МОЛЕКУЛЯРНОЙ СПЕКТРОСКОПИИ ДЛЯ РАСЧЕТА ТЕРМОДИНАМИЧЕСКИХ ФУНКЦИЙ. ОСНОВЫ СТАТИСТИЧЕСКОЙ ТЕРМОДИНАМИКИ
>
Сумма по состояниям и термодинамические функции двухатомного газа в идеальном состоянии. Изучение строения молекул и развитие статистической физики привели к созданию нового метода расчета термодинамических функций. Этот метод позволяет вычис- лять значения относительной* внутренней энергии U—Uq, относительной энтальпии Н—Но, теплоемкости, энтропии и приведенных энергий Гельмгольца ~ .......и Гиббса - т для газообразных
веществ в широком интервале температур, включая и те температуры, при которых экспериментальное определение этих функций практически невозможно. Связь термодинамических функций с мо-
* Относительно внутренней энергии при О К.
29
пекулярными константами осуществляется через сумму по состояниям:
(L81)
где gi — статистический вес энергетического уровня (вырождение); Ei — энергия энергетического уровня; k —константа Больцмана.
Сумма по состояниям обладает свойством мультипликативности
7 — 7 7 7 7
-- ^ПОСТ^Вр^КОЛ^
(1.82)
Эл >
где /пост, ^вр, 2кол, Лш — поступательная, вращательная, колебательная и электронная сумма по состояниям.
Поступательная сумма по состояниям является функцией массы молекулы, как молекулярной константы, и параметров состояния объема и температуры или давления и температуры:
ПОСТ --
(2nmkT)3^ve
(1.83)
где т — масса молекулы; NA — постоянная Авогадро; h — постоянная Планка; v — объем; е — основание натурального логарифма.
Если массу молекулы выразить через молекулярную массу и сгруппировать молекулярные константы и параметры состояния, то (1.83) будет иметь вид
Z,I0C.r = , (1.84)
9 С 1 V Д
где М — молекулярная масса; v—молярный объем газа.
После подстановки в уравнение (1.84) постоянных величин в единицах СИ и логарифмирования получим
In Zn0CT = 3,4539 lg М 4- 3,4539 lg Т +2,3023 lg v + 6,7433. . (1.85)
Если же молярный объем газа в идеальном состоянии выразить через давление и температуру, то уравнение (1.85) преобразуется:
In ZnoCT = 3,45394g М + 5,7565 lg Т - 2,3026Jg р + 8,8612, (1.86)
где р— давление, Па.
Вращательная сумма по состояниям является функцией молекулярных констант: момента инерции, коэффициента симметрии молекулы и температуры как параметра состояния системы. Тогда
8л2& вр = “лГ
(Е87)
где I — момент инерции молекулы; а — коэффициент симметрии, равный 1 для гетероядерных двухатомных молекул и 2 для гомо-ядерных молекул.
30
После подстановки в (1.87) постоянных величин в единицах СИ и логарифмирования получим
InZBp = 2,3026 lgI — 2,30261g a +2,30261gT 4- 104,5265. (1.88)
Колебательная .сумма по состояниям является функцией молекулярной константы — колебательной постоянной молекулы сое—и параметра состояния — температуры. Тогда
^кОл — ‘ 0 -89)
1 — Q
Величина hc^e/k называется характеристической температурой 0. Если сое выражена в см”1, то
6 = (heIk) = 1,4388a>. (1.90)
Тогда
1 е^т
кол " 1 _ е-е/г ~ еут _ j
(1.91)
Электронная сумма по состояниям является функцией только молекулярной константы — вырождения нулевого электронного уровня
'Эл — ^О^эл •
(1.92)
у у -я ь
Вырождение нулевого электронного уровня определяется суммарным спином молекулы:
gOi3jI-2S + l. (1.93)
Внутренняя энергия изолированной системы равна сумме произведений энергии каждого энергетического уровня на число молекул на энергетическом уровне:
(1.94)
Если в (1.94) подставить
Ni =
»
то получим
w
I uT-u^\
—E;lkT NK l!
(1.96)
I
I
Z-1
Производная от суммы по состояниям (1.81) постоянном объеме имеет вид
i i
по температуре при
dZ дТ
kT2
Яа
RT*
X J z 1
Z-l
(Е97)
31
дТ
(=1
Сопоставление правой части уравнения (1.97) с правой частью уравнения (1.96) дает
ит - fZ0 = RT2 . (1.98)
\ / V
Поступательная составляющая внутренней энергии из (1.98) соответствует
= 1э5/гг. (1.99)
(If-p ^ЛОпос'г — £Л1ос'г — /?7*2
дТ
Вращательная составляющая внутренней энергии из (1.98) и (1.87) составляет
(Ur - /70)вр = £4Р = ЯГ. (1.Ю0)
Колебательная составляющая внутренней энергии описывается уравнением
(Цт - £/о)к0л = RT2 (д 'П/кол ) = ЛТ (I• 101)
\ сП /» 1 — е 1
Из уравнения (1.101) видно, что колебательная составляющая внутренней энергии является функцией только 9 и Т. Величины [Uт — U§ А п /т’
ПрИ различных значениях 0/7 рассчитаны и сведены \ 7 } ко ;|
в таблицы термодинамических функций Эйнштейна для линейного гармонического осциллятора. Электронная составляющая внутренней энергии равна нулю, поскольку вырождение нулевого электронного \ ровня не зависит от температуры. Внутренняя энергия одного моля газа в-идеальном состоянии равна сумме поступательной, вращательной и колебательной составляющих внутренней энергии:
Up — U§ -= Uиост 4- Z7Bp + (Up — б^д)кол. (1.102)
Теплоемкость при постоянном объеме равна производной внутренней энергии по температуре:
Ж \ д (UT - Z70) дТ ~ дТ
(1.103)
Теплоемкость газа равна сумме составляющих поступательной, вращательной и колебательной теплоемкости:
Cv = Спост 4- СВр 4- Скол* (1.104)
Из (1.99) следует, что поступательная составляющая теплоемкости
Сиост - 1,5/?. (1.105)
Вращательная составляющая теплоемкости из (1.100) выражается
Свр = 1,0/?. (1.106)
Колебательная составляющая теплоемкости описывается равенством
r _d(Up — 7?о)л0;| д
Кол -
(0/Т)е-^т I _ X <е/г)2
1- (ее/г_1)2 • (1-107)
дТ
32
Величины Скол = Се рассчитаны как функции 0/Г и сведены в таблицы термодинамических функций Эйнштейна для линейного гармонического осциллятора.
Теплоемкость при постоянном давлении рассчитывают по уравнению
Ср = Ср 4- (1.108)
Энтропия газа в идеальном состоянии как функция суммы по состояниям выражается уравнением
дТ
(1.109)
Так как энтропия есть функция давления,
то первый член уравнения (1.109) рассчитывают по (1.86) при заданном давлении, а второй— при постоянных молярном объеме газа, давлении и температуре. Если от левой и правой частей уравнения A — U—TS вычесть Uq, то получим
At — Uq = Ut-Uq — TS. (1.110)
После подстановки в (1.110) уравнений (1.109) и (1.98) имеем
Др
II
it-ж'
Энтропию и функцию ляющих:
рассчитывают как суммы состав-
пост
КОЛ I °Эл >
иг
ил 12)
(1.111)
ж
Т I
* /пост
вр
/Эл
(1.113)
1|
1
Колебательные составляющие энтропии и функции ———- определяют по таблицам термодинамических функций Эйнштейна для линейного гармонического осциллятора, электронные составляющие— по уравнению (1.92). Приведенную энергию Гиббса рассчитывают по уравнению
(1-114)
Ж
Ж
И :ж
Сумма по состояниям и термодинамические функции многоатомного газа. Поступательную сумму по состояниям многоатомного газа вычисляют аналогично поступательной сумме по состояниям двухатомного газа по уравнению (1.86). Вращательную сум-5 му по состояниям линейных многоатомных молекул рассчитывают также, как и для двухатомных молекул, по уравнению (1.87). Для нелинейных многоатомных молекул, обладающих тремя степенями 2—2083 33
свободы, вращательную сумму по состояниям вычисляют по уравнению
(1.115)
где Iy} Iz — главные моменты инерции относительно взаимно перпендикулярных осей вращения, проходящих через центр массы молекулы, кг-м2; а — коэффициент симметрии.
После подстановки постоянных величин в (1.115) и логарифмирования получим
In ZBp^ 1,15131g (/Д/2) 4-3,45391g Г-2,3026 1g а -р 157,3621. (1.116)
Колебательную сумму по состояниям многоатомных молекул вычисляют как произведение сумм по состояниям всех колебательных степеней свободы молекулы. Для каждой из них 2кол=/(9/П* Электронную сумму по состояниям вычисляют по уравнению (1.92), Для газов, состоящих из линейных многоатомных молекул, внутренняя энергия равна
ЗА"—’
(1.117)
для нелинейных многоатомных молекул
Uf — Z7q ~ 3,0.RT -J- 7*
ЗЛА-6
Тогда энтальпия равна
Нт - = UT - Z70 + RT.
(1.118)
(1.119)
Теплоемкость (многоатомного газа вычисляют суммированием составляющих теплоемкости по всем 3N степеням свободы:
(7^ -- б*поСТ "4“ б*вр “Г б*кол •
(1.120)
Поступательную составляющую теплоемкости для многоатомного газа вычисляют по уравнению (1.105). Для линейных многоатомных молекул, обладающих двумя степенями свободы вращения, вращательную составляющую теплоемкости рассчитывают по уравнению (1.106), для нелинейных — по уравнению
(1.121)
Колебательную составляющую теплоемкости вычисляют как сумму составляющих по всем колебательным степеням свободы, число которых определяют по уравнению (1.68) или (1.69).
Энтропию многоатомного газа рассчитывают, суммируя составляющие энтропии по всем 3N степеням свободы. Поступательную составляющую определяют аналогично тому, как для двухатомного газа. Вращательную составляющую энтропии рассчитывают по уравнению (1.109). Логарифм вращательной суммы по состоянию
34
и производную по температуре вычисляют по уравнению (I.I16) или (1.115).
Колебательную составляющую энтропии и функции - ~ -
вычисляют суммированием колебательных энтропий и функций —z—— по всем колебательным степеням свободы. Электронную составляющую энтропии и функции -- U° рассчитывают по (Вырождению нулевого электронного уровня, которое определяется на основании суммарного спина молекулы.
Функцию °т-~ вычисляют по уравнению (1.114).
6. ПРИНЦИП УСТРОЙСТВА СПЕКТРАЛЬНЫХ ПРИБОРОВ*
Универсальный монохроматор УМ-2. Принцип работы универсального монохроматора основан на разложении светового потока, прошедшего через кювету с исследуемым веществом или раствором, в спектр при помощи призмы. Световой поток от электрической лампочки 12 (рис. 16) направляется на конденсор с диафрагмой 11. Параллельный пучок светового потока проходит поочередно через кювету с раствором и кювету с растворителем. Далее пучок светового потока собирается конденсором 10 на входную щель <8, защищенную стеклом 9. Изображение входной щели проектируется объективом коллиматора 6 на призму 5, которая разлагает световой поток в спектр и изменяет его направление на 90°. Объективом камеры 4 спектр проектируется на выходную щель 3, вырезающую монохроматический участок. Монохроматический световой поток проходит
через защитное стекло выходной щели 2 и попадает на фотоэлемент с запирающим слоем 1. Фотоэлемент связан с зеркальным гальванометром отсчетного приспособления. Неподвижная прозрачная шкала, освещаемая с одной стороны лампочкой, отражается зеркальцем гальванометра через ряд увеличительных линз на матовое стекло, где и производится отсчет. Отсчетное приспособление содержит три шкалы: миллиметровую и две логарифмические.
16. Оптическая схема универсального монохроматора УМ-2
* Здесь описаны только те приборы, на которых могут быть выполнены лабораторные работы.
2* 35
Первая шкала разбита на 1000 делений, логарифмические — от —оо до + оо и от 0 до оо.
Призма 5 (рис. 16) поворачивается при вращении барабана 10 (рис. 17). При помощи индекса на выдвижной рейке 3 по спиральной шкале, разделенной на 3600 делений, можно установить такое положение призмы, при котором сквозь выходную щель будет проходить монохроматический световой поток с определенной длиной волны. Ножи входной щели (рис. 16), закрытой защитным стеклом 2 (рис. 17), находятся в фокальной плоскости объектива коллиматора 6. Фокусное расстояние объектива коллиматора зависит от длины волны, поэтому предусмотрена фокусировка коллиматора
Рис. 17. Универсальный монохроматор УМ-2
при помощи маховичка 9 (рис. 17) и шкалы 4 по неподвижному индексу. Установку барабана длин волн производят по калибровочной кривой или по заранее составленной таблице. Расстояние между ножами во входной и в выходной щелях изменяется вращением микрометрических барабанов 1 и 7 с ценой делений 0,01 мм. Уменьшение ширины выходной щели повышает монохроматичность светового потока (уменьшает интервал длин волн). В трубе коллиматора 5 (рис. 17) между щелью и объективом коллиматора 6 помещен затвор 7 >(см. рис. 16) для прекращения доступа светового потока в прибор. Затвор закрывается рукояткой 8 (рис. 17).
Для градуирования шкалы длин волн спектральных приборов, работающих в видимом участке спектра, обычно используют спектр излучения ртути. Спектр излучения ртути и таблица длин волн приведены в приложении.
В комплекте универсального монохроматора имеется ртутнокварцевая лампа СВДШ-250 с питающим устройством. Лампу СВДШ-250 устанавливают на оптической скамье монохроматора вместо источника излучения. Выходную щель заменяют трубой с окуляром. В поле зрения окуляра имеется индекс, относительно ко
36
торого устанавливается спектральная линия -спектра излучения ртути. Вращением барабана длин волн устанавливают определенную спектральную линию против индекса и в таблицу записывают длину волны линии ртути и отсчет по барабану длин волн. Для получения более четкого изображения спектральной линии следует повернуть -маховичок 9 (рис. 17). После того как закончено составление таблицы по всем спектральным линиям, для которых указаны длины волн, трубу с окуляром заменяют выходной щелью, за которой помещается фотоэлемент. Барабан длин волн устанавливают на линию ртути № 16 с Х=435,84 нм, ширина выходной щели должна быть 0,01 мм. Медленным вращением барабана в сторону меньших длин волн добиваются максимального показания миллиметровой шкалы отсчетного приспособления. Операцию повторяют несколько раз, каждый раз -отмечая показание шкалы длин волн. Если наблюдается расхождение с отсчетом по индексу окуляра, то вводится постоянная положительная или отрицательная поправка по шкале на все отсчеты для линий ртути.
Последовательность выполнения работы. Включить лампочку прибора, отсчетное приспособление и установить корректором шкалу на ноль. Установить начальную длину волны по таблице или по калибровочной кривой на барабане длин волн. На пути светового потока установить кювету с растворителем. Открыть затвор и сделать отсчет по шкале отсчетного приспособления. Закрыть затвор. Осторожно переместить столик с кюветами, поставив на пути светового потока кювету с раствором, открыть затвор и произвести отсчет по шкале отсчетного приспособления. Закрыть затвор. Изменить положение шкалы длин волн и произвести измерение интенсивности светового потока, сначала прошедшего через кювету с раствором, а затем — через кювету с растворителем. Такая последовательность измерений уменьшает число перемещений столика с кюветами. Аналогично снять показание шкалы отсчетного приспособления при всех заданных длинах волн.
По окончании измерений рассчитать оптические плотности раствора при всех длинах волн. В зависимости от того, какая применялась шкала (миллиметровая или логарифмическая), необходимо произвести, логарифмирование или вычитание. Результаты измерений и вычислений занести в таблицу по образцу:
Раствор . . концентрация, моль/л . . .; длина кюветы, см . . .
Показание барабана Длина волны, нм Показание отсчетного приспособления Оптическая плотность
ДЛЯ растворителя для раствора
г
37
Если оптическая плотность при некоторых длинах волн выходит за пределы 0,1—2,0, то следует изменить толщину поглощающего слоя, заменив кювету так, чтобы оптическая плотность была в указанных пределах. Произвести вновь измерения при этих длинах волн. По полученным оптическим плотностям рассчитать коэффициенты погашения по уравнению (1.3), приведенную оптическую плотность — делением оптической плотности на толщину поглощающего слоя. При этом выбирать те измерения, при которых получены оптические плотности раствора в пределах от 0,1 до 2,0.
Спектрофотометр СФ-4. Спектрофотометр СФ-4 предназначен для изучения спектров поглощения жидких веществ или растворов в области от 220 до 1100 нм. Однако дисперсия прибора позволяет надежно вести работу лишь в ультрафиолетовой части спектра.
Световой поток от источника света 1 (рис. 18) проектируется конденсором 2 и плоским зеркалом 5 на входную щель прибора 6. Изображение входной щели сферическим зеркалом 7 фокусируется на кварцевую призму 8 с зеркальной гранью. Световой поток, разложенный в спектр, отражается от зеркальной грани, вновь проходит через призму и проектируется сферическим зеркалом 7 на нижнюю часть щели 6, которая
вырезает из спектра монохроматический участок. При повороте призмы на плоскости выходной щели смещается изображение спектра, а выходная щель таким образом, выделит другой участок спектра. Монохроматический световой поток,
Рис. 18. Оптическая схема слектрофотомет- вышедший ИЗ щели, попада-ров СФ-4 и СФ-5 ет на кювету 4 с поглощаю-
щим веществом и далее на фотоэлемент 3. На пути монохроматического светового потока между щелью и кюветой помещен светофильтр из стекла УФС-2, который применяется при работе в области 320—380 нм или фильтр из стекла ОС-14 при работе в области 590—700 нм.
В качестве приемника излучения применяются фотоэлементы: сурьмяно-цезиевый при работе в области 220—650 нм и кислород-но-цезиевый при работе в области 600—1100 нм. Фототок от фотоэлементов усиливается усилителем постоянного тока и подается на потенциометрическое отсчетное приспособление.
Последовательность выполнения работы. Включить водородную лампу, для чего проверить, находятся ли выключатели электронного стабилизатора ЭПС-86 «накал» и «высокое напряжение» в положениях «выключено», повернуть рукоятку в центре пульта стабилизатора по 'стрелке влево до упора, включить стабилизатор в сеть напряжением 220 В и поставить выключатель «накал» в положение «включено». Через 2 мин повернуть выключатель «высокое напряже
ние» в положение «включено». При этом должна загореться водородная лампа.
Заполнить одинаковые кюветы исследуемым раствором и растворителем. У|Стано.В1йть кюветы в кюветную часть спектрофотометра. Для этого открыть крышку 13 (рис. 19), отжать прижимные пружины и установить в левую часть каретки кювету с исследуемым раствором, а в правую — кювету с растворителем. Закрыть крышку кюветного отделения. Установить переключатель 6 в положение «включено», поставить потенциометр 4 в сред-
Рис. 19. Спектрофотометр СФ-4, СФ-5
нее положение (примерно четыре оборота от любого крайнего положения) . Установить длину волны 220 нм рукояткой длин волн 7 по шкале 8.
‘ Примечание. Устанавливать длину волны следует только в сторону увеличения показания шкалы. Если шкала повернется случайно на больший угол, то следует вернуться на 10—15 делений и вновь аккуратно подвести индекс к требуемому делению шкалы.
Установить сурьмяно-цезиевый фотоэлемент. Движок «фотоэлемент» 1 должен при этом быть полностью выдвинут. При закрытой шторке фотоэлемента (рукоятка 14) вывести рукояткой «темновой ток» 5 стрелку миллиамперметра 11 в нулевое положение. Установить каретку ,с кюветами движком 2 в такое положение, чтобы на пути света находилась кювета с растворителем. Движок 2 при этом должен быть выдвинут до положения, отмеченного цифрой «2 к». Открыть шторку фотоэлемента, повернув рукоятку 14 в положение «открыто». Рукояткой 12 (щель) установить стрелку миллиамперметра Ив пулевое положение. Более точно установить нулевое положение стрелки миллиамперметра 11 рукояткой «потенциометр чувствительности» 4,
Установить каретку с кюветами в такое положение, чтобы световой поток проходил через кювету с веществом. Установить переключатель 6 в положение, обозначенное «XI», и вращением руко
39
ятки 9 отсчетного потенциометра установить стрелку миллиамперметра И вновь в нулевое положение. По верхней шкале 10 снять отсчет оптической плотности раствора. Если оптическая плотность раствора больше единицы, то переключатель 6 следует установить в положение «ХО, 1», и вновь повторить установку нуля рукояткой отсчетного потенциометра 9. При этом к значению оптической плотности, полученному по шкале 10, следует прибавить единицу. После измерения закрыть шторку 14 фотоэлемента. Если в положении «закрыто» стрелка длительное время стоит на нуле, то каждый раз закрывать шторку и проверять «темновой ток» не обязательно. При работе в области 320—380 нм следует устанавливать светофильтр УФС-2. Для этого выдвинуть движок 3 до положения, когда на рамке светофильтра будет отметка УФС-2.
После каждого измерения оптической плотности изменять установку длины волны и вновь производить измерение оптической плотности. По окончании измерений оптической плотности в заданном диапазоне длин волн рассчитать коэффициенты погашения. Результаты измерений записать в таблицу по образцу:
Раствор . . .; концентрация, моль/л . . длина кюветы, см . . .
Длина волны, нм
Светофильтр
Оптическая плотность
Коэффициент погашения
По измерениям оптических плотностей при разных длинах волн и расчету коэффициентов погашения построить график зависимости оптической плотности или коэффициента погашения от длины волны. Для построения спектра поглощения использовать только те измерения, при которых оптические плотности 'были в пределах от 0,1 до 2,0.
Спектрофотометр СФ-5. Спектрофотометр СФ-5 предназначен для изучения спектров поглощения жидких или твердых веществ в области спектра от 380 до 1100 нм. Устройство прибора и порядок работы на нем в основном те же, что и прибора СФ-4.
Источником излучения в спектрофотометре служит лампа накаливания, питающаяся через стабилизатор. При работе на спектрофотометре применяются для определенных областей спектра светофильтры: для области 480—600 нм—светофильтр ЖС-17, для области 600—900 нм —светофильтр КС-11, для области от 900 до 1100 нм —светофильтр ОС-14. В области от 380 до 480 нм следует работать без светофильтра. П|еремещение светофильтров осуществляется рукояткой 3 (рис. 19).
Спектрофотометр СФД-2 (СФД-2 м). Спектрофотометр СФД-2 предназначен для изучения спектров поглощения жидких веществ, растворов и веществ в твердом состоянии в области спектра от 220 до 1000 нм. В приборе установлено два источника излучения—•во
40
дородная лампа ДВС-25 для работы в области 220—380 нм и лампа накаливания для работы в области 380—1000 нм. Диспергирующим элементом в приборе служит дифракционная решетка (реплика) с числом штрихов 600 на 1 мм. Обратная дисперсия решетки составляет 3,2 нм/мм. В качестве приемников излучения служат два фотоэлемента: сурьмяно-цезиевый для работы в области 220— 650 нм и кислородно-цезиевый для работы в области 650—1000 нм. Фототок усиливается двухкаскадным усилителем постоянного тока, в электрическую схему которого введено отсчетное устройство. Питание прибора осуществляется через стабилизатор от сети переменного тока напряжением 220 В. Точность отсчета по длинам волн составляет ±0,3 нм, по шкале процента пропускания — ±0,5—1.%.
Световой поток от источника излучения 1 (рис. 20) попадает на эллиптический зеркальный конденсор наружного серебрения 2, который фокусирует изображение, отраженное плоским зеркалом 7,
Рис. 20. Оптическая схема спектрофотометра СФД-2м
на входную щель 9, защищенную кварцевой линзой 8. Далее световой поток попадает на сферический зеркальный объектив 10, который направляет светойой поток параллельным пучкам на дифракционную решетку 11.' От дифракционной решетки 11 световой лоток отражается вновь на зеркальный объектив 10 и проектируется на плоскости выходной щели в виде спектра. Выходная щель является продолжением входной щели и располагается ниже ее. За выходной щелью помещаются сменные светофильтры 6.
Монохроматический световой поток из выходной щели попадает на кювету с растворителем или на кювету с раствором 5. Из кюветы световой поток попадает на фотоэлемент 3. Перед фотоэлементом помещена шторка затвора 4.
Последовательность выполнения работы. Включить прибор в электросеть напряжением 220 В. Включить водородную лампу или лампу накаливания, как это описано на с. 38. Заполнить кюветы одинаковой длины растворителем и раствором и, открыв крышку кюветного отделения спектрофотометра, установить обе кюветы на подвижную каретку. Отметить положения штока каретки, при ко
41
торых на пути светового потока находятся кюветы с растворителем и с раствором. Вращением рукоятки длин волн установить по шкале начальную длину волны. Устанавливать длину волны следует только в сторону увеличения. Если случайно установилось большее деление, то следует вернуться на 10—20 делений шкалы и вновь подвести индекс к заданному делению шкалы.
Рукоятками (грубой и плавной) «темнового тока» установить стрелку миллиамперметра на условный нуль. Установить на пути светового потока кювету с растворителем или со стандартным раствором, открыть шторку затвора фотоэлемента, установив ее в положение «откр», и с помощью микрометрического винта подобрать такую ширину щели, чтобы стрелка миллиамперметра вновь установилась в положение условного нуля. Штоком каретки переместить кюветы так, чтобы на пути светового потока была кювета с раствором или с исследуемым веществом, переключателем ввести в рабочее положение отсчетный потенциометр и вращением рукоятки отсчетного потенциометра установить на условный нуль стрелку миллиамперметра. После этого снять показание со шкалы оптической плотности или шкалы процента пропускания. Далее изменить длину волны и повторить определение оптической плотности или процента пропускания.
Спектрофотометр СФ-16. Спектрофотометр СФ-16 предназначен для изучения спектров поглощения твердых, жидких веществ и растворов в области спектра 185—1100 нм. Принцип устройства и оптическая схема спектрофотометра СФ-16 не отличаются от СФ-4. Отличие заключается лишь в том, что в спектрофотометре СФ-IS вместо водородной установлена дейтериевая лампа, обеспечивающая работу в диапазоне длин волн 185—200 нм. При работе в области длин волн от 185 до 200 нм необходимо работать в атмосфере азота.
Последовательность выполнения работы. Включить прибор в электросеть напряжением 220 В. Перед включением тумблера «сеть» установить выключатели электронного стабилизатора «накал» и «высокое напряжение» в положение «выключено». Повернуть рукоятку в центре стабилизатора по стрелке влево до упора, Включить тумблер «сеть». Поставить выключатель «накал» в положение «включено». Через 2 мин повернуть выключатель «высокое напряжение» в положение «включено». При этом загорается дейтериевая лампа.
Заполнить две кюветы одинаковой толщины растворителем и раствором. Открыть крышку кюветного отделения 5 (рис. 21), перед этим проверить, стоит ли шторка 8 фотоэлемента в положении «закрыто». Установить на подвижную каретку кювету с растворителем и кювету с раствором. Кювету с растворителем следует установить так, чтобы на штоке 9 была пометка «1», кюветы с раствором— пометка «2». Плотно закрыть крышку кюветного отделе-ления 5. Поставить рукоятку 11 в среднее положение, переключатель чувствительности 12 — в положение «2». Установить рукояткой длин волн 16 по шкале 1 начальное деление, соответствующее
42
длине водны. Установку длины волны следует производить только от меньших значений к большим.
Установить на пути светового потока кювету с растворителем, а на штоке 9 каретки кювет в положение «1». Поставить переключатель измерительного потенциометра 14 в положение «выключено». При закрытой шторке 8 фотоэлемента скомпенсировать темновой ток сначала грубо рукояткой 13. затем более точно рукояткой 6 так, чтобы стрелка миллиамперметра 3 установилась на нуль. Открыть шторку 8 фотоэлемента и вращением рукоятки 10 установить стрелку миллиамперметра 3 вновь в положение «О». Это будет соответствовать условному нулю или 100% пропускания эталона. Пе-
рис. 21. Общий вид спектрофотометра СФ-16
реместить шток 9 каретки с кюветами в положение «2». При этом на пути светового потока находится кювета с исследуемым раствором. Установить переключатель 14 измерительного потенциометра в положение «1» и, вращая ручку потенциометра 15. установить стрелку миллиамперметра 3 на нуль. Отсчет по шкале 2 записать в таблицу против установленной длины волны. На шкале 2 можно отсчитывать процент пропускания и оптическую плотность.
Переключатель /4 установить в положение «выключено», переместить штоком 9 каретку с кюветами в положение «1». При этом стрелка миллиамперметра должна установиться на нуле. Если стрелка миллиамперметра не устанавливается на нуль, то измерение произведено неверно и его следует повторить. Если же измерение произведено верно, то рукояткой 16 установить следующее значение длины волны и произвести аналогично измерение оптической плотности.
При длине 'Волны 320 нм следует перейти от дейтериевой лампы к лампе накаливания. Переключатель зеркала источника светового потока находится на задней стенке осветителя 4. При длине волны 600 нм следует заменить приемник излучения сурьмяно-цезиевый фотоэлемент на кислородно-цезиевый. Для этого рукоятку 7 следует повернуть из положения с меткой «Ф» в положение «К». По окончании измерений закрыть шторку 8 фотоэлемента, открыть крышку кюветного отделения, вылить растворитель и раствор из кювет, вымыть кюветы. Установить зеркало источника излучения
43
в положение работы дейтериевой лампы, установить переключатель фотоэлементов 7 в положение «Ф», переключатель измерительного потенциометра 14 поставить в положение «выключено». Выключить тумблер «сеть» на стабилизаторе.
Спектрофотометр СФ-26. Спектрофотометр предназначен для изучения спектров поглощения жидких, твердых и газообразных веществ в диапазоне длин волн от 186 до 1100 нм. Погрешность измерения на спектрофотометре СФ-26 в области спектра от 190 до 1100 нм по шкале длин волн составляет не более 1%. Погрешность градуировки шкалы длин волн:
Длина волны, нм 186—300 300—350 350—400 400--550 550—1000 1000—1100 Погрешность 0,1 0,2 0,3 0,5 1,0 5,0
Среднее квадратическое отклонение коэффициентов пропускания не более 0,25% по шкале стрелочного прибора и не более 0,1 % по шкалам с повышенной чувствительностью.
В монохроматический поток излучения поочередно вводятся стандартный и исследуемый образцы. В качестве стандартного образца чаще всего -используется растворитель. При введении стандартного образца стрелка измерительного прибора устанавливается на 100% регулировкой ширины входной и выходной щелей прибора. Величину светового потока при этом принимают за 100% пропускания. При введении в световой поток исследуемого образца стрелка измерительного прибора отклоняется пропорционально изменению интенсивности светового потока. Шкала измерительного
Рис. 22. Оптическая схема спектрофотометра СФ-26
прибора градуирована в процентах пропускания и в единицах оптической плотности.
Световой поток от источника 1 (рис. 22) попадает на вогнутое зеркало 2 наружного серебрения. Зеркало 2 направляет световой поток на плоское зеркало 8 и дает изображение источника излучения на плоскости входной щели 10. Перед входной щелью находится линза 9. Световой поток проходит через входную щель 10 и попадает на зеркальный объектив 11, отразившись от которого параллельным пучком, направляется на кварцевую призму 7. В призме световой поток разлагается в спектр. После отражения от зад
44
ней алюминированной зеркальной поверхности световой поток, разложенный в спектр, вновь проходит через призму, в результате чего дисперсия „возрастает. Световой поток попадает на зеркальный объектив 11, который фокусирует изображение спектра на плоскости выходной щели 10. Выходная щель является продолжением входной щели и расположена она в верхней ее части.
Выходная щель вырезает монохроматический световой поток, который попадает на кювету со стандартным веществом или на кювету с исследуемым веществом 7. Далее монохроматический световой поток проходит через кварцевую линзу 6 и, отразившись от поворотного зеркала 4, попадает на один из фотоэлементов 3 или 5 в зависимости от участка спектра. При повороте призмы 12 изображение спектра смещается относительно неподвижной выходной щели 10, чем изменяется длина волны монохроматического светового потока.
В качестве источника излучения применяется дейтериевая лам-пи, которая работает в диапазоне длин волн от 186 до 350 нм, и лампа накаливания, которая работает от 340 до 1100 нм. В качестве приемника излучения применяется сурьмяно-цезиевый фотоэле-
1
Н 121! ЮЗ 8 7 6 5
Рис. 23. Общий вид спектрофотометра СФ-26
мент для диапазона спектра от 186 до 700 им и кислородно-цезие-вый фотоэлемент для диапазона от 600 до 1100 нм. Переход от одного к другому фотоэлементу следует при длине волны 685 нм.
Последовательность выполнения работы. В зависимости от диапазона длин волн установить рукоятку переключения 1 (рис. 23) в положение «Ф» или «К». Положение «Ф» означает, что включен сурьмяно-цезиевый фотоэлемент, положение «К» — кислородно-це-зиевый. Переключателем 1 необходимо установить источник излучения 3 — дейтериевую лампу или лампу накаливания. Закрыть шторку 6 фотоэлемента, поставив ее в положение «закр». Рукояткой 8 установить ширину щели 0,1. Включить тумблер «сеть» 15, при этом должны загореться сигнальная лампа «сеть» и сигнальная лампа «Д» 13 или «Н» 14 в соответствии с выбранным источни
ком излучения. Стабильная работа спектрофотометра обеспечивается через 1 ч после его включения.
Заполнить две кюветы одинаковой длины растворителем (стандартным раствором) или исследуемым раствором. Открыть крышку кюветного отделения 2 и установить кюветы с растворителем и раствором в держатель. Отметить положения на штоке 7, когда на пути светового потока будут находиться кюветы с раствором и растворителем. Плотно закрыть крышку кюветного отделения. Установить рукоятку 4 в рабочее положение «1». Вращением рукоятки, 12 в сторону увеличения отсчета длины волны по шкале 16 установить начальную длину волны. Штоком 7 установить на пути светового потока кювету с растворителем. Рукояткой 5 установить стрелку измерительного прибора 9 на нуле. Установить рукоятку 10 в положение «1». Открыть шторку 6 фотоэлемента, установив ее в положение «откр». Установить стрелку измерительного прибора 9 вращением рукоятки 8 (изменение ширины щели) в положение 100%. Штоком 7 установить на пути светового потока кювету с раствором и снять показания по шкале измерительного прибора 9. Записать процент пропускания или оптическую плотность против соответствующей длины волны. Штоком 7 вновь установить кювету с растворителем на пути светового потока. При этом стрелка измерительного прибора 9 должна вернуться к делению 100%.
Установить следующее значение длины волны рукояткой 12 и повторить измерение. Таким образом измерить поглощение во всем диапазоне длин волн. Для переключения дейтериевой лампы на лампу накаливания повернуть вогнутое зеркало осветителя рукояткой /. После минутного прогрева автоматически загораются лампа накаливания и индикатор «Н» 14,
Для измерения процента пропускания с повышенной чувствительностью установить штоком 7 кювету с растворителем, изменением ширины щели рукояткой 8 добиться показания измерительного прибора 100%, установить кювету с раствором и вращением рукоятки 11 добиться показания измерительного прибора меньше 10%. Установить рукоятку 10 в положение «Х0,1» и снять отсчет с измерительного прибора 9. Для получения процента пропускания отсчет на измерительном приборе умножить на 0,1 и прибавить число процентов, соответствующее положению рукоятки компенсатора 11, Например, отсчет по шкале измерительного прибора 82,6, положение рукоятки компенсатора «40». Процент пропускания будет 82,6-0,1+40=48,3%.
Для измерения процента пропускания от 0 до 10% установить рукоятку 10 в положение «КАЛИБР», установить на пути светового потока кювету с растворителем, изменением ширины щели рукояткой 8 подвести стрелку измерительного прибора к 100%, установить на пути светового потока кювету с раствором и вращением рукоятки 11 добиться по измерительному прибору отсчета меньше 1%. Установить рукоятку 10 в положение «ХОД». Полученный по шкале отсчет умножить на 0,01 и прибавить показание рукоятки компенсатора 11, умноженное на 0,10. Например, отсчет по шкале 46
измерительного прибора 34,9%, положение рукоятки И «20», тогда величина пропускания будет 34,9 -0,01+20-0,1 = 2,35% *
По окончании работы закрыть шторку 6 фотоэлемента, открыть крышку 2 кюветного отделения, вынуть кюветы, вылить из них содержимое и вымыть их. Выключить тумблер «сеть».
Спектрофотометр СФ-14 (СФ-10). Регистрирующий спектрофотометр СФ-14 предназначен для съемки спектров поглощения и спектров диффузного и зеркального отражения веществ, находящихся в жидком и твердом состояниях в диапазоне длин волн от 400 до 750 нм и автоматической записи спектра на градуированный бланк.
Спектрофотометр состоит из источника светового потока, двойного призменного монохроматора, фотометра поляризационного типа, приемно-усилительного и записывающего устройств. Оптическая схема прибора (рис. 24) состоит из спектральной и фотометри-
Рис. 24. Оптическая схема спектрофотометра СФ-10
ческой частей. Световой поток от кинопроекционной лампы 1 через конденсор 2 и входную щель 3 попадает на первый монохроматор. Входная щель 3 проектируется объективом коллиматора 4 на дисперсионную призму 5, где световой поток разлагается в спектр и изображение спектра проектируется объективом 6 на плоскости Л—Л. В плоскости Л—Л расположена промежуточная щель. Промежуточная щель состоит из зеркала 7, плоскость которого перпендикулярна плоскости Л—Л, и ножа щели S, находящегося в плоскости А—А. Промежуточная щель вырезает из полученного на плоскости Л—Л спектра узкий его участок, который объективом 9 проектируется на призму 10 второго монохроматора, где узкий
47
участок спектра вновь разлагается в спектр. Объективом И спектр проектируется на выходную щель 12 второго монохроматора. Выходная щель вырезает монохроматический участок спектра определенной длины волны. Монохроматический световой поток, вышедший из щели /2, пройдя через систему линз 13 и призм 14, разделяется двоякопреломляющей призмой 14 на два взаимно перпендикулярно поляризованных световых потока. Один из этих пучков поглощается диафрагмой 15, <а другой направляется на вторую двоякопрелом ляющую призму 16, которая разделяет его на два поляризованных световых потока одинаковой 1интенС|Ивности. Световые потоки под некоторым углом проходят через линзы 17, находящиеся внутри прерывателя 18. Прерыватель представляет собой цилиндр, вращающийся вокруг оси и имеющий два окна во взаимно перпендикулярных и перпендикулярных оси вращения направлениях. Прерыватель 18 поочередно перекрывает световые потоки с частотой 50 Гц.
Световой поток, прошедший через кюветы 19 и 20, отражается зеркалом 21 внутрь интегрирующей сферы 23. После многократного отражения от внутренней диффузно отражающей поверхности интегрирующей сферы 21 рассеянный световой поток попадает на фотоэлемент 22. Фотоэлемент 22 связан с усилителем переменного тока. Если исследуемое вещество поглощает излучение, то интенсивности световых потоков, прошедших через кюветы с растворителем и с раствором, будут разные, что даст пульсирующий ток от фотоэлемента на усилитель. Переменный сигнал усиливается и подается на обмотку электродвигателя, который через систему передач вращает призму 16 в фотометрической части прибора. Призма 16 ослабляет интенсивность светового потока, направляющегося в кювету с растворителем. Вращение призмы 16 происходит до тех пор, пока интенсивности обоих световых потоков не станут одинаковыми. При этом от фотоэлемента на усилитель переменного тока будет поступать постоянный ток, который не будет усиливаться.
Движение пера записывающего приспособления связано с вращением призмы 16, которое через систему кулачков смещается пропорционально оптической плотности или проценту поглощения. Смещение пера записывающего приспособления происходит параллельно оси цилиндра, на который помещается градуированный бланк для записи спектра. Цилиндр вращается с помощью мотора, и его вращение обеспечивает перемещение промежуточной щели в плоскости А—А. Таким образом, угол поворота цилиндра с градуированным по длинам волн бланком соответствует длине волны монохроматического светового потока, выходящего из выходной щели 12. С цилиндром связана шкала длин волн, расположенная справа относительно цилиндра. Шкала длин волн линейная и градуирована через 1 нм.
Бланк для регистрации спектра имеет шкалу длин волн, градуированную также через 1 нм, и две ординаты. Шкала оптической плотности градуирована через 0,010=1 мм. Шкала процента пропускания 1°/о = 1 мм. Скорость вращения цилиндра и, следователь-48
но, скорость перемещения промежуточной щели может быть измерена при помощи переключателя в средней части панели прибора. Продолжительность записи спектра может изменяться от 2 до 12 (мин. 'Обратная линейная дисперсия монохроматора для разных длин волн различна и составляет при 400 нм 1,6, при 500 нм 4,0, при 600 нм 7,8 и при 750 нм 16,5 нм/мм. У спектрофотометра СФ-14 имеется два диапазона измерения пропускания: 0—100% и 0 — 10% и два диапазона для измерения оптической плотности в пределах от 0 до 2,5 и от 0 до 1,0, что позволяет повысить чувствительность и точность измерений.
Последовательность выполнения работы. Включить прибор в электросеть напряжением 220 В. Поставить выключатель «сеть» в положение «включено». При этом загорается сигнальная лампа «сеть». Поставить выключатель «отработка» в положение «выключено». Открыть крышку записывающего устройства и поднять перо, выдвинув кнопку справа от цилиндра. Установить 400 нм на шкале длин волн вращением маховичка на корпусе прибора. Подготовить бланк, для чего обрезать ножницами его по линии отреза вдоль шкалы длин волн. Укрепить бланк на цилиндре, для этого откинуть прижимную планку, подвести бланк снизу цилиндра вниз сеткой, шкалой длин волн справа соединить концы бланка так, чтобы закрывались отверстия на цилиндре, прижать к цилиндру прижимную планку. Бланк должен быть установлен так, чтобы его правая сторона плотно прижималась к бортику цилиндра. Опустить перо так, чтобы оно касалось диаграммного бланка. Для этого нажать на кнопку справа от цилиндра. Вращая рукой цилиндр с диаграммным бланком, установить его так, чтобы перо касалось линии, соответствующей длине волны 400 нм. Открыть крышку кюветного отделения.
Внимание! Крышку кюветного отделения можно открывать только при выключенном тумблере «отработка». Несоблюдение этого правила приведет к выходу из строя фотоэлемента.
Заполнить две кюветы одинаковой толщины растворителем и установить их в кюветное отделение. Закрыть крышку кюветного отделения. Поставить последовательно выключатели «прерыватель» и «отработка» в положение «включено». Поставить выключатель «лампа» в положение «включено». При этом перо должно двигаться вправо до начала координат. Если перо двигается влево, то тумблер «изменение направления» переключить. Установить переключатель «скорость записи» в положение «4». Поставить тумблер «развертка спектра» в положение «включено», при этом цилиндр будет вращаться, а перо — вычерчивать линию, соответствующую нулевому поглощению. Выключить тумблер «лампа», как только перо дойдет до конца шкалы длин волн 750 нм. Поставить тумблер «развертка спектра» в положение «выключено». Вручную продолжить вращение цилиндра до установки по шкале 400 нм. Поставить тумблер «отработка» в положение «выключено», открыть крышку кюветного отделения и заменить в левой кювете растворитель на
49
раствор. Плотно закрыть крышку кюветного отделения, включить «отработка», переключатель скорости записи установить в заданное положение. Поставить выключатели «лампа» и затем «развертка спектра» в положение «включено». При этом перо запишет спектр исследуемого вещества. По окончании работы выключить «отработку», «прерыватель», открыть крышку кюветного отделения, вылить растворитель и раствор из кювет, вымыть,их и выключить тумблер «сеть». Снять бланк с цилиндра, для чего откинуть прижимную планку с диаграммного бланка.
Спектрограф КС-55. Спектрограф КС-55 предназначен для съемки спектров в области от 200 до 1000 нм на фотографическую пластинку. Прибор снабжен сменными кварцевыми и стеклянными призмами и объективами. Это позволяет фотографировать спектры излучения и поглощения в видимой и в ультрафиолетовой части его. Прибор обладает высокой обратной дисперсией. Величина обратной дисперсии при разных длинах волн для кварцевой и стеклянной оптики приведена в табл. 3.
Таблица 3. Обратная дисперсия спектрографа КС-55
Обратная дисперсия, нм/мм
Обратная дисперсия, нм/мм
Длина волны, нм
Кварцевая Стеклянная
призма призма
Длина волны, нм
Кварцевая Стеклянная
призма призма
200
250
300
350
0,12 0,25 0,46 0,72
0,36
400
500
600
1,15
2,10
3,40
0,55
1,20
2,15
Световой поток от источника излучения 9 (рис. 25) кварцевым конденсором 8 проектируется на конденсор 7 с диафрагмой. Конденсор 7 проектирует световой поток при помощи зеркала 6 на конденсор 5, помещенный на оправе входной щели прибора 4. Изображение освещенной снаружи входной щели отражается плоским зеркалом 11 и проектируется сменным объективом 12 на сменную призму 13, При двойном прохождении светового потока через призму с зеркальной задней гранью излучение разлагается в спектр, который проектируется объективом 12 на фотопластинку 10, Вследствие большого расстояния хода луча близко расположенные спектральные линии на фотографической пластинке получаются раздельно.
Для выполнения экспозиций за входной щелью прибора помещен затвор, который открывается при помощи рукоятки «затвор», расположенной на передней панели спектрографа справа от барабана длин волн. Для фотографирования на спектрограмму милли-
метровой шкалы последняя прижимается к фотографической пластинке поворотом маховичка против часовой стрелки до упора в правой части кассетного отделения. Миллиметровая шкала освещается лампочкой /, которая включается переключателем на панели при-
50
бора между двумя красными индикаторными лампочками. Для нанесения на спектрограмму шкалы длин волн имеется прозрачная шкала длин волн 2, участок которой, соответствующий длине волны в средней части спектрограммы, освещается лампочкой 3 и проектируется в центр спектрограммы. Лампочка 3 также включается переключателем, расположенным на панели прибора между красными индикаторными лампочками. При включении должна загореться соответствующая индикаторная лампочка. Перед входной щелью помещается диафрагма с фигурным или ступенчатым вырезом, служащая для фотографирования спектров сравнения рядом со спектром исследуемого вещества. На фотопластинке получается лишь небольшая часть спектра. Участок спектра устанавливается при по-
Рис. 25. Оптическая схема кварцево-стеклянного спектрографа КС-55
мощи барабана длин волн с двумя шкалами, предназначенными для стеклянной «С» и кварцевой «К» оптики. При вращении барабана длин волн каретка с призмой /3, объективом 12 и шкалой длин волн 2 перемещаются и одновременно поворачиваются призма 13 и кассетная часть спектрографа с фотопластинкой 10. Все это необходимо для фокусирования изображения спектра.
Спектрограф ИСП-51 с фотоэлектроприставкой ФЭП-1. Трех-призменный стеклянный спектрограф ИСП-51 применяется для получения спектров от источников со слабым излучением в видимой области спектра. Этот спектрограф применяется для регистрации спектров комбинационного рассеяния.
Прибор состоит из источника излучения, конденсора, спектро-графа и фотоэлектроприставки. Источник излучения (рис. 26) предназначен для облучения исследуемого вещества монохроматичес-ним световым потоком. Источник излучения смонтирован на рейтере, который крепится на оптической скамье. Корпус осветителя 2 rf представляет собой отливку сферической формы. Внутри имеется | полость эллиптической формы, в фокальных осях которой разме-I 51
щаются «ртутно-кварцевая лампа 1 и кювета 3. Внутренняя поверхность источника излучения хромирована, за счет чего максимум освещенности концентрируется на фокальной оси, где помещена ци-
Рис. 26. Источник излучения для съемки спектров комбинационного рассеяния
линдрическая часть кюветы с веществом. Ртутно-кварцевая лампа ПРК-2 разогревается до красного каления кварца. Кювета с испытуемым веществом располагается близко к ртутной лампе. Нагревание вещества при съемке спектра комбинационного рассеяния нежелательно, а в большинстве случаев даже недопустимо. Для поглощения теплового излучения между лампой и кюветой помещают тепловой фильтр 4 в виде рамки с двумя стеклами, между которыми протекает вода. Для охлаждения источника излучения внутрь его в специальные полости поступает проточная вода.
Монохроматический световой поток из спектра излучения ртути, состоящего из значительного количест-
Таблица 4. Характеристика светофильтров для выделения монохроматического светового потока из спектра ртути
Пропускание светового потока при длинах волн (нм), %
Номер светофильтра
578
546
436
405
365
57,0 0,34 0
о
78,1 0 0 0
0
0
43,8
2,07 0
0 1,75 31,6
0 о
о о 0,2
ва отдельных спектральных линий, выделяется стеклянным светофильтром, который помещается за тепловым фильтром. В комплекте имеются светофильтры, характеристики которых приведены в табл. 4.
Световой поток от ртутно-кварцевой лампы 4 (рис. 27) проходит через тепловой 3 и световой 2 фильтры и попадает на кювету 1
52
диафрагмой с фигурными вырезами.
Рис. 27. Оптическая схема спектрографа
ИСП-51
с исследуемым веществом. Излучение, рассеянное веществом, кон* денсируется линзой 1конденсора 12 на щель спектрографа 11. На оправе конденсора крепятся два раздвижных кожуха, .предотвращающих попадание светового потока из помещения в спектрограф. Ширина входной щели регулируется от 0 до 0,3 мм при помощи микрометрического винта с ценой деления 0,001 мм. Щель находится в фокальной плоскости объектива коллиматора 10. Высота щели ограничивается специальной Объектив коллиматора направляет световой поток на призменную систему 9. Призменная система состоит из трех призм. Световой поток, разложенный призмами в спектр, фокусируется объективами камеры 8 и 7 на плоскость выходной щели С за которой располагается фотоумножитель 5, Входная щель 6 вырезает из спектра монохроматический участок. Монохроматический световой поток превращается в электрический сигнал в фотоумножителе. Электрический сигнал усиливается примерно в
105 раз усилителем постоянного тока. Фотоумножитель, выходная щель, усилитель в комплекте представляют фотоэлектрическую приставку ФЭП-1. В приставке имеется мотор, который поворачивает столик с призмами 9. Синхронный мотор через редуктор с одной стороны поворачивает столик с призмами, с другой стороны устанавливает объектив камеры в фокальное положение относительно выходной щели.
Сигнал от усилителя постоянного тока подается на регистрирующее приспособление потенциометра. Диаграммная лента регистрирующего приспособления двигается синхронным мотором, перо потенциометра перемещается пропорционально величине сигнала. Таким образом производится регистрация спектра комбинационного рассеяния. Перед приемником излучения (фотоумножитель ФЭУ-17) помещается зеркало, с помощью которого можно закрыть световой поток.
Примечание. Попадание излучения на фотокатод в виде интенсивного рассеяния линий излучения ртутно-кварцевой лампы может испортить фотоумножитель. Для предотвращения этого служит зеркальная заслонка, на которой указаны диапазоны длин волн, при которых она должна быть закрыта.
Последовательность выполнения работы. Включить прибор и | ФЭП-1 в электросеть напряжением 220 В. Включить воду в тепло
53
вой фильтр и осветитель. Закрыть зеркальную заслонку перед фотоумножителем, включить ртутно-кварцевую лампу, для чего включить тумблер ПРК-2 на распределительном щите прибора и коротким нажатием кнопки конденсатора в середине распределительного щита — лампу. Включить тумблер «сеть» на ФЭП-1, при этом загорается красная индикаторная лампочка. Через 2—3 мин включить тумблер «высокое напряжение», при этом загорается индикаторная лампочка. Открыть дверцу потенциометра и включить потенциометр. Включить тумблер «диаграмма». Установить начальное деление шкалы на барабане длин волн. Для этого при1 легком покачивании барабана длин волн левой рукой вывести из зацепления редуктор, переместив его рычаг в левое положение. Вращая рукой барабан длин волн, установить начальное деление. Установить кювету с веществом в осветитель и убедиться в устойчивом режиме работы ПРК-2. Ток и напряжение должны оставаться постоянными. Рукояткой «темновой ток» на ФЭП-1 установить перо потенциометра на делении 1. Открыть зеркальную заслонку перед приемником излучения. Рукояткой «чувствительность» установить перо на делении 3—4. Включить тумблер «диаграмма» на потенциометре. Поставить тумблер около барабана длин волн в положение «вкл», при этом должно начаться вращение барабана длин волн. Периодически, через определенное число делений барабана длин волн, .перо потенциометра резко смещается влево и возвращается в прежнее положение. Так фиксируется шкала длин волн на диаграммной ленте. Перо записывает спектр комбинационного рассеяния вещества, помещенного в кювету.
Градуировка шкалы прибора. Градуировку шкалы прибора производят по спектру комбинационного рассеяния вещества, у которого известны волновые числа линий комбинационного рассеяния. Съемку спектра см. на с. 53. Деления барабана длин волн определяют для всех линий спектра комбинационного рассеяния, волновые числа которых известны, (Интерполяцией между реперными отметками на диаграммной ленте, которые автоматически отмечаются при съемке спектра комбинационного рассеяния. На основании данных о волновых числах и делениях барабана длин волн строят градуировочный график или методом наименьших квадратов определяют коэффициенты зависимости волнового числа от деления барабана длин волн. Такой график применим для определения волновых чисел линий комбинационного рассеяния в спектре, снятом пр и любой скорости регистр ации.
Спектрофотометр ИКС-14. Двухлучевой спектрофотометр ИКС-14 предназначен для изучения спектров поглощения жидких, твердых и газообразных веществ в диапазоне волновых чисел от 400 до 13 300 см"1. Для обеспечения такой широкой области спектра в комплект входят четыре сменные призмы. Материал призм и диапазон волновых чисел применения приведены ниже:
Материал призмы КВг NaCl LiF Стекло Ф-1
Диапазон работы, см"1 4.00—670 650—2000 1820—5000 3850—13300
54
Время записи полного спектра (млн): 7, 22, 65, 200, 580, 990. Воспроизводимость пропускания от 10 до 100% составляет ±0,5%. Разрешающая способность в области 1000 см-1 составляет 3—4 см-1. Спектрофотометр состоит из двухлучевого источника излучения I, монохроматора II, усилителя /// и записывающего приспособления IV (рис. 28, а). Инфракрасное излучение от глобара 1 (силитового стержня) (рис. 28, б), нагретого до 1300— 1400°С, направляется на два плоских зер-
6 7 3 9 10 11 12- 13 1ч- 15
Рис. 28. Общий вид
(а)
и оптическая ИКС-14 (б)
схема спектрофотометра
кала 3 и 24, отразившись от которых, попадает на сферические зеркала 2 и 25. Сферические зеркала фокусируют изображение источника излучения на плоскость, где помещены линзы коллективы 5 и 22. Световой поток I проходит через кювету 4 с изучаемым веществом. Световой поток II — через кювету 23 сравнения. Световой поток I, отразившись от плоских зеркал 6 и 7, попадает на сферическое зеркало 8, которое направляет его на другое сферическое зеркало 19. Между зеркалами 8 и 19 находится прерыватель Р, вы
полненный в виде вращающегося диска с секторами, поверхности которого имеют наружное серебрение. Прерыватель 9 вращается с частотой вращения 4,5 об/с. Наличие двух секторов обеспечивает частоту прерывания светового потока 9 Гц. В то время, когда прерыватель перекрывает световой поток I, он отражает световой лоток II на то же сферическое зеркало 19, которое фокусирует изображение после отражения от плоского зеркала 20 через линзу 10 на плоскости входной щели 13. Входная щель автоматически изменяется от 0,01 до 3 мм в зависимости от диапазона длин волн спектра. Перед входной щелью помещена линза 18. Изображение входной щели параболическим зеркалом 15 направляется параллельным пучком на дисперсионную призму 16, при прохождении которой излучение разлагается в спектр и направляется на зеркало Литтрова 17. Отразившись от плоского зеркала 17, световой поток вновь проходит через призму 16, при этом возрастает дисперсия. Параболическое зеркало 15 фокусирует изображение входной щели 18, разложенное в спектр, на плоскость выходной щели 11. Выходная щель вырезает из изображения спектра монохроматический световой поток. Монохроматическое излучение отражается плоским зеркалом 12 и концентрируется эллиптическим зеркалом 14 на приемник инфракрасного излучения 13. При повороте зеркала 17 изображение спектра на выходной щели 11 перемещается, что приводит к изменению длины волны или волнового числа монохроматического светового потока.
Сигнал от приемника излучения направляется на усилитель переменного тока. Если на пути светового потока I поместить поглощающее вещество, то сигнал будет переменным. Усиленный ток подается на обмотку электродвигателя, который перемещает фотометрический клин 21, ослабляющий интенсивность светового потока II до такой степени, когда на приемник излучения будет попадать световой поток постоянной интенсивности, который будет вызывать постоянный ток в приемнике излучения и не будет усиливаться усилителем переменного тока. Движение фотометрического клина связано с перемещением пера регистрирующего приспособления. Запись спектра осуществляется на диаграммной ленте, которая двигается от синхронного мотора. Синхронным же мотором поворачивается и зеркало 17. Поворот зеркала 17 и движение диаграммной ленты осуществляются через редукторы, что позволяет изменять скорость развертки спектра (скорость сканирования) и скорость записи спектра.
Спектрофотометр ИКС-14 имеет равномерно градуированную шкалу на 2000 делений. Это требует предварительного построения калибровочной кривой зависимости делений шкалы от волновых чисел.
Последовательность выполнения работы. Проверить все тумблеры на приборе, которые должны стоять в положении «выключено». Включить питание прибора в электросеть напряжением 220 В. Включить тумблер «сеть» на усилителе и дать прогреться прибору примерно 20 мин. Пустить воду для охлаждения источника излуче-56
I
X-;
ЯвдЕй!;* г
v. ния. Включить источник излучения, выключатель которого находится в левой части основания прибора. Включить мотор отработки и мотор прерывателя. При включении мотора и прерыва^е-; ля необходимо нажать пусковую кнопку. Снять заслонки с осветителя и монохроматора для двух световых потоков I и II. При этом перо записывающего приспособления должно установиться на 100% пропускания. Поставить заслонку на пути светового потока I. Перо должно перемещаться влево и установиться на 0%.
Для построения калибровочной кривой на пути светового потока I помещают пленку из полистирола толщиной 0,025 мм -в картонной оправе. Спектр полистирола см. в приложении. Поставить переключатель скорости развертки спектра на неоцифрованное деление. При перемещении движка редуктора следует левой рукой слегка покачивать 'барабан длин волн. Установить примерно 10 делений до начального значения по барабану длин волн. Установить переключатель скоростей развертки спектра на заданное деление. Включить тумблер развертки спектра, при этом необходимо одновременно нажать на пусковую кнопку. Выключить тумблер, когда шкала длин волн установится на начальное деление. Установить заданную скорость движения диаграммной ленты переключателем скорости, расположенным в левой части прибора. При этом правой рукой следует слепка покачивать маховичок (ведущего валика диаграммной ленты, а левой рукой устанавливать «рычажок редуктора. Включить тумблер мотора записи спектра, нажав одновременно на пусковую кнопку. При движении диаграммной ленты перо будет записывать 100% пропускания. Когда перо совпадает с поперечной линией на диаграммной ленте, выключить мотор движения диаграммной _ленты. Закрыть заслонкой световой поток I. Перо установится на 0% пропускания и прочертит линию начала спектра. Линия начала спектра соответствует точно установленному делению на шкале длин волн. Место, где остановится перо, отметить карандашом и записать на диаграммной ленте скорость сканирования, скорость движения диаграммной ленты, начальное деление шкалы длин волн. Вынуть заслонку и вместо нее установить пленку из полистирола. Когда перо остановится, включить одновременно тумблер регистрации спектра и движения диаграммной ленты, нажав при этом обе пусковые кнопки. Следить за записью спектра. По достижении заданного конечного деления барабана длин волн выключить одновременно тумблеры развертки спектра и диаграммной ленты. Заменить пленку полистирола на заслонку. При этом перо прочертит линию конца спектра. Эта линия соответствует конечному делению барабана длин волн. Перо остановится на 0% пропускания. Записать у линии конца спектра деление барабана длин волн. Включить тумблер и нажать пусковую кнопку движения диаграммной ленты. Перо запишет 0% пропускания. Примерно через 5 мм движения диаграммы вынуть заслонку. Перо будет двигать-ся к 100% пропускания и через некоторое время запишет прямую линию, соответствующую 100%.
57
Выключить мотор диаграммной ленты, вывести из зацепления редуктор движения диаграммы. Вращая вручную маховичок справа от ведущего валика диаграммы, подвинуть ленту так, чтобы ее можно было бы отрезать. Построить калибровочную кривую зависимости делений шкалы от волновых чисел.
Записать волновые числа против максимумов соответствующих полос поглощения (см. спектр и табл, в приложении). Определить деление шкалы барабана длин волн, измерив миллиметровой линейкой расстояние от линии начала спектра до линии максимума поглощения. Это расстояние соответствует числу делений шкалы в 1 мм. Построить график зависимости волнового числа от деления шкалы или методом наименьших квадратов определить по программе ЭВМ (см. приложение) коэффициенты а и b в уравнении
v = а 4- Ьп,
где v — волновое число; п — деление шкалы "барабана длин волн.
Порядок съемки спектра поглощения. Порядок съемки спектра поглощения исследуемого вещества совершенно аналогичен порядку съемки спектра полистирола. Вместо пленки из полистирола установить кювету с исследуемым веществом. Если исследуемое вещество газообразное, то в комплекте прибора .имеются специальные кюветы. В один из световых потоков ставится кювета сравнения. Если нет кюветы с совершенно одинаковыми оптическими свойствами, то можно кювету не ставить. Если вещество жидкое, то его следует поместить между окнами, прозрачными для исследуемого участка спектра. Если исследуется раствор, то в луч сравнения II для учета поглощения излучения молекулами растворителя поместить кювету с растворителем, причем толщина поглощающего слоя должна быть аналогична толщине поглощающего слоя раствора. Твердые вещества снимаются в виде суспензии в вазелиновом масле или в виде таблетки, спрессованной с бромидом калия.
Спектрофотометр ИКС-21. Прибор предназначен для изучения спектров поглощения в области волновых чисел спектра от 667 до 5000 см”1. Если на приборе установлена призма, изготовленная из CsI, то обеспечивается работа в области волновых чисел от 200 до 500 см-1. Прибор работает по однолучевой схеме. Световой поток от источника инфракрасного излучения (силитовый стержень 1) (рис. 29), нагреваемого до 1300°С, направляется на защитное стекло 5. Между глобаром и защитным стеклом находится модулятор 2 с частотой прерывания потока излучения 9 Гц. Отразившись от плоского зеркала 4, излучение направляется на сферическое зеркало 19, отразившись от которого, проходит через защитное окно 18т кювету с исследуемым веществом 17, защитное окно щели 16 и входную щель 5. Перед входной щелью установлена диафрагма 15. Излучение, прошедшее через входную щель, попадает на параболическое зеркало 11, разлагается в спектр призмой 12, отражается от плоского зеркала Литтрова 13, вновь проходит через призму и, отразившись от сферического зеркала 11 и плоского зеркала 14„ 58
проектируется на плоскости выходной щели 7. Перед выходной щелью помещена диафрагма 8,
Выходная щель вырезает из спектра монохроматическое излучение, которое плоским зеркалом 6 и эллиптическим зеркалом 10 фокусируется .на приемник излучения 9. Электрический сигнал усиливается усилителем переменного тока и регистрируется потенциометром ЭПП-09. В результате поворота зеркала Литтрова 13 изображение спектра смещается относительно неподвижной выходной щели. При этом изменяется волновое число монохроматического светового потока, попадающего на приемник излучения. Зеркало
Рис. 29. Оптическая схема инфракрасного спектрофотометра
ИКС-21
Литтрова вращается синхронным мотором. Этот же мотор двигает диаграммную ленту на потенциометре ЭПП-09. Скорость развертки спектра может изменяться переключателем на панели прибора. Шкала длин волн равномерная и имеет 2000 делений. Для изучения спектров поглощения шкала предварительно должна быть прокалибрована п о спектру полистирола (см. приложение) .
Спектрофотометр ИКС-22. Спектрофотометр предназначен для изучения ИК-спектров поглощения в области волновых чисел от 650 до 5000 см"1. Прибор работает по двухлучевой схеме. Запись спектра производится на калиброванном бумажном бланке. На оси абсцисс отложена шкала волновых чисел (см-1), на оси ординат — процент пропускания. Точность градуировки шкалы волновых чисел при 1000 см-1 составляет ±5 см-1. Воспроизводимость по шкале пропускания ±1,5%. Разрешающая способность прибора 3— 4 см-1. Конструкция прибора позволяет регистрировать спектр с двумя скоростями. Полный спектр регистрируется в течение 15 или 120 мин.
Излучение от источника ИК-излучения (силитового стержня) глобара 1 (рис. 30) зеркалами 2, 3, 22, 23 направляется на кювету с исследуемым веществом 4 и кювету сравнения 2L Световой поток 1 отражается от зеркал 19 и 20 и направляется на модулятор 18, который выполнен в виде вращающегося диска с двумя секторами. Частота модуляции 9 Гц. Пройдя через сектор модулятора»
59
излучение попадает на сферическое зеркало 7. Световой поток II отражается от плоского зеркала 6 или проходит через сектор модулятора, или отражается от модулятора на зеркало 7. Излучение от зеркала 7 распространяется последовательно с чатотой 9 Гц (прошедшее через кювету с исследуемым веществом и через кювету сравнения).
Сферическое зеркало 7 направляет излучение на плоское зеркало 17 и на входную щель 16. Далее параболическое зеркало 13 на-
Рис. 30. Оптическая схема спектрофотометра ИКС-22
правляет его на призму 14, где излучение разлагается в спектр. Отразившись от зеркала Литтрова 15, световой поток вновь проходит через призму 14 и параболическим зеркалом 13 направляется на выходную щель 10. Из спектра вырезается монохроматическое излучение, которое отражается от зеркала 9 и попадает на вращающееся зеркало 8. Эллиптическое зеркало 12 фокусирует изображение выходной щели на приемник излучения 11.
Если исследуемое вещество поглощает излучение, то на приемник излучения будет попадать световой поток, пульсирующий с частотой 9 Гц. Это приведет к пульсирующему изменению сигнала от приемника излучения. Сигнал усиливается усилителем переменно-ного тока ЗУ-1 или ЭУ-2 и подается на обмотку электродвигателя, который перемещает оптический клин 5 в световом потоке II до тех пор, пока сигнал не станет постоянным, т. е. когда интенсивности световых потоков, прошедших через кювету с исследуемым веществом и кювету сравнения, не станут одинаковыми. Вместе с перемещением оптического клина 5 перемещается и перо записывающего приспособления. От синхронного мотора через редуктор смещается платформа с диаграммой. Перемещение диаграммы связано с поворотом зеркала Литтрова 15. Таким образом, смещение диаграммы пропорционально волновому числу, а перемещение пера — проценту пропускания.
60
Порядок работы на приборе. Прибор ИКС-22 для термостатиро-вания монохром,агора постоянно включен в электросеть напряжением 220 В. Включить правый тумблер на блоке питания, моторы отработки и модулятора (тумблеры находятся на панели прибора). Пустить воду для охлаждения*1ИСточн1И1Ка излучения. Вынуть четыре заслонки с осветителя и монохроматора. Включить источник излучения, установив средний переключатель на блоке питания в заданное положение. Закрепить диаграмму на платформе, для чего подвести бланк под приспособление с пером. Перо при этом должно быть приподнято, шкала волновых чисел на бланке должна быть со стороны наблюдателя.
Установить на шкале волновых чисел деление 4000 см-1. Если на шкале деление менее 4000 см"1, то третий тумблер слева установить в положение «обратный ход», включить правый тумблер и выключить его при показании шкалы более чем 4000 см-1. Включить тумблер «рабочий ход» и правый тумблер, выключить его, когда показание шкалы будет точно 4000 см-1. Согласовать диаграмму со шкалой прибора, для чего второй тумблер слева «масштаб» установить в нейтральное вертикальное положение. Маховичком, расположенным на основании прибора, установить платформу с диаграммой так, чтобы перо в опущенном положении показывало точно 4000 ом-1. Тумблер «масштаб» установить в положение «I». Поместить на пути светового потока I заслонку. При этом перо двигается влево и остановится на 0%., пропускания. Вынуть заслонку, перо двигается вправо и остановится на 100% пропускания. При движении перо должно прочертить линию начала съемки спектра. Поместить в прибор кювету с исследуемым веществом и кювету сравнения (кювету сравнения можно не ставить, если исследуется газ, твердое или чистое жидкое вещество). Когда перо остановится, включить тумблер слева на панели в положение «быстро» и тумблер справа развертки спектра в положение «вкл». Запись спектра началась. При 650 см-1 запись спектра автоматически прекращается. Поднять перо, поставить третий тумблер слева в положение «обратный ход», включить тумблер развертки спектра и установить по шкале 4000 см™1. Прибор подготовлен к съемке второго спектра. Второй спектр можно снять на том же бланке. По окончании съемки спектров выключить прибор. Выключить мотор отработки, модулятора, источник излучения и правый тумблер на блоке питания. Через некоторое время прекратить подачу воды.
Спектрометр UR-10. Спектрометр UR-10 — автоматически регистрирующий двухлучевой, призменный прибор для изучения ИК<пекторов поглощения твердых, жидких и газообразных веществ в области волновых чисел от 400 до 5000 см-1. Запись спектра осуществляется на специальной бумаге с восковым слоем, на которой вместе со спектром наносится сетка, градуированная в волновых числах через 10 см"1 и процентах пропускания через 2%.
Прибор выполнен в виде стола, на котором в массивном литом корпусе помещаются осветитель и монохроматор. Передняя стенка монохроматора представляет пульт управления прибором. Управ-
61
ление прибором программированное. В левой тумбе стола помещается блок питания прибора. Пульт управления блока питания расположен на передней стенке левой тумбы. На пульте имеются выключатели прибора, кондиционера монохроматора и источника инфракрасного излучения. Там же расположены предохранители и амперметр для измерения силы тока источника излучения. В правой тумбе стола размещена усилительная схема прибора и замедлитель, который регулирует скорость записи спектра при резком изменении поглощения. На передней панели правой тумбы выведены выключатели усилителя и замедлителя и рукоятки установки режима работы усилителя и замедлителя. В левой части прибора расположен источник излучения —силитовый стержень, нагревающийся до 1300°С. К этой части прибора примыкает кюветное отделение, закрывающееся двумя металлическими шторками. В центральной части прибора под круглой крышкой на вращающемся столике расположены три призмы, выполненные из монокристаллов КВг, NaCl и LiF. Призмы имеют рабочий диапазон волновых чисел, который определяется дисперсией материала призмы и прозрачностью:
Призма КВг NaCl LiF
Av, см’1 400—70'0: 700—1800 1800—500-0
Для предохранения призм и некоторых деталей оптической схемы прибора от действия влаги атмосферного воздуха прибор снабжен постоянно работающим кондиционером, который осушает воздух, поглощает из него СО2, подопревает воздух до некоторой постоянной температуры и поддерживает постоянной дисперсию прибора,
В правой части прибора под прозрачной крышкой находится записывающее устройство прибора. Запись спектра осуществляется агатовым тонким стержнем. Одновременно с записью спектра на диаграммную ленту наносится гребенкой из набора агатовых стержней, координатная сетка. На левом крае ленты печатается значение волновых чисел через интервал 200 см-1 и обозначение призмы, использованной при съемке спектра. Например, АН6 означает призма NaCl, волновое число 1600 см-1; К7— призма КВг, волновое число 700 см-1; L28 — призма LiF, волновое число 2800 см-1.
Прибор может записывать как весь спектр от 400 до 5000 см”1, так и отдельные его участки. Под шкалой волновых чисел на круглом диске в центре прибора расположено программное устройство. Вдвигая узкие пластинки, можно исключить из спектра соответствующий участок. Эти участки спектра не регистрируются. Волновое число, соответствующее каждому моменту времени записи спектра, .можно наблюдать через окуляр, расположенный ib средней ч а ст и м о н ох ром а тор а.
Оптическая схема спектрометра UR-10 представлена на рис. 31, Инфракрасное излучение от силитового стержня 1 направляется двумя зеркалами 2 и 22 на кювету с поглощающим веществом 3 и кювету сравнения 21, Зеркалами 4 и 18 оба световых потока на-62
пр являются на сферическое зеркало 8. Зеркало 4 вращается вокруг оси ,и имеет два .вырезанных сектора. Это зеркало пропускает поочередно на сферическое зеркало 8 то световой поток, прошедший через кювету сравнения, то световой поток, прошедший через кювету с исследуемым веществом. Сферическое зеркало 8 находится в фокусе источника излучения, поэтому оно направляет световой поток параллельным пучком на призму 7, которая вместе с двумя другими призмами находится на вращающемся столике 6.
Излучение, разложенное призмой 7 в спектр, отражается плоским зеркалом 17 и вторично проходит через призму 7. Сферическое
Рис. 31. Оптическая схема инфракрасного спектрометра UR-10
зеркало 8 отражает излучение на плоское зеркало 5 и изображение спектра получается на плоскости, где помещена неподвижная выходная щель 9. Через выходную щель проходит монохроматический участок спектра, который сферическим зеркалом 11 фокусируется на термоэлемент 10. Поскольку зеркало 4 поочередно пропускает потоки инфракрасного излучения разной интенсивности при поглощении его веществом, то в термоэлементе возникает пульсирующий ток, который подается на усилитель переменного тока 12. Увеличенное напряжение от усилителя 12 подается на мотор 16, который через механический привод 20 вращает оптический клин ' 19, ослабляющий поток излучения, прошедший через кювету сравнения 21, до интенсивности потока излучения, прошедшего через кювету с исследуемым веществом. При равенстве интенсивностей светового потока усилитель переменного тока не будет усиливать термоток. При этом напряжение на моторе станет равным нулю. Вместе с движением оптического клина 19 приспособлением 15 перемещается перо 13. Диаграммная лента 14 двигается синхронно с поворотом зеркала 17. Таким образом производится запись спектра. Если необходима смена призмы, то столик с тремя призмами быстро поворачивается. При быстром вращении зеркала 17 или столика призм 6 движения диаграммной ленты не происходит.
Последовательность выполнения работы. Пустить воду для охлаждения источника инфракрасного излучения, включить последо-
63
вательно выключатели «горелка», «усилитель», «маркировка», «предварительный разделитель», «спектрометр». Поместить кювету с исследуемым веществом .в кюветное отделение в положение 3 (рис. 31). Если есть необходимость, то в положение 21 поместить кювету сравнения, включить выключатель «текущее число волн» после остановки пера записывающего устройства. По окончании записи спектра выключить прибор. Выключать прибор строго в обратном порядке.
Компаратор ИЗА-2. Компаратор ИЗА-2 предназначен для точного измерения расстояний между спектральными линиями в спектрах, снятых на фотографическую пластинку. Точность определения расстояния составляет 0,0002 мм. Измерение расстояния между спектральными линиями -эталонного и исследуемого спектров дает возможность определять длины волн, волновые числа или частоты спектральных линий в спектрах излучения и комбинационного рассеяния.
Принцип работы компаратора основан на том, что расстояние между спектральными линиями на фотопластинке сравнивают со шкалой. Сравнение производится при помощи двух жестко связанных микроскопов. Левый микроскоп имеет в поле зрения окуляра перекрестие, которое наводится на исследуемую линию в спектре. Спектрограмма помещается на столике, она освещается зеркалом, расположенным под столиком; четкое изображение спектра обеспечивается маховичком наводки фокуса, а вращением муфты окуляра — четкое изображение перекрестия.
Столик компаратора может свободно перемещаться. Если необходимо незначительное перемещение, следует закрепить винт в левой нижней, части столика. После закрепления стопорного винта перемещение столика может производиться микрометрическим винтом, расположенным в правой части компаратора. Микрометрический винт может перемещать столик на 9 мм, поэтому им следует пользоваться только при точной наводке на линию, а перемещение от линии к линии производится смещением столика рукой. Правый микроскоп является отсчетным. Он жестко установлен на стойке станины и направлен на миллиметровую шкалу, нанесенную на матовое стекло и жестко скрепленную со столиком компаратора. Шкала освещается снизу зеркалом. В поле зрения микроскопа видны вертикальные штрихи с цифрами вверху (рис. 32). Это — увеличенное изображение миллиметровой шкалы. Далее имеется горизонтальная шкала с десятью делениями и круглая шкала в левой части поля зрения окуляра. Круглая шкала поворачивается при вращении маховичка справа на головке отсчетного микроскопа. Она имеет 100 делений. Для отсчета необходимо вращением маховичка добиться, чтобы штрих миллиметровой шкалы располагался -между линиями двойной спирали. После этого произвести отсчет. Записывают число целых миллиметров, десятые доли миллиметра отсчитывают на горизонтальной шкале микроскопа. Записывают число слева от штриха миллиметровой шкалы, который был совмещен с двойной спиралью. Сотые, тысячные и десятитысячные
64
доли миллиметра отсчитываются по круглой шкале против горизонтального индекса. Например, отсчет на рис. 32 — 84,3990 мм.
Последовательность выполнения работы. Поместить спектрограмму на столик компаратора вверх эмульсией, сфокусировать изображение спектра в левом микроскопе. Изучить спектр железа в требуемом диапазоне длин волн, сопоставляя все спектральные линии со спектром железа в атласе (см. приложение). Определить номера линий в спектре железа, между которыми расположена
Рис. 32. Поле зрения отсчетного микроскопа ИЗА-2 (а) и увеличенное изображение участка шкалы (б)
спектральная линия в изучаемом спектре. Установить столик компаратора так, чтобы левая пронумерованная в атласе линия железа совпала с перекрестием в левом микроскопе и сделать отсчет по шкале правого микроскопа. Переместить столик компаратора до совпадения спектральной линии в изучаемом спектре с перекрестием и сделать отсчет по шкале правого микроскопа. Переместить столик компаратора до совпадения правой пронумерованной в атласе линии железа с перекрестием, сделать отсчет по шкале правого микроскопа.
7. ОЦЕНКА ТОЧНОСТИ СПЕКТРАЛЬНЫХ ИЗМЕРЕНИЙ
Минимальная погрешность оптической плотности. Ошибки при измерении процента пропускания или оптической плотности могут возникать, например, за счет неопытности экспериментатора, некачественного проведения эксперимента (неправильная установка кювет в кюветное отделение, отсутствие стабилизации источника излучения, нестабильная работа усилителя и измерительного устройства прибора). Эти погрешности устранимы.
Рассмотрим возникновение погрешности, вытекающей из самой сущности законов поглощения. Теоретически оптическая плотность меняется от 0 до оог а процент пропускания—от 0 до 1ЮО%, однако не во всем диапазоне эти величины могут быть измерены с одинаковой точностью. Сделаем допущение, чта
3—2083
65
при ©течете оптической плотности и процента пропускания погрешности АР и АТ на всем диапазоне шкальг одинаковы. Из уравнения (1.3) следует:
(1.122)
Продифференцируем уравнение Z) = lg(/0/A), для чего сначала преобразуем его:
dD = d 1g Zo - d 1g Ii - 0 - 0,4343 (dZ/Zz). (1.123)
Относительная погрешность равна
d£> 0,4343^Z 0,4343d?
D = ~ DIi =~ DIO-0
(1.124)
Чтобы определить значение оптической плотности, при которой погрешность измерения будет минимальной, продифференцируем (1.124):
I AD \ ! 0,4343d/ \ 0,4343d/ /10° In 10 10° \ ,
----57“ (1Л25)
Zz ^ZqIO”^-
(1.126)
/'dD \
Приравняем I яулю и рассчитаем оптическую плотность, при кото-
рой погрешность будет минимальной:
DmIn = 1/1п Ю “ 0,4343.
(1.127)
Относительная погрешность определения концентрации зависит от оптической плотности. Она минимальная при D—9,4343. Зависимость Ас/с от D пред-
Рис. 33. Определение относительной погрешности оптической плотности
ставлена на рис. 33. Минимальная относительная погрешность D составляет +2,9% и наблюдается в пределах от 0,3 до 0,7. Удвоенная минимальная погрешность ±5,8% получается в пределах D от Ю,1 до 1,3.
Определение максимальной относительной погрешности концентрации. В общем виде зависимость D/l = f (с) выражается уравнением прямой:
D/1 ^£0 (1.128)
или
с = (D/L - 6)/е.
(1.129)
Подставив частные производные из
Предельная абсолютная погрешность будет
(1.129) в (1.13Ю), получим
de.
(1.130)
(1.131)
Вместо dP, dZ, dfr, ds подставим значения абсолютных погрешностей, предполагая наиболее неблагоприятный случай, все абсолютные погрешности возьмем со знаком «+»:
£
£2
(1.132)
Если предположить,, что систематическая 0, то
погрешность
b отсутствует, но
(1.133)
Все составляющие погрешности могут быть рассчитаны отдельно.
Погрешность определения волновых чисел или длин волн. Если шкала прибора калибрована в делениях волновых чисел или длин волн, то в паспорте прибора обычно указана погрешность измерения данных величин. Для призменных приборов погрешность измерения волновых чисел — величина переменная и зависит от волнового числа. Если же нет указаний на точность измерения волнового числа, то принимается за точность минимальный отсчет по шкале. Если шкала прибора не калибрована, то при определении каждого отсчета в спектре стандартного вещества, по которому производится калибрование шкалы, допускается погрешность. Значения волновых чисел или длин волн в спектре стандартного вещества принимаются за точные. Погрешности зависят от ширины спектральных линий или полос поглощения и от других факторов.
Все измерения по шкале прибора для известных волновых чисел обрабатьь ваются методом наименьших квадратов, т. е. рассчитывается значение v с наименьшей квадратичной ошибкой. По методу наименьших квадратов определяются коэффициенты а и b в уравнении
v = ап + b. (1.134)
Работа 1. Изучение колебательно-вращательного спектра поглощения двухатомных газообразных молекул
Изучение колебательно-вращательного спектра поглощения можно провести на спектрофотометрах ИКС-14, ИКС-21, ИКС-24, ИКС-29 и спектрометре UR-10.
Газовую кювету длиной 10 или 15 см заполнить газом, осушенным концентрированной серной кислотой. Для этого входной штуцер газовой кюветы соединить шлангом с установкой для получения осушенного газа. Выходной штуцер соединить шлангом с установкой для поглощения газа. Заполнение газовой кюветы следует производить в вытяжном шкафу. После длительного пропускания газа через кювету закрыть входной кран кюветы и затем — выходной, с тем чтобы в кювете не было избыточного давления.
Спектр поглощения снимать при самых малых скоростях сканирования, так как спектр состоит из узких, близко расположенных друг к другу спектральных линий. Скорость сканирования и скорость регистрации задается преподавателем. Отградуировать шкалу прибора по волновым числам, для чего снять спектр полистирола и построить график v=f(n) (см. с. 57). Если же шкала прибора или бланк, на которых снимается спектр, градуированы, то проверить шкалу по спектру полистирола. При отклонениях показаний шкалы от волновых чисел полос поглощения полистирола ввести поправку. Эта поправка может быть постоянной для всей шкалы.
Диапазон волновых чисел, в котором расположены колебательно-вращательные полосы поглощения для разных газов, приведен ниже.
Газ
V, СМ”1
HCl HI НВг . СО
2600—3100 2100—2500 2300—230*0 .2000—2251
3*
67
Работа 2. Определение равновесного межъядерного расстояния
Сделать анализ полученного колебательно-вращательного спектра поглощения газа (см. 67). На спектрограмме написать серию линий, которая относится к Р- и 7?-ветви. Против каждой линии написать вращательные квантовые числа исходного и конечного состояний молекулы.
Определить по калибровочному графику или непосредственным отсчетом по шкале волновых чисел волновые числа линий, соответствующих переходам 0<-1 и j'-*-/". Определить среднюю разность волновых чисел соседних линий в Р-ветви. Рассчитать вращательную постоянную по (1.38), момент инерции и равновесное межъядерное расстояние по (1.6). Повторить расчет для трех-четырех значений Для каждого значения Av рассчитать ге, найти относительную и абсолютную погрешности равновесного межъядерного расстояния.
Работа 3. Определение зависимости вращательной постоянной от колебательного квантового числа. Определение Ве и ге
Сделать анализ полученного колебательно-вращательного спектра поглощения. На спектрограмме пометить, какая полоса относится к Р~ и Р-ветви. Написать вращательные квантовые числа исходного и конечного состояний молекулы против каждой линии в спектре. Определить волновые числа линий, соответствующих одинаковым значениям /" в Р- и P-ветвях для нескольких значений Результаты записать в таблицу по образцу:
Р-ветви, см—1
для
Р-ветви, см—*
Решить совместно два уравнения (1.53) для двух разных значений /" и рассчитать Ве и а. Расчет повторить для трех-четырех пар уравнений. Взять средние арифметические значения Ве и а. Определить абсолютные и относительные погрешности величин В& и а. Записать зависимость Bv—i(u) [см. (1.39)] для исследуемого вещества. По значению вращательной постоянной Ве рассчитать момент инерции и равновесное межъядерное расстояние по (1.10) и (1.6), рассчитать погрешность межъядерного расстояния.
Работа 4. Определение распределения молекул по вращательным квантовым уровням
Снять спектр поглощения газа (см. с. 67). При съемке колебательно-вращательного спектра поглощения необходимо отметить температуру газа. Если термостатирование кюветы отсутствует, то
68
I
£ $
а:
температура исследуемого газа соответствует температуре окружающей среды. Произвести анализ полученного колебательно-враща* На спектрограмме пометать Р- и
0%
Рис. 34. Определение оптической плот* ности по максимумам лииий поглоще*
НИЯ
тельного спектра поглощения. R.-ветви. Написать против каждой линии вращательное квантовое число исходного и конечного состояний молекулы. Измерить оптические плотности всех заметных в ^-ветви колебательно-вращательных линий. Для измерения оптической плотности линии провести базовую линию А—А (рис. 34). Измерить миллиметровой линейкой расстояние от линии 0% пропускания через максимум поглощения до базовой линии а и от линии 0% пропускания до максимума поглощения Ь. Оп
тическую плотность рассчитать по уравнению D = \g(a/b). Оптические плотности всех линий в 7?-ветви записать в таблицу по образцу:
т
Я-ветвь
iff
If
Mo
Ny
№
[по уравнению
(1.15)]
в 7?-ветви
о _
каждой ли-
оптическая вращатель-
ь
Определить сумму оптических плотностей всех линий 2 Dj* и рассчитать отношение оптической плотности у’-О нии к сумме оптических плотностей. Предполагая, что плотность пропорциональна числу молекул на данном
ном квантовом уровне, рассчитать отношение числа молекул на уровне j" к числу молекул на нулевом вращательном квантовом уровне. Сопоставить полученные на основании оптических плотностей соотношения Nj'/Nq с рассчитанными по (1.15) при температуре Т.
Работа 5. Определение волнового числа основной полосы поглощения двухатомной молекулы
Снять спектр поглощения газа (см. с. 67). В колебательно-вращательном спектре поглощения отсутствует полоса, соответствующая Д/ —0. Однако по волновым числам линий вращательной струк
69
туры колебательно-вращательного спектра определить значение (1—2хе), Определить значения волновых чисел симметрично расположенных линий в Р- и /?-ветвях и взять среднее арифметическое значение. По (1.36) и (1.37) рассчитать <ое(1—2хе). Результаты записать в таблицу по образцу:
Г Vg , СМ — 1 Vp, СМ-1 + ''р , CM 1 2
Из трех-четырех значений о)е(1—2хе) взять среднее арифметическое м вычислить относительную и абсолютную погрешности.
Работа 6. Определение теплоемкости Ср и зависимости теплоемкости от температуры статистическим методом
Снять спектр газа (см. с. 67). Определить значение юе(1—2хе). Не допуская большой погрешности, можно принять, что 2хе<^1 и <ве(1—2хе)=(ое. Рассчитать 0 по (1.90) и Q/Т при 298 К и заданной температуре. Определить СЕ при 298 К и заданной температуре. Рассчитать Ср по (1.104), (1.105), (1.106) и (1.108). Чтобы установить зависимость теплоемкости от температуры в виде уравнения С°р = а + ЬТ + сТ’2 в диапазоне от 298 до 1000 К, следует вычислить 6/Т для температур от 300 до 1000 через 100 К и методом наименьших квадратов — коэффициенты а, b и с. Расчет С°р при заданных температурах можно произвести на ЭВМ по программе, приведенной в приложении.
Работа 7. Определение энтальпии (Н°т—Я°о) вещества статистическим методом и зависимости (Н°т—Н°о) от температуры
Снять спектр поглощения газа (см. с. 67) и определить сов(1 — —2хе). Не допуская большой ошибки, можно принять, что 2хе<С1 и ше=(ое(1—2хе). Рассчитать 0 по (1.90) и Q/Т при 298 К и заданной температуре. По таблице термодинамических функций Эйнштейна для линейного гармонического осциллятора определить при обеих температурах. Рассчитать (UT—£7о)кол и по
(1.102), (L99) и (1.100) (UT—С70); (Н°т—Н°0) при 298 К и заданной температуре.
Расчет (Я°т—Я°о) при нескольких температурах можно произвести на ЭВМ с применением программы, приведенной в приложении.
70
Г> « О Г» “ Ат—^0 ™ г)
Работа 8. Определение функции —----- и --------
Снять спектр поглощения (см. с. 67) и определить значение <0е (1—2хе). Не допуская большой ошибки, можно принять, что 2хе<С1 и ®е=Юе(1—2хе). Рассчитать 0 по (1.90) и 0/Т для 298К и заданной температуры. Колебательную составляющую функции - ~ — найти по таблице термодинамических функций Эйнштейна для линейного гармонического осциллятора. Для расчета In /пост, In ZBp и In Zэл воспользоваться уравнениями (1.86), (1.88), (1.92). Расчет произвести при давлении 1,0133* 105 Па. Рассчитать по (1.111) поступательную и вращательную составляющие функции
• Суммируя поступательную, вращательную, колебательную и электронную составляющие, определить — - и по (1.114)
G°T -H°Q
--- . Расчет функции —- при нескольких температурах можно провести с помощью ЭВМ по программе, приведенной в приложении.
Работа 9. Определение энтропии и зависимости энтропии от температуры
Снять спектр газа (см. с. 67) и определить значение «е(1— — 2хе). Не допуская большой ошибки, можно принять, что 2хе<Д. Рассчитать 0 по (1.90) и Q/T для 298 К и заданной температуры. По таблице термодинамических функций Эйнштейна для линейного гармонического осциллятора определить колебательную составляющую энтропии при обеих температурах. Логарифм поступательной, вращательной и электронной суммы по состояниям определить по уравнениям (1.86), (1.88), (1.92). По (1.84) и (1.87) рассчитать частную производную логарифма поступательной и вращательной суммы по состояниям при постоянном объеме. Расчет поступательной суммы по состояниям по уравнению (1.86) проводить для давления 1,0133* 105 Па. Таким образом, вычисленная энтропия будет стандартной энтропией вещества. По уравнению (1.109) вычислить поступательную, вращательную и электронную составляющие энтропии и сложить полученные величины с колебательной составляющей. Если требуется определить энтропию при нескольких температурах, то расчет следует произвести с помощью ЭВМ по программе, приведенной в приложении.
Работа 10. Изучение колебательно-вращательного спектра метана
Работу можно выполнять на приборах ИКС-14, ИКС-21, UR-10. Для съемки спектра поглощения метана заполнить газовую кювету
71
длиной 10 или 15 см газом, осушенным концентрированной серной кислотой. Заполнение кюветы производить медленным пропусканием газа через концентрированную серную кислоту. Вся установка должна размещаться в вытяжном шкафу, в котором не должно быть открытого огня или другого источника воспламенения метана.
Отградуировать шкалу прибора, для чего снять спектр полистирола и построить график зависимости волнового числа от делений шкалы длин волн прибора (см. с. 57). Если же шкала прибора или бланк, на который регистрируется спектр, градуированы в волновых числах, то произвести проверку шкалы по спектру полистирола. При отклонениях показаний шкалы от значений волновых чисел полос поглощения полистирола ввести поправку.
Работа 11. Определение вращательной постоянной, момента инерции и равновесного межъядерного расстояния гс-н молекулы метана
Снять колебательно-вращательный спектр поглощения метана в области 2700—3300 см-1. На спектрограмме написать отнесение линий к Р-, Q- и /?-ветвяш Против каждой линии написать вращательные квантовые числа исходного и конечного состояний молекулы. Определить волновые числа линий, соответствующих переходам 0<-1 и /'<-/" в Р-ветви асимметричного валентного колебания. Определить значения трех-четырех разностей волновых чисел соседних линий Av для трех-четырех разных значений По значениям Av рассчитать вращательную постоянную и момент инерции. Равновесное межъядерное расстояние /с-н рассчитать по моменту инерции и исходя из тетраэдрической структуры молекулы метана. Рассчитать относительные и абсолютные погрешности при определении / И Гс-Н.
Работа 12. Определение зависимости вращательной постоянной от колебательного квантового числа. Определение Ве и гс -п
Расшифровать полученный колебательно-вращательный спектр поглощения метана (см. выше). На спектрограмме пометить, какая ветвь в спектре относится к Р-, Q-и/?-ветвям. Написать вращательные квантовые числа исходного и конечного состояний молекулы против каждой линии в Р- и 7?-ветвлх. Определить волновые числа линий, соответствующих одинаковым значениям j/z в Р- и /?-ветвях для нескольких значений /" Результаты записать в таблицу по образцу:
Р-ветвь AJ-ветвь
Г 't см-1 v см-1 Г ( см 1
I
72
Рассчитать разности волновых чисел линий, соответствующих одинаковому значению вращательного квантового числа исходного состояния молекулы в Р- и 7?-ветвях. Решить совместно два уравнения (1.44) для двух разных значений /" и рассчитать Ве и г. Расчет повторить для трех-четырех пар уравнений. Взять среднее арифметическое значение Ве и г. Определить относительные и абсолютные погрешности величин Ве иг. Записать уравнение зависимости Bv=f(v) метана. На основании Ве рассчитать момент инерции СН4, равновесное межъядерное расстояние гс-н и погрешность вычисления гс-н.
Работа 13. Определение волновых чисел основных полос поглощения СН4
В колебательно-вращательном спектре поглощения многоатомных молекул наблюдаются Р-, Q- и /?-ветви. Это позволяет определить непосредственным измерением волновые числа основных полос поглощения валентного асимметричного, деформационных асимметричного и симметричного колебаний. Волновое число симметричного валентного колебания молекул метана не может быть определено по ИК-спектру поглощения, так как при дан-
\ дд /?=о ном колебании равно нулю и интенсивность полосы равна нулю. Эту величину можно определить по спектру комбинационного рассеяния. Она равна 2916,5 см”1. Отнесение полос поглощения в спектре поглощения метана можно сделать на основании значений (du. \
0> приведенных в приложении, при сопоставлении интенсивностей полос. Установить вырождение каждого колебания.
Работа 14. Определение изобарной теплоемкости Ср и зависимости Cp=f(T) статистическим методом
По волновым числам полос поглощения асимметричного валентного и деформационного асимметричного и симметричного колебаний и волновому числу симметричного валентного колебания, определенному по спектру комбинационного рассеяния, равному 2916,5 см *, рассчитать для всех четырех видов колебаний характеристическую температуру 0 по (1.90), приняв, что коэффициент ангармоничности хе<^1. Определить Q/Т при 298 К и заданной температуре. По таблице термодинамических функций Эйнштейна для линейного гармонического осциллятора найти СЕ для всех видов колебаний молекулы метана при 298 К и заданной температуре. Рассчитать Скол как сумму СЕ по всем девяти колебательным степеням свободы. При вычислении учесть вырождение каждого колебания (см. приложение). Рассчитать Ср по (1.108) и (1.121).
Если необходимо установить зависимость теплоемкости от температуры в виде уравнения С?Р=а+ЬТ+сТ2 в диапазоне от 298 до 1000 К, то вычислять 0/Т для температур от 300 до 1000 через
73
100 К. Определить С°р при указанных температурах на ЭВМ по программе, приведенной в приложении, и, применяя метод наименьших квадратов, рассчитать значения коэффициентов a, b и с.
Работа 15. Определение энтальпии (Я°т—Но0) метана статистическим методом и зависимости (Н°т—Я°о) от температуры
Определить значения волновых чисел всех колебаний молекул СН4 (см. с. 73). По волновым числам полос поглощения валентного асимметричного и деформационных симметричного и асимметричного колебаний, а также волновому числу симметричного валентного колебания, определенного по спектру комбинационного рассеяния, 2916,5 см-1, принимая, что хе<ёС1, рассчитать 0 для всех колебаний молекулы метана по (1.90). Рассчитать Q/T при 298 К и заданной температуре. По таблице термодинамических функций Эйнштейна для линейного гармонического осциллятора найти \ дЛЯ всех колебаний. Рассчитать (—----------------—как
\ 7* / кол \ ^ / кол
сумму по всем девяти колебательным степеням свободы. При вычислении учесть вырождение колебаний (см. приложение). Рассчитать Я°298—Я°0 И Н°т—Н°0.
Если требуется установить зависимость Н°т—H°Q от температуры, то можно воспользоваться ЭВМ. Программа приведена в приложении.
Работа 16. Определение энтропии метана статистическим методом и зависимости энтропии от температуры
Определить значения волновых чисел полос поглощения метана (см. с. 73). По волновым числам полос поглощения валентного асимметричного колебания и деформационных симметричного и асимметричного колебаний и определенному по спектру комбинационного рассеяния волновому числу симметричного валентного колебания 2916,5 см-1, принимая, что хе<С1, рассчитать 0 для всех колебаний по (1.90). Определить Q/T при 298 К и заданной температуре для всех колебаний молекулы метана.
По таблице термодинамических функций Эйнштейна для линейного гармонического осциллятора найти 5КОл для всех колебаний. Рассчитать колебательную составляющую энтропии, суммируя величины Зкол по всем девяти колебательным степеням свободы. По колебательно-вращательному спектру определить момент инерции метана (см. с. 72). Определить по значению момента инерции по (1.88) и (1.116) вращательную составляющую энтропии, а по (1.109) и (1.86)—поступательную составляющую энтропии при давлении 1,0132-105 Па. Рассчитать энтропию метана при 298 и заданной температуре и стандартном давлении. Если требуется определить энтропию при нескольких температурах, то расчет произвести на ЭВМ по программе, приведенной в приложении.
74
J' Q
Работа 17. Определение функции —~---------метана статистическим
методом
Определить значения волновых чисел полос поглощения метана (см. с. 73). По волновым числам полос поглощения асимметричного валентного и деформационных асимметричного и симметричного колебаний и волновому числу симметричного валентного колебания, найденному по спектру комбинационного рассеяния» 2916,5 см-1, принимая, что хеС1, рассчитать 6 для всех колебаний по (1.90). Определить 0/Т для 298 К и заданной температуры для всех колебаний молекулы метана.
По таблице термодинамических функций Эйнштейна для линей-о [ Ат — U\
ного гармонического осциллятора наити I—-———) для всех
\ 2 /КОЛ
колебаний. Суммированием (— ------- 1 по всем девяти коле-
\ Т /кол
бательным степеням свободы рассчитать колебательную составляющую функции — ~—Е дЛЯ 298 К и заданной температуры.
По колебательно-вращательному спектру поглощения метана определить момент инерции (см. с. 72). Рассчитать In ZBp по (1.116), InZnocT по (1.86) при давлении 1,0132-105 Па
От-^о
и
по уравнению (1.114) для 298 К и заданной
темпера-
туры.
Если требуется определить функции
0
о
при
несколь-
ких температурах, то расчет произвести на ЭВМ по программе, приведенной в приложении.
Работа 18. Изучение колебательных спектров комбинационного рассеяния веществ в жидком состоянии
Работу проводить на спектрографе ИСП-51 с фотоэлектроприставкой ФЭП-1. Провести градуировку шкалы прибора (описание см. на с. 54) и снять спектр комбинационного рассеяния в заданном диапазоне волновых чисел. По калибровочной кривой определить волновые числа линий комбинационного рассеяния изучаемого соединения. Пользуясь таблицами характеристических частот колебаний, сделать отнесение линий комбинационного рассеяния к колебаниям в определенных функциональных группах.
Работа 19. Изучение спектра комбинационного рассеяния молекул, обладающих тетраэдрической структурой
Работа описана на с. 53. В спектре комбинационного рассеяния тетраэдрических молекул разрешены все четыре колебания и
75
наблюдаются четыре стоксовые линии. В качестве объекта изучения предлагается взять одно из соединений: СС14, CBr4, SiCl4, GeCl4> SnCl4. Определить волновые числа всех колебаний в молекуле изучаемого вещества. Учитывая, что v2<v4<viO3 (см. приложение), указать, к какому типу относятся полученные колебания в спектре. Определить вырождение каждого колебания.
Работа 20. Определение термодинамических функций веществ, молекулы которых обладают тетраэдрической структурой, по спектрам комбинационного рассеяния
Снять спектр комбинационного рассеяния (см. с. 53), расшифровать спектр. По волновым числам всех колебаний рассчитать термодинамическую функцию при определенных условиях существования вещества в идеальном газообразном состоянии. Для вычисления вращательных составляющих термодинамических функций воспользоваться данными о равновесных межъядерных расстояниях между центральным и периферийным атомом.
Молекула ХУ4 СС14 CBr4 SiCl4 SnCl4 SiBr4 GeCl4 GeBr4 SnBr4
rx-Y-1010, м 1,766 1,942 2,01 2,31 2,15 2,08 2,29 2,44
Если требуется определить термодинамическую функцию при нескольких температурах, то работу выполнять на ЭВМ по программе, приведенной в приложении.
Работа 21. Изучение электронно-колебательно-вращательного спектра излучения радикала CN
Работу выполнять на кварцево-стеклянном спектрографе КС-55. Спектр излучения CN возникает при горении вольтовой дуги переменного тока между электродами из графита в атмосфере воздуха.
Последовательность выполнения работы. Приготовить два угольных электрода длиной 4—5 см. Концы электродов заточить на конус. Оба электрода закрепить в электрододержателе на специальном калибровочном столике, при помощи которого устанавливается заданное расстояние между электродами. Аналогично приготовить электрододержатель с железными электродами. По шкале длин волн установить деление 360 для стеклянной оптики (индекс «С»), Установить ширину щели 0,01 мм. Диафрагму с фигурным вырезом установить в положении «1» (рис. 35, а). Зарядить кассету фотопластинкой 9X12 эмульсией вниз в среднюю часть кассеты. Установить кассету в кассетную часть спектрографа, прижав ее двумя винтами сверху. Выдвинуть переднюю крышку кассеты. Маховичком слева от кассеты устанотить деление «20». Открыть, дверцу штатива и установить в специальные его гнезда вилку электродо-держателя с угольными электродами. Плотно закрыть дверцу штатива. Включить генератор дуги переменного тока ДГ-2 в электросеть напряжением 220 В. Индикаторная лампа в центральной части 76
панели должна светиться. Нажать на кнопку «пуск», расположенную на генераторе или на выносном кнопочном выключателе. При этом загорается дуга переменного тока между электродами.
Изображение дуги проектируется линзами на входную щель прибора. Произвести съемку спектра излучения CN с экспозицией 4 мин. Так как длительное горение дуги недопустимо, то экспозицию в 4 мин следует разделить на четыре минутные экспозиции. После того как включена дуга, открыть затвор на 1 мин. Закрыть
Рис. 35. Схема установки диафрагмы с фигурным вырезом перед щелью спектрографа
затвор и выключить дугу. Примерно через 2—3 мин повторить экспозицию.
Заменить электрододержатель с угольными электродами на
электрододержатель с железными электродами, закрыть дверцу
штатива, переместить диафрагму перед входной щелью в положе-
ние «2» (рис. 35, б). Это дает возможность получать спектр сравнения, необходимый для определения волновых чисел линий.
Снять спектр железа подобно съемке спектра CN с экспозицией 4 мин. Переместить кассету в положение «25». Установить барабан длин волн в положение «380». При этом на ту же среднюю часть
фотопластинки будет проектироваться новый участок спектра. Снять спектры железа и CN, но с экспозицией 1 мин. Переместить кассету в положение «30», установить барабан длин волн на деление «415» и снять спектры CN и железа с экспозицией 1 мин. Все
показания и экспозиции записать в лабораторный журнал по образцу:
Вещество
Длина волны, нм
Положение кассеты
Положение диафрагмы
Экспозиция, мни
CN Fe Fe
CN CN Fe
360
360
380
380
415
415
20
20
25
25
30
30
П
По окончании съемки всех спектров закрыть переднюю крышку кассеты, повернуть прижимные винты сверху кассеты и снять кассету со спектрографа. Проявить спектрограмму в фотокабине, для чего открыть заднюю крышку кассеты, вынуть фотопластинку и поместить ее в ванну с проявителем на 4 мин вверх эмульсией. Промыть фотопластинку и поместить ее в ванну с фиксажем на 8—10 мин. После фиксации фотопластинку тщательно промыть проточной водой и высушить на штативе в потоке теплого воздуха.
Расшифровать полученные спектры. Определение волновых чисел спектральных линий проводить на компараторе ИЗА-2. Поместить спектрограмму на столик компаратора вверх эмульсией так, чтобы линии в спектре излучения CN сходились влево. Переместить спектрограмму винтом с левой стороны "столика компаратора так, чтобы в поле зрения левого микроскопа была бы видна верхняя часть спектра. Ослабить винт под столиком компаратора в левой части и, перемещая столик вручную, проверить, не смещается ли по вертикали изображение спектра в левом микроскопе. Если наблюдается смещение спектра, то повернуть на небольшой угол планку, на которую опирается спектрограмма. Установить четкое изображение спектра в поле зрения левого микроскопа маховичком фокусировки. Установить четкое изображение индекса в поле зрения микроскопа вращением муфты окуляра.
Определить волновые числа спектральных линий в спектре излучения CN, соответствующих переходам 0~>1, 1~>2, 0->0 и 1~>1 (см. рис. 10). Для этого ознакомиться и разобраться в спектре железа, сопоставив его со спектром железа в атласе спектральных линий (см. приложение).
Примечание. В атласе спектральных линий железа некоторые спектральные линии пронумерованы. Для этих спектральных линий в таблице приложения приведены значения длин волн, волновых чисел и разностей волновых чисел соседних пронумерованных линий.
Сопоставлением изображения спектра на спектрограмме со спектром железа найти, между какими спектральными линиями расположена спектральная линия CN. Произвести отсчеты по микроскопу компаратора справа для обеих линий железа, между которыми находится измеряемая линия CN, и линии CN. Перед каждым отсчетом микрометрическим винтом столика компаратора справа* зажав винт снизу столика, переместить спектрограмму так, чтобы измеряемая линия совпала с индексом в поле зрения левого микроскопа. Линейной интерполяцией, зная волновые числа линий железа слева и справа, рассчитать волновое число линии CN. Повторить аналогичные измерения трижды для всех линий CN. Рассчитать колебательную постоянную сое и ангармоничность щехе радикала CN в электронно-невозбужденном состоянии и энергию химической связи.
Примечание. Линии в спектре излучения CN, соответствующие переходам Ду — —1, располагаются в области спектральных линий железа от № 6 до^ № 10; линии, соответствующие переходам Ау = @ и Ау= +1, — в области линиш железа от № 35 до № 42.
Работа 22. Определение колебательной постоянной и ангармоничности для радикала GN в электронно-возбужденном состоянии
Снять спектры излучения радикала CN и железа (см. с. 76). Определить волновые числа линий в спектре излучения CN, которые соответствуют переходам 1~>0, 2~>1, 0->0 и 1~>1; колебательную постоянную и ангармоничность для радикала, находящегося в электронно-возбужденном состоянии; энергию химической связи в радикале в электронно-возбужденном состоянии. На основании полученных данных 78 вычертить кривую потенциальной энергии как функцию межъядерного расстояния (кривую Морзе). Для этого рассчитать минимальное и максимальное межъядерные расстояния в радикале CN для энергетических уровней при с = 0, 5, 10 и 15 для нормального и электронно-возбужденного состояний.
Работа 23. Определение константы равновесия реакции образования комплекса по электронно-колебательному спектру поглощения
Работу можно выполнять на приборах: УМ-2, СФ-4, СФ-5, СФД-2, СФ-16, СФ-26, СФ-10 или СФ-14.
Определить константу равновесия реакции комплексообразования:
Со2+ + 4SCN- = [Со (SCN)4]2-
Приготовить восемь водноацетоновых растворов, содержащих 0,025 М Co(NO3)2 и 0,25 М NH4SCN в разных соотношениях:
Номер раствора 1 2 3 4 5 6 7 8
cCo(NO3)3 * моль/л 0,5 0,5 0,5 0,5 0,5 0,5 0,5 0,5
^NH4SCN'L°2’ моль/л °’5 2’° 3’° 4,0 20’0 0
Рассчитать объемы, необходимые для приготовления растворов заданных концентраций. Приготовить по 25 мл каждого раствора. Снять спектры поглощения растворов 7 и 8 и вычертить спектры на одном листе миллиметровой бумаги. Считая, что раствор 7 имеет большой избыток ионов SCN“, можно предположить, что в равновесной смеси все ионы Со2+ связаны в комплекс. Спектр поглощения раствора 8 соответствует спектру поглощения ионов Со2+. Выбрать длину волны, при которой коэффициент погашения ионов Со2+ практически равен нулю, а оптическая плотность комплексов (Со (SCN)4]2~ максимальна. Определить оптические плотности всех приготовленных растворов при выбранной длине волны и построить график зависимости D~ f (cSCN_).
По известным значениям оптических плотностей и коэффициенту погашения комплекса определить концентрации [Со(SCN)4)]2-и вычислить константу равновесия для трех-четырех растворов. Рассчитать среднюю арифметическую константы равновесия и по
79
грешность определения константы равновесия. При вычислении коэффициента погашения комплекса принять, что концентрация комплекса в растворе 7 равна концентрации ионов Со2+. Измерения оптической плотности и съемку спектров поглощения производить в кюветах длиной 1 мм.
Работа 24. Определение констант диссоциации слабых органических кислот по электронно-колебательным спектрам поглощения
Константа диссоциации слабой органической кислоты в растворе
НА^Н+4-А“ может быть выражена через активности ионов и молекулы в рас- . творе:
™ ан+ад-/аНА.
С учетом среднеионного коэффициента активности ионов и считая, что активность недиссоциированной кислоты равна концентрации, т. е. коэффициент активности слабой кислоты в разбавленном растворе равен единице, можно записать
pXa-pH-lg-^— — IgY-u* (1.135)
Среднеионный коэффициент активности для 1 — 1-электролита можно рассчитать по закону Дебая и Гюккеля.
Спектральный метод определения констант диссоциации слабых органических кислот основан на том, что спектры аниона и молекулы кислоты различаются, т. е. максимумы поглощения аниона и молекулы наблюдаются при разных длинах волн. По оптическим плотностям растворов при разных длинах волн можно определить равновесную концентрацию анионов и молекул. При постепенном изменении pH растворов оптические плотности максимумов полос поглощения изменяются. При а~1 в спектре наблюдается только одна полоса поглощения, соответствующая поглощению аниона А". При низком значении pH диссоциация кислоты практически подавлена и а~0, в спектре останется полоса, принадлежащая поглощению кислоты НА.
На рис. 36 приведено семейство спектров поглощения слабой кислоты при различных значениях pH. Спектры 1 и 2 принадлежат растворам, в которых диссоциация практически подавлена, а= = 0; для спектров 7 и 8 а=1; спектры 3—6 соответствуют а, отличной от 0 и 1. Все спектры пересекаются в одной точке, где поглощение не зависит от pH. Эта точка называется изобестической точкой. Коэффициенты погашения аниона и молекулы кислоты при этой длине волны одинаковы. Оптическая плотность при этой длине волны не зависит от соотношения концентраций молекулы кислоты и аниона. По числу изобестических точек можно судить о числе рав
80
новесий в растворах. Слева и справа от изобестической точки оптические плотности при различных значениях pH меняются. Поглощение складывается из двух величин:
D = £>на + Dk-
или
D — еНАСНА* + ед—ЙА—'
Если степень диссоциации кислоты а, то при длине волны, соответствующей полосе поглощения молекулы НА,
DHA = D1(l-a), a D = D2a,
Л
где Di — предельная оптическая плотность при низких значениях pH; D2— предельная оптическая плотность при высоких значениях pH. Окончательно получим уравнение
D = — а)
из решения которого по оптической плотности D рассчитаем а:
Ct — — Z?2) *
Все значения D, а также Di и D% должны быть определены при одной и той же длине волны X]. При другой длине волны отвеча-
ем
2%
Рис. 36. Спектры поглощения слабой органической кислоты при различных значениях pH растворов
Рис. 37. Зависимость оптической плотности раствора от pH
Л
ющей поглощению аниона А” степень диссоциации может быть определена по уравнению
а = (Z) — Dx)/(D2 - Рх).
Таким образом, зная степень диссоциации, среднеионный коэффициент активности и pH раствора, можно рассчитать р/<а. Если в первом приближении считать раствор идеальным и принять, что у±=1, то уравнение (1.135) примет вид
р/( = рН —lg[a/(l~a)].
(1.136)
81
По уравнению (1.136) получим кажущуюся константу диссоциации кислоты. Если а—0,5, то pK=pHi/2. Для определения рК строят график зависимости D = f (pH) при м и л2- По значению (Ь2— —Di)/2 определяют рК (рис. 37).
Последовательность выполнения работы. Работу выполнять на приборах: УМ-2, СФ-4, СФД-2, СФ-5, СФ-16, СФ-26, СФ-10 или СФ-14. Приготовить десять растворов слабой кислоты с одинаковой концентрацией, но разными значениями pH. Для приготовления растворов градуированной пипеткой на 5 мл в десять пробирок налить по 3 мл раствора кислоты. В каждую пробирку добавить из градуированной пипетки на 10 мл по 7 мл буферного раствора с определенным значением pH. Рассчитать концентрацию кислоты в полученных растворах.
Если работа производится на нерегистрирующем спектрофотометре, то снять спектры поглощения двух крайних растворов с максимальным и минимальным значениями pH в диапазоне длин волн от 400 до 700 нм. Толщина поглощающего слоя 1 см. Вычертить спектры поглощения обоих растворов на одном листе миллиметровой бумаги. Определить длины волн максимумов поглощения аниона и молекулы кислоты. При длинах волн максимумов поглощения аниона и кислоты измерить оптические плотности всех десяти растворов. Точками отметить максимумы поглощения на спектрах.
Если работа выполняется на саморегистрирующем спектрофотометре, то снять спектры поглощения всех десяти растворов в кювете с толщиной поглощающего слоя 1 см.
Указать на спектре, какая из полос поглощения относится к аниону и какая к молекуле кислоты. Построить график зависимости оптической плотности от pH для обеих длин волн максимумов поглощения. Результаты записать в таблицу по образцу:
м==
D а РКа
Рассчитать рКа для четырех-пяти растворов при средних значениях pH, среднее значение рАа, погрешность при определении величины рАа.
Определение константы диссоциации бромкрезолового зеленого. Бромкрезоловый зеленый представляет собой слабую кислоту. С точки зрения метода МО ЛКАО определить переходы электрона, в результате которых появляется полоса поглощения. Какова природа смещения полосы?
Концентрация раствора кислоты 1,25-10~4 моль/л. Буферные растворы взять в пределах pH от 3,5 до 8,0. Составы буферных растворов приведены в таблице.
82
Определение константы диссоциации крезолового красного. Крезоловый красный представляет собой слабую кислоту. С точки зрения метода МО ЛКАО определить переходы электрона, в результате которых появляется полоса поглощения. Какова природа смещения полосы?
Концентрация раствора кислоты 1,7-Г0~4 моль/л. Взять буферные растворы в пределах от 3 до 12 pH. Составы буферных растворов приведены в таблице.
Составы буферных растворов
Концентрация, моль/л
Na2HPO4 СзНЦОН) (СООН)3
Концентрация, моль/л
Na2HPO4 СзНЦОН) (СООН)3
3,64 0,0644 0,0675 5,20 0,1075 0,0464
3,85 0,0710 0,0643 5,35 0,1150 0,0443
4,00 0,0771 0,0614 5,53 0,1160 0,0420
4,18 0,0878 0,0586 5,75 0,1210 0,0396
4,40 0,0882 0,0559 6,15 0,1320 0,0340
4,60 0,0935 0,0532 6,90 0,1650 0,0175
4,78 0,0985 0,0507 7,55 0,1870 0,0065
5,00 0,1030 0,0485 х 7,75 0,1910 0,0043
pH
Концентрация, моль/л
Na2B4O7 NaOH НС1
pH
Концентрация, моль/л
N а2В4О7
NaOH
НС1
3,20
6,00
7,20
7,75
8,50
0,0300 0,0280 0,0300 0,0320 0,0394
0,0595 0,0600 0,0580 0,0530
0,0318
9,00
9,20
9,85
11,00
12,00
0,0500
0,0495
0,0312
* 0,0268 0,0236
0,0015 0,0564
0,0696 0,0792
Определение константы диссоциации бромкрезолового пурпурного. Бромкрезоловый пурпурный представляет собой слабую кислоту. С точки зрения метода МО ЛКАО определить переходы электрона, в результате которых появляется полоса поглощения. Какова природа смещения полосы? Концентрация кислоты 7,4-10-4 моль/л. Буферные растворы взять в пределах pH от 3,2 до 12. Составы буферных растворов приведены в таблице.
ГЛАВА II
ПОЛЯРИЗАЦИЯ МОЛЕКУЛ
1. ПОЛЯРИЗАЦИЯ И ПОЛЯРИЗУЕМОСТЬ МОЛЕКУЛ
Все молекулы могут быть разделены на два типа: полярные и неполярные. Если неполярная молекула помещена во внешнее электрическое поле или в полу соседней полярной молекулы или иона,
то легко подвижные электроны претерпевают некоторое смещение, в результате чего появляется наведенный или индуцированный электрический момент диполя. Смещение в электрическом поле претерпевают и атомы, входящие в состав молекулы. Полярные молекулы ориентируются в электрическом поле, и у них также происходит смещение атомов и электронов. Смещение электронов, атомов, ориентация молекулы в электрическом поле называется поляризацией. Величина поляризации Р зависит от способности к смещению центров тяжести зарядов молекулы в электрическом поле. Способность молекул к поляризации характеризуется поляризуемостью. Поляризация складывается из электронной РЭл, атомнс#сРат и ориентационной Рор:
Р = РЭл+Раг+Рор. (П.1)
Поляризуемость молекулы а равна сумме электронной, атомной и ориентационной поляризуемостей:
а ~ азл + аат 4~ аор* (П.2)
Электронная и атомная поляризуемости не зависят от температуры, ориентационная поляризуемость обратно пропорциональна температуре и пропорциональна квадрату электрического момента диполя. Поляризация диэлектрика, по Дебаю, описывается уравне-
нием
(П.З)
где в — диэлектрическая проницаемость, отнесенная к диэлектрической проницаемости вакуума; М — молекулярная масса; р — плотность.
Согласно формуле Клаузиуса — Моссоти поляризуемость, электрический момент диполя и диэлектрическая проницаемость связаны соотношением
р = АЯДГА (аэл + аат + d£-\ , (П.4)
где ц— электрический момент диполя, Кл*м.
При изменении направления внешнего электрического поля происходит переориентация полярных молекул и изменение направления вектора наведенного диполя. При увеличении частоты электрического поля сначала отпадает ориентационная поляризация. Полярные молекулы не успевают за сменой направления поля. При дальнейшем увеличении частоты отпадает атомная поляризация. Электронная поляризация сохраняется даже в переменном электрическом поле с частотой 1015 с-1, что соответствует частоте колебаний видимого света. При бесконечно большой длине волны диэлектрическая проницаемость диэлектрика е—
Рефракция молекул. Молярная рефракция представляет собой электронную поляризацию и рассчитывается по уравнению
Л2_ 1 М
РЭЛ==Р =-------—, (II.5)
п2-Ь2 р 4 '
где « — показатель преломления. Молярная рефракция не зависит ни от агрегатного состояния вещества, ни от температуры. Она обладает конститутивным свойством
— 2^,ат
i , i
(П.6)
где Rit ат, Яь ц, Ri, св — рефракция атомная, циклов и кратных связей; пи, щ. k — число атомов, циклов, кратных связей. По молярной рефракции можно установить структуру молекулы, для чего подбирают такую структурную формулу, для которой вычисленная молярная рефракция по уравнению (II.6) равна экспериментально полученному значению. Рефракция, отнесенная к молекулярной массе вещества, называется удельной рефракцией г:
(П.7)
Удельная рефракция используется при изучении свойств растворов. При условии отсутствия межмолекулярного взаимодействия между компонентами раствора
гав ~ гкха гв (I — (П.8)
где гав, Га, гв— удельные рефракции раствора, растворенного вещества и растворителя; ха— массовая доля растворенного вещества А.
Из уравнений (П.7) и (П.8) следует
^АВ — I 1 ЛА — I ХА — I I — ХА
tg — = -4—---------- 4- -4...........- , (II. 9)
пав + ?ав ла+2 Ра «B"b^ Рв
где пав, «а, пв— показатели преломления раствора, растворенного вещества и растворителя; рав, Ра, рв — плотности раствора, растворенного вещества и растворителя при температуре, при которой измерялся показатель преломления.
Электрический момент диполя молекул. Уравнение зависимости поляризации от температуры (II.4) можно записать
Р = а + (11.10)
где А= — лЛ7А (а9л-J- аат); В — лЛ/\ ~ •
Тангенс угла наклона прямой в координатах Р—1/7" равен В, Электрический момент диполя тогда будет
= 4,27-10-32/5,
(II.11)
где В — постоянная, см3 - К/моль.
Если в (ПЛ) пренебречь атомной поляризацией (она составляет 5—8% от молярной поляризации), то В—(Р—RM)T. Поляризацию измеряют, рефракцию рассчитывают по атомным рефракциям,
85
рефракциям кратных связей и циклов или определяют экспериментально.
Электрический момент диполя молекул полярных веществ в жидком состоянии, рассчитанный по уравнениям (11.11), отличается от электрического момента диполя молекул вещества в газообразном состоянии из-за поляризации молекул при межмолекулярном взаимодействии. Для уменьшения межмолекулярного взаимодействия полярное вещество растворяют в неполярном растворителе, определяют поляризацию при нескольких концентрациях и экстраполируют поляризацию на бесконечное разведение. Поляризацию растворенного вещества определяют через удельную поляризацию:
еАВ — 1 1 еА 1 * Х А £В “ 1 1 ~ ХА
________ ______ = --------- ----- |_ -------- -------- еав +2 Рав ’ еа + 2 Ра Ев +2 Рв
(II. 12)
где еав, еа, ев — диэлектрическая проницаемость раствора, раство-£ав 1— 1
ренного вещества и растворителя. Величины тту ------ й
Едв - Рав
Ев — 1
Ев + 2
1
— определяют экспериментально, причем первую из них Рв
при нескольких концентрациях.
Большим затруднением при определении поляризации растворенного вещества при бесконечном разведении в неполярном растворителе является экстраполяция. Либо должна быть выполнена большая серия определений удельной поляризации раствора при нескольких концентрациях и графической экстраполяцией получена величина Ра,<х>> либо можно воспользоваться одной из экстраполяционных формул
ев-1 УИА Л4В 31£в-3(ЕВ- 1)(ев + 2)
= ев+2 Рв + Рв (8в + 2)2 ’ (И’13)
еАВ — £В п ?АВ “ ?В
где а=---------; р =; хА—молярная доля раство-
ХА ХА
ренного вещества А. С учетом Ра,™ электрический момент диполя
(Кл-м) вычисляют по уравнению
[Л = 4,27• 10-32 у(Рк -R^T. (II. 14)
В уравнении (П.14) поляризация и молярная рефракция растворенного вещества должны быть выражены в см3/моль.
2. РЕФРАКТОМЕТРЫ
Назначение, устройство и принцип работы рефрактометра ИРФ-23. Рефрактометр ИРФ-23 предназначен для определения показателей преломления монохроматического света жидких и твер*
86
дых веществ в интервале 1,33—1,78 с точностью 10-5 при фиксированной температуре. В комплект прибора входят призмы для разных диапазонов измерения показателя преломления: призма № 1 для измерений в интервале от 1,33 до 1,59; призма № 2 для измерений в интервале от 1,57 до 1,72; призма № 3 для измерений в интервале от 1,65 до 1,78. Принцип устройства основан на точном измерении угла полного внутреннего отражения при переходе светового потока из более плотной в менее плотную среду.
Оптическая схема состоит из измерительной призмы I (рис. 38), осветительной системы //, зрительной трубы III и отсчетной системы IV. Источником монохроматического светового потока служат
Рис. 38. Оптическая схема рефрактометра ИРФ-23
трубка Гейслера или натриевая лампа 1. При использовании трубок Гейслера, наполненных водородом или гелием, применяется конденсор 2. При этом призма 3 отводится от оптической оси. При использовании натриевой лампы 4 призму 3 с приклеенным к ней конденсором 5 поворачивают так, чтобы луч света, отраженный от ее грани, прошел через конденсор 5 и попал на кювету с исследуемым веществом. В поле зрения зрительной трубы (правый окуляр) имеется перекрестие. Зрительную трубу III можно поворачивать вокруг оси лимба 8. Для грубой наводки следует ослабить винт и поворачивать зрительную трубу на нужный угол. Точная установка перекрестия зрительной трубы на верхнюю границу спек-
87
Рис. 39. Поле зрения отсчетного микроскопа рефрактометра ИРФ-23
тральной линии монохроматического излучения осуществляется микрометрическим винтом. При точной наводке зрительной трубы винт ее должен быть завернут до отказа. В зрительной трубе помещается призма 6, которая служит для определения нуля шкалы прибора. Призма 6 освещается через систему призм лампочкой 7. Для отсчета угла поворота зрительной трубы имеется лимб 8 со спиральным окуляром Р. Шкала спирального окуляра освещается лампочкой 7. Для отсчета угла наклона зрительной трубы необходимо маховичком, расположенным в нижней части окуляр-микрометра, повернуть диск с двойной спиралью до совмещения штриха градусного деления с двойной спиралью (рис. 39). Угол наклона на оис. 39 равен 12,2715°.
В правой части осветительного устройства имеется окно, которое открывается только для установки нуля прибора. Трубки Гейслера включаются через автотрансформатор, рукоятка которо-
го перед включением должна находиться в крайнем левом положении «меньше». После появления свечения трубки рукоятку повернуть до нормального режима свечения в сторону «больше». Перед включением водородной лампы на нее следует надеть печь, соединенную с трансформатором «подогрев», включить «подогрев» и через 2—3 мин включить ее.
Таблица 5. Длины волн спектральных линий и условные обозначения показателя преломления
Источник светового потока Цвет Длина волны, нм Обозначение показателя преломления
Лампа
водородная Красный 656,3 Пс
натриевая Желтый 589,3 Пи
гелиевая » 587,6 nd
ртутная Зеленый 546,8 ng
водородная Синий 486,1 Пг
ртутная » 435,8 пе
водородная » 434,1 tlG
88
а (Г
Рис. 40. Поле зрения зрительной трубы рефрактометра ИРФ-23 при установке нуля прибора (а) и при установке на спектральные линии натрия (О
Натриевая лампа включается через дроссель. Ее включение можно производить только при положении сердечника дросселя «ток меньше». После начала свечения лампы маховичок дросселя поворачивают на один оборот в сторону «ток больше».
Последовательность выполнения работы. Установить на рефрактометре призму с показателем преломления, большим, чем предполагаемый показатель преломления исследуемого вещества. Включить источник монохроматического света. Длины волн спектральных линий приведены в табл. 5.
Поместить 1,5 мл исследуемой жидкости в стакан, приклеенный к призме. Установить температуру в ультратермостате по термометру на рефрактометре. Надеть деревянную колодку на стакан с исследуемой жидкостью и опустить в нее обогревательное приспособление, через которое протекает вода из ультратермостата. Определить нуль шкалы рефрактометра, для чего открыть заслонку справа от лампы подсветки 7 (см. рис. 38), ослабить стопорный винт и установить светлый квадрат на правой части поля зрения между штрихами перекрестия, зажать стопорный винт
и точно совместить риски на квадрате с перекрестием (рис. 40, а). Произвести отсчет ф0 по спиральному окулярмикрометру. Измерить угол ф, для чего ослабить стопорный винт зрительной трубы, подвести перекрестие в поле окуляра к верхней части изображения спектральной линии, завернув стопорный винт, микрометрическим винтом установить перекрестие точно на верхнюю часть изображения спектральной линии (рис. 40, б). Определить показатель преломления по абсолютному значению угла ф1—фь, пользуясь таблицей, приложенной к прибору. Результаты измерений и вычислений показателя преломления записать в таблицу по образцу:
Вещество . . .; призма . . .; температура . . .; источник света . .
длина волны . . .
Показания прибора Отсчеты Среднее значение
1 2 3 4 5
Начало отсчёта по шкале <pQ Угол отсчета по шкале дч для исследуемого вещества Предельный угол <pi—ф0 Справочная величина показателя преломления 1
Назначение, устройство и принцип работы рефрактометра Аббе. Рефрактометр Аббе предназначен для измерения показателей преломления жидкостей в пределах от 1,33 до 1,70. Принцип работы основан на определении угла полного внутреннего отражения. Имеется приспособление для термостатирования исследуемой жидкости.
Исследуемое вещество помещают между двумя прямоугольными призмами 2 и 4 (рис. 41). Световой поток от зеркала 1 отра-
Рис. 41. Ход лучей света в рефрактометре Аббе
преломления призмы и вещества мы 2 есть величина постоянная.
жается на прямоугольную призму 2. Преломившись на границе раздела воздух — стекло, световой поток попадает на поверхность раздела 3 стекло — исследуемое вещество. Если постепенно увеличивать угол падения, то при некотором угле выходящий из призмы 2 световой поток направится вдоль грани призмы, т. е. наступит полное внутреннее отражение. Угол, при котором наступает полное внутреннее отражение, зависит от показателей 3. Показатель преломления приз-
13 приборе изменяют угол наклона призмы относительно зеркала до тех пор, пока граница раздела освещенной и темной половины поля зрения окуляра не будет установлена точно на перекрестие. На шкале рефрактометра непосредственно нанесены значения показателя преломления с точностью до 10-3. На рефрактометре Аббе показатель преломления измеряется в белом свете. При появлении радужной границы раздела необходимо изменить положение призм-компенсаторов, которые вращаются маховичком, расположенным справа от зрительной трубы.
Последовательность выполнения работы. Наклонить зрительную трубу вместе со шкалой прибора от себя. Открыть призму и протереть обе призмы растворителем. Нанести каплю исследуемой жидкости на верхнюю призму, находящуюся при этом горизонтально. Закрыть нижнюю призму, наклонить зрительную трубу со шкалой на себя и произвести измерение, для чего вращением маховичка слева от шкалы добиться расположения границы темного и светлого поля на перекрестии. Вращением маховичка призмы компенсатора добиться, чтобы граница между светлым и темным полями не была радужной. Откорректировать положение границы относительно перекрестия и сделать отсчет по шкале.
Назначение, устройство и принцип работы лабораторного рефрактометра типа РЛ. Рефрактометр РЛ предназначен для измерения показателя преломления растворов сахара и жидкости в пределах от 1,33 до 1,54. Измерения на приборе производят с использованием дневного света. Прибор имеет кроме шкалы показателя пре
90
ломления шкалу процентного содержания сахара в водном растворе. Принцип действия рефрактометра основан на определении угла полного внутреннего отражения. Вещество помещают между призмами. Световой поток, вышедший из нижней призмы, рассматривают в поле зрения окуляра. Перекрестие в поле зрения окуляра совмещают с границей темного и светлого поля поворотом рукоятки, на которой смонтирован окуляр. Показатель преломления исследуемой жидкости отсчитывают по шкале.
Последовательность выполнения работы. Открыть верхнюю призму с оправой. Нанести каплю исследуемого вещества на нижнюю призму. Навести зеркалом световой поток на окно нижней призмы. Перемещать рукоятку с окуляром до тех пор, пока перекрестие не совместится с границей полей. Добиться отсутствия радужной границы полей вращением маховичка призмы компенсатора на рукоятке окуляра. Уточнить установку перекрестия на границе полей. Произвести отсчет показателя преломления по левой шкале в поле зрения окуляра.
3. диэлькометры
Назначение, устройство и принцип работы диэлькометра типа Е8-1. Прибор Е8-1 предназначен для измерения межэлектродных емкостей. На приборе можно измерять емкости в пределах от 10-4 до 50 пФ (1 пФ=10~12 Ф). Прибор имеет пять поддиапазонов. Пределы измерения емкости от 10~4 до 5-10~3 пФ: первый поддиапазон (X0,0001), второй поддиапазон (X0,001) от 5-Ю-3 до 5-10-2 пФ, третий поддиапазон (Х0,01) от 5-10~2 до 5-10~! пФ, четвертый поддиапазон (Х0,1) от 0,5 до 5,0 пФ и пятый поддиапазон (Х1) от 5,0 до 50 пФ. Погрешность измерения возрастает с уменьшением измеряемой емкости. Она составляет от 0,0001 до 0,001 пФ±5%, от 0,001 до 0,1 пФ±2%, от 0,1 до 50 пФ±1%. Частота тока генератора 465 кГц±2%. Измерение емкости производится по мостовой схеме (рис. 42). В диагональ ВД моста подается напряжение от генератора Г высокой частоты 465 кГц. С диагонали АБ напряжение снимается через усилитель переменного тока У. При балансе схемы напряжение между точками АБ равно нулю. Это напряжение подается на сетку измерительной электронной лампы, что обеспечивает максимальный анодный ток, а следовательно, и максимальное отклонение стрелки прибора. При наличии напряжения между точками АБ отклонение стрелки прибора уменьшается. Следовательно, для получения баланса схемы необходимо добиваться максимального отклонения стрелки прибора. Изменение в балансе схемы производится конденсатором переменной емкости С, обеспечивающим линейную зависимость емкости от угла поворота подвижных пластин конденсатора, связанного с равномерной шкалой прибора. В два плеча схемы включены конденсатор измеряемой емкости Сх и эталонный конденсатор Со. В два других плеча включены сопротивления и Т?2 и конденсатор переменной емкости С.
91
Диэлектрическую проницаемость жидкости определяют измерением емкости конденсатора. Емкость конденсатора прямо пропорциональна диэлектрической проницаемости диэлектрика, помещенного между электродами. Емкость конденсатора с плоскопараллельными электродами равна
C = eS/4nZ, (П.15>
где S — площадь электродов, см2; I — расстояние между электродами, см. Соотношение емкостей конденсатора, заполненного разными диэлектриками, равно соотношению их диэлектрических прони-цаемостей:
СЛ/С1 = вх/*1- (П.16)
Таким образом, необходимо измерить емкость конденсатора, заполненного жидкостью с известной диэлектрической проницаемостью,
Рис. 42. Принципиальная схема измерения емкости конденсатора
Рис. 43. Сосуд для измерения дипольного момента молекул
и емкость этого же конденсатора, но заполненного исследуемой жидкостью. Тогда
или
СХ^К*Х,
К = S/4nZ.
(II.17)
(II. 18)
При измерении емкости в последнюю включается не только емкость конденсатора с диэлектриком, но и емкость всех проводников, соединяющих конденсатор с измерительной схемой Сп. Чтобы учесть емкость проводников Сп, необходимо измерить емкость измерительной ячейки, заполненной воздухом. Для этого тщательно продуть ячейку (рис. 43) теплым воздухом для удаления летучих веществ и произвести измерение емкости незаполненного конденсатора вместе с емкостью проводников:
Cq = Cq 4- Сп == 4~ Сп• (11.19)
92
Емкость конденсатора, заполненного диэлектриком с известным значением диэлектрической проницаемости 8i вместе с емкостью проводников, будет
— (7П — /С?! CQ —
(11.20)
Емкость конденсатора, заполненного диэлектриком с неизвестной диэлектрической проницаемостью еж вместе с емкостью проводников, будет
(11.21)
Совместное решение уравнений (11.20) и (11.21)
ех __ 1 ~р (ej -р 1) (СЛ — С0)/(С1 — Со).
(11.22)
Диэлектрическая проницаемость диэлектрика зависит от температуры. Поэтому ячейка для измерения должна быть термостати-рована. Ячейка (рис. 43) представляет сосуд 1 с впаянными в него двумя электродами 3, Для исключения проводимости между электродами последние покрыты тонким слоем стекла. Сосуд помещен в термостатирующую рубашку 2, через которую циркулирует вода из ультратермостата. Сосуд крепится непосредственно на приборе^ с тем чтобы по возможности снизить емкость подводящих проводников. Эталонное или исследуемое вещество заливают в сосуд через воронку, помещают термометр, включают диэлькометр в электросеть напряжением 220 В, включают ультратермостат и примерна через 10 мин, когда установится заданная температура, производится измерение.
Для измерения установить переключатель поддиапазонов в положение «XI». Поставить регулятор чувствительности в крайнее против часовой стрелки положение. Поставить регулятор компенсации потерь в крайнее левое положение. 'Вращением лимба «отсчет Сж» добиться максимального отклонения стрелки гальванометра. Увеличить чувствительность прибора поворотом рукоятки «чувствительность» по часовой стрелке. При этом показание гальванометра ? должно уменьшаться, а чувствительность возрасти. Вновь настроить рукояткой «отсчет Сх» на максимальное показание гальванометра. Увеличить по возможности показание гальванометра вращением ру-J коятки «компенсация потерь» и вновь добиться максимального по-; казания гальванометра вращением рукоятки «отсчет Сж». Произвес-j ти отсчет по барабану и лимбу. Измеряемая емкость равна сумме I показаний на лимбе и на барабане. Полученную величину емкости | следует умножить на показание переключателя «множитель». Про-I извести измерение емкости незаполненного конденсатора, конденса-i тора, заполненного жидкостью с известным значением диэлектри-1 ческой проницаемостью, и жидкостью, значение диэлектрической проницаемости которой необходимо измерить. По уравнению (II.22) вычислить диэлектрическую проницаемость еж.
93
Работа 1. Изучение зависимости показателя преломления1 жидкости от температуры и длины волны светового потока
Работа производится на рефрактометре ИРФ-23, на котором следует определить показатель преломления жидкости при трехчетырех температурах и при трех-четырех длинах волн светового потока.
Последовательность выполнения работы. Установить низшую заданную температуру при помощи контактного термометра на ультратермостате. Поместить исследуемую жидкость в сосуд на призме рефрактометра. Включить натриевую лампу и, когда установится температура по термометру на рефрактометре, произвести измерение угла преломления. Угол преломления необходимо измерять трижды, подводя перекрестие в поле зрения зрительной трубы к спектральной линии излучения натрия со стороны меньшего и большего отсчета угла. По таблице, приложенной к рефрактометру, для призмы, установленной на рефрактометре, определить показатели преломления для всех трех отсчетов угла. Определить среднее арифметическое значение показателя преломления и погрешность. Изменить температуру контактным термометром на ультратермостате. После того как установится постоянная температура по термометру, на рефрактометре повторить измерения углов преломления и определить показатель преломления для данной температуры. Произвести определение показателя преломления при всех заданных температурах.
Вычертить график зависимости показателя преломления жидкости при длине волны 589,3 нм от температуры. Рассчитать коэффициенты линейного уравнения зависимости показателя преломления от температуры методом наименьших квадратов, воспользовавшись ЭВМ и программой, приведенной в приложении. Определить производную dn/dT. Установить заданную температуру для изучения показателя преломления жидкости в зависимости от длины волны светового потока. Изменяя источники излучения, произвести измерение угла преломления светового потока в зависимости от длины волны. Каждое измерение произвести трижды. По таблице определить показатели преломления. Вычертить график зависимости показателя преломления от длины волны светового потока.
Работа 2. Определение концентрации растворенного вещества по показателю преломления раствора
Работа может быть выполнена на рефрактометре типа РЛ, Аббе или ИРФ-23.
Последовательность выполнения работы. Приготовить разбавлением стандартного раствора известной концентрации пять-шесть растворов с заданными концентрациями растворенного вещества. Измерить показатели преломления всех растворов при заданной температуре. Построить график зависимости показателя преломле
94
ния раствора от концентрации. При построении графика воспользоваться методом наименьших квадратов. Вычислить погрешность каждого измерения.
Работа 3. Изучение зависимости показателя преломления от концентрации растворов
Работа выполняется на рефрактометре типа РЛ, Аббе или ИРФ-23.
Последовательность выполнения работы. Измерить показатели преломления чистых жидкостей, из которых затем приготовить растворы. Приготовить смешением чистых жидкостей растворы с объемным содержанием первого компонента 0,2 (20%); 0,4 (40%); 0,6 (60%) и 0,8 (80%). Измерить показатели преломления приготовленных растворов. Построить график зависимости показателя преломления от содержания первого компонента. Определить плотности обеих жидкостей пикнометрическим способом (см. с. 100). Рассчитать молярное содержание в процентах всех приготовленных растворов. Построить график зависимости показателя преломления от концентрации. Сделать заключение о характере полученных зависимостей.
Работа 4. Определение молярной рефракции вещества и установление строения молекул
Работа выполняется на рефрактометре типа РЛ, Аббе или ИРФ-23.
Измерить показатель преломления и плотность (см. с. 100) жидкости при заданной температуре. Рассчитать молярную рефракцию вещества при заданной температуре по уравнению (И.5).
Написать все возможные структурные формулы соединения на основании эмпирической формулы. Рассчитать молярную рефракцию веществ со всеми возможными структурами молекул по уравнению (П.6). Сопоставить экспериментально полученную молярную рефракцию с рассчитанными и сделать заключение относительно структурной формулы исследуемого вещества.
Рассчитать погрешность определенного значения рефракции. Для этого прологарифмировать уравнение (II.5):
In - In (п2 — 1) - In (п2 + 2) 4- In М - In р. (И .23)
После дифференцирования (11.23) и приняв, что все относительные погрешности положительны, вычислить относительную погрешность молярной рефракции:
d d (и2 — 1) d (к2 4- 2) rd р
/?м = п2 — 1 + П2 + 2 + V ’
ИЛИ
d 2nd п 2п d п 2nd п dp п2—1~^п2 — l~f~n24-2~t~p*
§5
Относительная погрешность в определении плотности рассчитывается по уравнению
d р _ dg2 dg0 dgi dg0
Р g%— go g2 — go gl — go gl — gO
(11.24)
we go, gi и g% — масса пикнометра пустого, с водой и с веществом соответственно.
Относительная погрешность рефракции после замены дифференциалов конечными значениями абсолютных погрешностей будет
А#м _ 2/гДп 2пАп &g2 AgQ &gp
R ~п2 — 1 п2 +2g2 — g0 g2 — g0 gl — gQ gl —go’
f
Абсолютная погрешность молярной рефракции получается умножением относительной погрешности на молярную рефракцию.
Работа 5. Изучение свойства молярной рефракции вещества
Работа проводится на рефрактометре типа РЛ, Аббе или ИРФ-23. Определить показатель преломления жидкого вещества при трех-четырех температурах. Определить плотность того же вещества (см. с. 100) при тех же температурах, при которых измерены показатели преломления. Плотность удобно определять одновременно с определением показателя преломления, термостатируя пикнометр в том же ультратермостате, который использовался длятер-мостатирования рефрактометра. Рассчитать молярную рефракцию вещества при всех температурах. Определить погрешность молярной рефракции при всех температурах. Сделать заключение относительно зависимости молярной рефракции от температуры.
Работа 6. Определение электрического момента диполя молекул . вещества
Работа выполняется на диэлькометре типа Е 8-1. Приготовить несколько растворов полярного вещества в неполярном растворителе. Определить емкость конденсатора, заполненного воздухом, растворителем и каждым раствором. Рассчитать диэлектрические проницаемости всех растворов при заданной температуре по уравнению (11.22), используя справочное значение диэлектрической проницаемости растворителя.
Определить плотности всех растворов при той же температуре, при которой производилось измерение емкости. Рассчитать поляризацию. растворенного вещества по уравнению (11.12). Построить график зависимости поляризации растворенного вещества от концентрации раствора. Экстраполяцией до бесконечного разведения определить Л», показатели преломления всех растворов и растворителя. Рассчитать удельную рефракцию растворенного вещества по уравнению (II.9). Определить молярную рефракцию растворенного вещества. По уравнению (11.14) рассчитать электрический момент диполя молекул растворенного вещества.
96
ГЛАВА III
ЖИДКОЕ СОСТОЯНИЕ ВЕЩЕСТВА
ПОВЕРХНОСТНОЕ НАТЯЖЕНИЕ. ПАРАХОР
Поверхностное натяжение есть работа или изменение энергии Гиббса при увеличении единицы площади поверхности
<j=AG, (Ш.1)
где о — поверхностное натяжение, Н/м.
Д. И. Менделеев установил, что зависимость поверхностного натяжения от температуры линейна. Поверхностное натяжение уменьшается с ростом температуры и-становится равным нулю при критической температуре. Так как (d&G/dT)p = —AS, то по зависимости поверхностного натяжения от температуры можно вычислить AS при образовании единицы площади новой поверхности. Из линейного характера этой зависимости видно, что AS — величина постоянная. Между поверхностным натяжением и плотностями жидкости и пара, находящихся в равновесии, при данной температуре установлена зависимость
а = С (рж — рп)4, (Ш.2)
где С — коэффициент пропорциональности, зависящий от свойств вещества; рж— плотность жидкости; рп — плотность пара. Так как плотность жидкости при температурах, далеких от критической, значительно больше плотности насыщенного пара, то уравнение (III.2) можно записать
с = (Ш.З)
откуда
С’/^а’Л/рж. (Ш.4>
После умножения обеих частей (III.4)'на молекулярную массу получим величину, называемую парахором:
Парахор обладает конструктивным свойством, т. е.
Р ^/А\ат 4~ ^Z/A.cb 4~ ^/А,ц» (III.6)
Z I I
где А, ат А, св, А, ц — парахоры атомов, кратных связей, циклов; Л/, m-i, 1[ — число атомов, кратных связей, циклов.
Плотность жидкости. Плотностью называется масса единицы объема вещества. Плотность жидкости зависит от температуры. По Менделееву, эта зависимость описывается уравнением
= Рг0 [! — —Го)], (Ш.7)
где р — плотность; р— коэффициент объемного расширения;
1 / dv \
? = — Н— (III.8)
v0 V аТ /р
4—2083
97
Коэффициент р в широком интервале температур не зависит от температуры:
р = М-у, (Ш.9)
где Av— изменение объема при изменении температуры на Г, Коэффициент объемного расширения воды в интервале 273—310 К равен 2,07-10“4 К"1. Коэффициент объемного расширения большинства органических жидкостей в 2—5 раз больше. Следовательно, допустимая погрешность плотности не должна превышать ±0,001 X Х103 кг/м3; необходимо поддерживать постоянство температуры в пределах 1 К.
Рис. 44. Схема установки для измерения поверхностного натяжения жидкости
Работа 1. Определение поверхностного натяжения жидкости
Поверхностное натяжение жидкости определяют измерением наибольшего давления газовых пузырьков, образующихся в жидкости. Для этого капилляр трубки 4 (рис. 44) погружают вертикально в жидкость так, чтобы ее торец только касался поверхности жидкости. Если давление в трубке 4 больше, чем давление над поверхностью жидкости, то на конце капилляра образуется пузырек газа, который по мере уменьшения давления над поверхностью жидкости будет ра
сти до некоторого предела. Избыточное давление прямо пропорционально радиусу капилляра:
/? = Ла/r. (ШЛО)
Обычно для определения поверхностного натяжения пользуются стандартной жидкостью, поверхностное натяжение которой при заданной температуре известно. Для двух жидкостей с различными поверхностными натяжениями а и По при использовании одного и того же капилляра справедливо соотношение
^0-^0- (ПЕН)
Тогда поверхностное натяжение исследуемой жидкости будет
(III.12)
Последовательность выполнения работы. Налить в сосуд 5 (рис. 44) стандартное вещество так, чтобы капилляр трубки 4 касался поверхности жидкости. Налить немного больше требуемо
98
го количества жидкости и избыток ее отобрать капилляром. Если поверхностное натяжение жидкости измеряется при температуре более высокой, чем температура окружающей среды, то уровень жидкости следует установить после термостатирования. Сосуд 6 для термостатирования следует соединить с ультратермостатом и установить контактным термометром заданную температуру в ультратермостате. Температуру жидкости измерять термометром 3. Поместить трубку 4 в сосуд 5. При этом кран 2 должен быть открыт на атмосферу. Записать показание тягомера 7. Повернуть кран 2 в такое положение, чтобы сосуды 1 и 5 были соединены. Открыть кран 8 так, чтобы из сосуда 1 по каплям вытекала вода со скоростью не более одной капли в 3—4 с. При этом будет создаваться разрежение в сосуде /ив пространстве над жидкостью в сосуде 5. Давление внутри капилляра останется постоянным и равным атмосферному. Записать максимальное показание тягомера. При увеличении разрежения над жидкостью показание тягомера растет, так как давление над правым открытым коленом сообщающегося сосуда остается постоянным. При достижении определенного разрежения пузырек воздуха отрывается от капилляра. Измерения производят 10—20 раз. Стандартную жидкость заменяют на исследуемую. Замену произвести пипеткой. Сполоснуть сосуд 5 исследуемой жидкостью, удалить ее и вновь заполнить сосуд. Произвести измерения по тягомеру и рассчитать поверхностное натяжение по уравнению (III.12) и погрешность определения поверхностного натяжения.
Примечание. Стандартную жидкость желательно подбирать так, чтобы ее поверхностное натяжение было бы близко к поверхностному натяжению исследуемой жидкости.
Работа 2. Изучение зависимости поверхностного натяжения жидкости от температуры •
Поверхностное натяжение определяют аналогично описанному в работе /.
Последовательность выполнения работы. Залить в сосуд 5 (рис. 44) стандартную жидкость и измерить ее поверхностное натяжение при нескольких температурах. Температуру измерять термометром 3. Она может на 1—2 К отличаться от той, которую показывает термометр в ультратермостате. По окончании измерений поверхностного натяжения стандартной жидкости пипеткой извлечь ее из сосуда 5, высушить его и залить исследуемую жидкость. При замене жидкости в сосуде 5 через ультратермостат пустить холодную водопроводную воду для того, чтобы температура воды в ультратермостате снизилась до начальной. Измерить разрежение тягомером для исследуемой жидкости при всех заданных температурах.
Построить графики зависимости поверхностного натяжения стандартной и исследуемой жидкости от температуры. Рассчитать по
4*
99
верхностное натяжение. Результаты измерений обработать по методу наименьших квадратов, применяя ЭВМ и программу, приведенную в приложении. Экстраполяцией определить критическую температуру.
Работа 3. Изучение термодинамики поверхностных явлений
Измерить поверхностное натяжение исследуемой жидкости, как это было описано в работе 1. Построить график v=f(T). По тангенсу угла наклона прямой рассчитать изменение энтропии образования единицы поверхности жидкости. По уравнению (III.1) рассчитать АН образования единицы поверхности жидкости.
Работа 4. Определение парахора и установление структуры молекул
Измерить поверхностное натяжение (см. с. 98) жидкости с известной эмпирической формулой, но с неизвестной структурой. Измерить плотность исследуемой жидкости (см. ниже). Рассчитать парахор жидкости по уравнению (III.5). Сделать предположение относительно структуры исследуемого вещества. Рассчитать парахор для всех предполагаемых структур по уравнению (III.6). Сделать заключение относительно структуры молекулы исследуемой жидкости.
Работа 5. Определение плотности жидкости
Плотность жидкости определяют взвешиванием известного объема ее в пикнометре. Вместимость пикнометра устанавливается по массе заполняющей его воды. Зная массу воды и ее плотность при заданной температуре, можно рассчитать вместимость пикнометра. Даже при наиболее точных измерениях плотности не следует брать пикнометр, вместимость которого больше 30 мл.
Последовательность выполнения работы. Пикнометр промыть хромовой смесью, водой, высушить и взвесить с точностью до 0,0001 г. 3аполнить пикнометр водой до метки и поместить его в ультратермостат на 15—20 мин до установления заданной температуры жидкости. Если уровень жидкости за счет нагревания ее повысился, то капиллярной пипеткой следует удалить часть жидкости так, чтобы уровень совпадал с меткой. Если же он оказался ниже метки, то следует добавить небольшое количество жидкости. После термостатирования взвесить пикнометр с водой на аналитических весах, предварительно высушив его наружную поверхность фильтровальной бумагой. Вылить воду, высушить пикнометр и залить исследуемой жидкостью. Поместить пикнометр с жидкостью в ультратермостат на 15—20 мин, довести до метки уровень жидкости при
100
ее термостатировании. Вынуть, высушить наружную поверхность пикнометра и взвесить его на аналитических весах. Плотность жидкости рассчитать по уравнению
? = (т2 — — т0) рНа0,
(III.13)
где то — масса пустого пикнометра; т\— масса пикнометра с водой; m2 — масса пикнометра с исследуемой жидкостью; рн2о— плотность воды.
Обычно плотность определяют в двух пикнометрах параллельным взвешиванием. Погрешность определения плотности жидкости вычисляют по уравнению (11.24). Рассчитать относительную и абсолютную погрешность определения плотности жидкости.
Работа 6. Изучение зависимости плотности жидкости от температуры
Принцип определения плотности жидкости при различных температурах аналогичен описанному в работе 5. Плотность воды в
зависимости от температуры приведена в справочниках для широкого диапазона температур. Для установления зависимости плотности от температуры необходимо при заданных температурах взвесить пикнометр с водой и с исследуемой жидкостью. Масса пустого пикнометра не зависит от температуры. Работу проводить также в двух пикнометрах при параллельном взвешивании. Построить график зависимости плотности жидкости от температуры.
ГЛАВА IV
ЭЛЕКТРОНОГРАФИЧЕСКИЙ АНАЛИЗ
1. МЕТОД ЭЛЕКТРОНОГРАФИИ И ЕГО ПРИМЕНЕНИЕ
Электронооптический анализ основан на волновых свойствах электронов и делится на микроскопический, проводимый в электронном микроскопе, и дифракционный, изучающий атомно-кристаллическое строение вещества в электронографе или электронном микроскопе. В наиболее распространенных электронографах типа ЭГ-100 и электронных микроскопах типа ЭМВ-100 применяют электрические поля с ускоряющим напряжением V= = 404-100 кВ. На рис. 45 показана принципиальная оптическая схема электронографа. В соответствии с уравнением де Бройля длина волны движущегося электрона определяется по уравнению
— h/itiqU ,
(IV. 1)
где h — постоянная Планка; те — масса электрона; ve — скорость электрона.
101
Для ускорения электронов используют электрические поля. В зависимости от разности потенциалов длина волны (нм) без учета релятивистской поправки определяется по уравнению
= 1,2236/VV. (IV.2)
При ускоряющем фракция электронов,
напряжении 100 кВ Хе=3,7-10 3 нм. Ди-рассеянных кристаллической решеткой, как
Рис. 45. Принципи-
альная оптическая
и дифракция рентгеновского излучения, описывается уравнением Вульфа — Бреггов, которое для небольших длин волн и углов 0 имеет вид
г --- \eL!d
HKL
(IV.3)
где г — расстояние от рефлекса до следа первичного пучка электронов, м; L — расстояние от образца до фотографической пластинки, м.
Если объект состоит из множества беспорядочно ориентированных кристаллов, электронные волны, претерпевшие дифракцию на одинаковых кристаллических плоскостях, образуют конус пучка электронов, пересекающих экран (или фотопластинку) по кольцу, радиус которого г. Отсюда по (IV.3) вычисляют межплоскостное расстояние.
Обычно прибор калибруют по дифракционной картине, создаваемой известным веществом, при заданном ускоряющем напряжении. Калибрование сводится к определению посто-
явной прибора:
С =
(VL4)
схема электр оно-
графа:
1 — электронная пушка; 2 — катод; 3 — фокусирующий электрод; 4 — аиод; 5, 6 — электромагнитные
линзы; 7 — кристал-лодержатель; 8 — фотопластинка; L — расстояние от образца до фотопластинки; г — расстояние рефлекса от первичного пучка электронов
d =С/г. (IV.5)
ЛА Ь
Точность определения периодов кристаллической решетки по электронной дифракции по сравнению с рентгеновской дифракцией невелика. Однако преимуществом электронографии является то, что при помощи ее можно получить информацию для кристаллов вещества размером 2—20 нм и их субмикроколичеств. Это же предъявляет высокие требования к чис-
тоте проведения анализа, так как мельчайшие загрязнения на поверхности объекта дают собственную дифракционную картину. Электронографическим анализом решаются те же задачи, что и рентгенографическим анализом: определение фазового состава и
кристаллической структуры вещества, его текстуры, ориентировок и т. п. Метод электронографии применяют для анализа тонких
102
пленок и порошков, а также поверхностных слоев массивных образцов. В соответствии с этим существуют два метода работы: «на просвет» и «на отражение». На просвет исследуются тонкие порошки, оксидные пленки толщиной не более 102 нм при V—100 кВ. Пленки получают конден-
сацией в вакууме, отделением от металла или другими способами. При работе на просвет для анализа получают полную электронограмму (рис. 46, а). Съемку на отражение проводят в пучке электронов, скользящем вдоль поверхности объекта. При этом наиболее вероятно рассеяние электронов на мельчайших
выступах поверхности объекта. При работе на отражение получают половину электронограммы для анализа (рис. 46, б).
Рис. 46. Схема электрон огр амм поликри-сталлического образца на просвет (а) и поликристаллического массивного образца на отражение (б):
г — радиус кодец, D,, D2 — диаметры колец; 1 — след первичного пучка электронов (точка отсчета на электронограмме); 2 — ближние отражения;
3—дальние отражения
2. ПРИГОТОВЛЕНИЕ ОБРАЗЦОВ
ДЛЯ ЭЛЕКТРОНОГРАФИЧЕСКОГО АНАЛИЗА
Подготовка препаратов для электронографического анализа на просвет принципиально не отличается от подобной работы при электронно-микроскопическом анализе. Порошки для исследования на просвет наносят на пленку-подложку, которая помещается на металлическую сетку-держатель размером 2—6 мм. В качестве подложки применяют, например, пленку коллодия или углерода. Пленку коллодия готовят из раствора целлулоида (отмытую от эмульсии фотографическую или рентгеновскую пленку растворяют в амилацетате). Углеродную пленку получают на вакуумной установке типа ВУП испарением углерода из угольных спектральных электродов. Один из электродов затачивают на конус с углом 30— 45°, а на другом делают площадку под углом 45—60°.
При соприкосновении углеродных электродов и подаче тока силой до 100 А в течение 5—15 с на образце 1 образуется тонкий слой углерода—пленка-подложка. Обычно углерод осаждают на свежий скол, например, галита или слюды. Снимают пленку-подложку опусканием кусочка галита под небольшим углом в дистиллированную воду (рис. 47). Углеродная пленка всплывает и затем ее вынимают на сетку-держатель, которую подводят под плен
103
ку с помощью пинцета. Далее пленку на сетке высушивают, нарезают по размеру держателя электронографа или микроскопа. На подготовленную сетку-держатель с пленкой-подложкой наносят каплю жидкости с исследуемым порошком (рис. 48). Непосредственно на скол галита наносят исследуемый порошок (если он не взаимодействует с водой) и после напыления углеродной пленки, отделения ее от поверхности галита и вылавливания ее на сетку-держатель образец готов для исследований.
Для изготовления коллодиевых пленок используют поверхность дистиллированной воды. Рекомендуется брать сосуд диаметром
Рис. 47. Снятие углеродной пленки со скола:
1 — чашка Петри с дистиллиро-рованной водой; 2— кусок галита; 3— углеродная пленка
Рис. 49. Схема уста- . новки шлифа для
сушки:
Рис. 48. Нанесение образца на пленку-подложку, укрепленную на сетке-держателе:
1 — сетка-держатель; 2 — углеродная пленка-подложка; 3 — капля взвеси исследуемого порошка в спирте (или в воде); 4 — пипетка
/ — шлиф; 2 — полированная и затем травленая поверхность шлифа;
3 — слой коллодия
около 90 и глубиной не менее 60 мм. Для предварительной очистки поверхности воды от пыли и других загрязнений на нее наносят каплю коллодия (1%-ный раствор коллодия в амилацетате) и образовавшуюся пленку вынимают и выбрасывают. На очищенную поверхность воды снова наносят каплю коллодия и пленку вынимают на сетки-держатели. Далее образец наносят в виде капли взвеси (рис. 48).
При исследовании твердого вещества образец, например сплав в виде металлографического шлифа, травят по режиму, обеспечивающему разделение фаз. После образования на шлифе значительного осадка (при этом поверхность шлифа темнеет и начинает мазаться) на нее наносят каплю 5%-ного раствора коллодия. Образец ставят вертикально (рис. 49). После высыхания пленку нарезают на квадраты размером 3—5 мм и отделяют от поверхности шлифа погружением образца в тот же раствор, в котором происходило травление. Куски пленки вместе с вытравленной частью образца всплывают, их вылавливают на сетки-держатели, осторожно промывают в дистиллированной воде и спирте и высушивают под колпаком, предохраняя от пыли.
104
Конденсацией из пара в вакууме получают пленки из чистого металла или сплава. Сплавы готовят одновременным испарением отдельных компонентов со сплава из разных испарителей с последующей термической обработкой для диффузионного выравнивания состава. Структура пленки металла или сплава существенно зависит от ее толщины, скорости испарения, материала и температуры подложки. При конденсации образца на подогретую монокристаллическую подложку можно получить практически монокристаллическую пленку, пригодную для многих исследований. Толщину пленки рассчитывают по формулам:
Рсл — /?z/4nr2;
l р
(IV.6)
где рСл — плотность напыленного слоя, кг/м2 (рСл —0,75т/4лг2); т— масса напыленной навески, кг; р — плотность исходного материала, кг/м3; г — расстояние от испарителя до подложки, м; I — толщина слоя, м.
Работа. Получение и расчет электронограмм v
поликристаллических образцов
Электронограф состоит из колонны, вакуумной части, блоков электрического питания и управления (см. также техническую инструкцию на эксплуатацию прибора). В верхней части колонны расположены электронная пушка, электромагнитные линзы, кри-сталлодержатель образца, смотровые окна, проекционный тубус, фотокамера с фотопластинками.
Последовательность выполнения работы. Подготовить эталон* ный образец (MgO, NaCl, Al). Подготовить образец для исследования на просвет (металлическую, например, алюминиевую фольгу, приготовленную травлением). Подготовить образец для исследования на отражение (шлиф металла или сплава). Снять элек-тронограммы: эталонного вещества, изучаемого вещества на просвет или на отражение, изучаемого и эталонного вещества одновременно (последнее проводить нанесением эталонного вещества на готовый препарат, например каплю раствора NaCl на готовый образец, если он не растворяется в воде). Определить постоянную прибора, используя данные о межплоскостных расстояниях C^hkl) эталона из справочников. Провести качественный фазовый анализ образца по полученной электронограмме. Определить кристаллическую структуру одной из фаз объекта. Радиус колец измерить металлической линейкой или с помощью компаратора с возможной точностью в двух взаимно перпендикулярных направлениях. При использовании линейки со скошенным краем измерения записывать с точностью до 0,1 мм. На рис. 46 показана схема
105
измерений электронограмм. Результаты и определения постоянной прибора занести в таблицу по образцу:
Напряжение . . . (60, 80, 100 кВ)
Эталон . . .
Номер кольца fCp’ Н KL d„KL< нм ММ" НМ
и -
Постоянную прибора рассчитать по формуле (IV.4). Ошибка в определении постоянной прибора равна AC=dHKLAr; Аг — ошибка в измерении радиуса кольца (0,1 мм). Электронограммы поли-кристаллического образца определить по межплосткостным расстояниям по формуле (IV.5), а идентификацию исследуемого вещества— по справочникам. Результаты измерений, расчета и справочные данные записать в таблицу по образцу:
Исследуемое вещество . . .; напряжение, кВ . . . (40, 60, 80, 100); по-
стоянная прибора . . .
Номер кольца ч Интенсивность rcps мм ^HKL — н къ’ нм Справочные данные
фаза А фаза Б
KL> нм интен- сивность KL’ НМ интен- сивность
i
В справочниках указывают значения интенсивности, оцениваемые по рентгенограммам. В связи с существенными различиями рентгеновских и электронографических факторов интенсивности приведенные справочные данные об интенсивности интерференционных максимумов можно использовать лишь для грубой оценки соотношения интенсивности соседних линий. Также необходимо учитывать невысокую точность электронографических , определений межплоскостных расстояний (10~* нм).
Рассчитанные межплоскостные расстояния для кубической, гексагональной или тетрагональной сингоний позволяют производить индицирование отражений и определение периода решетки. При это используются те же закономерности, что и при расчете рентгенограмм. Для кубической сингонии
1
aHKL
-у к2-V
Д2
106
Следовательно, соотношение квадратов межплоскостных расстояний должно соответствовать соотношению целых чисел:
(h2 + k\ + l^-.{h2 + kI
2 • • • —
I2} : (Н2 + /<з +I2)...
В расчетую таблицу записать отношение квадрата межплоскостного расстояния первого отражения к квадрату межплоскостно-го расстояния каждого последующего отражения
а затем привести эти соотношения к целым числам, которые принять за сумму квадратов индексов отражений Я2 + 7(2 + £2. Период решетки определить по формуле
d — - р /j 2 -L- /^2 Ц-
(VI .7)
Результаты расчета электронограммы исследуемого вещества (кубической сингонии) записать в таблицу по образцу:
Исследуемое вещество . . напряжение, кВ . . . (60, 80, 100); постоянная прибора . . .
а (J
<
Результаты индицирования проверить анализом погасаний и оценкой интенсивности колец. Интенсивность рефлексов электронограмм определить по изменению почернений на негативе. При фазовом анализе или при определении простых структур ограничиться визуальной оценкой интенсивности, которую провести по девятибалльной шкале, аналогичной шкале для рентгенограмм, и которой можно приписать числовые соотношения:
О. о. с.............. 10*0 Ср. сл................ 8
О. с................... 70 Сл..................... 5
С .... ................. 36 Оч. сл................ 2
Ср. с.................... 18 Оч. оч. сл............ 1
При использовании этой шкалы ошибка может составлять 50—100%. Для точных определений интенсивности использовать микрофотомегрирование. При точных измерениях период решетки или межплоскостных расстояний для дальних линий имеет значение поправка А к измеренным радиусам дифракционных колец:
^испр — ^изм (I — Д) - - ГиЗМ — ГиЗМД.
107
Построить график зависимости поправки от отношения радиуса кольца г к расстоянию L. Ниже приведены поправки А в зависимости от r/L:
r/L д r/L Д
0,01 0,02 0,03 0,04 0,05 0,06
3,7-10-’ 1,5-10-4 3,37-1'0-* 6,0-10-* 9,35-10-* 13,45-10-*
0,07 0.08 0,09 0,10
18,27-10-* 23,90-10-* 30,20-10-* 37,26-10~*
ГЛАВА V
РЕНТГЕНОСТРУКТУРНЫЙ АНАЛИЗ
1. ПОЛУЧЕНИЕ РЕНТГЕНОВСКОГО ИЗЛУЧЕНИЯ
Рентгеновское излучение — электромагнитное излучение с длиной волны от 80 до 10-5 нм, возникающее в веществе при резком торможении электронов высокой энергии, бомбардирующих вещество. Рентгеновское излучение образуется в специальных электровакуумных приборах — рентгеновских трубках, представляющих собой вакуумированный стеклянный сосуд (вакуум 1,33-10~3— 1,33-10~5 Па). В противоположные концы сосуда впаяны катод и анод. Катод в виде спирали из вольфрамовой проволоки накаливают электрическим током, который является источником свободных электронов. Анод—массивный стержень, обращенный своим
з г 1
Рис. 50. Схема электронной рентгеновской трубки:
1 — катод; 2 — окно для выпуска рентгеновского излучения; 3 — анод;
4— защитный цилиндр; 5 — фокусирующий, колпачок
торцом к катоду, в торец которого впаивается пластина какого-либо металла (антикатод), называемая зеркалом анода. Схема рентгеновской трубки приведена на рис. 50. Под действием электрического поля, образованного напряжением, приложенным к электродам трубки, свободные электроны от катода в результате его нагревания с большой скоростью летят к аноду, наталкиваясь на который резко тормозятся. При этом большая часть энергии превращается в теплоту и только малая часть энергии (0,1 — 1%)
108
превращается в тормозное и характеристическое рентгеновское излучение.
Тормозной рентгеновский спектр, называемый также сплошным или белым, вызывается всегда при торможении электронов. Кинетическая энергия электрона при резком торможении переходит в энергию фотона:
= = (V.1)
где е — заряд электрона; Е —разность потенциалов; h — постоянная Планка; с —скорость света; у — частота электромагнитного колебания; X — длина волны.
Торможение электронов на аноде рентгеновской трубки происходит по-разному: одни из них тормозятся мгновенно на поверхности анода, что соответствует максимальной энергии фотона [(уравнение (V. 1)]; другие проникают в глубь анода, теряя свою энергию постепенно. Таким образом, при торможении электронов возникают фотоны разной энергии, количество их в единицу времени велико и тормозной спектр при этом состоит из непрерывного
Рис. 51. Распределение интенсивности рентгеновского тормозного излучения при различном ускоряющем напряжении на рентгеновской трубке (анод вольфрамовый)
Рис. 52. Тормозное и характеристическое излучение медного анода при напряжении 50 кВ (/) и зависимость коэффициента массового поглощения рентгеновского излучения фильтром из никеля от длины волны (2)
ряда длин волн с резкой границей в коротковолновой части. Характер распределения энергий в спектре торможения при различных напряжениях показан на рис. 51.
Характеристический рентгеновский спектр образуется, когда энергия электронов превосходит порог возбуждения, характерный для атомов анодного вещества (рис. 52). Длина волны однородного характеристического излучения зависит от вещества анода и не зависит от приложенного напряжения. Характеристический рентгеновский спектр состоит из нескольких групп линий (серий), значительно отличающихся друг от друга по длине волны. Для более тяжелых элементов таких серий четыре: К, L, М, N. Каждая'
109
серия состоит из определенного числа спектральных линий, которые обозначаются а, р, у с индексами 1, 2 и т. д. (например, kai. Лр). Характеристические спектры различных элементов примерно одинаковы по числу и взаимному расположению линий в сериях и отличаются друг от друга только длиной волны. С увеличением порядкового номера элемента спектр сдвигается в коротковолновую область. Все линии любой серии возникают одновременно, как только напряжение на электродах рентгеновской трубки (а следовательно, и энергия электронов) достигает определенной величины для этой серии.
Для возбуждения /(-серии рентгеновского спектра необходимо удалить электрон с самого внутреннего /(-уровня на периферию атома (рис. 53). Это удаление может быть произведено одним из электронов, бомбардирующих вещество анода рентгеновской трубки. Освободившееся место /(-уровня будет тотчас же заполнено
Рис. 53. Схема возникновения отдельных серий характеристического рентгеновского излучения
одним из электронов верхних уровней, в результате чего выделится квант излучения, энергия которого равна разности энергии уровня,, на который он перешел:
= (V.2)
где Е'\\— энергия электрона на более удаленной орбитали; Е\— энергия электрона на более близкой орбитали.
Так как процесс возбуждения /(-серии наблюдается одновременно в большом количестве атомов, а вероятность перехода электрона из вышерасположенных слоев на /(-уровень неодинакова,
ПО
то все спектральные линии серии возникнут одновременно, а их интенсивность (определяемая вероятностью переходов) будет различной. На рис. 53 показана схема возникновения К- и Л-серий рентгеновского излучения. Практическое значение в рентгено-структурном анализе имеют наиболее сильные линии /(-серии: kai, Анализ проводят на линии /С-серии с длиной вол-
ны Аа, которую определяют:
Линии /Qj-серии поглощаются селективным фильтром. В качестве фильтра берут вещество с порядковым номером на единицу меньше, чем порядковый номер вещества анода рентгеновской трубки. Если анод из кобальта, то фильтр должен быть из фольги железа, если анод из меди, то фильтр — из никеля и т. п.
2. КРАТКИЕ СВЕДЕНИЯ О СТРУКТУРЕ КРИСТАЛЛОВ
В кристаллическом веществе пространственное расположение атомов и молекул периодично. На-известных расстояниях и в определенных направлениях можно встретить одинаковые атомы, ионы, молекулы. Пространственную решетку можно представить себе состоящей из бесконечно большого числа совершенно одинаковых параллелепипедов, называемых элементарными ячейками. Форму и размер элементарной ячейки характеризуют шестью константами: длинами ребер а, Ь, с (период решетки или осевые
Рис. 54. Пространственная кристаллическая решетка
Рис. 55. Различные семейства атомных плоскостей
единицы) и углами между ними а, р, у (рис. 54). Для построения простой кубической решетки достаточно знать одну константу а, называемую постоянной решетки. Постоянная решетки характеризует расстояние между идентичными атомами по осям координат. Существуют и более сложные решетки, в которых частицы веще
111
ства расположены не только в вершинах параллелепипедов. Такие решетки можно рассматривать как совокупность нескольких простых решеток, сдвинутых определенным образом одна относительно другой. В металлах наиболее часто встречаются кубические решетки двух типов: объемноцентрированные и гранецентрированные.
Объемноцентрированную решетку можно представить как две простые решетки, вложенные одна в другую, причем узловые атомы одной из них находятся в центре объема элементарной ячейки другой. Гранецентрированную решетку можно представить как четыре простые решетки, расположенные так, что каждая вершина второй, третьей и четвертой решеток находится в центре граней куба первой решетки. Следовательно, количество атомов, приходящихся на одну элементарную ячейку, будет зависеть от ’•типа решетки. В простой решетке на одну элементарную ячейку приходится только один атом, так как каждый атом в вершине куба относится к восьми соседним ячейкам. Поэтому данной элементарной ячейке будет принадлежать только один атом (или узел), а остальные семь атомов — соответственно семи соседним ячейкам. Тогда в объемноцентрированной и гранецентрированной решетках на одну ячейку приходится соответственно два и четыре атома.
В пространственной решетке через отдельные группы атомов можно провести бесчисленное количество параллельных плоскостей. Совокупность параллельных атомных плоскостей называется семейством атомных плоскостей, а расстояние между ними — межплоскостным расстоянием d (рис. 55). Количество атомов, входящих в ту или иную плоскость, различно и тем меньше, чем меньше межплоскостное расстояние. Положение любой атомной плоскости в пространственной решетке определяется при помощи трех простых целых чисел. Эти числа называются индексами плоскости (индексы Миллера) и представляют собой величины, обратные величинам отрезков, отсекаемых плоскостью на осях координат. Индексы плоскости обозначаются буквами h, k, I и заключаются в скобки.
Преимущества определения положения атомных плоскостей при помощи индексов Л, k, Z, а не осевых отрезков, отсекаемых плоскостями на осях координат, будут очевидны, если учесть, что они всегда являются простыми целыми числами и величина их не зависит от внешних влияний (температура, растяжение, сжатие и т. п.), чего не наблюдается у осевых отрезков. Кроме того, индексы k, I наиболее просто определяют положение атомных плоскостей в кристаллической решетке. Постоянную кубической решетки можно рассчитать, если известны индексы плоскости и межплоскостные расстояния, по уравнению
а = dhkl /й2 + ^2 + /2. (V.4)
Здесь имеется в виду межплоскостное расстояние того семейства, индексы которого стоят под корнем.
112
3. ДИФРАКЦИЯ РЕНТГЕНОВСКОГО ИЗЛУЧЕНИЯ
Прохождение рентгеновского излучения сквозь кристаллическое вещество сопровождается отклонением его от первоначального направления. Это явление называется дифракцией рентгенов
ского излучения.
Под действием электромагнитного поля рентгеновского излучения возбуждаются колебания электронов атомов, входящих в кристаллическую решетку вещества. Частота этих колебаний равна частоте электромагнитного поля первичного пучка рентгеновского излучения. Колеблющийся атом становится источником электромагнитных волн, распространяющихся от него во все стороны с частотой, равной частоте первичного излучения. Расположение атомов в любой кристаллической решетке закономерно и расстоя-
ния между ними в данном направлении одинаковы, поэтому пучки излучения, рассеянные отдельными атомами, будут интерферировать между собой. Интенсивность их в одних направлениях будет получаться значительно больше, чем в других. Следовательно, для рентгеновского излучения кристалл является трехмерной дифрак
ционной решеткой.
Уравнение
Вульфа
— Бреггов. Русский физик Г. В. Вульф и
английские физики отец и сын Брегги почти одновременно дали
наглядное объяснение отклонению рентгеновского излучения при прохождении его сквозь кристаллическое вещество. Они показали,
Рис. 56. Дифракция рентгеновского излучения
что рассеивание его атомами можно рассматривать как отражение рентгеновского излучения от параллельных атомных плоскостей кристалла.
Пусть на кристалл падает пучок монохроматического рентгеновского излучения, образуя угол 0 с одним из семейств атомных плоскостей (рис. 56, а). Пучок излучения Si, попадая на атомную плоскость Р19 отразится от нее в направлении S. Второй пучок S2, пройдя первую атомную плоскость (на основании свойства рентгеновского излучения проникать сквозь вещество), отразится от плоскости Р2 и также выйдет в направлении S и т. п. Отраженные параллельными атомными плоскостями пучки излучения будут интерферировать между собой и в зависимости от их фазового со-
113
•отношения усиливать или ослаблять друг друга. Отсюда следует, что в направлении S интенсивность рентгеновского излучения будет максимальной, когда пучки излучения, отраженные отдельными атомными плоскостями, будут находиться в одной фазе, т. е. когда разность их хода будет кратна длине волны.
Фазовые соотношения отраженных пучков излучения будут зависеть от длины волны Л, межплоскостного расстояния d и угла скольжения 0 (рис. 56, б). Выразим математически эту зависимость. В точках В и С лучи Si и S2 находятся в одной фазе (так называемый фронт плоской волны). После отражения пучка Si в точке А и пучка S2 в точке С оба они пойдут в одном направлении CAS. Пучок S2 пройдет путь больший, чем пучок Si, на разность хода АС — АВ. Отражение рентгеновского излучения в направлении CAS наблюдается при разности хода, кратной длине волны, т. е. при условии АС—АВ = п\ где п — целое число:
пк — 2d sin 9. (V.5)
Таким образом, каждое семейство атомных плоскостей будет давать ряд отражений в зависимости от того,, какие значения принимает п (1, 2, 3 и т. д.), чтобы sin0 не превышал единицы. Соотношение (V.5), называемое уравнением Вульфа — Бреггов, является основным расчетным уравнением рентгеноструктурного и рентгеноспектрального анализов. Зная межплоскостные расстояния и углы скольжения, можно по уравнению (V.5) вычислить длины волн отраженного рентгеновского излучения (рентгеноспектральный анализ). А зная длину волны монохроматического или характеристического рентгеновских излучений и углы скольжения, можно вычислить межплоскостные расстояния (рентгеноструктурный анализ).
4. МЕТОД РЕНТГЕНОСТРУКТУРНОГО АНАЛИЗА
Метод Дебая — Шеррера. Данный метод позволяет выполнять рентгеноструктурные исследования с порошкообразным веществом. Пусть некоторое семейство плоскостей в кристалле образует с падающим пучком монохроматического рентгеновского излучения угол 0, удовлетворяющий уравнению Вульфа — Бреггов (рис. 57). Не меняя угла скольжения (т. е. сохраняя условие Вульфа — Брегга), будем вращать кристалл вокруг оси первичного пучка. Отраженный пучок излучения опишет в пространстве конус с углом при вершине, равным 40. Другое семейство плоскостей этого же кристалла даст такой же конус, но уже с иным углом при вершине и т. п. Если на пути отраженных пучков излучения перпендикулярно первичному пучку поставить фотопластинку, то на ней зафиксируется ряд концентрических колец по числу семейств атомных плоскостей, отражающих рентгеновское излучение.
В поликристаллическом веществе из-за беспорядочного расположения отдельных кристаллов всегда будут такие, у которых 114
рассматриваемое семейство плоскостей удовлетворяет уравнению Вульфа — Бреггов. Следовательно, отражения от данного семейства атомных плоскостей каждого кристалла, находящегося в пучке рентгеновского излучения, будут сливаться в одну, сплошную конусную поверхность (конус дифракции). Количество таких конусов будет соответствовать
количеству семейств атомных плоскостей (рис. 58). Зафиксировать положения дифракционных конусов можно только при применении гибкой фотопленки, так как телесный угол, в котором они наблюдаются, равен 360°. Расстояние между двумя симметричными линиями на пленке зависит от угла при вершине кону
Рис. 57. Схема образования конусов отраженного излучения:
1 ~ нормаль к отраженной плоскости;
2 — конус отраженного излучения; 3 — первичный пучок; 4 — отражающая плоскость
са и расстоянии пленки от иссле-
дуемого вещества. Эти величины связаны между собой следующим образом:
2/ = г49,
(V.6)
где 2/ — расстояние между симметричными линиями, измеренное по экваториальной линии на пленке, мм; г — радиус цилиндриче
Рис, 58. Образование симметричных линий на рентгенограмме
ской пленки, мм; 9 — угол скольжения, рад; угол скольжения в. градусах рассчитывают по формуле
О = (2//4г) 57,3. (V.7)
Если известен диаметр цилиндрической пленки и расстояние между симметричными линиями на ней, то определяют углы скольжения. Значения межплоскостных расстояний определяют по уравнению Вульфа — Бреггов (V.5).
115
5. АППАРАТУРА, ПРИМЕНЯЕМАЯ В РЕНТГЕНОСТРУКТУРНОМ АНАЛИЗЕ
Рентгеновские аппараты, применяемые в рентгеноструктурном анализе, выполнены по принципиально одинаковой схеме и содержат генератор рентгеновского излучения (рентгеновская трубка), блок питания рентгеновской трубки или высоковольтный блок, в который входят высоковольтный трансформатор и трансформаторы накала катода трубки; пульт управления, на котором установлены средства регулирования и контроля работы рентгеновской трубки. Во всех рентгеновских аппаратах рентгеновскую трубку помещают в специальный кожух, защищающий персонал от рентгеновского излучения. Также всегда принимают меры по защите персонала от поражения электрическим током.
В настоящее время применяют два метода регистрации рентгеновского излучения: фотографический метод, использующий специальную пленку типа РТ; и ионизационный или сцинтилляционный метод, использующий различные типы счетчиков рентгеновских квантов (детекторы). Фотографический метод предусматривает использование специальных камер, конструкция которых зависит от проводимого анализа.
Рентгеновская камера для съемки образцов-порошков по методу Дебая — Шеррера (рис. 59, а) имеет цилиндрический корпус 3,
Рис. 59. Камера для рентгенографического анализа порошков (а), коллиматор рентгеновской камеры (б):
1 — подставка; 2 — экран; 3 — корпус со столиком для крепления образца;
4 ~ коллиматор с диафрагмой; 5, 6 — щели коллиматора; 7— предохранительный колпачок, 8 — крышка с кассетой для крепления пленки
закрепленный на подставке 1. Рентгеновское излучение попадает на образец через коллиматор (рис. 59, б), находящийся в передней части камеры и предназначенный для ограничения пучка рентгеновского излучения. Коллиматор представляет собой латунную трубку, в которую вставлены диафрагмы. Коллиматор снабжен предохранительным колпачком 7, поглощающим рассеянное
116
щелью 6 излучение. Первичный пучок проходит сквозь отверстия колпачка, не задевая его стенок.
Образец устанавливают в центре камеры и юстируют. Для этого вынимают коллиматор, снимают с него крышку 8, колпачок и заменяют экран лупой. Камеру с коллиматором ставят так, чтобы можно было рассматривать образец через лупу. Образец, закрепленный на магнитной пленке, устанавливают на оси вращения столика и, осторожно вращая, подводят образец к оси камеры. Затем заряжают камеру фотопленкой. Трубчатым ножом по шаблону вырезают в пленке отверстие для выхода первичного пучка рентгеновского излучения и укладывают пленку на внутреннюю цилиндрическую поверхность корпуса камеры, следя за тем, чтобы концы пленки были направлены к коллиматору; нижняя часть пленки вводится в кольцевой паз корпуса, а верхняя ее часть закрепляется прижимным кольцом крышки камеры. Далее закрепляют крышку камеры, надевают колпачок на коллиматор и заменяют юстировочную лупу экраном. Приготовленную к съемке камеру устанавливают на пути рентгеновского излучения, выходящего из окошка рентгеновской трубки, так, чтобы в центре флюоресцирующего экрана был виден пучок рентгеновского излучения и посредине его тень от образца. Время экспозиции зависит от конструкции камеры, рентгеновской трубки, режима ее работы, рентгеновской пленки, природы образца и т. п. По окончании экспозиции пленку проявляют, фиксируют и высушивают.
Ионизационный или сцинтилляционный метод предусматривает использование специальных устройств — гониометров. Регистрация дифракционной картины с применением в качестве детектора ионизационного или сцинтилляционного счетчика имеет ряд преимуществ по сравнению с фотографической регистрацией. Это-быстрота получения рентгенограммы для фазового и структурного анализа и более простой ее расчет, возможность простого и точного определения интегральной интенсивности и диффузионного фона, более точное и быстрое определение ориентировки монокристаллов и т. и.
Поэтому рентгеновские дифрактометры получили широкое распространение. Преимущество фотографического метода по сравнению с дифрактометрическим методом состоит в возможности получения пространственного распределения дифрагированного излучения; это определяет специфику применения указанных методов. Если при фотографическом методе все отраженные от образца пучки излучения фиксируются фотопленкой, то при ионизационном методе установленный на гониометре счетчик излучения, непрерывно двигаясь по окружности, в центре которой установлен исследуемый образец, последовательно фиксирует дифракционные максимумы, встречающиеся на пути его движения. Электрический сигнал от счетчика через специальные устройства подается на электронный самопишущий потенциометр. Отклонение пера потенциометра прямо пропорционально мощности рентгеновского излучения, отраженного от образца.
117
Для определения точного углового положения дифракционного максимума гониометр снабжен соответствующим устройством, которое вырабатывает короткие импульсы. Эти импульсы передаются на потенциометр и фиксируются в виде тонких штрихов, наносимых непосредственно на записываемую диаграммную ленту. Скорости движения гониометра и бумажной ленты строго равномерны.
Исследуемый образец при ионизационном методе исследования берут в виде шлифа, поэтому и исследуемые порошки набивают в специальную кювету и устанавливают в держатель образцов на гониометре. Конструкция держателя такова, что плоская поверхность образца всегда совпадает с осью вращения счетчика излучения, а скорость его вращения вдвое меньше вращения счетчика.
Работа. Определение параметра и типа кубической кристаллической решетки
Приготовление поликристаллических образцов для съемки с
фотографической регистрацией. Образцы для камер Дебая изго
товляют из пластических металлов протяжкой их в проволоку диаметром 0,2—1,0 мм, а также обточкой на токарном станке или
выпиливанием напильником вручную; для устранения текстур волочения проводят отжиг образца. После обработки образца поверхностный слой стравляют, так как дебаевские кольца сильно деформированных металлов получаются размытыми. Толщина деформированного слоя достигает 0,2—0,3 мм при грубых обработках (сверловка, обдирка и т. п.). Монолитные образцы в форме шлифов изготовляют из исследуемого металла и подготавливают обычными механическими методами, перед съемкой подвергают электролитической полировке для снятия наклепа. Наиболее удобны образцы размером 10X10X4 мм.
Образцы из порошков приготовляют в виде тонкого цилиндра диаметром 3—5 мм. Исследуемое вещество после измельчения в агатовой ступке просеивают через сито № 0063 и набивают в тонкостенный капилляр из целлулоида. Капилляр изготовляют следующим образом. Металлическую проволоку диаметром 5 мм покрывают тонким слоем вазелина и опускают в цапонлак (раствор целлулоида в ацетоне) и медленно из него вынимают. На проволоке остается тонкий.слой лака, который высушивают и снимают. Набивку порошка в капилляр производят плотно и полностью, после этого на конец капилляра наносят каплю цапонлака и обра
зец готов к исследованию.
Последовательность выполнения работы. Приготовить в зависимости от природы образец одним из перечисленных способов. Получить дебаеграмму. (Съемку дебаеграммы проводит лаборант рентгеновской лаборатории.) Рассчитать рентгенограмму. Взять лист белой бумаги и положить на нее рентгенограмму, тушью или чернилами сделать надписи, как указано на рис. 60: над центральным отверстием символ элемента анода рентгеновской трубки (с индексом а при съемке с фильтром); в левом углу номер рентге-
118
нограммы; в верхней части диаметр камеры и толщину образца; под центральным отверстием режим работы аппарата; симметричные части колец нумеровать от центра пленки к краям; вдоль всей рентгенограммы, по середине, провести экваториальную линию. Оценить по пятибалльной шкале интенсивности линий. Интенсивность оценивать визуально или на денситометре. Самую яркую (черную) линию принять за 5 баллов (очень яркая), самую ела-
Рис. 60. Схема рентгенограммы поликристаллического образца
бую — за 1 балл (очень слабая). Остальные линии имеют промежуточные значения: очень яркая (о. я) 5; яркая (я.) 4; средняя (ср.) 3; слабая (сл.) 2; очень слабая (оч.сл.) 1.
Измерить на рентгенограмме расстояния 21. Измерение проводить металлической линейкой с миллиметровой шкалой. Более точные замеры проводить на компараторе. Расстояние 21 измерять в миллиметрах с точностью до десятых долей. Расстояния между широкими или размытыми линиями измерять по средним частям. Вычислить углы скольжения по уравнению (V.7), которое для данной камеры можно записать
0-2ZK, (V.8)
где К=57,3/4г и называется коэффициентом камеры. Коэффициент стандартной камеры равен 0,5 и угол 0 в градусах будет равен половине расстояния 21 в миллиметрах.
Ввести поправку на толщину образца. Любой образец имеет определенную толщину, и рентгеновское излучение, проходя сквозь него, всегда будет поглощаться тем больше, чем толще образец и мягче излучение. Если предположить (в первом приближении), что пучок рентгеновского излучения параллелен и полностью поглощается в поверхностном слое образца, то линии на (рентгенограмме должны получаться только за счет отражения от тонкого поверхностного слоя. Поэтому измеренные расстояния 21 будут всегда больше истинных. Поправку можно рассчитать по формуле
ДО = (24//4) (1 4-cos 2 6), (V.9)
где 2d — диаметр образца, мм. Тогда исправленный угол будет
0ЦСП — 6иЗМ - Аб* (V . 10)
Для вычисления поправки пользоваться табл. 6. Определить sin0
119
Таблица 6. Значения 1 +cos 29 для различных углов 0
6 l + cos 20 е l+cos 20 0 [ l + cos 20 1 0 l + cos 20 е l+cos 29
0,00 2,00 18,0 1,81 36,0 1,31 54,0 0,69 72,0 0,19
0,5 2,00 18,5 1,80 36,5 1,29 54,5 0,67 72,5 0,18
1,0 2,00 19,0 1,80 37,0 1,28 55,0 0,66 73,0 0,17
1,5 2,00 19,5 1,79 37,5 1,26 55,5 0,64 73,5 0,16
2,00 2,00 20,0 1,77 38,0 1,24 56,0 0,62 74,0 0,15
2,5 2,00 20,5 1,76 38,5 1,22 56,5 0,61 74,5 0,14
3,0 2,00 21,0 1,74 39,0 1,21 ; 57,0 0,59 75,0 0,13
3,5 1,99 21,5 1,73 39.5 1,19 1 57,5 0,58 75,5 0,12
4,0 1,99 22,0 1,72 40,0 1,17 58,0 0,56 76.0 0,12
4,5 1 89 22,5 1,71 40,5 1,16 58,5 0,55 76,5 0,11
5,0 1,98 23,0 1,70 41,0 1,14 59,0 0,53 77,0 1,10
5,5 1,98 23,5 1,68 41,5 1,12 59,5 0,52 77,5 0,09
6,0 1,98 24,0 1,67 42,0 1,10 60,0 0,50 78,0 0,09
6,5 1,97 24,5 1,66 42,5 1,09 60,5 0,48 78,5 0,08
7,0 1,97 25,0 1,64 43,0 1,07 61,0 0,47 I 79,0 0,07
7,5 1.97 25,5 1,63 43,5 1,05 61,5 0,46 j 79,5 0,07
8,0 1,96 26.0 1,62 44,0 1,04 62,0 0,44 ! 80,0 0,06
8,5 1,96 26,5 1,60 44,5 1,02 62,5 0,42 80,5 0,05
9,0 1,95 27,0 1,59 45,0 1,00 63,0 0,41 81,0 0,05
9,5 1,95 27,5 1,57 45,5 0,98 63,5 0,40 81,5 0,04
10,0 1,94 28,0 1,56 46,0 0,96 64,0 0,38 82,0 0,04
10,5 1,93 28,5 1,54 46,5 0,95 64,5 0,37 82,5 0,03
11,0 1,93 29,0 1,53 47,0 0,93 65,0 0,36 83,0 0,03
11,5 1,92 29,5 1,52 47,5 0,91 65,5 0,34 83,5 0,03
12,0 1,91 30,0 1,50 48,0 0,90 66,0 0,33 84,0 0,02
12,5 1,91 30,5 1,48 48,5 0,88 66,5 0,32 84,5 0,02
13,0 1,90 31,0 1,47 49,0 0,86 67,0 0,30 85,0 0,02
13,5 1,89 31,5 1,45 49,5 0,84 67,5 0,29 85,5 0,02
14,0 1,88 32,0 1,44 50,0 0,83 68,0 0,28 86,0 0,02
14,5 1,88 32,5 1,42 50,5 0,81 68,5 0,27 86,5 0,02
15,0 1,87 33,0 1,41 51,0 0,79 69,0 0,26 87,0 0,00
15,5 1,86 33,5 1,39 51,5 0,78 69,5 0,24 87,5 0,00
16,0 1,85 34,0 1,38 52,0 0,76 70,0 0,23 88,0 0,00
16,5 1,84 34,5 1,36 52,5 0,74 70,5 0,22 88,5 0,00
17,0 1,83 35,0 1,34 53,0 0,73 71,0 0,21 89,0 0,00
17,5 1,82 35,5 1,33 53,5 0,71 71,5 0,20 1 89,5 0,00
Таблица 7. Длины волн линий /(-серий некоторых металлов, применяемых в качестве анодов в рентгеновских трубках
Элемент Номер элемента в периодической системе Длина волн А,, нм Оптимальные рабочие напряжения, кВ Требуемый фильтр для поглощения излучения серии К
сильная а2 очень сильная средняя р
Сг 24 0,22935 0,22896 0,20848 20—25 Ванадий
Fe 26 0,19399 0,19360 0,17565 25—30 Марганец
Со 27 0,17928 0,17889 0,16208 25—30 Железо
Ni 28 0,16617 0,16578 0,15001 30—35 Кобальт
Си 29 0,15443 0,15405 0,13922 35—40 Никель
Мо 42 0,07135 0,07093 0,06323 55—65 Цирконий
120
по исправленному углу 0, пользуясь тригонометрическими таблицами с точностью до 0,001. Рассчитать межплоскостные расстояния при и=1 по уравнению (V.3). Длины волн К-серии некоторых металлов приведены в табл. 7.
По рентгенограмме определить вещество и соединения, из которых ОНО 'СОСТОИТ.
Произвести индицирование рентгенограммы, т. е. определить индексы атомных плоскостей, которые дали на рентгенограмме линии. Индицирование можно провести несколькими способами: 1) графически; 2) по соотношению sin20; 3) при помощи логарифмической линейки. Индицировать рентгенограмму графическим способом. Согласно уравнению (V.4) можно записать
X г______________
sin 6 = — У /г2 -р £2 /2,
2а
(V.11)
где Z/2&— постоянная для данного кристаллического вещества и применяемого излучения трубки; член У Л2-|-&2-|-/2 может принимать дискретные значения, зависящие только от типа кристаллической решетки (см. табл. 8).
Таблица 8. Квадратичные формы кубической сингонии
h, k, I
h, k, I
0,0,1 0,1,1
1,1,1 0,0,2
0,1,2 ,
1,1,2
0,2,2
0,0,3 и 1,2,2 0,1,3
2,2,2 0,2,3 1,2,3 0,0,4
0,1,4 и 2,2,3
1,1,4 и 0,3,3 1,3,3 0,2,4
1,2,4
2,2,3
1
2
3
4
5
6
8
9
10
12
13
14
16
17
18
19
20
21
22
2,2,4
0,0,5’и 0,3,4
0,1,5 и 1,3,4
1,1,5 и 3,3,3
0,2,5 и 2,3,4
1,2,5
0,4,4
2,2,5 и 1,4,4
0,3,5 и 3,3,4
1,3,5
0,0,6 и 2,5,6
0,1,6
1,1,6 и 2,3,5
0,2,6
1,2,6; 0,4,5 и 3,4,4
1,4,5
3,3,5
2,2,6
0,3,6 и 2,4,5
24
25
26
27
29
30
32
33
34
35
36
37
38
40
41
42
43
44
45
Зависимость sin0 от К/2а для всех возможных значений /г, k, I выражается прямыми линиями (рис. 61). Индексы плоскостей определяют по рис. 61. Для определения индексов плоскостей на график наложить полоску бумаги шириной 15—20 мм. Отметить
121
штрихами все вычисленные значения sin 0. Если предположить, что все атомные плоскости кубической решетки отражали рентгеновское излучение, то первое кольцо на рентгенограмме должно принадлежать первой возможной атомнбй плоскости (0,0, 1; 0, 1, О или 1, 0, 0). Затем переместить полоску бумаги так, чтобы нижний ее край совпадал с осью абсцисс и полоска была параллельна оси ординат до совпадения первого штриха с первой линией. Если принятое предположение справедливо, то при этом значении Х/2а все штрихи, отмеченные на полоске, совпадут с несколькими линиями. Индексы этих линий являются индексами атомных плоскостей исследуемого кристалла.
Несовпадение штрихов с прямыми указывает на то, что предположение сделано неверно. Тогда необходимо совместить первый
Рис. 61. Определение индексов плоскостей кубических кристаллических решеток
штрих на полоске со второй линией, соответствующей атомной плоскости с индексами (0, 1, 1). Если и в этом случае не наблюдается совпадения, то операцию повторяют до тех пор, пока все штрихи sin0 на полоске не совпадут с линиями. На рис. 61 приведен пример совмещения всех штрихов sin0 с линиями, причем первый штрих совмещен с линией, соответствующей атомной пло-
122
скости (1,1,1). Данные расшифровки рентгенограммы графическим способом записать в таблицу по образцу:
Определить тип кристаллической решетки. Если сумма индексов каждой отдельно взятой плоскости представляет собой числа с одинаковой четностью (нуль — четное число), например (1, 1, 1) (3,5,7) (0,0,2) и т. д., то первые индексы — все числа нечетные, вторые — все числа нечетные, третьи — все числа четные. Следовательно, индексы имеют одинаковую четность и решетка гранецентрированная. Неподчинение первым двум правилам указывает, что решетка кубическая примитивная. Определить параметр решетки по уравнению (V.4) для каждой линии отдельно, исключая те значения а, которые существенно отличаются от средних. Если расхождения в значениях больше 3-10-3 нм, то ошибку следует искать в определении 21.
Определить межплоскостные расстояния в кубической решетке. Сопоставить полученные межплоскостные расстояния с табличными данными и определить вещество. Определить тип кристаллической решетки. Провести индицирование графическим способом.
Пример расчета рентгенограммы. Условия съемки рентгенограммы: трубка с-Cu-анодом (применен фильтр для p-излучения), диаметр 2г — 57,3 мм, толщина образца 26—6,6 мм. Всего на рентгенограмме получилось восемь пар линий. Интенсивности приведены в таблице.
1. Измеряют на рентгенограмме (см. рис. 60) 21 логарифмической линейкой с точностью до десятых долей миллиметра.
2. Вычисляют углы скольжения, предварительно определив коэффициент камеры:
Умножив измеренные 21 на 0,5, получают значение Оизм-
3. Вводят поправку ДО на поглощение в образце:
2с
Д9 —----(1 + cos 26).
4
При толщине образца '0,6 мм 25/4—6,15. Значение l+cos20 находят по табл. 6, откуда 1+ cos 20 = 1,72 (для угла 0 — 22,0°) и ДО —0,15-1,72=6,26°. Далее вычисляют исправленный угол:
0ИСЛР = еизм“ АО =22,10° -0,26° = 21,84°.
4. Находят sin 0 по тригонометрическим таблицам с точностью третьего знака.
123
5. Вычисляют межплоскостные расстояния d по уравнению Вульфа — Брегга, переписав его следующим образом:
d = (zzk/2) (1/sin 0).
Считая, что все линии рентгенограммы первого порядка отражения принимают л—1. Далее определяют значение X* /2 для меди:
(2\и + М/3 0Д5418
2 ~ 2
0,0771
нм.
6. Определяют индексы у атомных плоскостей h, k, I, от которых получились отражения, и вычисляют значения sin20.
7. Определяют соотношения sin2 0n/sin2 0! делением каждого последующего sin2 On на sin2 0Ь
8. Анализируют полученный ряд соотношений, который показывает, что только для линий 1 и 5 получаются числа, близкие к целым. Следовательно, умножая sin2 0n/sin2 01 на 2, получают ряд чисел: 2; 2,66; 5,32; 7,30; 7,94; 10,6; 12,64; 13,24. Решетка непримитивная, и первая атомная плоскость не (0,0,1). Полученный ряд чисел не представляет целые числа. Следовательно, решетка не объемноцентрированная, а индексы первой плоскости не (0,1,1). Умножая sin3 0 ,.,/sin2 01 на 3, получают ряд чисел: 3; 3,99; 7,98; 1!0,95; 11,91; 15,93; 18,96; 19,86, которые можно написать в виде ряда целых чисел: 3; 4; 8; 11; 12; 16; 19; 20. По табл. 3 находят индексы атомных плоскостей, которые имеют одинаковую четность для всех атомных плоскостей; следовательно, решетка — кубическая, гранецеитрированная.
9. Вычисляют по уравнению (V.3) постоянную решетки для всех линий и определяют среднее значение. В данном случае а=0,3610 нм.
'0 . Выделяют три-четыре линии с наибольшей интенсивностью для определения исследуемого вещества и записывают значения их межплоскостных расстояний d в порядке убывания по абсолютной величине 2,07; 1,80; 1,27; 1,084. Ориентируясь на самую интенсивную линию, подбирают такое вещество, у которого примерно совпадали бы межплоскостные расстояния. Находят по справочнику ближайшие значения для первой линии = 0,207 нм). Это могут быть 0,206 нм для ВеО; 0,234 нм для р-Со; 0,2100 нм для Си; 0,209 нм для NiO; и 0,209 нм для Zn. Составляют следующую сравнительную таблицу для линий исследуемого и всех возможных веществ:
Межплоскостное расстояние веществ, нм
расстояние исследуемого вещества, нм ВеО З-Со Си Ni Zn
0,207 0,206 0,204 0,209 0,209 0,209
0,180 0,234 0,177 0,181 0,241 0,247
0,127 0,219 0,125 0,128 0,148 0,231
0,1084 — — "
Из таблицы видно, что ВеО, |3-Со, Ni, Zn не подходят, так как имеют линии, межплоскостные расстояния которых не совпадают с полученными. Остается более тщательно проверить медь. Из трех линий две примерно сходятся по своим межплоскостным расстояниям, но есть некоторое расхождение в оценке интенсивности линий. Поэтому в полном перечне межплоскостных расстояний находят медь и сверяют опытные данные с табличными. Из сопоставления межплоскостных расстояний видно, что все они приблизительно совпадают. Имеется лишь незначительная разница в оценке интенсивности линий, которой можно прене-
124
бречь. Итак, исследуемым веществом будет медь с кубической, гранецентрированной решеткой:
Исследуемое вещество Си Исследуемое вещество Си
d интенсивность d интенсивность d интенсивность d интенсивность
0,207 5 0,2088 100 0,1039 2 0,1044 5
0,180 5 0,1808 46 0,0900 1 0,0904 3
0,1270 4 0,1278 20 0,0825 2 0,0829 9
0,1084 5 0,1090 17 0,0806 2 0,0088 8
d Интенсивность d Интенсивность d Интенсивность
0,207 0,180 0,1270 0,1084 0,1039
5 4 4 5 2
0,0900 0,0825 0,0806 0,2088 0,1808 0,1278 0,1090
1 2 2 100 46 20 17
0,1044 0,0904 0,0829 0,00888
5 3 9 8
Полученные результаты записать в таблицу по образцу:
СМ
ф с со
ЕС
1
2
3
4
г* о
6
7
8
5
4
4
5
2
2
2
44,2
51,4
75,0
90,7
96,0
118,0
138,0
146,3
22,10 25,70 37,50 45,35
48,00 59,00 69,00 73,15
0,26 0,24
0,19 0,15 0,14 0,08 0,04 0,03
21,84
25,46
37,31
45,20 47,86 58,92 68,96
73,12
0,372 0,428 0,606 0,710 0,741 0,856 0,934 0,957
0,207 0,180
0,1270 0,1084 0,1039 0,0900 0,0825 0,0806
0,138 0,183 0,367 0,504 1,549 0,733 0,872 0,914
1,00 1,33 2,66 3,65 3,97 5,31
6,32 6,62
(1ЛЛ) (0,0,2) (0,2,2) (Ы,3) (2,2,2) (0,0,4) (1,3,3) (0,2,4)
0,359 0,360 0,360 0,360 0,360 0,360 0,360 0,361
S
ГЛАВА VI
ТЕРМОХИМИЯ
ТЕРМОХИМИЧЕСКИЕ ИЗМЕРЕНИЯ
Для большинства физико-химических расчетов необходимо знать теплоемкости веществ, участвующих в процессе, тепловые эффекты процессов растворения, фазовых превращений и химических реакций. Эти величины можно измерить экспериментально. При температурах, близких к комнатной (20—50°С), широко применяется калориметрический метод.
125
При калориметрических опытах величина и знак теплового эф-
<Ьекта Q процесса определяются по изменению температуры кало-
Л .......................................
риметра ет:
Q = ЩсЛ М = cw&t, \ i /
(VI. 1)
где mi — массы исследуемого вещества, калориметра и вспомогательных устройств (мешалки, ампулы, термометра); с — удельные ‘ теплоемкости исследуемого вещества, калориметра и вспомогательных устройств; cw — суммарная теплоемкость калориметрической системы. Уравнение (VI. 1) может быть записано
(VI.2) частей калори-
где д — константа ‘Калориметра, т. е. теплоемкость метра и вспомогательных устройств, участвующих в теплообмене, Дж/К; ci — теплоемкость содержимого калориметра; mi— масса содержимого калориметра; &t — изменение температуры процесса, протекающего в условиях отсутствия теплообмена калориметра с окружающей средой.
Калориметр с изотермической оболочкой (диатермический) позволяет учесть теплообмен его с окружающей средой, что дает возможность вычислить изменение температуры At, соответствующее опыту без теплообмена.
Теплоемкостью системы С называют производную dQ/dT. Теплоемкость газов и жидкостей зависит от температуры, а теплоемкость твердых веществ при средних и высоких температурах практически от нее не зависит. При расчетах часто используют среднюю теплоемкость.
Средней теплоемкостью однородного тела называют отношение подведенной теплоты к повышению температуры:
C = Q/(T2-Ti)^Q/\T. (VI.3)
Средняя теплоемкость С зависит от интервала температур (Тъ— —Т1). Зависимость между истинной и средней теплоемкостями выражается уравнением
(VI. 4)
При ДТ^5° даже на совершенных калориметрах (при измерении с точностью 0,05%) не удается установить различия между истинной и средней теплоемкостью. Поэтому теплоемкость, определенную в результате изменения температуры калориметра на 2— 3е, принимают за истинную и относят ее к температуре (Тг + Ti) /2. Теплоемкость однородного тела зависит от его массы:
С—ст или С—cm, (V 1.5)
где с — удельная теплоемкость вещества; m— масса вещества. Если масса равна молекулярной или атомной массе, то теплоемкость
126
будет соответственно -молярной или атомной. Если во время опыта давление в калориметрической системе остается постоянным (в калориметрах открытого типа оно равно атмосферному), то тепловой эффект процесса при постоянном давлении будет Qp, а теплоемкость Ср. При термохимических измерениях процессам, сопровождающимся выделением теплоты (экзотермическим процессам), приписывается положительный знак. При выделении системной теплоты ее энтальпия убывает. Откуда Qp——АН. Если в уравнениях тепловой эффект обозначен Q или q, то следует применять термохимическую систему знаков.
Калориметрическая установка (диатермический калориметр). Калориметрическая установка состоит из воздушного термостата и помещенного в нем калориметра, .Термостат прёДстаОяет**П5обо1Г бокс с застекленными стенками, в котором установлены нагреватель, вентилятор, термохимический и контактный термометры. Нагреватель выключается при помощи реле при достижении в боксе заданной температуры. В качестве нагревателя используется электрическая лампочка, обладающая малой тепловой инерцией. Температура в боксе поддерживается с точностью ±0,02°. Воздушная среда в боксе с постоянной температурой является изотермической оболочкой калориметра.
Калориметр состоит из калориметрического сосуда (полиэтиленовый стакан). Через отверстия в крышке бокса в калориметре крепятся стеклянная мешалка, термометр Бекмана (см. с. 181К электронагреватель и ампула с исследуемым веществом. Калориметр устанавливается в боксе на столике, перемещающемся вертикально. Электронагреватель питается от электросети через стабилизатор и трансформатор. Число оборотов вентилятора и мешалки регулируют лабораторными автотрансформаторами. Напряжение в электронагревателе регулируют реостатом. Отсчеты времени про^ изводятся с помощью звукового сигнализатора, подающего сигналы через каждые 30 с. Тепловой баланс процесса в калориметрическом опыте выражается уравнением
Q -= (2 mici\ “Ь (VI. 6>
\ / /
где q — теплообмен калориметра с окружающей средой за период калориметрического опыта.
Если бы исследуемый процесс и выравнивание температуры в калориметре происходили мгновенно, то теплообмен со средой был бы равен нулю В реальных условиях протекание процесса
и выравнивание температуры требует времени, в течение которого калориметр получает от среды или отдает ей некоторое количество теплоты q. Величину q не вычисляют, но опыт проводят в калориметре так, чтобы на основании полученных данных можно было вычислить изменение температуры At (отличное от At') того же процесса, но протекающего мгновенно без тепловых потерь. Калориметрический опыт следует начинать при условии, если система близка к состоянию теплового равновесия, характеризуемого не-
127
значительным температурным ходом (не более 0,04 пр ад/мин). Это условие можно выполнить, установив температуру содержимого калориметра при работающей мешалке на 1—2° ниже температуры воздуха в боксе. При такой разности температур скорость поступления теплоты в калориметр от воздуха становится равной скорости отдачи теплоты за счет испарения воды, находящейся в калориметрическом сосуде, что обеспечивает тепловое равновесие системы. Если в |И|Сследуемом процессе наблюдается выделение теплоты, то в начальном периоде температура калориметра должна повышаться. Если в процессе наблюдается поглощение теплоты, то температура калориметра должна понижаться. При постоянной скорости изменения температуры производят 10—12 отсчетов по термометру Бекмана через каждые 30 с. Это — начальный период калориметрического опыта. Затем проводят определение теплового эффекта процесса. Температуру по термометру Бекмана непрерывно продолжают отсчитывать через те же промежутки времени. За счет выделения или поглощения теплоты в процессе происходит резкое изменение температуры. Это — главный, период калориметрического опыта. По завершении главного периода вновь устанавливается равномерный ход температуры. Это—конечный период калориметрического опыта, в течение которого производят еще 12— 15 отсчетов по термометру Бекмана. (Если во время калориметрического опыта очередной отсчет показания термометра был пропущен, то следует прочеркнуть и записать следующий под своим порядковым номером.)
Вычисление At. Типичный вид температурной кривой правильно поставленного калорит-метрического опыта при измерении экзотермического эффекта показан на рис. 62. Величину At с учетом теплообмена можно рассчитать аналитическим или графическим способом.
При графическом определении At на миллиметровой бумаге на оси абсцисс откладывают время в масштабе 1 мин = == 1 см, на оси ординат — тем-
пературу, выбор масштаба которой зависит от величины At. При А/АсГ 1°= 10 см; А^^1° 1° — 5 см. После того как на график нанесены все экспериментальные точки, получается кривая ABCD, Участок АВ называется начальным периодом, ВС — главным, CD — конечным. Чтобы определить изменение температуры At, не искаженное теплообменом, происходящим в течение главного периода, продолжают АВ и CD до пересечения с вертикальной прямой EF. Для этого точки т и /г, соответствующие начальной и конечной
Время, мин
Рис. 62. Определение изменения температуры в ходе калориметрического опыта
128
температурам главного периода, наносят на ось ординат. Через середину отрезка тп проводят линию /СР. Пересечение этой линии с кривой ВС дает точку I, определяющую положение прямой EF. Отрезок ЕЕ и будет равен At, отрезок mn=At'. Чем меньше температурный ход в начальном и конечном периодах, тем меньше потери теплоты за счет теплообмена и тем ближе АГ к At. Если температурный ход содержимого калориметра при работающей мешалке равен нулю, то это состояние соответствует ^равн. Характер линии ВС зависит от условий протекания теплового процесса (например, от размешивания), наклон линий АВ и CD зависит от характера теплообмена с окружающей средой. Таким образом, по виду кривой ABCD можно судить о качестве проведенного опыта.
Так как определение поправки на теплообмен с внешней средой всегда связано с некоторой неточностью, то 'Надо выбирать условия, при которых значение q было мало по сравнению с величиной Это достигается, если в ходе опыта отклонения системы от состояния теплового равновесия невелики, что характеризуется соотношением m<YpaBH<n. Указанное условие соблюдается, если температурный ход в начальном и конечном периодах имеет противоположный знак, а по абсолютной величине ход температуры в начальном периоде должен быть несколько больше, чем в конечном периоде. Чем меньше At, тем меньше должен -быть ход температуры в начальном и конечном периодах.
Основной источник погрешности результатов калориметрических опытов. Работа производится на установке упрощенного типа, позволяющей при тщательном проведении калориметрических опытов и .правильно выбранных условиях (продолжительность опыта не должна превышать 5 мин) получать результаты с погрешностью около ±1%. Главными факторами, определяющими точность результата, будут погрешности At, так как ошибки взвешивания не превышают сотых долей процента. В калориметрической установке температуры измеряют при помощи термометра Бекмана, точность отсчета по которому составляет в данных условиях около ±0,005°, поэтому возможная погрешность в определении At составит ±0,01°. Относительная ошибка, вносимая .в результат за счет неточности измерения температур, выражается отношением погрешности к ДГш. Так, при указанной точности измерений по термометру Бекмана и At^l° погрешность АГш составляет ±1%, при Af=O,l° она равна ±10%.
Работа 1. Определение суммарной теплоемкости калориметрической системы электрическим методом
Определение суммарной теплоемкости составных частей калориметрической системы является обязательным для всех калориметрических опытов. Согласно уравнению (VI.1) cw рассчитывают, определив экспериментально Q и At.
lAemjxu определения Q в зависимости от поставленной задачи и требуемой точности различны, но наиболее распространенным яв-5—2083 129
ляется электрический метод. К калориметрической системе подводят известное количество электрической энергии, превращающейся в теплоту Q. Необходимо точно измерить время пропускания тока, силу тока и падение напряжения на нагревателе. Количество сообщенной системе теплоты вычисляют по закону Джоуля — Ленца
Q = /£r, (VI.7)
где I — сила тока, А; Е — падение напряжения на нагревателе, В; т— время, с.
Изменение температуры At3 калориметрической системы в ходе этого опыта определяют графически.
Последовательность выполнения работы. Включить вентилятор и термостат, установленный на температуру в пределах 24—26°С. Взвесить калориметрический сосуд на технических весах, залить в него 200 мл воды или исследуемой жидкости и снова взвесить. Установить калориметрический сосуд в термостат и закрепить его на такой высоте, чтобы ртутный резервуар термометра Бекмана был полностью покрыт водой (или исследуемым раствором). Лопасти мешалки должны быть расположены у дна сосуда. Включить мешалку, установить с помощью реостата предельную скорость вращения, при которой не происходит разбрызгивания исследуемой жидкости. Установить с помощью реостата силу тока в нагревателе 1—ЗА и отключить нагреватель. Проверить скорость изменения температуры содержимого калориметра, которое не должно превышать 0,04 град/мин. Начать запись показаний термометра Бекмана с точностью 0,005° через 30 с, если температурный ход постоянен (начальный период). После одиннадцатого отсчета включить нагреватель на время т, не прерывая записи показаний термометра через каждые 30 с. Температура в начале резко повышается (главный период), затем, после выключения нагревателя, начинает равномерно подать, приближаясь к равновесной температуре калориметра /равн. Отсчетом температуры, с которой начинается равномерное понижение, кончается главный период калориметрического опыта и начинается конечный период. Произвести 12—15 отсчетов после того, как установится постоянный ход температуры (конечный период). Выключить мешалку. Определить графически изменение температуры Вычислить суммарную теплоемкость cw по уравнению
(VI.8)
Р абота 2. Определение удельной интегральной теплоты растворения соли
В работе следует определить суммарную теплоемкость системы и удельную интегральную теплоту q растворения соли.
Тепловой эффект, сопровождающий растворение твердого вещества в жидкости и отнесенный к 1 г растворяемого вещества, называют удельной теплотой растворения. Тепловой эффект, отнесенный к 1 моль растворяемого вещества, называют молярной тепло-
130
той растворения. Теплота растворения зависит от концентрации раствора. Различают интегральную теплоту растворения — тепловой эффект, сопровождающий процесс растворения 1 моль (молярная) или 1 г (удельная) (вещества в данном количестве растворителя; и дифференциальную теплоту растворения — тепловой эффект, сопровождающий процесс растворения 1 моль вещества в бесконечно большом количестве раствора заданной концентрации. Интегральные теплоты растворения определяют экспериментально, а дифференциальные вычисляют по зависимости интегральных теп-лот растворения от концентрации раствора:
£даА/раств Я ------Z---
(VL9)
где g — навеска исследуемого вещества; Д^раств — изменение температуры при растворении.
Вариант А. Суммарная теплоемкость системы cw определяется электрическим методом.
Последовательность выполнения работы. 1. Определение изменения температуры Д/раств при растворении исследуемого вещества и продолжительности главного периода Дт (см. с. 127). В стакан калориметра залить 200 мл воды. Отвесить на технических весах 2 г тщательно измельченного исследуемого вещества, перенести его во взвешенную на аналитических весах ампулу и вновь взвесить ее. Укрепить ампулу в крышке термостата, погрузив в воду. После одиннадцатого отсчета разбить пробойником ампулу, не прерывая записи температуры через каждые 30 с. Температура воды при эндотермическом растворении сначала резко падает (главный период), затем начинает равномерно расти, приближаясь к средней температуре .системы. Отсчетом температуры, с которой начинается ее равномерное повышение, кончается главный период калориметрического опыта и начинается конечный период.
Определить графически изменение температуры Д£раств и продолжительность главного периода Дт.
2. Определение суммарной теплоемкости cw системы. Суммарная теплоемкость калориметрической системы зависит от условий проведения калориметрического опыта, поэтому ее следует определять при условиях, близких к условиям проведения калориметрического опыта при растворении соли (определение Д^раств). Наиболее важно добиться одинаковой продолжительности главного периода Дт и одинаковых абсолютных величин At в обоих опытах. Изменение температуры Д/э содержимого калориметра зависит от силы тока при пропускании его через нагреватель. Чтобы установить силу тока, при которой Д^э будет равно Д/раСтв, необходимо провести три опыта, пропуская в нагреватель, погруженный в раствор, ток силой Ii — 1 А, /2 = 2 А, /З = 3 А в течение времени т=Дт. Построить график и определить интерполяцией силу тока, при
котором Д^ = Д^раств.
Описание определения Д^э приведено на с. 130. Построить график Д£э—f(I): отложить по оси ординат температуру 0,1° = 10 мм, 5* 131
л о оси абсцисс-—силу тока 1 А=50 мм и определить силу тока при А^э=Л^Раств. Установить с помощью реостата силу тока, проходящего через нагреватель, записать соответствующее показание вольтметра Е. Вычислить cw по уравнению (VI.5), подставив в него найденные значения Л Е, &tQ, Ат. Вычислить q по уравнению (VI.9).
Вариант Б. Суммарная теплоемкость системы cw определяется по теплоте растворения хлорида калия.
Последовательность выполнения работы. 1. Определение суммарной теплоемкости cw системы. Ход опыта описан на с. 129. Суммарную теплоемкость cw калориметрической системы вычислить по уравнению
трасте
Q ~ cw--------М, (V 1.10)
где Q — теплота растворения КС1 (из справочника), Дж/моль; gi — навеска КО, г; М —молекулярная масса КС1.
Есликалориметрическая установка позволяет получать результаты с погрешностью менее 1 %, то из найденного значения cw следует вычислить константу калориметра К по уравнению (VI.2) : cw = K -F(m -Fgjc, (VI.11)
’ >
где m — масса 200 мл воды, г; gi— навеска КС1, г; с — теплоемкость раствора электролита, отличающаяся от значения 4,11 Дж/(г-К) для данной концентрации.
2. Определение теплоты растворения q исследуемого вещества. Ход опыта описан на с. 130. Вычислить удельную теплоту растворения q соли по уравнению
q ~ ^ад^^расгв/^2» О I 12)
где §2 — навеска исследуемого вещества. Чтобы более точно вычислить cw по (VI. И), нужно взять значение К из первого опыта, а навески воды m и исследуемого вещества gz из второго опыта.
Работа 3*. Определение интегральной теплоты растворения соли при образовании концентрированного раствора
В работе необходимо определить теплоту растворения соли с образованием раствора, концентрация которого близка к насыщению. Если конечная концентрация раствора близка к насыщению, то скорость растворения настолько замедляется в конце процесса, что прямое определение теплоты растворения Q становится невозможным. Это подтверждается уравнением
= (VI. 13)
d т
где dC/dr —скорость растворения; К — константа скорости растворения; Снас и Сх — концентрация соли в насыщенном растворе и в
* Работа выполняется при условии, если kt определяется с точностью до 0,001°.
132
момент времени т. Скорость растворения в конце процесса настолько замедляется (Снас—Сх->0), что прямое определение интегральной теплоты растворения Q становится невозможным. Теплоту растворения в этом случае определяют косвенным путем.
Теплоту образования концентрированного раствора определяют в две стадии. Каждая стадия протекает с достаточно большой скоростью. В первой стадии определяют теплоту растворения Qi соли при образовании раствора концентрации т2 меньшей, чем ть а во второй стадии — теплоту разбавления концентрированного раствора Q2 концентрации до концентрации т2. Тогда по закону Гесса
Q = Q1“Q2- CVI.14)
Величину Qi вычисляют по зависимости интегральных теплот растворения от концентрации, используя справочные данные. Значение Q2 определяют экспериментально и вычисляют по уравнению
, (VI .15)
g
где cw— суммарная теплоемкость системы; .Д/раств—изменение температуры в процессе разбавления; М — молекулярная масса соли; g— навеска соли, содержащейся в исходном объеме концентри-рованного раствора.
1. Определение Д£раСтв температуры изменения при растворении исследуемого вещества. Последовательность выполнения работы.
Приготовить 50 мл раствора концентрации mi (взвешивать на аналитических весах). Дальнейшее описание опыта см. на с. 131. Взвесить ампулу на аналитических весах, поместить в нее 8 мл раствора концентрации mi, снова взвесить ее и укрепить в крышке термостата, погрузив в воду.
2. Определение суммарной теплоемкости cw системы. Описание опыта см. на с. 129.
Условия опыта: сила тока 2 А; время 1 мин. Вычислить Q2 по уравнению (VI.15), a Q — по уравнению (VI.14).
Определение 1интегральной теплоты растворения соли при образовании концентрированного раствора можно провести по методике, описанной на с. 131.
Работа 4*. Определение теплоты образования твердого раствора из двух твердых компонентов
В работе следует определить теплоту образования твердого раствора KCl- KBr из КС1 и КВг. Бромид и хлорид калия неограниченно растворимы друг в друге как в жидком, так и в твердом состояниях. Диаграмма плавкости этой системы представлена на рис. 63. Величина и знак теплового эффекта при образовании твердого раствора из индивидуальных кристаллических веществ позво-
* Работа может быть выполнена при условии, если калориметрическая установка позволяет определить Д/ с точностью 0,001°.
133
ляют судить о характере взаимодействия их в твердом растворе. Если образование твердого раствора сопровождается выделением теплоты, то раствор обладает отрицательным отклонением от идеальности. Поглощение теплоты указывает на положительное откло
нение.
Теплоту образования твердого раствора QTB.p из кристаллических компонентов непосредственно в калориметрах обычного типа
определить нельзя, так как скорость процесса чрезвычайно мала. Однако Qtb.p можно определить калориметрически косвенным путем. Для этого следует предварительно приготовить твердый раствор при высокой температуре и определить теплоты растворения твердого раствора и механической смеси того же состава. По закону Гесса
Qib.d Qaj-B QaB’ (VI. 16)
Рис. 63. Диаграмма плавкости где Qtb.p — теплота образования системы КС1--КВг твердого раствора из А и В; Qab —
теплота растворения твердого раствора; Qa-j-b — теплота растворения механической смеси компонентов А и В;
Фд + В “ ("а£а + лв^в)
(VI. 17)
где Qa — молярная теплота растворения A; QB — молярная теплота растворения В; — содержание вещества А, моль; пв— содержание вещества В, моль.
Последовательность выполнения работы. Измельчить тщательно в ступке 5 г КС1 и 5 г КВг и перенести в фарфоровый тигель. Поместить тигель со смесью в сушильный шкаф и выдержать его 10— 15 мин при 100—150°С. Перенести тигель со смесью в печь, нагретую до 600°С, довести температуру печи до 750°С и выдержать при этой температуре 15 мин. Выключить печь, вынуть тигель через 10 мин и охладить его на (Воздухе. Разбить тигель, извлечь кристалл лы твердого раствора и тщательно растереть их в фарфоровой ступке. Поместить приготовленный твердый раствор в бюкс с притертой (Крышкой. Использовать полученный твердый раствор для калориметрического опыта можно не ранее чем через 0,5—1 ч. Определить Kt и Ат при растворении твердого раствора КО*КВг в воде (см. с. 130). Навеска твердого раствора должна быть 2 г. Определить суммарную теплоемкость калориметрической системы после растворения твердого раствора (см. с. 129). Рассчитать теплоту растворения твердого раствора по уравнению
(VI. 18)
134
где 2п/= 1/Л4кс1+1/Жвг; AfKCi и ЛГквг — молекулярные массы КС! и КВг. Рассчитать теплоту растворения механической смеси QA+B по уравнению (VI.11), используя справочные данные и учитывая состав смеси и концентрацию раствора, а теплоту образования твердого раствора Qtb.p — по уравнению (VL16).
Работа 5. Определение теплоты образования кристаллогидрата из соли и воды
f
В работе следует определить теплоту образования CuSO4-5H2O из CuSO4 и Н2О. Образование кристаллогидрата сульфата меди протекает по уравнению
CuSO4 (тв) + 5Н2О (ж) ~ CuSO4- 5Н2О (тв) -р Q
Теплота этого процесса не может быть измерена в калориметре непосредственно, так как скорость образования CuSO4*5H2O мала. При образовании устойчивого кристаллогидрата теплоту гидратации можно определить калориметрически. Практически теплоты образования кристаллогидратов определяют по разности теплот растворения безводной соли и кристаллогидрата в большом количестве воды. При интенсивном перемешивании раствора скорость растворения описывается уравнением (VI. 13). Если растворять тонкоиз-мельченный безводный сульфат меди в большом количестве раствора концентрации, близкой к насыщенному раствору, то при быстром перемешивании гидратация сульфата меди пройдет мгновенно, а растворение будет происходить медленно, так как (Снас—С) близко к нулю. Таким образом, измеренный тепловой эффект будет соответствовать только теплоте образования кристаллогидрата.
Последовательность выполнения работы. Приготовить 500 г 15%-ного раствора сульфата меди в расчете на CuSO4. Включить термостат и установить температуру в боксе 24—26°С. Залить 200 мл раствора сульфата меди в калориметрический сосуд. Взвесить ампулу на аналитических весах, поместить в нее примерно 1 г безводного сульфата меди и вновь взвесить. При взвешивании следует помнить, что безводный сульфат меди гигроскопичен. Опре-делить Д/, Дт (главного периода) и cw (см. с. 130). Вычислить теплоту образования пятиводного кристаллогидрата сульфата меди по уравнению (VI.9) и сопоставить полученную величину со справочной.
Работа 6. Определение теплоты нейтрализации
В работе следует определить теплоту нейтрализации соляной кислоты раствором едкого кали. При нейтрализации сильных кислот и оснований теплота нейтрализации почти одинакова. По мере разбавления реагентов теплота нейтрализации приближается к предельной величине —55 938 Дж/моль при 20°С. Эта величина
135
представляет собой теплоту образования молекул |воды из ионов водорода и гидроксила:
н+ 4- ОН" = Н2о 4- Qi (I)
Уравнение реакции нейтрализации, например раствора соляной кислоты раствором едкого натра,
н^Кн2о + ^ОН^що “ МаС1(Л.+0)Нао 4- 4- СС
(И)
где хи у — число молей воды на 1 моль реагента.
Теплоты реакций (I) и (II) отличаются на теплоту разбавления реагентов
Q- О1+29разб, (VI. 19)
где Qi — тепловой эффект реакции (I); 2Qpa36 — сумма теплот разбавления растворов NaOH и НО. Тепловой эффект реакции (II) определяется экспериментально, а 2Qpa36 вычисляется по данным справочника.
Последовательность выполнения работы. Включить термостат, установленный на заданную температуру (24—26°С). Залить в калориметрический сосуд 200 мл 0,1 н. КОН. Установить температуру раствора в калориметрическом сосуде на 2° ниже температуры воздуха в боксе. Взвесить ампулу на аналитических весах, залить в нее 5 мл 2н. НС1 и вновь взвесить. Укрепить ампулу в крышке термостата, погрузив ее в раствор КОН. Дальнейшее описание опыта см. на с. 131. Определить Д^ и время главного периода Дть Вылить раствор из калориметрического сосуда и высушить его. Залить в калориметрический сосуд 200 мл 0,1 н. НО. Установить температуру раствора в калориметрическом сосуде на 2° ниже температуры воздуха в боксе. Взвесить ампулу на аналитических весах, залить в нее 5 мл 2 н. КОН и вновь взвесить. Дальнейшее описание опыта см. на с. 131. Определить графически Д£2 и время главного периода. Определить суммарную теплоемкость калориметрической системы (см. с. 129). Так как процесс нейтрализации протекает практически мгновенно, т. е. Дт главного периода равно нулю, при определении cw принимают, что т=60 с [(см. уравнение (VI.8)]. Вычислить теплоты процессов, по уравнениям
--—----; Q" ---------
ПНС1 лкон
(VI. 20)
Рассчитать SQpa36, используя справочные данные. Вычислить Qi по уравнению (VI. 17), (VI.19). Оценить погрешность полученного значения теплоты нейтрализации по отношению к —55 938 Дж/моль.
Работа 7. Определение теплоты реакции окисления
В данной работе следует определять теплоту окисления щавелевой кислоты перманганатом калия в кислой среде и проверить степень полноты протекания реакции.
Тепловые эффекты химических реакций могут быть определены непосредственно при проведении их в калориметре, если-они про
136
текают достаточно быстро и доходят до .конца или до определенного равновесного состояния. В последнем случае после опыта необходимо определить степень превращения исходных веществ. Термодинамически все реакции обратимы; в растворах реакции доходят практически до конца, если один из продуктов выпадает в осадок или выделяется в виде газа. Примером такой .реакции может служить окисление щавелевой кислоты перманганатом калия:
С2О4Н2 • 2Н2О (тв) -h 0,4КМпО4 (р) + 0,6H2SO4 (р)
= 0,2K2SO4(р) 4- 0,4MnSO4 (р) + 2СО2 (г) + 3,бН2О (р) + Q
В результате реакции образующийся СО2 растворяется в воде, что приводит к выделению дополнительной теплоты растворения. Однако при интенсивном перемешивании системы и низком парциальном давлении СО2 в воздухе устанавливается равновесие раствора с газовой фазой, которое практически смещено вправо. Поэтому в пределах точности определений (1%) можно принять, что весь образующийся СО2 будет находиться в газообразном состоянии.
Примечание. Так как энтальпия растворенных веществ зависит от концентрации, то тепловой эффект исследуемой реакции также будет зависеть от концентрации реагентов.
Поэтому необходимо провести два калориметрических опыта. Один, когда щавелевая кислота для реакции взята в избытке; второй, когда в избытке взят перманганат калия. В обоих калориметрических опытах следует брать одинаковое количество перманганата калия.
Последовательность выполнения работы. Включить термостат, установленный на заданную температуру в пределах 24—26°С. Залить в калориметрический сосуд из бюретки 68 мл воды, 50 мл 30%-ного раствора серной кислоты и 32 мл 0,5 н. КМпО4. Установить температуру раствора на 2° ниже температуры воздуха в боксе. Взвесить ампулу на аналитических весах, поместить в нее 1,2 г щавелевой кислоты и вновь взвесить. Укрепить ампулу в крышке термостата. Далее провести работу так, как это описано в работе 2, вариант А. Вылить содержимое калориметрического стакана, вымыть и высушить его. Залить в калориметрический сосуд из бюретки 68 мл воды, 50 мл 30%-ного раствора серной кислоты и 32 мл 0,5 н. КМпО4. Установить температуру раствора на 2° ниже температуры воздуха в боксе. Взвесить ампулу на аналитических весах, внести в нее 0,8 г щавелевой кислоты и вновь взвесить. Укрепить ампулу в крышке термостата. Далее провести работу так же, как это описано в работе 2, вариант А. Определить тепловой эффект окисления щавелевой кислоты перманганатом калия для обоих опытов по уравнению
Рреак = ‘ ~ — Сраств ~~ > (VI. 21)
<8 <5
где М — молекулярная масса (СООН)2-2Н2О; g — навеска
137
(СООН)2-2Н2О, г; Ag— избыток (СООН)2>2Н2О (в первом опыте равен 0,4 г, во втором опыте — нулю); QpacTB — —35338,3 Дж/моль. Сделать вывод о полноте протекания реакции окисления, сопоста* вив тепловые эффекты обоих опытов.
Работа 8. Определение истинной теплоемкости вещества в жидком состоянии
Для определения истинной теплоемкости жидкости Сж уравнение (VI.2) используют для двух систем: калориметра, содержащего воду + и калориметра, содержащего исследуемую
жидкость cWi=Kгде К — постоянная калориметра; Гн2О” удельная теплоемкость воды; сж — удельная теплоемкость жидкости; ^н2о и gm — навески воды и исследуемой жидкости. Совместное решение данных уравнений приводит к выражению
(VI.22)
и определяются экспериментально (см. с. 130).
Для определения cWl в калориметр залить 200 мл воды, а для определения cWi —200 мл исследуемой жидкости. Ток через нагреватель пропускать 2 мин. Условия проведения опытов (начальная температура, ток и напряжение) должны быть при определении и одинаковы.
Работа 9. Определение средней теплоемкости твердых и жидких веществ методом смешения
В работе следует определить среднюю теплоемкость малого количества вещества.
Прибор для определения средней теплоемкости состоит из двух ультратермостатов, контейнера, пробирки для исследуемого вещества и калориметра (рис. 64). Термостат I представляет собой металлический сосуд с крышкой /, заполненный водой. В крышке закреплены электронагреватель 4, мешалка 3, контактный термометр 5, электронное реле 7, термохимический термометр 6, Кон-' тактный термометр и электронное реле соединены с электронагревателем и позволяют поддерживать в термостате заданную температуру (fi = 50°C) с точностью до 0,01°. В крышке термостата I закреплен также контейнер 2, представляющий собой пробирку емкостью 120 мл, на дно которой помещается слой ваты, пробирка 8 для исследуемого вещества и термометр 9, Сверху контейнер закрыт также ватой. Пробирка для исследуемого вещества — узкая, толстостенная, вместимостью 3 мл, закрывается стеклянной палочкой 10, на которую надета каучуковая трубка. В крышке термостата II (f^25°C) закреплен калориметр, состоящий из калориметри-
138
веского сосуда 8 (пробирка вместимостью 120 мл), помещенного в стеклянную оболочку 2, и термометра Бекмана 11.
Метод смешения заключается в том, что два вещества, имеющие разные температуры, приводятся в термический контакт. В результате теплообмена система приходит в тепловое равновесие, при котором температуры обоих веществ выравниваются. Если такой процесс провести в условиях, когда теплообмен с внешней средой исключен или может быть учтен, то к системе применимо уравнение теплового баланса
cw\t — (^1^1 + К)
где cw — суммарная теплоемкость системы.
Согласно уравнению (VI. 1)
= znCTcCTa vl ,92 + ^н2о^нао*
(VI.23)
(VI. 24)
где Wh2o, wCt’ т\~ массы воды, калориметрического сосуда, исследуемого вещества; Сщо, Ст теплоемкости воды, стекла, исследуемого вещества; сн2о=4,187; сст= 1,0837 Дж/(г-К); а — часть калориметрического /сосуда, заполненная водой; а=0,6; v —
Рис. 64. Схема прибора для определения средней теплоемкости твердых и жидких тел методом смешения
объем погруженной части термометра Бекмана; 1,92 — теплоемкость единицы объема, погруженной части термометра Бекмана (у 1,92^20,8 Дж/К); К — тепловая константа пробирки для исследуемого вещества; А/ — изменение температуры калориметра, полученное графически.
Методом смешения можно определять среднюю теплоемкость вещества с достаточно высокой точностью (±3%). Работа выполняется в три стадии: определение суммарной теплоемкости калориметра, тепловой константы К пробирки для исследуемого вещества и теплоемкости исследуемого /вещества.
Определение теплоемкости твердого вещества. 1. Определение
139
константы К калориметра. Последовательность выполнения работы, Включить ультратермостат I (рис. 64), установленный на температуру 50°С. Закрепить контейнер в крышке ультратермостата. Поместить пустую пробирку для исследуемого вещества в контейнер и-закрыть тампоном ваты. Включить ультратермостат II, установленный на температуру 25°С. Взвесить калориметрический сосуд (тСт) на технических весах, залить в калориметрический сосуд 50 мл воды и вновь взвесить. Поместить термометр Бекмана в калориметрический сосуд с водой так, чтобы надетое на термометр резиновое кольцо закрывало калориметрический сосуд. Отметить длину погруженной части термометра Бекмана и высоту заполненной части калориметрического сосуда. Погрузить термометр Бекмана в мерный цилиндр с водой и определить объем погружаемой части термометра. Закрепить калориметр в ультратермостате II, поместить термометр Бекмана в калориметрический сосуд и закрепить его в штативе. Начать калориметрический опыт через 40 мин после того, как в ультратермостате I установится температура 50°С и скорость изменения температуры воды в калориметре станет менее 0,04 град/мин. Измерять температуру по термометру Бекмана через каждые 30 с. После 11-го отсчета вынуть контейнер с пробиркой из ультратермостата I и быстро перенести пробирку в калориметрический сосуд. Перемешивать содержимое калориметрического сосуда пробиркой. Продолжать непрерывно измерять температуру по термометру Бекмана через каждые 30 с. Сначала будет наблюдаться быстрое повышение температуры, а затем — постепенное снижение. Сделать одиннадцать отсчетов температуры после того как установится равномерная скорость изменения температуры. Записать показания температуры t\ термометра 9 в контейнере, затем измерить этим же термометром температуру воды t% в калориметрическом сосуде. Определить графически АЛ Рассчитать суммарную теплоемкость калориметрической системы cw по уравнению (VI.24). Рассчитать К по уравнению
K = (VI. 25)
2. Определение средней теплоемкости с вещества. Взвесить пробирку для исследуемого вещества на аналитических весах. Заполнить ее на % объема исследуемым веществом и вновь взвесить. Поместить пробирку в контейнер и закрыть его ватным тампоном. Провести опыт, подобный определению К калориметра. Вычислить среднюю теплоемкость с исследуемого вещества по уравнению (VI .23).
Определение средней теплоемкости жидкого вещества. Последовательность выполнения работы описана на с. 139. Взвесить пробирку для исследуемого вещества на аналитических (весах, поместить в нее 2 мл воды и снова взвесить. Поместить пробирку в контейнер и закрыть его ватой. Рассчитать К по уравнению (VI.23) Поместить в пробирку для исследуемого вещества 2 мл исследуемой жидкости и взвесить на аналитических весах.
140
Работа 10. Определение средней теплоемкости твердых веществ методом смешения
В работе следует определить теплоемкость твердого вещества.
Устройство прибора описано в работе 9, но термостат I (см. рис.
64) заменяется на сосуд Дьюара с охлаждающей смесью, т. е. в этом варианте При работе с сосудом Дьюара обязательно надевать защитные очки, так как при неосторожном обращении с ним он может лопнуть. '
Последовательность выполнения работы. Приготовить охлаждающую смесь смешением 500 г измельченного льда и 150 г поваренной соли. Перенести приготовленную смесь в сосуд Дьюара и оп-/ ределить ее температуру Л. Поместить контейнер в охлаждающую смесь. Дальнейшее описание опыта см. на с. 140.
Начать калориметрический опыт через 40 мин после того, как пробирка для исследуемого вещества будет помещена в контейнер, а скорость изменения температуры воды в калориметре станет ^меньше 0,04 гр ад /мин. Начать отсчеты температуры по термометру Бекмана через каждые 30 с. Вынуть контейнер из сосуда Дьюара после 11-го отсчета температуры и быстро перенести пробирку в калориметрический сосуд. Перемешивать воду в калориметрическом сосуде пробиркой. Продолжение опыта описано на с. 140.
Р а бота 11. Определение истинной теплоемкости жидкости
^методом калорифера
В работе следует определить теплоемкость вещества в жидком состоянии методом калорифера. В данном определении известное ^количество теплоты подводят к системе или отнимают от нее при помощи тела, нагретого или охлажденного до определенной температуры. Если точно известно количество теплоты, отдаваемое или .получаемое калорифером от жидкости, и изменение температуры жидкости, то по уравнению теплового баланса можно вычислить ’теплоемкость жидкости.
Калорифер представляет собой пробирку с 3 мл бензола, закрытую пробкой с термометром, градуированным через 0,Г. Калорифер используется для 'Охлаждения содержимого калориметра. Температура плавления бензола 5,4°С, теплоемкость твердого бензола 1,71, жидкого 1,62 Дж/(г-К), теплота плавления 126,6 Дж/г. 'Если калорифер охлажден ниже температуры плавления бензола, то охлаждение калориметра происходит за счет процесса плавления бензола. От калориметра в двух опытах с водой и исследуемой жидкостью отбирается одно и то же количество теплоты Q; если начальная температура ^<?Пл и конечная температура ^2>/пл» то
Q = (zniCi + К)А^1 = (/7X2^2 + К) (VI.26)
тде К—постоянная калориметра; т\—масса воды; т2— масса исследуемой жидкости; С\— удельная теплоемкость воды 4,1 Дж/(гХ ЗХК); с2 —удельная теплоемкость исследуемой жидкости; &fi— по-
141
нижение температуры в главном периоде опыта с водой; А/2— понижение температуры в главном периоде опыта с исследуемой жидкостью. Из уравнения (VI.26) следует
С2 = ^1+К(Д^-^2) .
При определении теплоемкости методом калорифера используются те же приборы, что и при методе смешения (см. с. 139). Работа выполняется в две стадии: определение АЛ и А/2. Опыты по определению АЛ и А/2 отличаются только тем, что в первом случае в калориметр заливают 50 мл воды, во втором — 50 мл исследуемой жидкости.
Последовательность выполнения работы. Подготовить охлаждающую смесь смешением 400 г измельченного льда со 120 г охлажденного и измельченного хлорида калия. Перенести смесь в сосуд Дьюара (работать в защитных очках). Температура криогидратной смеси —11 °C. Поместить в сосуд Дьюара контейнер. Последовательность выполнения работы см. на с. 139. Начать измерения температуры по термометру Бекмана через каждые 30 с после того, как установится скорость изменения температуры не менее 0,04 град/мин. Вынуть контейнер с калорифером из сосуда Дьюара после 11-го отсчета температуры и быстро поместить калорифер в калометрический сосуд. Перемешивать содержимое калориметрического сосуда калорифером. (Калорифер переносить за термометр.) Записать начальную температуру калорифера. Продолжать непрерывно отсчеты температуры по термометру Бекмана. (Должно наблюдаться быстрое понижение температуры после помещения калорифера в калориметрический сосуд.) Извлечь калорифер из калориметрического сосуда после того, как его температура достигнет + Ю°С. Температура по термометру Бекмана некоторое время будет продолжать понижаться, а затем начнет повышаться. По установлении равномерной скорости изменения температуры по термометру Бекмана сделать еще 11 отсчетов. Определить графически АЛ или А/2. Перенести калорифер в контейнер в сосуде Дьюара. Вычислить К согласно уравнениям (VI.23), (VI.24):
К = mCTcCT H-vl,92. (VI.28)
Рассчитать удельную теплоемкость исследуемой жидкости по уравнению (VI.27).
Работа 12. Определение теплоты образования насыщенного раствора при ^290 К (полной энтальпии растворения АН)
Полную энтальпию растворения для насыщенного раствора определяют косвенным путем, используя законы Гесса (см. работа 3) или Кирхгофа:
= дЯ^298 _ дМ7 - 298), (VI.29}
&Hsm,T — интегральная теплота растворения для раствора с кон-142
цептрацией, равной концентрации насыщения s при температуре Т; А//3Ш!298 находят графически по зависимости АЯ/пгэв от концентрации (моль/1000 г), построенной по справочным данным:
\Ср = ^Н20^Н20 Сиссл.влиссл.в> (VI.30)
— удельная теплоемкость раствора, определяемая экспериментально; Сн2о и Сиссл.в — молярные теплоемкости воды и исследуемого вещества; пИСсл.в— 1 моль исследуемого вещества; пн2о — число молей воды, приходящееся на 1 моль исследуемого вещества; gp — масса такого количества раствора, в котором находится 1 моль исследуемого вещества и п моль воды.
По графику зависимости растворимости данного вещества от температуры найти концентрацию исследуемого насыщенного раствора, приготовить 25—30 г этого раствора, взвешивая исследуемое вещество и воду на аналитических весах. Теплоемкость раствора определить методом смешения -согласно работе 9. Значения Аср и AHsm,r вычислить по уравнениям (VI.30) и (VI.31).
Работа 13. Построение диаграммы плавкости неизоморфных
веществ
В работе следует построить часть диаграммы плавкости карбамид— вода. Зависимость растворимости xi от температуры начала кристаллизации 7\ описывается уравнением Шредера для систем,
состоящих из неизоморфно кристаллизующихся веществ и образующих идеальные жидкие растворы (рис. 65):
, (VI.31)
АЯПЛ I ( 1 I 2,3-8,31 к Г z ~ Tt
где Xi—-растворимость i-ro компонента или молярная доля насыщенного раствора, находящегося в равновесии с твердой фазой при температуре 7\; АЯпл г —теплота плавления i-ro компонента; Гпл L — температура плавления чистого i-ro компонента.
Уравнение (V.32) описывает кривые ликвидуса на диаграмме плавкости. Если известны АЯпл и Тпл обоих компо-
Рис. 65. Диаграмма плавкости иеизоморфной системы, образующей в жидком состоянии идеальный раствор
чентов, то, задаваясь Xi, можно вычис-
лить температуры начала кристаллизации. Уравнение (VI.31) справедливо только при условии А//пл = const (АЯпл i не зависит от температуры). Если при растворении один из компонентов меняет фазовое состояние (растворение твердого вещества в жидком растворе), то для идеальных растворов
(VI. 32) где АЛг — дифференциальная теплота растворения (относительная
143
энтальпря). Если компонент не меняет своего фазового состояния при растворении (растворение жидкой воды в растворе), то АНг = = 0. Тогда согласно уравнению Гиббса — Дюгема можно записать
= лТю(кн2)2л^со(хн2)2 + Л'н2од^н2О’ (VI.33)
где АНт— интегральная теплота растворения карбамида при образовании 1моль раствора. Интегральную теплоту растворения определяют экспериментально, что дает возможность вычислить теплоту плавления и построить часть диаграммы плавкости. Уравнение (VI.31) можно записать в более удобной для расчета форме:
lg XCO(NH2)2 ~~ Д Т,
где
А^пл CO(NHa)g г CO(NH2)2
0,3-8,31 ’ 2,3-8,31
(VI. 34)
Температура плавления карбамида 405 К-
Работа состоит из трех частей: 1) определение теплоты растворения карбамида АНт\ 2) построение диаграммы плавкости системы CO(NH2)2—НоО: а) по расчету с использованием значения АЯМ и уравнения (VI.34); б) по экспериментальным данным; 3) проверка предположения об идеальности раствора экспериментальным определением теплоты разбавления.
Последовательность выполнения опытов. Определить АНт при растворении 2 г карбамида в 200 г воды (см. с. 131). Интегральная теплота растворения определяется по уравнению
И/ , (VI .35)
где ^И/=йсо(?ш2)а + янао; ^co(nh2)2 = ^/A1co(nh2)2J #н2о=200/Л4нао;
A1co(nh2)3; 7Ин2о — молекулярные массы карбамида и воды; g — навеска карбамида.
Вычислить АЯплС0(кн2)2 по уравнению (VI.33). Построить диаграмму плавкости, использовав значение ДЯПл cO(Nh2)2, Гплсо(кн2ь и уравнение (VI.34). Построить диаграмму плавкости по экспериментальным и справочным данным.
Температура начала кристаллизации для некоторых составов системы карбамид — вода.
1,0Ю 0,97 0,95 0,93 0,99 0,80 0,70 0,60 0,50 0,40 0,30 0,20 0,10 0,00 т/к 273 271 268 2G3 263 283 306 328 346 362 375 387 396 405
Дать заключение о пределах концентраций, для которых применимо уравнение (VI.31). Определить теплоту разбавления 200 мл 20 %-кого (молярное содержание) раствора карбамида 6 мл воды. (Эту часть работы можно выполнить при условии, если At определяется с точностью не менее чем ±0,001.) Описание опыта см. на с. 1сю.
144
it Работа 14. Определение теплот сгорания органических веществ У
гтч
Теплота сгорания вещества — количество теплоты, которое вы-ф деляется при полном сгорании 1 г или 1 моль вещества и охлажде-/Ц нии продуктов сгорания до начальной температуры опыта. При пол-£ ном сгорании органического вещества углерод превращается в диоксид углерода, водород — в воду, сера — в SO2, галогены—в га-логеноводороды, азот выделяется в свободном состоянии. При вы-; числении теплоты сгорания необходимо учитывать, в каком агре-, гатном состоянии находится вода. При сжигании веществ в калории етрической бомбе (v=iconst) тепловой эффект будет равен Qv:
Qv = Qp + &vRTt (VI. 36)
где Qv — теплота сгорания при постоянном объеме; Qp — теплота сгорания при постоянном давлении; Av — разность числа молей га-за до и после реакции.
Если уравнение реакции горения органического вещества может быть записано в общем виде
C*HmOpNr + ( k + - £-] О2 = ЙСО2 + V Н2о + -J- N2 + (VI.37)
то изменение числа молей газообразных продуктов Av составит (т/2) — р — г Данные по теплотам сгорания при постоянном дав-
лении позволяют рассчитать теплоту образования А//°т соединения при стандартных условиях —AH°T = QP.
Для определения теплоты сгорания навеску исследуемого вещества помещают в калориметрическую бомбу. Калориметрическую бомбу заполняют кислородом до давления 25-105 — 45-105 Па и помещают в калориметр с водой. В начале главного периода вещество поджигают при помощи железной проволоки, через которую пропускают электрический ток. На крышке калориметрической бомбы расположены входной клапан для наполнения бомбы кислородом, выходной клапан для выпуска газов по окончании опыта, клемма токоведущего штифта.
Работу по определению теплоты сгорания выполняют в три стадии: 1) определение суммарной теплоемкости калориметрической бомбы и ее содержимого; 2) определение теплоты сгорания исследуемого вещества; 3) определение теплоты сгорания вспомогательных материалов. Суммарную теплоемкость определяют по At калориметра при сгорании определенного количества вещества с точно известной теплотой сгорания. Стандартным веществом для определения тепловых эффектов сгорания служит бензойная кислота (о. ч.). Для стандартных установок масса бензойной кислоты составляет 0,8—1,2 г, что обеспечивает в результате ее сгорания подъем температуры в калориметре на 2—3°.
1. Определение суммарной теплоемкости cw. Взвесить на аналитических весах 14 см запальной проволоки, вставить ее в виде петли в цилиндр пресса. Концы проволоки должны выходить наружу.
145
Взвесить г г исследуемого вещества на технических весах, всыпать навеску в цилиндр пресса, завернуть винт пресса до отказа, отодвинуть нижнюю пластинку и выдавить брикет с торчащими сверху концами проволоки. Если поверхность брикета загрязнена, то ее следует очистить бритвой. Взвесить брикет на аналитических весах. (После взвешивания брикет брать только за концы проволоки.) Налить пипеткой 10 мл дистиллированной воды для насыщения внутреннего пространства бомбы водяными парами и для растворения в ней образующихся при сгорании веществ оксидов азота. Установить на штатив с кольцом крышку калориметрической бомбы. Укрепить чашечку с навеской бензойной кислоты на конце токоведущего штифта. Присоединить один конец запальной проволоки к токоведущему штифту, другой — к трубке выходного клапана. Привязать хлопчатобумажную нить, концы которой опустить на дно чашечки таким образом, чтобы брикет прижал концы нити. Погрузить крышку с надетыми на нее резиновым и металлическим кольцами осторожно без перекосов в стакан. Надеть зажимное кольцо и завинтить крышку до отказа. Присоединить к входному клапану бомбы металлическую трубку от кислородного баллона с редуктором, отрегулированным на 30-105 Па. Открыть входной и выходной клапаны бомбы и осторожно, чтобы избежать разбрызгивания воды, налитой в бомбу, открыть вентиль баллона. Слабый ток кислорода пропускать 2—3 мин. Закрыть выходной клапан после вытеснения из бомбы воздуха кислородом и наблюдать за скоростью повышения давления в бомбе. Скорость не должна превышать 4-Ю5 — 5-Ю5 Па/мин. Закрыть вентиль баллона и входной клапан, когда давление в бомбе достигнет 25-105— 30-105 Па. Отделить металлическую трубку от бомбы. Погрузить бомбу в калориметрический сосуд, присоединить к клеммам на крышке провода, установить мешалку и вращением ее вручную убедиться в том, что она не задевает за стенки бомбы. Залить воду в калориметрический сосуд, определив ее массу по разности массы сосуда с водой и пустого, из которого заполняется калориметр. Закрепить термометр Бекмана, настроенный на 0,5—1,5° в начале опыта. Закрыть калориметр крышкой и включить мешалку, регулируя ее скорость реостатом. Для перемешивания воды в калориметрическом сосуде частота вращения мешалки должна быть 60—80 об/мин. При работе верхняя пластинка мешалки не должна выходить из воды и ударять об ее стенки. Приступить к проведению опыта через 5— 10 мин после установления бомбы в калориметр. За это время произойдет выравнивание температуры во всех частях калориметрической системы. О выравнивании температуры можно судить по отсчетам температуры термометром Бекмана через каждые 30 с. Только после того как скорость будет равномерной, можно начинать отсчеты. Отсчет показаний термометра Бекмана производить через каждые 30 с. Включить цепь зажигания образца после 11-го отсчета температуры. При этом должна загореться лампочка. После перегорания нити запала лампочка должна погаснуть. Если лампочка не загорелась, то это значит, что неисправны контакты и весь 146
опыт следует начать сначала. Если лампочка загорелась и не гаснет, а подъем температуры начался, это означает, что после зажигания произошло короткое замыкание. Необходимо выключить ток и продолжать измерения температуры. Главный период характеризуется резким подъемом температуры. Он считается законченным, если вновь наступает равномерная скорость изменения температуры, обусловленная теплообменом калориметра с окружающей средой. Главный период обычно длится 6—9 мин. Сделать не менее 11 отсчетов в конечном периоде калориметрического опыта. Открыть крышку калориметра, вынуть термометр, отключить от бомбы провода^ вынуть мешалку и осторожно перенести на стол бомбу. Открыть осторожно выходной клапан и в течение 4—5 мин выпустить газы из бомбы. При быстром выходе газов возможен унос капелек воды с растворенной ,в них кислотой. Отвинтить зажимное кольцо и вынуть крышку из бомбы по окончании выхода газов. Тщательно осмотреть внутренние части бомбы. (При наличии сажи или части веществ опыт следует повторить.) Вылить воду в химическийстакан и промыть дистиллированной водой внутреннюю поверхность бомбы, крышку, чашечку, трубку и токоведущий штифт, собирая промывные воды в тот же стакан. Объем промывных вод должен составить 150—200 мл. Собрать остатки несгоревшей проволоки и взвесить ее. Определить титрованием количество образовавшейся при горении азотной кислоты. Стакан и крышку тщательно вытереть и оставить открытыми до следующего опыта. Вычислить тепловую постоянную калориметрической бомбы cw по уравнению
cw ---------------------- , (VI. 38)
где а — масса бензойной кислоты, г; Qa — теплота сгорания 1 г бензойной кислоты, равная 26,455 кДж/г; b—- масса сгоревшей части проволоки, г; Qb — теплота сгорания 1 г проволоки, кДж/г (для железа 19,228, для никелина 3,239, для меди 2,508 кДж/г); с — масса хлопчатобумажной нити, г; Qc — теплота сгорания хлопчатобумажной нити, равная 76,720 кДж/г; d — количество образовавшейся кислоты*, моль; Qd — молярная теплота образования раствора кислоты, равная, 57,78 Дж/моль; А/— изменение температуры калориметра за счет теплоты, выделившейся в процессе сгорания, при отсутствии теплообмена с окружающей средой (вычисляется графически или аналитически).
2. Определение теплоты сгорания q твердого вещества. Провести опыт аналогично опыту 1, если из исследуемого вещества можно
* Для определения содержания кислоты необходимо прокипятить раствор в стакане, накрытом часовым стеклом, 4—5 мин для удаления растворенного диоксида углерода, охладить, добавить 2—3 капли раствора фенолфталеина и титровать 0,1 и. NaOH до появления неисчезающей розовой окраски. Количество молей кислоты составит а=--и\\у-\ где п — количество 0,1 н. NaOH, израсходованное на титрование, мл.
147
приготовить брикет. Теплоту сгорания q рассчитать по уравнению
q = (cwM — bQB — Qc-— QD)/a. (VI. 39)
Если же из вещества невозможно приготовить брикет, то вещество засыпать в мешочек из полиэтилена или тонкой резины. При расчете теплоты сгорания необходимо учесть теплоту сгорания мешочка и уравнение (VI.39) примет вид
q = — Z>QB — dQD — eQE)/a, (VI.40)
где a—масса исследуемого вещества; Qe — теплота сгорания вспомогательных материалов; е— масса вспомогательных материалов (полиэтилена или резины, для которых теплоты сгорания равны 39,8 и 40,4 кДж/г соответственно).
3. Определение теплот сгорания вспомогательных материалов Qe. Взять навеску органического вещества с известной теплотой сгорания. Вещество не брикетировать. Поместить в мешочек, опустить в мешочек запальную проволоку и проводить опыт. Рассчитать теплоту сгорания вспомогательных материалов по уравнению (VI.40).
Предостережение. При работе с калориметрической бомбой присутствие даже небольшого количества вазелина или любой смазки в бомбе может вызвать взрыв при заполнении ее кислородом.
Работа 15. Термометрическое титрование
Для определения концентрации вещества А можно применять термометрическое титрование, которое основано на использовании теплоты, выделяющейся (или поглощающейся) при химических реакциях. Для этого подбирают вещество В (титрант), который вступает в идущую до конца реакцию с веществом А. К титруемому раствору приливают постепенно титрант. В ходе приливания титранта в результате взаимодействия вещества А и В выделяется или поглощается теплота и температура реакционной смеси меняется до тех пор, пока вещество А не прореагирует полностью. По достижении стехиометрического соотношения А и В дальнейшее приливание титранта не изменяет температуры реакционной смеси и она остается постоянной или плавно меняется за счет теплообмена с окружающей средой. На кривой зависимости изменения температуры реакционной смеси от количества добавленного раствора В появляется точка перегиба, позволяющая определить стехиометрическое количество вещества В.
Термометрическое титрование применяется и тогда, когда другие методы титрования неудобны или неприменимы. При удачном подборе титрующего реагента метод можно применять для титрования окрашенных, вязких веществ и систем, содержащих смолистые или твердые примеси. Термометрическое титрование можно
143
1:
Sj! применять для следующих типов реакций: 1) взаимодействие кис-лот ц .оснований:
NaOH + НС1 = NaCl + Н2О; NH4OH + НС1 = NH4C1 + Н2О;
1 NH4C1 + NaOH = NH4OH + NaCl
Концентрация титруемого раствора не должна быть меньше ! *0,05—0,5 г-экв/л (при низких концентрациях мало изменение тем-; пературы). Концентрация титрующего раствора не должна превы-< шать 1 г-экв/л, так как смешение концентрированных растворов кислот и щелочей кроме теплоты нейтрализации сопровождается выделением теплоты разбавления;
2) реакции, при которых одним из конечных продуктов является практически нерастворимый осадок:
ВаС12 + Li2SO4 = 2LiCl + |BaSO4
Ni (NO3)2 + 2NaOH = |Ni (OH)2 -f- 2NaNO3
Чем меньше растворимость осадка, тем выше точность результатов титрования. Произведение растворимости гидроксидов при 25°С составляет (моль/л)
ПРВадо4 = 1 • 5 • 10—9; ПРМЦОН^ =2-10~16.
Незначительная растворимость этих осадков позволяет с достаточной точностью (±1%) определить концентрации ионов Li+ и Na2+ в 0,05—0,5 н. растворах;
3) реакции, при которых одним из продуктов является растворимое устойчивое комплексное соединение
Zn2+ 4- 4NH3 = (Zn (NH3)4]2+
Устойчивость комплексного соединения определяется константой //нестойкости:
[zn2+](NH3r
[Zn(NH3)^+] *
В данной работе следует определить 1концентрацию соляной кис-доты по тепловому эффекту реакции нейтрализации (Q):
Н+ 4- ОН- - Н2О 4- Q
Последовательность выполнения работы. Налить 20 мл титруемого раствора в пробирку со стеклянной оболочкой, закрепленную . на штативе, опустить в раствор стеклянную мешалку и термометр Бекмана. В течение 2—3 мин наблюдать за изменением температуры. Скорость изменения температуры не должна превышать 0,02 град/мин. С помощью обычного термометра сравнить температуру титруемого раствора /и титранта. Если различие в температурах не превышает 0,1°, титрант пылить в бюретку, закрепленную на том же штативе, что и пробирка. Конец бюретки опустить в пробирку таким образом, чтобы титрант не попадал на термометр, н приливать его к титруемому раствору непрерывно. Титруемый раст
149
вор все время размешивать мешалкой. Показания термометра Бекмана записывать после каждого добавленного объема титранта. На основании полученных данных построить график в координатах температура — объем титранта (рис. 66).
Точку эквивалентности титрования для первого предварительного опыта определить по графику и на основании полученных данных установить условия для проведения трех точных опытов исходя из следующих положений: точка эквивалентности титрования
О&ъем титрантс^мл
Рис. 66. Кривые термометрического титрования
Ооъем титранта^ ил
должна лежать в интервале 3—8 мл (расход титранта не должен превышать 8 мл, так как изменения высоты столба титранта в бюретке и объема титруемого раствора уменьшают скорость приливания и дают дополнительный излом на кривой, что затрудняет определение точки эквивалентности). Если точка эквивалентности лежит в интервале 3—8 мл, концентрации титруемого и титрующего растворов оставляют без изменения; для интервала 0—3 мл следует уменьшить концентрацию титранта; для интервала 8 мл и более следует уменьшить концентрацию титруемого раствора. При окончательном титровании записать показания термометра Бекмана после приливания каждых 0,5 мл раствора. Расхождение между результатами отдельных опытов при определении точки эквивалентности не должно превышать 0,2 мл. Чтобы результат анализа был более точным, необходимо прокалибровать пипетку для титруемого раствора и ту часть бюретки, объем которой равен объему жидкости, израсходованному на титрование.
После калибрования посуды рассчитать концентрацию А и В по уравнению
(VL41>
где vA —объем титруемого раствора, мл; Ад—концентрация титруемого раствора, г-экв/л; — объем раствора, израсходованный на титрование, мл; Ав— концентрация титранта, г-экв/л;
Ад = ^вАв/уд или сА = ^вАвЛЧ">
(VI.42>
г
F
—концентрация титруемого раствора, моль/л; п — количество молей титрующего вещества В, которое вступает в реакцию с J 1 моль титруемого вещества А.
Примечание. При выполнении работы надо помнить о соблюдении условий: ? а) приливать титрант к титруемому раствору надо непрерывно и с постоянной
I скоростью. Высота столба титранта в бюретке при титровании не должна спи-
жаться больше чем на одну треть по сравнению с начальной высотой; б) тер-мометр Бекмана обладает значительной тепловой инерцией, поэтому скорость J вытекания титранта из капилляра не должна превышать 1 мл/мин. '"го?’
Калибровка мерной посуды " 2 *
1. Калибровка пипетки. На технических весах с точностью до 0,01 г взвесить р коническую колбу вместимостью 50 мл. Пипетку заполнить дистиллированной s водой до метки, содержимое ее вылить во взвешенную колбу и взесить. Истин-у ный объем пипетки оп вычислить по уравнению
I va = g/p, . (VI.43)
где g— масса воды, г; р — плотность воды при данной температуре, г/мл. Плот-ность воды при разных температурах:
/ t, °C 15—18 19—23 24—25
। р, г/мл 0,999 0,998 0,997
2. Калибровка бюретки. Бюретку заполнить водой и из нее во взвешенную f коническую колбу спустить такой же объем воды, который был израсходован J на титрование. Воду взвесить, ее истинный объем определить так же, как объем •у пипетки.
( Работа 16. Определение константы скорости растворения КС1*
F Скорость растворения описывается уравнением
4^ = /<(ci-c)=/<oS(cs-c), (VI.44)
|к ' ат
где cs и с — концентрации насыщения и данного раствора соответственно, (г/мл); S — площадь поверхности растворяющегося образца хлорида калия, см2; Kv— константа скорости растворения, см/мин, /<=/(,-S.
f ПРИ растворении малого «количества хлорида калия в -большом объеме раствора хлорида калия с концентрацией с в начальном пе-у: риоде опыта (за 15—60 с) разность (cs—с) можно считать постоян-ной -величиной, и фур!/1' '
Ц" d х Ах
7,— — ~— -const. (VI. 45)
d т Ат
< Тогда при проведении растворения в калориметре скорость изменения температуры Д//Дт будет пропорциональна Дх/Дт, и вместо уравнения (VI.44) можно записать
м
К'(cs — с). (VI.46)
* Работа рассчитана на два занятия.
151
•S
Для выявления связи между Kv и К' используем уравнение теплового баланса (VI. 1), согласно которому
д — М — cw&t,
(VI. 47)
где Qkci — дифференциальная теплота растворения KCI; cw — суммарная теплоемкость калориметрической системы; М — молекулярная масса КО. Объединив уравнения (VI.44), (VI.45), (VI.46), (VI.47), получим
Ах — —----
Qkci
А/;
\tczz,M
Qkci^t
— Ку S(cs — с);
М
Дт
(cs — с);
К' — KySQ^Q}JcwM\
Ку — К' cwM/SQ^q\ ,
(VI.48)
(VI. 49)
По интегральным теплотам растворения рассчитать Qkci~ЛЯш (см. справочник), определить опытным путем S, cWi К'.
Последовательность выполнения опытов. 1. Определение /С. Изготовление таблетки хлорида описано на с. 146; масса таблетки должна быть около 2,0000 г. Для определения площади поверхности таблетки микрометром измерить диаметры на верхнем и нижнем торцах по нескольким направлениям и высоту по нескольким образующим цилиндрической 'части. Для вычисления взять средние значения диаметра и высоты.
Описание калориметра см. на с. 127. В калориметр поместить стакан, содержащий 200 мл водного раствора хлорида калия (в первой серии опытов концентрация 0,150—0,200, во второй серии— 0,200—0,300 г/мл); над раствором за проволоку подвесить таблетку хлорида калия.
Калориметрический опыт следует начинать тогда, когда установится скорость изменения температуры, не превышающая 0,005 град/мин. Частота вращения мешалки 190—210 об/мин. Температуру раствора перед началом растворения определить термохимическим термометром с точностью 0,02°. В начальном периоде через каждые 30 с произвести отсчеты температуры и записать показания термометра. После пятого отсчета погрузить таблетку в раствор, так чтобы она не касалась дна стенок сосуда, и продолжать запись показаний термометра через каждые 15 с в течение 1 мин. Затем опыт закончить, таблетку вынуть и высушить фильтровальной бумагой. Если поверхность таблетки и концентрация раствора изменились незначительно, то можно с ними провести еще три последовательных опыта. Ко второму и третьему опытам калориметр подготовить так же, как описано выше.
Следующие три опыта выполнить со второй таблеткой и с раствором другой концентрации, 152
На основании полученных данных построить графики в координатах температура — время в масштабе О,Г — 2 см, 30 с— 1 см. Из графиков определить тангенс угла наклона прямой Ах/тА (для первой минуты от начала растворения). Концентрацию насыщения Cs.kci для каждого опыта найти по данным:
t, °C 20 22 24 25 26 28 30
с„ г/мл 0,298 0,306 0,310 0,312 0,314 0,317 0,320
Вычислить К' по уравнению (VI.46).
2. Определение cw. Для обоих использованных растворов определить cw, как описано в работе 1.
Дифференциальную теплоту растворения хлорида калия найти графически по данным справочника для концентраций, соответствующих использованным растворам.
Вычислить Ко по уравнению (VI.49). Погрешность найденного значения константы скорости вычислить, использовав 6—8 определений. Результаты вычислений представить в виде таблицы по образцу:
ГЛАВА VII
ФАЗОВЫЕ РАВНОВЕСИЯ '
г
1. ОБЩИЕ УСЛОВИЯ РАВНОВЕСИЯ В ГЕТЕРОГЕННЫХ СИСТЕМАХ
Равновесия в гетерогенных системах, в которых не происходит химического взаимодействия между компонентами, а протекают лишь процессы перехода компонентов из одной фазы в другую (или в другие), называются фазовыми равновесиями. Кипение, замерзание и внезапное проявление ферромагнетизма — все это связано с изменением состояния системы без изменения химического состава. Любая гетерогенная система характеризуется определенным числом фаз, компонентов и числом степеней свободы.
Фаза —часть гетерогенной системы, ограниченная поверхностью раздела и характеризующаяся в отсутствие внешнего поля сил одинаковыми физическими свойствами во всех своих точках. Фаза может быть раздроблена на отдельные части, но это не увеличивает
153
числа фаз в системе. Например, в -насыщенном растворе хлорида натрия в присутствии любого количества кристаллов соли и пара над раствором будут одна кристаллическая, одна жидкая и одна парообразная фазы. Система, содержащая более одной фазы, называется гетерогенной.
Число составных частей—это число тех видов частиц, составляющих систему, которые могут существовать отдельно и вне системы. Так, в водном растворе хлорида натрия будет содержаться много видов частиц (молекулы соли и воды, гидратированные ионы Na+, Cl“, Н+). В действительности же в системе только две составные части: вода и поваренная соль, так как ни один из перечисленных ионов не может быть извлечен из данной системы в отдельности.
Независимые компоненты — вещества, наименьшее число которых необходимо и достаточно для образования всех возможных фаз данной системы, находящейся в равновесном состоянии.
Термодинамические степени свободы — независимые термодинамические параметры фаз системы, находящейся в равновесии, изменение которых в определяемых пределах не вызывает исчезновения одних и образования других фаз. К этим параметрам относятся температура, давление и концентрация вещества.
Вариантность системы—число степеней свободы равновесной термодинамической системы. Из какого бы числа компонентов и фаз ни состояла гетерогенная система, условием равновесия между фазами в ней является то, что химический потенциал любого компонента должен быть одинаковым во всех фазах системы при постоянных температуре и давлении. Условия равновесия гетерогенной системы подчиняются правилу фаз Гиббса. Уравнение правила фаз Гиббса устанавливает зависимость между числом степеней свободы, числом компонентов и числом фаз в данной равновесной системе:
/ - - К — Ф + 2 или f-Kr — q — Ф + 2, (VII.1)
где f — число степеней свободы; Ф —число фаз; К — число компонентов; q — число уравнений связи; К' — число составных частей системы. Так как то Ф=СК + 2. Поэтому невозможно подобрать такие значения давления и температуры, при которых существовали бы, например, сера ромбическая, монокристаллическая, жидкая и парообразная. То же самое исключает сосуществование пяти фаз в двухкомпонентной системе. Если никаких ограничений не наложено, то # = 0. Так, в системе Н20(ж) =Н2О(г) (рис. 67) f=l — 2 + 2=1.
Следовательно, каждой температуре отвечает единственное значение давления насыщенного пара; поэтому при фиксированной температуре давление насыщенного пара постоянно, т. е. оно не зависит от объема. Если изменить только один из параметров, например р (при T=const) или Т (при p=const), то равновесие нарушится, так как исчезнет одна из фаз. Это приводит к увеличению 154
числа степеней свободы на единицу, т. е. создается возможность
менять одновременно р и Г независимо друг от друга.
Если в системе протекают химические реакции, то величина f
равна числу независимых уравнений реакций. Так, в системе СаСО3 = = CaO Т* СО2 с/ — 1 и f~ 3— 1—3 4-2=1.
Равновесие при диссоциации СаСО3 определяется одним параметром, а именно температурой, т. е. каждой температуре соответствует определенное давление СО2. Если химического взаимодействия не происходит, но фазы имеют тождественный состав, то появляется дополнительное ограничение. Поэтому уравнение правила фаз (VII. 1) преобразуется:
/ = + (VII.2)
Рис. 67. Диаграмма фазового состояния воды
т. е. независимо от числа веществ система будет вести себя как однокомпонентная. В качестве примера можно привести систему жидкость— пар, в которой состав фаз совпадает (азеотропная смесь).
2. ЗАВИСИМОСТЬ ДАВЛЕНИЯ НАСЫЩЕННОГО ПАРА ОТ ТЕМПЕРАТУРЫ
Давление насыщенного пара индивидуальных жидкостей. Одним из важнейших свойств жидкости является давление ее насыщенного пара, характеризующее способность жидкости к испарению. Тепловое движение молекул ведет к отрыву их от поверхности жидкости и переходу в газовую фазу. Однако такой отрыв может произойти, если кинетическая энергия молекулы будет больше энергии взаимной связи с .молекулами жидкости. Часть молекул, оторвавшихся от поверхности жидкости, впоследствии снова конденсируется, другая же часть остается в газообразной фазе. Таким образом, на поверхности жидкости всегда происходит одновременно два процесса: испарение и конденсация. Если эти процессы осуществляют в,замкнутом пространстве, то скорости испарения и конденсации выравниваются, и между жидкой и газообразной фазами наступает состояние динамического равновесия. Давление, которое молекулы пара, находящегося в равновесии с жидкой фазой, оказывают на стенки сосуда и на поверхность жидкости, называется давлением насыщенного пара (для краткости давление пара жидкости). Давление пара является функцией кинетической энергии молекул и числа их в единице объема (т. е. плотности) и выражается основной формулой кинетической теории газов:
— (VII. 3)
где р—давление пара; v — объем пара; N— число молекул; т — масса молекулы; w — средняя квадратическая скорость молекул.
155
Согласно закону распределения Больцмана число молекул, обладающих энергией, большей некоторого заданного предела е, рассчитывают по уравнению
N z = N^ilkT, (VII. 4>
где Ni — число молекул, обладающих энергией — число молекул на нулевом уровне; Т — абсолютная температура; k— постоянная Больцмана.
Согласно уравнению (VII.4) количество молекул, обладающих кинетической энергией, достаточной для преодоления сил сцепления, с повышением температуры возрастает в экспоненциальной зависимости, и скорость испарения будет увеличиваться с повышением температуры. В то же время скорость конденсации определяется средней квадратичной скоростью молекул по уравнению
(VII.5)
Таким образом, скорость конденсации с повышением, температуры возрастает пропорционально корню квадратному из температуры» т. е. значительно медленнее, чем скорость испарения. Поэтому с повышением температуры сильно возрастает плотность газовой фазы» а следовательно, и давление пара. Согласно правилу фаз система с одним компонентом и двумя сосуществующими фазами имеет только одну степень свободы. Давление пара над плоской поверхностью стабильного химического вещества определяется только температурой и не зависит от количества взятой жидкости (твердого тела), от количества пара и от наличия и концентрации воздуха или другого газа, инертного по отношению к другому пару.. На давление пара, помимо температуры, оказывает влияние также форма (кривизна) поверхности жидкости (твердого тела) и наличие на нем электрического заряда. Термодинамика равновесных фазовых переходов приводит к уравнению Клапейрона — Клаузиуса (для плоской поверхности)
d р дя(1’2)
—Д’ = --------Г = "ДГ---- > (VI1.6)
d Т Т (£Д — ^i) ТAv
где — теплота фазового перехода, Дж/моль; Av— изменение молярного объема при переходе из фазы 1 в фазу 2. Производная dp/dl указывает на соотношение температуры и давления при сохранении равновесия между обеими фазами.
Для процессов испарения и сублимации уравнение (VII.6) связывает изменение давления пара с температурой dp/dT, изменением объема и тепловой эффект процесса, а для процессов плавления и полиморфного превращения — изменение температуры перехода с давлением и соответствующие изменения объема и тепловой эффект. При испарении или возгонке уравнение (VII.6) можно упростить, сделав следующие допущения: 1) (f — молярный
объем жидкости или кристалла), поэтому можно пренебречь в зна-
156
менателе величиной v, 2) пар подчиняется уравнению состояния идеальных газов:
pv = RT. (VII. 7>
С учетом данных допущений уравнение (VII.6) принимает вид
dlnp Д^(исп.возг) ,,7„
АТ = -----575---- (VII.8>
* а Г R12
(АН —теплота испарения или возгонки); 3) если теплота испарения не зависит от температуры.
Уравнение (VII.8) после интегрирования при постоянных АН и R имеет вид
Гпр = — &H/RT Ч-В' или IgP = — А/Т Ч-В, (VII.9>
где Л =АЯ/2,3037?=ДЯ/(2,303/8,314) =АЯ/19,15, а В=В'/2,303. Величина В зависит от размерности, в которой выражено давление (правильно .в паскалях, приемлемо в атм или в мм рт. ст.).
Уравнение (VII.9) отвечает линейной зависимости 1gр от 1/Т; по тангенсу угла наклона прямой можно определить теплоту перехода, так как АН=—2,3/?tga, где а — угол, образованный прямой и осью абсциссы. После интегрирования (VII.9) в пределах получаем
р2 Ш fl JJ.
А 2,3037? lr2 tJ’
(VII. 10)
что позволяет, зная давление лара при двух температурах, расечи-тать теплоту фазового перехода.
Наиболее грубым допущением из принятых при выводе уравнения (VIIЛ 0) является предположение о независимости АН от температуры. Эту неточность можно устранить, воспользовавшись уравнением Кирхгофа
d Л// .. . х
(VII.11)
где Ас — разность молярных теплоемкостей фаз. Величина Ас зависит от температуры. Эту зависимость обычно выражают уравнением
Ас = Ла Ч- кЬТ Ч- ДсП, (VII. 12)
где а, Ь, с — эмпирические коэффициенты.
Согласно (VII.11)
т т
\Н = Д//298.15 "h f АСр d Г = А//298.15 Ч~ f (A# "h A^/* 4“ ДсГ^) d T.
298,15 298,15
(VII. 13)
Выражение (VIL13) нужно подставить в уравнение (VII.8) вместо АН. После интегрирования полученного таким образом выражения
157
находим зависимость p = f(T) почти точно. В ряде случаев ограничиваются двумя членами в уравнении (11.12), что дает
d In p/d Т =
А ^298,15 f (\а + \ЬТ)
298,15
RT2
и
1g = —ДЯ298115/2,303/?Г + \а igT/R 4- Д^/2,303Л 4-const 7. (VII. 14)
В уравнении (VII.14) const называется химической константой индивидуального вещества. Ее можно определить, как эмпирическую величину по измерению давления пара.
Первые два допущения не вносят больших искажений в расчет, но следует иметь в виду, что пар ведет себя как идеальный газ лишь при достаточно низких давлениях. Молярные объемы жидкости и пара сближаются при увеличении давления.
Работа 1. Определение давления насыщенного пара индивидуальных жидкостей статическим методом
Статический метод основан на непосредственном измерении давления при заданной температуре. Пары исследуемого вещества, находящегося в вакуумированном закрытом приборе с короткой манометрической трубкой, заполненной наполовину ртутью, создают в ней некоторую разность давления. Эта разность давлений выравнивается введением воздуха во второе колено манометрической трубки. Давление пара исследуемого вещества отсчитывают непосредственно по ртутному манометру или вакуумметру, присоединенному <к установке.
Последовательность выполнения работы. Обычный стеклянный аквариум (рис. 68) залить дистиллированной водой (термостат), снабдить электронагревателем 5, мешалкой 2, контактным термометром 5, термометром 4 и держателем /О, в котором крепится прибор 1. Прибор через систему кранов 6, 7, 8 соединить с вакуумметром 9, вакуумным насосом (через ловушку, форвакуумный бу- , ферный баллон и трехходовой пран) и атмосферой.
Тщательно вымытый и высушенный прибор 1 укрепить в держателе 10, расположенном над поверхностью термостата. Перед началом опыта залить чистую ртуть в шарик D. Ртути налить столько, чтобы была заполнена до половины манометрическая трубка прибора /. В шарик Н через горлышко залить испытуемую жидкость на !/2 его объема. Горлышко смазать вакуумной смазкой и надеть нагретый пришлифованный колпачок (для более точных измерений лучше прибор запаять). После того как в прибор 1 залиты исследуемая жидкость и ртуть, его соединить вакуумным шлангом с кранами, кран 6 связывает прибор с атмосферой, кран 8—-с вакуумным насосом и кран 7—с вакуумметром 9. При закрытом кране 6 открыть кран 8 и откачать воздух из всей системы. Когда испарится приблизительно !/з жидкости, вакуумирование прекратить. Не
153
-снимая -каучуковой трубки, прибор 1 вынуть -из держателя 10, наклонить влево и перелить ртуть из шарика в манометрическую трубку прибора 1. Затем его вновь вставить в держатель и опустить в термостат. Кран 8 закрыть.
При вакуумировании температура жидкости в приборе становится ниже температуры термостата, поэтому после помещения прибора в термостат -будет изменяться уровень ртути в манометрической трубке, так как давление пара вещества увеличивается с повышением температуры. Чтобы установить ртуть в обоих коленах на одном уровне, через кран 6, соединяющий прибор с атмосферой, очень медленно впускают воздух. Если же воздуха введено боль-
Рис. 68. Схема установки для определения давления насыщенного пара над жидкостью (статический метод)
ше, чем это нужно, то для выравнивания давления в обоих коленах манометрической трубки нужно Осторожно открыть кран 8, соединяющий систему с вакуумным насосом (который в течение опыта находится в рабочем состоянии) и откачать лишний воздух. Когда уровни ртути в манометрической трубке будут уравнены, произвести отсчет давления пара по вакуумметру 9 или по открытому манометру и записать его как давление, соответствующее первой измеренной температуре. Затем электронагревателем 3, соединенным через реле с контактным термометром 5, нагреть термостат на несколько градусов (4—6°). И каждый раз по достижении установленной температуры впуском воздуха уравнять давление в системе и снять соответствующее показание вакуумметра. Таким образом, постепенно повышая температуру термостата, определить давление жидкости при нескольких температурах. Таких определений нео-б-
159
ходимо произвести шесть—восемь. Если давление пара измеряется открытым манометром, его рассчитывают по формуле
Рпар ~ Рбар
где Ао — показание открытого манометра (если же давление измеряется закрытым манометром, то оно будет равно показанию вакуумметра). После измерений следует выключить электронагреватель и мешалку. Держатель вместе с прибором приподнять из термостата и открыть кран 8. Прибор вынуть из держателя и наклоном вправо перелить ртуть в шарик D, кран 8 закрыть, а кран 6 открыть и прибор вновь поставить в держатель. Насос выключить.
Меры предосторожности при проведении опытов. 1. После вакуумирования (после выключения рубильника) необходимо повернуть трехходовой кран на атмосферу. Иначе масло может быть втянуто в систему.
2. При нагревании прибора быстрый впуск воздуха в него может перебросить ртуть в сосуд с веществом. Поэтому необходимо очень осторожно поворачивать кран 6, соединяющий систему с атмосферой.
Результаты измерений записать в таблицу по образцу:
Исследуемое вещество ...
№ п/п
Показания
термометра манометра, мм рт. ст. Барометрическое давление, мм рт. ст.
t, °C 1/Г К
^пар
Давление пара ^пар Гбар
II — II —^^Й|
1g ^пар
Давление в единицах СИ
На основании полученных данных: 1) построить график зависимости давления паров исследуемой жидкости от температуры в координатах р—FC и 1gр—1/7 К; 2) вывести эмпирическое уравнение прямой \g р=А + В/Т и, используя метод наименьших квадратов, определить значения коэффициентов А и В; 3) определить температуру кипения жидкости при атмосферном давлении по уравнению Клапейрона — Клаузиуса; 4) вычислить теплоту испарения жидкости по эмпирическому уравнению прямой; 5) рассчитать теплоты испарения для трех интервалов температуры по уравнению Клапейрона — Клаузиуса; 6) рассчитать изменение энтропии в процессе испарения 1 моль вещества.
Работа 2. Определение давления насыщенного пара индивидуальных жидкостей динамическим методом
Динамический метод основан на определении температур кипения вещества при разных давлениях. Кипение происходит при той температуре, при которой давление насыщенного пара равняется
160
внешнему давлению, поэтому измерения температур кипения при разных давлениях дают зависимость давления насыщенных паров от температуры.
Последовательность выполнения работы. Схема прибора приведена на рис. 69. Исследуемую жидкость (75—100 мл) налить в сосуд 1, Туда же для устранения -местных перегревов и облегчения образования новой фазы поместить несколько кусочков активированного угля или неглазурованного фарфора. Сосуд 1 закрыть пришлифованной пробкой с термометром 3 и погрузить в термостат с дистиллированной водой. В комплекте термостата необходимо иметь мешалку 6, контактный термометр 7 и кипятильник 2. Сосуд
Рис. 69. Схема установки для определения давления насыщенного пара над жидкостью (динамический метод)
1 соединить с вакуумной системой через змеевидный холодильник 4, в котором улавливаются пары исследуемой жидкости. Это необходимо для предупреждения конденсации паров на стенках соединительных трубок и в манометре 11. Холодильник 4 через краны 5 и 8 соединен с вакуум-насосом или насосом Комовского. Для предохранения насоса от попадания паров и загрязнения маслом между краном 8 и насосом поместить колонку 9 с активированным углем.
Для измерения давления насыщенного пара из прибора откачивать воздух до тех пор, пока жидкость в сосуде 1 не начнет интенсивно кипеть. После того как жидкость в сосуде 1 закипит, кипятильник 2 выключить, кран 5 закрыть, а кран 2 соединить с атмосферой. Через 5 мин после закрытия крана 5 отметить показания термометра и отсчитать давление по открытому манометру 11, который соединен с холодильником 4 через ловушку 10.
6-2083 161
Измерения начинать при комнатной температуре, а затем последовательно увеличивать каждый раз температуру на 4—5°, Таким образом, постепенно повышая температуру термостата, определить давление Даров жидкости при нескольких температурах. Таких измерениях необходимо сделать восемь —десять. После каждого измерений давления пара в прибор постепенно впускать воздух. Для этого при открытом на атмосферу трехходовом кране 8 осторожно приоткрыть кран 5 и привести давление в системе к атмосферному. Давление пара рассчитать по формуле
/’пар ~ Рбар fy)*
причем h.Q отсчитывать по открытому манометру 11 как разность высот ртути в двух коленах. (Если же давление измеряется закрытым манометром, то оно будет равно показанию вакуумметра.) Результаты измерений записать в таблицу по образцу:
№ п/п Показания . Барометрическое давление, мм рт. ст. Давление пара Гпар”Рбар Давление в единицах СИ
термометра манометра, мм рт. ст.
i, °C 1/Т К ^пар 1g ^пар
На основании полученных данных: 1) построить график зависимости давления паров исследуемой жидкости от температуры в координатах р — °C и 1g р—1/Т К; 2) вывести эмпирическое уравнение прямой lgp = a+&/T и, используя метод наименьших квадратов, определить значения коэффициентов а и &; 3) определить температуру кипения жидкости при атмосферном давлении по уравнению Клапейрона — Клаузиуса; 4) вычислить теплоту испарения жидкости по эмпирическому уравнению прямой; 5) рассчитать теплоты испарения для трех интервалов температуры по уравнению Клапейрона —Клаузиуса; 6) определить изменение энтропии в процессе испарения 1 моль вещества.
Работа 3. Определение давления насыщенного пара индивидуальных жидкостей по температурам кипения
Метод основан на определении температур кипения при разных давлениях. Кипение происходит при той температуре, при которой давление насыщенного пара равняется внешнему давлению. Измерение температур кипения при разных давлениях дает зависимость давления насыщенных паров от температуры. Особенностью данного метода является то, что давление над жидко-
162
стью автоматически поддерживается на необходимом уровне при помощи маностата.
Последовательность выполнения работы. Схема установки приведена на рис. 70. В стеклянный сосуд 5 налить исследуемую жидкость. Над жидкостью поместить термометр 8. Чтобы освободиться от местных перегревов и облегчить образование новой фазы, в сосуд 5 ввести электроподогреватель 7. Сосуд 5 заключен в стеклянную рубашку 6, которая служит термостатом. В рубашку 6 из ультратермостата поступает вода, что обеспечивает равномерность нагрева жидкости. На пути циркуляции на-
Рис. 70. Схема установки для определения давления насыщенного пара над жидкостью
гревающей воды поставлен второй термометр 9 для контроля температуры воды на выходе из рубанки. Следует добиваться такого положения, чтобы термометры 8 и 9 показывали примерно одинаковую температуру. Через холодильник 3, в котором конденсируются пары, образующиеся при кипении, сосуд 5 соединен с манометром 4, по которому производить отсчет давления. Разрежение в системе создается при помощи масляного насоса, который присоединяется к ней посредством трехходового крана 13 и крана 11. Для предохранения насоса от попадания в него паров и загрязнения системы ьдаслом между краном 13 6* 163
и насосом установить колонку 7 с активированным углем. Для поддержания постоянного давления в системе во время кипения служит маностат 2. Он представляет собой стеклянный цилиндр, внутри которого находится резиновый шарик. Через краны 12 и И шарик соединен отводной трубкой с насосом. Цилиндр соединен с атмосферой краном 10 и с насосом — краном 11. Принцип действия маностатйоснован на том, что вследствие разности давлений внутри резинового шарика и в системе шарик расширяется и закрывает отверстие; ведущее от системы к вакуумному насосу. Благодаря эластичности резинового шарика поддерживается постоянное давление в систему Чтобы после остановки насоса масло не втянуло в систему, необходимо кран 13 повернуть на атмосферу. \
При помощи ультратермостата установить в сосуде 5 постоянную температуру, при которой измерять давление насыщенных паров исследуемой жидкости. Затем, открыв кран 10, убедиться в том, что ртуть в обоих коленах манометра находится на одном уровне, после чего закрыть кран 10. Жидкость в сосуде 5 находится теперь под атмосферным давлением. Осторожно открыть краны 11 и 12 и постепенно откачать воздух из системы, при этом давления в резиновом шарике и цилиндре остаются одинаковыми, поэтому шарик будет сжат. Закрыть кран 12 и продолжать откачивание воздуха из сосуда 5. Так как давление в шарике становится большим, чем давление в системе, то шарик увеличивается в объеме и закрывает отверстие А, сообщающее сосуд 5 с насосом. Если жидкость в этот момент не закипит, то это значит, что давление в сосуде 5 слишком велико для кипения при данной температуре. В этом случае в системе необходимо создать большее разрежение. Для этого отверстие, сообщающее систему с вакуумным насосом, нужно открыть, чтобы шарик уменьшился в объеме. Это произойдет, если разность давлений внутри шарика и в системе уменьшится. Поэтому закрыв кран 11, приоткрыть кран 12, а затем снова его закрыть. Разность Давлений в шарике и в цилиндре становится меньше, за счет этого шарик уменьшается в объеме и открывает отверстие А, сообщающее систему с вакуумным насосом. Открыть кран 11 и продолжать вакуумирование системы. Регулируя таким образом давление в системе при помощи кранов И, 12, добиться того, чтобы жидкость закипела. Отметить показания термометра 8 и манометра. Давление паров при данной температуре определяется как разность между барометрическим давлением и показанием манометра:
/^пар Рбар
причем hQ отсчитывать по открытому манометру 4, как разность высот ртути в двух коленах. Эта разность составляется из высоты подъема ртути в левом колене и понижения ее в правом колене.
164
Производя отсчет, закрыть кран 11, выключить насос и повернуть кран 13 на атмосферу. Затем осторожно открыть сначала кран 12, а затем кран 10 и создать в системе атмосферное давление. После этого при помощи ультратермостата повысить температуру в сосуде 5 на 4—5° С и вновь проводить измерения. Таких измерений необходимо сделать шесть-семь, причем нужно строго следить за тем, чтобы исследуемая жидкость не перегревалась и кипела равномерно. Результаты измерений записать в таблицу по образцу:
Показания Барометрическое давление, мм рт. ст. Давление пара Спар~Сбар Давление в единицах СИ
термометра манометра, мм рт. ст.
t °C ЦТ к Рпар Рпар
На основании полученных данных 1) построить график зависимости давления паров исследуемой жидкости от температуры в координатах р — °C и р—\/Т К; 2) вывести эмпирическое уравнение прямой lgp=4+&/T и, используя метод наименьших квадратов, определить значения коэффициентов А и &; 3) определить температуру кипения жидкости при атмосферном давлении по уравнению Клапейрона—Клаузиуса; 4) вычислить теплоту испарения жидкости по эмпирическому уравнению прямой; 5) рассчитать теплоты испарения для трех интервалов температуры по уравнению Клапейрона — Клаузиуса; 6) рассчитать изменения энтропии в процессе испарения для 1 моль вещества; 7) рассчитать изменения AS, AG при испарении 1 моль исследуемой жидкости при атмосферном давлении. %.
Работа 4. Определение давления насыщенного пара твердых веществ и жидкостей методом увлечения
Метод увлечения заключается в том, что струя газа, индифферентного по отношению к испытуемому веществу, насыщается его парами. Для этого поток чистого газа медленно барботирует через известное количество жидкости или твердого вещества, давление насыщенных паров которых надо измерить. Температуру жидкости или твердого тела поддерживают постоянной. Количество испарившегося вещества определяют по убыли массы. Пары исследуемого вещества из газового потока улавливают с помощью адсорбента или охлаждаемой ловушки и затем взвешивают. Если объем газа v содержит Еш исследуемого вещест-
165
ва с молекулярной массой М, то парциальное давление равновесного пара при температуре Т будет равно
р — Vj mRT/(Mv). (VII. 15)
Последовательность выполнения работы. Схема установки приведена на рис. 71. Азот из газометра 1 через градуированный реометр 2, обеспечивающий постоянство скорости газового потока во время опыта, поступает в четыре последовательно соединенных сосуда 5, в которых газ насыщается веществом, например SO2 в растворе гидросульфита цинка и парами воды, до
Рнс. 71. Схема установки для определения давления насыщенного пара твердых веществ и жидкостей методом увлечения
равновесия. Сосуды 5 помещены в термостат, настроенный на определенную температуру. Комплект термостата должен быть снабжен мешалкой 6, контактным термометром 4, электронагревателем 3 и термометром 8. Азот, насыщенный SO2 и парами воды над исследуемым раствором гидросульфита цинка при данной температуре, проходит через два поглотительных сосуда Я 10, после чего поступает в аспиратор 11. В первый поглотительный сосуд налито 25 мл 0,1 н. раствора 12 для поглощения SO2, во второй—10 мл 0,1 н. Ыа2820з для улавливания паров иода, которые уносятся газом из первого поглотительного сосуда.
Газометр наполнить из баллона азотом или инертным газом, предварительно очищенным от кислорода. Очистку азота от кислорода производить пропусканием газа через три колонки, наполненные спиралями из металлической меди и насыщенные раствором NH4C1 в NH4OH. При поглощении кислорода металлическая медь окисляется, до Си2+ и раствор синеет. После подачи газа раствор быстро обесцвечивается, вследствие восстановления Си2+ до Си+ металлической медью. После того как температура в термостате достигнет определенного значения, в сосуды 5 залить раствор гидросульфита цинка определенной концентрации (исследуемое вещество). Заполненные сосуды поместить в термостат и включить в общую систему. В аспиратор залить воду. Проверить герметичность системы и после установ-166
ления определенной скорости газа по реометру начать опыт. Началом опыта считать открытие кранов у первого поглотительного сосуда с иодом или другим поглотителем и у аспиратора. За время опыта фиксировать: температуру воды в термостате, в последнем сосуде и в аспираторе, атмосферное давление, давление в системе и в аспираторе по манометрам 7. Газ пропускать через раствор с иодом в течение 30 мин, после чего избыток иода оттитровывать раствором гипосульфита и замерить объем вытекшей за это время воды из аспиратора цилиндром 12. Содержание SO2 в растворе определять иодометрически. Избыток иода оттитровать титрованным раствором гипосульфита. По количеству израсходованного иода (на соединение с SO2) определить количество SO2. Количество газа (N2+H2O), прошедшее за время опыта, определить по объему вытекшей из аспиратора 11 воды.
Навеску 25 г раствора, насыщенного SO2, из бюкса перенести в мерную колбу вместимостью 250 мл. Предварительно в колбу налить около 100 мл воды, чтобы концентрация раствора при разведении была невелика и SO2 не улетучивался. Затем в коническую колбу налить 25 мл 0,1 н. раствора 12 и сюда же перенести 10 мл раствора из мерной колбы. Диоксид серы взаимодействует с >иодом. Избыток иода оттитровать 0,1 н. Na2S2O3 в присутствии крахмала:
2Na2S2O3 4- I2 = Na2S4O6 4- 2NaI
Содержание SO2 в анализируемом растворе определить по количеству иода, израсходованного в реакции
H2SO3 4-12 + Н2О - H2SO4 4- 2HI
Анализ газа на содержание SO2 повторить три-четыре раза. Измерения начинать при комнатной температуре, а затем последовательно увеличивать каждый раз температуру на 4—6°. Таким образом, постепенно повышая температуру термостата, определить давление паров жидкости (раствора) при нескольких температурах. Таких измерений необходимо сделать восемь — десять.
Давление SO2 над раствором гидросульфита цинка рассчитать следующим образом. Через установку пропущено газа:
v = vS02 + ^'на0 + vn2
Давление газовой смеси, находящейся в равновесии с Zn(HSO3)2, будет
Р “ ^SOs + ^Н20 + Pns *
Опытным путем определить следующие величины: 1) р~ = рбаР—Pi, где рбар—атмосферное давление, мм рт. ст.; pi — разрежение в последнем сосуде, мм рт. ст.; 2) Рн2о— равновесное давление паров воды (вычисляется по уравнению Рауля);
167
3) ‘OsOj —объем сухого SO2 при н. у. (вычисляется по расходу иода);
^SO, — (Vl/u2 — t’Na1S203^Na2S2O3)>
где t»i2—объем иода, взятый для анализа; ‘i»Na2s,os — объем
гипосульфита, израсходованный на титрование избытка иода; Kit- поправочный коэффициент к 0,1 и. раствору I2; A3a2s2o2 — поправочный коэффициент к 0,1 н. Na2S2O3; 4) un2 — объем сухого азота при н. у. (вычисляется по объему воды, вытекшей из аспиратора);
Рбар — Расп — Р2 273 .
Vn» ~ 760 273+ / ’
Расп — разрежение в аспираторе; р2 — давление паров при температуре в аспираторе; t — температура газа в аспираторе; v — объем воды, вытекшей из аспиратора. Основываясь на зависимости
у _ vso2 __ vh2o _ vn2 р ~ Pso2 ~ Рн.о ~ Pn2 ’
Р — Рн20 .
можно написать, что /?so2=^’soa---------— • Отсюда формула для
v “ VH2O
определения давления SO2 над растворами гидросульфита цинка будет иметь вид
ysoa Pso2 — (Р — Рн2о) , yso3 + ^n2
Кроме давления SO2 над растворами можно рассчитать давление паров воды.
Результаты измерений записать в таблицу по образцу:
Л °C 1/Г К ^бар ^Н2О i PS02 Давление в единицах СИ
На основании полученных данных необходимо: 1) построить график зависимости давления паров исследуемой жидкости от температуры в координатах р — t и Igp—1/Т; 2) вывести эмпирическое уравнение прямой \gp = a-\-b/Т и рассчитать коэффициенты а и Ь\ 3) определить температуру кипения жидкости при атмосферном давлении по уравнению Клапейрона —• Клаузиуса; 4) вычислить теплоту испарения жидкости по эмпирическому уравнению прямой, теплоты испарения для трех интервалов температуры по уравнению Клапейрона — Клаузиуса; изменения энтропии в этом процессе для 1 моль.
168
Работа 5. Определение температур кипения микрометодами
Рис. 72. Схема установки для определения температуры кипения жидкости по методу Эмиха
В данной работе необходимо определить температуру кипения небольшого количества (менее 1 мл) ным давлением по методу Эмиха (рис. ния определяют, допуская равенство давления пара и атмосферного давления, не учитывая того, что пар, выделяющийся из некоторого объема жидкости, должен преодолеть давление верхних слоев жидкости.
Последовательность выполнения работы. Приготовить капилляры длиной 7— 8 см с внутренним диаметром 0,5—1 мм при толщине стенок 0,1 мм. Один из концов капилляра оттянуть в тонкое острие длиной 2 см. Острым концом набрать каплю исследуемой жидкости. Этот конец капилляра заплавить на газовой горелке. Чтобы образовался пузырек пара, необходимо в капилляре оставить небольшой пузырек воздуха. После этого проверить наличие пузырька воздуха, объем которого должен быть меньше объема жидкости. Если пузырек слишком велик или вовсе не обнаруживается, то капилляр непригоден. Правильно заполненные капилляры 3 и 4 (рис. 72) прикрепить к термометру 2 при помощи металлической скобы 6 и поместить в глицериновую баню 5. Вы
сота слоя жидкости в баце должна быть 4—5 см, и жидкость должна хорошо перемешиваться мешалкой 1, При приближении, к температуре кипения капля жидкости в капилляре начнет подниматься и достигнет уровня жидкости в бане. Показание термометра, соответствующее этому моменту, принимают за температуру кипения. Необходимо сделать два определения и взять среднее значение температуры кипения. Точность метода составляет ±1°. Полученные данные записать в таблицу по образцу:
вещества под атмосфер-72). Температуру кипе-
Вещество
Температура кипения при атмосферном давлении, °C
Температура кипения при нормальном давлении, К
Температура кипения по справочнику, °C
Ошибка опыта
Известное
Исследуемое
169
На основании полученных данных: 1) привести температуру кипения к нормальным условиям по уравнению
Д/кип = 0,00012 (760 - р) (273 4- *о),
где р — атмосферное давление, при котором проводится опыт; 2) рассчитать теплоту испарения исследуемой жидкости, применяя уравнение Трутона
Д/7ИСп = 8,9-10-2Ткип кДж/моль,
или более точное уравнение Кистяковского
ДЯИС11 - /?ГКИ1[ 1п(88,07Гкип) - 7\й1( (36,7 + 19,1 1g Гкйл)10-з кДж/моль.
Соотношение АЯисп/ГКИп выражает изменение энтропии при испарении и оба уравнения применимы только к неполярным жидкостям; 3) рассчитать эбуллиоскопическую постоянную и сравнить ее со справочными данными; 4) рассчитать для 1 моль исследуемого вещества изменения AS, АЯ, AG при температуре кипения.
ГЛАВА VIII
ЖИДКИЕ РАСТВОРЫ
Растворами называются фазы, состав которых можно изменять непрерывно (в известных пределах), т. е. фазы переменного состава. Растворы — это однородные смеси молекул, а также атомов, ионов двух или более веществ, между которыми имеются физические и нередко химические взаимодействия. Ассоциация молекул какого-либо соединения и сольватация (соединение молекул растворенного вещества и молекул растворителя в непрочные комплексы), не образующие особенно больших молекул, не нарушают однородности раствора. С термодинамической точки зрения вещества, составляющие раствор, равноценны и деление на «растворитель» и «растворенное вещество» не носит принципиального характера. Растворителем обычно называют тот компонент раствора, количество которого больше (если растворитель, растворенные вещества и раствор находятся в одинаковых агрегатных состояниях). Если агрегатные состояния веществ до образования раствора различны, то растворителем считают то вещество, которое при данных условиях является жидкостью. Состав раствора или его концентрацию выражают различными способами: молярная доля — отношение числа молей Гго вещества к общему числу молей всех компонентов в данном количестве раствора S/2/:
xi и/;
(VIII. 1)
моляльность т — число молей растворенного вещества на 1000 г растворителя; молярность с — число молей растворенного
170
вещества в 1000 мл раствора; концентрация (не имеет названия) обозначает число молей растворителя, приходящееся на 1 моль растворенного вещества.
Пересчет концентраций для двухкомпонентных растворов можно осуществить с помощью следующих уравнений:
п2 М\т<2 М2с2 .
х ------—----------- —----------------. (VIII .2)
1 4- Мхт2 р — ^2(^2 — Ml)
£2№с2 4
Mi (1 • лД Трасте Р — М2С2
_____Р£2______р/п2__п2
Mi 4- х2 (М2 — Mt) 1 + М2т2 Ураств ’
(VIII.3)
(VIII.4)
где х2— молярная доля компонента 2; т2 — моляльная концентрация растворенного вещества; с2— молярная концентрация растворенного вещества; ti\ и п2 — число молей первого и второго компонентов; Alj и Л12 — молекулярные массы первого и второго компонентов соответственно; индекс 1 относится к растворителю; индекс 2 — к растворенному веществу; р — плотность раствора; g — навеска растворителя, равная 1000 г; V — объем раствора, равный 1000 мл.
1. ИДЕАЛЬНЫЕ РАЗБАВЛЕННЫЕ РАСТВОРЫ
Раствор, активности компонентов которого совпадают с их молярными долями (за стандартное состояние принимается состояние чистого компонента), называется идеальным. Идеальные растворы подчиняются закону Рауля:
= 1 = (1 — *2)Роа> (VIII.5)
где индекс 1 относится к растворителю, а индекс 2 — к нелетучему растворенному веществу. Для неидеальных растворов уравнение (VII 1.5) применимо, если молярная доля растворенного вещества х2 достаточно мала, и поэтому влиянием растворенного вещества на растворитель можно пренебречь.
Уравнение (VIII.5) можно преобразовать следующим образом:
Ро, 1 ~ Pi Роа
«2
= Х2 =---------
«1 4-"2
(VIII. 6)
>
где Род—Pi = Ар — депрессия.
В разбавленном растворе неэлектролита число частиц совпадает с числом молекул, в то время как в разбавленном растворе электролита число частиц увеличивается в результате диссоциации и во столько же раз возрастает депрессия. Поэтому для растворов электролитов в уравнение вводится соответствующая поправка на диссоциацию, так называемый изотонический коэффи-
171
циент Вант-Гоффа
i. Тогда уравнение (VIII.6)
Р0,1—Р1_ ^2
Ж 1 /И2 4-И1
= 1X2,
принимает вид
(VIII.7)
где pi — давление пара над раствором электролита. Коэффициент i определяют по уравнению
/=14-a(v —1), (VIII.8)
где a — степень диссоциации; v — число ионов, образующихся из одной молекулы.
Применение закона Рауля. Давление пара растворителя понижается при растворении в нем нелетучего вещества. Чтобы давление пара раствора соответствовало давлению чистого растворителя, необходимо нагреть раствор выше температуры кипения чистого растворителя. На рис. 73 приведена зависимость
Рис. 73. Зависимость давления насыщен- Рис. 74. Повышение температуры ки-ного пара над растворителем и над рас- пения разбавленных растворов творами нелетучего растворенного вещества различных концентраций от температуры
давления насыщенного пара раствора и чистого растворителя от температуры. Температура, при которой жидкий раствор с данной концентрацией растворенного вещества (веществ) при равновесии образует пар, давление которого равно внешнему давлению, называется температурой кипения раствора. Температура кипения разбавленного раствора, содержащего нелетучее растворенное вещество, выше температуры кипения растворителя (рис. 74) и повышение температуры кипения пропорционально концентрации растворенного вещества. Таким образом,
А7\иг£ —
(VIII.9)
172
где ДТкип — повышение температуры кипения; Кэ — эбуллиоскопическая константа или моляльное повышение температуры кипения. Значение Кэ зависит от природы растворителя и рассчитывается по уравнению
рт% РТ^
кип кип
ДЯИСП “ 1000 Д/г
I ООО —
м
(VIII. 10)
где ГКип — температура кипения растворителя; Д/г — удельная теплота испарения растворителя. Уравнение (VIII.10) выведено из приближенного уравнения Клапейрона — Клаузиуса и закона Рауля.
Повышение температуры кипения, происходящее при добавлении к растворителю определенного количества растворяющегося вещества, позволяет рассчитать моляльность раствора и отсюда его молекулярную массу по уравнению
М2= ЮООКэ^/^ДГ^, (VIII. 11)
где М2 — молекулярная масса растворенного вещества; g{, g2 — масса растворенного вещества и растворителя соответственно.
Для разбавленных растворов электролитов уравнение (VIII.11) принимает вид
М2 —/1000Кэ^г2/^1Д7,кИ[г ИЛИ ДТ\И:Е ~ (VIII. 12)
Для раствора, который разбавлен, но не идеален, химический
потенциал записывается в виде
Pi ~ 4- RT In a-i = uj 4- RT In ~ 4- RT In X[ 4- RT la (VIII. 13)
где — рациональный коэффициент активности*.
Коэффициент активности растворителя рассчитывают по повышению температуры кипения по уравнению
— In
^сп , X ДГ \2- .
RT№ 2 R \ ^кип / J
(VIII. 14)
Для очень разбавленных растворов по повышению температуры кипения можно определить коэффициент активности растворенного вещества:
1Пу ^—2/,
(VIII.15)
где /--1 —(ДТкиП//(э).
Однако вычисление коэффициента активности можно рассчитать более точно по понижению температуры замерзания. Если растворенное вещество является электролитом, распадающимся
* Мерой отклонения свойств данного раствора от свойств идеального раствора той же концентрации является соотношение atixi, которое называется коэффициентом активности
173
на два иона, то коэффициент активности можно определить по уравнению
In у = — 3/,
(VIII. 16)
где /=1—
Температура, при которой из жидкого раствора с данной концентрацией растворенного вещества (веществ) начинают появляться при условии равновесия кристаллы твердой фазы, называется температурой кристаллизации раствора; Если растворенное вещество и растворитель не образуют твердого раствора, то температура кристаллизации раствора ниже температуры кристаллизации растворителя. Понижение температуры кристаллизации пропорционально концентрации растворенного вещества. Для определения понижения температуры замерзания справедливо соотношение
зам — KrW,
(VIII. 17)
где ДГзам— понижение температуры кристаллизации; Лк —криоскопическая константа растворителя или моляльное понижение температуры кристаллизации. Криоскопическую постоянную рассчитывают по уравнению
рт^ ВТ?1
х зам______ л зам
1000 (Д/Y/Af) = ЮООД/г *
(VIII. 18)
где Д/i— удельная теплота плавления растворителя; Тзам — температура замерзания растворителя.
Молекулярную массу рассчитывают по уравнению
1000КкШ£уЛГзам. (VIII.19)
Для разбавленных растворов электролитов уравнение (VIII.17) примет вид
ДГзам =lKRtn. (VIII.20)
2. ХИМИЧЕСКИЙ ПОТЕНЦИАЛ
При описании энергетических свойств растворов пользуются парциальной энергией Гиббса Gt компонента* к
{ дв \
(h = [-— , (VIII.21)
\ дп.
где G — общая энергия Гиббса системы. Энергия Гиббса Ch представляет собой общую реакцию ограниченной системы на прибавление бесконечно малого количества компонента при постоянных р, Т и rij. Химический потенциал является интенсивной переменной и зависит только от р и Т и не зависит от щ. Химический
* Обычно энергию Гиббса обозначают Цг.
174
потенциал компонента может быть определен как часть общей энергии Гиббса, относящаяся к данному компоненту в растворе. Химический потенциал компонента в жидкой смеси, пары над которой можно считать смесью идеальных газов, рассчитывают по уравнению
+ (VIII.22)
где pt — парциальное давление насыщенного пара i-ro компонента в жидкой смеси; — давление чистого компонента.
Для идеальных растворов, где выполняется закон Рауля для х-го компонента, уравнение для химического потенциала имеет вид
= и." +ЛГ1пх.. (VIII.23)
В реальных растворах соотношение p/pi,® не равно молярной доли жидкого компонента. Оно количественно отражает положительные или отрицательные отклонения от закона Рауля и называется термодинамической активностью компонента Таким образом, если пар над раствором можно считать смесью идеальных газов, активность компонента в растворе будет = PtlPi& Если пар нельзя считать идеальным газом, то давления надо заменить соответственно фугитивностями fi, и тогда
Для реального раствора химический потенциал рассчитывают по уравнению
р.. = р.’ +/?Т Ina,. (VIII.24)
I I
Выбор стандартного состояния. Вследствие неизвестности абсолютных значений термодинамических величин (кроме энтропии) химический потенциал отсчитывают от произвольно выбранного значения в так называемом стандартном состоянии. Стандартное состояние удобно выбирать так, чтобы для него выполнялось условие |1станд=|1° и в стандартном состоянии поведение реального раствора совпадало бы с поведением идеального раствора. Тогда активность совпадает с мольной долей, aL = Xi. Например, для жидкого компонента раствора в стандартном состоянии
р..-!л° = /?Г1п(р1./р;о) = /?7'1пх. = О. (VIII.25)
Это условие выполняется при х^1. Одновременно в бесконечно разбавленном растворе свойства реального и идеального растворов совпадают. Для жидкого растворителя стандартным состоянием будет растворитель в чистом виде. Активность нелетучего растворенного вещества совпадает с его концентрацией в бесконечно разбавленных растворах. Поэтому для раствора, пар которого является идеальным газом, постулируют
Пту2 = у2= Пт —=1, (VIII.26)
Хц-*0 лг2->0 х2
т. е. в предельно разбавленном растворе коэффициенты активности растворителя и растворенного вещества равны единице.
175
Зависимость активности от температуры. Введя в уравнение Гиббса—Гельмгольца интегрирующий множитель \/Т, получим
д(Gt/T) ]
ИЛИ
d(\G/T)
дТ
(VIII.27)
Энергия Гиббса раствора связана с химическими потенциалами компонентов первым уравнением Гиббса — Дюгема
= (VIII.28)
или с учетом выбора стандартного состояния
О/ “ G - = 2 (VIII.29)
Tit - Н] - 2 (VIII.30)
где &Ht — относительная парциальная молярная теплота растворения данного компонента.
Подставив выражения (VIII.24), (VIII.29) и (VIII.30) в уравнение Гиббса — Гельмгольца, получим для многокомпонентного раствора
ЯЛ
(VIII.31)
и для i-го компонента в растворе
\ /р,х = '
(VIII. 32)
При интегрировании уравнения (VIII.32) в широком интервале температур надо учитывать зависимость теплоты растворения ДЯ от температуры.
Определение активности по уравнению Гиббса — Дюгема. Интегрируя уравнение Гиббса — Дюгема
Xi d In Yi + х2 d In ?2 = 0
в пределах от x2 = 0 до некоторой концентрации х2 получим
in
Д1
X а
(VIII. 33)
так как (ai/ль) Ж2==о == 1-
Уравнение (VIII.33) решают, учитывая зависимость a2=f(xz) графическим интегрированием (рис. 75).
Определение а2 по а\ проводят по уравнению (VIII.33). Однако вычисления приводят к неточным результатам: если в расчете Ci по а2 кривая 1gуг = f(IgYi) экстраполируется в начало
176
координат (рис. 75), то при вычислении активности растворенного вещества кривая асимптотически приближается к вертикальной оси (так как по мере разбавления, т. е. увеличения соотношения Xi/хг до-оо, величина 1g (ai/xf) уменьшается до нуля);
это вносит значительную погрешность в графическое интегрирование. Поэтому был предложен искусственный прием расчета — построение графика условно выбранной функции от Gi, которая по мере разбавления приближается к нулю.
Для летучего растворителя расчет активности производят с помощью соответствующего графика, построенного на основании уравнения
Рис. 75. Определение активности растворителя по зависимости активности растворенного вещества от концент-
(VIII.34)
рации
Пример. Вычислите активность
ртути в амальгамах таллия при 25° С, если активность таллия в амальгамах разной концентрации при этой температуре
имеет следующие значения:
х2 0,01 0,05 0,1 0,2 0,3 0,4 0,5
а2 0,0115 0,090 0,284 0:996 1,98 3,93 3,99
Решение. Расчет производим по уравнению (VIII.34), для чего вычисляем значения x2/xi и Ig(Gs/x2), а затем на основании этих данных строим график в координатах x2/xi— lg(a2/x2) (рис. 75). Площадь, заключенная между кривой, осью абсцисс и соответствующей ординатой, отвечает значению a^/xi. Промежуточные вычисления и окончательный результат приведены ниже:
X2/Xj
Ig(^2/X2)
—lg(«l/Xi)
0,0101 0,061 0,ООЮ5 0,989
0,0526 0,255
0,0065 0,936
0,111 0,453
0,0220 0,855
0,250 0,697
0,0635 0,691
0,429 0,820 1,103 0,552
0,666 0,879 0,134 0,441
1,000 0,902 0,151
0,353
Работа 1. Определение активности и химических потенциалов воды и соли в растворе при 40°С
Последовательность выполнения работы. Установить термостат на температуру 40° С. Приготовить растворы соли (например, хлорида натрия) в воде с содержанием (моль/1000 г): 1; 2; 3; 4; 5. На установке, описанной на с. 162, измерить давление насыщенного пара чистой воды pi,0 и над приготовленными рас
творами, начиная с малых концентраций. На основании полученных экспериментальных данных: а) рассчитать активности воды в указанных растворах по формуле ^н2о = А/А,о; построить график в координатах пн2б—хн2о; б) рассчитать молярную долю и коэффициент активности воды по формуле Yh2o = ан2о/хн2о в растворах; В) построить График В координатах (1 — xNaCl/xNaCl~ и графическим интегрированием (см. 177) определить коэффициент активности соли указанной концентрации; г) рассчитать химические потенциалы воды и соли в растворе относительно стандартного состояния (бесконечно разбавленный раствор); д) рассчитать максимальную работу — AG образования 1 моль раствора данной концентрации по уравнению Гиббса — Дюгема
Л А(7 = «н2о^н2о ^NaClf^NaCl *
Результаты расчета свести в таблицу по образцу:
Работа 2. Определение парциальных молярных теплот и энтропии растворения
Последовательность выполнения работы. Приготовить растворы соли (например, хлорида натрия) в воде концентрации {моль/1000 г): 1; 2; 3; 4; 5. На установке, описанной на с. 162, измерить давление насыщенного пара чистой воды и над приготовленными растворами, начиная с меньших концентраций. Измерения провести при температурах (°C): 50, 60, 70. На основании полученных экспериментальных данных: а) рассчитать активность воды в указанных растворах при различных температурах по формуле Р з/Т’ол; б) построить график в координатах lg&H3o—1/Г и по тангенсу угла наклона кривой определить парциальную молярную теплоту растворения воды, ДЯ/, при разных концентрациях соли (ДЯН0= 2,37? tg а); в) рассчитать химические потенциалы воды^о при различных температурах; г) построить график в координатах Р-н3о~г и по тангенсу угла наклона кривой ^н2о/^ ~ Д5’н2о определить парциальную энтропию
растворения при указанной температуре (д'ф^/ДГ === — ash0). Результаты измерений и расчетов свести в таблицу по образцу: 173
РНзО
аН20
-VH2O
мт
Работа 3. Определение молекулярной массы растворенного вещества криоскопическим методом
Для криоскопических измерений применяют прибор (рис. 76)> который состоит из стеклянной широкой пробирки 1 для раство
рителя, имеющей в верхней части отросток 5 для внесения растворяемого вещества. Пробирку закрывают корковой пробкой 2?
в которую вставлены термометр Бекмана 3 и латунная мешалка 4. При помощи резиновой прокладки 6 пробирку помещают в воздушную стеклянную рубашку 7, которую погружают в криостат 8. Криостат представляет собой толстостенный стакан или металлическую баню, наполненную охлаждающей смесью (лед + -4-соль), внутри которой установлена мешалка 9 и термометр 10.
Предварительно многократно определяют температуру замерзания растворителя, затем после внесения определенного количества растворенного вещества в данный растворитель — температуру замерзания полученного раствора. Процесс кристаллизации чистого растворителя, начиная с появления первого кристалла до полного затвердевания всей жидкости, протекает при р = const, постоянной и единственной температуре.
Криоскопический метод применим к сильно разбавленным растворам бинарных неизоморфных систем. При затвердевании такого раствора сначала выпадают кристаллы чистого растворителя и раствор становится более концентрирован-
Рис. 76. Схема прибора для определения температуры замерзания
ным, а температура кристаллизации бо-
лее низкой. Поэтому при определении температуры затвердевания
раствора следует измерять температуру начала кристаллизации. Иногда жидкость переохлаждается, и кристаллизация начинается при более низкой температуре, что приводит к ошибке в измерении величины ДТзам. Для более точного определения истинной температуры начала кристаллизации нельзя допускать сильного переох
179
лаждения раствора. Количество растворителя (обычно около 20 мл), которое отмеряют из бюретки или отвешивают на аналитических весах, должно покрыть весь основной резервуар термометра Бекмана. На пробирку с растворителем надевают воздушную рубашку, после чего пробирку вставляют в приготовленную охлаждающую смесь и дают растворителю охладиться, а в это время настраивают термометр Бекмана.
Последовательность выполнения работы. Подготовленный термометр Бекмана вставить в прибор и начать наблюдать за температурой. Для равномерного охлаждения жидкость медленно помешивают вставленной в прибор мешалкой. Помешивание прекратить, когда температура на 0,5° станет выше ожидаемой температуры кристаллизации. После этого внимательно следить за понижением температуры. Без помешивания жидкость легко переохлаждается, о чем свидетельствуют показания термометра. Для чистого растворителя переохлаждение допустимо на 0,5—Г. Возобновление перемешивания переохлажденной жидкости вызывает кристаллизацию. При кристаллизации выделяется теплота и температура начинает заметно повышаться. Не прекращая равномерного помешивания, следить за температурой, отмечая максимальную температуру подъема (из переохлажденного состоя* ния), которая и будет истинной температурой кристаллизации данной жидкости. После этого пробирку вынуть из воздушной рубашки и, подогревая ее рукой, растворить образовавшиеся кристаллы. Затем пробирку вновь опустить в стеклянную рубашку, оставленную в охлаждающей смеси, и повторить переохлаждение с последующей кристаллизацией. Опыт следует повторить несколько раз, пока последние два определения температуры кристаллизации будут отличаться не более чем на 0,01°. Записав температуру кристаллизации растворителя, открыть боковой тубус (если его нет, приподнять пробку) и всыпать навеску исследуемого вещества. Навеска определяется по массе бюкса с исследуемым веществом и без него. После этого вынуть пробирку из рубашки, подогреть рукой раствор, вызывая расплавление кристаллов растворителя и растворение в нем навески. Вставить пробирку вновь в рубашку и провести процесс охлаждения, как и с растворителем. Надо помнить, что раствор переохлаждать более чем на 0,2° нельзя. Температуру кристаллизации раствора определять три-четыре раза; из полученных данных рассчитать среднюю температуру кристаллизации, а также разность средних температур кристаллизации растворителя и раствора. Рассчитать молекулярную массу по уравнению (VIII.19).
Температуры кристаллизации и криоскопические постоянные некоторых растворителей приведены ниже:
Растворитель 7\р, К
Кк, на б л
н?о C6H5NO2
273,2 278,9
1,86 6,90
с6н3 278,7 5,10
СбНдОН камфора
313,2 451,2
730 49,0
180
Результаты отдельных измерений и окончательный результат определения молекулярной массы записать в таблицу по образцу:
Растворитель . . растворенное вещество . . масса пустой пробирки gQ . . масса пробирки с растворителем g . . масса растворителя g[ (или объем растворителя V и плотность р) . . масса бкжса go' . . масса бюк-са с растворенным веществом g' . . масса растворенного вещества g2
Исследуемая система Температура кристаллизации, С Повышение температуры кристаллизации Молекулярная масса растворенного вещества
измеренная средняя определенная истинная
Чистый растворитель Раствор
Термометр Бекмана (метастатический). Метастатический термометр предназначен для измерений с достаточной точностью (10~3 К) небольших разностей температур в различных интервалах абсолютных температур. Особенность его устройства состоит в возможности изменять количество ртути в основном (нижнем) резервуаре в соответствии с областью измерений. Для этого термометр снабжен верхним (запасным) резервуаром, куда можно переводить ртуть из основного резервуара (для измерений при высоких температурах) или переводить ее в основной резервуар (для измерений при низких температурах). Шкала такого термометра, имеющего длину 25—30 см, градуирована всего на 5° (иногда на 2 или 6°) с отметкой между ними десятых и сотых долей. Переводя некоторое количество ртути из нижнего резервуара в верхний или . добавляя из верхнего в нижний, всегда можно настроить термометр так, чтобы температура замерзания данного растворителя попадала бы где-нибудь на середину этой условной шкалы (между 4 и 2°).
Чтобы настроить термометр, необходимо привести в, соприкосновение ртуть обоих резервуаров. Для этого, подогревая рукой (а если этого недостаточно, то опустив в теплую воду), вызывают расширение ртути в нижнем резервуаре с тем, чтобы заполнить ею весь капилляр доверху. Затем быстро перевернув термометр головкой вниз и, слегка постукивая по головке, добиваются того, чтобы ртуть в запасном резервуаре подошла вплотную к верхушке капилляра, наполненного ртутью из нижнего резервуара, и слилась с ней. После этого термометр осторожно (чтобы не разорвать ртуть) возвращают в прежнее вертикальное положение. Нижний резервуар с ртутью тотчас погружают в заранее подготовленный стакан с водой, температура которой должна соответствовать температуре замерзания чистого растворителя. Метал-
181
лическую головку термометра зажимают в штативе. Термометр ве должен касаться стенок и опираться на дно. Размешивая воду палочкой и добавляя в нее небольшие кусочки льда или снег, в течение 3—5 мин поддерживают ее температуру постоянной. Это обеспечивает то, что ртуть нижнего резервуара, охлаждаясь, сжимается и, будучи слита с ртутью верхнего резервуара (через капилляр), перетягивает недостающее количество ртути из верхнего резервуара в нижний. Через 5 мин, освободив из штатива головку термометра и плотно зажав основной стержень термометра посередине одной рукой, быстро его вынимают из воды и, энергично стукнув руку с зажатым термометром о другую, вызывают отрыв верхней ртути от капилляра. Таким образом заканчивается основная, но еще не полная настройка термометра, так как в рабочей части его (нижнем резервуаре с капилляром) имеется некоторый избыток ртути. Таким избытком является то небольшое количество ртути, находящееся в верхней половине капилляра, между намеченными делениями шкалы и местом, где был осуществлен отрыв ртути. Слегка подогрев рукой нижний резервуар, вызывают расширение ртути, что тотчас замечают по небольшой капельке ртути, которая появляется у конца капилляра в верхнем резервуаре. Эту капельку следует стряхнуть в верхний резервуар. Необходимо проверить, достаточно ли этого количества сброшенной ртути или нет, повторным погружением термометра в стакан с водой. Если температура, соответствующая температуре замерзания растворителя, попадает на шкалу выше желаемого деления (наиболее удобным для опыта является положение ртути между 2 и 4°), то встряхивание капельки ртути следует повторить и еще раз проверить показание термометра, опустив его в тот же стакан с водой. После этого необходимо записать, какая температура в градусах Цельсия соответствует делениям или градусам термометра Бекмана.
С настроенным термометром следует обращаться особенно осторожно, его нельзя класть на стол или оставлять длительное время при комнатной температуре. В нерабочие промежутки времени термометр должен быть укреплен вертикально за головку в штативе, а нижний резервуар должен быть опущен или непосредственно в жидкость, или в сухую воздушную рубашку, обязательно погруженную в охладительную смесь. Если растворителем служит вода, то установление нужной температуры в стакане осуществляется особенно легко. Смесь воды со снегом или кусочками льда является устойчивой равновесной системой, которая находится при 0°С, пока не растает последний кристалл льда или не замерзнет последняя капля воды.
Существует и другой способ настройки термометра. Он основан на том, что температура воды в стакане, куда опускают термометр после соединения ртути обоих резервуаров, берется не равной температуре замерзания растворителя, а на 2—3° выше. Например, если растворителем служит бензол, температура замерзания которого 5,5° С, воду в стакане поддерживают при 182
7,5—8,5° С, а если растворителем служит вода, то воду берут при 2—3° С. Такое положение ртути является окончательным, температуру воды в стакане следует поддерживать более тщательно и более 5—10 мин (вместо 3—5 мин). По истечении 10 мин ртуть обрывают, и термометр погружают в другой стакан, где температура воды должна равняться температуре замерзания растворителя, т. е. должна быть на 2—3° ниже, чем в первом стакане.
Чтобы настроить термометр Бекмана на 0°С, необходимо приготовить в ваннах охладительную смесь лед — вода и опустить в нее термометр Бекмана. Проследить, где остановится ртуть нижнего резервуара. Если ртуть нижнего резервуара остановилась на делении ниже 2°, то необходимо из верхнего резервуара добавить некоторое количество ртути. Для этого поворачивают термометр головкой вниз и слегка постукивают по головке, добиваясь, чтобы ртуть нижнего резервуара через капилляр соединилась с ртутью запасного резервуара. После этого осторожно опускают термометр в охлаждающую смесь лед — вода, наблюдая за тем, чтобы не произошло разрыва ртутного капилляра с шариком ртути в запасном резервуаре. Ориентировочно ртуть перетягивают из запасного резервуара до нулевого деления шкалы. Затем, вынув термометр из охлаждающей смеси, энергично постукивают рукой по верхней части термометра, добиваются отрыва ртути в шарике от капилляра, вновь опускают термометр в охлаждающую смесь лед —вода и следят за показателями ртутного капилляра.
Если ртуть нижнего резервуара остановилась выше 4° шкалы Бекмана, термометр вынимают из охлаждающей смеси, слегка подогрев рукой нижний резервуар, вызывают расширение ртути, которая появится у конца капилляра. Эту капельку следует стряхнуть в запасной резервуар, энергично стукнув рукой по верхней части термометра. Быстро опускают термометр в охлаждающую смесь и следят за показаниями. Термометр Бекмана настроен правильно, если ртуть в капилляре остановилась между 2—4°. Настроив термометр, укрепить его в штативе, не вынимая из охлаждающей смеси. Охлаждающую смесь перемешивают в течение 2—3 мин, записывают минимальное показание термометра.
Установка термометра Бекмана для эбуллиоскопии. Термометр Бекмана надо установить так, чтобы температура кипения чистого, растворителя соответствовала на шкале 2—3°. Для этого берут два стакана, один с чистым растворителем, другой с раствором, произвольной, но достаточно высокой концентрации, и нагревают оба до кипения. После этого обычным термометром определяют температуру кипения чистого растворителя и рас-твора. Температура кипения раствора должна быть на 2,0—2,5° выше температуры кипения чистого растворителя. Термометр Бекмана приводят в горизонтальное положение и, слегка постукивая по его верхней части, подгоняют ртуть верхнего резервуара
183
к верхнему концу капилляра, затем легким нагреванием рукой нижнего шарика термометра ртуть поднимается по капилляру и соединяется со ртутью верхнего резервуара. После этого термометр осторожно (чтобы не разорвать ртуть) приводят в вертикальное положение и ставят на 20 мин в стакан с кипящим раствором, укрепив его в штативе. Далее термометр вынимают из раствора и, держа его за середину шкалы, быстрым ударом по верхней части обрывают ртуть в верхнем резервуаре. Затем термометр ставят в стакан с кипящим чистым растворителем на 10—15 мин. Если температура кипения чистого растворителя будет соответствовать 2—3°, то термометр готов для работы.
Работа 4. Определение концентрации растворенного вещества криоскопическим методом
Устройство криостата с термоэлектрическим микрохолоднль-ником. Микрохолодильник типа ТЛМ состоит из рабочего стакана, двухкаскадной полупроводниковой термоэлектрической батареи, водяного теплообменника, корпуса. Питание микрохолодильника осуществляется от выпрямителя типа ВСП-33-5. Двухкас-Цадная термобатарея с последовательным питанием каскадов состоит из 29 термоэлементов на нижнем каскаде и'четырех на верхнем, соединенных металлическими теплопереходами. Для подачи и слива воды в теплообменник введены два штуцера. Корпус и стакан крепятся винтами к теплообменнику. Между стаканом и теплообменником расположена термобатарея. Для автоматического регулирования температуры в рабочем объеме на дне стакана установлен датчик-терморезистор.
Последовательность выполнения работы. Налить в пробирку 40 мл исследуемого растворителя и поместить в рабочий стакан микрохолодильника. Включить выпрямитель ВСП-33-5 в электросеть на 220 В. Установить переключатель трансформатора в положение «1». Для постепенного усиления охлаждения через 20 мин поставить переключатель в положение «2», еще через 22 мни — в положение «3». После замерзания растворителя перевести переключатель в положение «1> трансформатора, при котором следует выполнять работу. Настроить термометр Бекмана в отдельной охладительной смеси со льдом (см. с. 181). Положение столбика ртути в термометре должно быть между 2—4°. После настройки термометра Бекмана его нельзя оставлять при комнатной температуре длительное время, его следует быстро сполоснуть чистым растворителем, вытереть и перенести в рабочую пробирку с охлажденным исследуемым раствором. Определить температуру замерзания чистого растворителя. Для этого установить пробирку с чистым растворителем и погруженным в него термометром в рабочий стакан холодильника и, медленно помешивая растворитель в пробирке, следить за изменением его температуры. Помешивание прекратить при температуре на 0,5° выше температуры замерзания растворителя. После переохлаж-18-1
I дения растворителя на 0,5° ниже температуры его замерзания возобновить перемешивание жидкости. В качестве истинной температуры замерзания записать максимальную температуру после ее подъема при кристаллизации из переохлажденного состояния. Вынуть пробирку из холодильника, осторожно подогревая рукой, расплавить образовавшиеся кристаллы и повторить измерения не менее трех раз.
« Между измерениями при замене раствора и другими операциями настроенный термометр следует закрепить в штативе так, 1 чтобы его нижний резервуар со ртутью был погружен в охлади-j тельную смесь.
Определить температуру замерзания раствора. Для этого по-; вторить все операции с исследуемым раствором.
i Опыт 1. Определение концентрации раствора. Раствор налить
в сухую пробирку и поместить в стакан микрохолодильника. При j появлении первых кристаллов в пробирку быстро перенести термометр Бекмана. Подогревая рукой пробирку, расплавить кри-j сталлы, после чего снова поместить пробирку с термометром в j микрохолодильник. Процесс охлаждения раствора проводить при I помешивании. Помешивание прекратить, когда температура будет I на 0Д° ниже температуры кристаллизации растворителя. Раствор
I охладить без перемешивания на 0,2—0,3°. Снова начать переме-
шивание до установления температуры кристаллизации раствора. / Температуру замерзания раствора определять не менее трех раз, I добиваясь уменьшения разброса результатов измерения.
I Опыт 2. Определение активности и коэффициента активности неэлектролита в водном растворе. Приготовить шесть растворов неэлектролита (карбамид, сахар и др.) с различными концентрациями:
&
x.W
?.:.3g
J Раствор карбамида, моль/1000 г 0,1 0,2 0,3 0,4 0,5 0,7 0,9
’ Раствор сахара, моль/1000 г 0,05 *0,1 0,15 0,2 0,3 0,4
Определить ДТзам каждого раствора (см. с. 184). Вычислить вспомогательные величины / и j/m для каждого раствора и занести в таблицу. Построить график в координатах j/m—т и графическим интегрированием определить коэффициент активности и активность растворенного вещества при заданной концентрации раствора.
Опыт 3. Определение активности и коэффициента активности электролита в водном растворе. Приготовить шесть растворов электролита (КС1, NaCO3, CaSO< и др.) с концентрациями (моль/1000 г): 0,01; 0,02; 0,05; 0,1; 0,2; 0,5. Определить ДТзам каждого раствора (см. с. 184). Вычислить вспомогательные величины j и j/m для каждого раствора и занести в таблицу. Построить график в координатах j/m—т и графическим интегрированием определить средний ионный коэффициент активности и активность электролита в растворе при заданной концентрации.
- 185
ш
Рассчитать средний ионный коэффициент активности по формуле 1g у±=—0,509£iL2 и сравнить с экспериментальной величиной.
Техника безопасности в описанных работах
1. При неосторожном обращении с термометром Бекмана можно легко стряхнуть капельку ртути из капилляра в запасной резервуар. Это повлечет к повторению всех операций в работе.
2. Неправильные показания термометра могут быть следствием того, что термометр касался стенок пробирки или мешалки.
3. Если кристаллы вещества не будут полностью расплавлены, переохлаждения не произойдет. Надо обеспечить полное расплавление кристаллов на стенке термометра, которые могут образоваться выше уровня жидкости за счет попавших туда мелких брызг.
4. При очень устойчивом переохлаждении, когда кристаллизацию не удается вызвать помешиванием, надо внести затравку (кристалл растворителя).
5. Несовпадение результатов параллельных определений может быть вызвано неодинаковыми условиями при проведении опыта.
Для успешного проведения опыта необходимо соблюдать одинаковые условия каждого определения истинной температуры затвердевания. Так, охлаждение при помешивании должно прекращаться при одном и том же показании термометра; кристаллизацию начинать с одного и того же показания термометра.
Работа 5. Определение молекулярной массы вещества по методу Раста
Последовательность выполнения работы. В методе Раста необходимо брать растворитель, обладающий высокой криоскопической постоянной, например камфору. Для нагревания образцов следует пользоваться блоком, представляющим собой массивный кусок меди. Блок можно применять в интервале температуры от 273 до 1000 К- Блок имеет отверстия для термометра и капилляра, в котором плавится исследуемое вещество. Размеры отверстий таковы, что термометры и капилляры плотно входят в них, чем достигается хороший термический контакт с металлом. Чтобы видеть нижние концы капилляров, в блоке следует прорезать горизонтальные отверстия, которые закрыты стеклами для защиты от проникновения холодного воздуха. Через эти отверстия наблюдают за изучаемым веществом в капилляре. Блок покрыт слоем асбеста. При работе блок нагреть (рис. 77) на слабом огне горелки и произвести отсчет температуры плавления.
186
чистую камфору, а также смеси соотношении: 1:40, 1 :25; 1 : 18;
Рис. 77. Схема установки для определения молекулярной массы по методу
Плавление вещества в капилляре наблюдать при помощи бинокулярной лупы с увеличением в десять раз при боковом освещении. Около блока поставить осветительную лампу.
Для проведения опыта взять бензойной кислоты и камфоры в 1:16; 1 : 14; 1 :12; 1 :10. Смеси для анализа после взвешивания необходимо расплавить, затем тщательно растереть. Чистую камфору и смеси поместить в капилляры. Открытым концом капилляра набрать небольшое количество вещества. Затем эту порцию перевести на дно капилляра, для чего бросить капилляр внутри более широкой трубки высотой около 30 см на стол. Исследуемое вещество должно уплотниться на дне капилляра. Это обеспечит наилучшее соприкосновение пробы вещества со стенками капилляра. Высота слоя в капилляре должна быть 4— 5 мм. Меньшее количество затрудняет наблюдение температуры плавления: наличие более высокого слоя не повышает точности определения температуры плавления.
В отверстие блока поместить капилляр с веществом, термометр на 150—200° С вставить в расположенное рядом отверстие, а наблюдение за веществом в капилляре производить через сквозное боковое отверстие. Нагревание блока вести на газовой горелке или электрической плитке. Для плавного повышения температуры блока между ним и нагревателем нужно поместить асбестовые прокладки. Блок нагревать сначала очень быстро, но примерно за 30° до температуры плавления вещества скорость нагревания регулировать таким образом, чтобы она не превышала 1 град/мин. Сначала определить температуру плавления чистой камфоры, а затем температуры плавления смесей. Для получения более точных результатов каждое определение производить дважды и в качестве конечного результата взять среднюю величину из двух измерений. При нагревании внешний вид вещества в капилляре сначала не изменяется, а затем кристаллы начинают плавиться и становятся прозрачными.
В расплавленном состоянии смеси, а также чистая камфора представляют собой бесцветные жидкости, быстро мутнеющие при застывании, если капилляр вынуть из нагретого блока. При
187
плохом прессовании смесей по мере их плавления обнаруживаются пузырьки воздуха, которые вызывают повышение температуры плавления; поэтому от пузырьков воздуха следует избавиться легким постукиванием капилляра о дно блока. При хорошем прессовании порошки в капиллярах плавятся, начиная с верхних слоев. При наличии пузырьков воздуха плавление начинается в нижних слоях, вверху же оно замедляется, так как верхние кристаллы отделяются от нижнего расплавленного слоя воздушной прослойкой. После удаления пузырьков кристаллы опускаются вниз и тонут в расплавленном веществе. Температурой плавления следует считать ту температуру, при которой появляются первые признаки плавления вещества в капилляре. Экспериментальные и расчетные данные записать в таблицу по образцу:
Исследуемое вещество
Концентрация растворенного вещества, моль/1000 г
Температура плавления, °C
Понижение температуры замерзания
Растворитель Смеси:
1 : 40
1 : 25
На основании экспериментальных данных рассчитать молекулярную массу растворенного вещества.
Работа 6. Эбуллиоскопический метод определения молекулярной массы веществ
В работе применяют эбуллиоскоп Свентославского (рис. 78).
В сосуд 3 налить жидкость и нагреть ее, образующиеся пары уносят с собой жидкость по трубке 2 в широкую трубку 6, помещенную в пробирку 7. Жидкость тонкой струей должна стекать по термометру. В трубке 2 вся избыточная теплота, уносимая жидкостью вследствие ее перегрева, расходуется на превращение соответствующего количества жидкости в пар. Таким образом, температура, которая устанавливается в трубке 6, соответствует температуре сосуществования пара с жидкостью при данном давлении. Пар конденсируется в холодильнике 5, а образовавшаяся жидкость по трубке 4 возвращается в сосуд 3.
Последовательность выполнения работы. В прибор (рис. 78) налить исследуемую жидкость так, чтобы ее уровень находился на 2 см выше шейки сосуда 3. После этого прибор закрепить в штативе. В пробирку 7 вставить на пробке предварительно уста-
188
Рис. 78. Схема эбуллиоскопа Свентославского
новленный и тщательно вытертый фильтровальной бумагой термометр Бекмана 1. Пробка с термометром должна быть плотно вставлена в пробирку 7, а сам термометр не должен касаться стенок трубки 3. Затем в холодильник 5 пустить воду и к прибору подставить горелку. Жидкость рекомендуется нагревать при помощи маленькой горелки, дающей полукруглое пламя, охватывающее дно сосуда 3.
Температуру кипения растворителя определить два-три раза и взять среднее значение. После этого сосуд 3 освободить от растворителя.
Для определения молекулярной массы растворенного вещества в чистый и сухой сосуд 3 ввести отвешенное количество растворителя. Чтобы растворитель заполнил объем шарика 3 и трубку 2 по высоте на 1 см, следует брать его около 30—40 мл. Для этого стакан (или колбу) с растворителем взвесить. Затем из стакана в прибор залить небольшое количество растворителя. После этого ввести точно взвешенное (на аналитических весах) количество исследуемого вещества. Затем из стакана добавить растворитель, которым смыть со стенок исследуемое вещество. Взвесить стакан с оставшимся растворителем и по разности масс получить количество введенного раство
рителя. Таким образом определить температуру кипения полученного раствора известной концентрации.
Температуру кипения раствора определить три-четыре раза; из полученных данных рассчитать среднюю температуру кипения, а также разность средних температур кипения растворителя и раствора. Рассчитать молекулярную массу растворенного. вещества. Полученные экспериментальные данные записать в таблицу по образцу:
Растворитель Раствор Температура кипения, °C Повышение температуры кипения, At °C
при нормальном давлении при нормальных условиях
растворитель раствор растворитель раствор
1 1
189
Температуры кипения и эбуллиоскопические постоянные некоторых растворителей приведены ниже:
Растворитель
Т’квн, К
Тэ
н2о С2Н6ОН С2Н5-О-С2Н5 С6Нб СС14
373,2 351.7 307,8 353,3 350,0
0,520 1,20 2,1 2,6 5,0
Работа 7. Определение температуры кипения чистого вещества и раствора по методу Сиволобова
Последовательность выполнения работы. Схема прибора приведена на рис. 79. Трубку с внутренним диаметром 4—5 мм разрезать на части длиной 18—20 см. Середину каждой трубки вы-
тянуть до диаметра 2—3 мм и разрезать по узкому месту на равные части с тем, чтобы получить трубки длиной 6—8 см. Один конец этих капилляров запаять. В пробирку 4 налить примерно 0,5 мл исследуемой жидкости и ввести в нее очень тонкий капилляр 3 с запаянным концом, обращенным вверх. Пробирку 4 закрепить при помощи резинового кольца на термометре 1 так, чтобы иследуемая жидкость находилась на уровне ртутного шарика. Термометр с пробиркой погрузить в глицериновую баню, снабженную мешалкой 2, укрепленную на штативе на асбестовой сетке и, равномерно нагревать до тех пор, пока из капилляра 3 не начнут непрерывно выделяться пузырьки. Эта температура соответствует температу-
Рис. 79. Схема установки ре кипения исследуемой жидкости, для определения темпер ату- Опыт проводить дважды и взять сред-ры кипения нее значение температуры кипения
(расхождения параллельных опытов не должны превышать 0,5°). Опытные данные записать в таблицу
по образцу:
Параметры Растворитель Раствор Ошибка опыта
Температура кипения, °C при данном давлении при нормальном давлении Расчетное значение 7G Справочное значение Кэ
190
Необходимо привести температуры кипения к нормальным условиям. Пересчет произвести по уравнению
Д^кип = 0,00012 (760 — р) (273 + /),
где /Кип И t — температуры кипения при данном и нормальном давлениях, °C; р — атмосферное давление в момент проведения опыта. Рассчитать теплоту испарения (кДж/моль) исследуемой жидкости по уравнению Трутона А//°исп = 8,9-10~2 Ткип или по более точному уравнению Кистяковского:
^кип In (88,07 Гкип) - Гкнп (36,7 + 19,1 1g Гкнп) 10-з.
Соотношение ДН°Исп/7кип выражает изменение энтропии при испарении и оба уравнения применимы только к неполярным жидкостям. Рассчитать эбуллиоскопическую постоянную и сравнить ее с табличными данными. Рассчитать для процесса испарения на основании полученных данных для 1 моль исследуемого вещества величины A/f, AU, AG, АА при температуре кипения.
ГЛАВА IX
РАСТВОРЫ жидкостей в жидкостях
1. ТЕРМОДИНАМИКА РАСТВОРЕНИЯ
Процесс образования растворов на молекулярном уровне можно представить следующим образом. Каждая группа молекул чистого вещества должна сначала перестроиться таким образом, чтобы молекулы были удалены друг от друга на расстояния, соответствующие конечной концентрации раствора. (Например, в разбавленном растворе метанола в воде молекулы метанола очень удалены друг от друга, а молекулы воды находятся почти так же близко друг к другу, как в чистой воде.) Далее системы с удаленными молекулами должны сблизиться, образовав раствор конечной плотности. Сначала индивидуальные вещества должны поглотить энергию, чтобы произошло разделение частиц; однако при сближении частиц «раздвинутых» систем при образовании раствора энергия выделяется. Смешение частиц увеличивает статический «беспорядок» системы, что сопровождается увеличением энтропии.
Для процессов растворения характерна склонность к самопроизвольному протеканию к отрицательному изменению энергии Гиб-са в результате смешивания частиц растворителя и растворенного вещества.
Основные различия в поведении растворов зависят от баланса выделяемой и поглощаемой энергии, связанной с изменениями межчастичных расстояний. Величины А77 для процессов растворения отличаются друг от друга. Образование раствора является экзотермическим процессом (изменение энтальпии отрицательно), если
191
при смешении частиц освобождается больше энергии, чем необходимо для первоначального разделения частиц. Если же для разделения частиц требуется больше энергии, чем освобождается при смешении, процесс растворения эндотермичен (изменение энтальпии положительно). Поскольку энтальпийная функция дает вклад в изменение энергии Гиббса AG=AH—TAS, можно ожидать, что именно для эндотермического процесса наиболее вероятна ограниченная растворимость; это в действительности подтверждается термодинамическими измерениями. Однако на молекулярном уровне эндотермический ход процесса растворения обусловлен тем, что однородные частицы в чистых жидкостях притягивают друг друга в смеси в среднем сильнее по сравнению с разнородными. Следовательно, для разделения однородных частиц в систему необходимо ввести больше энергии, чем выделится при сближении разнородных частиц в процессе смешения.
Приведенные рассуждения включают ряд допущений, в действительности поведение растворов часто значительно сложнее. Например, конечный раствор представляет собой систему с совершенно случайным распределением частиц, при рассмотрении которой была исключена возможность существования в растворе некоторой упорядоченной структуры. Если наличие упорядоченной структуры вносит свой вклад в образование раствора, то изменение энтропии будет иметь меньшее положительное значение, которое трудно поддается анализу. Тем не менее корреляция положительных отклонений от поведения идеальных растворов и ограниченной растворимости с энергиями притяжения между однородными частицами является достаточно хорошим первым приближением.
Образование раствора может продолжаться до тех пор, пока химические потенциалы компонентов заметно понизятся при переходе в раствор. Теоретическая модель процесса растворения объясняет понижение химического потенциала как результат разупоря-дочения при смешении частиц и влияния энергетических эффектов за счет притяжения между частицами. Если энергия притяжения между однородными частицами больше энергии притяжения между разнородными частицами, то снижение,химического потенциала велико только для очень разбавленных растворов, а затем становится незначительным. Тогда можно предсказать, будут ли два вещества обладать высокой или низкой взаимной растворимостью; для этого необходимо только оценить степень притяжения между однородными частицами.
Теплоты испарения служат удобной и достаточно достоверной мерой энергий притяжения, хотя следует иметь в виду вклады других факторов, таких, как размеры молекул и специфические взаимодействия между частицами. В общем, два компонента будут обладать ограниченной взаимной растворимостью, если один из них характеризуется значительно большей теплотой испарения по сравнению с другим.
При смешении двух жидкостей могут наблюдаться всевозможные градации взаимной растворимости: от практически полной не
растворимости друг в друге (например, ртуть и вода) до смешения в любых соотношениях с образованием однородного раствора (например, этанол и вода). Промежуточное положение занимает смесь ограниченной взаимной растворимости. Смесь жидкостей А и В (например, анилин и вода) разделяется после взбалтывания на два слоя: насыщенный раствор А в В и насыщенный раствор В в А. Однако и в этом случае могут существовать области температуры и состава, в которых компоненты А и В образуют однородную смесь.
Ввиду разнообразия в поведении растворы классифицируют в соответствии с их термодинамическими свойствами. С этой точки зрения различают идеальные и неидеалъные растворы.
2. НЕОГРАНИЧЕННО СМЕШИВАЮЩИЕСЯ ЖИДКОСТИ
Давление пара над однородной жидкой смесью в зависимости от ее состава может быть представлено, как показал Коновалов, тремя основными типами кривых для идеальных и неидеальных растворов.
Идеальные растворы. Раствор является идеальным, если уравнение + применимо к нему во всем возможном ин-
тервале концентраций. Из закона Рауля следует, что давление пара над раствором есть линейная функция от молярной доли:
^’a = /’o,axA’ Pb = Pq,bxb- (IX1)
Эту зависимость можно представить графически (рис. 80). Точка Ро,а отвечает давлению пара над чистым компонентом А, а точка ро, в — давлению над чистым компонентом В. Прямая ро,хВ выражает зависимость парциального давления пара компонента А от состава, прямая ро, вА— зависимость парциального давления пара компонента В от состава. Общее давление, равное, согласно закону Дальтона, сумме парциальных давлений, выражается прямой ро, аРо, в. (Газовую фазу считаем смесью идеальных газов.)
Растворы подчиняются закону Рауля, если силы взаимодействия между частицами разных веществ (А—В) равны силам, действующим между частицами одного и того же вещества (А—А и В—В), причем смещение компонентов не сопровождается ни поглощением, ни выделением теплоты или изменением объема. Таким образом, &v=0; ДЯ=0.
Неидеальные растворы. К неидеальным растворам применимо уравнение
р, _ у,* д- рт 1п а. — уЛ + ВТ In х. + RT In у, - (IX.2)
4 4 * X 4
По отклонению свойств растворов от свойств идеальных растворов судят о силе взаимодействия между молекулами его компонентов. Отклонения эти растут с увеличением концентрации.
Положительные отклонения от закона Рауля характерны для растворов: ацетон — сероуглерод, этанол — этило
7—2083 193
вый эфир и др. (рис. 81). Положительные отклонения можно объяснить меньшими силами взаимодействия между молекулами разного типа (А—В), чем между молекулами одного и того же вещества (А—А и В—В). Положительные отклонения объясняются распадом (диссоциацией) ассоциированных молекул одного или обоих чистых компонентов при их смешении. Это повышает летучесть компонентов. Образование растворов такого типа, как правило, сопро-
Рис. 80. Зависимость парциальных и общего давлений от состава раствора (идеальный раствор) при / — = const
Рис. 81. Зависимость парциального и общего давлений от состава раствора (положительное отклонение от закона Рауля) при Z = const:
------ отклонение от закона Рауля; — ---—подчинение закону Рауля
вождается увеличением объема и поглощением теплоты, т. е. Д^> >0; Д/7>0; это уменьшает теплоту парообразования.
Отрицательные отклонения от закона Рауля характерны для растворов: вода — хлористый водород, вода —серная кислота и т. п. Для данных растворов наблюдается уменьшение давления пара по сравнению с идеальными растворами (рис. 82). Отрицательные отклонения обусловливаются большими силами притяжения между молекулами разных типов (взаимодействие А — В больше, чем А — А и В — В). Отрицательные отклонения наблюдаются у растворов, склонных к сольватации, в частности гидратации и т. п. Образование раствора такого типа, как правило, сопровождается уменьшением объема и выделением теплоты, т. е. Дг?<0; ДЯ<0, Поэтому теплота парообразования растворенного компонента оказывается больше, чем чистого компонента. Это затрудняет парообразование. Если отклонения от закона Рауля очень велики, кривая общего давления пара может иметь максимум или минимум, в зависимости от того, какие отклонения наблюдаются — положительные или отрицательные.
Состав пара смеси не всегда не совпадает с составом жидкости, находящейся в равновесии с паром. На различии составов жидкости
194
и пара основана перегонка смесей, имеющая большое практическое значение. На рис. 83 приведена зависимость температуры кипения от состава жидкости (кривая /дС7в) и пара (кривая /дс^в). Точка /д отвечает температуре кипения чистого компонента А, точка — температуре кипения компонента В. Область 1 относится к жидкости; область II — к пару. В условиях гомогенности двухкомпонентные системы имеют две степени свободы: состав и температуру. Точка а обозначает жидкость состава xq. При нагревании до температуры /н жидкая смесь начинает кипеть. В процессе кипения жидкость обогащается более высококипящим компонентом и при этом изменяется температура кипения. Когда вся жидкая смесь испарится, состав пара будет равен составу исходной смеси xQ. От-
пения раствора от состава жидкости и пара при р — const
рбщего давлений от состава раствора (отрицательное отклонение от закона Рауля) при / — const:
----- —- отклонение от закона Рауля; ----- — подчинение закону Рауля
сюда можно определить температуру кипения последней порции жидкой смеси, которая будет соответствовать Дальнейшее нагревание не приведет к изменению состава смеси, что и показано на рис. 83 вертикальными стрелками. Между кривыми /дС/в и £да£в находится область /// гетерогенного равновесия между жидкостью и паром. Любая точка внутри этой области характеризует систему, которая распадается на жидкую и парообразную фазы. Соотношение между количествами этих фаз при температуре кипения опре-пеляется по правилу рычага.
Экспериментально установлено, что пар обогащается более летучим компонентом. Эта закономерность впервые была выведена Ц. П. Коноваловым и известна как первый закон Коновалова: а) повышение относительного содержания данного компонента в жидкой фазе всегда увеличивает относительное содержание его и в парах; б) в двойной системе пар, по сравнению с находящейся у*
195
с ним в равновесии жидкостью, относительно богаче тем из компонентов, прибавление которого к системе повышает общее давление пара, т. е. понижает температуру кипения смеси при данном давле-
нии.
Первый закон Коновалова имеет большое значение для перегонки. При перегонке пар постепенно обогащается более летучим
Рис, 84. Зависимость температуры кипе-
няя от состава жидкости и пара при р—const
компонентом и на этом основано разделение компонентов или обогащение смеси каким-либо компонентом. Одним из видов перегонки является перегонка в равновесии, которая основана на том, что при испарении части жидкости пар находится в соприкосновении с жидкой смесью, а затем он конденсируется. Пусть имеется смесь состава Хо (рис. 84), при температуре t\ она закипит, при этом образуется пар состава Xi0. При отгонке пара, обогащенного более летучим компонентом, жидкость обо-
гащается менее летучим компонентом В. Состав жидкой
смеси будет Xi. Температура кипения раствора повысится и пар состава xj\ отвечающего составу раствора.Xi, будет содержать меньше летучего компонента А, чем исходный раствор. Такая перегонка не может полностью разделить
смесь.
Перегонку ведут, сохраняя контакт фаз, но при этом непрерывно компенсируется образующийся пар. Таким образом, и процесс проходит через непрерывный ряд возможных равновесий. Если бинарная система не обладает экстремумом, то перегонка позволяет выделить любой компонент, если экстремум существует, то только
один из них.
Практически наибольшее значение имеет фракционная перегонка. Пар, полученный при испарении раствора первоначального состава, конденсируют и полученную жидкую смесь вновь перегоняют. При этом полученный пар будет еще более обогащен летучим компонентом. Повторяя эту операцию несколько раз, можно в конденсате получить почти легколетучий компонент. Состав же жидкости по мере нагревания будет обогащаться менее летучим компонентом. Таким образом, можно осуществлять разделение смесей. Фракционную перегонку можно проводить непрерывно.
Для растворов, которые точно подчиняются закону Рауля, кривые температура — состав могут быть построены по расчетным данным. Если же смесь дает отклонения от закона Рауля, то кривая
196
может быть построена по опытным данным. Однако если отклонения эти очень велики, то на кривых давление пара —состав (или температура кипения — состав) может появиться максимум или минимум в зависимости от того, положительные или отрицательные отклонения проявляют эти растворы. В точках максимума или минимума кривая жидкости обязательно коснется кривой пара. Такая точка, в которой состав пара и состав жидкости одинаковы, называется азеотропной точкой. Смесь кипит как одно целое, и разделить ее на составные части путем перегонки оказывается невозможным.
Свойства смесей с максимумом или минимумом температур кипения описываются вторым законом Коновалова, согласно которому в точках экстремума давления пара (или температуры кипения) составы жидкости и пара совпадают. Максимум на кривой давления пара отвечает минимуму на кривой температуры кипения, а минимуму на кривой давления отвечает максимум на кривой температурь! кипения. На рис. 85 приведена зависимость давления и состава пара от состава жидкости при Z=const, а на рис. 86 — за-
Рис. 85. Зависимость давления от состава пара и жидкости (диаграмма с азеотропной смесью) при t = const
Рис. 86. Зависимость температуры кипения раствора от состава (диа-* грамма кипения с азеотропной смесью) при р=const
висимость температуры кипения и состава насыщенного пара от состава раствора при p=const. Смеси при разделении дают чистый компонент и азеотропную смесь. Прекращение процесса ректификации в точках экстремума объясняется совпадением состава жидкости с составом пара при определенном соотношении компонентов, вследствие чего движущая сила разгонки (различие в составе фаз) становится равной нулю. Поэтому при равновесии азеотропная смесь испаряется как чистое вещество. Состав азеотропной смеси можно изменить давлением. Это положение было сформулировано в законах Вревского.
Первый закон: при повышении температуры раствора заданного состава его пар обогащается тем компонентом, теплота испарения которого больше.
197
Рис. 87. Зависимость температуры кипения раствора от состава при р~ ~const
Второй закон: если компоненты образуют азеотропную смесь, то следует различать два случая: при наличии максимума на кривой давления пара с повышением температуры азеотропной смеси в ней возрастает концентрация компонента, обладающего большей теплотой испарения; при наличии минимума на кривой давления пара возрастает концентрация того компонента, у которого теплота испарения меньше.
Третий закон: с изменением температуры (или давления) состав пара и азеотропной смеси изменяется в одном и том же направлении при максимуме давления пара, и в противоположных направлениях— при минимуме давления пара.
Таким образом, для разделения азеотропных смесей можно использовать изменение температуры и давления.
Правило рычага. При расчетах фазовых равновесий необходимо определять, каким будет количество компонента в каждой из равновесных фаз при заданном общем составе. Для решения этих и других подобных задач (в том числе и задач, относящихся не только к фазовым равновесиям) удобно пользоваться правилом рычага и диаграммой. При равновесии жидкость — пар в двойной системе необходимо (рис. 87) определять количественное соотношение между жидкостью и паром при различных температурах. Рассмотрим исходную жидкую смесь состава х0 по компоненту В при Л; х0— содержание компонента В в паре; хж— содержание компонента В в жидкости. Пусть исходная жидкая смесь состоит из m моль обоих компонентов. Через у
обозначим количество образовавшегося пара, через tn—^/ — количество оставшейся жидкости, через х0— общее количество компонента В в исходной смеси (х— молярная доля). Составим материальный баланс по компоненту В:
ухп 4~ (т — у) хж.
После некоторых преобразований получим правило рычага:
количество жидкости а
количество пара b
Соотношение равновесных количеств пара и жидкости (сопряжен-198
них фаз) равно соотношению противолежащих отрезков конноды, которые она образует с линией, проведенной через суммарный состав смеси.
Работа. Изучение равновесий жидкость — пар в двойных жидких системах
Для определения температур кипения жидкостей служат приборы разных конструкций, называемые эбуллиоскопами.
На рис. 88 представлена схема простейшей установки для определения температур кипения и составов пара. Раствор помещают в колбу и осторожно нагревают. Когда раствор закипит, нагрев уменьшают, пока не будет достигнута температура кипения при
Рис. 88. Схема установки для изучения равновесия жидкость — пар в двойных системах
Рис. 89. Эбуллиоскоп Свенто-славского для изучения равновесия жидкость — пар в двойных системах:
1 — положение крана при определении температуры кипения смеси; 2, 3 — положения крана при накоплении конденсата
постоянном давлении (1 атм~ 1,032-105 Па). Небольшие количества испарившейся жидкости все время конденсируются в обратном холодильнике и возвращаются в перегонную колбу. Это обеспечивает равновесное состояние при исходном составе кипящей жидкости. По достижении температуры кипения отбирают для анализа небольшое количество жидкости. Аналитический метод определения состава пара и жидкости основан на измерениях показателя преломления или плотности. Эти измерения, проведенные многократно
199
для растворов различного состава, позволяют построить график зависимости температур кипения при постоянном давлении от состава жидкости (диаграмма Т — х). Фазовую диаграмму для состава пара можно получить, если отбирать для анализа небольшое количество конденсирующегося пара в углублении, находящемся непосредственно под холодильником (рис. 89). Источники ошибок измерений связаны с колебаниями температуры, отбором проб для анализов и изменением равновесных концентраций паров компонентов на пути от кипящей жидкости до холодильника.
Последовательность выполнения работы. В восьми конических колбах вместимостью 20 мл с притертыми пробками приготовить бинарные органические растворы с различным объемным содержанием:
Номер колбы 12 3
Объемное содер-
жание компонен-
та, %
А 1'00 95 90
М 100 95 90
4 5 6 7 8 9 1-0
85 80 70 60 45 30 0
75 60 50 35 20 10 0
Каждая колба должна содержать по 10 мл смеси, кроме первой и последней (100 и 0%). Смесь готовить смешением одной из высококипящей жидкости: СбНзСНз, СбНб, CeHgCl и др. с одной из жидкостей (СНз)гСО, СНС1з, СС14 или С4Н9ОН (смесь состава А) или смешением высококипящей жидкости с метанолом (смесь М).
Измерив показатели преломления чистых жидкостей и раство
ров известного состава, вычертить кривую зависимости показателя преломления от состава (калибровочная кривая). Зная плотности чистых жидкостей, можно перейти от объемного содержания, выраженного в процентах, к массовому или молярному содержанию, выраженному в процентах, н построить график зависимости показателя преломления от состава.
По показателю преломления раствора неизвестной концентрации по калибровочной кривой определить его состав. Затем определить температуры кипения смесей. Для определения температуры кипения смеси используют установку* (рис. 88), которая состоит из сосуда 1 для кипячения, термометра 2 и холодильника 3. Внутренняя трубка холодильника вставлена в пробку 4 так, чтобы холодильник можно было перевести в положение, необходимое для конденсации пара и отбора конденсата. Сосуд для кипячения с 10 мл смеси известного состава укрепить в штативе. Во избежание перегрева жидкости и для обеспечения равномерного кипения в сосуд помещают мелкие кусочки неглазурованного фарфора или стеклянные капилляры, запаянные с одного конца. После этого сосуд закрыть пробкой с термометром так, чтобы шарик термометра был погружен в жидкость. Затем соединить сосуд с холодильником, пустить воду в холодильник и медленно нагревать сосуд. После того как температура кипения жидкости установится, ее записать, за
* В этой работе можно применять эбуллиоскоп Свентославского (рис. 89).
200
тем холодильник повернуть в положение для отбора конденсата. В заранее приготовленную пробирку с пришлифованной пробкой отобрать 5—10 капель конденсата, пробирку немедленно закрыть. Чтобы состав пробы не изменился, пробирку погрузить в тающий лед или холодную воду (ни в коем случае не держать ее в руках). После этого холодильник вновь поставить в вертикальное положение, продолжать кипячение 2—3 мин и вновь записать температуру кипения, при этом температура не должна отличаться от температуры до отбора пробы более чем на Г. Затем нагревание прекратить, сосуд после охлаждения вынуть из прибора и содержимое вылить в колбу.
Такой же опыт провести с остальными смесями и чистыми компонентами. При определении температуры кипения чистых компонентов термометр поместить в паровую фазу. Перед каждым опытом кусочки фарфора (или капилляры) заменять новыми. Сосуд и холодильник перед каждым опытом продуть теплым воздухом. Состав пара определять по показателю преломления собранного конденсата и по калибровочной кривой найти его состав. Коэффициент преломления п конденсата определять при той же температуре, при которой определяли п исходных смесей. Призмы рефрактометра перед каждым определением осторожным прикосновением вытереть фильтровальной бумагой. Результаты опытов записать в таблицу по образцу:
Номер пробы 1 ь Температура кипения, °C Показатель преломления Состав, %
до отбора пробы после отбора пробы средняя жидкость конденсат жидкости пара
Построить калибровочную кривую. Построить график в координатах температура кипения — состав. Одну кривую построить по температурам кипения до отбора пробы и по процентному содержанию смесей. Вторую кривую построить по средним температурам кипения и процентному содержанию пара. Построить график в координатах состав пара — состав жидкости. Все три графика чертить на одном листе миллиметровой бумаги, один под другим, сохраняя абсциссу (состав) в одном и том же масштабе. Построить диаграммы состояния в координатах температура кипения — состав, состав пара — состав жидкости для двойной системы, которая может быть составлена из веществ, расположенных по температурам кипения от более высокой к более низкой: СбН3С1 (132°С); С4Н9ОН (117,7°С); С6Н5СН3 (110°С); Н2О (100°С); С3Н7ОН (97,2°С); С2Н4С12 (84° С); С6Н6 (80,2°С); СС14 (76,6°С); СН3ОН (64,7°С); СНС13 (61,2°С); (СН3)2СО (56°С).
Меры предосторожности. Настоящая работа проводится, как правило, с летучими и легковоспламеняющимися органическими жид-
201
костями. Во избежание воспламенения жидкостей или их паров следует, при работе соблюдать следующие правила: 1) приступая К работе, убедиться в наличии у рабочего места противопожарных средств; 2) нагревание прибора при определении температуры кипения должно производиться на пламени микрогорелки или'на закрытой электрической плитке; 3) кипение жидкости должно быть равномерным (что достигается введением пористой керамики в реакционную колбу) и не слишком бурным; 4) введение жидкости в прибор, отбор пробы конденсата и удаление остатка жидкости из прибора можно производить, лишь погасив микрогорелку или отключив электроплитку; 5) сушку прибора можно производить также только на достаточно большом расстоянии от горящих горелок; 6) работу следует вести под тягой.
ГЛАВАХ
ВЗАИМНАЯ РАСТВОРИМОСТЬ ЖИДКОСТЕЙ
1. ОГРАНИЧЕННАЯ РАСТВОРИМОСТЬ ЖИДКОСТЕЙ
Существует ограниченная растворимость, если переход молекул через поверхность раздела сопряжен с совершением работы, заметно превышающей среднюю энергию движения молекул жидкостей при данных условиях. Чем больше работа перехода, тем меньше в соответствии с законом распределения Максвелла доля молекул, способных осуществить эту работу, и тем меньше растворимость одной жидкости в другой. Работа перехода и ограниченная растворимость связаны с неодинаковой интенсивностью межмолекулярных взаимодействий обеих жидкостей.
Температура различно влияет на ограниченную растворимость жидкостей. Это объясняется тем, что жидкости или вступают в химическое взаимодействие, или нет. Если жидкости химически не взаимодействуют, то повышение температуры влияет на растворимость лишь постольку, поскольку оно вызывает перераспределение энергии поступательного движения молекул. С ростом температуры увеличивается доля молекул с повышенной энергией. Такие молекулы способны осуществить работу перехода, а следовательно, взаимная растворимость жидкостей будет увеличиваться. При этом составы сопряженных растворов будут все более и более сближаться и при некоторой температуре станут тождественными. Начиная с этой температуры и выше наблюдается неограниченная растворимость жидкостей друг в Друге. Это явление было впервые изучено В. Ф. Алексеевым. Температура, выше которой жидкости неограниченно смешиваются друг с другом, называется верхней критической температурой растворения. Когда ограниченно растворимые жидкости образуют молекулярные соединения, повышение температуры уменьшает их взаимную растворимость. Повышение температуры способствует диссоциации сложных молекул на более про-202
стые, плохо растворимые компоненты, что и уменьшает взаимную растворимость. Наоборот, при понижении температуры взаимная растворимость увеличивается. При понижении температуры составы сопряженных растворов могут стать одинаковыми. Температура, при которой это происходит, называется нижней критической температурой растворения.
Существуют и такие смеси, которые обладают двумя критическими температурами растворения — верхней и нижней. Примером системы, которая дает верхнюю критическую температуру, может служить система фенол — вода. Вода и фенол в жидком состоянии проявляют ограниченную растворимость, а в твердом состоянии полностью нерастворимы друг в Друге. Диаграмма состояния фенол— вода представлена на рис. 90. Точки а и b отвечают температурам плавления фенола и льда. Кривые аВ и Со отвечают процессу кристаллизации фенола при охлаждении. Кривая Ьо соответ
Рис. 90. Диаграмма фазового состояния системы фенол—вода
ствует процессу кристаллизации льда. Кривая ВКС — кривая расслоения; кривая ВК выражает состав фенольного раствора воды; кривая КС— состав водного раствора фенола. Над кривой аВКСоЬ находится устойчивая жидкая фаза. Области соответствуют aBg— смеси фенола с фенольным раствором; ВКС — смеси фенольного и водного растворов; gCod — смеси твердого фенола с водным раствором; obe — смеси льда с водным раствором. Ниже изотермы doe расположена область смеси кристаллического фенола и льда. Диаграмму эту можно рассматривать как диаграмму неизоморфной смеси, усложненную наличием области ограниченной растворимости.
Рассмотрим диаграмму с точки зрения правила фаз и проследим изменения, происходящие во времени с жидкостями различного состава при их охлаждении (рис. 90) с примерным учетом материального баланса по различным стадиям, приняв, что во всех десяти рассматриваемых случаях берется одинаковое количество вещества.
203
Точка /. Охлаждение фенола ведет к его кристаллизации в точке а (если не будет переохлаждения). При t>ta система обладает одной степенью свободы, поэтому температура понижается. При ta появляется твердый фенол; система становится двухфазной (инвариантной), что отвечает температурной остановке, длительность которой зависит от количества фенола и скорости отвода теплоты (отвердевание фенола). В момент исчезновения последней капли жидкого фенола система вновь станет одновариантной, а температура начнет понижаться, так как с исчезновением жидкой фазы исчерпался и источник теплоты кристаллизации. Процесс охлаждения воды (точка 10 и кривая охлаждения 10) аналогичен рассмотренному.
Точка 2. Это точка соответствует фенольному раствору. Система является бивариантной. Следовательно, можно изменять в известных пределах температуру и состав, не нарушая числа фаз в системе. При Л начинает кристаллизоваться фенол (точка й). Скорость охлаждения замедляется, что отразится на кривой охлаждения (см. кривую охлаждения 2). Система становится моновариант-ной, т. е. при t<t\ каждой концентрации раствора соответствует единственная температура кристаллизации фенола. Это может быть представлено графически (кривая аВ). Состав раствора, насыщенного фенолом, определяется пересечением изотермы с кривой аВ. Связь же между количеством твердого фенола и количеством раствора определяется правилом рычага; так, при температуре t2 количество твердого фенола отрезок
количество раствора отрезок III
Результатом кристаллизации фенола является обогащение фенольного раствора водой. В точке В раствор становится насыщенным относительно обоих компонентов; это приводит к выделению Н2О и при этом в жидком виде, так как t2>tb. При температуре tgH вода обладает определенной растворяющей способностью. При этой температуре соотношение фенола и воды равно CH/(gC) (концентрация фенола выражается точкой С); вода образует первую каплю сопряженного раствора состава В. Появление третьей фазы соответствует превращению системы из моновариантной в инвариантную, т. е. приводит к температурной остановке (см. кривую охлаждения 2). Фенольный раствор расслаивается на фенол и водный раствор состава С в пропорции, отвечающей точке В, а избыток фенола выделяется в твердом виде. Таким образом, по мере отвода теплоты раствор В распадается на твердый фенол и раствор состава С, т. е. равновесие
раствор В раствор С + фенол
сдвигается вправо. Точка на диаграмме, отвечающая суммарному составу жидких фаз, перемещается от точки В к точке С, и если в данный момент она соответствует точке Л!, то это значит, что соотношение между образующимися и исчезающими растворами равно отношению отрезка 57И к отрезку МС, В точке С исчезает по-204
следняя капля фенольного раствора, температура опять начинает понижаться, и фенол выделяется из водного раствора, т. е. возобновляется процесс, протекающий по кривой аВ и прерванный из-за ограниченной растворимости (область В КС). При достижении К раствор становится насыщенным не только фенолом, но и водой, т. е. начинается кристаллизация эвтектики в точке о без изменения состава жидкой фазы. Появление льда (третья фаза) вновь приводит ж температурной остановке. После замерзания последней капли жидкости температура падаем без изменений в системе. Длины отрезков на кривой охлаждения, которые отвечают температурным остановкам для состава 2 и составов 5, 4 и 5 (см. ниже), пропорциональны ' количеству кристаллизующегося вещества. Это можно определить исходя из общего количества первоначально взятой смеси и положения точек В, С и М.
f Точка 3. При охлаждении до в системе существует одна фаза. В точке /дн раствор начинает распадаться на твердый фенол и раствор состава С. Дальнейшие изменения уже рассмотрены при процессе охлаждения смеси состава 2. Так как в смеси состава 3 воды содержится больше, чем в смеси 2, и так как до /дн фенол не выпадает, то при прочих равных условиях длительность температурных остановок на кривой охлаждения будет больше, чем для состава 2.
Точка 4. При охлаждении раствора 4 при t2 начнет происходить расслоение его на водный и фенольный растворы, при /дн появится твердый фенол. На кривой охлаждения момент расслоения вследствие незначительности теплового эффекта почти незаметен.
Точки 5—7. Процессы охлаждения этих растворов можно не рассматривать. Первый из них аналогичен охлаждению раствора 4; процессы охлаждения растворов 6 и 7 повторяют процессы -охлаждения растворов 2—5 (начиная, соответственно, с точек С и п). Разница заключается в том, что иными будут температуры расслоения, соотношения между фазами и длительность температурных остановок.
Точка 8. При температуре tQ (точка о) происходит одновременная кристаллизация фенола и воды и образуется эвтектическая смесь.
Точка 9. Из раствора кристаллизуется лед: в точке о затвердевают все компоненты одновременно, т. е. кристаллизуется весь ос-дгавшийся раствор.
2. ТРЕТИЙ КОМПОНЕНТ В ДВУХФАЗНОЙ ЖИДКОЙ СИСТЕМЕ.
ЗАКОН РАСПРЕДЕЛЕНИЯ
Если в систему из двух компонентов, содержащую две равновесные жидкие фазы, ввести небольшое количество третьего компонента, то после установления равновесия он окажется в обеих фазах, но в разном количестве. Если концентрация третьего компонента невелика и состояние его частиц в обеих фазах одинаково, то
205
увеличение его количества в системе пропорционально увеличит его концентрации в обеих фазах.
Для каждой температуры отношение концентраций третьего компонента в двух равновесных жидких фазах при различных его концентрациях является величиной постоянной (закон распределения). Этот закон, характеризующий равновесное состояние системы, может быть выведен следующим образом. Предположим, что растворенное вещество в обоих растворителях находится в одинаковом состоянии, например, в виде мономерных молекул, и обозначим активность растворенного вещества в растворителе 1 через а/, 1, а в растворителе 2 — через Химические потенциалы веществ, находящихся в двух сопряженных фазах, должны быть одинаковыми. Если в первой фазе химический потенциал исследуемого вещества
(л 1па +Д(Г), * J А I J X
а во второй фазе
(л,.2==/?7’1па.2+/2(Г),
«.
где fi(Г) и /г(Т) —стандартные потенциалы z-ro компонента в фазах 1 и 2, т. е. ц°х, 1 и ц°х, 2,
Отсюда
(Х.1)
Величину К называют коэффициентом распределения. Он зависит от природы компонентов, составляющих систему, и температуры. В разбавленных растворах вместо соотношения активностей для расчета коэффициента распределения можно пользоваться соотношением концентраций растворенного вещества в обеих фазах:
сг,1/с,,2 = К. (Х.2)
В ряде случаев закон распределения может быть представлен уравнением
(Х.З)
где п— показатель при постоянной температуре, не зависящий от концентрации и характеризующийся свойствами всех трех компонентов, составляющих систему.
Отклонения от закона распределения наблюдаются при различных состояниях растворенных молекул в одной из фаз системы. Такими различными состояниями могут быть диссоциированные или ассоциированные молекулы растворенного вещества. По коэффициенту распределения можно определить степень ассоциации или диссоциации растворенного вещества в том или ином растворителе, константу равновесия реакции, протекающей в одной из фаз, активности растворенных веществ и другие свойства. Закон распределе-206
ния широко используется при экстрагировании вещества из раствора.
Примем следующие обозначения: gQ— начальное количество вещества, которое подвергается экстрагированию; Ui — объем раствора, в котором находится экстрагируемое вещество; — объем растворителя, при помощи которого производится одно экстрагирование; п — общее число экстрагирований; g^ ..., gn — количество вещества, остающееся в первоначальном растворе после 1, 2, ...» n-го экстрагирований; К — коэффициент распределения экстрагируемого вещества. Пусть после первого экстрагирования в исходном объеме раствора Vi останется g\ г растворенного вещества, а экстрагируется —gi г вещества, причем это количество эк-
страгировано в объеме п2. По закону распределения (Х.2)
С?0 — gl)/V2 '
откуда
+ V2)
(Х.4)
(Х.5)
После второго экстрагирования
К = --~C'V ; 82 = + v2). (Х.6)
(£1 —
Если вместо g\ подставим его значение из (Х.5), то получим
\2
+ v2 /
(Х.7)
После п экстрагирований в исходном растворе останется gn г исходного вещества:
/ Kv\
Sn — Sq I ~ i •
VKvi 4- v2 )
(Х.8)
А всего будет извлечено вещества g=gQ—gn, т. е.
(Х.9)
При экстракции коэффициентом распределения условились называть отношение концентрации раствора, из которого экстрагируется распределяющееся вещество, к концентрации раствора, которым производится экстрагирование.
3. ВЗАИМНАЯ РАСТВОРИМОСТЬ ТРЕХ ЖИДКОСТЕЙ
В трехкомпонентных системах существует большое число вариантов взаимной растворимости. Рассмотрим некоторые из них, наиболее характерные и часто встречающиеся на практике (рис.91).
1. Любые два из трех компонентов системы смешиваются друг с другом во всех соотношениях (рис. 91, а). В тройных системах
207
$
жидкости также смешиваются во всех соотношениях (например, в системе вода — спирт этиловый — ацетон). В тройных смесях в некоторой области концентраций наблюдается расслоение системы на две фазы (например, в системе вода — фенол — ацетон при 67— 90°С) (рис. 91, б).
2. Два компонента (А и С) ограниченно растворимы друг в друге, а остальные две пары компонентов (А — В и В —С) обнаруживают неограниченную взаимную растворимость (например, в системе вода — хлороформ — уксусная кислота) (рис. 91, в).
3. Два компонента (А и С) неограниченно растворимы друг в друге, а остальные две пары компонентов (А — В и В — С) обна-
Рис. 91. Основные типы диаграмм взаимной растворимости трех жидкостей
руживают ограниченную взаимную растворимость (например, в системе анилин — фенол — вода). Добавление третьего компонента к двухфазной двухкомпонентной системе увеличивает взаимную растворимость и приводит к образованию однофазовой системы (рис. 91, г). Добавление третьего компонента к двухфазной системе почти не увеличивает взаимной растворимости (или уменьшает ее). Возможен непрерывный переход от двухфазной смеси компонентов А и В к двухфазной смеси компонентов В и С (рис. 91, д).
4. Для всех трех пар компонентов (А —В, В — С и А — С) наблюдается ограниченная взаимная растворимость (например, в системе вода — анилин — гексан). Добавление к двухфазной системе третьего компонента резко увеличивает взаимную растворимость двух компонентов и приводит к образованию однофазной системы (рис. 91, е). Добавление к двухфазной системе третьего компонента существенно не увеличивает взаимной растворимости и может привести к образованию трехфазной системы (рис. 91, ж).
5. Все три компонента совершенно нерастворимы друг в Друге (например, в системе вода — ртуть — бензол) (рис. 91, з).
208
Отнести данную систему к тому или иному типу можно лишь с учетом условий (например, температуры), при которых она рассматривается. Изменение температуры влияет на взаимную растворимость жидкостей и иногда столь значительно, что может изменить даже тип диаграммы (изотерма взаимной растворимости).
Состав трехкомпонентной системы изображается на диаграмме, представляющей правильный треугольник (рис. 92). Вершины равностороннего треугольника соответствуют чистым веществам А, В и С. На сторонах, соединяющих вершины, откладываются составы двух компонентных систем, образованных веществами, находящимися в прилегающих вершинах треугольника. Все точки, расположенные внутри треугольника, выражают составы трехкомпонентных систем. Процентное содержание каждого из компонентов в системе тем больше, чем ближе расположена данная точка к соответствующей вершине.
Существуют два метода определения состава трехкомпонентной системы. В методе, предложенном Гиббсом, за 100% (или единицу) принимается высота правильного треугольника. Метод основан на том, что сумма длин перпендикуляров, опущенных из любой точки внутри треугольника на его стороны, есть величина постоянная и равная высоте этого треугольника. Процентное содержание каждого компонента определяется расстоянием от точки р, отражающей состав системы, до стороны треугольника, противолежащей вершине, соответствующей чистому компоненту (рис. 92, а).
В В
Рис. 92. Треугольники для изображения состава тройных смесей
Отрезок ра соответствует процентому содержанию компонента А, отрезок pb — процентному содержанию компонента В, отрезок рс — процентному содержанию компонента С. По методу Розебома за 100% (или единицу) принимается длина стороны правильного треугольника. Сумма отрезков, проведенных параллельно сторонам треугольника из точки р, отражающей состав системы (ра + + р& + рс), равна стороне треугольника: содержание компонента А (рис. 92, б)—длина отрезков pa=bc —b'b = ac\ содержание компонента В — длина отрезков pb = a'b=Ac=c'C, содержание ком-
209
лонента С — длина отрезков pc=Aaf=cbr=Ba. Оба метода приводят к одинаковым результатам.
На рис. 93 приведена треугольная диаграмма взаимной растворимости системы вода (В) —ацетон (Д) —-хлороформ (С) при постоянных температуре (25° С) и давлении.
Состояние такой системы есть функция четырех термодинамических параметров: температуры, давления и концентраций двух компонентов. Поскольку температура и давление заданы (298 К и 1,013-105 Па), то можно задавать в известных пределах произвольно значения еще двух параметров — концентраций, не изменяя числа фаз в системе. Это значит, что на треугольной диаграмме об-
Рис. 93. Зависимость растворимости от состава в тройных жидких системах
ласть существования однофазных систем должна быть двухмерной, занимая часть треугольника состава.
Если в системе появится вторая фаза, т. е. если система окажется состоящей из двух сопряженных фаз (фаз, находящихся в равновесии), то число степеней свободы уменьшится на единицу и станет равно трем. Значит, при определенных температуре и давлении в трехкомпонентной двухфазной системе произвольно может быть задано значение концентрации только одного компонента в одной из . фаз. Концентрации двух других компонентов при этом уже однозначно определяются. Таким образом, состав каждой из сопряженных фаз двухфазной трехкомпонентной системы есть функция одного параметра. Область существования каждой из сопряженных фаз должна, следовательно, выразиться на треугольной диаграмме некоторой линией. Эти линии представляют собой две ветви бинодальной кривой. Так, на рис. 93 ветвь ЬК отвечает рас-210
творам ацетона в водном слое, а ветвь аК— растворам ацетона в слое, богатом хлороформом. Прибавление к двухфазной смеси из двух компонентов (воды и хлороформа) третьего компонента (ацетона) увеличивает взаимную растворимость. В результате этого ветвн бинодальной кривой постепенно сближаются. Третий компонент распределяется между двумя фазами неравномерно в соответствии с законом распределения Нернста — Шилова.
Отметив на соответствующих ветвях бинодальной кривой точки, отвечающие концентрациям ацетона, укажем фазовые точки двух сопряженных растворов (точки а\ и &i). Соединив эти точки отрезком прямой, получим конноду а\Ь\. Любая точка конноды, например точка d, отвечает двухфазной системе, состоящей из тех же двух сопряженных растворов (&i и &i). При этом массы сопряженных растворов, в соответствии с правилом рычага, обратно пропорциональны расстояниям от фазовых точек этих растворов до точки системы d\
Если добавить к рассматриваемой системе новые порции треть-, его компонента (ацетона), то он будет неравномерно распределяться между двумя слоями. При этом на диаграмме наклон коннод будет возрастать. Определять направление коннод с помощью закона распределения становится невозможно, так как при больших концентрациях с\ и С2 и значительной взаимной растворимости самих растворителей закон распределения усложняется. Поскольку при добавлении к системе третьего компонента увеличивается взаимная растворимость первых двух компонентов (рис. 93), то длина конноды постепенно уменьшается и при определенном составе системы, отвечающем точке К, обе ветви бинодальной кривой сходятся, составы обоих сопряженных растворов совпадают и система становится однофазной.
Составы сопряженных растворов и направление коннод в области составов системы, близкой к критической точке, можно определить, пользуясь приближенным эмпирическим правилом Тарасенкова. Согласно этому правилу продолжения всех коннод для данной области расслоения пересекаются приблизительно в одной точке (точка Т на рис. 93). Это правило применимо лишь к простейшим системам взаимной растворимости трех жидкостей.
4. ЗАВИСИМОСТЬ ВЗАИМНОЙ РАСТВОРИМОСТИ
ТРЕХ ЖИДКОСТЕЙ ОТ ТЕМПЕРАТУРЫ
Взаимная растворимость жидкостей в тройных системах, как и в двойных, зависит от температуры. Для большинства систем с повышением температуры взаимная растворимость жидкостей увеличивается, однако встречаются и системы, в которых растворимость уменьшается при повышении температуры. Диаграмма, выражающая зависимость взаимной растворимости трех жидкостей
211
ют температуры, должна быть пространственной и иметь вид трехгранной призмы. В основании призмы лежит треугольник состава тройной системы, а вертикальное ребро является осью температур. Такие диаграммы могут быть очень сложными и разнообразными. Простейший тип такой диаграммы представлен на рис. 94, а. По мере повышения температуры и роста взаимной растворимости жидкостей область расслоения уменьшается и, наконец, при достижении верхней критической
Рис. 94. Простейший вид диаграммы — поли-термы взаимной растворимости трех жидкостей
температуры растворения исчезает. Критический состав системы при этом также меняется, причем критические точки для разных температур образуют критическую кривую dk, заканчивающуюся в верхней критической точке Верхняя критическая точка растворения
может быть как двойной, лежащей на грани призмы и отвечающей двой-
ной смеси, так и тройной, т. е. лежащей внутри призмы и отвечающей некоторой определенной тройной смеси. Тройная верхняя критическая точка наблюда-
ется, например, в системе вода — фенол — ацетон. Если рассечь пространственную диаграмму рядом горизонтальных плоскостей — изотерм и спроектировать полученные для каждой изотермы би-
нодальные кривые на основание призмы, то получится плоская треугольная диаграмма (рис. 94, б), на которой роль горизонталей играют изотермические бинодальные кривые.
Работа 1. Построение диаграммы для системы вода — фенол
Для изучения взаимной растворимости используют метод Алексеева, который основан на том, что приготовляют ряд ампул с известным содержанием исследуемых веществ и запаивают их. Ампулы помещают в водяную баню, температуру которой постепенно повышают, и непрерывно ампулы встряхивают для перемешивания смеси. Определяют температуру, при которой система становится однородной. Определение проверяют, медленно охлаждая систему до появления второй жидкой фазы (помутнение раствора в ампуле). Разница между показаниями термометра не должна превышать 1°. Среднее из полученных значений соответствует температуре, при которой исследуемые вещества растворимы в том соотношении, в котором они были взяты.
Для проведения работы нужно взять набор пронумерованных ампул, содержащих смеси фенола и воды различного состава. Со
212
ставы смеси и рекомендуемая температура среды, в которую погружают ампулы, указаны ниже.
Номер пробирки 1 2 3 4 5 6 7» 8 9 ГО 11 12 13 14 15
Фенол, % 1'00 90 80 79 70 60 50 40 30 20 10 7 6 5 3
Температура, °C 50 15 5 5 35 60 70 70 70 70 45 2 10 -3 —2
Данные определения можно проводить в пробирках.
Каждую пробирку снабжают термометром, причем для состава 11—1.5 желательно иметь точные термометры с температурным интервалом от +5 до —5°С.
Последовательность выполнения работы. Нагреть водяную баню примерно до 70° С и поочередно погружать в нее пробирки с двумя жидкими слоями. Температуру исследуемой смеси контролировать по находящемуся в пробирке термометру. При непрерывном встряхивании пробирки отмечать температуру, при которой мутная смесь внезапно становится прозрачной. Охлаждая жидкость, отмечать температуру, при которой вновь появляется помутнение. Повторить опыт с той же смесью несколько раз и найти среднее значение температуры. То же самое сделать и с другими смесями. Определенные таким путем температуры будут температурами, при которых оба компонента растворяются друг в друге в той пропорции, в которой они содержатся во взятой для опыта пробирке.
В тех пробирках, в которых находится раствор, представляющий собой одну фазу, опыт начинать с охлаждения и признаком гетерогенности следует считать выделение кристаллов фенола или льда (или помутнение раствора). При малых концентрациях фенола определение следует вести при помощи охладительной смеси (смесь соли со снегом). Если приходится работать с одним термометром, то рекомендуется менять состав исследуемой смеси, приливая воду к первоначально взятому количеству фенола. Результаты каждого опыта записывать в таблицу по образцу:
Температура, °C
Номер пробирки
Количество фенола, %
гомогенизации
гетерогенизации
Средняя температура, °C
Затем экспериментальные данные обработать графически. На ось абсцисс нанести состав, а на ось ординат — температуру (среднюю), отвечающую равновесию соответствующих фаз.
Изучение взаимной растворимости фенол — вода можно проводить в приборе, схема которого приведена на рис. 95.
Последовательность выполнения работы. Необходимо взять 25— 30 г фенола и поместить его в прибор /, куда уже должна быть опущена магнитная мешалка. Прибор 1 заключен в стеклянную рубашку 2, куда из водопровода через медный змеевик, нагреваемый газовой горелкой, поступает вода. Циркуляцией воды достигается
213
нагревание или охлаждение смеси исследуемого состава. Температура фиксируется термометром 4, который закреплен в пробке и опущен в исследуемую смесь. Смесь в продолжение всего опыта перемешивается электромагнитной мешалкой 3. Навеску фенола, помещенную в прибор /, нагревать и при помощи термометра 4 определить температуру исчезновения последнего кристалла: затем в прибор из бюретки прилить 2—3 мл воды, смесь охладить и снова
нагреть до исчезновения последнего кристалла и записать эту тем-
Рис. 95. Схема установки для изучения фазового состояния в системе с ограниченной растворимостью двух жидкостей
пературу, затем вновь прилить из бюретки 2—3 мл воды и повторить опыт.
Если при составе, при котором наступает ограниченная растворимость, т. е. при охлаждении, появляются не кристаллы, а эмульсия, смесь начать постепенно нагревать, непрерывно наблюдая за цветом. По достижении температуры, при которой наступает взаимное растворение, смесь становится прозрачной. В момент исчезновения мути зафиксировать температуру. Далее опыт следует вести в обратном направлении, т. е. смесь охлаждать, и в мо-
мент появления помутнения — появления первых капелек второй фазы — температуру снова зафиксировать. Разница температур не должна превышать 0,2°. Установ-
ленная температура есть температура взаимной растворимости фенола в воде. Далее смесь разбавить водой. В области, близкой к
чистой воде, свойства этой системы надо определять криоскопическим методом. Для этого в пробирку, заключенную в воздушную рубашку, поместить чистый растворитель, т. е. воду. Рубашку, в свою очередь, поместить в охладительную смесь. По термометру Бекмана отмечать температуру кристаллизации чистой воды. Затем в пробирку поместить водный раствор фенола (например, 3%-ный). По термометру Бекмана фиксировать температуру кристаллизации данного состава. По разности температур кристаллизации смеси и чистой воды (Д/=/н2о—Дм) определить температуру замерзания раствора. Таким способом определить температуру замерзания 3, 5 и 7%-ных водных растворов. Опытные данные записать в таблицу по образцу:
Количество фенола, г Количество воды, мл Массовое содержание смеси (по фенолу), % Температура, сС Средняя температура, °C
гомогенизации гегерогени-зации
214
На основании полученных данных построить диаграмму состояния температура — состав. Используя данные криоскопических определений и концентрацию фенола, рассчитать молекулярную массу растворенного фенола.
Работа 2. Определение коэффициента распределения
В данной работе определяется коэффициент распределения вещества между двумя различными растворителями. Одним из растворителей является вода. Исследуемый компонент, например уксусная кислота, должен быть хорошо растворим в воде. Изучение распределения уксусной кислоты между водой и органическим растворителем— бензолом, производится при двух температурах: при комнатной (которую необходимо определить и записать) и выше комнатной на 20°.
Последовательность выполнения работы. Приготовить четыре раствора уксусной кислоты в воде примерно следующих концентраций: 1,2; 0,9; 0,6 и 0,3 г-экв/л. Пипеткой отобрать 50 мл каждого раствора и поместить в отдельную колбу с притертой пробкой вместимостью 200—250 мл, туда же пипеткой добавить 50 мл бензола. Колбы поместить на аппарат для встряхивания и перемешивать в течение 40 мин. Затем содержимое колбы выдержать 20—25 мин для разделения слоев или смесь перелить в делительную воронку и через несколько минут отделить водный слой от неводного.
' При другой температуре поступить аналогично, только растворы и бензол в том же количестве поместить в делительную воронку, снабженную термостатирующей рубашкой, куда из ультратермостата поступает вода. Эти делительные воронки плотно закрыть пробками и закрепить на аппарате для встряхивания. Встряхивание растворов должно производиться в течение 30—40 мин. После встряхивания колбы или делительные воронки выдержать в течение 15—20 мин. После этого пипетками на 10 мл взять по три пробы из верхнего и нижнего слоев. При взятии проб нужно следить, чтобы не попали капельки из другого слоя. Для каждого слоя требуется отдельная пипетка. Чтобы отобрать пробу из нижнего слоя, необходимо закрыть пальцем верхнее отверстие пипетки и опустить ее через верхний слой; при этом капля верхнего слоя все же попадает в носик пипетки. Чтобы эту каплю удалить, необходимо, не открывая верхнего отверстия пипетки, нагреть ее рукой; расширяющийся воздух вытолкнет каплю. Если это не удается, тогда слегка выдуть каплю.
Каждый из растворов при достижении равновесия титровать по три раза и взять средний результат. На этом опыт закончить. Водный раствор титровать 0,5 н. раствором щелочи в присутствии фенолфталеина.
Пробы из органического слоя должны отбираться при помощи груши (ртом засасывать не разрешается!). Пробу поместить в колбу с притертой пробкой, туда добавить пипеткой 25 мл примерно 0,03 н. раствора щелочи и несколько капель фенолфталеина.
215
Смесь энергично встряхивать в течение 1—2 мин; при этом она должна приобрести малиновую окраску (избыток щелочи). Избыток щелочи оттитровать 0,02 н. НС1. Предварительно следует выяснить, какое количество соляной кислоты идет на титрование 25 мл щелочи. Нормальность кислоты в органическом слое рассчитать по формуле
ЛСН8СООН “ “ псм)
где Псм — количество кислоты, израсходованное на титрование смеси, мл; Л^нс1 — нормальность соляной кислоты; — количество НС1, израсходованное на нейтрализацию 25 мл щелочи, прибавляемой к пробе из бензольного слоя, мл. Полученные результаты свести в таблицу по обращу:
Раствор
Количество щелочи, израсходованное на титрование, мл
водного слоя
Концентрация СЩСООН в водном слое,
СН20
Количество щелочи, израсходованное на титрование, мл бензольного слоя
Концентрация СНзСООН в бензольном слое,
При расчетах следует учитывать, что уксусная кислота, как и другие карбоновые кислоты в неполярных растворителях, существует главным образом в виде двойных молекул
СН3С
образованных за счет водородных связей. В димерной молекуле уксусной кислоты есть две водородные связи.
В воде уксусная кислота не димеризуется, так как вода разрывает димерные молекулы и сама образует водородные связи с уксусной кислотой. В воде в незначительной степени молекулы уксусной кислоты диссоциируют. В данной работе диссоциацией уксусной кислоты можно пренебречь.
Пусть концентрация кислоты в водном слое с1, а в бензольном — с11, концентрация мономерных молекул с&, димерных саа. Равновесие между мономерными и димерными молекулами характеризуется константой равновесия
Ki = (V )7с”. (Х.Ю)
а равновесия между мономерными молекулами в водном' слое и органическом
К2 = с^1с\. (Х.11)
216
При титровании растворенной в бензоле кислоты щелочью оттит-ровываются обе ее формы:
^=^+2^. (Х.12)
Концентрация кислоты в водном слое составит с1=са. Разделив концентрации кислоты в водном и органическом слое друг на друга, получим
== (с!1 (Х.13)
Решив совместно уравнения (Х.11) и (Х.12), получим
(X-14)
По полученным данным необходимо: 1) определить константы распределения между водой и бензолом и теплоты димеризации уксусной кислоты в соответствующем неполярном растворителе; 2) определить константы распределения между водой и бензолом и теплоты димеризации бензойной кислоты в соответствующем неполярном растворителе.
Работа 3. Определение коэффициента распределения иода между органическими и неорганическими растворителями
В работе следует определить коэффициент распределения иода между водой и органическим растворителем. В качестве органического растворителя можно использовать бензол, толуол, ксилол, сероуглерод, эфир, хлороформ, дихлорэтан, четыреххлористый углерод и др.
Растворяемое вещество иод плохо растворяется в воде, но достаточно хорошо в другом растворителе. Изучение равновесия распределения иода между водой и органическим растворителем, например бензолом, производится при комнатной температуре (которую необходимо определить и записать) и при температуре выше комнатной на 20°.
Последовательность выполнения работы. Взять 0,05 н. раствор 12 в органическом растворителе. Приготовить четыре смеси из раствора, чистого растворителя и воды:
Номер смеси 1 2 3 4
Количество, мл раствора 12 в органическом растворителе 5 10 12 15
органического растворителя 15 10 8 5
воды 100 100 100 100
Если опыт проводится при комнатной температуре, смеси поместить в отдельные колбы вместимостью 200—250 мл с притертыми пробками, если опыт проводится при повышенных температурах, смеси поместить в делительные воронки с термостатными рубашками.
Приготовленные таким образом смеси, помещенные в колбы вместимостью 200—250 мл или в делительные воронки и закрытые
217
пробками, встряхивать на аппарате для встряхивания 30—40 мин. По окончании встряхивания колбы (или делительные воронки) оставить на 15—20 мин для расслоения жидкостей. Затем содержимое колб вылить в делительную воронку (без термостатной рубашки), отделить водный слой от неводного и определить содержание иода в обоих слоях титрованием гипосульфитом или, не отделяя водный слой от неводного, отобрать пробы пипеткой из водного слоя и органического слоя (как было описано в работе 2) и оттитровать.
Для определения концентрации иода в органическом слое взять пипеткой 1—5 мл пробы и перенести в колбу для титрования, содержащую 25 мл дистиллированной воды, и титровать 0,05 н. ЫагЗгОз в присутствии крахмала*. Для определения концентрации иода в водном слое пипеткой 20—25 мл отобрать пробы и титровать 0,001 н. Na^SsOs в присутствии крахмала. Каждый раствор титровать три раза и взять средний результат. Результаты опытов записать в таблицу по образцу:
Температура опыта . . .
Органический растворитель . . .
Водный слой
Неводный слой
Номер смеси
Объем взятой пробы для титрования, мл
Количество 0,001 н. NasSgOa, израсходованное на титрование, мл
Концентрация иода в водном слое с2
Объем взятой пробы для титрования, мл
Количество 0,001 н. Na2S2O3, израсходованное на титрование, мл
Концентрация иода в неводном слое с2
Поскольку трудно предсказать молекулярную массу растворенного вещества в обоих растворителях, для ее расчета следует пользоваться уравнением (Х.З).
Для расчета константы распределения К необходимо знать равновесные концентрации с2 в водном и с{ в органическом слоях и значение п. Концентрации рассчитывать по формуле cvi = v2N, гд$ N — нормальность титранта, г-экв/л; Vi — объем титруемого раствора, мл; v2 — объем раствора, израсходованный на титрование пробы, мл.
Если проведено несколько опытов (четыре-пять) с различными концентрациями иода, то экспериментальные данные нанести на
* Вода добавляется, так как гипосульфитом можно титровать только водный раствор. В процессе титрования колбу постоянно встряхивать, чтобы иод постепенно экстрагировался в водный слой. Титрование продолжать до момента обесцвечивания водного слоя.
218
график в координатах lg Ci—1g с2- Тангенс угла наклона полученной прямой будет равен п. Зная й, с2 и п, рассчитать коэффициент распределения /С для всех исследованных смесей и взять среднее значение.
В работе можно определить коэффициент распределения методом экстракции. Для этого проводят экстрагирование вещества несколькими равными порциями соответствующего растворителя. После последнего экстрагирования определяют титрованием количество оставшегося вещества и вычисляют по уравнению коэффициент распределения или исходное количество растворенного вещества:
где g0 — начальное количество экстрагируемого вещества; щ — объем раствора, из которого экстрагируется вещество; v2 — объем растворителя, потребляемый на одно экстрагирование; п — общее число экстрагирований; g — количество вещества, остающееся в первоначальном растворе после п экстрагирований; К — коэффициент распределения экстрагируемого вещества.
Определение коэффициента распределения какого-либо вещества между двумя несмешивающимися растворителями позволяет рассчитать активность этого вещества в соответствующем растворителе. На основании закона распределения можно определить активность:
/< = а1/а2,
где К — коэффициент распределения; ал и а2 — активности растворенного вещества в двух соприкасающихся растворителях. При бесконечном разбавлении раствора обе активности равны соответствующим концентрациям, по которым определяют /С Если значение коэффициента распределения велико, то при увеличении общего количества растворяемого вещества второй раствор может оставаться идеальным вплоть до насыщения. Тогда закон распределения можно записать так:
Работа 4. Изучение взаимной растворимости в трехкомпонентной системе
В данной работе необходимо построить треугольную диаграмму взаимной растворимости трех жидкостей, а также определить составы сопряженных растворов, направление коннод и критический состав трехкомпонентной жидкой системы. Взаимную растворимость в трехкомпонентной системе определяют методами титрования и анализа сопряженных растворов.
Метод титрования. Для построения диаграммы взаимной растворимости трех жидкостей при постоянной температуре часто пользуются методом титрования. Этот метод заключается в том, что,
219
в
Рис. 96. Диаграмма растворимости в трехкомпонентных системах
приготовив ряд смесей двух компонентов, например А и В, различного состава, поочередно титруют эти смеси, а также чистый компонент А третьим компонентом С до помутнения. Помутнение указывает на насыщение титруемой смеси третьим компонентом и на появление следов второй фазы. Изменение состава системы при титровании приведено на рис. 96. Если в точках bi происходит насыщение смесей компонентом'С, то линия ЬЬф^Ьз и т. д. дает кривую растворимости компонента С в смесях А и В. После этого готовят смеси другой пары компонентов (В и С) разного состава и титруют их, а также компонент С — компонентом А до помутнения. Изменение состава системы при таком титровании будет происходить по лучам с-А. Если насыщение смесей компонентом А наступит в точках йг-, то линия ddxdzdz ... di дает кривую раство-с римости компонента А в смесях В и С.
Метод титрования позволяет быстро и точно построить диаграмму растворимости трех жидкостей, но он не дает возможности установить направление коннод, состав сопряженных растворов и критический состав системы.
Метод анализа сопряженных растворов. Для изучения взаимной растворимости трех жидкостей методом анализа сопряженных растворов необходимо приготовить несколько двухфазных трехкомпонентных смесей разного состава, выдержать смеси до состояния равновесия и проанализировать сопряженные растворы в каждой из смесей.
Чтобы получить диаграмму взаимной растворимости трех жидкостей, наносят на треугольную диаграмму точки, соответствующие составам исследованных сопряженных растворов, и соединяют эти точки плавной кривой. Точки, выражающие суммарный состав каждой смеси и составы соответствующих двух сопряженных растворов, должны лежать на одной конноде (рис. 96).
Этот метод более трудоемок, чем метод титрования, но зато позволяет не только построить бинодальную кривую, но и определить направление коннод. Согласно правилу Тарасенкова продолжения коннод пересекаются приблизительно в одной точке. Пользуясь этим правилом, легко найти по диаграмме состав сопряженных растворов для любой двухфазной системы, а также и критический состав системы. Возьмем три вещества А, В и С, которые дают всего одну пару частично смешивающихся друг в друге жидкостей (например, В в С), а другие две пары (А в В и А в С) пол-
220
костью растворимы в любых соотношениях. Примером последней смеси могут служить следующие вещества:
Ацетон, метанол, этанол, уксусная кислота
Бензол, толуол, хлороформ, ксилол, четыреххлористый углерод
Вода
Если слить вместе бензол и воду, то получится два слоя, из которых верхний будет представлять собой насыщенный раствор воды в бензоле (при 20°С содержит сотые доли процента). Нижний— насыщенный раствор бензола в воде. Третий компонент — ацетон (или спирт, уксусная кислота и др.), в любом процентном содержании смешанный отдельно с водой или бензолом, полностью растворяется в них. Смешивают две полностью растворимые друг в друге жидкости, доливают к ним третью (которая в одном из двух также растворяется полностью, а в другом частично) до появления мути, что означает переход однофазной смеси (всех трех жидкостей) в двухфазную.
Последовательность выполнения работы. Приготовить восемь конических колб вместимостью 25 мл с притертыми пробками. Заполнить одну бюретку веществом А, другую — веществом В и приступить к составлению двухкомпонентных смесей А и В в следующем составе:
Номер пробирки
Объемное содержание компонентов, %
Объем исходных компонентов, мл
Объемное содержание;
С в смеси А+В-гС, %
2
3
4
5
6
7
8
30
45
60
70
80
85
90
95
70
55
40
30
20
15
10
5
Количество миллилитров А и В в каждой колбе следует рассчитать так, чтобы в сумме их всегда было 5 мл (А мл + В мл —5 мл). После этого приступить к главной и наиболее ответственной части работы: последовательно смесь каждой колбы титровать водой до появления мути. Для смесей в первых колбах иногда достаточно одной-двух капель воды, поэтому для точности титрование следует проводить из микробюретки. После каждой прилитой капли колбу закрывать пробкой и тщательно встряхивать, наблюдая появление
221
мути. Если муть, исчезающая при стоянии, вновь появляется при повторных встряхиваниях, значит титрование окончено. Количество израсходованной воды записать в таблицу (С мл).
Вычертить график и выделить гетерогенную зону (частично нерастворимую), расслаивающуюся при стоянии. Для этого следует пересчитать миллилитры воды на объемное содержание в процентах, отнесенных к трехкомпонентной системе, по уравнению
С х мл С-100
rl2° 5 мл (А + В) -{- х мл С
Данные расчета также записать в таблицу. При этом нет необходимости пересчитывать на объемное содержание в процентах количества А и В.
Для нанесения точек на график следует лишь каждую точку линии АВ (соответствующую смеси А и В различного процентного состава) соединить с вершиной треугольника С. На этих линиях следует откладывать объемное содержание Н2О в процентах. На линии АВ имеется количество С, равное 0%, и каждая следующая горизонтальная линия над стороной АВ соответствует приросту воды на 5%. Соединив точки, получить кривую линию, идущую от В к С и направленную выпуклостью к А. Область, лежащая в правой части треугольника и ограниченная линией, образованной точками, соответствущими концентрациям начала появления мути, является областью гетерогенной, т. е. той областью, где количество ацетона, например, в системе ацетон — бензол — вода недостаточно, чтобы он, растворив в себе бензол и воду, мог образовать однофазную прозрачную жидкость.
Область же, лежащая слева и примыкающая к точке А, является однофазной областью, т. е. областью полной растворимости всех трех компонентов друг в друге. Из-за различной растворимости А в В и С кривая может быть несимметричной и критическая точка смещения не всегда будет совпадать с максимумом кривой.
Поскольку работа выполняется при комнатной температуре, необходимо избегать резких колебаний температуры титруемых смесей. Для этого следует колбу при титровании держать только за горло, избегая нагревания содержимого колбы рукой. Часто при смешении жидкостей выделяется или поглощается значительное количество теплоты. Это может привести к заметному повышению или понижению температуры смеси при титровании, особенно если добавление титрующей жидкости производится быстро. В этих случаях необходимо титрование вести как можно медленнее, чтобы температура смеси могла достигнуть комнатной температуры. Для ускорения достижения комнатной температуры колбу перед концом титрования следует погрузить на несколько минут в большой сосуд с водой, имеющей комнатную температуру. Иногда конец титрования бывает недостаточно четким из-за малой скорости растворения добавляемой жидкости в почти насыщенной ею смеси. Чтобы добиться точного результата, надо, оттитровав смесь до появления
222
мути, выдержать несколько минут, интенсивно перемешивая содержимое колбы. Если при этом муть исчезнет, то надо добавить еще каплю титранта и снова выждать несколько минут, перемешивая смесь. Титрование считать законченным тогда, когда муть перестанет исчезать.
ГЛАВА XI
РАВНОВЕСИЕ ЖИДКОСТЬ— КРИСТАЛЛИЧЕСКАЯ ФАЗА
1. ПОСТРОЕНИЕ ДИАГРАММ СОСТОЯНИЯ !
КРИСТАЛЛИЧЕСКАЯ ФАЗА — ЖИДКОСТЬ
Если два вещества смешать друг с другом в определенных пропорциях и смесь нагреть до высокой температуры, то, как правило, образуется однородная жидкость, представляющая собой раствор одного компонента в другом. Некоторые системы образуют два жидких слоя взаимно насыщенных растворов, и только немногие вещества не будут растворяться друг в друге ни при каких условиях. Это относится к веществам, которые не разлагаются до температуры плавления. Если такой раствор или расплав охладить, то при некоторой температуре он начинает кристаллизоваться, поскольку растворимость веществ с понижением температуры уменьшается.
При кристаллизации жидких систем могут выделяться как чистые компоненты и образуемые ими химические соединения, так и твердые растворы на основе чистых компонентов и их соединений. Кристаллизацию чистых компонентов и их соединений из жидкого раствора, а также кристаллизацию из двух несмешиваю-щихся жидкостей с образованием твердых растворов следует считать предельными случаями.
В зависимости от того, какая фаза выделяется из раствора, двухкомпонентные системы с неограниченной взаимной растворимостью компонентов в жидком состоянии могут быть разделены на следующие типы: 1) без химических соединений и твердых растворов; 2) с образованием устойчивого химического соединения (плавящегося конгруэнтно); 3) с образованием неустойчивого химического соединения (плавящегося инконгруэнтно); 4) с неограниченной растворимостью компонентов в твердом состоянии; 5) с ограниченной растворимостью компонентов в твердом состоянии.
2. РАВНОВЕСИЕ ТВЕРДЫЙ КОМПОНЕНТ — РАСТВОР
Каждая фаза двухкомпонентной системы может быть охарактеризована тремя переменными: температурой Г, давлением р и концентрацией х одного из компонентов, выраженной в моляр-
223
ных долях. Для графического изображения функции f(T, р, х) необходима система координат в пространстве трех измерений. Откладывая по трем взаимно перпендикулярным осям экспериментальные значения температур, давлений и концентраций, можно построить полную диаграмму состояния данной системы. Однако построение полных диаграмм состояния сопряжено с большими экспериментальными трудностями и пока осуществимо лишь для немногих систем. Если система конденсирована, то небольшие изменения давления мало отражаются на равновесии. Поэтому при исследовании конденсированных систем при атмосферном давлении можно без ощутимой погрешности принять давление постоянным. Если рассматривать свойства системы при p=const и выбрать это давление большим, чем давление насыщенных паров смесей любого состава, то в системе не будет газообразной фазы. Число переменных уменьшается до двух. Графически зависимость между ними может быть выполнена на плоскости в координатах Т— х Построенную диаграмму называют диаграммой состояния (диаграммой плавкости).
Диаграммы состояния изображают равновесия между всеми жидкими и твердыми фазами, образуемыми компонентами системы при различных концентрациях и температурах и постоянном давлении. При построении этих диаграмм системы изучаются (поскольку это возможно) во всем интервале концентраций, т. е. ют чистого вещества А до чистого вещества В. Интервал температур, в котором проводится исследование, выбирается так, чтобы на диаграмме отражалось как равновесие жидких фаз с другими жидкими и твердыми фазами, так и превращения, протекающие в системе ниже температуры ее полного затвердевания.
3. МЕТОДЫ ИЗУЧЕНИЯ ГЕТЕРОГЕННЫХ СИСТЕМ
К методам, применяемым для построения диаграмм состояния, относятся методы растворимости, термического анализа, определения микроструктуры, рентгено-, электроно- и нейтронографии.
Метод растворимости основан на достижении состояния равновесия и аналитическом определении состава жидкой фазы, находящейся при постоянной температуре в равновесии с твердой фазой, и состава твердой фазы, выделенной из жидкости. По полученным данным строят диаграммы растворимости в координатах температура— состав насыщенных растворов.
Метод термического анализа является способом установления температур при равновесии между жидкими и твердыми фазами, который не требует ни механического разделения, ни химического анализа. Методы термического анализа в основном различаются способами регистрации температурных изменений, соответствующих фазовым переходом. Основные способы: 1) визуальный, при котором отмечается температура появления (или исчезновения)
224
кристаллов в прозрачном растворе; 2) построение кривых охлаждения (или нагревания).
В способе построения кривых через небольшие промежутки времени измеряют с помощью термометра или термопары температуру непрерывно охлаждающейся (или нагревающейся) системы. Результаты измерений наносят на график, откладывая по оси абсцисс время, а по оси ординат — температуру. Запись изменения температуры со временем может быть осуществлена автоматически. Если при непрерывном изменении температуры система не претерпевает никаких фазовых превращений, сопровождающихся выделением или поглощением теплоты, то ее температура является непрерывной функцией времени. Поэтому зависимость температу-
Бремя
Рис. 97. Кривые охлаждения:
1 — при отсутствии фазовых превращений; 2 — кристаллизация при постоянной температуре; 3, 4 — кристаллизация в температурном интервале;
5 — кристаллизация с переохлаждением
ры системы от времени изображается непрерывной кривой (рис. 97, кривая /). Если же при охлаждении (нагревании) в системе происходят какие-либо превращения, например выпадение твердой фазы из жидкости, переход одной полиморфной модификации в другую, то теплота, выделяющаяся (поглощающаяся) при превращении, изменяет скорость падения (роста) температуры системы, что выражается изменением углового коэффициента кривой температура — время. Поэтому в промежутки времени, отвечающие температурам фазовых превращений, на кривых температура— время появляются изломы или горизонтальные участки, которые позволяют определять температуры превращений, не видя и не выделяя фаз, образующихся или исчезающих при охлаждении (нагревании) системы.
Если какое-либо чистое вещество (металл, соль, органическое соединение) нагреть до температуры в точке а, лежащей выше точки его плавления, а затем записывать кривую охлаждения, то вначале падение температуры от времени будет выражаться плавной кривой ab (кривая 2). В некоторый момент времени t\ в точке b понижение температуры прекратится. На кривой появляется го
8—2083 225
ризонтальный участок be. Это указывает на то, что в. системе происходит процесс, сопровождающийся выделением теплоты. На горизонтальном участке be скорость охлаждения становится равной ’ нулю и сохраняет это значение до времени /2, начиная с которого кривая вновь плавно идет вниз до температуры в точке .d, при которой запись кривой охлаждения прекращается. Появление горизонтального участка Ьс объясняется тем, что происходит переход вещества из жидкого состояния в твердое. Выделяющаяся при этом теплота кристаллизации возмещает потерю теплоты в окружающую среду, вследствие чего до окончания затвердевания температура держится на одном уровне. Из правила фаз следует, что при постоянном давлении чистое вещество, распределенное между двумя фазами (твердой и жидкой), не имеет ни одной степени свободы, т. е. кристаллизация протекает при постоянной температуре.
Рассмотрим двухкомпонентные системы, когда на кривой охлаждения имеется одна горизонтальная остановка. Из правила фаз следует, что если при постоянном давлении в системе из двух компонентов в равновесии находятся три фазы, то система не имеет ни одной степени свободы. Таким образом, горизонтальный участок на кривой охлаждения двухкомпонентной системы указывает на то, что при температурной остановке в равновесии находятся три фазы (две твердые и одна жидкая). Если кристаллизующаяся твердая фаза (твердый раствор, чистый компонент или определенное соединение) отличается по составу от существующей с ней жидкости, то при охлаждении жидкой фазы от начальной температуры в точке а до температуры начала кристаллизации в точке b (кривая 3) кривая охлаждения плавно идет вниз. В момент появления твердой фазы, вследствие выделения теплоты кристаллизации, скорость охлаждения уменьшается. Поэтому на кривой охлаждения в точке b появляется излом, отвечающий температуре начала кристаллизации. При этом число степеней свободы уменьшается на единицу, система из дивариантной становится моновариантной. Если на протяжении всего процесса кристаллизации в равновесии с жидкой фазой находится только одна твердая фаза, то затвердевание заканчивается при температуре в точке Наблюдаемый при этой температуре второй излом на кривой охлаждения отвечает полному исчезновению жидкой фазы и, следовательно, приобретению одной степени свободы, система из моновариантной становится дивариантной. Однако если в конце кристаллизации появляется еще одна твердая фаза, кроме той, которая выделилась первично, то система теряет еще одну степень свободы и затвердевание заканчивается инвариантным равновесием, которому отвечает горизонтальный участок се (кривая 4). По окончании затвердевания система, состоящая из двух твердых фаз, имеет одну степень свободы, охлаждение ее идет по плавной кривой и заканчивается при температуре в точке d.
Система, отдавая теплоту окружающей среде, проходит ряд последовательных состояний равновесия и ее температура во всех
226
точках одинакова. Однако на практике эти условия никогда не могут быть полностью соблюдены, и экспериментально получаемые кривые охлаждения всегда в той или иной мере отклоняются от идеального хода. Одной из наиболее частых причин таких отклонений является переохлаждение. При переохлаждении температура в точке а плавно падает ниже точки равновесной кристаллизации bf, В точке bf наблюдается неустойчивое переохлаждение системы и поэтому вследствие самопроизвольного возникновения центра кристаллизации начинается выпадение твердой фазы. Выделяющаяся теплота быстро повышает температуру, которая при благоприятных условиях (больших теплоте кристаллизации и скорости роста кристаллов) поднимается до температуры равновесной кристаллизации, отвечающей остановке Ьс на кривой 5. При малой скорости кристаллизации температура может и не достигнуть уровня горизонтального участка Ьс (кривая 5).
Кроме переохлаждения одной из причин отклонения кривых охлаждения от идеального хода является неравномерность распределения температуры по объему застывающей среды. Вследствие температурного градиента линии Ьс отклоняются от горизонтального направления вниз. Поэтому на кривых охлаждения систем, затвердевающих в некотором температурном интервале, излом, отвечающий температуре конца затвердевания, нередко бывает выражен нечетко. Более достоверные данные получают с помощью кривых нагревания, так как твердое кристаллическое вещество нельзя перегревать выше температуры начала его плавления. На основании кривых охлаждения строятся диаграммы зависимости температуры того или иного фазового перехода от состава системы. На основании этих диаграмм делается заключение о характере химического взаимодействия между компонентами системы.
4. НЕИЗОМОРФНЫЕ СМЕСИ С ПРОСТОЙ ЭВТЕКТИКОЙ
Рассмотрим диаграмму плавкости двух неизоморфно кристаллизующихся веществ с простой эвтектикой и проследим за изменениями, происходящими с жидкостями различного состава при их охлаждении (рис. 98).
Вещества А и В неограниченно растворимы в жидком состоянии, не образуют химических соединений, не претерпевают полиморфных превращений и кристаллизуются из жидкости в виде чистых компонентов. Системы, лежащие выше линий АЕ и ВЕ, дивариантны, так как здесь два компонента и одна жидкая фаза. Кривые АЕ и ВЕ называются кривыми ликвидуса. На этих кривых системы моновариантны. Прямая CD называется линией солидуса. В точке пересечения кривых ликвидуса Е оба твердых компонента находятся в равновесии с жидким расплавом состава, отвечающего точке Е. Ниже температуры точки Е (ниже линии CD) могут существовать только смеси твердых компонентов. Среди в* 227
всех сочетаний компонентов А и В расплав состава, отвечающего точке Е, имеет наиболее низкую температуру кристаллизации. Точка Е называется эвтектической, а отвечающая ей смесь кристаллов А и В — эвтектической смесью или эвтектикой. Эвтектика— состав из двух (или нескольких) компонентов, имеющий определенную характерную структуру, образующий при плавлении расплав, насыщенный относительно всех входящих в него компонентов. Составные части эвтектики хорошо видны под микроскопом и могут быть отделены друг от друга механическим путем или с помощью растворителей.
В эвтектической точке Е и по всей линии CD (кроме крайних точек) система инвариантна, здесь три фазы: две твердые (А и В)
и одна жидкая (эвтектического состава). Область, лежащая ниже эвтектической температуры, отвечает твердым смесям кристаллов А и В, причем область АСЕО — смесям кристаллов А+эвтектика, область EDHG — смесям кристаллов В + эвтектика. Системы удовлетворяют условиям равновесия между фазой из чистого компонента и фазой раствора (рис. 98). Если раствор идеален, то граничные кривые ликвидуса будут подчиняться уравнению Шредера
где х — молярное содержание z-ro компонента, доля; АЯ,— теплота плавления z-го компонента; Т° — температура плавления г'-го компонента.
Кривые ликвидуса можно рассматривать как кривые зависимости растворимости каждого чистого компонента в растворе от температуры. Для идеальных растворов по таким кривым можно вычис-
лить теплоту плавления Л/Л каждого компонента. И, наоборот, если значения теплот плавления компонентов известны, можно рассчитать температуру кристаллизации эвтектики и ее состав. Для реальных растворов уравнение Шредера примет вид
Рассмотрим процессы охлаждения некоторых смесей и чистых компонентов.
Точка 1. Система выше точки А одновариантна, происходит непрерывное понижение температуры по закону Ньютона:
——- = /( (Т'сис Т'окр.Ср)* d t
Появление в точке А твердой фазы делает систему инвариантной. Это отвечает температурной остановке, затем идет опять охлаждение по закону Ньютона; так же ведет себя система, определяемая точкой 9,
Точка 2. Система выше точки Г двухвариантна, т. е. для характеристики подобной системы необходимо фиксировать температуру и состав.; В точке Г начинается кристаллизация вещества А. Выделение теплоты кристаллизации замедляет охлаждение системы. По мере увеличения количества твердого вещества А расплав обогащается веществом В, вследствие чего температура кристаллизации непрерывно понижается. Соотношение между количествами твердой и жидкой фаз определяется по правилу рычага. Поскольку с момента образования твердой фазы система стала одновариантной, то между температурой и составом насыщенных растворов будет существовать зависимость, которая и выражается кривой АЕ. Следовательно, отмечая температуру начала кристаллизации, тем самым устанавливают состав; наоборот, каждому составу отвечает единственная температура равновесия твердое вещество А—расплав. По достижении температуры эвтектики расплав будет насыщен обоими веществами; появляется новая фаза —твердое вещество В, и система становится инвариантной. При температуре эвтектики оба вещества выпадают в соотношении, отвечающем составу оставшейся жидкости, поэтому жидкость кристаллизуется без изменения состава. Кристаллизация эвтектического расплава изменяет состав твердой массы, так как последняя пополняется не только веществом А, но и веществом В. При исчезновении последней капли жидкости состав твердой массы совпадает с составом исходного расплава. После этого температура начинает падать, так как с исчезновением жидкости система становится одновариантной. Для точки 2 показаны процесс охлаждения, по линии ликвидуса — изменение состава жидкой фазы, по линии солидуса— изменение состава твердой массы. Твердая масса состоит из двух фаз: компонента А и компонента В. Для смеси 7 показан процесс нагревания.
229
Точка 4, Оба вещества будут кристаллизоваться одновременно, и длительность эвтектической остановки и тем самым величины горизонтального участка на кривой охлаждения будут максимальными.
Процессы охлаждения смесей, указанных точками 3, 5, 6, 7, 8, аналогичны процессу охлаждения смеси точки 2. В смеси состава точки 3 первоначально кристаллизуется компонент А; в смесях 5— 8 — компонент В.
5. НЕИЗОМОРФНЫЕ СМЕСИ, ОБРАЗУЮЩИЕ УСТОЙЧИВОЕ ХИМИЧЕСКОЕ СОЕДИНЕНИЕ
Вид диаграммы изменяется, если два компонента образуют между собой какое-либо химическое соединение (рис. 99). Диаграмму в координатах температура — состав для одного устойчивого химического соединения можно считать состоящей как бы из двух диаграмм с эвтектикой. Первая диаграмма отвечает системе
Рис. 99. Диаграмма плавкости неизоморфной бинарной системы, образующей устойчивое химическое соединение
вещество А — химическое соединение, вторая — системе химическое соединение — вещество В. В первой системе образутся эвтектика Е (А + АВ), во второй системе образуется эвтектика F (АВ + 4-А). Левее вертикали EL и правее вертикали FN происходит кристаллизация с выделением вещества А или соответственно вещества В и последующим выпадением эвтектических смесей Е и F. Правее вертикали EL и левее вертикали FN охлаждение распла-
230
bob приводит к кристаллизации химического соединения с последующим выделением эвтектик. Температура плавления химического соединения в точке С может быть выше температур плавления чистых компонентов или ниже температуры плавления более легкоплавкого компонента. Первый случай наблюдается тогда, когда молекулярное соединение образуется с большим выделением теплоты. По характеру максимума, отвечающего температуре плавления химического соединения АВ, можно судить о его прочности. Частичное разложение соединения в расплаве, происходящее при плавлении, понижает температуру плавления. Поэтому для неустойчивых соединений максимум становится менее острым. Состав химического соединения точно соответствует положению максимума на диаграмме плавкости.
6. НЕИЗОМОРФНЫЕ СМЕСИ, ОБРАЗУЮЩИЕ НЕУСТОЙЧИВОЕ ХИМИЧЕСКОЕ СОЕДИНЕНИЕ
Некоторые молекулярные соединения бывают столь неустойчивы, что разлагаются при температурах ниже температуры их плавления. Для таких соединений кривая ликвидуса не имеет максимума и в некоторой точке наблюдается перегиб. Температура в точке перегиба будет соответствовать той температуре, при которой химическое соединение разлагается. Состав соединения бу-
Рис. 100. Диаграмма плавкости неизоморфной бинарной системы, образующей неустойчивое химическое соединение
дет отвечать вертикали DM. Диаграмма плавкости со скрытым максимумом и кривые охлаждения приведены на рис. 100. В точке С присутствуют две твердые (компонент В и соединение АВ) и жидкая фазы. Излом на кривой ВЕ, определяемый точкой С, будет более или менее резко выражен. Это зависит от растворимости вещества В и соединения АВ в жидкой фазе: он будет тем глубже, чем больше разница в растворимости В и АВ. Если эти растворимости мало отличаются, то перегиб кривой на диаграмме будет
231
незаметен, и присутствие химического соединения может быть обнаружено только при процезионном проведении эксперимента.
Рассмотрим охлаждение смесей, составы которых отвечают точкам 3, 4, 5 и 6.
Точка 3. До линии СЕ охлаждение идет по закону Ньютона. На линии СЕ начинается кристаллизация химического соединения АВ из расплава, причем вследствие выделения теплоты кристаллизации охлаждение пойдет замедленно. Жидкая фаза по мере выпадения вещества АВ будет насыщаться компонентом А, и, наконец, наступит такой момент, когда она будет насыщена относительно компонента А и соединения АВ. При этом будет кристаллизоваться эвтектика на линии ЕН при постоянной температуре. После полной кристаллизации смеси охлаждение пойдет по закону Ньютона без каких-либо термических эффектов.
Точка 4. До линии ВС жидкая фаза охлаждается по закону Ньютона. На линии ВС начинается выпадение избыточного компонента В, которое снижает скорость охлаждения и продолжается до линии CG, соответствующей определенной температуре. При этой температуре возможно существование химического соединения АВ, поэтому здесь начинается его образование и выпадение из расплава. При этом твердый компонент В переходит в расплав, поддерживая его состав постоянным. В результате этого температура расплава и твердых фаз остается постоянной. Когда твердый компонент В полностью перейдет в расплав, дальнейшее выпадение химического соединения будет сопровождаться изменением состава жидкой фазы: она будет обедняться компонентом В и соответственно насыщаться компонентом А, Это вызовет падение температура до линии ЕН, на которой при постоянной температуре будет кристаллизоваться эвтектика химического соединения АВ и компонента А.
Точка 5. Состав смеси, обозначенной точкой 5, соответствует составу химического соединения АВ. Вначале процесс охлаждения идет аналогично процессу 4 до точки D. Здесь начинается образование и выпадение химического соединения АВ, обеднение жидкой фазы компонентом В и переход его в расплав. По мере того как выпадает соединение АВ, количество жидкой фазы уменьшается, при этом уменьшается также и количество твердого компонента В. В тот момент, когда исчезнут последние капли жидкости, весь твердый компонент В израсходуется, система будет состоять только из чистого твердого соединения АВ. Поэтому дальнейшее охлаждение ниже точки D пойдет по закону Ньютона без всяких термических изменений.
Точка 6. Здесь имеется большой избыток компонента В. Охлаждение до линии ВС идет по закону Ньютона, на ней и ниже ее выпадает компонент В. Компонента В выпадает большое количество и поэтому соответствующий участок кривой охлаждения длиннее, чем в процессах 4 и 5. При температуре, соответствующей линии CG, в системе будет большое количество твердого вещества В и малое количество жидкой фазы. При этой температуре начи-232
кается образование соединения АВ. При этом компонент В переходит в расплав, поддерживая постоянство состава жидкой фазы, и, следовательно, температура также остается постоянной. Когда исчезнут последние капли расплава, система будет содержать твердое соединение АВ и избыток выпавшего твердого компонента В. Образовавшийся конгломерат не является эвтектической смесью.
7. ИЗОМОРФНЫЕ СМЕСИ. СИСТЕМЫ С ОГРАНИЧЕННОЙ РАСТВОРИМОСТЬЮ В ТВЕРДОМ состоянии
Изоморфные смеси образуются веществами со сходным строением кристаллических решеток. Поэтому оба вещества при любом соотношении кристаллизуются совместно и образуют твердый раствор. Диаграмма плавкости изоморфной смеси приведена на рис. 101. На ней указаны фазовые составы, обозначенные соответствующими площадями. Все процессы охлаждения смесей по существу одинаковы. Рассмотрим один из них. При охлаждении
Рис. 101. Диаграмма плавкости изоморфной бинарной системы
смеси, соответствующей кривой 3, в точке появляется первый кристалл состава х2. Жидкость, теряя больше компонента В, чем А, обогащается легкоплавким компонентом; поэтому температура ее отвердевания понижается. Составы жидкой и твердой фаз изменяются соответственно по кривым ликвидуса и солидуса. По мере понижения температуры (начиная с охлаждение замедляется, процентное содержание твердой фазы растет (это можно определить по правилу рычага). При t% затвердевает последняя капля расплава (состав х3) с образованием кристалла состава х0; после этого происходит охлаждение твердого раствора. Процесс охлаждения на рис. 101 указан стрелками. Смеси составов, соответствующих кривым 2, 4, ведут себя аналогично.
233
Если система дает твердые растворы с ограниченной растворимостью, то, подобно жидким растворам, растворителем считается тот компонент, количество которого в растворе больше. Таким образом, для двух веществ возможны два типа растворов, называемых твердыми а- и p-растворами. Так, на диаграмме состояния (рис. 102) твердые растворы А в В будут называться р-кристалла-ми, а твердые растворы В в А — а-кристаллами.
Рассмотрим процессы охлаждения некоторых расплавов.
Точка 2. Система состоит из большого количества компонента А с незначительной примесью компонента В. До точки Ь2 охлажде-
Рис. 102. Диаграмма плавкости бинарной системы с ограниченной растворимостью в твердом состоянии
ние идет по закону Ньютона. В точке Ь2 начинается выпадение твердого раствора В в А (a-кристаллы), причем охлаждение идет с задержкой. Первые порции будут содержать лишь ничтожное количество компонента В, которым твердый раствор будет обогащаться по мере охлаждения. Так как количество компонента В мало, то раствор очень быстро затвердеет и ниже кривой АС никаких изменений не произойдет.
Точка 3. В точке Ь3 начинает выпадать твердый раствор В в А (а-кристаллы). На кривой АС процесс кристаллизации закончится, и ниже раствор будет охлаждаться по закону Ньютона. При охлаждении растворимость компонента В в А в твердой фазе уменьшается и твердый раствор в точке становится насыщенным. Он распадается на два раствора, насыщенных а- и р-кристал-лами, т. е. появляется новая твердая фаза. Чем сильнее охлаждается система, тем меньше взаимная растворимость компонентов друг в Друге в твердом состоянии, тем больше становится количество насыщенных p-кристаллов и меньше насыщенных а-кристал-лов. В твердом состоянии охлаждение идет по закону Ньютона, однако тепловые эффекты растворимости столь ничтожны, что на кривых охлаждения не проявляются.
234
Точка 4. В точке Ь4 начинается выпадение a-кристаллов, причем по мере охлаждения их состав изменяется по кривой АС, а состав жидкой фазы —по кривой Ь^Е, Когда температура упадет до линии СЕ, состав твердой фазы будет соответствовать точке С, т. е. составу насыщенных a-кристаллов, а состав жидкой фазы — точке Е, т. е. такой жидкости, из которой может выпадать только эвтектика из насыщенных а- и fj-кристаллов. Поэтому здесь эта эвтектика и выпадает при постоянной температуре. Ниже линии СЕ охлаждение пойдет по закону Ньютона с ничтожным отклонением вследствие понижения растворимости компонентов. Часто наблюдается независимость взаимной растворимости компонентов в твердом состоянии от температуры. Тогда прямые CF и DS идут строго вертикально. Площадь CFSD, соответствующая насыщенным растворам, зависит от того, хорошо или плохо растворяются компоненты друг в друге.
Наряду с методами термического анализа используется метод микроструктуры. Метод микроструктуры заключается в том, что соответствующим образом подготовленный образец рассматривается под микроскопом. Микроскопическое исследование позволяет установить число и взаимное расположение фаз и последовательность кристаллизации в системе. Сопоставляя эти данные с полученными методами термического анализа, можно проверить и дополнить заключения о характере взаимодействия компонентов системы и границах фазовых областей.
Другим современным методом, служащим для построения диаграмм состояния, является метод рентгеноструктурного анализа. Рентгеноструктурный анализ является одним из наиболее совершенных методов изучения всех превращений, сопровождающихся изменением кристаллической решетки. Поэтому он особенно полезен при исследовании полиморфных превращений, образования и распада твердых растворов, а также образования химических соединений. Методами рентгеноструктурного анализа изучают металлы, сплавы, минералы, неорганические и органические соединения. Рентгеноструктурный анализ применяется для качественного и количественного фазового анализа гетерогенных систем, для исследования изменений в твердых растворах, определения типа твердого раствора и границ растворимости. Рентгеноструктурный анализ является дифракционным структурным методом; он основан на взаимодействии рентгеновского излучения с электронами вещества, в результате которого возникает дифракция рентгеновского излучения. Основную информацию в рентгеноструктурном анализе получают из рентгенограмм. Типы рентгенограмм сильно зависят от природы и состава фаз. Между типом рентгенограммы и типом диаграммы состояния существует определенная связь. Особенно полезны рентгенографические данные для построения той части диаграмм, которые описывают равновесные процессы в твердом состоянии, где процессы установления равновесных состояний протекают очень медленно.
В настоящее время для физико-химического анализа широко
235
применяются электроно- и нейтронография. Электронография имеет ряд особенностей. Электронное излучение имеет малую длину пробега. В толще кристалла оно сильно поглощается. Поэтому электронография позволяет исследовать поверхностные слои кристаллов. Благодаря этой особенности электронографически можно исследовать очень мелкие кристаллы, зародыши новых фаз, не доступные рентгенографии. Электронография дает сведения о положении в решетке легких атомов (даже таких, как водород). Она также позволяет проводить исследования при значительно меньших экспозициях по сравнению с рентгенографическими.
Работа 1. Изучение кристаллизации из растворов при высоких температурах
В данной работе следует ознакомиться с методом термического анализа и построением диаграммы плавкости двойной системы, компоненты которой (и их химическое соединение, если оно существует) практически нерастворимы друг в друге в твердом состоянии.
Для изучения систем с высокими температурами плавления пользуются термопарой (рис. 103), которая состоит из двух металлических проволок 1 и 2 (например, медной и константановой),
Рис. 103. Схема термопары
4 Рис. 104. Калибровочная кривая термопары
т8
Температура
спаянных в точке 5 (горячий спай). Проволоки изолируют друг от друга фарфоровой или стеклянной трубками 7 и помещают в фарфоровый или металлический кожух 6 так, чтобы спай не касался дна чехла. Концы проволок выводят наружу, и холодный спай 4 (константан с медью) погружают в тающий лед 3. Возникающая э. д. с. пропорциональна разности температур между горячим и хо
236
л одним спаями и измеряется милливольтметром 8 или компенсационным методом.
Градуирование термопары производят по известным температурам плавления чистых веществ. При градуировании термопары строят калибровочный график. По оси абсцисс откладывают истинные температуры плавления чистых веществ, а по оси ординат— показания милливольтметра при их кристаллизации. Полученные точки соединяют прямой (рис. 104), которая служит для перевода показаний прибора в истинные значения температуры. Для калибрования часто применяют металлы, соли и органические вещества; температуры плавления некоторых из них приведены ниже.
Вещество Zn Pb Sn К2СГ2О7 KNO3 NaNO3 Нафталин
Т. пл., °C 419 327 238 398 337 310 80
Вещество Дифенил С6Н5ОН
- - Т. пл., °C 56 41
(• •
Выбрав подходящие вещества для нужного интервала температур, расплавляют их в тиглях или пробирках и записывают их г кривые охлаждения. На кривых охлаждения находят остановки, отвечающие температуре кристаллизации вещества, и строят по у ним калибровочную кривую.
; Последовательность выполнения работы. Измерение температу-
I ры при работе с солевыми или металлическими сплавами произ-
I вести при помощи термопары, присоединенной к гальванометру
или включенной в компенсационную схему. Исследуемую смесь L солей или металлов поместить в фарфоровый тигель. Тигель по-ставить в электрическую печь, включить ее и расплавить смесь, стараясь не перегревать ее выше температуры плавления. Перемешать сплав, выключить печь и опустить в сплав горячий спай термопары. Закрепить термопару в штативе. Конец термопары должен находиться в расплаве, почти у дна тигля и не касаться сте-нок тигля.
Когда начнется охлаждение, записать показания гальванометра или другого прибора через равные промежутки времени (15 с). Причем один из наблюдающих следит за временем и ведет запись по прибору. Затем вновь нагреть печь до расплавления сплава и вынуть из него термопару (это делается в том случае, если имеется одна термопара). После этого тигель со сплавом вынуть из печи и на его место поставить другой со следующей смесью. Далее все операции повторить в указанной последовательности. Кроме кривых охлаждения сплавов получить кривые охлаждения чистых компонентов.
По окончании исследования всех сплавов построить для каж-дого из них кривую охлаждения в координатах время — показа-; ние милливольтметра. На кривых охлаждения отметить точки, от-I вечающие кристаллизации сплава. Это будут горизонтальные или
[ слегка наклонные кривые для моновариантных процессов (кри-
I ' 237
УТ; •
сталлизация чистых веществ или кристаллизация эвтектики) и кривые с перегибами или изломами для дивариантных процессов. По калибровочной кривой термопары определить температуры точек, отвечающих кристаллизации каждого сплава.
По полученным данным построить диаграмму состояния системы. По оси абсцисс откладывать состав сплавов, а по оси ординат— температуру начала кристаллизации сплава и кристаллизации эвтектики.
Работа 2. Построение диаграммы плавкости системы
KNO3—NaNO3
Температуру измеряют медно-константановой термопарой. Э. д. с. для данной пары металлов пропорциональна разности температур горячего и холодного спаев; э. д. с. измеряется компенсационным методом (рис. 105). Э. д. с. термопары (термоэлектродвижущая сила) компенсируется реохордом. Питание реохорда осуществляется от аккумулятора; термопара присоединяется в боковую цепь так, чтобы направление тока было обратным направлению тока от аккумулятора. Перемещая подвижной контакт реохорда, находят то его положение, при котором исследуемая э. д. с. будет точно компенсироваться, а гальванометр покажет отсутствие тока. Ввиду чрезвычайно малой вели-
чины термо-э. д. с. последовательно к аккумулятору присоединяют ползунковый реостат и магазин сопротивления, при помощи которых изменяется э.д. с. на определенную величину, необходимую для компенсации.
Пользуясь реостатом (магазин сопротивления не включают), добиваются такого его положения, при котором
Рис. 105. Подключение термо- компенсация э. д. с. нормального эле-пары к компенсационной схеме мента осуществляется в самом начале реохорда. Э. д. с. нормального элемента постоянна и равна 1,018 В, следовательно, если она компенси-
руется в начале реохорда, то падение напряжения на реохорде будет равно 1,018 В. Добившись компенсации нормального эле-
мента в начале реохорда, положение движка на реостате оставляют фиксированным и приступают к измерению термо- э. д. с. системы KNO3—NaNO3. Для получения диаграммы плавкости не следует производить расчет э. д. с. для каждой точки, отмечают только положение движка на реохорде при компенсации.
Последовательность выполнения работы. Для проведения работы взять набор пробирок, содержащих смеси KNO3 и NaNO3 следующих составов:
Номер пробирки
NaNO3, %
1 2 3 4 5 6 7
0 10 30 50 70 90 100
238
Собрать устайрвку по схеме, приведенной на рис. 105. Опустить холодные концы термопары в тающий лед, компенсировать э. д. с. элемента Вестона и приступить к измерениям, начиная с калибрования термопары. Взять пробирку с KNO3, погрузить в нее термопару. Пробирку закрепить в штативе и опустить в тигельную печь, не касаясь дна и стенок последней. Включить печь. После расплавления твердой фазы пробирку закрыть асбестом для равномерного охлаждения, печь выключить и при помощи магазина сопротивления (положение ползунка на реостате не изменять) подобрать такое сопротивление, чтобы максимальная температура плавления KNO3 отвечала компенсации на реохорде порядка 80—90 делениям. Дальнейшие измерения производить через 15—30 с. Один из работающих отмечает время по секундомеру, другой компенсирует и записывает показания на реохорде. После температурной остановки, отвечающей кристаллизации соли (в это время точка компенсации не изменяется), произвести еще пять-шесть измерений, затем пробирку вынуть. Аналогичные опыты проделать с пробирками 4 и 7.
На основании полученных данных построить калибровочную кривую, по которой, пользуясь показаниями реохорда, найти температуру плавления смеси. Аналогичные опыты провести со смесями всех указанных составов. По окончании кристаллизации произвести еще четыре-пять отсчетов. Показания реохорда записать в таблицу по образцу.
Время от начала опыта Показания реохорда для чистых компонентов и смесей Контрольная задача
1 2 3 4 5 6 7 8
На основании этих данных построить на миллиметровой бумаге кривые охлаждения в координатах показания реохорда (ось ординат)— время (ось абсцисс). Рекомендуемая цена делений: по оси ибсцисс 1—2 мм соответствуют 15 с, по оси ординат 1 мм реохорда соответствует 1—2 мм. Затем по калибровочной кривой определить истинное значение температур, соответствующих характерным точкам на кривых охлаждения. На основании полученных данных построить диаграмму плавкости в координатах температура— состав. Начертить схему установки.
Работа 3. Построение диаграммы плавкости системы
KNO3—NaNO3 и изучение структуры сплава
Термический анализ системы KNO3—NaNO3 производят при помощи термопары и самопишущего гальванометра. Выбор металлов термопары зависит от области рабочих температур и чувстви-
239
дельности гальванометра (самопишущего или обычного) или потенциометра.
Последовательность выполнения работы. Подготовить шесть пронумерованных тиглей, пять пробирок и пять предметных стекол. Компоненты смеси следует тщательно перемешать и измельчить в ступке. Тигли плотно наполнить солями или их смесями, составы которых приведены ниже.
Номер тигля 1 2 3 4 5
Содержание, %
KNO3 100 — 50 75 65
NaNO3 — 100 50 25 35
В шестой тигель насыпать чистый песок. Кривая охлаждения песка, в котором в процессе охлаждения в интервале температур от 400°С до комнатной температуры не происходит термических превращений, является контрольной.
Записать номера тиглей и соответствующие им составы солей. Перед тем как тигли установить в печь, из каждого отсыпать небольшое количество смеси (на кончике ножа) в пробирку для дальнейшего изучения этих солей под микроскопом. Тигли установить в печь и в каждый из них опустить хромель-алюмелевую термопару, подключенную к самопишущему гальванометру (рис. 106). Холодные спаи термопар в стеклянных чехлах поместить в тающий лед, это обеспечит постоянную температуру холодных
горячий спой
хромель алюмель
пройирки.
холодный, спай
медь медь
к самопишущему гальванометру
Рис. 106. Схема подключения термопары к потенциометру
спаев 0° С (температура смеси воды со льдом измеряется обычным термометром). Самопишущий гальванометр включить в электрическую сеть. К одному самопишущему гальванометру типа СГ-39 можно подключить одновременно не более трех термопар (рис. 107). При работе с шестью тиглями термопары подключаются к двум гальванометрам. После того как тигли установлены в печь, закрыть ее крышкой и включить в электрическую сеть.
После выключения печи сплавы медленно охлаждаются и на ленте самопишущего гальванометра вычерчиваются кривые охлаждения. Кривые нагревания, которые вычерчивались перёд кривыми охлаждения, отражают процесс нагревания в неравновесных условиях и поэтому не нужны для обработки экспериментальных результатов.
Пока смеси охлаждаются, рассмотреть под микроскопом" структуру чистых солей и эвтектических смесей, полученных другим 240
методом (см. ниже). Содержимое пробирок растворить в дистиллированной воде. Воды налить 3/4 пробирки. Количество воды не имеет значения, но растворы не должны быть концентрированными. Каплю раствора нанести на чистое предметное стекло. На стекле должен быть тонкий слой раствора. Соли KNO3 и NaNO3 не образуют кристаллогидратов и после испарения влаги на стекле остаются их осадки. Необходимо зарисовать и описать осадки, наблюдаемые под микроскопом.
По окончании процесса охлаждения необходимо построить калибровочную кривую, соответствующую данной термопаре. Зави-
Рис. 107. Схема подключения нескольких термопар к потенциометру
симость термо-э. д. с. хромель-копелевой и хромель-алюмелевой термопар от температуры в первом приближении можно рассматривать как прямолинейную. Температуры плавления веществ определить по длине отрезка на кривой охлаждения. Когда оба спая термопары находятся при одной и той же температуре (например^ при 0°С), то термо-э. д. с. не возникает. Полученные экспериментальные данные записать в таблицу по образцу:
Номер тигля Содержание смеси, % Номер пробирки Показание термопары, мВ Температура кристаллизации, °C Вид под. микроскопом
массовое молярное начало конец
По полученным данным построить диаграмму плавкости системы.
Работа 4. Изучение кристаллизации веществ из растворов при низких температурах
В данной работе следует ознакомиться с методом термического анализа и построить диаграмму плавкости двойной системы, компоненты которой (и их химическое соединение, если оно существует) практически нерастворимы друг в друге в твердом состоянии.
241
Вариант А. Органические вещества обладают сравнительно низкими температурами плавления (нет необходимости пользоваться электрической печью). Для этих легкоплавких систем термический анализ можно производить при помощи термометра. Системы из органических веществ можно исследовать в стеклянных пробирках.
Последовательность выполнения работы. В несколько пробирок насыпать чистые вещества и их смеси. Масса каждой пробы должна быть равна 4—5 г. Пробирки закрыть пробками, в отверстия которых вставлены термометры со шкалой 50—200°С и мешалка; затем пробирки по очереди поместить в водяную или масляную баню, нагретую до необходимой температуры. После того как содержимое пробирки расплавится и несколько перегреется, пробирку со смесью перенести в другую более широкую пробирку (воздушная рубашка), и-через каждые 15—30 с записывать показания термометра. Один из работающих при помощи лупы следит за показаниями термометра, другой по секундомеру производит отсчеты времени и записывает результаты. Измерения вести при непрерывном перемешивании охлаждаемого вещества (мешалку из смеси не вынимать). После появления первых кристаллов перемешивание прекратить. Для чистых веществ наблюдения за температурой прекратить после температурной остановки; для смесей наблюдение прекратить вслед за отвердеванием эвтектики (рекомендуется после застывания всей массы произвести отсчет температуры еще три-четыре раза). Результаты измерения температуры записать в таблицу по образцу:
Температура смеси, °C
Время от начала опыта, мин
На основании полученных данных построить кривые охлаждения, по которым определить температуру начала кристаллизации, эвтектики и длительность эвтектической остановки. Эти результаты вместе с данными о составе записать в таблицу по образцу:
Номер смеси Содержание смеси, % Температура начала кристаллизации, °C Кристаллизация эвтектики
массовое молярное температура, °C продолжительность температурной остановки
242
Построить диаграмму в координатах температура — состав для одной из систем: фенол — нафталин, нафталин — азобензол, а-нафталин — нафталин, нафтол — нафталин, фенол — метиламин,, камфора — бензойная кислота и др.
Вариант Б. Термический анализ легкоплавких смесей может быть проведен при помощи термопары и автоматического самопишущего потенциометра КСП-4. Для этого один спай (горячий) помещают в исследуемую смесь, а другой (холодный) подключают к компенсационной схеме автоматического самопишущего потенциометра КСП-4. На концах термопары возникает термо-э. д. с.г величина которой прямо пропорциональна разности температур на концах спаев. Поэтому шкала потенциометра отградуирована в градусах Цельсия. В момент компенсации стрелка прибора показывает температуру исследуемой системы, а регистрирующее устройство печатает на диаграммной ленте точку с номером. Наличие в приборе переключателя позволяет с интервалом 20 с измерять и регистрировать температуры параллельно шести исследуемых смесей.
Последовательность выполнения работы. Включить нагреватель водяной бани в сеть. Поместить в водяную баню комплект пробирок с исследуемыми смесями и опущенными в них хромель-копе-левыми термопарами.
Включить автоматический потенциометр КСП-4 в сеть, тумблер «прибор» на передней панели КСП-4 перевести в положение «вкл.». В процессе нагревания смеси в пробирках расплавятся^ а указатель на шкале прибора переместится до температуры 9о°С. По достижении температуры 95°С тумблер «диаграмма» на передней панели прибора поставить в положение «Вкл.». Из водяной бани последовательно с интервалом 4 мин вынуть пробирки со смесями под номерами 1, 2, 3 и т. д. и поместить их в соответствующие гнезда подставки в стеклянном стакане. Нагреватель водяной бани выключить из сети. Когда пробирки с исследуемыми смесями охладятся до комнатной температуры, тумблеры «диаграмма» и «прибор» поставить в положение «откл.». Прибор КСП-4 отключить от сети. Отделить диаграмму с записью кривых охлаждения и по полученным кривым построить диаграмму плавкости. (На диаграмме кривые охлаждения записываются таким образом, что каждой точке с цифрой на диаграммной ленте соответствует температура пробирки с тем же номером). Определить и занести в таблицу температуры начала и конца кристаллизации всех изученных смесей, а также длительность температурной остановки в миллиметрах диаграммной ленты.
Номер Пробирки Содержание смеси, % Температура кристаллизации, °C Длительность температурной остановки
массовое молярное начало конец
243
На листе миллиметровой бумаги построить диаграмму плавкости в координатах температура — состав. Через точки температур конца кристаллизации провести линию солидуса. С помощью треугольника Таммана, построенного под диаграммой плавкости, определить состав эвтектической смеси и перенести его на линию солидуса. Провести через точки температур начала кристаллизации плавные кривые ликвидуса. Приняв, что изучаемая система идеальная, рассчитать теплоты плавления обоих веществ с помощью уравнения Шредера.
Работа 5. Изучение кристаллизации веществ из растворов
В работе следует ознакомиться с визуальным методом исследования кристаллизации двойных жидких систем, представленных небольшими количествами. (Массовые количества компонентов, необходимые для каждого опыта, выражаются в сотых долях грамма).
При визуальном методе сплав известного состава нагревают и отмечают температуру плавления, а потом его охлаждают и измеряют температуру появления твердой фазы. Этот метод применим только к прозрачным объектам. Медленным нагреванием и охлаждением сплавов можно добиться хорошего совпадения (до 0,1°) температур исчезновения и появления твердой фазы. Преимуществом этого метода является его простота и быстрота, а недостатком— возможность субъективных ошибок, непригодность для исследования непрозрачных или окрашенных объектов.
Этот метод основан на определении температуры плавления соединения или смеси веществ в виде очень мелкого порошка, который помещают в толстостенный капилляр (см. метод Раста). За плавлением вещества в капилляре наблюдают при помощи бинокулярной лупы с увеличением в 10 раз в боковом освещении.
Последовательность выполнения работы. Взвесить запаянный с одного конца капилляр, наполнить его первым компонентом и снова взвесить; туда же добавить второй компонент и опять взвесить. После этого запаять второй конец капилляра. Для хорошего перемешивания компонентов после их плавления капилляр вынуть из блока и расплав перемешать встряхиванием. Затем капилляр поместить в блок и отмечать температуру плавления, а не кристаллизации, так как при охлаждении наблюдается сильное переохлаждение. В массе кристаллов, видимых в поле зрения бинокуляра, наблюдать за одним из них, отмечая температуру в момент его расплавления. В качестве объектов можно использовать следующие системы: пикриновая кислота — антрацен, фенол — нафталин, бензойная кислота — камфора и др.
ГЛАВА XII
ХИМИЧЕСКОЕ РАВНОВЕСИЕ
1. ХИМИЧЕСКОЕ РАВНОВЕСИЕ В ГОМОГЕННЫХ
И ГЕТЕРОГЕННЫХ СИСТЕМАХ
Химические реакции делятся на гомогенные и гетерогенные. Гомогенными реакциями называются реакции, протекающие в пределах одной фазы. К гомогенным реакциям относятся многие химические процессы, протекающие в газовой фазе или в растворах. Примерами могут служить реакции:
СО (г) + Н2О (г) XСО2 (г) + Н2 (г)
СНзСООН (ж) + С2Н5ОН (ж) 2: СН3СООС2Н5 (ж) + Н2О (ж)
Химическая реакция, протекающая на границе раздела фаз, называется гетерогенной. Примерами гетерогенных реакций могут служить
СаО (тв) + СО2 (г) ДД СаСОз (тв); С (тв) + О2 (г)“£СО2 (г)
Реакция протекает до равновесия, при котором имеются как продукты, так и реагенты, и при этом не происходит изменения их концентраций. Иногда количество продукта значительно превышает количество оставшихся реагентов в равновесной смеси, и с практической точки зрения реакция завершается. Реакционная смесь при равновесии содержит значительные концентрации как реагентов, так и продуктов.
Для предсказания равновесных концентраций в любых условиях реакции применяют термодинамические расчеты. Естественным направлением химических реакций является направление к минимуму энергии Гиббса. Величина, количественно характеризующая термодинамическую возможность протекания данной химической реакции, равная — т- е- алгебраической сумме произведе-
*
ний химических потенциалов всех веществ, участвующих в реакции, на их стехиометрические коэффициенты в уравнении реакции с обратным знаком, называется химическим сродством. Допустим, что протекает реакция по уравнению А^В при постоянных температуре и давлении. Элементарное изменение энергии Гиббса можно записать в виде
d G — — d -j- нв d пв (р — и Т = const),
где Цг — химические потенциалы реагентов и продуктов реакций; dtii — изменения молярных количеств реагентов и продуктов реакции.
Условием термодинамического химического равновесия является соотношение концентраций продуктов реакции и исходных веществ, при котором в реакционной системе бС=0(2цДп/ —0), причем энергия Гиббса имеет минимальное значение.
245
Молекулярно-статистически химическое равновесие определяется как такое состояние, при котором скорости прямой и обратной реакций равны друг другу, при этом равновесие наступает тогда, когда состав смеси с течением времени при постоянных внешних условиях не меняется. Однако неизменяемость состава смеси с течением времени может служить признаком, достаточным для констатирования наступившего равновесия лишь в том случае, если эта неизменяемость была достигнута в итоге самой реакции, т. е. при условии, что состав смеси до некоторого времени менялся вследствие реакции, а потом перестал меняться.
Иногда состав смеси, в которой возможна химическая реакция, остается продолжительное время неизменным, но не йотому, что процесс уже закончился и наступило равновесие, а вследствие того, что без катализатора процесс протекает настолько медленно, что происходящие изменения не могут быть экспериментально обнаружены. Для установления равновесия можно воспользоваться вторым признаком равновесия — признаком его подвижности. Система, находящаяся в равновесии, может быть выведена из этого положения внешним воздействием. При прекращении воздействия система самопроизвольно возвращается в прежнее состояние. При изменении внешних условий (температура, давление и т. п.) состав смеси будет изменяться. При возвращении системы к первоначальным условиям она будет переходить к исходному состоянию. Это означает, что рассматриваемое состояние является равновесным. Если же этого нет, то система не достигла еще состояния равновесия. Система, состояние которой характеризуется двумя признаками—.неизменяемостью состава и подвижностью, называется равновесной системой, а состав ее — равновесным составом.
Состояние равновесия химической реакции характеризуется термодинамической константой равновесия Ка- Величина Ка выражает для данной химической реакции
Bt 4- V2 в2 4- •.. 4- . = Vj Bj 4- Vg В2 4- • • • 4- v- B-
соотношение между активностями сц участвующих в ней
веществ
при равновесии:
Аналогичное соотношение между молярно-объемными концентрациями выражает константу равновесия /<с. Когда к реакционной системе применимы законы идеальных смесей (идеальных газов, идеальных жидких растворов), Кс при данной температуре имеет постоянное значение, не зависящее от исходных концентраций реагентов.
Константу равновесия выражают также аналогичными соотношениями между фугитивностями (летучестями) или между парци-246
альными давлениями, или между молярными долями веществ (с теми или с другими ограничениями). Значения константы равновесия будут различны при разных способах ее выражения. Константу равновесия можно выразить через равновесные парциальные давления системы
V V v
1 «2 « i
(XII. 1)
Константу равновесия можно выразить через общее давление системы и равновесный состав. Например, при проведении реакции СО + 2Н2^СНзОН исходные вещества были взяты в стехиометрическом соотношении: 1 моль СО и 2 моль Н2. При равновесии получено х моль СНзОН. Тогда (1—х)—равновесное количество молей СО; 2(1—х)— равновесное количество молей Н2. Парциальное давление равно общему давлению, умноженному ную долю данного компонента:
РСО = ^С0*> Рн2 ~ РХН^ ^СНзОН = ^ХСНзОГР где p=%pi — общее давление в системе; х,— молярная
компонента. Для определения молярной доли необходимо рассчитать общее равновесное количество молей в системе: S^z’PaBH =
I
на моляр-
(XII. 2)
ДОЛЯ 1-ГО
I,paw / Zini,равв’ ХС0=<1 ~*)/(3 — 2х)> хН2~2<1 ~х)/(3~ 2х)>
(XIL3)
-^снзОН = -*7(3 — 2х).
Константа равновесия данной реакции равна
__ ^снзон______х (3 — 2х)2______х (3 — 2х)2
” р2(1_Х)(2-2х)2 ~4Р2(1-х)3
Равновесие системы может смещаться при изменении начальных концентраций реагирующих веществ, константа же равновесия остается неизменной. Поэтому, зная константу равновесия при каких-либо внешних условиях, начальные концентрации и общее давление, можно рассчитать равновесный выход.
Рассмотрим химические гетерогенные равновесия, например равновесие образования и разложения карбонатов металлов:
МеСОзЗ^МеО 4-СО2
Константа равновесия будет определяться величиной Рсоа:
“ РсО^Мео/РмеСО.»
где рмео и рмесо3 — давление насыщенного пара веществ. Но давление насыщенного пара вещества в присутствии его конденсированной фазы при постоянной температуре постоянно. Поэтому соотно
247
шение Рмео/^месо3 есть величина постоянная, которую можно ввести в константу равновесия. Таким образом, = РСОа const и Хр = = рсо2* Термодинамическая константа равновесия гетерогенных реакций зависит от равновесности формы, отступлений от стехиометрии твердого образца и поэтому может оказаться в опыте не определяемой.
2. УРАВНЕНИЕ ИЗОТЕРМЫ РЕАКЦИИ
При изучении химической реакции важно знать, будет ли она протекать, а если будет, то в каком направлении. На это можно ответить, применяя к химическим реакциям второй закон термодинамики, согласно которому всякий самопроизвольный процесс, в том числе и химическая реакция в изолированной системе, протекает в направлении увеличения энтропии. При р и Т= const реакция протекает самопроизвольно в направлении уменьшения энергии Гиббса. Если все реагирующие вещества подчиняются законам идеальных газов, то ее уменьшение, наблюдаемое при обратном протекании реакции до состояния равновесия, выражается уравнением
- дс? = RT InКр- RT In /> !Р ? / (/>М)
о - О / 4 1 z
(XII.4)
где Р3>, Р3'“‘ и ~ произвольные парциальные давления
исходных и конечных веществ. Для реальных систем в.уравнение (XII.4) вместо парциальных давлений следует подставлять фугитивности или активности. Уравнение (XII.4) позволяет установить влияние температуры, давления инертного газа и начальных концентраций реагентов на направленность химической реакции. В стандартном состоянии при всех парциальных давлениях, равных 1 атм,
— AGj, — RT In /\р.
(XII. 5)
3. ЗАВИСИМОСТЬ КОНСТАНТЫ РАВНОВЕСИЯ ОТ ТЕМПЕРАТУРЫ
Зависимость константы равновесия от температуры описывается уравнением Вант-Гоффа
dlnXp
НГ RT2
(XII. 6)
Уравнение (XII.6) выведено из уравнения Гиббса—Гельмголь-
и уравнения
-ДС^ЛПпКр, (ХИ.8)
где АН° — изменение энтальпии (тепловой эффект) реакции. Знак теплового эффекта определяет знак производной dlnKp/dT в уравнении изобары Вант-Гоффа (XII.6). Если ДН°т>>0 (эндотермическая реакция), знак производной положителен, следовательно, константа равновесия с повышением температуры возрастает (рис. 108). Если ДН°т<0 (экзотермическая реакция), то константа равновесия с повышением температуры уменьшается. Если ДЯГ - о (тепловой эффект весьма мал), то константа равновесия не зависит от температуры. Согласно закону Гесса для уравнения реакции V1B1+V2B2 = v/BiZ + V2ZB2/
ДЯ^Ж' + Zj В j
Д77 />в
(XII. 9)
где дя° ,, д//° , ... — теплоты образования одного моля соответст-Z,Bi /,в2 г
вующего вещества.
Теплоты образования должны быть взяты при тех же условиях,
при которых определялся тепловой эффект реакции. Если теплоты образования взяты при стандартных условиях, то и тепловой эффект реакции получается при этих же условиях. Затем тепловой эффект при базисной температуре пересчитывается по уравнению Кирхгофа на ту температуру, при которой рассчитывается энергия Гиббса реакции
Рис. 108. Зависимость константы равновесия реакции от температуры (а) и логарифма константы равновесия реакции от обратной температуры (б)
* Z
откуда +
298
(ХИЛО)
Чтобы вычислить константу равновесия при любой температуре, следует проинтегрировать уравнение Вант-Гоффа.
1. При допущении независимости теплового эффекта от температуры интегрирование уравнения (XII.6) дает приближенное выражение
д//° 1 А
lg/<p= “Тзбзя т + const: +
ИЛИ
____кН / 1 1 \ ~2>303* U1 ~ т2 /’
(ХИ.11)
(ХИ. 12)
249
2. Для учета зависимости теплового эффекта от температуры требуется знать температурные зависимости молекулярных теплоемкостей реагентов ср°(Т). Обычно эти зависимости представляют степенными рядами по Т:
с°— а ЬТ -р сТ% -р с' ГТ^. г
Тогда для реакции можно написать
Дс° =Ьа + &ЬТ + ЬсТ2 + й.с'/Т2, (XII. 13)
Л'
где Да, ДЬ, Дс и Дс'—алгебраические суммы, которые вычисляют йо соответственным коэффициентам для теплоемкостей индивидуальных веществ. Согласно уравнению (XII.10) получаем зависимость теплового эффекта от температуры:
Д?/^= Д//о + 4- 1/2А^+ 1/зАсГЗ —Дс7Г.
(XII-14)
Подставляя в уравнение Вант-Гоффа значение ДНо из уравнения (XII.14) после интегрирования и переходя от натуральных логарифмов к десятичным, получаем
2,303/?Г R
kb Ag , . Ag'f~2 1
2-2,303/? 6-2,303/?7 “ 2-2,303/? +
(ХП.15)
где I — постоянная интегрирования.
В уравнение (XII. 15) входит константа интегрирования ДН® уравнения Кирхгофа. Константу I можно вычислить, если константа равновесия при какой-либо температуре определена по уравнению (XII.15) или из химических констант индивидуальных веществ. Уравнение (ХП.15) является в отличие от уравнения (XII.11) принципиально точным.
Для вычисления постоянной интегрирования 1, а следовательно, и константы равновесия, существует несколько методов. Одним из таких методов расчета /<Р является метод, который основан на применении стандартных таблиц термодинамических функций-В качестве стандартных условий принимают давление р—\ атм и базисную температуру Т = 298,15 К- Стандартные таблицы содержат абсолютные значения энтропии S°298 для простых веществ и химических соединений, величины или Д0о/,298 для хими-
ческих соединений. Эти величины выражают изменения энтальпии и энергии Гиббса при реакции образования соединения из простых веществ. Для простых веществ, устойчивых при стандартных условиях, ДЯО;!298 и Дб°/)298 принимают равными нулю. При помощи данных, приведенных в стандартных таблицах, вычислим ДН°298 и ДО°298 данного химического процесса:
~ V1 ( Лf, 29б) в' + v2 (Д^/,298)В' ~ V1 29в) В х V2 (Д^/,298)ва» X 2
(XII. 16)
250
AG298 ™ vx (А^/,298)в' + v2 (Д^/,29з)в^ “ V1 (^/ДЭв^ “ Vs(^^/(29s)b J
(XII. 17)
AS2g8 = Vi (s298) л 4-V2 (S29g) / — vx (529з)В1 “ v2 (*^298)ва- (XII. 18) 1 2
Поскольку
298
(XII. 19)
д<7298= —/?Г1п/Ср.
Отсюда вычисляем при Г—298 К
lg/<p = AG298/(2,3/?-298).
Итак, для вычисления константы равновесия реакции необхо-димо знать для каждого из реагирующих веществ: 1) температур-| ную зависимость теплоемкости ср°; 2) изменение энтальпии обра-| ЗОВаНИЯ При СТаНДарТНЫХ УСЛОВИЯХ A/f°f,298; 3) ЭНТРОПИЮ S°298 или изменение энергии Гиббса при стандартных условиях.
4. ПРИНЦИП ЭКСПЕРИМЕНТАЛЬНОГО ИЗУЧЕНИЯ
Ж ХИМИЧЕСКОГО РАВНОВЕСИЯ
II
11г Основная задача экспериментального изучения химического
- равновесия — определение состава равновесной смеси. Для этого необходимо, сохраняя внешние условия постоянными, проследить #. за изменением состава реагирующей смеси с течением времени, f пока состав не перестанет изменяться. Постоянство температуры | осуществляется с помощью термостатов. Постоянство давления
| обеспечивается маностатом. Во избежание изменений равновесно-
го состава в ходе его измерений применяют физико-химические методы анализа, позволяющие анализировать смесь без наруше-| ния установившегося равновесия. Особенно удобны электрохими-1ческие и спектральные измерения (электрическая проводимость, э. д. с., поглощение света, ЯМР и ЭПР и др.). Если приходится производить анализ при температуре более низкой, чем при равновесии, то применяют метод «закаливания равновесия», основанный на резком уменьшении скорости реакции при низкой температуре.
* Работа 1. Определение химического равновесия в гомогенных
| системах (жидкая фаза)
| В данной работе следует изучить равновесие при образовании
1 сложного эфира по уравнению
I СНзСООН 4- С2Н5ОН qt СН3СООС2Н5 4- Н2О
| Реакция при комнатной температуре протекает медленно. Поэтому ее проводят при 70—80°С в присутствии катализатора соля-
251
ной кислоты в течение 2—3 ч. Чтобы низкокипящие вещества, участвующие в реакции, не улетучивались при нагревании, реакцию проводят в колбах, снабженных обратными холодильниками. Теплота реакции образования этилацетата очень мала, и поэтому константа равновесия этой реакции, как это следует из уравнения Вант-Гоффа (XII.6), почти не зависит от температуры. За изменением состава смеси следят по изменению в ней суммарной концентрации кислоты. Изучение равновесия осуществляют измерением скоростей прямой и обратной реакций при различных исходных концентрациях СН3СООН (для прямой реакции) и СНзСООС2Н5 (для обратной). За изменением концентрации реагирующих веществ следят до тех пор, пока не наступит равновесие.
Последовательность выполнения работы. Взять четыре сухие колбы вместимостью 50 мл и присоединить к ним на корковых пробках обратные холодильники, укрепленные в штативах. Убедившись в надежности соединений и работы холодильников, приступить к опыту. Пронумеровать колбы, налить в первую колбу из бюретки 15 мл 4 н. СН3СООН, 5 мл 0,5 н. НС1 и 5 мл этилового спирта. Соединив колбу с холодильником и пустив в него воду, поместить колбу в термостат, установленный на 70—80°С. (Строгое поддержание постоянной температуры необязательно). Затем во вторую колбу налить из бюретки 15 мл 2 н. СН3СООН, 5 мл 0,5 н. НС1 и 5 мл этилового спирта. После соединения второй колбы с холодильником поместить ее в термостат. Налить из бюретки в третью колбу 15 мл дистиллированной воды, 5 мл этилацетата и 5 мл 0,5 н. НС1. Соединив третью колбу с холодильником, погрузить ее в термостат. Налить в четвертую колбу 12 мл дистиллированной воды, 3 мл этилацетата и 5 мл 0,5 н. НС1. Присоединив последнюю колбу к холодильнику и пустив в него воду, поместить колбу в термостат. Для удобства работы колбы погружать в термостат одну за другой с промежутками 10—15 мин. Время погружения каждой колбы в термостат отмечать отдельно. Колбы со смесями выдержать в термостатах 2—3 ч. За это время произвести точное определение концентраций соляной и уксусной кислот, взятых для составления смесей. Для этого взять 1 мл уксусной кислоты и отдельно 5 мл соляной кислоты, титровать их 0,5 н. NaOH в присутствии фенолфталеина. После 2-часового нагревания колбы в термостате взять из нее первую пробу. Для этого колбу, не разъединяя с холодильником, вынуть из термостата и поместить для охлаждения в ледяную воду, чтобы понизить давление паров смеси и избежать потери вещества и изменения состава. После охлаждения смеси отсоединить холодильник и пипеткой отобрать 1 мл смеси для анализа. Взятую пробу поместить в колбу Эрленмейера, в которую предварительно налить примерно 50 мл ледяной дистиллированной воды (ледяная вода должна приостановить реакцию и фиксировать момент, к которому относится измерение скорости). Затем, присоединив холодильник к колбе, поместить ее обратно в термостат, а во взятой пробе определить суммарную концентрацию кислоты титрованием 0,05 н. раствором NaOH в присутствии 252
фенолфталеина. Через 30 мин после отбора первой пробы из этой же колбы взять вторую пробу и оттитровать ее. Пробы брать через каждые 30 мин до тех пор, пока результаты титрования последних двух проб не совпадут между собой в пределах 0,1—0,2 мл 0,05 н. NaOH. Тогда можно считать, что равновесие наступило, и опыт с данной смесью можно закончить. Точно так же поступить и с другими колбами. Первую пробу взять по истечение 2 ч с момента погружения данной колбы в термостат, последующие — с промежутками в 30 мин.
Для расчета константы равновесия требуются следующие данные: количества взятых веществ и суммарная концентрация кислоты в равновесной смеси. Ход расчета различен для опытов по образованию этилацетата из спирта и уксусной кислоты и для опытов по омылению этого эфира водой.
Расчет 1. Обозначим через *>сн3соон — объем уксусной кислоты, взятый для составления исходной смеси, мл; ?сн3соон—плотность уксусной кислоты; асн3соон — содержание уксусной кислоты в данном объеме (единицы массы).
Пусть нормальность исходного раствора уксусной кислоты, определенная титрованием, равна N, а общий объем смеси и, исходная концентрация уксусной кислоты (моль/’л) в смеси равна
ссн3соон ™ ^vch3cooh/v-
Исходная концентрация спирта (моль/л) равна
СС2Н5()Н^ vC8HbOHPcaH5OHaCaH5OH* 1000/(^C2H5OHV)’
где ^с,н5он“ молекулярная масса спирта. Исходная концентрация воды (моль/л)
6’н2О = [VCH3COOH?CH3COOH 0 “ асн3соон) + vC2H5OH?CaH5OH С1 “ ас2н5он) +
7 1000
+ VHC1PhC! (! “ аНС1)] м • HaOv
Так как концентрация соляной кислоты незначительна (0,5 моль/л), то без большой погрешности можно считать произведение Унарна(1—ана) равным Унарнсь
Пусть по окончании реакции суммарная концентрация кислоты в равновесной смеси равна с2, а концентрация взятой соляной кислоты— тогда концентрация уксусной кислоты с (моль/л) в равновесной смеси будет
Ссн3соон = С2 (clvHCl/v)*
Концентрация этилацетата в равновесной смеси, поскольку его не было в исходной смеси, равна уменьшению концентрации уксусной кислоты, т. е.
— сСН3СООН “ cCHsCOOH’
концентрация этанола в равновесной смеси соответственно равна
сс2н5он — Сс2н5он Сэ:
равновесная концентрация воды будет
fH2o — сн2о +
Константа равновесия
К с — ^н2о/(ссн3соонсс2н5он) •
Расчет Кс—производят отдельно для двух взятых растворов.
Расчет 2. Обозначим через — исходный объем этилацетата, мл; рэ — плотность этилацетата; Л4Э — молекулярная масса этилацетата ^н2о ~ объем воды в исходной смеси, мл.
Определяем концентрации (моль/л) этилацетата в исходной смеси
и воды
Равновесная концентрация уксусной кислоты будет
C1VGH3COOH cgh3gooh ~ с ~
равновесная концентрация спирта
СС2Н50Н ~ ссн3соон* концентрация этилацетата в равновесной смеси
с3 — е3 — ^снзСОон»
концентрация воды в равновесной смеси
сн2о — сн2о ССН3СООН'
Константу равновесия вычисляем по уравнению
/<е = сэсНз0/ (^сн3соонсс2н5он) •
Экспериментальные и расчетные данные записать в таблицы по образцам:
Номер пробы Колба 1 Колба 2
Время отбора пробы Количество 0,05 н. NaOH, израсходованное на титрование, мл Время отбора пробы Количество 0,05 н. NaOH, израсходо-* ванное на титрование
254
Номер опыта Начальные концентрации, моль/л Равновесные концентрации, моль/л
Колба I Колба 2 Колба 1 Колба 2
Суммарная концентрация в равновесной смеси: уксусной кислоты этилового спирта этилацетата воды
На основании экспериментальных данных рассчитать для исследуемой реакции среднее значение константы равновесия и энергию Гиббса реакции.
Работа 2. Изучение химического равновесия реакции 2Fe3++2I-^2Fe2++I2
Последовательность выполнения работы. Взять четыре сухие колбы с притертыми пробками вместимостью 100 мл. Пронумеровав колбы, налить в них следующие количества растворов точной концентрации:
Колба
Раствор, мл
0,03 М FeCl3
0,03 М кг
12 3 4
50 —— 55 —
— 50 — 45
Затем колбы поместить на 30 мин в термостат при температуре 25±0,2°С. Приготовить для титрования восемь конических колб вместимостью 100 мл. В каждую колбу налить 35—50 мл дистиллированной воды и поставить их для охлаждения в охладительную смесь (лед). Слить вместе содержимое колб 1 и 2, а через 10 мин после этого содержимое колб 3 и 4. Момент сливания растворов отметить по часам. Колбы плотно закрыть пробками и установить в термостат. Через 25 мин от момента смешения из каждой колбы, не вынимая ее из термостата, отобрать пипеткой 45 мл раствора и слить в сильно охлажденную колбу для титрования. За момент отбора пробы считать сливание раствора из пипетки в колбу для титрования. Время отбора пробы отмечать с точностью до 1 мин. Сразу после сливания выделившийся иод титровать 0,015 М Na2S2O3 с известным титром. Раствор гипосульфита прибавлять до образования бледно-желтой окраски раствора. Затем добавить несколько капель раствора крахмала и титровать раствором гипосульфита до
255
исчезновения синего окрашивания раствора. Светло-синяя окраска раствора, появляющаяся через некоторое время после титрования, не учитывается. Пипетку перед отбором пробы сполоснуть исследуемым раствором.
Через 30 мин после отбора пробы из каждой колбы взять снова 15 мл раствора и титровать гипосульфитом. Затем через 40 мин отобрать третью пробу и т. д. Одинаковое количество миллилитров гипосульфита, израсходованное на титрование иода в двух последовательно взятых пробах из каждой колбы, указывает на достижение равновесия.
Расчет.
1. Концентрация иода в состоянии равновесия равна
где <?Na2s2o3~ концентрация гипосульфита, моль/л; Vi — количество гипосульфита, израсходованное на титрование иода в момент равновесия, мл; — количество взятой пробы, мл.
2. Концентрация ионов Fe2+ равна удвоенной концентрации иода, так как по уравнению реакции образуется одна молекула иода и два иона Fe2+, следовательно, eFe24- = 2el2.
3. Концентрация ионов Fe3+ при равновесии равна разности начальной и равновесной концентрации ионов Fe2+, так как прирост концентрации Fe2+ равен убыли концентрации Fe3+:
CFe3+ ~ CFeCl3 ~ cFe2+ *
ИЛИ
cFe3+ = cFeCl3 “2Ч2-
Содержание FeCl3 вычисляют из концентрации исходного раствора и степени разбавления его при смешении растворов:
cFeCl3 = cFeCl3 1а/(а + ^)] >
где ересь— начальная концентрация раствора FeCl3, моль/л; а и b — количество растворов FeCl3 и KI соответственно.
4. Концентрацию ионов 1“ рассчитывают по уравнениям:
где с°к! — концентрация исходного раствора, моль/л.
В такой же последовательности провести опыт при другой температуре (например, при 40°С). Константу Кс рассчитать отдельно 256
для двух взятых растворов при двух температурах. Экспериментальные и расчетные данные записать в таблицу по образцу:
Колба 1 Колба 2 Вещество Колба 1 Колба 2 Колба 1 Колба 2
Номер пробы । Время отбора пробы Количество Na2S2O3, израсходованное иа титрование, мл Время отбора пробы Г Количество Na2S2O3, израсходованное на титрование, мл Начальная концентрация при t\ Равновесная концентрация при t2 Начальная концентрация при h Равновесная концентрация при Ц
На основании экспериментальных данных рассчитать для исследуемой реакции среднюю константу равновесия при двух температурах, средний тепловой эффект ЛЯ.
Работа 3. Изучение химического равновесия в растворах при образовании комплекса методом распределения
Растворимость иода в воде увеличивается при добавлении йодидов или бромидов. Это объясняется образованием комплексов (1з)“ и (Вг12)_. Используя данные по распределению иода между водой и органическим растворителем, можно рассчитать константу равновесия реакций образования комплексов при тех же температурах, при которых определяется константа распределения. Константу равновесия рассчитывают по уравнениям:
Кс = [1г]/([!2][I-]); Кс = [Вг17]/ф2][В-]).
Для этого изучают распределение иода между 0,05 н. КВг или 0,01 н. KI и органическим растворителем.
Последовательность выполнейия работы. Предварительно определить коэффициент распределения иода между водой и органическим растворителем (см. с 217). Затем провести опыты, используя вместо дистиллированной воды 0,01 н. KI или 0,05 н. КВг. При изучении равновесия реакции
12 4- КВг К (Вг 12)~
равновесные концентрации в органическом и водном слоях определяют титрованием пробы из органического слоя 0,05 н. Na2S2Cb и пробы из водного слоя 0,001 н. Na2S2O3. При изучении равновесия реакции
12 + к1:£К(1з)-
9—2083
257
пробы из органического слоя титруют 0,1 н. Na2S2O3 и пробы из водного слоя — 0,01 н. Na2S2O3. При титровании водного слоя раствором гипосульфита определяют весь растворенный иод, находящийся как в свободном виде 12, так и в виде комплексного иона (1з)“ или (12Вг)~. Концентрацию молекулярного кода 12 в водном растворе определяют по коэффициенту распределения и концентрации иода в сопряженном неводном слое из соотношения
Д' = [12]орг/[12]н2О ’
где К — коэффициент распределения иода между водой и органическим растворителем.
Концентрацию комплексного иона (13)~ или (Вг12)" рассчитывают по уравнению
Мсв + [(1з)“] - [12],
где [12] —суммарное содержание иода в водном слое.
Откуда
= [12] - [12]св.
Концентрацию свободного •[!“] или [Вг~] находят из равенств:
[Ь]св + №)“] - [KI],
[Вг~]св н- [(Вг12)-] = [КВг],
где [KI] — концентрация исходного раствора КК [КВг]—концентрация исходного раствора КВг.
Провести опыты при температурах и Т2, определить константы соответственно равновесия Kcj* при 72 и Кс,т\ при Ть используя уравнение изобары Вант-Гоффа:
Вычислить тепловой эффект химической реакции к AS.
Работа 4, Определение химического равновесия в гомогенных системах (газовая фаза)
Реакция разложения N2O4 протекает с изменением количества молекул по уравнению N2O4^2NO2, что позволяет определить степень термической диссоциации и константу равновесия при различных температурах по измерениям давления к объема реакционной смеси. Пусть при заданной температуре степень диссоциации N2O4 а, тогда из каждого исходного моля N2O4 остается (1—а) долей моля N2O4 и образуется 2а молей NO2. Общее число молей равно (1-ра). Общее давление при установившемся равновесии р, соответственно парциальные давления компонентов равны:
1 — а 2сс
Л\2о4 = ! , п Р‘> Аю2 7Т~ Р *
1 Lv 1 "г*
258
Подставив в уравнение (XII.1) равновесные парциальные давления, получим
Степень диссоциации а определяют по уравнению Клапейрона — Менделеева
pv — nRT или
(1 ос) RT,
(XII. 20)
где v — объем реакционного сосуда; Т — температура, К; g— навеска тетраоксида азота, г; молекулярная масса тетраок-
сида азота.
Из уравнения (ХП.20) видно, что при уменьшении давления в системе должна увеличиваться степень диссоциации тетраоксида азота. Реакция распада тетраоксида азота на диоксид азота протекает с положительным тепловым эффектом. Поэтому при повышении температуры равновесие реакции будет смещаться в сторону образования диоксида азота. Реакцию изучают в интервале температур 25—100°С и не выше давления 1,013-Ю5 Па, так как
2 /
Рис. 109. Схема установки для изучения химического равновесия в газовых системах при различных температурах
NO2 выше 100°С диссоциирует на оксид азота и кислород. Нижний предел температуры должен быть выше температуры кипения тетраоксида азота (21,2°С при р= 1,013-Ю5 Па).
Прежде чем приступить к опыту, необходимо по стандартным табличным данным рассчитать степень диссоциации при температурах 25 и 100°С и давлении 1,013-105 Па. Равновесие реакции N2CW2NO2 изучается статическим методом. Установка (рис. 109) состоит из реакционной колбы которая опущена в термостат 2, 9* 259
нуль-манометра 3, который представляет собой мембранный манометр; вакуумметра 9, манометра с открытым коленом 4, шлифа с запаянным отростком 10, колбы 5 для равномерного откачивания системы и напуска воздуха, кранов 8, 7 и 6. Кран 8 служит для сохранения вакуума в колбе /, кран 7 — для введения воздуха в систему и кран 6 — для откачиваний воздуха из системы.
Последовательность выполнения работы. Под тягой на специальной установке ампулу заполнить определенным количеством N2O4. Ампула представляет собой небольшой сосуд, оттянутый с обоих концов в тонкостенные капилляры. Один из капилляров изогнут таким образом, чтобы при опускании его в отросток шлифа 10 и при повороте этого отростка вокруг своей оси капилляр мог обломиться и жидкость могла стечь в трубку 11. Изготовив ампулу, поместить ее в отросток 10 таким образом, чтобы конец капилляра касался трубки 11 и ампула не скользила. Ампулу закрепить в специальной корковой пробке, после чего систему следует откачать. Для этого необходимо осторожно повернуть кран 6, причем кран 8 открыт, а кран 7 закрыт. После откачивания системы краны 6 и 8 закрыть. Катетометр навести на стрелку нуль-манометра, которая будет находиться в середине трубки. Затем отросток шлифа 10 повернуть вокруг собственной оси. Отросток ампулы обламывается и жидкость стекает в реакционный сосуд. При этом надо следить за стрелкой нуль-манометра. Стрелка отклоняется, так как давление в реакционном сосуде возрастает. Чтобы стрелка не отклонялась от среднего положения, открыть кран 7 и вводить воздух в нужном количестве. Температура в момент разбивания ампулы должна быть самой минимальной (порядка 10—15°С). Когда ампула разбита и температура в термостате доведена до той, при которой должна быть определена константа равновесия, выждать еще 10— 15 мин и замерить давление в реакционном сосуде.
Давление в момент компенсации отсчитывать при низких температурах по вакуумметру, а при высоких — по манометру. При компенсации стрелка нуль-манометра должна находиться в среднем положении. Затем температуру в термостате нужно поднять на 5°, снова повторить те же операции. Таких операций провести девять-десять. Полученные данные записать в таблицу по образцу:
Температура, °C Объем реакционного сосуда Навеска, г Давление а ЦТ
Объем реакционного сосуда указывается в задании.
На основании полученных данных построить графики в координатах Кр—Т и 1g КР—1,/Т и по тангенсу угла наклона прямой к оси 1/7 определить тепловой эффект реакции. Кроме того, опреде-
260
лить тепловой эффект для трех интервалов температур по уравнению
S Ki — 2,3/? Т2 ) ’
Полученное среднее значение сравнить с величиной, найденной графически. С помощью метода наименьших квадратов определить постоянные А и В в уравнении
Кр А + В/Т.
При помощи уравнения (XII.6) рассчитать температуру диссоциации (температура, при которой давление паров равно il,013Х ХЮ5 Па). При помощи уравнений (XIIJ16) и (XIIJ18) рассчитать изменение энтропии AS0, изменение энергии Гиббса AG0 для этой реакции и сравнить последние с табличными данными.
Работа 5. Определение химического равновесия в гетерогенных системах (исследование карбонатов)
Рис. ПО. Схема установки для изучения химического равновесия гетерогенных реакций при высоких температурах
При повышенных температурах карбонаты металлов диссоциируют по уравнению МеСО3^МеО + СО2. Константа Кр определяется давлением СО2: К?=Рсой.
Работа проводится на установке, схема которой приведена на рис. НО. Кварцевая ампула 2 с навеской карбоната кальция находится в печи 1. Печь снабжена регулятором скорости нагрева 8.
Температуру измеряют при помощи термопары 3, соединенной с милливольтметром 6. Холодные спаи термопары погружены в сосуд Дьюара 7 и находятся при 0°С.. Давление СО2 133,ЗХ ХЮ2 Па измеряют при помощи закрытого манометра 4, выше при помощи открытого манометра 5. Разность уровней в закрытом манометре показывает давление в системе, а разность уровней в открытом манометре — на сколько давление в системе меньше атмосферного.
Последовательность выполнения работы. Заполнить сосуд Дьюара смесью льда с водой. Включить печь. Измерения давления СО2 целесообразно начинать только после достижения системой темпера-
туры 600°С. После того как пирометр покажет 600°С, нагревание печи прекратить. В силу инерции стрелка пирометра продолжает двигаться и после прекращения нагревания. Записать максималь-
261
ную температуру, показанную пирометром, к максимальное давление по манометру. Когда измерения произведены, печь снова включить. Каждое последующее измерение производить через 10—20°С. Измерения прекратить, когда давление СО2 достигнет 400-102—500- 102 Па. Печь выключить и дать ей отстыть до 500сС. Для получения воспроизводимых результатов все измерения следует повторить один-два раза. Если давление СО2 измерить при непрерывном нагревании, то результаты получаются заниженными, так как скорость роста температуры печи превышает скорость установления равновесного давления СО2. Данным способом при ограниченном времени исполнения получают приближенные результаты.
Более точные данные можно получить по усложненной методике. Для этого работу следует начинать с предварительного разогревания печи до 500—600°С. Регулятор скорости нагрева печи поставить в положение «1». Предварительное медленное разогревание печи необходимо во избежание растрескивания керамических частей установки и порчи печи. По достижении 500°С можно начать более интенсивное нагревание, переключив регулятор скорости нагревания в положение «8». Предположим, что нужно провести измерение при 650°С. Печи дают остыть до 580—590сС, а затем снова ее включают. В момент прохождения стрелки пирометра через отметку 600°С включить секундомер и измерить время нагрева до 650°С. При достижении 650°С выключить ток и измерить давление СО2 в установке. Печи дают остыть до 600°С, установив регулятор на меньшую скорость нагрева, к повторяют измерения еще раз.
Чтобы определить равновесное давление при данной температуре, следует измерить давление при трех-четырех скоростях нагревания, так как с уменьшением скорости нагревания уменьшается разрыв между измеряемым и равновесным давлением. Когда скорость нагрева приближается к нулю, измеряемое давление приближается к равновесному.
Построив график в координатах скорость нагревания — давление СО2, рассчитать для данной температуры давление СО2 при скорости нагревания, равной нулю. После этого перейти к следующей температуре. Всего следует провести пять-шесть измерений в интервале температур 650—850°С, Результаты измерений записать в таблицу по образцу:
ir °C РСО2 = кр 1/Л к
I
На основании полученных данных построить графики в координатах Кр — t, °C и Кр—1/7" и по тангенсу угла наклона прямой к 262
оси 1/Т определить тепловой эффект реакции. Определить величину Д/7 в трех интервалах температур по уравнению
получить среднее значение АН и сравнить с величиной, найденной графически. С помощью метода наименьших квадратов определить постоянные Л и В в уравнении
1g/<р = А 4-В/Т.
При помощи данного уравнения рассчитать температуру диссоциации (температуру, при которой давление СО2 равно 1,013х Х105 Па). При помощи уравнений (XII.16) и (XII.18) рассчитать для этой реакции изменение энтропии Д3°, энергии Гиббса AG0, сравнить последние со справочными данными.
Работа 6. Исследование химического равновесия в
кристаллогидратах
Кристаллогидрат ферроцианида калия (желтая кровяная соль) при температурах выше комнатной теряет кристаллизационную воду. Каждой температуре соответствует определенное соотношение равновесия в системе
К4 [Fe (CN)3] ЗН2О X К4 [Fe (CN)6] -Ь ЗН2О
\ л
Рис. 111. Схема установки для изучения химического равновесия гетерогенных реакций (разложение кристаллогидратов) при средних температурах
Константа равновесия этой реакции равна /Ср=/Щ2о-
Скорость установления равновесия достаточно велика. Это позволяет использовать статический метод исследования, при котором система выдерживается в изотермических условиях до установления равновесия.
Последовательность выполнения работы. Работа проводится на установке, схема которой приведена на рис. 111. Тонкоизмельченный K4[Fe(CN) 6]-3H2O поместить в сосуд 10 с короткой манометрической трубкой 5, погруженной в термостат 6, Второй конец манометрической трубки соединен со стеклянным резервуаром 1, снабженным закрытым манометром 2. Кран 11 соединяет резервуар с вакуум-
установкой, кран 12 — с атмосферой. В шарик 4 манометрической трубки залить ртуть. Перед началом работы из прибора тщательно откачать воздух до остаточного давления 0,0133 Па, после чего закрыть кран И, перелить ртуть из шарика 4 в манометрическую
263
трубку и укрепить прибор в термостате 6 в вертикальном положении. Вследствие равенства давлений в сосуде 10 и резервуаре 1 ртуть в обоих коленах манометрической трубки будет на одном уровне.
Наблюдение за уровнем ртути вести через смотровое окно 3 термостата. После того как прибор установлен, включить обогрев термостата и мешалку 9. Повышение температуры вызывает диссоциацию. Давлением паров воды ртуть в левом колене манометрической трубки выдавливается. При достижении определенной температуры, задаваемой при помощи терморегулятора 7, произвести измерение давления. Осторожным открытием крана 12 впускать в резервуар 1 воздух до тех пор, пока уровни ртути в обоих коленах не сравняются. Записать показания манометра 2 к термометра 8. Систему выдерживать при постоянной температуре, периодически (через 5—10 мин) измеряя давление. После того как давление в сосуде 10 установится, терморегулятор перевести на более высокую температуру и повторить в той же последовательности новый опыт. При переходе от одной температуры к другой во избежание переброса ртути следует время от времени выравнивать уровни в манометрической трубке.
Опыт провести в температурном интервале 35~95°С. Через 5— 110° измерять давление. Результаты измерений записать в таблицу по образцу:
На основании полученных данных построить графики в координатах КР — t, °C; 1g КР— 1/Т; по тангенсу угла наклона прямой к оси 1/Т определить тепловой эффект реакции. Определить АЯ для трех интервалов температур по уравнению
Кг АЯ / 1 _ 1 \ g Ki ~2,ЗЯ \ Тг Т2) ’
получить среднее значение АЯ и сравнить с величиной, найденной графически. С помощью метода наименьших квадратов определить постоянные А и В в уравнении
lgKp-Лч-В/Г.
При помощи данного уравнения рассчитать температуру диссоциации (температуру, при которой давление паров равно 1,013Х Х105 Па). При помощи уравнений (XII.16) и (XII.18) найти изменение энтропии AS° и энергии Гиббса AG° для этой реакции к сравнить их с табличными данными.
264
Работа 7. Изучение равновесия реакций дегидрирования
спиртов в газовой фазе
Для более быстрого установления равновесия в системе
Rr - СНОН - R2 Z Rr - СО - R2 Н- Н2
где протекает реакция дегидрирования спиртов, применяют различные катализаторы. Равновесие реакции дегидрирования спиртов целесообразно исследовать в интервале температур 80—160°С. При этих условиях к-бутиловый, «зо-бутиловый, к-пропиловый, изопропиловый спирты дегидрируются без образования побочных продуктов. Кроме того, реакции дегидрирования спиртов сопровожда-
ются поглощением теплоты, поэтому повышение температуры приводит к увеличению константы равновесия и соответственно выхода кетона к водорода. Константа равновесия реакции дегидрирования, спирта при постоянном давлении описывается выражением
/Ср = f
где /?кет, /7Нз> Реи ~ равновесные парциальные давления кетона, водорода и спирта.
В данной работе изучение равновесия реакции дегидрирования спирта проводят динамическим методом.
Установка (рис. 112) состоит из электропечи 9, в которой при помощи термопары 8 к потенциометра 1 поддерживается заданная температура с точностью ±1,5°С. В печи помещена реакционная колонка 7 с катализатором. Через диафрагму 11, укрепленную в корпусе холодильника 10 втулкой, электродвигателем 14 и шприцем 12 в каталитическую колонку подается спирт для дегидрирования с постоянной
Рис. 112. Схема установки для изучения равновесия дегидрирования спиртов
скоростью. Продукты реакции проходят через холодильник 6. Жидкая фаза конденсируется и собирается в отборнике 5, а газообразный водород поступает в бюретку 5.
Последовательность выполнения работы. Перед работой установки необходимо сначала пустить воду в холодильники 10 и 6.. Затем нагреть печь до заданной температуры. Для этого тумбле-
265
ром 17 включить потенциометр. Вращая ручку потенциометра по часовой стрелке, установить против красной черты заданную температуру по верхней шкале. Вытеснить газ из бюретки 3. Для этого кран 2 поставить в соответствующее положение и поднимать вверх склянку 4. После заполнения бюретки водой кран 2 перевести в другое положение. Заполнить дозирующее устройство спиртом. Для этого шприц вынуть из колонки и ослабить винт 13. Иглу опустить в бюкс со спиртом и, вращая муфту 16 по часовой стрелке, наполнить шприц. Завинтить винт 13, иглу шприца опустить в отверстие холодильника 10 и тумблером 15 включить подачу спирта в систему.
Спирт дегидрируется на катализаторе, и выделяющийся водород поступает в газовую бюретку 3. Измерять объем выделяющегося газа через каждые 1—2 мин. При каждом измерении необходимо привести давление в бюретке к атмосферному. Это достигается опусканием уравнительной склянки до выравнивания уровня воды в бюретке. Результаты измерений занести в таблицу по образцу:
Время, мин
Уровень воды в бюретке, мл
Д/
и по ним рассчитать скорость выделения водорода. Если в течение 2,0—2,5 мин скорость выделения водорода остается постоянной, то можно считать, что в системе установилось состояние тер-
модинамического равновесия.
Из отборника 5 слить жидкую фазу, которая накопилась с мо-
мента пуска до момента установления равновесия. Работу продолжать в течение 5—10 мин. Затем в пробирку отобрать из отборника 5 пробу спирта и кетона равновесного состава для хроматографического анализа к установку выключить. Для этого тумблерами 15 и 17 отключить подачу спирта и нагревания печи. Перекрыть доступ воды в холодильники. Состав жидкой фазы анализировать
на хроматографе.
Хроматографический анализ системы спирт-кетон. Анализ про-
водить на газо-жидкостном хроматографе, блок-схема которого представлена на рис. ИЗ. Прибор состоит из четырех блоков: термостата 1, газораспределительного блока II, блока управления III, потенциометра IV. Для подготовки прибора к анализу необходимо выполнить следующие операции: 1) пустить воду в холодильник; 2) открыть редуктор на баллоне с газом-носителем и при помощи вентиля на манометре установить заданное давление; 3) включить питание прибора; 4) поставить ручку переключения рода работы в
положение «температура колонки» и при помощи регулятора установить заданную температуру. Температура фиксируется на шкале
266
потенциометра; 5) установить ток детектора. Для этого переключатель перевести в положение «ток детектора» к установить по шкале заданный ток; 6) перевести переключатель в положение «анализ» и установить стрелку прибора в положение «О»; 7) включить диаграммную ленту прибора. Прямая линия на диаграммной ленте означает готовность прибора к работе.
Прежде чем приступить к анализу, нужно провести идентификацию спирта и кетона, которая осуществляется для определения, к какому из веществ в анализируемой смеси следует отнести дан
Рис. 113. Блок-схема газо-жидкостного хроматографа
Рис. 114. Хроматограмма смеси
спирт — кетон
ный пик хроматограммы. Для этого ввести хроматографическим шприцем через испаритель пробу спирта. На диаграммной ленте через некоторое время получается хроматографический пик спирта. Отметить время удерживания спирта в колонке. Аналогичную операцию провести с кетоном. Сравнить время удерживания спирта и кетона в колонке и установить последовательность выхода продуктов на диаграммной ленте.
Анализируемую пробу спирта с кетоном шприцем ввести в испаритель. На диаграммной ленте (рис. 114) получаются два пика. Процентное содержание спирта и кетона рассчитать, измерив площадь каждого пика, которую определяют как произведение — — hibi, bi — ьысжъ пика; bi — ширина пика на половине высоты. Ширину bi измерять специальной лупой с точностью до ОД мм. Равновесный состав жидкой фазы рассчитать по формулам:
£сп ~ *^сп/(*^сп *^кет)» ^кет “ *^кет/(*^сп 4“ 5кет).
Для расчета взять среднее значение из результатов анализа трех хроматограмм. После снятия хроматограмм прибор выключить; для этого отключить газ из баллона, затем воду и электропитание.
Константу равновесия рассчитать по количеству выделившегося водорода, если в каталитическую колонку спирт подавался со скоростью моль/мин. При установившемся равновесии в газовой бюретке собирается водород с постоянной объемной скоростью W, моль/мин. Объемная скорость — объем выделившегося за ми-
267
нуту водорода, который приведен к нормальным условиям. Исходя из объемной скорости рассчитать равновесное число молей смеси:
водорода /22 400
кетона w^/22 400
спирта (n0 — w^)/22 400
S п1 = (по + ^н2)/22 400 •
Парциальные давления в условиях равновесия равны:
ра.-р.п- 22400», + ^, Г-
22 400/Zq — дащ
А:п " ’ о р *
22 4OOno * * S
где р — общее давление.
Константа равновесия будет равна
22 400—1 wHa )
Константу равновесия можно рассчитать на основании равновесного состава жидкой фазы. Данные, полученные на основе анализа хроматограмм, разделить на молекулярные массы:
спирт £СпЛИсп кетон ^кет/^кет водород ^кегЖкет
~ £спМ4ст 4" кет •
Тогда парциальные давления в условиях равновесия: _________________________________ ^кет/^кег____ ^сп/Л1ст + Р'
_ ^сп/^сп_______
^спМ^сп 4* 2£СП/Л1К8Т
Константа равновесия будет равна
„(ffкегМ^кет) (^сп^сп) (^ГспМ^сп) (-^кет/^кет 4“ 2)
Вычислить константу равновесия дегидрирования изучаемого спирта и энергию Гиббса реакции.
268
уГЛАВА XIII
РАСТВОРЫ ЭЛЕКТРОЛИТОВ
1. ЭЛЕКТРИЧЕСКАЯ ПРОВОДИМОСТЬ
Согласно теории Аррениуса некоторые вещества, называемые электролитами, обладают способностью при растворении в различных растворителях распадаться на ионы. Количество ионов, обра-зовавшихся в результате диссоциации одной молекулы, так же как величина и знак заряда этих конов, зависит от природы электролита. Ионы в растворе электролитов являются переносчиками электричества. Не все электролиты диссоциируют в одинаковой степени: сильные электролиты практически полностью диссоциированы и поэтому хорошо проводят^TQKr слабые электролкть^
тельно и, вследствие этого, проводят ток хуже. В растворе электролита коны обладают тепловым движением, т. е. беспорядочно движутся с самыми различными скоростями. Если раствор поместить в электрическое поле, то ионы, сохраняя свое тепловое движение, начнут смещаться по направлению силовых линий поля. При этом движение катионов будет происходить в направлении, прямо противоположном движению анионов. Так как ионы являются носителями зарядов, то их направленное перемещение представляет собой прохождение электрического тока через электролит. Чем больше зарядов имеет ион и чем большее количество конов пройдет, в секунду через сечение раствора, перпендикулярное силовым линиям поля, тем больше будет электрическая проводимость раствора. Для раствора электролита количество ионов, прошедших через данное сечение, определяется их концентрацией и скоростью движения по направлению, перпендикулярному этому сечению. Эта скорость пропорциональна при прочих одинаковых условиях градиенту потенциала
v+ = U+E' и = U-E',
(XIII. 1)
где и+ — скорость движения катиона, см/с; — скорость движения аниона, см/с; U±, U_ — коэффициенты пропорциональности; Ef— градиент потенциала, В/см.
При Е'= 1 В/см скорость движения катионов равна J7+, а ско-
рость движения анионов V-—LL. Коэффициенты U являются величи-
нами, характерными для каждого вида конов, и называются подвижностями. Величины подвижностей конов зависят от сопротивления, которое испытывают коны при своем движении в растворе; чем меньше сопротивление, тем больше подвижность. Сопротивление же увеличивается с увеличением размеров сольватированного (гидратированного) иона, электростатического взаимодействия между конами к вязкостью раствора.
Таким образом, электрическая проводимость является функцией количества зарядов ионов, концентрации ионов и их подвижности.
269
2. УДЕЛЬНАЯ И ЭКВИВАЛЕНТНАЯ ЭЛЕКТРИЧЕСКИЕ ПРОВОДИМОСТИ
Растворы электролитов характеризуются удельной и эквивалентной электрическими проводимостями.
Удельная электрическая проводимость х— величина, обратная удельному сопротивлению х = 1/р. Электрическое сопротивление проводника связано с удельным сопротивлением р уравнением
(XIII.2)
где I— длина проводника; S—-площадь поперечного сечения проводника.
Из уравнения (XIII.2) следует
1/р = (1//?) (Z/S). (XIII.3)
Единицей измерения удельной электрической проводимости является См/м (Ом-1-см-1). Так как растворы электролитов, подобно другим проводникам, подчиняются закону Ома R = V/I, то
х = (//П(//5). (XIII.4)
Таким образом, удельная электрическая проводимость равна силе тока 7, проходящего через слой раствора с поперечным сечением, равным единице, под действием градиента потенциала 1 В на единицу длины. Удельная электрическая проводимость характеризует электрическую проводимость раствора, заключенного между двумя параллельными инертными электродами площадью 1 см2 и расположенными на расстоянии !1 см друг от друга.
Эквивалентная электрическая проводимость представляет электрическую проводимость объема электролита, содержащего 1 г-экв растворенного вещества к находящегося между двумя параллельными электродами, расположенными на расстоянии 1 см друг от друга. Единицей измерения эквивалентной электрической проводимости является См-м2./моль (Ом”1-г-экв-1 -см2). Эквивалентная электрическая проводимость Л рассчитывается:
= * (XIII. 5)
где V — разведенке, объем раствора в см3, содержащего в 1 г-экв растворенного вещества, эта зависимость может быть выражена и через концентрацию:
%/с. (XIII.6)
Если концентрацию раствора выразить через число молей в едйни-це объема, то электрическую проводимость называют молярной электрической проводимостью Лт. Электрическая проводимость растворов электролитов дашсит от концентрации,давления, температуры, природы растворенного вещества и растворителя, вязкости, диэлектрической проницаемости.
270
ЗАВИСИМОСТЬ ЭЛЕКТРИЧЕСКОЙ ПРОВОДИМОСТИ
ЮТ КОНЦЕНТРАЦИИ \ л
Удельная и эквивалентная электрические проводимости растворов электролитов зависят от концентрации. При увеличении концентрации удельная электрическая проводимость сначала растет, а затем падает (рис. 115). В разбавленных растворах удельная электрическая проводимость с дальнейшим разбавлением всегда уменьшается.
Г Эквивалентная электрическая проводимость возрастает с увеличением разведения (рис. 116): у слабых электролитов — вследствие изменения.„степени диссоциации, у сильных электролитов — в ре-
Рис. 115. Зависимость удельной электрической проводимости от концентрации растворов электролитов
Рис. 116. Зависимость эквивалентной электрической проводимости от концентрации электролитов
зультате уменьшения электростатического взаимодействия между ионами. Эквивалентная электрическая проводимость растворов сильных электролитов с ростом разведения при с-М) стремится к предельному значению Ао.
Согласно закону независимого движения ионов в разбавленных растворах (закон Кольрауша)
Ao = Af+A7, (ХШ.7)
где Ло+ и Ло — ионные электрические проводимости при бесконечном разведении. Ионные электрические проводимости при беско-
271
вечном разведении пропорциональны абсолютным скоростям движения ионов:
= (ХШ.8)
где — абсолютные скорости движения ионов; F— постоянная Фарадея.
Для растворов сильных электролитов отношение Л к Ло равно некоторой величине, называемой коэффициентом электрической проводимости*
/Д = Л/ЛО. (ХШ.9)
Коэффициент электрической проводимости }л вносит поправку на межионное взаимодействие в растворах сильных электролитов при прохождении тока. Он уменьшается с повышением концентрации и приближается к Ло при бесконечном разведении. В растворах слабых электролитов /д= 1,а соотношение (VIII.9) позволяет рассчитать степень диссоциации а:
а~Л/Л0. (XIII. 10)
Согласно теории сильных электролитов Дебая — Хюккеля каждый ион полностью диссоциированного электролита окружен ионами, создающими поле противоположного знака. Такое распределение ионов в пространстве называется ионной атмосферой. При наложении внешнего пол я центр ал ьн ы й ” иЖТГионна1^^тмо с ф ер а, как обладающие зарядами, одинаковыми по величине, но обратными по знаку, движутся в противоположные направления. Силы межионного взаимодействия вызывают торможения, растущие с увеличением концентрации, и, следовательно, уменьшающие эквивалентную электрическую проводимость. Движение ионной атмосферы в сторону, противоположную центральному иону, вызывает электрофоретическое торможение, обусловленное движением сольватированного иона против потока сольватированных ионов ионной атмосферы. Второй эффект торможения обусловлен нарушением симметрии расположения ионной атмосферы вокруг центрального кона при его движении под действием поля. Движение приводит к разрушению ионной атмосферы позади иона и образование ее на новом месте. Для этого требуется время релаксации, и потому позади движущегося иона всегда находится некоторый избыток заряда противоположного знака, тормозящего его движение. Это торможение называют релаксационным. На скорость движения кона в растворе влияет вязкость, среды, создавая дополнительный эффект трения, который учитывается уравнением Стокса fT = 63iT)n7, где /т— сила трения; ц — вязкость растворителя; г —радиус кона; v— скорость движения иона.
Эффект торможения ионов зависит от концентрации электролита, поэтому эквивалентная электрическая проводимость может быть описана уравнением _ _
Л = Л0-Л/с, ХХШ.Н)
где АУ с — функция обоих торможений. Уравнение (XIIIJ11) пред
272
ставляет собой прямую с тангенсом угла наклона, равным А. Экстраполяцией зависимости A=f(j/ с) на нулевую концентрацию мойно рассчитать эквивалентную электрическую проводимость при
бесконечном разбавлении До (рис. 117). Для бинарного 1—1 -валентного электролита в разбавленных растворах постоянная А в (III.11) представляет сумму электрофоретического А* и релаксационного В взаимодействий: А—Д*+ВЛо. Подставив значения Л* и В в уравнение (XIII.11), получим
Рис. 117. Зависимость эквивалентной электрической проводимости от корня квадратного из концентрации растворов электролитов
(XIII. 12)
Для водного раствора 1—1-зарядного электролита, ес-
ли принять Т=298К, ен2о^78,3 и ц = 8.9-10 4 Па* с, уравнение (XIII.12) преобразуется к виду
Л=Л0— [60,64 + 0,2273Лд|Ус. (Х1П.13)
4. ЭЛЕКТРИЧЕСКАЯ ПРОВОДИМОСТЬ НЕВОДНЫХ РАСТВОРОВ ЭЛЕКТРОЛИТОВ
Электрической проводимостью обладают не только водные, но и неводные растворы электролитов. Электрическая проводимость неводных растворов также определяется концентрацией ионов и скоростью их движения; она зависит от вязкости и диэлектрической проницаемости среды.
Для неводных растворов электролитов характерно явление аномальной электрической проводимости, которое заключается в том, что при увеличении концентрации электролита эквивалентная электрическая проводимость в растворителях с низкими диэлектрическими постоянными проходит через минимум, за которым следует плавный подъем (рис. 118). Аномальная электрическая проводимость объясняется ассоциацией ионов электролитов. Ассоциация наиболее отчетливо проявляется в растворителях с низкой диэлектрической постоянной и приводит к появлению в растворе комплексных молекулярных и конных соединений. Появление в растворе ионных пар наряду с обычными молекулами приводит*к более быстрому падению электрической проводимости. В концентрированных растворах возможно образование ионных тройников. Представление об конных тройниках позволяет объяснить минимум на кривой
273
зависимости эквивалентной электрической проводимости от разве
дения, а также появление максимума.
Способность растворителя изменять степень диссоциации растворенного вещества зависит от его диэлектрической проницаемости: чем она выше, тем
’Л,Ом'1'см2
К/7
больше диссоциирует вещество в растворе (правило Каблукова — Нерн-ста — Томсена).
Разведение, отвечающее минимуму электрической проводимости, и диэлектрическая проницаемость 8 растворителя связаны соотношением з>—
гу V —const. Для неводных растворителей элек-
Рис. 118. Кривые аномальной электрической проводимости:
1 — диссоциация электролита; 2 — ассоциация; 3 — комплексная диссоциация
фект описывается уравнением (правило ского)
трическая проводимость зависит от вязкости растворителя, с ростом ее снижается электрическая проводимость. Этот эф-
Вальдена — Писсаржев-
Aoiq0 = const,
(ХШ.14)
где цо — вязкость чистого растворителя. Это правило соблюдается для ионов с большим радиусом, превышающим размеры молекул органического растворителя. Для ионов малых размеров это правило не соблюдается, так как в разных растворителях ионы по-разному сольватированы.
Влияние диэлектрической проницаемости и вязкости на подвижность ионов описывается формулой
Д0^0 = Ае"в/е, (ХШ.15)
где А, В — эмпирические константы.
Электрическая проводимость, включающая эффект вязкости среды, называется корригированной электрической проводимостью
Дк- Д(7)Л1о), (ХШ.16)
где ц, цо — вязкость при данном к бесконечном разбавлении.
5. ЗАВИСИМОСТЬ ЭЛЕКТРИЧЕСКОЙ ПРОВОДИМОСТИ ОТ ТЕМПЕРАТУРЫ
Электрическая проводимость растворов увеличивается с ростом температуры. С повышением температуры уменьшается вязкость растворов согласно уравнению
'It = ^еЕ^Т; (XIII. 17)
274
приэтом падает сопротивление, которое испытывают ионы при движении, и скорость перемещения ионов к электродам увеличивается. Зависимость электрической проводимости растворов электролитов от температуры выражается
(XIII. 18)
где А — константа, не зависящая от температуры; Е — энергия активации процесса, определяющего скорость движения ионов. Дифференцируя уравнение (XIII.18) по температуре, получаем
d In Др 1 d Ар
(XIII. 19)
dT Ло dF RT%'
Интегральная форма (XIIIJ19) имеет вид
4*const.
(XIII ,20)
При интегрировании в пределах от Г] до Г2 уравнение (ХШ.19) приобретает вид
Влияние температуры на электрическую проводимость при бесконечном разведении может быть представлено эмпирическим уравнением
Ар, Т = Лр.298 [1 4- а (Г - 298) -Ь ₽ (Г - 298)2]
(XIII. 22)
у. где Ло, т, До, 298 — электрическая проводимость при бесконечном
f разведении при Т к 298 К; а, р— константы для данного раствори-
v теля. В узком интервале температур константой р можно пренебречь; значение а для различных ионов, за исключением ионов Н+ и ОНу колеблется в пределах от 0,016 до 0,022 в зависимости от л типа электролита.
Зависимость удельной электрической проводимости от температуры проходит через максимум, который определяется концентрацией, типом электролита и природой растворителя.
Дифференцируя уравнение (ХШ.22) по температуре и пренебрегая значением р для узкого интервала температур, получаем д выражение
и, следовательно, энергия активации будет
= aRT 2.
(XIII. 24)
275
6. РАВНОВЕСИЕ В РАСТВОРАХ ЭЛЕКТРОЛИТОВ
Для слабых электролитов, диссоциирующих по схеме АВ^А+ч-Ч-В“, константа электролитической диссоциации выражается
или
[А+][В~] д2С
[АВ] = 1 - а
_____Л2С
СС=АО(ДО-Д) •
(XIII.25)
(XIII. 26)
Уравнение (XIII.26) известно как закон разведения Оствальда. Его легко можно преобразовать к виду
(XIII.27)
Тогда для слабых электролитов зависимость 1/A=f(Ac) будет выражена прямой (рис. 119) с тангенсом угла наклона, равным 1/(ЯсЛ02). Величину Ао находят
экстраполяцией линейной зависимости на нулевую концентрацию. Константу диссоциации слабого электролита рассчитывают по уравнению
Ke = tga/A2. (XIII.28)
Значения Ао и Кс постоянны, не зависят от концентрации раствора электролита, а зависят только
Рис. 119. Графический метод оп- от температуры и природы ве-ределения Хо и КдЙСс ществ.
Однако при точных расчетах даже для слабых электролитов не следует пренебрегать различием между активностью и концентрацией:
/ЙАВ»
(XIII.29)
ИЛИ
Ка = (Ч+С3_/С^ (Ya+Yb_/Y±ab) ,
(XIII. 30)
где Ка— термодинамическая константа равновесия реакции диссоциации; Ya+, Yb~> Y±ab — коэффициенты активности соответствующих ионов и недиссоциированных молекул электролита. Так как Ya+, Yb_=y2±ab, гдеу+дв — средний коэффициент активности ионов, выражение (ХШ.ЗО) можно преобразовать:
Ка = ^Y2±ab/Y±ab- (XIII. 31)
Для незаряженных частиц у±ав=1 и тогда
(XIII. 32)
276
или
pKa=PKr-21gY±AB.
(ХШ.ЗЗ)
Коэффициент активности рассчитывают по уравнению
lg Y±ab = > (XIII. 34)
где A—коэффициент пропорциональности, постоянный для данного растворителя при данной температуре и равный 1,823-106(е?)3/2; г — диэлектрическая постоянная растворителя; Z\ , z2 — заряды ионов; I — ионная сила раствора;
/ = 1/2 2^?, (XIII. 35)
где Ci — концентрация каждого вида кона, присутствующего в растворе; zi — заряд иона. При вычислении ионной силы раствора надо пользоваться истинной конной концентрацией; для слабого электролита эта величина равна g = clc.
7. ВЛИЯНИЕ ТЕМПЕРАТУРЫ НА КОНСТАНТУ ДИССОЦИАЦИИ РАСТВОРОВ ЭЛЕКТРОЛИТОВ
Зависимость константы равновесия диссоциации от температуры описывается уравнением изобары Вант-Гоффа. По температурной зависимости константы диссоциации можно рассчитать ряд термодинамических функций процесса диссоциации: энергию Гиббса, теплоту диссоциации, энтропию диссоциации (см. с. 70, 71). Эти зависимости можно использовать для изучения растворимости малорастворимых соединений. Зависимость растворимости от температуры выражается уравнением
d Inc А//
dZ ~ /?Г2 ’
(XIIL36)
где АЯ— дифференциальная теплота растворения; с — концентрация насыщенного раствора, моль/л.
Интегральная форма уравнения (XIII.36) позволяет рассчитывать дифференциальную теплоту растворения, если известна растворимость при двух температурах,
st2 ЬН Г 1 1 ]
(ХШ.37)
В насыщенном растворе существует равновесие между твердой солью к ее насыщенным раствором МА^М++А~. Для этого процесса константа равновесия Ка выражается
~ ~ /атв»
где к а~ — активности катиона и аниона; аТв— активность твердой соли. При данной температуре произведение — величина постоянная и называется произведением растворимости ПР = а+а_.
277
Учитывая, что а = у±с и что при малой растворимости соли коэффициенты активности у+ и у- равны единице, ПР может быть выражено:
ПР==СК+СА-’
где ск+, — концентрации катиона и аниона в насыщенном растворе.
Работа 1. Измерение электрической проводимости растворов электролитов
Рис. 120. Электрическая схема установки для измерения электрической проводимости
Измерение электрической проводимости растворов электролитов производят при помощи моста Кольрауша, питаемого переменным током высокой частоты (2-Ю3—3-103 Гц) от звукового генера-тора^Установка для измерения сопротивления раствора электролита состоит из барабанного реохорда с известным сопротивлением (курбельные магазины сопротивления от 0,1 до 105 Ом), сосуда для измерения электрической проводимости (рис. 120), нуль-инструмента (низкоомный телефон, индикатор нуля, осциллограф) . Компенсационная схема моста Кольрауша основана на применении закона Кирхгофа, согласно которому в точке А ток разветвляется и идет по проводникам АСВ и ADB. Обозначив силу тока в проводнике ADB через /1, а. в проводнике АСВ — че
рез /2 и выразив падение напряжения на участке цепи через произведение силы тока на соответствующее сопротивление, получим
[XRX = 1%АС,
(XIII. 38>
(XIII. 39)
Равенства (XIII.38) и (XIII.39) отвечают условию балансирования моста; отсутствие тока в проводнике CD достигается подбором соответствующего сопротивления и передвижного контакта С по реохорду АВ. Тогда
Rx (АСЮ В), (XII 1.40)
где Rx — сопротивление раствора. Из уравнение (XIIL3) следует, что
(XIII.41)
278
Если соотношение 1/S для данного сосуда (данной пары электродов) постоянно, то уравнение (XIII.41) преобразуется:
x = <p/£x. (ХШ.42)
В уравнение (ХШ.42) входят две неизвестные величины х и ф, следовательно, удельная электрическая проводимость может быть найдена лишь после того, как будет определена константа прибора ф. Так как электрическая проводимость раствора зависит от размеров электродов, расстояния между ними, их формы, взаимного расположения, степени погружения, то удельная электрическая проводимость х пропорциональна измеренной электрической проводимости, т. е. х=фхэ.
Последовательность выполнения работы. Для определения константы прибора ф применяют стандартные растворы, удельные электрические проводимости которых известны для широкого диапазона температур. К стандартным растворам относится 0,02 н. КС1. Данным раствором ополоснуть стакан с введенными в него электрода-шцналилИ^мОхш^^ на
Компенсационной схеме сопротивление раствора. При измерении1 следует подобрать три значения сопротивления 7<м (например 100, 200, 300 Ом), при которых точка компенсации находится в средней части шкалы реохорда, и для каждого значения /?.м рассчитать Rx по (XIII.40). Расхождение между параллельными измерениями не должно превышать 2—3 Ом. Константу прибора вычислить по уравнению
Т = хтабЛ/?Х(Ср. (XIII.43)
а
Рис. 121. Электроды для измерения электрической проводимости
Точность измерения электрической проводимости зависит от состояния поверхности электродов. Поверхность электродов платинируют, что значительно увеличивает их рабочую поверхность. Измерение электрической проводимости проводят в стеклянном сосуде с впаянными друг против друга платинированными электродами (рис. 121, а, б). При наполнении сосуда рабочей жидкостью не следует прикасаться к электродам пипеткой, так как можно повредить платинированную поверхность электрода, что исказит результат измерения.
Для платинирования электроды погрузить в раствор, содержащий в 100 мл воды 3 г тетрахлорида платины и 0,2—0,25 г ацетата свинца (ацетат свинца способствует осаждению мелкодисперсной платины, осадок платины при этом не содержит свинца). Через электро-
2 79
ды пропустить постоянный ток напряжением 2—4 В, направление тока менять через каждые 0,5— 1 мин. Для платинирования новых электродов требуется 10 мин, для использованных — 3 мин. По окончании платинирования электроды становятся бархатисто-черными. Электроды соединить последовательно и погрузить в 1О°/о-ный раствор серной кислоты, через который в течение нескольких минут пропускать ток. Катодом служат электроды, анодом — платиновая проволока. Ток пропускать для удаления хлора, адсорбировавшегося на платиновых электродах; хлор восстанавливается на катоде и переходит в раствор, образуя НС1. После этого электроды тщательно промыть и хранить в дистиллированной воде.
Основным инструментом в открытой схеме моста Кольрауша является барабанный реохорд, значение плеч которого используется при измерениях сопротивлений растворов электролитов. Для повышения точности измерения при расчете систематических и случайных ошибок следует производить калибрование проволоки реохорда. Толщина проволоки реохорда обычно неодинакова по всей
длине и, кроме того, она изменяется с течением времени за счет истирания при движении контакта АС (см. рис. 120). Вследствие это-
Показания реохорда, мм
Рис. 122. Калибровочная кривая
го сопротивление проволоки неодинаково на различных ее участках. Проволоку реохорда калибровать следующим образом: в разветвления моста AD и DB включить известные сопротивления такой величины, чтобы их соотношения были равны 9 : 1; 8 : 2; 7 :3; 6:4 и т. д. Длины плеч проволоки АС и СВ должны находиться в
таких же соотношениях.
При этом проволока будет проверена в девяти точках на расстоянии 100, 200, 300,..., 900 мм. Действительную величину плеча реохорда определить для каждого соотношения сопротивлений по по
ложению подвижного контакта, соответствующего состоянию компенсации моста.
На основании опытных данных определить поправку для плеча любой длины и построить калибровочный график на миллиметровой бумаге и использовать его при расчете сопротивлений (рис. 122).
Примечание. Для точного измерения сопротивлений растворов электролитов могут быть использованы реохордные мосты типа Р-568, Р-572 и др. с осциллографическим индикатором нуля. Питание моста осуществляется от генератора звуковой частоты, который позволяет варьировать частоту переменного тока от 0 до 3*105 Гц. В мосте предусматривается возможность измерения емкостной (реактивной) составляющей измерительной цепи. Реохордным мостом можно измерять сопротивление растворов до 105 Ом.
280
Работа 2. Изучение влияния температуры на электрическую проводимость и вязкость растворов электролитов в воде и водно-органических растворителях
Последовательность выполнения работы. В ячейке специальной конструкции (рис. 123) измерить константу прибора (см. с. 279). Затем тщательно промыть прибор и пипеткой внести в шарик трубки около 10 мл раствора электролита известной концентрации в воде или в органическом растворителе. Резиновым баллончиком через трубку и кран 1 засосать раствор в оба шарика вискозиметра 2 и 3 так, чтобы раствор полностью, без воздушных пузырьков, заполнил всю ячейку немного выше отметки а. Перекрыть кран 1 и приступить к измерению электрической проводимости раствора при различных температурах. Ячейку присоединить к термостату, контактным термометром установить нужную температуру опыта, в течение 5—7 мин выдержать раствор в данном температурном режиме и только после этого приступить к измерениям (см. с. 278). При исследовании водных растворов электролитов измерительным
прибором может служить мост сопротивлений Р-38. Интервал изучаемых температур 288— 360 к.
Одновременно при тех же температурах опыта измерить вяз- : кость раствора электролита. Для этого открыть кран 1 и отметить по секундомеру время истечения раствора из шарика 2 (от отметки а до отметки б), вновь засосать раствор в шарик 2 выше верхней отметки и повторить измерение. Расхождение в параллельных измерениях не должно превышать 1 с. Вязкость раствора вычислить по уравнению
V =^iPP1h2o/(z2P2), (XIII.44)
где ц — вязкость растворителя; /1 — время истечения раствора; /2 — время истечения чистого растворителя; pi — плотность раствора; р2—'Плотность чистого растворителя. Для расчета температурную зависимость плотности
Рис. 123. Прибор для измерения электрической проводимости и вязкости электролитов
растворителя заимствовать из
справочника, плотность раствора и время истечения чистого растворителя должны быть предварительно измерены и сведены в таблицы, температурную зависимость вязкости воды (растворителя) заимствовать из справочника.
281
По полученным экспериментальным данным рассчитать удельную и молярную электрические проводимости по уравнениям (XIII.4) и (XIII.5). По рассчитанным значениям вязкости раствора и воды при различных температурах рассчитать по уравнению (XIIL16) корригированную эквивалентную электрическую проводимость. Построить графики зависимости In ц от и по тангенсу угла наклона прямой рассчитать энергию активации вязкости. Вычислить по уравнению (XIII.21) энергию активации электрической проводимости и проверить расчет графически. Сопоставить полученное значение со значением этой величины, рассчитанной по уравнению (XIII.17). Результаты измерений занести в таблицы по образцам:
Электролит . . растворитель . . .; концентрация раствора . . .
т, к Сопротивление, Ом Электрическая проводимость Дж/моль
удельная, Ом-1 - см—1 эквивалентная, Ом-1-смг-г-экв—1
Время истечения, с
Вязкость, Па-с
раствора растворителя
относительная
раствора
-7], Дж/моль
Р а б о т а 3. Изучение зависимости электрической проводимости растворов электролитов от концентрации
Для измерения электрической проводимости растворов электролитов используется та же аппаратура и методика измерения, что и при определении константы прибора (см. с. 279).
Последовательность выполнения работы. Собрать установку по схеме, приведенной на рис. 120. Тщательно проверить плотность зажима контактов, проволоку барабанного реохорда протереть ватой, смоченной этанолом. При измерениях следить, чтобы подвижной контакт плотно прижимался к проволоке реохорда. Термостатированную ячейку перед измерениями залить раствором электролита и выдержать в течение 10 мин при температуре опыта. Для измерения удельной электрической проводимости раствора электролита необходимо знать константу прибора, которую измеряют по 0,02 н. КС1 (см. с. 279). Полученные экспериментальные данные записать в таблицу по образцу.
282
Раствор 0,02 н. КС1. Температура опыта .
Номер измерения р изм> Ом Плечи реохорда Я, Ом *сР, Ом X, Ом—1-см—1 Ч> Примечание
а 100 — а
\
После этого тщательно промыть ячейку дистиллированной водой и двух-, трехкратным погружением в воду промыть платиновые электроды. Затем ячейку заполнить дистиллированной водой и измерить ее электрическую проводимость, Для дважды перегнанной воды, хранящейся в кварцевой или серебряной посуде без доступа СО2, при 291 К х-4,4-10-8 Ом-1-см-1. В дистиллированной воде в результате растворения в ней СО2 и NH3 и выщелачивания стекла х=1 -10-6 Ом-1-см"1. Чтобы определить удельную электрическую проводимость электролита^надо определить удельную электрическую провбдць^тьГводы^и вычесть ее значение из электрической проводимости раствора. Электрическую проводимость воды изме-рятГШДЩОО Ом. Трехкратные измерения сопротивле-ний позволяют достаточно точно оценить удельную электрическую проводимость воды. Расчет производить по уравнению (XIII.42). Полученные данные записать в таблицу по образцу:
Температура опыта . . .
Номер измерения
^изм» Ом / Плечи реохорда /?, ом ^ср» Ом \абл > Ом—^Х Хсм—1 хНвО, Ом-IX Хсм—1
а 100 — а
Примечание
На степень диссоциации электролита влияют растворенные в воде угольная кислота и аммиак. Поэтому для определения электрической проводимости растворенного вещества следует вносить поправку. Чтобы эта поправка была наименьшей, применяют дважды перегнанную воду. Сначала перегонку дистиллированной воды осуществляют с КМпО4 для окисления органических примесей, а затем с Ва(ОН)2. Перегонку проводят в кварцевой установке (кварцевые приемник, холодильник и колба для перегонки). Воду хранят в емкостях из пирексового стекла.
При измерениях электрической проводимости раствора электролита в термостатированную ячейку налить 50 мл исследуемого раствора. Погрузить платиновые электроды и выдержать раствор при температуре опыта в течение 40 мин. Электроды присоединить к мостовой схеме и приступить к измерению. Известные сопротив-
283
ления подбирать так, чтобы при компенсации подвижной контакт находился в пределах четырех—шести делений шкалы реохорда. Измерения проводить при трех известных сопротивлениях. Последующие разбавления проводить пипеткой на 25 мл, отбирать 25 мл раствора и добавлять 25 мл дистиллированной воды. Раствор каждый раз тщательно перемешивать подъемом и погружением в раствор платиновых электродов. Вынимать электроды из ячейки при разбавлении исходного раствора категорически запрещается. Если исходная концентрация раствора 1/32 г-экв/л, то последующими раз-' бавлениями приготовить растворы содержанием (г-экв/л): 1/64/ 1/128, 1/256, 1/512, 1/1024, измерить их сопротивления и полученные э результаты записать в таблицу по образцу:
Исследуемый раствор . . .; температура опыта . . .
Номер измерения R хизм> Ом Плечи реохорда R, Ом *ср, Ом X, Ом — 1Х Хсм — 1 Ом-1Х Хсм2 Ao Клисе
а 100—а
На основании полученных данных рассчитать:^) константу прибор а/2/>удельную электрическую проводимость воды/J) удельную и эйм(валентную электрические проводимости исследуемого электролита для шести концентраций; 4) построить графики зависимости удельной и эквивалентной электрических проводимостей от разведения или концентрации, а также зависимость эквивалентной электрической проводимости от корня концентрации. Для раствора сильного электролита экстраполяцией до с = 0 определить значение До, сравнить его со значением, рассчитанным по подвижностям ионов;(ю) построить график 1/Д—f(Ac); если зависимость линейна, то экстраполяцией до с=0 определить для раствора слабого электролита величину До и сравнить ее значение с рассчитанным по уравнению (XI1L7) . При расчете учесть температурные коэффициенты подвижностей^ рассчитать степень диссоциации раствора слабого электролита noi(XIII. 10) для шести концентраций. Построить график зависимости степени диссоциации электролита от разведения или концентрации/?^ рассчитать для раствора слабого электролита для шести концентраций по уравнению (XIII.25) и графически константу диссоциации КДИсс; 8) графически определить для раствора сильного электролита угловой коэффициент прямой А и сравнить его с рассчитанным по уравнению (XIIIJ13); 9) оценить вклад релаксационного и электрофоретического эффектов в общий эффект торможения согласно (XIII.12); 10) рассчитать коэффициент электрической проводимости по (XIII.9) для всех полученных концен
284
траций и сравнить с табличным значением для 1—1-зарядных электролитов; оценить погрешности измерений и точность полученных расчетных данных.
Работа 4. Определение растворимости труднорастворимой соли при различных температурах методом электрической проводимости
Последовательность выполнения работы. Небольшое количество труднорастворимой соли растереть в ступке. Тонкоизмельченную соль поместить в коническую колбу с притертой пробкой и два-три раза промыть дистиллированной водой для удаления легкорастворимых примесей. Отмытую соль залить 100 мл дистиллированной воды и, плотно закрыв колбу пробкой, непрерывно взбалтывать ее в течение 20 мин. Полученную суспензию отфильтровать через стеклянный фильтр, а осадок употребить для приготовления насыщенного раствора. Приготовить еще такой же насыщенный раствор и полученную суспензию профильтровать через осадок, нанесенный на фильтр. Такое фильтрование предупреждает образование пересыщенного раствора труднорастворимой соли. Полученный раствор слить в сосуд для измерения электрической проводимости, погрузить его в термостат с определенной температурой и измерить сопротивление раствора.
Осадок в колбе вновь залить дистиллированной водой и. при непрерывном помешивании колбу погрузить в термостат, но при более высокой температуре. (Температура опыта должна быть задана.) Раствор с осадком выдержать в термостате в течение 20 мин, дать осадку отстояться, раствор слить в сосуд для измерения электрической проводимости и измерить его сопротивление. Результаты измерения записать в таблицу (см. с. 284). Затем измерить постоянную сосуда и электрическую проводимость воды, применяемой для приготовления насыщенных растворов. Электрическую проводимость воды измерять при температурах проведения опыта; значение температуры учитывать при определении электрической проводимости СОЛИ хСОли = ХРасТв— хн2о-
Эквивалентную электрическую проводимость насыщенного раствора труднорастворимой соли можно приравнять электрической проводимости при бесконечном разведении. Эквивалентную электрическую проводимость при бесконечном разведении рассчитать по уравнению (XIII.22) с учетом температурных коэффициентов подвижностей, значения которых взять из справочника. Затем рассчитать растворимость соли S (моль/л) по формуле
S ~ хс оли1000/Ад.
Определив растворимость при двух температурах, рассчитать AG°, АЯ° и AS°. Если определение растворимости труднорастворимых соединений приводилось при трех-четырех температурах, то дальнейший расчет проводить графически. Построить график lg (1/Т) и по тангенсу угла наклона прямой рассчитать ЛН°.
285
Полученное значение дифференциальной теплоты растворения труд-нерастворимой соли использовать для дальнейших термодинамических расчетов.
На основании полученных результатов рассчитать: 1) растворимость соли по значениям удельной электрической проводимости и электрической проводимости при бесконечном разведении; 2) ^дифференциальную теплоту Д//° растворения соли, 3) энергию Гиббса AG° растворения и изменение энтропии растворения. Построить график зависимости растворимости соли от температуры, если измерения растворимости проводили при нескольких температурах.
Полученные расчетные данные свести в таблицу по образцу:
Соль ПРГ1 ПРГ 1 2 А 6°, Дж/моль АЯ°, Дж/моль 1 AS0, Дж/(моль-К)
ГЛАВА XIV
ЭЛЕКТРОДВИЖУЩИЕ СИЛЫ ГАЛЬВАНИЧЕСКИХ ЦЕПЕЙ
1. ГАЛЬВАНИЧЕСКИЕ ЭЛЕМЕНТЫ
При соприкосновении двух проводников, различающихся физическими или химическими свойствами, происходит их разноименная .электризация, т. е. проводники оказываются при различных потенциалах. На границе раздела проводников возникает скачок потенциала, который равен разности потенциалов проводников:
Ег=Е'2-~Е[. (XIV.1)
Если образовать цепь из нескольких соприкасающихся проводников, то скачки потенциалов возникнут на всех границах разделов и сумма их составит электродвижущую силу (э.д. с.) цепи:
Ех 4-Е2-Н ... 4-Е/==Е. (XIV.2)
Из соотношений (XIV.1) и (XIV.2) вытекает, что э.д.с. цепи равна разности потенциалов между последним и первым проводниками, входящими в цепь:
Е'. (XIV.3)
При замыкании правильно разомкнутой цепи (первый и последний проводники цепи тождественны), состоящей только из металлов, в цепи не наблюдается химических процессов и ток не возникает; э. д. с. этих цепей равна нулю. Если же некоторые из проводников (хотя бы один), составляющих цепь, являются элек
2BQ
тролитами, то э.д.с. такой правильно разомкнутой цепи не равна нулю. При замыкании цепи в ней возникает электрический ток и на границе соприкосновения металлов и электролитов протекают химические процессы. Такие цепи .называются гальваническими. Часть гальванической цепи, состоящая из электролита и двух погруженных в него металлических электродов (или из нескольких соприкасающихся друг с другом электролитов и двух электродов,, погруженных в крайние электролиты), называется гальваническим элементом.
2. МЕХАНИЗМ ВОЗНИКНОВЕНИЯ Э. Д. С. *
Скачки потенциалов, из которых складывается э.д. с. гальванической цепи, можно разделить на три типа: 1) скачки потенциалов^ возникающие на границе соприкосновения двух металлов (скачки контактных потенциалов); 2) скачки потенциалов, возникающие на границе соприкосновения металла с электролитом (скачки электродных потенциалов); 3) скачки потенциалов, возникающие на границе соприкосновения двух электролитов (скачки диффузионных потенциалов). Механизм возникновения скачков потенциалов различен.
Контактные потенциалы. «Свободные» электроны металла могут выходить за границы ионной решетки; вследствие этого на поверхности металла возникает положительный (по отношению к потенциалу вакуума) потенциал. Величина этого потенциала зависит от природы металлов. Это обусловливает частично скачок потенциала на границе соприкосновения двух металлов. Скачок потенциала при соприкосновении двух металлов возникает за счет перехода свободных электронов из одного металла в другой. Так как энергетические уровни электронов в металлах не одинаковы, то из того металла^ где этот уровень выше, будет переходить в другой металл, где этот уровень ниже, большее количество электронов, чем в обратном направлении за то же самое время. Направленный процесс перехода прекратится, когда энергетические уровни электронов в обоих металлах сравняются. При этом металл, из которого преимущественно уходили электроны, зарядится положительно, а другой — отрицательно. Контактные потенциалы, как правило, малы, и при измерении э. д. с. гальванических элементов их можно не учитывать.
Электродные потенциалы. Возникновение электродных потенциалов обусловлено рядом сложных процессов, протекающих на границе соприкосновения металла с электролитом.
Если металл погружен в водный раствор своей соли, то ионьц находящиеся на поверхности кристаллической решетки металла, взаимодействуя с сильно полярными молекулами воды, гидратируются. Их связь с остальными ионами кристаллической решетки ослабляется, и те ионы, кинетическая энергия теплового движения которых достаточно велика, переходят в слой раствора, прилегаю-
287
Рис. 124. Схема двойного электрического слоя
щий к поверхности металла. Одновременно из раствора переходят на металл ионы, обладающие достаточно большой кинетической энергией. Если скорость перехода ионов в раствор больше, чем скорость перехода ионов из раствора на металл, то поверхность металла заряжается отрицательно за счет избыточных электронов. Возникает электростатическое притяжение между электронами металла и ионами, перешедшими в раствор. Скорость перехода ионов в раствор уменьшается, а скорость перехода ионов на металл увеличивается. Обе скорости сравниваются. Устанавливается подвижное равновесие, причем на границе соприкосновения металла с раствором образуется двойной электрический слой (рис, 124, а) и возникает определенный скачок потенциала.
Если начальная скорость перехода ионов из раствора на металл больше скорости пёрехода ионов с металла в раствор, металл заряжается положительно, а раствор у поверхности металла — отрицательно за счет избытка анионов. Вследст-уменьшается скорость пе-
•! 'L А.."
вие электростатического взаимодействия рехода ионов из раствора на металл и увеличивается скорость обратного провеса. Обе скорости сравниваются; образуется двойной электрический слой (рис. 124, б) и возникает определенный скачок потенциала. Двойной электрический слой способствует возникновению электродного потенциала.
Если начальная скорость перехода ионов из раствора на металл и начальная скорость перехода ионов из металла в раствор будут равны, двойной слой не образуется. Однако это не означает, что скачок потенциала на границе металл/электролит будет равен нулю. На поверхности м.еталла адсорбируются молекулы воды и их диполи обусловливают возникновение скачка потенциала. Скачок потенциала может возникнуть и за счет адсорбции поверхностью металла ионов, находящихся в растворе. Но даже когда нет адсорбированных на поверхности металла молекул или ионов, т. е. когда заряд поверхности равен нулю, то и тогда наблюдается скачок потенциала на границе металл/электролит, так называемый! потенциал нулевого заряда.
Электродные потенциалы, в основном, определяют э.д. с. гальванического элемента.
Диффузионные потенциалы. Диффузионный потенциал возникает на границе соприкосновения двух растворов разных электролитов или двух растворов, содержащих один и тог же электролит разной концентрации. Рассмотрим процесс, протекающий при сопри-
288
косновении двух растворов, содержащих один и тот же электролит разной концентрации.
В момент соприкосновения растворов ионы переходят из одного раствора в другой. Скорость перехода ионов из более концентрированного раствора в менее концентрированный будет больше, нежели скорость перехода ионов в обратном направлении. Так как подвижности катионов и анионов различны, то и.количество их, проходящее в начале диффузии через границу соприкосновения растворов, будет различно. Если подвижность катионов больше, то их больше перейдет в менее концентрированный раствор, чем анионов. Тогда менее концентрированный раствор у поверхности раздела зарядится положительно, а более концентрированный — отрицательно. Вследствие этого скорость движения катионов начнет уменьшаться, а скорость движения анионов — увеличиваться. Через некоторое время скорости катионов и анионов сравняются и количества их, переходящие границу раздела между растворами, станут равными. Образуется двойной электрический слой с определенным скачком диффузионного потенциала. Диффузионные потенциалы невелики; их величина не превышает нескольких сотых вольта. Точно измерить величину диффузионного потенциала трудно, так как она зависит не только от состава и концентрации соприкасающихся растворов, но и от других причин, например формы сосуда. Поэтому при измерениях э.д. с. нужно сделать диффузионный потенциал возможно малым. Это достигается соединением двух различных электролитов солевым мостиком. Последний представляет собой концентрированный раствор соли, ионы которой обладают примерно одинаковой подвижностью (КС1, KNO3).
Возникновение тока в гальванических цепях. Скачки потенциалов возникают и принимают равновесные значения через очень короткий промежуток времени после соприкосновения различных проводников до замыкания цепи. При замыкании цепи электроны от более отрицательного электрода потекут по соединительному проводу к электроду, обладающему более положительным потенциалом. Как только количество электронов на первом электроде начнет уменьшаться, нарушится равновесие в двойном электрическом слое и катионы с первого электрода станут переходить в раствор. Электроны, подойдя ко второму электроду, образуют с его катионами нейтральные атомы. Это нарушит равновесие двойного электрического слоя у второго электрода и катионы из раствора сейчас же начнут переходить на второй электрод. Таким образом, происходит непрерывное растворение одного электрода и выделение металла на другом электроде; одновременно во внешней цепи текут электроны и гальванический элемент непрерывно дает ток.
3. ТЕРМОДИНАМИКА ОБРАТИМЫХ ЭЛЕКТРОХИМИЧЕСКИХ СИСТЕМ
Когда контактные потенциалы очень малы, а диффузионные по-тенциалы уменьшены почти до нуля с помощью солевого мостика, ' .................................................. .... '..
э.д.с. гальванического элемента определяется разностью электрод-
10—2083
289
ных потенциалов. Ввиду сложной природы возникновения электродных потенциалов их величину и э.д. с. гальванических элементов не представляется пока возможным вычислить исходя из представлений о механизме электродных процессов. Расчет осуществляется с помощью термодинамических функций* Величина электрической энергии, полуШгнои от элемента, эквивалентна полезной максимальной работе протекающего в нем процесса:
nFE = -±G, (XIV.4)
или
E~—kG/nF, (XIV.5)
где Е — э.д.с.; —AG— максимальная полезная работа процесса; и— число электронов, участвующих в токообразующей реакции; F—1 Фарадей, равный 96 485 Кл/моль.
Если в элементе протекает реакция A + B^C + D, то величину полезной максимальной работы ее рассчитывают по уравнению
-AG = — RT \nKa + RT\n , (XIV.6)
где /? — универсальная газовая постоянная; Т — абсолютная температура; Ка — константа равновесия; ад, пв, ас, ац— активности всех веществ, входящих в состав раствора. Активность связана с концентрацией (моляльностью) соотношением а = ут, где у — коэффициент активности; m — моляльность. Это соотношение дает возможность вычислять активность исходных веществ и продуктов реакции, так как коэффициенты активности для ряда электролитов и ионов определяются экспериментально. Разделив обе части уравнения (XIV.5) на лЕ, получим
(XIV.7)
Согласно уравнению Гиббса — Гельмгольца
до = дя + г
(XIV.8)
где Д7/ — тепловой эффект реакции при постоянном давлении.
Решая совместно уравнения (XIV.7) и (XIV.8), получаем
ДЯ /дЕ \
Е=-—~4-Е — , nF \дТ )р
где дЕ/дТ — температурный коэффициент э. д. с. элемента.
Уравнение (XIV.9) имеет большое значение, так как, зная Е и дЕ/дТ, можно определить тепловой эффект реакции в элементе. Аналогично можно термодинамически вычислить и электродные потенциалы. Соотношение между AG =—nFE и Д/7 определяется знаком температурного коэффициента э.д. с.. Так, если (дЕ/ОТ) р = 0, то э.д. с. элемента не зависит от температуры, работа элемента происходит за счет убыли энтальпии, элемент работает без тепло-290
(XIV.9)
обмена. При отрицательном значений (дЕ1дТ)р, когда э.д. с. элемента уменьшается с повышением температуры, 7'ЛЗ<0, т. е. рабо-та становится метгьшетеплового эффекта соответствующего химического процесса. Избыток энергии элемент отдает в окружающую среду .При положительном значении (дЕ1дТ)р>0, когда э.д.с. элемента возрастает с повышением температуры, TAS>0, т. е. работа электрического тока становится больше теплового эффекта реакции. Теплота поглощается из окружающей среды, элемент работает с поглощением теплоты. Реакция, протекающая в гальваническом элементе,— эндотермическая. Величину температурного коэффициента dEfdT определяют экспериментально по графической зависимости э. д. с. от температуры, что позволяет рассчитать изменение энтропии реакции:
AS - 96 485/7 d £/d Т. (XIV. 10)
Например, на электроде протекает реакция восстановления по схеме
Максимальная полезная работа этой реакции будет:
-Д(7=/?Г1п/<а-/?Г In
(XIV. 11)
е
откуда
«Г , „ RT , «С ~ 1пКа — —Z" In —------п
nF nF а
е
(XIV. 12)
Так как активность электронов является постоянной то, переписав уравнение (XIV.11), получим
„ RT t Ка RT t
С* __ In „ "" In
nF
величиной
я— е
(XIV. 13)
Первый член правой части уравнения (XIV.13) при данной температуре— величина постоянная. Обозначим его через Е°:
ЕТ ка
—In
А
e
тогда уравнение Нернста преобразуется:
RT E = E° — In -------—
nF
Переходя от натуральных логарифмов к десятичным и подставляя числовые значения R и F, получим
„ 1,984-10-4Г f «с
Е Е° —------------- Ip-----
л а „ ,
ДГ4 Т"
(XIV. 14)
(XIV. 15)
10*
291
Когда каждая из активностей реагирующих веществ равна единице, то второй член правой части уравнения (XIV. 15) становится равным нулю и
е = е\ (XIV.16>
где Е° — стандартный (нормальный) потенциал. Величина стандартного потенциала зависит от температуры; обычно ее определяют при 25° С. Тогда уравнение (XIV.15) принимает вид
0,059 а™
Е = EQ — -— lg . (XIV. 17)
Л #исх
4. ЭЛЕКТРОДЫ
Электроды, составляющие гальванический элемент, делят на три группы: 1) электроды первого рода, обратимые относительно катиона; 2) электроды второго рода, обратимые относительно аниона; 3) окислительно-восстановительные электроды (редокс-электро-ды).
Электроды, обратимые относительно катионов. Металлические электроды. К данным электродам относятся металлы, погруженные в растворы своих солей, и водородный электрод. На границе металл— раствор протекает процесс по схеме
Мега+ +
Так как активность металла постоянна, то согласно уравнению (XIVJ17) для электродного потенциала металлического электрода имеем
^Ме = ^Ме + V5 ’я аме„+. (XIV. 18)
Водородный электрод (рис. 125) состоит из платиновой платинированной пластинки, наполовину погруженной в раствор, содержащий ионы водорода, и омываемой газообразным водородом. На поверхности раздела H2|H+ протекает реакция по уравнению
Н+ + е £. 1/2Н2
Электродный потенциал водородного электрода равен:
ан+
ещ = ен3 + °>0591§ —(XIV. 19) ан.
Водородный электрод при активности ионов водорода, равной единице, и при давлении газообразного водорода, равном 1 атм (при этом активность водорода равна его давлению), принят в качестве стандартного электрода сравнения; его потенциал условно считают при всех температурах равным нулю (£н.=0). Электродный потенциал водородного электрода при других условиях равен:
ан+
Ен =0,059 1g---Ру- , (XIV.20)
ан,
292
а если давление газообразного водорода равно единице, то
£На — 0,059 lg ан+ (XIV .21)
Поскольку пользоваться стандартным водородным электродом неудобно, его по мере возможности заменяют другими, более удоб-
Рис. 125. Водородно-каломельный гальванический элемент, применяемый для определения pH растворов
ными в работе электродами с постоянными скачками электродного потенциала. Такими электродами являются каломельный, хлоридсеребряный и хингидронный электроды.
Рис. 126. Каломельный электрод:
1 — ртуть; 2 — платина;
3 - Hg2CI2; 4 — КС1; 5-медь
Электроды, обратимые относительно анионов. К данным электродам относятся металлические электроды, покрытые малорастворимой солью соответствующего металла и погруженные в раствор другой, хорошо растворимой соли, содержащей тот же анион; примером такого электрода может служить каломельный электрод, хлоридсеребряный, кислородный и хлорный.
Каломельный электрод. Этот электрод (рис. 126) состоит из ртути, сверху которой находится паста из каломели, смешанной с чистой ртутью. Этот электрод соприкасается с раствором хлорида калия, предварительно насыщенным каломелью. На границе раздела Hg|Hg2C12| KCl протекает реакция по уравнению
l/2Hg2CI2 + e^Hg + CI-
Так как активности HgCb и Hg постоянны, то электродный потенциал каломельного электрода определяется уравнением
£к.э = <э - 0,059 lg а . (XIV.22)
U1
293
Так как активность ионов С1“ зависит от концентрации КС1, то скачок потенциала каломельного электрода зависит от концентрации хлорида калия в растворе. Применяются три вида каломельных электродов:
1. Насыщенный каломельный электрод, содержащий насыщенный раствор хлорида калия. Скачок его потенциала относительно стандартного водородного электрода при 25° С равен 0,2420 В; температурный коэффициент равен 0,00076 В/град.
2. Нормальный каломельный электрод, содержащий 1 н. КС1. Скачок его потенциала при 25°С равен 0,2810 В, температурный коэффициент— 0,00025 В/град.
3. Децинормальный каломельный электрод, содержащий 0,1 н. КО. Скачок потенциала этого электрода при 25°С равен 0,3358 В, температурный коэффициент — 0,00007 В/град.
Для приготовления каломельного электрода химически чистую каломель слегка растирают в фарфоровой чашке с ртутью. Смесь взбалтывают с заранее приготовленным раствором хлорида калия нужной концентрации. Образовавшуюся суспензию наливают в сосуд (рис. 126) на предварительно налитую туда ртуть. Электрический контакт осуществляется платиновой проволокой, впаянной в стеклянную трубку, проходящую через пробку сосуда.
Окислительно-восстановительные электроды. Процессы окисле-ние^восстановление протекают на всех электродах. Для одних электродов в этих процессах участвует материал электрода, для других электродов, называемых окислительно-восстановительными, материал электрода не участвует в реакции. Па поверхности электрода восстанавливаются и окисляются вещества, находящиеся в растворе, а сам электрод служит лишь передатчиком электронов. Примером окислительно-восстановительного электрода может служить платиновая пластинка, опущенная в раствор, содержащий FeCI2 и FeCl3. На электроде протекает процесс по уравнению
Fe3+ 4- е. y.Fe2+
Электродный потенциал этого электрода выражается уравнением
арР2+
£ з+/Р 2+ з+,р 2+ — 0,059 1g- -----, (XIV.23)
ГсЭ \/Fc-^ Fc^/I-cz^ a nt
Fe3 +
где £pe3+/Fe24- — стандартный электродный потенциал.
Для окислительно-восстановительных систем коэффициент активности для разбавленных растворов рассчитывается по уравнению
1g — = Л (4 - 4) У1- (XIV.24)
Yb
Подставив (XIV.24) в (XIV.23), получим
Е ~Е 4“ 1п 4“2,3 ^о) У *
пЕ пЕ v
(XIV. 25)
294
fi
I
Если растворитель вода, то при 298 К константа А равна 0,512 и уравнение (XIV.25) примет вид
„ Г.О 0.059 с
Е = EQ д----1g—
п с
0,03
п
(XIV ,26)
Ь'
4
до
о
Если окислительно-восстановительные реакции протекают при участии водородных или гидроксильных ионов, то окислительно-восстановительный потенциал системы зависит от концентрации (активности) ионов среды. Например, для системы МпГ |Мп2+ электродная реакция протекает в кислой среде: МпОГ4-8Н+ + + 5б^ ^Мп2++4Н2О и потенциал будет равен:
а8 .
МпО~
Л 94-Мп2+
К этому типу электродов относится и хингидронный электрод, который состоит из платиновой проволоки, опущенной в раствор, насыщенный хингидроном. Он применяется вместо водородного электрода, если pH раствора <8. На поверхности платины протекает реакция по уравнению
МпО-'/Мп2+ nF П
Электродный потенциал определяется выражением
2
о 0,059 хг = £хг 4- —
(XIV. 27)
&
В растворе хингидрона, когда соотношение активностей хинона и гидрохинона равно единице, электродный потенциал зависит только от активности ионов водорода:
17 Д’® в* 059 | 2
^хг = ^хг + “ЛГ" = £хг
(XIV. 28)
Стандартный потенциал хингидронного электрода при 25°С равен Есхг=+0,6994, его зависимость от температуры имеет вид
Е’г = 0,6994 - 0,00074 (t — 25). (XIV .29)
Для приготовления хингидронного электрода испытуемый раствор в стакане взбалтывают с небольшим количеством хингидрона (хингидрон набирают на кончик небольшого шпателя). Хингидрон оседает на одно стакана. Электродом служит гладкая слегка прокаленная платиновая проволока, впаянная в стеклянную трубку. Проволоку наполовину погружают в осадок хингидрона.
5. ГАЛЬВАНИЧЕСКИЕ ЭЛЕМЕНТЫ
Любой гальванический элемент состоит из двух электродов — катода и анода. На катоде протекает реакция восстановления, а на аноде — реакция окисления. Э. д. с. любого элемента является ве-
295
личиной положительной, так как токообразующий процесс идет самопроизвольно. Гальванические элементы можно разделить на два типа: 1) химические и 2) концентрационные.
Химические элементы состоят из двух различных электродов, например водородного и кислородного, каломельного и хингидронного. Примером химического элемента является элемент Якоби — Даниэля. При работе этого элемента у одного электрода протекает процесс по уравнению Cu2++2e^Cu°, а у другого — по уравнению Zn°-^Zn2++2e, суммарным результатом которых является уравнение реакции
ZnO 4- 012+ Zn2+ + CuO
Аналогичным образом протекают процессы и в других химических элементах. Таким образом, ток в этих элементах получается за счет химической реакции и э. д. с. их определяется максимальной полезной работой реакции. Э. д. с. может быть рассчитана как разность соответствующих электродных потенциалов. Например, э.д. с. элемента Якоби —Даниэля равна:
Е ~ ЕС^+/Си ~ £Zn2+/Zn - I £Cu2+/Cu + 2 ,g ЯСи2+
. 0,059 \
fiZn2+/Zn + 2 g Я2п2+ I ’
или
0,059 acu2+
Cu2+/Cu ~ Zn2+/Zn + 2 g a
(XIV.30)
Концентрационные элементы состоят из одинаковых электродов, погруженных в растворы с различной активностью соответствующих ионов, например:
Ag 1 AgNO3 (ад) || AgNO3 (а2) | Ag
где ai, а2—активности ионов Ag+ в растворе AgNO3 и AgNO3 соответственно, причем Различие в активностях обусловливает неравенство потенциалов электродов, что при замыкании цепи приводит к возникновению тока. При работе этого элемента у правого электрода протекает процесс по схеме Ag++e->Ag, а у левого — процесс, обратный первому, Ag-^Ag+4-е.
Очевидно, что никакой химической реакции в элементе не происходит. Однако при работе элемента активность ионов Ag+ в растворе AgNO3 (а2) будет уменьшаться, а в растворе AgNO3(ai) увеличиваться, т. е. активности ионов Ag+ будут выравниваться. Полезная максимальная работа этого процесса проявляется в виде работы тока, т. е. э.д. с. концентрационных элементов определяется максимальной работой изотермического выравнивания активностей. Этот процесс идет с поглощением теплоты из внешней среды. Та-296
ким образом, источником электрической энергии концентрационного элемента является теплота окружающей среды.
Э.д.с. концентрационного элемента равна:
— ©
~ ^2»
(XIV. 31)
поэтому
,059
(XIV. 32)
Работа 1. Измерение э. д. с. элемента Якоби— Даниэля
Рис. 127. Электрическая схема установки для измерения э. д. с. гальванических элементов
При работе гальванического элемента-его э.д.с. не сохраняет строго определенного значения вследствие изменений, происходящих у электродов. Поэтому точно э. д. с. измеряется методом ком-пенсации. Этот метод основан наизмерении э.д. с. элементу по раз-ности потенциалов в условиях обратимости.
На’1ШсГТ2/ приведена принципиальная схема измерения э.д.с. гальванического элемента. Компенсационная схема состоит из источника тока — аккумулятора Л разность потенциалов которого подается на реохорд АВ\ гальванометра 6 чувствительностью 10~6 А; элемента Вестона 4\ исследуемого элемента 3; переключателя 5; прерывателя 2 и подвижного контакта С, Метод основан на том, что измеряемая э. дГс. ^компенсируется э. д. с. аккумулятора. Аккумулятор 1 присоединен к концам сопротивления АВ (реохорд, барабанный реохорд, магазин сопротивления). С помощью прерывателя 2 с этого сопротивления снимается любая разность потенциалов ОТ нуля ДО ^ак ПОДВИЖНЫМ КОН-тактом С. Разность потенциалов аккумулятора направляется против из-меряедаи^^^с^мем^нтд. Аккумулятор и элемент включаются одноименными полюсами навстречу друг другу. Передвигая контакт С вдоль реохорда АВ при замыкании прерывателя 2,
можно найти такое положение, при котором падение напряжения аккумулятора на участке АС точно равно э. д. с. иссле.дуемрго эле-, мента. Чтобы правильно установить точку:=тойпёнгации71Геобходи-мо контакт передвигать вправо и влево на 1 мм от найденной точки компенсации. При этом стрелка гальванометра должна отклоняться от нулевого положения на одинаковую величину (приблизительно одно—три деления) в разные стороны. В момент компенсации
297
гальванометр покажет в боковой цепи отсутствие тока, а э. д. с. элемента будет равна
Ех = Елкк(АСх/АВ), (XIV. 33)
где Ех, £акк — э. д. с. исследуемого элемента и аккумулятора; АСХ — отрезок проволоки, на котором падение напряжения аккумулятора равно э.д.с. исследуемого элемента.
Непосредственно из уравнения (XIV.33) нельзя вычислить значение Ех, так как .неизвестно значение £акк. Для этого в боковую цепь вместо исследуемого элемента включается нормальный элемент Вестона, э. д. с. которого £в точно известна. Скомпенсировав э.д. с. нормального элемента, получим
Ев = £акк(ЛСв/ЛВ), (XIV.34)
где АСв — отрезок проволоки реохорда, на котором падение напряжения аккумулятора равно э.д.с. нормального элемента. Разделив уравнение (XIV.33) на (XIV.34), получим э.д.с. исследуемого элемента
Ех = Ев (АСЛ/АСВ), (XIV. 35)
где Ев/АСв — цена деления реохорда. Изменение напряжения на аккумуляторе влияет на цену деления, поэтому в процессе работы необходимо время от времени проверять ее.
Элемент Вестона. Для измерений э. д. с. гальванических элемен-ментов в компенсационных схемах применяется в качестве эталонного элемента химический элемент Вестона. Одним из электродов элемента Вестона является 42,5%-ная амальгама кадмия, находящаяся в контакте с насыщенным водным раствором сульфата кадмия CdSO4. Вторым электродом служат ртуть и твердый сульфат ртути (I) в растворе сульфата кадмия (рис. 128)
Pt, Hg — Cd | CdSO4 | Hg2SO4 ] Hg, Pt
Э. д. с. этого элемента отличается большим постоянством и малым температурным коэффициентом, что позволяег использовать данный элемент в качестве стандартного при потенциометрических измерениях. Уравнения реакций, протекающих на правом и левом электродах:
на катоде Hg2SO4 4- 2е 2 HgO 4- SO4~
на аноде Cd — 2е Cd2+
Суммарное уравнение токообразующей реакции в элементе
Hg2SO4 + Cdo 2 Hgo 4- SO^“ 4- Cd2+
Э. д. с. элемента Вестона вычисляют по уравнению
пт aHg°aso2-aCd2+
р__ Р о _2_'____________i-
nr aCd°aHgsSO4
298
учитывая, что активности твердых фаз постоянны и равны единице. Э. д. с. элемента Вестона равна
ЯГ .
й = £ ° -*cdS04 •
Э. д. с. нормального элемента Вестона зависит от температуры:
Ет = ^293 — 0,0000406 (Т — 293). (XIV. 36)
При измерении малых величин э. д. с. гальванических элементов в исследуемую цепь включают элемент Вестона и э.д.с. исследуемого элемента (рис. 129, а) рассчитывают по уравнению
Eb + Ex=Er(ACx/ACb),
откуда
Ех= Ев (АСх/АСв) — Ев-
При измерении больших величин э. д. с. (рис. 129, б) расчет ведут по уравнению
Ех-Ев=Ев(АСх/АСа),
откуда
Ех = Ев+Ев(АСх/АСв)-
При измерении э. д. с. необходимо соблюдать следующие правила: 1) замыкать цепь прерывателем тока только на короткое время,
Рис. 128. Элемент Вестона:
1 — раствор CdSC>4; 2 — кристаллы CdSO4-8/3 Н2О; 3 — амальгама кадмия; 4 -- кристаллы Hg2SO4; 5 —
ртуть
Рис. 129. Электрические схемы установок для измерения малых (а) и больших (б)
э. д. с.
так как при длительной работе элемента э. д. с. изменяется вследствие нарушения электрохимического равновесия; длительная работа нормального элемента может привести к его порче; 2) следить за тем, чтобы клеммы электродов не соприкасались с растворами;
299
3) провеРять после нескольких измерений э. д. с. исследуемого элемента постоянство э. д. с. аккумулятора по элементу Вестона. Колебание показаний при проверке аккумулятора указывает на плохой контакт в цепи. При отсутствии компенсации нужно проверить правильность включения полюсов испытуемого элемента и контакты аккумулятора. Следить, чтобы в электролитическом мостике не было воздушного пузырька, так как это нарушает контакт между растворами.
Измерения э. Д* с. при помощи потенциометра. Для измерения э. д. с. применяют высокоомный потенциометр постоянного тока (Р-37/1; Р-300; Р-375). Точность измерения на нем составляет несколько сотых милливольта.
ПослеД°вательность измерения э. д. с. при помощи потенциометра ППТВ-1.
1, Присоединить к соответствующим клеммам потенциометра вспомогательную батарею (аккумулятор на 1,3 и 2,2 В), гальванометр, элемент Вестона и исследуемый элемент. 2. Установить рабочий ток потенциометра. Для этого вычислить по формуле (XIV.36) э. д. с. нормального элемента при температуре опыта. 3. Установить переключателем нормативного элемента величину э. д. с. (если £в =1,0184 В, то переключатель «н. э.» ставят в положение «4». 4. Поставить переключатель в положение «н. э.» и поворотом ручек магазина сопротивления (грубо__точно) добиться компенсации элемента Вестона. 5. Поставить пере-
ключатель в положение «х» и поворотом рычагов декадных реостатов добиться компенсации. Значение э. д. с. исследуемого элемента отсчитывать в смотровых окошках потенциометра.
ПосЛеД°вательность выполнения работы. Для измерения э. д. с. элемент# Якоби — Даниэля применить компенсационную установ-
ку. ЭлеМент Якоби — Даниэля
Рис. 130- Элемент Якоби — Даниэля
состоит из медной пластинки, погруженной в раствор сульфата меди (II), и цинковой пластинки, погруженной в раствор сульфата цинка. Соединение между отдельными электродами осуществляется электролитическим ключом (солевой мостик) (рис. 130):
Си, Zn | ZnSO4 jj CuSO4 | Си
Медную пластинку перед погружением в раствор тщательно очистить наждачной бумагой, промыть дистиллированной водой и электролитически покрыть слоем меди; цинковый электрод амальгамировать погружением его на несколько секунд в раствор нитрата ртути (I), выделившуюся
капельКУ ртути на поверхности цинкового электрода растереть фильтрсмшльной бумагой до равномерного покрытия поверхности электро/и амальгамой цинка. Потенциал цинка при этом не меняется та^ как ПРИ одновременном присутствии двух металлов потенциал определяется потенциалом менее благородного металла (цинка) -
300
В разные стаканы налить раствор сульфата меди (II) и сульфата цинка с таким расчетом, чтобы V2 поверхности электродов была покрыта жидкостью; погрузить в растворы медный и цинковый электроды, вставить электролитический ключ и полученный гальванический элемент включить в измерительную схему. Э. д. с. элемента измерять компенсационным методом при различных концентрациях растворов. Полученные данные записать в таблицу по образцу:
Концентрация, г-экв/л АСв, СМ AC v см э. д. с., В Э. д. с. по Нернсту, В Ошибка измерения
С u SO* ZnSO*
1 1
1 0,01
0,01 1
0,01 0,01
Полученные данные сопоставить с рассчитанными величинами ио уравнению Нернста.
Для теоретического расчета э. д. с. все необходимые данные заимствовать из справочника (стандартные электродные потенциалы металлов, средние коэффициенты активности для растворов указанных концентраций).
Определение электродных потенциалов. Для измерения электродного потенциала медного и цинкового электродов используют стандартный каломельный электрод и составляют электрохимические цепи:
Си, Hg I Hg2Cl2 | КС1нас || CuSO4 I Си
CuZn | ZnSO4 || KClHac, Hg2Cl2 | Hg—Си
'Э. д. с. первой цепи очень мала, поэтому для точного измерения ее необходимо включить последовательно элемент Вестона. Измерив суммарную э. д. с. такой цепи и вычтя из нее э. д. с. элемента Вестона, рассчитать электродный потенциал. Во второй цепи э. д. с. достаточно велика и включения элемента Вестона не требуется. Отдельные электродные потенциалы определить по формулам:
E1 ~ £Сц2+/Сио ~ Е“ал 1 £Cu2+/Cu« - £1 + Ехал>
Е2 ~ £кал — EZa2+;z^ Eia2+lzno ~Е2~ Ew
301
Полученные данные записать в таблицу по образцу:
Элемент Концентрация раствора электролита, г«екв/л АСВ, см АСХ, см Э. д. с., в ^кал» В К* расч» В Ошибка измерения
1 0,01
1 0,01 И-
В данной работе следует; 1) измерить э. д. с. элемента Якоби — Даниэля при различных концентрациях (активностях) исходных солей; 2) измерить электродные потенциалы медного и цинкового электродов; 3) сопоставить полученные данные с. рассчитанными значениями э. д. с. по уравнению (XIV.30); 4) сопоставить разность электродных потенциалов для соответствующих концентраций растворов с ранее измеренными значениями э. д. с. элементов; 5) оценить погрешность измерений.
Работа 2. Измерение э. д. с. окислительно-восстановительных элементов, электродных потенциалов и изучение влияния добавок на окислительно-восстановительные потенциалы
Окислительно-восстановительные элементы составляют из различных окислительно-восстановительных электродов. В дайной работе могут быть использованы следующие электроды.
Pt|I“, 1~з — раствор является окислительно-восстановительной системой, содержащей 0,1 н. раствор KI и растворенный в нем 0,001 г-экв/л кристаллического иода.
Pt|Fe3-b, Fe2+ — для составления такого электрода используют растворы солей железа (II) и (III): сульфаты, хлориды, железоаммиачные квасцы и соль Мора FeSO4(NH4)2SO4-6H2O, ферро-фер-рицианидные комплексы железа. Растворы солей железа приготовляют в 0,1 н. растворах H2SO4, НО. При составлении окислительно-восстановительной системы в стакан наливают равные объемы (по 15 мл) растворов солей железа (II) и (III), в которые погружают инертный электрод (Pt, С, Au).
Pt|MnO4“, Mn2+ — раствор содержит соли марганца различной степени окисления. При составлении окислительно-восстановительной системы смешивают 25 мл 0,1 н. КМпО4 с 5 мл 0,01 М MnSO4.
Pt|CrO42“, Сг3+ — для приготовления окислительно-восстановительной системы смешивают 25 мл 0,1 М раствора КзСгО4 и 5 мл 0,01 т раствора хромовых квасцов.
302
Последовательность выполнения работы. Из полученных окислительно-восстановительных электродов составить гальванический элемент, компенсационным методом измерить его э. д. с. до постоянного значения (различие между параллельными значениями не должно превышать 2—3 мВ).
Определить электродные потенциалы гальванической цепи, состоящей из исследуемого электрода и стандартного каломельного электрода:
Pt, /Hg I HgaCl2 | КС1нас || Мех+ | Ме?+ | Pt
Измерить э. д./с. такого электрода методом компенсации. Если э. д. с. таких цепёй окажется слишком малой (при менее 0,1 В точка компенсации находится в крайнем положении на реохорде), необходимо включить последовательно элемент Вестона. Измерив суммарное значение э. д. с. такой цепи, вычислить электродный потенциал:
£=£мх+|м,+ -£кал, (XIV.37)
+ | М^+ + £кал •
Э. д. с., рассчитанная по разности окислительно-восстановительных потенциалов, должна совпадать с ранее определенной э. д. с. окислительно-восстановительной цепи (расхождение допускается до 10%). Если расхождение превышает 10%, необходимо определить еще раз э. д. с. элемента и вновь проверить потенциалы окислительно-восстановительных электродов.
Так как окислительно-восстановительный потенциал зависит от соотношения активностей окисленной и восстановленной форм ионов, то, варьируя это соотношение, изучить характер изменения электродного потенциала, окислительные и восстановительные свойства раствора. Это осуществить, вводя в раствор вещества, связывающие ту или иную форму ионов. Уменьшение концентрации окисленной формы при постоянной концентрации восстановленной формы снижает потенциал исследуемой системы, и, наоборот, всякое уменьшение концентрации восстановленной формы при постоянной концентрации окисленной увеличивает окислительно-восстановительный потенциал системы. При этом изменяются окислительные и восстановительные свойства раствора. Так, например, введение в раствор, содержащий ферро-ферри-ионы, ацетата натрия уменьшает окислительно-восстановительный потенциал, поскольку ионы Fe3+ связываются в комплекс. Это снижает окислительную способность раствора и повышает его восстановительное действие. Введение в эту же систему оксалата аммония, образующего комплекс с ионами F2+, увеличивает потенциал изучаемой системы; при этом возрастает окислительное свойство раствора и снижается восстановительная способность его. Для установления характера изменения окислительно-восстановительного потенциала систем составить гальванический элемент типа
Pt,Hg [ Hg2Cl2, КС1нас II Мех+ I Meff+ | Pt
303
э. д. с. которого измерять по мере введения различных добавок. Добавки вводить небольшими порциями; растворы добавлять из капельницы по каплям, твердые вещества вносить по 0,1—0,2 г. Каждую последующую порцию добавлять только после того, как установится постоянное значение потенциала электрода. Добавки вводить при непрерывном и тщательном размешивании, одну за другой в соответствующей последовательности.
Добавки к Pt | Fe3+| Ее2+-электроду: 1) концентрированная серная кислота; 2) кристаллы ацетата натрия (комплексообразова-тель); вводить до образования красной окраски [ацетат железа (Ш)]; 3) кристаллы оксалата аммония; вводить небольшими порциями до появления желтовато-зеленой окраски раствора.
Добавки к Pt | СгзО42“| Сг3+- и Pt |МпО4“|Мп2+-электродам: 1) концентрированная серная кислота, которую добавлять из микробюретки по каплям. После каждой капли кислоты измерить э. д. с.; вводить ее до тех пор, пока потенциал электрода не перестанет изменяться.
Чтобы выяснить характер влияния концентрации ионов водорода на электродный потенциал изучаемых систем, вычертить график зависимости потенциала электрода от количества прибавленной кислоты.
Изменение электродных потенциалов системы от pH раствора можно исследовать, применяя для измерения pH-метр со стеклянным электродом.
Добавки к Pt | Г-, 13“-электроду: 1) концентрированная серная кислота; кислоту вводить 8—10 капель до постоянства потенциала; 2) пероксид водорода; вводить до тех пор, пока раствор не приобретет красной окраски; 3) концентрированная азотная кислота; вводить постепенно до обесцвечивания раствора.
В данной работе необходимо: 1) измерить э. д. с. окислительно-восстановительного элемента; 2) измерить потенциалы окислительно-восстановительного электрода по отношению к каломельному электроду и сравнить измеренную э. д. с. элемента с величиной*, полученной по разности двух потенциалов; 3) изучить влияние добавок различных веществ на окислительно-восстановительный потенциал и объяснить наблюдаемые закономерности; 4) рассчитать по уравнению Нернста э.д.с. исследуемого гальванического элемента, используя данные справочника. Коэффициенты активности рассчитать по уравнению (XIV.24).
Полученные данные записать в таблицы по образцам:
К измерению э. д. с. окислительно-восстановительного элемента
Температура опыта ...
Окислительно-восстановительный элемент
анод катод Л CR D Э. д. с., В
Ошибка измерения
304
К определению окислительно-восстановительных потенциалов отдельных электродов
Электрод о АСХ Э. д. с., В Потенциал электрода
К влиянию добавок на окислительно-восстановительные потенциалы
Электрод Добавка лсв АС Э. д. с., В Потенциал электрода
- ч
Работа 3. Определение стандартного потенциала ферри-ферро-электрода, расчет константы равновесия электродной реакции, изучение окислительно-восстановительной способности раствора
Для измерения потенциала редокс-электрода собрать гальванический элемент Pt, Hg|Hg2Cl2, КС1Нас Н Fe3+, Fe2+|Pt и методом компенсации измерить его э. д. с.
Последовательность выполнения работы. Окислительно-восстановительные системы приготовить из 0,1 М растворов сульфатов, хлоридов железа разной степени окисления (II и III). Титрованием определить концентрации исходных растворов. Если концентрация растворов одинакова, то соотношение активности ионов Fe3+ и Fe2+ можно заменить соотношением их объемов. Составить смеси с различным соотношением окисленной и восстановленной форм ионов железа 9 : 1; 8 : 2; 7 : 3; 6 : 4; 5 : 5; 4 :6; 3 : 7; 2 :8; 1 :9. В сосуд для измерения налить 10 мл приготовленной смеси, погрузить платиновый электрод и с помощью каломелевого электрода измерить ре-докс-потенциал. В качестве компенсационной установки использовать потенциометр. Перед заполнением сосуда последующей смесью необходимо ополоснуть дистиллированной водой сосуд, платиновый электрод, солевой мостик. Измерения э.д.с. повторять до тех пор, пока расхождения не будут превышать 1—2 мВ.
При изучении окислительно-восстановительных свойств данных растворов исследовать влияние комплексообразователя фторида калия. Насыщенный раствор KF добавить по каплям в исследуемую редокс-систему, перемешать раствор и измерить редокс-потен-циал. Последующую порцию фторида калия добавлять в исследуемую смесь после установления постоянного значения э. д. с. По характеру изменения потенциала судят об окислительно-восстановительной способности раствора: чем положительнее потенциал
•3.
305
электрода, тем больше окислительное действие раствора, уменьшение потенциала системы указывает на увеличение восстановительного действия раствора. Окислительно-восстановительный потенциал электрода рассчитать по уравнению (XIV.25), коэффициенты активности— по уравнению (XIV.24). Ионная сила раствора равна Т=722^гд
Пример расчета ионной силы раствора: исследуемый раствор содержит Ю,01 М FeCl3, 0,01 М FeCl2, 0,05 М НС1 при соотношении редокс-форм 1:9. Ионная сила раствора равна:
Z = 1/2(0,001-32 4-0,001-3-12 +о,ОО9.22 4-0,009-2-12 _р
4-0,05-12 4-0,05-12) = 0,083.
В данной работе следует: 1) приготовить смеси с различным содержанием ионов Fe3+ и F2+; 2) измерить компенсационным методом для всех смесей э. д. с. и рассчитать стандартный окислительно-восстановительный потенциал исследуемой системы. По полученному значению Е° рассчитать константу равновесия и Абт° электродной реакции; 3) исследовать влияние комплексообразова-теля. на редокс-систему. Установить характер изменения редокс-потенциала. Построить график зависимости редокс-потенциала от состава изучаемой системы; 4) построить график зависимости редокс-потенциала от логарифма отношения активности окисленной и восстановленной форм ионов; определить экстраполяцией стандартный редокс-гютенциал Е°.
Все полученные экспериментальные и расчетные данные занести в таблицы по образцу:
Соотношение рсдоксформ Э. д. с„ В Э. д. с. с комплексо-образователем, В Е равн» В К* равн с комплексо-образователем, В
9 : 1 г
8:2
• • *
Соотношение редокс-форм Е равн» В СО 1g 1 / То 1g Тв я0, в ^равн AG0, кДж
5) сделать выводы из результатов работы и оценить погрешность измерений.
306
Работа 4. Определение коэффициента активности растворов хлористоводородной кислоты методом измерения э. д. с.
Коэффициент активности выражается отношением средней ионной активности к общей моляльной концентрации раствора электролита у±— а±1т. Активность выражает эффективную концентрацию какого-либо вида ионов. Наиболее точно среднеионный коэффициент активности определяют методом измерения э. д. с. Для этого применяют гальванический элемент без жидкостных границ— элемент без переноса (отсутствует диффузионный потенциал).
Гальванический элемент состоит из двух электродов, опущенных в раствор и обратимых как по катиону, так и по аниону. Для определения коэффициента активности соляной кислоты применяют элемент, составленный из водородного и хлоридсеребряного электродов
Pt, Н2 | НС! I AgCl I Ag
Э. д. с. такого элемента рассчитывают по уравнению
о ГRT RT
Е — Е , . п— ----In а 4------In а .
Ag + /Ag° nF пр
(XIV.38)
или
(XIV. 39)
Заменив ионные активности на среднюю активность и выразив ее как а± = у±т, получим
Е — Е*
~ Ag + /AgO
2RT nF
2RT
In т —----In
nF
In m
Для 25°C выражение (XIV.40) приобретает вид
(XIV.40)
2RTjnF
— (£ 4-0,1192 1g m)
0,1192
(XIV.41)
или
0,1192 1g у , = £° — (£ 4- 0,1192 1g m). (XIV.42)
Обозначив £+0,11921g m через £, получим
lgy±=(£°-0/0,1192. (XIV.43)
Для Ag/AgCl £°=0,2234 В при 298 К и, определив Е из опыта, можно рассчитать коэффициент активности. Значение в общем случае можно определить экстраполяцией э. д. с. к бесконечному разведению. Так как при /п—>-0 коэффициент активности у± стре
307
мится к единице, то Е°=Е-[-0,1192 Igm. Определение Е° проводят графически: на оси ординат откладывают экспериментальные значения Е+0,11931g т, а по оси абсцисс — значение т или Ут. Экст-
раполируя прямую до пересечения с
осью ординат, получают отрезок, равный £°. Коэффици-
Рис. 131. Зависимость lgy± от У I для
ент активности многих электролитов является сложной функцией от концентрации (ионной силы) (рис. 131), что объясняется влиянием на коэффициент активности диэлектрической проницаемости, величины ионного радиуса, заряда иона.
Приготовление хлоридсеребряного электрода. Для получения устойчивого потенциала хлоридсеребряного электрода электролитически нанести слой серебра на серебряную пластинку или сетку. Электрод, состоящий из
1—1-валентных электролитов серебряной сетки размером
3X2 см, погрузить в сосуд с
6—10%-ным раствором AgNO3. Электрод-сетка служит катодом, в
качестве анода применяют серебряные пластинки, расположенные по периферии электролитического сосуда. Электролиз ведут от ак-
кумулятора напряжением 4 В со следующим режимом:
Плотность тока, мА/см2 Время, мин
0,2 0,5 1 2 2,5 8,5
60 45 45 30 30 15
По окончании электролиза серебряный электрод вынуть, погрузить в 0,1 н. НС1 и продолжать электролиз. Использовать в качестве анода серебряный электрод, а в качестве катода — платиновую проволоку. Допустимая плотность тока 10—8 мА/см2. При пропускании тока в течение 10 мин металлическое серебро под действием образующегося хлора превращается в хлорид серебра. Затем поменять полюса: на электроде Ag/AgCl выделяется водород, который вытесняет ионы С1_ с поверхности электрода. После этого электрод тщательно промыть водой в течение 2 ч, меняя воду через каждые 20—30 мин, до полного удаления ионов СН
Хлоридсеребряный электрод можно изготовить нанесением хлорида серебра на серебряную проволоку. Для этого провести электролиз в 0,1 н. КС1 с использованием в качестве анода серебряной проволоки, а в качестве катода — платиновой проволоки. Электролиз ведут от аккумулятора напряжением 4 В в течение 10 мин, перед включением тока серебряный электрод должен быть катодом. Это предотвращает адсорбцию свободного хлора на 308
электроде. После электролиза электрод тщательно промыть водой до полного удаления ионов С1_. При большой площади поверхности серебряного электрода первая методика приготовления хлоридсеребряного электрода является более надежной. Хлоридсеребряные электроды изготовляются серийно и их можно использовать в работе. Однако вследствие малой рабочей площади поверхности серийного электрода увеличивается время установления равновесия на электроде.
Последовательность проведения работы. Для измерения коэффициентов активности 0,1 т раствор НС1 разбавить до 5*10~4 моль/1000 г. Концентрацию исходного раствора установить титрованием, Э. д. с. измерять в сосуде (рис. 132) диаметром 40 и высо-
Рис. 132. Схема установки для измерения коэффициента активности:
1 — электролизер для получения водорода; 2 — склянка с водой; 3 — буферный объем;
4 — сосуд для измерения э. д. с,; 5 — потенциометр
той 60 мм, в который вставить два электрода на шлифах, один из них водородный, а другой хлоридсеребряный. Исследуемая жидкость должна покрывать меньше половины поверхности платины водородного электрода. Водород получать на специальной установке. Перед началом опыта всю систему не менее 30 мин продуть водородом.
Питание водородного электролизера осуществить от выпрямителя. Измерительная часть установки состоит из потенциометра. Измерения проводить в растворах НС1 различной концентрации, начиная от наиболее разбавленного. Пропускать водород в течение 10 мин и измерять э. д. с. Если результаты измерений не совпадают, насыщение и измерения продолжать до тех пор, пока два последних отсчета не будут отличаться менее чем на 0,2—0,3 мВ. Затем сосуд отсоединить от установки, кислоту вылить, сосуд промыть более концентрированным раствором соляной кислоты, заполнить его этой кислотой и вновь повторить опыт. Таким образом
309
произвести измерение э. д. с. при всех концентрациях НО. Результаты измерения записать в таблицу по образцу:
Элемент Концентрация НС1, моль/л Э. д. с., В ЙО о ЕЦ Т ± эксн теор ат.
В данной работе следует: 1) измерить э. д. с. гальванических элементов с различной концентрацией НО; 2) определить графически по экспериментальным данным £°; 3) рассчитать коэффициенты активности растворов НО и построить график lgy± =
4) рассчитать активности растворов НО; 5) сопоставить полученные значения с табличными данными для НО и оценить погрешность измерений.
Работа 5. Определение термодинамических функций реакции, протекающей в окислительно-восстановительном элементе
Определение термодинамических характеристик реакций, протекающих в обратимых гальванических элементах, можно проводить как на системах, состоящих из органических соединений хи-нон-гидрохинон, так и на ряде окислительно-восстановительных систем, содержащих неорганические ионы в различных степенях окисления. В качестве примера обратимой реакции, используемой для определения термодинамических функций и протекающей в гальваническом элементе, состоящем из водородного и хингидронного электродов, рассмотрим восстановление хинона в гидрохинон. Реакция протекает в две стадии с образованием, в качестве промежуточного продукта хингидрона:
СбН4О2-Н 1/2Н2(г)^С6Н4О2.СзН4(ОН)2 (тв) (1)
1 /2 Н2 4- 1 /2 С6Н4О2 С5Н4 (ОН)2 X С3Н4 (ОН)2 (II)
Суммарное уравнение
Сйн4О2 + н2 х С6Н4 (ОН)2 (тв) (III)
Изменение энергии Гиббса реакции (III) равно сумме изменений AGi и Абл:
Величины AGi и AGn могут быть получены измерением э. д. с. элементов, в которых осуществляются эти реакции:
Pt, Н2 I Н+ I Н+, X, ХГ ] Pt (IV)
Pt, Н2 I Н+ I Н+, Г, ХГ I Pt (V)
где X — хинон; ХГ — хингидрон; Г — гидрохинон. 310
В данных элементах концентрация ионов Н+ достаточно велика (0,1 моль и более). При этом условии присутствие хинона и гидрохинона в растворе не влияет на активности ионов Н+. Ионы водорода могут быть введены в систему раствором любой кислоты, например серной, не взаимодействующей с хиноном, гидрохиноном и водородом. Для элемента (IV), т. е. для реакции (I),
ACrjy — — FEiy.
Для элемента (V), т. е. для реакции (II),
Отсюда
At?in — —Е(Е[у^Еу).
По измерению э. д. с. при разных температурах рассчитывают по уравнению (XIV.9) тепловой эффект реакции и по уравнению (XIV.10) — изменение энтропии суммарной реакции.
Такое же исследование может быть проведено на окислительно-восстановительных элементах, электроды которых содержат неорганические ионы с различной степенью окисления, например
Fe3+/Fe2+; NO^/NO^; [Fe (CN)6]3-/[Fe (CN)6]^; 1-/1“
a также на элементах, одним из электродов которых является каломельный электрод, другим — любая окислительно-восстановительная система, например,
Pt, Hg | Hg2CI2, КС1нас II FeCl3, FeCl2 | Pt
(a)
Pt | FeCI3/FeCl2l HC1 || H2SO4, NaNO3, NaNO2 | Pt
Pt | K4 [Fe (CN)3]4~/K3 [Fe (СМ)б]3“ I! KI, I2 I Pt (в)
Для расчета термодинамических функций Дб, ДЯ, AS реакций окисления — восстановления, протекающих в данных окислительно-восстановительных элементах (рис. 133), изучают зависимость э. д. с. от температуры.
Последовательность выполнения работы. Собрать гальванический элемент, работа которого основана на восстановлении хинона в гидрохинон. Собрать установку для измерения э. д. с., включить электролизер для получения водорода, которым продуть всю установку. В сосуд 1 (рис. 133, а) поместить около 0,2 г хингидрона и 0,2 г хинона или гидрохинона, вставить гладкий платиновый электрод так, чтобы платиновая проволока была погружена в осадок. Затем осадок залить 0,1 н. H2SO4 с таким расчетом, чтобы электролитический ключ 4 был заполнен кислотой. Операцию заполнения ключа надо производить осторожно, не взмучивая осадка. Сосуд 1 должен быть плотно закрыт, иначе жидкость из ключа 4 будет вытекать и нарушится контакт с водородным электродом.
Водородный полуэлемент представляет собой сосуд 2, в котором платинированная платиновая пластинка наполовину погружена в 0,1 н. H2SO4; он снабжен гидравлическим затвором 3. Сосуды
311
1 и 2 соединить ключом и погрузить в термостат. Измерения э. д. с. производить после установления постоянной температуры через каждые 10—15 мин до тех пор, пока четыре последующих измерения не покажут значения, совпадающие в пределах 0,5 мВ-Измерения э. д. с. производить с интервалом 5—6, не превышая 40°С.
Если опыт проводили при давлении, отличном от 1 атм, то необходимо при расчете потенциала внести поправку: £ = — 0,1х
Р ~ Лйо ,
X • 10-31g-----, где р — барометрическое давление; рн2о — давле-
700
ние паров 0,1 н. H2SO4 при температуре опыта.
По исправленным значениям э. д. с. построить график E = f(Г), рассчитать графически (дЕ/дТ)р, по (XIV.4), (XIV.9), (XIV.10) — AG, АЯ и AS реакцищ протекающей в гальваническом элементе.
Для приготовления окислительно-восстановительного гальванического элемента с неорганическими ионами типа (а), (б), (в) рабочий электролит составить из растворов, содержащих ионы различных степеней окисления, в равных объемных соотношениях (10 мл 0,1 н. NaNO3 и 10 мл 0,1 н. NaNO2). Если потенциал окисли-
Рис. 133. Схема прибора для измерения э. тельной системы хинон — гидрохинон {а) и окислительно-восстановительных систем, иоиы (б)
д. с. окислительно-восстанови-прибор для измерения э. д. с. содержащих неорганические
тельно-восстановительной системы зависит от pH среды, следует эти растворы подкислить 8—10 каплями концентрированной H2SO4, тщательно перемешать раствор и измерить потенциал системы по каломельному электроду сравнения. Измерения потенциала проводить до постоянного значения его, не меняющегося от последующего добавления кислоты. Затем растворы налить в специальную термостатированную ячейку (рис. 133, б), погрузить
312
платиновые электроды и, соблюдая правило знаков, присоединить элемент к потенциометрической измерительной схеме. Измерения э. д. с. проводить на потенциометре с точностью 10-4 В, с чувствительным гальванометром 10~8—10~9 A/деление, в пределах температур от 15 до 45°С с интервалом в 5—6°. При каждой температуре измерение э. д. с. проводить в течение 15—20 мин, пока расхождение в параллельных замерах не будет превышать 1—2 мВ. По зависимости E = f(T) графически определить dE/dT и по уравнениям (XIV.4), (XIV.9) и (XIV.10) рассчитать Дб, ДЯ, AS реакции.
По справочным данным рассчитать Е° исследуемых окислительно-восстановительных систем и по теплотам образования и стандартным энтропиям ионов — Дб°, ДЯ° и Д5°. Сравнить полученные результаты и объяснить причины расхождения расчетных и экспериментальных величин. Полученные данные занести в таблицу по образцу:
т, к Д В d£/dT -AG кДж дя, кДж AS, Дж
Работа 6. Определение термодинамических функций реакции окисления — восстановления методом потенциометрического титрования
Потенциометрическое титрование основано на скачкообразном изменении вблизи эквивалентной точки концентрации титруемого вещества при добавлении к нему небольшого количества титранта. В основе этого титрования лежит линейная зависимость электродного потенциала от логарифма активности ионов или логарифма отношения активности ионов окислителя к активности ионов восстановителя в титруемой системе. Если применяемый при титровании электрод обратим по отношению к ионам титруемого или титрующего вещества, то изменение потенциала такого электрода будет указывать на изменение концентрации ионов в растворе. Зависимость потенциала обратимого электрода от активности ионов выражается уравнением (XIV. 17). Всякому резкому изменению концентрации или активности ионов при титровании отвечает скачкообразное изменение потенциала электрода, обратимого по отношению к ионам титруемого вещества или титранта. Зависимость потенциала от количества прилитого титранта выражают графически (рис. 134). Полученная кривая называется потенциометрической кривой. Точка эквивалентности соответствует точке перегиба кривой.
Природа присутствующих ионов в растворе и тип протекающей реакции определяют выбор индикаторного электрода. Так, при ре-
313
акции нейтрализации индикаторным электродом служат электро-
ды, потенциал которых зависит от активности ионов водорода в растворе. Таковы водородный, хингидронный, сурьмяный, стеклянный электроды. Выбор электрода зависит от условий титрования: присутствия загрязнений, окислителей и восстановителей в растворе, интервала изменения pH во время титрования и т. п. При реакциях окисления — восстановления в качестве индикатор-
Рис. 134. Потенциометрическая кривая титрования окислительно-восстановительной системы
ионами Ag+ малорастворимые
ного электрода используют электроды из индифферентных металлов: платины, золота. Электрод погружают в окислительно-восстановительную систему, он осуществляет перенос электронов между компонентами, которые и обусловливают величину электродного потенциала.
При реакциях осаждений и комплексообразования индикаторным электродом служит электрод, потенциал которого является функцией активности ионов, участвующих в реакциях осаждения или комплексообразования. Например, серебряный электрод используется для определения серебра, а также ионов, дающих с соли или прочные комплексные
соединения. В качестве электродов сравнения используют каломельный или хлоридсеребряный электрод. Последний, если это допустимо, погружают непосредственно в исследуемый раствор
или соединяют при помощи электролитического ключа с титруемым раствором. Измерение возникающей э. д. с. можно проводить по компенсационному и некомпенсационному методам (измерение потенциала электрода, измерение силы тока).
Потенциометрические кривые могут быть использованы для количественной оценки результатов титрования и для получения физико-химических величин: константы диссоциации слабой кислоты, pH при титровании многоосновных кислот с учетом гидролиза, растворимости малорастворимых солей, константы нестойкости комплекса. Используя потенциометрические кривые реак-
ции окисления — восстановления, можно рассчитать стандартные окислительно-восстановительные потенциалы, константы равновесия электродных реакций, энергию Гиббса и т. п. Если проводить потенциометрическое титрование в небольшом интервале температур, то по кривым титрования можно определить температурный коэффициент э. д. с., энергию Гиббса, тепловой эффект и AS реакции.
Последовательность выполнения работы. Собрать установку для потенциометрического титрования. Для этого в реакционный со
314
суд— термостатированную ячейку налить 10 мл 0,1 н. FeSO4, добавить 20 мл 2 н. H2SO4 и в раствор погрузить платиновый электрод и электрод сравнения (каломельный или хлоридсеребряный электрод). Полученный гальванический элемент включить в потенциометрическую схему измерения (потенциометр, см. с. 300), соблюдая правило знаков. Титрование проводить при непрерывном перемешивании магнитной мешалкой. Спустя 5—10 мин, когда ис-
следуемый раствор примет температуру термостата, прилить из бюретки 0,05 мл соответствующего титранта [0,1 н. Н2О2; 0,1 н. КМпО4; 0,1 н. Ce2(SO4)3]. После каждой порции добавленного реагента измерять э.д.с. гальванического элемента. Приближаясь к точке эквивалентности, где потенциал системы меняется резко, добавлять по 0,2 мл реагента. После точки эквивалентности реагент добавлять по 0,5—1 мл. Титрование повторить, но при другой температуре (на 5° выше). Получен'ные данные нанести на график с координатами потенциал электрода — объем израсходованного титранта
(см. рис. 134). Точка эквивалентности отвечает точке х, в точке 0,5% оттитрована половина количества окислителя или восстановителя и соотношение их эквивалентностей равно единице, что отвечает стандартному окислительно-восстановительному потенциалу Ei0 данной системы. Если после точки эквивалентности к раствору прибавить эквивалентный избыток окислителя 2х, то стандартный окислительно-восстановительный потенциал титранта равен Е2°. По потенциометрической кривой рассчитать стандартные окислительно-восстановительные потенциалы Е°, константы равновесия электродных
Рис. 135. Определение стандартного окислительно-восстановительного потенциала и числа окислительно-восстановительных эквивалентов
реакций Да, изменение термодинамических функций реакции
Дб°, ДЯ°, Д5°, температурный коэффициент э. д. с. и число окисли-
тельно-восстановительных эквивалентов (рис. 135). Экспериментальные и расчетные данные занести в таблицу по образцу:
315
Полученные результаты сравнить с рассчитанными по справочным данным и сделать выводы. Оценить погрешность в измерениях.
Работа 7. Определение pH образования гидроксидов металлов
Для разбавленных растворов существует линейная зависимость между активностью металла в растворе и pH среды. Если предположить, что недиссоциированный гидроксид металла Ме(ОН)п в растворе находится в равновесии с ионами по схеме
Me (ОН)я^Ме"+ + (ОН)" то
ПР=[Меп+][ОН-]” = [Мея+](Ки/[Н+])я = КС. (XIV.44)
Активность ионов ОН- равна
он-' н+
(XIV. 45)
Переходя от концентрации к активностям и логарифмируя (XIV.44), получим
1g „ =nlga _ = nlg—, (XIV.46)
Л wf ОН d
Мега+ Н+
1g ПР —lg a =nlg/<w + npH. (XIV.47)
1
При п = 2
pH = 1/2 1g ПР - 1/2 1g аМе2+ - lgKw. (XIV.48)
Последовательность выполнения работы. Приготовить 0,2 н. растворы солей металлов (II) с одинаковым анионом. Последующие растворы готовить разведением исходного раствора до концентраций (г-экв/л): 0,1; 0,5; 0,025. В стакан налить 5 мл раствора соли и разбавить его водой до 50 мл. Погрузить в раствор стеклянный электрод так, чтобы шарик его был полностью покрыт жидкостью. Опустив в этот же раствор хлоридсеребряный электрод, включить собранный гальванический элемент в потенциометрическую схему. Прибором для измерения служит рН-метр. pH-Метр включить в сеть на 220 В, прогреть лампы прибора в течение 20 мин и приступить к калибровке стеклянного электрода по буферным растворам с известными значениями pH (см. инструкцию к прибору). После калибрования стеклянного электрода приступить к потенциометрическому титрованию приготовленных растворов. Из бюретки при непрерывном перемешивании магнитной мешалкой добавить в стакан по 0,1 мл 0,01 н. КОН, измеряя при этом pH раствора и э. д. с. исследуемого элемента. Количество прилитого титранта должно в два раза превышать количество взятого для исследования раствора. По кривым титрования определить pH начала образования гидроксида, по протяженности площадки кривой титрования определить концентрацию ионов металла. Зная анион, входящий в состав соли, и концентрацию ионов металла»
316
рассчитать ионную силу раствора, по таблице справочника определить средний коэффициент активности.
По полученным значениям pH образования гидроксида и активности ионов металла рассчитать произведение растворимости гидроксида. Построить график зависимости pH образования гидроксида от активности ионов металла. Полученные экспериментальные и расчетные данные занести в таблицу по образцу.
Соль ссоли> г-экв/л Объем КОН, мл Me * г-экв/л 1 т± Me ПР с соли» ₽^экв/л
Из результатов исследования сделать выводы и оценить погрешности рассчитываемых величин.
Работа 8. Определение буферной емкости растворов методом потенциометрического титрования
Существуют растворы, добавление к которым некоторого количества кислоты или основания не вызывает заметного изменения pH. Эту способность называют буферностью. Буферные растворы представляют собой смеси слабых кислот и оснований с какой-либо их солью, образованной сильным основанием или кислотой. Действие буферного раствора определяется его буферной емкостью, которая характеризуется числом грамм-эквивалентов кислоты или основания, добавляемых к 1 л раствора для изменения pH на единицу
(XIV.49)
Количество прибавленного реагента
d pH v где b — количество добавленного основания или кислоты при титровании, г-экв/л; v — объем раствора, израсходованный на титрование, мл. Представление о буферном действии раствора дают кривые изменения pH
ных кислот (рис. 136). Потенциал электрода при потенциометрическом титровании выражается уравнением
Cn — b
nF b
Рис. 136. Потенциометрические кривые буферной емкости:
1 — титрование сильной кислоты; 2 — титрование слабой кислоты
при нейтрализации различ-
(XIV. 50)
где Кдисс — константа диссоциации слабой кислоты; — началь-
317
ная концентрация кислоты, г-экв/л. Если оттитрована половина количества кислоты & = с0/2, то потенциал в этой точке будет
РТ RT
Е = Е° + — lg^HCC = £° +—Igc , (XIV.51)
nF nF и
и для этой точки кривой титрования рН^рК, при этом буферная
емкость раствора максимальна. При гидролизе в растворе концентрация ионов Н+ выражается уравнением
с
Kw^JlHCC /^,
(XIV.52)
где Kw — ионное произведение воды; А'дисс — константа диссоциации слабой кислоты. Из уравнения (XIV.52) можно рассчитать pH:
pH ~ 0,5 1g 6 — 0,5 1g/<да — 0,5 1g/Слисс.
(XIV. 53)
Буферная емкость может быть рассчитана аналитически. Буферное действие увеличивается при переходе к растворам слабых кислот или слабых оснований, особенно в присутствии соответствующих солей. Если к раствору, содержащему п г-экв слабой кислоты, добавлено т г-экв сильного основания (n>m) и
« = а ^ = са_4-сон„-сн+, (XIV.54)
то константу диссоциации слабой кислоты можно представить в виде
(XIV.55)
^-^ck_/incA
Решение уравнения (XIV.55) относительно с _ дает А
т= [л^на/(^на + сн+)] +%н_-сн+,
(XIV.56)
откуда можно найти буферную емкость:
(XIV. 57)
Буферная емкость раствора может быть определена графически. Если провести касательную к любой точке потенциометрической кривой, то величина, обратная тангенсу угла наклона касательной к оси абсцисс, будет соответствовать буферной емкости раствора.
Последовательность выполнения работы. В стакан для титрования налить 10 мл сильной или слабой кислоты известной концентрации, добавить 10—15 мл дистиллированной воды и тщательно перемешать раствор, затем добавить в раствор хингидрон и погрузить в стакан платиновый электрод. Стакан с раствором поместить на магнитную мешалку и непрерывно перемешивать раствор при титровании. Стакан с раствором соединить с помощью солевого мостика с электродом сравнения (каломельным или хлоридсеребряным электродом). Собранный гальванический элемент включить в потенциометрическую схему и провести потенциометрическое тит
318
рование. В начальный период титрант добавлять по 0,5 мл при тщательном размешивании раствора. После каждой порции прилитого титранта измерять на потенциометрической установке э.д. с. гальванической цепи компенсационным методом. Когда изменение э. д. с. от каждой порции добавленного титранта становится значительным, то количество прибавленного титранта уменьшить до 0,1 мл. После точки эквивалентности титрант добавлять по 0,5 мл до постоянного значения потенциала. Экспериментальные данные нанести на график и по полученной потенциометрической кривой, по точке эквивалентности вычислить концентрацию исследуемого раствора, определить графически буферную емкость раствора.
В работе следует оттитровать многоосновную кислоту или смесь слабой и сильной кислот, определить их концентрацию и буферную емкость.
ГЛАВА XV
КИНЕТИКА РЕАКЦИЙ В РАСТВОРАХ
1. СКОРОСТЬ ХИМИЧЕСКОЙ РЕАКЦИИ. КИНЕТИЧЕСКАЯ
КЛАССИФИКАЦИЯ РЕАКЦИЙ
Химическая кинетика изучает скорость и механизм протекания химических процессов, а также зависимость их от различных факторов.
Скорость реакции. Скорость химической реакции определяется изменением количества данного компонента в единицу времени в единице объеме:
1 d п
(XV. 1)
где п— число молей данного компонента в объеме v данной фазы в момент времени /. '
При условий постоянства объема выражение (XV.1) упрощается :
d (n/v) de
и скорость определяется изменением концентрации реагирующего вещества в единицу времени.
Скорость реакции всегда положительна. Однако при протека-________________________________________________________________ нии реакции во времени концентрации исходных веществ умень-шаются, а продуктов реакции возрастают (рис. 137). В результате соотношение (с2—Ci)/(t2—1\) и производная dejdt могут быть положительными или отрицательными в зависимости от того, изучают скорость реакции по изменению концентрации одного из
319
продуктов реакции (знак « + ») или одного из исходных веществ (знак «—»).
Скорость реакции выражается как производная от концентрации по времени для любого вещества, участвующего в реакции.
В общем случае скорость реакции, протекающей по уравнению
V1А 4- V2 В qt г* С 4- VgD,
(а)
где vi, V2, v/ и v%'— стехиометрические коэффициенты, может быть представлена несколькими выражениями
1 dcA 1 dcB 1 dcc 1 dcD
чяии -"-- " , 1 I - - jr
Vi d I V2 d t vj d t Vg d t
Исходные вещества расходуются, а продукты реакции образуются в эквивалентных количествах (соответственно стехиометриче-
Рис. 137. Кинетические кривые:
I — изменение концентрации одного из исходных веществ во времени; 2 — изменение концентрации одного из продуктов реакции {а) во времени
ским коэффициентам), поэтому при определении скорости реакции нет необходимости следить за изменением концентрации всех взаимодействующих веществ.
Скорость реакции можно опреде-
лить так же, как производную степени полноты реакции (глубина ее протекания) по времени. Скорость реакции зависит от природы реагирующих веществ, их..концентраций,
присутствия посторонних веществ (например, катализаторов) и их концентрации; среды, в которой проте-
кает реакция, и условии протекания реакции: температуры, давления (особенно для реакций с участием-газов), облучения (фотохимические реакции) и т. п.
Основным законом химической кинетики является постулат, выражающий зависимость скорости реакции от концентрации реагирующих веществ: скорость реакции в каждый момент времени про-
порциональна произведению возведенных в некоторую степень концентраций реагирующих веществ (закон действия масс). Так, для реакции (а) скорость может быть записана
w = k [А]а [В]*.
(XV.2)
Коэффициент пропорциональности k в уравнении (XV.2) называется константой скорости реакции. Она равняется скорости реакции при условии, если концентрация каждого из реагирующих веществ равна единице, поэтому ее называют также удельной скоростью реакции. Такой физический смысл константы скорости указывает на то, что величина ее должна зависеть от всех факторов, которые влияют на скорость реакции, за исключением изменения концентрации реагирующих веществ. Числовое значение константы скорости зависит также от выбора единиц времени и концентра-320
ции. Размерность ее определяется тем кинетическим уравнением, по которому производится ее расчет, т. е. зависит от порядка реакции.
Порядок реакции. Различают порядок реакции и ее молекуляр-
ствующих в одном элементжно^ЗКхе химического превращения. При этом число молекул образующихся веществ не имеет значения. В зависимости от этого различают реакции: 1) мономолеку-лярные, 2) бимолекулярные, 3) тримолекулярные.
Порядок реакции определяется показателем степени при концентрации в кинетическом уравнении реакции. Если порядок равен единице, то реакцию называют реакцией первого порядка, если двум — второго порядка, если трем — третьего порядка.
Уравнение (XV.2), связывающее скорость реакции с концентрациями реагирующих веществ, называется кинетическим уравнением. В зависимости от порядка реакции кинетические уравнения для расчета константы скорости реакции различны.
Порядок реакции является чисто эмпирической величиной. Только для элементарной реакции, протёкающей^в^одйн этап, он равен ее молекулярности, так как стехиометрическое уравнение правильно отражает истинный механизм такой реакции. Различают полный и частный порядок реакции. Каждый из показателей степени при концентрациях в дифференциальном уравнении скорости выражает частный порядок. Сумма показателей степени при концентрациях определяет полный (суммарный) порядок реакции.
Протекание реакции сложным путем, в несколько стадий, является одной из причин расхождения между порядком реакции и ее молекулярностью. Другой причиной расхождения может быть значительный избыток одного из реагентов в реакционной смеси. Тогда концентрация этого реагента остается практически" постоянной в ходе реакции, а порядок реакции будет меньше, чем определяемый по стехиометрическому уравнению. Например, б им о ле ку л я р ны е реакции: инверсия тростникового сахара или гидролиз'^уксусного ангидрида — кинетически оказываются реакциями первого поряд-&&~хак как конценхрацию воды здесь можно считать неизменной. Подобного рода реакции иногда называют псевдомономолекуляр-ными. Порядок реакции зависит от условий ее протекания. Его можно изменить, например, варьированием концентрации или давления.
Кинетическая классификация реакций. В химической кинетике реакции разделяют по следующим признакам: 1) по числу частиц, участвующих в реакции (молекулярность и порядок реакции); 2) по природе частиц, участвующих в элементарном акте реакции. Реакции, в которых участвуют молекулы, относятся к группе молекулярных реакций; реакции с участием атомов или свободных радикалов—к группе цепных реакций; реакции с участием ионов — к группе ионных реакций; 3) по числу фаз, участвующих в реакции. Реакции, протекающие в одной фазе, называют гомогенными. Реакции, протекающие на поверхности или у поверхности раздела фаз, 11—2083 32 у
называют гетерогенными; 4) по применимости катализаторов: ката литические, автокаталитические и некаталитические; 5) по степени сложности (по механизму протекания): а) обратимые и необратимые; б) изолированные и параллельные; в) последовательные (конструктивные или многоступенчатые); г) сопряженные.
2. ЗАВИСИМОСТЬ СКОРОСТИ РЕАКЦИИ
ОТ КОНЦЕНТРАЦИИ РЕАГИРУЮЩИХ ВЕЩЕСТВ
Простые односторонние реакции. Односторонними, или необратимыми, реакциями называются такие, в которых конечные "npi>^
дукты^ отсутствуют (в начальные моменты для любой реакции) или присутствуют лишь в очень небольших количествах (выпадают в осадок, выделяются в виде газа, образуют малодиссоциировагн-ное соединение), а также такие, в которых скорости прямой и обратной реакций несоизмеримы т. е. протекают в одном
направлений.
В тексте используются следующие обозначения: а и b — начальные концентрации исходных веществ А и В соответственно (с0А и Сов); (#—х) и (Ь—х) — концентрации этих веществ к моменту времени t от начала реакции (сА и св); х— концентрация* прореагировавшего вещества за истекший промежуток времени (от начала реакции до момента определения концентрации, т. е. концентрация продукта реакции, сх).
Реакции первого порядка. К этим реакциям относятся реакции изомеризации, термического разложения веществ, радиоактивного распада и многие бимолекулярные реакции при условии^ что концентрация одного из реагирующих веществ поддерживается постоянной. Для реакции типа А-^С скорость выражается уравнением
d (а — х) dx
г/ — —------- =---~k(a — x). (XV.3)
d/ dZ
Интегрируя уравнение (XV.3), получим
1п (а — х) = — kt Ц- const.
(XV.3a)
Интегрирование (XV.3a) в пределах от Zo=O до t приводит к уравнению
(XV. 4)
из которого видно, что константа скорости реакции первого поряд-
ка имеет размерность, обратную времени: —
время
Количественной характеристикой скорости реакции первого порядка кроме константы скорости могут служить период полупревращения (полураспада) Zi/s> а также среднее время жизни частицы. t. Среднее время жизни определяется как время, необходимое 322
для того, чтобы концентрация исходного вещества уменьшилась в е раз. Выражение (XV.4) можно представить в экспоненциальной форме
a — x — ae~kit (XV.5)
откуда следует, что (а—х) достигает а/е в момент времени т=1/й, т. е. среднее время жизни одной исходной молекулы (частицы) равно обратной величине константы скорости реакции.
Как ti/2 реакций первого порядка, так и т непосредственно связаны с константами скоростей и не зависят от начальной концентрации вещества.
Реакции второго порядка, .Примером такой реакции может служить омыление сложного эфира щелочью. Реакцию второго порядка представим схемой
А + В — продукты реакции
При равных начальных концентрациях исходных веществ (а = &) скорость реакции выражается dx/dt=^k(a—%)2, откуда после интегрирования
1
:----= kt 4- const. (XV. 6)
а — х
Взяв определенный интеграл (от t = 0 до t), получаем
L х
t а(а — х)'
(XV.7)
При Cqat^ob (а^=Ь) уравнение скорости реакции второго порядка в дифференциальной форме примет вид
— k (а — х)(Ь — х)
и после интегрирования
£ 2’3 ^(^ — X*)
t {а — Ь) * а (Ь — х)
(XV.8)
Разновидностью реакций второго порядка являются автокаталитические реакции, в которых катализатором служит один из продуктов реакции. В этих реакциях концентрация катализатора увеличивается со временем и дифференциальное уравнение скорости преобразуется:
= k {а + х) (6 — х)
и после интегрирования принимает вид
2,3 b (а -г х)
-........ jcr------------,
т (Л + b) а (b — х)
(XV.9)
(XV. 10)
Примером служит реакция иодирования ацетона. Уравнения (XV.7), (XV.8) и (XV.10) показывают, что при этом размерность
11»
323
k выражается величиной, обратной концентрации и времени, т. е. /-‘с-1.
Реакции третьего порядка. К ним относятся взаимодействия NO с Нг, Ог, С1г, Вгг, а также процессы рекомбинации атомов или
свободных радикалов в молекулы, реакции окисления — восстановления. При CoA=CoB=CoD=a для.реакции A+B+D^-продукты получаем дифференциальное уравнение скорости —~ = k(a — х)3
d t
и после интегрирования
1
—------------ kt + const
2 (а — х)2
*1 / 1 _ 1\
2t \(<Z — х)2 <Z2 J*
[(XV. И)
(XV Л 2)
Константа скорости k здесь выражается величиной, обратной времени и квадрату концентрации.
Реакции дробных порядков. Дробный порядок реакции указывает обычно на одновременное протекание нескольких этапов реакций, мало отличающихся друг от друга по скоростям, или на протекание обратимых реакций. Это может быть также следствием участия в реакциях атомов наряду с молекулами. Например, реакция превращения ортоводорода в пароводород
о-Н2 + Н^п-Н2 + Н
Здесь атомы Н получаются за счет термического разложения Н2. Скорость этой реакции выражается уравнением d с о/п _
--- = kCrx Стг = k’cA , /2=1,5.
d t На н На
Реакции нулевых порядков. Такой порядок получается при постоянстве скорости реакции, т. е. независимости ее от концентрации одного или нескольких реагирующих веществ. Тогда скорость будет —dc/d/=& и после интегрирования с=—+const. При / = 0 const=Co и
Сл С
k^------. (XV. 13)
i *
Это встречается главным образом среди гетерогенных реакций, происходящих на поверхности. Скорость реакции может определяться не только концентрацией, но и другими факторами, например количеством поглощенной световой энергии при фотохимических реакциях или количеством катализатора в каталитических реакциях. Каталитические реакции могут иметь первый порядок по катализатору и нулевой — по реагирующему веществу. Реакции общего нулевого порядка встречаются редко. Для реакции и-го порядка (кроме первого) при равенстве концентраций всех исходных веществ константа скорости выражается уравнением
1 г 1 ___1
(«—!)£ _ (а — х)”"1 ап~
(XV. 14)
324
Сложные реакции. Теоретическое изучение сложных реакций ос-
новывается на положении о независимом протекании элементарных реакций. Согласно этому положению при протекании в системе од
новременно нескольких реакций каждая из них идет независимо от
других и подчиняется закону действия масс (принцип независимости).
В обратимых реакциях скорости прямой и обратной реакций соизмеримы. Простейшей обратимой (двусторонней) реакцией является обратимая реакция первого порядка. Примерами могут служить процессы взаимного превращения изомеров-цис-транс-изомеризация или изомеризация цианида аммония в карбамид*. NHiCNO^ (NH2)2CO. Схематически такая реакция может быть представлена
уравнением
Кинетические кривые для такого типа реакций приведены на рис. 138. Наблюдаемая скорость здесь определяется разно-
Рис. 138. Кинетические кривые для обратимой реакции первого порядка:
/ — прямая реакция; 2 — обратная реакция
стью скоростей прямой и обратной реакций: шнабл = ^1—ом, а диф-
ференциальное уравнение скорости имеет вид
Для состояния равновесия dx/cW=0 и, следовательно, первое сла-kta — k—\b
гаемое в скобке ™-------— = х , где — изменение концентра-
i 00
ции, соответствующее равновесию. Таким образом, получим
После интегрирования
(XV. 15)
В то же время, в условиях равновесия, когда оц' = а>_1' и х=хоо,
(XV. 16)
где — константа равновесия, выраженная через концентрации. Уравнения (XV. 15) и (XV. 16) дают возможность рассчитать обе константы скорости и k_x. Из выражения (XV. 16) видно, что
325
константа равновесия представляет собой соотношение констант скоростей прямой и обратной реакций.
Изолированные реакции — реакции, в ходе которых образуется продукт только одного типа.
Параллельные реакции — такие, в которых взятые вещества одновременно реагируют в двух или более направлениях (в результате образуются разные продукты). Так, при хлорировании бензола получаются одновременно три изомера двузамещенных бензола: о-, м- и n-хлорбензол. Схематическая запись простейшей реакции этого типа:
Вещество А участвует одновременно в двух реакциях первого порядка с образованием продуктов В и С. Наблюдаемая скорость реакции (скорость расходования исходных веществ) равна сумме скоростей параллельных реакций:
^набл™ ^1 + ^2-
Обозначив через а—исходную концентрацию вещества А; через х — убыль его концентрации к моменту времени /, а через х^ и х2 — концентрации продуктов В и С (x=xi + x2), можно выразить скорость реакции в виде
d х2
— ’ х) —J- k2 (л —• х) — ^2) £)•
После интегрирования
*эксп = kx + k2 = 1g . (XV. 17)
Каждую из констант скоростей находят по соотношению концентраций продуктов:
kx[k2 = x^x2. (XV. IS)
Кинетические кривые для такой простейшей параллельной реакции приведены на рис. 139.
Последовательные (многоступенчатые, или консекутивные) реакции. К таким реакциям относятся реакции с промежуточными стадиями. Промежуточными продуктами в них могут быть обычные молекулы, вступающие далее в реакцию, или свободные атомы или радикалы. Большинство химических реакций протекает именно по такому пути. В простейшем случае это две последовательные односторонние реакции первого порядка, например термический крекинг нефти, где бензин является промежуточным продуктом, распадающимся далее на газообразные вещества. Уравнение простейшей последовательной реакции
ft1 ftjt А^В+С
326
Скорость накопления промежуточного вещества В будет шв = — —w2, а концентрацию этого вещества рассчитывают по уравнению
[В] = а ——---------
&2 —
где а — начальная концентрация вещества А. нечного продукта С вычисляют по уравнению
(XV. 19)
Концентрацию ко-
(XV.20)
На рис. 140 приведены кривые изменения во времени количеств веществ А, В и С (сА, св и сс). Чем меньше k2fkXy тем выше лежит максимум кривой для вещества В и тем дальше он от мо-
Рис. 140. Изменение концентраций веществ в ходе последовательной реакции с необратимыми элементарными стадиями при
Рис, 139. Кинетические кривые двух параллельных реакций первого порядка:
/ — убыль А в реакции (1); 2 — убыль А в реакции (2); 3— суммарная убыль А в параллельных реакциях (1) и (2)
мента начала реакции. В начале реакции вещество С вообще нельзя обнаружить. Это — скрытый период, называемый, периодом индукций /инд.
Если одна из составляющих реакций обладает явно меньшей скоростью, чем остальные, то общая скорость реакции определя-ется* именно этой наиболее замедленной (лимитирующей) стадией. Тогда в кинетических уравнениях быстро протекающие реакции могут не учитываться.
Многие реакции протекают в несколько последовательных и параллельных стадиях. Кинетический расчет таких реакций очень сложен. В химической кинетике широко используется еще один приближенный метод — метод стационарных концентраций (стационарного состояния). Этот метод применим к системам последовательных и последовательно-параллельных реакций, если промежуточные продукты в них характеризуются высокой реакционной способностью. Предполагается, что концентрация промежуточных соединений, находящихся в системе в незначительных количествах по сравнению с исходными веществами и продуктами ре
327
акции, не изменяется в ходе реакции. Вначале равная нулю концентрация быстро достигает постоянного (стационарного) значения на всем протяжении процесса. Такой режим процесса называется стационарным. Скорость изменения концентрации промежуточных частиц здесь считается равной нулю, а это позволяет заменить для них дифференциальные уравнения алгебраическими. Тогда весь комплекс дифференциальных уравнений при описании кинетики сложного химического процесса упрощается.
Сопряженные реакции. Это — системы как минимум двух протекающих одновременно в одной фазе реакций, из которых одна зависит от другой. Такие реакции часто протекают в растворах и весьма распространены в живых организмах. Наиболее изученными являются реакции окисления. Например, окисление FeSO4 и бензола пероксидом водорода. Сульфат железа (II) окисляется независимо от присутствия бензола (первичная реакция), но бензол окисляется лишь в присутствии FeSO4. Причина здесь в образовании общего промежуточного продукта, связывающего оба процесса,— радикала ОН. *
Таким образом, это совместные реакции вида
А 4- В *первичная реакция (1)
А 4- С -вторичная реакция (2)
Реакция (2) осуществляется лишь при протекании реакции (1), т. е. при условии, если взяты вещества А, В и С. Вещество В, необ- / ходимое для возбуждения реакции (2), является ее индуктором (Ге2+), вещество А, общее для обеих реакций, называется актором (Н2О2), вещество С, трудноокисляемое, вовлекается в реакцию под действием индуктора и называется акцептором (С6Н3). Сопряжение реакций возможно, если промежуточное вещество реакции (1) является исходным в реакции (2), т. е. связывает оба процесса (ОН). Такое явление передачи реакционной способности от одной реакции к другой называется химической индукцией. В сопряженных реакциях индуктор расходуется и не регенерируется.
3. ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ СКОРОСТИ И ПОРЯДКА РЕАКЦИИ
Измерение скорости реакции основано на определении концентрации одного из реагирующих веществ через различные промежутки времени от начала реакции. Для определения концентраций можно применять методы физико-химического анализа, основанные на зависимости физических свойств смеси от ее состава (например, определение показателя преломления, угла вращения плоскости поляризации, вязкости, электрической проводимости, объема, плотности, изменения температур замерзания и кипения, интенсивности окраски и т. п.), и методы аналитической химии (например, титрование). Поскольку концентрации по ходу реакции 328
непрерывно меняются, то необходимо или очень быстрое измерение концентрации (методы физико-химического анализа), или торможение реакции во взятой пробе (химический контроль). Торможение может быть достигнуто охлаждением, резким разбавленй-нием, устранением катализатора или совместным действием всех указанных факторов. Если реакция, протекающая в газовой фазе, сопровождается изменением числа молекул, то ее течение удобно контролировать по изменению давления смеси во времени.
К сравнительно медленным реакциям со временем полупревращения порядка получаса и более можно применять спектроскопию, масс-спектрометрию и хроматографию. Для исследования скоростей очень быстрых реакций (с периодом полупревращения до 10-7 и даже 10-9 с) используются специально разработанные методы и особая аппаратура.
Для определения порядка реакции необходимо иметь экспериментальные данные об изменении концентрации реагирующих веществ со временем. Если в реакции участвует несколько веществ, то пользуются методом изолирования Оствальда. Допустим, в реакцию вступают три вещества: А, В и D
V! А + V2 В + v3 D-продукты
Скорость этой реакции может быть выражена кинетическим уравнением
— — ~kraebrd
Сначала проводят реакцию с большими избытками веществ В и D (концентрация вещества А равняется от 0,1 до 0,001 начальной концентрации всех остальных веществ). Тогда
de „ -77=*1Са’
где k\ — kcBbCT)d. При таких условиях определяют а. Затем проводят второй опыт с большим избытком веществ А и В для определения d. В третьем опыте берут большой избыток веществ А и D и определяют значение Ь. Таким образом, порядок реакции каждый раз искусственно снижается и сводится к определению частных порядков а, b и d. Общий порядок реакции равен сумме a+'b-\-d. Все методы определения частных порядков реакций можно разделить на две группы — интегральные и дифференциальные.
Интегральные методы. Здесь используются кинетические уравнения для определения скорости реакции в интегральной форме (полученные после интегрирования дифференциального уравнения скорости реакции). Разновидности этой группы методов:
1) метод подбора уравнений, основанный на подстановке экспериментальных данных по концентрации вещества для каждого момента времени в кинетические уравнения реакций различных порядков. Определяемый порядок реакции соответствует тому урав-
329
нению, для которого при различных начальных концентрациях исходных веществ и в различные моменты времени при заданной температуре константа скорости будет оставаться постоянной;
2) графический метод, основанный на том, что определяют такую функцию от концентрации, которая на графике зависимости ее от времени дает прямую
Inc
Рис. 141. Зависимость различных функций концентрации исходных веществ от времени для реакций различных порядков:
? —реакция первого порядка; 2 — реакция второго порядка; 3 — реакция третьего порядка
линию. Для реакций первого порядка (прямая 1) такой функцией является '1g с (рис. 141); для реакций второго порядка (прямая 2) — 1/с (при Соа = сов);для реакций третьего, порядка (прямая 3) — 1/с2, что вытекает из кинетических уравнений (XV.3), (XV.6) и (XV.11). Если концентрации исходных веществ различны, то для выделения частного порядка реакции по одному из компонентов ад, Ьв, ... используется метод изолирования Оствальда с последующим определением а, 6.
По тангенсу угла наклона полученной прямой вычисляют константу скорости реакции k\
3) метод определения по периоду полупревращения п/г. Для реакций
первого порядка время превращения половины (или определенной доли) вещества не зависит от начальной концентрации с0 = а, при х = 0,5я t=Ti/2 и в соответствии с уравнением (XV.4)
(XV.21)
Для реакции гг-го порядка соотношение между временем разложения половины исходного вещества и начальной концентрацией в соответствии с уравнением (XV. 14) будет
1
J-Г* II»
т1/2 ~ „п-1 ’
где а — начальная концентрация исходного вещества; п— порядок реакции. Опыты проводят при двух различных начальных концентрациях й! и а2 (при этом все исходные вещества берут в эквивалентных количествах или используют метод изолирования Оствальда) :
(XV.22)
(XV. 23)
330
Уравнение (XV.22) делят на (XV.23) и логарифмируют, откуда
(т1/2/т1/2)
1g (^2/^1)
Те же соотношения сохраняются и при определении времени превращения любой доли исходной концентрации (т. е. не обязательно определять ti/2, можно определить, например, п/з или Т1/5) • Этот метод называют иногда методом Оствальда — Нойеса.
Дифференциальные методы. Эти методы основаны на использовании уравнения для скорости реакции в дифференциальной фор
ме.
Метод Вант-Гоффа состоит в том, что реакцию проводят с компонентами, взятыми при двух различных исходных концентрациях и а2. Тогда
(XV. 24)
(XV.25)
После деления вания получим
уравнения (XV.24) на (XV.25) и логарифмиро-
так как
/ &с \ ( Ас \
Графические варианты метода Вант-Гоффа пользовании уравнения
(XV.26)
S основаны на ис-
lg w = lg£ + nlgc,
которое получается при логарифмировании выражения для скорости реакции n-го порядка w=kcn. На графике в координатах lg w—1g с полученная зависимость изображается прямой линией с тангенсом угла наклона к оси 1g с, соответствующим порядку реакции (tgtp=n). Отрезок, отсекаемый на оси lgw, равен lgk. Скорость определяется тангенсом угла наклона касательной к кривой относительно оси времени. В зависимости от того, какая определяется скорость — в начальный момент реакции или в различные промежутки времени от начала реакции — различаются два графических варианта этого метода. Вариант I (рис. 142) дает возможность рассчитать концентрационный или истинный порядок «с реакции, так как скорость здесь определяется в условиях отсут-
331
ствия конечных или промежуточных продуктов, которые могут оказывать на нее влияние. В варианте II (рис. 143) скорость определяется в различные моменты времени от начала реакции. Тангенс угла наклона прямой на рис. 143, б соответствует порядку реакции, который называют временным nt. Таким образом, различие
Рис. 142. Определение порядка реакции по методу Вант-Гоффа (вариант I):
а — определение начальных скоростей; б — определение но-рядка реакции
Рис. 143. Определение порядка реакций по методу Вант-Гоффа (вариант II):
а — определение скорости реакции в разные моменты времени; б — определение порядка реакции nt
L
между пс и nt позволяет обнаружить влияние на скорость реакции ее конечных или промежуточных продуктов.
Интегральными методами можно определить суммарный временной порядок реакции, а не истинный, так как проводится обработка результатов только одного опыта.
Дифференциальные методы осложняются невозможностью точного определения тангенса угла наклона касательной к кривой в данной точке. (Применение тангиметров устраняет этот недостаток.)
332
4. ЗАВИСИМОСТЬ СКОРОСТИ РЕАКЦИИ ОТ ТЕМПЕРАТУРЫ
Скорость химических реакций с повышением температуры резко возрастает. Эта зависимость для гомогенной реакции приближенно описывается эмпирическим правилом Вант-Гоффа: при нагревании на 10° константа скорости увеличивается в два — четыре раза, т. е.
где —-константа скорости при температуре 1+10°; kt — кон-
станта скорости той же реакции при температуре t.
Соотношение ktwlkt называется температурным коэффициентом и обозначается у. Определив у, можно приближенно рассчитать константу скорости при любой температуре по уравнению
= (XV.27)
{в небольшом интервале относительно невысоких температур).
Более точно зависимость скорости реакции от температуры описывается уравнением Аррениуса:
dln&CKk Е
V к
ат ~ RT2 '
(XV. 28)
где feCT< — константа скорости реакции; Т — термодинамическая температура, К; R — универсальная газовая постоянная; Е — эмпирическая постоянная, имеющая размерность энергии (энергия активации). После интегрирования уравнение (XV.27)( принимает вид
Е
In k = —____+ const
RT
а в экспоненциальной форме
k = Ае~~Е^т,
(XV. 29)
(XV. 30)
где А — эмпирическая постоянная, называемая предэкспоненциаль-ным множителем и численно равная константе скорости при Т-^оа. л
Энергией активации’реакции согласно теории соударений называется минимальная энергия (в расчете на 1 моль), которой должны обладать реагирующие частицы, чтобы столкновение между ними привело к взаимодействию. Частицы, энергия которых больше или равна Е, называются активными. Эта энергия необходима для преодоления энергетического барьера реакции. Столкновение будет эффективным, если суммарная величина энергии сталкивающихся частиц равна или больше энергии активации Е, характерной для данной реакции. Для сложной реакции (протекающей в несколько стадий) параметр Е в уравнении Аррениуса не равен энергии активации лимитирующей стадии, а представля
333
ет некоторую функцию энергий активации отдельных стадий или вообще эмпирическую величину.
Стерический фактор. Согласно теории соударений пред-экспоненциальный множитель А (эмпирическая постоянная в уравнении Аррениуса) представляет собой фактор соударений Zo, который связан с общим числом двойных соударений z в 1 см3 в 1 с соотношением
= ^A^/fnSlOOO),
где п— число частиц (молекул) в 1 см3; Ад/1000 — множитель для молярной концентрации; Ад — постоянная Авогадро.
Величины z и z0 могут быть определены на основании молекулярно-кинетической теории. Поэтому, заменив А на Zo, константу скорости можно рассчитать по уравнению
k =
Однако такой расчет скорости реакции часто дает завышенные результаты, особенно для реакций в растворах. Для согласования расчетных данных с опытными в уравнение вводят дополнительный множитель Р, называемый стерическим или вероятностным фактором:
(XV.31>
Следовательно,
(XV.32>
Подстановка в уравнение (XV.32) выражения для z$ позволяет рассчитать константу скорости реакции. Для взаимодействия разнородных частиц константа скорости будет
А п2 /о _ Ш
АВ_ АВ I ЖЖ / 1000
\ А о /
0-Е/$г
с? 5
(XV.33>
где МА и Mb — молекулярные массы молекул; Одв= (оа+ов)/2 — усредненный диаметр (сгд и <гв — диаметры молекул А и В); £ — энергия активации данной реакции.
Стерический фактор Р зависит от природы реакции и может изменяться в пределах от 1 до 10-8; для некоторых ионных реакций, протекающих в растворах, стерический фактор может быть больше единицы. Стерический фактор учитывает вероятность того» что при столкновении частицы будут обладать необходимой для протекания реакции пространственной ориентацией.
Предэкспоненциальный множитель определяется экспериментально измерением констант скорости при нескольких температурах. Вычислив энергию активации, можно по значению k при некоторой температуре рассчитать Р.
Вопрос о возможности теоретического расчета скоростей реакций (без экспериментального их изучения) на основании свойств
реагирующих молекул решается теорией абсолютных скоростей реакций (теорией переходного состояния). Промежуточным состоянием любого элементарного акта химического превращения является образование активированного комплекса. Согласно этой теории активированный комплекс представляет собой особого рода неустойчивую молекулу с очень малой продолжительностью жизни.
Активированному комплексу отвечает максимальное значение потенциальной энергии при переходе от начального состояния си-
стемы к конечному. На рис. 144 показано изменение потенциальной энергии системы атомов А— —В—С вдоль координаты пути реакции (или координаты реакции) :
АВ 4- С->А — В — С->А + ВС
Координата реакции характеризует перемещение системы вдоль наиболее выгодного энергетически пути реакции.
Активированный комплекс отличается от обычных молекул тем, что частота его нормального колебания вдоль координаты разложения (в направлении линии валентных связей) имеет минимальное значение, приводящее к появлению особой степени свободы поступательного движения. Распад активированного комплекса с образованием продуктов
Координата реакции
Рис. 144. Энергетический профиль пути реакции:
Eq — энергия активации прямой (^изотермической) реакции; Eq' — энергия / активации обратной (эндотермической) реакции; б — область, отвечающая переходному состоянию
реакции происходит с частотой, не зависимой от его природы и определяемой только температурой.
Истинной энергией активации элементарного акта химического превращения согласно теории переходного состояния называется минимальная энергия, которой должна обладать исходная система сверх своей нулевой энергии, чтобы в ней могло произойти химическое превращение. Энергия активации равна разности нулевых энергий переходного и исходного состояний системы (рис. 144). Разность истинных энергий активации прямой и обратной реакций равна тепловому эффекту реакции при абсолютном пуле. Энергия активации Е (эффективная энергия активации) оп-
ределяется экспериментально.
Одной из важных особенностей теории переходного состояния
является возможность расчета некоторых характеристик, связанных с механизмом изучаемого процесса. Так, на основании экспериментальных констант скоростей и энергии активации можно рассчитать теплоту активации АН^ или AU^, энтропию активации 45^ и энергию активации Гиббса AG^.
335
Уравнение теории переходного состояния может быть записано в квазитермодинамической форме, так как в основе теории лежит предположение о существовании равновесия между активирован* ным комплексом и исходными веществами:
АТ
(XV.34)
h 1
где k — константа Больцмана; /г —постоянная Планка; К?г~— константа равновесия процесса образования активированного комплекса; х— трансмиссионный коэффициент, учитывающий вероятность того, что система, достигнув переходного состояния, пройдет через него в направлении образования продуктов реакции. Теоретического метода расчета этого коэффициента нет, но для большинства химических реакций его практически принимают равным единице. Однако х может быть и менее единицы в условиях, например, недостаточной стабилизации образующихся высокоэнергетических частиц или при изменении электронного состояния системы.
выражается
Для процесса активации константа равновесия уравнением
= / rt! я.
Преобразовав уравнение (XV.34), получим
kc = x2L6as0¥7/? е-ьн^1%т' ск h
При и T=const константа скорости выражается
ь _ х Л'СК — Л г & &
п
(XV. 35)
(XV. 36)
В уравнениях (XV.35) и (XV.36) ДЯ°*иД[/°*— теплота активации, т. е. стандартное изменение энтальпии и внутренней энергии в процессе перехода исходного состояния в состояние активированного комплекса; ASQ=^ — энтропия активации, т. е. изменение энтропии при образовании одного моля активированных комплексов в стандартных условиях (при концентрациях активированного комплекса и исходных частиц, равных 1 моль/л, или активностях, равных единице). Значения AUO¥= и ДЯО#= не могут быть определены экспериментально, но они связаны с энергией активации £Эксп> получаемой опытным путем. При независимых параметрах р и Т
+ (XV. 37)
Подстановка АНФ из выражения (XV.37) в (XV.35) приводит к уравнению
кР,ск = ^е'^-е^1Ке~Е^1КТ. (XV.38)
h
За стандартное состояние реагирующих веществ принято парциальное давление, равное 1 атм. Выбор стандартного состояния
336
определяет размерность скорости реакции. Так, при стандартном состоянии, равном 1 атм, (х==1),
kT
(XV. 39)
Применительно к бимолекулярной реакции типа АВ + С ^Л ~ В — С продукты
можно записать
AG* - -RTinKf + RT In
^АВ^С
или после потенцирования
Kt = g~AG0?"/j?r Ра~В~-. (XV.39a>
р Р^Рс
S*
Подставляя выражение (V.39a) в уравнение (XV.39), полу* чаем ;
kT Ра-в-с
«р,СК - , 6 1
А ^АВ^С
Для стандартных условий парциальные давления соответствуют 1 атм, поэтому единицей измерения константы скорости будет [&р]=атм“1-с”1.
Для стандартного состояния 1 моль/л единицей измерения константы скорости будет [£с, Ск]= (моль/л)“^с”1.
При независимых переменных и и Г
/ £Экс„ = Аб^ + RT. (XV.40)
Для растворов за стандартное состояние выбирают исходные и увечные концентрации (илу активности), равные единице. Тогда /замена ДС/^ в уравнении (XV.36) на ^эксп приводит к выражению
kT хе — h
tS°*/R _Е jot с ' _ х'эксп//о
(XV.41)
где константа скорости реакции второго порядка имеет размерность [концентрация-1-время-1]. Уравнение (XV.41) применимо и к мономолекулярным газофазным реакциям, причем ДЗ^ не зависит от выбора стандартного состояния, a kp и &с равны.
Если имеется несколько констант скоростей при разных температурах; то удобным методом расчета энтропии активации и одновременно энергии активации является графический метод: квази-термодинамическая форма уравнения теории переходного состояния записывается в виде линейного уравнения, которое и пред-
337
ставляется графически в виде прямой линии. Так, кулярной газофазной реакции или для реакции в х=1)
для мономоле-растворе (при
.... J
k^e^Le^lRe~E/RTt h
Обозначим
Подстановка у в уравнение дает yRT = — £эксп*
Окончательно уравнение прямой у— (AS^/jR) — (E/RT), которое можно представить в виде графика y—f(l/T). На рис. 145 tg<p= =—£/R; £=R tg<p. Отрезок, отсекаемый на оси ординат, Ь — =AS^/R; AS^=bR.
Рис. 145. Определение энтропии и энергии активации реакции
Рис. 146. Зависимость логарифма константы скорости реакции от обратной термодинамической температуры
Преобразование уравнений (XV.31), (XV.32) и (XV.38)' дает возможность рассчитать также стерический (энтропийный или вероятностный) фактор:
И При % = 1
Л
P = e^e^/Rt т. е. p~eis+/R "(XV.42)
йг0
Теория переходного состояния позволяет вычислить предэкспо-ненциальный множитель (а также Р и Д5*).
338
Способы расчета энергии активации. 1. Интегрирование уравнения Аррениуса (XV.28) в пределах от до Т2 дает
<хулз>
Отсюда—возможность расчета Е по константам скоростей при двух температурах.
2; Построение графической зависимости lg k от 1/Г (рис. 146)„ соответствующей уравнению (XV.29), когда известен ряд констант скоростей при разных температурах,
tg? = -£/(2,3/?).
3. Вычисление по основному уравнению теории соударений (XV.31). Этот способ возможен только для немногих простейших бимолекулярных реакций, для которых Р—\. Зная значение kCK для одной какой-либо температуры и рассчитав г0, можно определить Е.
4. Использование кинетических кривых, которые выражают изменение концентрации одного из реагентов со временем в ходе химического превращения. Такие кривые нужно получить при нескольких температурах (минимум при двух) и при постоянном исходном составе. Далее эти кривые трансформируют по осщ времени, т. е. совмещают их с какой-то выбранной стандартной кривой. Трансформирование осуществляется изменением масштаба
Рис. 147. Кинетические кривые реакции при трех температурах Т\ < Т 2 < Т з
Рис. 148. Трансформация кинетических кривых реакции на кинетическую кривую
графика по оси времени. Множитель, который при этом используется, называется коэффициентом трансформации х. Коэффициенты трансформации, например для трех кинетических кривых, представленных на рис. 147, определяются следующим образом. Если кривая /, полученная при температуре принята за стандартную,
339
коэффициенты трансформации и2 и хз рассчитываются как соотношения
х2 == /ст//2 и х3 = /ст/^з
при одной и той же ординате (в данном случае при c=ci). Соотношение /СТД по существу аналогично соотношению скоростей реакций
% = w]fC'ct “ ^ст/’
а скорости можно сопоставлять только при одинаковой глубине превращения, т. е. при равных концентрациях. Изменив значения абсцисс кинетических кривых 2 и 3 (при концентрациях с2, с3 и т. д.) умножением их соответственно на хг и х3, совмещают эти кривые со стандартной кривой 1 (рис. 148). Поскольку концентрация реагирующих веществ не зависит от температуры, скорость реакции может быть также выражена уравнением Аррениуса
где — постоянный параметр. Тогда
х = — = — Wr.e~E/^ = 4e-EW, WCT WCT "
где куст — постоянная величина; хо— постоянный параметр, не зависящий от температуры.
Таким образом, изменение коэффициента трансформации от температуры выражается уравнением Аррениуса. Это дает возможность, используя аррениусовские координаты \g%=f(l/T) определить графически энергию активации данной реакции по тангенсу угла наклона полученной прямой к оси 1/Г. Последний способ позволяет рассчитать энергию активации без определения порядка реакции. Кроме того, можно получать кинетические кривые по любому параметру системы (давлению, оптической плотности, электрической проводимости, вязкости и т. и.).
Ограничением в применении метода трансформации является обязательное условие совмещения кривых, что выполняется при
И'з/®! = х = const при Т = и с — const.
5. ОСОБЕННОСТИ КИНЕТИКИ РЕАКЦИЙ В РАСТВОРАХ
Элементарные химические реакции классифицируются по способам разрыва и образования связей, зависящим от химической природы реагентов и среды, в которой происходит реакция. По этому признаку различают реакции гомолитические (или радикальные) и гетеролитические (или ионные).
В гомолитических реакциях исходными веществами или конечными продуктами служат свободные радикалы (молекулы с неспаренными электронами). Возникновение связей здесь осуществляется путем спаривания электронов двух реагирующих ча-340
стиц, а разрыв связей — путем разделения спаренных электронов. Гомолитический разрыв связей происходит обычно в газофазных реакциях, в растворах же он наблюдается только при малой диэлектрической постоянной среды и для малополярных связей. Растворитель в таких реакциях действует лишь как заполнитель пространства и только таким образом влияет на скорость. Поэтому скорость гомолитических реакций почти не меняется при проведении их в растворе и в газовой фазе.
В гетеролит и че с ких реакциях не ’Происходит разрушения и возникновения электронных пар, а взаимодействие происходит за счет электронов, которые уже были спаренными до реакции. Такие реакции почти не наблюдаются в газах, но являются характерными для растворов — большие диэлектрические постоянные многих растворителей понижают электростатическое притяжение ионов, а электрические поля полярных молекул растворителя способствуют поляризации диссоциирующей связи. Таким образом, гетерополярные реакции протекают с существенным перераспределением зарядов между реагирующими частицами и образование активированного комплекса при этом сопровождается значительной перестройкой сольватных оболочек. Поэтому скорость гетеро-литических реакций, в противоположность гомолитическим, существенно зависит от природы растворителя, и известно множество реакций, которые вообще не идут без растворителя. Скорость реакции
АВ 4- С А.. .В.. .С продукты реакции (
в растворе можно сравнить со скоростью той же реакции в газовой фазе. Согласно уравнению Бренстеда — Бьеррума
£ск = MabYc/Y7"’ (XV.44)
где Аск — константа скорости реакции в растворе; Ао— константа скорости той же реакции в газовой фазе (или в бесконечно разбавленном растворе); удв, ус и уф — коэффициенты активности исходных частиц и активированного комплекса соответственно. В газовой фазе эти коэффициенты принимаются равными 1. Величину у* легко удается определить только для реакций между ионами, где требуется учет лишь сил электростатического взаимодействия. При этом коэффициенты активности взаимодействующих ионов и в*в и активного комплекса выражаются при помощи предельного закона Дебая и Гюккеляша заряд активного комплекса принимается равным сумме зарядов взаимодействующих ионов: zx=za + zb. Тогда уравнение (XV.44) принимает вид
ln£CK = 1п/г0 4-2А.?А(гв|Л/, (XV.45)
где I — ионная сила раствора; А — коэффициент пропорциональности, зависящий от природы растворителя и температуры. В вод
341
ном растворе при 25°С коэффициент 0,509 и уравнение (XV.45) можно переписать:
lg kCK = 1g + 1,02глгв/7. (XV,5)
Константа скорости в разбавленных растворах солей возрастает с увеличением ионной силы раствора, если взаимодействуют ионы одного знака, и тем больше, чем больше их заряд. Для ионов разного знака в тех же условиях константа скорости убывает. Если одно из реагирующих веществ является нейтральной молекулой, то произведение zaZb обращается в нуль и скорость реакции в разбавленном растворе не зависит от ионной силы среды.
Изменение скорости реакции от ионной силы раствора называется первичным солевым эффектом. Присутствие солей, кроме того, может влиять на концентрацию реагирующих ионов слабого электролита (воздействуя на его диссоциацию или гидролиз). Вызванное этим изменение скорости реакции называется вторичным солевым эффектом.
Степень электростатического взаимодействия ионов, полярных молекул и ионов с полярными молекулами определяется диэлектрической постоянной среды, поэтому и константа скорости реакции между этими частицами зависит от диэлектрической постоянной. Эта зависимость выражается уравнением
d lnfcCK
(XV. 47)
ИЛИ
In ^ск — ^0 "”*
g2^B
DdkBkT ’
(XV.48)
где zAc и — заряды ионов; D — диэлектрическая постоянная; k — константа Больцмана; ko— константа скорости в среде с бесконечно большой диэлектрической проницаемостью; t/дв — размер активного комплекса, который определяется принятой моделью его строения.
Уравнение (XV.48) показывает, что константа скорости реакции между одноименно заряженными ионами растет с увеличением D, а при взаимодействии разноименных уменьшается. Так как уравнение (XV.48) является приближенным, линейная зависимость In k от 1/D соблюдается не всегда.
Строение активного комплекса, его свойства определяют величину предэкспоненциального множителя в уравнении Аррениуса, изменяя энтропию активации
а следовательно, и значение стерического фактора Р. .Активный комплекс, образованный ионами одного знака, несет двойной заряд, поэтому электростатическое его воздействие на соседние мо
342
лекулы растворителя возрастает по сравнению с исходными ионами (эффект электронаправленности). В результате хаотичность в расположении молекул растворителя уменьшается и энтропия активации становится отрицательной (ASs#<0)., т. е. стерический фактор Р<1.
Активированный комплекс из ионов противоположного знака несет меньший заряд, чем у реагентов, поэтому создает вокруг себя менее мощное электрическое поле и хаотичность в расположении молекул растворителя увеличивается по сравнению с исходным состоянием. Поэтому энтропия активации здесь положительна (AS¥=>0) и стерический фактор Р>1.
Скорость реакции может изменяться также за счет сольватации исходных веществ или активированного комплекса. При сольватации исходных веществ энергия активации реакции возрастает (т. е. скорость убывает), при сольватации активированного комплекса энергия активации убывает, т. е. скорость реакции возрастает.
6. ЗАВИСИМОСТЬ СКОРОСТИ РЕАКЦИИ ОТ КАТАЛИЗАТОРА (ГОМОГЕННЫЙ КАТАЛИЗ)
В присутствии катализатора^ скорость реакции резко изменяется. Катализаторами называются вещества, которые не расходуются в реакции (если и расходуются на промежуточных ее этапах, то полностью регенерируются по завершении реакции) , но способствуют изменению ее скорости. Процесс изменения скорости химической реакции под действием катализаторов называется катализом. Различают катализ положительный (при котором скорость реакции увеличивается) и отрицательный (уменьшающий скорость реакции). Свойствами катализатора может обладать один из продуктов реакции (автокатализ, который также может быть положительным и отрицательным). В зависимости от агрегатного состояния веществ, участвующих в реакции^жатализ делят на гомр-генный и гетерогенный. Если все взаимодействующие вещества и катализатор находятся в одной фазе, то катализ называется гомогенным. Наибольшее распространение7 он имеет среди реакций в жидкой фазе. / о
Особенности каталитических реакций: 1) количество катализатора остается неизменным (при отсутствии побочных реакций с его участием); 2) катализатор не изменяется химически в ходе реакции; 3) ничтожно малое количество катализатора (по сравнению с количеством реагирующего вещества) ^значительно изменяет скорость реакции, причем ускорение реакции примерно пропорционально концентрации катализатора. Для многих гомогенных реакций
k = kQ + ac, (XV.49)
где k— константа скорости реакции в присутствии катализатора; k® — константа скорости реакции при отсутствии катализатора;
343
a — каталитический коэффициент (константа скорости катализируемой реакции); с — концентрация катализатора; 4) действие катализатора специфично; 5) катализатор в обратимой реакции не смещает равновесия, а в одинаковой мере ускоряет и прямую, и обратную реакции; 6) катализаторы чувствительны к присутствию некоторых посторонних веществ, которые могут увеличивать каталитическую активность катализатора и уменьшать или совсем прекратить его действие.
В присутствии катализатора снижается энергия активации — механизм реакции изменяется, и она протекает по другому пути, энергетически более выгодному. Этим и объясняется увеличение скорости реакции под влиянием катализатора.
Катализатор обычно участвует в образовании лабильных промежуточных соединений с исходным веществом. В гомогенном катализе—это промежуточные соединения стехиометрического состава. В процессе реакции осуществляется многократное повторение циклов, включающих поочередное связывание и регенерацию катализатора. Представления об образовании промежуточных соединений при катализе и использование метода стационарных концентраций позволяют вывести кинетические уравнения, которые хорошо согласуются с опытными данными для многих гомогенных каталитических реакций.
Основные положения теории промежуточных соединений в гомогенном катализе: 1) катализатор образует с реагирующим веществом реакционноспособное неустойчивое промежуточное соединение; 2) образование промежуточного соединения является относительно быстро протекающим обратимым процессом; 4) неустойчивое промежуточное соединение относительно медленно распадается на продукты реакции и молекулу катализатора; 5) общая скорость процесса пропорциональна концентрации промежуточного продукта. В соответствии с этими положениями скорость процесса определяется скоростью распада промежуточного соединения и прямо пропорциональна концентрации катализатора, а порядок реакции получается дробным (между нулевым и п-ным) в зависимости от использованного катализатора и температурного режима. Наиболее часто встречающиеся виды гомогенного катализа.
/. Окислительно-восстановительный катализ. Здесь катализаторами являются ионы, способные к различной степени окисления и восстановления.
2. Катализ термического разложения. Этот процесс часто называют «каталазным», так как он является модельным для определения активности некоторых ферментов, в частности каталазы.
3. Катализ комплексами переходных металлов.
4. Кислотно-основной катализ. Кислотно-основной катализ сводится к передаче протона или от молекулы катализатора (кислоты) к реагенту (субстрату), или от молекулы субстрата к молекуле катализатора (основанию). В первом случае промежуточным продуктом является протонизированный субстрат, а во втором — 344
анион субстрата. Затем промежуточное соединение претерпевает внутримолекулярное превращение с изменением характера и расположения связей и, наконец, протон отщепляется от другой точки молекулы и присоединяется или к молекуле растворителя, или к сопряженному основанию, т. е. катализатор регенерируется. Аналогично регенерируется и основание при основном катализе (анион субстрата после внутримолекулярного превращения присоединяет протон от сопряженной кислоты, освобождая основание).
Кислотно-основной катализ делят на «специфический» и «общий». Наиболее важным является специфический кислотно-основной катализ, который осуществляется ионами водорода и гидроксила. Он широко распространен в органической химии; например гидролиз эфиров, инверсия сахаров, этерификация кислот и спиртов и др. В сильнокислых растворах эти реакции катализируются только ионами НзО^ и скорость реакции может быть рассчитана по уравнению
^ = *н+[Н3О+][5],
где каталитический коэффициент; S — субстрат (реагирующее вещество). В щелочной среде скорость реакции пропорциональна концентрации ионов гидроксила:
w = *он- [°н~] [S] •
В средах, близких к нейтральным, когда катализ осуществляется ионами Н3О+ и ОН~, скорость реакции выражается уравнением
w [S] + [Н3О+] [S] + [ОН-] [S],
где — константа скорости реакции в отсутствие катализатора. Для реакции первого порядка скорость реакции w = fe[S], тогда экспериментальная константа скорости k рассчитывается по уравнению
k = ч- [Н3О+] + £ои_ [ОН-].
Если скорость реакции в отсутствие катализатора мала, то уравнение принимает вид \
^йн+[Н3О+] + йон_[ОН-].
Катализ может осуществляться не только непосредственно ионами Н3О+, но и веществами — донорами или акцепторами протонов, например Н2О, NH3, NO2 (кислоты и основания Брёнсте-да). Такой кислотно-основной катализ называется общим.
Работа 1. Изучение скорости инверсии тростникового сахара
Процесс инверсии сахара является гидролитическим расщеплением сахарозы С12Н220ц на глюкозу и фруктозу по уравнению
С12Н22О11 + Н2О с6н12о6 + c6Ht2o6
глюкоза фруктоза
345
Эта реакция бимолекулярна, однако в большом избытке воды (слишком велика разница "^^молекулярных массах компонентов) она протекает по первому порядку: —dc/dr=&cCaxap и константа скорости ее может быть рассчитана по уравнениющХУ.4).
Скорость инверсии сахара в нейтральной среде очень мала. Присутствие ионов всщорода жак катализатора ускоряет реакцию и делает ее дбступнсш для жаблюдёнЙ'Йщпрйче^ скорость инверсии пропорциональна концентрации ионов водорода в растворе.
Тростниковый сахар и продукты его разложения принадлежат к числу оптически активных веществ, т. е. веществ, способных из-менять положение плоскости поляризации проходящего через них поляризованного светового потока (светового потока, в котором колебания происходят в определенной плоскости). Оптическая активность таких веществ связана с наличием в их молекулах асимметричных атомов углерода. Угол поворота плоскости колебаний поляризованного луча называется углом вращения плоскости поляризации иобозначаетея, н. Его величина прямо пропорциональна толщине слоя d и концентрации активного вещества с:
a = adc, (XV.50)
где а — коэффициент пропорциональности (постоянная поляризации или удельное вращение), который зависит от природы вещества, длины волны, температуры и природы растворителя. Для гомогенных оптически активных жидкостей концентрация в уравнении (XV.50) заменяется на плотность. Вращательная способность приблизительно обратно пропорциональна квадрату длины волны (вращательная дисперсия), а с температурой изменяется незначительно.
Удельное вращение равно углу вращения (в градусах) в слое раствора толщиной 1 дм, содержащего 1 г вещества в 1 мл при 20°С, при определенной длине волны (например, при длине волны желтой линии спектра паров натрия 589,6 нм). Зная- у го л вр аще -ния, удельное вращение (см. справочник) и толщину слоя раство-ра, легко вычислить его концентрацию.
«« Скорость инверсии тростникового сахара удобно изучать по ; измерению меняющегося со временем угла вращения плоскости J поляризации исследуемого раствора. Тростниковый сахар вращает Р’Тктоскость поляризации вправо (а=66,55°), а смесь продуктов ин-| версии — влево, так как глюкоза вращает вправо (аг=52*5°), а фруктоза — влево (а$ = —91,9°). Поэтому по мере протекания реакции угол вращения плоскости поля^изации^уменьщается^ падает до нуля и затем становится отрицательным (инверсия вращения). Окончанию реакций соответствует предельное, неизменяющееся отрицательное значение угла вращения «оо.
Для измерения угла вращения" плоскости поляризации используются специальные оптические приборы— поляриметры. Чаще всего применяются так называемые полутеневые поляриметры (рис. 149). Поляризатор 3 состоит из двух призм Николя (при другой конструкции их может быть и три), причем меньшая по 346
размерам призма прикрывает половину поля зрения. Плоскости поляризации этих призм находятся под некоторым углом друг к Другу, поэтому поле зрения, рассматриваемое в окуляр 7, разделено на две части, отличающиеся по цвету и яркости освещения. Поляризатор неподвижен. Анализатор 8 (также призмы Николя) может вращаться вокруг оптической оси прибора. Вращением анализатора вокруг оси прибора можно достичь положения, при ко-
Рис. 149. Схема поляриметра:
1 — источник света; 2 — светофильтр; 3 — поляризатор; 4 — поляриметрическая трубка; 5 — зеркало; 6 — лупа; 7 — окуляр; 8 — анализатор; 9— указатель;
10 — лимб
тором призмы Николя оказывается скрещенными и проходящий световой поток гасится. Если между поляризатором и анализатором р а сп ол ожен ;рпти^е^s дкхивцщи J,.§£тв qgT то скрещены а я призма уже не будет гасить проходящий световой поток и для достижения темноты необходим дополнительный поворот анализатора на некоторый угол. В полутеневом поляриметре положение плоскости поляризации светового потока определяется не по затемнению. в окуляре прибора всего поля зрения, а по наступлению равной слабой освещенности (установка на полутень). В других положениях анализатора поле зрения в окуляре резко разделено на две части по освещенности или все освещено ярко. Такое устройство обеспечивает более высокую точности измерений, так как
чувствительность глаза к различию в освещенности значительно выше, чем к установлению полного затемнения. Если полутеневое положение найдено правильно (рис. 150, б), то малейший поворот анализатора вправо (рис. 150, а) или влево (рис. 150, в) нарушает равномерность освещения вплоть до резкого контраста в освещенности обеих половин поля зрения. При отсутствии поляриметрической трубки в желобке поляриметра полутеневое положение долж
347
но соответствовать нулю по шкале (нулевое положение поляриметра).
Для получения параллельных пучков светового потока в приборе имеется система линз. Источником 'светового потока служит электрическая лампа. В белом световом потоке определению равной освещенности полей мешает разноцветность (вращательная дисперсия). Поэтому пользуются примерно монохроматическим световым потоком, употребляя светофильтр. Угол поворота анализатора отсчитывается по шкале (лимбу), движущейся при его вращении и неподвижному нониусу с точностью до десятых долей градуса. Отсчеты производятся следующим образом. Число целых градусов определяют по последнему делению шкалы, которое оказывается слева от нуля (центральной метки) нониуса; десятые доли градуса определяют на правой части шкалы нониуса по‘делению, совпадающему в данном положении с каким-либо делением основной шкалы лим'ба. Так отсчитывают положительные углы вращения. Например, на рис. 151 угол вращения соответствует 20,3°. При вращении анализатора в противоположную сторону отсчитывают отрицательные углы вращения. Десятые доли градуса определяются по делениям в левой части шкалы нониуса. Для удобства наблюдений и измерений, связанных с освещенностью, поляриметр закрывают черным чехлом.
Последовательность выполнения работы*. Приготовить примерно 20%-ный раствор тростникового сахара, для этого отвесить на технических весах 10 г сахара и, поместив его в 50-миллилитровую мерную колбу, довести об^ем ее дистиллированной водой до метки. Если раствор мутный, его нужно отфильтровать. Затем пипеткой или мерным цилиндром отобрать в колбу 25 мл этого раствора и туда же влить 25 мл 6 н. НО. Смесь перёмешать и поместить в термостат при 40°С ч4**. Оставшиеся 25 мл раствора
сахара смешать таким же образом с 25 мл раствора кислоты за-данной_концентрации. Момент сливания кислоты с раствором сахара отметить по часам как момент начала реакции. Смесь тотчас же тщательно перемешать и быстро влить в хорошо вымытую поляриметрическую трубку, предварительно сполоснув ее дистиллированной водой и два раза небольшим количеством исследуемого раствора. При наполнении трубки нужно следить за тем, чтобы в нее не попал воздух. Пузырьки воздуха вызывают в поле зрения появление темных пятен. Чтобы быстро заполнить трубку без пузырьков воздуха, предварительно нужно научиться заполнять ее водой. Трубку наполнить до краев, чтобы жидкость образовала выпуклый мениск, затем осторожно сбоку надвинуть покровное стеклышко и навинтить кольцо, прижимающее стекло к торцу
* Прежде чем начать работу, следует ознакомиться с устройством поляриметра и работой на нем.
** Если в работе используются поляриметрические трубки малого объема, то вместо 25 мл раствора сахара и кислоты берут 10 мл (т. е. общий объем реакционной смеси соответствует не 50 мл, а 20 мл).
348
трубки. При этом проверить также, не подтекает ли трубка (не образуется ли пузырьков воздуха через некоторое время после заполнения). Наполненную трубку обтереть снаружи фильтровальной бумагой, обратив особое внимание на чистоту, сухость и прозрачность стекол, закрывающих торцы трубки, и поместить^ в желобок поляриметра в крайнее положение, ближайшее к окуляру.
Отсчеты следует проводить только после получения четкого изображения шкалы и поля зрения при вращении соответствующей муфты на зрительной трубке поляриметра. Все измерения проводить при комнатной температуре с обязательной записью ее до и после опыта. Если измерения проводятся при температурах, отличающихся от комнатной, следует пользоваться поляриметрическими трубками с рубашками для обогрева, подключаемыми к термостату. Угол вращения определяется по полутеневому положению поляриметра через различные промежутки времени от начала реакции. Чем больше концентрация катализатора (кислоты), тем чаще следует производить отсчеты углов вращения, постепенно увеличивая время между измерениями. Например, с катализатором 4 н. НС1 следует произвести два-три измерения через 3 мин, одно-два измерения — через 5 мин, одно-два измерения—через 10 мин и т. д. Всего следует сделать 10—12 определений, при этом записывать показание шкалы прибора и соответствующее ему время по часам. Все определения следует проводить по возможно-' сти быстро.
После всех измерений следует определить угол вращения а, соответствующий концу реакции. Для этого трубку, в^ которой находился рабочий раствор, заполнить смесью раствора сахара с 6 н. НС1, предварительно охлажденной до температуры опыта (сполоснуть этим раствором трубку перед заполнением). Отсчет повторить через 30 мин и, убедившись, что угол вращения не меняется, принять его за ctoo. По окончании работы рекомендуется проверить нулевое положение поляриметра, чтобы при необходимости ввести соответствующие поправки. Так как кислота разрушает металлическую оправу поляриметрической трубки, необходи-* мо тотчас же по окончании опыта трубку промыт^ водой и высушить. Результаты наблюдений свести в таблицу по образцу:
Температура опыта . . .; концентрация НС1 . . .
Номер измерения Время измерения Время от начала реакции, мин а/ «0—аоо аоо Константа скорости инверсии k (с указанием ее равномерности) ь ср
А > ..•Pfl
349
По результатам опыта вычислить константу скорости реакции при данной температуре для каждого момента, кроме /=0 и ,./=оо, по формуле (XV.4), где а — концентрация сахара в исходном растворе; (а—х) — концентрация сахара б данный момент; 1 — время, протекшее от начала реакции до момента данного измерения.
В выражение (XV.4) вместо концентраций можно подставить пропорциональные им разности соответствующих углов вращения. 1огда
2,3
1g
const («о — «»)
const (а0 — ам) — const (а0 — at)
2,3 «о-«оо
= — 1g--------
t «^-«о.
(XV.51)
где а0 — угол вращения в момент начала реакции; щ —угол вращения в данный момент от начала реакции; а» —угол вращения, соответствующий концу реакции.
Все значения углов вращения подставлять в уравнение с соответствующими знаками: Goo имеет отрицательный знак, следовательно, эту величину нужно суммировать с величиной а0. Угол ао» соответствующий моменту начала реакции, практически измерить
Воегля
/
не удается, так как от начала реакции до первого измерения проводит значительное время, поэтому а0 определять экстраполяцией.
На миллиметровой бумаге построить гра-
Agfa-d^) Фик в координатах }g(at- ’Ct со )—т (рис.
0 °° 152) и экстраполяцией полученной линии
до т = 0 определить lg(a0- -ctoo), а затем найти а0.
Вычислить константы скорости для каждого момента, рассчитать йср и сравнить полученное значение с йср, найденной на графике. Для подтверждения по-Рис. 152. Определение а0 рядка реакции использовать также график 1£У = /[1£(аг—ctoo)]. Скорость реак-
щии w для каждого промежутка времени от начала реакции определять с помощью кинетической кривой at—по тангенсу угла наклона касательной к соответствующей точке на кривой.
Если опыт проводился при двух температурах, то можно определить энергию активации реакции по значениям kcp с помощью уравнения Аррениуса (XV.43) и рассчитать энтропию и теплоту активации исследованной реакции ((XV.40), (XV.41)].
Работа 2. Изучение скорости мутаротации глюкозы
При стоянии свежеприготовленных растворов а- или р-глюко* зы в них накапливается второй изомер и постепенно устанавливается равновесие между этими стереоизомерами:
a-глюкоза р-глюкоза
350
При этом удельное вращение плоскости поляризации светового потока изменяется (при 20° С для а-глюкозы + 110,1° и для р-глюкозы +19,3°, для смеси +52,5°). Это превращение ускоряется в присутствии кислот и особенно оснований. Поскольку реакция обратима, наблюдаемая константа скорости реакции, рассчитываемая по уравнению реакции 1-го порядка, является суммой констант скорости прямой и обратной реакций, т. е. k=ki + k~i.
Скорость реакции может быть изучена без катализатора и с катализатором и определена по изменению/угла вращения плоскости поляризации светового потока при помощи поляриметра.
Последовательность выполнения работы. Отвесить на технических весах две навески по 5 г кристаллической глюкозы. Одну из них поместить в мерную колбу вместимостью 50 мл. Отметив время, ’быстро довести объем колбы водой до метки, перемешать и отметить момент полного растворения глюкозы. Время —среднее между началом и концом растворения глюкозы—принять за момент начала реакции.
реакции изучается при
Примечание. Если скорость
щейся от комнатной, то вода, в которой растворяется предварительно нагрета до температуры опыта.
температуре, отличаю-глюкоза, должна быть
и налить в поляри-
Раствор, если он мутный, отфильтровать метрическую трубку, предварительно дважды сполоснув ее этим же раствором. Затем измерить углы вращения плоскости поляризации. После первого измерения перенести вторую навеску в 50-миллилитровую мерную колбу, растворить ее в соляной кислоте заданной концентрации и довести водой объем раствора в колбе до метки. За начало реакции принимается время, среднее между началом и концом растворения глюкозы. Измерения углов\враще-ния плоскости поляризации для смеси с катализатором чередуют с измерениями для смеси, в которой идет самопроизвольнаяЧтута-ротация. \
Для определения конечного угла вращения плоскости поляризации ctoo часть раствора, оставшегося после заполнения трубкщ поместить в термостат при 40°С, где и выдержать в течение всего опыта. По окончании измерений определить угол вращения плоскости поляризации оставшегося раствора, охладив его до температуры опыта. Начальный угол вращения плоскости поляризации раствора а0 рассчитать экстраполяцией (см. с. 350). Обработку полученных данных произвести в той же последовательности, как и в работе 1. Затем вычислить константы скорости реакции без катализатора и в присутствии катализатора по уравнению-(XV.51).
Используя опытные данные, вычислить по уравнению (XV.49) каталитический коэффициент, предполагая, что действие катализатора пропорционально его концентрации.
Вычислить и аналитически, и графическим путем, как это указано на с. 350, средние константы скорости и k. Результаты наблюдений записать в таблицу по образцу:
351
Температура опыта . . .; концентрация НО . . .
Мутаротация в отсутствие катализатора Мутаротация в присутствии катализатора
Работа 3. Изучение скорости реакции иодирования ацетона
Реакция иодирования ацетона протекает по уравнению
СН3СОСН3 +12 сн3сосн21 + н+ +1-
Процесс идет автокаталитически, так как ускоряется одним из продуктов реакции (ионами водорода). В отсутствие ионов водорода (в нейтральном разбавленном водном растворе) он протекает очень медленно. Иодирование ацетона происходит в две стадии:
обратимая реакция энолизации ацетона
н+
СН3 - С - СН3 i СНо-С = СН2 (1)
И I
о он
взаимодействие иода с энольной формой
СНз - С = СН2 -Ы2 ->СН3 - С - СН21 + Н+ + I- (2)
I
ОН о
Реакция (1) протекает медленно, реакция (2) —быстро и практически до конца. Поэтому скорость процесса определяется скоростью энолизации ацетона, она пропорциональна концентрации ионов водорода, но не зависит от концентрации иода, т. е. в соответствии с уравнениями (XV.9) и (XV. 10)
(XV. 52)
где Со, ац — начальная концентрация ацетона; —начальная концентрация ионов водорода; сх — концентрация ацетона, подвергшегося превращению за время t (убыль концентрации); t — время от начала реакции до данного измерения. Ход реакции контролируется по анализу проб, периодически отбираемых из реакционной смеси.
Последовательность выполнения работы. Установить термостат на заданную температуру. В 250-миллилитровую мерную колбу налить 25 мл 0,1 н. раствора 12 в 4%-ном растворе KI, добавить 25 мл 1 н. НС1 (содержание кислоты может быть увеличено или уменьшено) и долить водой до объема ниже метки примерно на 30—35 мл. Колбу с рабочим раствором и другую колбу с дистиллированной водой погрузить в термостат. Спустя 15—20 мин в колбу с реакционной смесью добавить примерно 1,5 г ацетона (навеску ацетона отвесить на аналитических весах в закрытом сосуде) или влить 25 мл 1 н. водного раствора ацетона*. Момент вливания ацетона отметить по часам. После вливания ацетона объем раствора в колбе быстро довести до метки дистиллированной водой, выдержанной в термостате, тщательно взболтать и тотчас отобрать пипеткой 25 мл пробы, отмечая этот момент по часам.
За начало реакции (f='0) принять время вливания ацетона в реакционную смесь. Колбу с реакционной смесью закрыть пррб-кой во избежание улетучивания ацетона. Отобранную пробу влить в колбу для титрования, содержащую 25 мл 0,1 н. NaHCOs. Содержание иода определять титрованием 0,01 н. Na2S2O3 в присутствии крахмала. За ходом реакции наблюдать по результатам анализа проб через определенные промежутки времени. Во время отбора проб колбу из термостата не вынимать. В течение опыта рекомендуется взять не менее семи-восьми проб. Вторую пробу отобрать через 10 мин после первой и по результатам титрований определить время, через которое надлежит брать следующие пробы. Расход гипосульфита на каждую последующую пробу должен изменяться на 1—3 мл. ~ \
По ходу реакции время между последовательными титрованиями постепенно следует увеличивать (например, первые три титрования проводят через 10 мин, затем два титрования —через 15 мин, еще два титрования — через 20 мин и, наконец, одно титрование— через 25 мин). Чем выше температура, при которой производятся исследования и чем больше содержание кислоты в реакционной смеси, тем чаще следует отбирать пробы. Результаты измерений записать в таблицу по образцу:
Температура опыта . . .; количество 1 н. НС1 . . .
. — -- -.— ..... — - - —. .... - - - .. .... ._ - ._ - — - - .... —
Номер измерения
Время измерения (астрономическое)
Время от начала реакции, мин
Количество, 0,01 и.
ИягЭгОз, мл
ац» г-экв/л
г-экв/л
г-экв/л
k (с указанием ее размерности)
* Использование водного раствора ацетона предпочтительнее, так как при этом уменьшаются ошибки за счет его улетучивания и искажения температурного режима в процессе растворения при вливании ацетона в реакционную смесь.
4
12—2083
Концентрацию ацетона сх определить по уравнению
где nt — количество 0,01 н. Na2S2O3, израсходованное на титрование данной пробы; мл; по — количество 0,01 н. Na2S20s, которое должно было быть израсходовано на титрование в момент начала реакции, мл; -A^Nats,o»— нормальность раствора Na2S20s.
Значение «о практически определить не удается, так как от момента вливания ацетона до момента взятия первой пробы протекает значительное время. Поэтому по определять по графику. Для этого на миллиметровой бумаге построить график в координатах п—t и экстраполяцией полученной прямой до 0 определить по. Используя уравнение (XV.52) и значения с0,ац( cOjH+ » сх и t, вычислить константу скорости k. После этого определить величину kcp и вычислить температурный коэффициент и энергию активации (значение k при другой температуре взять из справочника), а также рассчитать энтропию активации данной реакции [см. (XV. 41)].
Работа 4. Изучение скорости разложения пероксида водорода газометрическим методом
Пероксид водорода в водных растворах самопроизвольно медленно разлагается по уравнению
Н2О2 -► Н2О + 0,5О2
Катализаторами здесь могут быть ионы Fe2+, Fe3+, Сг2О72-, СгО42-, WO42-, МоО42-, смешанные катализаторы: CuSO4 + + МоО42-, CuSO4 + NiSO4 и многие другие. При соответствующем подборе условий реакция может протекать по первому или близкому к первому порядку. За ходом реакции наблюдают по измерениям объема выделившегося кислорода через различные промежутки времени от начала реакции.
Последовательность выполнения работы. Термостатировать раствор катализатора. Для этого в настроенный на заданную температуру термостат поместить колбу вместимостью 200—300 мл с 90—95 мл раствора катализатора. Одновременно термостатировать реакционный сосуд 1 (рис. 153). После 30 мин термостатирования в колбу с раствором катализатора влить определенное количество пероксида водорода. Раствор тщательно размешать и поместить в предварительно термостатированный реакционный сосуд, заполнив его так, чтобы высота воздушного пространства между уровнем жидкости и пробкой не превышала 2 см. Сосуд с реакционной смесью поместить в термостат 4. Уравнительный сосуд 3 и бюретку 2 наполнить подкрашенной водой и, присоединив реакционный сосуд к бюретке, проверить установку на герметичность. Для этого опустить уравнительный сосуд ниже уровня воды -в бюретке 354
одинаковые уровни жидко-
г- -
Схема установки для скорости разложения
Рис. 153.
изучения
Н2О2 газометрическим методом
(уровень воды в бюретке не должен изменяться в течение 3 мин). Затем уровень воды в бюретке установить на уровне верхнего крана. Верхний кран в течение 1—2 мин держать открытым (для вытеснения воздуха выделяющимся кислородом из реакционного сосуда), затем его закрыть, соединив таким образом реакционный сосуд с газовой бюреткой. Установить сти в бюретке и уравнительном сосуде, произвести первое измерение и записать уровень по бюретке и время. Каждое следующее измерение сопровождается такой же записью, причем уровни жидкости в бюретке и уравнительном сосуде непрерывно поддерживаются одинаковыми. Целесообразно записывать уровень жидкости в бюретке через 2—5 мин (чем выше температура, тем меньше время между измерениями). После того как реакция практически прекратится, реакционный сосуд поместить в кипящую водяную баню и выдержать в ней до полного разло-
жения пероксида водорода (око- . ч
ло 30 мин). Реакция считается законченной, если уровень газ\в бюретке не изменяется. При кипячении на водяной бане уравнй\ тельный сосуд поддерживать в наиболее высоком положении.
После полного разложения пероксида водорода реакционный
сосуд охладить до температуры термостата, выдержать в нем в течение 25—30 мин и при равенстве уровней жидкости в бюретке и уравнительном сосуде измерить уровень жидкости в бюретке Вначале уровень жидкости в бюретке изменяется медленно, так как выделяющийся кислород растворяется в этой жидкости (воде или водном окрашенном растворе) до насыщения. Только после этого объем выделяющегося кислорода можно считать пропорциональным количеству разложившегося пероксида водорода. Поэтому при обработке экспериментальных данных за начало реакции следует принимать третье или четвертое измерение (т. е. измерение в условиях уже установившегося режима).
По опытным данным построить графики зависимости: 1) разности объемов кислорода от времени [гл»—(на оси абсцисс откладывать время в минутах);
2) логарифма скорости реакции через различные промежутки времени от логарифма разности объемов выделенного кислорода lg fllgC^'oo—у/)]; скорость определять по тангенсу угла накло-
на касательной к кривой Voo—vt=f (t) к оси времени;
3) логарифма разности объемов кислорода от времени lg(Voo— ^"^0=f(0- Последние два графика использовать для определе-
12*
355
ния порядка реакции. Далее рассчитать константу скорости реакции при помощи графика lg(Uoo—и аналитическим путем, используя уравнение (XV.4), где концентрации Н2О2 в начальный момент и в момент измерения t заменить разностью объемов кислорода:
оо
где Voo — объем кислорода, выделившийся после разложения всего пероксида водорода (определяется как разность уровней в бюретке в момент, принятый за начало реакции, и после кипячения Н2О2 до полного разложения). Результаты записать в таблицу по образцу:
Температура опыта, °C . . катализатор . . количество пероксида во-
дорода . . .
га о
(с указанием размерности)
Используя результаты расчета и полученные графики, установить, протекает ли исследуемая реакция по первому порядку. При значительных отклонениях от первого порядка рассчитать константу скорости с помощью графика lg
Работа 5. Изучение скорости гидролиза уксусного ангидрида методом электрической проводимости
Гидролиз уксусного ангидрида протекает по уравнению
(СН3СО)2 О + Н2О = 2СН3СООН
и является 'бимолекулярной реакцией в ацетоне или в уксусной кислоте. Однако в разбавленном водном растворе гидролиз идет как реакция первого порядка и практически до конца. Молярная концентрация воды здесь значительно больше молярной концентрации уксусного ангидрида, поэтому убыль воды за счет реакции мала и содержание ее в растворе можно считать постоянным, а скорость реакции —пропорциональной только концентрации уксусного ангидрида. При таких условиях эту реакцию можно отнести к классу псевдомономолекулярных. Кинетику этой реакции следует изучать методами физико-химического анализа, причем наиболее удобным является измерение электрической проводимо-356
сти, не требующее отбора проб для анализа. Электрическая проводимость системы со временем значительно возрастает вследствие образования уксусной кислоты. Концентрация образующейся уксусной кислоты определяется по электрической проводимости раствора, измеряемой при помощи мостика Кольрауша (см. с. 278). Сопротивление раствора 7? определяют по уравнению
Д//(1000 - dl} = Rf/RtAar i
где /?маг —сопротивление магазина; at и (1000—at)—длины плеч реохорда, мм.
Эти измерения могут быть выполнены также при помощи специального прибора — мостика для измерения емкостей и сопротивлений, принципиальная схема которого не отличается от схёмы мостика Кольрауша.
Последовательность выполнения работы. Установить термостат на указанную температуру, проверить постоянство температурного режима (допустимые колебания температуры 0,14-0,2°), собрать схему для измерения электрической проводимости. При работе с мостиком для измерения емкостей и сопротивлений включить прибор в электросеть. В 50-миллилитровую мерную колбу поместить 6 мл уксусного ангидрида и довести объем раствора дистиллированной (предварительно термостатированной) водой до метки. В момент начала растворения уксусного ангидрида включить секундомер и не выключать его до конца опыта (до установления постоянного значения электрической проводимости). Отметить по секундомеру время начала и конца растворения (при приливании воды четко видна граница раздела двух жидких слоев, после взбалтывания наблюдается помутнение; момент исчезновения мути принять за конец растворения). Среднее время принять за время начала реакции. Растворение проводить при энергичном перемешивании. Сосуд для измерения электрической проводимости, снабженный притертой крышкой, после двукратного ополаскивания исследуемым раствором заполнить этим же раствором. Электроды должны быть, погружены в раствор на 0,5—1 см ниже уровня раствора. Сосуд погрузить в термостат, в котором встряхивать его в течение 3 мин до установления постоянного температурного режима. Одновременно на магазине сопротивления мостика Кольрауша подобрать определенное постоянное сопротивление так, чтобы отсутствие тока на участке CD (см. с. 278) соответствовало положению движка С реохорда в середине шкалы. Все дальнейшие измерения выполнять, не меняя этого сопротивления.
В работе участвуют два исследователя. Один измеряет электрическую проводимость, другой следит за секундомером и записывает показания секундомера и реохорда. Измерение электрической проводимости проводится тем чаще, чем выше температура опыта. Два-три измерения произвести через 30 с, четыре-пять измерений — через 1 мин, два-три измерения —через 5 мин одно после другого, далее делать измерения через 10 мин, через 1 ч и
357
еще через 1 ч. Постоянство показаний реохорда указывает на конец реакции*. Результаты измерений записать в таблицу по образцу:
Температура опыта, °C . . концентрация раствора . . /?маг . . .
(с указанием размерности)
При обработке результатов измерений следует учитывать, что исследуемая реакция является реакцией первого порядка, поэтому расчеты вести по уравнению (XV.4). Пренебрегая неполной и меняющейся с концентрацией диссоциацией уксусной кислоты, можно принять в первом приближении, что увеличение электрической проводимости во времени пропорционально концентрации образующейся уксусной кислоты. Это— грубое допущение, однако при расчете константы скорости происходит компенсация ошибок и конечный результат получается удовлетворительным. Количество образовавшейся уксусной кислоты пропорционально количеству взятого уксусного ангидрида, поэтому с упомянутым допущением общее увеличение электрической проводимости в течение реакции можно считать пропорциональным начальной концентрации уксусного ангидрида.
Если обозначить электрическую проводимость раствора в момент начала реакции хо, в данный момент хг и соответствующую последнему измерению Хоо (когда она уже не меняется), тогда Со = const (хос—хо): •
(с0 — йх) = const (хоо ” хо) — const — хо) = const (х« —
Заменив в уравнении (XV.4) концентрации на разность электрических проводимостей,
или, учтя далее, что х=<р//?, где <р — постоянная сосуда (см. с. 279), получим
* Поскольку реакция протекает быстро, рекомендуется для выполнения измерений провести вначале предварительный опыт, во время которого следует подобрать нужное сопротивление на магазине сопротивлений.
Сопротивление раствора в момент начала реакции /?о экспериментально не определяется, так как от начала реакции до первого измерения проходит некоторое время. Поэтому Ro определяют 1 1 экстраполяцией, для чего строят график —— — -—=/(/)> откла-^00
дывая время в минутах по оси абсцисс. Поскольку исследуемая
реакция является реакцией первого порядка, экспериментальные
данные в этих координатах должны укладываться на прямую. Экстраполируя полученную прямую до / = 0, нахо-
< ( 1 1 \
дят 1g----------, а затем вычисля-
ют/?о (рис. 154). Вычислив константу скорости реакции для каждого момента, определяют среднее ее значение и сравнивают с йср, полученной графически (тангенс угла наклона прямой к оси t). По константам скорости при двух температурах (вторую константу взять из справочника) рассчитать по
время
Рйс. 154. Определение 7?0
уравнению (XV.43) энергию активации данной реакции и по уравнению (XV.30) — значение предэкспоненциального множителя. По
полученным экспериментальным данным рассчитать теплоту и энтропию активации [уравнения (XV.40) и (XV.41)] иследованной реакции.
Работа 6. Изучение скорости реакции разложения карбамида в водных растворах методом электрической проводимости
Карбамид в водных растворах при 50°С и выше изомеризуется, т. е. превращается в цианат аммония с последующим переходом из цианата в карбонат:
СО (NH2)2 = NH4CNO
NH4CNO + 2Н2О^-> (NH4)2 СОз 2NH3 + С02 + Н20
Реакция превращения цианата в карбонат протекает практически необратимо. В результате образования карбоната электрическая проводимость раствора со временем растет. Это позволяет измерять скорость реакции, поскольку увеличение электрической проводимости можно считать пропорциональным концентрации конечного продукта.
Последовательность выполнения работы. В термостат, установленный предварительно на определенную температуру, поместить на 20 мин колбу, содержащую 100 мл дистиллированной воды. Так как исследования проводятся при температурах выше 45°С, поверхность воды в термостате следует покрывать слоем парафина для уменьшения испарения. После проверки температурного режима термостата следует собрать схему для измерения электриче
359
ской проводимости или при работе с мостиком для измерения емкостей и сопротивлений, включить прибор в сеть. Затем приготовить раствор карбамида определенной концентрации. На аналитических весах отвесить навеску карбамида и поместить ее в мерную колбу вместимостью 50 мл и довести объем раствора в колбе до метки дистиллированной водой, выдержанной в термостате. Время начала и конца растворения карбамида фиксировать по часам и среднее время принять за время начала реакции. В момент конца растворения включить секундомер. Колбу с раствором закрыть пробкой и поместить в термостат.
Работа выполняется двумя экспериментаторами. Подготовить схему для измерения электрической проводимости. Сосуд для измерения электрической проводимости, снабженный притертой крышкой, быстро ополоснуть приготовленным раствором и заполнить этим раствором. Электроды должны быть погружены в раствор на 0,5—1 см ниже его уровня. Затем сосуд укрепить в термостате так, чтобы уровень воды в термостате был выше уровня раствора в сосуде. Затем на магазине подобрать такое сопротивление, чтобы при отсутствии тока на участке CD (см. с. 278) движок С реохорда был близок к концу шкалы. По ходу реакции сопротивление исследуемого раствора уменьшается, поэтому следует перемещать движок реохорда С так, чтобы на участке CD ток отсутствовал. Все последующие измерение выполнять при постоянном сопротивлении магазина. При работе с мостиком для измерения емкостей и сопротивлений следует подобрать чувствительность индикаторной лампы и сохранять ее постоянной в течение всего опыта.
Через 10—15 мин после начала реакции приступить к измерениям электрической проводимости, причем один из работающих производит измерения, а другой следит за секундомером и записывает показания его и реохорда. В течение работы необходимо произвести два-три измерения через 5 мин, четыре-пять измерений — через 10 мин, два-три измерения — через 20 мин, два-три измерения— через 30 мин и одно — через час. Одновременно с термоста-тированием основного раствора перелить оставшуюся часть исследуемого раствора в колбу, снабженную обратным холодильником, и поставить ее на водяную баню, с тем чтобы довести реакцию до конца. Через 3—4 ч раствор охладить до температуры опыта и измерить его электрическую проводимость. Для этого реакционный сосуд вынуть из термостата, раствор из него вылить и заполнить его охлажденным раствором (предварительно сосуд ополоснуть данным раствором), после чего снова погрузить на 20 мин в термостат до установления температуры опыта. Электрическую проводимость, полученную при этой температуре, записать, а оставшийся раствор сохранить для повторного кипячения, которое производится на следующий день.
На второй и третий день следует сделать по одному контрольному измерению электрической проводимости. Максимальное значение электрической проводимости (или минимальное значение 360
сопротивления) выражается через Хоо (или 2?<х>)., т. е. соответствует электрической проводимости конца реакции^
Результаты промежуточных измерений, полученные после первого и последующих кипячений, для расчета константы скорости не используются и нужны только для определения конца реакции, когда электрическая проводимость принимает максимальное значение Хоо.
Результаты измерений свести в таблицу и обработать точно так же, как и в работе 5. Рассчитать ЛСр аналитическим и гфафи-ческим путем
график 1g
Эти данные использовать для графика
для определения порядка реакции (скорость определить по кинетической кривой для 4—5 промежутков времени от начала реакции). По экспериментальным данным рассчитать теплоту и энтропию активации {уравнения (XV.40) и (XV.41)].
Работа 7. Изучение кинетики акватации комплексных ионов Со3+ (кислотный гидролиз) колориметрическим методом
Колориметрические определения основаны на сравнении поглощения или пропускания светового потока стандартным и исследуемым окрашенными растворами. В практике преобладает фотоколориметрия, где для измерений используются фотоэлементы, так как визуальные измерения менее объективны. В основе метода лежит объединенный закон Бугера —Ламберта —Бэра (см. с. 6). Полученная по экспериментальным данным зависимость A=f (c) в виде прямой или кривой (при отклонении от закона Бэра) может далее служить калибровочным графиком. При помощи этого графика по оптической плотности раствора определяется концентрация данного компонента в растворе. Недостаточная монохроматичность поглощаемого светового потока обычно вызывает отрицательные отклонения от закона Бэра тем большие, чем шире интервал длин волн поглощаемого светового потока. Поэтому для увеличения чувствительности и точности фотометрического определения на пути светового потока перед поглощающим раствором помещают избирательный светофильтр. Светофильтры (стекла, пленки, растворы) пропускают световой поток только в определенном интервале длин волн с полушириной пропускания ^]/2макс—Vi/2MaKc* Этот интервал характеризует размытость максимума пропускания (рис. 155). Чем он уже, тем выше избирательность применяемого светофильтра к данным длинам волн.
Для колориметрии светофильтры выбирают исходя из спектра поглощения определяемого вещества так, чтобы спектральная область максимального поглощения светового потока окрашенным
361
ис. 155. Спектр поглощения светофильтра
раствором совпадала с областью максимального пропускания светового потока светофильтром, т. е. чтобы максимум поглощения раствора соответствовал максимуму пропускания (минимуму поглощения) светофильтра.
Точность определения с неправильно подобранным светофильтром оказывается даже меньше, чем без применения светофильтра.
Если спектральные характеристики для окрашенного раствора и светофильтра отсутствуют, нужный светофильтр подбирают экспериментально. Для этого готовят две пробы исследуемого раствора различной концентрации и измеряют их оптические плотности со всеми имеющимися светофильтрами. Затем для каждого светофильтра находят разность оптических плотностей, соответствующую взятой разности концентраций Дс окрашенного раствора. Светофильтр, дающий наибольшую разность оптических плотностей ДА, является наиболее подходящим для колориметрирования данного окрашенного раствора. Поскольку степень погло-
щения светового потока раствором обычно для одних волн больше, чем для других, выходящий световой поток бывает окрашен. Кажущийся цвет раствора всегда является дополнительным к цвету поглощенного излучения. Поэтому раствор, поглощающий в синей области, будет казаться желтым, поглощающий в зеленом участке— пурпурным и т. п. Это дает точный, но более быстрый прием выбора светофильтра — по цвету исследуемого раствора (табл. 9).
Оптическую плотность растворов измеряют в кюветах, в которых можно изменять толщину поглощающего слоя от 1 до 50 мм. Кюветы выбирают в соответствии с интенсивностью окраски исследуемого раствора. Для интенсивно окрашенных растворов, как правило, применяют кюветы с толщиной слоя до 1 см. Слабо окрашен-
ные растворы, наоборот, колориметрируют в самых больших кюветах. Современные фотоколориметры ФЭК-М, ФЭК-Н-57, ФЭК-56 являются двухлучевыми приборами с двумя фотоэлементами и имеют одинаковые принципиальные схемы.
возможность использовать менее
Таблица 9. Окраска растворов и соответствующие им светофильтры
Окраска
раствора
соответствующего светофильтра
Область максимального поглощения светового потока*, ммк
Желтый Синий 400—480
Красный Зеленый 500—56»
Синий Желтый 580—650
Зеленый Кр асный 650—750
типа ФЭК-М.
* Для фотоколориметров
362
Фотоэлектрический колориметр ФЭК-М. Внешний вид фотоколориметра ФЭК-М и принципиальная схема прибора представлены на рис. 156 и 157.
Действие прибора основано на том, что световые потоки от лампы Л (рис. 157), отразившись от зеркал 31 и 32, проходят через светофильтры Ci и С2, кюветы Ai и А2 и попадают на фотоэлементы Ф1 и Ф2. Последние соединены с гальванометром Г по дифференциальной схеме так, что при равенстве интенсивностей ^опадающих на фотоэлемент световых пучков стрелка гальванометра
Рис. 156. Фотоколориметр ФЭК-М:
1 — кюветодержатели; 2 — шкалы 1 отсчетных барабанов; 3 — корректор; 4 — отсчетные барабаны; 5 — арретир гальванометра; б — гальванометр; 7 — переключатель светофильтров; 8, 9— фотометрические клинья; 10 — переключатель гальванометра
*«.
Рис. 157. Принципиальная схема фотоколориметра ФЭК-М
стоит на нуле. Щелевая диафрагма Д при вращении связанного с ней барабана меняет ширину и тем самым меняет величину светового потока, падающего на фотоэлемент Ф2. Фотометрический нейтральный клин К служит для ослабления светового потока, падающего на фотоэлемент Фь
Набор цветных светофильтров состоит из нейтрального, зеленого, синего и красного. Их устанавливают попарно на пути прохождения световых пучков рукояткой на передней панели прибора. Цифры 1, 2, 4 на рукоятке показывают, какие светофильтры
включены: 1—нейтральный, 2 — зеленый, 3—синий, 4 — красный.
Методика измерений на фотоколориметре. Прибор включить в электросеть переменного тока через стабилизатор. Прибор стабилизируется через 15—20 мин после включения в электросеть. Перед началом работы арретир гальванометра поставить в положение «открыто» и установить корректором стрелку гальванометра на нуль. Во избежание поломки гальванометра нельзя вращать корректор при положении арретира «закрыто».
363
Для переключения гальванометра на большую или меньшую чувствительность служит рукоятка на левой боковой стенке прибора. Цифры «1» указывают на малую чувствительность, «2» — на большую чувствительность, «О» — гальванометр выключен. Работа на приборе должна начинаться при включении гальванометра на малую чувствительность. Переход на большую чувствительность разрешается после уравнивания световых потоков на малой чувствительности.
На отсчетных барабанах имеются шкалы: черная — коэффициенты светопропускания и красная —оптической плотности. Шкала оптической плотности левого барабана градуирована от 0 до 2 (100 — 0% светопропускания). Шкала оптической плотности правого барабана имеет пределы измерений 0,00—6,52, причем точность измерений на участке шкалы 0,15—0,52 (по шкале светопропускания 30—70%) выше, чем при измерениях на левом барабане. Для определения концентрации раствора обычно пользуются шкалой оптической плотности. Измерения можно производить двумя способами: при помощи левого и правого барабанов.
1. Первый способ измерений (по левому барабану). На пути правого пучка светового потока поместить кювету с исследуемым раствором, а на пути левого пучка —кювету (того же размера) с раствором сравнения (с растворителем). Левый барабан установить на нулевое деление шкалы оптической плотности (100% по шкале светопропускания, при этом щелевая диафрагма полностью открывается).
Вращением круговых фотометрических клиньев стрелку гальванометра установить на нуль: сначала при положении переключателя чувствительности «1», а затем «2». При этом на пути обоих световых потоков ввести нужные светофильтры. Затем в правый пучок светового потока ввести кювету с раствором сравнения: при этом стрелка гальванометра отклоняется от нуля. Вращением измерительных барабанов стрелку гальванометра вновь установить на нуль. Оптическую плотность раствора отсчитывать по левому барабану. Измерения следует повторить несколько раз, подводя стрелку гальванометра к нулю то слева, то справа. Из полученных отсчетов вычислить среднее значение оптической плотности.
При измерении оптической плотности растворов с А^0,05 следует применить способ с «перестановкой образцов». На пути левого светового потока установить кювету с раствором сравнения, а на пути правого светового потока — кювету с исследуемым раствором, ввести нужные светофильтры, полностью открыть щелевую диафрагму (А = 0,00) и фотометрическими клиньями стрелку гальванометра установить на нуль. Затем кюветы с растворами поменять местами и добиться фотометрического равновесия вращением левого измерительного барабана. Оптическую плотность отсчитать по левому барабану: она равна половине величины, отсчитанной по шкале: АРаств=0,5АИзм.
2. Второй способ измерений (по правому барабану). На пути правого и левого световых потоков поместить кюветы с раствором 364
сравнения и растворителем. Ввести нужные светофильтры. Правый барабан установить на нуль, при этом щелевая диафрагма имеет минимальную ширину. Вращением круговых фотометрических клиньев установить стрелку гальванометра на нуль. Затем в правый световой поток ввести кювету с исследуемым раствором. Стрелка гальванометра при этом отклоняется от нуля. Вращением барабанов увеличить ширину щелевой диафрагмы и установить стрелку гальванометра снова на нуль. Оптическую плотность раствора отсчитывать по правому барабану. На протяжении всей работы с данным веществом следует придерживаться одного способа измерения.
Работа на приборе.
1. Необходимо убедиться до включения прибора в электросеть в том, что рукоятка переключения чувствительности стоит в положении «О».
2. Включать чувствительный предел гальванометра (рукоятка в положении «2») можно только в том случае, если стрелка гальванометра подведена к нулю при включенном грубом пределе (рукоятка в положении «1»). В процессе измерений (например, при смене кювет) необходимо каждый раз переключать рукоятку из положения «2» в положение «1».
3. Начинать измерения следует спустя 15—20 мин после включения лампы. При кратковременных перерывах в работе (10— 20 мин) световые потоки следует перекрывать шторкой.
8
Рис. 158. Фотоколориметр-нефелометр ФЭК-Н-57:
1 — рукоятка защитной шторки; 2 — кюветодержатель; 3 — механический корректор; 4 — арретир гальванометра; 5 — переключатель фотоколориметрических и нефелометрических измерений; 6 — шкала отсчетного барабана; 7—.гальванометр; 8, 9— фотометрические клинья; J0 переключатель гальванометра; 11 — переключатель светофильтров; 12— шкала отсчетного барабана; 13 — линзы или точечные диафрагмы; 14 — электрический корректор
Фотоэлектрический колориметр-нефелометр ФЭК-Н-57. Оптическая схема ФЭК-Н-57 (рис. 158) аналогична схеме ФЭК-М. Одна-
ко фотоколориметр ФЭК-Н-57 имеет некоторые усовершенствования по сравнению с ФЭК-М. Он снабжен набором из девяти узко-
365
полосных светофильтров, благодаря чему может быть использован как упрощенный спектрофотометр.
Селеновые фотоэлементы заменены в нем сурьмяно-цезиевыми> что позволяет использовать светофильтр с Л3фф=360 нм (ближняя ультрафиолетовая область). Фотоэлементы включены по дифференциальной схеме через усилитель на стрелочный нуль-гальванометр. Схема включения предусматривает компенсацию «темнового тока», т. е. установку электрического нуля. Прибор может быть использован и как нефелометр. Для этого необходимо линзы, расположенные непосредственно перед кюветным отделением, заменить на точечные диафрагмы «12» и переключатель поставить в положение «нефелометр». Для нефелометрических измерений используют три особых светофильтра «9», «10» и «11». В остальном принцип работы на этом приборе ничем не отличается от работы на фотоколориметре ФЭК-М.
С методикой работы на соответствующем колориметре следует ознакомиться по описанию, прилагаемому к прибору.
Исследуется кинетика активации (кислотный гидролиз^ транс-дихлордиэтилендиаминокобальт (III) хлорида транс-[Со(еп)2С12]+С1. Следует определить среднюю константу скорости аналитическим и графическим способами, время полупревращения и энергию активации реакции.
Простые реакции замещения в октаэдрических комплексных соединениях (координационное число 6) можно представить в виде
Co3+L5X + Y - [CoL5Y] 4- X
(где X и Y — обменивающиеся лиганды; L — монодентатные лиганды). Такие реакции являются двухстадийными процессами в водных растворах. Обычно первая стадия — процесс замещения X на молекулу Н2О, а вторая стадия—последующее замещение Н2О на Y. Существует лишь небольшое число реакций, в которых X замещается сначала другими лигандами, которые обычно входят в состав комплекса. Поэтому специфическая реакция аквата-ции (или гидролиза), когда Y представляет собой воду (или ОН“ при щелочном гидролизе), как промежуточная стадия при обмене лигандами, имеет особое значение. Скорость гидролиза аминных комплексов Со3+ зависит от pH и описывается уравнением
ш [(CoL5X)2+] 4- [(CoL5X)+] [ОН-], (XV.53>
где k.\ и kb — константы скорости кислотного и основного гидролиза соответственно. При гидролизе в кислой среде второе слагаемое (XV.53) становится пренебрежимо малым и выражение для скорости приобретает вид
w = [(CoL5X)2+].
Реакции типа
[Со (NH3)5 С1]2+ + Н2О [Со (Шз)5 Н2О]3+ + С1~ относятся к псевдомономолекулярным, поскольку они протекают в разбавленных растворах, в которых концентрация воды значитель
366
но превышает концентрацию комплекса и может считаться постоянной. Поэтому для них справедливо уравнение (ХУЛ). Такого типа реакции имеют первый порядок по комплексу и нулевой по воде.
Реакция акватации комплексов состава трпкс-[Со(АА) 2С12]+ протекает по стадиям:
[Со (АА)2 С12] + + Н2О = [Со (АА)2 Н2ОСф+ + С1— (I)
[Со (АА)2 Н2ОС1]2+ + Н2О = [Со (АА)2 (Н2О)2]3+ + Cl- (II)
-Н+1Ш+ —H+flH+
[Cq(AA)2OHC1]++Н2О = [Co(AA)2(H2O)OH]2+ + Clf (III) !
где АА—бидентатный лиганд — лиганд с координационной емкостью, равной двум. Таким лигандом может быть этилендиамин NHg—СН2—СН2—NH2, 1,2-пропилендиамин СН3—CH(NH2) — —СН2—NH2, триметилендиамин H2N (СН2) 3—NH2, 1,2-изобути-лендиамин NH2—СН2—С(СН3)2—NH2 и т. п.
Реакции (I) — (III) протекают с разной скоростью. Скорость замещения второго атома хлора в аквакомплексе стадии (II) очень мала. Однако при протекании стадии (III) в щелочной среде образуется гидроксокомплекс, который гидратируется гораздо быстрее. Чтобы выяснить кинетику стадии (I), необходимо затормозить стадию (Ш), используя сильнокислую среду (рН<3). В области длин волн от 520 до 560 нм исходный комплекс транс-(Со(еп)2С12]+ имеет минимум на кривой поглощения, в то время как конечный продукт аквакомплекс транс-[Со (еп) 2Н2ОС1]2+ — максимум. Следовательно, за скоростью реакции можно следить по концентрации продукта реакции, измеряя оптическую плотность раствора.
Последовательность выполнения работы. Одному из работающих подготовить колориметр — включить, подобрать светофильтр (зеленый с максимумом пропускания в интервале 520—560 нм) и настроить прибор. Второму работающему приготовить в 100-мл мерной колбе 0,005 М раствор комплексной соли [Со(еп)2С12]С1, подвергающейся гидролизу в 0,1 М растворе HNO3. Для этого соответствующую навеску соли, взятую на аналитических весах, растворить в 0,1 М HNO3. Если исследования проводятся не при комнатной температуре, раствор HNO3 предварительно термоста-тировать. Отметить по. секундомеру время начала и конца растворения соли. Среднее время принять за начало реакции. Проверить при помощи индикаторной бумаги pH раствора (рН<3).
Половину приготовленного раствора поместить на 3,5—4 ч в термостат при 38—40°С для скорейшего завершения реакции (если опыт ведется при 40°С и выше, этого делать не следует). Оставшуюся часть раствора использовать для измерений его оптической плотности во времени At. Для исследований использовать кювету с толщиной слоя 10 мм (размер кюветы может быть и другим при иной концентрации исходного раствора). Первые два-три измерения произвести через/3—5 мин, два-три измерения — через
367
10 мин, затем —через 30 мин, 1 ч и т. д., постепенно увеличивая время между измерениями. Всего выполнить 10—12 измерений. Для определения периода полупревращения исходного вещества в продукт реакции установить визуальное наблюдение за изменением окраски раствора, выдерживая его при температуре опыта. Исходный раствор имеет зеленую окраску, а продукт реакции окрашен в красный цвет, в момент полупревращения раствор становится буровато-серым.
После всех измерений определить оптическую плотность раствора, термостатированного при 40°С, охладив часть его до температуры опыта. Неизменность оптической плотности этого раствора при повторном измерении через 30—40 мин указывает на окончание реакции. Полученное значение оптической плотности соответствует Лоо-
Результаты наблюдений и расчетов записать в таблицу по об* разцу:
Температура опыта, °C . . .
Концентрация комплексной соли в растворе, моль/л . . .
Концентрация HNO3 в растворе, моль/л . .
pH раствора (по индикаторной бумаге) . . . Время начала растворения . . .
Время конца растворения . . .
Время измерения
Время от начала реакции, мии
1g Мео-
*ск
(с указанием размерности)
Изучаемая реакция имеет первый порядок, поэтому константу скорости рассчитывать по уравнению (XV.4), в котором начальную концентрацию исходного вещества следует заменить на (Лоо—До), а концентрацию в данный момент—на (Лоо—Д#)-
(XV.54>
Экспериментальные данные представить в виде графиков: (Ах— -4г)=Н0;1ёИ СО Де) =f(0- Поскольку Ло в момент начала реакции измерить не удается, эту величину определить по графику 1£(Лоо—экстраполяцией до t=Q, Определить среднюю константу скорости по этому же графику по тангенсу угла наклона прямой к оси t и сравнить ее со средней константой скорости,, рассчитанной по уравнению (XV.54). Рассчитать период полупревращения и сравнить с экспериментальной величиной.
368
Работа 8. Изучение кинетики реакции гидролиза уксусного ангидрида колориметрическим методом
При гидратации уксусного ангидрида образуется уксусная кислота:
(СН3СО)2 О + Н2О = 2СН3СООН (1>
Если в реакционную смесь ввести KI и КЮ3, то по мере образования СН3СООН в ходе реакции гидратации будет протекать и другая реакция — окисление иодида йодатом:
Ю~ + 81- + 6Н+ -> 31^ + ЗН2О (II)
которая может протекать только в кислой среде. Количество выделяющегося иода будет связано с образованием уксусной кислоты/ Одцако при расчете скорости гидратации ангидрида с использованием измерений по сопряженной реакции (II) следует еще учитывать неполную диссоциацию уксусной кислоты.
Скорость реакции (II) определяется концентрацией ионов Н+. Результаты исследований кинетики реакции (II) приведены на с. 368. Они позволяют рассчитать концентрацию ионов Н+ и далее определить концентрацию СН3СООН. Полученный результат используется при расчете скорости гидратации уксусного ангидрида.
Для удобства вводятся следующие обозначения: — константа скорости реакции (I); а — исходная концентрация уксусного ангидрида (определяется по его йавеске); у — общая концентрация образующейся по реакции (I) уксусной кислоты; k% — константа скорости реакции (II) (ее значения см. на с. 370); z— концентрация ионов Н+, образовавшихся при диссоциации СН3СООН; Ь — концентрация иодид-иодатной смеси (для упрощения расчета берутся эквивалентные концентрации 1“ и Ю3“); и — концентрация непро-диссоциировавшей СН3СООН; х — количество образовавшегося по реакции (II) свободного иода; К — константа диссоциации СНзСООН (см. с. 370).
Реакция (I) в водной среде является псевдомономолекулярной, поэтому ее скорость выражается уравнением
d у
-_2~=kl(a-y). (1)
Скорость реакции (II) характеризуется уравнением
d х
' = А?2<2*2 (£ — х)3.
a t
Интегрирование уравнения (2) приводит к выражению ____________________________1 f 1 _
Z ” 2Л2* I (Ь — х)2 £2 J '
Интегрирование уравнения (1) приводит к выражению
(2)
(3)
(4)
369
В уравнении (4) величина у включает содержание ацетат-ионов (х) и ыедиссоциированной уксусной кислоты (и):
у = [СН3СОО-] + [СНзСООН].
Количество свободного иода соответствует количеству ацетат-ионов х. Поэтому
у~х + и. (5)
Константу диссоциации уксусной кислоты Кдисс можно представить в следующем виде:
_ [Н+] [СН3СОО-]
Адисс [СНзСООН]
zx
|..д •
»
и
отсюда
и — zx/K
дисс •
(6)
Замена и в уравнении (5) через (6) приводит к выражению
У — X -р %Х/ К лисе •
Таким образом накопление иода (I, а не 12, для удобства расчета) позволяет судить о скорости реакции (I). Содержание иода в реакционной смеси в каждый момент от начала реакции можно определять колориметрически (см. с. 363).
Значения констант скорости реакции (II) для некоторых температур:
/°C 0 15 25
k2-10-10, мин-1- (г-экв/л)-2 2,54 4,8 6,0
Как видно, из приведенных данных, температурный коэффициент скорости этой реакции является одним из наиболее низких и составляет Л ,4.
Значения констант диссоциации уксусной кислоты при различных температурах:
°C
Кдисс-Ю5
0 10 20 25 30 35 40
1,66 1,73 1,75 1,75 1,75 1,73 1,7
Последовательность выполнения работы. Подготовить для измерений колориметр: включить его в электросеть, выдержать время, необходимое для стабилизации прибора, и настроить его (см. с. 363). В это же время приготовить серию стандартных растворов иода * разбавлением 0,1 М раствора 12: 5*10~4; М0~3; 2-10~3; 7,5-10~3; 1,2-10~2; 2,5-10~2.
Для колориметрирования использовать кювету толщиной слоя 10 мм. Концентрация исходного раствора уксусного ангидрида мо
* Растворы иода готовить растворением его в 1 М KI. Концентрацию иода, полученную по калибровочному графику, удваивать, учитывая, для удобства расчета, содержание иода атомарного в реакционной смеси.
.370
жет быть увеличена от 0,01 до 0,06 моль/л, однако тогда нужно готовить другие стандартные растворы иода и измерения проводить в кюветах меньшего размера. Результаты измерений оптической плотности стандартных растворов иода следует записать в таблицу по образцу:
Светофильтр . . размер кюветы . . температура . . .
Номер колбы
Концентрация 12, моль/л
Количество раствора 12 для разбавления
Оптическая плотность
На основании полученных данных построить калибровочный график, откладывая по оси абсцисс концентрацию растворов 12, а по оси ординат — оптическую плотность.
Приготовить 100 мл раствора, содержащего 0,02 М КЮ3 и 0,16 М. KI (или использовать уже имеющийся раствор). Взвесить на аналитических весах 0,06 мл уксусного ангидрида (его объем можно изменять в пределах от 0,2 до 0,01 мл) и перенести навеску в 50-миллилитровую мерную колбу, содержащую 30—40 мл раствора 0,02 М К1Оз+0,16 М KI, и довести объем этим же раствором до метки. Отметить по секундомеру время сливания уксусного ангидрида с раствором KJ+КЮз и время конца его растворения (момент гомогенизации раствора), не выключая секундомера до завершения реакции. Время, среднее между началом и концом растворения уксусного ангидрида, принять за начало реакции. Полученной реакционной смесью заполнить одну из кювет фотоэлектроколориметра, в качестве раствора сравнения использовать применяемый иодид-иодатный раствор. По мере протекания реакции измерять оптическую плотность раствора. При этом один из работающих измеряет оптическую плотность раствора, а другой записывает показания секундомера и колориметра. Два-три измерения произвести через 30 с, четыре-пять измерений — через 1 мин, два-три измерения — через 5 мин, по одному измерению — через 10 мин, 20 мин, 30 мин и 1 ч. Неизменность оптической плотности Аоо раствора в последних измерениях свидетельствует об окончании реакции.
При обработке экспериментальных данных следует учитывать, что реакция гидратации уксусного ангидрида псевдомономолеку-лярна, и рассчитывать константу скорости реакции по уравнению (1). Исходную концентрацию уксусного ангидрида а в растворе рассчитать по его навеске, а концентрацию атомарного иода к моменту измерения — по оптической плотности раствора при помощи
371
калибровочного графика. Результаты наблюдений и расчетов свести в таблицу по образцу:
Температура опыта, °C . . навеска уксусного ангидрида . . .;
я, моль/л, . . Ь, г-экв/л, . . k2f мин-1 • (г-экв/л)-2 . . .
Время измерения
Время от начала реакции, мин А *1, моль/л Ь—х, г-экв/л 2.10е У а—у
4 мин-1
9
Вычислив k реакции для каждого момента от начала реакции, определить ее среднее значение и сравнить с £Ср, полученной по графикам 1g (а—у) = как тангенс угла наклона прямой к оси Л
По константам скорости при двух температурах (вторую константу взять из справочника) рассчитать энергию активации данной реакции.
Работа 9. Изучение кинетики реакции окисления тиомочевины
и тиоацетамида гексацианоферрата (III) в щелочном растворе
Гексацианоферрат (III) (красная кровяная соль)—сильный окислитель в щелочной среде.
Стехиометрически реакция протекает по уравнениям:
для тиомочевины
NH2CSNH2 + 10 (ОН)- + 8 [Fe (CN)6]3“ =
= NH2CONH2 + SO^“ д- 8 [Fe (CN)6]4- + 5H2O (I)
для тиоацетамида
CH3CSNH2 + 11 (OH)- + 8 [Fe (CN)6p- =
=*= CH3COO- + SQ2- 4- NH3 4- 8 [Fe (CN)3]4- 4- 5H2O (II)
Скорости реакций (I) и (II) зависят от концентрации ионов ОН-в растворе, что указывает на образование промежуточного енольного аниона. Вещества, не способные енолизироваться (как бензальдегид), не окисляются Кз[Ре(СМ)б].
Предложен следующий механизм протекания рассматриваемых реакций:
bi
RCSNH2 4- он- RCSNH- + Н2О
&2
RCSNH— 4- [Fe (CN)g]3“ —комплекс (х)
(а)
(б)
&3
комплекс (х) + [Fe (CN)3]3— —> 2 [Fe [CN)^]4— 4- продукт (в)
372
Стадия (а) включает образование енольного промежуточного аниона субстрата и является обратимой. Скорость ее зависит от природы восстановителя. В стадии (б) промежуточный анион образует комплекс с окислителем, который в стадии (в) быстро превращается в конечный продукт реакции, или следующий промежуточный продукт, немедленно переходящий в конечный продукт реакции. Последнее превращение происходит значительно быстрее предыдущих.
Предполагая, что концентрация активного промежуточного соединения RCSNH~ стационарна, в соответствии с методом стационарных концентраций получаем
d [RCSNH-] = [RCSNH , [0H_j _ rRCSNH_j _
d t
— k2 [RCSNH-] [Fe (CN)|~1 =0.
Отсюда
[RCSNH-]
[RCSNH2] [OH-] k-г + k2 [Fe (CN)§~
(1)
Общую скорость реакции можно выразить через убыль гексацианоферрата (III):
d [Fe (CN)e
3—'
= k2 [RCSNH-] [Fe (CN)|
+ &з [комплекс (х)] Fe (CN)|
(2)
Подставляя [RCSNH-] из уравнения (1) в (2) и учитывая, что в соответствии с механизмами (а) — (в) при превращении одной молекулы RCSNH2 реагируют две молекулы Кз[Ге(СМ)б], получаем
d Fe(CN)g
2k^k2 [RCSNH2] [OH-] [Fe (CN)|~ k-t + k2 [Fe(CN)g—
(3)
При окислении тиомочевины стадия (б) является лимитирующей, поэтому k-i^>k2 и уравнение (3) можно записать
d [Fe (CN)?-] 2k, kn
------ТТ------------ = [NH2CSNH2] [OH-] [Fe (CN)|“] . (4) a t---------------------------------------------------/с—। L J
Следовательно, окисление тиомочевины щелочным раствором гексацианоферрата (III) имеет первый порядок по тиомочевине, гексацианоферрату (III) и гидроксильным ионам.
При окислении тиоацетамида скорость определяется стадией (а), поэтому k\~^>k2 и уравнение (3) приобретает вид
d [Fe (CN)g-dt
= [CH3CSNH2] [OH-].
(5)
373
Таким образом, скорость окисления тиоацетамида не зависит от концентрации окислителя, т. е. имеет нулевой порядок по [Fe(CN)6]3~ и первый порядок по тиоацетамиду и ионам ОН~ Однако при низких концентрациях окислителя лимитирует стадия (б), и тогда для расчета скорости реакции следует использовать уравнение (4). Лимитирующая стадия (б) включает взаимодействие двух отрицательно заряженных ионов, поэтому в соответствии с уравнением (XV.46) скорость этой реакции возрастает с увеличением ионной силы раствора (в частности, при введении КС1 в реакционную смесь), а также при увеличении диэлектрической постоянной среды (XV.48). Поскольку скорости окисления тиомочевины и тиоацетамида очень чувствительны к концентрации щелочи, кинетику этих реакций изучают при постоянном значении pH и реакции проводят в присутствии карбонат-бикарбонатной буферной смеси*, которая поддерживает pH И. Исследование кинетики окисления тиомочевины и тиоацетамида облегчается тем, что реакция идет с заметной скоростью при температурах выше 30°С и замедляется при охлаждении; ход реакции можно контролировать измерением концентрации гексацианоферрата (III) в растворе, используя фотоэлектроколориметр с синим светофильтром (400—450 нм); гексацианоферрат (II) не поглощает в этой области.
Последовательность выполнения работы. Подготовить фотоэлектроколориметр для измерений: включить его в электросеть, выдержать время, необходимое для стабилизации прибора, и настроить его (см. с. 365). Приготовить серию стандартных (эталонных) растворов Ks[Fe(CN)6] в мерных колбах вместимостью 50 мл. Готовить стандартные растворы следующих концентраций (моль/л) :Ы0~4; 2*10~4; 2,5-10~4; 3-10~4; 4• 10~4; 5-10”4; 6-10~4 нужно разбавлением 0,02 М раствора K3[Fe(CN)6] карбонат-бикарбонатной буферной смесью. Результаты измерений оптической плотности стандартный растворов записать в таблицу по образцу:
Светофильтр . . .; размер кюветы . . .
Номер колбы Концентрация K3[Fe(CN)s], моль Объем рабочего раствора для получения стандартного раствора, мл Оптическая плотность
«а
После настройки фотоэлектроколориметра приготовить и поместись в термостат с заданной температурой (выше 35° С) мерную колбу вместимостью 500 мл, содержащую от 10 до 40 мл 0,02 М K3[Fe(CN)6] в буферной смеси; колбу, содержащую от 100 до
* Смесь равных объемов 0,1 М Na2CO3 и ОД М NaHCO3.
374
400' мл 0,02 М раствора тиомочевины и буферной смеси; колбу с карбонат-бикарбонатным буферным раствором.
Концентрация тиомочевины в реакционной смеси должна в 10— 15 раз превышать концентрацию Кз(Ре(СЫ)в]. Содержание тиоацетамида может быть эквивалентным содержанию гексацианоферрата (III) в растворе. При высокой концентрации восстанавливаемого вещества раствор мутнеет из-за осаждения серы. Нижний предел концентрации гексацианоферрата (III) определяется чувствительностью фотоэлектроколориметра. При концентрации гексацианоферрата (III) в растворе, превышающей 6-10-3 моль/л, проб^ для колориметрирования следует разбавлять.
Через 20—25 мин термостатированный раствор мочевины перелить в колбу с раствором Кз[Ре(СЫ)б], отметив момент сливания растворов как время начала реакции. Объем раствора в мерной колбе довести до метки термостатированной буферной смесью. Мерную колбу с реакционной смесью поместить в термостат. Из реакционной смеси по ходу реакции отбирать пробы, быстро охлаждать их водой со льдом и колориметрировать. Концентрацию гексацианоферрата (III) определять по оптической плотности, используя калибровочный график. Первую пробу отобрать через 5 мин после начала реакции, вторую — через 10 мин, далее — в зависимости от темпа изменения оптической плотности раствора. Объем проб определяется размерами выбранной кюветы. Полученные результаты занести в таблицу по образцу:
Температура опыта, °C . . исходная концентрация Кз[Ее(СЫ)б], моль/л . . исходная концентрация тиомочевины, моль/л ...
Время измерения Время от начала реакции, мин At Концентрация K3[Fe(CN)d, моль/л Ig (Со/С z) *ск МИН — 1
Поскольку концентрация ионов ОН~ в ходе реакции не меняется, а концентрация тиомочевины на порядок больше концентрации гекса-тгианоферрата (III), содержание тиомочевины можно также считать постоянным и уравнение (4) приобретает вид
d [Fe (CN)|~]
= k [Fe ,
откуда видно, что константу скорости можно рассчитать по уравнению реакции первого порядка. Рассчитать по константам скорости при двух температурах (вторую константу взять из справочника) энергию активации реакции.
375
Работа 10. Изучение кинетики реакции восстановления
гексацианоферрата (III) аскорбиновой кислотой
Слабыми окислителями аскорбиновая кислота дегидрируется до дегидроаскорбиновой кислоты (СбН6Ой). Реакция протекает по уравнению
С6Н8О6 + 2 [Fe (CN)6]3- = С6Н6О6 + 2 [Fe (CN)6]4- + 2Н+
Более энергичное окисление приводит к глубокому разрушению аскорбиновой кислоты.
Механизм восстановления гексацианоферрата (III) аскорбиновой кислотой (для краткости АН2) в кислой среде включает промежуточную стадию образования аскорбат-иона АН-:
АН2 АН- + Н+ (а)
»-1
Й 2
AH- + [Fe(CN)4]3-->AN-+[Fe(CN)6]4- (б)
Стадия (б)—лимитирующая. Далее свободный радикал аскорбиновой кислоты АН» атакуется ионом Fe(CN)e3- с образованием продукта реакции — дегидроаскорбиновой кислоты:
АН • + [Fe (CN)6]3- -> А + [Fe [CN)6]4- + Н+ (в)
Скорость исчезновения гексацианоферрата (III) можно рассчитать методом стационарных концентраций:
- -у- [Fe (CN)|—] = k2 [АН-] [Fe (CN)3-] + k3 [AH ] [Fe (CN)|-]; (1)
d[AH~] di
= k2 [AH-] [Fe (CN)|_
k3 [AH ] [Fe (CN)|-
d [AH-] d t
= *i [AH2] - Й-! [АН-] [H+] - k2 (AH-][[Fe (CN)34-1 = 0.
(2)
(3)
Решая совместно уравнения (1) — (3), получаем
-77- [Fe (CN)g—] = k2 [AH-] [Fe (CN)M + kx [AH2] - k-il[AH~]фН+](4) 1 I— «J
Решение уравнений (4) и
(3) приводит к выражению
Fe (CN)3-’
2kxk2 [АН2] [Fe (CN)|— Ь-х [H+] + k2 [Fe (CN)3-
(5)
Так как стадия (б) является медленной и определяющей скорость всего процесса и k-1»&2{Fe(CN)при низкой концентрации [Fe(CN)|~J, то
d d/
Fe (CN)'g—
4kxk2 [AH2] [Fe (CN)3-A:_i [H+]
(6)
376
В соответствии с указанным механизмом реакции на одну молекулу АН2 расходуется две молекулы Кз[Ге(СЫ)6], поэтому
—77 [Fe (CN)H = -^7 [АН2] = k' [АН2] [Fe (CN)36~] t (7) u Z u J Q Г u J
где k'—Zkikzl (£-i[H+]).
Правильность предложенного механизма восстановления аскорбиновой кислотой подтверждается характером влияния ионной силы раствора на скорость процесса.
За скоростью реакции следят по определению количества неизрасходованного гексацианоферрата (III) со временем. Содержание Ре(СМ)б]3- определяется на фотоэлектроколориметре с синим светофильтром (см. с. 363) или на монохроматоре при 400—450 нм.
Благодаря высокому начальному содержанию серной кислоты (pH 14-2) концентрация ионов Н+ в течение реакции практически не меняется, поэтому ионную силу раствора можно считать примерно постоянной. Поскольку реакционная смесь содержит 5—10-кратный избыток аскорбиновой кислоты по сравнению с содержанием [Ре(СЫ)б]3~, реакция [в соответствии с уравнением (7)] должна иметь первый порядок по ;[Fe(CN)6p“. В связи с.этим кривая на графике lg[Fe(CN)e']=:/ (0 должна быть линейной вплоть до протекания реакции приблизительно на 80%. Если в этом интервале концентраций раствор подчиняется закону Буггера — Ламберта — Бэра, то вместо концентрации [Fe(CN)6]3“ на график можно наносить пропорциональную ей величину оптической плотности,
При эквивалентном содержании реагентов ([Fe(CN)|”] = = 4-10“4 моль/л и [АН2] = 2• 10^4 моль/л) согласно уравнению (7) реакция должна иметь второй порядок и зависимость l/[Fe (CN)|—]= =f(t) должна быть линейной. Уравнение (7) показывает, что скорость восстановления гексацианоферрата (III) должна падать с увеличением концентрации ионов Н+
Последовательность выполнения работы. Подготовить фотоэлектроколориметр (или монохроматор) для измерений (см. с. 363). Подготовить и термостатировать две колбы с водными сернокислыми растворами реагентов (pH 1 — 1,1): одну мерную колбу вместимостью 100 мл заполнить до метки 4-10~4 М раствором Ks[Fe(GN)6], в другую колбу вместимостью 200—250 мл поместить 100 мл 3- IO-3 М раствора аскорбиновой кислоты *.
После 20—25 мин термостатирования раствор Кз[Ре(СЫ)6] перелить в колбу, содержащую 100 мл аскорбиновой кислоты. Момент сливания растворов отметить по часам как время начала реакции. Колбу с реакционной смесью поместить в термостат. Из реакционной смеси по ходу реакции отбирать пробы по 10 мл (или большего объема в зависимости от используемой кюветы), быстро охлаждать их на ледяной бане и, добавив по 0,5 мл концентриро
* Концентрации этих растворов могут варьироваться в пределах 5-ь 10Х Х10~3 моль/л для аскорбиновой кислоты и 4—2-10—4 моль/л для К3[Ре(СЫ)б].
377
ванной серной кислоты (для большего торможения реакции), измерять их оптическую плотность. Первую пробу отбирать через 5 мин после начала реакции, вторую — через 10 мин, а далее —в соответствии с темпом изменения оптической плотности раствора.
Полученные результаты занести в таблицу по образцу.
Температура опыта, °C . . .; исходная концентрация Кз[Ге(СМ)6],
моль/л . . .; исходная концентрация АН2 моль/л . . .; pH . . .
Время измерения
Время от начала реакции, мин
Ао
k, мин-i
*'=Ман2ь МИН-Л-МОЛЬ-i
ъ
* В экспериментах с эквивалентным количеством реагентов (например, [Fe(CN)d3-~ =4-10-4 и (АН2]=2-10“4 моль/л) в таблице графу заменить иа 1/с j<3[Fe(CN) }
и Засчитать по уравнению второго порядка. Для графического определения k ск использо вать график 1/с (/).
♦* Эту графу в таблице заменить на — .
CKe[Fe(CN)e] % K3[Fe(CN).]
Рассчитывать kCK по уравнению первого порядка, заменив концентрацию значениями оптической плотности:
Значение До найти экстраполяцией по графику 1g At = Среднюю скорость реакции сравнить со средней, найденной по тангенсу угла наклона прямой к оси времени &CK=2,3tga.
Аналогичный опыт и расчеты повторить при той же температуре с той же реакционной смесью, но содержащей КС1 или K2SO4 при концентрациях от 0,02 до 0,15 моль/л. Для этого исходные растворы Кз[Ре(СМ)б] и аскорбиновой кислоты должны содержать данные соли в одинаковых концентрациях и иметь pH 1 или 2 (довести pH до нужного значения концентрированной H2SO4). Всю процедуру повторить, приняв за начало реакции также время сливания двух растворов. Полученные результаты занести в таблицу по указанному образцу, дополнив исходные условия значениями концентраций добавленной соли.
Проверить на основании полученных данных выполнение уравнения (XV.52), приняв величину ko равной средней константе скорости kCKf полученной в опыте без добавок соли, поскольку в этом опыте ионная сила меньше.
Объяснить причину отклонения от уравнения (XV.46).
378
ГЛАВА XVI
ФОТОХИМИЧЕСКИЕ И ЦЕПНЫЕ РЕАКЦИИ, РАДИКАЛЬНАЯ ПОЛИМЕРИЗАЦИЯ
1. ФОТОХИМИЧЕСКИЕ РЕАКЦИИ
Реакции, протекающие под действием светового излучения (видимого и ультрафиолетового), которое вызывает активацию частиц одного из реагирующих веществ, называются фотохимическими. Основным законом фотохимии является закон фотохимической эквивалентности Эйнштейна, согласно которому каждый поглощенный квант электромагнитного излучения вызывает изменение одной молекулы. Изменение может быть как энергетическим, так и химическим.
Количественно фотохимические реакции характеризуются квантовым выходом ф. Квантовым выходом называется отношение образовавшихся или прореагировавших молекул п к числу поглощенных квантов п0:
Во многих реакциях число фотохимически реагирующих молекул не равно числу поглощенных квантов, т. е. ф не равен единице. Объясняется это сложностью протекания фотохимического процесса, который йожет включать три стадии: 1) начальный акт поглощения светового потока; 2) первичный фотохимический процесс; 3) вторичные реакции («темновые»).
Первая стадия приводит к переходу молекулы (за время 10~16 с) в электронно-возбужденное состояние: Вторую
стадию можно объединить с первой, назвав их вместе первичным фотохимическим процессом. Во второй стадии возбужденные молекулы за время своего существования (10~8 с) претерпевают различные превращения: а) диссоциацию с образованием свободных атомов и радикалов (или ионов при гетеролитическом разрыве), которые вступают в дальнейшее взаимодействие — вторичные реакции (третья стадия); б) дезактивацию при столкновениях с другими молекулами; в) переход в основное электронное состояние с испусканием кванта светового излучения (флуоресценция или фосфоресценция) или внутримолекулярное превращение (конверсия) энергии электронного возбуждения в колебания. Изучение спектров поглощения помогает решить вопрос о характере первичного фотохимического превращения.
Закон эквивалентности Эйнштейна справедлив только по отношению к начальному акту поглощения светового потока, где один квант приводит к образованию одной возбужденной молекулы, значение же общего квантового выхода может быть разным. По величине квантового выхода реакции можно разделить на четыре группы: 1) реакции с ф=1, если продукты первичной диссоциации
379
являются устойчивыми молекулами и вторичные реакции отсутствуют; 2) реакции с ср< 1 при рекомбинации продуктов первичной диссоциации или дезактивации за счет соударений, а также при потерях энергии на излучение или ее рассеяние по многим связям у сложных молекул; 3) реакции с ср> 1, к которым относятся реакции быстрой и полной диссоциации на устойчивые молекулы или радикалы реагирующие далее не по цепному механизму; 4) реакции с 1, если вторичные реакции протекают по цепному механизму. Классическим примером данных реакций является взаимодействие молекулярных хлора и водорода под действием светового потока: H2+CI2—^HGL Здесь квантовый выход достигает 106, что свидетельствует о наличии вторичных цепных процессов.
Изучение зависимости квантового выхода от условий опыта дает возможность судить о механизме протекающего процесса. Квантовый выход зависит от концентрации или давления реагирующих веществ и инертных добавок, от интенсивности светового потока, от длины волны, от температуры, от размеров сосуда и материалов стенки. Квантовый выход с ростом температуры часто увеличивается, так как при этом уменьшается вероятность рекомбинации активных частиц, возникших в ходе первичного процесса. С увеличением длины волны квантовый выход может расти, так как вероятность возбуждения молекул растворителя падает с уменьшением энергии инициирующего возбуждения.
Важной особенностью фотохимических реакций является независимость их скорости от температуры. Первичная фотохимическая реакция обычно является лимитирующей, а энергия кванта, поглощенного в ней, много выше энергии теплового движения и изменения ее с температурой. Влияние температуры становится заметным при существенном влиянии «темновых» реакций на скорость суммарного процесса.
К первичным фотохимическим процессам близки фотосенсиби-лизированные реакции. Это реакции, в которых участвуют молекулы, не чувствительные к излучению данной частоты, но способные получать энергию от возбужденных световым потоком других молекул (фотосенсибилизаторов). Фотосенсибилизатором является ртуть, при облучении паров которой световым потоком ртутной разрядной лампы происходит возбуждение атомов ртути: Hg+^v—*
*Hg*. Возбужденные атомы Hg* могут при соударении с молекулами углеводородов RH вызывать их распад на свободные радикалы:
Hg* + RH->Hg4-R4-H
2. ЦЕПНЫЕ РЕАКЦИИ
Цепными называются сложные реакции, в которых превращение исходных веществ в продукты осуществляется путем регулярного чередования нескольких элементарных реакций. В элементарных реакциях происходит регенерация первоначально зародившихся ак-
380
тивных промежуточных частиц: атомов, свободных радикалов, ионов или реже молекул с повышенным запасом энергии (колебательно- или электронно-возбужденных молекул). К цепным процессам принадлежат гомогенные газовые реакции горения и медленного окисления, многие реакции крекинга, разложения и полимеризации углеводородов, разложения ряда твердых, жидких и газообразных органических соединений, синтеза НС1, НВг, реакции расщепления ядер урана и др. Различают неразветвленные и разветвленные цепные реакции. В неразветвленных цепных реакциях каждая исчезающая активная промежуточная частица вызывает появление одной новой активной частицы. Типичным примером не-разветвленной цепной реакции служит-образование хлористого водорода из хлора и водорода под действием светового потока: )
Н2 + Cl2 4- hv Н2 4- 2С1 • зарождение цепи
СЬ 4-Н2НС1 4-Н • )
H +Cl^HCl+CI- “е”
Н. 4- Н • 4- S -> Н2 4- S ,
СЬ 4-СЬ 4-S->CI24-S
Н- 4-СЬ 4-S-+HC1 4-S
> обрыв цепи
где S — участок поверхности реакционного сосуда или частица постороннего вещества; С1 * — индуктор; Н2 — актор; СЬ — акцептор.
Если в результате одного элементарного акта взаимодействия возникают две или больше химически активных частиц, процесс называется разветвленным цепным процессом. К таким процессам принадлежит окисление водорода:
Н2->-2Н- зарождение цепи
Н- 4-О2->ОН- 4-6 разветвление цепи
6 4-Н2->ОН. 4-Н- 1
• Г продолжение цепи
• ОН 4- Н2 =-> Н2О 4- Н • J
Н • 4- стенка 1/2Н2
• ОН + Н • + стенка Н2О • Н 4- О2 4- М —>• * НО2 4- М
- обрыв цепи
где Н-—индуктор (на отщепление атома кислорода от О2 затрачивается работа); О2—актор; Н2 — акцептор (исходное, трудно окисляемое вещество).
Всякое цепное превращение включает три этапа: 1) зарождение цепи (возбуждение или инициирование), 2) развитие цепи или ее рост (продолжение) и 3) обрыв цепи.
Зарождение цепи требует энергии и может быть вызвано поглощением квантов светового потока, особо благоприятными соударениями, термической диссоциацией, химическим взаимодействием с атомами или ионами (на поверхности стенок или в объеме
381
сосуда), действием ионизирующих излучений и т. п. Процессу зарождения способствует инициаторы, легко образующие свободные радикалы. Это чаще всего перекиси: бензоила, тетралина, легко распадающиеся по связи О—О, гидроперекиси ROOH, а также азо-создинения, некоторые химически активные газы с неспаренными электронами (NO, N02, N0C1).
Развитие ц е п и — последовательность идущих друг за другом элементарных актов —циклов регенерации. Так, под влиянием инициирующей реакции при образовании НС1 возникает цепь превращений Совокупность реакций, которая
начинается с данного радикала и приводит к его регенерации, называется звеном цепи. Среднее число таких звеньев (или циклов), которое приходится на один первичный радикал, полученный при зарождении цепи (т. е. число молекул исходного вещества, прореагировавших в одном акте зарождения цепи), называется длиной цепи. Средняя длина цепи зависит прежде всего от природы реакции, а также от условий ее протекания: температуры, давления, примесей, формы сосуда и состояния его стенок и т. п.
Обрыв цепи — группа реакций, приводящая к исчезновению активных частиц и способная вызвать прекращение цепного процесса. Обрыв может быть линейным, если скорость его пропорциональна концентрации активных промежуточных частиц в первой степени, К ним относятся взаимодействия активных промежуточных частиц с поверхностью (материалом стенки) сосуда; с соединениями металлов разной степени окисления, способными отдать или отобрать один электрон; с валентно-насыщенными молекулами (ингибиторами), следствием чего является образование малоактивного свободного радикала. Такой радикал не способен к продолжению цепей и погибает при рекомбинации с каким-либо другим радикалом. Примером обрыва цепей на молекулах ингибитора может служить окисление углеводородов в присутствии дифениламина:
RO2 • + NH (С6Н5)2 ROOH + • N (С6Н5)2 (I)
•N(C6H5)2 + R‘-*RN(C6H6)2 (2)
Стадия (1) здесь является лимитирующей, поэтому обрыв линейный.
Обрыв цепей может быть квадратичным, если скорость его пропорциональна произведению концентраций двух активных промежуточных частиц или квадрату концентрации одной из них. Таков обрыв в объеме сосуда, происходящий в результате насыщения свободной валентности при рекомбинации (взаимодействии) двух свободных радикалов^ атомов или других активных промежуточных частиц:
R.+R. 4-M^RR-pM.
где М — третья частица, которой отдается избыточная энергия, что необходимо для стабилизации полученной молекулы.
Вероятность тройного соударения увеличивается при высоких давлениях газа в реагирующей системе (или в цепных реакциях, 382
протекающих в жидкой фазе), когда диффузия к стенкам затруднена. Повышение давления увеличивает роль тройных соударений. ["Для неразветвленных цепей, в которых каждая активная частица дает начало одной цепи, скорость реакции может быть представлена как произведение длины цепи у на число активных частиц пОг зарождающихся в единице объема за единицу времени (т. е. о>о)
w = wqv, (XVI. 1>
где и>о — скорость зарождения цепи. Таким образом, для неразветвленных цепей остаются справедливыми обычные уравнения химической кинетики с константой скорости, увеличенной в v раз. Для всех цепных реакций наблюдается характерная зависимость скорости реакции от времени. На рис. 159 приведена сравнительная зависимость скорости реакции от времени для молекулярных, ион*
Рис. 160. Зависимость скорости реакции от давления для разветвленных цепных реакций:
Pi — нижний предел давлений; р2— верхний предел давлений
Рис. 159. Зависимость скорости реакции от времени:
* 1 — молекулярные реакции; 2 — ионные реакции;
3' — неразветвленные цепные реакции при уменьшении концентрации реагируюдцих веществ; 3" — разветвленные цепные реакции со скоростью обрыва больше скорости разветвления; 3'" — разветвленные цепные реакции, у которых скорость обрыва цепи меньше скорости разветвления; ti — время, в течение которого реакция протекает с неизмеримо малой скоростью (период индук-
ции)
ных и цепных реакций. Одним из наиболее характерных признаков цепного механизма является высокий квантовый выход при фотохимическом инициировании. Для разветвленных цепных процессов начальное изменение скорости реакции от времени выражается уравнением
№ = ^(^'-1), (XVI.2>
где ср — константа нарастания, характеризующая скорость разветвления; t — время.
Если в уравнении (XVI.2) (при достаточном времени протекания реакции), то оно приобретает вид
w = Ле?/7 (X VI. 3>
причем
Д = w0/(8 —?) и ? = (8 —
где 5 — скорость разветвления цепи в данном звене; р—скорость
383
обрыва цепи в данном звене; А/ — время между двумя последовательными реакциями в цепи, т. е. время жизни одного звена цепи. Величины S и (3 зависят от давления и температуры. Если обрывы сильно преобладают над разветвлениями (р>6), то реакция протекает стационарно, а в определенном интервале давлений или концентраций (р<6) она идет как взрывная. На рис. 160 приведена зависимость скорости реакции от давления (или концентрации) для разветвленных цепей. При переходе через пределы р\ и р% реакции резко переходят от медленных стационарных к самоускоря-ющимся быстрым реакциям горения или взрыва и обратно. Заштрихованная область между двумя пределами давлений — область холодного (изотермического), взрыва. Нижний предел pi мало зависит от температуры и в большой степени зависит от состава смеси (наличия посторонних частиц в сосуде), формы и размера сосуда. Верхний предел р2 в большей мере зависит от температуры и примесей и почти не зависит от формы сосуда и состояния его стенок.
Существование пределов давлений объясняется тем, что наряду с разветвлением цепей происходит и их обрыв. При большой вероятности обрывов реакция течет медленно и спокойно, как и при неразветвляющихся цепях. Это происходит при низких давлениях, так как диффузия активных частиц к стенкам идет без затруднений. С ростом давления вероятность обрывов цепей за счет соударений со стенками уменьшается и разветвление цепей увеличивается. Реакция протекает самоускоренно вплоть до воспламенения (при высоких температурах и давлениях — до взрыва). Переход совершается очень резко при прохождении через нижний предел. По достижении верхнего предела разветвление цепей снова затрудняется вследствие обрыва в объеме. Этот обрыв пронсхо-* дит в результате тройных столкновений или соударений с молекулами примесей, концентрация которых растет с давлением. Тогда наблюдаемая скорость процесса зависит от числа тройных соударений. Дальнейшее повышение давления постепенно увеличивает скорость реакции вплоть до наступления теплового взрыва. Сжатие имеет адиабатический характер, поэтому температура повышается, приводит к сильному увеличению скорости реакции и еще большему выделению теплоты. В результате наступает «тепловой взрыв».
3. РАДИКАЛЬНАЯ ПОЛИМЕРИЗАЦИЯ
Полимеризацией называют реакции образования высокомолекулярных соединений из мономеров. В таких реакциях рост макромолекул происходит путем присоединения мономеров к •исходным инициирующим частицам и далее — к реакционноспособным группам на концах образующихся полимеров. К полимеризации способны органические соединения, содержащие либо кратные (двойные, тройные) связи, либо циклы. Число мономерных звеньев в данной макромолекуле определяет степень ее полимеризации.
Механизм полимеризации может быть цепным, а может быть ступенчатым в зависимости от химической природы промежуточных продуктов. Обязательные стадии процесса цепной полимериза-
384
ции включают инициирование (зарождение активных центров — радикалов), развитие (рост) цепи и обрыв цепи. Однако при полимеризации развитие кинетической цепи сопровождается образованием материальной молекулярной цепи:
AM* + М-> АМг
AMj + M-^AMg
А(М)*+М-^А(М)*+1
где М — молекула мономера; А(М)„* — активный промежуточный продукт, представляющий собой нестабильные короткоживущие частицы — макрорадикалы.
При ступенчатом механизме промежуточные продукты полимеризации достаточно стабильны и имеют значительную продолжительность жизни. Схема такого процесса:
лМ —(М)2 + (п —2)М диаметр
(М)2 + М(М)3 й Т. д.
Это — последовательное ступенчатое присоединение, которое на каждой стадии представляет собой самостоятельную реакцию.
В соответствии с химической природой активных частиц, участвующих в цепной полимеризации, различают радикальную и ионную полимеризацию. Цепная ионная полимеризация может быть катионной и анионной — промежуточными продуктами здесь являются коны, ионные пары или поляризованные комплексы. Если в полимеризации одновременно участвуют два или несколько различных мономеров, то ее называют совместной или сополимеризаций.
Радикальная полимеризация всегда протекает по цепному механизму и активными промежуточными - продуктами здесь являются свободные радикалы. Инициирование сводится к созданию в среде мономера свободных радикалов, способных начать цепь:
R. 4-СН2 = СНХR — СН2 — СНХ
1. Пути инициирования: 1) термическое инициирование за счет гомолитического распада нестойких веществ — инициаторов (при температуре 50°С и выше с энергией активации 100—150 кДж/моль) и в результате энергичных столкновений молекул мономера друг с другом; 2) химическое инциирование — при введении в среду мономера веществ, способных распадаться с образованием свободных радикалов, например
Н2О2 4- FeSO4 ->
Fe2+ 4-H2O2->Fe3+ 4-ОН~ 4-ОН (при умеренных и даже низких температурах) или при введении в среду мономера нерастворимых в воде окислительно-восстановительных систем: органические перекиси — органические соли Fe2+; органическая перекись— амин; могут быть также 13-2083 385
использованы только перекиси, так как связь О—О непрочная; 3) фотохимическое инциирование — образование свободных радикалов при поглощении квантов светового потока молекулами мономера или специально вводимыми фотоинициаторами и фотосенсибилизаторами. Например, полимеризация метилметакрилата при УФ-излучении с Х = 250—360 нм; 4) радиационно-химическое инициирование, при котором используются рентгеновское, электронное, нейтронное или у-излучения. При этом любая молекула распадается с образованием свободных радикалов.
II. Рост цепи, т. е. образование макромолекул путем последовательного присоединения молекул мономера к радикалу, возникшему при инициировании:
R. 4-СН2=СНR—СН2—CH-f-CH2—СН2—СН—СН2—СН-> т. ж
X XX XX
Энергия активации здесь порядка 12—40 Дж/моль.
Этот процесс кинетически является типичной неразветвленной цепной реакцией, поскольку идет с образованием свободного радикала, т. е. с его регенерацией. Каждый акт присоединения к растущему свободному радикалу новой молекулы мономера дает звено цепи. Длина цепи показывает, сколько молекул мономера вступило в процесс полимеризации в расчете на один начальный свободный радикал. Это — кинетическая длина цепи в отличие от длины цепи образующегося полимера (степени полимеризации). Если процесс полимеризации не осложнен дополнительными элементарными стадиями (например, стадиями передачи цепи), то степень полимеризации равна кинетической длине цепи v при обрыве цепи диспропорционированием, и равна удвоенной кинетической длине цепи 2v при обрыве в результате рекомбинации.
III, Обрыв цепи происходит:
1. Путем взаимодействия двух растущих радикалов: а) в результате их рекомбинации
--сн2—СН+НС-СН2—- СН2-СН—сн-сн2-~
XX XX
б) или диспропорционирования
—СН2'“СН_|_ИС—СН2———*•СН =СН4-СН2—СН2— II I I
XX XX
Одна из этих молекул обязательно содержит двойную связь в конце. Так обрывается и кинетическая, и материальная цепи. Для начала процесса опять нужны новые радикалы.
Поскольку обрыв цепи может произойти на любой стадии роста радикала, н при полимеризации образуются макромолекулы разной длины, то полимер обычно характеризуют средней молекулярной массой.
386
2082
2. Путем взаимодействия радикалов с ингибитором, которым могут быть: а) малоактивные свободные радикалы, не инициирующие полимеризацию, но способные рекомбинировать (или диспро-порционировать) с растущим радикалом; б) молекулы, которые, взаимодействуя, насыщают свободные валентности радикалов, а сами превращаются в малоактивные радикалы. Так действуют многие хиноны (бензохинон), ароматические ди- и тринитросоединения, молекулярный кислород, соединения металлов переменной степени окисления (соли Fe3+, Cu2+ и др.). Здесь ингибирование сводится к передаче электрона:
---СН2 - СНХ 4- FeCl3^ - — СН2 — СНХС1 4- FeCl2
IV. Для радикальной полимеризации характерны реакции передачи цепи. Сущность состоит в отрыве растущим радикалом атома или группы атомов от какой-либо молекулы (передатчика цепи). В результате радикал превращается в валентно-насыщенную молекулу и образуется новый радикал, способный к продолжению кинетической цепи. Передача цепи может осуществляться: 1) через молекулу мономера
R-4-CH2 = CH------>rh+ch2 = ch
ОСОСНз ососн2.
винилацетат
Молекула мономера превращается в мономерный радикал, который дальше обеспечивает рост новой макромолекулы:
СН2 = СН + СН2 — СН СН2=СН - СН2 - СН
ОСОСН2 • ОСОСНз ОСОСНз ОСОСНз
2) через молекулу растворителя |
---сн2-СНХ 4- СС14-+— СН2-СНХС1+СС13
Так осуществляется прекращение роста данной материальной цепи, т. е. снижение молекулярной массы образующегося полимера. Присутствие больших количеств передатчиков цепи приводит к образованию очень коротких цепей (теломеризация);
3) через полимерные молекулы, уже успевшие образоваться в реакционной системе:
СН2—СН2~"~
--сн—снх+снх----СН-СНХ+ • сх—-?•
Новый макрорадикал присоединяет молекулы мономера
— —СНд СН2
--СХСН2=СНХ->-~—СХ—СН—СНХ и т. д.
13*
387
В результате образуется разветвленная макромолекула. Вероятность передачи реакции цепи через полимер возрастает, когда концентрация макромолекул в системе достаточно велика. Это — один из источников возникновения разветвлений в полимерных цепях.
4. КИНЕТИКА РАДИКАЛЬНОЙ ПОЛИМЕРИЗАЦИИ
Скорость инициирования при термическом распаде инициаторов ^ИИ~ ИИ» (XVI.4)
где &ин—константа скорости; f — эффективность инициирования (или фактор инициирования)—величина, показывающая, какая доля радикалов, образовавшихся при распаде инициаторов, начинает реакционные цепи; сип— концентрация инициатора. Обычно f<l (для многих систем 0,5<f<l,0), так как часть образующихся радикалов может рекомбинировать или участвовать в каком-либо постороннем процессе.
Суммарная скорость полимеризации практически совпадает со скоростью роста цепи, так как мономеры расходуются в основном на стадиях роста цепи. При допущении, что реакции передачи цепи в системе отсутствуют, скорость роста цепи можно выразить уравнением:
^ = 1 ^P[Rz][M] (XVI.5)
7=1
где ki# — константа скорости присоединения мономера к радикалу при степени полимеризации n = [R/] — концентрация радикалов при степени полимеризации n=i; (М]—концентрация мономера.
Различия в скорости роста при присоединении мономеров имеются только для первых двух-трех звеньев, поэтому с хорошим приближением можно принять, что не зависит от величины макрорадикала. Выражение (XVI.5) для упрощается:
®Р = -= MR • ] [М] • (XVI.6)
Д £
Константа скорости и энергия активации роста цепи зависят от химической природы мономера. Химическая природа растворителя не влияет на константы скоростей при радикальной полимеризации. Скорость обрыва цепи описывается уравнением
- = *обр [К • ] [R • J = [R • F. (X VI. 7)
где &обр — константа скорости обрыва (в предположении, что реакционная способность радикалов в реакциях обрыва не зависит от их степени полимеризации).
388
Изменение концентрации радикалов в ходе процесса при отсутствии ингибитора может быть представлено в виде разности скоростей инициирования и обрыва цепи:
^5г-=®ии-М[К]2- (XVI. 8)
аг
Трудно определяемую экспериментально концентрацию свободных радикалов можно исключить из уравнения (XVI.8), применив метод стационарных концентраций, т. е. приняв d(R«]/d^=0, что практически выполняется через несколько секунд после начала реакции. Отсюда №ин = &o6p[R-]2, или
= ((XVI.9)
Выразив в уравнении (XVI.9) wH с помощью уравнения (XVI.4) при f=l и подставив полученное значение [R*] в уравнение (XVI.6), получим
- [М] [Син]1/2. (XVI. 10)
Уравнение (XVI. 10) справедливо для радикальной полимеризации при небольших степенях превращения мономера в полимер (не выше 10—15%). При более глубокой полимеризации наблюдаются отклонения из-за возрастания вязкости реакционной смеси при увеличении в ней концентрации полимера. Диффузия макрорадикалов в таких условиях замедляется и резко уменьшается вероятность их рекомбинации или диспропорционирования, т. е. уменьшается эффективная константа скорости обрыва. Поэтому концентрация радикалов в системе возрастает и, соответственно, увеличивается скорость полимеризации. Это приводит к возникновению так называемого «гель-эффекта» (студнеобразования) при 25—30% степени полимеризации.
Работа 1. Изучение кинетики фотохимического разложения Н2О2
Фотохимическое разложение Н2О2 является типичным цепным процессом, механизм которого можно представить уравнениями реакций:
Н2О2 + ftv->2OH.зарождение цепи
•ОН4-Н2О2^Н2О4-НО2. )
но2.+2н2о.2-н2о+н2о4+он.)цикл РегенеРац™
Н2О4 4-ОН- 4-О2 разветвление цепи
•ОН + ОН — Н2О2 обрыв цепи
Выделение кислорода происходит в результате реакции
Н2О4+Н2О2 4- о2
которая относится к цепным реакциям с «вырожденными развет-
389
влениями» *. В данном процессе разветвление осуществляется фото-химически за счет подвода энергии извне. Относительно устойчивым промежуточным продуктом является свободный радикал ОН-. Кинетика этой реакции выражается уравнением (XVI.3), логарифмирование которого дает
= + (XVI.11)
Уравнение (XVI.И) —уравнение прямой в системе координат In W—t.
Последовательность выполнения работы. Кварцевую колбу 1 (рис. 161) с раствором Н2О2 соединить с обратным холодильником 2 и установить против отверстия 6 в защитном щите 7. Открыть
кран 3 и уровень жидкости в бюретке 5 при помощи уравнительного сосуда 4 установить на нуль. Пустить воду в холодильник и выждать момент, когда установится уровень жидкости в газовой бюретке при закрытом кране 3. Если уровень жидкости в бюретке изменился, установить его при помощи крана 3. Этим достигается постоянство давления, равное атмосферному. После выравнивания уровней при закрытом кране 3 включить одновременно ртутно-кварцевую лампу 8 и секундомер. Первый отсчет записать
Рис. 161. Схема установки для изучения после ТОГО, как выделится кинетики фотохимического разложения 1,0 1,5 см3 кислорода, за-
Н2О2
тем через 3 мин; когда скорость выделения газа достиг-
нет 1 см3/мин, отсчет вести через минуту. При отсчете объема газа ио2 необходимо выравнивать уровни жидкости в уравнительном сосуде и газовой бюретке.
Опыт прекратить, когда выделится 75—100 см3 газа. Опытные данные записать в таблицу по образцу:
Номер измерения
Время от начала реакции, мин
Показания бюретки
Объем выделившегося О2, см3
Скорость реакции. см3/мин
* Цепными реакциями с «вырожденными разветвлениями» называются такие, в которых в результате распада продуктов реакции образуются свободные радикалы . В данной реакции промежуточный продукт НО2- дает при взаимодействии с Н2О2 радикал ОН- и молекулу тетраоксида водорода, приводящую далее к разветвлению цепи.
390
При обработке экспериментальных данных построить график в координатах уоа “ ** Эта зависимость необходима для расчета скорости. Скорость в различные моменты времени определять по уравнению
Интервал времени —i\ взять равным 2 мин. Зависимость скорости от времени выражается уравнением (XVI.3). Для определения эмпирических констант А и ср построить график зависимости ш от t и по восходящей ветви полученной кривой взять значения w в различные моменты времени и вычертить второй график в координатах lg w—/. Величину ср определить как тангенс угла наклона полученной прямой к оси t, a IgA — как отрезок, отсекаемый прямой от оси lg w при t = 0. Поскольку А = ад0/ф, то рассчитать скорость зарождения wQ—А ср.
Работа 2. Определение квантового выхода при фотохимическом разложении Н2О2
Поскольку эта реакция является цепной с вырожденным разветвлением (см. работу 1), кванты светового потока в ней, условно названные «химическими», расходуются на две элементарные реакции:
Н2О2 д ftv-^2ОН- (1)
Н2О4 + Av->2 ОН- 4- О2 (2)
Распределение общего количества квантов по реакциям завис,йт от условий проведения реакции и от стадии, на которой она в данный момент находится, т. е.ют концентраций Н2О2, Н2О4 и Н2О (концентрациями ОН- и О2 можно пренебречь). На начальном этапе реакции, когда [Н2О4] мала, большая часть «химических» фотонов будет расходоваться на реакцию (1), при сильном же снижении [Н2О2] и значительном возрастании [Н2О4] — на реакцию (2). Большая часть квантов излучения в УФ-области захватывается молекулами воды, примесями, радикалами ОН- и т. п. с переходом энергии квантов в теплоту. Эти кванты условно названы «тепловыми».
Зная интенсивность /о начального светового потока, температуру начала То и конца Т реакции и интенсивность светового потока /, прошедшего через реакционную смесь, можно рассчитать количество теплоты, эквивалентное поглощенным тепловым квантам, количество «тепловых» и химических квантов и квантовый выход.
Число молей разложившейся Н2О2 при облучении можно определить двумя методами: газометрическим (по объему выделившегося кислорода) или титриметрическим. В связи с этим предлагает-
391
Рис. 162. Схема установки для определения квантового выхода:
1 — реакционный сосуд; 2 — источник УФ-излучения; 3, 7 — полупроводниковые датчики; 4 — полупроводниковая термопара; 5 — край; 6 — обратный холодильник; 8 — электронный блок светового потока; 9 — электронный блок температуры; 10 — измеритель светового потока и температуры
ся два варианта работы. Для определения количества поглощенных квантов используется установка, схема которой приведена на рис. 162.
Газометрическое определение разложившейся Н2О2 при облучении. Схема установки, представленная на рис. 161, дает возможность определять количество разложившегося пероксида водорода в процессе облучения. Для одновременного фиксирования интенсивности светового потока и измерения температуры раствора эта установка снабжена дополнительно термопарой, погруженной в реакционный сосуд /, и приспособлением для измерения интенсивности светового потока (рис. 162).
Последовательность выполнения работы. Кварцевую колбу 1 (пропускающую ультрафиолетовое излучение) с раствором Н2О2 и погруженной в него термопарой 4 соединить с обратным холодильником 6 и установить против отверстия в защитном щите 7 (рис. 161). Между этим отверстием и колбой установить один из полупроводниковых датчиков светового потока 3 (рис. 162), поместив второй датчик 7 за колбой. Открыть кран 5 и установить на нуль уровень жидкости в бюретке 5 (рис. 161) при помощи уравнительного сосуда 4. Пустить воду в холодильник и, закрыв кранЗ (5), выждать момент, когда'установится уровень жидкости в газовой бюретке. При изменении уровня установить его с помощью крана 3 (5), достигая таким образом давле-
ния, равного атмосферному. После выравнивания уровней при закрытом уровне 3(5) включить ртутно-кварцевую лампу 2 (рис. 162) и выждать установление стационарного режима источника света. После выделения 1—1,5 см3 кислорода включить датчики интенсивности светового потока 3 и 7 и одновременно секундомер и зафиксировать уровень жидкости в бюретке (с обязательным условием выравнивания уровней жидкости в бюретке и в уравнительном сосуде) . Этот момент принять за начало реакции. Далее с интервалом 5 мин регистрировать значения интенсивности светового потока и температуры в реакционном сосуде, для чего тумблер «регистрируемая величина» поочередно переключать в положения «1» и «т»* 392
Запись показаний производить через 5—10 с после установления стрелки и результаты измерений записать в таблицу по образцу: То, °C , . объем исходного раствора Н2О2, мл / . внешнее давление,
Если измерения светового потока производятся одним датчиком, то второе значение рассчитывается по экспериментальной формуле для данного прибора. После 15—20 измерений записать уровень жидкости в бюретке и занести в таблицу. Разница в уровнях в начале и в конце опыта соответствует всему объему кислорода, полученному за время опыта, за вычетом его количества, растворившегося в воде. Примерно учесть это количество или хотя бы оценить ошибку за счет неучета растворимости можно, используя приведенные ниже справочные данные по растворимости кислорода в воде при разных температурах.
Г, °C 15 20 25 30 40 50 60
Растворимость О2, по- 0,0341 0,0310 0;0283. 0,0276 0,0231 0,0209 0,0195 глощенного 1 мл воды при 0°С и 1,013*105 Па при парциальном давлении О2
1,013-Ю5 Па, см3
По полученным данным построить графики зависимости /0, Ц, I и AQ от времени (рис. 163, а, б), рассчитать кванты, поглощенные раствором, а также число молей Н2О2, разложившихся за время опыта.
1. Определить общее число поглощенных квантов путем суммирования усредненной в интервале 5 мин разницы интенсивностей начального и прошедшего через раствор Ц потоков с последующим делением полученной суммы на энергию кванта светового потока с длиной волны X:
«общ ==
I
2. Рассчитать количество теплоты, поглощенное при облучении реакционной смеси, по формулам
z
Дф/ = тДГ; Д<?обш = 2л^ь
где с — удельная теплоемкость реакционной смеси (ввиду малой концентрации раствора Н2О2 ее можно принять равной удельной теп-
393
лоемкости воды) Дж/(г*К), a tn — масса исследуемого раствора (принять равной массе воды в объеме, равном объему реакционной смеси), г.
3. Определить число тепловых квантов для отдельных периодов и, суммированием — общее число тепловых квантов /гт:
nr = AQz/(Av); п
4. Рассчитать общее число химических квантов: ^общ = пх •
5. Определить число молекул разложившегося пероксида водорода. По объему кислорода, выделившемуся за время опыта, с ис-
Рис. 163. К расчету химических (а) и тепловых (6) квантов, поглощенных раствором при фотохимическом разложении пероксида водорода
пользованием уравнения состояния идеального газа рассчитать число молей кислорода, которое соответствует удвоенному числу молей разложившегося Н2О2 (2/г). Отсюда определить пн2о2-
6. На основании полученных данных рассчитать квантовый выход:
? ” пН2О2/пх-
Титрометрическое определение разложившегося Н2О2 при облучении. Опыт выполнить на установке, схема которой представлена на рис. 162. Содержание Н2О2 до и после опыта определить иодометрически с использованием каталитического действия молибдата аммония:
Н2О2 4- 2 Н+ + 2 1-->12 4-2 Н2О
Пипеткой 25 мл исходного раствора перенести в коническую колбу вместимостью 250 мл, прибавить 10 мл 4 н. H2SO4, 1 г иодида калия (избыток), 3 капли нейтрального 30%-ного раствора (NH4)2MoO4 и титровать раствором Na2S2O3 в присутствии крахмала.
394
Аналогичное титрование выполнить по завершении опыта и по разности концентраций рассчитатьжоличество молей разложившегося пероксида водорода: \
где vp — объем исследуемого раствора, мЖ^
Все остальные расчеты аналогичны опй^анным в предыдущем опыте. х
Работа 3. Изучение скорости реакции сульфирования карбамида
При температуре выше 140° С взаимодействие карбамида с избытком концентрированной серной кислоты протекает по уравнению
СО (NH2)2 + 2 H2SO4^CO24-(NH4)2 S2O7
Характер изменения скорости реакции со временем зависит от соотношения количеств исходных веществ. Так, при концентрациях карбамида в кислоте бо^ее 4 моль/л через некоторый промежуток времени (период индукции) наблюдается резкое (более чем в пять раз) повышение скорости реакции, напоминающее своеобразный взрыв. Затем очень быстро скорость падает до нуля. Это — не тепловой взрыв, поскольку выделение теплоты при этом отсутствует, температура смеси понижается (на 5—10°). При меньших концентрациях карбамида (5/3 моль/л и менее) после короткого периода индукции скорость реакции снижается согласно уравнению реакции первого порядка. Взрывоподобный ход реакции исчезает при наличии следов воды в смеси. Наиболее воспроизводимые результаты получаются при содержании воды в серной кислоте, близком к содержанию азеотропной смеси (т. кип. 330°С). Бурное развитие процесса сульфирования карбамида вызывается активными промежуточными продуктами сульфиновыми кислотами, которые, накапливаясь в ходе реакции до определенной концентрации, затем разветвляют цепь. Реакция развивается по схеме: первая стадия — возбуждение цепи
СО (NH.2)2 4- 2 H2SO4^ NH2SO3H + NH4HSO4 4- CO2 амидосульфоновая кислота
вторая стадия — развитие цепи
NH2SO3H + H2SO4--, NH (SO3H)2 + H2O имидосульфоновая
CO (NH3)2 + NH (SO3H)2 + H2SO4->CO2 + 3NH2SO3H
где NH(SO3H)2— носитель цепи, образует три молекулы второго переносчика цепи NH2SO3H, что приводит к разветвлению цепи.
Третья стадия — обрыв цепи
NH (SO3H)2 + 2H2SO4-,NH4HS2O7 4- H2S2O7
NH2SO3H + H2SO4-,NH4HS2O7
395
Образующиеся побочные продукт^ реакции NH4HSO4, NH4HS2O7 и H2S2O7 дают при взаимодействии с водой (NH4)2S2O7 и H2SO4, приводя к обрыву цепи по следующим уравнениям:
2 NH4HS2O7 zH2O (NH4)2 S2O7 + 2 H2SO4
^2О7 + H2O->2 H2SO4
? NH4HSO4 (NH4)2 S2O7 + H2O
При определенных концентрациях переносчиков цепи скорость реакции соответствует скорости взрыва. Эта критическая концентрация переносчиков цепи не достигается, если карбамида в смеси мало. Цепное течение реакции сопровождается быстрым расходованием реагентов, обрывом цепей и падением скорости процесса до нуля.
Скорость сульфирования карбамида определяется по скорости выделения диоксида углерода из смеси. Реакция проводится в про
Рис. 164. Схема установки для изучения скорости реакции сульфирования карбамида
бирке, соединенной с газометрической установкой (рис. 164). Для исследования употребляются 1—6 М растворы карбамида в серной кислоте.
Последовательность выполнения работы. Пробирку 2 с холодным свежеприготовленным раствором карбамида (навеску карбамида рассчитать на 3 мл серной кислоты, пл. 1, 84) установить в термостат 1 при заданной температуре. За начало реакции считать момент, когда раствор примет температуру термостата. Температуру определять термометром 3. В этот момент включить секундомер и не выключать его до конца опыта. Газ из пробирки 2 пропускать для осушки и охлаждения через барботер 4 с серной кислотой, далее охладить и освободить его от капель кислоты в трубке 5 со стеклянной ватой, и направить в сосуд 6, из которого газ выпускать в атмосферу через калиброванный капилляр, защищенный снизу стеклянной ватой 7.
Небольшое избыточное давление в сосуде 6 зависит от скорости газового потока через капилляр. Манометрическим устройством 8 396
измерять скорость выделения газ\т. е. скорость реакции (измерения производить через каждые 15 с^ча во время максимальной скорости реакции чаще). Чтобы избежать случайных резких перепадов скорости потока, манометрическое устройство снабжено капилляром 9. Шкалу манометра отградуировать в миллилитрах в 1 с по пропусканию определенного объема газачза известное время при постоянном давлении. Манометр заполнить подкрашенным насыщенным раствором NaCl. Начальные измерения скорости выделения газа в течение первых 2—3 мин непригодны для\асчета (нагревание реагирующей смеси, установление режима течения газа), но необходимы для более точного выполнения последующих измерений и общей оценки опыта. Наибольшее внимание следует уделить измерениям в период максимального нарастания скорости реакции. Результаты опыта следует представить в виде таблицы по образцу:
Навеска карбамида на 3 мл H2SO4, г . . .; концентрация раствора карбамида, моль/л . . .; температура опыта, °C . . .
Номер измерения
Показания секундомера, с Время от начала реакции, мни Высота столба манометрической жидкости, см Скорость выделения СО2, мл/с lg w
Константа нарастания <р
По полученным данным: 1) вычертить график зависимости реакции от времени. Если исследуется несколько концентраций, то для всех вычертить такие кривые; 2) построить график = по скоростям реакции с восходящей ветвью кривой w = 3) оп-
ределить по графику lgw = f(Q константу нарастания ср как тангенс угла наклона полученной прямой к оси t (ф/2,3 = tg а); 4) представить результаты в виде графика если исследуется ско-
рость реакции для ряда концентраций карбамида. Значения скоростей найти по зависимости w = f(t) через одинаковый для всех концентраций промежуток времени от начала реакции.
Работа 4. Исследование кинетики радикальной полимеризации метилметакрилата
В работе исследуется кинетика радикальной полимеризации в массе (в блоке). Это — один из способов практического осуществления полимеризации, когда процесс проводится в конденсированной фазе в отсутствие растворителя. Длительность полимеризации 30 мин. Полная полимеризация мономера приводит к образованию монолита (блока), имеющего форму сосуда, в котором проводился процесс.
397
В качестве инициатора полимеризации используется перекись бензоила, которая распадаетей по непрочной связи О—О, давая два свободных радикала—^ерекисный и фенильный:
С6Н5СО-Ог6-СОС6Н5->СбН5СОО-4-СО2+С6Н5
Оба радикала на^нают цепь.
Реакцию роста цепи в общем виде можно выразить уравнением А(М)* + М-А(М)*+1
для метилметакрилата:
f СНз .О у СНз уО
R \ . сн2 — с — с — о — сн3;я + сн2 = с — с — о — сн3->
/ СИз у
->R \ — СН2 — С — С — О — СНз/п+1 где R — один из радикалов, образующихся при разложении перекиси бензоила.
Последовательность выполнения работы. Предварительно ознакомиться с катетометром, поскольку в работе используется дилатометрический метод анализа, т. е. за ходом процесса следят по изменению объема реакционной смеси.
Для работы использовать готовый раствор перекиси бензоила в метилметакрилате концентрации 0,005 г/мл; 100 мл этого раствора и 50 мл чистого метилметакрилата в течение 30 мин продуть аргоном или азотом. Предварительно продутый инертным газом дилатометр наполнить приготовленным раствором метилметакрилата с помощью шприца с длинной иглой так, чтобы мениск слегка возвышался над шариком дилатометра. Закрыть дилатометр пробкой и поместить в термостат, предварительно установленный на температуру 60° С. Через 5 мин зафиксировать уровень мениска с помощью катетометра. Последующие измерения выполнять через каждые 3 мин в течение 30 мин. Аналогичные измерения провести с растворами метилметакрилата при концентрациях перекиси бензоила 0,004, 0,003 и 0,002 г/мл. Эти растворы готовить из исходного раствора перекиси бензоила в метилметакрилате, разбавляя его по расчету чистым метилметакрилатом.
Результаты измерений занести в таблицу по образцу:
Температура заполнения дилатометра, °C . . температура опыта, °C . . .;
объем исходной смеси, мл . .
Время измерения по часам
Показания катетометра
1g
398
Ввести обозначения: \
аур — средняя скорость полимеризации, вычисленная по уравнению \
wp = (XVIЛ 2)
где А/ — время между последовательным^ измерениями; Av7 — изменение объема реакционной массы за время A/; v — первоначальный объем реакционной массы, измеренный при комнатной температуре (температуре заполнения дилатометра^ v7— первоначальный объем при температуре опыта, который рассчитывается по уравнению v7— u[l+dM(Tn—Гзап)], где dM=0,001 мл/^рад — коэффициент расширения метилметакрилата; Тп — температура полимери-_ \ 1 зации; Тзап — температура при заполнении дилатометра; — —
Рм
1 .
— — — разность удельных объемов мономера и полимера, рп, рм — Рп
плотности полимера и мономера; рп=0,937 г/см3 и рм=1,19 г/см3.
Подсчитать пять-шесть средних скоростей реакции по уравнению (XVI. 12) (при одной и той же концентрации инициатора) для разных измерений в течение 30 мин и найти из них среднюю величину w ip. Аналогично рассчитать значения средних скоростей реакции при других концентрациях инициатора (й>2р, й>зр и w<p). По полученным значениям средних скоростей при разных концентрациях инициатора построить график lgw=/(lgcHH) (син откладывать по оси абсцисс). Тангенс угла наклона полученной прямой к осн абсцисс равен порядку реакции по инициатору. В соответствии с уравнением (XVI.10) ьУр=&Эфф[Син]'/2, т. е. 1g®p=lg^^+0,51gсин. Проверить, соблюдается ли это уравнение, а следовательно, и механизм полимеризации. Определить константу скорости процесса полимеризации йЭфф (по графику как отрезок, отсекаемый полученной прямой на оси ординат).
Внимание! Никогда не пользоваться перекисью бензоила в сухом виде — опасно! Она способна к самопроизвольному разложению со взрывом при механическом воздействии — трении, ударе. Температура ее разложения 70—80° С (разлагается с выделением СОг). Использовать только растворы перекиси бензоила в спирте (растворимость 1,2 г в 100 г спирта), в хлороформе (растворимость 26,8 г в 100 г СНС1з) или суспензию ее в воде.
ГЛАВА XVII
КИНЕТИКА ГЕТЕРОГЕННЫХ ПРОЦЕССОВ
Физико-химические процессы, в которых участвуют вещества, находящиеся в разных фазах системы, называются гетерогенными. К гетерогенным относятся процессы: испарения, растворения твердых тел и газов, кристаллизация. Многие химические взаимодействия также являются гетерогенными — восстановление оксидов
399
металлов, электрохимические и Каталитические реакции, протекающие на поверхности твердых /тел. Особенность гетерогенных и гетерогенно-каталитических р^оцессов состоит в том, что акт химического превращения вещер^в происходит на поверхности раздела фаз системы. Особое энергетическое состояние поверхностных атомов или молекул, их геометрическое расположение обусловливает специфические кинетрйеские закономерности этих реакций. Поэтому скорость гетерогенных реакций зависит от площади поверхности раздела фаз и ее р^ироды. Гетерогенные процессы являются многостадийными. Трк, например, в гетерогенно-каталитических процессах выделяют пять стадий: диффузия реагирующих веществ к поверхности; адсорбция исходных веществ реакции; химическое превращение на поверхности; десорбция продуктов реакции; диффузия продуктов реакции от поверхности в объем.
Скорость гетерогенного процесса в целом зависит от скорости отдельных стадий и их соотношения. Возможен случай, когда скорость только одной из них окажется меньше скорости других стадий. Тогда в стационарных условиях эта стадия будет определять скорость и кинетические закономерности всего процесса. Такую стадию называют лимитирующей. Если лимитирующими являются первая и пятая стадии процесса, то скорость этого процесса зависит от диффузии; поэтому кинетические процессы такого типа называются диффузионными процессами.
1. ДИФФУЗИЯ
Диффузией называется перемещение вещества в результате хаотического теплового движения его частиц. Направление и интенсивность диффузии определяются градиентом концентраций дс/дх илщ точнее, градиентом химического потенциала Таким обра-
зом, движущей силой диффузии является стремление системы к термодинамическому равновесию путем выравнивания химического-потенциала при соответствующем перераспределении вещества.
Рассмотрим диффузию в изотропной среде, одномерную для стационарного и нестационарного потоков вещества. Потоком вещества / называется количество вещества i, перемещающегося через единицу площади S за единицу времени /. Движущая сила потока Fi выразится
Рi = —— I 1 j . (XVII. 1>
\ дх }f
Градиент потенциала (или обобщенная сила Fi) приводит к диффузионному перемещению частиц вещества с некоторой скоростью wir которая связана с движущей силой потока Fi соотношением
wz = (XVII.2>
где Bi — подвижность частиц вещества. Поток I, возникающий в однородной системе под действием силы Г/, равен средней скорости
400
частицы, умноженной на число частиц (или молей) в единице объема: • \
Л = Bi^ci
(XVII.3)
где Ci — число частиц (или молей) в единцпе объема.
В соответствии с (XVII. 1) величина потока определяется уравнением \
(XVII. 4>
Знак «—» указывает на то, что поток вещества i имеетнаправление, противоположное тому, в котором величина (дуц/дх^ положительна. Так как практически проще судить о потоке по градиенту концентраций (величина, зависящая от градиента химического потенциала), то тип потока обычно характеризуется особенностями градиента концентраций. Стационарным потоком называется такой поток, при котором градиент концентраций — величина постоянная
дс \ кс
дх jf Дх
= const
(XVII. 5>
и изменения концентрации в данном сечении со временем нет
(XVII. 6>
Нестационарным потоком называется поток, при котором градиент концентраций — величина переменная
/ дс
дх
const
(XVII. 7)
и концентрация со временем меняется
(XVII. 8)
Диффузия описывается двумя дифференциальными уравнениями Фика. Первое уравнение
второе уравнение
( дс \ д2с\ [ д! \
— — = - — , (XVII.10)
\ dt 1х \дх% }t \ дх Jt
где D — коэффициент диффузии — величина, показывающая количество вещества, которое при градиенте концентраций, равном единице, переходит по нормали в единицу времени через единичное сечение поверхности. Коэффициент диффузии имеет размерность (длина) 2/время. Коэффициент диффузии зависит от природы среды и диффундирующего вещества, а также термодинамических параметров процесса. При 0° С коэффициент диффузии в газах имеет обыч-
401
мо порядок Ю"1 см2 * */с, в жидкос/ях 10-5 см2/с, в твердых телах 1О~20 см2/с. Однако различие в/значениях для некоторых веществ, диффундирующих в массе другого вещества, могут достигать 10— 12 порядков. Так, например, коэффициенты диффузии атомов азота и водорода в железо/ при 20° С составляют соответственно 1,5-10"5 и 8,8-10~17 см2^/ Коэффициент диффузии зависит о массы и размеров молекул рёществ, участвующих в процессе диффузии, а также от температуры и вязкости среды. При данной температуре средняя кинетическая энергия молекул различных газов одинакова,
ти2]2 = const. (XVII.11)
Но средняя скорость теплового движения молекул различных газов при одинаковой температуре различна, причем она тем больше, чем меньше масса молекул (или молекулярная масса вещества). Но чем больше скорость теплового движения молекул, тем больше и скорость диффузии, а следовательно, и больше коэффициент диффузии вещества. Чем меньше масса молекул того вещества, в котором происходит диффузия, тем меньше при столкнове
нии с его молекулами меняются скорость и направление движения молекул диффундирующего вещества.
С повышением температуры растет средняя кинетическая энергия молекул и средняя скорость их теплового движения. Чем выше температура, тем больше коэффициент диффузии данного вещества. Зависимость D от температуры описывается уравнением
D = Dtf~ElRT9 (XVII. 12)
где Dq — предэкспоненциальный множитель; Е — энергия активации диффузии. Энергия активации диффузии — эта та минимальная избыточная (по сравнению со средней) энергия, которой должна обладать частица, чтобы перемещаться среди своих «соседей». Предэкспоненциальный множитель характеризует величину диффузии при высоких температурах, когда экспонента становится близкой к единице. При диффузии в твердых телах находится в пределах от 1 до 10~9 см2/с, а Е составляет обычно 90—300 кДж/моль.
Для некоторых растворов изменение концентрации в пределах 5—90% растворенного вещества приводит к изменению D более чем на три порядка. В области концентраций, где молярная доля xt составляет 0—0,1, коэффициент диффузии практически постоянен, но если он несколько изменяется, это изменение не превышает 10%. Таким образом, без грубой ошибки можно принять, что при постоянной температуре в разбавленных растворах коэффициент диффузии является постоянной величиной.
2. КИНЕТИКА ПРОЦЕССА ИСПАРЕНИЯ ЖИДКОСТИ
Скорость испарения жидкостей определяет процессы сушки, упа-
ривания, перегонки и т. п. Скорость испарения определяется ско-
ростью диффузии. На поверхности конденсированной (жидкой или
402
твердой) фазы всегда происходят\процессы перехода молекул и& конденсированной фазы в газовую ^испарение или возгонка) и из газовой фазы в конденсированную (конденсация). Из жидкости в пар переходят молекулы, которые находится в поверхностном слое: жидкости и обладают кинетической энергией, достаточной для преодоления сил взаимодействия с окружающими молекулами. Распределение молекул по энергии зависит толь^ц от температуры, поэтому при определенной температуре в поверхностном слое данного вещества долю молекул, имеющих энергию, достаточную для преодоления сил взаимодействия, можно считать постоянной. Следовательно, и скорость перехода молекул из жидкости в пар wQ при данной температуре постоянна. Скорость конденсации о>к зависит от концентрации (парциального давления) пара над жидкостью. Скорость испарения измеряют количеством жидкости, испарившейся в единицу времени:
= — wK.
В вакууме скорость испарения максимальна, так как скорость перехода молекул из пара в жидкость равна нулю: wH — 0 и ^n=w0~ Если объем свободного пространства над жидкостью замкнут, то по мере испарения жидкости в пространстве над ней растет давление ее паров. С появлением над жидкостью ее паров скорость испарения уменьшается, а скорость конденсации увеличивается. Когда скорости обоих процессов сравняются (аук=ау0), между жидкостью и паром установится динамическое равновесие. При этом количество жидкости будет оставаться постоянным во времени и видимая скорость испарения ее будет равна нулю.
Диффузия, даже в газах,— процесс относительно медленный. Это объясняется тем, что каждая молекула диффундирующего газа испытывает огромное число соударений и перемещается по очень сложной траектории, длина которой несоизмеримо велика по сравнению с расстоянием, проходимым молекулой в направлении диффузии. Вследствие небольшой скорости процесса диффузии лишь небольшая часть молекул, вырвавшихся из жидкости, успевает удалиться от ее поверхности на значительное расстояние. В тонком слое газа над самой поверхностью жидкости накапливаются молекулы испаряющегося вещества. Парциальное давление паров в этом слое растет до тех пор, пока не станет почти равным давлению насыщенного пара.
Для определения коэффициента диффузии экспериментально создают такие условия, в которых процесс испарения жидкости и диффузия ее паров в тот или иной газ будет протекать стационарно. Характерной особенностью стационарного процесса является то, что его скорость, а также состояние системы в любой ее точке не г- дс д%с
зависит от момента времени.-Тогда из -= D (с)--- следует при
di д
dc/dt—Q (стационарный режим): £) (с)= 0. Полагая в усло-Ч дх?
4@3
у
виях опыта £>(c)=const, получают выражение <52с/<5х2=0, которое применяют при предельных граничных условиях опыта.
Стационарный процесс диффузии легко рассчитать. Так, для описания скорости испар^ия жидкости в вертикальной цилиндрической трубке, у верхнего среза которой поддерживается постоянное парциальное давление паров р0 (изобарно-изотермические условия: сумма парциальных давлений инертного газа и пара постоянна), Стефан получил уравнение
hAhpRT
ID =
• ЛШр1п[(р — Ро)/(Р —р5)] *
(XVII .13)
где h — расстояние от поверхности жидкости до среза капилляра; ДА— изменение высоты уровня жидкости в результате испарения за время t\ р — плотность жидкости; 7? — универсальная газовая постоянная; М — молекулярная масса жидкости; р — общее давление в газовой фазе над жидкостью; ps —давление насыщенного пара жидкости при данной температуре.
Чем больше скорость испарения, тем больше и охлаждение поверхности жидкости и тем значительнее разница в температурах поверхностного слоя жидкости и остальной ее массы. Скорость процесса перехода вещества из жидкости в пар wQ и парциальное давление паров ps в слое, прилегающем к поверхности жидкости, будут соответствовать температуре ее поверхностного слоя, а не температуре основной массы жидкости. Температуру поверхности жидкости практически измерить трудно. Обычно ее не измеряют и считают равной температуре основной массы жидкости; однако это допустимо только тогда, когда скорость испарения невелика. Если скорость испарения значительна, то охлаждением поверхности нельзя пренебрегать, так как ошибка измерений достигает ±20% и более. Исходя из молекулярно-кинетической теории, Максвелл вывел уравнение для коэффициента диффузии
___!_______т/ kZ. (_L + _L
2-1Й2 («1 Ч-л2) <з2 зг \ т\ т2
(XVII. 14)
где Гц и п2 — потоки первого и второго газов, молекула/см3; mi и т2 — массы молекул каждого из газов; о — кинетический диаметр (сумма радиусов молекул первого и второго газов); k — константа Больцмана. В уравнении (XVII.14) не учтена температурная поправка Сезерленда. Для расчета коэффициента диффузии применяют также эмпирические формулы, например формулу Джиллиленда
0,0043Г3/2
“ л,1/3 _j_ 7?/з\2
(XVII. 15)
где гц и v2:~ молярные объемы диффундирующих молекул веществ в жидком состоянии при температурах кипения; М\ и М2 — молекулярные массы участвующих веществ; 0,0043 — коэффициент, зависящий от выбранных единиц измерений.
404
3. КИНЕТИКА ПРОЦЕССА РАСТВОРЕНИЯ
Растворение твердых веществ в жидкостях является сложным процессом и состоит из двух стадий. Перв^ стадия есть переход молекул или ионов, образующих кристаллическую решетку растворяемого вещества, из твердой фазы в жидкий раствор. Молекулы или ионы, находящиеся на поверхности кристалла,- обладают наибольшей кинетической энергией, и амплитуда их колебаний около положения равновесия является наибольшей; удалившись от соседних частиц кристаллической решетки, они приближаются к ближайшим молекулам растворителя. При этом силы, удерживающие данную ' частицу в кристаллической решетке, могут настолько уменьшиться, а силы взаимодействия этой частицы с молекулами растворителя настолько возрастают, что она покидает кристаллическую решетку и связывается с молекулами растворителя (сольватируется). Число частиц, переходящих в раствор с единицы площади поверхности кристалла в единицу времени, определяется частицами, обладающими достаточно большой для такого перехода кинетической энергией, и при постоянной температуре является постоянным. Скорость этого процесса очень велика и количество частиц растворенного вещества, находящихся в растворе вблизи поверхности кристалла, будет быстро увеличиваться. Частицы растворенного вещества в растворе сталкиваются с молекулами растворителя, направление движения при каждом столкновении меняется и движение их приобретает хаотический характер.
Многие из таких частиц будут возвращаться к поверхности кристалла. Если кинетическая энергия их в этот момент будет достаточно велика, чтобы оторваться от сольватирующихся молекул растворителя и приблизиться к поверхности кристалла, то силовое поле поверхности кристалла может эти частицы удерживать и они снова будут занимать места в кристаллической решетке растворяемого вещества. Скорость процесса возвращения частиц растворенного вещества из раствора в кристаллическую решетку прямо пропорциональна их концентрации в растворе и будет расти по мере накопления растворенного вещества в растворе. Когда скорости процессов перехода вещества из кристалла в раствор и из раствора в кристалл сравняются, концентрация вещества в растворе перестанет изменяться — раствор станет насыщенным и установится равновесие между двумя фазами. Если объем раствора велик, то такое растворение установится не скоро. Лишь тонкий слой раство-' ра, непосредственно прилегающий к поверхности кристалла, очень быстро станет почти насыщенным, в более же удаленных слоях рас-твора концентрация будет меньше или равна нулю (если растворение производится в чистом растворителе). При этом неизбежно возникает процесс диффузии, в результате которого растворимое вещество будет распространяться от поверхности кристалла в глубь раствора.
Процесс диффузии протекает гораздо медленнее, чем процессы межфазовых переходов, и скорость процесса растворения в боль-
405
шинстве случаев определяется скоростью диффузии. Действительно, слой раствора, граничащий с поверхностью кристалла, практически насыщен, дополнительно раствориться может только такое
Рис. 165. Распределение концентраций при растворении с перемешиванием
количество вещества, которое за то же время продиффундирует из пограничного слоя в глубь раствора.
Допустим, что кристалл растворяемого вещества, площадь поверхности которого S, погружен в ненасыщенный раствор этого вещества. Пусть объем раствора будет равен V, а концентрация растворенного вещества в растворе с. Через некоторое время прилегающий к поверхности кристалла слой раствора почти насытится и его концентрация станет практически равной максимальной сн. Из этого слоя вещество диффундирует в глубь раствора. Обозначим через б расстояние от поверхности кристалла, где концентрация максимальная сн, до ближайшей точ-
ки, в которой концентрация раствора еще не изменилась и остается равной концентрации исходного раствора с. Это расстояние называется толщиной диффузионного слоя (рис. 165). При перемешивании жидкости некоторый слой, прилегающий к поверхности твердого тела, остается в относительном покое. Градиент концентрации будет равен
дс —- с
дх Ъ
(XVII. 16)
Скорость растворения обычно количественно характеризуют значением производной от концентрации растворяемого вещества в растворе по времени:
^расг —
дс di
(XVII. 17)
Если в начальный момент растворение проводилось в чистом растворителе и концентрация растворенного вещества была равна нулю, то концентрацию вещества в растворе в любой момент рассчитывают как частное от деления количества растворенного вещества на объем раствора:
c—n/V. (XVII. 18)
При этом объем раствора остается практически постоянным. Таким образом, скорость растворения можно выразить
дс 1 дп
dt V dt
406
Поскольку скорость растворения определяется скоростью диффузии и равна ей, то dnldt можно определи^ по закону Фика
отсюда
(XVII. 19)
где k — константа скорости растворения; &=£>£/(V6).
Для определения константы скорости растворения необходимо проинтегрировать уравнение (XVII.19):
k = [1/0 In (сн/(сн “ О] • (XVII.20)
Интегрирование уравнения Щукарева — Нернста (XVIL19) осуществляется легко при условии, если константа k не меняется во времени. Между тем из уравнения (XVII. 19) видно, что k зависит от коэффициента диффузии растворяемого вещества, от площади поверхности, на которой происходит процесс растворения, от объема раствора и толщины диффузионного слоя б. Чтобы уравнение (XVII.20) точно соблюдалось, необходимо выполнение следующих условий: 1) температура во время опыта должна оставаться постоянной; 2) объем раствора должен оставаться постоянным; 3) площадь поверхности растворяющегося кристалла не должна заметно меняться за время опыта; 4) толщина диффузионного слоя б также должна оставаться постоянной.
Чем выше температура, при которой проводится опыт, тем больше коэффициент диффузии D и константа скорости растворения k. Температура влияет не только на константу скорости растворения k, но и на растворимость вещества, определяемую концентрацией насыщенного раствора сн. Чаще всего растворимость значительно увеличивается с повышением температуры. Однако для некоторых веществ, например для CaSO4, Li2SO4 и др., повышение температуры уменьшает растворимость. При этом скорость растворения в отличие от константы скорости растворения может почти не зависеть от температуры или даже уменьшается с ее повышением.
Толщина диффузионного слоя б зависит главным образом от того, находится ли раствор в покое или перемешивается, а также ют того, каков режим (ламинарный или турбулентный) и какова скорость движения жидкости по отношению к поверхности кристалла. В спокойном растворе толщина диффузионного слоя максимальна. Чем интенсивнее перемешивание, тем тоньше слой раствора, прилегающий к неподвижной поверхности кристалла и остающийся в покое благодаря сцеплению молекул растворителя с поверхностью кристалла. В этом неподвижном слое перемешивание отсутствует и растворяемое вещество распространяется от слоя, прилегающего к кристаллу, к его наружной поверхности только в результате диффузии. Чем тоньше этот слой, тем больше градиент
407
концентраций в нем и тем больше, в соответствии с законом Фика, скорость диффузии и скорость растворения.
В первом приближении можно считать, что скорость растворения прямо пропорциональна скорости перемешивания в степени п, причем п=2/з.
4. КИНЕТИКА ТОПОХИМИЧЕСКИХ РЕАКЦИЙ
Реакции типа Атв^Втв+СГаз, у которых химическое взаимодействие не осложнено диффузионными процессами на поверхности, называют топохимическими. Для топохимических реакций характерно нарастание скорости реакций в начальные моменты процесса
и достижение ею предельного значения с последующим постепенным спадом до нуля. Внешним признаком топохимических реакций служит S-образный вид кинетической кривой (рис. 166). Для топохимических реакций выражение
Рис. 166. Кинетические кривые топохимических реакций:
1 — зависимость глубины разложения вещества от времени; 2 — зависимость скорости реакции от времени
скорости реакции через концентрации непригодно, так как процесс идет не в объеме, а на поверхности раздела фаз. Поэтому принимают за скорость реакции изменение во времени степени превращения а вещества (глубины превращения);
w = —, (XVII.21) d t
где а= (М)—^)/Af0; Af0, N—исходное и текущее количества вещества на поверхности.
На рис. 166 приведены кривые топохимических реакций, участки которых соответствуют отдельным реакциям разложения твердых веществ с образованием газообразных продуктов. На кривой /
участок а соответствует быстрому начальному выделению газа, чему способствует десорбция газа, физически адсорбированного на поверхности вещества. При этом иногда происходит и начальное поверхностное разложение вещества. Участок b — это период ин
дукции, в котором реакция почти совсем не идет или идет очень медленно. После индукционного периода следует участок с ускорения, продолжающийся до точки аг-, перегиба кривой /, в которой глубина превращения часто достигает значения 0,5 и меньше. Участок d соответствует замедлению реакции, приближающейся к завершению. В целом можно сказать, что функция a==f(f) является сигмоидной кривой, характерной для автокаталитических реакций или для реакций, в которых конечный продукт образуется через
408
промежуточные стадии (последовательные реакции). На кривой 2 отчетливо видно нарастание скорости реакции, достигающей максимума В момент
Топохимические реакции начинаются обычно не на всей поверхности исходного твердого вещества, а на отдельных ее участках— зародышах ядер кристаллизации новой фазы (продукта), которые образуются на поверхности кристалла. Ядра кристаллизации появляются раньше всего в областях дефектов кристаллической решетки. В простейшем случае это могут быть, например, выходы дислокаций на поверхности, вакансии, расположение атомов (ионов) в междоузлиях и т. п. Таким точкам, или элементам кристаллической решетки, свойственна повышенная энергия Гиббса и, следовательно, более высокая реакционная способность. Зародыши ядер называют также потенциальными центрами образования ядер. На рис. 167 представлена схема распространения реакции в кристалле* Около поверхностных зародышей начинается рост сферических ядер.
В течение реакции увеличивается поверхность ядер, т. е. поверхность раздела фаз исходного вещества и твердого продукта, в связи с чем реакция ускоряется. Когда ядра сливаются, площадь поверхности достигает наибольшего значения, а скорость реакции — максимума. Далее наступает замедление. Эта картина может значительно усложнить
ся растрескиванием кристаллов, связанным, в первую очередь, с различием плотностей образующейся и исходной фаз. Для объяснения первоначального ускорения реакции была предложена модель линейных разветвленных цепей (а=ехр (&£)]• Реакция начинается на поверхности в отдельных точках, откуда растут «тонкие нити» продукта. Рост продолжается до встречи нити с одним из разрывов кристаллической решетки, что происходит только на поверхности, а в точках разветвления начинается рост компактных объемных ядер.
Кинетика топохимических реакций зависит от закономерностей образования и роста ядер. В связи с этим рассмотрим некоторые закономерности образования ядер.
Так, допустим мгновенное образование ядер в начальный момент реакции. Это означает, что в кристалле с самого начала существует ряд точек — зародышей, связанных с разупорядоченным строением, выходом дислокаций на поверхность, вакансиями, скоплениями ионов в.междоузлиях и т. п. Допускается неизменное исходное число зародышей. Кинетический закон должен определяться формой образующихся из зародышей ядер и скоростью их роста.
Количество прореагировавшего вещества в начальной стадии реакции при образовании сферических ядер будет 4/з^о^3Я^3> так
Начальные центры зарожд. реакции
Рис. 167. Развитие и слияние сферических ядер твердого продукта при топохимической реак* ции
409
что получается кубическая зависимость a—f (/). Но она не удовлетворяет опытным данным.
Второе допущение состоит в том, что образование зародышей идет по первому порядку. Вещество содержит ряд особых малых областей, химически менее устойчивых, чем остальная масса кристалла. Это еще не зародыши, но они могут перейти в зародыши путем некоторого активационного процесса. Если устойчивость вещества во всех точках одинакова, то вероятность активации каждой точки можно положить равной
/ \ vexp — •—— ,
\ RT )
где v — некоторый частотный множитель; AG5^— энергия активации Гиббса. Тогда скорость образования ядер будет пропорциональна числу областей, етде неактивных в данный момент времени. Если в начальный момент (/ —0) имеется всего No таких областей и потенциальные центры еще не захватываются продуктом, то число ядер к моменту t в первом порядке станет в каждый момент равным
= wo (1 - (XVII.22)
где k — константа скорости образования зародышей.
Функцию (XVII.22) можно разложить в ряд и ограничиться первым членом разложения. Тогда количество ядер, образовавшееся к моменту t, будет пропорционально времени:
= (XVII.23)
Решая выражение (XVIL23), получаем для начальной стадии разложение а~£4.
Приемлемое уравнение, описывающее кинетику топохимических реакций, было выведено Б. В. Ерофеевым. Оно было получено на основании вероятности взаимодействия молекул данной системы и не связано ни с какими предположениями об истинном механизме реакции:
t
Р=1-е 0 , (XVII.24)
где Р — вероятность, что молекулы данной системы прореагируют к моменту времени /; Р'— вероятность, что молекулы прореагируют в течение времени от t до Если система состоит из большого числа молекул, то Р можно приравнять к доле прореагировавших молекул а:
t - (Р'м
а=1-е 0 . (XVII.25)
Определив вероятность взаимодействия молекул в интервале времени от t до Н-d/, можно получить кинетическое уравнение соответствующего процесса. Если, например, считать вероятность взаимо-
419
действия величиной постоянной, то из выражения (XVII.25) получается уравнение первого порядка. Вероятность взаимодействия молекул в момент времени от t до пропорциональна суммарной площади поверхности реакции, т. е. Р=const -S. Поверхность реакции изменяется во времени, если принять, что ее изменение следует закону
S = const
(XVII. 26)
где т — эмпирическая константа, тогда
(XVII. 27)
где k' — константа.
Подставляя значение Р из уравнения (XVII.27) в (XVII.25), получаем
-мт
(XVII. 28)
где k —константа скорости.
Если площадь поверхности реакции постоянна (S = const), то из выражения (XVII.28) можно вывести уравнение первого порядка: для S = const—e~kti; для S = const-72 а— 1—e~ht2\ для S= = const-/3 а—1—e~kt3 и т. д.
На рис. 168 приведена кривая, рассчитанная по уравнению (XVII.28) (для конкретного процесса) в предположении, что за один
и тот же промежуток времени прореагировало 35% исходного вещества. Если поверхность реакции постоянна, то не наблюдается нарастания скорости. Показатель степени в уравнении (XVII.28) находят графическим путем. Для этого уравнение (XVII.28), дважды логарифмируя, преобразуют в равенство вида
Рис. 168. Зависимость скорости топохимических реакций от времени
lg I — lg (1 — а)] = Ig й + m 1g Л
(XVII.29)
По тангенсу угла наклона полученной прямой определяют т. При помощи уравнения (XVII.28) рассчитывают константу скорости k. Зная константы скорости k\ и k% для двух
температур (7\ и 72) при постоянном т, можно рассчитать энергию активации Е топохимического процесса по уравнению Арре
ниуса
k = Ae~E/RT,
(XVII.30)
или
(XVII.31)
411
Работа 1. Изучение кинетики испарения жидкостей и определение коэффициента диффузии паров жидкости в воздух (и другие газы) методом увлечения
Коэффициент диффузии паров в воздух определяют методом увлечения. Он заключается в измерении скорости испарения жидкости из трубки диаметром 3—6 мм, у конца которой пропускается непрерывно поток газа, диффузию в который изучают. Скорость га-
К насосу
Рис. 169. Схема установки для изучения кинетики испарения чистых жидкостей методом увлечения:
1а — колонка с СаС12; 16 — колонка, со щелочью; 2 — буферная склянка; 3 — реометр; 4 — прибор для измерения; 5 — ультратермостат; 6 — буферная склянка с манометром; 7 — маностат
Рис. 170. Определение высоты диффузии онного пространства
зового потока должна быть такой, чтобы у среза трубки обеспечивалась нулевая концентрация паров исследуемого вещества. Схема установки для измерения коэффициента диффузии паров в воздух приведена на рис. 169.
Последовательность выполнения работы. 1 Измерить катетометром (до 0,1 мм) рас-стояние Ло от среза трубки до конца имеющегося в трубке стеклянного столбика (рис. 170). Налить в тщательно вымытую сухую трубку для испарения необходимое количество исследуемой жидкости. Вставить трубку с жидкостью в чистый и сухой прибор, помещенный в стеклянную рубашку. Закрыть прибор крышкой, предварительно слегка смазав шлиф вазелином. Настроить термостат точно на нужную температуру
(устройство термостата описано на с. 424). Включить насос и при
помощи зажима установить разность уровней жидкости в реометре,
которая должна равняться скорости воздушного потока
412
300 см3/мин. Выдержать прибор 15 мин при заданной температуре, навести катетометр при помощи окуляра и приступить к измерениям (работа с катетометром описана на с. 422). Измерить расстояние от верхней точки поверхности стеклянного столбика до нижней точки мениска А. Отметить время, когда было сделано измерение, атмосферное давление — по барометру и разницу между атмосферным давлением и давлением внутри прибора — по манометру.
Опыт вести в течение 2 ч, повторяя измерение каждые 15 мин. Результаты измерений записать в таблицу по образцу:
Скорость испарения исследуемого вещества . . . при температуре, °C . .
/го, см . . цена деления окуляра а, см . . р, мм рт. ст. . .
л
р, г/см3 . . молекулярная масса вещества . . .
Время, мин
Число делений по окуляру
мм рт. ст.
Время, мин
Число делений по окуляру
Р. мм рт. ст.
W-ж
деление= см; Л, см . . .;
Д2 деление = см; рСр, мм рт. ст. . . .
Д/z, см . . .; Д/, с . . .
Введение жидкости в узкие трубки пипеткой без особых предосторожностей приводит к размазыванию части жидкости по стенкам трубки. Это может внести в измерения большую ошибку, так как испарение будет происходить не только с поверхности мениска, но и со стенок трубки, и скорость снижения уровня не будет соответствовать скорости стационарного процесса диффузии паров от уровня жидкости до верхнего среза трубки. Чтобы избежать при наполнении трубки размазывания жидкости по стенкам, необходимо (рис. 171) в трубку для испарения 1 вставить воронку 2, конец которой должен быть на 10 мм выше уровня жидкости. Трубка воронки в конце имеет сужение с внутренним диаметром 1—1,5 мм. Пипеткой 3 с оттянутым капилляром и резиновым баллончиком (кусок мягкой каучуковой трубки, закрытой с одной стороны пробкой) набрать достаточное количество жидкости (0,5—3,0 мл), следя за тем, чтобы на конце пипетки не осталось капли. Капилляр пипетки ввести через воронку в трубку. Конец капилляра должен оказаться на 10—12 мм ниже конца воронки. Медленно выдавить из пипетки исследуемую жидкость и, когда уровень жидкости достигнет желаемой высоты (на 2—3 мм выше стеклянного столбика,
413
впаянного в трубку), вынуть пипетку, предварительно сняв каплю. Удалить из трубки воронку и вставить трубку в прибор для изме
рения коэффициента диффузии.
При измерении А/г нужна очень большая точность, так как ис-
парение из трубок идет очень медленно, и эта величина выражается десятыми и даже сотыми долями миллиметра. Поэтому для из-
мерения Ah пользуются только окуляром, которым снабжен катето-
метр. Окуляр позволяет измерять расстояние 2— 6 мм с точностью до 0,002—0,008 мм. Чтобы снижение уровня в трубке можно было измерить окуляром, необходимо иметь в центре трубки метку, которая уменьшалась бы в поле зрения окуляра одновременно с нижней точкой мениска жидкости. Такой меткой служит вершина стеклянного столбика, впаянного в центре трубки.
Прежде чем начать опыты с данной трубкой, необходимо измерить катетометром расстояние h^ от вершины столбика до среза трубки. После этого налить в трубку исследуемую жидкость и поместить ее в прибор для измерения коэффициента диффузии и создать желаемые условия. Навести одно из неподвижных делений шкалы окуляра на вершину столбика, а подвижную часть — на нижнюю точку мениска жидкости. Измерить расстояние от столбика до мениска Д1 в делениях окуляра. Измерение Ai повторить через равные промежутки времени восемь— десять раз за время опыта. Опыт закончить, когда А уменьшится по сравнению с первоначальной величиной не менее чем на 50 малых делений окуляра, отсчитываемых по барабану. Определить понижение уровня жидкости в трубке А/i (см), равное изменению А за время опыта,
Рис. 171. При* ДЙ = ДХ —Д2.
способление
для заполнения Чтобы рассчитать коэффициент диффузии, необ-трубок ходимо Ah выразить в сантиметрах. Эту величину следует умножить на цену деления окуляра, выраженную в сантиметрах. Среднее значение высоты диффузионного пространства h для данного опыта рассчитать по соотношению
fa — h$ —
где Ai — расстояние от вершины столбика до нижней точки мениска в начале опыта, см; А2 — расстояние от вершины столбика до нижней точки мениска в конце опыта, см; h — средняя высота диффузионного пространства, по которой вычисляют коэффицент диффузии, см.
414
Коэффициент диффузии рассчитать по уравнению
ЛА&р7?Г76О
(XVII.32>
ЛШ1,0133-106р2,303 1g-
где,/? — универсальная газовая постоянная; р — плотность, г/см3; Д£—время испарения, с; р— давление в системе, мм рт. ст.; ps — давление насыщенного пара при температуре опыта; 1,0133х X106/760 — коэффициент пересчета давления из мм рт. ст. в дин/см2.
Уравнение (XVII.32) применимо лишь для стационарного процесса, для которого скорость испарения жидкости постоянна. Если построить график (рис. 172), отложив по оси абсцисс время, а по* оси ординат — расстояние от вершины столбика до мениска Д, то
для стационарного процесса и незначительно различающихся значений Д все точки должны лечь на прямую. Однако в действительности при проведении опыта в том или ином интервале времени условия испарения менялись и процесс не был стационарным. Так, например, в начале опыта после введения жидкости в трубку пространство над жидкостью еще не содержит паров. В начальном периоде скорость испарения будет велика, причем , она будет уменьшаться со временем, пока не достигнет величины, более или менее отвечающей стационарному процессу. Поэтому точки на графике, соответствующие отсчетам
Рис. 172. Изменение скорости испарения вещества в трубке
Д, выпадают и лежат выше
прямой. Всякое нарушение стационарности процесса отражается на кривой зависимости Д от времени искривлением или изломом. Таким образом, построение графика позволяет выявить ту часть опыта, для которой процесс диффузии можно считать стационарным, а измеренное значение коэффициента диффузии достаточно* надежным. Полученный коэффициент диффузии Dp рассчитан при давлении р, при котором проводилось измерение. Чтобы пересчитать коэффициент диффузии на атмосферное давление, следует воспользоваться соотношением
D 760
^760 Р
(XVIE33>
где £>760 — коэффициент диффузии при нормальном атмосферном давлении.
Для расчета £>760 формулу (XVII.32) можно записать
35,63/гДД d £/ £>760 = -----------
----— P — Ps
(XV1I.34>
415
Коэффициент диффузии возрастает с туры:
повышением темпера-
(XVII. 35)
Экспериментальные данные по коэффициентам диффузии паров и газов показывают, что п имеет значение от 1,5 до 2,5. Если пока-
затель в уравнении (XVII.35) для данного вещества неизвестен, то его принимают равным 2.
В работе необходимо: 1) измерить коэффициент диффузии данного вещества при двух заданных температурах; 2) определить по-
казатель п и коэффициент диффузии при нормальных условиях. Все полученные данные записать в таблицу по образцу:
Вещество . . .; коэффициент диффузии . . .
Температура, К мм рт. ст. Время At с h, см Ай, см D, см2/с ^теор> см4/с О a3 ja о Я м ГО ” Q п
"ь
Работа 2. Изучение кинетики испарения жидкости и определение коэффициента диффузии паров жидкости в воздух методом адсорбции
Если пары исследуемого вещества хорошо сорбируются активи-
рованным углем, то для измерения коэффициента диффузии можно применять адсорбционный метод. Этот метод основан на измерении скорости испарения из трубки жидкости, пары которой адсорбируются углем, расположенным над трубкой. Схема прибора для изме-
рения коэффициента диффузии паров адсорбционным методом приведена на рис. 173.
Последовательность выполнения работы. Для определения коэффициента диффузии необходимо измерить катетометром (описание катетометра см. на с. 422) расстояние hQ от среза трубки до верхней части стеклянного столбика. Налить в трубку 1 (рис. 173) необходимое количество исследуемой жидкости и вставить ее в прибор, укрепленный в стеклянной рубашке 2. Затем вставить в трубку 1 пробирку 3, изготовленную из мед-
Рис. 173. Схема установки для изучения кинетики испарения чистых веществ адсорбционным методом
416
ной сетки, и наполнить ее высушенным и просеянным активированным углем. Трубку наполнить натронной известью и хлоридом кальция для поглощения паров воды и диоксида углерода и вставить в крышку прибора; шлиф крышки смазать вазелином, закрыть прибор. Настроить термостат 4 (устройство термостата см. на с. 424) точно на желаемую температуру и выдержать прибор при этой температуре 15 мин. Измерить расстояние от верхней части стеклянного столбика до нижней точки мениска Д, отметить время» когда был сделан отсчет, и записать атмосферное давление (дальнейшее проведение измерений см. работу 1, с. 412).
Работа 3. Изучение кинетики растворения малорастворимых веществ
Рис. 174. Схема установки для изучения кинетики растворения
В работе определяют константу скорости растворения сульфата кальция при заданных температуре и скорости перемешивания раствора, а также растворимость CaSO4. Скорость растворения определяют по данным измерений удельной электрической проводимости водного раствора CaSO4 в различные моменты времени.
На рис. 174 представлена схема прибора для проведения опыта. Реактор 1 вместимостью около 300 мл для измерения электрической проводимости (см. с. 279) имеет в притертой крышке 3 со впаянными в нее платиновыми электродами 2 дополнительную пришлифованную пробку 4. Эта пробка кончается стеклянной палочкой с крючком для крепления пластинки растворяемого вещества. Сосуд 1 снабжен водяной рубашкой 5, через которую циркулирует вода из ультратермостата для поддержания постоянной температуры раствора. Сосуд 1 при работе ставится на столик магнитной мешалки 7, якорь 6 перемешивает раствор. Электрическую проводимость раствора измеряют мостиком Кольрауша (описание см. на с. 278). К опыту приступают после ус-
тановки термостата на указанную температуру (устройство термостата см. на с. 424). Во время нагревания термостата подготовляют и собирают реактор 1.
Последовательность выполнения работы. В реактор 1 налить 100—120 мл дистиллированной воды, включить магнитную мешалку 7 и закрыть крышкой 3. Затем укрепить пластинку растворяемо
14—2083
417
го вещества и вставить пробку 4 в сосуд так, чтобы пластинка находилась над поверхностью воды и не касалась ее.
Во время этих приготовлений следует очень осторожно обращаться с платиновыми электродами: не касаться их руками и во избежание дополнительной проверки емкостного сопротивления сосуда не нарушать их взаимного расположения. По истечении 30 мин (время, в течение которого вода принимает температуру термостата) прибор включить в схему мостика Кольрауша для измерения электрической проводимости. Включить генератор звуковой частоты и в магазине сопротивлений установить сопротивление 1000—2000 Ом. Опыт выполняют два экспериментатора: один измеряет электрическую проводимость, другой по секундомеру записывает время и показания реохорда. Секундомер включить сразу же после погружения в воду пластинки растворяемого вещества. Этот момент будет временем начала опыта. Через 5 мин сделать первое измерение сопротивления раствора и результаты записать в таблицу по образцу:
Растворяемое вещество . . .; температура опыта, °C . . .; частота перемешивания, об/мин . . сопротивление магазина, Ом ...
Время измерения по часам
Время от начала растворения
Плечи реохорда мм
1000—£2
Так как в течение первого часа электрическая проводимость сильно возрастает, первые десять измерений необходимо проводить через каждые 5 мин. Последующие измерения проводить через 10—20 мин. Измерения продолжать до тех пор, пока электрическая проводимость не достигнет постоянной величины, т. е. пока не будет достигнуто насыщение раствора растворяемым веществом. Для этого требуется около 6 ч; время, необходимое для выполнения опыта, менять в зависимости от температуры, условий перемешивания и природы растворяемого вещества.
Константу скорости растворения вычислить по уравнению реакции первого порядка. Поскольку для труднорастворимых солей удельная электрическая проводимость в первом приближении пропорциональна концентрации соли в растворе ut&Ct, уравнение (XVII.20) можно переписать в виде
н —
(XVII. 36)
где «н — удельная электрическая проводимость насыщенного раствора; и/ — электрическая проводимость к моменту времени/.
418
Поскольку Я = = -i- ср, где р — удельное сопротивление;
Ф — постоянная сосуда; I — расстояние между электродами; S — . площадь поверхности электрода; то
__ 2,3 %н _ 2,3 2,3
Следовательно, расчеты можно проводить по уравнению , 2,3 1//?н
(XVII.37)
t
Результаты опыта следует представить в виде графиков в координатах \/Rt—t и lg[(l/^H) — (1/^01—Последний график использовать для определения йСр по тангенсу угла наклона полученной прямой к оси /. Константу йСр сравнить с &Ср, вычисленной как среднее арифметическое из значений констант скоростей, рассчитанных для каждого момента времени.
Работа 4. Изучение кинетики термического разложения перманганата калия
В данной работе следует ознакомиться с одним из методов изучения топохимических реакций.
Первый вариант» Работу проводят на установке, схема которой приведена на рис. 175. Метод основан на измерении объема кисло-
рода, выделившегося при разложении перманганата:
2 КМпО4 == К2М11О4 + М11О2 4* О2
Последовательность выполнения работы. Установить термостат (см. с. 424) на определенную температуру. Взять навеску 0,2—0,4 г перманганата. Кран 2 измерительной (градуированной) бюретки 3 поставить в положение «1» и уровень Жидкости в уравнительном сосуде 4 привести к 0. Навеску перманганата калия в металлическом бюксе при помощи тонкой проволоки подвесить на крючок колпачка 5 и осторожно опустить в реакционную пробирку 1. Кран 2 измерительной бюретки поставить в положение «2» так, чтобы реакционная пробирка сообщалась с бюреткой, и тотчас включить
Рис. 175. Схема установки для изу-чения скорости топохимических реакций
14*
419
секундомер. Это время принимается за начало реакции. Измерения объема выделившегося кислорода проводить приведением жидкости в бюретке 3 и уравнительном сосуде 4 к одному уровню каждые 5 мин при температуре выше 235°С через каждые 2 мин. Опыт прекратить, когда объем в течение 10—15 мин перестанет изменяться. По окончании опыта бюкс извлечь из реакционной пробирки, охладить и вновь взвесить, определяя потерю в массе Лтоп в результате реакции. Полученное значение Дтоп сравнить с теоретическим значением A^Teop, рассчитанным при предположении, что реакция идет до конца.
На основании опытных данных рассчитать по уравнению (XVII.29) константы k и т, в котором a—vtlv^ где vt—-объем кислорода в данный момент времени v™—-конечный объем выделившегося кислорода. Рассчитать величину а и 1—а, записать в таблицу по образцу:
Навеска перманганата т . . Дтоп . . ДтГеор . . .; температура,
Время реакции
—lg (1—а)
lg [—Ig( 1—ct) ]
lg k
Построить график lg[—lg(l—а)] =f(lgt), из которого найти константу т и рассчитать lg k.
Второй вариант. Данную работу можно провести с помощью автоматической газовой бюретки (рис. 176). Выделяющийся в результате разложения перманганата калия кислород через отверстие в реакционной пробирке 1 и трехходовой кран 2 поступает в измерительную бюретку 5 и манометр 3. В результате этого уровень жидкости в левом колене манометра 3 повышается и замыкает контакты 4 электромагнитного реле. Электромагнитное реле включает реверсивный электродвигатель 8, который, вращая винт 6, опускает гайку 7 и связанный с ней уравнительный сосуд 9. Уровень жидкости в бюретке понижается, давление в ней падает и контакты 4 размыкаются. В дальнейшем процесс периодически повторяется. Связанное с уравнительным сосудом перо 10 вычерчивает на вращающемся барабане 11 кривую объем выделившегося кислорода — время. Таким образом, измерение объема выделившегося кислорода осуществляется при постоянном давлении, равном атмосферному.
Порядок выполнения работы. Установить термостат на определенную температуру. Трехходовой кран 2 установить в положение,
420
при котором измерительная бюретка 5 и реакционная пробирка 1 сообщаются с атмосферой. Включить автоматическую газовую бюретку и с помощью тумблеров «уровень» установить уровень жидкости в измерительной бюретке 5 на нуль, после чего прибор выключить. Закрепить на барабане И миллиметровую бумагу и привести в рабочее положение перо 10, Установить реакционную пробирку в термостат. В специальном металлическом бюксе отвесить 0,2—0,4 г КМпСЦ. Металлический бюкс с навеской пермангана
та калия при помощи тонкой проволоки подвесить на крючок пробки реакционной пробирки и (осторожно!) опустить в реакционную пробирку /, плотно закрыв пробку. Трехходовой кран 2 установить в положение, при котором реакционная пробирка сообщается только с измерительной бюреткой, и тотчас же включить автоматическую бюретку. Следить за ходом процесса по вычерчиваемой кривой. Опыт прекратить, когда объем в течение 10—15 мин перестанет изменяться. Для этого необходимо выключить газовую бюретку, трехходовой кран 2 установить в положение, при котором реакционная пробирка и измерительная бюретка сообщаются с атмосферой, извлечь реакционную пробирку из термостата и выключить термостат. После охлаждения про-
Рис. 176. Схема установки с автоматической газовой бюреткой для изучения скорости топохимических реакций
бирки осторожно открыть пробку, извлечь и взвесить бюкс и определить потерю массы КМпСЦ Дтоп в результате реакции. Получен-
ное значение Дтоп сравнить с теоретическим значением Д/^теор> рассчитанным из предположения, что реакция идет до конца. Ис
пользуя полученную кривую, рассчитать значение а в различные моменты времени:
а = vt/v„.
Определив а, (1—а), lg[-lg(l-a)]=f(lgO,
lg[—lg(l—а)] и lg t, построить график по которому определить постоянную п
421
и Ig/C Построить график в координатах Av/At—t. Расчетные величины представить в виде таблицы по образцу:
Время от начала опыта, мин 1g i vt Л — —— 1—а —2,3 1g (1—а) lg[—lg(l— а)] п ftCK
Работа с катетометром. Катетометр состоит из вертикального штатива на треножнике, измерительной каретки, зрительной трубы н отсчетного микроскопа (рис. 177).
Измерительная каретка /, несущая зрительную трубу 3 и отсчетный микроскоп 2, перемещается по колонке иа роликах. Грубое перемещение измерительной каретки по вертикали осуществляется от руки при открепленном винте 6.
Рис. 177. Схема катетометра
Рис. 178. Угловой биссектор
О 2 4 6 8 10
Рнс. 179. Масштабная сетка окуляра
точнее с помощью микрометрического винта 5 при закрепленном винте 6. Зрительная труба 3 укреплена на. каретке. Фокусировка трубы на выбранную точку объекта производится вращением маховичка 4. Перед измерением открепить винт 6, поднять измерительную каретку на уровень точки объекта. Установить окуляр зрительной трубы на резкое изображение сетки, а фокусирующую линзу — на резкое изображение объекта. После этого произвести точную наводку
422
зрительной трубы на выбранную точку объекта: в вертикальной плоскости с помощью винта 5 при закрепленном винте 6. Сетка зрительной трубы имеет перекрестие, правый горизонтальный штрих которого выполнен в виде углового биссектора. При наводке трубы выбранная точка объекта должна располагаться в правой половине сетки точно посередине углового биссектора (рис. 178). После этого снимают первый отсчет по масштабной сетке, затем, перемещая каретку по колонке, наводят зрительную трубу на вторую точку измеряемого объекта и снимают, второй отсчет. Разность между двумя отсчетами дает величину отрезка. В поле зрения отсчетного микроскопа (рис. 177) одновременно видны изображения двух штрихов миллиметровой шкалы, обозначенные крупными цифрами, и масштабная сетка. Индексом для отсчета целых миллиметров служит нулевой биссектор. На рис. 179 штрих «16’2» прошел нулевой биссектор, а ближайший штрих еще не дошел до нулевого биссектора. Отсчет будет равен 162 мм плюс отрезок от штриха «162» до нулевого биссектора. В этом отрезке число десятых долей миллиметра обозначено цифрой последнего пройденного биссектора десятых долей миллиметра, в данном случае цифрой 2. Отсчет сотых долей миллиметра производится в горизонтальном направлении сетки там, где миллиметровый штрнх находится точно посередине биссектора. На рис. 179 миллиметровый штрих находится на четвертом делении сетки, что соответствует 0,04. Окончательный отсчет 162,24 мм.
Оптическая схема катетометра состоит нз зрительной трубы и отсчетного микроскопа с осветительной системой (рис. 180). Изображение штрихов милли-
Рис. 180. Оптическая схема катетометра:
/ — объектив зрительной трубы; 2 — масштабная сетка; 3, 6 — окуляры; 4 — фокусирующая лииза; 5 — сетка; 7 — сменные насадочные линзы; 8, 14 — светофильтры; £ —входной конденсор; 10 микрообъектив отсчетного микроскопа;
11 — призма; /2 — зеркало; 15 — источник света
метровой шкалы проектируется на плоскость масштабной сетки, установленной в фокальной плоскости окуляра отсчетного микроскопа. Масштабная сетка раз-* делена в вертикальном и горизонтальном направлениях иа десять частей. От* счетный микроскоп установлен таким образом, что десять горизонтальных биссекторов сетки укладываются между двумя штрихами миллиметровой шкалы, следовательно, каждому биссектору в вертикальном направлении соответствует 0,1 мм. В горизонтальном направлении десятая часть биссектора равна 0,01 мм. Тысячные доли миллиметра оцениваются на глаз. Измерение отрезков {расстояние между двумя точками) производится с помощью зрительной трубы и отсчетного микроскопа путем сравнения измеряемой длины с миллиметровой шкалой.
423
Рис. 181. Контактный термометр с магнитной регулировкой:
А — магнитная головка; Б — колпачок; В — термометр с двойной шкалой; 1 — магнит; 2 — корпус с клеммами; 3 — крышка; 4— стеклянная оболочка контактного приспособления; 5 — микровинт с якорем; 6 — стопорное кольцо; 7 — гайка с контактором; 8 —- подпятник с контактирующей спиралью; 9— шкала; 10, 11 — капилляры;
12 — ртуть
Рис. 182. Схема реле
Зрительная труба и отсчетный микроскоп смонтированы в одной каретке; перемещая каретку по колонке вдоль миллиметровой шкалы, а также вращая колонку вокруг вертикальной оси, осуществляют визирование на выбранные точки объекта; соответствующие отсчеты определяют через окуляр отсчетного микроскопа по шкале и масштабной сетке. Длины вертикальных отрезков определяют как разность соответствующих отсчетов по шкале.
Устройство термостата. Многие работы в области физической химии проводят при постоянной температуре. Термостатирование необходимо для всех работ по кинетике, электрохимии, химическому и фазовому равновесию и др.
Существуют различные конструкции термостатов в зависимости от темпера*
424
турного интервала, длительности термостатирования и допустимых пределов колебаний температуры. Наиболее простым и распространенным является водяной термостат с устройством для автоматической стабилизации температуры. Он состоит из резервуара для воды, электрического нагревателя, мотора с мешалкой, контрольного термометра, контактного термометра с реле. Схема контактного термометра с магнитной регулировкой контакта приведена на рис. 18 L Этот термометр включается в схему реле при помощи двух винтовых клемм, расположенных на колпачке Б. Настройка термометра на заданную температуру производится вращением магнитной головки А, При вращении вннта против часовой стрелки контактор 7 опускается в капилляр 10. Соприкосновение его со ртутью замыкает реле и выключает нагреватель. Температура, на которую настраивается термометр, определяется по положению нижнего обреза овальной гайкн 7 на верхней шкале. Установленный контактор закрепляется стопорными винтами на магнитной головке. При повышении температуры ртуть нижнего резервуара в контактном термометре при соприкосновении с контактной нглой замыкает цепь электромагнита в реле (рнс. 182). При этом якорь 3 притягивается к магниту 2 и размыкает цепь нагревания посредством лампы 1. При охлаждении термостатирующей жидкости уровень ртути контактного термометра опускается и нагреватель включается.
ГЛАВА XVIII
АДСОРБЦИЯ
1. ТЕРМОДИНАМИКА АДСОРБЦИИ
Силы, действующие на поверхности твердого тела, ненасыщены. Поэтому всякий раз, когда свежая поверхность подвергается действию газа, на ней создается более высокая концентрация молекул газа, чем в объеме собственно газовой фазы. Такое преимущественное концентрирование молекул на поверхности называется адсорбцией. Прочность связи молекул адсорбата с поверхностью адсорбента, а также величина адсорбции могут сильно меняться от системы к системе. Процессы адсорбции можно разделить на два основных типа: физическую адсорбцию и хемосорбцию. Физическая адсорбция вызывается силами молекулярного взаимодействия, к которым относятся силы взаимодействия постоянных и индуцированных диполей, а также силы квадрупольного притяжения. Хемосорбция обусловлена перераспределением электронов взаимодействующих между собой газа и твердого тела с последующим образованием химических связей. Физическая адсорбция подобна конденсации паров с образованием жидкости или процессу сжижения газов, а хемосорбция может рассматриваться как химическая реакция, протекание которой ограничено поверхностным слоем адсорбента. Типы адсорбции различают по нескольким критериям: 1) по теплотам адсорбции. Количество выделившейся в процессе физической адсорбции теплоты, отнесенное к одному молю адсорбированного вещества, обычно изменяется в пределах 8—40 кДж. Как правило, теплота хемосорбции превышает 80 кДж/моль; 2) по скорости протекания процесса. Поскольку физическая адсорбция подобна процессу сжижения газа, то она не требует активации и протекает очень быстро. Хемосорбция же, аналогично большинству хи
425
мических процессов, должна требовать активацию; 3) по скорости десорбции. Энергия активации десорбции физически адсорбированных соединений (при условии низкой пористости адсорбента) редко превышает несколько кДж/моль. Энергия активации десорбции хемосорбированных соединений обычно превышает 80 кДж/моль, причем она почти всегда больше теплоты хемосорбции или равна €Й; 4) по интервалу температур, в пределах которого протекает адсорбция. Физическая адсорбция реализуется только при температурах, близких к температуре кипения адсорбата (при заданном давлении). Хемосорбция протекает при температурах, существенно превышающих температуру кипения адсорбата при соответствующем давлении; 5) по степени специфичности взаимодействия газ — твердое тело. Процессы хемосорбции, как и любые химические реакции, имеют специфический характер. Это значит, что какой-то газ хемосорбируется данным твердым веществом при некоторых условиях, но из этого не следует, что в аналогичных условиях тот же газ будет хемосорбироваться другим твердым телом, имеющим такую же степень чистоты поверхности. Возможность хемосорбции регулируется химическими потенциалами взаимодействующих веществ и вероятных поверхностных продуктов.
Физическая адсорбция фактически представляет собой процесс, в течение которого происходит конденсация газа, причем адсорбат может образовывать на поверхности инертного твердого тела один или большее количество слоев. Количественно адсорбция может быть выражена с помощью нескольких величин: 1) величиной а, представляющей собой количество адсорбтива, находящегося в объеме адсорбционного слоя, отвечающего единице массы адсорбента. Эту величину обычно измеряют в моль/г; 2) величиной а, показывающей количество адсорбированного вещества, приходящегося на единицу поверхности адсорбента, т. е. она характеризует поверхностную концентрацию адсорбтива. Размерность а моль/м2 и ммоль/см2; 3) величиной, введенной Гиббсом и представляющей собой избыток количества молей адсорбтива в объеме поверхностного слоя площадью 1 см2 по сравнению с числом его молей в том же объеме, если бы у межфазной границы не происходило изменения концентрации адсорбтива. При малых концентрациях адсорбтива гиббсовская адсорбция Г близка к поверхностной концентрации а; при больших концентрациях адсорбтива величина Г отличается от а. Если по тем или иным причинам концентрация адсорбтива в поверхностном слое меньше его концентрации в объеме, величина Г отрицательна, а само явление называется отрицательной адсорбцией.
Адсорбцию характеризуют: 1) зависимость количества адсорбированного вещества а от температуры при постоянных равновесных давлениях или концентрациях с; кривые на графиках a=f(T) при р = const называются изобарами, а при const— изопикнами адсорбции; 2) зависимость равновесного давления (или концентрации) от температуры при постоянном количестве адсорбированного вещества; кривые на графиках p = f(T) и c=f(T) при a = const на
426
зываются изостерами; 3) зависимость количества адсорбированного вещества а от равновесного давления (или концентрации) при постоянной температуре; кривые на графиках a=f (р) или a = f (с) при 7 = const называются изотермами адсорбции.
Лэнгмюр описал динамическое равновесие между молекулами адсорбата, находящимися в газовой фазе при давлении р, и адсорбированными молекулами в поверхностном слое при доле занятых центров поверхности, равной 0. Он вывел уравнение изотермы:
8 = ~Рк -, (XVIII. 1)
1 4- Ьр
где b = kjk^\ k\ и k-i — констаты скорости адсорбции и десорбции соответственно: д = а/аоо — степень заполнения. В основе вывода уравнения Лэнгмюра лежали следующие допущения: 1) адсорбированные частицы связаны с определенными локализованными центрами на поверхности адсорбента; 2) каждый центр может присоединять только одну адсорбирующуюся частицу; 3) энергия адсорбированных частиц на всех центрах поверхности одинакова и не зависит от присутствия или отсутствия других адсорбированных частиц на соседних центрах. Изотерму Лэнгмюра можно преобразовать:
1 1 1
я “ ажЬр ’
С увеличением степени заполнения поверхности адсорбентом уменьшается теплота адсорбции. Тогда изотерма адсорбции описывается уравнением Фрейндлиха
1
а — kpn (л > 1).
(XVIII. 2)
Бр.унауер, Эммет и Тейлер создали обобщенную теорию физической адсорбции. Они рассматривали процесс адсорбции как образование на адсорбенте многих молекулярных слоев, которые составляют общую толщину адсорбционной пленки и указывают, что при любом равновесии на адсорбенте имеются пленки различной толщины. Таким образом, на адсорбционные силы, исходящие от поверхности адсорбента, накладываются силы Ван-дер-Ваальса, действующие между молекулами адсорбированного вещества. На основании этого предположения выведено уравнение изотермы, называемое изотермой БЭТ:
-P-Ps, = (xvln• 3> а (1 — p/ps) атС amCps
где С — константа; ат — константа, соответствующая количеству вещества, адсорбированному 1 г адсорбента при мономолекулярном покрытии поверхности, моль; am=S/(o; ps — давление насыщенного пара; S — удельная площадь поверхности адсорбента; со — площадь, занимаемая 1 моль вещества на поверхности адсорбента в конденсированном мономолекулярном слое.
427
Чтобы вычислить изотерму адсорбции для различных паров по уравнению БЭТ, необходимо знать константы ат и С. Уравнение БЭТ позволяет предсказать количество адсорбированного вещества, если известны ат и С. Согласно теории БЭТ можно точно оценить величину площади поверхности, если изотерма адсорбции получена экспериментально и определены константы ат и С.
Итак, по полученным изотермам и по известной площади посадочной площадки (берут из справочника), занимаемой адсорбируемой молекулой, можно рассчитать удельные площади поверхности адсорбентов:
5уд = ^Ю-з.6,02-1023^10—20, (XVIII.4)
где 5Уд — удельная площадь поверхности адсорбента или катализатора, м2/г; s — площадь поверхности, занимаемая молекулой на адсорбенте.
Чтобы определить константы в уравнении БЭТ, необходимо экспериментальные данные обработать графически. Для этого строят график в координатах plps. Теплота адсорбции он-
«(1— PtPs) ределяется по экспериментальным данным, представленным в виде изостеры адсорбции, которая описывается уравнением
(XVIII. 5) А К / 1 1 2 /
Все приведенные закономерности применимы и для описания адсорбции веществ твердыми телами из растворов. Только вместо равновесного давления используется равновесная концентрация растворенного вещества.
2. КИНЕТИКА АДСОРБЦИИ
Скорость покрытия поверхности адсорбатом зависит от ряда факторов, к которым относятся энергия активации и частота столкновений. Когда молекула сталкивается с поверхностью, она может удержаться на поверхности только в том случае, если ее энергия рассеется с достаточно большой скоростью в термические колебания лежащей под ней решетки. Иначе молекула просто отскочит от поверхности или будет перекатываться по поверхности до тех пор, пока не дойдет до ребра кристаллической решетки, откуда она может вернуться в исходную фазу. Процесс столкновений с поверхностью, приводящих к адсорбции, называется вероятностью прилипания р:
скорость адсорбции молекул поверхностью
Р скорость столкновения молекул с поверхностью
Знаменатель этого выражения может быть рассчитан из кинетической теории, если известно давление (концентрация), числитель—из изменения скорости давления (концентрации). Значения
428
р меняются в широких пределах. Скорость адсорбции описывается уравнением
d w
(XVIII.6)
где w — количество вещества, адсорбированного за время t\ а и а — константы для каждой системы при определенной температуре. В интегральной форме уравнение (XVIII.6) имеет вид
1 *— !_ I н —.......
(XVIII.7)
где £0—1/(<ш). Уравнение (XVIII.6) выполняется, если зависимость lg(dw/dt) от w выражается прямой.
Работа 1. Статический метод изучения адсорбции
Скорости гетерогенных и гетерогенно-каталитических реакций пропорциональны площади поверхности раздела фаз. Поэтому удельная площадь поверхности катализаторов и сорбентов является их важнейшей характеристикой. Существуют методы определения адсорбции: статический и динамический.
Статический метод заключается в том, что адсорбент помещают в атмосферу газа или пара и после установления адсорбционного равновесия измеряют давление и количество поглощенного вещества. Адсорбция проводится в высоковакуумной установке, в которой из адсорбента перед опытом удаляются ранее адсорбированные газы.
В данной работе следует построить изотермы адсорбции толуола и определить удельную площадь поверхности катализатора статическим методом. Для определения удельной площади поверхности катализатора используют весы Мак-Бэна. Газ приводится в соприкосновение с адсорбентом и после установления равновесия отмечают показания манометра и количество адсорбированного газа при данном давлении на весах Мак-Бэна. Проведя такие измерения при различных давлениях, вычерчивают изотерму адсорбции. По изотерме адсорбции и поверхности, занимаемой адсорбированной молекулой вещества, определяют удельную площадь поверхности адсорбента.
Установка для изучения адсорбции (рис. 183) состоит из измерительной и насосной частей, которые могут быть отделены друг от друга краном. В измерительную часть входят адсорбционная трубка 4Ь, U-образный манометр S, манометрическая лампа 7 и гальванометр 6, буферный сосуд 5. Адсорбционная трубка общей вместимостью 600 см3 состоит из двух частей: головки с крючком 4а и нижней части, где расположены адсорбционные весы 4. Важнейшей частью адсорбционных весов является спиральная пружина 4с, изготовленная из молибденовой проволоки, отожженной в атмосфере водорода. Пружина оканчивается двумя крючками. Верхний крючок
429
I
пружины подвешен на стеклянном крючке. На нижнем крючке пружина несет корзиночку, содержащую адсорбент. Корзиночка массой 0,4 г сделана из тонкого стекла. Головка адсорбционной трубки жестко закреплена на стенде и присоединена к измерительной части прибора. Головка с остальной частью трубки соединена при. помощи шлифа.
Последовательность выполнения работы. Для проведения опытов предварительно нужно откалибровать пружинные весы. Калиб-
Рис. 183. Схема установки для определения
рование основано на измерении растяжения спирали при подвешивании корзиночки, а затем при добавлении в нее разновесов 0,01; 0,02; 0,05; 0,1 г и т. д. Удлинение спирали фиксируется катетометром при наложении разновесов и при снятии их. На основании полученных данных построить калибровочную кривую.
После этого 0,5—0,6 г угля, предварительно обработанного (обезгажен-ного), насыпать в корзиночку. Затем на весы надвинуть трубку и посред-
площади поверхности катализатора CTBOM шлифа соединить С
головкой. Крепление с головкой осуществляется при помощи двух пружин. После этого
прибор или систему вакуумировать, для чего осторожно открыть кран 2, а затем очень осторожно — кран 1 на вакуум-насосе, до установления постоянной массы адсорбента. Когда адсорбент обезга-жен, закрыть кран 2 и осторожно открыть кран 3 для обезгажива-ния жидкости, пары которой будут адсорбироваться. После обез-гаживания жидкости кран 3 закрыть и продолжать вакуумирование, пока термоманометр не покажет разрежения от 4 до 8 мВ; когда это давление установится, закрыть кран 1 и приступить к снятию показаний катетометра. Отметить показания катетометра, .соответствующие обезгаженному адсорбенту. По калибровочной кривой определить навеску обезгаженного угля.
Затем осторожно приоткрыть кран 3 так, чтобы давление повысилось по милливольметру на 0,5 мВ при закрытых кранах 2 и затем при закрытых кранах 1 и 3 открыть кран 2, т. е. соединить измерительную часть с адсорбционной и спустя 10—15 мин снять показания установившегося давления по милливольтметру и показания катетометра, а по калибровочной кривой рассчитать массу адсорбента после адсорбции при данном равновесном давлении. Разность между массой адсорбента после адсорбции и массой ад-
430
сорбента после обезгаживания указывает на количество адсорбированного вещества на данном количестве адсорбента при соответ* ствующем равновесном давлении. Затем определить количество ад-сорбированого пара (газа) при другом давлении, для этого, впуская газ порциями так, чтобы показание милливольтметра упало еще на 0,5 мВ. Опыт повторить.
Такие определения провести в интервале от самого большого разрежения до давления насыщения. Давление отсчитывать вначале по термоманометру через каждые 0,5 мВ, а затем снимать показания по ртутному вакуумметру через каждый 1 мм (по калибровочной кривой милливольты перевести в миллиметры ртутного столба). Полученные опытные данные записать в таблицу по образцу:
Навеска угля ...
Равновесное давление в системе
нВ
мм рт. ст.
Давление насыщенного пара при температуре опыта, мм рт. ст.
Колччество адсорбированного пара при соответствующем равновесном давлении, г
Количество адсорбированного пара 1 г угля
ммоль
р'р$ й ^-p/ps)
По экспериментальным данным построить график зависимости количества адсорбированного пара, выраженного в ммоль/г (а), от давления при T=const. Рассчитать удельную площадь поверхнос-сти твердого тела, для чего построить график зависимости—----
от p/pSj приняв площадь адсорбированной молекулы $ = 0,344 нм, константа ат выражена в миллимолях. При этом должна получиться прямая линия, которая соответствует уравнению БЭТ. Обозначив —— через р, a p/ps — через х, Щатс)—через В, a(l — p/ps)
(с—1)/(атс) — через Д', получим уравнение у = В-\-Кх. Полученная на данном графике прямая отсекает на ординате отрезок, который равен В, а тангенс угла наклона этой прямой к оси р/р3 будет равен /<.
Работа 2. Динамический метод изучения адсорбции f
Динамический метод заключается в пропускании сквозь слой адсорбента тока инертного газа-носителя, содержащего пары адсорбирующегося вещества, и измерения нарастания его концентрации в газе за слоем адсорбента. Одним из вариантов динамического метода определения величины адсорбции и удельной площади поверхности является проявительный метод, основанный на исполь
431
зовании уравнений нелинейной равновесной хроматографии. Метод ’Позволяет построить изотерму адсорбции по форме выходной кривой хроматографического пика. Согласно теории равновесной нелинейной хроматографии величину адсорбции а вычисляют по уравнению
с
f (t — to) d cf
c
(XVIII. 8)
z 1о0
Рис. 184. Графическое интегрирование хроматографической кривой:
Л — момент ввода пробы; 1 — хроматографический пик для недесорбирующегося вещества; 2 — хроматографический пик адсорбирующегося вещества; /0 — расстояние на ленте от момента ввода пробы до выхода неадсор-бирующегося газа
где w — объемная скорость газа-носителя; m — масса адсорбента; с — концентрация адсорбирующегося вещества; t — время выхода адсорбента из хроматографической колонки; to — время выхода неадсорбирующего-ся газа (метан, азот);
/ = (XVHI.9)
где I — расстояние на ленте от точки, соответствующей моменту to, до точки, соответствующей времени /; нл — скорость движения ленты.
Концентрацию вещества в газе-носителе определяют по сигналу детектора:
(XVIII.10)
где ц — постоянная детектора; п — отклонение пера самописца от нулевой линии.
Подставляя (XVIII.9) и (XVIII.10) в (XVIII.8), получаем
ni
a = (Z — Zg) d п. = ——(XVIII.11)
uAtn J 1 илт 1
о
где sni —площадь под кривой хроматографического пика на уровне rii.
На рис. 184 показана хроматограмма адсорбирующегося вещества. На оси абсцисс отложена длина I на ленте самописца, а по оси ординат — величина сигнала детектора. Заштрихованная площадь snl равна интегралу в уравнении (XVIII.И) и определяется методом взвешивания или с помощью планиметра. По значению 5 для различных значений рассчитывают величину адсорб-
* ** 4-
ции.
Парциальное давление вычисляют по концентрации вещества в газовой фазе:
р. = с22400/?ат , (XVIII. 12)
Где Ратм — атмосферное давление. Или с учетом уравнения (XVIII. 10) имеем
= ^22 400ратм1
(XVIII. 13)
Чтобы определить постоянную детектора ц, в поток газа-носителя ввести известное количество вещества Qa> измерить площадь Sa под хроматографическим пиком и рассчитать ее по формуле
(XVIIL14)
По рассчитанным значениям а и pt построить изотерму адсорбции и, применяя теорию БЭТ, определить удельную площадь поверхности.
Точность метода составляет 10—15% от общей площади поверхности.
Работа 3. Построение изотермы адсорбции бензола
и определение удельной площади поверхности катализаторов
методом проявительной хроматогра
ии
Для построения изотермы адсорбции и определения удельной площади поверхности используется хроматограф ХЛ-4, схема которого представлена на рис. 185.
Из баллона 1 через ^педуктоо 2 и вентиль тонкой регулировки 3 подается газ-носитель ’(Не, N2, Н2). Давление газа измеряют манометром 4. Поток газа проходит через испаритель 5, в который им-
пульсом вводится точное количество жидкого адсорбата (бензол, н-геп-тан, циклогексан). Жидкая проба испаряется и потоком газа-носителя вносится в хроматографическую колонку 6, заполненную адсорбентом, пло
щадь поверхности которого требуется определить. В хроматографической колонке происходит адсорбция вещества. Че
Рис. 185. Схема хроматографа
рез некоторое время, зависящее от величины и характера адсорбции, адсорбат выходит из колонки вместе с газом-носителем. Кон
центрация его в газе определяется с помощью детектора 7.
Из детектора газ-носитель проходит через ротаметр 9, с помощью которого определяется его объемная скорость. Сигналы детектора подаются на вход самопишущего потенциометра 10. Колонка и детектор помещены в термостат 8, с помощью которого
поддерживается постоянная температура в колонке в интервале от 40 до 300° С. В качестве детектора используют прибор, фиксирующий изменение теплопроводности газа-носителя за счет присутствия в нем второго компонента. Такие детекторы называются катаро-
15-2083
433
метрами (рис. 186). Катарометр состоит из двух камер с нагретыми вольфрамовыми нитями, по две в каждой камере. Через одну из этих камер (сравнительную) протекает газ-носитель, а через другую (измерительную)—газ, выходящий из колонки. Нагреваемые нити включены в мост Уитстона. Перед опытом через обе камеры
пропускают газ-носитель и с помощью реостата балансируют мост так, чтобы разность потенциалов в точках А и В была равна нулю. Если через камеру Si пропускать газ, выходящий из колонки, то
вследствие различной теплопроводности смеси газов и газа-носителя температура нитей в камере S2 изменяется и в соответствии с этим изменяется и их сопротивление. Баланс моста нарушается и на выходе возникает разность потенциалов, которая фиксируется на ленте самописца. Величина сигнала катарометра пропорциональна концентрации вещества в газе-носителе.
Для измерения скорости потока газа использовать пенный ротаметр (рис. 187). Основной частью ротаметра является калиброванная стеклянная трубка 2, снабженная штуцером 3 для ввода газа. К нижней части трубки присоединена резиновая груша 4 с водным раствором пенообразователя. Если с помощью груши поднять уровень жидкости несколько выше уровня штуцера, то газ захватывает часть жидкости, которая
Рис. 187. Пенный ротаметр
Рис. 186. Схема катарометра:
1 — источник питания; 2 — амперметр; 3 — реостат; 4 — мост Уитстона; Яь Я2 — сопротивления;
Si, S2 — камеры
образует тонкую «мыльную» пленку. Мыльную пленку улавливают баллоном 1. Определяя скорость движения этой пленки по трубке, можно рассчитать объемную скорость газа. Пенный ротаметр не создает дополнительного перепада, как это имеется у поплавковых ротаметров или капиллярных реометров.
Последовательность выполнения работы. Хроматографическую колонку, содержащую 2—3 г адсорбента, поместить в термостат хроматографа. Открыть вентиль 2 (см. рис. 185) на баллоне с газом-носителем 1 и с помощью вентиля тонкой регулировки 3 по
434
пенному ротаметру установить скорость .газа-носителя в пределах от 20—30 см3/мин. Оптимальная скорость газа зависит от характера пористости и навески адсорбента. Включить хроматограф и установить температуру термостата 60°С, а температуру испарителя 200°С.
После установления режима работы хроматографа ввести через испаритель с помощью хроматографического шприца 50—60 мкл воздуха, фиксируя момент введения пробы на ленте самописца. Затем в колонку ввести три различных объема бензола (например, 8, 12, 20 мкм). Каждый объем вводить трижды и взять для расчета пики, площади которых отличаются друг от друга не более чем на 5%. По площади одного из пиков для каждой пробы рассчитать
Рис. 188. Определение величины адсорбции и постоянной катарометра по хроматографическим кривым:
1 — хроматографический пик инертного газа; 2, 3 — хроматографические пики бензола; 0 — момент ввода пробы
по уравнению (XVIII.14) постоянную катарометра. Для этого вырезать хроматографический пик (рис. 188, пик 2) и взвесить его. Рядом вырезать квадрат площадью 25 см2, определить его массу и массу 1 см2 бумаги. Площадь пика рассчитать по формуле
Sa^ma/myiL, (XVIII.15)
где та — масса бумаги под хроматографическим пиком; туд — масса 1 см2 бумаги.
Скорость протяжения ленты самописца должна быть указана или определена в ходе опыта.
Количество бензола а в пробе определить по формуле
aa^va?/M, (XVIII. 16)
где va — объем пробы, мл; р —плотность бензола; М — молекулярная масса бензола.
Далее обработать другой пик пробы. На хроматограмме отложить от момента напуска пробы бензола длину /0, определяемую по времени выхода СбНб из колонки, и провести вертикальную линию (рис. 188), Отрезок линии от нулевой линии до вер
шины пика разделить на 5—7 частей и вырезать площади, ограниченные линией и хроматограммой, от точек по до Пь от до «2 и т. д. Площадь бумаги определять взвешиванием. По уравнению (XVIII. 11) рассчитать величину адсорбции, а по уравнению (XVIII.13)—парциальное давление бензола над адсорбентом. Величину адсорбции отнести к навеске адсорбента. Давление насыщенного пара бензола при температуре опыта найти по справочным таблицам.
Рассчитанные данные записать в таблицу по образцу:
Температура опыта, °C . . атмосферное давление . . объемная скорость газа, см3/мин . . скорость движения ленты, см/мин . . постоянная катарометра, моль/см4 . . давление насыщенных паров бензола . . .
Яр см
PlPs
а,
P^s
« В-p/ps1
По данным таблицы построить график согласно теории БЭТ и найти постоянные ат и с. Рассчитать удельную площадь поверхности твердого тела.
Работа 4. Изучение адсорбционного равновесия из водных растворов карбоновых кислот на активированном угле кондуктометрическим методом
В работе исследуют влияние концентрации растворенного вещества на величину адсорбции при постоянных температуре и количестве адсорбента, одном и том же растворителе (Н2О). Для исследования используют водные растворы карбоновых кислот, например щавелевой, янтарной, малеиновой, глутаровой и др. Величина адсорбции при достижении адсорбционного равновесия устанавливается методом измерения электрической проводимости растворов по времени.
Последовательность выполнения работы. Определить константу прибора (см. с. 279). Установить зависимость удельной электрической проводимости раствора органической кислоты (адсорбат) от разведения (концентрации). Удельную электрическую проводимость раствора измерить не менее, чем при 8—10 разведениях (см. с. 278).
Построить калибровочный график зависимости удельной электрической проводимости от концентрации адсорбата. График использовать для определения равновесных концентраций адсорбата при
достижении адсорбционного равновесия. Изучить кинетику адсорбции на активированном угле из раствора данной концентрации измерением удельной электрической проводимости, ее изменения с течением времени. На основе экспериментальных данных рассчитать постоянные изотерм Лэнгмюра и Фрейндлиха.
Адсорбционное равновесие изучать методом измерения электрической проводимости растворов электролитов на обычных компенсационных схемах, используя для этого открытые схемы с барабанным реохордом (см. с. 278).
В термостатированную ячейку, тщательно вымытую дистиллированной водой, налить 50 мл исследуемого раствора данной концентрации, погрузить платиновые электроды для измерения электрической проводимости, установить на контактном термометре термостата заданную температуру и выдержать ячейку в заданном температурном режиме не менее 10 мин при непрерывном перемешивании при помощи магнитной мешалки. Электроды подключить к схеме измерения и измерить сопротивление раствора. Последовательность разбавлений (не менее 8 раз) провести, отбирая 25 мл раствора пипеткой и этой же пипеткой, не ополаскивая ее, добавить 25 мл дистиллированной воды той же температуры. Тщательно перемешать раствор и измерить его сопротивление, которое пересчитать на удельную электрическую проводимость раствора х=ф//?. Полученные значения удельной электрической проводимости в зависимости от концентрации раствора нанести на график u = f(c).
Изучение адсорбционного равновесия провести в той же термостатированной ячейке при тех же условиях. После того как в растворе установится заданная температура, измерить еще раз сопротивление раствора и проверить его по калибровочному графику, внося соответствующие исправления. Затем в бюксе с крышкой быстро взвесить 0,5 г активированного угля и всыпать его в исследуемый раствор; при непрерывном перемешивании измерить сопротивление раствора через определенные промежутки времени (15— 30 с). Время отмечать по секундомеру. Постоянство значений сопротивления на реохорде Р-38 указывает на достижение адсорбционного равновесия в растворе данной концентрации. Подобные измерения провести для трех-четырех концентраций раствора при постоянной температуре. Полученные сопротивления пересчитать на удельную электрическую проводимость раствора и по калибровочному графику x = f(c) определить равновесные концентрации сравн адсорбата. Зная исходную концентрацию вещества в растворе с0, его начальный объем и количество адсорбента g, рассчитать количество адсорбированного вещества Г в ммоль/г по формуле (со~^равН)/(ЮОО^). Полученные значения нанести на график r = f(CpaBH). Полученную экспериментальную зависимость описать с помощью уравнения Лэнгмюра. Рассчитать постоянные Гоо и 6. Рассчитать теплоту адсорбции по уравнению
437
Полученные экспериментальные и расчетные данные занести в таблицы по образцам.
Зависимость удельной электрической проводимости от концентрации
Адсорбат . . адсорбент . . температура опыта, °C . . *
Концентрация адсорбата со, моль/л
Сопротивление раствора, Ом
Удельная электрическая проводимость, Ом-^см"1
Кинетика адсорбции
Температура опыта, °C . . .
Время, с
X, Cq, б? равн, Г,
Ом-1 -см-1 ммоль/л ммоль/л ммоль/г
Гсравн
/?, Ом
Константы адсорбционного равновесия
Адсорбат с0, моль/л
Адсорбент
Константа адсорбционного равновесия b
кН
аде
ГЛАВА XIX
КИНЕТИКА
ГЕТЕРОГЕННО-КАТАЛИТИЧЕСКИХ РЕАКЦИЙ
1. КАТАЛИЗ И КАТАЛИЗАТОРЫ
Катализ — это ускорение химических реакций веществами, которые регенерируются в конце превращения. Если наблюдается замедление реакции под действием веществ, в ней не участвующих, то этот процесс называется ингибированием. Каталитическое ускорение реакции достигается за счет изменения ее механизма. При этом катализатор входит в состав новых промежуточных продуктов, которые образуются и затем переходят в конечные вещества с большей скоростью, чем при отсутствии катализатора.
При реакции катализатор входит в соприкосновение, или контакт, с реагентами, поэтому катализаторы называют контактами, 438
а каталитические реакции — контактными. Катализаторами могут быть самые разнообразные вещества во всех трех агрегатных состояниях. Каталитическое действие могут оказывать примеси, пыль, поверхность стенок сосуда, промежуточные продукты реакции и т. п.
Если же катализуемая система и катализатор находятся_в разных агрегатньоГ состояниях, катализ называют'Тетерогенным. При гетерогеТном~катализе реакция ускоряется при протекании^ на границе фаз^ Механизм многих реакций ввиду их сложности еще недостаточно выяснен. Для различных химических реакций активны-ми являются катал2П^ррь1^разЖтаргс) химического состава. Так, например, оптимальный катализатор для окисления NH3 совершенно отличен от оптимального катализатора для окисления SO2.
При поисках подходящего катализатора для определенной реакции до сих пор руководствуются некоторыми эмпирическими правилами и в основном пользуются м,етодом_Д1ррД^ Каждая реакция, катализируемая твердым веществом, начинается с адсорбции или хемосорбции одного или нескольких участников реакции»
Тейлор постулировал наличие на поверхности катализаторов особых активных центров, на которых протекают химические реакции. Гипотеза Тейлора явилась первым шагом на пути к выяснению реальной структуры поверхности катализаторов. Но и эта гипотеза не дает ответа на существенный вопрос, почему именно определенный катализатор, и только он один, пригоден для ускорения определенной реакции.
Под действием различных ферментов или энзимов с исключительной избирательностью в животных и растительных организмах проходят многочисленные реакции. Ферменты являются органическими катализаторами белковой природы и обладают свойствами, переходными между гомогенными и гетерогенными катализаторами, приближаясь, однако, к свойствам гетерогенных. Ферментативные реакции называют микрогетерогенными.
Различные химические процессы всегда протекают в ограниченных емкостях: трубках, колбах, автоклавах. Поэтому реакция, протекающая гомогенно, может быть по существу гетерогенной вследствие каталитического влияния поверхности стенок. Чтобы не впасть в ошибку, определяют — влияют ли стенки на скорость реакции. Для этого в реагирующую систему вводят материал стенок в измельченном состоянии. Если ^скорость реакции при этом не изменяется, можно считать,,, дта испытание
можно проводить и другим способом? сравнивают скорости той же реакции в идентичных условиях либо в сосудах из разного материала, либо покрывая стенки данного сосуда слоем другого вещества (парафинирование, металлизация).
Катализированные и некатализированные процессы подчиняются одним и тем же законам и протекают лишь по термодинамически возможным направлениям, независимо от присутствия или отсутствия катализатора, отличаясь лишь скоростью достижения равновесия. Ускорение катализированной реакции может быть очень
439
значительным. Многие технически важные реакции протекают со столь малой скоростью, что без катализаторов они не имеют практического значения. В присутствии катализаторов они проходят быстро, этим и объясняется широкое развитие и практическое значение каталитической химии в технологии.
Роль катализаторов особенно велика потому, что они позволяют проводить многостадийные процессы как бы в одну стадию и в заданном направлении при более низких температурах, что дает экономию энергии и времени и подавляет вторичные процессы, снижающие выходы основных продуктов.
2. ГЕТЕРОГЕННЫЕ КАТАЛИЗАТОРЫ
В соответствии с механизмом протекающих реакций гетерогенные катализаторы подразделяют на следующие три группы: 1) ионные, под влиянием которых протекают реакции с ионным механизмом; 2) электронные, катализирующие гомолитические реакции; 3) бифункциональные, совмещающие ионный и электронный катализ.
1. Кислотно-основные катализаторы: а) оксиды некоторых металлов: А12О3, 'W2O3, ThO2:; б) нейтральные и кислые соли: Со3(РО4)2, СаНРО4, MgHPO4, а также природные синтетические алюмосиликаты (Al2O3)m* (SiO2)rt-(Н2О)Р; в) протонные и апротонные кислоты на носителях (Н3РО4 на А12О3 или кизельгуре, ВР3 на А12О3, гетерополикислоты) и ионообменные смолы, обычно представляющие собой сульфированные нерастворимые полимеры сетчатой (сшитой) структуры, например сополимер стирола с бутадиеном-1,3 или дивинилбензолом, фенолоформальдегидный полимер и др.
2. Комплексообразующие соли переходных металлов на носителях:
Cu^CI2, HgCI2, PdGI24-CuCI2, CuC = CCu, NiCI2.
Все катализаторы ионных реакций являются изоляторами или ионными проводниками электрического тока. Наиболее распространены катализаторы кислотного типа, являющиеся протонными (бренстедовскими) или апротонными (льюисовскими) кислотами.
Гетерогенные катализаторы гомолитических реакций всегда являются электронными проводниками тока или полупроводниками и включают следующие группы веществ.
1. Переходные металлы первой подгруппы (Си, Ag) и VIII группы (Fe, Ni, Со, Pt, Pd) периодической системы Д. И. Менделеева.
2. Оксиды металлов (MgO, LuO, CuO, Fe2O3, Cr2O3, WO3, MoO3, V2O5), сильфиды (WS3, M0S3) и смеси, содержащие один основной оксид с небольшими добавками других (модифицированные оксидные катализаторы).
3. Сложные оксидные и сульфидные соединения с соизмеримым соотношением компонентов, а также полупроводники: хроматы 440
(CuO-Cr2O3, ZnO-Cr2O3), вольфраматы (CoO-WO3), молибдаты (Bi2O3-MoO3, NiS-MoS3) и др.
В органическом синтезе металлы (кроме Ag) применяют в процессах гидрирования; для этих же процессов распространены и сложные катализаторы. Для реакций дегидрирования используются главным образом оксиды (MgO, ZnO, Fe2O3, Cr2O3), для процессов окисления — некоторые оксиды (CuO, V2O5), а также вольфраматы, молибдаты и металлическое серебро. В реакциях гидрирования наиболее активны платина и палладий, меньше никель; еще более мягким действием обладают Fe, Со и Си. При окислении и дегидрировании Pt, Pd и Ni способствуют глубокому превращению реагентов; это же относится и к хроматам. Мягкими катализаторами неполного окисления являются CuO, V2O5, вольфраматы, молибдаты. Оксиды и сульфиды молибдена и вольфрама нечувствительны к отравлению сернистыми соединениями.
Некоторые оксиды являются бифункциональными катализаторами. Бифункциональности катализатора можно также достичь, используя смеси оксидов разного типа. Примером может служить система оксид цинка, нанесенный на А12О3, успешно применяющая- ся для синтеза бутадиена из этанола, где одновременно протекают ионные и гомолитические реакции. Большое применение нашли бифункциональные катализаторы, состоящие из носителя кислотного типа‘(А12О3, алюмосиликаты) с нанесенным на него металлом (Pt, Pd) — катализатором гомолитических реакций.
г- Гетерогенные катализаторы должны обладать: 1) высокой ка-I талитической активностью; 2) большой селективностью (избирательностью) в отношении целевой реакции; 3) простотой получения, обеспечивающей воспроизводимость всех свойств катализатора; 4) высокой механической прочностью к сжатию, удару и истиранию; 5) стабильностью всех свойств катализатора на протяжении его службы и способностью к их восстановлению при том или ином методе регенерации. Обеспечение этих требований достигается главным образом при разработке состава катализатора и способа его получения.
Гетерогенные катализаторы сравнительно редко применяются в виде индивидуальных веществ и часто содержат различные добавки, так называемые модификаторы. Цели их введения очень разнообразны: повышение активности катализатора (промоторы), избирательности и стабильности работы, улучшение механических или структурных свойств. Фазовые и структурные модификаторы стабилизируют активную фазу твердого катализатора или пористую структуру его поверхности. Так, в медь-хромовых катализаторах гидрированный оксид хрома препятствует восстановлению оксида меди (II) с превращением его в неактивную форму. Добавление уже 1% А12О3 к железному катализатору увеличивает его площадь поверхности, препятствуя спеканию и закрытию пор и т. п. Некоторые модификаторы существенно повышают стабильность работы катализатора или сильно изменяют характер его каталитической ак
441
тивности. Например, добавки щелочей к цинк-оксидному катализатору для синтеза метанола ведет к образованию высших спиртов.
В смешанных катализаторах, в которых компоненты находятся в соизмеримых количествах, могут образоваться новые, более активные соединения. При этом свойства смешанного катализатора не являются простой суммой свойств его компонентов. К числу модификаторов можно отнести и носители (трегеры), особенно часто применяемые для получения дорогостоящих металлических катализаторов (Pt, Pd, Ni, Со). Роль носителей состоит в повышении активной поверхности, увеличении термостойкости и механической прочности катализатора и т. п. В качестве носителей используют алюмосиликаты, оксиды алюминия, хрома или кремния, активированный уголь, пемзу, кизельгур и другие природные и синтетические материалы. Так, например, дегидрирование метилциклопентана платиной, нанесенной на активированный уголь, ведет к образованию метилциклопентана и пентадиена, а при дегидрировании на РЬАЬОз образуются бензол и циклогексан. Носители могут изменять активность и избирательность катализатора и т. п. Следовательно, роль носителя как модификатора свойств катализатора может быть очень большой, и его выбор является существенным при создании оптимального катализатора для данного процесса.
На свойства катализатора влияет и способ его получения. Поскольку химическая реакция протекает на поверхности, очень важно получить катализатор с максимально развитой поверхностью, т. е. с большим количеством пор. Для разных реакций оптимальными могут быть узкие или, наоборот, более широкие поры, а также комбинация широких (транспортных) пор с более узкими. Не менее важным является форма и размер зерен катализатора. От этого зависят удельная производительность и гидравлическое сопротивление слоя катализатора. Различают следующие виды промышленных катализаторов: I) осажденные (солевые, оксидные) — монолитные, таблетированные или порошкообразные формованные; 2) катализаторы на носителях (солевые, оксидные, металлические)— зерненные, таблетированные, формованные; 3) природные (силикаты); 4) плавленые (металлические, оксидные), в том числе металлы в виде сеток, спиралей и т. п.; 5) скелетные (металлические).
Все способы получения катализаторов делятся на мокрые и сухие, из которых наиболее распространен первый. Он состоит в осаждении активной основы катализатора в виде геля из водного раствора соли, например,
AI (NO3)3 + 3 NH4OH->A1 (ОН)з + 3 NH4NO3
Осаждением двух или более гидроксидов из смеси солей легко готовить промотированные, смешанные или солевые катализаторы. Данным же способом получают синтетические носители.
На свойства получаемых катализаторов влияют как выбор реагентов, так и степень их чистоты. Активность и пористая структура катализатора (или носителя) существенно зависят от темпера-442
туры и скорости осаждения, концентрации растворов, времени созревания осадка, pH среды и т. п. Структура катализатора зависит и от последующей обработки геля, состоящей в его отмывке от посторонних ионов, фильтровании, сушке и прокаливании. При некоторых из этих операций и происходит образование пор за счет выщелачивания примесей и удаления влаги.
Для получения катализатора нужной формы и для гранулирования имеется несколько способов, которые обеспечивают ему необходимую прочность. Иногда это достигается прокаливанием высушенного геля, когда образуется монолит, который затем дробят до нужного размера.
Катализаторы на носителях готовят методом пропитки. Обычно носитель пропитывают водным раствором соли или другим веществом до тех пор, пока не будет достигнуто нужное содержание компонентов, после чего при необходимости сушат и прокаливают. Если активным центром катализатора является оксид металла, пропитку ведут термически нестабильными солями или их смесями, которые при последующем прокаливании превращаются в оксиды. Эти оксиды для металлических катализаторов на носителях затем восстанавливают водородом до свободного металла.
Менее распространены сухие методы приготовления катализаторов. Таким путем получают, например, плавленые катализаторы (оксидные и металлические)., К данным методам относится и получение диспергированных катализаторов путем термического разложения их солей.
Особую группу составляют очень активные скелетные катализаторы, из которых чаще всего применяется так называемый никель Ренея, который получают выщелачиванием никель-алюминиевого сплава избытком горячего едкого натра. Таким путем удаляется почти весь алюминий и остается очень пористая губчатая (скелетная) масса никеля, которую из-за ее пирофорности нужно хранить под слоем инертной жидкости. Более перспективен катализатор, получаемый неполным выщелачиванием алюминия только с поверхностного слоя. В отличие от никеля Ренея он способен к регенерации путем повторного выщелачивания более глубоких слоев.
Гетерогенные катализаторы характеризуются рядом физических свойств. Фракционный состав зерен катализатора определяют ситовым и седиментационным анализом, фазовый состав — рентгеноструктурным и электронно-микроскопическим методами. Важной характеристикой является удельная площадь поверхности, отнесенная к единице количества катализатора. Ее определяют адсорбционным путем или газохроматографическим способом. Средний радиус пор вычисляют делением удвоенного удельнокю объема пор, определяемого по истинной и кажущейся пористости катализатора, на удельную площадь поверхности. Имеет значение и распределение пор по радиусам, которые определяют капиллярной конденсацией какого-либо вещества. Значение этих характеристик необходимо при каждой исследовательской работе, и они обязательно содержатся в паспорте промышленного катализатора.
3. МУЛЬТИПЛЕТНАЯ ТЕОРИЯ КАТАЛИЗА
Специфическое дегидрирование в шестичленном кольце послужило толчком для создания мультиплетной теории, впервые поставившей и частично разрешившей вопрос о составе активного цент
443
ра гетерогенного катализатора. Основные положения мультиплетной теории Баландина:
1) понятия «адсорбционного и каталитически активного центра» неравноценны; 2) молекулы адсорбируются на нескольких адсорбционных центрах несколькими участками, в результате чего происходит ослабление и перераспределение связей; 3) каталитически
активным центром является совокупность определенного числа адсорбционных центров, расположенных в геометрическом соответствии с расположением атомов в катализируемой молекуле.
Мультиплетная теория ставит геометрическое строение актив-ного центра в прямое соответствийсостроением претерпевающей превращение молекулы. Главной основной предпосылкой гетерогенного катализа является интенсивная адсорбция реагирующего вещества на поверхности катализатора. Особенно энергично адсорбируется вещество, когда между расположением атомов в адсор-
Рис. 189. Расположение молекулы циклогексана на октаэдрических гранях металла
бируемой молекуле и атомов в кристаллической решетке катализатора существует определенное соответствие. Например, при адсорбции циклогексана на октаэдрических гранях металлов молекула располагается на кристалле (рис. 189). Кат а л и тическо& действие происходит тогда,.когда соответствующие связи...в реаги-
руютней молекуле ослабляются. Для такого ослабления связей необходимо удаление друг от друга соседних атомов в молекуле. Когда размеры постоянной решетки кристалла превышают расстояние между атомами в реагирующей
молекуле, связи ослабляются и происходит каталадичес^
ние реакции^Поверхностное соединение образуется из одной или нескольких молекул вещества и из нескольких атомов катализатора. Группа атомов катализатора, вступающих в поверхностное соединение, называется мультиплетом. Обычно эта группа состоит~из
двух-трех атомов.
Поверхностное соединение образуется не всегда, а только в том случае, ес л и вещество адсорбируется н а ^поверхности определен н ы -
ми атомами, причем те атомы, между которыми рвется связь, адсорбируются разными атомами мультиплета. Те атомы, между которыми связь возникает, адсорбируются одинаковыми атомами мультиплета. Например,
С2Н5ОН С2Н4+Н2О
н н
I I
Н-С—С—н сн2-сн2 + Н2О
н он
444
н н
I 1 I Н-—С~— • —С““Н
где • — атомы мультиплета, которые прочно связаны с кристаллической решеткой; гм— расстояние между атомами кристаллической решетки металла. Металлы, у которых гм<^с-н и гм<гс-о, не будут катализаторами для этой реакции.
Все процессы образования адсорбционного соединения на активном центре и перераспределение связей на нем идут с определенными энергетическими эффектами. Возникает вопрос, нельзя ли, исходя из термодинамических характеристик процесса (энергия связи катализатора с субстратом, тепловой эффект реакции и энергия связи катализатора с продуктами реакции), выбрать наиболее активный катализатор для данного процесса. Поэтому был выдвинут принцип энергетического соответствия. Следуя этому принципу, разберем энергетические эффекты реакции обменного каталитиче-
ского разложения:
АВ + CD
катализатор
> AD 4- ВС
Согласно мультиплетной теории первой стадией будет разрыв связей молекул и взаимодействие получившихся радикалов с адсорбционными центрами катализатора, которое приводит к образованию мультиплетного комплекса. При этом затрачивается энергия разрываемых связей (—Qab и —Qcd) и выигрывается энергия образования промежуточных соединений (Qak, Qbk, Qck, Qdk). Таким образом, первая стадия состоит из двух процессов:
АВ + К->АК4-ВК; CD 4-К СК 4-DK
Теплота образования промежуточного мультиплетного комплекса равна
“ Qab ~ Qcp + Qak + Qbk + Qck + Qdk “ Qab —* Qcd ~ $»
где <7 ~ Qak 4-Qbk 4“ Qck 4-QDr.
Заключительной стадией процесса является образование в результате перераспределения связей продуктов реакции и их десорбция с катализатора:
AK + DK->AD 4-К; ВК 4- СК->ВС 4- К
Теплота распада промежуточного мультиплетного комплекса равна
^2 — Qad + Qbc ” (Qad + Qbk + Qck + Qdk) = Qab + Qbc $ •
Следовательно, общий тепловой эффект процесса в целом выразится соотношением
Е = Ei 4- Е% ~ QAR — Qcd 4- QAD 4- QCB.
445
Отсюда получаем условия для экзотермического процесса
Qab + QcO Q AD + QbC
и для эндотермического процесса
Qab + Qcd > Qad + Qbc •
Если рассматривать действие различных катализаторов на один и тот же процесс, то их специфичность, с энергетической точки зрения, будет выражаться различной энергией образования промежуточных соединений АК, В К, СК и DK. Следовательно, q показывает степень энергетической выгодности каталитического процесса по сравнению с некаталитическим и степень энергетической выгодности того или иного катализатора.
4. ТЕОРИЯ АКТИВНЫХ АНСАМБЛЕЙ
Теория Тэйлора и мультиплетная теория Баландина постулировали существование активных центров, но их свойства они описывали лишь качественно, не дав аппарата для экспериментального определения количественного состава и свойств активных центров разных процессов, их абсолютной активности и их общего числа. Эти задачи поставлены в теории активных ансамблей.
В реальном кристалле всегда имеются дефекты. Частицы, из которых состоит кристалл, попадая между~узл.ами.41ещ^ дя на поверхность и достраивая решетку, оставляют вакантные места. Может быть нарушено, и стехиометрическое соотношение между частицами (инородные примеси, недостаток или избыток одного из компонентов). Кроме того, структура реадьцоххи-жристалла может иметь ряд макронарушений, трещин, разделяющих его на отдельные микрокристаллические блоки, скрепленные друг с другом. Трещины и другие нарушения поверхности резко увеличивают активную поверхностями, следовательно, увеличивают число адсорбционных и каталитических центров. Наличие микротрещин ограничивает возможность миграции атомов иной химической природы на поверхности кристалла.
Если нанести на поверхность идеального кристалла атомы металла, то они в результате теплового движения будут распространяться по всей поверхности и в итоге закристаллизуются в один каталитически неактивный или мало активный агрегат атомов. Для блочно-построенного кристалла атомам металла, попавшим на определенные участки поверхности кристалла, необходима избыточная энергия для преодоления геометрических (а следовательно, и энергетических) барьеров и для передвижения по всей поверхности. Таким образом, поверхность адсорбента оказывается разбитой на энергетически замкнутые области, в которых при данной температуре осуществляется безактивационное движение атомов нанесенного металла. Эти области были названы областями свободной миграции.
446
Следовательно, нанесенные атомы располагаются на поверхности носителя в виде обособленных агрегаций (ансамблей), состоящих из нескольких атомов, локализованных по областям свободной миграции. На основании этих представлений была создана теория активных ансамблей.
1. Носителем каталитической активности является атомная (докристаллическая) фаза катализатора. Кристаллическая фаза или поверхность носителя выполняет роль инертной подложки.
2. Поверхность носителя в соответствии с блочным строением реальных кристаллических тел представляет собой совокупность замкнутых ячеек (области свободной миграции), отделенных друг от друга энергетическими (и геометрическими) барьерами, непреодолимыми при данной температуре для нанесенных атомов.
3. Для каждого процесса активным центром является ансамбль из определенного числа атомов катализатора.
При любой степени заполнения носителя активным веществом существуют ансамбли всех видов, но каждому заполнению отвечает максимальное число ансамблей некоторого определенного состава. На основе теории активных ансамблей были изучены многие процессы. Состав активного центра определяется^_осно.внрм..не_гер-. метрией ка.тализируемых молекул, а числом и типом разрываемых связей в перъошгча.лЪцом2акте.щктив.ащии, от которого зависит дальнейшее течение процесса. i
5. РЕАКЦИИ В ПОТОКЕ
Химические процессы обычно осуществляют в потоке, т. е. струе газа, проходящей через реактор с заданной температурой. Последний может быть пустым или со слоем зернистого катализатора. Примерами реакций, осуществляемых в потоке, могут служить крекинг нефтепродуктов, гидрокрекинг, каталитическое алкилирование, полимеризация, гидро- и дегидрогенизация углеводородов, галогенирование, нитрование окислами азота, синтез аммиака, контактный способ получения серной кислоты, каталитический риформинг и т. п.
Проведение реакций в потоке обеспечивает непрерывность процесса, высокую эффективность использования реактивов, создает большие возможности для регулирования и автоматизации процесса. Многие технически важные реакции при изучении их кинетики в лабораторных условиях проводят также в потоке.
Реакции в потоке можно классифицировать по режимам проведения. Предельными являются режимы идеального вытеснения и идеального перемешивания. На практике могут реализоваться и промежуточные режимы. Режим идеального вытеснения, осуществляемый в трубчатых реакторах, характеризуется тем, что в потоке реагирующего газа отсутствует продольное и поперечное пере-
447
мешивание. Режим идеального перемешивания характеризуется тем, что в результате интенсивного перемешивания концентрации всех реагирующих веществ в любой точке реактора равны их концентрациям на выходе из реактора. Строгая реализация идеальных режимов очень затруднительна, и поэтому химические процессы в потоке в действительности протекают или по режимам, близким к идеальным, или по смешанным.
Непрерывное проведение химического процесса осуществляется при прохождении потока вещества через реактор. При установившемся процессе количество прореагировавших веществ на участке между началом и концом реактора представляет собой величину постоянную. Количество молей вещества, проходящих в единицу времени через любое сечение реактора, не зависит от продолжительности работы установки. Кинетическому исследованию должен предшествовать термодинамический расчет константы равновесия реакции при данных условиях. Этот расчет необходим для правильного определения степеней превращения. Кинетическое исследование реакции в потоке должно установить связь между выходом продукта реакции, степенью превращения исходных веществ и объемной скоростью подачи исходных веществ в реактор, в котором осуществляется реакция.
Пусть для реакции А->В при данной температуре константа равновесия КР = рв/рА. Если начальное давление вещества А равно 1,013-105 Па, то конечное равновесное рд=рв = 0,5-105 Па. Следовательно, система придет в равновесие, когда половина исходного вещества А превратится в В. С этой точки зрения казалось бы, что степень превращения равна У2. Но такая степень превращения является предельно возможной в заданных условиях, поэтому ее принимают за 1. Успешность проведения реакции следует оценивать по тому, насколько удалось приблизиться к предельному превращению. Если предельное значение принять за 1, то реально получаемые степени превращения будут дробными числами. Так, если исходное вещество А только на Vs превратится в вещество В, а равновесное состояние соответствует превращению на .Уз, то степень превращения будет V5: V2 = 2/s-
Рассмотрим общие закономерности протекания реакций в проточном реакторе. В гетерогенном процессе реагируют только те вещества, которые адсорбированы на поверхности. Скорость w гетерогенной химической реакции определяют как количество вещества, реагирующего в единицу времени на единице площади поверхности катализатора
где п — количество реагирующего вещества в момент времени S — общая площадь поверхности катализатора, на которой идет химический процесс.
448
Скорость гетерогенного химического процесса, согласно основному постулату химической кинетики, прямо пропорциональна поверхностной концентрации веществ:
d п
w = —— — Ж d t
(XIX. 2}
Допустим, что в реакторе с сечением S и длиной слоя катализатора I протекает необратимая мономолекулярная реакция по уравнению А->В. Выделим в реакторе слой катализатора толщиной dl. Если скорость подачи реагента vo моль/с, то на входе в слой dl количество молей реагента равно vo—v, а на выходе — у0—(v + dv). Изменение числа молей за счет реакции равно:
dy—wSdZ, (XIX. 3)
где w — скорость реакции, отнесенная к единице объема, занимаемой катализатором. Так как реакция мономолекулярная, то
w = . (XIX.4)
Если вещества А и В сильно адсорбируются на катализаторе и при протекании реакции устанавливается адсорбционное равновесие, то согласно изотерме Лэнгмюра уравнение (XIX.4) примет вид
9-^а/(*ра4-£'рв),
(XIX.5)
где b = ka/kn и b'^ksjkji — адсорбционные коэффициенты веществ
А и В соответственно. \
Если общее давление в системе равно 1 атм, то парциальное давление Z-го компонента можно выразить через молярную долю
(XIX.6)
При этом va = vo—v, vb = v, Svi=vo. Подставляя (XIX.6) в (XIX.5). получаем
—у)/Ур
Ь (v0 — у)/у0 + $'у/у0
(XIX.7)
С учетом уравнений (XIX.3), (XIX.4) и (XIX.7) выразим изменение числа молей вещества А в слое катализатора:
Ь (у0 — у)
£ (v0 — v) -h Ь' у
ASdZ,
(XIX.8)
где Sd/~VK — элемент объема катализатора.
Интегрируя уравнение (XIX.8) в пределах от 0 до V и проведя преобразования, получаем
У А
“ In [1/(1 - z/)] - а + (у0/Ук) . (XIX.9)
*
где v/vo; а и р — константы.
449
Работа 1. Изучение кинетики реакции распада (крекинга) ацетона в потоке f
Ацетон разлагается при температуре около 500°С по уравнению СН3СОСН3 = СО + С2Н6
Эта реакция первого порядка. При проведении ее в статической системе константа скорости выражается по уравнению мономолеку-лярной реакции
k —-----— -- — In----, (XIX. 10)
t с i t 1 — у
где t — время от начала реакции; с0, Ct— концентрация ацетона в начальный момент и к моменту /; у — степень превращения.
В условиях проточной системы временем от начала реакции считается время нахождения паров ацетона в горячей части реактора и определяется по формуле
^=Z/<n, (XIX. 11)
где I — длина горячего участка реактора; со — скорость прохождения фронта потока вещества через поперечное сечение реактора.
Если, в выражении (XIX.11) числитель и знаменатель умножить на сечение трубки реактора яг2, то числитель будет соответствовать объему реактора 1лг2=а, являющемуся константой его, а знаменатель— объему газа, проходящего через реактор за единицу времени, o)jtr2=v. (XIX. 12)
Тогда, определив время от начала реакции t, можно записать уравнение (XIX.10) в форме
k = — In—1. (XIX. 13)
а 1 — у
Зависимость же константы скорости реакции k от температуры выражается уравнением Аррениуса. Энергия активации может быть вычислена из опытных данных. Для этого необходимо определить константу скорости реакции при трех температурах.
Перед выполнением работы следует рассчитать /Ср и равновесную степень распада ацетона при помощи таблицы термодинамических функций веществ. Расчет проводится в первом приближении, причем принимают, что АЯ° и AS0 реакции не зависят от температуры. Константа равновесия для изучаемой реакции рассчитывается по формуле
Хр = РсоЛзащ/^сНзЪСо- (XIX. 14)
Обозначив термодинамическую степень превращения ацетона через а, получим
ХР= ct2j9/(l - а2), (XIX.15)
где р — измеряемое давление смеси исходного вещества и продуктов реакции. Из уравнения (XIX. 15) определяют а. Кинетическая 450
степень превращения у будет равна //а, где у' — наблюдаемая степень превращения.
Работа проводится на установке, схема которой приведена на рис. 190, а, В трубчатую печь 1 помещен кварцевый реактор 2. Центральная часть реактора, заполненная стеклянной насадкой Зт соединена с колбой 14. Постоянство температуры обеспечивают электропечью при помощи авторансформатора или реостата. Температуру измеряют по показаниям пирометра/5, соединенного с тер-
Рис. 190. Схема установки для изучения кинетики гетерогенных каталитических реакций
5
Положение!
насосу
Положение Ж /Г насосу
S
Положение Т /Г прибору
Положение Ж.
мопарой, которая помещена в кармане 13 реактора. Пока устанавливается постоянство температуры (температура устанавливается до начала опыта в течение 20—30 мин), необходимо проверить прибор на герметичность. Для этого газовую бюретку 8 нужно полностью заполнить водой при помощи водоструйного насоса, с которым она соединена через трехходовой кран. При заполнении трехходовой кран ставят в положение «I» (рис. 190, б), открывают кран 9 и включают водоструйный насос, который создает разрежение в газовой бюретке. Вода в сосуде 10 через кран 9 заполняет свободное пространство газовой бюретки. После заполнения бюретки трехходовой кран 7 ставят в положение «II» (рис. 190, б), краны 5 (и 6 закрыты, а трехходовой кран 12 ставят в положение «I» (рис. 190, в), и вновь открывают кран 9.
При герметичности установки через кран 9 вытекает небольшое количество воды, после чего ток воды прекращается. Газовую бюретку вновь полностью заполняют водой, кран 9 закрывают, бюретку 4 заполняют ацетоном (трехходовой кран 12 ставят в положение «II»), после чего открывают кран 6. При этом уровни манометра 11 устанавливают в нулевое положение, трехходовой кран 7 ставят в | положение «II», а трехходовой кран 12 — в положение «I». После J этого можно приступить к подаче ацетона в реактор, предваритель-у 451
< •
-V уг
Д •• •
но записав в рабочую таблицу необходимые начальные данные (уровень ацетона, температуру и т. п.).
Последовательность выполнения работы. Подачу ацетона производить осторожным поворотом крана 5 бюретки 4. Постоянство атмосферного давления в установке контролировать по манометру 11 и регулировать краном 9. В момент падения первой капли включить секундомер и наблюдать изменение уровней манометра. Открыть кран 9 с таким расчетом, чтобы количество вытекающей воды обеспечило нулевое положение уровней манометра. Опыт вести в течение 25—30 мин. Через каждые 3 мин, не прекращая подачи ацетона, отмечать изменения уровня ацетона и объема выделившегося газа в газовой бюретке. Уровни манометра регулировать на протяжении всего опыта краном 9, поддерживая, их нулевое положение истечением воды.
Скорость подачи ацетона в опыте поддерживать по возможности постоянной. Всего следует провести четыре-пять опытов. Соотношение скоростей подачи в них выражается как 4 : 3 : 2 : 1 и достигается отсчетом скорости падения капли по другому секундомеру. Например, первый опыт вести со скоростью подачи ацетона одна капля в 1 с. В следующем опыте необходимо установить скорость подачи одна капля в 2 с, в третьем — одна капля в 3 с, в четвертом— одна капля в 4 с и т. п. По истечении 25—30 мин кран 7 закрыть, газовую бюретку вновь заполнить водой и повторить опыт.
Опыты проводить при постоянной и различных скоростях подачи ацетона. Если опыты проводятся при различных температурах, то их должно быть не меньше трех. Опытные данные записать в таблицу по образцу:
Время, мнн
Уровень ацетона в бюретке, мл
Расход гдетона, мл
Объем газа в бюретке, см3
Прирост объема, см3
Температура, °C
(XIX. 16)
в газовой
При обработке результатов объем выделившихся СО и С2Нб следует привести к нормальным условиям по формуле
(Ратм " ^н2о) von273
760 (273 -F ^0Мн) ’
где рн2о — давление паров воды над водным зеркалом
бюретке при комнатной температуре (см. Справочник). Степень превращения у определяется как отношение числа молей образовавшегося СО или С2Н6 (так как они образуются в одинаковых количествах) к числу молей пропущенного ацетона:
У — псо//?(сн3)2со» (XIX. 17)
где
п(Сн3)аСО “ «со ~~ vo/(2‘22,4);
452
V — rt(CH3)2CO1(W’
где v — скорость подачи исходного вещества за единицу времени. Полученные данные записать в таблицу по образцу:
Построить график зависимости lg k от 1/Т и определить энергию активации реакции разложения ацетона.
Работа 2. Каталитическая дегидратация этилового спирта
Наибольшее практическое значение имеют гетерогенно-каталитические реакции, проводимые в условиях проточной системы. При этом фактор продолжительности реакции зависит не только от диаметра реактора и скорости подачи, но и от условий адсорбции реагирующих веществ на поверхности катализатхша^хсехо^дктив-ности.
^“Любая реакция между газообразными веществами, протекающая на поверхности катализатора, может быть расчленена на= пять последовательных стадий: 1) подвод вещества к поверхности катализатора; 2) адсорбция его; 3) реакция на поверхности; 4) десорбция продуктов реакции с поверхности; 5) отвод продуктов реакции от поверхности в объем газовой фазы. Общая скорость процесса определяется наиболее медленной его стадией. Если наиболее медленной стадией является сама реакция на поверхности, то для описания кинетической зависимости следует решить дифференциальное уравнение общего вида
д%п
w =-----
(XIX. 18)
где w — скорость реакции, выраженная количеством молей прореагировавшего вещества за время t на площади поверхности 3; k — константа скорости поверхностной реакции; bi — адсорбционные коэффициенты вещества; pi — парциальные давления веществ в различных местах реакционной зоны.
Решение уравнения (Х1ХЛ8) для мономолекулярных реакций в потоке при постоянном давлении предложено Фростом и Баланди-
ным
v In
= «4- pvz/,
(XIX. 19)
где v — скорость подачи исходного вещества за единицу времени, отнесенная к единице объема катализатора; у — степень превраще
453
ния; а — постоянная, прямо пропорциональная общей площади поверхности катализатора и константе скорости поверхностной реакции; р — постоянная, характеризующая адсорбционные коэффициенты веществ; а и р — кинетические характеристики реакции при данных условиях, зависящие только от температуры.
Уравнение Фроста — Баландина (XIX.19) может быть представлено графически. На оси абсцисс откладывают значения vy, а на оси ординат — значения тНп—-— и получают прямую линию. Тан-1 —
гене угла наклона прямой к оси vy определяет значение р, а отрезок на оси ординат, отсекаемый прямой при у=0,— значение а.
В работе необходимо при 350—400°С определить постоянные аир реакции каталитической дегидратации этилового спирта над AI2O3, протекающей по уравнению
С2Н5ОН = С2Н4 4- Н2О
Молекула С2Н5ОН, адсорбируясь на поверхности катализатора, располагается на мультиплете так, что разные ее части связываются с разными атомами мультиплета.. Согласно мультиплетной теории катализа Баландина связи молекул, адсорбированных на разных атомах мультиплета, могут разрываться.
Работа проводится накоторой приведена на рис,.. 190. В трубчатую печь 1 помещен кварцевый реактор 2. Центральная часть реактора, заполненная стеклянной насадкой 3, соединена с колбой 14. Постоянство температуры обеспечивается электропечью при помощи автотрансформатора или реостата. Температуру измеряют по показаниям пирометра /5, соединенного с термопарой, которая помещена в кармане 13 реактора. Пока устанавливается постоянство температуры (она должна быть постоянной до начала опыта приблизительно в течение 20—30 мин), необходимо проверить установку на герметичность. Для этого газовую бюретку 8 нужно полностью заполнить водой при помощи водоструйного насоса, с которым она соединена через трехходовой кран. При заполнении трехходовой кран ставят в положение «I» (рис. 190, б), открывают кран 9 и включают водоструйный насос, который создает разрежение в газовой бюретке. Вода в сосуд 10 через кран 9 стремится заполнить свободное пространство газовой бюретки. После заполнения газовой бюретки трехходовой кран 7 станят в положение «Н>> (рис. 190, б), краны 5 и 6 закрыты, а трехходовой кран 12 ставят в положение «I» (рис. 190, в) и вновь открывают кран 9.
При герметичности установки через кран 9 вытекает небольшое количество воды, после чего ток воды прекращается. Газовую бюретку вновь полностью заполняют водой, кран 9 закрывают, бюретку 4 вновь полностью заполняют водой, кран 9 закрывают, бюретку 4 заполняют раствором спирта (трехходовой кран 12 ставят в положение «II»), после чего открывают кран 6. При этом уровни манометра устанавливаются в нулевое положение, трехходовой кран 7 ставят в положение «II», а трехходовой кран 12 — в положе-454
ние «I». После этого можно приступить к подаче раствора спирта в реактор, предварительно записав в рабочую таблицу начальные данные (уровень раствора спирта, температуру и т. п.).
Последовательность выполнения работы. Подачу раствора спирта производить осторожным поворотом крана бюретки 4. Постоянство атмосферного давления в установке контролировать по манометру 11 и регулировать краном 9. В момент падения первой капли включить секундомер и наблюдать изменение уровней манометра. Открыть кран 9 с таким расчетом, чтобы количество вытекающей воды обеспечило нулевое положение уровней манометра. Опыт вести в течение 25—30 мин. Через каждые 3 мин, не прекращая подачи раствора спирта, отмечать изменения уровня раствора спирта и объема выделившегося газа в газовой бюретке. Уровни манометра регулировать на протяжении всего опыта краном 9, поддерживая их нулевое положение истечением воды. Скорость подачи раствора спирта в опыте поддерживать постоянной. Всего следует провести четыре-пять опытов. Соотношение скоростей подачи в них выражается как 1 :2 : 3: 4 и достигается отсчетом скорости падения капли по другому секундомеру. Например, первый опыт вести со скоростью подачи спирта одна капля в 1 с. В следующем опыте необходимо установить скорость подачи одна капля в 2 с, в третьем — одна капля в 3 с, в четвертом — одна капля в 4 с и т. п.
По истечении 25—30 мин кран 5 закрыть, газовую бюретку вновь заполнить водой и начать очередной опыт.
Опыты проводить при различных, скоростях подачи раствора спирта и постоянной температуре или при разных температурах, но при постоянной скорости подачи спирта. Если опыты проводят при разных температурах, то их должно быть не меньше трех. Опытные данные записать в таблицу по образцу:
Время, мин
Уровень спирта в бюретке, мл
Расход спирта, мл
Объем газа в газовой бюретке, см3
Прирост объема, см3
Показание пирометра
Количество молей спирта, прошедшего через реактор, рассчитывают по навеске спирта т и его молекулярной массе М или по числу миллилитров водно-спиртовой смеси g и ее плотности р:
zn
лсащон~ м - м •
При этом необходимо внести поправку на процентное содержание спирта
n^g^B/M,
где В — процентное содержание спирта, деленное на 100.
455
Степень превращения спирта определяется по формуле
У = пс2н4/пс2н5он-
При расчете числа молей п этилена следует учитывать весь объем выделившегося газа, так как пары воды, выйдя из реактора, конденсируются. Объем выделившегося газа следует приводить к нормальным условиям:
(Рати — Рн2о) von273
760 (273 + ZK0MH)
где рнао —давление паров воды над водным зеркалом газовой бюретки при комнатной температуре (зависимость рн2о от температуры см. в Справочнике).
Объемная скорость подачи v (моль/мин) определяется на единицу объема катализатора
^^^щон1000/^)-
где t — продолжительность опыта, мин; v— объем катализатора, см3.
Результаты расчетов записать в таблицу по образцу:
rtC2H5OH
V In “ 1-1/
На основании экспериментальных данных построить график в
координатах v In--------vy и определить константы а и В.
1 ~ У
Работа 3. Каталитическое разложение метилового спирта
В работе исследуется кинетика разложения метилового спирта по уравнению реакции
СН3ОН = СО + 2Н2
протекающей в потоке при 250—400°С и 1,013-105 Па над хромцинковым катализатором ZnO-Cr2O3.
Работа проводится на установке, схема которой приведена на рис. 190 (описание проведения опыта см. с. 455). Количество молей пропущенного спирта определяют или по навеске спирта т и его молекулярной массе А1, или по числу миллилитров спирта g и его плотности р: _ _ _ _ S ... к
Степень превращения равна
У = псо/псн3он*
456
При определении числа молей оксида углерода следует весь объем выделившегося газа разделить на три, так как образуются в реакции три моля газа. Объем выделившегося газа приводят к нормальным условиям:
(Ратм РН20) ^73
° 760 (273 + 40МН) ’
где рн2о —давление паров воды в газовой бюретке при комнатной температуре (зависимость рн2о от температуры см. в Справочнике). Объемная скорость подачи (моль/мин) определяется на единицу объема катализатора:
v = «СНзОНЮ00/(^) ’
где v — объем катализатора, см1 * 3; t—-продолжительность опыта, мин. Результаты расчетов записать в таблицу по образцу:
ЛСНзОН
На основании опытных данных построить график в координатах — I
v In-------<vy и определить константы а и В.
1 ~ У
ГЛАВА XX
КИНЕТИКА ЭЛЕКТРОХИМИЧЕСКИХ РЕАКЦИЙ
1. ОБЩИЕ СВЕДЕНИЯ О КИНЕТИКЕ ЭЛЕКТРОДНЫХ ПРОЦЕССОВ
Электролизом называются процессы, протекающие при прохождении тока через раствор электролита. Соотношение между количеством протекшего электричества и количеством вещества, выделившегося при электролизе, описывается законами Фарадея. Электролиз начнется только тогда, когда наложенная внешняя э. д. с. больше разности равновесных потенциалов обоих электродов — катода и анода. Нарушение равновесного состояния электродов, связанное с прохождением тока через раствор, называется электрохимической поляризацией, а электроды, выведенные из состояния равновесия, — поляризованными. Поляризация является следствием сложных процессов, происходящих на поверхности электродов при протекании электрохимической реакции разряда — отдачи электронов электроду, или переходу электронов от электрода к разряжающейся
частице. Разность потенциалов, возникающая при электролизе, называется перенапряжением или потенциалом поляризации =
457
= Еп—Ер. Катодное перенапряжение — величина, на которую катодный потенциал оказывается более отрицательным, чем его равновесное значение; анодное перенапряжение — величина, на которую анодный потенциал оказывается более положительным, чем его равновесное значение.
Любая электрохимическая реакция протекает на поверхности раздела фаз электрод — раствор и является гетерогенной. Как гетерогенная химическая реакция она также является стадийной,-текущей через ряд последовательных стадий: 1) транспорт вещества к электроду — к зоне реакции; 2) собственный электрохимический акт взаимодействия реагирующей частицы с электродом (стадия разряда — ионизация); 3) отвод образовавшихся продуктов реакции от поверхности электрода. Первая и третья стадии имеют одни и те же закономерности и называются стадиями мас-сопереноса, осуществляемыми за счет малых коэффициентов миграции и конвекции. Для всех электродных процессов наличие этих трех стадий обязательно. Однако наряду с этим ряд электрохимических процессов может осложняться предшествующими и последующими химическими реакциями, протекающими в объеме раствора или на поверхности электрода. Кроме того, в ходе электрохимической реакции может происходить передвижение частиц по поверхности электрода (стадия поверхностной диффузии). Скорость электрохимического процесса, состоящего из ряда последовательных стадий, определяется наиболее замедленной, лимитирующей стадией. Для установления природы лимитирующей стадии, скорости ее течения, механизма электродного процесса, необходимо знать закономерности, которым подчиняются поляризационные характеристики I и ДЕ.
Если свойства поверхностного слоя не изменяются во времени, то протекающий через электрод ток определяется только скоростью самого электродного процесса и размерами электрода. В этом случае плотность тока является мерой скорости электрохимической реакции. Если скорость наиболее замедленной стадии электрохимической реакции определяется стадией массопереноса, то поляризация называется концентрационной. Поляризация электрода, обусловленная медленной химической реакцией (в результате разряда или ионизации), называется химической поляризацией. Если скорость электролиза лимитируется процессами образования новой фазы, как, например, при катодном выделении металлов, то возникающая поляризация называется фазовой. Зависимость скорости процесса от потенциала поляризации, т. е. / = /(ДЕ), графически выражается поляризационной кривой. Она может состоять из нескольких ветвей (рис. 191), причем участки кривой (cd, ef и т. п.) отвечают возникновению нового электрохимического процесса. Участок кривой Ьс соответствует предельной (максимальной) скорости электрохимического процесса. Повышение скорости процесса (увеличение плотности тока, ветвь ab) приводит к возрастанию потенциала, при котором возможен новый электрохимический процесс (ветвь cd). Плотность тока, при которой начинается быст-458
рый рост потенциала поляризации, называется предельной плотностью тока и обозначается Id. В зависимости от характера протекающего процесса электролиз может сопровождаться изменением окраски раствора, выделением кристаллов, газов и т. п. Так, например, для процесса окисления Мп2+=Мп3+ на платиновом электроде и в сильнокислой среде раствор из бесцветного переходит в малиново-красный, что соответствует появлению ионов Мп3+. При реакции I--->1° раствор желтеет от выделяющегося иода (капля крахмала окрашивает раствор в синий цвет). Окисление гидрохинона в хинон С6Н4 (ОН)2 = СбН4О2-г 2Н+ + 2е сопровождается пожелтением раствора и выделением кристаллов хинона.
С ростом температуры растет скорость электрохимической реакции. Существенное влияние на скорость процессов оказывает и потенциал электрода, характеризующий энергетику текущего электрохимического процесса. Зависимость плотности тока от температуры
Рис. 192. Три типа поляризации:
а — концентрационная; b — химическая; с — фазовая
Рис. 191. Поляризационная кривая
может служить критерием для установления типа поляризации. Согласно температурно-кинетическому методу С. В. Горбачева в любых электрохимических процессах тип поляризации определяется абсолютной величиной эффективной энергии активации, т. е. той энергии, которая необходима, чтобы молекула или ион вступили в электрохимическое взаимодействие. Энергия активации зависит от потенциала поляризации. Эффективную энергию активации электрохимической реакции определяют при постоянном потенциале поляризации по линейной зависимости логарифма плотности тока от обратного значения абсолютной температуры. Такая линейная зависимость наблюдается при концентрационной и химической поляризациях и не соблюдается при фазовой поляризации. Это позволяет определять энергию активации электрохимической реакции по уравнению
lnZ= - Лэфф/(^Г) + В, (XX. 1)
где I — плотность тока; ЛЭфф — эффективная энергия активации.
459
Зависимость логарифма плотности тока от обратной температуры для трех видов поляризации приведена на рис. 192. Для процесса с концентрационной поляризацией прямые а, относящиеся к различным потенциалам, параллельны. Эффективная энергия активации не зависит от потенциала поляризации и равна 10— 12 кДж/моль. При химической поляризации прямые &, соответствующие различным потенциалам поляризации, располагаются веерообразно. Энергия активации электрохимической реакции понижается с ростом потенциала поляризации и при больших потенциалах, при большой скорости процесса приближается к энергии активации концентрационной поляризации. При химической поляризации энергия активации имеет тот же порядок, что и энергия активации химической реакции в растворах (40—80 кДж/моль). Действительно, при электрохимических реакциях потенциальный барьер, характеризуемый энергией активации, преодолевается не только за счет теплового движения молекул или ионов, но и за счет добавочной энергии, приобретаемой реагирующей частицей при ее прохождении через двойной электрический слой на поверхности электрода. Другим фактором, отличающим химическую поляризацию от концентрационной, является влияние перемешивания на скорость (плотность тока) электрохимического процесса. При концентрационной поляризации скорость процесса возрастает с перемешиванием особенно в области предельных токов, когда концентрация реагирующего вещества близка к нулю и лимитирующей стадией становится его доставка к электроду. Скорость электрохимических реакций с химической поляризацией не зависит от скорости перемешивания.
При фазовой поляризации, обычно сопровождающей процесс выделения металлов, наиболее медленной стадией, лимитирующей скорость процесса, является процесс образования зародышей кристаллов. При фазовой поляризации (см. рис. 192) зависимость логарифма плотности тока от обратной температуры выражается кривой, проходящей через максимум. Этот максимум соответствует наибольшей вероятности образования зародышей новой фазы.
2 . КИНЕТИКА ОБРАТИМЫХ
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫХ ПРОЦЕССОВ
В обратимых окислительно-восстановительных системах скорость процесса окисления и восстановления ионов различной степени окисления зависит от соотношения активностей окисленной и восстановленной форм ионов. Если наиболее замедленная стадия процесса электролиза имеет диффузионную природу (концентрационная поляризация), то зависимость скорости (силы или плотности тока) электролиза от состава исследуемой обратимой системы описывается уравнением
(XX.2)
460
где х — равновесная концентрация окисленной формы ионов; # — эмпирическая постоянная для анодного и катодного процессов и изменяющаяся от температуры; М — молярная концентрация раствора; М—х — равновесная концентрация восстановленной формы ионов; /и=еп2?Л£/(нт); у — эмпирическая постоянная. Постоянную у рассчитывают по уравнению при условии, если соотношение окисленной и восстановленной форм ионов равно единице:
(XX. 3>
где со-—начальная концентрация ионов окисленной или восстановленной форм в смеси.
Для водных растворов ферро-феррицианидов, ферро-феррисуль-фатов постоянная у может быть рассчитана по уравнению
У^Ае^т,
(XX. 4)
где А — постоянная, равная 9—10; В — постоянная, определяемая по тангенсу угла наклона прямой к оси абсцисс и равная 1900.
Работа 1. Изучение кинетики электрохимических реакций температурно-кинетическим методом
Изучение кинетики электрохимических реакций проводят методом поляризационных измерений. Простейшая схема установки для поляризационных измерений приведена на рис. 193. Установка состоит из двух контуров: поляризующего (электролизного) а и измерительного (потенциометрического) б. В поляризующем контуре источником тока служит аккумулятор 1. При помощи потенциометрически включенного реостата 2 на электроды подают определенное напряжение, измеряемое вольтметром 3; амперметром 4 измеряют силу тока. Электролизером 10 служит трехэлектродная электрохимическая ячейка с рубашкой для термостатирования. Измерительный контур представляет собой потенциометрическую схему 6, или потенциометр. Схема включает аккумулятор 8 и элемент Вестона 9. Исследования ведут в интервале температур 20—80°С. Точность регулировки температуры ±0,1°.
При исследовании окислительно-восстановительных систем для измерения потенциала поляризации используют электрод сравнения, в качестве которого служит платиновый электрод, погруженный в исследуемый раствор. В качестве рабочего электрода используют вращающийся дисковый электрод.
Поляризационные измерения можно проводить также на специальных установках (потенциостатах) потенциодинамическим и гальваностатическим методами с автоматической записью поляризующего тока при постоянном потенциале или потенциала при фиксированном токе. Сочетание обоих методов позволяет более глубоко* изучать поведение электрохимических систем.
461
Последовательность выполнения работы. Собрать установку для поляризационных измерений (рис. 193). В электролизер вместимостью 150 мл налить около 100 мл исследуемого раствора, погрузить в него платиновый рабочий электрод (стационарный или вращающийся дисковый), вспомогательный Pt-электрод и электрод сравнения. Перед погружением в раствор платиновые электроды обработать концентрированной азотной кислотой и затем промыть дистиллированной водой. Электроды, погруженные в раствор, замыкаются накоротко на 3—5 мин для установления стационарно-
7
Рис. 193. Схема установки для поляризационных измерений
го потенциала. Затем включить электролизер в установку, установить температуру опыта и через определенные промежутки времени с помощью рукоятки 7 подавать на электроды поляризующее напряжение, увеличивая его на 0,1—0,2 В. В конце указанного промежутка времени фиксировать показания приборов: в поляризующем контуре — амперметра и вольтметра, в измерительном—-потенциометра 5. Если поляризационные измерения проводятся с помощью потенциостата, то при работе следует выполнять указания, изложенные в инструкции к прибору. Поляризационные изме- Г рения провести при трех температурах. Каждый опыт проводить со свежим раствором. Полученные данные нанести на график за-висимости / = f(E) для всех исследованных температур. На поля- *
ризационные кривые нанести изопотенциальные прямые и опреде- §
.лить плотность тока при различных температурах и постоянных J потенциалах поляризации. Энергию активации рассчитать для пяти-шести значений потенциалов поляризации. На основании полученных значений энергии активации установить характер зависи-
462
мости ее величины от потенциала поляризации и сделать вывод о природе лимитирующей стадии электрохимического процесса.
Для исследования применять насыщенный раствор сульфата марганца в 4 н. серной кислоте; ОД н. раствор КД 0,01 М растворы ферри-ферроцианидов калия, насыщенные растворы хингидрона в растворах серной кислоты различной концентрации и др.
Работа 2. Изучение зависимости скорости электролиза от состава окислительно-восстановительной системы методом поляризационных измерений
Собрать установку для поляризационных измерений (рис. 193), В электролизер — трехколенную термостатированную электролитическую ячейку налить 100 мл ОД М раствора ферро-феррицианидов> с различным соотношением окисленной и восстановленной форм ионов 9:1, 7:3, 5 : 5, 3 : 7, 1:9. При приготовлении смесей тщательно перемешать растворы обеих форм ионов. В качестве рабочего электрода использовать вращающийся платиновый дисковый электрод, в качестве электрода сравнения — гладкую платиновую проволоку, погруженную в тот же раствор. Установить температуру опыта (20—25°С) и включить электролизер в установку для поляризационных измерений. С помощью реостата 2 подавать поляризующее напряжение на электроды, увеличивая его на 0,01—0,02 В. Через определенные промежутки времени (1—2 мин) измерять потенциал поляризации с помощью потенциометра и фиксировать показания амперметра и вольтметра. Исследовать катодные или анодные процессы для пяти различных соотношений окисленной и восстановленной форм ионов. Полученные экспериментальные данные нанести на график /=rf(A£‘) для всех изученных соотношений. На поляризационные кривые нанести изопотенци-альные прямые через 20—30 мВ и при данном потенциале поляри-зации-построить график I=f (с).
В данной работе следует построить графики зависимости /== = f(AE) для различных соотношений редокс-форм ионов; I = при различных потенциалах поляризации; рассчитать эмпирическую постоянную у по (XX.3) и сопоставить со значением у, рассчитанным по (XX.4); по (XX.3) и I = f(c) по (XX.2) и
сопоставить с экспериментальными кривыми оценить погрешности измерений. Из полученных экспериментальных результатов сделать выводы о природе лимитирующей стадии электрохимического процесса.
ГЛАВА XXI
МАТЕМАТИЧЕСКАЯ ОБРАБОТКА
ЭКСПЕРИМЕНТАЛЬНЫХ ДАННЫХ >
1. ОЦЕНКА ТОЧНОСТИ И НАДЕЖНОСТИ ИЗМЕРЕННЫХ И РАССЧИТАННЫХ ВЕЛИЧИН
Каждая величина как результат измерения содержит некоторое отклонение от истинного значения. В настоящее время физико-химический эксперимент оснащается все более и более точными и чувствительными приборами. Повышение точности и чувствительности прибора зависит от малейших изменений условий, что приводит к изменению показаний прибора. Незначительные изменения внешних условий не поддаются количественной оценке. Прибор будет давать изменение показаний за счет тех случайных воздействий на изучаемую систему и на сам прибор, которые пока нельзя учесть. Повторное измерение будет давать несколько отличающуюся величину. Строгая математическая теория ошибок измерений еще далека от своего завершения. Разные авторы используют разные методы оценки точности полученных ими результатов исследований. Сопоставимые величины могут быть получены только в том случае, если будут сопоставимы условия эксперимента и оценки точности его.
2. ПРИРОДА ПРОИСХОЖДЕНИЯ ПОГРЕШНОСТИ ИЗМЕРЕННЫХ ВЕЛИЧИН. СИСТЕМАТИЧЕСКИЕ ОШИБКИ И ИХ ВЫЯВЛЕНИЕ
Систематические ошибки возникают в основном от неточности шкал приборов. Современные приборы снабжаются паспортом, в котором указывается погрешность шкалы в разных ее диапазонах. Если же паспортные данные у прибора отсутствуют, то необходимо провести калибрование шкалы прибора. Калибрование шкал приборов проводится измерением показаний шкал для стандартных веществ. Например, шкала рефрактометра проверяется по показателю преломления воды, шкала термометра — по температурам плавления и замерзания воды или других веществ в зависимости от диапазона шкалы. В качестве стандартных веществ обычно берут такие, значения физических свойств которых известны с высокой точностью.
Если градуирование шкалы прибора показывает одинаковую погрешность во всех ее диапазонах, то поправка будет постоянной. Юна может быть отрицательной и положительной. Иногда конструкция прибора позволяет провести корректирование шкалы. Корректирование шкалы полезно только в том случае, если поправка постоянна.
Если же поправка переменная, то строится график зависимости поправки от показаний шкалы прибора и поправка каждого измерения производится по этому графику. Любое прецизионное физи-
464
ко-химическое исследование начинается именно с проверки и калибровки шкал приборов, проверки градуировки бюреток, мерных колб, разновесов и т. д.
Случайные ошибки возникают в основном от неточности отсчетов со шкал прибора и с невоспроизводимостью условий опыта, с субъективными особенностями экспериментатора. Эти ошибки невелики, но они неустранимы. Действительно, термостатиро-вание, например, не может обеспечить постоянство температуры со сколь угодно высокой степенью точности, да и исследователь не сможет произвести измерение температуры безукоризненно точно.
Случайные ошибки отличаются от систематических тем, что увеличением числа измерений можно уменьшить их величину. Эта особенность обусловлена тем, что значения случайных ошибок с одинаковой степенью вероятности могут быть положительными и отрицательными. Казалось бы, это позволяет осуществить количественную оценку случайных ошибок. Однако это не так: число повторных измерений, как правило, невелико, поэтому методы теории вероятности неприменимы. Как же следует обрабатывать результаты отдельных измерений (каждое из которых содержит случайную ошибку) для того, чтобы получить величину, более всего приближающуюся к точному значению? Приступая к решению этой задачи, предполагаем, что систематические ошибки исключены. Прежде всего следует определить абсолютную и относительную погрешности измерения данной величины.
3. АБСОЛЮТНАЯ И ОТНОСИТЕЛЬНАЯ ОШИБКИ
Если обозначить через аь #2, &п измеренные величины, а через х — ее действительное значение, то разности
Д । = х — , Д2 = х — ,..., Дд = х — (1ц
будут абсолютными ошибками отдельных измерений. Таким образом, для i-ro измерения абсолютная ошибка рассчитывается по формуле
Д/ = х — а;, /= 1, 2, 3,(XXI.1)
Относительная ошибка It определяется как отношение абсолютной ошибки к истинному значению величины. Так как последняя неизвестна, то приходится считать, что
Z;= Az/x = Az/az, i = 1, 2, 3,..., п. (XXI.2)
Это не приводит к большой неточности, потому что при правильно поставленном опыте Дг- по сравнению с х и az ничтожно мало. По значениям д2, ..., ап вычисляют среднее арифметическое отдельных измерений
х — а ~ (di ~г d2 * 4- an)Jn, (XXI. 3)
которое принимают за действительное значение измеряемой вели-16—2083 465
чины. Если отдельные значения а/ сильно отклоняются от остальных, то их следует исключать из уравнения (XXI.3) только в тех случаях, когда они обусловлены плохим качеством опытов (например, вследствие перерыва работы мешалки или прекращения подачи тока в середине измерения). Если в серии измерений наблюдается систематическое увеличение последующих значений аг-, то расчет по уравнению (XXI.3) теряет смысл. Это происходит, если, вопреки предположению о неизменности системы, внешних условий или прибора, они в действительности непостоянны. Так может случиться при измерении химического равновесия, когда система лишь приближается к состоянию равновесия или во время опыта медленно повышается температура окружающей среды, или же вследствие нагревания прибора меняется его электрическое сопротивление.
Согласно уравнению (XXI.3) среднее отклонение для каждого опыта равно _
ait i — 1,2,3, ..., п. (XXI.4)
В дальнейшем вместо (XXI. 1) будем пользоваться выражением (XXL4). Вся совокупность измерений характеризуется средней величиной ошибки. Ее оценивают по-разному. Наиболее правильно пользоваться формулой
п
У, lsil
ё = —------, i = 1,2,..., (XXI.5)
п
т. е. не учитывать знака отклонений. Таким образом, искомая величина равна а±е. Рассчитав по (XXI.5) значение 8, определяют относительную ошибку г)а. Небольшое значение 8 свидетельствует о высокой точности измерений, но не об их правильности, так как все измерения могут содержать одну и ту же, и при этом значительную систематическую ошибку (например, вследствие неисправности прибора). Экспериментатор должен заранее позаботиться о том, чтобы такая ошибка была исключена (например, устранением разрыва нити термометра Бекмана).
Теперь обратимся к более общему случаю, когда искомая величина вычисляется по формуле, содержащей несколько измеренных величин. Каким образом, зная ошибки каждой из последних, определить погрешность результатов вычисления? Пусть неточности в значениях величин а, (3, у, ..., входящих в расчетное уравнение, равны соответственно Да, Д(3, Ду, ..., а погрешность в искомой величине у равна Дг/. Так как ошибки несоизмеримо меньше самих величин, то заменим разности дифференциалами. Тогда, имея в виду, что
У = ¥>•••), (XXI.6)
получим формулу
&У = 3, Y, • • •) d а + /р (а, у,...) d р + ..., (XXI. 7)
позволяющую рассчитать абсолютную ошибку результатов по абсо
466
лютной ошибке измерений (здесь fa, f$, ...— частные производные по переменным, указанным в индексе). Переходя от абсолютной погрешности к относительной, получаем
dt/ /«(“,?>¥,•••) /»(а, у,---)
~ —Т,—d а + 5 Z----------------------------d ,3 Н- (XXI.8)
У /(а, ?>¥>•• •) /(а. ₽,¥>••)
или
d In у = d In f (а, p, у,...). (XXI.9)
Следовательно, для нахождения относительной ошибки надо взять натуральный логарифм выражения искомой величины и продифференцировать его по измеренным величинам, рассматривая их как переменные.
Разберем несколько примеров применения этого правила. При этом везде вместо дифференциала будем записывать конечное изменение. Функция (XXI.6) имеет вид одночлена
(XXI. 10)
где k, гц, п2, и /14 — постоянные. Из (XXL 10) следует, что
In у = In k + п\ In а 4-.Л2 In 3 — я3 In у — n4 In В, откуда ку Да Д^ Ду ДВ
= П\ -J- /О ~ — п3 — “ •
У а р у В
Так как знаки ошибок неизвестны, то их принимают одинаковыми, т. е. пользуются уравнением
Да Да ДЗ Ду ДВ
= щ + п2 ~~ + п3 + п4 ~ . (XXI .11)
у а 3 у в
Это дает максимальную относительную ошибку. В соответствии с (XXI.11) относительная ошибка равна сумме произведений относительных ошибок измеренных величин, умноженных на постоянные показатели степеней.
Функция (XXI.6) имеет вид суммы, т. е.
r/ = a + ₽. (XXI.12)
После логарифмирования и дифференцирования (XXL11) получим
Дг/ Да + ДЗ
У а + 3 ’
Функция (XXI.6) имеет вид разности, т. е.
У = а — р.
(XXI. 13)
(XXI. 14)
Аналогично (XXI.13) находим
(XXI. 15)
У
При изучении свойств разбавленных растворов (температуры кипения, температуры кристаллизации, давления пара и др.) требуется
16*
467
высокая точность измерений, так как искомое значение равно разности двух близких величин. Действительно, различие в температурах кипения растворителя и сильно разбавленного раствора очень мало; поэтому приходится пользоваться очень точным термометром. Так, для показательной функции i/=a<3 и логарифмической функции г/ = In а получаем соответственно
д ц Да
-------- = In аДр + р У--------------а
и
Дг/ Да у а In а
(XXI. 16? в
(XXI. 17)
4. РАСЧЕТ ОШИБОК
Вычислим относительные погрешности значений, получаемых в результате расчета по формуле, включающей несколько измеряемых величин, например определение молекулярной массы вещества М по повышению температуры кипения АЛжп разбавленного раствора, когда справедлива формула
М = ЮОО^э/пД/ИрДТкип), (XXI. 18)
где Кэ— эбуллиоскопическая постоянная; т — масса растворенного вещества; тр— масса растворителя. Если величина /(э заимствована из таблиц и является точной, то ошибка в определении молекулярной массы AM будет функцией ошибок A/n, Атр и А(АТКип) непосредственно измеряемых величин. Формула (XXI. 18) является частным примером (XXI. 10), поэтому в соответствии с выражением (XXI.11) и с тем, что ni=//2 = ^3= 1, находим
ДЛ4 _ Дт ЛтР Д (Д Гкин)
Л4 tn tn$ Д7'кИ(1
(XXI. 19)
т. е. искомая величина равна сумме относительных ошибок измеренных величин.
Пример. Понижение температуры кипения раствора, содержащего 0,217 г серы в 19,18 г сероуглерода относительно 7Кип CS2 равно 0,104 К. Эбуллиоскопическая константа сероуглерода равна 2,37.
Решение. Пусть масса серы взята с точностью 0,0002 г (аналитические весы), а сероуглерода — с точностью 0,05 г (технические весы) и АТКип определена с точностью 0,002° (термометр Бекмана). Следовательно,
Дт/т = 0,0002/0,2170 = 0,92 -10—з (XXI.20)
И
Дтр/тпр= 0,05/19,18 =2,60-10-3. (XXI.21)
Так как величина АГКИП представляет собой разность температур кипения раствора и растворителя, то ее следует рассчитать по формуле (XXI. 15), учтя, что Да и Ар — величины, практически совпадающие; поэтому (XXI.15) принимает вид
Да 2Да
— =--------(XXI.22)
У а — р
468
Согласно выражению (XXL22)
А (АГкип) АГкип
2-0,002 0,104
= 38,50-10—3,
(XXI.23)
отсюда
дм ~м~
0,92-10-3 4-2,60-10“3+ 38,50-10~з = 4,20-10-2,
(XXI. 24)
т. е. наибольшая относительная ошибка при определении молекулярной массы составляет ±4,2%. Так как в (XXL24) третье слагаемое является наибольшим, то это означает, что при определении молекулярной массы эбуллиоскопическим методом точность результатов будет ограничиваться не Am/m или а
А(АТкип)/ДТкип. Поэтому повышение точности взвешивания приведет только к непроизводительной затрате времени. Уменьшение же А(АГКИП)/АТКИП за счет значительного увеличения знаменателя в (XXL23) исключено, так как хотя работа с более концентрированным раствором и привела бы к повышению температуры кипения, однако формула (XXL 18) утратила бы свою точность. Таким образом, здесь уменьшение случайной ошибки вызывает появление систематической ошибки. В данном случае единственным способом повысить надежность результатов является увеличение точности измерения температуры.
5. ЗАПИСЬ РЕЗУЛЬТАТОВ ИЗМЕРЕНИЙ
Надежность результатов должна быть отражена и в их записи. Так, если измерение температуры произведено термохимическим термометром с точностью 0,1 К, то будет неправильной запись с точностью до 0,001 К. При отбрасывании ненужных цифр (выходящих за пределы точности измерения) обычно сохраняют одну дополнительную для характеристики порядка величин. Так, записи температур кипения кислорода 86,190 К и иода 88,8 К свидетельствуют о том, что температура кипения кислорода известна с точностью 0,01 К, а иода — с точностью 1 К. Поэтому, например, неправильно температуру кипения кислорода характеризовать 86 К, а иода — 88,800 К; оба эти числа не отражают действительной точности этих величин.
При записи целых чисел останавливаются на первый приближенной цифре, заменяя остальные нулями. Если количество последних велико, то целесообразно применять два сомножителя. Так, вместо того, чтобы записывать постоянную Авогадро в виде
= 602252000000000000000000, пишут 6,02252-1023,
это не только удобнее, но и подчеркивает, что шестая значащая цифра ненадежна. Поэтому при вычислении среднеарифметических значений их округляют таким образом, чтобы последняя цифра была бы первой сомнительной. При округлении чисел пользуются следующим правилом: если первая отбрасываемая цифра меньше пяти, то последнюю остающуюся не изменяют; если она равна или больше пяти, то последнюю остающуюся цифру увеличивают на единицу. Если величина равна пяти, а за ней следуют нули, число округляют до ближайшего четного значения (так, и 6,75 и 6,85 округляют до 6,8).
469
6. ЭМПИРИЧЕСКИЕ ФОРМУЛЫ
Часто возникает задача придать изученной опытным путем зависимости вид уравнения с тем, чтобы при помощи последнего производить различные вычисления. Такого рода уравнения называются эмпирическими формулами, так как в их основе лежит только экспериментальный материал. В эти формулы помимо изученных величин входят и коэффициенты, число которых зависит от точности опытных данных и от широты интервалов условий. Эмпирическая формула должна удовлетворять двум в сущности противоречивым требованиям: быть простой, т. е. содержать немного постоянных, и быть по возможности точной, т. е. хорошо воспроизводить результаты измерений. Эмпирические формулы являются интерполяционными, т. е. справедливы лишь в пределах измерений. Поэтому пользоваться ими для экстраполяции следует в тех пределах, для которых были произведены измерения.
Существует еще две особенности эмпирических формул. Во-первых, их разнообразие (даже применительно к одной и той же зависимости). Так, для описания зависимости теплоемкости Ср от температуры Т применяются уравнения
Ср ~
Ср = 61^2 4- 4“
Ср =• -г
Ср — 4- 4“ •
(XXI.25) (XXI.26) (XXI.27) (XXI.28)
Во-вторых, наличие одинаковых по структуре формул, но с различными значениями коэффициентов. Подобные несовпадения значений коэффициентов — результат обработки одним и тем же методом различных экспериментальных данных, либо различными методами одних и тех же данных.
7. ПРИНЦИП ПОДБОРА ТИПА ЭМПИРИЧЕСКОЙ ФОРМУЛЫ
Прежде всего экспериментальные данные наносят на график = если при этом получается прямая, значит, искомая зависимость имеет вид
у = ах 4- Ь, если прямая получается в координатах lg#=f(lgx), то
у = ахп 4- Ь, если же к линейности приводят координаты y=f(\gx), то у ~ аепх 4- b
(XXI.29)
(XXI. 30)
(XXI.31)
(в последних уравнениях b может оказаться равным нулю). Пред-
470
ставим, что ни в обычных, ни в логарифмических, ни в полулогарифмических шкалах экспериментальные точки не укладываются на прямую. Если точки ложатся на плавную кривую, следует продолжать подбор типа уравнения, испробовав другие преобразования.
Числовые значения коэффициентов эмпирических формул подбираются различными методами. Для простоты покажем их применение на примере зависимости (XXI.29). По методу наименьших квадратов кривая должна проходить между экспериментальными точками таким образом, чтобы сумма квадратов отклонений точек ют нее была бы минимальной. Примем, что эти отклонения наблюдаются только параллельно оси ординат, т. е. ошибке подвержены только значения функции. Это означает, что если обозначить через у г измеренные значения, а через у — величины, найденные из искомого уравнения, то следует подобрать такие значения а и Ь, чтобы сумма
Дг/2 = Хр/- г/;)2 = у (а ч- bxi — у-ft (XXI.32)
i i
была минимальной. После дифференцирования выражения (XXI.32) по а и b и приравнивания полученных частных производных нулю имеем
п п п п
2 (х;)2 У У1 — X Xi X
1________1_________1 1 _________
п / п \2 »
«X (х<)2 - X
1 \ 1 /
(XXI. 32а)
(XXI. 33)
где п — число измерений.
Расчет целесообразно осуществлять следующим образом: в таблицу наряду с опытными значениями Xi и yi ввести значения х? и произведения Xiyi для каждого значения х%. Далее произвести п п п
суммирование и найти значения У*?, Ух^., 1 1 1
по уравнениям (XXI.32а), (XXI.33) — числовые значения коэффициентов а и & и подставить их в уравнение (XXI.29). Для установления справедливости полученной эмпирической формулы следует подставить в нее последовательно все значения xi и, вычислив соответствующие значения сравнить их с экспериментальными данными.
Рассчитать значения коэффициентов а и b можно, обрабатывая экспериментальные данные на ЭВМ. По программе, введя в ЭВМ
471
экспериментальные данные, получим значения а, Ь, коэффициент корреляции г и дисперсию:
(XX 1.34)
(XXI. 35)
Коэффициент корреляции г показывает степень связи между переменными величинами. Если г=0, то переменные не коррелированье а при г±1 между переменными имеется полная связь. Более простым, но менее точным является метод средних значений. Для этого записывают все уравнения — axi + b и делят их на две равные части (или почти равные) в порядке возрастания переменной Xt или уг. Складывая уравнения каждой части, получают два уравнения, которые решают относительно а и Ь.
Если уравнение y=f(x) квадратное, расчет усложняется, так как приходится решать систему трех уравнений. Коэффициенты а, Ь, и с в уравнении
у — а + Ьхсх2 (XXI.36)
следует определять методом наименьших квадратов, так как каждое экспериментально полученное значение yi при разных значениях Xi содержит погрешность. Смысл метода наименьших квадратов заключается в том, что S(z/—а—Ьх—сх2)2 должна быть минимальной. Для определения минимального значения этой суммы необходимо взять частные производные по коэффициентам а, b и с и приравнять эти производные нулю:
д
— S (у—а—Ьх - сх2)2 = _ 2 У (у - а — Ьх - сх2) = О, (XXI.37)
— У С/ — а — 'Ьх — сх2)2 — —2 У {у — а — Ьх — 6х2) = 0, (XXI. 38) дЬ
—•— У (У “ а — Ьх—сх2)2 — —2 У {у — а — Ьх — сх2) — 0. (XXI.39) ОС **
Параметры а, b и с уравнения (XXI.36) определяются из системы уравнений:
У {у — а — Ьх — сх2) — 0,
(XXI.40)
472
— a — bx — cx%) x — 0, (XXI.41)
%(y — a — bx — cx2)x2 = 0, (XXI.42)
так как x=/=0. Отсюда следует
2У = па + б2|х + сХх2> (XXI.43)
2ХУ = л 2 х + 2х2 с 2 х3’ (XXI.44)
2х2^==аХх2 + А2х3 + сХх4, (XXI.45)
где 5а—па; а — число экспериментальных значений у.
ПРИЛОЖЕНИЕ
Таблица 1. Длины волн спектральных линий ртути (рис. 1)
22 2! 20 13 231716
151413 12 11 109 8 7 65 4
Номер линии Цвет Длина ВОЛНЕ!, нм Номер линии Цвет
Длина волны, нм
Красная » »
Оранжевая » »
Желтая » »
Зеленая
»
708,19 12 Зеленая
690,72 13 Голубая
671,62 14 »
623,44 15 »
612,33 16 Синяя
607,26 17
589,02 18 »
579,07 19 Фиолетовая
576,96 20 »
567,59 21 »
546,07 22 »
529,01 510,24
502,56 491,60 435,84 434,75 433,92 410,81 407,78 404,66
390,64
Таблица 2. Длины волн и волновые числа спектральных линий в спектре излучения железа (рис. 2)
Номер I линии Длина волны, нм Волновое число, см—1 Номер линии Длина волны, нм Волновое число, СМ”1 Номер линии | Длина волны, нм Волновое ЧИСЛО, СМ-1 Номер линии Длина волны, нм
6
7
8
9
10
11
12
37
38
39
40
41
385,99 386,72
387,80 388,88 389,97
415,48 415,88 417,09 418,18
419,14
420,20 421,03
25907 25854 25784
25714 25643 24067 24044 23975 23912 23858 23798 23751
42
56
58
59
60
61
62
163
64
421,93 435,37 436,76 437,59 439,09
440,13 441,51
442,73 443,32 444,32 445,44 446,17
23702
22968,9
22895.9
22852,4
22774,1
22720,6
22649,6'
22587,11
22557.1
22506,3
22449,7
22413,0
65
66
67
68
69
70
71
72
73
74
75
76
446,94 447,60 448,42
449,46
451,75 452,51 453,12
454,79
455,61
458,15 459,27 460,29
22374,4 22341,4 22300,5 22248,9| 22136,1 22099,0 22069,5 21988,4 21958,6 21826,9 21774,9 21725,4
77
78
79
80
81
82
83
84
85
86
87
461,13 462,51 463,75 464,74 465,46 466,75 467,89 469,14
470,73 472,74 473,68
21685,9
21621,3
21563,3
21517,9
21484,1
21425,0
21372,8
21315,6
21243,7
21153,3
21111,4
474
Атлас спектральных линий
Рис. 1.П. Спектр излучения ртути
Атлас спектральных линий
ЦОЗО 4040 • 4050 4050 4070 4080 4090 4100
4190 4200 4210 4220 4230 4240 4250 4260 4270 4280 4290 4300
Рис. 2.П. Спектр излучения железа
и?: " ' жад.|||ии!ШМ
..................................................................................................... ii„„b....
.: '..TJi.mm'A'.^•хч^'МЛм.--!"?Х!^.^л.Д!.!!..|и.... .^...i.
I . Л ...., I ,.Д|4ЖЩР ..1Щ.11 v.. .^.
Атлас спектральных линий
50 51 52 53 50 55 56 57 58 59
ЧЧЮ ЧЧ20 ЧЧЗО ЧЧЧО ЧЧ&О 1/460 4470 4480 4430 4500 45Ю 4520 4550 4540 4550
4830 4840 4850 4860 4870 4880 4890 4900 4910 4920 4930 4940 4950 4960 4970 4980 4990 5000
!.L1 !l11T!. J..U U .1».1..!!! J-L JU"
x:"i• awM'. ч'чнлцф.
Атлас спектральных линий
Таблица 3. Волновые числа полос поглощения в ИК-спектре полистирола
------
Номер полосы
Волновое число, см-<
Номер полосы
Волновое число, см-1
ЙММаМ1111Ь«ММ<И>М1>*^^"
Номер полосы
Волновое число, см—1
1
2
3
4
5
6
698,9
906,7
1028,0
1069,1
1154,3
1181,4
1495,0
1601,4
1801,6
1871,0
1944,0
2850,0 I
13
14
15
16
17
2924,2
3002,8
3026,5
3060,5
3082,6
Таблица 4. Программы для спецнализнрованного управляющего устройства 15ВСМ-5*
Программа линейной регрессии. Уравнение у^а + Ьх
Шаг Код Шаг Код Шаг Код Шаг Код и Шаг Код J Шаг Код
000 0408 030 0715 060 0414 090 а 0405 120 0405 150 0005
1 0000 1 0515 1 0007 1 0005 1 0004 151 0405
2 0700 2 0400 2 0415 2 0606 2 0602 152 0000
3 0404 3 0002 3 0001 ; 3 0407 3 0414 153 0602
4 0000 4 0604 4 0405 . 4 0100 4 0007 154 0405
5 0404 5 0713 5 0002 5 0408 5 0415 155 0002
6 0001 6 0400 6 0602 6 0700 6 0001 156 0713
7 0404 7 0000 7 0605 7 0415 7 0405 157 0601
8 0002 8 0405 8 0401 8 0007 8 Q002 158 0405
9 0404 9 0007 9 0007 9 0405 9 0602 i 159 0007
010 0003 040 0602 070 0415 . 100 1200 130 0605 160 0601
1 0404 1 0605 1 0005 1 0603 1 0401 161 0405
2 0004 2 0400 2 0405 2 0414 2 0007 ' 162 0005
3 0404 3 0004 3 0000 3 1200 3 0415 163 0713
4 0005 4 0415 4 0602 4 0405 4 0005 164 0603
5 0408 5 0,005 5 0405 5 0001 5 0405 165 0605
6 0100 6 0405 6 0002 6 0602 6 0003 166 0612
7 0515 7 0003 7 0713 7 0405 7 0602 167 0604
8 0404 8 0602 8 0601 8 0002 8 0405 168 0715
9 0007 9 0405 9 0405 9 0606 9 0001 169 0515
020 0400 050 0001 080 1200 ПО 0601 140 0713 170 0511
1 0001 1 0713 1 0602 1 0405 1 0601 171 0512
2 0713 2 0601 2 0405 2 0005 2 0405
3 0404 3 0414 3 0007 3 0603 3 0007
4 0003 4 1200 4 0606 4 0405 4 0713
5 0701 5 0415 5 0612 5 1200 5 0606 кп= = 1567
6 0400 6 0005 6 0603 6 0606 6 0603
7 0005 7 0405 7 0510 7 0515 7 0414
8 0415 8 0004 8 0405 8 0415 8 0007
9 0005 9 0602 9 0005 9 0005 9 0415
* Все программы в приложении составлены и отлажены физической химии ЛТИ им. Ленсовета Лизогубом А. В.
ст. преподавателем кафедры
480
Продолжение табл. 4
Обращение к программе > 00
Занести в память
Индикация у=п (объем выборки) х=г (коэффициент регрессии)
1) поиск > 0 У—Ь\ х=а
2) ,,S" y=Syx; х — погашен.
В программе вычисляются коэффициенты уравнения у=а-\-Ьх а и &, эмпирический коэффициент корреляции г и среднее квадратичное отклонение Sxy,
482
U у’—ZTq б/у’—— 77q
Таблица 5. Программа для вычисления термодинамических функций СР, ---------------, ---—----, Sr
для двухатомных молекул
Шаг Код Шаг Код Шаг Код Шаг Код Шаг Код Шаг Код Шаг Код Шаг Код Шаг Код Шаг
000 1 J 0408 0701 030 1 0008 0603 060 1 0405 0006 090 1 0405 0001 120 1 0004 0611
2 0415 2 0405 2 0602 2 0611 0604
3 0005 3 0006 3 0414 3 0604 3 0703
4 0405 4 0602 4 0008 4 0701 4 0712
5 0014 0414 5 0405 0712 5 0705
6 0602 6 0008 6 0007 6 0705 6 0602
7 0405 7 0405 7 0614 7 0602 7 0405
8 0004 8 0006 8 0604 8 0414 8 0008
9 0605 9 0604 9 0701 9 0008 9 0600
010 0605 040 0703 070 0601 100 0405 130 0414
1 0404 1 0712 1 0405 1 0002 0008
2 0007- 2 0705 2 0008 2 0611 2 0405
3 0713 3 0602 3 0606 3 0604 3 0009
4 0604 4 0405 4 0603 4 0405 4 0611
0405 0008 0414 0008 5 0415
6 0007 6 0600 6 0008 6 0600 6 0008
7 0614 7 0515 7 0405 7 0414 7 0601
8 0602 ' 8 0408 8 0006 8 0008 8 0414
9 0414 9 0702 9 0604 9 0405 9 0008
020 0008 050 0415 080 0702 НО 0003 140 0415
1 0405 1 0005 1 0712 1 0611 1 0005
2 0007 2 0405 2 0705 2 0604 2 0405
3 0614 3 0014 3 0602 3 0405 3 0014
4 0604 4 0602 4 0405 4 0008 4 0612
0701 0405 0008 0606 0405
6 0601 6 0004 6 0600 6 0601 6 0004
7 0605 7 0603 7 0515 .7 0414 7 0603
8 0713 8 0414 8 0408 8 0008 8 0605
9 0415 9 0007 9 0703 9 0405 9 0711
150 1 0614 0604 170 1 0600 0515 190 1 0414 0008 210 1 0600 0414 230 1
2 0701 2 0408 2 0405 2 0008 2
3 0606 3 0704 3 0003 3 0415 3
4 0601 4 0405 4 0611 4 0005 4
0605 0001 0415 0405
6 0611 6 0611 6 0008 6 0014 6
7 0415 7 0604 7 0601 7 0602 7
8 0008 8 0701 8 0414 8 0405 8
9 0601 9 0712 9 0008 9 0004 9
160 0405 180 0705 200 0405 220 0603 240
1 0015 1 0602 0004 1 0605
2 0600 2 0414 2 0611 2 0711 2
3 0405 3 0008 3 0604 3 0614 3
4 0006 4 0405 4 0703 4 0604 4
0602 0002 0712 0701
6 0605 6 0611 6 0705 6 0606 6
7 0711 7 0415 7 0602 7 0601 7
8 0415 8 0008 8 0405 8 0605 8
9 0006 9 0600 9 0008 9 0611 9
250
1
Занести в ячейки памяти:
Л1->01
/~>02 в кг-м2
Р-^03 в Па
7->04 К
СОе~>05 В СМ™1
/?-^06 he
0415 0008 0601
0405 0015 0600 0405 0006 0602 0414 0008 0415 0006 0702 0712 0705 0602 0405 0008 0600 0515 0512
113,387—15
«поиск» «1»->СР
U 'Tf—Uо
«поиск» «2»—>----------
Т
«поиск» «3»-> ---------
Т
«поиск» «4»~>5т
Продолжение табл. 6
Шаг
250
1
2
3
4
5
6
7
8
9
260
1
2
3
264
5
6
7
8
9
270
1
2
3
4
5
6
7
8
9
280
1
2
3
4
5
6
7
8
9
290
1
2
3
4
5
6
297
8
9
300
1
2
3
4
Код i Шаг 1 Код Шаг Код 1 1 Шаг Код Шаг Код
0601 0405
0008 0606
0603 0405 0013 0602
0405 000*0
0600 0414
0000 !
0415 0’011
0405 i
0014 | 0602
0405
0004 0603
0414
0007 0405
0006 0602
0414
0008 0405
0007 0614
0604
0701 0601
0405 0008
0606
0603 0405
0012 0602
0405 0000
0600
0414
0000
0415
0006 0703
0602 0405
0000 0600 0515 0408
5
6
7
8
9
310
1
2
3
4
5
6
7
8
9
320
1
2
3
5
6
7
8
9
330
1
2
3
4
5
6
7
8
9
340
1
2
3
4
5
6
7
8
9
350
1
2
3
4
5
6
7
8
9
0703 360
0405 1
0001 2
0611 363
0604 4
0701 5
0712 6
0705 7
0602 8
0414 9
0008 370
0405 1
0002 2
0611 3
0604 4
0405 5
0016 6
0602 7-
0405 8
0008 9
0600 380
0414 | 1
0008 1 2
0405 3
0003 4
0611 5
0415 6
0008 7
0601 8
0414 9
0008 390
0405 1
0004 2
0611 3
0604 4
0704 5
0602 396
0405 7
0008 8
0600 9
0405 400
0017 1
0611 2
0601 3
0414 4
0008 5
0415 6
0005 7
0405 8
0014 9
0602 410
0405 1
0004 2
0603 3
0605 4
0611 0614
0604 0701
0606
0601 0605
0611
0405
0007 | 0415
0009 0405
0014 0602
0405
0004
0603 0605
0711 | 0614
0604 !
0701
0606
0601 I
0605
0611
0415
0012 0602
0404 0007
0600
0414
0007 0415
0010 0405
0014
0602
0405 0004
0603
0605 0711
0614
0604
0701
0606
0601
0605
0611
0415 0013
0602
5
6
7
8
9 420
1
2
3
4
5
6
7
8 429 430
1
2
3
4
5
6
7
8
9
440
1
2
3
4
5
6
7
8
9
450
1
2
3
4
5
6
7
8
9 460
1 462
3
4
5
6
7
8
9
0405 0007 0600 0414 0007 0415 ООН 0405 0014 0602 0405 0004 0603 0605 0711 0614 0604 0701 0606 0601 0605 0611 0415 0012 0602 0405 0007 0600 0405 0008 0606 0601 0405 0015 0600 0405 0006 0602 0605 0711 0604 0405 0606 0600 0515 0408 0704 0405 0001 0611 0)604 0701 0712 0705 0602
2
3
4
5
6
7
8
9
490
1
2
3
495
0414 0008 0405 0002 0611 0604 0405 0016 0602 0405 0008 0600 0414 0008 0405 0003 0611 0415 0008 0601 0414 006'8 0405 00’04 0611 0604 0704 0602 0405 0008 0600 0405 0017 0611 0601 0414 0008 0415 0005 0405 0014 0602 0405 0004 0603 0605 0711 0614 0604 0701 0606 0601 0605 0611
0404
Продолжение табл. 6
Шаг Код Шаг Код 1 Шаг Код Шаг Код Шаг Код
5 0007 4 0012 3 0701 2 00'04 • 1 0606
6 0415 545 0602 4 0606 3 0603 2 0601
7 0009 6 0405 5 0601 4 0605 3 0405
8 0405 7 0007 6 0605 5 0711 4 0015
9 0Ю14 8 060'0 7 0611 6 0614 5 0600
536 0602 9 0414 8 0415 7 0604 6 0405
1 0405 550 00’07 9 0013 8 0701 7 0006
2 0004 1 0415 570 0602 9 0606 8 0602
3 0603 2 оою 1 0405 590 0601 9 0414
4 0605 3 0405 2 0007 1 0605 610 0008
5 0711 4 0014 3 0600 2 0611 1 0415
6 0614 5 0602 4 0414 3 0415 2 0006
7 0604 6 0405 5 0007 4 0012 3 0707
8 0701 7 0004 6 0415 595 0602 4 0602
9 0606 8 0603 7 ООП 6 0405 5 0405
540 0601 9 0605 8 0405 7 ' 0007 6 0008
1 0605 560 0711 9 0014 8 0600 7 0600
2 0611 1 0614 58?0 0602 9 0405 8 0515
3 0415 2 0604 1 0405 - 600 0008 9 0512
КП-6443
Занести в ячейку
Л4->01
/—>4)2
Р—>03
Т->04
Vi->05
£->06
V2—>09
1,4388—>14
166,223—^15 коэфф in /->16
а->17
v3->10
V4“>1 1 «3»->12 «2»—>13
«поиск» «1»->Ср «поиск» «2» «поиск» «3» «поиск» «4»->Г
485
Таблица 7. Вычисление величины ЛЯ по формуле ДЯ =
Т?1прг/Р1Г1Г2
Т2 — П
1. Перейдите в режим программирования на адрес 00, нажав клавиши
В/О
на индикаторе
00
2. Введите программу.
Программа для расчета ЛЯ в реакции разложения СаСО3
Адрес команд
Нажатая клавиша
Код операции
Содержание операции
01
02
03
04
05
06
07
08
09
10
II
12
13
14
15
16
62
ОЕ
63
13
18
0Е
64
12
ОЕ 65
12
ОЕ
66
Вызов значения р2 из ячейки 2
Занесение р2 в регистр У
Вызов значения р\ из ячейки 3
Вычисление p2/pi
Вычисление In р2/р{
Занесение In рг/P'i в-регистр У
Вызов /?==8?314 Дж/моль из ячейки 4
Вычисление In рг/pi
Занесение 7? In p2/pi в регистр У
Вызов Т2 из ячейки 5
Вычисление RT2 In p2/pi
Зависание Д72 1П pz/pi в регистр У
Вызов 71 нз ячейки 6
12 Вычисление R7\T2 In p2/pi
47 Запись R7\T2 In p2/pi в ячейку 7
65
Вызов Т2
486
Продолжение
Адрес команд
Нажатая клавиша
Код операции
Содержание операции
17
18
6
19
20
21
Если
9.
10.
ОЕ
66
Bf
ОЕ
67
12
50
программа введена верно, на
Занесение Т2 в регистр Y
Вызов Т
Вычисление
Вычисление
Занесение
Т2
в регистр Y
Вызов значения RT[T2 In p2/pi из ячейки 7
Вычисление АЯ
Остановка для индикации
индикаторе
50 12 67 24
Перейдите в режим «Автоматическая работа», нажав клавиши
Введите клавиши
Введите клавиши
Введите
Введите
Введите
Пустите
в
в
в
в
в
АВТ
ячейку 2
ячейку 3
на индикаторе
0
значение р2, набрав величину р2,
значение набрав величину рь
ячейку 4 значение R, набрав величину 8,314,
ячейку 5 значение Т2, набрав величину Т2,
ячейку 6 значение.Гь набрав величину Ть
и
и
и
и
и
программу на счет с адреса 00, нажав клавиши
Прочтите результат на индикаторе.
затем
затем
нажмите
нажмите
нажмите
В/О
нажмите
нажмите
клавиши
клавиши
клавиши
и С/П
487
Пример: £2=1227,1 Па
P! = 610,8 Па
/? = 8,314 Дж/моль
Г2=283,15 К
71 = 273,15 К
Ответ: &Н=44859 Дж/моль.
В случае неправильного набора нажать клавишу
И. Для дальнейших расчетов необходимо очистить клавиши
ячеикн
памяти,
нажав
О
6
н ввести новые данные.
Таблица 8. Плотность некоторых жидкостей при различных температурах
I Плотность, г/см3
Вещество
50° С
6О°С
Ацетон
Гексан
Гептан
Толуол Хлороформ
СС14
Этиловый спирт
0,7793 0,6505 0,675
0,8580 1,4706 1,5748 0,781
0,7682 0,6412 0,6665 0,8483 1,4509 1,5557
0,7722
0,756 0,6318 0,6579 0,8388 1,4334 1,5361
0,7632
0,7496 0,622
0,6491 0,8293 1,4114 1,5165
0,754
Таблица 9. Коэффициенты диффузии индивидуальных веществ (см2/с) прн различных температурах (К)
0,109
273
Ацетон
Бензол
283
303
323
0,0834 0,092
0,1075
488
__________________Продолжение
С2Н5ОН 340 °-147
Хлороформ 273 0Д9* 1 2 3 4 5
СС14 273 0,070
Гексан 303 0,0793
Гептан 313 0,0807
Порядок оформления лабораторных работ
1) изложение цели работы и краткое описание проведения эксперимента;
2) схемы приборов, с помощью которых выполнялась работа;
3) внешние условия (Г и р);
4) результаты исследования и расчеты (уравнения должны быть приведены в общем виде и с представленными данными; результаты должны быть сведены в таблицу);
5) графическая обработка экспериментальных данных (графики следует выполнять только на миллиметровой бумаге). По оси ординат откладывают функцию, по оси абсцисс — аргумент с указанием единиц измерения. На осях нанесена десятичная шкала согласно выбранному масштабу. Единицы масштаба должны быть выбраны сообразно точности отсчета при эксперименте.
Координаты экспериментальной точки наносят только на плоскости, причем очень тонко, н обводят кружком, треугольником, квадратом, ромбом или намечают крестом. По экспериментальным точкам строится усредняющая кривая, причем выпавшие экспериментальные точки не используются, но показываются.
На листе, где выполнен график, должны быть указаны: номер работы, наименование графика, объект исследования, а также фамилия студента, проводившего работу.
Зачетные работы
Зачетная работа является завершающим этапом в лабораторном практикуме по физической химии. Однако зачетная работа в условиях бюджета времени, отводимого для нее, не может претендовать на научное исследование, и было бы совершенно неправильно ориентировать студентов на выполнение научного исследования в столь непродолжительный отрезок времени.
Зачетная работа строится так, чтобы ознакомить студента со всеми элементами эксперимента исследовательского характера, начиная с работы над литературой и кончая оформлением отчета и защитой.
Рекомендуется следующий план выполнения зачетной работы. Тема работы с указанием ссылки на реферат в реферативном журнале, статью нли монографию выдается заблаговременно. Студенту предлагается ознакомиться не только с указанной литературой, но н с работами, на которые ссылается автор статьи. После обсуждения плана эксперимента с преподавателем студент собирает установку или прибор, производит подготовку реактивов (очистку, проверку на чистоту), подготавливают прибор (проверяет его работу по стандартам н определяет точнось показаний) и, наконец, выполняет эксперимент.
На основании полученных экспериментальных данных производится расчет и определение точности полученных величин. После завершения работы составляется отчет, который проверяет преподаватель, и осуществляется защита.
ОГЛАВЛЕНИЕ
Предисловие.......................................................... 3
Глава I. Молекулярная спектроскопия . .
1 ТЪйпмопрйетРие электромагнитного излучения с молекулами вещества
2. Молекулярные спектры двухатомных молекул.........................
3. Спектры комбинационного рассеяния................................
4. Молекулярные спектры многоатомных молекул.................
5 Применение молекулярной спектроскопии для расчета термодинамических функций Основы статистической термодинамики....................
6. Принцип устройства спектральных приборов.........................
7 Оценка точности спектр а льны х измерении........................
Работа 1 Изучение колебательно-вращательного спектра поглощения двухатомных газообразных молекул..........................
Работа 2. Определение равновесного межъядерного расстояния . . Работа 3. Определение зависимости вращательной постоянной от колебательного квантового числа. Определение Ве и ге . . . . . . . Работа 4. Определение распределения молекул по вращательным
квантовым уровням............................................
Работа 5. Определение волнового числа основной полосы поглощения двухатомной молекулы . . ................................
Работа 6 Определение теплоемкости СР и зависимости теплоемкости от температуры статистическим методом....................
Работа 7. Определение энтальпии (Нто—Н0°) вещества статистическим методом и зависимости (Нт°~~Но°) от температуры.........
Ar — U0 G°T—Но
Работа 8, Определение функций ~ и .............. ............
Работа Р. Определение энтропии и зависимости энтропии от темпе-
t*
V
ратуры . ......... ...... ...............
Работа 10. Изучение колебательно-вращательного спектра метана . Работа 11. Определение вращательной постоянной, момента инерции н равновесного межъядерного расстояния гс-н молекулы метана Работа 12. Определение зависимости вращательной постоянной от колебательного квантового числа. Определение Ве и гс-н.
Работа 13 Определение волновых чисел основных полос поглоще-
ния СН4 . •................ * * „............*.................
Работа 14. Определение изобарной теплоемкости СР и зависимости q ___f/ статистическим методом.................................
Работа 15 Определение энтальпии (Нт°—Но°) метана статистическим методом и зависимости (Яг°—Яо°) от температуры............
Работа 16 Определение энтропии метана статистическим методом и зависимости энтропии от температуры..........................
GT— Но
Работа 17. Определение функции.....-..... метана статистическим
Работа 18 Изучение колебательных спектров комбинационного рассеяния веществ в жидком состоянии............................
Работа 19 Изучение спектра комбинационного рассеяния молекул, обладающих тетраэдрической структурой .......................
Работа 20 Определение термодинамических функции веществ, молекулы которых обладают тетраэдрической структурой, по спектрам комбинационного рассеяния....................................
5
7
21
23
29
35
65
67
68
68
68
69
70
70
71
71
71
72
72
73
73
74
74
75
75
75
76
490
Работа 21. Изучение электронно-колебательно-вращательного спектра излучения радикала CN ........................................ 76
Работа 22. Определение колебательной постоянной и ангармонично-
сти для радикала CN в электронно-возбужденном состоянии .... 79
Работа 23. Определение константы равновесия реакции образования комплекса по электронно-колебательному спектру поглощения ... 79
Работа 24. Определение констант диссоциации слабых органических кислот по электронно-колебательным спектрам поглощения......... 80
Глава II. Поляризация молекул.......................................... 83
1. Поляризация и поляризуемость молекул................................ 83
2. Рефрактометры...................................................... 86
3. Дйэлькометры ....................................................... 91
Работа 1. Изучение зависимости показателя преломления жидкости от температуры и от длины волны светового потока................. 94
Работа 2. Определение концентрации растворенного вещества по показателю преломления раствора.................................. 94
Работа 3. Изучение зависимости показателя преломления от концентрации растворов ..................................... ..... 95
Работа 4. Определение молярной рефракции вещества н установление строения молекул............................................. 95
Работа 5. Изучение свойства молярной рефракции вещества .... 96
Работа 6. Определение электрического момента диполя молекул вещества......................................................... 96
Глава III. Жидкое состояние вещества................................... 97
Поверхностное натяжение. Парахор...................................... 97
Работа 1. Определение поверхностного натяжения жидкости .... 98
Работа 2. Изучение зависимости поверхностного натяжения жидкости от температуры.............................................. 99
Работа 3. Изучение термодинамики поверхностных явлений......... 100
Работа 4. Определение парахора и установление структуры молекулы 100
Работа 5. Определение плотности жидкости...................... 100
Работа 6. Изучение зависимости плотности жидкости от температуры ......................................................... 101
Глава IV. Электронографический анализ................................ 101
1. Метод электронографии и его применение............................ 101
2. Приготовление образцов для электронографического анализа........... 103
Работа. Получение н расчет электронограмм поликристаллических образцов ..................................................... 105
Глава V. Рентгеноструктурный анализ.................................. 108
1. Получение рентгеновского излучения............................... 108
2. Краткие сведения о структуре кристаллов............................ 111
3. Дифракция рентгеновского излучения................................. 113
4. Метод рентгеноструктурного анализа................................. 114
5. Аппаратура, применяемая в рентгеноструктурном анализе.............. 116
Работа. Определение параметра и типа кубической кристаллической решетки......................................................... 118
Глава VI. Термохимия................................................. 125
Термохимические измерения............................................ 125
Работа 1. Определение суммарной теплоемкости калориметрической системы электрическим методом................................... 129
Работа 2. Определение удельной интегральной теплоты растворения соли ’.......................................................... 130
Работа 3. Определение интегральной теплоты растворения солн при образовании концентрированного раствора .................. 132
491
Работа 4. Определение теплоты образования твердого раствора из двух твердых компонентов ? ............ 133
Работа 5. Определение теплоты образования кристаллогидрата из соли и воды ............................................... . . 135
Работа 6. Определение теплоты нейтрализации.............. # . . 135
Работа 7. Определение теплоты реакции окисления ..... . . . . . 136
Работа 8. Определение истинной теплоемкости вещества в жидком состоянии . . . ............................................... 138
Работа 9. Определение средней теплоемкости твердых и жидких веществ методом смешения ... . ................... ..... . . . . 138
Работа 10. Определение средней теплоемкости твердых веществ методом смешения...................................... . . . . . 141
Работа 11. Определение истинной теплоемкости жидкости методом калорифера...................................................... 141
Работа 12. Определеиие теплоты образования насыщенного раствора при ^290 К (полной энтальпии растворения Д/7)............ . . . 142
Работа 13. Построение диаграммы плавкости иеизоморфных веществ ................................................... .... 143
Работа 14. Определение теплот сгорания органических веществ . . 145
Работа 15. Термометрическое титрование.................... . . 148
Работа 16. Определение константы скорости растворения КС1 . . . 151
Глава VII. Фазовые равновесия........................................ 153
1. Общие условия равновесия в гетерогенных Системах................ . 153
2. Зависимость давления насыщенного пара от температуры . . . . . . . 155
Работа 1. Определение давления насыщенного пара индивидуальных жидкостей статическим методом............................... 158
Работа 2. Определение давления насыщенного пара индивидуальных жидкостей динамическим методом.............. ............. . 160
Работа 3. Определение давления насыщенного пара индивидуальных жидкостей по температурам кипения.......................... 162
Работа 4. Определение давления насыщенного пара твердых веществ и жидкостей методом -увлечения ................................. 165
Работа 5. Определение температур кипения мнкрометодамн ..... 169
Глава VIII. Жидкие растворы........................................... 170
I. Идеальные разбавленные растворы................................ 171
2. Химический потенциал ............................................ 174
Работа 1. Определение активностей и химических потенциалов воды и солн в растворе при 40°С................................. 177
Работа 2. Определение парциальных молярных теплот и энтропии растворения..................................................... 178
Работа 3. Определение молекулярной массы растворенного вещества криоскопическим методом..................................... 179
Работа 4. Определение концентрации растворенного вещества криоскопическим методом............................................. 184
Работа 5. Определение молекулярной массы вещества по методу Раста.......................................................... 186
Работа 6. Эбуллиоскопический метод определения молекулярной массы веществ.................................................. 188
Работа 7. Определение температуры кипения чистого вещества и раствора по методу Сиволобова..................................... 190
Глава IX. Растворы жидкостей в жидкостях............................. 191
1. Термодинамика растворения......................................... 191
2. Неограниченно смешивающиеся жидкости ............................ 193
Работа. Изучение равновесий жидкость — пар в двойных жидких системах ...................................................... 199
Глава X. Взаимная растворимость жидкостей ........................... 202
1. Ограниченная растворимость жидкостей............................... 202
2. Третий компонент в двухфазной жидкой системе. Закон распределения 205 492
3. Взаимная растворимость трех жидкостей.......................... 207
4. Зависимость взаимной растворимости трех жидкостей от температуры 211
Работа 1. Построение диаграммы для системы вода :—фенол .... 212
Работа 2. Определение коэффициента распределения.......... . 215
Работа 3. Определение коэффициента распределения иода между органическими и неорганическими растворителями . ............. 217
Работа 4. Изучение взаимной растворимости в трехкомпонентной системе ...................................................... 219
Глава XI. Равновесие жидкость — кристаллическая фаза ..... . . . 223
1. Построение диаграмм состояния кристаллическая фаза — жидкость . . 223
2. Равновесие твердый компонент — раствор . ...................... 223
3. Методы изучения гетерогенных систем . . .'..................... 224
4. Неизоморфные смеси с простой эвтектикой........................ 227
5. Неизоморфные смеси, образующие устойчивое химическое соединение 230 6. Неизоморфиые смеси, образующие неустойчивое химическое соединение . . ....................................... .’ч . 231
7. Изоморфные смеси. Системы с ограниченной растворимостью в твердом состоянии........................................................ 233
Работа 1. Изучение кристаллизации из растворов при высоких температурах .......................... . . ..................... 236
Работа 2. Построение диаграммы плавкости системы KNO3—NaNO3 238
Работа 3. Построение диаграммы плавкости системы KNO3—NaNO3 н изучение структуры сплава . ............... ;.......... . 239
Работа 4. Изучение кристаллизации веществ из растворов при низких температурах.............................................. 241
Работа 5. Изучение кристаллизации веществ из растворов........ 244
Глава XII. Химическое равновесие................................. 245
1. Химическое равновесие в гомогенных и гетерогенных системах .... 245
2. Уравнение изотермы реакции...................................... 248
3. Зависимость константы равновесия от температуры ................ 248
4. Принцип экспериментального изучения химического равновесия .... 251
Работа 1. Определение химического равновесия в гомогенных системах (жидкая фаза)............................................. 251
Работа 2. Изучение химического равновесия реакции 2Fe3++2I_=^ ^2Fe2+ + I2 ........................... 25*5
Работа 3. Изучение химического равновесия в растворах при образовании комплекса методом распределения . .................... 257
Работа 4. Определение химического равновесия в гомогенных системах (газовая фаза)......................................... 258
Работа 5. Определение химического равновесия в гетерогенных системах (исследование карбонатов)............................ 261
Работа 6. Исследование химического равновесия в кристаллогидратах ....................................................... 263
Работа 7. Изучение равновесия реакций дегидрирования спиртов в газовой фазе................................................. 265
Глава XIII. Растворы электролитов.................................. 269
1. Электрическая проводимость . . . .............................. 269
2. Удельная и эквивалентная электрические проводимости . .......... 270
3. Зависимость электрической проводимости от концентрации.......... 271
4. Электрическая проводимость неводных растворов электролитов .... 273
5. Зависимость электрической проводимости от температуры.......... 274
6. Равновесие в растворах электролитов............................. 276
7. Влияние температуры на коистаиту диссоциации растворов электролитов 277
Работа 1. Измерение электрической проводимости растворов электролитов .................................................... 278
Работа 2. Изучение влияния температуры иа электрическую проводимость и вязкость растворов электролитов в воде и водно-органических растворителях ..................................... 281
Работа 3. Изучение зависимости электрической проводимости растворов электролитов от концентрации............................. 282
Работа 4. Определение растворимости труднорастворимой соли при различных температурах методом электрической проводимости . . . 285
Глава XIV. Электродвижущие силы гальванических цепей............ . . 286
1. Гальванические элементы........................................... 286
2. Механизм возникновения э. д. с..................................... 287
3. Термодинамика обратимых электрохимических систем................... 289
4. Электроды........................................................ 292
5. Гальванические элементы .......................................... 295
Работа 1, Измерение э. д. с. элемента Якоби — Даниэля ...... 297
Работа 2. Измерение э. д. с. окислительно-восстановительных элементов, электродных потенциалов и изучение влияния добавок на окислительно-восстановительные потенциалы....................... 302
Работа 3. Определение стандартного потенциала ферри-ферро-элект-рода, расчет константы равновесия электродной реакции, изучение окислительно-восстановительной способности раствора . .......... 305
Работа 4. Определение коэффициента активности растворов хлористоводородной кислоты методом измерения э. д. с................. 307
Работа 5. Определение термодинамических функций реакции, протекающей в окислительно-восстановительном элементе ............... 310
Работа 6. Определение термодинамических функций реакции окисления— восстановления методом потенциометрического титрования 313
Работа 7. Определение pH образования гидроксидов металлов ... 316
Работа 8. Определение буферной емкости растворов методом потенциометрического титрования...................................... 317
Глава XV. Кинетика реакций в растворах................................ 319
1. Скорость химической реакции. Кинетическая классификация реакций . . 319
2. Зависимость скорости реакции от концентрации реагирующих веществ 322 3. Экспериментальные методы определения скорости и порядка реакции . 328
4. Зависимость скорости реакции от температуры........................ 333
5. Особенности кинетики реакций в растворах ......................... 340
6. Зависимость скорости реакции от катализатора (гомогенный катализ) 343
Работа 1. Изучение скорости инверсии тростникового сахара . . . 345
Работа 2. Изучение скорости мутаротации глюкозы................. 350
Работа 3. Изучение скорости реакции иодирования ацетона .... 352
Работа 4. Изучение скорости разложения пероксида водорода газометрическим методом............................................. 354
Работа 5. Изучение скорости гидролиза уксусного ангидрида методом электрической проводимости.................................. 356
Работа 6. Изучение скорости реакции разложения карбамида в водных растворах методом электрической проводимости................ 359
Работа 7. Изучение кинетики акватаЦии комплексных ионов Со3+ (кислотный гидролиз) колориметрическим методом ................. 361
Работа 8. Изучение кинетики реакции гидролиза уксусного ангидрида колориметрическим методом ................................... 369
Работа 9. Изучение кинетики реакции окисления тиомочевины и тиоацетамида гексацианоферрата (III) в щелочном растворе.......... 372
Работа 10. Изучение кинетики реакции восстановления гексацнано-феррата (III) аскорбиновой кислотой................................ 376
Глава XVI. Фотохимические и цепные реакции, радикальная полимеризация .......................................................... 379
1. Фотохимические реакции............................................. 379
2. Цепные реакции..................................................... 380
3. Радикальная полимеризация.......................................... 384
4. Кинетика радикальной полимеризации................................. 388
Работа 1. Изучение кинетики фотохимического разложения Н2О2 . . 389
Работа 2. Определение квантового выхода при фотохимическом разложении Н2О2...............................................* 391
494
Работа 3. Изучение скорости реакции сульфирования карбамида . . 395
Работа 4. Исследование кинетики радикальной полимеризации метилметакрилата ................................................. 397
Глава XVII. Кинетика гетерогенных процессов........................... 399
1. Диффузия........................................................... 400
2. Кинетика процесса испарения жидкости............................... 402
3. Кинетика процесса растворения...................................... 405
4. Кинетика топохимических реакций................................... 408
Работа 1. Изучение кинетики испарения жидкостей и определение коэффициента диффузии паров жидкости в воздух (и другие газы) методом увлечения............................................... 412
Работа 2. Изучение кинетики испарения жидкости и определение коэффициента диффузии паров жидкости в воздух методом адсорбции ........................................................... 416
Работа 3. Изучение кинетики растворения малорастворимых веществ .......................................................... 417
Работа 4. Изучение кинетики термического разложения перманганата калия........................................................ 419
Глава XVIII. Адсорбция................................................ 425
1. Термодинамика адсорбции............................................ 425
2. Кинетика адсорбции................................................. 428
Работа 1. Статический метод изучения адсорбции.................. 429
Работа 2. Динамический метод изучения адсорбции................ 431
Работа 3. Построение изотермы адсорбции бензола и определение удельной площади поверхности катализаторов методом проявительной хроматографии............................................... 433
Работа 4. Изучение адсорбционного равновесия из водных растворов карбоновых кислот на активированном угле кондуктометрическим методом.................................................... 436
Глава XIX. Кинетика гетерогенно-каталитических реакций................ 438
1. Катализ и катализаторы . . ...................................... 438
2. Гетерогенные катализаторы......................................... 440
3. Мультиплетная теория катализа...................................... 443
4. Теория активных ансамблей ...................................... 446
5. Реакции в потоке................................................... 447
Работа /. Изучение кинетики реакции распада (крекинга) ацетона в потоке.........................'...................,.......... 450’
Работа 2. Каталитическая дегидратация этилового спирта......... 453
Работа 3. Каталитическое разложение метилового спирта.......... 456
Глава XX. Кинетика электрохимических реакций.......................... 457
1. Общие сведения о кинетике электродных процессов ................... 457
2. Кинетика обратимых окислительно-восстановительных процессов .... 460
Работа 1. Изучение кинетики электрохимических реакций температурно-кинетическим методом ................................... 461
Работа 2. Изучение зависимости скорости электролиза от состава окислительно-восстановительной системы методом поляризационных измерений........................................................ 463
Глава XXI. Математическая обработка экспериментальных данных . . . 464
1. Оценка точности и надежности измеренных и рассчитанных величин 464 2. Природа происхождения погрешности измеренных величин. Систематические ошибки н их выявление.......................................... 464
3. Абсолютная и относительная ошибки................................. 465
4. Расчет ошибок...................................................... 468
5. Запись результатов измерений....................................... 469
6. Эмпирические формулы.............................................. 470
7. Принцип подбора типа эмпирической формулы......................... 470
495
.Ввод программы
Регрессионный анализ
00
10
№№ команд Клавиши
Пояснение
№№ команд
20
ПРГ
Код
61
П-Х9
/
П-Х2
П- Ха
п-х^
х-пь
60
10
10
6
10
61
6L
10
п- 2
Перевод машины в режим „Программирование11
Клавиши Код
С/П 1» 50
Пояснение
№№ команд
Клавиш и
В/О
Вызов значения
Вызов значения
Вызов значения
Вызов значения yt-
Накопление суммы »2
в ячейке „а11
Вызов значения X;
Вызов значения X
(х-х)2
Накопление суммы
х-,.. —12
Формулы для расчета
s2(b) =
экспериментальные точки
п -общее число экспериментальных точек
30
40
П—Х8
2
Х“Па
П-ХЬ
Х”ПЬ
П-Х8
С/П
S (а) = —
•J •
——-среднее значение аргумента (центр эксперимента)
68
13
6”
68
Вызов значения п
Вызов значения £(yt Y;) in / V 4/12
' Расчет и запись в ячейку „а11
t „2 Lfy,-y,)2
^значения о —-------—----
>| п - 2
|сводная дисперсия
/ ; оценка точности эксперимента
г
Перевод в регистр У
| Вызов £fx-x)
4V расчет и запись в ячейку „Ь
V О 2
X значения S2 (Ь) —
2
дисперсия оценки коэффициента (Ь)
|| 2
I Вызов S
ртвревод в регистр У
Вызов п
Аг
^Расчет и запись в ячейку „с"
I 2 S2
Означения S (а)= -рг сводная
J дисперсия коэффициента fa)
00
10
20
ПРГ
Ввод программы
Код
Пояснение
Х-Г18
П-Х4
Х“П5
П~Х8
гьхь
Х-П6
С/П
П-Х6
С/П
48
62
10
21
64
12
68
6L
10
46
10
50
66
11
Перевод машины в режим I
I *
„Программ и рован и е“ I
Вызов значения
Вызов значения
(х~~х)
Вызов значения
-12 «2/и ।
Вызов значения
Вызов значения
Вызов значения
Вызов значения bXj
Вызов значения
>ч >»
I
S2(b)
S2(a)
*1
b
4
Расчет и запись значения
Вызов значения
Расчет и индикация значения
T + 'i-asfy,j
Вызов значения Y;
Вызов значения t|_a-Sfy/)
Расчет и индикация значения
Рассчитываемые величины:
(I) Сводная дисперсия- S2, которая представляет оценку точности эксперимента в целому
(2) Дисперсия оценки коэффициента b — S2(b) :
(8) Дисперсия оценки коэффициента а — S2fa).
1 ..